User login
Treatment of chronic inflammatory diseases with implantable medical devices*
Implantable devices are increasingly used in the treatment of diseases which have historically been targeted only with small molecule and biological therapeutic agents. In addition to well-established products such as subcutaneous insulin pumps and intra-arterial chemotherapy pumps, where the implantable device merely serves as a more efficient means of delivering the drug, there are a number of recently developed therapeutic approaches in which the implanted device itself functions to directly treat the underlying medical condition. One particularly successful example of this strategy is cardiac resynchronization using biventricular pacing devices for congestive heart failure (CHF). These devices were approved for marketing after having been proved to prolong survival in patients whose disease had progressed despite medical management.1 Implantable device products are now approved or in late-stage development for many other traditional “medical” disorders such as hypertension, obesity, diabetes, Parkinson’s disease, and glaucoma. Recent advances in understanding the interplay between the central nervous system and the immune system have made possible a feasible implantable device approach that may similarly find use in the management of rheumatoid arthritis (RA) and other chronic inflammatory diseases.2
NEUROSTIMULATION OF THE CHOLINERGIC ANTIINFLAMMATORY PATHWAY
The vagus nerve mediates an important neural reflex which senses inflammation both peripherally and in the central nervous system, and downregulates the inflammation via efferent neural outflow to the reticuloendothelial system. The efferent arm of this reflex has been termed the “cholinergic antiinflammatory pathway” (CAP). The CAP serves as a physiological regulator of inflammation by responding to environmental injury, pathogens, and other external threats with an appropriate degree of immune system activation.3 An increasing body of evidence indicates that the CAP can also be harnessed to reduce pathological inflammation. Electrical neurostimulation of the vagus nerve (NCAP) in an appropriate manner with an implantable device is emerging as a novel and potentially feasible means of treating diseases characterized by excessive and dysregulated inflammation.
Our current understanding of the CAP began with the observations of Kevin Tracey and colleagues over a decade ago. They demonstrated that systemic, hepatic, and splenic tumor necrosis factor (TNF) production as well as the physiological manifestations of endotoxemic shock in rodents were worsened by vagotomy and ameliorated by electrical stimulation of the cervical vagus nerve (VNS). Further, based on in vitro experiments they postulated that this effect was mediated directly by acetylcholine acting through specific receptors on macrophages in the reticuloendothelial system.4 It was later demonstrated that reducing the response to endotoxemia using NCAP required an intact spleen, and selective anatomical lesion experiments showed that an intact neural pathway to the spleen from the cervical vagus through the celiac ganglion and the splenic nerve was also necessary for this effect.5 Within the spleen itself, nerve fiber synaptic vesicles are found in close apposition to TNF-secreting macrophages.6 The α-7 nicotinic acetylcholine receptor, expressed on the surface of macrophages, is essential for the NCAP effect as demonstrated by antisense oligonucleotide and targeted disruption experiments.7 In the macrophage, the α-7 nicotinic acetylcholine receptor does not appear to transduce signals through ion channels, as is the case in neuronal tissue. Rather, the NCAP effect is mediated at the subcellular level by alterations in the NF-κB and JAK/STAT/SOCS pathways.8,9

When taken together, these studies show that NCAP has a dual set of immunological effects: it reduces production of systemically active cytokines by resident spleen cells and also causes circulating cells which traverse the spleen to develop an altered phenotype with reduced expression of inflammatory mediators and adhesion molecules upon trafficking to inflamed tissue.
An important characteristic of NCAP delivered by VNS is that very brief episodes of stimulation result in a remarkably prolonged biological effect. Huston et al delivered a single 30-second electrical VNS or sham treatment in rats, and then induced endotoxemia with intraperitoneal lipopolysaccharide (LPS) at varying times after VNS. Interestingly, this brief VNS stimulation reduced production of serum TNF in response to systemic LPS exposure for up to 48 hours. Similarly, after only 60 minutes of exposure to acetylcholine, cultured human macrophages are changed in phenotype such that they become refractory to in vitro LPS stimulation for up to 48 hours thereafter.15 The consistency of this phenomenon across species is corroborated by preliminary data in a canine model where 60-second VNS treatment results in reduced LPS-inducible TNF production in a whole-blood in vitro release assay for several days after the VNS (M Faltys, personal communication). A duration of biological effect lasting hours to days after periods of stimulation lasting for only seconds to minutes implies that an implantable device will probably only need to operate with very short daily duty cycles to effectively elicit an NCAP response. This will in turn greatly reduce the necessary size and complexity of the device itself, and increase its functional lifespan, with resultant reductions in overall cost of the treatment.
NEUROSTIMULATION OF THE CAP IN ANIMAL MODELS OF DISEASE

In a canine model of CHF induced by rapid ventricular pacing, inflammation and ventricular remodeling with fibrosis are typically accompanied by marked increases in serum C-reactive protein (CRP) levels. In addition to improving the physiological manifestations of CHF, VNS resulted in 60% to 80% reductions in CRP for up to 8 weeks.17 In another canine CHF model induced by repetitive microembolization, which is similarly associated with systemic and myocardial inflammation, VNS markedly reduced circulating levels of interleukin 6 and TNF for up to 12 weeks.18 Importantly, both these studies show that rapid tachyphylaxis does not appear to occur with NCAP over periods of time that are relatively chronic by the typical standards of animal models.
VAGAL NERVE STIMULATION FOR EPILEPSY AND DEPRESSION: EXPERIENCE IN HUMANS
VNS delivered using a surgically implanted cuffed cervical vagus nerve lead and pacemaker-style pulse generator device has been approved for the treatment of refractory epilepsy in the United States since the mid-1990s and has more recently been approved for treatment of depression. Over 50,000 patients have been implanted with these devices world wide since that time. The safety profile of both surgical implantation and VNS delivery in this setting is well established.19 The major tolerability problem is laryngeal and pharyngeal symptoms, such as hoarseness and dysphonia, which are present almost solely during periods of active device stimulation. The frequency and severity of these treatments decreases after receiving treatment for an extended time.20 With growing experience in VNS delivery over the first 5 years of use, it also became apparent that reducing the active stimulation duty cycle from 40% to 10%, and keeping stimulation currents at ≤ 1.5 mA results in a marked reduction in these symptoms.21 Of note, the stimulation currents necessary to evoke NCAP in animals are well below the 1.5 mA level, and as above, NCAP is effective even with very brief, once-daily periods of stimulation (ie, a duty cycle of 0.07% if given for 1 min each day). Thus it is likely that the laryngeal and pharyngeal adverse event profile of VNS will not be problematic in the setting of NCAP delivery for inflammation.
A POTENTIAL ROLE FOR THERAPEUTIC NCAP USING IMPLANTABLE DEVICES IN HUMAN INFLAMMATORY DISEASES
Autonomic nervous system activity can be measured indirectly by recording cardiac R-R interval variability and subjecting the data to power spectral analysis. Such heart rate variability (HRV) measurements are influenced by the levels of vagus nerve activity and by balance in cardiac sympathetic–parasympathetic tone. Reduced HRV is indicative of decreased vagal tone, and reductions in HRV have a strong inverse correlation with CRP levels, progression of atherosclerosis, and risk of sudden death.22,23 HRV is also reduced relative to normal subjects in patients with RA, systemic lupus erythematosus, and Sjögren syndrome, and the extent of reduction in HRV within the patient groups correlates with disease severity.24–26 Although these associations are only correlative and do not provide firm evidence of causality, they do provide additional epidemiological support for the hypothesis that driving increased vagal activity using implantable devices may have a favorable effect on inflammatory disease.
Implantable neurostimulation devices have not yet been tested in human patients with RA. However, preliminary evidence from a small study carried out in normal volunteers demonstrated that the CAP reflex can be elicited by brief mechanical stimulation of the afferent auricular branch of the vagus nerve, as shown by reduction of in vitro LPS-inducible cytokine production (T van der Poll, personal communication). Clinical testing of NCAP using implantable VNS devices will begin in the near future. The devices to be used for these initial studies will be very similar in design to those currently in use for epilepsy treatment. However, prototype versions of the device which will be used in follow-on studies are miniaturized to the point where they will be directly implantable on the vagus nerve, without the need for a pulse generator unit on the chest and will use a small self-contained battery system which can be recharged using transcutaneous radiofrequency induction. Given the long lifespan, relatively low cost, and potential for increased safety over currently available treatments, NCAP delivered using an implantable device holds great promise as a novel potential therapeutic approach for patients with RA and other inflammatory diseases.
- Bristow MR, Saxon LA, Boehmer J, et al Comparison of Medical Therapy, Pacing, and Defibrillation in Heart Failure (COMPANION) Investigators. Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. N Engl J Med 2004; 350:2140–2150.
- van Maanen MA, Vervoordeldonk MJ, Tak PP. The cholinergic anti-inflammatory pathway: towards innovative treatment of rheumatoid arthritis. Nat Rev Rheumatol 2009; 5:229–232.
- Tracey KJ. Reflex control of immunity. Nat Rev Immunol 2009; 9:418–428.
- Borovikova LV, Ivanova S, Zhang M, et al Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000; 405:458–462.
- Huston JM, Ochani M, Rosas-Ballina M, et al Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med 2006; 203:1623–1628.
- Rosas-Ballina M, Ochani M, Parrish WR, et al Splenic nerve is required for cholinergic antiinflammtory pathway control of TNF in endotoxemia. Proc Natl Acad Sci 2008; 105:11008–11013.
- Wang H, Yu M, Ochani M, et al Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003; 421:384–388.
- Wang H, Liao H, Ochani M, et al Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med 2004; 10:1216–1221.
- de Jonge WJ, van der Zanden EP, The FO, et al Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol 2005; 6:844–851.
- Huston JM, Rosas-Ballina M, Xue X, et al Cholinergic neural signals to the spleen down-regulate leukocyte trafficking via CD11b. J Immunol 2009; 183:552–559.
- O’Mahony C, van der Kleij H, Bienenstock J, et al Loss of vagal anti-inflammatory effect: in vivo visualization and adoptive transfer. Am J Physiol Regul Integr Comp Physiol 2009; 297:R1118–R1126.
- Ghia JE, Blennerhassett P, Kumar-Ondiveeran H, et al The vagus nerve: a tonic inhibitory influence associated with inflammatory bowel disease in a murine model. Gastroenterology 2006; 131:1122–1130.
- Ghia JE, Blennerhassett P, Collins SM. Vagus nerve integrity and experimental colitis. Am J Physiol Gastrointest Liver Physiol 2007; 293:G560–G567.
- Karimi K, Bienenstock J, Wang L, et al The vagus nerve modulates CD4+ T cell activity. Brain Behav Immun 2010; 24:316–323.
- Huston JM, Gallowitsch-Puerta M, Ochani M, et al Transcutaneous vagus nerve stimulation reduces serum high mobility group box 1 levels and improves survival in murine sepsis. Crit Care Med 2007; 35:2762–2768.
- Levine Y, Faltys M, Black K, et al Neurostimulation of the cholinergic anti-inflammatory pathway (NCAP) ameliorates CIA in rats. Ann Rheum Dis 2010; 69( suppl 3):191.
- Zhang Y, Popovic ZB, Bibevski S, et al Chronic vagus nerve stimulation improves autonomic control and attenuates systemic inflammation and heart failure progression in a canine high-rate pacing model. Circ Heart Fail 2009; 2:692–699.
- Gupta RC, Imai M, Jiang AJ, et al Chronic therapy with selective vagus nerve stimulation normalizes plasma concentration of tumor necrosis factor alpha, interleukin-6, and B-type natriuretic peptide in dogs with heart failure. J Am Coll Cardiol 2006; 47:77A.
- Beekwilder JP, Beems T. Overview of the clinical applications of vagus nerve stimulation. J Clin Neurophysiol 2010; 27:130–138.
- Ben-Menachem E. Vagus nerve stimulation, side effects, and longterm safety. J Clin Neurophysiol 2001; 18:415–418.
- Heck C, Helmers SL, DeGiorgio CM. Vagus nerve stimulation therapy, epilepsy, and device parameters: scientific basis and recommendations for use. Neurology 2002; 59( 6 suppl 4):S31–S37.
- Huikuri HV, Jokinen V, Syvänne M, et al Heart rate variability and progression of coronary atherosclerosis. Arterioscler Thromb Vasc Biol 1999; 19:1979–1985.
- Sajadieh A, Nielsen OW, Rasmussen V, et al Increased heart rate and reduced heart-rate variability are associated with subclinical inflammation in middle-aged and elderly subjects with no apparent heart disease. Eur Heart J 2004; 25:363–370.
- Louthrenoo W, Ruttanaumpawan P, Aramrattana A, et al Cardio vascular autonomic nervous system dysfunction in patients with rheumatoid arthritis and systemic lupus erythematosus. QJM 1999; 92:97–102.
- Evrengül H, Dursunoglu D, Cobankara V, et al Heart rate variability in patients with rheumatoid arthritis. Rheumatol Int 2004; 24:198–202.
- Stojanovich L, Milovanovich B, de Luka SR, et al Cardiovascular autonomic dysfunction in systemic lupus, rheumatoid arthritis, primary Sjögren syndrome and other autoimmune diseases. Lupus 2007; 16:181–185.
Implantable devices are increasingly used in the treatment of diseases which have historically been targeted only with small molecule and biological therapeutic agents. In addition to well-established products such as subcutaneous insulin pumps and intra-arterial chemotherapy pumps, where the implantable device merely serves as a more efficient means of delivering the drug, there are a number of recently developed therapeutic approaches in which the implanted device itself functions to directly treat the underlying medical condition. One particularly successful example of this strategy is cardiac resynchronization using biventricular pacing devices for congestive heart failure (CHF). These devices were approved for marketing after having been proved to prolong survival in patients whose disease had progressed despite medical management.1 Implantable device products are now approved or in late-stage development for many other traditional “medical” disorders such as hypertension, obesity, diabetes, Parkinson’s disease, and glaucoma. Recent advances in understanding the interplay between the central nervous system and the immune system have made possible a feasible implantable device approach that may similarly find use in the management of rheumatoid arthritis (RA) and other chronic inflammatory diseases.2
NEUROSTIMULATION OF THE CHOLINERGIC ANTIINFLAMMATORY PATHWAY
The vagus nerve mediates an important neural reflex which senses inflammation both peripherally and in the central nervous system, and downregulates the inflammation via efferent neural outflow to the reticuloendothelial system. The efferent arm of this reflex has been termed the “cholinergic antiinflammatory pathway” (CAP). The CAP serves as a physiological regulator of inflammation by responding to environmental injury, pathogens, and other external threats with an appropriate degree of immune system activation.3 An increasing body of evidence indicates that the CAP can also be harnessed to reduce pathological inflammation. Electrical neurostimulation of the vagus nerve (NCAP) in an appropriate manner with an implantable device is emerging as a novel and potentially feasible means of treating diseases characterized by excessive and dysregulated inflammation.
Our current understanding of the CAP began with the observations of Kevin Tracey and colleagues over a decade ago. They demonstrated that systemic, hepatic, and splenic tumor necrosis factor (TNF) production as well as the physiological manifestations of endotoxemic shock in rodents were worsened by vagotomy and ameliorated by electrical stimulation of the cervical vagus nerve (VNS). Further, based on in vitro experiments they postulated that this effect was mediated directly by acetylcholine acting through specific receptors on macrophages in the reticuloendothelial system.4 It was later demonstrated that reducing the response to endotoxemia using NCAP required an intact spleen, and selective anatomical lesion experiments showed that an intact neural pathway to the spleen from the cervical vagus through the celiac ganglion and the splenic nerve was also necessary for this effect.5 Within the spleen itself, nerve fiber synaptic vesicles are found in close apposition to TNF-secreting macrophages.6 The α-7 nicotinic acetylcholine receptor, expressed on the surface of macrophages, is essential for the NCAP effect as demonstrated by antisense oligonucleotide and targeted disruption experiments.7 In the macrophage, the α-7 nicotinic acetylcholine receptor does not appear to transduce signals through ion channels, as is the case in neuronal tissue. Rather, the NCAP effect is mediated at the subcellular level by alterations in the NF-κB and JAK/STAT/SOCS pathways.8,9
When taken together, these studies show that NCAP has a dual set of immunological effects: it reduces production of systemically active cytokines by resident spleen cells and also causes circulating cells which traverse the spleen to develop an altered phenotype with reduced expression of inflammatory mediators and adhesion molecules upon trafficking to inflamed tissue.
An important characteristic of NCAP delivered by VNS is that very brief episodes of stimulation result in a remarkably prolonged biological effect. Huston et al delivered a single 30-second electrical VNS or sham treatment in rats, and then induced endotoxemia with intraperitoneal lipopolysaccharide (LPS) at varying times after VNS. Interestingly, this brief VNS stimulation reduced production of serum TNF in response to systemic LPS exposure for up to 48 hours. Similarly, after only 60 minutes of exposure to acetylcholine, cultured human macrophages are changed in phenotype such that they become refractory to in vitro LPS stimulation for up to 48 hours thereafter.15 The consistency of this phenomenon across species is corroborated by preliminary data in a canine model where 60-second VNS treatment results in reduced LPS-inducible TNF production in a whole-blood in vitro release assay for several days after the VNS (M Faltys, personal communication). A duration of biological effect lasting hours to days after periods of stimulation lasting for only seconds to minutes implies that an implantable device will probably only need to operate with very short daily duty cycles to effectively elicit an NCAP response. This will in turn greatly reduce the necessary size and complexity of the device itself, and increase its functional lifespan, with resultant reductions in overall cost of the treatment.
NEUROSTIMULATION OF THE CAP IN ANIMAL MODELS OF DISEASE
In a canine model of CHF induced by rapid ventricular pacing, inflammation and ventricular remodeling with fibrosis are typically accompanied by marked increases in serum C-reactive protein (CRP) levels. In addition to improving the physiological manifestations of CHF, VNS resulted in 60% to 80% reductions in CRP for up to 8 weeks.17 In another canine CHF model induced by repetitive microembolization, which is similarly associated with systemic and myocardial inflammation, VNS markedly reduced circulating levels of interleukin 6 and TNF for up to 12 weeks.18 Importantly, both these studies show that rapid tachyphylaxis does not appear to occur with NCAP over periods of time that are relatively chronic by the typical standards of animal models.
VAGAL NERVE STIMULATION FOR EPILEPSY AND DEPRESSION: EXPERIENCE IN HUMANS
VNS delivered using a surgically implanted cuffed cervical vagus nerve lead and pacemaker-style pulse generator device has been approved for the treatment of refractory epilepsy in the United States since the mid-1990s and has more recently been approved for treatment of depression. Over 50,000 patients have been implanted with these devices world wide since that time. The safety profile of both surgical implantation and VNS delivery in this setting is well established.19 The major tolerability problem is laryngeal and pharyngeal symptoms, such as hoarseness and dysphonia, which are present almost solely during periods of active device stimulation. The frequency and severity of these treatments decreases after receiving treatment for an extended time.20 With growing experience in VNS delivery over the first 5 years of use, it also became apparent that reducing the active stimulation duty cycle from 40% to 10%, and keeping stimulation currents at ≤ 1.5 mA results in a marked reduction in these symptoms.21 Of note, the stimulation currents necessary to evoke NCAP in animals are well below the 1.5 mA level, and as above, NCAP is effective even with very brief, once-daily periods of stimulation (ie, a duty cycle of 0.07% if given for 1 min each day). Thus it is likely that the laryngeal and pharyngeal adverse event profile of VNS will not be problematic in the setting of NCAP delivery for inflammation.
A POTENTIAL ROLE FOR THERAPEUTIC NCAP USING IMPLANTABLE DEVICES IN HUMAN INFLAMMATORY DISEASES
Autonomic nervous system activity can be measured indirectly by recording cardiac R-R interval variability and subjecting the data to power spectral analysis. Such heart rate variability (HRV) measurements are influenced by the levels of vagus nerve activity and by balance in cardiac sympathetic–parasympathetic tone. Reduced HRV is indicative of decreased vagal tone, and reductions in HRV have a strong inverse correlation with CRP levels, progression of atherosclerosis, and risk of sudden death.22,23 HRV is also reduced relative to normal subjects in patients with RA, systemic lupus erythematosus, and Sjögren syndrome, and the extent of reduction in HRV within the patient groups correlates with disease severity.24–26 Although these associations are only correlative and do not provide firm evidence of causality, they do provide additional epidemiological support for the hypothesis that driving increased vagal activity using implantable devices may have a favorable effect on inflammatory disease.
Implantable neurostimulation devices have not yet been tested in human patients with RA. However, preliminary evidence from a small study carried out in normal volunteers demonstrated that the CAP reflex can be elicited by brief mechanical stimulation of the afferent auricular branch of the vagus nerve, as shown by reduction of in vitro LPS-inducible cytokine production (T van der Poll, personal communication). Clinical testing of NCAP using implantable VNS devices will begin in the near future. The devices to be used for these initial studies will be very similar in design to those currently in use for epilepsy treatment. However, prototype versions of the device which will be used in follow-on studies are miniaturized to the point where they will be directly implantable on the vagus nerve, without the need for a pulse generator unit on the chest and will use a small self-contained battery system which can be recharged using transcutaneous radiofrequency induction. Given the long lifespan, relatively low cost, and potential for increased safety over currently available treatments, NCAP delivered using an implantable device holds great promise as a novel potential therapeutic approach for patients with RA and other inflammatory diseases.
Implantable devices are increasingly used in the treatment of diseases which have historically been targeted only with small molecule and biological therapeutic agents. In addition to well-established products such as subcutaneous insulin pumps and intra-arterial chemotherapy pumps, where the implantable device merely serves as a more efficient means of delivering the drug, there are a number of recently developed therapeutic approaches in which the implanted device itself functions to directly treat the underlying medical condition. One particularly successful example of this strategy is cardiac resynchronization using biventricular pacing devices for congestive heart failure (CHF). These devices were approved for marketing after having been proved to prolong survival in patients whose disease had progressed despite medical management.1 Implantable device products are now approved or in late-stage development for many other traditional “medical” disorders such as hypertension, obesity, diabetes, Parkinson’s disease, and glaucoma. Recent advances in understanding the interplay between the central nervous system and the immune system have made possible a feasible implantable device approach that may similarly find use in the management of rheumatoid arthritis (RA) and other chronic inflammatory diseases.2
NEUROSTIMULATION OF THE CHOLINERGIC ANTIINFLAMMATORY PATHWAY
The vagus nerve mediates an important neural reflex which senses inflammation both peripherally and in the central nervous system, and downregulates the inflammation via efferent neural outflow to the reticuloendothelial system. The efferent arm of this reflex has been termed the “cholinergic antiinflammatory pathway” (CAP). The CAP serves as a physiological regulator of inflammation by responding to environmental injury, pathogens, and other external threats with an appropriate degree of immune system activation.3 An increasing body of evidence indicates that the CAP can also be harnessed to reduce pathological inflammation. Electrical neurostimulation of the vagus nerve (NCAP) in an appropriate manner with an implantable device is emerging as a novel and potentially feasible means of treating diseases characterized by excessive and dysregulated inflammation.
Our current understanding of the CAP began with the observations of Kevin Tracey and colleagues over a decade ago. They demonstrated that systemic, hepatic, and splenic tumor necrosis factor (TNF) production as well as the physiological manifestations of endotoxemic shock in rodents were worsened by vagotomy and ameliorated by electrical stimulation of the cervical vagus nerve (VNS). Further, based on in vitro experiments they postulated that this effect was mediated directly by acetylcholine acting through specific receptors on macrophages in the reticuloendothelial system.4 It was later demonstrated that reducing the response to endotoxemia using NCAP required an intact spleen, and selective anatomical lesion experiments showed that an intact neural pathway to the spleen from the cervical vagus through the celiac ganglion and the splenic nerve was also necessary for this effect.5 Within the spleen itself, nerve fiber synaptic vesicles are found in close apposition to TNF-secreting macrophages.6 The α-7 nicotinic acetylcholine receptor, expressed on the surface of macrophages, is essential for the NCAP effect as demonstrated by antisense oligonucleotide and targeted disruption experiments.7 In the macrophage, the α-7 nicotinic acetylcholine receptor does not appear to transduce signals through ion channels, as is the case in neuronal tissue. Rather, the NCAP effect is mediated at the subcellular level by alterations in the NF-κB and JAK/STAT/SOCS pathways.8,9
When taken together, these studies show that NCAP has a dual set of immunological effects: it reduces production of systemically active cytokines by resident spleen cells and also causes circulating cells which traverse the spleen to develop an altered phenotype with reduced expression of inflammatory mediators and adhesion molecules upon trafficking to inflamed tissue.
An important characteristic of NCAP delivered by VNS is that very brief episodes of stimulation result in a remarkably prolonged biological effect. Huston et al delivered a single 30-second electrical VNS or sham treatment in rats, and then induced endotoxemia with intraperitoneal lipopolysaccharide (LPS) at varying times after VNS. Interestingly, this brief VNS stimulation reduced production of serum TNF in response to systemic LPS exposure for up to 48 hours. Similarly, after only 60 minutes of exposure to acetylcholine, cultured human macrophages are changed in phenotype such that they become refractory to in vitro LPS stimulation for up to 48 hours thereafter.15 The consistency of this phenomenon across species is corroborated by preliminary data in a canine model where 60-second VNS treatment results in reduced LPS-inducible TNF production in a whole-blood in vitro release assay for several days after the VNS (M Faltys, personal communication). A duration of biological effect lasting hours to days after periods of stimulation lasting for only seconds to minutes implies that an implantable device will probably only need to operate with very short daily duty cycles to effectively elicit an NCAP response. This will in turn greatly reduce the necessary size and complexity of the device itself, and increase its functional lifespan, with resultant reductions in overall cost of the treatment.
NEUROSTIMULATION OF THE CAP IN ANIMAL MODELS OF DISEASE
In a canine model of CHF induced by rapid ventricular pacing, inflammation and ventricular remodeling with fibrosis are typically accompanied by marked increases in serum C-reactive protein (CRP) levels. In addition to improving the physiological manifestations of CHF, VNS resulted in 60% to 80% reductions in CRP for up to 8 weeks.17 In another canine CHF model induced by repetitive microembolization, which is similarly associated with systemic and myocardial inflammation, VNS markedly reduced circulating levels of interleukin 6 and TNF for up to 12 weeks.18 Importantly, both these studies show that rapid tachyphylaxis does not appear to occur with NCAP over periods of time that are relatively chronic by the typical standards of animal models.
VAGAL NERVE STIMULATION FOR EPILEPSY AND DEPRESSION: EXPERIENCE IN HUMANS
VNS delivered using a surgically implanted cuffed cervical vagus nerve lead and pacemaker-style pulse generator device has been approved for the treatment of refractory epilepsy in the United States since the mid-1990s and has more recently been approved for treatment of depression. Over 50,000 patients have been implanted with these devices world wide since that time. The safety profile of both surgical implantation and VNS delivery in this setting is well established.19 The major tolerability problem is laryngeal and pharyngeal symptoms, such as hoarseness and dysphonia, which are present almost solely during periods of active device stimulation. The frequency and severity of these treatments decreases after receiving treatment for an extended time.20 With growing experience in VNS delivery over the first 5 years of use, it also became apparent that reducing the active stimulation duty cycle from 40% to 10%, and keeping stimulation currents at ≤ 1.5 mA results in a marked reduction in these symptoms.21 Of note, the stimulation currents necessary to evoke NCAP in animals are well below the 1.5 mA level, and as above, NCAP is effective even with very brief, once-daily periods of stimulation (ie, a duty cycle of 0.07% if given for 1 min each day). Thus it is likely that the laryngeal and pharyngeal adverse event profile of VNS will not be problematic in the setting of NCAP delivery for inflammation.
A POTENTIAL ROLE FOR THERAPEUTIC NCAP USING IMPLANTABLE DEVICES IN HUMAN INFLAMMATORY DISEASES
Autonomic nervous system activity can be measured indirectly by recording cardiac R-R interval variability and subjecting the data to power spectral analysis. Such heart rate variability (HRV) measurements are influenced by the levels of vagus nerve activity and by balance in cardiac sympathetic–parasympathetic tone. Reduced HRV is indicative of decreased vagal tone, and reductions in HRV have a strong inverse correlation with CRP levels, progression of atherosclerosis, and risk of sudden death.22,23 HRV is also reduced relative to normal subjects in patients with RA, systemic lupus erythematosus, and Sjögren syndrome, and the extent of reduction in HRV within the patient groups correlates with disease severity.24–26 Although these associations are only correlative and do not provide firm evidence of causality, they do provide additional epidemiological support for the hypothesis that driving increased vagal activity using implantable devices may have a favorable effect on inflammatory disease.
Implantable neurostimulation devices have not yet been tested in human patients with RA. However, preliminary evidence from a small study carried out in normal volunteers demonstrated that the CAP reflex can be elicited by brief mechanical stimulation of the afferent auricular branch of the vagus nerve, as shown by reduction of in vitro LPS-inducible cytokine production (T van der Poll, personal communication). Clinical testing of NCAP using implantable VNS devices will begin in the near future. The devices to be used for these initial studies will be very similar in design to those currently in use for epilepsy treatment. However, prototype versions of the device which will be used in follow-on studies are miniaturized to the point where they will be directly implantable on the vagus nerve, without the need for a pulse generator unit on the chest and will use a small self-contained battery system which can be recharged using transcutaneous radiofrequency induction. Given the long lifespan, relatively low cost, and potential for increased safety over currently available treatments, NCAP delivered using an implantable device holds great promise as a novel potential therapeutic approach for patients with RA and other inflammatory diseases.
- Bristow MR, Saxon LA, Boehmer J, et al Comparison of Medical Therapy, Pacing, and Defibrillation in Heart Failure (COMPANION) Investigators. Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. N Engl J Med 2004; 350:2140–2150.
- van Maanen MA, Vervoordeldonk MJ, Tak PP. The cholinergic anti-inflammatory pathway: towards innovative treatment of rheumatoid arthritis. Nat Rev Rheumatol 2009; 5:229–232.
- Tracey KJ. Reflex control of immunity. Nat Rev Immunol 2009; 9:418–428.
- Borovikova LV, Ivanova S, Zhang M, et al Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000; 405:458–462.
- Huston JM, Ochani M, Rosas-Ballina M, et al Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med 2006; 203:1623–1628.
- Rosas-Ballina M, Ochani M, Parrish WR, et al Splenic nerve is required for cholinergic antiinflammtory pathway control of TNF in endotoxemia. Proc Natl Acad Sci 2008; 105:11008–11013.
- Wang H, Yu M, Ochani M, et al Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003; 421:384–388.
- Wang H, Liao H, Ochani M, et al Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med 2004; 10:1216–1221.
- de Jonge WJ, van der Zanden EP, The FO, et al Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol 2005; 6:844–851.
- Huston JM, Rosas-Ballina M, Xue X, et al Cholinergic neural signals to the spleen down-regulate leukocyte trafficking via CD11b. J Immunol 2009; 183:552–559.
- O’Mahony C, van der Kleij H, Bienenstock J, et al Loss of vagal anti-inflammatory effect: in vivo visualization and adoptive transfer. Am J Physiol Regul Integr Comp Physiol 2009; 297:R1118–R1126.
- Ghia JE, Blennerhassett P, Kumar-Ondiveeran H, et al The vagus nerve: a tonic inhibitory influence associated with inflammatory bowel disease in a murine model. Gastroenterology 2006; 131:1122–1130.
- Ghia JE, Blennerhassett P, Collins SM. Vagus nerve integrity and experimental colitis. Am J Physiol Gastrointest Liver Physiol 2007; 293:G560–G567.
- Karimi K, Bienenstock J, Wang L, et al The vagus nerve modulates CD4+ T cell activity. Brain Behav Immun 2010; 24:316–323.
- Huston JM, Gallowitsch-Puerta M, Ochani M, et al Transcutaneous vagus nerve stimulation reduces serum high mobility group box 1 levels and improves survival in murine sepsis. Crit Care Med 2007; 35:2762–2768.
- Levine Y, Faltys M, Black K, et al Neurostimulation of the cholinergic anti-inflammatory pathway (NCAP) ameliorates CIA in rats. Ann Rheum Dis 2010; 69( suppl 3):191.
- Zhang Y, Popovic ZB, Bibevski S, et al Chronic vagus nerve stimulation improves autonomic control and attenuates systemic inflammation and heart failure progression in a canine high-rate pacing model. Circ Heart Fail 2009; 2:692–699.
- Gupta RC, Imai M, Jiang AJ, et al Chronic therapy with selective vagus nerve stimulation normalizes plasma concentration of tumor necrosis factor alpha, interleukin-6, and B-type natriuretic peptide in dogs with heart failure. J Am Coll Cardiol 2006; 47:77A.
- Beekwilder JP, Beems T. Overview of the clinical applications of vagus nerve stimulation. J Clin Neurophysiol 2010; 27:130–138.
- Ben-Menachem E. Vagus nerve stimulation, side effects, and longterm safety. J Clin Neurophysiol 2001; 18:415–418.
- Heck C, Helmers SL, DeGiorgio CM. Vagus nerve stimulation therapy, epilepsy, and device parameters: scientific basis and recommendations for use. Neurology 2002; 59( 6 suppl 4):S31–S37.
- Huikuri HV, Jokinen V, Syvänne M, et al Heart rate variability and progression of coronary atherosclerosis. Arterioscler Thromb Vasc Biol 1999; 19:1979–1985.
- Sajadieh A, Nielsen OW, Rasmussen V, et al Increased heart rate and reduced heart-rate variability are associated with subclinical inflammation in middle-aged and elderly subjects with no apparent heart disease. Eur Heart J 2004; 25:363–370.
- Louthrenoo W, Ruttanaumpawan P, Aramrattana A, et al Cardio vascular autonomic nervous system dysfunction in patients with rheumatoid arthritis and systemic lupus erythematosus. QJM 1999; 92:97–102.
- Evrengül H, Dursunoglu D, Cobankara V, et al Heart rate variability in patients with rheumatoid arthritis. Rheumatol Int 2004; 24:198–202.
- Stojanovich L, Milovanovich B, de Luka SR, et al Cardiovascular autonomic dysfunction in systemic lupus, rheumatoid arthritis, primary Sjögren syndrome and other autoimmune diseases. Lupus 2007; 16:181–185.
- Bristow MR, Saxon LA, Boehmer J, et al Comparison of Medical Therapy, Pacing, and Defibrillation in Heart Failure (COMPANION) Investigators. Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. N Engl J Med 2004; 350:2140–2150.
- van Maanen MA, Vervoordeldonk MJ, Tak PP. The cholinergic anti-inflammatory pathway: towards innovative treatment of rheumatoid arthritis. Nat Rev Rheumatol 2009; 5:229–232.
- Tracey KJ. Reflex control of immunity. Nat Rev Immunol 2009; 9:418–428.
- Borovikova LV, Ivanova S, Zhang M, et al Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000; 405:458–462.
- Huston JM, Ochani M, Rosas-Ballina M, et al Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med 2006; 203:1623–1628.
- Rosas-Ballina M, Ochani M, Parrish WR, et al Splenic nerve is required for cholinergic antiinflammtory pathway control of TNF in endotoxemia. Proc Natl Acad Sci 2008; 105:11008–11013.
- Wang H, Yu M, Ochani M, et al Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003; 421:384–388.
- Wang H, Liao H, Ochani M, et al Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med 2004; 10:1216–1221.
- de Jonge WJ, van der Zanden EP, The FO, et al Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol 2005; 6:844–851.
- Huston JM, Rosas-Ballina M, Xue X, et al Cholinergic neural signals to the spleen down-regulate leukocyte trafficking via CD11b. J Immunol 2009; 183:552–559.
- O’Mahony C, van der Kleij H, Bienenstock J, et al Loss of vagal anti-inflammatory effect: in vivo visualization and adoptive transfer. Am J Physiol Regul Integr Comp Physiol 2009; 297:R1118–R1126.
- Ghia JE, Blennerhassett P, Kumar-Ondiveeran H, et al The vagus nerve: a tonic inhibitory influence associated with inflammatory bowel disease in a murine model. Gastroenterology 2006; 131:1122–1130.
- Ghia JE, Blennerhassett P, Collins SM. Vagus nerve integrity and experimental colitis. Am J Physiol Gastrointest Liver Physiol 2007; 293:G560–G567.
- Karimi K, Bienenstock J, Wang L, et al The vagus nerve modulates CD4+ T cell activity. Brain Behav Immun 2010; 24:316–323.
- Huston JM, Gallowitsch-Puerta M, Ochani M, et al Transcutaneous vagus nerve stimulation reduces serum high mobility group box 1 levels and improves survival in murine sepsis. Crit Care Med 2007; 35:2762–2768.
- Levine Y, Faltys M, Black K, et al Neurostimulation of the cholinergic anti-inflammatory pathway (NCAP) ameliorates CIA in rats. Ann Rheum Dis 2010; 69( suppl 3):191.
- Zhang Y, Popovic ZB, Bibevski S, et al Chronic vagus nerve stimulation improves autonomic control and attenuates systemic inflammation and heart failure progression in a canine high-rate pacing model. Circ Heart Fail 2009; 2:692–699.
- Gupta RC, Imai M, Jiang AJ, et al Chronic therapy with selective vagus nerve stimulation normalizes plasma concentration of tumor necrosis factor alpha, interleukin-6, and B-type natriuretic peptide in dogs with heart failure. J Am Coll Cardiol 2006; 47:77A.
- Beekwilder JP, Beems T. Overview of the clinical applications of vagus nerve stimulation. J Clin Neurophysiol 2010; 27:130–138.
- Ben-Menachem E. Vagus nerve stimulation, side effects, and longterm safety. J Clin Neurophysiol 2001; 18:415–418.
- Heck C, Helmers SL, DeGiorgio CM. Vagus nerve stimulation therapy, epilepsy, and device parameters: scientific basis and recommendations for use. Neurology 2002; 59( 6 suppl 4):S31–S37.
- Huikuri HV, Jokinen V, Syvänne M, et al Heart rate variability and progression of coronary atherosclerosis. Arterioscler Thromb Vasc Biol 1999; 19:1979–1985.
- Sajadieh A, Nielsen OW, Rasmussen V, et al Increased heart rate and reduced heart-rate variability are associated with subclinical inflammation in middle-aged and elderly subjects with no apparent heart disease. Eur Heart J 2004; 25:363–370.
- Louthrenoo W, Ruttanaumpawan P, Aramrattana A, et al Cardio vascular autonomic nervous system dysfunction in patients with rheumatoid arthritis and systemic lupus erythematosus. QJM 1999; 92:97–102.
- Evrengül H, Dursunoglu D, Cobankara V, et al Heart rate variability in patients with rheumatoid arthritis. Rheumatol Int 2004; 24:198–202.
- Stojanovich L, Milovanovich B, de Luka SR, et al Cardiovascular autonomic dysfunction in systemic lupus, rheumatoid arthritis, primary Sjögren syndrome and other autoimmune diseases. Lupus 2007; 16:181–185.
New frontiers in cardiovascular behavioral medicine: Comparative effectiveness of exercise and medication in treating depression
I am fortunate to be the recipient of the 2010 Bakken Institute Pioneer Award and feel especially honored to have my work recognized in this way. When informed that I was this year’s recipient, it prompted me to reflect on the meaning of the term “pioneer,” and how it related to me.
WHAT IS A PIONEER?
According to Merriam-Webster’s Collegiate Dictionary, a pioneer is one who (a) ventures into unknown or unclaimed territory to settle; and (b) opens up new areas of thought, research, or development. One requirement for any pioneer is that there be a frontier to explore. Thirty years ago, my colleagues and I began our investigations into cardiac rehabilitation (CR), which at the time we considered to be a new frontier for behavioral medicine.1
EXERCISE-BASED CARDIAC REHABILITATION
Historically, patients who suffered an acute myocardial infarction (AMI) were often discouraged from engaging in physical activity; patients were initially prescribed prolonged bed rest and told to avoid strenuous exercise.2 In the early 1950s, armchair therapy was proposed3 as an initial attempt to mobilize patients after a coronary event. Over the years, the value of physical exercise has been increasingly recognized and exercise is now considered to be the cornerstone of CR.4–7 Today, exercise-based CR, involving aerobic exercise supplemented by resistance training, is offered by virtually all CR programs in the United States.8 Proper medical management is also emphasized, along with dietary modification and smoking cessation, but exercise is the centerpiece of treatment.
Exercise has been shown to reduce traditional risk factors such as hypertension and hyperlipidemia,8 attenuate cardiovascular responses to mental stress,9 and reduce myocardial ischemia.10–12 Although no single study has demonstrated definitively that exercise reduces morbidity in patients with coronary heart disease (CHD), pooling data across clinical trials has shown that exercise may reduce risk of fatal CHD events by 25%.13 A recent, comprehensive meta-analysis by Jolliffe et al14 reported a 27% reduction in all-cause mortality and 31% reduction in cardiac mortality.
Not only is exercise considered beneficial for medical outcomes, but is also recognized as an important factor in improved quality of life. Indeed, there has been increased interest in the value of exercise for improving not just physical health, but also mental health.15–17 The mental health benefits of exercise are especially relevant for cardiac patients, as there is a growing literature documenting the importance of mental health, and, in particular the prognostic significance of depression, in patients with CHD.
PSYCHOSOCIAL RISK FACTORS: THE ROLE OF DEPRESSION IN CORONARY HEART DISEASE
There has long been an interest in psychosocial factors that contribute to the development and progression of CHD. More than three decades ago, researchers identified the type A behavior pattern as a risk factor for CHD.18 When subsequent studies failed to confirm the association of type A with adverse health outcomes, researchers turned their attention to other possible psychosocial risk factors, including anger and hostility,19 low social support,20 and most recently, depression.21 Indeed, the most consistent and compelling evidence is that clinical depression or elevated depressive symptoms in the presence of CHD increase the risk of fatal and nonfatal cardiac events and of all-cause mortality.22
Major depressive disorder (MDD) is a common and often chronic condition. Lifetime incidence estimates for MDD are approximately 12% in men and 20% in women.23 In addition, MDD is marked by high rates of relapse, with 22% to 50% of patients suffering recurrent episodes within 6 months after recovery.24 Furthermore, MDD is underrecognized and undertreated in older adults,25 CHD patients, and, especially, minorities.26–28
Cross-sectional studies have documented a higher prevalence of depression in CHD patients than in the general population. Point estimates range from 14% to as high as 47%, with higher rates recorded most often in patients with unstable angina, heart failure (HF), and patients awaiting coronary artery bypass graft (CABG) surgery.29–36
Depression associated with poor outcomes
A number of prospective studies have found that depression is associated with increased risk for mortality or nonfatal cardiac events in a variety of CHD populations. The most compelling evidence for depression as a risk factor has come from studies in Montreal, Canada. Frasure-Smith and colleagues31 assessed the impact of depression in 222 AMI patients, of whom 35 were diagnosed with MDD at the time of hospitalization. There were 12 deaths (six depressed and six nondepressed) over an initial 6-month followup period, representing more than a fivefold increased risk of death for depressed patients compared with nondepressed patients (hazard ratio, 5.7; 95% confidence interval [CI], 4.6 to 6.9). In a subsequent report,36 in which 896 AMI patients were followed for 1 year, the presence of elevated depressive symptoms was associated with more than a threefold increased risk in cardiac mortality after controlling for other multivariate predictors of mortality (odds ratio, 3.29 for women; 3.05 for men).
Studies of patients with stable CHD also have reported significant associations between the presence of depression and worse clinical outcomes. For example, Barefoot et al37 assessed 1,250 patients with documented CHD using the Zung self-report depression scale at the time of diagnostic coronary angiography and followed patients for up to 19.4 years. Results showed that patients with moderate to severe depression were at 69% greater risk for cardiac death and 78% greater risk for all-cause death.
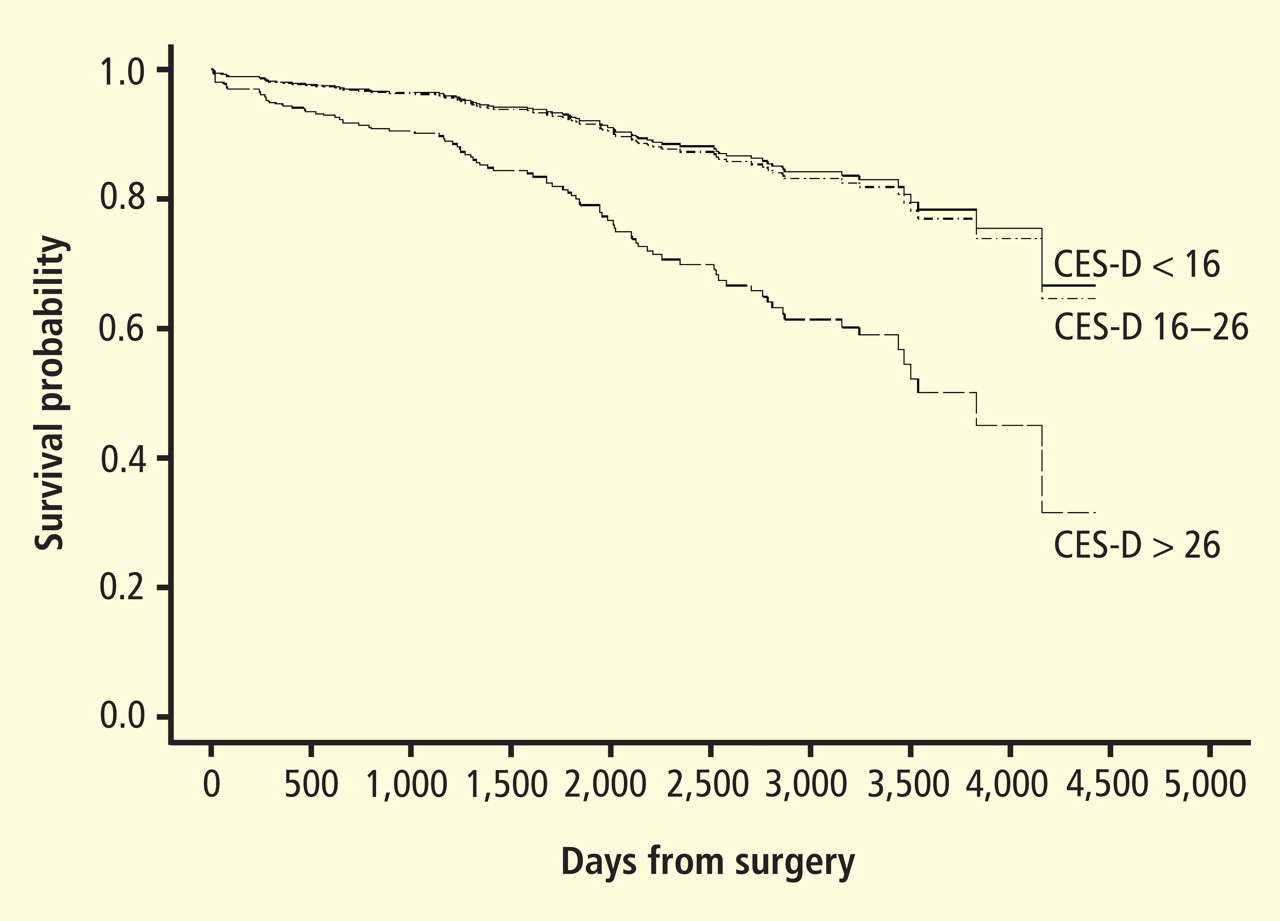
Depression and heart failure outcomes
Patients with HF represent a particularly vulnerable group; a meta-analysis of depression in HF patients suggested that one in five patients are clinically depressed (range, 9% to 60%).41 Not only is depression in HF patients associated with worse outcomes,42–46 but recent evidence suggests that worsening of depressive symptoms, independent of clinical status, is related to worse outcomes. Sherwood et al46 demonstrated that increased symptoms of depression, as indicated by higher scores on the Beck Depression Inventory (BDI) over a 1-year interval (BDI change [1-point] hazard ratio, 1.07; 95% CI, 1.02 to 1.12; P = .007), were associated with higher risk of death or cardiovascular hospitalization after controlling for baseline depression (baseline BDI hazard ratio, 1.1; 95% CI, 1.06 to 1.14, P < .001) and established risk factors, including HF etiology, age, ejection fraction, N-terminal pro-B-type natriuretic peptides, and prior hospitalizations. Consequently, strategies to reduce depressive symptoms and prevent the worsening of depression may have important implications for improving cardiac health as well as for enhancing quality of life.
MECHANISMS LINKING DEPRESSION AND CHD
CONVENTIONAL APPROACHES TO TREATMENT OF DEPRESSION
Treatment of depression has focused on reduction of symptoms and restoration of function. Antidepressant medications are generally considered the treatment of choice.56 In particular, second-generation antidepressants such as selective serotonin reuptake inhibitors (SSRIs) are widely prescribed.57 Current treatment guidelines suggest 6 to 12 weeks of acute treatment followed by a continuation phase of 3 to 9 months to maintain therapeutic benefit.58 However, meta-analyses of antidepressant medications have reported only modest benefits over placebo treatments.59,60 In particular, active drug–placebo differences in antidepressant efficacy are positively correlated with depression severity: antidepressants are often comparable with placebo in patients with low levels of depression but may be superior to placebo among patients with more severe depression. However, the explanation for this relationship may be that placebo is less effective for more depressed patients rather than antidepressants being more effective for more depressed patients.59
For acute treatment of MDD, approximately 60% of patients respond to second-generation antidepressants,61 with a 40% relapse rate after 1 year.62 A recent meta-analysis60 of second-generation antidepressants summarized four comparative trials and 23 placebo-controlled trials and found that second-generation antidepressants were generally comparable with each other. Interestingly, despite the modest benefit of antidepressants, the percentage of patients treated for depression in the United States increased from 0.73% in 1987 to 2.33% in 1997. The proportion of those treated who received antidepressants increased from 37.3% in 1987 to 74.5% in 1997.63 The percentage of treated outpatients who used antidepressants has not increased significantly since 1997, but the use of psycho therapy as a sole treatment declined from 53.6% in 1998 to 43.1% in 2007.64 Moreover, the national expenditure for the outpatient treatment of depression increased from $10.05 billion in 1998 to $12.45 billion in 2007, primarily driven by an increase in expenditures for antidepressant medications.
Uncertainty about value of antidepressant therapy
Despite compelling reasons for treating depression in cardiac patients, the clinical significance of treating depression remains uncertain. To date, only the Enhancing Recovery in CHD Patients (ENRICHD) trial has examined the impact of treating depression in post-MI patients on “hard” clinical end points.65 Although more than 2,400 patients were enrolled in the trial, the results were disappointing. There were only modest differences (ie, two points on the Hamilton Depression Rating Scale [HAM-D]) in reductions of depressive symptoms in the group receiving cognitive behavior therapy (CBT) relative to usual-care controls and there were no treatment group differences in the primary outcome—all-cause mortality and nonfatal cardiac events. By the end of the follow-up period, 28.0% of patients in the CBT group and 20.6% of patients in usual care had received antidepressant medication. Although a subsequent reanalysis of the ENRICHD study revealed that antidepressant use was associated with improved clinical outcomes,66 because patients were not randomized to pharmacologic treatment it could not be concluded that SSRI use was responsible for the improved outcomes.
In a randomized trial of patients with acute coronary syndrome (the Sertraline Antidepressant Heart Attack Randomized Trial, or SADHART),67 almost 400 patients were treated with the SSRI sertraline or with placebo. Reductions in depressive symptoms were similar for patients receiving sertraline compared with placebo in the full sample, although a subgroup analysis revealed that patients with more severe depression (ie, those patients who reported two or more previous episodes) benefited more from sertraline compared with placebo. Interestingly, patients receiving sertraline tended to have more favorable cardiac outcomes, including a composite measure of both “hard” and “soft” clinical events, compared with placebo controls. These results suggested that antidepressant medication may improve underlying physiologic processes, such as platelet function, independent of changes in depression.68 However, because SADHART was not powered to detect differences in clinical events, there remain unanswered questions about the clinical value of treating depression in cardiac patients with antidepressant medication.
In a second sertraline trial, SADHART-HF,69 469 men and women with MDD and chronic systolic HF were randomized to receive either sertraline or placebo for 12 weeks. Participants were followed for a minimum of 6 months. Results showed that while sertraline was safe, its use did not result in greater reductions in depressive symptoms compared with placebo (−7.1 ± 0.5 vs −6.8 ± 0.5) and there were no differences in clinical event rates between patients receiving sertraline compared with those receiving placebo.
In an observational study of patients with HF,44 use of antidepressant medication was associated with increased risk of mortality or hospitalization. Although the potential harmful effects of antidepressant medication could not be ruled out, a more likely interpretation is that antidepressant medication use was a marker for individuals with more severe depression, and that the underlying depression may have contributed to their higher risk. Further, patients who are depressed, despite receiving treatment, may represent a subset of treatment-resistant patients who may be especially vulnerable to further cardiac events. Indeed, worsening depression is associated with worse outcomes in HF patients46; this is consistent with data from the ENRICHD trial, which showed that patients receiving CBT (and, in some cases, antidepressant medication) who failed to improve with treatment had higher mortality rates compared with patients who exhibited a positive response to treatment.70
A fourth randomized trial of CHD patients, the Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial,71 used a modified “2 by 2” design; 284 CHD patients with MDD and HAM-D rating scores of 20 or greater were randomized to receive 12 weeks of (a) interpersonal therapy (IPT) plus clinical management (CM) or (b) CM only and citalopram or matching placebo. Because the same interventionists delivered the CM and IPT, patients assigned to IPT received IPT plus CM within the same (extended) session. Patients receiving citalopram had greater reductions in depressive symptoms compared with placebo, with a small to medium effect size of 0.33, and better remission rates (35.9%) compared with placebo (22.5%). Unexpectedly, patients who received just CM tended to have greater improvements in depressive symptoms compared with patients who received IPT plus CM (P < .07); no clinical CHD end points were assessed, however.
Alternative approaches needed
Taken together, these data illustrate that antidepressant medications may reduce depressive symptoms for some patients; for other patients, however, medication fails to adequately relieve depressive symptoms and may perform no better than placebo. Adverse effects also may affect a subgroup of patients and may be relatively more common or more problematic in older persons with CHD.72 Thus, a need remains to identify alternative approaches for treating depression in cardiac patients. We believe that aerobic exercise, the cornerstone of traditional CR, may be one such approach. Exercise is safe for most cardiac patients,73,74 including patients with HF,75 and, if proven effective as a treatment for depression, exercise would hold several potential advantages over traditional medical therapies: it is relatively inexpensive, improves cardiovascular functioning, and avoids the side effects sometimes associated with medication use.
EXERCISE THERAPY FOR DEPRESSION
Some studies of exercise treatment for CHD patients have tracked depressive symptoms and thus have provided information regarding the potential efficacy of exercise as a treatment for depression in this population.76 –81 Although most previous studies have reported significant improvements in depression after completion of an exercise program, many studies had important methodologic limitations, including the absence of a control group.
In one of the few controlled studies in this field, Stern et al82 randomized 106 male patients who had a recent history of AMI along with elevated depression and anxiety or low fitness to 12 weeks of exercise training, group therapy, or a usual-care control group. At 1-year followup, both the exercise and counseling groups showed improvements in depression relative to controls.
Cross-sectional studies of non-CHD samples have reported that active individuals obtain significantly lower depression scores on self-report measures than sedentary persons.83 Studies also have shown that aerobic exercise may reduce self-reported depressive symptoms in nonclinical populations and in patients diagnosed with MDD.83 In 2001, a meta-analysis evaluating 11 randomized controlled trials of non-CHD patients with MDD84 noted that studies were limited because of self-selection bias, absence of control groups or nonrandom controls, and inadequate assessment of exercise training effects; the authors concluded that “the effectiveness of exercise in reducing symptoms of depression cannot be determined because of a lack of good quality research on clinical populations with adequate followup.”
Randomized controlled trials needed
A subsequent meta-analysis85 included 25 studies; for 23 trials (907 participants) that compared exercise with no treatment or a control intervention, the pooled standardized mean difference (SMD) was −0.82 (95% CI, −1.12, −0.51), indicating a large effect size. However, when only the three trials (216 participants) with adequate allocation concealment, intention to treat analysis, and blinded outcome assessment were included, the pooled SMD was −0.43 (95% CI, −0.88, 0.03), with a point estimate that was half the size of that with all trials. As a result, the authors concluded that “exercise seems to improve depressive symptoms in people with a diagnosis of depression, but when only the methodologically robust trials are included, the effect size is only moderate.”
To date, no randomized clinical trials (RCTs) have examined the effects of exercise on clinical outcomes in depressed cardiac patients. However, data from the ENRICHD trial suggest that exercise may reduce the rates of mortality and nonfatal reinfarction in patients with depression or in post-MI patients who are socially isolated.86 Self-report data were used to categorize participants as exercising regularly or not exercising regularly. After controlling for medical and demographic variables, the magnitude of reduction in risk associated with regular exercise was nearly 40% for nonfatal reinfarction and 50% for mortality. The evidence that exercise mitigates depression, reduces CHD risk factors, and improves CHD outcomes suggests that exercise may be a particularly promising intervention for depressed CHD patients.
COMPARATIVE EFFECTIVENESS OF EXERCISE AND ANTIDEPRESSANT MEDICATION
In 2008, an Institute of Medicine (IOM) report called for a national initiative of research that would provide a basis for better decision-making about how to best treat various medical conditions, including depression. In 2009, the American Reinvestment Recovery Act provided a major boost in funding for comparative effectiveness research (CER). The act allotted $1.1 billion to support this form of research. CER refers to the generation and synthesis of evidence that compares the benefits and harms of alternative methods to prevent, diagnose, treat, and monitor a clinical condition, or to improve the delivery of care. The purpose of CER is to assist consumers, clinicians, purchasers, and policy makers in reaching informed decisions that will improve health care at both the individual and population levels.87
Two research categories inform decision-making
Two broad categories of research have been used to inform decision-making:
- Epidemiologic studies provide evidence linking various treatments with patient outcomes. These sources of data are limited because they seldom specify the basis for medical decisions and they fail to consider patient characteristics that affect both clinical decisions and clinical outcomes. Indeed, it has been suggested that “overcoming the limitations of observational research is the most important frontier of research on study methods.”88
- RCTs address these limitations by randomly assigning patients to different treatment conditions. While this design may eliminate some of the uncertainty and potential confounders that characterize purely observational studies, most RCTs are efficacy studies; patients are carefully selected and a treatment is usually compared with a placebo or usual care.
The RCT design addresses the question of whether a given treatment is effective, but it does not necessarily address questions that many physicians want answers to: namely, is this treatment better than that treatment? Further, physicians want to know if one treatment is more effective than another for a given patient. For example, Hlatky et al89 showed that mortality associated with percutaneous coronary interventions (PCIs) and CABG surgery was comparable; however, mortality with CABG surgery was significantly lower for patients older than 65 years while PCI was superior for patients younger than 55 years. Thus, examination of individual differences may also help to inform clinicians about the optimal therapy for their particular patients.
Treatment of depression not necessarily a research priority
The IOM committee sought advice from a broad range of stakeholders and prioritized areas for research. The top-ranked topic was comparison of treatment strategies for atrial fibrillation, including surgery, catheter ablation, and pharmacologic treatment. Coming in at #98 was comparison of the effectiveness of different treatment strategies (eg, psychotherapy, antidepressants, combination treatment with case management) for depression after MI and their impact on medication adherence, cardiovascular events, hospitalization, and death.
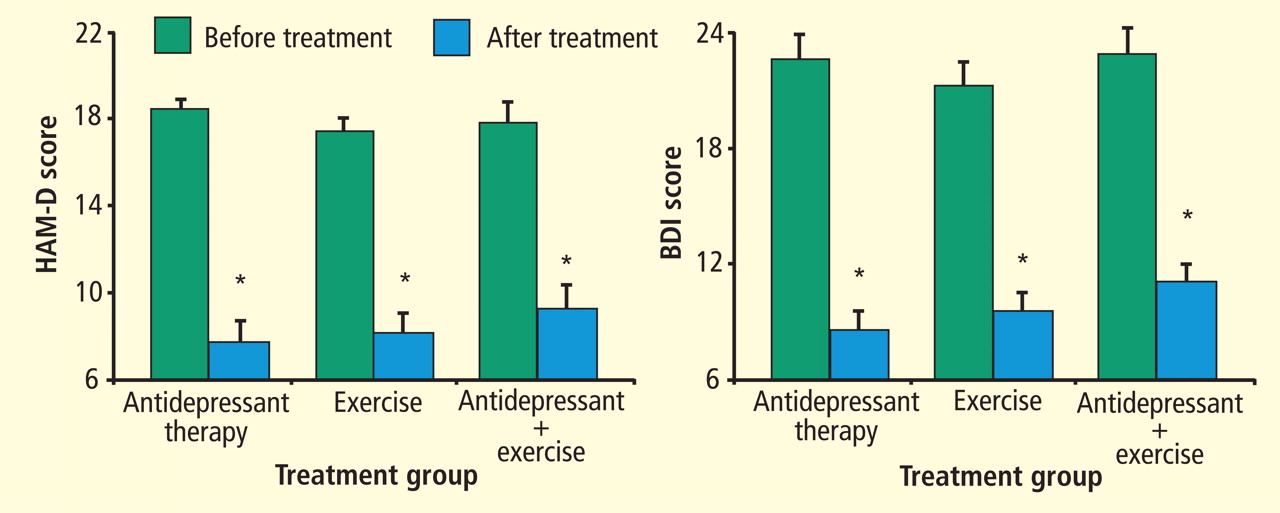
In a second Duke study that compared exercise and antidepressant medication,92 202 adults (153 women; 49 men) diagnosed with MDD were randomly assigned to one of four groups: supervised exercise in a group setting, home-based exercise, antidepressant medication (sertraline, 50 to 200 mg daily), or placebo pill for 16 weeks. Once again, patients underwent the Structured Clinical Interview for Depression and completed the HAM-D. After 4 months of treatment, 41% of participants achieved remission, defined as no longer meeting criteria for MDD and a HAM-D score of less than 8 points. Patients receiving active treatments tended to have higher remission rates than placebo controls: supervised exercise, 45%; home-based exercise, 40%; medication, 47%; placebo, 31% (P = .057). All treatment groups had lower HAM-D scores after treatment; scores for the active treatment groups were not significantly different from the placebo group (P = .23). However, when immediate responders (ie, those patients who reported more than 50% reduction in depressive symptoms after only 1 week of treatment) were excluded from the analysis, patients receiving active treatments (ie, either sertraline or exercise) had greater reductions in depressive symptoms compared with placebo controls (P = .048). There was no difference between the exercise and antidepressant groups. We concluded that the efficacy of exercise appears generally comparable with antidepressant medication and both tend to be better than placebo in patients with MDD. Placebo response rates were high, suggesting that a considerable portion of the therapeutic response could be determined by patient expectations, ongoing symptom monitoring, attention, and other nonspecific factors. Similar to our previous trial, participants who continued to exercise following the completion of the program were less likely to be depressed.93
Another RCT94 also demonstrated that exercise was associated with reduced depression, independent of group support. Participants exercised alone in a secluded setting, and the study included a no-treatment control group. Only 53 of 80 patients actually completed the 12-week trial, however, including only five of 13 no-treatment controls. Moreover, there was no active treatment comparison group, so that an estimate of comparative effectiveness could not be determined.
While these results are preliminary and should be interpreted with caution, it appears that exercise may be comparable with conventional antidepressant medication in reducing depressive symptoms, at least for patients who are willing to try it, and maintenance of exercise reduces the risk of relapse.
SUMMARY
Three decades ago, we recognized that CR was a new frontier for behavioral medicine. We now know that successful rehabilitation of patients with CHD involves modification of lifestyle behaviors, including smoking cessation, dietary modification, and exercise. Exercise is no longer considered unsafe for most cardiac patients, and exercise is currently the key component of CR services. Research also has provided strong evidence that depression is an important risk factor for CHD, although there is no consensus regarding the optimal way to treat depression in CHD patients.95 Research on comparative effectiveness of established and alternative treatments for depressed cardiac patients is a new frontier for future pioneers in heart-brain medicine.
- Blumenthal JA, Califf R, Williams RS, Hindman M. Cardiac rehabilitation: a new frontier for behavioral medicine. J Cardiac Rehabil 1983; 3:637–656.
- Lewis T. Diseases of the Heart. New York: Macmillan; 1933:41–49.
- Levine SA, Lown B. The “chair” treatment of acute thrombosis. Trans Assoc Am Physicians 1951; 64:316–327.
- Cain HD, Frasher WG, Stivelman R. Graded activity program for safe return to self-care after myocardial infarction. JAMA 1961; 177:111–115.
- Hellerstein HK, Ford AB. Rehabilitation of the cardiac patient. JAMA 1957; 14:225–231.
- Naughton J, Bruhn JG, Lategola MT. Effects of physical training on physiologic and behavioral characteristics of cardiac patients. Arch Phys Med Rehabil 1968; 49:131–137.
- O’Connor GT, Buring JE, Yusuf S, et al An overview of randomized trials of rehabilitation with exercise after myocardial infarction. Circulation 1989; 80:234–244.
- Wenger NK, Froelicher ES, Smith LK, et al Cardiac rehabilitation: clinical practice guideline no. 17. Rockville, MD: US Dept of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research, National Heart, Lung, and Blood Institute; October 1995; AHCPR publication 96-0672.
- Blumenthal JA, Emery CF, Cox DR, et al Exercise training in healthy type A middle-aged men: effects on behavioral and cardiovascular responses. Psychosom Med 1988; 50:418–433.
- Schuler G, Schlierf G, Wirth A, et al Low-fat diet and regular, supervised physical exercise in patients with symptomatic coronary artery disease: reduction of stress-induced myocardial ischemia. Circulation 1988; 77:172–181.
- Jiang W, Trauner MA, Coleman RE, et al Association of physical fitness and transient myocardial ischemia in patients with coronary artery disease. J Cardiopulm Rehabil 1995; 15:431–438.
- Blumenthal JA, Jiang W, Babyak MA, et al Stress management and exercise training in cardiac patients with myocardial ischemia: effects on prognosis and evaluation of mechanisms. Arch Int Med 1997; 157:2213–2223.
- Oldridge NB, Guyatt GH, Fisher ME, Rimm AA. Cardiac rehabilitation after myocardial infarction: combined experience of randomized clinical trials. JAMA 1988; 260:945–950.
- Jolliffe JA, Rees K, Taylor RS, Thompson D, Oldridge N, Ebrahim S. Exercise-based rehabilitation for coronary heart disease. Cochrane Database Syst Rev 2001:CD001800.
- Folkins CH, Sime WE. Physical fitness training and mental health. Am Psychol 1981; 36:373–389.
- Hughes JR. Psychological effects of habitual aerobic exercise: a critical review. Prev Med 1984; 13:66–78.
- Plante TG, Rodin J. Physical fitness and enhanced psychological health. Curr Psychol: Res Rev 1990; 9:3–24.
- Review Panel on Coronary-Prone Behavior and Coronary Heart Disease. Coronary-prone behavior and coronary heart disease: a critical review. Circulation 1981; 63:1199–1215.
- Smith TW. Hostility and health: current status of a psychosomatic hypothesis. Health Psychol 1992; 11:139–150.
- Lett HS, Blumenthal JA, Babyak MA, Strauman TJ, Robins C, Sherwood A. Social support and coronary heart disease: epidemiologic evidence and implications for treatment. Psychosom Med 2005; 67:869–878.
- Lett HS, Blumenthal JA, Babyak MA, et al Depression as a risk factor for coronary artery disease: evidence, mechanisms, and treatment. Psychosom Med 2004; 66:305–315.
- Barth J, Schumacher M, Herrmann-Lingen C. Depression as a risk factor for mortality in patients with coronary heart disease: a meta-analysis. Psychosom Med 2004; 66:802–813.
- Kessler RC, Berglund P, Demler O, et al The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCR-R). JAMA 2003; 289:3095–3105.
- Belsher G, Costello CG. Relapse after recovery from unipolar depression: a critical review. Psychol Bull 1988; 104:84–96.
- Strothers HS, Rust G, Minor P, Fresh E, Druss B, Satcher D. Disparities in antidepressant treatment in Medicaid elderly diagnosed with depression. J Am Geriatr Soc 2005; 53:456–461.
- Simpson SM, Krishnan LL, Kunik ME, Ruiz P. Racial disparities in diagnosis and treatment of depression: a literature review. Psychiatr Q 2007; 78:3–14.
- Sclar DA, Robison LM, Skaer TL. Ethnicity/race and the diagnosis of depression and use of antidepressants by adults in the United States. Int Clin Psychopharmacol 2008; 23:106–109.
- Waldman SV, Blumenthal JA, Babyak MA, et al Ethnic differences in the treatment of depression in patients with ischemic heart disease. Am Heart J 2009; 57:77–83.
- Carney RM, Rich MW, Freedland KE, et al Major depressive disorder predicts cardiac events in patients with coronary artery disease. Psychosom Med 1988; 50:627–633.
- Schleifer SJ, Macari-Hinson MM, Coyle DA, et al The nature and course of depression following myocardial infarction. Arch Intern Med 1989; 149:1785–1789.
- Frasure-Smith N, Lespérance F, Talajic M. Depression following myocardial infarction: impact on 6-month survival. JAMA 1993; 270:1819–1825.
- Gonzalez MB, Snyderman TB, Colket JT, et al Depression in patients with coronary artery disease. Depression 1996; 4:57–62.
- Connerney I, Shapiro PA, McLaughlin JS, Bagiella E, Sloan RP. Relation between depression after coronary artery bypass surgery and 12-month outcome: a prospective study. Lancet 2001; 358:1766–1771.
- Burker EJ, Blumenthal JA, Feldman M, et al Depression in male and female patients undergoing cardiac surgery. Br J Clin Psychol 1995; 34( Pt 1):119–128.
- Lespérance F, Frasure-Smith N, Juneau M, Théroux P. Depression and 1-year prognosis in unstable angina. Arch Intern Med 2000; 160:1354–1360.
- Frasure-Smith N, Lespérance F, Talajic M. Depression and 18-month prognosis after myocardial infarction. Circulation 1995; 91:999–1005.
- Barefoot JC, Helms MJ, Mark DB, et al Depression and long-term mortality risk in patients with coronary artery disease. Am J Cardiol 1996; 78:613–617.
- Burg MM, Benedetto C, Soufer R. Depressive symptoms and mortality two years after coronary artery bypass graft surgery (CABG) in men. Psychosom Med 2003; 65:508–510.
- Blumenthal JA, Lett HS, Babyak MA, et al Depression as a risk factor for mortality after coronary artery bypass surgery. Lancet 2003; 362:604–609.
- Connerney I, Sloan RP, Shapiro PA, Bagiella E, Seckman C. Depression is associated with increased mortality 10 years after coronary artery bypass surgery. Psychosom Med 2010; 72:874–881.
- Rutledge T, Reis VA, Linke SE, Greenberg BH, Mills PJ. Depression in heart failure: a meta analytic review of prevalence, intervention effects, and associations with clinical outcomes. J Am Coll Cardiol 2006; 48:1527–1537.
- Murberg TA, Furze G. Depressive symptoms and mortality in patients with congestive heart failure: a six-year follow-up study. Med Sci Monit 2004; 10:CR643–CR648.
- Jiang W, Alexander J, Christopher E, et al Relationship of depression to increased risk of mortality and rehospitalization in patients with congestive heart failure. Arch Intern Med 2001; 161:1849–1856.
- Sherwood A, Blumenthal JA, Trivedi R, et al Relationship of depression to death or hospitalization in patients with heart failure. Arch Int Med 2007; 167:367–373.
- Frasure-Smith N, Lespérance F, Habra M, et al Elevated depression symptoms predict long-term cardiovascular mortality in patients with atrial fibrillation and heart failure. Circulation 2009; 120:134–140.
- Sherwood A, Blumenthal JA, Hinderliter AL, et al Worsening depressive symptoms are associated with adverse clinical outcomes in patients with heart failure. J Am Coll Cardiol 2011; 57:418–423.
- Anderson RJ, Freedland KE, Clouse RE, Lustman PJ. The prevalence of comorbid depression in adults with diabetes: a meta-analysis. Diabetes Care 2001; 24:1069–1078.
- Thakore JH, Richards PJ, Reznek RH, Martin A, Dinan TG. Increased intra-abdominal fat deposition in patients with major depressive illness as measured by computed tomography. Biol Psychiatry 1997; 41:1140–1142.
- Musselman DL, Tomer A, Manatunga AK, et al Exaggerated platelet reactivity in major depression. Am J Psychiatry 1996; 153:1313–1317.
- Delgado PL, Moreno FA. Role of norepinephrine in depression. J Clin Psychiatry 2000; 61( suppl 1):5–12.
- Akil H, Haskett RF, Young EA, et al Multiple HPA profiles in endogenous depression: effect of age and sex on cortisol and beta-endorphin. Biol Psychiatry 1993; 33:73–85.
- Kop WJ, Gottdiener JS, Tangen CM, et al Inflammation and coagulation factors in persons > 65 years of age with symptoms of depression but without evidence of myocardial ischemia. Am J Cardiol 2002; 89:419–424.
- Carney RM, Freedland KE, Eisen SA, Rich MW, Jaffe AS. Major depression and medication adherence in elderly patients with coronary artery disease. Health Psychol 1995; 14:88–90.
- Lehto S, Koukkunen H, Hintikka J, Viinamäki H, Laakso M, Pyörälä K. Depression after coronary heart disease events. Scand Cardiovasc J 2000; 34:580–583.
- Camacho TC, Roberts RE, Lazarus NB, Kaplan GA, Cohen RD. Physical activity and depression: evidence from the Alameda County Study. Am J Epidemiol 1991; 134:220–231.
- Clinical Practice Guideline Number 5: Depression in Primary Care, 2: Treatment of Major Depression. Rockville, MD: U.S. Department of Health and Human Services, Agency for Health Care Policy and Research; 1993. AHCPR publication 93-0551.
- Anderson IM, Ferrier IN, Baldwin RC, et al Evidence-based guidelines for treating depressive disorders with antidepressants: a revision of the 2000 British Association for Psychopharmacology guidelines. J Psychopharmacol 2008; 22:343–396.
- American Psyciatric Association. Practice guideline for the treatment of patients with major depressive disorder (revision). Am J Psychiatry 2000; 157( suppl 4):1–45.
- Kirsch I, Deacon BJ, Huedo-Medina TB, Scoboria A, Moore TJ, Johnson BT. Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration. PLoS Med 2008; 5:e45.
- Hansen R, Gaynes B, Thieda P, et al Meta-analysis of major depressive disorder relapse and recurrence with second-generation antidepressants. Psychiatr Serv 2008; 59:1121–1129.
- Hansen RA, Gartlehner G, Lohr KN, Gaynes BN, Carey TS. Efficacy and safety of second-generation antidepressants in the treatment of major depressive disorder. Ann Intern Med 2005; 143:415–426.
- Rush AJ, Trivedi MH, Wisniewski SR, et al Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 2006; 163:1905–1917.
- Olfson M, Marcus SC, Druss B, Elinson L, Tanielian T, Pincus HA. National trends in the outpatient treatment of depression. JAMA 2002; 287:203–209.
- Marcus SC, Olfson M. National trends in the treatment of depression from 1998 to 2007. Arch Gen Psychiatry 2010; 67:1265–1273.
- Berkman LF, Blumenthal J, Burg M, et al Effects of treating depression and low perceived social support on clinical events after myocardial infarction: the Enhancing Recovery in Coronary Heart Disease Patients (ENRICHD) Randomized Trial. JAMA 2003; 289:3106–3116.
- Taylor CB, Youngblood ME, Catellier D, et al Effects of antidepressant medication on morbidity and mortality in depressed patients after myocardial infarction. Arch Gen Psychiatry 2005; 62:792–798.
- Glassman AH, O’Connor CM, Califf RM, et al Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709.
- Serebruany VL, Glassman AH, Malinin AI, et al Platelet/endothelial biomarkers in depressed patients treated with the selective serotonin reuptake inhibitor sertraline after acute coronary events: the Sertraline AntiDepressant Heart Attack Randomized Trial (SADHART) Platelet Substudy. Circulation 2003; 108:939–44.
- O’Connor CM, Jiang W, Kuchibhatla M, et al Safety and efficacy of sertraline for depression in patients with heart failure: results of the SADHART-CHF (Sertraline Against Depression and Heart Disease in Chronic Heart Failure) trial. J Am Coll Cardiol 2010; 56:692–699.
- Carney RM, Blumenthal JA, Freedland KE, et al Depression and late mortality after myocardial infarction in the Enhancing Recovery in Coronary Heart Disease (ENRICHD) study. Psychosom Med 2004; 66:466–474.
- Lespérance F, Frasure-Smith N, Koszycki D, et al Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) Trial. JAMA 2007; 297:367–379.
- Salzman C, Schneider L, Alexopoulos GS. Pharmacological treatment of depression in late life. In:Bloon F, Kupfer D, eds. Psychopharmacology: Fourth Generation of Progress. New York: Raven Press; 1995.
- Franklin BA, Bonzheim K, Gordon S, Timmis GC. Safety of medically supervised outpatient cardiac rehabilitation exercise therapy: a 16-year follow-up. Chest 1998; 114:902–906.
- Vongvanich P, Paul-Labrador MJ, Merz CN. Safety of medically supervised exercise in a cardiac rehabilitation center. Am J Cardiol 1996; 77:1383–1385.
- O’Connor CM, Whellan DJ, Lee KL, et al Efficacy and safety of exercise training in patients with chronic heart failure: H-F ACTION randomized controlled trial. JAMA 2009; 301:1439–1540.
- Milani RV, Lavie CJ, Cassidy MM. Effects of cardiac rehabilitation and exercise training programs on depression in patients after major coronary events. Am Heart J 1996; 132:726–732.
- Beniamini Y, Rubenstein JJ, Zaichkowsky LD, Crim MC. Effects of high-intensity strength training on quality-of-life parameters in cardiac rehabilitation patients. Am J Cardiol 1997; 80:841–846.
- Maines TY, Lavie CJ, Milani RV, Cassidy MM, Gilliland YE, Murgo JP. Effects of cardiac rehabilitation and exercise programs on exercise capacity, coronary risk factors, behavior, and quality of life in patients with coronary artery disease. South Med J 1997; 90:43–49.
- Milani RV, Lavie CJ. Prevalence and effects of cardiac rehabilitation on depression in the elderly with coronary heart disease. Am J Cardiol 1998; 81:1233–1236.
- Blumenthal JA, Emery CF, Rejeski WJ. The effects of exercise training on psychosocial functioning after myocardial infarction. J Cardiopulmonary Rehabil 1988; 8:183–193.
- Taylor CB, Houston-Miller N, Ahn DK, Haskell W, DeBusk RF. The effects of exercise training programs on psychosocial improvement in uncomplicated postmyocardial infarction patients. J Psychosom Res 1986; 30:581–587.
- Stern MJ, Gorman PA, Kaslow L. The group counseling v exercise therapy study: a controlled intervention with subjects following myocardial infarction. Arch Intern Med 1983; 143:1719–1725.
- Brosse AL, Sheets ES, Lett HS, Blumenthal JA. Exercise and the treatment of clinical depression in adults: recent findings and future directions. Sports Med 2002; 32:741–760.
- Lawlor DA, Hopker SW. The effectiveness of exercise as an intervention in the management of depression: systematic review and meta-regression analysis of randomised controlled trials. BMJ 2001; 322:763–767.
- Mead GE, Morley W, Campbell P, Greig CA, McMurdo M, Lawlor DA. Exercise for depression. Cochrane Database Syst Rev 2009:CD004366.
- Blumenthal JA, Babyak MA, Carney RM, et al Exercise, depression, and mortality after myocardial infarction in the ENRICHD trial. Med Sci Sports Exerc 2004; 36:746–755.
- Eden J, Wheatley B, McNeil B, Sox H, eds. Institute of Medicine. Knowing What Works in Health Care. A Roadmap for the Nation. Washington DC: National Academics Press; 2009.
- Sox HC, Greenfield S. Comparative effectiveness research: a report from the Institute of Medicine. Ann Intern Med 2009; 151:203–205.
- Hlatky MA, Boothroyd DB, Bravata DM, et al Coronary artery bypass surgery compared with percutaneous coronary interventions for multivessel disease: a collaborative analysis of individual patient data from ten randomised trials. Lancet 2009; 373:1190–1197.
- Blumenthal JA, Babyak MA, Moore KA, et al Effects of exercise training on older patients with major depression. Arch Intern Med 1999; 159:2349–2356.
- Babyak M, Blumenthal JA, Herman S. Exercise treatment for major depression: maintenance of therapeutic benefit at 10 months. Psychosom Med 2000; 62:633–638.
- Blumenthal JA, Babyak MA, Doraiswamy PM, et al Exercise and pharmacotherapy in the treatment of major depressive disorder. Psychosom Med 2007; 69:587–596.
- Hoffman B, Babyak M, Craighead WE, et al Exercise and pharmacotherapy in patients with major depression: one-year follow-up of the SMILE study. Psychosom Med 2010; 73:127–133.
- Dunn AL, Trivedi MH, Kampert JB, Clark CG, Chambliss HO. Exercise treatment for depression: efficacy and dose response. Am J Prev Med 2005; 28:1–8.
- Lichtman JH, Bigger JT, Blumenthal JA, et al Depression and coronary heart disease: recommendations for screening, referral, and treatment: a science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Psychiatric Association. Circulation 2008; 118:1768–1775.
I am fortunate to be the recipient of the 2010 Bakken Institute Pioneer Award and feel especially honored to have my work recognized in this way. When informed that I was this year’s recipient, it prompted me to reflect on the meaning of the term “pioneer,” and how it related to me.
WHAT IS A PIONEER?
According to Merriam-Webster’s Collegiate Dictionary, a pioneer is one who (a) ventures into unknown or unclaimed territory to settle; and (b) opens up new areas of thought, research, or development. One requirement for any pioneer is that there be a frontier to explore. Thirty years ago, my colleagues and I began our investigations into cardiac rehabilitation (CR), which at the time we considered to be a new frontier for behavioral medicine.1
EXERCISE-BASED CARDIAC REHABILITATION
Historically, patients who suffered an acute myocardial infarction (AMI) were often discouraged from engaging in physical activity; patients were initially prescribed prolonged bed rest and told to avoid strenuous exercise.2 In the early 1950s, armchair therapy was proposed3 as an initial attempt to mobilize patients after a coronary event. Over the years, the value of physical exercise has been increasingly recognized and exercise is now considered to be the cornerstone of CR.4–7 Today, exercise-based CR, involving aerobic exercise supplemented by resistance training, is offered by virtually all CR programs in the United States.8 Proper medical management is also emphasized, along with dietary modification and smoking cessation, but exercise is the centerpiece of treatment.
Exercise has been shown to reduce traditional risk factors such as hypertension and hyperlipidemia,8 attenuate cardiovascular responses to mental stress,9 and reduce myocardial ischemia.10–12 Although no single study has demonstrated definitively that exercise reduces morbidity in patients with coronary heart disease (CHD), pooling data across clinical trials has shown that exercise may reduce risk of fatal CHD events by 25%.13 A recent, comprehensive meta-analysis by Jolliffe et al14 reported a 27% reduction in all-cause mortality and 31% reduction in cardiac mortality.
Not only is exercise considered beneficial for medical outcomes, but is also recognized as an important factor in improved quality of life. Indeed, there has been increased interest in the value of exercise for improving not just physical health, but also mental health.15–17 The mental health benefits of exercise are especially relevant for cardiac patients, as there is a growing literature documenting the importance of mental health, and, in particular the prognostic significance of depression, in patients with CHD.
PSYCHOSOCIAL RISK FACTORS: THE ROLE OF DEPRESSION IN CORONARY HEART DISEASE
There has long been an interest in psychosocial factors that contribute to the development and progression of CHD. More than three decades ago, researchers identified the type A behavior pattern as a risk factor for CHD.18 When subsequent studies failed to confirm the association of type A with adverse health outcomes, researchers turned their attention to other possible psychosocial risk factors, including anger and hostility,19 low social support,20 and most recently, depression.21 Indeed, the most consistent and compelling evidence is that clinical depression or elevated depressive symptoms in the presence of CHD increase the risk of fatal and nonfatal cardiac events and of all-cause mortality.22
Major depressive disorder (MDD) is a common and often chronic condition. Lifetime incidence estimates for MDD are approximately 12% in men and 20% in women.23 In addition, MDD is marked by high rates of relapse, with 22% to 50% of patients suffering recurrent episodes within 6 months after recovery.24 Furthermore, MDD is underrecognized and undertreated in older adults,25 CHD patients, and, especially, minorities.26–28
Cross-sectional studies have documented a higher prevalence of depression in CHD patients than in the general population. Point estimates range from 14% to as high as 47%, with higher rates recorded most often in patients with unstable angina, heart failure (HF), and patients awaiting coronary artery bypass graft (CABG) surgery.29–36
Depression associated with poor outcomes
A number of prospective studies have found that depression is associated with increased risk for mortality or nonfatal cardiac events in a variety of CHD populations. The most compelling evidence for depression as a risk factor has come from studies in Montreal, Canada. Frasure-Smith and colleagues31 assessed the impact of depression in 222 AMI patients, of whom 35 were diagnosed with MDD at the time of hospitalization. There were 12 deaths (six depressed and six nondepressed) over an initial 6-month followup period, representing more than a fivefold increased risk of death for depressed patients compared with nondepressed patients (hazard ratio, 5.7; 95% confidence interval [CI], 4.6 to 6.9). In a subsequent report,36 in which 896 AMI patients were followed for 1 year, the presence of elevated depressive symptoms was associated with more than a threefold increased risk in cardiac mortality after controlling for other multivariate predictors of mortality (odds ratio, 3.29 for women; 3.05 for men).
Studies of patients with stable CHD also have reported significant associations between the presence of depression and worse clinical outcomes. For example, Barefoot et al37 assessed 1,250 patients with documented CHD using the Zung self-report depression scale at the time of diagnostic coronary angiography and followed patients for up to 19.4 years. Results showed that patients with moderate to severe depression were at 69% greater risk for cardiac death and 78% greater risk for all-cause death.
Depression and heart failure outcomes
Patients with HF represent a particularly vulnerable group; a meta-analysis of depression in HF patients suggested that one in five patients are clinically depressed (range, 9% to 60%).41 Not only is depression in HF patients associated with worse outcomes,42–46 but recent evidence suggests that worsening of depressive symptoms, independent of clinical status, is related to worse outcomes. Sherwood et al46 demonstrated that increased symptoms of depression, as indicated by higher scores on the Beck Depression Inventory (BDI) over a 1-year interval (BDI change [1-point] hazard ratio, 1.07; 95% CI, 1.02 to 1.12; P = .007), were associated with higher risk of death or cardiovascular hospitalization after controlling for baseline depression (baseline BDI hazard ratio, 1.1; 95% CI, 1.06 to 1.14, P < .001) and established risk factors, including HF etiology, age, ejection fraction, N-terminal pro-B-type natriuretic peptides, and prior hospitalizations. Consequently, strategies to reduce depressive symptoms and prevent the worsening of depression may have important implications for improving cardiac health as well as for enhancing quality of life.
MECHANISMS LINKING DEPRESSION AND CHD
CONVENTIONAL APPROACHES TO TREATMENT OF DEPRESSION
Treatment of depression has focused on reduction of symptoms and restoration of function. Antidepressant medications are generally considered the treatment of choice.56 In particular, second-generation antidepressants such as selective serotonin reuptake inhibitors (SSRIs) are widely prescribed.57 Current treatment guidelines suggest 6 to 12 weeks of acute treatment followed by a continuation phase of 3 to 9 months to maintain therapeutic benefit.58 However, meta-analyses of antidepressant medications have reported only modest benefits over placebo treatments.59,60 In particular, active drug–placebo differences in antidepressant efficacy are positively correlated with depression severity: antidepressants are often comparable with placebo in patients with low levels of depression but may be superior to placebo among patients with more severe depression. However, the explanation for this relationship may be that placebo is less effective for more depressed patients rather than antidepressants being more effective for more depressed patients.59
For acute treatment of MDD, approximately 60% of patients respond to second-generation antidepressants,61 with a 40% relapse rate after 1 year.62 A recent meta-analysis60 of second-generation antidepressants summarized four comparative trials and 23 placebo-controlled trials and found that second-generation antidepressants were generally comparable with each other. Interestingly, despite the modest benefit of antidepressants, the percentage of patients treated for depression in the United States increased from 0.73% in 1987 to 2.33% in 1997. The proportion of those treated who received antidepressants increased from 37.3% in 1987 to 74.5% in 1997.63 The percentage of treated outpatients who used antidepressants has not increased significantly since 1997, but the use of psycho therapy as a sole treatment declined from 53.6% in 1998 to 43.1% in 2007.64 Moreover, the national expenditure for the outpatient treatment of depression increased from $10.05 billion in 1998 to $12.45 billion in 2007, primarily driven by an increase in expenditures for antidepressant medications.
Uncertainty about value of antidepressant therapy
Despite compelling reasons for treating depression in cardiac patients, the clinical significance of treating depression remains uncertain. To date, only the Enhancing Recovery in CHD Patients (ENRICHD) trial has examined the impact of treating depression in post-MI patients on “hard” clinical end points.65 Although more than 2,400 patients were enrolled in the trial, the results were disappointing. There were only modest differences (ie, two points on the Hamilton Depression Rating Scale [HAM-D]) in reductions of depressive symptoms in the group receiving cognitive behavior therapy (CBT) relative to usual-care controls and there were no treatment group differences in the primary outcome—all-cause mortality and nonfatal cardiac events. By the end of the follow-up period, 28.0% of patients in the CBT group and 20.6% of patients in usual care had received antidepressant medication. Although a subsequent reanalysis of the ENRICHD study revealed that antidepressant use was associated with improved clinical outcomes,66 because patients were not randomized to pharmacologic treatment it could not be concluded that SSRI use was responsible for the improved outcomes.
In a randomized trial of patients with acute coronary syndrome (the Sertraline Antidepressant Heart Attack Randomized Trial, or SADHART),67 almost 400 patients were treated with the SSRI sertraline or with placebo. Reductions in depressive symptoms were similar for patients receiving sertraline compared with placebo in the full sample, although a subgroup analysis revealed that patients with more severe depression (ie, those patients who reported two or more previous episodes) benefited more from sertraline compared with placebo. Interestingly, patients receiving sertraline tended to have more favorable cardiac outcomes, including a composite measure of both “hard” and “soft” clinical events, compared with placebo controls. These results suggested that antidepressant medication may improve underlying physiologic processes, such as platelet function, independent of changes in depression.68 However, because SADHART was not powered to detect differences in clinical events, there remain unanswered questions about the clinical value of treating depression in cardiac patients with antidepressant medication.
In a second sertraline trial, SADHART-HF,69 469 men and women with MDD and chronic systolic HF were randomized to receive either sertraline or placebo for 12 weeks. Participants were followed for a minimum of 6 months. Results showed that while sertraline was safe, its use did not result in greater reductions in depressive symptoms compared with placebo (−7.1 ± 0.5 vs −6.8 ± 0.5) and there were no differences in clinical event rates between patients receiving sertraline compared with those receiving placebo.
In an observational study of patients with HF,44 use of antidepressant medication was associated with increased risk of mortality or hospitalization. Although the potential harmful effects of antidepressant medication could not be ruled out, a more likely interpretation is that antidepressant medication use was a marker for individuals with more severe depression, and that the underlying depression may have contributed to their higher risk. Further, patients who are depressed, despite receiving treatment, may represent a subset of treatment-resistant patients who may be especially vulnerable to further cardiac events. Indeed, worsening depression is associated with worse outcomes in HF patients46; this is consistent with data from the ENRICHD trial, which showed that patients receiving CBT (and, in some cases, antidepressant medication) who failed to improve with treatment had higher mortality rates compared with patients who exhibited a positive response to treatment.70
A fourth randomized trial of CHD patients, the Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial,71 used a modified “2 by 2” design; 284 CHD patients with MDD and HAM-D rating scores of 20 or greater were randomized to receive 12 weeks of (a) interpersonal therapy (IPT) plus clinical management (CM) or (b) CM only and citalopram or matching placebo. Because the same interventionists delivered the CM and IPT, patients assigned to IPT received IPT plus CM within the same (extended) session. Patients receiving citalopram had greater reductions in depressive symptoms compared with placebo, with a small to medium effect size of 0.33, and better remission rates (35.9%) compared with placebo (22.5%). Unexpectedly, patients who received just CM tended to have greater improvements in depressive symptoms compared with patients who received IPT plus CM (P < .07); no clinical CHD end points were assessed, however.
Alternative approaches needed
Taken together, these data illustrate that antidepressant medications may reduce depressive symptoms for some patients; for other patients, however, medication fails to adequately relieve depressive symptoms and may perform no better than placebo. Adverse effects also may affect a subgroup of patients and may be relatively more common or more problematic in older persons with CHD.72 Thus, a need remains to identify alternative approaches for treating depression in cardiac patients. We believe that aerobic exercise, the cornerstone of traditional CR, may be one such approach. Exercise is safe for most cardiac patients,73,74 including patients with HF,75 and, if proven effective as a treatment for depression, exercise would hold several potential advantages over traditional medical therapies: it is relatively inexpensive, improves cardiovascular functioning, and avoids the side effects sometimes associated with medication use.
EXERCISE THERAPY FOR DEPRESSION
Some studies of exercise treatment for CHD patients have tracked depressive symptoms and thus have provided information regarding the potential efficacy of exercise as a treatment for depression in this population.76 –81 Although most previous studies have reported significant improvements in depression after completion of an exercise program, many studies had important methodologic limitations, including the absence of a control group.
In one of the few controlled studies in this field, Stern et al82 randomized 106 male patients who had a recent history of AMI along with elevated depression and anxiety or low fitness to 12 weeks of exercise training, group therapy, or a usual-care control group. At 1-year followup, both the exercise and counseling groups showed improvements in depression relative to controls.
Cross-sectional studies of non-CHD samples have reported that active individuals obtain significantly lower depression scores on self-report measures than sedentary persons.83 Studies also have shown that aerobic exercise may reduce self-reported depressive symptoms in nonclinical populations and in patients diagnosed with MDD.83 In 2001, a meta-analysis evaluating 11 randomized controlled trials of non-CHD patients with MDD84 noted that studies were limited because of self-selection bias, absence of control groups or nonrandom controls, and inadequate assessment of exercise training effects; the authors concluded that “the effectiveness of exercise in reducing symptoms of depression cannot be determined because of a lack of good quality research on clinical populations with adequate followup.”
Randomized controlled trials needed
A subsequent meta-analysis85 included 25 studies; for 23 trials (907 participants) that compared exercise with no treatment or a control intervention, the pooled standardized mean difference (SMD) was −0.82 (95% CI, −1.12, −0.51), indicating a large effect size. However, when only the three trials (216 participants) with adequate allocation concealment, intention to treat analysis, and blinded outcome assessment were included, the pooled SMD was −0.43 (95% CI, −0.88, 0.03), with a point estimate that was half the size of that with all trials. As a result, the authors concluded that “exercise seems to improve depressive symptoms in people with a diagnosis of depression, but when only the methodologically robust trials are included, the effect size is only moderate.”
To date, no randomized clinical trials (RCTs) have examined the effects of exercise on clinical outcomes in depressed cardiac patients. However, data from the ENRICHD trial suggest that exercise may reduce the rates of mortality and nonfatal reinfarction in patients with depression or in post-MI patients who are socially isolated.86 Self-report data were used to categorize participants as exercising regularly or not exercising regularly. After controlling for medical and demographic variables, the magnitude of reduction in risk associated with regular exercise was nearly 40% for nonfatal reinfarction and 50% for mortality. The evidence that exercise mitigates depression, reduces CHD risk factors, and improves CHD outcomes suggests that exercise may be a particularly promising intervention for depressed CHD patients.
COMPARATIVE EFFECTIVENESS OF EXERCISE AND ANTIDEPRESSANT MEDICATION
In 2008, an Institute of Medicine (IOM) report called for a national initiative of research that would provide a basis for better decision-making about how to best treat various medical conditions, including depression. In 2009, the American Reinvestment Recovery Act provided a major boost in funding for comparative effectiveness research (CER). The act allotted $1.1 billion to support this form of research. CER refers to the generation and synthesis of evidence that compares the benefits and harms of alternative methods to prevent, diagnose, treat, and monitor a clinical condition, or to improve the delivery of care. The purpose of CER is to assist consumers, clinicians, purchasers, and policy makers in reaching informed decisions that will improve health care at both the individual and population levels.87
Two research categories inform decision-making
Two broad categories of research have been used to inform decision-making:
- Epidemiologic studies provide evidence linking various treatments with patient outcomes. These sources of data are limited because they seldom specify the basis for medical decisions and they fail to consider patient characteristics that affect both clinical decisions and clinical outcomes. Indeed, it has been suggested that “overcoming the limitations of observational research is the most important frontier of research on study methods.”88
- RCTs address these limitations by randomly assigning patients to different treatment conditions. While this design may eliminate some of the uncertainty and potential confounders that characterize purely observational studies, most RCTs are efficacy studies; patients are carefully selected and a treatment is usually compared with a placebo or usual care.
The RCT design addresses the question of whether a given treatment is effective, but it does not necessarily address questions that many physicians want answers to: namely, is this treatment better than that treatment? Further, physicians want to know if one treatment is more effective than another for a given patient. For example, Hlatky et al89 showed that mortality associated with percutaneous coronary interventions (PCIs) and CABG surgery was comparable; however, mortality with CABG surgery was significantly lower for patients older than 65 years while PCI was superior for patients younger than 55 years. Thus, examination of individual differences may also help to inform clinicians about the optimal therapy for their particular patients.
Treatment of depression not necessarily a research priority
The IOM committee sought advice from a broad range of stakeholders and prioritized areas for research. The top-ranked topic was comparison of treatment strategies for atrial fibrillation, including surgery, catheter ablation, and pharmacologic treatment. Coming in at #98 was comparison of the effectiveness of different treatment strategies (eg, psychotherapy, antidepressants, combination treatment with case management) for depression after MI and their impact on medication adherence, cardiovascular events, hospitalization, and death.
In a second Duke study that compared exercise and antidepressant medication,92 202 adults (153 women; 49 men) diagnosed with MDD were randomly assigned to one of four groups: supervised exercise in a group setting, home-based exercise, antidepressant medication (sertraline, 50 to 200 mg daily), or placebo pill for 16 weeks. Once again, patients underwent the Structured Clinical Interview for Depression and completed the HAM-D. After 4 months of treatment, 41% of participants achieved remission, defined as no longer meeting criteria for MDD and a HAM-D score of less than 8 points. Patients receiving active treatments tended to have higher remission rates than placebo controls: supervised exercise, 45%; home-based exercise, 40%; medication, 47%; placebo, 31% (P = .057). All treatment groups had lower HAM-D scores after treatment; scores for the active treatment groups were not significantly different from the placebo group (P = .23). However, when immediate responders (ie, those patients who reported more than 50% reduction in depressive symptoms after only 1 week of treatment) were excluded from the analysis, patients receiving active treatments (ie, either sertraline or exercise) had greater reductions in depressive symptoms compared with placebo controls (P = .048). There was no difference between the exercise and antidepressant groups. We concluded that the efficacy of exercise appears generally comparable with antidepressant medication and both tend to be better than placebo in patients with MDD. Placebo response rates were high, suggesting that a considerable portion of the therapeutic response could be determined by patient expectations, ongoing symptom monitoring, attention, and other nonspecific factors. Similar to our previous trial, participants who continued to exercise following the completion of the program were less likely to be depressed.93
Another RCT94 also demonstrated that exercise was associated with reduced depression, independent of group support. Participants exercised alone in a secluded setting, and the study included a no-treatment control group. Only 53 of 80 patients actually completed the 12-week trial, however, including only five of 13 no-treatment controls. Moreover, there was no active treatment comparison group, so that an estimate of comparative effectiveness could not be determined.
While these results are preliminary and should be interpreted with caution, it appears that exercise may be comparable with conventional antidepressant medication in reducing depressive symptoms, at least for patients who are willing to try it, and maintenance of exercise reduces the risk of relapse.
SUMMARY
Three decades ago, we recognized that CR was a new frontier for behavioral medicine. We now know that successful rehabilitation of patients with CHD involves modification of lifestyle behaviors, including smoking cessation, dietary modification, and exercise. Exercise is no longer considered unsafe for most cardiac patients, and exercise is currently the key component of CR services. Research also has provided strong evidence that depression is an important risk factor for CHD, although there is no consensus regarding the optimal way to treat depression in CHD patients.95 Research on comparative effectiveness of established and alternative treatments for depressed cardiac patients is a new frontier for future pioneers in heart-brain medicine.
I am fortunate to be the recipient of the 2010 Bakken Institute Pioneer Award and feel especially honored to have my work recognized in this way. When informed that I was this year’s recipient, it prompted me to reflect on the meaning of the term “pioneer,” and how it related to me.
WHAT IS A PIONEER?
According to Merriam-Webster’s Collegiate Dictionary, a pioneer is one who (a) ventures into unknown or unclaimed territory to settle; and (b) opens up new areas of thought, research, or development. One requirement for any pioneer is that there be a frontier to explore. Thirty years ago, my colleagues and I began our investigations into cardiac rehabilitation (CR), which at the time we considered to be a new frontier for behavioral medicine.1
EXERCISE-BASED CARDIAC REHABILITATION
Historically, patients who suffered an acute myocardial infarction (AMI) were often discouraged from engaging in physical activity; patients were initially prescribed prolonged bed rest and told to avoid strenuous exercise.2 In the early 1950s, armchair therapy was proposed3 as an initial attempt to mobilize patients after a coronary event. Over the years, the value of physical exercise has been increasingly recognized and exercise is now considered to be the cornerstone of CR.4–7 Today, exercise-based CR, involving aerobic exercise supplemented by resistance training, is offered by virtually all CR programs in the United States.8 Proper medical management is also emphasized, along with dietary modification and smoking cessation, but exercise is the centerpiece of treatment.
Exercise has been shown to reduce traditional risk factors such as hypertension and hyperlipidemia,8 attenuate cardiovascular responses to mental stress,9 and reduce myocardial ischemia.10–12 Although no single study has demonstrated definitively that exercise reduces morbidity in patients with coronary heart disease (CHD), pooling data across clinical trials has shown that exercise may reduce risk of fatal CHD events by 25%.13 A recent, comprehensive meta-analysis by Jolliffe et al14 reported a 27% reduction in all-cause mortality and 31% reduction in cardiac mortality.
Not only is exercise considered beneficial for medical outcomes, but is also recognized as an important factor in improved quality of life. Indeed, there has been increased interest in the value of exercise for improving not just physical health, but also mental health.15–17 The mental health benefits of exercise are especially relevant for cardiac patients, as there is a growing literature documenting the importance of mental health, and, in particular the prognostic significance of depression, in patients with CHD.
PSYCHOSOCIAL RISK FACTORS: THE ROLE OF DEPRESSION IN CORONARY HEART DISEASE
There has long been an interest in psychosocial factors that contribute to the development and progression of CHD. More than three decades ago, researchers identified the type A behavior pattern as a risk factor for CHD.18 When subsequent studies failed to confirm the association of type A with adverse health outcomes, researchers turned their attention to other possible psychosocial risk factors, including anger and hostility,19 low social support,20 and most recently, depression.21 Indeed, the most consistent and compelling evidence is that clinical depression or elevated depressive symptoms in the presence of CHD increase the risk of fatal and nonfatal cardiac events and of all-cause mortality.22
Major depressive disorder (MDD) is a common and often chronic condition. Lifetime incidence estimates for MDD are approximately 12% in men and 20% in women.23 In addition, MDD is marked by high rates of relapse, with 22% to 50% of patients suffering recurrent episodes within 6 months after recovery.24 Furthermore, MDD is underrecognized and undertreated in older adults,25 CHD patients, and, especially, minorities.26–28
Cross-sectional studies have documented a higher prevalence of depression in CHD patients than in the general population. Point estimates range from 14% to as high as 47%, with higher rates recorded most often in patients with unstable angina, heart failure (HF), and patients awaiting coronary artery bypass graft (CABG) surgery.29–36
Depression associated with poor outcomes
A number of prospective studies have found that depression is associated with increased risk for mortality or nonfatal cardiac events in a variety of CHD populations. The most compelling evidence for depression as a risk factor has come from studies in Montreal, Canada. Frasure-Smith and colleagues31 assessed the impact of depression in 222 AMI patients, of whom 35 were diagnosed with MDD at the time of hospitalization. There were 12 deaths (six depressed and six nondepressed) over an initial 6-month followup period, representing more than a fivefold increased risk of death for depressed patients compared with nondepressed patients (hazard ratio, 5.7; 95% confidence interval [CI], 4.6 to 6.9). In a subsequent report,36 in which 896 AMI patients were followed for 1 year, the presence of elevated depressive symptoms was associated with more than a threefold increased risk in cardiac mortality after controlling for other multivariate predictors of mortality (odds ratio, 3.29 for women; 3.05 for men).
Studies of patients with stable CHD also have reported significant associations between the presence of depression and worse clinical outcomes. For example, Barefoot et al37 assessed 1,250 patients with documented CHD using the Zung self-report depression scale at the time of diagnostic coronary angiography and followed patients for up to 19.4 years. Results showed that patients with moderate to severe depression were at 69% greater risk for cardiac death and 78% greater risk for all-cause death.
Depression and heart failure outcomes
Patients with HF represent a particularly vulnerable group; a meta-analysis of depression in HF patients suggested that one in five patients are clinically depressed (range, 9% to 60%).41 Not only is depression in HF patients associated with worse outcomes,42–46 but recent evidence suggests that worsening of depressive symptoms, independent of clinical status, is related to worse outcomes. Sherwood et al46 demonstrated that increased symptoms of depression, as indicated by higher scores on the Beck Depression Inventory (BDI) over a 1-year interval (BDI change [1-point] hazard ratio, 1.07; 95% CI, 1.02 to 1.12; P = .007), were associated with higher risk of death or cardiovascular hospitalization after controlling for baseline depression (baseline BDI hazard ratio, 1.1; 95% CI, 1.06 to 1.14, P < .001) and established risk factors, including HF etiology, age, ejection fraction, N-terminal pro-B-type natriuretic peptides, and prior hospitalizations. Consequently, strategies to reduce depressive symptoms and prevent the worsening of depression may have important implications for improving cardiac health as well as for enhancing quality of life.
MECHANISMS LINKING DEPRESSION AND CHD
CONVENTIONAL APPROACHES TO TREATMENT OF DEPRESSION
Treatment of depression has focused on reduction of symptoms and restoration of function. Antidepressant medications are generally considered the treatment of choice.56 In particular, second-generation antidepressants such as selective serotonin reuptake inhibitors (SSRIs) are widely prescribed.57 Current treatment guidelines suggest 6 to 12 weeks of acute treatment followed by a continuation phase of 3 to 9 months to maintain therapeutic benefit.58 However, meta-analyses of antidepressant medications have reported only modest benefits over placebo treatments.59,60 In particular, active drug–placebo differences in antidepressant efficacy are positively correlated with depression severity: antidepressants are often comparable with placebo in patients with low levels of depression but may be superior to placebo among patients with more severe depression. However, the explanation for this relationship may be that placebo is less effective for more depressed patients rather than antidepressants being more effective for more depressed patients.59
For acute treatment of MDD, approximately 60% of patients respond to second-generation antidepressants,61 with a 40% relapse rate after 1 year.62 A recent meta-analysis60 of second-generation antidepressants summarized four comparative trials and 23 placebo-controlled trials and found that second-generation antidepressants were generally comparable with each other. Interestingly, despite the modest benefit of antidepressants, the percentage of patients treated for depression in the United States increased from 0.73% in 1987 to 2.33% in 1997. The proportion of those treated who received antidepressants increased from 37.3% in 1987 to 74.5% in 1997.63 The percentage of treated outpatients who used antidepressants has not increased significantly since 1997, but the use of psycho therapy as a sole treatment declined from 53.6% in 1998 to 43.1% in 2007.64 Moreover, the national expenditure for the outpatient treatment of depression increased from $10.05 billion in 1998 to $12.45 billion in 2007, primarily driven by an increase in expenditures for antidepressant medications.
Uncertainty about value of antidepressant therapy
Despite compelling reasons for treating depression in cardiac patients, the clinical significance of treating depression remains uncertain. To date, only the Enhancing Recovery in CHD Patients (ENRICHD) trial has examined the impact of treating depression in post-MI patients on “hard” clinical end points.65 Although more than 2,400 patients were enrolled in the trial, the results were disappointing. There were only modest differences (ie, two points on the Hamilton Depression Rating Scale [HAM-D]) in reductions of depressive symptoms in the group receiving cognitive behavior therapy (CBT) relative to usual-care controls and there were no treatment group differences in the primary outcome—all-cause mortality and nonfatal cardiac events. By the end of the follow-up period, 28.0% of patients in the CBT group and 20.6% of patients in usual care had received antidepressant medication. Although a subsequent reanalysis of the ENRICHD study revealed that antidepressant use was associated with improved clinical outcomes,66 because patients were not randomized to pharmacologic treatment it could not be concluded that SSRI use was responsible for the improved outcomes.
In a randomized trial of patients with acute coronary syndrome (the Sertraline Antidepressant Heart Attack Randomized Trial, or SADHART),67 almost 400 patients were treated with the SSRI sertraline or with placebo. Reductions in depressive symptoms were similar for patients receiving sertraline compared with placebo in the full sample, although a subgroup analysis revealed that patients with more severe depression (ie, those patients who reported two or more previous episodes) benefited more from sertraline compared with placebo. Interestingly, patients receiving sertraline tended to have more favorable cardiac outcomes, including a composite measure of both “hard” and “soft” clinical events, compared with placebo controls. These results suggested that antidepressant medication may improve underlying physiologic processes, such as platelet function, independent of changes in depression.68 However, because SADHART was not powered to detect differences in clinical events, there remain unanswered questions about the clinical value of treating depression in cardiac patients with antidepressant medication.
In a second sertraline trial, SADHART-HF,69 469 men and women with MDD and chronic systolic HF were randomized to receive either sertraline or placebo for 12 weeks. Participants were followed for a minimum of 6 months. Results showed that while sertraline was safe, its use did not result in greater reductions in depressive symptoms compared with placebo (−7.1 ± 0.5 vs −6.8 ± 0.5) and there were no differences in clinical event rates between patients receiving sertraline compared with those receiving placebo.
In an observational study of patients with HF,44 use of antidepressant medication was associated with increased risk of mortality or hospitalization. Although the potential harmful effects of antidepressant medication could not be ruled out, a more likely interpretation is that antidepressant medication use was a marker for individuals with more severe depression, and that the underlying depression may have contributed to their higher risk. Further, patients who are depressed, despite receiving treatment, may represent a subset of treatment-resistant patients who may be especially vulnerable to further cardiac events. Indeed, worsening depression is associated with worse outcomes in HF patients46; this is consistent with data from the ENRICHD trial, which showed that patients receiving CBT (and, in some cases, antidepressant medication) who failed to improve with treatment had higher mortality rates compared with patients who exhibited a positive response to treatment.70
A fourth randomized trial of CHD patients, the Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial,71 used a modified “2 by 2” design; 284 CHD patients with MDD and HAM-D rating scores of 20 or greater were randomized to receive 12 weeks of (a) interpersonal therapy (IPT) plus clinical management (CM) or (b) CM only and citalopram or matching placebo. Because the same interventionists delivered the CM and IPT, patients assigned to IPT received IPT plus CM within the same (extended) session. Patients receiving citalopram had greater reductions in depressive symptoms compared with placebo, with a small to medium effect size of 0.33, and better remission rates (35.9%) compared with placebo (22.5%). Unexpectedly, patients who received just CM tended to have greater improvements in depressive symptoms compared with patients who received IPT plus CM (P < .07); no clinical CHD end points were assessed, however.
Alternative approaches needed
Taken together, these data illustrate that antidepressant medications may reduce depressive symptoms for some patients; for other patients, however, medication fails to adequately relieve depressive symptoms and may perform no better than placebo. Adverse effects also may affect a subgroup of patients and may be relatively more common or more problematic in older persons with CHD.72 Thus, a need remains to identify alternative approaches for treating depression in cardiac patients. We believe that aerobic exercise, the cornerstone of traditional CR, may be one such approach. Exercise is safe for most cardiac patients,73,74 including patients with HF,75 and, if proven effective as a treatment for depression, exercise would hold several potential advantages over traditional medical therapies: it is relatively inexpensive, improves cardiovascular functioning, and avoids the side effects sometimes associated with medication use.
EXERCISE THERAPY FOR DEPRESSION
Some studies of exercise treatment for CHD patients have tracked depressive symptoms and thus have provided information regarding the potential efficacy of exercise as a treatment for depression in this population.76 –81 Although most previous studies have reported significant improvements in depression after completion of an exercise program, many studies had important methodologic limitations, including the absence of a control group.
In one of the few controlled studies in this field, Stern et al82 randomized 106 male patients who had a recent history of AMI along with elevated depression and anxiety or low fitness to 12 weeks of exercise training, group therapy, or a usual-care control group. At 1-year followup, both the exercise and counseling groups showed improvements in depression relative to controls.
Cross-sectional studies of non-CHD samples have reported that active individuals obtain significantly lower depression scores on self-report measures than sedentary persons.83 Studies also have shown that aerobic exercise may reduce self-reported depressive symptoms in nonclinical populations and in patients diagnosed with MDD.83 In 2001, a meta-analysis evaluating 11 randomized controlled trials of non-CHD patients with MDD84 noted that studies were limited because of self-selection bias, absence of control groups or nonrandom controls, and inadequate assessment of exercise training effects; the authors concluded that “the effectiveness of exercise in reducing symptoms of depression cannot be determined because of a lack of good quality research on clinical populations with adequate followup.”
Randomized controlled trials needed
A subsequent meta-analysis85 included 25 studies; for 23 trials (907 participants) that compared exercise with no treatment or a control intervention, the pooled standardized mean difference (SMD) was −0.82 (95% CI, −1.12, −0.51), indicating a large effect size. However, when only the three trials (216 participants) with adequate allocation concealment, intention to treat analysis, and blinded outcome assessment were included, the pooled SMD was −0.43 (95% CI, −0.88, 0.03), with a point estimate that was half the size of that with all trials. As a result, the authors concluded that “exercise seems to improve depressive symptoms in people with a diagnosis of depression, but when only the methodologically robust trials are included, the effect size is only moderate.”
To date, no randomized clinical trials (RCTs) have examined the effects of exercise on clinical outcomes in depressed cardiac patients. However, data from the ENRICHD trial suggest that exercise may reduce the rates of mortality and nonfatal reinfarction in patients with depression or in post-MI patients who are socially isolated.86 Self-report data were used to categorize participants as exercising regularly or not exercising regularly. After controlling for medical and demographic variables, the magnitude of reduction in risk associated with regular exercise was nearly 40% for nonfatal reinfarction and 50% for mortality. The evidence that exercise mitigates depression, reduces CHD risk factors, and improves CHD outcomes suggests that exercise may be a particularly promising intervention for depressed CHD patients.
COMPARATIVE EFFECTIVENESS OF EXERCISE AND ANTIDEPRESSANT MEDICATION
In 2008, an Institute of Medicine (IOM) report called for a national initiative of research that would provide a basis for better decision-making about how to best treat various medical conditions, including depression. In 2009, the American Reinvestment Recovery Act provided a major boost in funding for comparative effectiveness research (CER). The act allotted $1.1 billion to support this form of research. CER refers to the generation and synthesis of evidence that compares the benefits and harms of alternative methods to prevent, diagnose, treat, and monitor a clinical condition, or to improve the delivery of care. The purpose of CER is to assist consumers, clinicians, purchasers, and policy makers in reaching informed decisions that will improve health care at both the individual and population levels.87
Two research categories inform decision-making
Two broad categories of research have been used to inform decision-making:
- Epidemiologic studies provide evidence linking various treatments with patient outcomes. These sources of data are limited because they seldom specify the basis for medical decisions and they fail to consider patient characteristics that affect both clinical decisions and clinical outcomes. Indeed, it has been suggested that “overcoming the limitations of observational research is the most important frontier of research on study methods.”88
- RCTs address these limitations by randomly assigning patients to different treatment conditions. While this design may eliminate some of the uncertainty and potential confounders that characterize purely observational studies, most RCTs are efficacy studies; patients are carefully selected and a treatment is usually compared with a placebo or usual care.
The RCT design addresses the question of whether a given treatment is effective, but it does not necessarily address questions that many physicians want answers to: namely, is this treatment better than that treatment? Further, physicians want to know if one treatment is more effective than another for a given patient. For example, Hlatky et al89 showed that mortality associated with percutaneous coronary interventions (PCIs) and CABG surgery was comparable; however, mortality with CABG surgery was significantly lower for patients older than 65 years while PCI was superior for patients younger than 55 years. Thus, examination of individual differences may also help to inform clinicians about the optimal therapy for their particular patients.
Treatment of depression not necessarily a research priority
The IOM committee sought advice from a broad range of stakeholders and prioritized areas for research. The top-ranked topic was comparison of treatment strategies for atrial fibrillation, including surgery, catheter ablation, and pharmacologic treatment. Coming in at #98 was comparison of the effectiveness of different treatment strategies (eg, psychotherapy, antidepressants, combination treatment with case management) for depression after MI and their impact on medication adherence, cardiovascular events, hospitalization, and death.
In a second Duke study that compared exercise and antidepressant medication,92 202 adults (153 women; 49 men) diagnosed with MDD were randomly assigned to one of four groups: supervised exercise in a group setting, home-based exercise, antidepressant medication (sertraline, 50 to 200 mg daily), or placebo pill for 16 weeks. Once again, patients underwent the Structured Clinical Interview for Depression and completed the HAM-D. After 4 months of treatment, 41% of participants achieved remission, defined as no longer meeting criteria for MDD and a HAM-D score of less than 8 points. Patients receiving active treatments tended to have higher remission rates than placebo controls: supervised exercise, 45%; home-based exercise, 40%; medication, 47%; placebo, 31% (P = .057). All treatment groups had lower HAM-D scores after treatment; scores for the active treatment groups were not significantly different from the placebo group (P = .23). However, when immediate responders (ie, those patients who reported more than 50% reduction in depressive symptoms after only 1 week of treatment) were excluded from the analysis, patients receiving active treatments (ie, either sertraline or exercise) had greater reductions in depressive symptoms compared with placebo controls (P = .048). There was no difference between the exercise and antidepressant groups. We concluded that the efficacy of exercise appears generally comparable with antidepressant medication and both tend to be better than placebo in patients with MDD. Placebo response rates were high, suggesting that a considerable portion of the therapeutic response could be determined by patient expectations, ongoing symptom monitoring, attention, and other nonspecific factors. Similar to our previous trial, participants who continued to exercise following the completion of the program were less likely to be depressed.93
Another RCT94 also demonstrated that exercise was associated with reduced depression, independent of group support. Participants exercised alone in a secluded setting, and the study included a no-treatment control group. Only 53 of 80 patients actually completed the 12-week trial, however, including only five of 13 no-treatment controls. Moreover, there was no active treatment comparison group, so that an estimate of comparative effectiveness could not be determined.
While these results are preliminary and should be interpreted with caution, it appears that exercise may be comparable with conventional antidepressant medication in reducing depressive symptoms, at least for patients who are willing to try it, and maintenance of exercise reduces the risk of relapse.
SUMMARY
Three decades ago, we recognized that CR was a new frontier for behavioral medicine. We now know that successful rehabilitation of patients with CHD involves modification of lifestyle behaviors, including smoking cessation, dietary modification, and exercise. Exercise is no longer considered unsafe for most cardiac patients, and exercise is currently the key component of CR services. Research also has provided strong evidence that depression is an important risk factor for CHD, although there is no consensus regarding the optimal way to treat depression in CHD patients.95 Research on comparative effectiveness of established and alternative treatments for depressed cardiac patients is a new frontier for future pioneers in heart-brain medicine.
- Blumenthal JA, Califf R, Williams RS, Hindman M. Cardiac rehabilitation: a new frontier for behavioral medicine. J Cardiac Rehabil 1983; 3:637–656.
- Lewis T. Diseases of the Heart. New York: Macmillan; 1933:41–49.
- Levine SA, Lown B. The “chair” treatment of acute thrombosis. Trans Assoc Am Physicians 1951; 64:316–327.
- Cain HD, Frasher WG, Stivelman R. Graded activity program for safe return to self-care after myocardial infarction. JAMA 1961; 177:111–115.
- Hellerstein HK, Ford AB. Rehabilitation of the cardiac patient. JAMA 1957; 14:225–231.
- Naughton J, Bruhn JG, Lategola MT. Effects of physical training on physiologic and behavioral characteristics of cardiac patients. Arch Phys Med Rehabil 1968; 49:131–137.
- O’Connor GT, Buring JE, Yusuf S, et al An overview of randomized trials of rehabilitation with exercise after myocardial infarction. Circulation 1989; 80:234–244.
- Wenger NK, Froelicher ES, Smith LK, et al Cardiac rehabilitation: clinical practice guideline no. 17. Rockville, MD: US Dept of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research, National Heart, Lung, and Blood Institute; October 1995; AHCPR publication 96-0672.
- Blumenthal JA, Emery CF, Cox DR, et al Exercise training in healthy type A middle-aged men: effects on behavioral and cardiovascular responses. Psychosom Med 1988; 50:418–433.
- Schuler G, Schlierf G, Wirth A, et al Low-fat diet and regular, supervised physical exercise in patients with symptomatic coronary artery disease: reduction of stress-induced myocardial ischemia. Circulation 1988; 77:172–181.
- Jiang W, Trauner MA, Coleman RE, et al Association of physical fitness and transient myocardial ischemia in patients with coronary artery disease. J Cardiopulm Rehabil 1995; 15:431–438.
- Blumenthal JA, Jiang W, Babyak MA, et al Stress management and exercise training in cardiac patients with myocardial ischemia: effects on prognosis and evaluation of mechanisms. Arch Int Med 1997; 157:2213–2223.
- Oldridge NB, Guyatt GH, Fisher ME, Rimm AA. Cardiac rehabilitation after myocardial infarction: combined experience of randomized clinical trials. JAMA 1988; 260:945–950.
- Jolliffe JA, Rees K, Taylor RS, Thompson D, Oldridge N, Ebrahim S. Exercise-based rehabilitation for coronary heart disease. Cochrane Database Syst Rev 2001:CD001800.
- Folkins CH, Sime WE. Physical fitness training and mental health. Am Psychol 1981; 36:373–389.
- Hughes JR. Psychological effects of habitual aerobic exercise: a critical review. Prev Med 1984; 13:66–78.
- Plante TG, Rodin J. Physical fitness and enhanced psychological health. Curr Psychol: Res Rev 1990; 9:3–24.
- Review Panel on Coronary-Prone Behavior and Coronary Heart Disease. Coronary-prone behavior and coronary heart disease: a critical review. Circulation 1981; 63:1199–1215.
- Smith TW. Hostility and health: current status of a psychosomatic hypothesis. Health Psychol 1992; 11:139–150.
- Lett HS, Blumenthal JA, Babyak MA, Strauman TJ, Robins C, Sherwood A. Social support and coronary heart disease: epidemiologic evidence and implications for treatment. Psychosom Med 2005; 67:869–878.
- Lett HS, Blumenthal JA, Babyak MA, et al Depression as a risk factor for coronary artery disease: evidence, mechanisms, and treatment. Psychosom Med 2004; 66:305–315.
- Barth J, Schumacher M, Herrmann-Lingen C. Depression as a risk factor for mortality in patients with coronary heart disease: a meta-analysis. Psychosom Med 2004; 66:802–813.
- Kessler RC, Berglund P, Demler O, et al The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCR-R). JAMA 2003; 289:3095–3105.
- Belsher G, Costello CG. Relapse after recovery from unipolar depression: a critical review. Psychol Bull 1988; 104:84–96.
- Strothers HS, Rust G, Minor P, Fresh E, Druss B, Satcher D. Disparities in antidepressant treatment in Medicaid elderly diagnosed with depression. J Am Geriatr Soc 2005; 53:456–461.
- Simpson SM, Krishnan LL, Kunik ME, Ruiz P. Racial disparities in diagnosis and treatment of depression: a literature review. Psychiatr Q 2007; 78:3–14.
- Sclar DA, Robison LM, Skaer TL. Ethnicity/race and the diagnosis of depression and use of antidepressants by adults in the United States. Int Clin Psychopharmacol 2008; 23:106–109.
- Waldman SV, Blumenthal JA, Babyak MA, et al Ethnic differences in the treatment of depression in patients with ischemic heart disease. Am Heart J 2009; 57:77–83.
- Carney RM, Rich MW, Freedland KE, et al Major depressive disorder predicts cardiac events in patients with coronary artery disease. Psychosom Med 1988; 50:627–633.
- Schleifer SJ, Macari-Hinson MM, Coyle DA, et al The nature and course of depression following myocardial infarction. Arch Intern Med 1989; 149:1785–1789.
- Frasure-Smith N, Lespérance F, Talajic M. Depression following myocardial infarction: impact on 6-month survival. JAMA 1993; 270:1819–1825.
- Gonzalez MB, Snyderman TB, Colket JT, et al Depression in patients with coronary artery disease. Depression 1996; 4:57–62.
- Connerney I, Shapiro PA, McLaughlin JS, Bagiella E, Sloan RP. Relation between depression after coronary artery bypass surgery and 12-month outcome: a prospective study. Lancet 2001; 358:1766–1771.
- Burker EJ, Blumenthal JA, Feldman M, et al Depression in male and female patients undergoing cardiac surgery. Br J Clin Psychol 1995; 34( Pt 1):119–128.
- Lespérance F, Frasure-Smith N, Juneau M, Théroux P. Depression and 1-year prognosis in unstable angina. Arch Intern Med 2000; 160:1354–1360.
- Frasure-Smith N, Lespérance F, Talajic M. Depression and 18-month prognosis after myocardial infarction. Circulation 1995; 91:999–1005.
- Barefoot JC, Helms MJ, Mark DB, et al Depression and long-term mortality risk in patients with coronary artery disease. Am J Cardiol 1996; 78:613–617.
- Burg MM, Benedetto C, Soufer R. Depressive symptoms and mortality two years after coronary artery bypass graft surgery (CABG) in men. Psychosom Med 2003; 65:508–510.
- Blumenthal JA, Lett HS, Babyak MA, et al Depression as a risk factor for mortality after coronary artery bypass surgery. Lancet 2003; 362:604–609.
- Connerney I, Sloan RP, Shapiro PA, Bagiella E, Seckman C. Depression is associated with increased mortality 10 years after coronary artery bypass surgery. Psychosom Med 2010; 72:874–881.
- Rutledge T, Reis VA, Linke SE, Greenberg BH, Mills PJ. Depression in heart failure: a meta analytic review of prevalence, intervention effects, and associations with clinical outcomes. J Am Coll Cardiol 2006; 48:1527–1537.
- Murberg TA, Furze G. Depressive symptoms and mortality in patients with congestive heart failure: a six-year follow-up study. Med Sci Monit 2004; 10:CR643–CR648.
- Jiang W, Alexander J, Christopher E, et al Relationship of depression to increased risk of mortality and rehospitalization in patients with congestive heart failure. Arch Intern Med 2001; 161:1849–1856.
- Sherwood A, Blumenthal JA, Trivedi R, et al Relationship of depression to death or hospitalization in patients with heart failure. Arch Int Med 2007; 167:367–373.
- Frasure-Smith N, Lespérance F, Habra M, et al Elevated depression symptoms predict long-term cardiovascular mortality in patients with atrial fibrillation and heart failure. Circulation 2009; 120:134–140.
- Sherwood A, Blumenthal JA, Hinderliter AL, et al Worsening depressive symptoms are associated with adverse clinical outcomes in patients with heart failure. J Am Coll Cardiol 2011; 57:418–423.
- Anderson RJ, Freedland KE, Clouse RE, Lustman PJ. The prevalence of comorbid depression in adults with diabetes: a meta-analysis. Diabetes Care 2001; 24:1069–1078.
- Thakore JH, Richards PJ, Reznek RH, Martin A, Dinan TG. Increased intra-abdominal fat deposition in patients with major depressive illness as measured by computed tomography. Biol Psychiatry 1997; 41:1140–1142.
- Musselman DL, Tomer A, Manatunga AK, et al Exaggerated platelet reactivity in major depression. Am J Psychiatry 1996; 153:1313–1317.
- Delgado PL, Moreno FA. Role of norepinephrine in depression. J Clin Psychiatry 2000; 61( suppl 1):5–12.
- Akil H, Haskett RF, Young EA, et al Multiple HPA profiles in endogenous depression: effect of age and sex on cortisol and beta-endorphin. Biol Psychiatry 1993; 33:73–85.
- Kop WJ, Gottdiener JS, Tangen CM, et al Inflammation and coagulation factors in persons > 65 years of age with symptoms of depression but without evidence of myocardial ischemia. Am J Cardiol 2002; 89:419–424.
- Carney RM, Freedland KE, Eisen SA, Rich MW, Jaffe AS. Major depression and medication adherence in elderly patients with coronary artery disease. Health Psychol 1995; 14:88–90.
- Lehto S, Koukkunen H, Hintikka J, Viinamäki H, Laakso M, Pyörälä K. Depression after coronary heart disease events. Scand Cardiovasc J 2000; 34:580–583.
- Camacho TC, Roberts RE, Lazarus NB, Kaplan GA, Cohen RD. Physical activity and depression: evidence from the Alameda County Study. Am J Epidemiol 1991; 134:220–231.
- Clinical Practice Guideline Number 5: Depression in Primary Care, 2: Treatment of Major Depression. Rockville, MD: U.S. Department of Health and Human Services, Agency for Health Care Policy and Research; 1993. AHCPR publication 93-0551.
- Anderson IM, Ferrier IN, Baldwin RC, et al Evidence-based guidelines for treating depressive disorders with antidepressants: a revision of the 2000 British Association for Psychopharmacology guidelines. J Psychopharmacol 2008; 22:343–396.
- American Psyciatric Association. Practice guideline for the treatment of patients with major depressive disorder (revision). Am J Psychiatry 2000; 157( suppl 4):1–45.
- Kirsch I, Deacon BJ, Huedo-Medina TB, Scoboria A, Moore TJ, Johnson BT. Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration. PLoS Med 2008; 5:e45.
- Hansen R, Gaynes B, Thieda P, et al Meta-analysis of major depressive disorder relapse and recurrence with second-generation antidepressants. Psychiatr Serv 2008; 59:1121–1129.
- Hansen RA, Gartlehner G, Lohr KN, Gaynes BN, Carey TS. Efficacy and safety of second-generation antidepressants in the treatment of major depressive disorder. Ann Intern Med 2005; 143:415–426.
- Rush AJ, Trivedi MH, Wisniewski SR, et al Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 2006; 163:1905–1917.
- Olfson M, Marcus SC, Druss B, Elinson L, Tanielian T, Pincus HA. National trends in the outpatient treatment of depression. JAMA 2002; 287:203–209.
- Marcus SC, Olfson M. National trends in the treatment of depression from 1998 to 2007. Arch Gen Psychiatry 2010; 67:1265–1273.
- Berkman LF, Blumenthal J, Burg M, et al Effects of treating depression and low perceived social support on clinical events after myocardial infarction: the Enhancing Recovery in Coronary Heart Disease Patients (ENRICHD) Randomized Trial. JAMA 2003; 289:3106–3116.
- Taylor CB, Youngblood ME, Catellier D, et al Effects of antidepressant medication on morbidity and mortality in depressed patients after myocardial infarction. Arch Gen Psychiatry 2005; 62:792–798.
- Glassman AH, O’Connor CM, Califf RM, et al Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709.
- Serebruany VL, Glassman AH, Malinin AI, et al Platelet/endothelial biomarkers in depressed patients treated with the selective serotonin reuptake inhibitor sertraline after acute coronary events: the Sertraline AntiDepressant Heart Attack Randomized Trial (SADHART) Platelet Substudy. Circulation 2003; 108:939–44.
- O’Connor CM, Jiang W, Kuchibhatla M, et al Safety and efficacy of sertraline for depression in patients with heart failure: results of the SADHART-CHF (Sertraline Against Depression and Heart Disease in Chronic Heart Failure) trial. J Am Coll Cardiol 2010; 56:692–699.
- Carney RM, Blumenthal JA, Freedland KE, et al Depression and late mortality after myocardial infarction in the Enhancing Recovery in Coronary Heart Disease (ENRICHD) study. Psychosom Med 2004; 66:466–474.
- Lespérance F, Frasure-Smith N, Koszycki D, et al Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) Trial. JAMA 2007; 297:367–379.
- Salzman C, Schneider L, Alexopoulos GS. Pharmacological treatment of depression in late life. In:Bloon F, Kupfer D, eds. Psychopharmacology: Fourth Generation of Progress. New York: Raven Press; 1995.
- Franklin BA, Bonzheim K, Gordon S, Timmis GC. Safety of medically supervised outpatient cardiac rehabilitation exercise therapy: a 16-year follow-up. Chest 1998; 114:902–906.
- Vongvanich P, Paul-Labrador MJ, Merz CN. Safety of medically supervised exercise in a cardiac rehabilitation center. Am J Cardiol 1996; 77:1383–1385.
- O’Connor CM, Whellan DJ, Lee KL, et al Efficacy and safety of exercise training in patients with chronic heart failure: H-F ACTION randomized controlled trial. JAMA 2009; 301:1439–1540.
- Milani RV, Lavie CJ, Cassidy MM. Effects of cardiac rehabilitation and exercise training programs on depression in patients after major coronary events. Am Heart J 1996; 132:726–732.
- Beniamini Y, Rubenstein JJ, Zaichkowsky LD, Crim MC. Effects of high-intensity strength training on quality-of-life parameters in cardiac rehabilitation patients. Am J Cardiol 1997; 80:841–846.
- Maines TY, Lavie CJ, Milani RV, Cassidy MM, Gilliland YE, Murgo JP. Effects of cardiac rehabilitation and exercise programs on exercise capacity, coronary risk factors, behavior, and quality of life in patients with coronary artery disease. South Med J 1997; 90:43–49.
- Milani RV, Lavie CJ. Prevalence and effects of cardiac rehabilitation on depression in the elderly with coronary heart disease. Am J Cardiol 1998; 81:1233–1236.
- Blumenthal JA, Emery CF, Rejeski WJ. The effects of exercise training on psychosocial functioning after myocardial infarction. J Cardiopulmonary Rehabil 1988; 8:183–193.
- Taylor CB, Houston-Miller N, Ahn DK, Haskell W, DeBusk RF. The effects of exercise training programs on psychosocial improvement in uncomplicated postmyocardial infarction patients. J Psychosom Res 1986; 30:581–587.
- Stern MJ, Gorman PA, Kaslow L. The group counseling v exercise therapy study: a controlled intervention with subjects following myocardial infarction. Arch Intern Med 1983; 143:1719–1725.
- Brosse AL, Sheets ES, Lett HS, Blumenthal JA. Exercise and the treatment of clinical depression in adults: recent findings and future directions. Sports Med 2002; 32:741–760.
- Lawlor DA, Hopker SW. The effectiveness of exercise as an intervention in the management of depression: systematic review and meta-regression analysis of randomised controlled trials. BMJ 2001; 322:763–767.
- Mead GE, Morley W, Campbell P, Greig CA, McMurdo M, Lawlor DA. Exercise for depression. Cochrane Database Syst Rev 2009:CD004366.
- Blumenthal JA, Babyak MA, Carney RM, et al Exercise, depression, and mortality after myocardial infarction in the ENRICHD trial. Med Sci Sports Exerc 2004; 36:746–755.
- Eden J, Wheatley B, McNeil B, Sox H, eds. Institute of Medicine. Knowing What Works in Health Care. A Roadmap for the Nation. Washington DC: National Academics Press; 2009.
- Sox HC, Greenfield S. Comparative effectiveness research: a report from the Institute of Medicine. Ann Intern Med 2009; 151:203–205.
- Hlatky MA, Boothroyd DB, Bravata DM, et al Coronary artery bypass surgery compared with percutaneous coronary interventions for multivessel disease: a collaborative analysis of individual patient data from ten randomised trials. Lancet 2009; 373:1190–1197.
- Blumenthal JA, Babyak MA, Moore KA, et al Effects of exercise training on older patients with major depression. Arch Intern Med 1999; 159:2349–2356.
- Babyak M, Blumenthal JA, Herman S. Exercise treatment for major depression: maintenance of therapeutic benefit at 10 months. Psychosom Med 2000; 62:633–638.
- Blumenthal JA, Babyak MA, Doraiswamy PM, et al Exercise and pharmacotherapy in the treatment of major depressive disorder. Psychosom Med 2007; 69:587–596.
- Hoffman B, Babyak M, Craighead WE, et al Exercise and pharmacotherapy in patients with major depression: one-year follow-up of the SMILE study. Psychosom Med 2010; 73:127–133.
- Dunn AL, Trivedi MH, Kampert JB, Clark CG, Chambliss HO. Exercise treatment for depression: efficacy and dose response. Am J Prev Med 2005; 28:1–8.
- Lichtman JH, Bigger JT, Blumenthal JA, et al Depression and coronary heart disease: recommendations for screening, referral, and treatment: a science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Psychiatric Association. Circulation 2008; 118:1768–1775.
- Blumenthal JA, Califf R, Williams RS, Hindman M. Cardiac rehabilitation: a new frontier for behavioral medicine. J Cardiac Rehabil 1983; 3:637–656.
- Lewis T. Diseases of the Heart. New York: Macmillan; 1933:41–49.
- Levine SA, Lown B. The “chair” treatment of acute thrombosis. Trans Assoc Am Physicians 1951; 64:316–327.
- Cain HD, Frasher WG, Stivelman R. Graded activity program for safe return to self-care after myocardial infarction. JAMA 1961; 177:111–115.
- Hellerstein HK, Ford AB. Rehabilitation of the cardiac patient. JAMA 1957; 14:225–231.
- Naughton J, Bruhn JG, Lategola MT. Effects of physical training on physiologic and behavioral characteristics of cardiac patients. Arch Phys Med Rehabil 1968; 49:131–137.
- O’Connor GT, Buring JE, Yusuf S, et al An overview of randomized trials of rehabilitation with exercise after myocardial infarction. Circulation 1989; 80:234–244.
- Wenger NK, Froelicher ES, Smith LK, et al Cardiac rehabilitation: clinical practice guideline no. 17. Rockville, MD: US Dept of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research, National Heart, Lung, and Blood Institute; October 1995; AHCPR publication 96-0672.
- Blumenthal JA, Emery CF, Cox DR, et al Exercise training in healthy type A middle-aged men: effects on behavioral and cardiovascular responses. Psychosom Med 1988; 50:418–433.
- Schuler G, Schlierf G, Wirth A, et al Low-fat diet and regular, supervised physical exercise in patients with symptomatic coronary artery disease: reduction of stress-induced myocardial ischemia. Circulation 1988; 77:172–181.
- Jiang W, Trauner MA, Coleman RE, et al Association of physical fitness and transient myocardial ischemia in patients with coronary artery disease. J Cardiopulm Rehabil 1995; 15:431–438.
- Blumenthal JA, Jiang W, Babyak MA, et al Stress management and exercise training in cardiac patients with myocardial ischemia: effects on prognosis and evaluation of mechanisms. Arch Int Med 1997; 157:2213–2223.
- Oldridge NB, Guyatt GH, Fisher ME, Rimm AA. Cardiac rehabilitation after myocardial infarction: combined experience of randomized clinical trials. JAMA 1988; 260:945–950.
- Jolliffe JA, Rees K, Taylor RS, Thompson D, Oldridge N, Ebrahim S. Exercise-based rehabilitation for coronary heart disease. Cochrane Database Syst Rev 2001:CD001800.
- Folkins CH, Sime WE. Physical fitness training and mental health. Am Psychol 1981; 36:373–389.
- Hughes JR. Psychological effects of habitual aerobic exercise: a critical review. Prev Med 1984; 13:66–78.
- Plante TG, Rodin J. Physical fitness and enhanced psychological health. Curr Psychol: Res Rev 1990; 9:3–24.
- Review Panel on Coronary-Prone Behavior and Coronary Heart Disease. Coronary-prone behavior and coronary heart disease: a critical review. Circulation 1981; 63:1199–1215.
- Smith TW. Hostility and health: current status of a psychosomatic hypothesis. Health Psychol 1992; 11:139–150.
- Lett HS, Blumenthal JA, Babyak MA, Strauman TJ, Robins C, Sherwood A. Social support and coronary heart disease: epidemiologic evidence and implications for treatment. Psychosom Med 2005; 67:869–878.
- Lett HS, Blumenthal JA, Babyak MA, et al Depression as a risk factor for coronary artery disease: evidence, mechanisms, and treatment. Psychosom Med 2004; 66:305–315.
- Barth J, Schumacher M, Herrmann-Lingen C. Depression as a risk factor for mortality in patients with coronary heart disease: a meta-analysis. Psychosom Med 2004; 66:802–813.
- Kessler RC, Berglund P, Demler O, et al The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCR-R). JAMA 2003; 289:3095–3105.
- Belsher G, Costello CG. Relapse after recovery from unipolar depression: a critical review. Psychol Bull 1988; 104:84–96.
- Strothers HS, Rust G, Minor P, Fresh E, Druss B, Satcher D. Disparities in antidepressant treatment in Medicaid elderly diagnosed with depression. J Am Geriatr Soc 2005; 53:456–461.
- Simpson SM, Krishnan LL, Kunik ME, Ruiz P. Racial disparities in diagnosis and treatment of depression: a literature review. Psychiatr Q 2007; 78:3–14.
- Sclar DA, Robison LM, Skaer TL. Ethnicity/race and the diagnosis of depression and use of antidepressants by adults in the United States. Int Clin Psychopharmacol 2008; 23:106–109.
- Waldman SV, Blumenthal JA, Babyak MA, et al Ethnic differences in the treatment of depression in patients with ischemic heart disease. Am Heart J 2009; 57:77–83.
- Carney RM, Rich MW, Freedland KE, et al Major depressive disorder predicts cardiac events in patients with coronary artery disease. Psychosom Med 1988; 50:627–633.
- Schleifer SJ, Macari-Hinson MM, Coyle DA, et al The nature and course of depression following myocardial infarction. Arch Intern Med 1989; 149:1785–1789.
- Frasure-Smith N, Lespérance F, Talajic M. Depression following myocardial infarction: impact on 6-month survival. JAMA 1993; 270:1819–1825.
- Gonzalez MB, Snyderman TB, Colket JT, et al Depression in patients with coronary artery disease. Depression 1996; 4:57–62.
- Connerney I, Shapiro PA, McLaughlin JS, Bagiella E, Sloan RP. Relation between depression after coronary artery bypass surgery and 12-month outcome: a prospective study. Lancet 2001; 358:1766–1771.
- Burker EJ, Blumenthal JA, Feldman M, et al Depression in male and female patients undergoing cardiac surgery. Br J Clin Psychol 1995; 34( Pt 1):119–128.
- Lespérance F, Frasure-Smith N, Juneau M, Théroux P. Depression and 1-year prognosis in unstable angina. Arch Intern Med 2000; 160:1354–1360.
- Frasure-Smith N, Lespérance F, Talajic M. Depression and 18-month prognosis after myocardial infarction. Circulation 1995; 91:999–1005.
- Barefoot JC, Helms MJ, Mark DB, et al Depression and long-term mortality risk in patients with coronary artery disease. Am J Cardiol 1996; 78:613–617.
- Burg MM, Benedetto C, Soufer R. Depressive symptoms and mortality two years after coronary artery bypass graft surgery (CABG) in men. Psychosom Med 2003; 65:508–510.
- Blumenthal JA, Lett HS, Babyak MA, et al Depression as a risk factor for mortality after coronary artery bypass surgery. Lancet 2003; 362:604–609.
- Connerney I, Sloan RP, Shapiro PA, Bagiella E, Seckman C. Depression is associated with increased mortality 10 years after coronary artery bypass surgery. Psychosom Med 2010; 72:874–881.
- Rutledge T, Reis VA, Linke SE, Greenberg BH, Mills PJ. Depression in heart failure: a meta analytic review of prevalence, intervention effects, and associations with clinical outcomes. J Am Coll Cardiol 2006; 48:1527–1537.
- Murberg TA, Furze G. Depressive symptoms and mortality in patients with congestive heart failure: a six-year follow-up study. Med Sci Monit 2004; 10:CR643–CR648.
- Jiang W, Alexander J, Christopher E, et al Relationship of depression to increased risk of mortality and rehospitalization in patients with congestive heart failure. Arch Intern Med 2001; 161:1849–1856.
- Sherwood A, Blumenthal JA, Trivedi R, et al Relationship of depression to death or hospitalization in patients with heart failure. Arch Int Med 2007; 167:367–373.
- Frasure-Smith N, Lespérance F, Habra M, et al Elevated depression symptoms predict long-term cardiovascular mortality in patients with atrial fibrillation and heart failure. Circulation 2009; 120:134–140.
- Sherwood A, Blumenthal JA, Hinderliter AL, et al Worsening depressive symptoms are associated with adverse clinical outcomes in patients with heart failure. J Am Coll Cardiol 2011; 57:418–423.
- Anderson RJ, Freedland KE, Clouse RE, Lustman PJ. The prevalence of comorbid depression in adults with diabetes: a meta-analysis. Diabetes Care 2001; 24:1069–1078.
- Thakore JH, Richards PJ, Reznek RH, Martin A, Dinan TG. Increased intra-abdominal fat deposition in patients with major depressive illness as measured by computed tomography. Biol Psychiatry 1997; 41:1140–1142.
- Musselman DL, Tomer A, Manatunga AK, et al Exaggerated platelet reactivity in major depression. Am J Psychiatry 1996; 153:1313–1317.
- Delgado PL, Moreno FA. Role of norepinephrine in depression. J Clin Psychiatry 2000; 61( suppl 1):5–12.
- Akil H, Haskett RF, Young EA, et al Multiple HPA profiles in endogenous depression: effect of age and sex on cortisol and beta-endorphin. Biol Psychiatry 1993; 33:73–85.
- Kop WJ, Gottdiener JS, Tangen CM, et al Inflammation and coagulation factors in persons > 65 years of age with symptoms of depression but without evidence of myocardial ischemia. Am J Cardiol 2002; 89:419–424.
- Carney RM, Freedland KE, Eisen SA, Rich MW, Jaffe AS. Major depression and medication adherence in elderly patients with coronary artery disease. Health Psychol 1995; 14:88–90.
- Lehto S, Koukkunen H, Hintikka J, Viinamäki H, Laakso M, Pyörälä K. Depression after coronary heart disease events. Scand Cardiovasc J 2000; 34:580–583.
- Camacho TC, Roberts RE, Lazarus NB, Kaplan GA, Cohen RD. Physical activity and depression: evidence from the Alameda County Study. Am J Epidemiol 1991; 134:220–231.
- Clinical Practice Guideline Number 5: Depression in Primary Care, 2: Treatment of Major Depression. Rockville, MD: U.S. Department of Health and Human Services, Agency for Health Care Policy and Research; 1993. AHCPR publication 93-0551.
- Anderson IM, Ferrier IN, Baldwin RC, et al Evidence-based guidelines for treating depressive disorders with antidepressants: a revision of the 2000 British Association for Psychopharmacology guidelines. J Psychopharmacol 2008; 22:343–396.
- American Psyciatric Association. Practice guideline for the treatment of patients with major depressive disorder (revision). Am J Psychiatry 2000; 157( suppl 4):1–45.
- Kirsch I, Deacon BJ, Huedo-Medina TB, Scoboria A, Moore TJ, Johnson BT. Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration. PLoS Med 2008; 5:e45.
- Hansen R, Gaynes B, Thieda P, et al Meta-analysis of major depressive disorder relapse and recurrence with second-generation antidepressants. Psychiatr Serv 2008; 59:1121–1129.
- Hansen RA, Gartlehner G, Lohr KN, Gaynes BN, Carey TS. Efficacy and safety of second-generation antidepressants in the treatment of major depressive disorder. Ann Intern Med 2005; 143:415–426.
- Rush AJ, Trivedi MH, Wisniewski SR, et al Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 2006; 163:1905–1917.
- Olfson M, Marcus SC, Druss B, Elinson L, Tanielian T, Pincus HA. National trends in the outpatient treatment of depression. JAMA 2002; 287:203–209.
- Marcus SC, Olfson M. National trends in the treatment of depression from 1998 to 2007. Arch Gen Psychiatry 2010; 67:1265–1273.
- Berkman LF, Blumenthal J, Burg M, et al Effects of treating depression and low perceived social support on clinical events after myocardial infarction: the Enhancing Recovery in Coronary Heart Disease Patients (ENRICHD) Randomized Trial. JAMA 2003; 289:3106–3116.
- Taylor CB, Youngblood ME, Catellier D, et al Effects of antidepressant medication on morbidity and mortality in depressed patients after myocardial infarction. Arch Gen Psychiatry 2005; 62:792–798.
- Glassman AH, O’Connor CM, Califf RM, et al Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709.
- Serebruany VL, Glassman AH, Malinin AI, et al Platelet/endothelial biomarkers in depressed patients treated with the selective serotonin reuptake inhibitor sertraline after acute coronary events: the Sertraline AntiDepressant Heart Attack Randomized Trial (SADHART) Platelet Substudy. Circulation 2003; 108:939–44.
- O’Connor CM, Jiang W, Kuchibhatla M, et al Safety and efficacy of sertraline for depression in patients with heart failure: results of the SADHART-CHF (Sertraline Against Depression and Heart Disease in Chronic Heart Failure) trial. J Am Coll Cardiol 2010; 56:692–699.
- Carney RM, Blumenthal JA, Freedland KE, et al Depression and late mortality after myocardial infarction in the Enhancing Recovery in Coronary Heart Disease (ENRICHD) study. Psychosom Med 2004; 66:466–474.
- Lespérance F, Frasure-Smith N, Koszycki D, et al Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) Trial. JAMA 2007; 297:367–379.
- Salzman C, Schneider L, Alexopoulos GS. Pharmacological treatment of depression in late life. In:Bloon F, Kupfer D, eds. Psychopharmacology: Fourth Generation of Progress. New York: Raven Press; 1995.
- Franklin BA, Bonzheim K, Gordon S, Timmis GC. Safety of medically supervised outpatient cardiac rehabilitation exercise therapy: a 16-year follow-up. Chest 1998; 114:902–906.
- Vongvanich P, Paul-Labrador MJ, Merz CN. Safety of medically supervised exercise in a cardiac rehabilitation center. Am J Cardiol 1996; 77:1383–1385.
- O’Connor CM, Whellan DJ, Lee KL, et al Efficacy and safety of exercise training in patients with chronic heart failure: H-F ACTION randomized controlled trial. JAMA 2009; 301:1439–1540.
- Milani RV, Lavie CJ, Cassidy MM. Effects of cardiac rehabilitation and exercise training programs on depression in patients after major coronary events. Am Heart J 1996; 132:726–732.
- Beniamini Y, Rubenstein JJ, Zaichkowsky LD, Crim MC. Effects of high-intensity strength training on quality-of-life parameters in cardiac rehabilitation patients. Am J Cardiol 1997; 80:841–846.
- Maines TY, Lavie CJ, Milani RV, Cassidy MM, Gilliland YE, Murgo JP. Effects of cardiac rehabilitation and exercise programs on exercise capacity, coronary risk factors, behavior, and quality of life in patients with coronary artery disease. South Med J 1997; 90:43–49.
- Milani RV, Lavie CJ. Prevalence and effects of cardiac rehabilitation on depression in the elderly with coronary heart disease. Am J Cardiol 1998; 81:1233–1236.
- Blumenthal JA, Emery CF, Rejeski WJ. The effects of exercise training on psychosocial functioning after myocardial infarction. J Cardiopulmonary Rehabil 1988; 8:183–193.
- Taylor CB, Houston-Miller N, Ahn DK, Haskell W, DeBusk RF. The effects of exercise training programs on psychosocial improvement in uncomplicated postmyocardial infarction patients. J Psychosom Res 1986; 30:581–587.
- Stern MJ, Gorman PA, Kaslow L. The group counseling v exercise therapy study: a controlled intervention with subjects following myocardial infarction. Arch Intern Med 1983; 143:1719–1725.
- Brosse AL, Sheets ES, Lett HS, Blumenthal JA. Exercise and the treatment of clinical depression in adults: recent findings and future directions. Sports Med 2002; 32:741–760.
- Lawlor DA, Hopker SW. The effectiveness of exercise as an intervention in the management of depression: systematic review and meta-regression analysis of randomised controlled trials. BMJ 2001; 322:763–767.
- Mead GE, Morley W, Campbell P, Greig CA, McMurdo M, Lawlor DA. Exercise for depression. Cochrane Database Syst Rev 2009:CD004366.
- Blumenthal JA, Babyak MA, Carney RM, et al Exercise, depression, and mortality after myocardial infarction in the ENRICHD trial. Med Sci Sports Exerc 2004; 36:746–755.
- Eden J, Wheatley B, McNeil B, Sox H, eds. Institute of Medicine. Knowing What Works in Health Care. A Roadmap for the Nation. Washington DC: National Academics Press; 2009.
- Sox HC, Greenfield S. Comparative effectiveness research: a report from the Institute of Medicine. Ann Intern Med 2009; 151:203–205.
- Hlatky MA, Boothroyd DB, Bravata DM, et al Coronary artery bypass surgery compared with percutaneous coronary interventions for multivessel disease: a collaborative analysis of individual patient data from ten randomised trials. Lancet 2009; 373:1190–1197.
- Blumenthal JA, Babyak MA, Moore KA, et al Effects of exercise training on older patients with major depression. Arch Intern Med 1999; 159:2349–2356.
- Babyak M, Blumenthal JA, Herman S. Exercise treatment for major depression: maintenance of therapeutic benefit at 10 months. Psychosom Med 2000; 62:633–638.
- Blumenthal JA, Babyak MA, Doraiswamy PM, et al Exercise and pharmacotherapy in the treatment of major depressive disorder. Psychosom Med 2007; 69:587–596.
- Hoffman B, Babyak M, Craighead WE, et al Exercise and pharmacotherapy in patients with major depression: one-year follow-up of the SMILE study. Psychosom Med 2010; 73:127–133.
- Dunn AL, Trivedi MH, Kampert JB, Clark CG, Chambliss HO. Exercise treatment for depression: efficacy and dose response. Am J Prev Med 2005; 28:1–8.
- Lichtman JH, Bigger JT, Blumenthal JA, et al Depression and coronary heart disease: recommendations for screening, referral, and treatment: a science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Psychiatric Association. Circulation 2008; 118:1768–1775.
Depression: A shared risk factor for cardiovascular and Alzheimer disease
The associations among depression, cardiovascular disease, and cognitive impairment are well known. Inflammation is increasingly recognized as playing an important role as well. However, the nature of their relationships and which may actually cause the other is not well understood. This article reviews studies over the past year that link depression with dementia and vascular disease. Desirable directions for future work are also explored.
DEPRESSION AND ALZHEIMER DISEASE
Several studies have shown significant correlations between depression and the risk of developing Alzheimer disease; the frequency of depressive episodes appears to be an important factor. Despite the risk relationship, however, depression and Alzheimer disease may not share a common pathology.
No shared pathology
Wilson and colleagues1 analyzed data from the Chicago Health and Aging Project, a longitudinal cohort study of risk factors for Alzheimer disease that involved two groups of people aged 65 years and older; one group was composed of people who developed dementia during the study, while the other group had already developed dementia or had some degree of cognitive impairment. The investigators reasoned that if pathologic changes are common to depression and dementia, then there would be evidence of change in depressive symptoms along with the progression of dementia. They found only a barely perceptible increase in depressive symptoms among people who developed Alzheimer disease and concluded that there is no shared pathology between depression and Alzheimer disease.
Degree of depression signals risk
Depression has been associated with nearly double the risk of developing dementia and Alzheimer disease. Saczynski et al2 evaluated 949 people in the Framingham study, mean age 79 years, using the 60-point Center for Epidemiologic Studies Depression Scale (depression defined as > 16 points). Individuals who had depression at baseline were 1.7 times more likely to develop dementia over the 17-year evaluation period. Results were similar when adjusted for major vascular risk factors (smoking, diabetes, hypertension, and cardiovascular disease). The correlation was slightly lower but still significant when subjects taking antidepressant medications were included in the depressed group.
The study also found a continuous relationship between the level of depression and the likelihood of developing dementia and Alzheimer disease: for every 10-point increase on the depression scale, the risk of developing dementia increased by nearly 50%. This study supports depression as a risk factor for dementia. One could also argue that depression as a simple prodrome to dementia seems unlikely because of the long followup between baseline assessment and the development of dementia.
Multiple episodes of depression increase risk
Dotson et al3 analyzed data from 1,239 older adults from the Baltimore Longitudinal Study of Aging who did not have depression, dementia, or mild cognitive impairment at baseline. Every 1 to 2 years for about 25 years, cognitive status and mood of the subjects were evaluated. About 10% of the participants developed dementia during the course of the study. Of those who developed dementia, 35% had at least one episode of depression; among those who did not develop dementia, only 23% had a depressive episode. Findings were similar when investigators controlled for vascular risks and vascular dementia.
One episode of depression was associated with an 87% increase in risk of dementia; at least two episodes of depression more than doubled the risk (108%). Overall, each episode of depression conferred an additional 14% risk of developing dementia. Among subjects who had had two or more episodes of depression, the median age of developing dementia was 85 years versus 95 years for those without an episode of depression.
This study had the advantages of being prospective for both depression and dementia and of having a long followup period. A dose-effect relationship was observed, with the “dose” being the number of depressive episodes (rather than severity of depression). Because the definition of a depressive episode included subsyndromal depression (not likely to meet the criteria of clinical depression, but still clinically significant), the findings suggest that even minor depression increases the risk of dementia.
Baseline depression predicts cognitive impairment
Rosenberg et al4 found depression to be associated with cognitive impairment in their evaluation of 436 women in their 70s; the women, who were participants in the Women’s Health and Aging Study, were evaluated for up to 9 years. To be included in the evaluation, subjects needed a Mini-Mental State Examination score of at least 24 points (out of 30 possible) and could not be impaired in more than one basic functional capacity: mobility and exercise tolerance, upper extremity, higher functioning (eg, shopping), and basic self-care). Baseline depressive symptoms were measured using the Geriatric Depression Scale.
Cognitive testing included Hopkins Verbal Learning Tests (for immediate and delayed word recall) and Trail Making Tests (for psychomotor speed and executive functioning). Those who were not impaired (ie, having a cognitive test score below the 10th percentile for age-adjusted norms) were followed with up to six examinations over the next 9 years.
Baseline depression was found to be highly associated with incident impairment in all cognitive areas, especially in executive functioning. For every point increase in the depression scale, a 6% to 7% increase was found in the annual risk of impairment in each cognitive domain.
DEPRESSION AND VASCULAR DISEASE LINKED
It is somewhat easier to assess the relationship between depression and vascular disease than between depression and cognitive impairment because of the availability of objective measures of cardiovascular function.
The International Stroke Study (INTERSTROKE),5 a case-control study in 22 countries with 3,000 cases of stroke and 3,000 age-, gender-, and ethnicity-matched controls, found nine risk factors that were correlated with 90% of ischemic stroke cases. Depression, with an odds ratio of 1.86, was found to be a more significant risk factor than physical activity, diet, or heavy drinking.
Paranthaman et al6 evaluated a number of measures of arterial anatomy and function in 25 subjects with depressive disorder and in 21 nondepressed controls (mean age, 72 years). They found that depressed subjects had significantly higher mean carotid intima media thickness, reduced dilation in response to acetyl choline in preconstricted small arteries, and more dilated Virchow-Robin spaces in the basal ganglia observed on magnetic resonance imaging. This study provides evidence that arterial structure and function may mediate the relationship between depression and vascular disease.
FUTURE DIRECTIONS
Future studies into depression as a risk factor should use very well-characterized cohorts that are controlled for blood pressure and other vascular risk factors. Finding biomarkers for depression would be useful, permitting its detection earlier and with more certainty than is now possible. Prospective studies to evaluate the relationship of depression to cognitive impairment and dementia are needed that start earlier than in middle or old age. The key question that needs study is whether treatment of depression can actually change the onset of cognitive impairment, Alzheimer disease, and vascular disease.
- Wilson RS, Hoganson GM, Rajan KB, Barnes LL, Mendes de Leon CF, Evans DA. Temporal course of depressive symptoms during the development of Alzheimer disease. Neurology 2010; 75:21–26.
- Saczynski JS, Beiser A, Seshadri S, Auerbach S, Wolf PA, Au R. Depressive symptoms and risk of dementia: the Framingham Heart Study. Neurology 2010; 75:35–41.
- Dotson VM, Beydoun MA, Zonderman AB. Recurrent depressive symptoms and the incidence of dementia and mild cognitive impairment. Neurology 2010; 75:27–34.
- Rosenberg PB, Mielke MM, Xue QL, Carlson MC. Depressive symptoms predict incident cognitive impairment in cognitive healthy older women. Am J Geriatr Psychiatry 2010; 18:204–211.
- O’Donnell MJ, Xavier D, Liu L, et al; INTERSTROKE investigators. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): a case-control study. Lancet 2010; 376:112–123. Epub 2010 Jun 17
- Paranthaman R, Greenstein AS, Burns AS, et al Vascular function in older adults with depressive disorder. Biol Psychiatry 2010; 68:133–139.
The associations among depression, cardiovascular disease, and cognitive impairment are well known. Inflammation is increasingly recognized as playing an important role as well. However, the nature of their relationships and which may actually cause the other is not well understood. This article reviews studies over the past year that link depression with dementia and vascular disease. Desirable directions for future work are also explored.
DEPRESSION AND ALZHEIMER DISEASE
Several studies have shown significant correlations between depression and the risk of developing Alzheimer disease; the frequency of depressive episodes appears to be an important factor. Despite the risk relationship, however, depression and Alzheimer disease may not share a common pathology.
No shared pathology
Wilson and colleagues1 analyzed data from the Chicago Health and Aging Project, a longitudinal cohort study of risk factors for Alzheimer disease that involved two groups of people aged 65 years and older; one group was composed of people who developed dementia during the study, while the other group had already developed dementia or had some degree of cognitive impairment. The investigators reasoned that if pathologic changes are common to depression and dementia, then there would be evidence of change in depressive symptoms along with the progression of dementia. They found only a barely perceptible increase in depressive symptoms among people who developed Alzheimer disease and concluded that there is no shared pathology between depression and Alzheimer disease.
Degree of depression signals risk
Depression has been associated with nearly double the risk of developing dementia and Alzheimer disease. Saczynski et al2 evaluated 949 people in the Framingham study, mean age 79 years, using the 60-point Center for Epidemiologic Studies Depression Scale (depression defined as > 16 points). Individuals who had depression at baseline were 1.7 times more likely to develop dementia over the 17-year evaluation period. Results were similar when adjusted for major vascular risk factors (smoking, diabetes, hypertension, and cardiovascular disease). The correlation was slightly lower but still significant when subjects taking antidepressant medications were included in the depressed group.
The study also found a continuous relationship between the level of depression and the likelihood of developing dementia and Alzheimer disease: for every 10-point increase on the depression scale, the risk of developing dementia increased by nearly 50%. This study supports depression as a risk factor for dementia. One could also argue that depression as a simple prodrome to dementia seems unlikely because of the long followup between baseline assessment and the development of dementia.
Multiple episodes of depression increase risk
Dotson et al3 analyzed data from 1,239 older adults from the Baltimore Longitudinal Study of Aging who did not have depression, dementia, or mild cognitive impairment at baseline. Every 1 to 2 years for about 25 years, cognitive status and mood of the subjects were evaluated. About 10% of the participants developed dementia during the course of the study. Of those who developed dementia, 35% had at least one episode of depression; among those who did not develop dementia, only 23% had a depressive episode. Findings were similar when investigators controlled for vascular risks and vascular dementia.
One episode of depression was associated with an 87% increase in risk of dementia; at least two episodes of depression more than doubled the risk (108%). Overall, each episode of depression conferred an additional 14% risk of developing dementia. Among subjects who had had two or more episodes of depression, the median age of developing dementia was 85 years versus 95 years for those without an episode of depression.
This study had the advantages of being prospective for both depression and dementia and of having a long followup period. A dose-effect relationship was observed, with the “dose” being the number of depressive episodes (rather than severity of depression). Because the definition of a depressive episode included subsyndromal depression (not likely to meet the criteria of clinical depression, but still clinically significant), the findings suggest that even minor depression increases the risk of dementia.
Baseline depression predicts cognitive impairment
Rosenberg et al4 found depression to be associated with cognitive impairment in their evaluation of 436 women in their 70s; the women, who were participants in the Women’s Health and Aging Study, were evaluated for up to 9 years. To be included in the evaluation, subjects needed a Mini-Mental State Examination score of at least 24 points (out of 30 possible) and could not be impaired in more than one basic functional capacity: mobility and exercise tolerance, upper extremity, higher functioning (eg, shopping), and basic self-care). Baseline depressive symptoms were measured using the Geriatric Depression Scale.
Cognitive testing included Hopkins Verbal Learning Tests (for immediate and delayed word recall) and Trail Making Tests (for psychomotor speed and executive functioning). Those who were not impaired (ie, having a cognitive test score below the 10th percentile for age-adjusted norms) were followed with up to six examinations over the next 9 years.
Baseline depression was found to be highly associated with incident impairment in all cognitive areas, especially in executive functioning. For every point increase in the depression scale, a 6% to 7% increase was found in the annual risk of impairment in each cognitive domain.
DEPRESSION AND VASCULAR DISEASE LINKED
It is somewhat easier to assess the relationship between depression and vascular disease than between depression and cognitive impairment because of the availability of objective measures of cardiovascular function.
The International Stroke Study (INTERSTROKE),5 a case-control study in 22 countries with 3,000 cases of stroke and 3,000 age-, gender-, and ethnicity-matched controls, found nine risk factors that were correlated with 90% of ischemic stroke cases. Depression, with an odds ratio of 1.86, was found to be a more significant risk factor than physical activity, diet, or heavy drinking.
Paranthaman et al6 evaluated a number of measures of arterial anatomy and function in 25 subjects with depressive disorder and in 21 nondepressed controls (mean age, 72 years). They found that depressed subjects had significantly higher mean carotid intima media thickness, reduced dilation in response to acetyl choline in preconstricted small arteries, and more dilated Virchow-Robin spaces in the basal ganglia observed on magnetic resonance imaging. This study provides evidence that arterial structure and function may mediate the relationship between depression and vascular disease.
FUTURE DIRECTIONS
Future studies into depression as a risk factor should use very well-characterized cohorts that are controlled for blood pressure and other vascular risk factors. Finding biomarkers for depression would be useful, permitting its detection earlier and with more certainty than is now possible. Prospective studies to evaluate the relationship of depression to cognitive impairment and dementia are needed that start earlier than in middle or old age. The key question that needs study is whether treatment of depression can actually change the onset of cognitive impairment, Alzheimer disease, and vascular disease.
The associations among depression, cardiovascular disease, and cognitive impairment are well known. Inflammation is increasingly recognized as playing an important role as well. However, the nature of their relationships and which may actually cause the other is not well understood. This article reviews studies over the past year that link depression with dementia and vascular disease. Desirable directions for future work are also explored.
DEPRESSION AND ALZHEIMER DISEASE
Several studies have shown significant correlations between depression and the risk of developing Alzheimer disease; the frequency of depressive episodes appears to be an important factor. Despite the risk relationship, however, depression and Alzheimer disease may not share a common pathology.
No shared pathology
Wilson and colleagues1 analyzed data from the Chicago Health and Aging Project, a longitudinal cohort study of risk factors for Alzheimer disease that involved two groups of people aged 65 years and older; one group was composed of people who developed dementia during the study, while the other group had already developed dementia or had some degree of cognitive impairment. The investigators reasoned that if pathologic changes are common to depression and dementia, then there would be evidence of change in depressive symptoms along with the progression of dementia. They found only a barely perceptible increase in depressive symptoms among people who developed Alzheimer disease and concluded that there is no shared pathology between depression and Alzheimer disease.
Degree of depression signals risk
Depression has been associated with nearly double the risk of developing dementia and Alzheimer disease. Saczynski et al2 evaluated 949 people in the Framingham study, mean age 79 years, using the 60-point Center for Epidemiologic Studies Depression Scale (depression defined as > 16 points). Individuals who had depression at baseline were 1.7 times more likely to develop dementia over the 17-year evaluation period. Results were similar when adjusted for major vascular risk factors (smoking, diabetes, hypertension, and cardiovascular disease). The correlation was slightly lower but still significant when subjects taking antidepressant medications were included in the depressed group.
The study also found a continuous relationship between the level of depression and the likelihood of developing dementia and Alzheimer disease: for every 10-point increase on the depression scale, the risk of developing dementia increased by nearly 50%. This study supports depression as a risk factor for dementia. One could also argue that depression as a simple prodrome to dementia seems unlikely because of the long followup between baseline assessment and the development of dementia.
Multiple episodes of depression increase risk
Dotson et al3 analyzed data from 1,239 older adults from the Baltimore Longitudinal Study of Aging who did not have depression, dementia, or mild cognitive impairment at baseline. Every 1 to 2 years for about 25 years, cognitive status and mood of the subjects were evaluated. About 10% of the participants developed dementia during the course of the study. Of those who developed dementia, 35% had at least one episode of depression; among those who did not develop dementia, only 23% had a depressive episode. Findings were similar when investigators controlled for vascular risks and vascular dementia.
One episode of depression was associated with an 87% increase in risk of dementia; at least two episodes of depression more than doubled the risk (108%). Overall, each episode of depression conferred an additional 14% risk of developing dementia. Among subjects who had had two or more episodes of depression, the median age of developing dementia was 85 years versus 95 years for those without an episode of depression.
This study had the advantages of being prospective for both depression and dementia and of having a long followup period. A dose-effect relationship was observed, with the “dose” being the number of depressive episodes (rather than severity of depression). Because the definition of a depressive episode included subsyndromal depression (not likely to meet the criteria of clinical depression, but still clinically significant), the findings suggest that even minor depression increases the risk of dementia.
Baseline depression predicts cognitive impairment
Rosenberg et al4 found depression to be associated with cognitive impairment in their evaluation of 436 women in their 70s; the women, who were participants in the Women’s Health and Aging Study, were evaluated for up to 9 years. To be included in the evaluation, subjects needed a Mini-Mental State Examination score of at least 24 points (out of 30 possible) and could not be impaired in more than one basic functional capacity: mobility and exercise tolerance, upper extremity, higher functioning (eg, shopping), and basic self-care). Baseline depressive symptoms were measured using the Geriatric Depression Scale.
Cognitive testing included Hopkins Verbal Learning Tests (for immediate and delayed word recall) and Trail Making Tests (for psychomotor speed and executive functioning). Those who were not impaired (ie, having a cognitive test score below the 10th percentile for age-adjusted norms) were followed with up to six examinations over the next 9 years.
Baseline depression was found to be highly associated with incident impairment in all cognitive areas, especially in executive functioning. For every point increase in the depression scale, a 6% to 7% increase was found in the annual risk of impairment in each cognitive domain.
DEPRESSION AND VASCULAR DISEASE LINKED
It is somewhat easier to assess the relationship between depression and vascular disease than between depression and cognitive impairment because of the availability of objective measures of cardiovascular function.
The International Stroke Study (INTERSTROKE),5 a case-control study in 22 countries with 3,000 cases of stroke and 3,000 age-, gender-, and ethnicity-matched controls, found nine risk factors that were correlated with 90% of ischemic stroke cases. Depression, with an odds ratio of 1.86, was found to be a more significant risk factor than physical activity, diet, or heavy drinking.
Paranthaman et al6 evaluated a number of measures of arterial anatomy and function in 25 subjects with depressive disorder and in 21 nondepressed controls (mean age, 72 years). They found that depressed subjects had significantly higher mean carotid intima media thickness, reduced dilation in response to acetyl choline in preconstricted small arteries, and more dilated Virchow-Robin spaces in the basal ganglia observed on magnetic resonance imaging. This study provides evidence that arterial structure and function may mediate the relationship between depression and vascular disease.
FUTURE DIRECTIONS
Future studies into depression as a risk factor should use very well-characterized cohorts that are controlled for blood pressure and other vascular risk factors. Finding biomarkers for depression would be useful, permitting its detection earlier and with more certainty than is now possible. Prospective studies to evaluate the relationship of depression to cognitive impairment and dementia are needed that start earlier than in middle or old age. The key question that needs study is whether treatment of depression can actually change the onset of cognitive impairment, Alzheimer disease, and vascular disease.
- Wilson RS, Hoganson GM, Rajan KB, Barnes LL, Mendes de Leon CF, Evans DA. Temporal course of depressive symptoms during the development of Alzheimer disease. Neurology 2010; 75:21–26.
- Saczynski JS, Beiser A, Seshadri S, Auerbach S, Wolf PA, Au R. Depressive symptoms and risk of dementia: the Framingham Heart Study. Neurology 2010; 75:35–41.
- Dotson VM, Beydoun MA, Zonderman AB. Recurrent depressive symptoms and the incidence of dementia and mild cognitive impairment. Neurology 2010; 75:27–34.
- Rosenberg PB, Mielke MM, Xue QL, Carlson MC. Depressive symptoms predict incident cognitive impairment in cognitive healthy older women. Am J Geriatr Psychiatry 2010; 18:204–211.
- O’Donnell MJ, Xavier D, Liu L, et al; INTERSTROKE investigators. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): a case-control study. Lancet 2010; 376:112–123. Epub 2010 Jun 17
- Paranthaman R, Greenstein AS, Burns AS, et al Vascular function in older adults with depressive disorder. Biol Psychiatry 2010; 68:133–139.
- Wilson RS, Hoganson GM, Rajan KB, Barnes LL, Mendes de Leon CF, Evans DA. Temporal course of depressive symptoms during the development of Alzheimer disease. Neurology 2010; 75:21–26.
- Saczynski JS, Beiser A, Seshadri S, Auerbach S, Wolf PA, Au R. Depressive symptoms and risk of dementia: the Framingham Heart Study. Neurology 2010; 75:35–41.
- Dotson VM, Beydoun MA, Zonderman AB. Recurrent depressive symptoms and the incidence of dementia and mild cognitive impairment. Neurology 2010; 75:27–34.
- Rosenberg PB, Mielke MM, Xue QL, Carlson MC. Depressive symptoms predict incident cognitive impairment in cognitive healthy older women. Am J Geriatr Psychiatry 2010; 18:204–211.
- O’Donnell MJ, Xavier D, Liu L, et al; INTERSTROKE investigators. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): a case-control study. Lancet 2010; 376:112–123. Epub 2010 Jun 17
- Paranthaman R, Greenstein AS, Burns AS, et al Vascular function in older adults with depressive disorder. Biol Psychiatry 2010; 68:133–139.
Inflammatory signaling in Alzheimer disease and depression
The relationships among inflammation, Alzheimer disease, and depression have been the subject of recent research at several centers. Alzheimer disease and depression appear to be linked by several genetic and inflammatory processes, although the exact nature of the relationship is not clearly understood. The two disorders also share risk factors for vascular disease. This article reviews the current state of knowledge about inflammation and its implications for Alzheimer disease and depression, and it presents recent findings from the Texas Alzheimer’s Research Consortium, which assessed an array of inflammatory markers in a cohort of patients with Alzheimer disease.
INFLAMMATION MAY MEDIATE DEPRESSION, COGNITIVE DECLINE, AND DEMENTIA
Alzheimer disease and depression share several vascular disease risk factors and appear to be linked through complex and integrated processes. The link may be mediated by long-term inflammatory processes. Hypothalamic-pituitary-adrenal (HPA) axis dysfunction, chronic inflammation, and a deficit in neurotrophin signaling all may play roles in the pathogenesis of depression and Alzheimer disease.1 Excessive release of glucocorticoids subsequent to HPA-axis dysfunction in chronic depression appears to damage the hippocampus: hippocampal atrophy is a feature in both depression and dementia, and recurrent depression is associated with greater atrophy. The direction of influence—whether depression leads to the factors that increase the risk of Alzheimer disease or the other way around—remains a controversial topic.
Symptoms of depression tend to appear early in Alzheimer disease and increase as dementia progresses to moderate severity. In advanced dementia, depression symptoms tend to decline, although this may reflect the difficulty in assessing depression at advanced stages of dementia.2
Numerous reports have linked inflammation to cognitive dysfunction or decline, as well as to the development of Alzheimer disease.3–5 Evidence suggests that inflammation is a key mediator between cardiovascular risk factors and Alzheimer disease, although this is also still controversial.
FINDINGS FROM THE TEXAS ALZHEIMER’S RESEARCH CONSORTIUM
The Texas Alzheimer’s Research Consortium, composed of five medical centers, is pursuing a longitudinal, multi-institutional study of Alzheimer disease. The group recently published the results of a study assessing whether inflammatory markers were over- or underexpressed in patients with Alzheimer disease, and whether biomarkers could predict Alzheimer disease status and the age at onset of the disease.4 The analysis included 197 patients with Alzheimer disease and 203 control subjects. The evaluation consisted of cognitive assessment, DNA analysis for human genome-wide association studies, and protein microarray analysis from blood. Cardiovascular risk factors were also measured, including serum lipids and blood factors for diabetes risk. The goal was to better understand the pathophysiology of cognitive decline and predict conversion of mild cognitive impairment to Alzheimer disease.
Significant differences were found in the study groups. For example, the median age in the Alzheimer group was significantly higher than in controls (79 vs 70 years, P < .0001), an issue that is being addressed as subjects are replaced due to attrition. The median educational level was higher in the control group (14 vs 16 years, P < .0001) than in the Alzheimer group. Subjects in the Alzheimer group were significantly more likely (P < .001) to carry at least one copy of the APOE ε4 allele.
Inflammation is associated with Alzheimer disease
Degree of inflammation also correlated with Mini-Mental State Examination (MMSE) scores. Subjects with a high inflammatory score had a more accelerated decline in MMSE scores over a 3-year period than those with a low inflammatory score. The association was significant, although not as dramatic as the association between inflammation and age at onset of Alzheimer disease.
The investigators concluded that their findings, while considered preliminary, suggest the existence of an inflammatory endophenotype associated with Alzheimer disease. The findings need to be validated in other populations, including ethnic groups other than Caucasian. The Consortium also will evaluate whether inflammatory biomarkers are associated with progression of mild cognitive impairment to Alzheimer disease.
Inflammation has a mixed association with depression
In a study whose results are not yet published, the Texas Consortium also examined the association between inflammatory markers and depression. Four subscales of depression were used, derived from the Geriatric Depression Scale (GDS) 30: dysphoria (consisting of 11 items), meaninglessness (seven items), apathy and withdrawal (six items), and cognitive impairment (six items).5
The GDS30 results as a whole suggested a trend toward an association between depression and inflammatory biomarkers, but the association was not significant. When the results were examined by subscale, however, striking differences were found between Alzheimer patients and the control group. For example, apathy was significantly associated with the C-reactive protein level, and the assocation was much stronger in patients with Alzheimer disease than in controls. Further, the association of apathy with C-reactive protein level was more significant in women than in men.
Other associations were found between several of the inflammatory and antiinflammatory cytokines and the various subscales; the relationship between inflammatory factors and depression appears to be complex and often gender-specific.
Inflammation-depression link is suggestive, not linear
Despite the relationships suggested by the data, no simple linear relationship was identified to indicate that more inflammation leads to more depression in Alzheimer disease. The relationship between inflammation and depression in Alzheimer disease appears to involve a complex interplay between many physiologic processes.
The effect of inflammation also varies with gender and with cognitive impairment. The mechanism that underlies these relationships remains to be determined and will be the focus of further studies with the Texas Alzheimer’s Research Consortium.
- Caraci F, Copani A, Nicoletti F, Drago F. Depression and Alzheimer’s disease: neurobiological links and common pharmacological targets. Eur J Pharmacol 2010; 626:64–71.
- Amore M, Tagariello P, Laterza C, Savoia EM. Subtypes of depression in dementia. Arch Gerontol Geriatr 2007; 44( suppl 1):23–33.
- O’Bryant SE, Xiao G, Barber R, et al., Texas Alzheimer’s Research Consortium. A serum protein-based algorithm for the detection of Alzheimer disease. Arch Neurol 2010; 67:1077–1081.
- Barber R, Xiao G, O’Bryant S, et al., Texas Alzheimer’s Research Consortium. An inflammatory endophenotype of Alzheimer’s disease. Alzheim Dement 2010; 6( suppl):S530.
- Hall JR, Davis TE. Factor structure of the Geriatric Depression Scale in cognitively impaired older adults. Clin Gerontol 2010; 33:39–48.
The relationships among inflammation, Alzheimer disease, and depression have been the subject of recent research at several centers. Alzheimer disease and depression appear to be linked by several genetic and inflammatory processes, although the exact nature of the relationship is not clearly understood. The two disorders also share risk factors for vascular disease. This article reviews the current state of knowledge about inflammation and its implications for Alzheimer disease and depression, and it presents recent findings from the Texas Alzheimer’s Research Consortium, which assessed an array of inflammatory markers in a cohort of patients with Alzheimer disease.
INFLAMMATION MAY MEDIATE DEPRESSION, COGNITIVE DECLINE, AND DEMENTIA
Alzheimer disease and depression share several vascular disease risk factors and appear to be linked through complex and integrated processes. The link may be mediated by long-term inflammatory processes. Hypothalamic-pituitary-adrenal (HPA) axis dysfunction, chronic inflammation, and a deficit in neurotrophin signaling all may play roles in the pathogenesis of depression and Alzheimer disease.1 Excessive release of glucocorticoids subsequent to HPA-axis dysfunction in chronic depression appears to damage the hippocampus: hippocampal atrophy is a feature in both depression and dementia, and recurrent depression is associated with greater atrophy. The direction of influence—whether depression leads to the factors that increase the risk of Alzheimer disease or the other way around—remains a controversial topic.
Symptoms of depression tend to appear early in Alzheimer disease and increase as dementia progresses to moderate severity. In advanced dementia, depression symptoms tend to decline, although this may reflect the difficulty in assessing depression at advanced stages of dementia.2
Numerous reports have linked inflammation to cognitive dysfunction or decline, as well as to the development of Alzheimer disease.3–5 Evidence suggests that inflammation is a key mediator between cardiovascular risk factors and Alzheimer disease, although this is also still controversial.
FINDINGS FROM THE TEXAS ALZHEIMER’S RESEARCH CONSORTIUM
The Texas Alzheimer’s Research Consortium, composed of five medical centers, is pursuing a longitudinal, multi-institutional study of Alzheimer disease. The group recently published the results of a study assessing whether inflammatory markers were over- or underexpressed in patients with Alzheimer disease, and whether biomarkers could predict Alzheimer disease status and the age at onset of the disease.4 The analysis included 197 patients with Alzheimer disease and 203 control subjects. The evaluation consisted of cognitive assessment, DNA analysis for human genome-wide association studies, and protein microarray analysis from blood. Cardiovascular risk factors were also measured, including serum lipids and blood factors for diabetes risk. The goal was to better understand the pathophysiology of cognitive decline and predict conversion of mild cognitive impairment to Alzheimer disease.
Significant differences were found in the study groups. For example, the median age in the Alzheimer group was significantly higher than in controls (79 vs 70 years, P < .0001), an issue that is being addressed as subjects are replaced due to attrition. The median educational level was higher in the control group (14 vs 16 years, P < .0001) than in the Alzheimer group. Subjects in the Alzheimer group were significantly more likely (P < .001) to carry at least one copy of the APOE ε4 allele.
Inflammation is associated with Alzheimer disease
Degree of inflammation also correlated with Mini-Mental State Examination (MMSE) scores. Subjects with a high inflammatory score had a more accelerated decline in MMSE scores over a 3-year period than those with a low inflammatory score. The association was significant, although not as dramatic as the association between inflammation and age at onset of Alzheimer disease.
The investigators concluded that their findings, while considered preliminary, suggest the existence of an inflammatory endophenotype associated with Alzheimer disease. The findings need to be validated in other populations, including ethnic groups other than Caucasian. The Consortium also will evaluate whether inflammatory biomarkers are associated with progression of mild cognitive impairment to Alzheimer disease.
Inflammation has a mixed association with depression
In a study whose results are not yet published, the Texas Consortium also examined the association between inflammatory markers and depression. Four subscales of depression were used, derived from the Geriatric Depression Scale (GDS) 30: dysphoria (consisting of 11 items), meaninglessness (seven items), apathy and withdrawal (six items), and cognitive impairment (six items).5
The GDS30 results as a whole suggested a trend toward an association between depression and inflammatory biomarkers, but the association was not significant. When the results were examined by subscale, however, striking differences were found between Alzheimer patients and the control group. For example, apathy was significantly associated with the C-reactive protein level, and the assocation was much stronger in patients with Alzheimer disease than in controls. Further, the association of apathy with C-reactive protein level was more significant in women than in men.
Other associations were found between several of the inflammatory and antiinflammatory cytokines and the various subscales; the relationship between inflammatory factors and depression appears to be complex and often gender-specific.
Inflammation-depression link is suggestive, not linear
Despite the relationships suggested by the data, no simple linear relationship was identified to indicate that more inflammation leads to more depression in Alzheimer disease. The relationship between inflammation and depression in Alzheimer disease appears to involve a complex interplay between many physiologic processes.
The effect of inflammation also varies with gender and with cognitive impairment. The mechanism that underlies these relationships remains to be determined and will be the focus of further studies with the Texas Alzheimer’s Research Consortium.
The relationships among inflammation, Alzheimer disease, and depression have been the subject of recent research at several centers. Alzheimer disease and depression appear to be linked by several genetic and inflammatory processes, although the exact nature of the relationship is not clearly understood. The two disorders also share risk factors for vascular disease. This article reviews the current state of knowledge about inflammation and its implications for Alzheimer disease and depression, and it presents recent findings from the Texas Alzheimer’s Research Consortium, which assessed an array of inflammatory markers in a cohort of patients with Alzheimer disease.
INFLAMMATION MAY MEDIATE DEPRESSION, COGNITIVE DECLINE, AND DEMENTIA
Alzheimer disease and depression share several vascular disease risk factors and appear to be linked through complex and integrated processes. The link may be mediated by long-term inflammatory processes. Hypothalamic-pituitary-adrenal (HPA) axis dysfunction, chronic inflammation, and a deficit in neurotrophin signaling all may play roles in the pathogenesis of depression and Alzheimer disease.1 Excessive release of glucocorticoids subsequent to HPA-axis dysfunction in chronic depression appears to damage the hippocampus: hippocampal atrophy is a feature in both depression and dementia, and recurrent depression is associated with greater atrophy. The direction of influence—whether depression leads to the factors that increase the risk of Alzheimer disease or the other way around—remains a controversial topic.
Symptoms of depression tend to appear early in Alzheimer disease and increase as dementia progresses to moderate severity. In advanced dementia, depression symptoms tend to decline, although this may reflect the difficulty in assessing depression at advanced stages of dementia.2
Numerous reports have linked inflammation to cognitive dysfunction or decline, as well as to the development of Alzheimer disease.3–5 Evidence suggests that inflammation is a key mediator between cardiovascular risk factors and Alzheimer disease, although this is also still controversial.
FINDINGS FROM THE TEXAS ALZHEIMER’S RESEARCH CONSORTIUM
The Texas Alzheimer’s Research Consortium, composed of five medical centers, is pursuing a longitudinal, multi-institutional study of Alzheimer disease. The group recently published the results of a study assessing whether inflammatory markers were over- or underexpressed in patients with Alzheimer disease, and whether biomarkers could predict Alzheimer disease status and the age at onset of the disease.4 The analysis included 197 patients with Alzheimer disease and 203 control subjects. The evaluation consisted of cognitive assessment, DNA analysis for human genome-wide association studies, and protein microarray analysis from blood. Cardiovascular risk factors were also measured, including serum lipids and blood factors for diabetes risk. The goal was to better understand the pathophysiology of cognitive decline and predict conversion of mild cognitive impairment to Alzheimer disease.
Significant differences were found in the study groups. For example, the median age in the Alzheimer group was significantly higher than in controls (79 vs 70 years, P < .0001), an issue that is being addressed as subjects are replaced due to attrition. The median educational level was higher in the control group (14 vs 16 years, P < .0001) than in the Alzheimer group. Subjects in the Alzheimer group were significantly more likely (P < .001) to carry at least one copy of the APOE ε4 allele.
Inflammation is associated with Alzheimer disease
Degree of inflammation also correlated with Mini-Mental State Examination (MMSE) scores. Subjects with a high inflammatory score had a more accelerated decline in MMSE scores over a 3-year period than those with a low inflammatory score. The association was significant, although not as dramatic as the association between inflammation and age at onset of Alzheimer disease.
The investigators concluded that their findings, while considered preliminary, suggest the existence of an inflammatory endophenotype associated with Alzheimer disease. The findings need to be validated in other populations, including ethnic groups other than Caucasian. The Consortium also will evaluate whether inflammatory biomarkers are associated with progression of mild cognitive impairment to Alzheimer disease.
Inflammation has a mixed association with depression
In a study whose results are not yet published, the Texas Consortium also examined the association between inflammatory markers and depression. Four subscales of depression were used, derived from the Geriatric Depression Scale (GDS) 30: dysphoria (consisting of 11 items), meaninglessness (seven items), apathy and withdrawal (six items), and cognitive impairment (six items).5
The GDS30 results as a whole suggested a trend toward an association between depression and inflammatory biomarkers, but the association was not significant. When the results were examined by subscale, however, striking differences were found between Alzheimer patients and the control group. For example, apathy was significantly associated with the C-reactive protein level, and the assocation was much stronger in patients with Alzheimer disease than in controls. Further, the association of apathy with C-reactive protein level was more significant in women than in men.
Other associations were found between several of the inflammatory and antiinflammatory cytokines and the various subscales; the relationship between inflammatory factors and depression appears to be complex and often gender-specific.
Inflammation-depression link is suggestive, not linear
Despite the relationships suggested by the data, no simple linear relationship was identified to indicate that more inflammation leads to more depression in Alzheimer disease. The relationship between inflammation and depression in Alzheimer disease appears to involve a complex interplay between many physiologic processes.
The effect of inflammation also varies with gender and with cognitive impairment. The mechanism that underlies these relationships remains to be determined and will be the focus of further studies with the Texas Alzheimer’s Research Consortium.
- Caraci F, Copani A, Nicoletti F, Drago F. Depression and Alzheimer’s disease: neurobiological links and common pharmacological targets. Eur J Pharmacol 2010; 626:64–71.
- Amore M, Tagariello P, Laterza C, Savoia EM. Subtypes of depression in dementia. Arch Gerontol Geriatr 2007; 44( suppl 1):23–33.
- O’Bryant SE, Xiao G, Barber R, et al., Texas Alzheimer’s Research Consortium. A serum protein-based algorithm for the detection of Alzheimer disease. Arch Neurol 2010; 67:1077–1081.
- Barber R, Xiao G, O’Bryant S, et al., Texas Alzheimer’s Research Consortium. An inflammatory endophenotype of Alzheimer’s disease. Alzheim Dement 2010; 6( suppl):S530.
- Hall JR, Davis TE. Factor structure of the Geriatric Depression Scale in cognitively impaired older adults. Clin Gerontol 2010; 33:39–48.
- Caraci F, Copani A, Nicoletti F, Drago F. Depression and Alzheimer’s disease: neurobiological links and common pharmacological targets. Eur J Pharmacol 2010; 626:64–71.
- Amore M, Tagariello P, Laterza C, Savoia EM. Subtypes of depression in dementia. Arch Gerontol Geriatr 2007; 44( suppl 1):23–33.
- O’Bryant SE, Xiao G, Barber R, et al., Texas Alzheimer’s Research Consortium. A serum protein-based algorithm for the detection of Alzheimer disease. Arch Neurol 2010; 67:1077–1081.
- Barber R, Xiao G, O’Bryant S, et al., Texas Alzheimer’s Research Consortium. An inflammatory endophenotype of Alzheimer’s disease. Alzheim Dement 2010; 6( suppl):S530.
- Hall JR, Davis TE. Factor structure of the Geriatric Depression Scale in cognitively impaired older adults. Clin Gerontol 2010; 33:39–48.
Vascular signaling abnormalities in Alzheimer disease
Alzheimer disease (AD) is a progressive, irreversible, neurodegenerative disease that affects more than 5.3 million people in the United States.1 This number is significantly higher than the previous estimate of 4.5 million and is projected to increase sharply to nearly 8 million by 2030.1 At present, the few agents that are approved by the US Food and Drug Administration for treatment of AD have demonstrated only modest effects in modifying clinical symptoms for relatively short periods; none has shown a clear effect on disease progression. New therapeutic approaches are desperately needed.
VASCULAR DISEASE AND ALZHEIMER DISEASE
Although AD is classified as a neurodegenerative dementia, there is epidemiologic and pathologic evidence of an association with vascular risk factors and vascular disease.2–6 Vascular disease appears to lower the threshold for the clinical presentation of dementia at a given level of AD-related pathology.7 The possible association of AD with vascular disease suggests that there are important pathogenic mechanisms common to both AD and vascular disease. For example, there is increasing evidence that perturbations in cerebral vascular structure and function occur in AD.8
It has been suggested that cerebral hypoperfusion/hypoxia triggers hypometabolic, cognitive, and degenerative changes in the brain and contributes to the pathologic processes of AD.9 A study by Roher and colleagues reveals an association between severe circle of Willis atherosclerosis and sporadic AD.10 These observations suggest that atherosclerosis-induced brain hypoperfusion contributes to the clinical and pathologic manifestations of AD.
Hypoxia is also known to stimulate angiogenesis, especially via upregulation of hypoxia-inducible genes such as vascular endothelial growth factor (VEGF).11,12 VEGF, a critical mediator of angiogenesis, is present in the AD brain in the walls of intra-parenchymal vessels, in diffuse perivascular deposits, and in clusters of reactive astrocytes.13 In addition, intrathecal levels of VEGF in AD are related to clinical severity and intrathecal levels of amyloid-beta (Aβ).14 Emerging data support the idea that factors and processes characteristic of angiogenesis are found in the AD brain.15,16
ENDOTHELIAL ACTIVATION AND ANGIOGENESIS
The angiogenic process is complex and involves several discrete steps, such as endothelial activation, extracellular matrix degradation, proliferation and migration of endothelial cells, and morphologic differentiation of endothelial cells to form tubes. Stimuli known to initiate angiogenesis, including hypoxia, inflammation, and mechanical factors such as shear stress and stretch,23 either directly or indirectly activate endothelial cells. Activated endothelial cells elaborate adhesion molecules, cytokines and chemokines, growth factors, vasoactive molecules, major histocompatibility complex molecules, procoagulant and anticoagulant moieties, and a variety of other gene products with biologic activity.24 The activated endothelium exerts direct local effects by producing at least 20 paracrine factors that act on adjacent cells.25
ANGIOGENIC SIGNALING MECHANISMS IN BRAIN MICROVESSELS
Signaling mechanisms that have been identified as important to endothelial cell viability and angiogenesis include PI3K/Akt, p38 kinase, ERK, and JNK. In this regard, intracellular Aβ accumulation is toxic to endothelial cells and decreases PI3K/Akt.26 Extracellular Aβ peptides decrease phosphorylation and thus activation of ERK and p38 kinase.26 VEGF promotes endothelial survival, proliferation, and migration through numerous pathways, including activation of ERK, p38 kinase, JNK, and Rho GTPase family members.23
VASCULAR ACTIVATION IN ALZHEIMER DISEASE
Despite increases in several proangiogenic factors in the AD brain, evidence for increased vascularity in AD is lacking. On the contrary, it has been suggested that the angiogenic process is delayed or impaired in aged tissues, with several studies showing decreased microvascular density in the AD brain.30–33 Paris et al showed that wild-type Aβ peptides have antiangiogenic effects in vitro and in vivo.34
How can the data showing antiangiogenic effects of Aβ be reconciled with the presence or expression of a large number of proangiogenic proteins by brain microvessels in AD? These conflicting observations suggest an imbalance between proangiogenic and antiangiogeneic processes in the AD brain.
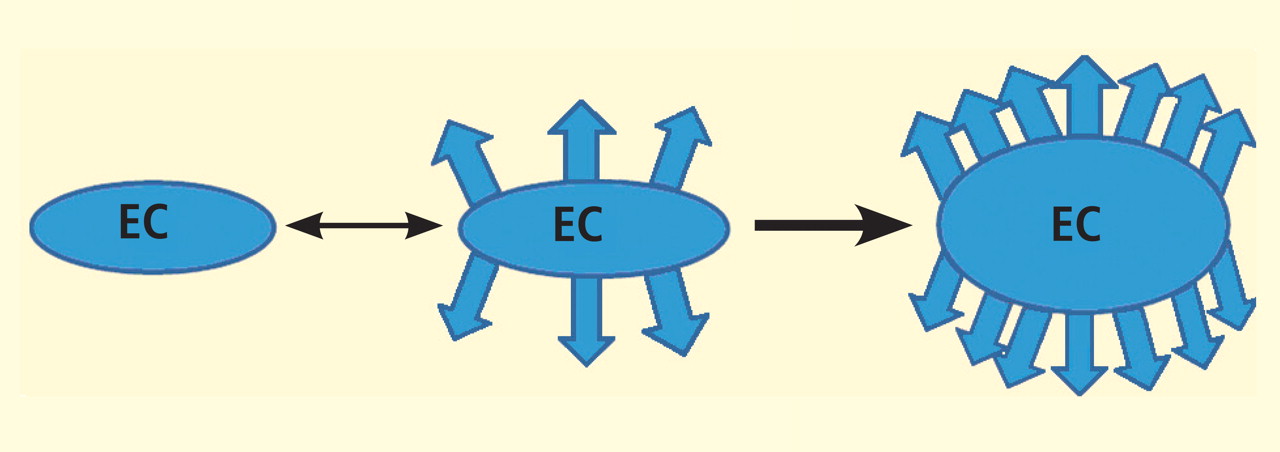
Preliminary experiments in our laboratory show that pharmacologic blockade of vascular activation improves cognitive function in an animal model of AD. Thus, “vascular activation” could be a novel, unexplored therapeutic target in AD.
Acknowledgment
The authors gratefully acknowledge the secretarial assistance of Terri Stahl.
- 2010 Alzheimer’s facts and figures. Alzheimer’s Association Web site. http://www.alz.org/alzheimers_disease_facts_and_figures.asp. Updated January 5, 2011. Accessed February 10, 2011.
- Stewart R, Prince M, Mann A. Vascular risk factors and Alzheimer’s disease. Aust N Z J Psychiatry 1999; 33:809–813.
- Schmidt R, Schmidt H, Fasekas F. Vascular risk factors in dementia. J Neurol 2000; 247:81–87.
- Shi J, Perry G, Smith MA, Friedland RP. Vascular abnormalities: the insidious pathogenesis of Alzheimer’s disease. Neurobiol Aging 2000; 21:357–361.
- Pansari K, Gupta A, Thomas P. Alzheimer’s disease and vascular factors: facts and theories. Int J Clin Pract 2002; 56:197–203.
- de la Torre JC. Alzheimer disease as a vascular disorder: nosological evidence. Stroke 2002; 33:1152–1162.
- Sadowski M, Pankiewicz J, Scholtzova H, et al Links between the pathology of Alzheimer’s disease and vascular dementia. Neurochem Res 2004; 29:1257–1266.
- Grammas P. A damaged microcirculation contributes to neuronal cell death in Alzheimer’s disease. Neurobiol Aging 2000; 21:199–205.
- de la Torre JC, Stefano GB. Evidence that Alzheimer’s disease is a microvascular disorder: the role of constitutive nitric oxide. Brain Res Rev 2000; 34:119–136.
- Roher AE, Esh C, Kokjohn TA, et al Circle of Willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler Thromb Vasc Biol 2003; 23:2055–2062.
- Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 2003; 9:677–684.
- Yamakawa M, Liu LX, Date T, et al Hypoxia-inducible factor-1 mediates activation of cultured vascular endothelial cells by inducing multiple angiogenic factors. Circ Res 2003; 93:664–673.
- Kalaria RN, Cohen DL, Premkumar DR, Nag S, LaManna JC, Lust WD. Vascular endothelial growth factor in Alzheimer’s disease and experimental ischemia. Brain Res Mol Brain Res 1998; 62:101–105.
- Tarkowski E, Issa R, Sjogren M, et al Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer’s disease and vascular dementia. Neurobiol Aging 2002; 23:237–243.
- Vagnucci AH, Li W. Alzheimer’s disease and angiogenesis. Lancet 2003; 361:605–608.
- Pogue AI, Lukiw WJ. Angiogenic signaling in Alzheimer’s disease. Neuroreport 2004; 15:1507–1510.
- Dorheim NA, Tracey WR, Pollock JS, Grammas P. Nitric oxide synthase activity is elevated in brain microvessels in Alzheimer’s disease. Biochem Biophys Res Commun 1994; 30:659–665.
- Grammas P, Ovase R. Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol Aging 2001; 22:837–842.
- Grammas P, Ovase R. Cerebrovascular TGF-β contributes to inflammation in the Alzheimer’s brain. Am J Pathol 2002; 160:1583–1587.
- Grammas P, Ghatreh-Samany P, Thirmangalakudi L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: implications for disease pathogenesis. J Alz Dis 2006; 9:51–58.
- Thirumangakudi L, Ghatreh-Samany P, Owoso A, Grammas P. Angiogenic proteins are expressed by brain blood vessels in Alzheimer’s disease. J Alz Dis 2006; 10:111–118.
- Yin X, Wright J, Wall T, Grammas P. Brain endothelial cells synthesize neurotoxic thrombin in Alzheimer’s disease. Am J Pathol 2010; 176:1600–1606.
- Milkiewicz M, Ispanovic E, Doyle JL, Haas TL. Regulators of angiogenesis and strategies for their therapeutic manipulation. Int J Biochem Cell Biol 2006; 38:333–357.
- Felmeden DC, Blann AD, Lip GYH. Angiogenesis: basic pathophysiology and implications for disease. Eur Heart J 2003; 24:586–603.
- Gimbrone MA, Topper JN, Nagel T, Anderson KR, Garcia-Cardeña G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann NY Acad Sci 2000; 902:230–240.
- Magrane J, Christensen RA, Rosen KM, Veereshwarayya V, Querfurth HW. Dissociation of ERK and Akt signaling in endothelial cell angiogenic responses to beta-amyloid. Exp Cell Res 2006; 312:996–1010.
- Wu Z, Guo H, Chow N, et al Role of the MEOX2 gene in neurovascular dysfunction in Alzheimer disease. Nat Med 2005; 11:959–965.
- Gorski DH, Leal AJ. Inhibition of endothelial cell activation by the homeobox gene Gax. J Surg Res 2003; 111:91–99.
- Patel S, Leal AD, Gorski DH. The homeobox gene Gax inhibits angiogenesis through inhibition of nuclear factor-kappaB-dependent endothelial cell gene expression. Cancer Res 2005; 65:1414–1424.
- Edelber JM, Reed MJ. Aging and angiogenesis. Front Biosci 2003; 8:s1199–s1209.
- Buee L, Hof PR, Bouras C, et al Pathological alterations of the cerebral microvasculature in Alzheimer’s disease and related dementing disorders. Acta Neuropathol 1994; 87:469–480.
- Buee L, Hof PR, Delacourte A. Brain microvascular changes in Alzheimer’s disease and other dementias. Ann NY Acad Sci 1997; 826:7–24.
- Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm 2002; 109:813–836.
- Paris D, Townsend K, Quadros A, et al Inhibition of angiogenesis by Aβ peptides. Angiogenesis 2004; 7:75–85.
Alzheimer disease (AD) is a progressive, irreversible, neurodegenerative disease that affects more than 5.3 million people in the United States.1 This number is significantly higher than the previous estimate of 4.5 million and is projected to increase sharply to nearly 8 million by 2030.1 At present, the few agents that are approved by the US Food and Drug Administration for treatment of AD have demonstrated only modest effects in modifying clinical symptoms for relatively short periods; none has shown a clear effect on disease progression. New therapeutic approaches are desperately needed.
VASCULAR DISEASE AND ALZHEIMER DISEASE
Although AD is classified as a neurodegenerative dementia, there is epidemiologic and pathologic evidence of an association with vascular risk factors and vascular disease.2–6 Vascular disease appears to lower the threshold for the clinical presentation of dementia at a given level of AD-related pathology.7 The possible association of AD with vascular disease suggests that there are important pathogenic mechanisms common to both AD and vascular disease. For example, there is increasing evidence that perturbations in cerebral vascular structure and function occur in AD.8
It has been suggested that cerebral hypoperfusion/hypoxia triggers hypometabolic, cognitive, and degenerative changes in the brain and contributes to the pathologic processes of AD.9 A study by Roher and colleagues reveals an association between severe circle of Willis atherosclerosis and sporadic AD.10 These observations suggest that atherosclerosis-induced brain hypoperfusion contributes to the clinical and pathologic manifestations of AD.
Hypoxia is also known to stimulate angiogenesis, especially via upregulation of hypoxia-inducible genes such as vascular endothelial growth factor (VEGF).11,12 VEGF, a critical mediator of angiogenesis, is present in the AD brain in the walls of intra-parenchymal vessels, in diffuse perivascular deposits, and in clusters of reactive astrocytes.13 In addition, intrathecal levels of VEGF in AD are related to clinical severity and intrathecal levels of amyloid-beta (Aβ).14 Emerging data support the idea that factors and processes characteristic of angiogenesis are found in the AD brain.15,16
ENDOTHELIAL ACTIVATION AND ANGIOGENESIS
The angiogenic process is complex and involves several discrete steps, such as endothelial activation, extracellular matrix degradation, proliferation and migration of endothelial cells, and morphologic differentiation of endothelial cells to form tubes. Stimuli known to initiate angiogenesis, including hypoxia, inflammation, and mechanical factors such as shear stress and stretch,23 either directly or indirectly activate endothelial cells. Activated endothelial cells elaborate adhesion molecules, cytokines and chemokines, growth factors, vasoactive molecules, major histocompatibility complex molecules, procoagulant and anticoagulant moieties, and a variety of other gene products with biologic activity.24 The activated endothelium exerts direct local effects by producing at least 20 paracrine factors that act on adjacent cells.25
ANGIOGENIC SIGNALING MECHANISMS IN BRAIN MICROVESSELS
Signaling mechanisms that have been identified as important to endothelial cell viability and angiogenesis include PI3K/Akt, p38 kinase, ERK, and JNK. In this regard, intracellular Aβ accumulation is toxic to endothelial cells and decreases PI3K/Akt.26 Extracellular Aβ peptides decrease phosphorylation and thus activation of ERK and p38 kinase.26 VEGF promotes endothelial survival, proliferation, and migration through numerous pathways, including activation of ERK, p38 kinase, JNK, and Rho GTPase family members.23
VASCULAR ACTIVATION IN ALZHEIMER DISEASE
Despite increases in several proangiogenic factors in the AD brain, evidence for increased vascularity in AD is lacking. On the contrary, it has been suggested that the angiogenic process is delayed or impaired in aged tissues, with several studies showing decreased microvascular density in the AD brain.30–33 Paris et al showed that wild-type Aβ peptides have antiangiogenic effects in vitro and in vivo.34
How can the data showing antiangiogenic effects of Aβ be reconciled with the presence or expression of a large number of proangiogenic proteins by brain microvessels in AD? These conflicting observations suggest an imbalance between proangiogenic and antiangiogeneic processes in the AD brain.
Preliminary experiments in our laboratory show that pharmacologic blockade of vascular activation improves cognitive function in an animal model of AD. Thus, “vascular activation” could be a novel, unexplored therapeutic target in AD.
Acknowledgment
The authors gratefully acknowledge the secretarial assistance of Terri Stahl.
Alzheimer disease (AD) is a progressive, irreversible, neurodegenerative disease that affects more than 5.3 million people in the United States.1 This number is significantly higher than the previous estimate of 4.5 million and is projected to increase sharply to nearly 8 million by 2030.1 At present, the few agents that are approved by the US Food and Drug Administration for treatment of AD have demonstrated only modest effects in modifying clinical symptoms for relatively short periods; none has shown a clear effect on disease progression. New therapeutic approaches are desperately needed.
VASCULAR DISEASE AND ALZHEIMER DISEASE
Although AD is classified as a neurodegenerative dementia, there is epidemiologic and pathologic evidence of an association with vascular risk factors and vascular disease.2–6 Vascular disease appears to lower the threshold for the clinical presentation of dementia at a given level of AD-related pathology.7 The possible association of AD with vascular disease suggests that there are important pathogenic mechanisms common to both AD and vascular disease. For example, there is increasing evidence that perturbations in cerebral vascular structure and function occur in AD.8
It has been suggested that cerebral hypoperfusion/hypoxia triggers hypometabolic, cognitive, and degenerative changes in the brain and contributes to the pathologic processes of AD.9 A study by Roher and colleagues reveals an association between severe circle of Willis atherosclerosis and sporadic AD.10 These observations suggest that atherosclerosis-induced brain hypoperfusion contributes to the clinical and pathologic manifestations of AD.
Hypoxia is also known to stimulate angiogenesis, especially via upregulation of hypoxia-inducible genes such as vascular endothelial growth factor (VEGF).11,12 VEGF, a critical mediator of angiogenesis, is present in the AD brain in the walls of intra-parenchymal vessels, in diffuse perivascular deposits, and in clusters of reactive astrocytes.13 In addition, intrathecal levels of VEGF in AD are related to clinical severity and intrathecal levels of amyloid-beta (Aβ).14 Emerging data support the idea that factors and processes characteristic of angiogenesis are found in the AD brain.15,16
ENDOTHELIAL ACTIVATION AND ANGIOGENESIS
The angiogenic process is complex and involves several discrete steps, such as endothelial activation, extracellular matrix degradation, proliferation and migration of endothelial cells, and morphologic differentiation of endothelial cells to form tubes. Stimuli known to initiate angiogenesis, including hypoxia, inflammation, and mechanical factors such as shear stress and stretch,23 either directly or indirectly activate endothelial cells. Activated endothelial cells elaborate adhesion molecules, cytokines and chemokines, growth factors, vasoactive molecules, major histocompatibility complex molecules, procoagulant and anticoagulant moieties, and a variety of other gene products with biologic activity.24 The activated endothelium exerts direct local effects by producing at least 20 paracrine factors that act on adjacent cells.25
ANGIOGENIC SIGNALING MECHANISMS IN BRAIN MICROVESSELS
Signaling mechanisms that have been identified as important to endothelial cell viability and angiogenesis include PI3K/Akt, p38 kinase, ERK, and JNK. In this regard, intracellular Aβ accumulation is toxic to endothelial cells and decreases PI3K/Akt.26 Extracellular Aβ peptides decrease phosphorylation and thus activation of ERK and p38 kinase.26 VEGF promotes endothelial survival, proliferation, and migration through numerous pathways, including activation of ERK, p38 kinase, JNK, and Rho GTPase family members.23
VASCULAR ACTIVATION IN ALZHEIMER DISEASE
Despite increases in several proangiogenic factors in the AD brain, evidence for increased vascularity in AD is lacking. On the contrary, it has been suggested that the angiogenic process is delayed or impaired in aged tissues, with several studies showing decreased microvascular density in the AD brain.30–33 Paris et al showed that wild-type Aβ peptides have antiangiogenic effects in vitro and in vivo.34
How can the data showing antiangiogenic effects of Aβ be reconciled with the presence or expression of a large number of proangiogenic proteins by brain microvessels in AD? These conflicting observations suggest an imbalance between proangiogenic and antiangiogeneic processes in the AD brain.
Preliminary experiments in our laboratory show that pharmacologic blockade of vascular activation improves cognitive function in an animal model of AD. Thus, “vascular activation” could be a novel, unexplored therapeutic target in AD.
Acknowledgment
The authors gratefully acknowledge the secretarial assistance of Terri Stahl.
- 2010 Alzheimer’s facts and figures. Alzheimer’s Association Web site. http://www.alz.org/alzheimers_disease_facts_and_figures.asp. Updated January 5, 2011. Accessed February 10, 2011.
- Stewart R, Prince M, Mann A. Vascular risk factors and Alzheimer’s disease. Aust N Z J Psychiatry 1999; 33:809–813.
- Schmidt R, Schmidt H, Fasekas F. Vascular risk factors in dementia. J Neurol 2000; 247:81–87.
- Shi J, Perry G, Smith MA, Friedland RP. Vascular abnormalities: the insidious pathogenesis of Alzheimer’s disease. Neurobiol Aging 2000; 21:357–361.
- Pansari K, Gupta A, Thomas P. Alzheimer’s disease and vascular factors: facts and theories. Int J Clin Pract 2002; 56:197–203.
- de la Torre JC. Alzheimer disease as a vascular disorder: nosological evidence. Stroke 2002; 33:1152–1162.
- Sadowski M, Pankiewicz J, Scholtzova H, et al Links between the pathology of Alzheimer’s disease and vascular dementia. Neurochem Res 2004; 29:1257–1266.
- Grammas P. A damaged microcirculation contributes to neuronal cell death in Alzheimer’s disease. Neurobiol Aging 2000; 21:199–205.
- de la Torre JC, Stefano GB. Evidence that Alzheimer’s disease is a microvascular disorder: the role of constitutive nitric oxide. Brain Res Rev 2000; 34:119–136.
- Roher AE, Esh C, Kokjohn TA, et al Circle of Willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler Thromb Vasc Biol 2003; 23:2055–2062.
- Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 2003; 9:677–684.
- Yamakawa M, Liu LX, Date T, et al Hypoxia-inducible factor-1 mediates activation of cultured vascular endothelial cells by inducing multiple angiogenic factors. Circ Res 2003; 93:664–673.
- Kalaria RN, Cohen DL, Premkumar DR, Nag S, LaManna JC, Lust WD. Vascular endothelial growth factor in Alzheimer’s disease and experimental ischemia. Brain Res Mol Brain Res 1998; 62:101–105.
- Tarkowski E, Issa R, Sjogren M, et al Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer’s disease and vascular dementia. Neurobiol Aging 2002; 23:237–243.
- Vagnucci AH, Li W. Alzheimer’s disease and angiogenesis. Lancet 2003; 361:605–608.
- Pogue AI, Lukiw WJ. Angiogenic signaling in Alzheimer’s disease. Neuroreport 2004; 15:1507–1510.
- Dorheim NA, Tracey WR, Pollock JS, Grammas P. Nitric oxide synthase activity is elevated in brain microvessels in Alzheimer’s disease. Biochem Biophys Res Commun 1994; 30:659–665.
- Grammas P, Ovase R. Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol Aging 2001; 22:837–842.
- Grammas P, Ovase R. Cerebrovascular TGF-β contributes to inflammation in the Alzheimer’s brain. Am J Pathol 2002; 160:1583–1587.
- Grammas P, Ghatreh-Samany P, Thirmangalakudi L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: implications for disease pathogenesis. J Alz Dis 2006; 9:51–58.
- Thirumangakudi L, Ghatreh-Samany P, Owoso A, Grammas P. Angiogenic proteins are expressed by brain blood vessels in Alzheimer’s disease. J Alz Dis 2006; 10:111–118.
- Yin X, Wright J, Wall T, Grammas P. Brain endothelial cells synthesize neurotoxic thrombin in Alzheimer’s disease. Am J Pathol 2010; 176:1600–1606.
- Milkiewicz M, Ispanovic E, Doyle JL, Haas TL. Regulators of angiogenesis and strategies for their therapeutic manipulation. Int J Biochem Cell Biol 2006; 38:333–357.
- Felmeden DC, Blann AD, Lip GYH. Angiogenesis: basic pathophysiology and implications for disease. Eur Heart J 2003; 24:586–603.
- Gimbrone MA, Topper JN, Nagel T, Anderson KR, Garcia-Cardeña G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann NY Acad Sci 2000; 902:230–240.
- Magrane J, Christensen RA, Rosen KM, Veereshwarayya V, Querfurth HW. Dissociation of ERK and Akt signaling in endothelial cell angiogenic responses to beta-amyloid. Exp Cell Res 2006; 312:996–1010.
- Wu Z, Guo H, Chow N, et al Role of the MEOX2 gene in neurovascular dysfunction in Alzheimer disease. Nat Med 2005; 11:959–965.
- Gorski DH, Leal AJ. Inhibition of endothelial cell activation by the homeobox gene Gax. J Surg Res 2003; 111:91–99.
- Patel S, Leal AD, Gorski DH. The homeobox gene Gax inhibits angiogenesis through inhibition of nuclear factor-kappaB-dependent endothelial cell gene expression. Cancer Res 2005; 65:1414–1424.
- Edelber JM, Reed MJ. Aging and angiogenesis. Front Biosci 2003; 8:s1199–s1209.
- Buee L, Hof PR, Bouras C, et al Pathological alterations of the cerebral microvasculature in Alzheimer’s disease and related dementing disorders. Acta Neuropathol 1994; 87:469–480.
- Buee L, Hof PR, Delacourte A. Brain microvascular changes in Alzheimer’s disease and other dementias. Ann NY Acad Sci 1997; 826:7–24.
- Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm 2002; 109:813–836.
- Paris D, Townsend K, Quadros A, et al Inhibition of angiogenesis by Aβ peptides. Angiogenesis 2004; 7:75–85.
- 2010 Alzheimer’s facts and figures. Alzheimer’s Association Web site. http://www.alz.org/alzheimers_disease_facts_and_figures.asp. Updated January 5, 2011. Accessed February 10, 2011.
- Stewart R, Prince M, Mann A. Vascular risk factors and Alzheimer’s disease. Aust N Z J Psychiatry 1999; 33:809–813.
- Schmidt R, Schmidt H, Fasekas F. Vascular risk factors in dementia. J Neurol 2000; 247:81–87.
- Shi J, Perry G, Smith MA, Friedland RP. Vascular abnormalities: the insidious pathogenesis of Alzheimer’s disease. Neurobiol Aging 2000; 21:357–361.
- Pansari K, Gupta A, Thomas P. Alzheimer’s disease and vascular factors: facts and theories. Int J Clin Pract 2002; 56:197–203.
- de la Torre JC. Alzheimer disease as a vascular disorder: nosological evidence. Stroke 2002; 33:1152–1162.
- Sadowski M, Pankiewicz J, Scholtzova H, et al Links between the pathology of Alzheimer’s disease and vascular dementia. Neurochem Res 2004; 29:1257–1266.
- Grammas P. A damaged microcirculation contributes to neuronal cell death in Alzheimer’s disease. Neurobiol Aging 2000; 21:199–205.
- de la Torre JC, Stefano GB. Evidence that Alzheimer’s disease is a microvascular disorder: the role of constitutive nitric oxide. Brain Res Rev 2000; 34:119–136.
- Roher AE, Esh C, Kokjohn TA, et al Circle of Willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler Thromb Vasc Biol 2003; 23:2055–2062.
- Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 2003; 9:677–684.
- Yamakawa M, Liu LX, Date T, et al Hypoxia-inducible factor-1 mediates activation of cultured vascular endothelial cells by inducing multiple angiogenic factors. Circ Res 2003; 93:664–673.
- Kalaria RN, Cohen DL, Premkumar DR, Nag S, LaManna JC, Lust WD. Vascular endothelial growth factor in Alzheimer’s disease and experimental ischemia. Brain Res Mol Brain Res 1998; 62:101–105.
- Tarkowski E, Issa R, Sjogren M, et al Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer’s disease and vascular dementia. Neurobiol Aging 2002; 23:237–243.
- Vagnucci AH, Li W. Alzheimer’s disease and angiogenesis. Lancet 2003; 361:605–608.
- Pogue AI, Lukiw WJ. Angiogenic signaling in Alzheimer’s disease. Neuroreport 2004; 15:1507–1510.
- Dorheim NA, Tracey WR, Pollock JS, Grammas P. Nitric oxide synthase activity is elevated in brain microvessels in Alzheimer’s disease. Biochem Biophys Res Commun 1994; 30:659–665.
- Grammas P, Ovase R. Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol Aging 2001; 22:837–842.
- Grammas P, Ovase R. Cerebrovascular TGF-β contributes to inflammation in the Alzheimer’s brain. Am J Pathol 2002; 160:1583–1587.
- Grammas P, Ghatreh-Samany P, Thirmangalakudi L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: implications for disease pathogenesis. J Alz Dis 2006; 9:51–58.
- Thirumangakudi L, Ghatreh-Samany P, Owoso A, Grammas P. Angiogenic proteins are expressed by brain blood vessels in Alzheimer’s disease. J Alz Dis 2006; 10:111–118.
- Yin X, Wright J, Wall T, Grammas P. Brain endothelial cells synthesize neurotoxic thrombin in Alzheimer’s disease. Am J Pathol 2010; 176:1600–1606.
- Milkiewicz M, Ispanovic E, Doyle JL, Haas TL. Regulators of angiogenesis and strategies for their therapeutic manipulation. Int J Biochem Cell Biol 2006; 38:333–357.
- Felmeden DC, Blann AD, Lip GYH. Angiogenesis: basic pathophysiology and implications for disease. Eur Heart J 2003; 24:586–603.
- Gimbrone MA, Topper JN, Nagel T, Anderson KR, Garcia-Cardeña G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann NY Acad Sci 2000; 902:230–240.
- Magrane J, Christensen RA, Rosen KM, Veereshwarayya V, Querfurth HW. Dissociation of ERK and Akt signaling in endothelial cell angiogenic responses to beta-amyloid. Exp Cell Res 2006; 312:996–1010.
- Wu Z, Guo H, Chow N, et al Role of the MEOX2 gene in neurovascular dysfunction in Alzheimer disease. Nat Med 2005; 11:959–965.
- Gorski DH, Leal AJ. Inhibition of endothelial cell activation by the homeobox gene Gax. J Surg Res 2003; 111:91–99.
- Patel S, Leal AD, Gorski DH. The homeobox gene Gax inhibits angiogenesis through inhibition of nuclear factor-kappaB-dependent endothelial cell gene expression. Cancer Res 2005; 65:1414–1424.
- Edelber JM, Reed MJ. Aging and angiogenesis. Front Biosci 2003; 8:s1199–s1209.
- Buee L, Hof PR, Bouras C, et al Pathological alterations of the cerebral microvasculature in Alzheimer’s disease and related dementing disorders. Acta Neuropathol 1994; 87:469–480.
- Buee L, Hof PR, Delacourte A. Brain microvascular changes in Alzheimer’s disease and other dementias. Ann NY Acad Sci 1997; 826:7–24.
- Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm 2002; 109:813–836.
- Paris D, Townsend K, Quadros A, et al Inhibition of angiogenesis by Aβ peptides. Angiogenesis 2004; 7:75–85.
Stress in medicine: Strategies for caregivers, patients, clinicians—The burdens of caregiver stress
The number of people in the United States who spend a significant part of each week working as unpaid caregivers is considerable, and the toll exacted for such work is high. Understanding the profi le of the caregiver, the nature of the duties performed, the stress imposed by such duties, and the consequences of the stress can assist the clinician in recognizing the caregiver in need of intervention.
A PROFILE OF THE CAREGIVER
A recent survey estimated that more than 65 million Americans provide unpaid assistance annually to older adults with disabilities.1 The value of that labor has been estimated at $306 billion annually, or nearly double the combined cost of home health care and nursing home care.2,3
The typical caregiver is a woman, about 48 years old, with some college education, who spends 20 hours or more each week providing unpaid care to someone aged 50 years or older.1 The recipients of care often have long-term physical disabilities; mental confusion or emotional problems frequently complicate care.
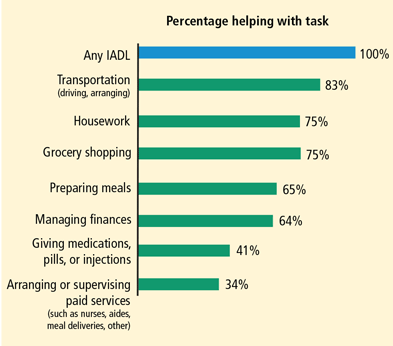
PSYCHOLOGIC AND PHYSICAL COSTS
Caregiving may take a toll on the caregiver in a variety of ways: behavioral, in the form of alcohol or substance use4; psychologic, in the form of depression or other mental health problems5; and physical, in the form of chronic health conditions and impaired immune response.6 About three-fifths of caregivers report fair or poor health, compared with one-third of noncaregivers, and caregivers have approximately twice as many chronic conditions, such as heart disease, cancer, arthritis, and diabetes, compared with noncaregivers.2,7 Caregiving also exacts a financial toll, as employees who are caregivers cost their employers $13.4 billion more per year in health care expenditures.8 In addition, absenteeism, workday interruptions, and shifts from full-time to part-time work by caregivers cost businesses between $17.1 and $33.6 billion per year.9
The cost of caregiving is higher for women, who exhibit higher levels of anxiety and depression and lower levels of subjective well-being, life satisfaction, and physical health.10,11 The stress of caregiving has also been identified as a risk factor for morbidity among older (66 to 96 years old) caregivers, who have a 63% greater mortality than noncaregivers of the same age.12
PSYCHOSOCIAL STRESS, UNHEALTHY BEHAVIORS, AND ILLNESS ARE LINKED
Psychosocial stress is a predictor of disease and can lead to unhealthy behaviors such as smoking, substance abuse, overeating, poor nutrition, and a sedentary lifestyle; these, in turn, can lead to physical and psychiatric illness. Behaviors adopted initially as coping skills may persist to become chronic, thereby promoting either continued wellness (in the case of healthy coping behaviors) or worsening levels of illness (in the case of unhealthy coping behaviors).
McEwen and Gianaros13 have suggested that these stress mechanisms arise from patterns of communication between the brain and the autonomic, cardiovascular, and immune systems, which mutually influenceone another. These so-called bidirectional stress processes affect cognition, experience, and behavior.
An integrated model of stress that maps the bidirectional causal pathways among psychosocial stressors, resulting unhealthy behaviors, and illness is needed. Although the steps from unhealthy behaviors to illness are fairly well understood, the links from psychosocial stress, such as those exhibited by caregivers, to unhealthy behaviors are not as clear. Several mediators are under study:
- Personality mediators can be either ameliorative (resilience, self-confi dence, self-control, optimism, high self-esteem, a sense of mastery, and finding meaning in life) or exacerbating (neuroticism and inhibition, which together form the so-called type D personality).
- Environmental mediators include social support, financial support, a history of a significant life change, and trauma early in life, which may increase one’s subsequent vulnerability to unhealthy behaviors.
- Biologic mediators may include prolonged sympathetic activation and enhanced platelet activation, caused by increased levels of depression and anxiety in chronically stressed caregivers.14
IMPLICATIONS FOR INTERVENTION
A significant percentage of caregivers do not need a clinician’s intervention to help them cope with stress or unhealthy coping skills. Among caregivers aged 50 years or older, 47% indicated in a recent study that the burden of caregiving is low (ie, 1 or 2 on a 5-point scale).1 Those who respond to stressors as challenges rather than threats tend to be resilient people who exert control over their lives, often through meditation or similar techniques, and have a strong social support network. Many report that caregiving provides them with an opportunity to act in accordance with their values and feel helpful rather than helpless.
Cognitive-behavioral interventions to alleviate stress-related symptoms appear to be more effective if offered as individual rather than group therapy. Teaching caregivers effective coping strategies, rather than merely providing social support, has been shown to improve caregiver psychologic health.15 Chief among the goals of intervention should be to alter brain function and instill optimism, a sense of control and self-esteem.13
- The National Alliance for Caregiving, in collaboration with the American Association of Retired Persons. Caregiving in the U.S. 2009. National Alliance for Caregiving Web site. http://www.caregiving.org/data/Caregiving_in_the_US_2009_full_report.pdf. Published November 2009. Accessed March 21, 2011.
- Family Caregiver Alliance. Caregiver health. A population at risk. National Alliance for Caregiving Web site. http://www.caregiver.org/caregiver/jsp/content_node.jsp?nodeid=1822. Published 2006. Accessed March 21, 2011.
- Family Caregiver Alliance. Prevalence, hours, and economic value of family caregiving, updated state-by-state analysis of 2004 national estimates. National Alliance for Caregiving Web site. http://www.caregiver.org/caregiver/jsp/content/pdfs/State_Caregiving_Data_Amo_20061107.pdf. Published 2006. Accessed March 21, 2011.
- Evercare. Study of caregivers in decline: a close-up look at the health risks of caring for a loved one. National Alliance for Caregiving Web site. http://www.caregiving.org/data/Caregivers%20in%20Decline%20Study-FINAL-lowres.pdf. Published 2006. Accessed March 21, 2011.
- Pinquart M, Sörensen S. Differences between caregivers and noncaregivers in psychological health and physical health: a metaanalysis. Psychol Aging 2003; 18:250–267.
- Vitaliano PP, Zhang J, Scanlan JM. Is caregiving hazardous to one’s physical health? A meta-analysis. Psychol Bull 2003; 129:946–972.
- Ho A, Collins S, Davis K, Doty M. A look at working-age caregivers’ roles, health concerns, and need for support (issue brief). New York, NY: The Commonwealth Fund; 2005.
- MetLife study of working caregivers and employer health care costs. MetLife Web site. http://www.metlife.com/assets/cao/mmi/publications/studies/2010/mmi-working-caregivers-employers-health-carecosts.pdf. Published July 2006. Accessed March 21, 2011.
- MetLife caregiving cost study: productivity losses to U.S. business. National Alliance for Caregiving Web site. http://www.caregiving. org/data/Caregiver%20Cost%20Study.pdf. Published July 2006. Accessed March 21, 2011.
- Pinquart M, Sörensen S. Gender differences in caregiver stressors, social resources, and health: an updated meta-analysis. J Gerontol B Psychol Sci Soc Sci 2006; 61:P33–P45.
- Johnson RW, Wiener JM. A profi le of frail older Americans and their caregivers. Urban Institute Web site. http://www.urban.org/UploadedPDF/311284_older_americans.pdf. Published February 2006. Accessed March 21, 2011.
- Schulz R, Beach SR. Caregiving as a risk factor for mortality: the caregiver health effects study. JAMA 1999; 282:2215–2219.
- McEwen BS, Gianaros PJ. Central role of the brain in stress and adaptation: links to socioeconomic status, health, and disease. Ann NY Acad Sci 2010; 1186:190–222.
- Aschbacher K, Mills PJ, von Känel R, et al. Effects of depressive and anxious symptoms on norepinephrine and platelet P-selectin responses to acute psychological stress among elderly caregivers. Brain Behav Immun 2008; 22:493–502.
- Selwood A, Johnston K, Katona C, Lyketsos C, Livingston G. Systematic review of the effect of psychological interventions on family caregivers of people with dementia. J Affect Disord 2007; 101:75–89.
The number of people in the United States who spend a significant part of each week working as unpaid caregivers is considerable, and the toll exacted for such work is high. Understanding the profi le of the caregiver, the nature of the duties performed, the stress imposed by such duties, and the consequences of the stress can assist the clinician in recognizing the caregiver in need of intervention.
A PROFILE OF THE CAREGIVER
A recent survey estimated that more than 65 million Americans provide unpaid assistance annually to older adults with disabilities.1 The value of that labor has been estimated at $306 billion annually, or nearly double the combined cost of home health care and nursing home care.2,3
The typical caregiver is a woman, about 48 years old, with some college education, who spends 20 hours or more each week providing unpaid care to someone aged 50 years or older.1 The recipients of care often have long-term physical disabilities; mental confusion or emotional problems frequently complicate care.
PSYCHOLOGIC AND PHYSICAL COSTS
Caregiving may take a toll on the caregiver in a variety of ways: behavioral, in the form of alcohol or substance use4; psychologic, in the form of depression or other mental health problems5; and physical, in the form of chronic health conditions and impaired immune response.6 About three-fifths of caregivers report fair or poor health, compared with one-third of noncaregivers, and caregivers have approximately twice as many chronic conditions, such as heart disease, cancer, arthritis, and diabetes, compared with noncaregivers.2,7 Caregiving also exacts a financial toll, as employees who are caregivers cost their employers $13.4 billion more per year in health care expenditures.8 In addition, absenteeism, workday interruptions, and shifts from full-time to part-time work by caregivers cost businesses between $17.1 and $33.6 billion per year.9
The cost of caregiving is higher for women, who exhibit higher levels of anxiety and depression and lower levels of subjective well-being, life satisfaction, and physical health.10,11 The stress of caregiving has also been identified as a risk factor for morbidity among older (66 to 96 years old) caregivers, who have a 63% greater mortality than noncaregivers of the same age.12
PSYCHOSOCIAL STRESS, UNHEALTHY BEHAVIORS, AND ILLNESS ARE LINKED
Psychosocial stress is a predictor of disease and can lead to unhealthy behaviors such as smoking, substance abuse, overeating, poor nutrition, and a sedentary lifestyle; these, in turn, can lead to physical and psychiatric illness. Behaviors adopted initially as coping skills may persist to become chronic, thereby promoting either continued wellness (in the case of healthy coping behaviors) or worsening levels of illness (in the case of unhealthy coping behaviors).
McEwen and Gianaros13 have suggested that these stress mechanisms arise from patterns of communication between the brain and the autonomic, cardiovascular, and immune systems, which mutually influenceone another. These so-called bidirectional stress processes affect cognition, experience, and behavior.
An integrated model of stress that maps the bidirectional causal pathways among psychosocial stressors, resulting unhealthy behaviors, and illness is needed. Although the steps from unhealthy behaviors to illness are fairly well understood, the links from psychosocial stress, such as those exhibited by caregivers, to unhealthy behaviors are not as clear. Several mediators are under study:
- Personality mediators can be either ameliorative (resilience, self-confi dence, self-control, optimism, high self-esteem, a sense of mastery, and finding meaning in life) or exacerbating (neuroticism and inhibition, which together form the so-called type D personality).
- Environmental mediators include social support, financial support, a history of a significant life change, and trauma early in life, which may increase one’s subsequent vulnerability to unhealthy behaviors.
- Biologic mediators may include prolonged sympathetic activation and enhanced platelet activation, caused by increased levels of depression and anxiety in chronically stressed caregivers.14
IMPLICATIONS FOR INTERVENTION
A significant percentage of caregivers do not need a clinician’s intervention to help them cope with stress or unhealthy coping skills. Among caregivers aged 50 years or older, 47% indicated in a recent study that the burden of caregiving is low (ie, 1 or 2 on a 5-point scale).1 Those who respond to stressors as challenges rather than threats tend to be resilient people who exert control over their lives, often through meditation or similar techniques, and have a strong social support network. Many report that caregiving provides them with an opportunity to act in accordance with their values and feel helpful rather than helpless.
Cognitive-behavioral interventions to alleviate stress-related symptoms appear to be more effective if offered as individual rather than group therapy. Teaching caregivers effective coping strategies, rather than merely providing social support, has been shown to improve caregiver psychologic health.15 Chief among the goals of intervention should be to alter brain function and instill optimism, a sense of control and self-esteem.13
The number of people in the United States who spend a significant part of each week working as unpaid caregivers is considerable, and the toll exacted for such work is high. Understanding the profi le of the caregiver, the nature of the duties performed, the stress imposed by such duties, and the consequences of the stress can assist the clinician in recognizing the caregiver in need of intervention.
A PROFILE OF THE CAREGIVER
A recent survey estimated that more than 65 million Americans provide unpaid assistance annually to older adults with disabilities.1 The value of that labor has been estimated at $306 billion annually, or nearly double the combined cost of home health care and nursing home care.2,3
The typical caregiver is a woman, about 48 years old, with some college education, who spends 20 hours or more each week providing unpaid care to someone aged 50 years or older.1 The recipients of care often have long-term physical disabilities; mental confusion or emotional problems frequently complicate care.
PSYCHOLOGIC AND PHYSICAL COSTS
Caregiving may take a toll on the caregiver in a variety of ways: behavioral, in the form of alcohol or substance use4; psychologic, in the form of depression or other mental health problems5; and physical, in the form of chronic health conditions and impaired immune response.6 About three-fifths of caregivers report fair or poor health, compared with one-third of noncaregivers, and caregivers have approximately twice as many chronic conditions, such as heart disease, cancer, arthritis, and diabetes, compared with noncaregivers.2,7 Caregiving also exacts a financial toll, as employees who are caregivers cost their employers $13.4 billion more per year in health care expenditures.8 In addition, absenteeism, workday interruptions, and shifts from full-time to part-time work by caregivers cost businesses between $17.1 and $33.6 billion per year.9
The cost of caregiving is higher for women, who exhibit higher levels of anxiety and depression and lower levels of subjective well-being, life satisfaction, and physical health.10,11 The stress of caregiving has also been identified as a risk factor for morbidity among older (66 to 96 years old) caregivers, who have a 63% greater mortality than noncaregivers of the same age.12
PSYCHOSOCIAL STRESS, UNHEALTHY BEHAVIORS, AND ILLNESS ARE LINKED
Psychosocial stress is a predictor of disease and can lead to unhealthy behaviors such as smoking, substance abuse, overeating, poor nutrition, and a sedentary lifestyle; these, in turn, can lead to physical and psychiatric illness. Behaviors adopted initially as coping skills may persist to become chronic, thereby promoting either continued wellness (in the case of healthy coping behaviors) or worsening levels of illness (in the case of unhealthy coping behaviors).
McEwen and Gianaros13 have suggested that these stress mechanisms arise from patterns of communication between the brain and the autonomic, cardiovascular, and immune systems, which mutually influenceone another. These so-called bidirectional stress processes affect cognition, experience, and behavior.
An integrated model of stress that maps the bidirectional causal pathways among psychosocial stressors, resulting unhealthy behaviors, and illness is needed. Although the steps from unhealthy behaviors to illness are fairly well understood, the links from psychosocial stress, such as those exhibited by caregivers, to unhealthy behaviors are not as clear. Several mediators are under study:
- Personality mediators can be either ameliorative (resilience, self-confi dence, self-control, optimism, high self-esteem, a sense of mastery, and finding meaning in life) or exacerbating (neuroticism and inhibition, which together form the so-called type D personality).
- Environmental mediators include social support, financial support, a history of a significant life change, and trauma early in life, which may increase one’s subsequent vulnerability to unhealthy behaviors.
- Biologic mediators may include prolonged sympathetic activation and enhanced platelet activation, caused by increased levels of depression and anxiety in chronically stressed caregivers.14
IMPLICATIONS FOR INTERVENTION
A significant percentage of caregivers do not need a clinician’s intervention to help them cope with stress or unhealthy coping skills. Among caregivers aged 50 years or older, 47% indicated in a recent study that the burden of caregiving is low (ie, 1 or 2 on a 5-point scale).1 Those who respond to stressors as challenges rather than threats tend to be resilient people who exert control over their lives, often through meditation or similar techniques, and have a strong social support network. Many report that caregiving provides them with an opportunity to act in accordance with their values and feel helpful rather than helpless.
Cognitive-behavioral interventions to alleviate stress-related symptoms appear to be more effective if offered as individual rather than group therapy. Teaching caregivers effective coping strategies, rather than merely providing social support, has been shown to improve caregiver psychologic health.15 Chief among the goals of intervention should be to alter brain function and instill optimism, a sense of control and self-esteem.13
- The National Alliance for Caregiving, in collaboration with the American Association of Retired Persons. Caregiving in the U.S. 2009. National Alliance for Caregiving Web site. http://www.caregiving.org/data/Caregiving_in_the_US_2009_full_report.pdf. Published November 2009. Accessed March 21, 2011.
- Family Caregiver Alliance. Caregiver health. A population at risk. National Alliance for Caregiving Web site. http://www.caregiver.org/caregiver/jsp/content_node.jsp?nodeid=1822. Published 2006. Accessed March 21, 2011.
- Family Caregiver Alliance. Prevalence, hours, and economic value of family caregiving, updated state-by-state analysis of 2004 national estimates. National Alliance for Caregiving Web site. http://www.caregiver.org/caregiver/jsp/content/pdfs/State_Caregiving_Data_Amo_20061107.pdf. Published 2006. Accessed March 21, 2011.
- Evercare. Study of caregivers in decline: a close-up look at the health risks of caring for a loved one. National Alliance for Caregiving Web site. http://www.caregiving.org/data/Caregivers%20in%20Decline%20Study-FINAL-lowres.pdf. Published 2006. Accessed March 21, 2011.
- Pinquart M, Sörensen S. Differences between caregivers and noncaregivers in psychological health and physical health: a metaanalysis. Psychol Aging 2003; 18:250–267.
- Vitaliano PP, Zhang J, Scanlan JM. Is caregiving hazardous to one’s physical health? A meta-analysis. Psychol Bull 2003; 129:946–972.
- Ho A, Collins S, Davis K, Doty M. A look at working-age caregivers’ roles, health concerns, and need for support (issue brief). New York, NY: The Commonwealth Fund; 2005.
- MetLife study of working caregivers and employer health care costs. MetLife Web site. http://www.metlife.com/assets/cao/mmi/publications/studies/2010/mmi-working-caregivers-employers-health-carecosts.pdf. Published July 2006. Accessed March 21, 2011.
- MetLife caregiving cost study: productivity losses to U.S. business. National Alliance for Caregiving Web site. http://www.caregiving. org/data/Caregiver%20Cost%20Study.pdf. Published July 2006. Accessed March 21, 2011.
- Pinquart M, Sörensen S. Gender differences in caregiver stressors, social resources, and health: an updated meta-analysis. J Gerontol B Psychol Sci Soc Sci 2006; 61:P33–P45.
- Johnson RW, Wiener JM. A profi le of frail older Americans and their caregivers. Urban Institute Web site. http://www.urban.org/UploadedPDF/311284_older_americans.pdf. Published February 2006. Accessed March 21, 2011.
- Schulz R, Beach SR. Caregiving as a risk factor for mortality: the caregiver health effects study. JAMA 1999; 282:2215–2219.
- McEwen BS, Gianaros PJ. Central role of the brain in stress and adaptation: links to socioeconomic status, health, and disease. Ann NY Acad Sci 2010; 1186:190–222.
- Aschbacher K, Mills PJ, von Känel R, et al. Effects of depressive and anxious symptoms on norepinephrine and platelet P-selectin responses to acute psychological stress among elderly caregivers. Brain Behav Immun 2008; 22:493–502.
- Selwood A, Johnston K, Katona C, Lyketsos C, Livingston G. Systematic review of the effect of psychological interventions on family caregivers of people with dementia. J Affect Disord 2007; 101:75–89.
- The National Alliance for Caregiving, in collaboration with the American Association of Retired Persons. Caregiving in the U.S. 2009. National Alliance for Caregiving Web site. http://www.caregiving.org/data/Caregiving_in_the_US_2009_full_report.pdf. Published November 2009. Accessed March 21, 2011.
- Family Caregiver Alliance. Caregiver health. A population at risk. National Alliance for Caregiving Web site. http://www.caregiver.org/caregiver/jsp/content_node.jsp?nodeid=1822. Published 2006. Accessed March 21, 2011.
- Family Caregiver Alliance. Prevalence, hours, and economic value of family caregiving, updated state-by-state analysis of 2004 national estimates. National Alliance for Caregiving Web site. http://www.caregiver.org/caregiver/jsp/content/pdfs/State_Caregiving_Data_Amo_20061107.pdf. Published 2006. Accessed March 21, 2011.
- Evercare. Study of caregivers in decline: a close-up look at the health risks of caring for a loved one. National Alliance for Caregiving Web site. http://www.caregiving.org/data/Caregivers%20in%20Decline%20Study-FINAL-lowres.pdf. Published 2006. Accessed March 21, 2011.
- Pinquart M, Sörensen S. Differences between caregivers and noncaregivers in psychological health and physical health: a metaanalysis. Psychol Aging 2003; 18:250–267.
- Vitaliano PP, Zhang J, Scanlan JM. Is caregiving hazardous to one’s physical health? A meta-analysis. Psychol Bull 2003; 129:946–972.
- Ho A, Collins S, Davis K, Doty M. A look at working-age caregivers’ roles, health concerns, and need for support (issue brief). New York, NY: The Commonwealth Fund; 2005.
- MetLife study of working caregivers and employer health care costs. MetLife Web site. http://www.metlife.com/assets/cao/mmi/publications/studies/2010/mmi-working-caregivers-employers-health-carecosts.pdf. Published July 2006. Accessed March 21, 2011.
- MetLife caregiving cost study: productivity losses to U.S. business. National Alliance for Caregiving Web site. http://www.caregiving. org/data/Caregiver%20Cost%20Study.pdf. Published July 2006. Accessed March 21, 2011.
- Pinquart M, Sörensen S. Gender differences in caregiver stressors, social resources, and health: an updated meta-analysis. J Gerontol B Psychol Sci Soc Sci 2006; 61:P33–P45.
- Johnson RW, Wiener JM. A profi le of frail older Americans and their caregivers. Urban Institute Web site. http://www.urban.org/UploadedPDF/311284_older_americans.pdf. Published February 2006. Accessed March 21, 2011.
- Schulz R, Beach SR. Caregiving as a risk factor for mortality: the caregiver health effects study. JAMA 1999; 282:2215–2219.
- McEwen BS, Gianaros PJ. Central role of the brain in stress and adaptation: links to socioeconomic status, health, and disease. Ann NY Acad Sci 2010; 1186:190–222.
- Aschbacher K, Mills PJ, von Känel R, et al. Effects of depressive and anxious symptoms on norepinephrine and platelet P-selectin responses to acute psychological stress among elderly caregivers. Brain Behav Immun 2008; 22:493–502.
- Selwood A, Johnston K, Katona C, Lyketsos C, Livingston G. Systematic review of the effect of psychological interventions on family caregivers of people with dementia. J Affect Disord 2007; 101:75–89.
Stress in medicine: Strategies for caregivers, patients, clinicians—Promoting better outcomes with stress and anxiety reduction
The traditional paradigm for cardiac care has emphasized the use of technology to treat disease. Our focus on technologies such as echocardiography, advanced imaging instrumentation, and cardiac catheterization mirrors the preoccupation of society as a whole with technologic advances.
Attention has only recently been given to the patient’s emotional experience and how this might relate to outcomes, recovery, and healing. An expanded paradigm of cardiac care incorporates pain relief, emotional support, spiritual healing, and a caring environment. These elements of patient-centered care aim to relieve stress and anxiety in order to achieve a better clinical outcome.
PATIENT-CENTERED CARE
The importance of patient-centered care is illustrated by the results of a 2007 survey in which 41% of patients cited elements of the patient experience as factors that most influenced their choice of hospital.1 Accepted wisdom on patient choice has historically centered on medical factors such as clinical reputation, physician recommendations, and hospital location, each of which was cited by 18% to 21% of the patients surveyed. Elements of the patient experience cited in the study include stress-reducing factors such as the appearance of the room, ease of scheduling, an environment that supports family needs, convenience and comfort of common areas, on-time performance, and simple registration procedures.
Székely et al2 found in a 4-year followup study that high levels of preoperative anxiety predicted greater mortality and cardiovascular morbidity following cardiac surgery. In a study by Tully et al,3 preoperative anxiety was also predictive of hospital readmission following cardiac surgery. Preoperative stress and anxiety are reliable predictors of postoperative distress.4
The variety and relative efficacy of interventions to reduce stress and anxiety are not well studied. Voss et al5 showed that cardiac surgery patients who were played soothing music experienced significantly reduced anxiety, pain, pain distress, and length of hospital stay. One Cleveland Clinic study of massage therapy, however, was unable to demonstrate a statistically significant therapeutic benefit, despite patient satisfaction with the therapy.6
THE ADVENT OF HEALING SERVICES
Identifying patients who exhibit significant preoperative stress and providing, as part of an expanded cardiac care paradigm, emotional care both pre- and postoperatively may ameliorate clinical outcomes. As such, the Heart and Vascular Institute at the Cleveland Clinic formed a healing services division, based on the concept that healing is more than simply physical recovery from a particular procedure. The division’s mission statement is: “To enhance the patient experience by promoting healing through a comprehensive set of coordinated services addressing the holistic needs of the patient.”

At the Cleveland Clinic, healing services are now integrated with standard services to enhance the cardiac care paradigm. Our standard medical services focus on areas of communication and pain control, both of which affect anxiety and stress. The need for enhanced communication is significant: 75% of patients admitted to a Chicago hospital were unable to name a single doctor assigned to their care, and of the remaining 25%, only 40% of responders were correct.7
It is worth noting that communicating more information to a patient is not necessarily better. Patients given detailed preoperative information about their disease and the potential complications of their cardiac surgery had levels of preoperative, perioperative, and postoperative stress, anxiety, and depression similar to those who received routine medical information.8,9 On the other hand, patients desire information about their postoperative plan of care while they are experiencing it, and value communication with physicians, nurses, healing services personnel, and other caregivers when it is presented in a calm and forthright manner. Communications should emphasize that the entire clinical team is there to help the patient get better.
THE FIFTH VITAL SIGN
Pain control is an aspect of care that was long ignored. The goal of the pain control task force at the Cleveland Clinic is the development of effective, efficient, and compassionate pain management.
The fifth vital sign, one that escapes the electronic medical record, can be addressed by this question: “How are you feeling?” Treating pain will reduce stress and anxiety. Before surgery, pain management priorities are discussed with patients, and at each daily encounter the goal is to set, refine, and exceed expectations for pain control through discussion and frequent pain assessments.
Reducing anxiety and stress is the goal of both standard care services and healing services, resulting in more satisfied patients with better clinical outcomes.
CASE: “YOU AND THE TEAM MADE ME GET OUTOF BED AND MOVE FORWARD”
Bobbi is a 78-year-old woman who was initially recovering well following cardiac surgery, including valve surgery, but had to return to the intensive care unit, which is difficult for patients. She was subsequently returned to the floor but was reluctant to walk and progressed slowly, despite normal electrocardiogram, radiographs, and blood panel results. We discovered that her husband was in hospice care in another state, causing Bobbi anxiety as she expressed concern over being her husband’s caregiver while being weakened physically herself. She was fearful of moving forward and her recovery stalled.
The primary care nurse referred her to the healing services team. The healing services team provided support for her anxiety and stress, and reviewed options for managing her husband’s care. She participated in Reiki, spiritual support, and social work services. During her admission her husband died, so the team provided appropriate support.
When asked about her experience upon leavingthe hospital, Bobbi did not mention her surgeon or the success of her heart valve procedure, but commented instead on the healing services team that enabled her to get through the experience.
- Grote KD, Newman JRS, Sutaria SS. A better hospital experience. The McKinsey Quarterly. November 2007.
- Székely A, Balog P, Benkö E, et al. Anxiety predicts mortality and morbidity after coronary artery and valve surgery—a 4-year followup study. Psychosom Med 2007; 69:625–631.
- Tully PJ, Baker RA, Turnbull D, Winefield H. The role of depression and anxiety symptoms in hospital readmissions after cardiac surgery. J Behav Med 2008; 31:281–290.
- Vingerhoets G. Perioperative anxiety and depression in open-heart surgery. Psychosomatics 1998; 39:30–37.
- Voss JA, Good M, Yates B, Baun MM, Thompson A, Hertzog M. Sedative music reduces anxiety and pain during chair rest after open-heart surgery. Pain 2004; 112:197–203.
- Albert NM, Gillinov AM, Lytle BW, Feng J, Cwynar R, Blackstone EH. A randomized trial of massage therapy after heart surgery. Heart Lung 2009; 38:480–490.
- Arora V, Gangireddy S, Mehrotra A, Ginde R, Tormey M, Meltzer D. Ability of hospitalized patients to identify their in-hospital physicians. Arch Intern Med 2009; 169:199–201.
- Ivarsson B, Larsson S, Lührs C, Sjöberg T. Extended written pre-operative information about possible complications at cardiac surgery—do the patients want to know? Eur J Cardiothorac Surg 2005; 28:407–414.
- Bergmann P, Huber S, Mächler H, et al. The influence of medical information on the perioperative course of stress in cardiac surgery patients. Anesth Analg 2001; 93:1093–1099.
The traditional paradigm for cardiac care has emphasized the use of technology to treat disease. Our focus on technologies such as echocardiography, advanced imaging instrumentation, and cardiac catheterization mirrors the preoccupation of society as a whole with technologic advances.
Attention has only recently been given to the patient’s emotional experience and how this might relate to outcomes, recovery, and healing. An expanded paradigm of cardiac care incorporates pain relief, emotional support, spiritual healing, and a caring environment. These elements of patient-centered care aim to relieve stress and anxiety in order to achieve a better clinical outcome.
PATIENT-CENTERED CARE
The importance of patient-centered care is illustrated by the results of a 2007 survey in which 41% of patients cited elements of the patient experience as factors that most influenced their choice of hospital.1 Accepted wisdom on patient choice has historically centered on medical factors such as clinical reputation, physician recommendations, and hospital location, each of which was cited by 18% to 21% of the patients surveyed. Elements of the patient experience cited in the study include stress-reducing factors such as the appearance of the room, ease of scheduling, an environment that supports family needs, convenience and comfort of common areas, on-time performance, and simple registration procedures.
Székely et al2 found in a 4-year followup study that high levels of preoperative anxiety predicted greater mortality and cardiovascular morbidity following cardiac surgery. In a study by Tully et al,3 preoperative anxiety was also predictive of hospital readmission following cardiac surgery. Preoperative stress and anxiety are reliable predictors of postoperative distress.4
The variety and relative efficacy of interventions to reduce stress and anxiety are not well studied. Voss et al5 showed that cardiac surgery patients who were played soothing music experienced significantly reduced anxiety, pain, pain distress, and length of hospital stay. One Cleveland Clinic study of massage therapy, however, was unable to demonstrate a statistically significant therapeutic benefit, despite patient satisfaction with the therapy.6
THE ADVENT OF HEALING SERVICES
Identifying patients who exhibit significant preoperative stress and providing, as part of an expanded cardiac care paradigm, emotional care both pre- and postoperatively may ameliorate clinical outcomes. As such, the Heart and Vascular Institute at the Cleveland Clinic formed a healing services division, based on the concept that healing is more than simply physical recovery from a particular procedure. The division’s mission statement is: “To enhance the patient experience by promoting healing through a comprehensive set of coordinated services addressing the holistic needs of the patient.”
At the Cleveland Clinic, healing services are now integrated with standard services to enhance the cardiac care paradigm. Our standard medical services focus on areas of communication and pain control, both of which affect anxiety and stress. The need for enhanced communication is significant: 75% of patients admitted to a Chicago hospital were unable to name a single doctor assigned to their care, and of the remaining 25%, only 40% of responders were correct.7
It is worth noting that communicating more information to a patient is not necessarily better. Patients given detailed preoperative information about their disease and the potential complications of their cardiac surgery had levels of preoperative, perioperative, and postoperative stress, anxiety, and depression similar to those who received routine medical information.8,9 On the other hand, patients desire information about their postoperative plan of care while they are experiencing it, and value communication with physicians, nurses, healing services personnel, and other caregivers when it is presented in a calm and forthright manner. Communications should emphasize that the entire clinical team is there to help the patient get better.
THE FIFTH VITAL SIGN
Pain control is an aspect of care that was long ignored. The goal of the pain control task force at the Cleveland Clinic is the development of effective, efficient, and compassionate pain management.
The fifth vital sign, one that escapes the electronic medical record, can be addressed by this question: “How are you feeling?” Treating pain will reduce stress and anxiety. Before surgery, pain management priorities are discussed with patients, and at each daily encounter the goal is to set, refine, and exceed expectations for pain control through discussion and frequent pain assessments.
Reducing anxiety and stress is the goal of both standard care services and healing services, resulting in more satisfied patients with better clinical outcomes.
CASE: “YOU AND THE TEAM MADE ME GET OUTOF BED AND MOVE FORWARD”
Bobbi is a 78-year-old woman who was initially recovering well following cardiac surgery, including valve surgery, but had to return to the intensive care unit, which is difficult for patients. She was subsequently returned to the floor but was reluctant to walk and progressed slowly, despite normal electrocardiogram, radiographs, and blood panel results. We discovered that her husband was in hospice care in another state, causing Bobbi anxiety as she expressed concern over being her husband’s caregiver while being weakened physically herself. She was fearful of moving forward and her recovery stalled.
The primary care nurse referred her to the healing services team. The healing services team provided support for her anxiety and stress, and reviewed options for managing her husband’s care. She participated in Reiki, spiritual support, and social work services. During her admission her husband died, so the team provided appropriate support.
When asked about her experience upon leavingthe hospital, Bobbi did not mention her surgeon or the success of her heart valve procedure, but commented instead on the healing services team that enabled her to get through the experience.
The traditional paradigm for cardiac care has emphasized the use of technology to treat disease. Our focus on technologies such as echocardiography, advanced imaging instrumentation, and cardiac catheterization mirrors the preoccupation of society as a whole with technologic advances.
Attention has only recently been given to the patient’s emotional experience and how this might relate to outcomes, recovery, and healing. An expanded paradigm of cardiac care incorporates pain relief, emotional support, spiritual healing, and a caring environment. These elements of patient-centered care aim to relieve stress and anxiety in order to achieve a better clinical outcome.
PATIENT-CENTERED CARE
The importance of patient-centered care is illustrated by the results of a 2007 survey in which 41% of patients cited elements of the patient experience as factors that most influenced their choice of hospital.1 Accepted wisdom on patient choice has historically centered on medical factors such as clinical reputation, physician recommendations, and hospital location, each of which was cited by 18% to 21% of the patients surveyed. Elements of the patient experience cited in the study include stress-reducing factors such as the appearance of the room, ease of scheduling, an environment that supports family needs, convenience and comfort of common areas, on-time performance, and simple registration procedures.
Székely et al2 found in a 4-year followup study that high levels of preoperative anxiety predicted greater mortality and cardiovascular morbidity following cardiac surgery. In a study by Tully et al,3 preoperative anxiety was also predictive of hospital readmission following cardiac surgery. Preoperative stress and anxiety are reliable predictors of postoperative distress.4
The variety and relative efficacy of interventions to reduce stress and anxiety are not well studied. Voss et al5 showed that cardiac surgery patients who were played soothing music experienced significantly reduced anxiety, pain, pain distress, and length of hospital stay. One Cleveland Clinic study of massage therapy, however, was unable to demonstrate a statistically significant therapeutic benefit, despite patient satisfaction with the therapy.6
THE ADVENT OF HEALING SERVICES
Identifying patients who exhibit significant preoperative stress and providing, as part of an expanded cardiac care paradigm, emotional care both pre- and postoperatively may ameliorate clinical outcomes. As such, the Heart and Vascular Institute at the Cleveland Clinic formed a healing services division, based on the concept that healing is more than simply physical recovery from a particular procedure. The division’s mission statement is: “To enhance the patient experience by promoting healing through a comprehensive set of coordinated services addressing the holistic needs of the patient.”
At the Cleveland Clinic, healing services are now integrated with standard services to enhance the cardiac care paradigm. Our standard medical services focus on areas of communication and pain control, both of which affect anxiety and stress. The need for enhanced communication is significant: 75% of patients admitted to a Chicago hospital were unable to name a single doctor assigned to their care, and of the remaining 25%, only 40% of responders were correct.7
It is worth noting that communicating more information to a patient is not necessarily better. Patients given detailed preoperative information about their disease and the potential complications of their cardiac surgery had levels of preoperative, perioperative, and postoperative stress, anxiety, and depression similar to those who received routine medical information.8,9 On the other hand, patients desire information about their postoperative plan of care while they are experiencing it, and value communication with physicians, nurses, healing services personnel, and other caregivers when it is presented in a calm and forthright manner. Communications should emphasize that the entire clinical team is there to help the patient get better.
THE FIFTH VITAL SIGN
Pain control is an aspect of care that was long ignored. The goal of the pain control task force at the Cleveland Clinic is the development of effective, efficient, and compassionate pain management.
The fifth vital sign, one that escapes the electronic medical record, can be addressed by this question: “How are you feeling?” Treating pain will reduce stress and anxiety. Before surgery, pain management priorities are discussed with patients, and at each daily encounter the goal is to set, refine, and exceed expectations for pain control through discussion and frequent pain assessments.
Reducing anxiety and stress is the goal of both standard care services and healing services, resulting in more satisfied patients with better clinical outcomes.
CASE: “YOU AND THE TEAM MADE ME GET OUTOF BED AND MOVE FORWARD”
Bobbi is a 78-year-old woman who was initially recovering well following cardiac surgery, including valve surgery, but had to return to the intensive care unit, which is difficult for patients. She was subsequently returned to the floor but was reluctant to walk and progressed slowly, despite normal electrocardiogram, radiographs, and blood panel results. We discovered that her husband was in hospice care in another state, causing Bobbi anxiety as she expressed concern over being her husband’s caregiver while being weakened physically herself. She was fearful of moving forward and her recovery stalled.
The primary care nurse referred her to the healing services team. The healing services team provided support for her anxiety and stress, and reviewed options for managing her husband’s care. She participated in Reiki, spiritual support, and social work services. During her admission her husband died, so the team provided appropriate support.
When asked about her experience upon leavingthe hospital, Bobbi did not mention her surgeon or the success of her heart valve procedure, but commented instead on the healing services team that enabled her to get through the experience.
- Grote KD, Newman JRS, Sutaria SS. A better hospital experience. The McKinsey Quarterly. November 2007.
- Székely A, Balog P, Benkö E, et al. Anxiety predicts mortality and morbidity after coronary artery and valve surgery—a 4-year followup study. Psychosom Med 2007; 69:625–631.
- Tully PJ, Baker RA, Turnbull D, Winefield H. The role of depression and anxiety symptoms in hospital readmissions after cardiac surgery. J Behav Med 2008; 31:281–290.
- Vingerhoets G. Perioperative anxiety and depression in open-heart surgery. Psychosomatics 1998; 39:30–37.
- Voss JA, Good M, Yates B, Baun MM, Thompson A, Hertzog M. Sedative music reduces anxiety and pain during chair rest after open-heart surgery. Pain 2004; 112:197–203.
- Albert NM, Gillinov AM, Lytle BW, Feng J, Cwynar R, Blackstone EH. A randomized trial of massage therapy after heart surgery. Heart Lung 2009; 38:480–490.
- Arora V, Gangireddy S, Mehrotra A, Ginde R, Tormey M, Meltzer D. Ability of hospitalized patients to identify their in-hospital physicians. Arch Intern Med 2009; 169:199–201.
- Ivarsson B, Larsson S, Lührs C, Sjöberg T. Extended written pre-operative information about possible complications at cardiac surgery—do the patients want to know? Eur J Cardiothorac Surg 2005; 28:407–414.
- Bergmann P, Huber S, Mächler H, et al. The influence of medical information on the perioperative course of stress in cardiac surgery patients. Anesth Analg 2001; 93:1093–1099.
- Grote KD, Newman JRS, Sutaria SS. A better hospital experience. The McKinsey Quarterly. November 2007.
- Székely A, Balog P, Benkö E, et al. Anxiety predicts mortality and morbidity after coronary artery and valve surgery—a 4-year followup study. Psychosom Med 2007; 69:625–631.
- Tully PJ, Baker RA, Turnbull D, Winefield H. The role of depression and anxiety symptoms in hospital readmissions after cardiac surgery. J Behav Med 2008; 31:281–290.
- Vingerhoets G. Perioperative anxiety and depression in open-heart surgery. Psychosomatics 1998; 39:30–37.
- Voss JA, Good M, Yates B, Baun MM, Thompson A, Hertzog M. Sedative music reduces anxiety and pain during chair rest after open-heart surgery. Pain 2004; 112:197–203.
- Albert NM, Gillinov AM, Lytle BW, Feng J, Cwynar R, Blackstone EH. A randomized trial of massage therapy after heart surgery. Heart Lung 2009; 38:480–490.
- Arora V, Gangireddy S, Mehrotra A, Ginde R, Tormey M, Meltzer D. Ability of hospitalized patients to identify their in-hospital physicians. Arch Intern Med 2009; 169:199–201.
- Ivarsson B, Larsson S, Lührs C, Sjöberg T. Extended written pre-operative information about possible complications at cardiac surgery—do the patients want to know? Eur J Cardiothorac Surg 2005; 28:407–414.
- Bergmann P, Huber S, Mächler H, et al. The influence of medical information on the perioperative course of stress in cardiac surgery patients. Anesth Analg 2001; 93:1093–1099.
Stress in medicine: Strategies for caregivers, patients, clinicians—Addressing the impact of clinician stress
The impact of clinician stress on the health care system is significant. It can adversely affect the patient experience, compromise patient safety, hinder the delivery of care in a manner that is inconsistent with producing quality outcomes, and increase the overall cost of care.
CLINICIAN STRESS IS PREVALENT
Models of health care that restore human interaction are desperately needed. Clinicians today are overwhelmed by performance assessments that are based on length of stay, use of evidence-based medication regimens, and morbidity and mortality outcomes. Yet clinicians have few opportunities to establish more than cursory relationships with their patients—relationships that would permit better understanding of patients’ emotional well-being and that would optimize the overall healing experience.
Shanafelt et al1 surveyed 7,905 surgeons and found that clinician stress is pervasive: 64% indicated that their work schedule left inadequate time for their personal or family life, 40% reported burnout, and 30% screened positive for symptoms of depression. Another survey of 763 practicing physicians in California found that 53% reported moderate to severe levels of stress.2 Nonphysician clinicians have significant levels of stress as well, with one survey of nurses finding that, of those who quit the profession, 26% cited stress as the cause.3
THE EFFECT OF CLINICIAN STRESSON QUALITY OF CARE
In the Shanafelt et al study, high levels of emotional exhaustion correlated positively with major medical errors over the previous 3 months.1 Nearly 9% of the surgeons surveyed reported making a stress-related major medical mistake in the past 3 months; among those surgeons with high levels of emotional exhaustion, that figure was nearly 15%. This study also found that every 1-point increase in the emotional exhaustion scale (range, 0 to 54) was associated with a 5% increase in the likelihood of reporting a medical error.1
In a study of internal medicine residents, fatigue and distress were associated with medical errors, which were reported by 39% of respondents.4
STRESS AND COMMUNICATION
Stress can damage the physician-nurse relationship, with a significant impact not only on clinicians, but also on delivery of care. The associated breakdowns in communication can negatively affect several areas, including critical care transitions and timely delivery of care. Stress also affects morale, job satisfaction, and job retention.5
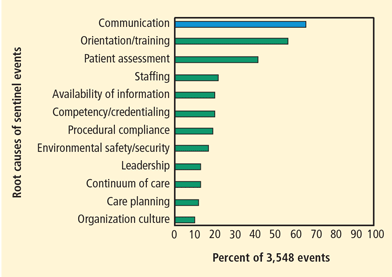
ADDRESSING THE IMPACT OF CLINICIAN STRESS
The traditional response to complaints registered by patients has been behavioral coaching, disruptive-behavior programs, and the punitive use of satisfaction metrics, which are incorporated into the physician’s annual evaluation. These approaches do little to address the cause of the stress and can inculcate cynicism instead.
A more useful approach is to define and strive for an optimal working environment for clinicians, thereby promoting an enhanced patient experience. This approach attempts to restore balance to both the business and art of medicine and may incorporate biofeedback and other healing services to clinicians as tools to minimize and manage stress.
The business of medicine may be restored by enhancing the culture and climate of the hospital, improving communication and collaboration, reducing administrative tasks, restoring authority and autonomy, and eliminating punitive practices. The art of medicine may be restored by valuing the sacred relationship between clinician and patient, learning to listen more carefully to the patient, creating better healing environments, providing emotional support, and supporting caregivers.
- Shanafelt TD, Balch CM, Bechamps G, et al. Burnout and medical errors among American surgeons. Ann Surg 2010; 251:995–1000.
- Beck M. Checking up on the doctor. What patients can learn from the ways physicians take care of themselves. Wall Street Journal. May 25, 2010. http://online.wsj.com/article/SB10001424052748704113504575264364125574500.html?KEYWORDS=Checking+up+on+the+doctor. Accessed April 27, 2011.
- Reineck C, Furino A. Nursing career fulfillment: statistics and statements from registered nurses. Nursing Economics 2005; 23:25–30.
- West CP, Tan AD, Habermann TM, Sloan JA, Shanafelt TD. Association of resident fatigue and distress with perceived medical errors. JAMA 2009; 302:1294–1300.
- Rosenstein AH. Nurse-physician relationships: Impact on nurses atisfaction and retention. Am J Nursing 2002; 102:26–34.
- Hickam DH, Severance S, Feldstein A, et al; Oregon Health & Science University Evidence-based Practice Center. The effect of health care working conditions on patient safety. Agency for Healthcare Research and Quality publication 03-E031. http://www.ahrq.gov/downloads/pub/evidence/pdf/work/work.pdf. Published May 2003. Accessed April 27, 2011.
The impact of clinician stress on the health care system is significant. It can adversely affect the patient experience, compromise patient safety, hinder the delivery of care in a manner that is inconsistent with producing quality outcomes, and increase the overall cost of care.
CLINICIAN STRESS IS PREVALENT
Models of health care that restore human interaction are desperately needed. Clinicians today are overwhelmed by performance assessments that are based on length of stay, use of evidence-based medication regimens, and morbidity and mortality outcomes. Yet clinicians have few opportunities to establish more than cursory relationships with their patients—relationships that would permit better understanding of patients’ emotional well-being and that would optimize the overall healing experience.
Shanafelt et al1 surveyed 7,905 surgeons and found that clinician stress is pervasive: 64% indicated that their work schedule left inadequate time for their personal or family life, 40% reported burnout, and 30% screened positive for symptoms of depression. Another survey of 763 practicing physicians in California found that 53% reported moderate to severe levels of stress.2 Nonphysician clinicians have significant levels of stress as well, with one survey of nurses finding that, of those who quit the profession, 26% cited stress as the cause.3
THE EFFECT OF CLINICIAN STRESSON QUALITY OF CARE
In the Shanafelt et al study, high levels of emotional exhaustion correlated positively with major medical errors over the previous 3 months.1 Nearly 9% of the surgeons surveyed reported making a stress-related major medical mistake in the past 3 months; among those surgeons with high levels of emotional exhaustion, that figure was nearly 15%. This study also found that every 1-point increase in the emotional exhaustion scale (range, 0 to 54) was associated with a 5% increase in the likelihood of reporting a medical error.1
In a study of internal medicine residents, fatigue and distress were associated with medical errors, which were reported by 39% of respondents.4
STRESS AND COMMUNICATION
Stress can damage the physician-nurse relationship, with a significant impact not only on clinicians, but also on delivery of care. The associated breakdowns in communication can negatively affect several areas, including critical care transitions and timely delivery of care. Stress also affects morale, job satisfaction, and job retention.5
ADDRESSING THE IMPACT OF CLINICIAN STRESS
The traditional response to complaints registered by patients has been behavioral coaching, disruptive-behavior programs, and the punitive use of satisfaction metrics, which are incorporated into the physician’s annual evaluation. These approaches do little to address the cause of the stress and can inculcate cynicism instead.
A more useful approach is to define and strive for an optimal working environment for clinicians, thereby promoting an enhanced patient experience. This approach attempts to restore balance to both the business and art of medicine and may incorporate biofeedback and other healing services to clinicians as tools to minimize and manage stress.
The business of medicine may be restored by enhancing the culture and climate of the hospital, improving communication and collaboration, reducing administrative tasks, restoring authority and autonomy, and eliminating punitive practices. The art of medicine may be restored by valuing the sacred relationship between clinician and patient, learning to listen more carefully to the patient, creating better healing environments, providing emotional support, and supporting caregivers.
The impact of clinician stress on the health care system is significant. It can adversely affect the patient experience, compromise patient safety, hinder the delivery of care in a manner that is inconsistent with producing quality outcomes, and increase the overall cost of care.
CLINICIAN STRESS IS PREVALENT
Models of health care that restore human interaction are desperately needed. Clinicians today are overwhelmed by performance assessments that are based on length of stay, use of evidence-based medication regimens, and morbidity and mortality outcomes. Yet clinicians have few opportunities to establish more than cursory relationships with their patients—relationships that would permit better understanding of patients’ emotional well-being and that would optimize the overall healing experience.
Shanafelt et al1 surveyed 7,905 surgeons and found that clinician stress is pervasive: 64% indicated that their work schedule left inadequate time for their personal or family life, 40% reported burnout, and 30% screened positive for symptoms of depression. Another survey of 763 practicing physicians in California found that 53% reported moderate to severe levels of stress.2 Nonphysician clinicians have significant levels of stress as well, with one survey of nurses finding that, of those who quit the profession, 26% cited stress as the cause.3
THE EFFECT OF CLINICIAN STRESSON QUALITY OF CARE
In the Shanafelt et al study, high levels of emotional exhaustion correlated positively with major medical errors over the previous 3 months.1 Nearly 9% of the surgeons surveyed reported making a stress-related major medical mistake in the past 3 months; among those surgeons with high levels of emotional exhaustion, that figure was nearly 15%. This study also found that every 1-point increase in the emotional exhaustion scale (range, 0 to 54) was associated with a 5% increase in the likelihood of reporting a medical error.1
In a study of internal medicine residents, fatigue and distress were associated with medical errors, which were reported by 39% of respondents.4
STRESS AND COMMUNICATION
Stress can damage the physician-nurse relationship, with a significant impact not only on clinicians, but also on delivery of care. The associated breakdowns in communication can negatively affect several areas, including critical care transitions and timely delivery of care. Stress also affects morale, job satisfaction, and job retention.5
ADDRESSING THE IMPACT OF CLINICIAN STRESS
The traditional response to complaints registered by patients has been behavioral coaching, disruptive-behavior programs, and the punitive use of satisfaction metrics, which are incorporated into the physician’s annual evaluation. These approaches do little to address the cause of the stress and can inculcate cynicism instead.
A more useful approach is to define and strive for an optimal working environment for clinicians, thereby promoting an enhanced patient experience. This approach attempts to restore balance to both the business and art of medicine and may incorporate biofeedback and other healing services to clinicians as tools to minimize and manage stress.
The business of medicine may be restored by enhancing the culture and climate of the hospital, improving communication and collaboration, reducing administrative tasks, restoring authority and autonomy, and eliminating punitive practices. The art of medicine may be restored by valuing the sacred relationship between clinician and patient, learning to listen more carefully to the patient, creating better healing environments, providing emotional support, and supporting caregivers.
- Shanafelt TD, Balch CM, Bechamps G, et al. Burnout and medical errors among American surgeons. Ann Surg 2010; 251:995–1000.
- Beck M. Checking up on the doctor. What patients can learn from the ways physicians take care of themselves. Wall Street Journal. May 25, 2010. http://online.wsj.com/article/SB10001424052748704113504575264364125574500.html?KEYWORDS=Checking+up+on+the+doctor. Accessed April 27, 2011.
- Reineck C, Furino A. Nursing career fulfillment: statistics and statements from registered nurses. Nursing Economics 2005; 23:25–30.
- West CP, Tan AD, Habermann TM, Sloan JA, Shanafelt TD. Association of resident fatigue and distress with perceived medical errors. JAMA 2009; 302:1294–1300.
- Rosenstein AH. Nurse-physician relationships: Impact on nurses atisfaction and retention. Am J Nursing 2002; 102:26–34.
- Hickam DH, Severance S, Feldstein A, et al; Oregon Health & Science University Evidence-based Practice Center. The effect of health care working conditions on patient safety. Agency for Healthcare Research and Quality publication 03-E031. http://www.ahrq.gov/downloads/pub/evidence/pdf/work/work.pdf. Published May 2003. Accessed April 27, 2011.
- Shanafelt TD, Balch CM, Bechamps G, et al. Burnout and medical errors among American surgeons. Ann Surg 2010; 251:995–1000.
- Beck M. Checking up on the doctor. What patients can learn from the ways physicians take care of themselves. Wall Street Journal. May 25, 2010. http://online.wsj.com/article/SB10001424052748704113504575264364125574500.html?KEYWORDS=Checking+up+on+the+doctor. Accessed April 27, 2011.
- Reineck C, Furino A. Nursing career fulfillment: statistics and statements from registered nurses. Nursing Economics 2005; 23:25–30.
- West CP, Tan AD, Habermann TM, Sloan JA, Shanafelt TD. Association of resident fatigue and distress with perceived medical errors. JAMA 2009; 302:1294–1300.
- Rosenstein AH. Nurse-physician relationships: Impact on nurses atisfaction and retention. Am J Nursing 2002; 102:26–34.
- Hickam DH, Severance S, Feldstein A, et al; Oregon Health & Science University Evidence-based Practice Center. The effect of health care working conditions on patient safety. Agency for Healthcare Research and Quality publication 03-E031. http://www.ahrq.gov/downloads/pub/evidence/pdf/work/work.pdf. Published May 2003. Accessed April 27, 2011.
Stress in medicine: Strategies for caregivers, patients, clinicians—Biofeedback in the treatment of stress
Traditionally, biofeedback was considered to be a stress management technique that targeted sympathetic nervous system (SNS) overdrive with an adrenal medullary system backup. Recent advances in autonomic physiology, however, have clarified that except in extreme situations, the SNS is not the key factor in day-to-day stress. Rather, the parasympathetic branch of the autonomic nervous system appears to be a more likely candidate for mediating routine stress because, unlike the SNS, which has slow-acting neurotransmitters (ie, catecholamines), the parasympathetic nervous system has the fast-acting transmitter acetylcholine.
VAGAL WITHDRAWAL: AN ALTERNATIVE TO SYMPATHETIC ACTIVATION
Porges1 first proposed the concept of vagal withdrawal as an indicator of stress and stress vulnerability; this contrasts with the idea that the stress response is a consequence of sympathetic activation and the hypothalamic-pituitary-adrenal axis response. In the vagal withdrawal model, the response to stress is stabilization of the sympathetic system followed by termination of parasympathetic activity, manifested as cardiac acceleration.
Respiratory sinus arrhythmia (RSA), or the variability in heart rate as it synchronizes with breathing, is considered an index of parasympathetic tone. In the laboratory, slow atropine infusion produces a transient paradoxical vagomimetic effect characterized by an initial increase in RSA, followed by a flattening and then a rise in the heart rate.2 This phenomenon has been measured in people during times of routine stress, such as when worrying about being late for an appointment. In such individuals, biofeedback training can result in recovery of normal RSA shortly after an episode of anxiety.
Historically, the focus of biofeedback was to cultivate low arousal, presumably reducing SNS activity, through the use of finger temperature, skin conductance training, and profound muscle relaxation. More sophisticated ways to look at both branches of the autonomic nervous system have since emerged that allow for sampling of the beat-by-beat changes in heart rate.
HEART RATE VARIABILITY BIOFEEDBACK
The concept of modifying the respiration rate (paced breathing) originated some 2,500 years ago as a component of meditation. It is being revisited today in the form of heart rate variability (HRV) biofeedback training, which is being used as a stress-management tool and a method to correct disorders in which autonomic regulation is thought to be important. HRV biofeedback involves training to increase the amplitude of HRV rhythms and thus improve autonomic homeostasis.
Normal HRV has a pattern of overlapping oscillatory frequency components, including:
- a high-frequency rhythm, 0.15 to 0.4 Hz, which is the RSA;
- a low-frequency rhythm, 0.05 to 0.15 Hz, associated with blood pressure oscillations; and
- a very-low-frequency rhythm, 0.005 to 0.05 Hz, which may regulate vascular tone and body temperature.
The goal of HRV biofeedback is to achieve respiratory rates at which resonance occurs between cardiac rhythms associated with respiration (RSA, or high-frequency oscillations) and those caused by baroreflex activity (low-frequency oscillations).
Spectral analysis has demonstrated that nearly all of the activity with HRV biofeedback occurs at a low-frequency band. The reason is that activity in the low-frequency band is related more to baroreflex activity than to HRV compared with other ranges of frequency. Breathing rates that correspond to baroreflex effects, called resonance frequency breathing, represent resonance in the cardiovascular system. Several devices are available whose mechanisms are based on the concept of achieving resonance frequency breathing. One such device is a slow-breathing monitor (Resp-e-rate) that has been approved by the US Food and Drug Administration for the adjunctive treatment of hypertension.
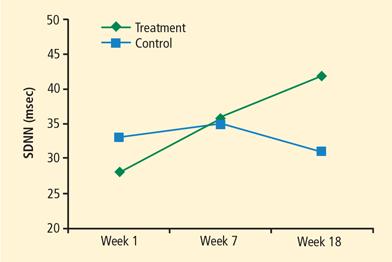
Improved HRV may suggest an improved risk status: Kleiger et al4 found that the relative risk of mortality was 5.3 times greater for people with SDNN of less than 50 msec compared with those whose SDNN was greater than 100 msec. In Del Pozo’s study, eight of 30 patients in the intervention group achieved an SDNN of greater than 50 msec (vs 0 at pretreatment) compared with three of 31 controls (vs two at pretreatment).3 As an additional benefit of HRV biofeedback, patients in the intervention group who entered the study with hypertension all became normotensive.
In a meta-analysis, van Dixhoorn and White5 found fewer cardiac events, fewer episodes of angina, and less occurrence of arrhythmia and exercise-induced ischemia from intensive supervised relaxation therapy in patients with ischemic heart disease. Improvements in scales of depression and anxiety were also observed with relaxation therapy.
Other studies have shown biofeedback to have beneficial effects based on the Posttraumatic Stress Disorder Checklist, the Hamilton Depression Rating Scale, and, in patients with mild to moderate heartfailure, the 6-minute walk test.6–8
The proposed mechanism for the beneficial effects of biofeedback found in clinical trials is improvement in baroreflex function, producing greater reflex efficiency and improved modulation of autonomic activity.
CONCLUSION
A shift in emphasis to vagal withdrawal has led to new forms of biofeedback that probably potentiate many of the same mechanisms thought to be present in Eastern practices such as yoga and tai chi. Results from small-scale trials have been promising for HRV biofeedback as a means of modifying responses to stress and promoting homeostatic processes that reduce the intensity of symptoms and improve surrogate markers associated with a number of disorders.
- Porges SW. Cardiac vagal tone: a physiological index of stress. Neurosci Biobehav Rev 1995; 19:225–233.
- Médigue C, Girard A, Laude D, Monti A, Wargon M, ElghoziJ-L. Relationship between pulse interval and respiratory sinusarrhythmia: a time- and frequency-domain analysis of the effects ofatropine. Eur J Physiol 2001; 441:650–655.
- Del Pozo JM, Gevirtz RN, Scher B, Guarneri E. Biofeedbacktreatment increases heart rate variability in patients withknown coronary artery disease. Am Heart J 2004; 147:e11. http://download.journals.elsevierhealth.com/pdfs/journals/0002-8703/PIIS0002870303007191.pdf. Accessed May 2, 2011.
- Kleiger RE, Miller JP, Bigger JT Jr, Moss AJ. Decreased heart ratevariability and its association with increased mortality after acutemyocardial infarciton. Am J Cardiol 1987; 59:256–262.
- van Dixhoorn JV, White A. Relaxation therapy for rehabilitationand prevention in ischaemic heart disease: a systematic review andmeta-analysis. Eur J Cardiovasc Prev Rehabil 2005; 12:193–202.
- Karavidas MK, Lehrer PM, Vaschillo E, et al. Preliminary resultsof an open label study of heart rate variability biofeedback for thetreatment of major depression. Appl Psychophysiol Biofeedback2007; 32:19–30.
- Zucker TL, Samuelson KW, Muench F, Greenberg MA, GevirtzRN. The effects of respiratory sinus arrhythmia biofeedback onheart rate variability and posttraumatic stress disorder symptoms: apilot study. Appl Psychophysiol Biofeedback 2009; 34:135–143.
- Swanson KS, Gevirtz RN, Brown M, Spira J, Guarneri E, StoletniyL. The effect of biofeedback on function in patients with heartfailure. Appl Psychophysiol Biofeedback 2009; 34:71–91.
Traditionally, biofeedback was considered to be a stress management technique that targeted sympathetic nervous system (SNS) overdrive with an adrenal medullary system backup. Recent advances in autonomic physiology, however, have clarified that except in extreme situations, the SNS is not the key factor in day-to-day stress. Rather, the parasympathetic branch of the autonomic nervous system appears to be a more likely candidate for mediating routine stress because, unlike the SNS, which has slow-acting neurotransmitters (ie, catecholamines), the parasympathetic nervous system has the fast-acting transmitter acetylcholine.
VAGAL WITHDRAWAL: AN ALTERNATIVE TO SYMPATHETIC ACTIVATION
Porges1 first proposed the concept of vagal withdrawal as an indicator of stress and stress vulnerability; this contrasts with the idea that the stress response is a consequence of sympathetic activation and the hypothalamic-pituitary-adrenal axis response. In the vagal withdrawal model, the response to stress is stabilization of the sympathetic system followed by termination of parasympathetic activity, manifested as cardiac acceleration.
Respiratory sinus arrhythmia (RSA), or the variability in heart rate as it synchronizes with breathing, is considered an index of parasympathetic tone. In the laboratory, slow atropine infusion produces a transient paradoxical vagomimetic effect characterized by an initial increase in RSA, followed by a flattening and then a rise in the heart rate.2 This phenomenon has been measured in people during times of routine stress, such as when worrying about being late for an appointment. In such individuals, biofeedback training can result in recovery of normal RSA shortly after an episode of anxiety.
Historically, the focus of biofeedback was to cultivate low arousal, presumably reducing SNS activity, through the use of finger temperature, skin conductance training, and profound muscle relaxation. More sophisticated ways to look at both branches of the autonomic nervous system have since emerged that allow for sampling of the beat-by-beat changes in heart rate.
HEART RATE VARIABILITY BIOFEEDBACK
The concept of modifying the respiration rate (paced breathing) originated some 2,500 years ago as a component of meditation. It is being revisited today in the form of heart rate variability (HRV) biofeedback training, which is being used as a stress-management tool and a method to correct disorders in which autonomic regulation is thought to be important. HRV biofeedback involves training to increase the amplitude of HRV rhythms and thus improve autonomic homeostasis.
Normal HRV has a pattern of overlapping oscillatory frequency components, including:
- a high-frequency rhythm, 0.15 to 0.4 Hz, which is the RSA;
- a low-frequency rhythm, 0.05 to 0.15 Hz, associated with blood pressure oscillations; and
- a very-low-frequency rhythm, 0.005 to 0.05 Hz, which may regulate vascular tone and body temperature.
The goal of HRV biofeedback is to achieve respiratory rates at which resonance occurs between cardiac rhythms associated with respiration (RSA, or high-frequency oscillations) and those caused by baroreflex activity (low-frequency oscillations).
Spectral analysis has demonstrated that nearly all of the activity with HRV biofeedback occurs at a low-frequency band. The reason is that activity in the low-frequency band is related more to baroreflex activity than to HRV compared with other ranges of frequency. Breathing rates that correspond to baroreflex effects, called resonance frequency breathing, represent resonance in the cardiovascular system. Several devices are available whose mechanisms are based on the concept of achieving resonance frequency breathing. One such device is a slow-breathing monitor (Resp-e-rate) that has been approved by the US Food and Drug Administration for the adjunctive treatment of hypertension.
Improved HRV may suggest an improved risk status: Kleiger et al4 found that the relative risk of mortality was 5.3 times greater for people with SDNN of less than 50 msec compared with those whose SDNN was greater than 100 msec. In Del Pozo’s study, eight of 30 patients in the intervention group achieved an SDNN of greater than 50 msec (vs 0 at pretreatment) compared with three of 31 controls (vs two at pretreatment).3 As an additional benefit of HRV biofeedback, patients in the intervention group who entered the study with hypertension all became normotensive.
In a meta-analysis, van Dixhoorn and White5 found fewer cardiac events, fewer episodes of angina, and less occurrence of arrhythmia and exercise-induced ischemia from intensive supervised relaxation therapy in patients with ischemic heart disease. Improvements in scales of depression and anxiety were also observed with relaxation therapy.
Other studies have shown biofeedback to have beneficial effects based on the Posttraumatic Stress Disorder Checklist, the Hamilton Depression Rating Scale, and, in patients with mild to moderate heartfailure, the 6-minute walk test.6–8
The proposed mechanism for the beneficial effects of biofeedback found in clinical trials is improvement in baroreflex function, producing greater reflex efficiency and improved modulation of autonomic activity.
CONCLUSION
A shift in emphasis to vagal withdrawal has led to new forms of biofeedback that probably potentiate many of the same mechanisms thought to be present in Eastern practices such as yoga and tai chi. Results from small-scale trials have been promising for HRV biofeedback as a means of modifying responses to stress and promoting homeostatic processes that reduce the intensity of symptoms and improve surrogate markers associated with a number of disorders.
Traditionally, biofeedback was considered to be a stress management technique that targeted sympathetic nervous system (SNS) overdrive with an adrenal medullary system backup. Recent advances in autonomic physiology, however, have clarified that except in extreme situations, the SNS is not the key factor in day-to-day stress. Rather, the parasympathetic branch of the autonomic nervous system appears to be a more likely candidate for mediating routine stress because, unlike the SNS, which has slow-acting neurotransmitters (ie, catecholamines), the parasympathetic nervous system has the fast-acting transmitter acetylcholine.
VAGAL WITHDRAWAL: AN ALTERNATIVE TO SYMPATHETIC ACTIVATION
Porges1 first proposed the concept of vagal withdrawal as an indicator of stress and stress vulnerability; this contrasts with the idea that the stress response is a consequence of sympathetic activation and the hypothalamic-pituitary-adrenal axis response. In the vagal withdrawal model, the response to stress is stabilization of the sympathetic system followed by termination of parasympathetic activity, manifested as cardiac acceleration.
Respiratory sinus arrhythmia (RSA), or the variability in heart rate as it synchronizes with breathing, is considered an index of parasympathetic tone. In the laboratory, slow atropine infusion produces a transient paradoxical vagomimetic effect characterized by an initial increase in RSA, followed by a flattening and then a rise in the heart rate.2 This phenomenon has been measured in people during times of routine stress, such as when worrying about being late for an appointment. In such individuals, biofeedback training can result in recovery of normal RSA shortly after an episode of anxiety.
Historically, the focus of biofeedback was to cultivate low arousal, presumably reducing SNS activity, through the use of finger temperature, skin conductance training, and profound muscle relaxation. More sophisticated ways to look at both branches of the autonomic nervous system have since emerged that allow for sampling of the beat-by-beat changes in heart rate.
HEART RATE VARIABILITY BIOFEEDBACK
The concept of modifying the respiration rate (paced breathing) originated some 2,500 years ago as a component of meditation. It is being revisited today in the form of heart rate variability (HRV) biofeedback training, which is being used as a stress-management tool and a method to correct disorders in which autonomic regulation is thought to be important. HRV biofeedback involves training to increase the amplitude of HRV rhythms and thus improve autonomic homeostasis.
Normal HRV has a pattern of overlapping oscillatory frequency components, including:
- a high-frequency rhythm, 0.15 to 0.4 Hz, which is the RSA;
- a low-frequency rhythm, 0.05 to 0.15 Hz, associated with blood pressure oscillations; and
- a very-low-frequency rhythm, 0.005 to 0.05 Hz, which may regulate vascular tone and body temperature.
The goal of HRV biofeedback is to achieve respiratory rates at which resonance occurs between cardiac rhythms associated with respiration (RSA, or high-frequency oscillations) and those caused by baroreflex activity (low-frequency oscillations).
Spectral analysis has demonstrated that nearly all of the activity with HRV biofeedback occurs at a low-frequency band. The reason is that activity in the low-frequency band is related more to baroreflex activity than to HRV compared with other ranges of frequency. Breathing rates that correspond to baroreflex effects, called resonance frequency breathing, represent resonance in the cardiovascular system. Several devices are available whose mechanisms are based on the concept of achieving resonance frequency breathing. One such device is a slow-breathing monitor (Resp-e-rate) that has been approved by the US Food and Drug Administration for the adjunctive treatment of hypertension.
Improved HRV may suggest an improved risk status: Kleiger et al4 found that the relative risk of mortality was 5.3 times greater for people with SDNN of less than 50 msec compared with those whose SDNN was greater than 100 msec. In Del Pozo’s study, eight of 30 patients in the intervention group achieved an SDNN of greater than 50 msec (vs 0 at pretreatment) compared with three of 31 controls (vs two at pretreatment).3 As an additional benefit of HRV biofeedback, patients in the intervention group who entered the study with hypertension all became normotensive.
In a meta-analysis, van Dixhoorn and White5 found fewer cardiac events, fewer episodes of angina, and less occurrence of arrhythmia and exercise-induced ischemia from intensive supervised relaxation therapy in patients with ischemic heart disease. Improvements in scales of depression and anxiety were also observed with relaxation therapy.
Other studies have shown biofeedback to have beneficial effects based on the Posttraumatic Stress Disorder Checklist, the Hamilton Depression Rating Scale, and, in patients with mild to moderate heartfailure, the 6-minute walk test.6–8
The proposed mechanism for the beneficial effects of biofeedback found in clinical trials is improvement in baroreflex function, producing greater reflex efficiency and improved modulation of autonomic activity.
CONCLUSION
A shift in emphasis to vagal withdrawal has led to new forms of biofeedback that probably potentiate many of the same mechanisms thought to be present in Eastern practices such as yoga and tai chi. Results from small-scale trials have been promising for HRV biofeedback as a means of modifying responses to stress and promoting homeostatic processes that reduce the intensity of symptoms and improve surrogate markers associated with a number of disorders.
- Porges SW. Cardiac vagal tone: a physiological index of stress. Neurosci Biobehav Rev 1995; 19:225–233.
- Médigue C, Girard A, Laude D, Monti A, Wargon M, ElghoziJ-L. Relationship between pulse interval and respiratory sinusarrhythmia: a time- and frequency-domain analysis of the effects ofatropine. Eur J Physiol 2001; 441:650–655.
- Del Pozo JM, Gevirtz RN, Scher B, Guarneri E. Biofeedbacktreatment increases heart rate variability in patients withknown coronary artery disease. Am Heart J 2004; 147:e11. http://download.journals.elsevierhealth.com/pdfs/journals/0002-8703/PIIS0002870303007191.pdf. Accessed May 2, 2011.
- Kleiger RE, Miller JP, Bigger JT Jr, Moss AJ. Decreased heart ratevariability and its association with increased mortality after acutemyocardial infarciton. Am J Cardiol 1987; 59:256–262.
- van Dixhoorn JV, White A. Relaxation therapy for rehabilitationand prevention in ischaemic heart disease: a systematic review andmeta-analysis. Eur J Cardiovasc Prev Rehabil 2005; 12:193–202.
- Karavidas MK, Lehrer PM, Vaschillo E, et al. Preliminary resultsof an open label study of heart rate variability biofeedback for thetreatment of major depression. Appl Psychophysiol Biofeedback2007; 32:19–30.
- Zucker TL, Samuelson KW, Muench F, Greenberg MA, GevirtzRN. The effects of respiratory sinus arrhythmia biofeedback onheart rate variability and posttraumatic stress disorder symptoms: apilot study. Appl Psychophysiol Biofeedback 2009; 34:135–143.
- Swanson KS, Gevirtz RN, Brown M, Spira J, Guarneri E, StoletniyL. The effect of biofeedback on function in patients with heartfailure. Appl Psychophysiol Biofeedback 2009; 34:71–91.
- Porges SW. Cardiac vagal tone: a physiological index of stress. Neurosci Biobehav Rev 1995; 19:225–233.
- Médigue C, Girard A, Laude D, Monti A, Wargon M, ElghoziJ-L. Relationship between pulse interval and respiratory sinusarrhythmia: a time- and frequency-domain analysis of the effects ofatropine. Eur J Physiol 2001; 441:650–655.
- Del Pozo JM, Gevirtz RN, Scher B, Guarneri E. Biofeedbacktreatment increases heart rate variability in patients withknown coronary artery disease. Am Heart J 2004; 147:e11. http://download.journals.elsevierhealth.com/pdfs/journals/0002-8703/PIIS0002870303007191.pdf. Accessed May 2, 2011.
- Kleiger RE, Miller JP, Bigger JT Jr, Moss AJ. Decreased heart ratevariability and its association with increased mortality after acutemyocardial infarciton. Am J Cardiol 1987; 59:256–262.
- van Dixhoorn JV, White A. Relaxation therapy for rehabilitationand prevention in ischaemic heart disease: a systematic review andmeta-analysis. Eur J Cardiovasc Prev Rehabil 2005; 12:193–202.
- Karavidas MK, Lehrer PM, Vaschillo E, et al. Preliminary resultsof an open label study of heart rate variability biofeedback for thetreatment of major depression. Appl Psychophysiol Biofeedback2007; 32:19–30.
- Zucker TL, Samuelson KW, Muench F, Greenberg MA, GevirtzRN. The effects of respiratory sinus arrhythmia biofeedback onheart rate variability and posttraumatic stress disorder symptoms: apilot study. Appl Psychophysiol Biofeedback 2009; 34:135–143.
- Swanson KS, Gevirtz RN, Brown M, Spira J, Guarneri E, StoletniyL. The effect of biofeedback on function in patients with heartfailure. Appl Psychophysiol Biofeedback 2009; 34:71–91.
Stress in medicine: Strategies for caregivers, patients, clinicians—Biofeedback for extreme stress: Wounded warriors
Posttraumatic stress disorder (PTSD) is a severe anxiety disorder whose symptoms emerge following exposure to extreme stress, such as those encountered in the battlefield or as a result of sexual abuse or natural disasters. The ability to employ coping mechanisms affects the disorder’s presentation as well as the frequency, intensity, and duration of the symptoms. The “Wounded Warrior” program at East Carolina University (Greenville, NC) was developed to promote the functional independence of US Marines, including those with PTSD.
STRESS RESPONSE: INTERACTION OF THE BRAIN AND IMMUNE SYSTEM
Walter Cannon coined the “flight or fight” response to stress in the early 20th century, in which he emphasized the importance of the parasympathetic system.1 In 1988, Folkow clarified the description as an immune response to stress.2 The stress response is now understood to be a neuroendocrine function that includes a feedback loop between the hypothalamus and the pituitary and adrenal glands; stimulation of the hypothalamus promotes secretion of corticotropin-releasing hormone (CRH) into the hypophyseal portal system, which supplies the anterior pituitarywith blood. CRH stimulates the secretion of adrenocorticotropic hormone into the bloodstream by the pituitary, prompting the adrenal glands to release the stress hormone cortisol.
Cortisol mobilizes the body’s defenses to meet the challenge of an adverse situation. It modulates the stress response by inhibiting the further release of CRH by the hypothalamus. Cortisol thus protects healthy cells and tissues by inhibiting an overreaction from the immune system. Without this protective effect, the interaction between the brain and the immune system can become dysregulated, increasing the risk of immune disorders.
THE CENTRAL AUTONOMIC NETWORK
The central nervous system that regulates the overall balance of the autonomic nervous system (ANS) has been called the central autonomic network (CAN).3 The CAN helps control executive, social, affective, attentional, and motivational functions. Therefore, the old paradigm of simply decreasing hyperarousal of the ANS to treat negative affective states and dispositions is inadequate. Instead, restoring the appropriate relationship between the ANS and the central nervous system is the aim behind interventions to treat PTSD.
Autonomic, cognitive, and affective functions assist humans in maintaining balance when confronted with external challenges. The CAN controls inhibitory or negative processes that permit specific behavior and redeploy resources needed elsewhere. When negative circuits are compromised, positive circuits develop, resulting in hypervigilance, the symptoms of which can be devastating and, if not ameliorated, can develop into permanent conditions. In one study,Vietnam veterans with PTSD had an 8% reduction in the volume of their right hippocampus compared with veterans without PTSD. Another study calculated a 26% reduction in the left hippocampus and a 22% reduction in the right in veterans with the most severe PTSD compared with veterans who were in combat but had no PTSD symptoms.4
A common subcortical neural system regulates defensive behavior, including autonomic, emotional, and cognitive behavior. When the prefrontal cortex is taken “off line” for whatever reason, parasympathetic inhibitory action is withdrawn, and relative sympathetic dominance, associated with defense, occurs.
CONFRONTING HYPERAROUSAL
The question then arises of how to train the ANS to avoid hypervigilance. Growing evidence supports the use of heart rate variability as a predictor of hypervigilance and inefficient allocation of attentional and cognitive resources.
The overall objective of heart rate variability training is to decrease ANS hyperarousal and to improve its balance. “Wounded warriors” learn to control ANS responses to stress-producing stimuli (eg, thoughts, memories, and images associated with combat). The goal of training is to decrease arousal and maintain ANS balance for increasing lengths of time.
Once it was observed that alpha waves were dysfunctional in vulnerable populations, protocols were developed to train alpha and theta waves as a method of improving function. Peniston and colleagues5–9 showed that increased alpha and theta brain wave production resulted in normalized personality measures and prolonged the period of time before relapse in alcoholics. This protocol has also shown efficacy as an intervention in depression and PTSD.
BIOFEEDBACK TRAINING PROGRAM
The US Department of Defense is studying a combination of central nervous system biofeedback with ANS biofeedback, with the goal of restoring and maintaining tone between the systems.
The training program used in the study lasts 1 month, and starts with a session for preassessment, 16 biofeedback sessions (four per week), a postprogram evaluation, and a 3-month followup. Each week, participants are exposed to stress-producing stimuli that increase in intensity:
- Week 1: Stroop Color Word Test, math stressor, talk stressor/everyday events
- Week 2: Talk stressor, combat experiences
- Week 3: Images and sounds of combat
- Week 4: Virtual Baghdad or Afghanistan (virtual reality exposure)
Each biofeedback session consists of 5 minutes of baseline evaluation; 5 minutes in which the veteran is subjected to the weekly stressor; 20 minutes of heart rate variability and neurofeedback training; 5 more minutes of training with the weekly stressor; 20 more minutes of heart rate variability and neurofeedback training; and finally 5 minutes of recovery.
SUMMARY
Dysfunction in the balance of both the ANS and central nervous system is associated with symptoms of PTSD in combat veterans. Methods that are designed to restore balance in these systems are needed to ameliorate these symptoms. Biofeedback and neurofeedback are safe methods with which to achieve these goals.
- Cannon WB. Bodily Changes in Pain, Hunger, Fear and Rage: An Account of Recent Researches into the Function of Emotional Excitement. 2nd ed. New York, NY: Appleton-Century-Crofts; 1929.
- Folkow B. Stress, hypothalamic function and neuroendocrine consequences. Acta Med Scand Suppl 1988; 723:61–69.
- Thayer JF, Brosschot JF. Psychosomatics and psychopathology: looking up and down from the brain. Psychoneuroendocrinology 2005; 30:1050–1058.
- van der Kolk BA. The psychobiology and psychopharmacology of PTSD. Hum Psychopharmacol 2001; 16:S49–S64.
- Peniston EG, Kulkosky PJ. Alpha-theta brainwave training and beta-endorphin levels in alcoholics. Alcohol Clin Exp Res 1989;13:271–279.
- Peniston EG, Kulkosky PJ. Alcoholic personality and alpha-thetabrainwave training. Medical Psychotherapy: An International Journal1990; 3:37–55.
- Peniston EG, Kulkosky PJ. Alpha-theta brainwave neurofeedbacktherapy for Vietnam veterans with combat-related posttraumaticstress disorder. Medical Psychotherapy: An International Journal1991; 4:47–60.
- Peniston EG, Kulkosky PJ. Alpha-theta EEG biofeedback trainingin alcoholism and posttraumatic stress disorder. The InternationalSociety for the Study of Subtle Energies and Energy Medicines1992; 2:5–7.
- Peniston EG, Marrinan DA, Deming WA, Kulkosky PJ. EEGalpha-theta brainwave synchronization in Vietnam theater veteranswith combat-related posttraumatic stress disorder and alcohol abuse.Medical Psychotherapy: An International Journal 1993; 6:37–50.
Posttraumatic stress disorder (PTSD) is a severe anxiety disorder whose symptoms emerge following exposure to extreme stress, such as those encountered in the battlefield or as a result of sexual abuse or natural disasters. The ability to employ coping mechanisms affects the disorder’s presentation as well as the frequency, intensity, and duration of the symptoms. The “Wounded Warrior” program at East Carolina University (Greenville, NC) was developed to promote the functional independence of US Marines, including those with PTSD.
STRESS RESPONSE: INTERACTION OF THE BRAIN AND IMMUNE SYSTEM
Walter Cannon coined the “flight or fight” response to stress in the early 20th century, in which he emphasized the importance of the parasympathetic system.1 In 1988, Folkow clarified the description as an immune response to stress.2 The stress response is now understood to be a neuroendocrine function that includes a feedback loop between the hypothalamus and the pituitary and adrenal glands; stimulation of the hypothalamus promotes secretion of corticotropin-releasing hormone (CRH) into the hypophyseal portal system, which supplies the anterior pituitarywith blood. CRH stimulates the secretion of adrenocorticotropic hormone into the bloodstream by the pituitary, prompting the adrenal glands to release the stress hormone cortisol.
Cortisol mobilizes the body’s defenses to meet the challenge of an adverse situation. It modulates the stress response by inhibiting the further release of CRH by the hypothalamus. Cortisol thus protects healthy cells and tissues by inhibiting an overreaction from the immune system. Without this protective effect, the interaction between the brain and the immune system can become dysregulated, increasing the risk of immune disorders.
THE CENTRAL AUTONOMIC NETWORK
The central nervous system that regulates the overall balance of the autonomic nervous system (ANS) has been called the central autonomic network (CAN).3 The CAN helps control executive, social, affective, attentional, and motivational functions. Therefore, the old paradigm of simply decreasing hyperarousal of the ANS to treat negative affective states and dispositions is inadequate. Instead, restoring the appropriate relationship between the ANS and the central nervous system is the aim behind interventions to treat PTSD.
Autonomic, cognitive, and affective functions assist humans in maintaining balance when confronted with external challenges. The CAN controls inhibitory or negative processes that permit specific behavior and redeploy resources needed elsewhere. When negative circuits are compromised, positive circuits develop, resulting in hypervigilance, the symptoms of which can be devastating and, if not ameliorated, can develop into permanent conditions. In one study,Vietnam veterans with PTSD had an 8% reduction in the volume of their right hippocampus compared with veterans without PTSD. Another study calculated a 26% reduction in the left hippocampus and a 22% reduction in the right in veterans with the most severe PTSD compared with veterans who were in combat but had no PTSD symptoms.4
A common subcortical neural system regulates defensive behavior, including autonomic, emotional, and cognitive behavior. When the prefrontal cortex is taken “off line” for whatever reason, parasympathetic inhibitory action is withdrawn, and relative sympathetic dominance, associated with defense, occurs.
CONFRONTING HYPERAROUSAL
The question then arises of how to train the ANS to avoid hypervigilance. Growing evidence supports the use of heart rate variability as a predictor of hypervigilance and inefficient allocation of attentional and cognitive resources.
The overall objective of heart rate variability training is to decrease ANS hyperarousal and to improve its balance. “Wounded warriors” learn to control ANS responses to stress-producing stimuli (eg, thoughts, memories, and images associated with combat). The goal of training is to decrease arousal and maintain ANS balance for increasing lengths of time.
Once it was observed that alpha waves were dysfunctional in vulnerable populations, protocols were developed to train alpha and theta waves as a method of improving function. Peniston and colleagues5–9 showed that increased alpha and theta brain wave production resulted in normalized personality measures and prolonged the period of time before relapse in alcoholics. This protocol has also shown efficacy as an intervention in depression and PTSD.
BIOFEEDBACK TRAINING PROGRAM
The US Department of Defense is studying a combination of central nervous system biofeedback with ANS biofeedback, with the goal of restoring and maintaining tone between the systems.
The training program used in the study lasts 1 month, and starts with a session for preassessment, 16 biofeedback sessions (four per week), a postprogram evaluation, and a 3-month followup. Each week, participants are exposed to stress-producing stimuli that increase in intensity:
- Week 1: Stroop Color Word Test, math stressor, talk stressor/everyday events
- Week 2: Talk stressor, combat experiences
- Week 3: Images and sounds of combat
- Week 4: Virtual Baghdad or Afghanistan (virtual reality exposure)
Each biofeedback session consists of 5 minutes of baseline evaluation; 5 minutes in which the veteran is subjected to the weekly stressor; 20 minutes of heart rate variability and neurofeedback training; 5 more minutes of training with the weekly stressor; 20 more minutes of heart rate variability and neurofeedback training; and finally 5 minutes of recovery.
SUMMARY
Dysfunction in the balance of both the ANS and central nervous system is associated with symptoms of PTSD in combat veterans. Methods that are designed to restore balance in these systems are needed to ameliorate these symptoms. Biofeedback and neurofeedback are safe methods with which to achieve these goals.
Posttraumatic stress disorder (PTSD) is a severe anxiety disorder whose symptoms emerge following exposure to extreme stress, such as those encountered in the battlefield or as a result of sexual abuse or natural disasters. The ability to employ coping mechanisms affects the disorder’s presentation as well as the frequency, intensity, and duration of the symptoms. The “Wounded Warrior” program at East Carolina University (Greenville, NC) was developed to promote the functional independence of US Marines, including those with PTSD.
STRESS RESPONSE: INTERACTION OF THE BRAIN AND IMMUNE SYSTEM
Walter Cannon coined the “flight or fight” response to stress in the early 20th century, in which he emphasized the importance of the parasympathetic system.1 In 1988, Folkow clarified the description as an immune response to stress.2 The stress response is now understood to be a neuroendocrine function that includes a feedback loop between the hypothalamus and the pituitary and adrenal glands; stimulation of the hypothalamus promotes secretion of corticotropin-releasing hormone (CRH) into the hypophyseal portal system, which supplies the anterior pituitarywith blood. CRH stimulates the secretion of adrenocorticotropic hormone into the bloodstream by the pituitary, prompting the adrenal glands to release the stress hormone cortisol.
Cortisol mobilizes the body’s defenses to meet the challenge of an adverse situation. It modulates the stress response by inhibiting the further release of CRH by the hypothalamus. Cortisol thus protects healthy cells and tissues by inhibiting an overreaction from the immune system. Without this protective effect, the interaction between the brain and the immune system can become dysregulated, increasing the risk of immune disorders.
THE CENTRAL AUTONOMIC NETWORK
The central nervous system that regulates the overall balance of the autonomic nervous system (ANS) has been called the central autonomic network (CAN).3 The CAN helps control executive, social, affective, attentional, and motivational functions. Therefore, the old paradigm of simply decreasing hyperarousal of the ANS to treat negative affective states and dispositions is inadequate. Instead, restoring the appropriate relationship between the ANS and the central nervous system is the aim behind interventions to treat PTSD.
Autonomic, cognitive, and affective functions assist humans in maintaining balance when confronted with external challenges. The CAN controls inhibitory or negative processes that permit specific behavior and redeploy resources needed elsewhere. When negative circuits are compromised, positive circuits develop, resulting in hypervigilance, the symptoms of which can be devastating and, if not ameliorated, can develop into permanent conditions. In one study,Vietnam veterans with PTSD had an 8% reduction in the volume of their right hippocampus compared with veterans without PTSD. Another study calculated a 26% reduction in the left hippocampus and a 22% reduction in the right in veterans with the most severe PTSD compared with veterans who were in combat but had no PTSD symptoms.4
A common subcortical neural system regulates defensive behavior, including autonomic, emotional, and cognitive behavior. When the prefrontal cortex is taken “off line” for whatever reason, parasympathetic inhibitory action is withdrawn, and relative sympathetic dominance, associated with defense, occurs.
CONFRONTING HYPERAROUSAL
The question then arises of how to train the ANS to avoid hypervigilance. Growing evidence supports the use of heart rate variability as a predictor of hypervigilance and inefficient allocation of attentional and cognitive resources.
The overall objective of heart rate variability training is to decrease ANS hyperarousal and to improve its balance. “Wounded warriors” learn to control ANS responses to stress-producing stimuli (eg, thoughts, memories, and images associated with combat). The goal of training is to decrease arousal and maintain ANS balance for increasing lengths of time.
Once it was observed that alpha waves were dysfunctional in vulnerable populations, protocols were developed to train alpha and theta waves as a method of improving function. Peniston and colleagues5–9 showed that increased alpha and theta brain wave production resulted in normalized personality measures and prolonged the period of time before relapse in alcoholics. This protocol has also shown efficacy as an intervention in depression and PTSD.
BIOFEEDBACK TRAINING PROGRAM
The US Department of Defense is studying a combination of central nervous system biofeedback with ANS biofeedback, with the goal of restoring and maintaining tone between the systems.
The training program used in the study lasts 1 month, and starts with a session for preassessment, 16 biofeedback sessions (four per week), a postprogram evaluation, and a 3-month followup. Each week, participants are exposed to stress-producing stimuli that increase in intensity:
- Week 1: Stroop Color Word Test, math stressor, talk stressor/everyday events
- Week 2: Talk stressor, combat experiences
- Week 3: Images and sounds of combat
- Week 4: Virtual Baghdad or Afghanistan (virtual reality exposure)
Each biofeedback session consists of 5 minutes of baseline evaluation; 5 minutes in which the veteran is subjected to the weekly stressor; 20 minutes of heart rate variability and neurofeedback training; 5 more minutes of training with the weekly stressor; 20 more minutes of heart rate variability and neurofeedback training; and finally 5 minutes of recovery.
SUMMARY
Dysfunction in the balance of both the ANS and central nervous system is associated with symptoms of PTSD in combat veterans. Methods that are designed to restore balance in these systems are needed to ameliorate these symptoms. Biofeedback and neurofeedback are safe methods with which to achieve these goals.
- Cannon WB. Bodily Changes in Pain, Hunger, Fear and Rage: An Account of Recent Researches into the Function of Emotional Excitement. 2nd ed. New York, NY: Appleton-Century-Crofts; 1929.
- Folkow B. Stress, hypothalamic function and neuroendocrine consequences. Acta Med Scand Suppl 1988; 723:61–69.
- Thayer JF, Brosschot JF. Psychosomatics and psychopathology: looking up and down from the brain. Psychoneuroendocrinology 2005; 30:1050–1058.
- van der Kolk BA. The psychobiology and psychopharmacology of PTSD. Hum Psychopharmacol 2001; 16:S49–S64.
- Peniston EG, Kulkosky PJ. Alpha-theta brainwave training and beta-endorphin levels in alcoholics. Alcohol Clin Exp Res 1989;13:271–279.
- Peniston EG, Kulkosky PJ. Alcoholic personality and alpha-thetabrainwave training. Medical Psychotherapy: An International Journal1990; 3:37–55.
- Peniston EG, Kulkosky PJ. Alpha-theta brainwave neurofeedbacktherapy for Vietnam veterans with combat-related posttraumaticstress disorder. Medical Psychotherapy: An International Journal1991; 4:47–60.
- Peniston EG, Kulkosky PJ. Alpha-theta EEG biofeedback trainingin alcoholism and posttraumatic stress disorder. The InternationalSociety for the Study of Subtle Energies and Energy Medicines1992; 2:5–7.
- Peniston EG, Marrinan DA, Deming WA, Kulkosky PJ. EEGalpha-theta brainwave synchronization in Vietnam theater veteranswith combat-related posttraumatic stress disorder and alcohol abuse.Medical Psychotherapy: An International Journal 1993; 6:37–50.
- Cannon WB. Bodily Changes in Pain, Hunger, Fear and Rage: An Account of Recent Researches into the Function of Emotional Excitement. 2nd ed. New York, NY: Appleton-Century-Crofts; 1929.
- Folkow B. Stress, hypothalamic function and neuroendocrine consequences. Acta Med Scand Suppl 1988; 723:61–69.
- Thayer JF, Brosschot JF. Psychosomatics and psychopathology: looking up and down from the brain. Psychoneuroendocrinology 2005; 30:1050–1058.
- van der Kolk BA. The psychobiology and psychopharmacology of PTSD. Hum Psychopharmacol 2001; 16:S49–S64.
- Peniston EG, Kulkosky PJ. Alpha-theta brainwave training and beta-endorphin levels in alcoholics. Alcohol Clin Exp Res 1989;13:271–279.
- Peniston EG, Kulkosky PJ. Alcoholic personality and alpha-thetabrainwave training. Medical Psychotherapy: An International Journal1990; 3:37–55.
- Peniston EG, Kulkosky PJ. Alpha-theta brainwave neurofeedbacktherapy for Vietnam veterans with combat-related posttraumaticstress disorder. Medical Psychotherapy: An International Journal1991; 4:47–60.
- Peniston EG, Kulkosky PJ. Alpha-theta EEG biofeedback trainingin alcoholism and posttraumatic stress disorder. The InternationalSociety for the Study of Subtle Energies and Energy Medicines1992; 2:5–7.
- Peniston EG, Marrinan DA, Deming WA, Kulkosky PJ. EEGalpha-theta brainwave synchronization in Vietnam theater veteranswith combat-related posttraumatic stress disorder and alcohol abuse.Medical Psychotherapy: An International Journal 1993; 6:37–50.