User login
Thrombotic thrombocytopenic purpura: The role of ADAMTS13
A breakthrough in understanding the pathogenesis of thrombotic thrombocytopenic purpura (TTP) came with the discovery of ADAMTS13 (an abbreviation for “a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13”), a plasma protein that cleaves von Willebrand factor, which interacts with platelets to promote blood clotting. If ADAMTS13 is lacking, unusually large multimers of von Willebrand factor can accumulate and trigger intravascular platelet aggregation and microthrombosis, causing the signs and symptoms of TTP.1–3
This knowledge has practical applications: we can now measure ADAMTS13 activity, ADAMTS13 inhibitor, and antibodies against ADAMTS13 to help us diagnose TTP and distinguish it from other forms of thrombotic microangiopathy, such as hemolytic-uremic syndrome, that have similar symptoms but require different treatment.
Using case studies, this article describes typical presentations of acute and relapsing TTP; the role of laboratory testing, including the ADAMTS13 assay; how to distinguish TTP from other conditions that present similarly; and how to manage this condition.
A HIGH RISK OF DEATH WITHOUT PLASMA EXCHANGE
TTP is characterized by disseminated microthrombi composed of agglutinated platelets and von Willebrand factor in small vessels. Tissue damage by microthrombi can cause thrombocytopenia (platelet deficiency), microangiopathic hemolytic anemia (loss of red blood cells caused by destructive conditions in small vessels), and multiorgan failure.1
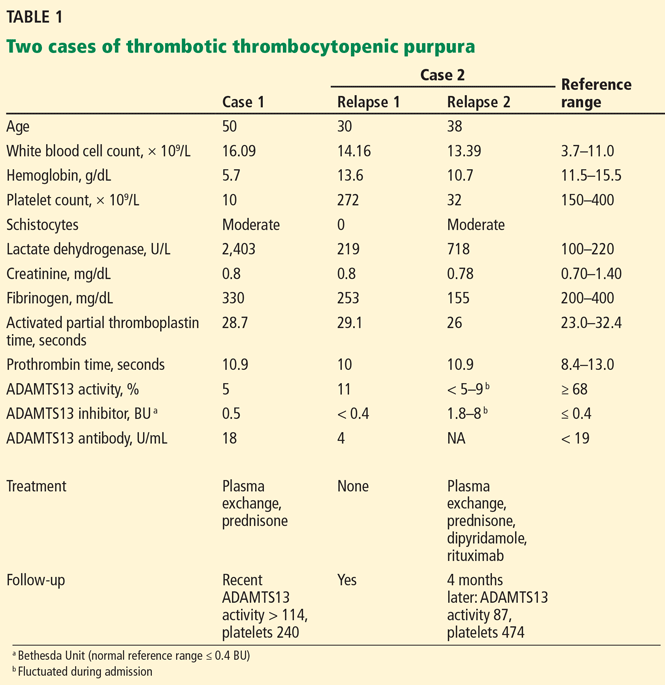
Untreated TTP has a mortality rate of about 90%.1 As shown in Case 1, Case 2, and Table 1, rapid diagnosis and prompt initiation of daily therapeutic plasma exchange can improve this grave outlook.4


ADAMTS13 DEFICIENCY CAN BE ACQUIRED OR CONGENITAL
Two major forms of TTP with ADAMTS13 deficiency and microvascular thrombosis are recognized:
Acquired TTP, the more common form, peaks in incidence between ages 30 and 50.2,5 It more often affects women, particularly during and after pregnancy (its estimated prevalence is 1 in 25,000 pregnancies), and African Americans.6 Acquired TTP may be:
- Primary (idiopathic or autoantibody-mediated), associated with severely decreased ADAMTS13 and the presence of ultra-large von Willebrand factor multimers, or
- Secondary (23%–67% of cases), arising from a variety of conditions, including autoimmune disorders (eg, systemic lupus erythematosus, rheumatoid arthritis), solid organ or hematopoietic cell transplant, malignancy, drugs, and pregnancy (Table 2).1,5–8 Secondary TTP has a worse prognosis than idiopathic TTP.5,9

Congenital TTP (Upshaw-Shulman syndrome) is a rare autosomal-recessive disease caused by compound heterozygous or homozygous mutations of the ADAMTS13 gene, producing nonfunctional ADAMTS13 protein. Patients have severely deficient ADAMTS13 activity but usually do not develop autoantibodies. There is a high risk of chronic, relapsing episodes; identified triggers include pregnancy and heavy alcohol intake.2,10 About half of patients with congenital TTP have an early onset, usually presenting with acute TTP between birth and age 5, and about half have a late onset, usually remaining without symptoms until age 20 to 40.
THE CLINICAL PICTURE OF TTP IS NOT ALWAYS CLASSIC
TTP is primarily diagnosed clinically, but diagnosis is often difficult because of various nonspecific symptoms. Typical TTP presents with the “classic pentad”:
- Severe thrombocytopenia (70%–100% of patients)
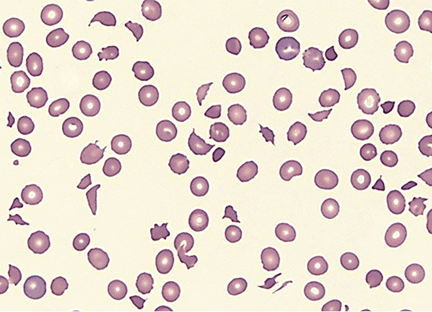
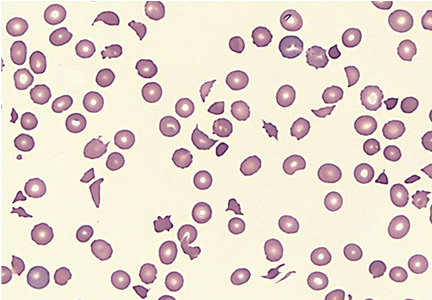
- Microangiopathic hemolytic anemia with multiple schistocytes (70%–100%) (Figure 1)
- Neurologic involvement (50%–90%)
- Renal abnormalities (about 50%)
- Fever (25%).
However, the entire picture often does not emerge in a single patient.2,6 Waiting for the entire pentad to develop before diagnosing TTP can have grave clinical consequences,1,2,5 and the presence of thrombocytopenia and unexplained microangiopathic hemolytic anemia are considered clinically sufficient to suspect TTP.5
Neurologic symptoms usually fluctuate. They can include mild abnormalities such as weakness, dizziness, headache, blurred vision, ataxia, and transient mental status changes, as well as severe abnormalities including stroke, seizure, and coma.2,6
Most patients have normal findings on computed tomography and magnetic resonance imaging at the onset of neurologic symptoms or with a history of TTP. Some patients (8%–39%) show reversible acute brain lesions, including ischemic changes.11–13
Other signs and symptoms may result from multiorgan failure due to microthrombosis; ischemia in retinal, coronary, and abdominal circulations; and unconjugated hyperbilirubinemia.2
Atypical presentations. About 18% of patients have cardiac involvement from microvascular occlusion, with arrhythmia, angina, or congestive heart failure. Abdominal pain and pancreatitis occur in 5% to 13%, and visual disturbances in 8% to 10%.
Patients with an atypical presentation may not have laboratory evidence of microangiopathic hemolytic anemia, but an ADAMTS13 assay will show severely decreased activity. Therapeutic plasma exchange can improve atypical symptoms.2,3,10,14,15
ADAMTS13 ASSAY IS KEY TO DIAGNOSIS
Laboratory evidence typically includes hemolytic anemia (reticulocytosis, schistocytes, elevated indirect bilirubin, reduced haptoglobin, elevated lactate dehydrogenase) and thrombocytopenia.3 There are no significant abnormalities in prothrombin time, international normalized ratio, activated partial thromboplastin time, fibrinogen, or D-dimer level.
Measuring the levels of ADAMTS13 activity, ADAMTS13 inhibitor, and ADAMTS13 antibody is becoming standard to confirm the diagnosis of TTP, to determine if it is congenital or acquired, and to distinguish it from thrombocytopenic conditions such as hemolytic-uremic syndrome, idiopathic thrombocytopenic purpura, and heparin-induced thrombocytopenia.4,5 A newer ADAMTS13 assay based on fluorescence energy transfer (FRET) technology with a synthetic amino acid-von Willebrand factor peptide substrate has a faster turnaround time and less test variability.6,16,17 This FRET assay can give the result of ADAMTS13 activity within 2 hours. In comparison, the assay based on multimeric von Willebrand factor takes 2 to 3 days, and mass spectrometry to measure the cleavage products of a synthetic von Willebrand factor molecule takes about 4 hours.3,10,16
About two-thirds of patients with the clinical diagnosis of idiopathic TTP have ADAMTS13 activity levels lower than 10%.5,14,18 In the appropriate clinical setting, this threshold level is highly sensitive (89%–100%) and specific (99%–100%) in differentiating TTP from other thrombotic angiopathies.2,3,18
Note: The ADAMTS13 assay was needed for early correct diagnosis in Case 1 and Case 2.
Inhibitors provide more clues
Autoantibodies can be classified according to whether they inhibit ADAMTS13 activity.
Neutralizing inhibitors. Most cases of acquired, idiopathic TTP with severe ADAMTS13 deficiency are related to circulating autoantibodies that neutralize ADAMTS13 activity. This ADAMTS13 inhibitor level is obtained by measuring residual ADAMTS13 activity after mixing equal amounts of patient plasma with normal pooled plasma. ADAMTS13 inhibitor is detectable in 44% to 93% of patients with severely deficient ADAMTS13 activity.3,6,19
Nonneutralizing inhibitors. From 10% to 15% of patients with TTP with severe ADAMTS13 deficiency lack ADAMTS13 autoantibodies measured by enzyme immunoassay but have nonneutralizing immunoglobulin G (IgG) or IgM autoantibodies. In such cases, ADAMTS13 deficiency may be related to increased antibody-mediated clearance or other unknown mechanisms.
Neutralizing inhibitors and nonneutralizing inhibitors may be present simultaneously in some patients.3,10,19,20
Blood factors affect ADAMTS13 activity
Specimen factors can affect ADAMTS13 activity and antibody levels.
Hemoglobin is a potent inhibitor of ADAMTS13, so an elevated plasma level of free hemoglobin (> 2 g/dL) can reduce ADAMTS13 activity, as can hyperbilirubinemia (> 15 mg/dL).
High levels of endogenous von Willebrand factor, lipids, thrombin, or other proteases that may cleave ADAMTS13 can also reduce ADAMTS13 activity.3 Conversely, recent plasma exchange or transfusion can mask the diagnosis of TTP because of false normalization of ADAMTS13 activity. In addition, ADAMTS13 autoantibody can be detected in other immune-mediated disorders (eg, systemic lupus erythematosus, antiphospholipid syndrome), and hypergammaglobulinemia, as well as in 10% to 15% of healthy individuals.19
CONSIDER OTHER CONDITIONS

Before diagnosing TTP, other conditions causing thrombocytopenia and hemolytic anemia should be excluded by taking a careful clinical, laboratory, and medication history (Table 2). Of these conditions, the most challenging to differentiate from TTP—and often indistinguishable from it at presentation—is hemolytic-uremic syndrome (Table 3).
Hemolytic-uremic syndrome
Hemolytic-uremic syndrome presents with a triad of thrombocytopenia, acute renal failure, and microangiopathic hemolytic anemia, with increased lactate dehydrogenase levels. Renal dysfunction from ischemia or tissue injury by microvascular thrombi predominates. Hemolytic-uremic syndrome most often occurs in children and is often related to hemorrhagic enterocolitis caused by infection with Escherichia coli O157:H7 or Shigella species (90%–95% of cases).1,2,5
From 5% to 10% of cases of hemolytic- uremic syndrome are atypical. These cases are not associated with diarrhea, and many are caused by genetic mutations that result in chronic excessive complement activation. Implicated genes regulate complement regulator factor H (20%–30% of cases) or CD46 (10%) and other cofactors, or autoantibodies against factor H (10%), which affect the alternate complement pathway.6,21–23
Initial therapeutic plasma exchange is commonly undertaken for atypical hemolytic- uremic syndrome, particularly for patients at risk of rapid progression to end-stage renal failure. But despite such treatment, about 60% of these patients die or develop permanent renal damage within 1 year.2,3,24
Eculizumab, a monoclonal antibody against complement component C5, has been approved by the US Food and Drug Administration for atypical hemolytic-uremic syndrome and may improve quality of life.25–27
PLASMA EXCHANGE IS THE MAINSTAY OF THERAPY
In 2012, the British Society for Haematology published revised guidelines for managing TTP and other thrombotic microangiopathies.28
Acquired idiopathic TTP with reduced ADAMTS13 activity requires immediate therapeutic plasma exchange. Daily plasma exchange combines plasmapheresis to remove circulating ultralarge von Willebrand factor-platelet strings and autoantibodies against ADAMTS13, and infusion of fresh-frozen plasma to replace ADAMTS13.18 This procedure is the mainstay of therapy and brings 70% to 90% of patients with idiopathic TTP to remission.1,2,5,6 However, the optimal duration of daily plasma exchange and the number of procedures required is highly variable according to clinical condition. Therapeutic plasma exchange can also cause plasma-related adverse reactions.9,28 Congenital TTP requires plasma infusion or exchange depending on the patient’s severity of ADAMTS13 deficiency.
Corticosteroids are used in combination with daily therapeutic plasma exchange, although evidence from controlled trials of their efficacy in this setting is lacking. Patients with severely decreased ADAMTS13 activity or low titers of ADAMTS13 autoantibodies tend to respond to the therapy.5,8,29
An ADAMTS13 assay with a short turn-around time can help guide the decision to initiate therapeutic plasma exchange. However, if there is a strong clinical suspicion of TTP, plasma exchange should be initiated immediately without waiting for test results.5,30 Monitoring ADAMTS13 activity or inhibitor during initial plasma exchange therapy has had conflicting results in several studies and is generally not recommended for patients with acquired TTP.8,30,31
RELAPSE IS COMMON
About 20% to 50% of patients with idiopathic TTP experience a relapse (Case 2). Most relapses occur within the first 2 years after the initial episode, with an estimated risk of 43% for relapse at 7.5 years.5,9
Factors that predict a higher risk of relapse include persistently severely decreased ADAMTS13 activity, positive inhibitor, and high titers of autoantibodies to ADAMTS13 during symptomatic TTP. During clinical remission, persistence of autoantibodies also indicates increased risk.1,3,5,6,9
Patients who have a relapse and whose disease is refractory to therapeutic plasma exchange (10%–20% of cases) have been treated with corticosteroids, splenectomy, or immunosuppressive agents (cyclosporine, azathioprine, or cyclophosphamide) with varying rates of success. Rituximab (monoclonal anti-CD20) has recently been used as second-line therapy in refractory or relapsing immune-mediated TTP or idiopathic TTP with neurologic or cardiac symptoms associated with a poor prognosis. Therapy including rituximab results in improved response and progression-free survival.32 Other potential therapies, including recombinant active ADAMTS13, are under investigation.9,23,28,30,33,34
- Sadler JE, Moake JL, Miyata T, George JN. Recent advances in thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program 2004; 1:407–423.
- Shenkman B, Einav Y. Thrombotic thrombocytopenic purpura and other thrombotic microangiopathic hemolytic anemias: diagnosis and classification. Autoimmun Rev 2014; 13:584–586.
- Shah N, Sarode R. Thrombotic thrombocytopenic purpura-what is new? J Clin Apher 2013; 28:30–35.
- Imanirad I, Rajasekhar A, Zumberg M. A case series of atypical presentations of thrombotic thrombocytopenic purpura. J Clin Apher 2012; 27:221–226.
- George JN, Al-Nouri ZL. Diagnostic and therapeutic challenges in the thrombotic thrombocytopenic purpura and hemolytic uremic syndromes. Hematology Am Soc Hematol Educ Program 2012; 1:604–609.
- Shah N, Rutherford C, Matevosyan K, Shen YM, Sarode R. Role of ADAMTS13 in the management of thrombotic microangiopathies including thrombotic thrombocytopenic purpura (TTP). Br J Haematol 2013; 163:514–519.
- Cataland SR, Yang S, Wu HM. The use of ADAMTS13 activity, platelet count, and serum creatinine to differentiate acquired thrombotic thrombocytopenic purpura from other thrombotic microangiopathies. Br J Haematol 2012; 157:501–503.
- Mannucci PM, Peyvandi F. TTP and ADAMTS13: when Is testing appropriate? Hematology Am Soc Hematol Educ Program 2007; 1:121–126.
- Chaturved S, Carcioppolo D, Zhang L, McCar KR. Management and outcomes of patients with TTP: analysis of 100 cases at a single institution. Am J Hematol 2013; 88:560–565.
- Peyvandi F, Palla R, Lotta LA, Mackie I, Scully MA, Machin SJ. ADAMTS-13 assays in thrombotic thrombocytopenic purpura. J Thromb Haemost 2010; 8:631–640.
- Cataland SR, Scully MA, Paskavitz J, et al. Evidence of persistent neurologic injury following thrombotic thrombocytopenic purpura. Am J Hematol 2011; 86:87–89.
- Meloni G, Proia A, Antonini G, et al. Thrombotic thrombocytopenic purpura: prospective neurologic, neuroimaging and neurophysiologic evaluation. Haematologica 2001; 86:1194–1199.
- Kwaan HC, Boggio LN. The clinical spectrum of thrombotic thrombocytopenic purpura. Semin Thromb Hemost 2005; 31:673–680.
- Sarode R. Atypical presentations of thrombotic thrombocytopenic purpura: a review. J Clin Apher 2009; 24:47–52.
- Volcy J, Nzerue CM, Oderinde A, Hewan-Iowe K. Cocaine-induced acute renal failure, hemolysis, and thrombocytopenia mimicking thrombotic thrombocytopenic purpura. Am J Kidney Dis 2000; 35:E3.
- Kremer Hovinga JA, Mottini M, Lammle B. Measurement of ADAMTS-13 activity in plasma by the FRETS-VWF73 assay: comparison with other assay methods. J Thromb Haemost 2006; 4:1146–1148.
- Groot E, Hulstein JJ, Rison CN, de Groot PG, Fijnheer R. FRETS-VWF73: a rapid and predictive tool for thrombotic thrombocytopenic purpura. J Thromb Haemost 2006; 4:698–699.
- Barrows BD, Teruya J. Use of the ADAMTS13 activity assay improved the accuracy and efficiency of the diagnosis and treatment of suspected acquired thrombotic thrombocytopenic purpura. Arch Pathol Lab Med 2014; 138:546–549.
- Rieger M, Mannucci PM, Kremer Hovinga JA, et al. ADAMTS13 autoantibodies in patients with thrombotic microangiopathies and other immunomediated diseases. Blood 2005; 106:1262–1267.
- Rogers HJ, Kottke-Marchant K. ADAMTS13 evaluation for thrombotic thrombocytopenic purpura. Pathology Innovations, Pathology and Laboratory Medicine Institute. Cleveland Clinic, Fall 2014:6–9.
- Józsi M, Licht C, Strobel S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 2008; 111:1512–1514.
- Diamante Chiodini B, Davin JC, Corazza F, et al. Eculizumab in anti-factor H antibodies associated with atypical hemolytic uremic syndrome. Pediatrics 2014; 133:e1764–e1768.
- Taylor CM, Machin S, Wigmore SJ, Goodship TH; working party from the Renal Association, the British Committee for Standards in Haematology and the British Transplantation Society. Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol 2009; 148:37–47.
- Loirat C, Garnier A, Sellier-Leclerc AL, Kwon T. Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost 2010; 36:673–681.
- Tsai HM, Kuo E. Eculizumab therapy leads to rapid resolution of thrombocytopenia in atypical hemolytic uremic syndrome. Adv Hematol 2014; 295323:1–7.
- Lapeyraque AL, Frémeaux-Bacchi V, Robitaille P. Efficacy of eculizumab in a patient with factor-H-associated atypical hemolytic uremic syndrome. Pediatr Nephrol 2011; 26:621–624.
- Baskin E, Gulleroglu K, Kantar A, Bayrakci U, Ozkaya O. Success of eculizumab in the treatment of atypical hemolytic uremic syndrome. Pediatr Nephrol 2015; 30:783–789.
- Scully M, Hunt BJ, Benjamin S, et al; British Committee for Standards in Haematology. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol 2012; 158:323–325.
- Abassi E, Yawn D, Leveque E, Nolasco L, Lopez J, Moake J. Correlation of ADAMTS-13 activity with response to plasma exchange in patients diagnosed with thrombotic thrombocytopenic purpura (Abstract #3921). Blood 2004; 104:242a.
- Blombery P, Scully M. Management of thrombocytic thrombocytopenic purpura: current perspectives. J Blood Med 2014; 5:15–23.
- Wu N, Liu J, Yang S, et al. Diagnostic and prognostic values of ADAMTS13 activity measured during daily plasma exchange therapy in patients with acquired thrombotic thrombocytopenic purpura. Transfusion 2015; 55:18–24.
- Cuker A. Adjuvant rituximab to prevent TTP relapse. Blood 2016; 127:2952–2953.
- Chapman K, Yuen S. Therapy for thrombotic thrombocytopenic purpura: past, present and future. Semin Thromb Hemost 2014; 40:34–40.
- Heidel F, Lipka DB, von Auer C, Huber C, Schrarrer I, Hess G. Addition of rituximab to standard therapy improves response rate and progression-free survival in relapsed or refractory thrombotic thrombocytopenic purpura and autoimmune haemolytic anaemia. Thromb Haemost 2007; 97:228–233.
A breakthrough in understanding the pathogenesis of thrombotic thrombocytopenic purpura (TTP) came with the discovery of ADAMTS13 (an abbreviation for “a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13”), a plasma protein that cleaves von Willebrand factor, which interacts with platelets to promote blood clotting. If ADAMTS13 is lacking, unusually large multimers of von Willebrand factor can accumulate and trigger intravascular platelet aggregation and microthrombosis, causing the signs and symptoms of TTP.1–3
This knowledge has practical applications: we can now measure ADAMTS13 activity, ADAMTS13 inhibitor, and antibodies against ADAMTS13 to help us diagnose TTP and distinguish it from other forms of thrombotic microangiopathy, such as hemolytic-uremic syndrome, that have similar symptoms but require different treatment.
Using case studies, this article describes typical presentations of acute and relapsing TTP; the role of laboratory testing, including the ADAMTS13 assay; how to distinguish TTP from other conditions that present similarly; and how to manage this condition.
A HIGH RISK OF DEATH WITHOUT PLASMA EXCHANGE
TTP is characterized by disseminated microthrombi composed of agglutinated platelets and von Willebrand factor in small vessels. Tissue damage by microthrombi can cause thrombocytopenia (platelet deficiency), microangiopathic hemolytic anemia (loss of red blood cells caused by destructive conditions in small vessels), and multiorgan failure.1
Untreated TTP has a mortality rate of about 90%.1 As shown in Case 1, Case 2, and Table 1, rapid diagnosis and prompt initiation of daily therapeutic plasma exchange can improve this grave outlook.4
ADAMTS13 DEFICIENCY CAN BE ACQUIRED OR CONGENITAL
Two major forms of TTP with ADAMTS13 deficiency and microvascular thrombosis are recognized:
Acquired TTP, the more common form, peaks in incidence between ages 30 and 50.2,5 It more often affects women, particularly during and after pregnancy (its estimated prevalence is 1 in 25,000 pregnancies), and African Americans.6 Acquired TTP may be:
- Primary (idiopathic or autoantibody-mediated), associated with severely decreased ADAMTS13 and the presence of ultra-large von Willebrand factor multimers, or
- Secondary (23%–67% of cases), arising from a variety of conditions, including autoimmune disorders (eg, systemic lupus erythematosus, rheumatoid arthritis), solid organ or hematopoietic cell transplant, malignancy, drugs, and pregnancy (Table 2).1,5–8 Secondary TTP has a worse prognosis than idiopathic TTP.5,9
Congenital TTP (Upshaw-Shulman syndrome) is a rare autosomal-recessive disease caused by compound heterozygous or homozygous mutations of the ADAMTS13 gene, producing nonfunctional ADAMTS13 protein. Patients have severely deficient ADAMTS13 activity but usually do not develop autoantibodies. There is a high risk of chronic, relapsing episodes; identified triggers include pregnancy and heavy alcohol intake.2,10 About half of patients with congenital TTP have an early onset, usually presenting with acute TTP between birth and age 5, and about half have a late onset, usually remaining without symptoms until age 20 to 40.
THE CLINICAL PICTURE OF TTP IS NOT ALWAYS CLASSIC
TTP is primarily diagnosed clinically, but diagnosis is often difficult because of various nonspecific symptoms. Typical TTP presents with the “classic pentad”:
- Severe thrombocytopenia (70%–100% of patients)
- Microangiopathic hemolytic anemia with multiple schistocytes (70%–100%) (Figure 1)
- Neurologic involvement (50%–90%)
- Renal abnormalities (about 50%)
- Fever (25%).
However, the entire picture often does not emerge in a single patient.2,6 Waiting for the entire pentad to develop before diagnosing TTP can have grave clinical consequences,1,2,5 and the presence of thrombocytopenia and unexplained microangiopathic hemolytic anemia are considered clinically sufficient to suspect TTP.5
Neurologic symptoms usually fluctuate. They can include mild abnormalities such as weakness, dizziness, headache, blurred vision, ataxia, and transient mental status changes, as well as severe abnormalities including stroke, seizure, and coma.2,6
Most patients have normal findings on computed tomography and magnetic resonance imaging at the onset of neurologic symptoms or with a history of TTP. Some patients (8%–39%) show reversible acute brain lesions, including ischemic changes.11–13
Other signs and symptoms may result from multiorgan failure due to microthrombosis; ischemia in retinal, coronary, and abdominal circulations; and unconjugated hyperbilirubinemia.2
Atypical presentations. About 18% of patients have cardiac involvement from microvascular occlusion, with arrhythmia, angina, or congestive heart failure. Abdominal pain and pancreatitis occur in 5% to 13%, and visual disturbances in 8% to 10%.
Patients with an atypical presentation may not have laboratory evidence of microangiopathic hemolytic anemia, but an ADAMTS13 assay will show severely decreased activity. Therapeutic plasma exchange can improve atypical symptoms.2,3,10,14,15
ADAMTS13 ASSAY IS KEY TO DIAGNOSIS
Laboratory evidence typically includes hemolytic anemia (reticulocytosis, schistocytes, elevated indirect bilirubin, reduced haptoglobin, elevated lactate dehydrogenase) and thrombocytopenia.3 There are no significant abnormalities in prothrombin time, international normalized ratio, activated partial thromboplastin time, fibrinogen, or D-dimer level.
Measuring the levels of ADAMTS13 activity, ADAMTS13 inhibitor, and ADAMTS13 antibody is becoming standard to confirm the diagnosis of TTP, to determine if it is congenital or acquired, and to distinguish it from thrombocytopenic conditions such as hemolytic-uremic syndrome, idiopathic thrombocytopenic purpura, and heparin-induced thrombocytopenia.4,5 A newer ADAMTS13 assay based on fluorescence energy transfer (FRET) technology with a synthetic amino acid-von Willebrand factor peptide substrate has a faster turnaround time and less test variability.6,16,17 This FRET assay can give the result of ADAMTS13 activity within 2 hours. In comparison, the assay based on multimeric von Willebrand factor takes 2 to 3 days, and mass spectrometry to measure the cleavage products of a synthetic von Willebrand factor molecule takes about 4 hours.3,10,16
About two-thirds of patients with the clinical diagnosis of idiopathic TTP have ADAMTS13 activity levels lower than 10%.5,14,18 In the appropriate clinical setting, this threshold level is highly sensitive (89%–100%) and specific (99%–100%) in differentiating TTP from other thrombotic angiopathies.2,3,18
Note: The ADAMTS13 assay was needed for early correct diagnosis in Case 1 and Case 2.
Inhibitors provide more clues
Autoantibodies can be classified according to whether they inhibit ADAMTS13 activity.
Neutralizing inhibitors. Most cases of acquired, idiopathic TTP with severe ADAMTS13 deficiency are related to circulating autoantibodies that neutralize ADAMTS13 activity. This ADAMTS13 inhibitor level is obtained by measuring residual ADAMTS13 activity after mixing equal amounts of patient plasma with normal pooled plasma. ADAMTS13 inhibitor is detectable in 44% to 93% of patients with severely deficient ADAMTS13 activity.3,6,19
Nonneutralizing inhibitors. From 10% to 15% of patients with TTP with severe ADAMTS13 deficiency lack ADAMTS13 autoantibodies measured by enzyme immunoassay but have nonneutralizing immunoglobulin G (IgG) or IgM autoantibodies. In such cases, ADAMTS13 deficiency may be related to increased antibody-mediated clearance or other unknown mechanisms.
Neutralizing inhibitors and nonneutralizing inhibitors may be present simultaneously in some patients.3,10,19,20
Blood factors affect ADAMTS13 activity
Specimen factors can affect ADAMTS13 activity and antibody levels.
Hemoglobin is a potent inhibitor of ADAMTS13, so an elevated plasma level of free hemoglobin (> 2 g/dL) can reduce ADAMTS13 activity, as can hyperbilirubinemia (> 15 mg/dL).
High levels of endogenous von Willebrand factor, lipids, thrombin, or other proteases that may cleave ADAMTS13 can also reduce ADAMTS13 activity.3 Conversely, recent plasma exchange or transfusion can mask the diagnosis of TTP because of false normalization of ADAMTS13 activity. In addition, ADAMTS13 autoantibody can be detected in other immune-mediated disorders (eg, systemic lupus erythematosus, antiphospholipid syndrome), and hypergammaglobulinemia, as well as in 10% to 15% of healthy individuals.19
CONSIDER OTHER CONDITIONS
Before diagnosing TTP, other conditions causing thrombocytopenia and hemolytic anemia should be excluded by taking a careful clinical, laboratory, and medication history (Table 2). Of these conditions, the most challenging to differentiate from TTP—and often indistinguishable from it at presentation—is hemolytic-uremic syndrome (Table 3).
Hemolytic-uremic syndrome
Hemolytic-uremic syndrome presents with a triad of thrombocytopenia, acute renal failure, and microangiopathic hemolytic anemia, with increased lactate dehydrogenase levels. Renal dysfunction from ischemia or tissue injury by microvascular thrombi predominates. Hemolytic-uremic syndrome most often occurs in children and is often related to hemorrhagic enterocolitis caused by infection with Escherichia coli O157:H7 or Shigella species (90%–95% of cases).1,2,5
From 5% to 10% of cases of hemolytic- uremic syndrome are atypical. These cases are not associated with diarrhea, and many are caused by genetic mutations that result in chronic excessive complement activation. Implicated genes regulate complement regulator factor H (20%–30% of cases) or CD46 (10%) and other cofactors, or autoantibodies against factor H (10%), which affect the alternate complement pathway.6,21–23
Initial therapeutic plasma exchange is commonly undertaken for atypical hemolytic- uremic syndrome, particularly for patients at risk of rapid progression to end-stage renal failure. But despite such treatment, about 60% of these patients die or develop permanent renal damage within 1 year.2,3,24
Eculizumab, a monoclonal antibody against complement component C5, has been approved by the US Food and Drug Administration for atypical hemolytic-uremic syndrome and may improve quality of life.25–27
PLASMA EXCHANGE IS THE MAINSTAY OF THERAPY
In 2012, the British Society for Haematology published revised guidelines for managing TTP and other thrombotic microangiopathies.28
Acquired idiopathic TTP with reduced ADAMTS13 activity requires immediate therapeutic plasma exchange. Daily plasma exchange combines plasmapheresis to remove circulating ultralarge von Willebrand factor-platelet strings and autoantibodies against ADAMTS13, and infusion of fresh-frozen plasma to replace ADAMTS13.18 This procedure is the mainstay of therapy and brings 70% to 90% of patients with idiopathic TTP to remission.1,2,5,6 However, the optimal duration of daily plasma exchange and the number of procedures required is highly variable according to clinical condition. Therapeutic plasma exchange can also cause plasma-related adverse reactions.9,28 Congenital TTP requires plasma infusion or exchange depending on the patient’s severity of ADAMTS13 deficiency.
Corticosteroids are used in combination with daily therapeutic plasma exchange, although evidence from controlled trials of their efficacy in this setting is lacking. Patients with severely decreased ADAMTS13 activity or low titers of ADAMTS13 autoantibodies tend to respond to the therapy.5,8,29
An ADAMTS13 assay with a short turn-around time can help guide the decision to initiate therapeutic plasma exchange. However, if there is a strong clinical suspicion of TTP, plasma exchange should be initiated immediately without waiting for test results.5,30 Monitoring ADAMTS13 activity or inhibitor during initial plasma exchange therapy has had conflicting results in several studies and is generally not recommended for patients with acquired TTP.8,30,31
RELAPSE IS COMMON
About 20% to 50% of patients with idiopathic TTP experience a relapse (Case 2). Most relapses occur within the first 2 years after the initial episode, with an estimated risk of 43% for relapse at 7.5 years.5,9
Factors that predict a higher risk of relapse include persistently severely decreased ADAMTS13 activity, positive inhibitor, and high titers of autoantibodies to ADAMTS13 during symptomatic TTP. During clinical remission, persistence of autoantibodies also indicates increased risk.1,3,5,6,9
Patients who have a relapse and whose disease is refractory to therapeutic plasma exchange (10%–20% of cases) have been treated with corticosteroids, splenectomy, or immunosuppressive agents (cyclosporine, azathioprine, or cyclophosphamide) with varying rates of success. Rituximab (monoclonal anti-CD20) has recently been used as second-line therapy in refractory or relapsing immune-mediated TTP or idiopathic TTP with neurologic or cardiac symptoms associated with a poor prognosis. Therapy including rituximab results in improved response and progression-free survival.32 Other potential therapies, including recombinant active ADAMTS13, are under investigation.9,23,28,30,33,34
A breakthrough in understanding the pathogenesis of thrombotic thrombocytopenic purpura (TTP) came with the discovery of ADAMTS13 (an abbreviation for “a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13”), a plasma protein that cleaves von Willebrand factor, which interacts with platelets to promote blood clotting. If ADAMTS13 is lacking, unusually large multimers of von Willebrand factor can accumulate and trigger intravascular platelet aggregation and microthrombosis, causing the signs and symptoms of TTP.1–3
This knowledge has practical applications: we can now measure ADAMTS13 activity, ADAMTS13 inhibitor, and antibodies against ADAMTS13 to help us diagnose TTP and distinguish it from other forms of thrombotic microangiopathy, such as hemolytic-uremic syndrome, that have similar symptoms but require different treatment.
Using case studies, this article describes typical presentations of acute and relapsing TTP; the role of laboratory testing, including the ADAMTS13 assay; how to distinguish TTP from other conditions that present similarly; and how to manage this condition.
A HIGH RISK OF DEATH WITHOUT PLASMA EXCHANGE
TTP is characterized by disseminated microthrombi composed of agglutinated platelets and von Willebrand factor in small vessels. Tissue damage by microthrombi can cause thrombocytopenia (platelet deficiency), microangiopathic hemolytic anemia (loss of red blood cells caused by destructive conditions in small vessels), and multiorgan failure.1
Untreated TTP has a mortality rate of about 90%.1 As shown in Case 1, Case 2, and Table 1, rapid diagnosis and prompt initiation of daily therapeutic plasma exchange can improve this grave outlook.4
ADAMTS13 DEFICIENCY CAN BE ACQUIRED OR CONGENITAL
Two major forms of TTP with ADAMTS13 deficiency and microvascular thrombosis are recognized:
Acquired TTP, the more common form, peaks in incidence between ages 30 and 50.2,5 It more often affects women, particularly during and after pregnancy (its estimated prevalence is 1 in 25,000 pregnancies), and African Americans.6 Acquired TTP may be:
- Primary (idiopathic or autoantibody-mediated), associated with severely decreased ADAMTS13 and the presence of ultra-large von Willebrand factor multimers, or
- Secondary (23%–67% of cases), arising from a variety of conditions, including autoimmune disorders (eg, systemic lupus erythematosus, rheumatoid arthritis), solid organ or hematopoietic cell transplant, malignancy, drugs, and pregnancy (Table 2).1,5–8 Secondary TTP has a worse prognosis than idiopathic TTP.5,9
Congenital TTP (Upshaw-Shulman syndrome) is a rare autosomal-recessive disease caused by compound heterozygous or homozygous mutations of the ADAMTS13 gene, producing nonfunctional ADAMTS13 protein. Patients have severely deficient ADAMTS13 activity but usually do not develop autoantibodies. There is a high risk of chronic, relapsing episodes; identified triggers include pregnancy and heavy alcohol intake.2,10 About half of patients with congenital TTP have an early onset, usually presenting with acute TTP between birth and age 5, and about half have a late onset, usually remaining without symptoms until age 20 to 40.
THE CLINICAL PICTURE OF TTP IS NOT ALWAYS CLASSIC
TTP is primarily diagnosed clinically, but diagnosis is often difficult because of various nonspecific symptoms. Typical TTP presents with the “classic pentad”:
- Severe thrombocytopenia (70%–100% of patients)
- Microangiopathic hemolytic anemia with multiple schistocytes (70%–100%) (Figure 1)
- Neurologic involvement (50%–90%)
- Renal abnormalities (about 50%)
- Fever (25%).
However, the entire picture often does not emerge in a single patient.2,6 Waiting for the entire pentad to develop before diagnosing TTP can have grave clinical consequences,1,2,5 and the presence of thrombocytopenia and unexplained microangiopathic hemolytic anemia are considered clinically sufficient to suspect TTP.5
Neurologic symptoms usually fluctuate. They can include mild abnormalities such as weakness, dizziness, headache, blurred vision, ataxia, and transient mental status changes, as well as severe abnormalities including stroke, seizure, and coma.2,6
Most patients have normal findings on computed tomography and magnetic resonance imaging at the onset of neurologic symptoms or with a history of TTP. Some patients (8%–39%) show reversible acute brain lesions, including ischemic changes.11–13
Other signs and symptoms may result from multiorgan failure due to microthrombosis; ischemia in retinal, coronary, and abdominal circulations; and unconjugated hyperbilirubinemia.2
Atypical presentations. About 18% of patients have cardiac involvement from microvascular occlusion, with arrhythmia, angina, or congestive heart failure. Abdominal pain and pancreatitis occur in 5% to 13%, and visual disturbances in 8% to 10%.
Patients with an atypical presentation may not have laboratory evidence of microangiopathic hemolytic anemia, but an ADAMTS13 assay will show severely decreased activity. Therapeutic plasma exchange can improve atypical symptoms.2,3,10,14,15
ADAMTS13 ASSAY IS KEY TO DIAGNOSIS
Laboratory evidence typically includes hemolytic anemia (reticulocytosis, schistocytes, elevated indirect bilirubin, reduced haptoglobin, elevated lactate dehydrogenase) and thrombocytopenia.3 There are no significant abnormalities in prothrombin time, international normalized ratio, activated partial thromboplastin time, fibrinogen, or D-dimer level.
Measuring the levels of ADAMTS13 activity, ADAMTS13 inhibitor, and ADAMTS13 antibody is becoming standard to confirm the diagnosis of TTP, to determine if it is congenital or acquired, and to distinguish it from thrombocytopenic conditions such as hemolytic-uremic syndrome, idiopathic thrombocytopenic purpura, and heparin-induced thrombocytopenia.4,5 A newer ADAMTS13 assay based on fluorescence energy transfer (FRET) technology with a synthetic amino acid-von Willebrand factor peptide substrate has a faster turnaround time and less test variability.6,16,17 This FRET assay can give the result of ADAMTS13 activity within 2 hours. In comparison, the assay based on multimeric von Willebrand factor takes 2 to 3 days, and mass spectrometry to measure the cleavage products of a synthetic von Willebrand factor molecule takes about 4 hours.3,10,16
About two-thirds of patients with the clinical diagnosis of idiopathic TTP have ADAMTS13 activity levels lower than 10%.5,14,18 In the appropriate clinical setting, this threshold level is highly sensitive (89%–100%) and specific (99%–100%) in differentiating TTP from other thrombotic angiopathies.2,3,18
Note: The ADAMTS13 assay was needed for early correct diagnosis in Case 1 and Case 2.
Inhibitors provide more clues
Autoantibodies can be classified according to whether they inhibit ADAMTS13 activity.
Neutralizing inhibitors. Most cases of acquired, idiopathic TTP with severe ADAMTS13 deficiency are related to circulating autoantibodies that neutralize ADAMTS13 activity. This ADAMTS13 inhibitor level is obtained by measuring residual ADAMTS13 activity after mixing equal amounts of patient plasma with normal pooled plasma. ADAMTS13 inhibitor is detectable in 44% to 93% of patients with severely deficient ADAMTS13 activity.3,6,19
Nonneutralizing inhibitors. From 10% to 15% of patients with TTP with severe ADAMTS13 deficiency lack ADAMTS13 autoantibodies measured by enzyme immunoassay but have nonneutralizing immunoglobulin G (IgG) or IgM autoantibodies. In such cases, ADAMTS13 deficiency may be related to increased antibody-mediated clearance or other unknown mechanisms.
Neutralizing inhibitors and nonneutralizing inhibitors may be present simultaneously in some patients.3,10,19,20
Blood factors affect ADAMTS13 activity
Specimen factors can affect ADAMTS13 activity and antibody levels.
Hemoglobin is a potent inhibitor of ADAMTS13, so an elevated plasma level of free hemoglobin (> 2 g/dL) can reduce ADAMTS13 activity, as can hyperbilirubinemia (> 15 mg/dL).
High levels of endogenous von Willebrand factor, lipids, thrombin, or other proteases that may cleave ADAMTS13 can also reduce ADAMTS13 activity.3 Conversely, recent plasma exchange or transfusion can mask the diagnosis of TTP because of false normalization of ADAMTS13 activity. In addition, ADAMTS13 autoantibody can be detected in other immune-mediated disorders (eg, systemic lupus erythematosus, antiphospholipid syndrome), and hypergammaglobulinemia, as well as in 10% to 15% of healthy individuals.19
CONSIDER OTHER CONDITIONS
Before diagnosing TTP, other conditions causing thrombocytopenia and hemolytic anemia should be excluded by taking a careful clinical, laboratory, and medication history (Table 2). Of these conditions, the most challenging to differentiate from TTP—and often indistinguishable from it at presentation—is hemolytic-uremic syndrome (Table 3).
Hemolytic-uremic syndrome
Hemolytic-uremic syndrome presents with a triad of thrombocytopenia, acute renal failure, and microangiopathic hemolytic anemia, with increased lactate dehydrogenase levels. Renal dysfunction from ischemia or tissue injury by microvascular thrombi predominates. Hemolytic-uremic syndrome most often occurs in children and is often related to hemorrhagic enterocolitis caused by infection with Escherichia coli O157:H7 or Shigella species (90%–95% of cases).1,2,5
From 5% to 10% of cases of hemolytic- uremic syndrome are atypical. These cases are not associated with diarrhea, and many are caused by genetic mutations that result in chronic excessive complement activation. Implicated genes regulate complement regulator factor H (20%–30% of cases) or CD46 (10%) and other cofactors, or autoantibodies against factor H (10%), which affect the alternate complement pathway.6,21–23
Initial therapeutic plasma exchange is commonly undertaken for atypical hemolytic- uremic syndrome, particularly for patients at risk of rapid progression to end-stage renal failure. But despite such treatment, about 60% of these patients die or develop permanent renal damage within 1 year.2,3,24
Eculizumab, a monoclonal antibody against complement component C5, has been approved by the US Food and Drug Administration for atypical hemolytic-uremic syndrome and may improve quality of life.25–27
PLASMA EXCHANGE IS THE MAINSTAY OF THERAPY
In 2012, the British Society for Haematology published revised guidelines for managing TTP and other thrombotic microangiopathies.28
Acquired idiopathic TTP with reduced ADAMTS13 activity requires immediate therapeutic plasma exchange. Daily plasma exchange combines plasmapheresis to remove circulating ultralarge von Willebrand factor-platelet strings and autoantibodies against ADAMTS13, and infusion of fresh-frozen plasma to replace ADAMTS13.18 This procedure is the mainstay of therapy and brings 70% to 90% of patients with idiopathic TTP to remission.1,2,5,6 However, the optimal duration of daily plasma exchange and the number of procedures required is highly variable according to clinical condition. Therapeutic plasma exchange can also cause plasma-related adverse reactions.9,28 Congenital TTP requires plasma infusion or exchange depending on the patient’s severity of ADAMTS13 deficiency.
Corticosteroids are used in combination with daily therapeutic plasma exchange, although evidence from controlled trials of their efficacy in this setting is lacking. Patients with severely decreased ADAMTS13 activity or low titers of ADAMTS13 autoantibodies tend to respond to the therapy.5,8,29
An ADAMTS13 assay with a short turn-around time can help guide the decision to initiate therapeutic plasma exchange. However, if there is a strong clinical suspicion of TTP, plasma exchange should be initiated immediately without waiting for test results.5,30 Monitoring ADAMTS13 activity or inhibitor during initial plasma exchange therapy has had conflicting results in several studies and is generally not recommended for patients with acquired TTP.8,30,31
RELAPSE IS COMMON
About 20% to 50% of patients with idiopathic TTP experience a relapse (Case 2). Most relapses occur within the first 2 years after the initial episode, with an estimated risk of 43% for relapse at 7.5 years.5,9
Factors that predict a higher risk of relapse include persistently severely decreased ADAMTS13 activity, positive inhibitor, and high titers of autoantibodies to ADAMTS13 during symptomatic TTP. During clinical remission, persistence of autoantibodies also indicates increased risk.1,3,5,6,9
Patients who have a relapse and whose disease is refractory to therapeutic plasma exchange (10%–20% of cases) have been treated with corticosteroids, splenectomy, or immunosuppressive agents (cyclosporine, azathioprine, or cyclophosphamide) with varying rates of success. Rituximab (monoclonal anti-CD20) has recently been used as second-line therapy in refractory or relapsing immune-mediated TTP or idiopathic TTP with neurologic or cardiac symptoms associated with a poor prognosis. Therapy including rituximab results in improved response and progression-free survival.32 Other potential therapies, including recombinant active ADAMTS13, are under investigation.9,23,28,30,33,34
- Sadler JE, Moake JL, Miyata T, George JN. Recent advances in thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program 2004; 1:407–423.
- Shenkman B, Einav Y. Thrombotic thrombocytopenic purpura and other thrombotic microangiopathic hemolytic anemias: diagnosis and classification. Autoimmun Rev 2014; 13:584–586.
- Shah N, Sarode R. Thrombotic thrombocytopenic purpura-what is new? J Clin Apher 2013; 28:30–35.
- Imanirad I, Rajasekhar A, Zumberg M. A case series of atypical presentations of thrombotic thrombocytopenic purpura. J Clin Apher 2012; 27:221–226.
- George JN, Al-Nouri ZL. Diagnostic and therapeutic challenges in the thrombotic thrombocytopenic purpura and hemolytic uremic syndromes. Hematology Am Soc Hematol Educ Program 2012; 1:604–609.
- Shah N, Rutherford C, Matevosyan K, Shen YM, Sarode R. Role of ADAMTS13 in the management of thrombotic microangiopathies including thrombotic thrombocytopenic purpura (TTP). Br J Haematol 2013; 163:514–519.
- Cataland SR, Yang S, Wu HM. The use of ADAMTS13 activity, platelet count, and serum creatinine to differentiate acquired thrombotic thrombocytopenic purpura from other thrombotic microangiopathies. Br J Haematol 2012; 157:501–503.
- Mannucci PM, Peyvandi F. TTP and ADAMTS13: when Is testing appropriate? Hematology Am Soc Hematol Educ Program 2007; 1:121–126.
- Chaturved S, Carcioppolo D, Zhang L, McCar KR. Management and outcomes of patients with TTP: analysis of 100 cases at a single institution. Am J Hematol 2013; 88:560–565.
- Peyvandi F, Palla R, Lotta LA, Mackie I, Scully MA, Machin SJ. ADAMTS-13 assays in thrombotic thrombocytopenic purpura. J Thromb Haemost 2010; 8:631–640.
- Cataland SR, Scully MA, Paskavitz J, et al. Evidence of persistent neurologic injury following thrombotic thrombocytopenic purpura. Am J Hematol 2011; 86:87–89.
- Meloni G, Proia A, Antonini G, et al. Thrombotic thrombocytopenic purpura: prospective neurologic, neuroimaging and neurophysiologic evaluation. Haematologica 2001; 86:1194–1199.
- Kwaan HC, Boggio LN. The clinical spectrum of thrombotic thrombocytopenic purpura. Semin Thromb Hemost 2005; 31:673–680.
- Sarode R. Atypical presentations of thrombotic thrombocytopenic purpura: a review. J Clin Apher 2009; 24:47–52.
- Volcy J, Nzerue CM, Oderinde A, Hewan-Iowe K. Cocaine-induced acute renal failure, hemolysis, and thrombocytopenia mimicking thrombotic thrombocytopenic purpura. Am J Kidney Dis 2000; 35:E3.
- Kremer Hovinga JA, Mottini M, Lammle B. Measurement of ADAMTS-13 activity in plasma by the FRETS-VWF73 assay: comparison with other assay methods. J Thromb Haemost 2006; 4:1146–1148.
- Groot E, Hulstein JJ, Rison CN, de Groot PG, Fijnheer R. FRETS-VWF73: a rapid and predictive tool for thrombotic thrombocytopenic purpura. J Thromb Haemost 2006; 4:698–699.
- Barrows BD, Teruya J. Use of the ADAMTS13 activity assay improved the accuracy and efficiency of the diagnosis and treatment of suspected acquired thrombotic thrombocytopenic purpura. Arch Pathol Lab Med 2014; 138:546–549.
- Rieger M, Mannucci PM, Kremer Hovinga JA, et al. ADAMTS13 autoantibodies in patients with thrombotic microangiopathies and other immunomediated diseases. Blood 2005; 106:1262–1267.
- Rogers HJ, Kottke-Marchant K. ADAMTS13 evaluation for thrombotic thrombocytopenic purpura. Pathology Innovations, Pathology and Laboratory Medicine Institute. Cleveland Clinic, Fall 2014:6–9.
- Józsi M, Licht C, Strobel S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 2008; 111:1512–1514.
- Diamante Chiodini B, Davin JC, Corazza F, et al. Eculizumab in anti-factor H antibodies associated with atypical hemolytic uremic syndrome. Pediatrics 2014; 133:e1764–e1768.
- Taylor CM, Machin S, Wigmore SJ, Goodship TH; working party from the Renal Association, the British Committee for Standards in Haematology and the British Transplantation Society. Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol 2009; 148:37–47.
- Loirat C, Garnier A, Sellier-Leclerc AL, Kwon T. Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost 2010; 36:673–681.
- Tsai HM, Kuo E. Eculizumab therapy leads to rapid resolution of thrombocytopenia in atypical hemolytic uremic syndrome. Adv Hematol 2014; 295323:1–7.
- Lapeyraque AL, Frémeaux-Bacchi V, Robitaille P. Efficacy of eculizumab in a patient with factor-H-associated atypical hemolytic uremic syndrome. Pediatr Nephrol 2011; 26:621–624.
- Baskin E, Gulleroglu K, Kantar A, Bayrakci U, Ozkaya O. Success of eculizumab in the treatment of atypical hemolytic uremic syndrome. Pediatr Nephrol 2015; 30:783–789.
- Scully M, Hunt BJ, Benjamin S, et al; British Committee for Standards in Haematology. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol 2012; 158:323–325.
- Abassi E, Yawn D, Leveque E, Nolasco L, Lopez J, Moake J. Correlation of ADAMTS-13 activity with response to plasma exchange in patients diagnosed with thrombotic thrombocytopenic purpura (Abstract #3921). Blood 2004; 104:242a.
- Blombery P, Scully M. Management of thrombocytic thrombocytopenic purpura: current perspectives. J Blood Med 2014; 5:15–23.
- Wu N, Liu J, Yang S, et al. Diagnostic and prognostic values of ADAMTS13 activity measured during daily plasma exchange therapy in patients with acquired thrombotic thrombocytopenic purpura. Transfusion 2015; 55:18–24.
- Cuker A. Adjuvant rituximab to prevent TTP relapse. Blood 2016; 127:2952–2953.
- Chapman K, Yuen S. Therapy for thrombotic thrombocytopenic purpura: past, present and future. Semin Thromb Hemost 2014; 40:34–40.
- Heidel F, Lipka DB, von Auer C, Huber C, Schrarrer I, Hess G. Addition of rituximab to standard therapy improves response rate and progression-free survival in relapsed or refractory thrombotic thrombocytopenic purpura and autoimmune haemolytic anaemia. Thromb Haemost 2007; 97:228–233.
- Sadler JE, Moake JL, Miyata T, George JN. Recent advances in thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program 2004; 1:407–423.
- Shenkman B, Einav Y. Thrombotic thrombocytopenic purpura and other thrombotic microangiopathic hemolytic anemias: diagnosis and classification. Autoimmun Rev 2014; 13:584–586.
- Shah N, Sarode R. Thrombotic thrombocytopenic purpura-what is new? J Clin Apher 2013; 28:30–35.
- Imanirad I, Rajasekhar A, Zumberg M. A case series of atypical presentations of thrombotic thrombocytopenic purpura. J Clin Apher 2012; 27:221–226.
- George JN, Al-Nouri ZL. Diagnostic and therapeutic challenges in the thrombotic thrombocytopenic purpura and hemolytic uremic syndromes. Hematology Am Soc Hematol Educ Program 2012; 1:604–609.
- Shah N, Rutherford C, Matevosyan K, Shen YM, Sarode R. Role of ADAMTS13 in the management of thrombotic microangiopathies including thrombotic thrombocytopenic purpura (TTP). Br J Haematol 2013; 163:514–519.
- Cataland SR, Yang S, Wu HM. The use of ADAMTS13 activity, platelet count, and serum creatinine to differentiate acquired thrombotic thrombocytopenic purpura from other thrombotic microangiopathies. Br J Haematol 2012; 157:501–503.
- Mannucci PM, Peyvandi F. TTP and ADAMTS13: when Is testing appropriate? Hematology Am Soc Hematol Educ Program 2007; 1:121–126.
- Chaturved S, Carcioppolo D, Zhang L, McCar KR. Management and outcomes of patients with TTP: analysis of 100 cases at a single institution. Am J Hematol 2013; 88:560–565.
- Peyvandi F, Palla R, Lotta LA, Mackie I, Scully MA, Machin SJ. ADAMTS-13 assays in thrombotic thrombocytopenic purpura. J Thromb Haemost 2010; 8:631–640.
- Cataland SR, Scully MA, Paskavitz J, et al. Evidence of persistent neurologic injury following thrombotic thrombocytopenic purpura. Am J Hematol 2011; 86:87–89.
- Meloni G, Proia A, Antonini G, et al. Thrombotic thrombocytopenic purpura: prospective neurologic, neuroimaging and neurophysiologic evaluation. Haematologica 2001; 86:1194–1199.
- Kwaan HC, Boggio LN. The clinical spectrum of thrombotic thrombocytopenic purpura. Semin Thromb Hemost 2005; 31:673–680.
- Sarode R. Atypical presentations of thrombotic thrombocytopenic purpura: a review. J Clin Apher 2009; 24:47–52.
- Volcy J, Nzerue CM, Oderinde A, Hewan-Iowe K. Cocaine-induced acute renal failure, hemolysis, and thrombocytopenia mimicking thrombotic thrombocytopenic purpura. Am J Kidney Dis 2000; 35:E3.
- Kremer Hovinga JA, Mottini M, Lammle B. Measurement of ADAMTS-13 activity in plasma by the FRETS-VWF73 assay: comparison with other assay methods. J Thromb Haemost 2006; 4:1146–1148.
- Groot E, Hulstein JJ, Rison CN, de Groot PG, Fijnheer R. FRETS-VWF73: a rapid and predictive tool for thrombotic thrombocytopenic purpura. J Thromb Haemost 2006; 4:698–699.
- Barrows BD, Teruya J. Use of the ADAMTS13 activity assay improved the accuracy and efficiency of the diagnosis and treatment of suspected acquired thrombotic thrombocytopenic purpura. Arch Pathol Lab Med 2014; 138:546–549.
- Rieger M, Mannucci PM, Kremer Hovinga JA, et al. ADAMTS13 autoantibodies in patients with thrombotic microangiopathies and other immunomediated diseases. Blood 2005; 106:1262–1267.
- Rogers HJ, Kottke-Marchant K. ADAMTS13 evaluation for thrombotic thrombocytopenic purpura. Pathology Innovations, Pathology and Laboratory Medicine Institute. Cleveland Clinic, Fall 2014:6–9.
- Józsi M, Licht C, Strobel S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 2008; 111:1512–1514.
- Diamante Chiodini B, Davin JC, Corazza F, et al. Eculizumab in anti-factor H antibodies associated with atypical hemolytic uremic syndrome. Pediatrics 2014; 133:e1764–e1768.
- Taylor CM, Machin S, Wigmore SJ, Goodship TH; working party from the Renal Association, the British Committee for Standards in Haematology and the British Transplantation Society. Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol 2009; 148:37–47.
- Loirat C, Garnier A, Sellier-Leclerc AL, Kwon T. Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost 2010; 36:673–681.
- Tsai HM, Kuo E. Eculizumab therapy leads to rapid resolution of thrombocytopenia in atypical hemolytic uremic syndrome. Adv Hematol 2014; 295323:1–7.
- Lapeyraque AL, Frémeaux-Bacchi V, Robitaille P. Efficacy of eculizumab in a patient with factor-H-associated atypical hemolytic uremic syndrome. Pediatr Nephrol 2011; 26:621–624.
- Baskin E, Gulleroglu K, Kantar A, Bayrakci U, Ozkaya O. Success of eculizumab in the treatment of atypical hemolytic uremic syndrome. Pediatr Nephrol 2015; 30:783–789.
- Scully M, Hunt BJ, Benjamin S, et al; British Committee for Standards in Haematology. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol 2012; 158:323–325.
- Abassi E, Yawn D, Leveque E, Nolasco L, Lopez J, Moake J. Correlation of ADAMTS-13 activity with response to plasma exchange in patients diagnosed with thrombotic thrombocytopenic purpura (Abstract #3921). Blood 2004; 104:242a.
- Blombery P, Scully M. Management of thrombocytic thrombocytopenic purpura: current perspectives. J Blood Med 2014; 5:15–23.
- Wu N, Liu J, Yang S, et al. Diagnostic and prognostic values of ADAMTS13 activity measured during daily plasma exchange therapy in patients with acquired thrombotic thrombocytopenic purpura. Transfusion 2015; 55:18–24.
- Cuker A. Adjuvant rituximab to prevent TTP relapse. Blood 2016; 127:2952–2953.
- Chapman K, Yuen S. Therapy for thrombotic thrombocytopenic purpura: past, present and future. Semin Thromb Hemost 2014; 40:34–40.
- Heidel F, Lipka DB, von Auer C, Huber C, Schrarrer I, Hess G. Addition of rituximab to standard therapy improves response rate and progression-free survival in relapsed or refractory thrombotic thrombocytopenic purpura and autoimmune haemolytic anaemia. Thromb Haemost 2007; 97:228–233.
KEY POINTS
- Symptoms of TTP are usually neurologic but can also be cardiac or abdominal. Thrombocytopenia and unexplained microangiopathic hemolytic anemia are sufficient to highly suspect the disease.
- In the appropriate clinical setting, an ADAMTS13 activity level lower than 10% is highly indicative of TTP.
- ADAMTS13 inhibitor and ADAMTS13 antibody assays provide more diagnostic clues. ADAMTS13 antibody is generally absent in the congenital form.
- The ADAMTS13 assay can help distinguish TTP from hemolytic-uremic syndrome, which presents similarly but typically involves normal or only mildly reduced ADAMTS13 activity.
- A strong clinical suspicion of TTP warrants immediate initiation of therapeutic plasma exchange without waiting for ADAMTS13 test results.
Ankle pain in a young woman with Gaucher disease
A 20-year-old woman with Gaucher disease presents with pain in her right ankle and in her back. She has had the ankle pain for the past 12 months and the back pain for the past 2 years. She describes the ankle pain as stabbing and moderately severe. It is constant, present both at rest and during physical activity, but aggravated by walking and twisting movements. She has noticed grinding and clicking sounds as she moves her ankle. The ankle pain has worsened over the past several months.
She says her back pain is similar to her ankle pain but less severe. She also reports generalized mild aches and bone pain. No other joints are involved. She has no history of fever, chills, or trauma.
A COMPLICATED MEDICAL HISTORY
Her Gaucher disease was diagnosed at age 4 when she presented with failure to thrive and with thrombocytopenia and splenomegaly. She and was found to have an N370S/IVS2+1 mutation of the GBA gene. She underwent removal of 90% of her spleen at the time of diagnosis and was on enzyme replacement therapy with imiglucerase until 3 years ago, when the treatment was stopped because the drug had become unavailable (because of a temporary closure of the manufacturing facility), and because she had developed neutralizing antibodies to it. Despite a dosage as high as 120 U/kg every 2 weeks (the recommended range is 2.5 U/kg three times a week up to 60 U/kg every 2 weeks), her anemia and thrombocytopenia worsened to the point that she became dependent on transfusion of red blood cells and platelets. She has also taken glucocorticoids at various times in the past as a premedication before enzyme replacement therapy.
About 3 years ago, she developed dryness of the skin, pruritus, shiny skin, hardening of the skin, and decreased oral aperture, which was diagnosed as scleroderma.
During the past 5 years, she has had multiple episodes of pale coloration of her skin on exposure to cold, suggestive of Raynaud phenomenon. And for the past 5 months, she has noticed a burning sensation in her throat and retrosternal pain, suggestive of gastroesophageal reflux disease.
She is a college student, with no history of smoking or use of alcohol or recreational drugs. She is sexually active, with no history of sexually transmitted disease, and she uses condoms and oral contraceptives for contraception.
Her father and mother are both carriers of Gaucher disease. She is not of Ashkenazi Jewish descent.
FINDINGS ON PHYSICAL EXAMINATION
On physical examination, her temperature, blood pressure, pulse, and respiratory rate are within normal limits. She has extensive tattooing on her upper chest to hide scarring from previous cannulation ports. The right ankle joint is moderately swollen but shows no other signs of inflammation; its range of motion is limited by severe pain. She has tenderness of the spinous processes and paraspinal area, in addition to multiple tender points in the thoracolumbar area. Palpation of the right hip reveals tenderness of the groin and trochanteric bursa.
No lymphadenopathy, hepatomegaly, splenomegaly, or abdominal masses are noted. Neurologic examination is essentially nonfocal.
Her current medications include omeprazole, ergocalciferol, calcium carbonate, gabapentin, citalopram, and celecoxib. She also takes a multivitamin daily.
1. Which is the most likely underlying cause of her ankle pain?
- Rheumatoid arthritis
- Gaucher disease
- Septic arthritis
- Avascular necrosis secondary to steroid use
Rheumatoid arthritis varies in its presentation. It is usually insidious in onset, migratory, and intermittent, with polyarticular or even monoarticular involvement, and it presents with pain, stiffness, and swelling of the joint.1 Most often affected are the metacarpophalangeal, proximal interphalangeal, wrist, and metatarsophalangeal joints. Involvement of large joints of the upper and lower limbs is also common.2 This is not the most likely cause of this patient’s symptoms, based on the history and the current presentation.
Gaucher disease is a lipidosis caused by accumulation of cellular glycolipids, especially glucocerebrosides, due to deficiency of the enzyme beta-glucosidase. Clinical manifestations include hepatomegaly, splenomegaly, and bone marrow disease presenting as anemia, thrombocytopenia, or skeletal disease.3 Skeletal involvement in Gaucher disease includes bone pain, bone infarcts, and lytic lesions.
Whether splenectomy predisposes the patient to bone manifestations is controversial. Some believe that splenectomy decreases the total body reservoir for the storage of glycolipids and predisposes to their deposition in bone, which in turn results in cortical thinning, impaired remodeling, and decreased intraosseous blood flow, leading to osteonecrosis and fractures.4 This is more common in patients with type 1 Gaucher disease who have undergone splenectomy. (Types 2 and 3 are much rarer, occurring mainly in children; central nervous system involvement is a key feature. A discussion of these types is beyond the focus of this paper.) However, some studies suggest that the increase in bone manifestations after splenectomy may be simply because of severe disease.5 It should be noted that, since the advent of enzyme replacement therapy for Gaucher disease, splenectomy is now rarely performed.6
Anemia is also considered an independent risk factor for the development of avascular necrosis in type 1 Gaucher disease.7 Osteonecrosis due to Gaucher disease is relatively common in the femur, tibia, and humerus and uncommon in the ankle joints.8
Septic arthritis is unlikely in this patient in the absence of fever or signs of inflammation of the joint. Her long-standing history of ankle pain would also be unusual for infection, but a superimposed infectious process should always be suspected in an arthritic joint.
Avascular necrosis secondary to steroid use. Glucocorticoids are notorious for their adverse effects on bone. They induce osteocyte apoptosis and a decrease in bone remodeling, potentially predisposing to osteonecrosis.9 There is a high incidence of osteoporosis, osteonecrosis, and fracture risk with glucocorticoid therapy, and the incidence is dose-dependent. Discontinuation of the drug only partially restores fracture risk to baseline levels.10,11
A meta-analysis of cohort studies with a total sample size of about 42,000 reported an increased risk of fracture at all ages with the use of glucocorticoids.12 Because the minimum dosage and duration of therapy to prevent glucocorticoid-induced osteoporosis are not known, the only recommendation is to keep the dosage as low as possible.13
Glucocorticoid therapy is the most common cause of nontraumatic avascular necrosis. The risk of osteonecrosis in patients on long-term glucocorticoid therapy may be as high as 40%.14 The risk is increased with prolonged treatment and with high doses, but it can also occur with short-term exposure to high doses. The increased risk has been shown to persist for as long as 2 years after the drugs are discontinued.15 Glucocorticoid-induced bone disease commonly affects the hip and vertebrae.
At this stage of the workup, we cannot completely rule out glucocorticoid use as the cause. However, after considering this patient’s presentation and the key features of the other diagnoses, her ankle pain and back pain are more likely caused by her preexisting Gaucher disease.
CONTINUED EVALUATION
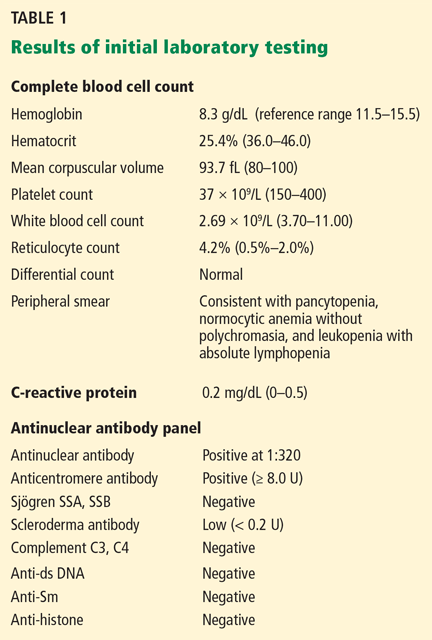
Initial laboratory tests (Table 1) reveal severe anemia and thrombocytopenia. Bone marrow biopsy of the iliac crest done as part of the workup for these conditions shows extensive bone marrow space replacement by histiocytic infiltrate, consistent with Gaucher disease. No other marrow process is observed.
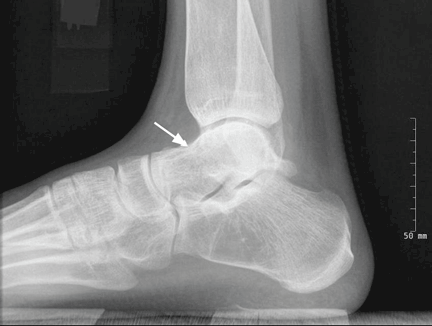
Radiography of the ankle (Figure 1) shows a subtle lucency in the talar dome with minimal subarticular collapse seen on the lateral view, suggestive of avascular necrosis and diffuse osteopenia. Joint spaces are maintained.
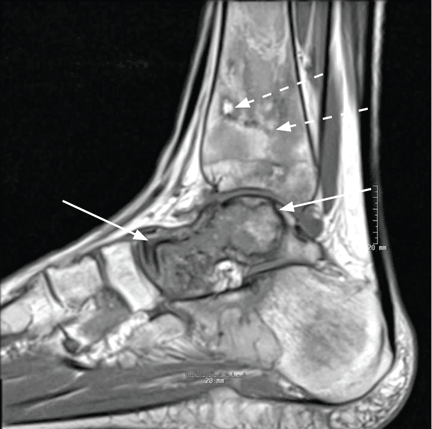
Magnetic resonance imaging (MRI) of the ankle shows numerous bone infarcts with an approximately 15-mm region of mild articular surface collapse in the central and lateral aspect of the talar dome.
MRI of the back shows extensive abnormal bone marrow signal intensity throughout the spine, compatible with a marrow replacement process. Patchy nonexpansile T2/stir hyperintensity with serpiginous enhancement within the T9, T11, T12, L2, and L3 vertebral bodies as well as throughout the entire sacrum is consistent with bone infarct.
2. Based on the results of radiographic studies, which is most likely the immediate cause of her ankle pain?
- Talar avascular necrosis secondary to rheumatoid arthritis
- Talar avascular necrosis secondary to Gaucher disease
- Trauma-induced fracture of the talus
- Plantar fasciitis
Of the bones of the feet, the talus is unique. It is the second largest of the tarsal bones and does not have muscular or tendinous attachments. Sixty percent of the talus bone is covered by articular cartilage,16 so only a limited area is available for penetration of blood vessels. Also, small nutrient vessels and variations of intraosseous anastomoses with a lack of collateral circulation predispose the talus to osteonecrosis when the vascular supply is compromised.16
Radiographic evidence of avascular necrosis is the presence of bone that is more radiopaque than normal bone; this is necrotic bone surrounded by osteopenic bone. Avascular necrosis causes hyperemia and resorption of bone. The resorption does not take place in necrotic bone because of the lack of a vascular supply, and so it appears radiopaque, whereas the bone surrounding the necrotic bone becomes osteopenic and radiolucent.
The sclerotic rim of a bone infarct is also enhanced by an attempted healing process in which new bone forms on the surface of necrotic trabeculae, a process known as “creeping substitution.” This gives a typical sclerotic picture of the talus.
MRI is the most sensitive technique for detecting osteonecrosis. A characteristic radiographic pattern is seen with osteonecrosis of the talus starting with talar dome opacity, followed by deformity and, in severe cases, articular collapse and bone fragmentation.17
The radiograph in our patient’s case is not consistent with features of rheumatoid arthritis or traumatic fracture of the talus. In plantar fasciitis, radiographs are used to rule out other pathologies of the foot, and the only finding may be a bone spur seen at the site of pain. The bone spur is not the cause of pain in plantar fasciitis but may be a result of the plantar fasciitis itself.
Therefore, avascular necrosis secondary to Gaucher disease is most likely the immediate cause of her ankle pain.
THE COURSE OF TREATMENT
The patient is started on enzyme replacement therapy with taliglucerase alfa (see discussion of enzyme replacement below). For the ankle pain, conservative management is prescribed, with application of a splint and a boot.
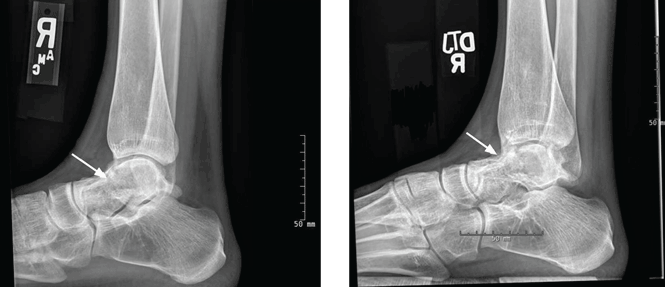
After 4 months of conservative management, radiography (Figure 2) and magnetic resonance imaging (Figure 3) show progressive deterioration of the talus body, and her ankle pain has worsened. A 6-week trial of an ankle brace also proves futile. Her pain continues to worsen and is not controllable with high doses of pain medication. She requests below-the-knee amputation.
Given the complexity of this patient’s medical condition, fusion of the ankle and hindfoot—which in some patients is preferable to amputation—is not considered because of her extensive bone involvement and ongoing thrombocytopenia, which would impede healing after the procedure. Below-the-knee amputation is performed without complications.
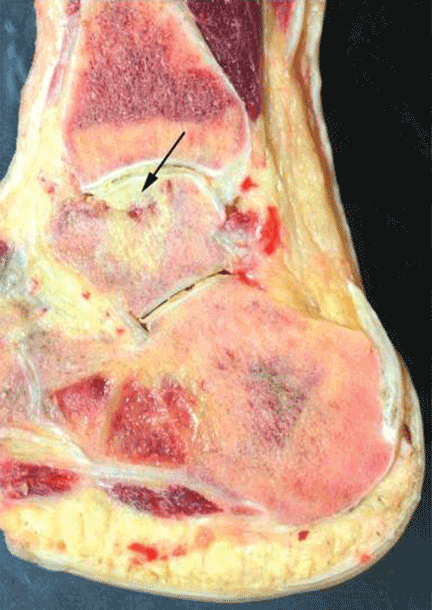
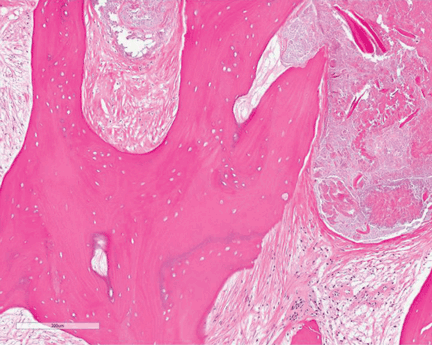
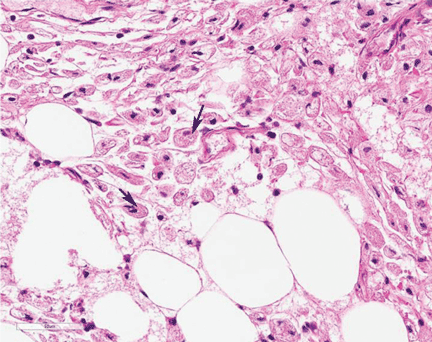
Study of the specimen after amputation reveals talar bone necrosis and bone marrow infiltration by foamy macrophages, consistent with Gaucher disease (Figures 4–6).
GAUCHER DISEASE
Pharmacologic treatments, effective only for type 1 Gaucher disease, target hepatosplenomegaly, cytopenia, and bone manifestations. Two approaches are enzyme replacement therapy—ie, to replace the defective enzyme—and substrate reduction therapy—ie, to reduce the production and thus the accumulation of glucocerebroside. Enzyme replacement is the first choice of therapy; substrate reduction is reserved for patients unable to tolerate enzyme replacement therapy.
Enzyme replacement
Current drugs for enzyme replacement therapy are imiglucerase, taliglucerase alfa, and velaglucerase alfa. The drugs are given by intravenous infusion over 1 to 2 hours in an outpatient clinic or office every 2 weeks.
These drugs are extremely expensive. Currently, the estimated cost of therapy for 1 year would be $432,978 for imiglucerase, $324,870 for taliglucerase alfa, and $368,550 for velaglucerase alfa. (The estimated costs are for 1 year of treatment for a 70-kg patient at 60 U/kg every 2 weeks.)18 Taliglucerase alfa is less expensive than the other two because it is plant-derived and thus can be more readily produced on a large scale.19
Substrate reduction
Current drugs for substrate reduction therapy are eliglustat and miglustat. They are given orally. Eliglustat is the first oral drug approved as a first-line treatment for Gaucher disease.20 Miglustat is approved only for mild to moderate disease when enzyme replacement fails or is not tolerated.
Patients can develop antibodies to any of the enzyme replacement drugs. It is not known whether this antibody response differs among the three drugs.21
Avascular necrosis of bone can occur in many clinical settings especially after a fracture, particularly of the head of the femur, which leads to interruption of blood supply to the area. Patients with sickle cell disease, those on corticosteroids or bisphosphonates (the latter causing osteonecrosis of the jaw), and those who have pancreatitis or human immunodeficiency virus infection are more prone to this bone complication.
In Gaucher disease, osteonecrosis is associated with splenectomy and severe disease and tends to occur at a younger age than in patients with other diagnoses.8 The plasma chitotriosidase activity and pulmonary and activation-regulated chemokines (PARC/CCL18), which are 10 to 40 times higher than normal in symptomatic patients with Gaucher disease, can be used as a biomarker of disease activity.8 Only plasma chitotriosidase is clinically available and used on a routine basis.
Bone involvement is seen in approximately 75% of the patients with type 1 Gaucher disease,22 and osteonecrosis is a severe form of bone involvement. Monitoring of patients for bone involvement is recommended. Enzyme replacement therapy for Gaucher disease needs to be started even if visceral disease is absent if the patient has evidence of bone involvement in the form of avascular necrosis.7 Prospective studies have shown that enzyme replacement therapy reduces the incidence of osteonecrosis.23
FOLLOW-UP MANAGEMENT OF OUR PATIENT
Avascular necrosis in Gaucher disease more typically involves the hips and shoulders. In the case of our patient, the talus was the most affected bone. Other contributing factors may have been the use of steroids as a premedication (often unnecessary) for her enzyme replacement therapy, as well as the coexistent scleroderma.24
The decision to switch from imiglucerase, to which she developed antibodies, to taliglucerase was made in the hope that the antibodies would not cross-react. After she started taliglucerase, her complete blood count values improved steadily. She did not require transfusions for more than 1 year. Her platelet count rose to 90 × 109/L, and her hemoglobin to 12 g/dL.
A multidisciplinary approach with regular monitoring and appropriate initiation of therapy is necessary to prevent disastrous complications in patients with Gaucher disease.
- Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet 2001; 358:903–911.
- Fleming A, Crown JM, Corbett M. Early rheumatoid disease. I. Onset. Ann Rheum Dis 1976; 35:357–360.
- Grabowski GA, Andria G, Baldellou A, et al. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis, and assessment. Consensus statements. Eur J Pediatr 2004; 163:58–66.
- Rodrigue SW, Rosenthal DI, Barton NW, Zurakowski D, Mankin HJ. Risk factors for osteonecrosis in patients with type 1 Gaucher’s disease. Clin Orthop Relat Res 1999; May (362):201–207.
- Lee RE. The pathology of Gaucher disease. Prog Clin Biol Res 1982; 95:177–217.
- Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis 2008; 31:319–336.
- Khan A, Hangartner T, Weinreb NJ, Taylor JS, Mistry PK. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. J Bone Miner Res 2012; 27:1839–1848.
- Deegan PB, Pavlova E, Tindall J, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine (Baltimore) 2011; 90:52–60.
- Weinstein RS. Glucocorticoid-induced osteonecrosis. Endocrine 2012; 41:183–190.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johnell O, Oden A, et al. The risk and burden of vertebral fractures in Sweden. Osteoporos Int 2004; 15:20–26.
- Seguro LP, Rosario C, Shoenfeld Y. Long-term complications of past glucocorticoid use. Autoimmun Rev 2013; 12:629–632.
- Weinstein RS. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol Metab Clin North Am 2012; 41:595–611.
- Cooper C, Steinbuch M, Stevenson R, Miday R, Watts NB. The epidemiology of osteonecrosis: findings from the GPRD and THIN databases in the UK. Osteoporos Int 2010; 21:569–577.
- Mulfinger GL, Trueta J. The blood supply of the talus. J Bone Joint Surg Br 1970; 52:160–167.
- Pearce DH, Mongiardi CN, Fornasier VL, Daniels TR. Avascular necrosis of the talus: a pictoral essay. Radiographics 2005; 25:399–410.
- In brief: Taliglucerase (Elelyso) for Gaucher disease. Med Lett Drugs Ther 2012 Jul 9; 54(1394):56.
- Hollak CE. An evidence-based review of the potential benefits of taliglucerase alfa in the treatment of patients with Gaucher disease. Core Evid 2012; 7:15–20.
- Poole RM. Eliglustat: first global approval. Drugs 2014; 74:1829–1836.
- Bennett LL, Mohan D. Gaucher disease and its treatment options. Ann Pharmacother 2013; 47:1182–1193.
- Germain DP. Gaucher’s disease: a paradigm for interventional genetics. Clin Genet 2004; 65:77–86.
- Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet 2008; 73:430–440.
- Rennie C, Britton J, Prouse P. Bilateral avascular necrosis of the lunate in a patient with severe Raynaud’s phenomenon and scleroderma. J Clin Rheumatol 1999; 5:165–168.
A 20-year-old woman with Gaucher disease presents with pain in her right ankle and in her back. She has had the ankle pain for the past 12 months and the back pain for the past 2 years. She describes the ankle pain as stabbing and moderately severe. It is constant, present both at rest and during physical activity, but aggravated by walking and twisting movements. She has noticed grinding and clicking sounds as she moves her ankle. The ankle pain has worsened over the past several months.
She says her back pain is similar to her ankle pain but less severe. She also reports generalized mild aches and bone pain. No other joints are involved. She has no history of fever, chills, or trauma.
A COMPLICATED MEDICAL HISTORY
Her Gaucher disease was diagnosed at age 4 when she presented with failure to thrive and with thrombocytopenia and splenomegaly. She and was found to have an N370S/IVS2+1 mutation of the GBA gene. She underwent removal of 90% of her spleen at the time of diagnosis and was on enzyme replacement therapy with imiglucerase until 3 years ago, when the treatment was stopped because the drug had become unavailable (because of a temporary closure of the manufacturing facility), and because she had developed neutralizing antibodies to it. Despite a dosage as high as 120 U/kg every 2 weeks (the recommended range is 2.5 U/kg three times a week up to 60 U/kg every 2 weeks), her anemia and thrombocytopenia worsened to the point that she became dependent on transfusion of red blood cells and platelets. She has also taken glucocorticoids at various times in the past as a premedication before enzyme replacement therapy.
About 3 years ago, she developed dryness of the skin, pruritus, shiny skin, hardening of the skin, and decreased oral aperture, which was diagnosed as scleroderma.
During the past 5 years, she has had multiple episodes of pale coloration of her skin on exposure to cold, suggestive of Raynaud phenomenon. And for the past 5 months, she has noticed a burning sensation in her throat and retrosternal pain, suggestive of gastroesophageal reflux disease.
She is a college student, with no history of smoking or use of alcohol or recreational drugs. She is sexually active, with no history of sexually transmitted disease, and she uses condoms and oral contraceptives for contraception.
Her father and mother are both carriers of Gaucher disease. She is not of Ashkenazi Jewish descent.
FINDINGS ON PHYSICAL EXAMINATION
On physical examination, her temperature, blood pressure, pulse, and respiratory rate are within normal limits. She has extensive tattooing on her upper chest to hide scarring from previous cannulation ports. The right ankle joint is moderately swollen but shows no other signs of inflammation; its range of motion is limited by severe pain. She has tenderness of the spinous processes and paraspinal area, in addition to multiple tender points in the thoracolumbar area. Palpation of the right hip reveals tenderness of the groin and trochanteric bursa.
No lymphadenopathy, hepatomegaly, splenomegaly, or abdominal masses are noted. Neurologic examination is essentially nonfocal.
Her current medications include omeprazole, ergocalciferol, calcium carbonate, gabapentin, citalopram, and celecoxib. She also takes a multivitamin daily.
1. Which is the most likely underlying cause of her ankle pain?
- Rheumatoid arthritis
- Gaucher disease
- Septic arthritis
- Avascular necrosis secondary to steroid use
Rheumatoid arthritis varies in its presentation. It is usually insidious in onset, migratory, and intermittent, with polyarticular or even monoarticular involvement, and it presents with pain, stiffness, and swelling of the joint.1 Most often affected are the metacarpophalangeal, proximal interphalangeal, wrist, and metatarsophalangeal joints. Involvement of large joints of the upper and lower limbs is also common.2 This is not the most likely cause of this patient’s symptoms, based on the history and the current presentation.
Gaucher disease is a lipidosis caused by accumulation of cellular glycolipids, especially glucocerebrosides, due to deficiency of the enzyme beta-glucosidase. Clinical manifestations include hepatomegaly, splenomegaly, and bone marrow disease presenting as anemia, thrombocytopenia, or skeletal disease.3 Skeletal involvement in Gaucher disease includes bone pain, bone infarcts, and lytic lesions.
Whether splenectomy predisposes the patient to bone manifestations is controversial. Some believe that splenectomy decreases the total body reservoir for the storage of glycolipids and predisposes to their deposition in bone, which in turn results in cortical thinning, impaired remodeling, and decreased intraosseous blood flow, leading to osteonecrosis and fractures.4 This is more common in patients with type 1 Gaucher disease who have undergone splenectomy. (Types 2 and 3 are much rarer, occurring mainly in children; central nervous system involvement is a key feature. A discussion of these types is beyond the focus of this paper.) However, some studies suggest that the increase in bone manifestations after splenectomy may be simply because of severe disease.5 It should be noted that, since the advent of enzyme replacement therapy for Gaucher disease, splenectomy is now rarely performed.6
Anemia is also considered an independent risk factor for the development of avascular necrosis in type 1 Gaucher disease.7 Osteonecrosis due to Gaucher disease is relatively common in the femur, tibia, and humerus and uncommon in the ankle joints.8
Septic arthritis is unlikely in this patient in the absence of fever or signs of inflammation of the joint. Her long-standing history of ankle pain would also be unusual for infection, but a superimposed infectious process should always be suspected in an arthritic joint.
Avascular necrosis secondary to steroid use. Glucocorticoids are notorious for their adverse effects on bone. They induce osteocyte apoptosis and a decrease in bone remodeling, potentially predisposing to osteonecrosis.9 There is a high incidence of osteoporosis, osteonecrosis, and fracture risk with glucocorticoid therapy, and the incidence is dose-dependent. Discontinuation of the drug only partially restores fracture risk to baseline levels.10,11
A meta-analysis of cohort studies with a total sample size of about 42,000 reported an increased risk of fracture at all ages with the use of glucocorticoids.12 Because the minimum dosage and duration of therapy to prevent glucocorticoid-induced osteoporosis are not known, the only recommendation is to keep the dosage as low as possible.13
Glucocorticoid therapy is the most common cause of nontraumatic avascular necrosis. The risk of osteonecrosis in patients on long-term glucocorticoid therapy may be as high as 40%.14 The risk is increased with prolonged treatment and with high doses, but it can also occur with short-term exposure to high doses. The increased risk has been shown to persist for as long as 2 years after the drugs are discontinued.15 Glucocorticoid-induced bone disease commonly affects the hip and vertebrae.
At this stage of the workup, we cannot completely rule out glucocorticoid use as the cause. However, after considering this patient’s presentation and the key features of the other diagnoses, her ankle pain and back pain are more likely caused by her preexisting Gaucher disease.
CONTINUED EVALUATION
Initial laboratory tests (Table 1) reveal severe anemia and thrombocytopenia. Bone marrow biopsy of the iliac crest done as part of the workup for these conditions shows extensive bone marrow space replacement by histiocytic infiltrate, consistent with Gaucher disease. No other marrow process is observed.
Radiography of the ankle (Figure 1) shows a subtle lucency in the talar dome with minimal subarticular collapse seen on the lateral view, suggestive of avascular necrosis and diffuse osteopenia. Joint spaces are maintained.
Magnetic resonance imaging (MRI) of the ankle shows numerous bone infarcts with an approximately 15-mm region of mild articular surface collapse in the central and lateral aspect of the talar dome.
MRI of the back shows extensive abnormal bone marrow signal intensity throughout the spine, compatible with a marrow replacement process. Patchy nonexpansile T2/stir hyperintensity with serpiginous enhancement within the T9, T11, T12, L2, and L3 vertebral bodies as well as throughout the entire sacrum is consistent with bone infarct.
2. Based on the results of radiographic studies, which is most likely the immediate cause of her ankle pain?
- Talar avascular necrosis secondary to rheumatoid arthritis
- Talar avascular necrosis secondary to Gaucher disease
- Trauma-induced fracture of the talus
- Plantar fasciitis
Of the bones of the feet, the talus is unique. It is the second largest of the tarsal bones and does not have muscular or tendinous attachments. Sixty percent of the talus bone is covered by articular cartilage,16 so only a limited area is available for penetration of blood vessels. Also, small nutrient vessels and variations of intraosseous anastomoses with a lack of collateral circulation predispose the talus to osteonecrosis when the vascular supply is compromised.16
Radiographic evidence of avascular necrosis is the presence of bone that is more radiopaque than normal bone; this is necrotic bone surrounded by osteopenic bone. Avascular necrosis causes hyperemia and resorption of bone. The resorption does not take place in necrotic bone because of the lack of a vascular supply, and so it appears radiopaque, whereas the bone surrounding the necrotic bone becomes osteopenic and radiolucent.
The sclerotic rim of a bone infarct is also enhanced by an attempted healing process in which new bone forms on the surface of necrotic trabeculae, a process known as “creeping substitution.” This gives a typical sclerotic picture of the talus.
MRI is the most sensitive technique for detecting osteonecrosis. A characteristic radiographic pattern is seen with osteonecrosis of the talus starting with talar dome opacity, followed by deformity and, in severe cases, articular collapse and bone fragmentation.17
The radiograph in our patient’s case is not consistent with features of rheumatoid arthritis or traumatic fracture of the talus. In plantar fasciitis, radiographs are used to rule out other pathologies of the foot, and the only finding may be a bone spur seen at the site of pain. The bone spur is not the cause of pain in plantar fasciitis but may be a result of the plantar fasciitis itself.
Therefore, avascular necrosis secondary to Gaucher disease is most likely the immediate cause of her ankle pain.
THE COURSE OF TREATMENT
The patient is started on enzyme replacement therapy with taliglucerase alfa (see discussion of enzyme replacement below). For the ankle pain, conservative management is prescribed, with application of a splint and a boot.
After 4 months of conservative management, radiography (Figure 2) and magnetic resonance imaging (Figure 3) show progressive deterioration of the talus body, and her ankle pain has worsened. A 6-week trial of an ankle brace also proves futile. Her pain continues to worsen and is not controllable with high doses of pain medication. She requests below-the-knee amputation.
Given the complexity of this patient’s medical condition, fusion of the ankle and hindfoot—which in some patients is preferable to amputation—is not considered because of her extensive bone involvement and ongoing thrombocytopenia, which would impede healing after the procedure. Below-the-knee amputation is performed without complications.
Study of the specimen after amputation reveals talar bone necrosis and bone marrow infiltration by foamy macrophages, consistent with Gaucher disease (Figures 4–6).
GAUCHER DISEASE
Pharmacologic treatments, effective only for type 1 Gaucher disease, target hepatosplenomegaly, cytopenia, and bone manifestations. Two approaches are enzyme replacement therapy—ie, to replace the defective enzyme—and substrate reduction therapy—ie, to reduce the production and thus the accumulation of glucocerebroside. Enzyme replacement is the first choice of therapy; substrate reduction is reserved for patients unable to tolerate enzyme replacement therapy.
Enzyme replacement
Current drugs for enzyme replacement therapy are imiglucerase, taliglucerase alfa, and velaglucerase alfa. The drugs are given by intravenous infusion over 1 to 2 hours in an outpatient clinic or office every 2 weeks.
These drugs are extremely expensive. Currently, the estimated cost of therapy for 1 year would be $432,978 for imiglucerase, $324,870 for taliglucerase alfa, and $368,550 for velaglucerase alfa. (The estimated costs are for 1 year of treatment for a 70-kg patient at 60 U/kg every 2 weeks.)18 Taliglucerase alfa is less expensive than the other two because it is plant-derived and thus can be more readily produced on a large scale.19
Substrate reduction
Current drugs for substrate reduction therapy are eliglustat and miglustat. They are given orally. Eliglustat is the first oral drug approved as a first-line treatment for Gaucher disease.20 Miglustat is approved only for mild to moderate disease when enzyme replacement fails or is not tolerated.
Patients can develop antibodies to any of the enzyme replacement drugs. It is not known whether this antibody response differs among the three drugs.21
Avascular necrosis of bone can occur in many clinical settings especially after a fracture, particularly of the head of the femur, which leads to interruption of blood supply to the area. Patients with sickle cell disease, those on corticosteroids or bisphosphonates (the latter causing osteonecrosis of the jaw), and those who have pancreatitis or human immunodeficiency virus infection are more prone to this bone complication.
In Gaucher disease, osteonecrosis is associated with splenectomy and severe disease and tends to occur at a younger age than in patients with other diagnoses.8 The plasma chitotriosidase activity and pulmonary and activation-regulated chemokines (PARC/CCL18), which are 10 to 40 times higher than normal in symptomatic patients with Gaucher disease, can be used as a biomarker of disease activity.8 Only plasma chitotriosidase is clinically available and used on a routine basis.
Bone involvement is seen in approximately 75% of the patients with type 1 Gaucher disease,22 and osteonecrosis is a severe form of bone involvement. Monitoring of patients for bone involvement is recommended. Enzyme replacement therapy for Gaucher disease needs to be started even if visceral disease is absent if the patient has evidence of bone involvement in the form of avascular necrosis.7 Prospective studies have shown that enzyme replacement therapy reduces the incidence of osteonecrosis.23
FOLLOW-UP MANAGEMENT OF OUR PATIENT
Avascular necrosis in Gaucher disease more typically involves the hips and shoulders. In the case of our patient, the talus was the most affected bone. Other contributing factors may have been the use of steroids as a premedication (often unnecessary) for her enzyme replacement therapy, as well as the coexistent scleroderma.24
The decision to switch from imiglucerase, to which she developed antibodies, to taliglucerase was made in the hope that the antibodies would not cross-react. After she started taliglucerase, her complete blood count values improved steadily. She did not require transfusions for more than 1 year. Her platelet count rose to 90 × 109/L, and her hemoglobin to 12 g/dL.
A multidisciplinary approach with regular monitoring and appropriate initiation of therapy is necessary to prevent disastrous complications in patients with Gaucher disease.
A 20-year-old woman with Gaucher disease presents with pain in her right ankle and in her back. She has had the ankle pain for the past 12 months and the back pain for the past 2 years. She describes the ankle pain as stabbing and moderately severe. It is constant, present both at rest and during physical activity, but aggravated by walking and twisting movements. She has noticed grinding and clicking sounds as she moves her ankle. The ankle pain has worsened over the past several months.
She says her back pain is similar to her ankle pain but less severe. She also reports generalized mild aches and bone pain. No other joints are involved. She has no history of fever, chills, or trauma.
A COMPLICATED MEDICAL HISTORY
Her Gaucher disease was diagnosed at age 4 when she presented with failure to thrive and with thrombocytopenia and splenomegaly. She and was found to have an N370S/IVS2+1 mutation of the GBA gene. She underwent removal of 90% of her spleen at the time of diagnosis and was on enzyme replacement therapy with imiglucerase until 3 years ago, when the treatment was stopped because the drug had become unavailable (because of a temporary closure of the manufacturing facility), and because she had developed neutralizing antibodies to it. Despite a dosage as high as 120 U/kg every 2 weeks (the recommended range is 2.5 U/kg three times a week up to 60 U/kg every 2 weeks), her anemia and thrombocytopenia worsened to the point that she became dependent on transfusion of red blood cells and platelets. She has also taken glucocorticoids at various times in the past as a premedication before enzyme replacement therapy.
About 3 years ago, she developed dryness of the skin, pruritus, shiny skin, hardening of the skin, and decreased oral aperture, which was diagnosed as scleroderma.
During the past 5 years, she has had multiple episodes of pale coloration of her skin on exposure to cold, suggestive of Raynaud phenomenon. And for the past 5 months, she has noticed a burning sensation in her throat and retrosternal pain, suggestive of gastroesophageal reflux disease.
She is a college student, with no history of smoking or use of alcohol or recreational drugs. She is sexually active, with no history of sexually transmitted disease, and she uses condoms and oral contraceptives for contraception.
Her father and mother are both carriers of Gaucher disease. She is not of Ashkenazi Jewish descent.
FINDINGS ON PHYSICAL EXAMINATION
On physical examination, her temperature, blood pressure, pulse, and respiratory rate are within normal limits. She has extensive tattooing on her upper chest to hide scarring from previous cannulation ports. The right ankle joint is moderately swollen but shows no other signs of inflammation; its range of motion is limited by severe pain. She has tenderness of the spinous processes and paraspinal area, in addition to multiple tender points in the thoracolumbar area. Palpation of the right hip reveals tenderness of the groin and trochanteric bursa.
No lymphadenopathy, hepatomegaly, splenomegaly, or abdominal masses are noted. Neurologic examination is essentially nonfocal.
Her current medications include omeprazole, ergocalciferol, calcium carbonate, gabapentin, citalopram, and celecoxib. She also takes a multivitamin daily.
1. Which is the most likely underlying cause of her ankle pain?
- Rheumatoid arthritis
- Gaucher disease
- Septic arthritis
- Avascular necrosis secondary to steroid use
Rheumatoid arthritis varies in its presentation. It is usually insidious in onset, migratory, and intermittent, with polyarticular or even monoarticular involvement, and it presents with pain, stiffness, and swelling of the joint.1 Most often affected are the metacarpophalangeal, proximal interphalangeal, wrist, and metatarsophalangeal joints. Involvement of large joints of the upper and lower limbs is also common.2 This is not the most likely cause of this patient’s symptoms, based on the history and the current presentation.
Gaucher disease is a lipidosis caused by accumulation of cellular glycolipids, especially glucocerebrosides, due to deficiency of the enzyme beta-glucosidase. Clinical manifestations include hepatomegaly, splenomegaly, and bone marrow disease presenting as anemia, thrombocytopenia, or skeletal disease.3 Skeletal involvement in Gaucher disease includes bone pain, bone infarcts, and lytic lesions.
Whether splenectomy predisposes the patient to bone manifestations is controversial. Some believe that splenectomy decreases the total body reservoir for the storage of glycolipids and predisposes to their deposition in bone, which in turn results in cortical thinning, impaired remodeling, and decreased intraosseous blood flow, leading to osteonecrosis and fractures.4 This is more common in patients with type 1 Gaucher disease who have undergone splenectomy. (Types 2 and 3 are much rarer, occurring mainly in children; central nervous system involvement is a key feature. A discussion of these types is beyond the focus of this paper.) However, some studies suggest that the increase in bone manifestations after splenectomy may be simply because of severe disease.5 It should be noted that, since the advent of enzyme replacement therapy for Gaucher disease, splenectomy is now rarely performed.6
Anemia is also considered an independent risk factor for the development of avascular necrosis in type 1 Gaucher disease.7 Osteonecrosis due to Gaucher disease is relatively common in the femur, tibia, and humerus and uncommon in the ankle joints.8
Septic arthritis is unlikely in this patient in the absence of fever or signs of inflammation of the joint. Her long-standing history of ankle pain would also be unusual for infection, but a superimposed infectious process should always be suspected in an arthritic joint.
Avascular necrosis secondary to steroid use. Glucocorticoids are notorious for their adverse effects on bone. They induce osteocyte apoptosis and a decrease in bone remodeling, potentially predisposing to osteonecrosis.9 There is a high incidence of osteoporosis, osteonecrosis, and fracture risk with glucocorticoid therapy, and the incidence is dose-dependent. Discontinuation of the drug only partially restores fracture risk to baseline levels.10,11
A meta-analysis of cohort studies with a total sample size of about 42,000 reported an increased risk of fracture at all ages with the use of glucocorticoids.12 Because the minimum dosage and duration of therapy to prevent glucocorticoid-induced osteoporosis are not known, the only recommendation is to keep the dosage as low as possible.13
Glucocorticoid therapy is the most common cause of nontraumatic avascular necrosis. The risk of osteonecrosis in patients on long-term glucocorticoid therapy may be as high as 40%.14 The risk is increased with prolonged treatment and with high doses, but it can also occur with short-term exposure to high doses. The increased risk has been shown to persist for as long as 2 years after the drugs are discontinued.15 Glucocorticoid-induced bone disease commonly affects the hip and vertebrae.
At this stage of the workup, we cannot completely rule out glucocorticoid use as the cause. However, after considering this patient’s presentation and the key features of the other diagnoses, her ankle pain and back pain are more likely caused by her preexisting Gaucher disease.
CONTINUED EVALUATION
Initial laboratory tests (Table 1) reveal severe anemia and thrombocytopenia. Bone marrow biopsy of the iliac crest done as part of the workup for these conditions shows extensive bone marrow space replacement by histiocytic infiltrate, consistent with Gaucher disease. No other marrow process is observed.
Radiography of the ankle (Figure 1) shows a subtle lucency in the talar dome with minimal subarticular collapse seen on the lateral view, suggestive of avascular necrosis and diffuse osteopenia. Joint spaces are maintained.
Magnetic resonance imaging (MRI) of the ankle shows numerous bone infarcts with an approximately 15-mm region of mild articular surface collapse in the central and lateral aspect of the talar dome.
MRI of the back shows extensive abnormal bone marrow signal intensity throughout the spine, compatible with a marrow replacement process. Patchy nonexpansile T2/stir hyperintensity with serpiginous enhancement within the T9, T11, T12, L2, and L3 vertebral bodies as well as throughout the entire sacrum is consistent with bone infarct.
2. Based on the results of radiographic studies, which is most likely the immediate cause of her ankle pain?
- Talar avascular necrosis secondary to rheumatoid arthritis
- Talar avascular necrosis secondary to Gaucher disease
- Trauma-induced fracture of the talus
- Plantar fasciitis
Of the bones of the feet, the talus is unique. It is the second largest of the tarsal bones and does not have muscular or tendinous attachments. Sixty percent of the talus bone is covered by articular cartilage,16 so only a limited area is available for penetration of blood vessels. Also, small nutrient vessels and variations of intraosseous anastomoses with a lack of collateral circulation predispose the talus to osteonecrosis when the vascular supply is compromised.16
Radiographic evidence of avascular necrosis is the presence of bone that is more radiopaque than normal bone; this is necrotic bone surrounded by osteopenic bone. Avascular necrosis causes hyperemia and resorption of bone. The resorption does not take place in necrotic bone because of the lack of a vascular supply, and so it appears radiopaque, whereas the bone surrounding the necrotic bone becomes osteopenic and radiolucent.
The sclerotic rim of a bone infarct is also enhanced by an attempted healing process in which new bone forms on the surface of necrotic trabeculae, a process known as “creeping substitution.” This gives a typical sclerotic picture of the talus.
MRI is the most sensitive technique for detecting osteonecrosis. A characteristic radiographic pattern is seen with osteonecrosis of the talus starting with talar dome opacity, followed by deformity and, in severe cases, articular collapse and bone fragmentation.17
The radiograph in our patient’s case is not consistent with features of rheumatoid arthritis or traumatic fracture of the talus. In plantar fasciitis, radiographs are used to rule out other pathologies of the foot, and the only finding may be a bone spur seen at the site of pain. The bone spur is not the cause of pain in plantar fasciitis but may be a result of the plantar fasciitis itself.
Therefore, avascular necrosis secondary to Gaucher disease is most likely the immediate cause of her ankle pain.
THE COURSE OF TREATMENT
The patient is started on enzyme replacement therapy with taliglucerase alfa (see discussion of enzyme replacement below). For the ankle pain, conservative management is prescribed, with application of a splint and a boot.
After 4 months of conservative management, radiography (Figure 2) and magnetic resonance imaging (Figure 3) show progressive deterioration of the talus body, and her ankle pain has worsened. A 6-week trial of an ankle brace also proves futile. Her pain continues to worsen and is not controllable with high doses of pain medication. She requests below-the-knee amputation.
Given the complexity of this patient’s medical condition, fusion of the ankle and hindfoot—which in some patients is preferable to amputation—is not considered because of her extensive bone involvement and ongoing thrombocytopenia, which would impede healing after the procedure. Below-the-knee amputation is performed without complications.
Study of the specimen after amputation reveals talar bone necrosis and bone marrow infiltration by foamy macrophages, consistent with Gaucher disease (Figures 4–6).
GAUCHER DISEASE
Pharmacologic treatments, effective only for type 1 Gaucher disease, target hepatosplenomegaly, cytopenia, and bone manifestations. Two approaches are enzyme replacement therapy—ie, to replace the defective enzyme—and substrate reduction therapy—ie, to reduce the production and thus the accumulation of glucocerebroside. Enzyme replacement is the first choice of therapy; substrate reduction is reserved for patients unable to tolerate enzyme replacement therapy.
Enzyme replacement
Current drugs for enzyme replacement therapy are imiglucerase, taliglucerase alfa, and velaglucerase alfa. The drugs are given by intravenous infusion over 1 to 2 hours in an outpatient clinic or office every 2 weeks.
These drugs are extremely expensive. Currently, the estimated cost of therapy for 1 year would be $432,978 for imiglucerase, $324,870 for taliglucerase alfa, and $368,550 for velaglucerase alfa. (The estimated costs are for 1 year of treatment for a 70-kg patient at 60 U/kg every 2 weeks.)18 Taliglucerase alfa is less expensive than the other two because it is plant-derived and thus can be more readily produced on a large scale.19
Substrate reduction
Current drugs for substrate reduction therapy are eliglustat and miglustat. They are given orally. Eliglustat is the first oral drug approved as a first-line treatment for Gaucher disease.20 Miglustat is approved only for mild to moderate disease when enzyme replacement fails or is not tolerated.
Patients can develop antibodies to any of the enzyme replacement drugs. It is not known whether this antibody response differs among the three drugs.21
Avascular necrosis of bone can occur in many clinical settings especially after a fracture, particularly of the head of the femur, which leads to interruption of blood supply to the area. Patients with sickle cell disease, those on corticosteroids or bisphosphonates (the latter causing osteonecrosis of the jaw), and those who have pancreatitis or human immunodeficiency virus infection are more prone to this bone complication.
In Gaucher disease, osteonecrosis is associated with splenectomy and severe disease and tends to occur at a younger age than in patients with other diagnoses.8 The plasma chitotriosidase activity and pulmonary and activation-regulated chemokines (PARC/CCL18), which are 10 to 40 times higher than normal in symptomatic patients with Gaucher disease, can be used as a biomarker of disease activity.8 Only plasma chitotriosidase is clinically available and used on a routine basis.
Bone involvement is seen in approximately 75% of the patients with type 1 Gaucher disease,22 and osteonecrosis is a severe form of bone involvement. Monitoring of patients for bone involvement is recommended. Enzyme replacement therapy for Gaucher disease needs to be started even if visceral disease is absent if the patient has evidence of bone involvement in the form of avascular necrosis.7 Prospective studies have shown that enzyme replacement therapy reduces the incidence of osteonecrosis.23
FOLLOW-UP MANAGEMENT OF OUR PATIENT
Avascular necrosis in Gaucher disease more typically involves the hips and shoulders. In the case of our patient, the talus was the most affected bone. Other contributing factors may have been the use of steroids as a premedication (often unnecessary) for her enzyme replacement therapy, as well as the coexistent scleroderma.24
The decision to switch from imiglucerase, to which she developed antibodies, to taliglucerase was made in the hope that the antibodies would not cross-react. After she started taliglucerase, her complete blood count values improved steadily. She did not require transfusions for more than 1 year. Her platelet count rose to 90 × 109/L, and her hemoglobin to 12 g/dL.
A multidisciplinary approach with regular monitoring and appropriate initiation of therapy is necessary to prevent disastrous complications in patients with Gaucher disease.
- Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet 2001; 358:903–911.
- Fleming A, Crown JM, Corbett M. Early rheumatoid disease. I. Onset. Ann Rheum Dis 1976; 35:357–360.
- Grabowski GA, Andria G, Baldellou A, et al. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis, and assessment. Consensus statements. Eur J Pediatr 2004; 163:58–66.
- Rodrigue SW, Rosenthal DI, Barton NW, Zurakowski D, Mankin HJ. Risk factors for osteonecrosis in patients with type 1 Gaucher’s disease. Clin Orthop Relat Res 1999; May (362):201–207.
- Lee RE. The pathology of Gaucher disease. Prog Clin Biol Res 1982; 95:177–217.
- Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis 2008; 31:319–336.
- Khan A, Hangartner T, Weinreb NJ, Taylor JS, Mistry PK. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. J Bone Miner Res 2012; 27:1839–1848.
- Deegan PB, Pavlova E, Tindall J, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine (Baltimore) 2011; 90:52–60.
- Weinstein RS. Glucocorticoid-induced osteonecrosis. Endocrine 2012; 41:183–190.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johnell O, Oden A, et al. The risk and burden of vertebral fractures in Sweden. Osteoporos Int 2004; 15:20–26.
- Seguro LP, Rosario C, Shoenfeld Y. Long-term complications of past glucocorticoid use. Autoimmun Rev 2013; 12:629–632.
- Weinstein RS. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol Metab Clin North Am 2012; 41:595–611.
- Cooper C, Steinbuch M, Stevenson R, Miday R, Watts NB. The epidemiology of osteonecrosis: findings from the GPRD and THIN databases in the UK. Osteoporos Int 2010; 21:569–577.
- Mulfinger GL, Trueta J. The blood supply of the talus. J Bone Joint Surg Br 1970; 52:160–167.
- Pearce DH, Mongiardi CN, Fornasier VL, Daniels TR. Avascular necrosis of the talus: a pictoral essay. Radiographics 2005; 25:399–410.
- In brief: Taliglucerase (Elelyso) for Gaucher disease. Med Lett Drugs Ther 2012 Jul 9; 54(1394):56.
- Hollak CE. An evidence-based review of the potential benefits of taliglucerase alfa in the treatment of patients with Gaucher disease. Core Evid 2012; 7:15–20.
- Poole RM. Eliglustat: first global approval. Drugs 2014; 74:1829–1836.
- Bennett LL, Mohan D. Gaucher disease and its treatment options. Ann Pharmacother 2013; 47:1182–1193.
- Germain DP. Gaucher’s disease: a paradigm for interventional genetics. Clin Genet 2004; 65:77–86.
- Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet 2008; 73:430–440.
- Rennie C, Britton J, Prouse P. Bilateral avascular necrosis of the lunate in a patient with severe Raynaud’s phenomenon and scleroderma. J Clin Rheumatol 1999; 5:165–168.
- Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet 2001; 358:903–911.
- Fleming A, Crown JM, Corbett M. Early rheumatoid disease. I. Onset. Ann Rheum Dis 1976; 35:357–360.
- Grabowski GA, Andria G, Baldellou A, et al. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis, and assessment. Consensus statements. Eur J Pediatr 2004; 163:58–66.
- Rodrigue SW, Rosenthal DI, Barton NW, Zurakowski D, Mankin HJ. Risk factors for osteonecrosis in patients with type 1 Gaucher’s disease. Clin Orthop Relat Res 1999; May (362):201–207.
- Lee RE. The pathology of Gaucher disease. Prog Clin Biol Res 1982; 95:177–217.
- Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis 2008; 31:319–336.
- Khan A, Hangartner T, Weinreb NJ, Taylor JS, Mistry PK. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. J Bone Miner Res 2012; 27:1839–1848.
- Deegan PB, Pavlova E, Tindall J, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine (Baltimore) 2011; 90:52–60.
- Weinstein RS. Glucocorticoid-induced osteonecrosis. Endocrine 2012; 41:183–190.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johnell O, Oden A, et al. The risk and burden of vertebral fractures in Sweden. Osteoporos Int 2004; 15:20–26.
- Seguro LP, Rosario C, Shoenfeld Y. Long-term complications of past glucocorticoid use. Autoimmun Rev 2013; 12:629–632.
- Weinstein RS. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol Metab Clin North Am 2012; 41:595–611.
- Cooper C, Steinbuch M, Stevenson R, Miday R, Watts NB. The epidemiology of osteonecrosis: findings from the GPRD and THIN databases in the UK. Osteoporos Int 2010; 21:569–577.
- Mulfinger GL, Trueta J. The blood supply of the talus. J Bone Joint Surg Br 1970; 52:160–167.
- Pearce DH, Mongiardi CN, Fornasier VL, Daniels TR. Avascular necrosis of the talus: a pictoral essay. Radiographics 2005; 25:399–410.
- In brief: Taliglucerase (Elelyso) for Gaucher disease. Med Lett Drugs Ther 2012 Jul 9; 54(1394):56.
- Hollak CE. An evidence-based review of the potential benefits of taliglucerase alfa in the treatment of patients with Gaucher disease. Core Evid 2012; 7:15–20.
- Poole RM. Eliglustat: first global approval. Drugs 2014; 74:1829–1836.
- Bennett LL, Mohan D. Gaucher disease and its treatment options. Ann Pharmacother 2013; 47:1182–1193.
- Germain DP. Gaucher’s disease: a paradigm for interventional genetics. Clin Genet 2004; 65:77–86.
- Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet 2008; 73:430–440.
- Rennie C, Britton J, Prouse P. Bilateral avascular necrosis of the lunate in a patient with severe Raynaud’s phenomenon and scleroderma. J Clin Rheumatol 1999; 5:165–168.
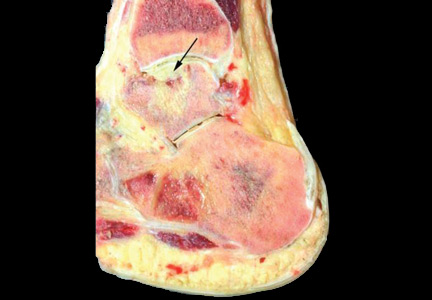
A 54-year-old woman with pancytopenia
A 54-year-old woman with a 1-month history of progressive weakness was transported to the emergency department of a local hospital when a family member found her unresponsive. Before this event, the patient had said she had been feeling tired and cold and looking pale for several weeks.
In the emergency department, her temperature was low. Cableomputed tomography (CT) of the head showed a 1.4-cm hyperdense extraaxial mass. Imaging of the chest showed focal consolidations within the anterior segment of the right upper lobe and the left and right lower lobes.
A urine toxicology screen was positive for acetaminophen (Tylenol), opiates, and benzodiazepines. She was given three doses of naloxone (Narcan), which raised her level of arousal; however, she later became obtunded again and was intubated and transferred to Cleveland Clinic.
A new CT scan of the head confirmed a small left temporal, extradural, calcified lesion with no mass effect or overt bleeding; it appeared most compatible with a solitary calcified meningioma—a likely benign finding.
Her medical history includes hypertension, type 2 diabetes (controlled with diet), and osteoarthritis of the spine. In 1999, she had undergone a hysterectomy that necessitated a blood transfusion. She has never smoked tobacco and does not consume alcohol or use illicit drugs. In the past she worked as a nurse’s aid in a nursing home. However, for the past several years she has stayed at home. Her only avocation of note is gardening.
Initial physical examination
The patient is intubated and sedated. Her temperature is 35.3°C (95.5°F), blood pressure 122/81 mm Hg, heart rate 83 beats per minute, and respiratory rate 14 on assist-controlled ventilator settings with an Fio2 of 100% and a positive end-expiratory pressure of 5 cm H2O.
Her pupils are round, equal, and reactive to light. Her face is symmetric and notable for hirsutism over the chin. Her neck is supple and without lymphadenopathy or thyromegaly.
Rhonchi can be heard at both lung bases. She has normal bowel sounds, and her abdomen is soft and nondistended, with no masses or palpable hepatosplenomegaly. She has no pedal edema on either side, and no clubbing or cyanosis. Her skin is intact, without rashes, lesions, or tattoos. She is able to withdraw from painful stimuli in all four extremities.
INITIAL TESTS PROVIDE A CLUE
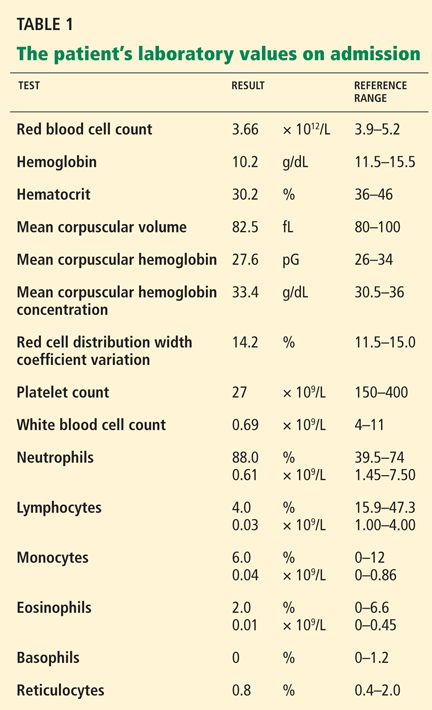
1. Which of the following is the likely cause of this patient’s pancytopenia?
- Folate deficiency
- Gastrointestinal bleeding secondary to colon cancer
- Paroxysmal nocturnal hemoglobinuria
- Myelophthisis
- Other

Causes of pancytopenia are listed in Table 2.
Folate deficiency
Folate is necessary for thymidylate synthesis, a rate-limiting step in DNA synthesis. The minimum daily requirement for dietary folate intake is 50 μg.
Severe deficiency of folate has been reported to cause pancytopenia in alcoholics.1 Abuse of alcohol leads to an abrupt decrease in serum folate (within 2 to 4 days of ceasing intake of proper amounts of folate, as in an alcoholic binge) by inhibiting its absorption in the proximal jejunum as well as its metabolism in the liver.2 The resulting folate deficiency, if sustained, can develop into megaloblastosis in 5 to 10 weeks.
The duration of weakness and pallor reported by this patient would raise suspicion of folate deficiency if she had a history of malnutrition or of alcohol abuse, but she has neither. Further, her mean corpuscular volume is 82.5 fL, red blood cell folate 391 ng/mL (reference range 257–800 ng/mL), and serum vitamin B12 1,886 pg/mL (22–700 pg/mL), and she has no macro-ovalocytes or hypersegmented neutrophils on a peripheral blood smear. This makes folate or vitamin B12 deficiency less likely.
Gastrointestinal bleeding due to colon cancer
Iron-deficiency anemia, hematochezia, melena, a change in bowel habits, and abdominal pain may be manifestations of colon cancer. Cancers of the colon originate from adenomatous polyps arising from the colonic mucosa.
The quantity of occult blood loss depends on the site of the tumor. Patients with tumors in the cecum or ascending colon lose an average of 9 mL/day, whereas those with tumors in the transverse, descending, or sigmoid colon or rectum lose less than 2 mL/day.3
Pertinent laboratory findings in iron-deficiency anemia are a low iron concentration, a low transferrin saturation, a depleted serum ferritin, and a normal to high total iron-binding capacity. An initial microcytic normochromic anemia eventually progresses to a microcytic hypochromic anemia that has a tendency to increasingly demonstrate anisocytosis and poikilocytosis.
Our patient’s symptoms, signs, and laboratory values (with normocytic normochromic anemia) are inconsistent with symptomatic colon cancer leading to iron-deficiency anemia.
Acute myeloid leukemia
Acute myeloid leukemia generally manifests with symptoms related to pancytopenia, with weakness and fatigability being the most common.4
In this condition, genetic alterations in hematopoietic precursor cells result in reduced differentiation capacity and accumulation of leukemic blasts in the bone marrow, peripheral blood, and other tissues.
Peripheral blood analysis usually reveals normocytic normochromic anemia with blasts. To establish a diagnosis of acute myeloid leukemia, one must observe at least 20% myeloblasts in the blood, the bone marrow, or both.
No blasts are seen on our patient’s peripheral blood smear, making acute myeloid leukemia less likely.
Paroxysmal nocturnal hemoglobinuria
Paroxysmal nocturnal hemoglobinuria is a possibility in the setting of intravascular hemolytic anemia, bone marrow failure, and thrombosis.
These processes are due to a defect in the glycosyl phosphatidyl inositol (GPI) anchor caused by an abnormality in the PIG-A gene. Partial or complete absence of the GPI anchor allows for activation of complement-mediated hemolysis. A diminished rate of hematopoiesis is presumably responsible for reticulocytopenia, granulocytopenia, or thrombocytopenia, though reticulocytosis can also be seen.5,6 The highly thrombogenic state is believed to occur because of microparticles rich in phosphatidylserine.7
Our patient’s peripheral smear has rare fragmented red blood cells and lacks teardrop red cells. Although paroxysmal nocturnal hemoglobinuria does not have characteristic morphologic features in the peripheral blood, there are no signs of thrombosis in our patient. Her lactate dehydrogenase level is 395 U/L (reference range 100–220 U/L), and her haptoglobin level is less than 20 mg/dL (33–246). These findings could indicate a low level of intravascular hemolysis.
Myelophthisis
Myelophthisis refers to any disorder in which an abnormal cell process invades the bone marrow, damaging hematopoietic tissue. These processes include neoplastic diseases, storage disorders, and a variety of infections. A decrease in all three cell types may result, depending on the severity of invasion. Documented infectious causes include hepatitis viruses, Epstein-Barr virus, human immunodeficiency virus (HIV), mycobacteria, and fungi.
Our patient’s condition is likely due to a marrow-based process of uncertain etiology. In myelophthisic processes, one may see teardrop red cells, which are not seen in this patient’s smear. However, on her chest imaging, the finding of focal consolidations within the anterior segment of the right upper lobe and both lower lobes raises suspicion of an infectious cause.
CASE CONTINUED: SHE UNDERGOES DIAGNOSTIC TESTING
Let us recap some of the laboratory studies that document the extent of our patient’s pancytopenia and the pattern of her anemia:
- Hemoglobin 10.2 g/dL (reference range 11.5–15.5 g/dL)
- Platelet count 27 × 109/L (150–400)
- Leukopenia with profound T-cell lymphopenia
- Iron 59 μg/dL (30–140)
- Total iron-binding capacity 110 μg/dL (210–415)
- Ferritin 3,004 ng/mL (18–300)
- Transferrin saturation 54% (11%–46%).
2. Which of the following would be the best test to obtain next?
- Bone marrow examination
- Blood cultures
- Tuberculin skin test
- Liver biopsy
- Positron emission tomography and CT
Our patient has unexplained pancytopenia. While all the tests listed above might shed light on her condition, a bone marrow examination would be the best test to obtain next.
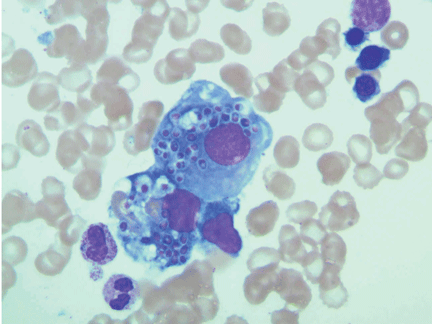
Urine histoplasma antigen studies are positive at greater than 39 ng/mL (normal 0, low positive < 0.6–3.9, moderate positive 4.0–19.9, high positive 20–39 ng/mL). A culture of the marrow subsequently grows this organism.
3. Which of the following tests would establish a definitive diagnosis in this patient?
- Methenamine silver stain of the marrow
- Serum antibody testing
- Fungal culture
- Peripheral blood smear
- Carbolfuchsin stain of marrow
- Urine histoplasma antigen
A prompt diagnosis is critical in patients with acute pulmonary histoplasmosis or progressive disseminated histoplasmosis because early treatment may shorten the clinical course and length of treatment and, in cases of disseminated histoplasmosis, prevent death.8–10
Histopathologic examination of the bone marrow gives the most rapid results, although biopsy to obtain the tissue is invasive. It can give a definitive diagnosis if it reveals the typical 2- to 4-μm yeast structures of H capsulatum. These are observed on an aspirate smear of the patient’s bone marrow biopsy (Figure 1) and can be confirmed by methenamine silver or periodic acid-Schiff staining of the tissue.
Antibody detection is less practical because the antibodies take 2 to 6 weeks after infection to form.11 Also, it is less useful in cases of disseminated infection because many of these patients are immunosuppressed.
Fungal culture remains the gold standard diagnostic test for histoplasmosis. However, results may take up to 1 month and may be falsely negative in less severe cases.
Histoplasma antigen testing is of greater utility in patients with severe disease, including cases of disseminated histoplasmosis. Rates of antigen detection approach 90% in urine specimens from non-AIDS patients with disseminated infection.12 The urine assay has a greater sensitivity and specificity than the serum assay. The rate of detection is lower (ie, around 82%) in patients with acute pulmonary histoplasmosis when both the serum and urine specimens are tested.13
The immunoassay for histoplasma antigen is particularly useful for monitoring the response to therapy. Antigen levels should be measured before treatment is started and at 2 weeks, 1 month, and then approximately every 3 months during therapy.14 If the treatment is effective, antigens should decline by at least 20% in the first month of treatment and by another 20% in each of the following 3-month intervals. Antigen testing should be done every 3 months until a negative antigen level is achieved. The antigen level should also be followed for at least 6 months after treatment has stopped.14
HISTOPLASMA IS INHALED
H capsulatum is the cause of one of the most common pulmonary and systemic mycotic infections in the world, with hundreds of thousands of new cases annually. In areas where the soil is contaminated by bird or bat guano, the fungus is inhaled, resulting in an asymptomatic or a self-limiting influenza-like syndrome in an immunocompetent individual.15
An antigen-specific CD4+ T lymphocytemediated immunity occurs. The immune response of the host is thought to be fungistatic rather than fungicidal, resulting in a persistent inactive infection capable of reactivation in the presence of a host-pathogen imbalance.16
Most infections are asymptomatic or self-limited. For every 2,000 acute infections there is one that results in severe and progressive dissemination, usually in an immunocompromised host.17,18
TREATMENT OF HISTOPLASMOSIS
4. What is the appropriate initial choice of treatment for a severe case of disseminated histoplasmosis?
- Amphotericin B in a lipid complex formulation (Abelcet)
- Itraconazole (Sporanox)
- Fluconazole (Diflucan)
- Ketoconazole (Nizoral)
Untreated, acute disseminated histoplasmosis can progress over a period of 2 to 12 weeks, ultimately killing the patient.17,19
The leading therapies include amphotericin B in a lipid formulation and azole drugs, in particular itraconazole. Fluconazole and ketoconazole are not first-line options in severe cases because they are less predictably effective, and ketoconazole has a higher rate of side effects.20–23 The current recommendation is to treat severely ill hospitalized patients with one of the liposomal formulations or the lipid complex formulation of amphotericin B. Itraconazole is used for patients who have mild to moderate symptoms and as a step-down therapy in patients who improve after initial use of amphotericin B.
CASE CONCLUDED: THE PATIENT RECOVERS
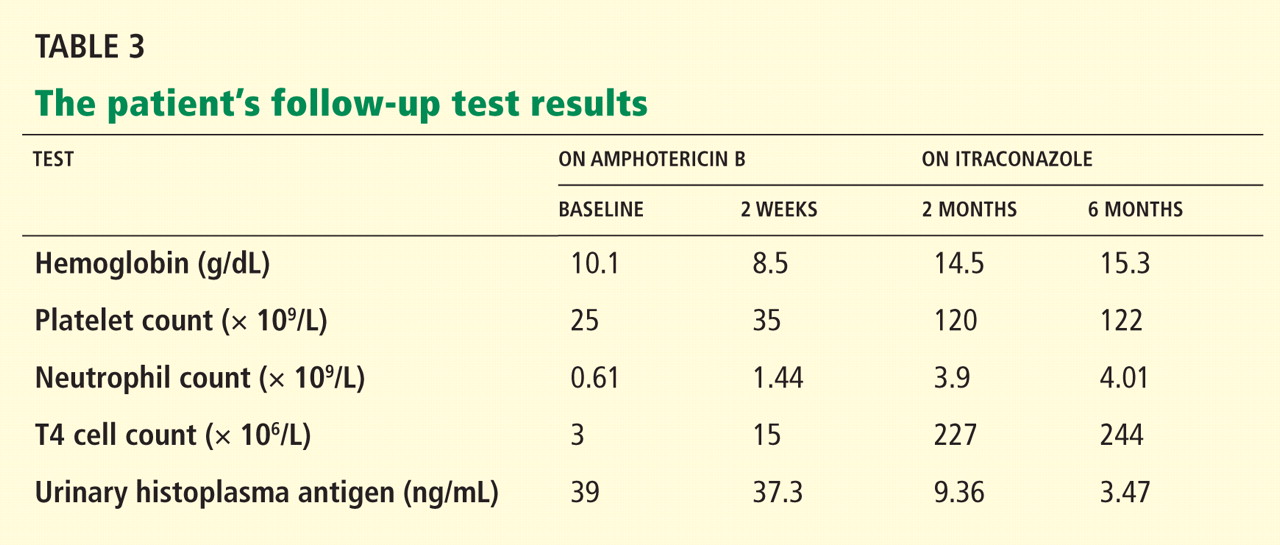
At the time of the initial patient encounter, there was no history of or obvious cause of immunosuppression in this patient. She was found to be HIV-negative and was subsequently diagnosed with “profound immunosuppression of unknown etiology” resulting in a low CD4 count.
The patient receives trimethoprim-sulfamethoxazole (Bactrim, Septra) and azithromycin (Zithromax) for prophylaxis against Pneumocystis carinii pneumonia and Mycobacterium avium intracellulare infection. Two months after the hospitalization, she recalls being at a corn maze 1 month before becoming ill.
- Clarke V, Weston-Smith S. Severe folate-deficiency pancytopenia. BMJ Case Reports 2010; published online.
- Anthony AC. Megaloblastic anemias. In:Hoffman R, Benz EJ, Shattil SJ, Furie B, Cohebn HJ, Silberstein LE, editors. Hematology: Basic Principles and Practice, 2nd ed. New York, NY: Churchill Livingston, 1995:552–586.
- Macrae FA, St John DJ. Relationship between patterns of bleeding and Hemoccult sensitivity in patients with colorectal cancers or adenomas. Gastroenterology 1982; 82:891–898.
- Meyers CA, Albitar M, Estey E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer 2005; 104:788–793.
- Parker CJ. Bone marrow failure syndromes: paroxysmal nocturnal hemoglobinuria. Hematol Oncol Clin North Am 2009; 23:333–346.
- Young NS, Maciejewski JP, Sloand E, et al. The relationship of aplastic anemia and PNH. Int J Hematol 2002; 76(suppl 2):168–172.
- Rosse W. A new way to prevent thrombosis? Blood 2007; 110:3821.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Meals LT, McKinney WP. Acute pulmonary histoplasmosis: progressive pneumonia resulting from high inoculum exposure. J Ky Med Assoc 1998; 96:258–260.
- Salomon J, Flament Saillour M, De Truchis P, et al. An outbreak of acute pulmonary histoplasmosis in members of a trekking trip in Martinique, French West Indies. J Travel Med 2003; 10:87–93.
- Joseph Wheat L. Current diagnosis of histoplasmosis. Trends Microbiol 2003; 11:488–494.
- Wheat LJ, Kauffman CA. Histoplasmosis. Infect Dis Clin North Am 2003; 17:1–19.
- Swartzentruber S, Rhodes L, Kurkjian K, et al. Diagnosis of acute pulmonary histoplasmosis by antigen detection. Clin Infect Dis 2009; 49:1878–1882.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Retallack DM, Woods JP. Molecular epidemiology, pathogenesis, and genetics of the dimorphic fungus Histoplasma capsulatum. Microbes Infect 1999; 1:817–825.
- Deepe GS. The immune response to Histoplasma capsulatum: unearthing its secrets. J Lab Clin Med 1994; 123:201–205.
- Goodwin RA, Shapiro JL, Thurman GH, Thurman SS, Des Prez RM. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore) 1980; 59:1–33.
- Wheat LJ, Connolly-Stringfield PA, Baker RL, et al. Disseminated histoplasmosis in the acquired immune deficiency syndrome: clinical findings, diagnosis and treatment, and review of the literature. Medicine (Baltimore) 1990; 69:361–374.
- Rubin H, Furcolow ML, Yates JL, Brasher CA. The course and prognosis of histoplasmosis. Am J Med 1959; 27:278–288.
- Wheat J, MaWhinney S, Hafner R, et al. Treatment of histoplasmosis with fluconazole in patients with acquired immunodeficiency syndrome. National Institute of Allergy and Infectious Diseases Acquired Immunodeficiency Syndrome Clinical Trials Group and Mycoses Study Group. Am J Med 1997; 103:223–232.
- McKinsey DS, Kauffman CA, Pappas PG, et al. Fluconazole therapy for histoplasmosis. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Clin Infect Dis 1996; 23:996–1001.
- Slama TG. Treatment of disseminated and progressive cavitary histoplasmosis with ketoconazole. Am J Med 1983; 74:70–73.
- Treatment of blastomycosis and histoplasmosis with ketoconazole. Results of a prospective randomized clinical trial. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Ann Intern Med 1985; 103:861–872.
A 54-year-old woman with a 1-month history of progressive weakness was transported to the emergency department of a local hospital when a family member found her unresponsive. Before this event, the patient had said she had been feeling tired and cold and looking pale for several weeks.
In the emergency department, her temperature was low. Cableomputed tomography (CT) of the head showed a 1.4-cm hyperdense extraaxial mass. Imaging of the chest showed focal consolidations within the anterior segment of the right upper lobe and the left and right lower lobes.
A urine toxicology screen was positive for acetaminophen (Tylenol), opiates, and benzodiazepines. She was given three doses of naloxone (Narcan), which raised her level of arousal; however, she later became obtunded again and was intubated and transferred to Cleveland Clinic.
A new CT scan of the head confirmed a small left temporal, extradural, calcified lesion with no mass effect or overt bleeding; it appeared most compatible with a solitary calcified meningioma—a likely benign finding.
Her medical history includes hypertension, type 2 diabetes (controlled with diet), and osteoarthritis of the spine. In 1999, she had undergone a hysterectomy that necessitated a blood transfusion. She has never smoked tobacco and does not consume alcohol or use illicit drugs. In the past she worked as a nurse’s aid in a nursing home. However, for the past several years she has stayed at home. Her only avocation of note is gardening.
Initial physical examination
The patient is intubated and sedated. Her temperature is 35.3°C (95.5°F), blood pressure 122/81 mm Hg, heart rate 83 beats per minute, and respiratory rate 14 on assist-controlled ventilator settings with an Fio2 of 100% and a positive end-expiratory pressure of 5 cm H2O.
Her pupils are round, equal, and reactive to light. Her face is symmetric and notable for hirsutism over the chin. Her neck is supple and without lymphadenopathy or thyromegaly.
Rhonchi can be heard at both lung bases. She has normal bowel sounds, and her abdomen is soft and nondistended, with no masses or palpable hepatosplenomegaly. She has no pedal edema on either side, and no clubbing or cyanosis. Her skin is intact, without rashes, lesions, or tattoos. She is able to withdraw from painful stimuli in all four extremities.
INITIAL TESTS PROVIDE A CLUE
1. Which of the following is the likely cause of this patient’s pancytopenia?
- Folate deficiency
- Gastrointestinal bleeding secondary to colon cancer
- Paroxysmal nocturnal hemoglobinuria
- Myelophthisis
- Other
Causes of pancytopenia are listed in Table 2.
Folate deficiency
Folate is necessary for thymidylate synthesis, a rate-limiting step in DNA synthesis. The minimum daily requirement for dietary folate intake is 50 μg.
Severe deficiency of folate has been reported to cause pancytopenia in alcoholics.1 Abuse of alcohol leads to an abrupt decrease in serum folate (within 2 to 4 days of ceasing intake of proper amounts of folate, as in an alcoholic binge) by inhibiting its absorption in the proximal jejunum as well as its metabolism in the liver.2 The resulting folate deficiency, if sustained, can develop into megaloblastosis in 5 to 10 weeks.
The duration of weakness and pallor reported by this patient would raise suspicion of folate deficiency if she had a history of malnutrition or of alcohol abuse, but she has neither. Further, her mean corpuscular volume is 82.5 fL, red blood cell folate 391 ng/mL (reference range 257–800 ng/mL), and serum vitamin B12 1,886 pg/mL (22–700 pg/mL), and she has no macro-ovalocytes or hypersegmented neutrophils on a peripheral blood smear. This makes folate or vitamin B12 deficiency less likely.
Gastrointestinal bleeding due to colon cancer
Iron-deficiency anemia, hematochezia, melena, a change in bowel habits, and abdominal pain may be manifestations of colon cancer. Cancers of the colon originate from adenomatous polyps arising from the colonic mucosa.
The quantity of occult blood loss depends on the site of the tumor. Patients with tumors in the cecum or ascending colon lose an average of 9 mL/day, whereas those with tumors in the transverse, descending, or sigmoid colon or rectum lose less than 2 mL/day.3
Pertinent laboratory findings in iron-deficiency anemia are a low iron concentration, a low transferrin saturation, a depleted serum ferritin, and a normal to high total iron-binding capacity. An initial microcytic normochromic anemia eventually progresses to a microcytic hypochromic anemia that has a tendency to increasingly demonstrate anisocytosis and poikilocytosis.
Our patient’s symptoms, signs, and laboratory values (with normocytic normochromic anemia) are inconsistent with symptomatic colon cancer leading to iron-deficiency anemia.
Acute myeloid leukemia
Acute myeloid leukemia generally manifests with symptoms related to pancytopenia, with weakness and fatigability being the most common.4
In this condition, genetic alterations in hematopoietic precursor cells result in reduced differentiation capacity and accumulation of leukemic blasts in the bone marrow, peripheral blood, and other tissues.
Peripheral blood analysis usually reveals normocytic normochromic anemia with blasts. To establish a diagnosis of acute myeloid leukemia, one must observe at least 20% myeloblasts in the blood, the bone marrow, or both.
No blasts are seen on our patient’s peripheral blood smear, making acute myeloid leukemia less likely.
Paroxysmal nocturnal hemoglobinuria
Paroxysmal nocturnal hemoglobinuria is a possibility in the setting of intravascular hemolytic anemia, bone marrow failure, and thrombosis.
These processes are due to a defect in the glycosyl phosphatidyl inositol (GPI) anchor caused by an abnormality in the PIG-A gene. Partial or complete absence of the GPI anchor allows for activation of complement-mediated hemolysis. A diminished rate of hematopoiesis is presumably responsible for reticulocytopenia, granulocytopenia, or thrombocytopenia, though reticulocytosis can also be seen.5,6 The highly thrombogenic state is believed to occur because of microparticles rich in phosphatidylserine.7
Our patient’s peripheral smear has rare fragmented red blood cells and lacks teardrop red cells. Although paroxysmal nocturnal hemoglobinuria does not have characteristic morphologic features in the peripheral blood, there are no signs of thrombosis in our patient. Her lactate dehydrogenase level is 395 U/L (reference range 100–220 U/L), and her haptoglobin level is less than 20 mg/dL (33–246). These findings could indicate a low level of intravascular hemolysis.
Myelophthisis
Myelophthisis refers to any disorder in which an abnormal cell process invades the bone marrow, damaging hematopoietic tissue. These processes include neoplastic diseases, storage disorders, and a variety of infections. A decrease in all three cell types may result, depending on the severity of invasion. Documented infectious causes include hepatitis viruses, Epstein-Barr virus, human immunodeficiency virus (HIV), mycobacteria, and fungi.
Our patient’s condition is likely due to a marrow-based process of uncertain etiology. In myelophthisic processes, one may see teardrop red cells, which are not seen in this patient’s smear. However, on her chest imaging, the finding of focal consolidations within the anterior segment of the right upper lobe and both lower lobes raises suspicion of an infectious cause.
CASE CONTINUED: SHE UNDERGOES DIAGNOSTIC TESTING
Let us recap some of the laboratory studies that document the extent of our patient’s pancytopenia and the pattern of her anemia:
- Hemoglobin 10.2 g/dL (reference range 11.5–15.5 g/dL)
- Platelet count 27 × 109/L (150–400)
- Leukopenia with profound T-cell lymphopenia
- Iron 59 μg/dL (30–140)
- Total iron-binding capacity 110 μg/dL (210–415)
- Ferritin 3,004 ng/mL (18–300)
- Transferrin saturation 54% (11%–46%).
2. Which of the following would be the best test to obtain next?
- Bone marrow examination
- Blood cultures
- Tuberculin skin test
- Liver biopsy
- Positron emission tomography and CT
Our patient has unexplained pancytopenia. While all the tests listed above might shed light on her condition, a bone marrow examination would be the best test to obtain next.
Urine histoplasma antigen studies are positive at greater than 39 ng/mL (normal 0, low positive < 0.6–3.9, moderate positive 4.0–19.9, high positive 20–39 ng/mL). A culture of the marrow subsequently grows this organism.
3. Which of the following tests would establish a definitive diagnosis in this patient?
- Methenamine silver stain of the marrow
- Serum antibody testing
- Fungal culture
- Peripheral blood smear
- Carbolfuchsin stain of marrow
- Urine histoplasma antigen
A prompt diagnosis is critical in patients with acute pulmonary histoplasmosis or progressive disseminated histoplasmosis because early treatment may shorten the clinical course and length of treatment and, in cases of disseminated histoplasmosis, prevent death.8–10
Histopathologic examination of the bone marrow gives the most rapid results, although biopsy to obtain the tissue is invasive. It can give a definitive diagnosis if it reveals the typical 2- to 4-μm yeast structures of H capsulatum. These are observed on an aspirate smear of the patient’s bone marrow biopsy (Figure 1) and can be confirmed by methenamine silver or periodic acid-Schiff staining of the tissue.
Antibody detection is less practical because the antibodies take 2 to 6 weeks after infection to form.11 Also, it is less useful in cases of disseminated infection because many of these patients are immunosuppressed.
Fungal culture remains the gold standard diagnostic test for histoplasmosis. However, results may take up to 1 month and may be falsely negative in less severe cases.
Histoplasma antigen testing is of greater utility in patients with severe disease, including cases of disseminated histoplasmosis. Rates of antigen detection approach 90% in urine specimens from non-AIDS patients with disseminated infection.12 The urine assay has a greater sensitivity and specificity than the serum assay. The rate of detection is lower (ie, around 82%) in patients with acute pulmonary histoplasmosis when both the serum and urine specimens are tested.13
The immunoassay for histoplasma antigen is particularly useful for monitoring the response to therapy. Antigen levels should be measured before treatment is started and at 2 weeks, 1 month, and then approximately every 3 months during therapy.14 If the treatment is effective, antigens should decline by at least 20% in the first month of treatment and by another 20% in each of the following 3-month intervals. Antigen testing should be done every 3 months until a negative antigen level is achieved. The antigen level should also be followed for at least 6 months after treatment has stopped.14
HISTOPLASMA IS INHALED
H capsulatum is the cause of one of the most common pulmonary and systemic mycotic infections in the world, with hundreds of thousands of new cases annually. In areas where the soil is contaminated by bird or bat guano, the fungus is inhaled, resulting in an asymptomatic or a self-limiting influenza-like syndrome in an immunocompetent individual.15
An antigen-specific CD4+ T lymphocytemediated immunity occurs. The immune response of the host is thought to be fungistatic rather than fungicidal, resulting in a persistent inactive infection capable of reactivation in the presence of a host-pathogen imbalance.16
Most infections are asymptomatic or self-limited. For every 2,000 acute infections there is one that results in severe and progressive dissemination, usually in an immunocompromised host.17,18
TREATMENT OF HISTOPLASMOSIS
4. What is the appropriate initial choice of treatment for a severe case of disseminated histoplasmosis?
- Amphotericin B in a lipid complex formulation (Abelcet)
- Itraconazole (Sporanox)
- Fluconazole (Diflucan)
- Ketoconazole (Nizoral)
Untreated, acute disseminated histoplasmosis can progress over a period of 2 to 12 weeks, ultimately killing the patient.17,19
The leading therapies include amphotericin B in a lipid formulation and azole drugs, in particular itraconazole. Fluconazole and ketoconazole are not first-line options in severe cases because they are less predictably effective, and ketoconazole has a higher rate of side effects.20–23 The current recommendation is to treat severely ill hospitalized patients with one of the liposomal formulations or the lipid complex formulation of amphotericin B. Itraconazole is used for patients who have mild to moderate symptoms and as a step-down therapy in patients who improve after initial use of amphotericin B.
CASE CONCLUDED: THE PATIENT RECOVERS
At the time of the initial patient encounter, there was no history of or obvious cause of immunosuppression in this patient. She was found to be HIV-negative and was subsequently diagnosed with “profound immunosuppression of unknown etiology” resulting in a low CD4 count.
The patient receives trimethoprim-sulfamethoxazole (Bactrim, Septra) and azithromycin (Zithromax) for prophylaxis against Pneumocystis carinii pneumonia and Mycobacterium avium intracellulare infection. Two months after the hospitalization, she recalls being at a corn maze 1 month before becoming ill.
A 54-year-old woman with a 1-month history of progressive weakness was transported to the emergency department of a local hospital when a family member found her unresponsive. Before this event, the patient had said she had been feeling tired and cold and looking pale for several weeks.
In the emergency department, her temperature was low. Cableomputed tomography (CT) of the head showed a 1.4-cm hyperdense extraaxial mass. Imaging of the chest showed focal consolidations within the anterior segment of the right upper lobe and the left and right lower lobes.
A urine toxicology screen was positive for acetaminophen (Tylenol), opiates, and benzodiazepines. She was given three doses of naloxone (Narcan), which raised her level of arousal; however, she later became obtunded again and was intubated and transferred to Cleveland Clinic.
A new CT scan of the head confirmed a small left temporal, extradural, calcified lesion with no mass effect or overt bleeding; it appeared most compatible with a solitary calcified meningioma—a likely benign finding.
Her medical history includes hypertension, type 2 diabetes (controlled with diet), and osteoarthritis of the spine. In 1999, she had undergone a hysterectomy that necessitated a blood transfusion. She has never smoked tobacco and does not consume alcohol or use illicit drugs. In the past she worked as a nurse’s aid in a nursing home. However, for the past several years she has stayed at home. Her only avocation of note is gardening.
Initial physical examination
The patient is intubated and sedated. Her temperature is 35.3°C (95.5°F), blood pressure 122/81 mm Hg, heart rate 83 beats per minute, and respiratory rate 14 on assist-controlled ventilator settings with an Fio2 of 100% and a positive end-expiratory pressure of 5 cm H2O.
Her pupils are round, equal, and reactive to light. Her face is symmetric and notable for hirsutism over the chin. Her neck is supple and without lymphadenopathy or thyromegaly.
Rhonchi can be heard at both lung bases. She has normal bowel sounds, and her abdomen is soft and nondistended, with no masses or palpable hepatosplenomegaly. She has no pedal edema on either side, and no clubbing or cyanosis. Her skin is intact, without rashes, lesions, or tattoos. She is able to withdraw from painful stimuli in all four extremities.
INITIAL TESTS PROVIDE A CLUE
1. Which of the following is the likely cause of this patient’s pancytopenia?
- Folate deficiency
- Gastrointestinal bleeding secondary to colon cancer
- Paroxysmal nocturnal hemoglobinuria
- Myelophthisis
- Other
Causes of pancytopenia are listed in Table 2.
Folate deficiency
Folate is necessary for thymidylate synthesis, a rate-limiting step in DNA synthesis. The minimum daily requirement for dietary folate intake is 50 μg.
Severe deficiency of folate has been reported to cause pancytopenia in alcoholics.1 Abuse of alcohol leads to an abrupt decrease in serum folate (within 2 to 4 days of ceasing intake of proper amounts of folate, as in an alcoholic binge) by inhibiting its absorption in the proximal jejunum as well as its metabolism in the liver.2 The resulting folate deficiency, if sustained, can develop into megaloblastosis in 5 to 10 weeks.
The duration of weakness and pallor reported by this patient would raise suspicion of folate deficiency if she had a history of malnutrition or of alcohol abuse, but she has neither. Further, her mean corpuscular volume is 82.5 fL, red blood cell folate 391 ng/mL (reference range 257–800 ng/mL), and serum vitamin B12 1,886 pg/mL (22–700 pg/mL), and she has no macro-ovalocytes or hypersegmented neutrophils on a peripheral blood smear. This makes folate or vitamin B12 deficiency less likely.
Gastrointestinal bleeding due to colon cancer
Iron-deficiency anemia, hematochezia, melena, a change in bowel habits, and abdominal pain may be manifestations of colon cancer. Cancers of the colon originate from adenomatous polyps arising from the colonic mucosa.
The quantity of occult blood loss depends on the site of the tumor. Patients with tumors in the cecum or ascending colon lose an average of 9 mL/day, whereas those with tumors in the transverse, descending, or sigmoid colon or rectum lose less than 2 mL/day.3
Pertinent laboratory findings in iron-deficiency anemia are a low iron concentration, a low transferrin saturation, a depleted serum ferritin, and a normal to high total iron-binding capacity. An initial microcytic normochromic anemia eventually progresses to a microcytic hypochromic anemia that has a tendency to increasingly demonstrate anisocytosis and poikilocytosis.
Our patient’s symptoms, signs, and laboratory values (with normocytic normochromic anemia) are inconsistent with symptomatic colon cancer leading to iron-deficiency anemia.
Acute myeloid leukemia
Acute myeloid leukemia generally manifests with symptoms related to pancytopenia, with weakness and fatigability being the most common.4
In this condition, genetic alterations in hematopoietic precursor cells result in reduced differentiation capacity and accumulation of leukemic blasts in the bone marrow, peripheral blood, and other tissues.
Peripheral blood analysis usually reveals normocytic normochromic anemia with blasts. To establish a diagnosis of acute myeloid leukemia, one must observe at least 20% myeloblasts in the blood, the bone marrow, or both.
No blasts are seen on our patient’s peripheral blood smear, making acute myeloid leukemia less likely.
Paroxysmal nocturnal hemoglobinuria
Paroxysmal nocturnal hemoglobinuria is a possibility in the setting of intravascular hemolytic anemia, bone marrow failure, and thrombosis.
These processes are due to a defect in the glycosyl phosphatidyl inositol (GPI) anchor caused by an abnormality in the PIG-A gene. Partial or complete absence of the GPI anchor allows for activation of complement-mediated hemolysis. A diminished rate of hematopoiesis is presumably responsible for reticulocytopenia, granulocytopenia, or thrombocytopenia, though reticulocytosis can also be seen.5,6 The highly thrombogenic state is believed to occur because of microparticles rich in phosphatidylserine.7
Our patient’s peripheral smear has rare fragmented red blood cells and lacks teardrop red cells. Although paroxysmal nocturnal hemoglobinuria does not have characteristic morphologic features in the peripheral blood, there are no signs of thrombosis in our patient. Her lactate dehydrogenase level is 395 U/L (reference range 100–220 U/L), and her haptoglobin level is less than 20 mg/dL (33–246). These findings could indicate a low level of intravascular hemolysis.
Myelophthisis
Myelophthisis refers to any disorder in which an abnormal cell process invades the bone marrow, damaging hematopoietic tissue. These processes include neoplastic diseases, storage disorders, and a variety of infections. A decrease in all three cell types may result, depending on the severity of invasion. Documented infectious causes include hepatitis viruses, Epstein-Barr virus, human immunodeficiency virus (HIV), mycobacteria, and fungi.
Our patient’s condition is likely due to a marrow-based process of uncertain etiology. In myelophthisic processes, one may see teardrop red cells, which are not seen in this patient’s smear. However, on her chest imaging, the finding of focal consolidations within the anterior segment of the right upper lobe and both lower lobes raises suspicion of an infectious cause.
CASE CONTINUED: SHE UNDERGOES DIAGNOSTIC TESTING
Let us recap some of the laboratory studies that document the extent of our patient’s pancytopenia and the pattern of her anemia:
- Hemoglobin 10.2 g/dL (reference range 11.5–15.5 g/dL)
- Platelet count 27 × 109/L (150–400)
- Leukopenia with profound T-cell lymphopenia
- Iron 59 μg/dL (30–140)
- Total iron-binding capacity 110 μg/dL (210–415)
- Ferritin 3,004 ng/mL (18–300)
- Transferrin saturation 54% (11%–46%).
2. Which of the following would be the best test to obtain next?
- Bone marrow examination
- Blood cultures
- Tuberculin skin test
- Liver biopsy
- Positron emission tomography and CT
Our patient has unexplained pancytopenia. While all the tests listed above might shed light on her condition, a bone marrow examination would be the best test to obtain next.
Urine histoplasma antigen studies are positive at greater than 39 ng/mL (normal 0, low positive < 0.6–3.9, moderate positive 4.0–19.9, high positive 20–39 ng/mL). A culture of the marrow subsequently grows this organism.
3. Which of the following tests would establish a definitive diagnosis in this patient?
- Methenamine silver stain of the marrow
- Serum antibody testing
- Fungal culture
- Peripheral blood smear
- Carbolfuchsin stain of marrow
- Urine histoplasma antigen
A prompt diagnosis is critical in patients with acute pulmonary histoplasmosis or progressive disseminated histoplasmosis because early treatment may shorten the clinical course and length of treatment and, in cases of disseminated histoplasmosis, prevent death.8–10
Histopathologic examination of the bone marrow gives the most rapid results, although biopsy to obtain the tissue is invasive. It can give a definitive diagnosis if it reveals the typical 2- to 4-μm yeast structures of H capsulatum. These are observed on an aspirate smear of the patient’s bone marrow biopsy (Figure 1) and can be confirmed by methenamine silver or periodic acid-Schiff staining of the tissue.
Antibody detection is less practical because the antibodies take 2 to 6 weeks after infection to form.11 Also, it is less useful in cases of disseminated infection because many of these patients are immunosuppressed.
Fungal culture remains the gold standard diagnostic test for histoplasmosis. However, results may take up to 1 month and may be falsely negative in less severe cases.
Histoplasma antigen testing is of greater utility in patients with severe disease, including cases of disseminated histoplasmosis. Rates of antigen detection approach 90% in urine specimens from non-AIDS patients with disseminated infection.12 The urine assay has a greater sensitivity and specificity than the serum assay. The rate of detection is lower (ie, around 82%) in patients with acute pulmonary histoplasmosis when both the serum and urine specimens are tested.13
The immunoassay for histoplasma antigen is particularly useful for monitoring the response to therapy. Antigen levels should be measured before treatment is started and at 2 weeks, 1 month, and then approximately every 3 months during therapy.14 If the treatment is effective, antigens should decline by at least 20% in the first month of treatment and by another 20% in each of the following 3-month intervals. Antigen testing should be done every 3 months until a negative antigen level is achieved. The antigen level should also be followed for at least 6 months after treatment has stopped.14
HISTOPLASMA IS INHALED
H capsulatum is the cause of one of the most common pulmonary and systemic mycotic infections in the world, with hundreds of thousands of new cases annually. In areas where the soil is contaminated by bird or bat guano, the fungus is inhaled, resulting in an asymptomatic or a self-limiting influenza-like syndrome in an immunocompetent individual.15
An antigen-specific CD4+ T lymphocytemediated immunity occurs. The immune response of the host is thought to be fungistatic rather than fungicidal, resulting in a persistent inactive infection capable of reactivation in the presence of a host-pathogen imbalance.16
Most infections are asymptomatic or self-limited. For every 2,000 acute infections there is one that results in severe and progressive dissemination, usually in an immunocompromised host.17,18
TREATMENT OF HISTOPLASMOSIS
4. What is the appropriate initial choice of treatment for a severe case of disseminated histoplasmosis?
- Amphotericin B in a lipid complex formulation (Abelcet)
- Itraconazole (Sporanox)
- Fluconazole (Diflucan)
- Ketoconazole (Nizoral)
Untreated, acute disseminated histoplasmosis can progress over a period of 2 to 12 weeks, ultimately killing the patient.17,19
The leading therapies include amphotericin B in a lipid formulation and azole drugs, in particular itraconazole. Fluconazole and ketoconazole are not first-line options in severe cases because they are less predictably effective, and ketoconazole has a higher rate of side effects.20–23 The current recommendation is to treat severely ill hospitalized patients with one of the liposomal formulations or the lipid complex formulation of amphotericin B. Itraconazole is used for patients who have mild to moderate symptoms and as a step-down therapy in patients who improve after initial use of amphotericin B.
CASE CONCLUDED: THE PATIENT RECOVERS
At the time of the initial patient encounter, there was no history of or obvious cause of immunosuppression in this patient. She was found to be HIV-negative and was subsequently diagnosed with “profound immunosuppression of unknown etiology” resulting in a low CD4 count.
The patient receives trimethoprim-sulfamethoxazole (Bactrim, Septra) and azithromycin (Zithromax) for prophylaxis against Pneumocystis carinii pneumonia and Mycobacterium avium intracellulare infection. Two months after the hospitalization, she recalls being at a corn maze 1 month before becoming ill.
- Clarke V, Weston-Smith S. Severe folate-deficiency pancytopenia. BMJ Case Reports 2010; published online.
- Anthony AC. Megaloblastic anemias. In:Hoffman R, Benz EJ, Shattil SJ, Furie B, Cohebn HJ, Silberstein LE, editors. Hematology: Basic Principles and Practice, 2nd ed. New York, NY: Churchill Livingston, 1995:552–586.
- Macrae FA, St John DJ. Relationship between patterns of bleeding and Hemoccult sensitivity in patients with colorectal cancers or adenomas. Gastroenterology 1982; 82:891–898.
- Meyers CA, Albitar M, Estey E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer 2005; 104:788–793.
- Parker CJ. Bone marrow failure syndromes: paroxysmal nocturnal hemoglobinuria. Hematol Oncol Clin North Am 2009; 23:333–346.
- Young NS, Maciejewski JP, Sloand E, et al. The relationship of aplastic anemia and PNH. Int J Hematol 2002; 76(suppl 2):168–172.
- Rosse W. A new way to prevent thrombosis? Blood 2007; 110:3821.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Meals LT, McKinney WP. Acute pulmonary histoplasmosis: progressive pneumonia resulting from high inoculum exposure. J Ky Med Assoc 1998; 96:258–260.
- Salomon J, Flament Saillour M, De Truchis P, et al. An outbreak of acute pulmonary histoplasmosis in members of a trekking trip in Martinique, French West Indies. J Travel Med 2003; 10:87–93.
- Joseph Wheat L. Current diagnosis of histoplasmosis. Trends Microbiol 2003; 11:488–494.
- Wheat LJ, Kauffman CA. Histoplasmosis. Infect Dis Clin North Am 2003; 17:1–19.
- Swartzentruber S, Rhodes L, Kurkjian K, et al. Diagnosis of acute pulmonary histoplasmosis by antigen detection. Clin Infect Dis 2009; 49:1878–1882.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Retallack DM, Woods JP. Molecular epidemiology, pathogenesis, and genetics of the dimorphic fungus Histoplasma capsulatum. Microbes Infect 1999; 1:817–825.
- Deepe GS. The immune response to Histoplasma capsulatum: unearthing its secrets. J Lab Clin Med 1994; 123:201–205.
- Goodwin RA, Shapiro JL, Thurman GH, Thurman SS, Des Prez RM. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore) 1980; 59:1–33.
- Wheat LJ, Connolly-Stringfield PA, Baker RL, et al. Disseminated histoplasmosis in the acquired immune deficiency syndrome: clinical findings, diagnosis and treatment, and review of the literature. Medicine (Baltimore) 1990; 69:361–374.
- Rubin H, Furcolow ML, Yates JL, Brasher CA. The course and prognosis of histoplasmosis. Am J Med 1959; 27:278–288.
- Wheat J, MaWhinney S, Hafner R, et al. Treatment of histoplasmosis with fluconazole in patients with acquired immunodeficiency syndrome. National Institute of Allergy and Infectious Diseases Acquired Immunodeficiency Syndrome Clinical Trials Group and Mycoses Study Group. Am J Med 1997; 103:223–232.
- McKinsey DS, Kauffman CA, Pappas PG, et al. Fluconazole therapy for histoplasmosis. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Clin Infect Dis 1996; 23:996–1001.
- Slama TG. Treatment of disseminated and progressive cavitary histoplasmosis with ketoconazole. Am J Med 1983; 74:70–73.
- Treatment of blastomycosis and histoplasmosis with ketoconazole. Results of a prospective randomized clinical trial. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Ann Intern Med 1985; 103:861–872.
- Clarke V, Weston-Smith S. Severe folate-deficiency pancytopenia. BMJ Case Reports 2010; published online.
- Anthony AC. Megaloblastic anemias. In:Hoffman R, Benz EJ, Shattil SJ, Furie B, Cohebn HJ, Silberstein LE, editors. Hematology: Basic Principles and Practice, 2nd ed. New York, NY: Churchill Livingston, 1995:552–586.
- Macrae FA, St John DJ. Relationship between patterns of bleeding and Hemoccult sensitivity in patients with colorectal cancers or adenomas. Gastroenterology 1982; 82:891–898.
- Meyers CA, Albitar M, Estey E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer 2005; 104:788–793.
- Parker CJ. Bone marrow failure syndromes: paroxysmal nocturnal hemoglobinuria. Hematol Oncol Clin North Am 2009; 23:333–346.
- Young NS, Maciejewski JP, Sloand E, et al. The relationship of aplastic anemia and PNH. Int J Hematol 2002; 76(suppl 2):168–172.
- Rosse W. A new way to prevent thrombosis? Blood 2007; 110:3821.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Meals LT, McKinney WP. Acute pulmonary histoplasmosis: progressive pneumonia resulting from high inoculum exposure. J Ky Med Assoc 1998; 96:258–260.
- Salomon J, Flament Saillour M, De Truchis P, et al. An outbreak of acute pulmonary histoplasmosis in members of a trekking trip in Martinique, French West Indies. J Travel Med 2003; 10:87–93.
- Joseph Wheat L. Current diagnosis of histoplasmosis. Trends Microbiol 2003; 11:488–494.
- Wheat LJ, Kauffman CA. Histoplasmosis. Infect Dis Clin North Am 2003; 17:1–19.
- Swartzentruber S, Rhodes L, Kurkjian K, et al. Diagnosis of acute pulmonary histoplasmosis by antigen detection. Clin Infect Dis 2009; 49:1878–1882.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Retallack DM, Woods JP. Molecular epidemiology, pathogenesis, and genetics of the dimorphic fungus Histoplasma capsulatum. Microbes Infect 1999; 1:817–825.
- Deepe GS. The immune response to Histoplasma capsulatum: unearthing its secrets. J Lab Clin Med 1994; 123:201–205.
- Goodwin RA, Shapiro JL, Thurman GH, Thurman SS, Des Prez RM. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore) 1980; 59:1–33.
- Wheat LJ, Connolly-Stringfield PA, Baker RL, et al. Disseminated histoplasmosis in the acquired immune deficiency syndrome: clinical findings, diagnosis and treatment, and review of the literature. Medicine (Baltimore) 1990; 69:361–374.
- Rubin H, Furcolow ML, Yates JL, Brasher CA. The course and prognosis of histoplasmosis. Am J Med 1959; 27:278–288.
- Wheat J, MaWhinney S, Hafner R, et al. Treatment of histoplasmosis with fluconazole in patients with acquired immunodeficiency syndrome. National Institute of Allergy and Infectious Diseases Acquired Immunodeficiency Syndrome Clinical Trials Group and Mycoses Study Group. Am J Med 1997; 103:223–232.
- McKinsey DS, Kauffman CA, Pappas PG, et al. Fluconazole therapy for histoplasmosis. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Clin Infect Dis 1996; 23:996–1001.
- Slama TG. Treatment of disseminated and progressive cavitary histoplasmosis with ketoconazole. Am J Med 1983; 74:70–73.
- Treatment of blastomycosis and histoplasmosis with ketoconazole. Results of a prospective randomized clinical trial. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Ann Intern Med 1985; 103:861–872.
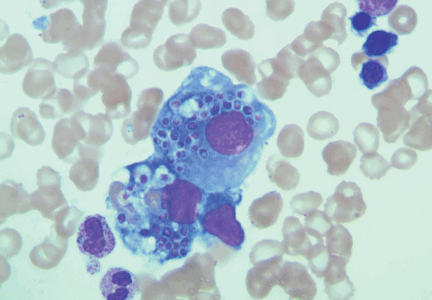
How safe are erythropoiesis-stimulating agents?
The year 2007 was a rough one for erythropoiesis-stimulating agents (ESAs). Increasing concerns about their safety were raised in important meta-analyses of previously published data, specifically, the possibility that these agents increase the risk of venous thromboembolic phenomenona and shorten survival. These trends were seen primarily in studies of cancer patients.1 Meanwhile, front-page headlines in The New York Times were unkind: “Doctors reaping millions for use of anemia drugs.”2 However, the signals came earlier than 2007.
THE RISE AND POSSIBLE FALL OF ESAs
1989—These costly drugs are introduced and start their ascent to becoming one of the most widely used drug classes, helped along by direct-to-consumer advertising. (In one ad, Grandpa can run after the grandchildren despite being on chemotherapy because he uses erythropoietin!)
2001—A study declares that the higher the hemoglobin level rises in response to ESAs, the better the quality of life. It also hints that these agents improve survival.3
2002—The American Society of Hematology/American Society of Clinical Oncology Practice Guideline Writing Committee reviews the medical literature, performs a systematic review, and recommends that patients with low-risk myelodysplasia and those on chemotherapy who become anemic (with a hemoglobin level approaching 10 g/dL) have the option of receiving ESAs to raise their hemoglobin and decrease the need for transfusion.4
2003—Henke et al5 report that anemic patients with head and neck cancer who underwent radiotherapy and received erythropoietin in a randomized study had poorer survival and progression-free survival.
2005—Leyland-Jones et al,6 in another randomized study, report that patients with metastatic breast cancer receiving first-line chemotherapy (most of whom were not anemic) had a higher mortality rate if they received epoetin alfa.
2006—The guideline authors meet again to start the process of writing an update. A meta-analysis shows the thromboembolic risk and survival problems in a more systematic way, covering multiple studies.7
2007—The Centers for Medicare and Medicaid Services cuts back the reimbursement for the use of erythropoietin. The US Food and Drug Administration (FDA) publishes a black box warning suggesting that any hemoglobin level greater than 12 g/dL would be detrimental to a patient. The Guideline Writing Committee works on its document with this backdrop of turmoil.
2008—The updated guidelines are published. They recommend continuing to use ESAs for patients with low-risk myelodysplasia, and as an option to raise hemoglobin levels and prevent the need for transfusion in cancer patients undergoing chemotherapy whose hemoglobin level falls to 10 g/dL or less.8 The document includes stronger language against the use of ESAs in patients with anemia from cancer who are not undergoing chemotherapy. Meanwhile, some editorialists have suggested that it may be time to abandon ESAs because these drugs may promote more rapid tumor growth or pose a prohibitive risk of thromboembolic disease.9,10
In mid-March 2008, after reviewing the most recent data, an FDA panel calls for new limits on the use of ESAs: cancer patients who are undergoing treatment that could cure their cancer should not receive them, and neither should patients with advanced breast cancer or head and neck cancer. Furthermore, the FDA panel stipulates that when doctors do prescribe these drugs, they should warn patients of the possible dangers and seek their informed consent.
WHAT HAPPENS NOW?
Will the FDA take ESAs off the market? That is unlikely. Nephrologists need these drugs to avoid the need for transfusion in dialysis patients (see the accompanying article by Drs. Demirjian and Nurko on the use of ESAs in patients with chronic kidney disease on page 353 of this issue of the Journal). Some of the very first signals of harm with raising the hemoglobin too high came from the nephrology field.
Hematologists should still have the option of using ESAs in some settings, particularly in patients with low-risk myelodysplasia who are becoming more and more anemic and in those who have comorbid conditions in which lower hemoglobin levels are unsafe, particularly if they have coronary artery disease. They also should still be able to use ESAs for selected patients who develop severe chemotherapy-induced anemia and who become so weakened from their anemic state that their life and quality of life are threatened. If ESAs are taken away from the formulary of hematologists and oncologists, these specialists will rely on transfusions to treat their anemic patients, whether the anemia be due to myelodysplasia or to chemotherapy. Some say that the blood supply is so safe that we should not have the same worries about using blood transfusions as we did in the late 1980s. However, we all have seen patients who adamantly do not want transfusions.
Certainly, more events will transpire in the next year with the ESAs. There will probably be more data on erythropoietin receptors on the surface of tumor cells and what happens when pharmacologic doses of erythropoietin interact with these receptors. Patient-specific meta-analyses will probably shed more light on individual patients’ risk of thromboembolic disease and early death from the use of these agents.
The ESA story is far from over.
- Bohlius J, Wilson J, Seidenfeld J, et al. Erythropoietin or darbopoietin for patients with cancer. Cochrane Database Syst Rev 2007; 2:CD 003407.
- Berenson A, Pollack A. Doctors reaping millions for use of anemia drugs. New York Times, May 9, 2007, page1A.
- Littlewood TJ, Bajette E, Nortier JW, et al. Effects of epoetin alfa on hematologic parameters & quality of life in cancer patients receiving nonplatinum chemotherapy: results of a randomized, double-blind, placebo-controlled trial. J Clin Oncol 2001; 19:2865–2874.
- Rizzo JD, Lichtin AE, Woolf SH, et al. Use of epoetin in patients with cancer: evidence based clinical practice guidelines of the American Society of Clinical Oncology and the American Society of Hematology. J Clin Oncol 2002; 20:4083–4107.
- Henke M, Laszig R, Rube C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double–blind, placebo-controlled trial. Lancet 2003; 362:1255–1260.
- Leyland-Jones B, Semiglazov V, Pawlicki M, et al. Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol 2005; 23:5960–5972.
- Bohlius J, Wilson J, Seidenfeld J, et al. Recombinant human erythropoietins and cancer patients: updated meta-analysis of 57 studies including 9353 patients. J Natl Cancer Inst 2006; 98:708–714.
- Rizzo JD, Somerfield MR, Hagerty K, et al. Use of epoetin and darbepoetin in patients with cancer: 2007 American Society of Hematology/American Society of Clinical Oncology clinical practice guideline update. Blood 2008; 111:25–41.
- Tefferi A. Pharmaceutical erythropoietin use in patients with cancer: is it time to abandon ship or just drop anchor? [editorial] Mayo Clin Proc 2007; 82:1316–1318.
- Anonymous. Erythropoietin analogues: an unnecessary class of drugs [editorial]. Lancet Oncol 2008; 9:81.
The year 2007 was a rough one for erythropoiesis-stimulating agents (ESAs). Increasing concerns about their safety were raised in important meta-analyses of previously published data, specifically, the possibility that these agents increase the risk of venous thromboembolic phenomenona and shorten survival. These trends were seen primarily in studies of cancer patients.1 Meanwhile, front-page headlines in The New York Times were unkind: “Doctors reaping millions for use of anemia drugs.”2 However, the signals came earlier than 2007.
THE RISE AND POSSIBLE FALL OF ESAs
1989—These costly drugs are introduced and start their ascent to becoming one of the most widely used drug classes, helped along by direct-to-consumer advertising. (In one ad, Grandpa can run after the grandchildren despite being on chemotherapy because he uses erythropoietin!)
2001—A study declares that the higher the hemoglobin level rises in response to ESAs, the better the quality of life. It also hints that these agents improve survival.3
2002—The American Society of Hematology/American Society of Clinical Oncology Practice Guideline Writing Committee reviews the medical literature, performs a systematic review, and recommends that patients with low-risk myelodysplasia and those on chemotherapy who become anemic (with a hemoglobin level approaching 10 g/dL) have the option of receiving ESAs to raise their hemoglobin and decrease the need for transfusion.4
2003—Henke et al5 report that anemic patients with head and neck cancer who underwent radiotherapy and received erythropoietin in a randomized study had poorer survival and progression-free survival.
2005—Leyland-Jones et al,6 in another randomized study, report that patients with metastatic breast cancer receiving first-line chemotherapy (most of whom were not anemic) had a higher mortality rate if they received epoetin alfa.
2006—The guideline authors meet again to start the process of writing an update. A meta-analysis shows the thromboembolic risk and survival problems in a more systematic way, covering multiple studies.7
2007—The Centers for Medicare and Medicaid Services cuts back the reimbursement for the use of erythropoietin. The US Food and Drug Administration (FDA) publishes a black box warning suggesting that any hemoglobin level greater than 12 g/dL would be detrimental to a patient. The Guideline Writing Committee works on its document with this backdrop of turmoil.
2008—The updated guidelines are published. They recommend continuing to use ESAs for patients with low-risk myelodysplasia, and as an option to raise hemoglobin levels and prevent the need for transfusion in cancer patients undergoing chemotherapy whose hemoglobin level falls to 10 g/dL or less.8 The document includes stronger language against the use of ESAs in patients with anemia from cancer who are not undergoing chemotherapy. Meanwhile, some editorialists have suggested that it may be time to abandon ESAs because these drugs may promote more rapid tumor growth or pose a prohibitive risk of thromboembolic disease.9,10
In mid-March 2008, after reviewing the most recent data, an FDA panel calls for new limits on the use of ESAs: cancer patients who are undergoing treatment that could cure their cancer should not receive them, and neither should patients with advanced breast cancer or head and neck cancer. Furthermore, the FDA panel stipulates that when doctors do prescribe these drugs, they should warn patients of the possible dangers and seek their informed consent.
WHAT HAPPENS NOW?
Will the FDA take ESAs off the market? That is unlikely. Nephrologists need these drugs to avoid the need for transfusion in dialysis patients (see the accompanying article by Drs. Demirjian and Nurko on the use of ESAs in patients with chronic kidney disease on page 353 of this issue of the Journal). Some of the very first signals of harm with raising the hemoglobin too high came from the nephrology field.
Hematologists should still have the option of using ESAs in some settings, particularly in patients with low-risk myelodysplasia who are becoming more and more anemic and in those who have comorbid conditions in which lower hemoglobin levels are unsafe, particularly if they have coronary artery disease. They also should still be able to use ESAs for selected patients who develop severe chemotherapy-induced anemia and who become so weakened from their anemic state that their life and quality of life are threatened. If ESAs are taken away from the formulary of hematologists and oncologists, these specialists will rely on transfusions to treat their anemic patients, whether the anemia be due to myelodysplasia or to chemotherapy. Some say that the blood supply is so safe that we should not have the same worries about using blood transfusions as we did in the late 1980s. However, we all have seen patients who adamantly do not want transfusions.
Certainly, more events will transpire in the next year with the ESAs. There will probably be more data on erythropoietin receptors on the surface of tumor cells and what happens when pharmacologic doses of erythropoietin interact with these receptors. Patient-specific meta-analyses will probably shed more light on individual patients’ risk of thromboembolic disease and early death from the use of these agents.
The ESA story is far from over.
The year 2007 was a rough one for erythropoiesis-stimulating agents (ESAs). Increasing concerns about their safety were raised in important meta-analyses of previously published data, specifically, the possibility that these agents increase the risk of venous thromboembolic phenomenona and shorten survival. These trends were seen primarily in studies of cancer patients.1 Meanwhile, front-page headlines in The New York Times were unkind: “Doctors reaping millions for use of anemia drugs.”2 However, the signals came earlier than 2007.
THE RISE AND POSSIBLE FALL OF ESAs
1989—These costly drugs are introduced and start their ascent to becoming one of the most widely used drug classes, helped along by direct-to-consumer advertising. (In one ad, Grandpa can run after the grandchildren despite being on chemotherapy because he uses erythropoietin!)
2001—A study declares that the higher the hemoglobin level rises in response to ESAs, the better the quality of life. It also hints that these agents improve survival.3
2002—The American Society of Hematology/American Society of Clinical Oncology Practice Guideline Writing Committee reviews the medical literature, performs a systematic review, and recommends that patients with low-risk myelodysplasia and those on chemotherapy who become anemic (with a hemoglobin level approaching 10 g/dL) have the option of receiving ESAs to raise their hemoglobin and decrease the need for transfusion.4
2003—Henke et al5 report that anemic patients with head and neck cancer who underwent radiotherapy and received erythropoietin in a randomized study had poorer survival and progression-free survival.
2005—Leyland-Jones et al,6 in another randomized study, report that patients with metastatic breast cancer receiving first-line chemotherapy (most of whom were not anemic) had a higher mortality rate if they received epoetin alfa.
2006—The guideline authors meet again to start the process of writing an update. A meta-analysis shows the thromboembolic risk and survival problems in a more systematic way, covering multiple studies.7
2007—The Centers for Medicare and Medicaid Services cuts back the reimbursement for the use of erythropoietin. The US Food and Drug Administration (FDA) publishes a black box warning suggesting that any hemoglobin level greater than 12 g/dL would be detrimental to a patient. The Guideline Writing Committee works on its document with this backdrop of turmoil.
2008—The updated guidelines are published. They recommend continuing to use ESAs for patients with low-risk myelodysplasia, and as an option to raise hemoglobin levels and prevent the need for transfusion in cancer patients undergoing chemotherapy whose hemoglobin level falls to 10 g/dL or less.8 The document includes stronger language against the use of ESAs in patients with anemia from cancer who are not undergoing chemotherapy. Meanwhile, some editorialists have suggested that it may be time to abandon ESAs because these drugs may promote more rapid tumor growth or pose a prohibitive risk of thromboembolic disease.9,10
In mid-March 2008, after reviewing the most recent data, an FDA panel calls for new limits on the use of ESAs: cancer patients who are undergoing treatment that could cure their cancer should not receive them, and neither should patients with advanced breast cancer or head and neck cancer. Furthermore, the FDA panel stipulates that when doctors do prescribe these drugs, they should warn patients of the possible dangers and seek their informed consent.
WHAT HAPPENS NOW?
Will the FDA take ESAs off the market? That is unlikely. Nephrologists need these drugs to avoid the need for transfusion in dialysis patients (see the accompanying article by Drs. Demirjian and Nurko on the use of ESAs in patients with chronic kidney disease on page 353 of this issue of the Journal). Some of the very first signals of harm with raising the hemoglobin too high came from the nephrology field.
Hematologists should still have the option of using ESAs in some settings, particularly in patients with low-risk myelodysplasia who are becoming more and more anemic and in those who have comorbid conditions in which lower hemoglobin levels are unsafe, particularly if they have coronary artery disease. They also should still be able to use ESAs for selected patients who develop severe chemotherapy-induced anemia and who become so weakened from their anemic state that their life and quality of life are threatened. If ESAs are taken away from the formulary of hematologists and oncologists, these specialists will rely on transfusions to treat their anemic patients, whether the anemia be due to myelodysplasia or to chemotherapy. Some say that the blood supply is so safe that we should not have the same worries about using blood transfusions as we did in the late 1980s. However, we all have seen patients who adamantly do not want transfusions.
Certainly, more events will transpire in the next year with the ESAs. There will probably be more data on erythropoietin receptors on the surface of tumor cells and what happens when pharmacologic doses of erythropoietin interact with these receptors. Patient-specific meta-analyses will probably shed more light on individual patients’ risk of thromboembolic disease and early death from the use of these agents.
The ESA story is far from over.
- Bohlius J, Wilson J, Seidenfeld J, et al. Erythropoietin or darbopoietin for patients with cancer. Cochrane Database Syst Rev 2007; 2:CD 003407.
- Berenson A, Pollack A. Doctors reaping millions for use of anemia drugs. New York Times, May 9, 2007, page1A.
- Littlewood TJ, Bajette E, Nortier JW, et al. Effects of epoetin alfa on hematologic parameters & quality of life in cancer patients receiving nonplatinum chemotherapy: results of a randomized, double-blind, placebo-controlled trial. J Clin Oncol 2001; 19:2865–2874.
- Rizzo JD, Lichtin AE, Woolf SH, et al. Use of epoetin in patients with cancer: evidence based clinical practice guidelines of the American Society of Clinical Oncology and the American Society of Hematology. J Clin Oncol 2002; 20:4083–4107.
- Henke M, Laszig R, Rube C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double–blind, placebo-controlled trial. Lancet 2003; 362:1255–1260.
- Leyland-Jones B, Semiglazov V, Pawlicki M, et al. Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol 2005; 23:5960–5972.
- Bohlius J, Wilson J, Seidenfeld J, et al. Recombinant human erythropoietins and cancer patients: updated meta-analysis of 57 studies including 9353 patients. J Natl Cancer Inst 2006; 98:708–714.
- Rizzo JD, Somerfield MR, Hagerty K, et al. Use of epoetin and darbepoetin in patients with cancer: 2007 American Society of Hematology/American Society of Clinical Oncology clinical practice guideline update. Blood 2008; 111:25–41.
- Tefferi A. Pharmaceutical erythropoietin use in patients with cancer: is it time to abandon ship or just drop anchor? [editorial] Mayo Clin Proc 2007; 82:1316–1318.
- Anonymous. Erythropoietin analogues: an unnecessary class of drugs [editorial]. Lancet Oncol 2008; 9:81.
- Bohlius J, Wilson J, Seidenfeld J, et al. Erythropoietin or darbopoietin for patients with cancer. Cochrane Database Syst Rev 2007; 2:CD 003407.
- Berenson A, Pollack A. Doctors reaping millions for use of anemia drugs. New York Times, May 9, 2007, page1A.
- Littlewood TJ, Bajette E, Nortier JW, et al. Effects of epoetin alfa on hematologic parameters & quality of life in cancer patients receiving nonplatinum chemotherapy: results of a randomized, double-blind, placebo-controlled trial. J Clin Oncol 2001; 19:2865–2874.
- Rizzo JD, Lichtin AE, Woolf SH, et al. Use of epoetin in patients with cancer: evidence based clinical practice guidelines of the American Society of Clinical Oncology and the American Society of Hematology. J Clin Oncol 2002; 20:4083–4107.
- Henke M, Laszig R, Rube C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double–blind, placebo-controlled trial. Lancet 2003; 362:1255–1260.
- Leyland-Jones B, Semiglazov V, Pawlicki M, et al. Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol 2005; 23:5960–5972.
- Bohlius J, Wilson J, Seidenfeld J, et al. Recombinant human erythropoietins and cancer patients: updated meta-analysis of 57 studies including 9353 patients. J Natl Cancer Inst 2006; 98:708–714.
- Rizzo JD, Somerfield MR, Hagerty K, et al. Use of epoetin and darbepoetin in patients with cancer: 2007 American Society of Hematology/American Society of Clinical Oncology clinical practice guideline update. Blood 2008; 111:25–41.
- Tefferi A. Pharmaceutical erythropoietin use in patients with cancer: is it time to abandon ship or just drop anchor? [editorial] Mayo Clin Proc 2007; 82:1316–1318.
- Anonymous. Erythropoietin analogues: an unnecessary class of drugs [editorial]. Lancet Oncol 2008; 9:81.