User login
FDA approves Taltz for treatment of ankylosing spondylitis
(AS), according to a press release from Eli Lilly.

AS is the third indication for ixekizumab, along with moderate to severe plaque psoriasis in adult patients who are candidates for systemic therapy or phototherapy and active psoriatic arthritis in adults.
Approval of the humanized interleukin-17A antagonist was based on results from a pair of randomized, double-blind, placebo-controlled, phase 3 studies involving 657 adult patients with active AS: the COAST-V trial in those naive to biologic disease-modifying antirheumatic drugs (bDMARDs) and the COAST-W trial in those who were intolerant or had inadequate response to tumor necrosis factor (TNF) inhibitors. The primary endpoint in both trials was achievement of 40% improvement in Assessment of Spondyloarthritis International Society criteria (ASAS40) at 16 weeks, compared with placebo.
In COAST-V, 48% of patients who received ixekizumab achieved ASAS40, compared with 18% of controls (P less than .0001). In COAST-W, 25% of patients who received ixekizumab achieved ASAS40 versus 13% of controls (P less than .05). The adverse events reported during both trials were consistent with the safety profile in patients who receive ixekizumab for the treatment of plaque psoriasis, including injection-site reactions, upper respiratory tract infections, nausea, and tinea infections.
“Results from the phase 3 clinical trial program in ankylosing spondylitis show that Taltz helped reduce pain and inflammation and improve function in patients who had never been treated with a bDMARD as well as those who previously failed TNF inhibitors. This approval is an important milestone for patients and physicians who are looking for a much-needed alternative to address symptoms of AS,” said Philip Mease, MD, of Providence St. Joseph Health and the University of Washington, both in Seattle.
(AS), according to a press release from Eli Lilly.
AS is the third indication for ixekizumab, along with moderate to severe plaque psoriasis in adult patients who are candidates for systemic therapy or phototherapy and active psoriatic arthritis in adults.
Approval of the humanized interleukin-17A antagonist was based on results from a pair of randomized, double-blind, placebo-controlled, phase 3 studies involving 657 adult patients with active AS: the COAST-V trial in those naive to biologic disease-modifying antirheumatic drugs (bDMARDs) and the COAST-W trial in those who were intolerant or had inadequate response to tumor necrosis factor (TNF) inhibitors. The primary endpoint in both trials was achievement of 40% improvement in Assessment of Spondyloarthritis International Society criteria (ASAS40) at 16 weeks, compared with placebo.
In COAST-V, 48% of patients who received ixekizumab achieved ASAS40, compared with 18% of controls (P less than .0001). In COAST-W, 25% of patients who received ixekizumab achieved ASAS40 versus 13% of controls (P less than .05). The adverse events reported during both trials were consistent with the safety profile in patients who receive ixekizumab for the treatment of plaque psoriasis, including injection-site reactions, upper respiratory tract infections, nausea, and tinea infections.
“Results from the phase 3 clinical trial program in ankylosing spondylitis show that Taltz helped reduce pain and inflammation and improve function in patients who had never been treated with a bDMARD as well as those who previously failed TNF inhibitors. This approval is an important milestone for patients and physicians who are looking for a much-needed alternative to address symptoms of AS,” said Philip Mease, MD, of Providence St. Joseph Health and the University of Washington, both in Seattle.
(AS), according to a press release from Eli Lilly.
AS is the third indication for ixekizumab, along with moderate to severe plaque psoriasis in adult patients who are candidates for systemic therapy or phototherapy and active psoriatic arthritis in adults.
Approval of the humanized interleukin-17A antagonist was based on results from a pair of randomized, double-blind, placebo-controlled, phase 3 studies involving 657 adult patients with active AS: the COAST-V trial in those naive to biologic disease-modifying antirheumatic drugs (bDMARDs) and the COAST-W trial in those who were intolerant or had inadequate response to tumor necrosis factor (TNF) inhibitors. The primary endpoint in both trials was achievement of 40% improvement in Assessment of Spondyloarthritis International Society criteria (ASAS40) at 16 weeks, compared with placebo.
In COAST-V, 48% of patients who received ixekizumab achieved ASAS40, compared with 18% of controls (P less than .0001). In COAST-W, 25% of patients who received ixekizumab achieved ASAS40 versus 13% of controls (P less than .05). The adverse events reported during both trials were consistent with the safety profile in patients who receive ixekizumab for the treatment of plaque psoriasis, including injection-site reactions, upper respiratory tract infections, nausea, and tinea infections.
“Results from the phase 3 clinical trial program in ankylosing spondylitis show that Taltz helped reduce pain and inflammation and improve function in patients who had never been treated with a bDMARD as well as those who previously failed TNF inhibitors. This approval is an important milestone for patients and physicians who are looking for a much-needed alternative to address symptoms of AS,” said Philip Mease, MD, of Providence St. Joseph Health and the University of Washington, both in Seattle.
Class I recall issued for Sapien 3 balloon
The Food and Drug Administration has issued a class I correction recall for Edward Lifescience’s Sapien 3 balloon system, which is used to deploy transcatheter aortic valve replacements, according to a release from the agency. A class I recall is the most serious the agency issues and indicates risk of severe injury or even death.

Rather than indicate removal of the device from market, this recall provides details on how to use the device cautiously and safely and instructs physicians on proper technique to safely retract the delivery system into the sheath in cases of a suspected balloon burst. Failure to observe these recommendations can result in vascular injury, bleeding, or surgical intervention.
These recommendations and instructions reiterate those issued in an Urgent Field Safety Notice that was provided by Edward Lifesciences on July 9, 2019. Edward Lifesciences could not be reached for comment.
The Food and Drug Administration has issued a class I correction recall for Edward Lifescience’s Sapien 3 balloon system, which is used to deploy transcatheter aortic valve replacements, according to a release from the agency. A class I recall is the most serious the agency issues and indicates risk of severe injury or even death.
Rather than indicate removal of the device from market, this recall provides details on how to use the device cautiously and safely and instructs physicians on proper technique to safely retract the delivery system into the sheath in cases of a suspected balloon burst. Failure to observe these recommendations can result in vascular injury, bleeding, or surgical intervention.
These recommendations and instructions reiterate those issued in an Urgent Field Safety Notice that was provided by Edward Lifesciences on July 9, 2019. Edward Lifesciences could not be reached for comment.
The Food and Drug Administration has issued a class I correction recall for Edward Lifescience’s Sapien 3 balloon system, which is used to deploy transcatheter aortic valve replacements, according to a release from the agency. A class I recall is the most serious the agency issues and indicates risk of severe injury or even death.
Rather than indicate removal of the device from market, this recall provides details on how to use the device cautiously and safely and instructs physicians on proper technique to safely retract the delivery system into the sheath in cases of a suspected balloon burst. Failure to observe these recommendations can result in vascular injury, bleeding, or surgical intervention.
These recommendations and instructions reiterate those issued in an Urgent Field Safety Notice that was provided by Edward Lifesciences on July 9, 2019. Edward Lifesciences could not be reached for comment.
Fifty-one percent of U.S. adolescents fully vaccinated against HPV
according to a report published in Morbidity and Mortality Weekly Report.
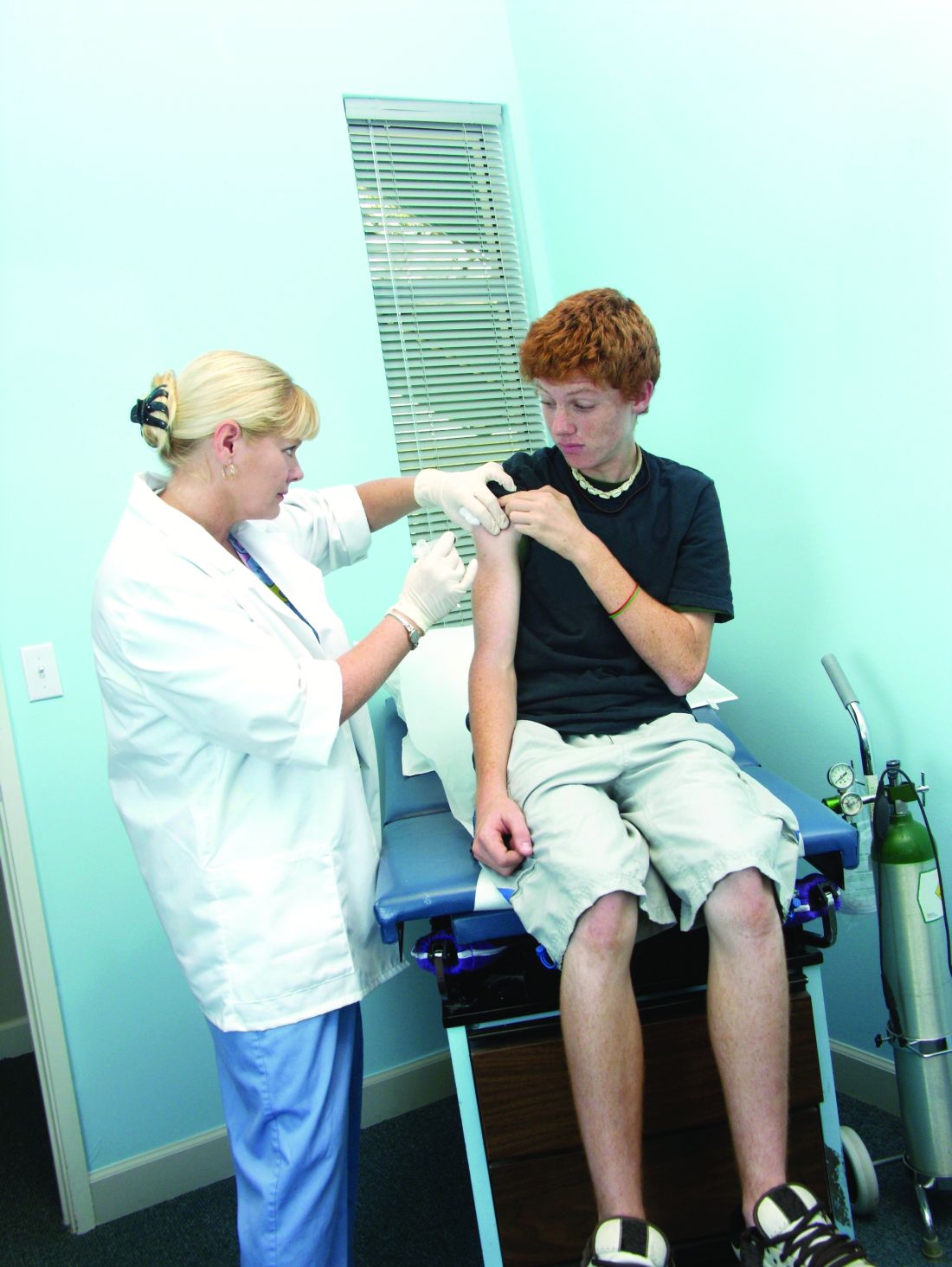
Researchers analyzed data from 18,700 adolescents aged 13-17 years – 48% of whom were female – in the 2018 National Immunization Survey–Teen to discover that 51% of adolescents were up to date with the human papillomavirus (HPV) vaccine, and 68% had received at least one dose of the vaccine.
There was an increase in HPV vaccination coverage from 2017 to 2018, but this was attributable to a 4.4 percentage point increase in males who were up to date, compared with a 0.6 percentage point increase in females.
“Although HPV vaccination coverage improved, increases among all adolescents were modest compared with increases in previous years and were observed only among males,” wrote Tanja Y. Walker of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention, and coauthors.![]()
The number of adolescents who had at least one dose of the quadrivalent meningococcal conjugate (4MenB) vaccine increased by 1.5 percentage points to 86.6%, while among individuals aged 17 years, coverage with two or more doses of 4MenB vaccine increased by 6.5 percentage points to 50.8%. Tdap coverage remained the same at 89% (MMWR 2019;68(33):718-23).
However, the study saw no significant increases in coverage with three or more hepatitis B vaccine doses, two or more MMR vaccine doses, or with one or more varicella vaccine doses in adolescents without a history of varicella disease.
Adolescents with Medicaid had higher HPV vaccination coverage than did adolescents with private health insurance. Uninsured adolescents had lower coverage overall, ranging from 4 percentage points lower for one or more varicella vaccine doses to 19 percentage points lower for two or more 4MenB vaccines, compared with adolescents with private health insurance.
Vaccination rates were lower among adolescents outside metropolitan areas, particularly when it came to being up to date with HPV vaccination, where there was a 15 percentage point difference, and with two or more doses of the quadrivalent meningococcal conjugate vaccine, where there was a 20 percentage point difference.
Provider recommendations to parents were associated with a higher rate of coverage with one or more doses of the HPV vaccine, but the prevalence of provider recommendations varied significantly from state to state. Overall, 78% of parents said they received a provider recommendation for the adolescent HPV vaccine, but that figure was as low as 60% in Mississippi and as high as 91% in Massachusetts.
Parents living in nonmetropolitan areas were less likely to report receiving a provider recommendation than were those in metropolitan principal cities.
“Equipping providers with the tools they need to give strong recommendations that emphasize the importance of HPV vaccination in preventing cancer and effectively address parental concerns is a priority, especially in states where provider recommendations were less commonly reported,” Ms. Walker and associates said.
No conflicts of interest were declared.
according to a report published in Morbidity and Mortality Weekly Report.
Researchers analyzed data from 18,700 adolescents aged 13-17 years – 48% of whom were female – in the 2018 National Immunization Survey–Teen to discover that 51% of adolescents were up to date with the human papillomavirus (HPV) vaccine, and 68% had received at least one dose of the vaccine.
There was an increase in HPV vaccination coverage from 2017 to 2018, but this was attributable to a 4.4 percentage point increase in males who were up to date, compared with a 0.6 percentage point increase in females.
“Although HPV vaccination coverage improved, increases among all adolescents were modest compared with increases in previous years and were observed only among males,” wrote Tanja Y. Walker of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention, and coauthors.![]()
The number of adolescents who had at least one dose of the quadrivalent meningococcal conjugate (4MenB) vaccine increased by 1.5 percentage points to 86.6%, while among individuals aged 17 years, coverage with two or more doses of 4MenB vaccine increased by 6.5 percentage points to 50.8%. Tdap coverage remained the same at 89% (MMWR 2019;68(33):718-23).
However, the study saw no significant increases in coverage with three or more hepatitis B vaccine doses, two or more MMR vaccine doses, or with one or more varicella vaccine doses in adolescents without a history of varicella disease.
Adolescents with Medicaid had higher HPV vaccination coverage than did adolescents with private health insurance. Uninsured adolescents had lower coverage overall, ranging from 4 percentage points lower for one or more varicella vaccine doses to 19 percentage points lower for two or more 4MenB vaccines, compared with adolescents with private health insurance.
Vaccination rates were lower among adolescents outside metropolitan areas, particularly when it came to being up to date with HPV vaccination, where there was a 15 percentage point difference, and with two or more doses of the quadrivalent meningococcal conjugate vaccine, where there was a 20 percentage point difference.
Provider recommendations to parents were associated with a higher rate of coverage with one or more doses of the HPV vaccine, but the prevalence of provider recommendations varied significantly from state to state. Overall, 78% of parents said they received a provider recommendation for the adolescent HPV vaccine, but that figure was as low as 60% in Mississippi and as high as 91% in Massachusetts.
Parents living in nonmetropolitan areas were less likely to report receiving a provider recommendation than were those in metropolitan principal cities.
“Equipping providers with the tools they need to give strong recommendations that emphasize the importance of HPV vaccination in preventing cancer and effectively address parental concerns is a priority, especially in states where provider recommendations were less commonly reported,” Ms. Walker and associates said.
No conflicts of interest were declared.
according to a report published in Morbidity and Mortality Weekly Report.
Researchers analyzed data from 18,700 adolescents aged 13-17 years – 48% of whom were female – in the 2018 National Immunization Survey–Teen to discover that 51% of adolescents were up to date with the human papillomavirus (HPV) vaccine, and 68% had received at least one dose of the vaccine.
There was an increase in HPV vaccination coverage from 2017 to 2018, but this was attributable to a 4.4 percentage point increase in males who were up to date, compared with a 0.6 percentage point increase in females.
“Although HPV vaccination coverage improved, increases among all adolescents were modest compared with increases in previous years and were observed only among males,” wrote Tanja Y. Walker of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention, and coauthors.![]()
The number of adolescents who had at least one dose of the quadrivalent meningococcal conjugate (4MenB) vaccine increased by 1.5 percentage points to 86.6%, while among individuals aged 17 years, coverage with two or more doses of 4MenB vaccine increased by 6.5 percentage points to 50.8%. Tdap coverage remained the same at 89% (MMWR 2019;68(33):718-23).
However, the study saw no significant increases in coverage with three or more hepatitis B vaccine doses, two or more MMR vaccine doses, or with one or more varicella vaccine doses in adolescents without a history of varicella disease.
Adolescents with Medicaid had higher HPV vaccination coverage than did adolescents with private health insurance. Uninsured adolescents had lower coverage overall, ranging from 4 percentage points lower for one or more varicella vaccine doses to 19 percentage points lower for two or more 4MenB vaccines, compared with adolescents with private health insurance.
Vaccination rates were lower among adolescents outside metropolitan areas, particularly when it came to being up to date with HPV vaccination, where there was a 15 percentage point difference, and with two or more doses of the quadrivalent meningococcal conjugate vaccine, where there was a 20 percentage point difference.
Provider recommendations to parents were associated with a higher rate of coverage with one or more doses of the HPV vaccine, but the prevalence of provider recommendations varied significantly from state to state. Overall, 78% of parents said they received a provider recommendation for the adolescent HPV vaccine, but that figure was as low as 60% in Mississippi and as high as 91% in Massachusetts.
Parents living in nonmetropolitan areas were less likely to report receiving a provider recommendation than were those in metropolitan principal cities.
“Equipping providers with the tools they need to give strong recommendations that emphasize the importance of HPV vaccination in preventing cancer and effectively address parental concerns is a priority, especially in states where provider recommendations were less commonly reported,” Ms. Walker and associates said.
No conflicts of interest were declared.
FROM MMWR
Key clinical point: Slightly more than half of adolescents in the United States are fully vaccinated with the HPV vaccine.
Major finding: Rates of full HPV vaccination are 51% among adolescents aged 13-17 years.
Study details: Analysis of data from 18,700 adolescents aged 13-17 years in the 2018 National Immunization Survey–Teen.
Disclosures: No conflicts of interest were declared.
Source: Walker T et al. MMWR 2019 Aug 23;68(33):718-23.
FDA approves Xenleta for community-acquired bacterial pneumonia treatment
The Food and Drug Administration has announced its approval of lefamulin (Xenleta) for the treatment of community-acquired bacterial pneumonia in adults.
Approval was based on results of two clinical trials assessing a total of 1,289 people with community-acquired bacterial pneumonia. In these trials, lefamulin was compared with moxifloxacin with and without linezolid. Patients who received lefamulin had similar rates of treatment success as those taking moxifloxacin alone or moxifloxacin plus linezolid.
The most common adverse reactions associated with lefamulin include diarrhea, nausea, reactions at the injection site, elevated liver enzymes, and vomiting. Patients with prolonged QT interval, patients with arrhythmias, patients receiving treatment with antiarrhythmic agents, and patients receiving other drugs that prolong the QT interval are contraindicated. In addition, because of evidence of fetal harm in animal studies, pregnant women should be advised of potential risks before receiving lefamulin.
“This new drug provides another option for the treatment of patients with community-acquired bacterial pneumonia, a serious disease. For managing this serious disease, it is important for physicians and patients to have treatment options,” Ed Cox, MD, MPH, director of the FDA’s Office of Antimicrobial Products, said in the press release.
The Food and Drug Administration has announced its approval of lefamulin (Xenleta) for the treatment of community-acquired bacterial pneumonia in adults.
Approval was based on results of two clinical trials assessing a total of 1,289 people with community-acquired bacterial pneumonia. In these trials, lefamulin was compared with moxifloxacin with and without linezolid. Patients who received lefamulin had similar rates of treatment success as those taking moxifloxacin alone or moxifloxacin plus linezolid.
The most common adverse reactions associated with lefamulin include diarrhea, nausea, reactions at the injection site, elevated liver enzymes, and vomiting. Patients with prolonged QT interval, patients with arrhythmias, patients receiving treatment with antiarrhythmic agents, and patients receiving other drugs that prolong the QT interval are contraindicated. In addition, because of evidence of fetal harm in animal studies, pregnant women should be advised of potential risks before receiving lefamulin.
“This new drug provides another option for the treatment of patients with community-acquired bacterial pneumonia, a serious disease. For managing this serious disease, it is important for physicians and patients to have treatment options,” Ed Cox, MD, MPH, director of the FDA’s Office of Antimicrobial Products, said in the press release.
The Food and Drug Administration has announced its approval of lefamulin (Xenleta) for the treatment of community-acquired bacterial pneumonia in adults.
Approval was based on results of two clinical trials assessing a total of 1,289 people with community-acquired bacterial pneumonia. In these trials, lefamulin was compared with moxifloxacin with and without linezolid. Patients who received lefamulin had similar rates of treatment success as those taking moxifloxacin alone or moxifloxacin plus linezolid.
The most common adverse reactions associated with lefamulin include diarrhea, nausea, reactions at the injection site, elevated liver enzymes, and vomiting. Patients with prolonged QT interval, patients with arrhythmias, patients receiving treatment with antiarrhythmic agents, and patients receiving other drugs that prolong the QT interval are contraindicated. In addition, because of evidence of fetal harm in animal studies, pregnant women should be advised of potential risks before receiving lefamulin.
“This new drug provides another option for the treatment of patients with community-acquired bacterial pneumonia, a serious disease. For managing this serious disease, it is important for physicians and patients to have treatment options,” Ed Cox, MD, MPH, director of the FDA’s Office of Antimicrobial Products, said in the press release.
FDA approves baroreflex activation for advanced HF
The Food and Drug Administration has approved the Barostim Neo System, an electronic carotid sinus baroreceptor stimulator, for advanced heart failure patients who have a regular heart rhythm, an ejection fraction of 35% or less, and who are not candidates for cardiac resynchronization.

A tiny, unilateral electrode delivers a pulse that decreases sympathetic but increases parasympathetic tone. The effect is that blood vessels relax and production of stress hormones drops. The device is powered by a small generator implanted under the collarbone.
Approval was based on BeAT-HF, a randomized trial with 408 patients on guideline-directed medical therapy with left ventricular ejection fractions at or below 35% and New York Heart Association class III disease.
At 6 months, 125 patients implanted with the device had improved about 14 points more than controls on a quality of life scale, walked about 60 meters farther in 6 minutes, and were more likely to have improved a functional class or two. The benefits corresponded with a drop in the N-terminal of the prohormone brain natriuretic peptide.
Possible complications include infection, low blood pressure, nerve damage, arterial damage, heart failure exacerbation, stroke, and death. Contraindications include certain nervous system disorders, uncontrolled and symptomatic bradycardia, and atherosclerosis or ulcerative carotid plaques near the implant zone, the FDA said.
The system, from CRVx in Minneapolis, received priority review as a breakthrough device. The agency is requiring a phase 4 investigation of its potential to reduce hospitalizations and prolong life.
The Food and Drug Administration has approved the Barostim Neo System, an electronic carotid sinus baroreceptor stimulator, for advanced heart failure patients who have a regular heart rhythm, an ejection fraction of 35% or less, and who are not candidates for cardiac resynchronization.
A tiny, unilateral electrode delivers a pulse that decreases sympathetic but increases parasympathetic tone. The effect is that blood vessels relax and production of stress hormones drops. The device is powered by a small generator implanted under the collarbone.
Approval was based on BeAT-HF, a randomized trial with 408 patients on guideline-directed medical therapy with left ventricular ejection fractions at or below 35% and New York Heart Association class III disease.
At 6 months, 125 patients implanted with the device had improved about 14 points more than controls on a quality of life scale, walked about 60 meters farther in 6 minutes, and were more likely to have improved a functional class or two. The benefits corresponded with a drop in the N-terminal of the prohormone brain natriuretic peptide.
Possible complications include infection, low blood pressure, nerve damage, arterial damage, heart failure exacerbation, stroke, and death. Contraindications include certain nervous system disorders, uncontrolled and symptomatic bradycardia, and atherosclerosis or ulcerative carotid plaques near the implant zone, the FDA said.
The system, from CRVx in Minneapolis, received priority review as a breakthrough device. The agency is requiring a phase 4 investigation of its potential to reduce hospitalizations and prolong life.
The Food and Drug Administration has approved the Barostim Neo System, an electronic carotid sinus baroreceptor stimulator, for advanced heart failure patients who have a regular heart rhythm, an ejection fraction of 35% or less, and who are not candidates for cardiac resynchronization.
A tiny, unilateral electrode delivers a pulse that decreases sympathetic but increases parasympathetic tone. The effect is that blood vessels relax and production of stress hormones drops. The device is powered by a small generator implanted under the collarbone.
Approval was based on BeAT-HF, a randomized trial with 408 patients on guideline-directed medical therapy with left ventricular ejection fractions at or below 35% and New York Heart Association class III disease.
At 6 months, 125 patients implanted with the device had improved about 14 points more than controls on a quality of life scale, walked about 60 meters farther in 6 minutes, and were more likely to have improved a functional class or two. The benefits corresponded with a drop in the N-terminal of the prohormone brain natriuretic peptide.
Possible complications include infection, low blood pressure, nerve damage, arterial damage, heart failure exacerbation, stroke, and death. Contraindications include certain nervous system disorders, uncontrolled and symptomatic bradycardia, and atherosclerosis or ulcerative carotid plaques near the implant zone, the FDA said.
The system, from CRVx in Minneapolis, received priority review as a breakthrough device. The agency is requiring a phase 4 investigation of its potential to reduce hospitalizations and prolong life.
New transfusion guidelines for thalassemia
Fresher blood products are not necessarily better for patients with beta thalassemia, according to a pair of experts.

Red blood cell units stored less than 2 weeks are ideal, but older units are acceptable, and phenotype matching should take priority over unit age, advised Ashutosh Lal, MD, and Elliott Vichinsky, MD, both of UCSF Benioff Children’s Hospital Oakland (Calif.). They discussed these and other recommendations for transfusing patients with thalassemia during a webinar hosted by the Centers for Disease Control and Prevention.
Indications for transfusion
Dr. Lal said patients with beta thalassemia major should be transfused if their hemoglobin is less than 7 g/dL on two occasions 2 weeks apart at baseline, or if their hemoglobin is greater than 7 g/dL and they have symptoms of anemia.
Patients with hemoglobin E beta thalassemia major should be transfused only if they have symptoms of anemia.
“The rationale is that, in beta thalassemia major, it is well established that, once the hemoglobin levels fall below 7 g/dL in young children, there is going to be massive bone marrow expansion, and there will be severe symptoms from anemia,” Dr. Lal said. “But the relationship of hemoglobin with symptoms in E beta thalassemia is less precise.”
The symptoms that should prompt transfusion include slowed growth, skeletal facial changes, splenomegaly, symptomatic or moderate to severe extramedullary hematopoiesis, cerebrovascular events, venous thromboembolism, pulmonary hypertension, osteoporotic fracture, and impaired quality of life in adults.
Dr. Lal said physicians should consider a 6-month trial of transfusions if the indication is unclear. He also noted that red cell antigen genotyping should be performed in all patients who may need transfusions.
Blood products
Dr. Lal said beta thalassemia patients should receive packed red blood cells that are leukoreduced prior to storage. The storage solution can be citrate-phosphate-dextrose solution with adenine (hematocrit 75%) or additive solution (hematocrit 60%).
“It’s important to note that the hematocrit of the two is quite different, and that needs to be inculcated into the decisions on how much volume to transfuse to younger children,” Dr. Lal said.
He noted that units should not be irradiated, as this damages the red cell membrane. And patients with severe allergic reactions should receive washed red blood cells because washing units removes residual donor plasma proteins.
Finally, units should be less than 2 weeks old if possible. Dr. Lal said using fresh units increases the survival of red blood cells post transfusion. However, he and Dr. Vichinsky both stressed that older units are acceptable, and phenotype matching is more important than the age of the unit.
Phenotype matching
Beta thalassemia patients who do not have preexisting alloantibodies or have transient autoantibodies should be matched to Rh and Kell, according to Dr. Lal.
Patients with preexisting alloantibodies should be matched to Rh, Kell, Duffy, Kidd, S, and the specific alloantibody. Patients with persistent autoantibodies should be matched to Rh, Kell, Duffy, Kidd, S, and any alloantibody.
Patients who start transfusions after 5 years of age should be matched to Rh, Kell, Duffy, Kidd, and S. Pregnant patients should be matched to Rh, Kell, Duffy, Kidd, and S, and units should be cytomegalovirus negative.
How to transfuse
Dr. Lal said the pretransfusion hemoglobin target is 10 g/dL, with a range of 9.5-10.5 g/dL in beta thalassemia major and a range of 9.0-10.5 g/dL for E beta thalassemia. A target of 10 g/dL is adequate for most individuals, Dr. Lal said, but he recommends individualization of hemoglobin target for patients with E beta thalassemia.
In general, patients should be transfused every 3 weeks, although 4 weeks is acceptable in younger children and those with hemoglobin E beta thalassemia.
As for the volume of a transfusion, children should receive 4 mL per kg of body weight, per gram increase in hemoglobin desired. Partial units can be used to avoid undertransfusion.
For adults, in general, those with pretransfusion hemoglobin less than 10 g/dL should receive three units, and those with pretransfusion hemoglobin of 10 g/dL or greater should receive two units.
The hemoglobin threshold should be adjusted based on fatigue or bone pain, Dr. Lal said. He also noted that patients with intact spleens have higher transfusion needs.
The rate of transfusion should be 5 mL/kg/hour in children and 200-300 mL/hour in adults, based on tolerance. Patients with impaired cardiac function should receive a reduced blood volume at a reduced rate.
Non–transfusion dependent thalassemia
Dr. Vichinsky discussed recommendations for non–transfusion dependent thalassemia (NTDT), noting that these patients may need transient transfusions to prevent morbidity.

Hemoglobin should not be the sole determinant of transfusion need in NTDT patients, he said. Their well-being – activity level, growth, and skeletal changes – is more important than hemoglobin levels. However, patients with hemoglobin levels less than 7 g/dL often have severe morbidity, and those with levels of 10 g/dL or greater are usually protected from severe morbidity.
Indications for transfusion in NTDT patients include:
- Growth failure.
- Hematopoietic tumors.
- Pulmonary hypertension.
- Silent brain infarcts.
- Skin ulcers.
- Severe bone pain.
- Poor quality of life.
- Frequent hemolytic crises.
- Marked and enlarging spleen.
- Failure of secondary sex development.
- Cosmetic and facial changes.
- Pregnancy.
“There is a risk to transfusing this population,” Dr. Vichinsky said. “They’re older, and when you transfuse them, they can get iron overloaded.”
He added that splenectomized NTDT patients have a high risk of alloimmunization, and the transfusion duration should be serially reevaluated in NTDT patients.
Alpha thalassemia major
For alpha thalassemia major, Dr. Vichinsky discussed the importance of prevention, screening, and fetal therapy. He said couples with a fetus at risk of alpha thalassemia major should be identified early and offered, in addition to termination, the option of early fetal transfusion.
Dr. Vichinsky recommended prenatal testing and monitoring of at-risk pregnancies with ultrasound. If the fetus requires a transfusion, monitoring hemoglobin Barts and hemoglobin A is necessary.
A fetus that requires a transfusion should receive packed red blood cells that are cytomegalovirus negative, are less than 7 days old, have been irradiated, have a hemoglobin mass greater than 75%, and have been optimally cross matched with the mother first.
“These babies appear, with serial transfusions, to survive and have a relatively normal neonatal period,” Dr. Vichinsky said.
He added, however, that postnatal management of alpha thalassemia major involves an aggressive transfusion protocol. These patients should be transfused to a higher hemoglobin level than patients with beta thalassemia – roughly 12 g/dL versus 10 g/dL.
These and Dr. Lal’s recommendations are based on information in the Standards of Care Guidelines for Thalassemia – Oakland 2011, the Thalassemia International Federation Guidelines – 2014, the Thalassemia Management Checklists: United States – 2018, the Thalassemia Western Consortium Consensus: US – 2019, and the International Collaboration for Transfusion Medicine Guidelines – 2019.
Dr. Lal and Dr. Vichinsky did not disclose any conflicts of interest.
Fresher blood products are not necessarily better for patients with beta thalassemia, according to a pair of experts.
Red blood cell units stored less than 2 weeks are ideal, but older units are acceptable, and phenotype matching should take priority over unit age, advised Ashutosh Lal, MD, and Elliott Vichinsky, MD, both of UCSF Benioff Children’s Hospital Oakland (Calif.). They discussed these and other recommendations for transfusing patients with thalassemia during a webinar hosted by the Centers for Disease Control and Prevention.
Indications for transfusion
Dr. Lal said patients with beta thalassemia major should be transfused if their hemoglobin is less than 7 g/dL on two occasions 2 weeks apart at baseline, or if their hemoglobin is greater than 7 g/dL and they have symptoms of anemia.
Patients with hemoglobin E beta thalassemia major should be transfused only if they have symptoms of anemia.
“The rationale is that, in beta thalassemia major, it is well established that, once the hemoglobin levels fall below 7 g/dL in young children, there is going to be massive bone marrow expansion, and there will be severe symptoms from anemia,” Dr. Lal said. “But the relationship of hemoglobin with symptoms in E beta thalassemia is less precise.”
The symptoms that should prompt transfusion include slowed growth, skeletal facial changes, splenomegaly, symptomatic or moderate to severe extramedullary hematopoiesis, cerebrovascular events, venous thromboembolism, pulmonary hypertension, osteoporotic fracture, and impaired quality of life in adults.
Dr. Lal said physicians should consider a 6-month trial of transfusions if the indication is unclear. He also noted that red cell antigen genotyping should be performed in all patients who may need transfusions.
Blood products
Dr. Lal said beta thalassemia patients should receive packed red blood cells that are leukoreduced prior to storage. The storage solution can be citrate-phosphate-dextrose solution with adenine (hematocrit 75%) or additive solution (hematocrit 60%).
“It’s important to note that the hematocrit of the two is quite different, and that needs to be inculcated into the decisions on how much volume to transfuse to younger children,” Dr. Lal said.
He noted that units should not be irradiated, as this damages the red cell membrane. And patients with severe allergic reactions should receive washed red blood cells because washing units removes residual donor plasma proteins.
Finally, units should be less than 2 weeks old if possible. Dr. Lal said using fresh units increases the survival of red blood cells post transfusion. However, he and Dr. Vichinsky both stressed that older units are acceptable, and phenotype matching is more important than the age of the unit.
Phenotype matching
Beta thalassemia patients who do not have preexisting alloantibodies or have transient autoantibodies should be matched to Rh and Kell, according to Dr. Lal.
Patients with preexisting alloantibodies should be matched to Rh, Kell, Duffy, Kidd, S, and the specific alloantibody. Patients with persistent autoantibodies should be matched to Rh, Kell, Duffy, Kidd, S, and any alloantibody.
Patients who start transfusions after 5 years of age should be matched to Rh, Kell, Duffy, Kidd, and S. Pregnant patients should be matched to Rh, Kell, Duffy, Kidd, and S, and units should be cytomegalovirus negative.
How to transfuse
Dr. Lal said the pretransfusion hemoglobin target is 10 g/dL, with a range of 9.5-10.5 g/dL in beta thalassemia major and a range of 9.0-10.5 g/dL for E beta thalassemia. A target of 10 g/dL is adequate for most individuals, Dr. Lal said, but he recommends individualization of hemoglobin target for patients with E beta thalassemia.
In general, patients should be transfused every 3 weeks, although 4 weeks is acceptable in younger children and those with hemoglobin E beta thalassemia.
As for the volume of a transfusion, children should receive 4 mL per kg of body weight, per gram increase in hemoglobin desired. Partial units can be used to avoid undertransfusion.
For adults, in general, those with pretransfusion hemoglobin less than 10 g/dL should receive three units, and those with pretransfusion hemoglobin of 10 g/dL or greater should receive two units.
The hemoglobin threshold should be adjusted based on fatigue or bone pain, Dr. Lal said. He also noted that patients with intact spleens have higher transfusion needs.
The rate of transfusion should be 5 mL/kg/hour in children and 200-300 mL/hour in adults, based on tolerance. Patients with impaired cardiac function should receive a reduced blood volume at a reduced rate.
Non–transfusion dependent thalassemia
Dr. Vichinsky discussed recommendations for non–transfusion dependent thalassemia (NTDT), noting that these patients may need transient transfusions to prevent morbidity.
Hemoglobin should not be the sole determinant of transfusion need in NTDT patients, he said. Their well-being – activity level, growth, and skeletal changes – is more important than hemoglobin levels. However, patients with hemoglobin levels less than 7 g/dL often have severe morbidity, and those with levels of 10 g/dL or greater are usually protected from severe morbidity.
Indications for transfusion in NTDT patients include:
- Growth failure.
- Hematopoietic tumors.
- Pulmonary hypertension.
- Silent brain infarcts.
- Skin ulcers.
- Severe bone pain.
- Poor quality of life.
- Frequent hemolytic crises.
- Marked and enlarging spleen.
- Failure of secondary sex development.
- Cosmetic and facial changes.
- Pregnancy.
“There is a risk to transfusing this population,” Dr. Vichinsky said. “They’re older, and when you transfuse them, they can get iron overloaded.”
He added that splenectomized NTDT patients have a high risk of alloimmunization, and the transfusion duration should be serially reevaluated in NTDT patients.
Alpha thalassemia major
For alpha thalassemia major, Dr. Vichinsky discussed the importance of prevention, screening, and fetal therapy. He said couples with a fetus at risk of alpha thalassemia major should be identified early and offered, in addition to termination, the option of early fetal transfusion.
Dr. Vichinsky recommended prenatal testing and monitoring of at-risk pregnancies with ultrasound. If the fetus requires a transfusion, monitoring hemoglobin Barts and hemoglobin A is necessary.
A fetus that requires a transfusion should receive packed red blood cells that are cytomegalovirus negative, are less than 7 days old, have been irradiated, have a hemoglobin mass greater than 75%, and have been optimally cross matched with the mother first.
“These babies appear, with serial transfusions, to survive and have a relatively normal neonatal period,” Dr. Vichinsky said.
He added, however, that postnatal management of alpha thalassemia major involves an aggressive transfusion protocol. These patients should be transfused to a higher hemoglobin level than patients with beta thalassemia – roughly 12 g/dL versus 10 g/dL.
These and Dr. Lal’s recommendations are based on information in the Standards of Care Guidelines for Thalassemia – Oakland 2011, the Thalassemia International Federation Guidelines – 2014, the Thalassemia Management Checklists: United States – 2018, the Thalassemia Western Consortium Consensus: US – 2019, and the International Collaboration for Transfusion Medicine Guidelines – 2019.
Dr. Lal and Dr. Vichinsky did not disclose any conflicts of interest.
Fresher blood products are not necessarily better for patients with beta thalassemia, according to a pair of experts.
Red blood cell units stored less than 2 weeks are ideal, but older units are acceptable, and phenotype matching should take priority over unit age, advised Ashutosh Lal, MD, and Elliott Vichinsky, MD, both of UCSF Benioff Children’s Hospital Oakland (Calif.). They discussed these and other recommendations for transfusing patients with thalassemia during a webinar hosted by the Centers for Disease Control and Prevention.
Indications for transfusion
Dr. Lal said patients with beta thalassemia major should be transfused if their hemoglobin is less than 7 g/dL on two occasions 2 weeks apart at baseline, or if their hemoglobin is greater than 7 g/dL and they have symptoms of anemia.
Patients with hemoglobin E beta thalassemia major should be transfused only if they have symptoms of anemia.
“The rationale is that, in beta thalassemia major, it is well established that, once the hemoglobin levels fall below 7 g/dL in young children, there is going to be massive bone marrow expansion, and there will be severe symptoms from anemia,” Dr. Lal said. “But the relationship of hemoglobin with symptoms in E beta thalassemia is less precise.”
The symptoms that should prompt transfusion include slowed growth, skeletal facial changes, splenomegaly, symptomatic or moderate to severe extramedullary hematopoiesis, cerebrovascular events, venous thromboembolism, pulmonary hypertension, osteoporotic fracture, and impaired quality of life in adults.
Dr. Lal said physicians should consider a 6-month trial of transfusions if the indication is unclear. He also noted that red cell antigen genotyping should be performed in all patients who may need transfusions.
Blood products
Dr. Lal said beta thalassemia patients should receive packed red blood cells that are leukoreduced prior to storage. The storage solution can be citrate-phosphate-dextrose solution with adenine (hematocrit 75%) or additive solution (hematocrit 60%).
“It’s important to note that the hematocrit of the two is quite different, and that needs to be inculcated into the decisions on how much volume to transfuse to younger children,” Dr. Lal said.
He noted that units should not be irradiated, as this damages the red cell membrane. And patients with severe allergic reactions should receive washed red blood cells because washing units removes residual donor plasma proteins.
Finally, units should be less than 2 weeks old if possible. Dr. Lal said using fresh units increases the survival of red blood cells post transfusion. However, he and Dr. Vichinsky both stressed that older units are acceptable, and phenotype matching is more important than the age of the unit.
Phenotype matching
Beta thalassemia patients who do not have preexisting alloantibodies or have transient autoantibodies should be matched to Rh and Kell, according to Dr. Lal.
Patients with preexisting alloantibodies should be matched to Rh, Kell, Duffy, Kidd, S, and the specific alloantibody. Patients with persistent autoantibodies should be matched to Rh, Kell, Duffy, Kidd, S, and any alloantibody.
Patients who start transfusions after 5 years of age should be matched to Rh, Kell, Duffy, Kidd, and S. Pregnant patients should be matched to Rh, Kell, Duffy, Kidd, and S, and units should be cytomegalovirus negative.
How to transfuse
Dr. Lal said the pretransfusion hemoglobin target is 10 g/dL, with a range of 9.5-10.5 g/dL in beta thalassemia major and a range of 9.0-10.5 g/dL for E beta thalassemia. A target of 10 g/dL is adequate for most individuals, Dr. Lal said, but he recommends individualization of hemoglobin target for patients with E beta thalassemia.
In general, patients should be transfused every 3 weeks, although 4 weeks is acceptable in younger children and those with hemoglobin E beta thalassemia.
As for the volume of a transfusion, children should receive 4 mL per kg of body weight, per gram increase in hemoglobin desired. Partial units can be used to avoid undertransfusion.
For adults, in general, those with pretransfusion hemoglobin less than 10 g/dL should receive three units, and those with pretransfusion hemoglobin of 10 g/dL or greater should receive two units.
The hemoglobin threshold should be adjusted based on fatigue or bone pain, Dr. Lal said. He also noted that patients with intact spleens have higher transfusion needs.
The rate of transfusion should be 5 mL/kg/hour in children and 200-300 mL/hour in adults, based on tolerance. Patients with impaired cardiac function should receive a reduced blood volume at a reduced rate.
Non–transfusion dependent thalassemia
Dr. Vichinsky discussed recommendations for non–transfusion dependent thalassemia (NTDT), noting that these patients may need transient transfusions to prevent morbidity.
Hemoglobin should not be the sole determinant of transfusion need in NTDT patients, he said. Their well-being – activity level, growth, and skeletal changes – is more important than hemoglobin levels. However, patients with hemoglobin levels less than 7 g/dL often have severe morbidity, and those with levels of 10 g/dL or greater are usually protected from severe morbidity.
Indications for transfusion in NTDT patients include:
- Growth failure.
- Hematopoietic tumors.
- Pulmonary hypertension.
- Silent brain infarcts.
- Skin ulcers.
- Severe bone pain.
- Poor quality of life.
- Frequent hemolytic crises.
- Marked and enlarging spleen.
- Failure of secondary sex development.
- Cosmetic and facial changes.
- Pregnancy.
“There is a risk to transfusing this population,” Dr. Vichinsky said. “They’re older, and when you transfuse them, they can get iron overloaded.”
He added that splenectomized NTDT patients have a high risk of alloimmunization, and the transfusion duration should be serially reevaluated in NTDT patients.
Alpha thalassemia major
For alpha thalassemia major, Dr. Vichinsky discussed the importance of prevention, screening, and fetal therapy. He said couples with a fetus at risk of alpha thalassemia major should be identified early and offered, in addition to termination, the option of early fetal transfusion.
Dr. Vichinsky recommended prenatal testing and monitoring of at-risk pregnancies with ultrasound. If the fetus requires a transfusion, monitoring hemoglobin Barts and hemoglobin A is necessary.
A fetus that requires a transfusion should receive packed red blood cells that are cytomegalovirus negative, are less than 7 days old, have been irradiated, have a hemoglobin mass greater than 75%, and have been optimally cross matched with the mother first.
“These babies appear, with serial transfusions, to survive and have a relatively normal neonatal period,” Dr. Vichinsky said.
He added, however, that postnatal management of alpha thalassemia major involves an aggressive transfusion protocol. These patients should be transfused to a higher hemoglobin level than patients with beta thalassemia – roughly 12 g/dL versus 10 g/dL.
These and Dr. Lal’s recommendations are based on information in the Standards of Care Guidelines for Thalassemia – Oakland 2011, the Thalassemia International Federation Guidelines – 2014, the Thalassemia Management Checklists: United States – 2018, the Thalassemia Western Consortium Consensus: US – 2019, and the International Collaboration for Transfusion Medicine Guidelines – 2019.
Dr. Lal and Dr. Vichinsky did not disclose any conflicts of interest.
FDA approves upadacitinib for rheumatoid arthritis
according to a release from its developer. The indication specifies that patients must have shown inadequate response or intolerance to methotrexate.
The approval is based on the SELECT program, which included 4,400 patients across five studies that tested the oral Janus kinase inhibitor in various settings, such as alone or with methotrexate. All primary and secondary endpoints were met in these trials. For example, among patients with inadequate response to methotrexate in one study, 68% of those treated with 15-mg upadacitinib monotherapy achieved 20% improvement in American College of Rheumatology response criteria (ACR20) at week 14 versus 41% of those who had continued on methotrexate. In another study of patients with in adequate response to methotrexate, 71% of those treated with upadacitinib/methotrexate combination therapy achieved ACR20 at week 12 versus 36% of those receiving placebo and methotrexate. Compared with other treatments, better rates of clinical remission and radiographic inhibition were seen with upadacitinib-based therapies, too.
Upadacitinib carries a boxed warning – the most serious warning the FDA issues – for risks of serious infection, malignancy, and thrombosis; these serious risks should be weighed against treatment benefits in any patients with increased risk for these problems or currently experiencing any of them. Concomitant use with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants is not recommended; its use also is not recommended for patients with severe hepatic impairment. The most common adverse reactions are upper respiratory tract infection, nausea, cough, and pyrexia. Live vaccines should be avoided with patients taking this drug, and patients who are breastfeeding should be advised against use of it.
More prescribing information can be found in the drug’s label, which can be found on the FDA’s website.
Upadacitinib is also being evaluated for treatment of other immune-mediated diseases.
according to a release from its developer. The indication specifies that patients must have shown inadequate response or intolerance to methotrexate.
The approval is based on the SELECT program, which included 4,400 patients across five studies that tested the oral Janus kinase inhibitor in various settings, such as alone or with methotrexate. All primary and secondary endpoints were met in these trials. For example, among patients with inadequate response to methotrexate in one study, 68% of those treated with 15-mg upadacitinib monotherapy achieved 20% improvement in American College of Rheumatology response criteria (ACR20) at week 14 versus 41% of those who had continued on methotrexate. In another study of patients with in adequate response to methotrexate, 71% of those treated with upadacitinib/methotrexate combination therapy achieved ACR20 at week 12 versus 36% of those receiving placebo and methotrexate. Compared with other treatments, better rates of clinical remission and radiographic inhibition were seen with upadacitinib-based therapies, too.
Upadacitinib carries a boxed warning – the most serious warning the FDA issues – for risks of serious infection, malignancy, and thrombosis; these serious risks should be weighed against treatment benefits in any patients with increased risk for these problems or currently experiencing any of them. Concomitant use with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants is not recommended; its use also is not recommended for patients with severe hepatic impairment. The most common adverse reactions are upper respiratory tract infection, nausea, cough, and pyrexia. Live vaccines should be avoided with patients taking this drug, and patients who are breastfeeding should be advised against use of it.
More prescribing information can be found in the drug’s label, which can be found on the FDA’s website.
Upadacitinib is also being evaluated for treatment of other immune-mediated diseases.
according to a release from its developer. The indication specifies that patients must have shown inadequate response or intolerance to methotrexate.
The approval is based on the SELECT program, which included 4,400 patients across five studies that tested the oral Janus kinase inhibitor in various settings, such as alone or with methotrexate. All primary and secondary endpoints were met in these trials. For example, among patients with inadequate response to methotrexate in one study, 68% of those treated with 15-mg upadacitinib monotherapy achieved 20% improvement in American College of Rheumatology response criteria (ACR20) at week 14 versus 41% of those who had continued on methotrexate. In another study of patients with in adequate response to methotrexate, 71% of those treated with upadacitinib/methotrexate combination therapy achieved ACR20 at week 12 versus 36% of those receiving placebo and methotrexate. Compared with other treatments, better rates of clinical remission and radiographic inhibition were seen with upadacitinib-based therapies, too.
Upadacitinib carries a boxed warning – the most serious warning the FDA issues – for risks of serious infection, malignancy, and thrombosis; these serious risks should be weighed against treatment benefits in any patients with increased risk for these problems or currently experiencing any of them. Concomitant use with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants is not recommended; its use also is not recommended for patients with severe hepatic impairment. The most common adverse reactions are upper respiratory tract infection, nausea, cough, and pyrexia. Live vaccines should be avoided with patients taking this drug, and patients who are breastfeeding should be advised against use of it.
More prescribing information can be found in the drug’s label, which can be found on the FDA’s website.
Upadacitinib is also being evaluated for treatment of other immune-mediated diseases.
TAVR valves now FDA approved for low-risk patients
The Food and Drug Administration has expanded the indication for the Sapien 3, Sapien 3 Ultra, CoreValve Evolut R, and CoreValve Evolut PRO transcatheter heart valves to include patients with severe aortic valve stenosis at low risk for death or major complications associated with open-heart surgery.
The announcement was based on results of a pair of clinical trials involving patients with severe aortic valve stenosis. In the first, 1,000 patients were randomly sorted to receive either transcatheter aortic valve replacement (TAVR) with the Edwards Lifescience’s Sapien 3 device or open-heart surgery. In the second, 1,468 patients received either Medtronic’s CoreValve Evolut R or CoreValve Evolut PRO or open heart surgery. In both studies, after an average follow-up time of 15-17 months, outcomes such as all-cause mortality and stroke were similar in patients who underwent open heart surgery and who received the transcatheter heart valve.
Serious adverse events associated with transcatheter heart valves include death, stroke, acute kidney injury, heart attack, bleeding, and the need for a permanent pacemaker. Patients who cannot tolerate blood-thinning medication or have an infection in the heart are contraindicated; in addition, the CoreValve devices should not be used in patients sensitive to titanium or nickel. Because the longevity of transcatheter heart valves, compared with open-heart surgery, has not been established, younger patients should discuss options with their health care provider.
“This new approval significantly expands the number of patients that can be treated with this less invasive procedure for aortic valve replacement and follows a thorough review of data demonstrating these devices are safe and effective for this larger population,” Bram Zuckerman, MD, director of the Office of Cardiovascular Devices in the FDA’s Center for Devices and Radiological Health, said in the press release.
Find the full press release on the FDA website.
The Food and Drug Administration has expanded the indication for the Sapien 3, Sapien 3 Ultra, CoreValve Evolut R, and CoreValve Evolut PRO transcatheter heart valves to include patients with severe aortic valve stenosis at low risk for death or major complications associated with open-heart surgery.
The announcement was based on results of a pair of clinical trials involving patients with severe aortic valve stenosis. In the first, 1,000 patients were randomly sorted to receive either transcatheter aortic valve replacement (TAVR) with the Edwards Lifescience’s Sapien 3 device or open-heart surgery. In the second, 1,468 patients received either Medtronic’s CoreValve Evolut R or CoreValve Evolut PRO or open heart surgery. In both studies, after an average follow-up time of 15-17 months, outcomes such as all-cause mortality and stroke were similar in patients who underwent open heart surgery and who received the transcatheter heart valve.
Serious adverse events associated with transcatheter heart valves include death, stroke, acute kidney injury, heart attack, bleeding, and the need for a permanent pacemaker. Patients who cannot tolerate blood-thinning medication or have an infection in the heart are contraindicated; in addition, the CoreValve devices should not be used in patients sensitive to titanium or nickel. Because the longevity of transcatheter heart valves, compared with open-heart surgery, has not been established, younger patients should discuss options with their health care provider.
“This new approval significantly expands the number of patients that can be treated with this less invasive procedure for aortic valve replacement and follows a thorough review of data demonstrating these devices are safe and effective for this larger population,” Bram Zuckerman, MD, director of the Office of Cardiovascular Devices in the FDA’s Center for Devices and Radiological Health, said in the press release.
Find the full press release on the FDA website.
The Food and Drug Administration has expanded the indication for the Sapien 3, Sapien 3 Ultra, CoreValve Evolut R, and CoreValve Evolut PRO transcatheter heart valves to include patients with severe aortic valve stenosis at low risk for death or major complications associated with open-heart surgery.
The announcement was based on results of a pair of clinical trials involving patients with severe aortic valve stenosis. In the first, 1,000 patients were randomly sorted to receive either transcatheter aortic valve replacement (TAVR) with the Edwards Lifescience’s Sapien 3 device or open-heart surgery. In the second, 1,468 patients received either Medtronic’s CoreValve Evolut R or CoreValve Evolut PRO or open heart surgery. In both studies, after an average follow-up time of 15-17 months, outcomes such as all-cause mortality and stroke were similar in patients who underwent open heart surgery and who received the transcatheter heart valve.
Serious adverse events associated with transcatheter heart valves include death, stroke, acute kidney injury, heart attack, bleeding, and the need for a permanent pacemaker. Patients who cannot tolerate blood-thinning medication or have an infection in the heart are contraindicated; in addition, the CoreValve devices should not be used in patients sensitive to titanium or nickel. Because the longevity of transcatheter heart valves, compared with open-heart surgery, has not been established, younger patients should discuss options with their health care provider.
“This new approval significantly expands the number of patients that can be treated with this less invasive procedure for aortic valve replacement and follows a thorough review of data demonstrating these devices are safe and effective for this larger population,” Bram Zuckerman, MD, director of the Office of Cardiovascular Devices in the FDA’s Center for Devices and Radiological Health, said in the press release.
Find the full press release on the FDA website.
FDA approves fedratinib for myelofibrosis
The Food and Drug Administration has approved fedratinib (Inrebic), an oral JAK2/FLT3 inhibitor, to treat myelofibrosis.
Fedratinib is approved to treat adults with intermediate-2 or high-risk primary or secondary (post–polycythemia vera or post–essential thrombocythemia) myelofibrosis.
The prescribing information for fedratinib includes a boxed warning detailing the risk of serious and fatal encephalopathy, including Wernicke’s.
The encephalopathy risk prompted Sanofi to stop developing fedratinib in 2013. The FDA placed a clinical hold on all trials of fedratinib after potential cases of Wernicke’s encephalopathy were observed in eight patients.
The FDA lifted the clinical hold in 2017, and Celgene Corporation decided to develop fedratinib when the company acquired Impact Biomedicines in 2018.
In the phase 3 JAKARTA trial, fedratinib significantly reduced splenomegaly and symptom burden in patients with primary or secondary myelofibrosis (JAMA Oncol. 2015 Aug;1[5]:643-51). In the phase 2 JAKARTA2 trial, fedratinib produced responses in myelofibrosis patients previously treated with ruxolitinib (Lancet Haematol. 2017 Jul;4[7]:e317-e324).
Fedratinib received orphan drug designation from the FDA, and the application for fedratinib received priority review.
The FDA granted approval of fedratinib to Impact Biomedicines, a wholly owned subsidiary of Celgene.
The Food and Drug Administration has approved fedratinib (Inrebic), an oral JAK2/FLT3 inhibitor, to treat myelofibrosis.
Fedratinib is approved to treat adults with intermediate-2 or high-risk primary or secondary (post–polycythemia vera or post–essential thrombocythemia) myelofibrosis.
The prescribing information for fedratinib includes a boxed warning detailing the risk of serious and fatal encephalopathy, including Wernicke’s.
The encephalopathy risk prompted Sanofi to stop developing fedratinib in 2013. The FDA placed a clinical hold on all trials of fedratinib after potential cases of Wernicke’s encephalopathy were observed in eight patients.
The FDA lifted the clinical hold in 2017, and Celgene Corporation decided to develop fedratinib when the company acquired Impact Biomedicines in 2018.
In the phase 3 JAKARTA trial, fedratinib significantly reduced splenomegaly and symptom burden in patients with primary or secondary myelofibrosis (JAMA Oncol. 2015 Aug;1[5]:643-51). In the phase 2 JAKARTA2 trial, fedratinib produced responses in myelofibrosis patients previously treated with ruxolitinib (Lancet Haematol. 2017 Jul;4[7]:e317-e324).
Fedratinib received orphan drug designation from the FDA, and the application for fedratinib received priority review.
The FDA granted approval of fedratinib to Impact Biomedicines, a wholly owned subsidiary of Celgene.
The Food and Drug Administration has approved fedratinib (Inrebic), an oral JAK2/FLT3 inhibitor, to treat myelofibrosis.
Fedratinib is approved to treat adults with intermediate-2 or high-risk primary or secondary (post–polycythemia vera or post–essential thrombocythemia) myelofibrosis.
The prescribing information for fedratinib includes a boxed warning detailing the risk of serious and fatal encephalopathy, including Wernicke’s.
The encephalopathy risk prompted Sanofi to stop developing fedratinib in 2013. The FDA placed a clinical hold on all trials of fedratinib after potential cases of Wernicke’s encephalopathy were observed in eight patients.
The FDA lifted the clinical hold in 2017, and Celgene Corporation decided to develop fedratinib when the company acquired Impact Biomedicines in 2018.
In the phase 3 JAKARTA trial, fedratinib significantly reduced splenomegaly and symptom burden in patients with primary or secondary myelofibrosis (JAMA Oncol. 2015 Aug;1[5]:643-51). In the phase 2 JAKARTA2 trial, fedratinib produced responses in myelofibrosis patients previously treated with ruxolitinib (Lancet Haematol. 2017 Jul;4[7]:e317-e324).
Fedratinib received orphan drug designation from the FDA, and the application for fedratinib received priority review.
The FDA granted approval of fedratinib to Impact Biomedicines, a wholly owned subsidiary of Celgene.
FDA takes another swing at updating cigarette pack warnings
illustrating the harms of smoking, but this could be subjected to legal challenge.

Several years ago, tobacco companies filed a lawsuit, which ultimately shut down a similar proposal.
The warnings focus on lesser-known complications – including diabetes, cataracts, gangrene, stroke, bladder cancer, erectile dysfunction, and obstructive pulmonary disease – and would take up the top half of the front and back of cigarette packs, and at least the top 20% of print advertisements. Each pack and ad would be required to carry 1 of the 13 proposed warnings, according to the announcement.
The approach would be similar to, but not as aggressive as Canada’s. For years, cigarettes packs sold in Canada have included disturbing photographs of diseased lungs, rotted teeth, and dying patients. The lasting impact of such imagery has been demonstrated in the literature (for example, Am J Prev Med. 2007 Mar;32[3]:202-9).
The new proposal is the FDA’s second attempt to enact something comparable in the United States, after being directed to do so by the Tobacco Control Act of 2009.
The first effort to add strong, illustrated warnings to cigarette packs was widely backed by medical groups, but challenged in the courts by R.J. Reynolds and other tobacco companies, and blocked on appeal in 2012 as an abridgment of commercial free speech. The federal government dropped the case in 2013.
The American Lung Association and other public health groups subsequently sued the FDA in 2016 to enact the Tobacco Act mandate. Subsequently, a federal judge ordered the agency to publish a new rule by August 2019, and issue a final rule in March 2020.
This time around, the FDA “took the necessary time to get these new proposed warnings right ... based on – and within the limits of – both science and the law,” the agency said. The new images, though graphic, are less disturbing than those used in Canada and the agency’s previous proposals, which included an apparent corpse with a sternotomy. The 1-800-Quit-Now cessation hotline number, which was a sticking point in the 2012 ruling, has also been dropped.
When asked about the new efforts, R.J. Reynolds spokesperson Kaelan Hollon said, “We are carefully reviewing FDA’s latest proposal for graphic warnings on cigarettes. We firmly support public awareness of the harms of smoking cigarettes, but the manner in which those messages are delivered to the public cannot run afoul of the First Amendment protections that apply to all speakers, including cigarette manufacturers.”
Warnings on U.S. cigarettes haven’t changed since 1984, when the risks of lung cancer, heart disease, emphysema, and pregnancy complications were added to the side of cigarette packs. With time, the FDA said the surgeon general’s warnings have become “virtually invisible” to consumers.
The American Lung Association, American Academy of Pediatrics, and other plaintiffs in the 2016 suit called the new proposal a “dramatic improvement” over the current situation and “long overdue” in a joint statement on Aug. 15.
Although rates have declined substantially in recent decades, about 34.3 million U.S. adults and almost 1.4 million teenagers still smoke. The habit kills about a half million Americans every year, at a health cost of more than $300 billion, the FDA said.
Comments on the proposed rule are being accepted through Oct. 15. The agency is open to suggestions for alternative text and images.
illustrating the harms of smoking, but this could be subjected to legal challenge.
Several years ago, tobacco companies filed a lawsuit, which ultimately shut down a similar proposal.
The warnings focus on lesser-known complications – including diabetes, cataracts, gangrene, stroke, bladder cancer, erectile dysfunction, and obstructive pulmonary disease – and would take up the top half of the front and back of cigarette packs, and at least the top 20% of print advertisements. Each pack and ad would be required to carry 1 of the 13 proposed warnings, according to the announcement.
The approach would be similar to, but not as aggressive as Canada’s. For years, cigarettes packs sold in Canada have included disturbing photographs of diseased lungs, rotted teeth, and dying patients. The lasting impact of such imagery has been demonstrated in the literature (for example, Am J Prev Med. 2007 Mar;32[3]:202-9).
The new proposal is the FDA’s second attempt to enact something comparable in the United States, after being directed to do so by the Tobacco Control Act of 2009.
The first effort to add strong, illustrated warnings to cigarette packs was widely backed by medical groups, but challenged in the courts by R.J. Reynolds and other tobacco companies, and blocked on appeal in 2012 as an abridgment of commercial free speech. The federal government dropped the case in 2013.
The American Lung Association and other public health groups subsequently sued the FDA in 2016 to enact the Tobacco Act mandate. Subsequently, a federal judge ordered the agency to publish a new rule by August 2019, and issue a final rule in March 2020.
This time around, the FDA “took the necessary time to get these new proposed warnings right ... based on – and within the limits of – both science and the law,” the agency said. The new images, though graphic, are less disturbing than those used in Canada and the agency’s previous proposals, which included an apparent corpse with a sternotomy. The 1-800-Quit-Now cessation hotline number, which was a sticking point in the 2012 ruling, has also been dropped.
When asked about the new efforts, R.J. Reynolds spokesperson Kaelan Hollon said, “We are carefully reviewing FDA’s latest proposal for graphic warnings on cigarettes. We firmly support public awareness of the harms of smoking cigarettes, but the manner in which those messages are delivered to the public cannot run afoul of the First Amendment protections that apply to all speakers, including cigarette manufacturers.”
Warnings on U.S. cigarettes haven’t changed since 1984, when the risks of lung cancer, heart disease, emphysema, and pregnancy complications were added to the side of cigarette packs. With time, the FDA said the surgeon general’s warnings have become “virtually invisible” to consumers.
The American Lung Association, American Academy of Pediatrics, and other plaintiffs in the 2016 suit called the new proposal a “dramatic improvement” over the current situation and “long overdue” in a joint statement on Aug. 15.
Although rates have declined substantially in recent decades, about 34.3 million U.S. adults and almost 1.4 million teenagers still smoke. The habit kills about a half million Americans every year, at a health cost of more than $300 billion, the FDA said.
Comments on the proposed rule are being accepted through Oct. 15. The agency is open to suggestions for alternative text and images.
illustrating the harms of smoking, but this could be subjected to legal challenge.
Several years ago, tobacco companies filed a lawsuit, which ultimately shut down a similar proposal.
The warnings focus on lesser-known complications – including diabetes, cataracts, gangrene, stroke, bladder cancer, erectile dysfunction, and obstructive pulmonary disease – and would take up the top half of the front and back of cigarette packs, and at least the top 20% of print advertisements. Each pack and ad would be required to carry 1 of the 13 proposed warnings, according to the announcement.
The approach would be similar to, but not as aggressive as Canada’s. For years, cigarettes packs sold in Canada have included disturbing photographs of diseased lungs, rotted teeth, and dying patients. The lasting impact of such imagery has been demonstrated in the literature (for example, Am J Prev Med. 2007 Mar;32[3]:202-9).
The new proposal is the FDA’s second attempt to enact something comparable in the United States, after being directed to do so by the Tobacco Control Act of 2009.
The first effort to add strong, illustrated warnings to cigarette packs was widely backed by medical groups, but challenged in the courts by R.J. Reynolds and other tobacco companies, and blocked on appeal in 2012 as an abridgment of commercial free speech. The federal government dropped the case in 2013.
The American Lung Association and other public health groups subsequently sued the FDA in 2016 to enact the Tobacco Act mandate. Subsequently, a federal judge ordered the agency to publish a new rule by August 2019, and issue a final rule in March 2020.
This time around, the FDA “took the necessary time to get these new proposed warnings right ... based on – and within the limits of – both science and the law,” the agency said. The new images, though graphic, are less disturbing than those used in Canada and the agency’s previous proposals, which included an apparent corpse with a sternotomy. The 1-800-Quit-Now cessation hotline number, which was a sticking point in the 2012 ruling, has also been dropped.
When asked about the new efforts, R.J. Reynolds spokesperson Kaelan Hollon said, “We are carefully reviewing FDA’s latest proposal for graphic warnings on cigarettes. We firmly support public awareness of the harms of smoking cigarettes, but the manner in which those messages are delivered to the public cannot run afoul of the First Amendment protections that apply to all speakers, including cigarette manufacturers.”
Warnings on U.S. cigarettes haven’t changed since 1984, when the risks of lung cancer, heart disease, emphysema, and pregnancy complications were added to the side of cigarette packs. With time, the FDA said the surgeon general’s warnings have become “virtually invisible” to consumers.
The American Lung Association, American Academy of Pediatrics, and other plaintiffs in the 2016 suit called the new proposal a “dramatic improvement” over the current situation and “long overdue” in a joint statement on Aug. 15.
Although rates have declined substantially in recent decades, about 34.3 million U.S. adults and almost 1.4 million teenagers still smoke. The habit kills about a half million Americans every year, at a health cost of more than $300 billion, the FDA said.
Comments on the proposed rule are being accepted through Oct. 15. The agency is open to suggestions for alternative text and images.