User login
Light Brown and Pink Macule on the Upper Arm
The Diagnosis: Desmoplastic Spitz Nevus
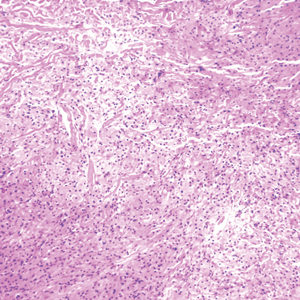
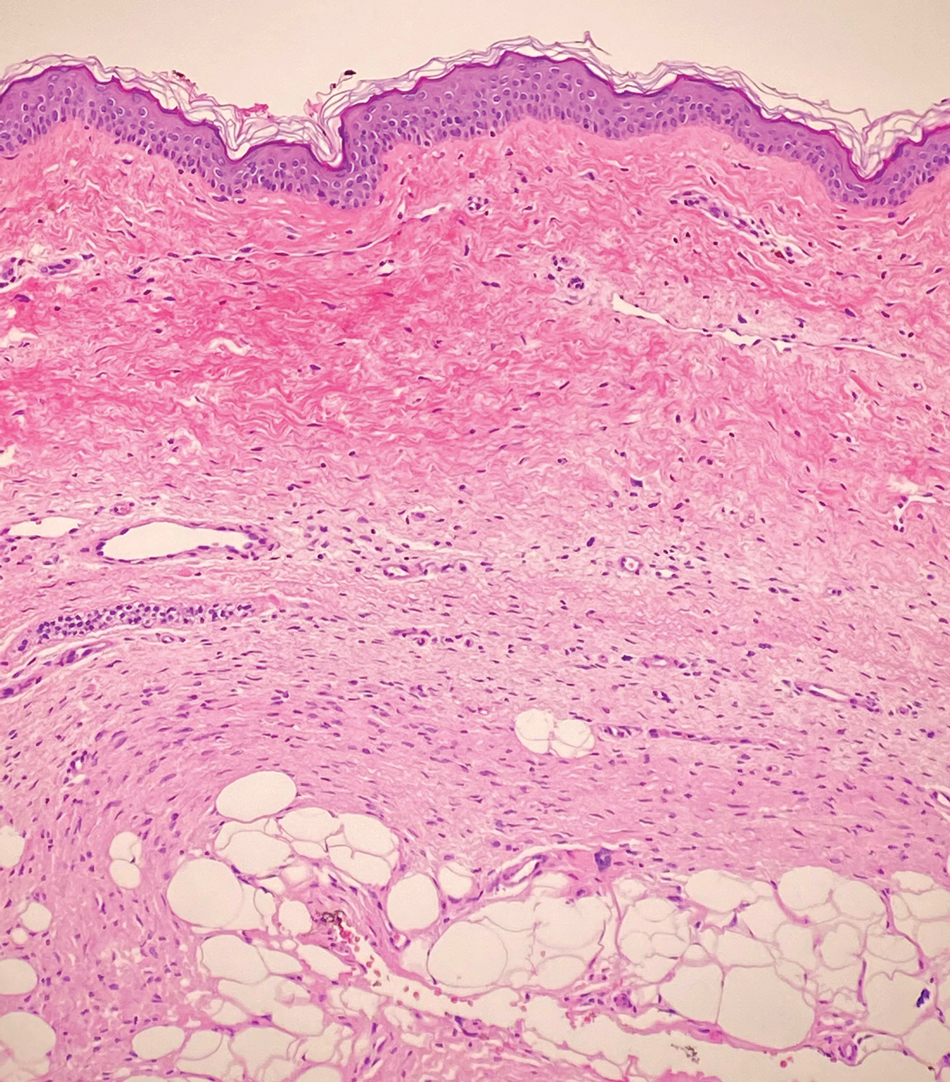
Desmoplastic Spitz nevus is a rare variant of Spitz nevus that commonly presents as a red to brown papule on the head, neck, or extremities. It is pertinent to review the histologic features of this neoplasm, as it can be confused with other more sinister entities such as spitzoid melanoma. Histologically, there is a dermal infiltrate of melanocytes containing eosinophilic cytoplasm and vesicular nuclei. Junctional involvement is rare, and there should be no pagetoid spread.1 This entity features abundant stromal fibrosis formed by dense collagen bundles, low cellular density, and polygonal-shaped melanocytes, which helps to differentiate it from melanoma.2,3 In a retrospective study comparing the characteristics of desmoplastic Spitz nevi with desmoplastic melanoma, desmoplastic Spitz nevi histologically were more symmetric and circumscribed with greater melanocytic maturation and adnexal structure involvement.3 Although this entity demonstrates maturation from the superficial to the deep dermis, it also may feature deep dermal vascular proliferation.4 S-100 and SRY-related HMG box 10, SOX-10, are noted to be positive in desmoplastic Spitz nevi, which can help to differentiate it from nonmelanocytic entities (Figure 1).
 positivity (original magnification ×40).")
Although spitzoid lesions can be ambiguous and difficult even for experts to classify, spitzoid melanoma tends to have a high Breslow thickness, high cell density, marked atypia, and an increased nucleus to cytoplasm ratio.5 Additionally, desmoplastic melanoma was found to more often display “melanocytic junctional nests associated with discohesive cells, variations in size and shape of the nests, lentiginous melanocytic proliferation, actinic elastosis, pagetoid spread, dermal mitosis, perineural involvement and brisk inflammatory infiltrate.”3 Given the challenge of histologically separating desmoplastic Spitz nevi from melanoma, immunostaining can be useful. For example, Hilliard et al6 used a p16 antibody to differentiate desmoplastic Spitz nevi from desmoplastic melanoma, finding that most desmoplastic melanomas (81.8%; n=11) were negative for p16, whereas all desmoplastic Spitz nevi were at least moderately positive. However, another study re-evaluated the utility of p16 in desmoplastic melanoma and found that 72.7% (16/22) were at least focally reactive for the immunostain.7 Thus, caution must be exercised when using p16.
PReferentially expressed Antigen in MElanoma (PRAME) is a newer nuclear immunohistochemical marker that tends to be positive in melanomas and negative in nevi. Desmoplastic Spitz nevi would be expected to be negative for PRAME, while desmoplastic melanoma may be positive; however, this marker seems to be less effective in desmoplastic melanoma than in most other subtypes of the malignancy. In one study, only 35% (n=20) of desmoplastic melanomas were positive for PRAME.8 Likewise, another study showed that some benign Spitz nevi may diffusely express PRAME.9 As such, PRAME should be used prudently.
For cases in which immunohistochemistry is equivocal, molecular testing may aid in differentiating Spitz nevi from melanoma. For example, comparative genomic hybridization has revealed an increased copy number of chromosome 11p in approximately 20% of Spitz nevi cases10; this finding is not seen in melanoma. Mutation analyses of HRas proto-oncogene, GTPase, HRAS; B-Raf proto-oncogene, serine/threonine kinase, BRAF; and NRAS proto-oncogene, GTPase, NRAS, also have shown some promise in distinguishing spitzoid lesions from melanoma, but these analyses may be oversimplified.11 Fluorescence in situ hybridization (FISH) is another diagnostic modality that has been studied to differentiate benign nevi from melanoma. One study challenged the utility of FISH, reporting 7 of 15 desmoplastic melanomas tested positive compared to 0 of 15 sclerotic melanocytic nevi.12 Thus, negative FISH cannot reliably rule out melanoma. Ultimately, a combination of immunostains along with FISH or another genetic study would prove to be most effective in ruling out melanoma in difficult cases. Even then, a dermatopathologist may be faced with a degree of uncertainty.
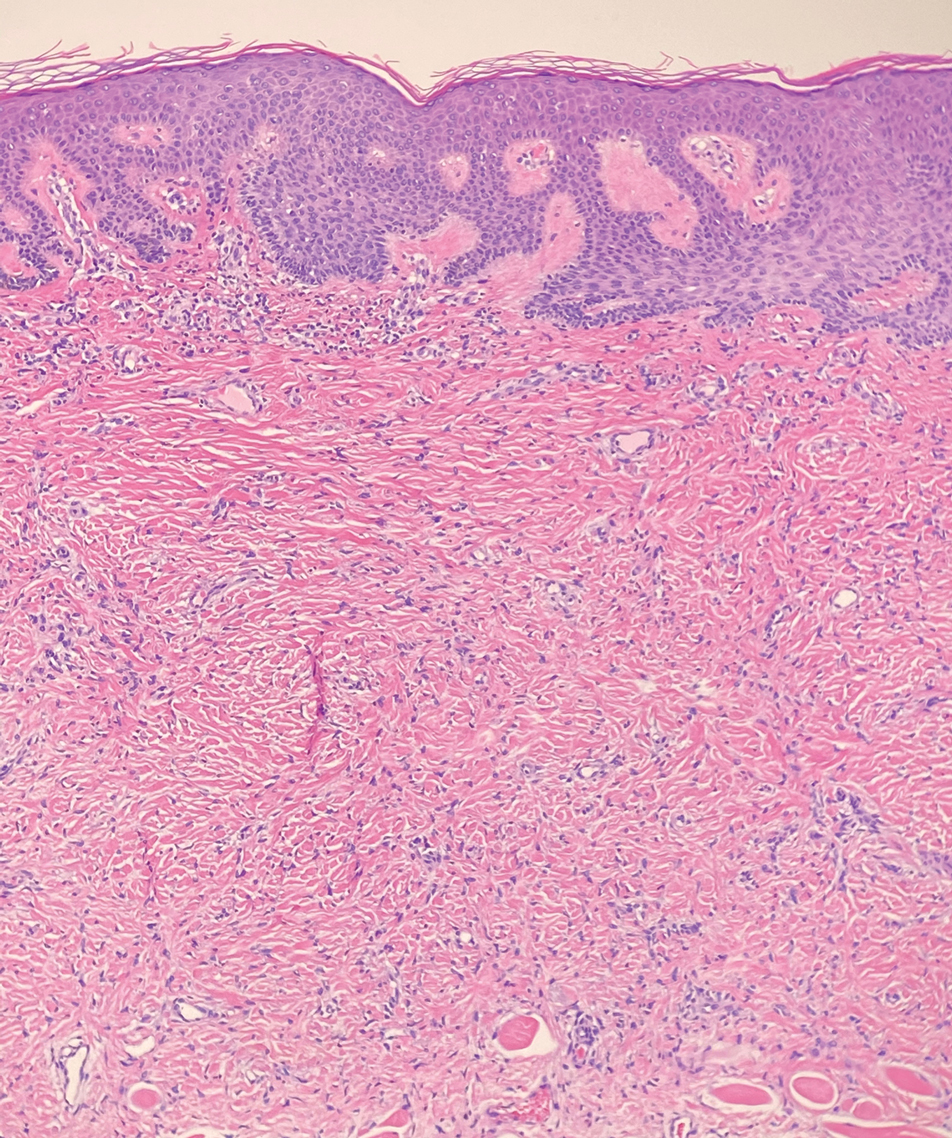
Cellular blue nevi predominantly affect adults younger than 40 years and commonly are seen on the buttocks.13 This benign neoplasm demonstrates areas that are distinctly sclerotic as well as those that are cellular in nature.14 This entity demonstrates a well-circumscribed dermal growth pattern with 2 main populations of cells. The sclerotic portion of the cellular blue nevus mimics that of the blue nevus in that it is noted superficially with irregular margins. The cellular aspect of the nevus features spindle cells contained within well-circumscribed nodules (Figure 2). Stromal melanophages are not uncommon, and some can be observed adjacent to nerve fibers. Although this blue nevus variant displays features of the common blue nevus, its melanocytes track along adnexal and neurovascular structures similar to the deep penetrating nevus and the desmoplastic Spitz nevus. However, these melanocytes are variable in morphology and can appear on a spectrum spanning from pale and lightly pigmented to clear.15

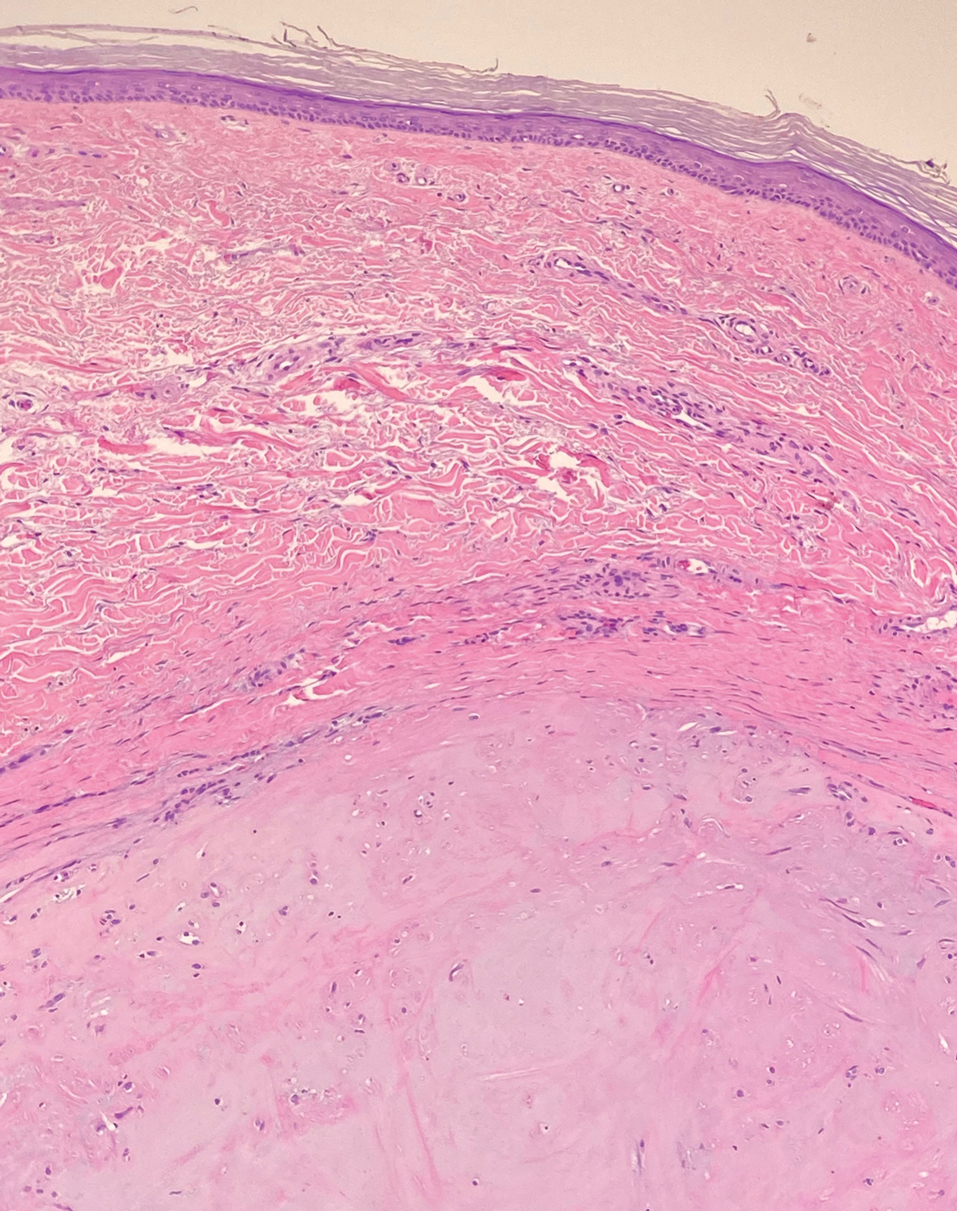
The breast is the most common site of origin of tumor metastasis to the skin. These cutaneous metastases can vary in both their clinical and histological presentations. For example, cutaneous metastatic breast adenocarcinoma often can present clinically as pink-violaceous papules and plaques on the breast or on other parts of the body. Histologically, it can demonstrate a varying degree of patterns such as collagen infiltration by single cells, cords, tubules, and sheets of atypical cells (Figure 3) that can be observed together in areas of mucin or can form glandular structures.16 Metastatic breast carcinoma is noted to be positive for gross cystic disease fluid protein-15, estrogen receptor, and cytokeratin 7, which can help differentiate this entity from other tumors of glandular origin.16 Although rare, primary melanoma of the breast has been reported in the literature.17,18 These malignant melanocytic lesions easily could be differentiated from other breast tumors such as adenocarcinoma using immunohistochemical staining patterns.

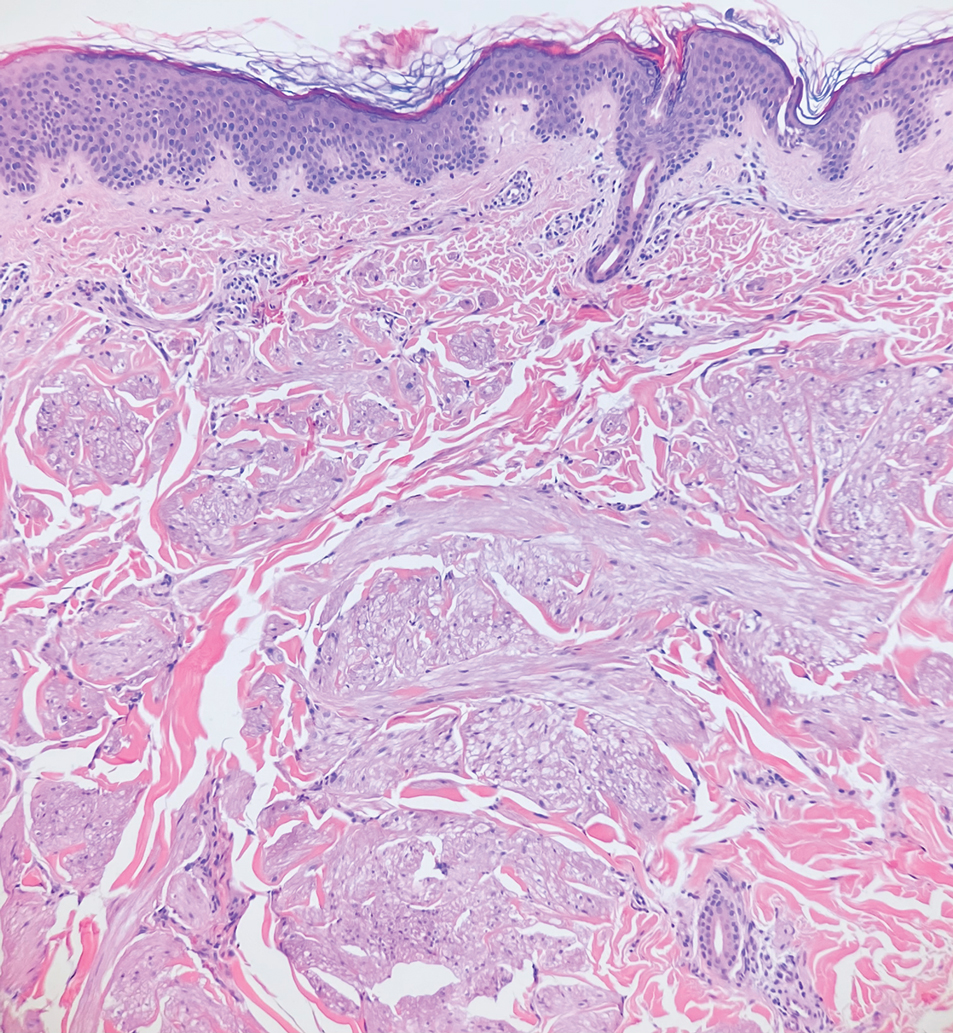
Deep penetrating nevi most often are observed clinically as blue, brown, or black papules or nodules on the head or neck.19 Histologically, this lesion features a wedge-shaped infiltrate of deep dermal melanocytes with oval nuclei. It commonly extends to the reticular dermis or further into the subcutis (Figure 4).20,21 This neoplasm frequently tracks along adnexal and neurovascular structures, resulting in a plexiform appearance.22 The adnexal involvement of deep penetrating nevi is a shared feature with desmoplastic Spitz nevi. The presence of any number of melanophages is characteristic of this lesion.23 Lastly, there is a well-documented association between β-catenin mutations and deep penetrating nevi.24 Multicentric reticulohistiocytosis (MRH) is a rare form of non-Langerhans cell histiocytosis that has the pathognomonic clinical finding of pink-red papules (coral beading) with a predilection for acral surfaces. Histology of affected skin reveals a dermal infiltrate of ground glass as well as eosinophilic histiocytes that most often stain positive for CD68 and human alveolar macrophage 56 but negative for S-100 and CD1a (Figure 5).25 Although MRH is rare, negative staining for S-100 could serve as a useful diagnostic clue to differentiate it from other entities that are positive for S-100, such as the desmoplastic Spitz nevus. Arthritis mutilans is a potential complication of MRH, but a reported association with an underlying malignancy is seen in approximately 25% of cases.26 Thus, the cutaneous, rheumatologic, and oncologic implications of this disease help to distinguish it from other differential diagnoses that may be considered.


- Luzar B, Bastian BC, North JP, et al. Melanocytic nevi. In: Calonje E, Brenn T, Lazar AJ, et al, eds. McKee’s Pathology of the Skin. 5th ed. Elsevier; 2020:1275-1280.
- Busam KJ, Gerami P. Spitz nevi. In: Busam KJ, Gerami P, Scolyer RA, eds. Pathology of Melanocytic Tumors. Elsevier; 2019:37-60.
- Nojavan H, Cribier B, Mehregan DR. Desmoplastic Spitz nevus: a histopathological review and comparison with desmoplastic melanoma [in French]. Ann Dermatol Venereol. 2009;136:689-695.
- Tomizawa K. Desmoplastic Spitz nevus showing vascular proliferation more prominently in the deep portion. Am J Dermatopathol. 2002;24:184-185.
- Requena C, Botella R, Nagore E, et al. Characteristics of spitzoid melanoma and clues for differential diagnosis with Spitz nevus. Am J Dermatopathol. 2012;34:478-486.
- Hilliard NJ, Krahl D, Sellheyer K. p16 expression differentiates between desmoplastic Spitz nevus and desmoplastic melanoma. J Cutan Pathol. 2009;36:753-759.
- Blokhin E, Pulitzer M, Busam KJ. Immunohistochemical expression of p16 in desmoplastic melanoma. J Cutan Pathol. 2013;40:796-800.
- Lezcano C, Jungbluth AA, Nehal KS, et al. PRAME expression in melanocytic tumors. Am J Surg Pathol. 2018;42:1456-1465.
- Raghavan SS, Wang JY, Kwok S, et al. PRAME expression in melanocytic proliferations with intermediate histopathologic or spitzoid features. J Cutan Pathol. 2020;47:1123-1131.
- Bauer J, Bastian BC. DNA copy number changes in the diagnosis of melanocytic tumors [in German]. Pathologe. 2007;28:464-473.
- Luo S, Sepehr A, Tsao H. Spitz nevi and other spitzoid lesions part I. background and diagnoses. J Am Acad Dermatol. 2011;65:1073-1084.
- Gerami P, Beilfuss B, Haghighat Z, et al. Fluorescence in situ hybridization as an ancillary method for the distinction of desmoplastic melanomas from sclerosing melanocytic nevi. J Cutan Pathol. 2011;38:329-334.
- Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2017; 37:401-415.
- Rodriguez HA, Ackerman LV. Cellular blue nevus. clinicopathologic study of forty-five cases. Cancer. 1968;21:393-405.
- Phadke PA, Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2011;31:345-358.
- Ko CJ. Metastatic tumors and simulators. In: Elston DM, Ferringer T, eds. Dermatopathology. 3rd ed. Elsevier Limited; 2019:496-504.
- Drueppel D, Schultheis B, Solass W, et al. Primary malignant melanoma of the breast: case report and review of the literature. Anticancer Res. 2015;35:1709-1713.
- Kurul S, Tas¸ F, Büyükbabani N, et al. Different manifestations of malignant melanoma in the breast: a report of 12 cases and a review of the literature. Jpn J Clin Oncol. 2005;35:202-206.
- Strazzula L, Senna MM, Yasuda M, et al. The deep penetrating nevus. J Am Acad Dermatol. 2014;71:1234-1240.
- Mehregan DA, Mehregan AH. Deep penetrating nevus. Arch Dermatol. 1993;129:328-331.
- Robson A, Morley-Quante M, Hempel H, et al. Deep penetrating naevus: clinicopathological study of 31 cases with further delineation of histological features allowing distinction from other pigmented benign melanocytic lesions and melanoma. Histopathology. 2003;43:529-537.
- Luzar B, Calonje E. Deep penetrating nevus: a review. Arch Pathol Lab Med. 2011;135:321-326.
- Cooper PH. Deep penetrating (plexiform spindle cell) nevus. a frequent participant in combined nevus. J Cutan Pathol. 1992;19:172-180.
- de la Fouchardière A, Caillot C, Jacquemus J, et al. β-Catenin nuclear expression discriminates deep penetrating nevi from other cutaneous melanocytic tumors. Virchows Arch. 2019;474:539-550.
- Gorman JD, Danning C, Schumacher HR, et al. Multicentric reticulohistiocytosis: case report with immunohistochemical analysis and literature review. Arthritis Rheum. 2000;43:930-938.
- Selmi C, Greenspan A, Huntley A, et al. Multicentric reticulohistiocytosis: a critical review. Curr Rheumatol Rep. 2015;17:511.
The Diagnosis: Desmoplastic Spitz Nevus
Desmoplastic Spitz nevus is a rare variant of Spitz nevus that commonly presents as a red to brown papule on the head, neck, or extremities. It is pertinent to review the histologic features of this neoplasm, as it can be confused with other more sinister entities such as spitzoid melanoma. Histologically, there is a dermal infiltrate of melanocytes containing eosinophilic cytoplasm and vesicular nuclei. Junctional involvement is rare, and there should be no pagetoid spread.1 This entity features abundant stromal fibrosis formed by dense collagen bundles, low cellular density, and polygonal-shaped melanocytes, which helps to differentiate it from melanoma.2,3 In a retrospective study comparing the characteristics of desmoplastic Spitz nevi with desmoplastic melanoma, desmoplastic Spitz nevi histologically were more symmetric and circumscribed with greater melanocytic maturation and adnexal structure involvement.3 Although this entity demonstrates maturation from the superficial to the deep dermis, it also may feature deep dermal vascular proliferation.4 S-100 and SRY-related HMG box 10, SOX-10, are noted to be positive in desmoplastic Spitz nevi, which can help to differentiate it from nonmelanocytic entities (Figure 1).
Although spitzoid lesions can be ambiguous and difficult even for experts to classify, spitzoid melanoma tends to have a high Breslow thickness, high cell density, marked atypia, and an increased nucleus to cytoplasm ratio.5 Additionally, desmoplastic melanoma was found to more often display “melanocytic junctional nests associated with discohesive cells, variations in size and shape of the nests, lentiginous melanocytic proliferation, actinic elastosis, pagetoid spread, dermal mitosis, perineural involvement and brisk inflammatory infiltrate.”3 Given the challenge of histologically separating desmoplastic Spitz nevi from melanoma, immunostaining can be useful. For example, Hilliard et al6 used a p16 antibody to differentiate desmoplastic Spitz nevi from desmoplastic melanoma, finding that most desmoplastic melanomas (81.8%; n=11) were negative for p16, whereas all desmoplastic Spitz nevi were at least moderately positive. However, another study re-evaluated the utility of p16 in desmoplastic melanoma and found that 72.7% (16/22) were at least focally reactive for the immunostain.7 Thus, caution must be exercised when using p16.
PReferentially expressed Antigen in MElanoma (PRAME) is a newer nuclear immunohistochemical marker that tends to be positive in melanomas and negative in nevi. Desmoplastic Spitz nevi would be expected to be negative for PRAME, while desmoplastic melanoma may be positive; however, this marker seems to be less effective in desmoplastic melanoma than in most other subtypes of the malignancy. In one study, only 35% (n=20) of desmoplastic melanomas were positive for PRAME.8 Likewise, another study showed that some benign Spitz nevi may diffusely express PRAME.9 As such, PRAME should be used prudently.
For cases in which immunohistochemistry is equivocal, molecular testing may aid in differentiating Spitz nevi from melanoma. For example, comparative genomic hybridization has revealed an increased copy number of chromosome 11p in approximately 20% of Spitz nevi cases10; this finding is not seen in melanoma. Mutation analyses of HRas proto-oncogene, GTPase, HRAS; B-Raf proto-oncogene, serine/threonine kinase, BRAF; and NRAS proto-oncogene, GTPase, NRAS, also have shown some promise in distinguishing spitzoid lesions from melanoma, but these analyses may be oversimplified.11 Fluorescence in situ hybridization (FISH) is another diagnostic modality that has been studied to differentiate benign nevi from melanoma. One study challenged the utility of FISH, reporting 7 of 15 desmoplastic melanomas tested positive compared to 0 of 15 sclerotic melanocytic nevi.12 Thus, negative FISH cannot reliably rule out melanoma. Ultimately, a combination of immunostains along with FISH or another genetic study would prove to be most effective in ruling out melanoma in difficult cases. Even then, a dermatopathologist may be faced with a degree of uncertainty.
Cellular blue nevi predominantly affect adults younger than 40 years and commonly are seen on the buttocks.13 This benign neoplasm demonstrates areas that are distinctly sclerotic as well as those that are cellular in nature.14 This entity demonstrates a well-circumscribed dermal growth pattern with 2 main populations of cells. The sclerotic portion of the cellular blue nevus mimics that of the blue nevus in that it is noted superficially with irregular margins. The cellular aspect of the nevus features spindle cells contained within well-circumscribed nodules (Figure 2). Stromal melanophages are not uncommon, and some can be observed adjacent to nerve fibers. Although this blue nevus variant displays features of the common blue nevus, its melanocytes track along adnexal and neurovascular structures similar to the deep penetrating nevus and the desmoplastic Spitz nevus. However, these melanocytes are variable in morphology and can appear on a spectrum spanning from pale and lightly pigmented to clear.15
The breast is the most common site of origin of tumor metastasis to the skin. These cutaneous metastases can vary in both their clinical and histological presentations. For example, cutaneous metastatic breast adenocarcinoma often can present clinically as pink-violaceous papules and plaques on the breast or on other parts of the body. Histologically, it can demonstrate a varying degree of patterns such as collagen infiltration by single cells, cords, tubules, and sheets of atypical cells (Figure 3) that can be observed together in areas of mucin or can form glandular structures.16 Metastatic breast carcinoma is noted to be positive for gross cystic disease fluid protein-15, estrogen receptor, and cytokeratin 7, which can help differentiate this entity from other tumors of glandular origin.16 Although rare, primary melanoma of the breast has been reported in the literature.17,18 These malignant melanocytic lesions easily could be differentiated from other breast tumors such as adenocarcinoma using immunohistochemical staining patterns.
Deep penetrating nevi most often are observed clinically as blue, brown, or black papules or nodules on the head or neck.19 Histologically, this lesion features a wedge-shaped infiltrate of deep dermal melanocytes with oval nuclei. It commonly extends to the reticular dermis or further into the subcutis (Figure 4).20,21 This neoplasm frequently tracks along adnexal and neurovascular structures, resulting in a plexiform appearance.22 The adnexal involvement of deep penetrating nevi is a shared feature with desmoplastic Spitz nevi. The presence of any number of melanophages is characteristic of this lesion.23 Lastly, there is a well-documented association between β-catenin mutations and deep penetrating nevi.24 Multicentric reticulohistiocytosis (MRH) is a rare form of non-Langerhans cell histiocytosis that has the pathognomonic clinical finding of pink-red papules (coral beading) with a predilection for acral surfaces. Histology of affected skin reveals a dermal infiltrate of ground glass as well as eosinophilic histiocytes that most often stain positive for CD68 and human alveolar macrophage 56 but negative for S-100 and CD1a (Figure 5).25 Although MRH is rare, negative staining for S-100 could serve as a useful diagnostic clue to differentiate it from other entities that are positive for S-100, such as the desmoplastic Spitz nevus. Arthritis mutilans is a potential complication of MRH, but a reported association with an underlying malignancy is seen in approximately 25% of cases.26 Thus, the cutaneous, rheumatologic, and oncologic implications of this disease help to distinguish it from other differential diagnoses that may be considered.
The Diagnosis: Desmoplastic Spitz Nevus
Desmoplastic Spitz nevus is a rare variant of Spitz nevus that commonly presents as a red to brown papule on the head, neck, or extremities. It is pertinent to review the histologic features of this neoplasm, as it can be confused with other more sinister entities such as spitzoid melanoma. Histologically, there is a dermal infiltrate of melanocytes containing eosinophilic cytoplasm and vesicular nuclei. Junctional involvement is rare, and there should be no pagetoid spread.1 This entity features abundant stromal fibrosis formed by dense collagen bundles, low cellular density, and polygonal-shaped melanocytes, which helps to differentiate it from melanoma.2,3 In a retrospective study comparing the characteristics of desmoplastic Spitz nevi with desmoplastic melanoma, desmoplastic Spitz nevi histologically were more symmetric and circumscribed with greater melanocytic maturation and adnexal structure involvement.3 Although this entity demonstrates maturation from the superficial to the deep dermis, it also may feature deep dermal vascular proliferation.4 S-100 and SRY-related HMG box 10, SOX-10, are noted to be positive in desmoplastic Spitz nevi, which can help to differentiate it from nonmelanocytic entities (Figure 1).
Although spitzoid lesions can be ambiguous and difficult even for experts to classify, spitzoid melanoma tends to have a high Breslow thickness, high cell density, marked atypia, and an increased nucleus to cytoplasm ratio.5 Additionally, desmoplastic melanoma was found to more often display “melanocytic junctional nests associated with discohesive cells, variations in size and shape of the nests, lentiginous melanocytic proliferation, actinic elastosis, pagetoid spread, dermal mitosis, perineural involvement and brisk inflammatory infiltrate.”3 Given the challenge of histologically separating desmoplastic Spitz nevi from melanoma, immunostaining can be useful. For example, Hilliard et al6 used a p16 antibody to differentiate desmoplastic Spitz nevi from desmoplastic melanoma, finding that most desmoplastic melanomas (81.8%; n=11) were negative for p16, whereas all desmoplastic Spitz nevi were at least moderately positive. However, another study re-evaluated the utility of p16 in desmoplastic melanoma and found that 72.7% (16/22) were at least focally reactive for the immunostain.7 Thus, caution must be exercised when using p16.
PReferentially expressed Antigen in MElanoma (PRAME) is a newer nuclear immunohistochemical marker that tends to be positive in melanomas and negative in nevi. Desmoplastic Spitz nevi would be expected to be negative for PRAME, while desmoplastic melanoma may be positive; however, this marker seems to be less effective in desmoplastic melanoma than in most other subtypes of the malignancy. In one study, only 35% (n=20) of desmoplastic melanomas were positive for PRAME.8 Likewise, another study showed that some benign Spitz nevi may diffusely express PRAME.9 As such, PRAME should be used prudently.
For cases in which immunohistochemistry is equivocal, molecular testing may aid in differentiating Spitz nevi from melanoma. For example, comparative genomic hybridization has revealed an increased copy number of chromosome 11p in approximately 20% of Spitz nevi cases10; this finding is not seen in melanoma. Mutation analyses of HRas proto-oncogene, GTPase, HRAS; B-Raf proto-oncogene, serine/threonine kinase, BRAF; and NRAS proto-oncogene, GTPase, NRAS, also have shown some promise in distinguishing spitzoid lesions from melanoma, but these analyses may be oversimplified.11 Fluorescence in situ hybridization (FISH) is another diagnostic modality that has been studied to differentiate benign nevi from melanoma. One study challenged the utility of FISH, reporting 7 of 15 desmoplastic melanomas tested positive compared to 0 of 15 sclerotic melanocytic nevi.12 Thus, negative FISH cannot reliably rule out melanoma. Ultimately, a combination of immunostains along with FISH or another genetic study would prove to be most effective in ruling out melanoma in difficult cases. Even then, a dermatopathologist may be faced with a degree of uncertainty.
Cellular blue nevi predominantly affect adults younger than 40 years and commonly are seen on the buttocks.13 This benign neoplasm demonstrates areas that are distinctly sclerotic as well as those that are cellular in nature.14 This entity demonstrates a well-circumscribed dermal growth pattern with 2 main populations of cells. The sclerotic portion of the cellular blue nevus mimics that of the blue nevus in that it is noted superficially with irregular margins. The cellular aspect of the nevus features spindle cells contained within well-circumscribed nodules (Figure 2). Stromal melanophages are not uncommon, and some can be observed adjacent to nerve fibers. Although this blue nevus variant displays features of the common blue nevus, its melanocytes track along adnexal and neurovascular structures similar to the deep penetrating nevus and the desmoplastic Spitz nevus. However, these melanocytes are variable in morphology and can appear on a spectrum spanning from pale and lightly pigmented to clear.15
The breast is the most common site of origin of tumor metastasis to the skin. These cutaneous metastases can vary in both their clinical and histological presentations. For example, cutaneous metastatic breast adenocarcinoma often can present clinically as pink-violaceous papules and plaques on the breast or on other parts of the body. Histologically, it can demonstrate a varying degree of patterns such as collagen infiltration by single cells, cords, tubules, and sheets of atypical cells (Figure 3) that can be observed together in areas of mucin or can form glandular structures.16 Metastatic breast carcinoma is noted to be positive for gross cystic disease fluid protein-15, estrogen receptor, and cytokeratin 7, which can help differentiate this entity from other tumors of glandular origin.16 Although rare, primary melanoma of the breast has been reported in the literature.17,18 These malignant melanocytic lesions easily could be differentiated from other breast tumors such as adenocarcinoma using immunohistochemical staining patterns.
Deep penetrating nevi most often are observed clinically as blue, brown, or black papules or nodules on the head or neck.19 Histologically, this lesion features a wedge-shaped infiltrate of deep dermal melanocytes with oval nuclei. It commonly extends to the reticular dermis or further into the subcutis (Figure 4).20,21 This neoplasm frequently tracks along adnexal and neurovascular structures, resulting in a plexiform appearance.22 The adnexal involvement of deep penetrating nevi is a shared feature with desmoplastic Spitz nevi. The presence of any number of melanophages is characteristic of this lesion.23 Lastly, there is a well-documented association between β-catenin mutations and deep penetrating nevi.24 Multicentric reticulohistiocytosis (MRH) is a rare form of non-Langerhans cell histiocytosis that has the pathognomonic clinical finding of pink-red papules (coral beading) with a predilection for acral surfaces. Histology of affected skin reveals a dermal infiltrate of ground glass as well as eosinophilic histiocytes that most often stain positive for CD68 and human alveolar macrophage 56 but negative for S-100 and CD1a (Figure 5).25 Although MRH is rare, negative staining for S-100 could serve as a useful diagnostic clue to differentiate it from other entities that are positive for S-100, such as the desmoplastic Spitz nevus. Arthritis mutilans is a potential complication of MRH, but a reported association with an underlying malignancy is seen in approximately 25% of cases.26 Thus, the cutaneous, rheumatologic, and oncologic implications of this disease help to distinguish it from other differential diagnoses that may be considered.
- Luzar B, Bastian BC, North JP, et al. Melanocytic nevi. In: Calonje E, Brenn T, Lazar AJ, et al, eds. McKee’s Pathology of the Skin. 5th ed. Elsevier; 2020:1275-1280.
- Busam KJ, Gerami P. Spitz nevi. In: Busam KJ, Gerami P, Scolyer RA, eds. Pathology of Melanocytic Tumors. Elsevier; 2019:37-60.
- Nojavan H, Cribier B, Mehregan DR. Desmoplastic Spitz nevus: a histopathological review and comparison with desmoplastic melanoma [in French]. Ann Dermatol Venereol. 2009;136:689-695.
- Tomizawa K. Desmoplastic Spitz nevus showing vascular proliferation more prominently in the deep portion. Am J Dermatopathol. 2002;24:184-185.
- Requena C, Botella R, Nagore E, et al. Characteristics of spitzoid melanoma and clues for differential diagnosis with Spitz nevus. Am J Dermatopathol. 2012;34:478-486.
- Hilliard NJ, Krahl D, Sellheyer K. p16 expression differentiates between desmoplastic Spitz nevus and desmoplastic melanoma. J Cutan Pathol. 2009;36:753-759.
- Blokhin E, Pulitzer M, Busam KJ. Immunohistochemical expression of p16 in desmoplastic melanoma. J Cutan Pathol. 2013;40:796-800.
- Lezcano C, Jungbluth AA, Nehal KS, et al. PRAME expression in melanocytic tumors. Am J Surg Pathol. 2018;42:1456-1465.
- Raghavan SS, Wang JY, Kwok S, et al. PRAME expression in melanocytic proliferations with intermediate histopathologic or spitzoid features. J Cutan Pathol. 2020;47:1123-1131.
- Bauer J, Bastian BC. DNA copy number changes in the diagnosis of melanocytic tumors [in German]. Pathologe. 2007;28:464-473.
- Luo S, Sepehr A, Tsao H. Spitz nevi and other spitzoid lesions part I. background and diagnoses. J Am Acad Dermatol. 2011;65:1073-1084.
- Gerami P, Beilfuss B, Haghighat Z, et al. Fluorescence in situ hybridization as an ancillary method for the distinction of desmoplastic melanomas from sclerosing melanocytic nevi. J Cutan Pathol. 2011;38:329-334.
- Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2017; 37:401-415.
- Rodriguez HA, Ackerman LV. Cellular blue nevus. clinicopathologic study of forty-five cases. Cancer. 1968;21:393-405.
- Phadke PA, Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2011;31:345-358.
- Ko CJ. Metastatic tumors and simulators. In: Elston DM, Ferringer T, eds. Dermatopathology. 3rd ed. Elsevier Limited; 2019:496-504.
- Drueppel D, Schultheis B, Solass W, et al. Primary malignant melanoma of the breast: case report and review of the literature. Anticancer Res. 2015;35:1709-1713.
- Kurul S, Tas¸ F, Büyükbabani N, et al. Different manifestations of malignant melanoma in the breast: a report of 12 cases and a review of the literature. Jpn J Clin Oncol. 2005;35:202-206.
- Strazzula L, Senna MM, Yasuda M, et al. The deep penetrating nevus. J Am Acad Dermatol. 2014;71:1234-1240.
- Mehregan DA, Mehregan AH. Deep penetrating nevus. Arch Dermatol. 1993;129:328-331.
- Robson A, Morley-Quante M, Hempel H, et al. Deep penetrating naevus: clinicopathological study of 31 cases with further delineation of histological features allowing distinction from other pigmented benign melanocytic lesions and melanoma. Histopathology. 2003;43:529-537.
- Luzar B, Calonje E. Deep penetrating nevus: a review. Arch Pathol Lab Med. 2011;135:321-326.
- Cooper PH. Deep penetrating (plexiform spindle cell) nevus. a frequent participant in combined nevus. J Cutan Pathol. 1992;19:172-180.
- de la Fouchardière A, Caillot C, Jacquemus J, et al. β-Catenin nuclear expression discriminates deep penetrating nevi from other cutaneous melanocytic tumors. Virchows Arch. 2019;474:539-550.
- Gorman JD, Danning C, Schumacher HR, et al. Multicentric reticulohistiocytosis: case report with immunohistochemical analysis and literature review. Arthritis Rheum. 2000;43:930-938.
- Selmi C, Greenspan A, Huntley A, et al. Multicentric reticulohistiocytosis: a critical review. Curr Rheumatol Rep. 2015;17:511.
- Luzar B, Bastian BC, North JP, et al. Melanocytic nevi. In: Calonje E, Brenn T, Lazar AJ, et al, eds. McKee’s Pathology of the Skin. 5th ed. Elsevier; 2020:1275-1280.
- Busam KJ, Gerami P. Spitz nevi. In: Busam KJ, Gerami P, Scolyer RA, eds. Pathology of Melanocytic Tumors. Elsevier; 2019:37-60.
- Nojavan H, Cribier B, Mehregan DR. Desmoplastic Spitz nevus: a histopathological review and comparison with desmoplastic melanoma [in French]. Ann Dermatol Venereol. 2009;136:689-695.
- Tomizawa K. Desmoplastic Spitz nevus showing vascular proliferation more prominently in the deep portion. Am J Dermatopathol. 2002;24:184-185.
- Requena C, Botella R, Nagore E, et al. Characteristics of spitzoid melanoma and clues for differential diagnosis with Spitz nevus. Am J Dermatopathol. 2012;34:478-486.
- Hilliard NJ, Krahl D, Sellheyer K. p16 expression differentiates between desmoplastic Spitz nevus and desmoplastic melanoma. J Cutan Pathol. 2009;36:753-759.
- Blokhin E, Pulitzer M, Busam KJ. Immunohistochemical expression of p16 in desmoplastic melanoma. J Cutan Pathol. 2013;40:796-800.
- Lezcano C, Jungbluth AA, Nehal KS, et al. PRAME expression in melanocytic tumors. Am J Surg Pathol. 2018;42:1456-1465.
- Raghavan SS, Wang JY, Kwok S, et al. PRAME expression in melanocytic proliferations with intermediate histopathologic or spitzoid features. J Cutan Pathol. 2020;47:1123-1131.
- Bauer J, Bastian BC. DNA copy number changes in the diagnosis of melanocytic tumors [in German]. Pathologe. 2007;28:464-473.
- Luo S, Sepehr A, Tsao H. Spitz nevi and other spitzoid lesions part I. background and diagnoses. J Am Acad Dermatol. 2011;65:1073-1084.
- Gerami P, Beilfuss B, Haghighat Z, et al. Fluorescence in situ hybridization as an ancillary method for the distinction of desmoplastic melanomas from sclerosing melanocytic nevi. J Cutan Pathol. 2011;38:329-334.
- Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2017; 37:401-415.
- Rodriguez HA, Ackerman LV. Cellular blue nevus. clinicopathologic study of forty-five cases. Cancer. 1968;21:393-405.
- Phadke PA, Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2011;31:345-358.
- Ko CJ. Metastatic tumors and simulators. In: Elston DM, Ferringer T, eds. Dermatopathology. 3rd ed. Elsevier Limited; 2019:496-504.
- Drueppel D, Schultheis B, Solass W, et al. Primary malignant melanoma of the breast: case report and review of the literature. Anticancer Res. 2015;35:1709-1713.
- Kurul S, Tas¸ F, Büyükbabani N, et al. Different manifestations of malignant melanoma in the breast: a report of 12 cases and a review of the literature. Jpn J Clin Oncol. 2005;35:202-206.
- Strazzula L, Senna MM, Yasuda M, et al. The deep penetrating nevus. J Am Acad Dermatol. 2014;71:1234-1240.
- Mehregan DA, Mehregan AH. Deep penetrating nevus. Arch Dermatol. 1993;129:328-331.
- Robson A, Morley-Quante M, Hempel H, et al. Deep penetrating naevus: clinicopathological study of 31 cases with further delineation of histological features allowing distinction from other pigmented benign melanocytic lesions and melanoma. Histopathology. 2003;43:529-537.
- Luzar B, Calonje E. Deep penetrating nevus: a review. Arch Pathol Lab Med. 2011;135:321-326.
- Cooper PH. Deep penetrating (plexiform spindle cell) nevus. a frequent participant in combined nevus. J Cutan Pathol. 1992;19:172-180.
- de la Fouchardière A, Caillot C, Jacquemus J, et al. β-Catenin nuclear expression discriminates deep penetrating nevi from other cutaneous melanocytic tumors. Virchows Arch. 2019;474:539-550.
- Gorman JD, Danning C, Schumacher HR, et al. Multicentric reticulohistiocytosis: case report with immunohistochemical analysis and literature review. Arthritis Rheum. 2000;43:930-938.
- Selmi C, Greenspan A, Huntley A, et al. Multicentric reticulohistiocytosis: a critical review. Curr Rheumatol Rep. 2015;17:511.
A 37-year-old woman with a history of fibrocystic breast disease and a family history of breast cancer presented with a light brown macule on the right upper arm of 10 years’ duration. The patient first noticed this macule 10 years prior; however, within the last 4 months she noticed a small amount of homogenous darkening and occasional pruritus. Physical examination revealed a 4.0-mm, light brown and pink macule on the right upper arm. Dermoscopy showed a homogenous pigment network with reticular lines and branched streaks centrally. No crystalline structures, milky red globules, or pseudopods were appreciated. A tangential shave biopsy was obtained and submitted for hematoxylin and eosin staining.
.")
.")
Enlarging Nodule on the Back
The Diagnosis: Cutaneous Myxoma
Microscopic analysis showed features of cutaneous myxoma (quiz images). The epidermis was essentially unremarkable. Stellate to spindle cells with bland nuclear chromatin were present in the dermis with abundant pools of myxoid stroma. Colloidal iron staining highlighted the markedly increased dermal mucin.
Cutaneous myxomas (also referred to as superficial angiomyxomas) are rare, well-demarcated tumors of the dermis and subcutis.1,2 They can present as solitary, fleshcolored nodules on the trunk, lower extremities, head, or neck, and they often measure between 1 and 5 cm.2,3 Histologically, cutaneous myxomas are hypocellular with some stellate fibroblasts, occasional epithelial structures, and an abundant myxoid stroma, with notable thinwalled small blood vessels.2,4 These lesions contain pools of mucin and are positive for mesenchymal mucin stains such as colloidal iron and Alcian blue.1 Moreover, perivascular neutrophils are a distinguishing characteristic of cutaneous myxomas.4
Multiple cutaneous myxomas should raise concern for Carney complex,1,5 a genodermatologic syndrome that arises due to a mutation in the protein kinase CAMP-dependent type I regulatory subunit alpha gene, PRKAR1A, on chromosome 2.1,5 Additional cutaneous manifestations include blue nevi, lentigines, and café-aulait macules.5 Carney complex also is known for endocrine overactivity and cardiac myxomas, which can cause serious embolic complications.1
Recommended management is complete excision with close follow-up, as these lesions may recur in up to one-third of cases. Although there is a potential for recurrence, metastases are uncommon.3 Even without recurrence in the presenting location, follow-up should include screening for manifestations of Carney complex.1,3
The clinical and histological differential for cutaneous myxoma may include nerve sheath myxoma or neurofibroma. A nerve sheath myxoma is a dermal tumor that manifests as a solitary, flesh-colored nodule, measuring less than 2 cm. These lesions commonly present on the head, neck, and upper body.6 Cutaneous myxomas can grow larger than 2 cm, but these two lesions have a great deal of overlap in their other features.3,6 Thus, histology can be used to distinguish them.

Nerve sheath myxomas are circumscribed nonencapsulated tumors of the dermis composed of multilobular aggregates of spindle to epithelioid cells in a mucinous matrix (Figure 1). Clefts often are present around the cell aggregates. Despite previously being termed myxoid neurothekeomas, nerve sheath myxomas are S-100 positive, whereas cellular neurothekeomas are S-100 negative and likely not of neural origin. Cutaneous myxomas, in contrast to nerve sheath myxomas, are S-100 negative. Nerve sheath myxomas are more cellular and lack the characteristic mucin pools compared with cutaneous myxomas.1,2,6 Neurofibromas frequently are flesh colored and pedunculated, as was the lesion in our patient, yet they are vastly different microscopically. The stroma of neurofibromas can vary, but cellularity typically is greater than a cutaneous myxoma and consists of increased numbers of bland spindle cells with wavy nuclei (Schwann cells) and fibrillar cytoplasm as well as mast cells and fibroblasts (Figure 2). Neurofibromas stain positively for S-100 and SOX-10 (Sry-related HMg-box 10).2,7 In addition to café-au-lait macules, axillary freckling, optic gliomas, and positive family history, neurofibromas are associated with neurofibromatosis type 1, which is linked to a defect in a tumor suppressor gene that codes for neurofibromin.7

Nodular fasciitis is a self-limited myofibroblastic neoplasm that contains fusion genes, with the most common being myosin-9–ubiquitin specific peptidase 6, MYH9-USP6, which leads to overexpression of USP6. Nodular fasciitis presents as a solitary, rapidly enlarging nodule affecting the subcutaneous tissue, muscles, or fascia.8,9 It usually presents in the third or fourth decades of life.8 The arms are the most common location in adults, while the most commonly affected site in children is the head or neck. Histopathology reveals a characteristic tissue culture pattern with a proliferation of plump spindle and stellate fibroblasts as well as myofibroblasts (Figure 3). Early lesions have haphazard spindle cells with a proliferation of small blood vessels and extravasated erythrocytes. Despite increased mitotic figures, cellular atypia is rare. The fibroblasts and myofibroblasts react positively for vimentin and muscle-specific actin.8 This lesion is highly cellular comparatively and notably lacks the perivascular neutrophils and epithelial structures that would be expected in a cutaneous myxoma.4,8

Spindle cell lipomas, solitary subcutaneous masses commonly presenting on the upper back in middle-aged men, also can mimic cutaneous myxomas.4 Histologically, these lesions may contain short bundles of spindle cells arranged in a school of fish–like pattern, mature adipocytes, or myxoid stroma and characteristic CD34 positivity (Figure 4). Spindle cell lipomas often will present with ropey collagen, which can easily distinguish them from cutaneous myxomas.4

- Lanjewar DN, Bhatia VO, Lanjewar SD, et al. Cutaneous myxoma: an important clue to Carney complex. Indian J Pathol Microbiol. 2014;57:460-462.
- Choi HJ, Kim YJ, Yim JH, et al. Unusual presentation of solitary cutaneous myxoma. J Eur Acad Dermatol Venereol. 2007;21:403-404. doi:10.1111/j.1468-3083.2006.01881.x
- Kura MM, Jindal SR. Solitary superficial acral angiomyxoma: an infrequently reported soft tissue tumor. Indian J Dermatol. 2014;59:1-3. doi:10.4103/0019-5154.139893
- Zou Y, Billings SD. Myxoid cutaneous tumors: a review. J Cutan Pathol. 2016;43:903-918.
- Sarfo A, Helm K, Flamm A. Cutaneous myxomas and a psammomatous melanotic schwannoma in a patient with Carney complex. J Cutan Pathol. 2019;46:93-96. doi:10.1111/cup.13385
- Gill P, Abi Daoud MS. Multiple cellular neurothekeomas in a middleaged woman including the lower extremity: a case report and review of the current literature. J Cutan Pathol. 2019;46:67-73. doi:10.1111/ cup.13366
- Ohgaki H, Kim Y, Steinbach JP. Nervous system tumors associated with familial tumor syndromes. Curr Opin Neurol. 2010;23:583-591. doi:10.1097/WCO.0b013e3283405b5f
- Luna A, Molinari L, Bollea Garlatti LA, et al. Nodular fasciitis, a forgotten entity. Int J Dermatol. 2019;58:190-193. doi:10.1111/ijd.14219
- Patel N, Chrisinger J, Demicco E, et al. USP6 activation in nodular fasciitis by promoter-swapping gene fusions. Mod Pathol. 2017; 30:1577-1588.
The Diagnosis: Cutaneous Myxoma
Microscopic analysis showed features of cutaneous myxoma (quiz images). The epidermis was essentially unremarkable. Stellate to spindle cells with bland nuclear chromatin were present in the dermis with abundant pools of myxoid stroma. Colloidal iron staining highlighted the markedly increased dermal mucin.
Cutaneous myxomas (also referred to as superficial angiomyxomas) are rare, well-demarcated tumors of the dermis and subcutis.1,2 They can present as solitary, fleshcolored nodules on the trunk, lower extremities, head, or neck, and they often measure between 1 and 5 cm.2,3 Histologically, cutaneous myxomas are hypocellular with some stellate fibroblasts, occasional epithelial structures, and an abundant myxoid stroma, with notable thinwalled small blood vessels.2,4 These lesions contain pools of mucin and are positive for mesenchymal mucin stains such as colloidal iron and Alcian blue.1 Moreover, perivascular neutrophils are a distinguishing characteristic of cutaneous myxomas.4
Multiple cutaneous myxomas should raise concern for Carney complex,1,5 a genodermatologic syndrome that arises due to a mutation in the protein kinase CAMP-dependent type I regulatory subunit alpha gene, PRKAR1A, on chromosome 2.1,5 Additional cutaneous manifestations include blue nevi, lentigines, and café-aulait macules.5 Carney complex also is known for endocrine overactivity and cardiac myxomas, which can cause serious embolic complications.1
Recommended management is complete excision with close follow-up, as these lesions may recur in up to one-third of cases. Although there is a potential for recurrence, metastases are uncommon.3 Even without recurrence in the presenting location, follow-up should include screening for manifestations of Carney complex.1,3
The clinical and histological differential for cutaneous myxoma may include nerve sheath myxoma or neurofibroma. A nerve sheath myxoma is a dermal tumor that manifests as a solitary, flesh-colored nodule, measuring less than 2 cm. These lesions commonly present on the head, neck, and upper body.6 Cutaneous myxomas can grow larger than 2 cm, but these two lesions have a great deal of overlap in their other features.3,6 Thus, histology can be used to distinguish them.
Nerve sheath myxomas are circumscribed nonencapsulated tumors of the dermis composed of multilobular aggregates of spindle to epithelioid cells in a mucinous matrix (Figure 1). Clefts often are present around the cell aggregates. Despite previously being termed myxoid neurothekeomas, nerve sheath myxomas are S-100 positive, whereas cellular neurothekeomas are S-100 negative and likely not of neural origin. Cutaneous myxomas, in contrast to nerve sheath myxomas, are S-100 negative. Nerve sheath myxomas are more cellular and lack the characteristic mucin pools compared with cutaneous myxomas.1,2,6 Neurofibromas frequently are flesh colored and pedunculated, as was the lesion in our patient, yet they are vastly different microscopically. The stroma of neurofibromas can vary, but cellularity typically is greater than a cutaneous myxoma and consists of increased numbers of bland spindle cells with wavy nuclei (Schwann cells) and fibrillar cytoplasm as well as mast cells and fibroblasts (Figure 2). Neurofibromas stain positively for S-100 and SOX-10 (Sry-related HMg-box 10).2,7 In addition to café-au-lait macules, axillary freckling, optic gliomas, and positive family history, neurofibromas are associated with neurofibromatosis type 1, which is linked to a defect in a tumor suppressor gene that codes for neurofibromin.7
Nodular fasciitis is a self-limited myofibroblastic neoplasm that contains fusion genes, with the most common being myosin-9–ubiquitin specific peptidase 6, MYH9-USP6, which leads to overexpression of USP6. Nodular fasciitis presents as a solitary, rapidly enlarging nodule affecting the subcutaneous tissue, muscles, or fascia.8,9 It usually presents in the third or fourth decades of life.8 The arms are the most common location in adults, while the most commonly affected site in children is the head or neck. Histopathology reveals a characteristic tissue culture pattern with a proliferation of plump spindle and stellate fibroblasts as well as myofibroblasts (Figure 3). Early lesions have haphazard spindle cells with a proliferation of small blood vessels and extravasated erythrocytes. Despite increased mitotic figures, cellular atypia is rare. The fibroblasts and myofibroblasts react positively for vimentin and muscle-specific actin.8 This lesion is highly cellular comparatively and notably lacks the perivascular neutrophils and epithelial structures that would be expected in a cutaneous myxoma.4,8
Spindle cell lipomas, solitary subcutaneous masses commonly presenting on the upper back in middle-aged men, also can mimic cutaneous myxomas.4 Histologically, these lesions may contain short bundles of spindle cells arranged in a school of fish–like pattern, mature adipocytes, or myxoid stroma and characteristic CD34 positivity (Figure 4). Spindle cell lipomas often will present with ropey collagen, which can easily distinguish them from cutaneous myxomas.4
The Diagnosis: Cutaneous Myxoma
Microscopic analysis showed features of cutaneous myxoma (quiz images). The epidermis was essentially unremarkable. Stellate to spindle cells with bland nuclear chromatin were present in the dermis with abundant pools of myxoid stroma. Colloidal iron staining highlighted the markedly increased dermal mucin.
Cutaneous myxomas (also referred to as superficial angiomyxomas) are rare, well-demarcated tumors of the dermis and subcutis.1,2 They can present as solitary, fleshcolored nodules on the trunk, lower extremities, head, or neck, and they often measure between 1 and 5 cm.2,3 Histologically, cutaneous myxomas are hypocellular with some stellate fibroblasts, occasional epithelial structures, and an abundant myxoid stroma, with notable thinwalled small blood vessels.2,4 These lesions contain pools of mucin and are positive for mesenchymal mucin stains such as colloidal iron and Alcian blue.1 Moreover, perivascular neutrophils are a distinguishing characteristic of cutaneous myxomas.4
Multiple cutaneous myxomas should raise concern for Carney complex,1,5 a genodermatologic syndrome that arises due to a mutation in the protein kinase CAMP-dependent type I regulatory subunit alpha gene, PRKAR1A, on chromosome 2.1,5 Additional cutaneous manifestations include blue nevi, lentigines, and café-aulait macules.5 Carney complex also is known for endocrine overactivity and cardiac myxomas, which can cause serious embolic complications.1
Recommended management is complete excision with close follow-up, as these lesions may recur in up to one-third of cases. Although there is a potential for recurrence, metastases are uncommon.3 Even without recurrence in the presenting location, follow-up should include screening for manifestations of Carney complex.1,3
The clinical and histological differential for cutaneous myxoma may include nerve sheath myxoma or neurofibroma. A nerve sheath myxoma is a dermal tumor that manifests as a solitary, flesh-colored nodule, measuring less than 2 cm. These lesions commonly present on the head, neck, and upper body.6 Cutaneous myxomas can grow larger than 2 cm, but these two lesions have a great deal of overlap in their other features.3,6 Thus, histology can be used to distinguish them.
Nerve sheath myxomas are circumscribed nonencapsulated tumors of the dermis composed of multilobular aggregates of spindle to epithelioid cells in a mucinous matrix (Figure 1). Clefts often are present around the cell aggregates. Despite previously being termed myxoid neurothekeomas, nerve sheath myxomas are S-100 positive, whereas cellular neurothekeomas are S-100 negative and likely not of neural origin. Cutaneous myxomas, in contrast to nerve sheath myxomas, are S-100 negative. Nerve sheath myxomas are more cellular and lack the characteristic mucin pools compared with cutaneous myxomas.1,2,6 Neurofibromas frequently are flesh colored and pedunculated, as was the lesion in our patient, yet they are vastly different microscopically. The stroma of neurofibromas can vary, but cellularity typically is greater than a cutaneous myxoma and consists of increased numbers of bland spindle cells with wavy nuclei (Schwann cells) and fibrillar cytoplasm as well as mast cells and fibroblasts (Figure 2). Neurofibromas stain positively for S-100 and SOX-10 (Sry-related HMg-box 10).2,7 In addition to café-au-lait macules, axillary freckling, optic gliomas, and positive family history, neurofibromas are associated with neurofibromatosis type 1, which is linked to a defect in a tumor suppressor gene that codes for neurofibromin.7
Nodular fasciitis is a self-limited myofibroblastic neoplasm that contains fusion genes, with the most common being myosin-9–ubiquitin specific peptidase 6, MYH9-USP6, which leads to overexpression of USP6. Nodular fasciitis presents as a solitary, rapidly enlarging nodule affecting the subcutaneous tissue, muscles, or fascia.8,9 It usually presents in the third or fourth decades of life.8 The arms are the most common location in adults, while the most commonly affected site in children is the head or neck. Histopathology reveals a characteristic tissue culture pattern with a proliferation of plump spindle and stellate fibroblasts as well as myofibroblasts (Figure 3). Early lesions have haphazard spindle cells with a proliferation of small blood vessels and extravasated erythrocytes. Despite increased mitotic figures, cellular atypia is rare. The fibroblasts and myofibroblasts react positively for vimentin and muscle-specific actin.8 This lesion is highly cellular comparatively and notably lacks the perivascular neutrophils and epithelial structures that would be expected in a cutaneous myxoma.4,8
Spindle cell lipomas, solitary subcutaneous masses commonly presenting on the upper back in middle-aged men, also can mimic cutaneous myxomas.4 Histologically, these lesions may contain short bundles of spindle cells arranged in a school of fish–like pattern, mature adipocytes, or myxoid stroma and characteristic CD34 positivity (Figure 4). Spindle cell lipomas often will present with ropey collagen, which can easily distinguish them from cutaneous myxomas.4
- Lanjewar DN, Bhatia VO, Lanjewar SD, et al. Cutaneous myxoma: an important clue to Carney complex. Indian J Pathol Microbiol. 2014;57:460-462.
- Choi HJ, Kim YJ, Yim JH, et al. Unusual presentation of solitary cutaneous myxoma. J Eur Acad Dermatol Venereol. 2007;21:403-404. doi:10.1111/j.1468-3083.2006.01881.x
- Kura MM, Jindal SR. Solitary superficial acral angiomyxoma: an infrequently reported soft tissue tumor. Indian J Dermatol. 2014;59:1-3. doi:10.4103/0019-5154.139893
- Zou Y, Billings SD. Myxoid cutaneous tumors: a review. J Cutan Pathol. 2016;43:903-918.
- Sarfo A, Helm K, Flamm A. Cutaneous myxomas and a psammomatous melanotic schwannoma in a patient with Carney complex. J Cutan Pathol. 2019;46:93-96. doi:10.1111/cup.13385
- Gill P, Abi Daoud MS. Multiple cellular neurothekeomas in a middleaged woman including the lower extremity: a case report and review of the current literature. J Cutan Pathol. 2019;46:67-73. doi:10.1111/ cup.13366
- Ohgaki H, Kim Y, Steinbach JP. Nervous system tumors associated with familial tumor syndromes. Curr Opin Neurol. 2010;23:583-591. doi:10.1097/WCO.0b013e3283405b5f
- Luna A, Molinari L, Bollea Garlatti LA, et al. Nodular fasciitis, a forgotten entity. Int J Dermatol. 2019;58:190-193. doi:10.1111/ijd.14219
- Patel N, Chrisinger J, Demicco E, et al. USP6 activation in nodular fasciitis by promoter-swapping gene fusions. Mod Pathol. 2017; 30:1577-1588.
- Lanjewar DN, Bhatia VO, Lanjewar SD, et al. Cutaneous myxoma: an important clue to Carney complex. Indian J Pathol Microbiol. 2014;57:460-462.
- Choi HJ, Kim YJ, Yim JH, et al. Unusual presentation of solitary cutaneous myxoma. J Eur Acad Dermatol Venereol. 2007;21:403-404. doi:10.1111/j.1468-3083.2006.01881.x
- Kura MM, Jindal SR. Solitary superficial acral angiomyxoma: an infrequently reported soft tissue tumor. Indian J Dermatol. 2014;59:1-3. doi:10.4103/0019-5154.139893
- Zou Y, Billings SD. Myxoid cutaneous tumors: a review. J Cutan Pathol. 2016;43:903-918.
- Sarfo A, Helm K, Flamm A. Cutaneous myxomas and a psammomatous melanotic schwannoma in a patient with Carney complex. J Cutan Pathol. 2019;46:93-96. doi:10.1111/cup.13385
- Gill P, Abi Daoud MS. Multiple cellular neurothekeomas in a middleaged woman including the lower extremity: a case report and review of the current literature. J Cutan Pathol. 2019;46:67-73. doi:10.1111/ cup.13366
- Ohgaki H, Kim Y, Steinbach JP. Nervous system tumors associated with familial tumor syndromes. Curr Opin Neurol. 2010;23:583-591. doi:10.1097/WCO.0b013e3283405b5f
- Luna A, Molinari L, Bollea Garlatti LA, et al. Nodular fasciitis, a forgotten entity. Int J Dermatol. 2019;58:190-193. doi:10.1111/ijd.14219
- Patel N, Chrisinger J, Demicco E, et al. USP6 activation in nodular fasciitis by promoter-swapping gene fusions. Mod Pathol. 2017; 30:1577-1588.
A 43-year-old man with an unremarkable medical history presented to our clinic with an enlarging painful nodule on the upper back that was present for years without bleeding or ulceration. He denied prior treatment or any similar lesions. Physical examination was notable for a 2×1.5-cm, pedunculated, flesh-colored nodule on the left upper back. A shave excision of the lesion was performed.



Tender Subcutaneous Nodule in a Prepubescent Boy
The Diagnosis: Dermatomyofibroma
Dermatomyofibroma is an uncommon, benign, cutaneous mesenchymal neoplasm composed of fibroblasts and myofibroblasts.1-3 This skin tumor was first described in 1991 by Hugel4 in the German literature as plaquelike fibromatosis. Pediatric dermatomyofibromas are exceedingly rare, with pediatric patients ranging in age from infants to teenagers.1
Clinically, dermatomyofibromas appear as long-standing, isolated, ill-demarcated, flesh-colored, slightly hyperpigmented or erythematous nodules or plaques that may be raised or indurated.1 Dermatomyofibromas may present with constant mild pain or pruritus, though in most cases the lesions are asymptomatic.1,3 The clinical presentation of dermatomyofibroma has a few distinct differences in children compared to adults. In adulthood, dermatomyofibroma has a strong female predominance and most commonly is located on the shoulder and adjacent upper body regions, including the axilla, neck, upper arm, and upper trunk.1-3 In childhood, the majority of dermatomyofibromas occur in young boys and usually are located on the neck with other upper body regions occurring less frequently.1,2 A shared characteristic includes the tendency for dermatomyofibromas to have an initial period of enlargement followed by stabilization or slow growth.1 Reported pediatric lesions have ranged in size from 4 to 60 mm with an average size of 14.9 mm (median, 12 mm).2
The diagnosis of dermatomyofibroma is based on histopathologic features in addition to clinical presentation. Histology from punch biopsy usually reveals a noninvasive dermal proliferation of bland, uniform, slender spindle cells oriented parallel to the overlying epidermis with increased and fragmented elastic fibers.1,3 Infiltration into the mid or deep dermis is common. The adnexal structures usually are spared; the stroma contains collagen and increased small blood vessels; and there typically is no inflammatory infiltrate, except for occasional scattered mast cells.2 Cytologically, the monomorphic spindleshaped tumor cells have an ill-defined, pale, eosinophilic cytoplasm and nuclei that are elongated with tapered edges.3 Dermatomyofibroma has a variable immunohistochemical profile, as it may stain focally positive for CD34 or smooth muscle actin, with occasional staining of factor XIIIa, desmin, calponin, or vimentin.1-3 Normal to increased levels of often fragmented elastic fibers is a helpful clue in distinguishing dermatomyofibroma from dermatofibroma, hypertrophic scar, dermatofibrosarcoma protuberans, and pilar leiomyoma, in which elastic fibers typically are reduced.3 Differential diagnoses of mesenchymal tumors in children include desmoid fibromatosis, connective tissue nevus, myofibromatosis, and smooth muscle hamartoma.1
A punch biopsy with clinical observation and followup is recommended for the management of lesions in cosmetically sensitive areas or in very young children who may not tolerate surgery. In symptomatic or cosmetically unappealing cases of dermatomyofibroma, simple surgical excision remains a viable treatment option. Recurrence is uncommon, even if only partially excised, and no instances of metastasis have been reported.1-5
Dermatomyofibromas may be mistaken for several other entities both benign and malignant. For example, the benign dermatofibroma is the second most common fibrohistiocytic tumor of the skin and presents as a firm, nontender, minimally elevated to dome-shaped papule that usually measures less than or equal to 1 cm in diameter with or without overlying skin changes.5,6 It primarily is seen in adults with a slight female predominance and favors the lower extremities.5 Patients usually are asymptomatic but often report a history of local trauma at the lesion site.6 Histologically, dermatofibroma is characterized by a nodular dermal proliferation of spindleshaped fibrous cells and histiocytes in a storiform pattern (Figure 1).6 Epidermal induction with acanthosis overlying the tumor often is found with occasional basilar hyperpigmentation.5 Dermatofibroma also characteristically has trapped collagen (“collagen balls”) seen at the periphery.5,6
Piloleiomyomas are benign smooth muscle tumors arising from arrector pili muscles that may be solitary or multiple.5 Clinically, they typically present as firm, reddish-brown to flesh-colored papules or nodules that develop more commonly in adulthood.5,7 Piloleiomyomas favor the extremities and trunk, particularly the shoulder, and can be associated with spontaneous or induced pain. Histologically, piloleiomyomas are well circumscribed and centered within the reticular dermis situated closely to hair follicles (Figure 2).5 They are composed of numerous interlacing fascicles or whorls of smooth muscle cells with abundant eosinophilic cytoplasm and blunt-ended, cigar-shaped nuclei.5,7
Solitary cutaneous myofibroma is a benign fibrous tumor found in adolescents and adults and is the counterpart to infantile myofibromatosis.8 Clinically, myofibromas typically present as painless, slow-growing, firm nodules with an occasional bluish hue. Histologically, solitary cutaneous myofibromas appear in a biphasic pattern, with hemangiopericytomatous components as well as spindle cells arranged in short bundles and fascicles resembling leiomyoma (Figure 3). The spindle cells also have abundant eosinophilic cytoplasm with short plump nuclei; the random, irregularly intersecting angles can be used to help differentiate myofibromas from smooth muscle lesions.8 Solitary cutaneous myofibroma is in the differential diagnosis for dermatomyofibroma because of their shared myofibroblastic nature.9
Dermatofibrosarcoma protuberans (DFSP) is an uncommon, locally invasive sarcoma with a high recurrence rate that favors young to middle-aged adults, with rare childhood onset reported.5,10,11 Clinically, DFSP typically presents as an asymptomatic, slow-growing, firm, flesh-colored, indurated plaque that develops into a violaceous to reddish-brown nodule.5 The atrophic variant of DFSP is characterized by a nonprotuberant lesion and can be especially difficult to distinguish from other entities such as dermatomyofibroma.11 The majority of DFSP lesions occur on the trunk, particularly in the shoulder or pelvic region.5 Histologically, early plaque lesions are comprised of monomorphic spindle cells arranged in long fascicles (parallel to the skin surface), infiltrating adnexal structures, and subcutaneous adipocytes in a multilayered honeycomb pattern; the spindle cells of late nodular lesions are arranged in short fascicles in a matted or storiform pattern (Figure 4).5,10 Early stages of DFSP as well as variations in childhood-onset DFSP can easily be misdiagnosed and incompletely excised.5
- Ma JE, Wieland CN, Tollefson MM. Dermatomyofibromas arising in children: report of two new cases and review of the literature. Pediatr Dermatol. 2017;34:347-351.
- Tardio JC, Azorin D, Hernandez-Nunez A, et al. Dermatomyofibromas presenting in pediatric patients: clinicopathologic characteristics and differential diagnosis. J Cutan Pathol. 2011;38:967-972.
- Mentzel T, Kutzner H. Dermatomyofibroma: clinicopathologic and immunohistochemical analysis of 56 cases and reappraisal of a rare and distinct cutaneous neoplasm. Am J Dermatopathol. 2009;31:44-49.
- Hugel H. Plaque-like dermal fibromatosis. Hautarzt. 1991;42:223-226.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. WB Saunders Co; 2012.
- Myers DJ, Fillman EP. Dermatofibroma. StatPearls [Internet]. StatPearls Publishing; 2020.
- Dilek N, Yuksel D, Sehitoglu I, et al. Cutaneous leiomyoma in a child: a case report. Oncol Lett. 2013;5:1163-1164.
- Roh HS, Paek JO, Yu HJ, et al. Solitary cutaneous myofibroma on the sole: an unusual localization. Ann Dermatol. 2012;24:220-222.
- Weedon D, Strutton G, Rubin AI, et al. Weedon’s Skin Pathology. Churchill Livingstone/Elsevier; 2010.
- Mendenhall WM, Zlotecki RA, Scarborough MT. Dermatofibrosarcoma protuberans. Cancer. 2004;101:2503-2508.
- Akay BN, Unlu E, Erdem C, et al. Dermatoscopic findings of atrophic dermatofibrosarcoma protuberans. Dermatol Pract Concept. 2015;5:71-73.
The Diagnosis: Dermatomyofibroma
Dermatomyofibroma is an uncommon, benign, cutaneous mesenchymal neoplasm composed of fibroblasts and myofibroblasts.1-3 This skin tumor was first described in 1991 by Hugel4 in the German literature as plaquelike fibromatosis. Pediatric dermatomyofibromas are exceedingly rare, with pediatric patients ranging in age from infants to teenagers.1
Clinically, dermatomyofibromas appear as long-standing, isolated, ill-demarcated, flesh-colored, slightly hyperpigmented or erythematous nodules or plaques that may be raised or indurated.1 Dermatomyofibromas may present with constant mild pain or pruritus, though in most cases the lesions are asymptomatic.1,3 The clinical presentation of dermatomyofibroma has a few distinct differences in children compared to adults. In adulthood, dermatomyofibroma has a strong female predominance and most commonly is located on the shoulder and adjacent upper body regions, including the axilla, neck, upper arm, and upper trunk.1-3 In childhood, the majority of dermatomyofibromas occur in young boys and usually are located on the neck with other upper body regions occurring less frequently.1,2 A shared characteristic includes the tendency for dermatomyofibromas to have an initial period of enlargement followed by stabilization or slow growth.1 Reported pediatric lesions have ranged in size from 4 to 60 mm with an average size of 14.9 mm (median, 12 mm).2
The diagnosis of dermatomyofibroma is based on histopathologic features in addition to clinical presentation. Histology from punch biopsy usually reveals a noninvasive dermal proliferation of bland, uniform, slender spindle cells oriented parallel to the overlying epidermis with increased and fragmented elastic fibers.1,3 Infiltration into the mid or deep dermis is common. The adnexal structures usually are spared; the stroma contains collagen and increased small blood vessels; and there typically is no inflammatory infiltrate, except for occasional scattered mast cells.2 Cytologically, the monomorphic spindleshaped tumor cells have an ill-defined, pale, eosinophilic cytoplasm and nuclei that are elongated with tapered edges.3 Dermatomyofibroma has a variable immunohistochemical profile, as it may stain focally positive for CD34 or smooth muscle actin, with occasional staining of factor XIIIa, desmin, calponin, or vimentin.1-3 Normal to increased levels of often fragmented elastic fibers is a helpful clue in distinguishing dermatomyofibroma from dermatofibroma, hypertrophic scar, dermatofibrosarcoma protuberans, and pilar leiomyoma, in which elastic fibers typically are reduced.3 Differential diagnoses of mesenchymal tumors in children include desmoid fibromatosis, connective tissue nevus, myofibromatosis, and smooth muscle hamartoma.1
A punch biopsy with clinical observation and followup is recommended for the management of lesions in cosmetically sensitive areas or in very young children who may not tolerate surgery. In symptomatic or cosmetically unappealing cases of dermatomyofibroma, simple surgical excision remains a viable treatment option. Recurrence is uncommon, even if only partially excised, and no instances of metastasis have been reported.1-5
Dermatomyofibromas may be mistaken for several other entities both benign and malignant. For example, the benign dermatofibroma is the second most common fibrohistiocytic tumor of the skin and presents as a firm, nontender, minimally elevated to dome-shaped papule that usually measures less than or equal to 1 cm in diameter with or without overlying skin changes.5,6 It primarily is seen in adults with a slight female predominance and favors the lower extremities.5 Patients usually are asymptomatic but often report a history of local trauma at the lesion site.6 Histologically, dermatofibroma is characterized by a nodular dermal proliferation of spindleshaped fibrous cells and histiocytes in a storiform pattern (Figure 1).6 Epidermal induction with acanthosis overlying the tumor often is found with occasional basilar hyperpigmentation.5 Dermatofibroma also characteristically has trapped collagen (“collagen balls”) seen at the periphery.5,6
Piloleiomyomas are benign smooth muscle tumors arising from arrector pili muscles that may be solitary or multiple.5 Clinically, they typically present as firm, reddish-brown to flesh-colored papules or nodules that develop more commonly in adulthood.5,7 Piloleiomyomas favor the extremities and trunk, particularly the shoulder, and can be associated with spontaneous or induced pain. Histologically, piloleiomyomas are well circumscribed and centered within the reticular dermis situated closely to hair follicles (Figure 2).5 They are composed of numerous interlacing fascicles or whorls of smooth muscle cells with abundant eosinophilic cytoplasm and blunt-ended, cigar-shaped nuclei.5,7
Solitary cutaneous myofibroma is a benign fibrous tumor found in adolescents and adults and is the counterpart to infantile myofibromatosis.8 Clinically, myofibromas typically present as painless, slow-growing, firm nodules with an occasional bluish hue. Histologically, solitary cutaneous myofibromas appear in a biphasic pattern, with hemangiopericytomatous components as well as spindle cells arranged in short bundles and fascicles resembling leiomyoma (Figure 3). The spindle cells also have abundant eosinophilic cytoplasm with short plump nuclei; the random, irregularly intersecting angles can be used to help differentiate myofibromas from smooth muscle lesions.8 Solitary cutaneous myofibroma is in the differential diagnosis for dermatomyofibroma because of their shared myofibroblastic nature.9
Dermatofibrosarcoma protuberans (DFSP) is an uncommon, locally invasive sarcoma with a high recurrence rate that favors young to middle-aged adults, with rare childhood onset reported.5,10,11 Clinically, DFSP typically presents as an asymptomatic, slow-growing, firm, flesh-colored, indurated plaque that develops into a violaceous to reddish-brown nodule.5 The atrophic variant of DFSP is characterized by a nonprotuberant lesion and can be especially difficult to distinguish from other entities such as dermatomyofibroma.11 The majority of DFSP lesions occur on the trunk, particularly in the shoulder or pelvic region.5 Histologically, early plaque lesions are comprised of monomorphic spindle cells arranged in long fascicles (parallel to the skin surface), infiltrating adnexal structures, and subcutaneous adipocytes in a multilayered honeycomb pattern; the spindle cells of late nodular lesions are arranged in short fascicles in a matted or storiform pattern (Figure 4).5,10 Early stages of DFSP as well as variations in childhood-onset DFSP can easily be misdiagnosed and incompletely excised.5
The Diagnosis: Dermatomyofibroma
Dermatomyofibroma is an uncommon, benign, cutaneous mesenchymal neoplasm composed of fibroblasts and myofibroblasts.1-3 This skin tumor was first described in 1991 by Hugel4 in the German literature as plaquelike fibromatosis. Pediatric dermatomyofibromas are exceedingly rare, with pediatric patients ranging in age from infants to teenagers.1
Clinically, dermatomyofibromas appear as long-standing, isolated, ill-demarcated, flesh-colored, slightly hyperpigmented or erythematous nodules or plaques that may be raised or indurated.1 Dermatomyofibromas may present with constant mild pain or pruritus, though in most cases the lesions are asymptomatic.1,3 The clinical presentation of dermatomyofibroma has a few distinct differences in children compared to adults. In adulthood, dermatomyofibroma has a strong female predominance and most commonly is located on the shoulder and adjacent upper body regions, including the axilla, neck, upper arm, and upper trunk.1-3 In childhood, the majority of dermatomyofibromas occur in young boys and usually are located on the neck with other upper body regions occurring less frequently.1,2 A shared characteristic includes the tendency for dermatomyofibromas to have an initial period of enlargement followed by stabilization or slow growth.1 Reported pediatric lesions have ranged in size from 4 to 60 mm with an average size of 14.9 mm (median, 12 mm).2
The diagnosis of dermatomyofibroma is based on histopathologic features in addition to clinical presentation. Histology from punch biopsy usually reveals a noninvasive dermal proliferation of bland, uniform, slender spindle cells oriented parallel to the overlying epidermis with increased and fragmented elastic fibers.1,3 Infiltration into the mid or deep dermis is common. The adnexal structures usually are spared; the stroma contains collagen and increased small blood vessels; and there typically is no inflammatory infiltrate, except for occasional scattered mast cells.2 Cytologically, the monomorphic spindleshaped tumor cells have an ill-defined, pale, eosinophilic cytoplasm and nuclei that are elongated with tapered edges.3 Dermatomyofibroma has a variable immunohistochemical profile, as it may stain focally positive for CD34 or smooth muscle actin, with occasional staining of factor XIIIa, desmin, calponin, or vimentin.1-3 Normal to increased levels of often fragmented elastic fibers is a helpful clue in distinguishing dermatomyofibroma from dermatofibroma, hypertrophic scar, dermatofibrosarcoma protuberans, and pilar leiomyoma, in which elastic fibers typically are reduced.3 Differential diagnoses of mesenchymal tumors in children include desmoid fibromatosis, connective tissue nevus, myofibromatosis, and smooth muscle hamartoma.1
A punch biopsy with clinical observation and followup is recommended for the management of lesions in cosmetically sensitive areas or in very young children who may not tolerate surgery. In symptomatic or cosmetically unappealing cases of dermatomyofibroma, simple surgical excision remains a viable treatment option. Recurrence is uncommon, even if only partially excised, and no instances of metastasis have been reported.1-5
Dermatomyofibromas may be mistaken for several other entities both benign and malignant. For example, the benign dermatofibroma is the second most common fibrohistiocytic tumor of the skin and presents as a firm, nontender, minimally elevated to dome-shaped papule that usually measures less than or equal to 1 cm in diameter with or without overlying skin changes.5,6 It primarily is seen in adults with a slight female predominance and favors the lower extremities.5 Patients usually are asymptomatic but often report a history of local trauma at the lesion site.6 Histologically, dermatofibroma is characterized by a nodular dermal proliferation of spindleshaped fibrous cells and histiocytes in a storiform pattern (Figure 1).6 Epidermal induction with acanthosis overlying the tumor often is found with occasional basilar hyperpigmentation.5 Dermatofibroma also characteristically has trapped collagen (“collagen balls”) seen at the periphery.5,6
Piloleiomyomas are benign smooth muscle tumors arising from arrector pili muscles that may be solitary or multiple.5 Clinically, they typically present as firm, reddish-brown to flesh-colored papules or nodules that develop more commonly in adulthood.5,7 Piloleiomyomas favor the extremities and trunk, particularly the shoulder, and can be associated with spontaneous or induced pain. Histologically, piloleiomyomas are well circumscribed and centered within the reticular dermis situated closely to hair follicles (Figure 2).5 They are composed of numerous interlacing fascicles or whorls of smooth muscle cells with abundant eosinophilic cytoplasm and blunt-ended, cigar-shaped nuclei.5,7
Solitary cutaneous myofibroma is a benign fibrous tumor found in adolescents and adults and is the counterpart to infantile myofibromatosis.8 Clinically, myofibromas typically present as painless, slow-growing, firm nodules with an occasional bluish hue. Histologically, solitary cutaneous myofibromas appear in a biphasic pattern, with hemangiopericytomatous components as well as spindle cells arranged in short bundles and fascicles resembling leiomyoma (Figure 3). The spindle cells also have abundant eosinophilic cytoplasm with short plump nuclei; the random, irregularly intersecting angles can be used to help differentiate myofibromas from smooth muscle lesions.8 Solitary cutaneous myofibroma is in the differential diagnosis for dermatomyofibroma because of their shared myofibroblastic nature.9
Dermatofibrosarcoma protuberans (DFSP) is an uncommon, locally invasive sarcoma with a high recurrence rate that favors young to middle-aged adults, with rare childhood onset reported.5,10,11 Clinically, DFSP typically presents as an asymptomatic, slow-growing, firm, flesh-colored, indurated plaque that develops into a violaceous to reddish-brown nodule.5 The atrophic variant of DFSP is characterized by a nonprotuberant lesion and can be especially difficult to distinguish from other entities such as dermatomyofibroma.11 The majority of DFSP lesions occur on the trunk, particularly in the shoulder or pelvic region.5 Histologically, early plaque lesions are comprised of monomorphic spindle cells arranged in long fascicles (parallel to the skin surface), infiltrating adnexal structures, and subcutaneous adipocytes in a multilayered honeycomb pattern; the spindle cells of late nodular lesions are arranged in short fascicles in a matted or storiform pattern (Figure 4).5,10 Early stages of DFSP as well as variations in childhood-onset DFSP can easily be misdiagnosed and incompletely excised.5
- Ma JE, Wieland CN, Tollefson MM. Dermatomyofibromas arising in children: report of two new cases and review of the literature. Pediatr Dermatol. 2017;34:347-351.
- Tardio JC, Azorin D, Hernandez-Nunez A, et al. Dermatomyofibromas presenting in pediatric patients: clinicopathologic characteristics and differential diagnosis. J Cutan Pathol. 2011;38:967-972.
- Mentzel T, Kutzner H. Dermatomyofibroma: clinicopathologic and immunohistochemical analysis of 56 cases and reappraisal of a rare and distinct cutaneous neoplasm. Am J Dermatopathol. 2009;31:44-49.
- Hugel H. Plaque-like dermal fibromatosis. Hautarzt. 1991;42:223-226.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. WB Saunders Co; 2012.
- Myers DJ, Fillman EP. Dermatofibroma. StatPearls [Internet]. StatPearls Publishing; 2020.
- Dilek N, Yuksel D, Sehitoglu I, et al. Cutaneous leiomyoma in a child: a case report. Oncol Lett. 2013;5:1163-1164.
- Roh HS, Paek JO, Yu HJ, et al. Solitary cutaneous myofibroma on the sole: an unusual localization. Ann Dermatol. 2012;24:220-222.
- Weedon D, Strutton G, Rubin AI, et al. Weedon’s Skin Pathology. Churchill Livingstone/Elsevier; 2010.
- Mendenhall WM, Zlotecki RA, Scarborough MT. Dermatofibrosarcoma protuberans. Cancer. 2004;101:2503-2508.
- Akay BN, Unlu E, Erdem C, et al. Dermatoscopic findings of atrophic dermatofibrosarcoma protuberans. Dermatol Pract Concept. 2015;5:71-73.
- Ma JE, Wieland CN, Tollefson MM. Dermatomyofibromas arising in children: report of two new cases and review of the literature. Pediatr Dermatol. 2017;34:347-351.
- Tardio JC, Azorin D, Hernandez-Nunez A, et al. Dermatomyofibromas presenting in pediatric patients: clinicopathologic characteristics and differential diagnosis. J Cutan Pathol. 2011;38:967-972.
- Mentzel T, Kutzner H. Dermatomyofibroma: clinicopathologic and immunohistochemical analysis of 56 cases and reappraisal of a rare and distinct cutaneous neoplasm. Am J Dermatopathol. 2009;31:44-49.
- Hugel H. Plaque-like dermal fibromatosis. Hautarzt. 1991;42:223-226.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. WB Saunders Co; 2012.
- Myers DJ, Fillman EP. Dermatofibroma. StatPearls [Internet]. StatPearls Publishing; 2020.
- Dilek N, Yuksel D, Sehitoglu I, et al. Cutaneous leiomyoma in a child: a case report. Oncol Lett. 2013;5:1163-1164.
- Roh HS, Paek JO, Yu HJ, et al. Solitary cutaneous myofibroma on the sole: an unusual localization. Ann Dermatol. 2012;24:220-222.
- Weedon D, Strutton G, Rubin AI, et al. Weedon’s Skin Pathology. Churchill Livingstone/Elsevier; 2010.
- Mendenhall WM, Zlotecki RA, Scarborough MT. Dermatofibrosarcoma protuberans. Cancer. 2004;101:2503-2508.
- Akay BN, Unlu E, Erdem C, et al. Dermatoscopic findings of atrophic dermatofibrosarcoma protuberans. Dermatol Pract Concept. 2015;5:71-73.

A 12-year-old boy with olive skin presented with a tender subcutaneous nodule on the back of 6 months’ duration. He reported the lesion initially grew rapidly with increasing pain for approximately 3 months with subsequent stabilization in size and modest resolution of his symptoms. Physical examination revealed a solitary, 15-mm, ill-defined, indurated, tender, subcutaneous nodule with subtle overlying hyperpigmentation on the left side of the upper back. Hematoxylin and eosin staining of a 4-mm punch biopsy revealed a nonencapsulated mass of monomorphic eosinophilic spindle cells organized into fascicles arranged predominantly parallel to the skin surface. The mass extended from the mid reticular dermis to the upper subcutis, sparing adnexal structures.
Firm Digital Papulonodules in an Infant
The Diagnosis: Infantile Digital Fibromatosis
Infantile digital fibromatosis (IDF) is a rare benign neoplasm of infancy prone to recurrence after resection but not to metastasis. It usually is limited to the fingers and toes.1 One-third of cases occur at birth. Most patients develop clinical symptoms within the first year of life, but presentation can occur in adolescents and adults. The exact etiology and pathogenesis of IDF remain unclear, but trauma is thought to be a trigger.
Physical examination reveals single or multiple smooth, round, pink papules or nodules confined to the sides and backs of the fingers, sparing the thumb and first toe.2,3 The nodules typically are firm, less than 2 cm in diameter, and often painless. Infantile digital fibromatosis exhibits an indolent progression followed by a rapid growth phase during several months, which may lead to functional impairment and joint deformities.4,5 Histopathology displays spindle cells with eosinophilic cytoplasmic inclusions that range from round to oval with uneven distribution, lack of refraction, and a large size difference (3–15 μm).6 The inclusions are deep red with Masson trichrome staining and can express smooth muscle actin and calponin. Tumor cells usually express vimentin, smooth muscle actin, calponin, and desmin but fail to express S-100 protein. The Ki67 proliferation index is 2% to 15%.6,7
Nonsurgical treatments for IDF include topical imiquimod, topical or intradermal injection of glucocorticoids, and intradermal injection of 5-fluorouracil. Complete resection should be reserved for cases with invasive growth that may lead to joint deformities, tendon or ligament involvement, digit or contracture deformity, and complications such as decreased joint mobility. Although there is a recurrence rate of up to 50% after excision, most lesions eventually will spontaneously regress and will leave no scar.8-10
The clinical and histopathologic differential diagnoses of IDF include other cutaneous diseases that occur in the digits. A dermatofibroma is a round, firm, fibrohistiocytic nodule that mainly occurs on the extensor limbs. Histopathology includes both fibrous and cellular types.11 Histologic analysis shows an ill-defined dermal proliferation of spindled fibroblasts with pale eosinophilic cytoplasm and bland fusiform nuclei growing in bands or fascicles that trap collagen fibers at the periphery (Figure 1). Generally, dermatofibromas have marked epidermal hyperplasia, which differs from IDF.
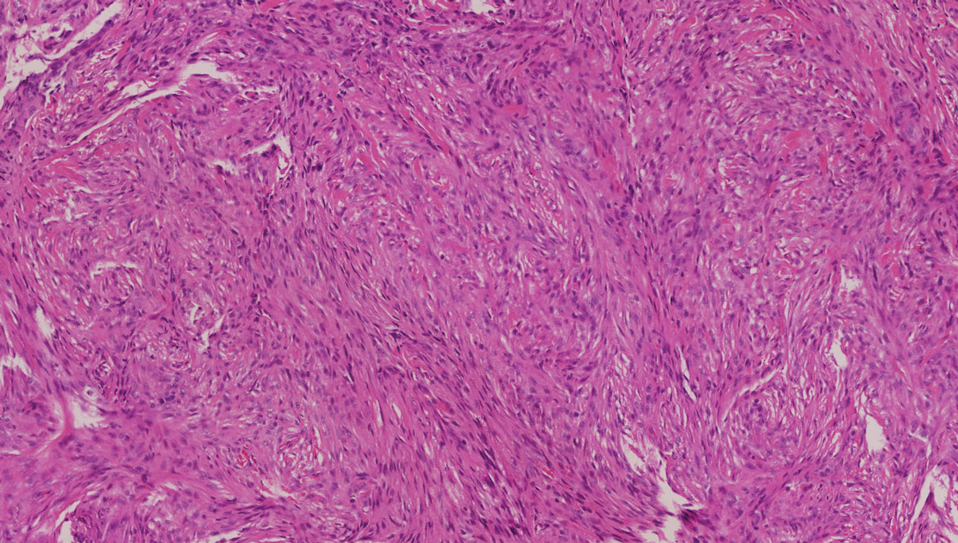
A digital myxoid cyst is characterized by a fleshcolored, hemispherical, and translucent cystic nodule that arises from the dorsum of the distal interphalangeal joint.12 It commonly is associated with injury and chronic pressure. Translucent viscous liquid may flow out when the cyst is punctured, a hallmark feature of this entity. Clinical variants of myxoid cyst include myxomatous and ganglion types. Histopathology reveals excessive mucin deposited in the dermis, and the surrounding collagen is compressed to form the pseudocyst (Figure 2).
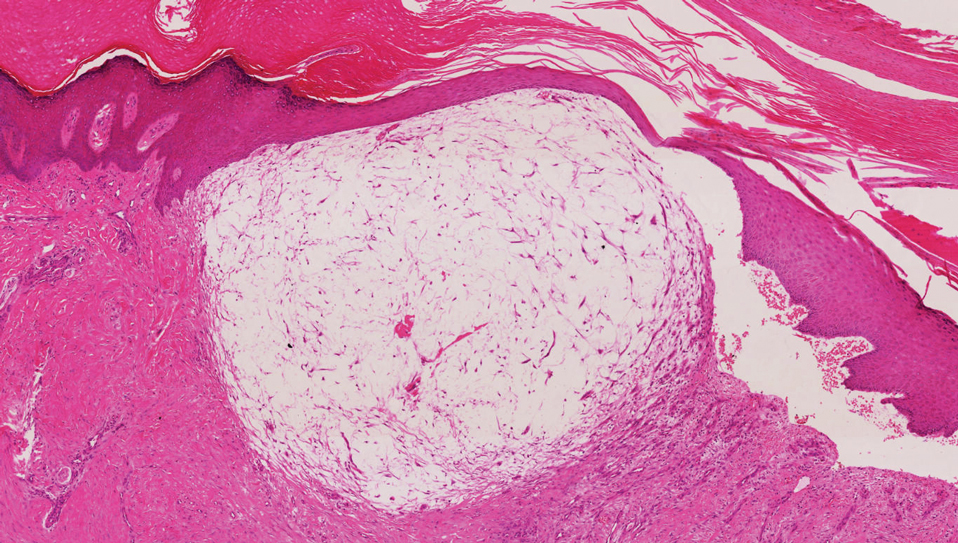
A giant cell tumor of the tendon sheath presents with asymptomatic nodules or lumps. Lesions frequently are localized to the tendon sheath, especially on the fingers and wrists, with no malignant tendency or propensity for spontaneous regression.13 The local recurrence rate is as high as 45%, which is related to surgical resection insufficiency.14 Histopathologic examination shows lobulated tumor tissue surrounded by dense fibrosis. The tumor cells are histiocytic with scattered giant cells (Figure 3). The characteristic osteoclastlike giant cells have eosinophilic cytoplasm and irregularly arranged nuclei in varying numbers.
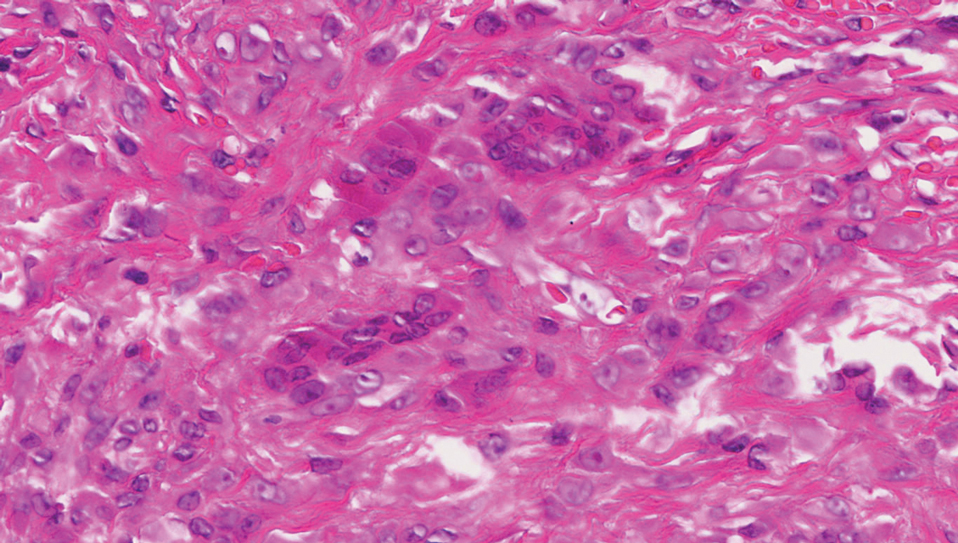
Keloids are connective tissue hyperplasias caused by skin injury. Histopathologically, keloids are characterized by nodules of thick hyalinized collagen bundles and whorled fibroblasts (Figure 4). No inclusions in the fibroblasts and a history of trauma can differentiate keloids from IDF.

- Marks E, Ewart M. Infantile digital fibroma: a rare fibromatosis. Arch Pathol Lab Med. 2016;140:1153‐1156.
- Botelho LF, Matsushigue T, Enokihara MM, et al. Case for diagnosis. An Bras Dermatol. 2012;87:493-494.
- Paloni G, Mattei I, Salmaso R, et al. Infantile digital fibromatosis. Arch Dis Child. 2013;98:308.
- Girgenti V, Restano L, Arcangeli F, et al. Infantile digital fibromatosis: a rare tumour of infancy. report of five cases. Australas J Dermatol. 2012;53:285-287.
- Eypper EH, Lee JC, Tarasen AJ, et al. An algorithmic approach to the management of infantile digital fibromatosis: review of literature and a case report. Eplasty. 2018;18:E19.
- Laskin WB, Miettinen M, Fetsch JF. Infantile digital fibroma /fibromatosis: a clinicopathologic and immunohistochemical study of 69 tumors from 57 patients with long-term follow-up. Am J Surg Pathol. 2009;33:1-13.
- Henderson H, Peng YJ, Salter DM. Anti-calponin 1 antibodies highlight intracytoplasmic inclusions of infantile digital fibromatosis. Histopathology. 2014,64:752-755.
- Campbell LB, Petrick MG. Mohs micrographic surgery for a problematic infantile digital fibroma. Dermatol Surg. 2007;33:385-387.
- Ochi H, Puhaindran ME, Tan KW. Firm digital papulonodules in a young boy. Int J Dermatol. 2019;58:91-92.
- Albertini JG, Welsch MJ, Conger LA, et al. Infantile digital fibroma treated with Mohs micrography surgery. Dermatol Surg. 2002;28:959-961.
- Alves JV, Matos DM, Barreiros HF, et al. Variants of dermatofibroma— a histopathological study. An Bras Dermatol. 2014;89:472-477.
- Meyers AL, Fallahi AKM. Digital Mucous Cyst. StatPearls Publishing; 2020.
- Zhao Q, Lu H. Giant cell tumor of tendon sheath in the wrist that damaged the extensor indicis proprius tendon: a case report and literature review. BMC Cancer. 2019;19:1057.
- DiGrazia S, Succi G, Fragetta F, et al. Giant cell tumor of tendon sheath: study of 64 cases and review of literature. G Chir. 2013;34:149-152.
The Diagnosis: Infantile Digital Fibromatosis
Infantile digital fibromatosis (IDF) is a rare benign neoplasm of infancy prone to recurrence after resection but not to metastasis. It usually is limited to the fingers and toes.1 One-third of cases occur at birth. Most patients develop clinical symptoms within the first year of life, but presentation can occur in adolescents and adults. The exact etiology and pathogenesis of IDF remain unclear, but trauma is thought to be a trigger.
Physical examination reveals single or multiple smooth, round, pink papules or nodules confined to the sides and backs of the fingers, sparing the thumb and first toe.2,3 The nodules typically are firm, less than 2 cm in diameter, and often painless. Infantile digital fibromatosis exhibits an indolent progression followed by a rapid growth phase during several months, which may lead to functional impairment and joint deformities.4,5 Histopathology displays spindle cells with eosinophilic cytoplasmic inclusions that range from round to oval with uneven distribution, lack of refraction, and a large size difference (3–15 μm).6 The inclusions are deep red with Masson trichrome staining and can express smooth muscle actin and calponin. Tumor cells usually express vimentin, smooth muscle actin, calponin, and desmin but fail to express S-100 protein. The Ki67 proliferation index is 2% to 15%.6,7
Nonsurgical treatments for IDF include topical imiquimod, topical or intradermal injection of glucocorticoids, and intradermal injection of 5-fluorouracil. Complete resection should be reserved for cases with invasive growth that may lead to joint deformities, tendon or ligament involvement, digit or contracture deformity, and complications such as decreased joint mobility. Although there is a recurrence rate of up to 50% after excision, most lesions eventually will spontaneously regress and will leave no scar.8-10
The clinical and histopathologic differential diagnoses of IDF include other cutaneous diseases that occur in the digits. A dermatofibroma is a round, firm, fibrohistiocytic nodule that mainly occurs on the extensor limbs. Histopathology includes both fibrous and cellular types.11 Histologic analysis shows an ill-defined dermal proliferation of spindled fibroblasts with pale eosinophilic cytoplasm and bland fusiform nuclei growing in bands or fascicles that trap collagen fibers at the periphery (Figure 1). Generally, dermatofibromas have marked epidermal hyperplasia, which differs from IDF.
A digital myxoid cyst is characterized by a fleshcolored, hemispherical, and translucent cystic nodule that arises from the dorsum of the distal interphalangeal joint.12 It commonly is associated with injury and chronic pressure. Translucent viscous liquid may flow out when the cyst is punctured, a hallmark feature of this entity. Clinical variants of myxoid cyst include myxomatous and ganglion types. Histopathology reveals excessive mucin deposited in the dermis, and the surrounding collagen is compressed to form the pseudocyst (Figure 2).
A giant cell tumor of the tendon sheath presents with asymptomatic nodules or lumps. Lesions frequently are localized to the tendon sheath, especially on the fingers and wrists, with no malignant tendency or propensity for spontaneous regression.13 The local recurrence rate is as high as 45%, which is related to surgical resection insufficiency.14 Histopathologic examination shows lobulated tumor tissue surrounded by dense fibrosis. The tumor cells are histiocytic with scattered giant cells (Figure 3). The characteristic osteoclastlike giant cells have eosinophilic cytoplasm and irregularly arranged nuclei in varying numbers.
Keloids are connective tissue hyperplasias caused by skin injury. Histopathologically, keloids are characterized by nodules of thick hyalinized collagen bundles and whorled fibroblasts (Figure 4). No inclusions in the fibroblasts and a history of trauma can differentiate keloids from IDF.
The Diagnosis: Infantile Digital Fibromatosis
Infantile digital fibromatosis (IDF) is a rare benign neoplasm of infancy prone to recurrence after resection but not to metastasis. It usually is limited to the fingers and toes.1 One-third of cases occur at birth. Most patients develop clinical symptoms within the first year of life, but presentation can occur in adolescents and adults. The exact etiology and pathogenesis of IDF remain unclear, but trauma is thought to be a trigger.
Physical examination reveals single or multiple smooth, round, pink papules or nodules confined to the sides and backs of the fingers, sparing the thumb and first toe.2,3 The nodules typically are firm, less than 2 cm in diameter, and often painless. Infantile digital fibromatosis exhibits an indolent progression followed by a rapid growth phase during several months, which may lead to functional impairment and joint deformities.4,5 Histopathology displays spindle cells with eosinophilic cytoplasmic inclusions that range from round to oval with uneven distribution, lack of refraction, and a large size difference (3–15 μm).6 The inclusions are deep red with Masson trichrome staining and can express smooth muscle actin and calponin. Tumor cells usually express vimentin, smooth muscle actin, calponin, and desmin but fail to express S-100 protein. The Ki67 proliferation index is 2% to 15%.6,7
Nonsurgical treatments for IDF include topical imiquimod, topical or intradermal injection of glucocorticoids, and intradermal injection of 5-fluorouracil. Complete resection should be reserved for cases with invasive growth that may lead to joint deformities, tendon or ligament involvement, digit or contracture deformity, and complications such as decreased joint mobility. Although there is a recurrence rate of up to 50% after excision, most lesions eventually will spontaneously regress and will leave no scar.8-10
The clinical and histopathologic differential diagnoses of IDF include other cutaneous diseases that occur in the digits. A dermatofibroma is a round, firm, fibrohistiocytic nodule that mainly occurs on the extensor limbs. Histopathology includes both fibrous and cellular types.11 Histologic analysis shows an ill-defined dermal proliferation of spindled fibroblasts with pale eosinophilic cytoplasm and bland fusiform nuclei growing in bands or fascicles that trap collagen fibers at the periphery (Figure 1). Generally, dermatofibromas have marked epidermal hyperplasia, which differs from IDF.
A digital myxoid cyst is characterized by a fleshcolored, hemispherical, and translucent cystic nodule that arises from the dorsum of the distal interphalangeal joint.12 It commonly is associated with injury and chronic pressure. Translucent viscous liquid may flow out when the cyst is punctured, a hallmark feature of this entity. Clinical variants of myxoid cyst include myxomatous and ganglion types. Histopathology reveals excessive mucin deposited in the dermis, and the surrounding collagen is compressed to form the pseudocyst (Figure 2).
A giant cell tumor of the tendon sheath presents with asymptomatic nodules or lumps. Lesions frequently are localized to the tendon sheath, especially on the fingers and wrists, with no malignant tendency or propensity for spontaneous regression.13 The local recurrence rate is as high as 45%, which is related to surgical resection insufficiency.14 Histopathologic examination shows lobulated tumor tissue surrounded by dense fibrosis. The tumor cells are histiocytic with scattered giant cells (Figure 3). The characteristic osteoclastlike giant cells have eosinophilic cytoplasm and irregularly arranged nuclei in varying numbers.
Keloids are connective tissue hyperplasias caused by skin injury. Histopathologically, keloids are characterized by nodules of thick hyalinized collagen bundles and whorled fibroblasts (Figure 4). No inclusions in the fibroblasts and a history of trauma can differentiate keloids from IDF.
- Marks E, Ewart M. Infantile digital fibroma: a rare fibromatosis. Arch Pathol Lab Med. 2016;140:1153‐1156.
- Botelho LF, Matsushigue T, Enokihara MM, et al. Case for diagnosis. An Bras Dermatol. 2012;87:493-494.
- Paloni G, Mattei I, Salmaso R, et al. Infantile digital fibromatosis. Arch Dis Child. 2013;98:308.
- Girgenti V, Restano L, Arcangeli F, et al. Infantile digital fibromatosis: a rare tumour of infancy. report of five cases. Australas J Dermatol. 2012;53:285-287.
- Eypper EH, Lee JC, Tarasen AJ, et al. An algorithmic approach to the management of infantile digital fibromatosis: review of literature and a case report. Eplasty. 2018;18:E19.
- Laskin WB, Miettinen M, Fetsch JF. Infantile digital fibroma /fibromatosis: a clinicopathologic and immunohistochemical study of 69 tumors from 57 patients with long-term follow-up. Am J Surg Pathol. 2009;33:1-13.
- Henderson H, Peng YJ, Salter DM. Anti-calponin 1 antibodies highlight intracytoplasmic inclusions of infantile digital fibromatosis. Histopathology. 2014,64:752-755.
- Campbell LB, Petrick MG. Mohs micrographic surgery for a problematic infantile digital fibroma. Dermatol Surg. 2007;33:385-387.
- Ochi H, Puhaindran ME, Tan KW. Firm digital papulonodules in a young boy. Int J Dermatol. 2019;58:91-92.
- Albertini JG, Welsch MJ, Conger LA, et al. Infantile digital fibroma treated with Mohs micrography surgery. Dermatol Surg. 2002;28:959-961.
- Alves JV, Matos DM, Barreiros HF, et al. Variants of dermatofibroma— a histopathological study. An Bras Dermatol. 2014;89:472-477.
- Meyers AL, Fallahi AKM. Digital Mucous Cyst. StatPearls Publishing; 2020.
- Zhao Q, Lu H. Giant cell tumor of tendon sheath in the wrist that damaged the extensor indicis proprius tendon: a case report and literature review. BMC Cancer. 2019;19:1057.
- DiGrazia S, Succi G, Fragetta F, et al. Giant cell tumor of tendon sheath: study of 64 cases and review of literature. G Chir. 2013;34:149-152.
- Marks E, Ewart M. Infantile digital fibroma: a rare fibromatosis. Arch Pathol Lab Med. 2016;140:1153‐1156.
- Botelho LF, Matsushigue T, Enokihara MM, et al. Case for diagnosis. An Bras Dermatol. 2012;87:493-494.
- Paloni G, Mattei I, Salmaso R, et al. Infantile digital fibromatosis. Arch Dis Child. 2013;98:308.
- Girgenti V, Restano L, Arcangeli F, et al. Infantile digital fibromatosis: a rare tumour of infancy. report of five cases. Australas J Dermatol. 2012;53:285-287.
- Eypper EH, Lee JC, Tarasen AJ, et al. An algorithmic approach to the management of infantile digital fibromatosis: review of literature and a case report. Eplasty. 2018;18:E19.
- Laskin WB, Miettinen M, Fetsch JF. Infantile digital fibroma /fibromatosis: a clinicopathologic and immunohistochemical study of 69 tumors from 57 patients with long-term follow-up. Am J Surg Pathol. 2009;33:1-13.
- Henderson H, Peng YJ, Salter DM. Anti-calponin 1 antibodies highlight intracytoplasmic inclusions of infantile digital fibromatosis. Histopathology. 2014,64:752-755.
- Campbell LB, Petrick MG. Mohs micrographic surgery for a problematic infantile digital fibroma. Dermatol Surg. 2007;33:385-387.
- Ochi H, Puhaindran ME, Tan KW. Firm digital papulonodules in a young boy. Int J Dermatol. 2019;58:91-92.
- Albertini JG, Welsch MJ, Conger LA, et al. Infantile digital fibroma treated with Mohs micrography surgery. Dermatol Surg. 2002;28:959-961.
- Alves JV, Matos DM, Barreiros HF, et al. Variants of dermatofibroma— a histopathological study. An Bras Dermatol. 2014;89:472-477.
- Meyers AL, Fallahi AKM. Digital Mucous Cyst. StatPearls Publishing; 2020.
- Zhao Q, Lu H. Giant cell tumor of tendon sheath in the wrist that damaged the extensor indicis proprius tendon: a case report and literature review. BMC Cancer. 2019;19:1057.
- DiGrazia S, Succi G, Fragetta F, et al. Giant cell tumor of tendon sheath: study of 64 cases and review of literature. G Chir. 2013;34:149-152.
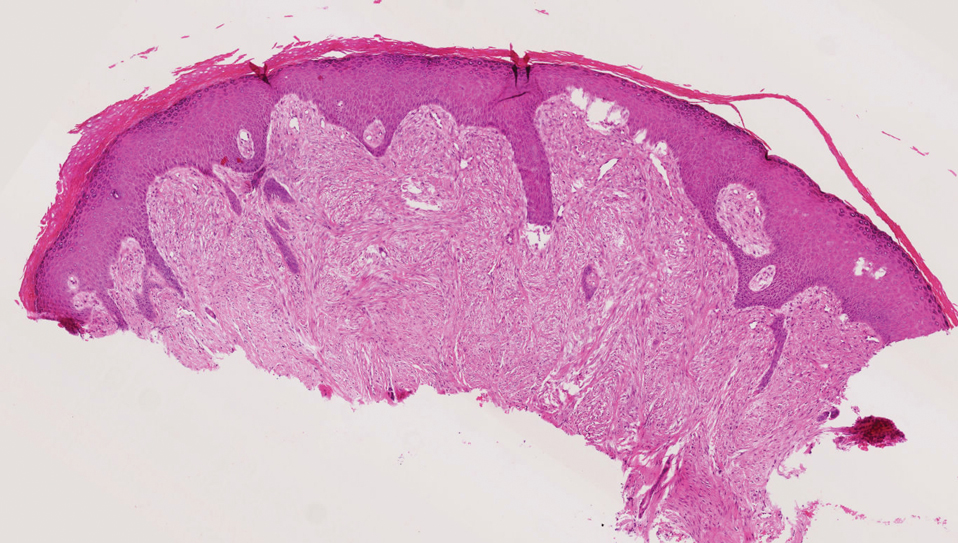
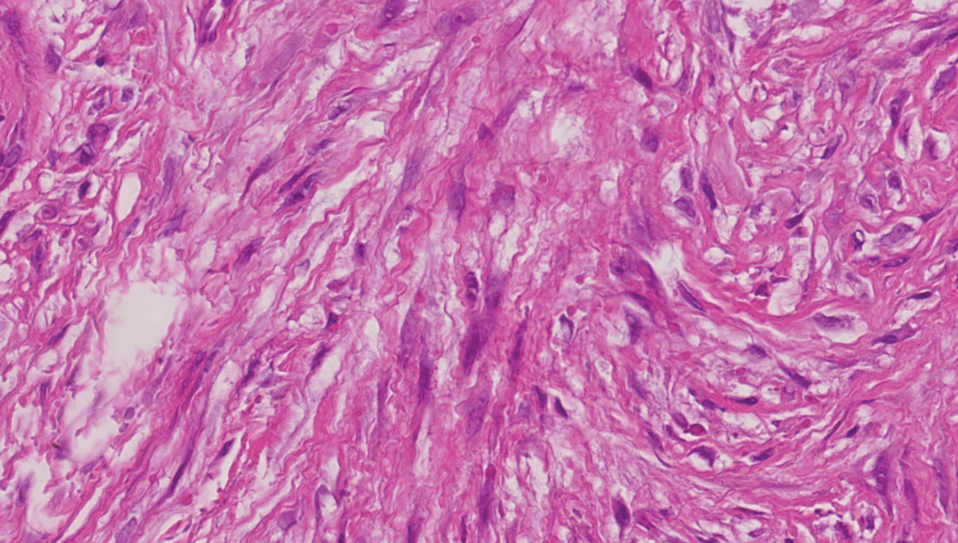
A 3-month-old girl presented with papulonodules on the distal left ring finger. Initially the lesions were thought to be insect bites but became firm over the course of 3 weeks and then gradually increased in size over 2 months. Physical examination revealed a 0.5×0.5-cm firm nodule and a 0.2×0.3-cm firm papule on the radial aspect of the left ring finger over the distal interphalangeal joint. There was no deformity or dysfunction of the finger. Radiography showed soft tissue swelling without bony abnormalities. The lesions were excised; however, a new fleshy nodule reappeared 1 month postoperatively on the radial aspect of the left ring finger over the distal interphalangeal joint. The patient did not seem bothered by the lesions and was in good general health.
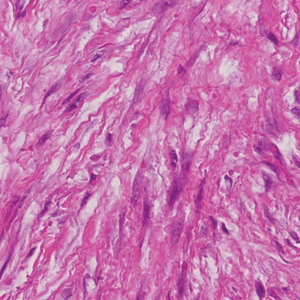
Soft Nodule on the Forearm
The Diagnosis: Schwannoma
Schwannoma, also known as neurilemmoma, is a benign encapsulated neoplasm of the peripheral nerve sheath that presents as a subcutaneous nodule.1 It also may present in the retroperitoneum, mediastinum, and viscera (eg, gastrointestinal tract, bone, upper respiratory tract, lymph nodes). It may occur as multiple lesions when associated with certain syndromes. It usually is an asymptomatic indolent tumor with neurologic symptoms, such as pain and tenderness, in the lesions that are deeper, larger, or closer in proximity to nearby structures.2,3
Histologically, a schwannoma is encapsulated by the perineurium of the nerve bundle from which it originates (quiz image [top]). The tumor consists of hypercellular (Antoni type A) and hypocellular (Antoni type B) areas. Antoni type A areas consist of tightly packed, spindleshaped cells with elongated wavy nuclei and indistinct cytoplasmic borders. These nuclei tend to align into parallel rows with intervening anuclear zones forming Verocay bodies (quiz image [bottom]).4 Verocay bodies are not seen in all schwannomas, and similar formations may be seen in other tumors as well. Solitary circumscribed neuromas also have Verocay bodies, whereas dermatofibromas and leiomyomas have Verocay-like bodies. Antoni type B areas have scattered spindled or ovoid cells in an edematous or myxoid matrix interspersed with inflammatory cells such as lymphocytes and histiocytes. Vessels with thick hyalinized walls are a helpful feature in diagnosis.2 Schwann cells of a schwannoma stain diffusely positive with S-100 protein. The capsule stains positively with epithelial membrane antigen due to the presence of perineurial cells.2
The morphologic variants of this entity include conventional (common, solitary), cellular, plexiform, ancient, melanotic, epithelioid, pseudoglandular, neuroblastomalike, and microcystic/reticular schwannomas. There are additional variants that are associated with genetic syndromes, such as multiple cutaneous plexiform schwannomas linked with neurofibromatosis type 2, psammomatous melanotic schwannoma presenting in Carney complex, schwannomatosis, and segmental schwannomatosis (a distinct form of neurofibromatosis characterized by multiple schwannomas localized to one limb). Either presentation may have alteration or deletion of the neurofibromatosis type 2 gene, NF2, on chromosome 22.2,5
Nodular fasciitis is a benign tumor of fibroblasts and myofibroblasts that usually arises in the subcutaneous tissues. It most commonly occurs in the upper extremities, trunk, head, and neck. It presents as a single, often painful, rapidly growing, subcutaneous nodule. Histologically, lesions mostly are well circumscribed yet unencapsulated, in contrast to schwannomas. They may be hypocellular or hypercellular and are composed of uniform spindle cells with a feathery or fascicular (tissue culture–like) appearance in a loose, myxoid to collagenous stroma. There may be foci of hemorrhage and conspicuous mitoses but not atypical figures (Figure 1). Immunohistochemically, the cells stain positively for smooth muscle actin and negatively for S-100 protein, which sets it apart from a schwannoma. Most cases contain fusion genes, with myosin heavy chain 9 ubiquitin-specific peptidase 6, MYH9-USP6, being the most common fusion product.6
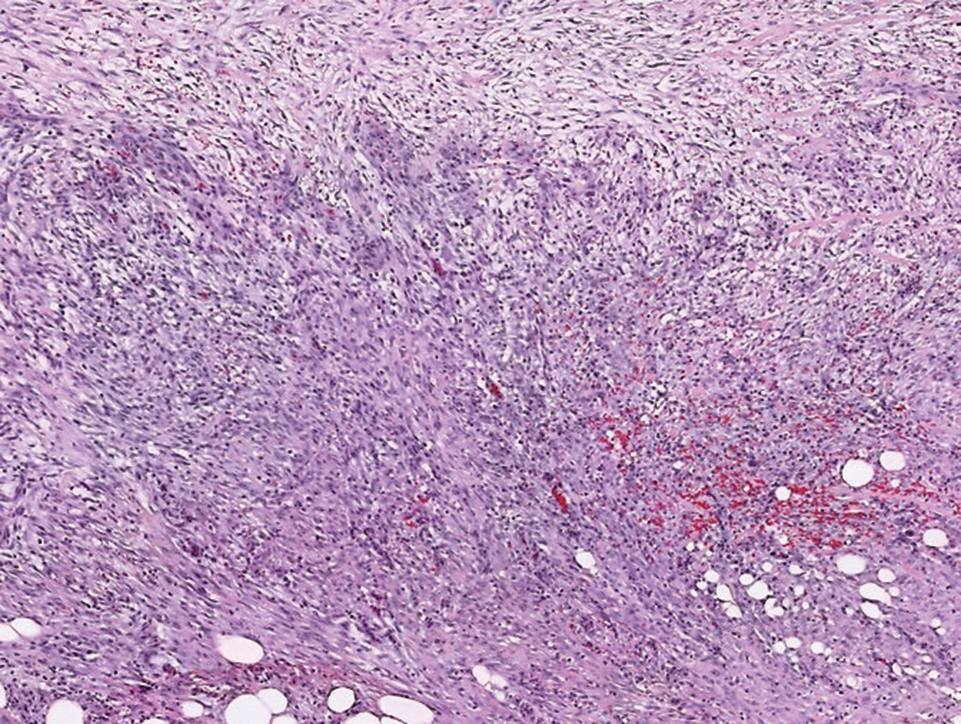
Solitary circumscribed neuroma (palisaded encapsulated neuroma) is a benign, usually solitary dermal lesion. It most commonly occurs in middle-aged to elderly adults as a small (<1 cm), firm, flesh-colored to pink papule on the face (ie, cheeks, nose, nasolabial folds) and less commonly in the oral and acral regions and on the eyelids and penis. The lesion usually is unilobular; however, other growth patterns such as plexiform, multilobular, and fungating variants have been identified. Histologically, it is a well-circumscribed nodule with a thin capsule of perineurium that is composed of interlacing bundles of Schwann cells with a characteristic clefting artifact (Figure 2). Cells have wavy dark nuclei with scant cytoplasm that occasionally form palisades or Verocay bodies causing these lesions to be confused with schwannomas. Immunohistochemically, the Schwann cells stain positively with S-100 protein, and the perineurium stains positively with epithelial membrane antigen, Claudin-1, and Glut-1. Neurofilament protein stains axons throughout neuromas, whereas in schwannoma, the expression often is limited to entrapped axons at the periphery of the tumor.7
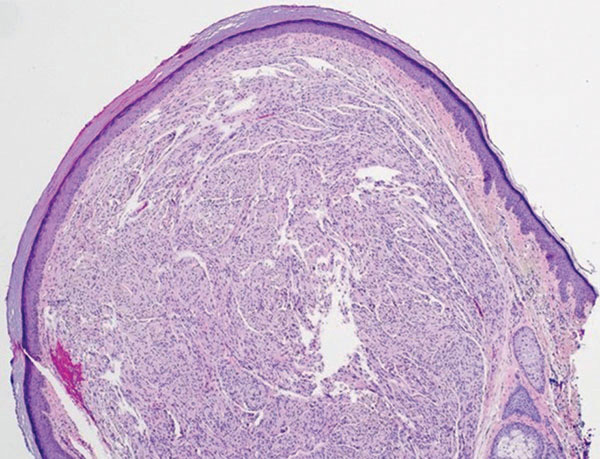
Angioleiomyoma is an uncommon, benign, smooth muscle neoplasm of the skin and subcutaneous tissue that originates from vascular smooth muscle. It most commonly presents in adult females aged 30 to 60 years, with a predilection for the lower limbs. These tumors typically are solitary, slow growing, and less than 2 cm in diameter and may be painful upon compression. Similar to schwannoma, angioleiomyoma is an encapsulated lesion composed of interlacing, uniform, smooth muscle bundles distributed around vessels (Figure 3). Smooth muscle cells have oval- or cigar-shaped nuclei with a small perinuclear vacuole of glycogen. Immunohistochemically, there is strong diffuse staining for smooth muscle actin and h-caldesmon. Recurrence after excision is rare.2,8
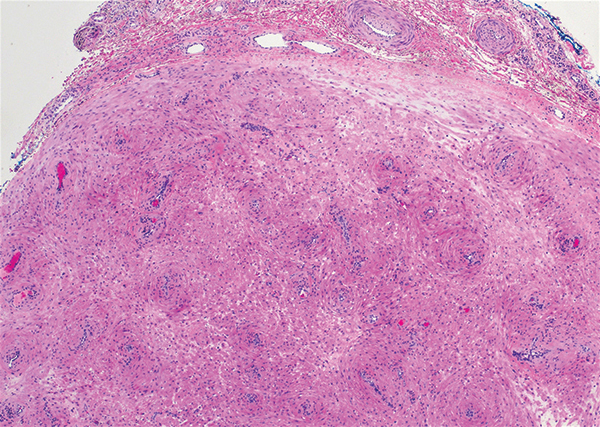
Neurofibroma is a common, mostly sporadic, benign tumor of nerve sheath origin. The solitary type may be localized (well circumscribed, unencapsulated) or diffuse. The presence of multiple, deep, and plexiform lesions is associated with neurofibromatosis type 1 (von Recklinghausen disease) that is caused by germline mutations in the NF1 gene. Histologically, the tumor is composed of Schwann cells, fibroblasts, perineurial cells, and nerve axons within an extracellular myxoid to collagenous matrix (Figure 4). The diffuse type is an ill-defined proliferation that entraps adnexal structures. The plexiform type is defined by multinodular serpentine fascicles. Immunohistochemically, the Schwann cells stain positive for S-100 protein and SOX10 (SRY-Box Transcription Factor 10). Epithelial membrane antigen stains admixed perineurial cells. Neurofilament protein highlights intratumoral axons, which generally are not found throughout schwannomas. Transformation to a malignant peripheral nerve sheath tumor occurs in up to 10% of patients with neurofibromatosis type 1, usually in plexiform neurofibromas, and is characterized by increased cellularity, atypia, mitotic activity, and necrosis.9
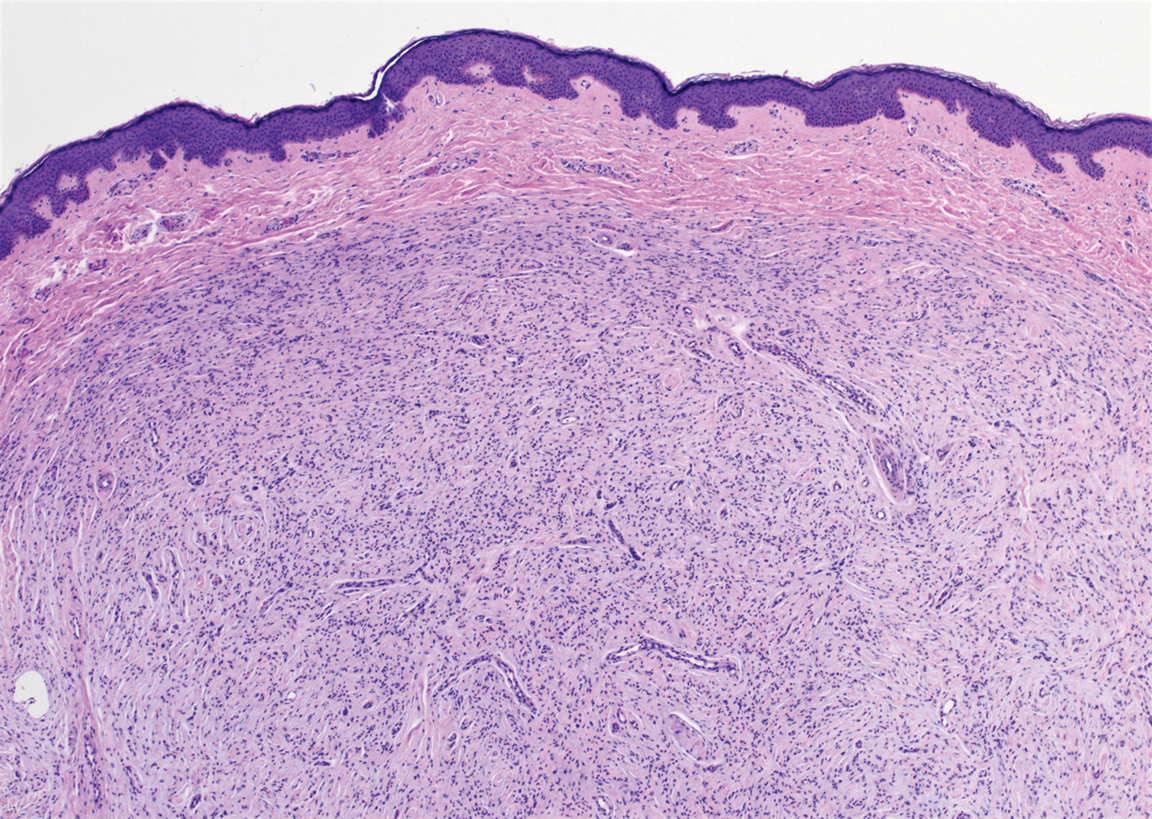
- Ritter SE, Elston DM. Cutaneous schwannoma of the foot. Cutis. 2001;67:127-129.
- Calonje E, Damaskou V, Lazar AJ. Connective tissue tumors. In: Calonje E, Brenn T, Lazar AJ, et al, eds. McKee’s Pathology of the Skin. 5th ed. Vol 2. Elsevier Saunders; 2020:1698-1894.
- Knight DM, Birch R, Pringle J. Benign solitary schwannomas: a review of 234 cases. J Bone Joint Surg Br. 2007;89:382-387.
- Lespi PJ, Smit R. Verocay body—prominent cutaneous leiomyoma. Am J Dermatopathol. 1999;21:110-111.
- Kurtkaya-Yapicier O, Scheithauer B, Woodruff JM. The pathobiologic spectrum of schwannomas. Histol Histopathol. 2003;18:925-934.
- Erickson-Johnson MR, Chou MM, Evers BR, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Invest. 2011;91:1427-1433.
- Leblebici C, Savli TC, Yeni B, et al. Palisaded encapsulated (solitary circumscribed) neuroma: a review of 30 cases. Int J Surg Pathol. 2019;27:506-514.
- Yeung CM, Moore L, Lans J, et al. Angioleiomyoma of the hand: a case series and review of the literature. Arch Bone Jt Surg. 2020; 8:373-377.
- Skovronsky DM, Oberholtzer JC. Pathologic classification of peripheral nerve tumors. Neurosurg Clin North Am. 2004;15:157-166.
The Diagnosis: Schwannoma
Schwannoma, also known as neurilemmoma, is a benign encapsulated neoplasm of the peripheral nerve sheath that presents as a subcutaneous nodule.1 It also may present in the retroperitoneum, mediastinum, and viscera (eg, gastrointestinal tract, bone, upper respiratory tract, lymph nodes). It may occur as multiple lesions when associated with certain syndromes. It usually is an asymptomatic indolent tumor with neurologic symptoms, such as pain and tenderness, in the lesions that are deeper, larger, or closer in proximity to nearby structures.2,3
Histologically, a schwannoma is encapsulated by the perineurium of the nerve bundle from which it originates (quiz image [top]). The tumor consists of hypercellular (Antoni type A) and hypocellular (Antoni type B) areas. Antoni type A areas consist of tightly packed, spindleshaped cells with elongated wavy nuclei and indistinct cytoplasmic borders. These nuclei tend to align into parallel rows with intervening anuclear zones forming Verocay bodies (quiz image [bottom]).4 Verocay bodies are not seen in all schwannomas, and similar formations may be seen in other tumors as well. Solitary circumscribed neuromas also have Verocay bodies, whereas dermatofibromas and leiomyomas have Verocay-like bodies. Antoni type B areas have scattered spindled or ovoid cells in an edematous or myxoid matrix interspersed with inflammatory cells such as lymphocytes and histiocytes. Vessels with thick hyalinized walls are a helpful feature in diagnosis.2 Schwann cells of a schwannoma stain diffusely positive with S-100 protein. The capsule stains positively with epithelial membrane antigen due to the presence of perineurial cells.2
The morphologic variants of this entity include conventional (common, solitary), cellular, plexiform, ancient, melanotic, epithelioid, pseudoglandular, neuroblastomalike, and microcystic/reticular schwannomas. There are additional variants that are associated with genetic syndromes, such as multiple cutaneous plexiform schwannomas linked with neurofibromatosis type 2, psammomatous melanotic schwannoma presenting in Carney complex, schwannomatosis, and segmental schwannomatosis (a distinct form of neurofibromatosis characterized by multiple schwannomas localized to one limb). Either presentation may have alteration or deletion of the neurofibromatosis type 2 gene, NF2, on chromosome 22.2,5
Nodular fasciitis is a benign tumor of fibroblasts and myofibroblasts that usually arises in the subcutaneous tissues. It most commonly occurs in the upper extremities, trunk, head, and neck. It presents as a single, often painful, rapidly growing, subcutaneous nodule. Histologically, lesions mostly are well circumscribed yet unencapsulated, in contrast to schwannomas. They may be hypocellular or hypercellular and are composed of uniform spindle cells with a feathery or fascicular (tissue culture–like) appearance in a loose, myxoid to collagenous stroma. There may be foci of hemorrhage and conspicuous mitoses but not atypical figures (Figure 1). Immunohistochemically, the cells stain positively for smooth muscle actin and negatively for S-100 protein, which sets it apart from a schwannoma. Most cases contain fusion genes, with myosin heavy chain 9 ubiquitin-specific peptidase 6, MYH9-USP6, being the most common fusion product.6
Solitary circumscribed neuroma (palisaded encapsulated neuroma) is a benign, usually solitary dermal lesion. It most commonly occurs in middle-aged to elderly adults as a small (<1 cm), firm, flesh-colored to pink papule on the face (ie, cheeks, nose, nasolabial folds) and less commonly in the oral and acral regions and on the eyelids and penis. The lesion usually is unilobular; however, other growth patterns such as plexiform, multilobular, and fungating variants have been identified. Histologically, it is a well-circumscribed nodule with a thin capsule of perineurium that is composed of interlacing bundles of Schwann cells with a characteristic clefting artifact (Figure 2). Cells have wavy dark nuclei with scant cytoplasm that occasionally form palisades or Verocay bodies causing these lesions to be confused with schwannomas. Immunohistochemically, the Schwann cells stain positively with S-100 protein, and the perineurium stains positively with epithelial membrane antigen, Claudin-1, and Glut-1. Neurofilament protein stains axons throughout neuromas, whereas in schwannoma, the expression often is limited to entrapped axons at the periphery of the tumor.7
Angioleiomyoma is an uncommon, benign, smooth muscle neoplasm of the skin and subcutaneous tissue that originates from vascular smooth muscle. It most commonly presents in adult females aged 30 to 60 years, with a predilection for the lower limbs. These tumors typically are solitary, slow growing, and less than 2 cm in diameter and may be painful upon compression. Similar to schwannoma, angioleiomyoma is an encapsulated lesion composed of interlacing, uniform, smooth muscle bundles distributed around vessels (Figure 3). Smooth muscle cells have oval- or cigar-shaped nuclei with a small perinuclear vacuole of glycogen. Immunohistochemically, there is strong diffuse staining for smooth muscle actin and h-caldesmon. Recurrence after excision is rare.2,8
Neurofibroma is a common, mostly sporadic, benign tumor of nerve sheath origin. The solitary type may be localized (well circumscribed, unencapsulated) or diffuse. The presence of multiple, deep, and plexiform lesions is associated with neurofibromatosis type 1 (von Recklinghausen disease) that is caused by germline mutations in the NF1 gene. Histologically, the tumor is composed of Schwann cells, fibroblasts, perineurial cells, and nerve axons within an extracellular myxoid to collagenous matrix (Figure 4). The diffuse type is an ill-defined proliferation that entraps adnexal structures. The plexiform type is defined by multinodular serpentine fascicles. Immunohistochemically, the Schwann cells stain positive for S-100 protein and SOX10 (SRY-Box Transcription Factor 10). Epithelial membrane antigen stains admixed perineurial cells. Neurofilament protein highlights intratumoral axons, which generally are not found throughout schwannomas. Transformation to a malignant peripheral nerve sheath tumor occurs in up to 10% of patients with neurofibromatosis type 1, usually in plexiform neurofibromas, and is characterized by increased cellularity, atypia, mitotic activity, and necrosis.9
The Diagnosis: Schwannoma
Schwannoma, also known as neurilemmoma, is a benign encapsulated neoplasm of the peripheral nerve sheath that presents as a subcutaneous nodule.1 It also may present in the retroperitoneum, mediastinum, and viscera (eg, gastrointestinal tract, bone, upper respiratory tract, lymph nodes). It may occur as multiple lesions when associated with certain syndromes. It usually is an asymptomatic indolent tumor with neurologic symptoms, such as pain and tenderness, in the lesions that are deeper, larger, or closer in proximity to nearby structures.2,3
Histologically, a schwannoma is encapsulated by the perineurium of the nerve bundle from which it originates (quiz image [top]). The tumor consists of hypercellular (Antoni type A) and hypocellular (Antoni type B) areas. Antoni type A areas consist of tightly packed, spindleshaped cells with elongated wavy nuclei and indistinct cytoplasmic borders. These nuclei tend to align into parallel rows with intervening anuclear zones forming Verocay bodies (quiz image [bottom]).4 Verocay bodies are not seen in all schwannomas, and similar formations may be seen in other tumors as well. Solitary circumscribed neuromas also have Verocay bodies, whereas dermatofibromas and leiomyomas have Verocay-like bodies. Antoni type B areas have scattered spindled or ovoid cells in an edematous or myxoid matrix interspersed with inflammatory cells such as lymphocytes and histiocytes. Vessels with thick hyalinized walls are a helpful feature in diagnosis.2 Schwann cells of a schwannoma stain diffusely positive with S-100 protein. The capsule stains positively with epithelial membrane antigen due to the presence of perineurial cells.2
The morphologic variants of this entity include conventional (common, solitary), cellular, plexiform, ancient, melanotic, epithelioid, pseudoglandular, neuroblastomalike, and microcystic/reticular schwannomas. There are additional variants that are associated with genetic syndromes, such as multiple cutaneous plexiform schwannomas linked with neurofibromatosis type 2, psammomatous melanotic schwannoma presenting in Carney complex, schwannomatosis, and segmental schwannomatosis (a distinct form of neurofibromatosis characterized by multiple schwannomas localized to one limb). Either presentation may have alteration or deletion of the neurofibromatosis type 2 gene, NF2, on chromosome 22.2,5
Nodular fasciitis is a benign tumor of fibroblasts and myofibroblasts that usually arises in the subcutaneous tissues. It most commonly occurs in the upper extremities, trunk, head, and neck. It presents as a single, often painful, rapidly growing, subcutaneous nodule. Histologically, lesions mostly are well circumscribed yet unencapsulated, in contrast to schwannomas. They may be hypocellular or hypercellular and are composed of uniform spindle cells with a feathery or fascicular (tissue culture–like) appearance in a loose, myxoid to collagenous stroma. There may be foci of hemorrhage and conspicuous mitoses but not atypical figures (Figure 1). Immunohistochemically, the cells stain positively for smooth muscle actin and negatively for S-100 protein, which sets it apart from a schwannoma. Most cases contain fusion genes, with myosin heavy chain 9 ubiquitin-specific peptidase 6, MYH9-USP6, being the most common fusion product.6
Solitary circumscribed neuroma (palisaded encapsulated neuroma) is a benign, usually solitary dermal lesion. It most commonly occurs in middle-aged to elderly adults as a small (<1 cm), firm, flesh-colored to pink papule on the face (ie, cheeks, nose, nasolabial folds) and less commonly in the oral and acral regions and on the eyelids and penis. The lesion usually is unilobular; however, other growth patterns such as plexiform, multilobular, and fungating variants have been identified. Histologically, it is a well-circumscribed nodule with a thin capsule of perineurium that is composed of interlacing bundles of Schwann cells with a characteristic clefting artifact (Figure 2). Cells have wavy dark nuclei with scant cytoplasm that occasionally form palisades or Verocay bodies causing these lesions to be confused with schwannomas. Immunohistochemically, the Schwann cells stain positively with S-100 protein, and the perineurium stains positively with epithelial membrane antigen, Claudin-1, and Glut-1. Neurofilament protein stains axons throughout neuromas, whereas in schwannoma, the expression often is limited to entrapped axons at the periphery of the tumor.7
Angioleiomyoma is an uncommon, benign, smooth muscle neoplasm of the skin and subcutaneous tissue that originates from vascular smooth muscle. It most commonly presents in adult females aged 30 to 60 years, with a predilection for the lower limbs. These tumors typically are solitary, slow growing, and less than 2 cm in diameter and may be painful upon compression. Similar to schwannoma, angioleiomyoma is an encapsulated lesion composed of interlacing, uniform, smooth muscle bundles distributed around vessels (Figure 3). Smooth muscle cells have oval- or cigar-shaped nuclei with a small perinuclear vacuole of glycogen. Immunohistochemically, there is strong diffuse staining for smooth muscle actin and h-caldesmon. Recurrence after excision is rare.2,8
Neurofibroma is a common, mostly sporadic, benign tumor of nerve sheath origin. The solitary type may be localized (well circumscribed, unencapsulated) or diffuse. The presence of multiple, deep, and plexiform lesions is associated with neurofibromatosis type 1 (von Recklinghausen disease) that is caused by germline mutations in the NF1 gene. Histologically, the tumor is composed of Schwann cells, fibroblasts, perineurial cells, and nerve axons within an extracellular myxoid to collagenous matrix (Figure 4). The diffuse type is an ill-defined proliferation that entraps adnexal structures. The plexiform type is defined by multinodular serpentine fascicles. Immunohistochemically, the Schwann cells stain positive for S-100 protein and SOX10 (SRY-Box Transcription Factor 10). Epithelial membrane antigen stains admixed perineurial cells. Neurofilament protein highlights intratumoral axons, which generally are not found throughout schwannomas. Transformation to a malignant peripheral nerve sheath tumor occurs in up to 10% of patients with neurofibromatosis type 1, usually in plexiform neurofibromas, and is characterized by increased cellularity, atypia, mitotic activity, and necrosis.9
- Ritter SE, Elston DM. Cutaneous schwannoma of the foot. Cutis. 2001;67:127-129.
- Calonje E, Damaskou V, Lazar AJ. Connective tissue tumors. In: Calonje E, Brenn T, Lazar AJ, et al, eds. McKee’s Pathology of the Skin. 5th ed. Vol 2. Elsevier Saunders; 2020:1698-1894.
- Knight DM, Birch R, Pringle J. Benign solitary schwannomas: a review of 234 cases. J Bone Joint Surg Br. 2007;89:382-387.
- Lespi PJ, Smit R. Verocay body—prominent cutaneous leiomyoma. Am J Dermatopathol. 1999;21:110-111.
- Kurtkaya-Yapicier O, Scheithauer B, Woodruff JM. The pathobiologic spectrum of schwannomas. Histol Histopathol. 2003;18:925-934.
- Erickson-Johnson MR, Chou MM, Evers BR, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Invest. 2011;91:1427-1433.
- Leblebici C, Savli TC, Yeni B, et al. Palisaded encapsulated (solitary circumscribed) neuroma: a review of 30 cases. Int J Surg Pathol. 2019;27:506-514.
- Yeung CM, Moore L, Lans J, et al. Angioleiomyoma of the hand: a case series and review of the literature. Arch Bone Jt Surg. 2020; 8:373-377.
- Skovronsky DM, Oberholtzer JC. Pathologic classification of peripheral nerve tumors. Neurosurg Clin North Am. 2004;15:157-166.
- Ritter SE, Elston DM. Cutaneous schwannoma of the foot. Cutis. 2001;67:127-129.
- Calonje E, Damaskou V, Lazar AJ. Connective tissue tumors. In: Calonje E, Brenn T, Lazar AJ, et al, eds. McKee’s Pathology of the Skin. 5th ed. Vol 2. Elsevier Saunders; 2020:1698-1894.
- Knight DM, Birch R, Pringle J. Benign solitary schwannomas: a review of 234 cases. J Bone Joint Surg Br. 2007;89:382-387.
- Lespi PJ, Smit R. Verocay body—prominent cutaneous leiomyoma. Am J Dermatopathol. 1999;21:110-111.
- Kurtkaya-Yapicier O, Scheithauer B, Woodruff JM. The pathobiologic spectrum of schwannomas. Histol Histopathol. 2003;18:925-934.
- Erickson-Johnson MR, Chou MM, Evers BR, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Invest. 2011;91:1427-1433.
- Leblebici C, Savli TC, Yeni B, et al. Palisaded encapsulated (solitary circumscribed) neuroma: a review of 30 cases. Int J Surg Pathol. 2019;27:506-514.
- Yeung CM, Moore L, Lans J, et al. Angioleiomyoma of the hand: a case series and review of the literature. Arch Bone Jt Surg. 2020; 8:373-377.
- Skovronsky DM, Oberholtzer JC. Pathologic classification of peripheral nerve tumors. Neurosurg Clin North Am. 2004;15:157-166.

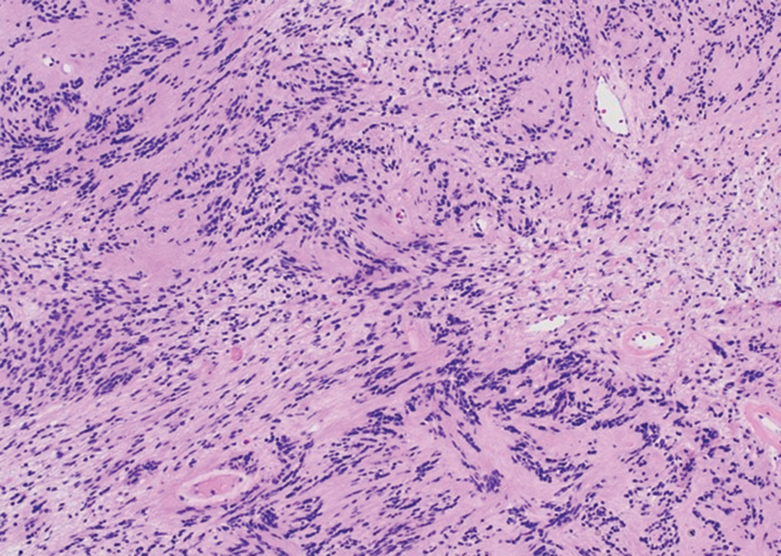
A 54-year-old woman presented with an enlarging mass on the right volar forearm. Physical examination revealed a 1-cm, soft, mobile, subcutaneous nodule. Excision revealed tan-pink, indurated, fibrous, nodular tissue.
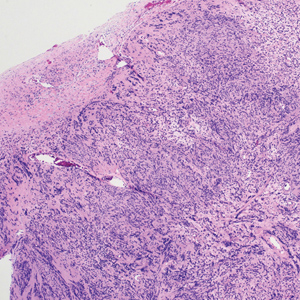
Erythematous and Ulcerated Plaque on the Left Temple
The Diagnosis: Primary Cutaneous Carcinosarcoma
The immunohistochemical findings supported an epithelial component consistent with moderately differentiated squamous cell carcinoma (SCC) and a mesenchymal component with features consistent with a sarcoma. Consequently, the lesion was diagnosed as a primary cutaneous carcinosarcoma (PCCS).
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm consisting of malignant epithelial (carcinoma) and mesenchymal (sarcoma) components.1 Primary cutaneous carcinosarcomas are uncommon, poorly understood, primary cutaneous tumors.2,3 Characteristic of this tumor, cytokeratins highlight the epithelial component while vimentin highlights the mesenchymal component.4 Histologically, the sarcomatous components of PCCS often are highly variable, with an absence of transitional areas within the epithelial component, which frequently resembles basal cell carcinoma and/ or SCC.5-7 Primary cutaneous carcinosarcoma favors areas of chronic UV radiation exposure, particularly on the head and neck. Most tumors present with a slowly growing, polypoid, flesh-colored to erythematous nodule due to the infiltrative mesenchymal component.7 Primary cutaneous carcinosarcoma primarily is diagnosed in elderly patients, with the majority of cases diagnosed in the eighth or ninth decades of life (range, 32–98 years).1,8 Men appear to be twice as likely to be diagnosed with a PCCS compared to women.1 Primary cutaneous carcinosarcomas are recognized as aggressive tumors with a high propensity to metastasize and recur locally, necessitating early diagnosis and treatment.4 Accurate diagnosis of PCCSs can be challenging due to the biphasic nature of the neoplasm as well as poor differentiation or unequal proportions of the epithelial and mesenchymal components.5 Additionally, overlapping diagnostic criteria coupled with vague demarcation between soft-tissue sarcomas and distinct carcinomas also may contribute to a delay in diagnosis.9 Treatment is achieved surgically by complete wide resection, with no evidence to support the use of adjuvant or neoadjuvant external beam radiation therapy. Due to the small number of reported cases, no treatment recommendations currently exist.1
Surgical management with wide local excision has been disappointing, with recurrence rates reported as high as 33%.6 Primary cutaneous carcinosarcoma has an estimated overall recurrence rate of 19% and a 5-year disease-free rate of 50%.10 Risk factors associated with poorer prognosis include tumors with adnexal subtype, age less than 65 years, rapid tumor growth, a tumor greater than 20 mm at presentation, and a long-standing tumor lasting up to 30 years.2,4 Although wide local excision and Mohs micrographic surgery (MMS) both have been utilized successfully, MMS has been shown to result in a cure rate of greater than 98%.6
Atypical fibroxanthoma (AFX) is a cutaneous tumor of fibrohistiocytic mesenchymal origin that typically manifests on sun-damaged skin in elderly individuals. Clinically, it presents as a rapidly growing neoplasm that often ulcerates and bleeds. These heterogenous neoplasms have several distinct characteristics, including dense cellularity with disorganized, large, pleomorphic, and atypical-appearing spindle-shaped cells arising in the upper layers of the dermis, often disseminating into the reticular dermis and occasionally into the subcutaneous fat (Figure 1). The neoplastic cells often exhibit hyperchromic and irregular nuclei, multinucleated giant cells, and atypical mitotic figures. In most cases, negative immunohistochemical staining with SOX-10, S-100, cytokeratins, desmin, and caldesmon will allow pathologists to differentiate between AFX and other common tumors on the differential diagnosis, such as SCC, melanoma, and leiomyosarcoma. CD10 and procollagen type 1 are positive antigenic markers in AFX, but they are not specific. The standard treatment of AFX includes wide local excision or MMS for superior margin control.11
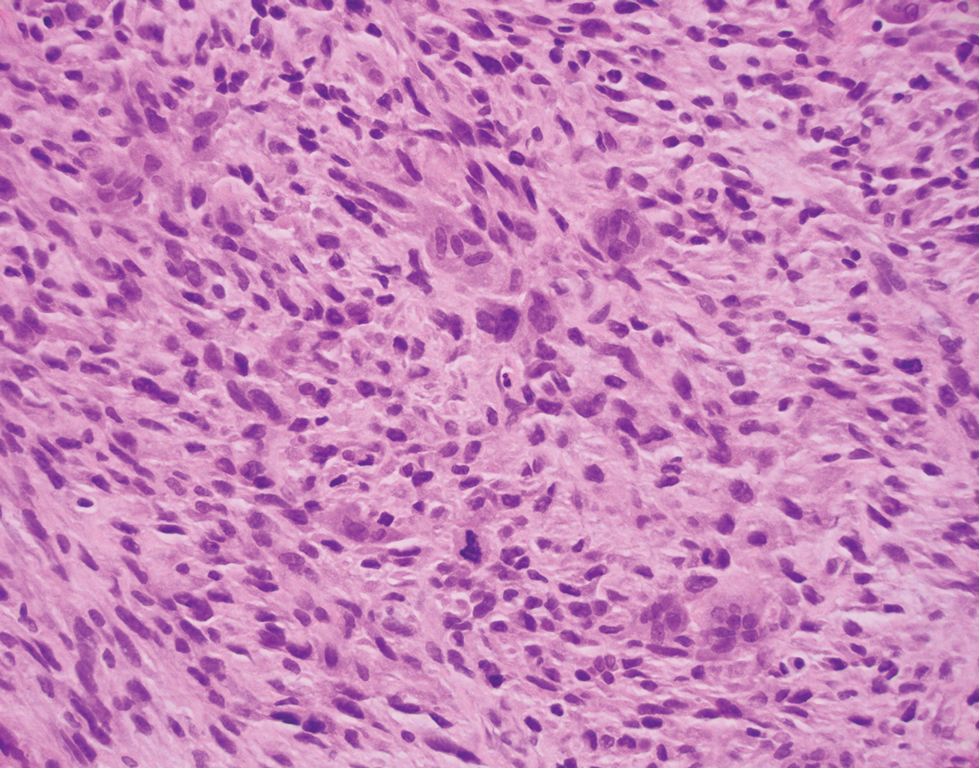
Spindle cell SCC presents as a raised or exophytic nodule, often with spontaneous bleeding and central ulceration. It usually presents on sun-damaged skin or in individuals with a history of ionizing radiation. Histologically, it is characterized by atypical spindleshaped keratinocytes in the dermis existing as single cells or cohesive nests along with keratin pearls (Figure 2). The atypical spindle cells may comprise the entire tumor or only a small portion. The use of immunohistochemical markers often is required to establish a definitive diagnosis. Spindle cell SCC stains positively, albeit frequently focally, for p63, p40, and high-molecular-weight cytokeratins such as cytokeratin 5/6, while S-100 protein, SOX-10, MART-1/Melan-A, and muscle-specific actin stains typically are negative. Wide local excision or MMS is recommended for treatment of these lesions.12
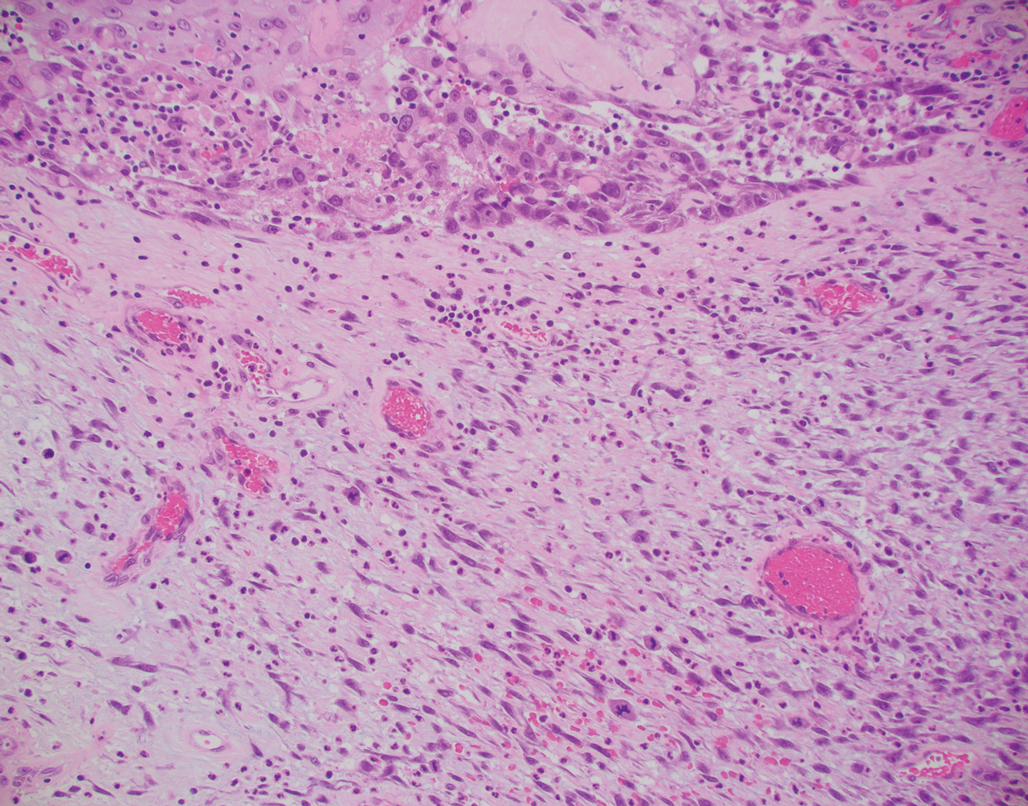
Primary cutaneous myoepithelial carcinomas are uncommon neoplasms of myoepithelial differentiation. Clinically, they often arise as soft nodular lesions on the head, neck, and lower extremities with a bimodal age distribution (50 years). Histologically cutaneous myoepithelial tumors are well-differentiated, dermal-based nodules without connection to the overlying epidermis (Figure 3). The myoepithelial cells can exhibit spindled, epithelioid, plasmacytoid, or clear cell morphologic features and show variability in cell growth patterns. One of the most common growth patterns is oval to round cells forming cords and chains in a chondromyxoid stroma. Most cases display an immunophenotyped co-expression of an epithelial cytokeratin and S-100 protein. Myoepithelial markers also may be present, including keratins, smooth muscle actin, calponin, glial fibrillary acidic protein, p63, and desmin. Surgical removal with wide local excision or MMS is essential.13
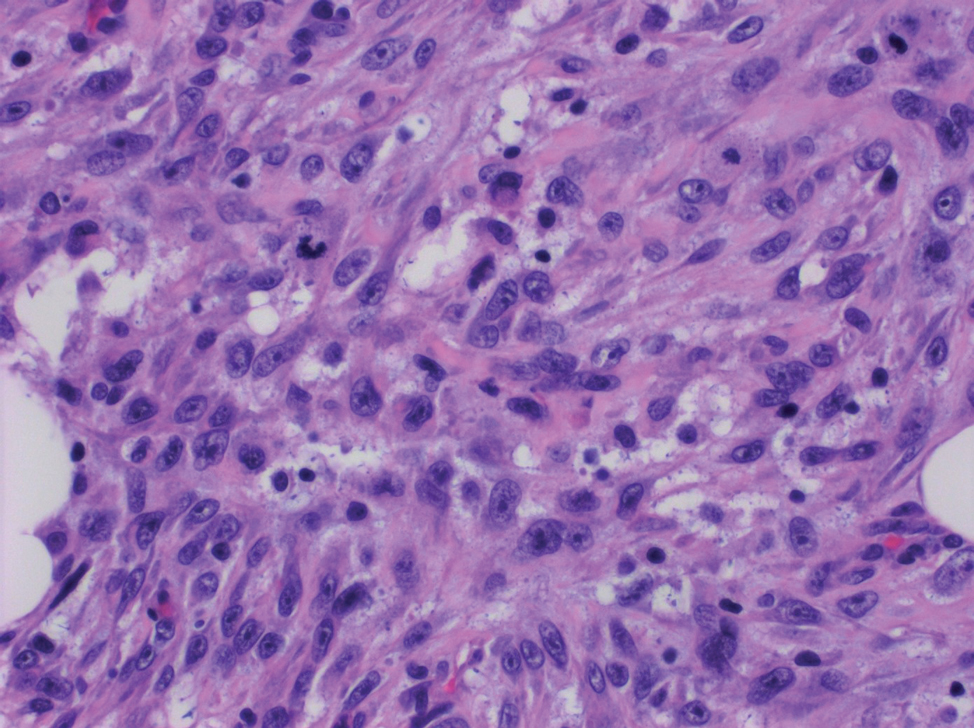
Leiomyosarcoma (LMS) is a tumor that originates from smooth muscle and rarely develops in the dermis.14 Pleomorphic LMS is a morphologic variant of LMS that has a low propensity to metastasize but commonly exhibits local recurrence.15 Leiomyosarcoma can present in any age group but most commonly manifests in individuals aged 50 to 70 years. Clinically, LMS presents as a firm solitary nodule with a smooth pink surface or a more exophytic tumor with a reddish or brown color on the extensor surface of the lower limbs; it is less common on the scalp and face.14 Histologically, most cases of pleomorphic LMS show small foci of fascicles consisting of smooth muscle tumor cells in addition to cellular pleomorphism (Figure 4).15 Many of these cells demonstrate a clear perinuclear vacuole that generally is appreciated in neoplastic smooth muscle cells.14 Pleomorphic LMS typically stains positively for at least one smooth muscle marker including desmin, h-caldesmon, muscle-specific actin, α-smooth muscle actin, or smooth muscle myosin in the leiomyosarcomatous fascicular areas.16 Complete surgical excision is the treatment of choice, and the best results are obtained with MMS.14
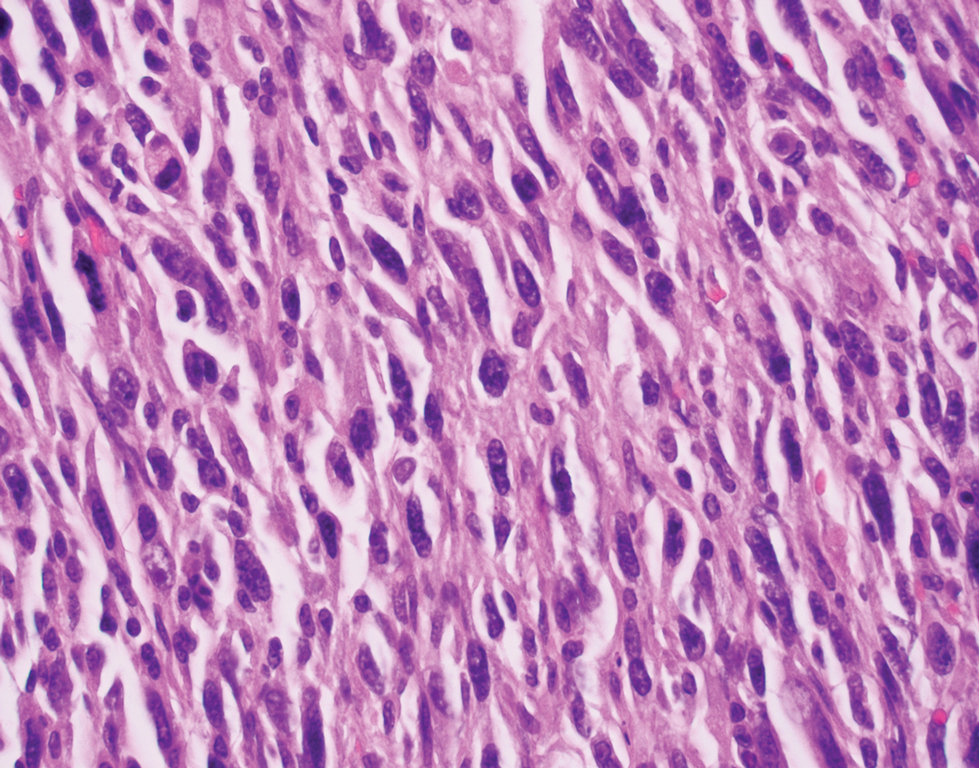
- Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
- Bourgeault E, Alain J, Gagne E. Primary cutaneous carcinosarcoma of the basal cell subtype should be treated as a high-risk basal cell carcinoma. J Cutan Med Surg. 2015;19:407-411.
- West L, Srivastava D. Cutaneous carcinosarcoma of the medial canthus discovered on Mohs debulk analysis. Dermatol Surg. 2019;45:1700-1702.
- Kwan JM, Satter EK. Carcinosarcoma: a primary cutaneous tumor with biphasic differentiation. Cutis. 2013;92:247-249.
- Suh KY, Lacouture M, Gerami P. p63 in primary cutaneous carcinosarcoma. Am J Dermatopathol. 2007;29:374‐377.
- Ruiz-Villaverde R, Aneiros-Fernandez J. Primary cutaneous carcinosarcoma: a cutaneous neoplasm with an exceptional presentation. Sultan Qaboos Univ Med J. 2018;18:E114-E115.
- Smart CN, Pucci RA, Binder SW, et al. Cutaneous carcinosarcoma with myoepithelial differentiation: immunohistochemical and cytogenetic analysis of a case presenting in an unusual location. Am J Dermatopathol. 2009;31:715‐717.
- Clark JJ, Bowen AR, Bowen GM, et al. Cutaneous carcinosarcoma: a series of six cases and a review of the literature. J Cutan Pathol. 2017;44:34‐44.
- Müller CS, Pföhler C, Schiekofer C, et al. Primary cutaneous carcinosarcomas: a morphological histogenetic concept revisited. Am J Dermatopathol. 2014;36:328‐339.
- Bellew S, Del Rosso JQ, Mobini N. Primary carcinosarcoma of the ear: case report and review of the literature. J Clin Aesthet Dermatol. 2009;2:33‐35.
- Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
- Soleymani T, Aasi SZ, Novoa R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: updates on classification and management. Dermatol Clin. 2019;37:253-259.
- Parekh V, Seykora JT. Cutaneous squamous cell carcinoma. Clin Lab Med. 2017;37:503-525.
- Johnson GE, Stevens K, Morrison AO, et al. Cutaneous myoepithelial carcinoma with disseminated metastases. Cutis. 2017;99:E19-E26.
- Llombart B, Serra-Guillén C, Requena C, et al. Leiomyosarcoma and pleomorphic dermal sarcoma: guidelines for diagnosis and treatment. Actas Dermosifiliogr. 2019;110:4-11.
- Oda Y, Miyajima K, Kawaguchi K, et al. Pleomorphic leiomyosarcoma: clinicopathologic and immunohistochemical study with special emphasis on its distinction from ordinary leiomyosarcoma and malignant fibrous histiocytoma. Am J Surg Pathol. 2001;25:1030-1038.
The Diagnosis: Primary Cutaneous Carcinosarcoma
The immunohistochemical findings supported an epithelial component consistent with moderately differentiated squamous cell carcinoma (SCC) and a mesenchymal component with features consistent with a sarcoma. Consequently, the lesion was diagnosed as a primary cutaneous carcinosarcoma (PCCS).
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm consisting of malignant epithelial (carcinoma) and mesenchymal (sarcoma) components.1 Primary cutaneous carcinosarcomas are uncommon, poorly understood, primary cutaneous tumors.2,3 Characteristic of this tumor, cytokeratins highlight the epithelial component while vimentin highlights the mesenchymal component.4 Histologically, the sarcomatous components of PCCS often are highly variable, with an absence of transitional areas within the epithelial component, which frequently resembles basal cell carcinoma and/ or SCC.5-7 Primary cutaneous carcinosarcoma favors areas of chronic UV radiation exposure, particularly on the head and neck. Most tumors present with a slowly growing, polypoid, flesh-colored to erythematous nodule due to the infiltrative mesenchymal component.7 Primary cutaneous carcinosarcoma primarily is diagnosed in elderly patients, with the majority of cases diagnosed in the eighth or ninth decades of life (range, 32–98 years).1,8 Men appear to be twice as likely to be diagnosed with a PCCS compared to women.1 Primary cutaneous carcinosarcomas are recognized as aggressive tumors with a high propensity to metastasize and recur locally, necessitating early diagnosis and treatment.4 Accurate diagnosis of PCCSs can be challenging due to the biphasic nature of the neoplasm as well as poor differentiation or unequal proportions of the epithelial and mesenchymal components.5 Additionally, overlapping diagnostic criteria coupled with vague demarcation between soft-tissue sarcomas and distinct carcinomas also may contribute to a delay in diagnosis.9 Treatment is achieved surgically by complete wide resection, with no evidence to support the use of adjuvant or neoadjuvant external beam radiation therapy. Due to the small number of reported cases, no treatment recommendations currently exist.1
Surgical management with wide local excision has been disappointing, with recurrence rates reported as high as 33%.6 Primary cutaneous carcinosarcoma has an estimated overall recurrence rate of 19% and a 5-year disease-free rate of 50%.10 Risk factors associated with poorer prognosis include tumors with adnexal subtype, age less than 65 years, rapid tumor growth, a tumor greater than 20 mm at presentation, and a long-standing tumor lasting up to 30 years.2,4 Although wide local excision and Mohs micrographic surgery (MMS) both have been utilized successfully, MMS has been shown to result in a cure rate of greater than 98%.6
Atypical fibroxanthoma (AFX) is a cutaneous tumor of fibrohistiocytic mesenchymal origin that typically manifests on sun-damaged skin in elderly individuals. Clinically, it presents as a rapidly growing neoplasm that often ulcerates and bleeds. These heterogenous neoplasms have several distinct characteristics, including dense cellularity with disorganized, large, pleomorphic, and atypical-appearing spindle-shaped cells arising in the upper layers of the dermis, often disseminating into the reticular dermis and occasionally into the subcutaneous fat (Figure 1). The neoplastic cells often exhibit hyperchromic and irregular nuclei, multinucleated giant cells, and atypical mitotic figures. In most cases, negative immunohistochemical staining with SOX-10, S-100, cytokeratins, desmin, and caldesmon will allow pathologists to differentiate between AFX and other common tumors on the differential diagnosis, such as SCC, melanoma, and leiomyosarcoma. CD10 and procollagen type 1 are positive antigenic markers in AFX, but they are not specific. The standard treatment of AFX includes wide local excision or MMS for superior margin control.11
Spindle cell SCC presents as a raised or exophytic nodule, often with spontaneous bleeding and central ulceration. It usually presents on sun-damaged skin or in individuals with a history of ionizing radiation. Histologically, it is characterized by atypical spindleshaped keratinocytes in the dermis existing as single cells or cohesive nests along with keratin pearls (Figure 2). The atypical spindle cells may comprise the entire tumor or only a small portion. The use of immunohistochemical markers often is required to establish a definitive diagnosis. Spindle cell SCC stains positively, albeit frequently focally, for p63, p40, and high-molecular-weight cytokeratins such as cytokeratin 5/6, while S-100 protein, SOX-10, MART-1/Melan-A, and muscle-specific actin stains typically are negative. Wide local excision or MMS is recommended for treatment of these lesions.12
Primary cutaneous myoepithelial carcinomas are uncommon neoplasms of myoepithelial differentiation. Clinically, they often arise as soft nodular lesions on the head, neck, and lower extremities with a bimodal age distribution (50 years). Histologically cutaneous myoepithelial tumors are well-differentiated, dermal-based nodules without connection to the overlying epidermis (Figure 3). The myoepithelial cells can exhibit spindled, epithelioid, plasmacytoid, or clear cell morphologic features and show variability in cell growth patterns. One of the most common growth patterns is oval to round cells forming cords and chains in a chondromyxoid stroma. Most cases display an immunophenotyped co-expression of an epithelial cytokeratin and S-100 protein. Myoepithelial markers also may be present, including keratins, smooth muscle actin, calponin, glial fibrillary acidic protein, p63, and desmin. Surgical removal with wide local excision or MMS is essential.13
Leiomyosarcoma (LMS) is a tumor that originates from smooth muscle and rarely develops in the dermis.14 Pleomorphic LMS is a morphologic variant of LMS that has a low propensity to metastasize but commonly exhibits local recurrence.15 Leiomyosarcoma can present in any age group but most commonly manifests in individuals aged 50 to 70 years. Clinically, LMS presents as a firm solitary nodule with a smooth pink surface or a more exophytic tumor with a reddish or brown color on the extensor surface of the lower limbs; it is less common on the scalp and face.14 Histologically, most cases of pleomorphic LMS show small foci of fascicles consisting of smooth muscle tumor cells in addition to cellular pleomorphism (Figure 4).15 Many of these cells demonstrate a clear perinuclear vacuole that generally is appreciated in neoplastic smooth muscle cells.14 Pleomorphic LMS typically stains positively for at least one smooth muscle marker including desmin, h-caldesmon, muscle-specific actin, α-smooth muscle actin, or smooth muscle myosin in the leiomyosarcomatous fascicular areas.16 Complete surgical excision is the treatment of choice, and the best results are obtained with MMS.14
The Diagnosis: Primary Cutaneous Carcinosarcoma
The immunohistochemical findings supported an epithelial component consistent with moderately differentiated squamous cell carcinoma (SCC) and a mesenchymal component with features consistent with a sarcoma. Consequently, the lesion was diagnosed as a primary cutaneous carcinosarcoma (PCCS).
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm consisting of malignant epithelial (carcinoma) and mesenchymal (sarcoma) components.1 Primary cutaneous carcinosarcomas are uncommon, poorly understood, primary cutaneous tumors.2,3 Characteristic of this tumor, cytokeratins highlight the epithelial component while vimentin highlights the mesenchymal component.4 Histologically, the sarcomatous components of PCCS often are highly variable, with an absence of transitional areas within the epithelial component, which frequently resembles basal cell carcinoma and/ or SCC.5-7 Primary cutaneous carcinosarcoma favors areas of chronic UV radiation exposure, particularly on the head and neck. Most tumors present with a slowly growing, polypoid, flesh-colored to erythematous nodule due to the infiltrative mesenchymal component.7 Primary cutaneous carcinosarcoma primarily is diagnosed in elderly patients, with the majority of cases diagnosed in the eighth or ninth decades of life (range, 32–98 years).1,8 Men appear to be twice as likely to be diagnosed with a PCCS compared to women.1 Primary cutaneous carcinosarcomas are recognized as aggressive tumors with a high propensity to metastasize and recur locally, necessitating early diagnosis and treatment.4 Accurate diagnosis of PCCSs can be challenging due to the biphasic nature of the neoplasm as well as poor differentiation or unequal proportions of the epithelial and mesenchymal components.5 Additionally, overlapping diagnostic criteria coupled with vague demarcation between soft-tissue sarcomas and distinct carcinomas also may contribute to a delay in diagnosis.9 Treatment is achieved surgically by complete wide resection, with no evidence to support the use of adjuvant or neoadjuvant external beam radiation therapy. Due to the small number of reported cases, no treatment recommendations currently exist.1
Surgical management with wide local excision has been disappointing, with recurrence rates reported as high as 33%.6 Primary cutaneous carcinosarcoma has an estimated overall recurrence rate of 19% and a 5-year disease-free rate of 50%.10 Risk factors associated with poorer prognosis include tumors with adnexal subtype, age less than 65 years, rapid tumor growth, a tumor greater than 20 mm at presentation, and a long-standing tumor lasting up to 30 years.2,4 Although wide local excision and Mohs micrographic surgery (MMS) both have been utilized successfully, MMS has been shown to result in a cure rate of greater than 98%.6
Atypical fibroxanthoma (AFX) is a cutaneous tumor of fibrohistiocytic mesenchymal origin that typically manifests on sun-damaged skin in elderly individuals. Clinically, it presents as a rapidly growing neoplasm that often ulcerates and bleeds. These heterogenous neoplasms have several distinct characteristics, including dense cellularity with disorganized, large, pleomorphic, and atypical-appearing spindle-shaped cells arising in the upper layers of the dermis, often disseminating into the reticular dermis and occasionally into the subcutaneous fat (Figure 1). The neoplastic cells often exhibit hyperchromic and irregular nuclei, multinucleated giant cells, and atypical mitotic figures. In most cases, negative immunohistochemical staining with SOX-10, S-100, cytokeratins, desmin, and caldesmon will allow pathologists to differentiate between AFX and other common tumors on the differential diagnosis, such as SCC, melanoma, and leiomyosarcoma. CD10 and procollagen type 1 are positive antigenic markers in AFX, but they are not specific. The standard treatment of AFX includes wide local excision or MMS for superior margin control.11
Spindle cell SCC presents as a raised or exophytic nodule, often with spontaneous bleeding and central ulceration. It usually presents on sun-damaged skin or in individuals with a history of ionizing radiation. Histologically, it is characterized by atypical spindleshaped keratinocytes in the dermis existing as single cells or cohesive nests along with keratin pearls (Figure 2). The atypical spindle cells may comprise the entire tumor or only a small portion. The use of immunohistochemical markers often is required to establish a definitive diagnosis. Spindle cell SCC stains positively, albeit frequently focally, for p63, p40, and high-molecular-weight cytokeratins such as cytokeratin 5/6, while S-100 protein, SOX-10, MART-1/Melan-A, and muscle-specific actin stains typically are negative. Wide local excision or MMS is recommended for treatment of these lesions.12
Primary cutaneous myoepithelial carcinomas are uncommon neoplasms of myoepithelial differentiation. Clinically, they often arise as soft nodular lesions on the head, neck, and lower extremities with a bimodal age distribution (50 years). Histologically cutaneous myoepithelial tumors are well-differentiated, dermal-based nodules without connection to the overlying epidermis (Figure 3). The myoepithelial cells can exhibit spindled, epithelioid, plasmacytoid, or clear cell morphologic features and show variability in cell growth patterns. One of the most common growth patterns is oval to round cells forming cords and chains in a chondromyxoid stroma. Most cases display an immunophenotyped co-expression of an epithelial cytokeratin and S-100 protein. Myoepithelial markers also may be present, including keratins, smooth muscle actin, calponin, glial fibrillary acidic protein, p63, and desmin. Surgical removal with wide local excision or MMS is essential.13
Leiomyosarcoma (LMS) is a tumor that originates from smooth muscle and rarely develops in the dermis.14 Pleomorphic LMS is a morphologic variant of LMS that has a low propensity to metastasize but commonly exhibits local recurrence.15 Leiomyosarcoma can present in any age group but most commonly manifests in individuals aged 50 to 70 years. Clinically, LMS presents as a firm solitary nodule with a smooth pink surface or a more exophytic tumor with a reddish or brown color on the extensor surface of the lower limbs; it is less common on the scalp and face.14 Histologically, most cases of pleomorphic LMS show small foci of fascicles consisting of smooth muscle tumor cells in addition to cellular pleomorphism (Figure 4).15 Many of these cells demonstrate a clear perinuclear vacuole that generally is appreciated in neoplastic smooth muscle cells.14 Pleomorphic LMS typically stains positively for at least one smooth muscle marker including desmin, h-caldesmon, muscle-specific actin, α-smooth muscle actin, or smooth muscle myosin in the leiomyosarcomatous fascicular areas.16 Complete surgical excision is the treatment of choice, and the best results are obtained with MMS.14
- Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
- Bourgeault E, Alain J, Gagne E. Primary cutaneous carcinosarcoma of the basal cell subtype should be treated as a high-risk basal cell carcinoma. J Cutan Med Surg. 2015;19:407-411.
- West L, Srivastava D. Cutaneous carcinosarcoma of the medial canthus discovered on Mohs debulk analysis. Dermatol Surg. 2019;45:1700-1702.
- Kwan JM, Satter EK. Carcinosarcoma: a primary cutaneous tumor with biphasic differentiation. Cutis. 2013;92:247-249.
- Suh KY, Lacouture M, Gerami P. p63 in primary cutaneous carcinosarcoma. Am J Dermatopathol. 2007;29:374‐377.
- Ruiz-Villaverde R, Aneiros-Fernandez J. Primary cutaneous carcinosarcoma: a cutaneous neoplasm with an exceptional presentation. Sultan Qaboos Univ Med J. 2018;18:E114-E115.
- Smart CN, Pucci RA, Binder SW, et al. Cutaneous carcinosarcoma with myoepithelial differentiation: immunohistochemical and cytogenetic analysis of a case presenting in an unusual location. Am J Dermatopathol. 2009;31:715‐717.
- Clark JJ, Bowen AR, Bowen GM, et al. Cutaneous carcinosarcoma: a series of six cases and a review of the literature. J Cutan Pathol. 2017;44:34‐44.
- Müller CS, Pföhler C, Schiekofer C, et al. Primary cutaneous carcinosarcomas: a morphological histogenetic concept revisited. Am J Dermatopathol. 2014;36:328‐339.
- Bellew S, Del Rosso JQ, Mobini N. Primary carcinosarcoma of the ear: case report and review of the literature. J Clin Aesthet Dermatol. 2009;2:33‐35.
- Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
- Soleymani T, Aasi SZ, Novoa R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: updates on classification and management. Dermatol Clin. 2019;37:253-259.
- Parekh V, Seykora JT. Cutaneous squamous cell carcinoma. Clin Lab Med. 2017;37:503-525.
- Johnson GE, Stevens K, Morrison AO, et al. Cutaneous myoepithelial carcinoma with disseminated metastases. Cutis. 2017;99:E19-E26.
- Llombart B, Serra-Guillén C, Requena C, et al. Leiomyosarcoma and pleomorphic dermal sarcoma: guidelines for diagnosis and treatment. Actas Dermosifiliogr. 2019;110:4-11.
- Oda Y, Miyajima K, Kawaguchi K, et al. Pleomorphic leiomyosarcoma: clinicopathologic and immunohistochemical study with special emphasis on its distinction from ordinary leiomyosarcoma and malignant fibrous histiocytoma. Am J Surg Pathol. 2001;25:1030-1038.
- Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
- Bourgeault E, Alain J, Gagne E. Primary cutaneous carcinosarcoma of the basal cell subtype should be treated as a high-risk basal cell carcinoma. J Cutan Med Surg. 2015;19:407-411.
- West L, Srivastava D. Cutaneous carcinosarcoma of the medial canthus discovered on Mohs debulk analysis. Dermatol Surg. 2019;45:1700-1702.
- Kwan JM, Satter EK. Carcinosarcoma: a primary cutaneous tumor with biphasic differentiation. Cutis. 2013;92:247-249.
- Suh KY, Lacouture M, Gerami P. p63 in primary cutaneous carcinosarcoma. Am J Dermatopathol. 2007;29:374‐377.
- Ruiz-Villaverde R, Aneiros-Fernandez J. Primary cutaneous carcinosarcoma: a cutaneous neoplasm with an exceptional presentation. Sultan Qaboos Univ Med J. 2018;18:E114-E115.
- Smart CN, Pucci RA, Binder SW, et al. Cutaneous carcinosarcoma with myoepithelial differentiation: immunohistochemical and cytogenetic analysis of a case presenting in an unusual location. Am J Dermatopathol. 2009;31:715‐717.
- Clark JJ, Bowen AR, Bowen GM, et al. Cutaneous carcinosarcoma: a series of six cases and a review of the literature. J Cutan Pathol. 2017;44:34‐44.
- Müller CS, Pföhler C, Schiekofer C, et al. Primary cutaneous carcinosarcomas: a morphological histogenetic concept revisited. Am J Dermatopathol. 2014;36:328‐339.
- Bellew S, Del Rosso JQ, Mobini N. Primary carcinosarcoma of the ear: case report and review of the literature. J Clin Aesthet Dermatol. 2009;2:33‐35.
- Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
- Soleymani T, Aasi SZ, Novoa R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: updates on classification and management. Dermatol Clin. 2019;37:253-259.
- Parekh V, Seykora JT. Cutaneous squamous cell carcinoma. Clin Lab Med. 2017;37:503-525.
- Johnson GE, Stevens K, Morrison AO, et al. Cutaneous myoepithelial carcinoma with disseminated metastases. Cutis. 2017;99:E19-E26.
- Llombart B, Serra-Guillén C, Requena C, et al. Leiomyosarcoma and pleomorphic dermal sarcoma: guidelines for diagnosis and treatment. Actas Dermosifiliogr. 2019;110:4-11.
- Oda Y, Miyajima K, Kawaguchi K, et al. Pleomorphic leiomyosarcoma: clinicopathologic and immunohistochemical study with special emphasis on its distinction from ordinary leiomyosarcoma and malignant fibrous histiocytoma. Am J Surg Pathol. 2001;25:1030-1038.
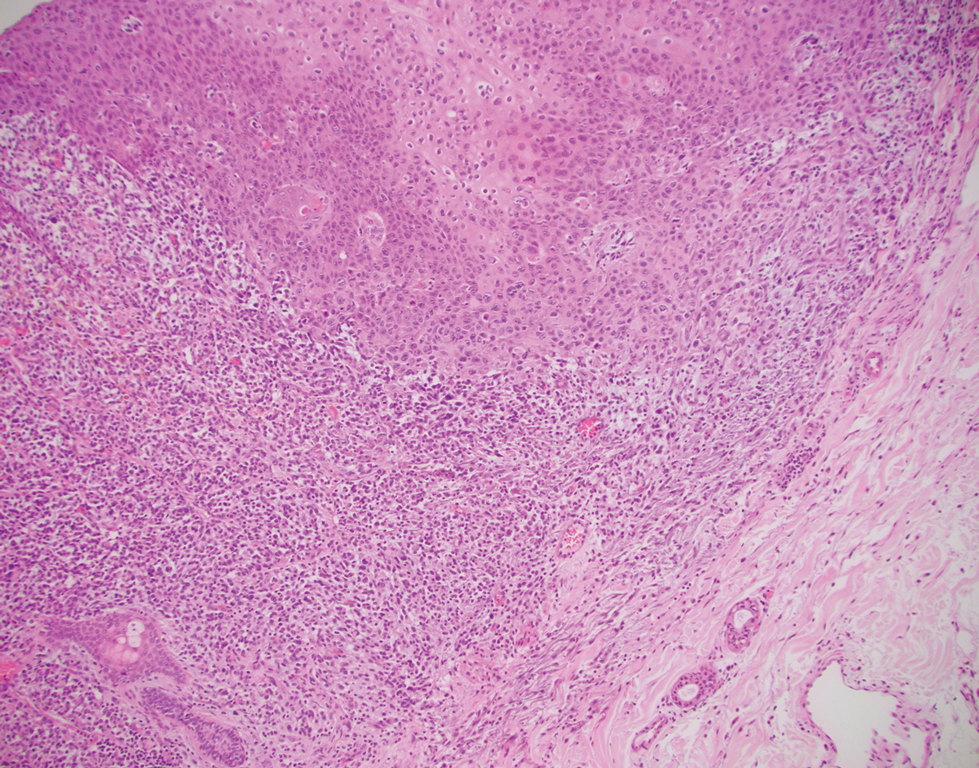
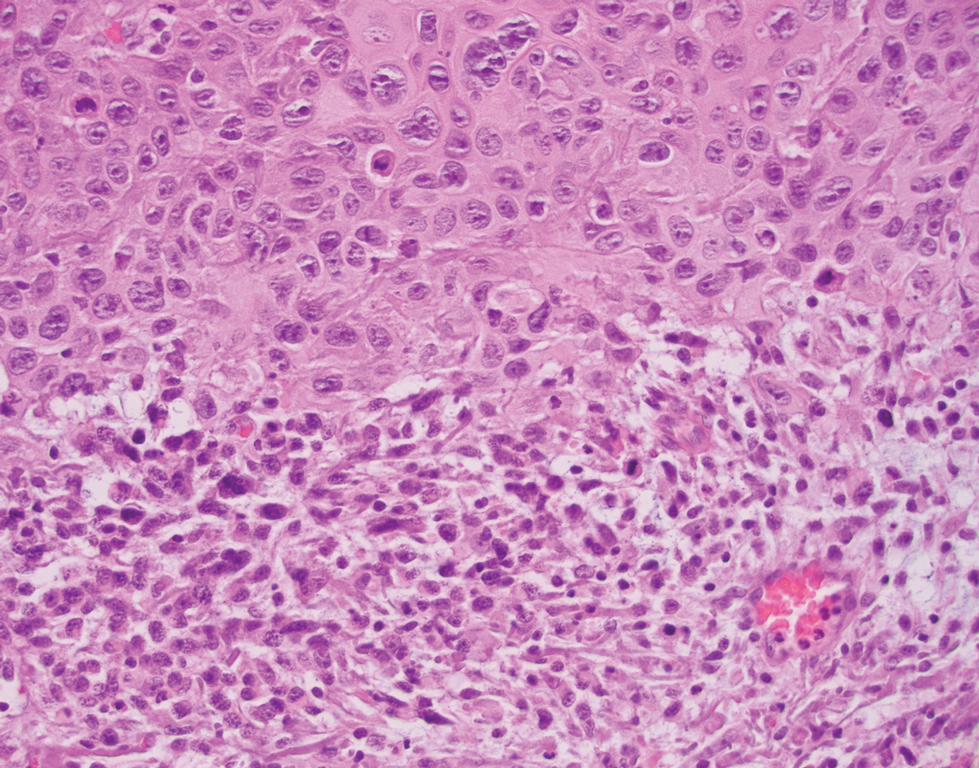
A 72-year-old man with a history of nonmelanoma skin cancer and lung transplant maintained on stable doses of prednisone and tacrolimus presented with a 1.3×1.8-cm, slow-growing, well-demarcated, ulcerated, erythematous plaque with overlying serous crust on the left temple of 6 months’ duration. No cervical or axillary lymphadenopathy was appreciated on physical examination. A biopsy was performed followed by Mohs micrographic surgery. Microscopic examination of the debulking specimen revealed atypical spindle cells in the papillary and reticular dermis radiating from a central focus of a moderately differentiated squamous cell carcinoma. The squamous cells stained positive for cytokeratin 5/6, pankeratin, and p40, while the spindle cells stained positive only for vimentin.
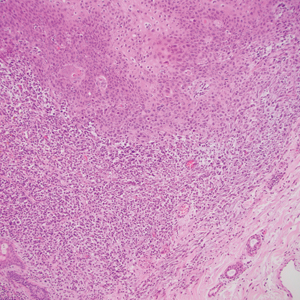
Bullous Retiform Purpura on the Ears and Legs
The Diagnosis: Levamisole-Induced Vasculopathy
Biopsy of one of the bullous retiform purpura on the leg (Figure 1) revealed a combined leukocytoclastic vasculitis and thrombotic vasculopathy (quiz images). Periodic acid-Schiff and Gram stains, with adequate controls, were negative for pathogenic fungal and bacterial organisms. Although this reaction pattern has an extensive differential, in this clinical setting with associated cocaine-positive urine toxicologic analysis, perinuclear antineutrophil cytoplasmic antibodies (p-ANCA), and leukopenia, the histopathologic findings were consistent with levamisole-induced vasculopathy (LIV).1,2 Although not specific, leukocytoclastic vasculitis and thrombotic vasculopathy have been reported as the classic histopathologic findings of LIV. In addition, interstitial and perivascular neovascularization have been reported as a potential histopathologic finding associated with this entity but was not seen in our case.3
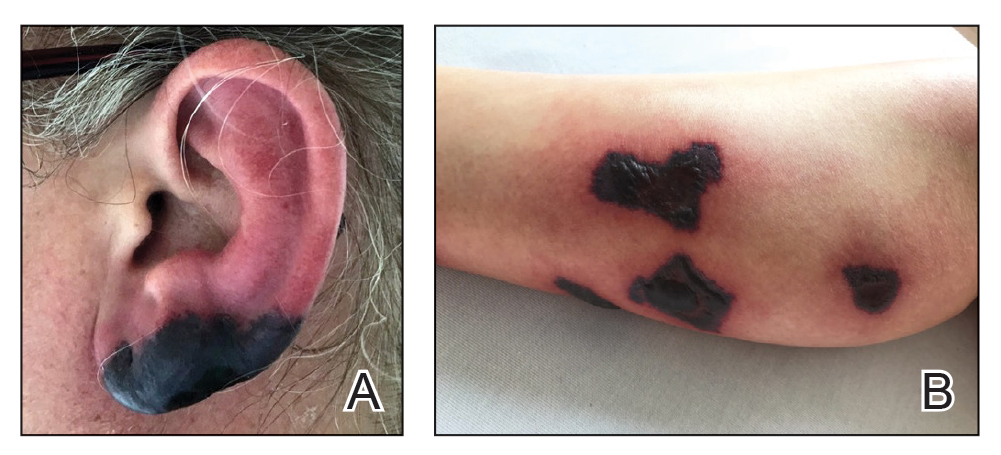
Levamisole is an anthelminthic agent used to adulterate cocaine, a practice first noted in 2003 with increasing incidence.1 Both levamisole and cocaine stimulate the sympathetic nervous system by increasing dopamine in the euphoric areas of the brain.1,3 By combining the 2 substances, preparation costs are reduced and stimulant effects are enhanced. It is estimated that 69% to 80% of cocaine in the United States is contaminated with levamisole.2,4,5 The constellation of findings seen in patients abusing levamisole-contaminated cocaine include agranulocytosis; p-ANCA; and a tender, vasculitic, retiform purpura presentation. The most common sites for the purpura include the cheeks and ears. The purpura can progress to bullous lesions, as seen in our patient, followed by necrosis.4,6 Recurrent use of levamisole-contaminated cocaine is associated with recurrent agranulocytosis and classic skin findings, which is suggestive of a causal relationship.6
Serologic testing for levamisole exposure presents a challenge. The half-life of levamisole is relatively short (estimated at 5.6 hours) and is found in urine samples approximately 3% of the time.1,3,6 The volatile diagnostic characteristics of levamisole make concrete laboratory confirmation difficult. Although a skin biopsy can be helpful to rule out other causes of vasculitislike presentations, it is not specific for LIV. Therefore, clinical suspicion for LIV should remain high in patients who present with the cutaneous findings described as well as agranulocytosis, positive p-ANCA, and a history of cocaine use with a skin biopsy showing leukocytoclastic vasculitis and thrombotic vasculopathy.
The differential diagnosis for LIV with retiform bullous lesions includes several other vasculitides and vesiculobullous diseases. Eosinophilic granulomatosis with polyangiitis (EGPA) is a multisystem vasculitis that is characterized by eosinophilia, asthma, and rhinosinusitis. Eosinophilic granulomatosis with polyangiitis primarily affects small and medium arteries in the skin and respiratory tract and occurs in 3 stages: prodromal, eosinophilic, and vasculitic. These stages are characterized by mild asthma or rhinitis, eosinophilia with multiorgan infiltration, and vasculitis with extravascular granulomatosis, respectively. Diagnosis often is clinical based on these findings and laboratory evaluation. Eosinophilic granulomatosis with polyangiitis presents with positive p-ANCA in 40% to 60% of patients.7 The vasculitis stage of EGPA presents with cutaneous findings in 60% of cases, including palpable purpura, infiltrated papules and plaques, urticaria, necrotizing lesions, and rarely vesicles and bullae.8 Classic histopathologic features include leukocytoclastic or eosinophilic vasculitis, an eosinophilic infiltrate, granuloma formation, and eosinophilic granule deposition onto collagen fibrils (otherwise known as flame figures)(Figure 2). Biopsy of these lesions with the aforementioned findings, in constellation with the described systemic signs and symptoms, can aid in diagnosis of EGPA.
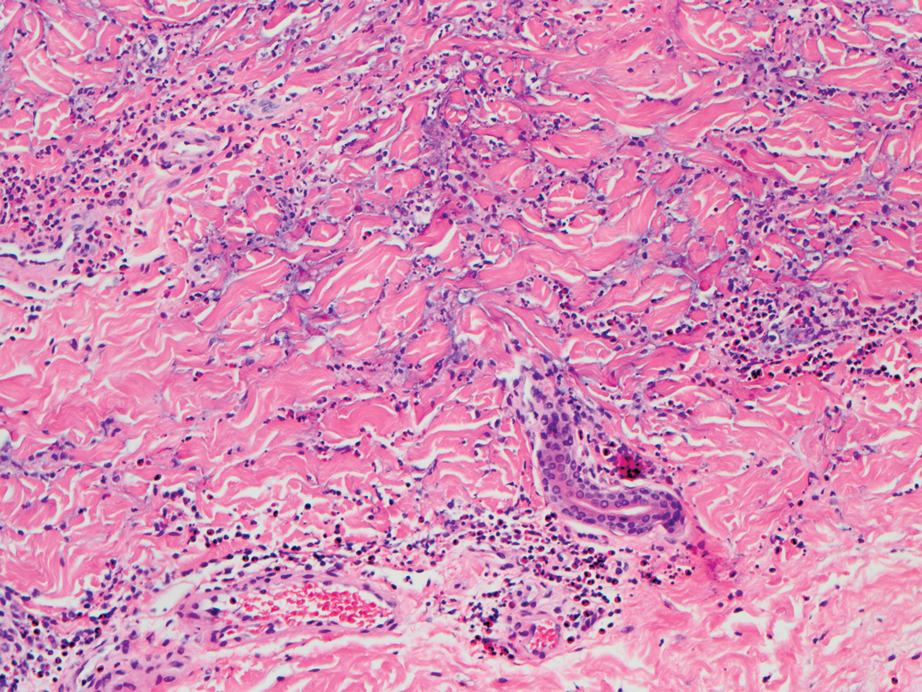
Polyarteritis nodosa (PAN) is a vasculitis that can be either multisystem or limited to one organ. Classic PAN affects the small- to medium-sized vessels. When there is multisystem involvement, it most often affects the skin, gastrointestinal tract, and kidneys. It presents with subcutaneous or dermal nodules, necrotic lesions, livedo reticularis, hypertension, abdominal pain, and an acute abdomen.9 When PAN is in its limited form, it most commonly occurs in the skin. The cutaneous manifestations of skin-limited PAN are identical to classic PAN, most commonly occurring on the legs and arms and less often on the trunk, head, and neck.10 To aid in diagnosis, biopsies of cutaneous lesions are beneficial. Dermatopathologic examination of PAN reveals fibrinoid necrosis of small and medium vessels with a perivascular mononuclear inflammatory infiltrate (Figure 3). Cutaneous PAN rarely progresses to multisystem classic PAN and carries a more favorable prognosis.
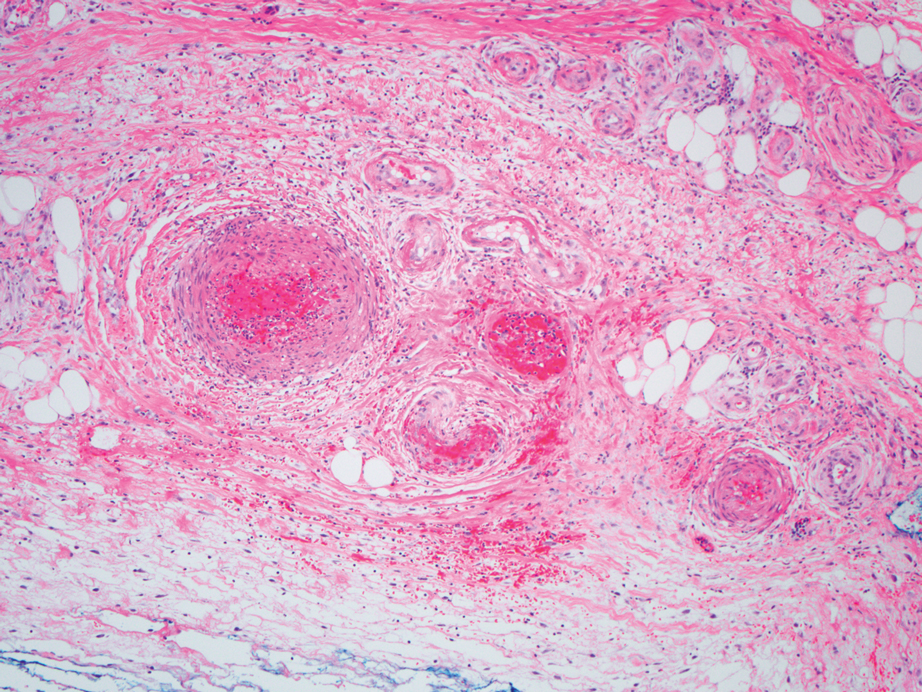
Microvascular occlusion syndromes can result in clinical presentations that resemble LIV. Idiopathic thrombocytopenic purpura is a hematologic autoimmune condition resulting in destruction of platelets and subsequent thrombocytopenia. Idiopathic thrombocytopenic purpura can be either primary or secondary to infections, drugs, malignancy, or other autoimmune conditions. Clinically, it presents as mucosal or cutaneous bleeding, epistaxis, hematochezia, or hematuria and can result in substantial hemorrhage. On the skin, it can appear as petechiae and ecchymoses in dependent areas and rarely hemorrhagic bullae of the skin and mucous membranes in cases of severe thrombocytopenia.11,12 Biopsies of these lesions will show notable extravasation of red blood cells with incipient hemorrhagic bullae formation (Figure 4). Recognition of hemorrhagic bullae as a presentation of idiopathic thrombocytopenic purpura is critical to identifying severe underlying disease.
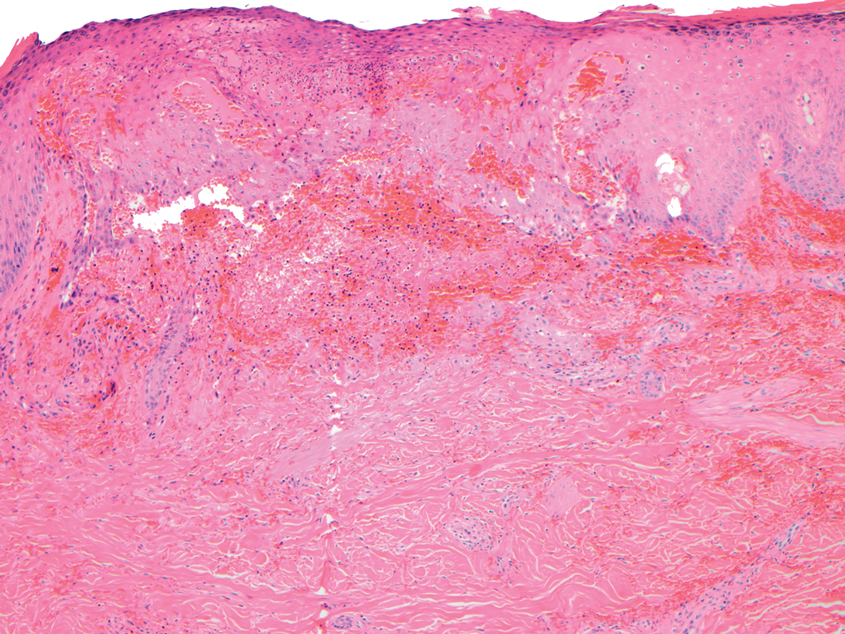
Beyond other vasculitides and microvascular occlusion syndromes, vessel-invasive microorganisms can result in similar histopathologic and clinical presentations to LIV. Ecthyma gangrenosum (EG) is a septic vasculitis, often caused by Pseudomonas aeruginosa, usually affecting immunocompromised patients. Ecthyma gangrenosum presents with vesiculobullous lesions with erythematous violaceous borders that develop into hemorrhagic bullae with necrotic centers.13 Biopsy of EG will show vascular occlusion and basophilic granular material within or around vessels, suggestive of bacterial sepsis (Figure 5). The detection of an infectious agent on histopathology allows one to easily distinguish between EG and LIV.
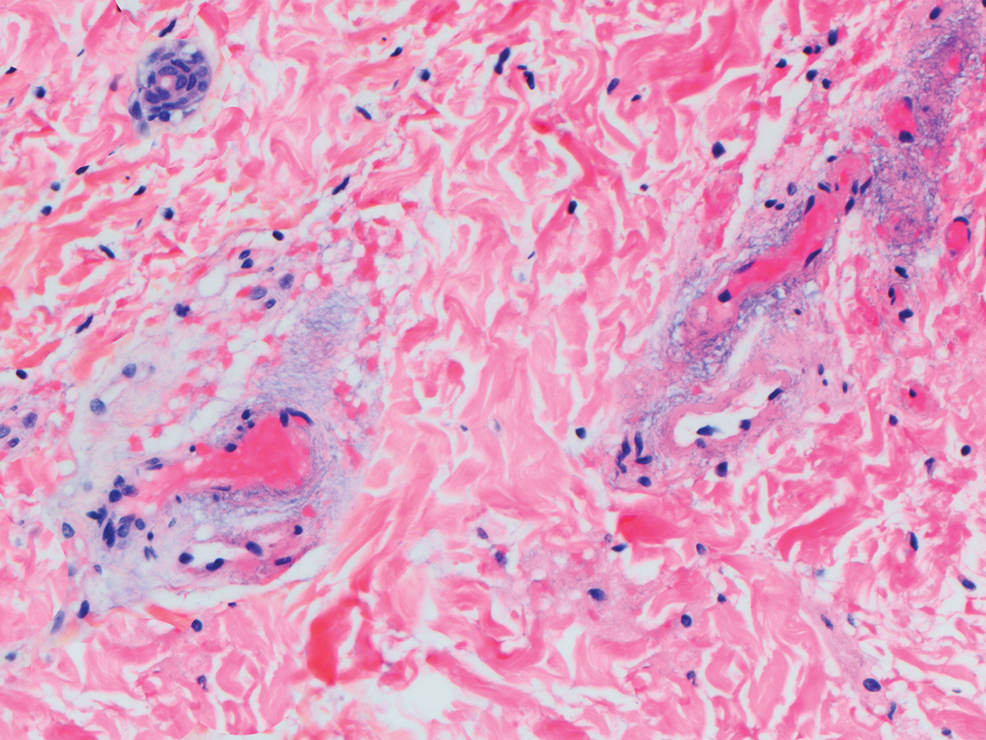
- Bajaj S, Hibler B, Rossi A. Painful violaceous purpura on a 44-year-old woman. Am J Med. 2016;129:E5-E7.
- Munoz-Vahos CH, Herrera-Uribe S, Arbelaez-Cortes A, et al. Clinical profile of levamisole-adulterated cocaine-induced vasculitis/vasculopathy. J Clin Rheumatol. 2019;25:E16-E26.
- Jacob RS, Silva CY, Powers JG, et al. Levamisole-induced vasculopathy: a report of 2 cases and a novel histopathologic finding. Am J Dermatopathol. 2012;34:208-213.
- Gillis JA, Green P, Williams J. Levamisole-induced vasculopathy: staging and management. J Plast Reconstr Aesthet Surg. 2014;67:E29-E31.
- Farhat EK, Muirhead TT, Chafins ML, et al. Levamisole-induced cutaneous necrosis mimicking coagulopathy. Arch Dermatol. 2010;146:1320-1321.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia-a potential public health epidemic associated with levamisole-adulterated cocaine. J Am Acad Dermatol. 2010;65:722-725.
- Negbenebor NA, Khalifian S, Foreman RK, et al. A 92-year-old male with eosinophilic asthma presenting with recurrent palpable purpuric plaques. Dermatopathology (Basel). 2018;5:44-48.
- Sherman S, Gal N, Didkovsky E, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) relapsing as bullous eruption. Acta Derm Venereol. 2017;97:406-407.
- Braungart S, Campbell A, Besarovic S. Atypical Henoch-Schonlein purpura? consider polyarteritis nodosa! BMJ Case Rep. 2014. doi:10.1136/bcr-2013-201764
- Alquorain NAA, Aljabr ASH, Alghamdi NJ. Cutaneous polyarteritis nodosa treated with pentoxifylline and clobetasol propionate: a case report. Saudi J Med Sci. 2018;6:104-107.
- Helms AE, Schaffer RI. Idiopathic thrombocytopenic purpura with black oral mucosal lesions. Cutis. 2007;79:456-458.
- Lountzis N, Maroon M, Tyler W. Mucocutaneous hemorrhagic bullae in idiopathic thrombocytopenic purpura. J Am Acad Dermatol. 2009;60:AB124.
- Llamas-Velasco M, Alegeria V, Santos-Briz A, et al. Occlusive nonvasculitic vasculopathy. Am J Dermatopathol. 2017;39:637-662.
The Diagnosis: Levamisole-Induced Vasculopathy
Biopsy of one of the bullous retiform purpura on the leg (Figure 1) revealed a combined leukocytoclastic vasculitis and thrombotic vasculopathy (quiz images). Periodic acid-Schiff and Gram stains, with adequate controls, were negative for pathogenic fungal and bacterial organisms. Although this reaction pattern has an extensive differential, in this clinical setting with associated cocaine-positive urine toxicologic analysis, perinuclear antineutrophil cytoplasmic antibodies (p-ANCA), and leukopenia, the histopathologic findings were consistent with levamisole-induced vasculopathy (LIV).1,2 Although not specific, leukocytoclastic vasculitis and thrombotic vasculopathy have been reported as the classic histopathologic findings of LIV. In addition, interstitial and perivascular neovascularization have been reported as a potential histopathologic finding associated with this entity but was not seen in our case.3
Levamisole is an anthelminthic agent used to adulterate cocaine, a practice first noted in 2003 with increasing incidence.1 Both levamisole and cocaine stimulate the sympathetic nervous system by increasing dopamine in the euphoric areas of the brain.1,3 By combining the 2 substances, preparation costs are reduced and stimulant effects are enhanced. It is estimated that 69% to 80% of cocaine in the United States is contaminated with levamisole.2,4,5 The constellation of findings seen in patients abusing levamisole-contaminated cocaine include agranulocytosis; p-ANCA; and a tender, vasculitic, retiform purpura presentation. The most common sites for the purpura include the cheeks and ears. The purpura can progress to bullous lesions, as seen in our patient, followed by necrosis.4,6 Recurrent use of levamisole-contaminated cocaine is associated with recurrent agranulocytosis and classic skin findings, which is suggestive of a causal relationship.6
Serologic testing for levamisole exposure presents a challenge. The half-life of levamisole is relatively short (estimated at 5.6 hours) and is found in urine samples approximately 3% of the time.1,3,6 The volatile diagnostic characteristics of levamisole make concrete laboratory confirmation difficult. Although a skin biopsy can be helpful to rule out other causes of vasculitislike presentations, it is not specific for LIV. Therefore, clinical suspicion for LIV should remain high in patients who present with the cutaneous findings described as well as agranulocytosis, positive p-ANCA, and a history of cocaine use with a skin biopsy showing leukocytoclastic vasculitis and thrombotic vasculopathy.
The differential diagnosis for LIV with retiform bullous lesions includes several other vasculitides and vesiculobullous diseases. Eosinophilic granulomatosis with polyangiitis (EGPA) is a multisystem vasculitis that is characterized by eosinophilia, asthma, and rhinosinusitis. Eosinophilic granulomatosis with polyangiitis primarily affects small and medium arteries in the skin and respiratory tract and occurs in 3 stages: prodromal, eosinophilic, and vasculitic. These stages are characterized by mild asthma or rhinitis, eosinophilia with multiorgan infiltration, and vasculitis with extravascular granulomatosis, respectively. Diagnosis often is clinical based on these findings and laboratory evaluation. Eosinophilic granulomatosis with polyangiitis presents with positive p-ANCA in 40% to 60% of patients.7 The vasculitis stage of EGPA presents with cutaneous findings in 60% of cases, including palpable purpura, infiltrated papules and plaques, urticaria, necrotizing lesions, and rarely vesicles and bullae.8 Classic histopathologic features include leukocytoclastic or eosinophilic vasculitis, an eosinophilic infiltrate, granuloma formation, and eosinophilic granule deposition onto collagen fibrils (otherwise known as flame figures)(Figure 2). Biopsy of these lesions with the aforementioned findings, in constellation with the described systemic signs and symptoms, can aid in diagnosis of EGPA.
Polyarteritis nodosa (PAN) is a vasculitis that can be either multisystem or limited to one organ. Classic PAN affects the small- to medium-sized vessels. When there is multisystem involvement, it most often affects the skin, gastrointestinal tract, and kidneys. It presents with subcutaneous or dermal nodules, necrotic lesions, livedo reticularis, hypertension, abdominal pain, and an acute abdomen.9 When PAN is in its limited form, it most commonly occurs in the skin. The cutaneous manifestations of skin-limited PAN are identical to classic PAN, most commonly occurring on the legs and arms and less often on the trunk, head, and neck.10 To aid in diagnosis, biopsies of cutaneous lesions are beneficial. Dermatopathologic examination of PAN reveals fibrinoid necrosis of small and medium vessels with a perivascular mononuclear inflammatory infiltrate (Figure 3). Cutaneous PAN rarely progresses to multisystem classic PAN and carries a more favorable prognosis.
Microvascular occlusion syndromes can result in clinical presentations that resemble LIV. Idiopathic thrombocytopenic purpura is a hematologic autoimmune condition resulting in destruction of platelets and subsequent thrombocytopenia. Idiopathic thrombocytopenic purpura can be either primary or secondary to infections, drugs, malignancy, or other autoimmune conditions. Clinically, it presents as mucosal or cutaneous bleeding, epistaxis, hematochezia, or hematuria and can result in substantial hemorrhage. On the skin, it can appear as petechiae and ecchymoses in dependent areas and rarely hemorrhagic bullae of the skin and mucous membranes in cases of severe thrombocytopenia.11,12 Biopsies of these lesions will show notable extravasation of red blood cells with incipient hemorrhagic bullae formation (Figure 4). Recognition of hemorrhagic bullae as a presentation of idiopathic thrombocytopenic purpura is critical to identifying severe underlying disease.
Beyond other vasculitides and microvascular occlusion syndromes, vessel-invasive microorganisms can result in similar histopathologic and clinical presentations to LIV. Ecthyma gangrenosum (EG) is a septic vasculitis, often caused by Pseudomonas aeruginosa, usually affecting immunocompromised patients. Ecthyma gangrenosum presents with vesiculobullous lesions with erythematous violaceous borders that develop into hemorrhagic bullae with necrotic centers.13 Biopsy of EG will show vascular occlusion and basophilic granular material within or around vessels, suggestive of bacterial sepsis (Figure 5). The detection of an infectious agent on histopathology allows one to easily distinguish between EG and LIV.
The Diagnosis: Levamisole-Induced Vasculopathy
Biopsy of one of the bullous retiform purpura on the leg (Figure 1) revealed a combined leukocytoclastic vasculitis and thrombotic vasculopathy (quiz images). Periodic acid-Schiff and Gram stains, with adequate controls, were negative for pathogenic fungal and bacterial organisms. Although this reaction pattern has an extensive differential, in this clinical setting with associated cocaine-positive urine toxicologic analysis, perinuclear antineutrophil cytoplasmic antibodies (p-ANCA), and leukopenia, the histopathologic findings were consistent with levamisole-induced vasculopathy (LIV).1,2 Although not specific, leukocytoclastic vasculitis and thrombotic vasculopathy have been reported as the classic histopathologic findings of LIV. In addition, interstitial and perivascular neovascularization have been reported as a potential histopathologic finding associated with this entity but was not seen in our case.3
Levamisole is an anthelminthic agent used to adulterate cocaine, a practice first noted in 2003 with increasing incidence.1 Both levamisole and cocaine stimulate the sympathetic nervous system by increasing dopamine in the euphoric areas of the brain.1,3 By combining the 2 substances, preparation costs are reduced and stimulant effects are enhanced. It is estimated that 69% to 80% of cocaine in the United States is contaminated with levamisole.2,4,5 The constellation of findings seen in patients abusing levamisole-contaminated cocaine include agranulocytosis; p-ANCA; and a tender, vasculitic, retiform purpura presentation. The most common sites for the purpura include the cheeks and ears. The purpura can progress to bullous lesions, as seen in our patient, followed by necrosis.4,6 Recurrent use of levamisole-contaminated cocaine is associated with recurrent agranulocytosis and classic skin findings, which is suggestive of a causal relationship.6
Serologic testing for levamisole exposure presents a challenge. The half-life of levamisole is relatively short (estimated at 5.6 hours) and is found in urine samples approximately 3% of the time.1,3,6 The volatile diagnostic characteristics of levamisole make concrete laboratory confirmation difficult. Although a skin biopsy can be helpful to rule out other causes of vasculitislike presentations, it is not specific for LIV. Therefore, clinical suspicion for LIV should remain high in patients who present with the cutaneous findings described as well as agranulocytosis, positive p-ANCA, and a history of cocaine use with a skin biopsy showing leukocytoclastic vasculitis and thrombotic vasculopathy.
The differential diagnosis for LIV with retiform bullous lesions includes several other vasculitides and vesiculobullous diseases. Eosinophilic granulomatosis with polyangiitis (EGPA) is a multisystem vasculitis that is characterized by eosinophilia, asthma, and rhinosinusitis. Eosinophilic granulomatosis with polyangiitis primarily affects small and medium arteries in the skin and respiratory tract and occurs in 3 stages: prodromal, eosinophilic, and vasculitic. These stages are characterized by mild asthma or rhinitis, eosinophilia with multiorgan infiltration, and vasculitis with extravascular granulomatosis, respectively. Diagnosis often is clinical based on these findings and laboratory evaluation. Eosinophilic granulomatosis with polyangiitis presents with positive p-ANCA in 40% to 60% of patients.7 The vasculitis stage of EGPA presents with cutaneous findings in 60% of cases, including palpable purpura, infiltrated papules and plaques, urticaria, necrotizing lesions, and rarely vesicles and bullae.8 Classic histopathologic features include leukocytoclastic or eosinophilic vasculitis, an eosinophilic infiltrate, granuloma formation, and eosinophilic granule deposition onto collagen fibrils (otherwise known as flame figures)(Figure 2). Biopsy of these lesions with the aforementioned findings, in constellation with the described systemic signs and symptoms, can aid in diagnosis of EGPA.
Polyarteritis nodosa (PAN) is a vasculitis that can be either multisystem or limited to one organ. Classic PAN affects the small- to medium-sized vessels. When there is multisystem involvement, it most often affects the skin, gastrointestinal tract, and kidneys. It presents with subcutaneous or dermal nodules, necrotic lesions, livedo reticularis, hypertension, abdominal pain, and an acute abdomen.9 When PAN is in its limited form, it most commonly occurs in the skin. The cutaneous manifestations of skin-limited PAN are identical to classic PAN, most commonly occurring on the legs and arms and less often on the trunk, head, and neck.10 To aid in diagnosis, biopsies of cutaneous lesions are beneficial. Dermatopathologic examination of PAN reveals fibrinoid necrosis of small and medium vessels with a perivascular mononuclear inflammatory infiltrate (Figure 3). Cutaneous PAN rarely progresses to multisystem classic PAN and carries a more favorable prognosis.
Microvascular occlusion syndromes can result in clinical presentations that resemble LIV. Idiopathic thrombocytopenic purpura is a hematologic autoimmune condition resulting in destruction of platelets and subsequent thrombocytopenia. Idiopathic thrombocytopenic purpura can be either primary or secondary to infections, drugs, malignancy, or other autoimmune conditions. Clinically, it presents as mucosal or cutaneous bleeding, epistaxis, hematochezia, or hematuria and can result in substantial hemorrhage. On the skin, it can appear as petechiae and ecchymoses in dependent areas and rarely hemorrhagic bullae of the skin and mucous membranes in cases of severe thrombocytopenia.11,12 Biopsies of these lesions will show notable extravasation of red blood cells with incipient hemorrhagic bullae formation (Figure 4). Recognition of hemorrhagic bullae as a presentation of idiopathic thrombocytopenic purpura is critical to identifying severe underlying disease.
Beyond other vasculitides and microvascular occlusion syndromes, vessel-invasive microorganisms can result in similar histopathologic and clinical presentations to LIV. Ecthyma gangrenosum (EG) is a septic vasculitis, often caused by Pseudomonas aeruginosa, usually affecting immunocompromised patients. Ecthyma gangrenosum presents with vesiculobullous lesions with erythematous violaceous borders that develop into hemorrhagic bullae with necrotic centers.13 Biopsy of EG will show vascular occlusion and basophilic granular material within or around vessels, suggestive of bacterial sepsis (Figure 5). The detection of an infectious agent on histopathology allows one to easily distinguish between EG and LIV.
- Bajaj S, Hibler B, Rossi A. Painful violaceous purpura on a 44-year-old woman. Am J Med. 2016;129:E5-E7.
- Munoz-Vahos CH, Herrera-Uribe S, Arbelaez-Cortes A, et al. Clinical profile of levamisole-adulterated cocaine-induced vasculitis/vasculopathy. J Clin Rheumatol. 2019;25:E16-E26.
- Jacob RS, Silva CY, Powers JG, et al. Levamisole-induced vasculopathy: a report of 2 cases and a novel histopathologic finding. Am J Dermatopathol. 2012;34:208-213.
- Gillis JA, Green P, Williams J. Levamisole-induced vasculopathy: staging and management. J Plast Reconstr Aesthet Surg. 2014;67:E29-E31.
- Farhat EK, Muirhead TT, Chafins ML, et al. Levamisole-induced cutaneous necrosis mimicking coagulopathy. Arch Dermatol. 2010;146:1320-1321.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia-a potential public health epidemic associated with levamisole-adulterated cocaine. J Am Acad Dermatol. 2010;65:722-725.
- Negbenebor NA, Khalifian S, Foreman RK, et al. A 92-year-old male with eosinophilic asthma presenting with recurrent palpable purpuric plaques. Dermatopathology (Basel). 2018;5:44-48.
- Sherman S, Gal N, Didkovsky E, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) relapsing as bullous eruption. Acta Derm Venereol. 2017;97:406-407.
- Braungart S, Campbell A, Besarovic S. Atypical Henoch-Schonlein purpura? consider polyarteritis nodosa! BMJ Case Rep. 2014. doi:10.1136/bcr-2013-201764
- Alquorain NAA, Aljabr ASH, Alghamdi NJ. Cutaneous polyarteritis nodosa treated with pentoxifylline and clobetasol propionate: a case report. Saudi J Med Sci. 2018;6:104-107.
- Helms AE, Schaffer RI. Idiopathic thrombocytopenic purpura with black oral mucosal lesions. Cutis. 2007;79:456-458.
- Lountzis N, Maroon M, Tyler W. Mucocutaneous hemorrhagic bullae in idiopathic thrombocytopenic purpura. J Am Acad Dermatol. 2009;60:AB124.
- Llamas-Velasco M, Alegeria V, Santos-Briz A, et al. Occlusive nonvasculitic vasculopathy. Am J Dermatopathol. 2017;39:637-662.
- Bajaj S, Hibler B, Rossi A. Painful violaceous purpura on a 44-year-old woman. Am J Med. 2016;129:E5-E7.
- Munoz-Vahos CH, Herrera-Uribe S, Arbelaez-Cortes A, et al. Clinical profile of levamisole-adulterated cocaine-induced vasculitis/vasculopathy. J Clin Rheumatol. 2019;25:E16-E26.
- Jacob RS, Silva CY, Powers JG, et al. Levamisole-induced vasculopathy: a report of 2 cases and a novel histopathologic finding. Am J Dermatopathol. 2012;34:208-213.
- Gillis JA, Green P, Williams J. Levamisole-induced vasculopathy: staging and management. J Plast Reconstr Aesthet Surg. 2014;67:E29-E31.
- Farhat EK, Muirhead TT, Chafins ML, et al. Levamisole-induced cutaneous necrosis mimicking coagulopathy. Arch Dermatol. 2010;146:1320-1321.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia-a potential public health epidemic associated with levamisole-adulterated cocaine. J Am Acad Dermatol. 2010;65:722-725.
- Negbenebor NA, Khalifian S, Foreman RK, et al. A 92-year-old male with eosinophilic asthma presenting with recurrent palpable purpuric plaques. Dermatopathology (Basel). 2018;5:44-48.
- Sherman S, Gal N, Didkovsky E, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) relapsing as bullous eruption. Acta Derm Venereol. 2017;97:406-407.
- Braungart S, Campbell A, Besarovic S. Atypical Henoch-Schonlein purpura? consider polyarteritis nodosa! BMJ Case Rep. 2014. doi:10.1136/bcr-2013-201764
- Alquorain NAA, Aljabr ASH, Alghamdi NJ. Cutaneous polyarteritis nodosa treated with pentoxifylline and clobetasol propionate: a case report. Saudi J Med Sci. 2018;6:104-107.
- Helms AE, Schaffer RI. Idiopathic thrombocytopenic purpura with black oral mucosal lesions. Cutis. 2007;79:456-458.
- Lountzis N, Maroon M, Tyler W. Mucocutaneous hemorrhagic bullae in idiopathic thrombocytopenic purpura. J Am Acad Dermatol. 2009;60:AB124.
- Llamas-Velasco M, Alegeria V, Santos-Briz A, et al. Occlusive nonvasculitic vasculopathy. Am J Dermatopathol. 2017;39:637-662.
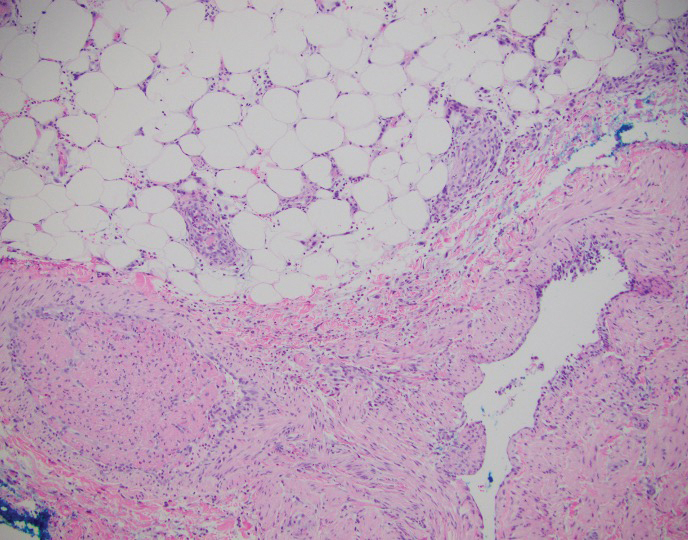
A 40-year-old woman presented with a progressive painful rash on the ears and legs of 2 weeks’ duration. She described the rash as initially red and nonpainful; it started on the right leg and progressed to the left leg, eventually involving the earlobes 4 days prior to presentation. Physical examination revealed edematous purpura of the earlobes and bullous retiform purpura on the lower extremities. Laboratory studies revealed leukopenia (3.6×103 /cm2 [reference range, 4.0–10.5×103 /cm2 ]) and elevated antineutrophil cytoplasmic antibodies (1:320 titer [reference range, <1:40]) in a perinuclear pattern (perinuclear antineutrophil cytoplasmic antibodies). Urine toxicology screening was positive for cocaine and opiates. A punch biopsy of a bullous retiform purpura on the right thigh was obtained for standard hematoxylin and eosin staining.
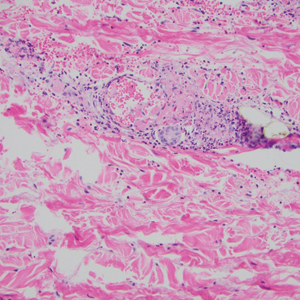
Subcutaneous Nodule on the Chest
The Diagnosis: Cystic Panfolliculoma
Panfolliculoma is a rare tumor of follicular origin.1 Clinical examination can reveal a papule, nodule, or tumor that typically is mistaken for an epidermal inclusion cyst, trichoepithelioma, or basal cell carcinoma (BCC).2 As with other benign follicular neoplasms, it often exhibits a protracted growth pattern.3,4 Most cases reported in the literature have been shown to occur in the head or neck region. One hypothesis is that separation into the various components of the hair follicle occurs at a higher frequency in areas with a higher hair density such as the face and scalp.4 The lesion typically presents in patients aged 20 to 70 years, as in our patient, with cases equally distributed among males and females.4,5 Neill et al1 reported a rare case of cystic panfolliculoma occurring on the right forearm of a 64-year-old woman.
As its name suggests, panfolliculoma is exceptional in that it displays features of all segments of the hair follicle, including the infundibulum, isthmus, stem, and bulb.6 Although not necessary for diagnosis, immunohistochemical staining can be utilized to identify each hair follicle component on histopathologic examination. Panfolliculoma stains positive for 34βE12 and cytokeratin 5/6, highlighting infundibular and isthmus keratinocytes and the outer root sheath, respectively. Additionally, Ber-EP4 labels germinative cells, while CD34 highlights contiguous fibrotic stroma and trichilemmal areas.3,4
In our patient, histopathology revealed a cystic structure that was lined by an infundibular epithelium with a prominent granular layer. Solid collections of basaloid germinative cells that demonstrated peripheral palisading were observed (quiz image [top]). Cells with trichohyalin granules, indicative of inner root sheath differentiation, were encased by matrical cells (quiz image [bottom]).
Historically, panfolliculomas characteristically have been known to reside in the dermis, with only focal connection to the epidermis, if at all present. Nevertheless, Harris et al7 detailed 2 cases that displayed predominant epidermal involvement, defined by the term epidermal panfolliculoma. In a study performed by Shan and Guo,2 an additional 9 cases (19 panfolliculomas) were found to have similar findings, for which the term superficial panfolliculoma was suggested. In cases that display a primary epidermal component, common mimickers include tumor of the follicular infundibulum and the reactive process of follicular induction.7
Cystic panfolliculoma is a rare subtype further characterized as a lesion with distinctive features of a panfolliculoma that arises from a cyst wall composed of the follicular infundibulum.2,6 The origin of cystic panfolliculoma has not been fully elucidated. It has been hypothesized that the formation of such lesions may arise due to epithelial-mesenchymal interactions. One explanation is that basal cells with stem cell capability may progress into hair follicle structures after communication with underlying dermal cells during invagination of the epidermis, while the epithelial cells not in close proximity to dermal cells maintain stem cell capability.8
The histologic differential diagnosis of cystic panfolliculoma includes dilated pore of Winer, epidermal inclusion cyst, pilar cyst, trichofolliculoma, folliculosebaceous cystic hamartoma, cystic trichoblastoma, and BCC.5 Panfolliculoma can mimic both trichoblastoma and trichoepithelioma on a low-power field; however, the latter follicular tumors lack differentiation to the infundibulum, isthmus, outer root sheath, or hair shaft, as in a panfolliculoma.4 Trichoblastoma is composed of germinative hair follicle cells, with differentiation limited to the hair germ and papilla (Figure 1).9 Panfolliculoma additionally differs from trichoblastoma by having a more prevalent epithelial factor compared to a more pronounced stromal factor in trichoblastoma.1 The cystic subtype of trichoblastoma differs from cystic panfolliculoma in that the cyst wall develops from the infundibulum only and has germinative cells protruding outwards from the cyst wall.

Although BCCs may arise in cystic structures, panfolliculomas can be discerned from this entity by their sharp demarcation, lack of peritumoral clefting, and presence of cytokeratin 20-positive Merkel cells.5 Unlike panfolliculoma, the tumor islands in BCC commonly display peripheral palisading of nuclei with a surrounding fibromyxoid stroma (Figure 2). Additionally, BCCs can exhibit crowding of nuclei, atypia, and mitoses.6

Folliculosebaceous cystic hamartomas and cystic panfolliculomas both contain a cystic structure with differentiation of the cyst wall to the hair follicle. However, folliculosebaceous cystic hamartomas are dilated infundibulocystic configurations that contain sebaceous glands emanating from the cyst wall (Figure 3). Kimura et al10 described defining features of the mesenchymal component of this follicular tumor, including an increase in fibroplasia, vascularity, and adipose tissue. In addition, the epithelial aspect exhibits clefting among the stroma and uninvolved dermis.6

Dilated pore of Winer consists of a cystic opening with connection to the epidermis. The cyst wall resembles the follicular infundibulum, and the cavity is filled with lamellar orthokeratosis (Figure 4).5,11 Epidermal inclusion cysts also contain a cyst wall that resembles the infundibular epithelium, without differentiation to all segments of the hair follicle. They are lined by a stratified squamous epithelium, retain a granular layer, and contain lamellar keratin within the cyst cavity.5,12

In summary, panfolliculoma is a rare benign neoplasm that demonstrates differentiation to each component of the hair follicle structure. Our case demonstrates a unique subtype showcasing cystic changes that infrequently has been described in the literature.
- Neill B, Bingham C, Braudis K, et al. A rare cutaneous adnexal neoplasm: cystic panfolliculoma. J Cutan Pathol. 2016;43:1183-1185.
- Shan SJ, Guo Y. Panfolliculoma and histopathologic variants: a study of 19 cases. Am J Dermatopathol. 2014;36:965-971.
- Hoang MP, Levenson BM. Cystic panfolliculoma. Arch Pathol Lab Med. 2006;130:389-392.
- Huang CY, Wu YH. Panfolliculoma: report of two cases. Dermatol Sínica. 2010;28:73-76.
- Alkhalidi HM, Alhumaidy AA. Cystic panfolliculoma of the scalp: report of a very rare case and brief review. Indian J Pathol Microbiol. 2013;56:437-439.
- López-Takegami JC, Wolter M, Löser C, et al. Classification of cysts with follicular germinative differentiation. J Cutan Pathol. 2016;43:191-199.
- Harris A, Faulkner-Jones B, Zimarowski MJ. Epidermal panfolliculoma: a report of 2 cases. Am J Dermatopathol. 2011;33:E7-E10.
- Fukuyama M, Sato Y, Yamazaki Y, et al. Immunohistochemical dissection of cystic panfolliculoma focusing on the expression of multiple hair follicle lineage markers with an insight into the pathogenesis. J Cutan Pathol. 2017;44:861-866.
- Tellechea O, Cardoso JC, Reis JP, et al. Benign follicular tumors. An Bras Dermatol. 2015;90:780-796; quiz 797-788.
- Kimura T, Miyazawa H, Aoyagi T, et al. Folliculosebaceous cystic hamartoma. a distinctive malformation of the skin. Am J Dermatopathol. 1991;13:213-220.
- Misago N, Inoue T, Narisawa Y. Cystic trichoblastoma: a report of two cases with an immunohistochemical study. J Dermatol. 2015;42:305-310.
- Weir CB, St. Hilaire NJ. Epidermal inclusion cyst. StatPearls. StatPearls Publishing; 2020.
The Diagnosis: Cystic Panfolliculoma
Panfolliculoma is a rare tumor of follicular origin.1 Clinical examination can reveal a papule, nodule, or tumor that typically is mistaken for an epidermal inclusion cyst, trichoepithelioma, or basal cell carcinoma (BCC).2 As with other benign follicular neoplasms, it often exhibits a protracted growth pattern.3,4 Most cases reported in the literature have been shown to occur in the head or neck region. One hypothesis is that separation into the various components of the hair follicle occurs at a higher frequency in areas with a higher hair density such as the face and scalp.4 The lesion typically presents in patients aged 20 to 70 years, as in our patient, with cases equally distributed among males and females.4,5 Neill et al1 reported a rare case of cystic panfolliculoma occurring on the right forearm of a 64-year-old woman.
As its name suggests, panfolliculoma is exceptional in that it displays features of all segments of the hair follicle, including the infundibulum, isthmus, stem, and bulb.6 Although not necessary for diagnosis, immunohistochemical staining can be utilized to identify each hair follicle component on histopathologic examination. Panfolliculoma stains positive for 34βE12 and cytokeratin 5/6, highlighting infundibular and isthmus keratinocytes and the outer root sheath, respectively. Additionally, Ber-EP4 labels germinative cells, while CD34 highlights contiguous fibrotic stroma and trichilemmal areas.3,4
In our patient, histopathology revealed a cystic structure that was lined by an infundibular epithelium with a prominent granular layer. Solid collections of basaloid germinative cells that demonstrated peripheral palisading were observed (quiz image [top]). Cells with trichohyalin granules, indicative of inner root sheath differentiation, were encased by matrical cells (quiz image [bottom]).
Historically, panfolliculomas characteristically have been known to reside in the dermis, with only focal connection to the epidermis, if at all present. Nevertheless, Harris et al7 detailed 2 cases that displayed predominant epidermal involvement, defined by the term epidermal panfolliculoma. In a study performed by Shan and Guo,2 an additional 9 cases (19 panfolliculomas) were found to have similar findings, for which the term superficial panfolliculoma was suggested. In cases that display a primary epidermal component, common mimickers include tumor of the follicular infundibulum and the reactive process of follicular induction.7
Cystic panfolliculoma is a rare subtype further characterized as a lesion with distinctive features of a panfolliculoma that arises from a cyst wall composed of the follicular infundibulum.2,6 The origin of cystic panfolliculoma has not been fully elucidated. It has been hypothesized that the formation of such lesions may arise due to epithelial-mesenchymal interactions. One explanation is that basal cells with stem cell capability may progress into hair follicle structures after communication with underlying dermal cells during invagination of the epidermis, while the epithelial cells not in close proximity to dermal cells maintain stem cell capability.8
The histologic differential diagnosis of cystic panfolliculoma includes dilated pore of Winer, epidermal inclusion cyst, pilar cyst, trichofolliculoma, folliculosebaceous cystic hamartoma, cystic trichoblastoma, and BCC.5 Panfolliculoma can mimic both trichoblastoma and trichoepithelioma on a low-power field; however, the latter follicular tumors lack differentiation to the infundibulum, isthmus, outer root sheath, or hair shaft, as in a panfolliculoma.4 Trichoblastoma is composed of germinative hair follicle cells, with differentiation limited to the hair germ and papilla (Figure 1).9 Panfolliculoma additionally differs from trichoblastoma by having a more prevalent epithelial factor compared to a more pronounced stromal factor in trichoblastoma.1 The cystic subtype of trichoblastoma differs from cystic panfolliculoma in that the cyst wall develops from the infundibulum only and has germinative cells protruding outwards from the cyst wall.
Although BCCs may arise in cystic structures, panfolliculomas can be discerned from this entity by their sharp demarcation, lack of peritumoral clefting, and presence of cytokeratin 20-positive Merkel cells.5 Unlike panfolliculoma, the tumor islands in BCC commonly display peripheral palisading of nuclei with a surrounding fibromyxoid stroma (Figure 2). Additionally, BCCs can exhibit crowding of nuclei, atypia, and mitoses.6
Folliculosebaceous cystic hamartomas and cystic panfolliculomas both contain a cystic structure with differentiation of the cyst wall to the hair follicle. However, folliculosebaceous cystic hamartomas are dilated infundibulocystic configurations that contain sebaceous glands emanating from the cyst wall (Figure 3). Kimura et al10 described defining features of the mesenchymal component of this follicular tumor, including an increase in fibroplasia, vascularity, and adipose tissue. In addition, the epithelial aspect exhibits clefting among the stroma and uninvolved dermis.6
Dilated pore of Winer consists of a cystic opening with connection to the epidermis. The cyst wall resembles the follicular infundibulum, and the cavity is filled with lamellar orthokeratosis (Figure 4).5,11 Epidermal inclusion cysts also contain a cyst wall that resembles the infundibular epithelium, without differentiation to all segments of the hair follicle. They are lined by a stratified squamous epithelium, retain a granular layer, and contain lamellar keratin within the cyst cavity.5,12
In summary, panfolliculoma is a rare benign neoplasm that demonstrates differentiation to each component of the hair follicle structure. Our case demonstrates a unique subtype showcasing cystic changes that infrequently has been described in the literature.
The Diagnosis: Cystic Panfolliculoma
Panfolliculoma is a rare tumor of follicular origin.1 Clinical examination can reveal a papule, nodule, or tumor that typically is mistaken for an epidermal inclusion cyst, trichoepithelioma, or basal cell carcinoma (BCC).2 As with other benign follicular neoplasms, it often exhibits a protracted growth pattern.3,4 Most cases reported in the literature have been shown to occur in the head or neck region. One hypothesis is that separation into the various components of the hair follicle occurs at a higher frequency in areas with a higher hair density such as the face and scalp.4 The lesion typically presents in patients aged 20 to 70 years, as in our patient, with cases equally distributed among males and females.4,5 Neill et al1 reported a rare case of cystic panfolliculoma occurring on the right forearm of a 64-year-old woman.
As its name suggests, panfolliculoma is exceptional in that it displays features of all segments of the hair follicle, including the infundibulum, isthmus, stem, and bulb.6 Although not necessary for diagnosis, immunohistochemical staining can be utilized to identify each hair follicle component on histopathologic examination. Panfolliculoma stains positive for 34βE12 and cytokeratin 5/6, highlighting infundibular and isthmus keratinocytes and the outer root sheath, respectively. Additionally, Ber-EP4 labels germinative cells, while CD34 highlights contiguous fibrotic stroma and trichilemmal areas.3,4
In our patient, histopathology revealed a cystic structure that was lined by an infundibular epithelium with a prominent granular layer. Solid collections of basaloid germinative cells that demonstrated peripheral palisading were observed (quiz image [top]). Cells with trichohyalin granules, indicative of inner root sheath differentiation, were encased by matrical cells (quiz image [bottom]).
Historically, panfolliculomas characteristically have been known to reside in the dermis, with only focal connection to the epidermis, if at all present. Nevertheless, Harris et al7 detailed 2 cases that displayed predominant epidermal involvement, defined by the term epidermal panfolliculoma. In a study performed by Shan and Guo,2 an additional 9 cases (19 panfolliculomas) were found to have similar findings, for which the term superficial panfolliculoma was suggested. In cases that display a primary epidermal component, common mimickers include tumor of the follicular infundibulum and the reactive process of follicular induction.7
Cystic panfolliculoma is a rare subtype further characterized as a lesion with distinctive features of a panfolliculoma that arises from a cyst wall composed of the follicular infundibulum.2,6 The origin of cystic panfolliculoma has not been fully elucidated. It has been hypothesized that the formation of such lesions may arise due to epithelial-mesenchymal interactions. One explanation is that basal cells with stem cell capability may progress into hair follicle structures after communication with underlying dermal cells during invagination of the epidermis, while the epithelial cells not in close proximity to dermal cells maintain stem cell capability.8
The histologic differential diagnosis of cystic panfolliculoma includes dilated pore of Winer, epidermal inclusion cyst, pilar cyst, trichofolliculoma, folliculosebaceous cystic hamartoma, cystic trichoblastoma, and BCC.5 Panfolliculoma can mimic both trichoblastoma and trichoepithelioma on a low-power field; however, the latter follicular tumors lack differentiation to the infundibulum, isthmus, outer root sheath, or hair shaft, as in a panfolliculoma.4 Trichoblastoma is composed of germinative hair follicle cells, with differentiation limited to the hair germ and papilla (Figure 1).9 Panfolliculoma additionally differs from trichoblastoma by having a more prevalent epithelial factor compared to a more pronounced stromal factor in trichoblastoma.1 The cystic subtype of trichoblastoma differs from cystic panfolliculoma in that the cyst wall develops from the infundibulum only and has germinative cells protruding outwards from the cyst wall.
Although BCCs may arise in cystic structures, panfolliculomas can be discerned from this entity by their sharp demarcation, lack of peritumoral clefting, and presence of cytokeratin 20-positive Merkel cells.5 Unlike panfolliculoma, the tumor islands in BCC commonly display peripheral palisading of nuclei with a surrounding fibromyxoid stroma (Figure 2). Additionally, BCCs can exhibit crowding of nuclei, atypia, and mitoses.6
Folliculosebaceous cystic hamartomas and cystic panfolliculomas both contain a cystic structure with differentiation of the cyst wall to the hair follicle. However, folliculosebaceous cystic hamartomas are dilated infundibulocystic configurations that contain sebaceous glands emanating from the cyst wall (Figure 3). Kimura et al10 described defining features of the mesenchymal component of this follicular tumor, including an increase in fibroplasia, vascularity, and adipose tissue. In addition, the epithelial aspect exhibits clefting among the stroma and uninvolved dermis.6
Dilated pore of Winer consists of a cystic opening with connection to the epidermis. The cyst wall resembles the follicular infundibulum, and the cavity is filled with lamellar orthokeratosis (Figure 4).5,11 Epidermal inclusion cysts also contain a cyst wall that resembles the infundibular epithelium, without differentiation to all segments of the hair follicle. They are lined by a stratified squamous epithelium, retain a granular layer, and contain lamellar keratin within the cyst cavity.5,12
In summary, panfolliculoma is a rare benign neoplasm that demonstrates differentiation to each component of the hair follicle structure. Our case demonstrates a unique subtype showcasing cystic changes that infrequently has been described in the literature.
- Neill B, Bingham C, Braudis K, et al. A rare cutaneous adnexal neoplasm: cystic panfolliculoma. J Cutan Pathol. 2016;43:1183-1185.
- Shan SJ, Guo Y. Panfolliculoma and histopathologic variants: a study of 19 cases. Am J Dermatopathol. 2014;36:965-971.
- Hoang MP, Levenson BM. Cystic panfolliculoma. Arch Pathol Lab Med. 2006;130:389-392.
- Huang CY, Wu YH. Panfolliculoma: report of two cases. Dermatol Sínica. 2010;28:73-76.
- Alkhalidi HM, Alhumaidy AA. Cystic panfolliculoma of the scalp: report of a very rare case and brief review. Indian J Pathol Microbiol. 2013;56:437-439.
- López-Takegami JC, Wolter M, Löser C, et al. Classification of cysts with follicular germinative differentiation. J Cutan Pathol. 2016;43:191-199.
- Harris A, Faulkner-Jones B, Zimarowski MJ. Epidermal panfolliculoma: a report of 2 cases. Am J Dermatopathol. 2011;33:E7-E10.
- Fukuyama M, Sato Y, Yamazaki Y, et al. Immunohistochemical dissection of cystic panfolliculoma focusing on the expression of multiple hair follicle lineage markers with an insight into the pathogenesis. J Cutan Pathol. 2017;44:861-866.
- Tellechea O, Cardoso JC, Reis JP, et al. Benign follicular tumors. An Bras Dermatol. 2015;90:780-796; quiz 797-788.
- Kimura T, Miyazawa H, Aoyagi T, et al. Folliculosebaceous cystic hamartoma. a distinctive malformation of the skin. Am J Dermatopathol. 1991;13:213-220.
- Misago N, Inoue T, Narisawa Y. Cystic trichoblastoma: a report of two cases with an immunohistochemical study. J Dermatol. 2015;42:305-310.
- Weir CB, St. Hilaire NJ. Epidermal inclusion cyst. StatPearls. StatPearls Publishing; 2020.
- Neill B, Bingham C, Braudis K, et al. A rare cutaneous adnexal neoplasm: cystic panfolliculoma. J Cutan Pathol. 2016;43:1183-1185.
- Shan SJ, Guo Y. Panfolliculoma and histopathologic variants: a study of 19 cases. Am J Dermatopathol. 2014;36:965-971.
- Hoang MP, Levenson BM. Cystic panfolliculoma. Arch Pathol Lab Med. 2006;130:389-392.
- Huang CY, Wu YH. Panfolliculoma: report of two cases. Dermatol Sínica. 2010;28:73-76.
- Alkhalidi HM, Alhumaidy AA. Cystic panfolliculoma of the scalp: report of a very rare case and brief review. Indian J Pathol Microbiol. 2013;56:437-439.
- López-Takegami JC, Wolter M, Löser C, et al. Classification of cysts with follicular germinative differentiation. J Cutan Pathol. 2016;43:191-199.
- Harris A, Faulkner-Jones B, Zimarowski MJ. Epidermal panfolliculoma: a report of 2 cases. Am J Dermatopathol. 2011;33:E7-E10.
- Fukuyama M, Sato Y, Yamazaki Y, et al. Immunohistochemical dissection of cystic panfolliculoma focusing on the expression of multiple hair follicle lineage markers with an insight into the pathogenesis. J Cutan Pathol. 2017;44:861-866.
- Tellechea O, Cardoso JC, Reis JP, et al. Benign follicular tumors. An Bras Dermatol. 2015;90:780-796; quiz 797-788.
- Kimura T, Miyazawa H, Aoyagi T, et al. Folliculosebaceous cystic hamartoma. a distinctive malformation of the skin. Am J Dermatopathol. 1991;13:213-220.
- Misago N, Inoue T, Narisawa Y. Cystic trichoblastoma: a report of two cases with an immunohistochemical study. J Dermatol. 2015;42:305-310.
- Weir CB, St. Hilaire NJ. Epidermal inclusion cyst. StatPearls. StatPearls Publishing; 2020.


A healthy 45-year-old man presented to the dermatology clinic with a slow-growing subcutaneous nodule on the left chest that had been present for years.
Atrophic Lesions in a Pregnant Woman
The Diagnosis: Degos Disease
The pathophysiology of Degos disease (malignant atrophic papulosis) is unknown.1 Histopathology demonstrates a wedge-shaped area of dermal necrosis with edema and mucin deposition extending from the papillary dermis to the deep reticular dermis. Occluded vessels, thrombosis, and perivascular lymphocytic infiltrates also may be seen, particularly at the dermal subcutaneous junction and at the periphery of the wedge-shaped infarction. The vascular damage that occurs may be the result of vasculitis, coagulopathy, or endothelial cell dysfunction.1
Patients typically present with small, round, erythematous papules that eventually develop atrophic porcelain white centers and telangiectatic rims. These lesions most commonly occur on the trunk and arms. In the benign form of atrophic papulosis, only the skin is involved; however, systemic involvement of the gastrointestinal tract and central nervous system can occur, resulting in bowel perforation and stroke, respectively.1 Although there is no definitive treatment of Degos disease, successful therapy with aspirin or dipyridamole has been reported.1 Eculizumab, a monoclonal antibody that binds C5, and treprostinil, a prostacyclin analog, are emerging treatment options.2,3 The differential diagnosis of Degos disease may include granuloma annulare, guttate extragenital lichen sclerosus, livedoid vasculopathy, and lymphomatoid papulosis.
Granuloma annulare may clinically mimic the erythematous papules seen in early Degos disease, and histopathology can be used to distinguish between these two disease processes. Localized granuloma annulare is the most common variant and clinically presents as pink papules and plaques in an annular configuration.4 Histopathology demonstrates an unremarkable epidermis; however, the dermis contains degenerated collagen surrounded by palisading histiocytes as well as lymphocytes. Similar to Degos disease, increased mucin is seen within these areas of degeneration, but occluded vessels and thrombosis typically are not seen (Figure 1).4,5
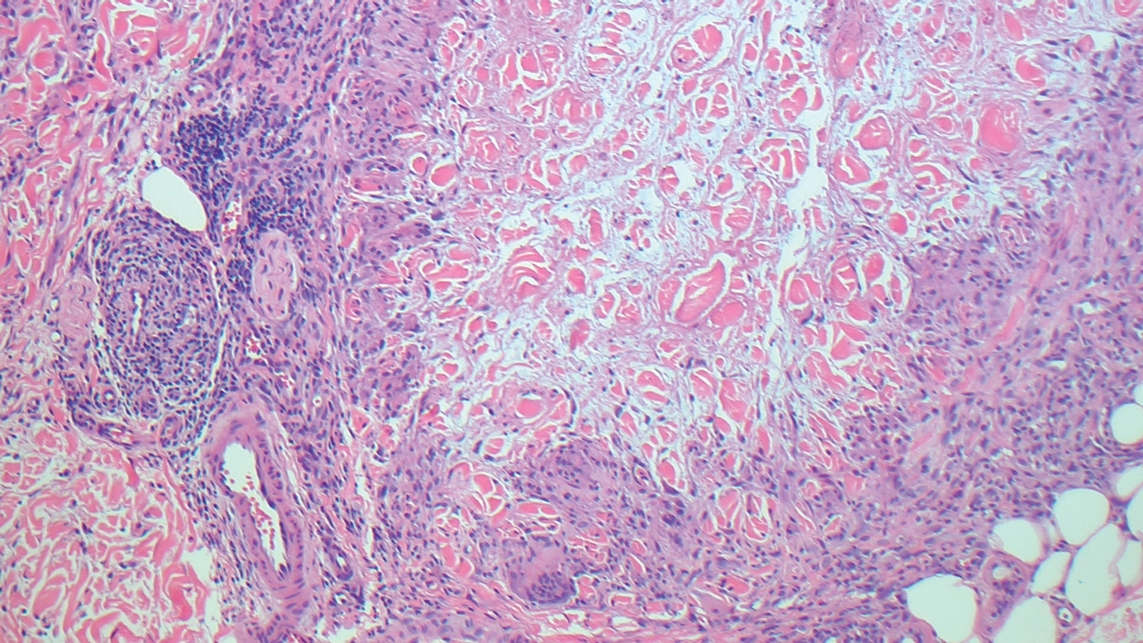
Guttate extragenital lichen sclerosus initially presents as polygonal, bluish white papules that coalesce into plaques.6 Over time, these lesions become more atrophic and may mimic Degos disease but appear differently on histopathology. Histopathology of lichen sclerosus classically demonstrates atrophy of the epidermis with loss of the rete ridges and vacuolar surface changes. Homogenization of the superficial/papillary dermis with an underlying bandlike lymphocytic infiltrate also is seen (Figure 2).6
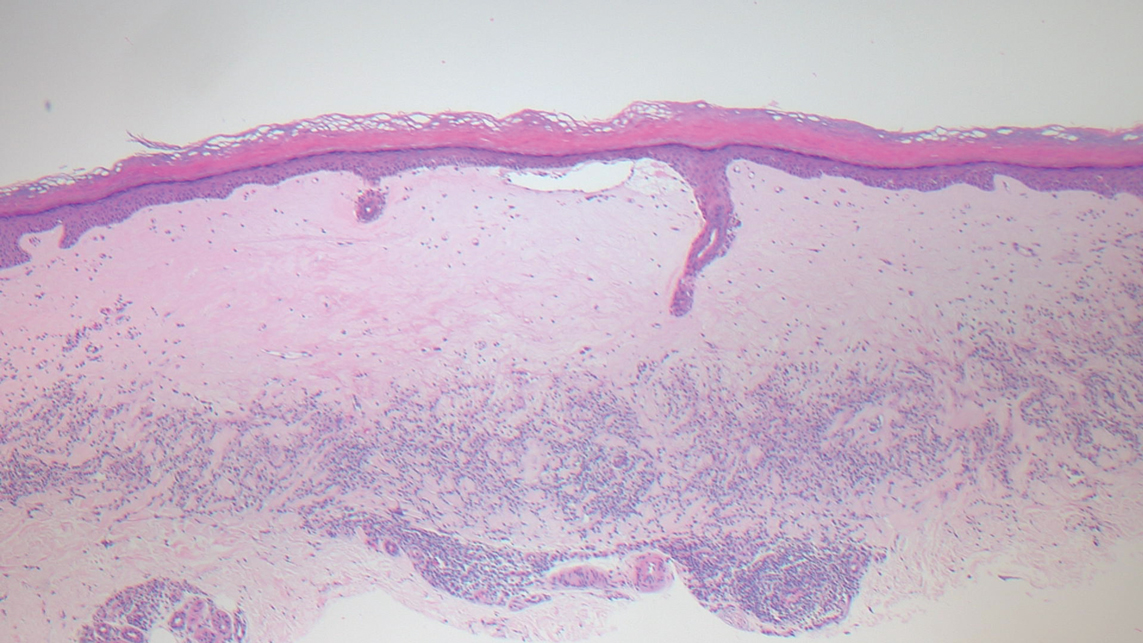
Livedoid vasculopathy is characterized by chronic recurrent ulceration of the legs secondary to thrombosis and subsequent ischemia. In the initial phase of this disease, livedo reticularis is seen followed by the development of ulcerations. As these ulcerations heal, they leave behind porcelain white scars referred to as atrophie blanche.7 The areas of scarring in livedoid vasculopathy are broad and angulated, differentiating them from the small, round, porcelain white macules in end-stage Degos disease. Histopathology demonstrates thrombosis and fibrin occlusion of the upper and mid dermal vessels. Very minimal perivascular infiltrate typically is seen, but when it is present, the infiltrate mostly is lymphocytic. Hyalinization of the vessel walls also is seen, particularly in the atrophie blanche stage (Figure 3).7
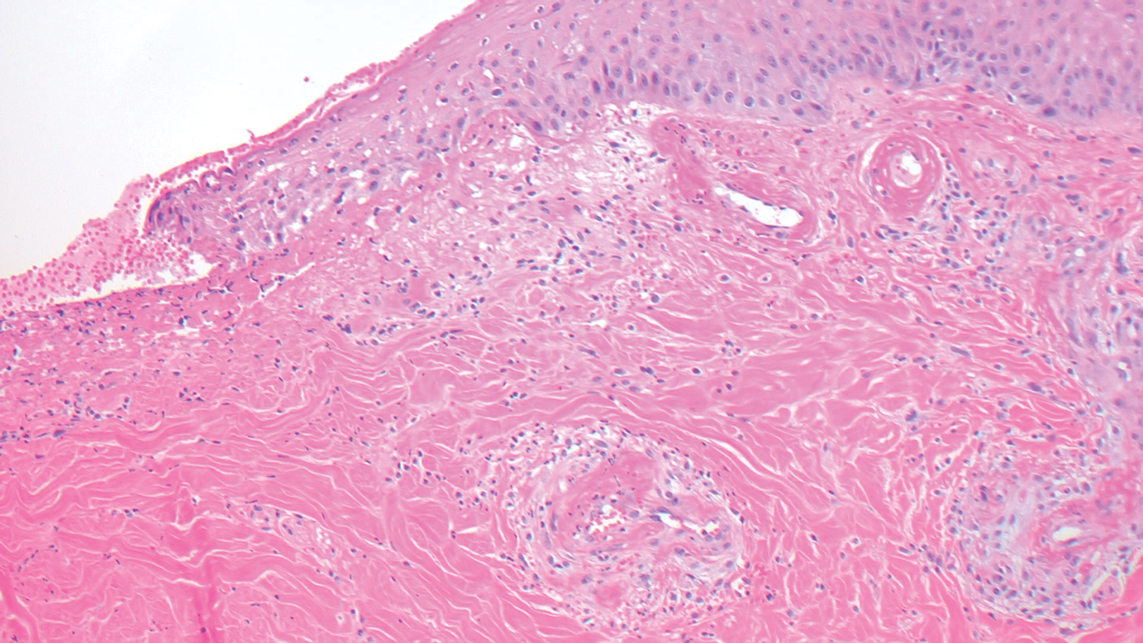
Lymphomatoid papulosis classically presents with pruritic red papules that often spontaneously involute. After resolution of the primary lesions, atrophic varioliform scars may be left behind that can resemble Degos disease.8 Classically, there are 5 histopathologic subtypes: A, B, C, D, and E. Type A is the most common type of lymphomatoid papulosis, and histopathology demonstrates a dermal lymphocytic infiltrate that consists of cells arranged in small clusters. Numerous medium- to large-sized atypical lymphocytes with prominent nucleoli and abundant cytoplasm are seen, and mitotic figures are common (Figure 4).8
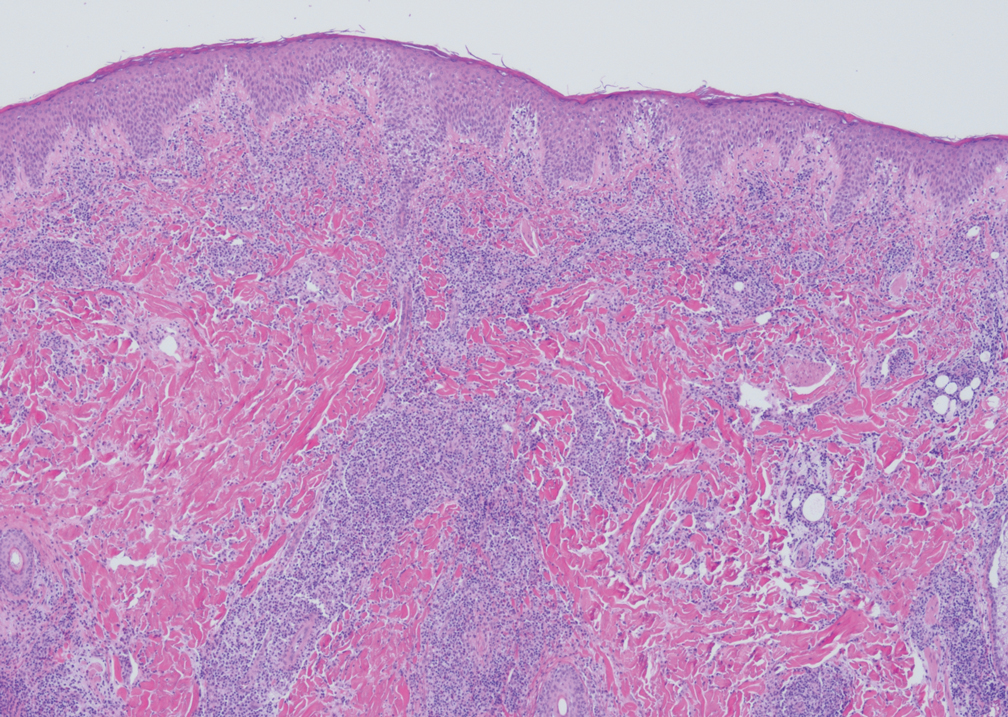
Our case was particularly interesting because the patient was 2 to 3 weeks pregnant. Degos disease in pregnancy appears to be quite exceptional. A PubMed search of articles indexed for MEDLINE using the terms Degos disease and pregnancy revealed only 4 other cases reported in the literature.9-12 With the exception of a single case that was complicated by severe abdominal pain requiring labor induction, the other reported cases resulted in uncomplicated pregnancies.9-12 Conversely, our patient's pregnancy was complicated by gestational hypertension and fetal hydrops requiring a preterm cesarean delivery. Furthermore, the infant had multiple complications, which were attributed to both placental insufficiency and a coagulopathic state.
Our patient also was found to have a heterozygous factor V Leiden mutation on workup. A PubMed search using the terms factor V Leiden mutation and Degos disease revealed 2 other cases of factor V Leiden mutation-associated Degos disease.13,14 The importance of factor V Leiden mutations in patients with Degos disease currently is unclear.
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)--a review. Orphanet J Rare Dis. 2013;8:10.
- Oliver B, Boehm M, Rosing DR, et al. Diffuse atrophic papules and plaques, intermittent abdominal pain, paresthesias, and cardiac abnormalities in a 55-year-old woman. J Am Acad Dermatol. 2016;75:1274-1277.
- Magro CM, Wang X, Garrett-Bakelman F, et al. The effects of eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis. 2013;8:185.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Tronnier M, Mitteldorf C. Histologic features of granulomatous skin diseases. part 1: non-infectious granulomatous disorders. J Dtsch Dermatol Ges. 2015;13:211-216.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478‐488.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73.
- Moulin G, Barrut D, Franc MP, et al. Familial Degos' atrophic papulosis (mother-daughter). Ann Dermatol Venereol. 1984;111:149-155.
- Bogenrieder T, Kuske M, Landthaler M, et al. Benign Degos' disease developing during pregnancy and followed for 10 years. Acta Derm Venereol. 2002;82:284-287.
- Sharma S, Brennan B, Naden R, et al. A case of Degos disease in pregnancy. Obstet Med. 2016;9:167-168.
- Zhao Q, Zhang S, Dong A. An unusual case of abdominal pain. Gastroenterology. 2018;154:E1-E2.
- Darwich E, Guilabert A, Mascaró JM Jr, et al. Dermoscopic description of a patient with thrombocythemia and factor V Leiden mutation-associated Degos' disease. Int J Dermatol. 2011;50:604-606.
- Hohwy T, Jensen MG, Tøttrup A, et al. A fatal case of malignant atrophic papulosis (Degos' disease) in a man with factor V Leiden mutation and lupus anticoagulant. Acta Derm Venereol. 2006;86:245-247.
The Diagnosis: Degos Disease
The pathophysiology of Degos disease (malignant atrophic papulosis) is unknown.1 Histopathology demonstrates a wedge-shaped area of dermal necrosis with edema and mucin deposition extending from the papillary dermis to the deep reticular dermis. Occluded vessels, thrombosis, and perivascular lymphocytic infiltrates also may be seen, particularly at the dermal subcutaneous junction and at the periphery of the wedge-shaped infarction. The vascular damage that occurs may be the result of vasculitis, coagulopathy, or endothelial cell dysfunction.1
Patients typically present with small, round, erythematous papules that eventually develop atrophic porcelain white centers and telangiectatic rims. These lesions most commonly occur on the trunk and arms. In the benign form of atrophic papulosis, only the skin is involved; however, systemic involvement of the gastrointestinal tract and central nervous system can occur, resulting in bowel perforation and stroke, respectively.1 Although there is no definitive treatment of Degos disease, successful therapy with aspirin or dipyridamole has been reported.1 Eculizumab, a monoclonal antibody that binds C5, and treprostinil, a prostacyclin analog, are emerging treatment options.2,3 The differential diagnosis of Degos disease may include granuloma annulare, guttate extragenital lichen sclerosus, livedoid vasculopathy, and lymphomatoid papulosis.
Granuloma annulare may clinically mimic the erythematous papules seen in early Degos disease, and histopathology can be used to distinguish between these two disease processes. Localized granuloma annulare is the most common variant and clinically presents as pink papules and plaques in an annular configuration.4 Histopathology demonstrates an unremarkable epidermis; however, the dermis contains degenerated collagen surrounded by palisading histiocytes as well as lymphocytes. Similar to Degos disease, increased mucin is seen within these areas of degeneration, but occluded vessels and thrombosis typically are not seen (Figure 1).4,5
Guttate extragenital lichen sclerosus initially presents as polygonal, bluish white papules that coalesce into plaques.6 Over time, these lesions become more atrophic and may mimic Degos disease but appear differently on histopathology. Histopathology of lichen sclerosus classically demonstrates atrophy of the epidermis with loss of the rete ridges and vacuolar surface changes. Homogenization of the superficial/papillary dermis with an underlying bandlike lymphocytic infiltrate also is seen (Figure 2).6
Livedoid vasculopathy is characterized by chronic recurrent ulceration of the legs secondary to thrombosis and subsequent ischemia. In the initial phase of this disease, livedo reticularis is seen followed by the development of ulcerations. As these ulcerations heal, they leave behind porcelain white scars referred to as atrophie blanche.7 The areas of scarring in livedoid vasculopathy are broad and angulated, differentiating them from the small, round, porcelain white macules in end-stage Degos disease. Histopathology demonstrates thrombosis and fibrin occlusion of the upper and mid dermal vessels. Very minimal perivascular infiltrate typically is seen, but when it is present, the infiltrate mostly is lymphocytic. Hyalinization of the vessel walls also is seen, particularly in the atrophie blanche stage (Figure 3).7
Lymphomatoid papulosis classically presents with pruritic red papules that often spontaneously involute. After resolution of the primary lesions, atrophic varioliform scars may be left behind that can resemble Degos disease.8 Classically, there are 5 histopathologic subtypes: A, B, C, D, and E. Type A is the most common type of lymphomatoid papulosis, and histopathology demonstrates a dermal lymphocytic infiltrate that consists of cells arranged in small clusters. Numerous medium- to large-sized atypical lymphocytes with prominent nucleoli and abundant cytoplasm are seen, and mitotic figures are common (Figure 4).8
Our case was particularly interesting because the patient was 2 to 3 weeks pregnant. Degos disease in pregnancy appears to be quite exceptional. A PubMed search of articles indexed for MEDLINE using the terms Degos disease and pregnancy revealed only 4 other cases reported in the literature.9-12 With the exception of a single case that was complicated by severe abdominal pain requiring labor induction, the other reported cases resulted in uncomplicated pregnancies.9-12 Conversely, our patient's pregnancy was complicated by gestational hypertension and fetal hydrops requiring a preterm cesarean delivery. Furthermore, the infant had multiple complications, which were attributed to both placental insufficiency and a coagulopathic state.
Our patient also was found to have a heterozygous factor V Leiden mutation on workup. A PubMed search using the terms factor V Leiden mutation and Degos disease revealed 2 other cases of factor V Leiden mutation-associated Degos disease.13,14 The importance of factor V Leiden mutations in patients with Degos disease currently is unclear.
The Diagnosis: Degos Disease
The pathophysiology of Degos disease (malignant atrophic papulosis) is unknown.1 Histopathology demonstrates a wedge-shaped area of dermal necrosis with edema and mucin deposition extending from the papillary dermis to the deep reticular dermis. Occluded vessels, thrombosis, and perivascular lymphocytic infiltrates also may be seen, particularly at the dermal subcutaneous junction and at the periphery of the wedge-shaped infarction. The vascular damage that occurs may be the result of vasculitis, coagulopathy, or endothelial cell dysfunction.1
Patients typically present with small, round, erythematous papules that eventually develop atrophic porcelain white centers and telangiectatic rims. These lesions most commonly occur on the trunk and arms. In the benign form of atrophic papulosis, only the skin is involved; however, systemic involvement of the gastrointestinal tract and central nervous system can occur, resulting in bowel perforation and stroke, respectively.1 Although there is no definitive treatment of Degos disease, successful therapy with aspirin or dipyridamole has been reported.1 Eculizumab, a monoclonal antibody that binds C5, and treprostinil, a prostacyclin analog, are emerging treatment options.2,3 The differential diagnosis of Degos disease may include granuloma annulare, guttate extragenital lichen sclerosus, livedoid vasculopathy, and lymphomatoid papulosis.
Granuloma annulare may clinically mimic the erythematous papules seen in early Degos disease, and histopathology can be used to distinguish between these two disease processes. Localized granuloma annulare is the most common variant and clinically presents as pink papules and plaques in an annular configuration.4 Histopathology demonstrates an unremarkable epidermis; however, the dermis contains degenerated collagen surrounded by palisading histiocytes as well as lymphocytes. Similar to Degos disease, increased mucin is seen within these areas of degeneration, but occluded vessels and thrombosis typically are not seen (Figure 1).4,5
Guttate extragenital lichen sclerosus initially presents as polygonal, bluish white papules that coalesce into plaques.6 Over time, these lesions become more atrophic and may mimic Degos disease but appear differently on histopathology. Histopathology of lichen sclerosus classically demonstrates atrophy of the epidermis with loss of the rete ridges and vacuolar surface changes. Homogenization of the superficial/papillary dermis with an underlying bandlike lymphocytic infiltrate also is seen (Figure 2).6
Livedoid vasculopathy is characterized by chronic recurrent ulceration of the legs secondary to thrombosis and subsequent ischemia. In the initial phase of this disease, livedo reticularis is seen followed by the development of ulcerations. As these ulcerations heal, they leave behind porcelain white scars referred to as atrophie blanche.7 The areas of scarring in livedoid vasculopathy are broad and angulated, differentiating them from the small, round, porcelain white macules in end-stage Degos disease. Histopathology demonstrates thrombosis and fibrin occlusion of the upper and mid dermal vessels. Very minimal perivascular infiltrate typically is seen, but when it is present, the infiltrate mostly is lymphocytic. Hyalinization of the vessel walls also is seen, particularly in the atrophie blanche stage (Figure 3).7
Lymphomatoid papulosis classically presents with pruritic red papules that often spontaneously involute. After resolution of the primary lesions, atrophic varioliform scars may be left behind that can resemble Degos disease.8 Classically, there are 5 histopathologic subtypes: A, B, C, D, and E. Type A is the most common type of lymphomatoid papulosis, and histopathology demonstrates a dermal lymphocytic infiltrate that consists of cells arranged in small clusters. Numerous medium- to large-sized atypical lymphocytes with prominent nucleoli and abundant cytoplasm are seen, and mitotic figures are common (Figure 4).8
Our case was particularly interesting because the patient was 2 to 3 weeks pregnant. Degos disease in pregnancy appears to be quite exceptional. A PubMed search of articles indexed for MEDLINE using the terms Degos disease and pregnancy revealed only 4 other cases reported in the literature.9-12 With the exception of a single case that was complicated by severe abdominal pain requiring labor induction, the other reported cases resulted in uncomplicated pregnancies.9-12 Conversely, our patient's pregnancy was complicated by gestational hypertension and fetal hydrops requiring a preterm cesarean delivery. Furthermore, the infant had multiple complications, which were attributed to both placental insufficiency and a coagulopathic state.
Our patient also was found to have a heterozygous factor V Leiden mutation on workup. A PubMed search using the terms factor V Leiden mutation and Degos disease revealed 2 other cases of factor V Leiden mutation-associated Degos disease.13,14 The importance of factor V Leiden mutations in patients with Degos disease currently is unclear.
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)--a review. Orphanet J Rare Dis. 2013;8:10.
- Oliver B, Boehm M, Rosing DR, et al. Diffuse atrophic papules and plaques, intermittent abdominal pain, paresthesias, and cardiac abnormalities in a 55-year-old woman. J Am Acad Dermatol. 2016;75:1274-1277.
- Magro CM, Wang X, Garrett-Bakelman F, et al. The effects of eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis. 2013;8:185.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Tronnier M, Mitteldorf C. Histologic features of granulomatous skin diseases. part 1: non-infectious granulomatous disorders. J Dtsch Dermatol Ges. 2015;13:211-216.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478‐488.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73.
- Moulin G, Barrut D, Franc MP, et al. Familial Degos' atrophic papulosis (mother-daughter). Ann Dermatol Venereol. 1984;111:149-155.
- Bogenrieder T, Kuske M, Landthaler M, et al. Benign Degos' disease developing during pregnancy and followed for 10 years. Acta Derm Venereol. 2002;82:284-287.
- Sharma S, Brennan B, Naden R, et al. A case of Degos disease in pregnancy. Obstet Med. 2016;9:167-168.
- Zhao Q, Zhang S, Dong A. An unusual case of abdominal pain. Gastroenterology. 2018;154:E1-E2.
- Darwich E, Guilabert A, Mascaró JM Jr, et al. Dermoscopic description of a patient with thrombocythemia and factor V Leiden mutation-associated Degos' disease. Int J Dermatol. 2011;50:604-606.
- Hohwy T, Jensen MG, Tøttrup A, et al. A fatal case of malignant atrophic papulosis (Degos' disease) in a man with factor V Leiden mutation and lupus anticoagulant. Acta Derm Venereol. 2006;86:245-247.
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)--a review. Orphanet J Rare Dis. 2013;8:10.
- Oliver B, Boehm M, Rosing DR, et al. Diffuse atrophic papules and plaques, intermittent abdominal pain, paresthesias, and cardiac abnormalities in a 55-year-old woman. J Am Acad Dermatol. 2016;75:1274-1277.
- Magro CM, Wang X, Garrett-Bakelman F, et al. The effects of eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis. 2013;8:185.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Tronnier M, Mitteldorf C. Histologic features of granulomatous skin diseases. part 1: non-infectious granulomatous disorders. J Dtsch Dermatol Ges. 2015;13:211-216.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478‐488.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73.
- Moulin G, Barrut D, Franc MP, et al. Familial Degos' atrophic papulosis (mother-daughter). Ann Dermatol Venereol. 1984;111:149-155.
- Bogenrieder T, Kuske M, Landthaler M, et al. Benign Degos' disease developing during pregnancy and followed for 10 years. Acta Derm Venereol. 2002;82:284-287.
- Sharma S, Brennan B, Naden R, et al. A case of Degos disease in pregnancy. Obstet Med. 2016;9:167-168.
- Zhao Q, Zhang S, Dong A. An unusual case of abdominal pain. Gastroenterology. 2018;154:E1-E2.
- Darwich E, Guilabert A, Mascaró JM Jr, et al. Dermoscopic description of a patient with thrombocythemia and factor V Leiden mutation-associated Degos' disease. Int J Dermatol. 2011;50:604-606.
- Hohwy T, Jensen MG, Tøttrup A, et al. A fatal case of malignant atrophic papulosis (Degos' disease) in a man with factor V Leiden mutation and lupus anticoagulant. Acta Derm Venereol. 2006;86:245-247.
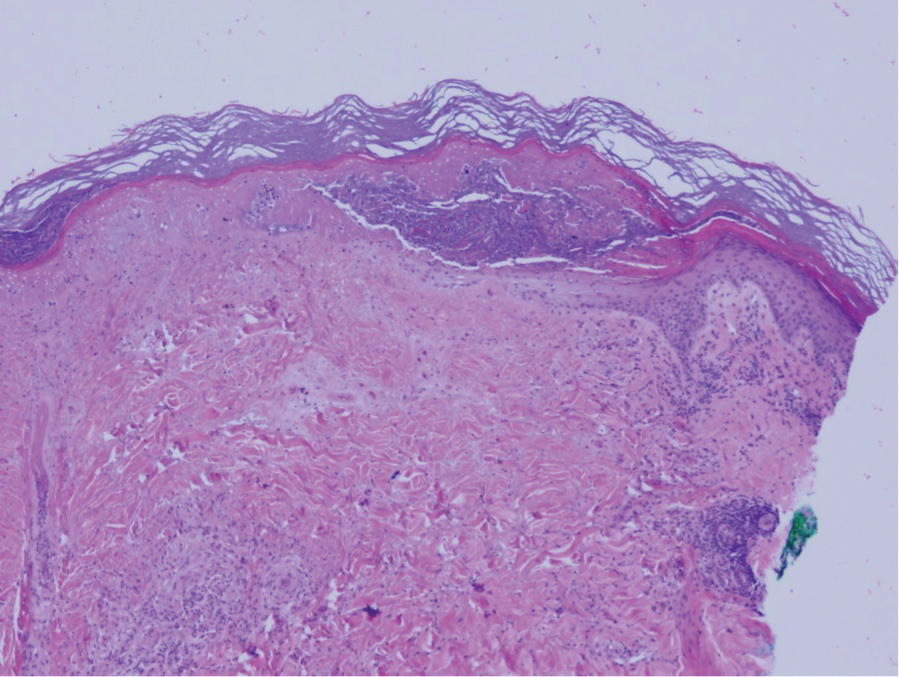
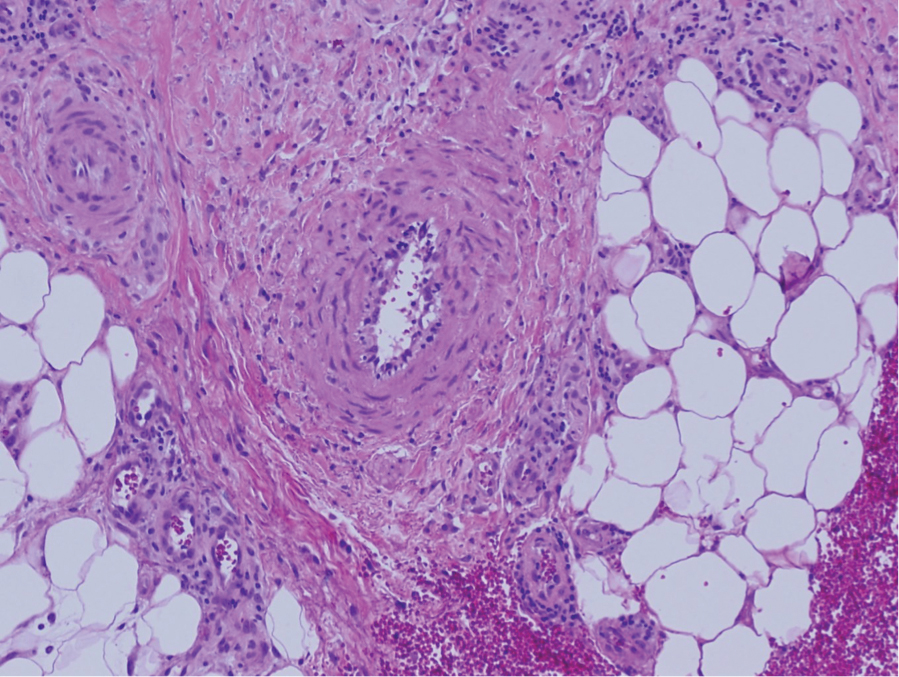
A 36-year-old pregnant woman presented with painful erythematous papules on the palms and fingers of 2 months’ duration. Similar lesions developed on the thighs and feet several weeks later. Two tender macules with central areas of porcelain white scarring rimmed by telangiectases on the right foot also were present. A punch biopsy of these lesions demonstrated a wedge-shaped area of ischemic necrosis associated with dermal mucin without associated necrobiosis. Fibrin thrombi were seen within several small dermal vessels and were associated with a perivascular lymphocytic infiltrate. Endotheliitis was observed within a deep dermal vessel. Laboratory workup including syphilis IgG, antinuclear antibodies, extractable nuclear antigen antibodies, anti–double-stranded DNA, antistreptolysin O antibodies, Russell viper venom time, cryoglobulin, hepatitis screening, perinuclear antineutrophil cytoplasmic antibodies (ANCA), and cytoplasmic ANCA was unremarkable. Hypercoagulable studies including prothrombin gene mutation, factor V Leiden, plasminogen, proteins C and S, antithrombin III, homocysteine, and antiphospholipid IgM and IgG antibodies were notable only for heterozygosity for factor V Leiden.
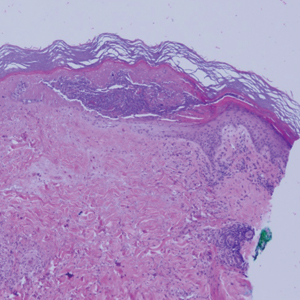
Subcutaneous, Mucocutaneous, and Mucous Membrane Tumors
The Diagnosis: Granular Cell Tumor
Histopathologic analysis from the axillary excision demonstrated cords and sheets of large polygonal cells in the dermis with uniform, oval, hyperchromatic nuclei and ample pink granular-staining cytoplasm (quiz images). An infiltrative growth pattern was noted; however, there was no evidence of conspicuous mitoses, nuclear pleomorphism, or necrosis. These results in conjunction with the immunohistochemistry findings were consistent with a benign granular cell tumor (GCT), a rare neoplasm considered to have neural/Schwann cell origin.1-3
Our case demonstrates the difficulty in clinically diagnosing cutaneous GCTs. The tumor often presents as a solitary, 0.5- to 3-cm, asymptomatic, firm nodule4,5; however, GCTs also can appear verrucous, eroded, or with other variable morphologies, which can create diagnostic challenges.5,6 Accordingly, a 1980 study of 110 patients with GCTs found that the preoperative clinical diagnosis was incorrect in all but 3 cases,7 emphasizing the need for histologic evaluation. Benign GCTs tend to exhibit sheets of polygonal tumor cells with eosinophilic granular cytoplasm and small central nuclei.3,5 The cytoplasmic granules are periodic acid-Schiff positive and diastase resistant.6 Many cases feature pseudoepitheliomatous hyperplasia, which can misleadingly resemble squamous cell carcinoma.3,5,6 Of note, invasive growth patterns on histology can occur with benign GCTs, as in our patient's case, and do not impact prognosis.3,4 On immunohistochemistry, benign, atypical, and malignant GCTs often stain positive for S-100 protein, vimentin, neuron-specific enolase, SOX10, and CD68.1,3
Although our patient's GCTs were benign, an estimated 1% to 2% are malignant.1,4 In 1998, Fanburg-Smith et al1 defined 6 histologic criteria that characterize malignant GCTs: necrosis, tumor cell spindling, vesicular nuclei with large nucleoli, high nuclear to cytoplasmic ratio, increased mitosis, and pleomorphism. Neoplasms with 3 or more of these features are classified as malignant, those with 1 or 2 are considered atypical, and those with only pleomorphism or no other criteria met are diagnosed as benign.1
Multiple GCTs have been reported in 10% to 25% of cases and, as highlighted in our case, can occur in both a metachronous and synchronous manner.2-4,6 Our patient developed a solitary GCT on the inferior lip 3 years prior to the appearance of 2 additional GCTs within 6 months of each other. The presence of multiple GCTs has been associated with genetic syndromes, such as neurofibromatosis type 1 and Noonan syndrome with multiple lentigines3,8; however, as our case demonstrates, multiple GCTs can occur in nonsyndromic patients as well. When multiple GCTs develop at distant sites, they can resemble metastasis.3 To differentiate these clinical scenarios, Machado et al3 proposed utilizing histology and anatomic location. Multiple tumors with benign characteristics on histology likely represent multiple GCTs, whereas tumors arising at sites common to GCT metastasis, such as lymph node, bone, or viscera, are more concerning for metastatic disease. It has been suggested that patients with multiple GCTs should be monitored with physical examination and repeat magnetic resonance imaging or computed tomography every 6 to 12 months.2 Given our patient's presentation with new tumors arising within 6 months of one another, we recommended a 6-month follow-up interval rather than 1 year. Due to the rarity of GCTs, clinical trials to define treatment guidelines and recommendations have not been performed.3 However, the most commonly utilized treatment modality is wide local excision, as performed in our patient.2,4
Melanoma, atypical fibroxanthoma (AFX), xanthoma, and leiomyosarcoma may be difficult to distinguish from GCT.1,3,4 Melanoma incidence has increased dramatically over the last several decades, with rates in the United States rising from 6.8 cases per 100,000 individuals in the 1970s to 20.1 in the early 2000s. Risk factors for its development include UV radiation exposure and particularly severe sunburns during childhood, along with a number of host risk factors such as total number of melanocytic nevi, family history, and fair complexion.9 Histologically, it often demonstrates irregularly distributed, poorly defined melanocytes with pagetoid spread and dyscohesive nests (Figure 1).10 Melanoma metastasis occasionally can present as a soft-tissue mass and often stains positive for S-100 and vimentin, thus resembling GCT1,4; however, unlike melanoma, GCTs lack melanosomes and stain negative for more specific melanocyte markers, such as melanoma antigen recognized by T cells 1 (MART-1).1,3,4
Atypical fibroxanthoma is a cutaneous neoplasm with fibrohistiocytic mesenchymal origin.11 These tumors typically arise on the head and neck in elderly individuals, particularly men with sun-damaged skin. They often present as superficial, rapidly growing nodules with the potential to ulcerate and bleed.11,12 Histologic features include pleomorphic spindle and epithelioid cells, whose nuclei appear hyperchromatic with atypical mitoses (Figure 2).12 Granular cell changes occur infrequently with AFXs, but in such cases immunohistochemistry can readily distinguish AFX from GCT. Although both tend to stain positive for CD68 and vimentin, AFXs lack S-100 protein and SOX10 expression that frequently is observed in GCTs.3,12
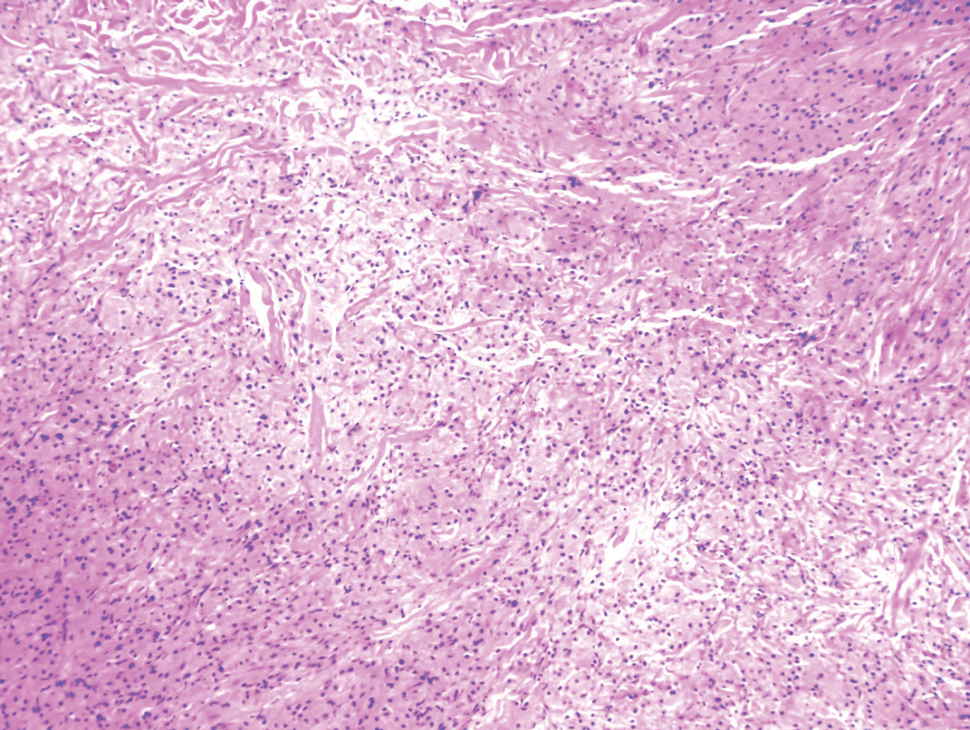

Xanthomas are localized lipid deposits in the connective tissue of the skin that often arise in association with dyslipidemia.13 They typically present as soft to semisolid yellow papules, plaques, or nodules. Their clinical appearance can resemble GCTs; however, histologic analysis enables differentiation with ease, as xanthomas demonstrate characteristic foam cells, consisting of lipid-laden macrophages (Figure 3).13
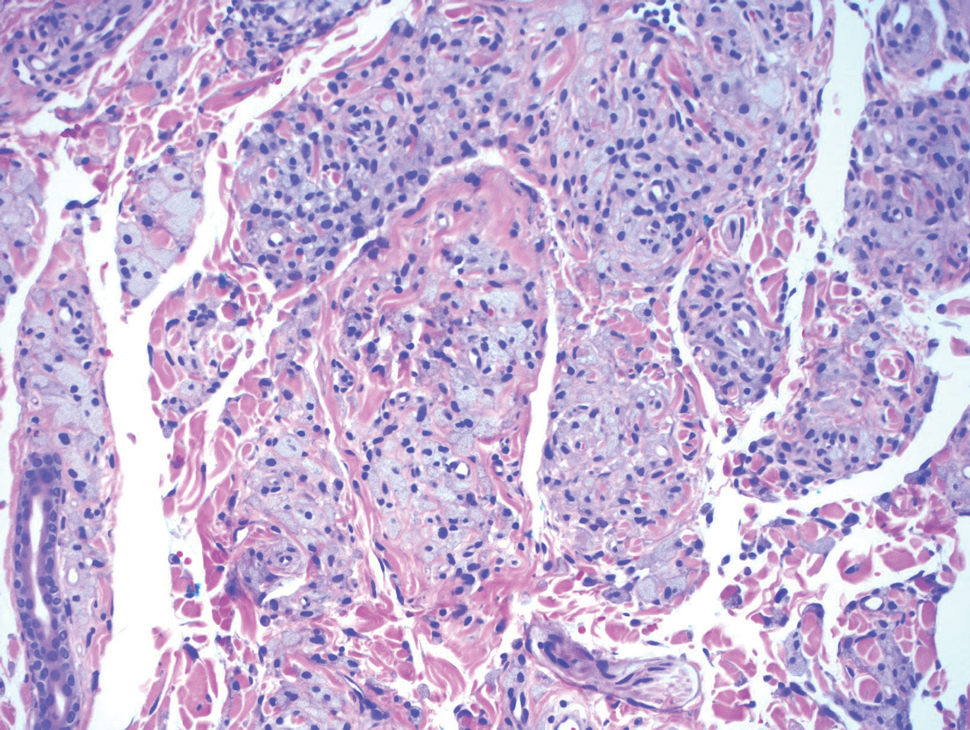
Cutaneous leiomyosarcoma is a rare dermal neoplasm, accounting for 2% to 3% of all sarcomas.14 They typically occur in White males during the fifth to seventh decades of life and often present as asymptomatic lesions on the lower extremities. They frequently arise from pilar smooth muscle. Unlike uterine and soft-tissue leiomyosarcoma, cutaneous leiomyosarcoma tends to follow an indolent course and rarely metastasizes.14 Histologically, these tumors display intersecting, well-defined, spindle-cell fascicles with abundant eosinophilic cytoplasm and cigar-shaped, blunt-ended nuclei (Figure 4).15 Occasionally, leiomyosarcomas can demonstrate cytoplasmic granularity due to lysosome accumulation4; nevertheless, the diagnosis usually can be elucidated by examining more typical histologic areas and utilizing immunohistochemistry, which often stains positive for α-smooth muscle actin, desmin, and h-caldesmon.4,15
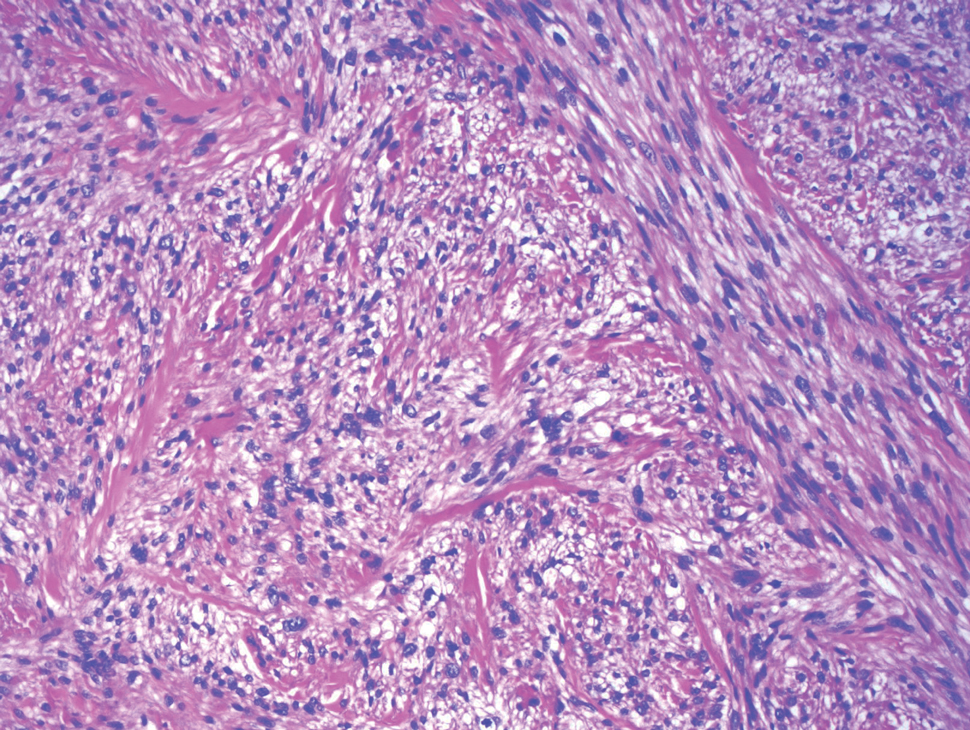
- Fanburg-Smith JC, Meis-Kindblom JM, Fante R, et al. Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol. 1998;22:779-794.
- Moten AS, Movva S, von Mehren M, et al. Granular cell tumor experience at a comprehensive cancer center. J Surg Res. 2018;226:1-7.
- Machado I, Cruz J, Lavernia J, et al. Solitary, multiple, benign, atypical, or malignant: the "granular cell tumor" puzzle. Virchows Arch. 2016;468:527-538.
- Ordóñez NG. Granular cell tumor: a review and update. Adv Anat Pathol. 1999;6:186-203.
- Vaughan V, Ferringer T. Granular cell tumor. Cutis. 2014;94:275, 279-280.
- Van L, Parker SR. Multiple morphologically distinct cutaneous granular cell tumors occurring in a single patient. Cutis. 2016;97:E26-E29.
- Lack EE, Worsham GF, Callihan MD, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
- Bamps S, Oyen T, Legius E, et al. Multiple granular cell tumors in a child with Noonan syndrome. Eur J Pediatr Surg. 2013;23:257-259.
- Rastrelli M, Tropea S, Rossi CR, et al. Melanoma: epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo. 2014;28:1005-1011.
- Smoller BR. Histologic criteria for diagnosing primary cutaneousmalignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40.
- Soleymani T, Aasi SZ, Novoa R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: updates on classification and management. Dermatol Clin. 2019;37:253-259.
- Cardis MA, Ni J, Bhawan J. Granular cell differentiation: a review of the published work. J Dermatol. 2017;44:251-258.
- Zak A, Zeman M, Slaby A, et al. Xanthomas: clinical and pathophysiological relations [published online April 29, 2014]. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:181-188.
- Sandhu N, Sauvageau AP, Groman A, et al. Cutaneous leiomyosarcoma: a SEER database analysis. Dermatol Surg. 2020;46:159-164.
- George S, Serrano C, Hensley ML, et al. Soft tissue and uterine leiomyosarcoma. J Clin Oncol. 2018;36:144-150.
The Diagnosis: Granular Cell Tumor
Histopathologic analysis from the axillary excision demonstrated cords and sheets of large polygonal cells in the dermis with uniform, oval, hyperchromatic nuclei and ample pink granular-staining cytoplasm (quiz images). An infiltrative growth pattern was noted; however, there was no evidence of conspicuous mitoses, nuclear pleomorphism, or necrosis. These results in conjunction with the immunohistochemistry findings were consistent with a benign granular cell tumor (GCT), a rare neoplasm considered to have neural/Schwann cell origin.1-3
Our case demonstrates the difficulty in clinically diagnosing cutaneous GCTs. The tumor often presents as a solitary, 0.5- to 3-cm, asymptomatic, firm nodule4,5; however, GCTs also can appear verrucous, eroded, or with other variable morphologies, which can create diagnostic challenges.5,6 Accordingly, a 1980 study of 110 patients with GCTs found that the preoperative clinical diagnosis was incorrect in all but 3 cases,7 emphasizing the need for histologic evaluation. Benign GCTs tend to exhibit sheets of polygonal tumor cells with eosinophilic granular cytoplasm and small central nuclei.3,5 The cytoplasmic granules are periodic acid-Schiff positive and diastase resistant.6 Many cases feature pseudoepitheliomatous hyperplasia, which can misleadingly resemble squamous cell carcinoma.3,5,6 Of note, invasive growth patterns on histology can occur with benign GCTs, as in our patient's case, and do not impact prognosis.3,4 On immunohistochemistry, benign, atypical, and malignant GCTs often stain positive for S-100 protein, vimentin, neuron-specific enolase, SOX10, and CD68.1,3
Although our patient's GCTs were benign, an estimated 1% to 2% are malignant.1,4 In 1998, Fanburg-Smith et al1 defined 6 histologic criteria that characterize malignant GCTs: necrosis, tumor cell spindling, vesicular nuclei with large nucleoli, high nuclear to cytoplasmic ratio, increased mitosis, and pleomorphism. Neoplasms with 3 or more of these features are classified as malignant, those with 1 or 2 are considered atypical, and those with only pleomorphism or no other criteria met are diagnosed as benign.1
Multiple GCTs have been reported in 10% to 25% of cases and, as highlighted in our case, can occur in both a metachronous and synchronous manner.2-4,6 Our patient developed a solitary GCT on the inferior lip 3 years prior to the appearance of 2 additional GCTs within 6 months of each other. The presence of multiple GCTs has been associated with genetic syndromes, such as neurofibromatosis type 1 and Noonan syndrome with multiple lentigines3,8; however, as our case demonstrates, multiple GCTs can occur in nonsyndromic patients as well. When multiple GCTs develop at distant sites, they can resemble metastasis.3 To differentiate these clinical scenarios, Machado et al3 proposed utilizing histology and anatomic location. Multiple tumors with benign characteristics on histology likely represent multiple GCTs, whereas tumors arising at sites common to GCT metastasis, such as lymph node, bone, or viscera, are more concerning for metastatic disease. It has been suggested that patients with multiple GCTs should be monitored with physical examination and repeat magnetic resonance imaging or computed tomography every 6 to 12 months.2 Given our patient's presentation with new tumors arising within 6 months of one another, we recommended a 6-month follow-up interval rather than 1 year. Due to the rarity of GCTs, clinical trials to define treatment guidelines and recommendations have not been performed.3 However, the most commonly utilized treatment modality is wide local excision, as performed in our patient.2,4
Melanoma, atypical fibroxanthoma (AFX), xanthoma, and leiomyosarcoma may be difficult to distinguish from GCT.1,3,4 Melanoma incidence has increased dramatically over the last several decades, with rates in the United States rising from 6.8 cases per 100,000 individuals in the 1970s to 20.1 in the early 2000s. Risk factors for its development include UV radiation exposure and particularly severe sunburns during childhood, along with a number of host risk factors such as total number of melanocytic nevi, family history, and fair complexion.9 Histologically, it often demonstrates irregularly distributed, poorly defined melanocytes with pagetoid spread and dyscohesive nests (Figure 1).10 Melanoma metastasis occasionally can present as a soft-tissue mass and often stains positive for S-100 and vimentin, thus resembling GCT1,4; however, unlike melanoma, GCTs lack melanosomes and stain negative for more specific melanocyte markers, such as melanoma antigen recognized by T cells 1 (MART-1).1,3,4
Atypical fibroxanthoma is a cutaneous neoplasm with fibrohistiocytic mesenchymal origin.11 These tumors typically arise on the head and neck in elderly individuals, particularly men with sun-damaged skin. They often present as superficial, rapidly growing nodules with the potential to ulcerate and bleed.11,12 Histologic features include pleomorphic spindle and epithelioid cells, whose nuclei appear hyperchromatic with atypical mitoses (Figure 2).12 Granular cell changes occur infrequently with AFXs, but in such cases immunohistochemistry can readily distinguish AFX from GCT. Although both tend to stain positive for CD68 and vimentin, AFXs lack S-100 protein and SOX10 expression that frequently is observed in GCTs.3,12
Xanthomas are localized lipid deposits in the connective tissue of the skin that often arise in association with dyslipidemia.13 They typically present as soft to semisolid yellow papules, plaques, or nodules. Their clinical appearance can resemble GCTs; however, histologic analysis enables differentiation with ease, as xanthomas demonstrate characteristic foam cells, consisting of lipid-laden macrophages (Figure 3).13
Cutaneous leiomyosarcoma is a rare dermal neoplasm, accounting for 2% to 3% of all sarcomas.14 They typically occur in White males during the fifth to seventh decades of life and often present as asymptomatic lesions on the lower extremities. They frequently arise from pilar smooth muscle. Unlike uterine and soft-tissue leiomyosarcoma, cutaneous leiomyosarcoma tends to follow an indolent course and rarely metastasizes.14 Histologically, these tumors display intersecting, well-defined, spindle-cell fascicles with abundant eosinophilic cytoplasm and cigar-shaped, blunt-ended nuclei (Figure 4).15 Occasionally, leiomyosarcomas can demonstrate cytoplasmic granularity due to lysosome accumulation4; nevertheless, the diagnosis usually can be elucidated by examining more typical histologic areas and utilizing immunohistochemistry, which often stains positive for α-smooth muscle actin, desmin, and h-caldesmon.4,15
The Diagnosis: Granular Cell Tumor
Histopathologic analysis from the axillary excision demonstrated cords and sheets of large polygonal cells in the dermis with uniform, oval, hyperchromatic nuclei and ample pink granular-staining cytoplasm (quiz images). An infiltrative growth pattern was noted; however, there was no evidence of conspicuous mitoses, nuclear pleomorphism, or necrosis. These results in conjunction with the immunohistochemistry findings were consistent with a benign granular cell tumor (GCT), a rare neoplasm considered to have neural/Schwann cell origin.1-3
Our case demonstrates the difficulty in clinically diagnosing cutaneous GCTs. The tumor often presents as a solitary, 0.5- to 3-cm, asymptomatic, firm nodule4,5; however, GCTs also can appear verrucous, eroded, or with other variable morphologies, which can create diagnostic challenges.5,6 Accordingly, a 1980 study of 110 patients with GCTs found that the preoperative clinical diagnosis was incorrect in all but 3 cases,7 emphasizing the need for histologic evaluation. Benign GCTs tend to exhibit sheets of polygonal tumor cells with eosinophilic granular cytoplasm and small central nuclei.3,5 The cytoplasmic granules are periodic acid-Schiff positive and diastase resistant.6 Many cases feature pseudoepitheliomatous hyperplasia, which can misleadingly resemble squamous cell carcinoma.3,5,6 Of note, invasive growth patterns on histology can occur with benign GCTs, as in our patient's case, and do not impact prognosis.3,4 On immunohistochemistry, benign, atypical, and malignant GCTs often stain positive for S-100 protein, vimentin, neuron-specific enolase, SOX10, and CD68.1,3
Although our patient's GCTs were benign, an estimated 1% to 2% are malignant.1,4 In 1998, Fanburg-Smith et al1 defined 6 histologic criteria that characterize malignant GCTs: necrosis, tumor cell spindling, vesicular nuclei with large nucleoli, high nuclear to cytoplasmic ratio, increased mitosis, and pleomorphism. Neoplasms with 3 or more of these features are classified as malignant, those with 1 or 2 are considered atypical, and those with only pleomorphism or no other criteria met are diagnosed as benign.1
Multiple GCTs have been reported in 10% to 25% of cases and, as highlighted in our case, can occur in both a metachronous and synchronous manner.2-4,6 Our patient developed a solitary GCT on the inferior lip 3 years prior to the appearance of 2 additional GCTs within 6 months of each other. The presence of multiple GCTs has been associated with genetic syndromes, such as neurofibromatosis type 1 and Noonan syndrome with multiple lentigines3,8; however, as our case demonstrates, multiple GCTs can occur in nonsyndromic patients as well. When multiple GCTs develop at distant sites, they can resemble metastasis.3 To differentiate these clinical scenarios, Machado et al3 proposed utilizing histology and anatomic location. Multiple tumors with benign characteristics on histology likely represent multiple GCTs, whereas tumors arising at sites common to GCT metastasis, such as lymph node, bone, or viscera, are more concerning for metastatic disease. It has been suggested that patients with multiple GCTs should be monitored with physical examination and repeat magnetic resonance imaging or computed tomography every 6 to 12 months.2 Given our patient's presentation with new tumors arising within 6 months of one another, we recommended a 6-month follow-up interval rather than 1 year. Due to the rarity of GCTs, clinical trials to define treatment guidelines and recommendations have not been performed.3 However, the most commonly utilized treatment modality is wide local excision, as performed in our patient.2,4
Melanoma, atypical fibroxanthoma (AFX), xanthoma, and leiomyosarcoma may be difficult to distinguish from GCT.1,3,4 Melanoma incidence has increased dramatically over the last several decades, with rates in the United States rising from 6.8 cases per 100,000 individuals in the 1970s to 20.1 in the early 2000s. Risk factors for its development include UV radiation exposure and particularly severe sunburns during childhood, along with a number of host risk factors such as total number of melanocytic nevi, family history, and fair complexion.9 Histologically, it often demonstrates irregularly distributed, poorly defined melanocytes with pagetoid spread and dyscohesive nests (Figure 1).10 Melanoma metastasis occasionally can present as a soft-tissue mass and often stains positive for S-100 and vimentin, thus resembling GCT1,4; however, unlike melanoma, GCTs lack melanosomes and stain negative for more specific melanocyte markers, such as melanoma antigen recognized by T cells 1 (MART-1).1,3,4
Atypical fibroxanthoma is a cutaneous neoplasm with fibrohistiocytic mesenchymal origin.11 These tumors typically arise on the head and neck in elderly individuals, particularly men with sun-damaged skin. They often present as superficial, rapidly growing nodules with the potential to ulcerate and bleed.11,12 Histologic features include pleomorphic spindle and epithelioid cells, whose nuclei appear hyperchromatic with atypical mitoses (Figure 2).12 Granular cell changes occur infrequently with AFXs, but in such cases immunohistochemistry can readily distinguish AFX from GCT. Although both tend to stain positive for CD68 and vimentin, AFXs lack S-100 protein and SOX10 expression that frequently is observed in GCTs.3,12
Xanthomas are localized lipid deposits in the connective tissue of the skin that often arise in association with dyslipidemia.13 They typically present as soft to semisolid yellow papules, plaques, or nodules. Their clinical appearance can resemble GCTs; however, histologic analysis enables differentiation with ease, as xanthomas demonstrate characteristic foam cells, consisting of lipid-laden macrophages (Figure 3).13
Cutaneous leiomyosarcoma is a rare dermal neoplasm, accounting for 2% to 3% of all sarcomas.14 They typically occur in White males during the fifth to seventh decades of life and often present as asymptomatic lesions on the lower extremities. They frequently arise from pilar smooth muscle. Unlike uterine and soft-tissue leiomyosarcoma, cutaneous leiomyosarcoma tends to follow an indolent course and rarely metastasizes.14 Histologically, these tumors display intersecting, well-defined, spindle-cell fascicles with abundant eosinophilic cytoplasm and cigar-shaped, blunt-ended nuclei (Figure 4).15 Occasionally, leiomyosarcomas can demonstrate cytoplasmic granularity due to lysosome accumulation4; nevertheless, the diagnosis usually can be elucidated by examining more typical histologic areas and utilizing immunohistochemistry, which often stains positive for α-smooth muscle actin, desmin, and h-caldesmon.4,15
- Fanburg-Smith JC, Meis-Kindblom JM, Fante R, et al. Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol. 1998;22:779-794.
- Moten AS, Movva S, von Mehren M, et al. Granular cell tumor experience at a comprehensive cancer center. J Surg Res. 2018;226:1-7.
- Machado I, Cruz J, Lavernia J, et al. Solitary, multiple, benign, atypical, or malignant: the "granular cell tumor" puzzle. Virchows Arch. 2016;468:527-538.
- Ordóñez NG. Granular cell tumor: a review and update. Adv Anat Pathol. 1999;6:186-203.
- Vaughan V, Ferringer T. Granular cell tumor. Cutis. 2014;94:275, 279-280.
- Van L, Parker SR. Multiple morphologically distinct cutaneous granular cell tumors occurring in a single patient. Cutis. 2016;97:E26-E29.
- Lack EE, Worsham GF, Callihan MD, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
- Bamps S, Oyen T, Legius E, et al. Multiple granular cell tumors in a child with Noonan syndrome. Eur J Pediatr Surg. 2013;23:257-259.
- Rastrelli M, Tropea S, Rossi CR, et al. Melanoma: epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo. 2014;28:1005-1011.
- Smoller BR. Histologic criteria for diagnosing primary cutaneousmalignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40.
- Soleymani T, Aasi SZ, Novoa R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: updates on classification and management. Dermatol Clin. 2019;37:253-259.
- Cardis MA, Ni J, Bhawan J. Granular cell differentiation: a review of the published work. J Dermatol. 2017;44:251-258.
- Zak A, Zeman M, Slaby A, et al. Xanthomas: clinical and pathophysiological relations [published online April 29, 2014]. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:181-188.
- Sandhu N, Sauvageau AP, Groman A, et al. Cutaneous leiomyosarcoma: a SEER database analysis. Dermatol Surg. 2020;46:159-164.
- George S, Serrano C, Hensley ML, et al. Soft tissue and uterine leiomyosarcoma. J Clin Oncol. 2018;36:144-150.
- Fanburg-Smith JC, Meis-Kindblom JM, Fante R, et al. Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol. 1998;22:779-794.
- Moten AS, Movva S, von Mehren M, et al. Granular cell tumor experience at a comprehensive cancer center. J Surg Res. 2018;226:1-7.
- Machado I, Cruz J, Lavernia J, et al. Solitary, multiple, benign, atypical, or malignant: the "granular cell tumor" puzzle. Virchows Arch. 2016;468:527-538.
- Ordóñez NG. Granular cell tumor: a review and update. Adv Anat Pathol. 1999;6:186-203.
- Vaughan V, Ferringer T. Granular cell tumor. Cutis. 2014;94:275, 279-280.
- Van L, Parker SR. Multiple morphologically distinct cutaneous granular cell tumors occurring in a single patient. Cutis. 2016;97:E26-E29.
- Lack EE, Worsham GF, Callihan MD, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
- Bamps S, Oyen T, Legius E, et al. Multiple granular cell tumors in a child with Noonan syndrome. Eur J Pediatr Surg. 2013;23:257-259.
- Rastrelli M, Tropea S, Rossi CR, et al. Melanoma: epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo. 2014;28:1005-1011.
- Smoller BR. Histologic criteria for diagnosing primary cutaneousmalignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40.
- Soleymani T, Aasi SZ, Novoa R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: updates on classification and management. Dermatol Clin. 2019;37:253-259.
- Cardis MA, Ni J, Bhawan J. Granular cell differentiation: a review of the published work. J Dermatol. 2017;44:251-258.
- Zak A, Zeman M, Slaby A, et al. Xanthomas: clinical and pathophysiological relations [published online April 29, 2014]. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:181-188.
- Sandhu N, Sauvageau AP, Groman A, et al. Cutaneous leiomyosarcoma: a SEER database analysis. Dermatol Surg. 2020;46:159-164.
- George S, Serrano C, Hensley ML, et al. Soft tissue and uterine leiomyosarcoma. J Clin Oncol. 2018;36:144-150.

A 26-year-old woman with a history of dysplastic nevi with severe atypia presented with a growth on the lower lip of 3 years’ duration. She denied any inciting event, such as prior trauma to the area, and reported that the lesion had been asymptomatic without a notable change in size. Physical examination revealed a translucent, soft, compressible cystic papule on the left inferior vermilion lip. Wide local excision following incisional biopsy was performed. Six months later, the patient returned to our clinic with a lesion on the right lateral tongue of 6 weeks’ duration as well as a 1-cm subcutaneous cyst in the left axilla of 6 months’ duration. Excisional biopsies of both lesions were performed for histopathologic analysis. Pathology results were similar among the lip, tongue, and axillary lesions. Immunohistochemistry revealed strong positive staining with antibodies to S-100 protein, SOX10, and CD68.