User login
Why Hospitalists Remain Outside Malpractice Insurers' High-Risk Categories, For Now

Source: The Doctors Company
Ten years ago, the national headlines on malpractice insurance were staggering. Media reports catalogued OB-GYNs who proclaimed they were shutting down their private practices in the face of runaway premiums. Surgeons and other proceduralists decried payments tied to lawsuits they’d argue were arbitrary and capricious. And the American Medical Association (AMA) made announcement after announcement about states being in a “malpractice crisis.”
In recent years, premiums have actually dropped and stabilized at levels that most physicians agree are manageable for bottom lines. But, in that time, there has been scant discussion about hospital medicine’s relationship with malpractice. It’s not because the issue isn’t omnipresent for all healthcare practitioners, including the relatively nascent specialty that is HM.
Practice management experts say anecdotally that delayed diagnosis of, or treatment for, a spinal epidural abscess (SEA) is likely to get more than a few hospitalists sued. And, the proliferation of co-management of other specialties—particularly those with higher risk of incidence and higher premiums than internal medicine—open up hospitalists to further liability.
The issue is that at less than 20 years as a specialty, HM is in its infancy when it comes to its interaction with malpractice premiums. Health insurance companies and trade groups that track the insurance industry are just beginning to have enough data on claims, premiums, and payouts to make recommendations on risk factors, risk mitigation, and potential trends.
Still, even in a landscape of limited information, there are a few rules of thumb hospitalist group leaders should live by when it comes to managing exposure to malpractice cases, according to interviews with a half dozen healthcare professionals:
- Know how your coverage works. Is there “tail coverage” that ensures you have protection for incidents that happened at an institution where you no longer practice? Even though hospital-employed physicians rarely have rate discussions directly (the hospital typically covers premiums as part of the compensation package), take the time to learn the basic details.
- Be diligent in documentation. Note concerns in charts when appropriate, and stand up for your point of view. There’s a fine line between picking fights with other physicians involved in a patient’s care and making your concerns known, but don’t be afraid to put your clinical view on the record.
- Avoid the practice of “defensive medicine.” Ordering tests and procedures that aren’t clinically necessary might seem like it can serve as a protection from later lawsuits, but it adds to healthcare costs and is just not the right thing to do, says hospitalist Allen Kachalia, MD, JD, of Brigham and Women’s Hospital in Boston, who has studied the phenomenon (see “Culture Shift Necessary to Defeat “Defensive” Medicine,” on p. 38).
- Recognize the risks associated with co-management. Caring for neurology, cardiology, and other subspecialty patients is a revenue boost for HM groups, but when some of those complex cases have adverse events, the hospitalist who interacted with the patient daily could be included in a lawsuit.
- Focus on communication skills. An analysis of claims data by The Doctors Company (TDC) (www.thedoctors.com), a medical malpractice insurance company exclusively endorsed by SHM, reports that the second most common factor contributing to patient injury by hospitalists is “communication breakdown among healthcare professionals.”
- Manage workloads to avoid burnout. Don’t take on too many patients at the expense of being involved in hospital committees or quality initiatives.
To be sure, many of the same tenets of being a productive hospitalist with high patient satisfaction scores—maintain manageable censuses; focus on patient centeredness; and use checklists, technology, and regimented protocols to reduce adverse events—translate very well to being a lower-risk hospitalist in relation to malpractice cases.
When you’re “thinking of patient satisfaction strategies, also think of them as risk mitigation strategies,” says John Nelson, MD, MHM, FACP, medical director of the hospitalist practice at Overlake Hospital Medical Center in Bellevue, Wash., an SHM co-founder and practice management columnist for The Hospitalist. “They overlap tremendously.”
A History Lesson
Medical malpractice has been around for centuries and has two prevailing goals: 1) to provide monetary remuneration to patients who have been injured via substandard care and 2) to deter that poor treatment through fiscal punishment.
Malpractice lawsuits were not prevalent enough to be a major medical concern until the early 1800s. By the middle of the 19th century, the country hit its first periods of crisis.1 Cycles ebbed and flowed from there, with malpractice premiums causing crises in the 1980s and again in the early 2000s.
continued below...
Now, rates for medical professional liability insurance have been dropping for seven years, and an eighth straight annual decline is expected this year, according to Mike Matray, the editor of trade publication Medical Liability Monitor and the chief content officer of its associated website, www.mymedicalmalpracticeinsurance.com.
“We are in the longest, deepest soft market that the malpractice insurance industry has ever been in,” he says. “Right now, things are really good for the doctors, as far as rates coming down.”
Matray says he understands that declining rates may seem immaterial to a physician who receives an insurance bill that eats into the bottom line. For some specialties, that premium can be as high as $200,000 per physician, per year—or more.
“I’m not saying it isn’t expensive,” he adds. “It’s expensive to run a medical practice. At the same time, medical malpractice insurance is less expensive in today’s dollars than it was in 2005.”
The reduction in rates is multi-faceted. Prominently, state-level tort reforms like non-economic damage caps, health courts, and arbitration hearings are making it harder to bring cases to trial, particularly for lawyers who take cases on contingency. Second, frivolous lawsuits “are making an impression on jury pools,” Matray says, which means fewer filed claims and fewer cases that make it to trial. Third, this soft cycle has outlasted the typical pattern of rates falling for three to four years before rebounding.
“A lot of smart actuaries keep saying this has to change soon, because in a soft market there is a lot of competition,” he says, noting that in order to compete for low rates, insurance companies offer credits to clients and use their own reserve cash piles. “So things are really going to change in the next couple of years.”

—Robin Diamond, senior vice president and chief patient safety officer, The Doctors Company
In Need of Data, Patience
So what does it all mean for hospitalists and HM group leaders looking to be proactive about medical malpractice liability insurance? Patience is required.
For starters, there is no designated premium category for hospitalists. Much like the situation that exists for coding issues, the closest proxy for HM is internal medicine. According to Medical Liability Monitor, the premium paid by internal medicine physicians as of July 1, 2012, varied widely across the country. In South Florida, internal medicine insurance premiums in Miami, Dade, and Broward counties were between $42,000 and $46,000 per year. In South Dakota, one insurer reported rates of just under $4,000 per year. There is no average or median figure available, and Matray notes that actual rates paid can vary from county to county.
Moreover, it is difficult for group leaders or hospital executives to use past history to negotiate rates with insurers because of a shortage of reliable data. In its spring 2013 newsletter, the PIAA (formerly known as the Physician Insurers Association of America) published its first report on hospitalist claims reported to its Data Sharing Project. Of the 92,868 closed claims reported from 2002-2011, just 312, or 0.3%, named hospitalists as the defendant.
The data also showed that, of those claims, 20% were settled through insurance company payments. Those payments totaled $17.1 million, with an average payout to a claimant (known as the indemnity) of $272,553 per claim. Overall, hospitalists had a 20% paid-to-closed ratio, totaling more than $17.1 million. By comparison, the percent of paid-to-closed claims for all physicians was 29.3%, according to PIAA.
In a separate data set compiled this year by TDC, 34% of allegations against hospitalists were related to missed or failed diagnoses, with 28% tied to “improper management of treatment.” Twelve percent of allegations were the result of either improper medication management or ordering errors.
Robin Diamond, TDC’s senior vice president and chief patient safety officer, says that teasing out trends from the initial data can be challenging. Hospitalists, she says, can deal with so many different patients, diseases, and severity levels that it is difficult to draw conclusions.
“Hospital medicine is different than other specialties, because the hospitalist treats a broad range of patients in an acute care setting—from a pediatric patient to an adult patient with many chronic illnesses,” she says.
Divya Parikh, PIAA’s director of research and loss prevention, says HM group leaders should avoid reading too much into the first batch of data, because it’s a small sample size.
“A big part of that is we feel that a lot of hospitalists are intermingled into the other medical specialties,” she says. “So this becomes a very small subset where they are distinctly identified as hospitalists. And that’s the challenge.”
In particular, Parikh is curious to see whether HM’s rate of claims paid through insurance payments drops from 20% (already below the overall healthcare industry average). “It will be interesting as we proceed...to see if they begin to mitigate areas of risk where we used to see a lot of claims,” she adds. “If you look at a hospital setting, there has been some shift change in what the errors are. And, what you’d hope with hospitalists within these environments who are really owning this specialty, is that you’d see a decrease in that. There would be that connective care. There would be the patient that felt that they had an individual who was their go-to individual throughout their care at a hospital.”
A Peek at the Future
Insurers have begun compiling claims data on hospitalists and are taking a longer-term view of the specialty. TDC, for example, has analyzed its data and identified characteristics it says make a low-risk hospitalist, an analysis the company says is the first of its kind (see Figure 1). The insurer adds that it sees its responsibility as making sure everyone understands the hospitalist’s role within the acute care setting so that its pricing is commensurate with the liability risk.
Source: The Doctors Company
“We’re looking at the systems within the hospitalist group, as well as how well that group is integrating with the hospital where they’re practicing,” Diamond says. “What kind of patient mix is this particular hospitalist group seeing in that particular hospital, because it can be different in a large healthcare corporation in Manhattan, New York, from a community hospital in rural Texas.”
The growing popularity of hospitalists taking on co-management responsibilities for other specialties is another trend to keep an eye on, as it creates what insurers call “vicarious liability.” Working together in teams with other specialties can improve communication, reduce errors during transitions of care, and create better outcomes. However, in instances where there are problems, being on a care team means hospitalists can open themselves to liability. To mitigate that risk, hospitalists can look to other groups that have dealt with shared liability issues in the past, Parikh says.
“Historically, you would have seen it with anesthesiology,” she explains. “And one huge improvement anesthesiologists have made when a patient comes in for a surgery now is they come out, introduce themselves, say hello, and tell you what’s going on. They put a face to the name, so that it’s not just a no-name anesthesiologist who gets included in the lawsuit as well because they’re naming everybody in the group.”
But, holistically, the best long-term mitigation strategy appears to be tort reform and new ways of looking at the way in which healthcare liability issues are handled in the U.S., says Anupam Jena, MD, PhD, assistant professor of healthcare policy and medicine at Harvard Medical School, and an internist at Massachusetts General Hospital, both in Boston. Dr. Jena says that there is limited evidence that enacted malpractice reforms have produced more than a 2% to 5% reduction in healthcare spending compared to states that have not.2 Instead, healthcare leaders should push for the elimination of defensive medicine, which he says contributes the lion’s share of the estimated $50 billion annual cost of malpractice liability across the country.
“Do I think the country is in a malpractice crisis? No,” he says. “Do I think that defensive medicine is larger than we think it is? Yes.
“If physicians practice as they felt they should practice without ordering extra tests and procedures, my guess would be you could reduce healthcare spending by substantially more than $50 billion.”
Richard Quinn is a freelance writer in New Jersey.
References
- Spiegel AD, Kavaler F. America’s first medical malpractice crisis, 1835-1865. J Community Health. 1997;22:283-308.
- Chandra A, Jena A, Seabury, S. Defensive medicine may be costlier than it seems. The Wall Street Journal website. http://online.wsj.com/article/SB10001424127887323701904578280112638373302.html. Accessed September 21, 2013.
Source: The Doctors Company
Ten years ago, the national headlines on malpractice insurance were staggering. Media reports catalogued OB-GYNs who proclaimed they were shutting down their private practices in the face of runaway premiums. Surgeons and other proceduralists decried payments tied to lawsuits they’d argue were arbitrary and capricious. And the American Medical Association (AMA) made announcement after announcement about states being in a “malpractice crisis.”
In recent years, premiums have actually dropped and stabilized at levels that most physicians agree are manageable for bottom lines. But, in that time, there has been scant discussion about hospital medicine’s relationship with malpractice. It’s not because the issue isn’t omnipresent for all healthcare practitioners, including the relatively nascent specialty that is HM.
Practice management experts say anecdotally that delayed diagnosis of, or treatment for, a spinal epidural abscess (SEA) is likely to get more than a few hospitalists sued. And, the proliferation of co-management of other specialties—particularly those with higher risk of incidence and higher premiums than internal medicine—open up hospitalists to further liability.
The issue is that at less than 20 years as a specialty, HM is in its infancy when it comes to its interaction with malpractice premiums. Health insurance companies and trade groups that track the insurance industry are just beginning to have enough data on claims, premiums, and payouts to make recommendations on risk factors, risk mitigation, and potential trends.
Still, even in a landscape of limited information, there are a few rules of thumb hospitalist group leaders should live by when it comes to managing exposure to malpractice cases, according to interviews with a half dozen healthcare professionals:
- Know how your coverage works. Is there “tail coverage” that ensures you have protection for incidents that happened at an institution where you no longer practice? Even though hospital-employed physicians rarely have rate discussions directly (the hospital typically covers premiums as part of the compensation package), take the time to learn the basic details.
- Be diligent in documentation. Note concerns in charts when appropriate, and stand up for your point of view. There’s a fine line between picking fights with other physicians involved in a patient’s care and making your concerns known, but don’t be afraid to put your clinical view on the record.
- Avoid the practice of “defensive medicine.” Ordering tests and procedures that aren’t clinically necessary might seem like it can serve as a protection from later lawsuits, but it adds to healthcare costs and is just not the right thing to do, says hospitalist Allen Kachalia, MD, JD, of Brigham and Women’s Hospital in Boston, who has studied the phenomenon (see “Culture Shift Necessary to Defeat “Defensive” Medicine,” on p. 38).
- Recognize the risks associated with co-management. Caring for neurology, cardiology, and other subspecialty patients is a revenue boost for HM groups, but when some of those complex cases have adverse events, the hospitalist who interacted with the patient daily could be included in a lawsuit.
- Focus on communication skills. An analysis of claims data by The Doctors Company (TDC) (www.thedoctors.com), a medical malpractice insurance company exclusively endorsed by SHM, reports that the second most common factor contributing to patient injury by hospitalists is “communication breakdown among healthcare professionals.”
- Manage workloads to avoid burnout. Don’t take on too many patients at the expense of being involved in hospital committees or quality initiatives.
To be sure, many of the same tenets of being a productive hospitalist with high patient satisfaction scores—maintain manageable censuses; focus on patient centeredness; and use checklists, technology, and regimented protocols to reduce adverse events—translate very well to being a lower-risk hospitalist in relation to malpractice cases.
When you’re “thinking of patient satisfaction strategies, also think of them as risk mitigation strategies,” says John Nelson, MD, MHM, FACP, medical director of the hospitalist practice at Overlake Hospital Medical Center in Bellevue, Wash., an SHM co-founder and practice management columnist for The Hospitalist. “They overlap tremendously.”
A History Lesson
Medical malpractice has been around for centuries and has two prevailing goals: 1) to provide monetary remuneration to patients who have been injured via substandard care and 2) to deter that poor treatment through fiscal punishment.
Malpractice lawsuits were not prevalent enough to be a major medical concern until the early 1800s. By the middle of the 19th century, the country hit its first periods of crisis.1 Cycles ebbed and flowed from there, with malpractice premiums causing crises in the 1980s and again in the early 2000s.
continued below...
Now, rates for medical professional liability insurance have been dropping for seven years, and an eighth straight annual decline is expected this year, according to Mike Matray, the editor of trade publication Medical Liability Monitor and the chief content officer of its associated website, www.mymedicalmalpracticeinsurance.com.
“We are in the longest, deepest soft market that the malpractice insurance industry has ever been in,” he says. “Right now, things are really good for the doctors, as far as rates coming down.”
Matray says he understands that declining rates may seem immaterial to a physician who receives an insurance bill that eats into the bottom line. For some specialties, that premium can be as high as $200,000 per physician, per year—or more.
“I’m not saying it isn’t expensive,” he adds. “It’s expensive to run a medical practice. At the same time, medical malpractice insurance is less expensive in today’s dollars than it was in 2005.”
The reduction in rates is multi-faceted. Prominently, state-level tort reforms like non-economic damage caps, health courts, and arbitration hearings are making it harder to bring cases to trial, particularly for lawyers who take cases on contingency. Second, frivolous lawsuits “are making an impression on jury pools,” Matray says, which means fewer filed claims and fewer cases that make it to trial. Third, this soft cycle has outlasted the typical pattern of rates falling for three to four years before rebounding.
“A lot of smart actuaries keep saying this has to change soon, because in a soft market there is a lot of competition,” he says, noting that in order to compete for low rates, insurance companies offer credits to clients and use their own reserve cash piles. “So things are really going to change in the next couple of years.”
—Robin Diamond, senior vice president and chief patient safety officer, The Doctors Company
In Need of Data, Patience
So what does it all mean for hospitalists and HM group leaders looking to be proactive about medical malpractice liability insurance? Patience is required.
For starters, there is no designated premium category for hospitalists. Much like the situation that exists for coding issues, the closest proxy for HM is internal medicine. According to Medical Liability Monitor, the premium paid by internal medicine physicians as of July 1, 2012, varied widely across the country. In South Florida, internal medicine insurance premiums in Miami, Dade, and Broward counties were between $42,000 and $46,000 per year. In South Dakota, one insurer reported rates of just under $4,000 per year. There is no average or median figure available, and Matray notes that actual rates paid can vary from county to county.
Moreover, it is difficult for group leaders or hospital executives to use past history to negotiate rates with insurers because of a shortage of reliable data. In its spring 2013 newsletter, the PIAA (formerly known as the Physician Insurers Association of America) published its first report on hospitalist claims reported to its Data Sharing Project. Of the 92,868 closed claims reported from 2002-2011, just 312, or 0.3%, named hospitalists as the defendant.
The data also showed that, of those claims, 20% were settled through insurance company payments. Those payments totaled $17.1 million, with an average payout to a claimant (known as the indemnity) of $272,553 per claim. Overall, hospitalists had a 20% paid-to-closed ratio, totaling more than $17.1 million. By comparison, the percent of paid-to-closed claims for all physicians was 29.3%, according to PIAA.
In a separate data set compiled this year by TDC, 34% of allegations against hospitalists were related to missed or failed diagnoses, with 28% tied to “improper management of treatment.” Twelve percent of allegations were the result of either improper medication management or ordering errors.
Robin Diamond, TDC’s senior vice president and chief patient safety officer, says that teasing out trends from the initial data can be challenging. Hospitalists, she says, can deal with so many different patients, diseases, and severity levels that it is difficult to draw conclusions.
“Hospital medicine is different than other specialties, because the hospitalist treats a broad range of patients in an acute care setting—from a pediatric patient to an adult patient with many chronic illnesses,” she says.
Divya Parikh, PIAA’s director of research and loss prevention, says HM group leaders should avoid reading too much into the first batch of data, because it’s a small sample size.
“A big part of that is we feel that a lot of hospitalists are intermingled into the other medical specialties,” she says. “So this becomes a very small subset where they are distinctly identified as hospitalists. And that’s the challenge.”
In particular, Parikh is curious to see whether HM’s rate of claims paid through insurance payments drops from 20% (already below the overall healthcare industry average). “It will be interesting as we proceed...to see if they begin to mitigate areas of risk where we used to see a lot of claims,” she adds. “If you look at a hospital setting, there has been some shift change in what the errors are. And, what you’d hope with hospitalists within these environments who are really owning this specialty, is that you’d see a decrease in that. There would be that connective care. There would be the patient that felt that they had an individual who was their go-to individual throughout their care at a hospital.”
A Peek at the Future
Insurers have begun compiling claims data on hospitalists and are taking a longer-term view of the specialty. TDC, for example, has analyzed its data and identified characteristics it says make a low-risk hospitalist, an analysis the company says is the first of its kind (see Figure 1). The insurer adds that it sees its responsibility as making sure everyone understands the hospitalist’s role within the acute care setting so that its pricing is commensurate with the liability risk.
Source: The Doctors Company
“We’re looking at the systems within the hospitalist group, as well as how well that group is integrating with the hospital where they’re practicing,” Diamond says. “What kind of patient mix is this particular hospitalist group seeing in that particular hospital, because it can be different in a large healthcare corporation in Manhattan, New York, from a community hospital in rural Texas.”
The growing popularity of hospitalists taking on co-management responsibilities for other specialties is another trend to keep an eye on, as it creates what insurers call “vicarious liability.” Working together in teams with other specialties can improve communication, reduce errors during transitions of care, and create better outcomes. However, in instances where there are problems, being on a care team means hospitalists can open themselves to liability. To mitigate that risk, hospitalists can look to other groups that have dealt with shared liability issues in the past, Parikh says.
“Historically, you would have seen it with anesthesiology,” she explains. “And one huge improvement anesthesiologists have made when a patient comes in for a surgery now is they come out, introduce themselves, say hello, and tell you what’s going on. They put a face to the name, so that it’s not just a no-name anesthesiologist who gets included in the lawsuit as well because they’re naming everybody in the group.”
But, holistically, the best long-term mitigation strategy appears to be tort reform and new ways of looking at the way in which healthcare liability issues are handled in the U.S., says Anupam Jena, MD, PhD, assistant professor of healthcare policy and medicine at Harvard Medical School, and an internist at Massachusetts General Hospital, both in Boston. Dr. Jena says that there is limited evidence that enacted malpractice reforms have produced more than a 2% to 5% reduction in healthcare spending compared to states that have not.2 Instead, healthcare leaders should push for the elimination of defensive medicine, which he says contributes the lion’s share of the estimated $50 billion annual cost of malpractice liability across the country.
“Do I think the country is in a malpractice crisis? No,” he says. “Do I think that defensive medicine is larger than we think it is? Yes.
“If physicians practice as they felt they should practice without ordering extra tests and procedures, my guess would be you could reduce healthcare spending by substantially more than $50 billion.”
Richard Quinn is a freelance writer in New Jersey.
References
- Spiegel AD, Kavaler F. America’s first medical malpractice crisis, 1835-1865. J Community Health. 1997;22:283-308.
- Chandra A, Jena A, Seabury, S. Defensive medicine may be costlier than it seems. The Wall Street Journal website. http://online.wsj.com/article/SB10001424127887323701904578280112638373302.html. Accessed September 21, 2013.
Source: The Doctors Company
Ten years ago, the national headlines on malpractice insurance were staggering. Media reports catalogued OB-GYNs who proclaimed they were shutting down their private practices in the face of runaway premiums. Surgeons and other proceduralists decried payments tied to lawsuits they’d argue were arbitrary and capricious. And the American Medical Association (AMA) made announcement after announcement about states being in a “malpractice crisis.”
In recent years, premiums have actually dropped and stabilized at levels that most physicians agree are manageable for bottom lines. But, in that time, there has been scant discussion about hospital medicine’s relationship with malpractice. It’s not because the issue isn’t omnipresent for all healthcare practitioners, including the relatively nascent specialty that is HM.
Practice management experts say anecdotally that delayed diagnosis of, or treatment for, a spinal epidural abscess (SEA) is likely to get more than a few hospitalists sued. And, the proliferation of co-management of other specialties—particularly those with higher risk of incidence and higher premiums than internal medicine—open up hospitalists to further liability.
The issue is that at less than 20 years as a specialty, HM is in its infancy when it comes to its interaction with malpractice premiums. Health insurance companies and trade groups that track the insurance industry are just beginning to have enough data on claims, premiums, and payouts to make recommendations on risk factors, risk mitigation, and potential trends.
Still, even in a landscape of limited information, there are a few rules of thumb hospitalist group leaders should live by when it comes to managing exposure to malpractice cases, according to interviews with a half dozen healthcare professionals:
- Know how your coverage works. Is there “tail coverage” that ensures you have protection for incidents that happened at an institution where you no longer practice? Even though hospital-employed physicians rarely have rate discussions directly (the hospital typically covers premiums as part of the compensation package), take the time to learn the basic details.
- Be diligent in documentation. Note concerns in charts when appropriate, and stand up for your point of view. There’s a fine line between picking fights with other physicians involved in a patient’s care and making your concerns known, but don’t be afraid to put your clinical view on the record.
- Avoid the practice of “defensive medicine.” Ordering tests and procedures that aren’t clinically necessary might seem like it can serve as a protection from later lawsuits, but it adds to healthcare costs and is just not the right thing to do, says hospitalist Allen Kachalia, MD, JD, of Brigham and Women’s Hospital in Boston, who has studied the phenomenon (see “Culture Shift Necessary to Defeat “Defensive” Medicine,” on p. 38).
- Recognize the risks associated with co-management. Caring for neurology, cardiology, and other subspecialty patients is a revenue boost for HM groups, but when some of those complex cases have adverse events, the hospitalist who interacted with the patient daily could be included in a lawsuit.
- Focus on communication skills. An analysis of claims data by The Doctors Company (TDC) (www.thedoctors.com), a medical malpractice insurance company exclusively endorsed by SHM, reports that the second most common factor contributing to patient injury by hospitalists is “communication breakdown among healthcare professionals.”
- Manage workloads to avoid burnout. Don’t take on too many patients at the expense of being involved in hospital committees or quality initiatives.
To be sure, many of the same tenets of being a productive hospitalist with high patient satisfaction scores—maintain manageable censuses; focus on patient centeredness; and use checklists, technology, and regimented protocols to reduce adverse events—translate very well to being a lower-risk hospitalist in relation to malpractice cases.
When you’re “thinking of patient satisfaction strategies, also think of them as risk mitigation strategies,” says John Nelson, MD, MHM, FACP, medical director of the hospitalist practice at Overlake Hospital Medical Center in Bellevue, Wash., an SHM co-founder and practice management columnist for The Hospitalist. “They overlap tremendously.”
A History Lesson
Medical malpractice has been around for centuries and has two prevailing goals: 1) to provide monetary remuneration to patients who have been injured via substandard care and 2) to deter that poor treatment through fiscal punishment.
Malpractice lawsuits were not prevalent enough to be a major medical concern until the early 1800s. By the middle of the 19th century, the country hit its first periods of crisis.1 Cycles ebbed and flowed from there, with malpractice premiums causing crises in the 1980s and again in the early 2000s.
continued below...
Now, rates for medical professional liability insurance have been dropping for seven years, and an eighth straight annual decline is expected this year, according to Mike Matray, the editor of trade publication Medical Liability Monitor and the chief content officer of its associated website, www.mymedicalmalpracticeinsurance.com.
“We are in the longest, deepest soft market that the malpractice insurance industry has ever been in,” he says. “Right now, things are really good for the doctors, as far as rates coming down.”
Matray says he understands that declining rates may seem immaterial to a physician who receives an insurance bill that eats into the bottom line. For some specialties, that premium can be as high as $200,000 per physician, per year—or more.
“I’m not saying it isn’t expensive,” he adds. “It’s expensive to run a medical practice. At the same time, medical malpractice insurance is less expensive in today’s dollars than it was in 2005.”
The reduction in rates is multi-faceted. Prominently, state-level tort reforms like non-economic damage caps, health courts, and arbitration hearings are making it harder to bring cases to trial, particularly for lawyers who take cases on contingency. Second, frivolous lawsuits “are making an impression on jury pools,” Matray says, which means fewer filed claims and fewer cases that make it to trial. Third, this soft cycle has outlasted the typical pattern of rates falling for three to four years before rebounding.
“A lot of smart actuaries keep saying this has to change soon, because in a soft market there is a lot of competition,” he says, noting that in order to compete for low rates, insurance companies offer credits to clients and use their own reserve cash piles. “So things are really going to change in the next couple of years.”
—Robin Diamond, senior vice president and chief patient safety officer, The Doctors Company
In Need of Data, Patience
So what does it all mean for hospitalists and HM group leaders looking to be proactive about medical malpractice liability insurance? Patience is required.
For starters, there is no designated premium category for hospitalists. Much like the situation that exists for coding issues, the closest proxy for HM is internal medicine. According to Medical Liability Monitor, the premium paid by internal medicine physicians as of July 1, 2012, varied widely across the country. In South Florida, internal medicine insurance premiums in Miami, Dade, and Broward counties were between $42,000 and $46,000 per year. In South Dakota, one insurer reported rates of just under $4,000 per year. There is no average or median figure available, and Matray notes that actual rates paid can vary from county to county.
Moreover, it is difficult for group leaders or hospital executives to use past history to negotiate rates with insurers because of a shortage of reliable data. In its spring 2013 newsletter, the PIAA (formerly known as the Physician Insurers Association of America) published its first report on hospitalist claims reported to its Data Sharing Project. Of the 92,868 closed claims reported from 2002-2011, just 312, or 0.3%, named hospitalists as the defendant.
The data also showed that, of those claims, 20% were settled through insurance company payments. Those payments totaled $17.1 million, with an average payout to a claimant (known as the indemnity) of $272,553 per claim. Overall, hospitalists had a 20% paid-to-closed ratio, totaling more than $17.1 million. By comparison, the percent of paid-to-closed claims for all physicians was 29.3%, according to PIAA.
In a separate data set compiled this year by TDC, 34% of allegations against hospitalists were related to missed or failed diagnoses, with 28% tied to “improper management of treatment.” Twelve percent of allegations were the result of either improper medication management or ordering errors.
Robin Diamond, TDC’s senior vice president and chief patient safety officer, says that teasing out trends from the initial data can be challenging. Hospitalists, she says, can deal with so many different patients, diseases, and severity levels that it is difficult to draw conclusions.
“Hospital medicine is different than other specialties, because the hospitalist treats a broad range of patients in an acute care setting—from a pediatric patient to an adult patient with many chronic illnesses,” she says.
Divya Parikh, PIAA’s director of research and loss prevention, says HM group leaders should avoid reading too much into the first batch of data, because it’s a small sample size.
“A big part of that is we feel that a lot of hospitalists are intermingled into the other medical specialties,” she says. “So this becomes a very small subset where they are distinctly identified as hospitalists. And that’s the challenge.”
In particular, Parikh is curious to see whether HM’s rate of claims paid through insurance payments drops from 20% (already below the overall healthcare industry average). “It will be interesting as we proceed...to see if they begin to mitigate areas of risk where we used to see a lot of claims,” she adds. “If you look at a hospital setting, there has been some shift change in what the errors are. And, what you’d hope with hospitalists within these environments who are really owning this specialty, is that you’d see a decrease in that. There would be that connective care. There would be the patient that felt that they had an individual who was their go-to individual throughout their care at a hospital.”
A Peek at the Future
Insurers have begun compiling claims data on hospitalists and are taking a longer-term view of the specialty. TDC, for example, has analyzed its data and identified characteristics it says make a low-risk hospitalist, an analysis the company says is the first of its kind (see Figure 1). The insurer adds that it sees its responsibility as making sure everyone understands the hospitalist’s role within the acute care setting so that its pricing is commensurate with the liability risk.
Source: The Doctors Company
“We’re looking at the systems within the hospitalist group, as well as how well that group is integrating with the hospital where they’re practicing,” Diamond says. “What kind of patient mix is this particular hospitalist group seeing in that particular hospital, because it can be different in a large healthcare corporation in Manhattan, New York, from a community hospital in rural Texas.”
The growing popularity of hospitalists taking on co-management responsibilities for other specialties is another trend to keep an eye on, as it creates what insurers call “vicarious liability.” Working together in teams with other specialties can improve communication, reduce errors during transitions of care, and create better outcomes. However, in instances where there are problems, being on a care team means hospitalists can open themselves to liability. To mitigate that risk, hospitalists can look to other groups that have dealt with shared liability issues in the past, Parikh says.
“Historically, you would have seen it with anesthesiology,” she explains. “And one huge improvement anesthesiologists have made when a patient comes in for a surgery now is they come out, introduce themselves, say hello, and tell you what’s going on. They put a face to the name, so that it’s not just a no-name anesthesiologist who gets included in the lawsuit as well because they’re naming everybody in the group.”
But, holistically, the best long-term mitigation strategy appears to be tort reform and new ways of looking at the way in which healthcare liability issues are handled in the U.S., says Anupam Jena, MD, PhD, assistant professor of healthcare policy and medicine at Harvard Medical School, and an internist at Massachusetts General Hospital, both in Boston. Dr. Jena says that there is limited evidence that enacted malpractice reforms have produced more than a 2% to 5% reduction in healthcare spending compared to states that have not.2 Instead, healthcare leaders should push for the elimination of defensive medicine, which he says contributes the lion’s share of the estimated $50 billion annual cost of malpractice liability across the country.
“Do I think the country is in a malpractice crisis? No,” he says. “Do I think that defensive medicine is larger than we think it is? Yes.
“If physicians practice as they felt they should practice without ordering extra tests and procedures, my guess would be you could reduce healthcare spending by substantially more than $50 billion.”
Richard Quinn is a freelance writer in New Jersey.
References
- Spiegel AD, Kavaler F. America’s first medical malpractice crisis, 1835-1865. J Community Health. 1997;22:283-308.
- Chandra A, Jena A, Seabury, S. Defensive medicine may be costlier than it seems. The Wall Street Journal website. http://online.wsj.com/article/SB10001424127887323701904578280112638373302.html. Accessed September 21, 2013.
Surgeons' Perception of Fluoroscopic Radiation Hazards to Vision
Strategies for Treating Scoliosis in Children With Spinal Muscular Atrophy

Consensus Recommendations From the American Acne & Rosacea Society on the Management of Rosacea, Part 1: A Status Report on the Disease State, General Measures, and Adjunctive Skin Care
Test your knowledge on consensus recommendations for rosacea with MD-IQ: the medical intelligence quiz. Click here to answer 5 questions.
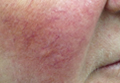
Superficial Plantar Fibromatosis
PSA screening: The USPSTF got it wrong

Prostate cancer is an important disease. It is the second leading cause of cancer death in men who don’t smoke, and in many cases it is detectable early and curable. The rates of both diagnosis and death from prostate cancer in men are similar to the rates of breast cancer in women.1
The current practical screening test for prostate cancer is the prostate specific antigen (PSA). Making routine use of it, as we know, however, is controversial. The false positive rate for PSA testing is high, for example, in men with chronic prostatitis and benign prostatic hypertrophy.2 In addition, many prostate cancers are diagnosed that will never harm the patient. Treatment for prostate cancer may result in complications, such as incontinence and impotence. Because of these facts, the US Preventive Services Task Force (USPSTF) has recommended against routine screening.2
The PSA test itself never hurt anyone
It is just a lab value, a piece of information. What doctors do with the information is the issue. Physicians may cause more harm than good by being overly aggressive with elevated PSA levels and indolent or low-grade prostate cancer—and 75% of prostate cancer is considered indolent (Gleason score of 6 on biopsy).3 Patients with such a finding can be watched, using active surveillance. The majority will never need treatment.3
Common sense tells us we must screen for prostate cancer. Not doing so on the basis of evidence-based medicine is not a defense when advanced cancer is diagnosed and screening was not offered to the patient.4 Rather than using the data from past physician behavior and recommending against screening with PSA, the USPSTF should have criticized the response to PSA test results and recommended a better way. I see this change rapidly becoming current practice.
PSA testing saves lives
Since the early 1990s, when PSA testing became widespread, there has been a 40% decline in prostate cancer mortality.5 A randomized trial in 7 countries in Europe clearly showed a survival benefit from screening for prostate cancer.6 Clinical trials in the United States have been ambiguous.
Not screening for prostate cancer with PSA is unacceptable to many physicians and patients. Most physicians have seen preventable prostate cancer deaths. Two patients in my practice illustrate this point. The PSA of one of them—a 62-year-old man—went from 2.4 to 24 in 2 years. The PSA of another, age 56, went from 2.6 to 34 in one year. Both men had no symptoms, and their prostate cancer was found on routine screening. Both had a high Gleason score and locally invasive prostate cancer. Now, years after undergoing cancer treatment, both have undetectable PSA levels and full function. They think the USPSTF’s recommendation not to screen is evidence of the government’s attempt to save money, reinforcing the notion that the government cannot be trusted.
Patients are increasingly savvy
With all the controversy around prostate cancer screening and the adverse effects of treatment, patients are getting savvier. Shared decision making between doctor and patient is becoming the standard of care, and physicians can meet their professional obligations by offering screening and answering any questions the patient may have. I find that most men with low-grade disease are happy to avoid surgery and radiation if active surveillance is offered and explained.
The American Academy of Family Physicians adopted the recommendation of the USPSTF to advise against screening for prostate cancer.7 The American College of Physicians recommends that men ages 50 to 69 be given the opportunity for informed decision making before screening.8 The American Urological Association recently recommended that men ages 55 to 69 be offered screening, with a discussion about the risks and benefits9; and the American Cancer Society recommends screening starting at age 50, and earlier for high-risk men.10
Not satisfied that any of these organizations really knows what is best and aware that the data are confusing and evolving, I continue to follow my overall practice approach: Start routine cancer screening at age 50 in the general population and at age 40 for high-risk groups. This works for colon, breast, and prostate cancer, the big 3 that are common, sometimes fatal, and often curable with early detection.
Men in my practice are offered a PSA test starting at age 50, and every one to 2 years thereafter based on both patient preference and the results. Black men and those with a family history of prostate cancer before age 60 are offered screening starting at age 40. I suggest that screening be stopped at age 80, or earlier if the patient has a serious chronic illness with a life expectancy of less than 10 years.
Active surveillance for low-grade disease
What is done with elevated or rising PSA levels is most controversial, with lots of room for doing harm. Dramatic rises in PSA, like those of the patients I described earlier, are easy: Go right to biopsy and usually, treatment. Gleason 6 prostate cancer is likely to remain localized and indolent, and not threaten life. I work with urologists who are not aggressive and are willing to follow patients with PSA levels up to 10. Noninvasive options are available, such as fractionating the PSA (free and total) and imaging such as MRI. Genetic testing is available and can add to the evaluation of the patient’s risk.
Active surveillance has become a standard of care in monitoring patients with low-grade disease. The outcomes for survival with active surveillance are as good as radical prostatectomy.11 The goal is to be aggressive in treatment only with patients who have life-threatening disease. A collaboration among the patient, the primary care physician, and the urologist is crucial to optimizing patient outcomes.
Recommending against screening for prostate cancer is not tenable. The responsible approach is to continuously improve cancer detection and therapy to maximize good and minimize harm. This approach is available today.
1. Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277-300.
2. US Preventive Services Task Force. Screening for prostate cancer: Ann Int Med. 2008;149:185-191.
3. Klotz L. Active surveillance: current and future directions. Curr Opin Urol. 2013;23:237-238.
4. Merenstein D. A piece of my mind: Winners and losers. JAMA. 2004;291:15-16.
5. Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;64:220-241.
6. Schroder FH, et al; ERSPC investigators. Screening and prostate cancer mortality in a randomized European study. N Engl J Med. 2009;360:1320-1328.
7. American Academy of Family Physicians Web site. Prostate cancer. Available at: http://www.aafp.org/patient-care/clinical-recommendations/all/prostate-cancer.html. Accessed October 16, 2013.
8. Qaseem A, Barry MJ, Denberg TD, et al. Screening for prostate cancer: a guideline statement from the clinical guidelines of the American College of Physicians. Ann Int Med. 2013;158:761-769.
9. Carter HB, Albertsen PC, Barry MJ, et al. Early detection of prostate cancer: AUA guideline. American Urological Association. 2013;1-28. Available at: http://www.auanet.org/common/pdf/education/clinical-guidance/Prostate-Cancer-Detection.pdf. Accessed October 16, 2013.
10. Wolf AM, Wender RC, Etzioni RB, et al. American Cancer Society guideline for the early detection of prostate cancer: update 2010. CA Cancer J Clin. 2010;60:70-98.
11. Wilt TJ, Brawer MK, Jones KM, et al. Radical prostatectomy versus observation for localized prostate cancer. N Engl J Med. 2012;367:203-213.
Prostate cancer is an important disease. It is the second leading cause of cancer death in men who don’t smoke, and in many cases it is detectable early and curable. The rates of both diagnosis and death from prostate cancer in men are similar to the rates of breast cancer in women.1
The current practical screening test for prostate cancer is the prostate specific antigen (PSA). Making routine use of it, as we know, however, is controversial. The false positive rate for PSA testing is high, for example, in men with chronic prostatitis and benign prostatic hypertrophy.2 In addition, many prostate cancers are diagnosed that will never harm the patient. Treatment for prostate cancer may result in complications, such as incontinence and impotence. Because of these facts, the US Preventive Services Task Force (USPSTF) has recommended against routine screening.2
The PSA test itself never hurt anyone
It is just a lab value, a piece of information. What doctors do with the information is the issue. Physicians may cause more harm than good by being overly aggressive with elevated PSA levels and indolent or low-grade prostate cancer—and 75% of prostate cancer is considered indolent (Gleason score of 6 on biopsy).3 Patients with such a finding can be watched, using active surveillance. The majority will never need treatment.3
Common sense tells us we must screen for prostate cancer. Not doing so on the basis of evidence-based medicine is not a defense when advanced cancer is diagnosed and screening was not offered to the patient.4 Rather than using the data from past physician behavior and recommending against screening with PSA, the USPSTF should have criticized the response to PSA test results and recommended a better way. I see this change rapidly becoming current practice.
PSA testing saves lives
Since the early 1990s, when PSA testing became widespread, there has been a 40% decline in prostate cancer mortality.5 A randomized trial in 7 countries in Europe clearly showed a survival benefit from screening for prostate cancer.6 Clinical trials in the United States have been ambiguous.
Not screening for prostate cancer with PSA is unacceptable to many physicians and patients. Most physicians have seen preventable prostate cancer deaths. Two patients in my practice illustrate this point. The PSA of one of them—a 62-year-old man—went from 2.4 to 24 in 2 years. The PSA of another, age 56, went from 2.6 to 34 in one year. Both men had no symptoms, and their prostate cancer was found on routine screening. Both had a high Gleason score and locally invasive prostate cancer. Now, years after undergoing cancer treatment, both have undetectable PSA levels and full function. They think the USPSTF’s recommendation not to screen is evidence of the government’s attempt to save money, reinforcing the notion that the government cannot be trusted.
Patients are increasingly savvy
With all the controversy around prostate cancer screening and the adverse effects of treatment, patients are getting savvier. Shared decision making between doctor and patient is becoming the standard of care, and physicians can meet their professional obligations by offering screening and answering any questions the patient may have. I find that most men with low-grade disease are happy to avoid surgery and radiation if active surveillance is offered and explained.
The American Academy of Family Physicians adopted the recommendation of the USPSTF to advise against screening for prostate cancer.7 The American College of Physicians recommends that men ages 50 to 69 be given the opportunity for informed decision making before screening.8 The American Urological Association recently recommended that men ages 55 to 69 be offered screening, with a discussion about the risks and benefits9; and the American Cancer Society recommends screening starting at age 50, and earlier for high-risk men.10
Not satisfied that any of these organizations really knows what is best and aware that the data are confusing and evolving, I continue to follow my overall practice approach: Start routine cancer screening at age 50 in the general population and at age 40 for high-risk groups. This works for colon, breast, and prostate cancer, the big 3 that are common, sometimes fatal, and often curable with early detection.
Men in my practice are offered a PSA test starting at age 50, and every one to 2 years thereafter based on both patient preference and the results. Black men and those with a family history of prostate cancer before age 60 are offered screening starting at age 40. I suggest that screening be stopped at age 80, or earlier if the patient has a serious chronic illness with a life expectancy of less than 10 years.
Active surveillance for low-grade disease
What is done with elevated or rising PSA levels is most controversial, with lots of room for doing harm. Dramatic rises in PSA, like those of the patients I described earlier, are easy: Go right to biopsy and usually, treatment. Gleason 6 prostate cancer is likely to remain localized and indolent, and not threaten life. I work with urologists who are not aggressive and are willing to follow patients with PSA levels up to 10. Noninvasive options are available, such as fractionating the PSA (free and total) and imaging such as MRI. Genetic testing is available and can add to the evaluation of the patient’s risk.
Active surveillance has become a standard of care in monitoring patients with low-grade disease. The outcomes for survival with active surveillance are as good as radical prostatectomy.11 The goal is to be aggressive in treatment only with patients who have life-threatening disease. A collaboration among the patient, the primary care physician, and the urologist is crucial to optimizing patient outcomes.
Recommending against screening for prostate cancer is not tenable. The responsible approach is to continuously improve cancer detection and therapy to maximize good and minimize harm. This approach is available today.
Prostate cancer is an important disease. It is the second leading cause of cancer death in men who don’t smoke, and in many cases it is detectable early and curable. The rates of both diagnosis and death from prostate cancer in men are similar to the rates of breast cancer in women.1
The current practical screening test for prostate cancer is the prostate specific antigen (PSA). Making routine use of it, as we know, however, is controversial. The false positive rate for PSA testing is high, for example, in men with chronic prostatitis and benign prostatic hypertrophy.2 In addition, many prostate cancers are diagnosed that will never harm the patient. Treatment for prostate cancer may result in complications, such as incontinence and impotence. Because of these facts, the US Preventive Services Task Force (USPSTF) has recommended against routine screening.2
The PSA test itself never hurt anyone
It is just a lab value, a piece of information. What doctors do with the information is the issue. Physicians may cause more harm than good by being overly aggressive with elevated PSA levels and indolent or low-grade prostate cancer—and 75% of prostate cancer is considered indolent (Gleason score of 6 on biopsy).3 Patients with such a finding can be watched, using active surveillance. The majority will never need treatment.3
Common sense tells us we must screen for prostate cancer. Not doing so on the basis of evidence-based medicine is not a defense when advanced cancer is diagnosed and screening was not offered to the patient.4 Rather than using the data from past physician behavior and recommending against screening with PSA, the USPSTF should have criticized the response to PSA test results and recommended a better way. I see this change rapidly becoming current practice.
PSA testing saves lives
Since the early 1990s, when PSA testing became widespread, there has been a 40% decline in prostate cancer mortality.5 A randomized trial in 7 countries in Europe clearly showed a survival benefit from screening for prostate cancer.6 Clinical trials in the United States have been ambiguous.
Not screening for prostate cancer with PSA is unacceptable to many physicians and patients. Most physicians have seen preventable prostate cancer deaths. Two patients in my practice illustrate this point. The PSA of one of them—a 62-year-old man—went from 2.4 to 24 in 2 years. The PSA of another, age 56, went from 2.6 to 34 in one year. Both men had no symptoms, and their prostate cancer was found on routine screening. Both had a high Gleason score and locally invasive prostate cancer. Now, years after undergoing cancer treatment, both have undetectable PSA levels and full function. They think the USPSTF’s recommendation not to screen is evidence of the government’s attempt to save money, reinforcing the notion that the government cannot be trusted.
Patients are increasingly savvy
With all the controversy around prostate cancer screening and the adverse effects of treatment, patients are getting savvier. Shared decision making between doctor and patient is becoming the standard of care, and physicians can meet their professional obligations by offering screening and answering any questions the patient may have. I find that most men with low-grade disease are happy to avoid surgery and radiation if active surveillance is offered and explained.
The American Academy of Family Physicians adopted the recommendation of the USPSTF to advise against screening for prostate cancer.7 The American College of Physicians recommends that men ages 50 to 69 be given the opportunity for informed decision making before screening.8 The American Urological Association recently recommended that men ages 55 to 69 be offered screening, with a discussion about the risks and benefits9; and the American Cancer Society recommends screening starting at age 50, and earlier for high-risk men.10
Not satisfied that any of these organizations really knows what is best and aware that the data are confusing and evolving, I continue to follow my overall practice approach: Start routine cancer screening at age 50 in the general population and at age 40 for high-risk groups. This works for colon, breast, and prostate cancer, the big 3 that are common, sometimes fatal, and often curable with early detection.
Men in my practice are offered a PSA test starting at age 50, and every one to 2 years thereafter based on both patient preference and the results. Black men and those with a family history of prostate cancer before age 60 are offered screening starting at age 40. I suggest that screening be stopped at age 80, or earlier if the patient has a serious chronic illness with a life expectancy of less than 10 years.
Active surveillance for low-grade disease
What is done with elevated or rising PSA levels is most controversial, with lots of room for doing harm. Dramatic rises in PSA, like those of the patients I described earlier, are easy: Go right to biopsy and usually, treatment. Gleason 6 prostate cancer is likely to remain localized and indolent, and not threaten life. I work with urologists who are not aggressive and are willing to follow patients with PSA levels up to 10. Noninvasive options are available, such as fractionating the PSA (free and total) and imaging such as MRI. Genetic testing is available and can add to the evaluation of the patient’s risk.
Active surveillance has become a standard of care in monitoring patients with low-grade disease. The outcomes for survival with active surveillance are as good as radical prostatectomy.11 The goal is to be aggressive in treatment only with patients who have life-threatening disease. A collaboration among the patient, the primary care physician, and the urologist is crucial to optimizing patient outcomes.
Recommending against screening for prostate cancer is not tenable. The responsible approach is to continuously improve cancer detection and therapy to maximize good and minimize harm. This approach is available today.
1. Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277-300.
2. US Preventive Services Task Force. Screening for prostate cancer: Ann Int Med. 2008;149:185-191.
3. Klotz L. Active surveillance: current and future directions. Curr Opin Urol. 2013;23:237-238.
4. Merenstein D. A piece of my mind: Winners and losers. JAMA. 2004;291:15-16.
5. Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;64:220-241.
6. Schroder FH, et al; ERSPC investigators. Screening and prostate cancer mortality in a randomized European study. N Engl J Med. 2009;360:1320-1328.
7. American Academy of Family Physicians Web site. Prostate cancer. Available at: http://www.aafp.org/patient-care/clinical-recommendations/all/prostate-cancer.html. Accessed October 16, 2013.
8. Qaseem A, Barry MJ, Denberg TD, et al. Screening for prostate cancer: a guideline statement from the clinical guidelines of the American College of Physicians. Ann Int Med. 2013;158:761-769.
9. Carter HB, Albertsen PC, Barry MJ, et al. Early detection of prostate cancer: AUA guideline. American Urological Association. 2013;1-28. Available at: http://www.auanet.org/common/pdf/education/clinical-guidance/Prostate-Cancer-Detection.pdf. Accessed October 16, 2013.
10. Wolf AM, Wender RC, Etzioni RB, et al. American Cancer Society guideline for the early detection of prostate cancer: update 2010. CA Cancer J Clin. 2010;60:70-98.
11. Wilt TJ, Brawer MK, Jones KM, et al. Radical prostatectomy versus observation for localized prostate cancer. N Engl J Med. 2012;367:203-213.
1. Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277-300.
2. US Preventive Services Task Force. Screening for prostate cancer: Ann Int Med. 2008;149:185-191.
3. Klotz L. Active surveillance: current and future directions. Curr Opin Urol. 2013;23:237-238.
4. Merenstein D. A piece of my mind: Winners and losers. JAMA. 2004;291:15-16.
5. Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;64:220-241.
6. Schroder FH, et al; ERSPC investigators. Screening and prostate cancer mortality in a randomized European study. N Engl J Med. 2009;360:1320-1328.
7. American Academy of Family Physicians Web site. Prostate cancer. Available at: http://www.aafp.org/patient-care/clinical-recommendations/all/prostate-cancer.html. Accessed October 16, 2013.
8. Qaseem A, Barry MJ, Denberg TD, et al. Screening for prostate cancer: a guideline statement from the clinical guidelines of the American College of Physicians. Ann Int Med. 2013;158:761-769.
9. Carter HB, Albertsen PC, Barry MJ, et al. Early detection of prostate cancer: AUA guideline. American Urological Association. 2013;1-28. Available at: http://www.auanet.org/common/pdf/education/clinical-guidance/Prostate-Cancer-Detection.pdf. Accessed October 16, 2013.
10. Wolf AM, Wender RC, Etzioni RB, et al. American Cancer Society guideline for the early detection of prostate cancer: update 2010. CA Cancer J Clin. 2010;60:70-98.
11. Wilt TJ, Brawer MK, Jones KM, et al. Radical prostatectomy versus observation for localized prostate cancer. N Engl J Med. 2012;367:203-213.
Construction Worker Falls From Scaffolding
ANSWER
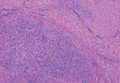
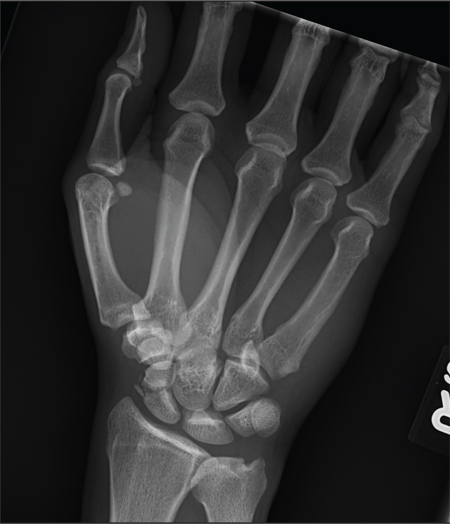
The radiograph shows an acute, comminuted fracture of the scaphoid bone. The patient was placed in a thumb spica splint and sling. He was instructed to follow up in one to two days with the hand surgeon who was on call, with anticipation of subsequent open reduction and internal fixation.
ANSWER
The radiograph shows an acute, comminuted fracture of the scaphoid bone. The patient was placed in a thumb spica splint and sling. He was instructed to follow up in one to two days with the hand surgeon who was on call, with anticipation of subsequent open reduction and internal fixation.
ANSWER
The radiograph shows an acute, comminuted fracture of the scaphoid bone. The patient was placed in a thumb spica splint and sling. He was instructed to follow up in one to two days with the hand surgeon who was on call, with anticipation of subsequent open reduction and internal fixation.
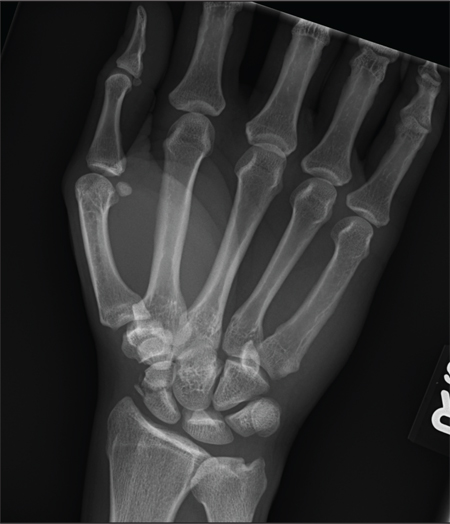
A 45-year-old construction worker is brought to your facility for evaluation following a fall. He was at a job site, standing on scaffolding approximately 20 feet above the ground, when he accidentally fell. He does not remember for sure, but he thinks he landed on his face. He did briefly lose consciousness. He is complaining of right-side facial pain and right wrist pain. His medical history is unremarkable. The physical exam reveals stable vital signs. The patient appears somewhat uncomfortable but is in no obvious distress. There is a moderate amount of periorbital soft-tissue swelling around his right eye, with moderate associated tenderness. Pupils are equal and react well bilaterally. Examination of the right wrist shows a moderate amount of soft-tissue swelling. The patient is unable to flex or extend his wrist due to pain. Good pulses and capillary refill of the nail beds are noted. There is also moderate tenderness along the base of the first metacarpal. Radiograph of the right wrist is shown. What is your impression?
Now Insured, Patient Wants to “Get Checked Out”
ANSWER
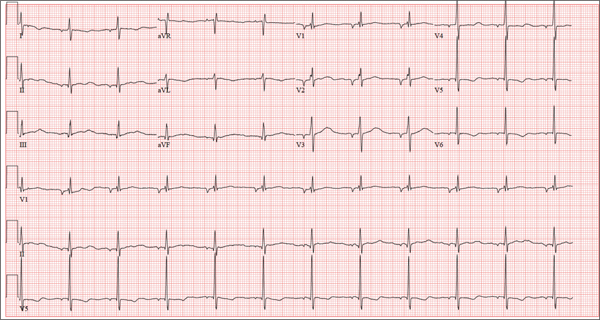
There are three findings on this ECG: unusual P waves consistent with a possible ectopic atrial rhythm, a prolonged QT interval, and T-wave abnormalities in the lateral leads.
Note that the P waves are negative in leads I and II, as well as in all chest leads. This is highly suggestive of an ectopic atrial rhythm originating low in the atria, conducting retrograde into the atria, and overriding the sinoatrial node. Limb lead reversal would result in negative P waves in lead I, but not in other leads.
A prolonged QT interval is determined by consulting any of the standard charts that correlate maximum heart rates with QT intervals and gender. In men, the QT interval is considered “prolonged” when it exceeds 440 ms, unless the heart rate is extremely slow.
Finally, T-wave inversions are present in the lateral leads (V5, V6). Although this may be an indication of lateral ischemia, there is no clinical correlation in this patient.
ANSWER
There are three findings on this ECG: unusual P waves consistent with a possible ectopic atrial rhythm, a prolonged QT interval, and T-wave abnormalities in the lateral leads.
Note that the P waves are negative in leads I and II, as well as in all chest leads. This is highly suggestive of an ectopic atrial rhythm originating low in the atria, conducting retrograde into the atria, and overriding the sinoatrial node. Limb lead reversal would result in negative P waves in lead I, but not in other leads.
A prolonged QT interval is determined by consulting any of the standard charts that correlate maximum heart rates with QT intervals and gender. In men, the QT interval is considered “prolonged” when it exceeds 440 ms, unless the heart rate is extremely slow.
Finally, T-wave inversions are present in the lateral leads (V5, V6). Although this may be an indication of lateral ischemia, there is no clinical correlation in this patient.
ANSWER
There are three findings on this ECG: unusual P waves consistent with a possible ectopic atrial rhythm, a prolonged QT interval, and T-wave abnormalities in the lateral leads.
Note that the P waves are negative in leads I and II, as well as in all chest leads. This is highly suggestive of an ectopic atrial rhythm originating low in the atria, conducting retrograde into the atria, and overriding the sinoatrial node. Limb lead reversal would result in negative P waves in lead I, but not in other leads.
A prolonged QT interval is determined by consulting any of the standard charts that correlate maximum heart rates with QT intervals and gender. In men, the QT interval is considered “prolonged” when it exceeds 440 ms, unless the heart rate is extremely slow.
Finally, T-wave inversions are present in the lateral leads (V5, V6). Although this may be an indication of lateral ischemia, there is no clinical correlation in this patient.
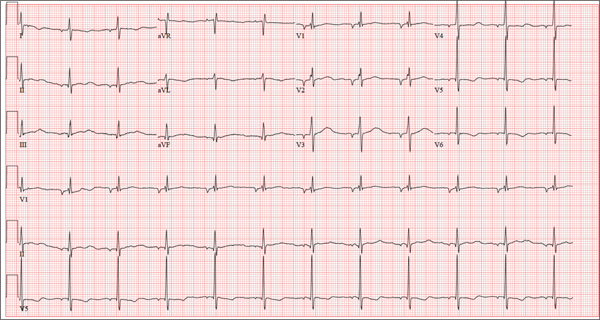
A 37-year-old man presents to your office to establish care. After being unemployed for two years, he recently obtained a position with a local manufacturing company and, as a result, has health benefits. He wants to “get checked out.” He has not seen a health care provider since having his tonsils removed at age 14. He says he is rarely ill, aside from an occasional cold. Besides the tonsillectomy, medical history is positive for a right clavicular fracture at age 6 and a left inguinal hernia repair at age 9. He had chickenpox and recalls that his immunizations were up to date until he graduated high school. His only medication is ibuprofen as needed for aches and pains. He has no known drug allergies. He uses two herbal supplements, fenugreek seed and horny goat weed, daily. He admits to recreational marijuana use. Family history is remarkable for coronary artery disease (father), diabetes (mother), and depression (sister). He consumes one six-pack of beer weekly and has smoked one pack of cigarettes per day for the past 23 years. He isn’t interested in quitting smoking. The patient is divorced, without children. He has been collecting unemployment since his last position was terminated due to budget constraints. A 20-point comprehensive review of systems is negative, with the exception of occasional palpitations and a productive morning smoker’s cough that quickly resolves. He states he’s “as healthy as a horse.” The physical exam reveals a thin, healthy-appearing middle-aged male. He is 72 in tall and weighs 167 lb. His blood pressure is 108/66 mm Hg; pulse, 70 beats/min and regular; respiratory rate, 14 breaths/min-1; and temperature, 98.4°F. The head, eyes, ears, nose, and throat (HEENT) exam is remarkable for poor dentition, with multiple caries readily visible. The tonsils are absent. Coarse expiratory crackles are present in both bases and clear with vigorous coughing. The abdominal exam is positive for a well-healed scar in the left inguinal crease. The remainder of the physical exam is normal. As part of a new patient visit, a chest x-ray and ECG are obtained. The ECG shows the following: a ventricular rate of 69 beats/min; PR interval, 178 ms; QRS duration, 90ms; QT/QTc interval, 442/473 ms; P axis, 231°; R axis, 84°; and T axis, 93°. What is your interpretation of this ECG?
A Purplish Rash on the Instep
ANSWER
The correct answer is lichen planus (choice “d”), an inflammatory condition marked by pathognomic histologic changes such as those described. Hardly on the tip of most primary care providers’ tongues, lichen planus is nonetheless quite commonly occuring.
Tinea pedis (choice “a”) certainly belongs in the differential, but it would be unusual in a woman of this age. The biopsy effectively ruled it out, as it did psoriasis (choice “b”) and contact dermatitis (choice “c”).
DISCUSSION
Lichen planus (LP) is the prototypical lichenoid interface dermatitis; an inflammatory infiltrate erases the normally well-defined dermoepidermal junction, replacing it with a jagged “sawtooth” band of lymphocytes. In the process, the cells it kills off (keratinocytes) collect at this interface and are incorporated as necrotic keratinocytes into the papillary dermis.
Classically, the recognition of LP is taught with “The Ps.” LP is said to be:
• Papular
• Planar
• Purple
• Polygonal
• Pruritic
• Plaquish
• Penile
• Puzzling
The word “puzzling” may sound nebulous, but it is actually quite useful for dermatology providers. It comes into play whenever a condition stumps us—as in, “What in the world is this?” This puzzlement causes us to at least consider LP.
LP is often papular, and these papules often have flat (planar) tops. The purple color is more striking in lighter-skinned patients. (Almost all inflammatory conditions are more difficult to diagnose in those with darker skin.) More typically, LP lesions are multi-angular or polygonal, often plaquish, and almost invariably pruritic.
LP is common on the legs and/or trunk, favoring the sacral area. It can affect the scalp (being in the differential for hair loss), can cause nail dystrophy, and is relatively common in the mouth, where it presents as a lacy, reticular, slightly erosive process, usually affecting the buccal mucosae. It may be found on the penis, where it is usually confined to the distal shaft and proximal coronal areas.
Like many otherwise benign conditions, LP can present with bullae. In anterior tibial areas, especially on darker-skinned persons, it can be remarkably hypertrophic.
The cause is usually unknown, although certain drugs—especially the antimalarials, gold salts, and penicillamine—have been known to cause LP-like eruptions. It’s been my observation that flares of LP are prompted by stress (certainly present in this patient’s case). LP may have a connection to hepatitis, although no convincing connection has been established.
TREATMENT
This patient’s condition was treated with topical class I corticosteroid ointment (halobetasol). For her, having a certain diagnosis was almost as important as successful treatment.
ANSWER
The correct answer is lichen planus (choice “d”), an inflammatory condition marked by pathognomic histologic changes such as those described. Hardly on the tip of most primary care providers’ tongues, lichen planus is nonetheless quite commonly occuring.
Tinea pedis (choice “a”) certainly belongs in the differential, but it would be unusual in a woman of this age. The biopsy effectively ruled it out, as it did psoriasis (choice “b”) and contact dermatitis (choice “c”).
DISCUSSION
Lichen planus (LP) is the prototypical lichenoid interface dermatitis; an inflammatory infiltrate erases the normally well-defined dermoepidermal junction, replacing it with a jagged “sawtooth” band of lymphocytes. In the process, the cells it kills off (keratinocytes) collect at this interface and are incorporated as necrotic keratinocytes into the papillary dermis.
Classically, the recognition of LP is taught with “The Ps.” LP is said to be:
• Papular
• Planar
• Purple
• Polygonal
• Pruritic
• Plaquish
• Penile
• Puzzling
The word “puzzling” may sound nebulous, but it is actually quite useful for dermatology providers. It comes into play whenever a condition stumps us—as in, “What in the world is this?” This puzzlement causes us to at least consider LP.
LP is often papular, and these papules often have flat (planar) tops. The purple color is more striking in lighter-skinned patients. (Almost all inflammatory conditions are more difficult to diagnose in those with darker skin.) More typically, LP lesions are multi-angular or polygonal, often plaquish, and almost invariably pruritic.
LP is common on the legs and/or trunk, favoring the sacral area. It can affect the scalp (being in the differential for hair loss), can cause nail dystrophy, and is relatively common in the mouth, where it presents as a lacy, reticular, slightly erosive process, usually affecting the buccal mucosae. It may be found on the penis, where it is usually confined to the distal shaft and proximal coronal areas.
Like many otherwise benign conditions, LP can present with bullae. In anterior tibial areas, especially on darker-skinned persons, it can be remarkably hypertrophic.
The cause is usually unknown, although certain drugs—especially the antimalarials, gold salts, and penicillamine—have been known to cause LP-like eruptions. It’s been my observation that flares of LP are prompted by stress (certainly present in this patient’s case). LP may have a connection to hepatitis, although no convincing connection has been established.
TREATMENT
This patient’s condition was treated with topical class I corticosteroid ointment (halobetasol). For her, having a certain diagnosis was almost as important as successful treatment.
ANSWER
The correct answer is lichen planus (choice “d”), an inflammatory condition marked by pathognomic histologic changes such as those described. Hardly on the tip of most primary care providers’ tongues, lichen planus is nonetheless quite commonly occuring.
Tinea pedis (choice “a”) certainly belongs in the differential, but it would be unusual in a woman of this age. The biopsy effectively ruled it out, as it did psoriasis (choice “b”) and contact dermatitis (choice “c”).
DISCUSSION
Lichen planus (LP) is the prototypical lichenoid interface dermatitis; an inflammatory infiltrate erases the normally well-defined dermoepidermal junction, replacing it with a jagged “sawtooth” band of lymphocytes. In the process, the cells it kills off (keratinocytes) collect at this interface and are incorporated as necrotic keratinocytes into the papillary dermis.
Classically, the recognition of LP is taught with “The Ps.” LP is said to be:
• Papular
• Planar
• Purple
• Polygonal
• Pruritic
• Plaquish
• Penile
• Puzzling
The word “puzzling” may sound nebulous, but it is actually quite useful for dermatology providers. It comes into play whenever a condition stumps us—as in, “What in the world is this?” This puzzlement causes us to at least consider LP.
LP is often papular, and these papules often have flat (planar) tops. The purple color is more striking in lighter-skinned patients. (Almost all inflammatory conditions are more difficult to diagnose in those with darker skin.) More typically, LP lesions are multi-angular or polygonal, often plaquish, and almost invariably pruritic.
LP is common on the legs and/or trunk, favoring the sacral area. It can affect the scalp (being in the differential for hair loss), can cause nail dystrophy, and is relatively common in the mouth, where it presents as a lacy, reticular, slightly erosive process, usually affecting the buccal mucosae. It may be found on the penis, where it is usually confined to the distal shaft and proximal coronal areas.
Like many otherwise benign conditions, LP can present with bullae. In anterior tibial areas, especially on darker-skinned persons, it can be remarkably hypertrophic.
The cause is usually unknown, although certain drugs—especially the antimalarials, gold salts, and penicillamine—have been known to cause LP-like eruptions. It’s been my observation that flares of LP are prompted by stress (certainly present in this patient’s case). LP may have a connection to hepatitis, although no convincing connection has been established.
TREATMENT
This patient’s condition was treated with topical class I corticosteroid ointment (halobetasol). For her, having a certain diagnosis was almost as important as successful treatment.
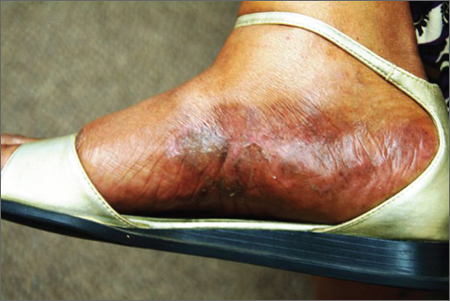
Several months ago, an itchy rash appeared on the foot of this 61-year-old African-American woman. She tried using a variety of OTC and prescription products, including tolnaftate cream, hydrocortisone 1% cream, and triamcinolone cream, but the rash persisted. She reports that the triamcinolone cream did relieve the itching for a few minutes after application, but the symptoms always returned. The rash has gradually grown. The patient, a retired schoolteacher, denies any other skin problems. She does have several relatively minor health problems, including hypertension (well controlled with metoprolol) and mild reactive airway disease (related to a 20-year history of smoking). The rash manifested around the time she was forced to retire in an unforeseen downsizing effort by her school district. As a result, she lost her health care coverage and could not afford private insurance for herself and her husband (neither of whom are old enough to qualify for Medicare). The rash, which covers a roughly 12 × 8–cm area, begins on her left instep and spills onto the lower ankle. It is quite dark, as would be expected in a person with type V skin, but there is a slightly purplish tinge to it. The surface of the affected area is a bit shiny and atrophic, and the margins are well defined and annular. There is almost no scaling, and the rest of the skin on her feet and legs is well within normal limits. A 4-mm punch biopsy is performed on the lesion. The pathology report shows obliteration of the dermoepidermal junction by a bandlike lymphocytic infiltrate in the papillary dermis, associated with vacuolar changes and the accumulation of necrotic keratinocytes in the basal layer.

Renal risk stratification with the new oral anticoagulants
To the Editor: I read with interest the review of the new oral anticoagulants by Fawole et al1 and agree with their comments on the prevention of bleeding and the importance of monitoring renal function in managing patients on the new classes of oral anticoagulants. However, no specifics were given on how to proceed. Thus, I recommend that renal risk stratification be done before and 1 week after starting these new drugs.
Originally, the US Food and Drug Administration approved dabigatran (Pradaxa) at a dose of 150 mg orally twice daily in patients with a creatinine clearance of 15 to 30 mL/min/1.73 m2. This dosing corresponded to the estimated glomerular filtration rate (eGFR) in patients with stage 4 chronic kidney disease, but this dosing is contraindicated in other guidelines worldwide (Canada, Europe, the United Kingdom, Japan, Australia, and New Zealand).2 Not unexpectedly, 3,781 serious adverse effects were noted in the 2011 US postmarketing experience with dabigatran. These included death (542 cases), hemorrhage (2,367 cases), acute renal failure (291 cases), stroke (644 cases), and suspected liver failure (15 cases).3 Thirteen months after dabigatran’s approval in the United States, Boehringer Ingelheim changed the dosage and product guidelines.2–4 The new dosage4 is 75 mg twice daily for patients with a creatinine clearance of 15 to 30 mL/min/1.73 m2.
Therefore, I suggest a nephrologic “way out”5 when using the new oral anticoagulants to avoid the problems with dabigatran noted above.
First, if these drugs are to be used in nonvalvular atrial fibrillation, risk factors should be determined using the CHADS2 or the CHADS2-VASc score. Special attention should be given to patients age 75 and older, women, and patients with a history of stroke, transient ischemic attack, or systemic embolism. All of these have been noted to be major risk factors.6,7
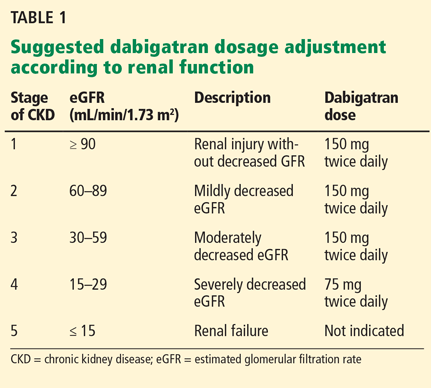
Second, renal risk stratification8 should be done using a comprehensive metabolic panel before and 1 week after starting new oral anticoagulants, or if there is a change in the patient’s clinical condition. Most US laboratories now provide an eGFR and the stage of chronic kidney disease.3,5 For example (Table 1), if dabigatran is used, one should follow current dosing guidelines for chronic kidney disease stages 1 through 3, ie, 150 mg twice daily. If stage 4 chronic kidney disease is detected (creatinine clearance 15–29 mL/min/1.73 m2), the updated recommended dosage is 75 mg twice daily. If stage 5 is noted (eGFR ≤ 15 mL/min/1.73 m2), dabigatran is not indicated. Similar steps can be done using current guidelines for the other new oral anticoagulants.
This simple renal risk stratification guideline should help avoid some of the problems noted in the dabigatran postmarketing experience, which were aggravated by the lack of approval of a 110-mg dose and by misleading advertising, claiming that no blood monitoring was required.2–5 Thus, the new oral anticoagulants should be a welcome addition to our armamentarium in patients who need them, and we hope to avoid the risks, morbidity, mortality, and expense of trying to reverse adverse effects.
- Fawole A, Daw HA, Crowther MA. Practical management of bleeding due to the anticoagulants, dabigatran, rivaroxaban, and apixaban. Cleve Clin J Med 2013, 80:443–451.
- Pazmiño PA. Dabigatran associated acute renal failure (DAARF). El Paso Physician 2011; 34:7–9.
- Pazmiño PA. Adverse effects of dabigatran (Letter). Ann Intern Med 2012; 157:916.
- Pradaxa (prescribing information). Ridgefield, CT. Boehringer Ingelheim Pharmaceuticals 2011.
- Pazmiño PA. Dabigatran: a nephrological way out. Am J Med 2013; 126;e21–e22.
- Lip GY, Nieuwlaat R, Pisters R, Lane DA, Crijns HJ. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach. The Euro Heart Survey on Atrial Fibrillation. Chest 2010; 137:263–272.
- Reinecke H, Brand E, Mesters R, et al. Dilemmas in the management of atrial fibrillation in chronic kidney disease. J Am Soc Nephrol 2009; 20:705–711.
- National Kidney Foundation. K/DOQI clinical practice guidelines for chronic disease: evaluation, classification and stratification. Am J Kidney Dis 2002; 39(suppl 1):S1–S266.
To the Editor: I read with interest the review of the new oral anticoagulants by Fawole et al1 and agree with their comments on the prevention of bleeding and the importance of monitoring renal function in managing patients on the new classes of oral anticoagulants. However, no specifics were given on how to proceed. Thus, I recommend that renal risk stratification be done before and 1 week after starting these new drugs.
Originally, the US Food and Drug Administration approved dabigatran (Pradaxa) at a dose of 150 mg orally twice daily in patients with a creatinine clearance of 15 to 30 mL/min/1.73 m2. This dosing corresponded to the estimated glomerular filtration rate (eGFR) in patients with stage 4 chronic kidney disease, but this dosing is contraindicated in other guidelines worldwide (Canada, Europe, the United Kingdom, Japan, Australia, and New Zealand).2 Not unexpectedly, 3,781 serious adverse effects were noted in the 2011 US postmarketing experience with dabigatran. These included death (542 cases), hemorrhage (2,367 cases), acute renal failure (291 cases), stroke (644 cases), and suspected liver failure (15 cases).3 Thirteen months after dabigatran’s approval in the United States, Boehringer Ingelheim changed the dosage and product guidelines.2–4 The new dosage4 is 75 mg twice daily for patients with a creatinine clearance of 15 to 30 mL/min/1.73 m2.
Therefore, I suggest a nephrologic “way out”5 when using the new oral anticoagulants to avoid the problems with dabigatran noted above.
First, if these drugs are to be used in nonvalvular atrial fibrillation, risk factors should be determined using the CHADS2 or the CHADS2-VASc score. Special attention should be given to patients age 75 and older, women, and patients with a history of stroke, transient ischemic attack, or systemic embolism. All of these have been noted to be major risk factors.6,7
Second, renal risk stratification8 should be done using a comprehensive metabolic panel before and 1 week after starting new oral anticoagulants, or if there is a change in the patient’s clinical condition. Most US laboratories now provide an eGFR and the stage of chronic kidney disease.3,5 For example (Table 1), if dabigatran is used, one should follow current dosing guidelines for chronic kidney disease stages 1 through 3, ie, 150 mg twice daily. If stage 4 chronic kidney disease is detected (creatinine clearance 15–29 mL/min/1.73 m2), the updated recommended dosage is 75 mg twice daily. If stage 5 is noted (eGFR ≤ 15 mL/min/1.73 m2), dabigatran is not indicated. Similar steps can be done using current guidelines for the other new oral anticoagulants.
This simple renal risk stratification guideline should help avoid some of the problems noted in the dabigatran postmarketing experience, which were aggravated by the lack of approval of a 110-mg dose and by misleading advertising, claiming that no blood monitoring was required.2–5 Thus, the new oral anticoagulants should be a welcome addition to our armamentarium in patients who need them, and we hope to avoid the risks, morbidity, mortality, and expense of trying to reverse adverse effects.
To the Editor: I read with interest the review of the new oral anticoagulants by Fawole et al1 and agree with their comments on the prevention of bleeding and the importance of monitoring renal function in managing patients on the new classes of oral anticoagulants. However, no specifics were given on how to proceed. Thus, I recommend that renal risk stratification be done before and 1 week after starting these new drugs.
Originally, the US Food and Drug Administration approved dabigatran (Pradaxa) at a dose of 150 mg orally twice daily in patients with a creatinine clearance of 15 to 30 mL/min/1.73 m2. This dosing corresponded to the estimated glomerular filtration rate (eGFR) in patients with stage 4 chronic kidney disease, but this dosing is contraindicated in other guidelines worldwide (Canada, Europe, the United Kingdom, Japan, Australia, and New Zealand).2 Not unexpectedly, 3,781 serious adverse effects were noted in the 2011 US postmarketing experience with dabigatran. These included death (542 cases), hemorrhage (2,367 cases), acute renal failure (291 cases), stroke (644 cases), and suspected liver failure (15 cases).3 Thirteen months after dabigatran’s approval in the United States, Boehringer Ingelheim changed the dosage and product guidelines.2–4 The new dosage4 is 75 mg twice daily for patients with a creatinine clearance of 15 to 30 mL/min/1.73 m2.
Therefore, I suggest a nephrologic “way out”5 when using the new oral anticoagulants to avoid the problems with dabigatran noted above.
First, if these drugs are to be used in nonvalvular atrial fibrillation, risk factors should be determined using the CHADS2 or the CHADS2-VASc score. Special attention should be given to patients age 75 and older, women, and patients with a history of stroke, transient ischemic attack, or systemic embolism. All of these have been noted to be major risk factors.6,7
Second, renal risk stratification8 should be done using a comprehensive metabolic panel before and 1 week after starting new oral anticoagulants, or if there is a change in the patient’s clinical condition. Most US laboratories now provide an eGFR and the stage of chronic kidney disease.3,5 For example (Table 1), if dabigatran is used, one should follow current dosing guidelines for chronic kidney disease stages 1 through 3, ie, 150 mg twice daily. If stage 4 chronic kidney disease is detected (creatinine clearance 15–29 mL/min/1.73 m2), the updated recommended dosage is 75 mg twice daily. If stage 5 is noted (eGFR ≤ 15 mL/min/1.73 m2), dabigatran is not indicated. Similar steps can be done using current guidelines for the other new oral anticoagulants.
This simple renal risk stratification guideline should help avoid some of the problems noted in the dabigatran postmarketing experience, which were aggravated by the lack of approval of a 110-mg dose and by misleading advertising, claiming that no blood monitoring was required.2–5 Thus, the new oral anticoagulants should be a welcome addition to our armamentarium in patients who need them, and we hope to avoid the risks, morbidity, mortality, and expense of trying to reverse adverse effects.
- Fawole A, Daw HA, Crowther MA. Practical management of bleeding due to the anticoagulants, dabigatran, rivaroxaban, and apixaban. Cleve Clin J Med 2013, 80:443–451.
- Pazmiño PA. Dabigatran associated acute renal failure (DAARF). El Paso Physician 2011; 34:7–9.
- Pazmiño PA. Adverse effects of dabigatran (Letter). Ann Intern Med 2012; 157:916.
- Pradaxa (prescribing information). Ridgefield, CT. Boehringer Ingelheim Pharmaceuticals 2011.
- Pazmiño PA. Dabigatran: a nephrological way out. Am J Med 2013; 126;e21–e22.
- Lip GY, Nieuwlaat R, Pisters R, Lane DA, Crijns HJ. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach. The Euro Heart Survey on Atrial Fibrillation. Chest 2010; 137:263–272.
- Reinecke H, Brand E, Mesters R, et al. Dilemmas in the management of atrial fibrillation in chronic kidney disease. J Am Soc Nephrol 2009; 20:705–711.
- National Kidney Foundation. K/DOQI clinical practice guidelines for chronic disease: evaluation, classification and stratification. Am J Kidney Dis 2002; 39(suppl 1):S1–S266.
- Fawole A, Daw HA, Crowther MA. Practical management of bleeding due to the anticoagulants, dabigatran, rivaroxaban, and apixaban. Cleve Clin J Med 2013, 80:443–451.
- Pazmiño PA. Dabigatran associated acute renal failure (DAARF). El Paso Physician 2011; 34:7–9.
- Pazmiño PA. Adverse effects of dabigatran (Letter). Ann Intern Med 2012; 157:916.
- Pradaxa (prescribing information). Ridgefield, CT. Boehringer Ingelheim Pharmaceuticals 2011.
- Pazmiño PA. Dabigatran: a nephrological way out. Am J Med 2013; 126;e21–e22.
- Lip GY, Nieuwlaat R, Pisters R, Lane DA, Crijns HJ. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach. The Euro Heart Survey on Atrial Fibrillation. Chest 2010; 137:263–272.
- Reinecke H, Brand E, Mesters R, et al. Dilemmas in the management of atrial fibrillation in chronic kidney disease. J Am Soc Nephrol 2009; 20:705–711.
- National Kidney Foundation. K/DOQI clinical practice guidelines for chronic disease: evaluation, classification and stratification. Am J Kidney Dis 2002; 39(suppl 1):S1–S266.