User login
Digestive Disease Week (DDW 2014)
Study takes air out of pre–bypass intragastric balloon
CHICAGO – Treatment with an intragastric balloon before bariatric surgery produces significant weight loss in the morbidly obese, but at the cost of more surgical complications, according to the prospective randomized BIGPOM trial.
Preoperative weight loss is recommended, and in some cases mandated by third-party payer organizations, to improve comorbidities and postoperative outcomes in the morbidly obese. It’s also been suggested to be a predictive factor of postoperative weight loss, but overall the data are limited and methodological quality of prospective studies in this area poor, Dr. Benoit Coffin said at the annual Digestive Disease Week.
The prospective BIGPOM (Balloon Intra Gastrique Préopératoire Pour Obésité Morbide) study randomly assigned 60 patients to standard medical care and 55 to an air- or water-filled intragastric balloon (IGB) for 6 months before laparoscopic bypass surgery. Their average body mass index was 54.7 kg/m2 and 53.9 kg/m2, respectively. The average balloon insertion time was 17 minutes and average age in both groups was 40 years.
Weight loss in months 0-6 was significantly higher in the IGB group, compared with standard care (–10.3 kg vs. –3.0 kg; P less than .0001), but did not differ from months 6-12 (–30.4 kg vs. –35.1 kg) or overall from months 0-12 (–40.7 kg vs. –38.1 kg), said Dr. Coffin, of Hôpital Louis Mourier, Colombes, France.
There was no significant difference in weight loss between those treated with water- or air-filled balloons (P = .0.96).
"Preoperative weight loss is not a predictive factor of postoperative weight loss," he said.
Treatment with an IGB, compared with standard care, did not modify operative time (188 minutes vs. 175 minutes), ICU stays greater than 24 hours (71.4% vs. 76%), mean hospital stay (6.8 vs. 6.3 days), or need for laparotomy (2 vs. 1).
Surgical complications, however, were significantly higher with IGB (5 vs. 0; P = .02), and included digestive hemorrhage, intraperitoneal abscess, fistula, peritonitis, occlusion, and reintervention, Dr. Coffin said.
It is not possible to conclude if the increase in complications noted in the group of patients treated by intragastric balloon is the consequence of the balloon itself or the weight loss it induced, he said.
There were no deaths or medical complications in the IGB group, although two patients experienced complications during IGB removal and one patient required surgical removal of the balloon.
Dr. Coffin reported financial relationships with Shire Pharmaceuticals, Cephalon, Almirall, Gerson Lehrman Group, Mayoly-Spindler, and Mundipharma.
CHICAGO – Treatment with an intragastric balloon before bariatric surgery produces significant weight loss in the morbidly obese, but at the cost of more surgical complications, according to the prospective randomized BIGPOM trial.
Preoperative weight loss is recommended, and in some cases mandated by third-party payer organizations, to improve comorbidities and postoperative outcomes in the morbidly obese. It’s also been suggested to be a predictive factor of postoperative weight loss, but overall the data are limited and methodological quality of prospective studies in this area poor, Dr. Benoit Coffin said at the annual Digestive Disease Week.
The prospective BIGPOM (Balloon Intra Gastrique Préopératoire Pour Obésité Morbide) study randomly assigned 60 patients to standard medical care and 55 to an air- or water-filled intragastric balloon (IGB) for 6 months before laparoscopic bypass surgery. Their average body mass index was 54.7 kg/m2 and 53.9 kg/m2, respectively. The average balloon insertion time was 17 minutes and average age in both groups was 40 years.
Weight loss in months 0-6 was significantly higher in the IGB group, compared with standard care (–10.3 kg vs. –3.0 kg; P less than .0001), but did not differ from months 6-12 (–30.4 kg vs. –35.1 kg) or overall from months 0-12 (–40.7 kg vs. –38.1 kg), said Dr. Coffin, of Hôpital Louis Mourier, Colombes, France.
There was no significant difference in weight loss between those treated with water- or air-filled balloons (P = .0.96).
"Preoperative weight loss is not a predictive factor of postoperative weight loss," he said.
Treatment with an IGB, compared with standard care, did not modify operative time (188 minutes vs. 175 minutes), ICU stays greater than 24 hours (71.4% vs. 76%), mean hospital stay (6.8 vs. 6.3 days), or need for laparotomy (2 vs. 1).
Surgical complications, however, were significantly higher with IGB (5 vs. 0; P = .02), and included digestive hemorrhage, intraperitoneal abscess, fistula, peritonitis, occlusion, and reintervention, Dr. Coffin said.
It is not possible to conclude if the increase in complications noted in the group of patients treated by intragastric balloon is the consequence of the balloon itself or the weight loss it induced, he said.
There were no deaths or medical complications in the IGB group, although two patients experienced complications during IGB removal and one patient required surgical removal of the balloon.
Dr. Coffin reported financial relationships with Shire Pharmaceuticals, Cephalon, Almirall, Gerson Lehrman Group, Mayoly-Spindler, and Mundipharma.
CHICAGO – Treatment with an intragastric balloon before bariatric surgery produces significant weight loss in the morbidly obese, but at the cost of more surgical complications, according to the prospective randomized BIGPOM trial.
Preoperative weight loss is recommended, and in some cases mandated by third-party payer organizations, to improve comorbidities and postoperative outcomes in the morbidly obese. It’s also been suggested to be a predictive factor of postoperative weight loss, but overall the data are limited and methodological quality of prospective studies in this area poor, Dr. Benoit Coffin said at the annual Digestive Disease Week.
The prospective BIGPOM (Balloon Intra Gastrique Préopératoire Pour Obésité Morbide) study randomly assigned 60 patients to standard medical care and 55 to an air- or water-filled intragastric balloon (IGB) for 6 months before laparoscopic bypass surgery. Their average body mass index was 54.7 kg/m2 and 53.9 kg/m2, respectively. The average balloon insertion time was 17 minutes and average age in both groups was 40 years.
Weight loss in months 0-6 was significantly higher in the IGB group, compared with standard care (–10.3 kg vs. –3.0 kg; P less than .0001), but did not differ from months 6-12 (–30.4 kg vs. –35.1 kg) or overall from months 0-12 (–40.7 kg vs. –38.1 kg), said Dr. Coffin, of Hôpital Louis Mourier, Colombes, France.
There was no significant difference in weight loss between those treated with water- or air-filled balloons (P = .0.96).
"Preoperative weight loss is not a predictive factor of postoperative weight loss," he said.
Treatment with an IGB, compared with standard care, did not modify operative time (188 minutes vs. 175 minutes), ICU stays greater than 24 hours (71.4% vs. 76%), mean hospital stay (6.8 vs. 6.3 days), or need for laparotomy (2 vs. 1).
Surgical complications, however, were significantly higher with IGB (5 vs. 0; P = .02), and included digestive hemorrhage, intraperitoneal abscess, fistula, peritonitis, occlusion, and reintervention, Dr. Coffin said.
It is not possible to conclude if the increase in complications noted in the group of patients treated by intragastric balloon is the consequence of the balloon itself or the weight loss it induced, he said.
There were no deaths or medical complications in the IGB group, although two patients experienced complications during IGB removal and one patient required surgical removal of the balloon.
Dr. Coffin reported financial relationships with Shire Pharmaceuticals, Cephalon, Almirall, Gerson Lehrman Group, Mayoly-Spindler, and Mundipharma.
AT DDW 2014
Key clinical point: A temporary intragastric balloon helps reduce weight in the morbidly obese awaiting bypass surgery, but induces morbidity after surgery.
Major finding: IGB patients lost more weight in months 0-6 than did controls (–10.3 kg vs. –3.0 kg; P less than .0001), but had more surgical complications (5 vs. 0; P = .02).
Data source: A prospective study in 115 morbidly obese patients.
Disclosures: Dr. Coffin reported financial relationships with Shire Pharmaceuticals, Cephalon, Almirall, Gerson Lehrman Group, Mayoly-Spindler, and Mundipharma.
Early endoscopic follow-up nets dysplasia in 9.5% of Barrett’s
CHICAGO – Early endoscopic follow-up within 24 months detected dysplasia in nearly one in 10 patients with nondysplastic or low-grade Barrett’s esophagus in a retrospective study at the Mayo Clinic.
Initial endoscopy missed four cases of high-grade dysplasia or esophageal adenocarcinoma (1.9%) and 16 cases of low-grade dysplasia (7.6%) for an overall miss-rate of 9.5%.
Patients on proton pump inhibitors were less likely to have dysplasia missed than were those off PPIs (20% vs. 52.6%, P = .008).
Those with long- versus short-segment Barrett’s esophagus were more likely to have dysplasia overlooked (85% vs. 53.6%; P = .008; mean 6 mm vs. 4 mm; P = .006), Dr. Kavel Visrodia said at the annual Digestive Disease Week.

Current American College of Gastroenterology (ACG) guidelines recommend early repeat esophagogastroduodenoscopy (EGD) to exclude the presence of missed dysplasia in newly diagnosed nondysplastic Barrett’s esophagus (BE), while the ACG and American Society for Gastrointestinal Endoscopy call for repeat EGD within 6 months for those with low-grade dysplasia.
The yield for repeat EGD has not been established, and only one study exists in the literature, said Dr. Visrodia of the department of medicine, Mayo Clinic, Rochester, Minn.
That study (Dis. Esophagus 2012 Sept. 28. [doi:10.1111/j.1442-2050.2012.01431.x]) showed a miss-rate of 8.2% among 146 patients with newly diagnosed nondysplastic BE. Long-segment BE was the only significant predictor of dysplasia on follow-up (odds ratio, 9.18; P = .008).
The cohort was relatively small and had no long-term follow-up, and with an interval to follow-up of 36 months, "it’s possible that some of these were actually incident cases of dysplasia and not prevalent cases," he said.
To address these gaps, Dr. Visrodia and his colleagues identified 488 BE cases from 1977 to 2011 in the Rochester Epidemiology Project in Olmsted County, Minn. A total of 278 patients were excluded because of high-grade dysplasia (HGD) or esophageal cancer on index endoscopy or repeat endoscopy after 24 months, leaving 181 patients with nondysplastic BE and 29 with low-grade dysplasia (LGD).
Repeat endoscopy within 24 months revealed 2 cases of HGD or cancer and 16 cases of LGD in the nondysplastic BE group, and 2 cases of HGD or cancer in the LGD group, Dr. Visrodia said.
Three of the four HGD/cancer cases were in patients with long-segment BE, defined as at least 3 cm of columnar mucosa.
Biopsies were insufficient in 63% of patients with missed dysplasia, compared with 55% in the group without missed dysplasia. Biopsies were considered adequate if the number of biopsies divided by the BE length was at least 2, indicating that samples were taken every 2 cm in accordance with guidelines. This risk factor is noteworthy, although the difference between groups was not statistically significant, possibly because of the small sample size, he said.
Finally, after a median of 6.8 years of follow-up, 30 asymptomatic, prevalent HGDs or cancers were detected within 24 months, compared with 22 incident cases detected after 24 months. This suggests that "a greater number of high-grade dysplasias and cancers were detected up front rather than during long-term careful surveillance," Dr. Visrodia said.
During a discussion of the study, one attendee asked whether the results make a better case for aggressive ablation up front rather than for surveillance, while others expressed surprise at the high miss rate at an institution such as the Mayo Clinic.
Dr. Visrodia replied that the results do give them pause, and suggested that tighter early endoscopic surveillance may be warranted, particularly in those with long-segment BE.
Dr. Visrodia and his coauthors reported no financial disclosures.
CHICAGO – Early endoscopic follow-up within 24 months detected dysplasia in nearly one in 10 patients with nondysplastic or low-grade Barrett’s esophagus in a retrospective study at the Mayo Clinic.
Initial endoscopy missed four cases of high-grade dysplasia or esophageal adenocarcinoma (1.9%) and 16 cases of low-grade dysplasia (7.6%) for an overall miss-rate of 9.5%.
Patients on proton pump inhibitors were less likely to have dysplasia missed than were those off PPIs (20% vs. 52.6%, P = .008).
Those with long- versus short-segment Barrett’s esophagus were more likely to have dysplasia overlooked (85% vs. 53.6%; P = .008; mean 6 mm vs. 4 mm; P = .006), Dr. Kavel Visrodia said at the annual Digestive Disease Week.
Current American College of Gastroenterology (ACG) guidelines recommend early repeat esophagogastroduodenoscopy (EGD) to exclude the presence of missed dysplasia in newly diagnosed nondysplastic Barrett’s esophagus (BE), while the ACG and American Society for Gastrointestinal Endoscopy call for repeat EGD within 6 months for those with low-grade dysplasia.
The yield for repeat EGD has not been established, and only one study exists in the literature, said Dr. Visrodia of the department of medicine, Mayo Clinic, Rochester, Minn.
That study (Dis. Esophagus 2012 Sept. 28. [doi:10.1111/j.1442-2050.2012.01431.x]) showed a miss-rate of 8.2% among 146 patients with newly diagnosed nondysplastic BE. Long-segment BE was the only significant predictor of dysplasia on follow-up (odds ratio, 9.18; P = .008).
The cohort was relatively small and had no long-term follow-up, and with an interval to follow-up of 36 months, "it’s possible that some of these were actually incident cases of dysplasia and not prevalent cases," he said.
To address these gaps, Dr. Visrodia and his colleagues identified 488 BE cases from 1977 to 2011 in the Rochester Epidemiology Project in Olmsted County, Minn. A total of 278 patients were excluded because of high-grade dysplasia (HGD) or esophageal cancer on index endoscopy or repeat endoscopy after 24 months, leaving 181 patients with nondysplastic BE and 29 with low-grade dysplasia (LGD).
Repeat endoscopy within 24 months revealed 2 cases of HGD or cancer and 16 cases of LGD in the nondysplastic BE group, and 2 cases of HGD or cancer in the LGD group, Dr. Visrodia said.
Three of the four HGD/cancer cases were in patients with long-segment BE, defined as at least 3 cm of columnar mucosa.
Biopsies were insufficient in 63% of patients with missed dysplasia, compared with 55% in the group without missed dysplasia. Biopsies were considered adequate if the number of biopsies divided by the BE length was at least 2, indicating that samples were taken every 2 cm in accordance with guidelines. This risk factor is noteworthy, although the difference between groups was not statistically significant, possibly because of the small sample size, he said.
Finally, after a median of 6.8 years of follow-up, 30 asymptomatic, prevalent HGDs or cancers were detected within 24 months, compared with 22 incident cases detected after 24 months. This suggests that "a greater number of high-grade dysplasias and cancers were detected up front rather than during long-term careful surveillance," Dr. Visrodia said.
During a discussion of the study, one attendee asked whether the results make a better case for aggressive ablation up front rather than for surveillance, while others expressed surprise at the high miss rate at an institution such as the Mayo Clinic.
Dr. Visrodia replied that the results do give them pause, and suggested that tighter early endoscopic surveillance may be warranted, particularly in those with long-segment BE.
Dr. Visrodia and his coauthors reported no financial disclosures.
CHICAGO – Early endoscopic follow-up within 24 months detected dysplasia in nearly one in 10 patients with nondysplastic or low-grade Barrett’s esophagus in a retrospective study at the Mayo Clinic.
Initial endoscopy missed four cases of high-grade dysplasia or esophageal adenocarcinoma (1.9%) and 16 cases of low-grade dysplasia (7.6%) for an overall miss-rate of 9.5%.
Patients on proton pump inhibitors were less likely to have dysplasia missed than were those off PPIs (20% vs. 52.6%, P = .008).
Those with long- versus short-segment Barrett’s esophagus were more likely to have dysplasia overlooked (85% vs. 53.6%; P = .008; mean 6 mm vs. 4 mm; P = .006), Dr. Kavel Visrodia said at the annual Digestive Disease Week.
Current American College of Gastroenterology (ACG) guidelines recommend early repeat esophagogastroduodenoscopy (EGD) to exclude the presence of missed dysplasia in newly diagnosed nondysplastic Barrett’s esophagus (BE), while the ACG and American Society for Gastrointestinal Endoscopy call for repeat EGD within 6 months for those with low-grade dysplasia.
The yield for repeat EGD has not been established, and only one study exists in the literature, said Dr. Visrodia of the department of medicine, Mayo Clinic, Rochester, Minn.
That study (Dis. Esophagus 2012 Sept. 28. [doi:10.1111/j.1442-2050.2012.01431.x]) showed a miss-rate of 8.2% among 146 patients with newly diagnosed nondysplastic BE. Long-segment BE was the only significant predictor of dysplasia on follow-up (odds ratio, 9.18; P = .008).
The cohort was relatively small and had no long-term follow-up, and with an interval to follow-up of 36 months, "it’s possible that some of these were actually incident cases of dysplasia and not prevalent cases," he said.
To address these gaps, Dr. Visrodia and his colleagues identified 488 BE cases from 1977 to 2011 in the Rochester Epidemiology Project in Olmsted County, Minn. A total of 278 patients were excluded because of high-grade dysplasia (HGD) or esophageal cancer on index endoscopy or repeat endoscopy after 24 months, leaving 181 patients with nondysplastic BE and 29 with low-grade dysplasia (LGD).
Repeat endoscopy within 24 months revealed 2 cases of HGD or cancer and 16 cases of LGD in the nondysplastic BE group, and 2 cases of HGD or cancer in the LGD group, Dr. Visrodia said.
Three of the four HGD/cancer cases were in patients with long-segment BE, defined as at least 3 cm of columnar mucosa.
Biopsies were insufficient in 63% of patients with missed dysplasia, compared with 55% in the group without missed dysplasia. Biopsies were considered adequate if the number of biopsies divided by the BE length was at least 2, indicating that samples were taken every 2 cm in accordance with guidelines. This risk factor is noteworthy, although the difference between groups was not statistically significant, possibly because of the small sample size, he said.
Finally, after a median of 6.8 years of follow-up, 30 asymptomatic, prevalent HGDs or cancers were detected within 24 months, compared with 22 incident cases detected after 24 months. This suggests that "a greater number of high-grade dysplasias and cancers were detected up front rather than during long-term careful surveillance," Dr. Visrodia said.
During a discussion of the study, one attendee asked whether the results make a better case for aggressive ablation up front rather than for surveillance, while others expressed surprise at the high miss rate at an institution such as the Mayo Clinic.
Dr. Visrodia replied that the results do give them pause, and suggested that tighter early endoscopic surveillance may be warranted, particularly in those with long-segment BE.
Dr. Visrodia and his coauthors reported no financial disclosures.
AT DDW 2014
Key clinical point: Early endoscopic follow-up catches 9.5% of dysplasia missed on initial exam, but is not universally recommended by medical societies.
Major finding: Index endoscopy missed 1.9% of high-grade dysplasia or esophageal cancer and 7.6% of low-grade dysplasia.
Data source: A retrospective study in 181 patients with Barrett’s esophagus.
Disclosures: Dr. Visrodia and his coauthors reported no financial disclosures.

Fecal transplant falls short in UC, but may not be the end
CHICAGO – Results were negative from the first randomized placebo-controlled trial of fecal microbiota transplant in ulcerative colitis, but enthusiasm remains for this newly regulated and trendy therapy.
"We need to get more data and we need to understand how better to use this approach, but I don’t think this study is telling us we should stop exploring. I still think it is an interesting avenue that we need to evaluate," study author Dr. Paul Moayyedi said during a late-breaking abstract session at Digestive Disease Week.

Part of the enthusiasm for what was described as "the new kid on the block for altering gut flora," has been fueled by a roughly 90% success rate for fecal transplant in treating Clostridium difficile infection.
Dr. Moayyedi also showcased a success story from the trial that "typifies a few patients in this study." The patient had ulcerative colitis for almost 20 years that was unresponsive to steroids and 5-aminosalicylic acid for 2 years before the study and so severe it caused bloody diarrhea 10-20 times per day. No improvement was seen after 6 weeks of placebo therapy and his Mayo Clinic score was "about as bad as it can be" at 12.
After crossing over to 6 weeks of open-label fecal microbiota transplantation (FMT), symptoms were much improved and his Mayo score dropped to 5. After 20 weeks, mucosa healed throughout the colon, his Mayo score reached 0, and he was "fine" on no medication.
"What we’re finding is that 6 weeks is usually not enough and that if you continue longer, you can get remission in some patients," said Dr. Moayyedi, the Richard Hunt-Astra Zeneca Chair in Gastroenterology, McMaster University, Hamilton, Ontario.
He went on to say, "I’m a very big proponent of evidenced-based medicine, but cases like these make you think something must be going on in some patients, but we don’t have the funding to do the 500-patient trial you need to get that signal."
Dr. Moayyedi and his associates enrolled ambulatory patients with active ulcerative colitis, defined as a Mayo score of at least 4 and an endoscopic Mayo score of at least 1, who tested negative for the C. difficile gene. Patients could be on ulcerative colitis medications, if doses were stable for at least 12 weeks but had to be off antibiotics for 30 days.
Patients were randomly assigned to receive a 50-mL retention enema containing fecal microbiota from an anonymous donor or water, once per week for 6 weeks. The primary outcome was remission of ulcerative colitis, defined as a Mayo score of 2 or less and an endoscopic Mayo score of 0 at week 7. Patients, clinicians, and investigators were blinded to therapy.
The 31 FMT and 30 placebo patients were well matched at baseline, except for significantly more pancolitis in the FMT group (64% vs. 36%).
Remission was achieved by seven FMT patients (23%) and two placebo patients (7%), which was not significantly different (P = .15), Dr. Moayyedi said.
There also were no differences between the FMT and placebo groups in any of the secondary outcomes: 6-week Mayo score (6.36 vs. 6.30; P = .95), 6-week Inflammatory Bowel Disease Questionnaire (148.4 vs. 146.4; P = .85), and 6-week EQ-5D health questionnaire (61.0 vs. 66.2; P = .34).
Based on these findings, the study is being stopped for futility, he said.
There were no major adverse events, although the diagnosis changed to Crohn’s colitis for two patients given FMT and one on placebo.
This was "much bigger than you’d expect," Dr. Moayyedi said. "We’re not sure why, and of course the worry is that FMT may change the phenotype, which would not be good."
If FMT is to succeed in ulcerative colitis, he suggested more data will be needed on the fecal microbiome and the best approach to administer FMT, including donor selection, timing, preparation, and duration of treatment. The group has not done an analysis of enema dwell time and response, and no signal was seen that FMT is more effective in left-sided disease.
More detailed microbiome analyses of the patient highlighted during the talk, however, revealed the man had a "very diverse and unstable" microbiome at baseline that gradually became more stable, "with a loss of Ruminococcus that seems to be a feature of improvement and a movement toward the phenotype of the donor," Dr. Moayyedi said.
During a discussion of the results, some audience members expressed concern that FMT might change the phenotype, while others were more enthusiastic about the therapy.
Dr. Scott Harris, a gastroenterologist and professor of medicine, Georgetown University Medical Center, Washington, said the trial was likely underpowered, and that by including patients with less severe disease, it may have been more difficult to see a treatment effect. Other trials have also shown that 6 weeks of therapy may not be enough for refractory patients.
"I’m very optimistic, I wouldn’t stop at this point," he said.
Session cochair Dr. John M. Inadomi, professor of medicine and head of gastroenterology, University of Washington School of Medicine, Seattle, agreed that the length of treatment as well as the study’s use of anonymous donors could have affected results. One of the big questions is whether the donor feces actually grafted and thus affected the recipient.
"If you use the wrong donor stool, if the stool didn’t graft, these kinds of things can obviously make a negative result, even if the concept is potentially fine," he said in an interview.
Last year, the Food and Drug Administration moved to require an investigational new drug permit to treat C. difficile with fecal microbiota, but changed course within weeks citing public pressure. While researchers are studying the potential to deliver feces via capsule, Dr. Moayyedi observed that some enthusiasts are offering Internet advice on how to mix your own FMT at home.
Dr. Moayyedi reported financial ties with AstraZeneca Pharmaceuticals, Forest Laboratories, and Shire Canada. Dr. Harris reported no conflicting interests. Dr. Inadomi reported financial relationships with Given Imaging, ChemImage, Cernostics, and Epigenomics.
CHICAGO – Results were negative from the first randomized placebo-controlled trial of fecal microbiota transplant in ulcerative colitis, but enthusiasm remains for this newly regulated and trendy therapy.
"We need to get more data and we need to understand how better to use this approach, but I don’t think this study is telling us we should stop exploring. I still think it is an interesting avenue that we need to evaluate," study author Dr. Paul Moayyedi said during a late-breaking abstract session at Digestive Disease Week.
Part of the enthusiasm for what was described as "the new kid on the block for altering gut flora," has been fueled by a roughly 90% success rate for fecal transplant in treating Clostridium difficile infection.
Dr. Moayyedi also showcased a success story from the trial that "typifies a few patients in this study." The patient had ulcerative colitis for almost 20 years that was unresponsive to steroids and 5-aminosalicylic acid for 2 years before the study and so severe it caused bloody diarrhea 10-20 times per day. No improvement was seen after 6 weeks of placebo therapy and his Mayo Clinic score was "about as bad as it can be" at 12.
After crossing over to 6 weeks of open-label fecal microbiota transplantation (FMT), symptoms were much improved and his Mayo score dropped to 5. After 20 weeks, mucosa healed throughout the colon, his Mayo score reached 0, and he was "fine" on no medication.
"What we’re finding is that 6 weeks is usually not enough and that if you continue longer, you can get remission in some patients," said Dr. Moayyedi, the Richard Hunt-Astra Zeneca Chair in Gastroenterology, McMaster University, Hamilton, Ontario.
He went on to say, "I’m a very big proponent of evidenced-based medicine, but cases like these make you think something must be going on in some patients, but we don’t have the funding to do the 500-patient trial you need to get that signal."
Dr. Moayyedi and his associates enrolled ambulatory patients with active ulcerative colitis, defined as a Mayo score of at least 4 and an endoscopic Mayo score of at least 1, who tested negative for the C. difficile gene. Patients could be on ulcerative colitis medications, if doses were stable for at least 12 weeks but had to be off antibiotics for 30 days.
Patients were randomly assigned to receive a 50-mL retention enema containing fecal microbiota from an anonymous donor or water, once per week for 6 weeks. The primary outcome was remission of ulcerative colitis, defined as a Mayo score of 2 or less and an endoscopic Mayo score of 0 at week 7. Patients, clinicians, and investigators were blinded to therapy.
The 31 FMT and 30 placebo patients were well matched at baseline, except for significantly more pancolitis in the FMT group (64% vs. 36%).
Remission was achieved by seven FMT patients (23%) and two placebo patients (7%), which was not significantly different (P = .15), Dr. Moayyedi said.
There also were no differences between the FMT and placebo groups in any of the secondary outcomes: 6-week Mayo score (6.36 vs. 6.30; P = .95), 6-week Inflammatory Bowel Disease Questionnaire (148.4 vs. 146.4; P = .85), and 6-week EQ-5D health questionnaire (61.0 vs. 66.2; P = .34).
Based on these findings, the study is being stopped for futility, he said.
There were no major adverse events, although the diagnosis changed to Crohn’s colitis for two patients given FMT and one on placebo.
This was "much bigger than you’d expect," Dr. Moayyedi said. "We’re not sure why, and of course the worry is that FMT may change the phenotype, which would not be good."
If FMT is to succeed in ulcerative colitis, he suggested more data will be needed on the fecal microbiome and the best approach to administer FMT, including donor selection, timing, preparation, and duration of treatment. The group has not done an analysis of enema dwell time and response, and no signal was seen that FMT is more effective in left-sided disease.
More detailed microbiome analyses of the patient highlighted during the talk, however, revealed the man had a "very diverse and unstable" microbiome at baseline that gradually became more stable, "with a loss of Ruminococcus that seems to be a feature of improvement and a movement toward the phenotype of the donor," Dr. Moayyedi said.
During a discussion of the results, some audience members expressed concern that FMT might change the phenotype, while others were more enthusiastic about the therapy.
Dr. Scott Harris, a gastroenterologist and professor of medicine, Georgetown University Medical Center, Washington, said the trial was likely underpowered, and that by including patients with less severe disease, it may have been more difficult to see a treatment effect. Other trials have also shown that 6 weeks of therapy may not be enough for refractory patients.
"I’m very optimistic, I wouldn’t stop at this point," he said.
Session cochair Dr. John M. Inadomi, professor of medicine and head of gastroenterology, University of Washington School of Medicine, Seattle, agreed that the length of treatment as well as the study’s use of anonymous donors could have affected results. One of the big questions is whether the donor feces actually grafted and thus affected the recipient.
"If you use the wrong donor stool, if the stool didn’t graft, these kinds of things can obviously make a negative result, even if the concept is potentially fine," he said in an interview.
Last year, the Food and Drug Administration moved to require an investigational new drug permit to treat C. difficile with fecal microbiota, but changed course within weeks citing public pressure. While researchers are studying the potential to deliver feces via capsule, Dr. Moayyedi observed that some enthusiasts are offering Internet advice on how to mix your own FMT at home.
Dr. Moayyedi reported financial ties with AstraZeneca Pharmaceuticals, Forest Laboratories, and Shire Canada. Dr. Harris reported no conflicting interests. Dr. Inadomi reported financial relationships with Given Imaging, ChemImage, Cernostics, and Epigenomics.
CHICAGO – Results were negative from the first randomized placebo-controlled trial of fecal microbiota transplant in ulcerative colitis, but enthusiasm remains for this newly regulated and trendy therapy.
"We need to get more data and we need to understand how better to use this approach, but I don’t think this study is telling us we should stop exploring. I still think it is an interesting avenue that we need to evaluate," study author Dr. Paul Moayyedi said during a late-breaking abstract session at Digestive Disease Week.
Part of the enthusiasm for what was described as "the new kid on the block for altering gut flora," has been fueled by a roughly 90% success rate for fecal transplant in treating Clostridium difficile infection.
Dr. Moayyedi also showcased a success story from the trial that "typifies a few patients in this study." The patient had ulcerative colitis for almost 20 years that was unresponsive to steroids and 5-aminosalicylic acid for 2 years before the study and so severe it caused bloody diarrhea 10-20 times per day. No improvement was seen after 6 weeks of placebo therapy and his Mayo Clinic score was "about as bad as it can be" at 12.
After crossing over to 6 weeks of open-label fecal microbiota transplantation (FMT), symptoms were much improved and his Mayo score dropped to 5. After 20 weeks, mucosa healed throughout the colon, his Mayo score reached 0, and he was "fine" on no medication.
"What we’re finding is that 6 weeks is usually not enough and that if you continue longer, you can get remission in some patients," said Dr. Moayyedi, the Richard Hunt-Astra Zeneca Chair in Gastroenterology, McMaster University, Hamilton, Ontario.
He went on to say, "I’m a very big proponent of evidenced-based medicine, but cases like these make you think something must be going on in some patients, but we don’t have the funding to do the 500-patient trial you need to get that signal."
Dr. Moayyedi and his associates enrolled ambulatory patients with active ulcerative colitis, defined as a Mayo score of at least 4 and an endoscopic Mayo score of at least 1, who tested negative for the C. difficile gene. Patients could be on ulcerative colitis medications, if doses were stable for at least 12 weeks but had to be off antibiotics for 30 days.
Patients were randomly assigned to receive a 50-mL retention enema containing fecal microbiota from an anonymous donor or water, once per week for 6 weeks. The primary outcome was remission of ulcerative colitis, defined as a Mayo score of 2 or less and an endoscopic Mayo score of 0 at week 7. Patients, clinicians, and investigators were blinded to therapy.
The 31 FMT and 30 placebo patients were well matched at baseline, except for significantly more pancolitis in the FMT group (64% vs. 36%).
Remission was achieved by seven FMT patients (23%) and two placebo patients (7%), which was not significantly different (P = .15), Dr. Moayyedi said.
There also were no differences between the FMT and placebo groups in any of the secondary outcomes: 6-week Mayo score (6.36 vs. 6.30; P = .95), 6-week Inflammatory Bowel Disease Questionnaire (148.4 vs. 146.4; P = .85), and 6-week EQ-5D health questionnaire (61.0 vs. 66.2; P = .34).
Based on these findings, the study is being stopped for futility, he said.
There were no major adverse events, although the diagnosis changed to Crohn’s colitis for two patients given FMT and one on placebo.
This was "much bigger than you’d expect," Dr. Moayyedi said. "We’re not sure why, and of course the worry is that FMT may change the phenotype, which would not be good."
If FMT is to succeed in ulcerative colitis, he suggested more data will be needed on the fecal microbiome and the best approach to administer FMT, including donor selection, timing, preparation, and duration of treatment. The group has not done an analysis of enema dwell time and response, and no signal was seen that FMT is more effective in left-sided disease.
More detailed microbiome analyses of the patient highlighted during the talk, however, revealed the man had a "very diverse and unstable" microbiome at baseline that gradually became more stable, "with a loss of Ruminococcus that seems to be a feature of improvement and a movement toward the phenotype of the donor," Dr. Moayyedi said.
During a discussion of the results, some audience members expressed concern that FMT might change the phenotype, while others were more enthusiastic about the therapy.
Dr. Scott Harris, a gastroenterologist and professor of medicine, Georgetown University Medical Center, Washington, said the trial was likely underpowered, and that by including patients with less severe disease, it may have been more difficult to see a treatment effect. Other trials have also shown that 6 weeks of therapy may not be enough for refractory patients.
"I’m very optimistic, I wouldn’t stop at this point," he said.
Session cochair Dr. John M. Inadomi, professor of medicine and head of gastroenterology, University of Washington School of Medicine, Seattle, agreed that the length of treatment as well as the study’s use of anonymous donors could have affected results. One of the big questions is whether the donor feces actually grafted and thus affected the recipient.
"If you use the wrong donor stool, if the stool didn’t graft, these kinds of things can obviously make a negative result, even if the concept is potentially fine," he said in an interview.
Last year, the Food and Drug Administration moved to require an investigational new drug permit to treat C. difficile with fecal microbiota, but changed course within weeks citing public pressure. While researchers are studying the potential to deliver feces via capsule, Dr. Moayyedi observed that some enthusiasts are offering Internet advice on how to mix your own FMT at home.
Dr. Moayyedi reported financial ties with AstraZeneca Pharmaceuticals, Forest Laboratories, and Shire Canada. Dr. Harris reported no conflicting interests. Dr. Inadomi reported financial relationships with Given Imaging, ChemImage, Cernostics, and Epigenomics.
AT DDW 2014
Key clinical finding: Fecal microbiota transplant is not ready for ulcerative colitis yet.
Major finding: Remission was achieved by seven FMT patients (23%) and two placebo patients (7%) (P = .15).
Data source: A double-blind, prospective study of 61 patients with ulcerative colitis.
Disclosures: Dr. Moayyedi reported financial ties with AstraZeneca Pharmaceuticals, Forest Laboratories, and Shire Canada. Dr. Harris reported no conflicting interests. Dr. Inadomi reported financial relationships with Given Imaging, ChemImage, Cernostics, and Epigenomics.

Infliximab monitoring during remission limits IBD flare-ups
CHICAGO – Infliximab discontinuation rates were lower in a pilot study of inflammatory bowel disease patients who were in remission and had their serum drug levels measured.
Trough infliximab concentrations were measured in patients who were in clinical remission at about 1 year on maintenance therapy. When trough concentrations fell below 5 mcg/mL, patients got a dose increase. The goal was to prevent secondary loss of response due to recurrence of symptoms or antibody-mediated side effects, such as infusion reactions, Dr. Byron P. Vaughn explained at the annual Digestive Disease Week.

In a retrospective, nonrandomized proof-of-concept study, 48 inflammatory bowel disease patients in remission on infliximab were proactively monitored, and 78 matched controls were conventionally managed. The infliximab discontinuation rate during up to 5 years of follow-up was 10% in the proactively monitored group and 31% in the controls. Nearly 90% of treatment discontinuations in the control group were caused by loss of response or development of acute infusion reactions; in contrast, no one in the proactively monitored group stopped infliximab for those reasons.
"We think this is exciting information. We now recommend dose optimization to a trough level of at least 3 mcg/mL, and our current clinical practice is to target a range of 5-10 mcg/mL," said Dr. Vaughn, of Beth Israel Deaconess Medical Center and Harvard University, Boston. Of course, the findings certainly need to be validated in a prospective study, he added.
For about the first year of infliximab therapy, the treatment continuation curves were similar for the two groups. After 1 year, the curves separated and the benefit of proactive trough monitoring could be seen.
The main reasons for stopping infliximab in the control group were a flare of symptoms (15 patients) and acute infusion reactions (6 patients). Neither of these events occurred in the proactively monitored patients. The reasons for treatment discontinuation in the proactively monitored group included drug-induced lupus (1 patient), psoriasis (1 patient), a delayed infusion reaction (1 patient), and a reason unrelated to the medication (1 patient).
Three-quarters of study participants had Crohn’s disease, and the rest had ulcerative colitis. Other investigators have previously shown that an undetectable serum infliximab trough concentration in the setting of Crohn’s disease often heralds a loss of response.
Dr. Vaughn emphasized that fully one in four patients had an undetectable trough level at the first measurement, and nearly two-thirds had levels below 5 mcg/mL. The infliximab dose was increased in those patients. Patients with a trough level of 5-10 mcg/mL received no change in their regimen, while those with a level of more than 10 mcg/mL on two occasions got either a dose reduction or an increase in the interval between doses if they were on 5 mg/kg.
In gastroenterology, serum infliximab concentrations are typically measured when patients stop responding or have side effects. So the costs of proactive infliximab monitoring and dose escalation are going to be an impediment to widespread implementation of this strategy under many health plans, at least until there is confirmatory data, he said.
Proactive trough concentration monitoring and dose titration are typical in recipients of solid organ transplants receiving cyclosporine, mycophenolate mofetil, and mTOR (mammalian target of rapamycin) inhibitors, and are often performed in sepsis patients receiving vancomycin and gentamicin.
When asked how often he recommends proactive infliximab trough testing, Dr. Vaughn replied, "I think when people are in a steady state – they’re not ill and they’re not flaring – you could probably check once every 6 months or maybe once a year. After a few stable troughs, that could be enough unless there’s been a major change like a big weight change, a change in other medications, or an illness."
Dr. Vaughn reported having no financial conflicts of interest regarding this study.
CHICAGO – Infliximab discontinuation rates were lower in a pilot study of inflammatory bowel disease patients who were in remission and had their serum drug levels measured.
Trough infliximab concentrations were measured in patients who were in clinical remission at about 1 year on maintenance therapy. When trough concentrations fell below 5 mcg/mL, patients got a dose increase. The goal was to prevent secondary loss of response due to recurrence of symptoms or antibody-mediated side effects, such as infusion reactions, Dr. Byron P. Vaughn explained at the annual Digestive Disease Week.
In a retrospective, nonrandomized proof-of-concept study, 48 inflammatory bowel disease patients in remission on infliximab were proactively monitored, and 78 matched controls were conventionally managed. The infliximab discontinuation rate during up to 5 years of follow-up was 10% in the proactively monitored group and 31% in the controls. Nearly 90% of treatment discontinuations in the control group were caused by loss of response or development of acute infusion reactions; in contrast, no one in the proactively monitored group stopped infliximab for those reasons.
"We think this is exciting information. We now recommend dose optimization to a trough level of at least 3 mcg/mL, and our current clinical practice is to target a range of 5-10 mcg/mL," said Dr. Vaughn, of Beth Israel Deaconess Medical Center and Harvard University, Boston. Of course, the findings certainly need to be validated in a prospective study, he added.
For about the first year of infliximab therapy, the treatment continuation curves were similar for the two groups. After 1 year, the curves separated and the benefit of proactive trough monitoring could be seen.
The main reasons for stopping infliximab in the control group were a flare of symptoms (15 patients) and acute infusion reactions (6 patients). Neither of these events occurred in the proactively monitored patients. The reasons for treatment discontinuation in the proactively monitored group included drug-induced lupus (1 patient), psoriasis (1 patient), a delayed infusion reaction (1 patient), and a reason unrelated to the medication (1 patient).
Three-quarters of study participants had Crohn’s disease, and the rest had ulcerative colitis. Other investigators have previously shown that an undetectable serum infliximab trough concentration in the setting of Crohn’s disease often heralds a loss of response.
Dr. Vaughn emphasized that fully one in four patients had an undetectable trough level at the first measurement, and nearly two-thirds had levels below 5 mcg/mL. The infliximab dose was increased in those patients. Patients with a trough level of 5-10 mcg/mL received no change in their regimen, while those with a level of more than 10 mcg/mL on two occasions got either a dose reduction or an increase in the interval between doses if they were on 5 mg/kg.
In gastroenterology, serum infliximab concentrations are typically measured when patients stop responding or have side effects. So the costs of proactive infliximab monitoring and dose escalation are going to be an impediment to widespread implementation of this strategy under many health plans, at least until there is confirmatory data, he said.
Proactive trough concentration monitoring and dose titration are typical in recipients of solid organ transplants receiving cyclosporine, mycophenolate mofetil, and mTOR (mammalian target of rapamycin) inhibitors, and are often performed in sepsis patients receiving vancomycin and gentamicin.
When asked how often he recommends proactive infliximab trough testing, Dr. Vaughn replied, "I think when people are in a steady state – they’re not ill and they’re not flaring – you could probably check once every 6 months or maybe once a year. After a few stable troughs, that could be enough unless there’s been a major change like a big weight change, a change in other medications, or an illness."
Dr. Vaughn reported having no financial conflicts of interest regarding this study.
CHICAGO – Infliximab discontinuation rates were lower in a pilot study of inflammatory bowel disease patients who were in remission and had their serum drug levels measured.
Trough infliximab concentrations were measured in patients who were in clinical remission at about 1 year on maintenance therapy. When trough concentrations fell below 5 mcg/mL, patients got a dose increase. The goal was to prevent secondary loss of response due to recurrence of symptoms or antibody-mediated side effects, such as infusion reactions, Dr. Byron P. Vaughn explained at the annual Digestive Disease Week.
In a retrospective, nonrandomized proof-of-concept study, 48 inflammatory bowel disease patients in remission on infliximab were proactively monitored, and 78 matched controls were conventionally managed. The infliximab discontinuation rate during up to 5 years of follow-up was 10% in the proactively monitored group and 31% in the controls. Nearly 90% of treatment discontinuations in the control group were caused by loss of response or development of acute infusion reactions; in contrast, no one in the proactively monitored group stopped infliximab for those reasons.
"We think this is exciting information. We now recommend dose optimization to a trough level of at least 3 mcg/mL, and our current clinical practice is to target a range of 5-10 mcg/mL," said Dr. Vaughn, of Beth Israel Deaconess Medical Center and Harvard University, Boston. Of course, the findings certainly need to be validated in a prospective study, he added.
For about the first year of infliximab therapy, the treatment continuation curves were similar for the two groups. After 1 year, the curves separated and the benefit of proactive trough monitoring could be seen.
The main reasons for stopping infliximab in the control group were a flare of symptoms (15 patients) and acute infusion reactions (6 patients). Neither of these events occurred in the proactively monitored patients. The reasons for treatment discontinuation in the proactively monitored group included drug-induced lupus (1 patient), psoriasis (1 patient), a delayed infusion reaction (1 patient), and a reason unrelated to the medication (1 patient).
Three-quarters of study participants had Crohn’s disease, and the rest had ulcerative colitis. Other investigators have previously shown that an undetectable serum infliximab trough concentration in the setting of Crohn’s disease often heralds a loss of response.
Dr. Vaughn emphasized that fully one in four patients had an undetectable trough level at the first measurement, and nearly two-thirds had levels below 5 mcg/mL. The infliximab dose was increased in those patients. Patients with a trough level of 5-10 mcg/mL received no change in their regimen, while those with a level of more than 10 mcg/mL on two occasions got either a dose reduction or an increase in the interval between doses if they were on 5 mg/kg.
In gastroenterology, serum infliximab concentrations are typically measured when patients stop responding or have side effects. So the costs of proactive infliximab monitoring and dose escalation are going to be an impediment to widespread implementation of this strategy under many health plans, at least until there is confirmatory data, he said.
Proactive trough concentration monitoring and dose titration are typical in recipients of solid organ transplants receiving cyclosporine, mycophenolate mofetil, and mTOR (mammalian target of rapamycin) inhibitors, and are often performed in sepsis patients receiving vancomycin and gentamicin.
When asked how often he recommends proactive infliximab trough testing, Dr. Vaughn replied, "I think when people are in a steady state – they’re not ill and they’re not flaring – you could probably check once every 6 months or maybe once a year. After a few stable troughs, that could be enough unless there’s been a major change like a big weight change, a change in other medications, or an illness."
Dr. Vaughn reported having no financial conflicts of interest regarding this study.
AT DDW 2014
Key clinical point: Infliximab dose adjustment based on trough concentration measures may avert flares of inflammatory bowel disease.
Major finding: The long-term infliximab discontinuation rate in patients on maintenance therapy was 10% if serum infliximab trough levels were measured with dose optimization based upon the results; the discontinuation rate was 31% in conventionally managed controls.
Data source: A retrospective case-control study of patients in clinical remission; 48 had proactive infliximab trough concentration measures and dose optimization, and 78 patients had standard care.
Disclosures: This study was supported using institutional funds. The presenter reported having no financial conflicts.

PEARL-II: Oral ‘3D’ regimen effective in hepatitis C with prior treatment failure
CHICAGO – The investigational "3D" combination of oral antiviral drugs achieved a cure rate that was nothing short of sensational in the phase III PEARL-II trial involving noncirrhotic patients with hepatitis C genotype 1b who had previously failed pegylated interferon–based therapy.
Moreover, these results were achieved without the use of daily oral ribavirin, formerly a standard part of hepatitis C therapy, and one responsible for a substantial burden of side effects.

Genotype 1b is the most prevalent form of hepatitis C disease worldwide. While it is especially common in Europe and Japan, genotype 1b also accounts for about one-third of cases of HCV in the United States.
The so-called 3D regimen is an all-oral combination of three direct-acting antiviral agents: ombitasvir, dasabuvir, and ABT-450. Each of these agents inhibits the viral life cycle at a different point, Dr. Peter Ferenci explained in presenting the PEARL-II results at the annual Digestive Disease Week.
PEARL-II was an open-label, 12-week trial which asked the question, what added benefit does daily oral ribavirin provide in conjunction with 3D therapy in noncirrhotic HCV genotype 1b patients previously treated unsuccessfully with a pegylated interferon/ribavirin regimen? The answer, as it turns out, is none.
The 186 participants were randomized to the 3D regimen with or without oral ribavirin. The oral 3D regimen consists of ABT-450 (150 mg)/ritonavir (100 mg)/ombitasvir (25 mg), formulated as a single once-daily pill plus dasabuvir at 250 mg twice daily.
The primary endpoint – a sustained virologic response 12 weeks after completion of the 12-week course of treatment – was achieved in 96.6% of the ribavirin group and 100% who didn’t receive ribavirin. Both rates are superior to the historical sustained virologic response rate seen in studies of the former state-of-the-art regimen of telaprevir plus pegylated interferon and ribavirin, noted Dr. Ferenci of the Medical University of Vienna.
There were no virologic failures in PEARL-II. Two patients stopped the study drugs: one who developed pancreatitis deemed unrelated to treatment and another with anxiety, tachycardia, shortness of breath, and fever considered possibly treatment related.
Results of 3D therapy were equally good in men and women and in former relapsers, nonresponders, and partial responders to pegylated interferon-based regimens.
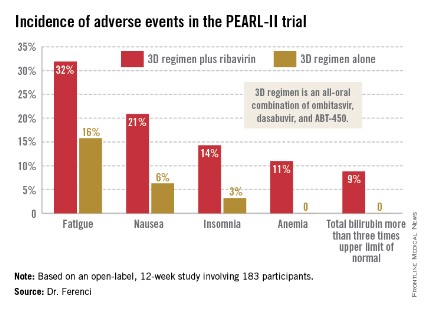
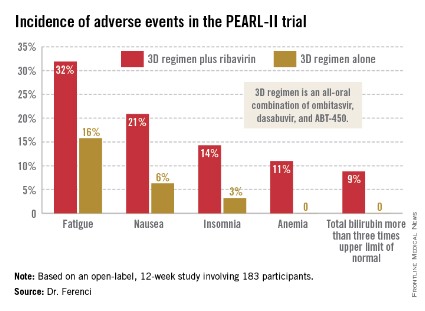
Treatment-related adverse events were more frequent in the ribavirin study arm (see graphic), but were generally mild and manageable.
Asked if in light of the impressive study results he thought physicians should hold off on treating PEARL-II–type patients until the 3D regimen receives marketing approval, Dr. Ferenci replied, "I think if you really need to treat a patient now, you have to use what exists. But I think in half a year your options will change."
PEARL-II was a companion trial to PEARL-III, conducted in previously untreated patients with HCV 1b infection (N. Engl. J. Med. 2014 [doi:10.1056/NEJMoa1402338]). Both studies were sponsored by AbbVie. Dr. Ferenci reported serving on advisory panels for roughly a dozen pharmaceutical companies.
daily oral ribavirin, formerly a standard part of hepatitis C therapy, Europe and Japan, HCV, ombitasvir, dasabuvir, ABT-450, Dr. Peter Ferenci, annual Digestive Disease Week,
CHICAGO – The investigational "3D" combination of oral antiviral drugs achieved a cure rate that was nothing short of sensational in the phase III PEARL-II trial involving noncirrhotic patients with hepatitis C genotype 1b who had previously failed pegylated interferon–based therapy.
Moreover, these results were achieved without the use of daily oral ribavirin, formerly a standard part of hepatitis C therapy, and one responsible for a substantial burden of side effects.
Genotype 1b is the most prevalent form of hepatitis C disease worldwide. While it is especially common in Europe and Japan, genotype 1b also accounts for about one-third of cases of HCV in the United States.
The so-called 3D regimen is an all-oral combination of three direct-acting antiviral agents: ombitasvir, dasabuvir, and ABT-450. Each of these agents inhibits the viral life cycle at a different point, Dr. Peter Ferenci explained in presenting the PEARL-II results at the annual Digestive Disease Week.
PEARL-II was an open-label, 12-week trial which asked the question, what added benefit does daily oral ribavirin provide in conjunction with 3D therapy in noncirrhotic HCV genotype 1b patients previously treated unsuccessfully with a pegylated interferon/ribavirin regimen? The answer, as it turns out, is none.
The 186 participants were randomized to the 3D regimen with or without oral ribavirin. The oral 3D regimen consists of ABT-450 (150 mg)/ritonavir (100 mg)/ombitasvir (25 mg), formulated as a single once-daily pill plus dasabuvir at 250 mg twice daily.
The primary endpoint – a sustained virologic response 12 weeks after completion of the 12-week course of treatment – was achieved in 96.6% of the ribavirin group and 100% who didn’t receive ribavirin. Both rates are superior to the historical sustained virologic response rate seen in studies of the former state-of-the-art regimen of telaprevir plus pegylated interferon and ribavirin, noted Dr. Ferenci of the Medical University of Vienna.
There were no virologic failures in PEARL-II. Two patients stopped the study drugs: one who developed pancreatitis deemed unrelated to treatment and another with anxiety, tachycardia, shortness of breath, and fever considered possibly treatment related.
Results of 3D therapy were equally good in men and women and in former relapsers, nonresponders, and partial responders to pegylated interferon-based regimens.
Treatment-related adverse events were more frequent in the ribavirin study arm (see graphic), but were generally mild and manageable.
Asked if in light of the impressive study results he thought physicians should hold off on treating PEARL-II–type patients until the 3D regimen receives marketing approval, Dr. Ferenci replied, "I think if you really need to treat a patient now, you have to use what exists. But I think in half a year your options will change."
PEARL-II was a companion trial to PEARL-III, conducted in previously untreated patients with HCV 1b infection (N. Engl. J. Med. 2014 [doi:10.1056/NEJMoa1402338]). Both studies were sponsored by AbbVie. Dr. Ferenci reported serving on advisory panels for roughly a dozen pharmaceutical companies.
CHICAGO – The investigational "3D" combination of oral antiviral drugs achieved a cure rate that was nothing short of sensational in the phase III PEARL-II trial involving noncirrhotic patients with hepatitis C genotype 1b who had previously failed pegylated interferon–based therapy.
Moreover, these results were achieved without the use of daily oral ribavirin, formerly a standard part of hepatitis C therapy, and one responsible for a substantial burden of side effects.
Genotype 1b is the most prevalent form of hepatitis C disease worldwide. While it is especially common in Europe and Japan, genotype 1b also accounts for about one-third of cases of HCV in the United States.
The so-called 3D regimen is an all-oral combination of three direct-acting antiviral agents: ombitasvir, dasabuvir, and ABT-450. Each of these agents inhibits the viral life cycle at a different point, Dr. Peter Ferenci explained in presenting the PEARL-II results at the annual Digestive Disease Week.
PEARL-II was an open-label, 12-week trial which asked the question, what added benefit does daily oral ribavirin provide in conjunction with 3D therapy in noncirrhotic HCV genotype 1b patients previously treated unsuccessfully with a pegylated interferon/ribavirin regimen? The answer, as it turns out, is none.
The 186 participants were randomized to the 3D regimen with or without oral ribavirin. The oral 3D regimen consists of ABT-450 (150 mg)/ritonavir (100 mg)/ombitasvir (25 mg), formulated as a single once-daily pill plus dasabuvir at 250 mg twice daily.
The primary endpoint – a sustained virologic response 12 weeks after completion of the 12-week course of treatment – was achieved in 96.6% of the ribavirin group and 100% who didn’t receive ribavirin. Both rates are superior to the historical sustained virologic response rate seen in studies of the former state-of-the-art regimen of telaprevir plus pegylated interferon and ribavirin, noted Dr. Ferenci of the Medical University of Vienna.
There were no virologic failures in PEARL-II. Two patients stopped the study drugs: one who developed pancreatitis deemed unrelated to treatment and another with anxiety, tachycardia, shortness of breath, and fever considered possibly treatment related.
Results of 3D therapy were equally good in men and women and in former relapsers, nonresponders, and partial responders to pegylated interferon-based regimens.
Treatment-related adverse events were more frequent in the ribavirin study arm (see graphic), but were generally mild and manageable.
Asked if in light of the impressive study results he thought physicians should hold off on treating PEARL-II–type patients until the 3D regimen receives marketing approval, Dr. Ferenci replied, "I think if you really need to treat a patient now, you have to use what exists. But I think in half a year your options will change."
PEARL-II was a companion trial to PEARL-III, conducted in previously untreated patients with HCV 1b infection (N. Engl. J. Med. 2014 [doi:10.1056/NEJMoa1402338]). Both studies were sponsored by AbbVie. Dr. Ferenci reported serving on advisory panels for roughly a dozen pharmaceutical companies.
daily oral ribavirin, formerly a standard part of hepatitis C therapy, Europe and Japan, HCV, ombitasvir, dasabuvir, ABT-450, Dr. Peter Ferenci, annual Digestive Disease Week,
daily oral ribavirin, formerly a standard part of hepatitis C therapy, Europe and Japan, HCV, ombitasvir, dasabuvir, ABT-450, Dr. Peter Ferenci, annual Digestive Disease Week,
AT DDW 2014
Major finding: Noncirrhotic patients with hepatitis C genotype 1b disease who had failed previous pegylated interferon–based therapy had a 100% sustained virologic response rate 12 weeks after the conclusion of a 12-week course of all-oral therapy with three direct-acting antiviral agents without daily ribavirin and a 96.6% rate if they did get ribavirin.
Data source: This was an open-label, 12-week, phase III trial including 186 patients.
Disclosures: The study was sponsored by AbbVie. The presenter reported serving on advisory panels for about a dozen pharmaceutical companies.
Oral PPI therapy gets nod following bleeding ulcer repair
CHICAGO – Oral proton pump inhibitor therapy on the medical ward following endoscopic treatment of patients with a bleeding ulcer is equally effective as the current guideline-recommended practice of administering a PPI bolus followed by a 72-hour continuous intravenous infusion – and a lot less resource intensive.
That’s the highly practical message from a meta-analysis of randomized trials addressing the issue that Dr. Hamita Sachar presented at the annual Digestive Disease Week.

Current national and international guidelines recommend an intravenous 80-mg bolus of PPI followed by a 72-hour infusion at 8 mg/hr after successful endoscopic therapy of a high-risk bleeding ulcer. These guidelines need to be revisited in light of the new data, according to Dr. Sachar, a fellow in digestive diseases at Yale University, New Haven, Conn.
"This is practice changing. Intermittent oral therapy is less expensive. Also, it’s quite easy to administer. If you can get away with twice-daily oral dosing, it’s preferred over an IV infusion by patients, it requires less nursing time, and there’s less pharmacy personnel time. As long as patients don’t have nausea and vomiting, I think it’s reasonable for them to get oral therapy because equivalent doses of IV and oral drug have exactly the same pharmacokinetic profiles," she said in an interview.
The meta-analysis, which featured a noninferiority design, included a dozen randomized trials totaling more than 1,600 patients. The primary outcome was the rate of rebleeding within 7 days. It turned out to be virtually identical in the two groups.
Moreover, there was no significant difference between patients who received intermittent oral PPI therapy at standard doses and those who got the guideline-recommended bolus, continuous-infusion regimen in terms of any of the secondary endpoints in the meta-analysis, which included rebleeding within 30 days, rebleeding within 3 days, need for blood transfusion, urgent surgical or endoscopic interventions, and length of hospital stay.
Dr. Sachar and her coinvestigators performed the meta-analysis because their physiologic model suggested that intravenous therapy was safely avoidable. They noted that several randomized trials in the literature concluded that the two treatment strategies might yield comparable outcomes; however, the individual studies were too small to allow any firm conclusions to be drawn.
The study was supported by institutional funds. Dr. Sachar reported having no financial conflicts regarding this study.
Sachar and colleagues presented an interesting systematic review and meta-analysis at DDW 2014 (Gastroenterology 2014;146(5 Suppl 1):S-75). This presentation has fueled discussions on whether high-risk patients with peptic ulcer bleeding (those who had successful endoscopic hemostasis for high-risk stigmata) should receive PPIs intermittently (intravenously or even orally) as opposed to the current practice of high-dose continuous intravenous infusion.

|
| Dr. Grigorios I. Leontiadis |
In conclusion, until the quality of the evidence can be fully assessed, it would be prudent to adhere to the recommendations from the existing practice guidelines and treat high-risk patients with peptic ulcer bleeding with high-dose intravenous infusion PPI treatment, since this is the only dose that has been proven to reduce not only rebleeding but also mortality.
Dr. Grigorios I. Leontiadis is assistant professor of medicine, division of gastroenterology, McMaster University, Hamilton, Ont. He was a consultant to a PPI manufacturer more than 5 years ago.
Sachar and colleagues presented an interesting systematic review and meta-analysis at DDW 2014 (Gastroenterology 2014;146(5 Suppl 1):S-75). This presentation has fueled discussions on whether high-risk patients with peptic ulcer bleeding (those who had successful endoscopic hemostasis for high-risk stigmata) should receive PPIs intermittently (intravenously or even orally) as opposed to the current practice of high-dose continuous intravenous infusion.
|
|
| Dr. Grigorios I. Leontiadis |
In conclusion, until the quality of the evidence can be fully assessed, it would be prudent to adhere to the recommendations from the existing practice guidelines and treat high-risk patients with peptic ulcer bleeding with high-dose intravenous infusion PPI treatment, since this is the only dose that has been proven to reduce not only rebleeding but also mortality.
Dr. Grigorios I. Leontiadis is assistant professor of medicine, division of gastroenterology, McMaster University, Hamilton, Ont. He was a consultant to a PPI manufacturer more than 5 years ago.
Sachar and colleagues presented an interesting systematic review and meta-analysis at DDW 2014 (Gastroenterology 2014;146(5 Suppl 1):S-75). This presentation has fueled discussions on whether high-risk patients with peptic ulcer bleeding (those who had successful endoscopic hemostasis for high-risk stigmata) should receive PPIs intermittently (intravenously or even orally) as opposed to the current practice of high-dose continuous intravenous infusion.
|
|
| Dr. Grigorios I. Leontiadis |
In conclusion, until the quality of the evidence can be fully assessed, it would be prudent to adhere to the recommendations from the existing practice guidelines and treat high-risk patients with peptic ulcer bleeding with high-dose intravenous infusion PPI treatment, since this is the only dose that has been proven to reduce not only rebleeding but also mortality.
Dr. Grigorios I. Leontiadis is assistant professor of medicine, division of gastroenterology, McMaster University, Hamilton, Ont. He was a consultant to a PPI manufacturer more than 5 years ago.
CHICAGO – Oral proton pump inhibitor therapy on the medical ward following endoscopic treatment of patients with a bleeding ulcer is equally effective as the current guideline-recommended practice of administering a PPI bolus followed by a 72-hour continuous intravenous infusion – and a lot less resource intensive.
That’s the highly practical message from a meta-analysis of randomized trials addressing the issue that Dr. Hamita Sachar presented at the annual Digestive Disease Week.
Current national and international guidelines recommend an intravenous 80-mg bolus of PPI followed by a 72-hour infusion at 8 mg/hr after successful endoscopic therapy of a high-risk bleeding ulcer. These guidelines need to be revisited in light of the new data, according to Dr. Sachar, a fellow in digestive diseases at Yale University, New Haven, Conn.
"This is practice changing. Intermittent oral therapy is less expensive. Also, it’s quite easy to administer. If you can get away with twice-daily oral dosing, it’s preferred over an IV infusion by patients, it requires less nursing time, and there’s less pharmacy personnel time. As long as patients don’t have nausea and vomiting, I think it’s reasonable for them to get oral therapy because equivalent doses of IV and oral drug have exactly the same pharmacokinetic profiles," she said in an interview.
The meta-analysis, which featured a noninferiority design, included a dozen randomized trials totaling more than 1,600 patients. The primary outcome was the rate of rebleeding within 7 days. It turned out to be virtually identical in the two groups.
Moreover, there was no significant difference between patients who received intermittent oral PPI therapy at standard doses and those who got the guideline-recommended bolus, continuous-infusion regimen in terms of any of the secondary endpoints in the meta-analysis, which included rebleeding within 30 days, rebleeding within 3 days, need for blood transfusion, urgent surgical or endoscopic interventions, and length of hospital stay.
Dr. Sachar and her coinvestigators performed the meta-analysis because their physiologic model suggested that intravenous therapy was safely avoidable. They noted that several randomized trials in the literature concluded that the two treatment strategies might yield comparable outcomes; however, the individual studies were too small to allow any firm conclusions to be drawn.
The study was supported by institutional funds. Dr. Sachar reported having no financial conflicts regarding this study.
CHICAGO – Oral proton pump inhibitor therapy on the medical ward following endoscopic treatment of patients with a bleeding ulcer is equally effective as the current guideline-recommended practice of administering a PPI bolus followed by a 72-hour continuous intravenous infusion – and a lot less resource intensive.
That’s the highly practical message from a meta-analysis of randomized trials addressing the issue that Dr. Hamita Sachar presented at the annual Digestive Disease Week.
Current national and international guidelines recommend an intravenous 80-mg bolus of PPI followed by a 72-hour infusion at 8 mg/hr after successful endoscopic therapy of a high-risk bleeding ulcer. These guidelines need to be revisited in light of the new data, according to Dr. Sachar, a fellow in digestive diseases at Yale University, New Haven, Conn.
"This is practice changing. Intermittent oral therapy is less expensive. Also, it’s quite easy to administer. If you can get away with twice-daily oral dosing, it’s preferred over an IV infusion by patients, it requires less nursing time, and there’s less pharmacy personnel time. As long as patients don’t have nausea and vomiting, I think it’s reasonable for them to get oral therapy because equivalent doses of IV and oral drug have exactly the same pharmacokinetic profiles," she said in an interview.
The meta-analysis, which featured a noninferiority design, included a dozen randomized trials totaling more than 1,600 patients. The primary outcome was the rate of rebleeding within 7 days. It turned out to be virtually identical in the two groups.
Moreover, there was no significant difference between patients who received intermittent oral PPI therapy at standard doses and those who got the guideline-recommended bolus, continuous-infusion regimen in terms of any of the secondary endpoints in the meta-analysis, which included rebleeding within 30 days, rebleeding within 3 days, need for blood transfusion, urgent surgical or endoscopic interventions, and length of hospital stay.
Dr. Sachar and her coinvestigators performed the meta-analysis because their physiologic model suggested that intravenous therapy was safely avoidable. They noted that several randomized trials in the literature concluded that the two treatment strategies might yield comparable outcomes; however, the individual studies were too small to allow any firm conclusions to be drawn.
The study was supported by institutional funds. Dr. Sachar reported having no financial conflicts regarding this study.
AT DDW 2014
Major finding: Oral proton pump inhibitor therapy can safely and effectively replace the guideline-recommended standard regimen consisting of a PPI bolus followed by 72 hours of continuous intravenous infusion after endoscopic treatment of a bleeding ulcer.
Data source: This was a meta-analysis with a noninferiority design. It included 12 randomized trials totaling 1,653 patients who had undergone endoscopic hemostasis for a bleeding ulcer.
Disclosures: The study was supported by institutional funds. Dr. Sachar reported having no financial conflicts regarding this study.

Think ‘celiac disease’ in patients requiring high-dose levothyroxine
CHICAGO – Hypothyroid patients who need either at least 125 mcg or 1.5 mcg/kg of levothyroxine per day in order to remain euthyroid should routinely be tested for celiac disease, Dr. Richard S. Zubarik asserted at the annual Digestive Disease Week.
In his cross-sectional study of 400 consecutive patients with hypothyroidism who underwent testing for celiac disease, 5% of those on a levothyroxine dose at or above that threshold were found to have biopsy-confirmed celiac disease.
Mass screening for celiac disease in the broad U.S. population isn’t recommended at present because the prevalence – 0.75% is deemed too low to justify such a practice. However, current national and international guidelines do recommend routine testing for case finding in selected populations known to be at increased risk of celiac disease. These include, for example, patients with asymptomatic iron-deficiency anemia, who have a celiac disease prevalence of 2.3%-5%. Thus, the 5% prevalence of celiac disease in hypothyroid patients requiring high-dose levothyroxine in order to maintain a euthyroid state is at least as high as, and perhaps higher than, the prevalence in groups having a guideline-recommended indication for testing, noted Dr. Zubarik, professor of medicine and director of GI endoscopy at the University of Vermont, Burlington.

Testing involves a simple serologic test for tissue transglutaminase. An elevated level triggers endoscopy with duodenal biopsies. A positive biopsy confirms the diagnosis and warrants initiation of a gluten-free diet.
The 400 consecutive patients on treatment for hypothyroidism in Dr. Zubarik’s study averaged 59 years of age, and 82% were women. Thirty percent of participants required 125 mcg or more of levothyroxine, and 29% needed at least 1.5 mcg/kg daily in order to maintain euthyroid status. Dr. Zubarik and his coinvestigators selected those doses as their threshold in the cross-sectional study because their earlier retrospective study had suggested patients requiring that much levothyroxine might have an increased prevalence of concomitant celiac disease (Am. J. Med. 2012;125:278-82). The group requiring an elevated dose and those who were able to remain euthyroid on lower doses were similar in age, sex, weight, body mass index, and the prevalence of diabetes.
All subjects took a serologic tissue transglutaminase test, and those with an elevated level underwent endoscopy with biopsies.
Eight of the 400 patients had an elevated serum tissue transglutaminase level, and seven of the eight were subsequently confirmed as having biopsy-proven celiac disease. Six of the seven patients with celiac disease met or exceeded the levothyroxine dose threshold.
Gastrointestinal symptoms weren’t helpful in differentiating the hypothyroid patients with and without celiac disease. Scores on the Gastrointestinal Symptom Rating Scale weren’t significantly different between the two groups.
Malabsorption of vitamins, minerals, nutrients, and medications is not uncommon in patients with celiac disease, which accounts for the increased disease prevalence seen among patients with iron-deficiency anemia. Whether the increased prevalence of celiac disease among hypothyroid patients requiring elevated doses of levothyroxine is the result of levothyroxine malabsorption or perhaps a consequence of more severe hypothyroidism being present in patients with concomitant celiac disease is an unanswered question Dr. Zubarik plans to study.
The association between thyroid disease and celiac disease is independent of gluten exposure and most likely stems from a common genetic predisposition, the gastroenterologist said.
This study was carried out free of commercial support. Dr. Zubarik reported having no financial conflicts.
CHICAGO – Hypothyroid patients who need either at least 125 mcg or 1.5 mcg/kg of levothyroxine per day in order to remain euthyroid should routinely be tested for celiac disease, Dr. Richard S. Zubarik asserted at the annual Digestive Disease Week.
In his cross-sectional study of 400 consecutive patients with hypothyroidism who underwent testing for celiac disease, 5% of those on a levothyroxine dose at or above that threshold were found to have biopsy-confirmed celiac disease.
Mass screening for celiac disease in the broad U.S. population isn’t recommended at present because the prevalence – 0.75% is deemed too low to justify such a practice. However, current national and international guidelines do recommend routine testing for case finding in selected populations known to be at increased risk of celiac disease. These include, for example, patients with asymptomatic iron-deficiency anemia, who have a celiac disease prevalence of 2.3%-5%. Thus, the 5% prevalence of celiac disease in hypothyroid patients requiring high-dose levothyroxine in order to maintain a euthyroid state is at least as high as, and perhaps higher than, the prevalence in groups having a guideline-recommended indication for testing, noted Dr. Zubarik, professor of medicine and director of GI endoscopy at the University of Vermont, Burlington.
Testing involves a simple serologic test for tissue transglutaminase. An elevated level triggers endoscopy with duodenal biopsies. A positive biopsy confirms the diagnosis and warrants initiation of a gluten-free diet.
The 400 consecutive patients on treatment for hypothyroidism in Dr. Zubarik’s study averaged 59 years of age, and 82% were women. Thirty percent of participants required 125 mcg or more of levothyroxine, and 29% needed at least 1.5 mcg/kg daily in order to maintain euthyroid status. Dr. Zubarik and his coinvestigators selected those doses as their threshold in the cross-sectional study because their earlier retrospective study had suggested patients requiring that much levothyroxine might have an increased prevalence of concomitant celiac disease (Am. J. Med. 2012;125:278-82). The group requiring an elevated dose and those who were able to remain euthyroid on lower doses were similar in age, sex, weight, body mass index, and the prevalence of diabetes.
All subjects took a serologic tissue transglutaminase test, and those with an elevated level underwent endoscopy with biopsies.
Eight of the 400 patients had an elevated serum tissue transglutaminase level, and seven of the eight were subsequently confirmed as having biopsy-proven celiac disease. Six of the seven patients with celiac disease met or exceeded the levothyroxine dose threshold.
Gastrointestinal symptoms weren’t helpful in differentiating the hypothyroid patients with and without celiac disease. Scores on the Gastrointestinal Symptom Rating Scale weren’t significantly different between the two groups.
Malabsorption of vitamins, minerals, nutrients, and medications is not uncommon in patients with celiac disease, which accounts for the increased disease prevalence seen among patients with iron-deficiency anemia. Whether the increased prevalence of celiac disease among hypothyroid patients requiring elevated doses of levothyroxine is the result of levothyroxine malabsorption or perhaps a consequence of more severe hypothyroidism being present in patients with concomitant celiac disease is an unanswered question Dr. Zubarik plans to study.
The association between thyroid disease and celiac disease is independent of gluten exposure and most likely stems from a common genetic predisposition, the gastroenterologist said.
This study was carried out free of commercial support. Dr. Zubarik reported having no financial conflicts.
CHICAGO – Hypothyroid patients who need either at least 125 mcg or 1.5 mcg/kg of levothyroxine per day in order to remain euthyroid should routinely be tested for celiac disease, Dr. Richard S. Zubarik asserted at the annual Digestive Disease Week.
In his cross-sectional study of 400 consecutive patients with hypothyroidism who underwent testing for celiac disease, 5% of those on a levothyroxine dose at or above that threshold were found to have biopsy-confirmed celiac disease.
Mass screening for celiac disease in the broad U.S. population isn’t recommended at present because the prevalence – 0.75% is deemed too low to justify such a practice. However, current national and international guidelines do recommend routine testing for case finding in selected populations known to be at increased risk of celiac disease. These include, for example, patients with asymptomatic iron-deficiency anemia, who have a celiac disease prevalence of 2.3%-5%. Thus, the 5% prevalence of celiac disease in hypothyroid patients requiring high-dose levothyroxine in order to maintain a euthyroid state is at least as high as, and perhaps higher than, the prevalence in groups having a guideline-recommended indication for testing, noted Dr. Zubarik, professor of medicine and director of GI endoscopy at the University of Vermont, Burlington.
Testing involves a simple serologic test for tissue transglutaminase. An elevated level triggers endoscopy with duodenal biopsies. A positive biopsy confirms the diagnosis and warrants initiation of a gluten-free diet.
The 400 consecutive patients on treatment for hypothyroidism in Dr. Zubarik’s study averaged 59 years of age, and 82% were women. Thirty percent of participants required 125 mcg or more of levothyroxine, and 29% needed at least 1.5 mcg/kg daily in order to maintain euthyroid status. Dr. Zubarik and his coinvestigators selected those doses as their threshold in the cross-sectional study because their earlier retrospective study had suggested patients requiring that much levothyroxine might have an increased prevalence of concomitant celiac disease (Am. J. Med. 2012;125:278-82). The group requiring an elevated dose and those who were able to remain euthyroid on lower doses were similar in age, sex, weight, body mass index, and the prevalence of diabetes.
All subjects took a serologic tissue transglutaminase test, and those with an elevated level underwent endoscopy with biopsies.
Eight of the 400 patients had an elevated serum tissue transglutaminase level, and seven of the eight were subsequently confirmed as having biopsy-proven celiac disease. Six of the seven patients with celiac disease met or exceeded the levothyroxine dose threshold.
Gastrointestinal symptoms weren’t helpful in differentiating the hypothyroid patients with and without celiac disease. Scores on the Gastrointestinal Symptom Rating Scale weren’t significantly different between the two groups.
Malabsorption of vitamins, minerals, nutrients, and medications is not uncommon in patients with celiac disease, which accounts for the increased disease prevalence seen among patients with iron-deficiency anemia. Whether the increased prevalence of celiac disease among hypothyroid patients requiring elevated doses of levothyroxine is the result of levothyroxine malabsorption or perhaps a consequence of more severe hypothyroidism being present in patients with concomitant celiac disease is an unanswered question Dr. Zubarik plans to study.
The association between thyroid disease and celiac disease is independent of gluten exposure and most likely stems from a common genetic predisposition, the gastroenterologist said.
This study was carried out free of commercial support. Dr. Zubarik reported having no financial conflicts.
AT DDW 2014
Major finding: Five percent of hypothyroid patients who needed 125 mcg or more of levothyroxine daily in order to remain euthyroid proved to have previously undiagnosed celiac disease, a prevalence deemed high enough to warrant routine testing for the GI disease in that population.
Data source: This was a cross-sectional study involving 400 consecutive patients being treated for hypothyroidism, all of whom underwent serologic testing for celiac disease via the serum tissue transglutaminase test.
Disclosures: This study was carried out free of commercial support. Dr. Zubarik reported having no financial conflicts.

Polyp, adenoma detection rises with Endocuff device
CHICAGO – German investigators reported on their experience using the disposable Endocuff device in a prospective, randomized two-center trial involving 498 patients undergoing colonoscopy. The flexible Endocuff attaches to the tip of the colonoscope and features two rows of small flexible, hinged wings, Dr. Tobias Meister said at the annual Digestive Disease Week.
The polyp detection rate was 56% for Endocuff-assisted colonoscopy and 42% for standard colonoscopy (P = .001), reported Dr. Meister of Helios Albert-Schweitzer-Klinik, Northeim, Germany.

Compared with standard colonoscopy, Endocuff-assisted colonoscopy detected significantly more polyps per patient (mean 1.58 vs. 0.97; P less than .0001), more sigmoid polyps less than 1 cm in size (120 vs. 42; P = .001), and more cecum polyps less than 1 cm (37 vs. 14; P = .002).
The adenoma detection rate was 36% with Endocuff colonoscopy and 28% with standard colonoscopy patients (P = .043).
The number of carcinomas detected was similar in the two groups (2 vs. 3; P = .68), but the Endocuff outperformed standard colonoscopy for low-grade adenomas (208 vs. 112; P = .008), mean adenomas per patient (0.91 vs. 0.49; P =.011), and hyperplastic polyps (33% vs. 21%; P = .003), Dr. Meister said.
Withdrawal time was not measured, although there was a slight increase in total procedure time in the Endocuff group (23.11 minutes vs. 21.51 minutes; P = .04).

Most patients (96%) had no complications, but minor mucosal lacerations were more common with the Endocuff (9 vs. 2; P = .028). The cuff was lost during six colonoscopies, but endoscopically retrieved.
"The Endocuff is feasible, safe, reliable, and easy to handle, and might have the potential to reduce the incidence of colorectal cancer," Dr. Meister concluded. The Endocuff is available in Europe and the United States; it was approved by the Food and Drug Administration in 2012.
The study was supported by Helios Albert-Schweitzer-Klinik. Dr. Meister reported no conflicting interests.
CHICAGO – German investigators reported on their experience using the disposable Endocuff device in a prospective, randomized two-center trial involving 498 patients undergoing colonoscopy. The flexible Endocuff attaches to the tip of the colonoscope and features two rows of small flexible, hinged wings, Dr. Tobias Meister said at the annual Digestive Disease Week.
The polyp detection rate was 56% for Endocuff-assisted colonoscopy and 42% for standard colonoscopy (P = .001), reported Dr. Meister of Helios Albert-Schweitzer-Klinik, Northeim, Germany.
Compared with standard colonoscopy, Endocuff-assisted colonoscopy detected significantly more polyps per patient (mean 1.58 vs. 0.97; P less than .0001), more sigmoid polyps less than 1 cm in size (120 vs. 42; P = .001), and more cecum polyps less than 1 cm (37 vs. 14; P = .002).
The adenoma detection rate was 36% with Endocuff colonoscopy and 28% with standard colonoscopy patients (P = .043).
The number of carcinomas detected was similar in the two groups (2 vs. 3; P = .68), but the Endocuff outperformed standard colonoscopy for low-grade adenomas (208 vs. 112; P = .008), mean adenomas per patient (0.91 vs. 0.49; P =.011), and hyperplastic polyps (33% vs. 21%; P = .003), Dr. Meister said.
Withdrawal time was not measured, although there was a slight increase in total procedure time in the Endocuff group (23.11 minutes vs. 21.51 minutes; P = .04).
Most patients (96%) had no complications, but minor mucosal lacerations were more common with the Endocuff (9 vs. 2; P = .028). The cuff was lost during six colonoscopies, but endoscopically retrieved.
"The Endocuff is feasible, safe, reliable, and easy to handle, and might have the potential to reduce the incidence of colorectal cancer," Dr. Meister concluded. The Endocuff is available in Europe and the United States; it was approved by the Food and Drug Administration in 2012.
The study was supported by Helios Albert-Schweitzer-Klinik. Dr. Meister reported no conflicting interests.
CHICAGO – German investigators reported on their experience using the disposable Endocuff device in a prospective, randomized two-center trial involving 498 patients undergoing colonoscopy. The flexible Endocuff attaches to the tip of the colonoscope and features two rows of small flexible, hinged wings, Dr. Tobias Meister said at the annual Digestive Disease Week.
The polyp detection rate was 56% for Endocuff-assisted colonoscopy and 42% for standard colonoscopy (P = .001), reported Dr. Meister of Helios Albert-Schweitzer-Klinik, Northeim, Germany.
Compared with standard colonoscopy, Endocuff-assisted colonoscopy detected significantly more polyps per patient (mean 1.58 vs. 0.97; P less than .0001), more sigmoid polyps less than 1 cm in size (120 vs. 42; P = .001), and more cecum polyps less than 1 cm (37 vs. 14; P = .002).
The adenoma detection rate was 36% with Endocuff colonoscopy and 28% with standard colonoscopy patients (P = .043).
The number of carcinomas detected was similar in the two groups (2 vs. 3; P = .68), but the Endocuff outperformed standard colonoscopy for low-grade adenomas (208 vs. 112; P = .008), mean adenomas per patient (0.91 vs. 0.49; P =.011), and hyperplastic polyps (33% vs. 21%; P = .003), Dr. Meister said.
Withdrawal time was not measured, although there was a slight increase in total procedure time in the Endocuff group (23.11 minutes vs. 21.51 minutes; P = .04).
Most patients (96%) had no complications, but minor mucosal lacerations were more common with the Endocuff (9 vs. 2; P = .028). The cuff was lost during six colonoscopies, but endoscopically retrieved.
"The Endocuff is feasible, safe, reliable, and easy to handle, and might have the potential to reduce the incidence of colorectal cancer," Dr. Meister concluded. The Endocuff is available in Europe and the United States; it was approved by the Food and Drug Administration in 2012.
The study was supported by Helios Albert-Schweitzer-Klinik. Dr. Meister reported no conflicting interests.
AT DDW 2014
Major finding: The polyp detection rate was 56% for Endocuff-assisted colonoscopy and 42% for standard colonoscopy (P = .001).
Data source: A prospective, two-center trial in 498 patients undergoing colonoscopy.
Disclosures: The study was supported by Helios Albert-Schweitzer-Klinik. Dr. Meister reported no conflicting interests.

Eluxadoline scores in phase III for irritable bowel syndrome
CHICAGO – A first-in-class oral drug targeting diarrhea-predominant irritable bowel syndrome achieved its primary and secondary endpoints in a pair of large phase III trials presented at the annual Digestive Disease Week.
Eluxadoline is a locally active mixed mu-opioid receptor agonist and delta-opioid receptor antagonist. It was compared with placebo at doses of 75 mg and 100 mg b.i.d. in the two double-blind phase III trials, which totaled 2,427 patients with diarrhea-predominant irritable bowel syndrome (IBS-D) by Rome III criteria. The trials featured identical outcome measures; however, one study entailed 12 weeks of double-blind therapy in accord with a Food and Drug Administration request, while the other involved 26 weeks as stipulated by the European Medicines Agency, explained Dr. Anthony Lembo, director of the GI motility center at Beth Israel Deaconess Medical Center, Boston.

The primary endpoint was a composite requiring at least 30% improvement in daily abdominal pain, compared with baseline, and improved stool consistency as reflected in a Bristol Stool Score of less than 5. In the 12-week trial, this was achieved by 27% of patients on eluxadoline at 100 mg b.i.d. and by 26.2% on 75 mg b.i.d., both significantly better rates than the 16.7% in placebo-treated controls. In the 26-week study, the rates were 31%, 26.7%, and 19.5%, respectively. Improvement in the eluxadoline group was seen within the first several days of treatment.
The drug was equally effective in men and women.
Turning to secondary endpoints, Dr. Lembo highlighted the finding that 42% of the eluxadoline 100 mg b.i.d. group reported more than 50% of their days during weeks 1-12 as being urgency free, compared with 21% of controls. Twenty-nine percent of those on the higher dose reported more than 75% of days were urgency free, compared with 12% on placebo.
Bowel movement frequency improved from a baseline average of 4.9 per day to 2.9 per day at week 26 in the group on eluxadoline 100 mg b.i.d., significantly better than the 3.3 movements per day in controls. A 40% or greater improvement in abdominal pain score was reported at week 12 by 43.2% of patients on eluxadoline 100 mg b.i.d. compared with 35.8% of controls.
Safety concerns arose. Eight confirmed cases of hepatobiliary sphincter of Oddi spasm occurred, all in patients on eluxadoline, and all in patients with prior cholecystectomy. Seven of the eight patients were in the high-dose therapy arm. Six cases occurred during the first week of therapy. All eight cases were rapidly reversed upon prompt drug discontinuation; seven were managed on an outpatient basis.
In addition, there were five confirmed cases of pancreatitis, again all in the eluxadoline group. All five cases were mild, and all occurred in patients with pancreatitis risk factors: Three were chronic heavy alcohol abusers, one had biliary sludge, and one had a history of cholecystectomy with sphincter of Oddi spasm. These five patients had brief hospitalizations with no sequelae.
Several audience members greeted this news as a potential roadblock in the drug’s development. Dr. Lembo took a different view.
"These risks can be mitigated through appropriate patient selection and education," the gastroenterologist said. "There does seem to be a group of patients that seem to be particularly at risk: those with prior cholecystectomy or chronic alcohol use. If that’s true and the drug is relatively safe in the remainder of the population, then I think the risk/benefit ratio would be acceptable, especially since there are relatively few other treatment options for these patients."
Other audience members were critical of eluxadoline’s efficacy. "You have a drug that has at best extremely modest effects, with about a 10% absolute overall improvement over placebo, which is statistically significant primarily because you had so many patients in the studies," one gastroenterologist asserted.
Dr. Lembo replied that this magnitude of benefit is similar to what’s seen with drugs now approved for the common and vexing problem of IBS-D.
The phase III trials were sponsored by Furiex Pharmaceuticals. Dr. Lembo reported serving as an adviser or consultant to five pharmaceutical companies, not including Furiex.
CHICAGO – A first-in-class oral drug targeting diarrhea-predominant irritable bowel syndrome achieved its primary and secondary endpoints in a pair of large phase III trials presented at the annual Digestive Disease Week.
Eluxadoline is a locally active mixed mu-opioid receptor agonist and delta-opioid receptor antagonist. It was compared with placebo at doses of 75 mg and 100 mg b.i.d. in the two double-blind phase III trials, which totaled 2,427 patients with diarrhea-predominant irritable bowel syndrome (IBS-D) by Rome III criteria. The trials featured identical outcome measures; however, one study entailed 12 weeks of double-blind therapy in accord with a Food and Drug Administration request, while the other involved 26 weeks as stipulated by the European Medicines Agency, explained Dr. Anthony Lembo, director of the GI motility center at Beth Israel Deaconess Medical Center, Boston.
The primary endpoint was a composite requiring at least 30% improvement in daily abdominal pain, compared with baseline, and improved stool consistency as reflected in a Bristol Stool Score of less than 5. In the 12-week trial, this was achieved by 27% of patients on eluxadoline at 100 mg b.i.d. and by 26.2% on 75 mg b.i.d., both significantly better rates than the 16.7% in placebo-treated controls. In the 26-week study, the rates were 31%, 26.7%, and 19.5%, respectively. Improvement in the eluxadoline group was seen within the first several days of treatment.
The drug was equally effective in men and women.
Turning to secondary endpoints, Dr. Lembo highlighted the finding that 42% of the eluxadoline 100 mg b.i.d. group reported more than 50% of their days during weeks 1-12 as being urgency free, compared with 21% of controls. Twenty-nine percent of those on the higher dose reported more than 75% of days were urgency free, compared with 12% on placebo.
Bowel movement frequency improved from a baseline average of 4.9 per day to 2.9 per day at week 26 in the group on eluxadoline 100 mg b.i.d., significantly better than the 3.3 movements per day in controls. A 40% or greater improvement in abdominal pain score was reported at week 12 by 43.2% of patients on eluxadoline 100 mg b.i.d. compared with 35.8% of controls.
Safety concerns arose. Eight confirmed cases of hepatobiliary sphincter of Oddi spasm occurred, all in patients on eluxadoline, and all in patients with prior cholecystectomy. Seven of the eight patients were in the high-dose therapy arm. Six cases occurred during the first week of therapy. All eight cases were rapidly reversed upon prompt drug discontinuation; seven were managed on an outpatient basis.
In addition, there were five confirmed cases of pancreatitis, again all in the eluxadoline group. All five cases were mild, and all occurred in patients with pancreatitis risk factors: Three were chronic heavy alcohol abusers, one had biliary sludge, and one had a history of cholecystectomy with sphincter of Oddi spasm. These five patients had brief hospitalizations with no sequelae.
Several audience members greeted this news as a potential roadblock in the drug’s development. Dr. Lembo took a different view.
"These risks can be mitigated through appropriate patient selection and education," the gastroenterologist said. "There does seem to be a group of patients that seem to be particularly at risk: those with prior cholecystectomy or chronic alcohol use. If that’s true and the drug is relatively safe in the remainder of the population, then I think the risk/benefit ratio would be acceptable, especially since there are relatively few other treatment options for these patients."
Other audience members were critical of eluxadoline’s efficacy. "You have a drug that has at best extremely modest effects, with about a 10% absolute overall improvement over placebo, which is statistically significant primarily because you had so many patients in the studies," one gastroenterologist asserted.
Dr. Lembo replied that this magnitude of benefit is similar to what’s seen with drugs now approved for the common and vexing problem of IBS-D.
The phase III trials were sponsored by Furiex Pharmaceuticals. Dr. Lembo reported serving as an adviser or consultant to five pharmaceutical companies, not including Furiex.
CHICAGO – A first-in-class oral drug targeting diarrhea-predominant irritable bowel syndrome achieved its primary and secondary endpoints in a pair of large phase III trials presented at the annual Digestive Disease Week.
Eluxadoline is a locally active mixed mu-opioid receptor agonist and delta-opioid receptor antagonist. It was compared with placebo at doses of 75 mg and 100 mg b.i.d. in the two double-blind phase III trials, which totaled 2,427 patients with diarrhea-predominant irritable bowel syndrome (IBS-D) by Rome III criteria. The trials featured identical outcome measures; however, one study entailed 12 weeks of double-blind therapy in accord with a Food and Drug Administration request, while the other involved 26 weeks as stipulated by the European Medicines Agency, explained Dr. Anthony Lembo, director of the GI motility center at Beth Israel Deaconess Medical Center, Boston.
The primary endpoint was a composite requiring at least 30% improvement in daily abdominal pain, compared with baseline, and improved stool consistency as reflected in a Bristol Stool Score of less than 5. In the 12-week trial, this was achieved by 27% of patients on eluxadoline at 100 mg b.i.d. and by 26.2% on 75 mg b.i.d., both significantly better rates than the 16.7% in placebo-treated controls. In the 26-week study, the rates were 31%, 26.7%, and 19.5%, respectively. Improvement in the eluxadoline group was seen within the first several days of treatment.
The drug was equally effective in men and women.
Turning to secondary endpoints, Dr. Lembo highlighted the finding that 42% of the eluxadoline 100 mg b.i.d. group reported more than 50% of their days during weeks 1-12 as being urgency free, compared with 21% of controls. Twenty-nine percent of those on the higher dose reported more than 75% of days were urgency free, compared with 12% on placebo.
Bowel movement frequency improved from a baseline average of 4.9 per day to 2.9 per day at week 26 in the group on eluxadoline 100 mg b.i.d., significantly better than the 3.3 movements per day in controls. A 40% or greater improvement in abdominal pain score was reported at week 12 by 43.2% of patients on eluxadoline 100 mg b.i.d. compared with 35.8% of controls.
Safety concerns arose. Eight confirmed cases of hepatobiliary sphincter of Oddi spasm occurred, all in patients on eluxadoline, and all in patients with prior cholecystectomy. Seven of the eight patients were in the high-dose therapy arm. Six cases occurred during the first week of therapy. All eight cases were rapidly reversed upon prompt drug discontinuation; seven were managed on an outpatient basis.
In addition, there were five confirmed cases of pancreatitis, again all in the eluxadoline group. All five cases were mild, and all occurred in patients with pancreatitis risk factors: Three were chronic heavy alcohol abusers, one had biliary sludge, and one had a history of cholecystectomy with sphincter of Oddi spasm. These five patients had brief hospitalizations with no sequelae.
Several audience members greeted this news as a potential roadblock in the drug’s development. Dr. Lembo took a different view.
"These risks can be mitigated through appropriate patient selection and education," the gastroenterologist said. "There does seem to be a group of patients that seem to be particularly at risk: those with prior cholecystectomy or chronic alcohol use. If that’s true and the drug is relatively safe in the remainder of the population, then I think the risk/benefit ratio would be acceptable, especially since there are relatively few other treatment options for these patients."
Other audience members were critical of eluxadoline’s efficacy. "You have a drug that has at best extremely modest effects, with about a 10% absolute overall improvement over placebo, which is statistically significant primarily because you had so many patients in the studies," one gastroenterologist asserted.
Dr. Lembo replied that this magnitude of benefit is similar to what’s seen with drugs now approved for the common and vexing problem of IBS-D.
The phase III trials were sponsored by Furiex Pharmaceuticals. Dr. Lembo reported serving as an adviser or consultant to five pharmaceutical companies, not including Furiex.
AT DDW 2014
Major finding: Thirty-one percent of patients with diarrhea-predominant irritable bowel syndrome on 100 mg b.i.d. of the investigational oral agent eluxadoline for 26 weeks met the primary composite study efficacy endpoint, compared with 19.5% on placebo.
Data source: These two phase III, double-blind, randomized, placebo-controlled trials included 2,427 patients with diarrhea-predominant irritable bowel syndrome.
Disclosures: The phase III trials were sponsored by Furiex Pharmaceuticals. Dr. Lembo reported serving as an adviser or consultant to five pharmaceutical companies, not including Furiex.

Larazotide eased symptoms in phase II celiac trial
CHICAGO – Larazotide acetate, a first-in-class oral medication developed specifically to treat celiac disease, reduced both gastrointestinal and non-GI symptoms in patients who were symptomatic despite being on a gluten-free diet in a 74-site, randomized, double-blind phase II study.
"Safety and tolerability were comparable to placebo, setting this drug up, we believe, for phase III trials," Dr. Joseph A. Murray said at the annual Digestive Disease Week.
The study included 342 adult patients with celiac disease who had been on a gluten-free diet (GFD) for at least 12 months and continued on the GFD throughout the 12-week treatment period. Thus, this was a study of larazotide as an adjunct to the GFD, not as an alternative to it.

A GFD is of well-established efficacy as the sole proven therapy for celiac disease. However, the diet can be challenging. Surveys indicate 70% of celiac disease patients on a GFD experience recurrent symptoms due to inadvertent exposure or lapses in the strict adherence required. Thus, there is a significant unmet need for an effective pharmacologic therapy for this common disease, observed Dr. Murray, professor of medicine at the Mayo Clinic in Rochester, Minn.
Study participants were randomized to 12 weeks of double-blind treatment with larazotide at 0.5, 1, or 2 mg three times daily or to placebo. The study provided a successful opportunity to validate a new instrument designed for evaluation of celiac disease symptoms both in clinical trials and in daily practice. The instrument, known as the Celiac Disease Patient Reported Outcome (CeD PRO), entails daily assessment of symptoms in three key domains: abdominal symptoms such as cramping, bloating, and pain; diarrhea and loose stools; and the non-GI symptoms of headache and tiredness, which figure prominently in the disease but aren’t included in the older Celiac Disease Gastrointestinal Symptom Rating Scale.
Patients in the larazotide 0.5-mg t.i.d. group showed a significant 26% reduction in the average weekly number of CeD PRO symptomatic days, compared with placebo-treated controls. Twenty-nine percent of patients in the larazotide 0.5-mg group also achieved at least a 50% decrease from baseline in the weekly average CeD PRO abdominal symptom domain scores for at least 6 of the 12 weeks of treatment, significantly greater than the 14% rate with placebo. In addition, the larazotide 0.5-mg t.i.d. group had a 31% increase in the average number of CeD PRO days with minimal or no symptoms, compared with controls. Moreover, patients on the lowest dose of larazotide showed a significant reduction in CeD PRO non-GI symptoms, the only study arm to do so.
Of note, patients on the two higher doses of larazotide didn’t significantly outperform the control group in any of these study endpoints, although favorable trends were seen.
No changes in serially measured levels of anti–tissue transglutaminase antibodies or anti–deaminated gliadin antibodies were seen in any treatment arm.
Larazotide is an oral peptide that prevents the opening of epithelial tight junctions in the small bowel, an event that occurs in response to gluten exposure and inflammatory cytokines in patients with celiac disease. When these tight junctions are open inappropriately, the result is increased intestinal permeability and inflammation.
One audience member expressed reservations about the finding that only the lowest tested dose in a dose-ranging study proved effective.
"I’m troubled by drugs that don’t show a dose-response curve. This positive result could just be the play of chance," he cautioned.

Dr. Murray replied that he believes the consistency of the positive results with the 0.5-mg dose across multiple endpoints reduces that likelihood. Also, the lowest tested dose was the one that was effective in all the prior challenge studies. In any case, he added, the larger patient numbers planned for the phase III trials will sort out this issue.
Dr. Benjamin Lebwohl was enthusiastic about the larazotide results.
"It’s a very exciting time in the celiac disease community. Larazotide is one of several nondietary treatments currently in the pipeline," noted Dr. Lebwohl of the celiac disease center at Columbia University, New York.
The time is at hand, he added, to think big in terms of celiac disease research questions.
"Is it possible to prevent celiac disease in those individuals at increased risk? And among those individuals who come to us having been diagnosed with celiac disease, can we stop it in its tracks? Can we tolerize patients to gluten, allowing them to eat gluten either intermittently or even resume a full gluten-containing diet? Will there be a cure for celiac disease?" Dr. Lebwohl asked.
The study was supported by Alba Therapeutics. Dr. Murray reported serving as a consultant to or on the advisory boards of nearly a dozen pharmaceutical companies, Alba not among them. Dr. Lebwohl reported having no financial conflicts.
CHICAGO – Larazotide acetate, a first-in-class oral medication developed specifically to treat celiac disease, reduced both gastrointestinal and non-GI symptoms in patients who were symptomatic despite being on a gluten-free diet in a 74-site, randomized, double-blind phase II study.
"Safety and tolerability were comparable to placebo, setting this drug up, we believe, for phase III trials," Dr. Joseph A. Murray said at the annual Digestive Disease Week.
The study included 342 adult patients with celiac disease who had been on a gluten-free diet (GFD) for at least 12 months and continued on the GFD throughout the 12-week treatment period. Thus, this was a study of larazotide as an adjunct to the GFD, not as an alternative to it.
A GFD is of well-established efficacy as the sole proven therapy for celiac disease. However, the diet can be challenging. Surveys indicate 70% of celiac disease patients on a GFD experience recurrent symptoms due to inadvertent exposure or lapses in the strict adherence required. Thus, there is a significant unmet need for an effective pharmacologic therapy for this common disease, observed Dr. Murray, professor of medicine at the Mayo Clinic in Rochester, Minn.
Study participants were randomized to 12 weeks of double-blind treatment with larazotide at 0.5, 1, or 2 mg three times daily or to placebo. The study provided a successful opportunity to validate a new instrument designed for evaluation of celiac disease symptoms both in clinical trials and in daily practice. The instrument, known as the Celiac Disease Patient Reported Outcome (CeD PRO), entails daily assessment of symptoms in three key domains: abdominal symptoms such as cramping, bloating, and pain; diarrhea and loose stools; and the non-GI symptoms of headache and tiredness, which figure prominently in the disease but aren’t included in the older Celiac Disease Gastrointestinal Symptom Rating Scale.
Patients in the larazotide 0.5-mg t.i.d. group showed a significant 26% reduction in the average weekly number of CeD PRO symptomatic days, compared with placebo-treated controls. Twenty-nine percent of patients in the larazotide 0.5-mg group also achieved at least a 50% decrease from baseline in the weekly average CeD PRO abdominal symptom domain scores for at least 6 of the 12 weeks of treatment, significantly greater than the 14% rate with placebo. In addition, the larazotide 0.5-mg t.i.d. group had a 31% increase in the average number of CeD PRO days with minimal or no symptoms, compared with controls. Moreover, patients on the lowest dose of larazotide showed a significant reduction in CeD PRO non-GI symptoms, the only study arm to do so.
Of note, patients on the two higher doses of larazotide didn’t significantly outperform the control group in any of these study endpoints, although favorable trends were seen.
No changes in serially measured levels of anti–tissue transglutaminase antibodies or anti–deaminated gliadin antibodies were seen in any treatment arm.
Larazotide is an oral peptide that prevents the opening of epithelial tight junctions in the small bowel, an event that occurs in response to gluten exposure and inflammatory cytokines in patients with celiac disease. When these tight junctions are open inappropriately, the result is increased intestinal permeability and inflammation.
One audience member expressed reservations about the finding that only the lowest tested dose in a dose-ranging study proved effective.
"I’m troubled by drugs that don’t show a dose-response curve. This positive result could just be the play of chance," he cautioned.
Dr. Murray replied that he believes the consistency of the positive results with the 0.5-mg dose across multiple endpoints reduces that likelihood. Also, the lowest tested dose was the one that was effective in all the prior challenge studies. In any case, he added, the larger patient numbers planned for the phase III trials will sort out this issue.
Dr. Benjamin Lebwohl was enthusiastic about the larazotide results.
"It’s a very exciting time in the celiac disease community. Larazotide is one of several nondietary treatments currently in the pipeline," noted Dr. Lebwohl of the celiac disease center at Columbia University, New York.
The time is at hand, he added, to think big in terms of celiac disease research questions.
"Is it possible to prevent celiac disease in those individuals at increased risk? And among those individuals who come to us having been diagnosed with celiac disease, can we stop it in its tracks? Can we tolerize patients to gluten, allowing them to eat gluten either intermittently or even resume a full gluten-containing diet? Will there be a cure for celiac disease?" Dr. Lebwohl asked.
The study was supported by Alba Therapeutics. Dr. Murray reported serving as a consultant to or on the advisory boards of nearly a dozen pharmaceutical companies, Alba not among them. Dr. Lebwohl reported having no financial conflicts.
CHICAGO – Larazotide acetate, a first-in-class oral medication developed specifically to treat celiac disease, reduced both gastrointestinal and non-GI symptoms in patients who were symptomatic despite being on a gluten-free diet in a 74-site, randomized, double-blind phase II study.
"Safety and tolerability were comparable to placebo, setting this drug up, we believe, for phase III trials," Dr. Joseph A. Murray said at the annual Digestive Disease Week.
The study included 342 adult patients with celiac disease who had been on a gluten-free diet (GFD) for at least 12 months and continued on the GFD throughout the 12-week treatment period. Thus, this was a study of larazotide as an adjunct to the GFD, not as an alternative to it.
A GFD is of well-established efficacy as the sole proven therapy for celiac disease. However, the diet can be challenging. Surveys indicate 70% of celiac disease patients on a GFD experience recurrent symptoms due to inadvertent exposure or lapses in the strict adherence required. Thus, there is a significant unmet need for an effective pharmacologic therapy for this common disease, observed Dr. Murray, professor of medicine at the Mayo Clinic in Rochester, Minn.
Study participants were randomized to 12 weeks of double-blind treatment with larazotide at 0.5, 1, or 2 mg three times daily or to placebo. The study provided a successful opportunity to validate a new instrument designed for evaluation of celiac disease symptoms both in clinical trials and in daily practice. The instrument, known as the Celiac Disease Patient Reported Outcome (CeD PRO), entails daily assessment of symptoms in three key domains: abdominal symptoms such as cramping, bloating, and pain; diarrhea and loose stools; and the non-GI symptoms of headache and tiredness, which figure prominently in the disease but aren’t included in the older Celiac Disease Gastrointestinal Symptom Rating Scale.
Patients in the larazotide 0.5-mg t.i.d. group showed a significant 26% reduction in the average weekly number of CeD PRO symptomatic days, compared with placebo-treated controls. Twenty-nine percent of patients in the larazotide 0.5-mg group also achieved at least a 50% decrease from baseline in the weekly average CeD PRO abdominal symptom domain scores for at least 6 of the 12 weeks of treatment, significantly greater than the 14% rate with placebo. In addition, the larazotide 0.5-mg t.i.d. group had a 31% increase in the average number of CeD PRO days with minimal or no symptoms, compared with controls. Moreover, patients on the lowest dose of larazotide showed a significant reduction in CeD PRO non-GI symptoms, the only study arm to do so.
Of note, patients on the two higher doses of larazotide didn’t significantly outperform the control group in any of these study endpoints, although favorable trends were seen.
No changes in serially measured levels of anti–tissue transglutaminase antibodies or anti–deaminated gliadin antibodies were seen in any treatment arm.
Larazotide is an oral peptide that prevents the opening of epithelial tight junctions in the small bowel, an event that occurs in response to gluten exposure and inflammatory cytokines in patients with celiac disease. When these tight junctions are open inappropriately, the result is increased intestinal permeability and inflammation.
One audience member expressed reservations about the finding that only the lowest tested dose in a dose-ranging study proved effective.
"I’m troubled by drugs that don’t show a dose-response curve. This positive result could just be the play of chance," he cautioned.
Dr. Murray replied that he believes the consistency of the positive results with the 0.5-mg dose across multiple endpoints reduces that likelihood. Also, the lowest tested dose was the one that was effective in all the prior challenge studies. In any case, he added, the larger patient numbers planned for the phase III trials will sort out this issue.
Dr. Benjamin Lebwohl was enthusiastic about the larazotide results.
"It’s a very exciting time in the celiac disease community. Larazotide is one of several nondietary treatments currently in the pipeline," noted Dr. Lebwohl of the celiac disease center at Columbia University, New York.
The time is at hand, he added, to think big in terms of celiac disease research questions.
"Is it possible to prevent celiac disease in those individuals at increased risk? And among those individuals who come to us having been diagnosed with celiac disease, can we stop it in its tracks? Can we tolerize patients to gluten, allowing them to eat gluten either intermittently or even resume a full gluten-containing diet? Will there be a cure for celiac disease?" Dr. Lebwohl asked.
The study was supported by Alba Therapeutics. Dr. Murray reported serving as a consultant to or on the advisory boards of nearly a dozen pharmaceutical companies, Alba not among them. Dr. Lebwohl reported having no financial conflicts.
AT DDW 2014
Major finding: Patients with symptomatic celiac disease despite being on a gluten-free diet showed a 26% greater reduction in a composite symptom score combining GI and non-GI symptoms in response to oral larazotide acetate, compared with placebo-treated controls.
Data source: This was a randomized, double-blind, placebo-controlled, 74-site, phase II study including 342 adult celiac disease patients who were symptomatic despite being on a gluten-free diet.
Disclosures: The study was sponsored by Alba Therapeutics. Dr. Murray reported having no financial relationship with that company.
