User login
Cre8 EVO stent loses sweet spot in diabetes at 2 years: SUGAR
BOSTON – Despite a promising start, extended follow-up from the SUGAR trial found that the Cre8 EVO drug-eluting stent could not maintain superiority over the Resolute Onyx DES at 2 years in patients with diabetes undergoing revascularization for coronary artery disease.
The Cre8 EVO stent (Alvimedica) is not available in the United States but, as previously reported, caused a stir last year after demonstrating a 35% relative risk reduction in the primary endpoint of target lesion failure (TLF) at 1 year in a prespecified superiority analysis.
At 2 years, however, the TLF rate was 10.4% with the polymer-free Cre8 EVO amphilimus-eluting stent and 12.1% with the durable polymer Resolute Onyx (Medtronic) zotarolimus-eluting stent, which did not achieve superiority (hazard ratio, 0.84; 95% confidence interval, 0.60-1.19).
Rates were numerically lower with the Cre8 EVO stent for the endpoint’s individual components of cardiac death (3.1% vs. 3.4%), target vessel MI (6.6% vs. 7.6%), and target lesion revascularization (4.3% vs. 4.6%).
Results were also similar between the Cre8 EVO and Resolute Onyx stents for all-cause mortality (7.1% vs. 6.8%), any MI (9.0% vs. 9.2%), target vessel revascularization (5.5% vs. 5.1%), all new revascularizations (7.6% vs. 9.4%), definite stent thrombosis (1.0% vs. 1.2%), and major adverse cardiac events (18.3% vs. 20.8%), Pablo Salinas, MD, PhD, of Hospital Clinico San Carlos, Madrid, reported at the Transcatheter Cardiovascular Therapeutics annual meeting.
He noted that all-cause mortality was 7% in just 2 years in the diabetic cohort, or twice the number of cardiac deaths. “In other words, these patients had the same chance of dying from cardiac causes and noncardiac causes, so we need a more comprehensive approach to the disease. Also, if you look at all new revascularizations, roughly 50% were off target, so there is disease progression at 2 years in this population.”
Among the 586 Cre8 EVO and 589 Resolute Onyx patients who underwent percutaneous coronary intervention (PCI), roughly half had multivessel coronary artery disease, 83% had hypertension, 81% had dyslipidemia, and 21% were current smokers. Nearly all patients had diabetes type 2 for an average of 10.6 years for Cre8 EVO and 11.4 years for Resolute Onyx, with hemoglobin A1c levels of 7.4% and 7.5%, respectively.
Although there is “insufficient evidence” the Cre8 EVO stent is superior to the Resolute Onyx stent with regard to TLF, Dr. Salinas concluded extended follow-up until 5 years is warranted.
During a discussion of the results, Dr. Salinas said he expects the 5-year results will “probably go parallel” but that it’s worth following this very valuable cohort. “There are not so many trials with 1,000 diabetic patients. We always speak about how complex they are, the results are bad, but we don’t use the diabetic population in trials,” he said at the meeting sponsored by the Cardiovascular Research Foundation.
Asked during a TCT press conference what could have caused the catch-up in TLF at 2 years, Dr. Salinas said there were only 25 primary events from years 1 to 2, driven primarily by periprocedural MI, but that the timing of restenosis was different. Events accrued “drop by drop” with the Cre8 EVO, whereas with the Resolute Onyx there was a “bump in restenosis” after 6 months “but then it is very nice to see it is flat, which means that durable polymers are also safe because we have not seen late events.”

Press conference discussant Carlo Di Mario, MD, from Careggi University Hospital, Florence, Italy, who was not involved in the study, said the reversal of superiority for the Cre8 EVO might be a “bitter note” for the investigators but “maybe it is not bitter for us because overall, the percentage of figures are so low that it’s very difficult to find a difference” between the two stents.
Roxana Mehran, MD, of Icahn School of Medicine at Mount Sinai, New York, who previously described the 1-year results as “almost too good to be true,” commented to this news organization, “We just saw in this trial, no benefit whatsoever at 2 years in terms of target lesion failure. So it’s very important for us to evaluate this going forward.”
She continued, “We’ve always been talking about these biodegradable polymers and then going back to the bare metal stent – oh that’s great because polymers aren’t so good – but now we’re seeing durable polymers may be okay, especially with the current technology.”
Asked whether Cre8 EVO, which is CE mark certified in Europe, remains an option in light of the new results, Dr. Mehran said, “I don’t think it kills it. It’s not worse; it’s another stent that’s available.”
Nevertheless, “what we’re looking for is some efficacious benefit for diabetic patients. We don’t have one yet,” observed Dr. Mehran, who is leading the ABILITY Diabetes Global trial, which just finished enrolling 3,000 patients with diabetes and is testing PCI with the Abluminus DES+ sirolimus-eluting stent system vs. the Xience everolimus-eluting stent. The study is estimated to be complete in August 2024.
The study was funded by the Spanish Society of Cardiology. Dr. Salinas reported consulting fees/honoraria from Boston Scientific, Abbott Vascular, Biomenco, and Medtronic.
A version of this article first appeared on Medscape.com.
BOSTON – Despite a promising start, extended follow-up from the SUGAR trial found that the Cre8 EVO drug-eluting stent could not maintain superiority over the Resolute Onyx DES at 2 years in patients with diabetes undergoing revascularization for coronary artery disease.
The Cre8 EVO stent (Alvimedica) is not available in the United States but, as previously reported, caused a stir last year after demonstrating a 35% relative risk reduction in the primary endpoint of target lesion failure (TLF) at 1 year in a prespecified superiority analysis.
At 2 years, however, the TLF rate was 10.4% with the polymer-free Cre8 EVO amphilimus-eluting stent and 12.1% with the durable polymer Resolute Onyx (Medtronic) zotarolimus-eluting stent, which did not achieve superiority (hazard ratio, 0.84; 95% confidence interval, 0.60-1.19).
Rates were numerically lower with the Cre8 EVO stent for the endpoint’s individual components of cardiac death (3.1% vs. 3.4%), target vessel MI (6.6% vs. 7.6%), and target lesion revascularization (4.3% vs. 4.6%).
Results were also similar between the Cre8 EVO and Resolute Onyx stents for all-cause mortality (7.1% vs. 6.8%), any MI (9.0% vs. 9.2%), target vessel revascularization (5.5% vs. 5.1%), all new revascularizations (7.6% vs. 9.4%), definite stent thrombosis (1.0% vs. 1.2%), and major adverse cardiac events (18.3% vs. 20.8%), Pablo Salinas, MD, PhD, of Hospital Clinico San Carlos, Madrid, reported at the Transcatheter Cardiovascular Therapeutics annual meeting.
He noted that all-cause mortality was 7% in just 2 years in the diabetic cohort, or twice the number of cardiac deaths. “In other words, these patients had the same chance of dying from cardiac causes and noncardiac causes, so we need a more comprehensive approach to the disease. Also, if you look at all new revascularizations, roughly 50% were off target, so there is disease progression at 2 years in this population.”
Among the 586 Cre8 EVO and 589 Resolute Onyx patients who underwent percutaneous coronary intervention (PCI), roughly half had multivessel coronary artery disease, 83% had hypertension, 81% had dyslipidemia, and 21% were current smokers. Nearly all patients had diabetes type 2 for an average of 10.6 years for Cre8 EVO and 11.4 years for Resolute Onyx, with hemoglobin A1c levels of 7.4% and 7.5%, respectively.
Although there is “insufficient evidence” the Cre8 EVO stent is superior to the Resolute Onyx stent with regard to TLF, Dr. Salinas concluded extended follow-up until 5 years is warranted.
During a discussion of the results, Dr. Salinas said he expects the 5-year results will “probably go parallel” but that it’s worth following this very valuable cohort. “There are not so many trials with 1,000 diabetic patients. We always speak about how complex they are, the results are bad, but we don’t use the diabetic population in trials,” he said at the meeting sponsored by the Cardiovascular Research Foundation.
Asked during a TCT press conference what could have caused the catch-up in TLF at 2 years, Dr. Salinas said there were only 25 primary events from years 1 to 2, driven primarily by periprocedural MI, but that the timing of restenosis was different. Events accrued “drop by drop” with the Cre8 EVO, whereas with the Resolute Onyx there was a “bump in restenosis” after 6 months “but then it is very nice to see it is flat, which means that durable polymers are also safe because we have not seen late events.”
Press conference discussant Carlo Di Mario, MD, from Careggi University Hospital, Florence, Italy, who was not involved in the study, said the reversal of superiority for the Cre8 EVO might be a “bitter note” for the investigators but “maybe it is not bitter for us because overall, the percentage of figures are so low that it’s very difficult to find a difference” between the two stents.
Roxana Mehran, MD, of Icahn School of Medicine at Mount Sinai, New York, who previously described the 1-year results as “almost too good to be true,” commented to this news organization, “We just saw in this trial, no benefit whatsoever at 2 years in terms of target lesion failure. So it’s very important for us to evaluate this going forward.”
She continued, “We’ve always been talking about these biodegradable polymers and then going back to the bare metal stent – oh that’s great because polymers aren’t so good – but now we’re seeing durable polymers may be okay, especially with the current technology.”
Asked whether Cre8 EVO, which is CE mark certified in Europe, remains an option in light of the new results, Dr. Mehran said, “I don’t think it kills it. It’s not worse; it’s another stent that’s available.”
Nevertheless, “what we’re looking for is some efficacious benefit for diabetic patients. We don’t have one yet,” observed Dr. Mehran, who is leading the ABILITY Diabetes Global trial, which just finished enrolling 3,000 patients with diabetes and is testing PCI with the Abluminus DES+ sirolimus-eluting stent system vs. the Xience everolimus-eluting stent. The study is estimated to be complete in August 2024.
The study was funded by the Spanish Society of Cardiology. Dr. Salinas reported consulting fees/honoraria from Boston Scientific, Abbott Vascular, Biomenco, and Medtronic.
A version of this article first appeared on Medscape.com.
BOSTON – Despite a promising start, extended follow-up from the SUGAR trial found that the Cre8 EVO drug-eluting stent could not maintain superiority over the Resolute Onyx DES at 2 years in patients with diabetes undergoing revascularization for coronary artery disease.
The Cre8 EVO stent (Alvimedica) is not available in the United States but, as previously reported, caused a stir last year after demonstrating a 35% relative risk reduction in the primary endpoint of target lesion failure (TLF) at 1 year in a prespecified superiority analysis.
At 2 years, however, the TLF rate was 10.4% with the polymer-free Cre8 EVO amphilimus-eluting stent and 12.1% with the durable polymer Resolute Onyx (Medtronic) zotarolimus-eluting stent, which did not achieve superiority (hazard ratio, 0.84; 95% confidence interval, 0.60-1.19).
Rates were numerically lower with the Cre8 EVO stent for the endpoint’s individual components of cardiac death (3.1% vs. 3.4%), target vessel MI (6.6% vs. 7.6%), and target lesion revascularization (4.3% vs. 4.6%).
Results were also similar between the Cre8 EVO and Resolute Onyx stents for all-cause mortality (7.1% vs. 6.8%), any MI (9.0% vs. 9.2%), target vessel revascularization (5.5% vs. 5.1%), all new revascularizations (7.6% vs. 9.4%), definite stent thrombosis (1.0% vs. 1.2%), and major adverse cardiac events (18.3% vs. 20.8%), Pablo Salinas, MD, PhD, of Hospital Clinico San Carlos, Madrid, reported at the Transcatheter Cardiovascular Therapeutics annual meeting.
He noted that all-cause mortality was 7% in just 2 years in the diabetic cohort, or twice the number of cardiac deaths. “In other words, these patients had the same chance of dying from cardiac causes and noncardiac causes, so we need a more comprehensive approach to the disease. Also, if you look at all new revascularizations, roughly 50% were off target, so there is disease progression at 2 years in this population.”
Among the 586 Cre8 EVO and 589 Resolute Onyx patients who underwent percutaneous coronary intervention (PCI), roughly half had multivessel coronary artery disease, 83% had hypertension, 81% had dyslipidemia, and 21% were current smokers. Nearly all patients had diabetes type 2 for an average of 10.6 years for Cre8 EVO and 11.4 years for Resolute Onyx, with hemoglobin A1c levels of 7.4% and 7.5%, respectively.
Although there is “insufficient evidence” the Cre8 EVO stent is superior to the Resolute Onyx stent with regard to TLF, Dr. Salinas concluded extended follow-up until 5 years is warranted.
During a discussion of the results, Dr. Salinas said he expects the 5-year results will “probably go parallel” but that it’s worth following this very valuable cohort. “There are not so many trials with 1,000 diabetic patients. We always speak about how complex they are, the results are bad, but we don’t use the diabetic population in trials,” he said at the meeting sponsored by the Cardiovascular Research Foundation.
Asked during a TCT press conference what could have caused the catch-up in TLF at 2 years, Dr. Salinas said there were only 25 primary events from years 1 to 2, driven primarily by periprocedural MI, but that the timing of restenosis was different. Events accrued “drop by drop” with the Cre8 EVO, whereas with the Resolute Onyx there was a “bump in restenosis” after 6 months “but then it is very nice to see it is flat, which means that durable polymers are also safe because we have not seen late events.”
Press conference discussant Carlo Di Mario, MD, from Careggi University Hospital, Florence, Italy, who was not involved in the study, said the reversal of superiority for the Cre8 EVO might be a “bitter note” for the investigators but “maybe it is not bitter for us because overall, the percentage of figures are so low that it’s very difficult to find a difference” between the two stents.
Roxana Mehran, MD, of Icahn School of Medicine at Mount Sinai, New York, who previously described the 1-year results as “almost too good to be true,” commented to this news organization, “We just saw in this trial, no benefit whatsoever at 2 years in terms of target lesion failure. So it’s very important for us to evaluate this going forward.”
She continued, “We’ve always been talking about these biodegradable polymers and then going back to the bare metal stent – oh that’s great because polymers aren’t so good – but now we’re seeing durable polymers may be okay, especially with the current technology.”
Asked whether Cre8 EVO, which is CE mark certified in Europe, remains an option in light of the new results, Dr. Mehran said, “I don’t think it kills it. It’s not worse; it’s another stent that’s available.”
Nevertheless, “what we’re looking for is some efficacious benefit for diabetic patients. We don’t have one yet,” observed Dr. Mehran, who is leading the ABILITY Diabetes Global trial, which just finished enrolling 3,000 patients with diabetes and is testing PCI with the Abluminus DES+ sirolimus-eluting stent system vs. the Xience everolimus-eluting stent. The study is estimated to be complete in August 2024.
The study was funded by the Spanish Society of Cardiology. Dr. Salinas reported consulting fees/honoraria from Boston Scientific, Abbott Vascular, Biomenco, and Medtronic.
A version of this article first appeared on Medscape.com.
AT TCT 2022

Add PCSK9 inhibitor to high-intensity statin at primary PCI, proposes sham-controlled EPIC-STEMI
It’s best to have patients on aggressive lipid-lowering therapy before discharge after an acute ST-segment elevation myocardial infarction (STEMI), so why not start it right away – even in the cath lab – using some of the most potent LDL cholesterol–lowering agents available?
That was a main idea behind the randomized, sham-controlled EPIC-STEMI trial, in which STEMI patients were started on a PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitor immediately before direct percutaneous coronary intervention (PCI) and on top of high-intensity statins.
Those in the trial getting the active agent showed a 22% drop in LDL cholesterol levels by 6 weeks, compared with the control group given a sham injection along with high-intensity statins. They were also more likely to meet LDL cholesterol goals specified in some guidelines, including reduction by at least 50%. And those outcomes were achieved regardless of baseline LDL cholesterol levels or prior statin use.
Adoption of the trial’s early, aggressive LDL cholesterolreduction strategy in practice “has the potential to substantially reduce morbidity and mortality” in such cases “by further reducing LDL beyond statins in a much greater number of high-risk patients than are currently being treated with these agents,” suggested principal investigator Shamir R. Mehta, MD, MSc, when presenting the findings at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
Adherence to secondary prevention measures in patients with acute coronary syndromes (ACS) is much better if they are started before hospital discharge, explained Dr. Mehta, senior scientist with Population Health Research Institute and professor of medicine at McMaster University, Hamilton, Ont. But “as soon as the patient has left the hospital, it is much more difficult to get these therapies on board.”
Routine adoption of such aggressive in-hospital, lipid-lowering therapy for the vast population with ACS would likely mean far fewer deaths and cardiovascular events “across a broader patient population.”
EPIC-STEMI is among the first studies to explore the strategy. “I think that’s the point of the trial that we wanted to make, that we don’t yet have data on this. We’re treading very carefully with PCSK9 inhibitors, and it’s just inching forward in populations. And I think we need a bold trial to see whether or not this changes things.”
The PCSK9 inhibitor alirocumab (Praluent) was used in EPIC-STEMI, which was published in EuroIntervention, with Dr. Mehta as lead author, the same day as his presentation. The drug and its sham injection were given on top of either atorvastatin 40-80 mg or rosuvastatin 40 mg.
Early initiation of statins in patients with acute STEMI has become standard, but there’s good evidence from intracoronary imaging studies suggesting that the addition of PCSK9 inhibitors might promote further stabilization of plaques that could potentially cause recurrent ischemic events.
Treatment with the injectable drugs plus statins led to significant coronary lesion regression in the GLAGOV trial of patients with stable coronary disease. And initiation of PCSK9 inhibitors with high-intensity statins soon after PCI for ACS improved atheroma shrinkage in non–infarct-related arteries over 1 year in the recent, placebo-controlled PACMAN-AMI trial.
Dr. Mehta pointed out that LDL reductions on PCSK9 inhibition, compared with the sham control, weren’t necessarily as impressive as might be expected from the major trials of long-term therapy with the drugs.
“You need longer [therapy] in order to see a difference in LDL levels when you use a PCSK9 inhibitor acutely. This is shown also on measures of infarct size.” There was no difference between treatment groups in infarct size as measured by levels of the MB fraction of creatine kinase, he reported.
“What this is telling us is that the acute use of a PCSK9 inhibitor did not modify the size or the severity of the baseline STEMI event.”
And EPIC-STEMI was too small and never intended to assess clinical outcomes; it was more about feasibility and what degree of LDL cholesterol lowering might be expected.
The trial was needed, Dr. Mehta said, because the PCSK9 inhibitors haven’t been extensively adopted into clinical practice and are not getting to the patients who could most benefit. One of the reasons for that is quite clear to him. “We are missing the high-risk patients because we are not treating them acutely,” Dr. Mehta said in an interview.
The strategy “has not yet been evaluated, and there have been barriers,” he observed. “Cost has been a barrier. Access to the drug has been a barrier. But in terms of the science, in terms of reducing cardiovascular events, this is a strategy that has to be tested.”

The aggressive, early LDL cholesterol reduction strategy should be evaluated for its effect on long-term outcomes, “especially knowing that in the first 30 days to 6 months post STEMI there’s a tremendous uptick in ischemic events, including recurrent myocardial infarction,” Roxana Mehran, MD, said at a media briefing on EPIC-STEMI held before Dr. Mehta’s formal presentation.
The “fantastic reduction acutely” with a PCSK9 inhibitor on top of statins, “hopefully reducing inflammation” similarly to what’s been observed in past trials, “absolutely warrants” a STEMI clinical outcomes trial, said Dr. Mehran, Icahn School of Medicine at Mount Sinai, New York, who isn’t connected with EPIC-STEMI.
If better post-discharge medication adherence is one of the acute strategy’s goals, it will be important to consider the potential influence of prescribing a periodically injected drug, proposed Eric A. Cohen, MD, Sunnybrook Health Sciences Center, Toronto, at the press conference.
“Keep in mind that STEMI patients typically come to the hospital on zero medications and leave 2 days later on five medications,” Dr. Cohen observed. “I’m curious whether having one of those as a sub-Q injection every 2 weeks, and reducing the pill burden, will help or deter adherence to therapy. I think it’s worth studying.”
The trial originally included 97 patients undergoing PCI for STEMI who were randomly assigned to receive the PCSK9 inhibitor or a sham injection on top of high-intensity statins, without regard to LDL cholesterol levels. Randomization took place after diagnostic angiography but before PCI.
The analysis, however, subsequently excluded 29 patients who could not continue with the study, “mainly because of hospital research clinic closure due to the COVID-19 pandemic,” the published report states.
That left 68 patients who had received at least one dose of PCSK9 inhibitor, alirocumab 150 mg subcutaneously, or the sham injection, and had at least one blood draw for LDL cholesterol response which, Dr. Mehta said, still left adequate statistical power for the LDL cholesterol–based primary endpoint.
By 6 weeks, LDL cholesterol levels had fallen 72.9% in the active-therapy group and by 48.1% in the control group (P < .001). Also, 92.1% and 56.7% of patients, respectively (P = .002), had achieved levels below the 1.4 mmol/L (54 mg/dL) goal in the European guidelines, Dr. Mehta reported.
Levels fell more than 50% compared with baseline in 89.5% of alirocumab patients and 60% (P = .007) of controls, respectively.
There was no significant difference in rates of attaining LDL cholesterol levels below the 70 mg/dL (1.8 mmol/L) threshold specified in U.S. guidelines for very high-risk patients: 94.7% of alirocumab patients and 83.4% of controls (P = .26).
Nor did the groups differ significantly in natriuretic peptide levels, which reflect ventricular remodeling; or in 6-week change in the inflammatory biomarker high-sensitivity C-reactive protein.
An open-label, randomized trial scheduled to launch before the end of 2022 will explore similarly early initiation of a PCSK9 inhibitor, compared with standard lipid management, in an estimated 4,000 patients hospitalized with STEMI or non-STEMI.
The EVOLVE MI trial is looking at the monoclonal antibody evolocumab (Repatha) for its effect on the primary endpoint of myocardial infarction, ischemic stroke, arterial revascularization, or death from any cause over an expected 3-4 years.
EPIC-STEMI was supported in part by Sanofi. Dr. Mehta reported an unrestricted grant from Sanofi to Hamilton Health Sciences for the present study and consulting fees from Amgen, Sanofi, and Novartis. Dr. Cohen disclosed receiving grant support from and holding research contracts with Abbott Vascular; and receiving fees for consulting, honoraria, or serving on a speaker’s bureau for Abbott Vascular, Medtronic, and Baylis. Dr. Mehran disclosed receiving grants or research support from numerous pharmaceutical companies; receiving consultant fee or honoraria or serving on a speaker’s bureau for Novartis, Abbott Vascular, Janssen, Medtronic, Medscape/WebMD, and Cine-Med Research; and holding equity, stock, or stock options with Control Rad, Applied Therapeutics, and Elixir Medical.
A version of this article first appeared on Medscape.com.
It’s best to have patients on aggressive lipid-lowering therapy before discharge after an acute ST-segment elevation myocardial infarction (STEMI), so why not start it right away – even in the cath lab – using some of the most potent LDL cholesterol–lowering agents available?
That was a main idea behind the randomized, sham-controlled EPIC-STEMI trial, in which STEMI patients were started on a PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitor immediately before direct percutaneous coronary intervention (PCI) and on top of high-intensity statins.
Those in the trial getting the active agent showed a 22% drop in LDL cholesterol levels by 6 weeks, compared with the control group given a sham injection along with high-intensity statins. They were also more likely to meet LDL cholesterol goals specified in some guidelines, including reduction by at least 50%. And those outcomes were achieved regardless of baseline LDL cholesterol levels or prior statin use.
Adoption of the trial’s early, aggressive LDL cholesterolreduction strategy in practice “has the potential to substantially reduce morbidity and mortality” in such cases “by further reducing LDL beyond statins in a much greater number of high-risk patients than are currently being treated with these agents,” suggested principal investigator Shamir R. Mehta, MD, MSc, when presenting the findings at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
Adherence to secondary prevention measures in patients with acute coronary syndromes (ACS) is much better if they are started before hospital discharge, explained Dr. Mehta, senior scientist with Population Health Research Institute and professor of medicine at McMaster University, Hamilton, Ont. But “as soon as the patient has left the hospital, it is much more difficult to get these therapies on board.”
Routine adoption of such aggressive in-hospital, lipid-lowering therapy for the vast population with ACS would likely mean far fewer deaths and cardiovascular events “across a broader patient population.”
EPIC-STEMI is among the first studies to explore the strategy. “I think that’s the point of the trial that we wanted to make, that we don’t yet have data on this. We’re treading very carefully with PCSK9 inhibitors, and it’s just inching forward in populations. And I think we need a bold trial to see whether or not this changes things.”
The PCSK9 inhibitor alirocumab (Praluent) was used in EPIC-STEMI, which was published in EuroIntervention, with Dr. Mehta as lead author, the same day as his presentation. The drug and its sham injection were given on top of either atorvastatin 40-80 mg or rosuvastatin 40 mg.
Early initiation of statins in patients with acute STEMI has become standard, but there’s good evidence from intracoronary imaging studies suggesting that the addition of PCSK9 inhibitors might promote further stabilization of plaques that could potentially cause recurrent ischemic events.
Treatment with the injectable drugs plus statins led to significant coronary lesion regression in the GLAGOV trial of patients with stable coronary disease. And initiation of PCSK9 inhibitors with high-intensity statins soon after PCI for ACS improved atheroma shrinkage in non–infarct-related arteries over 1 year in the recent, placebo-controlled PACMAN-AMI trial.
Dr. Mehta pointed out that LDL reductions on PCSK9 inhibition, compared with the sham control, weren’t necessarily as impressive as might be expected from the major trials of long-term therapy with the drugs.
“You need longer [therapy] in order to see a difference in LDL levels when you use a PCSK9 inhibitor acutely. This is shown also on measures of infarct size.” There was no difference between treatment groups in infarct size as measured by levels of the MB fraction of creatine kinase, he reported.
“What this is telling us is that the acute use of a PCSK9 inhibitor did not modify the size or the severity of the baseline STEMI event.”
And EPIC-STEMI was too small and never intended to assess clinical outcomes; it was more about feasibility and what degree of LDL cholesterol lowering might be expected.
The trial was needed, Dr. Mehta said, because the PCSK9 inhibitors haven’t been extensively adopted into clinical practice and are not getting to the patients who could most benefit. One of the reasons for that is quite clear to him. “We are missing the high-risk patients because we are not treating them acutely,” Dr. Mehta said in an interview.
The strategy “has not yet been evaluated, and there have been barriers,” he observed. “Cost has been a barrier. Access to the drug has been a barrier. But in terms of the science, in terms of reducing cardiovascular events, this is a strategy that has to be tested.”
The aggressive, early LDL cholesterol reduction strategy should be evaluated for its effect on long-term outcomes, “especially knowing that in the first 30 days to 6 months post STEMI there’s a tremendous uptick in ischemic events, including recurrent myocardial infarction,” Roxana Mehran, MD, said at a media briefing on EPIC-STEMI held before Dr. Mehta’s formal presentation.
The “fantastic reduction acutely” with a PCSK9 inhibitor on top of statins, “hopefully reducing inflammation” similarly to what’s been observed in past trials, “absolutely warrants” a STEMI clinical outcomes trial, said Dr. Mehran, Icahn School of Medicine at Mount Sinai, New York, who isn’t connected with EPIC-STEMI.
If better post-discharge medication adherence is one of the acute strategy’s goals, it will be important to consider the potential influence of prescribing a periodically injected drug, proposed Eric A. Cohen, MD, Sunnybrook Health Sciences Center, Toronto, at the press conference.
“Keep in mind that STEMI patients typically come to the hospital on zero medications and leave 2 days later on five medications,” Dr. Cohen observed. “I’m curious whether having one of those as a sub-Q injection every 2 weeks, and reducing the pill burden, will help or deter adherence to therapy. I think it’s worth studying.”
The trial originally included 97 patients undergoing PCI for STEMI who were randomly assigned to receive the PCSK9 inhibitor or a sham injection on top of high-intensity statins, without regard to LDL cholesterol levels. Randomization took place after diagnostic angiography but before PCI.
The analysis, however, subsequently excluded 29 patients who could not continue with the study, “mainly because of hospital research clinic closure due to the COVID-19 pandemic,” the published report states.
That left 68 patients who had received at least one dose of PCSK9 inhibitor, alirocumab 150 mg subcutaneously, or the sham injection, and had at least one blood draw for LDL cholesterol response which, Dr. Mehta said, still left adequate statistical power for the LDL cholesterol–based primary endpoint.
By 6 weeks, LDL cholesterol levels had fallen 72.9% in the active-therapy group and by 48.1% in the control group (P < .001). Also, 92.1% and 56.7% of patients, respectively (P = .002), had achieved levels below the 1.4 mmol/L (54 mg/dL) goal in the European guidelines, Dr. Mehta reported.
Levels fell more than 50% compared with baseline in 89.5% of alirocumab patients and 60% (P = .007) of controls, respectively.
There was no significant difference in rates of attaining LDL cholesterol levels below the 70 mg/dL (1.8 mmol/L) threshold specified in U.S. guidelines for very high-risk patients: 94.7% of alirocumab patients and 83.4% of controls (P = .26).
Nor did the groups differ significantly in natriuretic peptide levels, which reflect ventricular remodeling; or in 6-week change in the inflammatory biomarker high-sensitivity C-reactive protein.
An open-label, randomized trial scheduled to launch before the end of 2022 will explore similarly early initiation of a PCSK9 inhibitor, compared with standard lipid management, in an estimated 4,000 patients hospitalized with STEMI or non-STEMI.
The EVOLVE MI trial is looking at the monoclonal antibody evolocumab (Repatha) for its effect on the primary endpoint of myocardial infarction, ischemic stroke, arterial revascularization, or death from any cause over an expected 3-4 years.
EPIC-STEMI was supported in part by Sanofi. Dr. Mehta reported an unrestricted grant from Sanofi to Hamilton Health Sciences for the present study and consulting fees from Amgen, Sanofi, and Novartis. Dr. Cohen disclosed receiving grant support from and holding research contracts with Abbott Vascular; and receiving fees for consulting, honoraria, or serving on a speaker’s bureau for Abbott Vascular, Medtronic, and Baylis. Dr. Mehran disclosed receiving grants or research support from numerous pharmaceutical companies; receiving consultant fee or honoraria or serving on a speaker’s bureau for Novartis, Abbott Vascular, Janssen, Medtronic, Medscape/WebMD, and Cine-Med Research; and holding equity, stock, or stock options with Control Rad, Applied Therapeutics, and Elixir Medical.
A version of this article first appeared on Medscape.com.
It’s best to have patients on aggressive lipid-lowering therapy before discharge after an acute ST-segment elevation myocardial infarction (STEMI), so why not start it right away – even in the cath lab – using some of the most potent LDL cholesterol–lowering agents available?
That was a main idea behind the randomized, sham-controlled EPIC-STEMI trial, in which STEMI patients were started on a PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitor immediately before direct percutaneous coronary intervention (PCI) and on top of high-intensity statins.
Those in the trial getting the active agent showed a 22% drop in LDL cholesterol levels by 6 weeks, compared with the control group given a sham injection along with high-intensity statins. They were also more likely to meet LDL cholesterol goals specified in some guidelines, including reduction by at least 50%. And those outcomes were achieved regardless of baseline LDL cholesterol levels or prior statin use.
Adoption of the trial’s early, aggressive LDL cholesterolreduction strategy in practice “has the potential to substantially reduce morbidity and mortality” in such cases “by further reducing LDL beyond statins in a much greater number of high-risk patients than are currently being treated with these agents,” suggested principal investigator Shamir R. Mehta, MD, MSc, when presenting the findings at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
Adherence to secondary prevention measures in patients with acute coronary syndromes (ACS) is much better if they are started before hospital discharge, explained Dr. Mehta, senior scientist with Population Health Research Institute and professor of medicine at McMaster University, Hamilton, Ont. But “as soon as the patient has left the hospital, it is much more difficult to get these therapies on board.”
Routine adoption of such aggressive in-hospital, lipid-lowering therapy for the vast population with ACS would likely mean far fewer deaths and cardiovascular events “across a broader patient population.”
EPIC-STEMI is among the first studies to explore the strategy. “I think that’s the point of the trial that we wanted to make, that we don’t yet have data on this. We’re treading very carefully with PCSK9 inhibitors, and it’s just inching forward in populations. And I think we need a bold trial to see whether or not this changes things.”
The PCSK9 inhibitor alirocumab (Praluent) was used in EPIC-STEMI, which was published in EuroIntervention, with Dr. Mehta as lead author, the same day as his presentation. The drug and its sham injection were given on top of either atorvastatin 40-80 mg or rosuvastatin 40 mg.
Early initiation of statins in patients with acute STEMI has become standard, but there’s good evidence from intracoronary imaging studies suggesting that the addition of PCSK9 inhibitors might promote further stabilization of plaques that could potentially cause recurrent ischemic events.
Treatment with the injectable drugs plus statins led to significant coronary lesion regression in the GLAGOV trial of patients with stable coronary disease. And initiation of PCSK9 inhibitors with high-intensity statins soon after PCI for ACS improved atheroma shrinkage in non–infarct-related arteries over 1 year in the recent, placebo-controlled PACMAN-AMI trial.
Dr. Mehta pointed out that LDL reductions on PCSK9 inhibition, compared with the sham control, weren’t necessarily as impressive as might be expected from the major trials of long-term therapy with the drugs.
“You need longer [therapy] in order to see a difference in LDL levels when you use a PCSK9 inhibitor acutely. This is shown also on measures of infarct size.” There was no difference between treatment groups in infarct size as measured by levels of the MB fraction of creatine kinase, he reported.
“What this is telling us is that the acute use of a PCSK9 inhibitor did not modify the size or the severity of the baseline STEMI event.”
And EPIC-STEMI was too small and never intended to assess clinical outcomes; it was more about feasibility and what degree of LDL cholesterol lowering might be expected.
The trial was needed, Dr. Mehta said, because the PCSK9 inhibitors haven’t been extensively adopted into clinical practice and are not getting to the patients who could most benefit. One of the reasons for that is quite clear to him. “We are missing the high-risk patients because we are not treating them acutely,” Dr. Mehta said in an interview.
The strategy “has not yet been evaluated, and there have been barriers,” he observed. “Cost has been a barrier. Access to the drug has been a barrier. But in terms of the science, in terms of reducing cardiovascular events, this is a strategy that has to be tested.”
The aggressive, early LDL cholesterol reduction strategy should be evaluated for its effect on long-term outcomes, “especially knowing that in the first 30 days to 6 months post STEMI there’s a tremendous uptick in ischemic events, including recurrent myocardial infarction,” Roxana Mehran, MD, said at a media briefing on EPIC-STEMI held before Dr. Mehta’s formal presentation.
The “fantastic reduction acutely” with a PCSK9 inhibitor on top of statins, “hopefully reducing inflammation” similarly to what’s been observed in past trials, “absolutely warrants” a STEMI clinical outcomes trial, said Dr. Mehran, Icahn School of Medicine at Mount Sinai, New York, who isn’t connected with EPIC-STEMI.
If better post-discharge medication adherence is one of the acute strategy’s goals, it will be important to consider the potential influence of prescribing a periodically injected drug, proposed Eric A. Cohen, MD, Sunnybrook Health Sciences Center, Toronto, at the press conference.
“Keep in mind that STEMI patients typically come to the hospital on zero medications and leave 2 days later on five medications,” Dr. Cohen observed. “I’m curious whether having one of those as a sub-Q injection every 2 weeks, and reducing the pill burden, will help or deter adherence to therapy. I think it’s worth studying.”
The trial originally included 97 patients undergoing PCI for STEMI who were randomly assigned to receive the PCSK9 inhibitor or a sham injection on top of high-intensity statins, without regard to LDL cholesterol levels. Randomization took place after diagnostic angiography but before PCI.
The analysis, however, subsequently excluded 29 patients who could not continue with the study, “mainly because of hospital research clinic closure due to the COVID-19 pandemic,” the published report states.
That left 68 patients who had received at least one dose of PCSK9 inhibitor, alirocumab 150 mg subcutaneously, or the sham injection, and had at least one blood draw for LDL cholesterol response which, Dr. Mehta said, still left adequate statistical power for the LDL cholesterol–based primary endpoint.
By 6 weeks, LDL cholesterol levels had fallen 72.9% in the active-therapy group and by 48.1% in the control group (P < .001). Also, 92.1% and 56.7% of patients, respectively (P = .002), had achieved levels below the 1.4 mmol/L (54 mg/dL) goal in the European guidelines, Dr. Mehta reported.
Levels fell more than 50% compared with baseline in 89.5% of alirocumab patients and 60% (P = .007) of controls, respectively.
There was no significant difference in rates of attaining LDL cholesterol levels below the 70 mg/dL (1.8 mmol/L) threshold specified in U.S. guidelines for very high-risk patients: 94.7% of alirocumab patients and 83.4% of controls (P = .26).
Nor did the groups differ significantly in natriuretic peptide levels, which reflect ventricular remodeling; or in 6-week change in the inflammatory biomarker high-sensitivity C-reactive protein.
An open-label, randomized trial scheduled to launch before the end of 2022 will explore similarly early initiation of a PCSK9 inhibitor, compared with standard lipid management, in an estimated 4,000 patients hospitalized with STEMI or non-STEMI.
The EVOLVE MI trial is looking at the monoclonal antibody evolocumab (Repatha) for its effect on the primary endpoint of myocardial infarction, ischemic stroke, arterial revascularization, or death from any cause over an expected 3-4 years.
EPIC-STEMI was supported in part by Sanofi. Dr. Mehta reported an unrestricted grant from Sanofi to Hamilton Health Sciences for the present study and consulting fees from Amgen, Sanofi, and Novartis. Dr. Cohen disclosed receiving grant support from and holding research contracts with Abbott Vascular; and receiving fees for consulting, honoraria, or serving on a speaker’s bureau for Abbott Vascular, Medtronic, and Baylis. Dr. Mehran disclosed receiving grants or research support from numerous pharmaceutical companies; receiving consultant fee or honoraria or serving on a speaker’s bureau for Novartis, Abbott Vascular, Janssen, Medtronic, Medscape/WebMD, and Cine-Med Research; and holding equity, stock, or stock options with Control Rad, Applied Therapeutics, and Elixir Medical.
A version of this article first appeared on Medscape.com.
FROM TCT 2022

Legacy of neutral renal denervation trial recast by long-term outcomes: SYMPLICITY HTN-3
BOSTON – There’s an intriguing plot twist in the story of SYMPLICITY HTN-3, the sham-controlled clinical trial that nearly put the kibosh on renal denervation (RDN) therapy as a promising approach to treatment-resistant hypertension (HTN).
The trial famously showed no benefit for systolic blood pressure (BP) from the invasive procedure at 6 months and 12 months, dampening enthusiasm for RDN in HTN for both physicians and industry. But it turns out that disappointment in the study may have been premature.

The procedure led to significant improvements in systolic BP, whether in-office or ambulatory, compared with a sham control procedure, in a new analysis that followed the trial’s patients out to 3 years. Those who underwent RDN also required less intense antihypertensive drug therapy.
“These findings support that durable blood pressure reductions with radiofrequency renal artery denervation, in the presence of lifestyle modification and maximal medical therapy, are safely achievable,” Deepak L. Bhatt, MD, said in a Sept. 18 presentation at the Transcatheter Cardiovascular Therapeutics annual meeting, which was sponsored by the Cardiovascular Research Foundation.
Dr. Bhatt, of Boston’s Brigham and Women’s Hospital and Harvard Medical School, is lead author on the report published in The Lancet simultaneously with his presentation.
Strides in RDN technology and trial design since the neutral primary SYMPLICITY HTN-3 results were reported in 2014 have long since restored faith in the procedure, which is currently in advanced stages of clinical trials and expected to eventually make a mark on practice.
But Roxana Mehran, MD, not connected to SYMPLICITY HTN-3, expressed caution in interpreting the current analysis based on secondary endpoints and extended follow-up time.
And elsewhere at the TCT sessions, observers of the trial as well as Dr. Bhatt urged similar cautions interpreting “positive” secondary results from trials that were “negative” in their primary analyses.
Still, “I believe there is no question that we have now enough evidence to say that renal denervation on top of medications is probably something that we’re going to be seeing in the future,” Dr. Mehran, of the Icahn School of Medicine at Mount Sinai, New York, told this news organization.
Importantly, and a bit controversially, the RDN group in the 36-month SYMPLICITY HTN-3 analysis includes patients originally assigned to the sham control group who crossed over to receive RDN after the trial was unblinded. Their “control” BP responses were thereafter imputed by accepted statistical methodology that Dr. Bhatt characterized as “last observation carried forward.”
That’s another reason to be circumspect about the current results, observed Naomi Fisher, MD, also of Brigham and Women’s and Harvard Medical School, as a panelist following Dr. Bhatt’s formal presentation.
“With all the missing data and imputational calculations,” she said, “I think we have to apply caution in the interpretation.”
She also pointed out that blinding in the trial was lifted at 6 months, allowing patients to learn their treatment assignment, and potentially influencing subsequent changes to medications.
They were prescribed, on average, about five antihypertensive meds, Dr. Fisher noted, and “that’s already a red flag. Patients taking that many medications generally aren’t universally taking them. There’s very high likelihood that there could have been variable adherence.”
Patients who learned they were in the sham control group, for example, could have “fallen off” taking their medications, potentially worsening outcomes and amplifying the apparent benefit of RDN. Such an effect, Dr. Fisher said, “could have contributed” to the study’s long-term results.
As previously reported, the single-blind SYMPLICITY HTN-3 had randomly assigned 535 patients to either RDN or a sham control procedure, 364 and 171 patients respectively, at 88 U.S. centers. The trial used the Symplicity Flex RDN radiofrequency ablation catheter (Medtronic).
For study entry, patients were required to have office systolic BP of at least 160 mm Hg and 24-hour ambulatory systolic BP of at least 135 mm Hg despite stable, maximally tolerated dosages of a diuretic plus at least two other antihypertensive agents.
Blinding was lifted at 6 months, per protocol, after which patients in the sham control group who still met the trial’s BP entry criteria were allowed to cross over and undergo RDN. The 101 controls who crossed over were combined with the original active-therapy cohort for the current analysis.
From baseline to 36 months, mean number of medication classes per patient maintained between 4.5 and 5, with no significant difference between groups at any point.
However, medication burden expressed as number of doses daily held steady between 9.7 to 10.2 for controls while the RDN group showed a steady decline from 10.2 to 8.4. Differences between RDN patients and controls were significant at both 24 months (P = .01) and 36 months (P = .005), Dr. Bhatt reported.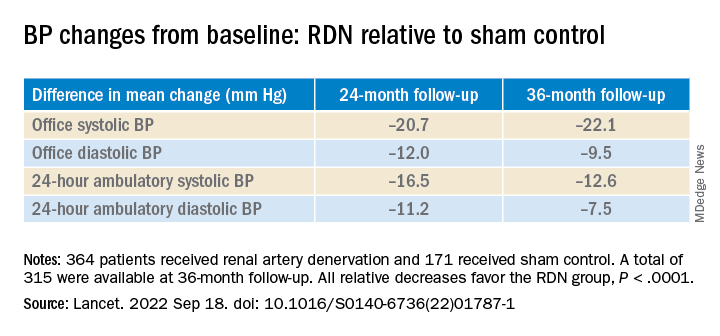
All relative decreases favor the RDN group, P < .0001
The RDN group spent a longer percentage of time with systolic BP at goal compared to those in the sham control group in an analysis that did not involve imputation of data, Dr. Bhatt reported. The proportions of time in therapeutic range were 18% for RDN patients and 9% for controls (P < .0001).
As in the 6- and 12-month analyses, there was no adverse safety signal associated with RDN in follow-up out to both 36 and 48 months. As Dr. Bhatt reported, the rates of the composite safety endpoint in RDN patients, crossovers, and noncrossover controls were 15%, 14%, and 14%, respectively.
The safety endpoint included death, new end-stage renal disease, significant embolic events causing end-organ damage, vascular complications, renal-artery reintervention, and “hypertensive emergency unrelated to nonadherence to medications,” Dr. Bhatt reported.
There are many patients with “out of control” HTN “who cannot remain compliant on their medications,” Dr. Mehran observed for this news organization. “I believe having an adjunct to medical management of these patients,” that is RDN, “is going to be tremendously important.”
SYMPLICITY HTN-3 was funded by Medtronic. Dr. Bhatt has disclosed ties with many companies, as well as WebMD, Medscape Cardiology, and other publications or organizations. Dr. Mehran disclosed ties to Abbott Vascular, AstraZeneca, Bayer, Bristol-Myers Squibb, CSL Behring, Daiichi-Sankyo/Eli Lilly, Medtronic, Novartis, OrbusNeich, Abiomed; Boston Scientific, Alleviant, Amgen, AM-Pharma, Applied Therapeutics, Arena, BAIM, Biosensors, Biotronik, CardiaWave, CellAegis, Concept Medical, CeloNova, CERC, Chiesi, Cytosorbents, Duke University, Element Science, Faraday, Humacyte, Idorsia, Insel Gruppe, Philips, RenalPro, Vivasure, and Zoll; as well as Medscape/WebMD, and Cine-Med Research; and holding equity, stock, or stock options with Control Rad, Applied Therapeutics, and Elixir Medical. Dr. Fisher disclosed ties to Medtronic, Recor Medical, and Aktiia; and receiving grants or hold research contracts with Recor Medical and Aktiia.
A version of this article first appeared on Medscape.com.
BOSTON – There’s an intriguing plot twist in the story of SYMPLICITY HTN-3, the sham-controlled clinical trial that nearly put the kibosh on renal denervation (RDN) therapy as a promising approach to treatment-resistant hypertension (HTN).
The trial famously showed no benefit for systolic blood pressure (BP) from the invasive procedure at 6 months and 12 months, dampening enthusiasm for RDN in HTN for both physicians and industry. But it turns out that disappointment in the study may have been premature.
The procedure led to significant improvements in systolic BP, whether in-office or ambulatory, compared with a sham control procedure, in a new analysis that followed the trial’s patients out to 3 years. Those who underwent RDN also required less intense antihypertensive drug therapy.
“These findings support that durable blood pressure reductions with radiofrequency renal artery denervation, in the presence of lifestyle modification and maximal medical therapy, are safely achievable,” Deepak L. Bhatt, MD, said in a Sept. 18 presentation at the Transcatheter Cardiovascular Therapeutics annual meeting, which was sponsored by the Cardiovascular Research Foundation.
Dr. Bhatt, of Boston’s Brigham and Women’s Hospital and Harvard Medical School, is lead author on the report published in The Lancet simultaneously with his presentation.
Strides in RDN technology and trial design since the neutral primary SYMPLICITY HTN-3 results were reported in 2014 have long since restored faith in the procedure, which is currently in advanced stages of clinical trials and expected to eventually make a mark on practice.
But Roxana Mehran, MD, not connected to SYMPLICITY HTN-3, expressed caution in interpreting the current analysis based on secondary endpoints and extended follow-up time.
And elsewhere at the TCT sessions, observers of the trial as well as Dr. Bhatt urged similar cautions interpreting “positive” secondary results from trials that were “negative” in their primary analyses.
Still, “I believe there is no question that we have now enough evidence to say that renal denervation on top of medications is probably something that we’re going to be seeing in the future,” Dr. Mehran, of the Icahn School of Medicine at Mount Sinai, New York, told this news organization.
Importantly, and a bit controversially, the RDN group in the 36-month SYMPLICITY HTN-3 analysis includes patients originally assigned to the sham control group who crossed over to receive RDN after the trial was unblinded. Their “control” BP responses were thereafter imputed by accepted statistical methodology that Dr. Bhatt characterized as “last observation carried forward.”
That’s another reason to be circumspect about the current results, observed Naomi Fisher, MD, also of Brigham and Women’s and Harvard Medical School, as a panelist following Dr. Bhatt’s formal presentation.
“With all the missing data and imputational calculations,” she said, “I think we have to apply caution in the interpretation.”
She also pointed out that blinding in the trial was lifted at 6 months, allowing patients to learn their treatment assignment, and potentially influencing subsequent changes to medications.
They were prescribed, on average, about five antihypertensive meds, Dr. Fisher noted, and “that’s already a red flag. Patients taking that many medications generally aren’t universally taking them. There’s very high likelihood that there could have been variable adherence.”
Patients who learned they were in the sham control group, for example, could have “fallen off” taking their medications, potentially worsening outcomes and amplifying the apparent benefit of RDN. Such an effect, Dr. Fisher said, “could have contributed” to the study’s long-term results.
As previously reported, the single-blind SYMPLICITY HTN-3 had randomly assigned 535 patients to either RDN or a sham control procedure, 364 and 171 patients respectively, at 88 U.S. centers. The trial used the Symplicity Flex RDN radiofrequency ablation catheter (Medtronic).
For study entry, patients were required to have office systolic BP of at least 160 mm Hg and 24-hour ambulatory systolic BP of at least 135 mm Hg despite stable, maximally tolerated dosages of a diuretic plus at least two other antihypertensive agents.
Blinding was lifted at 6 months, per protocol, after which patients in the sham control group who still met the trial’s BP entry criteria were allowed to cross over and undergo RDN. The 101 controls who crossed over were combined with the original active-therapy cohort for the current analysis.
From baseline to 36 months, mean number of medication classes per patient maintained between 4.5 and 5, with no significant difference between groups at any point.
However, medication burden expressed as number of doses daily held steady between 9.7 to 10.2 for controls while the RDN group showed a steady decline from 10.2 to 8.4. Differences between RDN patients and controls were significant at both 24 months (P = .01) and 36 months (P = .005), Dr. Bhatt reported.
All relative decreases favor the RDN group, P < .0001
The RDN group spent a longer percentage of time with systolic BP at goal compared to those in the sham control group in an analysis that did not involve imputation of data, Dr. Bhatt reported. The proportions of time in therapeutic range were 18% for RDN patients and 9% for controls (P < .0001).
As in the 6- and 12-month analyses, there was no adverse safety signal associated with RDN in follow-up out to both 36 and 48 months. As Dr. Bhatt reported, the rates of the composite safety endpoint in RDN patients, crossovers, and noncrossover controls were 15%, 14%, and 14%, respectively.
The safety endpoint included death, new end-stage renal disease, significant embolic events causing end-organ damage, vascular complications, renal-artery reintervention, and “hypertensive emergency unrelated to nonadherence to medications,” Dr. Bhatt reported.
There are many patients with “out of control” HTN “who cannot remain compliant on their medications,” Dr. Mehran observed for this news organization. “I believe having an adjunct to medical management of these patients,” that is RDN, “is going to be tremendously important.”
SYMPLICITY HTN-3 was funded by Medtronic. Dr. Bhatt has disclosed ties with many companies, as well as WebMD, Medscape Cardiology, and other publications or organizations. Dr. Mehran disclosed ties to Abbott Vascular, AstraZeneca, Bayer, Bristol-Myers Squibb, CSL Behring, Daiichi-Sankyo/Eli Lilly, Medtronic, Novartis, OrbusNeich, Abiomed; Boston Scientific, Alleviant, Amgen, AM-Pharma, Applied Therapeutics, Arena, BAIM, Biosensors, Biotronik, CardiaWave, CellAegis, Concept Medical, CeloNova, CERC, Chiesi, Cytosorbents, Duke University, Element Science, Faraday, Humacyte, Idorsia, Insel Gruppe, Philips, RenalPro, Vivasure, and Zoll; as well as Medscape/WebMD, and Cine-Med Research; and holding equity, stock, or stock options with Control Rad, Applied Therapeutics, and Elixir Medical. Dr. Fisher disclosed ties to Medtronic, Recor Medical, and Aktiia; and receiving grants or hold research contracts with Recor Medical and Aktiia.
A version of this article first appeared on Medscape.com.
BOSTON – There’s an intriguing plot twist in the story of SYMPLICITY HTN-3, the sham-controlled clinical trial that nearly put the kibosh on renal denervation (RDN) therapy as a promising approach to treatment-resistant hypertension (HTN).
The trial famously showed no benefit for systolic blood pressure (BP) from the invasive procedure at 6 months and 12 months, dampening enthusiasm for RDN in HTN for both physicians and industry. But it turns out that disappointment in the study may have been premature.
The procedure led to significant improvements in systolic BP, whether in-office or ambulatory, compared with a sham control procedure, in a new analysis that followed the trial’s patients out to 3 years. Those who underwent RDN also required less intense antihypertensive drug therapy.
“These findings support that durable blood pressure reductions with radiofrequency renal artery denervation, in the presence of lifestyle modification and maximal medical therapy, are safely achievable,” Deepak L. Bhatt, MD, said in a Sept. 18 presentation at the Transcatheter Cardiovascular Therapeutics annual meeting, which was sponsored by the Cardiovascular Research Foundation.
Dr. Bhatt, of Boston’s Brigham and Women’s Hospital and Harvard Medical School, is lead author on the report published in The Lancet simultaneously with his presentation.
Strides in RDN technology and trial design since the neutral primary SYMPLICITY HTN-3 results were reported in 2014 have long since restored faith in the procedure, which is currently in advanced stages of clinical trials and expected to eventually make a mark on practice.
But Roxana Mehran, MD, not connected to SYMPLICITY HTN-3, expressed caution in interpreting the current analysis based on secondary endpoints and extended follow-up time.
And elsewhere at the TCT sessions, observers of the trial as well as Dr. Bhatt urged similar cautions interpreting “positive” secondary results from trials that were “negative” in their primary analyses.
Still, “I believe there is no question that we have now enough evidence to say that renal denervation on top of medications is probably something that we’re going to be seeing in the future,” Dr. Mehran, of the Icahn School of Medicine at Mount Sinai, New York, told this news organization.
Importantly, and a bit controversially, the RDN group in the 36-month SYMPLICITY HTN-3 analysis includes patients originally assigned to the sham control group who crossed over to receive RDN after the trial was unblinded. Their “control” BP responses were thereafter imputed by accepted statistical methodology that Dr. Bhatt characterized as “last observation carried forward.”
That’s another reason to be circumspect about the current results, observed Naomi Fisher, MD, also of Brigham and Women’s and Harvard Medical School, as a panelist following Dr. Bhatt’s formal presentation.
“With all the missing data and imputational calculations,” she said, “I think we have to apply caution in the interpretation.”
She also pointed out that blinding in the trial was lifted at 6 months, allowing patients to learn their treatment assignment, and potentially influencing subsequent changes to medications.
They were prescribed, on average, about five antihypertensive meds, Dr. Fisher noted, and “that’s already a red flag. Patients taking that many medications generally aren’t universally taking them. There’s very high likelihood that there could have been variable adherence.”
Patients who learned they were in the sham control group, for example, could have “fallen off” taking their medications, potentially worsening outcomes and amplifying the apparent benefit of RDN. Such an effect, Dr. Fisher said, “could have contributed” to the study’s long-term results.
As previously reported, the single-blind SYMPLICITY HTN-3 had randomly assigned 535 patients to either RDN or a sham control procedure, 364 and 171 patients respectively, at 88 U.S. centers. The trial used the Symplicity Flex RDN radiofrequency ablation catheter (Medtronic).
For study entry, patients were required to have office systolic BP of at least 160 mm Hg and 24-hour ambulatory systolic BP of at least 135 mm Hg despite stable, maximally tolerated dosages of a diuretic plus at least two other antihypertensive agents.
Blinding was lifted at 6 months, per protocol, after which patients in the sham control group who still met the trial’s BP entry criteria were allowed to cross over and undergo RDN. The 101 controls who crossed over were combined with the original active-therapy cohort for the current analysis.
From baseline to 36 months, mean number of medication classes per patient maintained between 4.5 and 5, with no significant difference between groups at any point.
However, medication burden expressed as number of doses daily held steady between 9.7 to 10.2 for controls while the RDN group showed a steady decline from 10.2 to 8.4. Differences between RDN patients and controls were significant at both 24 months (P = .01) and 36 months (P = .005), Dr. Bhatt reported.
All relative decreases favor the RDN group, P < .0001
The RDN group spent a longer percentage of time with systolic BP at goal compared to those in the sham control group in an analysis that did not involve imputation of data, Dr. Bhatt reported. The proportions of time in therapeutic range were 18% for RDN patients and 9% for controls (P < .0001).
As in the 6- and 12-month analyses, there was no adverse safety signal associated with RDN in follow-up out to both 36 and 48 months. As Dr. Bhatt reported, the rates of the composite safety endpoint in RDN patients, crossovers, and noncrossover controls were 15%, 14%, and 14%, respectively.
The safety endpoint included death, new end-stage renal disease, significant embolic events causing end-organ damage, vascular complications, renal-artery reintervention, and “hypertensive emergency unrelated to nonadherence to medications,” Dr. Bhatt reported.
There are many patients with “out of control” HTN “who cannot remain compliant on their medications,” Dr. Mehran observed for this news organization. “I believe having an adjunct to medical management of these patients,” that is RDN, “is going to be tremendously important.”
SYMPLICITY HTN-3 was funded by Medtronic. Dr. Bhatt has disclosed ties with many companies, as well as WebMD, Medscape Cardiology, and other publications or organizations. Dr. Mehran disclosed ties to Abbott Vascular, AstraZeneca, Bayer, Bristol-Myers Squibb, CSL Behring, Daiichi-Sankyo/Eli Lilly, Medtronic, Novartis, OrbusNeich, Abiomed; Boston Scientific, Alleviant, Amgen, AM-Pharma, Applied Therapeutics, Arena, BAIM, Biosensors, Biotronik, CardiaWave, CellAegis, Concept Medical, CeloNova, CERC, Chiesi, Cytosorbents, Duke University, Element Science, Faraday, Humacyte, Idorsia, Insel Gruppe, Philips, RenalPro, Vivasure, and Zoll; as well as Medscape/WebMD, and Cine-Med Research; and holding equity, stock, or stock options with Control Rad, Applied Therapeutics, and Elixir Medical. Dr. Fisher disclosed ties to Medtronic, Recor Medical, and Aktiia; and receiving grants or hold research contracts with Recor Medical and Aktiia.
A version of this article first appeared on Medscape.com.
AT TCT 2022
Post-PCI FFR in multivessel disease predicts target vessel failure: FAME 3 analysis
Risk by FFR is continuous variable
In a new analysis of the previously published FAME 3 trial, which compared fractional flow reserve–guided percutaneous coronary interventions to coronary artery bypass surgery (CABG) in patients with three-vessel disease, post-PCI FFR was shown to predict both target vessel failure (TVF) and risk of cardiac events.
“We found that the post-PCI FFR had prognostic value both for the vessel and for the patient,” reported Zsolt Piroth, MD, PhD, deputy head, adult cardiology, György Gottsegen Institute of Cardiology, Budapest.

In this post hoc analysis, which was not a prespecified FAME 3 substudy, the goal was to look at the prognostic value of both post-PCI FFR and intravascular ultrasound, which were recommended in the study protocol. Several studies have addressed the value of these measures previously, according to Dr. Piroth, but he said the clinical value “has remained poorly defined” despite the currently available data.
The FAME 3 trial, published in the New England Journal of Medicine, was negative. It failed to confirm the study hypothesis that FFR-guided PCI is noninferior to CABG for the outcome of major adverse cardiac events (MACE) at 12 months.
However, this multinational trial has generated a large body of data with which to explore other issues relevant to revascularization. In this analysis, the goal was to evaluate whether post-PCI FFR predicted outcomes in complex multivessel revascularizations as it has been shown previously to do in single-vessel disease.
Presented at the Transcatheter Cardiovascular Therapeutics annual meeting, the focus of this analysis was on the 461 (61%) of patients in the 757-patient PCI arm of FAME 3 who underwent post-PCI FFR. The authors also looked at the predictive value of intravascular ultrasound, even though this was performed in just 11% of this group of trial participants.
As a continuous value, each 0.1-unit change in the post-PCI FFR was found to be prognostically significant for the outcome of TVF, defined as a composite of cardiac death, target vessel myocardial infarction, and target vessel revascularization (only postprocedural events were counted in this analysis). Specifically, for each 0.1-unit increase on a univariate analysis, the risk of TVF was reduced by about one-third (hazard ratio, 0.67; P = .0165).
On a patient level, a 0.1-unit increase in lowest post-PCI FFR of any assessed vessel was also associated with the same relative risk reduction (HR, 0.65; P = .0074) in the outcomes of cardiac death, target vessel MI, or target vessel revascularization, according to Dr. Piroth. On a receiver operating characteristic curve analysis, a value of 0.88 or below was predictive of TVF.
Although several other patient characteristics were also risk predictors of TVF on univariate analysis, only renal disease and the single lowest post-PCI FFR (as a continuous variable) emerged as predictors of TVF on multivariable analysis after adjustment for key clinical parameters, Dr. Piroth reported.
The reason why post-PCI FFR was not performed in almost 40% of patients randomized to PCI is unclear, but Dr. Piroth reported that the baseline characteristics of those who were or were not assessed with FFR after their procedure did not differ to any major degree.
Despite “a trend for improved outcomes in those who underwent post-PCI FFR,” Dr. Piroth, whose substudy was published in Circulation: Cardiovascular Interventions simultaneously with his TCT presentation, acknowledged that the reasons for a potential benefit cannot be derived from this post hoc analysis.
As for the prognostic value of IVUS, any conclusions are limited by the small proportion of patients who underwent this form of imaging. Overall, IVUS imaging was associated with longer procedures and more stents and “if anything, a signal for harm” in this analysis, but Dr. Piroth cautioned against any conclusions because of the small data pool.
The prognostic value of post-PCI FFR in complex multivessel disease is supported by these data, but the analysis was not designed to determine whether post-PCI FFR has relevance to intervention.
According to J. Dawn Abbott, MD, an FFR analysis conducted to identify lesions that are candidates for treatment should not be confused with FFR for physiologically guided PCI to optimize outcomes.
Noting that post-PCI FFR was encouraged in this study but not mandated and that these FFR values did not typically or necessarily lead to a change in management, take home messages about the value of post-PCI FFR in multivessel disease remain limited, said Dr. Abbott, director of interventional cardiology fellowship training, Brown University, Providence, R.I.
“There was a trend toward improved outcomes in patients who had this measurement done, but, unfortunately, we do not have data regarding whether these patients had further interventions performed,” Dr. Piroth acknowledged.
The post-PCI FFR values were made available to the treating physicians, but Dr. Piroth reiterated that it is unknown whether the physicians considered this information actionable. Moreover, “the vast majority had a nonsignificant post-PCI FFR” result, and “all of the patients had an angiographically successful PCI,” Dr. Piroth added.
Dr. Piroth has financial relationships with Abbott Vascular and Boston Scientific. Dr. Abbott reports financial relationships with Abbott Vascular, Boston Scientific, Medtronic, Microport, Philips, Penumbra, Recor, and Shockwave.
Risk by FFR is continuous variable
Risk by FFR is continuous variable
In a new analysis of the previously published FAME 3 trial, which compared fractional flow reserve–guided percutaneous coronary interventions to coronary artery bypass surgery (CABG) in patients with three-vessel disease, post-PCI FFR was shown to predict both target vessel failure (TVF) and risk of cardiac events.
“We found that the post-PCI FFR had prognostic value both for the vessel and for the patient,” reported Zsolt Piroth, MD, PhD, deputy head, adult cardiology, György Gottsegen Institute of Cardiology, Budapest.
In this post hoc analysis, which was not a prespecified FAME 3 substudy, the goal was to look at the prognostic value of both post-PCI FFR and intravascular ultrasound, which were recommended in the study protocol. Several studies have addressed the value of these measures previously, according to Dr. Piroth, but he said the clinical value “has remained poorly defined” despite the currently available data.
The FAME 3 trial, published in the New England Journal of Medicine, was negative. It failed to confirm the study hypothesis that FFR-guided PCI is noninferior to CABG for the outcome of major adverse cardiac events (MACE) at 12 months.
However, this multinational trial has generated a large body of data with which to explore other issues relevant to revascularization. In this analysis, the goal was to evaluate whether post-PCI FFR predicted outcomes in complex multivessel revascularizations as it has been shown previously to do in single-vessel disease.
Presented at the Transcatheter Cardiovascular Therapeutics annual meeting, the focus of this analysis was on the 461 (61%) of patients in the 757-patient PCI arm of FAME 3 who underwent post-PCI FFR. The authors also looked at the predictive value of intravascular ultrasound, even though this was performed in just 11% of this group of trial participants.
As a continuous value, each 0.1-unit change in the post-PCI FFR was found to be prognostically significant for the outcome of TVF, defined as a composite of cardiac death, target vessel myocardial infarction, and target vessel revascularization (only postprocedural events were counted in this analysis). Specifically, for each 0.1-unit increase on a univariate analysis, the risk of TVF was reduced by about one-third (hazard ratio, 0.67; P = .0165).
On a patient level, a 0.1-unit increase in lowest post-PCI FFR of any assessed vessel was also associated with the same relative risk reduction (HR, 0.65; P = .0074) in the outcomes of cardiac death, target vessel MI, or target vessel revascularization, according to Dr. Piroth. On a receiver operating characteristic curve analysis, a value of 0.88 or below was predictive of TVF.
Although several other patient characteristics were also risk predictors of TVF on univariate analysis, only renal disease and the single lowest post-PCI FFR (as a continuous variable) emerged as predictors of TVF on multivariable analysis after adjustment for key clinical parameters, Dr. Piroth reported.
The reason why post-PCI FFR was not performed in almost 40% of patients randomized to PCI is unclear, but Dr. Piroth reported that the baseline characteristics of those who were or were not assessed with FFR after their procedure did not differ to any major degree.
Despite “a trend for improved outcomes in those who underwent post-PCI FFR,” Dr. Piroth, whose substudy was published in Circulation: Cardiovascular Interventions simultaneously with his TCT presentation, acknowledged that the reasons for a potential benefit cannot be derived from this post hoc analysis.
As for the prognostic value of IVUS, any conclusions are limited by the small proportion of patients who underwent this form of imaging. Overall, IVUS imaging was associated with longer procedures and more stents and “if anything, a signal for harm” in this analysis, but Dr. Piroth cautioned against any conclusions because of the small data pool.
The prognostic value of post-PCI FFR in complex multivessel disease is supported by these data, but the analysis was not designed to determine whether post-PCI FFR has relevance to intervention.
According to J. Dawn Abbott, MD, an FFR analysis conducted to identify lesions that are candidates for treatment should not be confused with FFR for physiologically guided PCI to optimize outcomes.
Noting that post-PCI FFR was encouraged in this study but not mandated and that these FFR values did not typically or necessarily lead to a change in management, take home messages about the value of post-PCI FFR in multivessel disease remain limited, said Dr. Abbott, director of interventional cardiology fellowship training, Brown University, Providence, R.I.
“There was a trend toward improved outcomes in patients who had this measurement done, but, unfortunately, we do not have data regarding whether these patients had further interventions performed,” Dr. Piroth acknowledged.
The post-PCI FFR values were made available to the treating physicians, but Dr. Piroth reiterated that it is unknown whether the physicians considered this information actionable. Moreover, “the vast majority had a nonsignificant post-PCI FFR” result, and “all of the patients had an angiographically successful PCI,” Dr. Piroth added.
Dr. Piroth has financial relationships with Abbott Vascular and Boston Scientific. Dr. Abbott reports financial relationships with Abbott Vascular, Boston Scientific, Medtronic, Microport, Philips, Penumbra, Recor, and Shockwave.
In a new analysis of the previously published FAME 3 trial, which compared fractional flow reserve–guided percutaneous coronary interventions to coronary artery bypass surgery (CABG) in patients with three-vessel disease, post-PCI FFR was shown to predict both target vessel failure (TVF) and risk of cardiac events.
“We found that the post-PCI FFR had prognostic value both for the vessel and for the patient,” reported Zsolt Piroth, MD, PhD, deputy head, adult cardiology, György Gottsegen Institute of Cardiology, Budapest.
In this post hoc analysis, which was not a prespecified FAME 3 substudy, the goal was to look at the prognostic value of both post-PCI FFR and intravascular ultrasound, which were recommended in the study protocol. Several studies have addressed the value of these measures previously, according to Dr. Piroth, but he said the clinical value “has remained poorly defined” despite the currently available data.
The FAME 3 trial, published in the New England Journal of Medicine, was negative. It failed to confirm the study hypothesis that FFR-guided PCI is noninferior to CABG for the outcome of major adverse cardiac events (MACE) at 12 months.
However, this multinational trial has generated a large body of data with which to explore other issues relevant to revascularization. In this analysis, the goal was to evaluate whether post-PCI FFR predicted outcomes in complex multivessel revascularizations as it has been shown previously to do in single-vessel disease.
Presented at the Transcatheter Cardiovascular Therapeutics annual meeting, the focus of this analysis was on the 461 (61%) of patients in the 757-patient PCI arm of FAME 3 who underwent post-PCI FFR. The authors also looked at the predictive value of intravascular ultrasound, even though this was performed in just 11% of this group of trial participants.
As a continuous value, each 0.1-unit change in the post-PCI FFR was found to be prognostically significant for the outcome of TVF, defined as a composite of cardiac death, target vessel myocardial infarction, and target vessel revascularization (only postprocedural events were counted in this analysis). Specifically, for each 0.1-unit increase on a univariate analysis, the risk of TVF was reduced by about one-third (hazard ratio, 0.67; P = .0165).
On a patient level, a 0.1-unit increase in lowest post-PCI FFR of any assessed vessel was also associated with the same relative risk reduction (HR, 0.65; P = .0074) in the outcomes of cardiac death, target vessel MI, or target vessel revascularization, according to Dr. Piroth. On a receiver operating characteristic curve analysis, a value of 0.88 or below was predictive of TVF.
Although several other patient characteristics were also risk predictors of TVF on univariate analysis, only renal disease and the single lowest post-PCI FFR (as a continuous variable) emerged as predictors of TVF on multivariable analysis after adjustment for key clinical parameters, Dr. Piroth reported.
The reason why post-PCI FFR was not performed in almost 40% of patients randomized to PCI is unclear, but Dr. Piroth reported that the baseline characteristics of those who were or were not assessed with FFR after their procedure did not differ to any major degree.
Despite “a trend for improved outcomes in those who underwent post-PCI FFR,” Dr. Piroth, whose substudy was published in Circulation: Cardiovascular Interventions simultaneously with his TCT presentation, acknowledged that the reasons for a potential benefit cannot be derived from this post hoc analysis.
As for the prognostic value of IVUS, any conclusions are limited by the small proportion of patients who underwent this form of imaging. Overall, IVUS imaging was associated with longer procedures and more stents and “if anything, a signal for harm” in this analysis, but Dr. Piroth cautioned against any conclusions because of the small data pool.
The prognostic value of post-PCI FFR in complex multivessel disease is supported by these data, but the analysis was not designed to determine whether post-PCI FFR has relevance to intervention.
According to J. Dawn Abbott, MD, an FFR analysis conducted to identify lesions that are candidates for treatment should not be confused with FFR for physiologically guided PCI to optimize outcomes.
Noting that post-PCI FFR was encouraged in this study but not mandated and that these FFR values did not typically or necessarily lead to a change in management, take home messages about the value of post-PCI FFR in multivessel disease remain limited, said Dr. Abbott, director of interventional cardiology fellowship training, Brown University, Providence, R.I.
“There was a trend toward improved outcomes in patients who had this measurement done, but, unfortunately, we do not have data regarding whether these patients had further interventions performed,” Dr. Piroth acknowledged.
The post-PCI FFR values were made available to the treating physicians, but Dr. Piroth reiterated that it is unknown whether the physicians considered this information actionable. Moreover, “the vast majority had a nonsignificant post-PCI FFR” result, and “all of the patients had an angiographically successful PCI,” Dr. Piroth added.
Dr. Piroth has financial relationships with Abbott Vascular and Boston Scientific. Dr. Abbott reports financial relationships with Abbott Vascular, Boston Scientific, Medtronic, Microport, Philips, Penumbra, Recor, and Shockwave.
FROM TCT 2022
Amulet, Watchman 2.5 LAAO outcomes neck and neck at 3 years
The Amplatzer Amulet (Abbott) and first-generation Watchman 2.5 (Boston Scientific) devices provide relatively comparable results out to 3 years after left atrial appendage occlusion (LAAO), longer follow-up from the Amplatzer Amulet Left Atrial Appendage Occluder Versus Watchman Device for Stroke Prophylaxis (Amulet IDE) trial shows.

“The dual-seal Amplatzer Amulet left atrial appendage occluder continued to demonstrate safety and effectiveness through 3 years,” principal investigator Dhanunjaya Lakkireddy, MD, said in a late-breaking session at the recent Transcatheter Cardiovascular Therapeutics annual meeting.
Preliminary results, reported last year, showed that procedural complications were higher with the Amplatzer but that it provided superior closure of the left atrial appendage (LAA) at 45 days and was noninferior with respect to safety at 12 months and efficacy at 18 months.
Amulet IDE is the largest head-to-head comparison of the two devices, enrolling 1,878 high-risk patients with nonvalvular atrial fibrillation undergoing LAA closure to reduce the risk of stroke.
Three-year follow-up was higher with the Amulet device than with the Watchman, at 721 vs. 659 patients, driven by increased deaths (85 vs. 63) and withdrawals (50 vs. 23) in the Watchman group within 18 months, noted Dr. Lakkireddy, Kansas City Heart Rhythm Institute and Research Foundation, Overland Park, Kan.
Use of oral anticoagulation was higher in the Watchman group at 6 months (2.8% vs. 4.7%; P = .04), 18 months (3.1% vs. 5.6%; P = .01), and 3 years (3.7% vs. 7.3%; P < .01).
This was primarily driven by more late device-related thrombus (DRT) after 6 months with the Watchman device than with the Amulet occluder (23 vs. 10). “Perhaps the dual-closure mechanism of the Amulet explains this fundamental difference, where you have a nice smooth disc that covers the ostium,” he posited.
At 3 years, rates of cardiovascular death trended lower with Amulet than with Watchman (6.6% vs. 8.5%; P = .14), as did all-cause deaths (14.6% vs. 17.9%; P = .07).
Most cardiovascular deaths in the Amulet group were not preceded by a device factor, whereas DRT (1 vs. 4) and peridevice leak 3 mm or more (5 vs. 15) frequently preceded these deaths in the Watchman group, Dr. Lakkireddy observed. No pericardial effusion-related deaths occurred in either group.
Major bleeding, however, trended higher for the Amulet, at 16.1%, compared with 14.7% for the Watchman (P = .46). Ischemic stroke and systemic embolic rates also trended higher for Amulet, at 5%, and 4.6% for Watchman.
The protocol recommended aspirin only for both groups after 6 months. None of the 29 Amulet and 3 of the 29 Watchman patients with an ischemic stroke were on oral anticoagulation at the time of the stroke.
Device factors, however, frequently preceded ischemic strokes in the Watchman group, Dr. Lakkireddy said. DRT occurred in 1 patient with Amulet and 2 patients with Watchman and peridevice leak in 3 with Amulet and 15 with Watchman. “Again, the peridevice leak issue really stands out as an important factor,” he said at the meeting, which was sponsored by the Cardiovascular Research Foundation.
Based on “data from the large trials, it’s clearly evident that the presence of peridevice leak significantly raises the risk of stroke in follow-up,” he said. “So, attention has to be paid to the choice of the device and how we can mitigate the risk of peridevice leaks in these patients.”
The composite of stroke, systemic embolism, and cardiovascular death occurred in 11.1% of patients with Amulet and 12.7% with Watchman (P = .31).
Asked following the formal presentation whether the results justify use of one device over the other for LAA occlusion, Dr. Lakkireddy said he likes the dual closure mechanism of the Amulet and is more likely to use it in patients with proximal lobes, very large appendages, or a relatively shallow appendage. “In the rest of the cases, I think it’s a toss-up.”
As for how generalizable the results are, he noted that the study tested the Amulet against the legacy Watchman 2.5 but that the second-generation Watchman FLX is available in a larger size and has shown improved performance.
The Amplatzer Amulet does not require oral anticoagulants at discharge. However, the indication for the Watchman FLX was recently expanded to include 45-day dual antiplatelet therapy as a postprocedure alternative to oral anticoagulation plus aspirin.
Going forward, the “next evolution” is to test the Watchman FLX and Amulet on either single antiplatelet or a dual antiplatelet regimen without oral anticoagulation, he suggested.
Results from SWISS APERO, the first randomized trial to compare the Amulet and Watchman FLX (and a handful of 2.5 devices) in 221 patients, showed that the devices are not interchangeable for rates of complications or leaks.
During a press conference prior to the presentation, discussant Federico Asch, MD, MedStar Health Research Institute, Washington, said, “the most exciting thing here is that we have good options. We now can start to tease out which patients will benefit best from one or the other because we actually have two options.”
The Amulet IDE trial was funded by Abbott. Dr. Lakkireddy reports that he or his spouse/partner have received grant/research support from Abbott, AtriCure, Alta Thera, Medtronic, Biosense Webster, Biotronik, and Boston Scientific; and speaker honoraria from Abbott, Medtronic, Biotronik, and Boston Scientific.
A version of this article first appeared on Medscape.com.
The Amplatzer Amulet (Abbott) and first-generation Watchman 2.5 (Boston Scientific) devices provide relatively comparable results out to 3 years after left atrial appendage occlusion (LAAO), longer follow-up from the Amplatzer Amulet Left Atrial Appendage Occluder Versus Watchman Device for Stroke Prophylaxis (Amulet IDE) trial shows.
“The dual-seal Amplatzer Amulet left atrial appendage occluder continued to demonstrate safety and effectiveness through 3 years,” principal investigator Dhanunjaya Lakkireddy, MD, said in a late-breaking session at the recent Transcatheter Cardiovascular Therapeutics annual meeting.
Preliminary results, reported last year, showed that procedural complications were higher with the Amplatzer but that it provided superior closure of the left atrial appendage (LAA) at 45 days and was noninferior with respect to safety at 12 months and efficacy at 18 months.
Amulet IDE is the largest head-to-head comparison of the two devices, enrolling 1,878 high-risk patients with nonvalvular atrial fibrillation undergoing LAA closure to reduce the risk of stroke.
Three-year follow-up was higher with the Amulet device than with the Watchman, at 721 vs. 659 patients, driven by increased deaths (85 vs. 63) and withdrawals (50 vs. 23) in the Watchman group within 18 months, noted Dr. Lakkireddy, Kansas City Heart Rhythm Institute and Research Foundation, Overland Park, Kan.
Use of oral anticoagulation was higher in the Watchman group at 6 months (2.8% vs. 4.7%; P = .04), 18 months (3.1% vs. 5.6%; P = .01), and 3 years (3.7% vs. 7.3%; P < .01).
This was primarily driven by more late device-related thrombus (DRT) after 6 months with the Watchman device than with the Amulet occluder (23 vs. 10). “Perhaps the dual-closure mechanism of the Amulet explains this fundamental difference, where you have a nice smooth disc that covers the ostium,” he posited.
At 3 years, rates of cardiovascular death trended lower with Amulet than with Watchman (6.6% vs. 8.5%; P = .14), as did all-cause deaths (14.6% vs. 17.9%; P = .07).
Most cardiovascular deaths in the Amulet group were not preceded by a device factor, whereas DRT (1 vs. 4) and peridevice leak 3 mm or more (5 vs. 15) frequently preceded these deaths in the Watchman group, Dr. Lakkireddy observed. No pericardial effusion-related deaths occurred in either group.
Major bleeding, however, trended higher for the Amulet, at 16.1%, compared with 14.7% for the Watchman (P = .46). Ischemic stroke and systemic embolic rates also trended higher for Amulet, at 5%, and 4.6% for Watchman.
The protocol recommended aspirin only for both groups after 6 months. None of the 29 Amulet and 3 of the 29 Watchman patients with an ischemic stroke were on oral anticoagulation at the time of the stroke.
Device factors, however, frequently preceded ischemic strokes in the Watchman group, Dr. Lakkireddy said. DRT occurred in 1 patient with Amulet and 2 patients with Watchman and peridevice leak in 3 with Amulet and 15 with Watchman. “Again, the peridevice leak issue really stands out as an important factor,” he said at the meeting, which was sponsored by the Cardiovascular Research Foundation.
Based on “data from the large trials, it’s clearly evident that the presence of peridevice leak significantly raises the risk of stroke in follow-up,” he said. “So, attention has to be paid to the choice of the device and how we can mitigate the risk of peridevice leaks in these patients.”
The composite of stroke, systemic embolism, and cardiovascular death occurred in 11.1% of patients with Amulet and 12.7% with Watchman (P = .31).
Asked following the formal presentation whether the results justify use of one device over the other for LAA occlusion, Dr. Lakkireddy said he likes the dual closure mechanism of the Amulet and is more likely to use it in patients with proximal lobes, very large appendages, or a relatively shallow appendage. “In the rest of the cases, I think it’s a toss-up.”
As for how generalizable the results are, he noted that the study tested the Amulet against the legacy Watchman 2.5 but that the second-generation Watchman FLX is available in a larger size and has shown improved performance.
The Amplatzer Amulet does not require oral anticoagulants at discharge. However, the indication for the Watchman FLX was recently expanded to include 45-day dual antiplatelet therapy as a postprocedure alternative to oral anticoagulation plus aspirin.
Going forward, the “next evolution” is to test the Watchman FLX and Amulet on either single antiplatelet or a dual antiplatelet regimen without oral anticoagulation, he suggested.
Results from SWISS APERO, the first randomized trial to compare the Amulet and Watchman FLX (and a handful of 2.5 devices) in 221 patients, showed that the devices are not interchangeable for rates of complications or leaks.
During a press conference prior to the presentation, discussant Federico Asch, MD, MedStar Health Research Institute, Washington, said, “the most exciting thing here is that we have good options. We now can start to tease out which patients will benefit best from one or the other because we actually have two options.”
The Amulet IDE trial was funded by Abbott. Dr. Lakkireddy reports that he or his spouse/partner have received grant/research support from Abbott, AtriCure, Alta Thera, Medtronic, Biosense Webster, Biotronik, and Boston Scientific; and speaker honoraria from Abbott, Medtronic, Biotronik, and Boston Scientific.
A version of this article first appeared on Medscape.com.
The Amplatzer Amulet (Abbott) and first-generation Watchman 2.5 (Boston Scientific) devices provide relatively comparable results out to 3 years after left atrial appendage occlusion (LAAO), longer follow-up from the Amplatzer Amulet Left Atrial Appendage Occluder Versus Watchman Device for Stroke Prophylaxis (Amulet IDE) trial shows.
“The dual-seal Amplatzer Amulet left atrial appendage occluder continued to demonstrate safety and effectiveness through 3 years,” principal investigator Dhanunjaya Lakkireddy, MD, said in a late-breaking session at the recent Transcatheter Cardiovascular Therapeutics annual meeting.
Preliminary results, reported last year, showed that procedural complications were higher with the Amplatzer but that it provided superior closure of the left atrial appendage (LAA) at 45 days and was noninferior with respect to safety at 12 months and efficacy at 18 months.
Amulet IDE is the largest head-to-head comparison of the two devices, enrolling 1,878 high-risk patients with nonvalvular atrial fibrillation undergoing LAA closure to reduce the risk of stroke.
Three-year follow-up was higher with the Amulet device than with the Watchman, at 721 vs. 659 patients, driven by increased deaths (85 vs. 63) and withdrawals (50 vs. 23) in the Watchman group within 18 months, noted Dr. Lakkireddy, Kansas City Heart Rhythm Institute and Research Foundation, Overland Park, Kan.
Use of oral anticoagulation was higher in the Watchman group at 6 months (2.8% vs. 4.7%; P = .04), 18 months (3.1% vs. 5.6%; P = .01), and 3 years (3.7% vs. 7.3%; P < .01).
This was primarily driven by more late device-related thrombus (DRT) after 6 months with the Watchman device than with the Amulet occluder (23 vs. 10). “Perhaps the dual-closure mechanism of the Amulet explains this fundamental difference, where you have a nice smooth disc that covers the ostium,” he posited.
At 3 years, rates of cardiovascular death trended lower with Amulet than with Watchman (6.6% vs. 8.5%; P = .14), as did all-cause deaths (14.6% vs. 17.9%; P = .07).
Most cardiovascular deaths in the Amulet group were not preceded by a device factor, whereas DRT (1 vs. 4) and peridevice leak 3 mm or more (5 vs. 15) frequently preceded these deaths in the Watchman group, Dr. Lakkireddy observed. No pericardial effusion-related deaths occurred in either group.
Major bleeding, however, trended higher for the Amulet, at 16.1%, compared with 14.7% for the Watchman (P = .46). Ischemic stroke and systemic embolic rates also trended higher for Amulet, at 5%, and 4.6% for Watchman.
The protocol recommended aspirin only for both groups after 6 months. None of the 29 Amulet and 3 of the 29 Watchman patients with an ischemic stroke were on oral anticoagulation at the time of the stroke.
Device factors, however, frequently preceded ischemic strokes in the Watchman group, Dr. Lakkireddy said. DRT occurred in 1 patient with Amulet and 2 patients with Watchman and peridevice leak in 3 with Amulet and 15 with Watchman. “Again, the peridevice leak issue really stands out as an important factor,” he said at the meeting, which was sponsored by the Cardiovascular Research Foundation.
Based on “data from the large trials, it’s clearly evident that the presence of peridevice leak significantly raises the risk of stroke in follow-up,” he said. “So, attention has to be paid to the choice of the device and how we can mitigate the risk of peridevice leaks in these patients.”
The composite of stroke, systemic embolism, and cardiovascular death occurred in 11.1% of patients with Amulet and 12.7% with Watchman (P = .31).
Asked following the formal presentation whether the results justify use of one device over the other for LAA occlusion, Dr. Lakkireddy said he likes the dual closure mechanism of the Amulet and is more likely to use it in patients with proximal lobes, very large appendages, or a relatively shallow appendage. “In the rest of the cases, I think it’s a toss-up.”
As for how generalizable the results are, he noted that the study tested the Amulet against the legacy Watchman 2.5 but that the second-generation Watchman FLX is available in a larger size and has shown improved performance.
The Amplatzer Amulet does not require oral anticoagulants at discharge. However, the indication for the Watchman FLX was recently expanded to include 45-day dual antiplatelet therapy as a postprocedure alternative to oral anticoagulation plus aspirin.
Going forward, the “next evolution” is to test the Watchman FLX and Amulet on either single antiplatelet or a dual antiplatelet regimen without oral anticoagulation, he suggested.
Results from SWISS APERO, the first randomized trial to compare the Amulet and Watchman FLX (and a handful of 2.5 devices) in 221 patients, showed that the devices are not interchangeable for rates of complications or leaks.
During a press conference prior to the presentation, discussant Federico Asch, MD, MedStar Health Research Institute, Washington, said, “the most exciting thing here is that we have good options. We now can start to tease out which patients will benefit best from one or the other because we actually have two options.”
The Amulet IDE trial was funded by Abbott. Dr. Lakkireddy reports that he or his spouse/partner have received grant/research support from Abbott, AtriCure, Alta Thera, Medtronic, Biosense Webster, Biotronik, and Boston Scientific; and speaker honoraria from Abbott, Medtronic, Biotronik, and Boston Scientific.
A version of this article first appeared on Medscape.com.
FROM TCT 2022
Angiography in patients with prior CABG does better when planned with CT
BOSTON – Coronary angiography in patients who have previously undergone cardiac artery bypass grafting (CABG) is challenging, but the procedure can be streamlined and made safer when preprocedural CT coronary angiography (CTCA) is performed to plan the intervention, according to a randomized controlled trial.
In this study, all three endpoints, including a reduction in the incidence of contrast-induced nephropathy (CIN) and duration of the procedure, were met, according to Daniel Jones, MBBS, PhD.

Preprocedural CTCA was also associated with about a 40% improvement in patient satisfaction.
“When logistically possible, CTCA should be considered for any stable postbypass patient undergoing coronary angiography,” said Dr. Jones, who supported this assertion with data presented at the Transcatheter Cardiovascular Therapeutics annual meeting.
In this study, called BYPASS-CTCA, 688 patients with a prior CABG scheduled for invasive coronary angiography were randomized to a preprocedural CTCA or no preprocedural CTCA. Patients with stable angina and those with a non–ST elevated acute coronary syndrome were eligible. Those with ST-segment elevated MI or severe renal impairment (eGFR < 20 mL/min) were excluded.
All three co–primary endpoints favor CTCA
CTCA relative to no CTCA provided a significant advantage for all three of the coprimary endpoints, which were procedure duration, CIN as defined by KDIGO criteria, and patient satisfaction as measured by questionnaire.
The procedure duration was reduced by almost 21 minutes, cutting the time from nearly 39 minutes to less than 18 minutes (P < .001). This relative reduction was of similar magnitude across groups, such as those with or without acute coronary syndrome and procedures performed by a senior or a junior operator.
“Even when you include the preprocedural CTCA evaluation time, there was still a significant reduction [P < .001] in duration for those in the CTCA arm,” reported Dr. Jones, honorary consultant cardiologist, Barts Heart Centre, Queen Mary University, London.
The rates of CIN following the procedure in this study, which had a follow-up of 12 months, were 3.4% versus 27.9% (P < .0001) in the preprocedural CTCA and non-CTCA groups, respectively. Again, a sensitivity analysis showed a similar magnitude of risk reduction across all subgroups evaluated.
CTCA planning reduced contrast exposure
The reduced risk of CIN was consistent with a large reduction in contrast exposure for those in the CTCA group (77.4 vs. 173.0 mL; P < .001). The advantage narrowed substantially when adding in contrast exposure from CTCA, but still remained statistically significant (148.9 vs. 173.0 mL; P < .001).
Dr. Jones did not speculate about the specific reasons for the 40% improvement in patient satisfaction among those who underwent preprocedural CTCA relative to those who did not, but, again, a sensitivity analysis showed consistency across subgroups defined by race, operator experience, and underlying diagnosis.
Numerous secondary endpoints also favored CTCA over no CTCA. This included fewer catheters used to complete the procedure (three vs. four; P < .001), a greater likelihood that the procedure was performed with radial access (76.9% vs. 56.7%), and lower rates of procedural complications (2.3% vs. 10.8%; P < .001). This latter category included fewer vascular access complications such as bleeding (0.6 % vs. 4.4%; P = .007) and periprocedural MI (0.6% vs. 6.4%; P < 0.001).
In a graph of time to first major adverse cardiovascular event (MACE), the curves separated almost immediately with a consistently lower rate maintained in the CTCA arm over the 12 months of follow-up, but this is observational. Dr. Jones acknowledged that this trial was not powered to show a difference in MACE.
Study intriguing but not definitive
In a panel discussion that followed the presentation of these results at the meeting, sponsored by the Cardiovascular Research Foundation, some reservations with this study were expressed. In particular, several of the panelists, including Jeffrey W. Moses, MD, director of interventional cardiovascular therapeutics, Columbia University Medical Center, New York, expressed surprise at the 27% rate of CIN, which he considered uncommonly high even in a high-risk population.
The unusual rate of CIN was also considered problematic given that it was the most significant clinical outcome among the three co–primary endpoints. Procedural times and patient satisfaction, while valid endpoints, are important subjects of study, but Dr. Moses was not alone in suggesting this study deserves validation.
In particular, there appeared to be a consensus among panelists that a larger multicenter study looking at hard endpoints, such as MACE, would be more compelling. They indicated that even if CTCA poses a very low risk of meaningful complications, it does add expense and an extra step.
Dr. Jones reported no potential conflicts of interest. Dr. Moses reported financial relationships with Covanos, Orchestra Biomed, Ostial, and Xenter.
BOSTON – Coronary angiography in patients who have previously undergone cardiac artery bypass grafting (CABG) is challenging, but the procedure can be streamlined and made safer when preprocedural CT coronary angiography (CTCA) is performed to plan the intervention, according to a randomized controlled trial.
In this study, all three endpoints, including a reduction in the incidence of contrast-induced nephropathy (CIN) and duration of the procedure, were met, according to Daniel Jones, MBBS, PhD.
Preprocedural CTCA was also associated with about a 40% improvement in patient satisfaction.
“When logistically possible, CTCA should be considered for any stable postbypass patient undergoing coronary angiography,” said Dr. Jones, who supported this assertion with data presented at the Transcatheter Cardiovascular Therapeutics annual meeting.
In this study, called BYPASS-CTCA, 688 patients with a prior CABG scheduled for invasive coronary angiography were randomized to a preprocedural CTCA or no preprocedural CTCA. Patients with stable angina and those with a non–ST elevated acute coronary syndrome were eligible. Those with ST-segment elevated MI or severe renal impairment (eGFR < 20 mL/min) were excluded.
All three co–primary endpoints favor CTCA
CTCA relative to no CTCA provided a significant advantage for all three of the coprimary endpoints, which were procedure duration, CIN as defined by KDIGO criteria, and patient satisfaction as measured by questionnaire.
The procedure duration was reduced by almost 21 minutes, cutting the time from nearly 39 minutes to less than 18 minutes (P < .001). This relative reduction was of similar magnitude across groups, such as those with or without acute coronary syndrome and procedures performed by a senior or a junior operator.
“Even when you include the preprocedural CTCA evaluation time, there was still a significant reduction [P < .001] in duration for those in the CTCA arm,” reported Dr. Jones, honorary consultant cardiologist, Barts Heart Centre, Queen Mary University, London.
The rates of CIN following the procedure in this study, which had a follow-up of 12 months, were 3.4% versus 27.9% (P < .0001) in the preprocedural CTCA and non-CTCA groups, respectively. Again, a sensitivity analysis showed a similar magnitude of risk reduction across all subgroups evaluated.
CTCA planning reduced contrast exposure
The reduced risk of CIN was consistent with a large reduction in contrast exposure for those in the CTCA group (77.4 vs. 173.0 mL; P < .001). The advantage narrowed substantially when adding in contrast exposure from CTCA, but still remained statistically significant (148.9 vs. 173.0 mL; P < .001).
Dr. Jones did not speculate about the specific reasons for the 40% improvement in patient satisfaction among those who underwent preprocedural CTCA relative to those who did not, but, again, a sensitivity analysis showed consistency across subgroups defined by race, operator experience, and underlying diagnosis.
Numerous secondary endpoints also favored CTCA over no CTCA. This included fewer catheters used to complete the procedure (three vs. four; P < .001), a greater likelihood that the procedure was performed with radial access (76.9% vs. 56.7%), and lower rates of procedural complications (2.3% vs. 10.8%; P < .001). This latter category included fewer vascular access complications such as bleeding (0.6 % vs. 4.4%; P = .007) and periprocedural MI (0.6% vs. 6.4%; P < 0.001).
In a graph of time to first major adverse cardiovascular event (MACE), the curves separated almost immediately with a consistently lower rate maintained in the CTCA arm over the 12 months of follow-up, but this is observational. Dr. Jones acknowledged that this trial was not powered to show a difference in MACE.
Study intriguing but not definitive
In a panel discussion that followed the presentation of these results at the meeting, sponsored by the Cardiovascular Research Foundation, some reservations with this study were expressed. In particular, several of the panelists, including Jeffrey W. Moses, MD, director of interventional cardiovascular therapeutics, Columbia University Medical Center, New York, expressed surprise at the 27% rate of CIN, which he considered uncommonly high even in a high-risk population.
The unusual rate of CIN was also considered problematic given that it was the most significant clinical outcome among the three co–primary endpoints. Procedural times and patient satisfaction, while valid endpoints, are important subjects of study, but Dr. Moses was not alone in suggesting this study deserves validation.
In particular, there appeared to be a consensus among panelists that a larger multicenter study looking at hard endpoints, such as MACE, would be more compelling. They indicated that even if CTCA poses a very low risk of meaningful complications, it does add expense and an extra step.
Dr. Jones reported no potential conflicts of interest. Dr. Moses reported financial relationships with Covanos, Orchestra Biomed, Ostial, and Xenter.
BOSTON – Coronary angiography in patients who have previously undergone cardiac artery bypass grafting (CABG) is challenging, but the procedure can be streamlined and made safer when preprocedural CT coronary angiography (CTCA) is performed to plan the intervention, according to a randomized controlled trial.
In this study, all three endpoints, including a reduction in the incidence of contrast-induced nephropathy (CIN) and duration of the procedure, were met, according to Daniel Jones, MBBS, PhD.
Preprocedural CTCA was also associated with about a 40% improvement in patient satisfaction.
“When logistically possible, CTCA should be considered for any stable postbypass patient undergoing coronary angiography,” said Dr. Jones, who supported this assertion with data presented at the Transcatheter Cardiovascular Therapeutics annual meeting.
In this study, called BYPASS-CTCA, 688 patients with a prior CABG scheduled for invasive coronary angiography were randomized to a preprocedural CTCA or no preprocedural CTCA. Patients with stable angina and those with a non–ST elevated acute coronary syndrome were eligible. Those with ST-segment elevated MI or severe renal impairment (eGFR < 20 mL/min) were excluded.
All three co–primary endpoints favor CTCA
CTCA relative to no CTCA provided a significant advantage for all three of the coprimary endpoints, which were procedure duration, CIN as defined by KDIGO criteria, and patient satisfaction as measured by questionnaire.
The procedure duration was reduced by almost 21 minutes, cutting the time from nearly 39 minutes to less than 18 minutes (P < .001). This relative reduction was of similar magnitude across groups, such as those with or without acute coronary syndrome and procedures performed by a senior or a junior operator.
“Even when you include the preprocedural CTCA evaluation time, there was still a significant reduction [P < .001] in duration for those in the CTCA arm,” reported Dr. Jones, honorary consultant cardiologist, Barts Heart Centre, Queen Mary University, London.
The rates of CIN following the procedure in this study, which had a follow-up of 12 months, were 3.4% versus 27.9% (P < .0001) in the preprocedural CTCA and non-CTCA groups, respectively. Again, a sensitivity analysis showed a similar magnitude of risk reduction across all subgroups evaluated.
CTCA planning reduced contrast exposure
The reduced risk of CIN was consistent with a large reduction in contrast exposure for those in the CTCA group (77.4 vs. 173.0 mL; P < .001). The advantage narrowed substantially when adding in contrast exposure from CTCA, but still remained statistically significant (148.9 vs. 173.0 mL; P < .001).
Dr. Jones did not speculate about the specific reasons for the 40% improvement in patient satisfaction among those who underwent preprocedural CTCA relative to those who did not, but, again, a sensitivity analysis showed consistency across subgroups defined by race, operator experience, and underlying diagnosis.
Numerous secondary endpoints also favored CTCA over no CTCA. This included fewer catheters used to complete the procedure (three vs. four; P < .001), a greater likelihood that the procedure was performed with radial access (76.9% vs. 56.7%), and lower rates of procedural complications (2.3% vs. 10.8%; P < .001). This latter category included fewer vascular access complications such as bleeding (0.6 % vs. 4.4%; P = .007) and periprocedural MI (0.6% vs. 6.4%; P < 0.001).
In a graph of time to first major adverse cardiovascular event (MACE), the curves separated almost immediately with a consistently lower rate maintained in the CTCA arm over the 12 months of follow-up, but this is observational. Dr. Jones acknowledged that this trial was not powered to show a difference in MACE.
Study intriguing but not definitive
In a panel discussion that followed the presentation of these results at the meeting, sponsored by the Cardiovascular Research Foundation, some reservations with this study were expressed. In particular, several of the panelists, including Jeffrey W. Moses, MD, director of interventional cardiovascular therapeutics, Columbia University Medical Center, New York, expressed surprise at the 27% rate of CIN, which he considered uncommonly high even in a high-risk population.
The unusual rate of CIN was also considered problematic given that it was the most significant clinical outcome among the three co–primary endpoints. Procedural times and patient satisfaction, while valid endpoints, are important subjects of study, but Dr. Moses was not alone in suggesting this study deserves validation.
In particular, there appeared to be a consensus among panelists that a larger multicenter study looking at hard endpoints, such as MACE, would be more compelling. They indicated that even if CTCA poses a very low risk of meaningful complications, it does add expense and an extra step.
Dr. Jones reported no potential conflicts of interest. Dr. Moses reported financial relationships with Covanos, Orchestra Biomed, Ostial, and Xenter.
AT TCT 2022
PASCAL for MV repair noninferior to MitraClip in CLASP IID; FDA took notice
A newly available transcatheter device for edge-to-edge mitral valve (MV) repair, named for a famed scientist-inventor, is similar to the long-available MitraClip (Abbott) for short-term efficacy and safety, suggests an interim but prespecified analysis from a randomized trial.
In its comparison with MitraClip, the PASCAL transcatheter valve repair system (Edwards Lifesciences) was noninferior with respect to 30-day major adverse events and to success at achieving mitral regurgitation (MR) of no more than moderate severity within 6 months. The trial had entered patients with significant, symptomatic degenerative MR considered too high-risk for surgical repair or replacement.
The interim analysis covers 180 of the 300 patients followed in the study, of whom 117 received the PASCAL device and 63 were given MitraClip. Both groups showed significant gains in functional class, symptom status, and quality of life over 6 months, reported D. Scott Lim, MD, University of Virginia Health System Hospital, Charlottesville, and Konstantinos Koulogiannis, MD, Morristown Medical Center, N.J., jointly on Sept. 17 at the Transcatheter Cardiovascular Therapeutics (TCT) 2022 annual meeting in Boston.
Dr. Lim, one of the trial’s principal investigators, is also lead author on its same-day publication in JACC: Cardiovascular Interventions.
Based largely on those results from the CLASP IID pivotal trial, the U.S. Food and Drug Administration recently approved the PASCAL system for use in patients with degenerative MR, Edwards announced on Sept. 15. The device was approved in the European Union on Aug. 17.
MitraClip has been available in various iterations in the United States since 2013 and in Europe since 2008.
“It’s good for the field to be able to say we have two devices that are comparable,” giving clinicians more options, Vinod H. Thourani, MD, Piedmont Heart Institute, Atlanta, told this news organization.
The current analysis shows that “we’ve yet to figure out what patient pathologies will be beneficial” for each of the devices, Dr. Thourani said. “The goal will be to find out if there are certain anatomical considerations where one device is better than the other.”
It will be necessary to study “more patients, a larger cohort, with longer follow-up to allow us to see their true benefits,” he said, as well as to conduct more subgroup analyses. For now, the choice of device will probably be “operator-specific, which they feel comfortable with.”
Dr. Thourani, not an author on the current study, is the U.S. principal investigator for the CLASP IIF study looking at clinical outcomes with the two devices and says he consults for both Edwards and Abbott.
The findings are “preliminary for now,” said Michael Young, MD, Dartmouth-Hitchcock Medical Center, Lebanon, N.H., in part because, like most randomized trials, CLASP IID entered a select, not broadly representative population.
“They want to make, as best as they could, an apples-to-apples comparison, without confounding that might make it more difficult to interpret it afterwards,” Dr. Young, not associated with the trial, told this news organization.
But CLASP IID “did enroll patients that we do see and treat, so undoubtedly it’s a compelling study. We now have another device that is shown to be safe and effective. How we’re going to extrapolate it to all the patients that are being referred to our practices will, I think, be under debate and deliberation.”
The PASCAL and MitraClip devices each may be more suitable for different patients with varying mitral valve pathologies because of differences in their designs, Dr. Lim said. The PASCAL’s relative flexibility might make it preferable in patients with smaller mitral valves, and its ability to elongate during delivery could make it more suitable for patients with chordal-dense areas around the valve, he speculated.
MitraClip, Dr. Lim told this news organization, has a mechanical closure system for anchoring that may make it more appropriate for “more complicated, thicker leaflets with calcium.”
CLASP IID enrolled patients with grade 3+ or 4+ degenerative MR considered to be “at prohibitive surgical risk” at 43 sites in North America and Europe. It randomly assigned them 2-to-1 to receive the PASCAL device or MitraClip.
Either of two PASCAL versions were used, the original device or the “smaller, narrower” PASCAL Ace, Dr. Lim observed. Both versions are covered by the PASCAL Precision System FDA approval. About 40% of patients assigned to MitraClip received older versions of the device and about 60%, more recent versions, as they were entered into practice.
The mean procedure times were 88 minutes for PASCAL and 79 minutes for MitraClip (P = .023), with much of the difference attributable to the earliest PASCAL procedures. Procedure times for the device declined with greater operator experience, the published report states.
Rates of the primary safety endpoint of major adverse events at 30 days were 3.4% for PASCAL and 4.8% for MitraClip. The endpoint was a composite of cardiovascular mortality, stroke, myocardial infarction, new need for renal replacement therapy, severe bleeding, or nonelective MV reintervention.
The proportion of patients with MR grade 2+ or lower at 6 months, the primary effectiveness endpoint, assessed at a core laboratory, was 96.5% for the PASCAL group over a median follow-up of 179.5 days and 96.8% over a median of 184.5 days for those who received MitraClip.
Comparisons for both primary endpoints met the prespecified criteria for PASCAL noninferiority.
In a secondary analysis, the proportion of PASCAL patients with MR grade 1+ or less held about steady from postprocedure discharge out to 6 months, at 87.2% and 83.7%, respectively (P = .317).
But whereas 88.5% of MitraClip patients had MR grade 1+ or better at discharge, 71.2% were at that grade by 6 months (P = .003). That apparent hemodynamic deterioration raised some eyebrows at the TCT sessions as a potential sign that PASCAL functional results are more durable.
That sort of judgment is premature, offered Anita W. Asgar, MD, MSc, Montreal Heart Institute, Quebec City, as an invited discussant after the CLASP IID trial’s formal presentation at the meeting, which was sponsored by the Cardiovascular Research Foundation.
The trial is notable in part for “showing how safe this procedure is and how successful it is for these patients – this is phenomenal,” she said, but “I would caution comparing one device being better than another with such a small number of patients.”
MitraClip, Dr. Young observed, “has been, up to this point, our only option for edge-to-edge repair of the mitral valve. And many of us have years of experience and a lot of patients that we treat with that device.” His center hasn’t yet used PASCAL, but that may change as the field gains more familiarity with the device. Operators may use either device in different cases, he said.
“Depending on the program, and depending on the volume of mitral patients that you see and edge-to-edge repair that you do, it could be that you stick with one, or switch to another, or you integrate both of them and try to decide which patients might be better suited for one or the other.”
CLASP IID was sponsored by Edwards Lifesciences. Dr. Lim discloses consulting for Philips, Venus, and Valgen and receiving research grants from Abbott, Boston Scientific, Edwards Lifesciences, and Medtronic. Dr. Koulogiannis discloses consulting and serving on an advisory board for Edwards Lifesciences and as a speaker for Abbott and discloses holding equity, stocks, or stock options in 4C. Dr. Thourani discloses serving as a consultant to both Abbott and Edwards Lifesciences. Dr. Young discloses receiving consulting fees or honoraria or serving on a speaker’s bureau for Medtronic. Dr. Asgar discloses receiving research support from or holding a research contract with Abbott Vascular and receiving consulting fees or honoraria or serving on a speaker’s bureau for Medtronic, Edwards Lifesciences, and W. Gore & Associates.
A version of this article first appeared on Medscape.com.
A newly available transcatheter device for edge-to-edge mitral valve (MV) repair, named for a famed scientist-inventor, is similar to the long-available MitraClip (Abbott) for short-term efficacy and safety, suggests an interim but prespecified analysis from a randomized trial.
In its comparison with MitraClip, the PASCAL transcatheter valve repair system (Edwards Lifesciences) was noninferior with respect to 30-day major adverse events and to success at achieving mitral regurgitation (MR) of no more than moderate severity within 6 months. The trial had entered patients with significant, symptomatic degenerative MR considered too high-risk for surgical repair or replacement.
The interim analysis covers 180 of the 300 patients followed in the study, of whom 117 received the PASCAL device and 63 were given MitraClip. Both groups showed significant gains in functional class, symptom status, and quality of life over 6 months, reported D. Scott Lim, MD, University of Virginia Health System Hospital, Charlottesville, and Konstantinos Koulogiannis, MD, Morristown Medical Center, N.J., jointly on Sept. 17 at the Transcatheter Cardiovascular Therapeutics (TCT) 2022 annual meeting in Boston.
Dr. Lim, one of the trial’s principal investigators, is also lead author on its same-day publication in JACC: Cardiovascular Interventions.
Based largely on those results from the CLASP IID pivotal trial, the U.S. Food and Drug Administration recently approved the PASCAL system for use in patients with degenerative MR, Edwards announced on Sept. 15. The device was approved in the European Union on Aug. 17.
MitraClip has been available in various iterations in the United States since 2013 and in Europe since 2008.
“It’s good for the field to be able to say we have two devices that are comparable,” giving clinicians more options, Vinod H. Thourani, MD, Piedmont Heart Institute, Atlanta, told this news organization.
The current analysis shows that “we’ve yet to figure out what patient pathologies will be beneficial” for each of the devices, Dr. Thourani said. “The goal will be to find out if there are certain anatomical considerations where one device is better than the other.”
It will be necessary to study “more patients, a larger cohort, with longer follow-up to allow us to see their true benefits,” he said, as well as to conduct more subgroup analyses. For now, the choice of device will probably be “operator-specific, which they feel comfortable with.”
Dr. Thourani, not an author on the current study, is the U.S. principal investigator for the CLASP IIF study looking at clinical outcomes with the two devices and says he consults for both Edwards and Abbott.
The findings are “preliminary for now,” said Michael Young, MD, Dartmouth-Hitchcock Medical Center, Lebanon, N.H., in part because, like most randomized trials, CLASP IID entered a select, not broadly representative population.
“They want to make, as best as they could, an apples-to-apples comparison, without confounding that might make it more difficult to interpret it afterwards,” Dr. Young, not associated with the trial, told this news organization.
But CLASP IID “did enroll patients that we do see and treat, so undoubtedly it’s a compelling study. We now have another device that is shown to be safe and effective. How we’re going to extrapolate it to all the patients that are being referred to our practices will, I think, be under debate and deliberation.”
The PASCAL and MitraClip devices each may be more suitable for different patients with varying mitral valve pathologies because of differences in their designs, Dr. Lim said. The PASCAL’s relative flexibility might make it preferable in patients with smaller mitral valves, and its ability to elongate during delivery could make it more suitable for patients with chordal-dense areas around the valve, he speculated.
MitraClip, Dr. Lim told this news organization, has a mechanical closure system for anchoring that may make it more appropriate for “more complicated, thicker leaflets with calcium.”
CLASP IID enrolled patients with grade 3+ or 4+ degenerative MR considered to be “at prohibitive surgical risk” at 43 sites in North America and Europe. It randomly assigned them 2-to-1 to receive the PASCAL device or MitraClip.
Either of two PASCAL versions were used, the original device or the “smaller, narrower” PASCAL Ace, Dr. Lim observed. Both versions are covered by the PASCAL Precision System FDA approval. About 40% of patients assigned to MitraClip received older versions of the device and about 60%, more recent versions, as they were entered into practice.
The mean procedure times were 88 minutes for PASCAL and 79 minutes for MitraClip (P = .023), with much of the difference attributable to the earliest PASCAL procedures. Procedure times for the device declined with greater operator experience, the published report states.
Rates of the primary safety endpoint of major adverse events at 30 days were 3.4% for PASCAL and 4.8% for MitraClip. The endpoint was a composite of cardiovascular mortality, stroke, myocardial infarction, new need for renal replacement therapy, severe bleeding, or nonelective MV reintervention.
The proportion of patients with MR grade 2+ or lower at 6 months, the primary effectiveness endpoint, assessed at a core laboratory, was 96.5% for the PASCAL group over a median follow-up of 179.5 days and 96.8% over a median of 184.5 days for those who received MitraClip.
Comparisons for both primary endpoints met the prespecified criteria for PASCAL noninferiority.
In a secondary analysis, the proportion of PASCAL patients with MR grade 1+ or less held about steady from postprocedure discharge out to 6 months, at 87.2% and 83.7%, respectively (P = .317).
But whereas 88.5% of MitraClip patients had MR grade 1+ or better at discharge, 71.2% were at that grade by 6 months (P = .003). That apparent hemodynamic deterioration raised some eyebrows at the TCT sessions as a potential sign that PASCAL functional results are more durable.
That sort of judgment is premature, offered Anita W. Asgar, MD, MSc, Montreal Heart Institute, Quebec City, as an invited discussant after the CLASP IID trial’s formal presentation at the meeting, which was sponsored by the Cardiovascular Research Foundation.
The trial is notable in part for “showing how safe this procedure is and how successful it is for these patients – this is phenomenal,” she said, but “I would caution comparing one device being better than another with such a small number of patients.”
MitraClip, Dr. Young observed, “has been, up to this point, our only option for edge-to-edge repair of the mitral valve. And many of us have years of experience and a lot of patients that we treat with that device.” His center hasn’t yet used PASCAL, but that may change as the field gains more familiarity with the device. Operators may use either device in different cases, he said.
“Depending on the program, and depending on the volume of mitral patients that you see and edge-to-edge repair that you do, it could be that you stick with one, or switch to another, or you integrate both of them and try to decide which patients might be better suited for one or the other.”
CLASP IID was sponsored by Edwards Lifesciences. Dr. Lim discloses consulting for Philips, Venus, and Valgen and receiving research grants from Abbott, Boston Scientific, Edwards Lifesciences, and Medtronic. Dr. Koulogiannis discloses consulting and serving on an advisory board for Edwards Lifesciences and as a speaker for Abbott and discloses holding equity, stocks, or stock options in 4C. Dr. Thourani discloses serving as a consultant to both Abbott and Edwards Lifesciences. Dr. Young discloses receiving consulting fees or honoraria or serving on a speaker’s bureau for Medtronic. Dr. Asgar discloses receiving research support from or holding a research contract with Abbott Vascular and receiving consulting fees or honoraria or serving on a speaker’s bureau for Medtronic, Edwards Lifesciences, and W. Gore & Associates.
A version of this article first appeared on Medscape.com.
A newly available transcatheter device for edge-to-edge mitral valve (MV) repair, named for a famed scientist-inventor, is similar to the long-available MitraClip (Abbott) for short-term efficacy and safety, suggests an interim but prespecified analysis from a randomized trial.
In its comparison with MitraClip, the PASCAL transcatheter valve repair system (Edwards Lifesciences) was noninferior with respect to 30-day major adverse events and to success at achieving mitral regurgitation (MR) of no more than moderate severity within 6 months. The trial had entered patients with significant, symptomatic degenerative MR considered too high-risk for surgical repair or replacement.
The interim analysis covers 180 of the 300 patients followed in the study, of whom 117 received the PASCAL device and 63 were given MitraClip. Both groups showed significant gains in functional class, symptom status, and quality of life over 6 months, reported D. Scott Lim, MD, University of Virginia Health System Hospital, Charlottesville, and Konstantinos Koulogiannis, MD, Morristown Medical Center, N.J., jointly on Sept. 17 at the Transcatheter Cardiovascular Therapeutics (TCT) 2022 annual meeting in Boston.
Dr. Lim, one of the trial’s principal investigators, is also lead author on its same-day publication in JACC: Cardiovascular Interventions.
Based largely on those results from the CLASP IID pivotal trial, the U.S. Food and Drug Administration recently approved the PASCAL system for use in patients with degenerative MR, Edwards announced on Sept. 15. The device was approved in the European Union on Aug. 17.
MitraClip has been available in various iterations in the United States since 2013 and in Europe since 2008.
“It’s good for the field to be able to say we have two devices that are comparable,” giving clinicians more options, Vinod H. Thourani, MD, Piedmont Heart Institute, Atlanta, told this news organization.
The current analysis shows that “we’ve yet to figure out what patient pathologies will be beneficial” for each of the devices, Dr. Thourani said. “The goal will be to find out if there are certain anatomical considerations where one device is better than the other.”
It will be necessary to study “more patients, a larger cohort, with longer follow-up to allow us to see their true benefits,” he said, as well as to conduct more subgroup analyses. For now, the choice of device will probably be “operator-specific, which they feel comfortable with.”
Dr. Thourani, not an author on the current study, is the U.S. principal investigator for the CLASP IIF study looking at clinical outcomes with the two devices and says he consults for both Edwards and Abbott.
The findings are “preliminary for now,” said Michael Young, MD, Dartmouth-Hitchcock Medical Center, Lebanon, N.H., in part because, like most randomized trials, CLASP IID entered a select, not broadly representative population.
“They want to make, as best as they could, an apples-to-apples comparison, without confounding that might make it more difficult to interpret it afterwards,” Dr. Young, not associated with the trial, told this news organization.
But CLASP IID “did enroll patients that we do see and treat, so undoubtedly it’s a compelling study. We now have another device that is shown to be safe and effective. How we’re going to extrapolate it to all the patients that are being referred to our practices will, I think, be under debate and deliberation.”
The PASCAL and MitraClip devices each may be more suitable for different patients with varying mitral valve pathologies because of differences in their designs, Dr. Lim said. The PASCAL’s relative flexibility might make it preferable in patients with smaller mitral valves, and its ability to elongate during delivery could make it more suitable for patients with chordal-dense areas around the valve, he speculated.
MitraClip, Dr. Lim told this news organization, has a mechanical closure system for anchoring that may make it more appropriate for “more complicated, thicker leaflets with calcium.”
CLASP IID enrolled patients with grade 3+ or 4+ degenerative MR considered to be “at prohibitive surgical risk” at 43 sites in North America and Europe. It randomly assigned them 2-to-1 to receive the PASCAL device or MitraClip.
Either of two PASCAL versions were used, the original device or the “smaller, narrower” PASCAL Ace, Dr. Lim observed. Both versions are covered by the PASCAL Precision System FDA approval. About 40% of patients assigned to MitraClip received older versions of the device and about 60%, more recent versions, as they were entered into practice.
The mean procedure times were 88 minutes for PASCAL and 79 minutes for MitraClip (P = .023), with much of the difference attributable to the earliest PASCAL procedures. Procedure times for the device declined with greater operator experience, the published report states.
Rates of the primary safety endpoint of major adverse events at 30 days were 3.4% for PASCAL and 4.8% for MitraClip. The endpoint was a composite of cardiovascular mortality, stroke, myocardial infarction, new need for renal replacement therapy, severe bleeding, or nonelective MV reintervention.
The proportion of patients with MR grade 2+ or lower at 6 months, the primary effectiveness endpoint, assessed at a core laboratory, was 96.5% for the PASCAL group over a median follow-up of 179.5 days and 96.8% over a median of 184.5 days for those who received MitraClip.
Comparisons for both primary endpoints met the prespecified criteria for PASCAL noninferiority.
In a secondary analysis, the proportion of PASCAL patients with MR grade 1+ or less held about steady from postprocedure discharge out to 6 months, at 87.2% and 83.7%, respectively (P = .317).
But whereas 88.5% of MitraClip patients had MR grade 1+ or better at discharge, 71.2% were at that grade by 6 months (P = .003). That apparent hemodynamic deterioration raised some eyebrows at the TCT sessions as a potential sign that PASCAL functional results are more durable.
That sort of judgment is premature, offered Anita W. Asgar, MD, MSc, Montreal Heart Institute, Quebec City, as an invited discussant after the CLASP IID trial’s formal presentation at the meeting, which was sponsored by the Cardiovascular Research Foundation.
The trial is notable in part for “showing how safe this procedure is and how successful it is for these patients – this is phenomenal,” she said, but “I would caution comparing one device being better than another with such a small number of patients.”
MitraClip, Dr. Young observed, “has been, up to this point, our only option for edge-to-edge repair of the mitral valve. And many of us have years of experience and a lot of patients that we treat with that device.” His center hasn’t yet used PASCAL, but that may change as the field gains more familiarity with the device. Operators may use either device in different cases, he said.
“Depending on the program, and depending on the volume of mitral patients that you see and edge-to-edge repair that you do, it could be that you stick with one, or switch to another, or you integrate both of them and try to decide which patients might be better suited for one or the other.”
CLASP IID was sponsored by Edwards Lifesciences. Dr. Lim discloses consulting for Philips, Venus, and Valgen and receiving research grants from Abbott, Boston Scientific, Edwards Lifesciences, and Medtronic. Dr. Koulogiannis discloses consulting and serving on an advisory board for Edwards Lifesciences and as a speaker for Abbott and discloses holding equity, stocks, or stock options in 4C. Dr. Thourani discloses serving as a consultant to both Abbott and Edwards Lifesciences. Dr. Young discloses receiving consulting fees or honoraria or serving on a speaker’s bureau for Medtronic. Dr. Asgar discloses receiving research support from or holding a research contract with Abbott Vascular and receiving consulting fees or honoraria or serving on a speaker’s bureau for Medtronic, Edwards Lifesciences, and W. Gore & Associates.
A version of this article first appeared on Medscape.com.
FROM TCT 2022
A week of anticoagulation halves post-PCI radial occlusion rate
Serious bleeding is not increased
BOSTON – Following transradial access for angiography or a percutaneous coronary intervention (PCI), a low dose of the factor Xa inhibitor rivaroxaban for 7 days reduces the risk of an access-site occlusion by 50%, according to results of the randomized RIVARAD trial.
Of two multicenter, randomized trials to address this question it is the larger, according to Rania Hammami, MD, who presented the results at the Transcatheter Cardiovascular Therapeutics annual meeting.

In the open-label RIVARAD trial, 538 patients were randomized to 10 mg rivaroxaban or standard care alone. Standard care at the beginning of the procedure included unfractionated heparin in a dose of 50 IU/kg for angiography and up to 100 IU/kg for PCI. Manual compression was applied at the end of the procedure followed by an evaluation for complications, such as hematoma or aneurysm.
For the primary outcome of radial access occlusion at 30 days, the lower rate in the rivaroxaban arm (6.9% vs. 13.0%) translated into a statistically significant 50% reduction (odds ratio, 0.50; P = .011).
Rivaroxaban preserves radial pulse
Rivaroxaban was also favored for the endpoint of inability at 30 days to find a radial pulse (5.8% vs. 12.2%; P = .01). Interestingly, there was some disparity for this endpoint for clinical examination and ultrasound.
“In 12 patients, we were able to palpate a radial pulse, but the ultrasound showed an occlusion of the vessel, while in 7 patients we could not find a radial pulse even though the radial artery was patent on ultrasound,” Dr. Hammami, of the department of cardiology, Hedi Chaker Hospital, Sfax, Tunisia, said at the meeting, sponsored by the Cardiovascular Research Foundation.
The incidence of hemorrhagic complications was higher in the rivaroxaban group (2.7% vs. 1.9%), but the difference did not approach statistical significance (OR, 1.5; P = .54). Moreover, all of the bleeding complications were minor (Bleeding Academic Research Consortium level 1), and none of the bleeding complications were observed in patients receiving rivaroxaban alone. Rather, all patients with bleeding were taking one or more antiplatelet drugs along with rivaroxaban.
On univariate analysis, several baseline characteristics were associated with subsequent radial occlusion, including female sex (P = .02), current smoking (P = .03), renal failure (P = .004), and PCI for acute coronary syndrome (P = .02). On multivariate analysis, female sex (P = .001) and current smoking (P < .0001) became even stronger predictors of occlusion on a statistical basis, while a prior procedure involving radial access was also a significant predictor (P = .029).
“One woman out of two developed radial access occlusion if she had a history of smoking and had a history of a transradial puncture,” Dr. Hammami reported.
In a subgroup analysis, relative protection from radial artery occlusion from a 7-day course of rivaroxaban was particularly pronounced in those with diabetes, renal failure, or hypertension relative to those without these conditions, but the protective effect appeared to be about the same regardless of body mass index, age, sheath size, or current use of statins.
These findings are consistent with other studies evaluating the risk of radial access occlusion, according to Dr. Hammami. While different studies she cited reported incidences ranging from less than 1% to more than 30%, the risk has typically been highest in populations with increased susceptibility for thrombus formation, such as smokers and patients with diabetes.
Preventing radial artery occlusion has several benefits, not least of which is preserving this access point for future interventions, according to Dr. Hammami.
RIVARAD is the largest study to evaluate an anticoagulant for the prevention of radial artery occlusion, but it is not the first. Earlier in 2022, a Chinese trial called RESTORE was published in Circulation: Cardiovascular Interventions. In that placebo-controlled study of 382 patients, 7 days of 10 mg rivaroxaban was also linked to a significant reduction in radial artery occlusion at 30 days (3.8% vs. 11.5%; P = .011).
“We don’t know if a higher dose of rivaroxaban would be more effective and equally safe,” said Dr. Hammami, but added that a Canadian trial called CAPITAL RAPTOR will test this premise. In this trial, there is a planned enrollment of 1,800 patients who will be randomized to 15 mg rivaroxaban or standard treatment.
Occlusion risk appears underappreciated
The risk of radial artery occlusion might be underappreciated. According to data cited by Dr. Hammami, only about half of interventionalists in the United States and fewer than 10% outside of the United States routinely assess radial artery patency in conjunction with radial-access PCI. The data from this trial suggest that the risk can be substantially reduced, particularly in high-risk patients, with anticoagulant therapy.
Agreeing that this is a potentially avoidable complication, Roxanna Mehran, MD, director of interventional cardiovascular research and clinical trials, Icahn School of Medicine at Mount Sinai, New York, called the RIVARAD study “a clinically meaningful trial,” and valuable for identifying risk factors as well as for showing a treatment effect and acceptable safety from a short course of a factor Xa inhibitor.
“This is very important work,” said Dr. Mehran, who praised the quality of the study and the contribution it makes for considering how and when prophylaxis is needed.
Dr. Hammami reported no potential conflicts of interest. Dr. Mehran has financial relationships with more than 25 pharmaceutical companies but none with the sponsor of this trial, which was funded by Philadelphia Pharma, a drug company based in Tunisia.
Serious bleeding is not increased
Serious bleeding is not increased
BOSTON – Following transradial access for angiography or a percutaneous coronary intervention (PCI), a low dose of the factor Xa inhibitor rivaroxaban for 7 days reduces the risk of an access-site occlusion by 50%, according to results of the randomized RIVARAD trial.
Of two multicenter, randomized trials to address this question it is the larger, according to Rania Hammami, MD, who presented the results at the Transcatheter Cardiovascular Therapeutics annual meeting.
In the open-label RIVARAD trial, 538 patients were randomized to 10 mg rivaroxaban or standard care alone. Standard care at the beginning of the procedure included unfractionated heparin in a dose of 50 IU/kg for angiography and up to 100 IU/kg for PCI. Manual compression was applied at the end of the procedure followed by an evaluation for complications, such as hematoma or aneurysm.
For the primary outcome of radial access occlusion at 30 days, the lower rate in the rivaroxaban arm (6.9% vs. 13.0%) translated into a statistically significant 50% reduction (odds ratio, 0.50; P = .011).
Rivaroxaban preserves radial pulse
Rivaroxaban was also favored for the endpoint of inability at 30 days to find a radial pulse (5.8% vs. 12.2%; P = .01). Interestingly, there was some disparity for this endpoint for clinical examination and ultrasound.
“In 12 patients, we were able to palpate a radial pulse, but the ultrasound showed an occlusion of the vessel, while in 7 patients we could not find a radial pulse even though the radial artery was patent on ultrasound,” Dr. Hammami, of the department of cardiology, Hedi Chaker Hospital, Sfax, Tunisia, said at the meeting, sponsored by the Cardiovascular Research Foundation.
The incidence of hemorrhagic complications was higher in the rivaroxaban group (2.7% vs. 1.9%), but the difference did not approach statistical significance (OR, 1.5; P = .54). Moreover, all of the bleeding complications were minor (Bleeding Academic Research Consortium level 1), and none of the bleeding complications were observed in patients receiving rivaroxaban alone. Rather, all patients with bleeding were taking one or more antiplatelet drugs along with rivaroxaban.
On univariate analysis, several baseline characteristics were associated with subsequent radial occlusion, including female sex (P = .02), current smoking (P = .03), renal failure (P = .004), and PCI for acute coronary syndrome (P = .02). On multivariate analysis, female sex (P = .001) and current smoking (P < .0001) became even stronger predictors of occlusion on a statistical basis, while a prior procedure involving radial access was also a significant predictor (P = .029).
“One woman out of two developed radial access occlusion if she had a history of smoking and had a history of a transradial puncture,” Dr. Hammami reported.
In a subgroup analysis, relative protection from radial artery occlusion from a 7-day course of rivaroxaban was particularly pronounced in those with diabetes, renal failure, or hypertension relative to those without these conditions, but the protective effect appeared to be about the same regardless of body mass index, age, sheath size, or current use of statins.
These findings are consistent with other studies evaluating the risk of radial access occlusion, according to Dr. Hammami. While different studies she cited reported incidences ranging from less than 1% to more than 30%, the risk has typically been highest in populations with increased susceptibility for thrombus formation, such as smokers and patients with diabetes.
Preventing radial artery occlusion has several benefits, not least of which is preserving this access point for future interventions, according to Dr. Hammami.
RIVARAD is the largest study to evaluate an anticoagulant for the prevention of radial artery occlusion, but it is not the first. Earlier in 2022, a Chinese trial called RESTORE was published in Circulation: Cardiovascular Interventions. In that placebo-controlled study of 382 patients, 7 days of 10 mg rivaroxaban was also linked to a significant reduction in radial artery occlusion at 30 days (3.8% vs. 11.5%; P = .011).
“We don’t know if a higher dose of rivaroxaban would be more effective and equally safe,” said Dr. Hammami, but added that a Canadian trial called CAPITAL RAPTOR will test this premise. In this trial, there is a planned enrollment of 1,800 patients who will be randomized to 15 mg rivaroxaban or standard treatment.
Occlusion risk appears underappreciated
The risk of radial artery occlusion might be underappreciated. According to data cited by Dr. Hammami, only about half of interventionalists in the United States and fewer than 10% outside of the United States routinely assess radial artery patency in conjunction with radial-access PCI. The data from this trial suggest that the risk can be substantially reduced, particularly in high-risk patients, with anticoagulant therapy.
Agreeing that this is a potentially avoidable complication, Roxanna Mehran, MD, director of interventional cardiovascular research and clinical trials, Icahn School of Medicine at Mount Sinai, New York, called the RIVARAD study “a clinically meaningful trial,” and valuable for identifying risk factors as well as for showing a treatment effect and acceptable safety from a short course of a factor Xa inhibitor.
“This is very important work,” said Dr. Mehran, who praised the quality of the study and the contribution it makes for considering how and when prophylaxis is needed.
Dr. Hammami reported no potential conflicts of interest. Dr. Mehran has financial relationships with more than 25 pharmaceutical companies but none with the sponsor of this trial, which was funded by Philadelphia Pharma, a drug company based in Tunisia.
BOSTON – Following transradial access for angiography or a percutaneous coronary intervention (PCI), a low dose of the factor Xa inhibitor rivaroxaban for 7 days reduces the risk of an access-site occlusion by 50%, according to results of the randomized RIVARAD trial.
Of two multicenter, randomized trials to address this question it is the larger, according to Rania Hammami, MD, who presented the results at the Transcatheter Cardiovascular Therapeutics annual meeting.
In the open-label RIVARAD trial, 538 patients were randomized to 10 mg rivaroxaban or standard care alone. Standard care at the beginning of the procedure included unfractionated heparin in a dose of 50 IU/kg for angiography and up to 100 IU/kg for PCI. Manual compression was applied at the end of the procedure followed by an evaluation for complications, such as hematoma or aneurysm.
For the primary outcome of radial access occlusion at 30 days, the lower rate in the rivaroxaban arm (6.9% vs. 13.0%) translated into a statistically significant 50% reduction (odds ratio, 0.50; P = .011).
Rivaroxaban preserves radial pulse
Rivaroxaban was also favored for the endpoint of inability at 30 days to find a radial pulse (5.8% vs. 12.2%; P = .01). Interestingly, there was some disparity for this endpoint for clinical examination and ultrasound.
“In 12 patients, we were able to palpate a radial pulse, but the ultrasound showed an occlusion of the vessel, while in 7 patients we could not find a radial pulse even though the radial artery was patent on ultrasound,” Dr. Hammami, of the department of cardiology, Hedi Chaker Hospital, Sfax, Tunisia, said at the meeting, sponsored by the Cardiovascular Research Foundation.
The incidence of hemorrhagic complications was higher in the rivaroxaban group (2.7% vs. 1.9%), but the difference did not approach statistical significance (OR, 1.5; P = .54). Moreover, all of the bleeding complications were minor (Bleeding Academic Research Consortium level 1), and none of the bleeding complications were observed in patients receiving rivaroxaban alone. Rather, all patients with bleeding were taking one or more antiplatelet drugs along with rivaroxaban.
On univariate analysis, several baseline characteristics were associated with subsequent radial occlusion, including female sex (P = .02), current smoking (P = .03), renal failure (P = .004), and PCI for acute coronary syndrome (P = .02). On multivariate analysis, female sex (P = .001) and current smoking (P < .0001) became even stronger predictors of occlusion on a statistical basis, while a prior procedure involving radial access was also a significant predictor (P = .029).
“One woman out of two developed radial access occlusion if she had a history of smoking and had a history of a transradial puncture,” Dr. Hammami reported.
In a subgroup analysis, relative protection from radial artery occlusion from a 7-day course of rivaroxaban was particularly pronounced in those with diabetes, renal failure, or hypertension relative to those without these conditions, but the protective effect appeared to be about the same regardless of body mass index, age, sheath size, or current use of statins.
These findings are consistent with other studies evaluating the risk of radial access occlusion, according to Dr. Hammami. While different studies she cited reported incidences ranging from less than 1% to more than 30%, the risk has typically been highest in populations with increased susceptibility for thrombus formation, such as smokers and patients with diabetes.
Preventing radial artery occlusion has several benefits, not least of which is preserving this access point for future interventions, according to Dr. Hammami.
RIVARAD is the largest study to evaluate an anticoagulant for the prevention of radial artery occlusion, but it is not the first. Earlier in 2022, a Chinese trial called RESTORE was published in Circulation: Cardiovascular Interventions. In that placebo-controlled study of 382 patients, 7 days of 10 mg rivaroxaban was also linked to a significant reduction in radial artery occlusion at 30 days (3.8% vs. 11.5%; P = .011).
“We don’t know if a higher dose of rivaroxaban would be more effective and equally safe,” said Dr. Hammami, but added that a Canadian trial called CAPITAL RAPTOR will test this premise. In this trial, there is a planned enrollment of 1,800 patients who will be randomized to 15 mg rivaroxaban or standard treatment.
Occlusion risk appears underappreciated
The risk of radial artery occlusion might be underappreciated. According to data cited by Dr. Hammami, only about half of interventionalists in the United States and fewer than 10% outside of the United States routinely assess radial artery patency in conjunction with radial-access PCI. The data from this trial suggest that the risk can be substantially reduced, particularly in high-risk patients, with anticoagulant therapy.
Agreeing that this is a potentially avoidable complication, Roxanna Mehran, MD, director of interventional cardiovascular research and clinical trials, Icahn School of Medicine at Mount Sinai, New York, called the RIVARAD study “a clinically meaningful trial,” and valuable for identifying risk factors as well as for showing a treatment effect and acceptable safety from a short course of a factor Xa inhibitor.
“This is very important work,” said Dr. Mehran, who praised the quality of the study and the contribution it makes for considering how and when prophylaxis is needed.
Dr. Hammami reported no potential conflicts of interest. Dr. Mehran has financial relationships with more than 25 pharmaceutical companies but none with the sponsor of this trial, which was funded by Philadelphia Pharma, a drug company based in Tunisia.
AT TCT 2022
Ultrasonic renal denervation passes 2-month test in uncontrolled HTN: RADIANCE II
Systolic blood pressure went down safely and consistently 2 months after renal denervation achieved by ultrasound ablation in patients with uncontrolled, mild to moderate hypertension (HTN) in a key sham-controlled test of the balloon-equipped catheter.
The BP reductions were significant almost regardless of how they were measured – at home, in the office, during the day, at night, or over 24 hours – and weren’t dependent on baseline BP levels.
The 224-patient RADIANCE II Pivotal Study follows two earlier successful sham-controlled trials that used the same renal denervation catheter in other types of patients with HTN. They were RADIANCE-HTN SOLO, which entered patients with mild to moderate HTN not taking medication, and RADIANCE-HTN TRIO, which included patients with HTN despite fixed-dose, single-pill, triple-antihypertensive therapy.

The consistent results of all three trials suggest that the ultrasound renal denervation (uRDN) technique “lowers blood pressure across the spectrum of hypertension,” concluded co–principal investigator Ajay J. Kirtane, MD, SM, Columbia University Irving Medical Center, New York–Presbyterian Hospital, when presenting RADIANCE II at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
RADIANCE II, the largest of the three studies, met its prespecified primary efficacy endpoint of change in daytime ambulatory systolic BP at 2 months by showing a significant 6.3–mm Hg greater reduction in the uRDN group, compared with the sham-control group. There were no major adverse events at 30 days in either group.
The trial was similarly successful for the secondary endpoints of change in systolic BP measured in various other settings, including over 24 hours. Reductions after uRDN averaged 5-7 mm Hg greater than in the control group.
Sparse top-line results of the RADIANCE II pivotal trial were announced in July by the study’s sponsor, ReCor Medical.
Dr. Kirtane stressed in an interview that uRDN and likely any form of HTN renal denervation therapy is not a substitute for standard management. “This is really for patients in whom you’ve made best efforts to do the traditional things – lifestyle modification, medications, all of that – and yet they’re still uncontrolled.” At that point, assuming denervation therapy is available in practice, “it would be something to potentially consider.”
As a panelist after Dr. Kirtane’s formal presentation of RADIANCE II at the conference, Naomi D. Fisher, MD, who was a RADIANCE-HTN TRIO investigator, described how the treatment’s perceived intended patient population evolved over time.
“We all began with the idea that we were going to treat patients with resistant hypertension, that was going to be the first target. We have learned that those patients are far fewer than we thought,” said Dr. Fisher, who directs the hypertension service at Brigham and Women’s Hospital, Boston.
Initial estimates were that such patients with the resistant form, “meaning they require more than three drugs to control their blood pressure,” would represent 15%-20% of patients with HTN.
“We learned from our TRIO data that if you give these patients one single combined pill, lo and behold, many of them become controlled,” she said. “There is so much nonadherence out there in the world, about 50% of our patients aren’t taking their pills. It’s a hard and true fact.”
Exclude patients who aren’t adherent and “our true resistance population becomes minuscule. So, I don’t think that’s going to be the main population” for renal denervation therapy.
More likely, she said, it would be “patients who are uncontrolled and unable to take their medications. So that is going to include nonadherence, intolerance. It’s a very large category of patients. And the priorities can be stacked in favor of those who have higher cardiovascular risk.”
RADIANCE II can show the persistence of uRDN’s BP-lowering effect only out to 2 months so far, but the effect’s durability based on the RADIANCE program’s combined experience appears to be at least 2 years, Dr. Kirtane said in an interview.
“The RADIANCE II pivotal trial is a powerful, well-designed study attesting to the efficacy of renal denervation in BP lowering,” Franz H. Messerli, MD, Swiss Cardiovascular Center, University Hospital Bern, said in an interview.
The trial “shows the well-known unpredictability of antihypertensive response. We cannot predict who responds to renal denervation and who does not, and who even has a paradoxical increase in BP,” Dr. Messerli, an international hypertension expert not associated with the trial, said in an interview.
“As long as we cannot predict the antihypertensive response to renal denervation therapy, potential synergism/antagonism with drug therapy remains an educated guess,” he said.
“Hypertension is a disease that lasts years and decades. As impressive as RADIANCE II’s 2-month snapshot is, I look forward to similar or better BP data 12 and 24 months after renal denervation,” Dr. Messerli added.
RADIANCE II entered patients with mild to moderate uncontrolled HTN, that is, a systolic BP at least 140/90 mm Hg and less than 180/120 mm Hg, who were receiving no more than two antihypertensive medications. They could have no history of cardiovascular or cerebrovascular events or uncontrolled diabetes, and their estimated glomerular filtration rate (eGFR) had to be at least 40 mL/min per 1.73 m2.
After a 4-week drug washout period, patients who were clinically stable with an ambulatory BP of at least 135/85 mm Hg and less than 170/105 mm Hg underwent CT and renal angiography. Then, the 224 patients still anatomically eligible for the procedure were randomly assigned 2:1 to uRDN or a sham-control procedure: 150 and 74 patients, respectively.
At 2 months, daytime ambulatory systolic BP on average fell 7.9 mm Hg in the uRDN group and 1.8 mm Hg in the sham-control group, for a drop that was steeper by 6.3 mm Hg (P < .0001) after uRDN.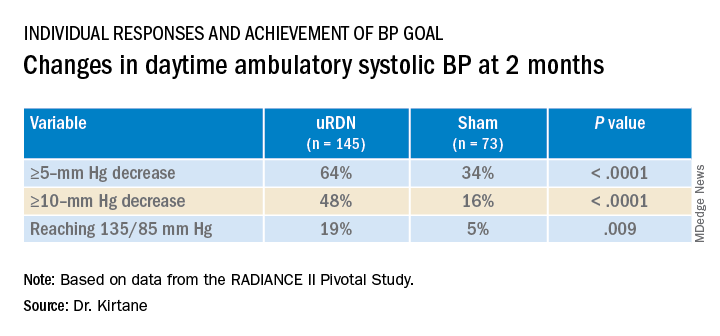
Also in the uRDN group, there was a 6.2–mm Hg larger decrease in 24-hour ambulatory systolic BP (P < .0001), a 5.8–mm Hg greater decline in nighttime ambulatory systolic BP (P < .0004), a 7.6–mm Hg steeper drop in mean home systolic BP (P < .0001), and 5.4 mm Hg more of a decrease in office-based systolic BP (P = .0035).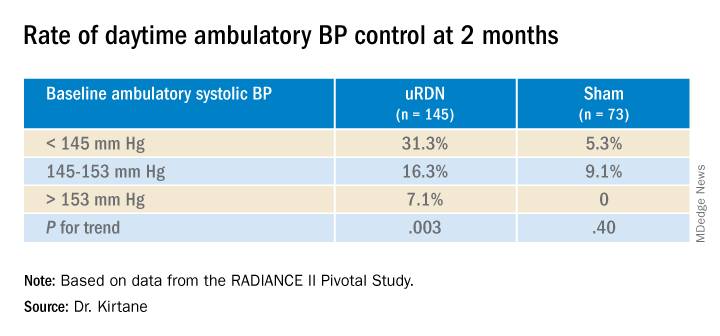
No significant differences were seen in subgroup analyses by sex, age, higher versus lower baseline systolic pressures, high versus low baseline eGFR, degree of abdominal obesity, U.S. versus European site, or whether patients entered before or during the COVID pandemic
Regulators have been accepting change in systolic BP as a surrogate for clinical endpoints in trials of antihypertensive therapy, whether pharmacologic or interventional, under consideration for approval. “That’s why safety endpoints are important to investigate” in these clinical trials, especially for invasive therapies like renal denervation, Dr. Kirtane observed.
That said, “in the longer-term follow-ups of the renal denervation therapies that are out there, including this one, there does not appear to be an appreciable decline in glomerular filtration rate, or any adverse safety signals that we see to date,” Dr. Kirtane said in an interview. “But we know that these are low-frequency events, so we have to be very vigilant, and we can’t get complacent about it.”
In RADIANCE II, there were zero adverse events within 30 days in both groups; the endpoint included death, new myocardial infarction, renal artery complications requiring invasive intervention, and hospitalization for major cardiovascular or hemodynamic-related events. Nor were there instances of new-onset renal artery stenosis greater than 70% documented by imaging at 6 months.
The ReCor uRDN catheter uses ultrasound energy to disrupt renal nerve signaling, a technology thought to deliver safer “burns,” compared with other renal denervation catheter technologies. It features an axially stabilizing balloon that transmits ultrasound energy – two to three sonications, each lasting 7 seconds, Dr. Kirtane said – outward through the arterial wall. The design is intended to ensure consistently circumferential ablation. Circulating saline within the balloon, Kirtane noted, directly cools the adjacent vessel wall to help it avoid thermal damage.
Dr. Kirtane reported receiving institutional funding from Medtronic, Boston Scientific, Abbott Vascular, Amgen, CSI, Philips, ReCor Medical, Neurotronic, Biotronik, Chiesi, Bolt Medical, Magenta Medical, Canon, SoniVie, Shockwave Medical, and Merck; consulting for IMDS; and receiving travel and meal expenses from Medtronic, Boston Scientific, Abbott Vascular, CSI, Siemens, Philips, ReCor Medical, Chiesi, OpSens, Zoll, and Regeneron. Dr. Fisher disclosed receiving honoraria or fees for consulting or serving on a speaker’s bureau for Medtronic, ReCor Medical, and Aktiia and receiving grant support or holding research contracts for Recor Medical and Aktiia. Dr. Messerli disclosed receiving honoraria from Medtronic, Menarini, Krka, and Ipca.
A version of this article first appeared on Medscape.com.
Systolic blood pressure went down safely and consistently 2 months after renal denervation achieved by ultrasound ablation in patients with uncontrolled, mild to moderate hypertension (HTN) in a key sham-controlled test of the balloon-equipped catheter.
The BP reductions were significant almost regardless of how they were measured – at home, in the office, during the day, at night, or over 24 hours – and weren’t dependent on baseline BP levels.
The 224-patient RADIANCE II Pivotal Study follows two earlier successful sham-controlled trials that used the same renal denervation catheter in other types of patients with HTN. They were RADIANCE-HTN SOLO, which entered patients with mild to moderate HTN not taking medication, and RADIANCE-HTN TRIO, which included patients with HTN despite fixed-dose, single-pill, triple-antihypertensive therapy.
The consistent results of all three trials suggest that the ultrasound renal denervation (uRDN) technique “lowers blood pressure across the spectrum of hypertension,” concluded co–principal investigator Ajay J. Kirtane, MD, SM, Columbia University Irving Medical Center, New York–Presbyterian Hospital, when presenting RADIANCE II at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
RADIANCE II, the largest of the three studies, met its prespecified primary efficacy endpoint of change in daytime ambulatory systolic BP at 2 months by showing a significant 6.3–mm Hg greater reduction in the uRDN group, compared with the sham-control group. There were no major adverse events at 30 days in either group.
The trial was similarly successful for the secondary endpoints of change in systolic BP measured in various other settings, including over 24 hours. Reductions after uRDN averaged 5-7 mm Hg greater than in the control group.
Sparse top-line results of the RADIANCE II pivotal trial were announced in July by the study’s sponsor, ReCor Medical.
Dr. Kirtane stressed in an interview that uRDN and likely any form of HTN renal denervation therapy is not a substitute for standard management. “This is really for patients in whom you’ve made best efforts to do the traditional things – lifestyle modification, medications, all of that – and yet they’re still uncontrolled.” At that point, assuming denervation therapy is available in practice, “it would be something to potentially consider.”
As a panelist after Dr. Kirtane’s formal presentation of RADIANCE II at the conference, Naomi D. Fisher, MD, who was a RADIANCE-HTN TRIO investigator, described how the treatment’s perceived intended patient population evolved over time.
“We all began with the idea that we were going to treat patients with resistant hypertension, that was going to be the first target. We have learned that those patients are far fewer than we thought,” said Dr. Fisher, who directs the hypertension service at Brigham and Women’s Hospital, Boston.
Initial estimates were that such patients with the resistant form, “meaning they require more than three drugs to control their blood pressure,” would represent 15%-20% of patients with HTN.
“We learned from our TRIO data that if you give these patients one single combined pill, lo and behold, many of them become controlled,” she said. “There is so much nonadherence out there in the world, about 50% of our patients aren’t taking their pills. It’s a hard and true fact.”
Exclude patients who aren’t adherent and “our true resistance population becomes minuscule. So, I don’t think that’s going to be the main population” for renal denervation therapy.
More likely, she said, it would be “patients who are uncontrolled and unable to take their medications. So that is going to include nonadherence, intolerance. It’s a very large category of patients. And the priorities can be stacked in favor of those who have higher cardiovascular risk.”
RADIANCE II can show the persistence of uRDN’s BP-lowering effect only out to 2 months so far, but the effect’s durability based on the RADIANCE program’s combined experience appears to be at least 2 years, Dr. Kirtane said in an interview.
“The RADIANCE II pivotal trial is a powerful, well-designed study attesting to the efficacy of renal denervation in BP lowering,” Franz H. Messerli, MD, Swiss Cardiovascular Center, University Hospital Bern, said in an interview.
The trial “shows the well-known unpredictability of antihypertensive response. We cannot predict who responds to renal denervation and who does not, and who even has a paradoxical increase in BP,” Dr. Messerli, an international hypertension expert not associated with the trial, said in an interview.
“As long as we cannot predict the antihypertensive response to renal denervation therapy, potential synergism/antagonism with drug therapy remains an educated guess,” he said.
“Hypertension is a disease that lasts years and decades. As impressive as RADIANCE II’s 2-month snapshot is, I look forward to similar or better BP data 12 and 24 months after renal denervation,” Dr. Messerli added.
RADIANCE II entered patients with mild to moderate uncontrolled HTN, that is, a systolic BP at least 140/90 mm Hg and less than 180/120 mm Hg, who were receiving no more than two antihypertensive medications. They could have no history of cardiovascular or cerebrovascular events or uncontrolled diabetes, and their estimated glomerular filtration rate (eGFR) had to be at least 40 mL/min per 1.73 m2.
After a 4-week drug washout period, patients who were clinically stable with an ambulatory BP of at least 135/85 mm Hg and less than 170/105 mm Hg underwent CT and renal angiography. Then, the 224 patients still anatomically eligible for the procedure were randomly assigned 2:1 to uRDN or a sham-control procedure: 150 and 74 patients, respectively.
At 2 months, daytime ambulatory systolic BP on average fell 7.9 mm Hg in the uRDN group and 1.8 mm Hg in the sham-control group, for a drop that was steeper by 6.3 mm Hg (P < .0001) after uRDN.
Also in the uRDN group, there was a 6.2–mm Hg larger decrease in 24-hour ambulatory systolic BP (P < .0001), a 5.8–mm Hg greater decline in nighttime ambulatory systolic BP (P < .0004), a 7.6–mm Hg steeper drop in mean home systolic BP (P < .0001), and 5.4 mm Hg more of a decrease in office-based systolic BP (P = .0035).
No significant differences were seen in subgroup analyses by sex, age, higher versus lower baseline systolic pressures, high versus low baseline eGFR, degree of abdominal obesity, U.S. versus European site, or whether patients entered before or during the COVID pandemic
Regulators have been accepting change in systolic BP as a surrogate for clinical endpoints in trials of antihypertensive therapy, whether pharmacologic or interventional, under consideration for approval. “That’s why safety endpoints are important to investigate” in these clinical trials, especially for invasive therapies like renal denervation, Dr. Kirtane observed.
That said, “in the longer-term follow-ups of the renal denervation therapies that are out there, including this one, there does not appear to be an appreciable decline in glomerular filtration rate, or any adverse safety signals that we see to date,” Dr. Kirtane said in an interview. “But we know that these are low-frequency events, so we have to be very vigilant, and we can’t get complacent about it.”
In RADIANCE II, there were zero adverse events within 30 days in both groups; the endpoint included death, new myocardial infarction, renal artery complications requiring invasive intervention, and hospitalization for major cardiovascular or hemodynamic-related events. Nor were there instances of new-onset renal artery stenosis greater than 70% documented by imaging at 6 months.
The ReCor uRDN catheter uses ultrasound energy to disrupt renal nerve signaling, a technology thought to deliver safer “burns,” compared with other renal denervation catheter technologies. It features an axially stabilizing balloon that transmits ultrasound energy – two to three sonications, each lasting 7 seconds, Dr. Kirtane said – outward through the arterial wall. The design is intended to ensure consistently circumferential ablation. Circulating saline within the balloon, Kirtane noted, directly cools the adjacent vessel wall to help it avoid thermal damage.
Dr. Kirtane reported receiving institutional funding from Medtronic, Boston Scientific, Abbott Vascular, Amgen, CSI, Philips, ReCor Medical, Neurotronic, Biotronik, Chiesi, Bolt Medical, Magenta Medical, Canon, SoniVie, Shockwave Medical, and Merck; consulting for IMDS; and receiving travel and meal expenses from Medtronic, Boston Scientific, Abbott Vascular, CSI, Siemens, Philips, ReCor Medical, Chiesi, OpSens, Zoll, and Regeneron. Dr. Fisher disclosed receiving honoraria or fees for consulting or serving on a speaker’s bureau for Medtronic, ReCor Medical, and Aktiia and receiving grant support or holding research contracts for Recor Medical and Aktiia. Dr. Messerli disclosed receiving honoraria from Medtronic, Menarini, Krka, and Ipca.
A version of this article first appeared on Medscape.com.
Systolic blood pressure went down safely and consistently 2 months after renal denervation achieved by ultrasound ablation in patients with uncontrolled, mild to moderate hypertension (HTN) in a key sham-controlled test of the balloon-equipped catheter.
The BP reductions were significant almost regardless of how they were measured – at home, in the office, during the day, at night, or over 24 hours – and weren’t dependent on baseline BP levels.
The 224-patient RADIANCE II Pivotal Study follows two earlier successful sham-controlled trials that used the same renal denervation catheter in other types of patients with HTN. They were RADIANCE-HTN SOLO, which entered patients with mild to moderate HTN not taking medication, and RADIANCE-HTN TRIO, which included patients with HTN despite fixed-dose, single-pill, triple-antihypertensive therapy.
The consistent results of all three trials suggest that the ultrasound renal denervation (uRDN) technique “lowers blood pressure across the spectrum of hypertension,” concluded co–principal investigator Ajay J. Kirtane, MD, SM, Columbia University Irving Medical Center, New York–Presbyterian Hospital, when presenting RADIANCE II at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
RADIANCE II, the largest of the three studies, met its prespecified primary efficacy endpoint of change in daytime ambulatory systolic BP at 2 months by showing a significant 6.3–mm Hg greater reduction in the uRDN group, compared with the sham-control group. There were no major adverse events at 30 days in either group.
The trial was similarly successful for the secondary endpoints of change in systolic BP measured in various other settings, including over 24 hours. Reductions after uRDN averaged 5-7 mm Hg greater than in the control group.
Sparse top-line results of the RADIANCE II pivotal trial were announced in July by the study’s sponsor, ReCor Medical.
Dr. Kirtane stressed in an interview that uRDN and likely any form of HTN renal denervation therapy is not a substitute for standard management. “This is really for patients in whom you’ve made best efforts to do the traditional things – lifestyle modification, medications, all of that – and yet they’re still uncontrolled.” At that point, assuming denervation therapy is available in practice, “it would be something to potentially consider.”
As a panelist after Dr. Kirtane’s formal presentation of RADIANCE II at the conference, Naomi D. Fisher, MD, who was a RADIANCE-HTN TRIO investigator, described how the treatment’s perceived intended patient population evolved over time.
“We all began with the idea that we were going to treat patients with resistant hypertension, that was going to be the first target. We have learned that those patients are far fewer than we thought,” said Dr. Fisher, who directs the hypertension service at Brigham and Women’s Hospital, Boston.
Initial estimates were that such patients with the resistant form, “meaning they require more than three drugs to control their blood pressure,” would represent 15%-20% of patients with HTN.
“We learned from our TRIO data that if you give these patients one single combined pill, lo and behold, many of them become controlled,” she said. “There is so much nonadherence out there in the world, about 50% of our patients aren’t taking their pills. It’s a hard and true fact.”
Exclude patients who aren’t adherent and “our true resistance population becomes minuscule. So, I don’t think that’s going to be the main population” for renal denervation therapy.
More likely, she said, it would be “patients who are uncontrolled and unable to take their medications. So that is going to include nonadherence, intolerance. It’s a very large category of patients. And the priorities can be stacked in favor of those who have higher cardiovascular risk.”
RADIANCE II can show the persistence of uRDN’s BP-lowering effect only out to 2 months so far, but the effect’s durability based on the RADIANCE program’s combined experience appears to be at least 2 years, Dr. Kirtane said in an interview.
“The RADIANCE II pivotal trial is a powerful, well-designed study attesting to the efficacy of renal denervation in BP lowering,” Franz H. Messerli, MD, Swiss Cardiovascular Center, University Hospital Bern, said in an interview.
The trial “shows the well-known unpredictability of antihypertensive response. We cannot predict who responds to renal denervation and who does not, and who even has a paradoxical increase in BP,” Dr. Messerli, an international hypertension expert not associated with the trial, said in an interview.
“As long as we cannot predict the antihypertensive response to renal denervation therapy, potential synergism/antagonism with drug therapy remains an educated guess,” he said.
“Hypertension is a disease that lasts years and decades. As impressive as RADIANCE II’s 2-month snapshot is, I look forward to similar or better BP data 12 and 24 months after renal denervation,” Dr. Messerli added.
RADIANCE II entered patients with mild to moderate uncontrolled HTN, that is, a systolic BP at least 140/90 mm Hg and less than 180/120 mm Hg, who were receiving no more than two antihypertensive medications. They could have no history of cardiovascular or cerebrovascular events or uncontrolled diabetes, and their estimated glomerular filtration rate (eGFR) had to be at least 40 mL/min per 1.73 m2.
After a 4-week drug washout period, patients who were clinically stable with an ambulatory BP of at least 135/85 mm Hg and less than 170/105 mm Hg underwent CT and renal angiography. Then, the 224 patients still anatomically eligible for the procedure were randomly assigned 2:1 to uRDN or a sham-control procedure: 150 and 74 patients, respectively.
At 2 months, daytime ambulatory systolic BP on average fell 7.9 mm Hg in the uRDN group and 1.8 mm Hg in the sham-control group, for a drop that was steeper by 6.3 mm Hg (P < .0001) after uRDN.
Also in the uRDN group, there was a 6.2–mm Hg larger decrease in 24-hour ambulatory systolic BP (P < .0001), a 5.8–mm Hg greater decline in nighttime ambulatory systolic BP (P < .0004), a 7.6–mm Hg steeper drop in mean home systolic BP (P < .0001), and 5.4 mm Hg more of a decrease in office-based systolic BP (P = .0035).
No significant differences were seen in subgroup analyses by sex, age, higher versus lower baseline systolic pressures, high versus low baseline eGFR, degree of abdominal obesity, U.S. versus European site, or whether patients entered before or during the COVID pandemic
Regulators have been accepting change in systolic BP as a surrogate for clinical endpoints in trials of antihypertensive therapy, whether pharmacologic or interventional, under consideration for approval. “That’s why safety endpoints are important to investigate” in these clinical trials, especially for invasive therapies like renal denervation, Dr. Kirtane observed.
That said, “in the longer-term follow-ups of the renal denervation therapies that are out there, including this one, there does not appear to be an appreciable decline in glomerular filtration rate, or any adverse safety signals that we see to date,” Dr. Kirtane said in an interview. “But we know that these are low-frequency events, so we have to be very vigilant, and we can’t get complacent about it.”
In RADIANCE II, there were zero adverse events within 30 days in both groups; the endpoint included death, new myocardial infarction, renal artery complications requiring invasive intervention, and hospitalization for major cardiovascular or hemodynamic-related events. Nor were there instances of new-onset renal artery stenosis greater than 70% documented by imaging at 6 months.
The ReCor uRDN catheter uses ultrasound energy to disrupt renal nerve signaling, a technology thought to deliver safer “burns,” compared with other renal denervation catheter technologies. It features an axially stabilizing balloon that transmits ultrasound energy – two to three sonications, each lasting 7 seconds, Dr. Kirtane said – outward through the arterial wall. The design is intended to ensure consistently circumferential ablation. Circulating saline within the balloon, Kirtane noted, directly cools the adjacent vessel wall to help it avoid thermal damage.
Dr. Kirtane reported receiving institutional funding from Medtronic, Boston Scientific, Abbott Vascular, Amgen, CSI, Philips, ReCor Medical, Neurotronic, Biotronik, Chiesi, Bolt Medical, Magenta Medical, Canon, SoniVie, Shockwave Medical, and Merck; consulting for IMDS; and receiving travel and meal expenses from Medtronic, Boston Scientific, Abbott Vascular, CSI, Siemens, Philips, ReCor Medical, Chiesi, OpSens, Zoll, and Regeneron. Dr. Fisher disclosed receiving honoraria or fees for consulting or serving on a speaker’s bureau for Medtronic, ReCor Medical, and Aktiia and receiving grant support or holding research contracts for Recor Medical and Aktiia. Dr. Messerli disclosed receiving honoraria from Medtronic, Menarini, Krka, and Ipca.
A version of this article first appeared on Medscape.com.
FROM TCT 2022
TAVR now used in almost 50% of younger severe aortic stenosis patients
Among patients with severe isolated aortic stenosis younger than 65, the rate of transcatheter aortic valve replacement (TAVR) now almost matches that of surgical aortic valve replacement (SAVR), despite guideline recommendations to the contrary, a study in a national U.S. population shows.
The 2020 American Heart Association/American College of Cardiology (AHA/ACC) valve guideline recommends SAVR for patients younger than 65 with severe aortic stenosis, the researchers note, but their study showed “near equal utilization between TAVR and SAVR in these younger patients by 2021,” at 48% and 52% respectively.
Toishi Sharma, MD, and colleagues presented these findings in an oral poster session at Transcatheter Cardiovascular Therapeutics 2022, and the study was simultaneously published as a Research Letter in the Journal of the American College of Cardiology (JACC).
“To our knowledge, the current findings represent the first national temporal trends study stratifying [aortic stenosis] therapies according to guideline-recommended age groups: our observations demonstrate the dramatic growth of TAVR in all age groups, including young patients,” the researchers conclude.
They analyzed changes in rates of TAVR and SAVR in a U.S. sample stratified by age: younger than 65 years, 65-80, and older than 80 years.
These findings have implications for lifetime management of younger patients who undergo TAVR, they write, “including issues related to lifetime coronary access, valve durability, and the potential for subsequent TAVR procedures over time.”
Three age groups
In a study published in JACC, this group examined changes in uptake of TAVR versus SAVR in 4,161 patients with aortic stenosis in Vermont, New Hampshire, and Maine, senior author Harold L. Dauerman, MD, said in an interview.
The greatest rate of rise of TAVR was in the group younger than 65, but that study ended in 2019, said Dr. Dauerman, from the University of Vermont Health Network, Burlington.
The 2020 guideline stratifies TAVR and SAVR recommendations such that “less than 65 should primarily be a surgical approach and greater than 80 primarily a TAVR approach, while 65 to 80 is a gray zone, and shared decision-making becomes important,” he noted.
The group hypothesized that recent trials and technology have led to a national increase in TAVR in people younger than 65.
From the Vizient clinical database, including more than 250 U.S. academic centers that perform both TAVR and SAVR, the researchers identified 142,953 patients who underwent TAVR or SAVR for isolated aortic stenosis from Oct. 1, 2015, to Dec. 31, 2021. From 2015 to 2021, the valve replacement rates in the three age groups changed as follows:
- Age less than 65: TAVR rose from 17% to 48%; SAVR fell from 83% to 52%.
- Age 65-80: TAVR rose from 46% to 87%; SAVR fell from 54% to 12%.
- Age greater than 80: TAVR rose from 83% to 99%; SAVR fell from 16% to 1%.
“All ages have grown in the last 7 years in TAVR,” Dr. Dauerman summarized. “The one that’s surprising, and in contradiction to the guideline, is the growth of TAVR in young patients less than 65.”
Among patients younger than 65, prior bypass surgery and congestive heart failure predicted the use of TAVR instead of surgery, whereas bicuspid aortic valve disease was the biggest predictor of surgery instead of TAVR.
Most studies on TAVR valve durability are limited to patients in the randomized trials who were primarily in their mid-70s to mid-80s, some of whom died before a 10-year follow-up, Dr. Dauerman noted.
European guidelines recommend surgery for patients younger than 70, and it would be interesting to see if clinicians there follow this recommendation or if TAVR is now the preferred approach, he added.
There is a need for further, longer study of TAVR in younger patients, he said, to determine whether there are long-term clinical issues of concern.
Strategy depends on more than age
The “findings are not too surprising,” John Carroll, MD, who was not involved in this research, said in an email.

“Age is only one of multiple patient characteristics that enter into consideration of TAVR versus SAVR,” said Dr. Carroll, from Anschutz Medical Campus, University of Colorado, Aurora.
“As the article reports,” he noted, “those less than 65 having TAVR are more likely to have comorbid conditions that likely made the risk of SAVR higher.”
Dr. Carroll was lead author of a review article published in 2020 based on data from the ACC–Society of Thoracic Surgeons (STS)–Transcatheter Valve Therapy (TVT) registry on 276,316 patients who had TAVR in the United States from 2011-2019.
He pointed out that Figure 2 in that review shows that “SAVR is often performed in conjunction with other surgical procedures – another major reason why SAVR remains an important treatment for valvular heart disease.”
“We are awaiting long-term data comparing TAVR to SAVR durability,” Dr. Carroll added, echoing Dr. Dauerman. “So far [there are] no major differences, but it remains a key need to fully understand TAVR and the various models in commercial use.”
“Both TAVR and SAVR used in adults are tissue valves (SAVR with mechanical valves is used in younger patients),” Dr. Carroll noted, “and all tissue valves will eventually fail if the patient lives long enough.”
Patient management strategies need to consider what treatment options exist when the first valve fails. “If the first valve is SAVR, there is now extensive experience with placing a TAVR valve inside a failing SAVR valve, so called Valve-in-Valve or TAVR-in-SAVR. This is the preferred treatment in most patients with failing SAVR valves,” he said.
“On the other hand,” he continued, “we are just beginning to see more and more patients with failing TAVR valves, and the TAVR-in-TAVR procedure is less well understood.”
“Issues such as acute coronary occlusion and long-term difficulty in accessing coronary arteries are being encountered in some patients having TAVR-in-TAVR,” Dr. Carroll noted, which he discusses in a recent editorial he coauthored about the complexities of redo TAVR, published in JACC: Cardiovascular Interventions.
The study received no funding. Dr. Dauerman has research grants and is a consultant for Medtronic and Boston Scientific. Dr. Carroll is a local principal investigator in trials sponsored by Medtronic, Abbott, and Edwards Lifesciences.
A version of this article first appeared on Medscape.com.
Among patients with severe isolated aortic stenosis younger than 65, the rate of transcatheter aortic valve replacement (TAVR) now almost matches that of surgical aortic valve replacement (SAVR), despite guideline recommendations to the contrary, a study in a national U.S. population shows.
The 2020 American Heart Association/American College of Cardiology (AHA/ACC) valve guideline recommends SAVR for patients younger than 65 with severe aortic stenosis, the researchers note, but their study showed “near equal utilization between TAVR and SAVR in these younger patients by 2021,” at 48% and 52% respectively.
Toishi Sharma, MD, and colleagues presented these findings in an oral poster session at Transcatheter Cardiovascular Therapeutics 2022, and the study was simultaneously published as a Research Letter in the Journal of the American College of Cardiology (JACC).
“To our knowledge, the current findings represent the first national temporal trends study stratifying [aortic stenosis] therapies according to guideline-recommended age groups: our observations demonstrate the dramatic growth of TAVR in all age groups, including young patients,” the researchers conclude.
They analyzed changes in rates of TAVR and SAVR in a U.S. sample stratified by age: younger than 65 years, 65-80, and older than 80 years.
These findings have implications for lifetime management of younger patients who undergo TAVR, they write, “including issues related to lifetime coronary access, valve durability, and the potential for subsequent TAVR procedures over time.”
Three age groups
In a study published in JACC, this group examined changes in uptake of TAVR versus SAVR in 4,161 patients with aortic stenosis in Vermont, New Hampshire, and Maine, senior author Harold L. Dauerman, MD, said in an interview.
The greatest rate of rise of TAVR was in the group younger than 65, but that study ended in 2019, said Dr. Dauerman, from the University of Vermont Health Network, Burlington.
The 2020 guideline stratifies TAVR and SAVR recommendations such that “less than 65 should primarily be a surgical approach and greater than 80 primarily a TAVR approach, while 65 to 80 is a gray zone, and shared decision-making becomes important,” he noted.
The group hypothesized that recent trials and technology have led to a national increase in TAVR in people younger than 65.
From the Vizient clinical database, including more than 250 U.S. academic centers that perform both TAVR and SAVR, the researchers identified 142,953 patients who underwent TAVR or SAVR for isolated aortic stenosis from Oct. 1, 2015, to Dec. 31, 2021. From 2015 to 2021, the valve replacement rates in the three age groups changed as follows:
- Age less than 65: TAVR rose from 17% to 48%; SAVR fell from 83% to 52%.
- Age 65-80: TAVR rose from 46% to 87%; SAVR fell from 54% to 12%.
- Age greater than 80: TAVR rose from 83% to 99%; SAVR fell from 16% to 1%.
“All ages have grown in the last 7 years in TAVR,” Dr. Dauerman summarized. “The one that’s surprising, and in contradiction to the guideline, is the growth of TAVR in young patients less than 65.”
Among patients younger than 65, prior bypass surgery and congestive heart failure predicted the use of TAVR instead of surgery, whereas bicuspid aortic valve disease was the biggest predictor of surgery instead of TAVR.
Most studies on TAVR valve durability are limited to patients in the randomized trials who were primarily in their mid-70s to mid-80s, some of whom died before a 10-year follow-up, Dr. Dauerman noted.
European guidelines recommend surgery for patients younger than 70, and it would be interesting to see if clinicians there follow this recommendation or if TAVR is now the preferred approach, he added.
There is a need for further, longer study of TAVR in younger patients, he said, to determine whether there are long-term clinical issues of concern.
Strategy depends on more than age
The “findings are not too surprising,” John Carroll, MD, who was not involved in this research, said in an email.
“Age is only one of multiple patient characteristics that enter into consideration of TAVR versus SAVR,” said Dr. Carroll, from Anschutz Medical Campus, University of Colorado, Aurora.
“As the article reports,” he noted, “those less than 65 having TAVR are more likely to have comorbid conditions that likely made the risk of SAVR higher.”
Dr. Carroll was lead author of a review article published in 2020 based on data from the ACC–Society of Thoracic Surgeons (STS)–Transcatheter Valve Therapy (TVT) registry on 276,316 patients who had TAVR in the United States from 2011-2019.
He pointed out that Figure 2 in that review shows that “SAVR is often performed in conjunction with other surgical procedures – another major reason why SAVR remains an important treatment for valvular heart disease.”
“We are awaiting long-term data comparing TAVR to SAVR durability,” Dr. Carroll added, echoing Dr. Dauerman. “So far [there are] no major differences, but it remains a key need to fully understand TAVR and the various models in commercial use.”
“Both TAVR and SAVR used in adults are tissue valves (SAVR with mechanical valves is used in younger patients),” Dr. Carroll noted, “and all tissue valves will eventually fail if the patient lives long enough.”
Patient management strategies need to consider what treatment options exist when the first valve fails. “If the first valve is SAVR, there is now extensive experience with placing a TAVR valve inside a failing SAVR valve, so called Valve-in-Valve or TAVR-in-SAVR. This is the preferred treatment in most patients with failing SAVR valves,” he said.
“On the other hand,” he continued, “we are just beginning to see more and more patients with failing TAVR valves, and the TAVR-in-TAVR procedure is less well understood.”
“Issues such as acute coronary occlusion and long-term difficulty in accessing coronary arteries are being encountered in some patients having TAVR-in-TAVR,” Dr. Carroll noted, which he discusses in a recent editorial he coauthored about the complexities of redo TAVR, published in JACC: Cardiovascular Interventions.
The study received no funding. Dr. Dauerman has research grants and is a consultant for Medtronic and Boston Scientific. Dr. Carroll is a local principal investigator in trials sponsored by Medtronic, Abbott, and Edwards Lifesciences.
A version of this article first appeared on Medscape.com.
Among patients with severe isolated aortic stenosis younger than 65, the rate of transcatheter aortic valve replacement (TAVR) now almost matches that of surgical aortic valve replacement (SAVR), despite guideline recommendations to the contrary, a study in a national U.S. population shows.
The 2020 American Heart Association/American College of Cardiology (AHA/ACC) valve guideline recommends SAVR for patients younger than 65 with severe aortic stenosis, the researchers note, but their study showed “near equal utilization between TAVR and SAVR in these younger patients by 2021,” at 48% and 52% respectively.
Toishi Sharma, MD, and colleagues presented these findings in an oral poster session at Transcatheter Cardiovascular Therapeutics 2022, and the study was simultaneously published as a Research Letter in the Journal of the American College of Cardiology (JACC).
“To our knowledge, the current findings represent the first national temporal trends study stratifying [aortic stenosis] therapies according to guideline-recommended age groups: our observations demonstrate the dramatic growth of TAVR in all age groups, including young patients,” the researchers conclude.
They analyzed changes in rates of TAVR and SAVR in a U.S. sample stratified by age: younger than 65 years, 65-80, and older than 80 years.
These findings have implications for lifetime management of younger patients who undergo TAVR, they write, “including issues related to lifetime coronary access, valve durability, and the potential for subsequent TAVR procedures over time.”
Three age groups
In a study published in JACC, this group examined changes in uptake of TAVR versus SAVR in 4,161 patients with aortic stenosis in Vermont, New Hampshire, and Maine, senior author Harold L. Dauerman, MD, said in an interview.
The greatest rate of rise of TAVR was in the group younger than 65, but that study ended in 2019, said Dr. Dauerman, from the University of Vermont Health Network, Burlington.
The 2020 guideline stratifies TAVR and SAVR recommendations such that “less than 65 should primarily be a surgical approach and greater than 80 primarily a TAVR approach, while 65 to 80 is a gray zone, and shared decision-making becomes important,” he noted.
The group hypothesized that recent trials and technology have led to a national increase in TAVR in people younger than 65.
From the Vizient clinical database, including more than 250 U.S. academic centers that perform both TAVR and SAVR, the researchers identified 142,953 patients who underwent TAVR or SAVR for isolated aortic stenosis from Oct. 1, 2015, to Dec. 31, 2021. From 2015 to 2021, the valve replacement rates in the three age groups changed as follows:
- Age less than 65: TAVR rose from 17% to 48%; SAVR fell from 83% to 52%.
- Age 65-80: TAVR rose from 46% to 87%; SAVR fell from 54% to 12%.
- Age greater than 80: TAVR rose from 83% to 99%; SAVR fell from 16% to 1%.
“All ages have grown in the last 7 years in TAVR,” Dr. Dauerman summarized. “The one that’s surprising, and in contradiction to the guideline, is the growth of TAVR in young patients less than 65.”
Among patients younger than 65, prior bypass surgery and congestive heart failure predicted the use of TAVR instead of surgery, whereas bicuspid aortic valve disease was the biggest predictor of surgery instead of TAVR.
Most studies on TAVR valve durability are limited to patients in the randomized trials who were primarily in their mid-70s to mid-80s, some of whom died before a 10-year follow-up, Dr. Dauerman noted.
European guidelines recommend surgery for patients younger than 70, and it would be interesting to see if clinicians there follow this recommendation or if TAVR is now the preferred approach, he added.
There is a need for further, longer study of TAVR in younger patients, he said, to determine whether there are long-term clinical issues of concern.
Strategy depends on more than age
The “findings are not too surprising,” John Carroll, MD, who was not involved in this research, said in an email.
“Age is only one of multiple patient characteristics that enter into consideration of TAVR versus SAVR,” said Dr. Carroll, from Anschutz Medical Campus, University of Colorado, Aurora.
“As the article reports,” he noted, “those less than 65 having TAVR are more likely to have comorbid conditions that likely made the risk of SAVR higher.”
Dr. Carroll was lead author of a review article published in 2020 based on data from the ACC–Society of Thoracic Surgeons (STS)–Transcatheter Valve Therapy (TVT) registry on 276,316 patients who had TAVR in the United States from 2011-2019.
He pointed out that Figure 2 in that review shows that “SAVR is often performed in conjunction with other surgical procedures – another major reason why SAVR remains an important treatment for valvular heart disease.”
“We are awaiting long-term data comparing TAVR to SAVR durability,” Dr. Carroll added, echoing Dr. Dauerman. “So far [there are] no major differences, but it remains a key need to fully understand TAVR and the various models in commercial use.”
“Both TAVR and SAVR used in adults are tissue valves (SAVR with mechanical valves is used in younger patients),” Dr. Carroll noted, “and all tissue valves will eventually fail if the patient lives long enough.”
Patient management strategies need to consider what treatment options exist when the first valve fails. “If the first valve is SAVR, there is now extensive experience with placing a TAVR valve inside a failing SAVR valve, so called Valve-in-Valve or TAVR-in-SAVR. This is the preferred treatment in most patients with failing SAVR valves,” he said.
“On the other hand,” he continued, “we are just beginning to see more and more patients with failing TAVR valves, and the TAVR-in-TAVR procedure is less well understood.”
“Issues such as acute coronary occlusion and long-term difficulty in accessing coronary arteries are being encountered in some patients having TAVR-in-TAVR,” Dr. Carroll noted, which he discusses in a recent editorial he coauthored about the complexities of redo TAVR, published in JACC: Cardiovascular Interventions.
The study received no funding. Dr. Dauerman has research grants and is a consultant for Medtronic and Boston Scientific. Dr. Carroll is a local principal investigator in trials sponsored by Medtronic, Abbott, and Edwards Lifesciences.
A version of this article first appeared on Medscape.com.
FROM TCT 2022