User login
Lithoplasty tames heavily calcified coronary lesions
PARIS – A novel therapeutic ultrasound-based technology known as lithoplasty is turning heads in interventional cardiology and vascular medicine because it addresses the bane of interventionalists’ existence: complex, heavily calcified coronary and peripheral artery lesions.
“Calcification is something we deal with every day in interventional cardiology. It makes the procedures more expensive, longer, and in fact several recent studies have shown that the complication rate for calcified lesions is higher than for any other lesion subtype. Calcification is the next big thing that we’re trying to take on in interventional cardiology,” Todd J. Brinton, MD, observed at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.

At EuroPCR, he presented the results of DISRUPT CAD, a seven-center study in which 60 patients with heavily calcified coronary lesions underwent lithoplasty in order to facilitate stent placement. The study met all of its safety and performance endpoints. As a result, the week prior to EuroPCR the European regulatory agency granted marketing approval for Shockwave Medical’s coronary lithoplasty system; the indication is for coronary vessel preparation prior to stenting. A large phase III U.S. trial aimed at gaining FDA approval is planned.
Moreover, on the basis of the earlier favorable DISRUPT PAD trial, lithoplasty has already been approved for treatment of peripheral artery disease (PAD) in Europe since late 2015 and by the FDA since September 2016. Now underway is DISRUPT PAD III, a large postmarketing randomized trial comparing lithoplasty with conventional balloon angioplasty in patients with heavily calcified PAD, added Dr. Brinton, an interventional cardiologist at Stanford (Calif.) University and cofounder of Shockwave Medical.
Lithoplasty is a potentially transformative technology which he described as “lithotripsy inside a balloon.” Lithotripsy has an established 30-year track record for the safe treatment of kidney stones. However, lithotripsy utilizes focused ultrasound, while lithoplasty relies upon circumferential unfocused therapeutic ultrasound delivered by miniaturized emitters placed inside a 12-mm intravascular balloon. The balloon is crossed to the target lesion, inflated to a modest pressure of 4 atmospheres, then the operator delivers lithoplasty pulses lasting over 1 microsec in duration at a rate of 1/sec for 10 seconds in order to fracture the thick intramedial calcium plaque, allowing the lesion to open up and thereby normalize vessel compliance.
“Once you’ve cracked the calcium you can easily dilate the lesion. It’s the calcium that’s restricting the ability to dilate. The real fundamental need here is to maximize acute gain to get really good stent apposition. We’re trying to get expansion,” the cardiologist explained.
That was readily achieved in the DISRUPT CAD study. The 60 participants had reference vessel diameters of 2.5-4.0 mm, with an average target lesion length of 20 mm. The calcification was heavy, covering on average 270 degrees of the vessel circumference as measured by optical coherence tomography, with an average calcium thickness of 0.97 mm and a calcified segment length of 22.3 mm.
The mean stent expansion was 112%. The minimum luminal diameter improved from 0.9 mm pretreatment to 2.6 mm post treatment, for an acute gain of 1.7 mm. The amount of acute gain was similar across the full range of vessel diameters.
The mean diameter stenosis went from 68% pretreatment to 13% post-treatment.
The primary safety endpoint was the 30-day rate of MACE, defined as cardiac death, MI, or target vessel revascularization. The rate was 5%, consisting of 3 patients with mild non–Q-wave MI defined by creatine kinase–MB elevations more than three times the upper limit of normal. The 6-month MACE rate was 8.5%, which included the three non–Q-wave MIs plus two cardiac deaths not related to the procedure or technology.
Final angiographic results adjudicated in a central core laboratory showed no perforations, abrupt closures, slow or no reflow events, or residual dissections. These are complications commonly seen with debulking devices such as rotational or orbital atherectomy, Dr. Brinton noted.
The primary performance endpoint in DISRUPT CAD was clinical success, defined as a residual stenosis of less than 50% post PCI with no in-hospital MACE. This was achieved in 57 of 60 patients, or 95%. The device was successfully delivered to the target lesion with subsequent performance of lithoplasty in 59 of 60 patients. An even more flexible and deliverable device will be released in the coming year, according to the cardiologist.
“I’d say the take-home is that the disease has changed,” Dr. Brinton commented. “It’s not the same disease that we had when Gruentzig did his first balloon angioplasty. These lesions are more calcified, more complex, yet for the most part we use the same balloon we’ve been using for the last 40 years. So lithoplasty is really an attempt to modernize the therapy in a new patient subset we now take care of who are much more complicated than the patients we originally took care of.”
“The reality is, we’re having difficulty taking care of these patients. For myself as an interventionalist, it’s not uncommon to look around the table and see a massive amount of tools when we’re doing these complex cases. Lithoplasty is intended to bring the simplicity. I would say it’s not necessarily to make the best operators better, it’s to bring all operators up to the ability to take on these complex lesions that are now usually reserved for high-volume centers that can do debulking,” he added.
Session cochair David R. Holmes Jr., MD, of the Mayo Clinic in Rochester, Minn., pronounced lithoplasty “tremendously exciting.” He and the other panelists focused on questions of safety and potential collateral damage: Where does the calcified debris go? What are the effects of the unfocused sonic pressure waves on noncalcified plaque? How hot does the vessel get?
Dr. Brinton replied that thick calcium plaque is located mostly in the medial vessel wall and stays there after fracturing. That’s why distal embolization wasn’t an issue in DISRUPT CAD. In animal studies, even at 20 times the energy dose used in clinical practice, lithoplasty had no effect on softer, noncalcified plaque or normal tissue. Vessel temperature increases by about 1.2 degrees C during lithoplasty, which isn’t sufficient to cause injury or drive restenosis.
Elsewhere at EuroPCR, Alberto Cremonesi, MD, who chaired a press conference where Dr. Brinton presented highlights of DISRUPT CAD, declared lithoplasty is “in my mind a real breakthrough, not only for coronary disease but also for PAD.”
Is it possible that stand-alone lithoplasty could reduce the need for multiple stents in longer coronary lesions, instead making possible more focal stenting? asked Dr. Cremonesi of Maria Cecilia Hospital in Cotignola, Italy.
That’s one of several possibilities worthy of future investigation, Dr. Brinton replied. Lithoplasty might also facilitate the results obtainable with bioresorbable coronary scaffolds or drug-coated balloons, he added.
He noted that as cofounder of and a consultant to Shockwave Medical, he has a sizable financial involvement with the company.
PARIS – A novel therapeutic ultrasound-based technology known as lithoplasty is turning heads in interventional cardiology and vascular medicine because it addresses the bane of interventionalists’ existence: complex, heavily calcified coronary and peripheral artery lesions.
“Calcification is something we deal with every day in interventional cardiology. It makes the procedures more expensive, longer, and in fact several recent studies have shown that the complication rate for calcified lesions is higher than for any other lesion subtype. Calcification is the next big thing that we’re trying to take on in interventional cardiology,” Todd J. Brinton, MD, observed at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
At EuroPCR, he presented the results of DISRUPT CAD, a seven-center study in which 60 patients with heavily calcified coronary lesions underwent lithoplasty in order to facilitate stent placement. The study met all of its safety and performance endpoints. As a result, the week prior to EuroPCR the European regulatory agency granted marketing approval for Shockwave Medical’s coronary lithoplasty system; the indication is for coronary vessel preparation prior to stenting. A large phase III U.S. trial aimed at gaining FDA approval is planned.
Moreover, on the basis of the earlier favorable DISRUPT PAD trial, lithoplasty has already been approved for treatment of peripheral artery disease (PAD) in Europe since late 2015 and by the FDA since September 2016. Now underway is DISRUPT PAD III, a large postmarketing randomized trial comparing lithoplasty with conventional balloon angioplasty in patients with heavily calcified PAD, added Dr. Brinton, an interventional cardiologist at Stanford (Calif.) University and cofounder of Shockwave Medical.
Lithoplasty is a potentially transformative technology which he described as “lithotripsy inside a balloon.” Lithotripsy has an established 30-year track record for the safe treatment of kidney stones. However, lithotripsy utilizes focused ultrasound, while lithoplasty relies upon circumferential unfocused therapeutic ultrasound delivered by miniaturized emitters placed inside a 12-mm intravascular balloon. The balloon is crossed to the target lesion, inflated to a modest pressure of 4 atmospheres, then the operator delivers lithoplasty pulses lasting over 1 microsec in duration at a rate of 1/sec for 10 seconds in order to fracture the thick intramedial calcium plaque, allowing the lesion to open up and thereby normalize vessel compliance.
“Once you’ve cracked the calcium you can easily dilate the lesion. It’s the calcium that’s restricting the ability to dilate. The real fundamental need here is to maximize acute gain to get really good stent apposition. We’re trying to get expansion,” the cardiologist explained.
That was readily achieved in the DISRUPT CAD study. The 60 participants had reference vessel diameters of 2.5-4.0 mm, with an average target lesion length of 20 mm. The calcification was heavy, covering on average 270 degrees of the vessel circumference as measured by optical coherence tomography, with an average calcium thickness of 0.97 mm and a calcified segment length of 22.3 mm.
The mean stent expansion was 112%. The minimum luminal diameter improved from 0.9 mm pretreatment to 2.6 mm post treatment, for an acute gain of 1.7 mm. The amount of acute gain was similar across the full range of vessel diameters.
The mean diameter stenosis went from 68% pretreatment to 13% post-treatment.
The primary safety endpoint was the 30-day rate of MACE, defined as cardiac death, MI, or target vessel revascularization. The rate was 5%, consisting of 3 patients with mild non–Q-wave MI defined by creatine kinase–MB elevations more than three times the upper limit of normal. The 6-month MACE rate was 8.5%, which included the three non–Q-wave MIs plus two cardiac deaths not related to the procedure or technology.
Final angiographic results adjudicated in a central core laboratory showed no perforations, abrupt closures, slow or no reflow events, or residual dissections. These are complications commonly seen with debulking devices such as rotational or orbital atherectomy, Dr. Brinton noted.
The primary performance endpoint in DISRUPT CAD was clinical success, defined as a residual stenosis of less than 50% post PCI with no in-hospital MACE. This was achieved in 57 of 60 patients, or 95%. The device was successfully delivered to the target lesion with subsequent performance of lithoplasty in 59 of 60 patients. An even more flexible and deliverable device will be released in the coming year, according to the cardiologist.
“I’d say the take-home is that the disease has changed,” Dr. Brinton commented. “It’s not the same disease that we had when Gruentzig did his first balloon angioplasty. These lesions are more calcified, more complex, yet for the most part we use the same balloon we’ve been using for the last 40 years. So lithoplasty is really an attempt to modernize the therapy in a new patient subset we now take care of who are much more complicated than the patients we originally took care of.”
“The reality is, we’re having difficulty taking care of these patients. For myself as an interventionalist, it’s not uncommon to look around the table and see a massive amount of tools when we’re doing these complex cases. Lithoplasty is intended to bring the simplicity. I would say it’s not necessarily to make the best operators better, it’s to bring all operators up to the ability to take on these complex lesions that are now usually reserved for high-volume centers that can do debulking,” he added.
Session cochair David R. Holmes Jr., MD, of the Mayo Clinic in Rochester, Minn., pronounced lithoplasty “tremendously exciting.” He and the other panelists focused on questions of safety and potential collateral damage: Where does the calcified debris go? What are the effects of the unfocused sonic pressure waves on noncalcified plaque? How hot does the vessel get?
Dr. Brinton replied that thick calcium plaque is located mostly in the medial vessel wall and stays there after fracturing. That’s why distal embolization wasn’t an issue in DISRUPT CAD. In animal studies, even at 20 times the energy dose used in clinical practice, lithoplasty had no effect on softer, noncalcified plaque or normal tissue. Vessel temperature increases by about 1.2 degrees C during lithoplasty, which isn’t sufficient to cause injury or drive restenosis.
Elsewhere at EuroPCR, Alberto Cremonesi, MD, who chaired a press conference where Dr. Brinton presented highlights of DISRUPT CAD, declared lithoplasty is “in my mind a real breakthrough, not only for coronary disease but also for PAD.”
Is it possible that stand-alone lithoplasty could reduce the need for multiple stents in longer coronary lesions, instead making possible more focal stenting? asked Dr. Cremonesi of Maria Cecilia Hospital in Cotignola, Italy.
That’s one of several possibilities worthy of future investigation, Dr. Brinton replied. Lithoplasty might also facilitate the results obtainable with bioresorbable coronary scaffolds or drug-coated balloons, he added.
He noted that as cofounder of and a consultant to Shockwave Medical, he has a sizable financial involvement with the company.
PARIS – A novel therapeutic ultrasound-based technology known as lithoplasty is turning heads in interventional cardiology and vascular medicine because it addresses the bane of interventionalists’ existence: complex, heavily calcified coronary and peripheral artery lesions.
“Calcification is something we deal with every day in interventional cardiology. It makes the procedures more expensive, longer, and in fact several recent studies have shown that the complication rate for calcified lesions is higher than for any other lesion subtype. Calcification is the next big thing that we’re trying to take on in interventional cardiology,” Todd J. Brinton, MD, observed at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
At EuroPCR, he presented the results of DISRUPT CAD, a seven-center study in which 60 patients with heavily calcified coronary lesions underwent lithoplasty in order to facilitate stent placement. The study met all of its safety and performance endpoints. As a result, the week prior to EuroPCR the European regulatory agency granted marketing approval for Shockwave Medical’s coronary lithoplasty system; the indication is for coronary vessel preparation prior to stenting. A large phase III U.S. trial aimed at gaining FDA approval is planned.
Moreover, on the basis of the earlier favorable DISRUPT PAD trial, lithoplasty has already been approved for treatment of peripheral artery disease (PAD) in Europe since late 2015 and by the FDA since September 2016. Now underway is DISRUPT PAD III, a large postmarketing randomized trial comparing lithoplasty with conventional balloon angioplasty in patients with heavily calcified PAD, added Dr. Brinton, an interventional cardiologist at Stanford (Calif.) University and cofounder of Shockwave Medical.
Lithoplasty is a potentially transformative technology which he described as “lithotripsy inside a balloon.” Lithotripsy has an established 30-year track record for the safe treatment of kidney stones. However, lithotripsy utilizes focused ultrasound, while lithoplasty relies upon circumferential unfocused therapeutic ultrasound delivered by miniaturized emitters placed inside a 12-mm intravascular balloon. The balloon is crossed to the target lesion, inflated to a modest pressure of 4 atmospheres, then the operator delivers lithoplasty pulses lasting over 1 microsec in duration at a rate of 1/sec for 10 seconds in order to fracture the thick intramedial calcium plaque, allowing the lesion to open up and thereby normalize vessel compliance.
“Once you’ve cracked the calcium you can easily dilate the lesion. It’s the calcium that’s restricting the ability to dilate. The real fundamental need here is to maximize acute gain to get really good stent apposition. We’re trying to get expansion,” the cardiologist explained.
That was readily achieved in the DISRUPT CAD study. The 60 participants had reference vessel diameters of 2.5-4.0 mm, with an average target lesion length of 20 mm. The calcification was heavy, covering on average 270 degrees of the vessel circumference as measured by optical coherence tomography, with an average calcium thickness of 0.97 mm and a calcified segment length of 22.3 mm.
The mean stent expansion was 112%. The minimum luminal diameter improved from 0.9 mm pretreatment to 2.6 mm post treatment, for an acute gain of 1.7 mm. The amount of acute gain was similar across the full range of vessel diameters.
The mean diameter stenosis went from 68% pretreatment to 13% post-treatment.
The primary safety endpoint was the 30-day rate of MACE, defined as cardiac death, MI, or target vessel revascularization. The rate was 5%, consisting of 3 patients with mild non–Q-wave MI defined by creatine kinase–MB elevations more than three times the upper limit of normal. The 6-month MACE rate was 8.5%, which included the three non–Q-wave MIs plus two cardiac deaths not related to the procedure or technology.
Final angiographic results adjudicated in a central core laboratory showed no perforations, abrupt closures, slow or no reflow events, or residual dissections. These are complications commonly seen with debulking devices such as rotational or orbital atherectomy, Dr. Brinton noted.
The primary performance endpoint in DISRUPT CAD was clinical success, defined as a residual stenosis of less than 50% post PCI with no in-hospital MACE. This was achieved in 57 of 60 patients, or 95%. The device was successfully delivered to the target lesion with subsequent performance of lithoplasty in 59 of 60 patients. An even more flexible and deliverable device will be released in the coming year, according to the cardiologist.
“I’d say the take-home is that the disease has changed,” Dr. Brinton commented. “It’s not the same disease that we had when Gruentzig did his first balloon angioplasty. These lesions are more calcified, more complex, yet for the most part we use the same balloon we’ve been using for the last 40 years. So lithoplasty is really an attempt to modernize the therapy in a new patient subset we now take care of who are much more complicated than the patients we originally took care of.”
“The reality is, we’re having difficulty taking care of these patients. For myself as an interventionalist, it’s not uncommon to look around the table and see a massive amount of tools when we’re doing these complex cases. Lithoplasty is intended to bring the simplicity. I would say it’s not necessarily to make the best operators better, it’s to bring all operators up to the ability to take on these complex lesions that are now usually reserved for high-volume centers that can do debulking,” he added.
Session cochair David R. Holmes Jr., MD, of the Mayo Clinic in Rochester, Minn., pronounced lithoplasty “tremendously exciting.” He and the other panelists focused on questions of safety and potential collateral damage: Where does the calcified debris go? What are the effects of the unfocused sonic pressure waves on noncalcified plaque? How hot does the vessel get?
Dr. Brinton replied that thick calcium plaque is located mostly in the medial vessel wall and stays there after fracturing. That’s why distal embolization wasn’t an issue in DISRUPT CAD. In animal studies, even at 20 times the energy dose used in clinical practice, lithoplasty had no effect on softer, noncalcified plaque or normal tissue. Vessel temperature increases by about 1.2 degrees C during lithoplasty, which isn’t sufficient to cause injury or drive restenosis.
Elsewhere at EuroPCR, Alberto Cremonesi, MD, who chaired a press conference where Dr. Brinton presented highlights of DISRUPT CAD, declared lithoplasty is “in my mind a real breakthrough, not only for coronary disease but also for PAD.”
Is it possible that stand-alone lithoplasty could reduce the need for multiple stents in longer coronary lesions, instead making possible more focal stenting? asked Dr. Cremonesi of Maria Cecilia Hospital in Cotignola, Italy.
That’s one of several possibilities worthy of future investigation, Dr. Brinton replied. Lithoplasty might also facilitate the results obtainable with bioresorbable coronary scaffolds or drug-coated balloons, he added.
He noted that as cofounder of and a consultant to Shockwave Medical, he has a sizable financial involvement with the company.
AT EUROPCR
Key clinical point:
Major finding: Lithoplasty of heavily calcified coronary lesions improved the minimum luminal diameter from 0.9 mm pretreatment to 2.6 mm post-treatment, for an immediate gain of 1.7 mm prior to stent placement.
Data source: This study featured 6-month follow-up of 60 patients with heavily calcified coronary lesions who underwent lithoplasty followed by stenting.
Disclosures: The DISRUPT CAD study was sponsored by Shockwave Medical, which is developing lithoplasty. The presenter cofounded the company.
Amplatzer devices outperform oral anticoagulation in atrial fib
PARIS – Percutaneous left atrial appendage closure with an Amplatzer device in patients with nonvalvular atrial fibrillation was associated with significantly lower rates of all-cause and cardiovascular mortality, compared with oral anticoagulation, in a large propensity score–matched observational registry study.
Left atrial appendage closure (LAAC) also bested oral anticoagulation (OAC) with warfarin or a novel oral anticoagulant (NOAC) in terms of net clinical benefit on the basis of the device therapy’s greater protection against stroke and systemic embolism coupled with a trend, albeit not statistically significant, for fewer bleeding events, Steffen Gloekler, MD, reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
The Watchman LAAC device, commercially available both in Europe and the United States, has previously been shown to be superior to OAC in terms of efficacy and noninferior regarding safety. But there have been no randomized trials of an Amplatzer device versus OAC. This lack of data was the impetus for Dr. Gloekler and his coinvestigators to create a meticulously propensity-matched observational registry.
Five hundred consecutive patients with AF who received an Amplatzer Cardiac Plug or its second-generation version, the Amplatzer Amulet, during 2009-2014 were tightly matched to an equal number of AF patients on OAC based on age, sex, body mass index, left ventricular ejection fraction, renal function, coronary artery disease status, hemoglobin level, CHA2DS2-VASc score, and HAS-BLED score. During a mean 2.7 years, or 2,645 patient-years, of follow-up, the composite primary efficacy endpoint, composed of stroke, systemic embolism, and cardiovascular or unexplained death occurred in 5.6% of the LAAC group, compared with 7.8% of controls in the OAC arm, for a statistically significant 30% relative risk reduction. Disabling stroke occurred in 0.7% of Amplatzer patients versus 1.5% of controls. The ischemic stroke rate was 1.5% in the device therapy group and 2% in the OAC arm.
All-cause mortality occurred in 8.3% of Amplatzer patients and 11.6% of the OAC group, for a 28% relative risk reduction. The cardiovascular death rate was 4% in the Amplatzer group, compared with 6.5% of controls, for a 36% risk reduction.
The composite safety endpoint, comprising all major procedural adverse events and major or life-threatening bleeding during follow-up, occurred in 3.6% of the Amplatzer group and 4.6% of the OAC group, for a 20% relative risk reduction that is not significant at this point because of the low number of events. Major, life-threatening, or fatal bleeding occurred in 2% of Amplatzer recipients versus 5.5% of controls, added Dr. Gloekler of University Hospital in Bern, Switzerland.
The net clinical benefit, a composite of death, bleeding, or stroke, occurred in 8.1% of the Amplatzer group, compared with 10.9% of controls, for a significant 24% reduction in relative risk in favor of device therapy.
Of note, at 2.7 years of follow-up only 55% of the OAC group were still taking an anticoagulant: 38% of the original 500 patients were on warfarin, and 17% were taking a NOAC. At that point, 8% of the Amplatzer group were on any anticoagulation therapy.
Discussion of the study focused on that low rate of medication adherence in the OAC arm. Dr. Gloekler’s response was that, after looking at the literature, he was no longer surprised by the finding that only 55% of the control group were on OAC at follow-up.
“If you look in the literature, that’s exactly the real-world adherence for OACs. Even in all four certification trials for the NOACs, the rate of discontinuation was 30% after 2 years – and these were controlled studies. Ours was observational, and it depicts a good deal of the problem with any OAC in my eyes,” Dr. Gloekler said.
Patients on warfarin in the real-world Amplatzer registry study spent on average a mere 30% of time in the therapeutic international normalized ratio range of 2-3.
“That means 70% of the time patients are higher and have an increased bleeding risk or they are lower and don’t have adequate stroke protection,” he noted.
This prompted one observer to comment, “We either have to do a better job in our clinics with OAC or we have to occlude more appendages.”
A large pivotal U.S. trial aimed at winning FDA approval for the Amplatzer Amulet for LAAC is underway. Patients with AF are being randomized to the approved Watchman or investigational Amulet at roughly 100 U.S. and 50 foreign sites.
Dr. Gloekler reported receiving research funds for the registry from the Swiss Heart Foundation and Abbott.
PARIS – Percutaneous left atrial appendage closure with an Amplatzer device in patients with nonvalvular atrial fibrillation was associated with significantly lower rates of all-cause and cardiovascular mortality, compared with oral anticoagulation, in a large propensity score–matched observational registry study.
Left atrial appendage closure (LAAC) also bested oral anticoagulation (OAC) with warfarin or a novel oral anticoagulant (NOAC) in terms of net clinical benefit on the basis of the device therapy’s greater protection against stroke and systemic embolism coupled with a trend, albeit not statistically significant, for fewer bleeding events, Steffen Gloekler, MD, reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
The Watchman LAAC device, commercially available both in Europe and the United States, has previously been shown to be superior to OAC in terms of efficacy and noninferior regarding safety. But there have been no randomized trials of an Amplatzer device versus OAC. This lack of data was the impetus for Dr. Gloekler and his coinvestigators to create a meticulously propensity-matched observational registry.
Five hundred consecutive patients with AF who received an Amplatzer Cardiac Plug or its second-generation version, the Amplatzer Amulet, during 2009-2014 were tightly matched to an equal number of AF patients on OAC based on age, sex, body mass index, left ventricular ejection fraction, renal function, coronary artery disease status, hemoglobin level, CHA2DS2-VASc score, and HAS-BLED score. During a mean 2.7 years, or 2,645 patient-years, of follow-up, the composite primary efficacy endpoint, composed of stroke, systemic embolism, and cardiovascular or unexplained death occurred in 5.6% of the LAAC group, compared with 7.8% of controls in the OAC arm, for a statistically significant 30% relative risk reduction. Disabling stroke occurred in 0.7% of Amplatzer patients versus 1.5% of controls. The ischemic stroke rate was 1.5% in the device therapy group and 2% in the OAC arm.
All-cause mortality occurred in 8.3% of Amplatzer patients and 11.6% of the OAC group, for a 28% relative risk reduction. The cardiovascular death rate was 4% in the Amplatzer group, compared with 6.5% of controls, for a 36% risk reduction.
The composite safety endpoint, comprising all major procedural adverse events and major or life-threatening bleeding during follow-up, occurred in 3.6% of the Amplatzer group and 4.6% of the OAC group, for a 20% relative risk reduction that is not significant at this point because of the low number of events. Major, life-threatening, or fatal bleeding occurred in 2% of Amplatzer recipients versus 5.5% of controls, added Dr. Gloekler of University Hospital in Bern, Switzerland.
The net clinical benefit, a composite of death, bleeding, or stroke, occurred in 8.1% of the Amplatzer group, compared with 10.9% of controls, for a significant 24% reduction in relative risk in favor of device therapy.
Of note, at 2.7 years of follow-up only 55% of the OAC group were still taking an anticoagulant: 38% of the original 500 patients were on warfarin, and 17% were taking a NOAC. At that point, 8% of the Amplatzer group were on any anticoagulation therapy.
Discussion of the study focused on that low rate of medication adherence in the OAC arm. Dr. Gloekler’s response was that, after looking at the literature, he was no longer surprised by the finding that only 55% of the control group were on OAC at follow-up.
“If you look in the literature, that’s exactly the real-world adherence for OACs. Even in all four certification trials for the NOACs, the rate of discontinuation was 30% after 2 years – and these were controlled studies. Ours was observational, and it depicts a good deal of the problem with any OAC in my eyes,” Dr. Gloekler said.
Patients on warfarin in the real-world Amplatzer registry study spent on average a mere 30% of time in the therapeutic international normalized ratio range of 2-3.
“That means 70% of the time patients are higher and have an increased bleeding risk or they are lower and don’t have adequate stroke protection,” he noted.
This prompted one observer to comment, “We either have to do a better job in our clinics with OAC or we have to occlude more appendages.”
A large pivotal U.S. trial aimed at winning FDA approval for the Amplatzer Amulet for LAAC is underway. Patients with AF are being randomized to the approved Watchman or investigational Amulet at roughly 100 U.S. and 50 foreign sites.
Dr. Gloekler reported receiving research funds for the registry from the Swiss Heart Foundation and Abbott.
PARIS – Percutaneous left atrial appendage closure with an Amplatzer device in patients with nonvalvular atrial fibrillation was associated with significantly lower rates of all-cause and cardiovascular mortality, compared with oral anticoagulation, in a large propensity score–matched observational registry study.
Left atrial appendage closure (LAAC) also bested oral anticoagulation (OAC) with warfarin or a novel oral anticoagulant (NOAC) in terms of net clinical benefit on the basis of the device therapy’s greater protection against stroke and systemic embolism coupled with a trend, albeit not statistically significant, for fewer bleeding events, Steffen Gloekler, MD, reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
The Watchman LAAC device, commercially available both in Europe and the United States, has previously been shown to be superior to OAC in terms of efficacy and noninferior regarding safety. But there have been no randomized trials of an Amplatzer device versus OAC. This lack of data was the impetus for Dr. Gloekler and his coinvestigators to create a meticulously propensity-matched observational registry.
Five hundred consecutive patients with AF who received an Amplatzer Cardiac Plug or its second-generation version, the Amplatzer Amulet, during 2009-2014 were tightly matched to an equal number of AF patients on OAC based on age, sex, body mass index, left ventricular ejection fraction, renal function, coronary artery disease status, hemoglobin level, CHA2DS2-VASc score, and HAS-BLED score. During a mean 2.7 years, or 2,645 patient-years, of follow-up, the composite primary efficacy endpoint, composed of stroke, systemic embolism, and cardiovascular or unexplained death occurred in 5.6% of the LAAC group, compared with 7.8% of controls in the OAC arm, for a statistically significant 30% relative risk reduction. Disabling stroke occurred in 0.7% of Amplatzer patients versus 1.5% of controls. The ischemic stroke rate was 1.5% in the device therapy group and 2% in the OAC arm.
All-cause mortality occurred in 8.3% of Amplatzer patients and 11.6% of the OAC group, for a 28% relative risk reduction. The cardiovascular death rate was 4% in the Amplatzer group, compared with 6.5% of controls, for a 36% risk reduction.
The composite safety endpoint, comprising all major procedural adverse events and major or life-threatening bleeding during follow-up, occurred in 3.6% of the Amplatzer group and 4.6% of the OAC group, for a 20% relative risk reduction that is not significant at this point because of the low number of events. Major, life-threatening, or fatal bleeding occurred in 2% of Amplatzer recipients versus 5.5% of controls, added Dr. Gloekler of University Hospital in Bern, Switzerland.
The net clinical benefit, a composite of death, bleeding, or stroke, occurred in 8.1% of the Amplatzer group, compared with 10.9% of controls, for a significant 24% reduction in relative risk in favor of device therapy.
Of note, at 2.7 years of follow-up only 55% of the OAC group were still taking an anticoagulant: 38% of the original 500 patients were on warfarin, and 17% were taking a NOAC. At that point, 8% of the Amplatzer group were on any anticoagulation therapy.
Discussion of the study focused on that low rate of medication adherence in the OAC arm. Dr. Gloekler’s response was that, after looking at the literature, he was no longer surprised by the finding that only 55% of the control group were on OAC at follow-up.
“If you look in the literature, that’s exactly the real-world adherence for OACs. Even in all four certification trials for the NOACs, the rate of discontinuation was 30% after 2 years – and these were controlled studies. Ours was observational, and it depicts a good deal of the problem with any OAC in my eyes,” Dr. Gloekler said.
Patients on warfarin in the real-world Amplatzer registry study spent on average a mere 30% of time in the therapeutic international normalized ratio range of 2-3.
“That means 70% of the time patients are higher and have an increased bleeding risk or they are lower and don’t have adequate stroke protection,” he noted.
This prompted one observer to comment, “We either have to do a better job in our clinics with OAC or we have to occlude more appendages.”
A large pivotal U.S. trial aimed at winning FDA approval for the Amplatzer Amulet for LAAC is underway. Patients with AF are being randomized to the approved Watchman or investigational Amulet at roughly 100 U.S. and 50 foreign sites.
Dr. Gloekler reported receiving research funds for the registry from the Swiss Heart Foundation and Abbott.
AT EUROPCR
Key clinical point:
Major finding: The primary composite efficacy endpoint of stroke, systemic embolism, or cardiovascular or unexplained death during a mean 2.7 years of follow-up occurred in 5.6% of Amplatzer device recipients, a 30% reduction, compared with the 7.8% rate in the oral anticoagulation group.
Data source: This observational registry included 500 patients with atrial fibrillation who received an Amplatzer left atrial appendage closure device and an equal number of carefully matched AF patients on oral anticoagulation.
Disclosures: The study presenter reported receiving research funds for the registry from the Swiss Heart Foundation and Abbott.
Bad news keeps piling up for Absorb coronary scaffold
PARIS – Device thrombosis occurred nearly four times more frequently in recipients of the Absorb everolimus-eluting bioresorbable vascular scaffold than with the Xience everolimus-eluting metallic stent during 2 years of prospective follow-up in the randomized AIDA trial.
AIDA (the Amsterdam Investigator-Initiated Absorb Strategy All-Comers Trial) was the first randomized trial designed to compare the Absorb scaffold to a drug-eluting metallic stent in a broad patient population reflecting routine real-world clinical practice. The disturbing AIDA finding follows upon earlier serious concerns raised regarding an increased risk of scaffold thrombosis – and the particularly worrisome complication of late thrombosis – in the ABSORB Japan and ABSORB II trials, Joanna J. Wykrzykowska, MD, reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.

The device was approved by the Food and Drug Administration in July 2016. In March 2017 the agency issued a safety alert regarding the Absorb scaffold after release of the 2-year data from the 2,008-patient ABSORB III trial showing a significantly higher rate of target-lesion failure than with the Xience stent. Both devices are marketed by Abbott Vascular.
AIDA was a single-blind multicenter Dutch trial that randomized 1,845 patients undergoing PCI, 55% of whom presented with acute coronary syndrome and 26% of whom had ST-elevation MI. The primary endpoint was target vessel failure, a composite of cardiac death, target vessel MI, or target vessel revascularization. The 2-year cumulative rate did not differ significantly between the two study arms: 11.7% in the scaffold group and 10.7% in the metallic stent recipients.
However, definite or probable device thrombosis occurred in 3.5% of the scaffold group compared with 0.9% of metallic stent recipients, for a highly significant 3.9-fold increased risk. This was associated with a significantly increased 2-year cumulative risk of MI: 5.5% versus 3.2%.
On the basis of this unsettling finding, coupled with the fact that ABSORB II investigators did not find any instance of very late scaffold thrombosis among 63 patients who remained on dual-antiplatelet therapy (DAPT) continuously for up to 3 years, Dr. Wykrzykowska and her coinvestigators have informed AIDA participants of their treatment assignment. They have also recommended that the Absorb recipients go on extended DAPT, even though there is no high-grade evidence as yet that this will prevent late scaffold thrombosis or that the drug-induced increased bleeding risk of prolonged DAPT might cancel or perhaps even outweigh the potential protection against device thrombosis.
On top of all this, implantation of the scaffold entails a longer procedure time and a greater volume of contrast material.
Discussant Mahmoud Hashemian, MD, observed that while bioresorbable vascular scaffolds are “physiologically ideal” because, unlike metallic stents, theoretically they leave no permanent implant to impede vasomotion and serve as a nidus for neoatherosclerosis, to date they have shown no real-world benefits over current-generation drug-eluting metallic stents, but only disadvantages.
“This doesn’t mean we have to feel hopeless. I’m not hopeless at all,” said Dr. Hashemian, an interventional cardiologist at Day General Hospital in Tehran. “I’m sure this [bioresorbable scaffolds] will be the future of our stents. But it needs more work. The company tells me they are going to launch a newer one, maybe next year, with thinner struts and more expandability.”
Asked about the likely mechanism of prolonged thrombosis risk with Absorb, Dr. Wykrzykowska was quick to say no one really knows at this point.
“Technique [predilation at a 1:1 balloon-to-artery ratio with an appropriately sized balloon] can obviously improve things in the short term for early events, but I don’t think we understand the biology of late events. We don’t understand the interaction between the device and the vessel. It’s extremely complex,” she said.
AIDA was funded by an unrestricted educational grant from Abbott Vascular. Dr. Wykrzykowska reported receiving consulting and lecture fees from the company.
PARIS – Device thrombosis occurred nearly four times more frequently in recipients of the Absorb everolimus-eluting bioresorbable vascular scaffold than with the Xience everolimus-eluting metallic stent during 2 years of prospective follow-up in the randomized AIDA trial.
AIDA (the Amsterdam Investigator-Initiated Absorb Strategy All-Comers Trial) was the first randomized trial designed to compare the Absorb scaffold to a drug-eluting metallic stent in a broad patient population reflecting routine real-world clinical practice. The disturbing AIDA finding follows upon earlier serious concerns raised regarding an increased risk of scaffold thrombosis – and the particularly worrisome complication of late thrombosis – in the ABSORB Japan and ABSORB II trials, Joanna J. Wykrzykowska, MD, reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
The device was approved by the Food and Drug Administration in July 2016. In March 2017 the agency issued a safety alert regarding the Absorb scaffold after release of the 2-year data from the 2,008-patient ABSORB III trial showing a significantly higher rate of target-lesion failure than with the Xience stent. Both devices are marketed by Abbott Vascular.
AIDA was a single-blind multicenter Dutch trial that randomized 1,845 patients undergoing PCI, 55% of whom presented with acute coronary syndrome and 26% of whom had ST-elevation MI. The primary endpoint was target vessel failure, a composite of cardiac death, target vessel MI, or target vessel revascularization. The 2-year cumulative rate did not differ significantly between the two study arms: 11.7% in the scaffold group and 10.7% in the metallic stent recipients.
However, definite or probable device thrombosis occurred in 3.5% of the scaffold group compared with 0.9% of metallic stent recipients, for a highly significant 3.9-fold increased risk. This was associated with a significantly increased 2-year cumulative risk of MI: 5.5% versus 3.2%.
On the basis of this unsettling finding, coupled with the fact that ABSORB II investigators did not find any instance of very late scaffold thrombosis among 63 patients who remained on dual-antiplatelet therapy (DAPT) continuously for up to 3 years, Dr. Wykrzykowska and her coinvestigators have informed AIDA participants of their treatment assignment. They have also recommended that the Absorb recipients go on extended DAPT, even though there is no high-grade evidence as yet that this will prevent late scaffold thrombosis or that the drug-induced increased bleeding risk of prolonged DAPT might cancel or perhaps even outweigh the potential protection against device thrombosis.
On top of all this, implantation of the scaffold entails a longer procedure time and a greater volume of contrast material.
Discussant Mahmoud Hashemian, MD, observed that while bioresorbable vascular scaffolds are “physiologically ideal” because, unlike metallic stents, theoretically they leave no permanent implant to impede vasomotion and serve as a nidus for neoatherosclerosis, to date they have shown no real-world benefits over current-generation drug-eluting metallic stents, but only disadvantages.
“This doesn’t mean we have to feel hopeless. I’m not hopeless at all,” said Dr. Hashemian, an interventional cardiologist at Day General Hospital in Tehran. “I’m sure this [bioresorbable scaffolds] will be the future of our stents. But it needs more work. The company tells me they are going to launch a newer one, maybe next year, with thinner struts and more expandability.”
Asked about the likely mechanism of prolonged thrombosis risk with Absorb, Dr. Wykrzykowska was quick to say no one really knows at this point.
“Technique [predilation at a 1:1 balloon-to-artery ratio with an appropriately sized balloon] can obviously improve things in the short term for early events, but I don’t think we understand the biology of late events. We don’t understand the interaction between the device and the vessel. It’s extremely complex,” she said.
AIDA was funded by an unrestricted educational grant from Abbott Vascular. Dr. Wykrzykowska reported receiving consulting and lecture fees from the company.
PARIS – Device thrombosis occurred nearly four times more frequently in recipients of the Absorb everolimus-eluting bioresorbable vascular scaffold than with the Xience everolimus-eluting metallic stent during 2 years of prospective follow-up in the randomized AIDA trial.
AIDA (the Amsterdam Investigator-Initiated Absorb Strategy All-Comers Trial) was the first randomized trial designed to compare the Absorb scaffold to a drug-eluting metallic stent in a broad patient population reflecting routine real-world clinical practice. The disturbing AIDA finding follows upon earlier serious concerns raised regarding an increased risk of scaffold thrombosis – and the particularly worrisome complication of late thrombosis – in the ABSORB Japan and ABSORB II trials, Joanna J. Wykrzykowska, MD, reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
The device was approved by the Food and Drug Administration in July 2016. In March 2017 the agency issued a safety alert regarding the Absorb scaffold after release of the 2-year data from the 2,008-patient ABSORB III trial showing a significantly higher rate of target-lesion failure than with the Xience stent. Both devices are marketed by Abbott Vascular.
AIDA was a single-blind multicenter Dutch trial that randomized 1,845 patients undergoing PCI, 55% of whom presented with acute coronary syndrome and 26% of whom had ST-elevation MI. The primary endpoint was target vessel failure, a composite of cardiac death, target vessel MI, or target vessel revascularization. The 2-year cumulative rate did not differ significantly between the two study arms: 11.7% in the scaffold group and 10.7% in the metallic stent recipients.
However, definite or probable device thrombosis occurred in 3.5% of the scaffold group compared with 0.9% of metallic stent recipients, for a highly significant 3.9-fold increased risk. This was associated with a significantly increased 2-year cumulative risk of MI: 5.5% versus 3.2%.
On the basis of this unsettling finding, coupled with the fact that ABSORB II investigators did not find any instance of very late scaffold thrombosis among 63 patients who remained on dual-antiplatelet therapy (DAPT) continuously for up to 3 years, Dr. Wykrzykowska and her coinvestigators have informed AIDA participants of their treatment assignment. They have also recommended that the Absorb recipients go on extended DAPT, even though there is no high-grade evidence as yet that this will prevent late scaffold thrombosis or that the drug-induced increased bleeding risk of prolonged DAPT might cancel or perhaps even outweigh the potential protection against device thrombosis.
On top of all this, implantation of the scaffold entails a longer procedure time and a greater volume of contrast material.
Discussant Mahmoud Hashemian, MD, observed that while bioresorbable vascular scaffolds are “physiologically ideal” because, unlike metallic stents, theoretically they leave no permanent implant to impede vasomotion and serve as a nidus for neoatherosclerosis, to date they have shown no real-world benefits over current-generation drug-eluting metallic stents, but only disadvantages.
“This doesn’t mean we have to feel hopeless. I’m not hopeless at all,” said Dr. Hashemian, an interventional cardiologist at Day General Hospital in Tehran. “I’m sure this [bioresorbable scaffolds] will be the future of our stents. But it needs more work. The company tells me they are going to launch a newer one, maybe next year, with thinner struts and more expandability.”
Asked about the likely mechanism of prolonged thrombosis risk with Absorb, Dr. Wykrzykowska was quick to say no one really knows at this point.
“Technique [predilation at a 1:1 balloon-to-artery ratio with an appropriately sized balloon] can obviously improve things in the short term for early events, but I don’t think we understand the biology of late events. We don’t understand the interaction between the device and the vessel. It’s extremely complex,” she said.
AIDA was funded by an unrestricted educational grant from Abbott Vascular. Dr. Wykrzykowska reported receiving consulting and lecture fees from the company.
AT EUROPCR
Key clinical point:
Major finding: During 2 years of prospective follow-up, definite or probable device thrombosis occurred in 3.5% of recipients of a bioresorbable vascular scaffold, compared with 0.9% of metallic stent recipients, for a highly significant 3.9-fold increased risk.
Data source: AIDA, a single-blind multicenter Dutch trial that randomized a broadly representative group of 1,845 patients undergoing PCI to the Absorb bioresorbable vascular scaffold or the Xience everolimus-eluting metallic stent.
Disclosures: The AIDA study was funded by an unrestricted educational grant from Abbott Vascular. The presenter reported receiving consulting and lecture fees from the company.
FFR stumbles in revascularization deferral decisions for ACS
Paris – One-year outcomes were significantly worse in patients with acute coronary syndrome whose revascularization was deferred based upon the results of fractional flow reserve than with instantaneous wave-free ratio, in the largest-ever study of patients whose revascularization decision was guided by physiologic measurements obtained via a pressure guidewire.
“The hypothesis that some authors have put forth – that in an ACS the hyperemic response of the myocardium is blunted by the ACS, and that this will affect the FFR hyperemic index – is now strengthened,” Javier Escaned, MD, said in presenting the study results at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
The study was a pooled, patient-level meta-analysis of the 4,529 participants with angiographically determined intermediate-risk stenoses in the previously reported randomized DEFINE FLAIR (N Engl J Med. 2017 May 11;376[19]:1824-34) and iFR SWEDEHEART (N Engl J Med. 2017 May 11;376[19]:1813-23) studies. The primary endpoint was the composite of death, nonfatal MI, or unplanned coronary revascularization within 12 months. And while the analysis brought unwelcome news for proponents of FFR with regard to the subset of patients with ACS, such patients comprised only 17% of the total study population.

“I think that overall these results are very reassuring. The big finding is that we have dramatically improved the safety of deferral of revascularization using pressure guidewires. If you look at the MACE [major adverse cardiovascular event] rate in the deferred ACS group, it was about 6%, which is much less than the event rate at 1 year with deferral in patients with stable coronary disease in the pivotal DEFER trial [Circulation. 2001 Jun 19;103(24):2928-34], which was our former standard,” observed Dr. Escaned, an interventional cardiologist at San Carlos Hospital in Madrid and a DEFER coinvestigator.
He attributed these greatly improved outcomes of physiologically guided revascularization during the past 15 years to vastly improved stent technology and more effective optimal medical management.
Among the key findings of the combined analysis of DEFINE FLAIR and iFR SWEDEHEART:
• More patients were deferred from PCI when iFR was used for decision-making: 50%, compared with 45% in the FFR arm. Yet 1-year outcomes were as good in the deferred iFR group as in the FFR group overall, and better than with FFR in the deferred ACS patients.
• Event rates were significantly higher in deferred ACS patients overall than in deferred patients with stable coronary disease: 5.9% versus 3.6%. But the deferral tool made a difference: When iFR was utilized, the 1-year event rate was 5.4% in deferred ACS patients, not significantly different from the 3.8% rate in deferred patients with stable coronary disease. In contrast, the event rate in ACS patients with FFR-based deferral was 6.4%, significantly higher than the 3.4% rate in FFR-deferred patients with stable coronary disease.
Dr. Escaned noted that this finding is consistent with the cautionary results of several recent studies, including one, albeit tenfold smaller, in which ACS patients in whom revascularization was deferred based on FFR had a 25% rate of major adverse cardiovascular events at 3.4 years, compared with a 12% rate in patients with stable coronary disease (J Am Coll Cardiol. 2016 Sep 13;68[11]:1181-91).
Discussant Peter Jüni, MD, professor of medicine at the University of Toronto, said “the main results of your study show in a completely waterproof fashion that there is no signal of harm with the experimental strategy” of deferred revascularization based on physiologic measurements, at least in patients with stable ischemic coronary disease.
The results, however, also raise the question of whether physiology-based revascularization decision-making in ACS patients is the best strategy.
“Considering that the event rate in the deferred ACS group was nearly twice as high compared with stable patients, my question to you is: Should we ignore any functional testing in ACS patients and just say, ‘Let’s move forward with revascularization because this clinical presentation is a very good clinical characteristic for risk stratification?’ ”
Dr. Escaned rejected that option. He noted that both the European and U.S. guidelines now state that it’s inappropriate to base a revascularization decision solely on a coronary vessel’s angiographic appearance, because that has been shown to result in unnecessary treatment, which causes harm. Adoption of pressure guidewires to assist in revascularization decision making, whether by FFR or iFR, is still limited in interventional cardiology. The priority in the field now should be to encourage more widespread use of this technology, regardless of which method is selected, he argued.
“The biggest room in the world is the room for improvement,” the cardiologist mused.
“I think one of the real problems that’s impeding adoption of physiologic testing is that many physicians are still afraid of leaving a stenosis without treatment,” he continued. “It’s strange: If you perform angioplasty and it wasn’t indicated and there is a complication, physicians seem to have some type of peace of mind that they did their best and they were trying to help the patient. That’s why it’s so important to establish that deferring revascularization – not treating when it is not needed – is safe.”
The DEFINE FLAIR and iFR SWEDEHEART studies were funded by unrestricted grants from Philips Volcano. Dr. Escaned reported serving as a consultant to Abbott, AstraZeneca, Biosensors, Boston Scientific, Medtronic, OrbusNeich, and Philips Healthcare.
Paris – One-year outcomes were significantly worse in patients with acute coronary syndrome whose revascularization was deferred based upon the results of fractional flow reserve than with instantaneous wave-free ratio, in the largest-ever study of patients whose revascularization decision was guided by physiologic measurements obtained via a pressure guidewire.
“The hypothesis that some authors have put forth – that in an ACS the hyperemic response of the myocardium is blunted by the ACS, and that this will affect the FFR hyperemic index – is now strengthened,” Javier Escaned, MD, said in presenting the study results at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
The study was a pooled, patient-level meta-analysis of the 4,529 participants with angiographically determined intermediate-risk stenoses in the previously reported randomized DEFINE FLAIR (N Engl J Med. 2017 May 11;376[19]:1824-34) and iFR SWEDEHEART (N Engl J Med. 2017 May 11;376[19]:1813-23) studies. The primary endpoint was the composite of death, nonfatal MI, or unplanned coronary revascularization within 12 months. And while the analysis brought unwelcome news for proponents of FFR with regard to the subset of patients with ACS, such patients comprised only 17% of the total study population.
“I think that overall these results are very reassuring. The big finding is that we have dramatically improved the safety of deferral of revascularization using pressure guidewires. If you look at the MACE [major adverse cardiovascular event] rate in the deferred ACS group, it was about 6%, which is much less than the event rate at 1 year with deferral in patients with stable coronary disease in the pivotal DEFER trial [Circulation. 2001 Jun 19;103(24):2928-34], which was our former standard,” observed Dr. Escaned, an interventional cardiologist at San Carlos Hospital in Madrid and a DEFER coinvestigator.
He attributed these greatly improved outcomes of physiologically guided revascularization during the past 15 years to vastly improved stent technology and more effective optimal medical management.
Among the key findings of the combined analysis of DEFINE FLAIR and iFR SWEDEHEART:
• More patients were deferred from PCI when iFR was used for decision-making: 50%, compared with 45% in the FFR arm. Yet 1-year outcomes were as good in the deferred iFR group as in the FFR group overall, and better than with FFR in the deferred ACS patients.
• Event rates were significantly higher in deferred ACS patients overall than in deferred patients with stable coronary disease: 5.9% versus 3.6%. But the deferral tool made a difference: When iFR was utilized, the 1-year event rate was 5.4% in deferred ACS patients, not significantly different from the 3.8% rate in deferred patients with stable coronary disease. In contrast, the event rate in ACS patients with FFR-based deferral was 6.4%, significantly higher than the 3.4% rate in FFR-deferred patients with stable coronary disease.
Dr. Escaned noted that this finding is consistent with the cautionary results of several recent studies, including one, albeit tenfold smaller, in which ACS patients in whom revascularization was deferred based on FFR had a 25% rate of major adverse cardiovascular events at 3.4 years, compared with a 12% rate in patients with stable coronary disease (J Am Coll Cardiol. 2016 Sep 13;68[11]:1181-91).
Discussant Peter Jüni, MD, professor of medicine at the University of Toronto, said “the main results of your study show in a completely waterproof fashion that there is no signal of harm with the experimental strategy” of deferred revascularization based on physiologic measurements, at least in patients with stable ischemic coronary disease.
The results, however, also raise the question of whether physiology-based revascularization decision-making in ACS patients is the best strategy.
“Considering that the event rate in the deferred ACS group was nearly twice as high compared with stable patients, my question to you is: Should we ignore any functional testing in ACS patients and just say, ‘Let’s move forward with revascularization because this clinical presentation is a very good clinical characteristic for risk stratification?’ ”
Dr. Escaned rejected that option. He noted that both the European and U.S. guidelines now state that it’s inappropriate to base a revascularization decision solely on a coronary vessel’s angiographic appearance, because that has been shown to result in unnecessary treatment, which causes harm. Adoption of pressure guidewires to assist in revascularization decision making, whether by FFR or iFR, is still limited in interventional cardiology. The priority in the field now should be to encourage more widespread use of this technology, regardless of which method is selected, he argued.
“The biggest room in the world is the room for improvement,” the cardiologist mused.
“I think one of the real problems that’s impeding adoption of physiologic testing is that many physicians are still afraid of leaving a stenosis without treatment,” he continued. “It’s strange: If you perform angioplasty and it wasn’t indicated and there is a complication, physicians seem to have some type of peace of mind that they did their best and they were trying to help the patient. That’s why it’s so important to establish that deferring revascularization – not treating when it is not needed – is safe.”
The DEFINE FLAIR and iFR SWEDEHEART studies were funded by unrestricted grants from Philips Volcano. Dr. Escaned reported serving as a consultant to Abbott, AstraZeneca, Biosensors, Boston Scientific, Medtronic, OrbusNeich, and Philips Healthcare.
Paris – One-year outcomes were significantly worse in patients with acute coronary syndrome whose revascularization was deferred based upon the results of fractional flow reserve than with instantaneous wave-free ratio, in the largest-ever study of patients whose revascularization decision was guided by physiologic measurements obtained via a pressure guidewire.
“The hypothesis that some authors have put forth – that in an ACS the hyperemic response of the myocardium is blunted by the ACS, and that this will affect the FFR hyperemic index – is now strengthened,” Javier Escaned, MD, said in presenting the study results at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
The study was a pooled, patient-level meta-analysis of the 4,529 participants with angiographically determined intermediate-risk stenoses in the previously reported randomized DEFINE FLAIR (N Engl J Med. 2017 May 11;376[19]:1824-34) and iFR SWEDEHEART (N Engl J Med. 2017 May 11;376[19]:1813-23) studies. The primary endpoint was the composite of death, nonfatal MI, or unplanned coronary revascularization within 12 months. And while the analysis brought unwelcome news for proponents of FFR with regard to the subset of patients with ACS, such patients comprised only 17% of the total study population.
“I think that overall these results are very reassuring. The big finding is that we have dramatically improved the safety of deferral of revascularization using pressure guidewires. If you look at the MACE [major adverse cardiovascular event] rate in the deferred ACS group, it was about 6%, which is much less than the event rate at 1 year with deferral in patients with stable coronary disease in the pivotal DEFER trial [Circulation. 2001 Jun 19;103(24):2928-34], which was our former standard,” observed Dr. Escaned, an interventional cardiologist at San Carlos Hospital in Madrid and a DEFER coinvestigator.
He attributed these greatly improved outcomes of physiologically guided revascularization during the past 15 years to vastly improved stent technology and more effective optimal medical management.
Among the key findings of the combined analysis of DEFINE FLAIR and iFR SWEDEHEART:
• More patients were deferred from PCI when iFR was used for decision-making: 50%, compared with 45% in the FFR arm. Yet 1-year outcomes were as good in the deferred iFR group as in the FFR group overall, and better than with FFR in the deferred ACS patients.
• Event rates were significantly higher in deferred ACS patients overall than in deferred patients with stable coronary disease: 5.9% versus 3.6%. But the deferral tool made a difference: When iFR was utilized, the 1-year event rate was 5.4% in deferred ACS patients, not significantly different from the 3.8% rate in deferred patients with stable coronary disease. In contrast, the event rate in ACS patients with FFR-based deferral was 6.4%, significantly higher than the 3.4% rate in FFR-deferred patients with stable coronary disease.
Dr. Escaned noted that this finding is consistent with the cautionary results of several recent studies, including one, albeit tenfold smaller, in which ACS patients in whom revascularization was deferred based on FFR had a 25% rate of major adverse cardiovascular events at 3.4 years, compared with a 12% rate in patients with stable coronary disease (J Am Coll Cardiol. 2016 Sep 13;68[11]:1181-91).
Discussant Peter Jüni, MD, professor of medicine at the University of Toronto, said “the main results of your study show in a completely waterproof fashion that there is no signal of harm with the experimental strategy” of deferred revascularization based on physiologic measurements, at least in patients with stable ischemic coronary disease.
The results, however, also raise the question of whether physiology-based revascularization decision-making in ACS patients is the best strategy.
“Considering that the event rate in the deferred ACS group was nearly twice as high compared with stable patients, my question to you is: Should we ignore any functional testing in ACS patients and just say, ‘Let’s move forward with revascularization because this clinical presentation is a very good clinical characteristic for risk stratification?’ ”
Dr. Escaned rejected that option. He noted that both the European and U.S. guidelines now state that it’s inappropriate to base a revascularization decision solely on a coronary vessel’s angiographic appearance, because that has been shown to result in unnecessary treatment, which causes harm. Adoption of pressure guidewires to assist in revascularization decision making, whether by FFR or iFR, is still limited in interventional cardiology. The priority in the field now should be to encourage more widespread use of this technology, regardless of which method is selected, he argued.
“The biggest room in the world is the room for improvement,” the cardiologist mused.
“I think one of the real problems that’s impeding adoption of physiologic testing is that many physicians are still afraid of leaving a stenosis without treatment,” he continued. “It’s strange: If you perform angioplasty and it wasn’t indicated and there is a complication, physicians seem to have some type of peace of mind that they did their best and they were trying to help the patient. That’s why it’s so important to establish that deferring revascularization – not treating when it is not needed – is safe.”
The DEFINE FLAIR and iFR SWEDEHEART studies were funded by unrestricted grants from Philips Volcano. Dr. Escaned reported serving as a consultant to Abbott, AstraZeneca, Biosensors, Boston Scientific, Medtronic, OrbusNeich, and Philips Healthcare.
AT EUROPCR
Key clinical point:
Major finding: In patients with acute coronary syndrome, the 1-year adverse event rate in patients with FFR-based deferral was 6.4%, significantly higher than the 3.4% rate in patients with FFR-based deferral with stable coronary disease.
Data source: A pooled patient-level meta-analysis of the 4,529 participants with angiographically intermediate-risk stenoses in two previously reported randomized trials of physiologic assessment of lesions by fractional flow reserve or instantaneous wave-free ratio.
Disclosures: The DEFINE FLAIR and iFR SWEDEHEART studies were funded by unrestricted grants from Philips Volcano. The presenter reported serving as a consultant to Abbott, AstraZeneca, Biosensors, Boston Scientific, Medtronic, OrbusNeich, and Philips Healthcare.
Fewer early neurologic complications with TAVR than SAVR
PARIS – Patients with severe aortic stenosis at intermediate operative risk have a significantly lower 30-day risk of stroke and other neurologic complications with transcatheter aortic valve replacement than with surgical replacement, according to new results from the landmark SURTAVI trial.
“This is the first time the stroke rate has been shown to be lower with TAVR than with surgery,” A. Pieter Kappetein, MD, noted in presenting the results at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
It’s a finding that adds to the momentum for studying TAVR in low-surgical-risk patients, he added.
“As we move toward lower-risk patients it will become even more important to see whether there is a difference in stroke. Suppose the stroke rate in SURTAVI had been a little higher with TAVR than SAVR? It would really make us more cautious about moving toward lower-risk patients. Now that we see that in intermediate-risk patients the stroke rate is actually a little bit lower than with surgery, I think we’ll feel more comfortable moving toward lower-risk patients,” according to Dr. Kappetein, professor of cardiothoracic surgery at Erasmus University Medical Center in Rotterdam.
SURTAVI (Surgical Replacement and Transcatheter Aortic Valve Implantation) involved randomization of 1,660 patients with severe symptomatic aortic stenosis to TAVR or SAVR. All participants were deemed to be at intermediate operative risk based upon a predicted surgical mortality of 3%-15%. The primary outcome -- a composite of all-cause mortality and disabling stroke at 2 years -- was presented at the 2017 meeting of the American College of Cardiology and simultaneously published (N Engl J Med. 2017 Apr 6;376(14):1321-1331). The rate was 12.6% with TAVR using the self-expanding CoreValve or Evolut R bioprosthesis and noninferior at 14% with SAVR.
Dr. Kappetein presented a prespecified secondary analysis of the 30-day rate of all neurologic complications, including nondisabling strokes and encephalopathy. He and the other SURTAVI organizers felt this was an important outcome because these early neurologic events have a major impact upon quality of life, including whether a patient will be discharged home or to a rehabilitation clinic or skilled nursing facility following aortic valve replacement.
The 30-day incidence of all stroke was 3.3% in the TAVR patients, significantly lower than the 5.4% rate in the SAVR group. By the 2-year mark, however, the difference was no longer statistically significant, with a rate of 6.3% in the TAVR group compared with 8.0% with SAVR.
Ninety-five percent of the early strokes were ischemic.
The 30-day incidence of disabling stroke was 1.2% with TAVR and 2.4% with SAVR, a difference that was not significant (P=0.057). The 2-year rate was 2.4% in the TAVR arm and 4.5% with SAVR, again not significantly different.
Half of the strokes in the TAVR group had a modified Rankin score of 0-1 at 30 days, meaning no or only minimal signs of stroke. In contrast, most of the strokes in the SAVR group were disabling, with a modified Rankin score of 2-6.
Only 36% of patients who had an early stroke were discharged home, compared with 87% of patients without a stroke. Not surprisingly, quality of life as assessed using the SF-36 physical summary was significantly worse in the early-stroke group. However, with or without stroke, TAVR patients recovered quality of life faster than SAVR patients.
He noted that the timing of the early strokes differed between the two groups. The great majority of both disabling and nondisabling strokes in the TAVR patients were periprocedural, occurring on the day of TAVR or the next day. Strokes in the SAVR group occurred then as well, but also on days 2-6.
One reason why SURTAVI is the first study to show a lower stroke risk with TAVR is that it was the first TAVR-versus-SAVR study to feature comprehensive neurologic testing pre- and post-procedure, along with evaluation of all suspected events by a neurologist or stroke specialist, according to Dr. Kappetein.
“As surgeons we all have said the stroke rate after SAVR is 1%-1.5%, but only when the patient wasn’t waving to us the next morning would we say, ‘Oh, that patient may have a stroke.’ Then we would call a neurologist. So there were many more subtle strokes that we never actually detected. If you do a proper examination of the patient before and after the procedure you’ll find many more strokes,” he said.
He and his coinvestigators systematically searched in vain for predictors of increased stroke risk among the TAVR and SAVR patients.
“Actually, the stroke risk is present for every patient we treat with TAVR or SAVR,” the surgeon continued.
However, discussant Adnan Kastrati, MD, chief physician at the German Heart Center in Munich, thought he spied in the SURTAVI data a potential opportunity to reduce early strokes in SAVR patients. He noted that new-onset atrial fibrillation is consistently more common in SAVR than TAVR patients, and that many of the strokes in the SAVR group occurred on days 2-6. When do heart surgeons typically start oral anticoagulation in their patients with postsurgical atrial fibrillation? he asked.
Not until after 48 hours, Dr. Kappetein replied.
“Those strokes on days 4, 5, and 6 might have to do with atrial fibrillation, and we may need to be more aggressive as surgeons in anticoagulating patients with atrial fibrillation after surgery,” he said.
Dr. Kappetein reported receiving research grant support from Medtronic, sponsor of SURTAVI.
This article was updated July 28, 2107.
PARIS – Patients with severe aortic stenosis at intermediate operative risk have a significantly lower 30-day risk of stroke and other neurologic complications with transcatheter aortic valve replacement than with surgical replacement, according to new results from the landmark SURTAVI trial.
“This is the first time the stroke rate has been shown to be lower with TAVR than with surgery,” A. Pieter Kappetein, MD, noted in presenting the results at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
It’s a finding that adds to the momentum for studying TAVR in low-surgical-risk patients, he added.
“As we move toward lower-risk patients it will become even more important to see whether there is a difference in stroke. Suppose the stroke rate in SURTAVI had been a little higher with TAVR than SAVR? It would really make us more cautious about moving toward lower-risk patients. Now that we see that in intermediate-risk patients the stroke rate is actually a little bit lower than with surgery, I think we’ll feel more comfortable moving toward lower-risk patients,” according to Dr. Kappetein, professor of cardiothoracic surgery at Erasmus University Medical Center in Rotterdam.
SURTAVI (Surgical Replacement and Transcatheter Aortic Valve Implantation) involved randomization of 1,660 patients with severe symptomatic aortic stenosis to TAVR or SAVR. All participants were deemed to be at intermediate operative risk based upon a predicted surgical mortality of 3%-15%. The primary outcome -- a composite of all-cause mortality and disabling stroke at 2 years -- was presented at the 2017 meeting of the American College of Cardiology and simultaneously published (N Engl J Med. 2017 Apr 6;376(14):1321-1331). The rate was 12.6% with TAVR using the self-expanding CoreValve or Evolut R bioprosthesis and noninferior at 14% with SAVR.
Dr. Kappetein presented a prespecified secondary analysis of the 30-day rate of all neurologic complications, including nondisabling strokes and encephalopathy. He and the other SURTAVI organizers felt this was an important outcome because these early neurologic events have a major impact upon quality of life, including whether a patient will be discharged home or to a rehabilitation clinic or skilled nursing facility following aortic valve replacement.
The 30-day incidence of all stroke was 3.3% in the TAVR patients, significantly lower than the 5.4% rate in the SAVR group. By the 2-year mark, however, the difference was no longer statistically significant, with a rate of 6.3% in the TAVR group compared with 8.0% with SAVR.
Ninety-five percent of the early strokes were ischemic.
The 30-day incidence of disabling stroke was 1.2% with TAVR and 2.4% with SAVR, a difference that was not significant (P=0.057). The 2-year rate was 2.4% in the TAVR arm and 4.5% with SAVR, again not significantly different.
Half of the strokes in the TAVR group had a modified Rankin score of 0-1 at 30 days, meaning no or only minimal signs of stroke. In contrast, most of the strokes in the SAVR group were disabling, with a modified Rankin score of 2-6.
Only 36% of patients who had an early stroke were discharged home, compared with 87% of patients without a stroke. Not surprisingly, quality of life as assessed using the SF-36 physical summary was significantly worse in the early-stroke group. However, with or without stroke, TAVR patients recovered quality of life faster than SAVR patients.
He noted that the timing of the early strokes differed between the two groups. The great majority of both disabling and nondisabling strokes in the TAVR patients were periprocedural, occurring on the day of TAVR or the next day. Strokes in the SAVR group occurred then as well, but also on days 2-6.
One reason why SURTAVI is the first study to show a lower stroke risk with TAVR is that it was the first TAVR-versus-SAVR study to feature comprehensive neurologic testing pre- and post-procedure, along with evaluation of all suspected events by a neurologist or stroke specialist, according to Dr. Kappetein.
“As surgeons we all have said the stroke rate after SAVR is 1%-1.5%, but only when the patient wasn’t waving to us the next morning would we say, ‘Oh, that patient may have a stroke.’ Then we would call a neurologist. So there were many more subtle strokes that we never actually detected. If you do a proper examination of the patient before and after the procedure you’ll find many more strokes,” he said.
He and his coinvestigators systematically searched in vain for predictors of increased stroke risk among the TAVR and SAVR patients.
“Actually, the stroke risk is present for every patient we treat with TAVR or SAVR,” the surgeon continued.
However, discussant Adnan Kastrati, MD, chief physician at the German Heart Center in Munich, thought he spied in the SURTAVI data a potential opportunity to reduce early strokes in SAVR patients. He noted that new-onset atrial fibrillation is consistently more common in SAVR than TAVR patients, and that many of the strokes in the SAVR group occurred on days 2-6. When do heart surgeons typically start oral anticoagulation in their patients with postsurgical atrial fibrillation? he asked.
Not until after 48 hours, Dr. Kappetein replied.
“Those strokes on days 4, 5, and 6 might have to do with atrial fibrillation, and we may need to be more aggressive as surgeons in anticoagulating patients with atrial fibrillation after surgery,” he said.
Dr. Kappetein reported receiving research grant support from Medtronic, sponsor of SURTAVI.
This article was updated July 28, 2107.
PARIS – Patients with severe aortic stenosis at intermediate operative risk have a significantly lower 30-day risk of stroke and other neurologic complications with transcatheter aortic valve replacement than with surgical replacement, according to new results from the landmark SURTAVI trial.
“This is the first time the stroke rate has been shown to be lower with TAVR than with surgery,” A. Pieter Kappetein, MD, noted in presenting the results at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
It’s a finding that adds to the momentum for studying TAVR in low-surgical-risk patients, he added.
“As we move toward lower-risk patients it will become even more important to see whether there is a difference in stroke. Suppose the stroke rate in SURTAVI had been a little higher with TAVR than SAVR? It would really make us more cautious about moving toward lower-risk patients. Now that we see that in intermediate-risk patients the stroke rate is actually a little bit lower than with surgery, I think we’ll feel more comfortable moving toward lower-risk patients,” according to Dr. Kappetein, professor of cardiothoracic surgery at Erasmus University Medical Center in Rotterdam.
SURTAVI (Surgical Replacement and Transcatheter Aortic Valve Implantation) involved randomization of 1,660 patients with severe symptomatic aortic stenosis to TAVR or SAVR. All participants were deemed to be at intermediate operative risk based upon a predicted surgical mortality of 3%-15%. The primary outcome -- a composite of all-cause mortality and disabling stroke at 2 years -- was presented at the 2017 meeting of the American College of Cardiology and simultaneously published (N Engl J Med. 2017 Apr 6;376(14):1321-1331). The rate was 12.6% with TAVR using the self-expanding CoreValve or Evolut R bioprosthesis and noninferior at 14% with SAVR.
Dr. Kappetein presented a prespecified secondary analysis of the 30-day rate of all neurologic complications, including nondisabling strokes and encephalopathy. He and the other SURTAVI organizers felt this was an important outcome because these early neurologic events have a major impact upon quality of life, including whether a patient will be discharged home or to a rehabilitation clinic or skilled nursing facility following aortic valve replacement.
The 30-day incidence of all stroke was 3.3% in the TAVR patients, significantly lower than the 5.4% rate in the SAVR group. By the 2-year mark, however, the difference was no longer statistically significant, with a rate of 6.3% in the TAVR group compared with 8.0% with SAVR.
Ninety-five percent of the early strokes were ischemic.
The 30-day incidence of disabling stroke was 1.2% with TAVR and 2.4% with SAVR, a difference that was not significant (P=0.057). The 2-year rate was 2.4% in the TAVR arm and 4.5% with SAVR, again not significantly different.
Half of the strokes in the TAVR group had a modified Rankin score of 0-1 at 30 days, meaning no or only minimal signs of stroke. In contrast, most of the strokes in the SAVR group were disabling, with a modified Rankin score of 2-6.
Only 36% of patients who had an early stroke were discharged home, compared with 87% of patients without a stroke. Not surprisingly, quality of life as assessed using the SF-36 physical summary was significantly worse in the early-stroke group. However, with or without stroke, TAVR patients recovered quality of life faster than SAVR patients.
He noted that the timing of the early strokes differed between the two groups. The great majority of both disabling and nondisabling strokes in the TAVR patients were periprocedural, occurring on the day of TAVR or the next day. Strokes in the SAVR group occurred then as well, but also on days 2-6.
One reason why SURTAVI is the first study to show a lower stroke risk with TAVR is that it was the first TAVR-versus-SAVR study to feature comprehensive neurologic testing pre- and post-procedure, along with evaluation of all suspected events by a neurologist or stroke specialist, according to Dr. Kappetein.
“As surgeons we all have said the stroke rate after SAVR is 1%-1.5%, but only when the patient wasn’t waving to us the next morning would we say, ‘Oh, that patient may have a stroke.’ Then we would call a neurologist. So there were many more subtle strokes that we never actually detected. If you do a proper examination of the patient before and after the procedure you’ll find many more strokes,” he said.
He and his coinvestigators systematically searched in vain for predictors of increased stroke risk among the TAVR and SAVR patients.
“Actually, the stroke risk is present for every patient we treat with TAVR or SAVR,” the surgeon continued.
However, discussant Adnan Kastrati, MD, chief physician at the German Heart Center in Munich, thought he spied in the SURTAVI data a potential opportunity to reduce early strokes in SAVR patients. He noted that new-onset atrial fibrillation is consistently more common in SAVR than TAVR patients, and that many of the strokes in the SAVR group occurred on days 2-6. When do heart surgeons typically start oral anticoagulation in their patients with postsurgical atrial fibrillation? he asked.
Not until after 48 hours, Dr. Kappetein replied.
“Those strokes on days 4, 5, and 6 might have to do with atrial fibrillation, and we may need to be more aggressive as surgeons in anticoagulating patients with atrial fibrillation after surgery,” he said.
Dr. Kappetein reported receiving research grant support from Medtronic, sponsor of SURTAVI.
This article was updated July 28, 2107.
AT EUROPCR
Key clinical point:
Major finding: The combined incidence of disabling and nondisabling stroke within 30 days of TAVR was 3.3%, significantly better than the 5.4% rate in patients who underwent SAVR.
Data source: SURTAVI was a multicenter trial which included 1,660 patients with severe aortic stenosis who were at intermediate operative risk and were randomized to TAVR or SAVR.
Disclosures: The study presenter reported receiving research grant support from Medtronic, sponsor of the SURTAVI trial.
Low-dose aspirin bests dual-antiplatelet therapy in TAVR
PARIS – Single-antiplatelet therapy with low-dose aspirin following transcatheter aortic valve replacement (TAVR) reduced the occurrence of major adverse events, compared with guideline-recommended dual-antiplatelet therapy (DAPT), in the randomized ARTE trial.
The TAVR guideline recommendation for DAPT with low-dose aspirin plus clopidogrel is not based on evidence. It relies on expert opinion. ARTE (Aspirin Versus Aspirin + Clopidogrel Following TAVR) is the first sizable randomized trial to address the safety and efficacy of aspirin alone versus DAPT in the setting of TAVR, Josep Rodés-Cabau, MD, noted in presenting the ARTE results at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.

ARTE was a multicenter, prospective, international open-label study of 222 TAVR patients who were randomized to 3 months of single-antiplatelet therapy (SAPT) with aspirin at 80-100 mg/day or to DAPT with aspirin at 80-100 mg/day plus clopidogrel at 75 mg/day after a single 300-mg loading dose. Participants had a mean Society of Thoracic Surgery Predicted Risk of Mortality score of 6.3%. The vast majority of participants received the balloon-expandable Edwards Lifesciences Sapien XT valve. The remainder got the Sapien 3 valve.
The primary outcome was the 3-month composite of death, MI, major or life-threatening bleeding, or stroke or transient ischemic attack. It occurred in 15.3% of the DAPT group and 7.2% on SAPT, a difference that didn’t reach statistical significance (P = .065) because of small patient numbers.
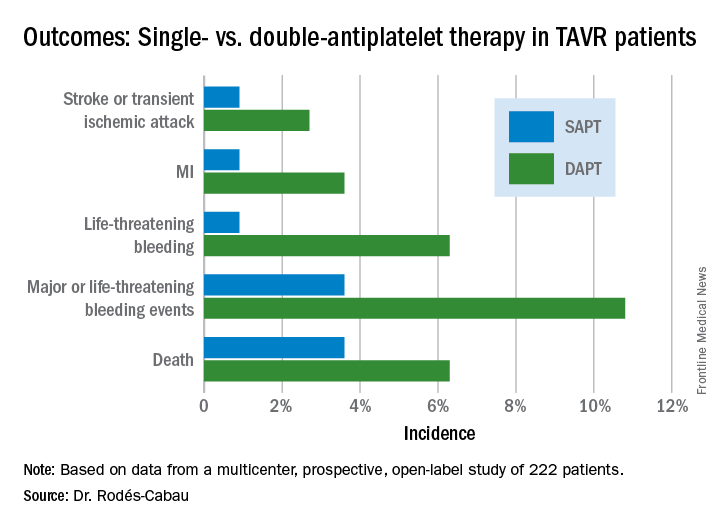
All subjects were on a proton pump inhibitor. The type, timing, and severity of bleeding events differed between the two study arms. All 4 bleeding events in the SAPT group were vascular in nature, while 5 of the 12 in the DAPT group were gastrointestinal. All the bleeding events in the SAPT group occurred within 72 hours after TAVR, whereas 5 of 12 in the DAPT recipients occurred later. Only one patient on SAPT experienced life-threatening bleeding, compared with seven DAPT patients who did.
“There were two prior smaller studies before ours,” according to Dr. Rodés-Cabau of Laval University in Quebec City. “One showed no differences, and an Italian one showed a tendency toward more bleeding with DAPT. So, I think there has been no sign to date that adding clopidogrel protects this group of patients from anything.”
Discussant Luis Nombela-Franco, MD, an interventional cardiologist at San Carlos Hospital in Madrid, pronounced the ARTE trial guideline-changing despite its limitations.
ARTE was supported by grants from Edwards Lifesciences and the Quebec Heart and Lung Institute.
Simultaneous with Dr. Rodés-Cabau’s presentation in Paris, the ARTE trial was published online (JACC Cardiovasc Interv. 2017 May 11. pii: S1936-8798[17]30812-9).

PARIS – Single-antiplatelet therapy with low-dose aspirin following transcatheter aortic valve replacement (TAVR) reduced the occurrence of major adverse events, compared with guideline-recommended dual-antiplatelet therapy (DAPT), in the randomized ARTE trial.
The TAVR guideline recommendation for DAPT with low-dose aspirin plus clopidogrel is not based on evidence. It relies on expert opinion. ARTE (Aspirin Versus Aspirin + Clopidogrel Following TAVR) is the first sizable randomized trial to address the safety and efficacy of aspirin alone versus DAPT in the setting of TAVR, Josep Rodés-Cabau, MD, noted in presenting the ARTE results at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
ARTE was a multicenter, prospective, international open-label study of 222 TAVR patients who were randomized to 3 months of single-antiplatelet therapy (SAPT) with aspirin at 80-100 mg/day or to DAPT with aspirin at 80-100 mg/day plus clopidogrel at 75 mg/day after a single 300-mg loading dose. Participants had a mean Society of Thoracic Surgery Predicted Risk of Mortality score of 6.3%. The vast majority of participants received the balloon-expandable Edwards Lifesciences Sapien XT valve. The remainder got the Sapien 3 valve.
The primary outcome was the 3-month composite of death, MI, major or life-threatening bleeding, or stroke or transient ischemic attack. It occurred in 15.3% of the DAPT group and 7.2% on SAPT, a difference that didn’t reach statistical significance (P = .065) because of small patient numbers.
All subjects were on a proton pump inhibitor. The type, timing, and severity of bleeding events differed between the two study arms. All 4 bleeding events in the SAPT group were vascular in nature, while 5 of the 12 in the DAPT group were gastrointestinal. All the bleeding events in the SAPT group occurred within 72 hours after TAVR, whereas 5 of 12 in the DAPT recipients occurred later. Only one patient on SAPT experienced life-threatening bleeding, compared with seven DAPT patients who did.
“There were two prior smaller studies before ours,” according to Dr. Rodés-Cabau of Laval University in Quebec City. “One showed no differences, and an Italian one showed a tendency toward more bleeding with DAPT. So, I think there has been no sign to date that adding clopidogrel protects this group of patients from anything.”
Discussant Luis Nombela-Franco, MD, an interventional cardiologist at San Carlos Hospital in Madrid, pronounced the ARTE trial guideline-changing despite its limitations.
ARTE was supported by grants from Edwards Lifesciences and the Quebec Heart and Lung Institute.
Simultaneous with Dr. Rodés-Cabau’s presentation in Paris, the ARTE trial was published online (JACC Cardiovasc Interv. 2017 May 11. pii: S1936-8798[17]30812-9).
PARIS – Single-antiplatelet therapy with low-dose aspirin following transcatheter aortic valve replacement (TAVR) reduced the occurrence of major adverse events, compared with guideline-recommended dual-antiplatelet therapy (DAPT), in the randomized ARTE trial.
The TAVR guideline recommendation for DAPT with low-dose aspirin plus clopidogrel is not based on evidence. It relies on expert opinion. ARTE (Aspirin Versus Aspirin + Clopidogrel Following TAVR) is the first sizable randomized trial to address the safety and efficacy of aspirin alone versus DAPT in the setting of TAVR, Josep Rodés-Cabau, MD, noted in presenting the ARTE results at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
ARTE was a multicenter, prospective, international open-label study of 222 TAVR patients who were randomized to 3 months of single-antiplatelet therapy (SAPT) with aspirin at 80-100 mg/day or to DAPT with aspirin at 80-100 mg/day plus clopidogrel at 75 mg/day after a single 300-mg loading dose. Participants had a mean Society of Thoracic Surgery Predicted Risk of Mortality score of 6.3%. The vast majority of participants received the balloon-expandable Edwards Lifesciences Sapien XT valve. The remainder got the Sapien 3 valve.
The primary outcome was the 3-month composite of death, MI, major or life-threatening bleeding, or stroke or transient ischemic attack. It occurred in 15.3% of the DAPT group and 7.2% on SAPT, a difference that didn’t reach statistical significance (P = .065) because of small patient numbers.
All subjects were on a proton pump inhibitor. The type, timing, and severity of bleeding events differed between the two study arms. All 4 bleeding events in the SAPT group were vascular in nature, while 5 of the 12 in the DAPT group were gastrointestinal. All the bleeding events in the SAPT group occurred within 72 hours after TAVR, whereas 5 of 12 in the DAPT recipients occurred later. Only one patient on SAPT experienced life-threatening bleeding, compared with seven DAPT patients who did.
“There were two prior smaller studies before ours,” according to Dr. Rodés-Cabau of Laval University in Quebec City. “One showed no differences, and an Italian one showed a tendency toward more bleeding with DAPT. So, I think there has been no sign to date that adding clopidogrel protects this group of patients from anything.”
Discussant Luis Nombela-Franco, MD, an interventional cardiologist at San Carlos Hospital in Madrid, pronounced the ARTE trial guideline-changing despite its limitations.
ARTE was supported by grants from Edwards Lifesciences and the Quebec Heart and Lung Institute.
Simultaneous with Dr. Rodés-Cabau’s presentation in Paris, the ARTE trial was published online (JACC Cardiovasc Interv. 2017 May 11. pii: S1936-8798[17]30812-9).
AT EUROPCR
Key clinical point:
Major finding: The 3-month composite of death, MI, major or life-threatening bleeding, or stroke or transient ischemic attack occurred in 15.3% of TAVR patients randomized to DAPT with low-dose aspirin plus clopidogrel, compared with 7.2% on aspirin only.
Data source: A randomized, multicenter, international, prospective open-label trial in 222 TAVR patients.
Disclosures: The presenter reported receiving research grants from Edwards Lifesciences and the Quebec Heart and Lung Institute, which supported the ARTE trial.
Transradial PCI in acute coronary syndrome causes less kidney damage
PARIS – Transradial-access percutaneous coronary intervention (PCI) in patients with acute coronary syndrome (ACS) results in a significantly lower risk of acute kidney injury (AKI), compared with the transfemoral approach, according to a new analysis from the large randomized MATRIX trial.
The results of this prespecified secondary subgroup analysis of MATRIX suggest it’s time to update the classic “five golden rules” for reduction of contrast medium–induced AKI by adding a sixth. “Use a transradial approach,” Bernardo Cortese, MD, said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.

He reported on 8,210 participants in the MATRIX trial (Minimizing Adverse Haemorrhagic Events by Transradial Access Site and Systemic Implementation of Angiox) who were randomized to transradial- or transfemoral-access PCI for non–ST-elevation MI or ST-elevation MI.
The primary results of the 78-site, four-country European study, previously published, showed that transradial PCI reduced the composite risk of death, MI, stroke, or major bleeding by 17%, compared with transfemoral PCI, a benefit mainly driven by a marked reduction in clinically important bleeding (Lancet. 2015 Jun 20;385[9986]:2465-76).
Left unanswered by the primary analysis was the question of whether transradial PCI in ACS patients also reduced AKI risk, as had previously been suggested by a meta-analysis of observational studies (Int J Cardiol. 2015 Jan 20;179:309-11). In designing the MATRIX trial, Dr. Cortese and the other investigators decided to address that issue separately in a prespecified secondary analysis known as AKI-MATRIX. For this purpose, AKI was defined as either a post-PCI in-hospital increase in serum creatinine level of more than 25%, compared with the preangiography baseline, or an absolute increase in serum creatinine of greater than 0.5 mg/dL.
AKI occurred in 15.4% of ACS patients who underwent PCI with transradial access and 17.3% of those randomized to transfemoral access, for a significant 13% relative risk reduction. This was accomplished without any increase in the volume of contrast media required. The average was 200 mL in both study groups.
The reduction in AKI achieved with transradial-access PCI was seen in all patient subgroups, including those at increased AKI risk because of an estimated glomerular filtration rate below 60 mL/min, age 75 or older, Killup class III or IV, or a Mehran score greater than 10.
Dr. Cortese proposed several possible mechanisms for the observed reduction in AKI seen with transradial-access PCI. The major factor in his view is that the transradial approach entails less bleeding, as earlier demonstrated in the primary analysis – and bleeding has been associated with impaired renal perfusion in several prior studies. Also, it’s plausible that the passage of the catheter across the renal arteries during the transfemoral approach dislodges atherosclerotic debris, which then travels down the renal vessels.
The five golden rules for preventing contrast media–induced AKI, he noted, are
1. Discontinue nephrotoxic drugs before the procedure.
2. Identify high-risk patients.
3. Hydrate them.
4. Choose an ideal contrast medium.
5. Adapt the dose of contrast medium to the patient’s specific situation.
Discussant Jacek Legutko, MD, PhD, of Jagiellonian University in Krakow, Poland, said the primary results of the MATRIX trial published in 2015 have had a major impact on Polish interventional cardiology, where transradial PCI is now used in 80% of PCIs. The AKI study results will reinforce this trend, he added.
“You have shown something opposite to what we’ve thought in the past, that maybe, with a radial approach, we would use more contrast medium, which is a risk factor for AKI. In your study – at least in ACS with very experienced transradial operators – there was no increase in contrast volume, and the risk of AKI decreased,” Dr. Legutko said.
Asked about the possibility that transradial PCI might be associated with an increased risk of embolization to the brain, much as the transfemoral approach might cause embolization to the kidneys, Dr. Cortese said there was no significant difference between the two AKI-MATRIX study arms in rates of transient ischemic attack or stroke.
“I did my first transradial PCI in 2003, and I haven’t seen any increase in these events or later dementia,” he added.
The prespecified secondary analysis of the MATRIX trial was conducted without commercial support. The presenter reported serving as a consultant to Abbott, AstraZeneca, Daiichi Sankyo, Eli Lilly, and Stentys.
Simultaneous with his presentation in Paris, the AKI-MATRIX study was published online at www.sciencedirect.com/science/article/pii/S0735109717368973.
PARIS – Transradial-access percutaneous coronary intervention (PCI) in patients with acute coronary syndrome (ACS) results in a significantly lower risk of acute kidney injury (AKI), compared with the transfemoral approach, according to a new analysis from the large randomized MATRIX trial.
The results of this prespecified secondary subgroup analysis of MATRIX suggest it’s time to update the classic “five golden rules” for reduction of contrast medium–induced AKI by adding a sixth. “Use a transradial approach,” Bernardo Cortese, MD, said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
He reported on 8,210 participants in the MATRIX trial (Minimizing Adverse Haemorrhagic Events by Transradial Access Site and Systemic Implementation of Angiox) who were randomized to transradial- or transfemoral-access PCI for non–ST-elevation MI or ST-elevation MI.
The primary results of the 78-site, four-country European study, previously published, showed that transradial PCI reduced the composite risk of death, MI, stroke, or major bleeding by 17%, compared with transfemoral PCI, a benefit mainly driven by a marked reduction in clinically important bleeding (Lancet. 2015 Jun 20;385[9986]:2465-76).
Left unanswered by the primary analysis was the question of whether transradial PCI in ACS patients also reduced AKI risk, as had previously been suggested by a meta-analysis of observational studies (Int J Cardiol. 2015 Jan 20;179:309-11). In designing the MATRIX trial, Dr. Cortese and the other investigators decided to address that issue separately in a prespecified secondary analysis known as AKI-MATRIX. For this purpose, AKI was defined as either a post-PCI in-hospital increase in serum creatinine level of more than 25%, compared with the preangiography baseline, or an absolute increase in serum creatinine of greater than 0.5 mg/dL.
AKI occurred in 15.4% of ACS patients who underwent PCI with transradial access and 17.3% of those randomized to transfemoral access, for a significant 13% relative risk reduction. This was accomplished without any increase in the volume of contrast media required. The average was 200 mL in both study groups.
The reduction in AKI achieved with transradial-access PCI was seen in all patient subgroups, including those at increased AKI risk because of an estimated glomerular filtration rate below 60 mL/min, age 75 or older, Killup class III or IV, or a Mehran score greater than 10.
Dr. Cortese proposed several possible mechanisms for the observed reduction in AKI seen with transradial-access PCI. The major factor in his view is that the transradial approach entails less bleeding, as earlier demonstrated in the primary analysis – and bleeding has been associated with impaired renal perfusion in several prior studies. Also, it’s plausible that the passage of the catheter across the renal arteries during the transfemoral approach dislodges atherosclerotic debris, which then travels down the renal vessels.
The five golden rules for preventing contrast media–induced AKI, he noted, are
1. Discontinue nephrotoxic drugs before the procedure.
2. Identify high-risk patients.
3. Hydrate them.
4. Choose an ideal contrast medium.
5. Adapt the dose of contrast medium to the patient’s specific situation.
Discussant Jacek Legutko, MD, PhD, of Jagiellonian University in Krakow, Poland, said the primary results of the MATRIX trial published in 2015 have had a major impact on Polish interventional cardiology, where transradial PCI is now used in 80% of PCIs. The AKI study results will reinforce this trend, he added.
“You have shown something opposite to what we’ve thought in the past, that maybe, with a radial approach, we would use more contrast medium, which is a risk factor for AKI. In your study – at least in ACS with very experienced transradial operators – there was no increase in contrast volume, and the risk of AKI decreased,” Dr. Legutko said.
Asked about the possibility that transradial PCI might be associated with an increased risk of embolization to the brain, much as the transfemoral approach might cause embolization to the kidneys, Dr. Cortese said there was no significant difference between the two AKI-MATRIX study arms in rates of transient ischemic attack or stroke.
“I did my first transradial PCI in 2003, and I haven’t seen any increase in these events or later dementia,” he added.
The prespecified secondary analysis of the MATRIX trial was conducted without commercial support. The presenter reported serving as a consultant to Abbott, AstraZeneca, Daiichi Sankyo, Eli Lilly, and Stentys.
Simultaneous with his presentation in Paris, the AKI-MATRIX study was published online at www.sciencedirect.com/science/article/pii/S0735109717368973.
PARIS – Transradial-access percutaneous coronary intervention (PCI) in patients with acute coronary syndrome (ACS) results in a significantly lower risk of acute kidney injury (AKI), compared with the transfemoral approach, according to a new analysis from the large randomized MATRIX trial.
The results of this prespecified secondary subgroup analysis of MATRIX suggest it’s time to update the classic “five golden rules” for reduction of contrast medium–induced AKI by adding a sixth. “Use a transradial approach,” Bernardo Cortese, MD, said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
He reported on 8,210 participants in the MATRIX trial (Minimizing Adverse Haemorrhagic Events by Transradial Access Site and Systemic Implementation of Angiox) who were randomized to transradial- or transfemoral-access PCI for non–ST-elevation MI or ST-elevation MI.
The primary results of the 78-site, four-country European study, previously published, showed that transradial PCI reduced the composite risk of death, MI, stroke, or major bleeding by 17%, compared with transfemoral PCI, a benefit mainly driven by a marked reduction in clinically important bleeding (Lancet. 2015 Jun 20;385[9986]:2465-76).
Left unanswered by the primary analysis was the question of whether transradial PCI in ACS patients also reduced AKI risk, as had previously been suggested by a meta-analysis of observational studies (Int J Cardiol. 2015 Jan 20;179:309-11). In designing the MATRIX trial, Dr. Cortese and the other investigators decided to address that issue separately in a prespecified secondary analysis known as AKI-MATRIX. For this purpose, AKI was defined as either a post-PCI in-hospital increase in serum creatinine level of more than 25%, compared with the preangiography baseline, or an absolute increase in serum creatinine of greater than 0.5 mg/dL.
AKI occurred in 15.4% of ACS patients who underwent PCI with transradial access and 17.3% of those randomized to transfemoral access, for a significant 13% relative risk reduction. This was accomplished without any increase in the volume of contrast media required. The average was 200 mL in both study groups.
The reduction in AKI achieved with transradial-access PCI was seen in all patient subgroups, including those at increased AKI risk because of an estimated glomerular filtration rate below 60 mL/min, age 75 or older, Killup class III or IV, or a Mehran score greater than 10.
Dr. Cortese proposed several possible mechanisms for the observed reduction in AKI seen with transradial-access PCI. The major factor in his view is that the transradial approach entails less bleeding, as earlier demonstrated in the primary analysis – and bleeding has been associated with impaired renal perfusion in several prior studies. Also, it’s plausible that the passage of the catheter across the renal arteries during the transfemoral approach dislodges atherosclerotic debris, which then travels down the renal vessels.
The five golden rules for preventing contrast media–induced AKI, he noted, are
1. Discontinue nephrotoxic drugs before the procedure.
2. Identify high-risk patients.
3. Hydrate them.
4. Choose an ideal contrast medium.
5. Adapt the dose of contrast medium to the patient’s specific situation.
Discussant Jacek Legutko, MD, PhD, of Jagiellonian University in Krakow, Poland, said the primary results of the MATRIX trial published in 2015 have had a major impact on Polish interventional cardiology, where transradial PCI is now used in 80% of PCIs. The AKI study results will reinforce this trend, he added.
“You have shown something opposite to what we’ve thought in the past, that maybe, with a radial approach, we would use more contrast medium, which is a risk factor for AKI. In your study – at least in ACS with very experienced transradial operators – there was no increase in contrast volume, and the risk of AKI decreased,” Dr. Legutko said.
Asked about the possibility that transradial PCI might be associated with an increased risk of embolization to the brain, much as the transfemoral approach might cause embolization to the kidneys, Dr. Cortese said there was no significant difference between the two AKI-MATRIX study arms in rates of transient ischemic attack or stroke.
“I did my first transradial PCI in 2003, and I haven’t seen any increase in these events or later dementia,” he added.
The prespecified secondary analysis of the MATRIX trial was conducted without commercial support. The presenter reported serving as a consultant to Abbott, AstraZeneca, Daiichi Sankyo, Eli Lilly, and Stentys.
Simultaneous with his presentation in Paris, the AKI-MATRIX study was published online at www.sciencedirect.com/science/article/pii/S0735109717368973.
AT EUROPCR
Key clinical point:
Major finding: Transradial-access PCI for ACS resulted in a 13% lower risk of acute kidney injury than the transfemoral approach.
Data source: A four-country European randomized trial of transradial- vs. transfemoral-access PCI in more than 8,200 patients with ACS.
Disclosures: This prespecified secondary analysis of the MATRIX trial was conducted without commercial support. The presenter reported serving as a consultant to Abbott, AstraZeneca, Daiichi Sankyo, Eli Lilly, and Stentys.
BIO-RESORT: A mandate to prescreen PCI patients for silent diabetes
PARIS – Undetected diabetes and prediabetes are pervasive in patients undergoing percutaneous coronary intervention, and they’re associated with a sharply increased risk of major adverse cardiovascular events, according to the results of the potentially practice-changing BIO-RESORT Silent Diabetes Study, Clemens von Birgelen, MD, PhD, reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
“Our data support screening PCI all-comers for silent diabetes, which may help identify patients with an increased event risk and improve their therapy,” said Dr. von Birgelen, professor of cardiology at the Thoraxcentrum of Twente, a high-volume center for cardiac interventions in Enschede, the Netherlands.

A substantial one-third of subjects turned out to have abnormal glucose tolerance according to World Health Organization criteria and an International Expert Committee Report (Diabetes Care. 2009 Jul;32[7]:1327-34). In a multivariate analysis, their 1-year rate of the primary study endpoint – target vessel failure, a composite of cardiac death, target vessel-related MI, or target vessel revascularization – was an adjusted 2.2 times greater than in the 788 normoglycemic patients.
Moreover, among the 7% of study participants who met diagnostic criteria for silent diabetes, the risk of target vessel failure was more than 4.4 times greater than in the normoglycemic group.
“To a very great extent, periprocedural MI is the driving force behind this difference that we saw. From a biological point of view, I think that the vulnerability of the vessel in the diabetic or prediabetic patient features more brittle plaque with a higher risk of cholesterol embolization, and with more plaque mass that can be pushed to the side so that side branch vessels can become occluded, leading to periprocedural MI,” he observed.
Glucose metabolism was assessed in all participants by two methods using the conventional cutoffs: a 2-hour oral glucose tolerance test (OGTT), and the combination of fasting plasma glucose and hemoglobin A1c. By OGTT, 7% of patients had silent, previously unrecognized diabetes and another 13% had prediabetes. Using the combination of fasting plasma glucose and HbA1c, a total of 25% of subjects had silent diabetes or prediabetes. Fully 33% of participants had abnormal glucose metabolism by one yardstick or the other.
“What we have seen is there is a group of patients that are missed with either. With the OGTT you don’t see all the diabetics, and with HbA1c and fasting blood glucose you also miss some patients,” said Dr. von Birgelen.
The 1-year cumulative incidence of target vessel failure was 13.2% in patients with silent diabetes as identified by the OGTT and 12.1% in those detected by the alternative method, compared with rates of 2.8% and 3.1%, respectively, in normoglycemic PCI patients. The event rate was 6.1% in patients with prediabetes by OGTT and similar at 5.5% in those found to be prediabetic based on fasting blood glucose and HbA1c, versus rates of 2.8% and 3.1%, respectively, in normoglycemic patients.
“The findings of this study suggest that post-PCI event risk associated with hyperglycemia is a continuum without a clear threshold effect, extending well beyond the threshold that currently defines diabetes,” Dr. von Birgelen said.
Once again, it’s worth emphasizing that the elevated target vessel failure rates seen in patients with abnormal glucose metabolism were due mostly to increased rates of acute MI within the first 24 hours after PCI. The target vessel–related MI rate was 10.3% in patients with silent diabetes, compared with just 1.8% in normoglycemic controls.
Asked what the take-home message for clinicians is from this study, he noted that the Netherlands has a relatively low prevalence of diabetes, and a highly developed primary care medicine system.
“We have a very good one-to-one relationship between the patient and the GP. So if we find 7% silent diabetes and up to one-third of patients with undetected abnormal glucose tolerance in a country with a relatively low prevalence of diabetes, you may expect that in other countries with a higher prevalence and perhaps a less developed primary care system the rate may be much, much higher,” Dr. von Birgelen cautioned.
The implications for the daily clinical practice of interventional cardiology are clear, he continued: “We’ve seen in several trials that the new stents are doing a fantastic job. So if we want to further improve the outcomes in our patients we have to do something else. We should look for subgroups of our PCI patients who have a particularly high risk. And we all realize that diabetics are such a problem, but I think we have shown that the prediabetic patients are also important. So we should identify and pretreat these patients, perhaps with aggressive lipid-lowering therapy during the weeks before a scheduled elective PCI.”
“There are data showing that with aggressive lipid-lowering you might reduce the risk of periprocedural MI,” the cardiologist noted.
As a practical matter, screening via fasting blood glucose and HbA1c is probably the way to go in clinical practice, according to Dr. von Birgelen.
“In this study, we performed the OGTT because it is still considered by many the gold standard. But there is increasing evidence favoring HbA1c data and fasting blood glucose,” he said.
Other possible pre-PCI interventions worthy of consideration in patients found to have previously unsuspected abnormal glucose tolerance might include medical therapy aimed at normalizing glucose metabolism, as well as perhaps resorting to the most potent forms of dual-antiplatelet therapy in patients with stable angina who have impaired glucose tolerance. However, these are possibilities that should be tested in randomized controlled trials before widespread adoption, he added.
The BIO-RESORT Silent Diabetes Study, which will continue for 5 years of post-PCI follow-up, is a prespecified substudy of the previously reported BIO-RESORT trial, which addressed another issue entirely. It was a three-arm, patient-blinded clinical trial comparing 1-year safety and efficacy outcomes in nearly 3,500 PCI patients randomized to PCI with very thin strut biodegradable polymer everolimus- or sirolimus-eluting stents or a durable polymer zotarolimus-eluting stent. Outcomes proved noninferior across the three treatment groups (Lancet. 2016 Nov 26;388[10060]:2607-17).
Dr. von Birgelen observed that the silent diabetes study broke new ground. Prior studies of PCI outcomes in patients with unrecognized diabetes were limited to recipients of plain old balloon angioplasty, bare metal, or first-generation drug-eluting stents. And studies of PCI in patients with unrecognized prediabetes are virtually nonexistent.
As the principal investigator for both the parent BIO-RESORT trial and the silent diabetes substudy, Dr. von Birgelen received research grants from Biotronik, Boston Scientific, and Medtronic, the cosponsors. He applauded the three companies for funding the silent diabetes substudy in the interest of science even though it had no commercial relevance to their stent businesses.
PARIS – Undetected diabetes and prediabetes are pervasive in patients undergoing percutaneous coronary intervention, and they’re associated with a sharply increased risk of major adverse cardiovascular events, according to the results of the potentially practice-changing BIO-RESORT Silent Diabetes Study, Clemens von Birgelen, MD, PhD, reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
“Our data support screening PCI all-comers for silent diabetes, which may help identify patients with an increased event risk and improve their therapy,” said Dr. von Birgelen, professor of cardiology at the Thoraxcentrum of Twente, a high-volume center for cardiac interventions in Enschede, the Netherlands.
A substantial one-third of subjects turned out to have abnormal glucose tolerance according to World Health Organization criteria and an International Expert Committee Report (Diabetes Care. 2009 Jul;32[7]:1327-34). In a multivariate analysis, their 1-year rate of the primary study endpoint – target vessel failure, a composite of cardiac death, target vessel-related MI, or target vessel revascularization – was an adjusted 2.2 times greater than in the 788 normoglycemic patients.
Moreover, among the 7% of study participants who met diagnostic criteria for silent diabetes, the risk of target vessel failure was more than 4.4 times greater than in the normoglycemic group.
“To a very great extent, periprocedural MI is the driving force behind this difference that we saw. From a biological point of view, I think that the vulnerability of the vessel in the diabetic or prediabetic patient features more brittle plaque with a higher risk of cholesterol embolization, and with more plaque mass that can be pushed to the side so that side branch vessels can become occluded, leading to periprocedural MI,” he observed.
Glucose metabolism was assessed in all participants by two methods using the conventional cutoffs: a 2-hour oral glucose tolerance test (OGTT), and the combination of fasting plasma glucose and hemoglobin A1c. By OGTT, 7% of patients had silent, previously unrecognized diabetes and another 13% had prediabetes. Using the combination of fasting plasma glucose and HbA1c, a total of 25% of subjects had silent diabetes or prediabetes. Fully 33% of participants had abnormal glucose metabolism by one yardstick or the other.
“What we have seen is there is a group of patients that are missed with either. With the OGTT you don’t see all the diabetics, and with HbA1c and fasting blood glucose you also miss some patients,” said Dr. von Birgelen.
The 1-year cumulative incidence of target vessel failure was 13.2% in patients with silent diabetes as identified by the OGTT and 12.1% in those detected by the alternative method, compared with rates of 2.8% and 3.1%, respectively, in normoglycemic PCI patients. The event rate was 6.1% in patients with prediabetes by OGTT and similar at 5.5% in those found to be prediabetic based on fasting blood glucose and HbA1c, versus rates of 2.8% and 3.1%, respectively, in normoglycemic patients.
“The findings of this study suggest that post-PCI event risk associated with hyperglycemia is a continuum without a clear threshold effect, extending well beyond the threshold that currently defines diabetes,” Dr. von Birgelen said.
Once again, it’s worth emphasizing that the elevated target vessel failure rates seen in patients with abnormal glucose metabolism were due mostly to increased rates of acute MI within the first 24 hours after PCI. The target vessel–related MI rate was 10.3% in patients with silent diabetes, compared with just 1.8% in normoglycemic controls.
Asked what the take-home message for clinicians is from this study, he noted that the Netherlands has a relatively low prevalence of diabetes, and a highly developed primary care medicine system.
“We have a very good one-to-one relationship between the patient and the GP. So if we find 7% silent diabetes and up to one-third of patients with undetected abnormal glucose tolerance in a country with a relatively low prevalence of diabetes, you may expect that in other countries with a higher prevalence and perhaps a less developed primary care system the rate may be much, much higher,” Dr. von Birgelen cautioned.
The implications for the daily clinical practice of interventional cardiology are clear, he continued: “We’ve seen in several trials that the new stents are doing a fantastic job. So if we want to further improve the outcomes in our patients we have to do something else. We should look for subgroups of our PCI patients who have a particularly high risk. And we all realize that diabetics are such a problem, but I think we have shown that the prediabetic patients are also important. So we should identify and pretreat these patients, perhaps with aggressive lipid-lowering therapy during the weeks before a scheduled elective PCI.”
“There are data showing that with aggressive lipid-lowering you might reduce the risk of periprocedural MI,” the cardiologist noted.
As a practical matter, screening via fasting blood glucose and HbA1c is probably the way to go in clinical practice, according to Dr. von Birgelen.
“In this study, we performed the OGTT because it is still considered by many the gold standard. But there is increasing evidence favoring HbA1c data and fasting blood glucose,” he said.
Other possible pre-PCI interventions worthy of consideration in patients found to have previously unsuspected abnormal glucose tolerance might include medical therapy aimed at normalizing glucose metabolism, as well as perhaps resorting to the most potent forms of dual-antiplatelet therapy in patients with stable angina who have impaired glucose tolerance. However, these are possibilities that should be tested in randomized controlled trials before widespread adoption, he added.
The BIO-RESORT Silent Diabetes Study, which will continue for 5 years of post-PCI follow-up, is a prespecified substudy of the previously reported BIO-RESORT trial, which addressed another issue entirely. It was a three-arm, patient-blinded clinical trial comparing 1-year safety and efficacy outcomes in nearly 3,500 PCI patients randomized to PCI with very thin strut biodegradable polymer everolimus- or sirolimus-eluting stents or a durable polymer zotarolimus-eluting stent. Outcomes proved noninferior across the three treatment groups (Lancet. 2016 Nov 26;388[10060]:2607-17).
Dr. von Birgelen observed that the silent diabetes study broke new ground. Prior studies of PCI outcomes in patients with unrecognized diabetes were limited to recipients of plain old balloon angioplasty, bare metal, or first-generation drug-eluting stents. And studies of PCI in patients with unrecognized prediabetes are virtually nonexistent.
As the principal investigator for both the parent BIO-RESORT trial and the silent diabetes substudy, Dr. von Birgelen received research grants from Biotronik, Boston Scientific, and Medtronic, the cosponsors. He applauded the three companies for funding the silent diabetes substudy in the interest of science even though it had no commercial relevance to their stent businesses.
PARIS – Undetected diabetes and prediabetes are pervasive in patients undergoing percutaneous coronary intervention, and they’re associated with a sharply increased risk of major adverse cardiovascular events, according to the results of the potentially practice-changing BIO-RESORT Silent Diabetes Study, Clemens von Birgelen, MD, PhD, reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
“Our data support screening PCI all-comers for silent diabetes, which may help identify patients with an increased event risk and improve their therapy,” said Dr. von Birgelen, professor of cardiology at the Thoraxcentrum of Twente, a high-volume center for cardiac interventions in Enschede, the Netherlands.
A substantial one-third of subjects turned out to have abnormal glucose tolerance according to World Health Organization criteria and an International Expert Committee Report (Diabetes Care. 2009 Jul;32[7]:1327-34). In a multivariate analysis, their 1-year rate of the primary study endpoint – target vessel failure, a composite of cardiac death, target vessel-related MI, or target vessel revascularization – was an adjusted 2.2 times greater than in the 788 normoglycemic patients.
Moreover, among the 7% of study participants who met diagnostic criteria for silent diabetes, the risk of target vessel failure was more than 4.4 times greater than in the normoglycemic group.
“To a very great extent, periprocedural MI is the driving force behind this difference that we saw. From a biological point of view, I think that the vulnerability of the vessel in the diabetic or prediabetic patient features more brittle plaque with a higher risk of cholesterol embolization, and with more plaque mass that can be pushed to the side so that side branch vessels can become occluded, leading to periprocedural MI,” he observed.
Glucose metabolism was assessed in all participants by two methods using the conventional cutoffs: a 2-hour oral glucose tolerance test (OGTT), and the combination of fasting plasma glucose and hemoglobin A1c. By OGTT, 7% of patients had silent, previously unrecognized diabetes and another 13% had prediabetes. Using the combination of fasting plasma glucose and HbA1c, a total of 25% of subjects had silent diabetes or prediabetes. Fully 33% of participants had abnormal glucose metabolism by one yardstick or the other.
“What we have seen is there is a group of patients that are missed with either. With the OGTT you don’t see all the diabetics, and with HbA1c and fasting blood glucose you also miss some patients,” said Dr. von Birgelen.
The 1-year cumulative incidence of target vessel failure was 13.2% in patients with silent diabetes as identified by the OGTT and 12.1% in those detected by the alternative method, compared with rates of 2.8% and 3.1%, respectively, in normoglycemic PCI patients. The event rate was 6.1% in patients with prediabetes by OGTT and similar at 5.5% in those found to be prediabetic based on fasting blood glucose and HbA1c, versus rates of 2.8% and 3.1%, respectively, in normoglycemic patients.
“The findings of this study suggest that post-PCI event risk associated with hyperglycemia is a continuum without a clear threshold effect, extending well beyond the threshold that currently defines diabetes,” Dr. von Birgelen said.
Once again, it’s worth emphasizing that the elevated target vessel failure rates seen in patients with abnormal glucose metabolism were due mostly to increased rates of acute MI within the first 24 hours after PCI. The target vessel–related MI rate was 10.3% in patients with silent diabetes, compared with just 1.8% in normoglycemic controls.
Asked what the take-home message for clinicians is from this study, he noted that the Netherlands has a relatively low prevalence of diabetes, and a highly developed primary care medicine system.
“We have a very good one-to-one relationship between the patient and the GP. So if we find 7% silent diabetes and up to one-third of patients with undetected abnormal glucose tolerance in a country with a relatively low prevalence of diabetes, you may expect that in other countries with a higher prevalence and perhaps a less developed primary care system the rate may be much, much higher,” Dr. von Birgelen cautioned.
The implications for the daily clinical practice of interventional cardiology are clear, he continued: “We’ve seen in several trials that the new stents are doing a fantastic job. So if we want to further improve the outcomes in our patients we have to do something else. We should look for subgroups of our PCI patients who have a particularly high risk. And we all realize that diabetics are such a problem, but I think we have shown that the prediabetic patients are also important. So we should identify and pretreat these patients, perhaps with aggressive lipid-lowering therapy during the weeks before a scheduled elective PCI.”
“There are data showing that with aggressive lipid-lowering you might reduce the risk of periprocedural MI,” the cardiologist noted.
As a practical matter, screening via fasting blood glucose and HbA1c is probably the way to go in clinical practice, according to Dr. von Birgelen.
“In this study, we performed the OGTT because it is still considered by many the gold standard. But there is increasing evidence favoring HbA1c data and fasting blood glucose,” he said.
Other possible pre-PCI interventions worthy of consideration in patients found to have previously unsuspected abnormal glucose tolerance might include medical therapy aimed at normalizing glucose metabolism, as well as perhaps resorting to the most potent forms of dual-antiplatelet therapy in patients with stable angina who have impaired glucose tolerance. However, these are possibilities that should be tested in randomized controlled trials before widespread adoption, he added.
The BIO-RESORT Silent Diabetes Study, which will continue for 5 years of post-PCI follow-up, is a prespecified substudy of the previously reported BIO-RESORT trial, which addressed another issue entirely. It was a three-arm, patient-blinded clinical trial comparing 1-year safety and efficacy outcomes in nearly 3,500 PCI patients randomized to PCI with very thin strut biodegradable polymer everolimus- or sirolimus-eluting stents or a durable polymer zotarolimus-eluting stent. Outcomes proved noninferior across the three treatment groups (Lancet. 2016 Nov 26;388[10060]:2607-17).
Dr. von Birgelen observed that the silent diabetes study broke new ground. Prior studies of PCI outcomes in patients with unrecognized diabetes were limited to recipients of plain old balloon angioplasty, bare metal, or first-generation drug-eluting stents. And studies of PCI in patients with unrecognized prediabetes are virtually nonexistent.
As the principal investigator for both the parent BIO-RESORT trial and the silent diabetes substudy, Dr. von Birgelen received research grants from Biotronik, Boston Scientific, and Medtronic, the cosponsors. He applauded the three companies for funding the silent diabetes substudy in the interest of science even though it had no commercial relevance to their stent businesses.
AT EUROPCR
Key clinical point:
Major finding: One-third of patients undergoing PCI have unsuspected silent diabetes or prediabetes, placing them at increased risk for major adverse cardiac events.
Data source: This prospective observational study included 988 patients not known to have diabetes who underwent screening for abnormal glucose tolerance 6 weeks after PCI with stenting.
Disclosures: The study was cosponsored by Biotronik, Boston Scientific, and Medtronic.
New DES hailed for smallest coronary vessels
Paris – The first multicenter, prospective trial of a drug-eluting stent designed specifically to treat lesions in coronary vessels less than 2.25 mm in diameter showed excellent outcomes, with a 1-year target lesion failure rate of 5% for the Resolute Onyx 2.0 mm diameter zotarolimus-eluting stent.
This result in the pivotal trial easily surpassed the prespecified performance goal of a 19% target lesion failure rate, Matthew J. Price, MD, reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.

Hemodynamically significant lesions in such small vessels are “not uncommon, particularly in diabetic patients,” Dr. Price said in an interview. Indeed, 47% of patients in the clinical trial had diabetes.
At present, the only ways to treat coronary disease in arteries having a reference vessel diameter less than 2.25 mm are off-label placement of an oversized stent, with its attendant risk of complications; standard balloon angioplasty, which entails a particularly high restenosis rate in this setting; or medical management, the cardiologist noted.
He presented a multicenter, prospective, open-label, single-arm trial of 101 patients with documented ischemia-producing obstructions in coronary arteries having a reference vessel diameter less than 2.25 mm, a lesion length less than 27 mm, and evidence of ischemia attributable to the lesion, typically via fractional flow reserve. The mean diameter by quantitative coronary angiography was 1.91 mm.
The primary endpoint was the rate of target lesion failure at 12 months, a composite comprising cardiac death, target vessel MI, or clinically driven target lesion revascularization. This endpoint occurred in 5% of patients. There was a 3% target vessel MI rate and a 2% target lesion revascularization rate. There were no cardiac deaths.
“Importantly, the stent thrombosis rate in these patients with extremely small vessels was zero,” the cardiologist emphasized.
The mean angiographic in-stent late lumen loss at 13 months was 0.26 mm, which Dr. Price characterized as “quite good.” The in-segment binary angiographic restenosis rate was 20%.
“That’s slightly higher than you would expect to see in vessels with larger reference diameters. I think that’s because of the lack of headroom. You have a very small vessel, and, even with a very small stent, even a small amount of late loss will give you a larger percent diameter restenosis over time,” he explained.
The 19% target lesion failure rate selected as a performance goal in the trial was set somewhat arbitrarily. It wasn’t possible to randomize patients to a comparator arm because there are no approved stents for vessels less than 2.25 mm in diameter. The 19% figure was arrived at in discussion with the Food and Drug Administration on the basis of similarity to the performance goal used in clinical trials to gain approval of 2.25-mm, drug-eluting stents. Because the Onyx 2.0-mm-diameter trial was developed in collaboration with the FDA and the stent aced its primary endpoint and showed excellent clinical outcomes, Dr. Price anticipates the device will readily gain regulatory approval. In April 2017, the FDA approved the Resolute Onyx in sizes of 2.25- to 5.0-mm diameter.
The study met with an enthusiastic reception.
“That was terrific. It’s clearly an incredibly important unmet clinical need,” commented session cochair David R. Holmes Jr., MD, of the Mayo Clinic in Rochester, Minn.
Assuming the stent is approved, how should interventionalists put it into practice? he asked.
Dr. Price replied that, first, it’s important to step back and ask if percutaneous coronary intervention of a particular lesion in a very small coronary artery is clinically indicated. The stent itself is readily manipulatable. It is a thin-strut device constructed of a single strand of a cobalt alloy with enhanced radiopacity.
Investigators in the trial used the standard approach to dual antiplatelet therapy – at least 6 months, with 12 months preferable.
The 20% in-segment binary restenosis rate at 13 months provides a clear message for interventionalists, he continued. “What this tells me is that, while this is a very good stent, we can’t forget to treat the patient aggressively with medical therapy to stop the progression of prediabetes, diabetes, and small vessel disease in addition to treating obstructive lesions with a small stent.”
Asked if the lack of headroom in these extra-small arteries warrants liberal use of intraprocedural imaging to make sure the stent is perfectly apposed, Dr. Price replied that he doesn’t think so. He noted that intravascular ultrasound and optical coherence tomography were seldom used in the trial, yet the results were reassuringly excellent.
The study results were published simultaneously with Dr. Price’s presentation (JACC Cardiovasc Interv. 2017 May 17. doi: 10.1016/j.jcin.2017.05.004). The trial was sponsored by Medtronic. Dr. Price reported serving as a consultant and paid speaker on behalf of that company, as well as AstraZeneca, Boston Scientific, St. Jude Medical, and The Medicines Company.
Paris – The first multicenter, prospective trial of a drug-eluting stent designed specifically to treat lesions in coronary vessels less than 2.25 mm in diameter showed excellent outcomes, with a 1-year target lesion failure rate of 5% for the Resolute Onyx 2.0 mm diameter zotarolimus-eluting stent.
This result in the pivotal trial easily surpassed the prespecified performance goal of a 19% target lesion failure rate, Matthew J. Price, MD, reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
Hemodynamically significant lesions in such small vessels are “not uncommon, particularly in diabetic patients,” Dr. Price said in an interview. Indeed, 47% of patients in the clinical trial had diabetes.
At present, the only ways to treat coronary disease in arteries having a reference vessel diameter less than 2.25 mm are off-label placement of an oversized stent, with its attendant risk of complications; standard balloon angioplasty, which entails a particularly high restenosis rate in this setting; or medical management, the cardiologist noted.
He presented a multicenter, prospective, open-label, single-arm trial of 101 patients with documented ischemia-producing obstructions in coronary arteries having a reference vessel diameter less than 2.25 mm, a lesion length less than 27 mm, and evidence of ischemia attributable to the lesion, typically via fractional flow reserve. The mean diameter by quantitative coronary angiography was 1.91 mm.
The primary endpoint was the rate of target lesion failure at 12 months, a composite comprising cardiac death, target vessel MI, or clinically driven target lesion revascularization. This endpoint occurred in 5% of patients. There was a 3% target vessel MI rate and a 2% target lesion revascularization rate. There were no cardiac deaths.
“Importantly, the stent thrombosis rate in these patients with extremely small vessels was zero,” the cardiologist emphasized.
The mean angiographic in-stent late lumen loss at 13 months was 0.26 mm, which Dr. Price characterized as “quite good.” The in-segment binary angiographic restenosis rate was 20%.
“That’s slightly higher than you would expect to see in vessels with larger reference diameters. I think that’s because of the lack of headroom. You have a very small vessel, and, even with a very small stent, even a small amount of late loss will give you a larger percent diameter restenosis over time,” he explained.
The 19% target lesion failure rate selected as a performance goal in the trial was set somewhat arbitrarily. It wasn’t possible to randomize patients to a comparator arm because there are no approved stents for vessels less than 2.25 mm in diameter. The 19% figure was arrived at in discussion with the Food and Drug Administration on the basis of similarity to the performance goal used in clinical trials to gain approval of 2.25-mm, drug-eluting stents. Because the Onyx 2.0-mm-diameter trial was developed in collaboration with the FDA and the stent aced its primary endpoint and showed excellent clinical outcomes, Dr. Price anticipates the device will readily gain regulatory approval. In April 2017, the FDA approved the Resolute Onyx in sizes of 2.25- to 5.0-mm diameter.
The study met with an enthusiastic reception.
“That was terrific. It’s clearly an incredibly important unmet clinical need,” commented session cochair David R. Holmes Jr., MD, of the Mayo Clinic in Rochester, Minn.
Assuming the stent is approved, how should interventionalists put it into practice? he asked.
Dr. Price replied that, first, it’s important to step back and ask if percutaneous coronary intervention of a particular lesion in a very small coronary artery is clinically indicated. The stent itself is readily manipulatable. It is a thin-strut device constructed of a single strand of a cobalt alloy with enhanced radiopacity.
Investigators in the trial used the standard approach to dual antiplatelet therapy – at least 6 months, with 12 months preferable.
The 20% in-segment binary restenosis rate at 13 months provides a clear message for interventionalists, he continued. “What this tells me is that, while this is a very good stent, we can’t forget to treat the patient aggressively with medical therapy to stop the progression of prediabetes, diabetes, and small vessel disease in addition to treating obstructive lesions with a small stent.”
Asked if the lack of headroom in these extra-small arteries warrants liberal use of intraprocedural imaging to make sure the stent is perfectly apposed, Dr. Price replied that he doesn’t think so. He noted that intravascular ultrasound and optical coherence tomography were seldom used in the trial, yet the results were reassuringly excellent.
The study results were published simultaneously with Dr. Price’s presentation (JACC Cardiovasc Interv. 2017 May 17. doi: 10.1016/j.jcin.2017.05.004). The trial was sponsored by Medtronic. Dr. Price reported serving as a consultant and paid speaker on behalf of that company, as well as AstraZeneca, Boston Scientific, St. Jude Medical, and The Medicines Company.
Paris – The first multicenter, prospective trial of a drug-eluting stent designed specifically to treat lesions in coronary vessels less than 2.25 mm in diameter showed excellent outcomes, with a 1-year target lesion failure rate of 5% for the Resolute Onyx 2.0 mm diameter zotarolimus-eluting stent.
This result in the pivotal trial easily surpassed the prespecified performance goal of a 19% target lesion failure rate, Matthew J. Price, MD, reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
Hemodynamically significant lesions in such small vessels are “not uncommon, particularly in diabetic patients,” Dr. Price said in an interview. Indeed, 47% of patients in the clinical trial had diabetes.
At present, the only ways to treat coronary disease in arteries having a reference vessel diameter less than 2.25 mm are off-label placement of an oversized stent, with its attendant risk of complications; standard balloon angioplasty, which entails a particularly high restenosis rate in this setting; or medical management, the cardiologist noted.
He presented a multicenter, prospective, open-label, single-arm trial of 101 patients with documented ischemia-producing obstructions in coronary arteries having a reference vessel diameter less than 2.25 mm, a lesion length less than 27 mm, and evidence of ischemia attributable to the lesion, typically via fractional flow reserve. The mean diameter by quantitative coronary angiography was 1.91 mm.
The primary endpoint was the rate of target lesion failure at 12 months, a composite comprising cardiac death, target vessel MI, or clinically driven target lesion revascularization. This endpoint occurred in 5% of patients. There was a 3% target vessel MI rate and a 2% target lesion revascularization rate. There were no cardiac deaths.
“Importantly, the stent thrombosis rate in these patients with extremely small vessels was zero,” the cardiologist emphasized.
The mean angiographic in-stent late lumen loss at 13 months was 0.26 mm, which Dr. Price characterized as “quite good.” The in-segment binary angiographic restenosis rate was 20%.
“That’s slightly higher than you would expect to see in vessels with larger reference diameters. I think that’s because of the lack of headroom. You have a very small vessel, and, even with a very small stent, even a small amount of late loss will give you a larger percent diameter restenosis over time,” he explained.
The 19% target lesion failure rate selected as a performance goal in the trial was set somewhat arbitrarily. It wasn’t possible to randomize patients to a comparator arm because there are no approved stents for vessels less than 2.25 mm in diameter. The 19% figure was arrived at in discussion with the Food and Drug Administration on the basis of similarity to the performance goal used in clinical trials to gain approval of 2.25-mm, drug-eluting stents. Because the Onyx 2.0-mm-diameter trial was developed in collaboration with the FDA and the stent aced its primary endpoint and showed excellent clinical outcomes, Dr. Price anticipates the device will readily gain regulatory approval. In April 2017, the FDA approved the Resolute Onyx in sizes of 2.25- to 5.0-mm diameter.
The study met with an enthusiastic reception.
“That was terrific. It’s clearly an incredibly important unmet clinical need,” commented session cochair David R. Holmes Jr., MD, of the Mayo Clinic in Rochester, Minn.
Assuming the stent is approved, how should interventionalists put it into practice? he asked.
Dr. Price replied that, first, it’s important to step back and ask if percutaneous coronary intervention of a particular lesion in a very small coronary artery is clinically indicated. The stent itself is readily manipulatable. It is a thin-strut device constructed of a single strand of a cobalt alloy with enhanced radiopacity.
Investigators in the trial used the standard approach to dual antiplatelet therapy – at least 6 months, with 12 months preferable.
The 20% in-segment binary restenosis rate at 13 months provides a clear message for interventionalists, he continued. “What this tells me is that, while this is a very good stent, we can’t forget to treat the patient aggressively with medical therapy to stop the progression of prediabetes, diabetes, and small vessel disease in addition to treating obstructive lesions with a small stent.”
Asked if the lack of headroom in these extra-small arteries warrants liberal use of intraprocedural imaging to make sure the stent is perfectly apposed, Dr. Price replied that he doesn’t think so. He noted that intravascular ultrasound and optical coherence tomography were seldom used in the trial, yet the results were reassuringly excellent.
The study results were published simultaneously with Dr. Price’s presentation (JACC Cardiovasc Interv. 2017 May 17. doi: 10.1016/j.jcin.2017.05.004). The trial was sponsored by Medtronic. Dr. Price reported serving as a consultant and paid speaker on behalf of that company, as well as AstraZeneca, Boston Scientific, St. Jude Medical, and The Medicines Company.
AT EUROPCR
Key clinical point:
Major finding: At 12 months’ follow-up, the key outcomes were a 3% rate of target vessel MI, a 2% rate of clinically driven target lesion revascularization, no stent thrombosis, and no cardiac deaths.
Data source: A prospective, multicenter, open-label trial in 101 patients who underwent percutaneous coronary intervention for coronary lesions with a reference vessel diameter of less than 2.25 mm.
Disclosures: The trial was sponsored by Medtronic. Dr. Price reported serving as a consultant to and paid speaker on behalf of that company as well as AstraZeneca, Boston Scientific, St. Jude Medical, and The Medicines Company.

Novel Lotus valve outperforms CoreValve in REPRISE III
PARIS – The investigational mechanically expandable Lotus valve system for transcatheter aortic valve replacement proved significantly more effective than the commercially available CoreValve platform in patients with severe aortic stenosis deemed at high or extreme surgical risk in the randomized pivotal phase III REPRISE III trial, Ted E. Feldman, MD, reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
The 1-year composite primary effectiveness endpoint comprised of all-cause mortality, disabling stroke, and moderate or greater paravalvular leak (PVL) occurred in 17% of patients randomized to the Lotus transcatheter aortic valve replacement (TAVR) device, compared with 29% of those in the CoreValve group, said Dr. Feldman, director of the cardiac catheterization laboratory at NorthShore University HealthSystem in Evanston, Ill.

A key finding was that the Lotus valve group had a 1-year rate of moderate or greater PVL of just 2% as assessed in a central core lab, compared with an 11% rate in patients randomized to the classic CoreValve or the subsequent-generation Evolut R device, he observed.
“With the Lotus valve there was no or only trace PVL in over 85% of patients. This is probably even more important than the low rate of moderate or severe PVL. The valve really does result in virtually no PVL in the vast majority of patients. That’s unique to this platform,” the cardiologist said in an interview.
The unprecedented low rate of moderate or severe PVL at 1 year postprocedure is attributable to the polymer seal delivered via the Lotus system for that express purpose, he explained.
REPRISE III was the first large randomized comparative clinical trial featuring two TAVR valves, an event that reflects the rapid expansion of the field. All previous major trials had compared TAVR with surgical aortic valve replacement.
REPRISE III randomized 912 TAVR patients at 55 centers 2:1 to the Lotus valve in its 23-, 25-, or 27-mm configurations or to a CoreValve at 26, 29, or 31 mm. Roughly half of the CoreValve group got the newer repositionable and retrievable Evolut R valve, while the earlier enrollees received the nonrepositionable classic CoreValve.
The Lotus valve group proved noninferior to the CoreValve recipients for the primary safety endpoint, a 30-day composite of all-cause mortality, stroke, major or life-threatening bleeding, major vascular complications, and stage 2 or 3 acute kidney injury. The rates were 20.3% in the Lotus arm and 17.2% with CoreValve.
The 1-year rate of disabling stroke was 3.6% in the Lotus group versus 7.3% in the CoreValve group. Dr. Feldman downplayed the importance of this difference, even though it was statistically significant. The Lotus valve performed as expected, but the disabling stroke rate in the CoreValve group was higher than in earlier studies for reasons unknown, most likely simply the play of chance, he said.
“I think the real message here is that the Lotus valve performed very well,” the cardiologist said. “There have been concerns that repositioning the valve into a better position during the deployment process might create excess stroke. It appears clear that’s not the case.”
The ability to reposition the Lotus device resulted in a significantly lower rate of repeat procedures at 1 year: 0.2% versus 2% with the CoreValve, as well as zero cases of aortic valve malposition and valve-in-valve deployment.
The need for a new pacemaker within 30 days after TAVR was strikingly more common in the Lotus valve group: 36%, compared with 20% with the CoreValve. Dr. Feldman attributed the high new pacemaker rate in the Lotus arm partly to the operators’ limited experience with the novel valve along with the fact that REPRISE III used a first-iteration device deployment mechanism. An improved deployment mechanism designed to minimize problematic contact with the left ventricular outflow tract was developed too late for inclusion in the trial. But in a recent European study using this proprietary deployment system, known as Depth Guard, the new pacemaker rate was below 20%.
The learning curve for the new Lotus valve system is “not at all challenging,” according to the cardiologist. He noted that U.S. operators participating in REPRISE III, who had no prior experience with the device, were allowed only two initial cases in order to gain experience; after that, every patient counted in the clinical trial results.
The REPRISE III results will be offered to the Food and Drug Administration to support regulatory approval of the device in high-surgical-risk patients. Dr. Feldman said Boston Scientific plans to conduct an additional clinical trial of the Lotus valve, this time in intermediate-risk patients, with the goal of gaining an expanded indication. This, too, will be a head-to-head comparison with a commercially available TAVR valve, probably the Edwards Sapien 3 valve.
REPRISE III was sponsored by Boston Scientific. Dr. Feldman reported serving as a consultant to that company, Abbott, and Edwards Lifesciences, and having received institutional research grants from those companies as well.
bjancin@frontlinemedcom.com
PARIS – The investigational mechanically expandable Lotus valve system for transcatheter aortic valve replacement proved significantly more effective than the commercially available CoreValve platform in patients with severe aortic stenosis deemed at high or extreme surgical risk in the randomized pivotal phase III REPRISE III trial, Ted E. Feldman, MD, reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
The 1-year composite primary effectiveness endpoint comprised of all-cause mortality, disabling stroke, and moderate or greater paravalvular leak (PVL) occurred in 17% of patients randomized to the Lotus transcatheter aortic valve replacement (TAVR) device, compared with 29% of those in the CoreValve group, said Dr. Feldman, director of the cardiac catheterization laboratory at NorthShore University HealthSystem in Evanston, Ill.
A key finding was that the Lotus valve group had a 1-year rate of moderate or greater PVL of just 2% as assessed in a central core lab, compared with an 11% rate in patients randomized to the classic CoreValve or the subsequent-generation Evolut R device, he observed.
“With the Lotus valve there was no or only trace PVL in over 85% of patients. This is probably even more important than the low rate of moderate or severe PVL. The valve really does result in virtually no PVL in the vast majority of patients. That’s unique to this platform,” the cardiologist said in an interview.
The unprecedented low rate of moderate or severe PVL at 1 year postprocedure is attributable to the polymer seal delivered via the Lotus system for that express purpose, he explained.
REPRISE III was the first large randomized comparative clinical trial featuring two TAVR valves, an event that reflects the rapid expansion of the field. All previous major trials had compared TAVR with surgical aortic valve replacement.
REPRISE III randomized 912 TAVR patients at 55 centers 2:1 to the Lotus valve in its 23-, 25-, or 27-mm configurations or to a CoreValve at 26, 29, or 31 mm. Roughly half of the CoreValve group got the newer repositionable and retrievable Evolut R valve, while the earlier enrollees received the nonrepositionable classic CoreValve.
The Lotus valve group proved noninferior to the CoreValve recipients for the primary safety endpoint, a 30-day composite of all-cause mortality, stroke, major or life-threatening bleeding, major vascular complications, and stage 2 or 3 acute kidney injury. The rates were 20.3% in the Lotus arm and 17.2% with CoreValve.
The 1-year rate of disabling stroke was 3.6% in the Lotus group versus 7.3% in the CoreValve group. Dr. Feldman downplayed the importance of this difference, even though it was statistically significant. The Lotus valve performed as expected, but the disabling stroke rate in the CoreValve group was higher than in earlier studies for reasons unknown, most likely simply the play of chance, he said.
“I think the real message here is that the Lotus valve performed very well,” the cardiologist said. “There have been concerns that repositioning the valve into a better position during the deployment process might create excess stroke. It appears clear that’s not the case.”
The ability to reposition the Lotus device resulted in a significantly lower rate of repeat procedures at 1 year: 0.2% versus 2% with the CoreValve, as well as zero cases of aortic valve malposition and valve-in-valve deployment.
The need for a new pacemaker within 30 days after TAVR was strikingly more common in the Lotus valve group: 36%, compared with 20% with the CoreValve. Dr. Feldman attributed the high new pacemaker rate in the Lotus arm partly to the operators’ limited experience with the novel valve along with the fact that REPRISE III used a first-iteration device deployment mechanism. An improved deployment mechanism designed to minimize problematic contact with the left ventricular outflow tract was developed too late for inclusion in the trial. But in a recent European study using this proprietary deployment system, known as Depth Guard, the new pacemaker rate was below 20%.
The learning curve for the new Lotus valve system is “not at all challenging,” according to the cardiologist. He noted that U.S. operators participating in REPRISE III, who had no prior experience with the device, were allowed only two initial cases in order to gain experience; after that, every patient counted in the clinical trial results.
The REPRISE III results will be offered to the Food and Drug Administration to support regulatory approval of the device in high-surgical-risk patients. Dr. Feldman said Boston Scientific plans to conduct an additional clinical trial of the Lotus valve, this time in intermediate-risk patients, with the goal of gaining an expanded indication. This, too, will be a head-to-head comparison with a commercially available TAVR valve, probably the Edwards Sapien 3 valve.
REPRISE III was sponsored by Boston Scientific. Dr. Feldman reported serving as a consultant to that company, Abbott, and Edwards Lifesciences, and having received institutional research grants from those companies as well.
bjancin@frontlinemedcom.com
PARIS – The investigational mechanically expandable Lotus valve system for transcatheter aortic valve replacement proved significantly more effective than the commercially available CoreValve platform in patients with severe aortic stenosis deemed at high or extreme surgical risk in the randomized pivotal phase III REPRISE III trial, Ted E. Feldman, MD, reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
The 1-year composite primary effectiveness endpoint comprised of all-cause mortality, disabling stroke, and moderate or greater paravalvular leak (PVL) occurred in 17% of patients randomized to the Lotus transcatheter aortic valve replacement (TAVR) device, compared with 29% of those in the CoreValve group, said Dr. Feldman, director of the cardiac catheterization laboratory at NorthShore University HealthSystem in Evanston, Ill.
A key finding was that the Lotus valve group had a 1-year rate of moderate or greater PVL of just 2% as assessed in a central core lab, compared with an 11% rate in patients randomized to the classic CoreValve or the subsequent-generation Evolut R device, he observed.
“With the Lotus valve there was no or only trace PVL in over 85% of patients. This is probably even more important than the low rate of moderate or severe PVL. The valve really does result in virtually no PVL in the vast majority of patients. That’s unique to this platform,” the cardiologist said in an interview.
The unprecedented low rate of moderate or severe PVL at 1 year postprocedure is attributable to the polymer seal delivered via the Lotus system for that express purpose, he explained.
REPRISE III was the first large randomized comparative clinical trial featuring two TAVR valves, an event that reflects the rapid expansion of the field. All previous major trials had compared TAVR with surgical aortic valve replacement.
REPRISE III randomized 912 TAVR patients at 55 centers 2:1 to the Lotus valve in its 23-, 25-, or 27-mm configurations or to a CoreValve at 26, 29, or 31 mm. Roughly half of the CoreValve group got the newer repositionable and retrievable Evolut R valve, while the earlier enrollees received the nonrepositionable classic CoreValve.
The Lotus valve group proved noninferior to the CoreValve recipients for the primary safety endpoint, a 30-day composite of all-cause mortality, stroke, major or life-threatening bleeding, major vascular complications, and stage 2 or 3 acute kidney injury. The rates were 20.3% in the Lotus arm and 17.2% with CoreValve.
The 1-year rate of disabling stroke was 3.6% in the Lotus group versus 7.3% in the CoreValve group. Dr. Feldman downplayed the importance of this difference, even though it was statistically significant. The Lotus valve performed as expected, but the disabling stroke rate in the CoreValve group was higher than in earlier studies for reasons unknown, most likely simply the play of chance, he said.
“I think the real message here is that the Lotus valve performed very well,” the cardiologist said. “There have been concerns that repositioning the valve into a better position during the deployment process might create excess stroke. It appears clear that’s not the case.”
The ability to reposition the Lotus device resulted in a significantly lower rate of repeat procedures at 1 year: 0.2% versus 2% with the CoreValve, as well as zero cases of aortic valve malposition and valve-in-valve deployment.
The need for a new pacemaker within 30 days after TAVR was strikingly more common in the Lotus valve group: 36%, compared with 20% with the CoreValve. Dr. Feldman attributed the high new pacemaker rate in the Lotus arm partly to the operators’ limited experience with the novel valve along with the fact that REPRISE III used a first-iteration device deployment mechanism. An improved deployment mechanism designed to minimize problematic contact with the left ventricular outflow tract was developed too late for inclusion in the trial. But in a recent European study using this proprietary deployment system, known as Depth Guard, the new pacemaker rate was below 20%.
The learning curve for the new Lotus valve system is “not at all challenging,” according to the cardiologist. He noted that U.S. operators participating in REPRISE III, who had no prior experience with the device, were allowed only two initial cases in order to gain experience; after that, every patient counted in the clinical trial results.
The REPRISE III results will be offered to the Food and Drug Administration to support regulatory approval of the device in high-surgical-risk patients. Dr. Feldman said Boston Scientific plans to conduct an additional clinical trial of the Lotus valve, this time in intermediate-risk patients, with the goal of gaining an expanded indication. This, too, will be a head-to-head comparison with a commercially available TAVR valve, probably the Edwards Sapien 3 valve.
REPRISE III was sponsored by Boston Scientific. Dr. Feldman reported serving as a consultant to that company, Abbott, and Edwards Lifesciences, and having received institutional research grants from those companies as well.
bjancin@frontlinemedcom.com
AT EUROPCR
Key clinical point:
Major finding: The rate of the 1-year composite primary effectiveness endpoint comprised of all-cause mortality, disabling stroke, and moderate or greater paravalvular leak was 17% in patients randomized to the investigational Lotus transcatheter aortic valve replacement system, compared with 29% in recipients of the CoreValve.
Data source: REPRISE III, a prospective, multicenter, international clinical trial randomized 912 patients with severe aortic stenosis who were at high surgical risk to TAVR with the investigational Lotus valve or a commercially available CoreValve.
Disclosures: REPRISE III was sponsored by Boston Scientific. The study presenter reported serving as a consultant to that company as well as for Abbott and Edwards Lifesciences. He has also received institutional research grants from those companies.