User login
Verrucous Carcinoma in a Wounded Military Amputee
To the Editor:
Verrucous carcinoma is a rare, well-differentiated, locally aggressive squamous cell carcinoma first described by Ackerman in 1948.1 There are 4 main clinicopathologic types: oral florid papillomatosis or Ackerman tumor, giant condyloma acuminatum or Buschke-Lowenstein tumor, plantar verrucous carcinoma, and cutaneous verrucous carcinoma.2,3 Historically, most patients are older white men. The lesion commonly occurs in sites of inflammation4 or chronic irritation/trauma. Clinically, patients present with a slowly enlarging, exophytic, verrucous plaque violating the skin, fascia, and occasionally bone. Although these lesions have little tendency to metastasize, substantial morbidity can be seen due to local invasion. Despite surgical excision, recurrence is not uncommon and is associated with a poor prognosis and higher infiltrative potential.5
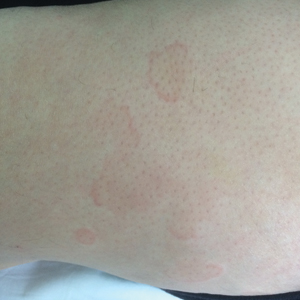
A 45-year-old male veteran initially presented to our dermatology clinic with a 4-cm, macerated, verrucous plaque on the left lateral ankle in the area of a skin graft placed during a prior limb salvage surgery (Figure 1). The patient experienced a traumatic blast injury while deployed 7 years prior with a subsequent right-sided below-the-knee amputation and left lower limb salvage. The lesion was clinically diagnosed as verruca vulgaris and treated with daily salicylic acid. Six weeks after the initial presentation, the lesion remained largely unchanged. A biopsy subsequently was obtained to confirm the diagnosis. At that time, the histopathology was consistent with verruca vulgaris without evidence of carcinoma. Due to the persistence of the lesion, lack of improvement with topical treatment, and overall size, the patient opted for surgical excision.
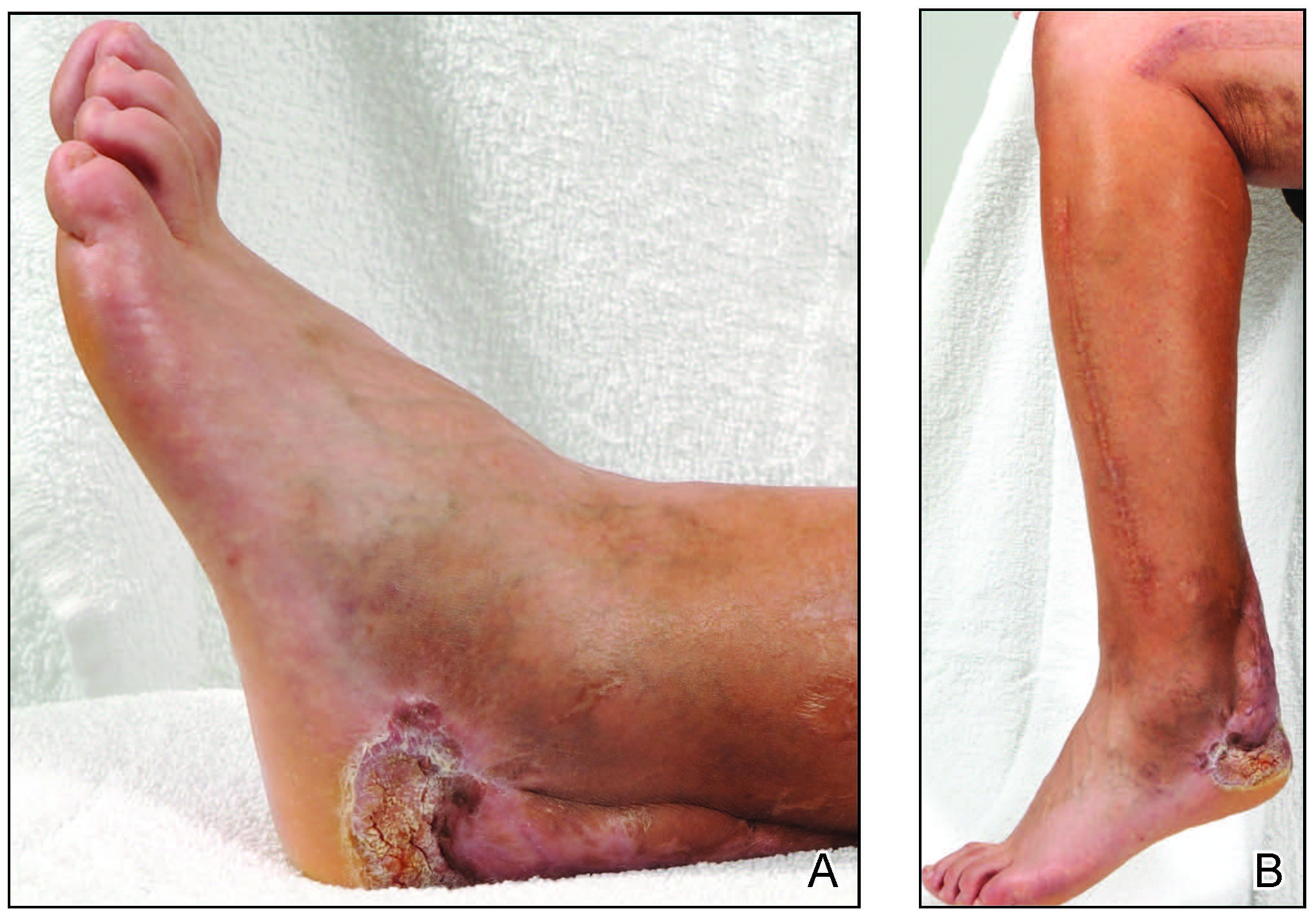
A year later, the lesion was excised again by orthopedic surgery, and the tissue was submitted for histopathologic evaluation, which was suggestive of a verrucous neoplasm with some disagreement on whether it was consistent with verrucous hyperplasia or verrucous carcinoma. Following excision, the patient sustained a nonhealing chronic ulcer that required wound care for a total of 6 months. The lesion recurred a year later and was surgically excised a third time. A split-thickness skin graft was utilized to repair the defect. Histopathology again was consistent with verrucous carcinoma. With a fourth and final recurrence of the verrucous plaque 6 months later, the patient elected to undergo a left-sided below-the-knee amputation.
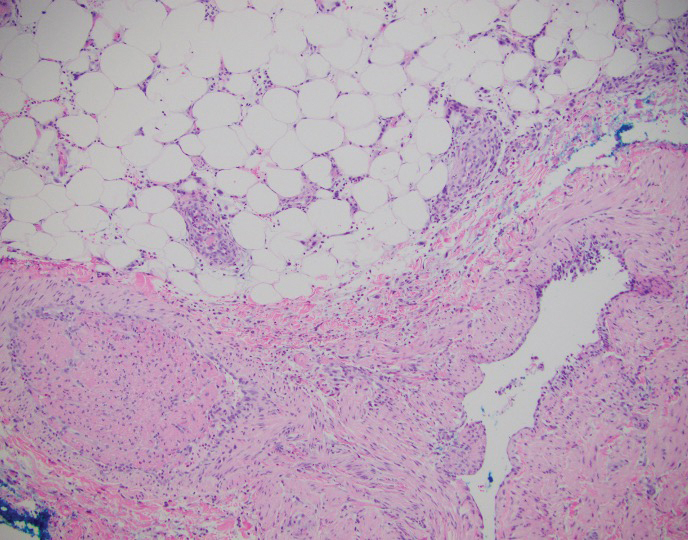
Verrucous carcinoma can represent a diagnostic dilemma, as histologic sections may mimic benign entities. The features of a well-differentiated squamous epithelium with hyperkeratosis, papillomatosis, and acanthosis can be mistaken for verruca vulgaris, keratoacanthoma, and pseudoepitheliomatous hyperplasia,6 which are characteristic of verrucous hyperplasia. Accurate diagnosis can be difficult with a superficial biopsy because of the mature appearance of the epithelium,7 prompting the need for multiple and deeper biopsies8 to include sampling of the base of the hyperplastic epithelium in which the characteristic bulbous pushing growth pattern of the rete ridges is visualized. Precise histologic diagnosis can be further confounded by external mechanical factors, such as pressure, which can distort the classic histopathology.7 The histopathologic features leading to the diagnosis of verrucous carcinoma in our specimen were minimal squamous atypia present in a predominantly exophytic squamous proliferation with human papillomavirus cytopathic effect and focal endophytic pushing borders by rounded bulbous rete ridges into the mid and deep dermis (Figure 2).

Diagnostic uncertainty can delay surgical excision and lead to progression of verrucous carcinoma. Unfortunately, even with appropriate surgical intervention, recurrence has been documented; therefore, close clinical follow-up is recommended. The tumor spreads by local invasion and may follow the path of least resistance.4 In our patient, the frequent tissue manipulation may have facilitated aggressive infiltration of the tumor, ultimately resulting in the loss of his remaining leg. Therefore, it is important for clinicians to recognize that verrucous carcinoma, especially one that develops on a refractory ulcer or scar tissue, may be a complex malignant neoplasm that requires extensive treatment at onset to prevent the amputation of a limb.
- Ackerman LV. Verrucous carcinoma of the oral cavity. Surgery. 1948;23:670-678.
- Yoshitasu S, Takagi T, Ohata C, et al. Plantar verrucous carcinoma: report of a case treated with Boyd amputation followed by reconstruction with a free forearm flap. J Dermatol. 2001;28:226-230.
- Schwartz R. Verrucous carcinoma of the skin and mucosa. J Am Acad Dermatol. 1995;32:1-14.
- Bernstein SC, Lim KK, Brodland DG, et al. The many faces of squamous cell carcinoma. Dermatol Surg. 1996;22:243-254.
- Costache M, Tatiana D, Mitrache L, et al. Cutaneous verrucous carcinoma—report of three cases with review of literature. Rom J Morphol Embryol. 2014;55:383-388.
- Shenoy A, Waghmare R, Kavishwar V, et al. Carcinoma cuniculatum of foot. Foot. 2011;21:207-208.
- Klima M, Kurtis B, Jordan P. Verrucous carcinoma of skin. J Cutan Pathol.1980;7:88-98.
- Pleat J, Sacks L, Rigby H. Cutaneous verrucous carcinoma. Br J Plast Surg. 2001;54:554-555.
To the Editor:
Verrucous carcinoma is a rare, well-differentiated, locally aggressive squamous cell carcinoma first described by Ackerman in 1948.1 There are 4 main clinicopathologic types: oral florid papillomatosis or Ackerman tumor, giant condyloma acuminatum or Buschke-Lowenstein tumor, plantar verrucous carcinoma, and cutaneous verrucous carcinoma.2,3 Historically, most patients are older white men. The lesion commonly occurs in sites of inflammation4 or chronic irritation/trauma. Clinically, patients present with a slowly enlarging, exophytic, verrucous plaque violating the skin, fascia, and occasionally bone. Although these lesions have little tendency to metastasize, substantial morbidity can be seen due to local invasion. Despite surgical excision, recurrence is not uncommon and is associated with a poor prognosis and higher infiltrative potential.5
A 45-year-old male veteran initially presented to our dermatology clinic with a 4-cm, macerated, verrucous plaque on the left lateral ankle in the area of a skin graft placed during a prior limb salvage surgery (Figure 1). The patient experienced a traumatic blast injury while deployed 7 years prior with a subsequent right-sided below-the-knee amputation and left lower limb salvage. The lesion was clinically diagnosed as verruca vulgaris and treated with daily salicylic acid. Six weeks after the initial presentation, the lesion remained largely unchanged. A biopsy subsequently was obtained to confirm the diagnosis. At that time, the histopathology was consistent with verruca vulgaris without evidence of carcinoma. Due to the persistence of the lesion, lack of improvement with topical treatment, and overall size, the patient opted for surgical excision.
A year later, the lesion was excised again by orthopedic surgery, and the tissue was submitted for histopathologic evaluation, which was suggestive of a verrucous neoplasm with some disagreement on whether it was consistent with verrucous hyperplasia or verrucous carcinoma. Following excision, the patient sustained a nonhealing chronic ulcer that required wound care for a total of 6 months. The lesion recurred a year later and was surgically excised a third time. A split-thickness skin graft was utilized to repair the defect. Histopathology again was consistent with verrucous carcinoma. With a fourth and final recurrence of the verrucous plaque 6 months later, the patient elected to undergo a left-sided below-the-knee amputation.
Verrucous carcinoma can represent a diagnostic dilemma, as histologic sections may mimic benign entities. The features of a well-differentiated squamous epithelium with hyperkeratosis, papillomatosis, and acanthosis can be mistaken for verruca vulgaris, keratoacanthoma, and pseudoepitheliomatous hyperplasia,6 which are characteristic of verrucous hyperplasia. Accurate diagnosis can be difficult with a superficial biopsy because of the mature appearance of the epithelium,7 prompting the need for multiple and deeper biopsies8 to include sampling of the base of the hyperplastic epithelium in which the characteristic bulbous pushing growth pattern of the rete ridges is visualized. Precise histologic diagnosis can be further confounded by external mechanical factors, such as pressure, which can distort the classic histopathology.7 The histopathologic features leading to the diagnosis of verrucous carcinoma in our specimen were minimal squamous atypia present in a predominantly exophytic squamous proliferation with human papillomavirus cytopathic effect and focal endophytic pushing borders by rounded bulbous rete ridges into the mid and deep dermis (Figure 2).
Diagnostic uncertainty can delay surgical excision and lead to progression of verrucous carcinoma. Unfortunately, even with appropriate surgical intervention, recurrence has been documented; therefore, close clinical follow-up is recommended. The tumor spreads by local invasion and may follow the path of least resistance.4 In our patient, the frequent tissue manipulation may have facilitated aggressive infiltration of the tumor, ultimately resulting in the loss of his remaining leg. Therefore, it is important for clinicians to recognize that verrucous carcinoma, especially one that develops on a refractory ulcer or scar tissue, may be a complex malignant neoplasm that requires extensive treatment at onset to prevent the amputation of a limb.
To the Editor:
Verrucous carcinoma is a rare, well-differentiated, locally aggressive squamous cell carcinoma first described by Ackerman in 1948.1 There are 4 main clinicopathologic types: oral florid papillomatosis or Ackerman tumor, giant condyloma acuminatum or Buschke-Lowenstein tumor, plantar verrucous carcinoma, and cutaneous verrucous carcinoma.2,3 Historically, most patients are older white men. The lesion commonly occurs in sites of inflammation4 or chronic irritation/trauma. Clinically, patients present with a slowly enlarging, exophytic, verrucous plaque violating the skin, fascia, and occasionally bone. Although these lesions have little tendency to metastasize, substantial morbidity can be seen due to local invasion. Despite surgical excision, recurrence is not uncommon and is associated with a poor prognosis and higher infiltrative potential.5
A 45-year-old male veteran initially presented to our dermatology clinic with a 4-cm, macerated, verrucous plaque on the left lateral ankle in the area of a skin graft placed during a prior limb salvage surgery (Figure 1). The patient experienced a traumatic blast injury while deployed 7 years prior with a subsequent right-sided below-the-knee amputation and left lower limb salvage. The lesion was clinically diagnosed as verruca vulgaris and treated with daily salicylic acid. Six weeks after the initial presentation, the lesion remained largely unchanged. A biopsy subsequently was obtained to confirm the diagnosis. At that time, the histopathology was consistent with verruca vulgaris without evidence of carcinoma. Due to the persistence of the lesion, lack of improvement with topical treatment, and overall size, the patient opted for surgical excision.
A year later, the lesion was excised again by orthopedic surgery, and the tissue was submitted for histopathologic evaluation, which was suggestive of a verrucous neoplasm with some disagreement on whether it was consistent with verrucous hyperplasia or verrucous carcinoma. Following excision, the patient sustained a nonhealing chronic ulcer that required wound care for a total of 6 months. The lesion recurred a year later and was surgically excised a third time. A split-thickness skin graft was utilized to repair the defect. Histopathology again was consistent with verrucous carcinoma. With a fourth and final recurrence of the verrucous plaque 6 months later, the patient elected to undergo a left-sided below-the-knee amputation.
Verrucous carcinoma can represent a diagnostic dilemma, as histologic sections may mimic benign entities. The features of a well-differentiated squamous epithelium with hyperkeratosis, papillomatosis, and acanthosis can be mistaken for verruca vulgaris, keratoacanthoma, and pseudoepitheliomatous hyperplasia,6 which are characteristic of verrucous hyperplasia. Accurate diagnosis can be difficult with a superficial biopsy because of the mature appearance of the epithelium,7 prompting the need for multiple and deeper biopsies8 to include sampling of the base of the hyperplastic epithelium in which the characteristic bulbous pushing growth pattern of the rete ridges is visualized. Precise histologic diagnosis can be further confounded by external mechanical factors, such as pressure, which can distort the classic histopathology.7 The histopathologic features leading to the diagnosis of verrucous carcinoma in our specimen were minimal squamous atypia present in a predominantly exophytic squamous proliferation with human papillomavirus cytopathic effect and focal endophytic pushing borders by rounded bulbous rete ridges into the mid and deep dermis (Figure 2).
Diagnostic uncertainty can delay surgical excision and lead to progression of verrucous carcinoma. Unfortunately, even with appropriate surgical intervention, recurrence has been documented; therefore, close clinical follow-up is recommended. The tumor spreads by local invasion and may follow the path of least resistance.4 In our patient, the frequent tissue manipulation may have facilitated aggressive infiltration of the tumor, ultimately resulting in the loss of his remaining leg. Therefore, it is important for clinicians to recognize that verrucous carcinoma, especially one that develops on a refractory ulcer or scar tissue, may be a complex malignant neoplasm that requires extensive treatment at onset to prevent the amputation of a limb.
- Ackerman LV. Verrucous carcinoma of the oral cavity. Surgery. 1948;23:670-678.
- Yoshitasu S, Takagi T, Ohata C, et al. Plantar verrucous carcinoma: report of a case treated with Boyd amputation followed by reconstruction with a free forearm flap. J Dermatol. 2001;28:226-230.
- Schwartz R. Verrucous carcinoma of the skin and mucosa. J Am Acad Dermatol. 1995;32:1-14.
- Bernstein SC, Lim KK, Brodland DG, et al. The many faces of squamous cell carcinoma. Dermatol Surg. 1996;22:243-254.
- Costache M, Tatiana D, Mitrache L, et al. Cutaneous verrucous carcinoma—report of three cases with review of literature. Rom J Morphol Embryol. 2014;55:383-388.
- Shenoy A, Waghmare R, Kavishwar V, et al. Carcinoma cuniculatum of foot. Foot. 2011;21:207-208.
- Klima M, Kurtis B, Jordan P. Verrucous carcinoma of skin. J Cutan Pathol.1980;7:88-98.
- Pleat J, Sacks L, Rigby H. Cutaneous verrucous carcinoma. Br J Plast Surg. 2001;54:554-555.
- Ackerman LV. Verrucous carcinoma of the oral cavity. Surgery. 1948;23:670-678.
- Yoshitasu S, Takagi T, Ohata C, et al. Plantar verrucous carcinoma: report of a case treated with Boyd amputation followed by reconstruction with a free forearm flap. J Dermatol. 2001;28:226-230.
- Schwartz R. Verrucous carcinoma of the skin and mucosa. J Am Acad Dermatol. 1995;32:1-14.
- Bernstein SC, Lim KK, Brodland DG, et al. The many faces of squamous cell carcinoma. Dermatol Surg. 1996;22:243-254.
- Costache M, Tatiana D, Mitrache L, et al. Cutaneous verrucous carcinoma—report of three cases with review of literature. Rom J Morphol Embryol. 2014;55:383-388.
- Shenoy A, Waghmare R, Kavishwar V, et al. Carcinoma cuniculatum of foot. Foot. 2011;21:207-208.
- Klima M, Kurtis B, Jordan P. Verrucous carcinoma of skin. J Cutan Pathol.1980;7:88-98.
- Pleat J, Sacks L, Rigby H. Cutaneous verrucous carcinoma. Br J Plast Surg. 2001;54:554-555.
Practice Points
- Verrucous carcinoma is a rare, well-differentiated, locally aggressive squamous cell carcinoma that commonly occurs in sites of inflammation or chronic irritation.
- Histologically, verrucous carcinoma can be mistaken for other entities including verruca vulgaris, keratoacanthoma, and pseudoepitheliomatous hyperplasia, often delaying the appropriate diagnosis and treatment.
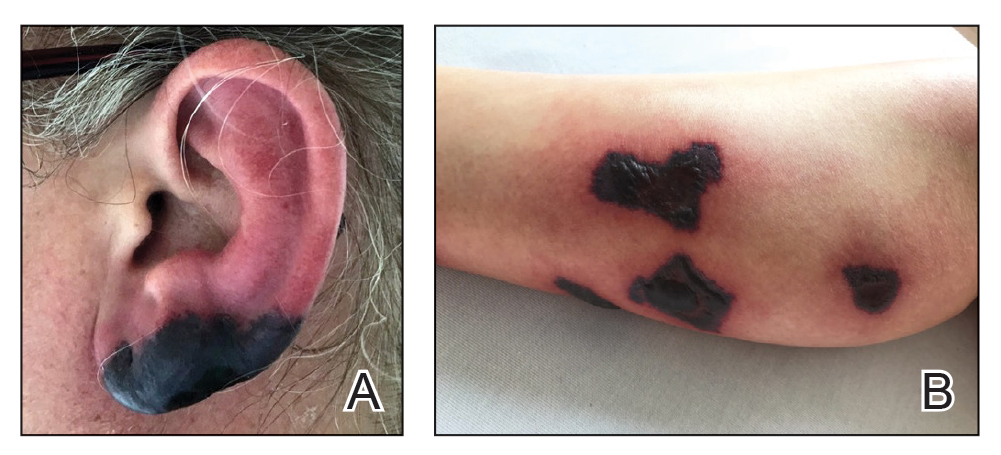
Bullous Retiform Purpura on the Ears and Legs
The Diagnosis: Levamisole-Induced Vasculopathy
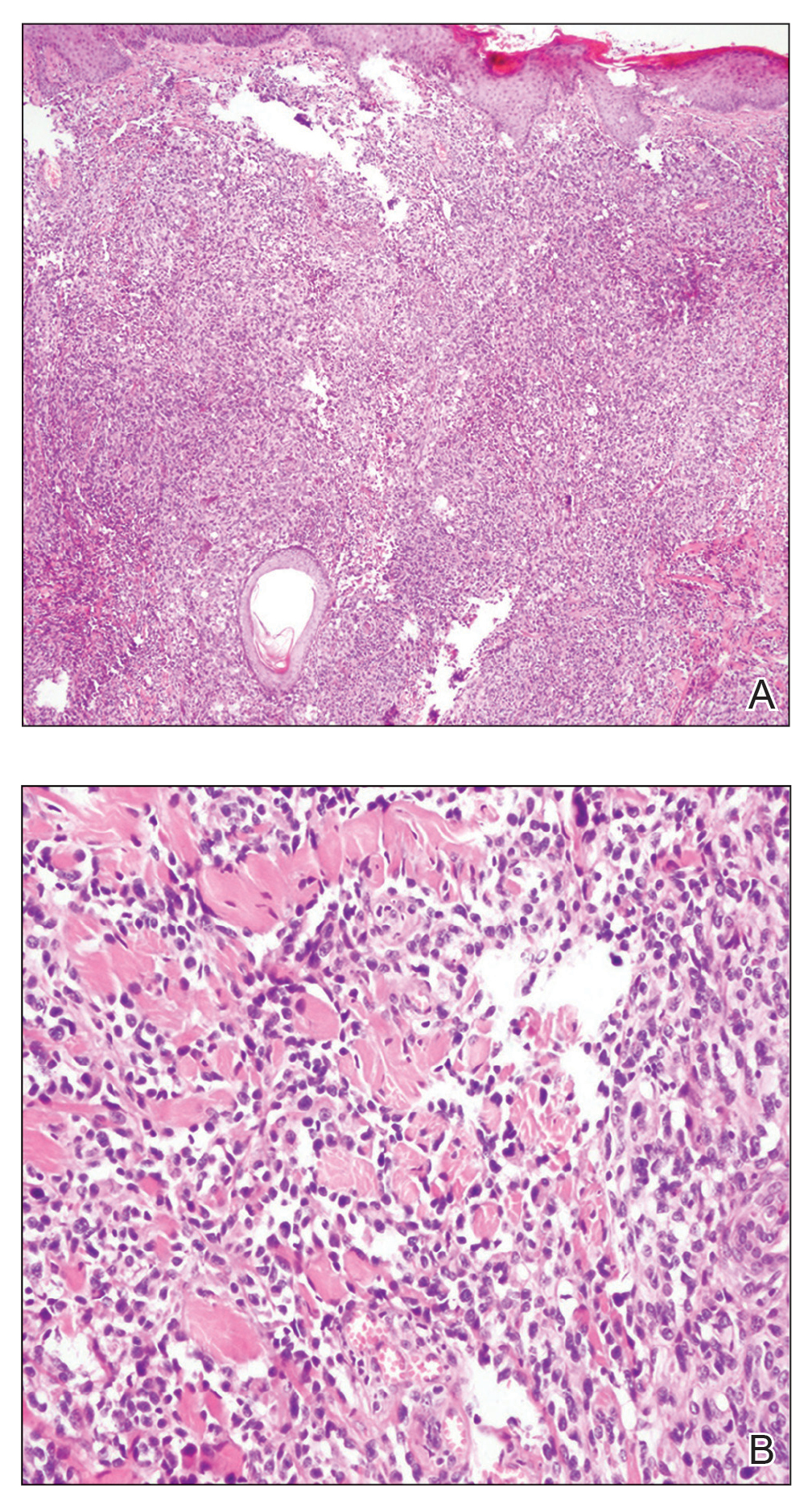
Biopsy of one of the bullous retiform purpura on the leg (Figure 1) revealed a combined leukocytoclastic vasculitis and thrombotic vasculopathy (quiz images). Periodic acid-Schiff and Gram stains, with adequate controls, were negative for pathogenic fungal and bacterial organisms. Although this reaction pattern has an extensive differential, in this clinical setting with associated cocaine-positive urine toxicologic analysis, perinuclear antineutrophil cytoplasmic antibodies (p-ANCA), and leukopenia, the histopathologic findings were consistent with levamisole-induced vasculopathy (LIV).1,2 Although not specific, leukocytoclastic vasculitis and thrombotic vasculopathy have been reported as the classic histopathologic findings of LIV. In addition, interstitial and perivascular neovascularization have been reported as a potential histopathologic finding associated with this entity but was not seen in our case.3
Levamisole is an anthelminthic agent used to adulterate cocaine, a practice first noted in 2003 with increasing incidence.1 Both levamisole and cocaine stimulate the sympathetic nervous system by increasing dopamine in the euphoric areas of the brain.1,3 By combining the 2 substances, preparation costs are reduced and stimulant effects are enhanced. It is estimated that 69% to 80% of cocaine in the United States is contaminated with levamisole.2,4,5 The constellation of findings seen in patients abusing levamisole-contaminated cocaine include agranulocytosis; p-ANCA; and a tender, vasculitic, retiform purpura presentation. The most common sites for the purpura include the cheeks and ears. The purpura can progress to bullous lesions, as seen in our patient, followed by necrosis.4,6 Recurrent use of levamisole-contaminated cocaine is associated with recurrent agranulocytosis and classic skin findings, which is suggestive of a causal relationship.6
Serologic testing for levamisole exposure presents a challenge. The half-life of levamisole is relatively short (estimated at 5.6 hours) and is found in urine samples approximately 3% of the time.1,3,6 The volatile diagnostic characteristics of levamisole make concrete laboratory confirmation difficult. Although a skin biopsy can be helpful to rule out other causes of vasculitislike presentations, it is not specific for LIV. Therefore, clinical suspicion for LIV should remain high in patients who present with the cutaneous findings described as well as agranulocytosis, positive p-ANCA, and a history of cocaine use with a skin biopsy showing leukocytoclastic vasculitis and thrombotic vasculopathy.
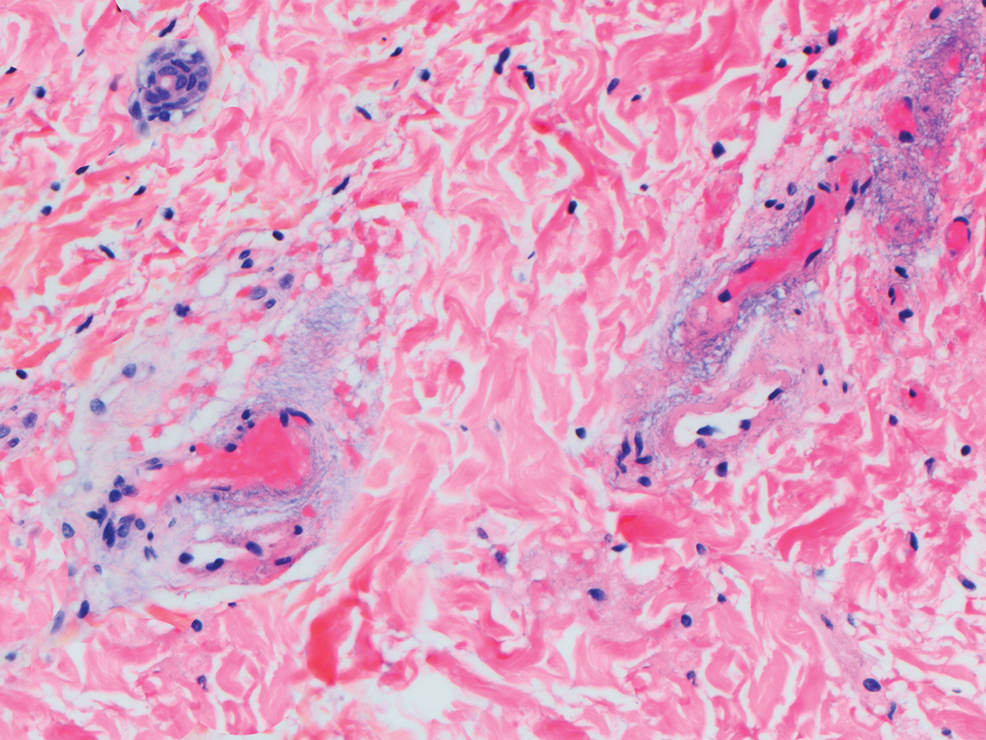
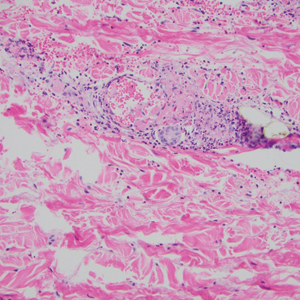
The differential diagnosis for LIV with retiform bullous lesions includes several other vasculitides and vesiculobullous diseases. Eosinophilic granulomatosis with polyangiitis (EGPA) is a multisystem vasculitis that is characterized by eosinophilia, asthma, and rhinosinusitis. Eosinophilic granulomatosis with polyangiitis primarily affects small and medium arteries in the skin and respiratory tract and occurs in 3 stages: prodromal, eosinophilic, and vasculitic. These stages are characterized by mild asthma or rhinitis, eosinophilia with multiorgan infiltration, and vasculitis with extravascular granulomatosis, respectively. Diagnosis often is clinical based on these findings and laboratory evaluation. Eosinophilic granulomatosis with polyangiitis presents with positive p-ANCA in 40% to 60% of patients.7 The vasculitis stage of EGPA presents with cutaneous findings in 60% of cases, including palpable purpura, infiltrated papules and plaques, urticaria, necrotizing lesions, and rarely vesicles and bullae.8 Classic histopathologic features include leukocytoclastic or eosinophilic vasculitis, an eosinophilic infiltrate, granuloma formation, and eosinophilic granule deposition onto collagen fibrils (otherwise known as flame figures)(Figure 2). Biopsy of these lesions with the aforementioned findings, in constellation with the described systemic signs and symptoms, can aid in diagnosis of EGPA.
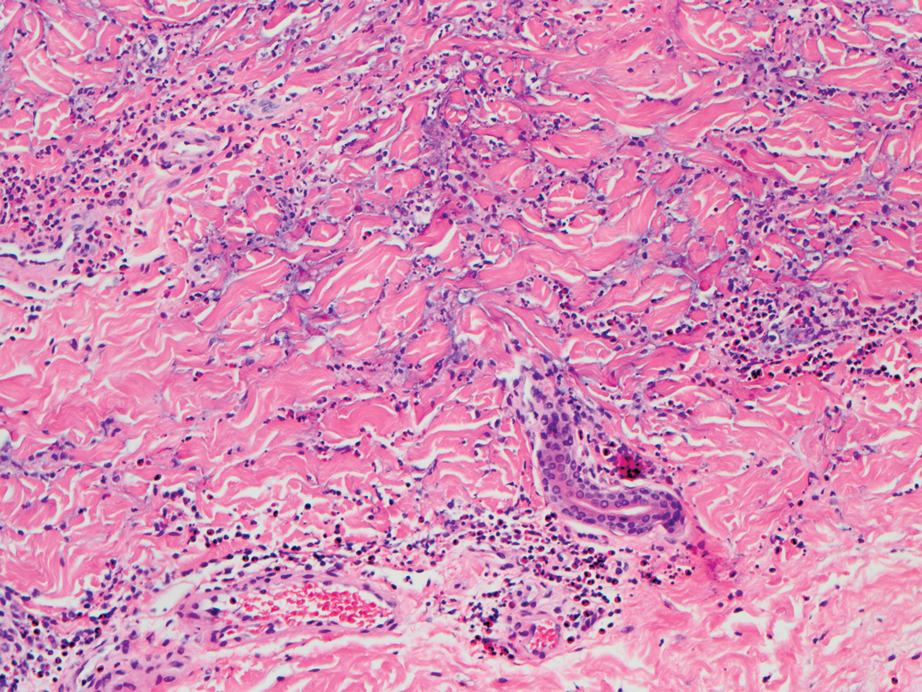
Polyarteritis nodosa (PAN) is a vasculitis that can be either multisystem or limited to one organ. Classic PAN affects the small- to medium-sized vessels. When there is multisystem involvement, it most often affects the skin, gastrointestinal tract, and kidneys. It presents with subcutaneous or dermal nodules, necrotic lesions, livedo reticularis, hypertension, abdominal pain, and an acute abdomen.9 When PAN is in its limited form, it most commonly occurs in the skin. The cutaneous manifestations of skin-limited PAN are identical to classic PAN, most commonly occurring on the legs and arms and less often on the trunk, head, and neck.10 To aid in diagnosis, biopsies of cutaneous lesions are beneficial. Dermatopathologic examination of PAN reveals fibrinoid necrosis of small and medium vessels with a perivascular mononuclear inflammatory infiltrate (Figure 3). Cutaneous PAN rarely progresses to multisystem classic PAN and carries a more favorable prognosis.
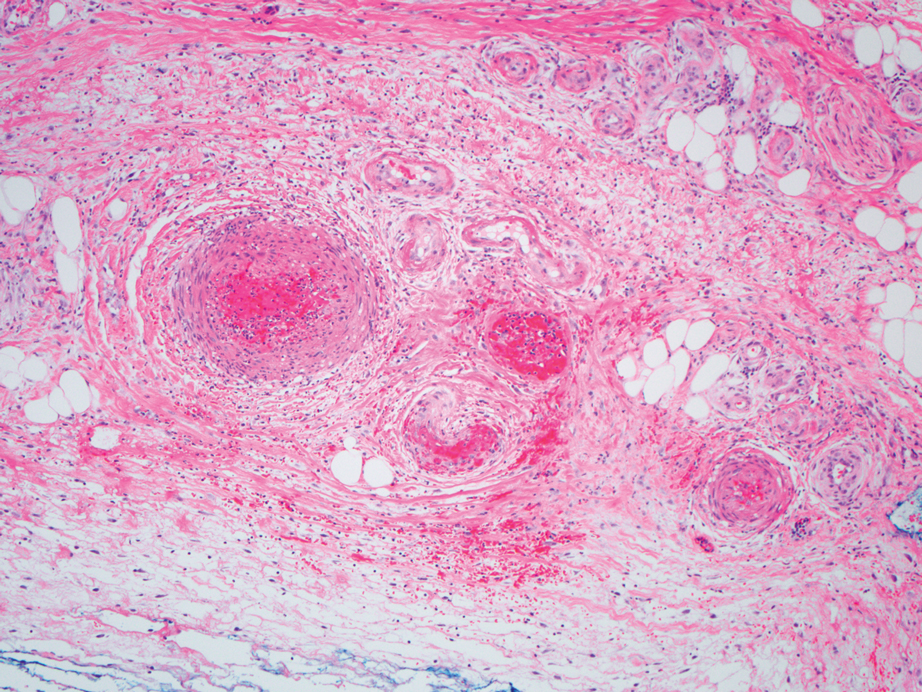
Microvascular occlusion syndromes can result in clinical presentations that resemble LIV. Idiopathic thrombocytopenic purpura is a hematologic autoimmune condition resulting in destruction of platelets and subsequent thrombocytopenia. Idiopathic thrombocytopenic purpura can be either primary or secondary to infections, drugs, malignancy, or other autoimmune conditions. Clinically, it presents as mucosal or cutaneous bleeding, epistaxis, hematochezia, or hematuria and can result in substantial hemorrhage. On the skin, it can appear as petechiae and ecchymoses in dependent areas and rarely hemorrhagic bullae of the skin and mucous membranes in cases of severe thrombocytopenia.11,12 Biopsies of these lesions will show notable extravasation of red blood cells with incipient hemorrhagic bullae formation (Figure 4). Recognition of hemorrhagic bullae as a presentation of idiopathic thrombocytopenic purpura is critical to identifying severe underlying disease.
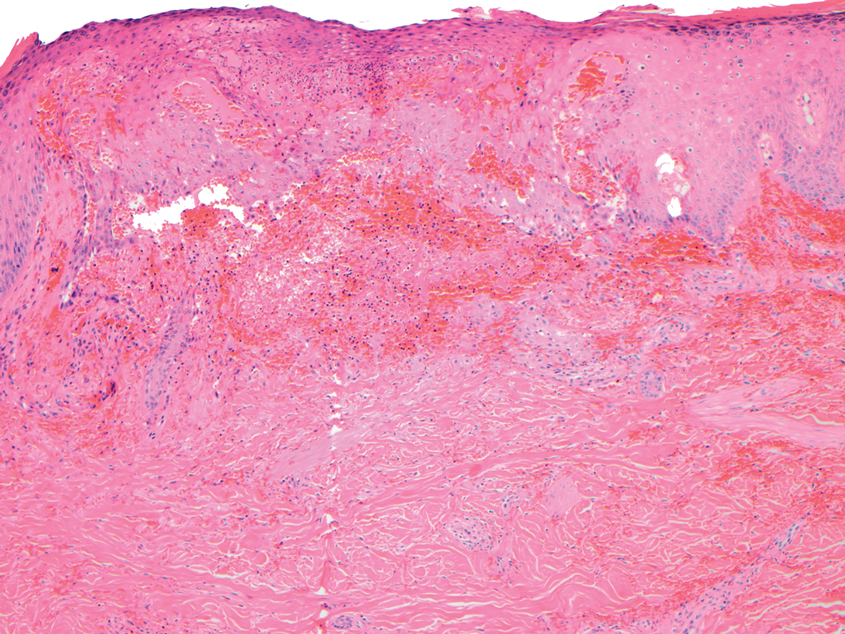
Beyond other vasculitides and microvascular occlusion syndromes, vessel-invasive microorganisms can result in similar histopathologic and clinical presentations to LIV. Ecthyma gangrenosum (EG) is a septic vasculitis, often caused by Pseudomonas aeruginosa, usually affecting immunocompromised patients. Ecthyma gangrenosum presents with vesiculobullous lesions with erythematous violaceous borders that develop into hemorrhagic bullae with necrotic centers.13 Biopsy of EG will show vascular occlusion and basophilic granular material within or around vessels, suggestive of bacterial sepsis (Figure 5). The detection of an infectious agent on histopathology allows one to easily distinguish between EG and LIV.
- Bajaj S, Hibler B, Rossi A. Painful violaceous purpura on a 44-year-old woman. Am J Med. 2016;129:E5-E7.
- Munoz-Vahos CH, Herrera-Uribe S, Arbelaez-Cortes A, et al. Clinical profile of levamisole-adulterated cocaine-induced vasculitis/vasculopathy. J Clin Rheumatol. 2019;25:E16-E26.
- Jacob RS, Silva CY, Powers JG, et al. Levamisole-induced vasculopathy: a report of 2 cases and a novel histopathologic finding. Am J Dermatopathol. 2012;34:208-213.
- Gillis JA, Green P, Williams J. Levamisole-induced vasculopathy: staging and management. J Plast Reconstr Aesthet Surg. 2014;67:E29-E31.
- Farhat EK, Muirhead TT, Chafins ML, et al. Levamisole-induced cutaneous necrosis mimicking coagulopathy. Arch Dermatol. 2010;146:1320-1321.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia-a potential public health epidemic associated with levamisole-adulterated cocaine. J Am Acad Dermatol. 2010;65:722-725.
- Negbenebor NA, Khalifian S, Foreman RK, et al. A 92-year-old male with eosinophilic asthma presenting with recurrent palpable purpuric plaques. Dermatopathology (Basel). 2018;5:44-48.
- Sherman S, Gal N, Didkovsky E, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) relapsing as bullous eruption. Acta Derm Venereol. 2017;97:406-407.
- Braungart S, Campbell A, Besarovic S. Atypical Henoch-Schonlein purpura? consider polyarteritis nodosa! BMJ Case Rep. 2014. doi:10.1136/bcr-2013-201764
- Alquorain NAA, Aljabr ASH, Alghamdi NJ. Cutaneous polyarteritis nodosa treated with pentoxifylline and clobetasol propionate: a case report. Saudi J Med Sci. 2018;6:104-107.
- Helms AE, Schaffer RI. Idiopathic thrombocytopenic purpura with black oral mucosal lesions. Cutis. 2007;79:456-458.
- Lountzis N, Maroon M, Tyler W. Mucocutaneous hemorrhagic bullae in idiopathic thrombocytopenic purpura. J Am Acad Dermatol. 2009;60:AB124.
- Llamas-Velasco M, Alegeria V, Santos-Briz A, et al. Occlusive nonvasculitic vasculopathy. Am J Dermatopathol. 2017;39:637-662.
The Diagnosis: Levamisole-Induced Vasculopathy
Biopsy of one of the bullous retiform purpura on the leg (Figure 1) revealed a combined leukocytoclastic vasculitis and thrombotic vasculopathy (quiz images). Periodic acid-Schiff and Gram stains, with adequate controls, were negative for pathogenic fungal and bacterial organisms. Although this reaction pattern has an extensive differential, in this clinical setting with associated cocaine-positive urine toxicologic analysis, perinuclear antineutrophil cytoplasmic antibodies (p-ANCA), and leukopenia, the histopathologic findings were consistent with levamisole-induced vasculopathy (LIV).1,2 Although not specific, leukocytoclastic vasculitis and thrombotic vasculopathy have been reported as the classic histopathologic findings of LIV. In addition, interstitial and perivascular neovascularization have been reported as a potential histopathologic finding associated with this entity but was not seen in our case.3
Levamisole is an anthelminthic agent used to adulterate cocaine, a practice first noted in 2003 with increasing incidence.1 Both levamisole and cocaine stimulate the sympathetic nervous system by increasing dopamine in the euphoric areas of the brain.1,3 By combining the 2 substances, preparation costs are reduced and stimulant effects are enhanced. It is estimated that 69% to 80% of cocaine in the United States is contaminated with levamisole.2,4,5 The constellation of findings seen in patients abusing levamisole-contaminated cocaine include agranulocytosis; p-ANCA; and a tender, vasculitic, retiform purpura presentation. The most common sites for the purpura include the cheeks and ears. The purpura can progress to bullous lesions, as seen in our patient, followed by necrosis.4,6 Recurrent use of levamisole-contaminated cocaine is associated with recurrent agranulocytosis and classic skin findings, which is suggestive of a causal relationship.6
Serologic testing for levamisole exposure presents a challenge. The half-life of levamisole is relatively short (estimated at 5.6 hours) and is found in urine samples approximately 3% of the time.1,3,6 The volatile diagnostic characteristics of levamisole make concrete laboratory confirmation difficult. Although a skin biopsy can be helpful to rule out other causes of vasculitislike presentations, it is not specific for LIV. Therefore, clinical suspicion for LIV should remain high in patients who present with the cutaneous findings described as well as agranulocytosis, positive p-ANCA, and a history of cocaine use with a skin biopsy showing leukocytoclastic vasculitis and thrombotic vasculopathy.
The differential diagnosis for LIV with retiform bullous lesions includes several other vasculitides and vesiculobullous diseases. Eosinophilic granulomatosis with polyangiitis (EGPA) is a multisystem vasculitis that is characterized by eosinophilia, asthma, and rhinosinusitis. Eosinophilic granulomatosis with polyangiitis primarily affects small and medium arteries in the skin and respiratory tract and occurs in 3 stages: prodromal, eosinophilic, and vasculitic. These stages are characterized by mild asthma or rhinitis, eosinophilia with multiorgan infiltration, and vasculitis with extravascular granulomatosis, respectively. Diagnosis often is clinical based on these findings and laboratory evaluation. Eosinophilic granulomatosis with polyangiitis presents with positive p-ANCA in 40% to 60% of patients.7 The vasculitis stage of EGPA presents with cutaneous findings in 60% of cases, including palpable purpura, infiltrated papules and plaques, urticaria, necrotizing lesions, and rarely vesicles and bullae.8 Classic histopathologic features include leukocytoclastic or eosinophilic vasculitis, an eosinophilic infiltrate, granuloma formation, and eosinophilic granule deposition onto collagen fibrils (otherwise known as flame figures)(Figure 2). Biopsy of these lesions with the aforementioned findings, in constellation with the described systemic signs and symptoms, can aid in diagnosis of EGPA.
Polyarteritis nodosa (PAN) is a vasculitis that can be either multisystem or limited to one organ. Classic PAN affects the small- to medium-sized vessels. When there is multisystem involvement, it most often affects the skin, gastrointestinal tract, and kidneys. It presents with subcutaneous or dermal nodules, necrotic lesions, livedo reticularis, hypertension, abdominal pain, and an acute abdomen.9 When PAN is in its limited form, it most commonly occurs in the skin. The cutaneous manifestations of skin-limited PAN are identical to classic PAN, most commonly occurring on the legs and arms and less often on the trunk, head, and neck.10 To aid in diagnosis, biopsies of cutaneous lesions are beneficial. Dermatopathologic examination of PAN reveals fibrinoid necrosis of small and medium vessels with a perivascular mononuclear inflammatory infiltrate (Figure 3). Cutaneous PAN rarely progresses to multisystem classic PAN and carries a more favorable prognosis.
Microvascular occlusion syndromes can result in clinical presentations that resemble LIV. Idiopathic thrombocytopenic purpura is a hematologic autoimmune condition resulting in destruction of platelets and subsequent thrombocytopenia. Idiopathic thrombocytopenic purpura can be either primary or secondary to infections, drugs, malignancy, or other autoimmune conditions. Clinically, it presents as mucosal or cutaneous bleeding, epistaxis, hematochezia, or hematuria and can result in substantial hemorrhage. On the skin, it can appear as petechiae and ecchymoses in dependent areas and rarely hemorrhagic bullae of the skin and mucous membranes in cases of severe thrombocytopenia.11,12 Biopsies of these lesions will show notable extravasation of red blood cells with incipient hemorrhagic bullae formation (Figure 4). Recognition of hemorrhagic bullae as a presentation of idiopathic thrombocytopenic purpura is critical to identifying severe underlying disease.
Beyond other vasculitides and microvascular occlusion syndromes, vessel-invasive microorganisms can result in similar histopathologic and clinical presentations to LIV. Ecthyma gangrenosum (EG) is a septic vasculitis, often caused by Pseudomonas aeruginosa, usually affecting immunocompromised patients. Ecthyma gangrenosum presents with vesiculobullous lesions with erythematous violaceous borders that develop into hemorrhagic bullae with necrotic centers.13 Biopsy of EG will show vascular occlusion and basophilic granular material within or around vessels, suggestive of bacterial sepsis (Figure 5). The detection of an infectious agent on histopathology allows one to easily distinguish between EG and LIV.
The Diagnosis: Levamisole-Induced Vasculopathy
Biopsy of one of the bullous retiform purpura on the leg (Figure 1) revealed a combined leukocytoclastic vasculitis and thrombotic vasculopathy (quiz images). Periodic acid-Schiff and Gram stains, with adequate controls, were negative for pathogenic fungal and bacterial organisms. Although this reaction pattern has an extensive differential, in this clinical setting with associated cocaine-positive urine toxicologic analysis, perinuclear antineutrophil cytoplasmic antibodies (p-ANCA), and leukopenia, the histopathologic findings were consistent with levamisole-induced vasculopathy (LIV).1,2 Although not specific, leukocytoclastic vasculitis and thrombotic vasculopathy have been reported as the classic histopathologic findings of LIV. In addition, interstitial and perivascular neovascularization have been reported as a potential histopathologic finding associated with this entity but was not seen in our case.3
Levamisole is an anthelminthic agent used to adulterate cocaine, a practice first noted in 2003 with increasing incidence.1 Both levamisole and cocaine stimulate the sympathetic nervous system by increasing dopamine in the euphoric areas of the brain.1,3 By combining the 2 substances, preparation costs are reduced and stimulant effects are enhanced. It is estimated that 69% to 80% of cocaine in the United States is contaminated with levamisole.2,4,5 The constellation of findings seen in patients abusing levamisole-contaminated cocaine include agranulocytosis; p-ANCA; and a tender, vasculitic, retiform purpura presentation. The most common sites for the purpura include the cheeks and ears. The purpura can progress to bullous lesions, as seen in our patient, followed by necrosis.4,6 Recurrent use of levamisole-contaminated cocaine is associated with recurrent agranulocytosis and classic skin findings, which is suggestive of a causal relationship.6
Serologic testing for levamisole exposure presents a challenge. The half-life of levamisole is relatively short (estimated at 5.6 hours) and is found in urine samples approximately 3% of the time.1,3,6 The volatile diagnostic characteristics of levamisole make concrete laboratory confirmation difficult. Although a skin biopsy can be helpful to rule out other causes of vasculitislike presentations, it is not specific for LIV. Therefore, clinical suspicion for LIV should remain high in patients who present with the cutaneous findings described as well as agranulocytosis, positive p-ANCA, and a history of cocaine use with a skin biopsy showing leukocytoclastic vasculitis and thrombotic vasculopathy.
The differential diagnosis for LIV with retiform bullous lesions includes several other vasculitides and vesiculobullous diseases. Eosinophilic granulomatosis with polyangiitis (EGPA) is a multisystem vasculitis that is characterized by eosinophilia, asthma, and rhinosinusitis. Eosinophilic granulomatosis with polyangiitis primarily affects small and medium arteries in the skin and respiratory tract and occurs in 3 stages: prodromal, eosinophilic, and vasculitic. These stages are characterized by mild asthma or rhinitis, eosinophilia with multiorgan infiltration, and vasculitis with extravascular granulomatosis, respectively. Diagnosis often is clinical based on these findings and laboratory evaluation. Eosinophilic granulomatosis with polyangiitis presents with positive p-ANCA in 40% to 60% of patients.7 The vasculitis stage of EGPA presents with cutaneous findings in 60% of cases, including palpable purpura, infiltrated papules and plaques, urticaria, necrotizing lesions, and rarely vesicles and bullae.8 Classic histopathologic features include leukocytoclastic or eosinophilic vasculitis, an eosinophilic infiltrate, granuloma formation, and eosinophilic granule deposition onto collagen fibrils (otherwise known as flame figures)(Figure 2). Biopsy of these lesions with the aforementioned findings, in constellation with the described systemic signs and symptoms, can aid in diagnosis of EGPA.
Polyarteritis nodosa (PAN) is a vasculitis that can be either multisystem or limited to one organ. Classic PAN affects the small- to medium-sized vessels. When there is multisystem involvement, it most often affects the skin, gastrointestinal tract, and kidneys. It presents with subcutaneous or dermal nodules, necrotic lesions, livedo reticularis, hypertension, abdominal pain, and an acute abdomen.9 When PAN is in its limited form, it most commonly occurs in the skin. The cutaneous manifestations of skin-limited PAN are identical to classic PAN, most commonly occurring on the legs and arms and less often on the trunk, head, and neck.10 To aid in diagnosis, biopsies of cutaneous lesions are beneficial. Dermatopathologic examination of PAN reveals fibrinoid necrosis of small and medium vessels with a perivascular mononuclear inflammatory infiltrate (Figure 3). Cutaneous PAN rarely progresses to multisystem classic PAN and carries a more favorable prognosis.
Microvascular occlusion syndromes can result in clinical presentations that resemble LIV. Idiopathic thrombocytopenic purpura is a hematologic autoimmune condition resulting in destruction of platelets and subsequent thrombocytopenia. Idiopathic thrombocytopenic purpura can be either primary or secondary to infections, drugs, malignancy, or other autoimmune conditions. Clinically, it presents as mucosal or cutaneous bleeding, epistaxis, hematochezia, or hematuria and can result in substantial hemorrhage. On the skin, it can appear as petechiae and ecchymoses in dependent areas and rarely hemorrhagic bullae of the skin and mucous membranes in cases of severe thrombocytopenia.11,12 Biopsies of these lesions will show notable extravasation of red blood cells with incipient hemorrhagic bullae formation (Figure 4). Recognition of hemorrhagic bullae as a presentation of idiopathic thrombocytopenic purpura is critical to identifying severe underlying disease.
Beyond other vasculitides and microvascular occlusion syndromes, vessel-invasive microorganisms can result in similar histopathologic and clinical presentations to LIV. Ecthyma gangrenosum (EG) is a septic vasculitis, often caused by Pseudomonas aeruginosa, usually affecting immunocompromised patients. Ecthyma gangrenosum presents with vesiculobullous lesions with erythematous violaceous borders that develop into hemorrhagic bullae with necrotic centers.13 Biopsy of EG will show vascular occlusion and basophilic granular material within or around vessels, suggestive of bacterial sepsis (Figure 5). The detection of an infectious agent on histopathology allows one to easily distinguish between EG and LIV.
- Bajaj S, Hibler B, Rossi A. Painful violaceous purpura on a 44-year-old woman. Am J Med. 2016;129:E5-E7.
- Munoz-Vahos CH, Herrera-Uribe S, Arbelaez-Cortes A, et al. Clinical profile of levamisole-adulterated cocaine-induced vasculitis/vasculopathy. J Clin Rheumatol. 2019;25:E16-E26.
- Jacob RS, Silva CY, Powers JG, et al. Levamisole-induced vasculopathy: a report of 2 cases and a novel histopathologic finding. Am J Dermatopathol. 2012;34:208-213.
- Gillis JA, Green P, Williams J. Levamisole-induced vasculopathy: staging and management. J Plast Reconstr Aesthet Surg. 2014;67:E29-E31.
- Farhat EK, Muirhead TT, Chafins ML, et al. Levamisole-induced cutaneous necrosis mimicking coagulopathy. Arch Dermatol. 2010;146:1320-1321.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia-a potential public health epidemic associated with levamisole-adulterated cocaine. J Am Acad Dermatol. 2010;65:722-725.
- Negbenebor NA, Khalifian S, Foreman RK, et al. A 92-year-old male with eosinophilic asthma presenting with recurrent palpable purpuric plaques. Dermatopathology (Basel). 2018;5:44-48.
- Sherman S, Gal N, Didkovsky E, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) relapsing as bullous eruption. Acta Derm Venereol. 2017;97:406-407.
- Braungart S, Campbell A, Besarovic S. Atypical Henoch-Schonlein purpura? consider polyarteritis nodosa! BMJ Case Rep. 2014. doi:10.1136/bcr-2013-201764
- Alquorain NAA, Aljabr ASH, Alghamdi NJ. Cutaneous polyarteritis nodosa treated with pentoxifylline and clobetasol propionate: a case report. Saudi J Med Sci. 2018;6:104-107.
- Helms AE, Schaffer RI. Idiopathic thrombocytopenic purpura with black oral mucosal lesions. Cutis. 2007;79:456-458.
- Lountzis N, Maroon M, Tyler W. Mucocutaneous hemorrhagic bullae in idiopathic thrombocytopenic purpura. J Am Acad Dermatol. 2009;60:AB124.
- Llamas-Velasco M, Alegeria V, Santos-Briz A, et al. Occlusive nonvasculitic vasculopathy. Am J Dermatopathol. 2017;39:637-662.
- Bajaj S, Hibler B, Rossi A. Painful violaceous purpura on a 44-year-old woman. Am J Med. 2016;129:E5-E7.
- Munoz-Vahos CH, Herrera-Uribe S, Arbelaez-Cortes A, et al. Clinical profile of levamisole-adulterated cocaine-induced vasculitis/vasculopathy. J Clin Rheumatol. 2019;25:E16-E26.
- Jacob RS, Silva CY, Powers JG, et al. Levamisole-induced vasculopathy: a report of 2 cases and a novel histopathologic finding. Am J Dermatopathol. 2012;34:208-213.
- Gillis JA, Green P, Williams J. Levamisole-induced vasculopathy: staging and management. J Plast Reconstr Aesthet Surg. 2014;67:E29-E31.
- Farhat EK, Muirhead TT, Chafins ML, et al. Levamisole-induced cutaneous necrosis mimicking coagulopathy. Arch Dermatol. 2010;146:1320-1321.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia-a potential public health epidemic associated with levamisole-adulterated cocaine. J Am Acad Dermatol. 2010;65:722-725.
- Negbenebor NA, Khalifian S, Foreman RK, et al. A 92-year-old male with eosinophilic asthma presenting with recurrent palpable purpuric plaques. Dermatopathology (Basel). 2018;5:44-48.
- Sherman S, Gal N, Didkovsky E, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) relapsing as bullous eruption. Acta Derm Venereol. 2017;97:406-407.
- Braungart S, Campbell A, Besarovic S. Atypical Henoch-Schonlein purpura? consider polyarteritis nodosa! BMJ Case Rep. 2014. doi:10.1136/bcr-2013-201764
- Alquorain NAA, Aljabr ASH, Alghamdi NJ. Cutaneous polyarteritis nodosa treated with pentoxifylline and clobetasol propionate: a case report. Saudi J Med Sci. 2018;6:104-107.
- Helms AE, Schaffer RI. Idiopathic thrombocytopenic purpura with black oral mucosal lesions. Cutis. 2007;79:456-458.
- Lountzis N, Maroon M, Tyler W. Mucocutaneous hemorrhagic bullae in idiopathic thrombocytopenic purpura. J Am Acad Dermatol. 2009;60:AB124.
- Llamas-Velasco M, Alegeria V, Santos-Briz A, et al. Occlusive nonvasculitic vasculopathy. Am J Dermatopathol. 2017;39:637-662.
A 40-year-old woman presented with a progressive painful rash on the ears and legs of 2 weeks’ duration. She described the rash as initially red and nonpainful; it started on the right leg and progressed to the left leg, eventually involving the earlobes 4 days prior to presentation. Physical examination revealed edematous purpura of the earlobes and bullous retiform purpura on the lower extremities. Laboratory studies revealed leukopenia (3.6×103 /cm2 [reference range, 4.0–10.5×103 /cm2 ]) and elevated antineutrophil cytoplasmic antibodies (1:320 titer [reference range, <1:40]) in a perinuclear pattern (perinuclear antineutrophil cytoplasmic antibodies). Urine toxicology screening was positive for cocaine and opiates. A punch biopsy of a bullous retiform purpura on the right thigh was obtained for standard hematoxylin and eosin staining.
Subcutaneous Nodule on the Chest
The Diagnosis: Cystic Panfolliculoma
Panfolliculoma is a rare tumor of follicular origin.1 Clinical examination can reveal a papule, nodule, or tumor that typically is mistaken for an epidermal inclusion cyst, trichoepithelioma, or basal cell carcinoma (BCC).2 As with other benign follicular neoplasms, it often exhibits a protracted growth pattern.3,4 Most cases reported in the literature have been shown to occur in the head or neck region. One hypothesis is that separation into the various components of the hair follicle occurs at a higher frequency in areas with a higher hair density such as the face and scalp.4 The lesion typically presents in patients aged 20 to 70 years, as in our patient, with cases equally distributed among males and females.4,5 Neill et al1 reported a rare case of cystic panfolliculoma occurring on the right forearm of a 64-year-old woman.
As its name suggests, panfolliculoma is exceptional in that it displays features of all segments of the hair follicle, including the infundibulum, isthmus, stem, and bulb.6 Although not necessary for diagnosis, immunohistochemical staining can be utilized to identify each hair follicle component on histopathologic examination. Panfolliculoma stains positive for 34βE12 and cytokeratin 5/6, highlighting infundibular and isthmus keratinocytes and the outer root sheath, respectively. Additionally, Ber-EP4 labels germinative cells, while CD34 highlights contiguous fibrotic stroma and trichilemmal areas.3,4
In our patient, histopathology revealed a cystic structure that was lined by an infundibular epithelium with a prominent granular layer. Solid collections of basaloid germinative cells that demonstrated peripheral palisading were observed (quiz image [top]). Cells with trichohyalin granules, indicative of inner root sheath differentiation, were encased by matrical cells (quiz image [bottom]).
Historically, panfolliculomas characteristically have been known to reside in the dermis, with only focal connection to the epidermis, if at all present. Nevertheless, Harris et al7 detailed 2 cases that displayed predominant epidermal involvement, defined by the term epidermal panfolliculoma. In a study performed by Shan and Guo,2 an additional 9 cases (19 panfolliculomas) were found to have similar findings, for which the term superficial panfolliculoma was suggested. In cases that display a primary epidermal component, common mimickers include tumor of the follicular infundibulum and the reactive process of follicular induction.7
Cystic panfolliculoma is a rare subtype further characterized as a lesion with distinctive features of a panfolliculoma that arises from a cyst wall composed of the follicular infundibulum.2,6 The origin of cystic panfolliculoma has not been fully elucidated. It has been hypothesized that the formation of such lesions may arise due to epithelial-mesenchymal interactions. One explanation is that basal cells with stem cell capability may progress into hair follicle structures after communication with underlying dermal cells during invagination of the epidermis, while the epithelial cells not in close proximity to dermal cells maintain stem cell capability.8
The histologic differential diagnosis of cystic panfolliculoma includes dilated pore of Winer, epidermal inclusion cyst, pilar cyst, trichofolliculoma, folliculosebaceous cystic hamartoma, cystic trichoblastoma, and BCC.5 Panfolliculoma can mimic both trichoblastoma and trichoepithelioma on a low-power field; however, the latter follicular tumors lack differentiation to the infundibulum, isthmus, outer root sheath, or hair shaft, as in a panfolliculoma.4 Trichoblastoma is composed of germinative hair follicle cells, with differentiation limited to the hair germ and papilla (Figure 1).9 Panfolliculoma additionally differs from trichoblastoma by having a more prevalent epithelial factor compared to a more pronounced stromal factor in trichoblastoma.1 The cystic subtype of trichoblastoma differs from cystic panfolliculoma in that the cyst wall develops from the infundibulum only and has germinative cells protruding outwards from the cyst wall.

Although BCCs may arise in cystic structures, panfolliculomas can be discerned from this entity by their sharp demarcation, lack of peritumoral clefting, and presence of cytokeratin 20-positive Merkel cells.5 Unlike panfolliculoma, the tumor islands in BCC commonly display peripheral palisading of nuclei with a surrounding fibromyxoid stroma (Figure 2). Additionally, BCCs can exhibit crowding of nuclei, atypia, and mitoses.6

Folliculosebaceous cystic hamartomas and cystic panfolliculomas both contain a cystic structure with differentiation of the cyst wall to the hair follicle. However, folliculosebaceous cystic hamartomas are dilated infundibulocystic configurations that contain sebaceous glands emanating from the cyst wall (Figure 3). Kimura et al10 described defining features of the mesenchymal component of this follicular tumor, including an increase in fibroplasia, vascularity, and adipose tissue. In addition, the epithelial aspect exhibits clefting among the stroma and uninvolved dermis.6

Dilated pore of Winer consists of a cystic opening with connection to the epidermis. The cyst wall resembles the follicular infundibulum, and the cavity is filled with lamellar orthokeratosis (Figure 4).5,11 Epidermal inclusion cysts also contain a cyst wall that resembles the infundibular epithelium, without differentiation to all segments of the hair follicle. They are lined by a stratified squamous epithelium, retain a granular layer, and contain lamellar keratin within the cyst cavity.5,12

In summary, panfolliculoma is a rare benign neoplasm that demonstrates differentiation to each component of the hair follicle structure. Our case demonstrates a unique subtype showcasing cystic changes that infrequently has been described in the literature.
- Neill B, Bingham C, Braudis K, et al. A rare cutaneous adnexal neoplasm: cystic panfolliculoma. J Cutan Pathol. 2016;43:1183-1185.
- Shan SJ, Guo Y. Panfolliculoma and histopathologic variants: a study of 19 cases. Am J Dermatopathol. 2014;36:965-971.
- Hoang MP, Levenson BM. Cystic panfolliculoma. Arch Pathol Lab Med. 2006;130:389-392.
- Huang CY, Wu YH. Panfolliculoma: report of two cases. Dermatol Sínica. 2010;28:73-76.
- Alkhalidi HM, Alhumaidy AA. Cystic panfolliculoma of the scalp: report of a very rare case and brief review. Indian J Pathol Microbiol. 2013;56:437-439.
- López-Takegami JC, Wolter M, Löser C, et al. Classification of cysts with follicular germinative differentiation. J Cutan Pathol. 2016;43:191-199.
- Harris A, Faulkner-Jones B, Zimarowski MJ. Epidermal panfolliculoma: a report of 2 cases. Am J Dermatopathol. 2011;33:E7-E10.
- Fukuyama M, Sato Y, Yamazaki Y, et al. Immunohistochemical dissection of cystic panfolliculoma focusing on the expression of multiple hair follicle lineage markers with an insight into the pathogenesis. J Cutan Pathol. 2017;44:861-866.
- Tellechea O, Cardoso JC, Reis JP, et al. Benign follicular tumors. An Bras Dermatol. 2015;90:780-796; quiz 797-788.
- Kimura T, Miyazawa H, Aoyagi T, et al. Folliculosebaceous cystic hamartoma. a distinctive malformation of the skin. Am J Dermatopathol. 1991;13:213-220.
- Misago N, Inoue T, Narisawa Y. Cystic trichoblastoma: a report of two cases with an immunohistochemical study. J Dermatol. 2015;42:305-310.
- Weir CB, St. Hilaire NJ. Epidermal inclusion cyst. StatPearls. StatPearls Publishing; 2020.
The Diagnosis: Cystic Panfolliculoma
Panfolliculoma is a rare tumor of follicular origin.1 Clinical examination can reveal a papule, nodule, or tumor that typically is mistaken for an epidermal inclusion cyst, trichoepithelioma, or basal cell carcinoma (BCC).2 As with other benign follicular neoplasms, it often exhibits a protracted growth pattern.3,4 Most cases reported in the literature have been shown to occur in the head or neck region. One hypothesis is that separation into the various components of the hair follicle occurs at a higher frequency in areas with a higher hair density such as the face and scalp.4 The lesion typically presents in patients aged 20 to 70 years, as in our patient, with cases equally distributed among males and females.4,5 Neill et al1 reported a rare case of cystic panfolliculoma occurring on the right forearm of a 64-year-old woman.
As its name suggests, panfolliculoma is exceptional in that it displays features of all segments of the hair follicle, including the infundibulum, isthmus, stem, and bulb.6 Although not necessary for diagnosis, immunohistochemical staining can be utilized to identify each hair follicle component on histopathologic examination. Panfolliculoma stains positive for 34βE12 and cytokeratin 5/6, highlighting infundibular and isthmus keratinocytes and the outer root sheath, respectively. Additionally, Ber-EP4 labels germinative cells, while CD34 highlights contiguous fibrotic stroma and trichilemmal areas.3,4
In our patient, histopathology revealed a cystic structure that was lined by an infundibular epithelium with a prominent granular layer. Solid collections of basaloid germinative cells that demonstrated peripheral palisading were observed (quiz image [top]). Cells with trichohyalin granules, indicative of inner root sheath differentiation, were encased by matrical cells (quiz image [bottom]).
Historically, panfolliculomas characteristically have been known to reside in the dermis, with only focal connection to the epidermis, if at all present. Nevertheless, Harris et al7 detailed 2 cases that displayed predominant epidermal involvement, defined by the term epidermal panfolliculoma. In a study performed by Shan and Guo,2 an additional 9 cases (19 panfolliculomas) were found to have similar findings, for which the term superficial panfolliculoma was suggested. In cases that display a primary epidermal component, common mimickers include tumor of the follicular infundibulum and the reactive process of follicular induction.7
Cystic panfolliculoma is a rare subtype further characterized as a lesion with distinctive features of a panfolliculoma that arises from a cyst wall composed of the follicular infundibulum.2,6 The origin of cystic panfolliculoma has not been fully elucidated. It has been hypothesized that the formation of such lesions may arise due to epithelial-mesenchymal interactions. One explanation is that basal cells with stem cell capability may progress into hair follicle structures after communication with underlying dermal cells during invagination of the epidermis, while the epithelial cells not in close proximity to dermal cells maintain stem cell capability.8
The histologic differential diagnosis of cystic panfolliculoma includes dilated pore of Winer, epidermal inclusion cyst, pilar cyst, trichofolliculoma, folliculosebaceous cystic hamartoma, cystic trichoblastoma, and BCC.5 Panfolliculoma can mimic both trichoblastoma and trichoepithelioma on a low-power field; however, the latter follicular tumors lack differentiation to the infundibulum, isthmus, outer root sheath, or hair shaft, as in a panfolliculoma.4 Trichoblastoma is composed of germinative hair follicle cells, with differentiation limited to the hair germ and papilla (Figure 1).9 Panfolliculoma additionally differs from trichoblastoma by having a more prevalent epithelial factor compared to a more pronounced stromal factor in trichoblastoma.1 The cystic subtype of trichoblastoma differs from cystic panfolliculoma in that the cyst wall develops from the infundibulum only and has germinative cells protruding outwards from the cyst wall.
Although BCCs may arise in cystic structures, panfolliculomas can be discerned from this entity by their sharp demarcation, lack of peritumoral clefting, and presence of cytokeratin 20-positive Merkel cells.5 Unlike panfolliculoma, the tumor islands in BCC commonly display peripheral palisading of nuclei with a surrounding fibromyxoid stroma (Figure 2). Additionally, BCCs can exhibit crowding of nuclei, atypia, and mitoses.6
Folliculosebaceous cystic hamartomas and cystic panfolliculomas both contain a cystic structure with differentiation of the cyst wall to the hair follicle. However, folliculosebaceous cystic hamartomas are dilated infundibulocystic configurations that contain sebaceous glands emanating from the cyst wall (Figure 3). Kimura et al10 described defining features of the mesenchymal component of this follicular tumor, including an increase in fibroplasia, vascularity, and adipose tissue. In addition, the epithelial aspect exhibits clefting among the stroma and uninvolved dermis.6
Dilated pore of Winer consists of a cystic opening with connection to the epidermis. The cyst wall resembles the follicular infundibulum, and the cavity is filled with lamellar orthokeratosis (Figure 4).5,11 Epidermal inclusion cysts also contain a cyst wall that resembles the infundibular epithelium, without differentiation to all segments of the hair follicle. They are lined by a stratified squamous epithelium, retain a granular layer, and contain lamellar keratin within the cyst cavity.5,12
In summary, panfolliculoma is a rare benign neoplasm that demonstrates differentiation to each component of the hair follicle structure. Our case demonstrates a unique subtype showcasing cystic changes that infrequently has been described in the literature.
The Diagnosis: Cystic Panfolliculoma
Panfolliculoma is a rare tumor of follicular origin.1 Clinical examination can reveal a papule, nodule, or tumor that typically is mistaken for an epidermal inclusion cyst, trichoepithelioma, or basal cell carcinoma (BCC).2 As with other benign follicular neoplasms, it often exhibits a protracted growth pattern.3,4 Most cases reported in the literature have been shown to occur in the head or neck region. One hypothesis is that separation into the various components of the hair follicle occurs at a higher frequency in areas with a higher hair density such as the face and scalp.4 The lesion typically presents in patients aged 20 to 70 years, as in our patient, with cases equally distributed among males and females.4,5 Neill et al1 reported a rare case of cystic panfolliculoma occurring on the right forearm of a 64-year-old woman.
As its name suggests, panfolliculoma is exceptional in that it displays features of all segments of the hair follicle, including the infundibulum, isthmus, stem, and bulb.6 Although not necessary for diagnosis, immunohistochemical staining can be utilized to identify each hair follicle component on histopathologic examination. Panfolliculoma stains positive for 34βE12 and cytokeratin 5/6, highlighting infundibular and isthmus keratinocytes and the outer root sheath, respectively. Additionally, Ber-EP4 labels germinative cells, while CD34 highlights contiguous fibrotic stroma and trichilemmal areas.3,4
In our patient, histopathology revealed a cystic structure that was lined by an infundibular epithelium with a prominent granular layer. Solid collections of basaloid germinative cells that demonstrated peripheral palisading were observed (quiz image [top]). Cells with trichohyalin granules, indicative of inner root sheath differentiation, were encased by matrical cells (quiz image [bottom]).
Historically, panfolliculomas characteristically have been known to reside in the dermis, with only focal connection to the epidermis, if at all present. Nevertheless, Harris et al7 detailed 2 cases that displayed predominant epidermal involvement, defined by the term epidermal panfolliculoma. In a study performed by Shan and Guo,2 an additional 9 cases (19 panfolliculomas) were found to have similar findings, for which the term superficial panfolliculoma was suggested. In cases that display a primary epidermal component, common mimickers include tumor of the follicular infundibulum and the reactive process of follicular induction.7
Cystic panfolliculoma is a rare subtype further characterized as a lesion with distinctive features of a panfolliculoma that arises from a cyst wall composed of the follicular infundibulum.2,6 The origin of cystic panfolliculoma has not been fully elucidated. It has been hypothesized that the formation of such lesions may arise due to epithelial-mesenchymal interactions. One explanation is that basal cells with stem cell capability may progress into hair follicle structures after communication with underlying dermal cells during invagination of the epidermis, while the epithelial cells not in close proximity to dermal cells maintain stem cell capability.8
The histologic differential diagnosis of cystic panfolliculoma includes dilated pore of Winer, epidermal inclusion cyst, pilar cyst, trichofolliculoma, folliculosebaceous cystic hamartoma, cystic trichoblastoma, and BCC.5 Panfolliculoma can mimic both trichoblastoma and trichoepithelioma on a low-power field; however, the latter follicular tumors lack differentiation to the infundibulum, isthmus, outer root sheath, or hair shaft, as in a panfolliculoma.4 Trichoblastoma is composed of germinative hair follicle cells, with differentiation limited to the hair germ and papilla (Figure 1).9 Panfolliculoma additionally differs from trichoblastoma by having a more prevalent epithelial factor compared to a more pronounced stromal factor in trichoblastoma.1 The cystic subtype of trichoblastoma differs from cystic panfolliculoma in that the cyst wall develops from the infundibulum only and has germinative cells protruding outwards from the cyst wall.
Although BCCs may arise in cystic structures, panfolliculomas can be discerned from this entity by their sharp demarcation, lack of peritumoral clefting, and presence of cytokeratin 20-positive Merkel cells.5 Unlike panfolliculoma, the tumor islands in BCC commonly display peripheral palisading of nuclei with a surrounding fibromyxoid stroma (Figure 2). Additionally, BCCs can exhibit crowding of nuclei, atypia, and mitoses.6
Folliculosebaceous cystic hamartomas and cystic panfolliculomas both contain a cystic structure with differentiation of the cyst wall to the hair follicle. However, folliculosebaceous cystic hamartomas are dilated infundibulocystic configurations that contain sebaceous glands emanating from the cyst wall (Figure 3). Kimura et al10 described defining features of the mesenchymal component of this follicular tumor, including an increase in fibroplasia, vascularity, and adipose tissue. In addition, the epithelial aspect exhibits clefting among the stroma and uninvolved dermis.6
Dilated pore of Winer consists of a cystic opening with connection to the epidermis. The cyst wall resembles the follicular infundibulum, and the cavity is filled with lamellar orthokeratosis (Figure 4).5,11 Epidermal inclusion cysts also contain a cyst wall that resembles the infundibular epithelium, without differentiation to all segments of the hair follicle. They are lined by a stratified squamous epithelium, retain a granular layer, and contain lamellar keratin within the cyst cavity.5,12
In summary, panfolliculoma is a rare benign neoplasm that demonstrates differentiation to each component of the hair follicle structure. Our case demonstrates a unique subtype showcasing cystic changes that infrequently has been described in the literature.
- Neill B, Bingham C, Braudis K, et al. A rare cutaneous adnexal neoplasm: cystic panfolliculoma. J Cutan Pathol. 2016;43:1183-1185.
- Shan SJ, Guo Y. Panfolliculoma and histopathologic variants: a study of 19 cases. Am J Dermatopathol. 2014;36:965-971.
- Hoang MP, Levenson BM. Cystic panfolliculoma. Arch Pathol Lab Med. 2006;130:389-392.
- Huang CY, Wu YH. Panfolliculoma: report of two cases. Dermatol Sínica. 2010;28:73-76.
- Alkhalidi HM, Alhumaidy AA. Cystic panfolliculoma of the scalp: report of a very rare case and brief review. Indian J Pathol Microbiol. 2013;56:437-439.
- López-Takegami JC, Wolter M, Löser C, et al. Classification of cysts with follicular germinative differentiation. J Cutan Pathol. 2016;43:191-199.
- Harris A, Faulkner-Jones B, Zimarowski MJ. Epidermal panfolliculoma: a report of 2 cases. Am J Dermatopathol. 2011;33:E7-E10.
- Fukuyama M, Sato Y, Yamazaki Y, et al. Immunohistochemical dissection of cystic panfolliculoma focusing on the expression of multiple hair follicle lineage markers with an insight into the pathogenesis. J Cutan Pathol. 2017;44:861-866.
- Tellechea O, Cardoso JC, Reis JP, et al. Benign follicular tumors. An Bras Dermatol. 2015;90:780-796; quiz 797-788.
- Kimura T, Miyazawa H, Aoyagi T, et al. Folliculosebaceous cystic hamartoma. a distinctive malformation of the skin. Am J Dermatopathol. 1991;13:213-220.
- Misago N, Inoue T, Narisawa Y. Cystic trichoblastoma: a report of two cases with an immunohistochemical study. J Dermatol. 2015;42:305-310.
- Weir CB, St. Hilaire NJ. Epidermal inclusion cyst. StatPearls. StatPearls Publishing; 2020.
- Neill B, Bingham C, Braudis K, et al. A rare cutaneous adnexal neoplasm: cystic panfolliculoma. J Cutan Pathol. 2016;43:1183-1185.
- Shan SJ, Guo Y. Panfolliculoma and histopathologic variants: a study of 19 cases. Am J Dermatopathol. 2014;36:965-971.
- Hoang MP, Levenson BM. Cystic panfolliculoma. Arch Pathol Lab Med. 2006;130:389-392.
- Huang CY, Wu YH. Panfolliculoma: report of two cases. Dermatol Sínica. 2010;28:73-76.
- Alkhalidi HM, Alhumaidy AA. Cystic panfolliculoma of the scalp: report of a very rare case and brief review. Indian J Pathol Microbiol. 2013;56:437-439.
- López-Takegami JC, Wolter M, Löser C, et al. Classification of cysts with follicular germinative differentiation. J Cutan Pathol. 2016;43:191-199.
- Harris A, Faulkner-Jones B, Zimarowski MJ. Epidermal panfolliculoma: a report of 2 cases. Am J Dermatopathol. 2011;33:E7-E10.
- Fukuyama M, Sato Y, Yamazaki Y, et al. Immunohistochemical dissection of cystic panfolliculoma focusing on the expression of multiple hair follicle lineage markers with an insight into the pathogenesis. J Cutan Pathol. 2017;44:861-866.
- Tellechea O, Cardoso JC, Reis JP, et al. Benign follicular tumors. An Bras Dermatol. 2015;90:780-796; quiz 797-788.
- Kimura T, Miyazawa H, Aoyagi T, et al. Folliculosebaceous cystic hamartoma. a distinctive malformation of the skin. Am J Dermatopathol. 1991;13:213-220.
- Misago N, Inoue T, Narisawa Y. Cystic trichoblastoma: a report of two cases with an immunohistochemical study. J Dermatol. 2015;42:305-310.
- Weir CB, St. Hilaire NJ. Epidermal inclusion cyst. StatPearls. StatPearls Publishing; 2020.


A healthy 45-year-old man presented to the dermatology clinic with a slow-growing subcutaneous nodule on the left chest that had been present for years.
Agminated Nodules on the Scalp
The Diagnosis: Cutaneous Angiosarcoma
Biopsy revealed a cellular neoplasm consisting of atypical polygonal cells with a hobnailed appearance, vasoformative characteristics, and rare extravasated erythrocytes. The tumor had an infiltrative growth pattern as demonstrated by dissecting dermal collagen and a poorly defined border with adjacent normal tissue (Figure 1). Immunohistochemistry revealed that the lesion was positive for CD31 and D2-40 (Figure 2) but negative for cytokeratin, CD10, CD68, human herpesvirus 8, CD34, and Melan A, thus confirming the endothelial origin of the tumor cells and the diagnosis of cutaneous angiosarcoma (CAS). The patient was treated with extended surgical excision and radiation therapy. No recurrence or metastasis was found throughout 2 years of follow-up.
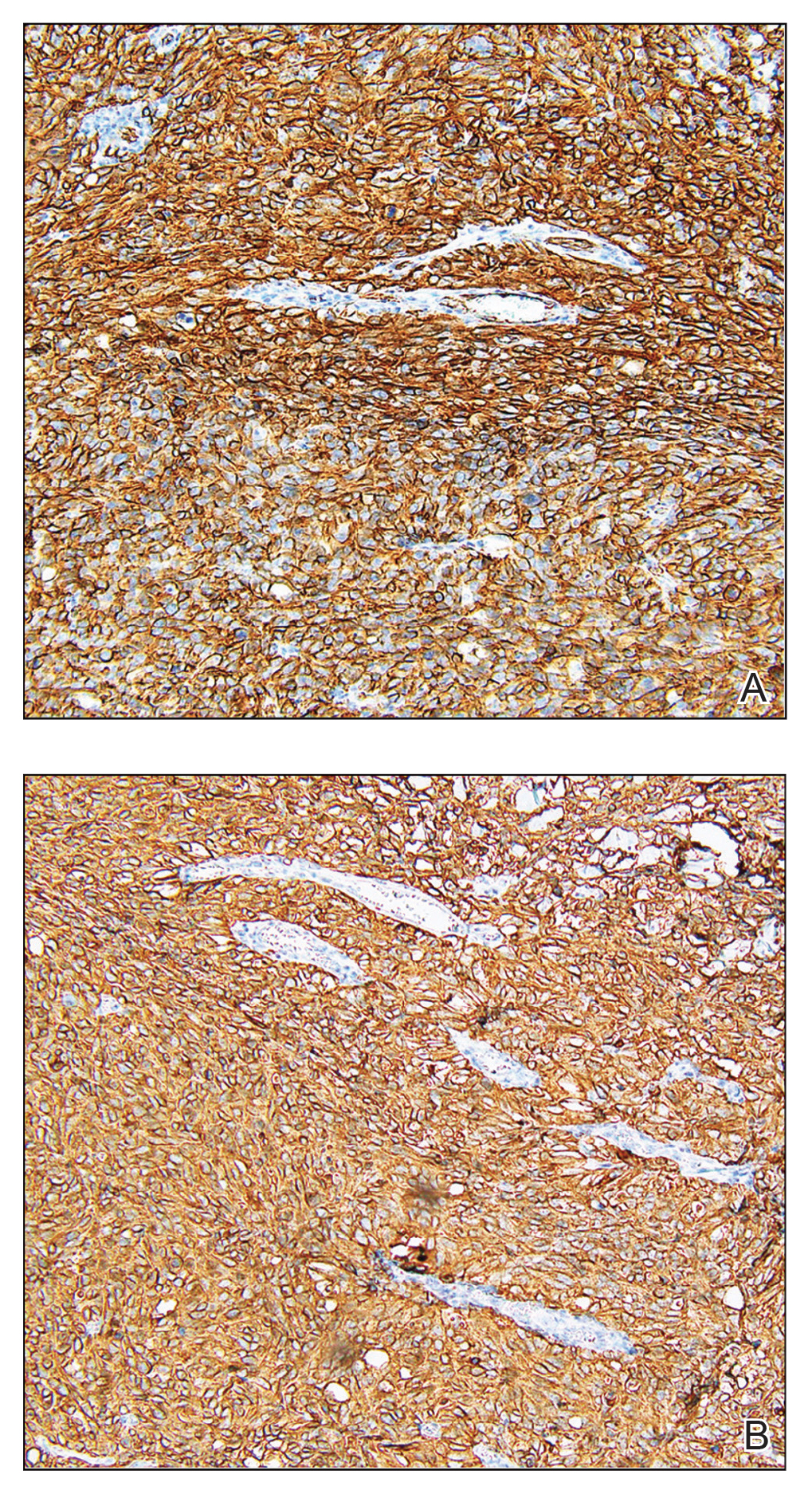
Angiosarcoma is a highly aggressive malignant neoplasm derived from vascular endothelial cells, most commonly involving the skin and superficial soft tissue. Angiosarcoma can be subdivided into CAS and visceral angiosarcoma according to the primary site of the tumor.1 Accurate and timely diagnosis of CAS is paramount due to its poor prognostic outcomes despite aggressive treatments. Clinically, CAS most frequently presents asymptomatically as an enlarging purple-red or bruiselike lesion with poorly defined margins. Cutaneous angiosarcoma often is misdiagnosed as an ecchymosis or hematoma due to its initial subtle presentation. It also may resemble eczema, hemangioma, and cellulitis; advanced lesions can mimic epithelial or mesenchymal neoplasms, including squamous cell carcinoma, keratoacanthoma, basal cell carcinoma, atypical fibroxanthoma (AFX), and malignant melanoma.2 Our patient lacked the classic clinical presentation of a hematomalike lesion and characteristic histologic features of anastomosing vascular structures with abundant extravasated erythrocytes at low magnification. However, the presence of erythrocytes in vascular channels along with CD31 and D2-40 immunoreactivity confirmed its vascular origin.
The prognosis of CAS is poor even with localized lesions. Age is a substantial prognostic factor, as a near 50% reduction of overall survival rate has been observed in patients older than 50 years.3 Other reported poor predictors for prognosis include male sex, the presence of cardiovascular diseases, location on the scalp, history of smoking, tumor size larger than 5 cm, and the presence of satellite lesions. Distant metastases are common, primarily affecting the lungs but also the bones and liver.4
Radical resection with a negative margin is considered the first-line treatment of choice. Although there is a paucity of studies assessing the specific width of surgical margins, application of no less than a 3-cm peripheral margin as well as a clear deep margin is recommended.5 Adjuvant radiation therapy also is essential to prevent local recurrence. Patients receiving combination therapy have a superior overall survival rate when compared to those undergoing surgery or radiation therapy alone.4
Cutaneous follicle center lymphoma also may present as 1 or more localized erythematous papules, plaques, and/or nodules, commonly arising on the scalp/forehead or trunk of middle-aged men. Despite being a low-grade lymphoma with a favorable prognosis, it may have a relatively fast growth and locally aggressive course if left untreated. The distinguishing histologic feature is a dense proliferation of neoplastic infiltrates in the dermis, which is separated from the epidermis by the grenz zone.6
The clinical presentation of cutaneous metastatic carcinomas varies greatly, with 1 or multiple localized or widespread lesions commonly involving the abdominal wall, scalp, and face. The lesions also may mimic benign dermatologic conditions, thus potentially resulting in erroneous clinical diagnosis and delayed therapy of the primary malignancy. Obtaining clinical history is crucial; however, a precise diagnosis may require histologic examination.7
Atypical fibroxanthoma is a rare superficial cutaneous sarcoma that typically occurs on the head and neck in sun-damaged elderly individuals. Clinically, AFX presents as well-circumscribed red or pink nodules or plaques with or without ulceration, crust, or scale.8 Atypical fibroxanthoma lesions usually are small, with a median diameter of 1 cm, while those greater than 2 cm reportedly account for less than 5% of cases.9 Atypical fibroxanthoma typically grows rapidly with no pain or discomfort. Histologically, AFX is characterized by a well-circumscribed dermal nodule consisting of pleomorphic spindle cells and multinucleated giant cells that can stain positively for CD10 and procollagen 1.10
Cutaneous pseudolymphoma is a benign inflammatory response process that stimulates polyclonal T- or B-cell lymphoproliferation. The clinical presentation may appear as localized or disseminated flesh-colored or red papules, infiltrated plaques, and nodules.11 Histopathology will show mixtures of B and T cells along with dendritic cells and macrophages, but irregular vascular structure and dissecting dermal collagen are not involved.
We present an unusual case of CAS with multiple pink nodules on the scalp. Early biopsy of these lesions is important to reach a correct diagnosis and to initiate appropriate treatment.
- Ishida Y, Otsuka A, Kabashima K. Cutaneous angiosarcoma: update on biology and latest treatment. Curr Opin Oncol. 2018;30:107-112.
- Dossett LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- Albores-Saavedra J, Schwartz AM, Henson DE, et al. Cutaneous angiosarcoma. analysis of 434 cases from the surveillance, epidemiology, and end results program, 1973-2007. Ann Diagn Pathol. 2011;15:93-97.
- Guadagnolo BA, Zagars GK, Araujo D, et al. Outcomes after definitive treatment for cutaneous angiosarcoma of the face and scalp. Head Neck. 2011;33:661-667.
- Lindford A, Böhling T, Vaalavirta L, et al. Surgical management of radiation-associated cutaneous breast angiosarcoma. J Plast Reconstr Aesthet Surg. 2011;64:1036-1042.
- Costa EPW, Lu.0cena BD, Amin GA, et al. Primary cutaneous follicle center lymphoma. An Bras Dermatol. 2017;92:701-703.
- Menon AR, Thomas AS, Suresh N, et al. Cutaneous metastasis: an unusual presenting feature of urologic malignancies. Urol Ann. 2016;8:377-380.
- Iorizzo LJ 3rd, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Kolb L, Schmieder GJ. Atypical fibroxanthoma. StatPearls. StatPearls Publishing; 2020.
- Sarac E, Yuksel M, Turkmen IC, et al. Case for diagnosis. atypical fibroxanthoma. An Bras Dermatol. 2019;94:239-241.
- Miguel D, Peckruhn M, Elsner P. Treatment of cutaneous pseudolymphoma: a systematic review. Acta Derm Venereol. 2018;98:310-317.
The Diagnosis: Cutaneous Angiosarcoma
Biopsy revealed a cellular neoplasm consisting of atypical polygonal cells with a hobnailed appearance, vasoformative characteristics, and rare extravasated erythrocytes. The tumor had an infiltrative growth pattern as demonstrated by dissecting dermal collagen and a poorly defined border with adjacent normal tissue (Figure 1). Immunohistochemistry revealed that the lesion was positive for CD31 and D2-40 (Figure 2) but negative for cytokeratin, CD10, CD68, human herpesvirus 8, CD34, and Melan A, thus confirming the endothelial origin of the tumor cells and the diagnosis of cutaneous angiosarcoma (CAS). The patient was treated with extended surgical excision and radiation therapy. No recurrence or metastasis was found throughout 2 years of follow-up.
Angiosarcoma is a highly aggressive malignant neoplasm derived from vascular endothelial cells, most commonly involving the skin and superficial soft tissue. Angiosarcoma can be subdivided into CAS and visceral angiosarcoma according to the primary site of the tumor.1 Accurate and timely diagnosis of CAS is paramount due to its poor prognostic outcomes despite aggressive treatments. Clinically, CAS most frequently presents asymptomatically as an enlarging purple-red or bruiselike lesion with poorly defined margins. Cutaneous angiosarcoma often is misdiagnosed as an ecchymosis or hematoma due to its initial subtle presentation. It also may resemble eczema, hemangioma, and cellulitis; advanced lesions can mimic epithelial or mesenchymal neoplasms, including squamous cell carcinoma, keratoacanthoma, basal cell carcinoma, atypical fibroxanthoma (AFX), and malignant melanoma.2 Our patient lacked the classic clinical presentation of a hematomalike lesion and characteristic histologic features of anastomosing vascular structures with abundant extravasated erythrocytes at low magnification. However, the presence of erythrocytes in vascular channels along with CD31 and D2-40 immunoreactivity confirmed its vascular origin.
The prognosis of CAS is poor even with localized lesions. Age is a substantial prognostic factor, as a near 50% reduction of overall survival rate has been observed in patients older than 50 years.3 Other reported poor predictors for prognosis include male sex, the presence of cardiovascular diseases, location on the scalp, history of smoking, tumor size larger than 5 cm, and the presence of satellite lesions. Distant metastases are common, primarily affecting the lungs but also the bones and liver.4
Radical resection with a negative margin is considered the first-line treatment of choice. Although there is a paucity of studies assessing the specific width of surgical margins, application of no less than a 3-cm peripheral margin as well as a clear deep margin is recommended.5 Adjuvant radiation therapy also is essential to prevent local recurrence. Patients receiving combination therapy have a superior overall survival rate when compared to those undergoing surgery or radiation therapy alone.4
Cutaneous follicle center lymphoma also may present as 1 or more localized erythematous papules, plaques, and/or nodules, commonly arising on the scalp/forehead or trunk of middle-aged men. Despite being a low-grade lymphoma with a favorable prognosis, it may have a relatively fast growth and locally aggressive course if left untreated. The distinguishing histologic feature is a dense proliferation of neoplastic infiltrates in the dermis, which is separated from the epidermis by the grenz zone.6
The clinical presentation of cutaneous metastatic carcinomas varies greatly, with 1 or multiple localized or widespread lesions commonly involving the abdominal wall, scalp, and face. The lesions also may mimic benign dermatologic conditions, thus potentially resulting in erroneous clinical diagnosis and delayed therapy of the primary malignancy. Obtaining clinical history is crucial; however, a precise diagnosis may require histologic examination.7
Atypical fibroxanthoma is a rare superficial cutaneous sarcoma that typically occurs on the head and neck in sun-damaged elderly individuals. Clinically, AFX presents as well-circumscribed red or pink nodules or plaques with or without ulceration, crust, or scale.8 Atypical fibroxanthoma lesions usually are small, with a median diameter of 1 cm, while those greater than 2 cm reportedly account for less than 5% of cases.9 Atypical fibroxanthoma typically grows rapidly with no pain or discomfort. Histologically, AFX is characterized by a well-circumscribed dermal nodule consisting of pleomorphic spindle cells and multinucleated giant cells that can stain positively for CD10 and procollagen 1.10
Cutaneous pseudolymphoma is a benign inflammatory response process that stimulates polyclonal T- or B-cell lymphoproliferation. The clinical presentation may appear as localized or disseminated flesh-colored or red papules, infiltrated plaques, and nodules.11 Histopathology will show mixtures of B and T cells along with dendritic cells and macrophages, but irregular vascular structure and dissecting dermal collagen are not involved.
We present an unusual case of CAS with multiple pink nodules on the scalp. Early biopsy of these lesions is important to reach a correct diagnosis and to initiate appropriate treatment.
The Diagnosis: Cutaneous Angiosarcoma
Biopsy revealed a cellular neoplasm consisting of atypical polygonal cells with a hobnailed appearance, vasoformative characteristics, and rare extravasated erythrocytes. The tumor had an infiltrative growth pattern as demonstrated by dissecting dermal collagen and a poorly defined border with adjacent normal tissue (Figure 1). Immunohistochemistry revealed that the lesion was positive for CD31 and D2-40 (Figure 2) but negative for cytokeratin, CD10, CD68, human herpesvirus 8, CD34, and Melan A, thus confirming the endothelial origin of the tumor cells and the diagnosis of cutaneous angiosarcoma (CAS). The patient was treated with extended surgical excision and radiation therapy. No recurrence or metastasis was found throughout 2 years of follow-up.
Angiosarcoma is a highly aggressive malignant neoplasm derived from vascular endothelial cells, most commonly involving the skin and superficial soft tissue. Angiosarcoma can be subdivided into CAS and visceral angiosarcoma according to the primary site of the tumor.1 Accurate and timely diagnosis of CAS is paramount due to its poor prognostic outcomes despite aggressive treatments. Clinically, CAS most frequently presents asymptomatically as an enlarging purple-red or bruiselike lesion with poorly defined margins. Cutaneous angiosarcoma often is misdiagnosed as an ecchymosis or hematoma due to its initial subtle presentation. It also may resemble eczema, hemangioma, and cellulitis; advanced lesions can mimic epithelial or mesenchymal neoplasms, including squamous cell carcinoma, keratoacanthoma, basal cell carcinoma, atypical fibroxanthoma (AFX), and malignant melanoma.2 Our patient lacked the classic clinical presentation of a hematomalike lesion and characteristic histologic features of anastomosing vascular structures with abundant extravasated erythrocytes at low magnification. However, the presence of erythrocytes in vascular channels along with CD31 and D2-40 immunoreactivity confirmed its vascular origin.
The prognosis of CAS is poor even with localized lesions. Age is a substantial prognostic factor, as a near 50% reduction of overall survival rate has been observed in patients older than 50 years.3 Other reported poor predictors for prognosis include male sex, the presence of cardiovascular diseases, location on the scalp, history of smoking, tumor size larger than 5 cm, and the presence of satellite lesions. Distant metastases are common, primarily affecting the lungs but also the bones and liver.4
Radical resection with a negative margin is considered the first-line treatment of choice. Although there is a paucity of studies assessing the specific width of surgical margins, application of no less than a 3-cm peripheral margin as well as a clear deep margin is recommended.5 Adjuvant radiation therapy also is essential to prevent local recurrence. Patients receiving combination therapy have a superior overall survival rate when compared to those undergoing surgery or radiation therapy alone.4
Cutaneous follicle center lymphoma also may present as 1 or more localized erythematous papules, plaques, and/or nodules, commonly arising on the scalp/forehead or trunk of middle-aged men. Despite being a low-grade lymphoma with a favorable prognosis, it may have a relatively fast growth and locally aggressive course if left untreated. The distinguishing histologic feature is a dense proliferation of neoplastic infiltrates in the dermis, which is separated from the epidermis by the grenz zone.6
The clinical presentation of cutaneous metastatic carcinomas varies greatly, with 1 or multiple localized or widespread lesions commonly involving the abdominal wall, scalp, and face. The lesions also may mimic benign dermatologic conditions, thus potentially resulting in erroneous clinical diagnosis and delayed therapy of the primary malignancy. Obtaining clinical history is crucial; however, a precise diagnosis may require histologic examination.7
Atypical fibroxanthoma is a rare superficial cutaneous sarcoma that typically occurs on the head and neck in sun-damaged elderly individuals. Clinically, AFX presents as well-circumscribed red or pink nodules or plaques with or without ulceration, crust, or scale.8 Atypical fibroxanthoma lesions usually are small, with a median diameter of 1 cm, while those greater than 2 cm reportedly account for less than 5% of cases.9 Atypical fibroxanthoma typically grows rapidly with no pain or discomfort. Histologically, AFX is characterized by a well-circumscribed dermal nodule consisting of pleomorphic spindle cells and multinucleated giant cells that can stain positively for CD10 and procollagen 1.10
Cutaneous pseudolymphoma is a benign inflammatory response process that stimulates polyclonal T- or B-cell lymphoproliferation. The clinical presentation may appear as localized or disseminated flesh-colored or red papules, infiltrated plaques, and nodules.11 Histopathology will show mixtures of B and T cells along with dendritic cells and macrophages, but irregular vascular structure and dissecting dermal collagen are not involved.
We present an unusual case of CAS with multiple pink nodules on the scalp. Early biopsy of these lesions is important to reach a correct diagnosis and to initiate appropriate treatment.
- Ishida Y, Otsuka A, Kabashima K. Cutaneous angiosarcoma: update on biology and latest treatment. Curr Opin Oncol. 2018;30:107-112.
- Dossett LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- Albores-Saavedra J, Schwartz AM, Henson DE, et al. Cutaneous angiosarcoma. analysis of 434 cases from the surveillance, epidemiology, and end results program, 1973-2007. Ann Diagn Pathol. 2011;15:93-97.
- Guadagnolo BA, Zagars GK, Araujo D, et al. Outcomes after definitive treatment for cutaneous angiosarcoma of the face and scalp. Head Neck. 2011;33:661-667.
- Lindford A, Böhling T, Vaalavirta L, et al. Surgical management of radiation-associated cutaneous breast angiosarcoma. J Plast Reconstr Aesthet Surg. 2011;64:1036-1042.
- Costa EPW, Lu.0cena BD, Amin GA, et al. Primary cutaneous follicle center lymphoma. An Bras Dermatol. 2017;92:701-703.
- Menon AR, Thomas AS, Suresh N, et al. Cutaneous metastasis: an unusual presenting feature of urologic malignancies. Urol Ann. 2016;8:377-380.
- Iorizzo LJ 3rd, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Kolb L, Schmieder GJ. Atypical fibroxanthoma. StatPearls. StatPearls Publishing; 2020.
- Sarac E, Yuksel M, Turkmen IC, et al. Case for diagnosis. atypical fibroxanthoma. An Bras Dermatol. 2019;94:239-241.
- Miguel D, Peckruhn M, Elsner P. Treatment of cutaneous pseudolymphoma: a systematic review. Acta Derm Venereol. 2018;98:310-317.
- Ishida Y, Otsuka A, Kabashima K. Cutaneous angiosarcoma: update on biology and latest treatment. Curr Opin Oncol. 2018;30:107-112.
- Dossett LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- Albores-Saavedra J, Schwartz AM, Henson DE, et al. Cutaneous angiosarcoma. analysis of 434 cases from the surveillance, epidemiology, and end results program, 1973-2007. Ann Diagn Pathol. 2011;15:93-97.
- Guadagnolo BA, Zagars GK, Araujo D, et al. Outcomes after definitive treatment for cutaneous angiosarcoma of the face and scalp. Head Neck. 2011;33:661-667.
- Lindford A, Böhling T, Vaalavirta L, et al. Surgical management of radiation-associated cutaneous breast angiosarcoma. J Plast Reconstr Aesthet Surg. 2011;64:1036-1042.
- Costa EPW, Lu.0cena BD, Amin GA, et al. Primary cutaneous follicle center lymphoma. An Bras Dermatol. 2017;92:701-703.
- Menon AR, Thomas AS, Suresh N, et al. Cutaneous metastasis: an unusual presenting feature of urologic malignancies. Urol Ann. 2016;8:377-380.
- Iorizzo LJ 3rd, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Kolb L, Schmieder GJ. Atypical fibroxanthoma. StatPearls. StatPearls Publishing; 2020.
- Sarac E, Yuksel M, Turkmen IC, et al. Case for diagnosis. atypical fibroxanthoma. An Bras Dermatol. 2019;94:239-241.
- Miguel D, Peckruhn M, Elsner P. Treatment of cutaneous pseudolymphoma: a systematic review. Acta Derm Venereol. 2018;98:310-317.

A 67-year-old man presented with pink nodules on the scalp that were enlarging and increasing over the course of 2 months. The patient was otherwise healthy, had no constitutional symptoms such as fever or weight loss, and did not note pruritus or pain. His medications included telmisartan and Salvia miltiorrhiza for hypertension and coronary heart disease, respectively. He had been a heavy smoker for 44 years. Physical examination revealed several dome-shaped, pink nodules with smooth surfaces distributed in an agminated appearance on the scalp. The lesions were indurated and ranged from 1 to 5 cm in diameter.
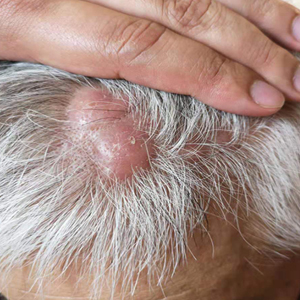
Progressive Axillary Hyperpigmentation
The Diagnosis: Dowling-Degos Disease
Histopathology demonstrated elongation of the epidermal rete ridges with increased basal pigmentation, suprapapillary epithelial thinning, dermal melanophages, and a mild lymphocytic infiltrate (Figure). Given the clinical and histologic findings, a diagnosis of Dowling-Degos disease (DDD) was made. The patient was counseled on the increased risk for her children developing DDD. Treatment with the erbium:YAG (Er:YAG) laser subsequently was initiated.

Dowling-Degos disease (also known as reticulate pigmented anomaly of the flexures) is an uncommon autosomal-dominant condition characterized by reticular hyperpigmentation involving the flexural and intertriginous sites. Classic DDD commonly is caused by lossof-function mutations in the keratin 5 gene, KRT51; however, DDD also may result from loss-of-function mutations in the protein O-fucosyltransferase 1, POFUT1, and protein O-glucosyltransferase 1, POGLUT1, genes.2
Rare cases of DDD associated with hidradenitis suppurativa are caused by mutations in the presenilin enhancer protein 2 gene, PSENEN.3
Of note, a missense mutation in KRT5 is implicated in epidermolysis bullosa simplex with mottled pigmentation. Onset of DDD typically occurs during the third to fourth decades of life. Reticulated hyperpigmented macules initially occur in the axillae and groin and progressively increase over time to involve the neck, inframammary folds, trunk, and flexural surfaces of the arms and thighs. Patients additionally may present with pitted perioral scars, comedolike lesions on the back and neck, epidermoid cysts, and hidradenitis suppurativa. Keratoacanthoma and squamous cell carcinoma rarely have been reported in association with classic DDD.4,5
Dowling-Degos disease usually is asymptomatic, though pruritus seldom may occur in the affected flexural areas. Histologically, the epidermal rete ridges are elongated in a filiform or antlerlike pattern with increased pigmentation of the basal layer and thinning of the suprapapillary epithelium. Dermal melanosis and a mild perivascular lymphohistiocytic infiltrate also are present with no increase in the number of melanocytes.6,7 Galli-Galli disease is a variant of DDD that shares similar clinical and histologic features of DDD but is distinguished from DDD by suprabasilar nondyskeratotic acantholysis on histology.8
Regarding other differential diagnoses for our patient, acanthosis nigricans may be distinguished clinically by the presence of velvety and/or verrucous plaques, commonly in the neck folds and axillae. Histologically, acanthosis nigricans is distinct from DDD and involves hyperkeratosis, acanthosis, and epidermal papillomatosis. Our patient had no history of diabetes mellitus or insulin resistance. Granular parakeratosis presents with hyperpigmented hyperkeratotic papules and plaques classically confined to the axillary region; however, the involvement of other intertriginous areas may occur. Histologically, granular parakeratosis demonstrates compact parakeratosis with small bluish keratohyalin granules within the stratum corneum. Confluent and reticulated papillomatosis presents with red-brown keratotic papules that initially appear in the intermammary region and spread laterally forming a reticulated pattern. Histology is similar to acanthosis nigricans and demonstrates hyperkeratosis, acanthosis, and papillomatosis. Inverse psoriasis presents with symmetric and sharply demarcated, erythematous, nonscaly plaques in the intertriginous areas. The plaques of inverse psoriasis may be pruritic and/or sore and occasionally may become macerated. Inverse psoriasis shares similar histologic findings compared to classic plaque psoriasis but may have less confluent parakeratosis.
Treatment of DDD essentially is reserved for cosmetic reasons. Topical hydroquinone, tretinoin, and corticosteroids have been used with limited to no success.5,9 Beneficial results after treatment with the Er:YAG laser have been reported.10
- Betz RC, Planko L, Eigelshoven S, et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet. 2006;78:510-519.
- Basmanav FB, Oprisoreanu AM, Pasternack SM, et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomaldominant Dowling-Degos disease. Am J Hum Genet. 2014;94:135-143.
- Pavlovsky M, Sarig O, Eskin-Schwartz M, et al. A phenotype combining hidradenitis suppurativa with Dowling-Degos disease caused by a founder mutation in PSENEN. Br J Dermatol. 2018;178:502-508.
- Ujihara M, Kamakura T, Ikeda M, et al. Dowling-Degos disease associated with squamous cell carcinomas on the dappled pigmentation. Br J Dermatol. 2002;147:568-571.
- Weber LA, Kantor GR, Bergfeld WF. Reticulate pigmented anomaly of the flexures (Dowling-Degos disease): a case report associated with hidradenitis suppurativa and squamous cell carcinoma. Cutis. 1990;45:446-450.
- Jones EW, Grice K. Reticulate pigmented anomaly of the flexures. Dowing Degos disease, a new genodermatosis. Arch Dermatol. 1978;114:1150-1157.
- Kim YC, Davis MD, Schanbacher CF, et al. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): a clinical and histopathologic study of 6 cases. J Am Acad Dermatol. 1999; 40:462-467.
- Reisenauer AK, Wordingham SV, York J, et al. Heterozygous frameshift mutation in keratin 5 in a family with Galli-Galli disease. Br J Dermatol. 2014;170:1362-1365.
- Oppolzer G, Schwarz T, Duschet P, et al. Dowling-Degos disease: unsuccessful therapeutic trial with retinoids [in German]. Hautarzt. 1987;38:615-618.
- Wenzel G, Petrow W, Tappe K, et al. Treatment of Dowling-Degos disease with Er:YAG-laser: results after 2.5 years. Dermatol Surg. 2003;29:1161-1162.
The Diagnosis: Dowling-Degos Disease
Histopathology demonstrated elongation of the epidermal rete ridges with increased basal pigmentation, suprapapillary epithelial thinning, dermal melanophages, and a mild lymphocytic infiltrate (Figure). Given the clinical and histologic findings, a diagnosis of Dowling-Degos disease (DDD) was made. The patient was counseled on the increased risk for her children developing DDD. Treatment with the erbium:YAG (Er:YAG) laser subsequently was initiated.
Dowling-Degos disease (also known as reticulate pigmented anomaly of the flexures) is an uncommon autosomal-dominant condition characterized by reticular hyperpigmentation involving the flexural and intertriginous sites. Classic DDD commonly is caused by lossof-function mutations in the keratin 5 gene, KRT51; however, DDD also may result from loss-of-function mutations in the protein O-fucosyltransferase 1, POFUT1, and protein O-glucosyltransferase 1, POGLUT1, genes.2
Rare cases of DDD associated with hidradenitis suppurativa are caused by mutations in the presenilin enhancer protein 2 gene, PSENEN.3
Of note, a missense mutation in KRT5 is implicated in epidermolysis bullosa simplex with mottled pigmentation. Onset of DDD typically occurs during the third to fourth decades of life. Reticulated hyperpigmented macules initially occur in the axillae and groin and progressively increase over time to involve the neck, inframammary folds, trunk, and flexural surfaces of the arms and thighs. Patients additionally may present with pitted perioral scars, comedolike lesions on the back and neck, epidermoid cysts, and hidradenitis suppurativa. Keratoacanthoma and squamous cell carcinoma rarely have been reported in association with classic DDD.4,5
Dowling-Degos disease usually is asymptomatic, though pruritus seldom may occur in the affected flexural areas. Histologically, the epidermal rete ridges are elongated in a filiform or antlerlike pattern with increased pigmentation of the basal layer and thinning of the suprapapillary epithelium. Dermal melanosis and a mild perivascular lymphohistiocytic infiltrate also are present with no increase in the number of melanocytes.6,7 Galli-Galli disease is a variant of DDD that shares similar clinical and histologic features of DDD but is distinguished from DDD by suprabasilar nondyskeratotic acantholysis on histology.8
Regarding other differential diagnoses for our patient, acanthosis nigricans may be distinguished clinically by the presence of velvety and/or verrucous plaques, commonly in the neck folds and axillae. Histologically, acanthosis nigricans is distinct from DDD and involves hyperkeratosis, acanthosis, and epidermal papillomatosis. Our patient had no history of diabetes mellitus or insulin resistance. Granular parakeratosis presents with hyperpigmented hyperkeratotic papules and plaques classically confined to the axillary region; however, the involvement of other intertriginous areas may occur. Histologically, granular parakeratosis demonstrates compact parakeratosis with small bluish keratohyalin granules within the stratum corneum. Confluent and reticulated papillomatosis presents with red-brown keratotic papules that initially appear in the intermammary region and spread laterally forming a reticulated pattern. Histology is similar to acanthosis nigricans and demonstrates hyperkeratosis, acanthosis, and papillomatosis. Inverse psoriasis presents with symmetric and sharply demarcated, erythematous, nonscaly plaques in the intertriginous areas. The plaques of inverse psoriasis may be pruritic and/or sore and occasionally may become macerated. Inverse psoriasis shares similar histologic findings compared to classic plaque psoriasis but may have less confluent parakeratosis.
Treatment of DDD essentially is reserved for cosmetic reasons. Topical hydroquinone, tretinoin, and corticosteroids have been used with limited to no success.5,9 Beneficial results after treatment with the Er:YAG laser have been reported.10
The Diagnosis: Dowling-Degos Disease
Histopathology demonstrated elongation of the epidermal rete ridges with increased basal pigmentation, suprapapillary epithelial thinning, dermal melanophages, and a mild lymphocytic infiltrate (Figure). Given the clinical and histologic findings, a diagnosis of Dowling-Degos disease (DDD) was made. The patient was counseled on the increased risk for her children developing DDD. Treatment with the erbium:YAG (Er:YAG) laser subsequently was initiated.
Dowling-Degos disease (also known as reticulate pigmented anomaly of the flexures) is an uncommon autosomal-dominant condition characterized by reticular hyperpigmentation involving the flexural and intertriginous sites. Classic DDD commonly is caused by lossof-function mutations in the keratin 5 gene, KRT51; however, DDD also may result from loss-of-function mutations in the protein O-fucosyltransferase 1, POFUT1, and protein O-glucosyltransferase 1, POGLUT1, genes.2
Rare cases of DDD associated with hidradenitis suppurativa are caused by mutations in the presenilin enhancer protein 2 gene, PSENEN.3
Of note, a missense mutation in KRT5 is implicated in epidermolysis bullosa simplex with mottled pigmentation. Onset of DDD typically occurs during the third to fourth decades of life. Reticulated hyperpigmented macules initially occur in the axillae and groin and progressively increase over time to involve the neck, inframammary folds, trunk, and flexural surfaces of the arms and thighs. Patients additionally may present with pitted perioral scars, comedolike lesions on the back and neck, epidermoid cysts, and hidradenitis suppurativa. Keratoacanthoma and squamous cell carcinoma rarely have been reported in association with classic DDD.4,5
Dowling-Degos disease usually is asymptomatic, though pruritus seldom may occur in the affected flexural areas. Histologically, the epidermal rete ridges are elongated in a filiform or antlerlike pattern with increased pigmentation of the basal layer and thinning of the suprapapillary epithelium. Dermal melanosis and a mild perivascular lymphohistiocytic infiltrate also are present with no increase in the number of melanocytes.6,7 Galli-Galli disease is a variant of DDD that shares similar clinical and histologic features of DDD but is distinguished from DDD by suprabasilar nondyskeratotic acantholysis on histology.8
Regarding other differential diagnoses for our patient, acanthosis nigricans may be distinguished clinically by the presence of velvety and/or verrucous plaques, commonly in the neck folds and axillae. Histologically, acanthosis nigricans is distinct from DDD and involves hyperkeratosis, acanthosis, and epidermal papillomatosis. Our patient had no history of diabetes mellitus or insulin resistance. Granular parakeratosis presents with hyperpigmented hyperkeratotic papules and plaques classically confined to the axillary region; however, the involvement of other intertriginous areas may occur. Histologically, granular parakeratosis demonstrates compact parakeratosis with small bluish keratohyalin granules within the stratum corneum. Confluent and reticulated papillomatosis presents with red-brown keratotic papules that initially appear in the intermammary region and spread laterally forming a reticulated pattern. Histology is similar to acanthosis nigricans and demonstrates hyperkeratosis, acanthosis, and papillomatosis. Inverse psoriasis presents with symmetric and sharply demarcated, erythematous, nonscaly plaques in the intertriginous areas. The plaques of inverse psoriasis may be pruritic and/or sore and occasionally may become macerated. Inverse psoriasis shares similar histologic findings compared to classic plaque psoriasis but may have less confluent parakeratosis.
Treatment of DDD essentially is reserved for cosmetic reasons. Topical hydroquinone, tretinoin, and corticosteroids have been used with limited to no success.5,9 Beneficial results after treatment with the Er:YAG laser have been reported.10
- Betz RC, Planko L, Eigelshoven S, et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet. 2006;78:510-519.
- Basmanav FB, Oprisoreanu AM, Pasternack SM, et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomaldominant Dowling-Degos disease. Am J Hum Genet. 2014;94:135-143.
- Pavlovsky M, Sarig O, Eskin-Schwartz M, et al. A phenotype combining hidradenitis suppurativa with Dowling-Degos disease caused by a founder mutation in PSENEN. Br J Dermatol. 2018;178:502-508.
- Ujihara M, Kamakura T, Ikeda M, et al. Dowling-Degos disease associated with squamous cell carcinomas on the dappled pigmentation. Br J Dermatol. 2002;147:568-571.
- Weber LA, Kantor GR, Bergfeld WF. Reticulate pigmented anomaly of the flexures (Dowling-Degos disease): a case report associated with hidradenitis suppurativa and squamous cell carcinoma. Cutis. 1990;45:446-450.
- Jones EW, Grice K. Reticulate pigmented anomaly of the flexures. Dowing Degos disease, a new genodermatosis. Arch Dermatol. 1978;114:1150-1157.
- Kim YC, Davis MD, Schanbacher CF, et al. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): a clinical and histopathologic study of 6 cases. J Am Acad Dermatol. 1999; 40:462-467.
- Reisenauer AK, Wordingham SV, York J, et al. Heterozygous frameshift mutation in keratin 5 in a family with Galli-Galli disease. Br J Dermatol. 2014;170:1362-1365.
- Oppolzer G, Schwarz T, Duschet P, et al. Dowling-Degos disease: unsuccessful therapeutic trial with retinoids [in German]. Hautarzt. 1987;38:615-618.
- Wenzel G, Petrow W, Tappe K, et al. Treatment of Dowling-Degos disease with Er:YAG-laser: results after 2.5 years. Dermatol Surg. 2003;29:1161-1162.
- Betz RC, Planko L, Eigelshoven S, et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet. 2006;78:510-519.
- Basmanav FB, Oprisoreanu AM, Pasternack SM, et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomaldominant Dowling-Degos disease. Am J Hum Genet. 2014;94:135-143.
- Pavlovsky M, Sarig O, Eskin-Schwartz M, et al. A phenotype combining hidradenitis suppurativa with Dowling-Degos disease caused by a founder mutation in PSENEN. Br J Dermatol. 2018;178:502-508.
- Ujihara M, Kamakura T, Ikeda M, et al. Dowling-Degos disease associated with squamous cell carcinomas on the dappled pigmentation. Br J Dermatol. 2002;147:568-571.
- Weber LA, Kantor GR, Bergfeld WF. Reticulate pigmented anomaly of the flexures (Dowling-Degos disease): a case report associated with hidradenitis suppurativa and squamous cell carcinoma. Cutis. 1990;45:446-450.
- Jones EW, Grice K. Reticulate pigmented anomaly of the flexures. Dowing Degos disease, a new genodermatosis. Arch Dermatol. 1978;114:1150-1157.
- Kim YC, Davis MD, Schanbacher CF, et al. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): a clinical and histopathologic study of 6 cases. J Am Acad Dermatol. 1999; 40:462-467.
- Reisenauer AK, Wordingham SV, York J, et al. Heterozygous frameshift mutation in keratin 5 in a family with Galli-Galli disease. Br J Dermatol. 2014;170:1362-1365.
- Oppolzer G, Schwarz T, Duschet P, et al. Dowling-Degos disease: unsuccessful therapeutic trial with retinoids [in German]. Hautarzt. 1987;38:615-618.
- Wenzel G, Petrow W, Tappe K, et al. Treatment of Dowling-Degos disease with Er:YAG-laser: results after 2.5 years. Dermatol Surg. 2003;29:1161-1162.

A 50-year-old Hispanic woman presented with asymptomatic, progressive, brown hyperpigmentation involving the axillae, neck, upper back, and inframammary areas of 5 years’ duration. She had no other notable medical history; family history was unremarkable. She had been treated with topical hydroquinone and tretinoin by an outside physician without improvement. Physical examination revealed reticulated hyperpigmented macules and patches involving the inverse regions of the neck, axillae, and inframammary regions. Additionally, acneform pitted scars involving the perioral region were seen. A 4.0-mm punch biopsy of the right axilla was performed.
Dermatopathology Etiquette 101
The Accreditation Council for Graduate Medical Education has established core competencies to serve as a foundation for the training received in a dermatology residency program.1 Although programs are required to have the same concentrations—patient care, medical knowledge, practice-based learning and improvement, interpersonal and communication skills, professionalism, and systems-based practice—no specific guidelines are in place regarding how each of these competencies should be reached within a training period.2 Instead, it remains the responsibility of each program to formulate an individualized curriculum to facilitate proficiency in the multiple areas encompassed by a residency.
In many dermatology residency programs, dermatopathology is a substantial component of educational objectives and the curriculum.1 Residents may spend as much as 25% of their training on dermatopathology. However, there is great variability among programs in methods of teaching dermatopathology. When Hinshaw3 surveyed 52 of 109 dermatology residency programs, they identified differences in dermatopathology teaching that included, but was not limited to, utilization of problem-based learning (in 40.4% of programs), integration of journal reviews (53.8%), and computer-based learning (19.2%). In addition, differences were identified in the recommended primary textbook and the makeup of faculty who taught dermatopathology.3
Although residency programs vary in their methods of teaching this important component of dermatology, most use a multiheaded microscope in some capacity for didactics or sign-out. For most trainees, the dermatopathology laboratory is a new environment compared to the clinical space that medical students and residents become accustomed to throughout their education, thus creating a knowledge gap for trainees on proper dermatopathology etiquette and universal guidelines.
With medical students, residents, and fellows in mind, we have prepared a basic “dermatopathology etiquette” reference for trainees. Just as there are universal rules in the operating room for surgery (eg, sterile technique), we want to establish a code of conduct at the microscope. We hope that these 10 tips will, first, be useful to those who are unsure how to approach their first experience with dermatopathology and, second, serve as a guideline to aid development of appropriate communication skills and functioning within this novel setting. This list also can serve as a resource for dermatopathology attendings to provide to rotating residents and students.
1. New to pathology? It’s okay to ask. Do not hesitate to ask upper-year residents, fellows, and attendings for instructions on such matters as how to adjust your eyepiece to get the best resolution.
2. If a slide drops on the floor, do not move! Your first instinct might be to move your chair to look for the dropped slide, but you might roll over it and break it.
3. When the attending is looking through the scope, you look through the scope. Dermatopathology is a visual exercise. Getting in your “optic mileage” is best done under the guidance of an experienced dermatopathologist.
4. Rules regarding food and drink at the microscope vary by pathologist. It’s best to ask what each attending prefers. Safe advice is to avoid foods that make noise, such as chewing gum and chips, and food that has a strong odor, such as microwaved leftovers.
5. Limit use of a laptop, cell phone, and smartwatch. If you think that using any of these is necessary, it generally is best to announce that you are looking up something related to the case and then share your findings (but not the most recent post on your Facebook News Feed).
6. If you notice that something needs correcting on the report, speak up! We are all human; we all make typos. Do not hesitate to mention this as soon as possible, especially before the case is signed out. You will likely be thanked by your attending because it is harder to rectify once the report has been signed out.
7. Small talk often is welcome during large excisions. This is a great time to ask what others are doing next weekend or what happened in clinic earlier that day, or just to tell a good (clean) joke that is making the rounds. Conversely, if the case is complex, it often is best to wait until it is completed before asking questions.
8. When participating in a roundtable diagnosis, you are welcome to directly state the diagnosis for bread-and-butter cases, such as basal cell carcinomas and seborrheic keratoses. It is appropriate to be more descriptive and methodical in more complex cases. When evaluating a rash, give the general inflammatory pattern first. For example, is it spongiotic? Psoriasiform? Interface? Or a mixed pattern?
9. Extra points for identifying special sites! These include mucosal, genital, and acral sites. You might even get bonus points if you can determine something about the patient (child or adult) based on the pathologic features, such as variation in collagen patterns.
10. Whenever you are in doubt, just describe what you see. You can use the traditional top-down approach or start with stating the most evident finding, then proceed to a top-down description. If it is a neoplasm, describe the overall architecture; then, what you see at a cellular level will get you some points as well.
We acknowledge that this list of 10 tips is not comprehensive and might vary by attending and each institution’s distinctive training format. We are hopeful, however, that these 10 points of etiquette can serve as a guideline.
- Hinshaw M, Hsu P, Lee L-Y, et al. The current state of dermatopathology education: a survey of the Association of Professors of Dermatology. J Cutan Pathol. 2009;36:620-628. doi:10.1111/j.1600-0560.2008.01128.x
- Hinshaw MA, Stratman EJ. Core competencies in dermatopathology. J Cutan Pathol. 2006;33:160-165. doi:10.1111/j.0303-6987.2006.00442.x
- Hinshaw MA. Dermatopathology education: an update. Dermatol Clin. 2012;30:815-826. doi:10.1016/j.det.2012.06.003
The Accreditation Council for Graduate Medical Education has established core competencies to serve as a foundation for the training received in a dermatology residency program.1 Although programs are required to have the same concentrations—patient care, medical knowledge, practice-based learning and improvement, interpersonal and communication skills, professionalism, and systems-based practice—no specific guidelines are in place regarding how each of these competencies should be reached within a training period.2 Instead, it remains the responsibility of each program to formulate an individualized curriculum to facilitate proficiency in the multiple areas encompassed by a residency.
In many dermatology residency programs, dermatopathology is a substantial component of educational objectives and the curriculum.1 Residents may spend as much as 25% of their training on dermatopathology. However, there is great variability among programs in methods of teaching dermatopathology. When Hinshaw3 surveyed 52 of 109 dermatology residency programs, they identified differences in dermatopathology teaching that included, but was not limited to, utilization of problem-based learning (in 40.4% of programs), integration of journal reviews (53.8%), and computer-based learning (19.2%). In addition, differences were identified in the recommended primary textbook and the makeup of faculty who taught dermatopathology.3
Although residency programs vary in their methods of teaching this important component of dermatology, most use a multiheaded microscope in some capacity for didactics or sign-out. For most trainees, the dermatopathology laboratory is a new environment compared to the clinical space that medical students and residents become accustomed to throughout their education, thus creating a knowledge gap for trainees on proper dermatopathology etiquette and universal guidelines.
With medical students, residents, and fellows in mind, we have prepared a basic “dermatopathology etiquette” reference for trainees. Just as there are universal rules in the operating room for surgery (eg, sterile technique), we want to establish a code of conduct at the microscope. We hope that these 10 tips will, first, be useful to those who are unsure how to approach their first experience with dermatopathology and, second, serve as a guideline to aid development of appropriate communication skills and functioning within this novel setting. This list also can serve as a resource for dermatopathology attendings to provide to rotating residents and students.
1. New to pathology? It’s okay to ask. Do not hesitate to ask upper-year residents, fellows, and attendings for instructions on such matters as how to adjust your eyepiece to get the best resolution.
2. If a slide drops on the floor, do not move! Your first instinct might be to move your chair to look for the dropped slide, but you might roll over it and break it.
3. When the attending is looking through the scope, you look through the scope. Dermatopathology is a visual exercise. Getting in your “optic mileage” is best done under the guidance of an experienced dermatopathologist.
4. Rules regarding food and drink at the microscope vary by pathologist. It’s best to ask what each attending prefers. Safe advice is to avoid foods that make noise, such as chewing gum and chips, and food that has a strong odor, such as microwaved leftovers.
5. Limit use of a laptop, cell phone, and smartwatch. If you think that using any of these is necessary, it generally is best to announce that you are looking up something related to the case and then share your findings (but not the most recent post on your Facebook News Feed).
6. If you notice that something needs correcting on the report, speak up! We are all human; we all make typos. Do not hesitate to mention this as soon as possible, especially before the case is signed out. You will likely be thanked by your attending because it is harder to rectify once the report has been signed out.
7. Small talk often is welcome during large excisions. This is a great time to ask what others are doing next weekend or what happened in clinic earlier that day, or just to tell a good (clean) joke that is making the rounds. Conversely, if the case is complex, it often is best to wait until it is completed before asking questions.
8. When participating in a roundtable diagnosis, you are welcome to directly state the diagnosis for bread-and-butter cases, such as basal cell carcinomas and seborrheic keratoses. It is appropriate to be more descriptive and methodical in more complex cases. When evaluating a rash, give the general inflammatory pattern first. For example, is it spongiotic? Psoriasiform? Interface? Or a mixed pattern?
9. Extra points for identifying special sites! These include mucosal, genital, and acral sites. You might even get bonus points if you can determine something about the patient (child or adult) based on the pathologic features, such as variation in collagen patterns.
10. Whenever you are in doubt, just describe what you see. You can use the traditional top-down approach or start with stating the most evident finding, then proceed to a top-down description. If it is a neoplasm, describe the overall architecture; then, what you see at a cellular level will get you some points as well.
We acknowledge that this list of 10 tips is not comprehensive and might vary by attending and each institution’s distinctive training format. We are hopeful, however, that these 10 points of etiquette can serve as a guideline.
The Accreditation Council for Graduate Medical Education has established core competencies to serve as a foundation for the training received in a dermatology residency program.1 Although programs are required to have the same concentrations—patient care, medical knowledge, practice-based learning and improvement, interpersonal and communication skills, professionalism, and systems-based practice—no specific guidelines are in place regarding how each of these competencies should be reached within a training period.2 Instead, it remains the responsibility of each program to formulate an individualized curriculum to facilitate proficiency in the multiple areas encompassed by a residency.
In many dermatology residency programs, dermatopathology is a substantial component of educational objectives and the curriculum.1 Residents may spend as much as 25% of their training on dermatopathology. However, there is great variability among programs in methods of teaching dermatopathology. When Hinshaw3 surveyed 52 of 109 dermatology residency programs, they identified differences in dermatopathology teaching that included, but was not limited to, utilization of problem-based learning (in 40.4% of programs), integration of journal reviews (53.8%), and computer-based learning (19.2%). In addition, differences were identified in the recommended primary textbook and the makeup of faculty who taught dermatopathology.3
Although residency programs vary in their methods of teaching this important component of dermatology, most use a multiheaded microscope in some capacity for didactics or sign-out. For most trainees, the dermatopathology laboratory is a new environment compared to the clinical space that medical students and residents become accustomed to throughout their education, thus creating a knowledge gap for trainees on proper dermatopathology etiquette and universal guidelines.
With medical students, residents, and fellows in mind, we have prepared a basic “dermatopathology etiquette” reference for trainees. Just as there are universal rules in the operating room for surgery (eg, sterile technique), we want to establish a code of conduct at the microscope. We hope that these 10 tips will, first, be useful to those who are unsure how to approach their first experience with dermatopathology and, second, serve as a guideline to aid development of appropriate communication skills and functioning within this novel setting. This list also can serve as a resource for dermatopathology attendings to provide to rotating residents and students.
1. New to pathology? It’s okay to ask. Do not hesitate to ask upper-year residents, fellows, and attendings for instructions on such matters as how to adjust your eyepiece to get the best resolution.
2. If a slide drops on the floor, do not move! Your first instinct might be to move your chair to look for the dropped slide, but you might roll over it and break it.
3. When the attending is looking through the scope, you look through the scope. Dermatopathology is a visual exercise. Getting in your “optic mileage” is best done under the guidance of an experienced dermatopathologist.
4. Rules regarding food and drink at the microscope vary by pathologist. It’s best to ask what each attending prefers. Safe advice is to avoid foods that make noise, such as chewing gum and chips, and food that has a strong odor, such as microwaved leftovers.
5. Limit use of a laptop, cell phone, and smartwatch. If you think that using any of these is necessary, it generally is best to announce that you are looking up something related to the case and then share your findings (but not the most recent post on your Facebook News Feed).
6. If you notice that something needs correcting on the report, speak up! We are all human; we all make typos. Do not hesitate to mention this as soon as possible, especially before the case is signed out. You will likely be thanked by your attending because it is harder to rectify once the report has been signed out.
7. Small talk often is welcome during large excisions. This is a great time to ask what others are doing next weekend or what happened in clinic earlier that day, or just to tell a good (clean) joke that is making the rounds. Conversely, if the case is complex, it often is best to wait until it is completed before asking questions.
8. When participating in a roundtable diagnosis, you are welcome to directly state the diagnosis for bread-and-butter cases, such as basal cell carcinomas and seborrheic keratoses. It is appropriate to be more descriptive and methodical in more complex cases. When evaluating a rash, give the general inflammatory pattern first. For example, is it spongiotic? Psoriasiform? Interface? Or a mixed pattern?
9. Extra points for identifying special sites! These include mucosal, genital, and acral sites. You might even get bonus points if you can determine something about the patient (child or adult) based on the pathologic features, such as variation in collagen patterns.
10. Whenever you are in doubt, just describe what you see. You can use the traditional top-down approach or start with stating the most evident finding, then proceed to a top-down description. If it is a neoplasm, describe the overall architecture; then, what you see at a cellular level will get you some points as well.
We acknowledge that this list of 10 tips is not comprehensive and might vary by attending and each institution’s distinctive training format. We are hopeful, however, that these 10 points of etiquette can serve as a guideline.
- Hinshaw M, Hsu P, Lee L-Y, et al. The current state of dermatopathology education: a survey of the Association of Professors of Dermatology. J Cutan Pathol. 2009;36:620-628. doi:10.1111/j.1600-0560.2008.01128.x
- Hinshaw MA, Stratman EJ. Core competencies in dermatopathology. J Cutan Pathol. 2006;33:160-165. doi:10.1111/j.0303-6987.2006.00442.x
- Hinshaw MA. Dermatopathology education: an update. Dermatol Clin. 2012;30:815-826. doi:10.1016/j.det.2012.06.003
- Hinshaw M, Hsu P, Lee L-Y, et al. The current state of dermatopathology education: a survey of the Association of Professors of Dermatology. J Cutan Pathol. 2009;36:620-628. doi:10.1111/j.1600-0560.2008.01128.x
- Hinshaw MA, Stratman EJ. Core competencies in dermatopathology. J Cutan Pathol. 2006;33:160-165. doi:10.1111/j.0303-6987.2006.00442.x
- Hinshaw MA. Dermatopathology education: an update. Dermatol Clin. 2012;30:815-826. doi:10.1016/j.det.2012.06.003
Atrophic Lesions in a Pregnant Woman
The Diagnosis: Degos Disease
The pathophysiology of Degos disease (malignant atrophic papulosis) is unknown.1 Histopathology demonstrates a wedge-shaped area of dermal necrosis with edema and mucin deposition extending from the papillary dermis to the deep reticular dermis. Occluded vessels, thrombosis, and perivascular lymphocytic infiltrates also may be seen, particularly at the dermal subcutaneous junction and at the periphery of the wedge-shaped infarction. The vascular damage that occurs may be the result of vasculitis, coagulopathy, or endothelial cell dysfunction.1
Patients typically present with small, round, erythematous papules that eventually develop atrophic porcelain white centers and telangiectatic rims. These lesions most commonly occur on the trunk and arms. In the benign form of atrophic papulosis, only the skin is involved; however, systemic involvement of the gastrointestinal tract and central nervous system can occur, resulting in bowel perforation and stroke, respectively.1 Although there is no definitive treatment of Degos disease, successful therapy with aspirin or dipyridamole has been reported.1 Eculizumab, a monoclonal antibody that binds C5, and treprostinil, a prostacyclin analog, are emerging treatment options.2,3 The differential diagnosis of Degos disease may include granuloma annulare, guttate extragenital lichen sclerosus, livedoid vasculopathy, and lymphomatoid papulosis.
Granuloma annulare may clinically mimic the erythematous papules seen in early Degos disease, and histopathology can be used to distinguish between these two disease processes. Localized granuloma annulare is the most common variant and clinically presents as pink papules and plaques in an annular configuration.4 Histopathology demonstrates an unremarkable epidermis; however, the dermis contains degenerated collagen surrounded by palisading histiocytes as well as lymphocytes. Similar to Degos disease, increased mucin is seen within these areas of degeneration, but occluded vessels and thrombosis typically are not seen (Figure 1).4,5
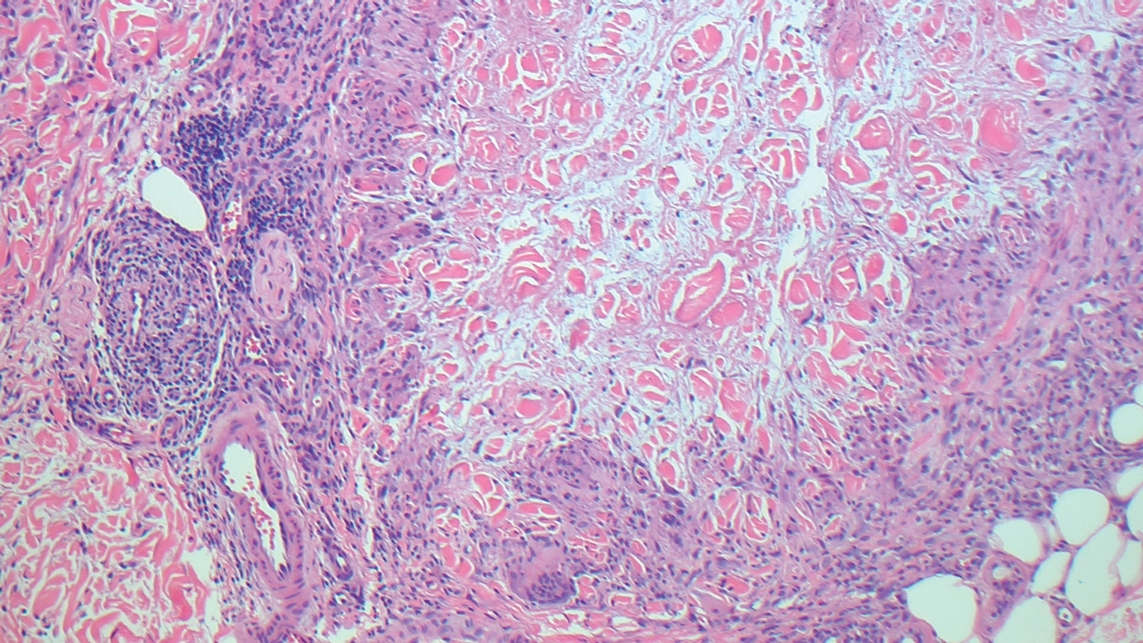
Guttate extragenital lichen sclerosus initially presents as polygonal, bluish white papules that coalesce into plaques.6 Over time, these lesions become more atrophic and may mimic Degos disease but appear differently on histopathology. Histopathology of lichen sclerosus classically demonstrates atrophy of the epidermis with loss of the rete ridges and vacuolar surface changes. Homogenization of the superficial/papillary dermis with an underlying bandlike lymphocytic infiltrate also is seen (Figure 2).6
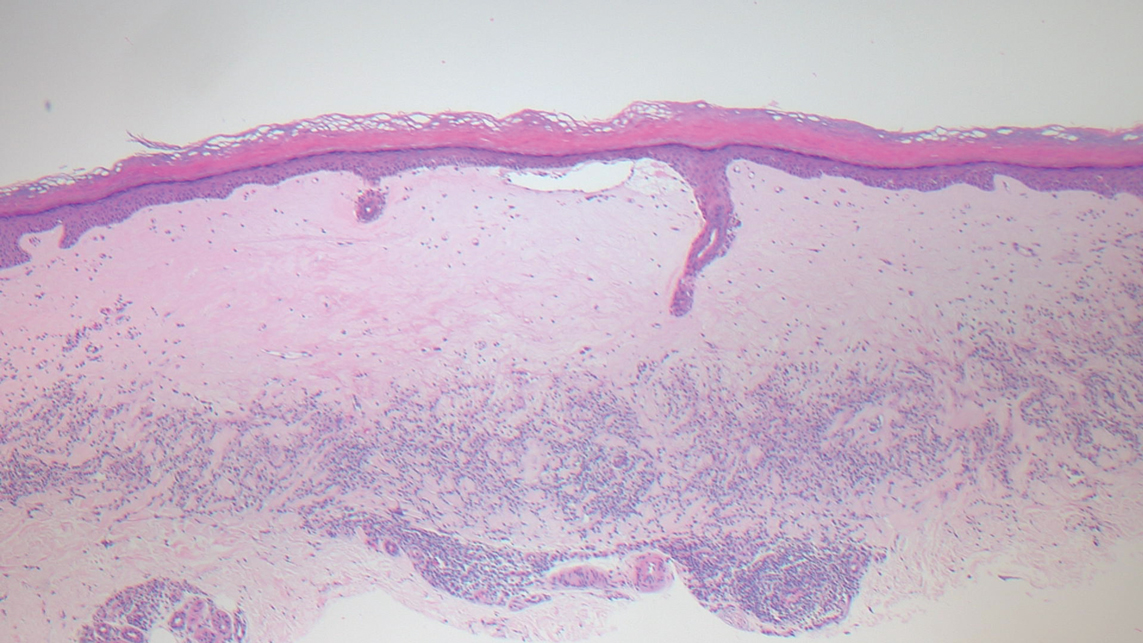
Livedoid vasculopathy is characterized by chronic recurrent ulceration of the legs secondary to thrombosis and subsequent ischemia. In the initial phase of this disease, livedo reticularis is seen followed by the development of ulcerations. As these ulcerations heal, they leave behind porcelain white scars referred to as atrophie blanche.7 The areas of scarring in livedoid vasculopathy are broad and angulated, differentiating them from the small, round, porcelain white macules in end-stage Degos disease. Histopathology demonstrates thrombosis and fibrin occlusion of the upper and mid dermal vessels. Very minimal perivascular infiltrate typically is seen, but when it is present, the infiltrate mostly is lymphocytic. Hyalinization of the vessel walls also is seen, particularly in the atrophie blanche stage (Figure 3).7
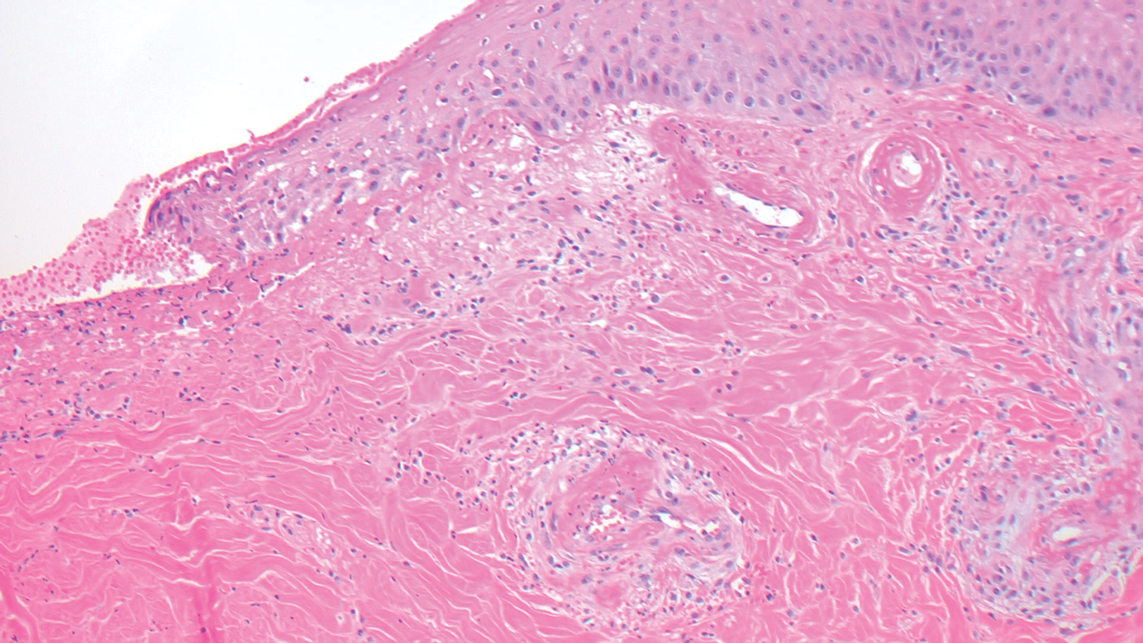
Lymphomatoid papulosis classically presents with pruritic red papules that often spontaneously involute. After resolution of the primary lesions, atrophic varioliform scars may be left behind that can resemble Degos disease.8 Classically, there are 5 histopathologic subtypes: A, B, C, D, and E. Type A is the most common type of lymphomatoid papulosis, and histopathology demonstrates a dermal lymphocytic infiltrate that consists of cells arranged in small clusters. Numerous medium- to large-sized atypical lymphocytes with prominent nucleoli and abundant cytoplasm are seen, and mitotic figures are common (Figure 4).8
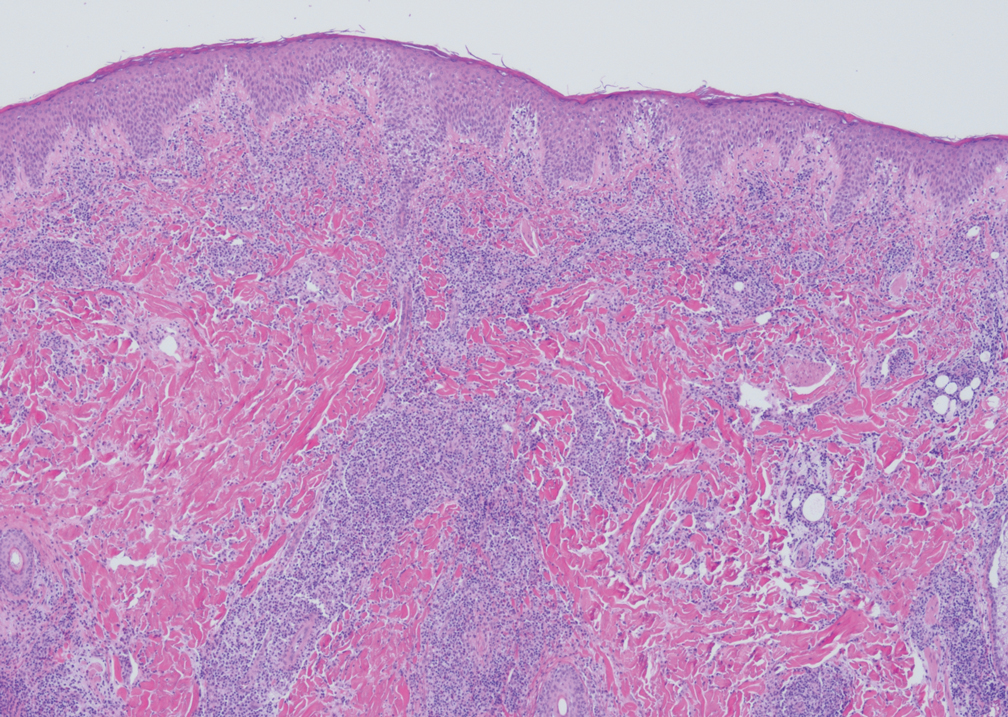
Our case was particularly interesting because the patient was 2 to 3 weeks pregnant. Degos disease in pregnancy appears to be quite exceptional. A PubMed search of articles indexed for MEDLINE using the terms Degos disease and pregnancy revealed only 4 other cases reported in the literature.9-12 With the exception of a single case that was complicated by severe abdominal pain requiring labor induction, the other reported cases resulted in uncomplicated pregnancies.9-12 Conversely, our patient's pregnancy was complicated by gestational hypertension and fetal hydrops requiring a preterm cesarean delivery. Furthermore, the infant had multiple complications, which were attributed to both placental insufficiency and a coagulopathic state.
Our patient also was found to have a heterozygous factor V Leiden mutation on workup. A PubMed search using the terms factor V Leiden mutation and Degos disease revealed 2 other cases of factor V Leiden mutation-associated Degos disease.13,14 The importance of factor V Leiden mutations in patients with Degos disease currently is unclear.
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)--a review. Orphanet J Rare Dis. 2013;8:10.
- Oliver B, Boehm M, Rosing DR, et al. Diffuse atrophic papules and plaques, intermittent abdominal pain, paresthesias, and cardiac abnormalities in a 55-year-old woman. J Am Acad Dermatol. 2016;75:1274-1277.
- Magro CM, Wang X, Garrett-Bakelman F, et al. The effects of eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis. 2013;8:185.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Tronnier M, Mitteldorf C. Histologic features of granulomatous skin diseases. part 1: non-infectious granulomatous disorders. J Dtsch Dermatol Ges. 2015;13:211-216.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478‐488.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73.
- Moulin G, Barrut D, Franc MP, et al. Familial Degos' atrophic papulosis (mother-daughter). Ann Dermatol Venereol. 1984;111:149-155.
- Bogenrieder T, Kuske M, Landthaler M, et al. Benign Degos' disease developing during pregnancy and followed for 10 years. Acta Derm Venereol. 2002;82:284-287.
- Sharma S, Brennan B, Naden R, et al. A case of Degos disease in pregnancy. Obstet Med. 2016;9:167-168.
- Zhao Q, Zhang S, Dong A. An unusual case of abdominal pain. Gastroenterology. 2018;154:E1-E2.
- Darwich E, Guilabert A, Mascaró JM Jr, et al. Dermoscopic description of a patient with thrombocythemia and factor V Leiden mutation-associated Degos' disease. Int J Dermatol. 2011;50:604-606.
- Hohwy T, Jensen MG, Tøttrup A, et al. A fatal case of malignant atrophic papulosis (Degos' disease) in a man with factor V Leiden mutation and lupus anticoagulant. Acta Derm Venereol. 2006;86:245-247.
The Diagnosis: Degos Disease
The pathophysiology of Degos disease (malignant atrophic papulosis) is unknown.1 Histopathology demonstrates a wedge-shaped area of dermal necrosis with edema and mucin deposition extending from the papillary dermis to the deep reticular dermis. Occluded vessels, thrombosis, and perivascular lymphocytic infiltrates also may be seen, particularly at the dermal subcutaneous junction and at the periphery of the wedge-shaped infarction. The vascular damage that occurs may be the result of vasculitis, coagulopathy, or endothelial cell dysfunction.1
Patients typically present with small, round, erythematous papules that eventually develop atrophic porcelain white centers and telangiectatic rims. These lesions most commonly occur on the trunk and arms. In the benign form of atrophic papulosis, only the skin is involved; however, systemic involvement of the gastrointestinal tract and central nervous system can occur, resulting in bowel perforation and stroke, respectively.1 Although there is no definitive treatment of Degos disease, successful therapy with aspirin or dipyridamole has been reported.1 Eculizumab, a monoclonal antibody that binds C5, and treprostinil, a prostacyclin analog, are emerging treatment options.2,3 The differential diagnosis of Degos disease may include granuloma annulare, guttate extragenital lichen sclerosus, livedoid vasculopathy, and lymphomatoid papulosis.
Granuloma annulare may clinically mimic the erythematous papules seen in early Degos disease, and histopathology can be used to distinguish between these two disease processes. Localized granuloma annulare is the most common variant and clinically presents as pink papules and plaques in an annular configuration.4 Histopathology demonstrates an unremarkable epidermis; however, the dermis contains degenerated collagen surrounded by palisading histiocytes as well as lymphocytes. Similar to Degos disease, increased mucin is seen within these areas of degeneration, but occluded vessels and thrombosis typically are not seen (Figure 1).4,5
Guttate extragenital lichen sclerosus initially presents as polygonal, bluish white papules that coalesce into plaques.6 Over time, these lesions become more atrophic and may mimic Degos disease but appear differently on histopathology. Histopathology of lichen sclerosus classically demonstrates atrophy of the epidermis with loss of the rete ridges and vacuolar surface changes. Homogenization of the superficial/papillary dermis with an underlying bandlike lymphocytic infiltrate also is seen (Figure 2).6
Livedoid vasculopathy is characterized by chronic recurrent ulceration of the legs secondary to thrombosis and subsequent ischemia. In the initial phase of this disease, livedo reticularis is seen followed by the development of ulcerations. As these ulcerations heal, they leave behind porcelain white scars referred to as atrophie blanche.7 The areas of scarring in livedoid vasculopathy are broad and angulated, differentiating them from the small, round, porcelain white macules in end-stage Degos disease. Histopathology demonstrates thrombosis and fibrin occlusion of the upper and mid dermal vessels. Very minimal perivascular infiltrate typically is seen, but when it is present, the infiltrate mostly is lymphocytic. Hyalinization of the vessel walls also is seen, particularly in the atrophie blanche stage (Figure 3).7
Lymphomatoid papulosis classically presents with pruritic red papules that often spontaneously involute. After resolution of the primary lesions, atrophic varioliform scars may be left behind that can resemble Degos disease.8 Classically, there are 5 histopathologic subtypes: A, B, C, D, and E. Type A is the most common type of lymphomatoid papulosis, and histopathology demonstrates a dermal lymphocytic infiltrate that consists of cells arranged in small clusters. Numerous medium- to large-sized atypical lymphocytes with prominent nucleoli and abundant cytoplasm are seen, and mitotic figures are common (Figure 4).8
Our case was particularly interesting because the patient was 2 to 3 weeks pregnant. Degos disease in pregnancy appears to be quite exceptional. A PubMed search of articles indexed for MEDLINE using the terms Degos disease and pregnancy revealed only 4 other cases reported in the literature.9-12 With the exception of a single case that was complicated by severe abdominal pain requiring labor induction, the other reported cases resulted in uncomplicated pregnancies.9-12 Conversely, our patient's pregnancy was complicated by gestational hypertension and fetal hydrops requiring a preterm cesarean delivery. Furthermore, the infant had multiple complications, which were attributed to both placental insufficiency and a coagulopathic state.
Our patient also was found to have a heterozygous factor V Leiden mutation on workup. A PubMed search using the terms factor V Leiden mutation and Degos disease revealed 2 other cases of factor V Leiden mutation-associated Degos disease.13,14 The importance of factor V Leiden mutations in patients with Degos disease currently is unclear.
The Diagnosis: Degos Disease
The pathophysiology of Degos disease (malignant atrophic papulosis) is unknown.1 Histopathology demonstrates a wedge-shaped area of dermal necrosis with edema and mucin deposition extending from the papillary dermis to the deep reticular dermis. Occluded vessels, thrombosis, and perivascular lymphocytic infiltrates also may be seen, particularly at the dermal subcutaneous junction and at the periphery of the wedge-shaped infarction. The vascular damage that occurs may be the result of vasculitis, coagulopathy, or endothelial cell dysfunction.1
Patients typically present with small, round, erythematous papules that eventually develop atrophic porcelain white centers and telangiectatic rims. These lesions most commonly occur on the trunk and arms. In the benign form of atrophic papulosis, only the skin is involved; however, systemic involvement of the gastrointestinal tract and central nervous system can occur, resulting in bowel perforation and stroke, respectively.1 Although there is no definitive treatment of Degos disease, successful therapy with aspirin or dipyridamole has been reported.1 Eculizumab, a monoclonal antibody that binds C5, and treprostinil, a prostacyclin analog, are emerging treatment options.2,3 The differential diagnosis of Degos disease may include granuloma annulare, guttate extragenital lichen sclerosus, livedoid vasculopathy, and lymphomatoid papulosis.
Granuloma annulare may clinically mimic the erythematous papules seen in early Degos disease, and histopathology can be used to distinguish between these two disease processes. Localized granuloma annulare is the most common variant and clinically presents as pink papules and plaques in an annular configuration.4 Histopathology demonstrates an unremarkable epidermis; however, the dermis contains degenerated collagen surrounded by palisading histiocytes as well as lymphocytes. Similar to Degos disease, increased mucin is seen within these areas of degeneration, but occluded vessels and thrombosis typically are not seen (Figure 1).4,5
Guttate extragenital lichen sclerosus initially presents as polygonal, bluish white papules that coalesce into plaques.6 Over time, these lesions become more atrophic and may mimic Degos disease but appear differently on histopathology. Histopathology of lichen sclerosus classically demonstrates atrophy of the epidermis with loss of the rete ridges and vacuolar surface changes. Homogenization of the superficial/papillary dermis with an underlying bandlike lymphocytic infiltrate also is seen (Figure 2).6
Livedoid vasculopathy is characterized by chronic recurrent ulceration of the legs secondary to thrombosis and subsequent ischemia. In the initial phase of this disease, livedo reticularis is seen followed by the development of ulcerations. As these ulcerations heal, they leave behind porcelain white scars referred to as atrophie blanche.7 The areas of scarring in livedoid vasculopathy are broad and angulated, differentiating them from the small, round, porcelain white macules in end-stage Degos disease. Histopathology demonstrates thrombosis and fibrin occlusion of the upper and mid dermal vessels. Very minimal perivascular infiltrate typically is seen, but when it is present, the infiltrate mostly is lymphocytic. Hyalinization of the vessel walls also is seen, particularly in the atrophie blanche stage (Figure 3).7
Lymphomatoid papulosis classically presents with pruritic red papules that often spontaneously involute. After resolution of the primary lesions, atrophic varioliform scars may be left behind that can resemble Degos disease.8 Classically, there are 5 histopathologic subtypes: A, B, C, D, and E. Type A is the most common type of lymphomatoid papulosis, and histopathology demonstrates a dermal lymphocytic infiltrate that consists of cells arranged in small clusters. Numerous medium- to large-sized atypical lymphocytes with prominent nucleoli and abundant cytoplasm are seen, and mitotic figures are common (Figure 4).8
Our case was particularly interesting because the patient was 2 to 3 weeks pregnant. Degos disease in pregnancy appears to be quite exceptional. A PubMed search of articles indexed for MEDLINE using the terms Degos disease and pregnancy revealed only 4 other cases reported in the literature.9-12 With the exception of a single case that was complicated by severe abdominal pain requiring labor induction, the other reported cases resulted in uncomplicated pregnancies.9-12 Conversely, our patient's pregnancy was complicated by gestational hypertension and fetal hydrops requiring a preterm cesarean delivery. Furthermore, the infant had multiple complications, which were attributed to both placental insufficiency and a coagulopathic state.
Our patient also was found to have a heterozygous factor V Leiden mutation on workup. A PubMed search using the terms factor V Leiden mutation and Degos disease revealed 2 other cases of factor V Leiden mutation-associated Degos disease.13,14 The importance of factor V Leiden mutations in patients with Degos disease currently is unclear.
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)--a review. Orphanet J Rare Dis. 2013;8:10.
- Oliver B, Boehm M, Rosing DR, et al. Diffuse atrophic papules and plaques, intermittent abdominal pain, paresthesias, and cardiac abnormalities in a 55-year-old woman. J Am Acad Dermatol. 2016;75:1274-1277.
- Magro CM, Wang X, Garrett-Bakelman F, et al. The effects of eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis. 2013;8:185.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Tronnier M, Mitteldorf C. Histologic features of granulomatous skin diseases. part 1: non-infectious granulomatous disorders. J Dtsch Dermatol Ges. 2015;13:211-216.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478‐488.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73.
- Moulin G, Barrut D, Franc MP, et al. Familial Degos' atrophic papulosis (mother-daughter). Ann Dermatol Venereol. 1984;111:149-155.
- Bogenrieder T, Kuske M, Landthaler M, et al. Benign Degos' disease developing during pregnancy and followed for 10 years. Acta Derm Venereol. 2002;82:284-287.
- Sharma S, Brennan B, Naden R, et al. A case of Degos disease in pregnancy. Obstet Med. 2016;9:167-168.
- Zhao Q, Zhang S, Dong A. An unusual case of abdominal pain. Gastroenterology. 2018;154:E1-E2.
- Darwich E, Guilabert A, Mascaró JM Jr, et al. Dermoscopic description of a patient with thrombocythemia and factor V Leiden mutation-associated Degos' disease. Int J Dermatol. 2011;50:604-606.
- Hohwy T, Jensen MG, Tøttrup A, et al. A fatal case of malignant atrophic papulosis (Degos' disease) in a man with factor V Leiden mutation and lupus anticoagulant. Acta Derm Venereol. 2006;86:245-247.
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)--a review. Orphanet J Rare Dis. 2013;8:10.
- Oliver B, Boehm M, Rosing DR, et al. Diffuse atrophic papules and plaques, intermittent abdominal pain, paresthesias, and cardiac abnormalities in a 55-year-old woman. J Am Acad Dermatol. 2016;75:1274-1277.
- Magro CM, Wang X, Garrett-Bakelman F, et al. The effects of eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis. 2013;8:185.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Tronnier M, Mitteldorf C. Histologic features of granulomatous skin diseases. part 1: non-infectious granulomatous disorders. J Dtsch Dermatol Ges. 2015;13:211-216.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478‐488.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73.
- Moulin G, Barrut D, Franc MP, et al. Familial Degos' atrophic papulosis (mother-daughter). Ann Dermatol Venereol. 1984;111:149-155.
- Bogenrieder T, Kuske M, Landthaler M, et al. Benign Degos' disease developing during pregnancy and followed for 10 years. Acta Derm Venereol. 2002;82:284-287.
- Sharma S, Brennan B, Naden R, et al. A case of Degos disease in pregnancy. Obstet Med. 2016;9:167-168.
- Zhao Q, Zhang S, Dong A. An unusual case of abdominal pain. Gastroenterology. 2018;154:E1-E2.
- Darwich E, Guilabert A, Mascaró JM Jr, et al. Dermoscopic description of a patient with thrombocythemia and factor V Leiden mutation-associated Degos' disease. Int J Dermatol. 2011;50:604-606.
- Hohwy T, Jensen MG, Tøttrup A, et al. A fatal case of malignant atrophic papulosis (Degos' disease) in a man with factor V Leiden mutation and lupus anticoagulant. Acta Derm Venereol. 2006;86:245-247.
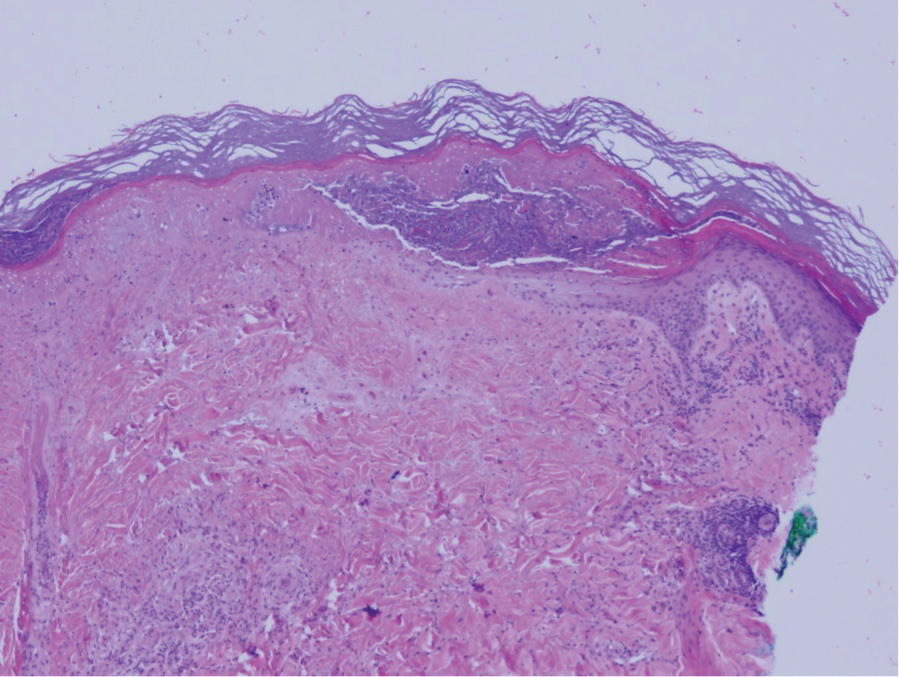
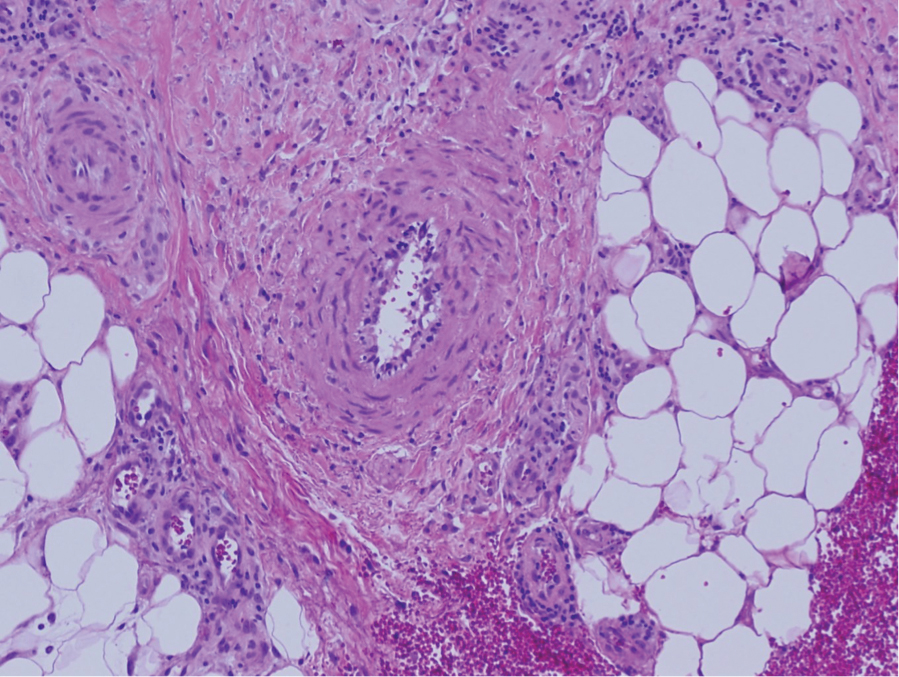
A 36-year-old pregnant woman presented with painful erythematous papules on the palms and fingers of 2 months’ duration. Similar lesions developed on the thighs and feet several weeks later. Two tender macules with central areas of porcelain white scarring rimmed by telangiectases on the right foot also were present. A punch biopsy of these lesions demonstrated a wedge-shaped area of ischemic necrosis associated with dermal mucin without associated necrobiosis. Fibrin thrombi were seen within several small dermal vessels and were associated with a perivascular lymphocytic infiltrate. Endotheliitis was observed within a deep dermal vessel. Laboratory workup including syphilis IgG, antinuclear antibodies, extractable nuclear antigen antibodies, anti–double-stranded DNA, antistreptolysin O antibodies, Russell viper venom time, cryoglobulin, hepatitis screening, perinuclear antineutrophil cytoplasmic antibodies (ANCA), and cytoplasmic ANCA was unremarkable. Hypercoagulable studies including prothrombin gene mutation, factor V Leiden, plasminogen, proteins C and S, antithrombin III, homocysteine, and antiphospholipid IgM and IgG antibodies were notable only for heterozygosity for factor V Leiden.
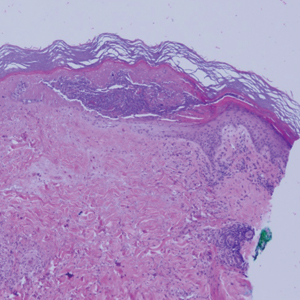
Oral Verrucous Plaques in a Patient With Urothelial Cancer
The Diagnosis: Paraneoplastic Acanthosis Nigricans
Histopathologic examination demonstrated verrucous epidermal hyperplasia (Figure, A). Fungal organisms were identified with an Alcian blue and periodic acid-Schiff stain (Figure, B). The organisms demonstrated a vertical orientation in relation to the mucosal surface, which was consistent with candidal organisms.
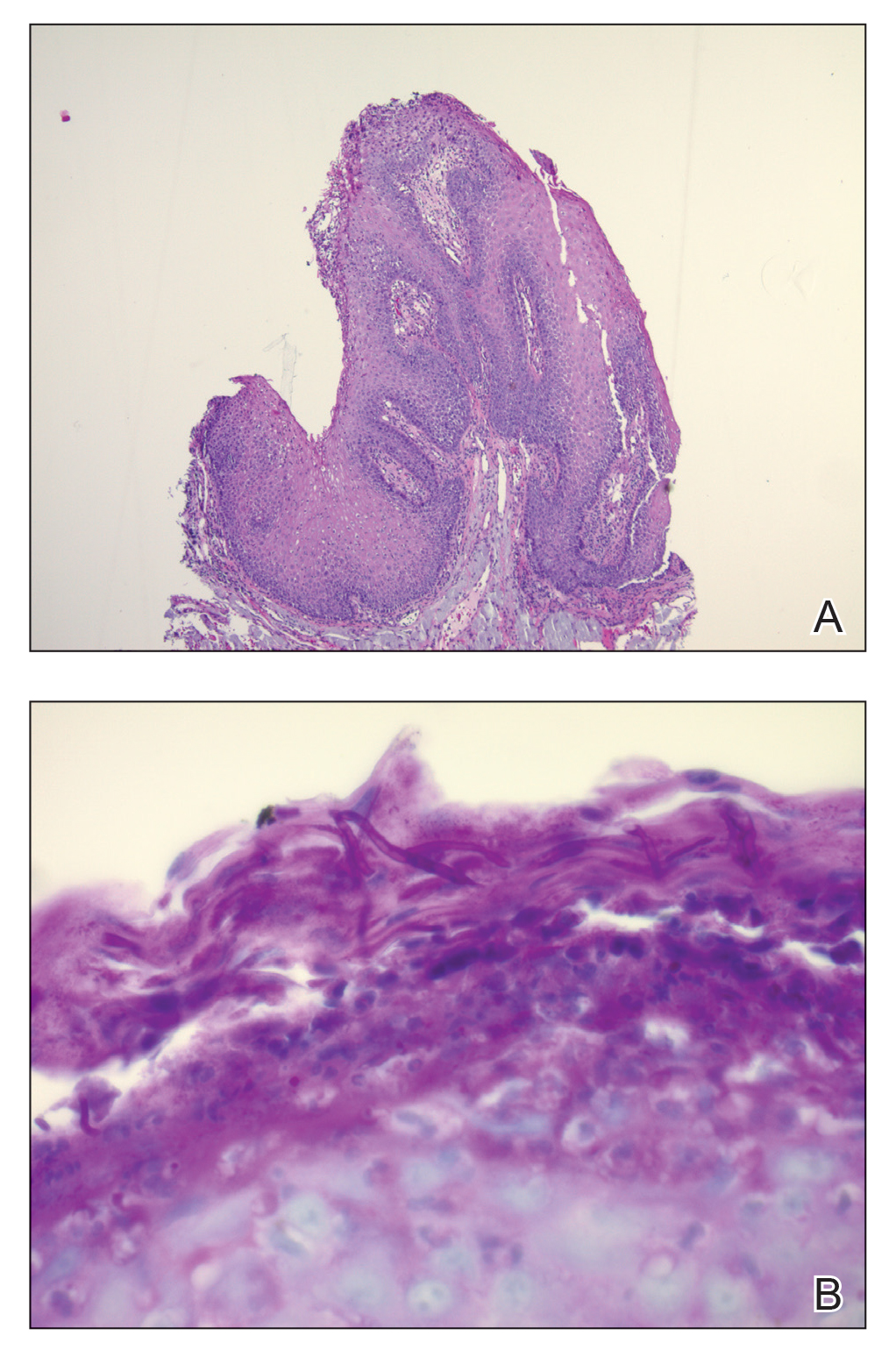
Given the rapid eruption of these plaques, the distribution on the oral and palmar surfaces (tripe palms), and the minimal improvement with both systemic steroids and antifungal treatment, a diagnosis of paraneoplastic acanthosis nigricans with secondary candidal infection was made. Drug-induced cheilitis was considered; however, improvement with discontinuation of the suspected offending drug would have been expected. Although chronic mucocutaneous candidiasis was possible, more prompt improvement upon initiation of systemic antifungal therapy would have been observed. Oral Crohn disease should be included in the differential, but it was unlikely given the lack of granulomas on pathology and absence of history of gastrointestinal tract symptoms. Melkersson-Rosenthal syndrome also was unlikely given the lack of facial nerve palsy as well as the lack of granulomas on pathology. Furthermore, none of these options would be associated with tripe palms, as seen in our patient.
Acanthosis nigricans is a localized skin disorder characterized by hyperpigmented velvety plaques arising in flexural and intertriginous regions. Although most cases (80%) are associated with idiopathic or benign conditions, the link between acanthosis nigricans and an underlying malignancy has been well documented.1-3 Most commonly associated with an underlying intra-abdominal malignancy (often gastric carcinoma), the lesions of paraneoplastic acanthosis nigricans are indistinguishable from their benign counterparts.1,4 When the condition presents abruptly and extensively in a nonobese patient, prompt workup for malignancy should be initiated. Rapid onset and atypical distribution (ie, palmar, perioral, or mucosal) more commonly is associated with a paraneoplastic etiology.5,6
Histopathology for acanthosis nigricans shows hyperkeratosis and epidermal papillomatosis. Horn pseudocyst formation is possible, but usually no hyperpigmentation is observed. The findings typically are indistinguishable from seborrheic keratoses, epidermal nevi, or lesions of confluent and reticulated papillomatosis of Gougerot and Carteaud.2
The underlying pathogenesis of acanthosis nigricans is poorly understood. In the benign subtype, insulin resistance commonly has been described. In the paraneoplastic subtype, it is proposed that the tumor produces a transforming growth factor that mimics epidermal growth factor and leads to keratinocyte proliferation.7,8 Paraneoplastic acanthosis nigricans has the potential to arise at any point of tumor development, further contributing to the diagnostic challenge. Treatment of the skin lesions involves management of the underlying malignancy. Unfortunately, many such malignancies often are at an advanced stage, and subsequent prognosis is poor.2
- Shah A, Jack A, Liu H, et al. Neoplastic/paraneoplastic dermatitis, fasciitis, and panniculitis. Rheum Dis Clin North Am. 2011;37:573-592.
- Chairatchaneeboon M, Kim EJ. Cutaneous paraneoplastic syndromes. In: Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick's Dermatology. 9th ed. McGraw-Hill Education; 2019:2441-2464.
- Lee HC, Ker KJ, Chong WS. Oral malignant acanthosis nigricans and tripe palms associated with renal urothelial carcinoma. JAMA Dermatol. 2015;151:1381-1383.
- Yu Q, Li XL, Ji G, et al. Malignant acanthosis nigricans: an early diagnostic clue for gastric adenocarcinoma. World J Surg Oncol. 2017;15:208.
- Mohrenschlager M, Vocks E, Wessner DB, et al. Tripe palms and malignant acanthosis nigricans: cutaneous signs of imminent metastasis in bladder cancer? J Urol. 2001;165:1629-1630.
- Cohen PR, Grossman ME, Almeida L, et al. Tripe palms and malignancy. J Clin Oncol. 1989;7:669-678.
- Higgins SP, Freemark M, Prose NS. Acanthosis nigricans: a practical approach to evaluation and management. Dermatol Online J. 2008;14:2.
- Torley D, Bellus GA, Munro CS. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096-1101.
The Diagnosis: Paraneoplastic Acanthosis Nigricans
Histopathologic examination demonstrated verrucous epidermal hyperplasia (Figure, A). Fungal organisms were identified with an Alcian blue and periodic acid-Schiff stain (Figure, B). The organisms demonstrated a vertical orientation in relation to the mucosal surface, which was consistent with candidal organisms.
Given the rapid eruption of these plaques, the distribution on the oral and palmar surfaces (tripe palms), and the minimal improvement with both systemic steroids and antifungal treatment, a diagnosis of paraneoplastic acanthosis nigricans with secondary candidal infection was made. Drug-induced cheilitis was considered; however, improvement with discontinuation of the suspected offending drug would have been expected. Although chronic mucocutaneous candidiasis was possible, more prompt improvement upon initiation of systemic antifungal therapy would have been observed. Oral Crohn disease should be included in the differential, but it was unlikely given the lack of granulomas on pathology and absence of history of gastrointestinal tract symptoms. Melkersson-Rosenthal syndrome also was unlikely given the lack of facial nerve palsy as well as the lack of granulomas on pathology. Furthermore, none of these options would be associated with tripe palms, as seen in our patient.
Acanthosis nigricans is a localized skin disorder characterized by hyperpigmented velvety plaques arising in flexural and intertriginous regions. Although most cases (80%) are associated with idiopathic or benign conditions, the link between acanthosis nigricans and an underlying malignancy has been well documented.1-3 Most commonly associated with an underlying intra-abdominal malignancy (often gastric carcinoma), the lesions of paraneoplastic acanthosis nigricans are indistinguishable from their benign counterparts.1,4 When the condition presents abruptly and extensively in a nonobese patient, prompt workup for malignancy should be initiated. Rapid onset and atypical distribution (ie, palmar, perioral, or mucosal) more commonly is associated with a paraneoplastic etiology.5,6
Histopathology for acanthosis nigricans shows hyperkeratosis and epidermal papillomatosis. Horn pseudocyst formation is possible, but usually no hyperpigmentation is observed. The findings typically are indistinguishable from seborrheic keratoses, epidermal nevi, or lesions of confluent and reticulated papillomatosis of Gougerot and Carteaud.2
The underlying pathogenesis of acanthosis nigricans is poorly understood. In the benign subtype, insulin resistance commonly has been described. In the paraneoplastic subtype, it is proposed that the tumor produces a transforming growth factor that mimics epidermal growth factor and leads to keratinocyte proliferation.7,8 Paraneoplastic acanthosis nigricans has the potential to arise at any point of tumor development, further contributing to the diagnostic challenge. Treatment of the skin lesions involves management of the underlying malignancy. Unfortunately, many such malignancies often are at an advanced stage, and subsequent prognosis is poor.2
The Diagnosis: Paraneoplastic Acanthosis Nigricans
Histopathologic examination demonstrated verrucous epidermal hyperplasia (Figure, A). Fungal organisms were identified with an Alcian blue and periodic acid-Schiff stain (Figure, B). The organisms demonstrated a vertical orientation in relation to the mucosal surface, which was consistent with candidal organisms.
Given the rapid eruption of these plaques, the distribution on the oral and palmar surfaces (tripe palms), and the minimal improvement with both systemic steroids and antifungal treatment, a diagnosis of paraneoplastic acanthosis nigricans with secondary candidal infection was made. Drug-induced cheilitis was considered; however, improvement with discontinuation of the suspected offending drug would have been expected. Although chronic mucocutaneous candidiasis was possible, more prompt improvement upon initiation of systemic antifungal therapy would have been observed. Oral Crohn disease should be included in the differential, but it was unlikely given the lack of granulomas on pathology and absence of history of gastrointestinal tract symptoms. Melkersson-Rosenthal syndrome also was unlikely given the lack of facial nerve palsy as well as the lack of granulomas on pathology. Furthermore, none of these options would be associated with tripe palms, as seen in our patient.
Acanthosis nigricans is a localized skin disorder characterized by hyperpigmented velvety plaques arising in flexural and intertriginous regions. Although most cases (80%) are associated with idiopathic or benign conditions, the link between acanthosis nigricans and an underlying malignancy has been well documented.1-3 Most commonly associated with an underlying intra-abdominal malignancy (often gastric carcinoma), the lesions of paraneoplastic acanthosis nigricans are indistinguishable from their benign counterparts.1,4 When the condition presents abruptly and extensively in a nonobese patient, prompt workup for malignancy should be initiated. Rapid onset and atypical distribution (ie, palmar, perioral, or mucosal) more commonly is associated with a paraneoplastic etiology.5,6
Histopathology for acanthosis nigricans shows hyperkeratosis and epidermal papillomatosis. Horn pseudocyst formation is possible, but usually no hyperpigmentation is observed. The findings typically are indistinguishable from seborrheic keratoses, epidermal nevi, or lesions of confluent and reticulated papillomatosis of Gougerot and Carteaud.2
The underlying pathogenesis of acanthosis nigricans is poorly understood. In the benign subtype, insulin resistance commonly has been described. In the paraneoplastic subtype, it is proposed that the tumor produces a transforming growth factor that mimics epidermal growth factor and leads to keratinocyte proliferation.7,8 Paraneoplastic acanthosis nigricans has the potential to arise at any point of tumor development, further contributing to the diagnostic challenge. Treatment of the skin lesions involves management of the underlying malignancy. Unfortunately, many such malignancies often are at an advanced stage, and subsequent prognosis is poor.2
- Shah A, Jack A, Liu H, et al. Neoplastic/paraneoplastic dermatitis, fasciitis, and panniculitis. Rheum Dis Clin North Am. 2011;37:573-592.
- Chairatchaneeboon M, Kim EJ. Cutaneous paraneoplastic syndromes. In: Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick's Dermatology. 9th ed. McGraw-Hill Education; 2019:2441-2464.
- Lee HC, Ker KJ, Chong WS. Oral malignant acanthosis nigricans and tripe palms associated with renal urothelial carcinoma. JAMA Dermatol. 2015;151:1381-1383.
- Yu Q, Li XL, Ji G, et al. Malignant acanthosis nigricans: an early diagnostic clue for gastric adenocarcinoma. World J Surg Oncol. 2017;15:208.
- Mohrenschlager M, Vocks E, Wessner DB, et al. Tripe palms and malignant acanthosis nigricans: cutaneous signs of imminent metastasis in bladder cancer? J Urol. 2001;165:1629-1630.
- Cohen PR, Grossman ME, Almeida L, et al. Tripe palms and malignancy. J Clin Oncol. 1989;7:669-678.
- Higgins SP, Freemark M, Prose NS. Acanthosis nigricans: a practical approach to evaluation and management. Dermatol Online J. 2008;14:2.
- Torley D, Bellus GA, Munro CS. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096-1101.
- Shah A, Jack A, Liu H, et al. Neoplastic/paraneoplastic dermatitis, fasciitis, and panniculitis. Rheum Dis Clin North Am. 2011;37:573-592.
- Chairatchaneeboon M, Kim EJ. Cutaneous paraneoplastic syndromes. In: Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick's Dermatology. 9th ed. McGraw-Hill Education; 2019:2441-2464.
- Lee HC, Ker KJ, Chong WS. Oral malignant acanthosis nigricans and tripe palms associated with renal urothelial carcinoma. JAMA Dermatol. 2015;151:1381-1383.
- Yu Q, Li XL, Ji G, et al. Malignant acanthosis nigricans: an early diagnostic clue for gastric adenocarcinoma. World J Surg Oncol. 2017;15:208.
- Mohrenschlager M, Vocks E, Wessner DB, et al. Tripe palms and malignant acanthosis nigricans: cutaneous signs of imminent metastasis in bladder cancer? J Urol. 2001;165:1629-1630.
- Cohen PR, Grossman ME, Almeida L, et al. Tripe palms and malignancy. J Clin Oncol. 1989;7:669-678.
- Higgins SP, Freemark M, Prose NS. Acanthosis nigricans: a practical approach to evaluation and management. Dermatol Online J. 2008;14:2.
- Torley D, Bellus GA, Munro CS. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096-1101.
A 75-year-old nonobese man with metastatic urothelial carcinoma presented for evaluation and treatment of swollen lips. The patient stated that his lips began to swell and crack shortly after beginning pembrolizumab approximately 5 months prior. The swelling had progressively worsened, prompting discontinuation of the pembrolizumab by oncology about 2 months prior to presentation to our dermatology clinic. He reported slight improvement after the discontinuation of pembrolizumab, and he had since been started on carboplatin and gemcitabine. He previously was treated with oral corticosteroids without improvement. His oncologist started him on oral fluconazole for treatment of oral thrush on the day of presentation to our clinic. Physical examination revealed diffuse papillomatous and verrucous plaques of the upper and lower lips with involvement of the buccal mucosa. He also had deep fissures and white plaques on the tongue. Velvety hyperpigmented plaques were noted in the axillae, and he had confluent thickening of the palms. A 3-mm punch biopsy from the lower lip was performed. The patient subsequently was evaluated 2 weeks after the initial appointment, and minor improvement in the oral verrucous hyperplasia was noted following antifungal therapy, with resolution of the candidiasis.
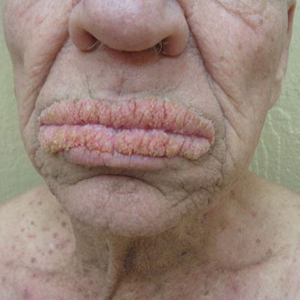
How to Save a Limb: Identification of Pyoderma Gangrenosum
Case Report
A 67-year-old woman presented with a painful expanding ulcer on the left leg and a new nearby ulcer of 2 months’ duration. She initially was seen 2 months prior for a wound on the left knee due to a fall as well as cellulitis, which was treated with intravenous vancomycin and ceftriaxone. Wound cultures were negative for bacteria, and she was discharged without antibiotics. She presented to the emergency department 1 month later for malodorous discharge of the first ulcer with zero systemic inflammatory response syndrome criteria; no fever; and no abnormal heart rate, respiratory rate, or leukocyte count. She was discharged with wound care. After 3 weeks, she returned with a second ulcer and worsening drainage but zero systemic inflammatory response syndrome criteria. She had a medical history of Crohn disease with 9-year remission, atrial fibrillation, pacemaker, mitral valve replacement, chronic obstructive pulmonary disease, and a 51 pack-year smoking history.
Physical examination of the left leg revealed a 3×3-cm deep lesion (ulcer A) on the distal left thigh located superomedial to the knee (Figure 1) as well as a 2×1-cm deep lesion (ulcer B) on the anteromedial knee with undermining and tunneling (Figure 2). A large amount of malodorous tan bloody discharge was present on both ulcers. There were no signs of induration or crepitus.Due to concerns of skin and soft tissue infection (SSTI) or osteomyelitis, a bone scan and wound and blood cultures were ordered. The patient was started on vancomycin and piperacillin-tazobactam in the emergency department, which later was augmented with cefepime. Trauma surgery scheduled debridement for the following morning with suspicion of necrotizing fasciitis. Additional consultations were requested, including infectious disease, wound care, and dermatology. Dermatology evaluated the wound, performed a punch biopsy, and canceled debridement due to unclear diagnosis. The clinical differential at that time included pyoderma gangrenosum (PG), atypical vasculitis, or infection. Additional workup revealed positive antineutrophil cytoplasmic antibodies but negative proteinase 3 and myeloperoxidase, disfavoring vasculitis. Wound cultures grew Staphylococcus aureus and Pseudomonas aeruginosa.
Histologic evaluation revealed deep dermal necrosis with a mixed inflammatory infiltrate (Figure 3) and no organisms or vasculitis. Antibiotics were discontinued, and she was discharged on a 14-day course of prednisone 60 mg daily for empirical treatment of PG with dermatology follow-up. Medical management included a 6-month course of dapsone that was extended to 7 months because of an intensive care unit stay for a cerebrovascular accident. Daily dosing was as follows: 100 mg for 5 months, 50 mg for 1 month, and 25 mg for 1 month, then stopped. She was followed with serial complete blood cell count every 1 to 2 months and home-health wound care. One month after dapsone initiation, the ulcers decreased in size. Ulcer B was fully healed after 4 months, and ulcer A was nearly closed at 6 months without any new flares.
Comment
Pyoderma gangrenosum is a rare inflammatory skin condition that classically presents as tender papules or pustules evolving into painful ulcers, most commonly on the lower extremities. Pyoderma gangrenosum has a propensity to exhibit pathergy, the hyperreactivity of the skin in response to minor trauma. This phenomenon in PG manifests as the rapid evolution from pustule to ulceration with violaceous undermining borders.
Diagnosis of PG
Pyoderma gangrenosum has been described as a diagnosis of exclusion, as its findings frequently mimic SSTIs. Important findings to obtain are histology, history, ulcer morphology, and response to treatment.
In 2018, Maverakis et al1 proposed diagnostic criteria for classic ulcerative PG (Table 1). A diagnosis of PG can be made if the patient meets 1 major criterion and 4 minor criteria. Our case met 0 major criteria and 5 minor criteria: history of inflammatory bowel disease (IBD); history of pustule ulcerating within 4 days of appearing; peripheral erythema, undermining border, and tenderness at ulceration site; multiple ulcerations, with at least 1 on an anterior lower leg; and decreased ulcer size within 1 month of initiating immunosuppressive medication(s). Although our patient’s biopsy demonstrated a mixed infiltrate, PG was not excluded due to spontaneous resolution at the time of biopsy, emphasizing the need to biopsy subsequent new lesions if neutrophils are not initially seen.1 Pyoderma gangrenosum frequently is associated with IBD, most often Crohn disease, as seen in our patient.2-4 Although IBD classically is associated with smoking, studies have yet to conclude if smoking is a predictive factor of PG.5 Our patient presented with an initial ulcer that evolved into 2 ulcers, similar to a case of bilateral ulcers.6
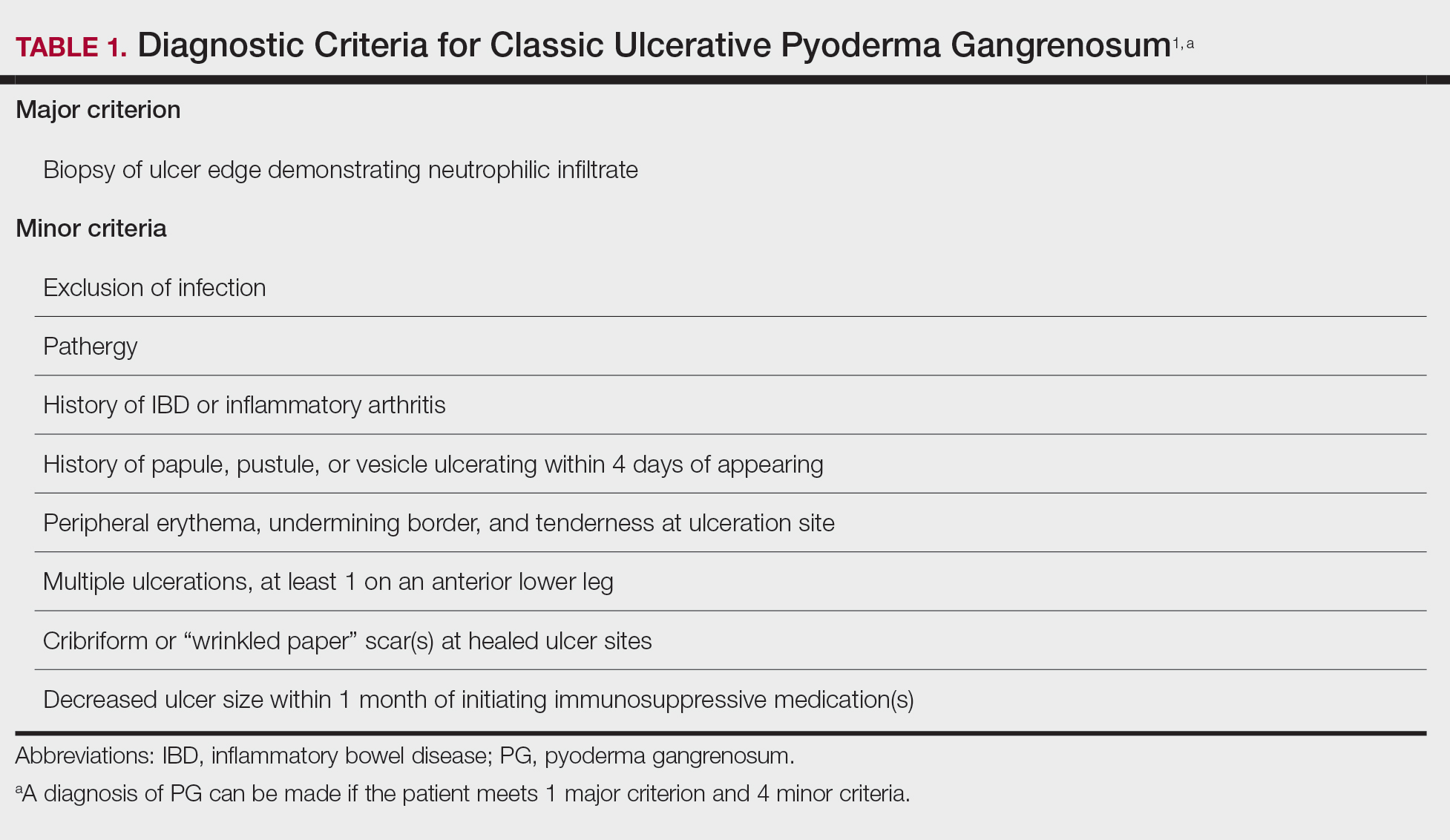
Differential Diagnosis of PG
Other possible diagnoses to consider are SSTI and vasculitis, the latter being disfavored by no evidence of vasculitis on biopsy and negative titers for proteinase 3 and myeloperoxidase antibodies. However, the presence of either, similar to a mixed infiltrate, does not exclude a diagnosis of PG, as they can occur simultaneously. Consequently, superinfection of a chronically open wound can occur due to underlying PG.7 The differences between PG and SSTI are listed in Table 2.
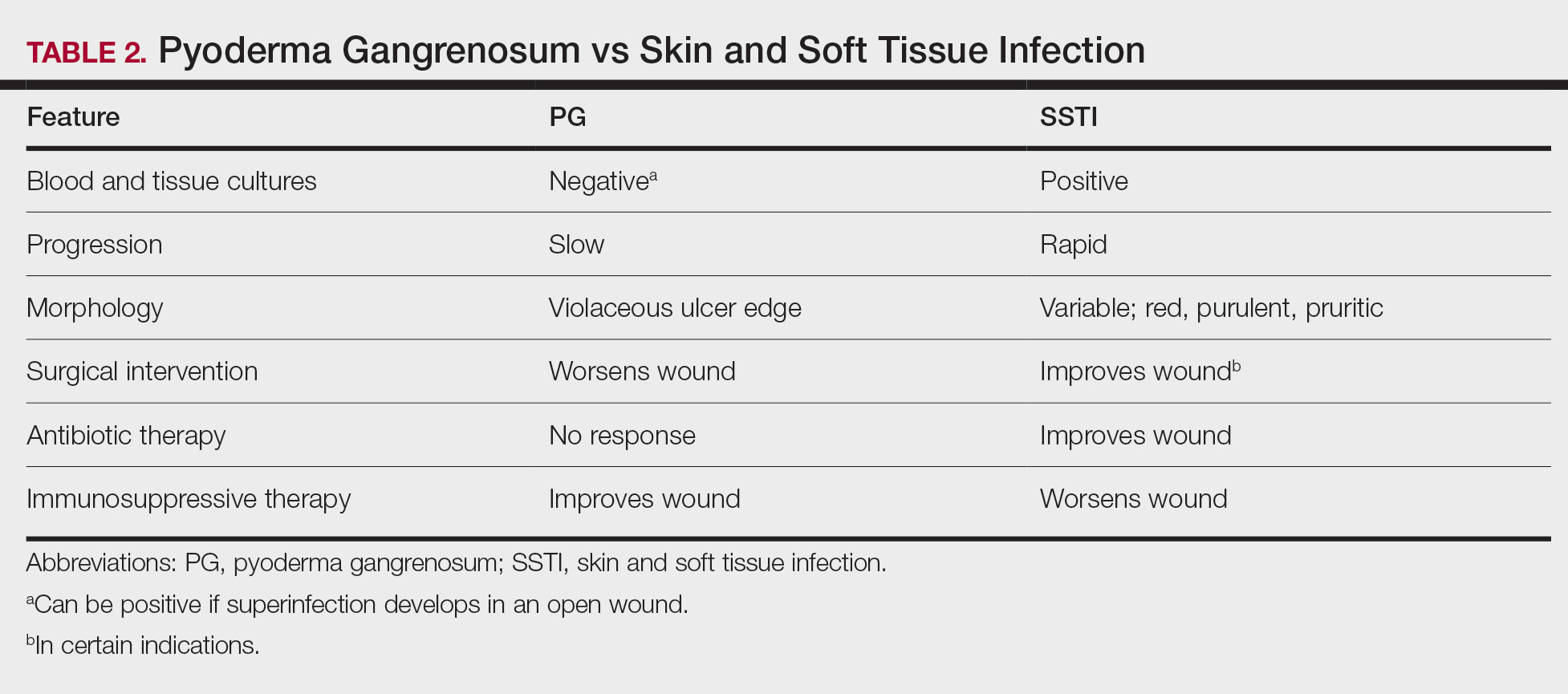
Although we know PG involves neutrophilic dysfunction, the pathophysiology remains poorly understood, contributing to the lack of clinical guidelines.8 Therefore, the diagnosis of PG often is delayed and is associated with severe consequences such as necrotizing fasciitis, osteomyelitis, cosmetic morbidity, and limb amputation.9,10 Dermatologic consultation can aid in early diagnosis and avoid amputation.7,10 Amputation has been used as a last resort to preserve optimal outcomes in patients with severe PG.11
Management of PG
A gold standard of treatment of PG does not exist, but the goal is to promote wound healing. Patients with limited disease typically can be managed with wound care and topical steroids or calcineurin inhibitors, though data on efficacy are limited. However, our patient had more extensive disease and needed to be treated with systemic therapy. First-line therapy for extensive disease includes oral prednisone or cyclosporine for patients who cannot tolerate systemic corticosteroids.12 Second-line and adjunctive therapy options include dapsone, minocycline, methotrexate, and infliximab. Our patient was prescribed a 7-month course of dapsone with outpatient dermatology and demonstrated resolution of both ulcers. Dapsone was tapered from a daily dose of 100 mg to 50 mg to 25 mg to none over the course of 2 to 3 months. Close monitoring with wound care is recommended, and petroleum jelly can be used for dry skin around the lesion for comfort.
Conclusion
The diagnosis of PG is challenging because it relies heavily on clinical signs and often mimics SSTI. Gathering a detailed medical history is critical to make the diagnosis of PG. In a patient with associated features of PG, dermatologic consultation and biopsy of skin lesions should be considered. Physicians should evaluate for suspected PG prior to proceeding with surgical intervention to avoid unnecessary amputation. The diagnostic criteria for classic ulcerative PG are gaining wider acceptance and are a useful tool for clinicians.
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatol. 2018;154:461-466.
- Bisarya K, Azzopardi S, Lye G, et al. Necrotizing fasciitis versus pyoderma gangrenosum: securing the correct diagnosis! a case report and literature review. Eplasty. 2011;11:E24.
- Perricone G, Vangeli M. Pyoderma gangrenosum in ulcerative colitis. N Engl J Med. 2018;379:E7.
- Ashchyan HJ, Butler DC, Nelson CA, et al. The association of age with clinical presentation and comorbidities of pyoderma gangrenosum. JAMA Dermatol. 2018;154:409-413.
- Ampuero J, Rojas-Feria M, Castro-Fernández M, et al. Predictive factors for erythema nodosum and pyoderma gangrenosum in inflammatory bowel disease. J Gastroenterol Hepatol. 2014;29:291-295.
- Ebner DW, Hu M, Poterucha TH. 29-year-old woman with fever and bilateral lower extremity lesions. Mayo Clin Proc. 2018;93:1659-1663.
- Marzak H, Von Hunolstein JJ, Lipsker D, et al. Management of a superinfected pyoderma gangrenosum after pacemaker implant. HeartRhythm Case Rep. 2018;5:63-65.
- Braswell SF, Kostopoulos TC, Ortega-Loayza AG. Pathophysiology of pyoderma gangrenosum (PG): an updated review. J Am Acad Dermatol. 2015;73:691-698.
- Saffie MG, Shroff A. A case of pyoderma gangrenosum misdiagnosed as necrotizing infection: a potential diagnostic catastrophe. Case Rep Infect Dis. 2018;2018:8907542.
- Haag CK, Nutan F, Cyrus JW, et al. Pyoderma gangrenosum misdiagnosis resulting in amputation: a review. J Trauma Acute Care Surg. 2019;86:307-313.
- Sanchez IM, Lowenstein S, Johnson KA, et al. Clinical features of neutrophilic dermatosis variants resembling necrotizing fasciitis. JAMA Dermatol. 2019;155:79-84.
- Alavi A, French LE, Davis MD, et al. Pyoderma gangrenosum: an update on pathophysiology, diagnosis and treatment. Am J Clin Dermatol. 2017;18:355-372.
Case Report
A 67-year-old woman presented with a painful expanding ulcer on the left leg and a new nearby ulcer of 2 months’ duration. She initially was seen 2 months prior for a wound on the left knee due to a fall as well as cellulitis, which was treated with intravenous vancomycin and ceftriaxone. Wound cultures were negative for bacteria, and she was discharged without antibiotics. She presented to the emergency department 1 month later for malodorous discharge of the first ulcer with zero systemic inflammatory response syndrome criteria; no fever; and no abnormal heart rate, respiratory rate, or leukocyte count. She was discharged with wound care. After 3 weeks, she returned with a second ulcer and worsening drainage but zero systemic inflammatory response syndrome criteria. She had a medical history of Crohn disease with 9-year remission, atrial fibrillation, pacemaker, mitral valve replacement, chronic obstructive pulmonary disease, and a 51 pack-year smoking history.
Physical examination of the left leg revealed a 3×3-cm deep lesion (ulcer A) on the distal left thigh located superomedial to the knee (Figure 1) as well as a 2×1-cm deep lesion (ulcer B) on the anteromedial knee with undermining and tunneling (Figure 2). A large amount of malodorous tan bloody discharge was present on both ulcers. There were no signs of induration or crepitus.Due to concerns of skin and soft tissue infection (SSTI) or osteomyelitis, a bone scan and wound and blood cultures were ordered. The patient was started on vancomycin and piperacillin-tazobactam in the emergency department, which later was augmented with cefepime. Trauma surgery scheduled debridement for the following morning with suspicion of necrotizing fasciitis. Additional consultations were requested, including infectious disease, wound care, and dermatology. Dermatology evaluated the wound, performed a punch biopsy, and canceled debridement due to unclear diagnosis. The clinical differential at that time included pyoderma gangrenosum (PG), atypical vasculitis, or infection. Additional workup revealed positive antineutrophil cytoplasmic antibodies but negative proteinase 3 and myeloperoxidase, disfavoring vasculitis. Wound cultures grew Staphylococcus aureus and Pseudomonas aeruginosa.
Histologic evaluation revealed deep dermal necrosis with a mixed inflammatory infiltrate (Figure 3) and no organisms or vasculitis. Antibiotics were discontinued, and she was discharged on a 14-day course of prednisone 60 mg daily for empirical treatment of PG with dermatology follow-up. Medical management included a 6-month course of dapsone that was extended to 7 months because of an intensive care unit stay for a cerebrovascular accident. Daily dosing was as follows: 100 mg for 5 months, 50 mg for 1 month, and 25 mg for 1 month, then stopped. She was followed with serial complete blood cell count every 1 to 2 months and home-health wound care. One month after dapsone initiation, the ulcers decreased in size. Ulcer B was fully healed after 4 months, and ulcer A was nearly closed at 6 months without any new flares.
Comment
Pyoderma gangrenosum is a rare inflammatory skin condition that classically presents as tender papules or pustules evolving into painful ulcers, most commonly on the lower extremities. Pyoderma gangrenosum has a propensity to exhibit pathergy, the hyperreactivity of the skin in response to minor trauma. This phenomenon in PG manifests as the rapid evolution from pustule to ulceration with violaceous undermining borders.
Diagnosis of PG
Pyoderma gangrenosum has been described as a diagnosis of exclusion, as its findings frequently mimic SSTIs. Important findings to obtain are histology, history, ulcer morphology, and response to treatment.
In 2018, Maverakis et al1 proposed diagnostic criteria for classic ulcerative PG (Table 1). A diagnosis of PG can be made if the patient meets 1 major criterion and 4 minor criteria. Our case met 0 major criteria and 5 minor criteria: history of inflammatory bowel disease (IBD); history of pustule ulcerating within 4 days of appearing; peripheral erythema, undermining border, and tenderness at ulceration site; multiple ulcerations, with at least 1 on an anterior lower leg; and decreased ulcer size within 1 month of initiating immunosuppressive medication(s). Although our patient’s biopsy demonstrated a mixed infiltrate, PG was not excluded due to spontaneous resolution at the time of biopsy, emphasizing the need to biopsy subsequent new lesions if neutrophils are not initially seen.1 Pyoderma gangrenosum frequently is associated with IBD, most often Crohn disease, as seen in our patient.2-4 Although IBD classically is associated with smoking, studies have yet to conclude if smoking is a predictive factor of PG.5 Our patient presented with an initial ulcer that evolved into 2 ulcers, similar to a case of bilateral ulcers.6
Differential Diagnosis of PG
Other possible diagnoses to consider are SSTI and vasculitis, the latter being disfavored by no evidence of vasculitis on biopsy and negative titers for proteinase 3 and myeloperoxidase antibodies. However, the presence of either, similar to a mixed infiltrate, does not exclude a diagnosis of PG, as they can occur simultaneously. Consequently, superinfection of a chronically open wound can occur due to underlying PG.7 The differences between PG and SSTI are listed in Table 2.
Although we know PG involves neutrophilic dysfunction, the pathophysiology remains poorly understood, contributing to the lack of clinical guidelines.8 Therefore, the diagnosis of PG often is delayed and is associated with severe consequences such as necrotizing fasciitis, osteomyelitis, cosmetic morbidity, and limb amputation.9,10 Dermatologic consultation can aid in early diagnosis and avoid amputation.7,10 Amputation has been used as a last resort to preserve optimal outcomes in patients with severe PG.11
Management of PG
A gold standard of treatment of PG does not exist, but the goal is to promote wound healing. Patients with limited disease typically can be managed with wound care and topical steroids or calcineurin inhibitors, though data on efficacy are limited. However, our patient had more extensive disease and needed to be treated with systemic therapy. First-line therapy for extensive disease includes oral prednisone or cyclosporine for patients who cannot tolerate systemic corticosteroids.12 Second-line and adjunctive therapy options include dapsone, minocycline, methotrexate, and infliximab. Our patient was prescribed a 7-month course of dapsone with outpatient dermatology and demonstrated resolution of both ulcers. Dapsone was tapered from a daily dose of 100 mg to 50 mg to 25 mg to none over the course of 2 to 3 months. Close monitoring with wound care is recommended, and petroleum jelly can be used for dry skin around the lesion for comfort.
Conclusion
The diagnosis of PG is challenging because it relies heavily on clinical signs and often mimics SSTI. Gathering a detailed medical history is critical to make the diagnosis of PG. In a patient with associated features of PG, dermatologic consultation and biopsy of skin lesions should be considered. Physicians should evaluate for suspected PG prior to proceeding with surgical intervention to avoid unnecessary amputation. The diagnostic criteria for classic ulcerative PG are gaining wider acceptance and are a useful tool for clinicians.
Case Report
A 67-year-old woman presented with a painful expanding ulcer on the left leg and a new nearby ulcer of 2 months’ duration. She initially was seen 2 months prior for a wound on the left knee due to a fall as well as cellulitis, which was treated with intravenous vancomycin and ceftriaxone. Wound cultures were negative for bacteria, and she was discharged without antibiotics. She presented to the emergency department 1 month later for malodorous discharge of the first ulcer with zero systemic inflammatory response syndrome criteria; no fever; and no abnormal heart rate, respiratory rate, or leukocyte count. She was discharged with wound care. After 3 weeks, she returned with a second ulcer and worsening drainage but zero systemic inflammatory response syndrome criteria. She had a medical history of Crohn disease with 9-year remission, atrial fibrillation, pacemaker, mitral valve replacement, chronic obstructive pulmonary disease, and a 51 pack-year smoking history.
Physical examination of the left leg revealed a 3×3-cm deep lesion (ulcer A) on the distal left thigh located superomedial to the knee (Figure 1) as well as a 2×1-cm deep lesion (ulcer B) on the anteromedial knee with undermining and tunneling (Figure 2). A large amount of malodorous tan bloody discharge was present on both ulcers. There were no signs of induration or crepitus.Due to concerns of skin and soft tissue infection (SSTI) or osteomyelitis, a bone scan and wound and blood cultures were ordered. The patient was started on vancomycin and piperacillin-tazobactam in the emergency department, which later was augmented with cefepime. Trauma surgery scheduled debridement for the following morning with suspicion of necrotizing fasciitis. Additional consultations were requested, including infectious disease, wound care, and dermatology. Dermatology evaluated the wound, performed a punch biopsy, and canceled debridement due to unclear diagnosis. The clinical differential at that time included pyoderma gangrenosum (PG), atypical vasculitis, or infection. Additional workup revealed positive antineutrophil cytoplasmic antibodies but negative proteinase 3 and myeloperoxidase, disfavoring vasculitis. Wound cultures grew Staphylococcus aureus and Pseudomonas aeruginosa.
Histologic evaluation revealed deep dermal necrosis with a mixed inflammatory infiltrate (Figure 3) and no organisms or vasculitis. Antibiotics were discontinued, and she was discharged on a 14-day course of prednisone 60 mg daily for empirical treatment of PG with dermatology follow-up. Medical management included a 6-month course of dapsone that was extended to 7 months because of an intensive care unit stay for a cerebrovascular accident. Daily dosing was as follows: 100 mg for 5 months, 50 mg for 1 month, and 25 mg for 1 month, then stopped. She was followed with serial complete blood cell count every 1 to 2 months and home-health wound care. One month after dapsone initiation, the ulcers decreased in size. Ulcer B was fully healed after 4 months, and ulcer A was nearly closed at 6 months without any new flares.
Comment
Pyoderma gangrenosum is a rare inflammatory skin condition that classically presents as tender papules or pustules evolving into painful ulcers, most commonly on the lower extremities. Pyoderma gangrenosum has a propensity to exhibit pathergy, the hyperreactivity of the skin in response to minor trauma. This phenomenon in PG manifests as the rapid evolution from pustule to ulceration with violaceous undermining borders.
Diagnosis of PG
Pyoderma gangrenosum has been described as a diagnosis of exclusion, as its findings frequently mimic SSTIs. Important findings to obtain are histology, history, ulcer morphology, and response to treatment.
In 2018, Maverakis et al1 proposed diagnostic criteria for classic ulcerative PG (Table 1). A diagnosis of PG can be made if the patient meets 1 major criterion and 4 minor criteria. Our case met 0 major criteria and 5 minor criteria: history of inflammatory bowel disease (IBD); history of pustule ulcerating within 4 days of appearing; peripheral erythema, undermining border, and tenderness at ulceration site; multiple ulcerations, with at least 1 on an anterior lower leg; and decreased ulcer size within 1 month of initiating immunosuppressive medication(s). Although our patient’s biopsy demonstrated a mixed infiltrate, PG was not excluded due to spontaneous resolution at the time of biopsy, emphasizing the need to biopsy subsequent new lesions if neutrophils are not initially seen.1 Pyoderma gangrenosum frequently is associated with IBD, most often Crohn disease, as seen in our patient.2-4 Although IBD classically is associated with smoking, studies have yet to conclude if smoking is a predictive factor of PG.5 Our patient presented with an initial ulcer that evolved into 2 ulcers, similar to a case of bilateral ulcers.6
Differential Diagnosis of PG
Other possible diagnoses to consider are SSTI and vasculitis, the latter being disfavored by no evidence of vasculitis on biopsy and negative titers for proteinase 3 and myeloperoxidase antibodies. However, the presence of either, similar to a mixed infiltrate, does not exclude a diagnosis of PG, as they can occur simultaneously. Consequently, superinfection of a chronically open wound can occur due to underlying PG.7 The differences between PG and SSTI are listed in Table 2.
Although we know PG involves neutrophilic dysfunction, the pathophysiology remains poorly understood, contributing to the lack of clinical guidelines.8 Therefore, the diagnosis of PG often is delayed and is associated with severe consequences such as necrotizing fasciitis, osteomyelitis, cosmetic morbidity, and limb amputation.9,10 Dermatologic consultation can aid in early diagnosis and avoid amputation.7,10 Amputation has been used as a last resort to preserve optimal outcomes in patients with severe PG.11
Management of PG
A gold standard of treatment of PG does not exist, but the goal is to promote wound healing. Patients with limited disease typically can be managed with wound care and topical steroids or calcineurin inhibitors, though data on efficacy are limited. However, our patient had more extensive disease and needed to be treated with systemic therapy. First-line therapy for extensive disease includes oral prednisone or cyclosporine for patients who cannot tolerate systemic corticosteroids.12 Second-line and adjunctive therapy options include dapsone, minocycline, methotrexate, and infliximab. Our patient was prescribed a 7-month course of dapsone with outpatient dermatology and demonstrated resolution of both ulcers. Dapsone was tapered from a daily dose of 100 mg to 50 mg to 25 mg to none over the course of 2 to 3 months. Close monitoring with wound care is recommended, and petroleum jelly can be used for dry skin around the lesion for comfort.
Conclusion
The diagnosis of PG is challenging because it relies heavily on clinical signs and often mimics SSTI. Gathering a detailed medical history is critical to make the diagnosis of PG. In a patient with associated features of PG, dermatologic consultation and biopsy of skin lesions should be considered. Physicians should evaluate for suspected PG prior to proceeding with surgical intervention to avoid unnecessary amputation. The diagnostic criteria for classic ulcerative PG are gaining wider acceptance and are a useful tool for clinicians.
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatol. 2018;154:461-466.
- Bisarya K, Azzopardi S, Lye G, et al. Necrotizing fasciitis versus pyoderma gangrenosum: securing the correct diagnosis! a case report and literature review. Eplasty. 2011;11:E24.
- Perricone G, Vangeli M. Pyoderma gangrenosum in ulcerative colitis. N Engl J Med. 2018;379:E7.
- Ashchyan HJ, Butler DC, Nelson CA, et al. The association of age with clinical presentation and comorbidities of pyoderma gangrenosum. JAMA Dermatol. 2018;154:409-413.
- Ampuero J, Rojas-Feria M, Castro-Fernández M, et al. Predictive factors for erythema nodosum and pyoderma gangrenosum in inflammatory bowel disease. J Gastroenterol Hepatol. 2014;29:291-295.
- Ebner DW, Hu M, Poterucha TH. 29-year-old woman with fever and bilateral lower extremity lesions. Mayo Clin Proc. 2018;93:1659-1663.
- Marzak H, Von Hunolstein JJ, Lipsker D, et al. Management of a superinfected pyoderma gangrenosum after pacemaker implant. HeartRhythm Case Rep. 2018;5:63-65.
- Braswell SF, Kostopoulos TC, Ortega-Loayza AG. Pathophysiology of pyoderma gangrenosum (PG): an updated review. J Am Acad Dermatol. 2015;73:691-698.
- Saffie MG, Shroff A. A case of pyoderma gangrenosum misdiagnosed as necrotizing infection: a potential diagnostic catastrophe. Case Rep Infect Dis. 2018;2018:8907542.
- Haag CK, Nutan F, Cyrus JW, et al. Pyoderma gangrenosum misdiagnosis resulting in amputation: a review. J Trauma Acute Care Surg. 2019;86:307-313.
- Sanchez IM, Lowenstein S, Johnson KA, et al. Clinical features of neutrophilic dermatosis variants resembling necrotizing fasciitis. JAMA Dermatol. 2019;155:79-84.
- Alavi A, French LE, Davis MD, et al. Pyoderma gangrenosum: an update on pathophysiology, diagnosis and treatment. Am J Clin Dermatol. 2017;18:355-372.
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatol. 2018;154:461-466.
- Bisarya K, Azzopardi S, Lye G, et al. Necrotizing fasciitis versus pyoderma gangrenosum: securing the correct diagnosis! a case report and literature review. Eplasty. 2011;11:E24.
- Perricone G, Vangeli M. Pyoderma gangrenosum in ulcerative colitis. N Engl J Med. 2018;379:E7.
- Ashchyan HJ, Butler DC, Nelson CA, et al. The association of age with clinical presentation and comorbidities of pyoderma gangrenosum. JAMA Dermatol. 2018;154:409-413.
- Ampuero J, Rojas-Feria M, Castro-Fernández M, et al. Predictive factors for erythema nodosum and pyoderma gangrenosum in inflammatory bowel disease. J Gastroenterol Hepatol. 2014;29:291-295.
- Ebner DW, Hu M, Poterucha TH. 29-year-old woman with fever and bilateral lower extremity lesions. Mayo Clin Proc. 2018;93:1659-1663.
- Marzak H, Von Hunolstein JJ, Lipsker D, et al. Management of a superinfected pyoderma gangrenosum after pacemaker implant. HeartRhythm Case Rep. 2018;5:63-65.
- Braswell SF, Kostopoulos TC, Ortega-Loayza AG. Pathophysiology of pyoderma gangrenosum (PG): an updated review. J Am Acad Dermatol. 2015;73:691-698.
- Saffie MG, Shroff A. A case of pyoderma gangrenosum misdiagnosed as necrotizing infection: a potential diagnostic catastrophe. Case Rep Infect Dis. 2018;2018:8907542.
- Haag CK, Nutan F, Cyrus JW, et al. Pyoderma gangrenosum misdiagnosis resulting in amputation: a review. J Trauma Acute Care Surg. 2019;86:307-313.
- Sanchez IM, Lowenstein S, Johnson KA, et al. Clinical features of neutrophilic dermatosis variants resembling necrotizing fasciitis. JAMA Dermatol. 2019;155:79-84.
- Alavi A, French LE, Davis MD, et al. Pyoderma gangrenosum: an update on pathophysiology, diagnosis and treatment. Am J Clin Dermatol. 2017;18:355-372.
Practice Points
- Pyoderma gangrenosum (PG) frequently is misdiagnosed due to its similar presentation to other skin and soft tissue infections (SSTIs). Patients with known risk factors for PG should be evaluated with a high index of suspicion to ensure early diagnosis and avoid serious complications. Common associations include inflammatory bowel disease (IBD), hematologic malignancies, and rheumatologic disorders.
- Response to treatment may be used to guide management when the diagnosis of SSTIs vs PG cannot be distinguished with clinical and histologic findings alone. In a worsening ulcer that has failed antibiotic therapy, clinicians should consider the diagnosis of PG and the risk of pathergy prior to surgical intervention such as debridement.
- Although typically a diagnosis of exclusion, clinicians can consider the use of diagnostic criteria for PG in patients of high clinical suspicion. A trial of immunosuppressants can be considered after infection has been ruled out.
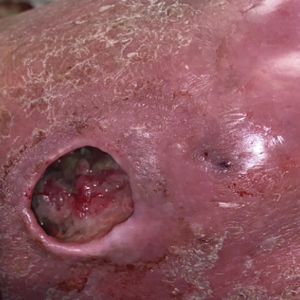
Urticarial Vasculitis Successfully Treated With Omalizumab
To the Editor:
Urticarial vasculitis (UV) is a clinicopathologic entity. It manifests as an eruption of erythematous wheals that clinically resemble urticaria, but the lesions of UV last longer, may leave residual hyperpigmentation, and may or may not be pruritic.1 Therapies most often employed include oral antihistamines and systemic immunosuppressant drugs such as corticosteroids, dapsone, colchicine, or hydroxychloroquine.2 We present a woman with UV who successfully was treated with omalizumab.
A 49-year-old woman presented to our outpatient clinic with generalized pruritic skin rashes of 2 years’ duration. She also described swelling on the upper eyelids 2 times monthly. She used several antihistamines (up to 4 times daily) and was taking systemic corticosteroids and antidepressants. Physical examination revealed generalized erythematous and edematous papules and plaques on the trunk and extremities (Figure 1). At follow-up a few days later, we observed that the lesions were lasting for more than 24 hours, but there was no residual pigmentation. According to clinical concerns and the association with angioedema, we initially thought the diagnosis was chronic urticaria and angioedema. The patient had no extracutaneous manifestations such as fever, arthralgia, or lymphadenopathy. Routine laboratory examinations including antinuclear antibodies were within reference range. She had normal C3 and C4 levels and an elevated total IgE level (344 IU/mL [reference range, 0–170 IU/mL]). Because the IgE level was elevated and she had no response to the highest dosages of antihistamines, we decided to start omalizumab therapy. Prior to starting omalizumab, we performed a skin biopsy for histopathologic and direct immunofluorescence examinations for UV, as the duration of the lesions was more than 24 hours. Histopathologic examination revealed lymphocytes within the vessel wall and perivascular lymphocytic infiltration with eosinophils (Figure 2). On direct immunofluorescence, perivascular IgA deposition was observed (Figure 3). Histopathologic findings were associated with lymphocytic vasculitis. Systemic involvement was not detected on detailed laboratory and radiologic examinations.
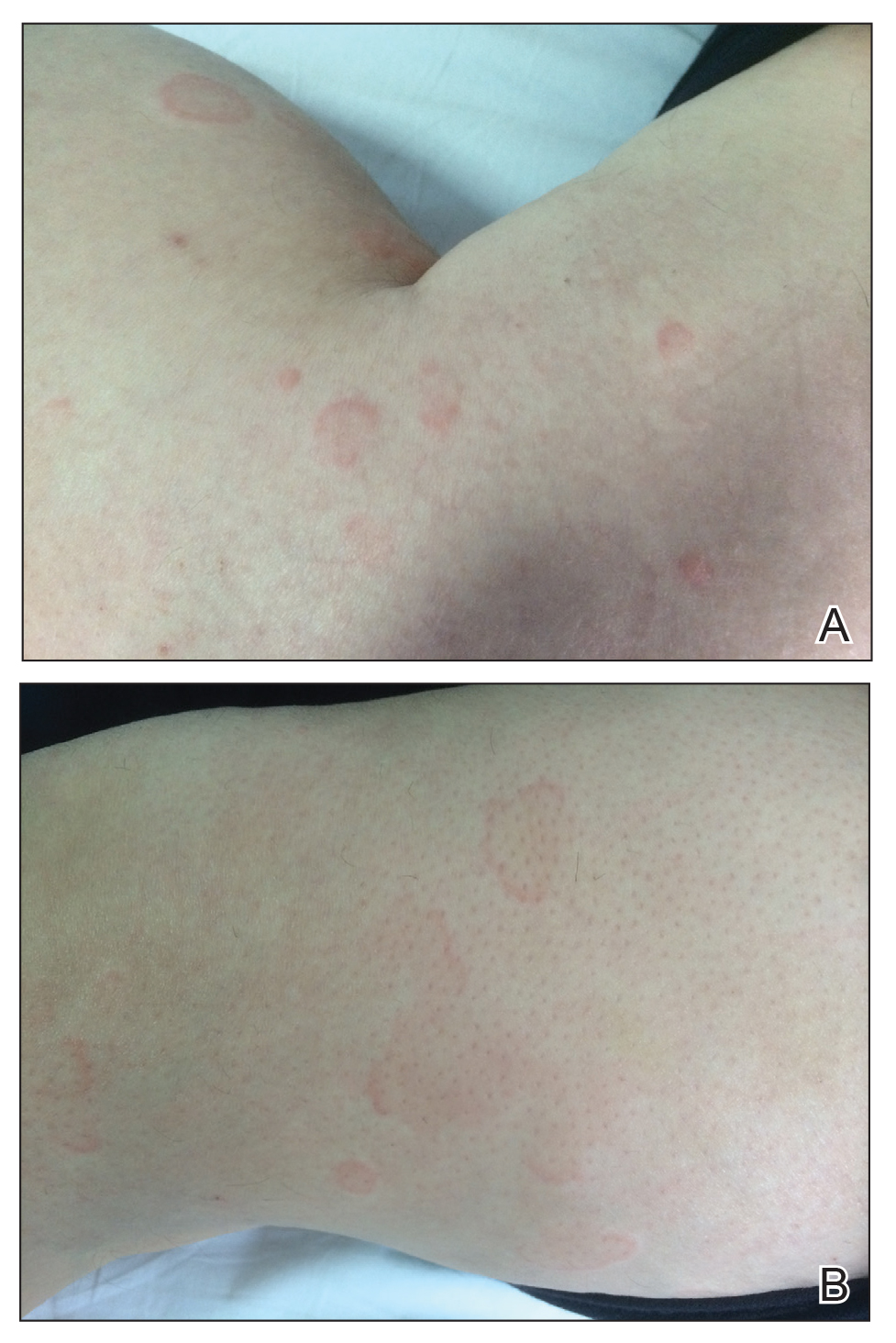


After the first application of omalizumab, the lesions disappeared within a few days. She was treated with subcutaneous omalizumab 300 mg every 4 weeks for 6 months, and we did not observe any adverse effects related to the drug. There was no relapse after therapy cessation.
Omalizumab is a recombinant humanized anti-IgE monoclonal antibody that is approved by the US Food and Drug Administration for treatment of chronic idiopathic urticaria.3-5 Studies have suggested that omalizumab might play an important role in the treatment of other potentially IgE-mediated disease processes including allergic asthma, atopic dermatitis, allergic rhinitis, nasal polyposis, and severe ocular allergies.6 The proposed mechanism of action of omalizumab includes reduction of free IgE through the reversible formation of tiny, biologically inert complexes; targeting IgE-expressing B cells; and inhibiting production of IgE. Because it reduces free IgE, omalizumab has been used in normal IgE or hyper-IgE situations. Omalizumab also induces eosinophil apoptosis; increases IL-2, IL-3, tumor necrosis factor α, and IFN-γ; and reduces IL-4.7 A number of off-label uses have been described such as atopic dermatitis, bullous pemphigoid, hyper-IgE syndrome, cutaneous mastocytosis, toxic epidermal necrolysis, and eosinophilic granulomatosis with polyangitis.8 There are no clinical studies of omalizumab for UV, and only a few case reports have shown that omalizumab also might be beneficial for this condition.2-4 Diez et al4 reported 3 cases of women aged 28, 51, and 54 years with spontaneous chronic urticaria with autoimmune and pressure components as well as vasculitis whose symptoms completely improved after starting omalizumab. Kai et al3 successfully treated a patient with normocomplementemic UV with omalizumab and suggested that omalizumab markedly improved the patient’s quality of life with chronic urticaria and UV. Ghazanfar and Thomsen2 reported the case of a 68-year-old man diagnosed with histopathologically confirmed leukocytoclastic vasculitis. He had used systemic corticosteroid therapy and dapsone without notable improvement. The patient was switched to subcutaneous omalizumab 300 mg once every 4 weeks; after 1 month, he observed complete remission of the UV and symptoms.2
Our case suggests that omalizumab has a beneficial effect on patients with UV. Omalizumab may be effective in UV through its reduction of IgE, as in chronic urticaria, and through downstream effects on cellular activation mechanisms (possibly a reduction in chemotaxis or immune complex formation). However, the mechanism of action of omalizumab for UV remains, in part, unresolved. It is not known whether omalizumab is efficacious against both normocomplementemic and hypocomplementemic UV. Further studies with a greater number of patients are needed to confirm the effects of omalizumab for vasculitic patients.
- Chang S, Carr W. Urticarial vasculitis. Allergy Asthma Proc. 2007;28:97-100.
- Ghazanfar MN, Thomsen SF. Omalizumab for urticarial vasculitis: case report and review of the literature. Case Rep Dermatol Med. 2015:576893.
- Kai AC, Flohr C, Grattan CE. Improvement in quality of life impairment followed by relapse with 6-monthly periodic administration of omalizumab for severe treatment-refractory chronic urticaria and urticarial vasculitis. Clin Exp Dermatol. 2014;39:651-652.
- Diez LS, Tamayo LM, Cardona R. Omalizumab: therapeutic option in chronic spontaneous urticaria difficult to control with associated vasculitis, report of three cases. Biomedica. 2013;33:503-512.
- Maurer M, Rosen K, Hsieh HJ. Omalizumab for chronic urticaria. N Engl J Med. 2013;368:2530.
- Ben Shoshan M. Omalizumab: not only for asthma. Recent Pat Inflamm Allergy Drug Discov. 2008;2:191-201.
- Fueyo-Casado A, Campos-Munoz L, Gonzalez-Guerra E, et al. Effectiveness of omalizumab in a case of urticarial vasculitis. Clin Exp Dermatol. Published March 1, 2017. doi:10.1111/ced.13076
- Chia JC, Mydlarski PR. Dermatologic uses of omalizumab. J Dermatol Treat. Published November 7, 2016. doi:10.1080/09546634.2016.1249819
To the Editor:
Urticarial vasculitis (UV) is a clinicopathologic entity. It manifests as an eruption of erythematous wheals that clinically resemble urticaria, but the lesions of UV last longer, may leave residual hyperpigmentation, and may or may not be pruritic.1 Therapies most often employed include oral antihistamines and systemic immunosuppressant drugs such as corticosteroids, dapsone, colchicine, or hydroxychloroquine.2 We present a woman with UV who successfully was treated with omalizumab.
A 49-year-old woman presented to our outpatient clinic with generalized pruritic skin rashes of 2 years’ duration. She also described swelling on the upper eyelids 2 times monthly. She used several antihistamines (up to 4 times daily) and was taking systemic corticosteroids and antidepressants. Physical examination revealed generalized erythematous and edematous papules and plaques on the trunk and extremities (Figure 1). At follow-up a few days later, we observed that the lesions were lasting for more than 24 hours, but there was no residual pigmentation. According to clinical concerns and the association with angioedema, we initially thought the diagnosis was chronic urticaria and angioedema. The patient had no extracutaneous manifestations such as fever, arthralgia, or lymphadenopathy. Routine laboratory examinations including antinuclear antibodies were within reference range. She had normal C3 and C4 levels and an elevated total IgE level (344 IU/mL [reference range, 0–170 IU/mL]). Because the IgE level was elevated and she had no response to the highest dosages of antihistamines, we decided to start omalizumab therapy. Prior to starting omalizumab, we performed a skin biopsy for histopathologic and direct immunofluorescence examinations for UV, as the duration of the lesions was more than 24 hours. Histopathologic examination revealed lymphocytes within the vessel wall and perivascular lymphocytic infiltration with eosinophils (Figure 2). On direct immunofluorescence, perivascular IgA deposition was observed (Figure 3). Histopathologic findings were associated with lymphocytic vasculitis. Systemic involvement was not detected on detailed laboratory and radiologic examinations.
After the first application of omalizumab, the lesions disappeared within a few days. She was treated with subcutaneous omalizumab 300 mg every 4 weeks for 6 months, and we did not observe any adverse effects related to the drug. There was no relapse after therapy cessation.
Omalizumab is a recombinant humanized anti-IgE monoclonal antibody that is approved by the US Food and Drug Administration for treatment of chronic idiopathic urticaria.3-5 Studies have suggested that omalizumab might play an important role in the treatment of other potentially IgE-mediated disease processes including allergic asthma, atopic dermatitis, allergic rhinitis, nasal polyposis, and severe ocular allergies.6 The proposed mechanism of action of omalizumab includes reduction of free IgE through the reversible formation of tiny, biologically inert complexes; targeting IgE-expressing B cells; and inhibiting production of IgE. Because it reduces free IgE, omalizumab has been used in normal IgE or hyper-IgE situations. Omalizumab also induces eosinophil apoptosis; increases IL-2, IL-3, tumor necrosis factor α, and IFN-γ; and reduces IL-4.7 A number of off-label uses have been described such as atopic dermatitis, bullous pemphigoid, hyper-IgE syndrome, cutaneous mastocytosis, toxic epidermal necrolysis, and eosinophilic granulomatosis with polyangitis.8 There are no clinical studies of omalizumab for UV, and only a few case reports have shown that omalizumab also might be beneficial for this condition.2-4 Diez et al4 reported 3 cases of women aged 28, 51, and 54 years with spontaneous chronic urticaria with autoimmune and pressure components as well as vasculitis whose symptoms completely improved after starting omalizumab. Kai et al3 successfully treated a patient with normocomplementemic UV with omalizumab and suggested that omalizumab markedly improved the patient’s quality of life with chronic urticaria and UV. Ghazanfar and Thomsen2 reported the case of a 68-year-old man diagnosed with histopathologically confirmed leukocytoclastic vasculitis. He had used systemic corticosteroid therapy and dapsone without notable improvement. The patient was switched to subcutaneous omalizumab 300 mg once every 4 weeks; after 1 month, he observed complete remission of the UV and symptoms.2
Our case suggests that omalizumab has a beneficial effect on patients with UV. Omalizumab may be effective in UV through its reduction of IgE, as in chronic urticaria, and through downstream effects on cellular activation mechanisms (possibly a reduction in chemotaxis or immune complex formation). However, the mechanism of action of omalizumab for UV remains, in part, unresolved. It is not known whether omalizumab is efficacious against both normocomplementemic and hypocomplementemic UV. Further studies with a greater number of patients are needed to confirm the effects of omalizumab for vasculitic patients.
To the Editor:
Urticarial vasculitis (UV) is a clinicopathologic entity. It manifests as an eruption of erythematous wheals that clinically resemble urticaria, but the lesions of UV last longer, may leave residual hyperpigmentation, and may or may not be pruritic.1 Therapies most often employed include oral antihistamines and systemic immunosuppressant drugs such as corticosteroids, dapsone, colchicine, or hydroxychloroquine.2 We present a woman with UV who successfully was treated with omalizumab.
A 49-year-old woman presented to our outpatient clinic with generalized pruritic skin rashes of 2 years’ duration. She also described swelling on the upper eyelids 2 times monthly. She used several antihistamines (up to 4 times daily) and was taking systemic corticosteroids and antidepressants. Physical examination revealed generalized erythematous and edematous papules and plaques on the trunk and extremities (Figure 1). At follow-up a few days later, we observed that the lesions were lasting for more than 24 hours, but there was no residual pigmentation. According to clinical concerns and the association with angioedema, we initially thought the diagnosis was chronic urticaria and angioedema. The patient had no extracutaneous manifestations such as fever, arthralgia, or lymphadenopathy. Routine laboratory examinations including antinuclear antibodies were within reference range. She had normal C3 and C4 levels and an elevated total IgE level (344 IU/mL [reference range, 0–170 IU/mL]). Because the IgE level was elevated and she had no response to the highest dosages of antihistamines, we decided to start omalizumab therapy. Prior to starting omalizumab, we performed a skin biopsy for histopathologic and direct immunofluorescence examinations for UV, as the duration of the lesions was more than 24 hours. Histopathologic examination revealed lymphocytes within the vessel wall and perivascular lymphocytic infiltration with eosinophils (Figure 2). On direct immunofluorescence, perivascular IgA deposition was observed (Figure 3). Histopathologic findings were associated with lymphocytic vasculitis. Systemic involvement was not detected on detailed laboratory and radiologic examinations.
After the first application of omalizumab, the lesions disappeared within a few days. She was treated with subcutaneous omalizumab 300 mg every 4 weeks for 6 months, and we did not observe any adverse effects related to the drug. There was no relapse after therapy cessation.
Omalizumab is a recombinant humanized anti-IgE monoclonal antibody that is approved by the US Food and Drug Administration for treatment of chronic idiopathic urticaria.3-5 Studies have suggested that omalizumab might play an important role in the treatment of other potentially IgE-mediated disease processes including allergic asthma, atopic dermatitis, allergic rhinitis, nasal polyposis, and severe ocular allergies.6 The proposed mechanism of action of omalizumab includes reduction of free IgE through the reversible formation of tiny, biologically inert complexes; targeting IgE-expressing B cells; and inhibiting production of IgE. Because it reduces free IgE, omalizumab has been used in normal IgE or hyper-IgE situations. Omalizumab also induces eosinophil apoptosis; increases IL-2, IL-3, tumor necrosis factor α, and IFN-γ; and reduces IL-4.7 A number of off-label uses have been described such as atopic dermatitis, bullous pemphigoid, hyper-IgE syndrome, cutaneous mastocytosis, toxic epidermal necrolysis, and eosinophilic granulomatosis with polyangitis.8 There are no clinical studies of omalizumab for UV, and only a few case reports have shown that omalizumab also might be beneficial for this condition.2-4 Diez et al4 reported 3 cases of women aged 28, 51, and 54 years with spontaneous chronic urticaria with autoimmune and pressure components as well as vasculitis whose symptoms completely improved after starting omalizumab. Kai et al3 successfully treated a patient with normocomplementemic UV with omalizumab and suggested that omalizumab markedly improved the patient’s quality of life with chronic urticaria and UV. Ghazanfar and Thomsen2 reported the case of a 68-year-old man diagnosed with histopathologically confirmed leukocytoclastic vasculitis. He had used systemic corticosteroid therapy and dapsone without notable improvement. The patient was switched to subcutaneous omalizumab 300 mg once every 4 weeks; after 1 month, he observed complete remission of the UV and symptoms.2
Our case suggests that omalizumab has a beneficial effect on patients with UV. Omalizumab may be effective in UV through its reduction of IgE, as in chronic urticaria, and through downstream effects on cellular activation mechanisms (possibly a reduction in chemotaxis or immune complex formation). However, the mechanism of action of omalizumab for UV remains, in part, unresolved. It is not known whether omalizumab is efficacious against both normocomplementemic and hypocomplementemic UV. Further studies with a greater number of patients are needed to confirm the effects of omalizumab for vasculitic patients.
- Chang S, Carr W. Urticarial vasculitis. Allergy Asthma Proc. 2007;28:97-100.
- Ghazanfar MN, Thomsen SF. Omalizumab for urticarial vasculitis: case report and review of the literature. Case Rep Dermatol Med. 2015:576893.
- Kai AC, Flohr C, Grattan CE. Improvement in quality of life impairment followed by relapse with 6-monthly periodic administration of omalizumab for severe treatment-refractory chronic urticaria and urticarial vasculitis. Clin Exp Dermatol. 2014;39:651-652.
- Diez LS, Tamayo LM, Cardona R. Omalizumab: therapeutic option in chronic spontaneous urticaria difficult to control with associated vasculitis, report of three cases. Biomedica. 2013;33:503-512.
- Maurer M, Rosen K, Hsieh HJ. Omalizumab for chronic urticaria. N Engl J Med. 2013;368:2530.
- Ben Shoshan M. Omalizumab: not only for asthma. Recent Pat Inflamm Allergy Drug Discov. 2008;2:191-201.
- Fueyo-Casado A, Campos-Munoz L, Gonzalez-Guerra E, et al. Effectiveness of omalizumab in a case of urticarial vasculitis. Clin Exp Dermatol. Published March 1, 2017. doi:10.1111/ced.13076
- Chia JC, Mydlarski PR. Dermatologic uses of omalizumab. J Dermatol Treat. Published November 7, 2016. doi:10.1080/09546634.2016.1249819
- Chang S, Carr W. Urticarial vasculitis. Allergy Asthma Proc. 2007;28:97-100.
- Ghazanfar MN, Thomsen SF. Omalizumab for urticarial vasculitis: case report and review of the literature. Case Rep Dermatol Med. 2015:576893.
- Kai AC, Flohr C, Grattan CE. Improvement in quality of life impairment followed by relapse with 6-monthly periodic administration of omalizumab for severe treatment-refractory chronic urticaria and urticarial vasculitis. Clin Exp Dermatol. 2014;39:651-652.
- Diez LS, Tamayo LM, Cardona R. Omalizumab: therapeutic option in chronic spontaneous urticaria difficult to control with associated vasculitis, report of three cases. Biomedica. 2013;33:503-512.
- Maurer M, Rosen K, Hsieh HJ. Omalizumab for chronic urticaria. N Engl J Med. 2013;368:2530.
- Ben Shoshan M. Omalizumab: not only for asthma. Recent Pat Inflamm Allergy Drug Discov. 2008;2:191-201.
- Fueyo-Casado A, Campos-Munoz L, Gonzalez-Guerra E, et al. Effectiveness of omalizumab in a case of urticarial vasculitis. Clin Exp Dermatol. Published March 1, 2017. doi:10.1111/ced.13076
- Chia JC, Mydlarski PR. Dermatologic uses of omalizumab. J Dermatol Treat. Published November 7, 2016. doi:10.1080/09546634.2016.1249819
Practice Points
- The differential diagnosis of urticaria and urticarial vasculitis may be complicated.
- Omalizumab is an effective urticaria treatment and also can be an alternative treatment choice in resistant urticarial vasculitis.