User login
Pulmonary infarction due to pulmonary embolism
A 76-year-old man whose history included abdominal aortic aneurysm repair, bilateral femoral artery bypass for popliteal artery aneurysm, hypertension, and peptic ulcer disease was admitted to a community hospital with pleuritic chest pain and shortness of breath. Two days earlier, he had undergone repair of a ventral hernia.
At the time of that admission, he reported no fever, chills, night sweats, cough, or history of heart or lung disease. His vital signs were normal, and physical examination had revealed no apparent respiratory distress, no jugular venous distention, normal heart sounds, and no pedal edema; however, decreased air entry was noted in the right lung base. Initial serum levels of troponin and N-terminal pro-B-type natriuretic peptide were normal.
At that time, computed tomographic angiography of the chest showed segmental pulmonary emboli in the left upper and right lower lobes of the lungs and right pleural effusion. Transthoracic echocardiography showed normal atrial and ventricular sizes with no right or left ventricular systolic dysfunction and a left ventricular ejection fraction of 59%.
Treatment with intravenous heparin was started, and the patient was transferred to our hospital.
PLEURAL EFFUSION AND PULMONARY EMBOLISM
1. Which of the following is true about pleural effusion?
- It is rarely, if ever, associated with pulmonary embolism
- Most patients with pleural effusion due to pulmonary embolism do not have pleuritic chest pain
- Pulmonary embolism should be excluded in all cases of pleural effusion without a clear cause
Pulmonary embolism should be excluded in all cases of pleural effusion that do not have a clear cause. As for the other answer choices:
- Pulmonary embolism is the fourth leading cause of pleural effusion in the United States, after heart failure, pneumonia, and malignancy.1
- About 75% of patients who develop pleural effusion in the setting of pulmonary embolism complain of pleuritic chest pain on the side of the effusion.2 Most effusions are unilateral, small, and usually exudative.3
EVALUATION BEGINS: RESULTS OF THORACENTESIS
Our patient continued to receive intravenous heparin.
He underwent thoracentesis on hospital day 3, and 1,000 mL of turbid sanguineous pleural fluid was removed. Analysis of the fluid showed pH 7.27, white blood cell count 3.797 × 109/L with 80% neutrophils, and lactate dehydrogenase (LDH) concentration 736 U/L (a ratio of pleural fluid LDH to a concurrent serum LDH > 0.6 is suggestive of an exudate); the fluid was also sent for culture and cytology. Thoracentesis was terminated early due to cough, and follow-up chest radiography showed a moderate-sized pneumothorax.

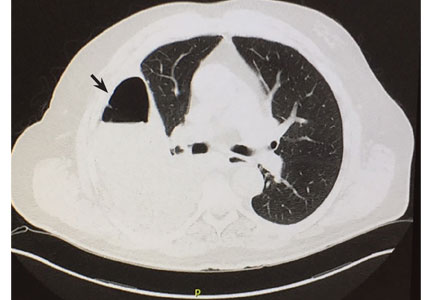
Computed tomography (CT) of the chest at this time showed a small wedge-shaped area of lung consolidation in the right lower lobe (also seen on CT done 1 day before admission to our hospital), with an intrinsic air-fluid level suggesting a focal infarct or lung abscess, now obscured by adjacent consolidation and atelectasis. In the interval since the previous CT, the multiloculated right pleural effusion had increased in size (Figure 1).
THE NEXT STEP
2. What is the most appropriate next step for this patient?
- Consult an interventional radiologist for chest tube placement
- Start empiric antibiotic therapy and ask an interventional radiologist to place a chest tube
- Start empiric antibiotic therapy, withhold anticoagulation, and consult a thoracic surgeon
- Start empiric antibiotic therapy and consult a thoracic surgeon while continuing anticoagulation
The most appropriate next step is to start empiric antibiotic therapy and consult a thoracic surgeon while continuing anticoagulation.
In this patient, it is appropriate to initiate antibiotics empirically on the basis of his significant pleural loculations, a wedge-shaped consolidation, and 80% neutrophils in the pleural fluid, all of which suggest infection. The unmasking of a wedge-shaped consolidation after thoracentesis, with a previously noted air-fluid level and an interval increase in multiloculated pleural fluid, raises suspicion of a necrotic infection that may have ruptured into the pleural space, a possible lung infarct, or a malignancy. Hence, simply placing a chest tube may not be enough.
Blood in the pleural fluid does not necessitate withholding anticoagulation unless the bleeding is heavy. A pleural fluid hematocrit greater than 50% of the peripheral blood hematocrit suggests hemothorax and is an indication to withhold anticoagulation.1 Our patient’s pleural fluid was qualitatively sanguineous but not frankly bloody, and therefore we judged that it was not necessary to stop his heparin.
HOW DOES PULMONARY INFARCTION PRESENT CLINICALLY?
3. Which of the following statements about pulmonary infarction is incorrect?
- Cavitation and infarction are more common with larger emboli
- Cavitation occurs in fewer than 10% of pulmonary infarctions
- Lung abscess develops in more than 50% of pulmonary infarctions
- Pulmonary thromboembolism is the most common cause of pulmonary infarction
Lung abscess develops in far fewer than 50% of cases of pulmonary infarction. The rest of the statements are correct.
Cavitation complicates about 4% to 7% of infarctions and is more common when the infarction is 4 cm or greater in diameter.4 These cavities are usually single and predominantly on the right side in the apical or posterior segment of the upper lobe or the apical segment of the right lower lobe, as in our patient.5–8 CT demonstrating scalloped inner margins and cross-cavity band shadows suggests a cavitary pulmonary infarction.9,10
Infection and abscess in pulmonary infarction are poorly understood but have been linked to larger infarctions, coexistent congestion or atelectasis, and dental or oropharyngeal infection. In an early series of 550 cases of pulmonary infarction, 23 patients (4.2%) developed lung abscess and 6 (1.1%) developed empyema.11 The mean time to cavitation for an infected pulmonary infarction has been reported to be 18 days.12
A reversed halo sign, generally described as a focal, rounded area of ground-glass opacity surrounded by a nearly complete ring of consolidation, has been reported to be more frequent with pulmonary infarction than with other diseases, especially when in the lower lobes.13
CASE CONTINUED: THORACOSCOPY
A cardiothoracic surgeon was consulted, intravenous heparin was discontinued, an inferior vena cava filter was placed, and the patient underwent video-assisted thoracoscopy.
Purulent fluid was noted on the lateral aspect of right lower lobe; this appeared to be the ruptured cavitary lesion functioning like an uncontrolled bronchopleural fistula. Two chest tubes, sizes 32F and 28F, were placed after decortication, resection of the lung abscess, and closure of the bronchopleural fistula. No significant air leak was noted after resection of this segment of lung.

Pathologic study showed acute organizing pneumonia with abscess formation; no malignant cells or granulomas were seen (Figure 2). Pleural fluid cultures grew Streptococcus intermedius, while the tissue culture was negative for any growth, including acid-fast bacilli and fungi.
On 3 different occasions, both chest tubes were shortened, backed out 2 cm, and resecured with sutures and pins, and Heimlich valves were applied before the patient was discharged.
Intravenous piperacillin-tazobactam was started on the fifth hospital day. On discharge, the patient was advised to continue this treatment for 3 weeks at home.
The patient was receiving enoxaparin subcutaneously in prophylactic doses; 72 hours after the thorascopic procedure this was increased to therapeutic doses, continuing after discharge. Bridging to warfarin was not advised in view of his chest tubes.
Our patient appeared to have developed a right lower lobe infarction that cavitated and ruptured into the pleural space, causing a bronchopleural fistula with empyema after a recent pulmonary embolism. Other reported causes of pulmonary infarction in pulmonary embolism are malignancy and heavy clot burden,6 but these have not been confirmed in subsequent studies.5 Malignancy was ruled out by biopsy of the resected portion of the lung, and our patient did not have a history of heart failure. A clear cavity was not noted (because it ruptured into the pleura), but an air-fluid level was described in a wedge-shaped consolidation, suggesting infarction.
How common is pulmonary infarction after pulmonary embolism?
Pulmonary infarction occurs in few patients with pulmonary embolism.13 Since the lungs receive oxygen from the airways and have a dual blood supply from the pulmonary and bronchial arteries, they are not particularly vulnerable to ischemia. However, the reported incidence of pulmonary infarction in patients with pulmonary embolism has ranged from 10% to higher than 30%.5,14,15
The reasons behind pulmonary infarction with complications after pulmonary embolism have varied in different case series in different eras. CT, biopsy, or autopsy studies reveal pulmonary infarction after pulmonary embolism to be more common than suspected by clinical symptoms.
In a Mayo Clinic series of 43 cases of pulmonary infarction diagnosed over a 6-year period by surgical lung biopsy, 18 (42%) of the patients had underlying pulmonary thromboembolism, which was the most common cause.16
RISK FACTORS FOR PULMONARY INFARCTION
4. Which statement about risk factors for pulmonary infarction in pulmonary embolism is incorrect?
- Heart failure may be a risk factor for pulmonary infarction
- Pulmonary hemorrhage is a risk factor for pulmonary infarction
- Pulmonary infarction is more common with more proximal sites of pulmonary embolism
- Collateral circulation may protect against pulmonary infarction
Infarction is more common with emboli that are distal rather than proximal.
Dalen et al15 suggested that after pulmonary embolism, pulmonary hemorrhage is an important contributor to the development of pulmonary infarction independent of the presence or absence of associated cardiac or pulmonary disease, but that the effect depends on the site of obstruction.
This idea was first proposed in 1913, when Karsner and Ghoreyeb17 showed that when pulmonary arteries are completely obstructed, the bronchial arteries take over, except when the embolism is present in a small branch of the pulmonary artery. This is because the physiologic anastomosis between the pulmonary artery and the bronchial arteries is located at the precapillary level of the pulmonary artery, and the bronchial circulation does not take over until the pulmonary arterial pressure in the area of the embolism drops to zero.
Using CT data, Kirchner et al5 confirmed that the risk of pulmonary infarction is higher if the obstruction is peripheral, ie, distal.
Using autopsy data, Tsao et al18 reported a higher risk of pulmonary infarction in embolic occlusion of pulmonary vessels less than 3 mm in diameter.
Collateral circulation has been shown to protect against pulmonary infarction. For example, Miniati et al14 showed that healthy young patients with pulmonary embolism were more prone to develop pulmonary infarction, probably because they had less efficient collateral systems in the peripheral lung fields. In lung transplant recipients, it has been shown that the risk of infarction decreased with development of collateral circulation.19
Dalen et al,15 however, attributed delayed resolution of pulmonary hemorrhage (as measured by resolution of infiltrate on chest radiography) to higher underlying pulmonary venous pressure in patients with heart failure and consequent pulmonary infarction. In comparison, healthy patients without cardiac or pulmonary disease have faster resolution of pulmonary hemorrhage when present, and less likelihood of pulmonary infarction (and death in submassive pulmonary embolism).
Data on the management of infected pulmonary infarction are limited. Mortality rates have been as high as 41% with noninfected and 73% with infected cavitary infarctions.4 Some authors have advocated early surgical resection in view of high rates of failure of medical treatment due to lack of blood supply within the cavity and continued risk of infection.
KEY POINTS
In patients with a recently diagnosed pulmonary embolism and concurrent symptoms of bacterial pneumonia, a diagnosis of cavitary pulmonary infarction should be considered.
Consolidations that are pleural-based with sharp, rounded margins and with focal areas of central hyperlucencies representing hemorrhage on the mediastinal windows on CT are more likely to represent a pulmonary infarct.20
- Light RW. Pleural Diseases. 4th ed. Baltimore, MD: Lippincott, Williams & Wilkins; 2001.
- Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100(3):598–603. pmid:1909617
- Light RW. Pleural effusion due to pulmonary emboli. Curr Opin Pulm Med 2001; 7(4):198–201. pmid:11470974
- Libby LS, King TE, LaForce FM, Schwarz MI. Pulmonary cavitation following pulmonary infarction. Medicine (Baltimore) 1985; 64(5):342–348. pmid:4033411
- Kirchner J, Obermann A, Stuckradt S, et al. Lung infarction following pulmonary embolism: a comparative study on clinical conditions and CT findings to identify predisposing factors. Rofo 2015; 187(6):440–444. doi:10.1055/s-0034-1399006
- He H, Stein MW, Zalta B, Haramati LB. Pulmonary infarction: spectrum of findings on multidetector helical CT. J Thorac Imaging 2006; 21(1):1–7. doi:10.1097/01.rti.0000187433.06762.fb
- Scharf J, Nahir AM, Munk J, Lichtig C. Aseptic cavitation in pulmonary infarction. Chest 1971; 59(4):456–458. pmid:5551596
- Wilson AG, Joseph AE, Butland RJ. The radiology of aseptic cavitation in pulmonary infarction. Clin Radiol 1986; 37(4):327–333. pmid:3731699
- Butler MD, Biscardi FH, Schain DC, Humphries JE, Blow O, Spotnitz WD. Pulmonary resection for treatment of cavitary pulmonary infarction. Ann Thorac Surg 1997; 63(3):849–850. pmid:9066420
- Koroscil MT, Hauser TR. Acute pulmonary embolism leading to cavitation and large pulmonary abscess: a rare complication of pulmonary infarction. Respir Med Case Rep 2016; 20:72–74. doi:10.1016/j.rmcr.2016.12.001
- Levin L, Kernohan JW, Moersch HJ. Pulmonary abscess secondary to bland pulmonary infarction. Dis Chest 1948; 14(2):218–232. pmid:18904835
- Marchiori E, Menna Barreto M, Pereira Freitas HM, et al. Morphological characteristics of the reversed halo sign that may strongly suggest pulmonary infarction. Clin Radiol 2018; 73(5):503.e7–503.e13. doi:10.1016/j.crad.2017.11.022
- Smith GT, Dexter L, Dammin GJ. Postmortem quantitative studies in pulmonary embolism. In: Sasahara AA, Stein M, eds. Pulmonary Embolic Disease. New York, NY: Grune & Stratton, Inc; 1965:120–126.
- Miniati M, Bottai M, Ciccotosto C, Roberto L, Monti S. Predictors of pulmonary infarction. Medicine (Baltimore) 2015; 94(41):e1488. doi:10.1097/MD.0000000000001488
- Dalen JE, Haffajee CI, Alpert JS, Howe JP, Ockene IS, Paraskos JA. Pulmonary embolism, pulmonary hemorrhage and pulmonary infarction. N Engl J Med 1977; 296(25):1431–1435. doi:10.1056/NEJM197706232962503
- Parambil JG, Savci CD, Tazelaar HD, Ryu JH. Causes and presenting features of pulmonary infarctions in 43 cases identified by surgical lung biopsy. Chest 2005; 127(4):1178–1183. doi:10.1378/chest.127.4.1178
- Karsner HT, Ghoreyeb AA. Studies in infarction: III. The circulation in experimental pulmonary embolism. J Exp Med 1913; 18(5):507–511. pmid:19867725
- Tsao MS, Schraufnagel D, Wang NS. Pathogenesis of pulmonary infarction. Am J Med 1982; 72(4):599–606. pmid:6462058
- Burns KE, Iacono AT. Incidence of clinically unsuspected pulmonary embolism in mechanically ventilated lung transplant recipients. Transplantation 2003; 76(6):964–968. doi:10.1097/01.TP.0000084523.58610.BA
- Yousem SA. The surgical pathology of pulmonary infarcts: diagnostic confusion with granulomatous disease, vasculitis, and neoplasia. Mod Pathol 2009; 22(5):679–685. doi:10.1038/modpathol.2009.20
A 76-year-old man whose history included abdominal aortic aneurysm repair, bilateral femoral artery bypass for popliteal artery aneurysm, hypertension, and peptic ulcer disease was admitted to a community hospital with pleuritic chest pain and shortness of breath. Two days earlier, he had undergone repair of a ventral hernia.
At the time of that admission, he reported no fever, chills, night sweats, cough, or history of heart or lung disease. His vital signs were normal, and physical examination had revealed no apparent respiratory distress, no jugular venous distention, normal heart sounds, and no pedal edema; however, decreased air entry was noted in the right lung base. Initial serum levels of troponin and N-terminal pro-B-type natriuretic peptide were normal.
At that time, computed tomographic angiography of the chest showed segmental pulmonary emboli in the left upper and right lower lobes of the lungs and right pleural effusion. Transthoracic echocardiography showed normal atrial and ventricular sizes with no right or left ventricular systolic dysfunction and a left ventricular ejection fraction of 59%.
Treatment with intravenous heparin was started, and the patient was transferred to our hospital.
PLEURAL EFFUSION AND PULMONARY EMBOLISM
1. Which of the following is true about pleural effusion?
- It is rarely, if ever, associated with pulmonary embolism
- Most patients with pleural effusion due to pulmonary embolism do not have pleuritic chest pain
- Pulmonary embolism should be excluded in all cases of pleural effusion without a clear cause
Pulmonary embolism should be excluded in all cases of pleural effusion that do not have a clear cause. As for the other answer choices:
- Pulmonary embolism is the fourth leading cause of pleural effusion in the United States, after heart failure, pneumonia, and malignancy.1
- About 75% of patients who develop pleural effusion in the setting of pulmonary embolism complain of pleuritic chest pain on the side of the effusion.2 Most effusions are unilateral, small, and usually exudative.3
EVALUATION BEGINS: RESULTS OF THORACENTESIS
Our patient continued to receive intravenous heparin.
He underwent thoracentesis on hospital day 3, and 1,000 mL of turbid sanguineous pleural fluid was removed. Analysis of the fluid showed pH 7.27, white blood cell count 3.797 × 109/L with 80% neutrophils, and lactate dehydrogenase (LDH) concentration 736 U/L (a ratio of pleural fluid LDH to a concurrent serum LDH > 0.6 is suggestive of an exudate); the fluid was also sent for culture and cytology. Thoracentesis was terminated early due to cough, and follow-up chest radiography showed a moderate-sized pneumothorax.
Computed tomography (CT) of the chest at this time showed a small wedge-shaped area of lung consolidation in the right lower lobe (also seen on CT done 1 day before admission to our hospital), with an intrinsic air-fluid level suggesting a focal infarct or lung abscess, now obscured by adjacent consolidation and atelectasis. In the interval since the previous CT, the multiloculated right pleural effusion had increased in size (Figure 1).
THE NEXT STEP
2. What is the most appropriate next step for this patient?
- Consult an interventional radiologist for chest tube placement
- Start empiric antibiotic therapy and ask an interventional radiologist to place a chest tube
- Start empiric antibiotic therapy, withhold anticoagulation, and consult a thoracic surgeon
- Start empiric antibiotic therapy and consult a thoracic surgeon while continuing anticoagulation
The most appropriate next step is to start empiric antibiotic therapy and consult a thoracic surgeon while continuing anticoagulation.
In this patient, it is appropriate to initiate antibiotics empirically on the basis of his significant pleural loculations, a wedge-shaped consolidation, and 80% neutrophils in the pleural fluid, all of which suggest infection. The unmasking of a wedge-shaped consolidation after thoracentesis, with a previously noted air-fluid level and an interval increase in multiloculated pleural fluid, raises suspicion of a necrotic infection that may have ruptured into the pleural space, a possible lung infarct, or a malignancy. Hence, simply placing a chest tube may not be enough.
Blood in the pleural fluid does not necessitate withholding anticoagulation unless the bleeding is heavy. A pleural fluid hematocrit greater than 50% of the peripheral blood hematocrit suggests hemothorax and is an indication to withhold anticoagulation.1 Our patient’s pleural fluid was qualitatively sanguineous but not frankly bloody, and therefore we judged that it was not necessary to stop his heparin.
HOW DOES PULMONARY INFARCTION PRESENT CLINICALLY?
3. Which of the following statements about pulmonary infarction is incorrect?
- Cavitation and infarction are more common with larger emboli
- Cavitation occurs in fewer than 10% of pulmonary infarctions
- Lung abscess develops in more than 50% of pulmonary infarctions
- Pulmonary thromboembolism is the most common cause of pulmonary infarction
Lung abscess develops in far fewer than 50% of cases of pulmonary infarction. The rest of the statements are correct.
Cavitation complicates about 4% to 7% of infarctions and is more common when the infarction is 4 cm or greater in diameter.4 These cavities are usually single and predominantly on the right side in the apical or posterior segment of the upper lobe or the apical segment of the right lower lobe, as in our patient.5–8 CT demonstrating scalloped inner margins and cross-cavity band shadows suggests a cavitary pulmonary infarction.9,10
Infection and abscess in pulmonary infarction are poorly understood but have been linked to larger infarctions, coexistent congestion or atelectasis, and dental or oropharyngeal infection. In an early series of 550 cases of pulmonary infarction, 23 patients (4.2%) developed lung abscess and 6 (1.1%) developed empyema.11 The mean time to cavitation for an infected pulmonary infarction has been reported to be 18 days.12
A reversed halo sign, generally described as a focal, rounded area of ground-glass opacity surrounded by a nearly complete ring of consolidation, has been reported to be more frequent with pulmonary infarction than with other diseases, especially when in the lower lobes.13
CASE CONTINUED: THORACOSCOPY
A cardiothoracic surgeon was consulted, intravenous heparin was discontinued, an inferior vena cava filter was placed, and the patient underwent video-assisted thoracoscopy.
Purulent fluid was noted on the lateral aspect of right lower lobe; this appeared to be the ruptured cavitary lesion functioning like an uncontrolled bronchopleural fistula. Two chest tubes, sizes 32F and 28F, were placed after decortication, resection of the lung abscess, and closure of the bronchopleural fistula. No significant air leak was noted after resection of this segment of lung.
Pathologic study showed acute organizing pneumonia with abscess formation; no malignant cells or granulomas were seen (Figure 2). Pleural fluid cultures grew Streptococcus intermedius, while the tissue culture was negative for any growth, including acid-fast bacilli and fungi.
On 3 different occasions, both chest tubes were shortened, backed out 2 cm, and resecured with sutures and pins, and Heimlich valves were applied before the patient was discharged.
Intravenous piperacillin-tazobactam was started on the fifth hospital day. On discharge, the patient was advised to continue this treatment for 3 weeks at home.
The patient was receiving enoxaparin subcutaneously in prophylactic doses; 72 hours after the thorascopic procedure this was increased to therapeutic doses, continuing after discharge. Bridging to warfarin was not advised in view of his chest tubes.
Our patient appeared to have developed a right lower lobe infarction that cavitated and ruptured into the pleural space, causing a bronchopleural fistula with empyema after a recent pulmonary embolism. Other reported causes of pulmonary infarction in pulmonary embolism are malignancy and heavy clot burden,6 but these have not been confirmed in subsequent studies.5 Malignancy was ruled out by biopsy of the resected portion of the lung, and our patient did not have a history of heart failure. A clear cavity was not noted (because it ruptured into the pleura), but an air-fluid level was described in a wedge-shaped consolidation, suggesting infarction.
How common is pulmonary infarction after pulmonary embolism?
Pulmonary infarction occurs in few patients with pulmonary embolism.13 Since the lungs receive oxygen from the airways and have a dual blood supply from the pulmonary and bronchial arteries, they are not particularly vulnerable to ischemia. However, the reported incidence of pulmonary infarction in patients with pulmonary embolism has ranged from 10% to higher than 30%.5,14,15
The reasons behind pulmonary infarction with complications after pulmonary embolism have varied in different case series in different eras. CT, biopsy, or autopsy studies reveal pulmonary infarction after pulmonary embolism to be more common than suspected by clinical symptoms.
In a Mayo Clinic series of 43 cases of pulmonary infarction diagnosed over a 6-year period by surgical lung biopsy, 18 (42%) of the patients had underlying pulmonary thromboembolism, which was the most common cause.16
RISK FACTORS FOR PULMONARY INFARCTION
4. Which statement about risk factors for pulmonary infarction in pulmonary embolism is incorrect?
- Heart failure may be a risk factor for pulmonary infarction
- Pulmonary hemorrhage is a risk factor for pulmonary infarction
- Pulmonary infarction is more common with more proximal sites of pulmonary embolism
- Collateral circulation may protect against pulmonary infarction
Infarction is more common with emboli that are distal rather than proximal.
Dalen et al15 suggested that after pulmonary embolism, pulmonary hemorrhage is an important contributor to the development of pulmonary infarction independent of the presence or absence of associated cardiac or pulmonary disease, but that the effect depends on the site of obstruction.
This idea was first proposed in 1913, when Karsner and Ghoreyeb17 showed that when pulmonary arteries are completely obstructed, the bronchial arteries take over, except when the embolism is present in a small branch of the pulmonary artery. This is because the physiologic anastomosis between the pulmonary artery and the bronchial arteries is located at the precapillary level of the pulmonary artery, and the bronchial circulation does not take over until the pulmonary arterial pressure in the area of the embolism drops to zero.
Using CT data, Kirchner et al5 confirmed that the risk of pulmonary infarction is higher if the obstruction is peripheral, ie, distal.
Using autopsy data, Tsao et al18 reported a higher risk of pulmonary infarction in embolic occlusion of pulmonary vessels less than 3 mm in diameter.
Collateral circulation has been shown to protect against pulmonary infarction. For example, Miniati et al14 showed that healthy young patients with pulmonary embolism were more prone to develop pulmonary infarction, probably because they had less efficient collateral systems in the peripheral lung fields. In lung transplant recipients, it has been shown that the risk of infarction decreased with development of collateral circulation.19
Dalen et al,15 however, attributed delayed resolution of pulmonary hemorrhage (as measured by resolution of infiltrate on chest radiography) to higher underlying pulmonary venous pressure in patients with heart failure and consequent pulmonary infarction. In comparison, healthy patients without cardiac or pulmonary disease have faster resolution of pulmonary hemorrhage when present, and less likelihood of pulmonary infarction (and death in submassive pulmonary embolism).
Data on the management of infected pulmonary infarction are limited. Mortality rates have been as high as 41% with noninfected and 73% with infected cavitary infarctions.4 Some authors have advocated early surgical resection in view of high rates of failure of medical treatment due to lack of blood supply within the cavity and continued risk of infection.
KEY POINTS
In patients with a recently diagnosed pulmonary embolism and concurrent symptoms of bacterial pneumonia, a diagnosis of cavitary pulmonary infarction should be considered.
Consolidations that are pleural-based with sharp, rounded margins and with focal areas of central hyperlucencies representing hemorrhage on the mediastinal windows on CT are more likely to represent a pulmonary infarct.20
A 76-year-old man whose history included abdominal aortic aneurysm repair, bilateral femoral artery bypass for popliteal artery aneurysm, hypertension, and peptic ulcer disease was admitted to a community hospital with pleuritic chest pain and shortness of breath. Two days earlier, he had undergone repair of a ventral hernia.
At the time of that admission, he reported no fever, chills, night sweats, cough, or history of heart or lung disease. His vital signs were normal, and physical examination had revealed no apparent respiratory distress, no jugular venous distention, normal heart sounds, and no pedal edema; however, decreased air entry was noted in the right lung base. Initial serum levels of troponin and N-terminal pro-B-type natriuretic peptide were normal.
At that time, computed tomographic angiography of the chest showed segmental pulmonary emboli in the left upper and right lower lobes of the lungs and right pleural effusion. Transthoracic echocardiography showed normal atrial and ventricular sizes with no right or left ventricular systolic dysfunction and a left ventricular ejection fraction of 59%.
Treatment with intravenous heparin was started, and the patient was transferred to our hospital.
PLEURAL EFFUSION AND PULMONARY EMBOLISM
1. Which of the following is true about pleural effusion?
- It is rarely, if ever, associated with pulmonary embolism
- Most patients with pleural effusion due to pulmonary embolism do not have pleuritic chest pain
- Pulmonary embolism should be excluded in all cases of pleural effusion without a clear cause
Pulmonary embolism should be excluded in all cases of pleural effusion that do not have a clear cause. As for the other answer choices:
- Pulmonary embolism is the fourth leading cause of pleural effusion in the United States, after heart failure, pneumonia, and malignancy.1
- About 75% of patients who develop pleural effusion in the setting of pulmonary embolism complain of pleuritic chest pain on the side of the effusion.2 Most effusions are unilateral, small, and usually exudative.3
EVALUATION BEGINS: RESULTS OF THORACENTESIS
Our patient continued to receive intravenous heparin.
He underwent thoracentesis on hospital day 3, and 1,000 mL of turbid sanguineous pleural fluid was removed. Analysis of the fluid showed pH 7.27, white blood cell count 3.797 × 109/L with 80% neutrophils, and lactate dehydrogenase (LDH) concentration 736 U/L (a ratio of pleural fluid LDH to a concurrent serum LDH > 0.6 is suggestive of an exudate); the fluid was also sent for culture and cytology. Thoracentesis was terminated early due to cough, and follow-up chest radiography showed a moderate-sized pneumothorax.
Computed tomography (CT) of the chest at this time showed a small wedge-shaped area of lung consolidation in the right lower lobe (also seen on CT done 1 day before admission to our hospital), with an intrinsic air-fluid level suggesting a focal infarct or lung abscess, now obscured by adjacent consolidation and atelectasis. In the interval since the previous CT, the multiloculated right pleural effusion had increased in size (Figure 1).
THE NEXT STEP
2. What is the most appropriate next step for this patient?
- Consult an interventional radiologist for chest tube placement
- Start empiric antibiotic therapy and ask an interventional radiologist to place a chest tube
- Start empiric antibiotic therapy, withhold anticoagulation, and consult a thoracic surgeon
- Start empiric antibiotic therapy and consult a thoracic surgeon while continuing anticoagulation
The most appropriate next step is to start empiric antibiotic therapy and consult a thoracic surgeon while continuing anticoagulation.
In this patient, it is appropriate to initiate antibiotics empirically on the basis of his significant pleural loculations, a wedge-shaped consolidation, and 80% neutrophils in the pleural fluid, all of which suggest infection. The unmasking of a wedge-shaped consolidation after thoracentesis, with a previously noted air-fluid level and an interval increase in multiloculated pleural fluid, raises suspicion of a necrotic infection that may have ruptured into the pleural space, a possible lung infarct, or a malignancy. Hence, simply placing a chest tube may not be enough.
Blood in the pleural fluid does not necessitate withholding anticoagulation unless the bleeding is heavy. A pleural fluid hematocrit greater than 50% of the peripheral blood hematocrit suggests hemothorax and is an indication to withhold anticoagulation.1 Our patient’s pleural fluid was qualitatively sanguineous but not frankly bloody, and therefore we judged that it was not necessary to stop his heparin.
HOW DOES PULMONARY INFARCTION PRESENT CLINICALLY?
3. Which of the following statements about pulmonary infarction is incorrect?
- Cavitation and infarction are more common with larger emboli
- Cavitation occurs in fewer than 10% of pulmonary infarctions
- Lung abscess develops in more than 50% of pulmonary infarctions
- Pulmonary thromboembolism is the most common cause of pulmonary infarction
Lung abscess develops in far fewer than 50% of cases of pulmonary infarction. The rest of the statements are correct.
Cavitation complicates about 4% to 7% of infarctions and is more common when the infarction is 4 cm or greater in diameter.4 These cavities are usually single and predominantly on the right side in the apical or posterior segment of the upper lobe or the apical segment of the right lower lobe, as in our patient.5–8 CT demonstrating scalloped inner margins and cross-cavity band shadows suggests a cavitary pulmonary infarction.9,10
Infection and abscess in pulmonary infarction are poorly understood but have been linked to larger infarctions, coexistent congestion or atelectasis, and dental or oropharyngeal infection. In an early series of 550 cases of pulmonary infarction, 23 patients (4.2%) developed lung abscess and 6 (1.1%) developed empyema.11 The mean time to cavitation for an infected pulmonary infarction has been reported to be 18 days.12
A reversed halo sign, generally described as a focal, rounded area of ground-glass opacity surrounded by a nearly complete ring of consolidation, has been reported to be more frequent with pulmonary infarction than with other diseases, especially when in the lower lobes.13
CASE CONTINUED: THORACOSCOPY
A cardiothoracic surgeon was consulted, intravenous heparin was discontinued, an inferior vena cava filter was placed, and the patient underwent video-assisted thoracoscopy.
Purulent fluid was noted on the lateral aspect of right lower lobe; this appeared to be the ruptured cavitary lesion functioning like an uncontrolled bronchopleural fistula. Two chest tubes, sizes 32F and 28F, were placed after decortication, resection of the lung abscess, and closure of the bronchopleural fistula. No significant air leak was noted after resection of this segment of lung.
Pathologic study showed acute organizing pneumonia with abscess formation; no malignant cells or granulomas were seen (Figure 2). Pleural fluid cultures grew Streptococcus intermedius, while the tissue culture was negative for any growth, including acid-fast bacilli and fungi.
On 3 different occasions, both chest tubes were shortened, backed out 2 cm, and resecured with sutures and pins, and Heimlich valves were applied before the patient was discharged.
Intravenous piperacillin-tazobactam was started on the fifth hospital day. On discharge, the patient was advised to continue this treatment for 3 weeks at home.
The patient was receiving enoxaparin subcutaneously in prophylactic doses; 72 hours after the thorascopic procedure this was increased to therapeutic doses, continuing after discharge. Bridging to warfarin was not advised in view of his chest tubes.
Our patient appeared to have developed a right lower lobe infarction that cavitated and ruptured into the pleural space, causing a bronchopleural fistula with empyema after a recent pulmonary embolism. Other reported causes of pulmonary infarction in pulmonary embolism are malignancy and heavy clot burden,6 but these have not been confirmed in subsequent studies.5 Malignancy was ruled out by biopsy of the resected portion of the lung, and our patient did not have a history of heart failure. A clear cavity was not noted (because it ruptured into the pleura), but an air-fluid level was described in a wedge-shaped consolidation, suggesting infarction.
How common is pulmonary infarction after pulmonary embolism?
Pulmonary infarction occurs in few patients with pulmonary embolism.13 Since the lungs receive oxygen from the airways and have a dual blood supply from the pulmonary and bronchial arteries, they are not particularly vulnerable to ischemia. However, the reported incidence of pulmonary infarction in patients with pulmonary embolism has ranged from 10% to higher than 30%.5,14,15
The reasons behind pulmonary infarction with complications after pulmonary embolism have varied in different case series in different eras. CT, biopsy, or autopsy studies reveal pulmonary infarction after pulmonary embolism to be more common than suspected by clinical symptoms.
In a Mayo Clinic series of 43 cases of pulmonary infarction diagnosed over a 6-year period by surgical lung biopsy, 18 (42%) of the patients had underlying pulmonary thromboembolism, which was the most common cause.16
RISK FACTORS FOR PULMONARY INFARCTION
4. Which statement about risk factors for pulmonary infarction in pulmonary embolism is incorrect?
- Heart failure may be a risk factor for pulmonary infarction
- Pulmonary hemorrhage is a risk factor for pulmonary infarction
- Pulmonary infarction is more common with more proximal sites of pulmonary embolism
- Collateral circulation may protect against pulmonary infarction
Infarction is more common with emboli that are distal rather than proximal.
Dalen et al15 suggested that after pulmonary embolism, pulmonary hemorrhage is an important contributor to the development of pulmonary infarction independent of the presence or absence of associated cardiac or pulmonary disease, but that the effect depends on the site of obstruction.
This idea was first proposed in 1913, when Karsner and Ghoreyeb17 showed that when pulmonary arteries are completely obstructed, the bronchial arteries take over, except when the embolism is present in a small branch of the pulmonary artery. This is because the physiologic anastomosis between the pulmonary artery and the bronchial arteries is located at the precapillary level of the pulmonary artery, and the bronchial circulation does not take over until the pulmonary arterial pressure in the area of the embolism drops to zero.
Using CT data, Kirchner et al5 confirmed that the risk of pulmonary infarction is higher if the obstruction is peripheral, ie, distal.
Using autopsy data, Tsao et al18 reported a higher risk of pulmonary infarction in embolic occlusion of pulmonary vessels less than 3 mm in diameter.
Collateral circulation has been shown to protect against pulmonary infarction. For example, Miniati et al14 showed that healthy young patients with pulmonary embolism were more prone to develop pulmonary infarction, probably because they had less efficient collateral systems in the peripheral lung fields. In lung transplant recipients, it has been shown that the risk of infarction decreased with development of collateral circulation.19
Dalen et al,15 however, attributed delayed resolution of pulmonary hemorrhage (as measured by resolution of infiltrate on chest radiography) to higher underlying pulmonary venous pressure in patients with heart failure and consequent pulmonary infarction. In comparison, healthy patients without cardiac or pulmonary disease have faster resolution of pulmonary hemorrhage when present, and less likelihood of pulmonary infarction (and death in submassive pulmonary embolism).
Data on the management of infected pulmonary infarction are limited. Mortality rates have been as high as 41% with noninfected and 73% with infected cavitary infarctions.4 Some authors have advocated early surgical resection in view of high rates of failure of medical treatment due to lack of blood supply within the cavity and continued risk of infection.
KEY POINTS
In patients with a recently diagnosed pulmonary embolism and concurrent symptoms of bacterial pneumonia, a diagnosis of cavitary pulmonary infarction should be considered.
Consolidations that are pleural-based with sharp, rounded margins and with focal areas of central hyperlucencies representing hemorrhage on the mediastinal windows on CT are more likely to represent a pulmonary infarct.20
- Light RW. Pleural Diseases. 4th ed. Baltimore, MD: Lippincott, Williams & Wilkins; 2001.
- Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100(3):598–603. pmid:1909617
- Light RW. Pleural effusion due to pulmonary emboli. Curr Opin Pulm Med 2001; 7(4):198–201. pmid:11470974
- Libby LS, King TE, LaForce FM, Schwarz MI. Pulmonary cavitation following pulmonary infarction. Medicine (Baltimore) 1985; 64(5):342–348. pmid:4033411
- Kirchner J, Obermann A, Stuckradt S, et al. Lung infarction following pulmonary embolism: a comparative study on clinical conditions and CT findings to identify predisposing factors. Rofo 2015; 187(6):440–444. doi:10.1055/s-0034-1399006
- He H, Stein MW, Zalta B, Haramati LB. Pulmonary infarction: spectrum of findings on multidetector helical CT. J Thorac Imaging 2006; 21(1):1–7. doi:10.1097/01.rti.0000187433.06762.fb
- Scharf J, Nahir AM, Munk J, Lichtig C. Aseptic cavitation in pulmonary infarction. Chest 1971; 59(4):456–458. pmid:5551596
- Wilson AG, Joseph AE, Butland RJ. The radiology of aseptic cavitation in pulmonary infarction. Clin Radiol 1986; 37(4):327–333. pmid:3731699
- Butler MD, Biscardi FH, Schain DC, Humphries JE, Blow O, Spotnitz WD. Pulmonary resection for treatment of cavitary pulmonary infarction. Ann Thorac Surg 1997; 63(3):849–850. pmid:9066420
- Koroscil MT, Hauser TR. Acute pulmonary embolism leading to cavitation and large pulmonary abscess: a rare complication of pulmonary infarction. Respir Med Case Rep 2016; 20:72–74. doi:10.1016/j.rmcr.2016.12.001
- Levin L, Kernohan JW, Moersch HJ. Pulmonary abscess secondary to bland pulmonary infarction. Dis Chest 1948; 14(2):218–232. pmid:18904835
- Marchiori E, Menna Barreto M, Pereira Freitas HM, et al. Morphological characteristics of the reversed halo sign that may strongly suggest pulmonary infarction. Clin Radiol 2018; 73(5):503.e7–503.e13. doi:10.1016/j.crad.2017.11.022
- Smith GT, Dexter L, Dammin GJ. Postmortem quantitative studies in pulmonary embolism. In: Sasahara AA, Stein M, eds. Pulmonary Embolic Disease. New York, NY: Grune & Stratton, Inc; 1965:120–126.
- Miniati M, Bottai M, Ciccotosto C, Roberto L, Monti S. Predictors of pulmonary infarction. Medicine (Baltimore) 2015; 94(41):e1488. doi:10.1097/MD.0000000000001488
- Dalen JE, Haffajee CI, Alpert JS, Howe JP, Ockene IS, Paraskos JA. Pulmonary embolism, pulmonary hemorrhage and pulmonary infarction. N Engl J Med 1977; 296(25):1431–1435. doi:10.1056/NEJM197706232962503
- Parambil JG, Savci CD, Tazelaar HD, Ryu JH. Causes and presenting features of pulmonary infarctions in 43 cases identified by surgical lung biopsy. Chest 2005; 127(4):1178–1183. doi:10.1378/chest.127.4.1178
- Karsner HT, Ghoreyeb AA. Studies in infarction: III. The circulation in experimental pulmonary embolism. J Exp Med 1913; 18(5):507–511. pmid:19867725
- Tsao MS, Schraufnagel D, Wang NS. Pathogenesis of pulmonary infarction. Am J Med 1982; 72(4):599–606. pmid:6462058
- Burns KE, Iacono AT. Incidence of clinically unsuspected pulmonary embolism in mechanically ventilated lung transplant recipients. Transplantation 2003; 76(6):964–968. doi:10.1097/01.TP.0000084523.58610.BA
- Yousem SA. The surgical pathology of pulmonary infarcts: diagnostic confusion with granulomatous disease, vasculitis, and neoplasia. Mod Pathol 2009; 22(5):679–685. doi:10.1038/modpathol.2009.20
- Light RW. Pleural Diseases. 4th ed. Baltimore, MD: Lippincott, Williams & Wilkins; 2001.
- Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest 1991; 100(3):598–603. pmid:1909617
- Light RW. Pleural effusion due to pulmonary emboli. Curr Opin Pulm Med 2001; 7(4):198–201. pmid:11470974
- Libby LS, King TE, LaForce FM, Schwarz MI. Pulmonary cavitation following pulmonary infarction. Medicine (Baltimore) 1985; 64(5):342–348. pmid:4033411
- Kirchner J, Obermann A, Stuckradt S, et al. Lung infarction following pulmonary embolism: a comparative study on clinical conditions and CT findings to identify predisposing factors. Rofo 2015; 187(6):440–444. doi:10.1055/s-0034-1399006
- He H, Stein MW, Zalta B, Haramati LB. Pulmonary infarction: spectrum of findings on multidetector helical CT. J Thorac Imaging 2006; 21(1):1–7. doi:10.1097/01.rti.0000187433.06762.fb
- Scharf J, Nahir AM, Munk J, Lichtig C. Aseptic cavitation in pulmonary infarction. Chest 1971; 59(4):456–458. pmid:5551596
- Wilson AG, Joseph AE, Butland RJ. The radiology of aseptic cavitation in pulmonary infarction. Clin Radiol 1986; 37(4):327–333. pmid:3731699
- Butler MD, Biscardi FH, Schain DC, Humphries JE, Blow O, Spotnitz WD. Pulmonary resection for treatment of cavitary pulmonary infarction. Ann Thorac Surg 1997; 63(3):849–850. pmid:9066420
- Koroscil MT, Hauser TR. Acute pulmonary embolism leading to cavitation and large pulmonary abscess: a rare complication of pulmonary infarction. Respir Med Case Rep 2016; 20:72–74. doi:10.1016/j.rmcr.2016.12.001
- Levin L, Kernohan JW, Moersch HJ. Pulmonary abscess secondary to bland pulmonary infarction. Dis Chest 1948; 14(2):218–232. pmid:18904835
- Marchiori E, Menna Barreto M, Pereira Freitas HM, et al. Morphological characteristics of the reversed halo sign that may strongly suggest pulmonary infarction. Clin Radiol 2018; 73(5):503.e7–503.e13. doi:10.1016/j.crad.2017.11.022
- Smith GT, Dexter L, Dammin GJ. Postmortem quantitative studies in pulmonary embolism. In: Sasahara AA, Stein M, eds. Pulmonary Embolic Disease. New York, NY: Grune & Stratton, Inc; 1965:120–126.
- Miniati M, Bottai M, Ciccotosto C, Roberto L, Monti S. Predictors of pulmonary infarction. Medicine (Baltimore) 2015; 94(41):e1488. doi:10.1097/MD.0000000000001488
- Dalen JE, Haffajee CI, Alpert JS, Howe JP, Ockene IS, Paraskos JA. Pulmonary embolism, pulmonary hemorrhage and pulmonary infarction. N Engl J Med 1977; 296(25):1431–1435. doi:10.1056/NEJM197706232962503
- Parambil JG, Savci CD, Tazelaar HD, Ryu JH. Causes and presenting features of pulmonary infarctions in 43 cases identified by surgical lung biopsy. Chest 2005; 127(4):1178–1183. doi:10.1378/chest.127.4.1178
- Karsner HT, Ghoreyeb AA. Studies in infarction: III. The circulation in experimental pulmonary embolism. J Exp Med 1913; 18(5):507–511. pmid:19867725
- Tsao MS, Schraufnagel D, Wang NS. Pathogenesis of pulmonary infarction. Am J Med 1982; 72(4):599–606. pmid:6462058
- Burns KE, Iacono AT. Incidence of clinically unsuspected pulmonary embolism in mechanically ventilated lung transplant recipients. Transplantation 2003; 76(6):964–968. doi:10.1097/01.TP.0000084523.58610.BA
- Yousem SA. The surgical pathology of pulmonary infarcts: diagnostic confusion with granulomatous disease, vasculitis, and neoplasia. Mod Pathol 2009; 22(5):679–685. doi:10.1038/modpathol.2009.20
Antisynthetase syndrome: Not just an inflammatory myopathy
A 66-year-old man was initially seen in clinic in March 2004 with a 5-month history of polyarthritis (affecting the finger joints, wrists, and knees) and several hours of morning stiffness. He also had significant proximal muscle weakness, progressive exertional dyspnea, and a nonproductive cough. There was no history of fever, chills, rash, dysphagia, or sicca symptoms. Findings on initial tests:
- His creatine kinase level was 700 U/L (reference range 30–220), which later rose to 1,664 U/L.
- He was positive for antinuclear antibody with a 5.7 optical density ratio (normal < 1.5) and for anti-Jo-1 antibody.
- An electromyogram was consistent with a necrotizing myopathy. Left rectus femoris biopsy revealed scattered degenerating and regenerating muscle fibers but no evidence of endomysial inflammation.
- On pulmonary function testing, his forced vital capacity was 80% of predicted, and his carbon monoxide diffusion capacity was 67% of predicted.
- High-resolution computed tomography revealed evidence of interstitial lung disease, characterized by bilateral patchy ground-glass opacities suggestive of active alveolitis, most extensive at the lung bases.
- Bronchoalveolar lavage indicated alveolitis, and transbronchial biopsy revealed pathologic changes consistent with cryptogenic organizing pneumonia. All cultures were negative.
This constellation of clinical manifestations, including myositis, interstitial lung disease, and polyarthritis, along with positive anti-Jo-1 antibody, confirmed the diagnosis of antisynthetase syndrome.
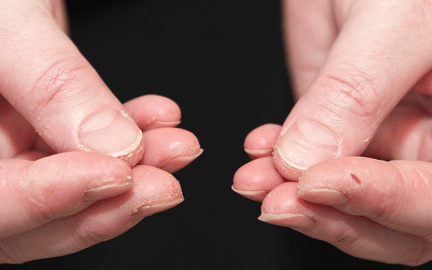
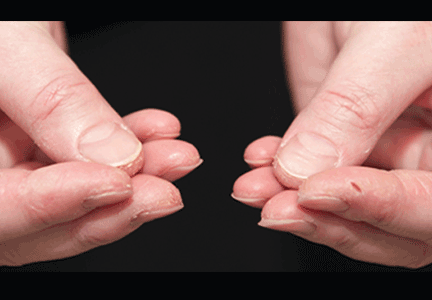
In June 2004, for his interstitial lung disease, he was started on daily oral cyclophosphamide along with high-dose oral prednisone. Three months later the skin of the tips and radial margins of his fingers started thickening and cracking, the appearance of which is classically described as “mechanic’s hands,” a well-described manifestation of antisynthetase syndrome (Figure 1).
Cyclophosphamide was continued for about a year. Subsequently, along with prednisone, he sequentially received various other immunosuppressive medications (methotrexate, tacrolimus, mycophenolate mofetil, and rituximab) over the next few years in an attempt to control his progressive interstitial lung disease. All of these agents were only partially and temporarily effective. Ultimately, despite all of these therapies, as his interstitial lung disease progressed, he needed supplemental oxygen and enrollment in a pulmonary rehabilitation program.
In March 2010, he was admitted with worsening dyspnea and significant peripheral edema and was found to have severe pulmonary arterial hypertension. He was started on bosentan. Eight months later sildenafil was added for progressive pulmonary arterial hypertension. However, his oxygenation status continued to decline.
In July 2011, he presented with chills, increasing shortness of breath, and a mild productive cough. As he was severely hypoxic, he was admitted to the intensive care unit and started on mechanical ventilation and broad-spectrum antibiotics. Despite escalation of oxygen therapy, his respiratory status rapidly deteriorated, and he developed hypotension requiring vasopressors. He ultimately died of cardiac arrest secondary to respiratory failure.
A CONSTELLATION OF MANIFESTATIONS
Antisynthetase syndrome, associated with anti-aminoacyl-transfer RNA (tRNA) synthetase antibodies, is characterized by a constellation of manifestations that include myositis, interstitial lung disease, mechanic’s hands, fever, Raynaud phenomenon, and nonerosive symmetric polyarthritis of the small joints.1
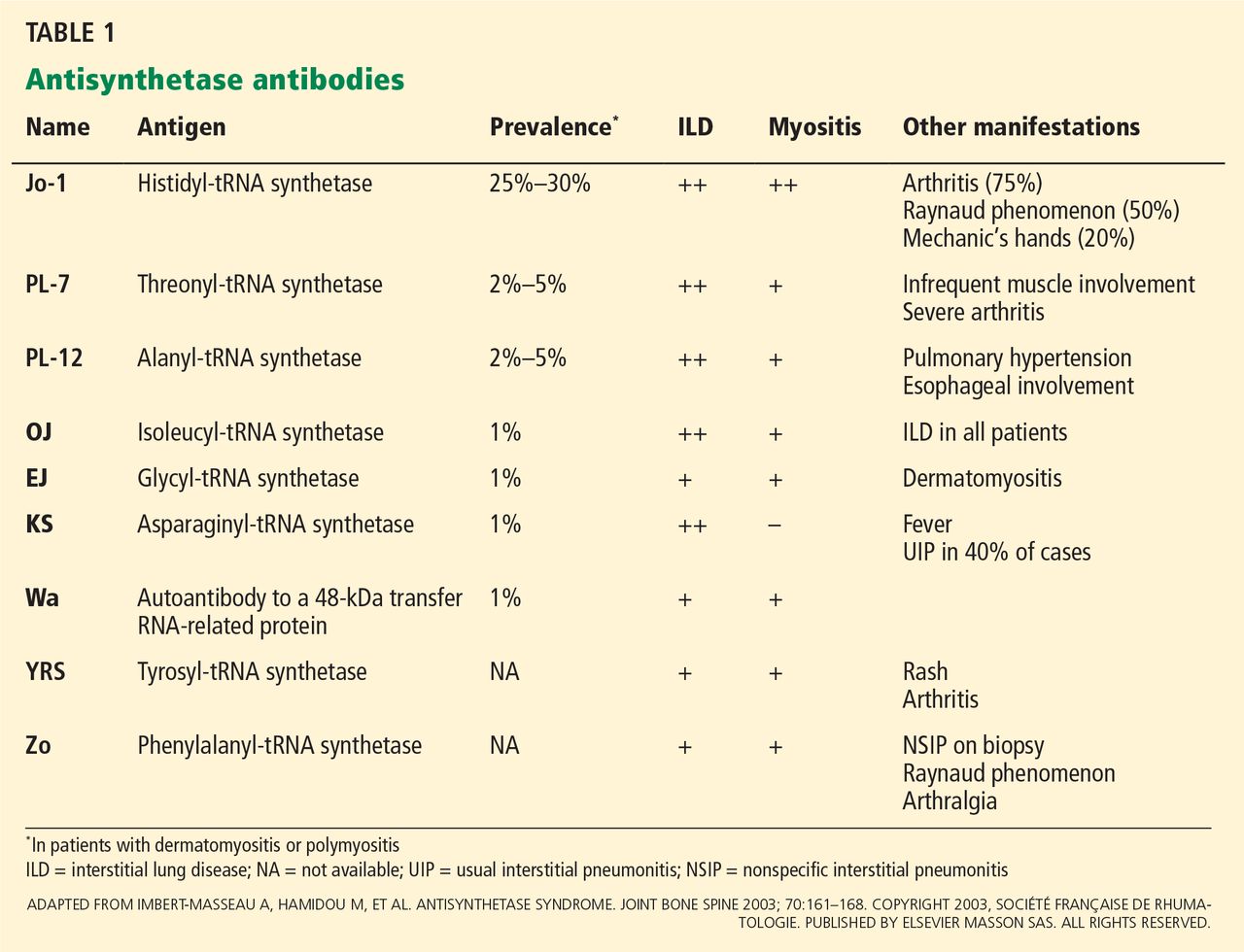
Anti-Jo-1 antibody (anti-histidyl-tRNA synthetase) is the most common of the antibodies and also was the first one to be identified (Table 1). It was named after John P, a patient with polymyositis and interstitial lung disease, in whom it was first detected in 1980.2 The onset of the syndrome associated with anti-Jo-1 antibody is often acute, and the myositis is usually steroid-responsive. However, not uncommonly, severe disease can develop over time, with a tendency to relapse and with a poor long-term prognosis.
RARE BUT UNDERRECOGNIZED
The true population prevalence of antisynthetase syndrome is unknown. Because this syndrome is rare, comprehensive epidemiologic studies are difficult to perform.
In several retrospective studies, the annual incidence of idiopathic inflammatory myopathies has been reported to be 2 to 10 new cases per million adults per year.3 Antisynthetase antibodies are detected in 20% to 40% of such cases.4–6 The disease is two to three times more common in women than in men.7
Early diagnosis is difficult because the clinical presentation is varied and often nonspecific, clinically milder disease may escape detection, and many general practitioners lack familiarity with this syndrome and consequently do not recognize it. Moreover, tests for myositis-specific antibodies (including antisynthetase antibodies) are often not ordered in the evaluation of myositis, and hence the diagnosis of antisynthetase syndrome cannot be substantiated. Furthermore, interstitial lung disease can predominate or can be the sole manifestation in the absence of clinically apparent myositis,8–10 and patients can be misdiagnosed as having idiopathic pulmonary fibrosis when underlying antisynthetase syndrome is not suspected. This distinction may be important because these conditions differ in their pathology and treatment. Histologically, the predominant pattern of lung injury in idiopathic pulmonary fibrosis is “usual interstitial pneumonia” which does not respond to immunosuppressive therapy, and hence lung transplantation is the only therapeutic option. On the other hand, in antisynthetase syndrome, the usual pattern of lung injury is “nonspecific interstitial pneumonia,” in which immunosuppressive therapy has a role.
Anti-Jo-1 antibody is detected in 15% to 25% of patients with polymyositis and in up to 70% of myositis patients with concomitant interstitial lung disease.11 Autoantibodies to seven other, less frequently targeted, aminoacyl tRNA synthetases have also been described in patients with polymyositis and interstitial lung disease (Table 1).11,12 In addition, an autoantibody to a 48-kDa transfer RNA-related protein (Wa) has been described.13 These non-Jo-1 antisynthetase antibodies are detected in only about 3% of myositis patients.14
ROLE OF ANTISYNTHETASE ANTIBODIES
Synthetases play a central role in protein synthesis by catalyzing the acetylation of tRNAs. The propensity of organ involvement in antisynthetase syndrome suggests that tissue-specific changes in muscle or lung lead to the production of unique forms of target autoantigens, the aminoacyl-tRNA synthetases. There is evidence that these enzymes themselves may be involved in recruiting both antigen-presenting and inflammatory cells to the site of muscle or lung injury.15 However, the molecular pathway that initiates and propagates this autoimmune response and the specific role of the antisynthetase antibodies in the pathogenesis of this syndrome are presently unknown.
SIX SALIENT CLINICAL FEATURES
There are six predominant clinical manifestations, which may be present at disease onset or appear later as the disease progresses:
- Fever
- Myositis
- Interstitial lung disease
- Mechanic’s hands
- Raynaud phenomenon
- Inflammatory polyarthritis.
There is considerable clinical heterogeneity, and one or other manifestation can predominate or can be the only expression of the syndrome. Furthermore, in the same patient, the individual features can prevail at different times and may develop years after onset of the disease. Therefore, in addition to patients with myositis, it would be important to suspect antisynthetase syndrome in patients presenting with isolated lung involvement (amyopathic interstitial lung disease), as there are therapeutic implications. Studies have demonstrated the efficacy of immunosuppressive agents in interstitial lung disease associated with antisynthetase syndrome (where the predominant pattern of lung injury is “nonspecific interstitial pneumonitis”), whereas lung transplantation has so far been the only treatment option in idiopathic pulmonary fibrosis.
Fever
About 20% of patients have a fever at disease onset or associated with relapses. Sometimes the fever can persist until treatment of antisynthetase syndrome is started.
Myositis
Muscle disease is seen in more than 90% of patients with anti-Jo-1 antisynthetase syndrome. It can be subclinical (in the absence of proximal myopathy), manifested by transient creatine kinase elevation only, which may normalize after therapy is initiated.
However, more commonly, patients develop profound proximal muscle weakness and sometimes muscle pain (Table 2). Weakness of the striated muscles of the upper esophagus, cricopharyngeus, and hypopharynx may cause dysphagia and makes these patients susceptible to aspiration pneumonia. Diaphragmatic and intercostal muscle weakness can contribute to shortness of breath in some patients. Myocarditis has also been reported.
Pulmonary disease
Interstitial lung disease develops in most patients with anti-Jo-1 antisynthetase syndrome, with a reported prevalence of about 90% in one series.16 Patients often present with acute, subacute, or insidious onset of exertional dyspnea. Sometimes there is an intractable nonproductive cough.
At the outset of antisynthetase syndrome, if the patient is profoundly weak because of myopathy or has inflammatory polyarthritis, mobility is significantly compromised, and exertional dyspnea may not be experienced. However, as the interstitial lung disease progresses, shortness of breath becomes overt, more so when the patient’s level of activity improves with treatment of myositis.
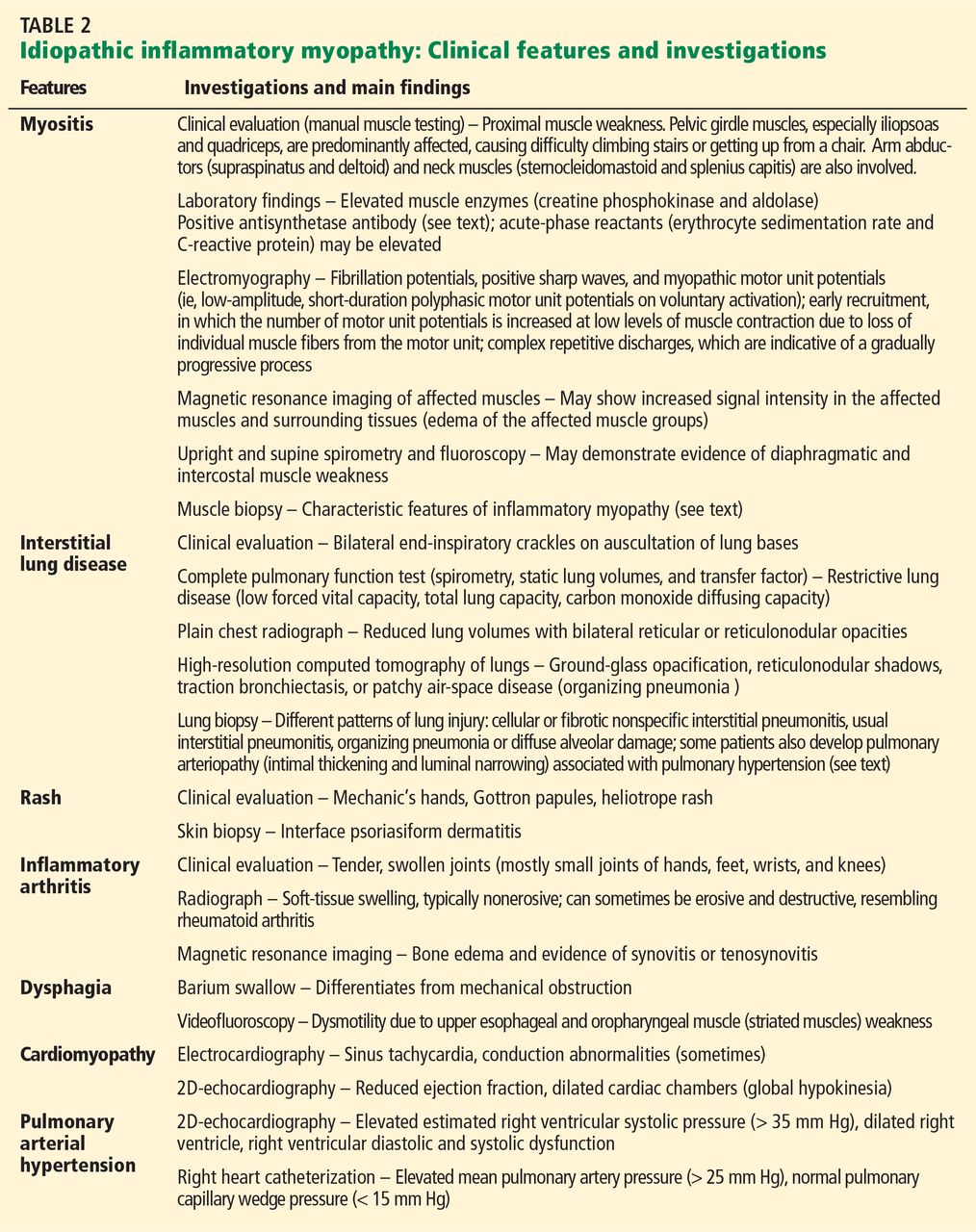
Inspiratory crackles on auscultation of the lung bases or changes on chest radiography are relatively insensitive findings and can miss early interstitial lung disease. Therefore, if antisynthetase syndrome is suspected or diagnosed, a baseline pulmonary function test (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease, and the diagnosis can then be confirmed with thoracic high-resolution computed tomography (Figure 2).
Pulmonary hypertension. Recent studies indicate that, similar to patients with other autoimmune rheumatic diseases, pulmonary hypertension can develop in patients with antisynthetase syndrome, with or without concomitant interstitial lung disease.17,18 This complication occurred in the case presented here. It has been found that when pulmonary hypertension coexists with interstitial lung disease, its degree may not correlate with the severity of the latter.17 Additionally, pulmonary hypertension, when present, has been found to contribute independently to prognosis and survival.
Mechanic’s hands
In about 30% of patients, the skin of the tips and margins of the fingers becomes thickened, hyperkeratotic, and fissured, the appearance of which is classically described as mechanic’s hands. It is a common manifestation of antisynthetase syndrome and is particularly prominent on the radial side of the index fingers (Figure 1). Biopsy of affected skin shows an interface psoriasiform dermatitis.19 In addition, some dermatomyositis patients with Gottron papules and a heliotrope rash have antisynthetase antibodies.
Raynaud phenomenon
Raynaud phenomenon develops in about 40% of patients. Some have nailfold capillary abnormalities.20 However, persistent or severe digital ischemia leading to digital ulceration or infarction is uncommon.21
Inflammatory arthritis
Arthralgias and arthritis are common (50%), the most common form being a symmetric polyarthritis of the small joints of the hands and feet. It is typically nonerosive but can sometimes be erosive and destructive.20
Because inflammatory arthritis mimics rheumatoid arthritis, antisynthetase syndrome should be considered in rheumatoid factor-negative patients presenting with polyarthritis.
ASSOCIATION WITH MALIGNANCY
Traditional teaching has been that antisynthetase antibody is protective against an underlying malignancy.22,23 However, several recently published case studies have reported various malignancies occurring within 6 to 12 months of the diagnosis of antisynthetase syndrome.7,24 The debate as to whether these are chance associations or causal (a paraneoplastic phenomenon) has not been resolved at this time.24
It is now recommended that patients with antisynthetase syndrome be screened for malignancies as appropriate for the patient’s age and sex. Screening should include a careful history and physical examination, complete blood cell count, comprehensive metabolic panel, chest radiography, mammography, and a gynecologic examination for women.25 If abnormalities are found, a more thorough evaluation for cancer is appropriate.
DIAGNOSIS
Muscle enzyme levels are often elevated
Muscle enzymes (creatine kinase and aldolase) are often elevated. Serum creatine kinase levels can range between 5 to 50 times the upper limit of normal. In an established case, creatine kinase levels along with careful manual muscle strength testing may help evaluate myositis activity. However, in chronic and advanced disease, creatine kinase may be within the normal range despite active myositis, partly because of extensive loss of muscle mass. In myositis, it may be prudent to check both creatine kinase and aldolase; sometimes only serum aldolase level rises, when immune-mediated injury predominantly affects the early regenerative myocytes.26
Judicious use of autoantibody testing
The characteristic clinical presentation is the initial clue to the diagnosis of antisynthetase syndrome, which is then supported by serologic testing.
Injudicious testing for a long list of antibodies should be avoided, as the cost is considerable and it does not influence further management. However, ordering an anti-Jo-1 antibody test in the correct clinical setting is appropriate, as it has high specificity,27,28 and thus can help establish or refute the clinical suspicion of antisynthetase syndrome.
Screening pulmonary function testing and thoracic high-resolution computed tomography for all patients with polymyositis or dermatomyositis is not considered “standard of care” and will likely not be reimbursed by third-party payers. However, in a patient with symptoms and signs of myositis, the presence of an antisynthetase antibody should prompt screening for occult interstitial lung disease, even in the absence of symptoms. As lung disease ultimately determines the prognosis in antisynthetase syndrome, early diagnosis and management is the key. Therefore, these tests would likely be approved to establish the diagnosis of interstitial lung disease and evaluate its severity.
If a myositis patient is also found to have interstitial lung disease or develops mechanic’s hands, the likely diagnosis is antisynthetase syndrome, which can be confirmed by serologic testing for antisynthetase antibodies. Interstitial lung disease in antisynthetase syndrome is often from “nonspecific interstitial pneumonitis”; therefore, medications tested and proven effective for this condition should be approved and reimbursed by payers.29–32
The coexistence of myositis and interstitial lung disease increases the sensitivity of anti-Jo-1 antibody.11 Thus, the clinician can have more confidence in early recognition and initiation of aggressive but targeted disease-modifying therapy.
Various methods can be used for detecting antisynthetase antibodies, with comparable results.28 Anti-Jo-1 antibody testing costs about $140. If that test is negative and antisynthetase syndrome is still suspected, then testing for the non-Jo-1 antisynthetase antibodies may be justified (Table 1). Though the cost of this panel of autoantibodies is about $300, it helps to confirm the diagnosis, and it influences the choice of second-line immunosuppressive agents such as tacrolimus29 and rituximab32 in patients resistant to conventional immunosuppressive agents such as azathioprine and methotrexate.
Often, anti-Ro52 SS-A antibodies are present concurrently in patients with anti-Jo-1 syndrome.33 In observational studies in patients with anti-Jo-1 antibody-associated interstitial lung disease, coexistence of anti-Ro52 SS-A antibody tended to predict a worse pulmonary outcome than in those with anti-Jo-1 antibody alone.34,35
Electromyography
Electromyography not only helps differentiate between myopathic and neuropathic weakness, but it may also support the diagnosis of “inflammatory” myopathy as suggested by prominent muscle membrane irritability (fibrillations, positive sharp waves) and abnormal motor unit action potentials (spontaneous activity showing small, short, polyphasic potentials and early recruitment). However, the findings can be nonspecific, and may even be normal in 10% to 15% of patients.36 Electromyographic abnormalities are most consistently observed in weak proximal muscles, and electromyography is also helpful in selecting a muscle for biopsy. Although no single electromyographic pattern is considered diagnostic for inflammatory myopathy, abnormalities are present in around 90% of patients (Table 2).3
Magnetic resonance imaging
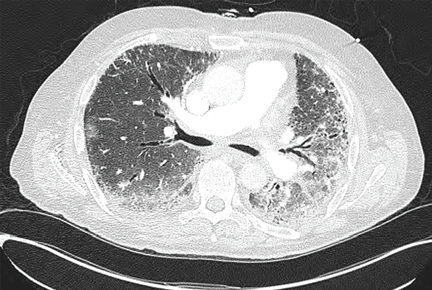
Magnetic resonance imaging may show increased signal intensity in the affected muscles and surrounding tissues (Figure 3).37 Because it lacks sensitivity and specificity, magnetic resonance imaging is not helpful in diagnosing the disease. However, in the correct clinical setting, it may be used to guide muscle biopsy, and it can help in monitoring the disease progress.38
Muscle histopathology
Muscle biopsy, though often helpful, is not always diagnostic, and antisynthetase syndrome should still be suspected in the right clinical context, even in the absence of characteristic pathologic changes.
Biopsy of sites recently studied by electromyography should be avoided, and if the patient has undergone electromyography recently, the contralateral side should be selected for biopsy.
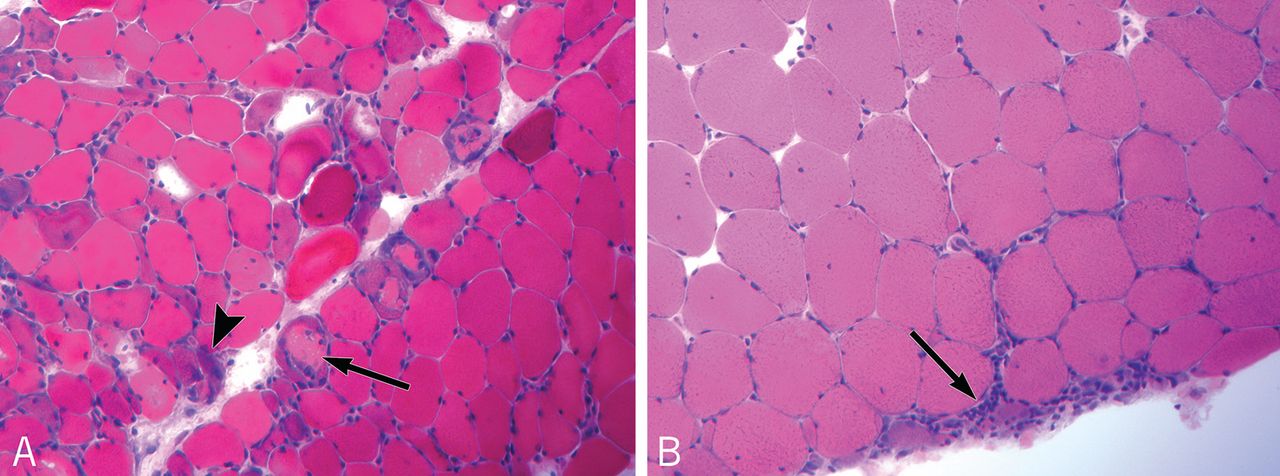
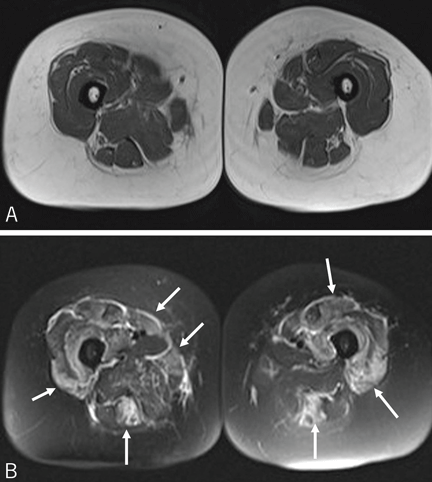
Reports of histopathologic findings in muscle biopsies in patients with antisynthetase syndrome document inflammatory myopathic features (Figure 4). In a series of patients with anti-Jo-1 syndrome, inflammation was noted in all cases, predominantly perimysial in location, with occasional endomysial and perivascular inflammation.39 Many of the inflammatory cells seen were macrophages and lymphocytes, in contrast to the predominantly lymphocytic infiltrates described in classic polymyositis and dermatomyositis. Perifascicular atrophy, similar to what is seen in dermatomyositis, was encountered; however, vascular changes, typical of dermatomyositis, were absent. Occasional degenerating and regenerating muscle fibers were also observed in most cases. Additionally, a characteristic perimysial connective tissue fragmentation was described, a feature less often seen in classic polymyositis and dermatomyositis.39
Pulmonary function testing
If antisynthetase syndrome is suspected or diagnosed, baseline pulmonary function testing (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease (reduced forced vital capacity and carbon monoxide diffusion capacity), and the diagnosis will be substantiated on thoracic high-resolution computed tomography. Respiratory muscle weakness can be detected with upright and supine spirometry.40 Weakness of these muscles contributes to shortness of breath, and patients may need ventilatory support.
Thoracic high-resolution computed tomography
Different patterns of lung injury can be seen in antisynthetase syndrome. Diffuse ground-glass opacification may suggest a nonspecific interstitial pneumonitis pattern, which is the most common form of interstitial lung disease (Figure 2), whereas coarse reticulation or honeycombing correlates with a usual interstitial pneumonitis pattern. Patchy consolidation or air-space disease can occur if cryptogenic organizing pneumonia is the predominant pattern of lung injury.
Swallowing evaluation
A comprehensive swallowing evaluation by a speech therapist may be necessary for evaluation of dysphagia (from oropharyngeal and striated esophageal muscle weakness) and determination of aspiration risk (Table 2).
Lung histopathology
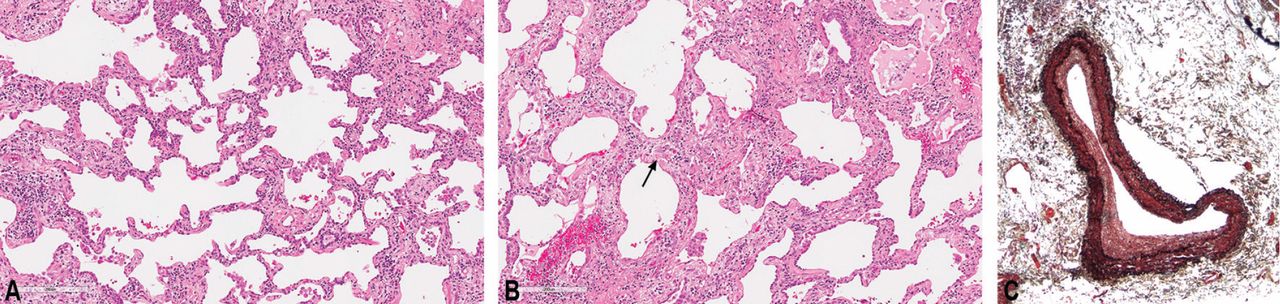
If necessary, a surgical lung biopsy is needed to document the pathologic pattern of injury, including the amount of fibrosis in the lung. Historically, in idiopathic inflammatory myopathy patients in general, this has taken the form of usual interstitial pneumonia, organizing pneumonia, or diffuse alveolar damage.41 With the emergence of the definition of nonspecific interstitial pneumonitis and fibrosis as a documented and accepted pattern, more studies have found this to be the most common pattern of lung injury.16 It is characterized by diffuse involvement of the lung by an interstitial chronic inflammatory infiltrate, a cellular type of nonspecific interstitial pneumonitis that progresses in a uniform pattern to a fibrotic type (Figure 5). This form of fibrosis rarely results in significant remodeling, so-called honeycomb changes. In addition, anti-Jo-1 antibody patients may also have an increased incidence of acute lung injury, including acute diffuse alveolar damage that is often superimposed on the underlying chronic lung disease.42
In patients with pulmonary hypertension, histopathologic studies of the muscular pulmonary arteries often show moderate intimal fibroplasia, suggesting that a pulmonary arteriopathy with intimal thickening and luminal narrowing develops in some of these patients (Figure 5), independent of chronic hypoxic pulmonary vasoconstriction or vascular obstruction due to its entrapment within fibrotic lung tissue.17
TREATMENT
Glucocorticoids are the mainstay
Glucocorticoids are considered the mainstay of treatment. Patients should be advised that long-term use of glucocorticoids is necessary, though the response is variable. It is also important to discuss possible side effects of long-term glucocorticoid use.
Standard practice is to initiate treatment with high doses for the first 4 to 6 weeks to achieve disease control, followed by a slow taper over the next 9 to 12 months to the lowest effective dose to maintain remission. If the patient is profoundly weak, especially with respiratory muscle weakness or significant dysphagia and aspiration risk, hospital admission for intravenous methylprednisolone 1,000 mg daily for 3 to 5 days may be necessary. Otherwise, oral prednisone 1 mg/kg/day would be the usual starting dose.
If the patient’s muscle strength initially improves and then declines weeks to months later despite adequate therapy, glucocorticoid-induced myopathy should be suspected, especially if the muscle enzymes are within the reference range. This is more likely to occur if high-dose prednisone is continued for more than 6 to 8 weeks.
Improvement in muscle strength, which can take several weeks to several months, is a more reliable indicator of response to therapy than the serum creatine kinase level, which may take much longer to normalize. Relying on normalization of the creatine kinase level alone may lead to unnecessary prolongation of high-dose glucocorticoid therapy. It may take several months for the muscle enzymes to normalize, and there is usually a time lag between normalization of muscle enzymes and complete recovery of muscle strength.
Long-term use of high-dose prednisone leads to glucocorticoid-induced osteoporosis. Therefore, patients should receive osteoporosis prophylaxis including antiresorptive therapy with a bisphosphonate. In addition, prophylaxis against Pneumocystis jirovecii is indicated for patients treated with high-dose glucocorticoids.
Additional immunosuppressive agents
Although glucocorticoids are considered the mainstay of treatment, additional immunosuppressive agents such as azathioprine and methotrexate are often required, both as glucocorticoid-sparing agents and to achieve adequate disease control.10 Addition of such agents from the outset is particularly necessary in patients with profound muscle weakness or those who have concomitant symptomatic interstitial lung disease.
No randomized controlled trial comparing azathioprine and methotrexate has been conducted to date. Therefore, the choice is based on patient preference, presence of coexisting interstitial lung disease or liver disease, commitment to limit alcohol consumption, and thiopurine methyltransferase status. Most patients need prolonged therapy.
In a randomized clinical trial, concomitant therapy with prednisone and azathioprine resulted in better functional outcomes and a significantly lower prednisone dose requirement for maintenance therapy at 3 years than with prednisone alone.43,44 Although no such randomized study has been conducted using methotrexate, several retrospective studies have demonstrated 70% to 80% response rates, including those for whom monotherapy with glucocorticoids had failed.45,46 The combination of methotrexate and azathioprine may be beneficial in patients who previously had inadequate responses to either of these agents alone.47
For severe pulmonary involvement associated with antisynthetase syndrome, monthly intravenous infusion of cyclophosphamide has been shown to be effective.48,49
Some recent studies established the role of tacrolimus in the treatment of both interstitial lung disease and myositis associated with antisynthetase syndrome.29 Cyclosporine has also been successfully used in a case of interstitial lung disease associated with anti Jo-1 syndrome.30
Rituximab, a monoclonal antibody to Blymphocyte antigen CD20, can also be used successfully in refractory disease,31 including refractory interstitial lung disease.32
In an open-label prospective study, polymyositis refractory to glucocorticoids and multiple conventional immunosuppressive agents responded well to high-dose intravenous immune globulin in the short term.50 However, the antisynthetase antibody status in this cohort was unknown; therefore, no definite conclusion could be drawn about the efficacy of intravenous immune globulin specifically in antisynthetase syndrome.
General measures
In patients with profound muscle weakness, physical therapy and rehabilitation should begin early. The goal is to reduce further muscle wasting from disuse and prevent muscle contractures. Patients with oropharyngeal and esophageal dysmotility should be advised about aspiration precautions and may need a swallow evaluation by a speech therapist; some may need temporary parenteral hyper-alimentation or J-tube insertion.
PROGNOSIS
If skeletal muscle involvement is the sole manifestation of antisynthetase syndrome, patients usually respond to glucocorticoids and immunosuppressive therapy and do fairly well. However, the outcome is not so promising when patients also develop interstitial lung disease, and the severity and type of lung injury usually determine the prognosis. As expected, patients with a progressive course of interstitial lung disease fare poorly, whereas those with a nonprogressive course tend to do relatively better. Older age at onset (> 60 years), presence of a malignancy, and a negative antinuclear antibody test are associated with a poor prognosis.7
Acknowledgment: The authors are grateful to Dr. Stephen Hatem, MD, staff radiologist, musculoskeletal radiology, Cleveland Clinic Imaging Institute, for help in the preparation of the magnetic resonance images. We also thank Dr. Steven Shook, MD, staff neurologist, Cleveland Clinic Neurological Institute, for help in summarizing the EMG findings.
- Katzap E, Barilla-LaBarca ML, Marder G. Antisynthetase syndrome. Curr Rheumatol Rep 2011; 13:175–181.
- Nishikai M, Reichlin M. Heterogeneity of precipitating antibodies in polymyositis and dermatomyositis. Characterization of the Jo-1 antibody system. Arthritis Rheum 1980; 23:881–888.
- Nagaraju K, Lundberg IE. Inflammatory diseases of muscle and other myopathies. In:Firestein GS, Budd RC, Harris ED, McInnes IB, Ruddy S, Sergent JS, editors. Kelley’s Textbook of Rheumatology. Philadelphia, PA: Saunders; 2008:1353–1380.
- Brouwer R, Hengstman GJ, Vree Egberts W, et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis 2001; 60:116–123.
- Vázquez-Abad D, Rothfield NF. Sensitivity and specificity of anti-Jo-1 antibodies in autoimmune diseases with myositis. Arthritis Rheum 1996; 39:292–296.
- Arnett FC, Targoff IN, Mimori T, Goldstein R, Warner NB, Reveille JD. Interrelationship of major histocompatibility complex class II alleles and autoantibodies in four ethnic groups with various forms of myositis. Arthritis Rheum 1996; 39:1507–1518.
- Dugar M, Cox S, Limaye V, Blumbergs P, Roberts-Thomson PJ. Clinical heterogeneity and prognostic features of South Australian patients with antisynthetase autoantibodies. Intern Med J 2011; 41:674–679.
- Friedman AW, Targoff IN, Arnett FC. Interstitial lung disease with autoantibodies against aminoacyl-tRNA synthetases in the absence of clinically apparent myositis. Semin Arthritis Rheum 1996; 26:459–467.
- Yoshifuji H, Fujii T, Kobayashi S, et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity 2006; 39:233–241.
- Tillie-Leblond I, Wislez M, Valeyre D, et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset. Thorax 2008; 63:53–59.
- Targoff IN. Update on myositis-specific and myositis-associated autoantibodies. Curr Opin Rheumatol 2000; 12:475–481.
- Ancuta CM, Ancuta E, Chirieac RM. Aminoacyl-tRNA synthetases in idiopathic inflammatory myopathies: an update on immunopathogenic significance, clinical and therapeutic implications. In:Gran JT, editor. Idiopathic Inflammatory Myopathies - Recent Developments. Rijeka, Croatia: InTech; 2011:77–90.
- Kajihara M, Kuwana M, Tokuda H, et al. Myositis and interstitial lung disease associated with autoantibody to a transfer RNA-related protein Wa. J Rheumatol 2000; 27:2707–2710.
- Yamasaki Y, Yamada H, Nozaki T, et al. Unusually high frequency of autoantibodies to PL-7 associated with milder muscle disease in Japanese patients with polymyositis/dermatomyositis. Arthritis Rheum 2006; 54:2004–2009.
- Ascherman DP. The role of Jo-1 in the immunopathogenesis of polymyositis: current hypotheses. Curr Rheumatol Rep 2003; 5:425–430.
- Yousem SA, Gibson K, Kaminski N, Oddis CV, Ascherman DP. The pulmonary histopathologic manifestations of the anti-Jo-1 tRNA synthetase syndrome. Mod Pathol 2010; 23:874–880.
- Chatterjee S, Farver C. Severe pulmonary hypertension in anti-Jo-1 syndrome. Arthritis Care Res (Hoboken) 2010; 62:425–429.
- Minai OA. Pulmonary hypertension in polymyositis-dermatomyositis: clinical and hemodynamic characteristics and response to vasoactive therapy. Lupus 2009; 18:1006–1010.
- Bugatti L, De Angelis R, Filosa G, Salaffi F. Bilateral, asymptomatic scaly and fissured cutaneous lesions of the fingers in a patient presenting with myositis. Indian J Dermatol Venereol Leprol 2005; 71:137–138.
- Mumm GE, McKown KM, Bell CL. Antisynthetase syndrome presenting as rheumatoid-like polyarthritis. J Clin Rheumatol 2010; 16:307–312.
- Hirakata M, Mimori T, Akizuki M, Craft J, Hardin JA, Homma M. Autoantibodies to small nuclear and cytoplasmic ribonucleoproteins in Japanese patients with inflammatory muscle disease. Arthritis Rheum 1992; 35:449–456.
- Love LA, Leff RL, Fraser DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991; 70:360–374.
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case-control study. Br J Dermatol 2001; 144:825–831.
- Legault D, McDermott J, Crous-Tsanaclis AM, Boire G. Cancer-associated myositis in the presence of anti-Jo1 autoantibodies and the antisynthetase syndrome. J Rheumatol 2008; 35:169–171.
- Selva-O’Callaghan A, Trallero-Araguás E, Grau-Junyent JM, Labrador-Horrillo M. Malignancy and myositis: novel autoantibodies and new insights. Curr Opin Rheumatol 2010; 22:627–632.
- Casciola-Rosen L, Hall JC, Mammen AL, Christopher-Stine L, Rosen A. Isolated elevation of aldolase in the serum of myositis patients: a potential biomarker of damaged early regenerating muscle cells. Clin Exp Rheumatol 2012; 30:548–553.
- Shovman O, Gilburd B, Barzilai O, et al. Evaluation of the BioPlex 2200 ANA screen: analysis of 510 healthy subjects: incidence of natural/predictive autoantibodies. Ann N Y Acad Sci 2005; 1050:380–388.
- Zampieri S, Ghirardello A, Iaccarino L, Tarricone E, Gambari PF, Doria A. Anti-Jo-1 antibodies. Autoimmunity 2005; 38:73–78.
- Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 2005; 52:2439–2446.
- Jankowska M, Butto B, Debska-Slizien A, Rutkowski B. Beneficial effect of treatment with cyclosporin A in a case of refractory antisynthetase syndrome. Rheumatol Int 2007; 27:775–780.
- Limaye V, Hissaria P, Liew CL, Koszyka B. Efficacy of rituximab in refractory antisynthetase syndrome. Intern Med J 2012; 42:e4–e7.
- Marie I, Dominique S, Janvresse A, Levesque H, Menard JF. Rituximab therapy for refractory interstitial lung disease related to antisynthetase syndrome. Respir Med 2012; 106:581–587.
- Rutjes SA, Vree Egberts WT, Jongen P, Van Den Hoogen F, Pruijn GJ, Van Venrooij WJ. Anti-Ro52 antibodies frequently co-occur with anti-Jo-1 antibodies in sera from patients with idiopathic inflammatory myopathy. Clin Exp Immunol 1997; 109:32–40.
- La Corte R, Lo Mo Naco A, Locaputo A, Dolzani F, Trotta F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity 2006; 39:249–253.
- Váncsa A, Csípo I, Németh J, Dévényi K, Gergely L, Dankó K. Characteristics of interstitial lung disease in SS-A positive/Jo-1 positive inflammatory myopathy patients. Rheumatol Int 2009; 29:989–994.
- Bohan A, Peter JB, Bowman RL, Pearson CM. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore) 1977; 56:255–286.
- Reimers CD, Finkenstaedt M. Muscle imaging in inflammatory myopathies. Curr Opin Rheumatol 1997; 9:475–485.
- O’Connell MJ. Whole-body MR imaging in the diagnosis of polymyositis. Am J Roentgenol 2002; 179:967–971.
- Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000; 68:472–478.
- Fromageot C, Lofaso F, Annane D, et al. Supine fall in lung volumes in the assessment of diaphragmatic weakness in neuromuscular disorders. Arch Phys Med Rehabil 2001; 82:123–128.
- Leslie KO. Historical perspective: a pathologic approach to the classification of idiopathic interstitial pneumonias. Chest 2005; 128(suppl 1):513S–519S.
- Nicholson AG, Colby TV, du Bois RM, Hansell DM, Wells AU. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 2000; 162:2213–2217.
- Bunch TW, Worthington JW, Combs JJ, Ilstrup DM, Engel AG. Azathioprine with prednisone for polymyositis. A controlled, clinical trial. Ann Intern Med 1980; 92:365–369.
- Bunch TW. Prednisone and azathioprine for polymyositis: long-term followup. Arthritis Rheum 1981; 24:45–48.
- Metzger AL, Bohan A, Goldberg LS, Bluestone R, Pearson CM. Polymyositis and dermatomyositis: combined methotrexate and corticosteroid therapy. Ann Intern Med 1974; 81:182–189.
- Joffe MM, Love LA, Leff RL, et al. Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med 1993; 94:379–387.
- Villalba L, Hicks JE, Adams EM, et al. Treatment of refractory myositis: a randomized crossover study of two new cytotoxic regimens. Arthritis Rheum 1998; 41:392–399.
- Yamasaki Y, Yamada H, Yamasaki M, et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford) 2007; 46:124–130.
- al-Janadi M, Smith CD, Karsh J. Cyclophosphamide treatment of interstitial pulmonary fibrosis in polymyositis/dermatomyositis. J Rheumatol 1989; 16:1592–1596.
- Cherin P, Pelletier S, Teixeira A, et al. Results and long-term followup of intravenous immunoglobulin infusions in chronic, refractory polymyositis: an open study with thirty-five adult patients. Arthritis Rheum 2002; 46:467–474.
A 66-year-old man was initially seen in clinic in March 2004 with a 5-month history of polyarthritis (affecting the finger joints, wrists, and knees) and several hours of morning stiffness. He also had significant proximal muscle weakness, progressive exertional dyspnea, and a nonproductive cough. There was no history of fever, chills, rash, dysphagia, or sicca symptoms. Findings on initial tests:
- His creatine kinase level was 700 U/L (reference range 30–220), which later rose to 1,664 U/L.
- He was positive for antinuclear antibody with a 5.7 optical density ratio (normal < 1.5) and for anti-Jo-1 antibody.
- An electromyogram was consistent with a necrotizing myopathy. Left rectus femoris biopsy revealed scattered degenerating and regenerating muscle fibers but no evidence of endomysial inflammation.
- On pulmonary function testing, his forced vital capacity was 80% of predicted, and his carbon monoxide diffusion capacity was 67% of predicted.
- High-resolution computed tomography revealed evidence of interstitial lung disease, characterized by bilateral patchy ground-glass opacities suggestive of active alveolitis, most extensive at the lung bases.
- Bronchoalveolar lavage indicated alveolitis, and transbronchial biopsy revealed pathologic changes consistent with cryptogenic organizing pneumonia. All cultures were negative.
This constellation of clinical manifestations, including myositis, interstitial lung disease, and polyarthritis, along with positive anti-Jo-1 antibody, confirmed the diagnosis of antisynthetase syndrome.
In June 2004, for his interstitial lung disease, he was started on daily oral cyclophosphamide along with high-dose oral prednisone. Three months later the skin of the tips and radial margins of his fingers started thickening and cracking, the appearance of which is classically described as “mechanic’s hands,” a well-described manifestation of antisynthetase syndrome (Figure 1).
Cyclophosphamide was continued for about a year. Subsequently, along with prednisone, he sequentially received various other immunosuppressive medications (methotrexate, tacrolimus, mycophenolate mofetil, and rituximab) over the next few years in an attempt to control his progressive interstitial lung disease. All of these agents were only partially and temporarily effective. Ultimately, despite all of these therapies, as his interstitial lung disease progressed, he needed supplemental oxygen and enrollment in a pulmonary rehabilitation program.
In March 2010, he was admitted with worsening dyspnea and significant peripheral edema and was found to have severe pulmonary arterial hypertension. He was started on bosentan. Eight months later sildenafil was added for progressive pulmonary arterial hypertension. However, his oxygenation status continued to decline.
In July 2011, he presented with chills, increasing shortness of breath, and a mild productive cough. As he was severely hypoxic, he was admitted to the intensive care unit and started on mechanical ventilation and broad-spectrum antibiotics. Despite escalation of oxygen therapy, his respiratory status rapidly deteriorated, and he developed hypotension requiring vasopressors. He ultimately died of cardiac arrest secondary to respiratory failure.
A CONSTELLATION OF MANIFESTATIONS
Antisynthetase syndrome, associated with anti-aminoacyl-transfer RNA (tRNA) synthetase antibodies, is characterized by a constellation of manifestations that include myositis, interstitial lung disease, mechanic’s hands, fever, Raynaud phenomenon, and nonerosive symmetric polyarthritis of the small joints.1
Anti-Jo-1 antibody (anti-histidyl-tRNA synthetase) is the most common of the antibodies and also was the first one to be identified (Table 1). It was named after John P, a patient with polymyositis and interstitial lung disease, in whom it was first detected in 1980.2 The onset of the syndrome associated with anti-Jo-1 antibody is often acute, and the myositis is usually steroid-responsive. However, not uncommonly, severe disease can develop over time, with a tendency to relapse and with a poor long-term prognosis.
RARE BUT UNDERRECOGNIZED
The true population prevalence of antisynthetase syndrome is unknown. Because this syndrome is rare, comprehensive epidemiologic studies are difficult to perform.
In several retrospective studies, the annual incidence of idiopathic inflammatory myopathies has been reported to be 2 to 10 new cases per million adults per year.3 Antisynthetase antibodies are detected in 20% to 40% of such cases.4–6 The disease is two to three times more common in women than in men.7
Early diagnosis is difficult because the clinical presentation is varied and often nonspecific, clinically milder disease may escape detection, and many general practitioners lack familiarity with this syndrome and consequently do not recognize it. Moreover, tests for myositis-specific antibodies (including antisynthetase antibodies) are often not ordered in the evaluation of myositis, and hence the diagnosis of antisynthetase syndrome cannot be substantiated. Furthermore, interstitial lung disease can predominate or can be the sole manifestation in the absence of clinically apparent myositis,8–10 and patients can be misdiagnosed as having idiopathic pulmonary fibrosis when underlying antisynthetase syndrome is not suspected. This distinction may be important because these conditions differ in their pathology and treatment. Histologically, the predominant pattern of lung injury in idiopathic pulmonary fibrosis is “usual interstitial pneumonia” which does not respond to immunosuppressive therapy, and hence lung transplantation is the only therapeutic option. On the other hand, in antisynthetase syndrome, the usual pattern of lung injury is “nonspecific interstitial pneumonia,” in which immunosuppressive therapy has a role.
Anti-Jo-1 antibody is detected in 15% to 25% of patients with polymyositis and in up to 70% of myositis patients with concomitant interstitial lung disease.11 Autoantibodies to seven other, less frequently targeted, aminoacyl tRNA synthetases have also been described in patients with polymyositis and interstitial lung disease (Table 1).11,12 In addition, an autoantibody to a 48-kDa transfer RNA-related protein (Wa) has been described.13 These non-Jo-1 antisynthetase antibodies are detected in only about 3% of myositis patients.14
ROLE OF ANTISYNTHETASE ANTIBODIES
Synthetases play a central role in protein synthesis by catalyzing the acetylation of tRNAs. The propensity of organ involvement in antisynthetase syndrome suggests that tissue-specific changes in muscle or lung lead to the production of unique forms of target autoantigens, the aminoacyl-tRNA synthetases. There is evidence that these enzymes themselves may be involved in recruiting both antigen-presenting and inflammatory cells to the site of muscle or lung injury.15 However, the molecular pathway that initiates and propagates this autoimmune response and the specific role of the antisynthetase antibodies in the pathogenesis of this syndrome are presently unknown.
SIX SALIENT CLINICAL FEATURES
There are six predominant clinical manifestations, which may be present at disease onset or appear later as the disease progresses:
- Fever
- Myositis
- Interstitial lung disease
- Mechanic’s hands
- Raynaud phenomenon
- Inflammatory polyarthritis.
There is considerable clinical heterogeneity, and one or other manifestation can predominate or can be the only expression of the syndrome. Furthermore, in the same patient, the individual features can prevail at different times and may develop years after onset of the disease. Therefore, in addition to patients with myositis, it would be important to suspect antisynthetase syndrome in patients presenting with isolated lung involvement (amyopathic interstitial lung disease), as there are therapeutic implications. Studies have demonstrated the efficacy of immunosuppressive agents in interstitial lung disease associated with antisynthetase syndrome (where the predominant pattern of lung injury is “nonspecific interstitial pneumonitis”), whereas lung transplantation has so far been the only treatment option in idiopathic pulmonary fibrosis.
Fever
About 20% of patients have a fever at disease onset or associated with relapses. Sometimes the fever can persist until treatment of antisynthetase syndrome is started.
Myositis
Muscle disease is seen in more than 90% of patients with anti-Jo-1 antisynthetase syndrome. It can be subclinical (in the absence of proximal myopathy), manifested by transient creatine kinase elevation only, which may normalize after therapy is initiated.
However, more commonly, patients develop profound proximal muscle weakness and sometimes muscle pain (Table 2). Weakness of the striated muscles of the upper esophagus, cricopharyngeus, and hypopharynx may cause dysphagia and makes these patients susceptible to aspiration pneumonia. Diaphragmatic and intercostal muscle weakness can contribute to shortness of breath in some patients. Myocarditis has also been reported.
Pulmonary disease
Interstitial lung disease develops in most patients with anti-Jo-1 antisynthetase syndrome, with a reported prevalence of about 90% in one series.16 Patients often present with acute, subacute, or insidious onset of exertional dyspnea. Sometimes there is an intractable nonproductive cough.
At the outset of antisynthetase syndrome, if the patient is profoundly weak because of myopathy or has inflammatory polyarthritis, mobility is significantly compromised, and exertional dyspnea may not be experienced. However, as the interstitial lung disease progresses, shortness of breath becomes overt, more so when the patient’s level of activity improves with treatment of myositis.
Inspiratory crackles on auscultation of the lung bases or changes on chest radiography are relatively insensitive findings and can miss early interstitial lung disease. Therefore, if antisynthetase syndrome is suspected or diagnosed, a baseline pulmonary function test (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease, and the diagnosis can then be confirmed with thoracic high-resolution computed tomography (Figure 2).
Pulmonary hypertension. Recent studies indicate that, similar to patients with other autoimmune rheumatic diseases, pulmonary hypertension can develop in patients with antisynthetase syndrome, with or without concomitant interstitial lung disease.17,18 This complication occurred in the case presented here. It has been found that when pulmonary hypertension coexists with interstitial lung disease, its degree may not correlate with the severity of the latter.17 Additionally, pulmonary hypertension, when present, has been found to contribute independently to prognosis and survival.
Mechanic’s hands
In about 30% of patients, the skin of the tips and margins of the fingers becomes thickened, hyperkeratotic, and fissured, the appearance of which is classically described as mechanic’s hands. It is a common manifestation of antisynthetase syndrome and is particularly prominent on the radial side of the index fingers (Figure 1). Biopsy of affected skin shows an interface psoriasiform dermatitis.19 In addition, some dermatomyositis patients with Gottron papules and a heliotrope rash have antisynthetase antibodies.
Raynaud phenomenon
Raynaud phenomenon develops in about 40% of patients. Some have nailfold capillary abnormalities.20 However, persistent or severe digital ischemia leading to digital ulceration or infarction is uncommon.21
Inflammatory arthritis
Arthralgias and arthritis are common (50%), the most common form being a symmetric polyarthritis of the small joints of the hands and feet. It is typically nonerosive but can sometimes be erosive and destructive.20
Because inflammatory arthritis mimics rheumatoid arthritis, antisynthetase syndrome should be considered in rheumatoid factor-negative patients presenting with polyarthritis.
ASSOCIATION WITH MALIGNANCY
Traditional teaching has been that antisynthetase antibody is protective against an underlying malignancy.22,23 However, several recently published case studies have reported various malignancies occurring within 6 to 12 months of the diagnosis of antisynthetase syndrome.7,24 The debate as to whether these are chance associations or causal (a paraneoplastic phenomenon) has not been resolved at this time.24
It is now recommended that patients with antisynthetase syndrome be screened for malignancies as appropriate for the patient’s age and sex. Screening should include a careful history and physical examination, complete blood cell count, comprehensive metabolic panel, chest radiography, mammography, and a gynecologic examination for women.25 If abnormalities are found, a more thorough evaluation for cancer is appropriate.
DIAGNOSIS
Muscle enzyme levels are often elevated
Muscle enzymes (creatine kinase and aldolase) are often elevated. Serum creatine kinase levels can range between 5 to 50 times the upper limit of normal. In an established case, creatine kinase levels along with careful manual muscle strength testing may help evaluate myositis activity. However, in chronic and advanced disease, creatine kinase may be within the normal range despite active myositis, partly because of extensive loss of muscle mass. In myositis, it may be prudent to check both creatine kinase and aldolase; sometimes only serum aldolase level rises, when immune-mediated injury predominantly affects the early regenerative myocytes.26
Judicious use of autoantibody testing
The characteristic clinical presentation is the initial clue to the diagnosis of antisynthetase syndrome, which is then supported by serologic testing.
Injudicious testing for a long list of antibodies should be avoided, as the cost is considerable and it does not influence further management. However, ordering an anti-Jo-1 antibody test in the correct clinical setting is appropriate, as it has high specificity,27,28 and thus can help establish or refute the clinical suspicion of antisynthetase syndrome.
Screening pulmonary function testing and thoracic high-resolution computed tomography for all patients with polymyositis or dermatomyositis is not considered “standard of care” and will likely not be reimbursed by third-party payers. However, in a patient with symptoms and signs of myositis, the presence of an antisynthetase antibody should prompt screening for occult interstitial lung disease, even in the absence of symptoms. As lung disease ultimately determines the prognosis in antisynthetase syndrome, early diagnosis and management is the key. Therefore, these tests would likely be approved to establish the diagnosis of interstitial lung disease and evaluate its severity.
If a myositis patient is also found to have interstitial lung disease or develops mechanic’s hands, the likely diagnosis is antisynthetase syndrome, which can be confirmed by serologic testing for antisynthetase antibodies. Interstitial lung disease in antisynthetase syndrome is often from “nonspecific interstitial pneumonitis”; therefore, medications tested and proven effective for this condition should be approved and reimbursed by payers.29–32
The coexistence of myositis and interstitial lung disease increases the sensitivity of anti-Jo-1 antibody.11 Thus, the clinician can have more confidence in early recognition and initiation of aggressive but targeted disease-modifying therapy.
Various methods can be used for detecting antisynthetase antibodies, with comparable results.28 Anti-Jo-1 antibody testing costs about $140. If that test is negative and antisynthetase syndrome is still suspected, then testing for the non-Jo-1 antisynthetase antibodies may be justified (Table 1). Though the cost of this panel of autoantibodies is about $300, it helps to confirm the diagnosis, and it influences the choice of second-line immunosuppressive agents such as tacrolimus29 and rituximab32 in patients resistant to conventional immunosuppressive agents such as azathioprine and methotrexate.
Often, anti-Ro52 SS-A antibodies are present concurrently in patients with anti-Jo-1 syndrome.33 In observational studies in patients with anti-Jo-1 antibody-associated interstitial lung disease, coexistence of anti-Ro52 SS-A antibody tended to predict a worse pulmonary outcome than in those with anti-Jo-1 antibody alone.34,35
Electromyography
Electromyography not only helps differentiate between myopathic and neuropathic weakness, but it may also support the diagnosis of “inflammatory” myopathy as suggested by prominent muscle membrane irritability (fibrillations, positive sharp waves) and abnormal motor unit action potentials (spontaneous activity showing small, short, polyphasic potentials and early recruitment). However, the findings can be nonspecific, and may even be normal in 10% to 15% of patients.36 Electromyographic abnormalities are most consistently observed in weak proximal muscles, and electromyography is also helpful in selecting a muscle for biopsy. Although no single electromyographic pattern is considered diagnostic for inflammatory myopathy, abnormalities are present in around 90% of patients (Table 2).3
Magnetic resonance imaging
Magnetic resonance imaging may show increased signal intensity in the affected muscles and surrounding tissues (Figure 3).37 Because it lacks sensitivity and specificity, magnetic resonance imaging is not helpful in diagnosing the disease. However, in the correct clinical setting, it may be used to guide muscle biopsy, and it can help in monitoring the disease progress.38
Muscle histopathology
Muscle biopsy, though often helpful, is not always diagnostic, and antisynthetase syndrome should still be suspected in the right clinical context, even in the absence of characteristic pathologic changes.
Biopsy of sites recently studied by electromyography should be avoided, and if the patient has undergone electromyography recently, the contralateral side should be selected for biopsy.
Reports of histopathologic findings in muscle biopsies in patients with antisynthetase syndrome document inflammatory myopathic features (Figure 4). In a series of patients with anti-Jo-1 syndrome, inflammation was noted in all cases, predominantly perimysial in location, with occasional endomysial and perivascular inflammation.39 Many of the inflammatory cells seen were macrophages and lymphocytes, in contrast to the predominantly lymphocytic infiltrates described in classic polymyositis and dermatomyositis. Perifascicular atrophy, similar to what is seen in dermatomyositis, was encountered; however, vascular changes, typical of dermatomyositis, were absent. Occasional degenerating and regenerating muscle fibers were also observed in most cases. Additionally, a characteristic perimysial connective tissue fragmentation was described, a feature less often seen in classic polymyositis and dermatomyositis.39
Pulmonary function testing
If antisynthetase syndrome is suspected or diagnosed, baseline pulmonary function testing (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease (reduced forced vital capacity and carbon monoxide diffusion capacity), and the diagnosis will be substantiated on thoracic high-resolution computed tomography. Respiratory muscle weakness can be detected with upright and supine spirometry.40 Weakness of these muscles contributes to shortness of breath, and patients may need ventilatory support.
Thoracic high-resolution computed tomography
Different patterns of lung injury can be seen in antisynthetase syndrome. Diffuse ground-glass opacification may suggest a nonspecific interstitial pneumonitis pattern, which is the most common form of interstitial lung disease (Figure 2), whereas coarse reticulation or honeycombing correlates with a usual interstitial pneumonitis pattern. Patchy consolidation or air-space disease can occur if cryptogenic organizing pneumonia is the predominant pattern of lung injury.
Swallowing evaluation
A comprehensive swallowing evaluation by a speech therapist may be necessary for evaluation of dysphagia (from oropharyngeal and striated esophageal muscle weakness) and determination of aspiration risk (Table 2).
Lung histopathology
If necessary, a surgical lung biopsy is needed to document the pathologic pattern of injury, including the amount of fibrosis in the lung. Historically, in idiopathic inflammatory myopathy patients in general, this has taken the form of usual interstitial pneumonia, organizing pneumonia, or diffuse alveolar damage.41 With the emergence of the definition of nonspecific interstitial pneumonitis and fibrosis as a documented and accepted pattern, more studies have found this to be the most common pattern of lung injury.16 It is characterized by diffuse involvement of the lung by an interstitial chronic inflammatory infiltrate, a cellular type of nonspecific interstitial pneumonitis that progresses in a uniform pattern to a fibrotic type (Figure 5). This form of fibrosis rarely results in significant remodeling, so-called honeycomb changes. In addition, anti-Jo-1 antibody patients may also have an increased incidence of acute lung injury, including acute diffuse alveolar damage that is often superimposed on the underlying chronic lung disease.42
In patients with pulmonary hypertension, histopathologic studies of the muscular pulmonary arteries often show moderate intimal fibroplasia, suggesting that a pulmonary arteriopathy with intimal thickening and luminal narrowing develops in some of these patients (Figure 5), independent of chronic hypoxic pulmonary vasoconstriction or vascular obstruction due to its entrapment within fibrotic lung tissue.17
TREATMENT
Glucocorticoids are the mainstay
Glucocorticoids are considered the mainstay of treatment. Patients should be advised that long-term use of glucocorticoids is necessary, though the response is variable. It is also important to discuss possible side effects of long-term glucocorticoid use.
Standard practice is to initiate treatment with high doses for the first 4 to 6 weeks to achieve disease control, followed by a slow taper over the next 9 to 12 months to the lowest effective dose to maintain remission. If the patient is profoundly weak, especially with respiratory muscle weakness or significant dysphagia and aspiration risk, hospital admission for intravenous methylprednisolone 1,000 mg daily for 3 to 5 days may be necessary. Otherwise, oral prednisone 1 mg/kg/day would be the usual starting dose.
If the patient’s muscle strength initially improves and then declines weeks to months later despite adequate therapy, glucocorticoid-induced myopathy should be suspected, especially if the muscle enzymes are within the reference range. This is more likely to occur if high-dose prednisone is continued for more than 6 to 8 weeks.
Improvement in muscle strength, which can take several weeks to several months, is a more reliable indicator of response to therapy than the serum creatine kinase level, which may take much longer to normalize. Relying on normalization of the creatine kinase level alone may lead to unnecessary prolongation of high-dose glucocorticoid therapy. It may take several months for the muscle enzymes to normalize, and there is usually a time lag between normalization of muscle enzymes and complete recovery of muscle strength.
Long-term use of high-dose prednisone leads to glucocorticoid-induced osteoporosis. Therefore, patients should receive osteoporosis prophylaxis including antiresorptive therapy with a bisphosphonate. In addition, prophylaxis against Pneumocystis jirovecii is indicated for patients treated with high-dose glucocorticoids.
Additional immunosuppressive agents
Although glucocorticoids are considered the mainstay of treatment, additional immunosuppressive agents such as azathioprine and methotrexate are often required, both as glucocorticoid-sparing agents and to achieve adequate disease control.10 Addition of such agents from the outset is particularly necessary in patients with profound muscle weakness or those who have concomitant symptomatic interstitial lung disease.
No randomized controlled trial comparing azathioprine and methotrexate has been conducted to date. Therefore, the choice is based on patient preference, presence of coexisting interstitial lung disease or liver disease, commitment to limit alcohol consumption, and thiopurine methyltransferase status. Most patients need prolonged therapy.
In a randomized clinical trial, concomitant therapy with prednisone and azathioprine resulted in better functional outcomes and a significantly lower prednisone dose requirement for maintenance therapy at 3 years than with prednisone alone.43,44 Although no such randomized study has been conducted using methotrexate, several retrospective studies have demonstrated 70% to 80% response rates, including those for whom monotherapy with glucocorticoids had failed.45,46 The combination of methotrexate and azathioprine may be beneficial in patients who previously had inadequate responses to either of these agents alone.47
For severe pulmonary involvement associated with antisynthetase syndrome, monthly intravenous infusion of cyclophosphamide has been shown to be effective.48,49
Some recent studies established the role of tacrolimus in the treatment of both interstitial lung disease and myositis associated with antisynthetase syndrome.29 Cyclosporine has also been successfully used in a case of interstitial lung disease associated with anti Jo-1 syndrome.30
Rituximab, a monoclonal antibody to Blymphocyte antigen CD20, can also be used successfully in refractory disease,31 including refractory interstitial lung disease.32
In an open-label prospective study, polymyositis refractory to glucocorticoids and multiple conventional immunosuppressive agents responded well to high-dose intravenous immune globulin in the short term.50 However, the antisynthetase antibody status in this cohort was unknown; therefore, no definite conclusion could be drawn about the efficacy of intravenous immune globulin specifically in antisynthetase syndrome.
General measures
In patients with profound muscle weakness, physical therapy and rehabilitation should begin early. The goal is to reduce further muscle wasting from disuse and prevent muscle contractures. Patients with oropharyngeal and esophageal dysmotility should be advised about aspiration precautions and may need a swallow evaluation by a speech therapist; some may need temporary parenteral hyper-alimentation or J-tube insertion.
PROGNOSIS
If skeletal muscle involvement is the sole manifestation of antisynthetase syndrome, patients usually respond to glucocorticoids and immunosuppressive therapy and do fairly well. However, the outcome is not so promising when patients also develop interstitial lung disease, and the severity and type of lung injury usually determine the prognosis. As expected, patients with a progressive course of interstitial lung disease fare poorly, whereas those with a nonprogressive course tend to do relatively better. Older age at onset (> 60 years), presence of a malignancy, and a negative antinuclear antibody test are associated with a poor prognosis.7
Acknowledgment: The authors are grateful to Dr. Stephen Hatem, MD, staff radiologist, musculoskeletal radiology, Cleveland Clinic Imaging Institute, for help in the preparation of the magnetic resonance images. We also thank Dr. Steven Shook, MD, staff neurologist, Cleveland Clinic Neurological Institute, for help in summarizing the EMG findings.
A 66-year-old man was initially seen in clinic in March 2004 with a 5-month history of polyarthritis (affecting the finger joints, wrists, and knees) and several hours of morning stiffness. He also had significant proximal muscle weakness, progressive exertional dyspnea, and a nonproductive cough. There was no history of fever, chills, rash, dysphagia, or sicca symptoms. Findings on initial tests:
- His creatine kinase level was 700 U/L (reference range 30–220), which later rose to 1,664 U/L.
- He was positive for antinuclear antibody with a 5.7 optical density ratio (normal < 1.5) and for anti-Jo-1 antibody.
- An electromyogram was consistent with a necrotizing myopathy. Left rectus femoris biopsy revealed scattered degenerating and regenerating muscle fibers but no evidence of endomysial inflammation.
- On pulmonary function testing, his forced vital capacity was 80% of predicted, and his carbon monoxide diffusion capacity was 67% of predicted.
- High-resolution computed tomography revealed evidence of interstitial lung disease, characterized by bilateral patchy ground-glass opacities suggestive of active alveolitis, most extensive at the lung bases.
- Bronchoalveolar lavage indicated alveolitis, and transbronchial biopsy revealed pathologic changes consistent with cryptogenic organizing pneumonia. All cultures were negative.
This constellation of clinical manifestations, including myositis, interstitial lung disease, and polyarthritis, along with positive anti-Jo-1 antibody, confirmed the diagnosis of antisynthetase syndrome.
In June 2004, for his interstitial lung disease, he was started on daily oral cyclophosphamide along with high-dose oral prednisone. Three months later the skin of the tips and radial margins of his fingers started thickening and cracking, the appearance of which is classically described as “mechanic’s hands,” a well-described manifestation of antisynthetase syndrome (Figure 1).
Cyclophosphamide was continued for about a year. Subsequently, along with prednisone, he sequentially received various other immunosuppressive medications (methotrexate, tacrolimus, mycophenolate mofetil, and rituximab) over the next few years in an attempt to control his progressive interstitial lung disease. All of these agents were only partially and temporarily effective. Ultimately, despite all of these therapies, as his interstitial lung disease progressed, he needed supplemental oxygen and enrollment in a pulmonary rehabilitation program.
In March 2010, he was admitted with worsening dyspnea and significant peripheral edema and was found to have severe pulmonary arterial hypertension. He was started on bosentan. Eight months later sildenafil was added for progressive pulmonary arterial hypertension. However, his oxygenation status continued to decline.
In July 2011, he presented with chills, increasing shortness of breath, and a mild productive cough. As he was severely hypoxic, he was admitted to the intensive care unit and started on mechanical ventilation and broad-spectrum antibiotics. Despite escalation of oxygen therapy, his respiratory status rapidly deteriorated, and he developed hypotension requiring vasopressors. He ultimately died of cardiac arrest secondary to respiratory failure.
A CONSTELLATION OF MANIFESTATIONS
Antisynthetase syndrome, associated with anti-aminoacyl-transfer RNA (tRNA) synthetase antibodies, is characterized by a constellation of manifestations that include myositis, interstitial lung disease, mechanic’s hands, fever, Raynaud phenomenon, and nonerosive symmetric polyarthritis of the small joints.1
Anti-Jo-1 antibody (anti-histidyl-tRNA synthetase) is the most common of the antibodies and also was the first one to be identified (Table 1). It was named after John P, a patient with polymyositis and interstitial lung disease, in whom it was first detected in 1980.2 The onset of the syndrome associated with anti-Jo-1 antibody is often acute, and the myositis is usually steroid-responsive. However, not uncommonly, severe disease can develop over time, with a tendency to relapse and with a poor long-term prognosis.
RARE BUT UNDERRECOGNIZED
The true population prevalence of antisynthetase syndrome is unknown. Because this syndrome is rare, comprehensive epidemiologic studies are difficult to perform.
In several retrospective studies, the annual incidence of idiopathic inflammatory myopathies has been reported to be 2 to 10 new cases per million adults per year.3 Antisynthetase antibodies are detected in 20% to 40% of such cases.4–6 The disease is two to three times more common in women than in men.7
Early diagnosis is difficult because the clinical presentation is varied and often nonspecific, clinically milder disease may escape detection, and many general practitioners lack familiarity with this syndrome and consequently do not recognize it. Moreover, tests for myositis-specific antibodies (including antisynthetase antibodies) are often not ordered in the evaluation of myositis, and hence the diagnosis of antisynthetase syndrome cannot be substantiated. Furthermore, interstitial lung disease can predominate or can be the sole manifestation in the absence of clinically apparent myositis,8–10 and patients can be misdiagnosed as having idiopathic pulmonary fibrosis when underlying antisynthetase syndrome is not suspected. This distinction may be important because these conditions differ in their pathology and treatment. Histologically, the predominant pattern of lung injury in idiopathic pulmonary fibrosis is “usual interstitial pneumonia” which does not respond to immunosuppressive therapy, and hence lung transplantation is the only therapeutic option. On the other hand, in antisynthetase syndrome, the usual pattern of lung injury is “nonspecific interstitial pneumonia,” in which immunosuppressive therapy has a role.
Anti-Jo-1 antibody is detected in 15% to 25% of patients with polymyositis and in up to 70% of myositis patients with concomitant interstitial lung disease.11 Autoantibodies to seven other, less frequently targeted, aminoacyl tRNA synthetases have also been described in patients with polymyositis and interstitial lung disease (Table 1).11,12 In addition, an autoantibody to a 48-kDa transfer RNA-related protein (Wa) has been described.13 These non-Jo-1 antisynthetase antibodies are detected in only about 3% of myositis patients.14
ROLE OF ANTISYNTHETASE ANTIBODIES
Synthetases play a central role in protein synthesis by catalyzing the acetylation of tRNAs. The propensity of organ involvement in antisynthetase syndrome suggests that tissue-specific changes in muscle or lung lead to the production of unique forms of target autoantigens, the aminoacyl-tRNA synthetases. There is evidence that these enzymes themselves may be involved in recruiting both antigen-presenting and inflammatory cells to the site of muscle or lung injury.15 However, the molecular pathway that initiates and propagates this autoimmune response and the specific role of the antisynthetase antibodies in the pathogenesis of this syndrome are presently unknown.
SIX SALIENT CLINICAL FEATURES
There are six predominant clinical manifestations, which may be present at disease onset or appear later as the disease progresses:
- Fever
- Myositis
- Interstitial lung disease
- Mechanic’s hands
- Raynaud phenomenon
- Inflammatory polyarthritis.
There is considerable clinical heterogeneity, and one or other manifestation can predominate or can be the only expression of the syndrome. Furthermore, in the same patient, the individual features can prevail at different times and may develop years after onset of the disease. Therefore, in addition to patients with myositis, it would be important to suspect antisynthetase syndrome in patients presenting with isolated lung involvement (amyopathic interstitial lung disease), as there are therapeutic implications. Studies have demonstrated the efficacy of immunosuppressive agents in interstitial lung disease associated with antisynthetase syndrome (where the predominant pattern of lung injury is “nonspecific interstitial pneumonitis”), whereas lung transplantation has so far been the only treatment option in idiopathic pulmonary fibrosis.
Fever
About 20% of patients have a fever at disease onset or associated with relapses. Sometimes the fever can persist until treatment of antisynthetase syndrome is started.
Myositis
Muscle disease is seen in more than 90% of patients with anti-Jo-1 antisynthetase syndrome. It can be subclinical (in the absence of proximal myopathy), manifested by transient creatine kinase elevation only, which may normalize after therapy is initiated.
However, more commonly, patients develop profound proximal muscle weakness and sometimes muscle pain (Table 2). Weakness of the striated muscles of the upper esophagus, cricopharyngeus, and hypopharynx may cause dysphagia and makes these patients susceptible to aspiration pneumonia. Diaphragmatic and intercostal muscle weakness can contribute to shortness of breath in some patients. Myocarditis has also been reported.
Pulmonary disease
Interstitial lung disease develops in most patients with anti-Jo-1 antisynthetase syndrome, with a reported prevalence of about 90% in one series.16 Patients often present with acute, subacute, or insidious onset of exertional dyspnea. Sometimes there is an intractable nonproductive cough.
At the outset of antisynthetase syndrome, if the patient is profoundly weak because of myopathy or has inflammatory polyarthritis, mobility is significantly compromised, and exertional dyspnea may not be experienced. However, as the interstitial lung disease progresses, shortness of breath becomes overt, more so when the patient’s level of activity improves with treatment of myositis.
Inspiratory crackles on auscultation of the lung bases or changes on chest radiography are relatively insensitive findings and can miss early interstitial lung disease. Therefore, if antisynthetase syndrome is suspected or diagnosed, a baseline pulmonary function test (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease, and the diagnosis can then be confirmed with thoracic high-resolution computed tomography (Figure 2).
Pulmonary hypertension. Recent studies indicate that, similar to patients with other autoimmune rheumatic diseases, pulmonary hypertension can develop in patients with antisynthetase syndrome, with or without concomitant interstitial lung disease.17,18 This complication occurred in the case presented here. It has been found that when pulmonary hypertension coexists with interstitial lung disease, its degree may not correlate with the severity of the latter.17 Additionally, pulmonary hypertension, when present, has been found to contribute independently to prognosis and survival.
Mechanic’s hands
In about 30% of patients, the skin of the tips and margins of the fingers becomes thickened, hyperkeratotic, and fissured, the appearance of which is classically described as mechanic’s hands. It is a common manifestation of antisynthetase syndrome and is particularly prominent on the radial side of the index fingers (Figure 1). Biopsy of affected skin shows an interface psoriasiform dermatitis.19 In addition, some dermatomyositis patients with Gottron papules and a heliotrope rash have antisynthetase antibodies.
Raynaud phenomenon
Raynaud phenomenon develops in about 40% of patients. Some have nailfold capillary abnormalities.20 However, persistent or severe digital ischemia leading to digital ulceration or infarction is uncommon.21
Inflammatory arthritis
Arthralgias and arthritis are common (50%), the most common form being a symmetric polyarthritis of the small joints of the hands and feet. It is typically nonerosive but can sometimes be erosive and destructive.20
Because inflammatory arthritis mimics rheumatoid arthritis, antisynthetase syndrome should be considered in rheumatoid factor-negative patients presenting with polyarthritis.
ASSOCIATION WITH MALIGNANCY
Traditional teaching has been that antisynthetase antibody is protective against an underlying malignancy.22,23 However, several recently published case studies have reported various malignancies occurring within 6 to 12 months of the diagnosis of antisynthetase syndrome.7,24 The debate as to whether these are chance associations or causal (a paraneoplastic phenomenon) has not been resolved at this time.24
It is now recommended that patients with antisynthetase syndrome be screened for malignancies as appropriate for the patient’s age and sex. Screening should include a careful history and physical examination, complete blood cell count, comprehensive metabolic panel, chest radiography, mammography, and a gynecologic examination for women.25 If abnormalities are found, a more thorough evaluation for cancer is appropriate.
DIAGNOSIS
Muscle enzyme levels are often elevated
Muscle enzymes (creatine kinase and aldolase) are often elevated. Serum creatine kinase levels can range between 5 to 50 times the upper limit of normal. In an established case, creatine kinase levels along with careful manual muscle strength testing may help evaluate myositis activity. However, in chronic and advanced disease, creatine kinase may be within the normal range despite active myositis, partly because of extensive loss of muscle mass. In myositis, it may be prudent to check both creatine kinase and aldolase; sometimes only serum aldolase level rises, when immune-mediated injury predominantly affects the early regenerative myocytes.26
Judicious use of autoantibody testing
The characteristic clinical presentation is the initial clue to the diagnosis of antisynthetase syndrome, which is then supported by serologic testing.
Injudicious testing for a long list of antibodies should be avoided, as the cost is considerable and it does not influence further management. However, ordering an anti-Jo-1 antibody test in the correct clinical setting is appropriate, as it has high specificity,27,28 and thus can help establish or refute the clinical suspicion of antisynthetase syndrome.
Screening pulmonary function testing and thoracic high-resolution computed tomography for all patients with polymyositis or dermatomyositis is not considered “standard of care” and will likely not be reimbursed by third-party payers. However, in a patient with symptoms and signs of myositis, the presence of an antisynthetase antibody should prompt screening for occult interstitial lung disease, even in the absence of symptoms. As lung disease ultimately determines the prognosis in antisynthetase syndrome, early diagnosis and management is the key. Therefore, these tests would likely be approved to establish the diagnosis of interstitial lung disease and evaluate its severity.
If a myositis patient is also found to have interstitial lung disease or develops mechanic’s hands, the likely diagnosis is antisynthetase syndrome, which can be confirmed by serologic testing for antisynthetase antibodies. Interstitial lung disease in antisynthetase syndrome is often from “nonspecific interstitial pneumonitis”; therefore, medications tested and proven effective for this condition should be approved and reimbursed by payers.29–32
The coexistence of myositis and interstitial lung disease increases the sensitivity of anti-Jo-1 antibody.11 Thus, the clinician can have more confidence in early recognition and initiation of aggressive but targeted disease-modifying therapy.
Various methods can be used for detecting antisynthetase antibodies, with comparable results.28 Anti-Jo-1 antibody testing costs about $140. If that test is negative and antisynthetase syndrome is still suspected, then testing for the non-Jo-1 antisynthetase antibodies may be justified (Table 1). Though the cost of this panel of autoantibodies is about $300, it helps to confirm the diagnosis, and it influences the choice of second-line immunosuppressive agents such as tacrolimus29 and rituximab32 in patients resistant to conventional immunosuppressive agents such as azathioprine and methotrexate.
Often, anti-Ro52 SS-A antibodies are present concurrently in patients with anti-Jo-1 syndrome.33 In observational studies in patients with anti-Jo-1 antibody-associated interstitial lung disease, coexistence of anti-Ro52 SS-A antibody tended to predict a worse pulmonary outcome than in those with anti-Jo-1 antibody alone.34,35
Electromyography
Electromyography not only helps differentiate between myopathic and neuropathic weakness, but it may also support the diagnosis of “inflammatory” myopathy as suggested by prominent muscle membrane irritability (fibrillations, positive sharp waves) and abnormal motor unit action potentials (spontaneous activity showing small, short, polyphasic potentials and early recruitment). However, the findings can be nonspecific, and may even be normal in 10% to 15% of patients.36 Electromyographic abnormalities are most consistently observed in weak proximal muscles, and electromyography is also helpful in selecting a muscle for biopsy. Although no single electromyographic pattern is considered diagnostic for inflammatory myopathy, abnormalities are present in around 90% of patients (Table 2).3
Magnetic resonance imaging
Magnetic resonance imaging may show increased signal intensity in the affected muscles and surrounding tissues (Figure 3).37 Because it lacks sensitivity and specificity, magnetic resonance imaging is not helpful in diagnosing the disease. However, in the correct clinical setting, it may be used to guide muscle biopsy, and it can help in monitoring the disease progress.38
Muscle histopathology
Muscle biopsy, though often helpful, is not always diagnostic, and antisynthetase syndrome should still be suspected in the right clinical context, even in the absence of characteristic pathologic changes.
Biopsy of sites recently studied by electromyography should be avoided, and if the patient has undergone electromyography recently, the contralateral side should be selected for biopsy.
Reports of histopathologic findings in muscle biopsies in patients with antisynthetase syndrome document inflammatory myopathic features (Figure 4). In a series of patients with anti-Jo-1 syndrome, inflammation was noted in all cases, predominantly perimysial in location, with occasional endomysial and perivascular inflammation.39 Many of the inflammatory cells seen were macrophages and lymphocytes, in contrast to the predominantly lymphocytic infiltrates described in classic polymyositis and dermatomyositis. Perifascicular atrophy, similar to what is seen in dermatomyositis, was encountered; however, vascular changes, typical of dermatomyositis, were absent. Occasional degenerating and regenerating muscle fibers were also observed in most cases. Additionally, a characteristic perimysial connective tissue fragmentation was described, a feature less often seen in classic polymyositis and dermatomyositis.39
Pulmonary function testing
If antisynthetase syndrome is suspected or diagnosed, baseline pulmonary function testing (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease (reduced forced vital capacity and carbon monoxide diffusion capacity), and the diagnosis will be substantiated on thoracic high-resolution computed tomography. Respiratory muscle weakness can be detected with upright and supine spirometry.40 Weakness of these muscles contributes to shortness of breath, and patients may need ventilatory support.
Thoracic high-resolution computed tomography
Different patterns of lung injury can be seen in antisynthetase syndrome. Diffuse ground-glass opacification may suggest a nonspecific interstitial pneumonitis pattern, which is the most common form of interstitial lung disease (Figure 2), whereas coarse reticulation or honeycombing correlates with a usual interstitial pneumonitis pattern. Patchy consolidation or air-space disease can occur if cryptogenic organizing pneumonia is the predominant pattern of lung injury.
Swallowing evaluation
A comprehensive swallowing evaluation by a speech therapist may be necessary for evaluation of dysphagia (from oropharyngeal and striated esophageal muscle weakness) and determination of aspiration risk (Table 2).
Lung histopathology
If necessary, a surgical lung biopsy is needed to document the pathologic pattern of injury, including the amount of fibrosis in the lung. Historically, in idiopathic inflammatory myopathy patients in general, this has taken the form of usual interstitial pneumonia, organizing pneumonia, or diffuse alveolar damage.41 With the emergence of the definition of nonspecific interstitial pneumonitis and fibrosis as a documented and accepted pattern, more studies have found this to be the most common pattern of lung injury.16 It is characterized by diffuse involvement of the lung by an interstitial chronic inflammatory infiltrate, a cellular type of nonspecific interstitial pneumonitis that progresses in a uniform pattern to a fibrotic type (Figure 5). This form of fibrosis rarely results in significant remodeling, so-called honeycomb changes. In addition, anti-Jo-1 antibody patients may also have an increased incidence of acute lung injury, including acute diffuse alveolar damage that is often superimposed on the underlying chronic lung disease.42
In patients with pulmonary hypertension, histopathologic studies of the muscular pulmonary arteries often show moderate intimal fibroplasia, suggesting that a pulmonary arteriopathy with intimal thickening and luminal narrowing develops in some of these patients (Figure 5), independent of chronic hypoxic pulmonary vasoconstriction or vascular obstruction due to its entrapment within fibrotic lung tissue.17
TREATMENT
Glucocorticoids are the mainstay
Glucocorticoids are considered the mainstay of treatment. Patients should be advised that long-term use of glucocorticoids is necessary, though the response is variable. It is also important to discuss possible side effects of long-term glucocorticoid use.
Standard practice is to initiate treatment with high doses for the first 4 to 6 weeks to achieve disease control, followed by a slow taper over the next 9 to 12 months to the lowest effective dose to maintain remission. If the patient is profoundly weak, especially with respiratory muscle weakness or significant dysphagia and aspiration risk, hospital admission for intravenous methylprednisolone 1,000 mg daily for 3 to 5 days may be necessary. Otherwise, oral prednisone 1 mg/kg/day would be the usual starting dose.
If the patient’s muscle strength initially improves and then declines weeks to months later despite adequate therapy, glucocorticoid-induced myopathy should be suspected, especially if the muscle enzymes are within the reference range. This is more likely to occur if high-dose prednisone is continued for more than 6 to 8 weeks.
Improvement in muscle strength, which can take several weeks to several months, is a more reliable indicator of response to therapy than the serum creatine kinase level, which may take much longer to normalize. Relying on normalization of the creatine kinase level alone may lead to unnecessary prolongation of high-dose glucocorticoid therapy. It may take several months for the muscle enzymes to normalize, and there is usually a time lag between normalization of muscle enzymes and complete recovery of muscle strength.
Long-term use of high-dose prednisone leads to glucocorticoid-induced osteoporosis. Therefore, patients should receive osteoporosis prophylaxis including antiresorptive therapy with a bisphosphonate. In addition, prophylaxis against Pneumocystis jirovecii is indicated for patients treated with high-dose glucocorticoids.
Additional immunosuppressive agents
Although glucocorticoids are considered the mainstay of treatment, additional immunosuppressive agents such as azathioprine and methotrexate are often required, both as glucocorticoid-sparing agents and to achieve adequate disease control.10 Addition of such agents from the outset is particularly necessary in patients with profound muscle weakness or those who have concomitant symptomatic interstitial lung disease.
No randomized controlled trial comparing azathioprine and methotrexate has been conducted to date. Therefore, the choice is based on patient preference, presence of coexisting interstitial lung disease or liver disease, commitment to limit alcohol consumption, and thiopurine methyltransferase status. Most patients need prolonged therapy.
In a randomized clinical trial, concomitant therapy with prednisone and azathioprine resulted in better functional outcomes and a significantly lower prednisone dose requirement for maintenance therapy at 3 years than with prednisone alone.43,44 Although no such randomized study has been conducted using methotrexate, several retrospective studies have demonstrated 70% to 80% response rates, including those for whom monotherapy with glucocorticoids had failed.45,46 The combination of methotrexate and azathioprine may be beneficial in patients who previously had inadequate responses to either of these agents alone.47
For severe pulmonary involvement associated with antisynthetase syndrome, monthly intravenous infusion of cyclophosphamide has been shown to be effective.48,49
Some recent studies established the role of tacrolimus in the treatment of both interstitial lung disease and myositis associated with antisynthetase syndrome.29 Cyclosporine has also been successfully used in a case of interstitial lung disease associated with anti Jo-1 syndrome.30
Rituximab, a monoclonal antibody to Blymphocyte antigen CD20, can also be used successfully in refractory disease,31 including refractory interstitial lung disease.32
In an open-label prospective study, polymyositis refractory to glucocorticoids and multiple conventional immunosuppressive agents responded well to high-dose intravenous immune globulin in the short term.50 However, the antisynthetase antibody status in this cohort was unknown; therefore, no definite conclusion could be drawn about the efficacy of intravenous immune globulin specifically in antisynthetase syndrome.
General measures
In patients with profound muscle weakness, physical therapy and rehabilitation should begin early. The goal is to reduce further muscle wasting from disuse and prevent muscle contractures. Patients with oropharyngeal and esophageal dysmotility should be advised about aspiration precautions and may need a swallow evaluation by a speech therapist; some may need temporary parenteral hyper-alimentation or J-tube insertion.
PROGNOSIS
If skeletal muscle involvement is the sole manifestation of antisynthetase syndrome, patients usually respond to glucocorticoids and immunosuppressive therapy and do fairly well. However, the outcome is not so promising when patients also develop interstitial lung disease, and the severity and type of lung injury usually determine the prognosis. As expected, patients with a progressive course of interstitial lung disease fare poorly, whereas those with a nonprogressive course tend to do relatively better. Older age at onset (> 60 years), presence of a malignancy, and a negative antinuclear antibody test are associated with a poor prognosis.7
Acknowledgment: The authors are grateful to Dr. Stephen Hatem, MD, staff radiologist, musculoskeletal radiology, Cleveland Clinic Imaging Institute, for help in the preparation of the magnetic resonance images. We also thank Dr. Steven Shook, MD, staff neurologist, Cleveland Clinic Neurological Institute, for help in summarizing the EMG findings.
- Katzap E, Barilla-LaBarca ML, Marder G. Antisynthetase syndrome. Curr Rheumatol Rep 2011; 13:175–181.
- Nishikai M, Reichlin M. Heterogeneity of precipitating antibodies in polymyositis and dermatomyositis. Characterization of the Jo-1 antibody system. Arthritis Rheum 1980; 23:881–888.
- Nagaraju K, Lundberg IE. Inflammatory diseases of muscle and other myopathies. In:Firestein GS, Budd RC, Harris ED, McInnes IB, Ruddy S, Sergent JS, editors. Kelley’s Textbook of Rheumatology. Philadelphia, PA: Saunders; 2008:1353–1380.
- Brouwer R, Hengstman GJ, Vree Egberts W, et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis 2001; 60:116–123.
- Vázquez-Abad D, Rothfield NF. Sensitivity and specificity of anti-Jo-1 antibodies in autoimmune diseases with myositis. Arthritis Rheum 1996; 39:292–296.
- Arnett FC, Targoff IN, Mimori T, Goldstein R, Warner NB, Reveille JD. Interrelationship of major histocompatibility complex class II alleles and autoantibodies in four ethnic groups with various forms of myositis. Arthritis Rheum 1996; 39:1507–1518.
- Dugar M, Cox S, Limaye V, Blumbergs P, Roberts-Thomson PJ. Clinical heterogeneity and prognostic features of South Australian patients with antisynthetase autoantibodies. Intern Med J 2011; 41:674–679.
- Friedman AW, Targoff IN, Arnett FC. Interstitial lung disease with autoantibodies against aminoacyl-tRNA synthetases in the absence of clinically apparent myositis. Semin Arthritis Rheum 1996; 26:459–467.
- Yoshifuji H, Fujii T, Kobayashi S, et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity 2006; 39:233–241.
- Tillie-Leblond I, Wislez M, Valeyre D, et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset. Thorax 2008; 63:53–59.
- Targoff IN. Update on myositis-specific and myositis-associated autoantibodies. Curr Opin Rheumatol 2000; 12:475–481.
- Ancuta CM, Ancuta E, Chirieac RM. Aminoacyl-tRNA synthetases in idiopathic inflammatory myopathies: an update on immunopathogenic significance, clinical and therapeutic implications. In:Gran JT, editor. Idiopathic Inflammatory Myopathies - Recent Developments. Rijeka, Croatia: InTech; 2011:77–90.
- Kajihara M, Kuwana M, Tokuda H, et al. Myositis and interstitial lung disease associated with autoantibody to a transfer RNA-related protein Wa. J Rheumatol 2000; 27:2707–2710.
- Yamasaki Y, Yamada H, Nozaki T, et al. Unusually high frequency of autoantibodies to PL-7 associated with milder muscle disease in Japanese patients with polymyositis/dermatomyositis. Arthritis Rheum 2006; 54:2004–2009.
- Ascherman DP. The role of Jo-1 in the immunopathogenesis of polymyositis: current hypotheses. Curr Rheumatol Rep 2003; 5:425–430.
- Yousem SA, Gibson K, Kaminski N, Oddis CV, Ascherman DP. The pulmonary histopathologic manifestations of the anti-Jo-1 tRNA synthetase syndrome. Mod Pathol 2010; 23:874–880.
- Chatterjee S, Farver C. Severe pulmonary hypertension in anti-Jo-1 syndrome. Arthritis Care Res (Hoboken) 2010; 62:425–429.
- Minai OA. Pulmonary hypertension in polymyositis-dermatomyositis: clinical and hemodynamic characteristics and response to vasoactive therapy. Lupus 2009; 18:1006–1010.
- Bugatti L, De Angelis R, Filosa G, Salaffi F. Bilateral, asymptomatic scaly and fissured cutaneous lesions of the fingers in a patient presenting with myositis. Indian J Dermatol Venereol Leprol 2005; 71:137–138.
- Mumm GE, McKown KM, Bell CL. Antisynthetase syndrome presenting as rheumatoid-like polyarthritis. J Clin Rheumatol 2010; 16:307–312.
- Hirakata M, Mimori T, Akizuki M, Craft J, Hardin JA, Homma M. Autoantibodies to small nuclear and cytoplasmic ribonucleoproteins in Japanese patients with inflammatory muscle disease. Arthritis Rheum 1992; 35:449–456.
- Love LA, Leff RL, Fraser DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991; 70:360–374.
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case-control study. Br J Dermatol 2001; 144:825–831.
- Legault D, McDermott J, Crous-Tsanaclis AM, Boire G. Cancer-associated myositis in the presence of anti-Jo1 autoantibodies and the antisynthetase syndrome. J Rheumatol 2008; 35:169–171.
- Selva-O’Callaghan A, Trallero-Araguás E, Grau-Junyent JM, Labrador-Horrillo M. Malignancy and myositis: novel autoantibodies and new insights. Curr Opin Rheumatol 2010; 22:627–632.
- Casciola-Rosen L, Hall JC, Mammen AL, Christopher-Stine L, Rosen A. Isolated elevation of aldolase in the serum of myositis patients: a potential biomarker of damaged early regenerating muscle cells. Clin Exp Rheumatol 2012; 30:548–553.
- Shovman O, Gilburd B, Barzilai O, et al. Evaluation of the BioPlex 2200 ANA screen: analysis of 510 healthy subjects: incidence of natural/predictive autoantibodies. Ann N Y Acad Sci 2005; 1050:380–388.
- Zampieri S, Ghirardello A, Iaccarino L, Tarricone E, Gambari PF, Doria A. Anti-Jo-1 antibodies. Autoimmunity 2005; 38:73–78.
- Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 2005; 52:2439–2446.
- Jankowska M, Butto B, Debska-Slizien A, Rutkowski B. Beneficial effect of treatment with cyclosporin A in a case of refractory antisynthetase syndrome. Rheumatol Int 2007; 27:775–780.
- Limaye V, Hissaria P, Liew CL, Koszyka B. Efficacy of rituximab in refractory antisynthetase syndrome. Intern Med J 2012; 42:e4–e7.
- Marie I, Dominique S, Janvresse A, Levesque H, Menard JF. Rituximab therapy for refractory interstitial lung disease related to antisynthetase syndrome. Respir Med 2012; 106:581–587.
- Rutjes SA, Vree Egberts WT, Jongen P, Van Den Hoogen F, Pruijn GJ, Van Venrooij WJ. Anti-Ro52 antibodies frequently co-occur with anti-Jo-1 antibodies in sera from patients with idiopathic inflammatory myopathy. Clin Exp Immunol 1997; 109:32–40.
- La Corte R, Lo Mo Naco A, Locaputo A, Dolzani F, Trotta F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity 2006; 39:249–253.
- Váncsa A, Csípo I, Németh J, Dévényi K, Gergely L, Dankó K. Characteristics of interstitial lung disease in SS-A positive/Jo-1 positive inflammatory myopathy patients. Rheumatol Int 2009; 29:989–994.
- Bohan A, Peter JB, Bowman RL, Pearson CM. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore) 1977; 56:255–286.
- Reimers CD, Finkenstaedt M. Muscle imaging in inflammatory myopathies. Curr Opin Rheumatol 1997; 9:475–485.
- O’Connell MJ. Whole-body MR imaging in the diagnosis of polymyositis. Am J Roentgenol 2002; 179:967–971.
- Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000; 68:472–478.
- Fromageot C, Lofaso F, Annane D, et al. Supine fall in lung volumes in the assessment of diaphragmatic weakness in neuromuscular disorders. Arch Phys Med Rehabil 2001; 82:123–128.
- Leslie KO. Historical perspective: a pathologic approach to the classification of idiopathic interstitial pneumonias. Chest 2005; 128(suppl 1):513S–519S.
- Nicholson AG, Colby TV, du Bois RM, Hansell DM, Wells AU. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 2000; 162:2213–2217.
- Bunch TW, Worthington JW, Combs JJ, Ilstrup DM, Engel AG. Azathioprine with prednisone for polymyositis. A controlled, clinical trial. Ann Intern Med 1980; 92:365–369.
- Bunch TW. Prednisone and azathioprine for polymyositis: long-term followup. Arthritis Rheum 1981; 24:45–48.
- Metzger AL, Bohan A, Goldberg LS, Bluestone R, Pearson CM. Polymyositis and dermatomyositis: combined methotrexate and corticosteroid therapy. Ann Intern Med 1974; 81:182–189.
- Joffe MM, Love LA, Leff RL, et al. Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med 1993; 94:379–387.
- Villalba L, Hicks JE, Adams EM, et al. Treatment of refractory myositis: a randomized crossover study of two new cytotoxic regimens. Arthritis Rheum 1998; 41:392–399.
- Yamasaki Y, Yamada H, Yamasaki M, et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford) 2007; 46:124–130.
- al-Janadi M, Smith CD, Karsh J. Cyclophosphamide treatment of interstitial pulmonary fibrosis in polymyositis/dermatomyositis. J Rheumatol 1989; 16:1592–1596.
- Cherin P, Pelletier S, Teixeira A, et al. Results and long-term followup of intravenous immunoglobulin infusions in chronic, refractory polymyositis: an open study with thirty-five adult patients. Arthritis Rheum 2002; 46:467–474.
- Katzap E, Barilla-LaBarca ML, Marder G. Antisynthetase syndrome. Curr Rheumatol Rep 2011; 13:175–181.
- Nishikai M, Reichlin M. Heterogeneity of precipitating antibodies in polymyositis and dermatomyositis. Characterization of the Jo-1 antibody system. Arthritis Rheum 1980; 23:881–888.
- Nagaraju K, Lundberg IE. Inflammatory diseases of muscle and other myopathies. In:Firestein GS, Budd RC, Harris ED, McInnes IB, Ruddy S, Sergent JS, editors. Kelley’s Textbook of Rheumatology. Philadelphia, PA: Saunders; 2008:1353–1380.
- Brouwer R, Hengstman GJ, Vree Egberts W, et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis 2001; 60:116–123.
- Vázquez-Abad D, Rothfield NF. Sensitivity and specificity of anti-Jo-1 antibodies in autoimmune diseases with myositis. Arthritis Rheum 1996; 39:292–296.
- Arnett FC, Targoff IN, Mimori T, Goldstein R, Warner NB, Reveille JD. Interrelationship of major histocompatibility complex class II alleles and autoantibodies in four ethnic groups with various forms of myositis. Arthritis Rheum 1996; 39:1507–1518.
- Dugar M, Cox S, Limaye V, Blumbergs P, Roberts-Thomson PJ. Clinical heterogeneity and prognostic features of South Australian patients with antisynthetase autoantibodies. Intern Med J 2011; 41:674–679.
- Friedman AW, Targoff IN, Arnett FC. Interstitial lung disease with autoantibodies against aminoacyl-tRNA synthetases in the absence of clinically apparent myositis. Semin Arthritis Rheum 1996; 26:459–467.
- Yoshifuji H, Fujii T, Kobayashi S, et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity 2006; 39:233–241.
- Tillie-Leblond I, Wislez M, Valeyre D, et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset. Thorax 2008; 63:53–59.
- Targoff IN. Update on myositis-specific and myositis-associated autoantibodies. Curr Opin Rheumatol 2000; 12:475–481.
- Ancuta CM, Ancuta E, Chirieac RM. Aminoacyl-tRNA synthetases in idiopathic inflammatory myopathies: an update on immunopathogenic significance, clinical and therapeutic implications. In:Gran JT, editor. Idiopathic Inflammatory Myopathies - Recent Developments. Rijeka, Croatia: InTech; 2011:77–90.
- Kajihara M, Kuwana M, Tokuda H, et al. Myositis and interstitial lung disease associated with autoantibody to a transfer RNA-related protein Wa. J Rheumatol 2000; 27:2707–2710.
- Yamasaki Y, Yamada H, Nozaki T, et al. Unusually high frequency of autoantibodies to PL-7 associated with milder muscle disease in Japanese patients with polymyositis/dermatomyositis. Arthritis Rheum 2006; 54:2004–2009.
- Ascherman DP. The role of Jo-1 in the immunopathogenesis of polymyositis: current hypotheses. Curr Rheumatol Rep 2003; 5:425–430.
- Yousem SA, Gibson K, Kaminski N, Oddis CV, Ascherman DP. The pulmonary histopathologic manifestations of the anti-Jo-1 tRNA synthetase syndrome. Mod Pathol 2010; 23:874–880.
- Chatterjee S, Farver C. Severe pulmonary hypertension in anti-Jo-1 syndrome. Arthritis Care Res (Hoboken) 2010; 62:425–429.
- Minai OA. Pulmonary hypertension in polymyositis-dermatomyositis: clinical and hemodynamic characteristics and response to vasoactive therapy. Lupus 2009; 18:1006–1010.
- Bugatti L, De Angelis R, Filosa G, Salaffi F. Bilateral, asymptomatic scaly and fissured cutaneous lesions of the fingers in a patient presenting with myositis. Indian J Dermatol Venereol Leprol 2005; 71:137–138.
- Mumm GE, McKown KM, Bell CL. Antisynthetase syndrome presenting as rheumatoid-like polyarthritis. J Clin Rheumatol 2010; 16:307–312.
- Hirakata M, Mimori T, Akizuki M, Craft J, Hardin JA, Homma M. Autoantibodies to small nuclear and cytoplasmic ribonucleoproteins in Japanese patients with inflammatory muscle disease. Arthritis Rheum 1992; 35:449–456.
- Love LA, Leff RL, Fraser DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991; 70:360–374.
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case-control study. Br J Dermatol 2001; 144:825–831.
- Legault D, McDermott J, Crous-Tsanaclis AM, Boire G. Cancer-associated myositis in the presence of anti-Jo1 autoantibodies and the antisynthetase syndrome. J Rheumatol 2008; 35:169–171.
- Selva-O’Callaghan A, Trallero-Araguás E, Grau-Junyent JM, Labrador-Horrillo M. Malignancy and myositis: novel autoantibodies and new insights. Curr Opin Rheumatol 2010; 22:627–632.
- Casciola-Rosen L, Hall JC, Mammen AL, Christopher-Stine L, Rosen A. Isolated elevation of aldolase in the serum of myositis patients: a potential biomarker of damaged early regenerating muscle cells. Clin Exp Rheumatol 2012; 30:548–553.
- Shovman O, Gilburd B, Barzilai O, et al. Evaluation of the BioPlex 2200 ANA screen: analysis of 510 healthy subjects: incidence of natural/predictive autoantibodies. Ann N Y Acad Sci 2005; 1050:380–388.
- Zampieri S, Ghirardello A, Iaccarino L, Tarricone E, Gambari PF, Doria A. Anti-Jo-1 antibodies. Autoimmunity 2005; 38:73–78.
- Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 2005; 52:2439–2446.
- Jankowska M, Butto B, Debska-Slizien A, Rutkowski B. Beneficial effect of treatment with cyclosporin A in a case of refractory antisynthetase syndrome. Rheumatol Int 2007; 27:775–780.
- Limaye V, Hissaria P, Liew CL, Koszyka B. Efficacy of rituximab in refractory antisynthetase syndrome. Intern Med J 2012; 42:e4–e7.
- Marie I, Dominique S, Janvresse A, Levesque H, Menard JF. Rituximab therapy for refractory interstitial lung disease related to antisynthetase syndrome. Respir Med 2012; 106:581–587.
- Rutjes SA, Vree Egberts WT, Jongen P, Van Den Hoogen F, Pruijn GJ, Van Venrooij WJ. Anti-Ro52 antibodies frequently co-occur with anti-Jo-1 antibodies in sera from patients with idiopathic inflammatory myopathy. Clin Exp Immunol 1997; 109:32–40.
- La Corte R, Lo Mo Naco A, Locaputo A, Dolzani F, Trotta F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity 2006; 39:249–253.
- Váncsa A, Csípo I, Németh J, Dévényi K, Gergely L, Dankó K. Characteristics of interstitial lung disease in SS-A positive/Jo-1 positive inflammatory myopathy patients. Rheumatol Int 2009; 29:989–994.
- Bohan A, Peter JB, Bowman RL, Pearson CM. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore) 1977; 56:255–286.
- Reimers CD, Finkenstaedt M. Muscle imaging in inflammatory myopathies. Curr Opin Rheumatol 1997; 9:475–485.
- O’Connell MJ. Whole-body MR imaging in the diagnosis of polymyositis. Am J Roentgenol 2002; 179:967–971.
- Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000; 68:472–478.
- Fromageot C, Lofaso F, Annane D, et al. Supine fall in lung volumes in the assessment of diaphragmatic weakness in neuromuscular disorders. Arch Phys Med Rehabil 2001; 82:123–128.
- Leslie KO. Historical perspective: a pathologic approach to the classification of idiopathic interstitial pneumonias. Chest 2005; 128(suppl 1):513S–519S.
- Nicholson AG, Colby TV, du Bois RM, Hansell DM, Wells AU. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 2000; 162:2213–2217.
- Bunch TW, Worthington JW, Combs JJ, Ilstrup DM, Engel AG. Azathioprine with prednisone for polymyositis. A controlled, clinical trial. Ann Intern Med 1980; 92:365–369.
- Bunch TW. Prednisone and azathioprine for polymyositis: long-term followup. Arthritis Rheum 1981; 24:45–48.
- Metzger AL, Bohan A, Goldberg LS, Bluestone R, Pearson CM. Polymyositis and dermatomyositis: combined methotrexate and corticosteroid therapy. Ann Intern Med 1974; 81:182–189.
- Joffe MM, Love LA, Leff RL, et al. Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med 1993; 94:379–387.
- Villalba L, Hicks JE, Adams EM, et al. Treatment of refractory myositis: a randomized crossover study of two new cytotoxic regimens. Arthritis Rheum 1998; 41:392–399.
- Yamasaki Y, Yamada H, Yamasaki M, et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford) 2007; 46:124–130.
- al-Janadi M, Smith CD, Karsh J. Cyclophosphamide treatment of interstitial pulmonary fibrosis in polymyositis/dermatomyositis. J Rheumatol 1989; 16:1592–1596.
- Cherin P, Pelletier S, Teixeira A, et al. Results and long-term followup of intravenous immunoglobulin infusions in chronic, refractory polymyositis: an open study with thirty-five adult patients. Arthritis Rheum 2002; 46:467–474.
KEY POINTS
- Antisynthetase syndrome can present with a wide variety of clinical manifestations, including myositis and interstitial lung disease.
- The type and severity of interstitial lung disease usually determine the long-term outcome.
- In the appropriate clinical setting, the diagnosis is usually confirmed by the detection of antibodies to various aminoacyl-transfer RNA synthetases, anti-Jo-1 antibody being the most common.
- Although glucocorticoids are considered the mainstay of treatment, additional immunosuppressive agents such as azathioprine or methotrexate are often required as steroid-sparing agents and also to achieve disease control.
- In the case of severe pulmonary involvement, cyclophosphamide is recommended.