User login
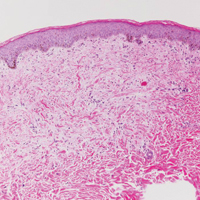
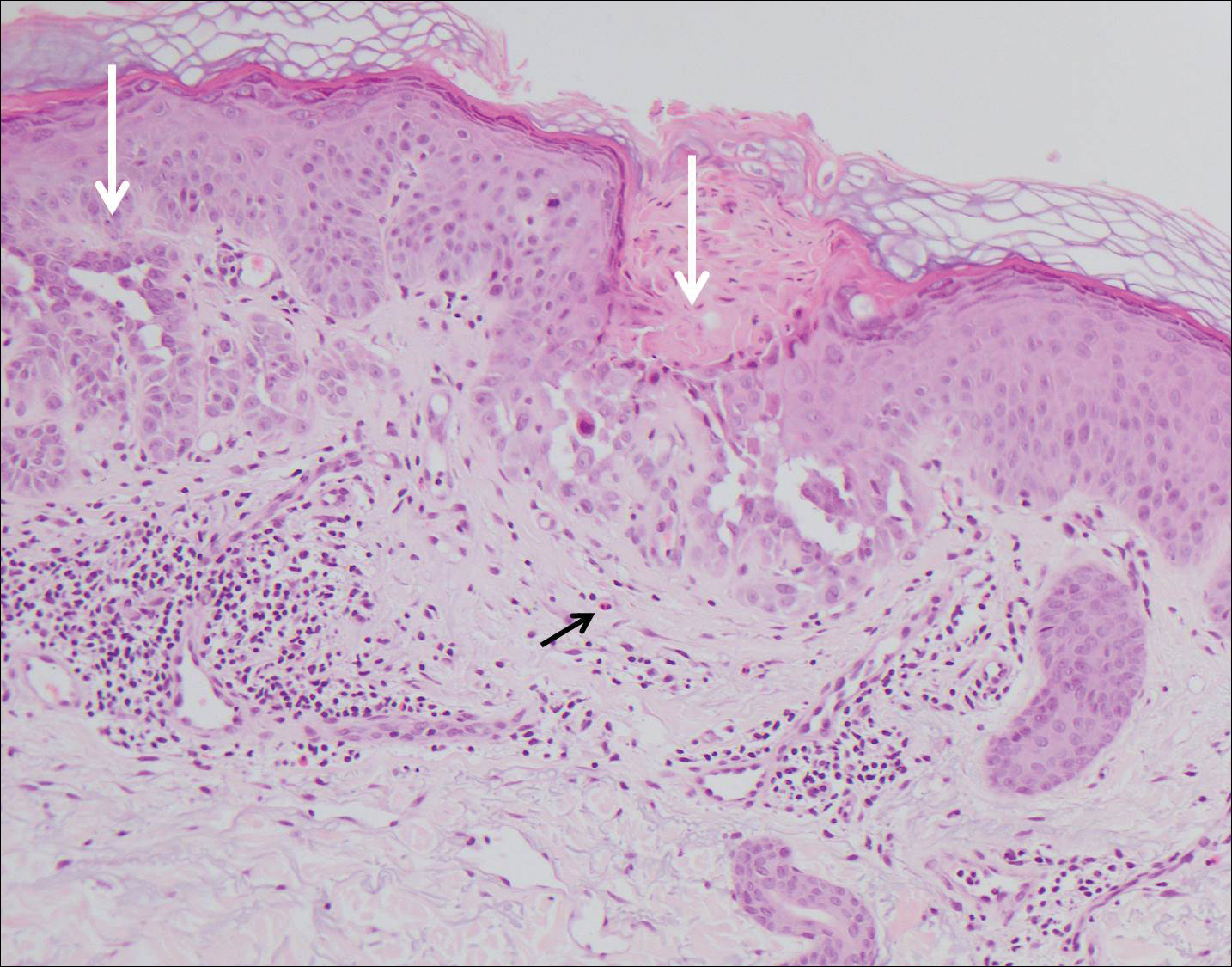
Verrucoid Lesion on the Eyelid
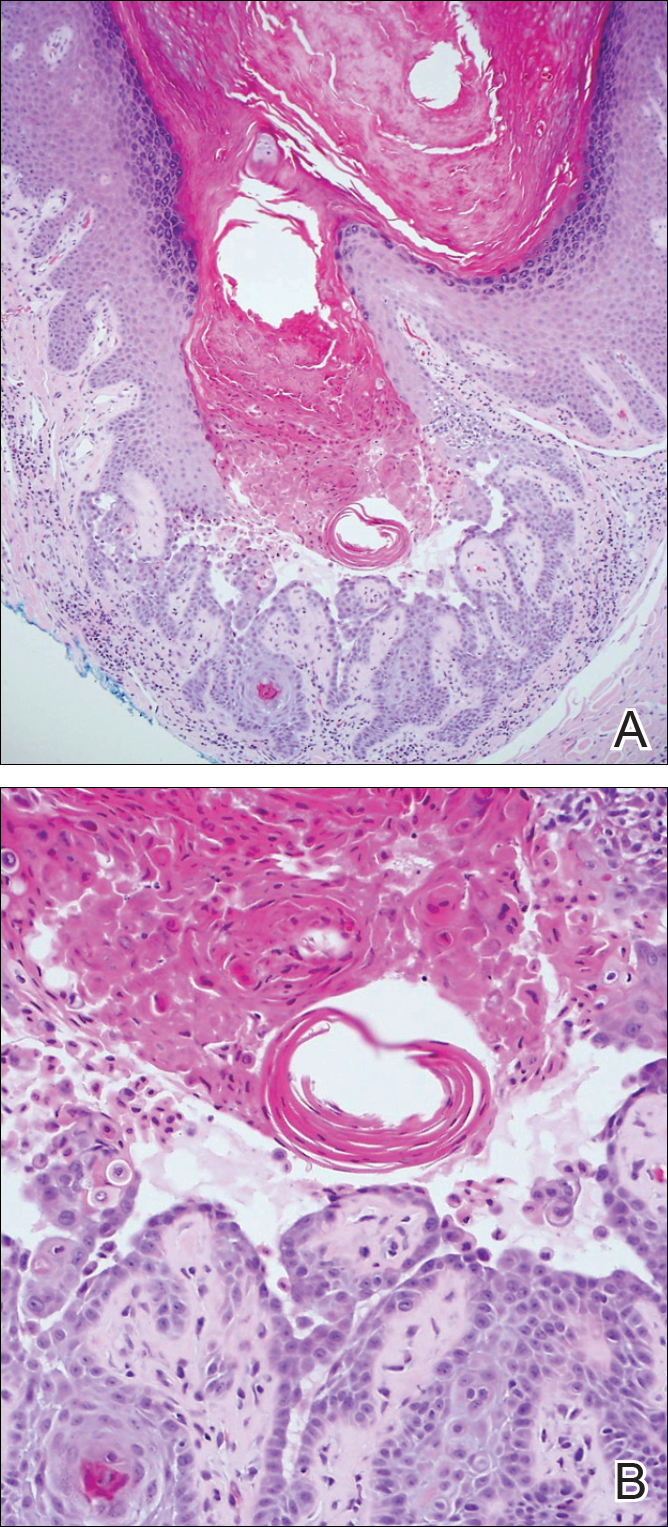
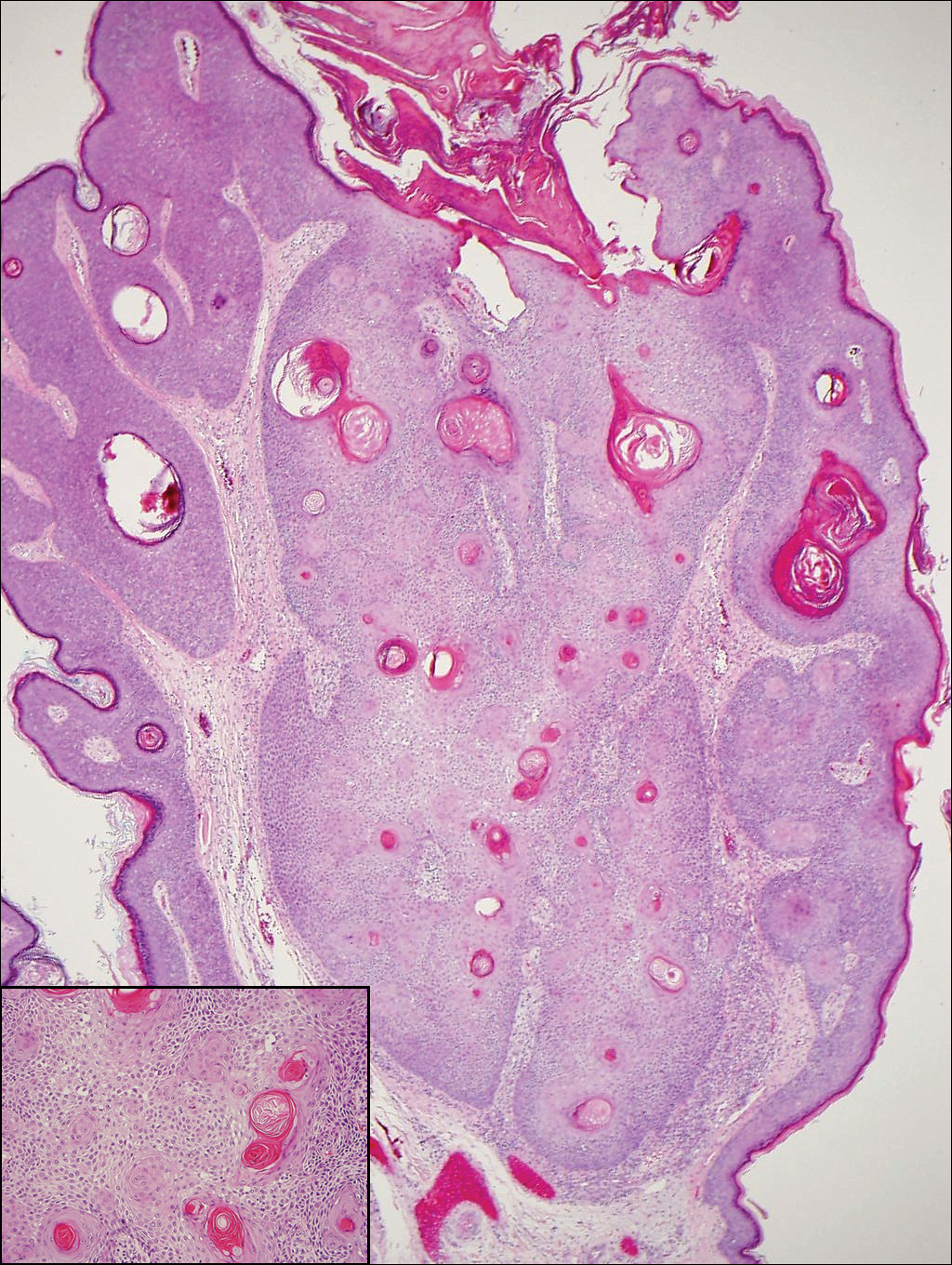
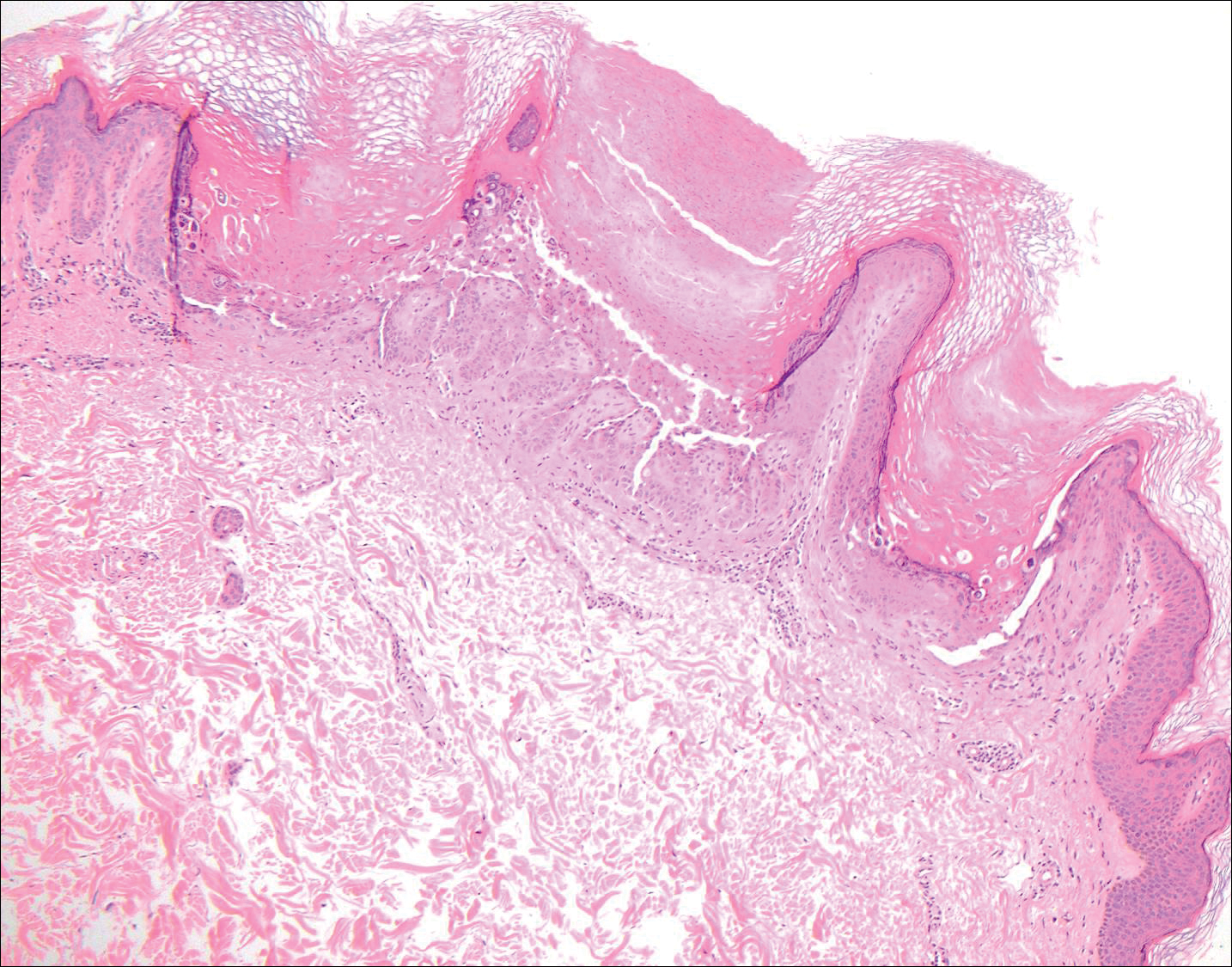
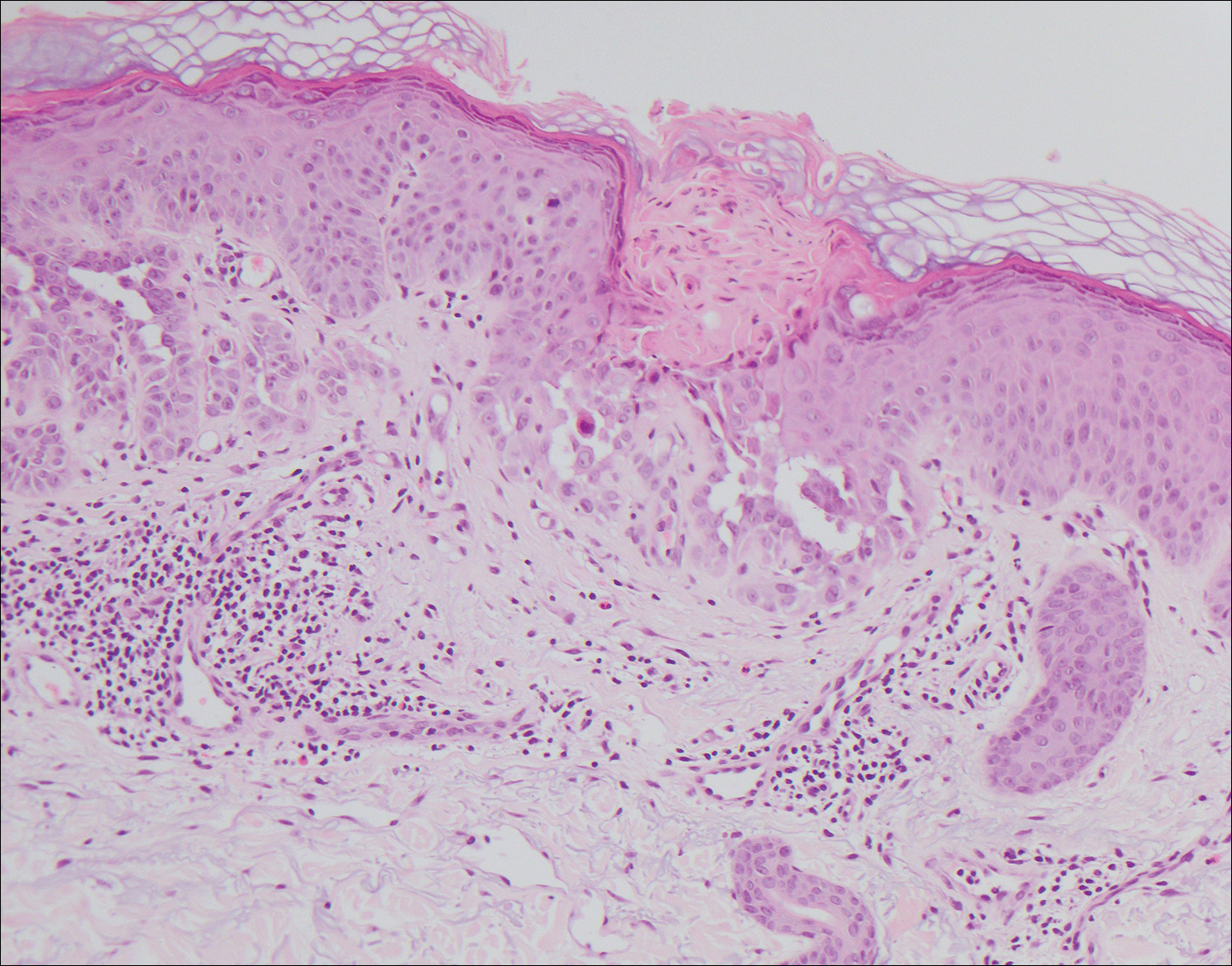
The Diagnosis: Inverted Follicular Keratosis
The differential diagnosis for endophytic squamous neoplasms encompasses benign and malignant entities. The histologic findings of our patient's lesion were compatible with the diagnosis of inverted follicular keratosis (IFK), a benign neoplasm that usually presents as a keratotic papule on the head or neck. Histologically, IFK is characterized by an endophytic growth pattern with squamous eddies (quiz images). Inverted follicular keratosis may represent an irritated seborrheic keratosis or a distinct neoplasm derived from the infundibular portion of the hair follicle; the exact etiology is uncertain.1,2 No relationship between IFK and human papillomavirus (HPV) has been established.3 Inverted follicular keratosis can mimic squamous cell carcinoma (SCC). Important clues to the diagnosis of IFK are the presence of squamous eddies and the lack of squamous pearls or cytologic atypia.4 Squamous eddies consist of whorled keratinocytes without keratinization or atypia. Superficial shave biopsies may fail to demonstrate the characteristic well-circumscribed architecture and may lead to an erroneous diagnosis.
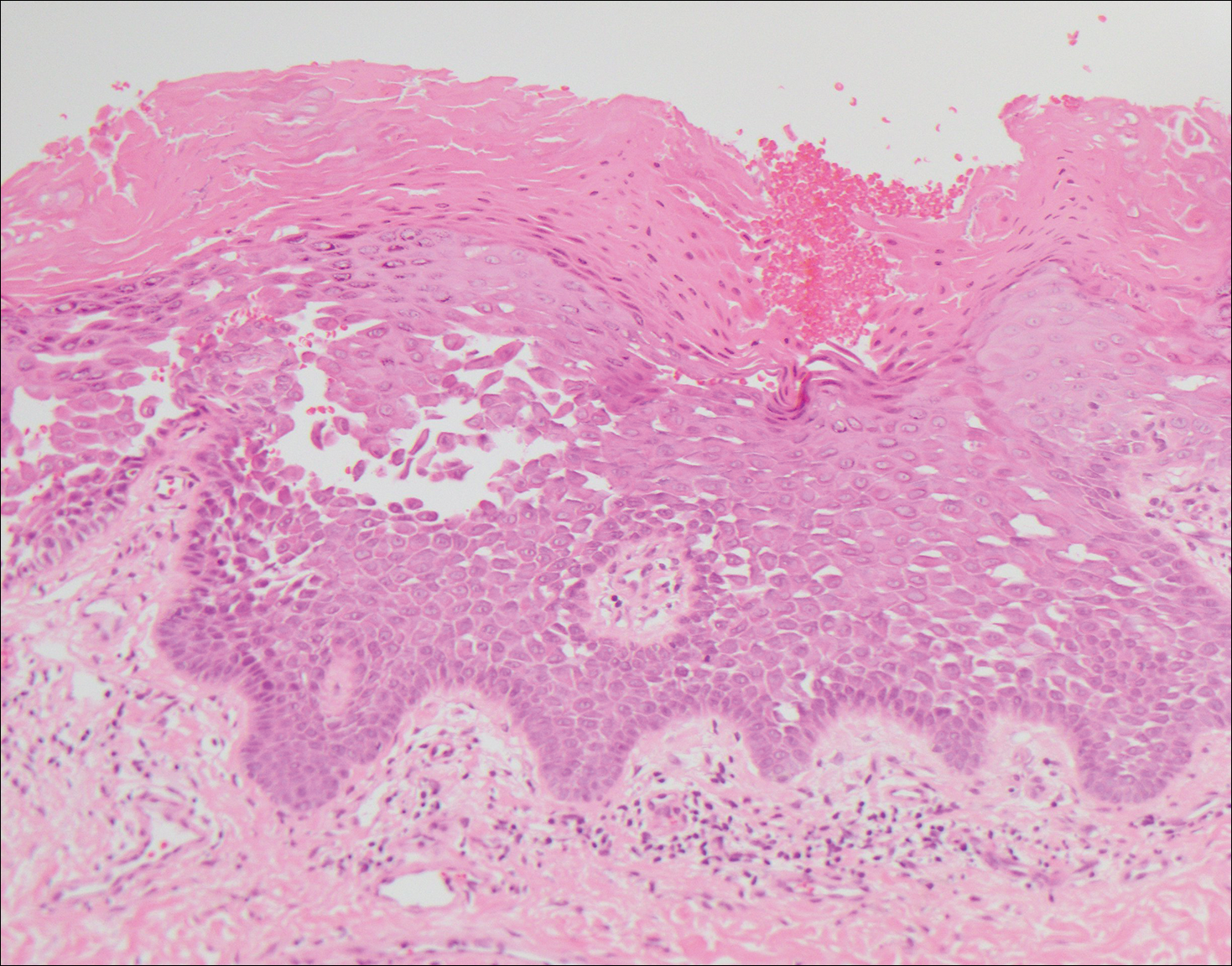
Acantholytic SCC is characterized by atypical keratinocytes that have lost cohesive properties, resulting in acantholysis (Figure 1).5 This histologic variant was once categorized as an aggressive variant of SCC, but studies have failed to support this assertion.5,6 Acantholytic SCC has a discohesive nature producing a pseudoglandular appearance sometimes mistaken for adenosquamous carcinoma or metastatic carcinoma. Recent literature has suggested that acantholytic SCCs, similar to IFKs, are derived from the follicular infundibulum.5,6 Also similar to IFKs, acantholytic SCCs often are located on the face. The invasive architecture and atypical cytology of acantholytic SCCs can differentiate them from IFKs. Acantholytic SCCs can contain keratin pearls with concentric keratinocytes showing incomplete keratinization centrally, often with retained nuclei, but rare to no squamous eddies unless irritated.

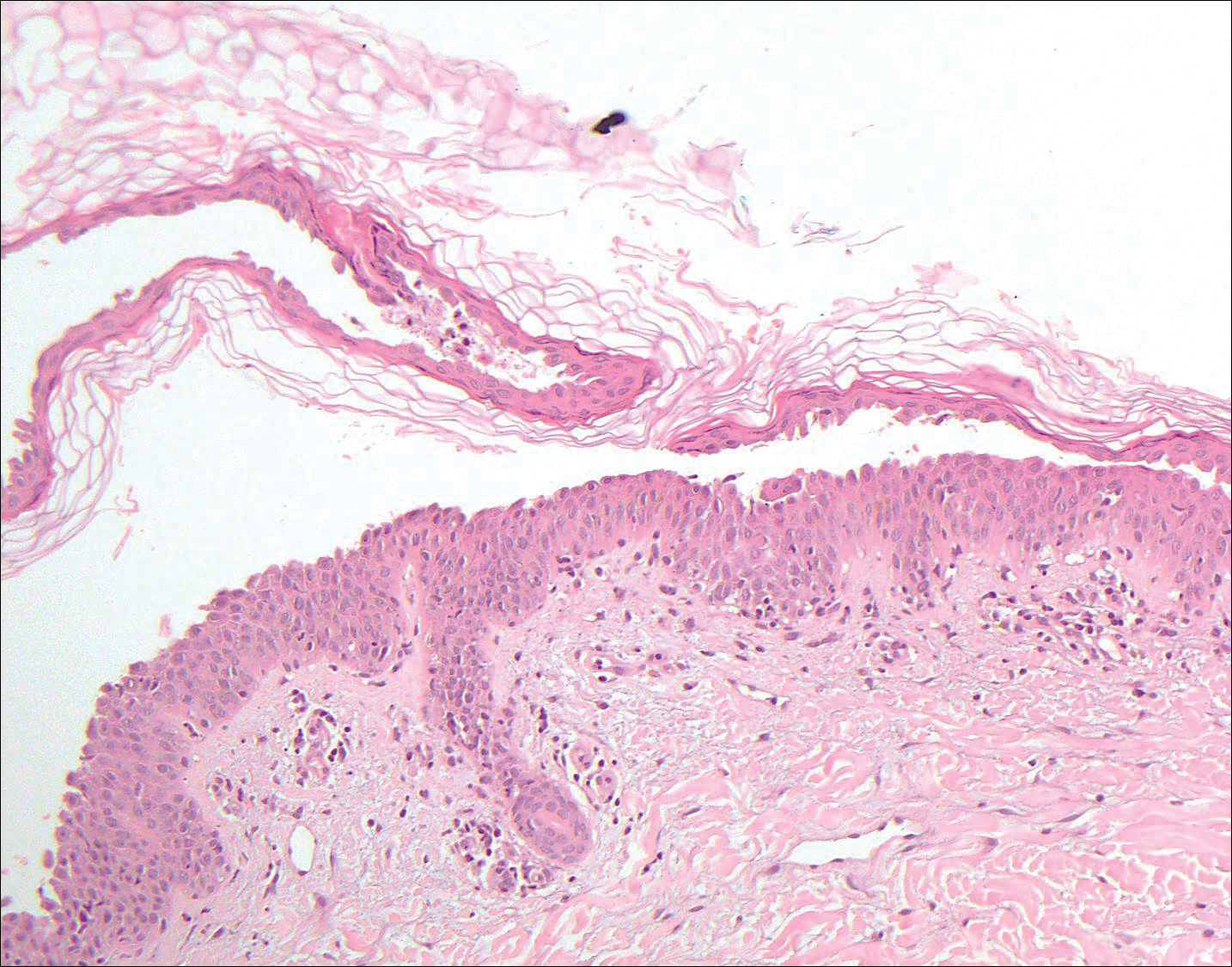
Trichilemmoma is an endophytic benign neoplasm derived from the outer sheath of the pilosebaceous follicle characterized by lobules of clear cells hanging from the epidermis.7 A study investigating the relationship between HPV and trichilemmomas failed to definitively detect HPV in trichilemmomas and this relationship remains unclear.8 Desmoplastic trichilemmoma is a subtype histologically characterized by jagged islands of epithelial cells separated by dense pink stroma and encased in a glassy basement membrane (Figure 2). The presence of desmoplasia and a jagged growth pattern can mimic invasive SCC, but the absence of cytologic atypia and the surrounding basement membrane differs from SCC.4,7 Trichilemmomas typically are solitary, but multiple lesions are associated with Cowden syndrome. Cowden syndrome is a rare autosomal-dominant condition characterized by the presence of benign hamartomas and a predisposition to the development of malignancies including breast, endometrial, and thyroid cancers.9,10 There is no such association with desmoplastic trichilemmomas.11

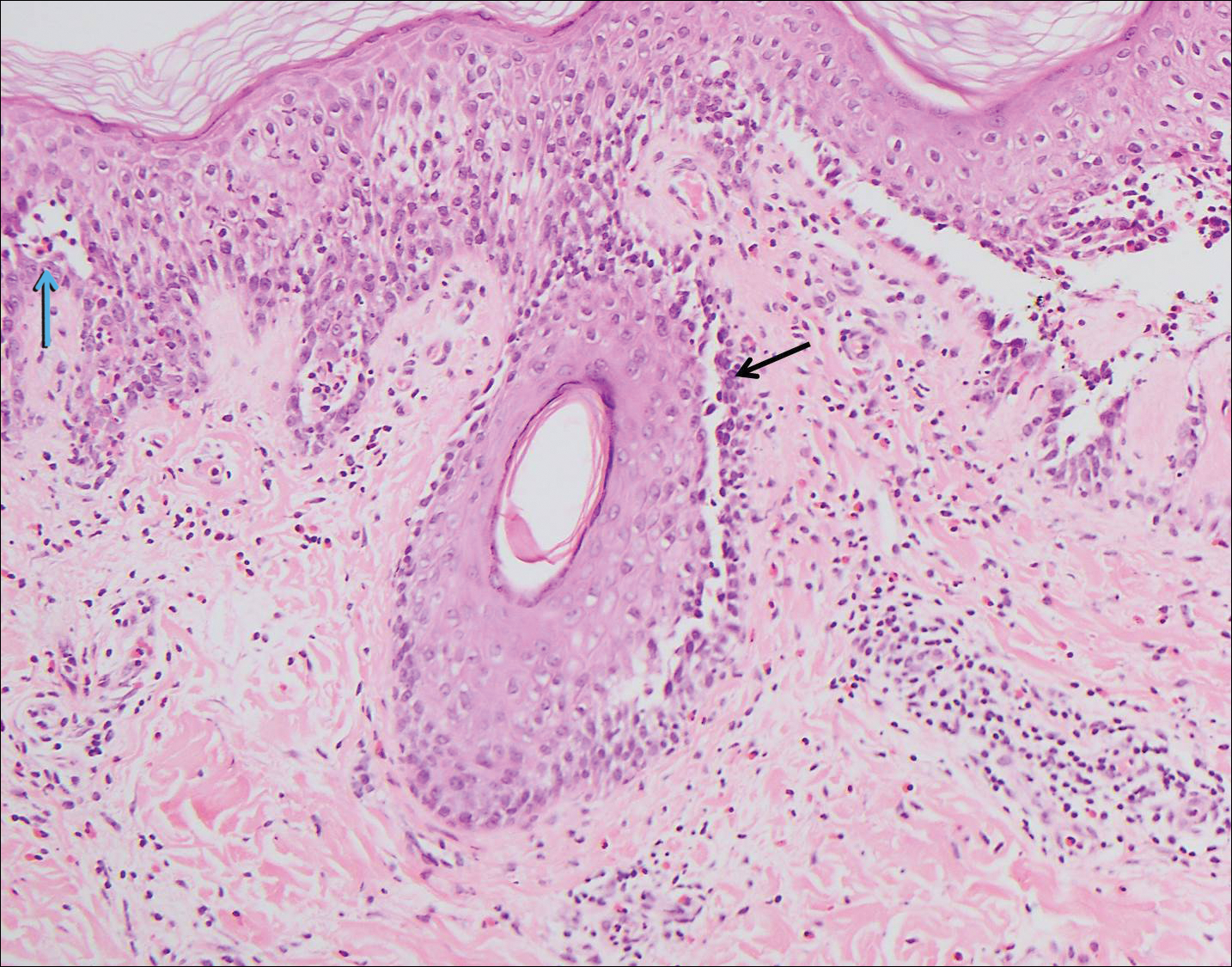
Pilar sheath acanthoma is a benign neoplasm that clinically presents as a solitary flesh-colored nodule with a central pore containing keratin.12 Histologically, pilar sheath acanthoma is similar to a dilated pore of Winer with the addition of acanthotic epidermal projections (Figure 3).

Warty dyskeratoma (WD) is a benign endophytic neoplasm traditionally seen as a solitary lesion histologically similar to Darier disease. Warty dyskeratomas are known to occur both on the skin and oral mucosa.13 Histologically, WD is characterized as a cup-shaped lesion with numerous villi at the base of the lesion along with acantholysis and dyskeratosis (Figure 4). The dyskeratotic cells in WD consist of corps ronds, which are cells with abundant pink cytoplasm, and small nuclei along with grains, which are flattened basophilic cells. These dyskeratotic cells help differentiate WD from IFK. Although they are endophytic neoplasms, WDs are well circumscribed and should not be confused with SCC. Despite this entity's name and histologic similarity to verrucae, no relationship with HPV has been established.14
- Ruhoy SM, Thomas D, Nuovo GJ. Multiple inverted follicular keratoses as a presenting sign of Cowden's syndrome: case report with human papillomavirus studies. J Am Acad Dermatol. 2004;51:411-415.
- Lever WF. Inverted follicular keratosis is an irritated seborrheic keratosis. Am J Dermatopathol. 1983;5:474.
- Kambiz KH, Kaveh D, Maede D, et al. Human papillomavirus deoxyribonucleic acid may not be detected in non-genital benign papillomatous skin lesions by polymerase chain reaction. Indian J Dermatol. 2014;59:334-338.
- Tan KB, Tan SH, Aw DC, et al. Simulators of squamous cell carcinoma of the skin: diagnostic challenges on small biopsies and clinicopathological correlation [published online June 25, 2013]. J Skin Cancer. 2013;2013:752864.
- Ogawa T, Kiuru M, Konia TH, et al. Acantholytic squamous cell carcinoma is usually associated with hair follicles, not acantholytic actinic keratosis, and is not "high risk": diagnosis, management, and clinical outcomes in a series of 115 cases. J Am Acad Dermatol. 2017;76:327-333.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer staging guidelines, prognostic factors, and histopathologic variants. Adv Anat Pathol. 2017;24:171-194.
- Sano DT, Yang JJ, Tebcherani AJ, et al. A rare clinical presentation of desmoplastic trichilemmoma mimicking invasive carcinoma. An Bras Dermatol. 2014;89:796-798.
- Stierman S, Chen S, Nuovo G, et al. Detection of human papillomavirus infection in trichilemmomas and verrucae using in situ hybridization. J Cutan Pathol. 2010;37:75-80.
- Ngeow J, Eng C. PTEN hamartoma tumor syndrome: clinical risk assessment and management protocol [published online October 22, 2014]. Methods. 2015;77-78:11-19.
- Molvi M, Sharma YK, Dash K. Cowden syndrome: case report, update and proposed diagnostic and surveillance routines. Indian J Dermatol. 2015;60:255-259.
- Jin M, Hampel H, Pilarski R, et al. Phosphatase and tensin homolog immunohistochemical staining and clinical criteria for Cowden syndrome in patients with trichilemmoma or associated lesions. Am J Dermatopathol. 2013;35:637-640.
- Mehregan AH, Brownstein MH. Pilar sheath acanthoma. Arch Dermatol. 1978;114:1495-1497.
- Newland JR, Leventon GS. Warty dyskeratoma of the oral mucosa. correlated light and electron microscopic study. Oral Surg Oral Med Oral Pathol. 1984;58:176-183.
- Kaddu S, Dong H, Mayer G, et al. Warty dyskeratoma--"follicular dyskeratoma": analysis of clinicopathologic features of a distinctive follicular adnexal neoplasm. J Am Acad Dermatol. 2002;47:423-428.
The Diagnosis: Inverted Follicular Keratosis
The differential diagnosis for endophytic squamous neoplasms encompasses benign and malignant entities. The histologic findings of our patient's lesion were compatible with the diagnosis of inverted follicular keratosis (IFK), a benign neoplasm that usually presents as a keratotic papule on the head or neck. Histologically, IFK is characterized by an endophytic growth pattern with squamous eddies (quiz images). Inverted follicular keratosis may represent an irritated seborrheic keratosis or a distinct neoplasm derived from the infundibular portion of the hair follicle; the exact etiology is uncertain.1,2 No relationship between IFK and human papillomavirus (HPV) has been established.3 Inverted follicular keratosis can mimic squamous cell carcinoma (SCC). Important clues to the diagnosis of IFK are the presence of squamous eddies and the lack of squamous pearls or cytologic atypia.4 Squamous eddies consist of whorled keratinocytes without keratinization or atypia. Superficial shave biopsies may fail to demonstrate the characteristic well-circumscribed architecture and may lead to an erroneous diagnosis.
Acantholytic SCC is characterized by atypical keratinocytes that have lost cohesive properties, resulting in acantholysis (Figure 1).5 This histologic variant was once categorized as an aggressive variant of SCC, but studies have failed to support this assertion.5,6 Acantholytic SCC has a discohesive nature producing a pseudoglandular appearance sometimes mistaken for adenosquamous carcinoma or metastatic carcinoma. Recent literature has suggested that acantholytic SCCs, similar to IFKs, are derived from the follicular infundibulum.5,6 Also similar to IFKs, acantholytic SCCs often are located on the face. The invasive architecture and atypical cytology of acantholytic SCCs can differentiate them from IFKs. Acantholytic SCCs can contain keratin pearls with concentric keratinocytes showing incomplete keratinization centrally, often with retained nuclei, but rare to no squamous eddies unless irritated.
Trichilemmoma is an endophytic benign neoplasm derived from the outer sheath of the pilosebaceous follicle characterized by lobules of clear cells hanging from the epidermis.7 A study investigating the relationship between HPV and trichilemmomas failed to definitively detect HPV in trichilemmomas and this relationship remains unclear.8 Desmoplastic trichilemmoma is a subtype histologically characterized by jagged islands of epithelial cells separated by dense pink stroma and encased in a glassy basement membrane (Figure 2). The presence of desmoplasia and a jagged growth pattern can mimic invasive SCC, but the absence of cytologic atypia and the surrounding basement membrane differs from SCC.4,7 Trichilemmomas typically are solitary, but multiple lesions are associated with Cowden syndrome. Cowden syndrome is a rare autosomal-dominant condition characterized by the presence of benign hamartomas and a predisposition to the development of malignancies including breast, endometrial, and thyroid cancers.9,10 There is no such association with desmoplastic trichilemmomas.11
Pilar sheath acanthoma is a benign neoplasm that clinically presents as a solitary flesh-colored nodule with a central pore containing keratin.12 Histologically, pilar sheath acanthoma is similar to a dilated pore of Winer with the addition of acanthotic epidermal projections (Figure 3).
Warty dyskeratoma (WD) is a benign endophytic neoplasm traditionally seen as a solitary lesion histologically similar to Darier disease. Warty dyskeratomas are known to occur both on the skin and oral mucosa.13 Histologically, WD is characterized as a cup-shaped lesion with numerous villi at the base of the lesion along with acantholysis and dyskeratosis (Figure 4). The dyskeratotic cells in WD consist of corps ronds, which are cells with abundant pink cytoplasm, and small nuclei along with grains, which are flattened basophilic cells. These dyskeratotic cells help differentiate WD from IFK. Although they are endophytic neoplasms, WDs are well circumscribed and should not be confused with SCC. Despite this entity's name and histologic similarity to verrucae, no relationship with HPV has been established.14
The Diagnosis: Inverted Follicular Keratosis
The differential diagnosis for endophytic squamous neoplasms encompasses benign and malignant entities. The histologic findings of our patient's lesion were compatible with the diagnosis of inverted follicular keratosis (IFK), a benign neoplasm that usually presents as a keratotic papule on the head or neck. Histologically, IFK is characterized by an endophytic growth pattern with squamous eddies (quiz images). Inverted follicular keratosis may represent an irritated seborrheic keratosis or a distinct neoplasm derived from the infundibular portion of the hair follicle; the exact etiology is uncertain.1,2 No relationship between IFK and human papillomavirus (HPV) has been established.3 Inverted follicular keratosis can mimic squamous cell carcinoma (SCC). Important clues to the diagnosis of IFK are the presence of squamous eddies and the lack of squamous pearls or cytologic atypia.4 Squamous eddies consist of whorled keratinocytes without keratinization or atypia. Superficial shave biopsies may fail to demonstrate the characteristic well-circumscribed architecture and may lead to an erroneous diagnosis.
Acantholytic SCC is characterized by atypical keratinocytes that have lost cohesive properties, resulting in acantholysis (Figure 1).5 This histologic variant was once categorized as an aggressive variant of SCC, but studies have failed to support this assertion.5,6 Acantholytic SCC has a discohesive nature producing a pseudoglandular appearance sometimes mistaken for adenosquamous carcinoma or metastatic carcinoma. Recent literature has suggested that acantholytic SCCs, similar to IFKs, are derived from the follicular infundibulum.5,6 Also similar to IFKs, acantholytic SCCs often are located on the face. The invasive architecture and atypical cytology of acantholytic SCCs can differentiate them from IFKs. Acantholytic SCCs can contain keratin pearls with concentric keratinocytes showing incomplete keratinization centrally, often with retained nuclei, but rare to no squamous eddies unless irritated.
Trichilemmoma is an endophytic benign neoplasm derived from the outer sheath of the pilosebaceous follicle characterized by lobules of clear cells hanging from the epidermis.7 A study investigating the relationship between HPV and trichilemmomas failed to definitively detect HPV in trichilemmomas and this relationship remains unclear.8 Desmoplastic trichilemmoma is a subtype histologically characterized by jagged islands of epithelial cells separated by dense pink stroma and encased in a glassy basement membrane (Figure 2). The presence of desmoplasia and a jagged growth pattern can mimic invasive SCC, but the absence of cytologic atypia and the surrounding basement membrane differs from SCC.4,7 Trichilemmomas typically are solitary, but multiple lesions are associated with Cowden syndrome. Cowden syndrome is a rare autosomal-dominant condition characterized by the presence of benign hamartomas and a predisposition to the development of malignancies including breast, endometrial, and thyroid cancers.9,10 There is no such association with desmoplastic trichilemmomas.11
Pilar sheath acanthoma is a benign neoplasm that clinically presents as a solitary flesh-colored nodule with a central pore containing keratin.12 Histologically, pilar sheath acanthoma is similar to a dilated pore of Winer with the addition of acanthotic epidermal projections (Figure 3).
Warty dyskeratoma (WD) is a benign endophytic neoplasm traditionally seen as a solitary lesion histologically similar to Darier disease. Warty dyskeratomas are known to occur both on the skin and oral mucosa.13 Histologically, WD is characterized as a cup-shaped lesion with numerous villi at the base of the lesion along with acantholysis and dyskeratosis (Figure 4). The dyskeratotic cells in WD consist of corps ronds, which are cells with abundant pink cytoplasm, and small nuclei along with grains, which are flattened basophilic cells. These dyskeratotic cells help differentiate WD from IFK. Although they are endophytic neoplasms, WDs are well circumscribed and should not be confused with SCC. Despite this entity's name and histologic similarity to verrucae, no relationship with HPV has been established.14
- Ruhoy SM, Thomas D, Nuovo GJ. Multiple inverted follicular keratoses as a presenting sign of Cowden's syndrome: case report with human papillomavirus studies. J Am Acad Dermatol. 2004;51:411-415.
- Lever WF. Inverted follicular keratosis is an irritated seborrheic keratosis. Am J Dermatopathol. 1983;5:474.
- Kambiz KH, Kaveh D, Maede D, et al. Human papillomavirus deoxyribonucleic acid may not be detected in non-genital benign papillomatous skin lesions by polymerase chain reaction. Indian J Dermatol. 2014;59:334-338.
- Tan KB, Tan SH, Aw DC, et al. Simulators of squamous cell carcinoma of the skin: diagnostic challenges on small biopsies and clinicopathological correlation [published online June 25, 2013]. J Skin Cancer. 2013;2013:752864.
- Ogawa T, Kiuru M, Konia TH, et al. Acantholytic squamous cell carcinoma is usually associated with hair follicles, not acantholytic actinic keratosis, and is not "high risk": diagnosis, management, and clinical outcomes in a series of 115 cases. J Am Acad Dermatol. 2017;76:327-333.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer staging guidelines, prognostic factors, and histopathologic variants. Adv Anat Pathol. 2017;24:171-194.
- Sano DT, Yang JJ, Tebcherani AJ, et al. A rare clinical presentation of desmoplastic trichilemmoma mimicking invasive carcinoma. An Bras Dermatol. 2014;89:796-798.
- Stierman S, Chen S, Nuovo G, et al. Detection of human papillomavirus infection in trichilemmomas and verrucae using in situ hybridization. J Cutan Pathol. 2010;37:75-80.
- Ngeow J, Eng C. PTEN hamartoma tumor syndrome: clinical risk assessment and management protocol [published online October 22, 2014]. Methods. 2015;77-78:11-19.
- Molvi M, Sharma YK, Dash K. Cowden syndrome: case report, update and proposed diagnostic and surveillance routines. Indian J Dermatol. 2015;60:255-259.
- Jin M, Hampel H, Pilarski R, et al. Phosphatase and tensin homolog immunohistochemical staining and clinical criteria for Cowden syndrome in patients with trichilemmoma or associated lesions. Am J Dermatopathol. 2013;35:637-640.
- Mehregan AH, Brownstein MH. Pilar sheath acanthoma. Arch Dermatol. 1978;114:1495-1497.
- Newland JR, Leventon GS. Warty dyskeratoma of the oral mucosa. correlated light and electron microscopic study. Oral Surg Oral Med Oral Pathol. 1984;58:176-183.
- Kaddu S, Dong H, Mayer G, et al. Warty dyskeratoma--"follicular dyskeratoma": analysis of clinicopathologic features of a distinctive follicular adnexal neoplasm. J Am Acad Dermatol. 2002;47:423-428.
- Ruhoy SM, Thomas D, Nuovo GJ. Multiple inverted follicular keratoses as a presenting sign of Cowden's syndrome: case report with human papillomavirus studies. J Am Acad Dermatol. 2004;51:411-415.
- Lever WF. Inverted follicular keratosis is an irritated seborrheic keratosis. Am J Dermatopathol. 1983;5:474.
- Kambiz KH, Kaveh D, Maede D, et al. Human papillomavirus deoxyribonucleic acid may not be detected in non-genital benign papillomatous skin lesions by polymerase chain reaction. Indian J Dermatol. 2014;59:334-338.
- Tan KB, Tan SH, Aw DC, et al. Simulators of squamous cell carcinoma of the skin: diagnostic challenges on small biopsies and clinicopathological correlation [published online June 25, 2013]. J Skin Cancer. 2013;2013:752864.
- Ogawa T, Kiuru M, Konia TH, et al. Acantholytic squamous cell carcinoma is usually associated with hair follicles, not acantholytic actinic keratosis, and is not "high risk": diagnosis, management, and clinical outcomes in a series of 115 cases. J Am Acad Dermatol. 2017;76:327-333.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer staging guidelines, prognostic factors, and histopathologic variants. Adv Anat Pathol. 2017;24:171-194.
- Sano DT, Yang JJ, Tebcherani AJ, et al. A rare clinical presentation of desmoplastic trichilemmoma mimicking invasive carcinoma. An Bras Dermatol. 2014;89:796-798.
- Stierman S, Chen S, Nuovo G, et al. Detection of human papillomavirus infection in trichilemmomas and verrucae using in situ hybridization. J Cutan Pathol. 2010;37:75-80.
- Ngeow J, Eng C. PTEN hamartoma tumor syndrome: clinical risk assessment and management protocol [published online October 22, 2014]. Methods. 2015;77-78:11-19.
- Molvi M, Sharma YK, Dash K. Cowden syndrome: case report, update and proposed diagnostic and surveillance routines. Indian J Dermatol. 2015;60:255-259.
- Jin M, Hampel H, Pilarski R, et al. Phosphatase and tensin homolog immunohistochemical staining and clinical criteria for Cowden syndrome in patients with trichilemmoma or associated lesions. Am J Dermatopathol. 2013;35:637-640.
- Mehregan AH, Brownstein MH. Pilar sheath acanthoma. Arch Dermatol. 1978;114:1495-1497.
- Newland JR, Leventon GS. Warty dyskeratoma of the oral mucosa. correlated light and electron microscopic study. Oral Surg Oral Med Oral Pathol. 1984;58:176-183.
- Kaddu S, Dong H, Mayer G, et al. Warty dyskeratoma--"follicular dyskeratoma": analysis of clinicopathologic features of a distinctive follicular adnexal neoplasm. J Am Acad Dermatol. 2002;47:423-428.
A 60-year-old man presented with a 3-mm verrucous papule on the right upper eyelid of 2 years' duration.
Orange Nodules on the Scalp
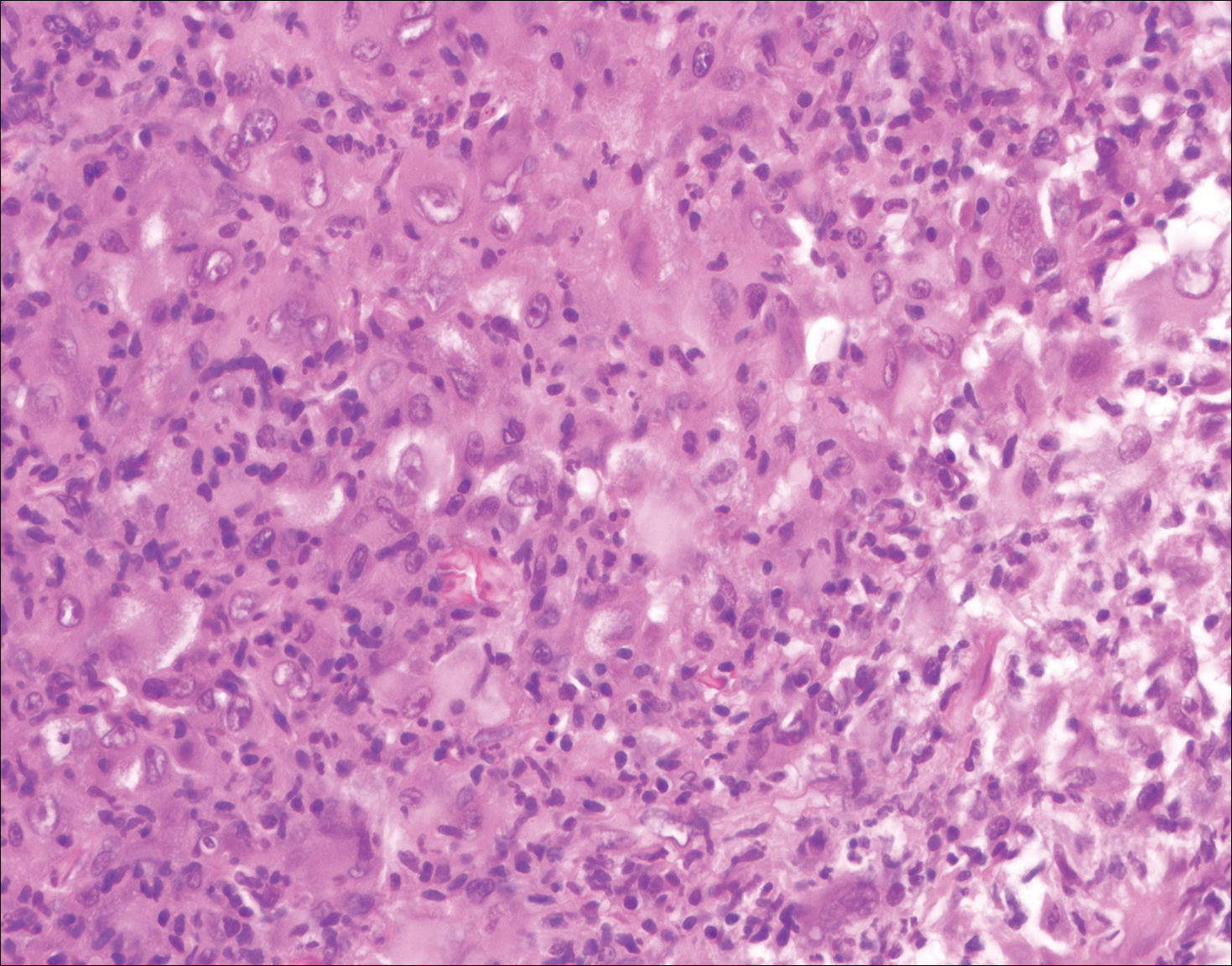
The Diagnosis: Rosai-Dorfman Disease
Rosai-Dorfman disease is a rare histiocytic proliferative disorder of unknown etiology. It has 2 forms: limited cutaneous and systemic. The systemic form, also known as sinus histiocytosis with massive lymphadenopathy, affects the lymph nodes and other organs at times. The disease is characterized by a proliferation of histiocytes in the lymph nodes, most commonly in the cervical basin1; however, the inguinal, axillary, mediastinal, or para-aortic nodes also may be affected.1,2 The skin is the most common site of extranodal disease, seen in approximately 10% of cases.1 Cutaneous involvement often is in the facial area but also can be found on the trunk, ears, neck, arms, legs, and genitals. Clinically, skin lesions appear as papules, plaques, and/or nodules.2
Histopathologic examination of Rosai-Dorfman disease generally shows a dense sheetlike dermal infiltrate of large polygonal histiocytes (Figure 1). Histiocytes may display pale pink or clear cytoplasm. The pathognomonic finding is emperipolesis, which consists of histiocytes with engulfed lymphocytes, erythrocytes, plasma cells, and/or granulocytes surrounded by a clear halo. Immunohistochemical staining also is characteristic, with lesional histiocytes showing expression of S-100 protein (Figure 1, inset) and CD68. The associated inflammatory infiltrate is mixed, containing primarily plasma cells but also lymphocytes, neutrophils, and eosinophils.
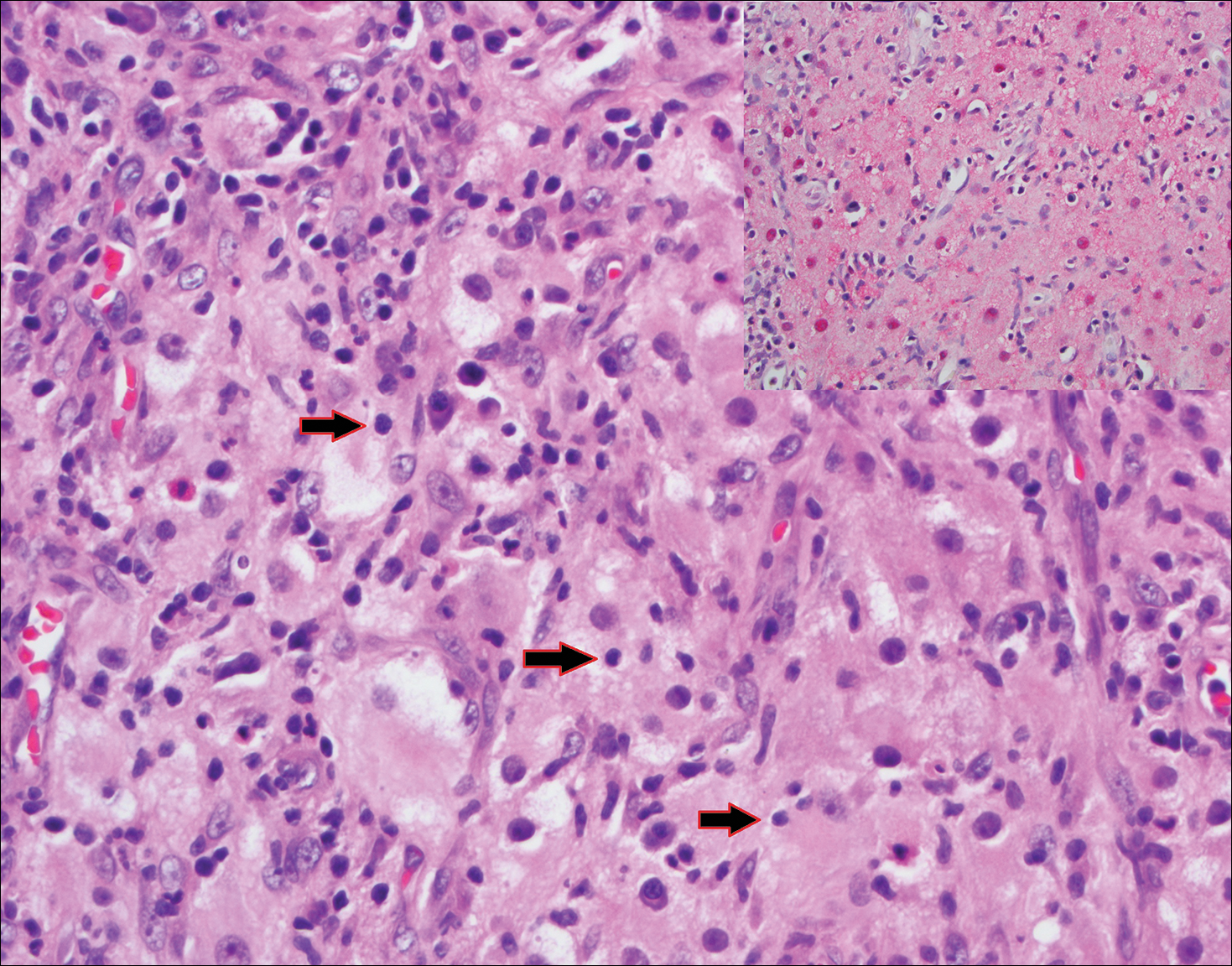
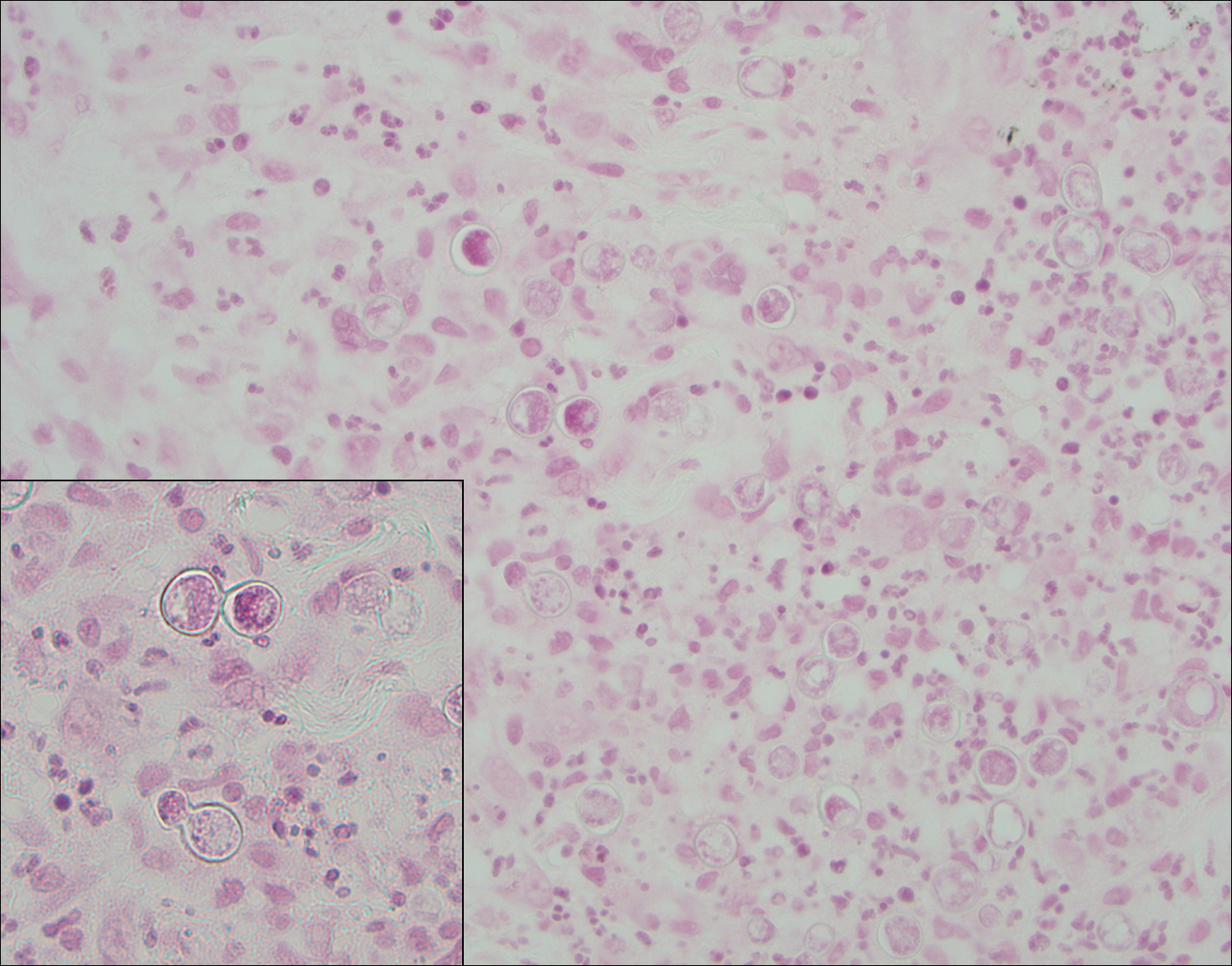
Blastomycosis (Figure 2) is a systemic infection due to inhalation of Blastomyces dermatitidis conidia. Primary infection occurs in the lungs, and with dissemination the skin is the most common subsequently involved organ.3 Cutaneous blastomycosis shows pseudoepitheliomatous hyperplasia with neutrophilic microabscesses and a dense dermal infiltrate containing suppurative granulomatous inflammation. The nonpigmented yeast phase typically is 8 to 15 µm in length with a refractile cell wall and characteristic single, broad-based budding.3
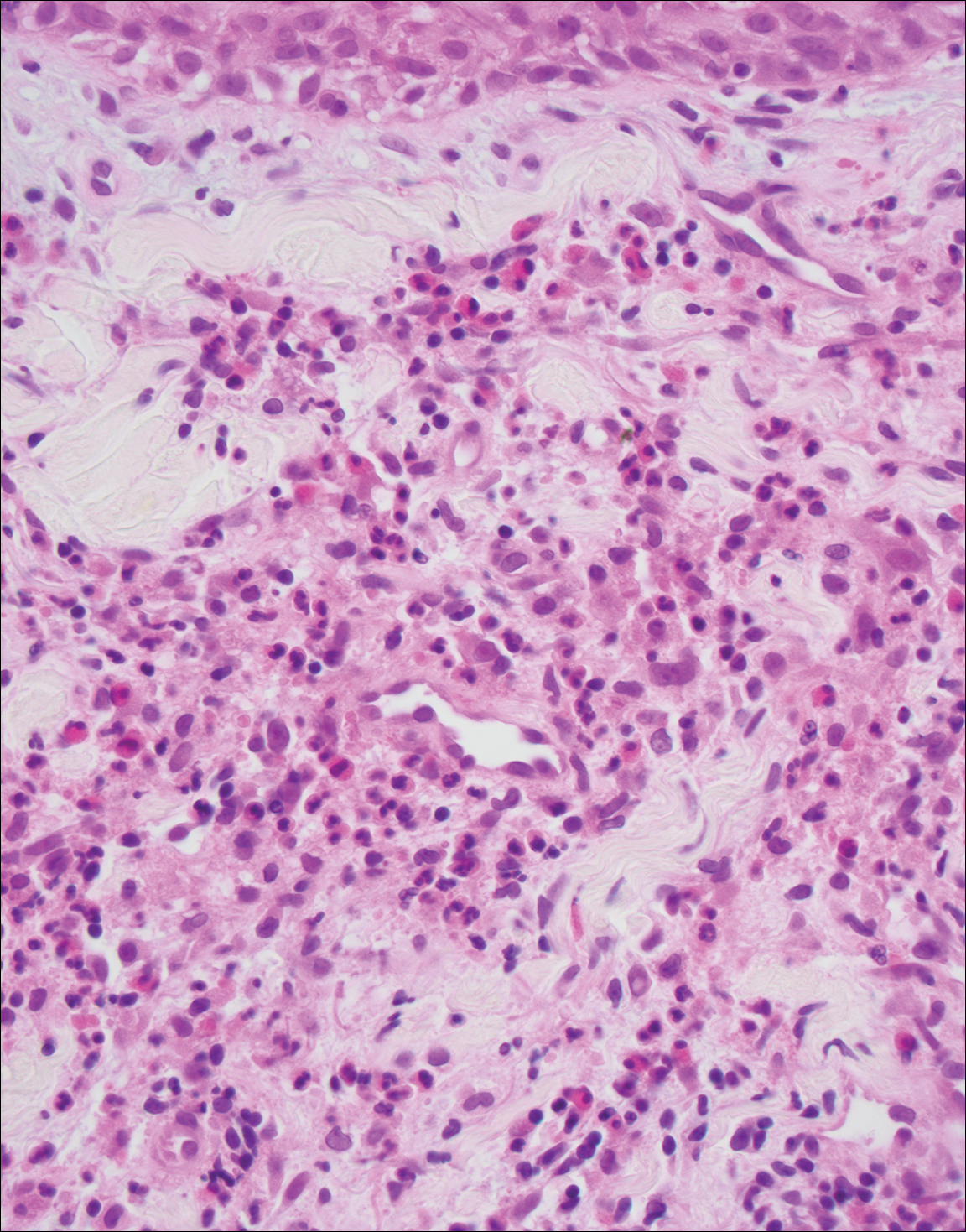
Granuloma faciale (Figure 3) is a rare disease with unknown etiology characterized by reddish brown plaques or nodules most commonly occurring on the face.4,5 Histology shows a dense nodular dermal infiltrate with a grenz zone. The infiltrate is mixed, containing mostly neutrophils with leukocytoclasis and eosinophils. Leukocytoclastic vasculitis is present with associated extravasated erythrocytes. In chronic fibrosing granuloma faciale, lesions can demonstrate fibrosis and hemosiderin deposition, similar to erythema elevatum diutinum.
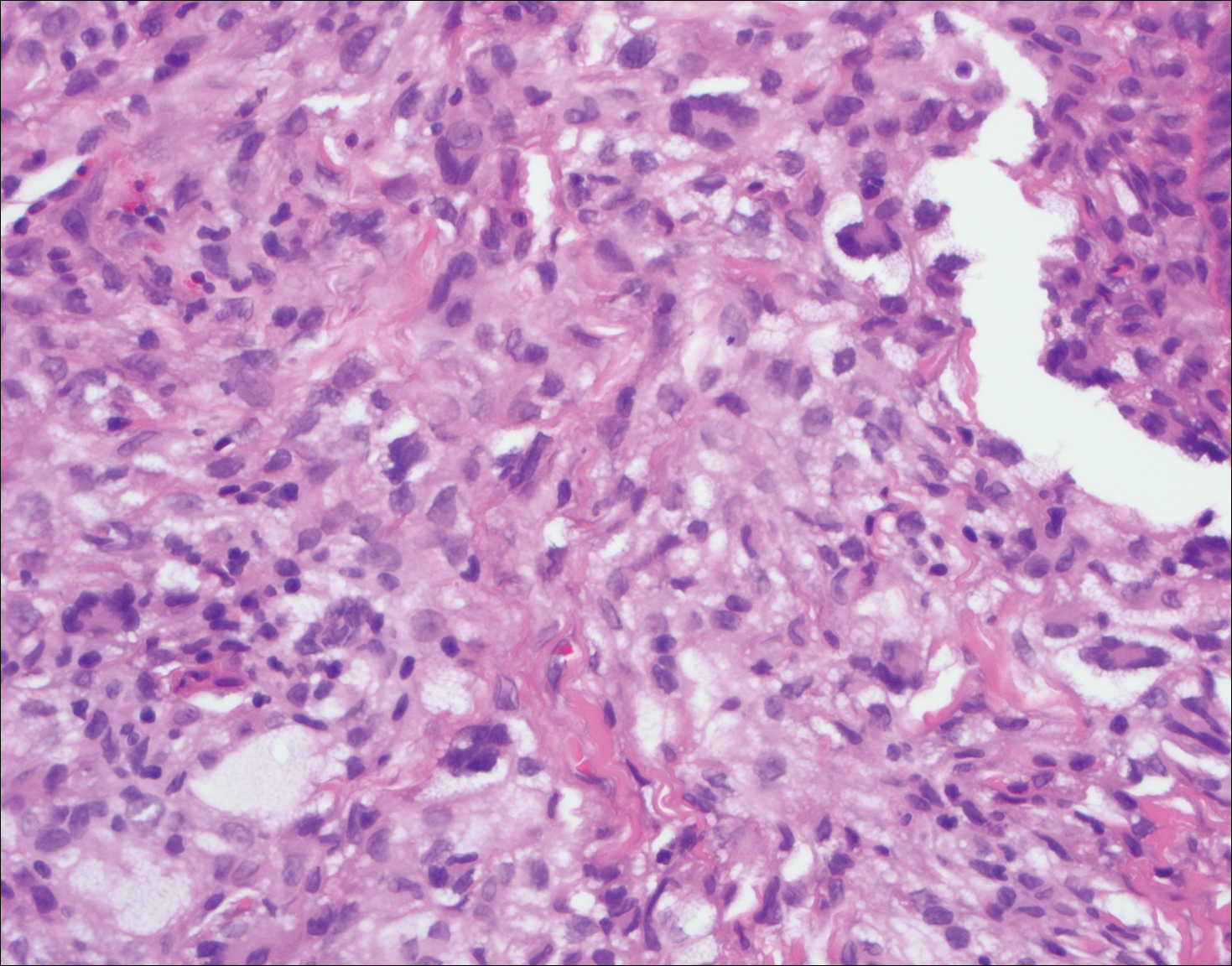
Juvenile xanthogranuloma (Figure 4) is a common histiocytic disease of early childhood, though adult cases have been reported.6 Tumors are found on the head and trunk and are typically firm, reddish yellow papules or nodules.6,7 Histologic examination shows a nodular infiltrate of foamy histiocytes in the superficial dermis. Touton-type multinucleated giant cells with a peripheral rim of xanthomatized foamy cytoplasm and a wreathlike arrangement of nuclei are characteristic. Associated eosinophils are seen. No emperipolesis is present.
Reticulohistiocytoma (Figure 5) is a benign dermal lesion that presents as solitary or less commonly multiple red-brown papules or nodules.8 Lesions consist of well-delineated nodular aggregates of histiocytes containing a finely granular eosinophilic ground glass cytoplasm. Few, if any, eosinophils are found. The lack of Touton multinucleated giant cells or emperipolesis and lack of expression of S-100 protein helps to distinguish reticulohistiocytoma from other entities in the differential diagnosis.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2015.
- Wolff K, Johnson R, Saavedra AP. Fitzpatrick's Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill; 2013.
- Marcoval J, Moreno A, Peyrí J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol. 2004;51:269-273.
- Rodriguez J, Ackerman AB. Xanthogranuloma in adults. Arch Dermatol. 1976;112:43-44.
- Tanz WS, Schwartz RA, Janniger CK. Juvenile xanthogranuloma. Cutis. 1994;54:241-245.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatology Online J. 2014;20. pii:doj_21725.
The Diagnosis: Rosai-Dorfman Disease
Rosai-Dorfman disease is a rare histiocytic proliferative disorder of unknown etiology. It has 2 forms: limited cutaneous and systemic. The systemic form, also known as sinus histiocytosis with massive lymphadenopathy, affects the lymph nodes and other organs at times. The disease is characterized by a proliferation of histiocytes in the lymph nodes, most commonly in the cervical basin1; however, the inguinal, axillary, mediastinal, or para-aortic nodes also may be affected.1,2 The skin is the most common site of extranodal disease, seen in approximately 10% of cases.1 Cutaneous involvement often is in the facial area but also can be found on the trunk, ears, neck, arms, legs, and genitals. Clinically, skin lesions appear as papules, plaques, and/or nodules.2
Histopathologic examination of Rosai-Dorfman disease generally shows a dense sheetlike dermal infiltrate of large polygonal histiocytes (Figure 1). Histiocytes may display pale pink or clear cytoplasm. The pathognomonic finding is emperipolesis, which consists of histiocytes with engulfed lymphocytes, erythrocytes, plasma cells, and/or granulocytes surrounded by a clear halo. Immunohistochemical staining also is characteristic, with lesional histiocytes showing expression of S-100 protein (Figure 1, inset) and CD68. The associated inflammatory infiltrate is mixed, containing primarily plasma cells but also lymphocytes, neutrophils, and eosinophils.
Blastomycosis (Figure 2) is a systemic infection due to inhalation of Blastomyces dermatitidis conidia. Primary infection occurs in the lungs, and with dissemination the skin is the most common subsequently involved organ.3 Cutaneous blastomycosis shows pseudoepitheliomatous hyperplasia with neutrophilic microabscesses and a dense dermal infiltrate containing suppurative granulomatous inflammation. The nonpigmented yeast phase typically is 8 to 15 µm in length with a refractile cell wall and characteristic single, broad-based budding.3
Granuloma faciale (Figure 3) is a rare disease with unknown etiology characterized by reddish brown plaques or nodules most commonly occurring on the face.4,5 Histology shows a dense nodular dermal infiltrate with a grenz zone. The infiltrate is mixed, containing mostly neutrophils with leukocytoclasis and eosinophils. Leukocytoclastic vasculitis is present with associated extravasated erythrocytes. In chronic fibrosing granuloma faciale, lesions can demonstrate fibrosis and hemosiderin deposition, similar to erythema elevatum diutinum.
Juvenile xanthogranuloma (Figure 4) is a common histiocytic disease of early childhood, though adult cases have been reported.6 Tumors are found on the head and trunk and are typically firm, reddish yellow papules or nodules.6,7 Histologic examination shows a nodular infiltrate of foamy histiocytes in the superficial dermis. Touton-type multinucleated giant cells with a peripheral rim of xanthomatized foamy cytoplasm and a wreathlike arrangement of nuclei are characteristic. Associated eosinophils are seen. No emperipolesis is present.
Reticulohistiocytoma (Figure 5) is a benign dermal lesion that presents as solitary or less commonly multiple red-brown papules or nodules.8 Lesions consist of well-delineated nodular aggregates of histiocytes containing a finely granular eosinophilic ground glass cytoplasm. Few, if any, eosinophils are found. The lack of Touton multinucleated giant cells or emperipolesis and lack of expression of S-100 protein helps to distinguish reticulohistiocytoma from other entities in the differential diagnosis.
The Diagnosis: Rosai-Dorfman Disease
Rosai-Dorfman disease is a rare histiocytic proliferative disorder of unknown etiology. It has 2 forms: limited cutaneous and systemic. The systemic form, also known as sinus histiocytosis with massive lymphadenopathy, affects the lymph nodes and other organs at times. The disease is characterized by a proliferation of histiocytes in the lymph nodes, most commonly in the cervical basin1; however, the inguinal, axillary, mediastinal, or para-aortic nodes also may be affected.1,2 The skin is the most common site of extranodal disease, seen in approximately 10% of cases.1 Cutaneous involvement often is in the facial area but also can be found on the trunk, ears, neck, arms, legs, and genitals. Clinically, skin lesions appear as papules, plaques, and/or nodules.2
Histopathologic examination of Rosai-Dorfman disease generally shows a dense sheetlike dermal infiltrate of large polygonal histiocytes (Figure 1). Histiocytes may display pale pink or clear cytoplasm. The pathognomonic finding is emperipolesis, which consists of histiocytes with engulfed lymphocytes, erythrocytes, plasma cells, and/or granulocytes surrounded by a clear halo. Immunohistochemical staining also is characteristic, with lesional histiocytes showing expression of S-100 protein (Figure 1, inset) and CD68. The associated inflammatory infiltrate is mixed, containing primarily plasma cells but also lymphocytes, neutrophils, and eosinophils.
Blastomycosis (Figure 2) is a systemic infection due to inhalation of Blastomyces dermatitidis conidia. Primary infection occurs in the lungs, and with dissemination the skin is the most common subsequently involved organ.3 Cutaneous blastomycosis shows pseudoepitheliomatous hyperplasia with neutrophilic microabscesses and a dense dermal infiltrate containing suppurative granulomatous inflammation. The nonpigmented yeast phase typically is 8 to 15 µm in length with a refractile cell wall and characteristic single, broad-based budding.3
Granuloma faciale (Figure 3) is a rare disease with unknown etiology characterized by reddish brown plaques or nodules most commonly occurring on the face.4,5 Histology shows a dense nodular dermal infiltrate with a grenz zone. The infiltrate is mixed, containing mostly neutrophils with leukocytoclasis and eosinophils. Leukocytoclastic vasculitis is present with associated extravasated erythrocytes. In chronic fibrosing granuloma faciale, lesions can demonstrate fibrosis and hemosiderin deposition, similar to erythema elevatum diutinum.
Juvenile xanthogranuloma (Figure 4) is a common histiocytic disease of early childhood, though adult cases have been reported.6 Tumors are found on the head and trunk and are typically firm, reddish yellow papules or nodules.6,7 Histologic examination shows a nodular infiltrate of foamy histiocytes in the superficial dermis. Touton-type multinucleated giant cells with a peripheral rim of xanthomatized foamy cytoplasm and a wreathlike arrangement of nuclei are characteristic. Associated eosinophils are seen. No emperipolesis is present.
Reticulohistiocytoma (Figure 5) is a benign dermal lesion that presents as solitary or less commonly multiple red-brown papules or nodules.8 Lesions consist of well-delineated nodular aggregates of histiocytes containing a finely granular eosinophilic ground glass cytoplasm. Few, if any, eosinophils are found. The lack of Touton multinucleated giant cells or emperipolesis and lack of expression of S-100 protein helps to distinguish reticulohistiocytoma from other entities in the differential diagnosis.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2015.
- Wolff K, Johnson R, Saavedra AP. Fitzpatrick's Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill; 2013.
- Marcoval J, Moreno A, Peyrí J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol. 2004;51:269-273.
- Rodriguez J, Ackerman AB. Xanthogranuloma in adults. Arch Dermatol. 1976;112:43-44.
- Tanz WS, Schwartz RA, Janniger CK. Juvenile xanthogranuloma. Cutis. 1994;54:241-245.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatology Online J. 2014;20. pii:doj_21725.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2015.
- Wolff K, Johnson R, Saavedra AP. Fitzpatrick's Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill; 2013.
- Marcoval J, Moreno A, Peyrí J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol. 2004;51:269-273.
- Rodriguez J, Ackerman AB. Xanthogranuloma in adults. Arch Dermatol. 1976;112:43-44.
- Tanz WS, Schwartz RA, Janniger CK. Juvenile xanthogranuloma. Cutis. 1994;54:241-245.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatology Online J. 2014;20. pii:doj_21725.
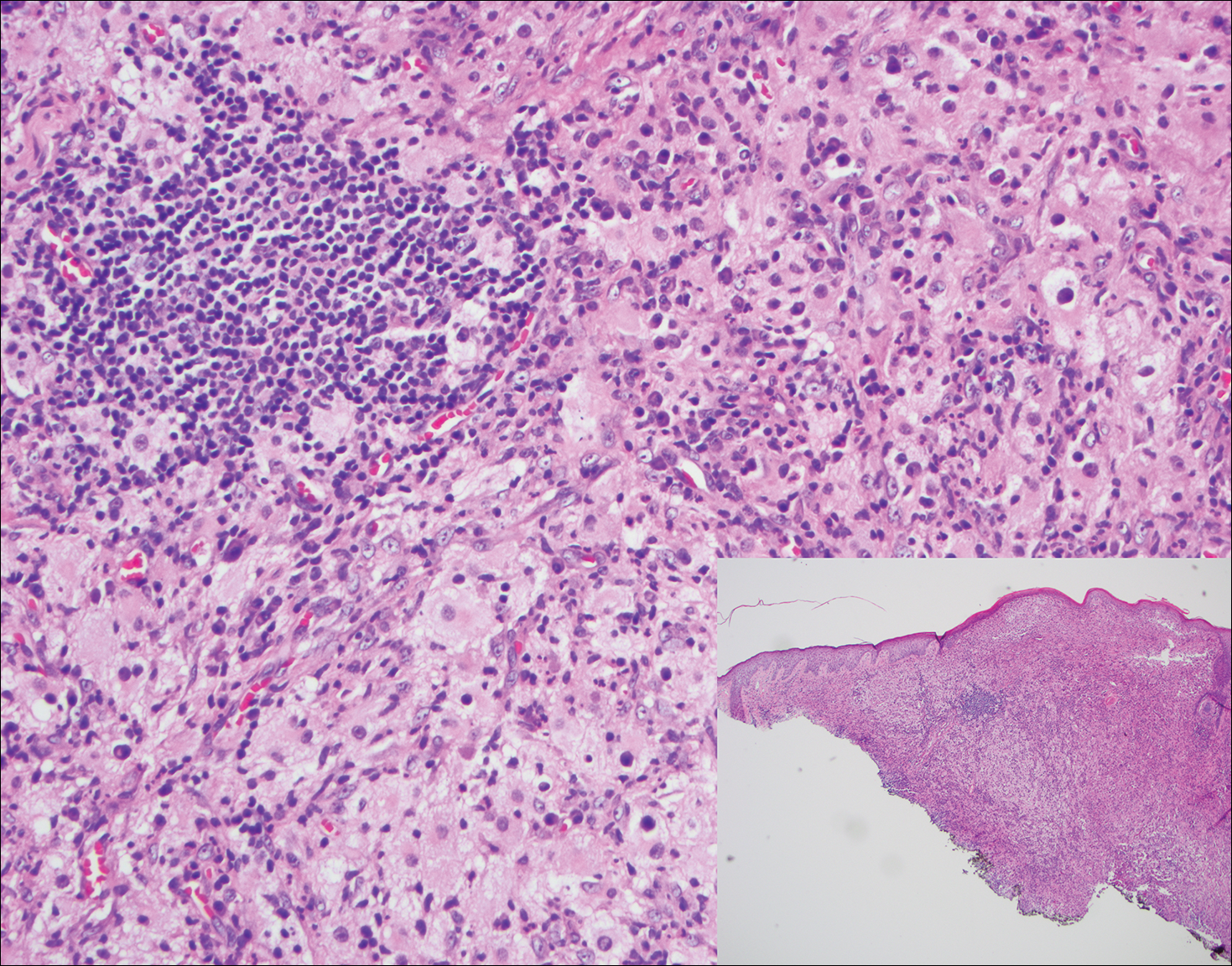
A 59-year-old man presented with itchy and mildly painful nodules on the head and neck of 7 months' duration. The patient denied fever, chills, unintentional weight loss, night sweats, and other systemic symptoms. Physical examination revealed multiple firm pink-orange nodules of varying sizes distributed on the scalp, face, and neck. Right-sided, painless, bulky cervical lymphadenopathy also was noted. An incisional biopsy was performed.
Pruritic Eruption on the Chest
The Diagnosis: Grover Disease
Grover disease (also known as transient acantholytic dermatosis) was first described by Ralph W. Grover in 1970 as an idiopathic, acquired, monomorphous, papulovesicular eruption. Although originally characterized by solely transient acantholytic dermatosis, over time the term Grover disease has been expanded to include persistent acantholytic dermatoses. Grover disease chiefly affects white adults older than 40 years and is more prevalent in males than females. Cases generally are self-limited but correlate with age, as older adults are more likely to have prolonged eruptions.1
Grover disease typically erupts with discrete, erythematous, edematous, acneform, red-brown or flesh-colored papules, papulovesicles, or keratotic papules that primarily are seen on the trunk and anterior portion of the chest. As the rash spreads, it can erupt on the neck and thighs. The etiology of Grover disease is unknown, but many factors have been associated with the condition in a limited number of patients, including exposure to UV radiation, excessive heat or sweating, use of sulfadoxine-pyrimethamine and recombinant human IL-4, and infection with Malassezia furfur and Demodex folliculorum.1 Grover disease also has been associated with other conditions such as asteatotic eczema, allergic contact dermatitis, and atopic dermatitis.2
Histologically, Grover disease (Figure 1) is an acantholytic process that can exhibit dyskeratosis (corps ronds and grains). Foci often are small and multiple foci are seen on shave biopsy. There also may be spongiotic changes when associated with an eczematous element. A perivascular lymphohistiocytic infiltrate with eosinophils usually is seen.3 Basket weave keratin may be seen; however, as the lesions cause pruritus, erosions and ulcerations often are present.4
Grover disease has multiple histologic variants that may resemble Darier disease, Hailey-Hailey disease, pemphigus foliaceus, pemphigus vulgaris, and spongiotic dermatitis and can present in combination.5
The variant of Grover disease that has a Darier-like pattern is difficult to distinguish from Darier disease, an autosomal-dominant-inherited disorder classified by small papules that emerge in seborrheic areas during childhood and adolescence. Histologically, Darier disease (Figure 2) shows broad areas of dyskeratosis and acantholysis that lead to suprabasal cleavage. Follicular extension may be present. In addition, there often is prominent vertical parakeratosis in Darier disease.6 Histologic features that favor Darier disease over the Darier-like variant of Grover disease include a broad focus of acanthotic dyskeratosis with follicular extension; the presence of a hyperkeratotic stratum corneum; and a lack of spongiosis and eosinophils, which are notably absent in Darier disease but may be present in Grover disease.4
Another variant of Grover disease has a Hailey-Hailey-like pattern, which is characterized by Hailey-Hailey disease's dilapidated brick wall appearance or the diffuse suprabasal acantholysis of all epidermal layers without notable dyskeratosis.4 Hailey-Hailey disease, also known as familial benign pemphigus, is an autosomal-dominant disorder that presents with erythematous vesicular plaques in flexural areas. The plaques progress to flaccid bullae with rupture and crusting and spread peripherally.7 Pathology shows suprabasilar clefts and numerous acantholytic cells (Figure 3). Dyskeratotic keratinocytes are rare with infrequent corps ronds and rare grains. The epidermis also is less hyperplastic in Grover disease than in Hailey-Hailey disease.1
Grover disease also may present histologically with a pemphiguslike pattern, mimicking pemphigus foliaceus and pemphigus vulgaris; however, direct immunofluorescence studies are negative in Grover disease.
Pemphigus foliaceus is an autoimmune disorder caused by autoantibodies to desmoglein 1, which are present on the surfaces of keratinocytes, and is characterized by scaly crusts and blisters.8 Histologically, pemphigus foliaceus (Figure 4) shows a superficial epidermal blistering process. The acantholysis may be subtle and is commonly localized to the stratum granulosum, extending into the stratum corneum. Complete loss of the stratum corneum can be seen, resulting in only scattered acantholytic cells. Spongiosis also may be seen. The dermis shows a perivascular infiltrate that often contains eosinophils. Pemphigus foliaceus is confirmed by direct immunofluorescence.9
Pemphigus vulgaris is an autoimmune blistering disorder that is characterized by IgG autoantibodies to desmoglein 3, a component of desmosomes that are involved in keratinocyte-to-keratinocyte adhesion. Clinically, patients present with flaccid fragile blisters on the skin and mucous membranes that rupture easily, leading to painful erosions.10 Intraepidermal blisters are seen histologically (Figure 5) with the loss of cohesion (acantholysis) seen classically in the lower portions of the epidermis where desmoglein 3 is most prominent. When only the basal layer remains, the histology has been likened to a tombstone row.11 Extension of the blister along the adnexa is common. The underlying dermis shows a perivascular infiltrate with eosinophils. Early lesions may show only eosinophilic spongiosis. Direct immunofluorescence studies show IgG and C3 in an intercellular pattern that resembles a fish net or chicken wire.4,11
The spongioticlike pattern of Grover disease is marked by epidermal edema with separation of the keratinocytes and the revelation of their intracellular bridges,4 which manifests as vesiculation in the stratum corneum or upper layers of the epidermis.12
Grover disease is self-limited and may spontaneously resolve; however, the disease may be responsive to topical and systemic steroids. Additionally, avoidance of aggravating factors such as sunlight, heat, and sweating can improve symptoms.2
- Parsons JM. Transient acantholytic dermatosis (Grover's disease): a global perspective. J Am Acad Dermatol. 1996;35(5, pt 1):653-666; quiz 667-670.
- Quirk CJ, Heenan PJ. Grover's disease: 34 years on. Australas J Dermatol. 2004;45:83-86.
- Davis MD, Dinneen AM, Landa N, et al. Grover's disease: clinicopathologic review of 72 cases. Mayo Clin Proc. 1999;74:229-234.
- Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133:1490-1494.
- Chalet M, Grover R, Ackerman AB. Transient acantholytic dermatosis: a reevaluation. Arch Dermatol. 1977;133:431-435.
- Takagi A, Kamijo M, Ikeda S. Darier disease. J Dermatol. 2016;43:275-279.
- Engin B, Kutlubay Z, Celik U, et al. Hailey-Hailey disease: a fold (intertriginous) dermatosis. Clin Dermatol. 2015;33:452-455.
- de Sena Nogueira Maehara L, Huizinga J, Jonkman MF. Rituximab therapy in pemphigus foliaceus: report of 12 cases and review of recent literature [published online March 31, 2015]. Br J Dermatol. 2015;172:1420-1423.
- James KA, Culton DA, Diaz LA. Diagnosis and clinical features of pemphigus foliaceus. Dermatol Clin. 2011;29:405-412.
- Black M, Mignogna MD, Scully C. Number II. pemphigus vulgaris. Oral Dis. 2005;11:119-130.
- Madke B, Doshi B, Khopkar U, et al. Appearances in dermatopathology: the diagnostic and the deceptive. Indian J Dermatol Venerol Leprol. 2013;79:338-348.
- Motaparthi K. Pseudoherpetic transient acantholytic dermatosis (Grover disease): case series and review of the literature [published online February 16, 2017]. J Cutan Pathol. 2017;44:486-489.
The Diagnosis: Grover Disease
Grover disease (also known as transient acantholytic dermatosis) was first described by Ralph W. Grover in 1970 as an idiopathic, acquired, monomorphous, papulovesicular eruption. Although originally characterized by solely transient acantholytic dermatosis, over time the term Grover disease has been expanded to include persistent acantholytic dermatoses. Grover disease chiefly affects white adults older than 40 years and is more prevalent in males than females. Cases generally are self-limited but correlate with age, as older adults are more likely to have prolonged eruptions.1
Grover disease typically erupts with discrete, erythematous, edematous, acneform, red-brown or flesh-colored papules, papulovesicles, or keratotic papules that primarily are seen on the trunk and anterior portion of the chest. As the rash spreads, it can erupt on the neck and thighs. The etiology of Grover disease is unknown, but many factors have been associated with the condition in a limited number of patients, including exposure to UV radiation, excessive heat or sweating, use of sulfadoxine-pyrimethamine and recombinant human IL-4, and infection with Malassezia furfur and Demodex folliculorum.1 Grover disease also has been associated with other conditions such as asteatotic eczema, allergic contact dermatitis, and atopic dermatitis.2
Histologically, Grover disease (Figure 1) is an acantholytic process that can exhibit dyskeratosis (corps ronds and grains). Foci often are small and multiple foci are seen on shave biopsy. There also may be spongiotic changes when associated with an eczematous element. A perivascular lymphohistiocytic infiltrate with eosinophils usually is seen.3 Basket weave keratin may be seen; however, as the lesions cause pruritus, erosions and ulcerations often are present.4
Grover disease has multiple histologic variants that may resemble Darier disease, Hailey-Hailey disease, pemphigus foliaceus, pemphigus vulgaris, and spongiotic dermatitis and can present in combination.5
The variant of Grover disease that has a Darier-like pattern is difficult to distinguish from Darier disease, an autosomal-dominant-inherited disorder classified by small papules that emerge in seborrheic areas during childhood and adolescence. Histologically, Darier disease (Figure 2) shows broad areas of dyskeratosis and acantholysis that lead to suprabasal cleavage. Follicular extension may be present. In addition, there often is prominent vertical parakeratosis in Darier disease.6 Histologic features that favor Darier disease over the Darier-like variant of Grover disease include a broad focus of acanthotic dyskeratosis with follicular extension; the presence of a hyperkeratotic stratum corneum; and a lack of spongiosis and eosinophils, which are notably absent in Darier disease but may be present in Grover disease.4
Another variant of Grover disease has a Hailey-Hailey-like pattern, which is characterized by Hailey-Hailey disease's dilapidated brick wall appearance or the diffuse suprabasal acantholysis of all epidermal layers without notable dyskeratosis.4 Hailey-Hailey disease, also known as familial benign pemphigus, is an autosomal-dominant disorder that presents with erythematous vesicular plaques in flexural areas. The plaques progress to flaccid bullae with rupture and crusting and spread peripherally.7 Pathology shows suprabasilar clefts and numerous acantholytic cells (Figure 3). Dyskeratotic keratinocytes are rare with infrequent corps ronds and rare grains. The epidermis also is less hyperplastic in Grover disease than in Hailey-Hailey disease.1
Grover disease also may present histologically with a pemphiguslike pattern, mimicking pemphigus foliaceus and pemphigus vulgaris; however, direct immunofluorescence studies are negative in Grover disease.
Pemphigus foliaceus is an autoimmune disorder caused by autoantibodies to desmoglein 1, which are present on the surfaces of keratinocytes, and is characterized by scaly crusts and blisters.8 Histologically, pemphigus foliaceus (Figure 4) shows a superficial epidermal blistering process. The acantholysis may be subtle and is commonly localized to the stratum granulosum, extending into the stratum corneum. Complete loss of the stratum corneum can be seen, resulting in only scattered acantholytic cells. Spongiosis also may be seen. The dermis shows a perivascular infiltrate that often contains eosinophils. Pemphigus foliaceus is confirmed by direct immunofluorescence.9
Pemphigus vulgaris is an autoimmune blistering disorder that is characterized by IgG autoantibodies to desmoglein 3, a component of desmosomes that are involved in keratinocyte-to-keratinocyte adhesion. Clinically, patients present with flaccid fragile blisters on the skin and mucous membranes that rupture easily, leading to painful erosions.10 Intraepidermal blisters are seen histologically (Figure 5) with the loss of cohesion (acantholysis) seen classically in the lower portions of the epidermis where desmoglein 3 is most prominent. When only the basal layer remains, the histology has been likened to a tombstone row.11 Extension of the blister along the adnexa is common. The underlying dermis shows a perivascular infiltrate with eosinophils. Early lesions may show only eosinophilic spongiosis. Direct immunofluorescence studies show IgG and C3 in an intercellular pattern that resembles a fish net or chicken wire.4,11
The spongioticlike pattern of Grover disease is marked by epidermal edema with separation of the keratinocytes and the revelation of their intracellular bridges,4 which manifests as vesiculation in the stratum corneum or upper layers of the epidermis.12
Grover disease is self-limited and may spontaneously resolve; however, the disease may be responsive to topical and systemic steroids. Additionally, avoidance of aggravating factors such as sunlight, heat, and sweating can improve symptoms.2
The Diagnosis: Grover Disease
Grover disease (also known as transient acantholytic dermatosis) was first described by Ralph W. Grover in 1970 as an idiopathic, acquired, monomorphous, papulovesicular eruption. Although originally characterized by solely transient acantholytic dermatosis, over time the term Grover disease has been expanded to include persistent acantholytic dermatoses. Grover disease chiefly affects white adults older than 40 years and is more prevalent in males than females. Cases generally are self-limited but correlate with age, as older adults are more likely to have prolonged eruptions.1
Grover disease typically erupts with discrete, erythematous, edematous, acneform, red-brown or flesh-colored papules, papulovesicles, or keratotic papules that primarily are seen on the trunk and anterior portion of the chest. As the rash spreads, it can erupt on the neck and thighs. The etiology of Grover disease is unknown, but many factors have been associated with the condition in a limited number of patients, including exposure to UV radiation, excessive heat or sweating, use of sulfadoxine-pyrimethamine and recombinant human IL-4, and infection with Malassezia furfur and Demodex folliculorum.1 Grover disease also has been associated with other conditions such as asteatotic eczema, allergic contact dermatitis, and atopic dermatitis.2
Histologically, Grover disease (Figure 1) is an acantholytic process that can exhibit dyskeratosis (corps ronds and grains). Foci often are small and multiple foci are seen on shave biopsy. There also may be spongiotic changes when associated with an eczematous element. A perivascular lymphohistiocytic infiltrate with eosinophils usually is seen.3 Basket weave keratin may be seen; however, as the lesions cause pruritus, erosions and ulcerations often are present.4
Grover disease has multiple histologic variants that may resemble Darier disease, Hailey-Hailey disease, pemphigus foliaceus, pemphigus vulgaris, and spongiotic dermatitis and can present in combination.5
The variant of Grover disease that has a Darier-like pattern is difficult to distinguish from Darier disease, an autosomal-dominant-inherited disorder classified by small papules that emerge in seborrheic areas during childhood and adolescence. Histologically, Darier disease (Figure 2) shows broad areas of dyskeratosis and acantholysis that lead to suprabasal cleavage. Follicular extension may be present. In addition, there often is prominent vertical parakeratosis in Darier disease.6 Histologic features that favor Darier disease over the Darier-like variant of Grover disease include a broad focus of acanthotic dyskeratosis with follicular extension; the presence of a hyperkeratotic stratum corneum; and a lack of spongiosis and eosinophils, which are notably absent in Darier disease but may be present in Grover disease.4
Another variant of Grover disease has a Hailey-Hailey-like pattern, which is characterized by Hailey-Hailey disease's dilapidated brick wall appearance or the diffuse suprabasal acantholysis of all epidermal layers without notable dyskeratosis.4 Hailey-Hailey disease, also known as familial benign pemphigus, is an autosomal-dominant disorder that presents with erythematous vesicular plaques in flexural areas. The plaques progress to flaccid bullae with rupture and crusting and spread peripherally.7 Pathology shows suprabasilar clefts and numerous acantholytic cells (Figure 3). Dyskeratotic keratinocytes are rare with infrequent corps ronds and rare grains. The epidermis also is less hyperplastic in Grover disease than in Hailey-Hailey disease.1
Grover disease also may present histologically with a pemphiguslike pattern, mimicking pemphigus foliaceus and pemphigus vulgaris; however, direct immunofluorescence studies are negative in Grover disease.
Pemphigus foliaceus is an autoimmune disorder caused by autoantibodies to desmoglein 1, which are present on the surfaces of keratinocytes, and is characterized by scaly crusts and blisters.8 Histologically, pemphigus foliaceus (Figure 4) shows a superficial epidermal blistering process. The acantholysis may be subtle and is commonly localized to the stratum granulosum, extending into the stratum corneum. Complete loss of the stratum corneum can be seen, resulting in only scattered acantholytic cells. Spongiosis also may be seen. The dermis shows a perivascular infiltrate that often contains eosinophils. Pemphigus foliaceus is confirmed by direct immunofluorescence.9
Pemphigus vulgaris is an autoimmune blistering disorder that is characterized by IgG autoantibodies to desmoglein 3, a component of desmosomes that are involved in keratinocyte-to-keratinocyte adhesion. Clinically, patients present with flaccid fragile blisters on the skin and mucous membranes that rupture easily, leading to painful erosions.10 Intraepidermal blisters are seen histologically (Figure 5) with the loss of cohesion (acantholysis) seen classically in the lower portions of the epidermis where desmoglein 3 is most prominent. When only the basal layer remains, the histology has been likened to a tombstone row.11 Extension of the blister along the adnexa is common. The underlying dermis shows a perivascular infiltrate with eosinophils. Early lesions may show only eosinophilic spongiosis. Direct immunofluorescence studies show IgG and C3 in an intercellular pattern that resembles a fish net or chicken wire.4,11
The spongioticlike pattern of Grover disease is marked by epidermal edema with separation of the keratinocytes and the revelation of their intracellular bridges,4 which manifests as vesiculation in the stratum corneum or upper layers of the epidermis.12
Grover disease is self-limited and may spontaneously resolve; however, the disease may be responsive to topical and systemic steroids. Additionally, avoidance of aggravating factors such as sunlight, heat, and sweating can improve symptoms.2
- Parsons JM. Transient acantholytic dermatosis (Grover's disease): a global perspective. J Am Acad Dermatol. 1996;35(5, pt 1):653-666; quiz 667-670.
- Quirk CJ, Heenan PJ. Grover's disease: 34 years on. Australas J Dermatol. 2004;45:83-86.
- Davis MD, Dinneen AM, Landa N, et al. Grover's disease: clinicopathologic review of 72 cases. Mayo Clin Proc. 1999;74:229-234.
- Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133:1490-1494.
- Chalet M, Grover R, Ackerman AB. Transient acantholytic dermatosis: a reevaluation. Arch Dermatol. 1977;133:431-435.
- Takagi A, Kamijo M, Ikeda S. Darier disease. J Dermatol. 2016;43:275-279.
- Engin B, Kutlubay Z, Celik U, et al. Hailey-Hailey disease: a fold (intertriginous) dermatosis. Clin Dermatol. 2015;33:452-455.
- de Sena Nogueira Maehara L, Huizinga J, Jonkman MF. Rituximab therapy in pemphigus foliaceus: report of 12 cases and review of recent literature [published online March 31, 2015]. Br J Dermatol. 2015;172:1420-1423.
- James KA, Culton DA, Diaz LA. Diagnosis and clinical features of pemphigus foliaceus. Dermatol Clin. 2011;29:405-412.
- Black M, Mignogna MD, Scully C. Number II. pemphigus vulgaris. Oral Dis. 2005;11:119-130.
- Madke B, Doshi B, Khopkar U, et al. Appearances in dermatopathology: the diagnostic and the deceptive. Indian J Dermatol Venerol Leprol. 2013;79:338-348.
- Motaparthi K. Pseudoherpetic transient acantholytic dermatosis (Grover disease): case series and review of the literature [published online February 16, 2017]. J Cutan Pathol. 2017;44:486-489.
- Parsons JM. Transient acantholytic dermatosis (Grover's disease): a global perspective. J Am Acad Dermatol. 1996;35(5, pt 1):653-666; quiz 667-670.
- Quirk CJ, Heenan PJ. Grover's disease: 34 years on. Australas J Dermatol. 2004;45:83-86.
- Davis MD, Dinneen AM, Landa N, et al. Grover's disease: clinicopathologic review of 72 cases. Mayo Clin Proc. 1999;74:229-234.
- Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133:1490-1494.
- Chalet M, Grover R, Ackerman AB. Transient acantholytic dermatosis: a reevaluation. Arch Dermatol. 1977;133:431-435.
- Takagi A, Kamijo M, Ikeda S. Darier disease. J Dermatol. 2016;43:275-279.
- Engin B, Kutlubay Z, Celik U, et al. Hailey-Hailey disease: a fold (intertriginous) dermatosis. Clin Dermatol. 2015;33:452-455.
- de Sena Nogueira Maehara L, Huizinga J, Jonkman MF. Rituximab therapy in pemphigus foliaceus: report of 12 cases and review of recent literature [published online March 31, 2015]. Br J Dermatol. 2015;172:1420-1423.
- James KA, Culton DA, Diaz LA. Diagnosis and clinical features of pemphigus foliaceus. Dermatol Clin. 2011;29:405-412.
- Black M, Mignogna MD, Scully C. Number II. pemphigus vulgaris. Oral Dis. 2005;11:119-130.
- Madke B, Doshi B, Khopkar U, et al. Appearances in dermatopathology: the diagnostic and the deceptive. Indian J Dermatol Venerol Leprol. 2013;79:338-348.
- Motaparthi K. Pseudoherpetic transient acantholytic dermatosis (Grover disease): case series and review of the literature [published online February 16, 2017]. J Cutan Pathol. 2017;44:486-489.
A 55-year-old man presented with small, erythematous, nonfollicular, pruritic papules on the mid chest.
Large Hyperpigmented Nodule on the Leg
The Diagnosis: Dermatofibroma
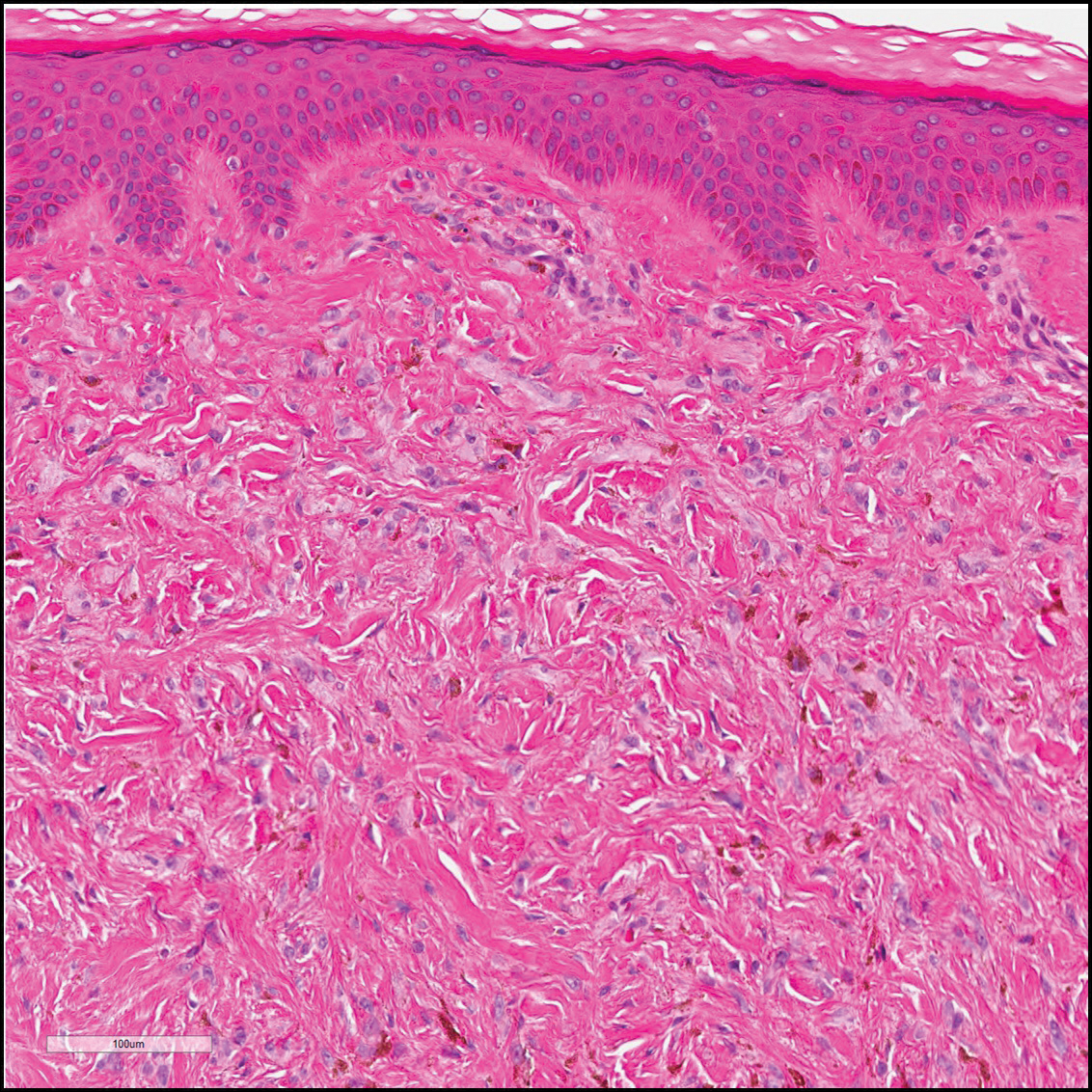
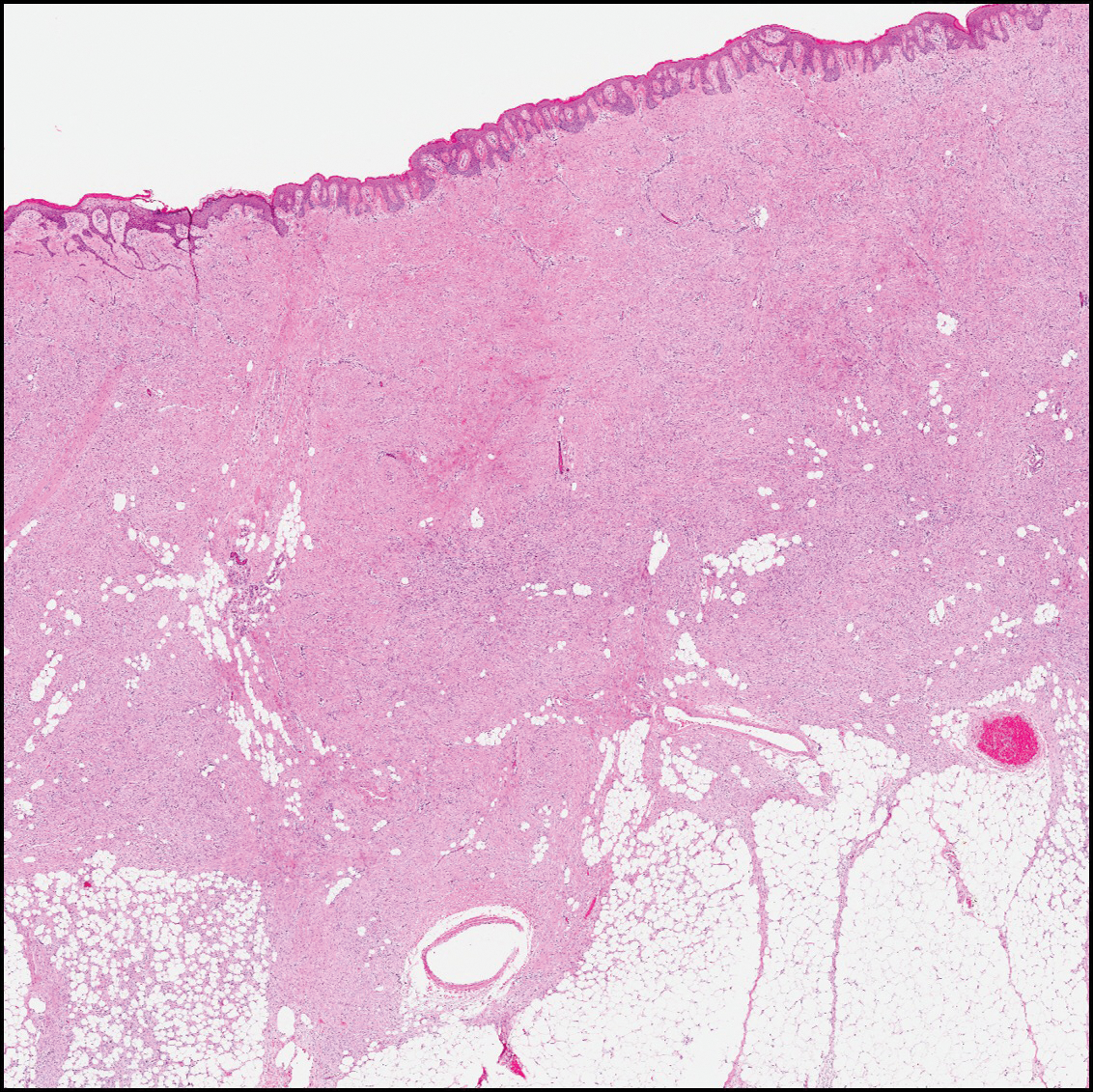
Dermatofibroma (DF) is a commonly encountered lesion. Although usually a straightforward clinical diagnosis, histopathological diagnosis is sometimes required. Conventional histologic findings of DF are hyperkeratosis, induction of the epidermis with acanthosis, and basal layer hyperpigmentation.1,2 Within the dermis there usually is proliferation of fibroblasts, histiocytes, and blood vessels that sometimes spares the overlying papillary dermis. Nomenclature of specific variants may be assigned based on the predominant component (eg, nodular subepidermal fibrosis, histiocytoma, sclerosing hemangioma) or histologic findings (eg, fibrocollagenous, sclerotic, cellular, histiocytic, lipidized, angiomatous, aneurysmal, clear cell, monster cell, myxoid, keloidal, palisading, osteoclastic, epithelioid).3-5 Of the histologic variants, fibrocollagenous is most common, but knowledge of other variants is important for accurate diagnosis, especially to exclude malignancy.
The sclerosing hemangioma variant of DF may pre-sent a diagnostic dilemma. In addition to typical features of DF, pseudovascular spaces, abundant hemosiderin, and reactive-appearing spindled cells are histologically demonstrated. The marked sclerosis and pigment deposition may mimic a blue nevus, and the dilated pseudovascular spaces may be reminiscent of a vascular neoplasm such as angiosarcoma or Kaposi sarcoma. However, the presence of characteristic features such as peripheral collagen trapping and overlying epidermal hyperplasia provide important clues for correct diagnosis.
Angiosarcomas (Figure 1) are malignant neoplasms with vascular differentiation. Cutaneous angiosarcomas present as purple plaques or nodules on the head and/or neck in elderly individuals as well as in patients with chronic lymphedema or prior radiation exposure.6-9 They are aggressive neoplasms with high rates of recurrence and metastases. Microscopically, the tumor is composed of anastomosing vascular channels lined by atypical endothelial cells with a multilayered appearance. There is frequent red blood cell extravasation, and substantial hemosiderin deposition may be noted in long-standing lesions. Neoplastic cells are positive for vascular markers (CD34, CD31, ETS-related gene transcription factor). Notably, cases associated with radiation exposure and chronic lymphedema are positive for MYC.10

Blue nevi (Figure 2) are benign melanocytic tumors that occur most frequently in children but may pre-sent in any age group. Clinical presentation is a blue to black, slightly raised papule that may be found on any site of the body. Biopsy typically shows a wedge-shaped infiltrate of spindled melanocytes with elongated dendritic processes in a sclerotic collagenous stroma. There frequently is a striking population of heavily pigmented melanophages. The melanocytes are positive for melanoma antigen recognized by T cells (MART-1)/melan-A, S-100, and transcription factor SOX-10. In contrast to other benign nevi, human melanoma black-45 will be positive in the dermal component.
Dermatofibrosarcoma protuberans (Figure 3) is a dermal-based tumor of intermediate malignant potential with a high rate of local recurrence and potential for sarcomatous transformation. Dermatofibrosarcoma protuberans most commonly presents in young adults as firm, pink to brown plaques and can occur on any site of the body. Histologically, they show a dermal proliferation of spindled cells that infiltrate in a storiform fashion into the subcutaneous adipose tissue,11 which imparts a honeycomb or Swiss cheese pattern. The tumor characteristically demonstrates positive staining for CD34. Loss of CD34 staining, increased mitoses, nuclear atypia, and fascicular growth are features suggestive of sarcomatous transformation.11,12 Dermatofibrosarcoma protuberans is associated with chromosomal abnormalities of chromosomes 17 and 22, resulting in COL1A1 (collagen type 1 alpha 1 chain) and PDGF-β (platelet-derived growth factor subunit B) gene fusion.13
Sclerotic fibromas (also known as storiform collagenomas)(Figure 4) may represent regressed DFs and are frequently associated with prior trauma to the affected area.14,15 They usually appear as flesh-colored papules or nodules on the face and trunk. The presence of multiple sclerotic fibromas is associated with Cowden syndrome.16,17 Histologically, the lesions present as well-demarcated, nonencapsulated, dermal nodules composed of a storiform or whorled arrangement of collagen with spindled fibroblasts. The sclerotic collagen bundles often are separated by small clefts imparting a plywoodlike pattern.16
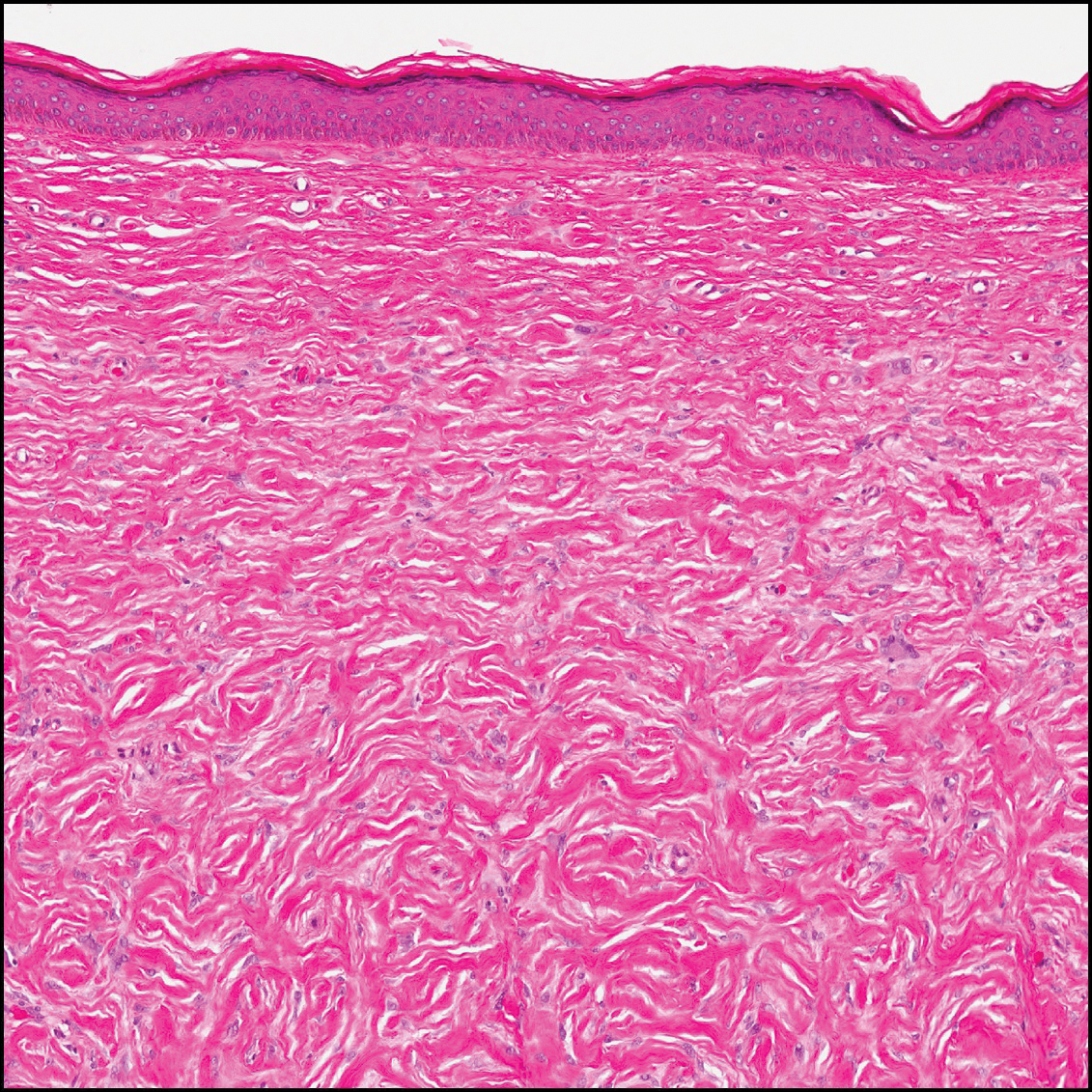
The differential diagnosis for DF expands once atypical clinical and histopathological findings are present. In this case, the nodule was much larger and darker than the usual appearance of DF (3-10 mm).2,4 Given the lesion's nodularity, the clinical dimple sign on lateral compression could not be seen. On biopsy, the predominance of blood vessels and sclerosis further complicated the diagnostic picture. In unusual cases such as this one, correlation of clinical history, histology, and immunophenotype is ever important.
- Zeidi M, North JP. Sebaceous induction in dermatofibroma: a common feature of dermatofibromas on the shoulder. J Cutan Pathol. 2015;42:400-405.
- Şenel E, Yuyucu Karabulut Y, Doğruer S¸enel S. Clinical, histopathological, dermatoscopic and digital microscopic features of dermatofibroma: a retrospective analysis of 200 lesions. J Eur Acad Dermatol Venereol. 2015;29:1958-1966.
- Vilanova JR, Flint A. The morphological variations of fibrous histiocytomas. J Cutan Pathol. 1974;1:155-164.
- Han TY, Chang HS, Lee JH, et al. A clinical and histopathological study of 122 cases of dermatofibroma (benign fibrous histiocytoma)[published online May 27, 2011]. Ann Dermatol. 2011;23:185-192.
- Alves JVP, Matos DM, Barreiros HF, et al. Variants of dermatofibroma--a histopathological study. An Bras Dermatol. 2014;89:472-477.
- Rosai J, Sumner HW, Major MC, et al. Angiosarcoma of the skin: a clinicopathologic and fine structural study. Hum Pathol. 1976;7:83-109.
- Haustein UF. Angiosarcoma of the face and scalp. Int J Dermatol. 1991;30:851-856.
- Stewart FW, Treves N. Lymphangiosarcoma in postmastectomy lymphedema: a report of six cases in elephantiasis chirurgica. Cancer. 1948;1:64-81.
- Goette DK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12(5 pt 2):922-926.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Voth H, Landsberg J, Hinz T, et al. Management of dermatofibrosarcoma protuberans with fibrosarcomatous transformation: an evidence-based review of the literature. J Eur Acad Dermatol Venereol. 2011;25:1385-1391.
- Goldblum JR. CD34 positivity in fibrosarcomas which arise in dermatofibrosarcoma protuberans. Arch Pathol Lab Med. 1995;119:238-241.
- Patel KU, Szabo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184-193.
- Sohn IB, Hwang SM, Lee SH, et al. Dermatofibroma with sclerotic areas resembling a sclerotic fibroma of the skin. J Cutan Pathol. 2002;29:44-47.
- Pujol RM, de Castro F, Schroeter AL, et al. Solitary sclerotic fibroma of the skin: a sclerotic dermatofibroma? Am J Dermatopathol. 1996;18:620-624.
- Requena L, Gutiérrez J, Sánchez Yus E. Multiple sclerotic fibromas of the skin: a cutaneous marker of Cowden's disease. J Cutan Pathol. 1992;19:346-351.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
The Diagnosis: Dermatofibroma
Dermatofibroma (DF) is a commonly encountered lesion. Although usually a straightforward clinical diagnosis, histopathological diagnosis is sometimes required. Conventional histologic findings of DF are hyperkeratosis, induction of the epidermis with acanthosis, and basal layer hyperpigmentation.1,2 Within the dermis there usually is proliferation of fibroblasts, histiocytes, and blood vessels that sometimes spares the overlying papillary dermis. Nomenclature of specific variants may be assigned based on the predominant component (eg, nodular subepidermal fibrosis, histiocytoma, sclerosing hemangioma) or histologic findings (eg, fibrocollagenous, sclerotic, cellular, histiocytic, lipidized, angiomatous, aneurysmal, clear cell, monster cell, myxoid, keloidal, palisading, osteoclastic, epithelioid).3-5 Of the histologic variants, fibrocollagenous is most common, but knowledge of other variants is important for accurate diagnosis, especially to exclude malignancy.
The sclerosing hemangioma variant of DF may pre-sent a diagnostic dilemma. In addition to typical features of DF, pseudovascular spaces, abundant hemosiderin, and reactive-appearing spindled cells are histologically demonstrated. The marked sclerosis and pigment deposition may mimic a blue nevus, and the dilated pseudovascular spaces may be reminiscent of a vascular neoplasm such as angiosarcoma or Kaposi sarcoma. However, the presence of characteristic features such as peripheral collagen trapping and overlying epidermal hyperplasia provide important clues for correct diagnosis.
Angiosarcomas (Figure 1) are malignant neoplasms with vascular differentiation. Cutaneous angiosarcomas present as purple plaques or nodules on the head and/or neck in elderly individuals as well as in patients with chronic lymphedema or prior radiation exposure.6-9 They are aggressive neoplasms with high rates of recurrence and metastases. Microscopically, the tumor is composed of anastomosing vascular channels lined by atypical endothelial cells with a multilayered appearance. There is frequent red blood cell extravasation, and substantial hemosiderin deposition may be noted in long-standing lesions. Neoplastic cells are positive for vascular markers (CD34, CD31, ETS-related gene transcription factor). Notably, cases associated with radiation exposure and chronic lymphedema are positive for MYC.10
Blue nevi (Figure 2) are benign melanocytic tumors that occur most frequently in children but may pre-sent in any age group. Clinical presentation is a blue to black, slightly raised papule that may be found on any site of the body. Biopsy typically shows a wedge-shaped infiltrate of spindled melanocytes with elongated dendritic processes in a sclerotic collagenous stroma. There frequently is a striking population of heavily pigmented melanophages. The melanocytes are positive for melanoma antigen recognized by T cells (MART-1)/melan-A, S-100, and transcription factor SOX-10. In contrast to other benign nevi, human melanoma black-45 will be positive in the dermal component.
Dermatofibrosarcoma protuberans (Figure 3) is a dermal-based tumor of intermediate malignant potential with a high rate of local recurrence and potential for sarcomatous transformation. Dermatofibrosarcoma protuberans most commonly presents in young adults as firm, pink to brown plaques and can occur on any site of the body. Histologically, they show a dermal proliferation of spindled cells that infiltrate in a storiform fashion into the subcutaneous adipose tissue,11 which imparts a honeycomb or Swiss cheese pattern. The tumor characteristically demonstrates positive staining for CD34. Loss of CD34 staining, increased mitoses, nuclear atypia, and fascicular growth are features suggestive of sarcomatous transformation.11,12 Dermatofibrosarcoma protuberans is associated with chromosomal abnormalities of chromosomes 17 and 22, resulting in COL1A1 (collagen type 1 alpha 1 chain) and PDGF-β (platelet-derived growth factor subunit B) gene fusion.13
Sclerotic fibromas (also known as storiform collagenomas)(Figure 4) may represent regressed DFs and are frequently associated with prior trauma to the affected area.14,15 They usually appear as flesh-colored papules or nodules on the face and trunk. The presence of multiple sclerotic fibromas is associated with Cowden syndrome.16,17 Histologically, the lesions present as well-demarcated, nonencapsulated, dermal nodules composed of a storiform or whorled arrangement of collagen with spindled fibroblasts. The sclerotic collagen bundles often are separated by small clefts imparting a plywoodlike pattern.16
The differential diagnosis for DF expands once atypical clinical and histopathological findings are present. In this case, the nodule was much larger and darker than the usual appearance of DF (3-10 mm).2,4 Given the lesion's nodularity, the clinical dimple sign on lateral compression could not be seen. On biopsy, the predominance of blood vessels and sclerosis further complicated the diagnostic picture. In unusual cases such as this one, correlation of clinical history, histology, and immunophenotype is ever important.
The Diagnosis: Dermatofibroma
Dermatofibroma (DF) is a commonly encountered lesion. Although usually a straightforward clinical diagnosis, histopathological diagnosis is sometimes required. Conventional histologic findings of DF are hyperkeratosis, induction of the epidermis with acanthosis, and basal layer hyperpigmentation.1,2 Within the dermis there usually is proliferation of fibroblasts, histiocytes, and blood vessels that sometimes spares the overlying papillary dermis. Nomenclature of specific variants may be assigned based on the predominant component (eg, nodular subepidermal fibrosis, histiocytoma, sclerosing hemangioma) or histologic findings (eg, fibrocollagenous, sclerotic, cellular, histiocytic, lipidized, angiomatous, aneurysmal, clear cell, monster cell, myxoid, keloidal, palisading, osteoclastic, epithelioid).3-5 Of the histologic variants, fibrocollagenous is most common, but knowledge of other variants is important for accurate diagnosis, especially to exclude malignancy.
The sclerosing hemangioma variant of DF may pre-sent a diagnostic dilemma. In addition to typical features of DF, pseudovascular spaces, abundant hemosiderin, and reactive-appearing spindled cells are histologically demonstrated. The marked sclerosis and pigment deposition may mimic a blue nevus, and the dilated pseudovascular spaces may be reminiscent of a vascular neoplasm such as angiosarcoma or Kaposi sarcoma. However, the presence of characteristic features such as peripheral collagen trapping and overlying epidermal hyperplasia provide important clues for correct diagnosis.
Angiosarcomas (Figure 1) are malignant neoplasms with vascular differentiation. Cutaneous angiosarcomas present as purple plaques or nodules on the head and/or neck in elderly individuals as well as in patients with chronic lymphedema or prior radiation exposure.6-9 They are aggressive neoplasms with high rates of recurrence and metastases. Microscopically, the tumor is composed of anastomosing vascular channels lined by atypical endothelial cells with a multilayered appearance. There is frequent red blood cell extravasation, and substantial hemosiderin deposition may be noted in long-standing lesions. Neoplastic cells are positive for vascular markers (CD34, CD31, ETS-related gene transcription factor). Notably, cases associated with radiation exposure and chronic lymphedema are positive for MYC.10
Blue nevi (Figure 2) are benign melanocytic tumors that occur most frequently in children but may pre-sent in any age group. Clinical presentation is a blue to black, slightly raised papule that may be found on any site of the body. Biopsy typically shows a wedge-shaped infiltrate of spindled melanocytes with elongated dendritic processes in a sclerotic collagenous stroma. There frequently is a striking population of heavily pigmented melanophages. The melanocytes are positive for melanoma antigen recognized by T cells (MART-1)/melan-A, S-100, and transcription factor SOX-10. In contrast to other benign nevi, human melanoma black-45 will be positive in the dermal component.
Dermatofibrosarcoma protuberans (Figure 3) is a dermal-based tumor of intermediate malignant potential with a high rate of local recurrence and potential for sarcomatous transformation. Dermatofibrosarcoma protuberans most commonly presents in young adults as firm, pink to brown plaques and can occur on any site of the body. Histologically, they show a dermal proliferation of spindled cells that infiltrate in a storiform fashion into the subcutaneous adipose tissue,11 which imparts a honeycomb or Swiss cheese pattern. The tumor characteristically demonstrates positive staining for CD34. Loss of CD34 staining, increased mitoses, nuclear atypia, and fascicular growth are features suggestive of sarcomatous transformation.11,12 Dermatofibrosarcoma protuberans is associated with chromosomal abnormalities of chromosomes 17 and 22, resulting in COL1A1 (collagen type 1 alpha 1 chain) and PDGF-β (platelet-derived growth factor subunit B) gene fusion.13
Sclerotic fibromas (also known as storiform collagenomas)(Figure 4) may represent regressed DFs and are frequently associated with prior trauma to the affected area.14,15 They usually appear as flesh-colored papules or nodules on the face and trunk. The presence of multiple sclerotic fibromas is associated with Cowden syndrome.16,17 Histologically, the lesions present as well-demarcated, nonencapsulated, dermal nodules composed of a storiform or whorled arrangement of collagen with spindled fibroblasts. The sclerotic collagen bundles often are separated by small clefts imparting a plywoodlike pattern.16
The differential diagnosis for DF expands once atypical clinical and histopathological findings are present. In this case, the nodule was much larger and darker than the usual appearance of DF (3-10 mm).2,4 Given the lesion's nodularity, the clinical dimple sign on lateral compression could not be seen. On biopsy, the predominance of blood vessels and sclerosis further complicated the diagnostic picture. In unusual cases such as this one, correlation of clinical history, histology, and immunophenotype is ever important.
- Zeidi M, North JP. Sebaceous induction in dermatofibroma: a common feature of dermatofibromas on the shoulder. J Cutan Pathol. 2015;42:400-405.
- Şenel E, Yuyucu Karabulut Y, Doğruer S¸enel S. Clinical, histopathological, dermatoscopic and digital microscopic features of dermatofibroma: a retrospective analysis of 200 lesions. J Eur Acad Dermatol Venereol. 2015;29:1958-1966.
- Vilanova JR, Flint A. The morphological variations of fibrous histiocytomas. J Cutan Pathol. 1974;1:155-164.
- Han TY, Chang HS, Lee JH, et al. A clinical and histopathological study of 122 cases of dermatofibroma (benign fibrous histiocytoma)[published online May 27, 2011]. Ann Dermatol. 2011;23:185-192.
- Alves JVP, Matos DM, Barreiros HF, et al. Variants of dermatofibroma--a histopathological study. An Bras Dermatol. 2014;89:472-477.
- Rosai J, Sumner HW, Major MC, et al. Angiosarcoma of the skin: a clinicopathologic and fine structural study. Hum Pathol. 1976;7:83-109.
- Haustein UF. Angiosarcoma of the face and scalp. Int J Dermatol. 1991;30:851-856.
- Stewart FW, Treves N. Lymphangiosarcoma in postmastectomy lymphedema: a report of six cases in elephantiasis chirurgica. Cancer. 1948;1:64-81.
- Goette DK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12(5 pt 2):922-926.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Voth H, Landsberg J, Hinz T, et al. Management of dermatofibrosarcoma protuberans with fibrosarcomatous transformation: an evidence-based review of the literature. J Eur Acad Dermatol Venereol. 2011;25:1385-1391.
- Goldblum JR. CD34 positivity in fibrosarcomas which arise in dermatofibrosarcoma protuberans. Arch Pathol Lab Med. 1995;119:238-241.
- Patel KU, Szabo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184-193.
- Sohn IB, Hwang SM, Lee SH, et al. Dermatofibroma with sclerotic areas resembling a sclerotic fibroma of the skin. J Cutan Pathol. 2002;29:44-47.
- Pujol RM, de Castro F, Schroeter AL, et al. Solitary sclerotic fibroma of the skin: a sclerotic dermatofibroma? Am J Dermatopathol. 1996;18:620-624.
- Requena L, Gutiérrez J, Sánchez Yus E. Multiple sclerotic fibromas of the skin: a cutaneous marker of Cowden's disease. J Cutan Pathol. 1992;19:346-351.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Zeidi M, North JP. Sebaceous induction in dermatofibroma: a common feature of dermatofibromas on the shoulder. J Cutan Pathol. 2015;42:400-405.
- Şenel E, Yuyucu Karabulut Y, Doğruer S¸enel S. Clinical, histopathological, dermatoscopic and digital microscopic features of dermatofibroma: a retrospective analysis of 200 lesions. J Eur Acad Dermatol Venereol. 2015;29:1958-1966.
- Vilanova JR, Flint A. The morphological variations of fibrous histiocytomas. J Cutan Pathol. 1974;1:155-164.
- Han TY, Chang HS, Lee JH, et al. A clinical and histopathological study of 122 cases of dermatofibroma (benign fibrous histiocytoma)[published online May 27, 2011]. Ann Dermatol. 2011;23:185-192.
- Alves JVP, Matos DM, Barreiros HF, et al. Variants of dermatofibroma--a histopathological study. An Bras Dermatol. 2014;89:472-477.
- Rosai J, Sumner HW, Major MC, et al. Angiosarcoma of the skin: a clinicopathologic and fine structural study. Hum Pathol. 1976;7:83-109.
- Haustein UF. Angiosarcoma of the face and scalp. Int J Dermatol. 1991;30:851-856.
- Stewart FW, Treves N. Lymphangiosarcoma in postmastectomy lymphedema: a report of six cases in elephantiasis chirurgica. Cancer. 1948;1:64-81.
- Goette DK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12(5 pt 2):922-926.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Voth H, Landsberg J, Hinz T, et al. Management of dermatofibrosarcoma protuberans with fibrosarcomatous transformation: an evidence-based review of the literature. J Eur Acad Dermatol Venereol. 2011;25:1385-1391.
- Goldblum JR. CD34 positivity in fibrosarcomas which arise in dermatofibrosarcoma protuberans. Arch Pathol Lab Med. 1995;119:238-241.
- Patel KU, Szabo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184-193.
- Sohn IB, Hwang SM, Lee SH, et al. Dermatofibroma with sclerotic areas resembling a sclerotic fibroma of the skin. J Cutan Pathol. 2002;29:44-47.
- Pujol RM, de Castro F, Schroeter AL, et al. Solitary sclerotic fibroma of the skin: a sclerotic dermatofibroma? Am J Dermatopathol. 1996;18:620-624.
- Requena L, Gutiérrez J, Sánchez Yus E. Multiple sclerotic fibromas of the skin: a cutaneous marker of Cowden's disease. J Cutan Pathol. 1992;19:346-351.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.

A 61-year-old woman presented with a 2.5-cm hyperpigmented exophytic nodule on the anterior aspect of the left shin of approximately 2 years' duration. The patient initially noticed a small lesion following a bee sting, but it subsequently grew over the ensuing 2 years. A shave biopsy was obtained.
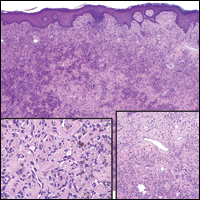
Enlarging Mass on the Lateral Neck
Branchial Cleft Cyst
Cystic lesions present in a myriad of ways and often require histopathologic examination for definitive diagnosis. Correct identification of the cells comprising the lining of the cyst and the composition of the surrounding tissue are utilized to classify these lesions.
Branchial cleft cysts (quiz image, Figure 1) most commonly present as a soft tissue swelling of the lateral neck anterior to the sternocleidomastoid; they also can present in the preauricular or mandibular region.1,2 Although the cyst is present at birth, it typically is not clinically apparent until the second or third decades of life. The origin of branchial cleft cysts is subject to some debate; however, the prevailing theory is that they result from failure of obliteration of the second branchial arch during development.1 Histopathologically, branchial cleft cysts are characterized by a stratified squamous epithelial lining and abundant lymphoid tissue with germinal centers.3,4 Infection is a common reason for presentation and excision is curative.
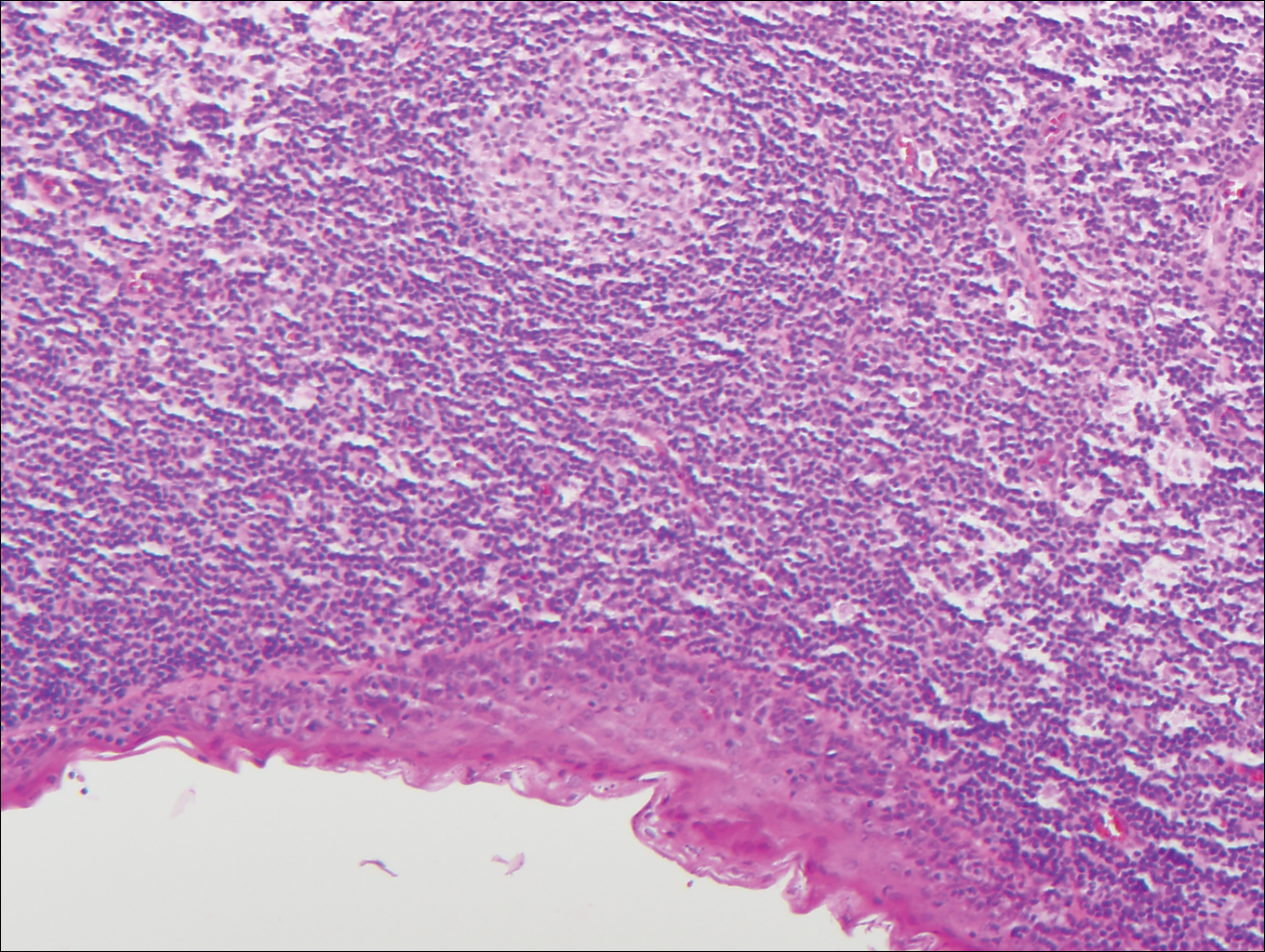
Bronchogenic cysts (Figure 2) present as midline lesions in the suprasternal notch and can present clinically due to compression of the airway.5 They develop as anomalies of the primitive foregut, budding off of the tracheobronchial tree. Similar to respiratory tissue, they are lined with a ciliated pseudostratified columnar epithelium and contain goblet cells. Concentric smooth muscle often surrounds the cyst and cartilage may be present.4 Excision is curative and recommended if the cyst encroaches on vital structures.
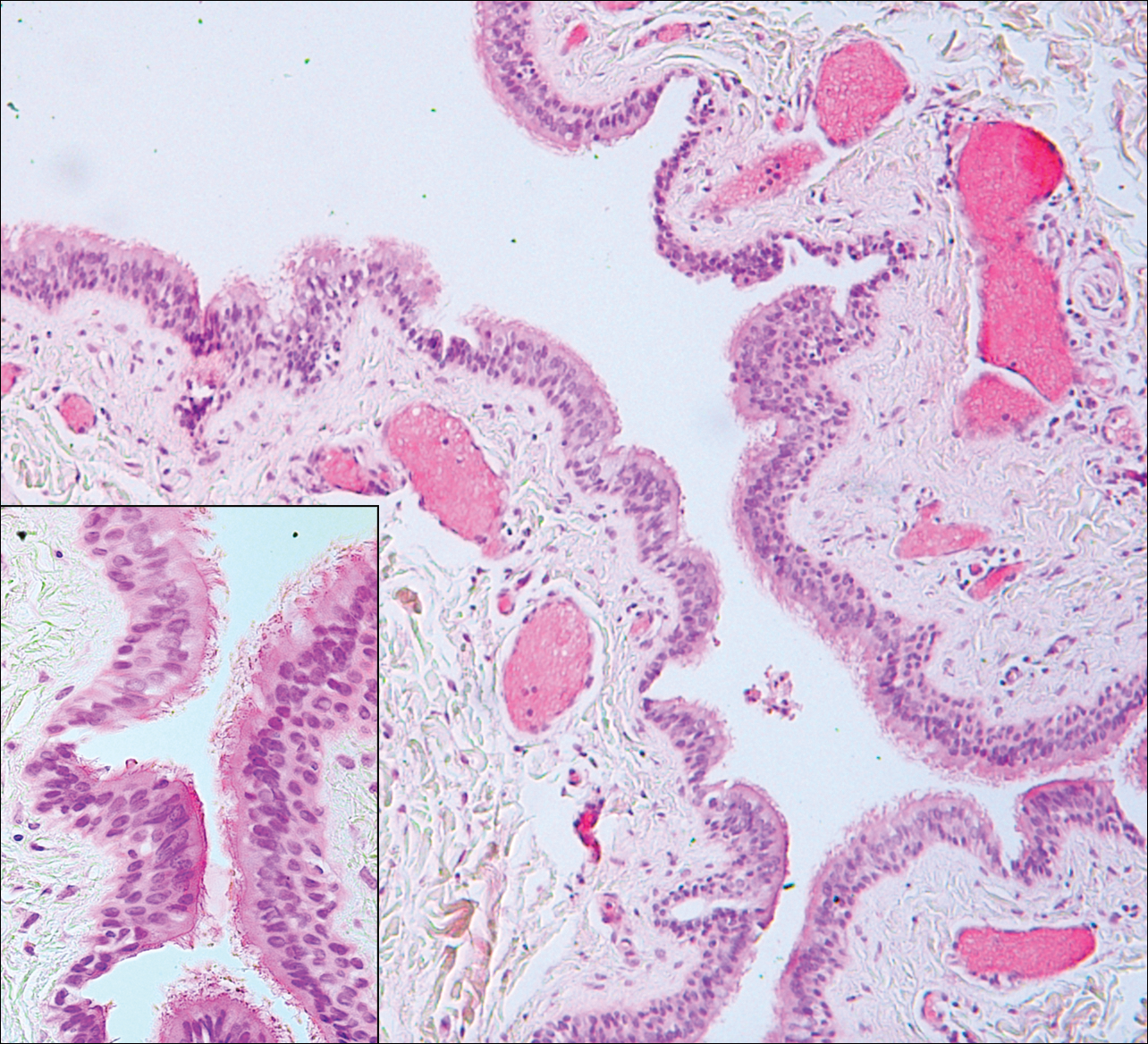
Median raphe cysts occur most commonly on the ventral surface of the penis on or near the glans (Figure 3). These cysts are thought to result from anomalous budding from the urethral epithelium, though they do not maintain contact with the urethra.3 The lining varies in thickness from 1 to 4 cell layers and mimics the transitional epithelium of the urethra. Amorphous debris often is seen within the cyst, and surrounding genital tissue can be appreciated by identification of delicate collagen, smooth muscle, and numerous small nerves and vessels.3,4 Excision is curative and often is sought when the cyst becomes irritated or cosmetically bothersome.
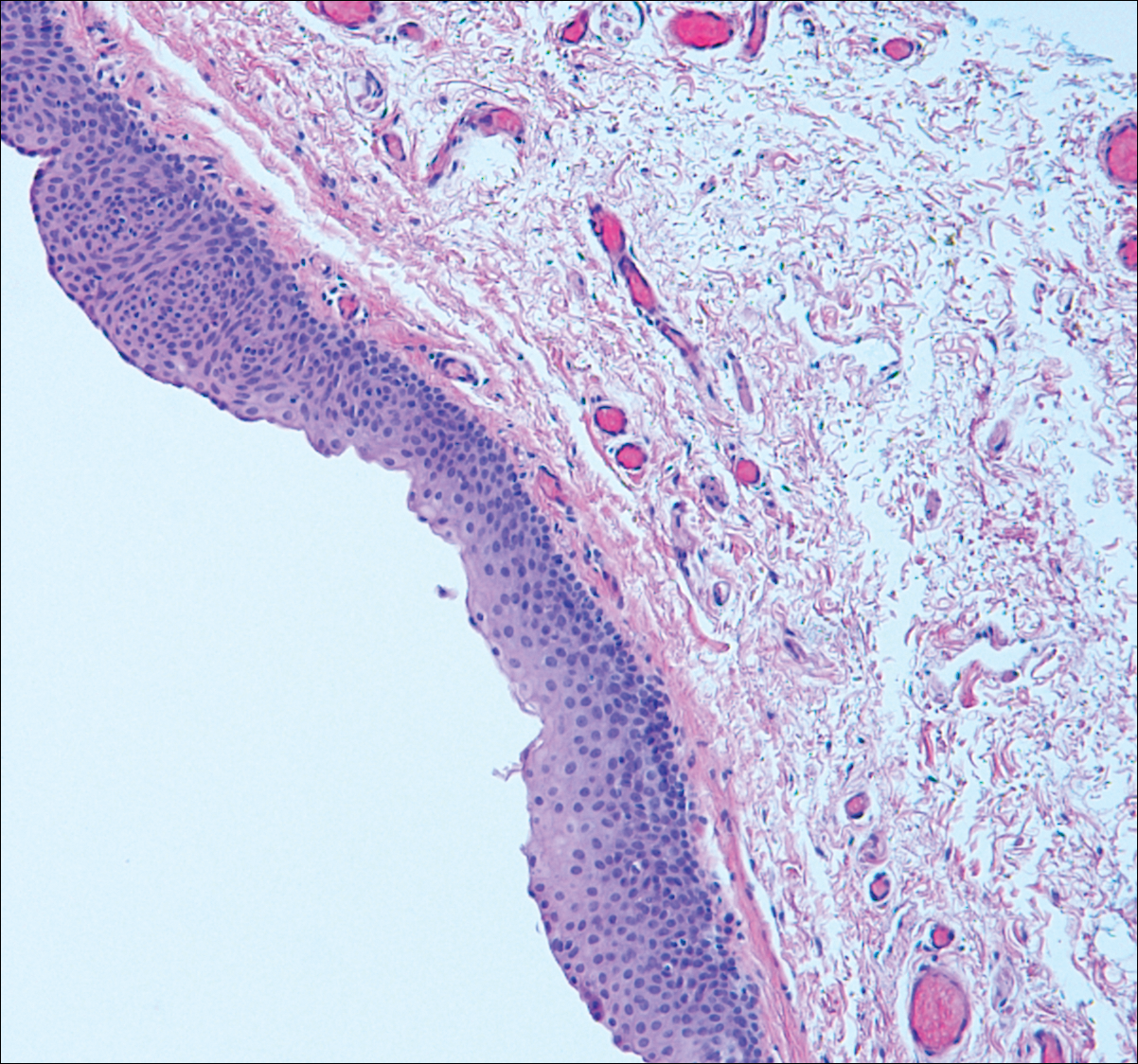
Steatocystomas can present as solitary (steatocystoma simplex) or multiple lesions (steatocystoma multiplex)(Figure 4). They present as small, well-defined, yellow cystic papules most commonly on the chest, axilla, or groin.2 Their lining is composed of a stratified squamous epithelium that lacks a granular layer and contains a distinct overlying corrugated "shark tooth" eosinophilic cuticle. Sebaceous lobules are characteristically present along or within the cyst wall.3,4 Excision is curative and treatment often is sought for cosmetic purposes.
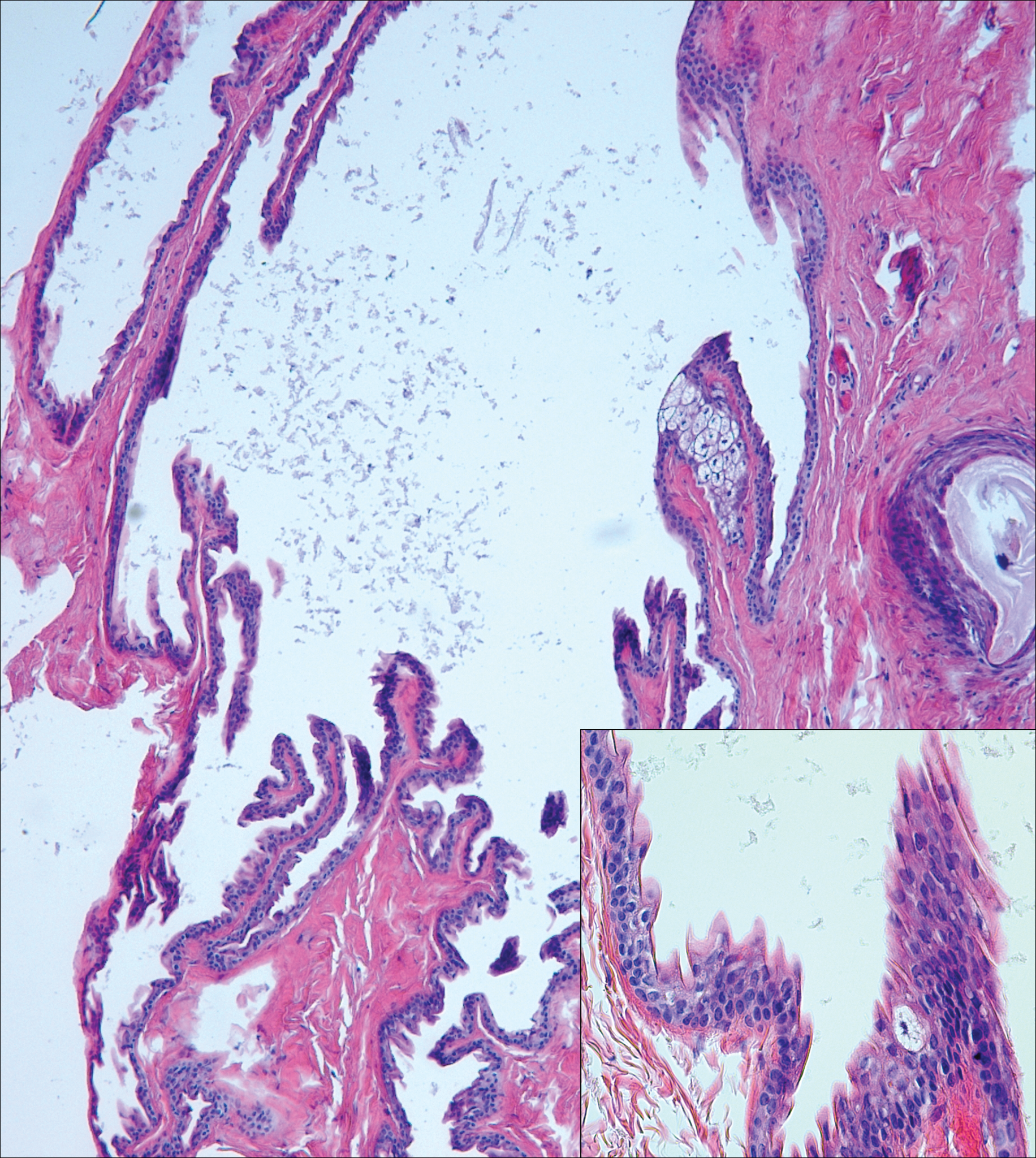
Similar to bronchogenic cysts, thyroglossal duct cysts (Figure 5) present on the midline neck, though they characteristically move with swallowing. The thyroglossal duct develops as the thyroid migrates from the floor of the pharynx to the anterior neck. Remnants of this duct result in the thyroglossal duct cyst.2 These cysts contain a respiratory-type epithelial lining and are distinguished by distinct thyroid follicles and lymphoid aggregates surrounding the cyst wall. Unlike bronchogenic cysts, they do not contain smooth muscle.3,4 Excision is curative.
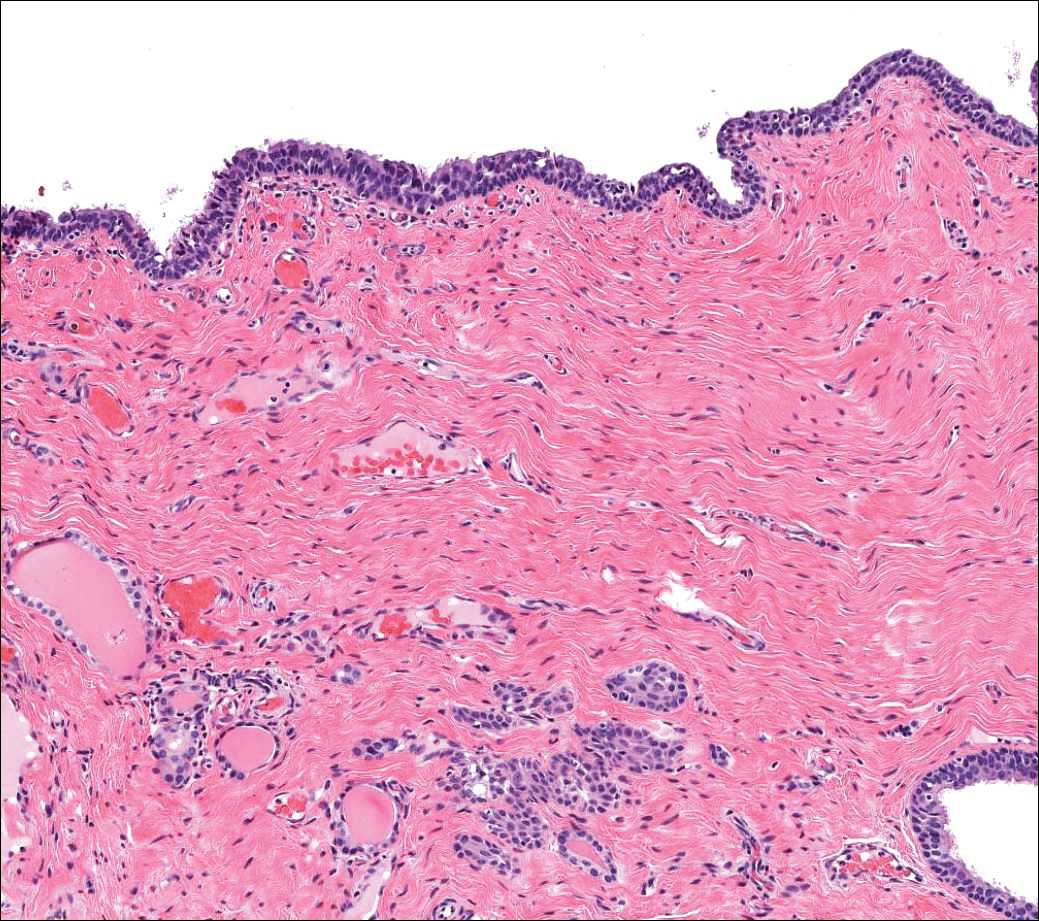
- Chavan S, Deshmukh R, Karande P, et al. Branchial cleft cyst: a case report and review of literature. J Oral Maxillofac Pathol. 2014;18:150.
- Stone MS. Cysts. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012:1817-1828.
- Kirkham N, Aljefri K. Tumors and cysts of the epidermis. In: Elder DE, Elenitsas R, Rosenbach M, et al, eds. Lever's Histopathology of the Skin. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015:969-1024.
- Elston DM. Benign tumors and cysts of the epidermis. In: Elston DM, Ferringer T, et al. Dermatopathology. 2nd ed. Philadelphia, PA: Elsevier/Saunders; 2014:49-55.
- Hsu CG, Heller M, Johnston GS, et al. An unusual cause of airway compromise in the emergency department: mediastinal bronchogenic cyst [published online December 13, 2016]. J Emerg Med. 2017;52:E91-E93.
Branchial Cleft Cyst
Cystic lesions present in a myriad of ways and often require histopathologic examination for definitive diagnosis. Correct identification of the cells comprising the lining of the cyst and the composition of the surrounding tissue are utilized to classify these lesions.
Branchial cleft cysts (quiz image, Figure 1) most commonly present as a soft tissue swelling of the lateral neck anterior to the sternocleidomastoid; they also can present in the preauricular or mandibular region.1,2 Although the cyst is present at birth, it typically is not clinically apparent until the second or third decades of life. The origin of branchial cleft cysts is subject to some debate; however, the prevailing theory is that they result from failure of obliteration of the second branchial arch during development.1 Histopathologically, branchial cleft cysts are characterized by a stratified squamous epithelial lining and abundant lymphoid tissue with germinal centers.3,4 Infection is a common reason for presentation and excision is curative.
Bronchogenic cysts (Figure 2) present as midline lesions in the suprasternal notch and can present clinically due to compression of the airway.5 They develop as anomalies of the primitive foregut, budding off of the tracheobronchial tree. Similar to respiratory tissue, they are lined with a ciliated pseudostratified columnar epithelium and contain goblet cells. Concentric smooth muscle often surrounds the cyst and cartilage may be present.4 Excision is curative and recommended if the cyst encroaches on vital structures.
Median raphe cysts occur most commonly on the ventral surface of the penis on or near the glans (Figure 3). These cysts are thought to result from anomalous budding from the urethral epithelium, though they do not maintain contact with the urethra.3 The lining varies in thickness from 1 to 4 cell layers and mimics the transitional epithelium of the urethra. Amorphous debris often is seen within the cyst, and surrounding genital tissue can be appreciated by identification of delicate collagen, smooth muscle, and numerous small nerves and vessels.3,4 Excision is curative and often is sought when the cyst becomes irritated or cosmetically bothersome.
Steatocystomas can present as solitary (steatocystoma simplex) or multiple lesions (steatocystoma multiplex)(Figure 4). They present as small, well-defined, yellow cystic papules most commonly on the chest, axilla, or groin.2 Their lining is composed of a stratified squamous epithelium that lacks a granular layer and contains a distinct overlying corrugated "shark tooth" eosinophilic cuticle. Sebaceous lobules are characteristically present along or within the cyst wall.3,4 Excision is curative and treatment often is sought for cosmetic purposes.
Similar to bronchogenic cysts, thyroglossal duct cysts (Figure 5) present on the midline neck, though they characteristically move with swallowing. The thyroglossal duct develops as the thyroid migrates from the floor of the pharynx to the anterior neck. Remnants of this duct result in the thyroglossal duct cyst.2 These cysts contain a respiratory-type epithelial lining and are distinguished by distinct thyroid follicles and lymphoid aggregates surrounding the cyst wall. Unlike bronchogenic cysts, they do not contain smooth muscle.3,4 Excision is curative.
Branchial Cleft Cyst
Cystic lesions present in a myriad of ways and often require histopathologic examination for definitive diagnosis. Correct identification of the cells comprising the lining of the cyst and the composition of the surrounding tissue are utilized to classify these lesions.
Branchial cleft cysts (quiz image, Figure 1) most commonly present as a soft tissue swelling of the lateral neck anterior to the sternocleidomastoid; they also can present in the preauricular or mandibular region.1,2 Although the cyst is present at birth, it typically is not clinically apparent until the second or third decades of life. The origin of branchial cleft cysts is subject to some debate; however, the prevailing theory is that they result from failure of obliteration of the second branchial arch during development.1 Histopathologically, branchial cleft cysts are characterized by a stratified squamous epithelial lining and abundant lymphoid tissue with germinal centers.3,4 Infection is a common reason for presentation and excision is curative.
Bronchogenic cysts (Figure 2) present as midline lesions in the suprasternal notch and can present clinically due to compression of the airway.5 They develop as anomalies of the primitive foregut, budding off of the tracheobronchial tree. Similar to respiratory tissue, they are lined with a ciliated pseudostratified columnar epithelium and contain goblet cells. Concentric smooth muscle often surrounds the cyst and cartilage may be present.4 Excision is curative and recommended if the cyst encroaches on vital structures.
Median raphe cysts occur most commonly on the ventral surface of the penis on or near the glans (Figure 3). These cysts are thought to result from anomalous budding from the urethral epithelium, though they do not maintain contact with the urethra.3 The lining varies in thickness from 1 to 4 cell layers and mimics the transitional epithelium of the urethra. Amorphous debris often is seen within the cyst, and surrounding genital tissue can be appreciated by identification of delicate collagen, smooth muscle, and numerous small nerves and vessels.3,4 Excision is curative and often is sought when the cyst becomes irritated or cosmetically bothersome.
Steatocystomas can present as solitary (steatocystoma simplex) or multiple lesions (steatocystoma multiplex)(Figure 4). They present as small, well-defined, yellow cystic papules most commonly on the chest, axilla, or groin.2 Their lining is composed of a stratified squamous epithelium that lacks a granular layer and contains a distinct overlying corrugated "shark tooth" eosinophilic cuticle. Sebaceous lobules are characteristically present along or within the cyst wall.3,4 Excision is curative and treatment often is sought for cosmetic purposes.
Similar to bronchogenic cysts, thyroglossal duct cysts (Figure 5) present on the midline neck, though they characteristically move with swallowing. The thyroglossal duct develops as the thyroid migrates from the floor of the pharynx to the anterior neck. Remnants of this duct result in the thyroglossal duct cyst.2 These cysts contain a respiratory-type epithelial lining and are distinguished by distinct thyroid follicles and lymphoid aggregates surrounding the cyst wall. Unlike bronchogenic cysts, they do not contain smooth muscle.3,4 Excision is curative.
- Chavan S, Deshmukh R, Karande P, et al. Branchial cleft cyst: a case report and review of literature. J Oral Maxillofac Pathol. 2014;18:150.
- Stone MS. Cysts. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012:1817-1828.
- Kirkham N, Aljefri K. Tumors and cysts of the epidermis. In: Elder DE, Elenitsas R, Rosenbach M, et al, eds. Lever's Histopathology of the Skin. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015:969-1024.
- Elston DM. Benign tumors and cysts of the epidermis. In: Elston DM, Ferringer T, et al. Dermatopathology. 2nd ed. Philadelphia, PA: Elsevier/Saunders; 2014:49-55.
- Hsu CG, Heller M, Johnston GS, et al. An unusual cause of airway compromise in the emergency department: mediastinal bronchogenic cyst [published online December 13, 2016]. J Emerg Med. 2017;52:E91-E93.
- Chavan S, Deshmukh R, Karande P, et al. Branchial cleft cyst: a case report and review of literature. J Oral Maxillofac Pathol. 2014;18:150.
- Stone MS. Cysts. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012:1817-1828.
- Kirkham N, Aljefri K. Tumors and cysts of the epidermis. In: Elder DE, Elenitsas R, Rosenbach M, et al, eds. Lever's Histopathology of the Skin. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015:969-1024.
- Elston DM. Benign tumors and cysts of the epidermis. In: Elston DM, Ferringer T, et al. Dermatopathology. 2nd ed. Philadelphia, PA: Elsevier/Saunders; 2014:49-55.
- Hsu CG, Heller M, Johnston GS, et al. An unusual cause of airway compromise in the emergency department: mediastinal bronchogenic cyst [published online December 13, 2016]. J Emerg Med. 2017;52:E91-E93.
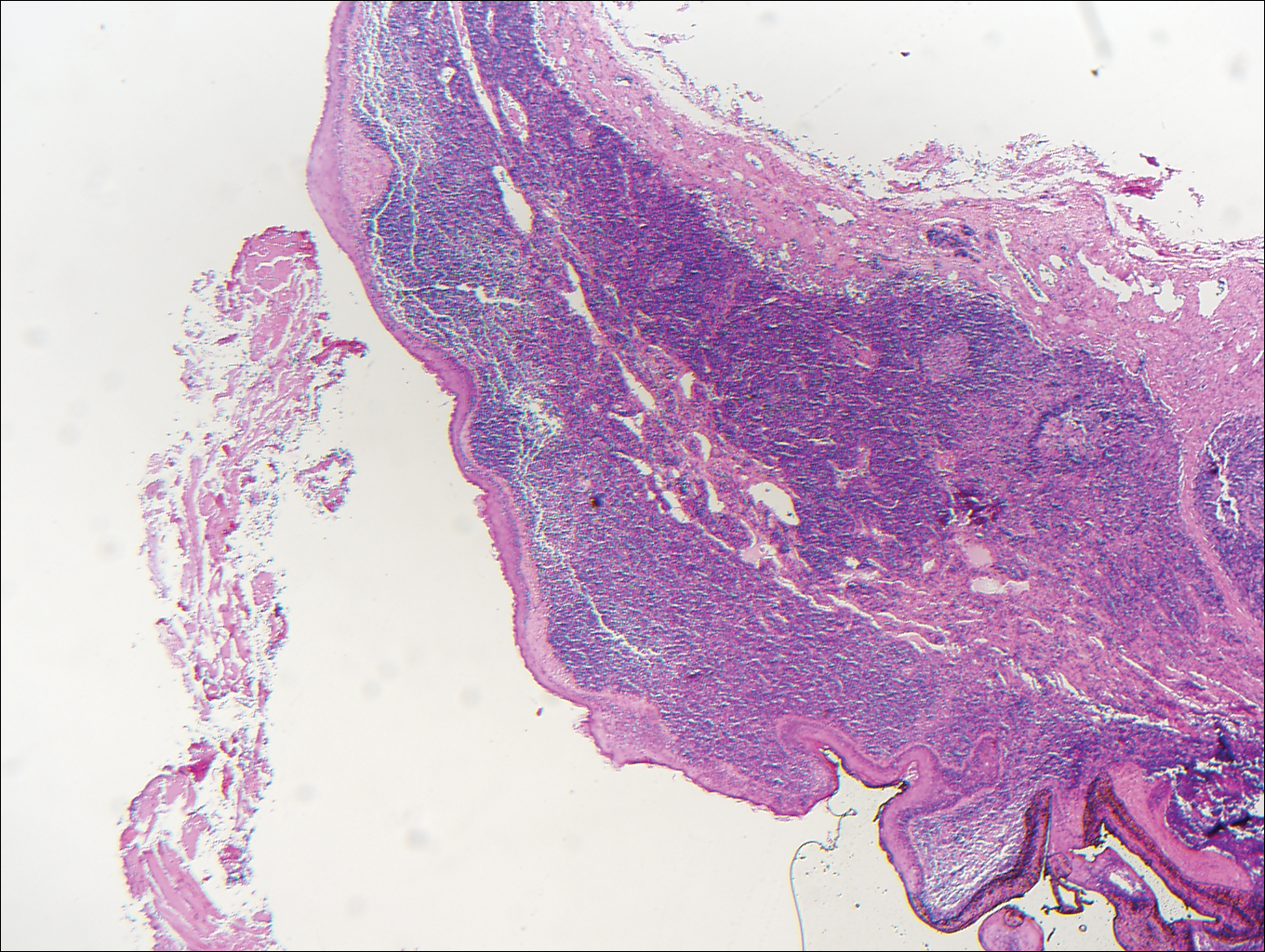
A 14-year-old adolescent boy presented with a nontender mass on the left lateral neck. The mass had been present since birth but had recently grown in size.
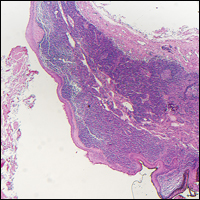
Pruritic Rash on the Buttock
Cutaneous Larva Migrans
Cutaneous larva migrans (CLM) is caused by the larval migration of animal hookworms. Ancylostoma braziliense, Ancylostoma ceylanicum, and Ancylostoma caninum are the species most commonly associated with the disease. The hookworm is endemic to tropical and subtropical climates in areas such as Africa, Southeast Asia, South America, and the southeastern United States.1 Although cats and dogs are most commonly affected, humans can be infected if they are exposed to sand or soil containing hookworm larvae, often due to contamination from animal feces.2 Cutaneous larva migrans is characterized by pruritic erythematous papules and linear or serpiginous, reddish brown, elevated tracks most commonly appearing on the feet, buttocks, thighs, and lower legs; however, lesions can appear anywhere. In human hosts, the larvae travel in the epidermis and are unable to invade the dermis; it is speculated that they lack the collagenase enzymes required to penetrate the basement membrane before invading the dermis.2
On histopathology, there typically are small cavities in the epidermis corresponding to the track of the larvae.3 There often is a spongiotic dermatitis with a mixed inflammatory infiltrate following the larvae with scattered eosinophils. The migrating larvae may be up to 1 mm in size and have bilateral double alae, or winglike projections, on the side of the body (Figure 1).4 The larvae are difficult to find on histopathology because they often travel beyond the areas that demonstrate clinical findings. The diagnosis of CLM is mostly clinical, but if a biopsy is performed, the specimen should be taken ahead of the track.
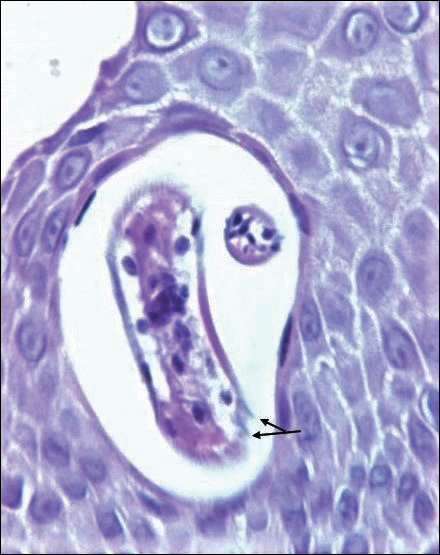
Disseminated strongyloidiasis is caused by Strongyloides stercoralis. When filariform larvae migrate out of the intestinal tract into the skin, they can cause an urticarial rash and serpiginous patterns on the skin that can move 5 to 15 cm per hour, a clinical condition known as larva currens. In immunocompromised individuals, there can be hyperinfection with diffuse petechial thumbprint purpura seen clinically, which characteristically radiate from the periumbilical area.1 On pathology, there may be numerous larvae found between the dermal collagen bundles, measuring 9 to 15 µm in diameter. Rarely, they also can be found in small blood vessels.3 They often are accompanied by extravasated red blood cells in the tissues (Figure 2).
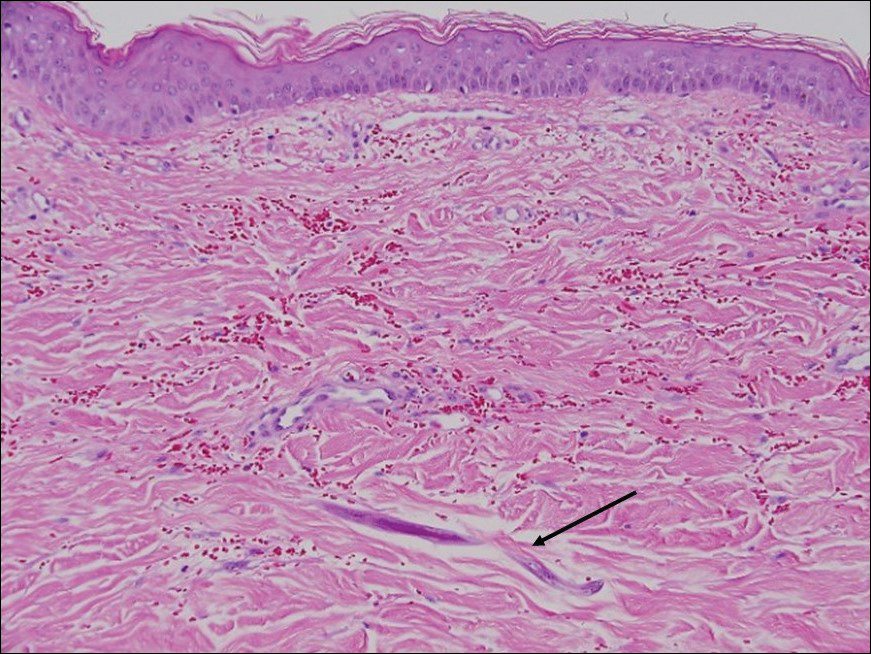
Myiasis represents the largest pathogen in the differential diagnosis for CLM. In myiasis, fly larvae will infest human tissue, usually by forming a small cavity in the dermis or subcutaneous tissue. The larvae are visible to the human eye and can be up to several centimeters in length. In the skin, the histology of myiasis usually is accompanied by a heavy mixed inflammatory cell infiltrate with many eosinophils. Fragments of the larvae are seen encased by a thick chitinous cuticle with widely spaced spines or pigmented setae (Figure 3) on the surface of the cuticle.5 Layers of striated muscle and internal organs may be seen beneath the cuticle.3

Onchocerciasis, or river blindness, is a parasitic disease caused by Onchocerca volvulus that is most often seen in sub-Saharan Africa. It may cause the skin finding of an onchocercoma, a subcutaneous nodule made up of Onchocerca nematodes. However, when the filaria disseminate, it may cause onchocerciasis with cutaneous findings of an eczematous dermatitis with itching and lichenification.1 In onchocercal dermatitis, microfilariae may be found in the dermis and there may be a mild dermal chronic inflammatory infiltrate with eosinophils.3 These microfilariae are smaller than Strongyloides larvae (Figure 4).
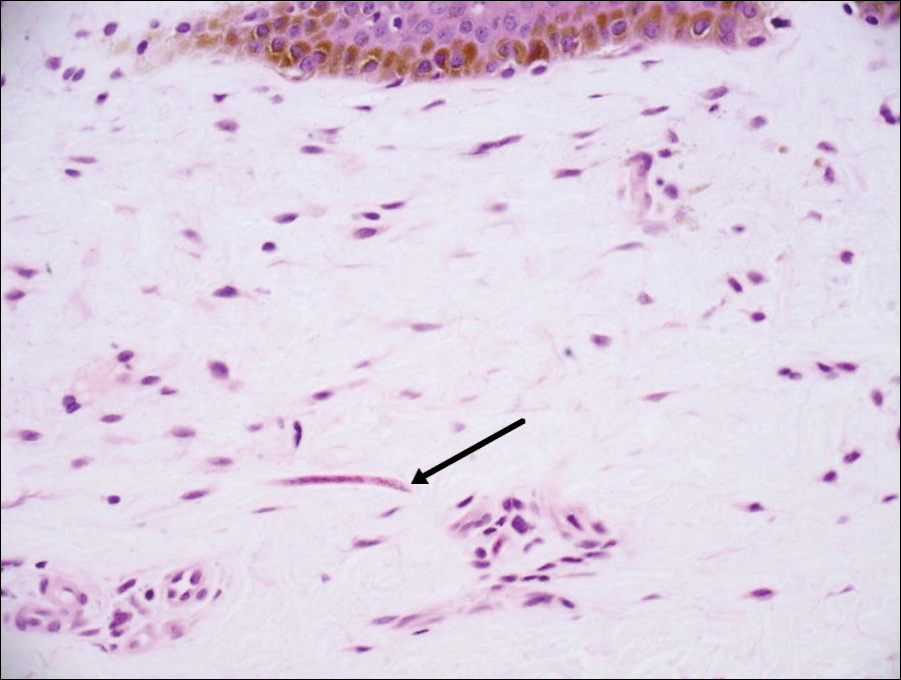
Sarcoptes scabiei are mites that are pathologically found limited to the stratum corneum. There often is a spongiotic dermatitis as the mite travels with an accompanying mixed cell inflammatory infiltrate with many eosinophils. One or more mites may be seen with or without eggs and excreta or scybala (Figure 5). Pink pigtails may be seen connected to the stratum corneum, representing egg fragments or casings left behind after the mite hatches.3 The female mite measures up to 0.4 mm in length.3
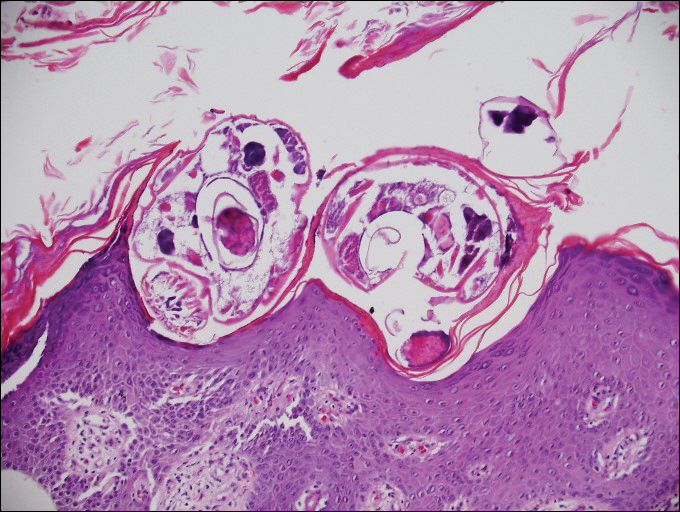
- Lupi O, Downing C, Lee M, et al. Mucocutaneous manifestations of helminth infections. J Am Acad Dermatol. 2015;73:929-944.
- James WD, Berger T, Elston D. Andrews' Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2016.
- Patterson J. Weedon's Skin Pathology. 4th ed. London, England: Churchill Livingstone Elsevier; 2016.
- Milner D. Diagnostic Pathology: Infectious Diseases. Philadelphia, PA: Elsevier; 2015.
- Ferringer T, Peckham S, Ko CJ, et al. Dermatopathology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2013.
Cutaneous Larva Migrans
Cutaneous larva migrans (CLM) is caused by the larval migration of animal hookworms. Ancylostoma braziliense, Ancylostoma ceylanicum, and Ancylostoma caninum are the species most commonly associated with the disease. The hookworm is endemic to tropical and subtropical climates in areas such as Africa, Southeast Asia, South America, and the southeastern United States.1 Although cats and dogs are most commonly affected, humans can be infected if they are exposed to sand or soil containing hookworm larvae, often due to contamination from animal feces.2 Cutaneous larva migrans is characterized by pruritic erythematous papules and linear or serpiginous, reddish brown, elevated tracks most commonly appearing on the feet, buttocks, thighs, and lower legs; however, lesions can appear anywhere. In human hosts, the larvae travel in the epidermis and are unable to invade the dermis; it is speculated that they lack the collagenase enzymes required to penetrate the basement membrane before invading the dermis.2
On histopathology, there typically are small cavities in the epidermis corresponding to the track of the larvae.3 There often is a spongiotic dermatitis with a mixed inflammatory infiltrate following the larvae with scattered eosinophils. The migrating larvae may be up to 1 mm in size and have bilateral double alae, or winglike projections, on the side of the body (Figure 1).4 The larvae are difficult to find on histopathology because they often travel beyond the areas that demonstrate clinical findings. The diagnosis of CLM is mostly clinical, but if a biopsy is performed, the specimen should be taken ahead of the track.
Disseminated strongyloidiasis is caused by Strongyloides stercoralis. When filariform larvae migrate out of the intestinal tract into the skin, they can cause an urticarial rash and serpiginous patterns on the skin that can move 5 to 15 cm per hour, a clinical condition known as larva currens. In immunocompromised individuals, there can be hyperinfection with diffuse petechial thumbprint purpura seen clinically, which characteristically radiate from the periumbilical area.1 On pathology, there may be numerous larvae found between the dermal collagen bundles, measuring 9 to 15 µm in diameter. Rarely, they also can be found in small blood vessels.3 They often are accompanied by extravasated red blood cells in the tissues (Figure 2).
Myiasis represents the largest pathogen in the differential diagnosis for CLM. In myiasis, fly larvae will infest human tissue, usually by forming a small cavity in the dermis or subcutaneous tissue. The larvae are visible to the human eye and can be up to several centimeters in length. In the skin, the histology of myiasis usually is accompanied by a heavy mixed inflammatory cell infiltrate with many eosinophils. Fragments of the larvae are seen encased by a thick chitinous cuticle with widely spaced spines or pigmented setae (Figure 3) on the surface of the cuticle.5 Layers of striated muscle and internal organs may be seen beneath the cuticle.3
Onchocerciasis, or river blindness, is a parasitic disease caused by Onchocerca volvulus that is most often seen in sub-Saharan Africa. It may cause the skin finding of an onchocercoma, a subcutaneous nodule made up of Onchocerca nematodes. However, when the filaria disseminate, it may cause onchocerciasis with cutaneous findings of an eczematous dermatitis with itching and lichenification.1 In onchocercal dermatitis, microfilariae may be found in the dermis and there may be a mild dermal chronic inflammatory infiltrate with eosinophils.3 These microfilariae are smaller than Strongyloides larvae (Figure 4).
Sarcoptes scabiei are mites that are pathologically found limited to the stratum corneum. There often is a spongiotic dermatitis as the mite travels with an accompanying mixed cell inflammatory infiltrate with many eosinophils. One or more mites may be seen with or without eggs and excreta or scybala (Figure 5). Pink pigtails may be seen connected to the stratum corneum, representing egg fragments or casings left behind after the mite hatches.3 The female mite measures up to 0.4 mm in length.3
Cutaneous Larva Migrans
Cutaneous larva migrans (CLM) is caused by the larval migration of animal hookworms. Ancylostoma braziliense, Ancylostoma ceylanicum, and Ancylostoma caninum are the species most commonly associated with the disease. The hookworm is endemic to tropical and subtropical climates in areas such as Africa, Southeast Asia, South America, and the southeastern United States.1 Although cats and dogs are most commonly affected, humans can be infected if they are exposed to sand or soil containing hookworm larvae, often due to contamination from animal feces.2 Cutaneous larva migrans is characterized by pruritic erythematous papules and linear or serpiginous, reddish brown, elevated tracks most commonly appearing on the feet, buttocks, thighs, and lower legs; however, lesions can appear anywhere. In human hosts, the larvae travel in the epidermis and are unable to invade the dermis; it is speculated that they lack the collagenase enzymes required to penetrate the basement membrane before invading the dermis.2
On histopathology, there typically are small cavities in the epidermis corresponding to the track of the larvae.3 There often is a spongiotic dermatitis with a mixed inflammatory infiltrate following the larvae with scattered eosinophils. The migrating larvae may be up to 1 mm in size and have bilateral double alae, or winglike projections, on the side of the body (Figure 1).4 The larvae are difficult to find on histopathology because they often travel beyond the areas that demonstrate clinical findings. The diagnosis of CLM is mostly clinical, but if a biopsy is performed, the specimen should be taken ahead of the track.
Disseminated strongyloidiasis is caused by Strongyloides stercoralis. When filariform larvae migrate out of the intestinal tract into the skin, they can cause an urticarial rash and serpiginous patterns on the skin that can move 5 to 15 cm per hour, a clinical condition known as larva currens. In immunocompromised individuals, there can be hyperinfection with diffuse petechial thumbprint purpura seen clinically, which characteristically radiate from the periumbilical area.1 On pathology, there may be numerous larvae found between the dermal collagen bundles, measuring 9 to 15 µm in diameter. Rarely, they also can be found in small blood vessels.3 They often are accompanied by extravasated red blood cells in the tissues (Figure 2).
Myiasis represents the largest pathogen in the differential diagnosis for CLM. In myiasis, fly larvae will infest human tissue, usually by forming a small cavity in the dermis or subcutaneous tissue. The larvae are visible to the human eye and can be up to several centimeters in length. In the skin, the histology of myiasis usually is accompanied by a heavy mixed inflammatory cell infiltrate with many eosinophils. Fragments of the larvae are seen encased by a thick chitinous cuticle with widely spaced spines or pigmented setae (Figure 3) on the surface of the cuticle.5 Layers of striated muscle and internal organs may be seen beneath the cuticle.3
Onchocerciasis, or river blindness, is a parasitic disease caused by Onchocerca volvulus that is most often seen in sub-Saharan Africa. It may cause the skin finding of an onchocercoma, a subcutaneous nodule made up of Onchocerca nematodes. However, when the filaria disseminate, it may cause onchocerciasis with cutaneous findings of an eczematous dermatitis with itching and lichenification.1 In onchocercal dermatitis, microfilariae may be found in the dermis and there may be a mild dermal chronic inflammatory infiltrate with eosinophils.3 These microfilariae are smaller than Strongyloides larvae (Figure 4).
Sarcoptes scabiei are mites that are pathologically found limited to the stratum corneum. There often is a spongiotic dermatitis as the mite travels with an accompanying mixed cell inflammatory infiltrate with many eosinophils. One or more mites may be seen with or without eggs and excreta or scybala (Figure 5). Pink pigtails may be seen connected to the stratum corneum, representing egg fragments or casings left behind after the mite hatches.3 The female mite measures up to 0.4 mm in length.3
- Lupi O, Downing C, Lee M, et al. Mucocutaneous manifestations of helminth infections. J Am Acad Dermatol. 2015;73:929-944.
- James WD, Berger T, Elston D. Andrews' Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2016.
- Patterson J. Weedon's Skin Pathology. 4th ed. London, England: Churchill Livingstone Elsevier; 2016.
- Milner D. Diagnostic Pathology: Infectious Diseases. Philadelphia, PA: Elsevier; 2015.
- Ferringer T, Peckham S, Ko CJ, et al. Dermatopathology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Lupi O, Downing C, Lee M, et al. Mucocutaneous manifestations of helminth infections. J Am Acad Dermatol. 2015;73:929-944.
- James WD, Berger T, Elston D. Andrews' Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2016.
- Patterson J. Weedon's Skin Pathology. 4th ed. London, England: Churchill Livingstone Elsevier; 2016.
- Milner D. Diagnostic Pathology: Infectious Diseases. Philadelphia, PA: Elsevier; 2015.
- Ferringer T, Peckham S, Ko CJ, et al. Dermatopathology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2013.
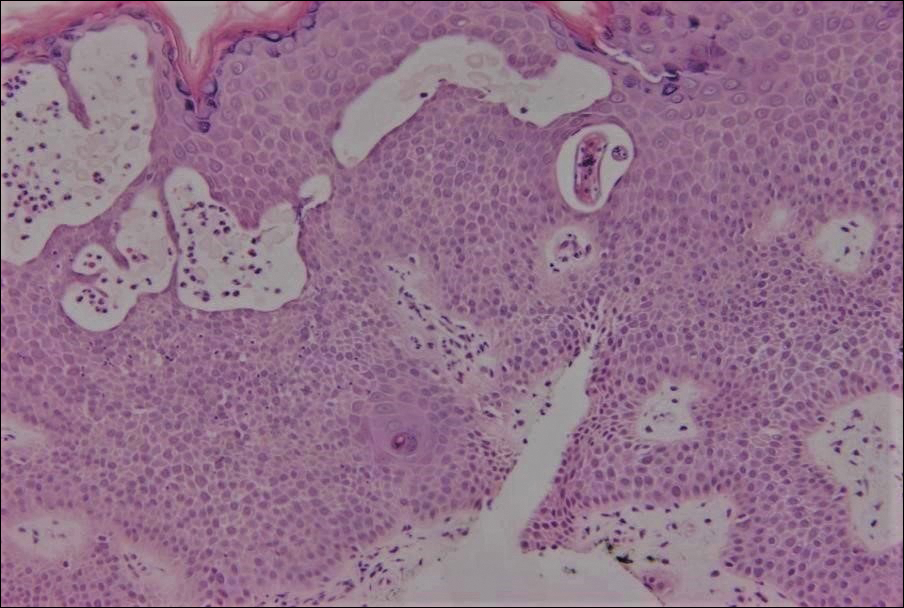
An 18-year-old man presented with a several-week history of an expanding pruritic serpiginous and linear eruption on the buttock. The patient recently had spent some time vacationing at the beach in the southeastern United States. Physical examination revealed erythematous linear papules and serpiginous raised tracks on the buttock. A biopsy of the lesion was performed.
Erythematous Pearly Papule on the Chest
Primary Cutaneous B-cell Lymphoma
Cutaneous B-cell lymphomas (CBCLs) are a diverse but rare group of cutaneous lymphoproliferative neoplasms that make up approximately 20% of the total number of hematolymphoid neoplasms primary to the skin.1 These lymphomas are comprised of neoplastic B cells in various stages of differentiation. As a whole, they are rare neoplasms that primarily involve the head, neck, trunk, arms, or legs.1 Clinically, patients present with nontender, compressible, solitary, red to violaceous papules or nodules. Most CBCLs are considered low-grade malignancies with nonaggressive behavior and excellent prognosis; however, the diffuse large B-cell lymphomas, including but not limited to intravascular and leg type; lymphomatoid granulomatosis; and B-cell lymphoblastic lymphoma can act more aggressively.1
Histopathologic examination of primary CBCL generally reveals a relatively normal epidermis accompanied by a nodular to diffuse monomorphic lymphocytic cellular infiltrate in the dermis that can occasionally extend into the subcutaneous tissue (quiz image). Although not specific for CBCLs, oftentimes there is an acellular portion of the superficial papillary dermis known as a grenz zone that can serve as a histopathologic clue to the diagnosis of a cutaneous lymphoproliferative disorder. The list of malignant B-cell neoplasms is extensive (eg, cutaneous marginal zone B-cell lymphoma, primary cutaneous follicle center lymphoma, diffuse large B-cell lymphoma, intravascular large B-cell lymphoma), and few are seen in the skin.
The most common type of CBCL is marginal zone B-cell lymphoma, which is considered to be a tumor of mucosa-associated (or skin-associated) lymphoid tissue. It is characterized by a monomorphous population of small mature lymphocytes showing characteristics of the B cells of the marginal zone of the lymph node. Some cells have the features of centrocytes/centroblasts (Figure 1) demonstrated by slightly irregular or indented nuclei and generous amounts of cytoplasm. Larger and more pleomorphic cells such as immunoblasts are similarly noted (Figure 1). The quiz image and Figure 1 demonstrate a cutaneous marginal zone B-cell lymphoma. A histomorphologic clue supporting a diagnosis of marginal zone B-cell lymphoma over reactive lymphoid hyperplasia is a B-cell predominate (B- to T-cell ratio of at least 3 to 1) infiltrate that is comprised of marginal zone-type cells. Immunohistochemistry demonstrating fewer differentiated B cells with light chain restriction may provide additional evidence that supports a clonal and potentially malignant process.
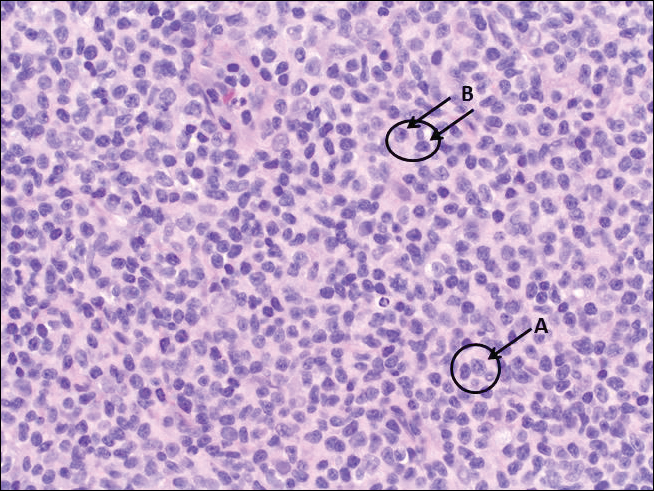
Erythematous to violaceous nodules on the head and neck of older individuals are characteristic of both granuloma faciale and CBCL. Histologically, granuloma faciale is characterized by a dense cellular infiltrate, often with a nodular outline, occupying the mid dermis.2 Granuloma faciale typically spares the immediate subepidermis and hair follicles, forming a grenz zone. The cellular infiltrate is polymorphic and consists of eosinophils and neutrophils with scattered plasma cells, mast cells, and lymphocytes in a vasculocentric distribution, eventually with chronic concentric fibrosis (Figure 2).
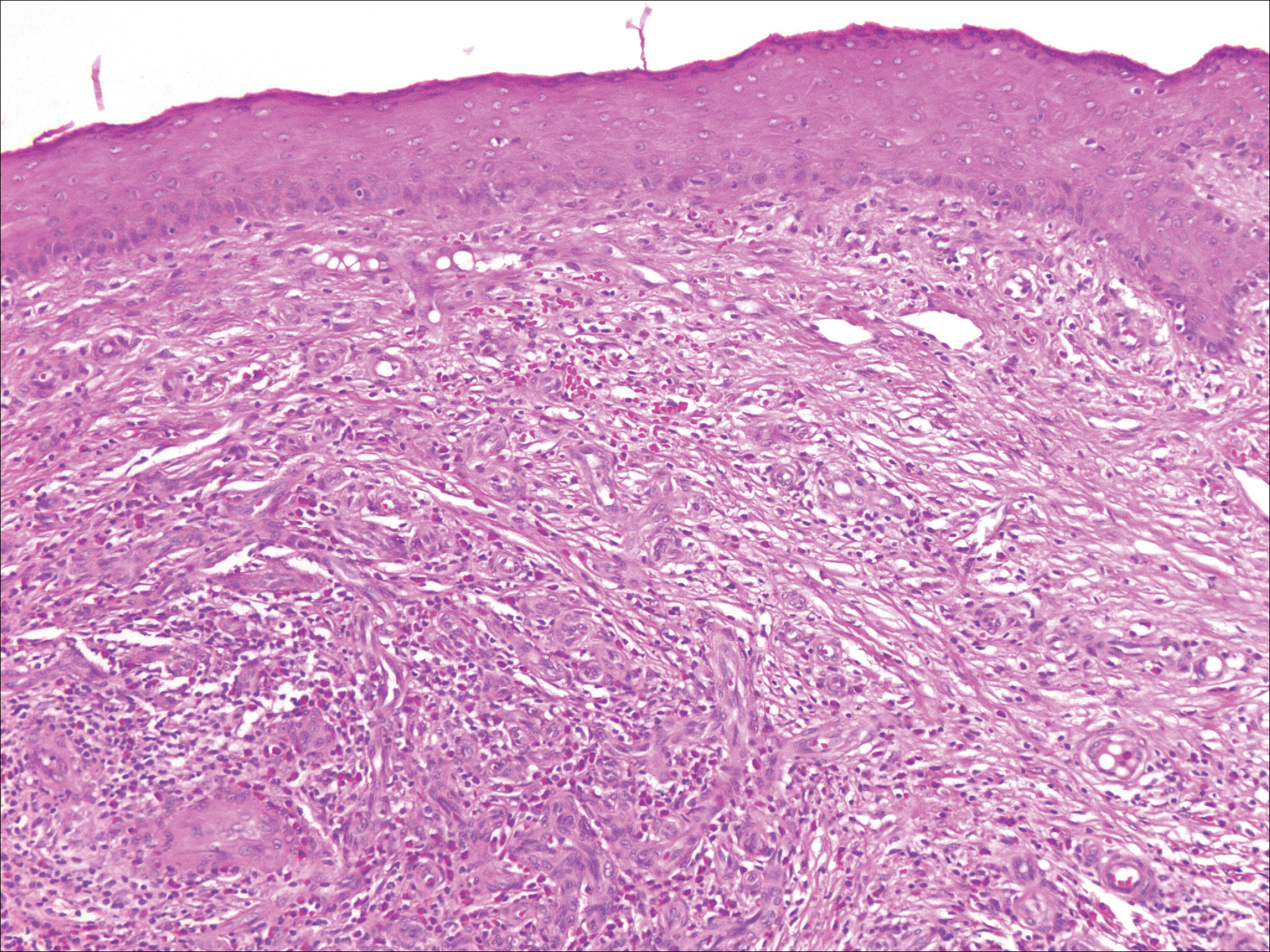
Leukemia cutis demonstrates a dermal infiltrate that contains atypical mononuclear cells (myeloblasts and myelocytes)(Figure 3).3 These markedly atypical mononuclear cells can have kidney bean-shaped nuclei and percolate through the dermal collagen, resembling single-file cells. They have increased nuclear to cytoplasmic ratios and occasionally have prominent nucleoli. Correlation with immunophenotypic and cytochemical studies is required for specific typing of the leukemic infiltrate.
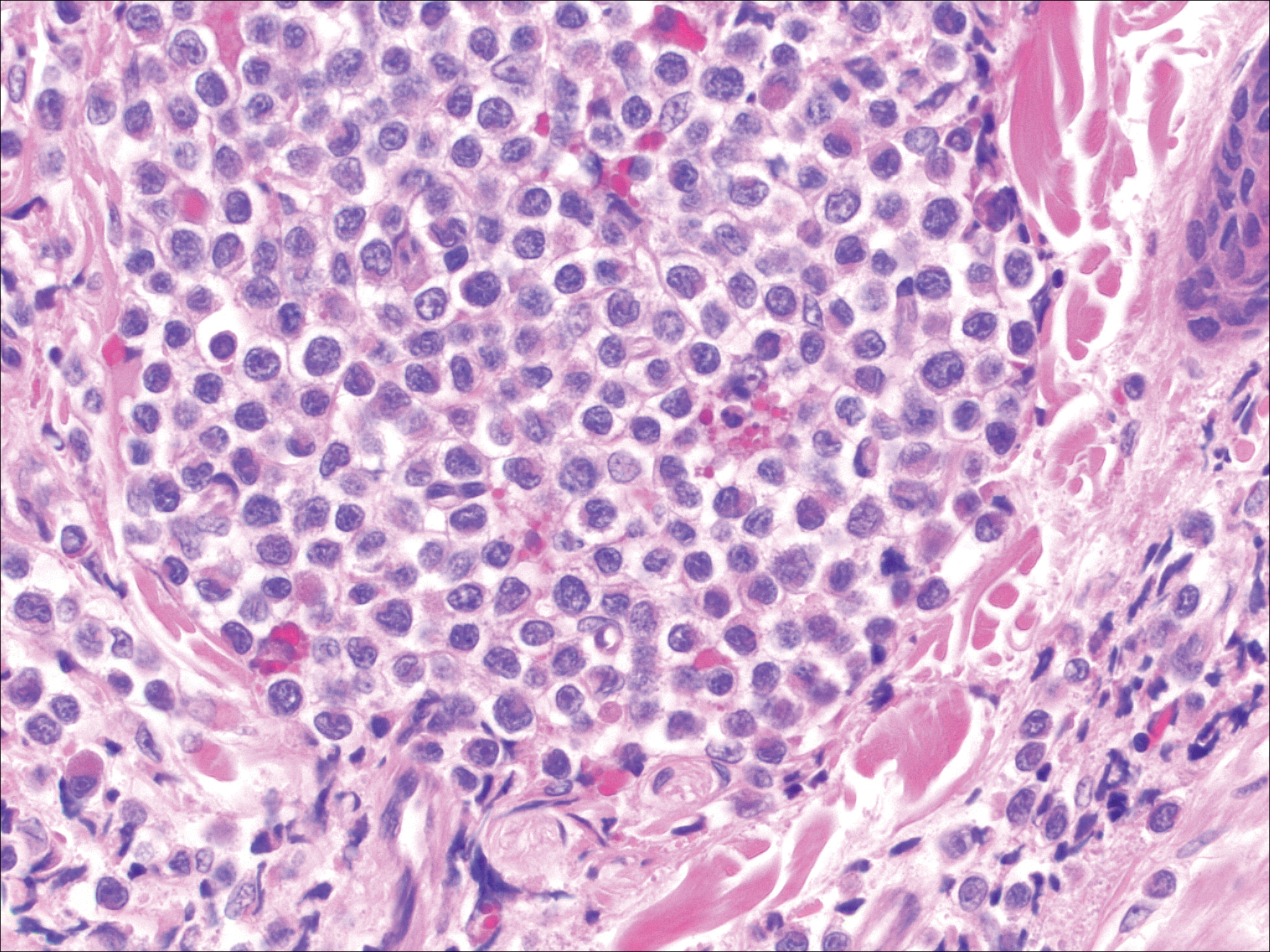
Similar to primary CBCL, lymphomatoid papulosis (LyP) consists of erythematous papules or nodules that can occur anywhere on the body. In contrast to CBCL, the lesions of LyP classically self-resolve. However, approximately 10% to 20% of patients develop a malignant lymphoma, with mycosis fungoides, Hodgkin disease, and anaplastic large cell lymphoma being the most commonly associated.
Histologic examination of lesions of LyP classically demonstrates a wedge-shaped dermal infiltrate with variable epidermal changes (Figure 4). The wedge-shaped infiltrate is composed of large atypical cells. Three main types of lesions have been delineated: types A, B, and C. Type A is characterized by an increased number of cells with large vesicular nuclei with clumped chromatin, prominent nucleoli, and pronounced cytoplasm. Reed-Sternberg-like cells with an admixture of inflammatory cells including small lymphocytes, macrophages, neutrophils, and eosinophils also are present. Type B neoplastic cells vary in size and feature hyperchromatic, convoluted, or cerebriform nuclei. The infiltrate can be dense and bandlike with fewer cells resembling mycosis fungoides; type B LyP has neoplastic cells, not inflammatory cells. Finally, type C demonstrates solid sheets of large atypical cells resembling anaplastic large cell lymphoma. Immunohistochemically, the atypical cells often are CD4+ and CD8- with variable loss of pan-T-cell antigens. The atypical cells of types A and C express CD30 reactivity.4
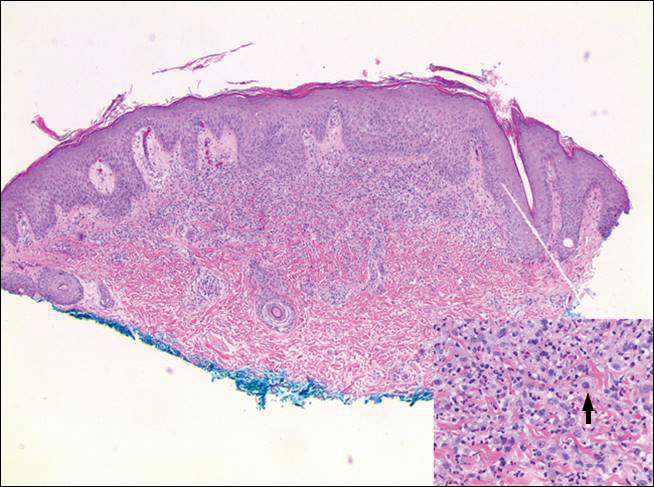
Merkel cell carcinoma (MCC) is a primary neuroendocrine carcinoma of the skin that usually arises on sun-exposed skin in elderly patients with lesions that histologically and clinically resemble cutaneous lymphoma.5 It classically is composed of small, round to oval, basophilic cells with a vesicular nucleus and multiple small nucleoli. Apoptotic cells and mitoses often are present.6 One key finding that helps to differentiate MCC from lymphoma is the presence of finely dispersed salt-and-pepper chromatin and molded nuclear contour in MCC (Figure 5).
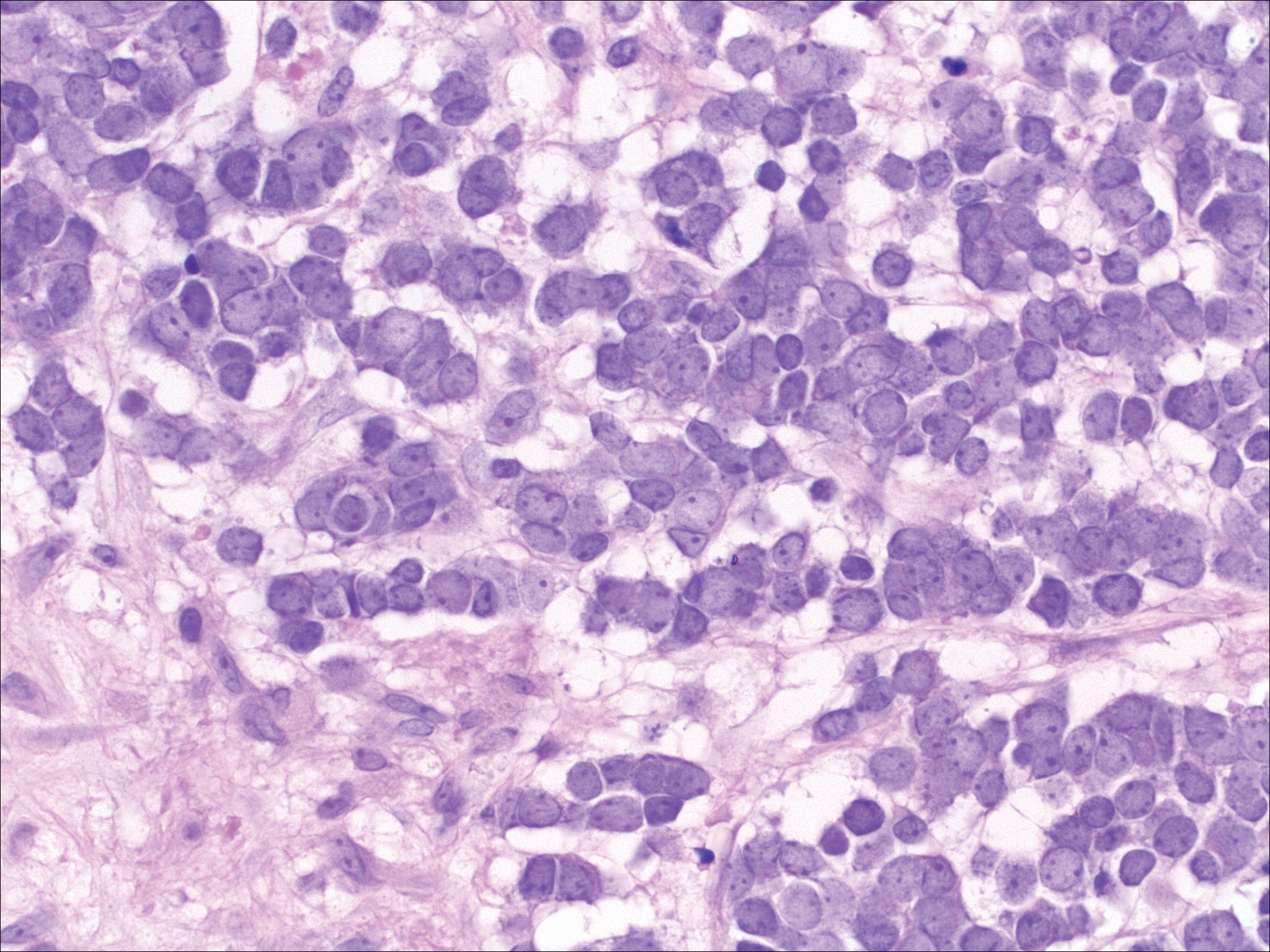
Immunophenotyping is important in the differentiation of these diagnoses. The atypical cells of LyP are positive for CD3, CD4, and CD30 but are negative for CD8. However, in type B LyP, the large CD30+ cells seen in the other types are not commonly seen. In contrast, MCC expresses reactivity with cytokeratins, in particular cytokeratin 20 and CAM5.2, classically in a paranuclear dotlike pattern. In keeping with MCC's neuroendocrine differentiation, the tumor cells will demonstrate reactivity with synaptophysin, chromogranin, and CD56. The immunohistochemistry for leukemia cutis varies depending on the type of leukemia. Acute myelomonocytic leukemia is positive for myeloperoxidase, CD13, CD33, and CD68. The immunophenotype of these marginal zone lymphoma cells is as follows: positive for CD20, CD79a, and Bcl-2; negative for Bcl-6, CD5, CD10, CD23, and cyclin D1 (Bcl-1).7
- Olsen EA. Evaluation, diagnosis, and staging of cutaneous lymphoma. Dermatol Clin. 2015;33:643-654.
- Ortonne N, Wechsler J, Bagot M, et al. Granuloma faciale: a clinicopathologic study of 66 patients. J Am Acad Dermatol. 2005;53:1002-1009.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Wieser I, Wohlmuth C, Nunez CA, et al. Lymphomatoid papulosis in children and adolescents: a systematic review. Am J Clin Dermatol. 2016;17:319-327.
- Sibley RK, Dehner LP, Rosai J. Primary neuroendocrine (Merkel cell?) carcinoma of the skin: I. a clinicopathologic and ultrastructural study of 43 cases. Am J Surg Pathol. 1985;9:95-108.
- Frigerio B, Capella C, Eusebi V, et al. Merkel cell carcinoma of the skin: the structure and origin of normal Merkel cells. Histopathology. 1983;7:229-249.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
Primary Cutaneous B-cell Lymphoma
Cutaneous B-cell lymphomas (CBCLs) are a diverse but rare group of cutaneous lymphoproliferative neoplasms that make up approximately 20% of the total number of hematolymphoid neoplasms primary to the skin.1 These lymphomas are comprised of neoplastic B cells in various stages of differentiation. As a whole, they are rare neoplasms that primarily involve the head, neck, trunk, arms, or legs.1 Clinically, patients present with nontender, compressible, solitary, red to violaceous papules or nodules. Most CBCLs are considered low-grade malignancies with nonaggressive behavior and excellent prognosis; however, the diffuse large B-cell lymphomas, including but not limited to intravascular and leg type; lymphomatoid granulomatosis; and B-cell lymphoblastic lymphoma can act more aggressively.1
Histopathologic examination of primary CBCL generally reveals a relatively normal epidermis accompanied by a nodular to diffuse monomorphic lymphocytic cellular infiltrate in the dermis that can occasionally extend into the subcutaneous tissue (quiz image). Although not specific for CBCLs, oftentimes there is an acellular portion of the superficial papillary dermis known as a grenz zone that can serve as a histopathologic clue to the diagnosis of a cutaneous lymphoproliferative disorder. The list of malignant B-cell neoplasms is extensive (eg, cutaneous marginal zone B-cell lymphoma, primary cutaneous follicle center lymphoma, diffuse large B-cell lymphoma, intravascular large B-cell lymphoma), and few are seen in the skin.
The most common type of CBCL is marginal zone B-cell lymphoma, which is considered to be a tumor of mucosa-associated (or skin-associated) lymphoid tissue. It is characterized by a monomorphous population of small mature lymphocytes showing characteristics of the B cells of the marginal zone of the lymph node. Some cells have the features of centrocytes/centroblasts (Figure 1) demonstrated by slightly irregular or indented nuclei and generous amounts of cytoplasm. Larger and more pleomorphic cells such as immunoblasts are similarly noted (Figure 1). The quiz image and Figure 1 demonstrate a cutaneous marginal zone B-cell lymphoma. A histomorphologic clue supporting a diagnosis of marginal zone B-cell lymphoma over reactive lymphoid hyperplasia is a B-cell predominate (B- to T-cell ratio of at least 3 to 1) infiltrate that is comprised of marginal zone-type cells. Immunohistochemistry demonstrating fewer differentiated B cells with light chain restriction may provide additional evidence that supports a clonal and potentially malignant process.
Erythematous to violaceous nodules on the head and neck of older individuals are characteristic of both granuloma faciale and CBCL. Histologically, granuloma faciale is characterized by a dense cellular infiltrate, often with a nodular outline, occupying the mid dermis.2 Granuloma faciale typically spares the immediate subepidermis and hair follicles, forming a grenz zone. The cellular infiltrate is polymorphic and consists of eosinophils and neutrophils with scattered plasma cells, mast cells, and lymphocytes in a vasculocentric distribution, eventually with chronic concentric fibrosis (Figure 2).
Leukemia cutis demonstrates a dermal infiltrate that contains atypical mononuclear cells (myeloblasts and myelocytes)(Figure 3).3 These markedly atypical mononuclear cells can have kidney bean-shaped nuclei and percolate through the dermal collagen, resembling single-file cells. They have increased nuclear to cytoplasmic ratios and occasionally have prominent nucleoli. Correlation with immunophenotypic and cytochemical studies is required for specific typing of the leukemic infiltrate.
Similar to primary CBCL, lymphomatoid papulosis (LyP) consists of erythematous papules or nodules that can occur anywhere on the body. In contrast to CBCL, the lesions of LyP classically self-resolve. However, approximately 10% to 20% of patients develop a malignant lymphoma, with mycosis fungoides, Hodgkin disease, and anaplastic large cell lymphoma being the most commonly associated.
Histologic examination of lesions of LyP classically demonstrates a wedge-shaped dermal infiltrate with variable epidermal changes (Figure 4). The wedge-shaped infiltrate is composed of large atypical cells. Three main types of lesions have been delineated: types A, B, and C. Type A is characterized by an increased number of cells with large vesicular nuclei with clumped chromatin, prominent nucleoli, and pronounced cytoplasm. Reed-Sternberg-like cells with an admixture of inflammatory cells including small lymphocytes, macrophages, neutrophils, and eosinophils also are present. Type B neoplastic cells vary in size and feature hyperchromatic, convoluted, or cerebriform nuclei. The infiltrate can be dense and bandlike with fewer cells resembling mycosis fungoides; type B LyP has neoplastic cells, not inflammatory cells. Finally, type C demonstrates solid sheets of large atypical cells resembling anaplastic large cell lymphoma. Immunohistochemically, the atypical cells often are CD4+ and CD8- with variable loss of pan-T-cell antigens. The atypical cells of types A and C express CD30 reactivity.4
Merkel cell carcinoma (MCC) is a primary neuroendocrine carcinoma of the skin that usually arises on sun-exposed skin in elderly patients with lesions that histologically and clinically resemble cutaneous lymphoma.5 It classically is composed of small, round to oval, basophilic cells with a vesicular nucleus and multiple small nucleoli. Apoptotic cells and mitoses often are present.6 One key finding that helps to differentiate MCC from lymphoma is the presence of finely dispersed salt-and-pepper chromatin and molded nuclear contour in MCC (Figure 5).
Immunophenotyping is important in the differentiation of these diagnoses. The atypical cells of LyP are positive for CD3, CD4, and CD30 but are negative for CD8. However, in type B LyP, the large CD30+ cells seen in the other types are not commonly seen. In contrast, MCC expresses reactivity with cytokeratins, in particular cytokeratin 20 and CAM5.2, classically in a paranuclear dotlike pattern. In keeping with MCC's neuroendocrine differentiation, the tumor cells will demonstrate reactivity with synaptophysin, chromogranin, and CD56. The immunohistochemistry for leukemia cutis varies depending on the type of leukemia. Acute myelomonocytic leukemia is positive for myeloperoxidase, CD13, CD33, and CD68. The immunophenotype of these marginal zone lymphoma cells is as follows: positive for CD20, CD79a, and Bcl-2; negative for Bcl-6, CD5, CD10, CD23, and cyclin D1 (Bcl-1).7
Primary Cutaneous B-cell Lymphoma
Cutaneous B-cell lymphomas (CBCLs) are a diverse but rare group of cutaneous lymphoproliferative neoplasms that make up approximately 20% of the total number of hematolymphoid neoplasms primary to the skin.1 These lymphomas are comprised of neoplastic B cells in various stages of differentiation. As a whole, they are rare neoplasms that primarily involve the head, neck, trunk, arms, or legs.1 Clinically, patients present with nontender, compressible, solitary, red to violaceous papules or nodules. Most CBCLs are considered low-grade malignancies with nonaggressive behavior and excellent prognosis; however, the diffuse large B-cell lymphomas, including but not limited to intravascular and leg type; lymphomatoid granulomatosis; and B-cell lymphoblastic lymphoma can act more aggressively.1
Histopathologic examination of primary CBCL generally reveals a relatively normal epidermis accompanied by a nodular to diffuse monomorphic lymphocytic cellular infiltrate in the dermis that can occasionally extend into the subcutaneous tissue (quiz image). Although not specific for CBCLs, oftentimes there is an acellular portion of the superficial papillary dermis known as a grenz zone that can serve as a histopathologic clue to the diagnosis of a cutaneous lymphoproliferative disorder. The list of malignant B-cell neoplasms is extensive (eg, cutaneous marginal zone B-cell lymphoma, primary cutaneous follicle center lymphoma, diffuse large B-cell lymphoma, intravascular large B-cell lymphoma), and few are seen in the skin.
The most common type of CBCL is marginal zone B-cell lymphoma, which is considered to be a tumor of mucosa-associated (or skin-associated) lymphoid tissue. It is characterized by a monomorphous population of small mature lymphocytes showing characteristics of the B cells of the marginal zone of the lymph node. Some cells have the features of centrocytes/centroblasts (Figure 1) demonstrated by slightly irregular or indented nuclei and generous amounts of cytoplasm. Larger and more pleomorphic cells such as immunoblasts are similarly noted (Figure 1). The quiz image and Figure 1 demonstrate a cutaneous marginal zone B-cell lymphoma. A histomorphologic clue supporting a diagnosis of marginal zone B-cell lymphoma over reactive lymphoid hyperplasia is a B-cell predominate (B- to T-cell ratio of at least 3 to 1) infiltrate that is comprised of marginal zone-type cells. Immunohistochemistry demonstrating fewer differentiated B cells with light chain restriction may provide additional evidence that supports a clonal and potentially malignant process.
Erythematous to violaceous nodules on the head and neck of older individuals are characteristic of both granuloma faciale and CBCL. Histologically, granuloma faciale is characterized by a dense cellular infiltrate, often with a nodular outline, occupying the mid dermis.2 Granuloma faciale typically spares the immediate subepidermis and hair follicles, forming a grenz zone. The cellular infiltrate is polymorphic and consists of eosinophils and neutrophils with scattered plasma cells, mast cells, and lymphocytes in a vasculocentric distribution, eventually with chronic concentric fibrosis (Figure 2).
Leukemia cutis demonstrates a dermal infiltrate that contains atypical mononuclear cells (myeloblasts and myelocytes)(Figure 3).3 These markedly atypical mononuclear cells can have kidney bean-shaped nuclei and percolate through the dermal collagen, resembling single-file cells. They have increased nuclear to cytoplasmic ratios and occasionally have prominent nucleoli. Correlation with immunophenotypic and cytochemical studies is required for specific typing of the leukemic infiltrate.
Similar to primary CBCL, lymphomatoid papulosis (LyP) consists of erythematous papules or nodules that can occur anywhere on the body. In contrast to CBCL, the lesions of LyP classically self-resolve. However, approximately 10% to 20% of patients develop a malignant lymphoma, with mycosis fungoides, Hodgkin disease, and anaplastic large cell lymphoma being the most commonly associated.
Histologic examination of lesions of LyP classically demonstrates a wedge-shaped dermal infiltrate with variable epidermal changes (Figure 4). The wedge-shaped infiltrate is composed of large atypical cells. Three main types of lesions have been delineated: types A, B, and C. Type A is characterized by an increased number of cells with large vesicular nuclei with clumped chromatin, prominent nucleoli, and pronounced cytoplasm. Reed-Sternberg-like cells with an admixture of inflammatory cells including small lymphocytes, macrophages, neutrophils, and eosinophils also are present. Type B neoplastic cells vary in size and feature hyperchromatic, convoluted, or cerebriform nuclei. The infiltrate can be dense and bandlike with fewer cells resembling mycosis fungoides; type B LyP has neoplastic cells, not inflammatory cells. Finally, type C demonstrates solid sheets of large atypical cells resembling anaplastic large cell lymphoma. Immunohistochemically, the atypical cells often are CD4+ and CD8- with variable loss of pan-T-cell antigens. The atypical cells of types A and C express CD30 reactivity.4
Merkel cell carcinoma (MCC) is a primary neuroendocrine carcinoma of the skin that usually arises on sun-exposed skin in elderly patients with lesions that histologically and clinically resemble cutaneous lymphoma.5 It classically is composed of small, round to oval, basophilic cells with a vesicular nucleus and multiple small nucleoli. Apoptotic cells and mitoses often are present.6 One key finding that helps to differentiate MCC from lymphoma is the presence of finely dispersed salt-and-pepper chromatin and molded nuclear contour in MCC (Figure 5).
Immunophenotyping is important in the differentiation of these diagnoses. The atypical cells of LyP are positive for CD3, CD4, and CD30 but are negative for CD8. However, in type B LyP, the large CD30+ cells seen in the other types are not commonly seen. In contrast, MCC expresses reactivity with cytokeratins, in particular cytokeratin 20 and CAM5.2, classically in a paranuclear dotlike pattern. In keeping with MCC's neuroendocrine differentiation, the tumor cells will demonstrate reactivity with synaptophysin, chromogranin, and CD56. The immunohistochemistry for leukemia cutis varies depending on the type of leukemia. Acute myelomonocytic leukemia is positive for myeloperoxidase, CD13, CD33, and CD68. The immunophenotype of these marginal zone lymphoma cells is as follows: positive for CD20, CD79a, and Bcl-2; negative for Bcl-6, CD5, CD10, CD23, and cyclin D1 (Bcl-1).7
- Olsen EA. Evaluation, diagnosis, and staging of cutaneous lymphoma. Dermatol Clin. 2015;33:643-654.
- Ortonne N, Wechsler J, Bagot M, et al. Granuloma faciale: a clinicopathologic study of 66 patients. J Am Acad Dermatol. 2005;53:1002-1009.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Wieser I, Wohlmuth C, Nunez CA, et al. Lymphomatoid papulosis in children and adolescents: a systematic review. Am J Clin Dermatol. 2016;17:319-327.
- Sibley RK, Dehner LP, Rosai J. Primary neuroendocrine (Merkel cell?) carcinoma of the skin: I. a clinicopathologic and ultrastructural study of 43 cases. Am J Surg Pathol. 1985;9:95-108.
- Frigerio B, Capella C, Eusebi V, et al. Merkel cell carcinoma of the skin: the structure and origin of normal Merkel cells. Histopathology. 1983;7:229-249.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
- Olsen EA. Evaluation, diagnosis, and staging of cutaneous lymphoma. Dermatol Clin. 2015;33:643-654.
- Ortonne N, Wechsler J, Bagot M, et al. Granuloma faciale: a clinicopathologic study of 66 patients. J Am Acad Dermatol. 2005;53:1002-1009.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Wieser I, Wohlmuth C, Nunez CA, et al. Lymphomatoid papulosis in children and adolescents: a systematic review. Am J Clin Dermatol. 2016;17:319-327.
- Sibley RK, Dehner LP, Rosai J. Primary neuroendocrine (Merkel cell?) carcinoma of the skin: I. a clinicopathologic and ultrastructural study of 43 cases. Am J Surg Pathol. 1985;9:95-108.
- Frigerio B, Capella C, Eusebi V, et al. Merkel cell carcinoma of the skin: the structure and origin of normal Merkel cells. Histopathology. 1983;7:229-249.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
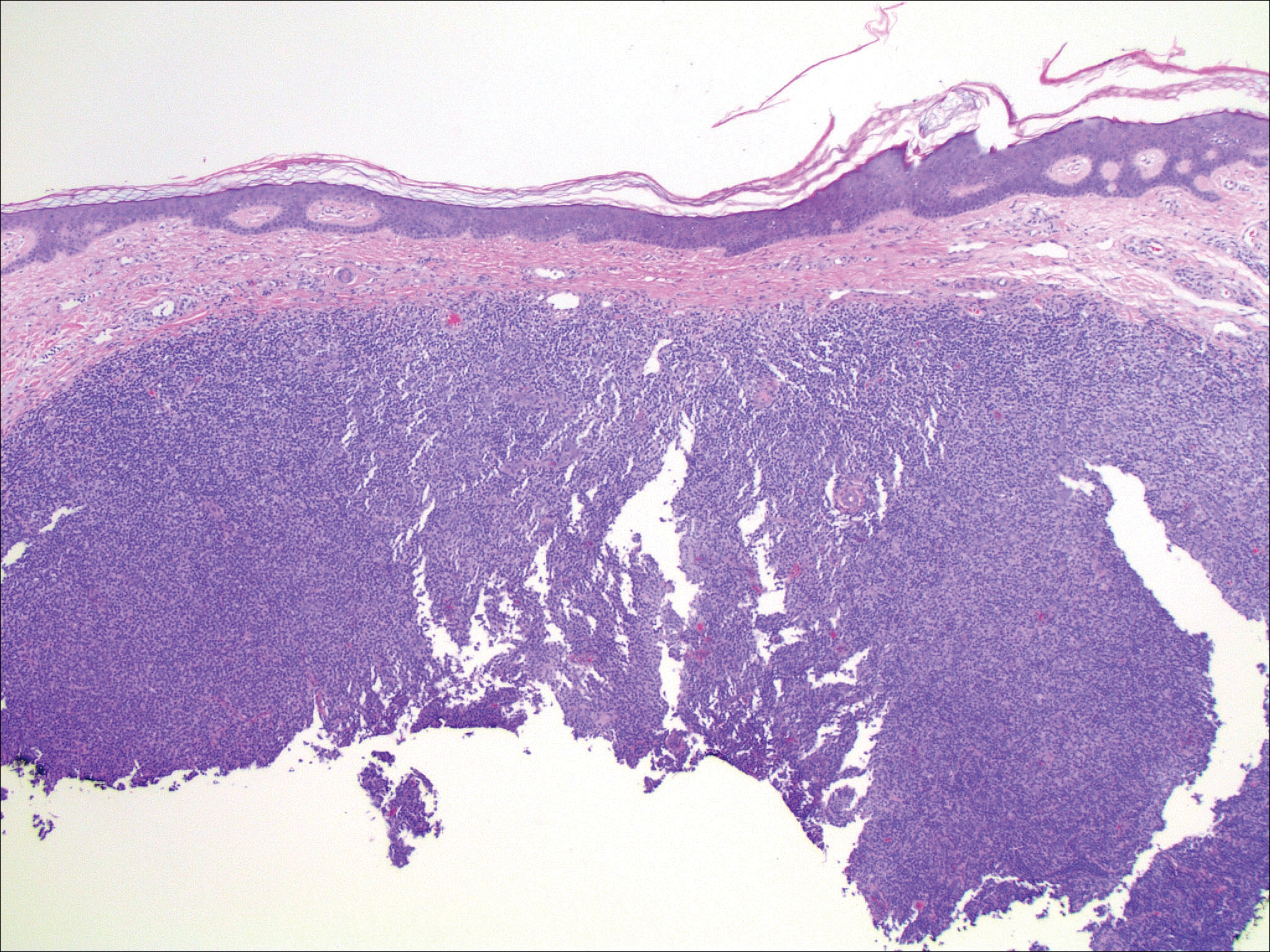
An 81-year-old man with a history of hyperthyroidism, paroxysmal atrial fibrillation, hypertension, and nonmelanoma skin cancer presented with an erythematous pearly papule on the right lateral chest of 1 year's duration. The patient reported no symptoms of pruritus, bleeding, or burning. He was otherwise asymptomatic, and a review of systems revealed no abnormalities. His current medications included aspirin, benazepril, finasteride, levothyroxine, tamsulosin, warfarin, and alprazolam. He denied any new medications, recent travel, or preceding trauma. He had a history of Agent Orange exposure. Physical examination revealed a 0.4-cm erythematous pearly papule on the right lateral chest. A shave biopsy was obtained.
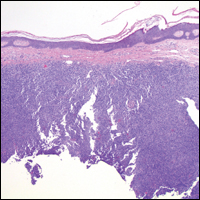
Progressive Widespread Warty Skin Growths
Epidermodysplasia Verruciformis
Epidermodysplasia verruciformis (EV) is a rare hereditary disorder that predisposes affected individuals to widespread infection with various forms of human papillomavirus (HPV). It is inherited in an autosomal-recessive pattern.1 The first manifestations generally are seen in childhood. The clinical appearance of lesions can vary, at times mimicking other disease processes. Patients can present with flat wartlike papules resembling verruca plana distributed in sun-exposed areas. Another distinct presentation is multiple salmon-colored, hyperpigmented, or hypopigmented macules, papules, or plaques with overlying scale that can resemble tinea versicolor.1,2 A large percentage of patients will go on to develop actinic keratosis and squamous cell carcinoma by 40 years of age.2 The malignancies most commonly develop in sun-exposed areas, suggesting UV radiation as an important contributor to development along with HPV infection. Mutations in the EVER1 and EVER2 genes that code for transmembrane proteins on the endoplasmic reticulum that are involved in zinc transport lead to EV. The mutations lead to decreased zinc movement into the cytoplasm, which is thought to play a role in preventing HPV infection. The decreased protection against HPV leads to infections from both common subtypes and those that immunocompetent individuals would be resistant to, namely the β-genus HPV-5, -8, -9 and -20.1,2 Immunosuppressed individuals, such as those with human immunodeficiency virus/AIDS, also are at an increased risk for infection with these HPV subtypes and generally have similar clinical and histological presentations.1 It is important to promote sunscreen use for preventive care in patients with EV due to the increased risk for squamous cell carcinoma.
Histologically, the lesions in EV are composed of acanthosis and hyperkeratosis with keratinocytes arranged in clusters.1,3 There is orthokeratosis and parakeratosis.1 Scattered or clustered keratinocytes in the granular layer or upper stratum spinosum appear swollen with foamy blue-gray cytoplasm (quiz image and Figure 1).1,4 The keratinocytes may become atypical and progress to squamous cell carcinoma, particularly in sun-exposed regions. Cell differentiation becomes disorganized and nuclei become enlarged and hyperchromatic.1

Condyloma acuminatum will have pronounced acanthosis and hyperkeratosis with exophytic growth. Rounded parakeratosis is visible. The characteristic cell is the koilocyte, a keratinocyte that has an enlarged nucleus with areas of surrounding clearing, increased dark color in the nucleus, and wrinkled nuclear membrane (Figure 2).1,3 True koilocytes may be rare in condyloma acuminatum.4 Other distinct features include coarse hypergranulosis and a compact stratum corneum.

Herpesvirus lesions typically demonstrate ballooning degeneration of keratinocytes.1 They will become pale and fuse to form multinucleated giant cells, a feature not found in verruca. The nuclei will be slate gray with margination of the chromatin, which can be identified due to its increased basophilic appearance (Figure 3).1,4 Inclusion bodies can be found, but unlike molluscum contagiosum (MC), these bodies are intranuclear as opposed to cytoplasmic.1
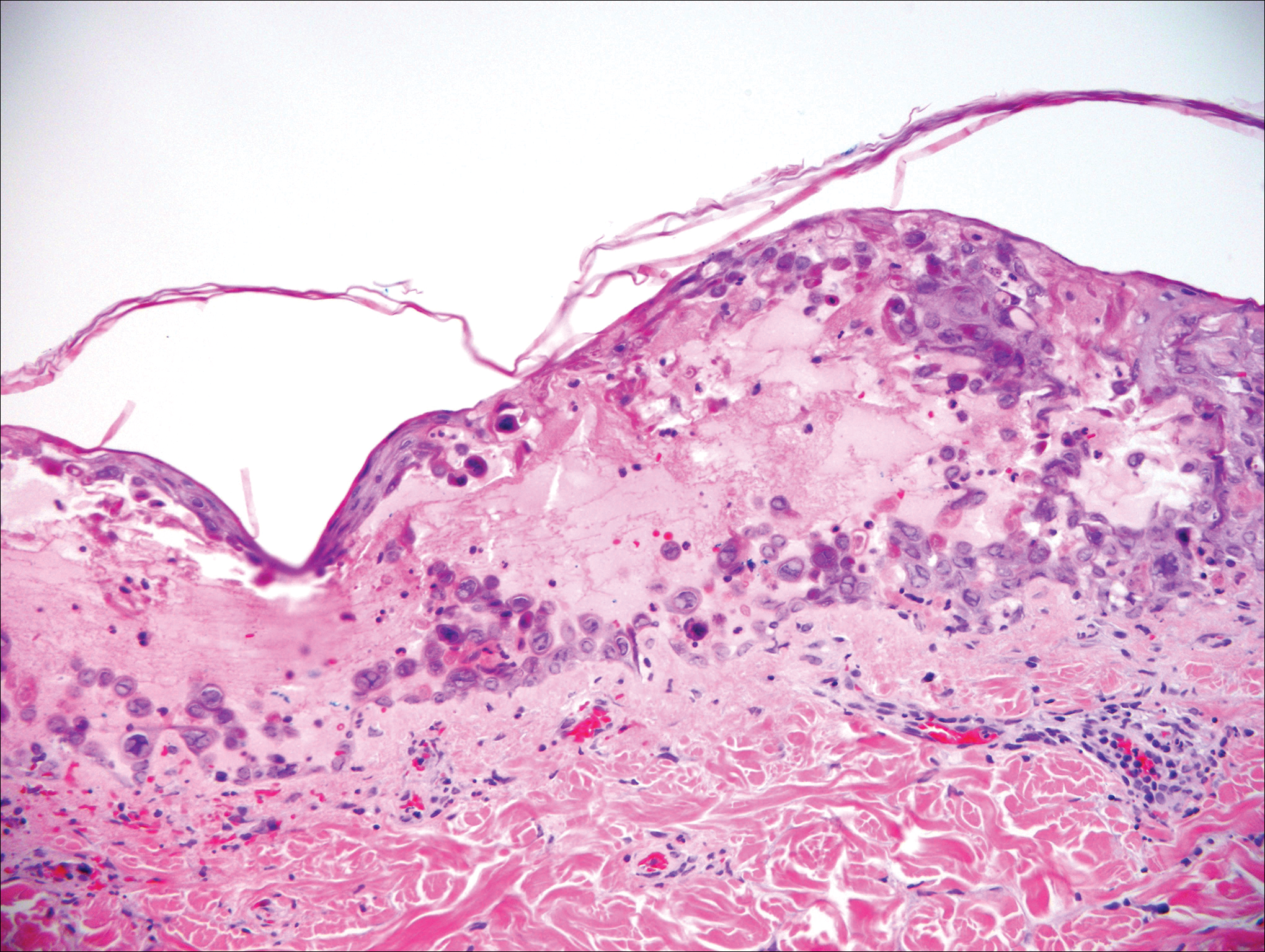
The telltale Henderson-Patterson (molluscum) bodies can identify MC histologically.4 Located within keratinocytes, these cytoplasmic inclusions can vary in both color and size as they mature. As the keratinocytes develop outward, the molluscum bodies grow larger and become more eosinophilic (Figure 4).1,4 Another feature of MC that can be used to differentiate it from EV is the scalloped borders located on lesions.4
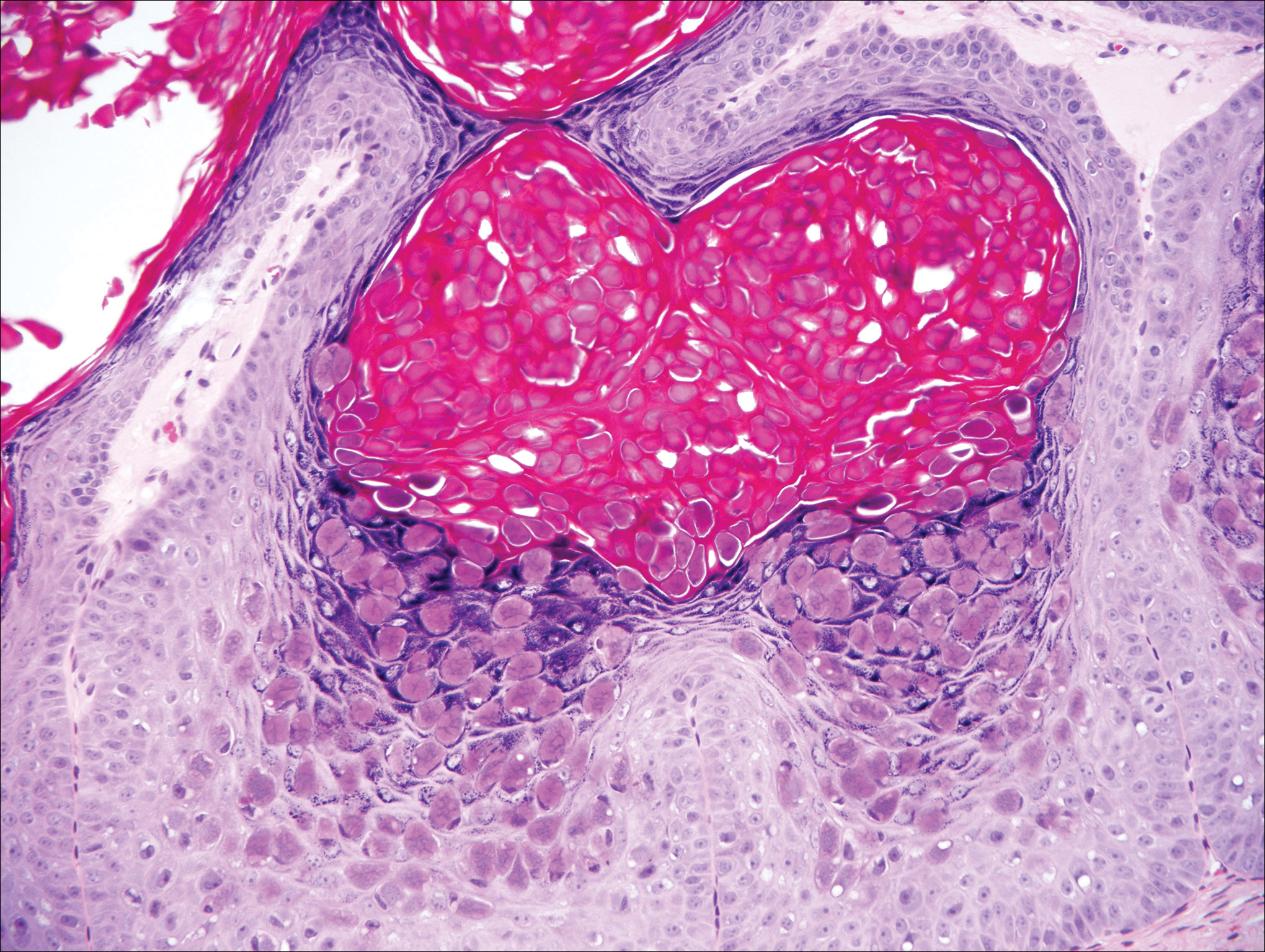
On histology, verruca vulgaris will have pronounced acanthosis with orthokeratosis and vertical tiers of parakeratosis.3,4 Growth is exophytic. The granular layer will have large irregular basophilic granules. Koilocytes may be seen. A distinctive feature is the papillomatosis with inward bending of rete ridges.3,4 It is common to see invasion of tortuous blood vessels into the exophytic projections.3 In myrmecia (palmoplantar warts) it is common to see thrombosis of these vessels and inclusions of red cytoplasmic bodies (Figure 5).1,4
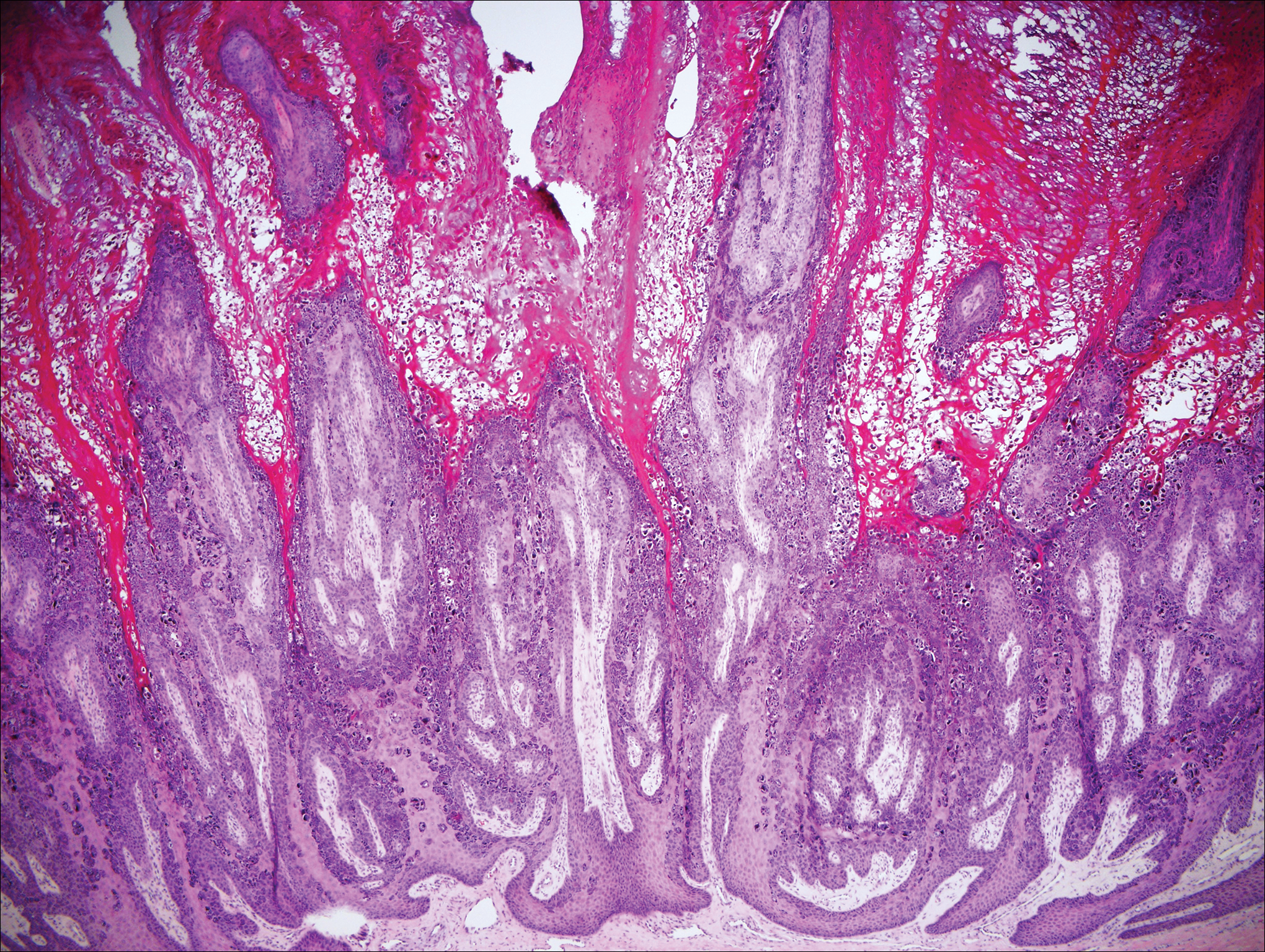
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012.
- Hunzeker CM, Soldano AC, Prystowsky S. Epidermodysplasia verruciformis. Dermatology Online J. 2008;14:2.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Elston DM, Ko CJ, Ferringer T. Dermatopathology. Edinburgh, Scotland: Saunders/Elsevier; 2009.
Epidermodysplasia Verruciformis
Epidermodysplasia verruciformis (EV) is a rare hereditary disorder that predisposes affected individuals to widespread infection with various forms of human papillomavirus (HPV). It is inherited in an autosomal-recessive pattern.1 The first manifestations generally are seen in childhood. The clinical appearance of lesions can vary, at times mimicking other disease processes. Patients can present with flat wartlike papules resembling verruca plana distributed in sun-exposed areas. Another distinct presentation is multiple salmon-colored, hyperpigmented, or hypopigmented macules, papules, or plaques with overlying scale that can resemble tinea versicolor.1,2 A large percentage of patients will go on to develop actinic keratosis and squamous cell carcinoma by 40 years of age.2 The malignancies most commonly develop in sun-exposed areas, suggesting UV radiation as an important contributor to development along with HPV infection. Mutations in the EVER1 and EVER2 genes that code for transmembrane proteins on the endoplasmic reticulum that are involved in zinc transport lead to EV. The mutations lead to decreased zinc movement into the cytoplasm, which is thought to play a role in preventing HPV infection. The decreased protection against HPV leads to infections from both common subtypes and those that immunocompetent individuals would be resistant to, namely the β-genus HPV-5, -8, -9 and -20.1,2 Immunosuppressed individuals, such as those with human immunodeficiency virus/AIDS, also are at an increased risk for infection with these HPV subtypes and generally have similar clinical and histological presentations.1 It is important to promote sunscreen use for preventive care in patients with EV due to the increased risk for squamous cell carcinoma.
Histologically, the lesions in EV are composed of acanthosis and hyperkeratosis with keratinocytes arranged in clusters.1,3 There is orthokeratosis and parakeratosis.1 Scattered or clustered keratinocytes in the granular layer or upper stratum spinosum appear swollen with foamy blue-gray cytoplasm (quiz image and Figure 1).1,4 The keratinocytes may become atypical and progress to squamous cell carcinoma, particularly in sun-exposed regions. Cell differentiation becomes disorganized and nuclei become enlarged and hyperchromatic.1
Condyloma acuminatum will have pronounced acanthosis and hyperkeratosis with exophytic growth. Rounded parakeratosis is visible. The characteristic cell is the koilocyte, a keratinocyte that has an enlarged nucleus with areas of surrounding clearing, increased dark color in the nucleus, and wrinkled nuclear membrane (Figure 2).1,3 True koilocytes may be rare in condyloma acuminatum.4 Other distinct features include coarse hypergranulosis and a compact stratum corneum.
Herpesvirus lesions typically demonstrate ballooning degeneration of keratinocytes.1 They will become pale and fuse to form multinucleated giant cells, a feature not found in verruca. The nuclei will be slate gray with margination of the chromatin, which can be identified due to its increased basophilic appearance (Figure 3).1,4 Inclusion bodies can be found, but unlike molluscum contagiosum (MC), these bodies are intranuclear as opposed to cytoplasmic.1
The telltale Henderson-Patterson (molluscum) bodies can identify MC histologically.4 Located within keratinocytes, these cytoplasmic inclusions can vary in both color and size as they mature. As the keratinocytes develop outward, the molluscum bodies grow larger and become more eosinophilic (Figure 4).1,4 Another feature of MC that can be used to differentiate it from EV is the scalloped borders located on lesions.4
On histology, verruca vulgaris will have pronounced acanthosis with orthokeratosis and vertical tiers of parakeratosis.3,4 Growth is exophytic. The granular layer will have large irregular basophilic granules. Koilocytes may be seen. A distinctive feature is the papillomatosis with inward bending of rete ridges.3,4 It is common to see invasion of tortuous blood vessels into the exophytic projections.3 In myrmecia (palmoplantar warts) it is common to see thrombosis of these vessels and inclusions of red cytoplasmic bodies (Figure 5).1,4
Epidermodysplasia Verruciformis
Epidermodysplasia verruciformis (EV) is a rare hereditary disorder that predisposes affected individuals to widespread infection with various forms of human papillomavirus (HPV). It is inherited in an autosomal-recessive pattern.1 The first manifestations generally are seen in childhood. The clinical appearance of lesions can vary, at times mimicking other disease processes. Patients can present with flat wartlike papules resembling verruca plana distributed in sun-exposed areas. Another distinct presentation is multiple salmon-colored, hyperpigmented, or hypopigmented macules, papules, or plaques with overlying scale that can resemble tinea versicolor.1,2 A large percentage of patients will go on to develop actinic keratosis and squamous cell carcinoma by 40 years of age.2 The malignancies most commonly develop in sun-exposed areas, suggesting UV radiation as an important contributor to development along with HPV infection. Mutations in the EVER1 and EVER2 genes that code for transmembrane proteins on the endoplasmic reticulum that are involved in zinc transport lead to EV. The mutations lead to decreased zinc movement into the cytoplasm, which is thought to play a role in preventing HPV infection. The decreased protection against HPV leads to infections from both common subtypes and those that immunocompetent individuals would be resistant to, namely the β-genus HPV-5, -8, -9 and -20.1,2 Immunosuppressed individuals, such as those with human immunodeficiency virus/AIDS, also are at an increased risk for infection with these HPV subtypes and generally have similar clinical and histological presentations.1 It is important to promote sunscreen use for preventive care in patients with EV due to the increased risk for squamous cell carcinoma.
Histologically, the lesions in EV are composed of acanthosis and hyperkeratosis with keratinocytes arranged in clusters.1,3 There is orthokeratosis and parakeratosis.1 Scattered or clustered keratinocytes in the granular layer or upper stratum spinosum appear swollen with foamy blue-gray cytoplasm (quiz image and Figure 1).1,4 The keratinocytes may become atypical and progress to squamous cell carcinoma, particularly in sun-exposed regions. Cell differentiation becomes disorganized and nuclei become enlarged and hyperchromatic.1
Condyloma acuminatum will have pronounced acanthosis and hyperkeratosis with exophytic growth. Rounded parakeratosis is visible. The characteristic cell is the koilocyte, a keratinocyte that has an enlarged nucleus with areas of surrounding clearing, increased dark color in the nucleus, and wrinkled nuclear membrane (Figure 2).1,3 True koilocytes may be rare in condyloma acuminatum.4 Other distinct features include coarse hypergranulosis and a compact stratum corneum.
Herpesvirus lesions typically demonstrate ballooning degeneration of keratinocytes.1 They will become pale and fuse to form multinucleated giant cells, a feature not found in verruca. The nuclei will be slate gray with margination of the chromatin, which can be identified due to its increased basophilic appearance (Figure 3).1,4 Inclusion bodies can be found, but unlike molluscum contagiosum (MC), these bodies are intranuclear as opposed to cytoplasmic.1
The telltale Henderson-Patterson (molluscum) bodies can identify MC histologically.4 Located within keratinocytes, these cytoplasmic inclusions can vary in both color and size as they mature. As the keratinocytes develop outward, the molluscum bodies grow larger and become more eosinophilic (Figure 4).1,4 Another feature of MC that can be used to differentiate it from EV is the scalloped borders located on lesions.4
On histology, verruca vulgaris will have pronounced acanthosis with orthokeratosis and vertical tiers of parakeratosis.3,4 Growth is exophytic. The granular layer will have large irregular basophilic granules. Koilocytes may be seen. A distinctive feature is the papillomatosis with inward bending of rete ridges.3,4 It is common to see invasion of tortuous blood vessels into the exophytic projections.3 In myrmecia (palmoplantar warts) it is common to see thrombosis of these vessels and inclusions of red cytoplasmic bodies (Figure 5).1,4
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012.
- Hunzeker CM, Soldano AC, Prystowsky S. Epidermodysplasia verruciformis. Dermatology Online J. 2008;14:2.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Elston DM, Ko CJ, Ferringer T. Dermatopathology. Edinburgh, Scotland: Saunders/Elsevier; 2009.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012.
- Hunzeker CM, Soldano AC, Prystowsky S. Epidermodysplasia verruciformis. Dermatology Online J. 2008;14:2.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Elston DM, Ko CJ, Ferringer T. Dermatopathology. Edinburgh, Scotland: Saunders/Elsevier; 2009.
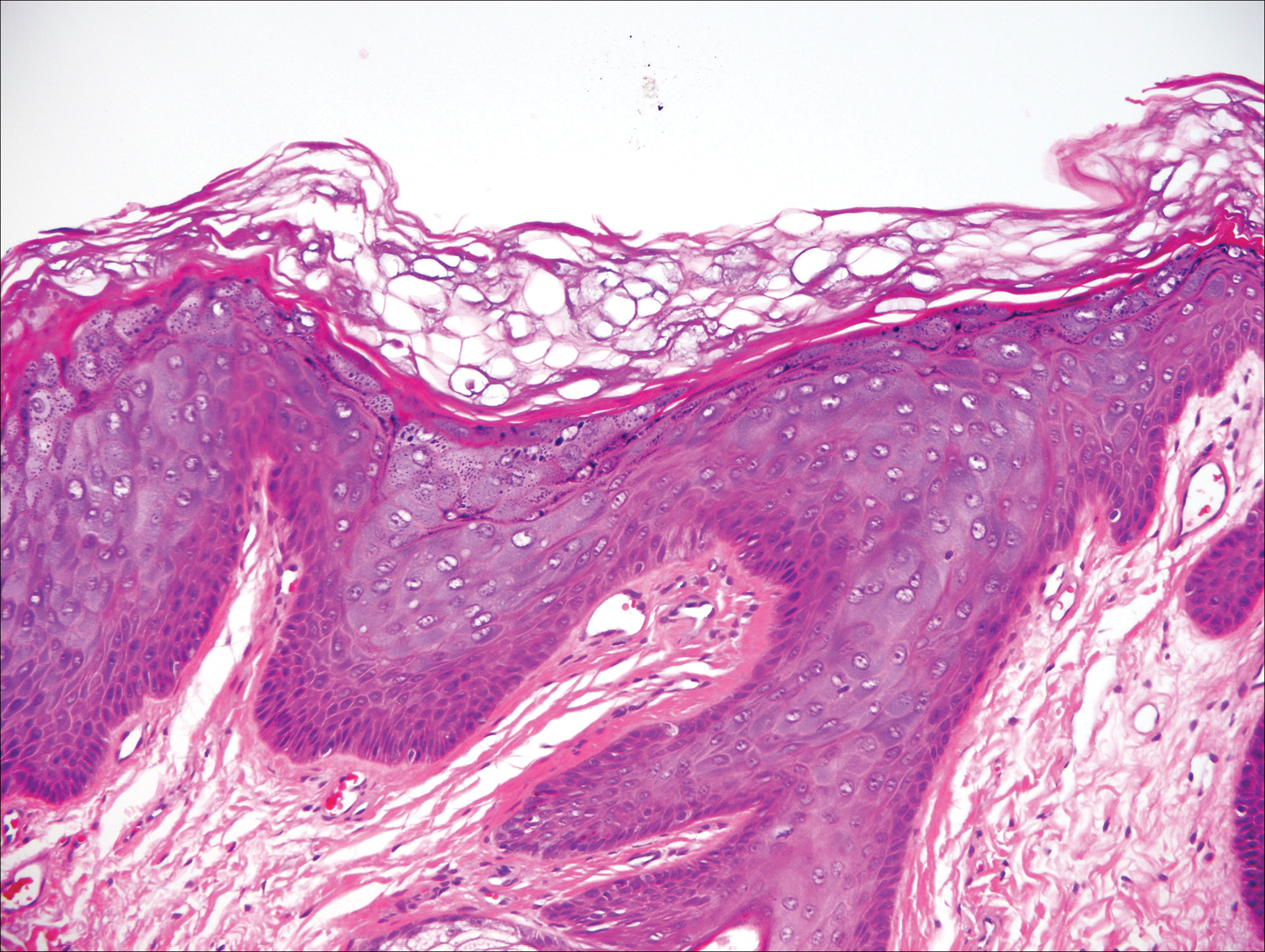
A 33-year-old man presented with progressive widespread warty skin growths that had been present since 6 years of age. Physical examination revealed numerous verrucous papules on the face and neck along with verrucous, tan-pink papules and plaques diffusely scattered on the trunk, arms, and legs. A biopsy of a lesion on the neck was performed.
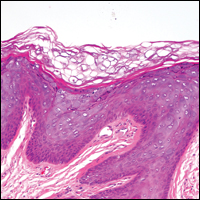
Red-Blue Nodule on the Scalp
Metastatic Clear Cell Renal Cell Carcinoma
The differential diagnosis of cutaneous neoplasms with clear cells is broad. Clear cell features can be seen in primary tumors arising from the epidermis and cutaneous adnexa as well as in mesenchymal and melanocytic neoplasms. Furthermore, metastatic disease should be considered in the histologic differential diagnosis, as many visceral malignancies have clear cell features. This patient was subsequently found to have a large renal mass with metastasis to the lungs, spleen, and bone. The histologic findings support the diagnosis of metastatic clear cell renal cell carcinoma (RCC) to the skin.
Approximately 30% of patients with clear cell RCC present with metastatic disease with approximately 8% of those involving the skin.1,2 Cutaneous RCC metastases show a predilection for the head, especially the scalp. The clinical presentation is variable, but there often is a history of a rapidly growing brown, black, or purple nodule or plaque. A thorough review of the patient's history should be conducted if metastatic RCC is in the differential diagnosis, as it has been reported to occur up to 20 years after initial diagnosis.3
Histologically, clear cell RCC (quiz image) is composed of nests of tumor cells with clear cytoplasm and centrally located nuclei with prominent nucleoli. The clear cell features result from abundant cytoplasmic glycogen and lipid but may not be present in every case. One of the most important histologic features is the presence of delicate branching blood vessels (Figure 1). Numerous extravasated red blood cells also may be present. Positive immunohistochemical staining for PAX8, CD10, and RCC antigens support the diagnosis.4
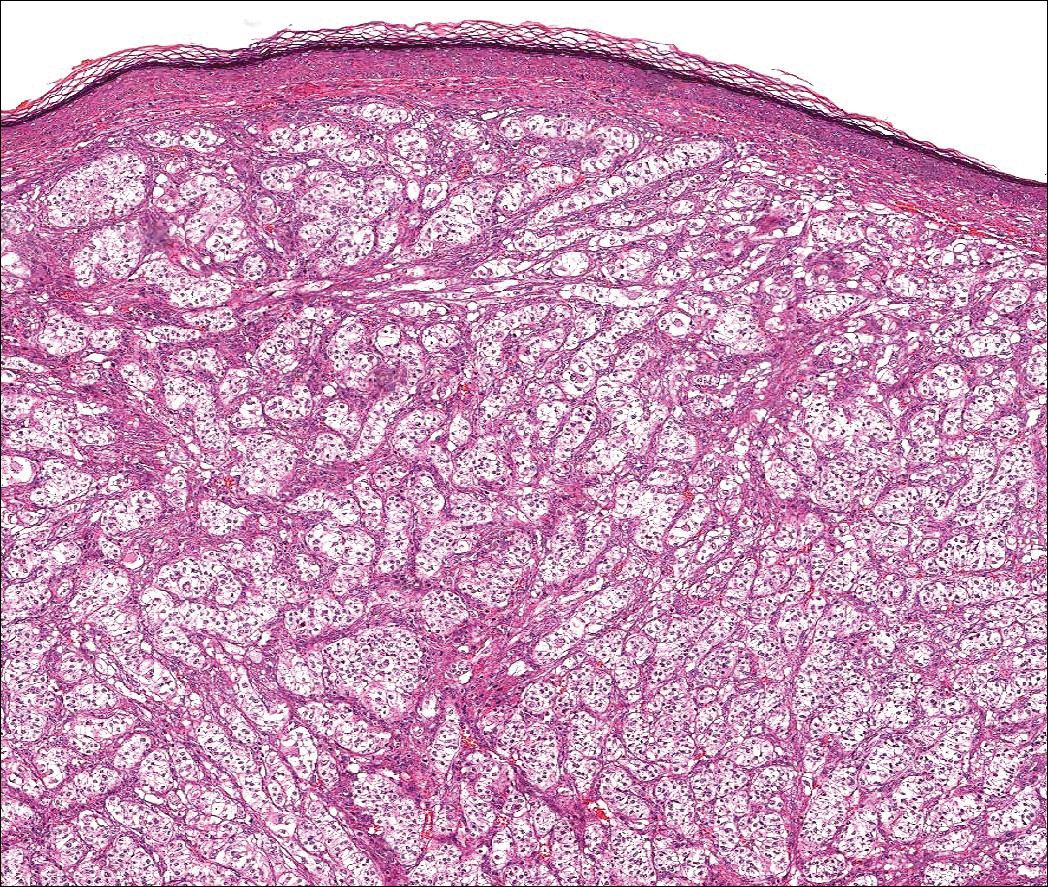
Balloon cell nevi (Figure 2) most commonly occur on the head and neck in adolescents and young adults but clinically are indistinguishable from other banal nevi. The nevus cells are large with foamy to finely vacuolated cytoplasm and lack atypia. The clear cell change is the result of melanosome degeneration and may be extensive. The presence of melanin pigment, nests of typical nevus cells, and positive staining with MART-1 can help distinguish the tumor from xanthomas and RCC.5
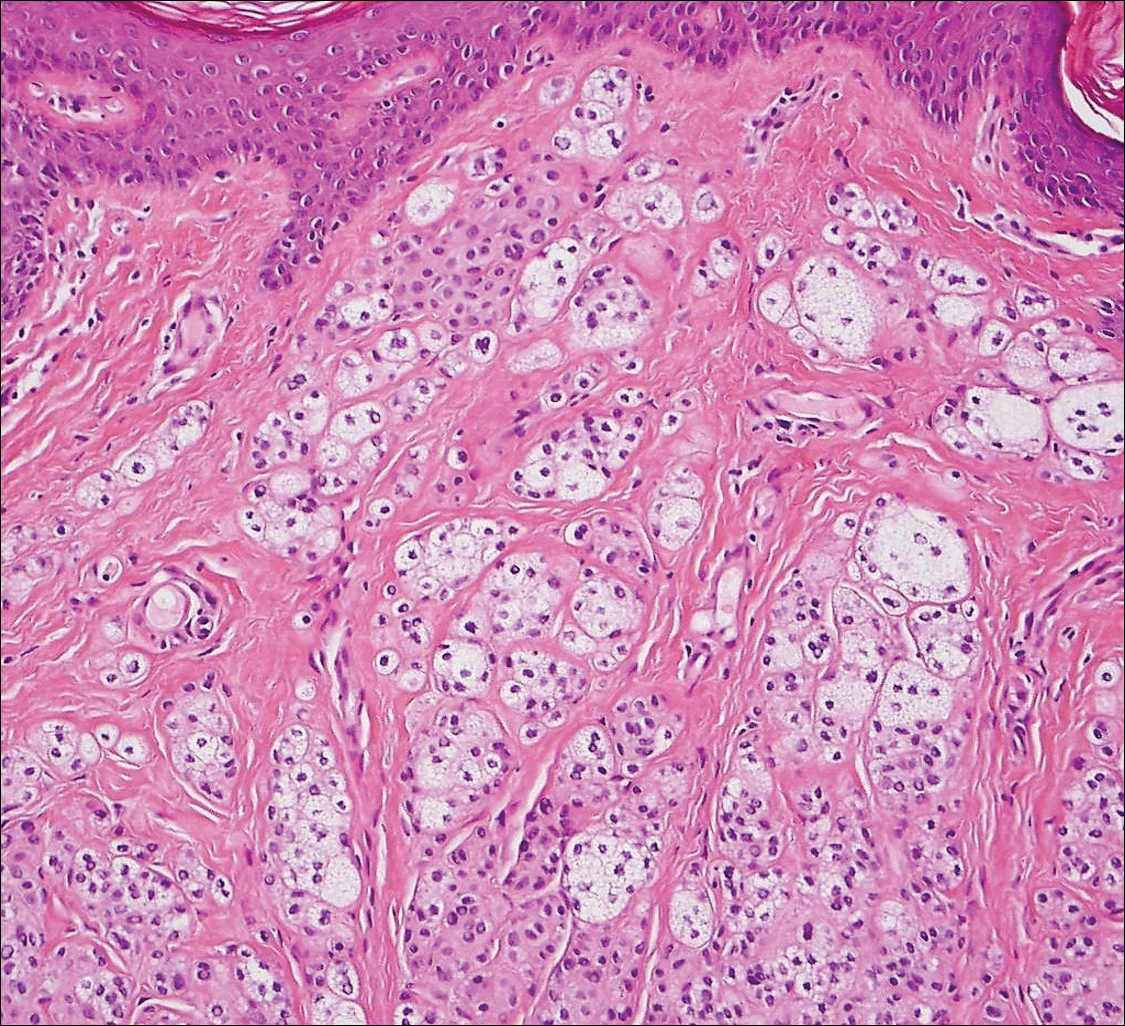
Clear cell hidradenoma (Figure 3) is a well-circumscribed tumor of sweat gland origin that arises in the dermis. The architecture usually is solid, cystic, or a combination of both. The cytology is classically bland with poroid, squamoid, or clear cell morphology. Clear cells that are positive on periodic acid-Schiff staining predominate in up to one-third of cases. Carcinoembryonic antigen and epithelial membrane antigen can be used to highlight the eosinophilic cuticles of ducts within solid areas.6

Sebaceous carcinoma (Figure 4) most frequently arises in a periorbital distribution, although extraocular lesions are known to occur. Histologically, there is a proliferation of both mature sebocytes and basaloid cells in the dermis, occasionally involving the epidermis. The mature sebocytes demonstrate clear cell features with foamy to vacuolated cytoplasm and large nuclei with scalloped borders. The clear cells may vary greatly in number and often are sparse in poorly differentiated tumors in which pleomorphic basaloid cells may predominate. The basaloid cells may resemble those of squamous or basal cell carcinoma, leading to a diagnostic dilemma in some cases. Special staining with Sudan black B and oil red O highlights the cytoplasmic lipid but must be performed on frozen section specimens. Although not entirely specific, immunohistochemical expression of epithelial membrane antigen, androgen receptor, and membranous vesicular adipophilin staining in sebaceous carcinoma can assist in the diagnosis.7
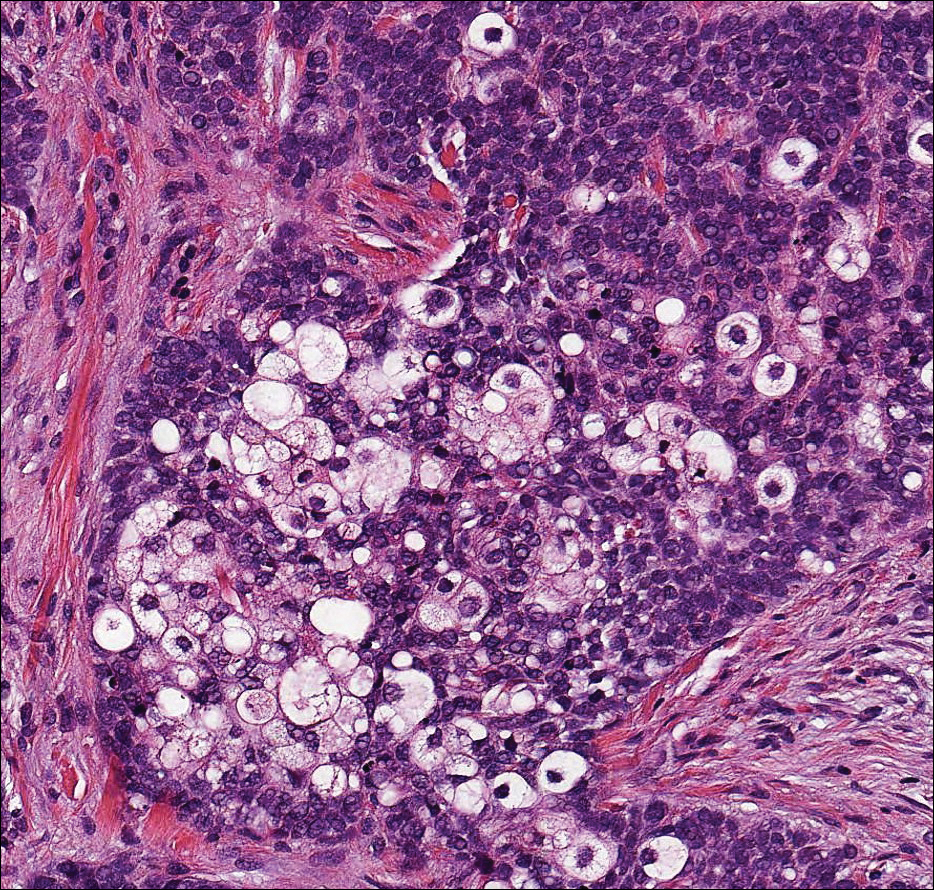
Cutaneous xanthomas (Figure 5) may arise in patients of any age and represent deposition of lipid-laden macrophages. Classification often is dependent on the clinical presentation; however, some subtypes demonstrate unique morphologic features (eg, verruciform xanthomas). Xanthomas classically arise in association with elevated serum lipids, but they also may occur in normolipemic patients. Individuals with Erdheim-Chester disease have an increased propensity to develop xanthelasma. Similarly, plane xanthomas have been associated with monoclonal gammopathy. Histologically, xanthomas are characterized by sheets of foamy macrophages within the dermis and subcutis. Positive immunohistochemical staining for CD68 highlighting the histiocytic nature of the cells and the absence of a delicate vascular network aid in the differentiation from RCC.
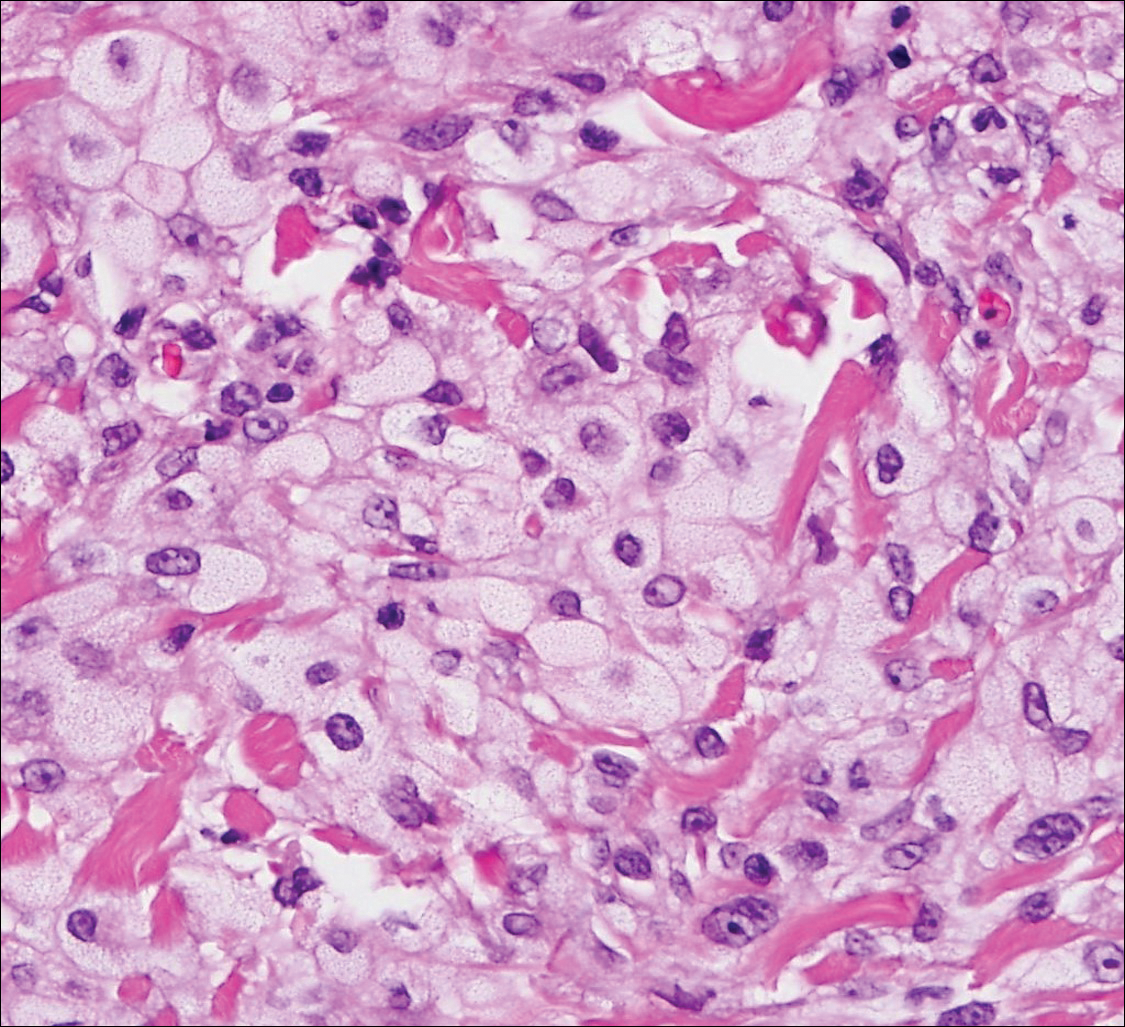
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Lin F, Prichard J. Handbook of Practical Immunohistochemistry: Frequently Asked Questions. 2nd ed. New York, NY: Springer; 2015.
- McKee PH, Calonje E. Diagnostic Atlas of Melanocytic Pathology. Edinburgh, Scotland: Mosby/Elsevier; 2009.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Ansai S, Takeichi H, Arase S, et al. Sebaceous carcinoma: an immunohistochemical reappraisal. Am J Dermatopathol. 2011;33:579-587.
Metastatic Clear Cell Renal Cell Carcinoma
The differential diagnosis of cutaneous neoplasms with clear cells is broad. Clear cell features can be seen in primary tumors arising from the epidermis and cutaneous adnexa as well as in mesenchymal and melanocytic neoplasms. Furthermore, metastatic disease should be considered in the histologic differential diagnosis, as many visceral malignancies have clear cell features. This patient was subsequently found to have a large renal mass with metastasis to the lungs, spleen, and bone. The histologic findings support the diagnosis of metastatic clear cell renal cell carcinoma (RCC) to the skin.
Approximately 30% of patients with clear cell RCC present with metastatic disease with approximately 8% of those involving the skin.1,2 Cutaneous RCC metastases show a predilection for the head, especially the scalp. The clinical presentation is variable, but there often is a history of a rapidly growing brown, black, or purple nodule or plaque. A thorough review of the patient's history should be conducted if metastatic RCC is in the differential diagnosis, as it has been reported to occur up to 20 years after initial diagnosis.3
Histologically, clear cell RCC (quiz image) is composed of nests of tumor cells with clear cytoplasm and centrally located nuclei with prominent nucleoli. The clear cell features result from abundant cytoplasmic glycogen and lipid but may not be present in every case. One of the most important histologic features is the presence of delicate branching blood vessels (Figure 1). Numerous extravasated red blood cells also may be present. Positive immunohistochemical staining for PAX8, CD10, and RCC antigens support the diagnosis.4
Balloon cell nevi (Figure 2) most commonly occur on the head and neck in adolescents and young adults but clinically are indistinguishable from other banal nevi. The nevus cells are large with foamy to finely vacuolated cytoplasm and lack atypia. The clear cell change is the result of melanosome degeneration and may be extensive. The presence of melanin pigment, nests of typical nevus cells, and positive staining with MART-1 can help distinguish the tumor from xanthomas and RCC.5
Clear cell hidradenoma (Figure 3) is a well-circumscribed tumor of sweat gland origin that arises in the dermis. The architecture usually is solid, cystic, or a combination of both. The cytology is classically bland with poroid, squamoid, or clear cell morphology. Clear cells that are positive on periodic acid-Schiff staining predominate in up to one-third of cases. Carcinoembryonic antigen and epithelial membrane antigen can be used to highlight the eosinophilic cuticles of ducts within solid areas.6
Sebaceous carcinoma (Figure 4) most frequently arises in a periorbital distribution, although extraocular lesions are known to occur. Histologically, there is a proliferation of both mature sebocytes and basaloid cells in the dermis, occasionally involving the epidermis. The mature sebocytes demonstrate clear cell features with foamy to vacuolated cytoplasm and large nuclei with scalloped borders. The clear cells may vary greatly in number and often are sparse in poorly differentiated tumors in which pleomorphic basaloid cells may predominate. The basaloid cells may resemble those of squamous or basal cell carcinoma, leading to a diagnostic dilemma in some cases. Special staining with Sudan black B and oil red O highlights the cytoplasmic lipid but must be performed on frozen section specimens. Although not entirely specific, immunohistochemical expression of epithelial membrane antigen, androgen receptor, and membranous vesicular adipophilin staining in sebaceous carcinoma can assist in the diagnosis.7
Cutaneous xanthomas (Figure 5) may arise in patients of any age and represent deposition of lipid-laden macrophages. Classification often is dependent on the clinical presentation; however, some subtypes demonstrate unique morphologic features (eg, verruciform xanthomas). Xanthomas classically arise in association with elevated serum lipids, but they also may occur in normolipemic patients. Individuals with Erdheim-Chester disease have an increased propensity to develop xanthelasma. Similarly, plane xanthomas have been associated with monoclonal gammopathy. Histologically, xanthomas are characterized by sheets of foamy macrophages within the dermis and subcutis. Positive immunohistochemical staining for CD68 highlighting the histiocytic nature of the cells and the absence of a delicate vascular network aid in the differentiation from RCC.
Metastatic Clear Cell Renal Cell Carcinoma
The differential diagnosis of cutaneous neoplasms with clear cells is broad. Clear cell features can be seen in primary tumors arising from the epidermis and cutaneous adnexa as well as in mesenchymal and melanocytic neoplasms. Furthermore, metastatic disease should be considered in the histologic differential diagnosis, as many visceral malignancies have clear cell features. This patient was subsequently found to have a large renal mass with metastasis to the lungs, spleen, and bone. The histologic findings support the diagnosis of metastatic clear cell renal cell carcinoma (RCC) to the skin.
Approximately 30% of patients with clear cell RCC present with metastatic disease with approximately 8% of those involving the skin.1,2 Cutaneous RCC metastases show a predilection for the head, especially the scalp. The clinical presentation is variable, but there often is a history of a rapidly growing brown, black, or purple nodule or plaque. A thorough review of the patient's history should be conducted if metastatic RCC is in the differential diagnosis, as it has been reported to occur up to 20 years after initial diagnosis.3
Histologically, clear cell RCC (quiz image) is composed of nests of tumor cells with clear cytoplasm and centrally located nuclei with prominent nucleoli. The clear cell features result from abundant cytoplasmic glycogen and lipid but may not be present in every case. One of the most important histologic features is the presence of delicate branching blood vessels (Figure 1). Numerous extravasated red blood cells also may be present. Positive immunohistochemical staining for PAX8, CD10, and RCC antigens support the diagnosis.4
Balloon cell nevi (Figure 2) most commonly occur on the head and neck in adolescents and young adults but clinically are indistinguishable from other banal nevi. The nevus cells are large with foamy to finely vacuolated cytoplasm and lack atypia. The clear cell change is the result of melanosome degeneration and may be extensive. The presence of melanin pigment, nests of typical nevus cells, and positive staining with MART-1 can help distinguish the tumor from xanthomas and RCC.5
Clear cell hidradenoma (Figure 3) is a well-circumscribed tumor of sweat gland origin that arises in the dermis. The architecture usually is solid, cystic, or a combination of both. The cytology is classically bland with poroid, squamoid, or clear cell morphology. Clear cells that are positive on periodic acid-Schiff staining predominate in up to one-third of cases. Carcinoembryonic antigen and epithelial membrane antigen can be used to highlight the eosinophilic cuticles of ducts within solid areas.6
Sebaceous carcinoma (Figure 4) most frequently arises in a periorbital distribution, although extraocular lesions are known to occur. Histologically, there is a proliferation of both mature sebocytes and basaloid cells in the dermis, occasionally involving the epidermis. The mature sebocytes demonstrate clear cell features with foamy to vacuolated cytoplasm and large nuclei with scalloped borders. The clear cells may vary greatly in number and often are sparse in poorly differentiated tumors in which pleomorphic basaloid cells may predominate. The basaloid cells may resemble those of squamous or basal cell carcinoma, leading to a diagnostic dilemma in some cases. Special staining with Sudan black B and oil red O highlights the cytoplasmic lipid but must be performed on frozen section specimens. Although not entirely specific, immunohistochemical expression of epithelial membrane antigen, androgen receptor, and membranous vesicular adipophilin staining in sebaceous carcinoma can assist in the diagnosis.7
Cutaneous xanthomas (Figure 5) may arise in patients of any age and represent deposition of lipid-laden macrophages. Classification often is dependent on the clinical presentation; however, some subtypes demonstrate unique morphologic features (eg, verruciform xanthomas). Xanthomas classically arise in association with elevated serum lipids, but they also may occur in normolipemic patients. Individuals with Erdheim-Chester disease have an increased propensity to develop xanthelasma. Similarly, plane xanthomas have been associated with monoclonal gammopathy. Histologically, xanthomas are characterized by sheets of foamy macrophages within the dermis and subcutis. Positive immunohistochemical staining for CD68 highlighting the histiocytic nature of the cells and the absence of a delicate vascular network aid in the differentiation from RCC.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Lin F, Prichard J. Handbook of Practical Immunohistochemistry: Frequently Asked Questions. 2nd ed. New York, NY: Springer; 2015.
- McKee PH, Calonje E. Diagnostic Atlas of Melanocytic Pathology. Edinburgh, Scotland: Mosby/Elsevier; 2009.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Ansai S, Takeichi H, Arase S, et al. Sebaceous carcinoma: an immunohistochemical reappraisal. Am J Dermatopathol. 2011;33:579-587.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Lin F, Prichard J. Handbook of Practical Immunohistochemistry: Frequently Asked Questions. 2nd ed. New York, NY: Springer; 2015.
- McKee PH, Calonje E. Diagnostic Atlas of Melanocytic Pathology. Edinburgh, Scotland: Mosby/Elsevier; 2009.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Ansai S, Takeichi H, Arase S, et al. Sebaceous carcinoma: an immunohistochemical reappraisal. Am J Dermatopathol. 2011;33:579-587.
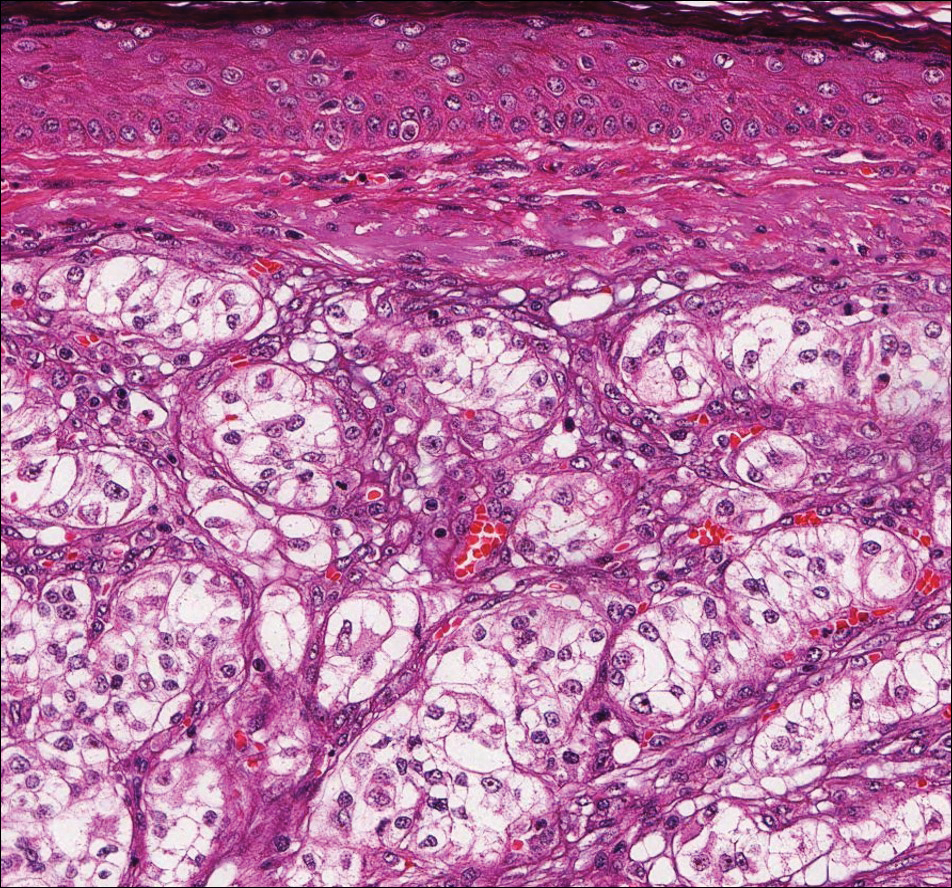
A 59-year-old man presented with a 1.5×1.0-cm asymptomatic, smooth, red-blue nodule on the left parietal scalp. The nodule had been rapidly enlarging over the last 3 weeks. After resection, the cut surface was golden yellow and focally hemorrhagic.
Flesh-Colored Papular Eruption
Papular Mucinosis/Scleromyxedema
Papular mucinosis/scleromyxedema, also known as generalized lichen myxedematosus, is a rare dermal mucinosis characterized by a papular eruption that can have an associated IgG λ paraproteinemia. The clinical presentation is gradual with the development of firm, flesh-colored, 2- to 3-mm papules often involving the hands, face, and neck that can progress to plaques that cover the entire body. Skin stiffening also can be seen.1 Extracutaneous symptoms are common and include dysphagia, arthralgia, myopathy, and cardiac dysfunction.2 Occasionally, central nervous system involvement can lead to the often fatal dermato-neuro syndrome.3,4
Histologically, papular mucinosis/scleromyxedema demonstrates increased, irregularly arranged fibroblasts in the reticular dermis with increased dermal mucin deposition (quiz image and Figure 1). The epidermis is normal or slightly thinned due to pressure from dermal changes. There may be a mild superficial perivascular lymphocytic infiltrate and atrophy of hair follicles.5 In this case, the clinical and histologic findings best supported a diagnosis of papular mucinosis/scleromyxedema.

Infundibulofolliculitis is a pruritic follicular papular eruption typically involving the neck, trunk, and proximal upper arms and shoulders. It is most common in black men who reside in hot and humid climates. Although infundibulofolliculitis would be included in the clinical differential diagnosis for the current patient, the histopathologic findings were quite distinct for the correct diagnosis of papular mucinosis/scleromyxedema. Infundibulofolliculitis shows widening of the upper part of the hair follicle (infundibulum) and infundibular inflammatory infiltrate with follicular spongiosis (Figure 2). Neither mucin deposition nor fibroblast proliferation is appreciated in infundibulofolliculitis.6,7
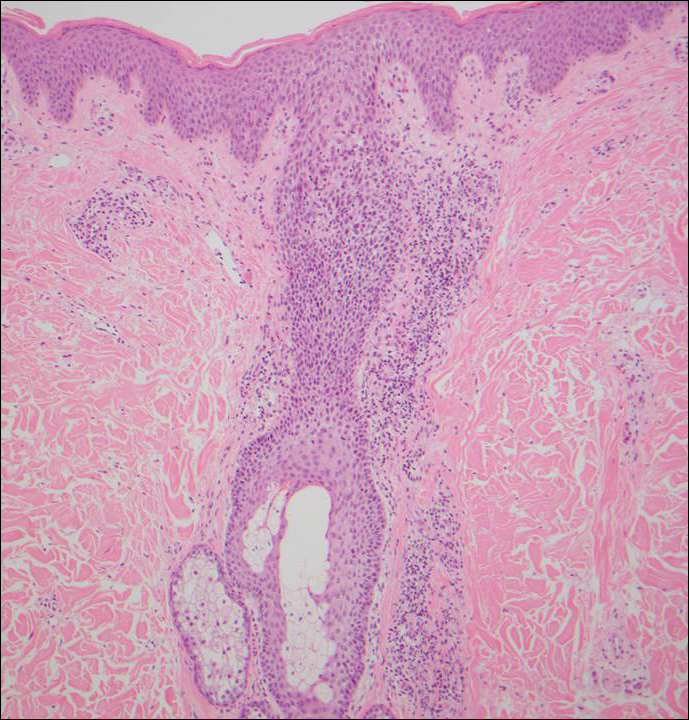
Granuloma annulare (GA) often can be distinguished clinically from papular mucinosis/scleromyxedema due to the annular appearance of papules and plaques in GA and the lack of stiffness of underlying skin. Interstitial granuloma annulare is a histologic variant of GA that can be included in the histologic differential diagnosis of papular mucinosis/scleromyxedema. Histologically, there is an interstitial infiltrate of cytologically bland histiocytes dissecting between collagen bundles in interstitial GA (Figure 3). Necrobiosis and collections of mucin often are inconspicuous. Occasionally, the presence of eosinophils can be a helpful clue.8 A fibroblast proliferation is not a feature of GA.
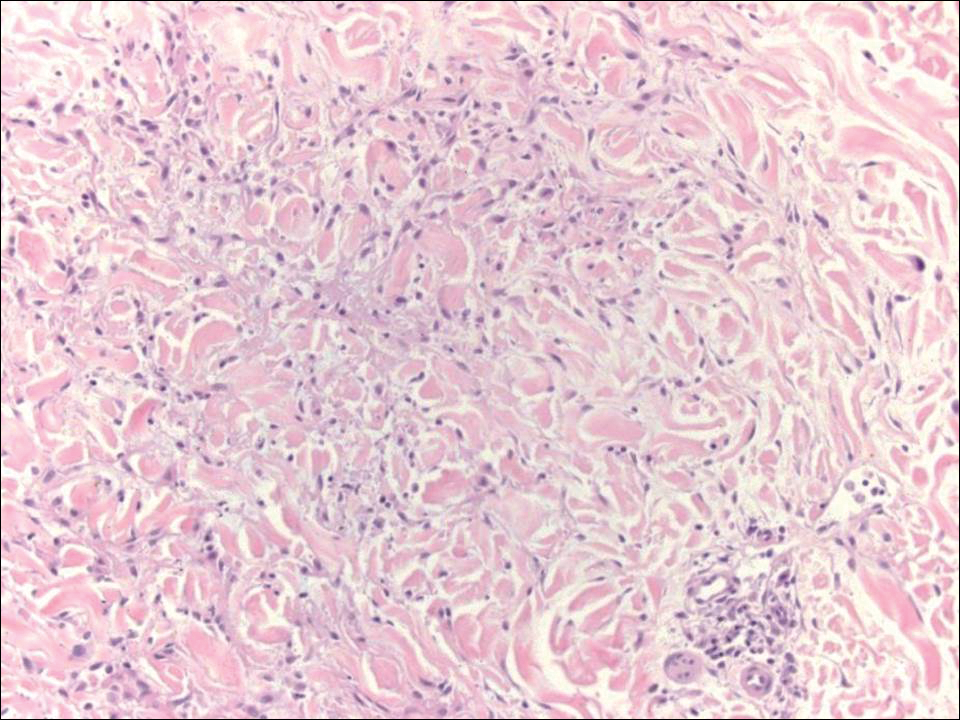
Reticular erythematous mucinosis also is a type of cutaneous mucinosis but with a classic clinical appearance of a reticulated erythematous plaque on the chest or back, making it clinically distinct from papular mucinosis/scleromyxedema and the presentation described in the current patient. Reticular erythematous mucinosis can be histologically distinguished from papular mucinosis/scleromyxedema by the presence of a superficial and deep perivascular lymphocytic infiltrate with increased dermal mucin deposition (Figure 4). It often shows a positive IgM deposition on the basement membrane on direct immunofluorescence.9

Similar to papular mucinosis/scleromyxedema, scleredema shows thickening of the skin with decreased movement of involved areas. Scleredema often involves the upper back, shoulders, and neck where affected areas often have a peau d'orange appearance. Scleredema is classified into 3 clinical forms based on clinical associations. Type 1 often is preceded by an infection, classically Streptococcus pyogenes. Type 2 is associated with a hematologic dyscrasia such as multiple myeloma, or it can have an associated paraproteinemia that is typically of the IgA κ type, which is distinct from papular mucinosis/scleromyxedema where IgG λ paraproteinemia typically is seen. Type 3 is associated with diabetes mellitus. Histologically, scleredema also is distinct from papular mucinosis/scleromyxedema. Although increased mucin is seen in the dermis, the mucin is classically more prominent in the deep reticular dermis as compared with papular mucinosis/scleromyxedema (Figure 5). Additionally, collagen bundles are thickened with clear separation between them. Hyperplasia of fibroblasts in the dermis that is a characteristic feature of papular mucinosis/scleromyxedema is not observed in scleredema.10
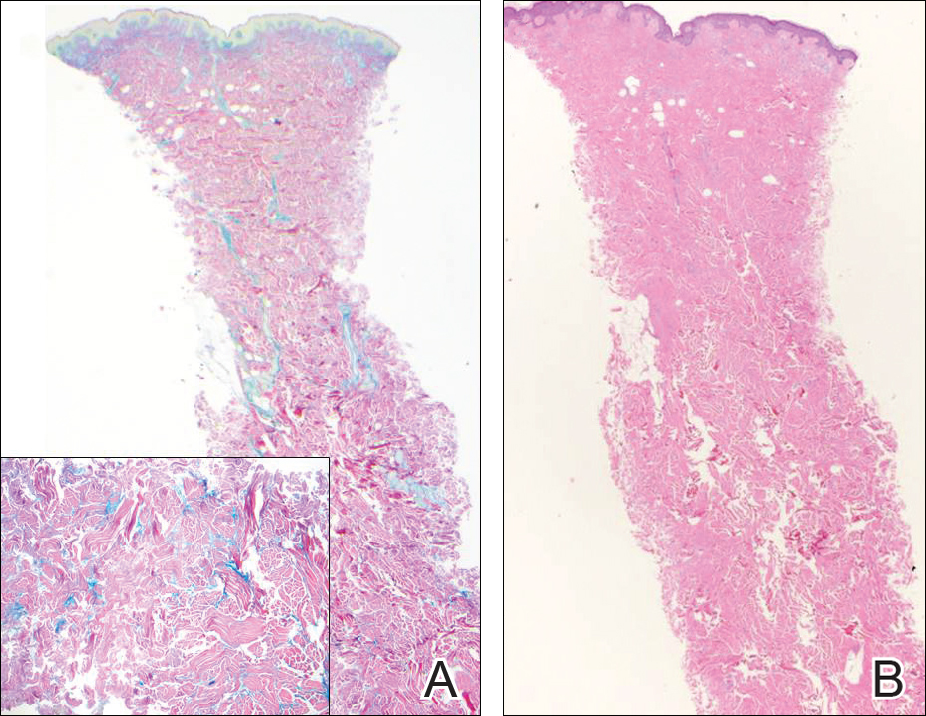
- Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Fleming KE, Virmani D, Sutton E, et al. Scleromyxedema and the dermato-neuro syndrome: case report and review of the literature. J Cutan Pathol. 2012;39:508-517.
- Hummers LK. Scleromyxedema. Curr Opin Rheumatol. 2014;26:658-662.
- Rongioleti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Owen WR, Wood C. Disseminate and recurrent infundibulofolliculitis. Arch Dermatol. 1979;5:174-175.
- Soyinka F. Recurrent disseminated infundibulofolliculitis. Int J Dermatol. 1973;12:314-317.
- Keimig EL. Granuloma annulare. Dermatol Clin. 2015;33:315-329.
- Thareja S, Paghdal K, Lein MH, et al. Reticular erythematous mucinosis--a review. Int J Dermatol. 2012;51:903-909.
- Beers WH, Ince AI, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
Papular Mucinosis/Scleromyxedema
Papular mucinosis/scleromyxedema, also known as generalized lichen myxedematosus, is a rare dermal mucinosis characterized by a papular eruption that can have an associated IgG λ paraproteinemia. The clinical presentation is gradual with the development of firm, flesh-colored, 2- to 3-mm papules often involving the hands, face, and neck that can progress to plaques that cover the entire body. Skin stiffening also can be seen.1 Extracutaneous symptoms are common and include dysphagia, arthralgia, myopathy, and cardiac dysfunction.2 Occasionally, central nervous system involvement can lead to the often fatal dermato-neuro syndrome.3,4
Histologically, papular mucinosis/scleromyxedema demonstrates increased, irregularly arranged fibroblasts in the reticular dermis with increased dermal mucin deposition (quiz image and Figure 1). The epidermis is normal or slightly thinned due to pressure from dermal changes. There may be a mild superficial perivascular lymphocytic infiltrate and atrophy of hair follicles.5 In this case, the clinical and histologic findings best supported a diagnosis of papular mucinosis/scleromyxedema.
Infundibulofolliculitis is a pruritic follicular papular eruption typically involving the neck, trunk, and proximal upper arms and shoulders. It is most common in black men who reside in hot and humid climates. Although infundibulofolliculitis would be included in the clinical differential diagnosis for the current patient, the histopathologic findings were quite distinct for the correct diagnosis of papular mucinosis/scleromyxedema. Infundibulofolliculitis shows widening of the upper part of the hair follicle (infundibulum) and infundibular inflammatory infiltrate with follicular spongiosis (Figure 2). Neither mucin deposition nor fibroblast proliferation is appreciated in infundibulofolliculitis.6,7
Granuloma annulare (GA) often can be distinguished clinically from papular mucinosis/scleromyxedema due to the annular appearance of papules and plaques in GA and the lack of stiffness of underlying skin. Interstitial granuloma annulare is a histologic variant of GA that can be included in the histologic differential diagnosis of papular mucinosis/scleromyxedema. Histologically, there is an interstitial infiltrate of cytologically bland histiocytes dissecting between collagen bundles in interstitial GA (Figure 3). Necrobiosis and collections of mucin often are inconspicuous. Occasionally, the presence of eosinophils can be a helpful clue.8 A fibroblast proliferation is not a feature of GA.
Reticular erythematous mucinosis also is a type of cutaneous mucinosis but with a classic clinical appearance of a reticulated erythematous plaque on the chest or back, making it clinically distinct from papular mucinosis/scleromyxedema and the presentation described in the current patient. Reticular erythematous mucinosis can be histologically distinguished from papular mucinosis/scleromyxedema by the presence of a superficial and deep perivascular lymphocytic infiltrate with increased dermal mucin deposition (Figure 4). It often shows a positive IgM deposition on the basement membrane on direct immunofluorescence.9
Similar to papular mucinosis/scleromyxedema, scleredema shows thickening of the skin with decreased movement of involved areas. Scleredema often involves the upper back, shoulders, and neck where affected areas often have a peau d'orange appearance. Scleredema is classified into 3 clinical forms based on clinical associations. Type 1 often is preceded by an infection, classically Streptococcus pyogenes. Type 2 is associated with a hematologic dyscrasia such as multiple myeloma, or it can have an associated paraproteinemia that is typically of the IgA κ type, which is distinct from papular mucinosis/scleromyxedema where IgG λ paraproteinemia typically is seen. Type 3 is associated with diabetes mellitus. Histologically, scleredema also is distinct from papular mucinosis/scleromyxedema. Although increased mucin is seen in the dermis, the mucin is classically more prominent in the deep reticular dermis as compared with papular mucinosis/scleromyxedema (Figure 5). Additionally, collagen bundles are thickened with clear separation between them. Hyperplasia of fibroblasts in the dermis that is a characteristic feature of papular mucinosis/scleromyxedema is not observed in scleredema.10
Papular Mucinosis/Scleromyxedema
Papular mucinosis/scleromyxedema, also known as generalized lichen myxedematosus, is a rare dermal mucinosis characterized by a papular eruption that can have an associated IgG λ paraproteinemia. The clinical presentation is gradual with the development of firm, flesh-colored, 2- to 3-mm papules often involving the hands, face, and neck that can progress to plaques that cover the entire body. Skin stiffening also can be seen.1 Extracutaneous symptoms are common and include dysphagia, arthralgia, myopathy, and cardiac dysfunction.2 Occasionally, central nervous system involvement can lead to the often fatal dermato-neuro syndrome.3,4
Histologically, papular mucinosis/scleromyxedema demonstrates increased, irregularly arranged fibroblasts in the reticular dermis with increased dermal mucin deposition (quiz image and Figure 1). The epidermis is normal or slightly thinned due to pressure from dermal changes. There may be a mild superficial perivascular lymphocytic infiltrate and atrophy of hair follicles.5 In this case, the clinical and histologic findings best supported a diagnosis of papular mucinosis/scleromyxedema.
Infundibulofolliculitis is a pruritic follicular papular eruption typically involving the neck, trunk, and proximal upper arms and shoulders. It is most common in black men who reside in hot and humid climates. Although infundibulofolliculitis would be included in the clinical differential diagnosis for the current patient, the histopathologic findings were quite distinct for the correct diagnosis of papular mucinosis/scleromyxedema. Infundibulofolliculitis shows widening of the upper part of the hair follicle (infundibulum) and infundibular inflammatory infiltrate with follicular spongiosis (Figure 2). Neither mucin deposition nor fibroblast proliferation is appreciated in infundibulofolliculitis.6,7
Granuloma annulare (GA) often can be distinguished clinically from papular mucinosis/scleromyxedema due to the annular appearance of papules and plaques in GA and the lack of stiffness of underlying skin. Interstitial granuloma annulare is a histologic variant of GA that can be included in the histologic differential diagnosis of papular mucinosis/scleromyxedema. Histologically, there is an interstitial infiltrate of cytologically bland histiocytes dissecting between collagen bundles in interstitial GA (Figure 3). Necrobiosis and collections of mucin often are inconspicuous. Occasionally, the presence of eosinophils can be a helpful clue.8 A fibroblast proliferation is not a feature of GA.
Reticular erythematous mucinosis also is a type of cutaneous mucinosis but with a classic clinical appearance of a reticulated erythematous plaque on the chest or back, making it clinically distinct from papular mucinosis/scleromyxedema and the presentation described in the current patient. Reticular erythematous mucinosis can be histologically distinguished from papular mucinosis/scleromyxedema by the presence of a superficial and deep perivascular lymphocytic infiltrate with increased dermal mucin deposition (Figure 4). It often shows a positive IgM deposition on the basement membrane on direct immunofluorescence.9
Similar to papular mucinosis/scleromyxedema, scleredema shows thickening of the skin with decreased movement of involved areas. Scleredema often involves the upper back, shoulders, and neck where affected areas often have a peau d'orange appearance. Scleredema is classified into 3 clinical forms based on clinical associations. Type 1 often is preceded by an infection, classically Streptococcus pyogenes. Type 2 is associated with a hematologic dyscrasia such as multiple myeloma, or it can have an associated paraproteinemia that is typically of the IgA κ type, which is distinct from papular mucinosis/scleromyxedema where IgG λ paraproteinemia typically is seen. Type 3 is associated with diabetes mellitus. Histologically, scleredema also is distinct from papular mucinosis/scleromyxedema. Although increased mucin is seen in the dermis, the mucin is classically more prominent in the deep reticular dermis as compared with papular mucinosis/scleromyxedema (Figure 5). Additionally, collagen bundles are thickened with clear separation between them. Hyperplasia of fibroblasts in the dermis that is a characteristic feature of papular mucinosis/scleromyxedema is not observed in scleredema.10
- Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Fleming KE, Virmani D, Sutton E, et al. Scleromyxedema and the dermato-neuro syndrome: case report and review of the literature. J Cutan Pathol. 2012;39:508-517.
- Hummers LK. Scleromyxedema. Curr Opin Rheumatol. 2014;26:658-662.
- Rongioleti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Owen WR, Wood C. Disseminate and recurrent infundibulofolliculitis. Arch Dermatol. 1979;5:174-175.
- Soyinka F. Recurrent disseminated infundibulofolliculitis. Int J Dermatol. 1973;12:314-317.
- Keimig EL. Granuloma annulare. Dermatol Clin. 2015;33:315-329.
- Thareja S, Paghdal K, Lein MH, et al. Reticular erythematous mucinosis--a review. Int J Dermatol. 2012;51:903-909.
- Beers WH, Ince AI, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
- Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Fleming KE, Virmani D, Sutton E, et al. Scleromyxedema and the dermato-neuro syndrome: case report and review of the literature. J Cutan Pathol. 2012;39:508-517.
- Hummers LK. Scleromyxedema. Curr Opin Rheumatol. 2014;26:658-662.
- Rongioleti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Owen WR, Wood C. Disseminate and recurrent infundibulofolliculitis. Arch Dermatol. 1979;5:174-175.
- Soyinka F. Recurrent disseminated infundibulofolliculitis. Int J Dermatol. 1973;12:314-317.
- Keimig EL. Granuloma annulare. Dermatol Clin. 2015;33:315-329.
- Thareja S, Paghdal K, Lein MH, et al. Reticular erythematous mucinosis--a review. Int J Dermatol. 2012;51:903-909.
- Beers WH, Ince AI, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
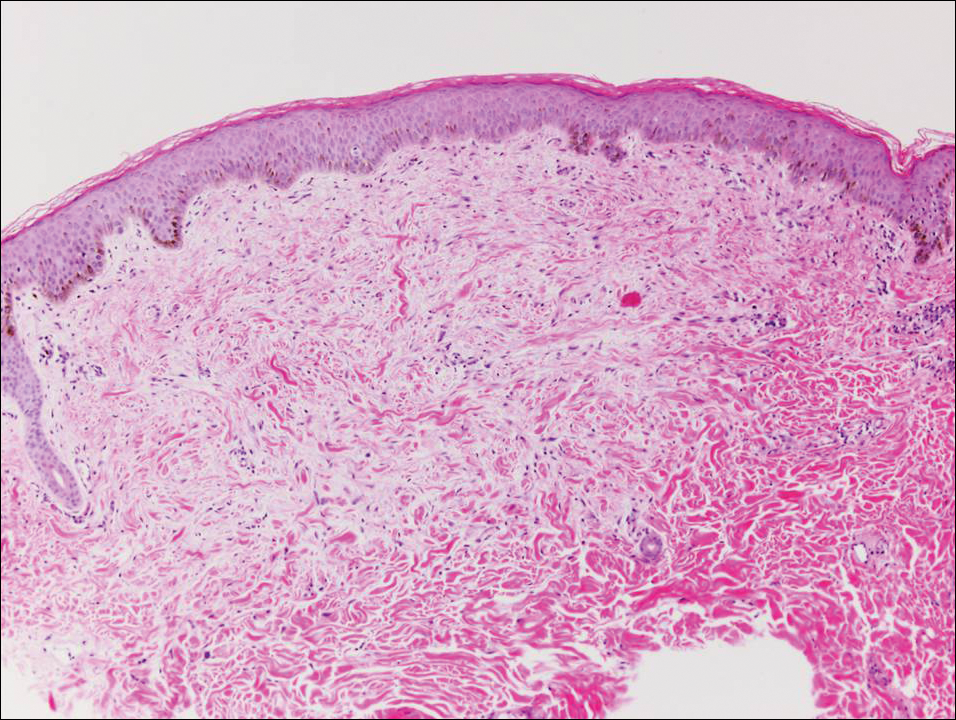
A 48-year-old black man presented with a rash of 7 months' duration that started on the face and spread to the body. He had extreme pruritus, increased stiffness in the hands and joints, and paresthesia. Physical examination revealed an eruption of 2- to 4-mm, flesh-colored papules with follicular accentuation on the face, neck, bilateral upper extremities, back, and thighs.