User login
Juvenile Dermatomyositis Is Easily Missed Diagnosis
NEW YORK – Because juvenile dermatomyositis is a rare disease and its symptoms often differ from those seen in adult dermatomyositis, physicians may not recognize it and diagnosis can be delayed, Dr. Brian Feldman said.
He described a recent practice survey that found variability in juvenile dermatomyositis (JDM) treatment and, in the absence of randomized controlled trials, urged clinicians to consult newly created consensus protocols developed by rheumatologists and to gather data that may help to optimize treatment and minimize side effects in the future for those with JDM.
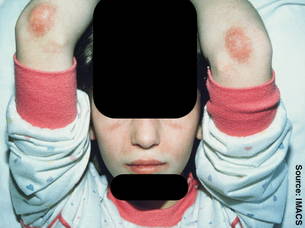
Dr. Feldman described a 15-year-old patient who had progressively deteriorated over a 3-year period despite being seen by pediatricians and dermatologists. His initial symptoms included fatigue and elevated muscle enzymes, including aspartate transaminase (AST), alanine transaminase (ALT), lactate dehydrogenase (LDH), and creatine phosphokinase (CPK).
Within a year, the patient developed a purple skin rash on his hands, elbows, and knees. He later developed Raynaud's syndrome and muscle weakness with decreased range of motion that interfered with participation in sports and other activities of daily living.
Upon examination, he was quite thin with a scaly rash over his knuckles (Gottron's papules), healing skin ulcerations on his hands (from severe Raynaud's), periungual erythema, and lipodystrophy and erythema of his forearms. Magnetic resonance imaging showed acute inflammation of his shoulder girdle, said Dr. Feldman, chief of pediatric rheumatology at the Hospital for Sick Children in Toronto.
Dr. Feldman said that one of the best diagnostic clues for JDM comes from microscopic examination of nail folds. In this case, the patient had tortuous, bushy nail folds, dilated or missing capillaries (capillary density of 3 per mm of nailfold length while normal ranges from 7-11 per mm), and cuticular overgrowth indicative of JDM.
The different presentation seen with children compared with adults may hinder a correct diagnosis. For instance, calcinosis is seen much more frequently in children than adults. Children are less likely to have some of the systemic symptoms, such as fever, poor weight gain, and pulmonary effects, and they almost never have cardiac problems. Unlike adults with dermatomyositis or polymyositis who appear to have a fourfold increased risk of malignancy, very few cases of malignancy have been associated with JDM. Children are more likely to experience dysphonia/dysphagia but are as likely to have arthritic symptoms (around 58% of each group). Myositis specific antibodies do not seem to play as important a diagnostic role for children as adults.
Children who develop JDM appear to have a better prognosis than adults. Although adults with myositis have appreciable mortality (about 10%) and progressive disability, which may be related to treatment of a long-term condition such as avascular necrosis or osteoporosis with compression fracture, findings from Dr. Feldman's group show that outcomes were often excellent in children. "We have not had a single death from JDM in 30 years," he said.
About two-thirds of children with JDM have a polycyclic, chronic, unremitting disease course, but about one-third follow a monocyclic course and have disease symptoms that last for 2-3 years and then go away, with, or potentially even without, treatment.
In a very small proportion of these patients, symptoms may return many years later. "The message here is that when the disease goes away, it almost certainly is gone forever," Dr. Feldman noted.
It is possible to predict which patients will follow a chronic course and which patients will have early remission. In his experience, the risk of persistent disease falls by half if Gottron's rash disappears within 3 months of diagnosis (Arthritis Rheum. 2008;58:3585-92).
The results of a recent practice survey by the Childhood Arthritis and Rheumatology Research Alliance (CARRA) of almost 200 pediatric rheumatologists in North America found both similarities and differences in treatment approaches to JDM (J. Rheumatol. 2010;37:1953-61).
Corticosteroids and methotrexate (MTX) are mainstays of care, but the route and pattern of corticosteroid administration varied: 82% of respondents initially pulsed with high-dose intravenous methylprednisolone (IVMP) while 18% prescribed oral prednisone. MTX was used concurrently with steroids 84% of the time. Sixteen percent reported also using IV immunoglobulin, with or without MTX, for more severe disease, refractory disease, and prominent cutaneous disease. Hydroxychloroquine was given for milder cases, especially for rash, while cyclophosphamide was prescribed for ulcerative disease or for patients with pulmonary symptoms.
While these findings reflect the current prescribing practices, there are almost no randomized controlled trials of medications for JDM, Dr. Feldman pointed out.
This reflects in part the rare nature of the disease, its complexity, the difficulty of conducting studies in children, and the high cost of randomized controlled trials. There have been studies using advanced analytic techniques to provide strong comparative data in lieu of an randomized controlled trial. One such study by Dr. Feldman and associates (Arthritis Rheum. 2008;59:989-95) showed that more aggressive corticosteroid therapy does not give better 3-year outcomes.
A recent randomized controlled trial presented at the annual meeting of the American College of Rheumatology looked at dermatomyositis in 76 adults and 48 children and found that 80% had a response to rituximab within a year of treatment. Interpretation of the study may have been limited by the design, which allowed patients to receive add-on medications during the trial.
In light of the CARRA study's findings of heterogeneity in the treatment of JDM and the absence of randomized controlled trials, a group of 12 pediatric rheumatologists met to study treatments in JDM using a new approach: by developing consensus treatment protocols (Arthritis Care Res. 2010;62:219-25).
"This is similar to what has been done in pediatric oncology," said Dr. Feldman, who was one of the participants. "We are hoping physicians throughout the world will take these protocols off the shelf, and by using standardized doses, follow-up, and measurements, we will be able to accumulate enough evidence over time to know which is the best therapy."
In brief, the group recommended three protocols for the treatment of patients with moderately severe JDM: pulse IVMP plus MTX; IVMP, MTX, plus IVIG; or oral prednisone plus MTX. The third protocol is the one followed most often at the Hospital for Sick Children. The treatment protocols are not intended as treatment recommendations, although it is hoped that a physician will choose to follow the standardized protocol that most closely reflects his or her preferred practice. It is presented as a "first step to allow comparison of different approaches to the treatment of JDM," he said.
Dr. Feldman has done contracted research with Bayer Healthcare Pharmaceuticals. He referenced unlabeled/unapproved uses of drugs or products in his presentation.
NEW YORK – Because juvenile dermatomyositis is a rare disease and its symptoms often differ from those seen in adult dermatomyositis, physicians may not recognize it and diagnosis can be delayed, Dr. Brian Feldman said.
He described a recent practice survey that found variability in juvenile dermatomyositis (JDM) treatment and, in the absence of randomized controlled trials, urged clinicians to consult newly created consensus protocols developed by rheumatologists and to gather data that may help to optimize treatment and minimize side effects in the future for those with JDM.
Dr. Feldman described a 15-year-old patient who had progressively deteriorated over a 3-year period despite being seen by pediatricians and dermatologists. His initial symptoms included fatigue and elevated muscle enzymes, including aspartate transaminase (AST), alanine transaminase (ALT), lactate dehydrogenase (LDH), and creatine phosphokinase (CPK).
Within a year, the patient developed a purple skin rash on his hands, elbows, and knees. He later developed Raynaud's syndrome and muscle weakness with decreased range of motion that interfered with participation in sports and other activities of daily living.
Upon examination, he was quite thin with a scaly rash over his knuckles (Gottron's papules), healing skin ulcerations on his hands (from severe Raynaud's), periungual erythema, and lipodystrophy and erythema of his forearms. Magnetic resonance imaging showed acute inflammation of his shoulder girdle, said Dr. Feldman, chief of pediatric rheumatology at the Hospital for Sick Children in Toronto.
Dr. Feldman said that one of the best diagnostic clues for JDM comes from microscopic examination of nail folds. In this case, the patient had tortuous, bushy nail folds, dilated or missing capillaries (capillary density of 3 per mm of nailfold length while normal ranges from 7-11 per mm), and cuticular overgrowth indicative of JDM.
The different presentation seen with children compared with adults may hinder a correct diagnosis. For instance, calcinosis is seen much more frequently in children than adults. Children are less likely to have some of the systemic symptoms, such as fever, poor weight gain, and pulmonary effects, and they almost never have cardiac problems. Unlike adults with dermatomyositis or polymyositis who appear to have a fourfold increased risk of malignancy, very few cases of malignancy have been associated with JDM. Children are more likely to experience dysphonia/dysphagia but are as likely to have arthritic symptoms (around 58% of each group). Myositis specific antibodies do not seem to play as important a diagnostic role for children as adults.
Children who develop JDM appear to have a better prognosis than adults. Although adults with myositis have appreciable mortality (about 10%) and progressive disability, which may be related to treatment of a long-term condition such as avascular necrosis or osteoporosis with compression fracture, findings from Dr. Feldman's group show that outcomes were often excellent in children. "We have not had a single death from JDM in 30 years," he said.
About two-thirds of children with JDM have a polycyclic, chronic, unremitting disease course, but about one-third follow a monocyclic course and have disease symptoms that last for 2-3 years and then go away, with, or potentially even without, treatment.
In a very small proportion of these patients, symptoms may return many years later. "The message here is that when the disease goes away, it almost certainly is gone forever," Dr. Feldman noted.
It is possible to predict which patients will follow a chronic course and which patients will have early remission. In his experience, the risk of persistent disease falls by half if Gottron's rash disappears within 3 months of diagnosis (Arthritis Rheum. 2008;58:3585-92).
The results of a recent practice survey by the Childhood Arthritis and Rheumatology Research Alliance (CARRA) of almost 200 pediatric rheumatologists in North America found both similarities and differences in treatment approaches to JDM (J. Rheumatol. 2010;37:1953-61).
Corticosteroids and methotrexate (MTX) are mainstays of care, but the route and pattern of corticosteroid administration varied: 82% of respondents initially pulsed with high-dose intravenous methylprednisolone (IVMP) while 18% prescribed oral prednisone. MTX was used concurrently with steroids 84% of the time. Sixteen percent reported also using IV immunoglobulin, with or without MTX, for more severe disease, refractory disease, and prominent cutaneous disease. Hydroxychloroquine was given for milder cases, especially for rash, while cyclophosphamide was prescribed for ulcerative disease or for patients with pulmonary symptoms.
While these findings reflect the current prescribing practices, there are almost no randomized controlled trials of medications for JDM, Dr. Feldman pointed out.
This reflects in part the rare nature of the disease, its complexity, the difficulty of conducting studies in children, and the high cost of randomized controlled trials. There have been studies using advanced analytic techniques to provide strong comparative data in lieu of an randomized controlled trial. One such study by Dr. Feldman and associates (Arthritis Rheum. 2008;59:989-95) showed that more aggressive corticosteroid therapy does not give better 3-year outcomes.
A recent randomized controlled trial presented at the annual meeting of the American College of Rheumatology looked at dermatomyositis in 76 adults and 48 children and found that 80% had a response to rituximab within a year of treatment. Interpretation of the study may have been limited by the design, which allowed patients to receive add-on medications during the trial.
In light of the CARRA study's findings of heterogeneity in the treatment of JDM and the absence of randomized controlled trials, a group of 12 pediatric rheumatologists met to study treatments in JDM using a new approach: by developing consensus treatment protocols (Arthritis Care Res. 2010;62:219-25).
"This is similar to what has been done in pediatric oncology," said Dr. Feldman, who was one of the participants. "We are hoping physicians throughout the world will take these protocols off the shelf, and by using standardized doses, follow-up, and measurements, we will be able to accumulate enough evidence over time to know which is the best therapy."
In brief, the group recommended three protocols for the treatment of patients with moderately severe JDM: pulse IVMP plus MTX; IVMP, MTX, plus IVIG; or oral prednisone plus MTX. The third protocol is the one followed most often at the Hospital for Sick Children. The treatment protocols are not intended as treatment recommendations, although it is hoped that a physician will choose to follow the standardized protocol that most closely reflects his or her preferred practice. It is presented as a "first step to allow comparison of different approaches to the treatment of JDM," he said.
Dr. Feldman has done contracted research with Bayer Healthcare Pharmaceuticals. He referenced unlabeled/unapproved uses of drugs or products in his presentation.
NEW YORK – Because juvenile dermatomyositis is a rare disease and its symptoms often differ from those seen in adult dermatomyositis, physicians may not recognize it and diagnosis can be delayed, Dr. Brian Feldman said.
He described a recent practice survey that found variability in juvenile dermatomyositis (JDM) treatment and, in the absence of randomized controlled trials, urged clinicians to consult newly created consensus protocols developed by rheumatologists and to gather data that may help to optimize treatment and minimize side effects in the future for those with JDM.
Dr. Feldman described a 15-year-old patient who had progressively deteriorated over a 3-year period despite being seen by pediatricians and dermatologists. His initial symptoms included fatigue and elevated muscle enzymes, including aspartate transaminase (AST), alanine transaminase (ALT), lactate dehydrogenase (LDH), and creatine phosphokinase (CPK).
Within a year, the patient developed a purple skin rash on his hands, elbows, and knees. He later developed Raynaud's syndrome and muscle weakness with decreased range of motion that interfered with participation in sports and other activities of daily living.
Upon examination, he was quite thin with a scaly rash over his knuckles (Gottron's papules), healing skin ulcerations on his hands (from severe Raynaud's), periungual erythema, and lipodystrophy and erythema of his forearms. Magnetic resonance imaging showed acute inflammation of his shoulder girdle, said Dr. Feldman, chief of pediatric rheumatology at the Hospital for Sick Children in Toronto.
Dr. Feldman said that one of the best diagnostic clues for JDM comes from microscopic examination of nail folds. In this case, the patient had tortuous, bushy nail folds, dilated or missing capillaries (capillary density of 3 per mm of nailfold length while normal ranges from 7-11 per mm), and cuticular overgrowth indicative of JDM.
The different presentation seen with children compared with adults may hinder a correct diagnosis. For instance, calcinosis is seen much more frequently in children than adults. Children are less likely to have some of the systemic symptoms, such as fever, poor weight gain, and pulmonary effects, and they almost never have cardiac problems. Unlike adults with dermatomyositis or polymyositis who appear to have a fourfold increased risk of malignancy, very few cases of malignancy have been associated with JDM. Children are more likely to experience dysphonia/dysphagia but are as likely to have arthritic symptoms (around 58% of each group). Myositis specific antibodies do not seem to play as important a diagnostic role for children as adults.
Children who develop JDM appear to have a better prognosis than adults. Although adults with myositis have appreciable mortality (about 10%) and progressive disability, which may be related to treatment of a long-term condition such as avascular necrosis or osteoporosis with compression fracture, findings from Dr. Feldman's group show that outcomes were often excellent in children. "We have not had a single death from JDM in 30 years," he said.
About two-thirds of children with JDM have a polycyclic, chronic, unremitting disease course, but about one-third follow a monocyclic course and have disease symptoms that last for 2-3 years and then go away, with, or potentially even without, treatment.
In a very small proportion of these patients, symptoms may return many years later. "The message here is that when the disease goes away, it almost certainly is gone forever," Dr. Feldman noted.
It is possible to predict which patients will follow a chronic course and which patients will have early remission. In his experience, the risk of persistent disease falls by half if Gottron's rash disappears within 3 months of diagnosis (Arthritis Rheum. 2008;58:3585-92).
The results of a recent practice survey by the Childhood Arthritis and Rheumatology Research Alliance (CARRA) of almost 200 pediatric rheumatologists in North America found both similarities and differences in treatment approaches to JDM (J. Rheumatol. 2010;37:1953-61).
Corticosteroids and methotrexate (MTX) are mainstays of care, but the route and pattern of corticosteroid administration varied: 82% of respondents initially pulsed with high-dose intravenous methylprednisolone (IVMP) while 18% prescribed oral prednisone. MTX was used concurrently with steroids 84% of the time. Sixteen percent reported also using IV immunoglobulin, with or without MTX, for more severe disease, refractory disease, and prominent cutaneous disease. Hydroxychloroquine was given for milder cases, especially for rash, while cyclophosphamide was prescribed for ulcerative disease or for patients with pulmonary symptoms.
While these findings reflect the current prescribing practices, there are almost no randomized controlled trials of medications for JDM, Dr. Feldman pointed out.
This reflects in part the rare nature of the disease, its complexity, the difficulty of conducting studies in children, and the high cost of randomized controlled trials. There have been studies using advanced analytic techniques to provide strong comparative data in lieu of an randomized controlled trial. One such study by Dr. Feldman and associates (Arthritis Rheum. 2008;59:989-95) showed that more aggressive corticosteroid therapy does not give better 3-year outcomes.
A recent randomized controlled trial presented at the annual meeting of the American College of Rheumatology looked at dermatomyositis in 76 adults and 48 children and found that 80% had a response to rituximab within a year of treatment. Interpretation of the study may have been limited by the design, which allowed patients to receive add-on medications during the trial.
In light of the CARRA study's findings of heterogeneity in the treatment of JDM and the absence of randomized controlled trials, a group of 12 pediatric rheumatologists met to study treatments in JDM using a new approach: by developing consensus treatment protocols (Arthritis Care Res. 2010;62:219-25).
"This is similar to what has been done in pediatric oncology," said Dr. Feldman, who was one of the participants. "We are hoping physicians throughout the world will take these protocols off the shelf, and by using standardized doses, follow-up, and measurements, we will be able to accumulate enough evidence over time to know which is the best therapy."
In brief, the group recommended three protocols for the treatment of patients with moderately severe JDM: pulse IVMP plus MTX; IVMP, MTX, plus IVIG; or oral prednisone plus MTX. The third protocol is the one followed most often at the Hospital for Sick Children. The treatment protocols are not intended as treatment recommendations, although it is hoped that a physician will choose to follow the standardized protocol that most closely reflects his or her preferred practice. It is presented as a "first step to allow comparison of different approaches to the treatment of JDM," he said.
Dr. Feldman has done contracted research with Bayer Healthcare Pharmaceuticals. He referenced unlabeled/unapproved uses of drugs or products in his presentation.
EXPERT ANALYSIS FROM A MEETING SPONSORED BY NEW YORK UNIVERSITY
Refining the Treatment of Juvenile Dermatomyositis
NEW YORK – Because juvenile dermatomyositis is a rare disease and its symptoms often differ from those seen in adult dermatomyositis, pediatricians and family physicians may not recognize it and diagnosis can be delayed, Dr. Brian Feldman said.
He described a recent practice survey that found variability in juvenile dermatomyositis (JDM) treatment and, in the absence of randomized controlled trials, urged clinicians to consult newly created consensus protocols developed by rheumatologists and to gather data that may help to optimize treatment and minimize side effects in the future for those with JDM.
Dr. Feldman described a 15-year-old patient who had progressively deteriorated over a 3-year period despite being seen by pediatricians and dermatologists. His initial symptoms included fatigue and elevated muscle enzymes, including aspartate transaminase (AST), alanine transaminase (ALT), lactate dehydrogenase (LDH), and creatine phosphokinase (CPK).
Within a year, the patient developed a purple skin rash on his hands, elbows, and knees. He later developed Raynaud’s syndrome and muscle weakness with decreased range of motion that interfered with participation in sports and other activities of daily living. Upon examination, he was quite thin with a scaly rash over his knuckles (Gottron’s papules), healing skin ulcerations on his hands (from severe Raynaud’s), periungual erythema, and lipodystrophy and erythema of his forearms. Magnetic resonance imaging showed acute inflammation of his shoulder girdle, said Dr. Feldman, chief of pediatric rheumatology at the Hospital for Sick Children in Toronto.
Dr. Feldman said that one of the best diagnostic clues for JDM comes from microscopic examination of nail folds. In this case, the patient had tortuous, bushy nail folds, dilated or missing capillaries (capillary density of 3 per mm of nailfold length while normal ranges from 7-11 per mm), and cuticular overgrowth indicative of JDM.
For rheumatologists, the different presentation seen with children compared with adults may hinder a correct diagnosis. For instance, calcinosis is seen much more frequently in children than adults. Children are less likely to have some of the systemic symptoms, such as fever, poor weight gain, and pulmonary effects, and they almost never have cardiac problems. Unlike adults with dermatomyositis or polymyositis who appear to have a fourfold increased risk of malignancy, very few cases of malignancy have been associated with JDM. Children are more likely to experience dysphonia/dysphagia but are as likely to have arthritic symptoms (around 58% of each group). Myositis specific antibodies do not seem to play as important a diagnostic role for children as adults.
Children who develop JDM appear to have a better prognosis than adults. Although adults with myositis have appreciable mortality (about 10%) and progressive disability, which may be related to treatment of a long-term condition such as avascular necrosis or osteoporosis with compression fracture, findings from Dr. Feldman’s group show that outcomes were often excellent in children. "We have not had a single death from JDM in 30 years," he said.
About two-thirds of children with JDM have a polycyclic, chronic, unremitting disease course, but about one-third follow a monocyclic course and have disease symptoms that last for 2-3 years and then go away, with, or potentially even without, treatment. In a very small proportion of these patients, symptoms may return many years later. "The message here is that when the disease goes away, it almost certainly is gone forever," Dr. Feldman noted.
It is possible to predict which patients will follow a chronic course and which patients will have early remission. In his experience, the risk of persistent disease falls by half if Gottron’s rash disappears within 3 months of diagnosis (odds ratio, 0.5; P = .0013) (Arthritis Rheum. 2008;58:3585-92).
The results of a recent practice survey by the Childhood Arthritis and Rheumatology Research Alliance (CARRA) of almost 200 pediatric rheumatologists in North America found both similarities and differences in treatment approaches to JDM (J. Rheumatol. 2010;37:1953-61).
Corticosteroids and methotrexate (MTX) are mainstays of care, but the route and pattern of corticosteroid administration varied: 82% of respondents initially pulsed with high-dose intravenous methylprednisolone (IVMP) while 18% prescribed oral prednisone. MTX was used concurrently with steroids 84% of the time. Sixteen percent reported also using IV immunoglobulin, with or without MTX, for more severe disease, refractory disease, and prominent cutaneous disease. Hydroxychloroquine was given for milder cases, especially for rash, while cyclophosphamide was prescribed for ulcerative disease or for patients with pulmonary symptoms.
While these findings reflect the current prescribing practices of rheumatologists, there are almost no randomized controlled trials of medications for JDM, Dr. Feldman pointed out.
This reflects in part the rare nature of the disease, its complexity, the difficulty of conducting studies in children, and the high cost of randomized controlled trials. There have been studies using advanced analytic techniques to provide strong comparative data in lieu of an randomized controlled trial. One such study by Dr. Feldman and associates (Arthritis Rheum. 2008;59:989-95) showed that more aggressive corticosteroid therapy does not give better 3-year outcomes.
A recent randomized controlled trial presented at the annual meeting of the American College of Rheumatology looked at dermatomyositis in 76 adults and 48 children and found that 80% had a response to rituximab within a year of treatment. Interpretation of the study may have been limited by the design, which allowed patients to receive add-on medications during the trial.
In light of the CARRA study’s findings of heterogeneity in the treatment of JDM and the absence of randomized controlled trials, a group of 12 pediatric rheumatologists met to study treatments in JDM using a new approach: by developing consensus treatment protocols (Arthritis Care Res. 2010;62:219-25).
"This is similar to what has been done in pediatric oncology," said Dr. Feldman, who was one of the participants. "We are hoping physicians throughout the world will take these protocols off the shelf, and by using standardized doses, follow-up, and measurements, we will be able to accumulate enough evidence over time to know which is the best therapy."
In brief, the group recommended three protocols for the treatment of patients with moderately severe JDM: pulse IVMP plus MTX; IVMP, MTX, plus IVIG; or oral prednisone plus MTX. The third protocol is the one followed most often at the Hospital for Sick Children. The treatment protocols are not intended as treatment recommendations, although it is hoped that a physician will choose to follow the standardized protocol that most closely reflects his or her preferred practice. It is presented as a "first step to allow comparison of different approaches to the treatment of JDM," he said.
Dr. Feldman has done contracted research with Bayer Healthcare Pharmaceuticals. He referenced unlabeled/unapproved uses of drugs or products in his presentation.
NEW YORK – Because juvenile dermatomyositis is a rare disease and its symptoms often differ from those seen in adult dermatomyositis, pediatricians and family physicians may not recognize it and diagnosis can be delayed, Dr. Brian Feldman said.
He described a recent practice survey that found variability in juvenile dermatomyositis (JDM) treatment and, in the absence of randomized controlled trials, urged clinicians to consult newly created consensus protocols developed by rheumatologists and to gather data that may help to optimize treatment and minimize side effects in the future for those with JDM.
Dr. Feldman described a 15-year-old patient who had progressively deteriorated over a 3-year period despite being seen by pediatricians and dermatologists. His initial symptoms included fatigue and elevated muscle enzymes, including aspartate transaminase (AST), alanine transaminase (ALT), lactate dehydrogenase (LDH), and creatine phosphokinase (CPK).
Within a year, the patient developed a purple skin rash on his hands, elbows, and knees. He later developed Raynaud’s syndrome and muscle weakness with decreased range of motion that interfered with participation in sports and other activities of daily living. Upon examination, he was quite thin with a scaly rash over his knuckles (Gottron’s papules), healing skin ulcerations on his hands (from severe Raynaud’s), periungual erythema, and lipodystrophy and erythema of his forearms. Magnetic resonance imaging showed acute inflammation of his shoulder girdle, said Dr. Feldman, chief of pediatric rheumatology at the Hospital for Sick Children in Toronto.
Dr. Feldman said that one of the best diagnostic clues for JDM comes from microscopic examination of nail folds. In this case, the patient had tortuous, bushy nail folds, dilated or missing capillaries (capillary density of 3 per mm of nailfold length while normal ranges from 7-11 per mm), and cuticular overgrowth indicative of JDM.
For rheumatologists, the different presentation seen with children compared with adults may hinder a correct diagnosis. For instance, calcinosis is seen much more frequently in children than adults. Children are less likely to have some of the systemic symptoms, such as fever, poor weight gain, and pulmonary effects, and they almost never have cardiac problems. Unlike adults with dermatomyositis or polymyositis who appear to have a fourfold increased risk of malignancy, very few cases of malignancy have been associated with JDM. Children are more likely to experience dysphonia/dysphagia but are as likely to have arthritic symptoms (around 58% of each group). Myositis specific antibodies do not seem to play as important a diagnostic role for children as adults.
Children who develop JDM appear to have a better prognosis than adults. Although adults with myositis have appreciable mortality (about 10%) and progressive disability, which may be related to treatment of a long-term condition such as avascular necrosis or osteoporosis with compression fracture, findings from Dr. Feldman’s group show that outcomes were often excellent in children. "We have not had a single death from JDM in 30 years," he said.
About two-thirds of children with JDM have a polycyclic, chronic, unremitting disease course, but about one-third follow a monocyclic course and have disease symptoms that last for 2-3 years and then go away, with, or potentially even without, treatment. In a very small proportion of these patients, symptoms may return many years later. "The message here is that when the disease goes away, it almost certainly is gone forever," Dr. Feldman noted.
It is possible to predict which patients will follow a chronic course and which patients will have early remission. In his experience, the risk of persistent disease falls by half if Gottron’s rash disappears within 3 months of diagnosis (odds ratio, 0.5; P = .0013) (Arthritis Rheum. 2008;58:3585-92).
The results of a recent practice survey by the Childhood Arthritis and Rheumatology Research Alliance (CARRA) of almost 200 pediatric rheumatologists in North America found both similarities and differences in treatment approaches to JDM (J. Rheumatol. 2010;37:1953-61).
Corticosteroids and methotrexate (MTX) are mainstays of care, but the route and pattern of corticosteroid administration varied: 82% of respondents initially pulsed with high-dose intravenous methylprednisolone (IVMP) while 18% prescribed oral prednisone. MTX was used concurrently with steroids 84% of the time. Sixteen percent reported also using IV immunoglobulin, with or without MTX, for more severe disease, refractory disease, and prominent cutaneous disease. Hydroxychloroquine was given for milder cases, especially for rash, while cyclophosphamide was prescribed for ulcerative disease or for patients with pulmonary symptoms.
While these findings reflect the current prescribing practices of rheumatologists, there are almost no randomized controlled trials of medications for JDM, Dr. Feldman pointed out.
This reflects in part the rare nature of the disease, its complexity, the difficulty of conducting studies in children, and the high cost of randomized controlled trials. There have been studies using advanced analytic techniques to provide strong comparative data in lieu of an randomized controlled trial. One such study by Dr. Feldman and associates (Arthritis Rheum. 2008;59:989-95) showed that more aggressive corticosteroid therapy does not give better 3-year outcomes.
A recent randomized controlled trial presented at the annual meeting of the American College of Rheumatology looked at dermatomyositis in 76 adults and 48 children and found that 80% had a response to rituximab within a year of treatment. Interpretation of the study may have been limited by the design, which allowed patients to receive add-on medications during the trial.
In light of the CARRA study’s findings of heterogeneity in the treatment of JDM and the absence of randomized controlled trials, a group of 12 pediatric rheumatologists met to study treatments in JDM using a new approach: by developing consensus treatment protocols (Arthritis Care Res. 2010;62:219-25).
"This is similar to what has been done in pediatric oncology," said Dr. Feldman, who was one of the participants. "We are hoping physicians throughout the world will take these protocols off the shelf, and by using standardized doses, follow-up, and measurements, we will be able to accumulate enough evidence over time to know which is the best therapy."
In brief, the group recommended three protocols for the treatment of patients with moderately severe JDM: pulse IVMP plus MTX; IVMP, MTX, plus IVIG; or oral prednisone plus MTX. The third protocol is the one followed most often at the Hospital for Sick Children. The treatment protocols are not intended as treatment recommendations, although it is hoped that a physician will choose to follow the standardized protocol that most closely reflects his or her preferred practice. It is presented as a "first step to allow comparison of different approaches to the treatment of JDM," he said.
Dr. Feldman has done contracted research with Bayer Healthcare Pharmaceuticals. He referenced unlabeled/unapproved uses of drugs or products in his presentation.
NEW YORK – Because juvenile dermatomyositis is a rare disease and its symptoms often differ from those seen in adult dermatomyositis, pediatricians and family physicians may not recognize it and diagnosis can be delayed, Dr. Brian Feldman said.
He described a recent practice survey that found variability in juvenile dermatomyositis (JDM) treatment and, in the absence of randomized controlled trials, urged clinicians to consult newly created consensus protocols developed by rheumatologists and to gather data that may help to optimize treatment and minimize side effects in the future for those with JDM.
Dr. Feldman described a 15-year-old patient who had progressively deteriorated over a 3-year period despite being seen by pediatricians and dermatologists. His initial symptoms included fatigue and elevated muscle enzymes, including aspartate transaminase (AST), alanine transaminase (ALT), lactate dehydrogenase (LDH), and creatine phosphokinase (CPK).
Within a year, the patient developed a purple skin rash on his hands, elbows, and knees. He later developed Raynaud’s syndrome and muscle weakness with decreased range of motion that interfered with participation in sports and other activities of daily living. Upon examination, he was quite thin with a scaly rash over his knuckles (Gottron’s papules), healing skin ulcerations on his hands (from severe Raynaud’s), periungual erythema, and lipodystrophy and erythema of his forearms. Magnetic resonance imaging showed acute inflammation of his shoulder girdle, said Dr. Feldman, chief of pediatric rheumatology at the Hospital for Sick Children in Toronto.
Dr. Feldman said that one of the best diagnostic clues for JDM comes from microscopic examination of nail folds. In this case, the patient had tortuous, bushy nail folds, dilated or missing capillaries (capillary density of 3 per mm of nailfold length while normal ranges from 7-11 per mm), and cuticular overgrowth indicative of JDM.
For rheumatologists, the different presentation seen with children compared with adults may hinder a correct diagnosis. For instance, calcinosis is seen much more frequently in children than adults. Children are less likely to have some of the systemic symptoms, such as fever, poor weight gain, and pulmonary effects, and they almost never have cardiac problems. Unlike adults with dermatomyositis or polymyositis who appear to have a fourfold increased risk of malignancy, very few cases of malignancy have been associated with JDM. Children are more likely to experience dysphonia/dysphagia but are as likely to have arthritic symptoms (around 58% of each group). Myositis specific antibodies do not seem to play as important a diagnostic role for children as adults.
Children who develop JDM appear to have a better prognosis than adults. Although adults with myositis have appreciable mortality (about 10%) and progressive disability, which may be related to treatment of a long-term condition such as avascular necrosis or osteoporosis with compression fracture, findings from Dr. Feldman’s group show that outcomes were often excellent in children. "We have not had a single death from JDM in 30 years," he said.
About two-thirds of children with JDM have a polycyclic, chronic, unremitting disease course, but about one-third follow a monocyclic course and have disease symptoms that last for 2-3 years and then go away, with, or potentially even without, treatment. In a very small proportion of these patients, symptoms may return many years later. "The message here is that when the disease goes away, it almost certainly is gone forever," Dr. Feldman noted.
It is possible to predict which patients will follow a chronic course and which patients will have early remission. In his experience, the risk of persistent disease falls by half if Gottron’s rash disappears within 3 months of diagnosis (odds ratio, 0.5; P = .0013) (Arthritis Rheum. 2008;58:3585-92).
The results of a recent practice survey by the Childhood Arthritis and Rheumatology Research Alliance (CARRA) of almost 200 pediatric rheumatologists in North America found both similarities and differences in treatment approaches to JDM (J. Rheumatol. 2010;37:1953-61).
Corticosteroids and methotrexate (MTX) are mainstays of care, but the route and pattern of corticosteroid administration varied: 82% of respondents initially pulsed with high-dose intravenous methylprednisolone (IVMP) while 18% prescribed oral prednisone. MTX was used concurrently with steroids 84% of the time. Sixteen percent reported also using IV immunoglobulin, with or without MTX, for more severe disease, refractory disease, and prominent cutaneous disease. Hydroxychloroquine was given for milder cases, especially for rash, while cyclophosphamide was prescribed for ulcerative disease or for patients with pulmonary symptoms.
While these findings reflect the current prescribing practices of rheumatologists, there are almost no randomized controlled trials of medications for JDM, Dr. Feldman pointed out.
This reflects in part the rare nature of the disease, its complexity, the difficulty of conducting studies in children, and the high cost of randomized controlled trials. There have been studies using advanced analytic techniques to provide strong comparative data in lieu of an randomized controlled trial. One such study by Dr. Feldman and associates (Arthritis Rheum. 2008;59:989-95) showed that more aggressive corticosteroid therapy does not give better 3-year outcomes.
A recent randomized controlled trial presented at the annual meeting of the American College of Rheumatology looked at dermatomyositis in 76 adults and 48 children and found that 80% had a response to rituximab within a year of treatment. Interpretation of the study may have been limited by the design, which allowed patients to receive add-on medications during the trial.
In light of the CARRA study’s findings of heterogeneity in the treatment of JDM and the absence of randomized controlled trials, a group of 12 pediatric rheumatologists met to study treatments in JDM using a new approach: by developing consensus treatment protocols (Arthritis Care Res. 2010;62:219-25).
"This is similar to what has been done in pediatric oncology," said Dr. Feldman, who was one of the participants. "We are hoping physicians throughout the world will take these protocols off the shelf, and by using standardized doses, follow-up, and measurements, we will be able to accumulate enough evidence over time to know which is the best therapy."
In brief, the group recommended three protocols for the treatment of patients with moderately severe JDM: pulse IVMP plus MTX; IVMP, MTX, plus IVIG; or oral prednisone plus MTX. The third protocol is the one followed most often at the Hospital for Sick Children. The treatment protocols are not intended as treatment recommendations, although it is hoped that a physician will choose to follow the standardized protocol that most closely reflects his or her preferred practice. It is presented as a "first step to allow comparison of different approaches to the treatment of JDM," he said.
Dr. Feldman has done contracted research with Bayer Healthcare Pharmaceuticals. He referenced unlabeled/unapproved uses of drugs or products in his presentation.
EXPERT ANALYSIS FROM A MEETING SPONSORED BY NEW YORK UNIVERSITY
Refining the Treatment of Juvenile Dermatomyositis
NEW YORK – Because juvenile dermatomyositis is a rare disease and its symptoms often differ from those seen in adult dermatomyositis, pediatricians and family physicians may not recognize it and diagnosis can be delayed, Dr. Brian Feldman said.
He described a recent practice survey that found variability in juvenile dermatomyositis (JDM) treatment and, in the absence of randomized controlled trials, urged clinicians to consult newly created consensus protocols developed by rheumatologists and to gather data that may help to optimize treatment and minimize side effects in the future for those with JDM.
Dr. Feldman described a 15-year-old patient who had progressively deteriorated over a 3-year period despite being seen by pediatricians and dermatologists. His initial symptoms included fatigue and elevated muscle enzymes, including aspartate transaminase (AST), alanine transaminase (ALT), lactate dehydrogenase (LDH), and creatine phosphokinase (CPK).
Within a year, the patient developed a purple skin rash on his hands, elbows, and knees. He later developed Raynaud’s syndrome and muscle weakness with decreased range of motion that interfered with participation in sports and other activities of daily living. Upon examination, he was quite thin with a scaly rash over his knuckles (Gottron’s papules), healing skin ulcerations on his hands (from severe Raynaud’s), periungual erythema, and lipodystrophy and erythema of his forearms. Magnetic resonance imaging showed acute inflammation of his shoulder girdle, said Dr. Feldman, chief of pediatric rheumatology at the Hospital for Sick Children in Toronto.
Dr. Feldman said that one of the best diagnostic clues for JDM comes from microscopic examination of nail folds. In this case, the patient had tortuous, bushy nail folds, dilated or missing capillaries (capillary density of 3 per mm of nailfold length while normal ranges from 7-11 per mm), and cuticular overgrowth indicative of JDM.
For rheumatologists, the different presentation seen with children compared with adults may hinder a correct diagnosis. For instance, calcinosis is seen much more frequently in children than adults. Children are less likely to have some of the systemic symptoms, such as fever, poor weight gain, and pulmonary effects, and they almost never have cardiac problems. Unlike adults with dermatomyositis or polymyositis who appear to have a fourfold increased risk of malignancy, very few cases of malignancy have been associated with JDM. Children are more likely to experience dysphonia/dysphagia but are as likely to have arthritic symptoms (around 58% of each group). Myositis specific antibodies do not seem to play as important a diagnostic role for children as adults.
Children who develop JDM appear to have a better prognosis than adults. Although adults with myositis have appreciable mortality (about 10%) and progressive disability, which may be related to treatment of a long-term condition such as avascular necrosis or osteoporosis with compression fracture, findings from Dr. Feldman’s group show that outcomes were often excellent in children. "We have not had a single death from JDM in 30 years," he said.
About two-thirds of children with JDM have a polycyclic, chronic, unremitting disease course, but about one-third follow a monocyclic course and have disease symptoms that last for 2-3 years and then go away, with, or potentially even without, treatment. In a very small proportion of these patients, symptoms may return many years later. "The message here is that when the disease goes away, it almost certainly is gone forever," Dr. Feldman noted.
It is possible to predict which patients will follow a chronic course and which patients will have early remission. In his experience, the risk of persistent disease falls by half if Gottron’s rash disappears within 3 months of diagnosis (odds ratio, 0.5; P = .0013) (Arthritis Rheum. 2008;58:3585-92).
The results of a recent practice survey by the Childhood Arthritis and Rheumatology Research Alliance (CARRA) of almost 200 pediatric rheumatologists in North America found both similarities and differences in treatment approaches to JDM (J. Rheumatol. 2010;37:1953-61).
Corticosteroids and methotrexate (MTX) are mainstays of care, but the route and pattern of corticosteroid administration varied: 82% of respondents initially pulsed with high-dose intravenous methylprednisolone (IVMP) while 18% prescribed oral prednisone. MTX was used concurrently with steroids 84% of the time. Sixteen percent reported also using IV immunoglobulin, with or without MTX, for more severe disease, refractory disease, and prominent cutaneous disease. Hydroxychloroquine was given for milder cases, especially for rash, while cyclophosphamide was prescribed for ulcerative disease or for patients with pulmonary symptoms.
While these findings reflect the current prescribing practices of rheumatologists, there are almost no randomized controlled trials of medications for JDM, Dr. Feldman pointed out.
This reflects in part the rare nature of the disease, its complexity, the difficulty of conducting studies in children, and the high cost of randomized controlled trials. There have been studies using advanced analytic techniques to provide strong comparative data in lieu of an randomized controlled trial. One such study by Dr. Feldman and associates (Arthritis Rheum. 2008;59:989-95) showed that more aggressive corticosteroid therapy does not give better 3-year outcomes.
A recent randomized controlled trial presented at the annual meeting of the American College of Rheumatology looked at dermatomyositis in 76 adults and 48 children and found that 80% had a response to rituximab within a year of treatment. Interpretation of the study may have been limited by the design, which allowed patients to receive add-on medications during the trial.
In light of the CARRA study’s findings of heterogeneity in the treatment of JDM and the absence of randomized controlled trials, a group of 12 pediatric rheumatologists met to study treatments in JDM using a new approach: by developing consensus treatment protocols (Arthritis Care Res. 2010;62:219-25).
"This is similar to what has been done in pediatric oncology," said Dr. Feldman, who was one of the participants. "We are hoping physicians throughout the world will take these protocols off the shelf, and by using standardized doses, follow-up, and measurements, we will be able to accumulate enough evidence over time to know which is the best therapy."
In brief, the group recommended three protocols for the treatment of patients with moderately severe JDM: pulse IVMP plus MTX; IVMP, MTX, plus IVIG; or oral prednisone plus MTX. The third protocol is the one followed most often at the Hospital for Sick Children. The treatment protocols are not intended as treatment recommendations, although it is hoped that a physician will choose to follow the standardized protocol that most closely reflects his or her preferred practice. It is presented as a "first step to allow comparison of different approaches to the treatment of JDM," he said.
Dr. Feldman has done contracted research with Bayer Healthcare Pharmaceuticals. He referenced unlabeled/unapproved uses of drugs or products in his presentation.
NEW YORK – Because juvenile dermatomyositis is a rare disease and its symptoms often differ from those seen in adult dermatomyositis, pediatricians and family physicians may not recognize it and diagnosis can be delayed, Dr. Brian Feldman said.
He described a recent practice survey that found variability in juvenile dermatomyositis (JDM) treatment and, in the absence of randomized controlled trials, urged clinicians to consult newly created consensus protocols developed by rheumatologists and to gather data that may help to optimize treatment and minimize side effects in the future for those with JDM.
Dr. Feldman described a 15-year-old patient who had progressively deteriorated over a 3-year period despite being seen by pediatricians and dermatologists. His initial symptoms included fatigue and elevated muscle enzymes, including aspartate transaminase (AST), alanine transaminase (ALT), lactate dehydrogenase (LDH), and creatine phosphokinase (CPK).
Within a year, the patient developed a purple skin rash on his hands, elbows, and knees. He later developed Raynaud’s syndrome and muscle weakness with decreased range of motion that interfered with participation in sports and other activities of daily living. Upon examination, he was quite thin with a scaly rash over his knuckles (Gottron’s papules), healing skin ulcerations on his hands (from severe Raynaud’s), periungual erythema, and lipodystrophy and erythema of his forearms. Magnetic resonance imaging showed acute inflammation of his shoulder girdle, said Dr. Feldman, chief of pediatric rheumatology at the Hospital for Sick Children in Toronto.
Dr. Feldman said that one of the best diagnostic clues for JDM comes from microscopic examination of nail folds. In this case, the patient had tortuous, bushy nail folds, dilated or missing capillaries (capillary density of 3 per mm of nailfold length while normal ranges from 7-11 per mm), and cuticular overgrowth indicative of JDM.
For rheumatologists, the different presentation seen with children compared with adults may hinder a correct diagnosis. For instance, calcinosis is seen much more frequently in children than adults. Children are less likely to have some of the systemic symptoms, such as fever, poor weight gain, and pulmonary effects, and they almost never have cardiac problems. Unlike adults with dermatomyositis or polymyositis who appear to have a fourfold increased risk of malignancy, very few cases of malignancy have been associated with JDM. Children are more likely to experience dysphonia/dysphagia but are as likely to have arthritic symptoms (around 58% of each group). Myositis specific antibodies do not seem to play as important a diagnostic role for children as adults.
Children who develop JDM appear to have a better prognosis than adults. Although adults with myositis have appreciable mortality (about 10%) and progressive disability, which may be related to treatment of a long-term condition such as avascular necrosis or osteoporosis with compression fracture, findings from Dr. Feldman’s group show that outcomes were often excellent in children. "We have not had a single death from JDM in 30 years," he said.
About two-thirds of children with JDM have a polycyclic, chronic, unremitting disease course, but about one-third follow a monocyclic course and have disease symptoms that last for 2-3 years and then go away, with, or potentially even without, treatment. In a very small proportion of these patients, symptoms may return many years later. "The message here is that when the disease goes away, it almost certainly is gone forever," Dr. Feldman noted.
It is possible to predict which patients will follow a chronic course and which patients will have early remission. In his experience, the risk of persistent disease falls by half if Gottron’s rash disappears within 3 months of diagnosis (odds ratio, 0.5; P = .0013) (Arthritis Rheum. 2008;58:3585-92).
The results of a recent practice survey by the Childhood Arthritis and Rheumatology Research Alliance (CARRA) of almost 200 pediatric rheumatologists in North America found both similarities and differences in treatment approaches to JDM (J. Rheumatol. 2010;37:1953-61).
Corticosteroids and methotrexate (MTX) are mainstays of care, but the route and pattern of corticosteroid administration varied: 82% of respondents initially pulsed with high-dose intravenous methylprednisolone (IVMP) while 18% prescribed oral prednisone. MTX was used concurrently with steroids 84% of the time. Sixteen percent reported also using IV immunoglobulin, with or without MTX, for more severe disease, refractory disease, and prominent cutaneous disease. Hydroxychloroquine was given for milder cases, especially for rash, while cyclophosphamide was prescribed for ulcerative disease or for patients with pulmonary symptoms.
While these findings reflect the current prescribing practices of rheumatologists, there are almost no randomized controlled trials of medications for JDM, Dr. Feldman pointed out.
This reflects in part the rare nature of the disease, its complexity, the difficulty of conducting studies in children, and the high cost of randomized controlled trials. There have been studies using advanced analytic techniques to provide strong comparative data in lieu of an randomized controlled trial. One such study by Dr. Feldman and associates (Arthritis Rheum. 2008;59:989-95) showed that more aggressive corticosteroid therapy does not give better 3-year outcomes.
A recent randomized controlled trial presented at the annual meeting of the American College of Rheumatology looked at dermatomyositis in 76 adults and 48 children and found that 80% had a response to rituximab within a year of treatment. Interpretation of the study may have been limited by the design, which allowed patients to receive add-on medications during the trial.
In light of the CARRA study’s findings of heterogeneity in the treatment of JDM and the absence of randomized controlled trials, a group of 12 pediatric rheumatologists met to study treatments in JDM using a new approach: by developing consensus treatment protocols (Arthritis Care Res. 2010;62:219-25).
"This is similar to what has been done in pediatric oncology," said Dr. Feldman, who was one of the participants. "We are hoping physicians throughout the world will take these protocols off the shelf, and by using standardized doses, follow-up, and measurements, we will be able to accumulate enough evidence over time to know which is the best therapy."
In brief, the group recommended three protocols for the treatment of patients with moderately severe JDM: pulse IVMP plus MTX; IVMP, MTX, plus IVIG; or oral prednisone plus MTX. The third protocol is the one followed most often at the Hospital for Sick Children. The treatment protocols are not intended as treatment recommendations, although it is hoped that a physician will choose to follow the standardized protocol that most closely reflects his or her preferred practice. It is presented as a "first step to allow comparison of different approaches to the treatment of JDM," he said.
Dr. Feldman has done contracted research with Bayer Healthcare Pharmaceuticals. He referenced unlabeled/unapproved uses of drugs or products in his presentation.
NEW YORK – Because juvenile dermatomyositis is a rare disease and its symptoms often differ from those seen in adult dermatomyositis, pediatricians and family physicians may not recognize it and diagnosis can be delayed, Dr. Brian Feldman said.
He described a recent practice survey that found variability in juvenile dermatomyositis (JDM) treatment and, in the absence of randomized controlled trials, urged clinicians to consult newly created consensus protocols developed by rheumatologists and to gather data that may help to optimize treatment and minimize side effects in the future for those with JDM.
Dr. Feldman described a 15-year-old patient who had progressively deteriorated over a 3-year period despite being seen by pediatricians and dermatologists. His initial symptoms included fatigue and elevated muscle enzymes, including aspartate transaminase (AST), alanine transaminase (ALT), lactate dehydrogenase (LDH), and creatine phosphokinase (CPK).
Within a year, the patient developed a purple skin rash on his hands, elbows, and knees. He later developed Raynaud’s syndrome and muscle weakness with decreased range of motion that interfered with participation in sports and other activities of daily living. Upon examination, he was quite thin with a scaly rash over his knuckles (Gottron’s papules), healing skin ulcerations on his hands (from severe Raynaud’s), periungual erythema, and lipodystrophy and erythema of his forearms. Magnetic resonance imaging showed acute inflammation of his shoulder girdle, said Dr. Feldman, chief of pediatric rheumatology at the Hospital for Sick Children in Toronto.
Dr. Feldman said that one of the best diagnostic clues for JDM comes from microscopic examination of nail folds. In this case, the patient had tortuous, bushy nail folds, dilated or missing capillaries (capillary density of 3 per mm of nailfold length while normal ranges from 7-11 per mm), and cuticular overgrowth indicative of JDM.
For rheumatologists, the different presentation seen with children compared with adults may hinder a correct diagnosis. For instance, calcinosis is seen much more frequently in children than adults. Children are less likely to have some of the systemic symptoms, such as fever, poor weight gain, and pulmonary effects, and they almost never have cardiac problems. Unlike adults with dermatomyositis or polymyositis who appear to have a fourfold increased risk of malignancy, very few cases of malignancy have been associated with JDM. Children are more likely to experience dysphonia/dysphagia but are as likely to have arthritic symptoms (around 58% of each group). Myositis specific antibodies do not seem to play as important a diagnostic role for children as adults.
Children who develop JDM appear to have a better prognosis than adults. Although adults with myositis have appreciable mortality (about 10%) and progressive disability, which may be related to treatment of a long-term condition such as avascular necrosis or osteoporosis with compression fracture, findings from Dr. Feldman’s group show that outcomes were often excellent in children. "We have not had a single death from JDM in 30 years," he said.
About two-thirds of children with JDM have a polycyclic, chronic, unremitting disease course, but about one-third follow a monocyclic course and have disease symptoms that last for 2-3 years and then go away, with, or potentially even without, treatment. In a very small proportion of these patients, symptoms may return many years later. "The message here is that when the disease goes away, it almost certainly is gone forever," Dr. Feldman noted.
It is possible to predict which patients will follow a chronic course and which patients will have early remission. In his experience, the risk of persistent disease falls by half if Gottron’s rash disappears within 3 months of diagnosis (odds ratio, 0.5; P = .0013) (Arthritis Rheum. 2008;58:3585-92).
The results of a recent practice survey by the Childhood Arthritis and Rheumatology Research Alliance (CARRA) of almost 200 pediatric rheumatologists in North America found both similarities and differences in treatment approaches to JDM (J. Rheumatol. 2010;37:1953-61).
Corticosteroids and methotrexate (MTX) are mainstays of care, but the route and pattern of corticosteroid administration varied: 82% of respondents initially pulsed with high-dose intravenous methylprednisolone (IVMP) while 18% prescribed oral prednisone. MTX was used concurrently with steroids 84% of the time. Sixteen percent reported also using IV immunoglobulin, with or without MTX, for more severe disease, refractory disease, and prominent cutaneous disease. Hydroxychloroquine was given for milder cases, especially for rash, while cyclophosphamide was prescribed for ulcerative disease or for patients with pulmonary symptoms.
While these findings reflect the current prescribing practices of rheumatologists, there are almost no randomized controlled trials of medications for JDM, Dr. Feldman pointed out.
This reflects in part the rare nature of the disease, its complexity, the difficulty of conducting studies in children, and the high cost of randomized controlled trials. There have been studies using advanced analytic techniques to provide strong comparative data in lieu of an randomized controlled trial. One such study by Dr. Feldman and associates (Arthritis Rheum. 2008;59:989-95) showed that more aggressive corticosteroid therapy does not give better 3-year outcomes.
A recent randomized controlled trial presented at the annual meeting of the American College of Rheumatology looked at dermatomyositis in 76 adults and 48 children and found that 80% had a response to rituximab within a year of treatment. Interpretation of the study may have been limited by the design, which allowed patients to receive add-on medications during the trial.
In light of the CARRA study’s findings of heterogeneity in the treatment of JDM and the absence of randomized controlled trials, a group of 12 pediatric rheumatologists met to study treatments in JDM using a new approach: by developing consensus treatment protocols (Arthritis Care Res. 2010;62:219-25).
"This is similar to what has been done in pediatric oncology," said Dr. Feldman, who was one of the participants. "We are hoping physicians throughout the world will take these protocols off the shelf, and by using standardized doses, follow-up, and measurements, we will be able to accumulate enough evidence over time to know which is the best therapy."
In brief, the group recommended three protocols for the treatment of patients with moderately severe JDM: pulse IVMP plus MTX; IVMP, MTX, plus IVIG; or oral prednisone plus MTX. The third protocol is the one followed most often at the Hospital for Sick Children. The treatment protocols are not intended as treatment recommendations, although it is hoped that a physician will choose to follow the standardized protocol that most closely reflects his or her preferred practice. It is presented as a "first step to allow comparison of different approaches to the treatment of JDM," he said.
Dr. Feldman has done contracted research with Bayer Healthcare Pharmaceuticals. He referenced unlabeled/unapproved uses of drugs or products in his presentation.
EXPERT ANALYSIS FROM A MEETING SPONSORED BY NEW YORK UNIVERSITY
Childhood Onset Lupus: Advances, Setbacks Change the Playing Field
NEW YORK – The good news in the management of childhood-onset systemic lupus erythematosus is that children who are diagnosed with the disease have a better prognosis and are living longer. The bad news is that over the course of a lifetime, affected children develop more morbidities and experience more medication-induced toxicities, which create additional management challenges, according to Dr. Phillip Kahn, who spoke at a meeting sponsored by New York University.
About 15%-20% of lupus cases present in childhood. Children younger than age 5 years rarely have the disease, but up to 40% of patients with childhood-onset SLE (cSLE) present prior to puberty, said Dr. Kahn of New York University. The incidence of cSLE is estimated to be 10-20 cases per 100,000 individuals, but the incidence is higher in blacks, at 200 cases per 100,000 individuals (Lupus 2007;16:546-9; 2005;14:83-8).
Often the diagnosis is delayed in children, as their clinical features do not fall within those defined by the 1997 American College of Rheumatology’s SLE criteria, which have not been validated in children. For example, Dr. Kahn described the case of a 13-year-old girl who presented with "constitutional symptoms" such as diffuse lymphadenopathy, malaise, weight loss, and fever – none of which are included in the 1997 criteria.
Mucocutaneous lesions are common in children, with up to 70% presenting with malar rash, 30% with alopecia, 66% with oral ulcers, and 10% with nasal ulcers. "Although nasal ulcers are less common in children than adults, they still may result in perforation of the septum," he said. Other common manifestations include painful polyarthritis, hepatosplenomegaly, headache, photosensitivity, and Raynaud’s phenomenon. Up to 40% of cSLE patients have neuropsychiatric symptoms, significantly more than do adults (Lupus 2008;17:314-22).
An important diagnostic tool for cSLE is a positive ANA (antinuclear antibody) finding, which is present in essentially all children with cSLE. In contrast, up to 10% of adults with SLE do not have a positive ANA, said Dr. Kahn.
Childhood-onset lupus may predict mortality in adults (Arthritis Care Res. [Hoboken] 2010;62:1152-9) and thus physicians must be vigilant about detecting, preventing, and aggressively treating some of its more dangerous manifestations. To prevent recurrent infection, Dr. Kahn recommends keeping a child up to date with immunizations, treating infections promptly when they occur, and considering PCP (pneumocystis carinii pneumonia) prophylaxis in the appropriate patient. Children with cSLE are also at increased risk for premature atherosclerosis, with dyslipidemia seen in 60%-85% of affected children (Nat. Clin. Pract. Rheumatol. 2008;4:258-65). Statin therapy may be appropriate in these children, especially to prevent long-term adverse events such as early MI. For example, adults – especially women – who had c SLE have increased incidence of MI when they reach their 30s (Arthritis Rheum. 2009;61:13-20). The ongoing APPLE (Atherosclerosis Prevention in Pediatric Lupus Erythematosus) trial is investigating, in greater detail, cardiovascular risk and the use of statins in cSLE (Lupus 2010;19:1315-25).
Because nephropathy is the main cause of mortality and morbidity in cSLE, it is important to monitor urinary markers of renal function. Nephropathy may take a greater toll on a pediatric patient, compared with adults; data show that children are more likely to have renal involvement, need hemodialysis and kidney transplantation, and require hospitalization and emergency department visits (Lupus 2008;17:314-22; Arthritis Rheum. 2009;61:13-20). To avoid end-stage renal disease, Dr. Kahn urges tight blood pressure control, as well as antihypertensive medication as necessary. "We know that 90% of the patients who ultimately develop renal disease will do so within the first 2 years of disease, so if you follow a patient for [more than 2] years and they do not develop the disease, the good news is they are much less likely to develop nephropathy," said Dr. Kahn.
In the 1950s, the 5-year survival rate for SLE in adults and children ranged from 18% to 70%; 30 years later, 5-year survival rose to 60%-93%, in large part because of the introduction of prednisone. Currently, prednisone is almost universally given to those with cSLE, and the proportion of children who are treated with prednisone is significantly higher than the proportion of adults with SLE (97% vs. 70%; P less than .0001) (Arthritis Rheum. 2008;58:556-62). The result is greater risk of osteoporosis, diabetes, and growth failure. In fact, up to 40% of cSLE patients have growth failure or short stature, in part because of chronic steroids.
That is why Dr. Kahn posed the question, "Can we move beyond steroids?" Until recently, the only Food and Drug Administration–approved medications for SLE were prednisone, hydroxychloroquine, and aspirin. Steroid-sparing immunomodulatory therapy is sometimes used off label when major organ (such as brain or kidney) involvement develops, but most evidence regarding the efficacy of these medications pertains to renal proliferative disease in adults.
Cyclophosphamide is often used for induction of nephritis, but 30% of patients may still develop end-stage renal disease despite treatment, said Dr. Kahn. Mycophenolate mofetil has been shown to have equal efficacy to cyclophosphamide in induction of remission of nephritis, but sufficient pediatric data are lacking. Azathioprine is effective for maintenance therapy of nephritis after the induction phase.
Other options that are sometimes used include intravenous immunoglobulin, methotrexate, calcineurin inhibitors (such as tacrolimus), plasmapheresis, and autologous stem cell transplantation/immunoablative therapy.
More recently, the focus has been on target-specific therapy for SLE with potentially fewer side effects. Rituximab, which targets B cells that have the CD20 surface antigen, is sometimes used to treat resistant patients, but there is no strong clinical evidence to demonstrate its effectiveness in cSLE (Arthritis Rheum. 2009;61:482-7; Clin. J. Am. Soc. Nephrol. 2009;4:579-87).
Abatacept, which targets T cells by binding to B7 peripheral membrane protein, has shown some clinical benefit in a phase IIB randomized clinical trial, and is undergoing clinical testing as an add-on induction therapy with cyclophosphamide (Arthritis Rheum. 2010;62:3077-87). Belimumab, a monoclonal antibody that targets BLyS (B-lymphocyte stimulator)/BAFF (B-cell activating factor), is the first FDA-approved medication for SLE in more than 50 years, based on recent clinical trials that demonstrate increased efficacy and fewer flares of disease, compared with placebo (Arthritis Rheum 2009;61:1168-78; Lancet 2011;377:721-31).
Although recent data regarding belimumab provide some hope, it "may be naive to think that a target-specific therapy exists in this complex, polygenic, heterogeneous disease," commented Dr. Kahn.
In concluding his talk, Dr. Kahn spoke about some barriers to the effective treatment of cSLE. These include physicians’ failure to detect the subtle changes of early disease, a lack of awareness regarding the neuropsychiatric manifestations of the disease, and physicians’ fear of being overly aggressive with treatment. He also warned about overt or covert adolescent noncompliance, and the danger of adolescents’ dropping out of the health care system once they need to transition to adult care.
NEW YORK – The good news in the management of childhood-onset systemic lupus erythematosus is that children who are diagnosed with the disease have a better prognosis and are living longer. The bad news is that over the course of a lifetime, affected children develop more morbidities and experience more medication-induced toxicities, which create additional management challenges, according to Dr. Phillip Kahn, who spoke at a meeting sponsored by New York University.
About 15%-20% of lupus cases present in childhood. Children younger than age 5 years rarely have the disease, but up to 40% of patients with childhood-onset SLE (cSLE) present prior to puberty, said Dr. Kahn of New York University. The incidence of cSLE is estimated to be 10-20 cases per 100,000 individuals, but the incidence is higher in blacks, at 200 cases per 100,000 individuals (Lupus 2007;16:546-9; 2005;14:83-8).
Often the diagnosis is delayed in children, as their clinical features do not fall within those defined by the 1997 American College of Rheumatology’s SLE criteria, which have not been validated in children. For example, Dr. Kahn described the case of a 13-year-old girl who presented with "constitutional symptoms" such as diffuse lymphadenopathy, malaise, weight loss, and fever – none of which are included in the 1997 criteria.
Mucocutaneous lesions are common in children, with up to 70% presenting with malar rash, 30% with alopecia, 66% with oral ulcers, and 10% with nasal ulcers. "Although nasal ulcers are less common in children than adults, they still may result in perforation of the septum," he said. Other common manifestations include painful polyarthritis, hepatosplenomegaly, headache, photosensitivity, and Raynaud’s phenomenon. Up to 40% of cSLE patients have neuropsychiatric symptoms, significantly more than do adults (Lupus 2008;17:314-22).
An important diagnostic tool for cSLE is a positive ANA (antinuclear antibody) finding, which is present in essentially all children with cSLE. In contrast, up to 10% of adults with SLE do not have a positive ANA, said Dr. Kahn.
Childhood-onset lupus may predict mortality in adults (Arthritis Care Res. [Hoboken] 2010;62:1152-9) and thus physicians must be vigilant about detecting, preventing, and aggressively treating some of its more dangerous manifestations. To prevent recurrent infection, Dr. Kahn recommends keeping a child up to date with immunizations, treating infections promptly when they occur, and considering PCP (pneumocystis carinii pneumonia) prophylaxis in the appropriate patient. Children with cSLE are also at increased risk for premature atherosclerosis, with dyslipidemia seen in 60%-85% of affected children (Nat. Clin. Pract. Rheumatol. 2008;4:258-65). Statin therapy may be appropriate in these children, especially to prevent long-term adverse events such as early MI. For example, adults – especially women – who had c SLE have increased incidence of MI when they reach their 30s (Arthritis Rheum. 2009;61:13-20). The ongoing APPLE (Atherosclerosis Prevention in Pediatric Lupus Erythematosus) trial is investigating, in greater detail, cardiovascular risk and the use of statins in cSLE (Lupus 2010;19:1315-25).
Because nephropathy is the main cause of mortality and morbidity in cSLE, it is important to monitor urinary markers of renal function. Nephropathy may take a greater toll on a pediatric patient, compared with adults; data show that children are more likely to have renal involvement, need hemodialysis and kidney transplantation, and require hospitalization and emergency department visits (Lupus 2008;17:314-22; Arthritis Rheum. 2009;61:13-20). To avoid end-stage renal disease, Dr. Kahn urges tight blood pressure control, as well as antihypertensive medication as necessary. "We know that 90% of the patients who ultimately develop renal disease will do so within the first 2 years of disease, so if you follow a patient for [more than 2] years and they do not develop the disease, the good news is they are much less likely to develop nephropathy," said Dr. Kahn.
In the 1950s, the 5-year survival rate for SLE in adults and children ranged from 18% to 70%; 30 years later, 5-year survival rose to 60%-93%, in large part because of the introduction of prednisone. Currently, prednisone is almost universally given to those with cSLE, and the proportion of children who are treated with prednisone is significantly higher than the proportion of adults with SLE (97% vs. 70%; P less than .0001) (Arthritis Rheum. 2008;58:556-62). The result is greater risk of osteoporosis, diabetes, and growth failure. In fact, up to 40% of cSLE patients have growth failure or short stature, in part because of chronic steroids.
That is why Dr. Kahn posed the question, "Can we move beyond steroids?" Until recently, the only Food and Drug Administration–approved medications for SLE were prednisone, hydroxychloroquine, and aspirin. Steroid-sparing immunomodulatory therapy is sometimes used off label when major organ (such as brain or kidney) involvement develops, but most evidence regarding the efficacy of these medications pertains to renal proliferative disease in adults.
Cyclophosphamide is often used for induction of nephritis, but 30% of patients may still develop end-stage renal disease despite treatment, said Dr. Kahn. Mycophenolate mofetil has been shown to have equal efficacy to cyclophosphamide in induction of remission of nephritis, but sufficient pediatric data are lacking. Azathioprine is effective for maintenance therapy of nephritis after the induction phase.
Other options that are sometimes used include intravenous immunoglobulin, methotrexate, calcineurin inhibitors (such as tacrolimus), plasmapheresis, and autologous stem cell transplantation/immunoablative therapy.
More recently, the focus has been on target-specific therapy for SLE with potentially fewer side effects. Rituximab, which targets B cells that have the CD20 surface antigen, is sometimes used to treat resistant patients, but there is no strong clinical evidence to demonstrate its effectiveness in cSLE (Arthritis Rheum. 2009;61:482-7; Clin. J. Am. Soc. Nephrol. 2009;4:579-87).
Abatacept, which targets T cells by binding to B7 peripheral membrane protein, has shown some clinical benefit in a phase IIB randomized clinical trial, and is undergoing clinical testing as an add-on induction therapy with cyclophosphamide (Arthritis Rheum. 2010;62:3077-87). Belimumab, a monoclonal antibody that targets BLyS (B-lymphocyte stimulator)/BAFF (B-cell activating factor), is the first FDA-approved medication for SLE in more than 50 years, based on recent clinical trials that demonstrate increased efficacy and fewer flares of disease, compared with placebo (Arthritis Rheum 2009;61:1168-78; Lancet 2011;377:721-31).
Although recent data regarding belimumab provide some hope, it "may be naive to think that a target-specific therapy exists in this complex, polygenic, heterogeneous disease," commented Dr. Kahn.
In concluding his talk, Dr. Kahn spoke about some barriers to the effective treatment of cSLE. These include physicians’ failure to detect the subtle changes of early disease, a lack of awareness regarding the neuropsychiatric manifestations of the disease, and physicians’ fear of being overly aggressive with treatment. He also warned about overt or covert adolescent noncompliance, and the danger of adolescents’ dropping out of the health care system once they need to transition to adult care.
NEW YORK – The good news in the management of childhood-onset systemic lupus erythematosus is that children who are diagnosed with the disease have a better prognosis and are living longer. The bad news is that over the course of a lifetime, affected children develop more morbidities and experience more medication-induced toxicities, which create additional management challenges, according to Dr. Phillip Kahn, who spoke at a meeting sponsored by New York University.
About 15%-20% of lupus cases present in childhood. Children younger than age 5 years rarely have the disease, but up to 40% of patients with childhood-onset SLE (cSLE) present prior to puberty, said Dr. Kahn of New York University. The incidence of cSLE is estimated to be 10-20 cases per 100,000 individuals, but the incidence is higher in blacks, at 200 cases per 100,000 individuals (Lupus 2007;16:546-9; 2005;14:83-8).
Often the diagnosis is delayed in children, as their clinical features do not fall within those defined by the 1997 American College of Rheumatology’s SLE criteria, which have not been validated in children. For example, Dr. Kahn described the case of a 13-year-old girl who presented with "constitutional symptoms" such as diffuse lymphadenopathy, malaise, weight loss, and fever – none of which are included in the 1997 criteria.
Mucocutaneous lesions are common in children, with up to 70% presenting with malar rash, 30% with alopecia, 66% with oral ulcers, and 10% with nasal ulcers. "Although nasal ulcers are less common in children than adults, they still may result in perforation of the septum," he said. Other common manifestations include painful polyarthritis, hepatosplenomegaly, headache, photosensitivity, and Raynaud’s phenomenon. Up to 40% of cSLE patients have neuropsychiatric symptoms, significantly more than do adults (Lupus 2008;17:314-22).
An important diagnostic tool for cSLE is a positive ANA (antinuclear antibody) finding, which is present in essentially all children with cSLE. In contrast, up to 10% of adults with SLE do not have a positive ANA, said Dr. Kahn.
Childhood-onset lupus may predict mortality in adults (Arthritis Care Res. [Hoboken] 2010;62:1152-9) and thus physicians must be vigilant about detecting, preventing, and aggressively treating some of its more dangerous manifestations. To prevent recurrent infection, Dr. Kahn recommends keeping a child up to date with immunizations, treating infections promptly when they occur, and considering PCP (pneumocystis carinii pneumonia) prophylaxis in the appropriate patient. Children with cSLE are also at increased risk for premature atherosclerosis, with dyslipidemia seen in 60%-85% of affected children (Nat. Clin. Pract. Rheumatol. 2008;4:258-65). Statin therapy may be appropriate in these children, especially to prevent long-term adverse events such as early MI. For example, adults – especially women – who had c SLE have increased incidence of MI when they reach their 30s (Arthritis Rheum. 2009;61:13-20). The ongoing APPLE (Atherosclerosis Prevention in Pediatric Lupus Erythematosus) trial is investigating, in greater detail, cardiovascular risk and the use of statins in cSLE (Lupus 2010;19:1315-25).
Because nephropathy is the main cause of mortality and morbidity in cSLE, it is important to monitor urinary markers of renal function. Nephropathy may take a greater toll on a pediatric patient, compared with adults; data show that children are more likely to have renal involvement, need hemodialysis and kidney transplantation, and require hospitalization and emergency department visits (Lupus 2008;17:314-22; Arthritis Rheum. 2009;61:13-20). To avoid end-stage renal disease, Dr. Kahn urges tight blood pressure control, as well as antihypertensive medication as necessary. "We know that 90% of the patients who ultimately develop renal disease will do so within the first 2 years of disease, so if you follow a patient for [more than 2] years and they do not develop the disease, the good news is they are much less likely to develop nephropathy," said Dr. Kahn.
In the 1950s, the 5-year survival rate for SLE in adults and children ranged from 18% to 70%; 30 years later, 5-year survival rose to 60%-93%, in large part because of the introduction of prednisone. Currently, prednisone is almost universally given to those with cSLE, and the proportion of children who are treated with prednisone is significantly higher than the proportion of adults with SLE (97% vs. 70%; P less than .0001) (Arthritis Rheum. 2008;58:556-62). The result is greater risk of osteoporosis, diabetes, and growth failure. In fact, up to 40% of cSLE patients have growth failure or short stature, in part because of chronic steroids.
That is why Dr. Kahn posed the question, "Can we move beyond steroids?" Until recently, the only Food and Drug Administration–approved medications for SLE were prednisone, hydroxychloroquine, and aspirin. Steroid-sparing immunomodulatory therapy is sometimes used off label when major organ (such as brain or kidney) involvement develops, but most evidence regarding the efficacy of these medications pertains to renal proliferative disease in adults.
Cyclophosphamide is often used for induction of nephritis, but 30% of patients may still develop end-stage renal disease despite treatment, said Dr. Kahn. Mycophenolate mofetil has been shown to have equal efficacy to cyclophosphamide in induction of remission of nephritis, but sufficient pediatric data are lacking. Azathioprine is effective for maintenance therapy of nephritis after the induction phase.
Other options that are sometimes used include intravenous immunoglobulin, methotrexate, calcineurin inhibitors (such as tacrolimus), plasmapheresis, and autologous stem cell transplantation/immunoablative therapy.
More recently, the focus has been on target-specific therapy for SLE with potentially fewer side effects. Rituximab, which targets B cells that have the CD20 surface antigen, is sometimes used to treat resistant patients, but there is no strong clinical evidence to demonstrate its effectiveness in cSLE (Arthritis Rheum. 2009;61:482-7; Clin. J. Am. Soc. Nephrol. 2009;4:579-87).
Abatacept, which targets T cells by binding to B7 peripheral membrane protein, has shown some clinical benefit in a phase IIB randomized clinical trial, and is undergoing clinical testing as an add-on induction therapy with cyclophosphamide (Arthritis Rheum. 2010;62:3077-87). Belimumab, a monoclonal antibody that targets BLyS (B-lymphocyte stimulator)/BAFF (B-cell activating factor), is the first FDA-approved medication for SLE in more than 50 years, based on recent clinical trials that demonstrate increased efficacy and fewer flares of disease, compared with placebo (Arthritis Rheum 2009;61:1168-78; Lancet 2011;377:721-31).
Although recent data regarding belimumab provide some hope, it "may be naive to think that a target-specific therapy exists in this complex, polygenic, heterogeneous disease," commented Dr. Kahn.
In concluding his talk, Dr. Kahn spoke about some barriers to the effective treatment of cSLE. These include physicians’ failure to detect the subtle changes of early disease, a lack of awareness regarding the neuropsychiatric manifestations of the disease, and physicians’ fear of being overly aggressive with treatment. He also warned about overt or covert adolescent noncompliance, and the danger of adolescents’ dropping out of the health care system once they need to transition to adult care.
FROM A MEETING SPONSORED BY NEW YORK UNIVERSITY
Major Finding: Childhood-onset SLE may be difficult to diagnose because its symptoms differ from that of adult-onset SLE. But cSLE is a predictor of mortality, and steps should be taken to control morbidities such as infection, premature atherosclerosis, and nephropathy. Prednisone remains a cornerstone of pharmacotherapy but has serious side effects, including osteoporosis and impaired growth. Additional nontargeted and targeted options are available.
Data Source: A clinical update presentation based on published reports and clinical experience.
Disclosures: Dr. Kahn reports no relevant disclosures.
Childhood Onset Lupus: Advances, Setbacks Change the Playing Field
NEW YORK – The good news in the management of childhood-onset systemic lupus erythematosus is that children who are diagnosed with the disease have a better prognosis and are living longer. The bad news is that over the course of a lifetime, affected children develop more morbidities and experience more medication-induced toxicities, which create additional management challenges, according to Dr. Phillip Kahn, who spoke at a meeting sponsored by New York University.
About 15%-20% of lupus cases present in childhood. Children younger than age 5 years rarely have the disease, but up to 40% of patients with childhood-onset SLE (cSLE) present prior to puberty, said Dr. Kahn of New York University. The incidence of cSLE is estimated to be 10-20 cases per 100,000 individuals, but the incidence is higher in blacks, at 200 cases per 100,000 individuals (Lupus 2007;16:546-9; 2005;14:83-8).
Often the diagnosis is delayed in children, as their clinical features do not fall within those defined by the 1997 American College of Rheumatology’s SLE criteria, which have not been validated in children. For example, Dr. Kahn described the case of a 13-year-old girl who presented with "constitutional symptoms" such as diffuse lymphadenopathy, malaise, weight loss, and fever – none of which are included in the 1997 criteria.
Mucocutaneous lesions are common in children, with up to 70% presenting with malar rash, 30% with alopecia, 66% with oral ulcers, and 10% with nasal ulcers. "Although nasal ulcers are less common in children than adults, they still may result in perforation of the septum," he said. Other common manifestations include painful polyarthritis, hepatosplenomegaly, headache, photosensitivity, and Raynaud’s phenomenon. Up to 40% of cSLE patients have neuropsychiatric symptoms, significantly more than do adults (Lupus 2008;17:314-22).
An important diagnostic tool for cSLE is a positive ANA (antinuclear antibody) finding, which is present in essentially all children with cSLE. In contrast, up to 10% of adults with SLE do not have a positive ANA, said Dr. Kahn.
Childhood-onset lupus may predict mortality in adults (Arthritis Care Res. [Hoboken] 2010;62:1152-9) and thus physicians must be vigilant about detecting, preventing, and aggressively treating some of its more dangerous manifestations. To prevent recurrent infection, Dr. Kahn recommends keeping a child up to date with immunizations, treating infections promptly when they occur, and considering PCP (pneumocystis carinii pneumonia) prophylaxis in the appropriate patient. Children with cSLE are also at increased risk for premature atherosclerosis, with dyslipidemia seen in 60%-85% of affected children (Nat. Clin. Pract. Rheumatol. 2008;4:258-65). Statin therapy may be appropriate in these children, especially to prevent long-term adverse events such as early MI. For example, adults – especially women – who had c SLE have increased incidence of MI when they reach their 30s (Arthritis Rheum. 2009;61:13-20). The ongoing APPLE (Atherosclerosis Prevention in Pediatric Lupus Erythematosus) trial is investigating, in greater detail, cardiovascular risk and the use of statins in cSLE (Lupus 2010;19:1315-25).
Because nephropathy is the main cause of mortality and morbidity in cSLE, it is important to monitor urinary markers of renal function. Nephropathy may take a greater toll on a pediatric patient, compared with adults; data show that children are more likely to have renal involvement, need hemodialysis and kidney transplantation, and require hospitalization and emergency department visits (Lupus 2008;17:314-22; Arthritis Rheum. 2009;61:13-20). To avoid end-stage renal disease, Dr. Kahn urges tight blood pressure control, as well as antihypertensive medication as necessary. "We know that 90% of the patients who ultimately develop renal disease will do so within the first 2 years of disease, so if you follow a patient for [more than 2] years and they do not develop the disease, the good news is they are much less likely to develop nephropathy," said Dr. Kahn.
In the 1950s, the 5-year survival rate for SLE in adults and children ranged from 18% to 70%; 30 years later, 5-year survival rose to 60%-93%, in large part because of the introduction of prednisone. Currently, prednisone is almost universally given to those with cSLE, and the proportion of children who are treated with prednisone is significantly higher than the proportion of adults with SLE (97% vs. 70%; P less than .0001) (Arthritis Rheum. 2008;58:556-62). The result is greater risk of osteoporosis, diabetes, and growth failure. In fact, up to 40% of cSLE patients have growth failure or short stature, in part because of chronic steroids.
That is why Dr. Kahn posed the question, "Can we move beyond steroids?" Until recently, the only Food and Drug Administration–approved medications for SLE were prednisone, hydroxychloroquine, and aspirin. Steroid-sparing immunomodulatory therapy is sometimes used off label when major organ (such as brain or kidney) involvement develops, but most evidence regarding the efficacy of these medications pertains to renal proliferative disease in adults.
Cyclophosphamide is often used for induction of nephritis, but 30% of patients may still develop end-stage renal disease despite treatment, said Dr. Kahn. Mycophenolate mofetil has been shown to have equal efficacy to cyclophosphamide in induction of remission of nephritis, but sufficient pediatric data are lacking. Azathioprine is effective for maintenance therapy of nephritis after the induction phase.
Other options that are sometimes used include intravenous immunoglobulin, methotrexate, calcineurin inhibitors (such as tacrolimus), plasmapheresis, and autologous stem cell transplantation/immunoablative therapy.
More recently, the focus has been on target-specific therapy for SLE with potentially fewer side effects. Rituximab, which targets B cells that have the CD20 surface antigen, is sometimes used to treat resistant patients, but there is no strong clinical evidence to demonstrate its effectiveness in cSLE (Arthritis Rheum. 2009;61:482-7; Clin. J. Am. Soc. Nephrol. 2009;4:579-87).
Abatacept, which targets T cells by binding to B7 peripheral membrane protein, has shown some clinical benefit in a phase IIB randomized clinical trial, and is undergoing clinical testing as an add-on induction therapy with cyclophosphamide (Arthritis Rheum. 2010;62:3077-87). Belimumab, a monoclonal antibody that targets BLyS (B-lymphocyte stimulator)/BAFF (B-cell activating factor), is the first FDA-approved medication for SLE in more than 50 years, based on recent clinical trials that demonstrate increased efficacy and fewer flares of disease, compared with placebo (Arthritis Rheum 2009;61:1168-78; Lancet 2011;377:721-31).
Although recent data regarding belimumab provide some hope, it "may be naive to think that a target-specific therapy exists in this complex, polygenic, heterogeneous disease," commented Dr. Kahn.
In concluding his talk, Dr. Kahn spoke about some barriers to the effective treatment of cSLE. These include physicians’ failure to detect the subtle changes of early disease, a lack of awareness regarding the neuropsychiatric manifestations of the disease, and physicians’ fear of being overly aggressive with treatment. He also warned about overt or covert adolescent noncompliance, and the danger of adolescents’ dropping out of the health care system once they need to transition to adult care.
NEW YORK – The good news in the management of childhood-onset systemic lupus erythematosus is that children who are diagnosed with the disease have a better prognosis and are living longer. The bad news is that over the course of a lifetime, affected children develop more morbidities and experience more medication-induced toxicities, which create additional management challenges, according to Dr. Phillip Kahn, who spoke at a meeting sponsored by New York University.
About 15%-20% of lupus cases present in childhood. Children younger than age 5 years rarely have the disease, but up to 40% of patients with childhood-onset SLE (cSLE) present prior to puberty, said Dr. Kahn of New York University. The incidence of cSLE is estimated to be 10-20 cases per 100,000 individuals, but the incidence is higher in blacks, at 200 cases per 100,000 individuals (Lupus 2007;16:546-9; 2005;14:83-8).
Often the diagnosis is delayed in children, as their clinical features do not fall within those defined by the 1997 American College of Rheumatology’s SLE criteria, which have not been validated in children. For example, Dr. Kahn described the case of a 13-year-old girl who presented with "constitutional symptoms" such as diffuse lymphadenopathy, malaise, weight loss, and fever – none of which are included in the 1997 criteria.
Mucocutaneous lesions are common in children, with up to 70% presenting with malar rash, 30% with alopecia, 66% with oral ulcers, and 10% with nasal ulcers. "Although nasal ulcers are less common in children than adults, they still may result in perforation of the septum," he said. Other common manifestations include painful polyarthritis, hepatosplenomegaly, headache, photosensitivity, and Raynaud’s phenomenon. Up to 40% of cSLE patients have neuropsychiatric symptoms, significantly more than do adults (Lupus 2008;17:314-22).
An important diagnostic tool for cSLE is a positive ANA (antinuclear antibody) finding, which is present in essentially all children with cSLE. In contrast, up to 10% of adults with SLE do not have a positive ANA, said Dr. Kahn.
Childhood-onset lupus may predict mortality in adults (Arthritis Care Res. [Hoboken] 2010;62:1152-9) and thus physicians must be vigilant about detecting, preventing, and aggressively treating some of its more dangerous manifestations. To prevent recurrent infection, Dr. Kahn recommends keeping a child up to date with immunizations, treating infections promptly when they occur, and considering PCP (pneumocystis carinii pneumonia) prophylaxis in the appropriate patient. Children with cSLE are also at increased risk for premature atherosclerosis, with dyslipidemia seen in 60%-85% of affected children (Nat. Clin. Pract. Rheumatol. 2008;4:258-65). Statin therapy may be appropriate in these children, especially to prevent long-term adverse events such as early MI. For example, adults – especially women – who had c SLE have increased incidence of MI when they reach their 30s (Arthritis Rheum. 2009;61:13-20). The ongoing APPLE (Atherosclerosis Prevention in Pediatric Lupus Erythematosus) trial is investigating, in greater detail, cardiovascular risk and the use of statins in cSLE (Lupus 2010;19:1315-25).
Because nephropathy is the main cause of mortality and morbidity in cSLE, it is important to monitor urinary markers of renal function. Nephropathy may take a greater toll on a pediatric patient, compared with adults; data show that children are more likely to have renal involvement, need hemodialysis and kidney transplantation, and require hospitalization and emergency department visits (Lupus 2008;17:314-22; Arthritis Rheum. 2009;61:13-20). To avoid end-stage renal disease, Dr. Kahn urges tight blood pressure control, as well as antihypertensive medication as necessary. "We know that 90% of the patients who ultimately develop renal disease will do so within the first 2 years of disease, so if you follow a patient for [more than 2] years and they do not develop the disease, the good news is they are much less likely to develop nephropathy," said Dr. Kahn.
In the 1950s, the 5-year survival rate for SLE in adults and children ranged from 18% to 70%; 30 years later, 5-year survival rose to 60%-93%, in large part because of the introduction of prednisone. Currently, prednisone is almost universally given to those with cSLE, and the proportion of children who are treated with prednisone is significantly higher than the proportion of adults with SLE (97% vs. 70%; P less than .0001) (Arthritis Rheum. 2008;58:556-62). The result is greater risk of osteoporosis, diabetes, and growth failure. In fact, up to 40% of cSLE patients have growth failure or short stature, in part because of chronic steroids.
That is why Dr. Kahn posed the question, "Can we move beyond steroids?" Until recently, the only Food and Drug Administration–approved medications for SLE were prednisone, hydroxychloroquine, and aspirin. Steroid-sparing immunomodulatory therapy is sometimes used off label when major organ (such as brain or kidney) involvement develops, but most evidence regarding the efficacy of these medications pertains to renal proliferative disease in adults.
Cyclophosphamide is often used for induction of nephritis, but 30% of patients may still develop end-stage renal disease despite treatment, said Dr. Kahn. Mycophenolate mofetil has been shown to have equal efficacy to cyclophosphamide in induction of remission of nephritis, but sufficient pediatric data are lacking. Azathioprine is effective for maintenance therapy of nephritis after the induction phase.
Other options that are sometimes used include intravenous immunoglobulin, methotrexate, calcineurin inhibitors (such as tacrolimus), plasmapheresis, and autologous stem cell transplantation/immunoablative therapy.
More recently, the focus has been on target-specific therapy for SLE with potentially fewer side effects. Rituximab, which targets B cells that have the CD20 surface antigen, is sometimes used to treat resistant patients, but there is no strong clinical evidence to demonstrate its effectiveness in cSLE (Arthritis Rheum. 2009;61:482-7; Clin. J. Am. Soc. Nephrol. 2009;4:579-87).
Abatacept, which targets T cells by binding to B7 peripheral membrane protein, has shown some clinical benefit in a phase IIB randomized clinical trial, and is undergoing clinical testing as an add-on induction therapy with cyclophosphamide (Arthritis Rheum. 2010;62:3077-87). Belimumab, a monoclonal antibody that targets BLyS (B-lymphocyte stimulator)/BAFF (B-cell activating factor), is the first FDA-approved medication for SLE in more than 50 years, based on recent clinical trials that demonstrate increased efficacy and fewer flares of disease, compared with placebo (Arthritis Rheum 2009;61:1168-78; Lancet 2011;377:721-31).
Although recent data regarding belimumab provide some hope, it "may be naive to think that a target-specific therapy exists in this complex, polygenic, heterogeneous disease," commented Dr. Kahn.
In concluding his talk, Dr. Kahn spoke about some barriers to the effective treatment of cSLE. These include physicians’ failure to detect the subtle changes of early disease, a lack of awareness regarding the neuropsychiatric manifestations of the disease, and physicians’ fear of being overly aggressive with treatment. He also warned about overt or covert adolescent noncompliance, and the danger of adolescents’ dropping out of the health care system once they need to transition to adult care.
NEW YORK – The good news in the management of childhood-onset systemic lupus erythematosus is that children who are diagnosed with the disease have a better prognosis and are living longer. The bad news is that over the course of a lifetime, affected children develop more morbidities and experience more medication-induced toxicities, which create additional management challenges, according to Dr. Phillip Kahn, who spoke at a meeting sponsored by New York University.
About 15%-20% of lupus cases present in childhood. Children younger than age 5 years rarely have the disease, but up to 40% of patients with childhood-onset SLE (cSLE) present prior to puberty, said Dr. Kahn of New York University. The incidence of cSLE is estimated to be 10-20 cases per 100,000 individuals, but the incidence is higher in blacks, at 200 cases per 100,000 individuals (Lupus 2007;16:546-9; 2005;14:83-8).
Often the diagnosis is delayed in children, as their clinical features do not fall within those defined by the 1997 American College of Rheumatology’s SLE criteria, which have not been validated in children. For example, Dr. Kahn described the case of a 13-year-old girl who presented with "constitutional symptoms" such as diffuse lymphadenopathy, malaise, weight loss, and fever – none of which are included in the 1997 criteria.
Mucocutaneous lesions are common in children, with up to 70% presenting with malar rash, 30% with alopecia, 66% with oral ulcers, and 10% with nasal ulcers. "Although nasal ulcers are less common in children than adults, they still may result in perforation of the septum," he said. Other common manifestations include painful polyarthritis, hepatosplenomegaly, headache, photosensitivity, and Raynaud’s phenomenon. Up to 40% of cSLE patients have neuropsychiatric symptoms, significantly more than do adults (Lupus 2008;17:314-22).
An important diagnostic tool for cSLE is a positive ANA (antinuclear antibody) finding, which is present in essentially all children with cSLE. In contrast, up to 10% of adults with SLE do not have a positive ANA, said Dr. Kahn.
Childhood-onset lupus may predict mortality in adults (Arthritis Care Res. [Hoboken] 2010;62:1152-9) and thus physicians must be vigilant about detecting, preventing, and aggressively treating some of its more dangerous manifestations. To prevent recurrent infection, Dr. Kahn recommends keeping a child up to date with immunizations, treating infections promptly when they occur, and considering PCP (pneumocystis carinii pneumonia) prophylaxis in the appropriate patient. Children with cSLE are also at increased risk for premature atherosclerosis, with dyslipidemia seen in 60%-85% of affected children (Nat. Clin. Pract. Rheumatol. 2008;4:258-65). Statin therapy may be appropriate in these children, especially to prevent long-term adverse events such as early MI. For example, adults – especially women – who had c SLE have increased incidence of MI when they reach their 30s (Arthritis Rheum. 2009;61:13-20). The ongoing APPLE (Atherosclerosis Prevention in Pediatric Lupus Erythematosus) trial is investigating, in greater detail, cardiovascular risk and the use of statins in cSLE (Lupus 2010;19:1315-25).
Because nephropathy is the main cause of mortality and morbidity in cSLE, it is important to monitor urinary markers of renal function. Nephropathy may take a greater toll on a pediatric patient, compared with adults; data show that children are more likely to have renal involvement, need hemodialysis and kidney transplantation, and require hospitalization and emergency department visits (Lupus 2008;17:314-22; Arthritis Rheum. 2009;61:13-20). To avoid end-stage renal disease, Dr. Kahn urges tight blood pressure control, as well as antihypertensive medication as necessary. "We know that 90% of the patients who ultimately develop renal disease will do so within the first 2 years of disease, so if you follow a patient for [more than 2] years and they do not develop the disease, the good news is they are much less likely to develop nephropathy," said Dr. Kahn.
In the 1950s, the 5-year survival rate for SLE in adults and children ranged from 18% to 70%; 30 years later, 5-year survival rose to 60%-93%, in large part because of the introduction of prednisone. Currently, prednisone is almost universally given to those with cSLE, and the proportion of children who are treated with prednisone is significantly higher than the proportion of adults with SLE (97% vs. 70%; P less than .0001) (Arthritis Rheum. 2008;58:556-62). The result is greater risk of osteoporosis, diabetes, and growth failure. In fact, up to 40% of cSLE patients have growth failure or short stature, in part because of chronic steroids.
That is why Dr. Kahn posed the question, "Can we move beyond steroids?" Until recently, the only Food and Drug Administration–approved medications for SLE were prednisone, hydroxychloroquine, and aspirin. Steroid-sparing immunomodulatory therapy is sometimes used off label when major organ (such as brain or kidney) involvement develops, but most evidence regarding the efficacy of these medications pertains to renal proliferative disease in adults.
Cyclophosphamide is often used for induction of nephritis, but 30% of patients may still develop end-stage renal disease despite treatment, said Dr. Kahn. Mycophenolate mofetil has been shown to have equal efficacy to cyclophosphamide in induction of remission of nephritis, but sufficient pediatric data are lacking. Azathioprine is effective for maintenance therapy of nephritis after the induction phase.
Other options that are sometimes used include intravenous immunoglobulin, methotrexate, calcineurin inhibitors (such as tacrolimus), plasmapheresis, and autologous stem cell transplantation/immunoablative therapy.
More recently, the focus has been on target-specific therapy for SLE with potentially fewer side effects. Rituximab, which targets B cells that have the CD20 surface antigen, is sometimes used to treat resistant patients, but there is no strong clinical evidence to demonstrate its effectiveness in cSLE (Arthritis Rheum. 2009;61:482-7; Clin. J. Am. Soc. Nephrol. 2009;4:579-87).
Abatacept, which targets T cells by binding to B7 peripheral membrane protein, has shown some clinical benefit in a phase IIB randomized clinical trial, and is undergoing clinical testing as an add-on induction therapy with cyclophosphamide (Arthritis Rheum. 2010;62:3077-87). Belimumab, a monoclonal antibody that targets BLyS (B-lymphocyte stimulator)/BAFF (B-cell activating factor), is the first FDA-approved medication for SLE in more than 50 years, based on recent clinical trials that demonstrate increased efficacy and fewer flares of disease, compared with placebo (Arthritis Rheum 2009;61:1168-78; Lancet 2011;377:721-31).
Although recent data regarding belimumab provide some hope, it "may be naive to think that a target-specific therapy exists in this complex, polygenic, heterogeneous disease," commented Dr. Kahn.
In concluding his talk, Dr. Kahn spoke about some barriers to the effective treatment of cSLE. These include physicians’ failure to detect the subtle changes of early disease, a lack of awareness regarding the neuropsychiatric manifestations of the disease, and physicians’ fear of being overly aggressive with treatment. He also warned about overt or covert adolescent noncompliance, and the danger of adolescents’ dropping out of the health care system once they need to transition to adult care.
FROM A MEETING SPONSORED BY NEW YORK UNIVERSITY
Guidelines Provide New Insights Into Systemic JIA Treatment
NEW YORK - Early and aggressive therapy is warranted in the management of systemic juvenile idiopathic arthritis, which accounts for 10% of arthritis cases in children and which can be devastating, according to Dr. Randy Q. Cron, who spoke about the condition at a meeting sponsored by New York University.
Dr. Cron, director of pediatric rheumatology at the University of Alabama at Birmingham, is a coauthor of the first ever 2011 ACR Recommendations for the Treatment of Juvenile Idiopathic Arthritis (JIA) that were published on April 1 (Arthritis Care Res. 2011;63:465-82) and which include guidelines for the treatment of systemic JIA. According to the publication, "the JIA category of systemic arthritis proved to be especially challenging to evaluate. Attempts to exhaustively depict the myriad possible clinical presentations of systemic arthritis were impractical." The result was that systemic JIA guidelines include two treatment algorithms, one for the active systemic features (for example, fever) and one for the active arthritis. Patients who have both concurrent active systemic features and active arthritis can be treated according to suggestions from both algorithms.
For treating the systemic features of systemic JIA, three first-line options are available: nonsteroidal anti-inflammatory drugs (NSAIDs), systemic corticosteroids, and anakinra (Kineret). It should be noted that interleukin-6 inhibitors and interleukin-1 inhibitors other than anakinra were not considered when developing the recommendations, since they were not widely available commercially at the time. TNF-alpha inhibitors were not considered because of their relatively poor effectiveness for the systemic manifestations.
While NSAID monotherapy might also help some children, the recommendations say that its use is inappropriate for patients with active fever and physician global assessment of overall disease activity (MD global) score equal to 7 of 10. It should also not be used for more than 1 month in patients with active fever.
Systemic glucocorticoids were recommended as initial therapy for patients with active fever and an MD global score of 7. Systemic glucocorticoids may also be added after 2 weeks of NSAID monotherapy in patients with active fever.
Anakinra was recommended for all patients with active fever who had poor prognostic features, no matter what they were currently taking. Anakinra was also recommended for patients who have or develop fever while on systemic glucocorticoids. Anakinra is a human recombinant interleukin-1 (IL-1) inhibitor that was approved to treat adults with moderate to severe arthritis who have not had an adequate response to conservative disease-modifying anti-rheumatic drug therapy. It is not currently approved for children with JIA.
Medications such as calcineurin inhibitors, intravenous immunoglobulin, methotrexate, and thalidomide were not formally recommended but might have roles in the treatment of systemic JIA.
For the treatment of the arthritic features of systemic JIA, initiation of NSAID monotherapy (with or without glucocorticoid joint injections) was recommended for all patients, regardless of the level of disease activity or presence of poor prognostic features. It was assumed that most patients with newly diagnosed systemic arthritis would have been started with NSAIDs. The next step would be methotrexate for all patients with active arthritis who were taking NSAID monotherapy for less than 1 month. If escalation of therapy was needed, the next choices would be either addition of a TNF-alpha inhibitor or anakinra. The recommendations indicate that initiation of anakinra should take place early rather than later in the disease course. For patients with moderate or high disease activity, regardless of features of poor prognosis, it was suggested that patients be switched from anakinra to a TNF-alpha inhibitor.
The last option is abatacept, a costimulatory blocker, for those who fail to be controlled by methotrexate or 4 months of a TNF-alpha inhibitor, and who have moderate to high disease activity.
During his presentation, Dr. Cron provided his perspective on the new systemic JIA recommendations. Most pediatric rheumatologists would probably begin with systemic corticosteroids at the onset of treatment, because that is what they are most familiar with and they are effective, he said.
He was pleased with the inclusion of anakinra in the guidelines. "With these guidelines, physicians can treat children with JIA from the start with anakinra." Inclusion of anakinra in the guidelines as a first option should help tremendously in helping patients receive reimbursement from insurance companies, he noted.
Dr. Cron cited evidence showing the effectiveness of anakinra as a first-line disease-modifying therapy in 46 patients with JIA, preventing refractory arthritis in almost 90% of patients (Arthritis Rheum. 2011;63:545-55). In this series of case studies, use of anakinra reduced elevated sedimentation rates and ferritin levels, decreased the number of active joint counts, and significantly lowered the dose of concomitant steroid therapy. "Kids can get remarkably better within days or months with anakinra. It can be a wonder drug for very sick children with systemic JIA," he said.
Although it was not discussed in the Recommendations, in his talk Dr. Cron said that anakinra is "revolutionizing" the treatment of macrophage activation syndrome (MAS) in systemic JIA (Rheumatology 2011;50:417-9) Dr. Cron reported that he has used anakinra as initial therapy to treat two children with MAS, and he found dramatic reductions in ferritin levels and liver enzymes within 2 days of initiation.
Systemic JIA is a relatively rare disease, with a prevalence of about 1 in 10,000. It has multiple systemic manifestations, including a high fever (which follows a unique pattern: typically once-daily, late-afternoon spikes in 30% of patients at presentation), rash, hepatosplenomegaly, lymphadenopathy, pericarditis, pleural effusion, pulmonary vasculitis, and even CNS stroke or seizure. Stroke or seizure might be related to MAS, a widespread coagulopathy that occurs in about 50% of patients and can be fatal.
Another common finding is asymmetry of the jaw. According to Dr. Cron, up to 80% of children with all forms of arthritis, including systemic JIA, can have arthritis of the temporomandibular joint (TMJ). This is often a subtle finding that can be overlooked until significant erosion has occurred. However, early TMJ arthritis can be detected with magnetic resonance screening.
Dr. Cron is a consultant for Genentech.
NEW YORK - Early and aggressive therapy is warranted in the management of systemic juvenile idiopathic arthritis, which accounts for 10% of arthritis cases in children and which can be devastating, according to Dr. Randy Q. Cron, who spoke about the condition at a meeting sponsored by New York University.
Dr. Cron, director of pediatric rheumatology at the University of Alabama at Birmingham, is a coauthor of the first ever 2011 ACR Recommendations for the Treatment of Juvenile Idiopathic Arthritis (JIA) that were published on April 1 (Arthritis Care Res. 2011;63:465-82) and which include guidelines for the treatment of systemic JIA. According to the publication, "the JIA category of systemic arthritis proved to be especially challenging to evaluate. Attempts to exhaustively depict the myriad possible clinical presentations of systemic arthritis were impractical." The result was that systemic JIA guidelines include two treatment algorithms, one for the active systemic features (for example, fever) and one for the active arthritis. Patients who have both concurrent active systemic features and active arthritis can be treated according to suggestions from both algorithms.
For treating the systemic features of systemic JIA, three first-line options are available: nonsteroidal anti-inflammatory drugs (NSAIDs), systemic corticosteroids, and anakinra (Kineret). It should be noted that interleukin-6 inhibitors and interleukin-1 inhibitors other than anakinra were not considered when developing the recommendations, since they were not widely available commercially at the time. TNF-alpha inhibitors were not considered because of their relatively poor effectiveness for the systemic manifestations.
While NSAID monotherapy might also help some children, the recommendations say that its use is inappropriate for patients with active fever and physician global assessment of overall disease activity (MD global) score equal to 7 of 10. It should also not be used for more than 1 month in patients with active fever.
Systemic glucocorticoids were recommended as initial therapy for patients with active fever and an MD global score of 7. Systemic glucocorticoids may also be added after 2 weeks of NSAID monotherapy in patients with active fever.
Anakinra was recommended for all patients with active fever who had poor prognostic features, no matter what they were currently taking. Anakinra was also recommended for patients who have or develop fever while on systemic glucocorticoids. Anakinra is a human recombinant interleukin-1 (IL-1) inhibitor that was approved to treat adults with moderate to severe arthritis who have not had an adequate response to conservative disease-modifying anti-rheumatic drug therapy. It is not currently approved for children with JIA.
Medications such as calcineurin inhibitors, intravenous immunoglobulin, methotrexate, and thalidomide were not formally recommended but might have roles in the treatment of systemic JIA.
For the treatment of the arthritic features of systemic JIA, initiation of NSAID monotherapy (with or without glucocorticoid joint injections) was recommended for all patients, regardless of the level of disease activity or presence of poor prognostic features. It was assumed that most patients with newly diagnosed systemic arthritis would have been started with NSAIDs. The next step would be methotrexate for all patients with active arthritis who were taking NSAID monotherapy for less than 1 month. If escalation of therapy was needed, the next choices would be either addition of a TNF-alpha inhibitor or anakinra. The recommendations indicate that initiation of anakinra should take place early rather than later in the disease course. For patients with moderate or high disease activity, regardless of features of poor prognosis, it was suggested that patients be switched from anakinra to a TNF-alpha inhibitor.
The last option is abatacept, a costimulatory blocker, for those who fail to be controlled by methotrexate or 4 months of a TNF-alpha inhibitor, and who have moderate to high disease activity.
During his presentation, Dr. Cron provided his perspective on the new systemic JIA recommendations. Most pediatric rheumatologists would probably begin with systemic corticosteroids at the onset of treatment, because that is what they are most familiar with and they are effective, he said.
He was pleased with the inclusion of anakinra in the guidelines. "With these guidelines, physicians can treat children with JIA from the start with anakinra." Inclusion of anakinra in the guidelines as a first option should help tremendously in helping patients receive reimbursement from insurance companies, he noted.
Dr. Cron cited evidence showing the effectiveness of anakinra as a first-line disease-modifying therapy in 46 patients with JIA, preventing refractory arthritis in almost 90% of patients (Arthritis Rheum. 2011;63:545-55). In this series of case studies, use of anakinra reduced elevated sedimentation rates and ferritin levels, decreased the number of active joint counts, and significantly lowered the dose of concomitant steroid therapy. "Kids can get remarkably better within days or months with anakinra. It can be a wonder drug for very sick children with systemic JIA," he said.
Although it was not discussed in the Recommendations, in his talk Dr. Cron said that anakinra is "revolutionizing" the treatment of macrophage activation syndrome (MAS) in systemic JIA (Rheumatology 2011;50:417-9) Dr. Cron reported that he has used anakinra as initial therapy to treat two children with MAS, and he found dramatic reductions in ferritin levels and liver enzymes within 2 days of initiation.
Systemic JIA is a relatively rare disease, with a prevalence of about 1 in 10,000. It has multiple systemic manifestations, including a high fever (which follows a unique pattern: typically once-daily, late-afternoon spikes in 30% of patients at presentation), rash, hepatosplenomegaly, lymphadenopathy, pericarditis, pleural effusion, pulmonary vasculitis, and even CNS stroke or seizure. Stroke or seizure might be related to MAS, a widespread coagulopathy that occurs in about 50% of patients and can be fatal.
Another common finding is asymmetry of the jaw. According to Dr. Cron, up to 80% of children with all forms of arthritis, including systemic JIA, can have arthritis of the temporomandibular joint (TMJ). This is often a subtle finding that can be overlooked until significant erosion has occurred. However, early TMJ arthritis can be detected with magnetic resonance screening.
Dr. Cron is a consultant for Genentech.
NEW YORK - Early and aggressive therapy is warranted in the management of systemic juvenile idiopathic arthritis, which accounts for 10% of arthritis cases in children and which can be devastating, according to Dr. Randy Q. Cron, who spoke about the condition at a meeting sponsored by New York University.
Dr. Cron, director of pediatric rheumatology at the University of Alabama at Birmingham, is a coauthor of the first ever 2011 ACR Recommendations for the Treatment of Juvenile Idiopathic Arthritis (JIA) that were published on April 1 (Arthritis Care Res. 2011;63:465-82) and which include guidelines for the treatment of systemic JIA. According to the publication, "the JIA category of systemic arthritis proved to be especially challenging to evaluate. Attempts to exhaustively depict the myriad possible clinical presentations of systemic arthritis were impractical." The result was that systemic JIA guidelines include two treatment algorithms, one for the active systemic features (for example, fever) and one for the active arthritis. Patients who have both concurrent active systemic features and active arthritis can be treated according to suggestions from both algorithms.
For treating the systemic features of systemic JIA, three first-line options are available: nonsteroidal anti-inflammatory drugs (NSAIDs), systemic corticosteroids, and anakinra (Kineret). It should be noted that interleukin-6 inhibitors and interleukin-1 inhibitors other than anakinra were not considered when developing the recommendations, since they were not widely available commercially at the time. TNF-alpha inhibitors were not considered because of their relatively poor effectiveness for the systemic manifestations.
While NSAID monotherapy might also help some children, the recommendations say that its use is inappropriate for patients with active fever and physician global assessment of overall disease activity (MD global) score equal to 7 of 10. It should also not be used for more than 1 month in patients with active fever.
Systemic glucocorticoids were recommended as initial therapy for patients with active fever and an MD global score of 7. Systemic glucocorticoids may also be added after 2 weeks of NSAID monotherapy in patients with active fever.
Anakinra was recommended for all patients with active fever who had poor prognostic features, no matter what they were currently taking. Anakinra was also recommended for patients who have or develop fever while on systemic glucocorticoids. Anakinra is a human recombinant interleukin-1 (IL-1) inhibitor that was approved to treat adults with moderate to severe arthritis who have not had an adequate response to conservative disease-modifying anti-rheumatic drug therapy. It is not currently approved for children with JIA.
Medications such as calcineurin inhibitors, intravenous immunoglobulin, methotrexate, and thalidomide were not formally recommended but might have roles in the treatment of systemic JIA.
For the treatment of the arthritic features of systemic JIA, initiation of NSAID monotherapy (with or without glucocorticoid joint injections) was recommended for all patients, regardless of the level of disease activity or presence of poor prognostic features. It was assumed that most patients with newly diagnosed systemic arthritis would have been started with NSAIDs. The next step would be methotrexate for all patients with active arthritis who were taking NSAID monotherapy for less than 1 month. If escalation of therapy was needed, the next choices would be either addition of a TNF-alpha inhibitor or anakinra. The recommendations indicate that initiation of anakinra should take place early rather than later in the disease course. For patients with moderate or high disease activity, regardless of features of poor prognosis, it was suggested that patients be switched from anakinra to a TNF-alpha inhibitor.
The last option is abatacept, a costimulatory blocker, for those who fail to be controlled by methotrexate or 4 months of a TNF-alpha inhibitor, and who have moderate to high disease activity.
During his presentation, Dr. Cron provided his perspective on the new systemic JIA recommendations. Most pediatric rheumatologists would probably begin with systemic corticosteroids at the onset of treatment, because that is what they are most familiar with and they are effective, he said.
He was pleased with the inclusion of anakinra in the guidelines. "With these guidelines, physicians can treat children with JIA from the start with anakinra." Inclusion of anakinra in the guidelines as a first option should help tremendously in helping patients receive reimbursement from insurance companies, he noted.
Dr. Cron cited evidence showing the effectiveness of anakinra as a first-line disease-modifying therapy in 46 patients with JIA, preventing refractory arthritis in almost 90% of patients (Arthritis Rheum. 2011;63:545-55). In this series of case studies, use of anakinra reduced elevated sedimentation rates and ferritin levels, decreased the number of active joint counts, and significantly lowered the dose of concomitant steroid therapy. "Kids can get remarkably better within days or months with anakinra. It can be a wonder drug for very sick children with systemic JIA," he said.
Although it was not discussed in the Recommendations, in his talk Dr. Cron said that anakinra is "revolutionizing" the treatment of macrophage activation syndrome (MAS) in systemic JIA (Rheumatology 2011;50:417-9) Dr. Cron reported that he has used anakinra as initial therapy to treat two children with MAS, and he found dramatic reductions in ferritin levels and liver enzymes within 2 days of initiation.
Systemic JIA is a relatively rare disease, with a prevalence of about 1 in 10,000. It has multiple systemic manifestations, including a high fever (which follows a unique pattern: typically once-daily, late-afternoon spikes in 30% of patients at presentation), rash, hepatosplenomegaly, lymphadenopathy, pericarditis, pleural effusion, pulmonary vasculitis, and even CNS stroke or seizure. Stroke or seizure might be related to MAS, a widespread coagulopathy that occurs in about 50% of patients and can be fatal.
Another common finding is asymmetry of the jaw. According to Dr. Cron, up to 80% of children with all forms of arthritis, including systemic JIA, can have arthritis of the temporomandibular joint (TMJ). This is often a subtle finding that can be overlooked until significant erosion has occurred. However, early TMJ arthritis can be detected with magnetic resonance screening.
Dr. Cron is a consultant for Genentech.
A MEETING SPONSORED BY NEW YORK UNIVERSITY
Guidelines Provide New Insights Into Systemic JIA Treatment
NEW YORK - Early and aggressive therapy is warranted in the management of systemic juvenile idiopathic arthritis, which accounts for 10% of arthritis cases in children and which can be devastating, according to Dr. Randy Q. Cron, who spoke about the condition at a meeting sponsored by New York University.
Dr. Cron, director of pediatric rheumatology at the University of Alabama at Birmingham, is a coauthor of the first ever 2011 ACR Recommendations for the Treatment of Juvenile Idiopathic Arthritis (JIA) that were published on April 1 (Arthritis Care Res. 2011;63:465-82) and which include guidelines for the treatment of systemic JIA. According to the publication, "the JIA category of systemic arthritis proved to be especially challenging to evaluate. Attempts to exhaustively depict the myriad possible clinical presentations of systemic arthritis were impractical." The result was that systemic JIA guidelines include two treatment algorithms, one for the active systemic features (for example, fever) and one for the active arthritis. Patients who have both concurrent active systemic features and active arthritis can be treated according to suggestions from both algorithms.
For treating the systemic features of systemic JIA, three first-line options are available: nonsteroidal anti-inflammatory drugs (NSAIDs), systemic corticosteroids, and anakinra (Kineret). It should be noted that interleukin-6 inhibitors and interleukin-1 inhibitors other than anakinra were not considered when developing the recommendations, since they were not widely available commercially at the time. TNF-alpha inhibitors were not considered because of their relatively poor effectiveness for the systemic manifestations.
While NSAID monotherapy might also help some children, the recommendations say that its use is inappropriate for patients with active fever and physician global assessment of overall disease activity (MD global) score equal to 7 of 10. It should also not be used for more than 1 month in patients with active fever.
Systemic glucocorticoids were recommended as initial therapy for patients with active fever and an MD global score of 7. Systemic glucocorticoids may also be added after 2 weeks of NSAID monotherapy in patients with active fever.
Anakinra was recommended for all patients with active fever who had poor prognostic features, no matter what they were currently taking. Anakinra was also recommended for patients who have or develop fever while on systemic glucocorticoids. Anakinra is a human recombinant interleukin-1 (IL-1) inhibitor that was approved to treat adults with moderate to severe arthritis who have not had an adequate response to conservative disease-modifying anti-rheumatic drug therapy. It is not currently approved for children with JIA.
Medications such as calcineurin inhibitors, intravenous immunoglobulin, methotrexate, and thalidomide were not formally recommended but might have roles in the treatment of systemic JIA.
For the treatment of the arthritic features of systemic JIA, initiation of NSAID monotherapy (with or without glucocorticoid joint injections) was recommended for all patients, regardless of the level of disease activity or presence of poor prognostic features. It was assumed that most patients with newly diagnosed systemic arthritis would have been started with NSAIDs. The next step would be methotrexate for all patients with active arthritis who were taking NSAID monotherapy for less than 1 month. If escalation of therapy was needed, the next choices would be either addition of a TNF-alpha inhibitor or anakinra. The recommendations indicate that initiation of anakinra should take place early rather than later in the disease course. For patients with moderate or high disease activity, regardless of features of poor prognosis, it was suggested that patients be switched from anakinra to a TNF-alpha inhibitor.
The last option is abatacept, a costimulatory blocker, for those who fail to be controlled by methotrexate or 4 months of a TNF-alpha inhibitor, and who have moderate to high disease activity.
During his presentation, Dr. Cron provided his perspective on the new systemic JIA recommendations. Most pediatric rheumatologists would probably begin with systemic corticosteroids at the onset of treatment, because that is what they are most familiar with and they are effective, he said.
He was pleased with the inclusion of anakinra in the guidelines. "With these guidelines, physicians can treat children with JIA from the start with anakinra." Inclusion of anakinra in the guidelines as a first option should help tremendously in helping patients receive reimbursement from insurance companies, he noted.
Dr. Cron cited evidence showing the effectiveness of anakinra as a first-line disease-modifying therapy in 46 patients with JIA, preventing refractory arthritis in almost 90% of patients (Arthritis Rheum. 2011;63:545-55). In this series of case studies, use of anakinra reduced elevated sedimentation rates and ferritin levels, decreased the number of active joint counts, and significantly lowered the dose of concomitant steroid therapy. "Kids can get remarkably better within days or months with anakinra. It can be a wonder drug for very sick children with systemic JIA," he said.
Although it was not discussed in the Recommendations, in his talk Dr. Cron said that anakinra is "revolutionizing" the treatment of macrophage activation syndrome (MAS) in systemic JIA (Rheumatology 2011;50:417-9) Dr. Cron reported that he has used anakinra as initial therapy to treat two children with MAS, and he found dramatic reductions in ferritin levels and liver enzymes within 2 days of initiation.
Systemic JIA is a relatively rare disease, with a prevalence of about 1 in 10,000. It has multiple systemic manifestations, including a high fever (which follows a unique pattern: typically once-daily, late-afternoon spikes in 30% of patients at presentation), rash, hepatosplenomegaly, lymphadenopathy, pericarditis, pleural effusion, pulmonary vasculitis, and even CNS stroke or seizure. Stroke or seizure might be related to MAS, a widespread coagulopathy that occurs in about 50% of patients and can be fatal.
Another common finding is asymmetry of the jaw. According to Dr. Cron, up to 80% of children with all forms of arthritis, including systemic JIA, can have arthritis of the temporomandibular joint (TMJ). This is often a subtle finding that can be overlooked until significant erosion has occurred. However, early TMJ arthritis can be detected with magnetic resonance screening.
Dr. Cron is a consultant for Genentech.
NEW YORK - Early and aggressive therapy is warranted in the management of systemic juvenile idiopathic arthritis, which accounts for 10% of arthritis cases in children and which can be devastating, according to Dr. Randy Q. Cron, who spoke about the condition at a meeting sponsored by New York University.
Dr. Cron, director of pediatric rheumatology at the University of Alabama at Birmingham, is a coauthor of the first ever 2011 ACR Recommendations for the Treatment of Juvenile Idiopathic Arthritis (JIA) that were published on April 1 (Arthritis Care Res. 2011;63:465-82) and which include guidelines for the treatment of systemic JIA. According to the publication, "the JIA category of systemic arthritis proved to be especially challenging to evaluate. Attempts to exhaustively depict the myriad possible clinical presentations of systemic arthritis were impractical." The result was that systemic JIA guidelines include two treatment algorithms, one for the active systemic features (for example, fever) and one for the active arthritis. Patients who have both concurrent active systemic features and active arthritis can be treated according to suggestions from both algorithms.
For treating the systemic features of systemic JIA, three first-line options are available: nonsteroidal anti-inflammatory drugs (NSAIDs), systemic corticosteroids, and anakinra (Kineret). It should be noted that interleukin-6 inhibitors and interleukin-1 inhibitors other than anakinra were not considered when developing the recommendations, since they were not widely available commercially at the time. TNF-alpha inhibitors were not considered because of their relatively poor effectiveness for the systemic manifestations.
While NSAID monotherapy might also help some children, the recommendations say that its use is inappropriate for patients with active fever and physician global assessment of overall disease activity (MD global) score equal to 7 of 10. It should also not be used for more than 1 month in patients with active fever.
Systemic glucocorticoids were recommended as initial therapy for patients with active fever and an MD global score of 7. Systemic glucocorticoids may also be added after 2 weeks of NSAID monotherapy in patients with active fever.
Anakinra was recommended for all patients with active fever who had poor prognostic features, no matter what they were currently taking. Anakinra was also recommended for patients who have or develop fever while on systemic glucocorticoids. Anakinra is a human recombinant interleukin-1 (IL-1) inhibitor that was approved to treat adults with moderate to severe arthritis who have not had an adequate response to conservative disease-modifying anti-rheumatic drug therapy. It is not currently approved for children with JIA.
Medications such as calcineurin inhibitors, intravenous immunoglobulin, methotrexate, and thalidomide were not formally recommended but might have roles in the treatment of systemic JIA.
For the treatment of the arthritic features of systemic JIA, initiation of NSAID monotherapy (with or without glucocorticoid joint injections) was recommended for all patients, regardless of the level of disease activity or presence of poor prognostic features. It was assumed that most patients with newly diagnosed systemic arthritis would have been started with NSAIDs. The next step would be methotrexate for all patients with active arthritis who were taking NSAID monotherapy for less than 1 month. If escalation of therapy was needed, the next choices would be either addition of a TNF-alpha inhibitor or anakinra. The recommendations indicate that initiation of anakinra should take place early rather than later in the disease course. For patients with moderate or high disease activity, regardless of features of poor prognosis, it was suggested that patients be switched from anakinra to a TNF-alpha inhibitor.
The last option is abatacept, a costimulatory blocker, for those who fail to be controlled by methotrexate or 4 months of a TNF-alpha inhibitor, and who have moderate to high disease activity.
During his presentation, Dr. Cron provided his perspective on the new systemic JIA recommendations. Most pediatric rheumatologists would probably begin with systemic corticosteroids at the onset of treatment, because that is what they are most familiar with and they are effective, he said.
He was pleased with the inclusion of anakinra in the guidelines. "With these guidelines, physicians can treat children with JIA from the start with anakinra." Inclusion of anakinra in the guidelines as a first option should help tremendously in helping patients receive reimbursement from insurance companies, he noted.
Dr. Cron cited evidence showing the effectiveness of anakinra as a first-line disease-modifying therapy in 46 patients with JIA, preventing refractory arthritis in almost 90% of patients (Arthritis Rheum. 2011;63:545-55). In this series of case studies, use of anakinra reduced elevated sedimentation rates and ferritin levels, decreased the number of active joint counts, and significantly lowered the dose of concomitant steroid therapy. "Kids can get remarkably better within days or months with anakinra. It can be a wonder drug for very sick children with systemic JIA," he said.
Although it was not discussed in the Recommendations, in his talk Dr. Cron said that anakinra is "revolutionizing" the treatment of macrophage activation syndrome (MAS) in systemic JIA (Rheumatology 2011;50:417-9) Dr. Cron reported that he has used anakinra as initial therapy to treat two children with MAS, and he found dramatic reductions in ferritin levels and liver enzymes within 2 days of initiation.
Systemic JIA is a relatively rare disease, with a prevalence of about 1 in 10,000. It has multiple systemic manifestations, including a high fever (which follows a unique pattern: typically once-daily, late-afternoon spikes in 30% of patients at presentation), rash, hepatosplenomegaly, lymphadenopathy, pericarditis, pleural effusion, pulmonary vasculitis, and even CNS stroke or seizure. Stroke or seizure might be related to MAS, a widespread coagulopathy that occurs in about 50% of patients and can be fatal.
Another common finding is asymmetry of the jaw. According to Dr. Cron, up to 80% of children with all forms of arthritis, including systemic JIA, can have arthritis of the temporomandibular joint (TMJ). This is often a subtle finding that can be overlooked until significant erosion has occurred. However, early TMJ arthritis can be detected with magnetic resonance screening.
Dr. Cron is a consultant for Genentech.
NEW YORK - Early and aggressive therapy is warranted in the management of systemic juvenile idiopathic arthritis, which accounts for 10% of arthritis cases in children and which can be devastating, according to Dr. Randy Q. Cron, who spoke about the condition at a meeting sponsored by New York University.
Dr. Cron, director of pediatric rheumatology at the University of Alabama at Birmingham, is a coauthor of the first ever 2011 ACR Recommendations for the Treatment of Juvenile Idiopathic Arthritis (JIA) that were published on April 1 (Arthritis Care Res. 2011;63:465-82) and which include guidelines for the treatment of systemic JIA. According to the publication, "the JIA category of systemic arthritis proved to be especially challenging to evaluate. Attempts to exhaustively depict the myriad possible clinical presentations of systemic arthritis were impractical." The result was that systemic JIA guidelines include two treatment algorithms, one for the active systemic features (for example, fever) and one for the active arthritis. Patients who have both concurrent active systemic features and active arthritis can be treated according to suggestions from both algorithms.
For treating the systemic features of systemic JIA, three first-line options are available: nonsteroidal anti-inflammatory drugs (NSAIDs), systemic corticosteroids, and anakinra (Kineret). It should be noted that interleukin-6 inhibitors and interleukin-1 inhibitors other than anakinra were not considered when developing the recommendations, since they were not widely available commercially at the time. TNF-alpha inhibitors were not considered because of their relatively poor effectiveness for the systemic manifestations.
While NSAID monotherapy might also help some children, the recommendations say that its use is inappropriate for patients with active fever and physician global assessment of overall disease activity (MD global) score equal to 7 of 10. It should also not be used for more than 1 month in patients with active fever.
Systemic glucocorticoids were recommended as initial therapy for patients with active fever and an MD global score of 7. Systemic glucocorticoids may also be added after 2 weeks of NSAID monotherapy in patients with active fever.
Anakinra was recommended for all patients with active fever who had poor prognostic features, no matter what they were currently taking. Anakinra was also recommended for patients who have or develop fever while on systemic glucocorticoids. Anakinra is a human recombinant interleukin-1 (IL-1) inhibitor that was approved to treat adults with moderate to severe arthritis who have not had an adequate response to conservative disease-modifying anti-rheumatic drug therapy. It is not currently approved for children with JIA.
Medications such as calcineurin inhibitors, intravenous immunoglobulin, methotrexate, and thalidomide were not formally recommended but might have roles in the treatment of systemic JIA.
For the treatment of the arthritic features of systemic JIA, initiation of NSAID monotherapy (with or without glucocorticoid joint injections) was recommended for all patients, regardless of the level of disease activity or presence of poor prognostic features. It was assumed that most patients with newly diagnosed systemic arthritis would have been started with NSAIDs. The next step would be methotrexate for all patients with active arthritis who were taking NSAID monotherapy for less than 1 month. If escalation of therapy was needed, the next choices would be either addition of a TNF-alpha inhibitor or anakinra. The recommendations indicate that initiation of anakinra should take place early rather than later in the disease course. For patients with moderate or high disease activity, regardless of features of poor prognosis, it was suggested that patients be switched from anakinra to a TNF-alpha inhibitor.
The last option is abatacept, a costimulatory blocker, for those who fail to be controlled by methotrexate or 4 months of a TNF-alpha inhibitor, and who have moderate to high disease activity.
During his presentation, Dr. Cron provided his perspective on the new systemic JIA recommendations. Most pediatric rheumatologists would probably begin with systemic corticosteroids at the onset of treatment, because that is what they are most familiar with and they are effective, he said.
He was pleased with the inclusion of anakinra in the guidelines. "With these guidelines, physicians can treat children with JIA from the start with anakinra." Inclusion of anakinra in the guidelines as a first option should help tremendously in helping patients receive reimbursement from insurance companies, he noted.
Dr. Cron cited evidence showing the effectiveness of anakinra as a first-line disease-modifying therapy in 46 patients with JIA, preventing refractory arthritis in almost 90% of patients (Arthritis Rheum. 2011;63:545-55). In this series of case studies, use of anakinra reduced elevated sedimentation rates and ferritin levels, decreased the number of active joint counts, and significantly lowered the dose of concomitant steroid therapy. "Kids can get remarkably better within days or months with anakinra. It can be a wonder drug for very sick children with systemic JIA," he said.
Although it was not discussed in the Recommendations, in his talk Dr. Cron said that anakinra is "revolutionizing" the treatment of macrophage activation syndrome (MAS) in systemic JIA (Rheumatology 2011;50:417-9) Dr. Cron reported that he has used anakinra as initial therapy to treat two children with MAS, and he found dramatic reductions in ferritin levels and liver enzymes within 2 days of initiation.
Systemic JIA is a relatively rare disease, with a prevalence of about 1 in 10,000. It has multiple systemic manifestations, including a high fever (which follows a unique pattern: typically once-daily, late-afternoon spikes in 30% of patients at presentation), rash, hepatosplenomegaly, lymphadenopathy, pericarditis, pleural effusion, pulmonary vasculitis, and even CNS stroke or seizure. Stroke or seizure might be related to MAS, a widespread coagulopathy that occurs in about 50% of patients and can be fatal.
Another common finding is asymmetry of the jaw. According to Dr. Cron, up to 80% of children with all forms of arthritis, including systemic JIA, can have arthritis of the temporomandibular joint (TMJ). This is often a subtle finding that can be overlooked until significant erosion has occurred. However, early TMJ arthritis can be detected with magnetic resonance screening.
Dr. Cron is a consultant for Genentech.
A MEETING SPONSORED BY NEW YORK UNIVERSITY