User login
Number of U.S. Zika-infected pregnancies tops 5,100
Almost 200 cases of pregnant women with Zika virus infection were reported in the United States during the 2 weeks ending March 28, with the number split evenly between the territories and the 50 states and Washington, D.C., according to the Centers for Disease Control and Prevention.
These latest 197 cases – 98 in the territories and 99 in the states/D.C. – bring the U.S. total since the beginning of 2016 to 5,177 pregnant women with laboratory evidence of Zika virus infection: 3,461 in the U.S. territories and 1,716 in the states/D.C., the CDC reported on April 6.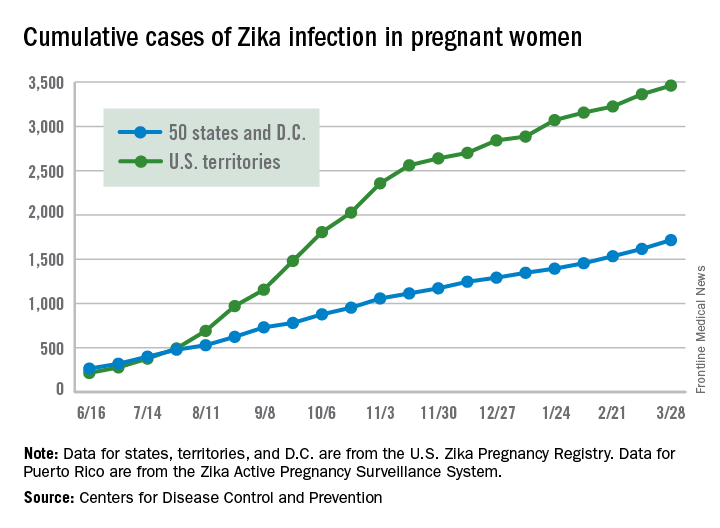
Since Jan. 1, 2015, a total of 41,701 cases of Zika virus infection have been reported among all Americans: 5,197 in the states/D.C. and 36,504 in the territories. Almost all of the territorial cases (97%) have occurred in Puerto Rico, while Florida (21%), New York (20%), and California (9%) together have accounted for half of the cases in the states/D.C., the CDC said.
These are not real-time data and reflect only pregnancy outcomes for women with any laboratory evidence of possible Zika virus infection, although it is not known if Zika virus was the cause of the poor outcomes. Zika-related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, or termination with evidence of birth defects.
Almost 200 cases of pregnant women with Zika virus infection were reported in the United States during the 2 weeks ending March 28, with the number split evenly between the territories and the 50 states and Washington, D.C., according to the Centers for Disease Control and Prevention.
These latest 197 cases – 98 in the territories and 99 in the states/D.C. – bring the U.S. total since the beginning of 2016 to 5,177 pregnant women with laboratory evidence of Zika virus infection: 3,461 in the U.S. territories and 1,716 in the states/D.C., the CDC reported on April 6.
Since Jan. 1, 2015, a total of 41,701 cases of Zika virus infection have been reported among all Americans: 5,197 in the states/D.C. and 36,504 in the territories. Almost all of the territorial cases (97%) have occurred in Puerto Rico, while Florida (21%), New York (20%), and California (9%) together have accounted for half of the cases in the states/D.C., the CDC said.
These are not real-time data and reflect only pregnancy outcomes for women with any laboratory evidence of possible Zika virus infection, although it is not known if Zika virus was the cause of the poor outcomes. Zika-related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, or termination with evidence of birth defects.
Almost 200 cases of pregnant women with Zika virus infection were reported in the United States during the 2 weeks ending March 28, with the number split evenly between the territories and the 50 states and Washington, D.C., according to the Centers for Disease Control and Prevention.
These latest 197 cases – 98 in the territories and 99 in the states/D.C. – bring the U.S. total since the beginning of 2016 to 5,177 pregnant women with laboratory evidence of Zika virus infection: 3,461 in the U.S. territories and 1,716 in the states/D.C., the CDC reported on April 6.
Since Jan. 1, 2015, a total of 41,701 cases of Zika virus infection have been reported among all Americans: 5,197 in the states/D.C. and 36,504 in the territories. Almost all of the territorial cases (97%) have occurred in Puerto Rico, while Florida (21%), New York (20%), and California (9%) together have accounted for half of the cases in the states/D.C., the CDC said.
These are not real-time data and reflect only pregnancy outcomes for women with any laboratory evidence of possible Zika virus infection, although it is not known if Zika virus was the cause of the poor outcomes. Zika-related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, or termination with evidence of birth defects.
FDA approves first home genetic health risk test
The Food and Drug Administration authorized 23andMe’s Personal Genome Service Genetic Health Risk (GHR) test, the first direct-to-consumer genetic screening test, according to a press release on Thursday, April 6.
FDA officials expect the product, which tests individuals for possible genetic predisposition for 10 diseases including Parkinson’s, late-onset Alzheimer’s, celiac disease, and hereditary hemochromatosis, to spur patients to consult with their physicians and make more informed lifestyle decisions.![]()
“Consumers can now have direct access to certain genetic risk information,” said Jeffrey Shuren, MD, director of the FDA’s Center for Devices and Radiological Health in the release. “But it is important that people understand that genetic risk is just one piece of the bigger puzzle, it does not mean they will or won’t ultimately develop a disease.”
The FDA has exempted all further GHR tests developed by 23andMe from premarket review, noting future GHR tests developed by other makers, excluding those used for diagnostic purposes, may also achieve this exemption after submitting their first premarket review.
For the full details, see the original announcement.
ezimmerman@frontlinemedcom.com
On Twitter @EAZTweets
The Food and Drug Administration authorized 23andMe’s Personal Genome Service Genetic Health Risk (GHR) test, the first direct-to-consumer genetic screening test, according to a press release on Thursday, April 6.
FDA officials expect the product, which tests individuals for possible genetic predisposition for 10 diseases including Parkinson’s, late-onset Alzheimer’s, celiac disease, and hereditary hemochromatosis, to spur patients to consult with their physicians and make more informed lifestyle decisions.![]()
“Consumers can now have direct access to certain genetic risk information,” said Jeffrey Shuren, MD, director of the FDA’s Center for Devices and Radiological Health in the release. “But it is important that people understand that genetic risk is just one piece of the bigger puzzle, it does not mean they will or won’t ultimately develop a disease.”
The FDA has exempted all further GHR tests developed by 23andMe from premarket review, noting future GHR tests developed by other makers, excluding those used for diagnostic purposes, may also achieve this exemption after submitting their first premarket review.
For the full details, see the original announcement.
ezimmerman@frontlinemedcom.com
On Twitter @EAZTweets
The Food and Drug Administration authorized 23andMe’s Personal Genome Service Genetic Health Risk (GHR) test, the first direct-to-consumer genetic screening test, according to a press release on Thursday, April 6.
FDA officials expect the product, which tests individuals for possible genetic predisposition for 10 diseases including Parkinson’s, late-onset Alzheimer’s, celiac disease, and hereditary hemochromatosis, to spur patients to consult with their physicians and make more informed lifestyle decisions.![]()
“Consumers can now have direct access to certain genetic risk information,” said Jeffrey Shuren, MD, director of the FDA’s Center for Devices and Radiological Health in the release. “But it is important that people understand that genetic risk is just one piece of the bigger puzzle, it does not mean they will or won’t ultimately develop a disease.”
The FDA has exempted all further GHR tests developed by 23andMe from premarket review, noting future GHR tests developed by other makers, excluding those used for diagnostic purposes, may also achieve this exemption after submitting their first premarket review.
For the full details, see the original announcement.
ezimmerman@frontlinemedcom.com
On Twitter @EAZTweets
Safety of N9-GP for hemophilia B needs further study, FDA committee agrees
Concerns about a possible safety issue with the investigational glycoPEGylated factor IX product nonacog beta pegol (N9-GP) for the treatment of hemophilia B left members of the Blood Products Advisory Committee of the Food and Drug Administration divided during an April 4 committee meeting about whether additional study should take place prior to FDA approval of a Biologics Licensing Application or in the postmarketing setting.
The committee was not asked to vote on a recommendation for approval of N9-GP. Committee members agreed that if N9-GP is approved, standardized postmarketing monitoring would be needed, particularly in very young and very old patients.

As a result, the FDA asked the advisory committee to consider the clinical significance of the preclinical findings, the nature and level of any safety concerns in various populations, the sufficiency of evidence from toxicology and clinical studies for intermittent and chronic use, clinical or laboratory assessments that might help ensure patient safety, and recommendations for additional studies to support the safety of the product.
Novo Nordisk is specifically seeking FDA marketing approval of N9-GP, which is administered weekly, for control and prevention of bleeding episodes, perioperative management, and routine prophylaxis in adults and children. The company submitted evidence for these indications from three phase III trials and a phase III extension study in adults and children.
“The Office of Tissues and Advanced Therapies – OTAT – appreciates that hemophilia B is a serious disease and we need improved therapies to benefit patients. We also appreciate that this is a rare disease, which limits the availability of data to address issues of safety and effectiveness,” Wilson Bryan, MD, director of OTAT, which is part of the FDA’s Center for Biologics Evaluation and Research, told the advisory committee.
The committee then heard from Novo Nordisk representatives who spoke about the unmet public health need for hemophilia B treatments, and N9-GP clinical efficacy, long-term safety, and risk/benefit analyses.
Shawn Hoskin, senior director of regulatory affairs for Novo Nordisk, noted that with weekly dosing of N9-GP, no adverse effects were seen in preclinical studies at doses up to 42 times the human dose. Further, no adverse effects were reported in clinical studies, in which high levels of factor IX were achieved.
“Our trials demonstrated that the higher factor IX levels achieved with N9-GP lead to better outcomes for patients, including reduced annual bleed rate, reduction in the number of spontaneous bleeds, and resolution of target joints,” he said.
Stephanie Seremetis, MD, chief medical officer and corporate vice president for hemophilia at Novo Nordisk, said the company has proposed a postapproval monitoring plan and safety study.
The advisory committee also heard from patients invited by Novo Nordisk to share their experiences, and from Maria Lehtinen, PhD, of Boston Children’s Hospital, who was invited to speak about choroid plexus biology.
The patients agreed that there is an urgent need and desire for effective, long-acting treatments. Ben Shuldiner, for example, said he was part of the clinical trials for N9-GP, has never had as much success sticking to a treatment regimen, and “is in much better shape, much less pain.”
“The [hemophilia] community needs choice,” said Mr. Shuldiner, a professor at Hunter College in New York, and an activist. He stressed that what works for one patient doesn’t necessarily work for another.
Committee members didn’t question the efficacy or value of the long-acting product, but did express concern about the unknown effects of PEG accumulation. One member questioned whether it might be feasible to restrict licensing to children over age 6 years and to adults under age 65 years pending additional study. Other members said that approach would restrict treatment for patients who might benefit the most from N9-GP.
Meera B. Chitlur, MD, noted that most children with hemophilia B are treated prophylactically by age 1, or at least by the time they are walking, and suggested it might be better to allow use of the product in those who need it, while collecting data going forward.
“Only over the last couple years have our patients finally had the opportunity to have something that has substantially changed how they are managed or what they can do. This class of drugs is one that has made it possible for these patients to lead a better life, so I think it is really important for us,” said Dr. Chitlur of Wayne State University and Children’s Hospital of Michigan, Detroit.
“The youngest children will probably benefit the most. Yes, there are concerns ... but at the same time I think I haven’t heard anything today, or read anything, that has made me want to say it is not safe for the patients that I’m going to take care of,” she added, noting that informed consent is possible and important.
Dr. Chitlur said she completely agrees with the need for systematic data collection, and feels there are already mechanisms in place to achieve that goal.
“I feel comfortable taking this to my patients and saying that here is another option for you,” she said.
Laura Manuelidis, MD, of Yale University, New Haven, Conn., a temporary voting member of the committee, said she was “less sanguine,” about immediate approval. Additional studies of the effects of PEG accumulation in animals are feasible, she said, and could be performed rapidly before approving treatment in children under age 2 years.
Michael Dobbs, MD, of the University of Kentucky, Lexington, also a temporary voting member, agreed it would be valuable to test available cerebrospinal fluid of animals from the preclinical studies. He had no recommendation for additional studies, but agreed on the importance of extensive postmarketing surveillance. He added that patients who undergo CSF testing or magnetic resonance imaging for other clinical reasons should be evaluated for the possible effects of PEG accumulation.
“I think we should follow cognitive outcomes in the postmarket data, probably requiring more neurocognitive data in pediatrics with some validated, standardized tests. It would be reasonable to require a full neurological exam pre- and post-[treatment], especially in those at fixed developmental states,” he said.
It would also be reasonable to monitor children for signs and symptoms of hydrocephalus and for papilledema and other visual disturbances, he added.
“But again, most of all ... whatever we really do recommend, we need standardization, validation – to do this the same [way] for all of the patients,” he said.
Dr. Bryan, of OTAT, said the input from members and guests will be considered as the FDA proceeds with its review of the Biologics Licensing Application for N9-GP. The FDA is not bound by the committee’s guidance.
All members of the advisory committee have been screened and found to be in compliance with respect to potential conflicts of interest. No conflict of interest waivers were issued. Patients who spoke in favor of N9-GP received travel and/or other support from Novo Nordisk.
Concerns about a possible safety issue with the investigational glycoPEGylated factor IX product nonacog beta pegol (N9-GP) for the treatment of hemophilia B left members of the Blood Products Advisory Committee of the Food and Drug Administration divided during an April 4 committee meeting about whether additional study should take place prior to FDA approval of a Biologics Licensing Application or in the postmarketing setting.
The committee was not asked to vote on a recommendation for approval of N9-GP. Committee members agreed that if N9-GP is approved, standardized postmarketing monitoring would be needed, particularly in very young and very old patients.
As a result, the FDA asked the advisory committee to consider the clinical significance of the preclinical findings, the nature and level of any safety concerns in various populations, the sufficiency of evidence from toxicology and clinical studies for intermittent and chronic use, clinical or laboratory assessments that might help ensure patient safety, and recommendations for additional studies to support the safety of the product.
Novo Nordisk is specifically seeking FDA marketing approval of N9-GP, which is administered weekly, for control and prevention of bleeding episodes, perioperative management, and routine prophylaxis in adults and children. The company submitted evidence for these indications from three phase III trials and a phase III extension study in adults and children.
“The Office of Tissues and Advanced Therapies – OTAT – appreciates that hemophilia B is a serious disease and we need improved therapies to benefit patients. We also appreciate that this is a rare disease, which limits the availability of data to address issues of safety and effectiveness,” Wilson Bryan, MD, director of OTAT, which is part of the FDA’s Center for Biologics Evaluation and Research, told the advisory committee.
The committee then heard from Novo Nordisk representatives who spoke about the unmet public health need for hemophilia B treatments, and N9-GP clinical efficacy, long-term safety, and risk/benefit analyses.
Shawn Hoskin, senior director of regulatory affairs for Novo Nordisk, noted that with weekly dosing of N9-GP, no adverse effects were seen in preclinical studies at doses up to 42 times the human dose. Further, no adverse effects were reported in clinical studies, in which high levels of factor IX were achieved.
“Our trials demonstrated that the higher factor IX levels achieved with N9-GP lead to better outcomes for patients, including reduced annual bleed rate, reduction in the number of spontaneous bleeds, and resolution of target joints,” he said.
Stephanie Seremetis, MD, chief medical officer and corporate vice president for hemophilia at Novo Nordisk, said the company has proposed a postapproval monitoring plan and safety study.
The advisory committee also heard from patients invited by Novo Nordisk to share their experiences, and from Maria Lehtinen, PhD, of Boston Children’s Hospital, who was invited to speak about choroid plexus biology.
The patients agreed that there is an urgent need and desire for effective, long-acting treatments. Ben Shuldiner, for example, said he was part of the clinical trials for N9-GP, has never had as much success sticking to a treatment regimen, and “is in much better shape, much less pain.”
“The [hemophilia] community needs choice,” said Mr. Shuldiner, a professor at Hunter College in New York, and an activist. He stressed that what works for one patient doesn’t necessarily work for another.
Committee members didn’t question the efficacy or value of the long-acting product, but did express concern about the unknown effects of PEG accumulation. One member questioned whether it might be feasible to restrict licensing to children over age 6 years and to adults under age 65 years pending additional study. Other members said that approach would restrict treatment for patients who might benefit the most from N9-GP.
Meera B. Chitlur, MD, noted that most children with hemophilia B are treated prophylactically by age 1, or at least by the time they are walking, and suggested it might be better to allow use of the product in those who need it, while collecting data going forward.
“Only over the last couple years have our patients finally had the opportunity to have something that has substantially changed how they are managed or what they can do. This class of drugs is one that has made it possible for these patients to lead a better life, so I think it is really important for us,” said Dr. Chitlur of Wayne State University and Children’s Hospital of Michigan, Detroit.
“The youngest children will probably benefit the most. Yes, there are concerns ... but at the same time I think I haven’t heard anything today, or read anything, that has made me want to say it is not safe for the patients that I’m going to take care of,” she added, noting that informed consent is possible and important.
Dr. Chitlur said she completely agrees with the need for systematic data collection, and feels there are already mechanisms in place to achieve that goal.
“I feel comfortable taking this to my patients and saying that here is another option for you,” she said.
Laura Manuelidis, MD, of Yale University, New Haven, Conn., a temporary voting member of the committee, said she was “less sanguine,” about immediate approval. Additional studies of the effects of PEG accumulation in animals are feasible, she said, and could be performed rapidly before approving treatment in children under age 2 years.
Michael Dobbs, MD, of the University of Kentucky, Lexington, also a temporary voting member, agreed it would be valuable to test available cerebrospinal fluid of animals from the preclinical studies. He had no recommendation for additional studies, but agreed on the importance of extensive postmarketing surveillance. He added that patients who undergo CSF testing or magnetic resonance imaging for other clinical reasons should be evaluated for the possible effects of PEG accumulation.
“I think we should follow cognitive outcomes in the postmarket data, probably requiring more neurocognitive data in pediatrics with some validated, standardized tests. It would be reasonable to require a full neurological exam pre- and post-[treatment], especially in those at fixed developmental states,” he said.
It would also be reasonable to monitor children for signs and symptoms of hydrocephalus and for papilledema and other visual disturbances, he added.
“But again, most of all ... whatever we really do recommend, we need standardization, validation – to do this the same [way] for all of the patients,” he said.
Dr. Bryan, of OTAT, said the input from members and guests will be considered as the FDA proceeds with its review of the Biologics Licensing Application for N9-GP. The FDA is not bound by the committee’s guidance.
All members of the advisory committee have been screened and found to be in compliance with respect to potential conflicts of interest. No conflict of interest waivers were issued. Patients who spoke in favor of N9-GP received travel and/or other support from Novo Nordisk.
Concerns about a possible safety issue with the investigational glycoPEGylated factor IX product nonacog beta pegol (N9-GP) for the treatment of hemophilia B left members of the Blood Products Advisory Committee of the Food and Drug Administration divided during an April 4 committee meeting about whether additional study should take place prior to FDA approval of a Biologics Licensing Application or in the postmarketing setting.
The committee was not asked to vote on a recommendation for approval of N9-GP. Committee members agreed that if N9-GP is approved, standardized postmarketing monitoring would be needed, particularly in very young and very old patients.
As a result, the FDA asked the advisory committee to consider the clinical significance of the preclinical findings, the nature and level of any safety concerns in various populations, the sufficiency of evidence from toxicology and clinical studies for intermittent and chronic use, clinical or laboratory assessments that might help ensure patient safety, and recommendations for additional studies to support the safety of the product.
Novo Nordisk is specifically seeking FDA marketing approval of N9-GP, which is administered weekly, for control and prevention of bleeding episodes, perioperative management, and routine prophylaxis in adults and children. The company submitted evidence for these indications from three phase III trials and a phase III extension study in adults and children.
“The Office of Tissues and Advanced Therapies – OTAT – appreciates that hemophilia B is a serious disease and we need improved therapies to benefit patients. We also appreciate that this is a rare disease, which limits the availability of data to address issues of safety and effectiveness,” Wilson Bryan, MD, director of OTAT, which is part of the FDA’s Center for Biologics Evaluation and Research, told the advisory committee.
The committee then heard from Novo Nordisk representatives who spoke about the unmet public health need for hemophilia B treatments, and N9-GP clinical efficacy, long-term safety, and risk/benefit analyses.
Shawn Hoskin, senior director of regulatory affairs for Novo Nordisk, noted that with weekly dosing of N9-GP, no adverse effects were seen in preclinical studies at doses up to 42 times the human dose. Further, no adverse effects were reported in clinical studies, in which high levels of factor IX were achieved.
“Our trials demonstrated that the higher factor IX levels achieved with N9-GP lead to better outcomes for patients, including reduced annual bleed rate, reduction in the number of spontaneous bleeds, and resolution of target joints,” he said.
Stephanie Seremetis, MD, chief medical officer and corporate vice president for hemophilia at Novo Nordisk, said the company has proposed a postapproval monitoring plan and safety study.
The advisory committee also heard from patients invited by Novo Nordisk to share their experiences, and from Maria Lehtinen, PhD, of Boston Children’s Hospital, who was invited to speak about choroid plexus biology.
The patients agreed that there is an urgent need and desire for effective, long-acting treatments. Ben Shuldiner, for example, said he was part of the clinical trials for N9-GP, has never had as much success sticking to a treatment regimen, and “is in much better shape, much less pain.”
“The [hemophilia] community needs choice,” said Mr. Shuldiner, a professor at Hunter College in New York, and an activist. He stressed that what works for one patient doesn’t necessarily work for another.
Committee members didn’t question the efficacy or value of the long-acting product, but did express concern about the unknown effects of PEG accumulation. One member questioned whether it might be feasible to restrict licensing to children over age 6 years and to adults under age 65 years pending additional study. Other members said that approach would restrict treatment for patients who might benefit the most from N9-GP.
Meera B. Chitlur, MD, noted that most children with hemophilia B are treated prophylactically by age 1, or at least by the time they are walking, and suggested it might be better to allow use of the product in those who need it, while collecting data going forward.
“Only over the last couple years have our patients finally had the opportunity to have something that has substantially changed how they are managed or what they can do. This class of drugs is one that has made it possible for these patients to lead a better life, so I think it is really important for us,” said Dr. Chitlur of Wayne State University and Children’s Hospital of Michigan, Detroit.
“The youngest children will probably benefit the most. Yes, there are concerns ... but at the same time I think I haven’t heard anything today, or read anything, that has made me want to say it is not safe for the patients that I’m going to take care of,” she added, noting that informed consent is possible and important.
Dr. Chitlur said she completely agrees with the need for systematic data collection, and feels there are already mechanisms in place to achieve that goal.
“I feel comfortable taking this to my patients and saying that here is another option for you,” she said.
Laura Manuelidis, MD, of Yale University, New Haven, Conn., a temporary voting member of the committee, said she was “less sanguine,” about immediate approval. Additional studies of the effects of PEG accumulation in animals are feasible, she said, and could be performed rapidly before approving treatment in children under age 2 years.
Michael Dobbs, MD, of the University of Kentucky, Lexington, also a temporary voting member, agreed it would be valuable to test available cerebrospinal fluid of animals from the preclinical studies. He had no recommendation for additional studies, but agreed on the importance of extensive postmarketing surveillance. He added that patients who undergo CSF testing or magnetic resonance imaging for other clinical reasons should be evaluated for the possible effects of PEG accumulation.
“I think we should follow cognitive outcomes in the postmarket data, probably requiring more neurocognitive data in pediatrics with some validated, standardized tests. It would be reasonable to require a full neurological exam pre- and post-[treatment], especially in those at fixed developmental states,” he said.
It would also be reasonable to monitor children for signs and symptoms of hydrocephalus and for papilledema and other visual disturbances, he added.
“But again, most of all ... whatever we really do recommend, we need standardization, validation – to do this the same [way] for all of the patients,” he said.
Dr. Bryan, of OTAT, said the input from members and guests will be considered as the FDA proceeds with its review of the Biologics Licensing Application for N9-GP. The FDA is not bound by the committee’s guidance.
All members of the advisory committee have been screened and found to be in compliance with respect to potential conflicts of interest. No conflict of interest waivers were issued. Patients who spoke in favor of N9-GP received travel and/or other support from Novo Nordisk.
Birth defects found in 10% of confirmed U.S. Zika pregnancies
FROM MMWR
About 1 in 10 pregnant women with confirmed Zika infection in the United States had a fetus or baby with Zika-related birth defects in 2016, officials at the Centers for Disease Control and Prevention reported.
Among 972 completed pregnancies with laboratory evidence of possible recent Zika virus infection – 895 liveborn infants and 77 pregnancy losses – there were 51 fetuses/infants with Zika-related birth defects (5%). Among 250 pregnancies with laboratory-confirmed Zika infection, there were 24 cases of birth defects (10%). Among the 60 pregnancies in which Zika infection was confirmed and onset occurred during the first trimester, there were nine cases of birth defects (15%).![]()
“We are seeing about 30-40 new Zika cases in pregnant women each week in the United States. With the current tally of more than 1,600 pregnant women with evidence of Zika” reported in at least 44 states, mostly from travel to endemic areas, “this devastating outbreak is far from over,” CDC Acting Director Anne Schuchat, MD, said during an April 4 press conference.
But current birth defect estimates may not reflect the full impact of Zika virus in pregnancy, since some Zika-related developmental problems may not become apparent until months after birth. “We recommend babies receive close developmental monitoring and follow-up,” she said.
Despite CDC recommendations, one in three infants with possible congenital Zika infection had no report of Zika testing at birth, and only 1 in 4 had brain imaging (Morb Mortal Wkly Rep. 2017 Apr 4. doi: 10.15585/mmwr.mm6613e1).
“Because the full clinical spectrum of congenital Zika virus infection is not yet known, all infants born to women with laboratory evidence of possible recent Zika virus infection during pregnancy should receive postnatal neuroimaging and Zika virus testing in addition to a comprehensive newborn physical exam and hearing screen,” the CDC officials wrote.
Birth defects – most commonly microcephaly or brain abnormalities – were reported in similar proportions of fetuses/infants whose mothers did and did not report Zika symptoms.
Dr. Schuchat noted that Zika virus can cause vision problems, hearing problems, and seizures. Some infants have little or no control over their arms or legs, and cannot reach out to touch things because of their constricted joints. There can be problems reaching developmental milestones, such as sitting up, as well as problems with feeding, swallowing, and breathing. Some babies cry inconsolably. Others may be born with a normal head size but experience slow head growth later on and develop microcephaly.
The study confirms the need for pregnant women to continue taking steps to prevent Zika virus exposure through mosquito bites and sexual transmission.
“We encourage [health care providers] to ask about possible Zika exposure when caring for both pregnant women and their babies, and to follow CDC guidance for evaluation and care of infants with possible Zika infection,” said Peggy Honein, PhD, senior investigator on the review, in a statement.
The mothers of the 51 fetuses or infants with birth defects were exposed to Zika during trips to 16 countries, including Barbados, Belize, Brazil, Cape Verde, Colombia, Dominican Republic, El Salvador, Guatemala, Guyana, Haiti, Honduras, Jamaica, Mexico, Puerto Rico, Republic of Marshall Islands, and Venezuela.
FROM MMWR
About 1 in 10 pregnant women with confirmed Zika infection in the United States had a fetus or baby with Zika-related birth defects in 2016, officials at the Centers for Disease Control and Prevention reported.
Among 972 completed pregnancies with laboratory evidence of possible recent Zika virus infection – 895 liveborn infants and 77 pregnancy losses – there were 51 fetuses/infants with Zika-related birth defects (5%). Among 250 pregnancies with laboratory-confirmed Zika infection, there were 24 cases of birth defects (10%). Among the 60 pregnancies in which Zika infection was confirmed and onset occurred during the first trimester, there were nine cases of birth defects (15%).![]()
“We are seeing about 30-40 new Zika cases in pregnant women each week in the United States. With the current tally of more than 1,600 pregnant women with evidence of Zika” reported in at least 44 states, mostly from travel to endemic areas, “this devastating outbreak is far from over,” CDC Acting Director Anne Schuchat, MD, said during an April 4 press conference.
But current birth defect estimates may not reflect the full impact of Zika virus in pregnancy, since some Zika-related developmental problems may not become apparent until months after birth. “We recommend babies receive close developmental monitoring and follow-up,” she said.
Despite CDC recommendations, one in three infants with possible congenital Zika infection had no report of Zika testing at birth, and only 1 in 4 had brain imaging (Morb Mortal Wkly Rep. 2017 Apr 4. doi: 10.15585/mmwr.mm6613e1).
“Because the full clinical spectrum of congenital Zika virus infection is not yet known, all infants born to women with laboratory evidence of possible recent Zika virus infection during pregnancy should receive postnatal neuroimaging and Zika virus testing in addition to a comprehensive newborn physical exam and hearing screen,” the CDC officials wrote.
Birth defects – most commonly microcephaly or brain abnormalities – were reported in similar proportions of fetuses/infants whose mothers did and did not report Zika symptoms.
Dr. Schuchat noted that Zika virus can cause vision problems, hearing problems, and seizures. Some infants have little or no control over their arms or legs, and cannot reach out to touch things because of their constricted joints. There can be problems reaching developmental milestones, such as sitting up, as well as problems with feeding, swallowing, and breathing. Some babies cry inconsolably. Others may be born with a normal head size but experience slow head growth later on and develop microcephaly.
The study confirms the need for pregnant women to continue taking steps to prevent Zika virus exposure through mosquito bites and sexual transmission.
“We encourage [health care providers] to ask about possible Zika exposure when caring for both pregnant women and their babies, and to follow CDC guidance for evaluation and care of infants with possible Zika infection,” said Peggy Honein, PhD, senior investigator on the review, in a statement.
The mothers of the 51 fetuses or infants with birth defects were exposed to Zika during trips to 16 countries, including Barbados, Belize, Brazil, Cape Verde, Colombia, Dominican Republic, El Salvador, Guatemala, Guyana, Haiti, Honduras, Jamaica, Mexico, Puerto Rico, Republic of Marshall Islands, and Venezuela.
FROM MMWR
About 1 in 10 pregnant women with confirmed Zika infection in the United States had a fetus or baby with Zika-related birth defects in 2016, officials at the Centers for Disease Control and Prevention reported.
Among 972 completed pregnancies with laboratory evidence of possible recent Zika virus infection – 895 liveborn infants and 77 pregnancy losses – there were 51 fetuses/infants with Zika-related birth defects (5%). Among 250 pregnancies with laboratory-confirmed Zika infection, there were 24 cases of birth defects (10%). Among the 60 pregnancies in which Zika infection was confirmed and onset occurred during the first trimester, there were nine cases of birth defects (15%).![]()
“We are seeing about 30-40 new Zika cases in pregnant women each week in the United States. With the current tally of more than 1,600 pregnant women with evidence of Zika” reported in at least 44 states, mostly from travel to endemic areas, “this devastating outbreak is far from over,” CDC Acting Director Anne Schuchat, MD, said during an April 4 press conference.
But current birth defect estimates may not reflect the full impact of Zika virus in pregnancy, since some Zika-related developmental problems may not become apparent until months after birth. “We recommend babies receive close developmental monitoring and follow-up,” she said.
Despite CDC recommendations, one in three infants with possible congenital Zika infection had no report of Zika testing at birth, and only 1 in 4 had brain imaging (Morb Mortal Wkly Rep. 2017 Apr 4. doi: 10.15585/mmwr.mm6613e1).
“Because the full clinical spectrum of congenital Zika virus infection is not yet known, all infants born to women with laboratory evidence of possible recent Zika virus infection during pregnancy should receive postnatal neuroimaging and Zika virus testing in addition to a comprehensive newborn physical exam and hearing screen,” the CDC officials wrote.
Birth defects – most commonly microcephaly or brain abnormalities – were reported in similar proportions of fetuses/infants whose mothers did and did not report Zika symptoms.
Dr. Schuchat noted that Zika virus can cause vision problems, hearing problems, and seizures. Some infants have little or no control over their arms or legs, and cannot reach out to touch things because of their constricted joints. There can be problems reaching developmental milestones, such as sitting up, as well as problems with feeding, swallowing, and breathing. Some babies cry inconsolably. Others may be born with a normal head size but experience slow head growth later on and develop microcephaly.
The study confirms the need for pregnant women to continue taking steps to prevent Zika virus exposure through mosquito bites and sexual transmission.
“We encourage [health care providers] to ask about possible Zika exposure when caring for both pregnant women and their babies, and to follow CDC guidance for evaluation and care of infants with possible Zika infection,” said Peggy Honein, PhD, senior investigator on the review, in a statement.
The mothers of the 51 fetuses or infants with birth defects were exposed to Zika during trips to 16 countries, including Barbados, Belize, Brazil, Cape Verde, Colombia, Dominican Republic, El Salvador, Guatemala, Guyana, Haiti, Honduras, Jamaica, Mexico, Puerto Rico, Republic of Marshall Islands, and Venezuela.
FDA approves deutetrabenazine for Huntington’s-associated chorea
The Food and Drug Administration has approved deutetrabenazine (Austedo) for the treatment of chorea associated with Huntington’s disease, according to an announcement by the drug’s manufacturer, Teva Pharmaceutical.
The random, sudden, involuntary twisting and writhing movements of chorea constitute perhaps the most striking symptom of Huntington’s disease and affect about 90% of the roughly 35,000 Americans with the fatal neurodegenerative disorder. Deutetrabenazine is only the second product approved to treat Huntington’s in any capacity, and it is the first in nearly a decade.
The most common side effects of deutetrabenazine were somnolence, diarrhea, dry mouth, and fatigue. Deutetrabenazine is contraindicated in patients with hepatic impairment; patients taking monoamine oxidase inhibitors (MAOIs), or within 14 days of discontinuing MAOI therapy; patients taking reserpine or within 20 days of discontinuing reserpine; and patients taking tetrabenazine. Deutetrabenazine can cause worsening of mood and is not recommended for suicidal patients or patients with improperly treated depression.
“Chorea associated with Huntington’s disease has a significant impact on those living with the disease and their families. The FDA’s approval of Austedo represents an important new treatment option for people with HD and highlights the need for more therapeutic resources for this underserved patient community,” Louise Vetter, CEO of the Huntington’s Disease Society of America, said in the company’s announcement.
The Food and Drug Administration has approved deutetrabenazine (Austedo) for the treatment of chorea associated with Huntington’s disease, according to an announcement by the drug’s manufacturer, Teva Pharmaceutical.
The random, sudden, involuntary twisting and writhing movements of chorea constitute perhaps the most striking symptom of Huntington’s disease and affect about 90% of the roughly 35,000 Americans with the fatal neurodegenerative disorder. Deutetrabenazine is only the second product approved to treat Huntington’s in any capacity, and it is the first in nearly a decade.
The most common side effects of deutetrabenazine were somnolence, diarrhea, dry mouth, and fatigue. Deutetrabenazine is contraindicated in patients with hepatic impairment; patients taking monoamine oxidase inhibitors (MAOIs), or within 14 days of discontinuing MAOI therapy; patients taking reserpine or within 20 days of discontinuing reserpine; and patients taking tetrabenazine. Deutetrabenazine can cause worsening of mood and is not recommended for suicidal patients or patients with improperly treated depression.
“Chorea associated with Huntington’s disease has a significant impact on those living with the disease and their families. The FDA’s approval of Austedo represents an important new treatment option for people with HD and highlights the need for more therapeutic resources for this underserved patient community,” Louise Vetter, CEO of the Huntington’s Disease Society of America, said in the company’s announcement.
The Food and Drug Administration has approved deutetrabenazine (Austedo) for the treatment of chorea associated with Huntington’s disease, according to an announcement by the drug’s manufacturer, Teva Pharmaceutical.
The random, sudden, involuntary twisting and writhing movements of chorea constitute perhaps the most striking symptom of Huntington’s disease and affect about 90% of the roughly 35,000 Americans with the fatal neurodegenerative disorder. Deutetrabenazine is only the second product approved to treat Huntington’s in any capacity, and it is the first in nearly a decade.
The most common side effects of deutetrabenazine were somnolence, diarrhea, dry mouth, and fatigue. Deutetrabenazine is contraindicated in patients with hepatic impairment; patients taking monoamine oxidase inhibitors (MAOIs), or within 14 days of discontinuing MAOI therapy; patients taking reserpine or within 20 days of discontinuing reserpine; and patients taking tetrabenazine. Deutetrabenazine can cause worsening of mood and is not recommended for suicidal patients or patients with improperly treated depression.
“Chorea associated with Huntington’s disease has a significant impact on those living with the disease and their families. The FDA’s approval of Austedo represents an important new treatment option for people with HD and highlights the need for more therapeutic resources for this underserved patient community,” Louise Vetter, CEO of the Huntington’s Disease Society of America, said in the company’s announcement.
FDA: Recall of select EpiPen products
Meridian Medical Technologies has issued a voluntary recall for 13 lots of EpiPen products, according to a press release from the U.S. Food and Drug Administration.
The voluntary recall includes EpiPen and EpiPen Jr. Auto-Injector distributed between Dec. 17, 2015, and July 1, 2016 by Mylan Specialty. Products included in the recall may contain a defective part which would prevent the device from activating.
Consumers who need their EpiPens should keep them until a they obtain a replacement, the FDA recommended.
Find the full press release on the FDA website.
Meridian Medical Technologies has issued a voluntary recall for 13 lots of EpiPen products, according to a press release from the U.S. Food and Drug Administration.
The voluntary recall includes EpiPen and EpiPen Jr. Auto-Injector distributed between Dec. 17, 2015, and July 1, 2016 by Mylan Specialty. Products included in the recall may contain a defective part which would prevent the device from activating.
Consumers who need their EpiPens should keep them until a they obtain a replacement, the FDA recommended.
Find the full press release on the FDA website.
Meridian Medical Technologies has issued a voluntary recall for 13 lots of EpiPen products, according to a press release from the U.S. Food and Drug Administration.
The voluntary recall includes EpiPen and EpiPen Jr. Auto-Injector distributed between Dec. 17, 2015, and July 1, 2016 by Mylan Specialty. Products included in the recall may contain a defective part which would prevent the device from activating.
Consumers who need their EpiPens should keep them until a they obtain a replacement, the FDA recommended.
Find the full press release on the FDA website.
Osimertinib receives full approval for advanced EGFR-mutated NSCLC
The Food and Drug Administration has converted accelerated approval of osimertinib to full approval for the treatment of patients with metastatic epidermal growth factor receptor (EGFR) T790M mutation–positive non–small cell lung cancer (NSCLC), as detected by an FDA-approved test.
Also included in the indication, disease must have progressed on or after EGFR tyrosine kinase inhibitor (TKI) therapy, the FDA said in a statement.
Full approval was based on an improvement in progression-free survival (PFS) in the phase III AURA3 study, which randomized 419 patients (2:1) to receive osimertinib (n = 279) 80 mg orally once daily or platinum-based doublet chemotherapy (n = 140). The hazard ratio for the investigator-assessed PFS was .30 (95% confidence interval: 0.23, 0.41; P less than .001).
The estimated median PFS was 10.1 months in the osimertinib arm and 4.4 months in the chemotherapy arm. Confirmed ORR was 65% (95% CI: 59%, 70%) and 29% (95% CI: 21%, 37%) in the osimertinib and chemotherapy arms, respectively (P less than .0001). Estimated median response durations were 11 months (95% CI: 8.6, 12.6) and 4.2 months (95% CI: 3.9, 5.9) in the osimertinib and chemotherapy arms, respectively, according to the FDA statement.
Overall survival data are immature, the FDA said.
All patients had metastatic EGFR T790M mutation–positive NSCLC, identified by the cobas EGFR mutation test performed in a central laboratory, and progressive disease following first-line EGFR TKI therapy. Patients in the chemotherapy arm received either pemetrexed, 500 mg/m2 with carboplatin AUC5, or pemetrexed, 500mg/m2 with cisplatin 75 mg/m2), on day 1 of every 21-day cycle for up to six cycles followed by pemetrexed maintenance therapy.
The most serious adverse reactions, evaluated in 833 patients receiving osimertinib, were interstitial lung disease/pneumonitis (3.5%), QTc interval prolongation (0.7%), cardiomyopathy (1.9%), and keratitis (0.7%). The most common adverse reactions were diarrhea, rash, dry skin, nail toxicity, and fatigue.
The recommended dose of osimertinib, to be marketed as Tagrisso by AstraZeneca, is 80 mg orally once daily, with or without food, until disease progression or unacceptable toxicity. The presence of an EGFR T790M mutation in a tumor specimen, or plasma specimen (if tumor tissue is unavailable), should be confirmed by an FDA-approved test prior to initiation of treatment.
Full prescribing information is available here.
The Food and Drug Administration has converted accelerated approval of osimertinib to full approval for the treatment of patients with metastatic epidermal growth factor receptor (EGFR) T790M mutation–positive non–small cell lung cancer (NSCLC), as detected by an FDA-approved test.
Also included in the indication, disease must have progressed on or after EGFR tyrosine kinase inhibitor (TKI) therapy, the FDA said in a statement.
Full approval was based on an improvement in progression-free survival (PFS) in the phase III AURA3 study, which randomized 419 patients (2:1) to receive osimertinib (n = 279) 80 mg orally once daily or platinum-based doublet chemotherapy (n = 140). The hazard ratio for the investigator-assessed PFS was .30 (95% confidence interval: 0.23, 0.41; P less than .001).
The estimated median PFS was 10.1 months in the osimertinib arm and 4.4 months in the chemotherapy arm. Confirmed ORR was 65% (95% CI: 59%, 70%) and 29% (95% CI: 21%, 37%) in the osimertinib and chemotherapy arms, respectively (P less than .0001). Estimated median response durations were 11 months (95% CI: 8.6, 12.6) and 4.2 months (95% CI: 3.9, 5.9) in the osimertinib and chemotherapy arms, respectively, according to the FDA statement.
Overall survival data are immature, the FDA said.
All patients had metastatic EGFR T790M mutation–positive NSCLC, identified by the cobas EGFR mutation test performed in a central laboratory, and progressive disease following first-line EGFR TKI therapy. Patients in the chemotherapy arm received either pemetrexed, 500 mg/m2 with carboplatin AUC5, or pemetrexed, 500mg/m2 with cisplatin 75 mg/m2), on day 1 of every 21-day cycle for up to six cycles followed by pemetrexed maintenance therapy.
The most serious adverse reactions, evaluated in 833 patients receiving osimertinib, were interstitial lung disease/pneumonitis (3.5%), QTc interval prolongation (0.7%), cardiomyopathy (1.9%), and keratitis (0.7%). The most common adverse reactions were diarrhea, rash, dry skin, nail toxicity, and fatigue.
The recommended dose of osimertinib, to be marketed as Tagrisso by AstraZeneca, is 80 mg orally once daily, with or without food, until disease progression or unacceptable toxicity. The presence of an EGFR T790M mutation in a tumor specimen, or plasma specimen (if tumor tissue is unavailable), should be confirmed by an FDA-approved test prior to initiation of treatment.
Full prescribing information is available here.
The Food and Drug Administration has converted accelerated approval of osimertinib to full approval for the treatment of patients with metastatic epidermal growth factor receptor (EGFR) T790M mutation–positive non–small cell lung cancer (NSCLC), as detected by an FDA-approved test.
Also included in the indication, disease must have progressed on or after EGFR tyrosine kinase inhibitor (TKI) therapy, the FDA said in a statement.
Full approval was based on an improvement in progression-free survival (PFS) in the phase III AURA3 study, which randomized 419 patients (2:1) to receive osimertinib (n = 279) 80 mg orally once daily or platinum-based doublet chemotherapy (n = 140). The hazard ratio for the investigator-assessed PFS was .30 (95% confidence interval: 0.23, 0.41; P less than .001).
The estimated median PFS was 10.1 months in the osimertinib arm and 4.4 months in the chemotherapy arm. Confirmed ORR was 65% (95% CI: 59%, 70%) and 29% (95% CI: 21%, 37%) in the osimertinib and chemotherapy arms, respectively (P less than .0001). Estimated median response durations were 11 months (95% CI: 8.6, 12.6) and 4.2 months (95% CI: 3.9, 5.9) in the osimertinib and chemotherapy arms, respectively, according to the FDA statement.
Overall survival data are immature, the FDA said.
All patients had metastatic EGFR T790M mutation–positive NSCLC, identified by the cobas EGFR mutation test performed in a central laboratory, and progressive disease following first-line EGFR TKI therapy. Patients in the chemotherapy arm received either pemetrexed, 500 mg/m2 with carboplatin AUC5, or pemetrexed, 500mg/m2 with cisplatin 75 mg/m2), on day 1 of every 21-day cycle for up to six cycles followed by pemetrexed maintenance therapy.
The most serious adverse reactions, evaluated in 833 patients receiving osimertinib, were interstitial lung disease/pneumonitis (3.5%), QTc interval prolongation (0.7%), cardiomyopathy (1.9%), and keratitis (0.7%). The most common adverse reactions were diarrhea, rash, dry skin, nail toxicity, and fatigue.
The recommended dose of osimertinib, to be marketed as Tagrisso by AstraZeneca, is 80 mg orally once daily, with or without food, until disease progression or unacceptable toxicity. The presence of an EGFR T790M mutation in a tumor specimen, or plasma specimen (if tumor tissue is unavailable), should be confirmed by an FDA-approved test prior to initiation of treatment.
Full prescribing information is available here.
Ocrelizumab gets first-ever FDA approval for primary progressive MS
The humanized monoclonal antibody ocrelizumab became the first drug to receive approval from the Food and Drug Administration for the treatment of primary progressive multiple sclerosis in adults, according to a March 29 announcement from the agency that also said it is approved for relapsing-remitting disease in adults.
The drug has a mechanism of action similar to rituximab (Rituxan) through its selective targeting of CD20-positive B cells, which depletes them from circulation. CD20-positive B cells are thought to be a key contributor to myelin and axonal damage.

Although the effect of ocrelizumab on primary progressive MS patients was modest, its stature as the only drug approved for the indication represents “a start for being able to treat progressive MS,” Fred Lublin, MD, a member of the steering committee that designed and monitored the phase III ocrelizumab trials, said in an interview.
In the ORATORIO trial that randomized 732 patients with primary progressive MS in a 2:1 ratio to infusions of either 600 mg ocrelizumab or placebo every 24 weeks for 120 weeks, ocrelizumab reduced the risk of progression in clinical disability by 24% at 12 weeks (the primary end point), 25% at 24 weeks, and 24% at 120 weeks, compared with placebo (N Engl J Med. 2017 Jan 19;376[3]:209-20).
Over 120 weeks, the antibody also reduced the volume of hyperintense T2 lesions by 3.4%, whereas patients taking placebo experienced a 7.4% increase. The rate of whole brain volume loss was also significantly reduced from week 24 to 120 (–0.9% with ocrelizumab vs. –1.1% for placebo).
The group of patients who participated in ORATORIO was younger and had more activity on MRI, but it’s unclear if there is a progressive MS patient population who will respond best to the biologic, said Dr. Lublin, director of the Corinne Goldsmith Dickinson Center for Multiple Sclerosis at Mount Sinai Hospital in New York.
“The safety was acceptable, and so I think it will get a lot of use. There are always some who like to wait until a drug has been out for a while, but there is considerable experience with this type of drug based on off-label use of rituximab” in relapsing-remitting patients, including phase II trial data (but not phase III), he said. There’s potential for ocrelizumab to be used as a first-line treatment for relapsing-remitting disease to the same extent as natalizumab (Tysabri), he added.
In two identical phase III trials, OPERA I and OPERA II, investigators randomized 1,656 relapsing-remitting MS patients to intravenous ocrelizumab 600 mg every 24 weeks plus placebo subcutaneous injections three times weekly or to subcutaneous interferon beta-1a 44 mcg (Rebif) three times weekly plus placebo IV infusions every 24 weeks over 96 weeks (N Engl J Med. 2017 Jan 19;376[3]:221-34).
Compared with interferon beta-1a, ocrelizumab reduced the annualized relapse rate by 46% in OPERA 1 and 47% in OPERA 2. In a pooled analysis of both trials, the risk of confirmed disability progression was 40% lower for ocrelizumab at both 12 and 24 weeks. Ocrelizumab conferred a 94% and 95% reduction in the total number of T1 gadolinium-enhancing lesions and a 77% and 83% reduction in the total number of new and/or enlarging hyperintense T2 lesions. An exploratory analysis also suggested that the drug reduced the rate of whole brain volume loss, compared with interferon beta-1a.
At 96 weeks, 47.9% and 47.5% of ocrelizumab patients versus 29.2% and 25.1% of interferon patients had no evidence of disease activity (NEDA) in the two studies. NEDA is a composite score defined as no relapses, no confirmed disability progression, and no new or enlarging T2 or gadolinium-enhancing T1 lesions.
Across both studies, relapses occurred in about 20% of ocrelizumab patients versus about 35% of interferon patients. About 10% of ocrelizumab patients had clinical disease progression, compared with about 15% of interferon patients. Similarly, about 10% of ocrelizumab patients developed new gadolinium-enhancing lesions, compared with about 35% in the interferon groups. New or enlarging T2 lesions were found in about 40% in the ocrelizumab groups but in more than 60% in the interferon arms.
Safety results at 24 weeks in the OPERA trials showed that infusion reactions were significantly more common with ocrelizumab than with interferon beta-1a (34% vs. 9.7%), and most of these were mild to moderate in severity. Otherwise, there were similar rates of serious adverse events, including serious infections.
In ORATORIO, infusion reaction occurred in 40% of ocrelizumab patients and 26% of placebo patients. Most of the reactions were mild to moderate in severity. Among all patients in ORATORIO, 13 malignancies occurred over 3 years, and they occurred more than twice as often in the ocrelizumab arm than in the placebo arm (2.3% vs. 0.8%). These included four breast cancers in the active arm and none in the placebo arm.
No cases of progressive multifocal leukoencephalopathy (PML) have been reported in association with ocrelizumab, although the risk of PML with long-term use is unknown.
The FDA said in its announcement that ocrelizumab should not be used in patients with hepatitis B infection or a history of life-threatening infusion-related reactions to the drug. Ocrelizumab is required to be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks. The agency also advised that vaccination with live or live attenuated vaccines is not recommended in patients receiving the drug.
Genentech says it plans to offer patient assistance programs through Genentech Access Solutions.
Dr. Lublin reported receiving fees for serving on an advisory board from Genentech/Roche and from many other companies investigating or marketing drugs for MS.
The humanized monoclonal antibody ocrelizumab became the first drug to receive approval from the Food and Drug Administration for the treatment of primary progressive multiple sclerosis in adults, according to a March 29 announcement from the agency that also said it is approved for relapsing-remitting disease in adults.
The drug has a mechanism of action similar to rituximab (Rituxan) through its selective targeting of CD20-positive B cells, which depletes them from circulation. CD20-positive B cells are thought to be a key contributor to myelin and axonal damage.
Although the effect of ocrelizumab on primary progressive MS patients was modest, its stature as the only drug approved for the indication represents “a start for being able to treat progressive MS,” Fred Lublin, MD, a member of the steering committee that designed and monitored the phase III ocrelizumab trials, said in an interview.
In the ORATORIO trial that randomized 732 patients with primary progressive MS in a 2:1 ratio to infusions of either 600 mg ocrelizumab or placebo every 24 weeks for 120 weeks, ocrelizumab reduced the risk of progression in clinical disability by 24% at 12 weeks (the primary end point), 25% at 24 weeks, and 24% at 120 weeks, compared with placebo (N Engl J Med. 2017 Jan 19;376[3]:209-20).
Over 120 weeks, the antibody also reduced the volume of hyperintense T2 lesions by 3.4%, whereas patients taking placebo experienced a 7.4% increase. The rate of whole brain volume loss was also significantly reduced from week 24 to 120 (–0.9% with ocrelizumab vs. –1.1% for placebo).
The group of patients who participated in ORATORIO was younger and had more activity on MRI, but it’s unclear if there is a progressive MS patient population who will respond best to the biologic, said Dr. Lublin, director of the Corinne Goldsmith Dickinson Center for Multiple Sclerosis at Mount Sinai Hospital in New York.
“The safety was acceptable, and so I think it will get a lot of use. There are always some who like to wait until a drug has been out for a while, but there is considerable experience with this type of drug based on off-label use of rituximab” in relapsing-remitting patients, including phase II trial data (but not phase III), he said. There’s potential for ocrelizumab to be used as a first-line treatment for relapsing-remitting disease to the same extent as natalizumab (Tysabri), he added.
In two identical phase III trials, OPERA I and OPERA II, investigators randomized 1,656 relapsing-remitting MS patients to intravenous ocrelizumab 600 mg every 24 weeks plus placebo subcutaneous injections three times weekly or to subcutaneous interferon beta-1a 44 mcg (Rebif) three times weekly plus placebo IV infusions every 24 weeks over 96 weeks (N Engl J Med. 2017 Jan 19;376[3]:221-34).
Compared with interferon beta-1a, ocrelizumab reduced the annualized relapse rate by 46% in OPERA 1 and 47% in OPERA 2. In a pooled analysis of both trials, the risk of confirmed disability progression was 40% lower for ocrelizumab at both 12 and 24 weeks. Ocrelizumab conferred a 94% and 95% reduction in the total number of T1 gadolinium-enhancing lesions and a 77% and 83% reduction in the total number of new and/or enlarging hyperintense T2 lesions. An exploratory analysis also suggested that the drug reduced the rate of whole brain volume loss, compared with interferon beta-1a.
At 96 weeks, 47.9% and 47.5% of ocrelizumab patients versus 29.2% and 25.1% of interferon patients had no evidence of disease activity (NEDA) in the two studies. NEDA is a composite score defined as no relapses, no confirmed disability progression, and no new or enlarging T2 or gadolinium-enhancing T1 lesions.
Across both studies, relapses occurred in about 20% of ocrelizumab patients versus about 35% of interferon patients. About 10% of ocrelizumab patients had clinical disease progression, compared with about 15% of interferon patients. Similarly, about 10% of ocrelizumab patients developed new gadolinium-enhancing lesions, compared with about 35% in the interferon groups. New or enlarging T2 lesions were found in about 40% in the ocrelizumab groups but in more than 60% in the interferon arms.
Safety results at 24 weeks in the OPERA trials showed that infusion reactions were significantly more common with ocrelizumab than with interferon beta-1a (34% vs. 9.7%), and most of these were mild to moderate in severity. Otherwise, there were similar rates of serious adverse events, including serious infections.
In ORATORIO, infusion reaction occurred in 40% of ocrelizumab patients and 26% of placebo patients. Most of the reactions were mild to moderate in severity. Among all patients in ORATORIO, 13 malignancies occurred over 3 years, and they occurred more than twice as often in the ocrelizumab arm than in the placebo arm (2.3% vs. 0.8%). These included four breast cancers in the active arm and none in the placebo arm.
No cases of progressive multifocal leukoencephalopathy (PML) have been reported in association with ocrelizumab, although the risk of PML with long-term use is unknown.
The FDA said in its announcement that ocrelizumab should not be used in patients with hepatitis B infection or a history of life-threatening infusion-related reactions to the drug. Ocrelizumab is required to be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks. The agency also advised that vaccination with live or live attenuated vaccines is not recommended in patients receiving the drug.
Genentech says it plans to offer patient assistance programs through Genentech Access Solutions.
Dr. Lublin reported receiving fees for serving on an advisory board from Genentech/Roche and from many other companies investigating or marketing drugs for MS.
The humanized monoclonal antibody ocrelizumab became the first drug to receive approval from the Food and Drug Administration for the treatment of primary progressive multiple sclerosis in adults, according to a March 29 announcement from the agency that also said it is approved for relapsing-remitting disease in adults.
The drug has a mechanism of action similar to rituximab (Rituxan) through its selective targeting of CD20-positive B cells, which depletes them from circulation. CD20-positive B cells are thought to be a key contributor to myelin and axonal damage.
Although the effect of ocrelizumab on primary progressive MS patients was modest, its stature as the only drug approved for the indication represents “a start for being able to treat progressive MS,” Fred Lublin, MD, a member of the steering committee that designed and monitored the phase III ocrelizumab trials, said in an interview.
In the ORATORIO trial that randomized 732 patients with primary progressive MS in a 2:1 ratio to infusions of either 600 mg ocrelizumab or placebo every 24 weeks for 120 weeks, ocrelizumab reduced the risk of progression in clinical disability by 24% at 12 weeks (the primary end point), 25% at 24 weeks, and 24% at 120 weeks, compared with placebo (N Engl J Med. 2017 Jan 19;376[3]:209-20).
Over 120 weeks, the antibody also reduced the volume of hyperintense T2 lesions by 3.4%, whereas patients taking placebo experienced a 7.4% increase. The rate of whole brain volume loss was also significantly reduced from week 24 to 120 (–0.9% with ocrelizumab vs. –1.1% for placebo).
The group of patients who participated in ORATORIO was younger and had more activity on MRI, but it’s unclear if there is a progressive MS patient population who will respond best to the biologic, said Dr. Lublin, director of the Corinne Goldsmith Dickinson Center for Multiple Sclerosis at Mount Sinai Hospital in New York.
“The safety was acceptable, and so I think it will get a lot of use. There are always some who like to wait until a drug has been out for a while, but there is considerable experience with this type of drug based on off-label use of rituximab” in relapsing-remitting patients, including phase II trial data (but not phase III), he said. There’s potential for ocrelizumab to be used as a first-line treatment for relapsing-remitting disease to the same extent as natalizumab (Tysabri), he added.
In two identical phase III trials, OPERA I and OPERA II, investigators randomized 1,656 relapsing-remitting MS patients to intravenous ocrelizumab 600 mg every 24 weeks plus placebo subcutaneous injections three times weekly or to subcutaneous interferon beta-1a 44 mcg (Rebif) three times weekly plus placebo IV infusions every 24 weeks over 96 weeks (N Engl J Med. 2017 Jan 19;376[3]:221-34).
Compared with interferon beta-1a, ocrelizumab reduced the annualized relapse rate by 46% in OPERA 1 and 47% in OPERA 2. In a pooled analysis of both trials, the risk of confirmed disability progression was 40% lower for ocrelizumab at both 12 and 24 weeks. Ocrelizumab conferred a 94% and 95% reduction in the total number of T1 gadolinium-enhancing lesions and a 77% and 83% reduction in the total number of new and/or enlarging hyperintense T2 lesions. An exploratory analysis also suggested that the drug reduced the rate of whole brain volume loss, compared with interferon beta-1a.
At 96 weeks, 47.9% and 47.5% of ocrelizumab patients versus 29.2% and 25.1% of interferon patients had no evidence of disease activity (NEDA) in the two studies. NEDA is a composite score defined as no relapses, no confirmed disability progression, and no new or enlarging T2 or gadolinium-enhancing T1 lesions.
Across both studies, relapses occurred in about 20% of ocrelizumab patients versus about 35% of interferon patients. About 10% of ocrelizumab patients had clinical disease progression, compared with about 15% of interferon patients. Similarly, about 10% of ocrelizumab patients developed new gadolinium-enhancing lesions, compared with about 35% in the interferon groups. New or enlarging T2 lesions were found in about 40% in the ocrelizumab groups but in more than 60% in the interferon arms.
Safety results at 24 weeks in the OPERA trials showed that infusion reactions were significantly more common with ocrelizumab than with interferon beta-1a (34% vs. 9.7%), and most of these were mild to moderate in severity. Otherwise, there were similar rates of serious adverse events, including serious infections.
In ORATORIO, infusion reaction occurred in 40% of ocrelizumab patients and 26% of placebo patients. Most of the reactions were mild to moderate in severity. Among all patients in ORATORIO, 13 malignancies occurred over 3 years, and they occurred more than twice as often in the ocrelizumab arm than in the placebo arm (2.3% vs. 0.8%). These included four breast cancers in the active arm and none in the placebo arm.
No cases of progressive multifocal leukoencephalopathy (PML) have been reported in association with ocrelizumab, although the risk of PML with long-term use is unknown.
The FDA said in its announcement that ocrelizumab should not be used in patients with hepatitis B infection or a history of life-threatening infusion-related reactions to the drug. Ocrelizumab is required to be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks. The agency also advised that vaccination with live or live attenuated vaccines is not recommended in patients receiving the drug.
Genentech says it plans to offer patient assistance programs through Genentech Access Solutions.
Dr. Lublin reported receiving fees for serving on an advisory board from Genentech/Roche and from many other companies investigating or marketing drugs for MS.
Dupilumab: FDA approves first biologic for atopic dermatitis
Dupilumab, a monoclonal antibody that targets both interleukin-4 and interleukin-13, has been approved for the treatment of moderate to severe atopic dermatitis in adults not adequately controlled with topical prescription therapies or for whom topicals are not appropriate.
The approval marks the first biologic approved for treating AD, according to a March 28 announcement from the Food and Drug Administration.![]()
Dupilumab “inhibits signaling of IL-4 and IL-13, two key cytokines required for the type 2 (including Th2) immune response, which is believed to be a major driver in the pathogenesis of the disease,” according to Regeneron, which will market dupilumab.
Approval was based on three phase III pivotal studies of adults with moderate to severe AD whose disease was not adequately controlled with topical prescription treatments: SOLO-1 and SOLO-2, which evaluated dupilumab as monotherapy, and the CHRONOS study, which compared dupilumab with topical corticosteroids to treatment with topical corticosteroids alone.
The 16-week data from the SOLO-1 and -2 studies were presented at the 2016 annual congress of the European Academy for Dermatology and Venereology.
In two phase III trials of identical design involving patients with atopic dermatitis, dupilumab, administered weekly or every 2 weeks, improved the signs and symptoms of atopic dermatitis, including pruritus, symptoms of anxiety and depression, and quality of life, as compared with placebo. Eczema Area and Severity Index was reported in significantly more patients who received each regimen of dupilumab than in patients who received placebo (P less than .001 for all comparisons).
Dupilumab also was associated with improvement in other clinical endpoints, including reductions in pruritus and symptoms of anxiety or depression, and an improvement in quality of life. Injection-site reactions and conjunctivitis were more frequent in the dupilumab groups than in the placebo groups. The results were published in the New England Journal of Medicine (2016;375:2335-48).
More recently, 52-week data from the CHRONOS study were reported at the annual meeting of the American Academy of Dermatology in March. In that study of 740 adults with moderate to severe AD that was not controlled with topical medications – including corticosteroids with or without calcineurin inhibitors – those randomized to 300 mg of dupilumab once a week, plus topical corticosteroids, showed significantly greater improvements in measures of overall disease severity at 16 weeks and at 52 weeks, compared with those treated with steroids alone. Measures used included Eczema Area and Severity Index and the Pruritus Numerical Rating Scale, Patient Oriented Eczema Measure, Dermatology Life Quality Index.
Adverse events experienced with dupilumab included injection site reactions, eye and eyelid inflammation, and cold sores on the mouth or lips, according to a Regeneron statement.
Dupilumab will be available “later this week,” according to the statement, which noted the wholesale acquisition cost of the medication is expected to be $37,000 annually.
The FDA approval announcement noted that the safety and efficacy of dupilumab had not been established in patients with asthma.
Dupilumab currently is being studied for children with AD in phase II studies and is being studied for other indications: eosinophilic esophagitis in phase II studies, and asthma and nasal polyps in phase III studies.
Dupilumab will be marketed as Dupixent by Regeneron.
Dupilumab, a monoclonal antibody that targets both interleukin-4 and interleukin-13, has been approved for the treatment of moderate to severe atopic dermatitis in adults not adequately controlled with topical prescription therapies or for whom topicals are not appropriate.
The approval marks the first biologic approved for treating AD, according to a March 28 announcement from the Food and Drug Administration.![]()
Dupilumab “inhibits signaling of IL-4 and IL-13, two key cytokines required for the type 2 (including Th2) immune response, which is believed to be a major driver in the pathogenesis of the disease,” according to Regeneron, which will market dupilumab.
Approval was based on three phase III pivotal studies of adults with moderate to severe AD whose disease was not adequately controlled with topical prescription treatments: SOLO-1 and SOLO-2, which evaluated dupilumab as monotherapy, and the CHRONOS study, which compared dupilumab with topical corticosteroids to treatment with topical corticosteroids alone.
The 16-week data from the SOLO-1 and -2 studies were presented at the 2016 annual congress of the European Academy for Dermatology and Venereology.
In two phase III trials of identical design involving patients with atopic dermatitis, dupilumab, administered weekly or every 2 weeks, improved the signs and symptoms of atopic dermatitis, including pruritus, symptoms of anxiety and depression, and quality of life, as compared with placebo. Eczema Area and Severity Index was reported in significantly more patients who received each regimen of dupilumab than in patients who received placebo (P less than .001 for all comparisons).
Dupilumab also was associated with improvement in other clinical endpoints, including reductions in pruritus and symptoms of anxiety or depression, and an improvement in quality of life. Injection-site reactions and conjunctivitis were more frequent in the dupilumab groups than in the placebo groups. The results were published in the New England Journal of Medicine (2016;375:2335-48).
More recently, 52-week data from the CHRONOS study were reported at the annual meeting of the American Academy of Dermatology in March. In that study of 740 adults with moderate to severe AD that was not controlled with topical medications – including corticosteroids with or without calcineurin inhibitors – those randomized to 300 mg of dupilumab once a week, plus topical corticosteroids, showed significantly greater improvements in measures of overall disease severity at 16 weeks and at 52 weeks, compared with those treated with steroids alone. Measures used included Eczema Area and Severity Index and the Pruritus Numerical Rating Scale, Patient Oriented Eczema Measure, Dermatology Life Quality Index.
Adverse events experienced with dupilumab included injection site reactions, eye and eyelid inflammation, and cold sores on the mouth or lips, according to a Regeneron statement.
Dupilumab will be available “later this week,” according to the statement, which noted the wholesale acquisition cost of the medication is expected to be $37,000 annually.
The FDA approval announcement noted that the safety and efficacy of dupilumab had not been established in patients with asthma.
Dupilumab currently is being studied for children with AD in phase II studies and is being studied for other indications: eosinophilic esophagitis in phase II studies, and asthma and nasal polyps in phase III studies.
Dupilumab will be marketed as Dupixent by Regeneron.
Dupilumab, a monoclonal antibody that targets both interleukin-4 and interleukin-13, has been approved for the treatment of moderate to severe atopic dermatitis in adults not adequately controlled with topical prescription therapies or for whom topicals are not appropriate.
The approval marks the first biologic approved for treating AD, according to a March 28 announcement from the Food and Drug Administration.![]()
Dupilumab “inhibits signaling of IL-4 and IL-13, two key cytokines required for the type 2 (including Th2) immune response, which is believed to be a major driver in the pathogenesis of the disease,” according to Regeneron, which will market dupilumab.
Approval was based on three phase III pivotal studies of adults with moderate to severe AD whose disease was not adequately controlled with topical prescription treatments: SOLO-1 and SOLO-2, which evaluated dupilumab as monotherapy, and the CHRONOS study, which compared dupilumab with topical corticosteroids to treatment with topical corticosteroids alone.
The 16-week data from the SOLO-1 and -2 studies were presented at the 2016 annual congress of the European Academy for Dermatology and Venereology.
In two phase III trials of identical design involving patients with atopic dermatitis, dupilumab, administered weekly or every 2 weeks, improved the signs and symptoms of atopic dermatitis, including pruritus, symptoms of anxiety and depression, and quality of life, as compared with placebo. Eczema Area and Severity Index was reported in significantly more patients who received each regimen of dupilumab than in patients who received placebo (P less than .001 for all comparisons).
Dupilumab also was associated with improvement in other clinical endpoints, including reductions in pruritus and symptoms of anxiety or depression, and an improvement in quality of life. Injection-site reactions and conjunctivitis were more frequent in the dupilumab groups than in the placebo groups. The results were published in the New England Journal of Medicine (2016;375:2335-48).
More recently, 52-week data from the CHRONOS study were reported at the annual meeting of the American Academy of Dermatology in March. In that study of 740 adults with moderate to severe AD that was not controlled with topical medications – including corticosteroids with or without calcineurin inhibitors – those randomized to 300 mg of dupilumab once a week, plus topical corticosteroids, showed significantly greater improvements in measures of overall disease severity at 16 weeks and at 52 weeks, compared with those treated with steroids alone. Measures used included Eczema Area and Severity Index and the Pruritus Numerical Rating Scale, Patient Oriented Eczema Measure, Dermatology Life Quality Index.
Adverse events experienced with dupilumab included injection site reactions, eye and eyelid inflammation, and cold sores on the mouth or lips, according to a Regeneron statement.
Dupilumab will be available “later this week,” according to the statement, which noted the wholesale acquisition cost of the medication is expected to be $37,000 annually.
The FDA approval announcement noted that the safety and efficacy of dupilumab had not been established in patients with asthma.
Dupilumab currently is being studied for children with AD in phase II studies and is being studied for other indications: eosinophilic esophagitis in phase II studies, and asthma and nasal polyps in phase III studies.
Dupilumab will be marketed as Dupixent by Regeneron.
PARP inhibitor approved as maintenance for recurrent ovarian cancer
The Food and Drug Administration has approved niraparib, a poly ADP-ribose polymerase (PARP) inhibitor, for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in complete or partial response to platinum-based chemotherapy.
The indication does not require a suspected or confirmed deleterious BRCA mutation.
Median PFS in women with germline BRCA mutations who received niraparib was 21 months, compared with 5.5 months for those on placebo (hazard ratio, 0.26; 95% confidence interval: 0.17-0.41; P less than .0001), according to a study presented at the 2016 European Society for Medical Oncology Congress and published simultaneously in the New England Journal of Medicine.
Among patients who did not have a germline BRCA mutation, median PFS for patients on niraparib was 9.3 months, compared with 3.9 months for those on placebo (HR, 0.45; 95% CI, 0.34-0.61; P less than .0001).
Patients enrolled in NOVA were randomized (2:1) within 8 weeks of the last therapy to either niraparib (300 mg orally daily) or matched placebo. Patients were assigned to one of two cohorts based on the BRACAnalysis CDx. Patients with deleterious or suspected deleterious germline BRCA mutations were assigned to the germline BRCA-mutated cohort (n = 203), and those without germline BRCA mutations were assigned to the nonmutated cohort (n = 350).
Niraparib’s safety was evaluated in 367 patients with platinum-sensitive recurrent ovarian, fallopian tube, and primary peritoneal cancer in the NOVA trial, according to a statement from the FDA.
The most common adverse reactions were thrombocytopenia, anemia, neutropenia, leukopenia, palpitations, nausea, constipation, vomiting, abdominal pain/distention, mucositis/stomatitis, diarrhea, dyspepsia, dry mouth, fatigue, decreased appetite, urinary tract infection, AST/ALT elevation, myalgia, back pain, arthralgia, headache, dizziness, dysgeusia, insomnia, anxiety, nasopharyngitis, dyspnea, cough, rash, and hypertension.
Myelodysplastic syndrome and/or acute myeloid leukemia occurred in 5 of 367 (1.4%) patients receiving niraparib and in 2 of 179 (1.1%) patients assigned to placebo. Grade 3-4 hypertension occurred in 9% of niraparib-treated patients, compared with 2% of patients assigned to placebo, according to the FDA.
Niraparib is the third PARP inhibitor to receive approval for ovarian cancer, following approval of olaparib (Lynparza) for patients with advanced ovarian cancer who have been treated with three or more prior lines of chemotherapy and whose tumors have a deleterious BRCA mutation as detected by an FDA-approved test and accelerated approval of rucaparib (Rubraca) for women with advanced ovarian cancer who have been treated with two or more chemotherapies and whose tumors have a deleterious BRCA mutation as identified by an FDA-approved companion diagnostic test.
The recommended dose and schedule of niraparib is 300 mg taken once daily with or without food.
Full prescribing information is available here.
Niraparib is being marketed as Zejula by Tesaro. Following FDA announcement of the approval, Tesaro announced an expanded development program for niraparib focused on the treatment of front-line metastatic ovarian and lung cancers and metastatic breast cancer.
The Food and Drug Administration has approved niraparib, a poly ADP-ribose polymerase (PARP) inhibitor, for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in complete or partial response to platinum-based chemotherapy.
The indication does not require a suspected or confirmed deleterious BRCA mutation.
Median PFS in women with germline BRCA mutations who received niraparib was 21 months, compared with 5.5 months for those on placebo (hazard ratio, 0.26; 95% confidence interval: 0.17-0.41; P less than .0001), according to a study presented at the 2016 European Society for Medical Oncology Congress and published simultaneously in the New England Journal of Medicine.
Among patients who did not have a germline BRCA mutation, median PFS for patients on niraparib was 9.3 months, compared with 3.9 months for those on placebo (HR, 0.45; 95% CI, 0.34-0.61; P less than .0001).
Patients enrolled in NOVA were randomized (2:1) within 8 weeks of the last therapy to either niraparib (300 mg orally daily) or matched placebo. Patients were assigned to one of two cohorts based on the BRACAnalysis CDx. Patients with deleterious or suspected deleterious germline BRCA mutations were assigned to the germline BRCA-mutated cohort (n = 203), and those without germline BRCA mutations were assigned to the nonmutated cohort (n = 350).
Niraparib’s safety was evaluated in 367 patients with platinum-sensitive recurrent ovarian, fallopian tube, and primary peritoneal cancer in the NOVA trial, according to a statement from the FDA.
The most common adverse reactions were thrombocytopenia, anemia, neutropenia, leukopenia, palpitations, nausea, constipation, vomiting, abdominal pain/distention, mucositis/stomatitis, diarrhea, dyspepsia, dry mouth, fatigue, decreased appetite, urinary tract infection, AST/ALT elevation, myalgia, back pain, arthralgia, headache, dizziness, dysgeusia, insomnia, anxiety, nasopharyngitis, dyspnea, cough, rash, and hypertension.
Myelodysplastic syndrome and/or acute myeloid leukemia occurred in 5 of 367 (1.4%) patients receiving niraparib and in 2 of 179 (1.1%) patients assigned to placebo. Grade 3-4 hypertension occurred in 9% of niraparib-treated patients, compared with 2% of patients assigned to placebo, according to the FDA.
Niraparib is the third PARP inhibitor to receive approval for ovarian cancer, following approval of olaparib (Lynparza) for patients with advanced ovarian cancer who have been treated with three or more prior lines of chemotherapy and whose tumors have a deleterious BRCA mutation as detected by an FDA-approved test and accelerated approval of rucaparib (Rubraca) for women with advanced ovarian cancer who have been treated with two or more chemotherapies and whose tumors have a deleterious BRCA mutation as identified by an FDA-approved companion diagnostic test.
The recommended dose and schedule of niraparib is 300 mg taken once daily with or without food.
Full prescribing information is available here.
Niraparib is being marketed as Zejula by Tesaro. Following FDA announcement of the approval, Tesaro announced an expanded development program for niraparib focused on the treatment of front-line metastatic ovarian and lung cancers and metastatic breast cancer.
The Food and Drug Administration has approved niraparib, a poly ADP-ribose polymerase (PARP) inhibitor, for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in complete or partial response to platinum-based chemotherapy.
The indication does not require a suspected or confirmed deleterious BRCA mutation.
Median PFS in women with germline BRCA mutations who received niraparib was 21 months, compared with 5.5 months for those on placebo (hazard ratio, 0.26; 95% confidence interval: 0.17-0.41; P less than .0001), according to a study presented at the 2016 European Society for Medical Oncology Congress and published simultaneously in the New England Journal of Medicine.
Among patients who did not have a germline BRCA mutation, median PFS for patients on niraparib was 9.3 months, compared with 3.9 months for those on placebo (HR, 0.45; 95% CI, 0.34-0.61; P less than .0001).
Patients enrolled in NOVA were randomized (2:1) within 8 weeks of the last therapy to either niraparib (300 mg orally daily) or matched placebo. Patients were assigned to one of two cohorts based on the BRACAnalysis CDx. Patients with deleterious or suspected deleterious germline BRCA mutations were assigned to the germline BRCA-mutated cohort (n = 203), and those without germline BRCA mutations were assigned to the nonmutated cohort (n = 350).
Niraparib’s safety was evaluated in 367 patients with platinum-sensitive recurrent ovarian, fallopian tube, and primary peritoneal cancer in the NOVA trial, according to a statement from the FDA.
The most common adverse reactions were thrombocytopenia, anemia, neutropenia, leukopenia, palpitations, nausea, constipation, vomiting, abdominal pain/distention, mucositis/stomatitis, diarrhea, dyspepsia, dry mouth, fatigue, decreased appetite, urinary tract infection, AST/ALT elevation, myalgia, back pain, arthralgia, headache, dizziness, dysgeusia, insomnia, anxiety, nasopharyngitis, dyspnea, cough, rash, and hypertension.
Myelodysplastic syndrome and/or acute myeloid leukemia occurred in 5 of 367 (1.4%) patients receiving niraparib and in 2 of 179 (1.1%) patients assigned to placebo. Grade 3-4 hypertension occurred in 9% of niraparib-treated patients, compared with 2% of patients assigned to placebo, according to the FDA.
Niraparib is the third PARP inhibitor to receive approval for ovarian cancer, following approval of olaparib (Lynparza) for patients with advanced ovarian cancer who have been treated with three or more prior lines of chemotherapy and whose tumors have a deleterious BRCA mutation as detected by an FDA-approved test and accelerated approval of rucaparib (Rubraca) for women with advanced ovarian cancer who have been treated with two or more chemotherapies and whose tumors have a deleterious BRCA mutation as identified by an FDA-approved companion diagnostic test.
The recommended dose and schedule of niraparib is 300 mg taken once daily with or without food.
Full prescribing information is available here.
Niraparib is being marketed as Zejula by Tesaro. Following FDA announcement of the approval, Tesaro announced an expanded development program for niraparib focused on the treatment of front-line metastatic ovarian and lung cancers and metastatic breast cancer.