User login
Cognitive Errors in Medical Injury
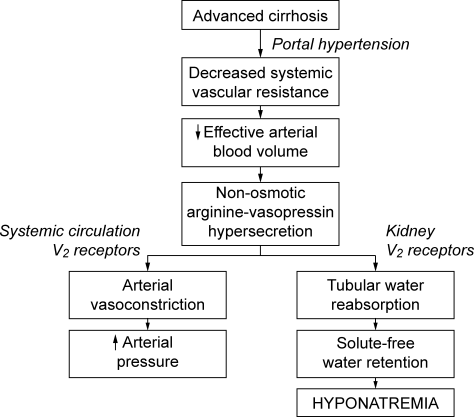
Promotion of safer healthcare by patient organizations has led to an expansion of studies aimed at understanding medical errors to minimize injury through systemic improvement. These efforts have focused on identifying patient‐related factors, reducing technology failures, and improving communication.1 In contrast, factors related to cognitive errors by healthcare providers have received relatively little attention, although such errors may be an important source of preventable harm.1, 2
Limited information is available on the types and prevalence of cognitive factors in cases of medical injury, although cognitive factors may be a major risk for medical injury. If these factors were confirmed to be important factors for medical injury, better educational strategies may be needed to reduce cognitive errors among physicians and to enhance quality improvement and patient safety. Better understanding of these cognitive factors may also help to implement educational programs aimed at the improvement of cognitive performance in medical schools or teaching hospital.35
Closed‐claim files for cases of medical injury contain valuable information for investigation of the factors involved in medical errors.3 In Japan, court claims were tried and closed orders were issued by judges without a jury system until 2009. Under this system, representatives for defense and plaintiffs can present medical experts. Courts can also appoint experts independent of either party. Court opinions in Japan are considered as neutral judgments for conflicts between plaintiffs and defendants. Usually there are 3 judges who are required to be involved with each judgment in Japanese courts.
Closed‐claim files in cases of medical injury contain information about the types and prevalence of cognitive factors suggested to be causally related to the injuries by verdicts in district courts. Thus, by analyzing these files, an unbiased description of the characteristics and epidemiology of cognitive factors can be obtained for cases of medical injury, with minimization of potentially biased claims indicated by both parties; ie, plaintiffs vs. hospitals. Therefore, in this study, by using information from closed claims files at district courts in Tokyo and Osaka, Japan, we aimed to determine the important cognitive factors associated with cases of medical injury from such factors as judgment, vigilance, memory, technical competence, or knowledge. Since we anticipated that cognitive factors would dominate among the causative factors, we also explored the association of these factors with cases in which a judgment of paid compensation was made.
Methods
Study Sample
The authors acknowledge that the methodologies are based on those from the Malpractice Insurers' Medical Errors Prevention Study.6 A claim was defined as a written demand for compensation for cases of medical injury, based on a similar approach in previous studies.7, 8 Reviews were performed for closed‐claim files for cases of medical injury involving physicians from 2001 to 2005. These files were published by the Division of the Tokyo‐Osaka Medical Malpractice Lawsuits, organized by district courts in Tokyo and Osaka. The files included all closed‐claim cases of medical injury involving physicians from 2001 to 2005 at district courts in Tokyo and Osaka. The locations of delivery of care were inpatients in this study. All patients in Japan were insured during the study period.
Data Collection
Reviews were conducted by 3 board‐certified Japanese physician‐investigators specializing in internal medicine (1 chief investigator and 2 coinvestigators). The chief investigator trained the coinvestigators in 1‐day sessions with regard to the content of claims files, data collection, and the confidentiality procedure. Reviews were first performed by 1 coinvestigator and then confirmed by the chief investigator.
Data were collected for patient demographics and characteristics of adverse events, including types, locations, clinical areas, and specialties involved in the claims. Classification of specialties was based on that of Singh et al.3 Types of adverse events included minor injury for cases with complete recovery within a year, significant injury for those with complete recovery requiring more than a year, major injury for those with incomplete recovery (any physical sequelae) after more than a year, and death. Clinical areas consisted of surgery, obstetrics, missed diagnosis, delayed diagnosis, medication, and fall. Data for litigation outcomes and the amounts of paid compensation in Japanese Yen (JY) were also collected for claims that received verdicts supporting the plaintiffs.
All factors identified in the verdicts as causally related to the medical injury were recorded for data analysis. Classification of these factors was based on that of Singh et al.3 Cognitive factors were drawn from a list of categories of physicians' tasks provided by the Occupational Information Network. This network is a database of occupational requirements and worker attributes and it describes occupations in terms of the skills and knowledge required, how the work is performed, and typical work settings. The list of cognitive factor categories of physicians' tasks included judgment, vigilance, memory, technical competence, or knowledge. Accordingly, the cognitive factor category list was considered to capture the work of clinicians across the entire range of specialties.3
An example concerning failure of judgment would be that a rapid respiratory rate in initial vital signs was missed or ignored in a patient who complained of upper abdominal pain, was sent home with a diagnosis of gastritis, and eventually died at home; and an autopsy diagnosis of myocardial infarction with congestive heart failure was later confirmed. A vigilance error example would be that, in an electronic ordering system, typing an incorrect medication that has the similar commercial name of a correct medication. An example of failure of memory as a cognitive error would be that a physician forgot a result of laboratory data (positive sputum cytology of lung cancer), and so the physician did not explain it to the patient and did not perform an appropriate subsequent treatment referral. A technical incompetence example would be an operative or procedural injury due to technical problems of physicians. An example of a knowledge error would be that a contraindicated drug combination was prescribed such as the use of both selective serotonin reuptake inhibitor and monoamine oxidase inhibitor.
For systemic factors, a teamwork problem (poor teamwork) was used to describe disruptive team behavior, based on the concept of teamwork described by the Agency for Healthcare Research and Quality and the British Medical Association.9, 10 Cases with teamwork problems were defined as those in which the original reviewer had judged that 1 or more of the following contributory factors played a role in the error: communication breakdowns, supervision problems, handoff problems, failures to establish clear lines of responsibility, and conflict among clinical staff. Technology failure indicated an error of commission or omission by devices, tools, or machines.
The Japanese courts analyze medical records but they do not open the records to the public and so we could not analyze the medical records of the cases in our study. Thus, we did not judge whether the adverse outcome could have been attributed to medical errors, while we analyzed the claims files and followed the conclusions reached by the end of the claims.
Statistical Analysis
Data are given as proportions for categorical variables and means or medians for continuous variables. Cognitive factors associated with cases receiving adjudication of a compensation payment by district courts (litigation outcomes) were analyzed using a logistic regression model including 5 types of cognitive errors. Analyses were conducted with the Stata SE 10.0 statistical software package (College Station, TX). All P values are 2‐sided and P < 0.05 was considered to be statistically significant. The study was approved by the ethics review board at the institution of the chief investigator.
Results
In a total of 274 closed cases of medical injury, the mean age of the patients was 49 years old and 45% were women (Table 1). The reviews performed by the coinvestigators were all confirmed by the chief investigator without discordance of the reviews between the coinvestigators and the chief investigator. The claims involved death of patients in 45% of cases; injuries that caused significant or major disability in 10% and 24%, respectively (a total of 34%); and minor adverse outcomes of medical care in 21% (57 cases). Closing verdicts supporting the plaintiffs (patients or family) by the district courts were given in 103 claims (38%), with compensation at a median of 8,000,000 JY (100 JY = $1 US in 2005). The compensation ranged from 20,000 JY to 222,710,251 JY. The highest compensation was ordered to be paid to a 36‐year‐old woman with an obstetrics‐related major injury and the court indicated the injury was causally related to the following 3 cognitive factors: error in judgment, failure of vigilance, and lack of technical competence.
| Characteristic | n (%) |
|---|---|
| |
| Demographic of patients | |
| Women | 121 (45) |
| Men | 153 (55) |
| Age, mean SD, year | 49 22 |
| Adverse outcome | |
| Minor | 57 (21) |
| Significant | 28 (10) |
| Major | 67 (24) |
| Death | 122 (45) |
| Operative | 36 |
| Delayed diagnosis | 35 |
| Medication | 26 |
| Missed diagnosis | 16 |
| Obstetrics | 8 |
| Clinical area | |
| Operative | 120 (44) |
| Delayed diagnosis | 54 (20) |
| Medication | 50 (18) |
| Missed diagnosis | 28 (10) |
| Obstetrics | 19 (7) |
| Fall | 3 (1) |
Operative injury was the most frequent reason for claims, followed by delayed diagnosis, medication error, and missed diagnosis. General surgery, orthopedics, internal medicine, and obstetrics/gynecology were the most frequently involved specialties, comprising 30% of all cases (Table 2). The verdicts suggested cognitive factors were the most prevalent factors associated with cases of medical injury: 73% of the injuries were judged to be the result of an error in judgment (Table 3), followed by failure of vigilance (65%), lack of technical competence (34%), and lack of knowledge (31%). Verdicts indicated systemic factors in only a few cases, including poor teamwork in 4% and technology failure in 2%. Patient‐related factors were suggested in 32% of the claims.
| Specialty | Cases, n (%) |
|---|---|
| General surgery | 27 (10) |
| Orthopedic surgery | 27 (10) |
| Internal medicine | 27 (10) |
| Obstetrics‐gynecology | 26 (9) |
| Neurosurgery | 19 (7) |
| Ear, nose, and throat | 18 (7) |
| Plastic surgery | 15 (5) |
| Psychiatry | 14 (5) |
| Cardiology | 13 (5) |
| Dental care | 13 (5) |
| Ophthalmology | 12 (4) |
| Hematology or oncology | 10 (4) |
| Adult primary care | 9 (3) |
| Pediatrics | 8 (3) |
| Urology | 8 (3) |
| Cardiothoracic surgery | 8 (3) |
| Neurology | 5 (2) |
| Anesthesiology | 4 (1) |
| Physical medicine or rehabilitation | 3 (1) |
| Emergency medicine | 2 (1) |
| Infectious disease | 2 (1) |
| Dermatology | 2 (1) |
| Radiology | 1 (<1) |
| Vascular surgery | 1 (<1) |
| Contributory Factor | n (%) |
|---|---|
| |
| Cognitive factors | |
| Error in judgment | 199 (73) |
| Failure of vigilance | 177 (65) |
| Lack of technical competence | 94 (34) |
| Lack of knowledge | 86 (31) |
| Failure of memory | 5 (2) |
| System factors | |
| Poor teamwork | 11 (4) |
| Technology failure | 5 (2) |
| Patient‐related factors | 87 (32) |
In a multivariable‐adjusted logistic regression analysis of cognitive factors with a potential association with the claims with paid compensation (Table 4), only error in judgment showed a significant association (odds ratio, 1.9; 95% confidence interval [CI], 1.01‐3.40). The other four cognitive factors in the model were not associated with these claims. The odds ratio for failure of memory was high (2.8), but this factor was identified by the courts in only 5 cases and was not significantly associated with claims with paid compensation.
| Cognitive Factor | Cases With No Compensation (n = 171), n (%) | Cases With Paid Compensation (n = 103), n (%) | Odds Ratio (95% CI)* |
|---|---|---|---|
| |||
| Error in judgment | 117 (68) | 82 (80) | 1.9 (1.03.4) |
| Failure of vigilance | 111 (65) | 66 (64) | 1.0 (0.61.7) |
| Failure of memory | 2 (1) | 3 (3) | 2.8 (0.518) |
| Lack of technical competence | 58 (34) | 36 (35) | 1.1 (0.61.8) |
| Lack of knowledge | 52 (30) | 34 (33) | 1.0 (0.61.7) |
Discussion
In this study of closed claims files, we identified 2 important cognitive factors involved in cases of medical injury. Error in judgment was the most common factor, comprising about 70% of all claims, and was significantly associated with cases with paid compensation for medical injury. The second cognitive factor was failure of vigilance, which was found in 65% of the claims. Other cognitive factors, such as lack of technical competence and knowledge or failure of memory, as well as systemic factors (poor teamwork and technology failure) were less frequently found to be causally related to cases with medical injury in the verdicts examined in the study.
Reasons for the low frequency of systemic factors involved in cases of medical injury in our study are unclear. This may be the cultural characteristics such as greater emphasis to working in teams and following rules of an organization in Japan. Another possibility is that plaintiffs might have tended to generate lawsuits in cases with suspected higher frequency of individual physicians' factors in Japan. Moreover, among cognitive factors, lack of technical competence and knowledge or failure of memory was also less frequently related to cases with medical injury in our study compared to those of the previous studies.3, 11
The study design of analyzing closed claims files of cases of medical injury is noteworthy for its methodology of error assessment and provides valuable information on errors related to medical injury.3, 7 Moreover, the system of court verdicts in Japan based on decisions by a professional judge allows elimination of potential bias from stakeholders (plaintiffs vs. hospitals) involved in cases of medical injury. Thus, probable causes related to adverse events can be determined from a neutral position. Previous studies of medical error have focused on medical record reviews, surveys, and interviews;12, 13 our study corroborates and extends the findings in these studies that cognitive errors are the most frequent source of medical injury.
Error in judgment is commonly made in the course of decision making in multiple clinical areas. This type of error is referred to recently as cognitive dispositions to respond,14 which is different from bias or heuristics, since not all heuristics are biased and not all errors in judgments come from bias. There is a well‐established value of heuristics in medical diagnosis. Moreover, the properties of this type of error are likely to be distinct from those associated with performance of procedures (lack of technical competence), such as operative injury, which are directly visible and can be prevented through rapid dissemination of information on safety procedures among a medical team. However, the consequences of error in judgment are important for patients, family, and healthcare providers, and these errors are also largely preventable by implementation of educational programs.15
Possible solutions for improving clinical judgment skills may be derived from recent education theory. The theory provides a means for minimizing errors in judgment through the process of meta‐cognition, in which cognitive forcing strategies can be developed through thinking that involves active control over the process of one's own thinking.14, 15 For example, reflective practice has been suggested to be an important instrument for improving clinical judgment and may particularly improve diagnoses in situations of uncertainty and uniqueness, thereby reducing diagnostic errors.16 The capability of critical reflection in real‐time practice (reflection‐in‐action) and on our own practice (reflection‐on‐action) appears to be a key requirement for developing and maintaining medical expertise.17, 18 For instance, case‐based discussion with clinician educators can be an opportunity for enhancing critical thinking skills of medical trainees.
Based on a context‐based approach that focuses on the nature of the clinical problem, potential systemic solutions have recently been proposed for reducing errors in judgment.1 These solutions utilize advanced technology, including symptom‐oriented diagnostic decision support, internet search engines for information on possible diagnoses, and automated reminders in electronic health records.1, 19 Previous studies have shown that long work hours and sleep deprivation can decrease cognitive function, leading to failure of vigilance and increased medical errors,20 and several systemic solutions provide models for avoidance of failure of vigilance. For instance, eliminating extended work shifts and reducing the number of work hours per week was shown to reduce serious medical errors through increased sleep and decreased failure of vigilance during night work in an intensive care unit.21, 22 Taking a brief nap during work hours has also been associated with decreased medical errors in a recent study conducted in Japan.23 Despite the well‐known importance of factors of physicians' workloads, our study did not analyze these factors and thus further studies are needed to confirm their importance in Japanese medical practice.
There were also 32% of patient‐related factors suggested as contributory factors to medical injury in verdicts of the closed claims. This finding may be also important in planning educational intervention strategies to reduce medical errors. Although our data did not include the relative frequency of components related to these factors, major components of patient‐related factors may include age, severity of illnesses, comorbidity, functional status, or mental status. Educational intervention programs may help healthcare providers to evaluate patients with these risk factors and to implement preventive strategies to avoid incidents among these patients.
General surgery, orthopedic surgery, internal medicine, and obstetrics‐gynecology were the most frequently involved specialties in our study. The reasons why these specialties were highly involved in the claims are unclear and our study could not analyze these issues. However, these specialties may be related to patients with greater clinical severity and thus they may have subsequently higher risk for receiving claims. Further, physicians in these specialties may be at higher risk for having various errors because of the complexity of care for patients.
Our study has several limitations. First, the closed claims are more likely to represent cases with severe injury.3 Therefore, it is unclear if we can generalize our findings beyond cases with severe injury.3 Second, certain contributory factors may not have been suggested by the verdicts, even though they played a role. Among these potential factors, poor teamwork and communication issues are unlikely to be identified as causative in verdicts, unless the allegation of the plaintiffs documented these issues. Moreover, the Japanese courts did not open the medical records to the public and so we could not analyze the medical records of the cases. Third, we only evaluated closed verdicts given by professional judges of district courts, who are unlikely to be medical experts. However, the closed verdicts underwent an extensive process involving testimony from medical professionals and academic societies. Fourth, we, as investigators, had few members with surgical backgrounds in this study so we might have underestimated issues related to technical competence among the claims. Finally, although a small percentage of closed‐ claim cases involving team performance were identified in our study, the plaintiffs might have indicated this point to the court claims, since it might have been difficult to describe this issue as a reason for requesting compensations from defendants. Thus, despite a low proportion of team performance involvement in the verdicts, we still believe that poor team performance is a factor related to most medical injuries.
In summary, causal factors obtained from closed claims files suggest the importance of cognitive factors in cases of medical injury. Among the cognitive factors, error in judgment and failure of vigilance were the most frequent. These findings may help leaders of medical schools and hospitals to allocate more resources for research into strategies to improve cognitive performance and thereby ensure patient safety. Further research is needed to better understand the cognitive mechanisms involved in medical errors and to translate this into educational strategies.
- ,.Diagnostic errors‐the next frontier for patient safety.JAMA.2009;301(10):1060–1062.
- ,,.Diagnostic error in internal medicine.Arch Intern Med.2005;165(13):1493–1499.
- ,,,.Medical errors involving trainees: a study of closed malpractice claims from 5 insurers.Arch Intern Med.2007;167(19):2030–2036.
- ,,.Understanding diagnostic errors in medicine: a lesson from aviation.Qual Saf Health Care.2006;15(3):159–164.
- .The importance of cognitive errors in diagnosis and strategies to minimize them.Acad Med.2003;78(8):775–780.
- ,,, et al.Claims, errors, and compensation payments in medical malpractice litigation.N Engl J Med.2006;354(19):2024–2033.
- ,,,,,.Negligent care and malpractice claiming behavior in Utah and Colorado.Med Care.2000;38(3):250–260.
- ,,, et al.Incidence and types of adverse events and negligent care in Utah and Colorado.Med Care.2000;38(3):261–271.
- ,,,,.Medical Teamwork and Patient Safety: The Evidence‐Based Relation.Rockville, MD:Agency for Healthcare Research and Quality;2005 [updated April 2005]; Available at: http://www.ahrq.gov/qual/medteam. Accessed June 2010.
- ,.Team working in Primary Health Care. Realising Shared Aims in Patient Care.London, UK:Royal Pharmaceutical Society and British Medical Association.2005.
- ,,,,.The nature and causes of unintended events reported at ten emergency departments.BMC Emerg Med.2009;9:16.
- ,,.To Err Is Human: Building a Safer Health System.Washington, USA:National Academy Press;2000.
- ,,,.Analysis of errors reported by surgeons at three teaching hospitals.Surgery.2003;133(6):614–621.
- .Achieving quality in clinical decision making: cognitive strategies and detection of bias.Acad Emerg Med.2002;9(11):1184–1204.
- .Cognitive forcing strategies in clinical decision making.Ann Emerg Med.2003;41(1):110–120.
- ,,.Effects of reflective practice on the accuracy of medical diagnoses.Med Educ.2008;42(5):468–475.
- .The Reflective Practitioner: How Professionals Think in Action.New York, NY:Basic Books;1983.
- ,,.Diagnostic errors and reflective practice in medicine.J Eval Clin Pract.2007;13(1):138–145.
- ,,,.Caught in the web: e‐diagnosis.J Hosp Med.2009;4(4):262–266.
- ,,, et al.Extended work duration and the risk of self‐reported percutaneous injuries in interns.JAMA.2006;296(9):1055–1062.
- ,,, et al.Effect of reducing interns' work hours on serious medical errors in intensive care units.N Engl J Med.2004;351(18):1838–1848.
- ,,, et al.Effect of reducing interns' weekly work hours on sleep and attentional failures.N Engl J Med.2004;351(18):1829–1837.
- ,,, et al.Influence of Residents' Workload, Mental State and Job Satisfaction on Procedural Error: a prospective daily questionnaire‐based study.General Medicine.2008;9(2):57–64.
Promotion of safer healthcare by patient organizations has led to an expansion of studies aimed at understanding medical errors to minimize injury through systemic improvement. These efforts have focused on identifying patient‐related factors, reducing technology failures, and improving communication.1 In contrast, factors related to cognitive errors by healthcare providers have received relatively little attention, although such errors may be an important source of preventable harm.1, 2
Limited information is available on the types and prevalence of cognitive factors in cases of medical injury, although cognitive factors may be a major risk for medical injury. If these factors were confirmed to be important factors for medical injury, better educational strategies may be needed to reduce cognitive errors among physicians and to enhance quality improvement and patient safety. Better understanding of these cognitive factors may also help to implement educational programs aimed at the improvement of cognitive performance in medical schools or teaching hospital.35
Closed‐claim files for cases of medical injury contain valuable information for investigation of the factors involved in medical errors.3 In Japan, court claims were tried and closed orders were issued by judges without a jury system until 2009. Under this system, representatives for defense and plaintiffs can present medical experts. Courts can also appoint experts independent of either party. Court opinions in Japan are considered as neutral judgments for conflicts between plaintiffs and defendants. Usually there are 3 judges who are required to be involved with each judgment in Japanese courts.
Closed‐claim files in cases of medical injury contain information about the types and prevalence of cognitive factors suggested to be causally related to the injuries by verdicts in district courts. Thus, by analyzing these files, an unbiased description of the characteristics and epidemiology of cognitive factors can be obtained for cases of medical injury, with minimization of potentially biased claims indicated by both parties; ie, plaintiffs vs. hospitals. Therefore, in this study, by using information from closed claims files at district courts in Tokyo and Osaka, Japan, we aimed to determine the important cognitive factors associated with cases of medical injury from such factors as judgment, vigilance, memory, technical competence, or knowledge. Since we anticipated that cognitive factors would dominate among the causative factors, we also explored the association of these factors with cases in which a judgment of paid compensation was made.
Methods
Study Sample
The authors acknowledge that the methodologies are based on those from the Malpractice Insurers' Medical Errors Prevention Study.6 A claim was defined as a written demand for compensation for cases of medical injury, based on a similar approach in previous studies.7, 8 Reviews were performed for closed‐claim files for cases of medical injury involving physicians from 2001 to 2005. These files were published by the Division of the Tokyo‐Osaka Medical Malpractice Lawsuits, organized by district courts in Tokyo and Osaka. The files included all closed‐claim cases of medical injury involving physicians from 2001 to 2005 at district courts in Tokyo and Osaka. The locations of delivery of care were inpatients in this study. All patients in Japan were insured during the study period.
Data Collection
Reviews were conducted by 3 board‐certified Japanese physician‐investigators specializing in internal medicine (1 chief investigator and 2 coinvestigators). The chief investigator trained the coinvestigators in 1‐day sessions with regard to the content of claims files, data collection, and the confidentiality procedure. Reviews were first performed by 1 coinvestigator and then confirmed by the chief investigator.
Data were collected for patient demographics and characteristics of adverse events, including types, locations, clinical areas, and specialties involved in the claims. Classification of specialties was based on that of Singh et al.3 Types of adverse events included minor injury for cases with complete recovery within a year, significant injury for those with complete recovery requiring more than a year, major injury for those with incomplete recovery (any physical sequelae) after more than a year, and death. Clinical areas consisted of surgery, obstetrics, missed diagnosis, delayed diagnosis, medication, and fall. Data for litigation outcomes and the amounts of paid compensation in Japanese Yen (JY) were also collected for claims that received verdicts supporting the plaintiffs.
All factors identified in the verdicts as causally related to the medical injury were recorded for data analysis. Classification of these factors was based on that of Singh et al.3 Cognitive factors were drawn from a list of categories of physicians' tasks provided by the Occupational Information Network. This network is a database of occupational requirements and worker attributes and it describes occupations in terms of the skills and knowledge required, how the work is performed, and typical work settings. The list of cognitive factor categories of physicians' tasks included judgment, vigilance, memory, technical competence, or knowledge. Accordingly, the cognitive factor category list was considered to capture the work of clinicians across the entire range of specialties.3
An example concerning failure of judgment would be that a rapid respiratory rate in initial vital signs was missed or ignored in a patient who complained of upper abdominal pain, was sent home with a diagnosis of gastritis, and eventually died at home; and an autopsy diagnosis of myocardial infarction with congestive heart failure was later confirmed. A vigilance error example would be that, in an electronic ordering system, typing an incorrect medication that has the similar commercial name of a correct medication. An example of failure of memory as a cognitive error would be that a physician forgot a result of laboratory data (positive sputum cytology of lung cancer), and so the physician did not explain it to the patient and did not perform an appropriate subsequent treatment referral. A technical incompetence example would be an operative or procedural injury due to technical problems of physicians. An example of a knowledge error would be that a contraindicated drug combination was prescribed such as the use of both selective serotonin reuptake inhibitor and monoamine oxidase inhibitor.
For systemic factors, a teamwork problem (poor teamwork) was used to describe disruptive team behavior, based on the concept of teamwork described by the Agency for Healthcare Research and Quality and the British Medical Association.9, 10 Cases with teamwork problems were defined as those in which the original reviewer had judged that 1 or more of the following contributory factors played a role in the error: communication breakdowns, supervision problems, handoff problems, failures to establish clear lines of responsibility, and conflict among clinical staff. Technology failure indicated an error of commission or omission by devices, tools, or machines.
The Japanese courts analyze medical records but they do not open the records to the public and so we could not analyze the medical records of the cases in our study. Thus, we did not judge whether the adverse outcome could have been attributed to medical errors, while we analyzed the claims files and followed the conclusions reached by the end of the claims.
Statistical Analysis
Data are given as proportions for categorical variables and means or medians for continuous variables. Cognitive factors associated with cases receiving adjudication of a compensation payment by district courts (litigation outcomes) were analyzed using a logistic regression model including 5 types of cognitive errors. Analyses were conducted with the Stata SE 10.0 statistical software package (College Station, TX). All P values are 2‐sided and P < 0.05 was considered to be statistically significant. The study was approved by the ethics review board at the institution of the chief investigator.
Results
In a total of 274 closed cases of medical injury, the mean age of the patients was 49 years old and 45% were women (Table 1). The reviews performed by the coinvestigators were all confirmed by the chief investigator without discordance of the reviews between the coinvestigators and the chief investigator. The claims involved death of patients in 45% of cases; injuries that caused significant or major disability in 10% and 24%, respectively (a total of 34%); and minor adverse outcomes of medical care in 21% (57 cases). Closing verdicts supporting the plaintiffs (patients or family) by the district courts were given in 103 claims (38%), with compensation at a median of 8,000,000 JY (100 JY = $1 US in 2005). The compensation ranged from 20,000 JY to 222,710,251 JY. The highest compensation was ordered to be paid to a 36‐year‐old woman with an obstetrics‐related major injury and the court indicated the injury was causally related to the following 3 cognitive factors: error in judgment, failure of vigilance, and lack of technical competence.
| Characteristic | n (%) |
|---|---|
| |
| Demographic of patients | |
| Women | 121 (45) |
| Men | 153 (55) |
| Age, mean SD, year | 49 22 |
| Adverse outcome | |
| Minor | 57 (21) |
| Significant | 28 (10) |
| Major | 67 (24) |
| Death | 122 (45) |
| Operative | 36 |
| Delayed diagnosis | 35 |
| Medication | 26 |
| Missed diagnosis | 16 |
| Obstetrics | 8 |
| Clinical area | |
| Operative | 120 (44) |
| Delayed diagnosis | 54 (20) |
| Medication | 50 (18) |
| Missed diagnosis | 28 (10) |
| Obstetrics | 19 (7) |
| Fall | 3 (1) |
Operative injury was the most frequent reason for claims, followed by delayed diagnosis, medication error, and missed diagnosis. General surgery, orthopedics, internal medicine, and obstetrics/gynecology were the most frequently involved specialties, comprising 30% of all cases (Table 2). The verdicts suggested cognitive factors were the most prevalent factors associated with cases of medical injury: 73% of the injuries were judged to be the result of an error in judgment (Table 3), followed by failure of vigilance (65%), lack of technical competence (34%), and lack of knowledge (31%). Verdicts indicated systemic factors in only a few cases, including poor teamwork in 4% and technology failure in 2%. Patient‐related factors were suggested in 32% of the claims.
| Specialty | Cases, n (%) |
|---|---|
| General surgery | 27 (10) |
| Orthopedic surgery | 27 (10) |
| Internal medicine | 27 (10) |
| Obstetrics‐gynecology | 26 (9) |
| Neurosurgery | 19 (7) |
| Ear, nose, and throat | 18 (7) |
| Plastic surgery | 15 (5) |
| Psychiatry | 14 (5) |
| Cardiology | 13 (5) |
| Dental care | 13 (5) |
| Ophthalmology | 12 (4) |
| Hematology or oncology | 10 (4) |
| Adult primary care | 9 (3) |
| Pediatrics | 8 (3) |
| Urology | 8 (3) |
| Cardiothoracic surgery | 8 (3) |
| Neurology | 5 (2) |
| Anesthesiology | 4 (1) |
| Physical medicine or rehabilitation | 3 (1) |
| Emergency medicine | 2 (1) |
| Infectious disease | 2 (1) |
| Dermatology | 2 (1) |
| Radiology | 1 (<1) |
| Vascular surgery | 1 (<1) |
| Contributory Factor | n (%) |
|---|---|
| |
| Cognitive factors | |
| Error in judgment | 199 (73) |
| Failure of vigilance | 177 (65) |
| Lack of technical competence | 94 (34) |
| Lack of knowledge | 86 (31) |
| Failure of memory | 5 (2) |
| System factors | |
| Poor teamwork | 11 (4) |
| Technology failure | 5 (2) |
| Patient‐related factors | 87 (32) |
In a multivariable‐adjusted logistic regression analysis of cognitive factors with a potential association with the claims with paid compensation (Table 4), only error in judgment showed a significant association (odds ratio, 1.9; 95% confidence interval [CI], 1.01‐3.40). The other four cognitive factors in the model were not associated with these claims. The odds ratio for failure of memory was high (2.8), but this factor was identified by the courts in only 5 cases and was not significantly associated with claims with paid compensation.
| Cognitive Factor | Cases With No Compensation (n = 171), n (%) | Cases With Paid Compensation (n = 103), n (%) | Odds Ratio (95% CI)* |
|---|---|---|---|
| |||
| Error in judgment | 117 (68) | 82 (80) | 1.9 (1.03.4) |
| Failure of vigilance | 111 (65) | 66 (64) | 1.0 (0.61.7) |
| Failure of memory | 2 (1) | 3 (3) | 2.8 (0.518) |
| Lack of technical competence | 58 (34) | 36 (35) | 1.1 (0.61.8) |
| Lack of knowledge | 52 (30) | 34 (33) | 1.0 (0.61.7) |
Discussion
In this study of closed claims files, we identified 2 important cognitive factors involved in cases of medical injury. Error in judgment was the most common factor, comprising about 70% of all claims, and was significantly associated with cases with paid compensation for medical injury. The second cognitive factor was failure of vigilance, which was found in 65% of the claims. Other cognitive factors, such as lack of technical competence and knowledge or failure of memory, as well as systemic factors (poor teamwork and technology failure) were less frequently found to be causally related to cases with medical injury in the verdicts examined in the study.
Reasons for the low frequency of systemic factors involved in cases of medical injury in our study are unclear. This may be the cultural characteristics such as greater emphasis to working in teams and following rules of an organization in Japan. Another possibility is that plaintiffs might have tended to generate lawsuits in cases with suspected higher frequency of individual physicians' factors in Japan. Moreover, among cognitive factors, lack of technical competence and knowledge or failure of memory was also less frequently related to cases with medical injury in our study compared to those of the previous studies.3, 11
The study design of analyzing closed claims files of cases of medical injury is noteworthy for its methodology of error assessment and provides valuable information on errors related to medical injury.3, 7 Moreover, the system of court verdicts in Japan based on decisions by a professional judge allows elimination of potential bias from stakeholders (plaintiffs vs. hospitals) involved in cases of medical injury. Thus, probable causes related to adverse events can be determined from a neutral position. Previous studies of medical error have focused on medical record reviews, surveys, and interviews;12, 13 our study corroborates and extends the findings in these studies that cognitive errors are the most frequent source of medical injury.
Error in judgment is commonly made in the course of decision making in multiple clinical areas. This type of error is referred to recently as cognitive dispositions to respond,14 which is different from bias or heuristics, since not all heuristics are biased and not all errors in judgments come from bias. There is a well‐established value of heuristics in medical diagnosis. Moreover, the properties of this type of error are likely to be distinct from those associated with performance of procedures (lack of technical competence), such as operative injury, which are directly visible and can be prevented through rapid dissemination of information on safety procedures among a medical team. However, the consequences of error in judgment are important for patients, family, and healthcare providers, and these errors are also largely preventable by implementation of educational programs.15
Possible solutions for improving clinical judgment skills may be derived from recent education theory. The theory provides a means for minimizing errors in judgment through the process of meta‐cognition, in which cognitive forcing strategies can be developed through thinking that involves active control over the process of one's own thinking.14, 15 For example, reflective practice has been suggested to be an important instrument for improving clinical judgment and may particularly improve diagnoses in situations of uncertainty and uniqueness, thereby reducing diagnostic errors.16 The capability of critical reflection in real‐time practice (reflection‐in‐action) and on our own practice (reflection‐on‐action) appears to be a key requirement for developing and maintaining medical expertise.17, 18 For instance, case‐based discussion with clinician educators can be an opportunity for enhancing critical thinking skills of medical trainees.
Based on a context‐based approach that focuses on the nature of the clinical problem, potential systemic solutions have recently been proposed for reducing errors in judgment.1 These solutions utilize advanced technology, including symptom‐oriented diagnostic decision support, internet search engines for information on possible diagnoses, and automated reminders in electronic health records.1, 19 Previous studies have shown that long work hours and sleep deprivation can decrease cognitive function, leading to failure of vigilance and increased medical errors,20 and several systemic solutions provide models for avoidance of failure of vigilance. For instance, eliminating extended work shifts and reducing the number of work hours per week was shown to reduce serious medical errors through increased sleep and decreased failure of vigilance during night work in an intensive care unit.21, 22 Taking a brief nap during work hours has also been associated with decreased medical errors in a recent study conducted in Japan.23 Despite the well‐known importance of factors of physicians' workloads, our study did not analyze these factors and thus further studies are needed to confirm their importance in Japanese medical practice.
There were also 32% of patient‐related factors suggested as contributory factors to medical injury in verdicts of the closed claims. This finding may be also important in planning educational intervention strategies to reduce medical errors. Although our data did not include the relative frequency of components related to these factors, major components of patient‐related factors may include age, severity of illnesses, comorbidity, functional status, or mental status. Educational intervention programs may help healthcare providers to evaluate patients with these risk factors and to implement preventive strategies to avoid incidents among these patients.
General surgery, orthopedic surgery, internal medicine, and obstetrics‐gynecology were the most frequently involved specialties in our study. The reasons why these specialties were highly involved in the claims are unclear and our study could not analyze these issues. However, these specialties may be related to patients with greater clinical severity and thus they may have subsequently higher risk for receiving claims. Further, physicians in these specialties may be at higher risk for having various errors because of the complexity of care for patients.
Our study has several limitations. First, the closed claims are more likely to represent cases with severe injury.3 Therefore, it is unclear if we can generalize our findings beyond cases with severe injury.3 Second, certain contributory factors may not have been suggested by the verdicts, even though they played a role. Among these potential factors, poor teamwork and communication issues are unlikely to be identified as causative in verdicts, unless the allegation of the plaintiffs documented these issues. Moreover, the Japanese courts did not open the medical records to the public and so we could not analyze the medical records of the cases. Third, we only evaluated closed verdicts given by professional judges of district courts, who are unlikely to be medical experts. However, the closed verdicts underwent an extensive process involving testimony from medical professionals and academic societies. Fourth, we, as investigators, had few members with surgical backgrounds in this study so we might have underestimated issues related to technical competence among the claims. Finally, although a small percentage of closed‐ claim cases involving team performance were identified in our study, the plaintiffs might have indicated this point to the court claims, since it might have been difficult to describe this issue as a reason for requesting compensations from defendants. Thus, despite a low proportion of team performance involvement in the verdicts, we still believe that poor team performance is a factor related to most medical injuries.
In summary, causal factors obtained from closed claims files suggest the importance of cognitive factors in cases of medical injury. Among the cognitive factors, error in judgment and failure of vigilance were the most frequent. These findings may help leaders of medical schools and hospitals to allocate more resources for research into strategies to improve cognitive performance and thereby ensure patient safety. Further research is needed to better understand the cognitive mechanisms involved in medical errors and to translate this into educational strategies.
Promotion of safer healthcare by patient organizations has led to an expansion of studies aimed at understanding medical errors to minimize injury through systemic improvement. These efforts have focused on identifying patient‐related factors, reducing technology failures, and improving communication.1 In contrast, factors related to cognitive errors by healthcare providers have received relatively little attention, although such errors may be an important source of preventable harm.1, 2
Limited information is available on the types and prevalence of cognitive factors in cases of medical injury, although cognitive factors may be a major risk for medical injury. If these factors were confirmed to be important factors for medical injury, better educational strategies may be needed to reduce cognitive errors among physicians and to enhance quality improvement and patient safety. Better understanding of these cognitive factors may also help to implement educational programs aimed at the improvement of cognitive performance in medical schools or teaching hospital.35
Closed‐claim files for cases of medical injury contain valuable information for investigation of the factors involved in medical errors.3 In Japan, court claims were tried and closed orders were issued by judges without a jury system until 2009. Under this system, representatives for defense and plaintiffs can present medical experts. Courts can also appoint experts independent of either party. Court opinions in Japan are considered as neutral judgments for conflicts between plaintiffs and defendants. Usually there are 3 judges who are required to be involved with each judgment in Japanese courts.
Closed‐claim files in cases of medical injury contain information about the types and prevalence of cognitive factors suggested to be causally related to the injuries by verdicts in district courts. Thus, by analyzing these files, an unbiased description of the characteristics and epidemiology of cognitive factors can be obtained for cases of medical injury, with minimization of potentially biased claims indicated by both parties; ie, plaintiffs vs. hospitals. Therefore, in this study, by using information from closed claims files at district courts in Tokyo and Osaka, Japan, we aimed to determine the important cognitive factors associated with cases of medical injury from such factors as judgment, vigilance, memory, technical competence, or knowledge. Since we anticipated that cognitive factors would dominate among the causative factors, we also explored the association of these factors with cases in which a judgment of paid compensation was made.
Methods
Study Sample
The authors acknowledge that the methodologies are based on those from the Malpractice Insurers' Medical Errors Prevention Study.6 A claim was defined as a written demand for compensation for cases of medical injury, based on a similar approach in previous studies.7, 8 Reviews were performed for closed‐claim files for cases of medical injury involving physicians from 2001 to 2005. These files were published by the Division of the Tokyo‐Osaka Medical Malpractice Lawsuits, organized by district courts in Tokyo and Osaka. The files included all closed‐claim cases of medical injury involving physicians from 2001 to 2005 at district courts in Tokyo and Osaka. The locations of delivery of care were inpatients in this study. All patients in Japan were insured during the study period.
Data Collection
Reviews were conducted by 3 board‐certified Japanese physician‐investigators specializing in internal medicine (1 chief investigator and 2 coinvestigators). The chief investigator trained the coinvestigators in 1‐day sessions with regard to the content of claims files, data collection, and the confidentiality procedure. Reviews were first performed by 1 coinvestigator and then confirmed by the chief investigator.
Data were collected for patient demographics and characteristics of adverse events, including types, locations, clinical areas, and specialties involved in the claims. Classification of specialties was based on that of Singh et al.3 Types of adverse events included minor injury for cases with complete recovery within a year, significant injury for those with complete recovery requiring more than a year, major injury for those with incomplete recovery (any physical sequelae) after more than a year, and death. Clinical areas consisted of surgery, obstetrics, missed diagnosis, delayed diagnosis, medication, and fall. Data for litigation outcomes and the amounts of paid compensation in Japanese Yen (JY) were also collected for claims that received verdicts supporting the plaintiffs.
All factors identified in the verdicts as causally related to the medical injury were recorded for data analysis. Classification of these factors was based on that of Singh et al.3 Cognitive factors were drawn from a list of categories of physicians' tasks provided by the Occupational Information Network. This network is a database of occupational requirements and worker attributes and it describes occupations in terms of the skills and knowledge required, how the work is performed, and typical work settings. The list of cognitive factor categories of physicians' tasks included judgment, vigilance, memory, technical competence, or knowledge. Accordingly, the cognitive factor category list was considered to capture the work of clinicians across the entire range of specialties.3
An example concerning failure of judgment would be that a rapid respiratory rate in initial vital signs was missed or ignored in a patient who complained of upper abdominal pain, was sent home with a diagnosis of gastritis, and eventually died at home; and an autopsy diagnosis of myocardial infarction with congestive heart failure was later confirmed. A vigilance error example would be that, in an electronic ordering system, typing an incorrect medication that has the similar commercial name of a correct medication. An example of failure of memory as a cognitive error would be that a physician forgot a result of laboratory data (positive sputum cytology of lung cancer), and so the physician did not explain it to the patient and did not perform an appropriate subsequent treatment referral. A technical incompetence example would be an operative or procedural injury due to technical problems of physicians. An example of a knowledge error would be that a contraindicated drug combination was prescribed such as the use of both selective serotonin reuptake inhibitor and monoamine oxidase inhibitor.
For systemic factors, a teamwork problem (poor teamwork) was used to describe disruptive team behavior, based on the concept of teamwork described by the Agency for Healthcare Research and Quality and the British Medical Association.9, 10 Cases with teamwork problems were defined as those in which the original reviewer had judged that 1 or more of the following contributory factors played a role in the error: communication breakdowns, supervision problems, handoff problems, failures to establish clear lines of responsibility, and conflict among clinical staff. Technology failure indicated an error of commission or omission by devices, tools, or machines.
The Japanese courts analyze medical records but they do not open the records to the public and so we could not analyze the medical records of the cases in our study. Thus, we did not judge whether the adverse outcome could have been attributed to medical errors, while we analyzed the claims files and followed the conclusions reached by the end of the claims.
Statistical Analysis
Data are given as proportions for categorical variables and means or medians for continuous variables. Cognitive factors associated with cases receiving adjudication of a compensation payment by district courts (litigation outcomes) were analyzed using a logistic regression model including 5 types of cognitive errors. Analyses were conducted with the Stata SE 10.0 statistical software package (College Station, TX). All P values are 2‐sided and P < 0.05 was considered to be statistically significant. The study was approved by the ethics review board at the institution of the chief investigator.
Results
In a total of 274 closed cases of medical injury, the mean age of the patients was 49 years old and 45% were women (Table 1). The reviews performed by the coinvestigators were all confirmed by the chief investigator without discordance of the reviews between the coinvestigators and the chief investigator. The claims involved death of patients in 45% of cases; injuries that caused significant or major disability in 10% and 24%, respectively (a total of 34%); and minor adverse outcomes of medical care in 21% (57 cases). Closing verdicts supporting the plaintiffs (patients or family) by the district courts were given in 103 claims (38%), with compensation at a median of 8,000,000 JY (100 JY = $1 US in 2005). The compensation ranged from 20,000 JY to 222,710,251 JY. The highest compensation was ordered to be paid to a 36‐year‐old woman with an obstetrics‐related major injury and the court indicated the injury was causally related to the following 3 cognitive factors: error in judgment, failure of vigilance, and lack of technical competence.
| Characteristic | n (%) |
|---|---|
| |
| Demographic of patients | |
| Women | 121 (45) |
| Men | 153 (55) |
| Age, mean SD, year | 49 22 |
| Adverse outcome | |
| Minor | 57 (21) |
| Significant | 28 (10) |
| Major | 67 (24) |
| Death | 122 (45) |
| Operative | 36 |
| Delayed diagnosis | 35 |
| Medication | 26 |
| Missed diagnosis | 16 |
| Obstetrics | 8 |
| Clinical area | |
| Operative | 120 (44) |
| Delayed diagnosis | 54 (20) |
| Medication | 50 (18) |
| Missed diagnosis | 28 (10) |
| Obstetrics | 19 (7) |
| Fall | 3 (1) |
Operative injury was the most frequent reason for claims, followed by delayed diagnosis, medication error, and missed diagnosis. General surgery, orthopedics, internal medicine, and obstetrics/gynecology were the most frequently involved specialties, comprising 30% of all cases (Table 2). The verdicts suggested cognitive factors were the most prevalent factors associated with cases of medical injury: 73% of the injuries were judged to be the result of an error in judgment (Table 3), followed by failure of vigilance (65%), lack of technical competence (34%), and lack of knowledge (31%). Verdicts indicated systemic factors in only a few cases, including poor teamwork in 4% and technology failure in 2%. Patient‐related factors were suggested in 32% of the claims.
| Specialty | Cases, n (%) |
|---|---|
| General surgery | 27 (10) |
| Orthopedic surgery | 27 (10) |
| Internal medicine | 27 (10) |
| Obstetrics‐gynecology | 26 (9) |
| Neurosurgery | 19 (7) |
| Ear, nose, and throat | 18 (7) |
| Plastic surgery | 15 (5) |
| Psychiatry | 14 (5) |
| Cardiology | 13 (5) |
| Dental care | 13 (5) |
| Ophthalmology | 12 (4) |
| Hematology or oncology | 10 (4) |
| Adult primary care | 9 (3) |
| Pediatrics | 8 (3) |
| Urology | 8 (3) |
| Cardiothoracic surgery | 8 (3) |
| Neurology | 5 (2) |
| Anesthesiology | 4 (1) |
| Physical medicine or rehabilitation | 3 (1) |
| Emergency medicine | 2 (1) |
| Infectious disease | 2 (1) |
| Dermatology | 2 (1) |
| Radiology | 1 (<1) |
| Vascular surgery | 1 (<1) |
| Contributory Factor | n (%) |
|---|---|
| |
| Cognitive factors | |
| Error in judgment | 199 (73) |
| Failure of vigilance | 177 (65) |
| Lack of technical competence | 94 (34) |
| Lack of knowledge | 86 (31) |
| Failure of memory | 5 (2) |
| System factors | |
| Poor teamwork | 11 (4) |
| Technology failure | 5 (2) |
| Patient‐related factors | 87 (32) |
In a multivariable‐adjusted logistic regression analysis of cognitive factors with a potential association with the claims with paid compensation (Table 4), only error in judgment showed a significant association (odds ratio, 1.9; 95% confidence interval [CI], 1.01‐3.40). The other four cognitive factors in the model were not associated with these claims. The odds ratio for failure of memory was high (2.8), but this factor was identified by the courts in only 5 cases and was not significantly associated with claims with paid compensation.
| Cognitive Factor | Cases With No Compensation (n = 171), n (%) | Cases With Paid Compensation (n = 103), n (%) | Odds Ratio (95% CI)* |
|---|---|---|---|
| |||
| Error in judgment | 117 (68) | 82 (80) | 1.9 (1.03.4) |
| Failure of vigilance | 111 (65) | 66 (64) | 1.0 (0.61.7) |
| Failure of memory | 2 (1) | 3 (3) | 2.8 (0.518) |
| Lack of technical competence | 58 (34) | 36 (35) | 1.1 (0.61.8) |
| Lack of knowledge | 52 (30) | 34 (33) | 1.0 (0.61.7) |
Discussion
In this study of closed claims files, we identified 2 important cognitive factors involved in cases of medical injury. Error in judgment was the most common factor, comprising about 70% of all claims, and was significantly associated with cases with paid compensation for medical injury. The second cognitive factor was failure of vigilance, which was found in 65% of the claims. Other cognitive factors, such as lack of technical competence and knowledge or failure of memory, as well as systemic factors (poor teamwork and technology failure) were less frequently found to be causally related to cases with medical injury in the verdicts examined in the study.
Reasons for the low frequency of systemic factors involved in cases of medical injury in our study are unclear. This may be the cultural characteristics such as greater emphasis to working in teams and following rules of an organization in Japan. Another possibility is that plaintiffs might have tended to generate lawsuits in cases with suspected higher frequency of individual physicians' factors in Japan. Moreover, among cognitive factors, lack of technical competence and knowledge or failure of memory was also less frequently related to cases with medical injury in our study compared to those of the previous studies.3, 11
The study design of analyzing closed claims files of cases of medical injury is noteworthy for its methodology of error assessment and provides valuable information on errors related to medical injury.3, 7 Moreover, the system of court verdicts in Japan based on decisions by a professional judge allows elimination of potential bias from stakeholders (plaintiffs vs. hospitals) involved in cases of medical injury. Thus, probable causes related to adverse events can be determined from a neutral position. Previous studies of medical error have focused on medical record reviews, surveys, and interviews;12, 13 our study corroborates and extends the findings in these studies that cognitive errors are the most frequent source of medical injury.
Error in judgment is commonly made in the course of decision making in multiple clinical areas. This type of error is referred to recently as cognitive dispositions to respond,14 which is different from bias or heuristics, since not all heuristics are biased and not all errors in judgments come from bias. There is a well‐established value of heuristics in medical diagnosis. Moreover, the properties of this type of error are likely to be distinct from those associated with performance of procedures (lack of technical competence), such as operative injury, which are directly visible and can be prevented through rapid dissemination of information on safety procedures among a medical team. However, the consequences of error in judgment are important for patients, family, and healthcare providers, and these errors are also largely preventable by implementation of educational programs.15
Possible solutions for improving clinical judgment skills may be derived from recent education theory. The theory provides a means for minimizing errors in judgment through the process of meta‐cognition, in which cognitive forcing strategies can be developed through thinking that involves active control over the process of one's own thinking.14, 15 For example, reflective practice has been suggested to be an important instrument for improving clinical judgment and may particularly improve diagnoses in situations of uncertainty and uniqueness, thereby reducing diagnostic errors.16 The capability of critical reflection in real‐time practice (reflection‐in‐action) and on our own practice (reflection‐on‐action) appears to be a key requirement for developing and maintaining medical expertise.17, 18 For instance, case‐based discussion with clinician educators can be an opportunity for enhancing critical thinking skills of medical trainees.
Based on a context‐based approach that focuses on the nature of the clinical problem, potential systemic solutions have recently been proposed for reducing errors in judgment.1 These solutions utilize advanced technology, including symptom‐oriented diagnostic decision support, internet search engines for information on possible diagnoses, and automated reminders in electronic health records.1, 19 Previous studies have shown that long work hours and sleep deprivation can decrease cognitive function, leading to failure of vigilance and increased medical errors,20 and several systemic solutions provide models for avoidance of failure of vigilance. For instance, eliminating extended work shifts and reducing the number of work hours per week was shown to reduce serious medical errors through increased sleep and decreased failure of vigilance during night work in an intensive care unit.21, 22 Taking a brief nap during work hours has also been associated with decreased medical errors in a recent study conducted in Japan.23 Despite the well‐known importance of factors of physicians' workloads, our study did not analyze these factors and thus further studies are needed to confirm their importance in Japanese medical practice.
There were also 32% of patient‐related factors suggested as contributory factors to medical injury in verdicts of the closed claims. This finding may be also important in planning educational intervention strategies to reduce medical errors. Although our data did not include the relative frequency of components related to these factors, major components of patient‐related factors may include age, severity of illnesses, comorbidity, functional status, or mental status. Educational intervention programs may help healthcare providers to evaluate patients with these risk factors and to implement preventive strategies to avoid incidents among these patients.
General surgery, orthopedic surgery, internal medicine, and obstetrics‐gynecology were the most frequently involved specialties in our study. The reasons why these specialties were highly involved in the claims are unclear and our study could not analyze these issues. However, these specialties may be related to patients with greater clinical severity and thus they may have subsequently higher risk for receiving claims. Further, physicians in these specialties may be at higher risk for having various errors because of the complexity of care for patients.
Our study has several limitations. First, the closed claims are more likely to represent cases with severe injury.3 Therefore, it is unclear if we can generalize our findings beyond cases with severe injury.3 Second, certain contributory factors may not have been suggested by the verdicts, even though they played a role. Among these potential factors, poor teamwork and communication issues are unlikely to be identified as causative in verdicts, unless the allegation of the plaintiffs documented these issues. Moreover, the Japanese courts did not open the medical records to the public and so we could not analyze the medical records of the cases. Third, we only evaluated closed verdicts given by professional judges of district courts, who are unlikely to be medical experts. However, the closed verdicts underwent an extensive process involving testimony from medical professionals and academic societies. Fourth, we, as investigators, had few members with surgical backgrounds in this study so we might have underestimated issues related to technical competence among the claims. Finally, although a small percentage of closed‐ claim cases involving team performance were identified in our study, the plaintiffs might have indicated this point to the court claims, since it might have been difficult to describe this issue as a reason for requesting compensations from defendants. Thus, despite a low proportion of team performance involvement in the verdicts, we still believe that poor team performance is a factor related to most medical injuries.
In summary, causal factors obtained from closed claims files suggest the importance of cognitive factors in cases of medical injury. Among the cognitive factors, error in judgment and failure of vigilance were the most frequent. These findings may help leaders of medical schools and hospitals to allocate more resources for research into strategies to improve cognitive performance and thereby ensure patient safety. Further research is needed to better understand the cognitive mechanisms involved in medical errors and to translate this into educational strategies.
- ,.Diagnostic errors‐the next frontier for patient safety.JAMA.2009;301(10):1060–1062.
- ,,.Diagnostic error in internal medicine.Arch Intern Med.2005;165(13):1493–1499.
- ,,,.Medical errors involving trainees: a study of closed malpractice claims from 5 insurers.Arch Intern Med.2007;167(19):2030–2036.
- ,,.Understanding diagnostic errors in medicine: a lesson from aviation.Qual Saf Health Care.2006;15(3):159–164.
- .The importance of cognitive errors in diagnosis and strategies to minimize them.Acad Med.2003;78(8):775–780.
- ,,, et al.Claims, errors, and compensation payments in medical malpractice litigation.N Engl J Med.2006;354(19):2024–2033.
- ,,,,,.Negligent care and malpractice claiming behavior in Utah and Colorado.Med Care.2000;38(3):250–260.
- ,,, et al.Incidence and types of adverse events and negligent care in Utah and Colorado.Med Care.2000;38(3):261–271.
- ,,,,.Medical Teamwork and Patient Safety: The Evidence‐Based Relation.Rockville, MD:Agency for Healthcare Research and Quality;2005 [updated April 2005]; Available at: http://www.ahrq.gov/qual/medteam. Accessed June 2010.
- ,.Team working in Primary Health Care. Realising Shared Aims in Patient Care.London, UK:Royal Pharmaceutical Society and British Medical Association.2005.
- ,,,,.The nature and causes of unintended events reported at ten emergency departments.BMC Emerg Med.2009;9:16.
- ,,.To Err Is Human: Building a Safer Health System.Washington, USA:National Academy Press;2000.
- ,,,.Analysis of errors reported by surgeons at three teaching hospitals.Surgery.2003;133(6):614–621.
- .Achieving quality in clinical decision making: cognitive strategies and detection of bias.Acad Emerg Med.2002;9(11):1184–1204.
- .Cognitive forcing strategies in clinical decision making.Ann Emerg Med.2003;41(1):110–120.
- ,,.Effects of reflective practice on the accuracy of medical diagnoses.Med Educ.2008;42(5):468–475.
- .The Reflective Practitioner: How Professionals Think in Action.New York, NY:Basic Books;1983.
- ,,.Diagnostic errors and reflective practice in medicine.J Eval Clin Pract.2007;13(1):138–145.
- ,,,.Caught in the web: e‐diagnosis.J Hosp Med.2009;4(4):262–266.
- ,,, et al.Extended work duration and the risk of self‐reported percutaneous injuries in interns.JAMA.2006;296(9):1055–1062.
- ,,, et al.Effect of reducing interns' work hours on serious medical errors in intensive care units.N Engl J Med.2004;351(18):1838–1848.
- ,,, et al.Effect of reducing interns' weekly work hours on sleep and attentional failures.N Engl J Med.2004;351(18):1829–1837.
- ,,, et al.Influence of Residents' Workload, Mental State and Job Satisfaction on Procedural Error: a prospective daily questionnaire‐based study.General Medicine.2008;9(2):57–64.
- ,.Diagnostic errors‐the next frontier for patient safety.JAMA.2009;301(10):1060–1062.
- ,,.Diagnostic error in internal medicine.Arch Intern Med.2005;165(13):1493–1499.
- ,,,.Medical errors involving trainees: a study of closed malpractice claims from 5 insurers.Arch Intern Med.2007;167(19):2030–2036.
- ,,.Understanding diagnostic errors in medicine: a lesson from aviation.Qual Saf Health Care.2006;15(3):159–164.
- .The importance of cognitive errors in diagnosis and strategies to minimize them.Acad Med.2003;78(8):775–780.
- ,,, et al.Claims, errors, and compensation payments in medical malpractice litigation.N Engl J Med.2006;354(19):2024–2033.
- ,,,,,.Negligent care and malpractice claiming behavior in Utah and Colorado.Med Care.2000;38(3):250–260.
- ,,, et al.Incidence and types of adverse events and negligent care in Utah and Colorado.Med Care.2000;38(3):261–271.
- ,,,,.Medical Teamwork and Patient Safety: The Evidence‐Based Relation.Rockville, MD:Agency for Healthcare Research and Quality;2005 [updated April 2005]; Available at: http://www.ahrq.gov/qual/medteam. Accessed June 2010.
- ,.Team working in Primary Health Care. Realising Shared Aims in Patient Care.London, UK:Royal Pharmaceutical Society and British Medical Association.2005.
- ,,,,.The nature and causes of unintended events reported at ten emergency departments.BMC Emerg Med.2009;9:16.
- ,,.To Err Is Human: Building a Safer Health System.Washington, USA:National Academy Press;2000.
- ,,,.Analysis of errors reported by surgeons at three teaching hospitals.Surgery.2003;133(6):614–621.
- .Achieving quality in clinical decision making: cognitive strategies and detection of bias.Acad Emerg Med.2002;9(11):1184–1204.
- .Cognitive forcing strategies in clinical decision making.Ann Emerg Med.2003;41(1):110–120.
- ,,.Effects of reflective practice on the accuracy of medical diagnoses.Med Educ.2008;42(5):468–475.
- .The Reflective Practitioner: How Professionals Think in Action.New York, NY:Basic Books;1983.
- ,,.Diagnostic errors and reflective practice in medicine.J Eval Clin Pract.2007;13(1):138–145.
- ,,,.Caught in the web: e‐diagnosis.J Hosp Med.2009;4(4):262–266.
- ,,, et al.Extended work duration and the risk of self‐reported percutaneous injuries in interns.JAMA.2006;296(9):1055–1062.
- ,,, et al.Effect of reducing interns' work hours on serious medical errors in intensive care units.N Engl J Med.2004;351(18):1838–1848.
- ,,, et al.Effect of reducing interns' weekly work hours on sleep and attentional failures.N Engl J Med.2004;351(18):1829–1837.
- ,,, et al.Influence of Residents' Workload, Mental State and Job Satisfaction on Procedural Error: a prospective daily questionnaire‐based study.General Medicine.2008;9(2):57–64.
Copyright © 2010 Society of Hospital Medicine
GAD Vaccine for Type 1 Diabetes Shows Continued Promise
KEYSTONE, Colo. – Right now, the Diamyd Medical’s GAD vaccine is in the sweet spot in the developmental pipeline – an interim period of enormous optimism that this novel autoantigen-based immunotherapy will safely prevent many cases of type 1 diabetes.
The results of three phase II studies are in and they look quite promising. Two large phase III clinical trials are well underway in Europe and the United States. The safety experience with the 65-kD isoform of GAD (glutamic acid decarboxylase-65) vaccine has been outstanding. The subcutaneous two-injection series is easy to administer. Acceptance of the vaccine by patients and their families is high. The vaccine targets a serious disease whose incidence is steadily climbing by 3%-5% per year in developed countries. And most patients with recently diagnosed type 1 diabetes possess GAD autoantibodies, so the Diamyd vaccine would be widely applicable.

All of that was good enough for Johnson and Johnson, which in June inked a huge development and marketing deal for the GAD vaccine with small Swedish biotech company Diamyd Medical. Under the deal, Diamyd receives $45 million upfront, milestone payments of up to $580 million, and tiered royalties after that. The Federal Trade Commission’s antitrust division has already approved the deal.
But during this blissful interlude, one key question remains: Is the Diamyd vaccine effective?
“It’s too early to say if this works. Absolutely too early. We have a phase III trial in Europe with results due next spring. And the TrialNet study [is] going on here in the U.S. So we will know in a year or 2,” Dr. Johnny L. Ludvigsson said at a conference on management of diabetes in youth sponsored by the Children’s Diabetes Foundation at Denver.
Dr. Ludvigsson, professor of pediatrics at the University of Linkoping (Sweden), led the phase III European trial evaluating whether the GAD vaccine preserves beta-cell function and residual insulin secretion in patients with type 1 diabetes diagnosed within 3 months of starting treatment. He also headed a phase II study that caused a favorable buzz within the diabetes research community (N. Engl. J. Med. 2008;359:1909-20) and for which he is now analyzing 5-year follow-up data.
And while the forthcoming phase III trial results will tell the tale as to clinical efficacy, at this time some useful interim observations can be made about the GAD vaccine, according to Dr. Ludvigsson:
• The vaccine has demonstrated excellent safety. Experience with the vaccine to date totals 850 patient-years in adults and 350 patient-years in children, with no adverse events reported. This is enormously reassuring because GAD transforms glutamate into GABA, an important neurotransmitter. Lack of GAD in the CNS leads to muscle rigidity and convulsions, while stimulation of CNS GAD results in inhibition of neurotransmission. The absence of any such adverse events indicates the vaccine is working, as designed, to affect only a very small part of the immune system: namely, the activated T cells that have targeted pancreatic beta-cells for destruction, Dr. Ludvigsson said.
• The vaccine has demonstrated prolonged immunologic effects. The immunologic response to the Diamyd vaccine lasts surprisingly long – approaching 5 years and still counting. It’s a GAD-specific, cell-mediated, and humoral immune response characterized by increased GAD autoantibodies, a Th2 shift marked by reduction in activated T cells and an increase in regulatory T cells, a sharp and sustained rise in levels of interleukins-2, -5, -10, -13, and -17, and GAD tolerance. “We see this response still after 4 years. The memory is there,” Dr. Ludvigsson observed.
• “The earlier we treat, the better the outcome.” That’s why the phase III European trial is restricted to patients diagnosed with type 1 diabetes within the past 3 months. It’s also the impetus for ongoing prevention trials in individuals at very high genetic risk for type 1 diabetes who have GAD autoantibodies but have not developed overt disease.
• The vaccine probably won’t work in diabetic patients without GAD autoantibodies. No studies have been carried out in such patients, but Dr. Ludvigsson said it’s his impression that the vaccine is more effective in individuals with higher than lower titers of GAD autoantibodies.
For the future, the GAD vaccine alone probably is not the solution to type 1 diabetes, Dr. Ludvigsson said candidly.
“I believe this opens the door to using different antigens, like in allergy. Allergists don’t use just cat antigen in patients who have cat, dog, and house dust mite allergies. I suppose we may also learn to combine autoantigens, together with possible stimulation of beta-cells in combination with drugs that promote beta-cell regeneration,” he continued.
Other autoantibodies commonly present in patients with type 1 diabetes, or at high risk for the disease, include insulin autoantibodies, islet cell autoantibodies, and antibodies to the zinc transporter. Combining the GAD vaccine with other major diabetes-specific autoantigens recognized by the immune system could provide synergistic benefits.

The likely necessity for a combined approach addressing multiple pathways was underscored in a separate presentation by Dr. Jay S. Skyler, chairman of the type 1 Diabetes TrialNet, a National Institutes of Health–funded international network of centers conducting clinical trials of diabetes therapies.
The GAD vaccine appears to have the same limitation as the other immunomodulatory therapies evaluated to date in clinical trials, including the B cell–depleting anti-CD20 agent rituximab, and the anti-CD3 biologics teplizumab and otelixizumab: namely, they preserve beta cell function for a while, but the effect is transient. Eventually fasting C-peptide levels start to fall off in parallel to the placebo group. That’s why combination therapy will probably be required in order to cure or prevent Type 1 diabetes, according to Dr. Skyler, a professor of medicine, pediatrics and psychology at the University of Miami.
Ideally, a combination therapy should be multipronged, with three goals: Stop immune destruction, preserve beta-cell mass, and replace or regenerate beta-cells. Such a regimen might start off with a potent anti-inflammatory therapy – perhaps an anti-interleukin-1beta agent or tumor necrosis factor inhibitor – to quell the metabolic stress surrounding the pancreatic islets. This might well need to be given on a continuing basis.
Next would come an immunomodulatory approach; for example, T-cell modulation with an anti-CD3 biologic or B cell depletion with rituximab. This could be followed up with an autoantigen-specific therapy such as the GAD vaccine or oral insulin. “Maybe it needs to be both,” Dr. Skyler continued.
The logical subsequent step would be to try to stimulate immunologic expansion of regulatory T cells, either with granulocyte colony–stimulating factor or by direct infusion of regulatory T cells themselves. This could be combined with beta-cell expansion via exenatide (Byetta) or the investigational HIP2B peptide.
“We could conceivably be doing some of these things even today,” Dr. Skyler said.
Dr. Ludvigsson reported receiving research grant support from Diamyd.
Dr. Skyler has served as a consultant to and/or received research grants from numerous pharmaceutical companies.
Type 1 diabetes (T1D) is an autoimmune disease caused by interplay of genetic and environmental factors. The incidence of childhood T1D has doubled worldwide over the past 20-25 years. Elimination of the environmental agent(s) responsible for this epidemic would be the most efficient approach to primary prevention; however, more work is needed to identify the environmental agents and to develop effective interventions.
Blocking progression from islet autoimmunity to clinical diabetes or secondary prevention has been attempted, so far to no avail, by a number of groups, including large randomized trials: the Diabetes Prevention Trial – Type 1, the European Nicotinamide Diabetes Intervention Trial, and the Type 1 Diabetes Prediction and Prevention Project.
Trials in patients with newly diagnosed T1D aim at tertiary prevention, such as preservation of remaining islet beta-cells to induce and prolong partial remission. Unfortunately, most islets have already been destroyed by the time diabetes is diagnosed and complete reversal of diabetes is highly unlikely. Benefits may include a simpler insulin regimen, lower HbA1c, and reduced risk of hypoglycemia and microvascular complications. The gain may be even greater if the intervention is applied as soon as the patient shows asymptomatic “dysglycemia,” detected by oral glucose tolerance test or A1c, before overt symptoms of diabetes.
While new interventions are often tested first in patients with established diabetes, and, when proven safe, applied to patients with pre-T1D, efficacy after diagnosis of diabetes is not to be a precondition to application in pre-T1D, as there may be a “point of no return” in the destruction of the islets, rendering some interventions effective only at the earlier stages of the process.
Antigen-specific vaccines
Among several approaches to prevention of T1D, “vaccination” using islet autoantigens (intact or altered peptides derived from insulin, GAD65 or other proteins) stands out as potentially inducing long-term tolerance by induction of regulatory T-cells that down-regulate immunity to autoantigens. Until recently, trials of insulin administered parenterally, orally, or intranasally have been unsuccessful. Therefore, the initial results from trials of the Diamyd vaccine, as reviewed here, were greeted with huge interest and excitement. The vaccine includes the whole recombinant human GAD65 (rhGAD65) molecule suspended in alum. The protective effect was most pronounced in patients treated within 3 months of diagnosis, and no serious side effects were observed.
Insulin-related molecules continue to attract great interest in vaccine development. Phase I studies have been completed or are nearing completion for a proinsulin peptide C19-A3, an insulin peptide with incomplete Freund adjuvant, and a plasmid encoding proinsulin.
Combination therapies may enhance efficacy while lowering risk of adverse events if utilizing therapies from different treatment pathways. While more targeted therapies are being employed, immunomodulatory agents are still relatively nonspecific and potentially toxic to some of the trial participants. Some may carry an unacceptable risk of long-term complications. This direction is important; however, multiple scientific and logistic issues remain, for example, the anticipated duration, toxicity, and complexity of immunotherapy.
In the long run, primary prevention will likely be the optimal approach to the prevention of T1D. Once more than one islet autoantibody is present, most individuals progress to diabetes in 5-10 years. The TrialNet consortium (www.diabetestrialnet.org) systematically evaluates therapies in new-onset patients as well as in pre-diabetic subjects, and invites proposals from the research community at large.
Marian Rewers, M.D., Ph.D., is professor of pediatrics and preventive medicine at the Barbara Davis Center for Childhood Diabetes, University of Colorado, Denver.
Type 1 diabetes (T1D) is an autoimmune disease caused by interplay of genetic and environmental factors. The incidence of childhood T1D has doubled worldwide over the past 20-25 years. Elimination of the environmental agent(s) responsible for this epidemic would be the most efficient approach to primary prevention; however, more work is needed to identify the environmental agents and to develop effective interventions.
Blocking progression from islet autoimmunity to clinical diabetes or secondary prevention has been attempted, so far to no avail, by a number of groups, including large randomized trials: the Diabetes Prevention Trial – Type 1, the European Nicotinamide Diabetes Intervention Trial, and the Type 1 Diabetes Prediction and Prevention Project.
Trials in patients with newly diagnosed T1D aim at tertiary prevention, such as preservation of remaining islet beta-cells to induce and prolong partial remission. Unfortunately, most islets have already been destroyed by the time diabetes is diagnosed and complete reversal of diabetes is highly unlikely. Benefits may include a simpler insulin regimen, lower HbA1c, and reduced risk of hypoglycemia and microvascular complications. The gain may be even greater if the intervention is applied as soon as the patient shows asymptomatic “dysglycemia,” detected by oral glucose tolerance test or A1c, before overt symptoms of diabetes.
While new interventions are often tested first in patients with established diabetes, and, when proven safe, applied to patients with pre-T1D, efficacy after diagnosis of diabetes is not to be a precondition to application in pre-T1D, as there may be a “point of no return” in the destruction of the islets, rendering some interventions effective only at the earlier stages of the process.
Antigen-specific vaccines
Among several approaches to prevention of T1D, “vaccination” using islet autoantigens (intact or altered peptides derived from insulin, GAD65 or other proteins) stands out as potentially inducing long-term tolerance by induction of regulatory T-cells that down-regulate immunity to autoantigens. Until recently, trials of insulin administered parenterally, orally, or intranasally have been unsuccessful. Therefore, the initial results from trials of the Diamyd vaccine, as reviewed here, were greeted with huge interest and excitement. The vaccine includes the whole recombinant human GAD65 (rhGAD65) molecule suspended in alum. The protective effect was most pronounced in patients treated within 3 months of diagnosis, and no serious side effects were observed.
Insulin-related molecules continue to attract great interest in vaccine development. Phase I studies have been completed or are nearing completion for a proinsulin peptide C19-A3, an insulin peptide with incomplete Freund adjuvant, and a plasmid encoding proinsulin.
Combination therapies may enhance efficacy while lowering risk of adverse events if utilizing therapies from different treatment pathways. While more targeted therapies are being employed, immunomodulatory agents are still relatively nonspecific and potentially toxic to some of the trial participants. Some may carry an unacceptable risk of long-term complications. This direction is important; however, multiple scientific and logistic issues remain, for example, the anticipated duration, toxicity, and complexity of immunotherapy.
In the long run, primary prevention will likely be the optimal approach to the prevention of T1D. Once more than one islet autoantibody is present, most individuals progress to diabetes in 5-10 years. The TrialNet consortium (www.diabetestrialnet.org) systematically evaluates therapies in new-onset patients as well as in pre-diabetic subjects, and invites proposals from the research community at large.
Marian Rewers, M.D., Ph.D., is professor of pediatrics and preventive medicine at the Barbara Davis Center for Childhood Diabetes, University of Colorado, Denver.
Type 1 diabetes (T1D) is an autoimmune disease caused by interplay of genetic and environmental factors. The incidence of childhood T1D has doubled worldwide over the past 20-25 years. Elimination of the environmental agent(s) responsible for this epidemic would be the most efficient approach to primary prevention; however, more work is needed to identify the environmental agents and to develop effective interventions.
Blocking progression from islet autoimmunity to clinical diabetes or secondary prevention has been attempted, so far to no avail, by a number of groups, including large randomized trials: the Diabetes Prevention Trial – Type 1, the European Nicotinamide Diabetes Intervention Trial, and the Type 1 Diabetes Prediction and Prevention Project.
Trials in patients with newly diagnosed T1D aim at tertiary prevention, such as preservation of remaining islet beta-cells to induce and prolong partial remission. Unfortunately, most islets have already been destroyed by the time diabetes is diagnosed and complete reversal of diabetes is highly unlikely. Benefits may include a simpler insulin regimen, lower HbA1c, and reduced risk of hypoglycemia and microvascular complications. The gain may be even greater if the intervention is applied as soon as the patient shows asymptomatic “dysglycemia,” detected by oral glucose tolerance test or A1c, before overt symptoms of diabetes.
While new interventions are often tested first in patients with established diabetes, and, when proven safe, applied to patients with pre-T1D, efficacy after diagnosis of diabetes is not to be a precondition to application in pre-T1D, as there may be a “point of no return” in the destruction of the islets, rendering some interventions effective only at the earlier stages of the process.
Antigen-specific vaccines
Among several approaches to prevention of T1D, “vaccination” using islet autoantigens (intact or altered peptides derived from insulin, GAD65 or other proteins) stands out as potentially inducing long-term tolerance by induction of regulatory T-cells that down-regulate immunity to autoantigens. Until recently, trials of insulin administered parenterally, orally, or intranasally have been unsuccessful. Therefore, the initial results from trials of the Diamyd vaccine, as reviewed here, were greeted with huge interest and excitement. The vaccine includes the whole recombinant human GAD65 (rhGAD65) molecule suspended in alum. The protective effect was most pronounced in patients treated within 3 months of diagnosis, and no serious side effects were observed.
Insulin-related molecules continue to attract great interest in vaccine development. Phase I studies have been completed or are nearing completion for a proinsulin peptide C19-A3, an insulin peptide with incomplete Freund adjuvant, and a plasmid encoding proinsulin.
Combination therapies may enhance efficacy while lowering risk of adverse events if utilizing therapies from different treatment pathways. While more targeted therapies are being employed, immunomodulatory agents are still relatively nonspecific and potentially toxic to some of the trial participants. Some may carry an unacceptable risk of long-term complications. This direction is important; however, multiple scientific and logistic issues remain, for example, the anticipated duration, toxicity, and complexity of immunotherapy.
In the long run, primary prevention will likely be the optimal approach to the prevention of T1D. Once more than one islet autoantibody is present, most individuals progress to diabetes in 5-10 years. The TrialNet consortium (www.diabetestrialnet.org) systematically evaluates therapies in new-onset patients as well as in pre-diabetic subjects, and invites proposals from the research community at large.
Marian Rewers, M.D., Ph.D., is professor of pediatrics and preventive medicine at the Barbara Davis Center for Childhood Diabetes, University of Colorado, Denver.
KEYSTONE, Colo. – Right now, the Diamyd Medical’s GAD vaccine is in the sweet spot in the developmental pipeline – an interim period of enormous optimism that this novel autoantigen-based immunotherapy will safely prevent many cases of type 1 diabetes.
The results of three phase II studies are in and they look quite promising. Two large phase III clinical trials are well underway in Europe and the United States. The safety experience with the 65-kD isoform of GAD (glutamic acid decarboxylase-65) vaccine has been outstanding. The subcutaneous two-injection series is easy to administer. Acceptance of the vaccine by patients and their families is high. The vaccine targets a serious disease whose incidence is steadily climbing by 3%-5% per year in developed countries. And most patients with recently diagnosed type 1 diabetes possess GAD autoantibodies, so the Diamyd vaccine would be widely applicable.
All of that was good enough for Johnson and Johnson, which in June inked a huge development and marketing deal for the GAD vaccine with small Swedish biotech company Diamyd Medical. Under the deal, Diamyd receives $45 million upfront, milestone payments of up to $580 million, and tiered royalties after that. The Federal Trade Commission’s antitrust division has already approved the deal.
But during this blissful interlude, one key question remains: Is the Diamyd vaccine effective?
“It’s too early to say if this works. Absolutely too early. We have a phase III trial in Europe with results due next spring. And the TrialNet study [is] going on here in the U.S. So we will know in a year or 2,” Dr. Johnny L. Ludvigsson said at a conference on management of diabetes in youth sponsored by the Children’s Diabetes Foundation at Denver.
Dr. Ludvigsson, professor of pediatrics at the University of Linkoping (Sweden), led the phase III European trial evaluating whether the GAD vaccine preserves beta-cell function and residual insulin secretion in patients with type 1 diabetes diagnosed within 3 months of starting treatment. He also headed a phase II study that caused a favorable buzz within the diabetes research community (N. Engl. J. Med. 2008;359:1909-20) and for which he is now analyzing 5-year follow-up data.
And while the forthcoming phase III trial results will tell the tale as to clinical efficacy, at this time some useful interim observations can be made about the GAD vaccine, according to Dr. Ludvigsson:
• The vaccine has demonstrated excellent safety. Experience with the vaccine to date totals 850 patient-years in adults and 350 patient-years in children, with no adverse events reported. This is enormously reassuring because GAD transforms glutamate into GABA, an important neurotransmitter. Lack of GAD in the CNS leads to muscle rigidity and convulsions, while stimulation of CNS GAD results in inhibition of neurotransmission. The absence of any such adverse events indicates the vaccine is working, as designed, to affect only a very small part of the immune system: namely, the activated T cells that have targeted pancreatic beta-cells for destruction, Dr. Ludvigsson said.
• The vaccine has demonstrated prolonged immunologic effects. The immunologic response to the Diamyd vaccine lasts surprisingly long – approaching 5 years and still counting. It’s a GAD-specific, cell-mediated, and humoral immune response characterized by increased GAD autoantibodies, a Th2 shift marked by reduction in activated T cells and an increase in regulatory T cells, a sharp and sustained rise in levels of interleukins-2, -5, -10, -13, and -17, and GAD tolerance. “We see this response still after 4 years. The memory is there,” Dr. Ludvigsson observed.
• “The earlier we treat, the better the outcome.” That’s why the phase III European trial is restricted to patients diagnosed with type 1 diabetes within the past 3 months. It’s also the impetus for ongoing prevention trials in individuals at very high genetic risk for type 1 diabetes who have GAD autoantibodies but have not developed overt disease.
• The vaccine probably won’t work in diabetic patients without GAD autoantibodies. No studies have been carried out in such patients, but Dr. Ludvigsson said it’s his impression that the vaccine is more effective in individuals with higher than lower titers of GAD autoantibodies.
For the future, the GAD vaccine alone probably is not the solution to type 1 diabetes, Dr. Ludvigsson said candidly.
“I believe this opens the door to using different antigens, like in allergy. Allergists don’t use just cat antigen in patients who have cat, dog, and house dust mite allergies. I suppose we may also learn to combine autoantigens, together with possible stimulation of beta-cells in combination with drugs that promote beta-cell regeneration,” he continued.
Other autoantibodies commonly present in patients with type 1 diabetes, or at high risk for the disease, include insulin autoantibodies, islet cell autoantibodies, and antibodies to the zinc transporter. Combining the GAD vaccine with other major diabetes-specific autoantigens recognized by the immune system could provide synergistic benefits.
The likely necessity for a combined approach addressing multiple pathways was underscored in a separate presentation by Dr. Jay S. Skyler, chairman of the type 1 Diabetes TrialNet, a National Institutes of Health–funded international network of centers conducting clinical trials of diabetes therapies.
The GAD vaccine appears to have the same limitation as the other immunomodulatory therapies evaluated to date in clinical trials, including the B cell–depleting anti-CD20 agent rituximab, and the anti-CD3 biologics teplizumab and otelixizumab: namely, they preserve beta cell function for a while, but the effect is transient. Eventually fasting C-peptide levels start to fall off in parallel to the placebo group. That’s why combination therapy will probably be required in order to cure or prevent Type 1 diabetes, according to Dr. Skyler, a professor of medicine, pediatrics and psychology at the University of Miami.
Ideally, a combination therapy should be multipronged, with three goals: Stop immune destruction, preserve beta-cell mass, and replace or regenerate beta-cells. Such a regimen might start off with a potent anti-inflammatory therapy – perhaps an anti-interleukin-1beta agent or tumor necrosis factor inhibitor – to quell the metabolic stress surrounding the pancreatic islets. This might well need to be given on a continuing basis.
Next would come an immunomodulatory approach; for example, T-cell modulation with an anti-CD3 biologic or B cell depletion with rituximab. This could be followed up with an autoantigen-specific therapy such as the GAD vaccine or oral insulin. “Maybe it needs to be both,” Dr. Skyler continued.
The logical subsequent step would be to try to stimulate immunologic expansion of regulatory T cells, either with granulocyte colony–stimulating factor or by direct infusion of regulatory T cells themselves. This could be combined with beta-cell expansion via exenatide (Byetta) or the investigational HIP2B peptide.
“We could conceivably be doing some of these things even today,” Dr. Skyler said.
Dr. Ludvigsson reported receiving research grant support from Diamyd.
Dr. Skyler has served as a consultant to and/or received research grants from numerous pharmaceutical companies.
KEYSTONE, Colo. – Right now, the Diamyd Medical’s GAD vaccine is in the sweet spot in the developmental pipeline – an interim period of enormous optimism that this novel autoantigen-based immunotherapy will safely prevent many cases of type 1 diabetes.
The results of three phase II studies are in and they look quite promising. Two large phase III clinical trials are well underway in Europe and the United States. The safety experience with the 65-kD isoform of GAD (glutamic acid decarboxylase-65) vaccine has been outstanding. The subcutaneous two-injection series is easy to administer. Acceptance of the vaccine by patients and their families is high. The vaccine targets a serious disease whose incidence is steadily climbing by 3%-5% per year in developed countries. And most patients with recently diagnosed type 1 diabetes possess GAD autoantibodies, so the Diamyd vaccine would be widely applicable.
All of that was good enough for Johnson and Johnson, which in June inked a huge development and marketing deal for the GAD vaccine with small Swedish biotech company Diamyd Medical. Under the deal, Diamyd receives $45 million upfront, milestone payments of up to $580 million, and tiered royalties after that. The Federal Trade Commission’s antitrust division has already approved the deal.
But during this blissful interlude, one key question remains: Is the Diamyd vaccine effective?
“It’s too early to say if this works. Absolutely too early. We have a phase III trial in Europe with results due next spring. And the TrialNet study [is] going on here in the U.S. So we will know in a year or 2,” Dr. Johnny L. Ludvigsson said at a conference on management of diabetes in youth sponsored by the Children’s Diabetes Foundation at Denver.
Dr. Ludvigsson, professor of pediatrics at the University of Linkoping (Sweden), led the phase III European trial evaluating whether the GAD vaccine preserves beta-cell function and residual insulin secretion in patients with type 1 diabetes diagnosed within 3 months of starting treatment. He also headed a phase II study that caused a favorable buzz within the diabetes research community (N. Engl. J. Med. 2008;359:1909-20) and for which he is now analyzing 5-year follow-up data.
And while the forthcoming phase III trial results will tell the tale as to clinical efficacy, at this time some useful interim observations can be made about the GAD vaccine, according to Dr. Ludvigsson:
• The vaccine has demonstrated excellent safety. Experience with the vaccine to date totals 850 patient-years in adults and 350 patient-years in children, with no adverse events reported. This is enormously reassuring because GAD transforms glutamate into GABA, an important neurotransmitter. Lack of GAD in the CNS leads to muscle rigidity and convulsions, while stimulation of CNS GAD results in inhibition of neurotransmission. The absence of any such adverse events indicates the vaccine is working, as designed, to affect only a very small part of the immune system: namely, the activated T cells that have targeted pancreatic beta-cells for destruction, Dr. Ludvigsson said.
• The vaccine has demonstrated prolonged immunologic effects. The immunologic response to the Diamyd vaccine lasts surprisingly long – approaching 5 years and still counting. It’s a GAD-specific, cell-mediated, and humoral immune response characterized by increased GAD autoantibodies, a Th2 shift marked by reduction in activated T cells and an increase in regulatory T cells, a sharp and sustained rise in levels of interleukins-2, -5, -10, -13, and -17, and GAD tolerance. “We see this response still after 4 years. The memory is there,” Dr. Ludvigsson observed.
• “The earlier we treat, the better the outcome.” That’s why the phase III European trial is restricted to patients diagnosed with type 1 diabetes within the past 3 months. It’s also the impetus for ongoing prevention trials in individuals at very high genetic risk for type 1 diabetes who have GAD autoantibodies but have not developed overt disease.
• The vaccine probably won’t work in diabetic patients without GAD autoantibodies. No studies have been carried out in such patients, but Dr. Ludvigsson said it’s his impression that the vaccine is more effective in individuals with higher than lower titers of GAD autoantibodies.
For the future, the GAD vaccine alone probably is not the solution to type 1 diabetes, Dr. Ludvigsson said candidly.
“I believe this opens the door to using different antigens, like in allergy. Allergists don’t use just cat antigen in patients who have cat, dog, and house dust mite allergies. I suppose we may also learn to combine autoantigens, together with possible stimulation of beta-cells in combination with drugs that promote beta-cell regeneration,” he continued.
Other autoantibodies commonly present in patients with type 1 diabetes, or at high risk for the disease, include insulin autoantibodies, islet cell autoantibodies, and antibodies to the zinc transporter. Combining the GAD vaccine with other major diabetes-specific autoantigens recognized by the immune system could provide synergistic benefits.
The likely necessity for a combined approach addressing multiple pathways was underscored in a separate presentation by Dr. Jay S. Skyler, chairman of the type 1 Diabetes TrialNet, a National Institutes of Health–funded international network of centers conducting clinical trials of diabetes therapies.
The GAD vaccine appears to have the same limitation as the other immunomodulatory therapies evaluated to date in clinical trials, including the B cell–depleting anti-CD20 agent rituximab, and the anti-CD3 biologics teplizumab and otelixizumab: namely, they preserve beta cell function for a while, but the effect is transient. Eventually fasting C-peptide levels start to fall off in parallel to the placebo group. That’s why combination therapy will probably be required in order to cure or prevent Type 1 diabetes, according to Dr. Skyler, a professor of medicine, pediatrics and psychology at the University of Miami.
Ideally, a combination therapy should be multipronged, with three goals: Stop immune destruction, preserve beta-cell mass, and replace or regenerate beta-cells. Such a regimen might start off with a potent anti-inflammatory therapy – perhaps an anti-interleukin-1beta agent or tumor necrosis factor inhibitor – to quell the metabolic stress surrounding the pancreatic islets. This might well need to be given on a continuing basis.
Next would come an immunomodulatory approach; for example, T-cell modulation with an anti-CD3 biologic or B cell depletion with rituximab. This could be followed up with an autoantigen-specific therapy such as the GAD vaccine or oral insulin. “Maybe it needs to be both,” Dr. Skyler continued.
The logical subsequent step would be to try to stimulate immunologic expansion of regulatory T cells, either with granulocyte colony–stimulating factor or by direct infusion of regulatory T cells themselves. This could be combined with beta-cell expansion via exenatide (Byetta) or the investigational HIP2B peptide.
“We could conceivably be doing some of these things even today,” Dr. Skyler said.
Dr. Ludvigsson reported receiving research grant support from Diamyd.
Dr. Skyler has served as a consultant to and/or received research grants from numerous pharmaceutical companies.
GAD Vaccine for Type 1 Diabetes Shows Continued Promise
KEYSTONE, Colo. – Right now, the Diamyd Medical’s GAD vaccine is in the sweet spot in the developmental pipeline – an interim period of enormous optimism that this novel autoantigen-based immunotherapy will safely prevent many cases of type 1 diabetes.
The results of three phase II studies are in and they look quite promising. Two large phase III clinical trials are well underway in Europe and the United States. The safety experience with the 65-kD isoform of GAD (glutamic acid decarboxylase-65) vaccine has been outstanding. The subcutaneous two-injection series is easy to administer. Acceptance of the vaccine by patients and their families is high. The vaccine targets a serious disease whose incidence is steadily climbing by 3%-5% per year in developed countries. And most patients with recently diagnosed type 1 diabetes possess GAD autoantibodies, so the Diamyd vaccine would be widely applicable.
All of that was good enough for Johnson and Johnson, which in June inked a huge development and marketing deal for the GAD vaccine with small Swedish biotech company Diamyd Medical. Under the deal, Diamyd receives $45 million upfront, milestone payments of up to $580 million, and tiered royalties after that. The Federal Trade Commission’s antitrust division has already approved the deal.
But during this blissful interlude, one key question remains: Is the Diamyd vaccine effective?
“It’s too early to say if this works. Absolutely too early. We have a phase III trial in Europe with results due next spring. And the TrialNet study [is] going on here in the U.S. So we will know in a year or 2,” Dr. Johnny L. Ludvigsson said at a conference on management of diabetes in youth sponsored by the Children’s Diabetes Foundation at Denver.
Dr. Ludvigsson, professor of pediatrics at the University of Linkoping (Sweden), led the phase III European trial evaluating whether the GAD vaccine preserves beta-cell function and residual insulin secretion in patients with type 1 diabetes diagnosed within 3 months of starting treatment. He also headed a phase II study that caused a favorable buzz within the diabetes research community (N. Engl. J. Med. 2008;359:1909-20) and for which he is now analyzing 5-year follow-up data.
And while the forthcoming phase III trial results will tell the tale as to clinical efficacy, at this time some useful interim observations can be made about the GAD vaccine, according to Dr. Ludvigsson:
• The vaccine has demonstrated excellent safety. Experience with the vaccine to date totals 850 patient-years in adults and 350 patient-years in children, with no adverse events reported. This is enormously reassuring because GAD transforms glutamate into GABA, an important neurotransmitter. Lack of GAD in the CNS leads to muscle rigidity and convulsions, while stimulation of CNS GAD results in inhibition of neurotransmission. The absence of any such adverse events indicates the vaccine is working, as designed, to affect only a very small part of the immune system: namely, the activated T cells that have targeted pancreatic beta-cells for destruction, Dr. Ludvigsson said.
• The vaccine has demonstrated prolonged immunologic effects. The immunologic response to the Diamyd vaccine lasts surprisingly long – approaching 5 years and still counting. It’s a GAD-specific, cell-mediated, and humoral immune response characterized by increased GAD autoantibodies, a Th2 shift marked by reduction in activated T cells and an increase in regulatory T cells, a sharp and sustained rise in levels of interleukins-2, -5, -10, -13, and -17, and GAD tolerance. “We see this response still after 4 years. The memory is there,” Dr. Ludvigsson observed.
• “The earlier we treat, the better the outcome.” That’s why the phase III European trial is restricted to patients diagnosed with type 1 diabetes within the past 3 months. It’s also the impetus for ongoing prevention trials in individuals at very high genetic risk for type 1 diabetes who have GAD autoantibodies but have not developed overt disease.
• The vaccine probably won’t work in diabetic patients without GAD autoantibodies. No studies have been carried out in such patients, but Dr. Ludvigsson said it’s his impression that the vaccine is more effective in individuals with higher than lower titers of GAD autoantibodies.
For the future, the GAD vaccine alone probably is not the solution to type 1 diabetes, Dr. Ludvigsson said candidly.
“I believe this opens the door to using different antigens, like in allergy. Allergists don’t use just cat antigen in patients who have cat, dog, and house dust mite allergies. I suppose we may also learn to combine autoantigens, together with possible stimulation of beta-cells in combination with drugs that promote beta-cell regeneration,” he continued.
Other autoantibodies commonly present in patients with type 1 diabetes, or at high risk for the disease, include insulin autoantibodies, islet cell autoantibodies, and antibodies to the zinc transporter. Combining the GAD vaccine with other major diabetes-specific autoantigens recognized by the immune system could provide synergistic benefits.
The likely necessity for a combined approach addressing multiple pathways was underscored in a separate presentation by Dr. Jay S. Skyler, chairman of the type 1 Diabetes TrialNet, a National Institutes of Health–funded international network of centers conducting clinical trials of diabetes therapies.
The GAD vaccine appears to have the same limitation as the other immunomodulatory therapies evaluated to date in clinical trials, including the B cell–depleting anti-CD20 agent rituximab, and the anti-CD3 biologics teplizumab and otelixizumab: namely, they preserve beta cell function for a while, but the effect is transient. Eventually fasting C-peptide levels start to fall off in parallel to the placebo group. That’s why combination therapy will probably be required in order to cure or prevent Type 1 diabetes, according to Dr. Skyler, a professor of medicine, pediatrics and psychology at the University of Miami.
Ideally, a combination therapy should be multipronged, with three goals: Stop immune destruction, preserve beta-cell mass, and replace or regenerate beta-cells. Such a regimen might start off with a potent anti-inflammatory therapy – perhaps an anti-interleukin-1beta agent or tumor necrosis factor inhibitor – to quell the metabolic stress surrounding the pancreatic islets. This might well need to be given on a continuing basis.
Next would come an immunomodulatory approach; for example, T-cell modulation with an anti-CD3 biologic or B cell depletion with rituximab. This could be followed up with an autoantigen-specific therapy such as the GAD vaccine or oral insulin. “Maybe it needs to be both,” Dr. Skyler continued.
The logical subsequent step would be to try to stimulate immunologic expansion of regulatory T cells, either with granulocyte colony–stimulating factor or by direct infusion of regulatory T cells themselves. This could be combined with beta-cell expansion via exenatide (Byetta) or the investigational HIP2B peptide.
“We could conceivably be doing some of these things even today,” Dr. Skyler said.
Dr. Ludvigsson reported receiving research grant support from Diamyd.
Dr. Skyler has served as a consultant to and/or received research grants from numerous pharmaceutical companies.
Type 1 diabetes (T1D) is an autoimmune disease caused by interplay of genetic and environmental factors. The incidence of childhood T1D has doubled worldwide over the past 20-25 years. Elimination of the environmental agent(s) responsible for this epidemic would be the most efficient approach to primary prevention; however, more work is needed to identify the environmental agents and to develop effective interventions.
Blocking progression from islet autoimmunity to clinical diabetes or secondary prevention has been attempted, so far to no avail, by a number of groups, including large randomized trials: the Diabetes Prevention Trial – Type 1, the European Nicotinamide Diabetes Intervention Trial, and the Type 1 Diabetes Prediction and Prevention Project.
Trials in patients with newly diagnosed T1D aim at tertiary prevention, such as preservation of remaining islet beta-cells to induce and prolong partial remission. Unfortunately, most islets have already been destroyed by the time diabetes is diagnosed and complete reversal of diabetes is highly unlikely. Benefits may include a simpler insulin regimen, lower HbA1c, and reduced risk of hypoglycemia and microvascular complications. The gain may be even greater if the intervention is applied as soon as the patient shows asymptomatic “dysglycemia,” detected by oral glucose tolerance test or A1c, before overt symptoms of diabetes.
While new interventions are often tested first in patients with established diabetes, and, when proven safe, applied to patients with pre-T1D, efficacy after diagnosis of diabetes is not to be a precondition to application in pre-T1D, as there may be a “point of no return” in the destruction of the islets, rendering some interventions effective only at the earlier stages of the process.
Antigen-specific vaccines
Among several approaches to prevention of T1D, “vaccination” using islet autoantigens (intact or altered peptides derived from insulin, GAD65 or other proteins) stands out as potentially inducing long-term tolerance by induction of regulatory T-cells that down-regulate immunity to autoantigens. Until recently, trials of insulin administered parenterally, orally, or intranasally have been unsuccessful. Therefore, the initial results from trials of the Diamyd vaccine, as reviewed here, were greeted with huge interest and excitement. The vaccine includes the whole recombinant human GAD65 (rhGAD65) molecule suspended in alum. The protective effect was most pronounced in patients treated within 3 months of diagnosis, and no serious side effects were observed.
Insulin-related molecules continue to attract great interest in vaccine development. Phase I studies have been completed or are nearing completion for a proinsulin peptide C19-A3, an insulin peptide with incomplete Freund adjuvant, and a plasmid encoding proinsulin.
Combination therapies may enhance efficacy while lowering risk of adverse events if utilizing therapies from different treatment pathways. While more targeted therapies are being employed, immunomodulatory agents are still relatively nonspecific and potentially toxic to some of the trial participants. Some may carry an unacceptable risk of long-term complications. This direction is important; however, multiple scientific and logistic issues remain, for example, the anticipated duration, toxicity, and complexity of immunotherapy.
In the long run, primary prevention will likely be the optimal approach to the prevention of T1D. Once more than one islet autoantibody is present, most individuals progress to diabetes in 5-10 years. The TrialNet consortium (www.diabetestrialnet.org) systematically evaluates therapies in new-onset patients as well as in pre-diabetic subjects, and invites proposals from the research community at large.
Marian Rewers, M.D., Ph.D., is professor of pediatrics and preventive medicine at the Barbara Davis Center for Childhood Diabetes, University of Colorado, Denver.
Type 1 diabetes (T1D) is an autoimmune disease caused by interplay of genetic and environmental factors. The incidence of childhood T1D has doubled worldwide over the past 20-25 years. Elimination of the environmental agent(s) responsible for this epidemic would be the most efficient approach to primary prevention; however, more work is needed to identify the environmental agents and to develop effective interventions.
Blocking progression from islet autoimmunity to clinical diabetes or secondary prevention has been attempted, so far to no avail, by a number of groups, including large randomized trials: the Diabetes Prevention Trial – Type 1, the European Nicotinamide Diabetes Intervention Trial, and the Type 1 Diabetes Prediction and Prevention Project.
Trials in patients with newly diagnosed T1D aim at tertiary prevention, such as preservation of remaining islet beta-cells to induce and prolong partial remission. Unfortunately, most islets have already been destroyed by the time diabetes is diagnosed and complete reversal of diabetes is highly unlikely. Benefits may include a simpler insulin regimen, lower HbA1c, and reduced risk of hypoglycemia and microvascular complications. The gain may be even greater if the intervention is applied as soon as the patient shows asymptomatic “dysglycemia,” detected by oral glucose tolerance test or A1c, before overt symptoms of diabetes.
While new interventions are often tested first in patients with established diabetes, and, when proven safe, applied to patients with pre-T1D, efficacy after diagnosis of diabetes is not to be a precondition to application in pre-T1D, as there may be a “point of no return” in the destruction of the islets, rendering some interventions effective only at the earlier stages of the process.
Antigen-specific vaccines
Among several approaches to prevention of T1D, “vaccination” using islet autoantigens (intact or altered peptides derived from insulin, GAD65 or other proteins) stands out as potentially inducing long-term tolerance by induction of regulatory T-cells that down-regulate immunity to autoantigens. Until recently, trials of insulin administered parenterally, orally, or intranasally have been unsuccessful. Therefore, the initial results from trials of the Diamyd vaccine, as reviewed here, were greeted with huge interest and excitement. The vaccine includes the whole recombinant human GAD65 (rhGAD65) molecule suspended in alum. The protective effect was most pronounced in patients treated within 3 months of diagnosis, and no serious side effects were observed.
Insulin-related molecules continue to attract great interest in vaccine development. Phase I studies have been completed or are nearing completion for a proinsulin peptide C19-A3, an insulin peptide with incomplete Freund adjuvant, and a plasmid encoding proinsulin.
Combination therapies may enhance efficacy while lowering risk of adverse events if utilizing therapies from different treatment pathways. While more targeted therapies are being employed, immunomodulatory agents are still relatively nonspecific and potentially toxic to some of the trial participants. Some may carry an unacceptable risk of long-term complications. This direction is important; however, multiple scientific and logistic issues remain, for example, the anticipated duration, toxicity, and complexity of immunotherapy.
In the long run, primary prevention will likely be the optimal approach to the prevention of T1D. Once more than one islet autoantibody is present, most individuals progress to diabetes in 5-10 years. The TrialNet consortium (www.diabetestrialnet.org) systematically evaluates therapies in new-onset patients as well as in pre-diabetic subjects, and invites proposals from the research community at large.
Marian Rewers, M.D., Ph.D., is professor of pediatrics and preventive medicine at the Barbara Davis Center for Childhood Diabetes, University of Colorado, Denver.
Type 1 diabetes (T1D) is an autoimmune disease caused by interplay of genetic and environmental factors. The incidence of childhood T1D has doubled worldwide over the past 20-25 years. Elimination of the environmental agent(s) responsible for this epidemic would be the most efficient approach to primary prevention; however, more work is needed to identify the environmental agents and to develop effective interventions.
Blocking progression from islet autoimmunity to clinical diabetes or secondary prevention has been attempted, so far to no avail, by a number of groups, including large randomized trials: the Diabetes Prevention Trial – Type 1, the European Nicotinamide Diabetes Intervention Trial, and the Type 1 Diabetes Prediction and Prevention Project.
Trials in patients with newly diagnosed T1D aim at tertiary prevention, such as preservation of remaining islet beta-cells to induce and prolong partial remission. Unfortunately, most islets have already been destroyed by the time diabetes is diagnosed and complete reversal of diabetes is highly unlikely. Benefits may include a simpler insulin regimen, lower HbA1c, and reduced risk of hypoglycemia and microvascular complications. The gain may be even greater if the intervention is applied as soon as the patient shows asymptomatic “dysglycemia,” detected by oral glucose tolerance test or A1c, before overt symptoms of diabetes.
While new interventions are often tested first in patients with established diabetes, and, when proven safe, applied to patients with pre-T1D, efficacy after diagnosis of diabetes is not to be a precondition to application in pre-T1D, as there may be a “point of no return” in the destruction of the islets, rendering some interventions effective only at the earlier stages of the process.
Antigen-specific vaccines
Among several approaches to prevention of T1D, “vaccination” using islet autoantigens (intact or altered peptides derived from insulin, GAD65 or other proteins) stands out as potentially inducing long-term tolerance by induction of regulatory T-cells that down-regulate immunity to autoantigens. Until recently, trials of insulin administered parenterally, orally, or intranasally have been unsuccessful. Therefore, the initial results from trials of the Diamyd vaccine, as reviewed here, were greeted with huge interest and excitement. The vaccine includes the whole recombinant human GAD65 (rhGAD65) molecule suspended in alum. The protective effect was most pronounced in patients treated within 3 months of diagnosis, and no serious side effects were observed.
Insulin-related molecules continue to attract great interest in vaccine development. Phase I studies have been completed or are nearing completion for a proinsulin peptide C19-A3, an insulin peptide with incomplete Freund adjuvant, and a plasmid encoding proinsulin.
Combination therapies may enhance efficacy while lowering risk of adverse events if utilizing therapies from different treatment pathways. While more targeted therapies are being employed, immunomodulatory agents are still relatively nonspecific and potentially toxic to some of the trial participants. Some may carry an unacceptable risk of long-term complications. This direction is important; however, multiple scientific and logistic issues remain, for example, the anticipated duration, toxicity, and complexity of immunotherapy.
In the long run, primary prevention will likely be the optimal approach to the prevention of T1D. Once more than one islet autoantibody is present, most individuals progress to diabetes in 5-10 years. The TrialNet consortium (www.diabetestrialnet.org) systematically evaluates therapies in new-onset patients as well as in pre-diabetic subjects, and invites proposals from the research community at large.
Marian Rewers, M.D., Ph.D., is professor of pediatrics and preventive medicine at the Barbara Davis Center for Childhood Diabetes, University of Colorado, Denver.
KEYSTONE, Colo. – Right now, the Diamyd Medical’s GAD vaccine is in the sweet spot in the developmental pipeline – an interim period of enormous optimism that this novel autoantigen-based immunotherapy will safely prevent many cases of type 1 diabetes.
The results of three phase II studies are in and they look quite promising. Two large phase III clinical trials are well underway in Europe and the United States. The safety experience with the 65-kD isoform of GAD (glutamic acid decarboxylase-65) vaccine has been outstanding. The subcutaneous two-injection series is easy to administer. Acceptance of the vaccine by patients and their families is high. The vaccine targets a serious disease whose incidence is steadily climbing by 3%-5% per year in developed countries. And most patients with recently diagnosed type 1 diabetes possess GAD autoantibodies, so the Diamyd vaccine would be widely applicable.
All of that was good enough for Johnson and Johnson, which in June inked a huge development and marketing deal for the GAD vaccine with small Swedish biotech company Diamyd Medical. Under the deal, Diamyd receives $45 million upfront, milestone payments of up to $580 million, and tiered royalties after that. The Federal Trade Commission’s antitrust division has already approved the deal.
But during this blissful interlude, one key question remains: Is the Diamyd vaccine effective?
“It’s too early to say if this works. Absolutely too early. We have a phase III trial in Europe with results due next spring. And the TrialNet study [is] going on here in the U.S. So we will know in a year or 2,” Dr. Johnny L. Ludvigsson said at a conference on management of diabetes in youth sponsored by the Children’s Diabetes Foundation at Denver.
Dr. Ludvigsson, professor of pediatrics at the University of Linkoping (Sweden), led the phase III European trial evaluating whether the GAD vaccine preserves beta-cell function and residual insulin secretion in patients with type 1 diabetes diagnosed within 3 months of starting treatment. He also headed a phase II study that caused a favorable buzz within the diabetes research community (N. Engl. J. Med. 2008;359:1909-20) and for which he is now analyzing 5-year follow-up data.
And while the forthcoming phase III trial results will tell the tale as to clinical efficacy, at this time some useful interim observations can be made about the GAD vaccine, according to Dr. Ludvigsson:
• The vaccine has demonstrated excellent safety. Experience with the vaccine to date totals 850 patient-years in adults and 350 patient-years in children, with no adverse events reported. This is enormously reassuring because GAD transforms glutamate into GABA, an important neurotransmitter. Lack of GAD in the CNS leads to muscle rigidity and convulsions, while stimulation of CNS GAD results in inhibition of neurotransmission. The absence of any such adverse events indicates the vaccine is working, as designed, to affect only a very small part of the immune system: namely, the activated T cells that have targeted pancreatic beta-cells for destruction, Dr. Ludvigsson said.
• The vaccine has demonstrated prolonged immunologic effects. The immunologic response to the Diamyd vaccine lasts surprisingly long – approaching 5 years and still counting. It’s a GAD-specific, cell-mediated, and humoral immune response characterized by increased GAD autoantibodies, a Th2 shift marked by reduction in activated T cells and an increase in regulatory T cells, a sharp and sustained rise in levels of interleukins-2, -5, -10, -13, and -17, and GAD tolerance. “We see this response still after 4 years. The memory is there,” Dr. Ludvigsson observed.
• “The earlier we treat, the better the outcome.” That’s why the phase III European trial is restricted to patients diagnosed with type 1 diabetes within the past 3 months. It’s also the impetus for ongoing prevention trials in individuals at very high genetic risk for type 1 diabetes who have GAD autoantibodies but have not developed overt disease.
• The vaccine probably won’t work in diabetic patients without GAD autoantibodies. No studies have been carried out in such patients, but Dr. Ludvigsson said it’s his impression that the vaccine is more effective in individuals with higher than lower titers of GAD autoantibodies.
For the future, the GAD vaccine alone probably is not the solution to type 1 diabetes, Dr. Ludvigsson said candidly.
“I believe this opens the door to using different antigens, like in allergy. Allergists don’t use just cat antigen in patients who have cat, dog, and house dust mite allergies. I suppose we may also learn to combine autoantigens, together with possible stimulation of beta-cells in combination with drugs that promote beta-cell regeneration,” he continued.
Other autoantibodies commonly present in patients with type 1 diabetes, or at high risk for the disease, include insulin autoantibodies, islet cell autoantibodies, and antibodies to the zinc transporter. Combining the GAD vaccine with other major diabetes-specific autoantigens recognized by the immune system could provide synergistic benefits.
The likely necessity for a combined approach addressing multiple pathways was underscored in a separate presentation by Dr. Jay S. Skyler, chairman of the type 1 Diabetes TrialNet, a National Institutes of Health–funded international network of centers conducting clinical trials of diabetes therapies.
The GAD vaccine appears to have the same limitation as the other immunomodulatory therapies evaluated to date in clinical trials, including the B cell–depleting anti-CD20 agent rituximab, and the anti-CD3 biologics teplizumab and otelixizumab: namely, they preserve beta cell function for a while, but the effect is transient. Eventually fasting C-peptide levels start to fall off in parallel to the placebo group. That’s why combination therapy will probably be required in order to cure or prevent Type 1 diabetes, according to Dr. Skyler, a professor of medicine, pediatrics and psychology at the University of Miami.
Ideally, a combination therapy should be multipronged, with three goals: Stop immune destruction, preserve beta-cell mass, and replace or regenerate beta-cells. Such a regimen might start off with a potent anti-inflammatory therapy – perhaps an anti-interleukin-1beta agent or tumor necrosis factor inhibitor – to quell the metabolic stress surrounding the pancreatic islets. This might well need to be given on a continuing basis.
Next would come an immunomodulatory approach; for example, T-cell modulation with an anti-CD3 biologic or B cell depletion with rituximab. This could be followed up with an autoantigen-specific therapy such as the GAD vaccine or oral insulin. “Maybe it needs to be both,” Dr. Skyler continued.
The logical subsequent step would be to try to stimulate immunologic expansion of regulatory T cells, either with granulocyte colony–stimulating factor or by direct infusion of regulatory T cells themselves. This could be combined with beta-cell expansion via exenatide (Byetta) or the investigational HIP2B peptide.
“We could conceivably be doing some of these things even today,” Dr. Skyler said.
Dr. Ludvigsson reported receiving research grant support from Diamyd.
Dr. Skyler has served as a consultant to and/or received research grants from numerous pharmaceutical companies.
KEYSTONE, Colo. – Right now, the Diamyd Medical’s GAD vaccine is in the sweet spot in the developmental pipeline – an interim period of enormous optimism that this novel autoantigen-based immunotherapy will safely prevent many cases of type 1 diabetes.
The results of three phase II studies are in and they look quite promising. Two large phase III clinical trials are well underway in Europe and the United States. The safety experience with the 65-kD isoform of GAD (glutamic acid decarboxylase-65) vaccine has been outstanding. The subcutaneous two-injection series is easy to administer. Acceptance of the vaccine by patients and their families is high. The vaccine targets a serious disease whose incidence is steadily climbing by 3%-5% per year in developed countries. And most patients with recently diagnosed type 1 diabetes possess GAD autoantibodies, so the Diamyd vaccine would be widely applicable.
All of that was good enough for Johnson and Johnson, which in June inked a huge development and marketing deal for the GAD vaccine with small Swedish biotech company Diamyd Medical. Under the deal, Diamyd receives $45 million upfront, milestone payments of up to $580 million, and tiered royalties after that. The Federal Trade Commission’s antitrust division has already approved the deal.
But during this blissful interlude, one key question remains: Is the Diamyd vaccine effective?
“It’s too early to say if this works. Absolutely too early. We have a phase III trial in Europe with results due next spring. And the TrialNet study [is] going on here in the U.S. So we will know in a year or 2,” Dr. Johnny L. Ludvigsson said at a conference on management of diabetes in youth sponsored by the Children’s Diabetes Foundation at Denver.
Dr. Ludvigsson, professor of pediatrics at the University of Linkoping (Sweden), led the phase III European trial evaluating whether the GAD vaccine preserves beta-cell function and residual insulin secretion in patients with type 1 diabetes diagnosed within 3 months of starting treatment. He also headed a phase II study that caused a favorable buzz within the diabetes research community (N. Engl. J. Med. 2008;359:1909-20) and for which he is now analyzing 5-year follow-up data.
And while the forthcoming phase III trial results will tell the tale as to clinical efficacy, at this time some useful interim observations can be made about the GAD vaccine, according to Dr. Ludvigsson:
• The vaccine has demonstrated excellent safety. Experience with the vaccine to date totals 850 patient-years in adults and 350 patient-years in children, with no adverse events reported. This is enormously reassuring because GAD transforms glutamate into GABA, an important neurotransmitter. Lack of GAD in the CNS leads to muscle rigidity and convulsions, while stimulation of CNS GAD results in inhibition of neurotransmission. The absence of any such adverse events indicates the vaccine is working, as designed, to affect only a very small part of the immune system: namely, the activated T cells that have targeted pancreatic beta-cells for destruction, Dr. Ludvigsson said.
• The vaccine has demonstrated prolonged immunologic effects. The immunologic response to the Diamyd vaccine lasts surprisingly long – approaching 5 years and still counting. It’s a GAD-specific, cell-mediated, and humoral immune response characterized by increased GAD autoantibodies, a Th2 shift marked by reduction in activated T cells and an increase in regulatory T cells, a sharp and sustained rise in levels of interleukins-2, -5, -10, -13, and -17, and GAD tolerance. “We see this response still after 4 years. The memory is there,” Dr. Ludvigsson observed.
• “The earlier we treat, the better the outcome.” That’s why the phase III European trial is restricted to patients diagnosed with type 1 diabetes within the past 3 months. It’s also the impetus for ongoing prevention trials in individuals at very high genetic risk for type 1 diabetes who have GAD autoantibodies but have not developed overt disease.
• The vaccine probably won’t work in diabetic patients without GAD autoantibodies. No studies have been carried out in such patients, but Dr. Ludvigsson said it’s his impression that the vaccine is more effective in individuals with higher than lower titers of GAD autoantibodies.
For the future, the GAD vaccine alone probably is not the solution to type 1 diabetes, Dr. Ludvigsson said candidly.
“I believe this opens the door to using different antigens, like in allergy. Allergists don’t use just cat antigen in patients who have cat, dog, and house dust mite allergies. I suppose we may also learn to combine autoantigens, together with possible stimulation of beta-cells in combination with drugs that promote beta-cell regeneration,” he continued.
Other autoantibodies commonly present in patients with type 1 diabetes, or at high risk for the disease, include insulin autoantibodies, islet cell autoantibodies, and antibodies to the zinc transporter. Combining the GAD vaccine with other major diabetes-specific autoantigens recognized by the immune system could provide synergistic benefits.
The likely necessity for a combined approach addressing multiple pathways was underscored in a separate presentation by Dr. Jay S. Skyler, chairman of the type 1 Diabetes TrialNet, a National Institutes of Health–funded international network of centers conducting clinical trials of diabetes therapies.
The GAD vaccine appears to have the same limitation as the other immunomodulatory therapies evaluated to date in clinical trials, including the B cell–depleting anti-CD20 agent rituximab, and the anti-CD3 biologics teplizumab and otelixizumab: namely, they preserve beta cell function for a while, but the effect is transient. Eventually fasting C-peptide levels start to fall off in parallel to the placebo group. That’s why combination therapy will probably be required in order to cure or prevent Type 1 diabetes, according to Dr. Skyler, a professor of medicine, pediatrics and psychology at the University of Miami.
Ideally, a combination therapy should be multipronged, with three goals: Stop immune destruction, preserve beta-cell mass, and replace or regenerate beta-cells. Such a regimen might start off with a potent anti-inflammatory therapy – perhaps an anti-interleukin-1beta agent or tumor necrosis factor inhibitor – to quell the metabolic stress surrounding the pancreatic islets. This might well need to be given on a continuing basis.
Next would come an immunomodulatory approach; for example, T-cell modulation with an anti-CD3 biologic or B cell depletion with rituximab. This could be followed up with an autoantigen-specific therapy such as the GAD vaccine or oral insulin. “Maybe it needs to be both,” Dr. Skyler continued.
The logical subsequent step would be to try to stimulate immunologic expansion of regulatory T cells, either with granulocyte colony–stimulating factor or by direct infusion of regulatory T cells themselves. This could be combined with beta-cell expansion via exenatide (Byetta) or the investigational HIP2B peptide.
“We could conceivably be doing some of these things even today,” Dr. Skyler said.
Dr. Ludvigsson reported receiving research grant support from Diamyd.
Dr. Skyler has served as a consultant to and/or received research grants from numerous pharmaceutical companies.
Tick, Tock, Tick, Tock
A new study in this month's Journal of Hospital Medicine that catalogues the daily routine of HM practitioners is a first step in helping streamline the hospitalist’s workflow for efficiency, say several people associated with the report.
The report, “Where Did the Day Go? A Time-Motion Study of Hospitalists,” attempted to capture the amount of time hospitalists spent on various activities, including interacting with electronic health records (EHR) (34.1%), communication with colleagues (25.9%), and direct care (7.4%) (J Hosp Med. 2010;5(6):323-328). But one of the report’s senior authors, as well as the co-author of an accompanying editorial, anticipate that the study will serve as a springboard for future research on how hospitalists can best use their time.
Hospitalists need to “lay the foundation to figure how not to just observe what the doctors are doing, but how, in the future, to what they should be doing,” says Mark Williams, MD, FHM, professor and chief of hospital medicine at Northwestern University's Feinberg School of Medicine in Chicago. “We’ve got to have a good understanding of what we’re doing every day to move forward.”
The research, which furthered a similar Northwestern study completed in 2006 found that 16% of all activities occurred simultaneously, meaning that the surveyed hospitalists spent about 9% of their average 10.3-hour shift multitasking.
“Sadly, we documented that the vast majority [of time] is away from the patient, not with the patient,” Dr. Williams says.
Dr. Williams and Amit Prachand, an administrator in the HM department at Northwestern, hope to see more research done to define the best workflow for a hospitalist. Both agree, though, that dedicated funding will have to be set aside, either by federal agencies or research institutions, to make that happen.
“We need to convince people the money is well spent in focusing on this,” says Prachand, co-author of the editorial “Hospitalists: Lean Leaders for Hospitals.” “I think the hospital is going to be the one with the most to gain by supporting these opportunities.”
A new study in this month's Journal of Hospital Medicine that catalogues the daily routine of HM practitioners is a first step in helping streamline the hospitalist’s workflow for efficiency, say several people associated with the report.
The report, “Where Did the Day Go? A Time-Motion Study of Hospitalists,” attempted to capture the amount of time hospitalists spent on various activities, including interacting with electronic health records (EHR) (34.1%), communication with colleagues (25.9%), and direct care (7.4%) (J Hosp Med. 2010;5(6):323-328). But one of the report’s senior authors, as well as the co-author of an accompanying editorial, anticipate that the study will serve as a springboard for future research on how hospitalists can best use their time.
Hospitalists need to “lay the foundation to figure how not to just observe what the doctors are doing, but how, in the future, to what they should be doing,” says Mark Williams, MD, FHM, professor and chief of hospital medicine at Northwestern University's Feinberg School of Medicine in Chicago. “We’ve got to have a good understanding of what we’re doing every day to move forward.”
The research, which furthered a similar Northwestern study completed in 2006 found that 16% of all activities occurred simultaneously, meaning that the surveyed hospitalists spent about 9% of their average 10.3-hour shift multitasking.
“Sadly, we documented that the vast majority [of time] is away from the patient, not with the patient,” Dr. Williams says.
Dr. Williams and Amit Prachand, an administrator in the HM department at Northwestern, hope to see more research done to define the best workflow for a hospitalist. Both agree, though, that dedicated funding will have to be set aside, either by federal agencies or research institutions, to make that happen.
“We need to convince people the money is well spent in focusing on this,” says Prachand, co-author of the editorial “Hospitalists: Lean Leaders for Hospitals.” “I think the hospital is going to be the one with the most to gain by supporting these opportunities.”
A new study in this month's Journal of Hospital Medicine that catalogues the daily routine of HM practitioners is a first step in helping streamline the hospitalist’s workflow for efficiency, say several people associated with the report.
The report, “Where Did the Day Go? A Time-Motion Study of Hospitalists,” attempted to capture the amount of time hospitalists spent on various activities, including interacting with electronic health records (EHR) (34.1%), communication with colleagues (25.9%), and direct care (7.4%) (J Hosp Med. 2010;5(6):323-328). But one of the report’s senior authors, as well as the co-author of an accompanying editorial, anticipate that the study will serve as a springboard for future research on how hospitalists can best use their time.
Hospitalists need to “lay the foundation to figure how not to just observe what the doctors are doing, but how, in the future, to what they should be doing,” says Mark Williams, MD, FHM, professor and chief of hospital medicine at Northwestern University's Feinberg School of Medicine in Chicago. “We’ve got to have a good understanding of what we’re doing every day to move forward.”
The research, which furthered a similar Northwestern study completed in 2006 found that 16% of all activities occurred simultaneously, meaning that the surveyed hospitalists spent about 9% of their average 10.3-hour shift multitasking.
“Sadly, we documented that the vast majority [of time] is away from the patient, not with the patient,” Dr. Williams says.
Dr. Williams and Amit Prachand, an administrator in the HM department at Northwestern, hope to see more research done to define the best workflow for a hospitalist. Both agree, though, that dedicated funding will have to be set aside, either by federal agencies or research institutions, to make that happen.
“We need to convince people the money is well spent in focusing on this,” says Prachand, co-author of the editorial “Hospitalists: Lean Leaders for Hospitals.” “I think the hospital is going to be the one with the most to gain by supporting these opportunities.”
In the Literature: Research You Need to Know
Clinical question: Do clinical outcomes differ with the use of dopamine and norepinephrine in the treatment of shock?
Background: Observational trials have suggested higher mortality among patients with shock who are treated with dopamine versus norepinephrine; however, there are limited data from randomized trials.
Study design: Randomized, double-blinded trial.
Setting: Eight ICUs in Europe.
Synopsis: The study enrolled 1,679 consecutive adult patients with shock despite intravenous fluids. Of these, 62.2% were classified as septic shock, 16.7% cardiogenic, and 15.7% hypovolemic. Clinicians titrated the blinded study drug (dopamine or norepinephrine) according to a pre-specified algorithm. If shock persisted despite titration of their study drug to a goal rate, then open-label norepinephrine was added, followed by epinephrine or vasopressin if necessary.
No difference in 28-day mortality between dopamine and norepinephrine (52% versus 48% of patients; odds ratio 1.17 (0.97-1.42); P=0.10) was detected. Patients receiving dopamine experienced more frequent (24% vs. 12%, P<0.001) and more severe arrhythmias (6.1% vs. 1.6%, P< 0.001).
In subgroup analysis, patients in cardiogenic shock had significantly increased 28-day mortality with dopamine (P=0.03).
Study limitations include the use of norepinephrine as an open-label treatment and the inclusion of patients in hypovolemic shock, who are not typically treated with vasopressors.
Bottom line: No mortality difference is detected between dopamine and norepinephrine in patients with shock. Dopamine results in increased rates of mortality in cardiogenic shock and serious arrhythmias in all patients.
Citation: De Backer D, Biston P, Devriendt J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med. 2010;362(9):779-789.
Reviewed for TH eWire by Robert Chang, MD, Anita Hart, MD, Hae-won Kim, MD, Robert Paretti, MD, Helena Pasieka, MD, and Matt Smitherman, MD, University of Michigan, Ann Arbor
For more physician reviews of HM-related research, visit our website.
Clinical question: Do clinical outcomes differ with the use of dopamine and norepinephrine in the treatment of shock?
Background: Observational trials have suggested higher mortality among patients with shock who are treated with dopamine versus norepinephrine; however, there are limited data from randomized trials.
Study design: Randomized, double-blinded trial.
Setting: Eight ICUs in Europe.
Synopsis: The study enrolled 1,679 consecutive adult patients with shock despite intravenous fluids. Of these, 62.2% were classified as septic shock, 16.7% cardiogenic, and 15.7% hypovolemic. Clinicians titrated the blinded study drug (dopamine or norepinephrine) according to a pre-specified algorithm. If shock persisted despite titration of their study drug to a goal rate, then open-label norepinephrine was added, followed by epinephrine or vasopressin if necessary.
No difference in 28-day mortality between dopamine and norepinephrine (52% versus 48% of patients; odds ratio 1.17 (0.97-1.42); P=0.10) was detected. Patients receiving dopamine experienced more frequent (24% vs. 12%, P<0.001) and more severe arrhythmias (6.1% vs. 1.6%, P< 0.001).
In subgroup analysis, patients in cardiogenic shock had significantly increased 28-day mortality with dopamine (P=0.03).
Study limitations include the use of norepinephrine as an open-label treatment and the inclusion of patients in hypovolemic shock, who are not typically treated with vasopressors.
Bottom line: No mortality difference is detected between dopamine and norepinephrine in patients with shock. Dopamine results in increased rates of mortality in cardiogenic shock and serious arrhythmias in all patients.
Citation: De Backer D, Biston P, Devriendt J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med. 2010;362(9):779-789.
Reviewed for TH eWire by Robert Chang, MD, Anita Hart, MD, Hae-won Kim, MD, Robert Paretti, MD, Helena Pasieka, MD, and Matt Smitherman, MD, University of Michigan, Ann Arbor
For more physician reviews of HM-related research, visit our website.
Clinical question: Do clinical outcomes differ with the use of dopamine and norepinephrine in the treatment of shock?
Background: Observational trials have suggested higher mortality among patients with shock who are treated with dopamine versus norepinephrine; however, there are limited data from randomized trials.
Study design: Randomized, double-blinded trial.
Setting: Eight ICUs in Europe.
Synopsis: The study enrolled 1,679 consecutive adult patients with shock despite intravenous fluids. Of these, 62.2% were classified as septic shock, 16.7% cardiogenic, and 15.7% hypovolemic. Clinicians titrated the blinded study drug (dopamine or norepinephrine) according to a pre-specified algorithm. If shock persisted despite titration of their study drug to a goal rate, then open-label norepinephrine was added, followed by epinephrine or vasopressin if necessary.
No difference in 28-day mortality between dopamine and norepinephrine (52% versus 48% of patients; odds ratio 1.17 (0.97-1.42); P=0.10) was detected. Patients receiving dopamine experienced more frequent (24% vs. 12%, P<0.001) and more severe arrhythmias (6.1% vs. 1.6%, P< 0.001).
In subgroup analysis, patients in cardiogenic shock had significantly increased 28-day mortality with dopamine (P=0.03).
Study limitations include the use of norepinephrine as an open-label treatment and the inclusion of patients in hypovolemic shock, who are not typically treated with vasopressors.
Bottom line: No mortality difference is detected between dopamine and norepinephrine in patients with shock. Dopamine results in increased rates of mortality in cardiogenic shock and serious arrhythmias in all patients.
Citation: De Backer D, Biston P, Devriendt J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med. 2010;362(9):779-789.
Reviewed for TH eWire by Robert Chang, MD, Anita Hart, MD, Hae-won Kim, MD, Robert Paretti, MD, Helena Pasieka, MD, and Matt Smitherman, MD, University of Michigan, Ann Arbor
For more physician reviews of HM-related research, visit our website.
CDC Recommends Gamma Release Assay (IGRA) for TB
New guidelines from the Centers for Disease Control and Prevention (CDC) that recommend interferon-gamma release assay (IGRA) blood tests over the century-old tuberculin skin test (TST) to detect Mycobacterium tuberculosis (TB) could help hospitalist groups save time and money, according to the head of a TB institute.
The guidelines, published in the CDC’s Morbidity & Mortality Weekly Report, recommend providers use IGRAs over TSTs for certain populations, including patients who historically are unlikely to return for a needed second visit to read the TST results and patients who have previously received Bacille Calmette-Guérin (BCG) as a vaccine or as a cancer therapy (MMWR Recomm Rep. 2010;59(RR-5):1-25).
TSTs remain the preferred test for children younger than 5 years old, although some research has suggested that using both tests in youngsters could increase diagnostic sensitivity for that population.
Lee B. Reichman, MD, MPH, FACP, FCCP, of New Jersey Medical School’s Global Tuberculosis Institute in Newark says the improved efficacy of IGRAs should help weed out the false positives associated with the TSTs. That should be a boon for hospitalists looking to boost cost efficiency by focusing care delivery on the most at-risk populations, he adds.
“The hospitalist is busy,” Dr. Reichman says. “So now he doesn’t have to worry about all those people who are turning out to be a false positive.”
Dr. Reichman hopes the new guidelines catch on quickly, particularly because IGRAs must be conducted in laboratory settings that help ensure better predictive results. He fears, however, that adherence to traditional methods like BCG vaccination, which is particularly popular in Europe, might stall widespread IGRA adoption.
“It will take time to get there,” he says. “The TB community is notoriously slow in adapting new technologies.”
New guidelines from the Centers for Disease Control and Prevention (CDC) that recommend interferon-gamma release assay (IGRA) blood tests over the century-old tuberculin skin test (TST) to detect Mycobacterium tuberculosis (TB) could help hospitalist groups save time and money, according to the head of a TB institute.
The guidelines, published in the CDC’s Morbidity & Mortality Weekly Report, recommend providers use IGRAs over TSTs for certain populations, including patients who historically are unlikely to return for a needed second visit to read the TST results and patients who have previously received Bacille Calmette-Guérin (BCG) as a vaccine or as a cancer therapy (MMWR Recomm Rep. 2010;59(RR-5):1-25).
TSTs remain the preferred test for children younger than 5 years old, although some research has suggested that using both tests in youngsters could increase diagnostic sensitivity for that population.
Lee B. Reichman, MD, MPH, FACP, FCCP, of New Jersey Medical School’s Global Tuberculosis Institute in Newark says the improved efficacy of IGRAs should help weed out the false positives associated with the TSTs. That should be a boon for hospitalists looking to boost cost efficiency by focusing care delivery on the most at-risk populations, he adds.
“The hospitalist is busy,” Dr. Reichman says. “So now he doesn’t have to worry about all those people who are turning out to be a false positive.”
Dr. Reichman hopes the new guidelines catch on quickly, particularly because IGRAs must be conducted in laboratory settings that help ensure better predictive results. He fears, however, that adherence to traditional methods like BCG vaccination, which is particularly popular in Europe, might stall widespread IGRA adoption.
“It will take time to get there,” he says. “The TB community is notoriously slow in adapting new technologies.”
New guidelines from the Centers for Disease Control and Prevention (CDC) that recommend interferon-gamma release assay (IGRA) blood tests over the century-old tuberculin skin test (TST) to detect Mycobacterium tuberculosis (TB) could help hospitalist groups save time and money, according to the head of a TB institute.
The guidelines, published in the CDC’s Morbidity & Mortality Weekly Report, recommend providers use IGRAs over TSTs for certain populations, including patients who historically are unlikely to return for a needed second visit to read the TST results and patients who have previously received Bacille Calmette-Guérin (BCG) as a vaccine or as a cancer therapy (MMWR Recomm Rep. 2010;59(RR-5):1-25).
TSTs remain the preferred test for children younger than 5 years old, although some research has suggested that using both tests in youngsters could increase diagnostic sensitivity for that population.
Lee B. Reichman, MD, MPH, FACP, FCCP, of New Jersey Medical School’s Global Tuberculosis Institute in Newark says the improved efficacy of IGRAs should help weed out the false positives associated with the TSTs. That should be a boon for hospitalists looking to boost cost efficiency by focusing care delivery on the most at-risk populations, he adds.
“The hospitalist is busy,” Dr. Reichman says. “So now he doesn’t have to worry about all those people who are turning out to be a false positive.”
Dr. Reichman hopes the new guidelines catch on quickly, particularly because IGRAs must be conducted in laboratory settings that help ensure better predictive results. He fears, however, that adherence to traditional methods like BCG vaccination, which is particularly popular in Europe, might stall widespread IGRA adoption.
“It will take time to get there,” he says. “The TB community is notoriously slow in adapting new technologies.”
What Leads to Lower-Quality Patient Care?
According to Evan Fieldston, MD, MBA, MSHP, the mismatches between a hospital staff’s workload and its workforce might predict periods of lower-quality care of patients. With a five-year research project in place, Dr. Fieldston is examining the impact of these mismatches on patient care at the Children’s Hospital of Philadelphia (CHOP), where he serves as an assistant professor in pediatrics. He is examining administrative data on approximately 40,500 retrospective cases and conducting more specific prospective validation on approximately 500 cases.
Part of his project is supported by SHM’s Junior Faculty Development Award, a two-year $50,000 grant awarded for the first time in April.
Dr. Fieldston explained to the TH eWire how he’s using the research funds.
Question: What have you done to organize the project?
Answer: I’ve put together an outstanding mentoring and advisory team to guide me through the design of these projects … the analysis and interpretation. I have also secured local support at the hospital and in the department of pediatrics. Now I’m starting to frame out the specifics and the logistics of each of the projects, and I’m preparing the applications for the institutional review board.
Q: How are you spending the grant?
A: The research grant is going to be spent primarily for two research assistants to work on data collection and validation. Frontline observations are important to patient care quality and patient flow work, so I am excited to have the funds to support that work. Other parts of funding are to support biostatistical programming and operations management expertise.
Q: How will you balance your time between research and hospital rounds?
A: Very fortunately, my faculty position here at the University of Pennsylvania and CHOP is primarily for research, so 75% of my time is dedicated to research purposes. As a hospitalist, I attend on the general pediatrics inpatient teaching service for about six to eight weeks a year. … On the weeks that I am on service, it’s a lot more challenging to do research work, but I still try to touch base with the various aspects of the project.
According to Evan Fieldston, MD, MBA, MSHP, the mismatches between a hospital staff’s workload and its workforce might predict periods of lower-quality care of patients. With a five-year research project in place, Dr. Fieldston is examining the impact of these mismatches on patient care at the Children’s Hospital of Philadelphia (CHOP), where he serves as an assistant professor in pediatrics. He is examining administrative data on approximately 40,500 retrospective cases and conducting more specific prospective validation on approximately 500 cases.
Part of his project is supported by SHM’s Junior Faculty Development Award, a two-year $50,000 grant awarded for the first time in April.
Dr. Fieldston explained to the TH eWire how he’s using the research funds.
Question: What have you done to organize the project?
Answer: I’ve put together an outstanding mentoring and advisory team to guide me through the design of these projects … the analysis and interpretation. I have also secured local support at the hospital and in the department of pediatrics. Now I’m starting to frame out the specifics and the logistics of each of the projects, and I’m preparing the applications for the institutional review board.
Q: How are you spending the grant?
A: The research grant is going to be spent primarily for two research assistants to work on data collection and validation. Frontline observations are important to patient care quality and patient flow work, so I am excited to have the funds to support that work. Other parts of funding are to support biostatistical programming and operations management expertise.
Q: How will you balance your time between research and hospital rounds?
A: Very fortunately, my faculty position here at the University of Pennsylvania and CHOP is primarily for research, so 75% of my time is dedicated to research purposes. As a hospitalist, I attend on the general pediatrics inpatient teaching service for about six to eight weeks a year. … On the weeks that I am on service, it’s a lot more challenging to do research work, but I still try to touch base with the various aspects of the project.
According to Evan Fieldston, MD, MBA, MSHP, the mismatches between a hospital staff’s workload and its workforce might predict periods of lower-quality care of patients. With a five-year research project in place, Dr. Fieldston is examining the impact of these mismatches on patient care at the Children’s Hospital of Philadelphia (CHOP), where he serves as an assistant professor in pediatrics. He is examining administrative data on approximately 40,500 retrospective cases and conducting more specific prospective validation on approximately 500 cases.
Part of his project is supported by SHM’s Junior Faculty Development Award, a two-year $50,000 grant awarded for the first time in April.
Dr. Fieldston explained to the TH eWire how he’s using the research funds.
Question: What have you done to organize the project?
Answer: I’ve put together an outstanding mentoring and advisory team to guide me through the design of these projects … the analysis and interpretation. I have also secured local support at the hospital and in the department of pediatrics. Now I’m starting to frame out the specifics and the logistics of each of the projects, and I’m preparing the applications for the institutional review board.
Q: How are you spending the grant?
A: The research grant is going to be spent primarily for two research assistants to work on data collection and validation. Frontline observations are important to patient care quality and patient flow work, so I am excited to have the funds to support that work. Other parts of funding are to support biostatistical programming and operations management expertise.
Q: How will you balance your time between research and hospital rounds?
A: Very fortunately, my faculty position here at the University of Pennsylvania and CHOP is primarily for research, so 75% of my time is dedicated to research purposes. As a hospitalist, I attend on the general pediatrics inpatient teaching service for about six to eight weeks a year. … On the weeks that I am on service, it’s a lot more challenging to do research work, but I still try to touch base with the various aspects of the project.
Managing Hyponatremia Patients With SIADH
Why is SIADH Important to Hospitalists?
Disorders of body fluids, and particularly hyponatremia, are among the most commonly encountered problems in clinical medicine, affecting up to 30% of hospitalized patients. In a study of 303,577 laboratory samples collected from 120,137 patients, the prevalence of hyponatremia (serum [Na+] 135 mmol/L) on initial presentation to a healthcare provider was 28.2% among those treated in an acute hospital care setting, 21% among those treated in an ambulatory hospital care setting, and 7.2% in community care centers.1 Numerous other studies have corroborated a high prevalence of hyponatremia in hospitalized patients,2 which reflects the increased vulnerability of this patient population to disruptions of body fluid homeostasis. Recognizing the many possible causes of hyponatremia in hospitalized patients and implementing appropriate treatment strategies therefore are critical steps toward optimizing care and improving outcomes in hospitalized patients with hyponatremia.
In addition to its frequency, hyponatremia is also important because it has been associated with worse clinical outcomes across the entire range of inpatient care, from the general hospital population to those treated in the intensive care unit (ICU). In a study of 4123 patients age 65 years or older who were admitted to a community hospital, 3.5% had clinically significant hyponatremia (serum [Na+] 130 mmol/L) at admission. Compared with nonhyponatremic patients, those with hyponatremia were twice as likely to die during their hospital stay (relative risk [RR], 1.95; P 0.05).3 In another study of 2188 patients admitted to a medical ICU over a 5‐year period, 13.7% had hyponatremia. The overall rate of in‐hospital mortality among all ICU patients was high at 37.7%. However, severe hyponatremia (serum [Na+] 125 mmol/L) more than doubled the risk of in‐hospital mortality (RR, 2.10; P 0.001).4 In addition to the general hospital population, in virtually every disease ever studied, the presence of hyponatremia has been found to be an independent risk factor for increased mortality, from congestive heart failure to tuberculosis to liver failure.2
What Causes Hyponatremia in Patients with SIADH?
Hyponatremia can be caused by 1 of 2 potential disruptions in fluid balance: dilution from retained water, or depletion from electrolyte losses in excess of water. Dilutional hyponatremias are associated with either a normal (euvolemic) or an increased (hypervolemic) extracellular fluid (ECF) volume, whereas depletional hyponatremias generally are associated with a decreased ECF volume (hypovolemic). Dilutional hyponatremia can arise from a primary defect in osmoregulation, such as in SIADH, or as a result of ECF volume expansion, as seen in conditions associated with concomitant secondary hyperaldosteronism such as heart failure, hepatic cirrhosis, or nephrotic syndrome. Among some hospitalized patient groups, euvolemic hyponatremia is the most common presentation of abnormally low serum [Na+]. In a study of patients who developed clinically significant postoperative hyponatremia (defined as a serum [Na+] 130 mmol/L) in a large teaching hospital, only 8% were hypovolemic, whereas 42% were euvolemic and 21% were hypervolemic.5
Euvolemic hyponatremia results from an increase in total body water, but with normal or near‐normal total body sodium. As a result, there is an absence of clinical manifestations of ECF volume expansion, such as subcutaneous edema or ascites. It is important to recognize that although SIADH clearly represents a state of volume expansion due to water retention, it rarely causes clinically recognizable hypervolemia since the retained water is distributed across the intracellular fluid (ICF) as well as the ECF, and because volume regulatory processes act to decrease the actual degree of ECF volume expansion.6 Euvolemic hyponatremia can accompany a wide variety of pathological processes, but the most common cause by far is SIADH. Normally, increased plasma osmolality activates osmoreceptors located in the anterior hypothalamus and stimulates the secretion of arginine vasopressin (AVP), also called antidiuretic hormone (ADH), a key neurohormone that regulates fluid homeostasis. In patients with euvolemic hyponatremia due to SIADH, plasma AVP levels are not suppressed despite normal or decreased plasma osmolality.7 This can be a result of ectopic production of AVP by tumors, or stimulation of endogenous pituitary AVP secretion as a result of nonosmotic stimuli that also stimulate vasopressinergic neurons, which include hypovolemia, hypotension, angiotensin II, nausea, hypoxia, hypercarbia, hypoglycemia, stress, and physical activity. Nonsuppressed AVP levels have been documented in the majority of hyponatremic patients, including those with SIADH8 and heart failure.9
SIADH can develop as the result of many different disease processes that disrupt the normal mechanisms that regulate AVP secretion, including pneumonias and other lung infections, thoracic and extrathoracic tumors, a variety of different central nervous system disorders, the postoperative state, human immunodeficiency virus (HIV), and many different drugs (Figure 1). Given the multiplicity of disorders and drugs that can cause disrupted AVP secretion, it is not surprising that hyponatremia is the most common electrolyte abnormality seen in clinical practice.

What Symptoms are Associated With SIADH?
Symptoms of hyponatremia correlate both with the degree of decrease in the serum [Na+] and with the chronicity of the hyponatremia. Acute hyponatremia, defined as 48 hours in duration, is often associated with life‐threatening clinical features such as obtundation, seizures, coma, and respiratory arrest. These symptoms can occur abruptly, sometimes with little warning.10 In the most severe cases, death can occur as a result of cerebral edema with tentorial herniation. Hypoxia secondary to neurogenic pulmonary edema can increase the severity of brain swelling.11
In contrast, chronic hyponatremia is much less symptomatic, and the reason for the profound differences between the symptoms of acute and chronic hyponatremia is now well understood to be due to the process of brain volume regulation.12 It is essential that this process be understood in order to understand the full spectrum of hyponatremic symptoms. As the ECF [Na+] decreases, regardless of whether due to a loss of sodium or a gain of water, there is an obligate movement of water into the brain along osmotic gradients. That water shift causes swelling of the brain, or cerebral edema. If the increased brain water reaches approximately 8% in adults, it exceeds the capacity of the skull to accommodate brain expansion, leading to tentorial herniation and death from respiratory arrest and/or ischemic brain damage. However, if the patient survives the initial hyponatremia, a very strong volume regulatory process follows, consisting of loss of electrolytes and small organic molecules called osmolytes from brain cells into brain ECF, and eventually the peripheral ECF.12, 13 As the solute content of the brain decreases, the water content is allowed to normalize, eventually reaching a state in which brain edema is virtually absent, and as a result symptoms are markedly less than with acute hyponatremia. Although the time required for the brain to acieve a volume‐regulated state varies across patients, this process is completed within 48 hours in experinmental animal studies, and probably follows a similar time course in humans.
Despite this powerful adaptation process, chronic hyponatremia is frequently associated with neurological symptomatology, albeit milder and more subtle in nature. A recent report found a fairly high incidence of symptoms in 223 patients with chronic hyponatremia as a result of thiazide administration: 49% had malaise/lethargy, 47% had dizzy spells, 35% had vomiting, 17% had confusion/obtundation, 17% experienced falls, 6% had headaches, and 0.9% had seizures.14 Although dizziness can potentially be attributed to a diuretic‐induced hypovolemia, symptoms such as confusion, obtundation and seizures are more consistent with hyponatremic symptomatology. Because thiazide‐induced hyponatremia can be readily corrected by stopping the thiazide and/or administering sodium, this represents an ideal situation in which to assess improvement in hyponatremia symptomatology with normalization of the serum [Na+]; in this study, all of these symptoms improved with correction of the hyponatremia. This represents one of the best examples demonstrating reversal of the symptoms associated with chronic hyponatremia by correction of the hyponatremia, because the patients in this study did not in general have severe underlying comorbidities that might complicate interpretation of their symptoms, as is often the case in patients with SIADH.
What Is Required for Making a Diagnosis of SIADH in Hospitalized Patients?
In patients with hypotonic hypoosmolality, ascertainment of their ECF volume status (ie, hypovolemic, euvolemic, or hypervolemic) is an essential first step, as this will segregate patients into different treatment paradigms. For example, in patients who are truly clinically hypovolemic with a decreased ECF volume by clinical parameters, treatment would generally consist of solute repletion with sodium, generally isotonic saline infusion with or without potassium, until the sodium levels normalize. In patients who are hypervolemic, treatment should focus first on the underlying disease rather than addressing the serum [Na+] directly. In patients with clinical euvolemia, the standard diagnostic pathway should be followed to confirm a diagnosis of SIADH as described below.
Assessing ECF volume status can be difficult, even for the most experienced clinicians. Physical signs such as orthostatic decreases in blood pressure and increases in pulse rate, dry mucus membranes, and skin tenting indicate hypovolemic hyponatremia, while signs such as subcutaneous edema, ascites, or anasarca indicate hypervolemic hyponatremia. Patients without any of these findings are generally considered to be euvolemic. However, in any situation these signs are only applicable if there are no other reasons to suspect an altered ECF volume. Along with a complete history and physical examination that includes a careful neurological evaluation, several laboratory tests can help to assess the etiology of the hyponatremia, once serum sodium concentrations have been shown to be below normal ([Na+] 135 mmol/L):
-
Urine osmolality. A urine osmolality (Uosm) less than 100 mOsm/kg H2O can indicate low dietary solute intake, primary polydipsia, or a reset osmostat after suppression of AVP release by a decrease in plasma osmolality below the osmotic threshold for AVP secretion, usually as a result of increased water loading.
-
Urine sodium concentration. Excretion of sodium, as measured by a spot urine [Na+] (UNa), can indicate depletional hyponatremia if the concentration is less than 30 mmol/L.15 A low UNa reflects a volume depleted state unless the patient has secondary hyperaldosteronism from heart failure or cirrhosis. Patients with a low UNa are more likely to respond to isotonic saline. Euvolemic patients who have a normal dietary sodium intake will generally have spot UNa 30 mmol/L and will not benefit from isotonic saline administration.15 In fact, in SIADH, these patients may respond to isotonic saline with a worsening of hyponatremia, since the sodium from the isotonic saline will be excreted in a concentrated urine while the free water is reabsorbed in the kidney collecting ducts. If the patient is on diuretic therapy, urine sodium values cannot always be accurately interpreted, since a UNa 30 mmol/L may reflect the natriuretic effect of the diuretic and not a volume replete state.
-
Blood tests. Additional indicators of volume status include serum blood nitrogen (BUN) and uric acid levels. A BUN 10 mg/dL and uric acid 4 mg/dL are generally consistent with a euvolemic state, particularly when there is glomerular hyperfiltration, which is often present in SIADH. Elevated serum BUN and uric acid levels (BUN >20 mg/dL and uric acid >6 mg/dL), especially if prior values are available for comparison, can also help to establish whether ineffective vascular volume status may be contributing to the pathophysiology of the hyponatremia. In certain clinical scenarios, the B‐type natriuretic protein (BNP) can be helpful to support a clinical impression of congestive heart failure.
The criteria necessary for a diagnosis of SIADH remain essentially as defined by Bartter and Schwartz16 in 1967 (Table 1), but several points deserve emphasis.17 First, true ECF hypoosmolality must be present and hyponatremia secondary to pseudohyponatremia or hyperglycemia excluded. Second, urinary osmolality must be inappropriate for plasma hypoosmolality (Posm). This does not require a Uosm>Posm, but simply that the urine osmolality is greater than maximally dilute (ie, Uosm>100 mOsm/kg H2O in adults). Furthermore, urine osmolality need not be inappropriately elevated at all levels of Posm but simply at some level under 275 mOsm/kg H2O, since in patients with a reset osmostat, AVP secretion can be suppressed at some level of osmolality resulting in maximal urinary dilution and free water excretion at plasma osmolalities below this level.18 Although some consider a reset osmostat to be a separate disorder rather than a variant of SIADH, such cases nonetheless illustrate that some hypoosmolar patients can exhibit an appropriately dilute urine at some, though not all, plasma osmolalities. Third, clinical euvolemia must be present to diagnose SIADH, and this diagnosis cannot be made in a hypovolemic or edematous patient. Importantly, this does not mean that patients with SIADH cannot become hypovolemic for other reasons, but in such cases it is impossible to diagnose the underlying SIADH until the patient is rendered euvolemic. The fourth criterion, renal salt wasting, has probably caused the most confusion in the diagnosis of SIADH. As noted above, the importance of this criterion lies in its usefulness in differentiating hypoosmolality caused by a decreased effective intravascular volume with high aldosterone levels in which case renal Na+ conservation occurs, from dilutional disorders in which urine Na+ excretion is normal or increased due to ECF volume expansion and a suppressed renin‐angiotensin‐aldosterone system. However, UNa can also be high in renal causes of solute depletion such as diuretic use or Addison's disease, and conversely patients with SIADH can have a low UNa if they subsequently become hypovolemic or solute depleted, conditions sometimes produced by imposed salt and water restriction. Consequently, although high urinary Na+ excretion is generally the rule in most patients with SIADH, its presence does not necessarily confirm this diagnosis, nor does its absence rule out the diagnosis. The final criterion emphasizes that SIADH remains a diagnosis of exclusion, and the absence of other potential causes of hypoosmolality must always be verified. Glucocorticoid deficiency and SIADH can be especially difficult to distinguish, since both primary and secondary hypocortisolism can cause elevated plasma AVP levels in addition to direct renal effects that prevent maximal urinary dilution.19 Therefore, no patient with chronic hyponatremia should be diagnosed as having SIADH without a thorough evaluation of adrenal function, preferably via a rapid adrenocorticotropic hormone (ACTH) stimulation test. Acute hyponatremia of obvious etiology, such as postoperatively or in association with pneumonitis, may be treated without adrenal testing as long as there are no other clinical signs or symptoms suggestive of adrenal dysfunction.20
|
| Essential |
| Decreased effective osmolality of the extracellular fluid (Posm 275 mOsm/kg H2O). |
| Inappropriate urinary concentration (Uosm >100 mOsm/kg H2O with normal renal function) at some level of hypoosmolality. |
| Clinical euvolemia, as defined by the absence of signs of hypovolemia (orthostasis, tachycardia, decreased skin turgor, dry mucous membranes) or hypervolemia (subcutaneous edema, ascites). |
| Elevated urinary sodium excretion while on a normal salt and water intake. |
| Absence of other potential causes of euvolemic hypoosmolality: hypothyroidism, hypocortisolism (Addison's disease or secondary adrenal insufficiency) and diuretic use. |
| Supplemental |
| Abnormal water load test (inability to excrete at least 90% of a 20 mL/kg water load in 4 hours and/or failure to dilute Uosm to 100 mOsm/kg H2O). |
| Plasma AVP level inappropriately elevated relative to plasma osmolality. |
| No significant correction of serum [Na+] with volume expansion but improvement after fluid restriction. |
Hyponatremia is a particularly common complication in elderly hospitalized patients, increasing in prevalence from approximately 7% in the general older population to 18% to 22% among elderly patients in chronic care facilities.21 Despite the many known causes of SIADH (Figure 1), hyponatremia is often associated with idiopathic SIADH in the elderly population. In a study of 119 nursing home residents aged 60 to 103 years, 53% had at least 1 episode of hyponatremia during the previous 12 months.22 Of these patients, 26% were diagnosed with idiopathic SIADH. In another study of elderly patients with hyponatremia and SIADH, 60% were diagnosed with idiopathic SIADH. Among remaining patients, the 2 main causes identified were pneumonia (9 cases/18%) and medications (6 cases/12%).23 Therefore, more than half of elderly patients who present with hyponatremia due to SIADH may have an idiopathic form, with no detectable underlying treatable disease.
Which Hospital Patients With SIADH are Candidates for Treatment of Hyponatremia?
Correction of hyponatremia is associated with markedly improved neurological outcomes in patients with severely symptomatic hyponatremia. In a retrospective review of patients who presented with severe neurological symptoms and serum [Na+] 125 mmol/L, prompt therapy with isotonic or hypertonic saline resulted in a correction in the range of 20 mmol/L over several days and neurological recovery in almost all cases. In contrast, in patients who were treated with fluid restriction alone, there was very little correction over the study period (5 mmol/L over 72 hours), and the neurological outcomes were much worse, with most of these patients either dying or entering a persistently vegetative state.24 Consequently, prompt therapy to rapidly increase the serum [Na+] represents the standard‐of‐care for treatment of patients presenting with severe life‐threatening symptoms of hyponatremia.
As discussed earlier, chronic hyponatremia is much less symptomatic as a result of the process of brain volume regulation. Because of this adaptation process, chronic hyponatremia is arguably a condition that clinicians feel they may not need to be as concerned about, and in some publications this has been called asymptomatic hyponatremia. However, such patients often do have neurological symptoms, even if milder and more subtle in nature, including headaches, nausea, mood disturbances, depression, difficulty concentrating, slowed reaction times, unstable gait, increased falls, confusion, and disorientation. Consequently, any patient with hyponatremia secondary to SIADH who manifests any neurological symptoms that could be related to the hyponatremia should be considered as appropriate candidates for treatment of the hyponatremia, regardless of the chronicity of the hyponatremia or the level of serum [Na+].
What Therapies are Currently Available to Manage SIADH in Hospitalized Patients?
Conventional management strategies for euvolemic hyponatremia range from saline infusion and fluid restriction to pharmacologic adjustment of fluid balance. Consideration of treatment options should include an evaluation of the benefits as well as the potential toxicities of any therapy (Table 2). Sometimes, simply stopping treatment with an agent that is associated with hyponatremia is sufficient to reverse a low serum [Na+].
| Therapy | Targets Underlying Pathophysiology | Limitations |
|---|---|---|
| ||
| Isotonic saline | Ineffective in dilutional hyponatremias; exacerbates the volume overload if used in edema‐forming disorders; no controlled safety database. | |
| Hypertonic saline | No consensus regarding appropriate infusion rates; overcorrection can cause osmotic demyelination; exacerbates the volume overload if used in edema‐forming disorders; no controlled safety database. | |
| Fluid restriction | Slow to correct over many days; poorly tolerated due to thirst; can not be used effectively in patients with high AVP levels and urine osmolalities. | |
| Demeclocycline | ✓ | Not FDA approved for hyponatremia; slow to correct; nephrotoxic in cirrhosis and heart failure. |
| Mineralocorticoids | Only one report in elderly patients with SIADH; no safety database; exacerbates the volume overload if used in edema‐forming disorders. | |
| Urea | Not FDA‐approved for hyponatremia; poor palatability. | |
| AVP receptor antagonists (vaptans) | ✓ | Conivaptan approved only for in‐hospital use secondary to CYP3A4 inhibition; infusion‐site reactions with intravenous use. Tolvaptan must be initiated and reinitiated in the hospital, as serum sodium needs to be monitored closely to avoid overly rapid correction of hyponatremia. |
Isotonic Saline
The treatment of choice for depletional hyponatremia (ie, hypovolemic hyponatremia) is isotonic saline ([Na+] = 154 mmol/L) to restore ECF volume and ensure adequate organ perfusion. This initial therapy is appropriate for patients who either have clinical signs of hypovolemia, or in whom a spot UNa+ is 30 mmol/L. However, this therapy is ineffective for dilutional hyponatremias such as SIADH,25 and continued inappropriate administration of isotonic saline to a euvolemic patient may worsen their hyponatremia,26 and/or cause fluid overload. Although isotonic saline may improve the serum [Na+] in patients with hypervolemic hyponatremia, their volume status will generally worsen with this therapy, so unless the hyponatremia is profound isotonic saline should be avoided.
Hypertonic Saline
Acute hyponatremia presenting with severe neurological symptoms is life‐threatening, and should be treated promptly with hypertonic solutions, typically 3% NaCl ([Na+] = 513 mmol/L), as this represents the most reliable method to quickly raise the serum [Na+]. A continuous infusion of hypertonic NaCl is usually utilized in inpatient settings. Various formulae have been suggested for calculating the initial rate of infusion of hypertonic solutions,27 but perhaps the simplest utilizes the following relationship:
- .Age and gender as risk factors for hyponatremia and hypernatremia.Clin Chim Acta.2003;337(1‐2):169–172.
- ,,.Incidence and prevalence of hyponatremia.Am J Med.2006;119(7 Suppl 1):S30–S35.
- ,,.Admission hyponatremia in the elderly: factors influencing prognosis.J Gen Intern Med1994;9:89–91.
- ,,, et al.[Incidence, causes and prognostic factors of hyponatremia in intensive care].Rev Med Interne.2003;24(4):224–229.
- ,,,.Postoperative hyponatremia. A prospective study.Arch Int Med.1986;146:333–336.
- .Whole‐body volume regulation and escape from antidiuresis.Am J Med.2006;119(7 Suppl 1):S21–S29.
- ,,.Neurogenic disorders of osmoregulation.Am J Med.1982;72:339–353.
- ,,.Vasopressin function in the syndrome of inappropriate antidiuresis.Annu Rev Med.1980;31:315–327.
- ,,,,,.Radioimmunoassay of plasma arginine vasopressin in hyponatremic patients with congestive heart failure.N Eng J Med.1981;305:263–266.
- ,.Epidemiology, pathophysiology, and management of hyponatremic encephalopathy.Am J Med.1997;102:67–77.
- ,.Pulmonary complications of hyponatremic encephalopathy. noncardiogenic pulmonary edema and hypercapnic respiratory failure [see comments].Chest.1995;107(2):517–521.
- ,.Control of brain volume during hyperosmolar and hypoosmolar conditions.Annu Rev Med.1993;44:289–301.
- .Control of brain volume during hypoosmolality and hyperosmolality.Adv Exp Med Biol.2006;576:113–129.
- ,,.Clinical studies of thiazide‐induced hyponatremia.J Natl Med Assoc.2004;96(10):1305–1308.
- ,,,.Clinical assessment of extracellular fluid volume in hyponatremia.Am J Med.1987;83:905–908.
- ,.The syndrome of inappropriate secretion of antidiuretic hormone.Am J Med.1967;42:790–806.
- .Hyponatremia and Hypo‐osmolar Disorders. In: Greenberg A, Cheung AK, Coffman TM, Falk RJ, Jennette JC, eds.Primer on Kidney Diseases.Philadelphia:Saunders Elsevier,2009:52–59.
- ,,,.Reset of osmoreceptors in association with normovolemic hyponatremia.Am J Med Sci.1974;267:267–273.
- .Hyponatremia and inappropriate secretion of vasopressin (antidiuretic hormone) in patients with hypopituitarism.N Eng J Med.1989;321:492–496.
- .The Syndrome of Inappropriate Antidiuretic Hormone Secretion and Other Hypoosmolar Disorders. In: Schrier RW, ed.Diseases of the Kidney and Urinary Tract.Philadelphia:Lippincott Williams 27:156–161.
- ,,.Hyponatremia in a nursing home population.J Am Geriatr Soc.1995;43(12):1410–1413.
- ,.The syndrome of inappropriate antidiuretic hormone secretion in the elderly.Am J Med.1997;103(4):270–273.
- .Diuretic‐induced hyponatremia [editorial].Arch Intern Med.1986;146(7):1295–1296.
- ,,,.A syndrome of renal sodium loss and hyponatremia probably resulting from inappropriate secretion of antidiuretic hormone.Am J Med.1957;23:529–542.
- ,,,,,.Postoperative hyponatremia despite near‐isotonic saline infusion: a phenomenon of desalination [see comments].Ann Intern Med.1997;126(1):20–25.
- ,.Hyponatremia.N Engl J Med.2000;342(21):1581–1589.
- ,,, et al.Statement of the Second International Exercise‐Associated Hyponatremia Consensus Development Conference, New Zealand, 2007.Clin J Sport Med.2008;18(2):111–121.
- ,,,.Neurologic sequelae after treatment of severe hyponatremia: a multicenter perspective.J Am Soc Nephrol.1994;4:1522–1530.
- ,,,.Randomized, controlled trial on the effect of a 20% mannitol solution and a 7.5% saline/6% dextran solution on increased intracranial pressure after brain injury.Crit Care Med.2005;33(1):196–202.
- ,,.Osmotic demyelination syndrome following correction of hyponatremia.N Engl J Med.1986;314:1535–1542.
- .Regulation of arginine vasopressin in the syndrome of inappropriate antidiuresis.Am J Med.2006;119(7 Suppl 1):S36–S42.
- .The syndrome of inappropriate secretion of antidiuretic hormone (SIADH).Semin Nephrol.2009;29(3):239–256.
- .Impact of solute intake on urine flow and water excretion.J Am Soc Nephrol.2008;19(6):1076–1078.
- ,.Demeclocycline‐induced nephrogenic diabetes insipidus. In‐vivo and in‐ vitro studies.Ann Intern Med.1973;79(5):679–683.
- ,,, et al.Involvement of arginine vasopressin and renal sodium handling in pathogenesis of hyponatremia in elderly patients.Endocr J.1996;43(1):101–108.
- ,.Urea for long‐term treatment of syndrome of inappropriate secretion of antidiuretic hormone.Br Med J (Clin Res Ed).1981;283:1081–1083.
- ,.Vasopressin receptor antagonists.Kidney Int.2006;69(12):2124–2130.
- ,,, et al.Potent aquaretic agent. A novel nonpeptide selective vasopressin 2 antagonist (OPC‐31260) in men.J Clin Invest.1993;92(6):2653–2659.
- ,,,,.Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia.Am J Nephrol.2007;27(5):447–457.
- ,,, et al.Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355(20):2099–2112.
- Vaprisol (conivaptan hydrochloride injection) prescribing information.Deerfield, IL:Astellas Pharma US, Inc.,2006.
- ,,,,.Hyponatremia treatment guidelines 2007: expert panel recommendations.Am J Med.2007;120(11 Suppl 1):S1–S21.
- Otsuka Pharmaceutical Co L, Tokyo J. Samsca (tolvaptan) prescribing information.2009.
- ,.Clinical practice. The syndrome of inappropriate antidiuresis.N Engl J Med.2007;356(20):2064–2072.
- ,,.The treatment of hyponatremia.Semin Nephrol.2009;29(3):282–299.
- .Hyponatremia and Hypo‐osmolar Disorders. In: Greenberg A, Cheung AK, Coffman TM, Falk RJ, Jennette JC, eds.Primer on Kidney Diseases.Philadelphia. PA:Saunders Elsevier;2009:52–59.
- ,,,.Risk factors for symptomatic hyponatraemia: the role of pre‐existing asymptomatic hyponatraemia.Intern Med J.2007;37(3):149–155.
- ,,,,.Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits.Am J Med.2006;119(1):71.
- ,,,,.Mild hyponatremia and risk of fracture in the ambulatory elderly.QJM.2008;101(7):583–588.
- ,,,,.Hyponatremia associated with large‐bone fracture in elderly patients.Int Urol Nephrol.2009;41(3):733–737.
- ,,, et al.Hyponatremia‐induced osteoporosis.J Bone Miner Res.2010;25(3):554–563.
Why is SIADH Important to Hospitalists?
Disorders of body fluids, and particularly hyponatremia, are among the most commonly encountered problems in clinical medicine, affecting up to 30% of hospitalized patients. In a study of 303,577 laboratory samples collected from 120,137 patients, the prevalence of hyponatremia (serum [Na+] 135 mmol/L) on initial presentation to a healthcare provider was 28.2% among those treated in an acute hospital care setting, 21% among those treated in an ambulatory hospital care setting, and 7.2% in community care centers.1 Numerous other studies have corroborated a high prevalence of hyponatremia in hospitalized patients,2 which reflects the increased vulnerability of this patient population to disruptions of body fluid homeostasis. Recognizing the many possible causes of hyponatremia in hospitalized patients and implementing appropriate treatment strategies therefore are critical steps toward optimizing care and improving outcomes in hospitalized patients with hyponatremia.
In addition to its frequency, hyponatremia is also important because it has been associated with worse clinical outcomes across the entire range of inpatient care, from the general hospital population to those treated in the intensive care unit (ICU). In a study of 4123 patients age 65 years or older who were admitted to a community hospital, 3.5% had clinically significant hyponatremia (serum [Na+] 130 mmol/L) at admission. Compared with nonhyponatremic patients, those with hyponatremia were twice as likely to die during their hospital stay (relative risk [RR], 1.95; P 0.05).3 In another study of 2188 patients admitted to a medical ICU over a 5‐year period, 13.7% had hyponatremia. The overall rate of in‐hospital mortality among all ICU patients was high at 37.7%. However, severe hyponatremia (serum [Na+] 125 mmol/L) more than doubled the risk of in‐hospital mortality (RR, 2.10; P 0.001).4 In addition to the general hospital population, in virtually every disease ever studied, the presence of hyponatremia has been found to be an independent risk factor for increased mortality, from congestive heart failure to tuberculosis to liver failure.2
What Causes Hyponatremia in Patients with SIADH?
Hyponatremia can be caused by 1 of 2 potential disruptions in fluid balance: dilution from retained water, or depletion from electrolyte losses in excess of water. Dilutional hyponatremias are associated with either a normal (euvolemic) or an increased (hypervolemic) extracellular fluid (ECF) volume, whereas depletional hyponatremias generally are associated with a decreased ECF volume (hypovolemic). Dilutional hyponatremia can arise from a primary defect in osmoregulation, such as in SIADH, or as a result of ECF volume expansion, as seen in conditions associated with concomitant secondary hyperaldosteronism such as heart failure, hepatic cirrhosis, or nephrotic syndrome. Among some hospitalized patient groups, euvolemic hyponatremia is the most common presentation of abnormally low serum [Na+]. In a study of patients who developed clinically significant postoperative hyponatremia (defined as a serum [Na+] 130 mmol/L) in a large teaching hospital, only 8% were hypovolemic, whereas 42% were euvolemic and 21% were hypervolemic.5
Euvolemic hyponatremia results from an increase in total body water, but with normal or near‐normal total body sodium. As a result, there is an absence of clinical manifestations of ECF volume expansion, such as subcutaneous edema or ascites. It is important to recognize that although SIADH clearly represents a state of volume expansion due to water retention, it rarely causes clinically recognizable hypervolemia since the retained water is distributed across the intracellular fluid (ICF) as well as the ECF, and because volume regulatory processes act to decrease the actual degree of ECF volume expansion.6 Euvolemic hyponatremia can accompany a wide variety of pathological processes, but the most common cause by far is SIADH. Normally, increased plasma osmolality activates osmoreceptors located in the anterior hypothalamus and stimulates the secretion of arginine vasopressin (AVP), also called antidiuretic hormone (ADH), a key neurohormone that regulates fluid homeostasis. In patients with euvolemic hyponatremia due to SIADH, plasma AVP levels are not suppressed despite normal or decreased plasma osmolality.7 This can be a result of ectopic production of AVP by tumors, or stimulation of endogenous pituitary AVP secretion as a result of nonosmotic stimuli that also stimulate vasopressinergic neurons, which include hypovolemia, hypotension, angiotensin II, nausea, hypoxia, hypercarbia, hypoglycemia, stress, and physical activity. Nonsuppressed AVP levels have been documented in the majority of hyponatremic patients, including those with SIADH8 and heart failure.9
SIADH can develop as the result of many different disease processes that disrupt the normal mechanisms that regulate AVP secretion, including pneumonias and other lung infections, thoracic and extrathoracic tumors, a variety of different central nervous system disorders, the postoperative state, human immunodeficiency virus (HIV), and many different drugs (Figure 1). Given the multiplicity of disorders and drugs that can cause disrupted AVP secretion, it is not surprising that hyponatremia is the most common electrolyte abnormality seen in clinical practice.
What Symptoms are Associated With SIADH?
Symptoms of hyponatremia correlate both with the degree of decrease in the serum [Na+] and with the chronicity of the hyponatremia. Acute hyponatremia, defined as 48 hours in duration, is often associated with life‐threatening clinical features such as obtundation, seizures, coma, and respiratory arrest. These symptoms can occur abruptly, sometimes with little warning.10 In the most severe cases, death can occur as a result of cerebral edema with tentorial herniation. Hypoxia secondary to neurogenic pulmonary edema can increase the severity of brain swelling.11
In contrast, chronic hyponatremia is much less symptomatic, and the reason for the profound differences between the symptoms of acute and chronic hyponatremia is now well understood to be due to the process of brain volume regulation.12 It is essential that this process be understood in order to understand the full spectrum of hyponatremic symptoms. As the ECF [Na+] decreases, regardless of whether due to a loss of sodium or a gain of water, there is an obligate movement of water into the brain along osmotic gradients. That water shift causes swelling of the brain, or cerebral edema. If the increased brain water reaches approximately 8% in adults, it exceeds the capacity of the skull to accommodate brain expansion, leading to tentorial herniation and death from respiratory arrest and/or ischemic brain damage. However, if the patient survives the initial hyponatremia, a very strong volume regulatory process follows, consisting of loss of electrolytes and small organic molecules called osmolytes from brain cells into brain ECF, and eventually the peripheral ECF.12, 13 As the solute content of the brain decreases, the water content is allowed to normalize, eventually reaching a state in which brain edema is virtually absent, and as a result symptoms are markedly less than with acute hyponatremia. Although the time required for the brain to acieve a volume‐regulated state varies across patients, this process is completed within 48 hours in experinmental animal studies, and probably follows a similar time course in humans.
Despite this powerful adaptation process, chronic hyponatremia is frequently associated with neurological symptomatology, albeit milder and more subtle in nature. A recent report found a fairly high incidence of symptoms in 223 patients with chronic hyponatremia as a result of thiazide administration: 49% had malaise/lethargy, 47% had dizzy spells, 35% had vomiting, 17% had confusion/obtundation, 17% experienced falls, 6% had headaches, and 0.9% had seizures.14 Although dizziness can potentially be attributed to a diuretic‐induced hypovolemia, symptoms such as confusion, obtundation and seizures are more consistent with hyponatremic symptomatology. Because thiazide‐induced hyponatremia can be readily corrected by stopping the thiazide and/or administering sodium, this represents an ideal situation in which to assess improvement in hyponatremia symptomatology with normalization of the serum [Na+]; in this study, all of these symptoms improved with correction of the hyponatremia. This represents one of the best examples demonstrating reversal of the symptoms associated with chronic hyponatremia by correction of the hyponatremia, because the patients in this study did not in general have severe underlying comorbidities that might complicate interpretation of their symptoms, as is often the case in patients with SIADH.
What Is Required for Making a Diagnosis of SIADH in Hospitalized Patients?
In patients with hypotonic hypoosmolality, ascertainment of their ECF volume status (ie, hypovolemic, euvolemic, or hypervolemic) is an essential first step, as this will segregate patients into different treatment paradigms. For example, in patients who are truly clinically hypovolemic with a decreased ECF volume by clinical parameters, treatment would generally consist of solute repletion with sodium, generally isotonic saline infusion with or without potassium, until the sodium levels normalize. In patients who are hypervolemic, treatment should focus first on the underlying disease rather than addressing the serum [Na+] directly. In patients with clinical euvolemia, the standard diagnostic pathway should be followed to confirm a diagnosis of SIADH as described below.
Assessing ECF volume status can be difficult, even for the most experienced clinicians. Physical signs such as orthostatic decreases in blood pressure and increases in pulse rate, dry mucus membranes, and skin tenting indicate hypovolemic hyponatremia, while signs such as subcutaneous edema, ascites, or anasarca indicate hypervolemic hyponatremia. Patients without any of these findings are generally considered to be euvolemic. However, in any situation these signs are only applicable if there are no other reasons to suspect an altered ECF volume. Along with a complete history and physical examination that includes a careful neurological evaluation, several laboratory tests can help to assess the etiology of the hyponatremia, once serum sodium concentrations have been shown to be below normal ([Na+] 135 mmol/L):
-
Urine osmolality. A urine osmolality (Uosm) less than 100 mOsm/kg H2O can indicate low dietary solute intake, primary polydipsia, or a reset osmostat after suppression of AVP release by a decrease in plasma osmolality below the osmotic threshold for AVP secretion, usually as a result of increased water loading.
-
Urine sodium concentration. Excretion of sodium, as measured by a spot urine [Na+] (UNa), can indicate depletional hyponatremia if the concentration is less than 30 mmol/L.15 A low UNa reflects a volume depleted state unless the patient has secondary hyperaldosteronism from heart failure or cirrhosis. Patients with a low UNa are more likely to respond to isotonic saline. Euvolemic patients who have a normal dietary sodium intake will generally have spot UNa 30 mmol/L and will not benefit from isotonic saline administration.15 In fact, in SIADH, these patients may respond to isotonic saline with a worsening of hyponatremia, since the sodium from the isotonic saline will be excreted in a concentrated urine while the free water is reabsorbed in the kidney collecting ducts. If the patient is on diuretic therapy, urine sodium values cannot always be accurately interpreted, since a UNa 30 mmol/L may reflect the natriuretic effect of the diuretic and not a volume replete state.
-
Blood tests. Additional indicators of volume status include serum blood nitrogen (BUN) and uric acid levels. A BUN 10 mg/dL and uric acid 4 mg/dL are generally consistent with a euvolemic state, particularly when there is glomerular hyperfiltration, which is often present in SIADH. Elevated serum BUN and uric acid levels (BUN >20 mg/dL and uric acid >6 mg/dL), especially if prior values are available for comparison, can also help to establish whether ineffective vascular volume status may be contributing to the pathophysiology of the hyponatremia. In certain clinical scenarios, the B‐type natriuretic protein (BNP) can be helpful to support a clinical impression of congestive heart failure.
The criteria necessary for a diagnosis of SIADH remain essentially as defined by Bartter and Schwartz16 in 1967 (Table 1), but several points deserve emphasis.17 First, true ECF hypoosmolality must be present and hyponatremia secondary to pseudohyponatremia or hyperglycemia excluded. Second, urinary osmolality must be inappropriate for plasma hypoosmolality (Posm). This does not require a Uosm>Posm, but simply that the urine osmolality is greater than maximally dilute (ie, Uosm>100 mOsm/kg H2O in adults). Furthermore, urine osmolality need not be inappropriately elevated at all levels of Posm but simply at some level under 275 mOsm/kg H2O, since in patients with a reset osmostat, AVP secretion can be suppressed at some level of osmolality resulting in maximal urinary dilution and free water excretion at plasma osmolalities below this level.18 Although some consider a reset osmostat to be a separate disorder rather than a variant of SIADH, such cases nonetheless illustrate that some hypoosmolar patients can exhibit an appropriately dilute urine at some, though not all, plasma osmolalities. Third, clinical euvolemia must be present to diagnose SIADH, and this diagnosis cannot be made in a hypovolemic or edematous patient. Importantly, this does not mean that patients with SIADH cannot become hypovolemic for other reasons, but in such cases it is impossible to diagnose the underlying SIADH until the patient is rendered euvolemic. The fourth criterion, renal salt wasting, has probably caused the most confusion in the diagnosis of SIADH. As noted above, the importance of this criterion lies in its usefulness in differentiating hypoosmolality caused by a decreased effective intravascular volume with high aldosterone levels in which case renal Na+ conservation occurs, from dilutional disorders in which urine Na+ excretion is normal or increased due to ECF volume expansion and a suppressed renin‐angiotensin‐aldosterone system. However, UNa can also be high in renal causes of solute depletion such as diuretic use or Addison's disease, and conversely patients with SIADH can have a low UNa if they subsequently become hypovolemic or solute depleted, conditions sometimes produced by imposed salt and water restriction. Consequently, although high urinary Na+ excretion is generally the rule in most patients with SIADH, its presence does not necessarily confirm this diagnosis, nor does its absence rule out the diagnosis. The final criterion emphasizes that SIADH remains a diagnosis of exclusion, and the absence of other potential causes of hypoosmolality must always be verified. Glucocorticoid deficiency and SIADH can be especially difficult to distinguish, since both primary and secondary hypocortisolism can cause elevated plasma AVP levels in addition to direct renal effects that prevent maximal urinary dilution.19 Therefore, no patient with chronic hyponatremia should be diagnosed as having SIADH without a thorough evaluation of adrenal function, preferably via a rapid adrenocorticotropic hormone (ACTH) stimulation test. Acute hyponatremia of obvious etiology, such as postoperatively or in association with pneumonitis, may be treated without adrenal testing as long as there are no other clinical signs or symptoms suggestive of adrenal dysfunction.20
|
| Essential |
| Decreased effective osmolality of the extracellular fluid (Posm 275 mOsm/kg H2O). |
| Inappropriate urinary concentration (Uosm >100 mOsm/kg H2O with normal renal function) at some level of hypoosmolality. |
| Clinical euvolemia, as defined by the absence of signs of hypovolemia (orthostasis, tachycardia, decreased skin turgor, dry mucous membranes) or hypervolemia (subcutaneous edema, ascites). |
| Elevated urinary sodium excretion while on a normal salt and water intake. |
| Absence of other potential causes of euvolemic hypoosmolality: hypothyroidism, hypocortisolism (Addison's disease or secondary adrenal insufficiency) and diuretic use. |
| Supplemental |
| Abnormal water load test (inability to excrete at least 90% of a 20 mL/kg water load in 4 hours and/or failure to dilute Uosm to 100 mOsm/kg H2O). |
| Plasma AVP level inappropriately elevated relative to plasma osmolality. |
| No significant correction of serum [Na+] with volume expansion but improvement after fluid restriction. |
Hyponatremia is a particularly common complication in elderly hospitalized patients, increasing in prevalence from approximately 7% in the general older population to 18% to 22% among elderly patients in chronic care facilities.21 Despite the many known causes of SIADH (Figure 1), hyponatremia is often associated with idiopathic SIADH in the elderly population. In a study of 119 nursing home residents aged 60 to 103 years, 53% had at least 1 episode of hyponatremia during the previous 12 months.22 Of these patients, 26% were diagnosed with idiopathic SIADH. In another study of elderly patients with hyponatremia and SIADH, 60% were diagnosed with idiopathic SIADH. Among remaining patients, the 2 main causes identified were pneumonia (9 cases/18%) and medications (6 cases/12%).23 Therefore, more than half of elderly patients who present with hyponatremia due to SIADH may have an idiopathic form, with no detectable underlying treatable disease.
Which Hospital Patients With SIADH are Candidates for Treatment of Hyponatremia?
Correction of hyponatremia is associated with markedly improved neurological outcomes in patients with severely symptomatic hyponatremia. In a retrospective review of patients who presented with severe neurological symptoms and serum [Na+] 125 mmol/L, prompt therapy with isotonic or hypertonic saline resulted in a correction in the range of 20 mmol/L over several days and neurological recovery in almost all cases. In contrast, in patients who were treated with fluid restriction alone, there was very little correction over the study period (5 mmol/L over 72 hours), and the neurological outcomes were much worse, with most of these patients either dying or entering a persistently vegetative state.24 Consequently, prompt therapy to rapidly increase the serum [Na+] represents the standard‐of‐care for treatment of patients presenting with severe life‐threatening symptoms of hyponatremia.
As discussed earlier, chronic hyponatremia is much less symptomatic as a result of the process of brain volume regulation. Because of this adaptation process, chronic hyponatremia is arguably a condition that clinicians feel they may not need to be as concerned about, and in some publications this has been called asymptomatic hyponatremia. However, such patients often do have neurological symptoms, even if milder and more subtle in nature, including headaches, nausea, mood disturbances, depression, difficulty concentrating, slowed reaction times, unstable gait, increased falls, confusion, and disorientation. Consequently, any patient with hyponatremia secondary to SIADH who manifests any neurological symptoms that could be related to the hyponatremia should be considered as appropriate candidates for treatment of the hyponatremia, regardless of the chronicity of the hyponatremia or the level of serum [Na+].
What Therapies are Currently Available to Manage SIADH in Hospitalized Patients?
Conventional management strategies for euvolemic hyponatremia range from saline infusion and fluid restriction to pharmacologic adjustment of fluid balance. Consideration of treatment options should include an evaluation of the benefits as well as the potential toxicities of any therapy (Table 2). Sometimes, simply stopping treatment with an agent that is associated with hyponatremia is sufficient to reverse a low serum [Na+].
| Therapy | Targets Underlying Pathophysiology | Limitations |
|---|---|---|
| ||
| Isotonic saline | Ineffective in dilutional hyponatremias; exacerbates the volume overload if used in edema‐forming disorders; no controlled safety database. | |
| Hypertonic saline | No consensus regarding appropriate infusion rates; overcorrection can cause osmotic demyelination; exacerbates the volume overload if used in edema‐forming disorders; no controlled safety database. | |
| Fluid restriction | Slow to correct over many days; poorly tolerated due to thirst; can not be used effectively in patients with high AVP levels and urine osmolalities. | |
| Demeclocycline | ✓ | Not FDA approved for hyponatremia; slow to correct; nephrotoxic in cirrhosis and heart failure. |
| Mineralocorticoids | Only one report in elderly patients with SIADH; no safety database; exacerbates the volume overload if used in edema‐forming disorders. | |
| Urea | Not FDA‐approved for hyponatremia; poor palatability. | |
| AVP receptor antagonists (vaptans) | ✓ | Conivaptan approved only for in‐hospital use secondary to CYP3A4 inhibition; infusion‐site reactions with intravenous use. Tolvaptan must be initiated and reinitiated in the hospital, as serum sodium needs to be monitored closely to avoid overly rapid correction of hyponatremia. |
Isotonic Saline
The treatment of choice for depletional hyponatremia (ie, hypovolemic hyponatremia) is isotonic saline ([Na+] = 154 mmol/L) to restore ECF volume and ensure adequate organ perfusion. This initial therapy is appropriate for patients who either have clinical signs of hypovolemia, or in whom a spot UNa+ is 30 mmol/L. However, this therapy is ineffective for dilutional hyponatremias such as SIADH,25 and continued inappropriate administration of isotonic saline to a euvolemic patient may worsen their hyponatremia,26 and/or cause fluid overload. Although isotonic saline may improve the serum [Na+] in patients with hypervolemic hyponatremia, their volume status will generally worsen with this therapy, so unless the hyponatremia is profound isotonic saline should be avoided.
Hypertonic Saline
Acute hyponatremia presenting with severe neurological symptoms is life‐threatening, and should be treated promptly with hypertonic solutions, typically 3% NaCl ([Na+] = 513 mmol/L), as this represents the most reliable method to quickly raise the serum [Na+]. A continuous infusion of hypertonic NaCl is usually utilized in inpatient settings. Various formulae have been suggested for calculating the initial rate of infusion of hypertonic solutions,27 but perhaps the simplest utilizes the following relationship:
Why is SIADH Important to Hospitalists?
Disorders of body fluids, and particularly hyponatremia, are among the most commonly encountered problems in clinical medicine, affecting up to 30% of hospitalized patients. In a study of 303,577 laboratory samples collected from 120,137 patients, the prevalence of hyponatremia (serum [Na+] 135 mmol/L) on initial presentation to a healthcare provider was 28.2% among those treated in an acute hospital care setting, 21% among those treated in an ambulatory hospital care setting, and 7.2% in community care centers.1 Numerous other studies have corroborated a high prevalence of hyponatremia in hospitalized patients,2 which reflects the increased vulnerability of this patient population to disruptions of body fluid homeostasis. Recognizing the many possible causes of hyponatremia in hospitalized patients and implementing appropriate treatment strategies therefore are critical steps toward optimizing care and improving outcomes in hospitalized patients with hyponatremia.
In addition to its frequency, hyponatremia is also important because it has been associated with worse clinical outcomes across the entire range of inpatient care, from the general hospital population to those treated in the intensive care unit (ICU). In a study of 4123 patients age 65 years or older who were admitted to a community hospital, 3.5% had clinically significant hyponatremia (serum [Na+] 130 mmol/L) at admission. Compared with nonhyponatremic patients, those with hyponatremia were twice as likely to die during their hospital stay (relative risk [RR], 1.95; P 0.05).3 In another study of 2188 patients admitted to a medical ICU over a 5‐year period, 13.7% had hyponatremia. The overall rate of in‐hospital mortality among all ICU patients was high at 37.7%. However, severe hyponatremia (serum [Na+] 125 mmol/L) more than doubled the risk of in‐hospital mortality (RR, 2.10; P 0.001).4 In addition to the general hospital population, in virtually every disease ever studied, the presence of hyponatremia has been found to be an independent risk factor for increased mortality, from congestive heart failure to tuberculosis to liver failure.2
What Causes Hyponatremia in Patients with SIADH?
Hyponatremia can be caused by 1 of 2 potential disruptions in fluid balance: dilution from retained water, or depletion from electrolyte losses in excess of water. Dilutional hyponatremias are associated with either a normal (euvolemic) or an increased (hypervolemic) extracellular fluid (ECF) volume, whereas depletional hyponatremias generally are associated with a decreased ECF volume (hypovolemic). Dilutional hyponatremia can arise from a primary defect in osmoregulation, such as in SIADH, or as a result of ECF volume expansion, as seen in conditions associated with concomitant secondary hyperaldosteronism such as heart failure, hepatic cirrhosis, or nephrotic syndrome. Among some hospitalized patient groups, euvolemic hyponatremia is the most common presentation of abnormally low serum [Na+]. In a study of patients who developed clinically significant postoperative hyponatremia (defined as a serum [Na+] 130 mmol/L) in a large teaching hospital, only 8% were hypovolemic, whereas 42% were euvolemic and 21% were hypervolemic.5
Euvolemic hyponatremia results from an increase in total body water, but with normal or near‐normal total body sodium. As a result, there is an absence of clinical manifestations of ECF volume expansion, such as subcutaneous edema or ascites. It is important to recognize that although SIADH clearly represents a state of volume expansion due to water retention, it rarely causes clinically recognizable hypervolemia since the retained water is distributed across the intracellular fluid (ICF) as well as the ECF, and because volume regulatory processes act to decrease the actual degree of ECF volume expansion.6 Euvolemic hyponatremia can accompany a wide variety of pathological processes, but the most common cause by far is SIADH. Normally, increased plasma osmolality activates osmoreceptors located in the anterior hypothalamus and stimulates the secretion of arginine vasopressin (AVP), also called antidiuretic hormone (ADH), a key neurohormone that regulates fluid homeostasis. In patients with euvolemic hyponatremia due to SIADH, plasma AVP levels are not suppressed despite normal or decreased plasma osmolality.7 This can be a result of ectopic production of AVP by tumors, or stimulation of endogenous pituitary AVP secretion as a result of nonosmotic stimuli that also stimulate vasopressinergic neurons, which include hypovolemia, hypotension, angiotensin II, nausea, hypoxia, hypercarbia, hypoglycemia, stress, and physical activity. Nonsuppressed AVP levels have been documented in the majority of hyponatremic patients, including those with SIADH8 and heart failure.9
SIADH can develop as the result of many different disease processes that disrupt the normal mechanisms that regulate AVP secretion, including pneumonias and other lung infections, thoracic and extrathoracic tumors, a variety of different central nervous system disorders, the postoperative state, human immunodeficiency virus (HIV), and many different drugs (Figure 1). Given the multiplicity of disorders and drugs that can cause disrupted AVP secretion, it is not surprising that hyponatremia is the most common electrolyte abnormality seen in clinical practice.
What Symptoms are Associated With SIADH?
Symptoms of hyponatremia correlate both with the degree of decrease in the serum [Na+] and with the chronicity of the hyponatremia. Acute hyponatremia, defined as 48 hours in duration, is often associated with life‐threatening clinical features such as obtundation, seizures, coma, and respiratory arrest. These symptoms can occur abruptly, sometimes with little warning.10 In the most severe cases, death can occur as a result of cerebral edema with tentorial herniation. Hypoxia secondary to neurogenic pulmonary edema can increase the severity of brain swelling.11
In contrast, chronic hyponatremia is much less symptomatic, and the reason for the profound differences between the symptoms of acute and chronic hyponatremia is now well understood to be due to the process of brain volume regulation.12 It is essential that this process be understood in order to understand the full spectrum of hyponatremic symptoms. As the ECF [Na+] decreases, regardless of whether due to a loss of sodium or a gain of water, there is an obligate movement of water into the brain along osmotic gradients. That water shift causes swelling of the brain, or cerebral edema. If the increased brain water reaches approximately 8% in adults, it exceeds the capacity of the skull to accommodate brain expansion, leading to tentorial herniation and death from respiratory arrest and/or ischemic brain damage. However, if the patient survives the initial hyponatremia, a very strong volume regulatory process follows, consisting of loss of electrolytes and small organic molecules called osmolytes from brain cells into brain ECF, and eventually the peripheral ECF.12, 13 As the solute content of the brain decreases, the water content is allowed to normalize, eventually reaching a state in which brain edema is virtually absent, and as a result symptoms are markedly less than with acute hyponatremia. Although the time required for the brain to acieve a volume‐regulated state varies across patients, this process is completed within 48 hours in experinmental animal studies, and probably follows a similar time course in humans.
Despite this powerful adaptation process, chronic hyponatremia is frequently associated with neurological symptomatology, albeit milder and more subtle in nature. A recent report found a fairly high incidence of symptoms in 223 patients with chronic hyponatremia as a result of thiazide administration: 49% had malaise/lethargy, 47% had dizzy spells, 35% had vomiting, 17% had confusion/obtundation, 17% experienced falls, 6% had headaches, and 0.9% had seizures.14 Although dizziness can potentially be attributed to a diuretic‐induced hypovolemia, symptoms such as confusion, obtundation and seizures are more consistent with hyponatremic symptomatology. Because thiazide‐induced hyponatremia can be readily corrected by stopping the thiazide and/or administering sodium, this represents an ideal situation in which to assess improvement in hyponatremia symptomatology with normalization of the serum [Na+]; in this study, all of these symptoms improved with correction of the hyponatremia. This represents one of the best examples demonstrating reversal of the symptoms associated with chronic hyponatremia by correction of the hyponatremia, because the patients in this study did not in general have severe underlying comorbidities that might complicate interpretation of their symptoms, as is often the case in patients with SIADH.
What Is Required for Making a Diagnosis of SIADH in Hospitalized Patients?
In patients with hypotonic hypoosmolality, ascertainment of their ECF volume status (ie, hypovolemic, euvolemic, or hypervolemic) is an essential first step, as this will segregate patients into different treatment paradigms. For example, in patients who are truly clinically hypovolemic with a decreased ECF volume by clinical parameters, treatment would generally consist of solute repletion with sodium, generally isotonic saline infusion with or without potassium, until the sodium levels normalize. In patients who are hypervolemic, treatment should focus first on the underlying disease rather than addressing the serum [Na+] directly. In patients with clinical euvolemia, the standard diagnostic pathway should be followed to confirm a diagnosis of SIADH as described below.
Assessing ECF volume status can be difficult, even for the most experienced clinicians. Physical signs such as orthostatic decreases in blood pressure and increases in pulse rate, dry mucus membranes, and skin tenting indicate hypovolemic hyponatremia, while signs such as subcutaneous edema, ascites, or anasarca indicate hypervolemic hyponatremia. Patients without any of these findings are generally considered to be euvolemic. However, in any situation these signs are only applicable if there are no other reasons to suspect an altered ECF volume. Along with a complete history and physical examination that includes a careful neurological evaluation, several laboratory tests can help to assess the etiology of the hyponatremia, once serum sodium concentrations have been shown to be below normal ([Na+] 135 mmol/L):
-
Urine osmolality. A urine osmolality (Uosm) less than 100 mOsm/kg H2O can indicate low dietary solute intake, primary polydipsia, or a reset osmostat after suppression of AVP release by a decrease in plasma osmolality below the osmotic threshold for AVP secretion, usually as a result of increased water loading.
-
Urine sodium concentration. Excretion of sodium, as measured by a spot urine [Na+] (UNa), can indicate depletional hyponatremia if the concentration is less than 30 mmol/L.15 A low UNa reflects a volume depleted state unless the patient has secondary hyperaldosteronism from heart failure or cirrhosis. Patients with a low UNa are more likely to respond to isotonic saline. Euvolemic patients who have a normal dietary sodium intake will generally have spot UNa 30 mmol/L and will not benefit from isotonic saline administration.15 In fact, in SIADH, these patients may respond to isotonic saline with a worsening of hyponatremia, since the sodium from the isotonic saline will be excreted in a concentrated urine while the free water is reabsorbed in the kidney collecting ducts. If the patient is on diuretic therapy, urine sodium values cannot always be accurately interpreted, since a UNa 30 mmol/L may reflect the natriuretic effect of the diuretic and not a volume replete state.
-
Blood tests. Additional indicators of volume status include serum blood nitrogen (BUN) and uric acid levels. A BUN 10 mg/dL and uric acid 4 mg/dL are generally consistent with a euvolemic state, particularly when there is glomerular hyperfiltration, which is often present in SIADH. Elevated serum BUN and uric acid levels (BUN >20 mg/dL and uric acid >6 mg/dL), especially if prior values are available for comparison, can also help to establish whether ineffective vascular volume status may be contributing to the pathophysiology of the hyponatremia. In certain clinical scenarios, the B‐type natriuretic protein (BNP) can be helpful to support a clinical impression of congestive heart failure.
The criteria necessary for a diagnosis of SIADH remain essentially as defined by Bartter and Schwartz16 in 1967 (Table 1), but several points deserve emphasis.17 First, true ECF hypoosmolality must be present and hyponatremia secondary to pseudohyponatremia or hyperglycemia excluded. Second, urinary osmolality must be inappropriate for plasma hypoosmolality (Posm). This does not require a Uosm>Posm, but simply that the urine osmolality is greater than maximally dilute (ie, Uosm>100 mOsm/kg H2O in adults). Furthermore, urine osmolality need not be inappropriately elevated at all levels of Posm but simply at some level under 275 mOsm/kg H2O, since in patients with a reset osmostat, AVP secretion can be suppressed at some level of osmolality resulting in maximal urinary dilution and free water excretion at plasma osmolalities below this level.18 Although some consider a reset osmostat to be a separate disorder rather than a variant of SIADH, such cases nonetheless illustrate that some hypoosmolar patients can exhibit an appropriately dilute urine at some, though not all, plasma osmolalities. Third, clinical euvolemia must be present to diagnose SIADH, and this diagnosis cannot be made in a hypovolemic or edematous patient. Importantly, this does not mean that patients with SIADH cannot become hypovolemic for other reasons, but in such cases it is impossible to diagnose the underlying SIADH until the patient is rendered euvolemic. The fourth criterion, renal salt wasting, has probably caused the most confusion in the diagnosis of SIADH. As noted above, the importance of this criterion lies in its usefulness in differentiating hypoosmolality caused by a decreased effective intravascular volume with high aldosterone levels in which case renal Na+ conservation occurs, from dilutional disorders in which urine Na+ excretion is normal or increased due to ECF volume expansion and a suppressed renin‐angiotensin‐aldosterone system. However, UNa can also be high in renal causes of solute depletion such as diuretic use or Addison's disease, and conversely patients with SIADH can have a low UNa if they subsequently become hypovolemic or solute depleted, conditions sometimes produced by imposed salt and water restriction. Consequently, although high urinary Na+ excretion is generally the rule in most patients with SIADH, its presence does not necessarily confirm this diagnosis, nor does its absence rule out the diagnosis. The final criterion emphasizes that SIADH remains a diagnosis of exclusion, and the absence of other potential causes of hypoosmolality must always be verified. Glucocorticoid deficiency and SIADH can be especially difficult to distinguish, since both primary and secondary hypocortisolism can cause elevated plasma AVP levels in addition to direct renal effects that prevent maximal urinary dilution.19 Therefore, no patient with chronic hyponatremia should be diagnosed as having SIADH without a thorough evaluation of adrenal function, preferably via a rapid adrenocorticotropic hormone (ACTH) stimulation test. Acute hyponatremia of obvious etiology, such as postoperatively or in association with pneumonitis, may be treated without adrenal testing as long as there are no other clinical signs or symptoms suggestive of adrenal dysfunction.20
|
| Essential |
| Decreased effective osmolality of the extracellular fluid (Posm 275 mOsm/kg H2O). |
| Inappropriate urinary concentration (Uosm >100 mOsm/kg H2O with normal renal function) at some level of hypoosmolality. |
| Clinical euvolemia, as defined by the absence of signs of hypovolemia (orthostasis, tachycardia, decreased skin turgor, dry mucous membranes) or hypervolemia (subcutaneous edema, ascites). |
| Elevated urinary sodium excretion while on a normal salt and water intake. |
| Absence of other potential causes of euvolemic hypoosmolality: hypothyroidism, hypocortisolism (Addison's disease or secondary adrenal insufficiency) and diuretic use. |
| Supplemental |
| Abnormal water load test (inability to excrete at least 90% of a 20 mL/kg water load in 4 hours and/or failure to dilute Uosm to 100 mOsm/kg H2O). |
| Plasma AVP level inappropriately elevated relative to plasma osmolality. |
| No significant correction of serum [Na+] with volume expansion but improvement after fluid restriction. |
Hyponatremia is a particularly common complication in elderly hospitalized patients, increasing in prevalence from approximately 7% in the general older population to 18% to 22% among elderly patients in chronic care facilities.21 Despite the many known causes of SIADH (Figure 1), hyponatremia is often associated with idiopathic SIADH in the elderly population. In a study of 119 nursing home residents aged 60 to 103 years, 53% had at least 1 episode of hyponatremia during the previous 12 months.22 Of these patients, 26% were diagnosed with idiopathic SIADH. In another study of elderly patients with hyponatremia and SIADH, 60% were diagnosed with idiopathic SIADH. Among remaining patients, the 2 main causes identified were pneumonia (9 cases/18%) and medications (6 cases/12%).23 Therefore, more than half of elderly patients who present with hyponatremia due to SIADH may have an idiopathic form, with no detectable underlying treatable disease.
Which Hospital Patients With SIADH are Candidates for Treatment of Hyponatremia?
Correction of hyponatremia is associated with markedly improved neurological outcomes in patients with severely symptomatic hyponatremia. In a retrospective review of patients who presented with severe neurological symptoms and serum [Na+] 125 mmol/L, prompt therapy with isotonic or hypertonic saline resulted in a correction in the range of 20 mmol/L over several days and neurological recovery in almost all cases. In contrast, in patients who were treated with fluid restriction alone, there was very little correction over the study period (5 mmol/L over 72 hours), and the neurological outcomes were much worse, with most of these patients either dying or entering a persistently vegetative state.24 Consequently, prompt therapy to rapidly increase the serum [Na+] represents the standard‐of‐care for treatment of patients presenting with severe life‐threatening symptoms of hyponatremia.
As discussed earlier, chronic hyponatremia is much less symptomatic as a result of the process of brain volume regulation. Because of this adaptation process, chronic hyponatremia is arguably a condition that clinicians feel they may not need to be as concerned about, and in some publications this has been called asymptomatic hyponatremia. However, such patients often do have neurological symptoms, even if milder and more subtle in nature, including headaches, nausea, mood disturbances, depression, difficulty concentrating, slowed reaction times, unstable gait, increased falls, confusion, and disorientation. Consequently, any patient with hyponatremia secondary to SIADH who manifests any neurological symptoms that could be related to the hyponatremia should be considered as appropriate candidates for treatment of the hyponatremia, regardless of the chronicity of the hyponatremia or the level of serum [Na+].
What Therapies are Currently Available to Manage SIADH in Hospitalized Patients?
Conventional management strategies for euvolemic hyponatremia range from saline infusion and fluid restriction to pharmacologic adjustment of fluid balance. Consideration of treatment options should include an evaluation of the benefits as well as the potential toxicities of any therapy (Table 2). Sometimes, simply stopping treatment with an agent that is associated with hyponatremia is sufficient to reverse a low serum [Na+].
| Therapy | Targets Underlying Pathophysiology | Limitations |
|---|---|---|
| ||
| Isotonic saline | Ineffective in dilutional hyponatremias; exacerbates the volume overload if used in edema‐forming disorders; no controlled safety database. | |
| Hypertonic saline | No consensus regarding appropriate infusion rates; overcorrection can cause osmotic demyelination; exacerbates the volume overload if used in edema‐forming disorders; no controlled safety database. | |
| Fluid restriction | Slow to correct over many days; poorly tolerated due to thirst; can not be used effectively in patients with high AVP levels and urine osmolalities. | |
| Demeclocycline | ✓ | Not FDA approved for hyponatremia; slow to correct; nephrotoxic in cirrhosis and heart failure. |
| Mineralocorticoids | Only one report in elderly patients with SIADH; no safety database; exacerbates the volume overload if used in edema‐forming disorders. | |
| Urea | Not FDA‐approved for hyponatremia; poor palatability. | |
| AVP receptor antagonists (vaptans) | ✓ | Conivaptan approved only for in‐hospital use secondary to CYP3A4 inhibition; infusion‐site reactions with intravenous use. Tolvaptan must be initiated and reinitiated in the hospital, as serum sodium needs to be monitored closely to avoid overly rapid correction of hyponatremia. |
Isotonic Saline
The treatment of choice for depletional hyponatremia (ie, hypovolemic hyponatremia) is isotonic saline ([Na+] = 154 mmol/L) to restore ECF volume and ensure adequate organ perfusion. This initial therapy is appropriate for patients who either have clinical signs of hypovolemia, or in whom a spot UNa+ is 30 mmol/L. However, this therapy is ineffective for dilutional hyponatremias such as SIADH,25 and continued inappropriate administration of isotonic saline to a euvolemic patient may worsen their hyponatremia,26 and/or cause fluid overload. Although isotonic saline may improve the serum [Na+] in patients with hypervolemic hyponatremia, their volume status will generally worsen with this therapy, so unless the hyponatremia is profound isotonic saline should be avoided.
Hypertonic Saline
Acute hyponatremia presenting with severe neurological symptoms is life‐threatening, and should be treated promptly with hypertonic solutions, typically 3% NaCl ([Na+] = 513 mmol/L), as this represents the most reliable method to quickly raise the serum [Na+]. A continuous infusion of hypertonic NaCl is usually utilized in inpatient settings. Various formulae have been suggested for calculating the initial rate of infusion of hypertonic solutions,27 but perhaps the simplest utilizes the following relationship:
- .Age and gender as risk factors for hyponatremia and hypernatremia.Clin Chim Acta.2003;337(1‐2):169–172.
- ,,.Incidence and prevalence of hyponatremia.Am J Med.2006;119(7 Suppl 1):S30–S35.
- ,,.Admission hyponatremia in the elderly: factors influencing prognosis.J Gen Intern Med1994;9:89–91.
- ,,, et al.[Incidence, causes and prognostic factors of hyponatremia in intensive care].Rev Med Interne.2003;24(4):224–229.
- ,,,.Postoperative hyponatremia. A prospective study.Arch Int Med.1986;146:333–336.
- .Whole‐body volume regulation and escape from antidiuresis.Am J Med.2006;119(7 Suppl 1):S21–S29.
- ,,.Neurogenic disorders of osmoregulation.Am J Med.1982;72:339–353.
- ,,.Vasopressin function in the syndrome of inappropriate antidiuresis.Annu Rev Med.1980;31:315–327.
- ,,,,,.Radioimmunoassay of plasma arginine vasopressin in hyponatremic patients with congestive heart failure.N Eng J Med.1981;305:263–266.
- ,.Epidemiology, pathophysiology, and management of hyponatremic encephalopathy.Am J Med.1997;102:67–77.
- ,.Pulmonary complications of hyponatremic encephalopathy. noncardiogenic pulmonary edema and hypercapnic respiratory failure [see comments].Chest.1995;107(2):517–521.
- ,.Control of brain volume during hyperosmolar and hypoosmolar conditions.Annu Rev Med.1993;44:289–301.
- .Control of brain volume during hypoosmolality and hyperosmolality.Adv Exp Med Biol.2006;576:113–129.
- ,,.Clinical studies of thiazide‐induced hyponatremia.J Natl Med Assoc.2004;96(10):1305–1308.
- ,,,.Clinical assessment of extracellular fluid volume in hyponatremia.Am J Med.1987;83:905–908.
- ,.The syndrome of inappropriate secretion of antidiuretic hormone.Am J Med.1967;42:790–806.
- .Hyponatremia and Hypo‐osmolar Disorders. In: Greenberg A, Cheung AK, Coffman TM, Falk RJ, Jennette JC, eds.Primer on Kidney Diseases.Philadelphia:Saunders Elsevier,2009:52–59.
- ,,,.Reset of osmoreceptors in association with normovolemic hyponatremia.Am J Med Sci.1974;267:267–273.
- .Hyponatremia and inappropriate secretion of vasopressin (antidiuretic hormone) in patients with hypopituitarism.N Eng J Med.1989;321:492–496.
- .The Syndrome of Inappropriate Antidiuretic Hormone Secretion and Other Hypoosmolar Disorders. In: Schrier RW, ed.Diseases of the Kidney and Urinary Tract.Philadelphia:Lippincott Williams 27:156–161.
- ,,.Hyponatremia in a nursing home population.J Am Geriatr Soc.1995;43(12):1410–1413.
- ,.The syndrome of inappropriate antidiuretic hormone secretion in the elderly.Am J Med.1997;103(4):270–273.
- .Diuretic‐induced hyponatremia [editorial].Arch Intern Med.1986;146(7):1295–1296.
- ,,,.A syndrome of renal sodium loss and hyponatremia probably resulting from inappropriate secretion of antidiuretic hormone.Am J Med.1957;23:529–542.
- ,,,,,.Postoperative hyponatremia despite near‐isotonic saline infusion: a phenomenon of desalination [see comments].Ann Intern Med.1997;126(1):20–25.
- ,.Hyponatremia.N Engl J Med.2000;342(21):1581–1589.
- ,,, et al.Statement of the Second International Exercise‐Associated Hyponatremia Consensus Development Conference, New Zealand, 2007.Clin J Sport Med.2008;18(2):111–121.
- ,,,.Neurologic sequelae after treatment of severe hyponatremia: a multicenter perspective.J Am Soc Nephrol.1994;4:1522–1530.
- ,,,.Randomized, controlled trial on the effect of a 20% mannitol solution and a 7.5% saline/6% dextran solution on increased intracranial pressure after brain injury.Crit Care Med.2005;33(1):196–202.
- ,,.Osmotic demyelination syndrome following correction of hyponatremia.N Engl J Med.1986;314:1535–1542.
- .Regulation of arginine vasopressin in the syndrome of inappropriate antidiuresis.Am J Med.2006;119(7 Suppl 1):S36–S42.
- .The syndrome of inappropriate secretion of antidiuretic hormone (SIADH).Semin Nephrol.2009;29(3):239–256.
- .Impact of solute intake on urine flow and water excretion.J Am Soc Nephrol.2008;19(6):1076–1078.
- ,.Demeclocycline‐induced nephrogenic diabetes insipidus. In‐vivo and in‐ vitro studies.Ann Intern Med.1973;79(5):679–683.
- ,,, et al.Involvement of arginine vasopressin and renal sodium handling in pathogenesis of hyponatremia in elderly patients.Endocr J.1996;43(1):101–108.
- ,.Urea for long‐term treatment of syndrome of inappropriate secretion of antidiuretic hormone.Br Med J (Clin Res Ed).1981;283:1081–1083.
- ,.Vasopressin receptor antagonists.Kidney Int.2006;69(12):2124–2130.
- ,,, et al.Potent aquaretic agent. A novel nonpeptide selective vasopressin 2 antagonist (OPC‐31260) in men.J Clin Invest.1993;92(6):2653–2659.
- ,,,,.Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia.Am J Nephrol.2007;27(5):447–457.
- ,,, et al.Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355(20):2099–2112.
- Vaprisol (conivaptan hydrochloride injection) prescribing information.Deerfield, IL:Astellas Pharma US, Inc.,2006.
- ,,,,.Hyponatremia treatment guidelines 2007: expert panel recommendations.Am J Med.2007;120(11 Suppl 1):S1–S21.
- Otsuka Pharmaceutical Co L, Tokyo J. Samsca (tolvaptan) prescribing information.2009.
- ,.Clinical practice. The syndrome of inappropriate antidiuresis.N Engl J Med.2007;356(20):2064–2072.
- ,,.The treatment of hyponatremia.Semin Nephrol.2009;29(3):282–299.
- .Hyponatremia and Hypo‐osmolar Disorders. In: Greenberg A, Cheung AK, Coffman TM, Falk RJ, Jennette JC, eds.Primer on Kidney Diseases.Philadelphia. PA:Saunders Elsevier;2009:52–59.
- ,,,.Risk factors for symptomatic hyponatraemia: the role of pre‐existing asymptomatic hyponatraemia.Intern Med J.2007;37(3):149–155.
- ,,,,.Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits.Am J Med.2006;119(1):71.
- ,,,,.Mild hyponatremia and risk of fracture in the ambulatory elderly.QJM.2008;101(7):583–588.
- ,,,,.Hyponatremia associated with large‐bone fracture in elderly patients.Int Urol Nephrol.2009;41(3):733–737.
- ,,, et al.Hyponatremia‐induced osteoporosis.J Bone Miner Res.2010;25(3):554–563.
- .Age and gender as risk factors for hyponatremia and hypernatremia.Clin Chim Acta.2003;337(1‐2):169–172.
- ,,.Incidence and prevalence of hyponatremia.Am J Med.2006;119(7 Suppl 1):S30–S35.
- ,,.Admission hyponatremia in the elderly: factors influencing prognosis.J Gen Intern Med1994;9:89–91.
- ,,, et al.[Incidence, causes and prognostic factors of hyponatremia in intensive care].Rev Med Interne.2003;24(4):224–229.
- ,,,.Postoperative hyponatremia. A prospective study.Arch Int Med.1986;146:333–336.
- .Whole‐body volume regulation and escape from antidiuresis.Am J Med.2006;119(7 Suppl 1):S21–S29.
- ,,.Neurogenic disorders of osmoregulation.Am J Med.1982;72:339–353.
- ,,.Vasopressin function in the syndrome of inappropriate antidiuresis.Annu Rev Med.1980;31:315–327.
- ,,,,,.Radioimmunoassay of plasma arginine vasopressin in hyponatremic patients with congestive heart failure.N Eng J Med.1981;305:263–266.
- ,.Epidemiology, pathophysiology, and management of hyponatremic encephalopathy.Am J Med.1997;102:67–77.
- ,.Pulmonary complications of hyponatremic encephalopathy. noncardiogenic pulmonary edema and hypercapnic respiratory failure [see comments].Chest.1995;107(2):517–521.
- ,.Control of brain volume during hyperosmolar and hypoosmolar conditions.Annu Rev Med.1993;44:289–301.
- .Control of brain volume during hypoosmolality and hyperosmolality.Adv Exp Med Biol.2006;576:113–129.
- ,,.Clinical studies of thiazide‐induced hyponatremia.J Natl Med Assoc.2004;96(10):1305–1308.
- ,,,.Clinical assessment of extracellular fluid volume in hyponatremia.Am J Med.1987;83:905–908.
- ,.The syndrome of inappropriate secretion of antidiuretic hormone.Am J Med.1967;42:790–806.
- .Hyponatremia and Hypo‐osmolar Disorders. In: Greenberg A, Cheung AK, Coffman TM, Falk RJ, Jennette JC, eds.Primer on Kidney Diseases.Philadelphia:Saunders Elsevier,2009:52–59.
- ,,,.Reset of osmoreceptors in association with normovolemic hyponatremia.Am J Med Sci.1974;267:267–273.
- .Hyponatremia and inappropriate secretion of vasopressin (antidiuretic hormone) in patients with hypopituitarism.N Eng J Med.1989;321:492–496.
- .The Syndrome of Inappropriate Antidiuretic Hormone Secretion and Other Hypoosmolar Disorders. In: Schrier RW, ed.Diseases of the Kidney and Urinary Tract.Philadelphia:Lippincott Williams 27:156–161.
- ,,.Hyponatremia in a nursing home population.J Am Geriatr Soc.1995;43(12):1410–1413.
- ,.The syndrome of inappropriate antidiuretic hormone secretion in the elderly.Am J Med.1997;103(4):270–273.
- .Diuretic‐induced hyponatremia [editorial].Arch Intern Med.1986;146(7):1295–1296.
- ,,,.A syndrome of renal sodium loss and hyponatremia probably resulting from inappropriate secretion of antidiuretic hormone.Am J Med.1957;23:529–542.
- ,,,,,.Postoperative hyponatremia despite near‐isotonic saline infusion: a phenomenon of desalination [see comments].Ann Intern Med.1997;126(1):20–25.
- ,.Hyponatremia.N Engl J Med.2000;342(21):1581–1589.
- ,,, et al.Statement of the Second International Exercise‐Associated Hyponatremia Consensus Development Conference, New Zealand, 2007.Clin J Sport Med.2008;18(2):111–121.
- ,,,.Neurologic sequelae after treatment of severe hyponatremia: a multicenter perspective.J Am Soc Nephrol.1994;4:1522–1530.
- ,,,.Randomized, controlled trial on the effect of a 20% mannitol solution and a 7.5% saline/6% dextran solution on increased intracranial pressure after brain injury.Crit Care Med.2005;33(1):196–202.
- ,,.Osmotic demyelination syndrome following correction of hyponatremia.N Engl J Med.1986;314:1535–1542.
- .Regulation of arginine vasopressin in the syndrome of inappropriate antidiuresis.Am J Med.2006;119(7 Suppl 1):S36–S42.
- .The syndrome of inappropriate secretion of antidiuretic hormone (SIADH).Semin Nephrol.2009;29(3):239–256.
- .Impact of solute intake on urine flow and water excretion.J Am Soc Nephrol.2008;19(6):1076–1078.
- ,.Demeclocycline‐induced nephrogenic diabetes insipidus. In‐vivo and in‐ vitro studies.Ann Intern Med.1973;79(5):679–683.
- ,,, et al.Involvement of arginine vasopressin and renal sodium handling in pathogenesis of hyponatremia in elderly patients.Endocr J.1996;43(1):101–108.
- ,.Urea for long‐term treatment of syndrome of inappropriate secretion of antidiuretic hormone.Br Med J (Clin Res Ed).1981;283:1081–1083.
- ,.Vasopressin receptor antagonists.Kidney Int.2006;69(12):2124–2130.
- ,,, et al.Potent aquaretic agent. A novel nonpeptide selective vasopressin 2 antagonist (OPC‐31260) in men.J Clin Invest.1993;92(6):2653–2659.
- ,,,,.Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia.Am J Nephrol.2007;27(5):447–457.
- ,,, et al.Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355(20):2099–2112.
- Vaprisol (conivaptan hydrochloride injection) prescribing information.Deerfield, IL:Astellas Pharma US, Inc.,2006.
- ,,,,.Hyponatremia treatment guidelines 2007: expert panel recommendations.Am J Med.2007;120(11 Suppl 1):S1–S21.
- Otsuka Pharmaceutical Co L, Tokyo J. Samsca (tolvaptan) prescribing information.2009.
- ,.Clinical practice. The syndrome of inappropriate antidiuresis.N Engl J Med.2007;356(20):2064–2072.
- ,,.The treatment of hyponatremia.Semin Nephrol.2009;29(3):282–299.
- .Hyponatremia and Hypo‐osmolar Disorders. In: Greenberg A, Cheung AK, Coffman TM, Falk RJ, Jennette JC, eds.Primer on Kidney Diseases.Philadelphia. PA:Saunders Elsevier;2009:52–59.
- ,,,.Risk factors for symptomatic hyponatraemia: the role of pre‐existing asymptomatic hyponatraemia.Intern Med J.2007;37(3):149–155.
- ,,,,.Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits.Am J Med.2006;119(1):71.
- ,,,,.Mild hyponatremia and risk of fracture in the ambulatory elderly.QJM.2008;101(7):583–588.
- ,,,,.Hyponatremia associated with large‐bone fracture in elderly patients.Int Urol Nephrol.2009;41(3):733–737.
- ,,, et al.Hyponatremia‐induced osteoporosis.J Bone Miner Res.2010;25(3):554–563.

V Receptor Antagonists for Treatment of Hyponatremia
Under normal circumstances, there is a balance between water intake and water excretion such that plasma osmolality and the serum sodium (Na+) concentration remain relatively constant. The principal mechanism responsible for prevention of hyponatremia and hyposmolality is renal water excretion. In all hyponatremic patients, water intake exceeds renal water excretion.
Excretion of water by the kidney is dependent on 3 factors. First, there must be adequate delivery of filtrate to the tip of the loop of Henle. Second, solute absorption in the ascending limb and the distal nephron must be preserved so that the tubular fluid will be diluted. Lastly, arginine vasopressin (AVP) levels must be low in the plasma. Of these 3 requirements for water excretion, the one which is most important in the genesis of hyponatremia is the failure to maximally suppress AVP levels. Given the central role of AVP in limiting renal water excretion, AVP receptor antagonists represent a physiologic and rational method to increase renal water excretion.
AVP in Regulation of Plasma Osmolality
AVP is synthesized in the supraoptic and paraventricular nucleus of the hypothalamus and then stored in the neurohypophysis (reviewed in the article Diagnostic Approach and Management of Inpatient Hyponatremia in this supplement). The release of AVP is exquisitely sensitive to changes in plasma osmolality. AVP is not detectable in the plasma at an osmolality below approximately 280 mOsm/kg but increases in a nearly linear fashion beginning with as little as a 2% to 3% increase in osmolality above this value. The extreme sensitivity of this system allows for plasma osmolality to be maintained within a narrow range.
A second major determinant of AVP release is the effective arterial blood volume. While AVP levels are very sensitive to plasma osmolality, small changes of 10% in blood pressure or blood volume have no effect on AVP levels. However, once decreases in volume or pressure exceed this value, baroreceptor‐mediated signals provide persistent stimuli for AVP secretion. Baroreceptor‐mediated AVP release will continue even when plasma osmolality falls below 280 mOsm/kg. Teleologically, this system can be viewed as an emergency mechanism to defend blood pressure. Thus, small decreases in blood volume and blood pressure will cause the body to retain NaCl which will raise osmolality and lead to water retention. However, if NaCl is not available and if blood pressure and volume are becoming dangerously low (down 10%), the body behaves as if defense of blood pressure is more important than defense of osmolality, and AVP is secreted.1 The specific compartment whose volume is sensed in order to determine AVP secretion in this setting is the effective arterial volume. This overriding effect of volume explains the persistence of high AVP levels in hyponatremic patients with conditions such as heart failure and cirrhosis.
Other stimuli for the release of AVP include pain, nausea, and hypoxia. Inappropriate release of AVP can occur with a variety of central nervous system and pulmonary diseases as well as with drugs, particularly those that act within the central nervous system.2 Certain tumors can synthesize and release AVP.
AVP exerts its effects on cells through 3 receptors (Table 1). The V1A receptor is expressed in a variety of tissues but is primarily found on vascular smooth muscle cells. Stimulation of this receptor results in vasoconstriction, platelet aggregation, inotropic stimulation and myocardial protein synthesis. The V1B receptor is expressed in cells of the anterior pituitary and throughout the brain. Stimulation of this receptor results in release of adrenocorticotropin stimulating hormone (ACTH). Stimulation of the V1A and V1B receptors activate phospholipase C leading to increases in inositol trisphosphate and diacylglycerol with secondary increases in cell calcium and activation of protein kinase C.
The V2 receptor is found on the basolateral surface of the renal collecting duct and vascular endothelium where it mediates the antidiuretic effects of AVP and stimulates the release of von Willebrand factor respectively. Unlike the V1A and V1B receptors, binding of AVP to the V2 receptor activates the GS‐coupled adenyl cyclase system causing increased intracellular levels of cyclic adenosine monophosphate (cAMP). In the kidney, generation of cAMP stimulates protein kinase A which then phosphorylates preformed aquaporin‐2 water channels causing trafficking and insertion of the channels into the luminal membrane of the tubular cells.3 The insertion of the aquaporin‐2 protein renders the collecting duct selectively permeable to water, which is then reabsorbed from the tubular lumen into the blood driven by the osmotic driving force of the hypertonic interstitium. In the absence of AVP, aquaporin membrane insertion and apical membrane water permeability are dramatically reduced.
Physiologic Rationale for Use of AVP Antagonists
AVP antagonists block the V2 receptor located on the basolateral surface of the collecting duct thereby antagonizing the ability of AVP to cause insertion of the aquaporin‐2 water channels into the luminal membrane. The increase in urine output is similar in quantity to diuretics but differs in content. V2 receptor antagonists increase water excretion with little to no change in urinary electrolytes. As a result, lowering of the serum K+ level, metabolic alkalosis, and increases in the serum creatinine and blood urea nitrogen concentration are avoided in contrast to diuretics such as furosemide and hydrochlorothiazide. In addition, orthostatic hypotension and activation of neurohumoral effectors such as angiotensin II, circulating catecholamines, and aldosterone are not features of V2 receptor blockade. These differences have lead to V2 receptor antagonists being characterized as aquaretic agents so as to distinguish them from diuretics.
The physiologic rationale for use of V2 receptor antagonists is best exemplified by considering the relationship between the serum Na+ concentration and the total body content of Na+, K+, and water approximated by the equation:
- ,.Integrated Response to Abnormalities in Tonicity. In: Seldin DW, Giebisch G, eds.Clinical Disturbances of Water Metabolism.New York, New York:Raven Press Ltd;1993:273–295.
- ,,.Causes and management of hyponatremia.Ann Pharmacother.2003;37:1694–1702.
- ,,,,,.Regulation of aquaporin‐2 trafficking.Handb Exp Pharmacol.2009;190:133–157.
- ,,.Evaluation and management of hyponatremia: an emerging role for vasopressin receptor antagonists.Nat Clin Pract Nephrol.2007;3:82–95.
- ,,,,.Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia.Am J Nephrol.2007;27:447–457.
- ,,, et al.Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355:2099–112.
- ,,, et al.V1a vasopressin receptors maintain normal blood pressure by regulating circulating blood volume and baroreflex sensitivity.Proc Natl Acad Sci.2006;103:7807–7812.
- .Pathogenesis of ascites and renal salt retention in cirrhosis.J Invest Med.1999;47:183–202.
- ,.Hyponatremia.N Engl J Med.2000;342:1581–1589.
- ,,.The treatment of hyponatremia.Semin Nephrol.2009;29:282–299.
- .Hyponatremia in the intensive care unit.Semin Nephrol.2009;29:257–270.
- ,,,,.Conivaptan and its role in the treatment of hyponatremia.Drug Des Devel Ther.2009;3:253–268.
- .Vaptans for the treatment of hyponatremia: how who when and why.Nephrol Self Assess Program.2007;6:199–209.
- .The syndrome of inappropriate secretion of antidiuretic hormone (SIADH).Semin Nephrol.2009;29(3):239–256.
- ,,,,.Mild hyponatremia and risk of fracture in the ambulatory elderly.Q J Med.2008;101:583–588.
- ,,,,.Hyponatremia independent of osteoporosis is associated with fracture occurrence.Clin J Am Soc Nephrol.2010 (in press).
- ,,, et al.Hyponatremia‐induced osteoporosis.J Bone Miner Res.2009;999:1–37.
- ,,,,.Impact of hospital‐associated hyponatremia on selected outcomes.Arch Intern Med.2010;170(3):294–302.
- ,,, et al.Mortality after hospitalization with mild, moderate, and severe hyponatremia.Am J Med.2009;122:857–865.
Under normal circumstances, there is a balance between water intake and water excretion such that plasma osmolality and the serum sodium (Na+) concentration remain relatively constant. The principal mechanism responsible for prevention of hyponatremia and hyposmolality is renal water excretion. In all hyponatremic patients, water intake exceeds renal water excretion.
Excretion of water by the kidney is dependent on 3 factors. First, there must be adequate delivery of filtrate to the tip of the loop of Henle. Second, solute absorption in the ascending limb and the distal nephron must be preserved so that the tubular fluid will be diluted. Lastly, arginine vasopressin (AVP) levels must be low in the plasma. Of these 3 requirements for water excretion, the one which is most important in the genesis of hyponatremia is the failure to maximally suppress AVP levels. Given the central role of AVP in limiting renal water excretion, AVP receptor antagonists represent a physiologic and rational method to increase renal water excretion.
AVP in Regulation of Plasma Osmolality
AVP is synthesized in the supraoptic and paraventricular nucleus of the hypothalamus and then stored in the neurohypophysis (reviewed in the article Diagnostic Approach and Management of Inpatient Hyponatremia in this supplement). The release of AVP is exquisitely sensitive to changes in plasma osmolality. AVP is not detectable in the plasma at an osmolality below approximately 280 mOsm/kg but increases in a nearly linear fashion beginning with as little as a 2% to 3% increase in osmolality above this value. The extreme sensitivity of this system allows for plasma osmolality to be maintained within a narrow range.
A second major determinant of AVP release is the effective arterial blood volume. While AVP levels are very sensitive to plasma osmolality, small changes of 10% in blood pressure or blood volume have no effect on AVP levels. However, once decreases in volume or pressure exceed this value, baroreceptor‐mediated signals provide persistent stimuli for AVP secretion. Baroreceptor‐mediated AVP release will continue even when plasma osmolality falls below 280 mOsm/kg. Teleologically, this system can be viewed as an emergency mechanism to defend blood pressure. Thus, small decreases in blood volume and blood pressure will cause the body to retain NaCl which will raise osmolality and lead to water retention. However, if NaCl is not available and if blood pressure and volume are becoming dangerously low (down 10%), the body behaves as if defense of blood pressure is more important than defense of osmolality, and AVP is secreted.1 The specific compartment whose volume is sensed in order to determine AVP secretion in this setting is the effective arterial volume. This overriding effect of volume explains the persistence of high AVP levels in hyponatremic patients with conditions such as heart failure and cirrhosis.
Other stimuli for the release of AVP include pain, nausea, and hypoxia. Inappropriate release of AVP can occur with a variety of central nervous system and pulmonary diseases as well as with drugs, particularly those that act within the central nervous system.2 Certain tumors can synthesize and release AVP.
AVP exerts its effects on cells through 3 receptors (Table 1). The V1A receptor is expressed in a variety of tissues but is primarily found on vascular smooth muscle cells. Stimulation of this receptor results in vasoconstriction, platelet aggregation, inotropic stimulation and myocardial protein synthesis. The V1B receptor is expressed in cells of the anterior pituitary and throughout the brain. Stimulation of this receptor results in release of adrenocorticotropin stimulating hormone (ACTH). Stimulation of the V1A and V1B receptors activate phospholipase C leading to increases in inositol trisphosphate and diacylglycerol with secondary increases in cell calcium and activation of protein kinase C.
The V2 receptor is found on the basolateral surface of the renal collecting duct and vascular endothelium where it mediates the antidiuretic effects of AVP and stimulates the release of von Willebrand factor respectively. Unlike the V1A and V1B receptors, binding of AVP to the V2 receptor activates the GS‐coupled adenyl cyclase system causing increased intracellular levels of cyclic adenosine monophosphate (cAMP). In the kidney, generation of cAMP stimulates protein kinase A which then phosphorylates preformed aquaporin‐2 water channels causing trafficking and insertion of the channels into the luminal membrane of the tubular cells.3 The insertion of the aquaporin‐2 protein renders the collecting duct selectively permeable to water, which is then reabsorbed from the tubular lumen into the blood driven by the osmotic driving force of the hypertonic interstitium. In the absence of AVP, aquaporin membrane insertion and apical membrane water permeability are dramatically reduced.
Physiologic Rationale for Use of AVP Antagonists
AVP antagonists block the V2 receptor located on the basolateral surface of the collecting duct thereby antagonizing the ability of AVP to cause insertion of the aquaporin‐2 water channels into the luminal membrane. The increase in urine output is similar in quantity to diuretics but differs in content. V2 receptor antagonists increase water excretion with little to no change in urinary electrolytes. As a result, lowering of the serum K+ level, metabolic alkalosis, and increases in the serum creatinine and blood urea nitrogen concentration are avoided in contrast to diuretics such as furosemide and hydrochlorothiazide. In addition, orthostatic hypotension and activation of neurohumoral effectors such as angiotensin II, circulating catecholamines, and aldosterone are not features of V2 receptor blockade. These differences have lead to V2 receptor antagonists being characterized as aquaretic agents so as to distinguish them from diuretics.
The physiologic rationale for use of V2 receptor antagonists is best exemplified by considering the relationship between the serum Na+ concentration and the total body content of Na+, K+, and water approximated by the equation:
Under normal circumstances, there is a balance between water intake and water excretion such that plasma osmolality and the serum sodium (Na+) concentration remain relatively constant. The principal mechanism responsible for prevention of hyponatremia and hyposmolality is renal water excretion. In all hyponatremic patients, water intake exceeds renal water excretion.
Excretion of water by the kidney is dependent on 3 factors. First, there must be adequate delivery of filtrate to the tip of the loop of Henle. Second, solute absorption in the ascending limb and the distal nephron must be preserved so that the tubular fluid will be diluted. Lastly, arginine vasopressin (AVP) levels must be low in the plasma. Of these 3 requirements for water excretion, the one which is most important in the genesis of hyponatremia is the failure to maximally suppress AVP levels. Given the central role of AVP in limiting renal water excretion, AVP receptor antagonists represent a physiologic and rational method to increase renal water excretion.
AVP in Regulation of Plasma Osmolality
AVP is synthesized in the supraoptic and paraventricular nucleus of the hypothalamus and then stored in the neurohypophysis (reviewed in the article Diagnostic Approach and Management of Inpatient Hyponatremia in this supplement). The release of AVP is exquisitely sensitive to changes in plasma osmolality. AVP is not detectable in the plasma at an osmolality below approximately 280 mOsm/kg but increases in a nearly linear fashion beginning with as little as a 2% to 3% increase in osmolality above this value. The extreme sensitivity of this system allows for plasma osmolality to be maintained within a narrow range.
A second major determinant of AVP release is the effective arterial blood volume. While AVP levels are very sensitive to plasma osmolality, small changes of 10% in blood pressure or blood volume have no effect on AVP levels. However, once decreases in volume or pressure exceed this value, baroreceptor‐mediated signals provide persistent stimuli for AVP secretion. Baroreceptor‐mediated AVP release will continue even when plasma osmolality falls below 280 mOsm/kg. Teleologically, this system can be viewed as an emergency mechanism to defend blood pressure. Thus, small decreases in blood volume and blood pressure will cause the body to retain NaCl which will raise osmolality and lead to water retention. However, if NaCl is not available and if blood pressure and volume are becoming dangerously low (down 10%), the body behaves as if defense of blood pressure is more important than defense of osmolality, and AVP is secreted.1 The specific compartment whose volume is sensed in order to determine AVP secretion in this setting is the effective arterial volume. This overriding effect of volume explains the persistence of high AVP levels in hyponatremic patients with conditions such as heart failure and cirrhosis.
Other stimuli for the release of AVP include pain, nausea, and hypoxia. Inappropriate release of AVP can occur with a variety of central nervous system and pulmonary diseases as well as with drugs, particularly those that act within the central nervous system.2 Certain tumors can synthesize and release AVP.
AVP exerts its effects on cells through 3 receptors (Table 1). The V1A receptor is expressed in a variety of tissues but is primarily found on vascular smooth muscle cells. Stimulation of this receptor results in vasoconstriction, platelet aggregation, inotropic stimulation and myocardial protein synthesis. The V1B receptor is expressed in cells of the anterior pituitary and throughout the brain. Stimulation of this receptor results in release of adrenocorticotropin stimulating hormone (ACTH). Stimulation of the V1A and V1B receptors activate phospholipase C leading to increases in inositol trisphosphate and diacylglycerol with secondary increases in cell calcium and activation of protein kinase C.
The V2 receptor is found on the basolateral surface of the renal collecting duct and vascular endothelium where it mediates the antidiuretic effects of AVP and stimulates the release of von Willebrand factor respectively. Unlike the V1A and V1B receptors, binding of AVP to the V2 receptor activates the GS‐coupled adenyl cyclase system causing increased intracellular levels of cyclic adenosine monophosphate (cAMP). In the kidney, generation of cAMP stimulates protein kinase A which then phosphorylates preformed aquaporin‐2 water channels causing trafficking and insertion of the channels into the luminal membrane of the tubular cells.3 The insertion of the aquaporin‐2 protein renders the collecting duct selectively permeable to water, which is then reabsorbed from the tubular lumen into the blood driven by the osmotic driving force of the hypertonic interstitium. In the absence of AVP, aquaporin membrane insertion and apical membrane water permeability are dramatically reduced.
Physiologic Rationale for Use of AVP Antagonists
AVP antagonists block the V2 receptor located on the basolateral surface of the collecting duct thereby antagonizing the ability of AVP to cause insertion of the aquaporin‐2 water channels into the luminal membrane. The increase in urine output is similar in quantity to diuretics but differs in content. V2 receptor antagonists increase water excretion with little to no change in urinary electrolytes. As a result, lowering of the serum K+ level, metabolic alkalosis, and increases in the serum creatinine and blood urea nitrogen concentration are avoided in contrast to diuretics such as furosemide and hydrochlorothiazide. In addition, orthostatic hypotension and activation of neurohumoral effectors such as angiotensin II, circulating catecholamines, and aldosterone are not features of V2 receptor blockade. These differences have lead to V2 receptor antagonists being characterized as aquaretic agents so as to distinguish them from diuretics.
The physiologic rationale for use of V2 receptor antagonists is best exemplified by considering the relationship between the serum Na+ concentration and the total body content of Na+, K+, and water approximated by the equation:
- ,.Integrated Response to Abnormalities in Tonicity. In: Seldin DW, Giebisch G, eds.Clinical Disturbances of Water Metabolism.New York, New York:Raven Press Ltd;1993:273–295.
- ,,.Causes and management of hyponatremia.Ann Pharmacother.2003;37:1694–1702.
- ,,,,,.Regulation of aquaporin‐2 trafficking.Handb Exp Pharmacol.2009;190:133–157.
- ,,.Evaluation and management of hyponatremia: an emerging role for vasopressin receptor antagonists.Nat Clin Pract Nephrol.2007;3:82–95.
- ,,,,.Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia.Am J Nephrol.2007;27:447–457.
- ,,, et al.Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355:2099–112.
- ,,, et al.V1a vasopressin receptors maintain normal blood pressure by regulating circulating blood volume and baroreflex sensitivity.Proc Natl Acad Sci.2006;103:7807–7812.
- .Pathogenesis of ascites and renal salt retention in cirrhosis.J Invest Med.1999;47:183–202.
- ,.Hyponatremia.N Engl J Med.2000;342:1581–1589.
- ,,.The treatment of hyponatremia.Semin Nephrol.2009;29:282–299.
- .Hyponatremia in the intensive care unit.Semin Nephrol.2009;29:257–270.
- ,,,,.Conivaptan and its role in the treatment of hyponatremia.Drug Des Devel Ther.2009;3:253–268.
- .Vaptans for the treatment of hyponatremia: how who when and why.Nephrol Self Assess Program.2007;6:199–209.
- .The syndrome of inappropriate secretion of antidiuretic hormone (SIADH).Semin Nephrol.2009;29(3):239–256.
- ,,,,.Mild hyponatremia and risk of fracture in the ambulatory elderly.Q J Med.2008;101:583–588.
- ,,,,.Hyponatremia independent of osteoporosis is associated with fracture occurrence.Clin J Am Soc Nephrol.2010 (in press).
- ,,, et al.Hyponatremia‐induced osteoporosis.J Bone Miner Res.2009;999:1–37.
- ,,,,.Impact of hospital‐associated hyponatremia on selected outcomes.Arch Intern Med.2010;170(3):294–302.
- ,,, et al.Mortality after hospitalization with mild, moderate, and severe hyponatremia.Am J Med.2009;122:857–865.
- ,.Integrated Response to Abnormalities in Tonicity. In: Seldin DW, Giebisch G, eds.Clinical Disturbances of Water Metabolism.New York, New York:Raven Press Ltd;1993:273–295.
- ,,.Causes and management of hyponatremia.Ann Pharmacother.2003;37:1694–1702.
- ,,,,,.Regulation of aquaporin‐2 trafficking.Handb Exp Pharmacol.2009;190:133–157.
- ,,.Evaluation and management of hyponatremia: an emerging role for vasopressin receptor antagonists.Nat Clin Pract Nephrol.2007;3:82–95.
- ,,,,.Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia.Am J Nephrol.2007;27:447–457.
- ,,, et al.Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355:2099–112.
- ,,, et al.V1a vasopressin receptors maintain normal blood pressure by regulating circulating blood volume and baroreflex sensitivity.Proc Natl Acad Sci.2006;103:7807–7812.
- .Pathogenesis of ascites and renal salt retention in cirrhosis.J Invest Med.1999;47:183–202.
- ,.Hyponatremia.N Engl J Med.2000;342:1581–1589.
- ,,.The treatment of hyponatremia.Semin Nephrol.2009;29:282–299.
- .Hyponatremia in the intensive care unit.Semin Nephrol.2009;29:257–270.
- ,,,,.Conivaptan and its role in the treatment of hyponatremia.Drug Des Devel Ther.2009;3:253–268.
- .Vaptans for the treatment of hyponatremia: how who when and why.Nephrol Self Assess Program.2007;6:199–209.
- .The syndrome of inappropriate secretion of antidiuretic hormone (SIADH).Semin Nephrol.2009;29(3):239–256.
- ,,,,.Mild hyponatremia and risk of fracture in the ambulatory elderly.Q J Med.2008;101:583–588.
- ,,,,.Hyponatremia independent of osteoporosis is associated with fracture occurrence.Clin J Am Soc Nephrol.2010 (in press).
- ,,, et al.Hyponatremia‐induced osteoporosis.J Bone Miner Res.2009;999:1–37.
- ,,,,.Impact of hospital‐associated hyponatremia on selected outcomes.Arch Intern Med.2010;170(3):294–302.
- ,,, et al.Mortality after hospitalization with mild, moderate, and severe hyponatremia.Am J Med.2009;122:857–865.
Managing Hyponatremia in Cirrhosis
The serum sodium (Na) level is the major determinant of serum osmolality. In normal physiologic states is tightly regulated between 135 mEq/L to 145 mEq/L despite variable intake of water and solute through the interaction of osmoreceptors in the hypothalamus where arginine vasopressin (AVP) is synthesized and then released by the posterior pituitary and the binding of AVP with V2 AVP receptors on the basolateral surface of the principal cells within the collecting duct of the kidney. Binding of AVP to the V2 receptors promotes the translocation and fusion of cytoplasmic vesicles which carry the water channel protein aquaporin 2 (AQP2) to the apical membrane of the cell and, in this manner, increases water permeability and absorption.1, 2, 3
Patients with hyponatremia, defined by a serum Na level 135 mEq/L, can be broadly classified by their volume status into those who are euvolemic, hypervolemic, and hypovolemic (Table 1). In patients with euvolemic hyponatremia such as those with Syndrome of Inappropriate Antidiuretic Hormone (SIADH), total body Na is nearly normal, but total body water is increased. In patients with hypervolemic hyponatremia, both total body Na and water are increased, but water to a much greater degree. These patients typically have increased extracellular fluid such as edema and/or ascites. The most common conditions associated with this condition are cirrhosis, congestive heart failure (CHF), and renal failure. In contrast, hypovolemic hyponatremia is associated with a reduction in both total body Na and water, but Na to a greater degree. This condition is encountered in patients with excessive fluid losses such as those with over‐diuresis, excessive gastrointestinal losses, burns, and pancreatitis.4
| Depletional Hyponatreima | Dilutional Hyponatremia | ||
|---|---|---|---|
| Euvolumic | Hypervolumic | ||
| Total body water | |||
| Total body Na | normal | ||
| Common etiologies | SIADH | cirrhosis/CHF | vomiting, diarrhea |
Hyponatremia is the most common electrolyte abnormality seen in general hospital patients.5 In a database of over 120,000 patients, a serum sodium level of 136mEq/L was observed in 28.2%.6 Hyponatremia is associated with selected medical conditions (especially cirrhosis and CHF), the extremes of age, and those receiving selected medications, including several that are commonly administered to cirrhotic patients (diuretics, selective serotonin reuptake inhibitors, opiates, proton‐pump inhibitors).7, 8 Hyponatremia is associated with increased total costs per hospital admission.5, 9 In an analysis of the effect of hyponatremia on length of stay in a retrospective cohort study of hospitalized patients derived from a large administrative database of 198,281 discharges from 39 US hospitals, mean length of stay was significantly greater among patients with hyponatremia than those with normal Na levels (8.6 8.0 vs. 7.2 8.2 days). After adjusting for confounders that may be associated with more severe disease and hyponatremia (age, gender, race, geographic region, teaching status of the hospital, admission source, principal payer, comorbidity index score and primary diagnosis), the presence of hyponatremia contributed an increase in length of stay of 1.0 day. Patients with hyponatremia are more frequently admitted to the intensive care unit (ICU) and require mechanical ventilation. In patients with CHF, the presence of hyponatremia at discharge is associated with increased risk for early mortality and rehospitalization.10
Although frequently asymptomatic, hyponatremia may be associated with a range of findings, from subtle and non‐specific complaints, including headache, fatigue, confusion, malaise, to severe and life‐threatening manifestations with lethargy, seizures, brainstem herniation, respiratory arrest and death.11 The most important complications are neurologic consequences related to cerebral edema. However, there is increased morbidity even in hyponatremic patients considered to be asymptomatic. Patients with low serum sodium have attention deficients, and falls are common. In a study of 122 patients who were considered to have chronic asymptomatic hyponatremia, the incidence of falls was significantly higher at 21.3% compared to only 5.3% in a control population.12
In hyponatremia, water enters into the cells to attain osmotic balance, resulting in cellular swelling.4 To avoid cerebral edema, the brain is capable of adapting to hyponatremia by regulating its volume to avoid swelling, especially when hyponatremia is chronic. In acute hyponatremia, astrocytes and neurons adapt through osmoregulatory mechanisms by extruding intracellular electrolytes such as potassium.13 Chronically, adaption occurs through the loss of low‐molecular weight organic compounds termed organic osmolytes including myoinsoitol, glutamine, choline and taurine. As a result, both the severity and the rate of its development are critical factors in determining the neurologic manifestation of hyponatremia in a given patient.14
Dilutional Hyponatremia and Cirrhosis
Patients with hyponatremia who are either euvolemic or hypervolemic are considered to have dilutional hyponatremia (DH). Management of these patients is distinct from those who are hypovolemic in whom appropriate therapy consists of the administration of normal saline. The remainder of this article addresses the pathogenesis, management and treatment of cirrhotic patients with DH.
Pathogenesis
The development of hyponatremia in cirrhosis is intimately related to the pathophysiology of portal hypertension and the non‐osmotic release of AVP3, 15 (Figure 1). In the early phases of cirrhosis, portal hypertension is the result of an increase in intrahepatic resistance. With the development of porto‐systemic collaterals, a hyperdynamic splanchnic circulation develops as a result of splanchnic arterial vasodilatation and increased vascular capacity. Nitric oxide, an endothelial derived relaxing factor, is the critical mediator of this process, and upregulation of its expression is pivotal in the pathogenesis of portal hypertension.
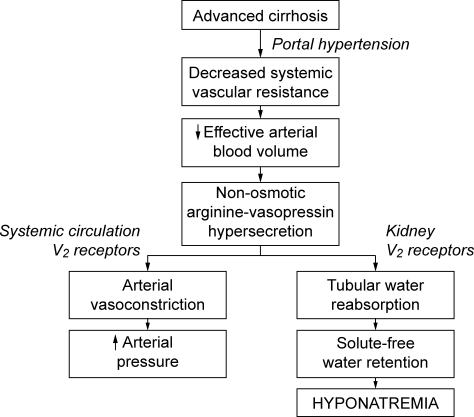
Multiple factors are related to the development of DH in cirrhosis. A reduction of effective central blood volume due to the development of porto‐venous collaterals and arterial splanchnic vasodilation, leading to baroreceptor‐mediated nonosmotic release of AVP, is considered the initiating and most important factor. Patients with cirrhosis and DH have higher plasma and urine vasopressin levels, higher plasma renin activity, and decreased plasma levels of atrial natriuretic factor than those with normal serum sodium concentrations, findings consistent with the presence of a decreased effective plasma volume.16 Arterial underfilling is sensed by baroreceptors located in the left ventricle, aortic arch, carotid sinus and renal afferent arterioles. Decreased activation leads to neurohumoral compensatory responses which include non‐osmotic release of vasopressin from the neurohypophysis and increased levels. Impaired catabolism of AVP that has been correlated with the severity of liver dysfunction may further contribute to increased levels.17 Initially, the increased AVP maintains arterial circulatory integrity by inducing splanchnic, peripheral and renal arterial vasoconstriction through its action on the V1a receptors and expansion of blood volume through renal water retention by its action on the V2 receptors located on the collecting ducts.
The initial adaptive response which leads to increased central blood volume can chronically result in detrimental effects, including the development of fluid overload with ascites, edema, and hyponatremia.16, 18 Additional factors that contribute to hyponatremia include decreased glomerular filtration rate (GFR) and/or increased proximal reabsorption of sodium (that reduce the distal delivery of filtrate and the potential for water reabsorption) and decreased cardiac function that further impairs effective central blood volume.19 In addition, urinary levels of AQP2 are increased in cirrhotic patients, especially those with decompensated disease with higher Child‐Pugh scores and ascites, and provide another potential mechanism to increase water reabsorption.20
Prevalence and Prognostic Significance
Hyponatremia in cirrhosis is a common finding. In a survey of 997 cirrhotic patients with ascites from 28 centers in Europe, North and South America, the prevalence of serum sodium concentration 135, 130, 125, 120 meq/L were 49.4%, 21.6%, 5.7%, and 1.2%, respectively.21 In a retrospective analysis of 188 inpatients, the prevalence of DH of 135, 130, and 125 were 20.8%, 14.9%, and 12.2%, respectively.22 The development of hyponatremia is a manifestation of increasing portal hypertension. In a natural history study of 263 patients hospitalized for first episode of significant ascites, 74 patients developed DH (Na level 130 mEq/L), including 11 patients in whom it appeared during the first episode and 63 cases during follow‐up (mean period of 40 3 months) with a 5‐year incidence of 37.1%.23
The presence of hyponatremia carries significant adverse prognostic significance. It is strongly associated with severity of liver function impairment as assessed by Child‐Pugh and model for end‐stage liver disease (MELD) scores.22 Even mild hyponatremia is associated with severe complications such as massive ascites, severe hepatic encephalopathy, spontaneous bacterial peritonitis (SBP), and hepatic hydrothorax, and the severity of hyponatremia is directly related to the severity of these complications.21, 22 (Figure 2). In a natural history study of patients presenting with large volume ascites, 1‐year survival after its development was reduced to only 25.6%.230
Hyponatremia is an especially poor prognostic sign for a hospitalized cirrhotic patient. In a retrospective analysis of 156 cirrhotic patients, hyponatremiapresent in 57 (29.8%) of admissionswas associated with increased hospital mortality (26.3% vs. 8.9% among those with normal Na levels), and the mortality rate was even higher (48%) among the 25 patients who developed severe hyponatremia during the hospital stay.24 In hospitalized patients, hyponatremia is predictive of the development of acute renal failure which is associated with substantially increased mortality (73% vs. 13%).25 Similarly, a low serum sodium level in critically ill cirrhotic patients admitted to the ICU is associated with complications, in‐hospital mortality, and poor short‐term prognosis.26
Whether hyponatremia should impact liver transplant prioritization remains an area of controversy. The United Network for Organ Sharing (UNOS) contracted by the Organ Procurement and Transplant Network (OPTN) to optimize the efficient use of deceased organs through fair and timely allocation, currently uses the MELD score, a formula that calculates the risk of death within three months from the bilirubin, creatinine, and International Normalized Ratio (INR) levels. Hyponatremia is an earlier and more sensitive marker than serum creatinine to detect renal impairment and/or circulatory dysfunction in patients with advanced cirrhosis and adds to MELD in predicting waitlist mortality.2729 In patients with a MELD score of 21, only low serum sodium and persistent ascites are independent predictors of mortality.28 To account for the importance of hyponatremia on survival, both modification of the MELD score in which the Na level is incorporated (MELD‐Na model) and the MELD to serum sodium ratio (MESO) have been developed. Adding hyponatremia to the MELD score is a better predictor of death than MELD alone, particularly in patients with low MELD scores.27, 2931 The OPTN/UNOS Liver and Intestinal Organ Transplantation Committee has discussed updating the liver allocation system to include the Na level. However, it was concluded that implementation of MELD‐Na would change the allocation status of only 4% of candidates. Further, based on the concerns about the ability to manipulate serum sodium levels and the utility of employing resources to change the system for a relatively small number of patients, it was decided to defer incorporating the Na level pending further analysis (Report of the OPTN/UNOS Liver and Intestinal Organ Transplantation Committee To the Board of Directors, Los Angeles, California, September 17‐18, 2007). At this time, the use of Na is a regional decision.32 However, the OPTN/UNOS Liver and Intestinal Organ Transplantation Committee has recently solicited feedback from the transplant community about including Na in allocation for review at a forum in April 2010.
Precipitating Factors
The most important factor related to development of hyponatremia in cirrhosis is increasing severity of portal hypertension that is associated with impaired central blood volume as a result of progressive splanchnic vasodilatation. In a study in which 170 patients with decompensated alcoholic cirrhosis were prospectively followed for 33.9 27.9 months, the initial hepatic venous pressure gradient (HVPG) was an independent predictive factor for the 20 patients who developed hyponatremia.22
Cirrhotic patients with ascites with hyponatremia have increased AVP secretion, higher levels of plasma renin activity, and higher serum concentrations of aldosterone and norepinephrine compared to those with normal Na levels.33 Diuretic therapy is associated with the development of DH by inducing volume depletion and arterial underfilling, further activating the renin‐angiotensin system (RAS) and increasing the non‐osmotic release of AVP.34 Although diuretics block the salt retention associated with the RAS activation, the water‐retaining effects of AVP persist, and DH develops. The process is further exacerbated by a low sodium intake and a frequent uncontrollable thirst. As a result, diuretic therapy is commonly associated with the development of hyponatremia in patients with ascites.24, 35 Similarly, paracentesis (particularly when performed without albumin) is often associated with an increase in blood urea nitrogen and marked elevations in plasma renin activity and plasma aldosterone concentration, which may exacerbate this physiology, leading to further reduction in serum sodium concentration.36 Tense ascites can contribute to DH by increasing baroreceptor mediated AVP release by increasing intrathoracic pressure.37 Finally, non‐steroidal anti‐inflammatory drugs (NSAIDs) can cause DH by inhibiting the synthesis of renal prostaglandins (which normally function to antagonize the tubular action of AVP and are important in the maintenance of appropriate renal tubular transport of fluid and electrolytes in states of renal hypoperfusion).38
Medical Impact of Hyponatremia: Marker of Severe Disease or Direct Pathophysiologic Role?
Hyponatremia is associated with severe ascites, impaired renal function, hepatic encephalopathy, SBP, and hepatorenal syndrome.3, 20 Because hyponatremia is frequently present in advanced liver failure, it is unclear whether it is only a marker of advanced disease or whether it plays a direct pathophysiologic role, or both. Until recently, it has not been possible to address this issue due to the inability to easily and rapidly correct the hyponatremia. However, there is increasing evidence that hyponatremia has direct impact on the severity of hepatic encephalopathy (see Hepatic Encephalopathy section). The recent introduction of tolvaptan for the treatment of hyponatremia in cirrhosis (discussed below) will allow this question to be directly answered.
Fluid Management and Diuresis
The typical cirrhotic patient with DH is characterized by expanded extracellular fluid with ascites and edema. The profound vasodilation of the splanchnic arterial circulation is associated with decreased effective arterial blood volume, leading to the non‐osmotic release of AVP. Diuretic therapy can further exacerbate this process. In addition, the increased water permeability induced by AVP results in reduced urine volume and fluid retention. As a result, hyponatremia directly adversely affects severity of fluid overload and limits and/or precludes diuretic treatment.
Hepatorenal Syndrome
Hyponatremia is an earlier and more sensitive marker than serum creatinine to detect renal impairment and/or circulatory dysfunction and is frequently a precursor to overt hepatorenal syndrome.27 Hyponatremia is predictive of the development of acute renal failure during hospitalization, and in‐hospital development of acute renal failure portends a high mortality.25 In patients admitted with SBP, the presence of hyponatremia is significantly associated with higher mortality and renal failure.39
Hepatic Encephalopathy
The neurologic manifestations of cerebral edema associated with hyponatremia closely mirror those of hepatic encephalopathy. In fact, a recently proposed pathogenic mechanism for hepatic encephalopathy is the development of low‐grade cerebral edema associated with astrocyte swelling in response to ammonia and other precipitating factors.40 DH is associated with a further reduction in brain organic osmolytes that probably reflects a compensatory osmoregulatory mechanism against cell swelling triggered by a combination of high intracellular glutamine and low extracellular osmolality.41 As a result, it has been proposed that hyponatremia contributes to the development of hepatic encephalopathy through the development or exacerbation of low‐grade cerebral edema. In this manner, low serum sodium acts as a second hit to the swelling produced by increased intracellular glutamine created by ammonia metabolism.42
Clinically, hyponatremia is a major risk factor for hepatic encephalopathy. Serum sodium and ammonia levels are the major factors that predict electroencephalographic abnormalities in cirrhotics who do not have hepatic encephalopathy.43 In a prospective study of 61 patients, hyponatremia was associated with a low brain concentration of organic osmolytes as assessed by proton magnetic resonance spectroscopy (1H‐MRS) and magnetic resonance imaging, and both conditions were major risk factors for the development of overt hepatic encephalopathy.44 Finally, hyponatremia is a risk factor for hepatic encephalopathy in patients undergoing TIPS.45
Adverse Effect on Outcome After Liver Transplantation
Hyponatremia before liver transplantation is associated with adverse post‐transplant outcomes. Among patients undergoing liver transplantation, the presence of hyponatremia is associated with abnormal cardiac response in patients after reperfusion.46 Pre‐transplant hyponatremia is associated with longer ICU and hospital stay, higher rates of delirium and neurologic disorders, acute renal failure, acute cellular rejection, infection, and in one study a reduced 3‐month survival compared to normonatremic recipients.32, 47, 48 In 1 retrospective study that compared post‐transplant outcomes of patients with corrected vs. uncorrected pre‐transplant hyponatremia, patients with pre‐operative correction of hyponatremia had a lower risk of prolonged post‐transplant hospitalization than those with uncorrected hyponatremia.32 However, both hyponatremic groups had more complicated post‐transplant courses compared to those without a history of hyponatremia. However, given the small sample size, retrospective design, and the potential for confounding, the impact of correction of pre‐transplant hyponatremia remains to be determined.
Management
Most patients with mild hypervolemic hyponatremia are asymptomatic. The initial recommended approach is fluid restriction and an Na‐restricted diet. For those with severe or progressive hyponatremia, diuretics should be minimized or discontinued to avoid intravascular volume depletion.49 For patients with tense ascites and severe DH, therapeutic paracentesis with plasma expanders is safe.33 Unfortunately, fluid restriction is limited in efficacy and often poorly tolerated. The use of hypertonic saline is generally not recommended unless severe neurologic symptoms are present as it leads to increased ascites and edema. When administered, it is important to avoid a rapid correction of the hyponatremia to prevent the development of central pontine myelinolysis and the osmotic demyelination syndrome.
Due to the pivotal role of AVP in the pathogenesis of DH, antagonism of its action has long been proposed to be the most rational approach, but until recently, effective and specific antagonism of AVP has remained elusive. Approaches that have been attempted include interference with its secretion and actions. Intravenous albumin has been reported to improve hyponatremia in patients with cirrhosis, ascites, and hyponatremia, presumably by decreasing AVP release by plasma volume expansion.50 An attempt at inhibition of central AVP release with the use of a kappa‐opioid receptor agonist, niravoline, was limited by loss of efficacy and potential adverse effects.51 Use of demeclocycline and lithium (which induce renal resistance to AVP and lead to a modest increase in urine volume with decreased urine osmolality and a corresponding rise in serum sodium) is limited by nephrotoxicity and hepatotoxicity.7, 52 Because of the important role played by prostaglandins in the maintenance of renal hemodynamics and water excretion in cirrhosis, oral misoprostol has also been evaluated but determined to be ineffective in inducing significant changes in free water clearance in patients with functional renal failure and/or DH.53
The recent introduction of vaptans, vasopressin receptor antagonists that block the physiologic action of vasopressin, represents a revolutionary and highly effective approach to the treatment of hyponatremia. Vaptans are antagonists of the V2 receptors of AVP in the principal cells of the collecting ducts. In healthy subjects, vaptans cause a dose‐dependent increase in urine volume and produce a dilute urine without causing natriuresis. To date, 2 AVP antagonists, conivaptan and tolvaptan, have been Food and Drug Administration (FDA)‐approved for the treatment of DH. Conivaptan, the first to be approved in 2005, is a mixed vasopressin V1a and V2 receptor antagonist that is administered intravenously for up to 4 days. In a randomized placebo‐controlled study of patients with euvolemic or hypervolemic hyponatremia, intravenous conivaptan treatment increased serum Na levels by >6 mEq/l or to a serum Na >135 mEq/l in 69 to 88.5% of subjects compared to 20.7% of those receiving placebo (Zeltser D, Rosansky S, Van Rensburg H, et al. Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia. American J Nephrology 2007;27:447457). In a pilot study involving 24 patients with end‐stage liver disease, an infusion of conivaptan over 1 to 4 days was associated with an increase of serum sodium by >5 mmol/L in 60% of patients not receiving diuretics and 67% of patients on concomitant diuretic therapy by the end of treatment (O'Leary and Davis, 2009). Despite a concern about the potential for conivaptan to increase portal hypertension due to inhibition of splanchnic V1a receptors, the brief treatment appeared to be well tolerated without significant changes in systolic blood pressure, serum creatinine, variceal bleeding or worsening of ascites during the infusion period. However, approval for only 4 days of therapy and requirement for intravenous use eliminate any potential for chronic use.54
Tolvaptan is an orally available, selective V2 receptor antagonist whose efficacy was assessed in two multicenter, prospective, randomized, placebo‐controlled trials, Study of Ascending Levels of Tolvaptan in Hyponatremia 1 and 2 (SALT‐1 and SALT‐2).55 In these trials, clinically stable patients with DH (Na 135meq/l) associated with cirrhosis (22.4% in SALT‐1, 30.5% in SALT‐2), CHF or SIADH were randomized in a hospital setting to receive tolvaptan 15mg daily or placebo. Repeat Na levels were obtained at 8 hours, 2, 3, and 4 days and then weekly at days 11, 18, 25 and 30 after which study drug was discontinued and follow‐up Na level was determined 7 days later. The dose was adjusted to 30 mg and then 60 mg in an attempt to achieve a Na level >135 in those in whom hyponatremia persisted. During the initial day of the titration phase, fluid restriction was not maintained, and the patients were encouraged to respond to thirst with increased water ingestion.
Tolvaptan use was associated with a prompt increase in Na level as early as 8 hours after administration of the first dose. Serum Na increased more among those receiving tolvaptan than among those receiving placebo during the first 4 days and throughout the study period regardless of baseline Na level but returned to baseline within 1 week after discontinuation (Figure 3). The main side effects were increased thirst, dry mouth and increased urination. Importantly, an increased incidence of renal failure was not observed. Based on these results, FDA approval for tolvaptan in patients with hyponatremia was obtained in May 2009 for patients with DH‐associated with cirrhosis, CHF or SIADH for patients with Na levels 125 or symptomatic patients with Na levels between 125 and 135 that have not responded to fluid restriction.
Management of the Hospitalized Cirrhotic Patient With Hyponatremia: Recommendations
Hyponatremia in hospitalized cirrhotic patients is a marker for severe disease and high risk of hospital mortality.24 As a result, prompt evaluation and treatment is imperative. The availability of tolvaptan potentially revolutionizes the manner in which these patients are treated. In the SALT trials, only clinically stable patients were enrolled. In this last section, a guideline for the evaluation and treatment of acutely ill, hospitalized cirrhotic patients with DH is presented.
Evaluation
Determination of volume status is paramount but frequently problematic in the hospitalized cirrhotic patient. Due to the vasodilated state present in severe portal hypertension that is characterized by a relative hypotension and resting tachycardia, the usual hemodynamic parameters of blood pressure and heart rate can be difficult to interpret. Although significant extravascular volume in the form of ascites and edema may be present, patients may be intravascularly depleted due to previous diuretic use and extra‐renal losses due to impaired oral intake, vomiting, lactulose‐induced diarrhea, and gastrointestinal bleeding. Infection is a commonly associated condition, and endotoxin mediated splanchnic vasodilatation, especially in the setting of SBP, can adversely effective central blood volume status in the presence of severe ascites. Also, due to the Na avidity of the kidney and previous diuretic use, renal electrolytes can be difficult to interpret.
For patients in whom there is strong clinical concern about intravascular depletion (history of impaired oral intake, excessive vomiting and/or diarrhea, rapid weight loss, small volume ascites with history of large volume, azotemia), administration of limited intravenous normal saline (0.5‐1 L) should be considered. Patients with severe neurological symptoms should receive normal saline or hypertonic saline. Unless severe neurologic symptoms associated with profound hyponatremia is present, however, intravenous normal saline should not be administered for the hyponatremia alone. Administration of salt poor albumin (25%), especially for those with marked fluid overload and ascites, is an effective means to expand the central blood volume without exacerbating ascites and edema. After evaluation for and/or treatment of hypovolemia, all patients should receive a Na restricted diet (2000 mg daily) and placed on fluid restriction (see below for liberalization of fluid restriction upon initiation of tolvaptan therapy).
Diagnostic paracentesis should be performed for those with ascites to rule out the presence of SBP, and antibiotics administered to those with evidence of infection. High dose intravenous salt poor albumin should also be administered, especially to those at high risk of renal failure as determined by the presence of azotemia (Cr > 1.0 mg/dL) or severe liver insufficiency (TBili > 4.0 mg/dL).56, 57 Finally, all medications should be reviewed, and those associated with hyponatremia (diuretics, selective serotonin reuptake inhibitors, opiates, proton‐pump inhibitors) discontinued if possible.
Tolvaptan for DH
Patient Selection
Appropriate patient selection for tolvaptan therapy is extremely important (Table 2). In the SALT trials, only clinically stable patients were enrolled. The presence of hyponatremia in a recently hospitalized cirrhotic patient, however, frequently indicates severe disease with a high risk of acute renal failure and hospital death. In the SALT trials, many received concomitant diuretic therapy. Because of the importance of avoiding tolvaptan administration to hypovolemic patients, discontinuation of diuretic therapy prior the initiation of tolvaptan therapy and/or reevaluation after limited volume expansion should be considered.
| Hospital setting |
| Euvolumia or hypervolumia |
| Absence of recent weight loss, decrease in ascites, edema |
| Absence of excessive vomiting, diarrhea |
| Consider discontinuation of diuretic therapy prior to initiation of tolvaptan |
| Consider evaluation after limited volume expansion, especially with salt poor albumin prior to initiation of tolvaptan |
| Presence of clinically significant hyponatremia: 125mEq/L or less severe but symptomatic hyponatremia (125 to 134 mEq/L) that has resisted fluid restriction |
| Absence of severe neurologic symptoms attributable to hyponatremia |
| No co‐administration with intravenous saline |
| Ability to respond to thirst |
| No co‐administration with strong CYP 3A inhibitors (ketoconazole) |
| Absence of kidney failure with anuria |
Tolvaptan is indicated for cirrhotic patients with DH in whom the serum sodium is 125 mEq/L and in those with less severe but symptomatic hyponatremia (125‐134 mEq/L) that has resisted fluid restriction. Although the definition of symptomatic was not specifically defined, possible considerations include symptoms of mild hepatic encephalopathy or inability to tolerate dieresis due to the presence of hyponatremia. According to FDA guidelines, tolvaptan therapy must be initiated and re‐initiated in a hospital setting. Patients with severe neurologic symptoms attributable to hyponatremia in whom rapid treatment is critical should not receive tolvaptan but should rather be treated with normal saline. Similarly, patients should not receive combination therapy with tolvaptan and normal saline due to potential for a too‐rapid correction of hyponatremia and the development of central pontine myelinolysis. If saline had been administered for treatment of possible hypovolemia, it should be discontinued and persistent hyponatremia confirmed before starting tolvaptan. Other factors that need to be considered before initiating tolvaptan include the ability of the patient to respond to thirst with increased water ingestion and recognition that the patient will experience increased urine volume and frequency, requiring easy access to toilet. Patients should not be fluid restricted during the first day of tolvaptan therapy, but should be instructed to respond to their thirst with increased water ingestion. As a result, caution should be exercised in administering tolvaptan to a confused, restrained, unresponsive and/or bed‐bound patient who is not able to respond appropriately to thirst or increased urination.
In the SALT trials, the incidence of hyperkalemia (5%) was similar in the tolvaptan and placebo treated patients.55 However, further analysis of all multiple‐dose, placebo‐controlled trials, demonstrated that the aggregate incidence of hyperkalemia was slightly higher for tolvaptan‐treated subjects compared with placebo‐treated subjects (Otsuka). Because treatment with tolvaptan is associated with an acute reduction of the extracellular fluid volume which could result in increased serum potassium through hemoconcentration, it is recommended that serum potassium levels be monitored after initiation of tolvaptan treatment in patients with a serum potassium > 5 mEq/L as well as those who are receiving drugs known to increase serum potassium levels such as angiotensin converting enzyme inhibitors, angiotensin receptor blockers, or potassium sparing diuretics (Samsca Package Insert, Otsuka). Because tolvaptan is metabolized by the cytochrome P 3A system, patients receiving strong inhibitors such as ketoconazole should not receive tolvaptan. Anuric patients will not respond to tolvaptan. Finally, it is extremely important to administer tolvaptan only to patients with true hyponatremia and not to those with pseudohyponatremia in whom the plasma osmolality is normal but the measured serum sodium concentration artificially low due to marked elevations of other substances, such as can be seen in severe hyperglycemia, marked hyperlipidemia, or hyperproteinemia (as in multiple myeloma).
Tolvaptan Administration
The initial dose of tolvaptan is 15 mg daily. After receiving tolvaptan, many patients will develop an increased sense of thirst and need to urinate. As a result, patients should not be fluid restricted during the first day of therapy, and it is important to monitor the hemodynamics and Na level closely after initiating therapy with a repeat Na level at approximately 8 hours after the first dose. As a result, it should probably be administered early in the day and not at bedtime. The dose should be increased to 30 mg, then 60 mg in patients who do not respond by at least 5 mEq/L over the previous 24 hours and remain hyponatremic. In those with an excessive response (more than 8 meq/L during the first 8 hours or 12 meq/L on any subsequent day), the patient should be encourage to either drink more water, or the dose should be held or reduced. After the appropriate dose has been identified, the patient may be discharged and continued on tolvaptan long‐term.
With the advent of this exciting therapy, practical issues will need to be addressed, most important of which is its cost at $250 per day (Otsuka). In addition, the current recommendation to initiate tolvaptan only in a hospital further limits its widespread use. Most important, long‐term clinical benefit will need to be demonstrated. Although the SALT trials only involved treatment for up to 1 month, a multicenter, open‐label extension study for a mean duration of 701 days demonstrated that prolonged administration of tolvaptan maintains an increased serum sodium level.58 However, at this time, tolvaptan can only be considered as one of the promising drugs whose long‐term cost‐effectiveness is yet to be proven. Proof will require showing that correction of the hyponatremia leads to improved clinical outcomes, such as a reduction in length of stay or frequency of hospitalization, decreased renal failure, improved hepatic encephalopathy, deceased mortality, and improved post‐transplant outcomes.
Unanswered Questions
The vaptans provide an important opportunity to clarify the role that hyponatremia plays in the pathogenesis of cirrhosis. In the past, DH in a cirrhotic patient represented a sign of advanced disease. With the availability of safe and effective therapy, we can now determine whether it also plays an important role in the pathophysiology of end‐stage liver disease and whether its treatment will have a beneficial effect on patient outcomes.
Specific clinical questions that will inevitably be addressed over the next few years to determine whether DH is only a marker for advanced disease or whether it plays a direct but modifiable role in the pathophysiology of cirrhosis will include:
-
Role of vaptans in the management of ascites: In a 14‐day randomized, trial of a satavaptan, another selective vasopressin V(2) receptor antagonist, vs. placebo with spironolactone, combination therapy was associated with improved control of ascites and improvements in serum sodium levels in hyponatremic patients with ascites.59 If future similar studies demonstrate more prolonged benefits, this would constitute an important advance in the treatment of ascites in cirrhosis.
-
Effect on renal function: Prolonged use of tolvaptan leads to a compensatory increase in endogenous levels of AVP and, potentially, increased stimulation of V1a receptors, which might be helpful in the setting of portal hypertension. In patients with hepatorenal syndrome, vasopressin stimulation of splanchnic V1a receptors leads to improved renal function, presumably by decreasing splanchnic blood flow and improving central blood volume.60 As a result, tolvaptan may indirectly improve kidney function in patients with advanced cirrhosis and refractory ascites. Whether long‐term tolvaptan therapy will help to prevent hepatorenal syndrome through this mechanism remains to be determined but is an exciting possibility.61
-
Effect on hepatic encephalopathy: Hepatic encephalopathy is associated with poor quality of life in patients with cirrhosis. Although hepatic encephalopathy was not directly assessed in the SALT trials, the mean mental component summary of the Short Form General Health Survey, a quality of life measure, improved in cirrhotic patients receiving tolvaptan to a greater degree that those receiving placebo.62 A possible explanation for this finding is a beneficial effect of tolvaptan on hepatic encephalopathy. Confirmation of this hypothesis, however, will require prospective studies in which hepatic encephalopathy is directly assessed.
-
Effect on medical economics: Based on retrospective reviews, hyponatremia has an adverse impact on length of stay and outcomes following liver transplantation. It will be important to demonstrate in prospective studies that correction of hyponatremia with tolvaptan reduces length of stay, complications, and costs.
- ,,, et al.The aquaporin family of water channels in kidney: an update on physiology and pathophysiology of aquaporin‐2.Kidney Int.1996;49:1718–1723.
- ,,, et al.Hyponatremia in cirrhosis: from pathogenesis to treatment.Hepatology.1998;28:851–864.
- ,.Hyponatremia in cirrhosis: pathogenesis, clinical significance, and management.Hepatology.2008;48:1002–1010.
- ,.Hyponatremia.N Engl J Med.2000;342:1581–1589.
- ,,,.Economic impact of hyponatremia in hospitalized patients: a retrospective cohort study.Postgrad Med.2009;121:186–191.
- .Age and gender as risk factors for hyponatremia and hypernatremia.Clin Chim Acta.2003;337:169–172.
- .Consequences of inadequate management of hyponatremia.Am J Nephrol.2005;25:240–249.
- ,,.A review of drug‐induced hyponatremia.Am J Kidney Dis.2008;52:144–153.
- ,,, et al.Epidemiology, clinical and economic outcomes of admission hyponatremia among hospitalized patients.Curr Med Res Opin.2008;24:1601–1608.
- ,,.Incidence and prevalence of hyponatremia.Am J Med.2006;119(7 Supple 1):S30–S35.
- ,,.Disorders of sodium and water balance in hospitalized patients.Can J Anesth.2009;56:151–167.
- ,,, et al.Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits.Am J Med.2006;119;71.e1‐8.
- ,.Brain volume regulation in response to hypo‐osmolality and its correction.Am J Med.2006;119 (7 Suppl 1):S12–S16.
- ,,.Neurological manifestations and morbidity of hyponatremia: correlation with brain water and electrolytes.Medicine.1976;55:121–129.
- ,.The hyperdynamic circulation of chronic liver diseases: from the patient to the molecule.Hepatology.2006;43:S121–S131.
- ,,, et al.Hyponatremia of cirrhosis: role of vasopressin and decreased “effective” plasma volume.Scand J Gastroenterol.1997;32:829–834.
- ,,,.Metabolic clearance rate of arginine vasopressin in patients with cirrhosis.Hepatology.1992;16:974–979.
- .Water and sodium retention in edematous disorders: role of vasopressin and aldosterone.Am J Med.2006;119:S47–S53.
- ,,, et al.Circulatory function and hepatorenal syndrome in cirrhosis.Hepatology.2005;42:439–447.
- ,,, et al.Aquaporin‐2 urinary excretion in cirrhosis: relationship to vasopressin and nitric oxide.Dig Dis Sci.2010;55(4):1135–1141.
- ,,, et al.Hyponatremia in cirrhosis: results of a patient population survey.Hepatology.2006;44:1535–1542.
- et al.Hyponatremia and mortality among patients on the liver‐transplant list.N Engl J Med.2009;359:1018–1026.
- ,,, et al.Natural history of patients hospitalized for management of cirrhotic ascites.Clin Gastroenterol Hepatol.2006;4:1385–1394.
- ,,, et al.Clinical relevance of hyponatremia for the hospital outcome of cirrhotic patieints.Dig Liver Dis.2000;32:605–610.
- ,,, et al.Incidence and factors predictive of acute renal failure in patients with advanced liver cirrhosis.Clin Nephrol.2006;65:28–33.
- ,,, et al.Serum sodium predicts prognosis in critically ill cirrhotic patients.J Clin Gastroenterol2010;44(3):220–226.
- ,,, et al.Addition of serum sodium into the MELD score predicts waiting list mortality better than MELD alone.Liver Transpl.2005;11:336–343.
- ,,, et al.Persistent ascites and low serum sodium identify patients with cirrhosis and low MELD scores who are at high risk for early death.Hepatology.2004;40:802–810.
- ,,, et al.,Validation of model for end‐stage liver disease score to serum sodium ratio index as a prognostic predictor in patients with cirrhosis.J Gastroenterol Hepatol.2009;24:1547–1553.
- ,,, et al.Hepatic venous pressure gradient can predict the development of hepatocellular carcinoma and hyponatremia in decompensated alcoholic cirrhosis.Eur J Gastroenterol Hepatol.2009;21:1241–1246.
- ,,, et al.Serum sodium predicts mortality in patients listed for liver transplantation.Hepatology.2005;41:32–39.
- ,,, et al.Effect of hyponatremia on outcomes following orthotopic liver transplantation.Liver Int.2009;29:1071–1077.
- ,,, et al.Total paracentesis in cirrhotic patients with tense ascites and dilutional hyponatremia.Am J Gastroenterol.1999;94:2219–2223.
- ,,, et al.Dilutional hyponatremia in patients with cirrhosis and ascites.Arch Intern Med.2002;162:323–328.
- ,.Therapeutic approaches to the treatment of edema and ascites: the use of diuretics.Am J Ther.2009;16:98–101.
- ,,, et al.Randomized comparative study of therapeutic paracentesis with and without intravenous albumin in cirrhosis.Gastroenterol.1988;94:1493–1502.
- ,,, et al.Effect of intrathoracic pressure on plasma arginine vasopressin levels.Gastroenterology.1991;101:607–617.
- .Nephrotoxicities of nonsteroidal anti‐inflammatory drugs.J Formos Med Assoc.1997;96:157–171.
- ,,, et al.Serum creatinine and bilirubin predict renal failure and mortality in patients with spontaneous bacterial peritonitis: a retrospective study.Liver Int.2009;29:415–419.
- ,.Pathogenic mechanisms of hepatic encephalopathy.Gut2008;57:1156–1165.
- ,,, et al.Effects of dilutional hyponatremia on brain organic osmolytes and water content in patients with cirrhosis.Hepatology.2004;39:1613–1622.
- .Low grade cerebral edema and the pathogenesis of hepatic encephalopathy in cirrhosis.Hepatology.2006;43:1187–1190.
- ,,, et al.Prevalence and prognostic value of quantified electroencephalogram (EEG) alterations in cirrhotic patients.J Hepatol.2001;35:37–45.
- ,,, et al.Hyponatremia is a risk factor of hepatic encephalopathy in patients with cirrhosis: a propective study with time‐dependent analysis.Am J Gastroenterol.2009;104:1382–1389.
- ,,, et al.Analysis of prognostic variables in the prediction of mortality, shunt failure, variceal rebleeding and encephalopathy following the transjugular intrahepatic portosystecim stent‐shunt for variceal haemorrhage.J Hepatol.1995;2:123–128.
- ,,, et al.Cardiac dysfunction during liver transplantation: incidence and preoperative predictors.Transplantation.2008;85:1766–1772.
- ,,, et al.,Impact of pretransplant hyponatremia on outcome following liver transplantation.Hepatology.2009;49:1610–1615.
- ,,, et al.Hyponatremia impairs early posttransplantation outcome in patients with cirrhosis undergoing liver transplantation.Gastroenterology.2006;130:1135–1143.
- ,,.Hyponatremia in cirrhosis: clinical features and management.Gastroenterol Clin Biol.2006;30:1144–1151.
- ,,, et al.Intravenous albumin infusion is an effective therapy for hyponatremia in cirrhotic patients with ascites.Gut.1990;31:204–207.
- ,,, et al.Comparison of two aquaretic drugs (niravoline and OPC‐31260) in cirrhotic rats with ascites and water retention.J Pharmacol Exp Ther.1999;289:194–201.
- ,,.Plasma demeclocycline levels and nephrotoxicity. Correlation in hyponatremic cirrhotic patients.JAMA.1980;243:2513–2515.
- ,,, et al.Oral misoprostol or intravenous prostaglandin E2 do not improve renal function in patients with cirrhosis and ascites with hyponatremia and renal failure.J Hepatol.1993;17:220–226.
- ,,, et al.Effect of the V1a/V2‐AVP receptor antagonist, Conivaptan, on renal water metabolism and systemic hemodynamics in rats with cirrhosis and ascites.J Hepatol.2003;38:755–761.
- ,,, et al.Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355:2099–2112.
- ,,, et al.Effect of intravenous albumin on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis.N Engl J Med.1999;341:403–409.
- ,,, et al.Restricted use of albumin for spontaneous bacterial peritonitis.Gut.2007;56:597–599.
- ,,, et al.Oral tolvaptan is safe and effective in chronic hypyonatremia.J Am Soc Nephrol.2010;21(4):705–712.
- ,,, et al.Effects of satavaptan, a selective vasopressin V(2) receptor antagonist, on ascites and serum sodium in cirrhosis with hyponatremia: a randomized trial.Hepatology.2008;48:204–213.
- ,,, et al.A randomized, prospective, double‐blind, placebo‐controlled trial of terlipressin for type 1 hepatorenal syndrome.Gastroenterology.2008;134:1360–1368.
- ,.Tolvapten and its potential in the treatment of hyponatremia.Ther Clin Risk Manag.2008;4:1149–11455.
- ,,, et al.The effects of vasopressin V2 receptor antagonist in the management of patients with cirrhosis and hyponatremia. Safety and efficacy of oral tolvaptan in the SALT trials.Hepatology.2009;50S:467A.
- ,,, et al.Treatment of hyponatremic cirrhosis with ascites resistant to diuretics by urea.Nephron.1986;44:337–343.
- ,,, et al.Aquaretic effects of niravoline, a kappa‐opioid agonist, in patients with cirrhosis.J Hepatol.2000;32:38–42.
- ,, et al.The association between the serum sodium level and the severity of complications in liver cirrhosis.Korean J Intern Med.2009;24:106–112.
- ,,, et al.Nitric oxide in ascitic fluid is an independent predictor of the development of renal impairment in patients with cirrhosis and spontaneous bacterial peritonitis.Eur J Gastroenterol Hepatol.2004;16:571–577.
The serum sodium (Na) level is the major determinant of serum osmolality. In normal physiologic states is tightly regulated between 135 mEq/L to 145 mEq/L despite variable intake of water and solute through the interaction of osmoreceptors in the hypothalamus where arginine vasopressin (AVP) is synthesized and then released by the posterior pituitary and the binding of AVP with V2 AVP receptors on the basolateral surface of the principal cells within the collecting duct of the kidney. Binding of AVP to the V2 receptors promotes the translocation and fusion of cytoplasmic vesicles which carry the water channel protein aquaporin 2 (AQP2) to the apical membrane of the cell and, in this manner, increases water permeability and absorption.1, 2, 3
Patients with hyponatremia, defined by a serum Na level 135 mEq/L, can be broadly classified by their volume status into those who are euvolemic, hypervolemic, and hypovolemic (Table 1). In patients with euvolemic hyponatremia such as those with Syndrome of Inappropriate Antidiuretic Hormone (SIADH), total body Na is nearly normal, but total body water is increased. In patients with hypervolemic hyponatremia, both total body Na and water are increased, but water to a much greater degree. These patients typically have increased extracellular fluid such as edema and/or ascites. The most common conditions associated with this condition are cirrhosis, congestive heart failure (CHF), and renal failure. In contrast, hypovolemic hyponatremia is associated with a reduction in both total body Na and water, but Na to a greater degree. This condition is encountered in patients with excessive fluid losses such as those with over‐diuresis, excessive gastrointestinal losses, burns, and pancreatitis.4
| Depletional Hyponatreima | Dilutional Hyponatremia | ||
|---|---|---|---|
| Euvolumic | Hypervolumic | ||
| Total body water | |||
| Total body Na | normal | ||
| Common etiologies | SIADH | cirrhosis/CHF | vomiting, diarrhea |
Hyponatremia is the most common electrolyte abnormality seen in general hospital patients.5 In a database of over 120,000 patients, a serum sodium level of 136mEq/L was observed in 28.2%.6 Hyponatremia is associated with selected medical conditions (especially cirrhosis and CHF), the extremes of age, and those receiving selected medications, including several that are commonly administered to cirrhotic patients (diuretics, selective serotonin reuptake inhibitors, opiates, proton‐pump inhibitors).7, 8 Hyponatremia is associated with increased total costs per hospital admission.5, 9 In an analysis of the effect of hyponatremia on length of stay in a retrospective cohort study of hospitalized patients derived from a large administrative database of 198,281 discharges from 39 US hospitals, mean length of stay was significantly greater among patients with hyponatremia than those with normal Na levels (8.6 8.0 vs. 7.2 8.2 days). After adjusting for confounders that may be associated with more severe disease and hyponatremia (age, gender, race, geographic region, teaching status of the hospital, admission source, principal payer, comorbidity index score and primary diagnosis), the presence of hyponatremia contributed an increase in length of stay of 1.0 day. Patients with hyponatremia are more frequently admitted to the intensive care unit (ICU) and require mechanical ventilation. In patients with CHF, the presence of hyponatremia at discharge is associated with increased risk for early mortality and rehospitalization.10
Although frequently asymptomatic, hyponatremia may be associated with a range of findings, from subtle and non‐specific complaints, including headache, fatigue, confusion, malaise, to severe and life‐threatening manifestations with lethargy, seizures, brainstem herniation, respiratory arrest and death.11 The most important complications are neurologic consequences related to cerebral edema. However, there is increased morbidity even in hyponatremic patients considered to be asymptomatic. Patients with low serum sodium have attention deficients, and falls are common. In a study of 122 patients who were considered to have chronic asymptomatic hyponatremia, the incidence of falls was significantly higher at 21.3% compared to only 5.3% in a control population.12
In hyponatremia, water enters into the cells to attain osmotic balance, resulting in cellular swelling.4 To avoid cerebral edema, the brain is capable of adapting to hyponatremia by regulating its volume to avoid swelling, especially when hyponatremia is chronic. In acute hyponatremia, astrocytes and neurons adapt through osmoregulatory mechanisms by extruding intracellular electrolytes such as potassium.13 Chronically, adaption occurs through the loss of low‐molecular weight organic compounds termed organic osmolytes including myoinsoitol, glutamine, choline and taurine. As a result, both the severity and the rate of its development are critical factors in determining the neurologic manifestation of hyponatremia in a given patient.14
Dilutional Hyponatremia and Cirrhosis
Patients with hyponatremia who are either euvolemic or hypervolemic are considered to have dilutional hyponatremia (DH). Management of these patients is distinct from those who are hypovolemic in whom appropriate therapy consists of the administration of normal saline. The remainder of this article addresses the pathogenesis, management and treatment of cirrhotic patients with DH.
Pathogenesis
The development of hyponatremia in cirrhosis is intimately related to the pathophysiology of portal hypertension and the non‐osmotic release of AVP3, 15 (Figure 1). In the early phases of cirrhosis, portal hypertension is the result of an increase in intrahepatic resistance. With the development of porto‐systemic collaterals, a hyperdynamic splanchnic circulation develops as a result of splanchnic arterial vasodilatation and increased vascular capacity. Nitric oxide, an endothelial derived relaxing factor, is the critical mediator of this process, and upregulation of its expression is pivotal in the pathogenesis of portal hypertension.
Multiple factors are related to the development of DH in cirrhosis. A reduction of effective central blood volume due to the development of porto‐venous collaterals and arterial splanchnic vasodilation, leading to baroreceptor‐mediated nonosmotic release of AVP, is considered the initiating and most important factor. Patients with cirrhosis and DH have higher plasma and urine vasopressin levels, higher plasma renin activity, and decreased plasma levels of atrial natriuretic factor than those with normal serum sodium concentrations, findings consistent with the presence of a decreased effective plasma volume.16 Arterial underfilling is sensed by baroreceptors located in the left ventricle, aortic arch, carotid sinus and renal afferent arterioles. Decreased activation leads to neurohumoral compensatory responses which include non‐osmotic release of vasopressin from the neurohypophysis and increased levels. Impaired catabolism of AVP that has been correlated with the severity of liver dysfunction may further contribute to increased levels.17 Initially, the increased AVP maintains arterial circulatory integrity by inducing splanchnic, peripheral and renal arterial vasoconstriction through its action on the V1a receptors and expansion of blood volume through renal water retention by its action on the V2 receptors located on the collecting ducts.
The initial adaptive response which leads to increased central blood volume can chronically result in detrimental effects, including the development of fluid overload with ascites, edema, and hyponatremia.16, 18 Additional factors that contribute to hyponatremia include decreased glomerular filtration rate (GFR) and/or increased proximal reabsorption of sodium (that reduce the distal delivery of filtrate and the potential for water reabsorption) and decreased cardiac function that further impairs effective central blood volume.19 In addition, urinary levels of AQP2 are increased in cirrhotic patients, especially those with decompensated disease with higher Child‐Pugh scores and ascites, and provide another potential mechanism to increase water reabsorption.20
Prevalence and Prognostic Significance
Hyponatremia in cirrhosis is a common finding. In a survey of 997 cirrhotic patients with ascites from 28 centers in Europe, North and South America, the prevalence of serum sodium concentration 135, 130, 125, 120 meq/L were 49.4%, 21.6%, 5.7%, and 1.2%, respectively.21 In a retrospective analysis of 188 inpatients, the prevalence of DH of 135, 130, and 125 were 20.8%, 14.9%, and 12.2%, respectively.22 The development of hyponatremia is a manifestation of increasing portal hypertension. In a natural history study of 263 patients hospitalized for first episode of significant ascites, 74 patients developed DH (Na level 130 mEq/L), including 11 patients in whom it appeared during the first episode and 63 cases during follow‐up (mean period of 40 3 months) with a 5‐year incidence of 37.1%.23
The presence of hyponatremia carries significant adverse prognostic significance. It is strongly associated with severity of liver function impairment as assessed by Child‐Pugh and model for end‐stage liver disease (MELD) scores.22 Even mild hyponatremia is associated with severe complications such as massive ascites, severe hepatic encephalopathy, spontaneous bacterial peritonitis (SBP), and hepatic hydrothorax, and the severity of hyponatremia is directly related to the severity of these complications.21, 22 (Figure 2). In a natural history study of patients presenting with large volume ascites, 1‐year survival after its development was reduced to only 25.6%.230
Hyponatremia is an especially poor prognostic sign for a hospitalized cirrhotic patient. In a retrospective analysis of 156 cirrhotic patients, hyponatremiapresent in 57 (29.8%) of admissionswas associated with increased hospital mortality (26.3% vs. 8.9% among those with normal Na levels), and the mortality rate was even higher (48%) among the 25 patients who developed severe hyponatremia during the hospital stay.24 In hospitalized patients, hyponatremia is predictive of the development of acute renal failure which is associated with substantially increased mortality (73% vs. 13%).25 Similarly, a low serum sodium level in critically ill cirrhotic patients admitted to the ICU is associated with complications, in‐hospital mortality, and poor short‐term prognosis.26
Whether hyponatremia should impact liver transplant prioritization remains an area of controversy. The United Network for Organ Sharing (UNOS) contracted by the Organ Procurement and Transplant Network (OPTN) to optimize the efficient use of deceased organs through fair and timely allocation, currently uses the MELD score, a formula that calculates the risk of death within three months from the bilirubin, creatinine, and International Normalized Ratio (INR) levels. Hyponatremia is an earlier and more sensitive marker than serum creatinine to detect renal impairment and/or circulatory dysfunction in patients with advanced cirrhosis and adds to MELD in predicting waitlist mortality.2729 In patients with a MELD score of 21, only low serum sodium and persistent ascites are independent predictors of mortality.28 To account for the importance of hyponatremia on survival, both modification of the MELD score in which the Na level is incorporated (MELD‐Na model) and the MELD to serum sodium ratio (MESO) have been developed. Adding hyponatremia to the MELD score is a better predictor of death than MELD alone, particularly in patients with low MELD scores.27, 2931 The OPTN/UNOS Liver and Intestinal Organ Transplantation Committee has discussed updating the liver allocation system to include the Na level. However, it was concluded that implementation of MELD‐Na would change the allocation status of only 4% of candidates. Further, based on the concerns about the ability to manipulate serum sodium levels and the utility of employing resources to change the system for a relatively small number of patients, it was decided to defer incorporating the Na level pending further analysis (Report of the OPTN/UNOS Liver and Intestinal Organ Transplantation Committee To the Board of Directors, Los Angeles, California, September 17‐18, 2007). At this time, the use of Na is a regional decision.32 However, the OPTN/UNOS Liver and Intestinal Organ Transplantation Committee has recently solicited feedback from the transplant community about including Na in allocation for review at a forum in April 2010.
Precipitating Factors
The most important factor related to development of hyponatremia in cirrhosis is increasing severity of portal hypertension that is associated with impaired central blood volume as a result of progressive splanchnic vasodilatation. In a study in which 170 patients with decompensated alcoholic cirrhosis were prospectively followed for 33.9 27.9 months, the initial hepatic venous pressure gradient (HVPG) was an independent predictive factor for the 20 patients who developed hyponatremia.22
Cirrhotic patients with ascites with hyponatremia have increased AVP secretion, higher levels of plasma renin activity, and higher serum concentrations of aldosterone and norepinephrine compared to those with normal Na levels.33 Diuretic therapy is associated with the development of DH by inducing volume depletion and arterial underfilling, further activating the renin‐angiotensin system (RAS) and increasing the non‐osmotic release of AVP.34 Although diuretics block the salt retention associated with the RAS activation, the water‐retaining effects of AVP persist, and DH develops. The process is further exacerbated by a low sodium intake and a frequent uncontrollable thirst. As a result, diuretic therapy is commonly associated with the development of hyponatremia in patients with ascites.24, 35 Similarly, paracentesis (particularly when performed without albumin) is often associated with an increase in blood urea nitrogen and marked elevations in plasma renin activity and plasma aldosterone concentration, which may exacerbate this physiology, leading to further reduction in serum sodium concentration.36 Tense ascites can contribute to DH by increasing baroreceptor mediated AVP release by increasing intrathoracic pressure.37 Finally, non‐steroidal anti‐inflammatory drugs (NSAIDs) can cause DH by inhibiting the synthesis of renal prostaglandins (which normally function to antagonize the tubular action of AVP and are important in the maintenance of appropriate renal tubular transport of fluid and electrolytes in states of renal hypoperfusion).38
Medical Impact of Hyponatremia: Marker of Severe Disease or Direct Pathophysiologic Role?
Hyponatremia is associated with severe ascites, impaired renal function, hepatic encephalopathy, SBP, and hepatorenal syndrome.3, 20 Because hyponatremia is frequently present in advanced liver failure, it is unclear whether it is only a marker of advanced disease or whether it plays a direct pathophysiologic role, or both. Until recently, it has not been possible to address this issue due to the inability to easily and rapidly correct the hyponatremia. However, there is increasing evidence that hyponatremia has direct impact on the severity of hepatic encephalopathy (see Hepatic Encephalopathy section). The recent introduction of tolvaptan for the treatment of hyponatremia in cirrhosis (discussed below) will allow this question to be directly answered.
Fluid Management and Diuresis
The typical cirrhotic patient with DH is characterized by expanded extracellular fluid with ascites and edema. The profound vasodilation of the splanchnic arterial circulation is associated with decreased effective arterial blood volume, leading to the non‐osmotic release of AVP. Diuretic therapy can further exacerbate this process. In addition, the increased water permeability induced by AVP results in reduced urine volume and fluid retention. As a result, hyponatremia directly adversely affects severity of fluid overload and limits and/or precludes diuretic treatment.
Hepatorenal Syndrome
Hyponatremia is an earlier and more sensitive marker than serum creatinine to detect renal impairment and/or circulatory dysfunction and is frequently a precursor to overt hepatorenal syndrome.27 Hyponatremia is predictive of the development of acute renal failure during hospitalization, and in‐hospital development of acute renal failure portends a high mortality.25 In patients admitted with SBP, the presence of hyponatremia is significantly associated with higher mortality and renal failure.39
Hepatic Encephalopathy
The neurologic manifestations of cerebral edema associated with hyponatremia closely mirror those of hepatic encephalopathy. In fact, a recently proposed pathogenic mechanism for hepatic encephalopathy is the development of low‐grade cerebral edema associated with astrocyte swelling in response to ammonia and other precipitating factors.40 DH is associated with a further reduction in brain organic osmolytes that probably reflects a compensatory osmoregulatory mechanism against cell swelling triggered by a combination of high intracellular glutamine and low extracellular osmolality.41 As a result, it has been proposed that hyponatremia contributes to the development of hepatic encephalopathy through the development or exacerbation of low‐grade cerebral edema. In this manner, low serum sodium acts as a second hit to the swelling produced by increased intracellular glutamine created by ammonia metabolism.42
Clinically, hyponatremia is a major risk factor for hepatic encephalopathy. Serum sodium and ammonia levels are the major factors that predict electroencephalographic abnormalities in cirrhotics who do not have hepatic encephalopathy.43 In a prospective study of 61 patients, hyponatremia was associated with a low brain concentration of organic osmolytes as assessed by proton magnetic resonance spectroscopy (1H‐MRS) and magnetic resonance imaging, and both conditions were major risk factors for the development of overt hepatic encephalopathy.44 Finally, hyponatremia is a risk factor for hepatic encephalopathy in patients undergoing TIPS.45
Adverse Effect on Outcome After Liver Transplantation
Hyponatremia before liver transplantation is associated with adverse post‐transplant outcomes. Among patients undergoing liver transplantation, the presence of hyponatremia is associated with abnormal cardiac response in patients after reperfusion.46 Pre‐transplant hyponatremia is associated with longer ICU and hospital stay, higher rates of delirium and neurologic disorders, acute renal failure, acute cellular rejection, infection, and in one study a reduced 3‐month survival compared to normonatremic recipients.32, 47, 48 In 1 retrospective study that compared post‐transplant outcomes of patients with corrected vs. uncorrected pre‐transplant hyponatremia, patients with pre‐operative correction of hyponatremia had a lower risk of prolonged post‐transplant hospitalization than those with uncorrected hyponatremia.32 However, both hyponatremic groups had more complicated post‐transplant courses compared to those without a history of hyponatremia. However, given the small sample size, retrospective design, and the potential for confounding, the impact of correction of pre‐transplant hyponatremia remains to be determined.
Management
Most patients with mild hypervolemic hyponatremia are asymptomatic. The initial recommended approach is fluid restriction and an Na‐restricted diet. For those with severe or progressive hyponatremia, diuretics should be minimized or discontinued to avoid intravascular volume depletion.49 For patients with tense ascites and severe DH, therapeutic paracentesis with plasma expanders is safe.33 Unfortunately, fluid restriction is limited in efficacy and often poorly tolerated. The use of hypertonic saline is generally not recommended unless severe neurologic symptoms are present as it leads to increased ascites and edema. When administered, it is important to avoid a rapid correction of the hyponatremia to prevent the development of central pontine myelinolysis and the osmotic demyelination syndrome.
Due to the pivotal role of AVP in the pathogenesis of DH, antagonism of its action has long been proposed to be the most rational approach, but until recently, effective and specific antagonism of AVP has remained elusive. Approaches that have been attempted include interference with its secretion and actions. Intravenous albumin has been reported to improve hyponatremia in patients with cirrhosis, ascites, and hyponatremia, presumably by decreasing AVP release by plasma volume expansion.50 An attempt at inhibition of central AVP release with the use of a kappa‐opioid receptor agonist, niravoline, was limited by loss of efficacy and potential adverse effects.51 Use of demeclocycline and lithium (which induce renal resistance to AVP and lead to a modest increase in urine volume with decreased urine osmolality and a corresponding rise in serum sodium) is limited by nephrotoxicity and hepatotoxicity.7, 52 Because of the important role played by prostaglandins in the maintenance of renal hemodynamics and water excretion in cirrhosis, oral misoprostol has also been evaluated but determined to be ineffective in inducing significant changes in free water clearance in patients with functional renal failure and/or DH.53
The recent introduction of vaptans, vasopressin receptor antagonists that block the physiologic action of vasopressin, represents a revolutionary and highly effective approach to the treatment of hyponatremia. Vaptans are antagonists of the V2 receptors of AVP in the principal cells of the collecting ducts. In healthy subjects, vaptans cause a dose‐dependent increase in urine volume and produce a dilute urine without causing natriuresis. To date, 2 AVP antagonists, conivaptan and tolvaptan, have been Food and Drug Administration (FDA)‐approved for the treatment of DH. Conivaptan, the first to be approved in 2005, is a mixed vasopressin V1a and V2 receptor antagonist that is administered intravenously for up to 4 days. In a randomized placebo‐controlled study of patients with euvolemic or hypervolemic hyponatremia, intravenous conivaptan treatment increased serum Na levels by >6 mEq/l or to a serum Na >135 mEq/l in 69 to 88.5% of subjects compared to 20.7% of those receiving placebo (Zeltser D, Rosansky S, Van Rensburg H, et al. Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia. American J Nephrology 2007;27:447457). In a pilot study involving 24 patients with end‐stage liver disease, an infusion of conivaptan over 1 to 4 days was associated with an increase of serum sodium by >5 mmol/L in 60% of patients not receiving diuretics and 67% of patients on concomitant diuretic therapy by the end of treatment (O'Leary and Davis, 2009). Despite a concern about the potential for conivaptan to increase portal hypertension due to inhibition of splanchnic V1a receptors, the brief treatment appeared to be well tolerated without significant changes in systolic blood pressure, serum creatinine, variceal bleeding or worsening of ascites during the infusion period. However, approval for only 4 days of therapy and requirement for intravenous use eliminate any potential for chronic use.54
Tolvaptan is an orally available, selective V2 receptor antagonist whose efficacy was assessed in two multicenter, prospective, randomized, placebo‐controlled trials, Study of Ascending Levels of Tolvaptan in Hyponatremia 1 and 2 (SALT‐1 and SALT‐2).55 In these trials, clinically stable patients with DH (Na 135meq/l) associated with cirrhosis (22.4% in SALT‐1, 30.5% in SALT‐2), CHF or SIADH were randomized in a hospital setting to receive tolvaptan 15mg daily or placebo. Repeat Na levels were obtained at 8 hours, 2, 3, and 4 days and then weekly at days 11, 18, 25 and 30 after which study drug was discontinued and follow‐up Na level was determined 7 days later. The dose was adjusted to 30 mg and then 60 mg in an attempt to achieve a Na level >135 in those in whom hyponatremia persisted. During the initial day of the titration phase, fluid restriction was not maintained, and the patients were encouraged to respond to thirst with increased water ingestion.
Tolvaptan use was associated with a prompt increase in Na level as early as 8 hours after administration of the first dose. Serum Na increased more among those receiving tolvaptan than among those receiving placebo during the first 4 days and throughout the study period regardless of baseline Na level but returned to baseline within 1 week after discontinuation (Figure 3). The main side effects were increased thirst, dry mouth and increased urination. Importantly, an increased incidence of renal failure was not observed. Based on these results, FDA approval for tolvaptan in patients with hyponatremia was obtained in May 2009 for patients with DH‐associated with cirrhosis, CHF or SIADH for patients with Na levels 125 or symptomatic patients with Na levels between 125 and 135 that have not responded to fluid restriction.
Management of the Hospitalized Cirrhotic Patient With Hyponatremia: Recommendations
Hyponatremia in hospitalized cirrhotic patients is a marker for severe disease and high risk of hospital mortality.24 As a result, prompt evaluation and treatment is imperative. The availability of tolvaptan potentially revolutionizes the manner in which these patients are treated. In the SALT trials, only clinically stable patients were enrolled. In this last section, a guideline for the evaluation and treatment of acutely ill, hospitalized cirrhotic patients with DH is presented.
Evaluation
Determination of volume status is paramount but frequently problematic in the hospitalized cirrhotic patient. Due to the vasodilated state present in severe portal hypertension that is characterized by a relative hypotension and resting tachycardia, the usual hemodynamic parameters of blood pressure and heart rate can be difficult to interpret. Although significant extravascular volume in the form of ascites and edema may be present, patients may be intravascularly depleted due to previous diuretic use and extra‐renal losses due to impaired oral intake, vomiting, lactulose‐induced diarrhea, and gastrointestinal bleeding. Infection is a commonly associated condition, and endotoxin mediated splanchnic vasodilatation, especially in the setting of SBP, can adversely effective central blood volume status in the presence of severe ascites. Also, due to the Na avidity of the kidney and previous diuretic use, renal electrolytes can be difficult to interpret.
For patients in whom there is strong clinical concern about intravascular depletion (history of impaired oral intake, excessive vomiting and/or diarrhea, rapid weight loss, small volume ascites with history of large volume, azotemia), administration of limited intravenous normal saline (0.5‐1 L) should be considered. Patients with severe neurological symptoms should receive normal saline or hypertonic saline. Unless severe neurologic symptoms associated with profound hyponatremia is present, however, intravenous normal saline should not be administered for the hyponatremia alone. Administration of salt poor albumin (25%), especially for those with marked fluid overload and ascites, is an effective means to expand the central blood volume without exacerbating ascites and edema. After evaluation for and/or treatment of hypovolemia, all patients should receive a Na restricted diet (2000 mg daily) and placed on fluid restriction (see below for liberalization of fluid restriction upon initiation of tolvaptan therapy).
Diagnostic paracentesis should be performed for those with ascites to rule out the presence of SBP, and antibiotics administered to those with evidence of infection. High dose intravenous salt poor albumin should also be administered, especially to those at high risk of renal failure as determined by the presence of azotemia (Cr > 1.0 mg/dL) or severe liver insufficiency (TBili > 4.0 mg/dL).56, 57 Finally, all medications should be reviewed, and those associated with hyponatremia (diuretics, selective serotonin reuptake inhibitors, opiates, proton‐pump inhibitors) discontinued if possible.
Tolvaptan for DH
Patient Selection
Appropriate patient selection for tolvaptan therapy is extremely important (Table 2). In the SALT trials, only clinically stable patients were enrolled. The presence of hyponatremia in a recently hospitalized cirrhotic patient, however, frequently indicates severe disease with a high risk of acute renal failure and hospital death. In the SALT trials, many received concomitant diuretic therapy. Because of the importance of avoiding tolvaptan administration to hypovolemic patients, discontinuation of diuretic therapy prior the initiation of tolvaptan therapy and/or reevaluation after limited volume expansion should be considered.
| Hospital setting |
| Euvolumia or hypervolumia |
| Absence of recent weight loss, decrease in ascites, edema |
| Absence of excessive vomiting, diarrhea |
| Consider discontinuation of diuretic therapy prior to initiation of tolvaptan |
| Consider evaluation after limited volume expansion, especially with salt poor albumin prior to initiation of tolvaptan |
| Presence of clinically significant hyponatremia: 125mEq/L or less severe but symptomatic hyponatremia (125 to 134 mEq/L) that has resisted fluid restriction |
| Absence of severe neurologic symptoms attributable to hyponatremia |
| No co‐administration with intravenous saline |
| Ability to respond to thirst |
| No co‐administration with strong CYP 3A inhibitors (ketoconazole) |
| Absence of kidney failure with anuria |
Tolvaptan is indicated for cirrhotic patients with DH in whom the serum sodium is 125 mEq/L and in those with less severe but symptomatic hyponatremia (125‐134 mEq/L) that has resisted fluid restriction. Although the definition of symptomatic was not specifically defined, possible considerations include symptoms of mild hepatic encephalopathy or inability to tolerate dieresis due to the presence of hyponatremia. According to FDA guidelines, tolvaptan therapy must be initiated and re‐initiated in a hospital setting. Patients with severe neurologic symptoms attributable to hyponatremia in whom rapid treatment is critical should not receive tolvaptan but should rather be treated with normal saline. Similarly, patients should not receive combination therapy with tolvaptan and normal saline due to potential for a too‐rapid correction of hyponatremia and the development of central pontine myelinolysis. If saline had been administered for treatment of possible hypovolemia, it should be discontinued and persistent hyponatremia confirmed before starting tolvaptan. Other factors that need to be considered before initiating tolvaptan include the ability of the patient to respond to thirst with increased water ingestion and recognition that the patient will experience increased urine volume and frequency, requiring easy access to toilet. Patients should not be fluid restricted during the first day of tolvaptan therapy, but should be instructed to respond to their thirst with increased water ingestion. As a result, caution should be exercised in administering tolvaptan to a confused, restrained, unresponsive and/or bed‐bound patient who is not able to respond appropriately to thirst or increased urination.
In the SALT trials, the incidence of hyperkalemia (5%) was similar in the tolvaptan and placebo treated patients.55 However, further analysis of all multiple‐dose, placebo‐controlled trials, demonstrated that the aggregate incidence of hyperkalemia was slightly higher for tolvaptan‐treated subjects compared with placebo‐treated subjects (Otsuka). Because treatment with tolvaptan is associated with an acute reduction of the extracellular fluid volume which could result in increased serum potassium through hemoconcentration, it is recommended that serum potassium levels be monitored after initiation of tolvaptan treatment in patients with a serum potassium > 5 mEq/L as well as those who are receiving drugs known to increase serum potassium levels such as angiotensin converting enzyme inhibitors, angiotensin receptor blockers, or potassium sparing diuretics (Samsca Package Insert, Otsuka). Because tolvaptan is metabolized by the cytochrome P 3A system, patients receiving strong inhibitors such as ketoconazole should not receive tolvaptan. Anuric patients will not respond to tolvaptan. Finally, it is extremely important to administer tolvaptan only to patients with true hyponatremia and not to those with pseudohyponatremia in whom the plasma osmolality is normal but the measured serum sodium concentration artificially low due to marked elevations of other substances, such as can be seen in severe hyperglycemia, marked hyperlipidemia, or hyperproteinemia (as in multiple myeloma).
Tolvaptan Administration
The initial dose of tolvaptan is 15 mg daily. After receiving tolvaptan, many patients will develop an increased sense of thirst and need to urinate. As a result, patients should not be fluid restricted during the first day of therapy, and it is important to monitor the hemodynamics and Na level closely after initiating therapy with a repeat Na level at approximately 8 hours after the first dose. As a result, it should probably be administered early in the day and not at bedtime. The dose should be increased to 30 mg, then 60 mg in patients who do not respond by at least 5 mEq/L over the previous 24 hours and remain hyponatremic. In those with an excessive response (more than 8 meq/L during the first 8 hours or 12 meq/L on any subsequent day), the patient should be encourage to either drink more water, or the dose should be held or reduced. After the appropriate dose has been identified, the patient may be discharged and continued on tolvaptan long‐term.
With the advent of this exciting therapy, practical issues will need to be addressed, most important of which is its cost at $250 per day (Otsuka). In addition, the current recommendation to initiate tolvaptan only in a hospital further limits its widespread use. Most important, long‐term clinical benefit will need to be demonstrated. Although the SALT trials only involved treatment for up to 1 month, a multicenter, open‐label extension study for a mean duration of 701 days demonstrated that prolonged administration of tolvaptan maintains an increased serum sodium level.58 However, at this time, tolvaptan can only be considered as one of the promising drugs whose long‐term cost‐effectiveness is yet to be proven. Proof will require showing that correction of the hyponatremia leads to improved clinical outcomes, such as a reduction in length of stay or frequency of hospitalization, decreased renal failure, improved hepatic encephalopathy, deceased mortality, and improved post‐transplant outcomes.
Unanswered Questions
The vaptans provide an important opportunity to clarify the role that hyponatremia plays in the pathogenesis of cirrhosis. In the past, DH in a cirrhotic patient represented a sign of advanced disease. With the availability of safe and effective therapy, we can now determine whether it also plays an important role in the pathophysiology of end‐stage liver disease and whether its treatment will have a beneficial effect on patient outcomes.
Specific clinical questions that will inevitably be addressed over the next few years to determine whether DH is only a marker for advanced disease or whether it plays a direct but modifiable role in the pathophysiology of cirrhosis will include:
-
Role of vaptans in the management of ascites: In a 14‐day randomized, trial of a satavaptan, another selective vasopressin V(2) receptor antagonist, vs. placebo with spironolactone, combination therapy was associated with improved control of ascites and improvements in serum sodium levels in hyponatremic patients with ascites.59 If future similar studies demonstrate more prolonged benefits, this would constitute an important advance in the treatment of ascites in cirrhosis.
-
Effect on renal function: Prolonged use of tolvaptan leads to a compensatory increase in endogenous levels of AVP and, potentially, increased stimulation of V1a receptors, which might be helpful in the setting of portal hypertension. In patients with hepatorenal syndrome, vasopressin stimulation of splanchnic V1a receptors leads to improved renal function, presumably by decreasing splanchnic blood flow and improving central blood volume.60 As a result, tolvaptan may indirectly improve kidney function in patients with advanced cirrhosis and refractory ascites. Whether long‐term tolvaptan therapy will help to prevent hepatorenal syndrome through this mechanism remains to be determined but is an exciting possibility.61
-
Effect on hepatic encephalopathy: Hepatic encephalopathy is associated with poor quality of life in patients with cirrhosis. Although hepatic encephalopathy was not directly assessed in the SALT trials, the mean mental component summary of the Short Form General Health Survey, a quality of life measure, improved in cirrhotic patients receiving tolvaptan to a greater degree that those receiving placebo.62 A possible explanation for this finding is a beneficial effect of tolvaptan on hepatic encephalopathy. Confirmation of this hypothesis, however, will require prospective studies in which hepatic encephalopathy is directly assessed.
-
Effect on medical economics: Based on retrospective reviews, hyponatremia has an adverse impact on length of stay and outcomes following liver transplantation. It will be important to demonstrate in prospective studies that correction of hyponatremia with tolvaptan reduces length of stay, complications, and costs.
The serum sodium (Na) level is the major determinant of serum osmolality. In normal physiologic states is tightly regulated between 135 mEq/L to 145 mEq/L despite variable intake of water and solute through the interaction of osmoreceptors in the hypothalamus where arginine vasopressin (AVP) is synthesized and then released by the posterior pituitary and the binding of AVP with V2 AVP receptors on the basolateral surface of the principal cells within the collecting duct of the kidney. Binding of AVP to the V2 receptors promotes the translocation and fusion of cytoplasmic vesicles which carry the water channel protein aquaporin 2 (AQP2) to the apical membrane of the cell and, in this manner, increases water permeability and absorption.1, 2, 3
Patients with hyponatremia, defined by a serum Na level 135 mEq/L, can be broadly classified by their volume status into those who are euvolemic, hypervolemic, and hypovolemic (Table 1). In patients with euvolemic hyponatremia such as those with Syndrome of Inappropriate Antidiuretic Hormone (SIADH), total body Na is nearly normal, but total body water is increased. In patients with hypervolemic hyponatremia, both total body Na and water are increased, but water to a much greater degree. These patients typically have increased extracellular fluid such as edema and/or ascites. The most common conditions associated with this condition are cirrhosis, congestive heart failure (CHF), and renal failure. In contrast, hypovolemic hyponatremia is associated with a reduction in both total body Na and water, but Na to a greater degree. This condition is encountered in patients with excessive fluid losses such as those with over‐diuresis, excessive gastrointestinal losses, burns, and pancreatitis.4
| Depletional Hyponatreima | Dilutional Hyponatremia | ||
|---|---|---|---|
| Euvolumic | Hypervolumic | ||
| Total body water | |||
| Total body Na | normal | ||
| Common etiologies | SIADH | cirrhosis/CHF | vomiting, diarrhea |
Hyponatremia is the most common electrolyte abnormality seen in general hospital patients.5 In a database of over 120,000 patients, a serum sodium level of 136mEq/L was observed in 28.2%.6 Hyponatremia is associated with selected medical conditions (especially cirrhosis and CHF), the extremes of age, and those receiving selected medications, including several that are commonly administered to cirrhotic patients (diuretics, selective serotonin reuptake inhibitors, opiates, proton‐pump inhibitors).7, 8 Hyponatremia is associated with increased total costs per hospital admission.5, 9 In an analysis of the effect of hyponatremia on length of stay in a retrospective cohort study of hospitalized patients derived from a large administrative database of 198,281 discharges from 39 US hospitals, mean length of stay was significantly greater among patients with hyponatremia than those with normal Na levels (8.6 8.0 vs. 7.2 8.2 days). After adjusting for confounders that may be associated with more severe disease and hyponatremia (age, gender, race, geographic region, teaching status of the hospital, admission source, principal payer, comorbidity index score and primary diagnosis), the presence of hyponatremia contributed an increase in length of stay of 1.0 day. Patients with hyponatremia are more frequently admitted to the intensive care unit (ICU) and require mechanical ventilation. In patients with CHF, the presence of hyponatremia at discharge is associated with increased risk for early mortality and rehospitalization.10
Although frequently asymptomatic, hyponatremia may be associated with a range of findings, from subtle and non‐specific complaints, including headache, fatigue, confusion, malaise, to severe and life‐threatening manifestations with lethargy, seizures, brainstem herniation, respiratory arrest and death.11 The most important complications are neurologic consequences related to cerebral edema. However, there is increased morbidity even in hyponatremic patients considered to be asymptomatic. Patients with low serum sodium have attention deficients, and falls are common. In a study of 122 patients who were considered to have chronic asymptomatic hyponatremia, the incidence of falls was significantly higher at 21.3% compared to only 5.3% in a control population.12
In hyponatremia, water enters into the cells to attain osmotic balance, resulting in cellular swelling.4 To avoid cerebral edema, the brain is capable of adapting to hyponatremia by regulating its volume to avoid swelling, especially when hyponatremia is chronic. In acute hyponatremia, astrocytes and neurons adapt through osmoregulatory mechanisms by extruding intracellular electrolytes such as potassium.13 Chronically, adaption occurs through the loss of low‐molecular weight organic compounds termed organic osmolytes including myoinsoitol, glutamine, choline and taurine. As a result, both the severity and the rate of its development are critical factors in determining the neurologic manifestation of hyponatremia in a given patient.14
Dilutional Hyponatremia and Cirrhosis
Patients with hyponatremia who are either euvolemic or hypervolemic are considered to have dilutional hyponatremia (DH). Management of these patients is distinct from those who are hypovolemic in whom appropriate therapy consists of the administration of normal saline. The remainder of this article addresses the pathogenesis, management and treatment of cirrhotic patients with DH.
Pathogenesis
The development of hyponatremia in cirrhosis is intimately related to the pathophysiology of portal hypertension and the non‐osmotic release of AVP3, 15 (Figure 1). In the early phases of cirrhosis, portal hypertension is the result of an increase in intrahepatic resistance. With the development of porto‐systemic collaterals, a hyperdynamic splanchnic circulation develops as a result of splanchnic arterial vasodilatation and increased vascular capacity. Nitric oxide, an endothelial derived relaxing factor, is the critical mediator of this process, and upregulation of its expression is pivotal in the pathogenesis of portal hypertension.
Multiple factors are related to the development of DH in cirrhosis. A reduction of effective central blood volume due to the development of porto‐venous collaterals and arterial splanchnic vasodilation, leading to baroreceptor‐mediated nonosmotic release of AVP, is considered the initiating and most important factor. Patients with cirrhosis and DH have higher plasma and urine vasopressin levels, higher plasma renin activity, and decreased plasma levels of atrial natriuretic factor than those with normal serum sodium concentrations, findings consistent with the presence of a decreased effective plasma volume.16 Arterial underfilling is sensed by baroreceptors located in the left ventricle, aortic arch, carotid sinus and renal afferent arterioles. Decreased activation leads to neurohumoral compensatory responses which include non‐osmotic release of vasopressin from the neurohypophysis and increased levels. Impaired catabolism of AVP that has been correlated with the severity of liver dysfunction may further contribute to increased levels.17 Initially, the increased AVP maintains arterial circulatory integrity by inducing splanchnic, peripheral and renal arterial vasoconstriction through its action on the V1a receptors and expansion of blood volume through renal water retention by its action on the V2 receptors located on the collecting ducts.
The initial adaptive response which leads to increased central blood volume can chronically result in detrimental effects, including the development of fluid overload with ascites, edema, and hyponatremia.16, 18 Additional factors that contribute to hyponatremia include decreased glomerular filtration rate (GFR) and/or increased proximal reabsorption of sodium (that reduce the distal delivery of filtrate and the potential for water reabsorption) and decreased cardiac function that further impairs effective central blood volume.19 In addition, urinary levels of AQP2 are increased in cirrhotic patients, especially those with decompensated disease with higher Child‐Pugh scores and ascites, and provide another potential mechanism to increase water reabsorption.20
Prevalence and Prognostic Significance
Hyponatremia in cirrhosis is a common finding. In a survey of 997 cirrhotic patients with ascites from 28 centers in Europe, North and South America, the prevalence of serum sodium concentration 135, 130, 125, 120 meq/L were 49.4%, 21.6%, 5.7%, and 1.2%, respectively.21 In a retrospective analysis of 188 inpatients, the prevalence of DH of 135, 130, and 125 were 20.8%, 14.9%, and 12.2%, respectively.22 The development of hyponatremia is a manifestation of increasing portal hypertension. In a natural history study of 263 patients hospitalized for first episode of significant ascites, 74 patients developed DH (Na level 130 mEq/L), including 11 patients in whom it appeared during the first episode and 63 cases during follow‐up (mean period of 40 3 months) with a 5‐year incidence of 37.1%.23
The presence of hyponatremia carries significant adverse prognostic significance. It is strongly associated with severity of liver function impairment as assessed by Child‐Pugh and model for end‐stage liver disease (MELD) scores.22 Even mild hyponatremia is associated with severe complications such as massive ascites, severe hepatic encephalopathy, spontaneous bacterial peritonitis (SBP), and hepatic hydrothorax, and the severity of hyponatremia is directly related to the severity of these complications.21, 22 (Figure 2). In a natural history study of patients presenting with large volume ascites, 1‐year survival after its development was reduced to only 25.6%.230
Hyponatremia is an especially poor prognostic sign for a hospitalized cirrhotic patient. In a retrospective analysis of 156 cirrhotic patients, hyponatremiapresent in 57 (29.8%) of admissionswas associated with increased hospital mortality (26.3% vs. 8.9% among those with normal Na levels), and the mortality rate was even higher (48%) among the 25 patients who developed severe hyponatremia during the hospital stay.24 In hospitalized patients, hyponatremia is predictive of the development of acute renal failure which is associated with substantially increased mortality (73% vs. 13%).25 Similarly, a low serum sodium level in critically ill cirrhotic patients admitted to the ICU is associated with complications, in‐hospital mortality, and poor short‐term prognosis.26
Whether hyponatremia should impact liver transplant prioritization remains an area of controversy. The United Network for Organ Sharing (UNOS) contracted by the Organ Procurement and Transplant Network (OPTN) to optimize the efficient use of deceased organs through fair and timely allocation, currently uses the MELD score, a formula that calculates the risk of death within three months from the bilirubin, creatinine, and International Normalized Ratio (INR) levels. Hyponatremia is an earlier and more sensitive marker than serum creatinine to detect renal impairment and/or circulatory dysfunction in patients with advanced cirrhosis and adds to MELD in predicting waitlist mortality.2729 In patients with a MELD score of 21, only low serum sodium and persistent ascites are independent predictors of mortality.28 To account for the importance of hyponatremia on survival, both modification of the MELD score in which the Na level is incorporated (MELD‐Na model) and the MELD to serum sodium ratio (MESO) have been developed. Adding hyponatremia to the MELD score is a better predictor of death than MELD alone, particularly in patients with low MELD scores.27, 2931 The OPTN/UNOS Liver and Intestinal Organ Transplantation Committee has discussed updating the liver allocation system to include the Na level. However, it was concluded that implementation of MELD‐Na would change the allocation status of only 4% of candidates. Further, based on the concerns about the ability to manipulate serum sodium levels and the utility of employing resources to change the system for a relatively small number of patients, it was decided to defer incorporating the Na level pending further analysis (Report of the OPTN/UNOS Liver and Intestinal Organ Transplantation Committee To the Board of Directors, Los Angeles, California, September 17‐18, 2007). At this time, the use of Na is a regional decision.32 However, the OPTN/UNOS Liver and Intestinal Organ Transplantation Committee has recently solicited feedback from the transplant community about including Na in allocation for review at a forum in April 2010.
Precipitating Factors
The most important factor related to development of hyponatremia in cirrhosis is increasing severity of portal hypertension that is associated with impaired central blood volume as a result of progressive splanchnic vasodilatation. In a study in which 170 patients with decompensated alcoholic cirrhosis were prospectively followed for 33.9 27.9 months, the initial hepatic venous pressure gradient (HVPG) was an independent predictive factor for the 20 patients who developed hyponatremia.22
Cirrhotic patients with ascites with hyponatremia have increased AVP secretion, higher levels of plasma renin activity, and higher serum concentrations of aldosterone and norepinephrine compared to those with normal Na levels.33 Diuretic therapy is associated with the development of DH by inducing volume depletion and arterial underfilling, further activating the renin‐angiotensin system (RAS) and increasing the non‐osmotic release of AVP.34 Although diuretics block the salt retention associated with the RAS activation, the water‐retaining effects of AVP persist, and DH develops. The process is further exacerbated by a low sodium intake and a frequent uncontrollable thirst. As a result, diuretic therapy is commonly associated with the development of hyponatremia in patients with ascites.24, 35 Similarly, paracentesis (particularly when performed without albumin) is often associated with an increase in blood urea nitrogen and marked elevations in plasma renin activity and plasma aldosterone concentration, which may exacerbate this physiology, leading to further reduction in serum sodium concentration.36 Tense ascites can contribute to DH by increasing baroreceptor mediated AVP release by increasing intrathoracic pressure.37 Finally, non‐steroidal anti‐inflammatory drugs (NSAIDs) can cause DH by inhibiting the synthesis of renal prostaglandins (which normally function to antagonize the tubular action of AVP and are important in the maintenance of appropriate renal tubular transport of fluid and electrolytes in states of renal hypoperfusion).38
Medical Impact of Hyponatremia: Marker of Severe Disease or Direct Pathophysiologic Role?
Hyponatremia is associated with severe ascites, impaired renal function, hepatic encephalopathy, SBP, and hepatorenal syndrome.3, 20 Because hyponatremia is frequently present in advanced liver failure, it is unclear whether it is only a marker of advanced disease or whether it plays a direct pathophysiologic role, or both. Until recently, it has not been possible to address this issue due to the inability to easily and rapidly correct the hyponatremia. However, there is increasing evidence that hyponatremia has direct impact on the severity of hepatic encephalopathy (see Hepatic Encephalopathy section). The recent introduction of tolvaptan for the treatment of hyponatremia in cirrhosis (discussed below) will allow this question to be directly answered.
Fluid Management and Diuresis
The typical cirrhotic patient with DH is characterized by expanded extracellular fluid with ascites and edema. The profound vasodilation of the splanchnic arterial circulation is associated with decreased effective arterial blood volume, leading to the non‐osmotic release of AVP. Diuretic therapy can further exacerbate this process. In addition, the increased water permeability induced by AVP results in reduced urine volume and fluid retention. As a result, hyponatremia directly adversely affects severity of fluid overload and limits and/or precludes diuretic treatment.
Hepatorenal Syndrome
Hyponatremia is an earlier and more sensitive marker than serum creatinine to detect renal impairment and/or circulatory dysfunction and is frequently a precursor to overt hepatorenal syndrome.27 Hyponatremia is predictive of the development of acute renal failure during hospitalization, and in‐hospital development of acute renal failure portends a high mortality.25 In patients admitted with SBP, the presence of hyponatremia is significantly associated with higher mortality and renal failure.39
Hepatic Encephalopathy
The neurologic manifestations of cerebral edema associated with hyponatremia closely mirror those of hepatic encephalopathy. In fact, a recently proposed pathogenic mechanism for hepatic encephalopathy is the development of low‐grade cerebral edema associated with astrocyte swelling in response to ammonia and other precipitating factors.40 DH is associated with a further reduction in brain organic osmolytes that probably reflects a compensatory osmoregulatory mechanism against cell swelling triggered by a combination of high intracellular glutamine and low extracellular osmolality.41 As a result, it has been proposed that hyponatremia contributes to the development of hepatic encephalopathy through the development or exacerbation of low‐grade cerebral edema. In this manner, low serum sodium acts as a second hit to the swelling produced by increased intracellular glutamine created by ammonia metabolism.42
Clinically, hyponatremia is a major risk factor for hepatic encephalopathy. Serum sodium and ammonia levels are the major factors that predict electroencephalographic abnormalities in cirrhotics who do not have hepatic encephalopathy.43 In a prospective study of 61 patients, hyponatremia was associated with a low brain concentration of organic osmolytes as assessed by proton magnetic resonance spectroscopy (1H‐MRS) and magnetic resonance imaging, and both conditions were major risk factors for the development of overt hepatic encephalopathy.44 Finally, hyponatremia is a risk factor for hepatic encephalopathy in patients undergoing TIPS.45
Adverse Effect on Outcome After Liver Transplantation
Hyponatremia before liver transplantation is associated with adverse post‐transplant outcomes. Among patients undergoing liver transplantation, the presence of hyponatremia is associated with abnormal cardiac response in patients after reperfusion.46 Pre‐transplant hyponatremia is associated with longer ICU and hospital stay, higher rates of delirium and neurologic disorders, acute renal failure, acute cellular rejection, infection, and in one study a reduced 3‐month survival compared to normonatremic recipients.32, 47, 48 In 1 retrospective study that compared post‐transplant outcomes of patients with corrected vs. uncorrected pre‐transplant hyponatremia, patients with pre‐operative correction of hyponatremia had a lower risk of prolonged post‐transplant hospitalization than those with uncorrected hyponatremia.32 However, both hyponatremic groups had more complicated post‐transplant courses compared to those without a history of hyponatremia. However, given the small sample size, retrospective design, and the potential for confounding, the impact of correction of pre‐transplant hyponatremia remains to be determined.
Management
Most patients with mild hypervolemic hyponatremia are asymptomatic. The initial recommended approach is fluid restriction and an Na‐restricted diet. For those with severe or progressive hyponatremia, diuretics should be minimized or discontinued to avoid intravascular volume depletion.49 For patients with tense ascites and severe DH, therapeutic paracentesis with plasma expanders is safe.33 Unfortunately, fluid restriction is limited in efficacy and often poorly tolerated. The use of hypertonic saline is generally not recommended unless severe neurologic symptoms are present as it leads to increased ascites and edema. When administered, it is important to avoid a rapid correction of the hyponatremia to prevent the development of central pontine myelinolysis and the osmotic demyelination syndrome.
Due to the pivotal role of AVP in the pathogenesis of DH, antagonism of its action has long been proposed to be the most rational approach, but until recently, effective and specific antagonism of AVP has remained elusive. Approaches that have been attempted include interference with its secretion and actions. Intravenous albumin has been reported to improve hyponatremia in patients with cirrhosis, ascites, and hyponatremia, presumably by decreasing AVP release by plasma volume expansion.50 An attempt at inhibition of central AVP release with the use of a kappa‐opioid receptor agonist, niravoline, was limited by loss of efficacy and potential adverse effects.51 Use of demeclocycline and lithium (which induce renal resistance to AVP and lead to a modest increase in urine volume with decreased urine osmolality and a corresponding rise in serum sodium) is limited by nephrotoxicity and hepatotoxicity.7, 52 Because of the important role played by prostaglandins in the maintenance of renal hemodynamics and water excretion in cirrhosis, oral misoprostol has also been evaluated but determined to be ineffective in inducing significant changes in free water clearance in patients with functional renal failure and/or DH.53
The recent introduction of vaptans, vasopressin receptor antagonists that block the physiologic action of vasopressin, represents a revolutionary and highly effective approach to the treatment of hyponatremia. Vaptans are antagonists of the V2 receptors of AVP in the principal cells of the collecting ducts. In healthy subjects, vaptans cause a dose‐dependent increase in urine volume and produce a dilute urine without causing natriuresis. To date, 2 AVP antagonists, conivaptan and tolvaptan, have been Food and Drug Administration (FDA)‐approved for the treatment of DH. Conivaptan, the first to be approved in 2005, is a mixed vasopressin V1a and V2 receptor antagonist that is administered intravenously for up to 4 days. In a randomized placebo‐controlled study of patients with euvolemic or hypervolemic hyponatremia, intravenous conivaptan treatment increased serum Na levels by >6 mEq/l or to a serum Na >135 mEq/l in 69 to 88.5% of subjects compared to 20.7% of those receiving placebo (Zeltser D, Rosansky S, Van Rensburg H, et al. Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia. American J Nephrology 2007;27:447457). In a pilot study involving 24 patients with end‐stage liver disease, an infusion of conivaptan over 1 to 4 days was associated with an increase of serum sodium by >5 mmol/L in 60% of patients not receiving diuretics and 67% of patients on concomitant diuretic therapy by the end of treatment (O'Leary and Davis, 2009). Despite a concern about the potential for conivaptan to increase portal hypertension due to inhibition of splanchnic V1a receptors, the brief treatment appeared to be well tolerated without significant changes in systolic blood pressure, serum creatinine, variceal bleeding or worsening of ascites during the infusion period. However, approval for only 4 days of therapy and requirement for intravenous use eliminate any potential for chronic use.54
Tolvaptan is an orally available, selective V2 receptor antagonist whose efficacy was assessed in two multicenter, prospective, randomized, placebo‐controlled trials, Study of Ascending Levels of Tolvaptan in Hyponatremia 1 and 2 (SALT‐1 and SALT‐2).55 In these trials, clinically stable patients with DH (Na 135meq/l) associated with cirrhosis (22.4% in SALT‐1, 30.5% in SALT‐2), CHF or SIADH were randomized in a hospital setting to receive tolvaptan 15mg daily or placebo. Repeat Na levels were obtained at 8 hours, 2, 3, and 4 days and then weekly at days 11, 18, 25 and 30 after which study drug was discontinued and follow‐up Na level was determined 7 days later. The dose was adjusted to 30 mg and then 60 mg in an attempt to achieve a Na level >135 in those in whom hyponatremia persisted. During the initial day of the titration phase, fluid restriction was not maintained, and the patients were encouraged to respond to thirst with increased water ingestion.
Tolvaptan use was associated with a prompt increase in Na level as early as 8 hours after administration of the first dose. Serum Na increased more among those receiving tolvaptan than among those receiving placebo during the first 4 days and throughout the study period regardless of baseline Na level but returned to baseline within 1 week after discontinuation (Figure 3). The main side effects were increased thirst, dry mouth and increased urination. Importantly, an increased incidence of renal failure was not observed. Based on these results, FDA approval for tolvaptan in patients with hyponatremia was obtained in May 2009 for patients with DH‐associated with cirrhosis, CHF or SIADH for patients with Na levels 125 or symptomatic patients with Na levels between 125 and 135 that have not responded to fluid restriction.
Management of the Hospitalized Cirrhotic Patient With Hyponatremia: Recommendations
Hyponatremia in hospitalized cirrhotic patients is a marker for severe disease and high risk of hospital mortality.24 As a result, prompt evaluation and treatment is imperative. The availability of tolvaptan potentially revolutionizes the manner in which these patients are treated. In the SALT trials, only clinically stable patients were enrolled. In this last section, a guideline for the evaluation and treatment of acutely ill, hospitalized cirrhotic patients with DH is presented.
Evaluation
Determination of volume status is paramount but frequently problematic in the hospitalized cirrhotic patient. Due to the vasodilated state present in severe portal hypertension that is characterized by a relative hypotension and resting tachycardia, the usual hemodynamic parameters of blood pressure and heart rate can be difficult to interpret. Although significant extravascular volume in the form of ascites and edema may be present, patients may be intravascularly depleted due to previous diuretic use and extra‐renal losses due to impaired oral intake, vomiting, lactulose‐induced diarrhea, and gastrointestinal bleeding. Infection is a commonly associated condition, and endotoxin mediated splanchnic vasodilatation, especially in the setting of SBP, can adversely effective central blood volume status in the presence of severe ascites. Also, due to the Na avidity of the kidney and previous diuretic use, renal electrolytes can be difficult to interpret.
For patients in whom there is strong clinical concern about intravascular depletion (history of impaired oral intake, excessive vomiting and/or diarrhea, rapid weight loss, small volume ascites with history of large volume, azotemia), administration of limited intravenous normal saline (0.5‐1 L) should be considered. Patients with severe neurological symptoms should receive normal saline or hypertonic saline. Unless severe neurologic symptoms associated with profound hyponatremia is present, however, intravenous normal saline should not be administered for the hyponatremia alone. Administration of salt poor albumin (25%), especially for those with marked fluid overload and ascites, is an effective means to expand the central blood volume without exacerbating ascites and edema. After evaluation for and/or treatment of hypovolemia, all patients should receive a Na restricted diet (2000 mg daily) and placed on fluid restriction (see below for liberalization of fluid restriction upon initiation of tolvaptan therapy).
Diagnostic paracentesis should be performed for those with ascites to rule out the presence of SBP, and antibiotics administered to those with evidence of infection. High dose intravenous salt poor albumin should also be administered, especially to those at high risk of renal failure as determined by the presence of azotemia (Cr > 1.0 mg/dL) or severe liver insufficiency (TBili > 4.0 mg/dL).56, 57 Finally, all medications should be reviewed, and those associated with hyponatremia (diuretics, selective serotonin reuptake inhibitors, opiates, proton‐pump inhibitors) discontinued if possible.
Tolvaptan for DH
Patient Selection
Appropriate patient selection for tolvaptan therapy is extremely important (Table 2). In the SALT trials, only clinically stable patients were enrolled. The presence of hyponatremia in a recently hospitalized cirrhotic patient, however, frequently indicates severe disease with a high risk of acute renal failure and hospital death. In the SALT trials, many received concomitant diuretic therapy. Because of the importance of avoiding tolvaptan administration to hypovolemic patients, discontinuation of diuretic therapy prior the initiation of tolvaptan therapy and/or reevaluation after limited volume expansion should be considered.
| Hospital setting |
| Euvolumia or hypervolumia |
| Absence of recent weight loss, decrease in ascites, edema |
| Absence of excessive vomiting, diarrhea |
| Consider discontinuation of diuretic therapy prior to initiation of tolvaptan |
| Consider evaluation after limited volume expansion, especially with salt poor albumin prior to initiation of tolvaptan |
| Presence of clinically significant hyponatremia: 125mEq/L or less severe but symptomatic hyponatremia (125 to 134 mEq/L) that has resisted fluid restriction |
| Absence of severe neurologic symptoms attributable to hyponatremia |
| No co‐administration with intravenous saline |
| Ability to respond to thirst |
| No co‐administration with strong CYP 3A inhibitors (ketoconazole) |
| Absence of kidney failure with anuria |
Tolvaptan is indicated for cirrhotic patients with DH in whom the serum sodium is 125 mEq/L and in those with less severe but symptomatic hyponatremia (125‐134 mEq/L) that has resisted fluid restriction. Although the definition of symptomatic was not specifically defined, possible considerations include symptoms of mild hepatic encephalopathy or inability to tolerate dieresis due to the presence of hyponatremia. According to FDA guidelines, tolvaptan therapy must be initiated and re‐initiated in a hospital setting. Patients with severe neurologic symptoms attributable to hyponatremia in whom rapid treatment is critical should not receive tolvaptan but should rather be treated with normal saline. Similarly, patients should not receive combination therapy with tolvaptan and normal saline due to potential for a too‐rapid correction of hyponatremia and the development of central pontine myelinolysis. If saline had been administered for treatment of possible hypovolemia, it should be discontinued and persistent hyponatremia confirmed before starting tolvaptan. Other factors that need to be considered before initiating tolvaptan include the ability of the patient to respond to thirst with increased water ingestion and recognition that the patient will experience increased urine volume and frequency, requiring easy access to toilet. Patients should not be fluid restricted during the first day of tolvaptan therapy, but should be instructed to respond to their thirst with increased water ingestion. As a result, caution should be exercised in administering tolvaptan to a confused, restrained, unresponsive and/or bed‐bound patient who is not able to respond appropriately to thirst or increased urination.
In the SALT trials, the incidence of hyperkalemia (5%) was similar in the tolvaptan and placebo treated patients.55 However, further analysis of all multiple‐dose, placebo‐controlled trials, demonstrated that the aggregate incidence of hyperkalemia was slightly higher for tolvaptan‐treated subjects compared with placebo‐treated subjects (Otsuka). Because treatment with tolvaptan is associated with an acute reduction of the extracellular fluid volume which could result in increased serum potassium through hemoconcentration, it is recommended that serum potassium levels be monitored after initiation of tolvaptan treatment in patients with a serum potassium > 5 mEq/L as well as those who are receiving drugs known to increase serum potassium levels such as angiotensin converting enzyme inhibitors, angiotensin receptor blockers, or potassium sparing diuretics (Samsca Package Insert, Otsuka). Because tolvaptan is metabolized by the cytochrome P 3A system, patients receiving strong inhibitors such as ketoconazole should not receive tolvaptan. Anuric patients will not respond to tolvaptan. Finally, it is extremely important to administer tolvaptan only to patients with true hyponatremia and not to those with pseudohyponatremia in whom the plasma osmolality is normal but the measured serum sodium concentration artificially low due to marked elevations of other substances, such as can be seen in severe hyperglycemia, marked hyperlipidemia, or hyperproteinemia (as in multiple myeloma).
Tolvaptan Administration
The initial dose of tolvaptan is 15 mg daily. After receiving tolvaptan, many patients will develop an increased sense of thirst and need to urinate. As a result, patients should not be fluid restricted during the first day of therapy, and it is important to monitor the hemodynamics and Na level closely after initiating therapy with a repeat Na level at approximately 8 hours after the first dose. As a result, it should probably be administered early in the day and not at bedtime. The dose should be increased to 30 mg, then 60 mg in patients who do not respond by at least 5 mEq/L over the previous 24 hours and remain hyponatremic. In those with an excessive response (more than 8 meq/L during the first 8 hours or 12 meq/L on any subsequent day), the patient should be encourage to either drink more water, or the dose should be held or reduced. After the appropriate dose has been identified, the patient may be discharged and continued on tolvaptan long‐term.
With the advent of this exciting therapy, practical issues will need to be addressed, most important of which is its cost at $250 per day (Otsuka). In addition, the current recommendation to initiate tolvaptan only in a hospital further limits its widespread use. Most important, long‐term clinical benefit will need to be demonstrated. Although the SALT trials only involved treatment for up to 1 month, a multicenter, open‐label extension study for a mean duration of 701 days demonstrated that prolonged administration of tolvaptan maintains an increased serum sodium level.58 However, at this time, tolvaptan can only be considered as one of the promising drugs whose long‐term cost‐effectiveness is yet to be proven. Proof will require showing that correction of the hyponatremia leads to improved clinical outcomes, such as a reduction in length of stay or frequency of hospitalization, decreased renal failure, improved hepatic encephalopathy, deceased mortality, and improved post‐transplant outcomes.
Unanswered Questions
The vaptans provide an important opportunity to clarify the role that hyponatremia plays in the pathogenesis of cirrhosis. In the past, DH in a cirrhotic patient represented a sign of advanced disease. With the availability of safe and effective therapy, we can now determine whether it also plays an important role in the pathophysiology of end‐stage liver disease and whether its treatment will have a beneficial effect on patient outcomes.
Specific clinical questions that will inevitably be addressed over the next few years to determine whether DH is only a marker for advanced disease or whether it plays a direct but modifiable role in the pathophysiology of cirrhosis will include:
-
Role of vaptans in the management of ascites: In a 14‐day randomized, trial of a satavaptan, another selective vasopressin V(2) receptor antagonist, vs. placebo with spironolactone, combination therapy was associated with improved control of ascites and improvements in serum sodium levels in hyponatremic patients with ascites.59 If future similar studies demonstrate more prolonged benefits, this would constitute an important advance in the treatment of ascites in cirrhosis.
-
Effect on renal function: Prolonged use of tolvaptan leads to a compensatory increase in endogenous levels of AVP and, potentially, increased stimulation of V1a receptors, which might be helpful in the setting of portal hypertension. In patients with hepatorenal syndrome, vasopressin stimulation of splanchnic V1a receptors leads to improved renal function, presumably by decreasing splanchnic blood flow and improving central blood volume.60 As a result, tolvaptan may indirectly improve kidney function in patients with advanced cirrhosis and refractory ascites. Whether long‐term tolvaptan therapy will help to prevent hepatorenal syndrome through this mechanism remains to be determined but is an exciting possibility.61
-
Effect on hepatic encephalopathy: Hepatic encephalopathy is associated with poor quality of life in patients with cirrhosis. Although hepatic encephalopathy was not directly assessed in the SALT trials, the mean mental component summary of the Short Form General Health Survey, a quality of life measure, improved in cirrhotic patients receiving tolvaptan to a greater degree that those receiving placebo.62 A possible explanation for this finding is a beneficial effect of tolvaptan on hepatic encephalopathy. Confirmation of this hypothesis, however, will require prospective studies in which hepatic encephalopathy is directly assessed.
-
Effect on medical economics: Based on retrospective reviews, hyponatremia has an adverse impact on length of stay and outcomes following liver transplantation. It will be important to demonstrate in prospective studies that correction of hyponatremia with tolvaptan reduces length of stay, complications, and costs.
- ,,, et al.The aquaporin family of water channels in kidney: an update on physiology and pathophysiology of aquaporin‐2.Kidney Int.1996;49:1718–1723.
- ,,, et al.Hyponatremia in cirrhosis: from pathogenesis to treatment.Hepatology.1998;28:851–864.
- ,.Hyponatremia in cirrhosis: pathogenesis, clinical significance, and management.Hepatology.2008;48:1002–1010.
- ,.Hyponatremia.N Engl J Med.2000;342:1581–1589.
- ,,,.Economic impact of hyponatremia in hospitalized patients: a retrospective cohort study.Postgrad Med.2009;121:186–191.
- .Age and gender as risk factors for hyponatremia and hypernatremia.Clin Chim Acta.2003;337:169–172.
- .Consequences of inadequate management of hyponatremia.Am J Nephrol.2005;25:240–249.
- ,,.A review of drug‐induced hyponatremia.Am J Kidney Dis.2008;52:144–153.
- ,,, et al.Epidemiology, clinical and economic outcomes of admission hyponatremia among hospitalized patients.Curr Med Res Opin.2008;24:1601–1608.
- ,,.Incidence and prevalence of hyponatremia.Am J Med.2006;119(7 Supple 1):S30–S35.
- ,,.Disorders of sodium and water balance in hospitalized patients.Can J Anesth.2009;56:151–167.
- ,,, et al.Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits.Am J Med.2006;119;71.e1‐8.
- ,.Brain volume regulation in response to hypo‐osmolality and its correction.Am J Med.2006;119 (7 Suppl 1):S12–S16.
- ,,.Neurological manifestations and morbidity of hyponatremia: correlation with brain water and electrolytes.Medicine.1976;55:121–129.
- ,.The hyperdynamic circulation of chronic liver diseases: from the patient to the molecule.Hepatology.2006;43:S121–S131.
- ,,, et al.Hyponatremia of cirrhosis: role of vasopressin and decreased “effective” plasma volume.Scand J Gastroenterol.1997;32:829–834.
- ,,,.Metabolic clearance rate of arginine vasopressin in patients with cirrhosis.Hepatology.1992;16:974–979.
- .Water and sodium retention in edematous disorders: role of vasopressin and aldosterone.Am J Med.2006;119:S47–S53.
- ,,, et al.Circulatory function and hepatorenal syndrome in cirrhosis.Hepatology.2005;42:439–447.
- ,,, et al.Aquaporin‐2 urinary excretion in cirrhosis: relationship to vasopressin and nitric oxide.Dig Dis Sci.2010;55(4):1135–1141.
- ,,, et al.Hyponatremia in cirrhosis: results of a patient population survey.Hepatology.2006;44:1535–1542.
- et al.Hyponatremia and mortality among patients on the liver‐transplant list.N Engl J Med.2009;359:1018–1026.
- ,,, et al.Natural history of patients hospitalized for management of cirrhotic ascites.Clin Gastroenterol Hepatol.2006;4:1385–1394.
- ,,, et al.Clinical relevance of hyponatremia for the hospital outcome of cirrhotic patieints.Dig Liver Dis.2000;32:605–610.
- ,,, et al.Incidence and factors predictive of acute renal failure in patients with advanced liver cirrhosis.Clin Nephrol.2006;65:28–33.
- ,,, et al.Serum sodium predicts prognosis in critically ill cirrhotic patients.J Clin Gastroenterol2010;44(3):220–226.
- ,,, et al.Addition of serum sodium into the MELD score predicts waiting list mortality better than MELD alone.Liver Transpl.2005;11:336–343.
- ,,, et al.Persistent ascites and low serum sodium identify patients with cirrhosis and low MELD scores who are at high risk for early death.Hepatology.2004;40:802–810.
- ,,, et al.,Validation of model for end‐stage liver disease score to serum sodium ratio index as a prognostic predictor in patients with cirrhosis.J Gastroenterol Hepatol.2009;24:1547–1553.
- ,,, et al.Hepatic venous pressure gradient can predict the development of hepatocellular carcinoma and hyponatremia in decompensated alcoholic cirrhosis.Eur J Gastroenterol Hepatol.2009;21:1241–1246.
- ,,, et al.Serum sodium predicts mortality in patients listed for liver transplantation.Hepatology.2005;41:32–39.
- ,,, et al.Effect of hyponatremia on outcomes following orthotopic liver transplantation.Liver Int.2009;29:1071–1077.
- ,,, et al.Total paracentesis in cirrhotic patients with tense ascites and dilutional hyponatremia.Am J Gastroenterol.1999;94:2219–2223.
- ,,, et al.Dilutional hyponatremia in patients with cirrhosis and ascites.Arch Intern Med.2002;162:323–328.
- ,.Therapeutic approaches to the treatment of edema and ascites: the use of diuretics.Am J Ther.2009;16:98–101.
- ,,, et al.Randomized comparative study of therapeutic paracentesis with and without intravenous albumin in cirrhosis.Gastroenterol.1988;94:1493–1502.
- ,,, et al.Effect of intrathoracic pressure on plasma arginine vasopressin levels.Gastroenterology.1991;101:607–617.
- .Nephrotoxicities of nonsteroidal anti‐inflammatory drugs.J Formos Med Assoc.1997;96:157–171.
- ,,, et al.Serum creatinine and bilirubin predict renal failure and mortality in patients with spontaneous bacterial peritonitis: a retrospective study.Liver Int.2009;29:415–419.
- ,.Pathogenic mechanisms of hepatic encephalopathy.Gut2008;57:1156–1165.
- ,,, et al.Effects of dilutional hyponatremia on brain organic osmolytes and water content in patients with cirrhosis.Hepatology.2004;39:1613–1622.
- .Low grade cerebral edema and the pathogenesis of hepatic encephalopathy in cirrhosis.Hepatology.2006;43:1187–1190.
- ,,, et al.Prevalence and prognostic value of quantified electroencephalogram (EEG) alterations in cirrhotic patients.J Hepatol.2001;35:37–45.
- ,,, et al.Hyponatremia is a risk factor of hepatic encephalopathy in patients with cirrhosis: a propective study with time‐dependent analysis.Am J Gastroenterol.2009;104:1382–1389.
- ,,, et al.Analysis of prognostic variables in the prediction of mortality, shunt failure, variceal rebleeding and encephalopathy following the transjugular intrahepatic portosystecim stent‐shunt for variceal haemorrhage.J Hepatol.1995;2:123–128.
- ,,, et al.Cardiac dysfunction during liver transplantation: incidence and preoperative predictors.Transplantation.2008;85:1766–1772.
- ,,, et al.,Impact of pretransplant hyponatremia on outcome following liver transplantation.Hepatology.2009;49:1610–1615.
- ,,, et al.Hyponatremia impairs early posttransplantation outcome in patients with cirrhosis undergoing liver transplantation.Gastroenterology.2006;130:1135–1143.
- ,,.Hyponatremia in cirrhosis: clinical features and management.Gastroenterol Clin Biol.2006;30:1144–1151.
- ,,, et al.Intravenous albumin infusion is an effective therapy for hyponatremia in cirrhotic patients with ascites.Gut.1990;31:204–207.
- ,,, et al.Comparison of two aquaretic drugs (niravoline and OPC‐31260) in cirrhotic rats with ascites and water retention.J Pharmacol Exp Ther.1999;289:194–201.
- ,,.Plasma demeclocycline levels and nephrotoxicity. Correlation in hyponatremic cirrhotic patients.JAMA.1980;243:2513–2515.
- ,,, et al.Oral misoprostol or intravenous prostaglandin E2 do not improve renal function in patients with cirrhosis and ascites with hyponatremia and renal failure.J Hepatol.1993;17:220–226.
- ,,, et al.Effect of the V1a/V2‐AVP receptor antagonist, Conivaptan, on renal water metabolism and systemic hemodynamics in rats with cirrhosis and ascites.J Hepatol.2003;38:755–761.
- ,,, et al.Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355:2099–2112.
- ,,, et al.Effect of intravenous albumin on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis.N Engl J Med.1999;341:403–409.
- ,,, et al.Restricted use of albumin for spontaneous bacterial peritonitis.Gut.2007;56:597–599.
- ,,, et al.Oral tolvaptan is safe and effective in chronic hypyonatremia.J Am Soc Nephrol.2010;21(4):705–712.
- ,,, et al.Effects of satavaptan, a selective vasopressin V(2) receptor antagonist, on ascites and serum sodium in cirrhosis with hyponatremia: a randomized trial.Hepatology.2008;48:204–213.
- ,,, et al.A randomized, prospective, double‐blind, placebo‐controlled trial of terlipressin for type 1 hepatorenal syndrome.Gastroenterology.2008;134:1360–1368.
- ,.Tolvapten and its potential in the treatment of hyponatremia.Ther Clin Risk Manag.2008;4:1149–11455.
- ,,, et al.The effects of vasopressin V2 receptor antagonist in the management of patients with cirrhosis and hyponatremia. Safety and efficacy of oral tolvaptan in the SALT trials.Hepatology.2009;50S:467A.
- ,,, et al.Treatment of hyponatremic cirrhosis with ascites resistant to diuretics by urea.Nephron.1986;44:337–343.
- ,,, et al.Aquaretic effects of niravoline, a kappa‐opioid agonist, in patients with cirrhosis.J Hepatol.2000;32:38–42.
- ,, et al.The association between the serum sodium level and the severity of complications in liver cirrhosis.Korean J Intern Med.2009;24:106–112.
- ,,, et al.Nitric oxide in ascitic fluid is an independent predictor of the development of renal impairment in patients with cirrhosis and spontaneous bacterial peritonitis.Eur J Gastroenterol Hepatol.2004;16:571–577.
- ,,, et al.The aquaporin family of water channels in kidney: an update on physiology and pathophysiology of aquaporin‐2.Kidney Int.1996;49:1718–1723.
- ,,, et al.Hyponatremia in cirrhosis: from pathogenesis to treatment.Hepatology.1998;28:851–864.
- ,.Hyponatremia in cirrhosis: pathogenesis, clinical significance, and management.Hepatology.2008;48:1002–1010.
- ,.Hyponatremia.N Engl J Med.2000;342:1581–1589.
- ,,,.Economic impact of hyponatremia in hospitalized patients: a retrospective cohort study.Postgrad Med.2009;121:186–191.
- .Age and gender as risk factors for hyponatremia and hypernatremia.Clin Chim Acta.2003;337:169–172.
- .Consequences of inadequate management of hyponatremia.Am J Nephrol.2005;25:240–249.
- ,,.A review of drug‐induced hyponatremia.Am J Kidney Dis.2008;52:144–153.
- ,,, et al.Epidemiology, clinical and economic outcomes of admission hyponatremia among hospitalized patients.Curr Med Res Opin.2008;24:1601–1608.
- ,,.Incidence and prevalence of hyponatremia.Am J Med.2006;119(7 Supple 1):S30–S35.
- ,,.Disorders of sodium and water balance in hospitalized patients.Can J Anesth.2009;56:151–167.
- ,,, et al.Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits.Am J Med.2006;119;71.e1‐8.
- ,.Brain volume regulation in response to hypo‐osmolality and its correction.Am J Med.2006;119 (7 Suppl 1):S12–S16.
- ,,.Neurological manifestations and morbidity of hyponatremia: correlation with brain water and electrolytes.Medicine.1976;55:121–129.
- ,.The hyperdynamic circulation of chronic liver diseases: from the patient to the molecule.Hepatology.2006;43:S121–S131.
- ,,, et al.Hyponatremia of cirrhosis: role of vasopressin and decreased “effective” plasma volume.Scand J Gastroenterol.1997;32:829–834.
- ,,,.Metabolic clearance rate of arginine vasopressin in patients with cirrhosis.Hepatology.1992;16:974–979.
- .Water and sodium retention in edematous disorders: role of vasopressin and aldosterone.Am J Med.2006;119:S47–S53.
- ,,, et al.Circulatory function and hepatorenal syndrome in cirrhosis.Hepatology.2005;42:439–447.
- ,,, et al.Aquaporin‐2 urinary excretion in cirrhosis: relationship to vasopressin and nitric oxide.Dig Dis Sci.2010;55(4):1135–1141.
- ,,, et al.Hyponatremia in cirrhosis: results of a patient population survey.Hepatology.2006;44:1535–1542.
- et al.Hyponatremia and mortality among patients on the liver‐transplant list.N Engl J Med.2009;359:1018–1026.
- ,,, et al.Natural history of patients hospitalized for management of cirrhotic ascites.Clin Gastroenterol Hepatol.2006;4:1385–1394.
- ,,, et al.Clinical relevance of hyponatremia for the hospital outcome of cirrhotic patieints.Dig Liver Dis.2000;32:605–610.
- ,,, et al.Incidence and factors predictive of acute renal failure in patients with advanced liver cirrhosis.Clin Nephrol.2006;65:28–33.
- ,,, et al.Serum sodium predicts prognosis in critically ill cirrhotic patients.J Clin Gastroenterol2010;44(3):220–226.
- ,,, et al.Addition of serum sodium into the MELD score predicts waiting list mortality better than MELD alone.Liver Transpl.2005;11:336–343.
- ,,, et al.Persistent ascites and low serum sodium identify patients with cirrhosis and low MELD scores who are at high risk for early death.Hepatology.2004;40:802–810.
- ,,, et al.,Validation of model for end‐stage liver disease score to serum sodium ratio index as a prognostic predictor in patients with cirrhosis.J Gastroenterol Hepatol.2009;24:1547–1553.
- ,,, et al.Hepatic venous pressure gradient can predict the development of hepatocellular carcinoma and hyponatremia in decompensated alcoholic cirrhosis.Eur J Gastroenterol Hepatol.2009;21:1241–1246.
- ,,, et al.Serum sodium predicts mortality in patients listed for liver transplantation.Hepatology.2005;41:32–39.
- ,,, et al.Effect of hyponatremia on outcomes following orthotopic liver transplantation.Liver Int.2009;29:1071–1077.
- ,,, et al.Total paracentesis in cirrhotic patients with tense ascites and dilutional hyponatremia.Am J Gastroenterol.1999;94:2219–2223.
- ,,, et al.Dilutional hyponatremia in patients with cirrhosis and ascites.Arch Intern Med.2002;162:323–328.
- ,.Therapeutic approaches to the treatment of edema and ascites: the use of diuretics.Am J Ther.2009;16:98–101.
- ,,, et al.Randomized comparative study of therapeutic paracentesis with and without intravenous albumin in cirrhosis.Gastroenterol.1988;94:1493–1502.
- ,,, et al.Effect of intrathoracic pressure on plasma arginine vasopressin levels.Gastroenterology.1991;101:607–617.
- .Nephrotoxicities of nonsteroidal anti‐inflammatory drugs.J Formos Med Assoc.1997;96:157–171.
- ,,, et al.Serum creatinine and bilirubin predict renal failure and mortality in patients with spontaneous bacterial peritonitis: a retrospective study.Liver Int.2009;29:415–419.
- ,.Pathogenic mechanisms of hepatic encephalopathy.Gut2008;57:1156–1165.
- ,,, et al.Effects of dilutional hyponatremia on brain organic osmolytes and water content in patients with cirrhosis.Hepatology.2004;39:1613–1622.
- .Low grade cerebral edema and the pathogenesis of hepatic encephalopathy in cirrhosis.Hepatology.2006;43:1187–1190.
- ,,, et al.Prevalence and prognostic value of quantified electroencephalogram (EEG) alterations in cirrhotic patients.J Hepatol.2001;35:37–45.
- ,,, et al.Hyponatremia is a risk factor of hepatic encephalopathy in patients with cirrhosis: a propective study with time‐dependent analysis.Am J Gastroenterol.2009;104:1382–1389.
- ,,, et al.Analysis of prognostic variables in the prediction of mortality, shunt failure, variceal rebleeding and encephalopathy following the transjugular intrahepatic portosystecim stent‐shunt for variceal haemorrhage.J Hepatol.1995;2:123–128.
- ,,, et al.Cardiac dysfunction during liver transplantation: incidence and preoperative predictors.Transplantation.2008;85:1766–1772.
- ,,, et al.,Impact of pretransplant hyponatremia on outcome following liver transplantation.Hepatology.2009;49:1610–1615.
- ,,, et al.Hyponatremia impairs early posttransplantation outcome in patients with cirrhosis undergoing liver transplantation.Gastroenterology.2006;130:1135–1143.
- ,,.Hyponatremia in cirrhosis: clinical features and management.Gastroenterol Clin Biol.2006;30:1144–1151.
- ,,, et al.Intravenous albumin infusion is an effective therapy for hyponatremia in cirrhotic patients with ascites.Gut.1990;31:204–207.
- ,,, et al.Comparison of two aquaretic drugs (niravoline and OPC‐31260) in cirrhotic rats with ascites and water retention.J Pharmacol Exp Ther.1999;289:194–201.
- ,,.Plasma demeclocycline levels and nephrotoxicity. Correlation in hyponatremic cirrhotic patients.JAMA.1980;243:2513–2515.
- ,,, et al.Oral misoprostol or intravenous prostaglandin E2 do not improve renal function in patients with cirrhosis and ascites with hyponatremia and renal failure.J Hepatol.1993;17:220–226.
- ,,, et al.Effect of the V1a/V2‐AVP receptor antagonist, Conivaptan, on renal water metabolism and systemic hemodynamics in rats with cirrhosis and ascites.J Hepatol.2003;38:755–761.
- ,,, et al.Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia.N Engl J Med.2006;355:2099–2112.
- ,,, et al.Effect of intravenous albumin on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis.N Engl J Med.1999;341:403–409.
- ,,, et al.Restricted use of albumin for spontaneous bacterial peritonitis.Gut.2007;56:597–599.
- ,,, et al.Oral tolvaptan is safe and effective in chronic hypyonatremia.J Am Soc Nephrol.2010;21(4):705–712.
- ,,, et al.Effects of satavaptan, a selective vasopressin V(2) receptor antagonist, on ascites and serum sodium in cirrhosis with hyponatremia: a randomized trial.Hepatology.2008;48:204–213.
- ,,, et al.A randomized, prospective, double‐blind, placebo‐controlled trial of terlipressin for type 1 hepatorenal syndrome.Gastroenterology.2008;134:1360–1368.
- ,.Tolvapten and its potential in the treatment of hyponatremia.Ther Clin Risk Manag.2008;4:1149–11455.
- ,,, et al.The effects of vasopressin V2 receptor antagonist in the management of patients with cirrhosis and hyponatremia. Safety and efficacy of oral tolvaptan in the SALT trials.Hepatology.2009;50S:467A.
- ,,, et al.Treatment of hyponatremic cirrhosis with ascites resistant to diuretics by urea.Nephron.1986;44:337–343.
- ,,, et al.Aquaretic effects of niravoline, a kappa‐opioid agonist, in patients with cirrhosis.J Hepatol.2000;32:38–42.
- ,, et al.The association between the serum sodium level and the severity of complications in liver cirrhosis.Korean J Intern Med.2009;24:106–112.
- ,,, et al.Nitric oxide in ascitic fluid is an independent predictor of the development of renal impairment in patients with cirrhosis and spontaneous bacterial peritonitis.Eur J Gastroenterol Hepatol.2004;16:571–577.