User login
The Official Newspaper of the American Association for Thoracic Surgery
Ex vivo lung perfusion device preserves donor organs
A device that preserves less-than-ideal donor lungs until they are cleared for transplantation has been approved, the Food and Drug Administration announced on Aug. 12.
The ex vivo perfusion device preserves donated lungs that initially do not meet all the criteria for a transplantable lung. The device does this by warming the donor lungs to "near normal body temperature," continuously flushing the lung with a sterile solution, and ventilating them, "which oxygenates the cells and makes it possible for the transplant team to examine the lung’s’ airways with a bronchoscope," according to the FDA statement.
The lungs can remain in the machine for up to 4 hours, providing time for the transplant team to evaluate the lungs to determine if they meet the criteria; donor lungs that meet the criteria are then transplanted into a patient.
The device, the XVIVO Perfusion System (XPS) with STEEN Solution, is manufactured by XVIVO Perfusion.
"With this approval, there may be more lungs available for transplant, which could allow more people with end-stage lung disease who have exhausted all other treatment options to be able to receive a lung transplant," Christy Foreman, director of the Office of Device Evaluation in the FDA’s Center for Devices and Radiological Health, Silver Spring, Md., said in the statement.
About 1 in 5 donor lungs meets the standard transplantation criteria. In the United States, 1,754 lung transplants were performed in 2012 and 1,616 potential recipients were on the lung transplant waiting list at the end of 2012, according to the FDA.
In two studies, outcomes for lung-transplant recipients were similar among those who received a donor lung preserved with the device and those who received donor lungs that were considered ideal and were preserved in cold storage.
"Both trials showed that recipients of the ideal and non-ideal lungs had similar survival rates up to 12 months after transplant and similar rates of organ rejection," the FDA statement said.
The manufacturer is required to conduct a long-term study of the effects of the device as a condition of approval.
emechcatie@frontlinemedcom.com

|
|
Dr. Jennifer Cox, FCCP, comments: This is exciting news given the shortage of available lungs that meet the current transplant criteria. Early studies showing similar 12-month survival rates and rates of organ rejection are encouraging. I would like to know if there were similar hospital lengths of stay and if there was a difference in post operative complications. Also, how significant will the financial impact be using the device. I look forward to the results of long- term studies. Hopefully this will be a viable option for our patients.
Dr. Jennifer D. Cox, FCCP, is an Assistant Professor of Pulmonary and Critical Care Medicine and clerkship director for the fourth-year medical student Critical Care Selective, Morsani College of Medicine, University of South Florida, in Tampa, Florida.
|
|
|
Dr. Jennifer Cox, FCCP, comments: This is exciting news given the shortage of available lungs that meet the current transplant criteria. Early studies showing similar 12-month survival rates and rates of organ rejection are encouraging. I would like to know if there were similar hospital lengths of stay and if there was a difference in post operative complications. Also, how significant will the financial impact be using the device. I look forward to the results of long- term studies. Hopefully this will be a viable option for our patients.
Dr. Jennifer D. Cox, FCCP, is an Assistant Professor of Pulmonary and Critical Care Medicine and clerkship director for the fourth-year medical student Critical Care Selective, Morsani College of Medicine, University of South Florida, in Tampa, Florida.
|
|
|
Dr. Jennifer Cox, FCCP, comments: This is exciting news given the shortage of available lungs that meet the current transplant criteria. Early studies showing similar 12-month survival rates and rates of organ rejection are encouraging. I would like to know if there were similar hospital lengths of stay and if there was a difference in post operative complications. Also, how significant will the financial impact be using the device. I look forward to the results of long- term studies. Hopefully this will be a viable option for our patients.
Dr. Jennifer D. Cox, FCCP, is an Assistant Professor of Pulmonary and Critical Care Medicine and clerkship director for the fourth-year medical student Critical Care Selective, Morsani College of Medicine, University of South Florida, in Tampa, Florida.
A device that preserves less-than-ideal donor lungs until they are cleared for transplantation has been approved, the Food and Drug Administration announced on Aug. 12.
The ex vivo perfusion device preserves donated lungs that initially do not meet all the criteria for a transplantable lung. The device does this by warming the donor lungs to "near normal body temperature," continuously flushing the lung with a sterile solution, and ventilating them, "which oxygenates the cells and makes it possible for the transplant team to examine the lung’s’ airways with a bronchoscope," according to the FDA statement.
The lungs can remain in the machine for up to 4 hours, providing time for the transplant team to evaluate the lungs to determine if they meet the criteria; donor lungs that meet the criteria are then transplanted into a patient.
The device, the XVIVO Perfusion System (XPS) with STEEN Solution, is manufactured by XVIVO Perfusion.
"With this approval, there may be more lungs available for transplant, which could allow more people with end-stage lung disease who have exhausted all other treatment options to be able to receive a lung transplant," Christy Foreman, director of the Office of Device Evaluation in the FDA’s Center for Devices and Radiological Health, Silver Spring, Md., said in the statement.
About 1 in 5 donor lungs meets the standard transplantation criteria. In the United States, 1,754 lung transplants were performed in 2012 and 1,616 potential recipients were on the lung transplant waiting list at the end of 2012, according to the FDA.
In two studies, outcomes for lung-transplant recipients were similar among those who received a donor lung preserved with the device and those who received donor lungs that were considered ideal and were preserved in cold storage.
"Both trials showed that recipients of the ideal and non-ideal lungs had similar survival rates up to 12 months after transplant and similar rates of organ rejection," the FDA statement said.
The manufacturer is required to conduct a long-term study of the effects of the device as a condition of approval.
emechcatie@frontlinemedcom.com
A device that preserves less-than-ideal donor lungs until they are cleared for transplantation has been approved, the Food and Drug Administration announced on Aug. 12.
The ex vivo perfusion device preserves donated lungs that initially do not meet all the criteria for a transplantable lung. The device does this by warming the donor lungs to "near normal body temperature," continuously flushing the lung with a sterile solution, and ventilating them, "which oxygenates the cells and makes it possible for the transplant team to examine the lung’s’ airways with a bronchoscope," according to the FDA statement.
The lungs can remain in the machine for up to 4 hours, providing time for the transplant team to evaluate the lungs to determine if they meet the criteria; donor lungs that meet the criteria are then transplanted into a patient.
The device, the XVIVO Perfusion System (XPS) with STEEN Solution, is manufactured by XVIVO Perfusion.
"With this approval, there may be more lungs available for transplant, which could allow more people with end-stage lung disease who have exhausted all other treatment options to be able to receive a lung transplant," Christy Foreman, director of the Office of Device Evaluation in the FDA’s Center for Devices and Radiological Health, Silver Spring, Md., said in the statement.
About 1 in 5 donor lungs meets the standard transplantation criteria. In the United States, 1,754 lung transplants were performed in 2012 and 1,616 potential recipients were on the lung transplant waiting list at the end of 2012, according to the FDA.
In two studies, outcomes for lung-transplant recipients were similar among those who received a donor lung preserved with the device and those who received donor lungs that were considered ideal and were preserved in cold storage.
"Both trials showed that recipients of the ideal and non-ideal lungs had similar survival rates up to 12 months after transplant and similar rates of organ rejection," the FDA statement said.
The manufacturer is required to conduct a long-term study of the effects of the device as a condition of approval.
emechcatie@frontlinemedcom.com
VIDEO: Rivaroxaban provides advantages for cardioversion in AF
BARCELONA – The first-ever prospective, randomized trial of a novel oral anticoagulant in patients with atrial fibrillation undergoing elective cardioversion showed oral rivaroxaban at 20 mg once daily to be an effective and safe alternative to standard-of-care warfarin. But the study, known as X-VeRT, also showed rivaroxaban offers something in addition: more expeditious cardioversion amenable to reliable scheduling.
Dr. Riccardo Cappato, who presented the X-VeRT results at the annual congress of the European Society of Cardiology, explains in this video interview.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
BARCELONA – The first-ever prospective, randomized trial of a novel oral anticoagulant in patients with atrial fibrillation undergoing elective cardioversion showed oral rivaroxaban at 20 mg once daily to be an effective and safe alternative to standard-of-care warfarin. But the study, known as X-VeRT, also showed rivaroxaban offers something in addition: more expeditious cardioversion amenable to reliable scheduling.
Dr. Riccardo Cappato, who presented the X-VeRT results at the annual congress of the European Society of Cardiology, explains in this video interview.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
BARCELONA – The first-ever prospective, randomized trial of a novel oral anticoagulant in patients with atrial fibrillation undergoing elective cardioversion showed oral rivaroxaban at 20 mg once daily to be an effective and safe alternative to standard-of-care warfarin. But the study, known as X-VeRT, also showed rivaroxaban offers something in addition: more expeditious cardioversion amenable to reliable scheduling.
Dr. Riccardo Cappato, who presented the X-VeRT results at the annual congress of the European Society of Cardiology, explains in this video interview.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT THE ESC CONGRESS 2014

Lung-MAP trial launched, matching patients to treatments based on tumor profiles
A phase II/III trial using genomic profiling to match patients with investigational treatments is currently recruiting patients with advanced squamous cell lung cancer.
The Lung-MAP (Lung Cancer Master Protocol) trial is a public-private collaboration among the National Cancer Institute (NCI), SWOG Cancer Research, Friends of Cancer Research, the Foundation for the National Institutes of Health, Foundation Medicine, and five pharmaceutical companies (Amgen, Genentech, Pfizer, AstraZeneca, and AstraZeneca’s global biologics R&D arm, MedImmune).
"Lung-MAP represents the first of several planned large, genomically driven treatment trials that will be conducted by NCI’s newly formed National Clinical Trials Network (NCTN)," Dr. Jeffrey S. Abrams, associate director of NCI’s Cancer Therapy Evaluation Program, said in a press release announcing the trial launch. "The restructuring and consolidation of NCI’s large trial treatment program, resulting in the formation of the NCTN, is quite timely, as it now can offer an ideal platform for bringing the benefits of more precise molecular diagnostics to cancer patients in communities large and small, " he said.
Lung-MAP is expected to enroll 10,000 patients and be completed in 2022. To be eligible, patients will have progressed after receiving exactly one front-line, platinum-containing metastatic chemotherapy regimen. Between 500 and 1,000 patients will be screened per year for more than 200 cancer-related gene alterations. Based on the tumor profiles, all patients will then be assigned to one of five initial treatment arms, testing four investigational targeted treatments and an anti-PD-L1 immunotherapy.
The trial will be conducted at more than 200 medical centers. As many as five to seven additional drugs may be evaluated over the next 5 years, the press release said. Information on enrolling patients can be found on the trial website.
A phase II/III trial using genomic profiling to match patients with investigational treatments is currently recruiting patients with advanced squamous cell lung cancer.
The Lung-MAP (Lung Cancer Master Protocol) trial is a public-private collaboration among the National Cancer Institute (NCI), SWOG Cancer Research, Friends of Cancer Research, the Foundation for the National Institutes of Health, Foundation Medicine, and five pharmaceutical companies (Amgen, Genentech, Pfizer, AstraZeneca, and AstraZeneca’s global biologics R&D arm, MedImmune).
"Lung-MAP represents the first of several planned large, genomically driven treatment trials that will be conducted by NCI’s newly formed National Clinical Trials Network (NCTN)," Dr. Jeffrey S. Abrams, associate director of NCI’s Cancer Therapy Evaluation Program, said in a press release announcing the trial launch. "The restructuring and consolidation of NCI’s large trial treatment program, resulting in the formation of the NCTN, is quite timely, as it now can offer an ideal platform for bringing the benefits of more precise molecular diagnostics to cancer patients in communities large and small, " he said.
Lung-MAP is expected to enroll 10,000 patients and be completed in 2022. To be eligible, patients will have progressed after receiving exactly one front-line, platinum-containing metastatic chemotherapy regimen. Between 500 and 1,000 patients will be screened per year for more than 200 cancer-related gene alterations. Based on the tumor profiles, all patients will then be assigned to one of five initial treatment arms, testing four investigational targeted treatments and an anti-PD-L1 immunotherapy.
The trial will be conducted at more than 200 medical centers. As many as five to seven additional drugs may be evaluated over the next 5 years, the press release said. Information on enrolling patients can be found on the trial website.
A phase II/III trial using genomic profiling to match patients with investigational treatments is currently recruiting patients with advanced squamous cell lung cancer.
The Lung-MAP (Lung Cancer Master Protocol) trial is a public-private collaboration among the National Cancer Institute (NCI), SWOG Cancer Research, Friends of Cancer Research, the Foundation for the National Institutes of Health, Foundation Medicine, and five pharmaceutical companies (Amgen, Genentech, Pfizer, AstraZeneca, and AstraZeneca’s global biologics R&D arm, MedImmune).
"Lung-MAP represents the first of several planned large, genomically driven treatment trials that will be conducted by NCI’s newly formed National Clinical Trials Network (NCTN)," Dr. Jeffrey S. Abrams, associate director of NCI’s Cancer Therapy Evaluation Program, said in a press release announcing the trial launch. "The restructuring and consolidation of NCI’s large trial treatment program, resulting in the formation of the NCTN, is quite timely, as it now can offer an ideal platform for bringing the benefits of more precise molecular diagnostics to cancer patients in communities large and small, " he said.
Lung-MAP is expected to enroll 10,000 patients and be completed in 2022. To be eligible, patients will have progressed after receiving exactly one front-line, platinum-containing metastatic chemotherapy regimen. Between 500 and 1,000 patients will be screened per year for more than 200 cancer-related gene alterations. Based on the tumor profiles, all patients will then be assigned to one of five initial treatment arms, testing four investigational targeted treatments and an anti-PD-L1 immunotherapy.
The trial will be conducted at more than 200 medical centers. As many as five to seven additional drugs may be evaluated over the next 5 years, the press release said. Information on enrolling patients can be found on the trial website.
The September issue of Thoracic Surgery News is now available online
Be sure to visit our interactive digital or PDF version of the September issue of Thoracic Surgery News. This month we are featuring stories ranging from the risks of aspirin resistance in pediatric cardiac surgery patients to the recent Centers for Medicare and Medicaid Services decision to cover transcatheter mitral valve repair (TMVR) procedures. Also, in our News from the AATS section there is a call for abstracts for AATS Week (comprising the 95th AATS Annual Meeting and the AATS Mitral Conclave), as well as several exciting cardiothoracic surgery fellowship opportunities.
To view our September PDF and interactive digital edition, click here.
Be sure to visit our interactive digital or PDF version of the September issue of Thoracic Surgery News. This month we are featuring stories ranging from the risks of aspirin resistance in pediatric cardiac surgery patients to the recent Centers for Medicare and Medicaid Services decision to cover transcatheter mitral valve repair (TMVR) procedures. Also, in our News from the AATS section there is a call for abstracts for AATS Week (comprising the 95th AATS Annual Meeting and the AATS Mitral Conclave), as well as several exciting cardiothoracic surgery fellowship opportunities.
To view our September PDF and interactive digital edition, click here.
Be sure to visit our interactive digital or PDF version of the September issue of Thoracic Surgery News. This month we are featuring stories ranging from the risks of aspirin resistance in pediatric cardiac surgery patients to the recent Centers for Medicare and Medicaid Services decision to cover transcatheter mitral valve repair (TMVR) procedures. Also, in our News from the AATS section there is a call for abstracts for AATS Week (comprising the 95th AATS Annual Meeting and the AATS Mitral Conclave), as well as several exciting cardiothoracic surgery fellowship opportunities.
To view our September PDF and interactive digital edition, click here.

VIDEO: Consider local anesthesia for transfemoral TAVR
BARCELONA – An analysis of FRANCE 2 registry showed that local anesthesia during the transfemoral aortic valve replacement procedure is a safe and effective option, compared with general anesthesia.
The study was performed based on data from 2010 and 2011, and at the annual congress of the European Society of Cardiology, Dr. Romain Chopard of the University Hospital of Besançon, Paris, and his coinvestigators reported that the practice is rather common – even routine in some centers – today.
But, performing TAVR under local anesthesia is not common practice in the United States, said Dr. Deepak L. Bhatt, executive director of interventional cardiovascular programs at Brigham and Women’s Hospital and professor of medicine at Harvard Medical School, both in Boston.
In a video, Dr. Bhatt shares his thoughts on performing local anesthesia and why it hasn’t taken hold in the United States. He also shares his advice with physicians who perform the TAVR procedure.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @naseemmiller
BARCELONA – An analysis of FRANCE 2 registry showed that local anesthesia during the transfemoral aortic valve replacement procedure is a safe and effective option, compared with general anesthesia.
The study was performed based on data from 2010 and 2011, and at the annual congress of the European Society of Cardiology, Dr. Romain Chopard of the University Hospital of Besançon, Paris, and his coinvestigators reported that the practice is rather common – even routine in some centers – today.
But, performing TAVR under local anesthesia is not common practice in the United States, said Dr. Deepak L. Bhatt, executive director of interventional cardiovascular programs at Brigham and Women’s Hospital and professor of medicine at Harvard Medical School, both in Boston.
In a video, Dr. Bhatt shares his thoughts on performing local anesthesia and why it hasn’t taken hold in the United States. He also shares his advice with physicians who perform the TAVR procedure.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @naseemmiller
BARCELONA – An analysis of FRANCE 2 registry showed that local anesthesia during the transfemoral aortic valve replacement procedure is a safe and effective option, compared with general anesthesia.
The study was performed based on data from 2010 and 2011, and at the annual congress of the European Society of Cardiology, Dr. Romain Chopard of the University Hospital of Besançon, Paris, and his coinvestigators reported that the practice is rather common – even routine in some centers – today.
But, performing TAVR under local anesthesia is not common practice in the United States, said Dr. Deepak L. Bhatt, executive director of interventional cardiovascular programs at Brigham and Women’s Hospital and professor of medicine at Harvard Medical School, both in Boston.
In a video, Dr. Bhatt shares his thoughts on performing local anesthesia and why it hasn’t taken hold in the United States. He also shares his advice with physicians who perform the TAVR procedure.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @naseemmiller
AT THE ESC CONGRESS 2014

VIDEO: Repositionable TAVR valve holds promise
BARCELONA – The transcatheter aortic valve replacement technology is changing rapidly, especially in Europe where the regulatory process is different and more types of valves are on the market.
One of the current focus areas in the development of TAVR devices is making retrievable and repositionable valves to improve the implantation process and patient outcomes. Late last year, Lotus Valve System reported favorable results for its valve in the REPRISE II study.
At the annual congress of the European Society of Cardiology, Dr. Stylianos A. Pyxaras presented another study showing that the repositionable Direct Flow Medical valve in elderly high-risk patients with severe aortic stenosis compared well in safety and efficacy with the results of the DISCOVER trial.
Dr. Deepak L. Bhatt, executive director of Interventional Cardiovascular Programs and professor of medicine at Harvard Medical School in Boston, shared his opinion about the findings and the future implications on practice. He was not involved in any of the mentioned studies.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Twitter: @naseemmiller
Lotus Valve System, REPRISE II, ESC
BARCELONA – The transcatheter aortic valve replacement technology is changing rapidly, especially in Europe where the regulatory process is different and more types of valves are on the market.
One of the current focus areas in the development of TAVR devices is making retrievable and repositionable valves to improve the implantation process and patient outcomes. Late last year, Lotus Valve System reported favorable results for its valve in the REPRISE II study.
At the annual congress of the European Society of Cardiology, Dr. Stylianos A. Pyxaras presented another study showing that the repositionable Direct Flow Medical valve in elderly high-risk patients with severe aortic stenosis compared well in safety and efficacy with the results of the DISCOVER trial.
Dr. Deepak L. Bhatt, executive director of Interventional Cardiovascular Programs and professor of medicine at Harvard Medical School in Boston, shared his opinion about the findings and the future implications on practice. He was not involved in any of the mentioned studies.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Twitter: @naseemmiller
BARCELONA – The transcatheter aortic valve replacement technology is changing rapidly, especially in Europe where the regulatory process is different and more types of valves are on the market.
One of the current focus areas in the development of TAVR devices is making retrievable and repositionable valves to improve the implantation process and patient outcomes. Late last year, Lotus Valve System reported favorable results for its valve in the REPRISE II study.
At the annual congress of the European Society of Cardiology, Dr. Stylianos A. Pyxaras presented another study showing that the repositionable Direct Flow Medical valve in elderly high-risk patients with severe aortic stenosis compared well in safety and efficacy with the results of the DISCOVER trial.
Dr. Deepak L. Bhatt, executive director of Interventional Cardiovascular Programs and professor of medicine at Harvard Medical School in Boston, shared his opinion about the findings and the future implications on practice. He was not involved in any of the mentioned studies.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Twitter: @naseemmiller
Lotus Valve System, REPRISE II, ESC
Lotus Valve System, REPRISE II, ESC
AT THE ESC CONGRESS 2014

Fractional flow reserve-guided PCI improves outcomes in stable heart disease
Fractional flow reserve-guided percutaneous coronary intervention plus best medical therapy improved outcomes when compared with medical therapy alone in patients with stable coronary artery disease in the Fractional Flow Reserve versus Angiography for Multivessel Evaluation 2 (FAME 2) trial.
A composite outcome of death from any cause, nonfatal myocardial infarction, or urgent revascularization within 2 years occurred in 8.1% of 447 patients who had at least one stenosis with a fractional flow reserve (FFR) of 0.80 and were randomized to undergo percutaneous coronary intervention (PCI) performed on the basis of the FFR, compared with 19.5% of 441 such patients who received medical therapy alone.
The difference was driven mainly by a 77% reduction in the need for urgent revascularization in interventional group as compared with the medical therapy group (4.0% vs. 16.3%, hazard ratio, 0.23). The overall rates of death and myocardial infarction did not differ significantly between the groups, Dr. Bernard D. Bruyne of the Cardiovascular Center Aalst, Belgium, and his colleagues reported at the annual congress of the European Society of Cardiology.
The findings of the open-label, randomized, multicenter trial were simultaneously published online Sept. 1 (N. Engl. J. Med. 2014 Sept. 1[doi:10.1056/NEJMoa1408758]).
The improved outcomes in the PCI group were the result of outcomes that occurred between 8 days and 2 years after randomization; in the first 7 days, more primary end-point events occurred in the PCI group than in the medical therapy group (2.2% vs. 0.9%, hazard ratio 2.49).
The rate of the primary endpoint was 9.0% among 332 additional patients who had an FFR of more than 0.80 in all stenoses, were enrolled into a registry, and received medical therapy alone, the researchers said.
The original FAME trial studied the procedure in patients who had already been selected for PCI. Compared with patients whose PCI was guided by angiography alone, those whose PCI was guided by FFR had significantly reduced rates of the composite end point of death, nonfatal myocardial infarction, and repeat revascularization at 1 year (N. Engl. J. Med. 2009;360:213-24).
FAME 2 evaluated use of FFR for improving the benefits of initial stenting as an alternative to noninvasive medical therapy. The trial was halted after a median of 7 months’ follow-up, when data safety and monitoring board found a highly statistically significant reduction in hospital readmission and urgent revascularization in the patients who received FFR-based stenting compared with those who received optimal medical therapy alone.
FAME 2 was supported by St. Jude Medical. Dr. De Bruyne reported that his institution receives grant support and consulting fees on his behalf from St. Jude Medical. Detailed disclosure information for several other authors is available with the full text of the article at www.NEJM.org.
The most impressive finding in the FAME 2 trial was a sustained reduced rate of urgent revascularization among patients undergoing early PCI.
Though the trigger in 60% of patients was based solely on clinical features and was potentially influenced by knowledge of the coronary anatomy, there was nonetheless a lower incidence of revascularization triggered by myocardial infarction or electrocardiographic changes in the PCI group than in the medical therapy group (3.4% versus 7.0%). This finding is consistent with recent evidence challenging the long-held view that acute coronary syndromes occur mainly at sites of noncritical stenoses.
It is plausible that stenting of vulnerable, hemodynamically significant lesions could prevent future coronary events.
The FAME 2 results show that early FFR-guided PCI in patients with stable coronary disease sustainably reduced the need for urgent revascularization. Given the continued improvement in the safety of stents and the procedures to implant them, PCI may eventually be shown to also have a favorable effect on other hard end points.
Dr. Jeffrey J. Rade is with the University of Massachusetts, Worcester. He made these comments in an accompanying editorial (N. Engl. J. Med. 2014 Sept. 1[doi:10.1056/NEJMe1410336]). He reported having no disclosures.
The most impressive finding in the FAME 2 trial was a sustained reduced rate of urgent revascularization among patients undergoing early PCI.
Though the trigger in 60% of patients was based solely on clinical features and was potentially influenced by knowledge of the coronary anatomy, there was nonetheless a lower incidence of revascularization triggered by myocardial infarction or electrocardiographic changes in the PCI group than in the medical therapy group (3.4% versus 7.0%). This finding is consistent with recent evidence challenging the long-held view that acute coronary syndromes occur mainly at sites of noncritical stenoses.
It is plausible that stenting of vulnerable, hemodynamically significant lesions could prevent future coronary events.
The FAME 2 results show that early FFR-guided PCI in patients with stable coronary disease sustainably reduced the need for urgent revascularization. Given the continued improvement in the safety of stents and the procedures to implant them, PCI may eventually be shown to also have a favorable effect on other hard end points.
Dr. Jeffrey J. Rade is with the University of Massachusetts, Worcester. He made these comments in an accompanying editorial (N. Engl. J. Med. 2014 Sept. 1[doi:10.1056/NEJMe1410336]). He reported having no disclosures.
The most impressive finding in the FAME 2 trial was a sustained reduced rate of urgent revascularization among patients undergoing early PCI.
Though the trigger in 60% of patients was based solely on clinical features and was potentially influenced by knowledge of the coronary anatomy, there was nonetheless a lower incidence of revascularization triggered by myocardial infarction or electrocardiographic changes in the PCI group than in the medical therapy group (3.4% versus 7.0%). This finding is consistent with recent evidence challenging the long-held view that acute coronary syndromes occur mainly at sites of noncritical stenoses.
It is plausible that stenting of vulnerable, hemodynamically significant lesions could prevent future coronary events.
The FAME 2 results show that early FFR-guided PCI in patients with stable coronary disease sustainably reduced the need for urgent revascularization. Given the continued improvement in the safety of stents and the procedures to implant them, PCI may eventually be shown to also have a favorable effect on other hard end points.
Dr. Jeffrey J. Rade is with the University of Massachusetts, Worcester. He made these comments in an accompanying editorial (N. Engl. J. Med. 2014 Sept. 1[doi:10.1056/NEJMe1410336]). He reported having no disclosures.
Fractional flow reserve-guided percutaneous coronary intervention plus best medical therapy improved outcomes when compared with medical therapy alone in patients with stable coronary artery disease in the Fractional Flow Reserve versus Angiography for Multivessel Evaluation 2 (FAME 2) trial.
A composite outcome of death from any cause, nonfatal myocardial infarction, or urgent revascularization within 2 years occurred in 8.1% of 447 patients who had at least one stenosis with a fractional flow reserve (FFR) of 0.80 and were randomized to undergo percutaneous coronary intervention (PCI) performed on the basis of the FFR, compared with 19.5% of 441 such patients who received medical therapy alone.
The difference was driven mainly by a 77% reduction in the need for urgent revascularization in interventional group as compared with the medical therapy group (4.0% vs. 16.3%, hazard ratio, 0.23). The overall rates of death and myocardial infarction did not differ significantly between the groups, Dr. Bernard D. Bruyne of the Cardiovascular Center Aalst, Belgium, and his colleagues reported at the annual congress of the European Society of Cardiology.
The findings of the open-label, randomized, multicenter trial were simultaneously published online Sept. 1 (N. Engl. J. Med. 2014 Sept. 1[doi:10.1056/NEJMoa1408758]).
The improved outcomes in the PCI group were the result of outcomes that occurred between 8 days and 2 years after randomization; in the first 7 days, more primary end-point events occurred in the PCI group than in the medical therapy group (2.2% vs. 0.9%, hazard ratio 2.49).
The rate of the primary endpoint was 9.0% among 332 additional patients who had an FFR of more than 0.80 in all stenoses, were enrolled into a registry, and received medical therapy alone, the researchers said.
The original FAME trial studied the procedure in patients who had already been selected for PCI. Compared with patients whose PCI was guided by angiography alone, those whose PCI was guided by FFR had significantly reduced rates of the composite end point of death, nonfatal myocardial infarction, and repeat revascularization at 1 year (N. Engl. J. Med. 2009;360:213-24).
FAME 2 evaluated use of FFR for improving the benefits of initial stenting as an alternative to noninvasive medical therapy. The trial was halted after a median of 7 months’ follow-up, when data safety and monitoring board found a highly statistically significant reduction in hospital readmission and urgent revascularization in the patients who received FFR-based stenting compared with those who received optimal medical therapy alone.
FAME 2 was supported by St. Jude Medical. Dr. De Bruyne reported that his institution receives grant support and consulting fees on his behalf from St. Jude Medical. Detailed disclosure information for several other authors is available with the full text of the article at www.NEJM.org.
Fractional flow reserve-guided percutaneous coronary intervention plus best medical therapy improved outcomes when compared with medical therapy alone in patients with stable coronary artery disease in the Fractional Flow Reserve versus Angiography for Multivessel Evaluation 2 (FAME 2) trial.
A composite outcome of death from any cause, nonfatal myocardial infarction, or urgent revascularization within 2 years occurred in 8.1% of 447 patients who had at least one stenosis with a fractional flow reserve (FFR) of 0.80 and were randomized to undergo percutaneous coronary intervention (PCI) performed on the basis of the FFR, compared with 19.5% of 441 such patients who received medical therapy alone.
The difference was driven mainly by a 77% reduction in the need for urgent revascularization in interventional group as compared with the medical therapy group (4.0% vs. 16.3%, hazard ratio, 0.23). The overall rates of death and myocardial infarction did not differ significantly between the groups, Dr. Bernard D. Bruyne of the Cardiovascular Center Aalst, Belgium, and his colleagues reported at the annual congress of the European Society of Cardiology.
The findings of the open-label, randomized, multicenter trial were simultaneously published online Sept. 1 (N. Engl. J. Med. 2014 Sept. 1[doi:10.1056/NEJMoa1408758]).
The improved outcomes in the PCI group were the result of outcomes that occurred between 8 days and 2 years after randomization; in the first 7 days, more primary end-point events occurred in the PCI group than in the medical therapy group (2.2% vs. 0.9%, hazard ratio 2.49).
The rate of the primary endpoint was 9.0% among 332 additional patients who had an FFR of more than 0.80 in all stenoses, were enrolled into a registry, and received medical therapy alone, the researchers said.
The original FAME trial studied the procedure in patients who had already been selected for PCI. Compared with patients whose PCI was guided by angiography alone, those whose PCI was guided by FFR had significantly reduced rates of the composite end point of death, nonfatal myocardial infarction, and repeat revascularization at 1 year (N. Engl. J. Med. 2009;360:213-24).
FAME 2 evaluated use of FFR for improving the benefits of initial stenting as an alternative to noninvasive medical therapy. The trial was halted after a median of 7 months’ follow-up, when data safety and monitoring board found a highly statistically significant reduction in hospital readmission and urgent revascularization in the patients who received FFR-based stenting compared with those who received optimal medical therapy alone.
FAME 2 was supported by St. Jude Medical. Dr. De Bruyne reported that his institution receives grant support and consulting fees on his behalf from St. Jude Medical. Detailed disclosure information for several other authors is available with the full text of the article at www.NEJM.org.
FROM THE ESC CONGRESS 2014
Key clinical point: Stenting cut the need for urgent revascularization in patients who had early PCI.
Major finding: The composite outcome occurred in 8.1% vs. 19.5% of intervention and medical therapy patients, respectively.
Data source: The open-label, randomized, multicenter FAME 2 study in 888 patients.
Disclosures: FAME 2 was supported by St. Jude Medical. Dr. De Bruyne reported that his institution receives grant support and consulting fees on his behalf from St. Jude Medical. Detailed disclosure information for several other authors is available with the full text of the article at www.NEJM.org.
Alirocumab sharply reduced major cardiovascular events
BARCELONA – When added to maximally tolerated statin therapy, the investigational PCSK9 inhibitor alirocumab resulted in a further 54% reduction in major cardiovascular events among high-cardiovascular-risk patients, based on a post-hoc analysis of a large randomized controlled Phase-3 trial.
The ODYSSEY LONG TERM trial is the largest and longest study of a PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitor to report results to date, with roughly 1,900 patient-years of double-blind exposure to alirocumab. And although the ongoing trial is primarily a safety study, it is also now the first PCSK9 trial to provide what everyone watching the development of this novel drug class has been eagerly awaiting: clinical outcomes data, albeit in this case from a post-hoc secondary analysis.
“This is the first trial with any of the PCSK9 inhibitors to suggest that there will be a further significant reduction in cardiovascular events when added on to maximized statin therapy,” Dr. Jennifer G. Robinson said in presenting interim results of ODYSSEY LONG TERM at the annual congress of the European Society of Cardiology.
“We’re on the right track in terms of trying to achieve further reduction in cardiovascular events through additional lipid lowering. But this is not the definitive evidence. We need the prospective outcomes trials to validate this data and also to establish the long-term safety of these drugs when added to the statins,” cautioned Dr. Robinson, professor of epidemiology and of medicine and director of the prevention intervention center at the University of Iowa, Iowa City.
Nonetheless, on the basis of the dramatic LDL-lowering and reassuring evidence of safety shown in ODYSSEY LONG TERM and the other double-blind phase III trials presented at the congress, Sanofi and Regeneron announced plans to file for U.S. and European Union marketing approval of alirocumab before the end of the year. The proposed indication will be for LDL lowering, which regulatory agencies have accepted as a surrogate endpoint for prevention of clinical events.
Meanwhile, the definitive ODYSSEY OUTCOMES trial is underway in 18,000 patients with acute coronary syndromes, with prospective evaluation of CV outcomes as its primary endpoint. The composite endpoint employed in the big OUTCOMES trial is identical to that used in the ODYSSEY LONG TERM post hoc analysis.
ODYSSEY LONG TERM includes 2,341 patients at high CV risk and an LDL level greater than 70 mg/dL despite maximally tolerated statin therapy. The patients fall into two categories: those with heterozygous familial hypercholesterolemia and others at very high risk because of known coronary heart disease. Participants were randomized 2:1 to 150 mg of alirocumab by self-administered subcutaneous injection at home every 2 weeks or placebo in addition to their statin.
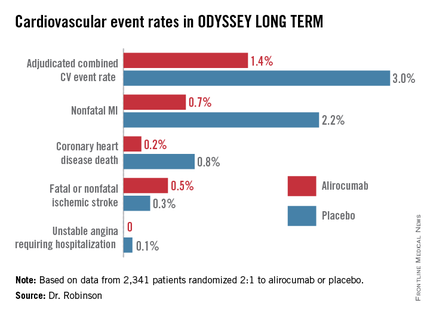
In the interim post-hoc efficacy analysis at 65 weeks, the combined rate of cardiac death, nonfatal MI, stroke, and unstable angina requiring hospitalization was 1.4% in the alirocumab arm compared to 3.0% in placebo-treated controls, for a highly significant 54% relative risk reduction (see graphic).
At 24 weeks, the alirocumab group showed a mean 62% reduction in LDL compared to placebo, a difference that remained constant at 52 weeks. The average LDL level at 52 weeks in the alirocumab group was 53 mg/dL, down from 123 mg/dL on active treatment at baseline; 79% of alirocumab-treated patients achieved an LDL below 70 mg/dL.
The incidence and types of adverse events in the alirocumab arm were essentially the same as with placebo, with no signal of problems in any domains, including neurocognitive function or allergic reactions.
ODYSSEY FH I and FH II
In a separate presentation, Dr. Michel Farnier reported on the alirocumab experience in 735 patients with heterozygous familial hypercholesterolemia in two phase III trials known as ODYSSEY FH I and FH II. At baseline, all were above their LDL goal despite maximally tolerated statin therapy, in two-thirds of cases with add-on ezetimibe. Participants were randomized 2:1 to add-on alirocumab at 75 mg every 2 weeks or to placebo.
The alirocumab-treated patients had 58% and 51% reductions in LDL, compared to actively treated controls at 24 weeks in the FH I and FH II trials. Of the alirocumab-treated patients, 72% and 81% achieved their prespecified LDL goal at 24 weeks, compared with 2% and 11% of controls.
“We have never before seen these kinds of percentages of patients with familial hypercholesterolemia reaching these LDL levels,” commented Dr. Farnier of Point Medical in Dijon, France.
ODYSSEY COMBO II
At the same hot-line clinical trials session, Dr. Christopher P. Cannon reported that alirocumab markedly outperformed ezetimibe as add-on therapy in the 720-patient, phase III, double-blind ODYSSEY COMBO II trial.
In this study, patients at very high CV risk who were unable to reach their desired goal of an LDL below 70 mg/dL despite maximum tolerated statin doses were randomized 2:1 to alirocumab at 75 mg once every 2 weeks or oral ezetimibe (Zetia) at its approved dose of 10 mg/day as an active comparator. Each participant also received placebo therapy.
By week 24, patients on alirocumab plus high-dose statin averaged a 51% reduction in LDL compared to baseline, versus a 21% reduction with ezetimibe plus statin. These effects were maintained at 1 year, with no evidence of tolerance.
Of patients on alirocumab, 77% achieved an LDL goal of less than 70 mg/dL at week 24, compared with 45% on ezetimibe. In addition, 60% of the alirocumab group had an LDL below 50 mg/dL, as did 15% on ezetimibe.
The study design called for patients in the alirocumab group who still had an LDL above 70 mg/dL at week 12 to be uptitrated from 75 mg to 150 mg every 2 weeks. But only 20% of patients needed to do so, according to Dr. Cannon, professor of medicine at Harvard University, Boston.
Will high-risk patient adhere long-term to treatment by self-injection? Dr. Cannon thinks so. He noted that 85% of patients in ODYSSEY COMBO II remained adherent to the biweekly self-injection protocol through 1 year.
“That has been a very pleasant surprise,” the cardiologist said. “The notion of injections for cholesterol management is foreign. It was a big surprise to us that patients really did it.”
‘Great news’ from alirocumab
The alirocumab results in the heterozygous familial hypercholesterolemia trials are “great news for patients with this disease,” discussant Dr. Robert M. Califf said. He noted that recent estimates put the prevalence of heterozygous familial hypercholesterolemia at roughly 1 in 250 persons in the general population, making the genetic disorder considerably more common than most physicians realize.
“These are people who have a terrible disease where a massive reduction in LDL, it seems to me, is clearly worthwhile,” commented Dr. Califf, professor of medicine and vice chancellor for clinical and translational research at Duke University in Durham, N.C.
As for Dr. Robinson’s ODYSSEY LONG TERM data showing a 54% reduction in major CV events, he said “It’s alluring. It looks great. It looks fantastic. And it sets up the large events trial. The only caution out of all of this from my perspective is that any study with less than 100 events is something that should be regarded with great interest but is not definitive.”
If approved, alirocumab could see widespread use, especially if the ODYSSEY OUTCOMES results prove positive. After all, heterozygous familial hypercholesterolemia is now recognized to be one of the most common of all inherited diseases. And Dr. Cannon said that in clinical trials in post-acute coronary syndrome patients placed on high-dose statins, it’s common for only about 40% to achieve an LDL level below 70 mg/dL. In clinical practice, the rate is probably even lower.*
“Down the line I think this level of LDL lowering is needed very badly. To get high-risk patients who have high cholesterol despite maximally dosed statins or statin-intolerance down to an LDL level of 50 mg/dL would be a pretty good thing. This will be applicable to millions of patients,” he predicted.*
Dr. Robinson said that at the American Heart Association meeting this November she and her ODYSSEY LONG TERM coinvestigators plan to present subgroup analyses looking at alirocumab efficacy and safety in patients with achieved LDL levels below 25 and even 15 mg/dL.
Dr. Robinson, Dr. Cannon, and Dr. Farnier reported receiving research grants and consultants’ fees from Sanofi and Regeneron as well as other pharmaceutical companies. Dr. Califf reported having no financial conflicts.
bjancin@frontlinemedcom.com
*CORRECTION: An earlier version of this story misattributed these statements.
BARCELONA – When added to maximally tolerated statin therapy, the investigational PCSK9 inhibitor alirocumab resulted in a further 54% reduction in major cardiovascular events among high-cardiovascular-risk patients, based on a post-hoc analysis of a large randomized controlled Phase-3 trial.
The ODYSSEY LONG TERM trial is the largest and longest study of a PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitor to report results to date, with roughly 1,900 patient-years of double-blind exposure to alirocumab. And although the ongoing trial is primarily a safety study, it is also now the first PCSK9 trial to provide what everyone watching the development of this novel drug class has been eagerly awaiting: clinical outcomes data, albeit in this case from a post-hoc secondary analysis.
“This is the first trial with any of the PCSK9 inhibitors to suggest that there will be a further significant reduction in cardiovascular events when added on to maximized statin therapy,” Dr. Jennifer G. Robinson said in presenting interim results of ODYSSEY LONG TERM at the annual congress of the European Society of Cardiology.
“We’re on the right track in terms of trying to achieve further reduction in cardiovascular events through additional lipid lowering. But this is not the definitive evidence. We need the prospective outcomes trials to validate this data and also to establish the long-term safety of these drugs when added to the statins,” cautioned Dr. Robinson, professor of epidemiology and of medicine and director of the prevention intervention center at the University of Iowa, Iowa City.
Nonetheless, on the basis of the dramatic LDL-lowering and reassuring evidence of safety shown in ODYSSEY LONG TERM and the other double-blind phase III trials presented at the congress, Sanofi and Regeneron announced plans to file for U.S. and European Union marketing approval of alirocumab before the end of the year. The proposed indication will be for LDL lowering, which regulatory agencies have accepted as a surrogate endpoint for prevention of clinical events.
Meanwhile, the definitive ODYSSEY OUTCOMES trial is underway in 18,000 patients with acute coronary syndromes, with prospective evaluation of CV outcomes as its primary endpoint. The composite endpoint employed in the big OUTCOMES trial is identical to that used in the ODYSSEY LONG TERM post hoc analysis.
ODYSSEY LONG TERM includes 2,341 patients at high CV risk and an LDL level greater than 70 mg/dL despite maximally tolerated statin therapy. The patients fall into two categories: those with heterozygous familial hypercholesterolemia and others at very high risk because of known coronary heart disease. Participants were randomized 2:1 to 150 mg of alirocumab by self-administered subcutaneous injection at home every 2 weeks or placebo in addition to their statin.
In the interim post-hoc efficacy analysis at 65 weeks, the combined rate of cardiac death, nonfatal MI, stroke, and unstable angina requiring hospitalization was 1.4% in the alirocumab arm compared to 3.0% in placebo-treated controls, for a highly significant 54% relative risk reduction (see graphic).
At 24 weeks, the alirocumab group showed a mean 62% reduction in LDL compared to placebo, a difference that remained constant at 52 weeks. The average LDL level at 52 weeks in the alirocumab group was 53 mg/dL, down from 123 mg/dL on active treatment at baseline; 79% of alirocumab-treated patients achieved an LDL below 70 mg/dL.
The incidence and types of adverse events in the alirocumab arm were essentially the same as with placebo, with no signal of problems in any domains, including neurocognitive function or allergic reactions.
ODYSSEY FH I and FH II
In a separate presentation, Dr. Michel Farnier reported on the alirocumab experience in 735 patients with heterozygous familial hypercholesterolemia in two phase III trials known as ODYSSEY FH I and FH II. At baseline, all were above their LDL goal despite maximally tolerated statin therapy, in two-thirds of cases with add-on ezetimibe. Participants were randomized 2:1 to add-on alirocumab at 75 mg every 2 weeks or to placebo.
The alirocumab-treated patients had 58% and 51% reductions in LDL, compared to actively treated controls at 24 weeks in the FH I and FH II trials. Of the alirocumab-treated patients, 72% and 81% achieved their prespecified LDL goal at 24 weeks, compared with 2% and 11% of controls.
“We have never before seen these kinds of percentages of patients with familial hypercholesterolemia reaching these LDL levels,” commented Dr. Farnier of Point Medical in Dijon, France.
ODYSSEY COMBO II
At the same hot-line clinical trials session, Dr. Christopher P. Cannon reported that alirocumab markedly outperformed ezetimibe as add-on therapy in the 720-patient, phase III, double-blind ODYSSEY COMBO II trial.
In this study, patients at very high CV risk who were unable to reach their desired goal of an LDL below 70 mg/dL despite maximum tolerated statin doses were randomized 2:1 to alirocumab at 75 mg once every 2 weeks or oral ezetimibe (Zetia) at its approved dose of 10 mg/day as an active comparator. Each participant also received placebo therapy.
By week 24, patients on alirocumab plus high-dose statin averaged a 51% reduction in LDL compared to baseline, versus a 21% reduction with ezetimibe plus statin. These effects were maintained at 1 year, with no evidence of tolerance.
Of patients on alirocumab, 77% achieved an LDL goal of less than 70 mg/dL at week 24, compared with 45% on ezetimibe. In addition, 60% of the alirocumab group had an LDL below 50 mg/dL, as did 15% on ezetimibe.
The study design called for patients in the alirocumab group who still had an LDL above 70 mg/dL at week 12 to be uptitrated from 75 mg to 150 mg every 2 weeks. But only 20% of patients needed to do so, according to Dr. Cannon, professor of medicine at Harvard University, Boston.
Will high-risk patient adhere long-term to treatment by self-injection? Dr. Cannon thinks so. He noted that 85% of patients in ODYSSEY COMBO II remained adherent to the biweekly self-injection protocol through 1 year.
“That has been a very pleasant surprise,” the cardiologist said. “The notion of injections for cholesterol management is foreign. It was a big surprise to us that patients really did it.”
‘Great news’ from alirocumab
The alirocumab results in the heterozygous familial hypercholesterolemia trials are “great news for patients with this disease,” discussant Dr. Robert M. Califf said. He noted that recent estimates put the prevalence of heterozygous familial hypercholesterolemia at roughly 1 in 250 persons in the general population, making the genetic disorder considerably more common than most physicians realize.
“These are people who have a terrible disease where a massive reduction in LDL, it seems to me, is clearly worthwhile,” commented Dr. Califf, professor of medicine and vice chancellor for clinical and translational research at Duke University in Durham, N.C.
As for Dr. Robinson’s ODYSSEY LONG TERM data showing a 54% reduction in major CV events, he said “It’s alluring. It looks great. It looks fantastic. And it sets up the large events trial. The only caution out of all of this from my perspective is that any study with less than 100 events is something that should be regarded with great interest but is not definitive.”
If approved, alirocumab could see widespread use, especially if the ODYSSEY OUTCOMES results prove positive. After all, heterozygous familial hypercholesterolemia is now recognized to be one of the most common of all inherited diseases. And Dr. Cannon said that in clinical trials in post-acute coronary syndrome patients placed on high-dose statins, it’s common for only about 40% to achieve an LDL level below 70 mg/dL. In clinical practice, the rate is probably even lower.*
“Down the line I think this level of LDL lowering is needed very badly. To get high-risk patients who have high cholesterol despite maximally dosed statins or statin-intolerance down to an LDL level of 50 mg/dL would be a pretty good thing. This will be applicable to millions of patients,” he predicted.*
Dr. Robinson said that at the American Heart Association meeting this November she and her ODYSSEY LONG TERM coinvestigators plan to present subgroup analyses looking at alirocumab efficacy and safety in patients with achieved LDL levels below 25 and even 15 mg/dL.
Dr. Robinson, Dr. Cannon, and Dr. Farnier reported receiving research grants and consultants’ fees from Sanofi and Regeneron as well as other pharmaceutical companies. Dr. Califf reported having no financial conflicts.
bjancin@frontlinemedcom.com
*CORRECTION: An earlier version of this story misattributed these statements.
BARCELONA – When added to maximally tolerated statin therapy, the investigational PCSK9 inhibitor alirocumab resulted in a further 54% reduction in major cardiovascular events among high-cardiovascular-risk patients, based on a post-hoc analysis of a large randomized controlled Phase-3 trial.
The ODYSSEY LONG TERM trial is the largest and longest study of a PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitor to report results to date, with roughly 1,900 patient-years of double-blind exposure to alirocumab. And although the ongoing trial is primarily a safety study, it is also now the first PCSK9 trial to provide what everyone watching the development of this novel drug class has been eagerly awaiting: clinical outcomes data, albeit in this case from a post-hoc secondary analysis.
“This is the first trial with any of the PCSK9 inhibitors to suggest that there will be a further significant reduction in cardiovascular events when added on to maximized statin therapy,” Dr. Jennifer G. Robinson said in presenting interim results of ODYSSEY LONG TERM at the annual congress of the European Society of Cardiology.
“We’re on the right track in terms of trying to achieve further reduction in cardiovascular events through additional lipid lowering. But this is not the definitive evidence. We need the prospective outcomes trials to validate this data and also to establish the long-term safety of these drugs when added to the statins,” cautioned Dr. Robinson, professor of epidemiology and of medicine and director of the prevention intervention center at the University of Iowa, Iowa City.
Nonetheless, on the basis of the dramatic LDL-lowering and reassuring evidence of safety shown in ODYSSEY LONG TERM and the other double-blind phase III trials presented at the congress, Sanofi and Regeneron announced plans to file for U.S. and European Union marketing approval of alirocumab before the end of the year. The proposed indication will be for LDL lowering, which regulatory agencies have accepted as a surrogate endpoint for prevention of clinical events.
Meanwhile, the definitive ODYSSEY OUTCOMES trial is underway in 18,000 patients with acute coronary syndromes, with prospective evaluation of CV outcomes as its primary endpoint. The composite endpoint employed in the big OUTCOMES trial is identical to that used in the ODYSSEY LONG TERM post hoc analysis.
ODYSSEY LONG TERM includes 2,341 patients at high CV risk and an LDL level greater than 70 mg/dL despite maximally tolerated statin therapy. The patients fall into two categories: those with heterozygous familial hypercholesterolemia and others at very high risk because of known coronary heart disease. Participants were randomized 2:1 to 150 mg of alirocumab by self-administered subcutaneous injection at home every 2 weeks or placebo in addition to their statin.
In the interim post-hoc efficacy analysis at 65 weeks, the combined rate of cardiac death, nonfatal MI, stroke, and unstable angina requiring hospitalization was 1.4% in the alirocumab arm compared to 3.0% in placebo-treated controls, for a highly significant 54% relative risk reduction (see graphic).
At 24 weeks, the alirocumab group showed a mean 62% reduction in LDL compared to placebo, a difference that remained constant at 52 weeks. The average LDL level at 52 weeks in the alirocumab group was 53 mg/dL, down from 123 mg/dL on active treatment at baseline; 79% of alirocumab-treated patients achieved an LDL below 70 mg/dL.
The incidence and types of adverse events in the alirocumab arm were essentially the same as with placebo, with no signal of problems in any domains, including neurocognitive function or allergic reactions.
ODYSSEY FH I and FH II
In a separate presentation, Dr. Michel Farnier reported on the alirocumab experience in 735 patients with heterozygous familial hypercholesterolemia in two phase III trials known as ODYSSEY FH I and FH II. At baseline, all were above their LDL goal despite maximally tolerated statin therapy, in two-thirds of cases with add-on ezetimibe. Participants were randomized 2:1 to add-on alirocumab at 75 mg every 2 weeks or to placebo.
The alirocumab-treated patients had 58% and 51% reductions in LDL, compared to actively treated controls at 24 weeks in the FH I and FH II trials. Of the alirocumab-treated patients, 72% and 81% achieved their prespecified LDL goal at 24 weeks, compared with 2% and 11% of controls.
“We have never before seen these kinds of percentages of patients with familial hypercholesterolemia reaching these LDL levels,” commented Dr. Farnier of Point Medical in Dijon, France.
ODYSSEY COMBO II
At the same hot-line clinical trials session, Dr. Christopher P. Cannon reported that alirocumab markedly outperformed ezetimibe as add-on therapy in the 720-patient, phase III, double-blind ODYSSEY COMBO II trial.
In this study, patients at very high CV risk who were unable to reach their desired goal of an LDL below 70 mg/dL despite maximum tolerated statin doses were randomized 2:1 to alirocumab at 75 mg once every 2 weeks or oral ezetimibe (Zetia) at its approved dose of 10 mg/day as an active comparator. Each participant also received placebo therapy.
By week 24, patients on alirocumab plus high-dose statin averaged a 51% reduction in LDL compared to baseline, versus a 21% reduction with ezetimibe plus statin. These effects were maintained at 1 year, with no evidence of tolerance.
Of patients on alirocumab, 77% achieved an LDL goal of less than 70 mg/dL at week 24, compared with 45% on ezetimibe. In addition, 60% of the alirocumab group had an LDL below 50 mg/dL, as did 15% on ezetimibe.
The study design called for patients in the alirocumab group who still had an LDL above 70 mg/dL at week 12 to be uptitrated from 75 mg to 150 mg every 2 weeks. But only 20% of patients needed to do so, according to Dr. Cannon, professor of medicine at Harvard University, Boston.
Will high-risk patient adhere long-term to treatment by self-injection? Dr. Cannon thinks so. He noted that 85% of patients in ODYSSEY COMBO II remained adherent to the biweekly self-injection protocol through 1 year.
“That has been a very pleasant surprise,” the cardiologist said. “The notion of injections for cholesterol management is foreign. It was a big surprise to us that patients really did it.”
‘Great news’ from alirocumab
The alirocumab results in the heterozygous familial hypercholesterolemia trials are “great news for patients with this disease,” discussant Dr. Robert M. Califf said. He noted that recent estimates put the prevalence of heterozygous familial hypercholesterolemia at roughly 1 in 250 persons in the general population, making the genetic disorder considerably more common than most physicians realize.
“These are people who have a terrible disease where a massive reduction in LDL, it seems to me, is clearly worthwhile,” commented Dr. Califf, professor of medicine and vice chancellor for clinical and translational research at Duke University in Durham, N.C.
As for Dr. Robinson’s ODYSSEY LONG TERM data showing a 54% reduction in major CV events, he said “It’s alluring. It looks great. It looks fantastic. And it sets up the large events trial. The only caution out of all of this from my perspective is that any study with less than 100 events is something that should be regarded with great interest but is not definitive.”
If approved, alirocumab could see widespread use, especially if the ODYSSEY OUTCOMES results prove positive. After all, heterozygous familial hypercholesterolemia is now recognized to be one of the most common of all inherited diseases. And Dr. Cannon said that in clinical trials in post-acute coronary syndrome patients placed on high-dose statins, it’s common for only about 40% to achieve an LDL level below 70 mg/dL. In clinical practice, the rate is probably even lower.*
“Down the line I think this level of LDL lowering is needed very badly. To get high-risk patients who have high cholesterol despite maximally dosed statins or statin-intolerance down to an LDL level of 50 mg/dL would be a pretty good thing. This will be applicable to millions of patients,” he predicted.*
Dr. Robinson said that at the American Heart Association meeting this November she and her ODYSSEY LONG TERM coinvestigators plan to present subgroup analyses looking at alirocumab efficacy and safety in patients with achieved LDL levels below 25 and even 15 mg/dL.
Dr. Robinson, Dr. Cannon, and Dr. Farnier reported receiving research grants and consultants’ fees from Sanofi and Regeneron as well as other pharmaceutical companies. Dr. Califf reported having no financial conflicts.
bjancin@frontlinemedcom.com
*CORRECTION: An earlier version of this story misattributed these statements.
AT THE ESC CONGRESS 2014
Key clinical point: The lower-is-better approach to LDL reduction gets a big boost from a study that showed adding alirocumab to maximally tolerated statin therapy was associated with 54% fewer major adverse cardiovascular events compared to statin plus placebo in high-cardiovascular-risk patients.
Major finding: The incidence of the composite cardiovascular event endpoint at 65 weeks was 1.4% in those on alirocumab plus maximally tolerated statin therapy and 3.0% in those on placebo plus statin.
Data source: This was an interim post hoc analysis from the Phase-3, double-blind, randomized, prospective ODYSSEY LONG TERM trial of more than 2,300 randomized patients.
Disclosures: The ODYSSEY clinical trials program is sponsored by Sanofi and Regeneron. The study presenters reported receiving research grants and consultants’ fees from those and other companies.

COPPS-2 curtails colchicine enthusiasm in cardiac surgery
Patients undergoing cardiac surgery who took colchicine had significantly less postpericardiotomy syndrome than did those on placebo, but this protective effect did not extend to postoperative atrial fibrillation and pericardial or pleural effusions in the double-blind COPPS-2 trial.
The failure of colchicine to prevent postoperative atrial fibrillation (AF) was probably due to more frequent adverse events (36 vs. 21 with placebo), especially gastrointestinal intolerance (26 vs. 12), and drug discontinuation (39 vs. 32), since a prespecified on-treatment analysis showed a significant reduction in AF in patients tolerating the drug, Dr. Massimo Imazio reported at the annual congress of the European Society of Cardiology.

"The high rate of adverse effects is a reason for concern and suggests that colchicine should be considered only in well-selected patients," Dr. Imazio and his associates wrote in an article on COPPS-2 simultaneously published online (JAMA 2014 [doi:10.1001/jama.2014.11026]).
Colchicine has been a promising strategy for postpericardiotomy syndrome prevention, besting methylprednisolone and aspirin in a large meta-analysis (Am. J. Cardiol. 2011;108:575-9).
In the largest trial, COPPS (Colchicine for the Prevention of the Postpericardiotomy Syndrome), Dr. Imazio reported that colchicine significantly reduced the incidence of postpericardiotomy syndrome (8.9% vs. 21.1%), postoperative pericardial effusions (relative risk reduction, 43.9%), and pleural effusions (RRR, 52.3%) at 12 months, compared with placebo (Am. Heart J. 2011;162:527-32 and Eur. Heart J. 2010;31:2749-54). Colchicine was given for 1 month, beginning on the third postoperative day with a 1-mg twice-daily loading dose.
In COPPS-2, the 360 consecutive candidates for cardiac surgery also were given colchicine or placebo for 1 month, but treatment was started 48-72 hours before surgery to pretreat patients and improve colchicine’s ability to prevent postoperative systemic inflammation and its complications.
Colchicine also was administered using weight-based dosing (0.5 mg twice daily in patients weighing at least 70 kg or 0.5 mg once daily in those under 70 kg), and they avoided the loading dose in an effort to improve adherence.
"However, we observed a 2-fold increase of adverse effects and study drug discontinuations compared with those reported in the COPPS trial, likely due to significant vulnerability of patients in the perioperative phase, when the use of antibiotics and proton pump inhibitors is common and also increases the risk of gastrointestinal effects (e.g., diarrhea)," explained Dr. Imazio of Maria Vittoria Hospital and the University of Torino (Italy).
Still, colchicine provided significant protection in the COPPS-2 primary outcome of postpericardiotomy syndrome, compared with placebo (19.4% vs. 29.4%; 95% confidence interval, 1.1%-18.7%). The number needed to treat was 10.
The outcome did not differ significantly among predetermined subgroups based on age, sex, and presence or absence of pericardial effusion, although colchicine was especially efficacious in the setting of systemic inflammation with elevated C-reactive protein, the authors noted.
The intention-to-treat analysis revealed no significant differences between the colchicine and placebo groups for postoperative AF (33.9% vs. 41.7%; 95% CI, –2.2%-17.6%) or postoperative pericardial/pleural effusion (57.2% vs. 58.9%; 95% CI, –8.5%-11.7%).
The prespecified on-treatment analysis, however, showed a 14.2% absolute difference in postoperative AF, favoring colchicine over placebo (27% vs. 41.2%; 95% CI, 3.3%-24.7%).
"While the efficacy of colchicine for postpericardiotomy syndrome prevention is confirmed, the extent of efficacy for postoperative AF needs to be further investigated in future trials," Dr. Imazio stated.
Ongoing studies also will better clarify the potential of colchicine using lower doses that may be better tolerated.
The 360 patients were evenly randomized from 11 centers in Italy between March 2012 and March 2014. Their mean age was 67.5 years, 69% were men, and 36% had planned valvular surgery. Key exclusion criteria were absence of sinus rhythm at enrollment, urgent cardiac surgery, cardiac transplantation, and contraindications to colchicine.
COPPS-2 was supported by the Italian National Health Service and FARGIM. Acarpia provided the study drug. Dr. Imazio reported no conflicts of interest. A coauthor reported consultancy for Servier, serving on an advisory board for Boehringer Ingelheim, and lecturer fees from Abbott, AstraZeneca, Merck, Serono, Richter Gedeon, and Teva.
Patients undergoing cardiac surgery who took colchicine had significantly less postpericardiotomy syndrome than did those on placebo, but this protective effect did not extend to postoperative atrial fibrillation and pericardial or pleural effusions in the double-blind COPPS-2 trial.
The failure of colchicine to prevent postoperative atrial fibrillation (AF) was probably due to more frequent adverse events (36 vs. 21 with placebo), especially gastrointestinal intolerance (26 vs. 12), and drug discontinuation (39 vs. 32), since a prespecified on-treatment analysis showed a significant reduction in AF in patients tolerating the drug, Dr. Massimo Imazio reported at the annual congress of the European Society of Cardiology.
"The high rate of adverse effects is a reason for concern and suggests that colchicine should be considered only in well-selected patients," Dr. Imazio and his associates wrote in an article on COPPS-2 simultaneously published online (JAMA 2014 [doi:10.1001/jama.2014.11026]).
Colchicine has been a promising strategy for postpericardiotomy syndrome prevention, besting methylprednisolone and aspirin in a large meta-analysis (Am. J. Cardiol. 2011;108:575-9).
In the largest trial, COPPS (Colchicine for the Prevention of the Postpericardiotomy Syndrome), Dr. Imazio reported that colchicine significantly reduced the incidence of postpericardiotomy syndrome (8.9% vs. 21.1%), postoperative pericardial effusions (relative risk reduction, 43.9%), and pleural effusions (RRR, 52.3%) at 12 months, compared with placebo (Am. Heart J. 2011;162:527-32 and Eur. Heart J. 2010;31:2749-54). Colchicine was given for 1 month, beginning on the third postoperative day with a 1-mg twice-daily loading dose.
In COPPS-2, the 360 consecutive candidates for cardiac surgery also were given colchicine or placebo for 1 month, but treatment was started 48-72 hours before surgery to pretreat patients and improve colchicine’s ability to prevent postoperative systemic inflammation and its complications.
Colchicine also was administered using weight-based dosing (0.5 mg twice daily in patients weighing at least 70 kg or 0.5 mg once daily in those under 70 kg), and they avoided the loading dose in an effort to improve adherence.
"However, we observed a 2-fold increase of adverse effects and study drug discontinuations compared with those reported in the COPPS trial, likely due to significant vulnerability of patients in the perioperative phase, when the use of antibiotics and proton pump inhibitors is common and also increases the risk of gastrointestinal effects (e.g., diarrhea)," explained Dr. Imazio of Maria Vittoria Hospital and the University of Torino (Italy).
Still, colchicine provided significant protection in the COPPS-2 primary outcome of postpericardiotomy syndrome, compared with placebo (19.4% vs. 29.4%; 95% confidence interval, 1.1%-18.7%). The number needed to treat was 10.
The outcome did not differ significantly among predetermined subgroups based on age, sex, and presence or absence of pericardial effusion, although colchicine was especially efficacious in the setting of systemic inflammation with elevated C-reactive protein, the authors noted.
The intention-to-treat analysis revealed no significant differences between the colchicine and placebo groups for postoperative AF (33.9% vs. 41.7%; 95% CI, –2.2%-17.6%) or postoperative pericardial/pleural effusion (57.2% vs. 58.9%; 95% CI, –8.5%-11.7%).
The prespecified on-treatment analysis, however, showed a 14.2% absolute difference in postoperative AF, favoring colchicine over placebo (27% vs. 41.2%; 95% CI, 3.3%-24.7%).
"While the efficacy of colchicine for postpericardiotomy syndrome prevention is confirmed, the extent of efficacy for postoperative AF needs to be further investigated in future trials," Dr. Imazio stated.
Ongoing studies also will better clarify the potential of colchicine using lower doses that may be better tolerated.
The 360 patients were evenly randomized from 11 centers in Italy between March 2012 and March 2014. Their mean age was 67.5 years, 69% were men, and 36% had planned valvular surgery. Key exclusion criteria were absence of sinus rhythm at enrollment, urgent cardiac surgery, cardiac transplantation, and contraindications to colchicine.
COPPS-2 was supported by the Italian National Health Service and FARGIM. Acarpia provided the study drug. Dr. Imazio reported no conflicts of interest. A coauthor reported consultancy for Servier, serving on an advisory board for Boehringer Ingelheim, and lecturer fees from Abbott, AstraZeneca, Merck, Serono, Richter Gedeon, and Teva.
Patients undergoing cardiac surgery who took colchicine had significantly less postpericardiotomy syndrome than did those on placebo, but this protective effect did not extend to postoperative atrial fibrillation and pericardial or pleural effusions in the double-blind COPPS-2 trial.
The failure of colchicine to prevent postoperative atrial fibrillation (AF) was probably due to more frequent adverse events (36 vs. 21 with placebo), especially gastrointestinal intolerance (26 vs. 12), and drug discontinuation (39 vs. 32), since a prespecified on-treatment analysis showed a significant reduction in AF in patients tolerating the drug, Dr. Massimo Imazio reported at the annual congress of the European Society of Cardiology.
"The high rate of adverse effects is a reason for concern and suggests that colchicine should be considered only in well-selected patients," Dr. Imazio and his associates wrote in an article on COPPS-2 simultaneously published online (JAMA 2014 [doi:10.1001/jama.2014.11026]).
Colchicine has been a promising strategy for postpericardiotomy syndrome prevention, besting methylprednisolone and aspirin in a large meta-analysis (Am. J. Cardiol. 2011;108:575-9).
In the largest trial, COPPS (Colchicine for the Prevention of the Postpericardiotomy Syndrome), Dr. Imazio reported that colchicine significantly reduced the incidence of postpericardiotomy syndrome (8.9% vs. 21.1%), postoperative pericardial effusions (relative risk reduction, 43.9%), and pleural effusions (RRR, 52.3%) at 12 months, compared with placebo (Am. Heart J. 2011;162:527-32 and Eur. Heart J. 2010;31:2749-54). Colchicine was given for 1 month, beginning on the third postoperative day with a 1-mg twice-daily loading dose.
In COPPS-2, the 360 consecutive candidates for cardiac surgery also were given colchicine or placebo for 1 month, but treatment was started 48-72 hours before surgery to pretreat patients and improve colchicine’s ability to prevent postoperative systemic inflammation and its complications.
Colchicine also was administered using weight-based dosing (0.5 mg twice daily in patients weighing at least 70 kg or 0.5 mg once daily in those under 70 kg), and they avoided the loading dose in an effort to improve adherence.
"However, we observed a 2-fold increase of adverse effects and study drug discontinuations compared with those reported in the COPPS trial, likely due to significant vulnerability of patients in the perioperative phase, when the use of antibiotics and proton pump inhibitors is common and also increases the risk of gastrointestinal effects (e.g., diarrhea)," explained Dr. Imazio of Maria Vittoria Hospital and the University of Torino (Italy).
Still, colchicine provided significant protection in the COPPS-2 primary outcome of postpericardiotomy syndrome, compared with placebo (19.4% vs. 29.4%; 95% confidence interval, 1.1%-18.7%). The number needed to treat was 10.
The outcome did not differ significantly among predetermined subgroups based on age, sex, and presence or absence of pericardial effusion, although colchicine was especially efficacious in the setting of systemic inflammation with elevated C-reactive protein, the authors noted.
The intention-to-treat analysis revealed no significant differences between the colchicine and placebo groups for postoperative AF (33.9% vs. 41.7%; 95% CI, –2.2%-17.6%) or postoperative pericardial/pleural effusion (57.2% vs. 58.9%; 95% CI, –8.5%-11.7%).
The prespecified on-treatment analysis, however, showed a 14.2% absolute difference in postoperative AF, favoring colchicine over placebo (27% vs. 41.2%; 95% CI, 3.3%-24.7%).
"While the efficacy of colchicine for postpericardiotomy syndrome prevention is confirmed, the extent of efficacy for postoperative AF needs to be further investigated in future trials," Dr. Imazio stated.
Ongoing studies also will better clarify the potential of colchicine using lower doses that may be better tolerated.
The 360 patients were evenly randomized from 11 centers in Italy between March 2012 and March 2014. Their mean age was 67.5 years, 69% were men, and 36% had planned valvular surgery. Key exclusion criteria were absence of sinus rhythm at enrollment, urgent cardiac surgery, cardiac transplantation, and contraindications to colchicine.
COPPS-2 was supported by the Italian National Health Service and FARGIM. Acarpia provided the study drug. Dr. Imazio reported no conflicts of interest. A coauthor reported consultancy for Servier, serving on an advisory board for Boehringer Ingelheim, and lecturer fees from Abbott, AstraZeneca, Merck, Serono, Richter Gedeon, and Teva.
FROM THE ESC CONGRESS 2014
Key clinical point: Perioperative use of colchicine should be considered only in well-selected patients.
Major finding: Perioperative colchicine use cut the incidence of postpericardiotomy syndrome, but not postoperative atrial fibrillation or pericardial/pleural effusion.
Data source: Double-blind, randomized clinical trial in 360 consecutive candidates for heart surgery.
Disclosures: COPPS-2 was supported by the Italian National Health Service and FARGIM. Acarpia provided the study drug. Dr. Imazio reported no conflicts of interest. A coauthor reported consultancy for Servier, serving on an advisory board for Boehringer Ingelheim, and lecturer fees from Abbott, AstraZeneca, Merck, Serono, Richter Gedeon, and Teva.

VEGF-A value may stratify risk in pediatric heart transplant recipients
SAN FRANCISCO – Monitoring plasma vascular endothelial growth factor A (VEGF-A) may help identify pediatric heart transplant patients who are at increased risk for poor outcomes, according to a study reported at the 2014 World Transplant Congress.
"Cardiac allograft vasculopathy [CAV] remains the leading cause of chronic allograft failure after heart transplantation. ... Therefore, it’s important for us to be able to anticipate the development of CAV and open up a therapeutic window," said Dr. Kevin P. Daly of Harvard Medical School and Boston Children’s Hospital.

"Our pilot data suggest that plasma VEGF-A levels below 90 pg/mL identify a low-risk patient population in whom a decreased frequency of coronary angiography can be considered. Future studies are needed to determine if using plasma VEGF-A levels to modify CAV screening frequency results in equivalent patient outcomes, with decreased resource utilization and improved quality of life," Dr. Daly commented at the congress, which was sponsored by the American Society of Transplant Surgeons.
As the vascular endothelium is the primary target of the immune response in CAV, the researchers hypothesized that VEGF-A likely contributes to an inflammatory cycle that leads to vascular damage and occlusion in the graft.
Participants in the single-center prospective cohort study were 44 consecutive children aged 2 years or older who were at least 18 months (median, 6 years) out from heart transplantation. They were scheduled for routine annual screening coronary angiography during 2009, and had no or mild CAV.
Moderate or severe CAV developed in 32% of patients who had VEGF-A values above the median value at baseline (90 pg/mL), compared with 5% of patients who had VEGF-A values below the median level (P = .02). Patients who developed this vasculopathy were more likely to die (38% vs. 0%), undergo retransplantation (38% vs. 0%), experience a myocardial infarction (12% vs. 0%), and be listed for retransplantation (12% vs. 0%).
"While this is a biomarker and we have shown it is associated with CAV, we have not shown that it is causal," Dr. Daly cautioned. Any treatment directed against VEGF would have to be conducted in the context of a clinical trial to assess its impact.
A subset of patients becomes nonadherent to therapy; a subset that is highly sensitized before transplant may have donor-specific antibody, Dr. Daly said. So "we don’t think we fully understand the inciting event, ... [but] VEGF-A has been shown before to be elevated in antibody-mediated rejection, so it’s not surprising to see this association."
"We didn’t have these data available clinically because it was all a research study, so we didn’t intervene on any of the patients in this cohort. But we have started to think about whether or not we could use VEGF-A levels at least in our ... patients who might not have arterial access, and it might be difficult to survey them for CAV. I think in order to really understand the appropriate way to use it, we would need a larger study," he remarked.
Dr. Daly disclosed that he had no conflicts of interest relevant to the study.
This was a very nice preliminary study but extremely limited in scope, as it has few patients and limited mechanistic studies. Most importantly, there was no validation cohort as is required to have confidence that a biomarker is predictive. A lot more work will be necessary before significance can be assigned to the use of VEGF-A as a potential biomarker.
Dr. Daniel R. Salomon is a professor and program medical director at the Scripps Center for Organ Transplantation, Scripps Research Institute, La Jolla, Calif. He was the cochair at the session where the research was presented, and made his remarks in an interview. He had no relevant conflicts of interest.
This was a very nice preliminary study but extremely limited in scope, as it has few patients and limited mechanistic studies. Most importantly, there was no validation cohort as is required to have confidence that a biomarker is predictive. A lot more work will be necessary before significance can be assigned to the use of VEGF-A as a potential biomarker.
Dr. Daniel R. Salomon is a professor and program medical director at the Scripps Center for Organ Transplantation, Scripps Research Institute, La Jolla, Calif. He was the cochair at the session where the research was presented, and made his remarks in an interview. He had no relevant conflicts of interest.
This was a very nice preliminary study but extremely limited in scope, as it has few patients and limited mechanistic studies. Most importantly, there was no validation cohort as is required to have confidence that a biomarker is predictive. A lot more work will be necessary before significance can be assigned to the use of VEGF-A as a potential biomarker.
Dr. Daniel R. Salomon is a professor and program medical director at the Scripps Center for Organ Transplantation, Scripps Research Institute, La Jolla, Calif. He was the cochair at the session where the research was presented, and made his remarks in an interview. He had no relevant conflicts of interest.
SAN FRANCISCO – Monitoring plasma vascular endothelial growth factor A (VEGF-A) may help identify pediatric heart transplant patients who are at increased risk for poor outcomes, according to a study reported at the 2014 World Transplant Congress.
"Cardiac allograft vasculopathy [CAV] remains the leading cause of chronic allograft failure after heart transplantation. ... Therefore, it’s important for us to be able to anticipate the development of CAV and open up a therapeutic window," said Dr. Kevin P. Daly of Harvard Medical School and Boston Children’s Hospital.
"Our pilot data suggest that plasma VEGF-A levels below 90 pg/mL identify a low-risk patient population in whom a decreased frequency of coronary angiography can be considered. Future studies are needed to determine if using plasma VEGF-A levels to modify CAV screening frequency results in equivalent patient outcomes, with decreased resource utilization and improved quality of life," Dr. Daly commented at the congress, which was sponsored by the American Society of Transplant Surgeons.
As the vascular endothelium is the primary target of the immune response in CAV, the researchers hypothesized that VEGF-A likely contributes to an inflammatory cycle that leads to vascular damage and occlusion in the graft.
Participants in the single-center prospective cohort study were 44 consecutive children aged 2 years or older who were at least 18 months (median, 6 years) out from heart transplantation. They were scheduled for routine annual screening coronary angiography during 2009, and had no or mild CAV.
Moderate or severe CAV developed in 32% of patients who had VEGF-A values above the median value at baseline (90 pg/mL), compared with 5% of patients who had VEGF-A values below the median level (P = .02). Patients who developed this vasculopathy were more likely to die (38% vs. 0%), undergo retransplantation (38% vs. 0%), experience a myocardial infarction (12% vs. 0%), and be listed for retransplantation (12% vs. 0%).
"While this is a biomarker and we have shown it is associated with CAV, we have not shown that it is causal," Dr. Daly cautioned. Any treatment directed against VEGF would have to be conducted in the context of a clinical trial to assess its impact.
A subset of patients becomes nonadherent to therapy; a subset that is highly sensitized before transplant may have donor-specific antibody, Dr. Daly said. So "we don’t think we fully understand the inciting event, ... [but] VEGF-A has been shown before to be elevated in antibody-mediated rejection, so it’s not surprising to see this association."
"We didn’t have these data available clinically because it was all a research study, so we didn’t intervene on any of the patients in this cohort. But we have started to think about whether or not we could use VEGF-A levels at least in our ... patients who might not have arterial access, and it might be difficult to survey them for CAV. I think in order to really understand the appropriate way to use it, we would need a larger study," he remarked.
Dr. Daly disclosed that he had no conflicts of interest relevant to the study.
SAN FRANCISCO – Monitoring plasma vascular endothelial growth factor A (VEGF-A) may help identify pediatric heart transplant patients who are at increased risk for poor outcomes, according to a study reported at the 2014 World Transplant Congress.
"Cardiac allograft vasculopathy [CAV] remains the leading cause of chronic allograft failure after heart transplantation. ... Therefore, it’s important for us to be able to anticipate the development of CAV and open up a therapeutic window," said Dr. Kevin P. Daly of Harvard Medical School and Boston Children’s Hospital.
"Our pilot data suggest that plasma VEGF-A levels below 90 pg/mL identify a low-risk patient population in whom a decreased frequency of coronary angiography can be considered. Future studies are needed to determine if using plasma VEGF-A levels to modify CAV screening frequency results in equivalent patient outcomes, with decreased resource utilization and improved quality of life," Dr. Daly commented at the congress, which was sponsored by the American Society of Transplant Surgeons.
As the vascular endothelium is the primary target of the immune response in CAV, the researchers hypothesized that VEGF-A likely contributes to an inflammatory cycle that leads to vascular damage and occlusion in the graft.
Participants in the single-center prospective cohort study were 44 consecutive children aged 2 years or older who were at least 18 months (median, 6 years) out from heart transplantation. They were scheduled for routine annual screening coronary angiography during 2009, and had no or mild CAV.
Moderate or severe CAV developed in 32% of patients who had VEGF-A values above the median value at baseline (90 pg/mL), compared with 5% of patients who had VEGF-A values below the median level (P = .02). Patients who developed this vasculopathy were more likely to die (38% vs. 0%), undergo retransplantation (38% vs. 0%), experience a myocardial infarction (12% vs. 0%), and be listed for retransplantation (12% vs. 0%).
"While this is a biomarker and we have shown it is associated with CAV, we have not shown that it is causal," Dr. Daly cautioned. Any treatment directed against VEGF would have to be conducted in the context of a clinical trial to assess its impact.
A subset of patients becomes nonadherent to therapy; a subset that is highly sensitized before transplant may have donor-specific antibody, Dr. Daly said. So "we don’t think we fully understand the inciting event, ... [but] VEGF-A has been shown before to be elevated in antibody-mediated rejection, so it’s not surprising to see this association."
"We didn’t have these data available clinically because it was all a research study, so we didn’t intervene on any of the patients in this cohort. But we have started to think about whether or not we could use VEGF-A levels at least in our ... patients who might not have arterial access, and it might be difficult to survey them for CAV. I think in order to really understand the appropriate way to use it, we would need a larger study," he remarked.
Dr. Daly disclosed that he had no conflicts of interest relevant to the study.
AT THE 2014 WORLD TRANSPLANT CONGRESS
Key clinical point: Plasma VEGF-A levels may be a biomarker of risk for pediatric heart transplant patients.
Major finding: Patients with plasma VEGF-A levels above the median value of 90 pg/mL had a 32% rate of moderate or severe cardiac allograft vasculopathy within 5 years.
Data source: A prospective cohort study of 44 consecutive children who had undergone heart transplantation.
Disclosures: Dr. Daly disclosed no relevant conflicts of interest.
