User login
VIDEO: Sun protection urged for Asian, Hispanic women
SAN FRANCISCO – Among Asian and Hispanic patients, women are more likely than men are to get nonmelanoma skin cancer, according to a review of 4,029 cases at the University of California, San Diego.
That’s a surprise, because the reverse is true in whites, and skin cancer is generally thought to be more common in men.
About 96% of the cases were in white patients, and two-thirds of those were in men. Among Hispanic and Asian patients, about two-thirds of the cases were in women.
The reason for the gender reversal is unclear, but the study has a clear message, according to study investigator Dr. Arisa Ortiz, director of laser and cosmetic dermatology at the university. She shared that message in an interview at the American Academy of Dermatology annual meeting.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN FRANCISCO – Among Asian and Hispanic patients, women are more likely than men are to get nonmelanoma skin cancer, according to a review of 4,029 cases at the University of California, San Diego.
That’s a surprise, because the reverse is true in whites, and skin cancer is generally thought to be more common in men.
About 96% of the cases were in white patients, and two-thirds of those were in men. Among Hispanic and Asian patients, about two-thirds of the cases were in women.
The reason for the gender reversal is unclear, but the study has a clear message, according to study investigator Dr. Arisa Ortiz, director of laser and cosmetic dermatology at the university. She shared that message in an interview at the American Academy of Dermatology annual meeting.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN FRANCISCO – Among Asian and Hispanic patients, women are more likely than men are to get nonmelanoma skin cancer, according to a review of 4,029 cases at the University of California, San Diego.
That’s a surprise, because the reverse is true in whites, and skin cancer is generally thought to be more common in men.
About 96% of the cases were in white patients, and two-thirds of those were in men. Among Hispanic and Asian patients, about two-thirds of the cases were in women.
The reason for the gender reversal is unclear, but the study has a clear message, according to study investigator Dr. Arisa Ortiz, director of laser and cosmetic dermatology at the university. She shared that message in an interview at the American Academy of Dermatology annual meeting.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT AAD 2015

Localized Argyria With Pseudo-ochronosis
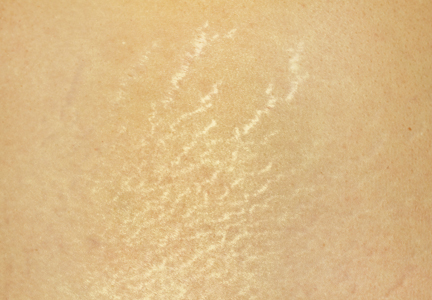
Localized cutaneous argyria often presents as asymptomatic black or blue-gray pigmented macules in areas of the skin exposed to silver-containing compounds.1 Silver may enter the skin by traumatic implantation or absorption via eccrine sweat glands.2 Our patient witnessed a gun fight several years ago while on a mission trip and sustained multiple shrapnel wounds.
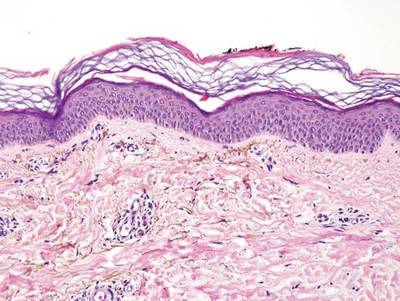
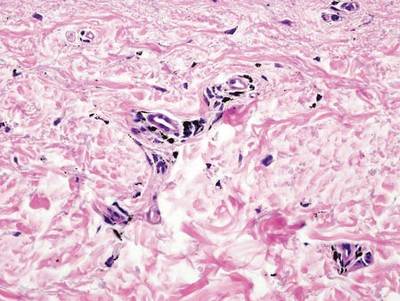
As in our patient, hyperpigmentation may appear years following initial exposure. Over time, incident light reduces colorless silver salts and compounds to black elemental silver.3 It also has been suggested that metallic silver granules stimulate tyrosine kinase activity, leading to locally increased melanin production.4 Together, these processes result in the clinical appearance of a blue-black macule. Despite its long-standing association with silver, this appearance also has been noted with deposition of other metals.5 Histologically, metal deposits can be seen as black granules surrounding eccrine glands, blood vessels, and elastic fibers on higher magnification.6 Granules also may be found in sebaceous glands and arrector pili muscle fibers. These findings do not distinguish from generalized argyria due to increased serum silver levels; however, some cases of localized cutaneous argyria have demonstrated spheroid black globules with surrounding collagen necrosis,1 which have not been reported with generalized disease. Localized cutaneous argyria also may be associated with ocher pigmentation of thickened collagen fibers, resembling changes typically found in alkaptonuria, an inherited deficiency of homogentisic acid oxidase (an enzyme involved in tyrosine metabolism).7 The resulting buildup of metabolic intermediates leads to ochronosis, a deposition of ocher-pigmented intermediates in connective tissue throughout the body. In the skin, ocher pigmentation occurs in elastic fibers of the reticular dermis.1 Grossly, these changes result in a blue-gray discoloration of the skin due to a light-scattering phenomenon known as the Tyndall effect. Exogenous ochronosis also can occur, most commonly from the topical application of hydroquinone or other skin-lightening compounds.1,5 Ocher pigmentation occurring in the setting of localized cutaneous argyria is referred to as pseudo-ochronosis, a finding first described by Robinson-Bostom et al.1 The etiology of this condition is poorly understood, but Robinson-Bostom et al1 noted the appearance of dark metal granules surrounding collagen bundles and hypothesized that metal aggregates surrounding collagen bundles in pseudo-ochronosis cause a homogenized appearance under light microscopy. Yellow-brown, swollen, homogenized collagen bundles can be visualized in the reticular dermis with surrounding deposition of metal granules (Figures 1 and 2).1 Typical patterns of granule deposition in localized argyria also are present.

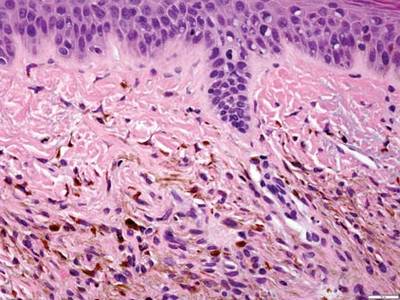
A blue nevus is a collection of proliferating dermal melanocytes. Many histologic subtypes exist and there may be extensive variability in the extent of sclerosis, cellular architecture, and tissue cellularity between each variant.8 Blue nevi commonly present as blue-black hyperpigmentation in the dermis and subcutaneous tissue.9 Histologically, they are characterized by slender, bipolar, dendritic melanocytes in a sclerotic stroma (Figure 3).8 Melanocytes are highly pigmented and contain small monomorphic nuclei. Lesions are relatively homogenous and typically are restricted to the dermis with epidermal sparing.9 Dark granules and ocher fibers are absent.
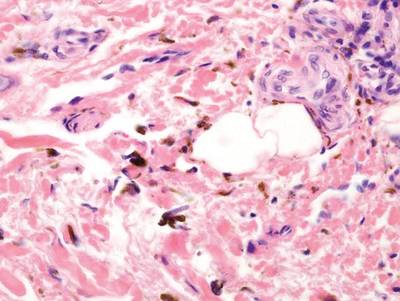
Long-term use of hydroxychloroquine or other antimalarials may cause a macular pattern of blue-gray hyperpigmentation.10 Biopsy specimens typically reveal coarse, yellow-brown pigment granules primarily affecting the superficial dermis (Figure 4). Granules are found both extracellularly and within macrophages. Fontana-Masson silver staining may identify melanin, as hydroxychloroquine-melanin binding may contribute to patterns of hyperpigmentation.10 Hemosiderin often is present in cases of hydroxychloroquine pigmentation. Preceding ecchymosis appears to favor the deposition of hydroxychloroquine in the skin.11 The absence of dark metal granules helps distinguish hydroxychloroquine pigmentation from argyria.
Regressed melanomas may appear clinically as gray macules. These lesions arise in cases of malignant melanoma that spontaneously regress without treatment. Spontaneous regression occurs in 10% to 35% of cases depending on tumor subtype.12 Lesions can have a variable appearance based on the degree of regression. Partial regression is demonstrated by mixed melanosis and fibrosis in the dermis (Figure 5).13,14 Melanin is housed within melanophages present in a variably expanded papillary dermis. Tumors in early stages of regression can be surrounded by an inflammatory infiltrate, which becomes diminished at later stages. However, a few exceptional cases have been noted with extensive inflammatory infiltrate and no residual tumor.14 Completely regressed lesions typically appear as a band of dermal melanophages in the absence of inflammation or melanocytic atypia.15 The finding of regressed melanoma should prompt further investigation including sentinel lymph node biopsy, as it may be associated with metastasis.
Tattooing occurs following traumatic penetration of the skin with impregnation of pigmented foreign material into deep dermal layers.16 Histologic examination usually reveals clumps of fine particulate material in the dermis (Figure 6). The color of the pigment depends on the agent used. For example, graphite appears as black particles that may be confused with localized cutaneous argyria. Distinction can be made using elemental identification techniques such as energy-dispersive X-ray spectroscopy.1 The intensity of the pigment in granules found in tattoos or localized cutaneous argyria will fail to diminish with the application of melanin bleach.6
- Robinson-Bostom L, Pomerantz D, Wilkel C, et al. Localized argyria with pseudo-ochronosis. J Am Acad Dermatol. 2002;46:222-227.
- Tajirian AL, Campbell RM, Robinson-Bostom L. Localized argyria after exposure to aerosolized solder. Cutis. 2006;78:305-308.
- Shelley WB, Shelley ED, Burmeister V. Argyria: the intradermal photograph, a manifestation of passive photosensitivity. J Am Acad Dermatol. 1987;16:211-217.
- Buckley WR, Terhaar CJ. The skin as an excretory organ in argyria. Trans St Johns Hosp Dermatol Soc. 1973;59:39-44.
- Shimizu I, Dill SW, McBean J, et al. Metal-induced granule deposition with pseudo-ochronosis. J Am Acad Dermatol. 2010;63:357-359.
- Rackoff EMJ, Benbenisty KM, Maize JC, et al. Localized cutaneous argyria from an acupuncture needle clini-cally concerning for metastatic melanoma. Cutis. 2007;80:423-426.
- Fernandez-Canon JM, Granadino B, Beltran-Valero de Bernabe D, et al. The molecular basis of alkaptonuria. Nat Genet. 1996;14:5-6.
- Busam KJ, Woodruff JM, Erlandson RA, et al. Large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2000;24:92-99.
- Granter SR, McKee PH, Calonje E, et al. Melanoma associated with blue nevus and melanoma mimicking cellular blue nevus: a clinicopathologic study of 10 cases on the spectrum of so-called ‘malignant blue nevus.’ Am J Surg Pathol. 2001;25:316.
- Puri PK, Lountzis NI, Tyler W, et al. Hydroxychloroquine-induced hyperpigmentation: the staining pattern. J Cutan Pathol. 2008;35:1134-1137.
- Jallouli M, Francès C, Piette JC, et al. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus: a case-control study. JAMA Dermatol. 2013;149:935-940.
- Blessing K, McLaren KM. Histological regression in primary cutaneous melanoma: recognition, prevalence and significance. Histopathology. 1992;20:315-322.
- LeBoit PE. Melanosis and its meanings. Am J Dermatopathol. 2002;24:369-372.
- Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181.
- Yang CH, Yeh JT, Shen SC, et al. Regressed subungual melanoma simulating cellular blue nevus: managed with sentinel lymph node biopsy. Dermatol Surg. 2006;32:577-581.
- Apfelberg DB, Manchester GH. Decorative and traumatic tattoo biophysics and removal. Clin Plast Surg. 1987;14:243-251.
Localized cutaneous argyria often presents as asymptomatic black or blue-gray pigmented macules in areas of the skin exposed to silver-containing compounds.1 Silver may enter the skin by traumatic implantation or absorption via eccrine sweat glands.2 Our patient witnessed a gun fight several years ago while on a mission trip and sustained multiple shrapnel wounds.
As in our patient, hyperpigmentation may appear years following initial exposure. Over time, incident light reduces colorless silver salts and compounds to black elemental silver.3 It also has been suggested that metallic silver granules stimulate tyrosine kinase activity, leading to locally increased melanin production.4 Together, these processes result in the clinical appearance of a blue-black macule. Despite its long-standing association with silver, this appearance also has been noted with deposition of other metals.5 Histologically, metal deposits can be seen as black granules surrounding eccrine glands, blood vessels, and elastic fibers on higher magnification.6 Granules also may be found in sebaceous glands and arrector pili muscle fibers. These findings do not distinguish from generalized argyria due to increased serum silver levels; however, some cases of localized cutaneous argyria have demonstrated spheroid black globules with surrounding collagen necrosis,1 which have not been reported with generalized disease. Localized cutaneous argyria also may be associated with ocher pigmentation of thickened collagen fibers, resembling changes typically found in alkaptonuria, an inherited deficiency of homogentisic acid oxidase (an enzyme involved in tyrosine metabolism).7 The resulting buildup of metabolic intermediates leads to ochronosis, a deposition of ocher-pigmented intermediates in connective tissue throughout the body. In the skin, ocher pigmentation occurs in elastic fibers of the reticular dermis.1 Grossly, these changes result in a blue-gray discoloration of the skin due to a light-scattering phenomenon known as the Tyndall effect. Exogenous ochronosis also can occur, most commonly from the topical application of hydroquinone or other skin-lightening compounds.1,5 Ocher pigmentation occurring in the setting of localized cutaneous argyria is referred to as pseudo-ochronosis, a finding first described by Robinson-Bostom et al.1 The etiology of this condition is poorly understood, but Robinson-Bostom et al1 noted the appearance of dark metal granules surrounding collagen bundles and hypothesized that metal aggregates surrounding collagen bundles in pseudo-ochronosis cause a homogenized appearance under light microscopy. Yellow-brown, swollen, homogenized collagen bundles can be visualized in the reticular dermis with surrounding deposition of metal granules (Figures 1 and 2).1 Typical patterns of granule deposition in localized argyria also are present.
A blue nevus is a collection of proliferating dermal melanocytes. Many histologic subtypes exist and there may be extensive variability in the extent of sclerosis, cellular architecture, and tissue cellularity between each variant.8 Blue nevi commonly present as blue-black hyperpigmentation in the dermis and subcutaneous tissue.9 Histologically, they are characterized by slender, bipolar, dendritic melanocytes in a sclerotic stroma (Figure 3).8 Melanocytes are highly pigmented and contain small monomorphic nuclei. Lesions are relatively homogenous and typically are restricted to the dermis with epidermal sparing.9 Dark granules and ocher fibers are absent.
Long-term use of hydroxychloroquine or other antimalarials may cause a macular pattern of blue-gray hyperpigmentation.10 Biopsy specimens typically reveal coarse, yellow-brown pigment granules primarily affecting the superficial dermis (Figure 4). Granules are found both extracellularly and within macrophages. Fontana-Masson silver staining may identify melanin, as hydroxychloroquine-melanin binding may contribute to patterns of hyperpigmentation.10 Hemosiderin often is present in cases of hydroxychloroquine pigmentation. Preceding ecchymosis appears to favor the deposition of hydroxychloroquine in the skin.11 The absence of dark metal granules helps distinguish hydroxychloroquine pigmentation from argyria.
Regressed melanomas may appear clinically as gray macules. These lesions arise in cases of malignant melanoma that spontaneously regress without treatment. Spontaneous regression occurs in 10% to 35% of cases depending on tumor subtype.12 Lesions can have a variable appearance based on the degree of regression. Partial regression is demonstrated by mixed melanosis and fibrosis in the dermis (Figure 5).13,14 Melanin is housed within melanophages present in a variably expanded papillary dermis. Tumors in early stages of regression can be surrounded by an inflammatory infiltrate, which becomes diminished at later stages. However, a few exceptional cases have been noted with extensive inflammatory infiltrate and no residual tumor.14 Completely regressed lesions typically appear as a band of dermal melanophages in the absence of inflammation or melanocytic atypia.15 The finding of regressed melanoma should prompt further investigation including sentinel lymph node biopsy, as it may be associated with metastasis.
Tattooing occurs following traumatic penetration of the skin with impregnation of pigmented foreign material into deep dermal layers.16 Histologic examination usually reveals clumps of fine particulate material in the dermis (Figure 6). The color of the pigment depends on the agent used. For example, graphite appears as black particles that may be confused with localized cutaneous argyria. Distinction can be made using elemental identification techniques such as energy-dispersive X-ray spectroscopy.1 The intensity of the pigment in granules found in tattoos or localized cutaneous argyria will fail to diminish with the application of melanin bleach.6
Localized cutaneous argyria often presents as asymptomatic black or blue-gray pigmented macules in areas of the skin exposed to silver-containing compounds.1 Silver may enter the skin by traumatic implantation or absorption via eccrine sweat glands.2 Our patient witnessed a gun fight several years ago while on a mission trip and sustained multiple shrapnel wounds.
As in our patient, hyperpigmentation may appear years following initial exposure. Over time, incident light reduces colorless silver salts and compounds to black elemental silver.3 It also has been suggested that metallic silver granules stimulate tyrosine kinase activity, leading to locally increased melanin production.4 Together, these processes result in the clinical appearance of a blue-black macule. Despite its long-standing association with silver, this appearance also has been noted with deposition of other metals.5 Histologically, metal deposits can be seen as black granules surrounding eccrine glands, blood vessels, and elastic fibers on higher magnification.6 Granules also may be found in sebaceous glands and arrector pili muscle fibers. These findings do not distinguish from generalized argyria due to increased serum silver levels; however, some cases of localized cutaneous argyria have demonstrated spheroid black globules with surrounding collagen necrosis,1 which have not been reported with generalized disease. Localized cutaneous argyria also may be associated with ocher pigmentation of thickened collagen fibers, resembling changes typically found in alkaptonuria, an inherited deficiency of homogentisic acid oxidase (an enzyme involved in tyrosine metabolism).7 The resulting buildup of metabolic intermediates leads to ochronosis, a deposition of ocher-pigmented intermediates in connective tissue throughout the body. In the skin, ocher pigmentation occurs in elastic fibers of the reticular dermis.1 Grossly, these changes result in a blue-gray discoloration of the skin due to a light-scattering phenomenon known as the Tyndall effect. Exogenous ochronosis also can occur, most commonly from the topical application of hydroquinone or other skin-lightening compounds.1,5 Ocher pigmentation occurring in the setting of localized cutaneous argyria is referred to as pseudo-ochronosis, a finding first described by Robinson-Bostom et al.1 The etiology of this condition is poorly understood, but Robinson-Bostom et al1 noted the appearance of dark metal granules surrounding collagen bundles and hypothesized that metal aggregates surrounding collagen bundles in pseudo-ochronosis cause a homogenized appearance under light microscopy. Yellow-brown, swollen, homogenized collagen bundles can be visualized in the reticular dermis with surrounding deposition of metal granules (Figures 1 and 2).1 Typical patterns of granule deposition in localized argyria also are present.
A blue nevus is a collection of proliferating dermal melanocytes. Many histologic subtypes exist and there may be extensive variability in the extent of sclerosis, cellular architecture, and tissue cellularity between each variant.8 Blue nevi commonly present as blue-black hyperpigmentation in the dermis and subcutaneous tissue.9 Histologically, they are characterized by slender, bipolar, dendritic melanocytes in a sclerotic stroma (Figure 3).8 Melanocytes are highly pigmented and contain small monomorphic nuclei. Lesions are relatively homogenous and typically are restricted to the dermis with epidermal sparing.9 Dark granules and ocher fibers are absent.
Long-term use of hydroxychloroquine or other antimalarials may cause a macular pattern of blue-gray hyperpigmentation.10 Biopsy specimens typically reveal coarse, yellow-brown pigment granules primarily affecting the superficial dermis (Figure 4). Granules are found both extracellularly and within macrophages. Fontana-Masson silver staining may identify melanin, as hydroxychloroquine-melanin binding may contribute to patterns of hyperpigmentation.10 Hemosiderin often is present in cases of hydroxychloroquine pigmentation. Preceding ecchymosis appears to favor the deposition of hydroxychloroquine in the skin.11 The absence of dark metal granules helps distinguish hydroxychloroquine pigmentation from argyria.
Regressed melanomas may appear clinically as gray macules. These lesions arise in cases of malignant melanoma that spontaneously regress without treatment. Spontaneous regression occurs in 10% to 35% of cases depending on tumor subtype.12 Lesions can have a variable appearance based on the degree of regression. Partial regression is demonstrated by mixed melanosis and fibrosis in the dermis (Figure 5).13,14 Melanin is housed within melanophages present in a variably expanded papillary dermis. Tumors in early stages of regression can be surrounded by an inflammatory infiltrate, which becomes diminished at later stages. However, a few exceptional cases have been noted with extensive inflammatory infiltrate and no residual tumor.14 Completely regressed lesions typically appear as a band of dermal melanophages in the absence of inflammation or melanocytic atypia.15 The finding of regressed melanoma should prompt further investigation including sentinel lymph node biopsy, as it may be associated with metastasis.
Tattooing occurs following traumatic penetration of the skin with impregnation of pigmented foreign material into deep dermal layers.16 Histologic examination usually reveals clumps of fine particulate material in the dermis (Figure 6). The color of the pigment depends on the agent used. For example, graphite appears as black particles that may be confused with localized cutaneous argyria. Distinction can be made using elemental identification techniques such as energy-dispersive X-ray spectroscopy.1 The intensity of the pigment in granules found in tattoos or localized cutaneous argyria will fail to diminish with the application of melanin bleach.6
- Robinson-Bostom L, Pomerantz D, Wilkel C, et al. Localized argyria with pseudo-ochronosis. J Am Acad Dermatol. 2002;46:222-227.
- Tajirian AL, Campbell RM, Robinson-Bostom L. Localized argyria after exposure to aerosolized solder. Cutis. 2006;78:305-308.
- Shelley WB, Shelley ED, Burmeister V. Argyria: the intradermal photograph, a manifestation of passive photosensitivity. J Am Acad Dermatol. 1987;16:211-217.
- Buckley WR, Terhaar CJ. The skin as an excretory organ in argyria. Trans St Johns Hosp Dermatol Soc. 1973;59:39-44.
- Shimizu I, Dill SW, McBean J, et al. Metal-induced granule deposition with pseudo-ochronosis. J Am Acad Dermatol. 2010;63:357-359.
- Rackoff EMJ, Benbenisty KM, Maize JC, et al. Localized cutaneous argyria from an acupuncture needle clini-cally concerning for metastatic melanoma. Cutis. 2007;80:423-426.
- Fernandez-Canon JM, Granadino B, Beltran-Valero de Bernabe D, et al. The molecular basis of alkaptonuria. Nat Genet. 1996;14:5-6.
- Busam KJ, Woodruff JM, Erlandson RA, et al. Large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2000;24:92-99.
- Granter SR, McKee PH, Calonje E, et al. Melanoma associated with blue nevus and melanoma mimicking cellular blue nevus: a clinicopathologic study of 10 cases on the spectrum of so-called ‘malignant blue nevus.’ Am J Surg Pathol. 2001;25:316.
- Puri PK, Lountzis NI, Tyler W, et al. Hydroxychloroquine-induced hyperpigmentation: the staining pattern. J Cutan Pathol. 2008;35:1134-1137.
- Jallouli M, Francès C, Piette JC, et al. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus: a case-control study. JAMA Dermatol. 2013;149:935-940.
- Blessing K, McLaren KM. Histological regression in primary cutaneous melanoma: recognition, prevalence and significance. Histopathology. 1992;20:315-322.
- LeBoit PE. Melanosis and its meanings. Am J Dermatopathol. 2002;24:369-372.
- Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181.
- Yang CH, Yeh JT, Shen SC, et al. Regressed subungual melanoma simulating cellular blue nevus: managed with sentinel lymph node biopsy. Dermatol Surg. 2006;32:577-581.
- Apfelberg DB, Manchester GH. Decorative and traumatic tattoo biophysics and removal. Clin Plast Surg. 1987;14:243-251.
- Robinson-Bostom L, Pomerantz D, Wilkel C, et al. Localized argyria with pseudo-ochronosis. J Am Acad Dermatol. 2002;46:222-227.
- Tajirian AL, Campbell RM, Robinson-Bostom L. Localized argyria after exposure to aerosolized solder. Cutis. 2006;78:305-308.
- Shelley WB, Shelley ED, Burmeister V. Argyria: the intradermal photograph, a manifestation of passive photosensitivity. J Am Acad Dermatol. 1987;16:211-217.
- Buckley WR, Terhaar CJ. The skin as an excretory organ in argyria. Trans St Johns Hosp Dermatol Soc. 1973;59:39-44.
- Shimizu I, Dill SW, McBean J, et al. Metal-induced granule deposition with pseudo-ochronosis. J Am Acad Dermatol. 2010;63:357-359.
- Rackoff EMJ, Benbenisty KM, Maize JC, et al. Localized cutaneous argyria from an acupuncture needle clini-cally concerning for metastatic melanoma. Cutis. 2007;80:423-426.
- Fernandez-Canon JM, Granadino B, Beltran-Valero de Bernabe D, et al. The molecular basis of alkaptonuria. Nat Genet. 1996;14:5-6.
- Busam KJ, Woodruff JM, Erlandson RA, et al. Large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2000;24:92-99.
- Granter SR, McKee PH, Calonje E, et al. Melanoma associated with blue nevus and melanoma mimicking cellular blue nevus: a clinicopathologic study of 10 cases on the spectrum of so-called ‘malignant blue nevus.’ Am J Surg Pathol. 2001;25:316.
- Puri PK, Lountzis NI, Tyler W, et al. Hydroxychloroquine-induced hyperpigmentation: the staining pattern. J Cutan Pathol. 2008;35:1134-1137.
- Jallouli M, Francès C, Piette JC, et al. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus: a case-control study. JAMA Dermatol. 2013;149:935-940.
- Blessing K, McLaren KM. Histological regression in primary cutaneous melanoma: recognition, prevalence and significance. Histopathology. 1992;20:315-322.
- LeBoit PE. Melanosis and its meanings. Am J Dermatopathol. 2002;24:369-372.
- Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181.
- Yang CH, Yeh JT, Shen SC, et al. Regressed subungual melanoma simulating cellular blue nevus: managed with sentinel lymph node biopsy. Dermatol Surg. 2006;32:577-581.
- Apfelberg DB, Manchester GH. Decorative and traumatic tattoo biophysics and removal. Clin Plast Surg. 1987;14:243-251.
Paclitaxel-Associated Melanonychia
To the Editor:
Taxane-based chemotherapy including paclitaxel and docetaxel is commonly used to treat solid tumor malignancies including lung, breast, ovarian, and bladder cancers.1 Taxanes work by interrupting normal microtubule function by inducing tubulin polymerization and inhibiting microtubule depolymerization, thereby leading to cell cycle arrest at the gap 2 (premitotic) and mitotic phase and the blockade of cell division.2
Cutaneous side effects have been reported with taxane-based therapies, including alopecia, skin rash and erythema, and desquamation of the hands and feet (hand-foot syndrome).3 Nail changes also have been reported to occur in 0% to 44% of treated patients,4 with one study reporting an incidence as high as 50.5%.5 Nail abnormalities that have been described primarily include onycholysis, and less frequently Beau lines, subungual hemorrhagic bullae, subungual hyperkeratosis, splinter hemorrhages, acute paronychia, and pigmentary changes such as nail bed dyschromia. Among the taxanes, nail abnormalities are more commonly seen with docetaxel; few reports address paclitaxel-induced nail changes.4 Onycholysis, diffuse fingernail orange discoloration, Beau lines, subungual distal hyperkeratosis, and brown discoloration of 3 fingernail beds sparing the lunula have been reported with paclitaxel.6-9 We report a unique case of paclitaxel-associated melanonychia.
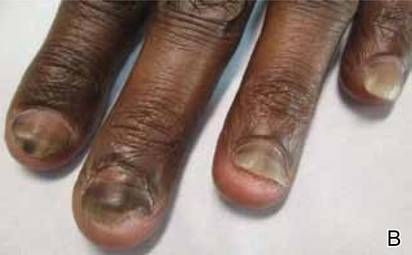
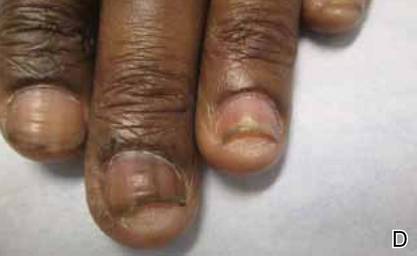
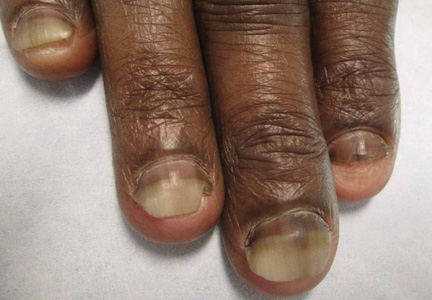
A 54-year-old black woman with a history of multiple myeloma and breast cancer who was being treated with paclitaxel for breast cancer presented with nail changes including nail darkening since initiating paclitaxel. She was diagnosed with multiple myeloma in 2010 and received bortezomib, dexamethasone, and an autologous stem cell transplant in August 2011. She never achieved complete remission but had been on lenalidomide with stable disease. She underwent a lumpectomy in December 2012, which revealed intraductal carcinoma with ductal carcinoma in situ that was estrogen receptor and progesterone receptor negative and ERBB2 (formerly HER2) positive. She was started on weekly paclitaxel (80 mg/m2) to complete 12 cycles and trastuzumab (6 mg/kg) every 3 weeks. While on paclitaxel, she developed grade 2 neuropathy of the hands, leading to subsequent dose reduction at week 9. She denied any other changes to her medications. On clinical examination she had diffuse and well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis involving all 20 nails (Figure, A and B). A nail clipping of the right hallux nail was sent for analysis. Pathology results showed evidence of scattered clusters of brown melanin pigment in the nail plate. Periodic acid–Schiff staining revealed numerous yeasts at the nail base but no infiltrating hyphae. Iron stain was negative for hemosiderin. The right index finger was injected with triamcinolone acetonide to treat the onycholysis. Four months after completing the paclitaxel, she began to notice lightening of the nails and improvement of the onycholysis in all nails (Figure, C and D).

| 
| |
| 
|
Initial appearance of diffuse, well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis in the nails on the right hand (A) and left hand (B). Four months after completing paclitaxel, the patient began to notice lightening of the nails and improvement of the onycholysis in the nails on the right hand (C) and left hand (D). |
The highly proliferating cells that comprise the nail matrix epithelium mature, differentiate, and keratinize to form the nail plate and are susceptible to the antimitotic effects of systemic chemotherapy. As a result, systemic chemotherapies may lead to abnormal nail plate production and keratinization of the nail plate, causing the clinical manifestations of Beau lines, onychomadesis, and leukonychia.10
Melanonychia is the development of melanin pigmentation of the nail plate and is typically caused by matrix melanin deposition through the activation of nail matrix melanocytes. There are 3 patterns of melanonychia: longitudinal, transverse, and diffuse. A single nail plate can involve more than one pattern of melanonychia and several nails may be affected. Longitudinal melanonychia typically develops from the activation of a group of melanocytes in the nail matrix, while diffuse pigmentation arises from diffuse melanocyte activation.11 Longitudinal melanonychia is common in darker-pigmented individuals12 and can be associated with systemic diseases.10 Transverse melanonychia has been reported in association with medications including many chemotherapy agents, and each band of transverse melanonychia may correspond to a cycle of therapy.11 Drug-induced melanonychia can affect several nails and tends to resolve after completion of therapy. Melanonychia has previously been described with vincristine, doxorubicin, hydroxyurea, cyclophosphamide, 5-fluorouracil, bleomycin, dacarbazine, methotrexate, and electron beam therapy.11 Nail pigmentation changes have been reported with docetaxel; a patient developed blue discoloration on the right and left thumb lunulae that improved 3 months after discontinuation of docetaxel therapy.13 While on docetaxel, another patient developed acral erythema, onycholysis, and longitudinal melanonychia in photoexposed areas, which was thought to be secondary to possible photosensitization.14 Possible explanations for paclitaxel-induced melanonychia include a direct toxic effect on the nail bed or nail matrix, focal stimulation of nail matrix melanocytes, or photosensitization. Drug-induced melanonychia commonly appears 3 to 8 weeks after drug intake and typically resolves 6 to 8 weeks after drug discontinuation.15
Predictors of taxane-related nail changes have been studied.5 Taxane-induced nail toxicity was more prevalent in patients who were female, had a history of diabetes mellitus, had received capecitabine with docetaxel, and had a diagnosis of breast or gynecological cancer. The nail changes increased with greater number of taxane cycles administered, body mass index, and severity of treatment-related neuropathy.5 Although nail changes often are temporary and typically resolve with drug withdrawal, they may persist in some patients.16 Possible measures have been proposed to prevent taxane-induced nail toxicity including frozen gloves,17 nail cutting, and avoiding potential fingernail irritants.18
It is possible that the nails of our darker-skinned patient may have been affected by some degree of melanonychia prior to starting the therapy, which cannot be ruled out. However, according to the patient, she only noticed the change after starting paclitaxel, raising the possibility of either new, worsening, or more diffuse involvement following initiation of paclitaxel therapy. Additionally, she was receiving weekly administration of paclitaxel and experienced severe neuropathy, both predictors of nail toxicity.5 No reports of melanonychia from lenalidomide have been reported in the literature indexed for MEDLINE. Although these nail changes are not life threatening, clinicians should be aware of these side effects, as they are cosmetically distressing to many patients and can impact quality of life.19
1. Crown J, O’Leary M. The taxanes: an update. Lancet. 2000;356:507-508.
2. Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by Taxol. Nature. 1979;277:665-667.
3. Heidary N, Naik H, Burgin S. Chemotherapeutic agents and the skin: an update. J Am Acad Dermatol. 2008;58:545-570.
4. Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;14:333-337.
5. Can G, Aydiner A, Cavdar I. Taxane-induced nail changes: predictors and efficacy of the use of frozen gloves and socks in the prevention of nail toxicity. Eur J Oncol Nurs. 2012;16:270-275.
6. Lüftner D, Flath B, Akrivakis C, et al. Dose-intensified weekly paclitaxel induces multiple nail disorders. Ann Oncol. 1998;9:1139-1141.
7. Hussain S, Anderson DN, Salvatti ME, et al. Onycholysis as a complication of systemic chemotherapy. report of five cases associated with prolonged weekly paclitaxel therapy and review of the literature. Cancer. 2000;88:2367-2371.
8. Almagro M, Del Pozo J, Garcia-Silva J, et al. Nail alterations secondary to paclitaxel therapy. Eur J Dermatol. 2000;10:146-147.
9. Flory SM, Solimando DA Jr, Webster GF, et al. Onycholysis associated with weekly administration of paclitaxel. Ann Pharmacother. 1999;33:584-586.
10. Hinds G, Thomas VD. Malignancy and cancer treatment-related hair and nail changes. Dermatol Clin. 2008;26:59-68.
11. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Practice. 2009;15:143-55.
12. Buka R, Friedman KA, Phelps RG, et al. Childhood longitudinal melanonychia: case reports and review of the literature. Mt Sinai J Med. 2001;68:331-335.
13. Halvorson CR, Erickson CL, Gaspari AA. A rare manifestation of nail changes with docetaxel therapy. Skinmed. 2010;8:179-180.
14. Ferreira O, Baudrier T, Mota A, et al. Docetaxel-induced acral erythema and nail changes distributed to photoexposed areas. Cutan Ocul Toxicol. 2010;29:296-299.
15. Piraccini BM, Iorizzo M. Drug reactions affecting the nail unit: diagnosis and management. Dermatol Clin. 2007;25:215-221.
16. Piraccini BM, Tosti A. Drug-induced nail disorders: incidence, management and prognosis. Drug Saf. 1999;21:187-201.
17. Scotté F, Tourani JM, Banu E, et al. Multicenter study of a frozen glove to prevent docetaxel-induced onycholysis and cutaneous toxicity of the hand. J Clin Oncol. 2005;23:4424-4429.
18. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Pract. 2009;15:143-155.
19. Hackbarth M, Haas N, Fotopoulou C, et al. Chemotherapy-induced dermatological toxicity: frequencies and impact on quality of life in women’s cancers. results of a prospective study. Support Care Cancer. 2008;16:267-273.
To the Editor:
Taxane-based chemotherapy including paclitaxel and docetaxel is commonly used to treat solid tumor malignancies including lung, breast, ovarian, and bladder cancers.1 Taxanes work by interrupting normal microtubule function by inducing tubulin polymerization and inhibiting microtubule depolymerization, thereby leading to cell cycle arrest at the gap 2 (premitotic) and mitotic phase and the blockade of cell division.2
Cutaneous side effects have been reported with taxane-based therapies, including alopecia, skin rash and erythema, and desquamation of the hands and feet (hand-foot syndrome).3 Nail changes also have been reported to occur in 0% to 44% of treated patients,4 with one study reporting an incidence as high as 50.5%.5 Nail abnormalities that have been described primarily include onycholysis, and less frequently Beau lines, subungual hemorrhagic bullae, subungual hyperkeratosis, splinter hemorrhages, acute paronychia, and pigmentary changes such as nail bed dyschromia. Among the taxanes, nail abnormalities are more commonly seen with docetaxel; few reports address paclitaxel-induced nail changes.4 Onycholysis, diffuse fingernail orange discoloration, Beau lines, subungual distal hyperkeratosis, and brown discoloration of 3 fingernail beds sparing the lunula have been reported with paclitaxel.6-9 We report a unique case of paclitaxel-associated melanonychia.
A 54-year-old black woman with a history of multiple myeloma and breast cancer who was being treated with paclitaxel for breast cancer presented with nail changes including nail darkening since initiating paclitaxel. She was diagnosed with multiple myeloma in 2010 and received bortezomib, dexamethasone, and an autologous stem cell transplant in August 2011. She never achieved complete remission but had been on lenalidomide with stable disease. She underwent a lumpectomy in December 2012, which revealed intraductal carcinoma with ductal carcinoma in situ that was estrogen receptor and progesterone receptor negative and ERBB2 (formerly HER2) positive. She was started on weekly paclitaxel (80 mg/m2) to complete 12 cycles and trastuzumab (6 mg/kg) every 3 weeks. While on paclitaxel, she developed grade 2 neuropathy of the hands, leading to subsequent dose reduction at week 9. She denied any other changes to her medications. On clinical examination she had diffuse and well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis involving all 20 nails (Figure, A and B). A nail clipping of the right hallux nail was sent for analysis. Pathology results showed evidence of scattered clusters of brown melanin pigment in the nail plate. Periodic acid–Schiff staining revealed numerous yeasts at the nail base but no infiltrating hyphae. Iron stain was negative for hemosiderin. The right index finger was injected with triamcinolone acetonide to treat the onycholysis. Four months after completing the paclitaxel, she began to notice lightening of the nails and improvement of the onycholysis in all nails (Figure, C and D).
|
|
| |
|
|
|
Initial appearance of diffuse, well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis in the nails on the right hand (A) and left hand (B). Four months after completing paclitaxel, the patient began to notice lightening of the nails and improvement of the onycholysis in the nails on the right hand (C) and left hand (D). |
The highly proliferating cells that comprise the nail matrix epithelium mature, differentiate, and keratinize to form the nail plate and are susceptible to the antimitotic effects of systemic chemotherapy. As a result, systemic chemotherapies may lead to abnormal nail plate production and keratinization of the nail plate, causing the clinical manifestations of Beau lines, onychomadesis, and leukonychia.10
Melanonychia is the development of melanin pigmentation of the nail plate and is typically caused by matrix melanin deposition through the activation of nail matrix melanocytes. There are 3 patterns of melanonychia: longitudinal, transverse, and diffuse. A single nail plate can involve more than one pattern of melanonychia and several nails may be affected. Longitudinal melanonychia typically develops from the activation of a group of melanocytes in the nail matrix, while diffuse pigmentation arises from diffuse melanocyte activation.11 Longitudinal melanonychia is common in darker-pigmented individuals12 and can be associated with systemic diseases.10 Transverse melanonychia has been reported in association with medications including many chemotherapy agents, and each band of transverse melanonychia may correspond to a cycle of therapy.11 Drug-induced melanonychia can affect several nails and tends to resolve after completion of therapy. Melanonychia has previously been described with vincristine, doxorubicin, hydroxyurea, cyclophosphamide, 5-fluorouracil, bleomycin, dacarbazine, methotrexate, and electron beam therapy.11 Nail pigmentation changes have been reported with docetaxel; a patient developed blue discoloration on the right and left thumb lunulae that improved 3 months after discontinuation of docetaxel therapy.13 While on docetaxel, another patient developed acral erythema, onycholysis, and longitudinal melanonychia in photoexposed areas, which was thought to be secondary to possible photosensitization.14 Possible explanations for paclitaxel-induced melanonychia include a direct toxic effect on the nail bed or nail matrix, focal stimulation of nail matrix melanocytes, or photosensitization. Drug-induced melanonychia commonly appears 3 to 8 weeks after drug intake and typically resolves 6 to 8 weeks after drug discontinuation.15
Predictors of taxane-related nail changes have been studied.5 Taxane-induced nail toxicity was more prevalent in patients who were female, had a history of diabetes mellitus, had received capecitabine with docetaxel, and had a diagnosis of breast or gynecological cancer. The nail changes increased with greater number of taxane cycles administered, body mass index, and severity of treatment-related neuropathy.5 Although nail changes often are temporary and typically resolve with drug withdrawal, they may persist in some patients.16 Possible measures have been proposed to prevent taxane-induced nail toxicity including frozen gloves,17 nail cutting, and avoiding potential fingernail irritants.18
It is possible that the nails of our darker-skinned patient may have been affected by some degree of melanonychia prior to starting the therapy, which cannot be ruled out. However, according to the patient, she only noticed the change after starting paclitaxel, raising the possibility of either new, worsening, or more diffuse involvement following initiation of paclitaxel therapy. Additionally, she was receiving weekly administration of paclitaxel and experienced severe neuropathy, both predictors of nail toxicity.5 No reports of melanonychia from lenalidomide have been reported in the literature indexed for MEDLINE. Although these nail changes are not life threatening, clinicians should be aware of these side effects, as they are cosmetically distressing to many patients and can impact quality of life.19
To the Editor:
Taxane-based chemotherapy including paclitaxel and docetaxel is commonly used to treat solid tumor malignancies including lung, breast, ovarian, and bladder cancers.1 Taxanes work by interrupting normal microtubule function by inducing tubulin polymerization and inhibiting microtubule depolymerization, thereby leading to cell cycle arrest at the gap 2 (premitotic) and mitotic phase and the blockade of cell division.2
Cutaneous side effects have been reported with taxane-based therapies, including alopecia, skin rash and erythema, and desquamation of the hands and feet (hand-foot syndrome).3 Nail changes also have been reported to occur in 0% to 44% of treated patients,4 with one study reporting an incidence as high as 50.5%.5 Nail abnormalities that have been described primarily include onycholysis, and less frequently Beau lines, subungual hemorrhagic bullae, subungual hyperkeratosis, splinter hemorrhages, acute paronychia, and pigmentary changes such as nail bed dyschromia. Among the taxanes, nail abnormalities are more commonly seen with docetaxel; few reports address paclitaxel-induced nail changes.4 Onycholysis, diffuse fingernail orange discoloration, Beau lines, subungual distal hyperkeratosis, and brown discoloration of 3 fingernail beds sparing the lunula have been reported with paclitaxel.6-9 We report a unique case of paclitaxel-associated melanonychia.
A 54-year-old black woman with a history of multiple myeloma and breast cancer who was being treated with paclitaxel for breast cancer presented with nail changes including nail darkening since initiating paclitaxel. She was diagnosed with multiple myeloma in 2010 and received bortezomib, dexamethasone, and an autologous stem cell transplant in August 2011. She never achieved complete remission but had been on lenalidomide with stable disease. She underwent a lumpectomy in December 2012, which revealed intraductal carcinoma with ductal carcinoma in situ that was estrogen receptor and progesterone receptor negative and ERBB2 (formerly HER2) positive. She was started on weekly paclitaxel (80 mg/m2) to complete 12 cycles and trastuzumab (6 mg/kg) every 3 weeks. While on paclitaxel, she developed grade 2 neuropathy of the hands, leading to subsequent dose reduction at week 9. She denied any other changes to her medications. On clinical examination she had diffuse and well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis involving all 20 nails (Figure, A and B). A nail clipping of the right hallux nail was sent for analysis. Pathology results showed evidence of scattered clusters of brown melanin pigment in the nail plate. Periodic acid–Schiff staining revealed numerous yeasts at the nail base but no infiltrating hyphae. Iron stain was negative for hemosiderin. The right index finger was injected with triamcinolone acetonide to treat the onycholysis. Four months after completing the paclitaxel, she began to notice lightening of the nails and improvement of the onycholysis in all nails (Figure, C and D).
|
|
| |
|
|
|
Initial appearance of diffuse, well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis in the nails on the right hand (A) and left hand (B). Four months after completing paclitaxel, the patient began to notice lightening of the nails and improvement of the onycholysis in the nails on the right hand (C) and left hand (D). |
The highly proliferating cells that comprise the nail matrix epithelium mature, differentiate, and keratinize to form the nail plate and are susceptible to the antimitotic effects of systemic chemotherapy. As a result, systemic chemotherapies may lead to abnormal nail plate production and keratinization of the nail plate, causing the clinical manifestations of Beau lines, onychomadesis, and leukonychia.10
Melanonychia is the development of melanin pigmentation of the nail plate and is typically caused by matrix melanin deposition through the activation of nail matrix melanocytes. There are 3 patterns of melanonychia: longitudinal, transverse, and diffuse. A single nail plate can involve more than one pattern of melanonychia and several nails may be affected. Longitudinal melanonychia typically develops from the activation of a group of melanocytes in the nail matrix, while diffuse pigmentation arises from diffuse melanocyte activation.11 Longitudinal melanonychia is common in darker-pigmented individuals12 and can be associated with systemic diseases.10 Transverse melanonychia has been reported in association with medications including many chemotherapy agents, and each band of transverse melanonychia may correspond to a cycle of therapy.11 Drug-induced melanonychia can affect several nails and tends to resolve after completion of therapy. Melanonychia has previously been described with vincristine, doxorubicin, hydroxyurea, cyclophosphamide, 5-fluorouracil, bleomycin, dacarbazine, methotrexate, and electron beam therapy.11 Nail pigmentation changes have been reported with docetaxel; a patient developed blue discoloration on the right and left thumb lunulae that improved 3 months after discontinuation of docetaxel therapy.13 While on docetaxel, another patient developed acral erythema, onycholysis, and longitudinal melanonychia in photoexposed areas, which was thought to be secondary to possible photosensitization.14 Possible explanations for paclitaxel-induced melanonychia include a direct toxic effect on the nail bed or nail matrix, focal stimulation of nail matrix melanocytes, or photosensitization. Drug-induced melanonychia commonly appears 3 to 8 weeks after drug intake and typically resolves 6 to 8 weeks after drug discontinuation.15
Predictors of taxane-related nail changes have been studied.5 Taxane-induced nail toxicity was more prevalent in patients who were female, had a history of diabetes mellitus, had received capecitabine with docetaxel, and had a diagnosis of breast or gynecological cancer. The nail changes increased with greater number of taxane cycles administered, body mass index, and severity of treatment-related neuropathy.5 Although nail changes often are temporary and typically resolve with drug withdrawal, they may persist in some patients.16 Possible measures have been proposed to prevent taxane-induced nail toxicity including frozen gloves,17 nail cutting, and avoiding potential fingernail irritants.18
It is possible that the nails of our darker-skinned patient may have been affected by some degree of melanonychia prior to starting the therapy, which cannot be ruled out. However, according to the patient, she only noticed the change after starting paclitaxel, raising the possibility of either new, worsening, or more diffuse involvement following initiation of paclitaxel therapy. Additionally, she was receiving weekly administration of paclitaxel and experienced severe neuropathy, both predictors of nail toxicity.5 No reports of melanonychia from lenalidomide have been reported in the literature indexed for MEDLINE. Although these nail changes are not life threatening, clinicians should be aware of these side effects, as they are cosmetically distressing to many patients and can impact quality of life.19
1. Crown J, O’Leary M. The taxanes: an update. Lancet. 2000;356:507-508.
2. Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by Taxol. Nature. 1979;277:665-667.
3. Heidary N, Naik H, Burgin S. Chemotherapeutic agents and the skin: an update. J Am Acad Dermatol. 2008;58:545-570.
4. Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;14:333-337.
5. Can G, Aydiner A, Cavdar I. Taxane-induced nail changes: predictors and efficacy of the use of frozen gloves and socks in the prevention of nail toxicity. Eur J Oncol Nurs. 2012;16:270-275.
6. Lüftner D, Flath B, Akrivakis C, et al. Dose-intensified weekly paclitaxel induces multiple nail disorders. Ann Oncol. 1998;9:1139-1141.
7. Hussain S, Anderson DN, Salvatti ME, et al. Onycholysis as a complication of systemic chemotherapy. report of five cases associated with prolonged weekly paclitaxel therapy and review of the literature. Cancer. 2000;88:2367-2371.
8. Almagro M, Del Pozo J, Garcia-Silva J, et al. Nail alterations secondary to paclitaxel therapy. Eur J Dermatol. 2000;10:146-147.
9. Flory SM, Solimando DA Jr, Webster GF, et al. Onycholysis associated with weekly administration of paclitaxel. Ann Pharmacother. 1999;33:584-586.
10. Hinds G, Thomas VD. Malignancy and cancer treatment-related hair and nail changes. Dermatol Clin. 2008;26:59-68.
11. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Practice. 2009;15:143-55.
12. Buka R, Friedman KA, Phelps RG, et al. Childhood longitudinal melanonychia: case reports and review of the literature. Mt Sinai J Med. 2001;68:331-335.
13. Halvorson CR, Erickson CL, Gaspari AA. A rare manifestation of nail changes with docetaxel therapy. Skinmed. 2010;8:179-180.
14. Ferreira O, Baudrier T, Mota A, et al. Docetaxel-induced acral erythema and nail changes distributed to photoexposed areas. Cutan Ocul Toxicol. 2010;29:296-299.
15. Piraccini BM, Iorizzo M. Drug reactions affecting the nail unit: diagnosis and management. Dermatol Clin. 2007;25:215-221.
16. Piraccini BM, Tosti A. Drug-induced nail disorders: incidence, management and prognosis. Drug Saf. 1999;21:187-201.
17. Scotté F, Tourani JM, Banu E, et al. Multicenter study of a frozen glove to prevent docetaxel-induced onycholysis and cutaneous toxicity of the hand. J Clin Oncol. 2005;23:4424-4429.
18. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Pract. 2009;15:143-155.
19. Hackbarth M, Haas N, Fotopoulou C, et al. Chemotherapy-induced dermatological toxicity: frequencies and impact on quality of life in women’s cancers. results of a prospective study. Support Care Cancer. 2008;16:267-273.
1. Crown J, O’Leary M. The taxanes: an update. Lancet. 2000;356:507-508.
2. Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by Taxol. Nature. 1979;277:665-667.
3. Heidary N, Naik H, Burgin S. Chemotherapeutic agents and the skin: an update. J Am Acad Dermatol. 2008;58:545-570.
4. Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;14:333-337.
5. Can G, Aydiner A, Cavdar I. Taxane-induced nail changes: predictors and efficacy of the use of frozen gloves and socks in the prevention of nail toxicity. Eur J Oncol Nurs. 2012;16:270-275.
6. Lüftner D, Flath B, Akrivakis C, et al. Dose-intensified weekly paclitaxel induces multiple nail disorders. Ann Oncol. 1998;9:1139-1141.
7. Hussain S, Anderson DN, Salvatti ME, et al. Onycholysis as a complication of systemic chemotherapy. report of five cases associated with prolonged weekly paclitaxel therapy and review of the literature. Cancer. 2000;88:2367-2371.
8. Almagro M, Del Pozo J, Garcia-Silva J, et al. Nail alterations secondary to paclitaxel therapy. Eur J Dermatol. 2000;10:146-147.
9. Flory SM, Solimando DA Jr, Webster GF, et al. Onycholysis associated with weekly administration of paclitaxel. Ann Pharmacother. 1999;33:584-586.
10. Hinds G, Thomas VD. Malignancy and cancer treatment-related hair and nail changes. Dermatol Clin. 2008;26:59-68.
11. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Practice. 2009;15:143-55.
12. Buka R, Friedman KA, Phelps RG, et al. Childhood longitudinal melanonychia: case reports and review of the literature. Mt Sinai J Med. 2001;68:331-335.
13. Halvorson CR, Erickson CL, Gaspari AA. A rare manifestation of nail changes with docetaxel therapy. Skinmed. 2010;8:179-180.
14. Ferreira O, Baudrier T, Mota A, et al. Docetaxel-induced acral erythema and nail changes distributed to photoexposed areas. Cutan Ocul Toxicol. 2010;29:296-299.
15. Piraccini BM, Iorizzo M. Drug reactions affecting the nail unit: diagnosis and management. Dermatol Clin. 2007;25:215-221.
16. Piraccini BM, Tosti A. Drug-induced nail disorders: incidence, management and prognosis. Drug Saf. 1999;21:187-201.
17. Scotté F, Tourani JM, Banu E, et al. Multicenter study of a frozen glove to prevent docetaxel-induced onycholysis and cutaneous toxicity of the hand. J Clin Oncol. 2005;23:4424-4429.
18. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Pract. 2009;15:143-155.
19. Hackbarth M, Haas N, Fotopoulou C, et al. Chemotherapy-induced dermatological toxicity: frequencies and impact on quality of life in women’s cancers. results of a prospective study. Support Care Cancer. 2008;16:267-273.
Verrucous Kaposi Sarcoma in an HIV-Positive Man
To the Editor:
Verrucous Kaposi sarcoma (VKS) is an uncommon variant of Kaposi sarcoma (KS) that rarely is seen in clinical practice or reported in the literature. It is strongly associated with lymphedema in patients with AIDS.1 We present a case of VKS in a human immunodeficiency virus (HIV)–positive man with cutaneous lesions that demonstrated minimal response to treatment with efavirenz-emtricitabine-tenofovir, doxorubicin, paclitaxel, and alitretinoin.
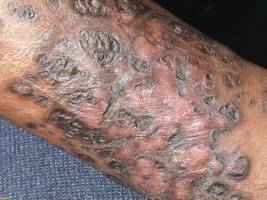
A 48-year-old man with a history of untreated HIV presented with a persistent eruption of heavily scaled, hyperpigmented, nonindurated, thin plaques in an ichthyosiform pattern on the bilateral lower legs and ankles of 4 years’ duration (Figure 1). He also had a number of soft, compressible, cystlike plaques without much overlying epidermal change on the lower extremities. He denied any prior episodes of skin breakdown, drainage, or secondary infection. Findings from the physical examination were otherwise unremarkable.
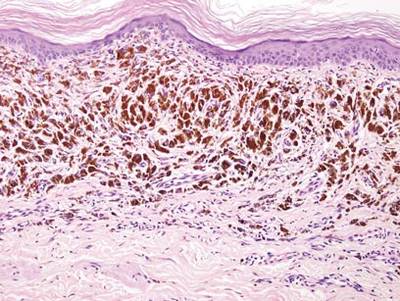
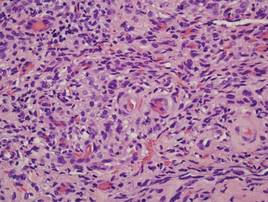
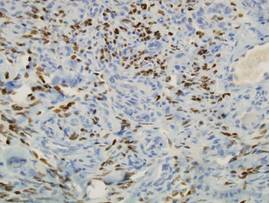
Two punch biopsies were performed on the lower legs, one from a scaly plaque and the other from a cystic area. The epidermis was hyperkeratotic and mildly hyperplastic with slitlike vascular spaces. A dense cellular proliferation of spindle-shaped cells was present in the dermis (Figure 2). Minimal cytologic atypia was noted. Immunohistochemical staining for human herpesvirus 8 (HHV-8) was strongly positive (Figure 3). Histologically, the cutaneous lesions were consistent with VKS.
At the current presentation, the CD4 count was 355 cells/mm3 and the viral load was 919,223 copies/mL. The CD4 count and viral load initially had been responsive to efavirenz-emtricitabine-tenofovir therapy; 17 months prior to the current presentation, the CD4 count was 692 cells/mm3 and the viral load was less than 50 copies/mL. However, the cutaneous lesions persisted despite therapy with efavirenz-emtricitabine-tenofovir, alitretinoin gel, and intralesional chemotherapeutic agents such as doxorubicin and paclitaxel.
Kaposi sarcoma, first described by Moritz Kaposi in 1872, represents a group of vascular neoplasms. Multiple subtypes have been described including classic, African endemic, transplant/AIDS associated, anaplastic, lymphedematous, hyperkeratotic/verrucous, keloidal, micronodular, pyogenic granulomalike, ecchymotic, and intravascular.1-3 Human herpesvirus 8 is associated with all clinical subtypes of KS.3 Immunohistochemical staining for HHV-8 latent nuclear antigen-1 has been shown in the literature to be highly sensitive and specific for KS and can potentially facilitate the diagnosis of KS among patients with similarly appearing dermatologic conditions, such as angiosarcoma, kaposiform hemangioendothelioma, or verrucous hemangioma.1,4 Human herpesvirus 8 infects endothelial cells and induces the proliferation of vascular spindle cells via the secretion of basic fibroblast growth factor and vascular endothelial growth factor.5 Human herpesvirus 8 also can lead to lymph vessel obstruction and lymph node enlargement by infecting cells within the lymphatic system. In addition, chronic lymphedema can itself lead to verruciform epidermal hyperplasia and hyperkeratosis, which has a clinical presentation similar to VKS.1
AIDS-associated KS typically starts as 1 or more purple-red macules that rapidly progress into papules, nodules, and plaques.1 These lesions have a predilection for the head, neck, trunk, and mucous membranes. Albeit a rare presentation, VKS is strongly associated with lymphedema in patients with AIDS.1,3,5 Previously, KS was often the presenting clinical manifestation of HIV infection, but since the use of highly active antiretroviral therapy (HAART) has become the standard of care, the incidence as well as the morbidity and mortality associated with KS has substantially decreased.1,5-7 Notably, in HIV patients who initially do not have signs or symptoms of KS, HHV-8 positivity is predictive of the development of KS within 2 to 4 years.6
In the literature, good prognostic indicators for KS include CD4 count greater than 150 cells/mm3, only cutaneous involvement, and negative B symptoms (eg, temperature >38°C, night sweats, unintentional weight loss >10% of normal body weight within 6 months).7 Kaposi sarcoma cannot be completely cured but can be appropriately managed with medical intervention. All KS subtypes are sensitive to radiation therapy; recalcitrant localized lesions can be treated with excision, cryotherapy, alitretinoin gel, laser ablation, or locally injected interferon or chemotherapeutic agents (eg, vincristine, vinblastine, actinomycin D).5,6 Liposomal anthracyclines (doxorubicin) and paclitaxel are first- and second-line agents for advanced KS, respectively.6
In HIV-associated KS, lesions frequently involute with the initiation of HAART; however, the cutaneous lesions in our patient persisted despite initiation of efavirenz-emtricitabine-tenofovir. He also was given intralesional doxorubicin andpaclitaxel as well as topical alitretinoin but did not experience complete resolution of the cutaneous lesions. It is possible that patients with VKS are recalcitrant to typical treatment modalities and therefore may require unconventional therapies to achieve maximal clearance of cutaneous lesions.
Verrucous Kaposi sarcoma is a rare presentation of KS that is infrequently seen in clinical practice or reported in the literature.3 A PubMed search of articles indexed for MEDLINE using the search term verrucous Kaposi sarcoma yielded 13 articles, one of which included a case series of 5 patients with AIDS and hyperkeratotic KS in Germany in the 1990s.5 Four of the articles were written in French, German, or Portuguese.8-11 The remainder of the articles discussed variants of KS other than VKS.
Although most patients with HIV and KS effectively respond to HAART, it may be possible that VKS is more difficult to treat. In addition, immunohistochemical staining for HHV-8, in particular HHV-8 latent nuclear antigen-1, may be useful to diagnose KS in HIV patients with uncharacteristic or indeterminate cutaneous lesions. Further research is needed to identify and delineate various efficacious therapeutic options for recalcitrant KS, particularly VKS.
Acknowledgment
We are indebted to Antoinette F. Hood, MD, Norfolk, Virginia, who digitized our patient’s histopathology slides.
1. Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
2. Amodio E, Goedert JJ, Barozzi P, et al. Differences in Kaposi sarcoma-associated herpesvirus-specific and herpesvirus-non-specific immune responses in classic Kaposi sarcoma cases and matched controls in Sicily. Cancer Sci. 2011;102:1769-1773.
3. Fagone S, Cavaleri A, Camuto M, et al. Hyperkeratotic Kaposi sarcoma with leg lymphedema after prolonged corticosteroid therapy for SLE. case report and review of the literature. Minerva Med. 2001;92:177-202.
4. Cheuk W, Wong KO, Wong CS, et al. Immunostaining for human herpesvirus 8 latent nuclear antigen-1 helps distinguish Kaposi sarcoma from its mimickers. Am J Clin Pathol. 2004;121:335-342.
5. Hengge UR, Stocks K, Goos M. Acquired immune deficiency syndrome-related hyperkeratotic Kaposi’s sarcoma with severe lymphedema: report of 5 cases. Br J Dermatol. 2000;142:501-505.
6. James WD, Berger TG, Elston DM, eds. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, PA: WB Saunders; 2006.
7. Thomas S, Sindhu CB, Sreekumar S, et al. AIDS associated Kaposi’s Sarcoma. J Assoc Physicians India. 2011;59:387-389.
8. Mukai MM, Chaves T, Caldas L, et al. Primary Kaposi’s sarcoma of the penis [in Portuguese]. An Bras Dermatol. 2009;84:524-526.
9. Weidauer H, Tilgen W, Adler D. Kaposi’s sarcoma of the larynx [in German]. Laryngol Rhinol Otol (Stuttg). 1986;65:389-391.
10. Basset A. Clinical aspects of Kaposi’s disease [in French]. Bull Soc Pathol Exot Filiales. 1984;77(4, pt 2):529-532.
11. Wlotzke U, Hohenleutner U, Landthaler M. Dermatoses in leg amputees [in German]. Hautarzt. 1996;47:493-501.
To the Editor:
Verrucous Kaposi sarcoma (VKS) is an uncommon variant of Kaposi sarcoma (KS) that rarely is seen in clinical practice or reported in the literature. It is strongly associated with lymphedema in patients with AIDS.1 We present a case of VKS in a human immunodeficiency virus (HIV)–positive man with cutaneous lesions that demonstrated minimal response to treatment with efavirenz-emtricitabine-tenofovir, doxorubicin, paclitaxel, and alitretinoin.
A 48-year-old man with a history of untreated HIV presented with a persistent eruption of heavily scaled, hyperpigmented, nonindurated, thin plaques in an ichthyosiform pattern on the bilateral lower legs and ankles of 4 years’ duration (Figure 1). He also had a number of soft, compressible, cystlike plaques without much overlying epidermal change on the lower extremities. He denied any prior episodes of skin breakdown, drainage, or secondary infection. Findings from the physical examination were otherwise unremarkable.
Two punch biopsies were performed on the lower legs, one from a scaly plaque and the other from a cystic area. The epidermis was hyperkeratotic and mildly hyperplastic with slitlike vascular spaces. A dense cellular proliferation of spindle-shaped cells was present in the dermis (Figure 2). Minimal cytologic atypia was noted. Immunohistochemical staining for human herpesvirus 8 (HHV-8) was strongly positive (Figure 3). Histologically, the cutaneous lesions were consistent with VKS.
At the current presentation, the CD4 count was 355 cells/mm3 and the viral load was 919,223 copies/mL. The CD4 count and viral load initially had been responsive to efavirenz-emtricitabine-tenofovir therapy; 17 months prior to the current presentation, the CD4 count was 692 cells/mm3 and the viral load was less than 50 copies/mL. However, the cutaneous lesions persisted despite therapy with efavirenz-emtricitabine-tenofovir, alitretinoin gel, and intralesional chemotherapeutic agents such as doxorubicin and paclitaxel.
Kaposi sarcoma, first described by Moritz Kaposi in 1872, represents a group of vascular neoplasms. Multiple subtypes have been described including classic, African endemic, transplant/AIDS associated, anaplastic, lymphedematous, hyperkeratotic/verrucous, keloidal, micronodular, pyogenic granulomalike, ecchymotic, and intravascular.1-3 Human herpesvirus 8 is associated with all clinical subtypes of KS.3 Immunohistochemical staining for HHV-8 latent nuclear antigen-1 has been shown in the literature to be highly sensitive and specific for KS and can potentially facilitate the diagnosis of KS among patients with similarly appearing dermatologic conditions, such as angiosarcoma, kaposiform hemangioendothelioma, or verrucous hemangioma.1,4 Human herpesvirus 8 infects endothelial cells and induces the proliferation of vascular spindle cells via the secretion of basic fibroblast growth factor and vascular endothelial growth factor.5 Human herpesvirus 8 also can lead to lymph vessel obstruction and lymph node enlargement by infecting cells within the lymphatic system. In addition, chronic lymphedema can itself lead to verruciform epidermal hyperplasia and hyperkeratosis, which has a clinical presentation similar to VKS.1
AIDS-associated KS typically starts as 1 or more purple-red macules that rapidly progress into papules, nodules, and plaques.1 These lesions have a predilection for the head, neck, trunk, and mucous membranes. Albeit a rare presentation, VKS is strongly associated with lymphedema in patients with AIDS.1,3,5 Previously, KS was often the presenting clinical manifestation of HIV infection, but since the use of highly active antiretroviral therapy (HAART) has become the standard of care, the incidence as well as the morbidity and mortality associated with KS has substantially decreased.1,5-7 Notably, in HIV patients who initially do not have signs or symptoms of KS, HHV-8 positivity is predictive of the development of KS within 2 to 4 years.6
In the literature, good prognostic indicators for KS include CD4 count greater than 150 cells/mm3, only cutaneous involvement, and negative B symptoms (eg, temperature >38°C, night sweats, unintentional weight loss >10% of normal body weight within 6 months).7 Kaposi sarcoma cannot be completely cured but can be appropriately managed with medical intervention. All KS subtypes are sensitive to radiation therapy; recalcitrant localized lesions can be treated with excision, cryotherapy, alitretinoin gel, laser ablation, or locally injected interferon or chemotherapeutic agents (eg, vincristine, vinblastine, actinomycin D).5,6 Liposomal anthracyclines (doxorubicin) and paclitaxel are first- and second-line agents for advanced KS, respectively.6
In HIV-associated KS, lesions frequently involute with the initiation of HAART; however, the cutaneous lesions in our patient persisted despite initiation of efavirenz-emtricitabine-tenofovir. He also was given intralesional doxorubicin andpaclitaxel as well as topical alitretinoin but did not experience complete resolution of the cutaneous lesions. It is possible that patients with VKS are recalcitrant to typical treatment modalities and therefore may require unconventional therapies to achieve maximal clearance of cutaneous lesions.
Verrucous Kaposi sarcoma is a rare presentation of KS that is infrequently seen in clinical practice or reported in the literature.3 A PubMed search of articles indexed for MEDLINE using the search term verrucous Kaposi sarcoma yielded 13 articles, one of which included a case series of 5 patients with AIDS and hyperkeratotic KS in Germany in the 1990s.5 Four of the articles were written in French, German, or Portuguese.8-11 The remainder of the articles discussed variants of KS other than VKS.
Although most patients with HIV and KS effectively respond to HAART, it may be possible that VKS is more difficult to treat. In addition, immunohistochemical staining for HHV-8, in particular HHV-8 latent nuclear antigen-1, may be useful to diagnose KS in HIV patients with uncharacteristic or indeterminate cutaneous lesions. Further research is needed to identify and delineate various efficacious therapeutic options for recalcitrant KS, particularly VKS.
Acknowledgment
We are indebted to Antoinette F. Hood, MD, Norfolk, Virginia, who digitized our patient’s histopathology slides.
To the Editor:
Verrucous Kaposi sarcoma (VKS) is an uncommon variant of Kaposi sarcoma (KS) that rarely is seen in clinical practice or reported in the literature. It is strongly associated with lymphedema in patients with AIDS.1 We present a case of VKS in a human immunodeficiency virus (HIV)–positive man with cutaneous lesions that demonstrated minimal response to treatment with efavirenz-emtricitabine-tenofovir, doxorubicin, paclitaxel, and alitretinoin.
A 48-year-old man with a history of untreated HIV presented with a persistent eruption of heavily scaled, hyperpigmented, nonindurated, thin plaques in an ichthyosiform pattern on the bilateral lower legs and ankles of 4 years’ duration (Figure 1). He also had a number of soft, compressible, cystlike plaques without much overlying epidermal change on the lower extremities. He denied any prior episodes of skin breakdown, drainage, or secondary infection. Findings from the physical examination were otherwise unremarkable.
Two punch biopsies were performed on the lower legs, one from a scaly plaque and the other from a cystic area. The epidermis was hyperkeratotic and mildly hyperplastic with slitlike vascular spaces. A dense cellular proliferation of spindle-shaped cells was present in the dermis (Figure 2). Minimal cytologic atypia was noted. Immunohistochemical staining for human herpesvirus 8 (HHV-8) was strongly positive (Figure 3). Histologically, the cutaneous lesions were consistent with VKS.
At the current presentation, the CD4 count was 355 cells/mm3 and the viral load was 919,223 copies/mL. The CD4 count and viral load initially had been responsive to efavirenz-emtricitabine-tenofovir therapy; 17 months prior to the current presentation, the CD4 count was 692 cells/mm3 and the viral load was less than 50 copies/mL. However, the cutaneous lesions persisted despite therapy with efavirenz-emtricitabine-tenofovir, alitretinoin gel, and intralesional chemotherapeutic agents such as doxorubicin and paclitaxel.
Kaposi sarcoma, first described by Moritz Kaposi in 1872, represents a group of vascular neoplasms. Multiple subtypes have been described including classic, African endemic, transplant/AIDS associated, anaplastic, lymphedematous, hyperkeratotic/verrucous, keloidal, micronodular, pyogenic granulomalike, ecchymotic, and intravascular.1-3 Human herpesvirus 8 is associated with all clinical subtypes of KS.3 Immunohistochemical staining for HHV-8 latent nuclear antigen-1 has been shown in the literature to be highly sensitive and specific for KS and can potentially facilitate the diagnosis of KS among patients with similarly appearing dermatologic conditions, such as angiosarcoma, kaposiform hemangioendothelioma, or verrucous hemangioma.1,4 Human herpesvirus 8 infects endothelial cells and induces the proliferation of vascular spindle cells via the secretion of basic fibroblast growth factor and vascular endothelial growth factor.5 Human herpesvirus 8 also can lead to lymph vessel obstruction and lymph node enlargement by infecting cells within the lymphatic system. In addition, chronic lymphedema can itself lead to verruciform epidermal hyperplasia and hyperkeratosis, which has a clinical presentation similar to VKS.1
AIDS-associated KS typically starts as 1 or more purple-red macules that rapidly progress into papules, nodules, and plaques.1 These lesions have a predilection for the head, neck, trunk, and mucous membranes. Albeit a rare presentation, VKS is strongly associated with lymphedema in patients with AIDS.1,3,5 Previously, KS was often the presenting clinical manifestation of HIV infection, but since the use of highly active antiretroviral therapy (HAART) has become the standard of care, the incidence as well as the morbidity and mortality associated with KS has substantially decreased.1,5-7 Notably, in HIV patients who initially do not have signs or symptoms of KS, HHV-8 positivity is predictive of the development of KS within 2 to 4 years.6
In the literature, good prognostic indicators for KS include CD4 count greater than 150 cells/mm3, only cutaneous involvement, and negative B symptoms (eg, temperature >38°C, night sweats, unintentional weight loss >10% of normal body weight within 6 months).7 Kaposi sarcoma cannot be completely cured but can be appropriately managed with medical intervention. All KS subtypes are sensitive to radiation therapy; recalcitrant localized lesions can be treated with excision, cryotherapy, alitretinoin gel, laser ablation, or locally injected interferon or chemotherapeutic agents (eg, vincristine, vinblastine, actinomycin D).5,6 Liposomal anthracyclines (doxorubicin) and paclitaxel are first- and second-line agents for advanced KS, respectively.6
In HIV-associated KS, lesions frequently involute with the initiation of HAART; however, the cutaneous lesions in our patient persisted despite initiation of efavirenz-emtricitabine-tenofovir. He also was given intralesional doxorubicin andpaclitaxel as well as topical alitretinoin but did not experience complete resolution of the cutaneous lesions. It is possible that patients with VKS are recalcitrant to typical treatment modalities and therefore may require unconventional therapies to achieve maximal clearance of cutaneous lesions.
Verrucous Kaposi sarcoma is a rare presentation of KS that is infrequently seen in clinical practice or reported in the literature.3 A PubMed search of articles indexed for MEDLINE using the search term verrucous Kaposi sarcoma yielded 13 articles, one of which included a case series of 5 patients with AIDS and hyperkeratotic KS in Germany in the 1990s.5 Four of the articles were written in French, German, or Portuguese.8-11 The remainder of the articles discussed variants of KS other than VKS.
Although most patients with HIV and KS effectively respond to HAART, it may be possible that VKS is more difficult to treat. In addition, immunohistochemical staining for HHV-8, in particular HHV-8 latent nuclear antigen-1, may be useful to diagnose KS in HIV patients with uncharacteristic or indeterminate cutaneous lesions. Further research is needed to identify and delineate various efficacious therapeutic options for recalcitrant KS, particularly VKS.
Acknowledgment
We are indebted to Antoinette F. Hood, MD, Norfolk, Virginia, who digitized our patient’s histopathology slides.
1. Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
2. Amodio E, Goedert JJ, Barozzi P, et al. Differences in Kaposi sarcoma-associated herpesvirus-specific and herpesvirus-non-specific immune responses in classic Kaposi sarcoma cases and matched controls in Sicily. Cancer Sci. 2011;102:1769-1773.
3. Fagone S, Cavaleri A, Camuto M, et al. Hyperkeratotic Kaposi sarcoma with leg lymphedema after prolonged corticosteroid therapy for SLE. case report and review of the literature. Minerva Med. 2001;92:177-202.
4. Cheuk W, Wong KO, Wong CS, et al. Immunostaining for human herpesvirus 8 latent nuclear antigen-1 helps distinguish Kaposi sarcoma from its mimickers. Am J Clin Pathol. 2004;121:335-342.
5. Hengge UR, Stocks K, Goos M. Acquired immune deficiency syndrome-related hyperkeratotic Kaposi’s sarcoma with severe lymphedema: report of 5 cases. Br J Dermatol. 2000;142:501-505.
6. James WD, Berger TG, Elston DM, eds. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, PA: WB Saunders; 2006.
7. Thomas S, Sindhu CB, Sreekumar S, et al. AIDS associated Kaposi’s Sarcoma. J Assoc Physicians India. 2011;59:387-389.
8. Mukai MM, Chaves T, Caldas L, et al. Primary Kaposi’s sarcoma of the penis [in Portuguese]. An Bras Dermatol. 2009;84:524-526.
9. Weidauer H, Tilgen W, Adler D. Kaposi’s sarcoma of the larynx [in German]. Laryngol Rhinol Otol (Stuttg). 1986;65:389-391.
10. Basset A. Clinical aspects of Kaposi’s disease [in French]. Bull Soc Pathol Exot Filiales. 1984;77(4, pt 2):529-532.
11. Wlotzke U, Hohenleutner U, Landthaler M. Dermatoses in leg amputees [in German]. Hautarzt. 1996;47:493-501.
1. Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
2. Amodio E, Goedert JJ, Barozzi P, et al. Differences in Kaposi sarcoma-associated herpesvirus-specific and herpesvirus-non-specific immune responses in classic Kaposi sarcoma cases and matched controls in Sicily. Cancer Sci. 2011;102:1769-1773.
3. Fagone S, Cavaleri A, Camuto M, et al. Hyperkeratotic Kaposi sarcoma with leg lymphedema after prolonged corticosteroid therapy for SLE. case report and review of the literature. Minerva Med. 2001;92:177-202.
4. Cheuk W, Wong KO, Wong CS, et al. Immunostaining for human herpesvirus 8 latent nuclear antigen-1 helps distinguish Kaposi sarcoma from its mimickers. Am J Clin Pathol. 2004;121:335-342.
5. Hengge UR, Stocks K, Goos M. Acquired immune deficiency syndrome-related hyperkeratotic Kaposi’s sarcoma with severe lymphedema: report of 5 cases. Br J Dermatol. 2000;142:501-505.
6. James WD, Berger TG, Elston DM, eds. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed. Philadelphia, PA: WB Saunders; 2006.
7. Thomas S, Sindhu CB, Sreekumar S, et al. AIDS associated Kaposi’s Sarcoma. J Assoc Physicians India. 2011;59:387-389.
8. Mukai MM, Chaves T, Caldas L, et al. Primary Kaposi’s sarcoma of the penis [in Portuguese]. An Bras Dermatol. 2009;84:524-526.
9. Weidauer H, Tilgen W, Adler D. Kaposi’s sarcoma of the larynx [in German]. Laryngol Rhinol Otol (Stuttg). 1986;65:389-391.
10. Basset A. Clinical aspects of Kaposi’s disease [in French]. Bull Soc Pathol Exot Filiales. 1984;77(4, pt 2):529-532.
11. Wlotzke U, Hohenleutner U, Landthaler M. Dermatoses in leg amputees [in German]. Hautarzt. 1996;47:493-501.
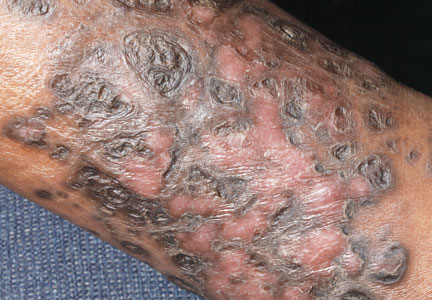
The Spectrum of Pigmented Purpuric Dermatosis and Mycosis Fungoides: Atypical T-Cell Dyscrasia
Case Report
A healthy 17-year-old adolescent boy with an unremarkable medical history presented with an asymptomatic fixed rash on the abdomen, buttocks, and legs. The rash initially developed in a small area on the right leg 2 years prior and had slowly progressed. He was not currently taking any medications and did not participate in intense physical activity. Multiple biopsies had previously been performed by an outside physician, the most recent one demonstrating an interface and superficial perivascular lymphocytic infiltrate with extravasated red blood cells consistent with pigmented purpura. He did not respond to treatment with intralesional corticosteroids, high-potency topical steroids, or high-dose oral prednisone.
Clinical examination revealed multiple annular purpuric patches on the abdomen, buttocks, and legs that covered approximately 20% of the body surface area (Figure 1). Over several follow-up visits, a few of the lesions evolved from patches to thin plaques. There was no adenopathy or hepatosplenomegaly. Three additional biopsies taken over the next 4 months demonstrated a mixture of small mature lymphocytes with some atypical lymphocytes in the dermis and epidermis exhibiting diminished CD7 staining and lymphocytes lining up at the dermoepidermal junction. T-cell receptor g gene rearrangements demonstrated the same clonal population in all 3 specimens. The patient was diagnosed with stage IB mycosis fungoides (MF) of the pigmented purpura–like variant. Marked improvement of the lesions was noted after 6 weeks of psoralen plus UVA therapy 3 times weekly (Figure 2). Treatment was continued for 6 months but was discontinued due to the international shortage of methoxsalen. Two months after discontinuation, most of the lesions had completely resolved (Figure 3).
|

Comment
Mycoses fungoides is a rare cutaneous lymphoma that affects approximately 2000 patients in the United States.1 Only 5% of all cases are known to occur in the first 2 decades of life,2 and even fewer cases pre-sent with pigmented purpura, usually of the lichenoid variant.3 Although the patches and plaques of MF can masquerade as many other dermatoses (eg, dermatophytosis, psoriasis, dermatitis), there have been few reports of patients presenting with lesions with the clinical appearance of pigmented purpuric dermatosis (PPD).4 As with the many cases of early MF, which are histologically indistinguishable from dermatitis, the pigmented purpura–like variant of MF initially may have the histologic appearance of pigmented purpura and generally evolves to the histologic appearance of MF over time.
Similar to our case, there have been reports of clinical and histologic diagnosis of PPD preceding the histologic diagnosis of MF. In a small cohort study of 3 young men, Barnhill and Braverman5 first demonstrated the progression of PPD to MF over a 12-year period. The age of onset ranged from 14 to 30 years, with a mean age of 24.3 years. Biopsies in all 3 patients were consistent with PPD for many years prior to the diagnosis of MF, with an average length of time to diagnosis of 8.4 years. Atypical from most cases of PPD, the patients in this study demonstrated extensive involvement of the trunk, arms, and legs.5 It has been suggested that atypical PPD is a variant of PPD that evolves into MF over many years; however, we believe that PPD is a variant of MF, similar to the way an indolent dermatitis may evolve to classical MF over time. If characterized by a T-cell clone, this period preceding the diagnosis of cutaneous T-cell lymphoma could be characterized as a cutaneous T-cell lymphoid dyscrasia.
Guitart and Magro6 noted multiple chronic conditions that are associated with T-cell clones, including PPD. These conditions occurred without a known trigger, were unresponsive to topical therapies, and often did not meet diagnostic criteria for MF. The investigators felt the criteria that may indicate a cutaneous T-cell lymphoid dyscrasia include widespread distribution, lymphocytic infiltrate, diminished CD7 and CD62L expression, and clonality. Lymphocytes may be small without notable atypia.6
In a study of 43 patients with PPD, Magro et al3 found monoclonality and diminished CD7 expression in 18 participants, correlating with large surface area involvement. Approximately 40% of patients had histologic findings consistent with MF, suggesting that T-cell gene rearrangement studies should be obtained for prognostic evaluation in patients with widespread disease.3

| 
|
To facilitate proper patient care, histopathology and molecular markers should be evaluated in conjunction with the clinical picture. A considerable increase in the size of the body surface area affected by purpuric patches combined with the presence of poikilodermatous changes and pruritus as well as disease lasting longer than 1 year should prompt an increased clinical suspicion of MF in patients with PPD.4,5 Histologically, the presence of Pautrier microabscesses, large cerebriform lymphocytes, and intraepidermal lymphocytic atypia extending beyond the dermis also would support a diagnosis of MF.3 Given the morphologic appearance and distribution of the lesions in our patient combined with epidermotropism, diminished CD7 expression, and monoclonality seen on pathology, we favored a diagnosis of MF. It would not be unreasonable to call this clonal variant of PPD a T-cell lymphoid dyscrasia. We appreciate that both PPD and MF will respond to phototherapy.7
Conclusion
We propose that there is a spectrum of disease presenting as PPD or MF sitting at either end of that spectrum and an intermediate stage, where not all criteria for cutaneous lymphoma are met, characterized as cutaneous T-cell lymphoid dyscrasia. Until the potential for evolution of PPD to malignant disease is better understood, patients with unusual presentations of pigmented purpura should be further evaluated for MF.
1. Criscione VD, Weinstock MA. Incidence of cutaneous T-cell lymphoma in the United States, 1973-2002. Arch Dermatol. 2007;143:854-859.
2. Koch SE, Zackheim HS, Williams ML, et al. Mycosis fungoides beginning in childhood and adolescence. J Am Acad Dermatol. 1987;17:563-570.
3. Magro CM, Schaefer JT, Crowson AN, et al. Pigmented purpuric dermatosis: classification by phenotypic and molecular profiles. Am J Clin Pathol. 2007;128:218-229.
4. Hanna S, Walsh N, D’Intino Y, et al. Mycosis fungoides presenting as pigmented purpuric dermatitis. Pediatr Dermatol. 2006;23:350-354.
5. Barnhill RL, Braverman IM. Progression of pigmented purpura-like eruptions to mycosis fungoides: report of three cases. J Am Acad Dermatol. 1988;19(1, pt 1):25-31.
6. Guitart J, Magro C. Cutaneous T-cell lymphoid dyscrasia: a unifying term for idiopathic chronic dermatoses with persistent T-cell clones. Arch Dermatol. 2007;143:921-932.
7. Seckin D, Yazici Z, Senol A, et al. A case of Schamberg’s disease responding dramatically to PUVA treatment. Photodermatol Photoimmunol Photomed. 2008;24:95-96.
Case Report
A healthy 17-year-old adolescent boy with an unremarkable medical history presented with an asymptomatic fixed rash on the abdomen, buttocks, and legs. The rash initially developed in a small area on the right leg 2 years prior and had slowly progressed. He was not currently taking any medications and did not participate in intense physical activity. Multiple biopsies had previously been performed by an outside physician, the most recent one demonstrating an interface and superficial perivascular lymphocytic infiltrate with extravasated red blood cells consistent with pigmented purpura. He did not respond to treatment with intralesional corticosteroids, high-potency topical steroids, or high-dose oral prednisone.
Clinical examination revealed multiple annular purpuric patches on the abdomen, buttocks, and legs that covered approximately 20% of the body surface area (Figure 1). Over several follow-up visits, a few of the lesions evolved from patches to thin plaques. There was no adenopathy or hepatosplenomegaly. Three additional biopsies taken over the next 4 months demonstrated a mixture of small mature lymphocytes with some atypical lymphocytes in the dermis and epidermis exhibiting diminished CD7 staining and lymphocytes lining up at the dermoepidermal junction. T-cell receptor g gene rearrangements demonstrated the same clonal population in all 3 specimens. The patient was diagnosed with stage IB mycosis fungoides (MF) of the pigmented purpura–like variant. Marked improvement of the lesions was noted after 6 weeks of psoralen plus UVA therapy 3 times weekly (Figure 2). Treatment was continued for 6 months but was discontinued due to the international shortage of methoxsalen. Two months after discontinuation, most of the lesions had completely resolved (Figure 3).
|
Comment
Mycoses fungoides is a rare cutaneous lymphoma that affects approximately 2000 patients in the United States.1 Only 5% of all cases are known to occur in the first 2 decades of life,2 and even fewer cases pre-sent with pigmented purpura, usually of the lichenoid variant.3 Although the patches and plaques of MF can masquerade as many other dermatoses (eg, dermatophytosis, psoriasis, dermatitis), there have been few reports of patients presenting with lesions with the clinical appearance of pigmented purpuric dermatosis (PPD).4 As with the many cases of early MF, which are histologically indistinguishable from dermatitis, the pigmented purpura–like variant of MF initially may have the histologic appearance of pigmented purpura and generally evolves to the histologic appearance of MF over time.
Similar to our case, there have been reports of clinical and histologic diagnosis of PPD preceding the histologic diagnosis of MF. In a small cohort study of 3 young men, Barnhill and Braverman5 first demonstrated the progression of PPD to MF over a 12-year period. The age of onset ranged from 14 to 30 years, with a mean age of 24.3 years. Biopsies in all 3 patients were consistent with PPD for many years prior to the diagnosis of MF, with an average length of time to diagnosis of 8.4 years. Atypical from most cases of PPD, the patients in this study demonstrated extensive involvement of the trunk, arms, and legs.5 It has been suggested that atypical PPD is a variant of PPD that evolves into MF over many years; however, we believe that PPD is a variant of MF, similar to the way an indolent dermatitis may evolve to classical MF over time. If characterized by a T-cell clone, this period preceding the diagnosis of cutaneous T-cell lymphoma could be characterized as a cutaneous T-cell lymphoid dyscrasia.
Guitart and Magro6 noted multiple chronic conditions that are associated with T-cell clones, including PPD. These conditions occurred without a known trigger, were unresponsive to topical therapies, and often did not meet diagnostic criteria for MF. The investigators felt the criteria that may indicate a cutaneous T-cell lymphoid dyscrasia include widespread distribution, lymphocytic infiltrate, diminished CD7 and CD62L expression, and clonality. Lymphocytes may be small without notable atypia.6
In a study of 43 patients with PPD, Magro et al3 found monoclonality and diminished CD7 expression in 18 participants, correlating with large surface area involvement. Approximately 40% of patients had histologic findings consistent with MF, suggesting that T-cell gene rearrangement studies should be obtained for prognostic evaluation in patients with widespread disease.3
|
|
|
To facilitate proper patient care, histopathology and molecular markers should be evaluated in conjunction with the clinical picture. A considerable increase in the size of the body surface area affected by purpuric patches combined with the presence of poikilodermatous changes and pruritus as well as disease lasting longer than 1 year should prompt an increased clinical suspicion of MF in patients with PPD.4,5 Histologically, the presence of Pautrier microabscesses, large cerebriform lymphocytes, and intraepidermal lymphocytic atypia extending beyond the dermis also would support a diagnosis of MF.3 Given the morphologic appearance and distribution of the lesions in our patient combined with epidermotropism, diminished CD7 expression, and monoclonality seen on pathology, we favored a diagnosis of MF. It would not be unreasonable to call this clonal variant of PPD a T-cell lymphoid dyscrasia. We appreciate that both PPD and MF will respond to phototherapy.7
Conclusion
We propose that there is a spectrum of disease presenting as PPD or MF sitting at either end of that spectrum and an intermediate stage, where not all criteria for cutaneous lymphoma are met, characterized as cutaneous T-cell lymphoid dyscrasia. Until the potential for evolution of PPD to malignant disease is better understood, patients with unusual presentations of pigmented purpura should be further evaluated for MF.
Case Report
A healthy 17-year-old adolescent boy with an unremarkable medical history presented with an asymptomatic fixed rash on the abdomen, buttocks, and legs. The rash initially developed in a small area on the right leg 2 years prior and had slowly progressed. He was not currently taking any medications and did not participate in intense physical activity. Multiple biopsies had previously been performed by an outside physician, the most recent one demonstrating an interface and superficial perivascular lymphocytic infiltrate with extravasated red blood cells consistent with pigmented purpura. He did not respond to treatment with intralesional corticosteroids, high-potency topical steroids, or high-dose oral prednisone.
Clinical examination revealed multiple annular purpuric patches on the abdomen, buttocks, and legs that covered approximately 20% of the body surface area (Figure 1). Over several follow-up visits, a few of the lesions evolved from patches to thin plaques. There was no adenopathy or hepatosplenomegaly. Three additional biopsies taken over the next 4 months demonstrated a mixture of small mature lymphocytes with some atypical lymphocytes in the dermis and epidermis exhibiting diminished CD7 staining and lymphocytes lining up at the dermoepidermal junction. T-cell receptor g gene rearrangements demonstrated the same clonal population in all 3 specimens. The patient was diagnosed with stage IB mycosis fungoides (MF) of the pigmented purpura–like variant. Marked improvement of the lesions was noted after 6 weeks of psoralen plus UVA therapy 3 times weekly (Figure 2). Treatment was continued for 6 months but was discontinued due to the international shortage of methoxsalen. Two months after discontinuation, most of the lesions had completely resolved (Figure 3).
|
Comment
Mycoses fungoides is a rare cutaneous lymphoma that affects approximately 2000 patients in the United States.1 Only 5% of all cases are known to occur in the first 2 decades of life,2 and even fewer cases pre-sent with pigmented purpura, usually of the lichenoid variant.3 Although the patches and plaques of MF can masquerade as many other dermatoses (eg, dermatophytosis, psoriasis, dermatitis), there have been few reports of patients presenting with lesions with the clinical appearance of pigmented purpuric dermatosis (PPD).4 As with the many cases of early MF, which are histologically indistinguishable from dermatitis, the pigmented purpura–like variant of MF initially may have the histologic appearance of pigmented purpura and generally evolves to the histologic appearance of MF over time.
Similar to our case, there have been reports of clinical and histologic diagnosis of PPD preceding the histologic diagnosis of MF. In a small cohort study of 3 young men, Barnhill and Braverman5 first demonstrated the progression of PPD to MF over a 12-year period. The age of onset ranged from 14 to 30 years, with a mean age of 24.3 years. Biopsies in all 3 patients were consistent with PPD for many years prior to the diagnosis of MF, with an average length of time to diagnosis of 8.4 years. Atypical from most cases of PPD, the patients in this study demonstrated extensive involvement of the trunk, arms, and legs.5 It has been suggested that atypical PPD is a variant of PPD that evolves into MF over many years; however, we believe that PPD is a variant of MF, similar to the way an indolent dermatitis may evolve to classical MF over time. If characterized by a T-cell clone, this period preceding the diagnosis of cutaneous T-cell lymphoma could be characterized as a cutaneous T-cell lymphoid dyscrasia.
Guitart and Magro6 noted multiple chronic conditions that are associated with T-cell clones, including PPD. These conditions occurred without a known trigger, were unresponsive to topical therapies, and often did not meet diagnostic criteria for MF. The investigators felt the criteria that may indicate a cutaneous T-cell lymphoid dyscrasia include widespread distribution, lymphocytic infiltrate, diminished CD7 and CD62L expression, and clonality. Lymphocytes may be small without notable atypia.6
In a study of 43 patients with PPD, Magro et al3 found monoclonality and diminished CD7 expression in 18 participants, correlating with large surface area involvement. Approximately 40% of patients had histologic findings consistent with MF, suggesting that T-cell gene rearrangement studies should be obtained for prognostic evaluation in patients with widespread disease.3
|
|
|
To facilitate proper patient care, histopathology and molecular markers should be evaluated in conjunction with the clinical picture. A considerable increase in the size of the body surface area affected by purpuric patches combined with the presence of poikilodermatous changes and pruritus as well as disease lasting longer than 1 year should prompt an increased clinical suspicion of MF in patients with PPD.4,5 Histologically, the presence of Pautrier microabscesses, large cerebriform lymphocytes, and intraepidermal lymphocytic atypia extending beyond the dermis also would support a diagnosis of MF.3 Given the morphologic appearance and distribution of the lesions in our patient combined with epidermotropism, diminished CD7 expression, and monoclonality seen on pathology, we favored a diagnosis of MF. It would not be unreasonable to call this clonal variant of PPD a T-cell lymphoid dyscrasia. We appreciate that both PPD and MF will respond to phototherapy.7
Conclusion
We propose that there is a spectrum of disease presenting as PPD or MF sitting at either end of that spectrum and an intermediate stage, where not all criteria for cutaneous lymphoma are met, characterized as cutaneous T-cell lymphoid dyscrasia. Until the potential for evolution of PPD to malignant disease is better understood, patients with unusual presentations of pigmented purpura should be further evaluated for MF.
1. Criscione VD, Weinstock MA. Incidence of cutaneous T-cell lymphoma in the United States, 1973-2002. Arch Dermatol. 2007;143:854-859.
2. Koch SE, Zackheim HS, Williams ML, et al. Mycosis fungoides beginning in childhood and adolescence. J Am Acad Dermatol. 1987;17:563-570.
3. Magro CM, Schaefer JT, Crowson AN, et al. Pigmented purpuric dermatosis: classification by phenotypic and molecular profiles. Am J Clin Pathol. 2007;128:218-229.
4. Hanna S, Walsh N, D’Intino Y, et al. Mycosis fungoides presenting as pigmented purpuric dermatitis. Pediatr Dermatol. 2006;23:350-354.
5. Barnhill RL, Braverman IM. Progression of pigmented purpura-like eruptions to mycosis fungoides: report of three cases. J Am Acad Dermatol. 1988;19(1, pt 1):25-31.
6. Guitart J, Magro C. Cutaneous T-cell lymphoid dyscrasia: a unifying term for idiopathic chronic dermatoses with persistent T-cell clones. Arch Dermatol. 2007;143:921-932.
7. Seckin D, Yazici Z, Senol A, et al. A case of Schamberg’s disease responding dramatically to PUVA treatment. Photodermatol Photoimmunol Photomed. 2008;24:95-96.
1. Criscione VD, Weinstock MA. Incidence of cutaneous T-cell lymphoma in the United States, 1973-2002. Arch Dermatol. 2007;143:854-859.
2. Koch SE, Zackheim HS, Williams ML, et al. Mycosis fungoides beginning in childhood and adolescence. J Am Acad Dermatol. 1987;17:563-570.
3. Magro CM, Schaefer JT, Crowson AN, et al. Pigmented purpuric dermatosis: classification by phenotypic and molecular profiles. Am J Clin Pathol. 2007;128:218-229.
4. Hanna S, Walsh N, D’Intino Y, et al. Mycosis fungoides presenting as pigmented purpuric dermatitis. Pediatr Dermatol. 2006;23:350-354.
5. Barnhill RL, Braverman IM. Progression of pigmented purpura-like eruptions to mycosis fungoides: report of three cases. J Am Acad Dermatol. 1988;19(1, pt 1):25-31.
6. Guitart J, Magro C. Cutaneous T-cell lymphoid dyscrasia: a unifying term for idiopathic chronic dermatoses with persistent T-cell clones. Arch Dermatol. 2007;143:921-932.
7. Seckin D, Yazici Z, Senol A, et al. A case of Schamberg’s disease responding dramatically to PUVA treatment. Photodermatol Photoimmunol Photomed. 2008;24:95-96.
Practice Points
- Pigmented purpuric dermatosis may lie on a spectrum with mycosis fungoides (MF).
- Pigmented purpuric dermatosis of MF should be closely followed and likely treated as MF.
- Pigmented purpuric dermatosis may have T-cell gene rearrangements that may or may not be associated with MF.
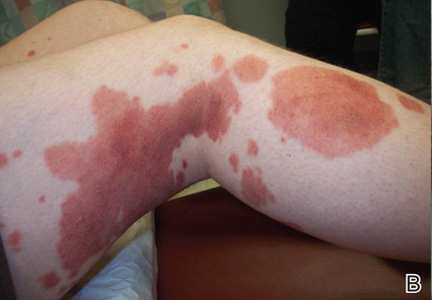
Hypopigmented Facial Papules on the Cheeks
The Diagnosis: Tumor of the Follicular Infundibulum
Histopathologic findings from a facial papule in our patient revealed multifocal hyperplasia of anastomosing follicular infundibular cells with multiple connections to the overlying epidermis (Figure). There was no atypia. Gomori methenamine-silver and periodic acid–Schiff stains for fungi were negative. The combined clinical presentation and histopathologic findings supported the diagnosis of multiple tumor of the follicular infundibulum (TFI).
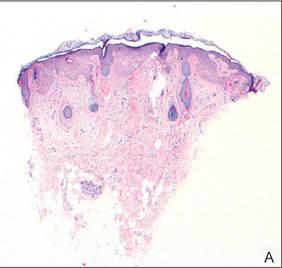
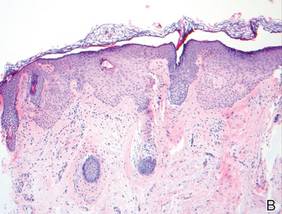
Tumor of the follicular infundibulum was diagnosed based on a biopsy from the right cheek that revealed multifocal hyperplasia of anastomosing follicular infundibular cells with multiple connections to the overlying epidermis (A and B)(H&E, original magnifications ×40 and ×100). |
Tumor of the follicular infundibulum is an uncommon benign neoplasm that was first described in 1961 by Mehregan and Butler.1 The reported frequency is 10 per 100,000 biopsies.2 The majority of cases have been reported as solitary lesions, and multiple TFI are rare.3 Tumor of the follicular infundibulum affects middle-aged and elderly individuals with a female predominance.4 Multiple lesions generally range in number from 10 to 20, but there are few reports of more than 100 lesions.2,3,5,6 The solitary tumors often are initially misdiagnosed as basal cell carcinomas (BCCs) or seborrheic keratosis. Multiple TFI have been described variably as hypopigmented, flesh-colored and pink, flat and slightly depressed macules and thin papules. Sites of predilection include the scalp, face, neck, and upper trunk.2,3,5
There is no histopathologic difference between solitary and multiple TFI. Tumor of the follicular infundibulum displays a characteristic pale platelike proliferation of keratinocytes within the upper dermis attached to the overlying epidermis. The proliferating cells stain positive with periodic acid–Schiff, diastase-digestible glycogen is present in the cells at the base of the tumor, and a thickened network or brushlike pattern of elastic fibers surrounds the periphery of the tumor.1 Tumor of the follicular infundibulum is occasionally discovered incidentally on biopsy and has been observed in the margin of wide excisions of a variety of neoplasms including BCC.7 Based on the close association of TFI and BCC in the same specimens, Weyers et al7 concluded that TFI may be a nonaggressive type of BCC. Cribier and Grosshans2 reported 2 cases of TFI overlying a nevus sebaceous and a fibroma.
Treatment of TFI includes topical keratolytics, topical retinoic acid,5 imiquimod,8 topical steroids, and oral etretinate,6 all of which result in minimal improvement or incomplete resolution. Destructive treatments include cryotherapy, curettage, electrosurgery, laser ablation, and surgical excision, but all may lead to an unacceptable cosmetic result.
1. Mehregan AH, Butler JD. A tumor of follicular infundibulum. Arch Dermatol. 1961;83:78-81.
2. Cribier B, Grosshans E. Tumor of the follicular infundibulum: a clinicopathologic study. J Am Acad Dermatol. 1995;33:979-984.
3. Kolenik SA 3rd, Bolognia JL, Castiglione FM Jr, et al. Multiple tumors of the follicular infundibulum. Int J Dermatol. 1996;35:282-284.
4. Ackerman AB, Reddy VB, Soyer HP. Neoplasms With Follicular Differentiation. New York, NY: Ardor Scribendi; 2001.
5. Kossard S, Finley AG, Poyzer K, et al. Eruptive infundibulomas. J Am Acad Dermatol. 1989;21:361-366.
6. Schnitzler L, Civatte J, Robin F, et al. Multiple tumors of the follicular infundibulum with basocellular degeneration. apropos of a case [in French]. Ann Dermatol Venereol. 1987;114:551-556.
7. Weyers W, Horster S, Diaz-Cascajo C. Tumor of follicular infundibulum is basal cell carcinoma. Am J Dermatopathol. 2009;31:634-641.
8. Martin JE, Hsu M, Wang LC. An unusual clinical presentation of multiple tumors of the follicular infundibulum. J Am Acad Dermatol. 2009;60:885-886.
The Diagnosis: Tumor of the Follicular Infundibulum
Histopathologic findings from a facial papule in our patient revealed multifocal hyperplasia of anastomosing follicular infundibular cells with multiple connections to the overlying epidermis (Figure). There was no atypia. Gomori methenamine-silver and periodic acid–Schiff stains for fungi were negative. The combined clinical presentation and histopathologic findings supported the diagnosis of multiple tumor of the follicular infundibulum (TFI).
|
Tumor of the follicular infundibulum was diagnosed based on a biopsy from the right cheek that revealed multifocal hyperplasia of anastomosing follicular infundibular cells with multiple connections to the overlying epidermis (A and B)(H&E, original magnifications ×40 and ×100). |
Tumor of the follicular infundibulum is an uncommon benign neoplasm that was first described in 1961 by Mehregan and Butler.1 The reported frequency is 10 per 100,000 biopsies.2 The majority of cases have been reported as solitary lesions, and multiple TFI are rare.3 Tumor of the follicular infundibulum affects middle-aged and elderly individuals with a female predominance.4 Multiple lesions generally range in number from 10 to 20, but there are few reports of more than 100 lesions.2,3,5,6 The solitary tumors often are initially misdiagnosed as basal cell carcinomas (BCCs) or seborrheic keratosis. Multiple TFI have been described variably as hypopigmented, flesh-colored and pink, flat and slightly depressed macules and thin papules. Sites of predilection include the scalp, face, neck, and upper trunk.2,3,5
There is no histopathologic difference between solitary and multiple TFI. Tumor of the follicular infundibulum displays a characteristic pale platelike proliferation of keratinocytes within the upper dermis attached to the overlying epidermis. The proliferating cells stain positive with periodic acid–Schiff, diastase-digestible glycogen is present in the cells at the base of the tumor, and a thickened network or brushlike pattern of elastic fibers surrounds the periphery of the tumor.1 Tumor of the follicular infundibulum is occasionally discovered incidentally on biopsy and has been observed in the margin of wide excisions of a variety of neoplasms including BCC.7 Based on the close association of TFI and BCC in the same specimens, Weyers et al7 concluded that TFI may be a nonaggressive type of BCC. Cribier and Grosshans2 reported 2 cases of TFI overlying a nevus sebaceous and a fibroma.
Treatment of TFI includes topical keratolytics, topical retinoic acid,5 imiquimod,8 topical steroids, and oral etretinate,6 all of which result in minimal improvement or incomplete resolution. Destructive treatments include cryotherapy, curettage, electrosurgery, laser ablation, and surgical excision, but all may lead to an unacceptable cosmetic result.
The Diagnosis: Tumor of the Follicular Infundibulum
Histopathologic findings from a facial papule in our patient revealed multifocal hyperplasia of anastomosing follicular infundibular cells with multiple connections to the overlying epidermis (Figure). There was no atypia. Gomori methenamine-silver and periodic acid–Schiff stains for fungi were negative. The combined clinical presentation and histopathologic findings supported the diagnosis of multiple tumor of the follicular infundibulum (TFI).
|
Tumor of the follicular infundibulum was diagnosed based on a biopsy from the right cheek that revealed multifocal hyperplasia of anastomosing follicular infundibular cells with multiple connections to the overlying epidermis (A and B)(H&E, original magnifications ×40 and ×100). |
Tumor of the follicular infundibulum is an uncommon benign neoplasm that was first described in 1961 by Mehregan and Butler.1 The reported frequency is 10 per 100,000 biopsies.2 The majority of cases have been reported as solitary lesions, and multiple TFI are rare.3 Tumor of the follicular infundibulum affects middle-aged and elderly individuals with a female predominance.4 Multiple lesions generally range in number from 10 to 20, but there are few reports of more than 100 lesions.2,3,5,6 The solitary tumors often are initially misdiagnosed as basal cell carcinomas (BCCs) or seborrheic keratosis. Multiple TFI have been described variably as hypopigmented, flesh-colored and pink, flat and slightly depressed macules and thin papules. Sites of predilection include the scalp, face, neck, and upper trunk.2,3,5
There is no histopathologic difference between solitary and multiple TFI. Tumor of the follicular infundibulum displays a characteristic pale platelike proliferation of keratinocytes within the upper dermis attached to the overlying epidermis. The proliferating cells stain positive with periodic acid–Schiff, diastase-digestible glycogen is present in the cells at the base of the tumor, and a thickened network or brushlike pattern of elastic fibers surrounds the periphery of the tumor.1 Tumor of the follicular infundibulum is occasionally discovered incidentally on biopsy and has been observed in the margin of wide excisions of a variety of neoplasms including BCC.7 Based on the close association of TFI and BCC in the same specimens, Weyers et al7 concluded that TFI may be a nonaggressive type of BCC. Cribier and Grosshans2 reported 2 cases of TFI overlying a nevus sebaceous and a fibroma.
Treatment of TFI includes topical keratolytics, topical retinoic acid,5 imiquimod,8 topical steroids, and oral etretinate,6 all of which result in minimal improvement or incomplete resolution. Destructive treatments include cryotherapy, curettage, electrosurgery, laser ablation, and surgical excision, but all may lead to an unacceptable cosmetic result.
1. Mehregan AH, Butler JD. A tumor of follicular infundibulum. Arch Dermatol. 1961;83:78-81.
2. Cribier B, Grosshans E. Tumor of the follicular infundibulum: a clinicopathologic study. J Am Acad Dermatol. 1995;33:979-984.
3. Kolenik SA 3rd, Bolognia JL, Castiglione FM Jr, et al. Multiple tumors of the follicular infundibulum. Int J Dermatol. 1996;35:282-284.
4. Ackerman AB, Reddy VB, Soyer HP. Neoplasms With Follicular Differentiation. New York, NY: Ardor Scribendi; 2001.
5. Kossard S, Finley AG, Poyzer K, et al. Eruptive infundibulomas. J Am Acad Dermatol. 1989;21:361-366.
6. Schnitzler L, Civatte J, Robin F, et al. Multiple tumors of the follicular infundibulum with basocellular degeneration. apropos of a case [in French]. Ann Dermatol Venereol. 1987;114:551-556.
7. Weyers W, Horster S, Diaz-Cascajo C. Tumor of follicular infundibulum is basal cell carcinoma. Am J Dermatopathol. 2009;31:634-641.
8. Martin JE, Hsu M, Wang LC. An unusual clinical presentation of multiple tumors of the follicular infundibulum. J Am Acad Dermatol. 2009;60:885-886.
1. Mehregan AH, Butler JD. A tumor of follicular infundibulum. Arch Dermatol. 1961;83:78-81.
2. Cribier B, Grosshans E. Tumor of the follicular infundibulum: a clinicopathologic study. J Am Acad Dermatol. 1995;33:979-984.
3. Kolenik SA 3rd, Bolognia JL, Castiglione FM Jr, et al. Multiple tumors of the follicular infundibulum. Int J Dermatol. 1996;35:282-284.
4. Ackerman AB, Reddy VB, Soyer HP. Neoplasms With Follicular Differentiation. New York, NY: Ardor Scribendi; 2001.
5. Kossard S, Finley AG, Poyzer K, et al. Eruptive infundibulomas. J Am Acad Dermatol. 1989;21:361-366.
6. Schnitzler L, Civatte J, Robin F, et al. Multiple tumors of the follicular infundibulum with basocellular degeneration. apropos of a case [in French]. Ann Dermatol Venereol. 1987;114:551-556.
7. Weyers W, Horster S, Diaz-Cascajo C. Tumor of follicular infundibulum is basal cell carcinoma. Am J Dermatopathol. 2009;31:634-641.
8. Martin JE, Hsu M, Wang LC. An unusual clinical presentation of multiple tumors of the follicular infundibulum. J Am Acad Dermatol. 2009;60:885-886.
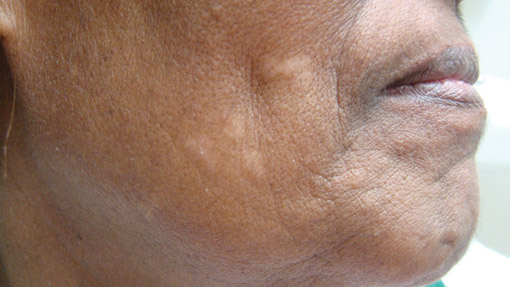
A 73-year-old woman presented with multiple mildly pruritic, hypopigmented, thin papules involving both cheeks of 5 months’ duration. The patient had no improvement with ketoconazole cream 2% and hydrocortisone cream 1% used daily for 1 month for presumed tinea versicolor. Physical examination revealed 10 ill-defined, 2- to 5-mm, round and oval, smooth hypopigmented, slightly raised papules located on the lower aspect of both cheeks.
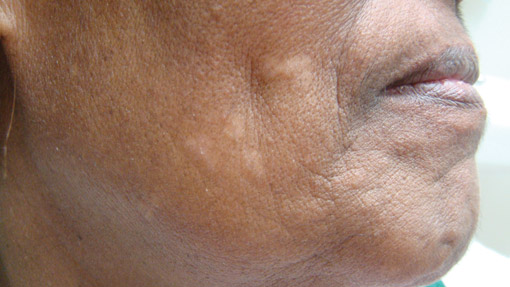
Cosmetic Corner: Dermatologists Weigh in on OTC Pigment Control Products
To improve patient care and outcomes, leading dermatologists offered their recommendations on the top OTC pigment control products. Consideration must be given to:
- Even Better
Clinique Laboratories, LLC
“It also is useful as prevention and offers many different options.”—Antonella Tosti, MD, Miami, Florida
“These OTC products have good clinical data to support use for hyperpigmentation. Patients tell me that they feel good on their skin and aren’t irritating.”—Gary Goldenberg, MD, New York, New York
- Lumixyl Brightening System
Envy Medical, Inc
“A great option for patients who may be experiencing modest issues with pigmentation. Use of a retinoid with this product also may enhance its efficacy.”—Joel Schlessinger, MD, Omaha, Nebraska
- Lytera Skin Brightening Complex
SkinMedica
“With key ingredients such as hexylresorcinol, retinol, and niacinamide, it has been clinically shown to lighten dark patches in its trials as well as adding luminosity to the skin.”—Anthony Rossi, MD, New York, New York
Recommended by Elizabeth K. Hale, MD, New York, New York
- Pigmentclar Serum
La-Roche Posay Laboratoire Dermatologique
“It attacks pigment production at every stage.”—Whitney Bowe, MD, Brooklyn, New York
Cutis invites readers to send us their recommendations. Mineral makeup, eyelash enhancers, and facial scrubs will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to cutis@frontlinemedcom.com.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on the top OTC pigment control products. Consideration must be given to:
- Even Better
Clinique Laboratories, LLC
“It also is useful as prevention and offers many different options.”—Antonella Tosti, MD, Miami, Florida
“These OTC products have good clinical data to support use for hyperpigmentation. Patients tell me that they feel good on their skin and aren’t irritating.”—Gary Goldenberg, MD, New York, New York
- Lumixyl Brightening System
Envy Medical, Inc
“A great option for patients who may be experiencing modest issues with pigmentation. Use of a retinoid with this product also may enhance its efficacy.”—Joel Schlessinger, MD, Omaha, Nebraska
- Lytera Skin Brightening Complex
SkinMedica
“With key ingredients such as hexylresorcinol, retinol, and niacinamide, it has been clinically shown to lighten dark patches in its trials as well as adding luminosity to the skin.”—Anthony Rossi, MD, New York, New York
Recommended by Elizabeth K. Hale, MD, New York, New York
- Pigmentclar Serum
La-Roche Posay Laboratoire Dermatologique
“It attacks pigment production at every stage.”—Whitney Bowe, MD, Brooklyn, New York
Cutis invites readers to send us their recommendations. Mineral makeup, eyelash enhancers, and facial scrubs will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to cutis@frontlinemedcom.com.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on the top OTC pigment control products. Consideration must be given to:
- Even Better
Clinique Laboratories, LLC
“It also is useful as prevention and offers many different options.”—Antonella Tosti, MD, Miami, Florida
“These OTC products have good clinical data to support use for hyperpigmentation. Patients tell me that they feel good on their skin and aren’t irritating.”—Gary Goldenberg, MD, New York, New York
- Lumixyl Brightening System
Envy Medical, Inc
“A great option for patients who may be experiencing modest issues with pigmentation. Use of a retinoid with this product also may enhance its efficacy.”—Joel Schlessinger, MD, Omaha, Nebraska
- Lytera Skin Brightening Complex
SkinMedica
“With key ingredients such as hexylresorcinol, retinol, and niacinamide, it has been clinically shown to lighten dark patches in its trials as well as adding luminosity to the skin.”—Anthony Rossi, MD, New York, New York
Recommended by Elizabeth K. Hale, MD, New York, New York
- Pigmentclar Serum
La-Roche Posay Laboratoire Dermatologique
“It attacks pigment production at every stage.”—Whitney Bowe, MD, Brooklyn, New York
Cutis invites readers to send us their recommendations. Mineral makeup, eyelash enhancers, and facial scrubs will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to cutis@frontlinemedcom.com.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
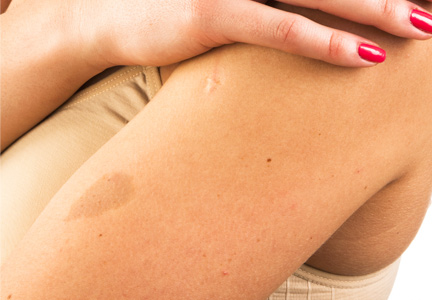
Granulomatous Changes Associated With Pigmented Purpuric Dermatosis
Pigmented purpuric dermatoses (PPDs) are a group of common chronic disorders characterized by speckled, cayenne pepper–like petechiae and orange-brown discoloration of the skin resulting from capillaritis.1 Pigmented purpuric dermatoses typically occur in the absence of underlying vascular insufficiency or other hematologic dysfunction. The 5 well-known clinicopathologic variants of PPD include Schamberg disease; purpura annularis telangiectodes of Majocchi; pigmented purpuric lichenoid dermatitis of Gougerot and Blum; eczematoidlike purpura of Doucas and Kapetanakis; and lichen aureus.2 All PPDs share common characteristic clinical and histologic features. Clinically, patients generally present with symmetric petechiae and/or pigmented macules. All 5 PPD variants share similar histologic findings, including a vasculocentric lymphocytic infiltrate in the papillary dermis, swelling of the endothelial cells, erythrocyte extravasation, and often hemosiderin-laden macrophages.1 Despite these clinical and histopathologic similarities, each variant contains additional distinctive features, such as telangiectasia (purpura annularis telangiectodes of Majocchi), a lichenoid infiltrate (pigmented purpuric lichenoid dermatitis of Gougerot and Blum), eczematous changes (eczematoidlike purpura of Doucas and Kapetanakis), and marked hemosiderin deposition (lichen aureus).
Granulomatous pigmented purpuric dermatosis (GPPD) is a rare variant of PPD.3-7 Clinically, these lesions appear similar to other PPDs; however, in addition to the characteristic changes associated with conventional PPD, histologic examination of GPPD reveals a granulomatous inflammatory reaction pattern. Although the pathogenesis of GPPD is not well understood, its association with hyperlipidemia may suggest a granulomatous response to capillaritis mediated by lipid deposition in the microvasculature.6,7
We present 3 cases of GPPD and provide a review of the literature. In all of our patients, biopsy specimens were fixed in 10% buffered formalin and embedded in paraffin by standard methodologies, and all stains were performed on sections by standard methodologies. Based on a PubMed search of articles indexed for MEDLINE using the terms granulomatous pigmented purpuric dermatosis, sarcoidosis, pigmented purpuric dermatosis, granulomas, and pigmented purpuric dermatosis, we review 5 additional reports describing 10 total patients.3-7
Case Reports
Patient 1
A 9-year-old white boy presented with a 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (Figure, A). The lesion appeared 3 to 4 years prior to presentation but had become progressively darker and centrally indurated over the last 2 years. The patient and his mother denied any history of trauma to the area. His medical history was unremarkable, and his current medications included fish oil and multivitamin tablets.
Histologic examination of a punch biopsy specimen taken from the center of the lesion revealed a lichenoid lymphohistiocytic infiltrate with marked red blood cell (RBC) extravasation and associated hemosiderin-laden macrophages. The lymphocytes comprising this infiltrate lacked cytologic atypia and exhibited minimal epidermotropism (Figure, B). Additionally, there was a superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised ofepithelioid histiocytes in the mid and deep dermis (Figure, C). Periodic acid–Schiff, acid-fast bacilli (AFB), and Fite stains were negative for organisms. Polarization was negative for refractile foreign material. Due to the patient’s age, no treatment was performed, and the lesion remains unchanged 1 year after biopsy.
Patient 2
A 49-year-old white woman presented with a 2-cm yellow-brown patch with a faint, nonblanchable, violaceous center on the right lateral thigh of 4 months’ duration. The patch initially appeared as a small asymptomatic purple papule. The patient denied any history of trauma to the area. A purified protein derivative (tuberculin) skin test was negative at the time of examination. The patient’s medical history was remarkable for renal calculi. Her current medications included progesterone; estradiol; lansoprazole; prenatal vitamins; vitamins C and E; zinc; and calcium. The patient had no family history of sarcoidosis. Complete blood cell count, urinalysis, liver function tests, and angiotensin-converting enzyme levels were unremarkable. Pulmonary function tests were normal, and there was no evidence of sarcoidosis on chest radiography. Initial biopsy of the lesion revealed a perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated RBCs in the papillary dermis (Figure, D). Similar to patient 1, the infiltrate lacked cytologic atypia and did not involve the overlying epidermis. There was perivascular granulomatous inflammation in the mid dermis (Figure, E). Periodic acid–Schiff, Warthin-Starry, and AFB stains were negative for organisms. Polarization was negative for refractile foreign material.
The lesion was treated with clobetasol propionate ointment 0.05% twice daily for 6 weeks with transient improvement, but the lesion recurred upon treatment cessation. Subsequent treatment with intralesional triamcinolone resulted in slight improvement of the lesion. A therapeutic trial of targeted pulsed dye laser treatment was ineffective. The lesion gradually increased in size over the next year with no therapy, and a repeat biopsy revealed a lichenoid lymphohistiocytic infiltrate with abundant extravasated RBCs consistent with persistent PPD. A granulomatous infiltrate was not evident in the superficial shave biopsy specimen.
Patient 3
A 75-year-old white woman presented with scattered, speckled, cayenne pepper–like, red-brown macules on the legs. Two years prior to presentation, a few scattered symmetrical macules appeared on the dorsal aspects of the feet, which gradually increased in number to form larger confluent patches that spread to the lower legs. The patient denied itching or burning but reported that the patches became painful when scratched and were aggravated by sun exposure. Her medical history was remarkable for asthma, chronic renal insufficiency, coronary artery disease, Barrett esophagus, obstructive sleep apnea, hypothyroidism, renal calculi, type 2 diabetes mellitus, and hyperlipidemia. Her current medications included carvedilol, valsartan, levothyroxine, aspirin, clopidogrel, furosemide, nitrofurantoin, temazepam, insulin, ezetimibe-simvastatin, and lansoprazole. Computed tomography of the chest revealed no signs of sarcoidosis. Pulmonary function tests revealed moderate obstructive lung disease. An ophthalmology examination showed no evidence of sarcoidosis. Laboratory results revealed an elevated glucose, blood urea nitrogen, creatinine, and triglyceride levels, as well as low hematocrit and vitamin D levels. Urinalysis, thyroid-stimulating hormone (thyrotropin) test, liver function tests, angiotensin-converting enzyme test, hepatitis B surface antigen, and IFN-g release assay were normal.
Histologic examination of a punch biopsy specimen revealed an inflammatory infiltrate confined to the papillary dermis. This infiltrate was comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated RBC extravasation and intimately associated granulomas (Figure, F). Additional inflammation in the deeper aspects of the dermis was not identified. Periodic acid–Schiff, AFB, and Fite stains were negative for organisms. Polarization was negative for refractile foreign material.

| 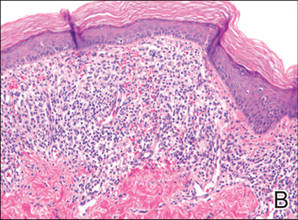
| 
| ||
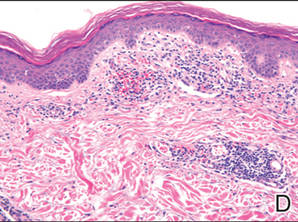
| 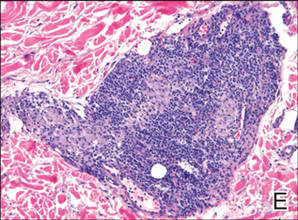
| 
| ||
| A 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (A). Lichenoid lymphohistiocytic infiltrate in the papillary dermis with marked red blood cell extravasation (B)(H&E, original magnification ×20). Superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised of epithelioid histiocytes in the mid and deep dermis (C)(H&E, original magnification ×20). Perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated red blood cells in the papillary dermis (D)(H&E, original magnification ×10). Perivascular lymphohistiocytic inflammation with epithelioid granulomas in the mid dermis (E)(H&E, original magnification ×20). Lymphohistiocytic inflammation in the papillary dermis comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated red blood cell extravasation and intimately associated granulomas (F)(H&E, original magnification ×20). | ||||
The patient was treated with topical steroids and minocycline 50 mg twice daily without improvement. The lesions improved after the patient underwent treatment with oral corticosteroids for pulmonary disease.
Comment
Pigmented purpuric dermatoses comprise a spectrum of clinical and pathologic conditions.1,2 Granulomatous PPD is a much less common variant, characterized by a granulomatous infiltrate admixed with PPD. We report 3 additional cases and review the literature on this rare and interesting variant of PPD.
We noted several unifying clinicopathologic features among our patients and those previously reported in the literature (Table).3-7 Including our cases, our review yielded 13 GPPD patients ranging in age from 9 to 75 years, with a mean age of 49.1 years. Two of our patients—patients 1 and 3—were the youngest and oldest patients, respectively, among the cases we reviewed. The majority of the cases we reviewed included patients of East Asian descent (6 Taiwanese; 2 Japanese; 1 Korean) as well as 4 white patients. No distinctive gender predilection was apparent, as our review included 8 females and 5 males.
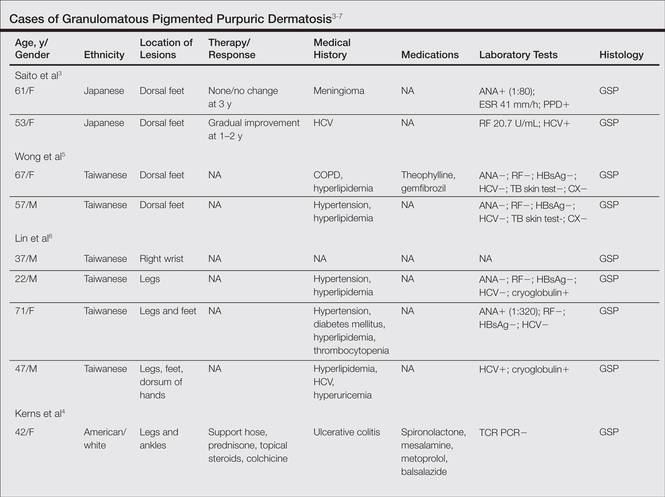
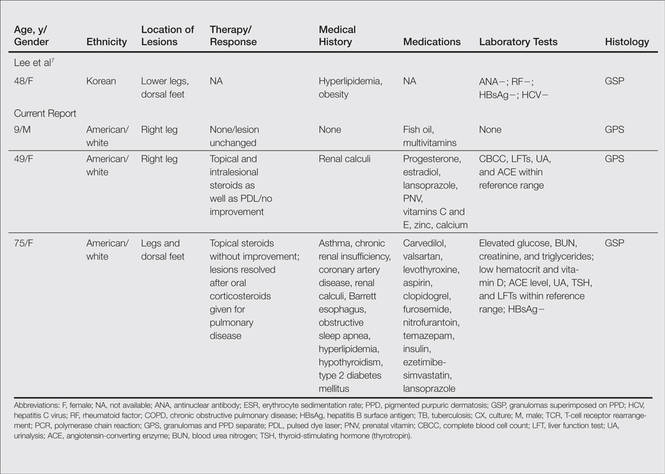
Our review revealed that GPPD lesions typically involve the lower extremities and usually are asymptomatic, with the exception of occasional pruritus. Additional lesions have been reported on the dorsal aspect of the hands, and 1 case noted exclusive involvement of the wrist.6 Lesions of GPPD can range in their clinical appearance. Three of 13 patients presented with purpuric papules and 2 had brown pigmentation with hemorrhagic papules3,4,6; the remaining 8 patients had erythematous or brown macules, papules, or plaques.5-7 The most commonly associated disease condition was hyperlipidemia, which was reported in 7 of 13 cases.5-7 Additional reported comorbidities included meningioma, renal calculi, obesity, hypertension, diabetes mellitus, chronic obstructive pulmonary disease, hepatitis C virus, ulcerative colitis, thrombocytopenia, and hyperuricemia. Reported serologic abnormalities included a rare positive antinuclear antibody, rheumatoid factor, and cryoglobulins.3,6 Therapeutic efficacy in the management of GPPD has not been well described; however, for the rare cases in which therapies were described, they were largely unsuccessful, with 1 patient exhibiting spontaneous improvement.3,4
Granulomatous PPD also appears to exhibit a range of histologic findings. All cases of GPPD shared fundamental components, such as a brisk perivascular infiltrate accompanied by RBC extravasation with variable hemosiderin-laden macrophages and a granulomatous infiltrate. All of the reports we reviewed described an intimate association between these components, with the granulomas being essentially superimposed on typical PPD. As for other types of PPD, obvious vasculitis characterized by a vasculocentric inflammatory infiltrate and evidence of vascular injury, such as fibrinoid necrosis of the vessel wall, has not been described in GPPD.3-7 Finally, histologic changes suggestive of a relationship with cutaneous T-cell lymphoma, cytologic atypia, and epidermotropism have been described for some forms of PPD but have not been described for GPPD.3-8
Our case reports expand the histologic spectrum of GPPD. Although patient 3 exhibited a relatively intimate association of granulomas and PPD, 2 of our cases (patients 1 and 2) demonstrated a granulomatous infiltrate in the mid to deep dermis, which is separate from the more superficially situated lichenoid lymphohistiocytic infiltrate, RBC extravasation, and hemosiderin-laden macrophages noted in the papillary dermis. Considered along with the absence of an obvious clinicopathologic explanation for the granulomatous infiltrates (eg, polarizable material, infectious organisms, systemic disease), these 2 cases (patients 1 and 2) suggest a composite form of PPD that combines the lichenoid pattern of PPD of Gougerot and Blum with a deep granulomatous component of GPPD. The importance of this distinction lies in the potential for physicians to overlook this potentially informative histologic pattern if only a superficial biopsy is performed. The clinical relevance is unclear; however, in our experience, it has been challenging to treat this relatively small subset of patients who have exhibited a limited response to treatment with topical steroids, intralesional steroids, pulsed dye laser, and vitamin supplementation.
The cause of the granulomatous infiltrate in GPPD is poorly understood. Seven of 13 cases included in our review occurred in patients with a history of hyperlipidemia.5-7 Some have postulated that the constellation of findings of GPPD in hyperlipidemic patients reflects an underlying vascular injury process induced by lipid deposition in the endothelial cells with subsequent RBC extravasation and a secondary granulomatous response to the lipid deposits.6,7 However, given the occurrence of GPPD in patients without hyperlipidemia, other mechanisms also must be considered in the pathogenesis of GPPD, including a reaction to medications, systemic diseases, and infectious etiologies (eg, hepatitis B virus).4,6 As additional cases are described in the literature, other unifying clinical etiologies for this histopathologic reaction pattern may emerge.
Conclusion
Granulomatous PPD may comprise an underrecognized variant of PPD in cases when only a superficial biopsy is evaluated. Clinicians and pathologists should be aware of this variant, and in refractory cases of PPD, deeper sampling may be warranted to identify granulomas.
1. Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
2. Piette WW. Purpura: mechanisms and differential diagnosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby; 2008:321-330.
3. Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551-555.
4. Kerns MJ, Mallatt BD, Shamma HN. Granulomatous pigmented purpura: an unusual histological variant. Am J Dermatopathol. 2009;31:77-80.
5. Wong WR, Kuo TT, Chen MJ, et al. Granulomatous variant of chronic pigmented purpuric dermatosis: report of two cases. Br J Dermatol. 2001;145:162-164.
6. Lin WL, Kou TT, Shih PY, et al. Granulomatous variant of chronic pigmented purpuric dermatoses: report of four new cases and an association with hyperlipidaemia [published online ahead of print May 29, 2007]. Clin Exp Dermatol. 2007;32:513-515.
7. Lee SH, Kwon JE, Lee KG, et al. Granulomatous variant of chronic pigmented purpuric dermatosis associated with hyperlipidemia. J Eur Acad Dermatol Venereol. 2010;24:1243-1245.
8. Toro JR, Sander CA, LeBoit PE. Persistent pigmented dermatoses and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
Pigmented purpuric dermatoses (PPDs) are a group of common chronic disorders characterized by speckled, cayenne pepper–like petechiae and orange-brown discoloration of the skin resulting from capillaritis.1 Pigmented purpuric dermatoses typically occur in the absence of underlying vascular insufficiency or other hematologic dysfunction. The 5 well-known clinicopathologic variants of PPD include Schamberg disease; purpura annularis telangiectodes of Majocchi; pigmented purpuric lichenoid dermatitis of Gougerot and Blum; eczematoidlike purpura of Doucas and Kapetanakis; and lichen aureus.2 All PPDs share common characteristic clinical and histologic features. Clinically, patients generally present with symmetric petechiae and/or pigmented macules. All 5 PPD variants share similar histologic findings, including a vasculocentric lymphocytic infiltrate in the papillary dermis, swelling of the endothelial cells, erythrocyte extravasation, and often hemosiderin-laden macrophages.1 Despite these clinical and histopathologic similarities, each variant contains additional distinctive features, such as telangiectasia (purpura annularis telangiectodes of Majocchi), a lichenoid infiltrate (pigmented purpuric lichenoid dermatitis of Gougerot and Blum), eczematous changes (eczematoidlike purpura of Doucas and Kapetanakis), and marked hemosiderin deposition (lichen aureus).
Granulomatous pigmented purpuric dermatosis (GPPD) is a rare variant of PPD.3-7 Clinically, these lesions appear similar to other PPDs; however, in addition to the characteristic changes associated with conventional PPD, histologic examination of GPPD reveals a granulomatous inflammatory reaction pattern. Although the pathogenesis of GPPD is not well understood, its association with hyperlipidemia may suggest a granulomatous response to capillaritis mediated by lipid deposition in the microvasculature.6,7
We present 3 cases of GPPD and provide a review of the literature. In all of our patients, biopsy specimens were fixed in 10% buffered formalin and embedded in paraffin by standard methodologies, and all stains were performed on sections by standard methodologies. Based on a PubMed search of articles indexed for MEDLINE using the terms granulomatous pigmented purpuric dermatosis, sarcoidosis, pigmented purpuric dermatosis, granulomas, and pigmented purpuric dermatosis, we review 5 additional reports describing 10 total patients.3-7
Case Reports
Patient 1
A 9-year-old white boy presented with a 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (Figure, A). The lesion appeared 3 to 4 years prior to presentation but had become progressively darker and centrally indurated over the last 2 years. The patient and his mother denied any history of trauma to the area. His medical history was unremarkable, and his current medications included fish oil and multivitamin tablets.
Histologic examination of a punch biopsy specimen taken from the center of the lesion revealed a lichenoid lymphohistiocytic infiltrate with marked red blood cell (RBC) extravasation and associated hemosiderin-laden macrophages. The lymphocytes comprising this infiltrate lacked cytologic atypia and exhibited minimal epidermotropism (Figure, B). Additionally, there was a superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised ofepithelioid histiocytes in the mid and deep dermis (Figure, C). Periodic acid–Schiff, acid-fast bacilli (AFB), and Fite stains were negative for organisms. Polarization was negative for refractile foreign material. Due to the patient’s age, no treatment was performed, and the lesion remains unchanged 1 year after biopsy.
Patient 2
A 49-year-old white woman presented with a 2-cm yellow-brown patch with a faint, nonblanchable, violaceous center on the right lateral thigh of 4 months’ duration. The patch initially appeared as a small asymptomatic purple papule. The patient denied any history of trauma to the area. A purified protein derivative (tuberculin) skin test was negative at the time of examination. The patient’s medical history was remarkable for renal calculi. Her current medications included progesterone; estradiol; lansoprazole; prenatal vitamins; vitamins C and E; zinc; and calcium. The patient had no family history of sarcoidosis. Complete blood cell count, urinalysis, liver function tests, and angiotensin-converting enzyme levels were unremarkable. Pulmonary function tests were normal, and there was no evidence of sarcoidosis on chest radiography. Initial biopsy of the lesion revealed a perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated RBCs in the papillary dermis (Figure, D). Similar to patient 1, the infiltrate lacked cytologic atypia and did not involve the overlying epidermis. There was perivascular granulomatous inflammation in the mid dermis (Figure, E). Periodic acid–Schiff, Warthin-Starry, and AFB stains were negative for organisms. Polarization was negative for refractile foreign material.
The lesion was treated with clobetasol propionate ointment 0.05% twice daily for 6 weeks with transient improvement, but the lesion recurred upon treatment cessation. Subsequent treatment with intralesional triamcinolone resulted in slight improvement of the lesion. A therapeutic trial of targeted pulsed dye laser treatment was ineffective. The lesion gradually increased in size over the next year with no therapy, and a repeat biopsy revealed a lichenoid lymphohistiocytic infiltrate with abundant extravasated RBCs consistent with persistent PPD. A granulomatous infiltrate was not evident in the superficial shave biopsy specimen.
Patient 3
A 75-year-old white woman presented with scattered, speckled, cayenne pepper–like, red-brown macules on the legs. Two years prior to presentation, a few scattered symmetrical macules appeared on the dorsal aspects of the feet, which gradually increased in number to form larger confluent patches that spread to the lower legs. The patient denied itching or burning but reported that the patches became painful when scratched and were aggravated by sun exposure. Her medical history was remarkable for asthma, chronic renal insufficiency, coronary artery disease, Barrett esophagus, obstructive sleep apnea, hypothyroidism, renal calculi, type 2 diabetes mellitus, and hyperlipidemia. Her current medications included carvedilol, valsartan, levothyroxine, aspirin, clopidogrel, furosemide, nitrofurantoin, temazepam, insulin, ezetimibe-simvastatin, and lansoprazole. Computed tomography of the chest revealed no signs of sarcoidosis. Pulmonary function tests revealed moderate obstructive lung disease. An ophthalmology examination showed no evidence of sarcoidosis. Laboratory results revealed an elevated glucose, blood urea nitrogen, creatinine, and triglyceride levels, as well as low hematocrit and vitamin D levels. Urinalysis, thyroid-stimulating hormone (thyrotropin) test, liver function tests, angiotensin-converting enzyme test, hepatitis B surface antigen, and IFN-g release assay were normal.
Histologic examination of a punch biopsy specimen revealed an inflammatory infiltrate confined to the papillary dermis. This infiltrate was comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated RBC extravasation and intimately associated granulomas (Figure, F). Additional inflammation in the deeper aspects of the dermis was not identified. Periodic acid–Schiff, AFB, and Fite stains were negative for organisms. Polarization was negative for refractile foreign material.
|
|
|
| ||
|
|
|
| ||
| A 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (A). Lichenoid lymphohistiocytic infiltrate in the papillary dermis with marked red blood cell extravasation (B)(H&E, original magnification ×20). Superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised of epithelioid histiocytes in the mid and deep dermis (C)(H&E, original magnification ×20). Perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated red blood cells in the papillary dermis (D)(H&E, original magnification ×10). Perivascular lymphohistiocytic inflammation with epithelioid granulomas in the mid dermis (E)(H&E, original magnification ×20). Lymphohistiocytic inflammation in the papillary dermis comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated red blood cell extravasation and intimately associated granulomas (F)(H&E, original magnification ×20). | ||||
The patient was treated with topical steroids and minocycline 50 mg twice daily without improvement. The lesions improved after the patient underwent treatment with oral corticosteroids for pulmonary disease.
Comment
Pigmented purpuric dermatoses comprise a spectrum of clinical and pathologic conditions.1,2 Granulomatous PPD is a much less common variant, characterized by a granulomatous infiltrate admixed with PPD. We report 3 additional cases and review the literature on this rare and interesting variant of PPD.
We noted several unifying clinicopathologic features among our patients and those previously reported in the literature (Table).3-7 Including our cases, our review yielded 13 GPPD patients ranging in age from 9 to 75 years, with a mean age of 49.1 years. Two of our patients—patients 1 and 3—were the youngest and oldest patients, respectively, among the cases we reviewed. The majority of the cases we reviewed included patients of East Asian descent (6 Taiwanese; 2 Japanese; 1 Korean) as well as 4 white patients. No distinctive gender predilection was apparent, as our review included 8 females and 5 males.
Our review revealed that GPPD lesions typically involve the lower extremities and usually are asymptomatic, with the exception of occasional pruritus. Additional lesions have been reported on the dorsal aspect of the hands, and 1 case noted exclusive involvement of the wrist.6 Lesions of GPPD can range in their clinical appearance. Three of 13 patients presented with purpuric papules and 2 had brown pigmentation with hemorrhagic papules3,4,6; the remaining 8 patients had erythematous or brown macules, papules, or plaques.5-7 The most commonly associated disease condition was hyperlipidemia, which was reported in 7 of 13 cases.5-7 Additional reported comorbidities included meningioma, renal calculi, obesity, hypertension, diabetes mellitus, chronic obstructive pulmonary disease, hepatitis C virus, ulcerative colitis, thrombocytopenia, and hyperuricemia. Reported serologic abnormalities included a rare positive antinuclear antibody, rheumatoid factor, and cryoglobulins.3,6 Therapeutic efficacy in the management of GPPD has not been well described; however, for the rare cases in which therapies were described, they were largely unsuccessful, with 1 patient exhibiting spontaneous improvement.3,4
Granulomatous PPD also appears to exhibit a range of histologic findings. All cases of GPPD shared fundamental components, such as a brisk perivascular infiltrate accompanied by RBC extravasation with variable hemosiderin-laden macrophages and a granulomatous infiltrate. All of the reports we reviewed described an intimate association between these components, with the granulomas being essentially superimposed on typical PPD. As for other types of PPD, obvious vasculitis characterized by a vasculocentric inflammatory infiltrate and evidence of vascular injury, such as fibrinoid necrosis of the vessel wall, has not been described in GPPD.3-7 Finally, histologic changes suggestive of a relationship with cutaneous T-cell lymphoma, cytologic atypia, and epidermotropism have been described for some forms of PPD but have not been described for GPPD.3-8
Our case reports expand the histologic spectrum of GPPD. Although patient 3 exhibited a relatively intimate association of granulomas and PPD, 2 of our cases (patients 1 and 2) demonstrated a granulomatous infiltrate in the mid to deep dermis, which is separate from the more superficially situated lichenoid lymphohistiocytic infiltrate, RBC extravasation, and hemosiderin-laden macrophages noted in the papillary dermis. Considered along with the absence of an obvious clinicopathologic explanation for the granulomatous infiltrates (eg, polarizable material, infectious organisms, systemic disease), these 2 cases (patients 1 and 2) suggest a composite form of PPD that combines the lichenoid pattern of PPD of Gougerot and Blum with a deep granulomatous component of GPPD. The importance of this distinction lies in the potential for physicians to overlook this potentially informative histologic pattern if only a superficial biopsy is performed. The clinical relevance is unclear; however, in our experience, it has been challenging to treat this relatively small subset of patients who have exhibited a limited response to treatment with topical steroids, intralesional steroids, pulsed dye laser, and vitamin supplementation.
The cause of the granulomatous infiltrate in GPPD is poorly understood. Seven of 13 cases included in our review occurred in patients with a history of hyperlipidemia.5-7 Some have postulated that the constellation of findings of GPPD in hyperlipidemic patients reflects an underlying vascular injury process induced by lipid deposition in the endothelial cells with subsequent RBC extravasation and a secondary granulomatous response to the lipid deposits.6,7 However, given the occurrence of GPPD in patients without hyperlipidemia, other mechanisms also must be considered in the pathogenesis of GPPD, including a reaction to medications, systemic diseases, and infectious etiologies (eg, hepatitis B virus).4,6 As additional cases are described in the literature, other unifying clinical etiologies for this histopathologic reaction pattern may emerge.
Conclusion
Granulomatous PPD may comprise an underrecognized variant of PPD in cases when only a superficial biopsy is evaluated. Clinicians and pathologists should be aware of this variant, and in refractory cases of PPD, deeper sampling may be warranted to identify granulomas.
Pigmented purpuric dermatoses (PPDs) are a group of common chronic disorders characterized by speckled, cayenne pepper–like petechiae and orange-brown discoloration of the skin resulting from capillaritis.1 Pigmented purpuric dermatoses typically occur in the absence of underlying vascular insufficiency or other hematologic dysfunction. The 5 well-known clinicopathologic variants of PPD include Schamberg disease; purpura annularis telangiectodes of Majocchi; pigmented purpuric lichenoid dermatitis of Gougerot and Blum; eczematoidlike purpura of Doucas and Kapetanakis; and lichen aureus.2 All PPDs share common characteristic clinical and histologic features. Clinically, patients generally present with symmetric petechiae and/or pigmented macules. All 5 PPD variants share similar histologic findings, including a vasculocentric lymphocytic infiltrate in the papillary dermis, swelling of the endothelial cells, erythrocyte extravasation, and often hemosiderin-laden macrophages.1 Despite these clinical and histopathologic similarities, each variant contains additional distinctive features, such as telangiectasia (purpura annularis telangiectodes of Majocchi), a lichenoid infiltrate (pigmented purpuric lichenoid dermatitis of Gougerot and Blum), eczematous changes (eczematoidlike purpura of Doucas and Kapetanakis), and marked hemosiderin deposition (lichen aureus).
Granulomatous pigmented purpuric dermatosis (GPPD) is a rare variant of PPD.3-7 Clinically, these lesions appear similar to other PPDs; however, in addition to the characteristic changes associated with conventional PPD, histologic examination of GPPD reveals a granulomatous inflammatory reaction pattern. Although the pathogenesis of GPPD is not well understood, its association with hyperlipidemia may suggest a granulomatous response to capillaritis mediated by lipid deposition in the microvasculature.6,7
We present 3 cases of GPPD and provide a review of the literature. In all of our patients, biopsy specimens were fixed in 10% buffered formalin and embedded in paraffin by standard methodologies, and all stains were performed on sections by standard methodologies. Based on a PubMed search of articles indexed for MEDLINE using the terms granulomatous pigmented purpuric dermatosis, sarcoidosis, pigmented purpuric dermatosis, granulomas, and pigmented purpuric dermatosis, we review 5 additional reports describing 10 total patients.3-7
Case Reports
Patient 1
A 9-year-old white boy presented with a 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (Figure, A). The lesion appeared 3 to 4 years prior to presentation but had become progressively darker and centrally indurated over the last 2 years. The patient and his mother denied any history of trauma to the area. His medical history was unremarkable, and his current medications included fish oil and multivitamin tablets.
Histologic examination of a punch biopsy specimen taken from the center of the lesion revealed a lichenoid lymphohistiocytic infiltrate with marked red blood cell (RBC) extravasation and associated hemosiderin-laden macrophages. The lymphocytes comprising this infiltrate lacked cytologic atypia and exhibited minimal epidermotropism (Figure, B). Additionally, there was a superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised ofepithelioid histiocytes in the mid and deep dermis (Figure, C). Periodic acid–Schiff, acid-fast bacilli (AFB), and Fite stains were negative for organisms. Polarization was negative for refractile foreign material. Due to the patient’s age, no treatment was performed, and the lesion remains unchanged 1 year after biopsy.
Patient 2
A 49-year-old white woman presented with a 2-cm yellow-brown patch with a faint, nonblanchable, violaceous center on the right lateral thigh of 4 months’ duration. The patch initially appeared as a small asymptomatic purple papule. The patient denied any history of trauma to the area. A purified protein derivative (tuberculin) skin test was negative at the time of examination. The patient’s medical history was remarkable for renal calculi. Her current medications included progesterone; estradiol; lansoprazole; prenatal vitamins; vitamins C and E; zinc; and calcium. The patient had no family history of sarcoidosis. Complete blood cell count, urinalysis, liver function tests, and angiotensin-converting enzyme levels were unremarkable. Pulmonary function tests were normal, and there was no evidence of sarcoidosis on chest radiography. Initial biopsy of the lesion revealed a perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated RBCs in the papillary dermis (Figure, D). Similar to patient 1, the infiltrate lacked cytologic atypia and did not involve the overlying epidermis. There was perivascular granulomatous inflammation in the mid dermis (Figure, E). Periodic acid–Schiff, Warthin-Starry, and AFB stains were negative for organisms. Polarization was negative for refractile foreign material.
The lesion was treated with clobetasol propionate ointment 0.05% twice daily for 6 weeks with transient improvement, but the lesion recurred upon treatment cessation. Subsequent treatment with intralesional triamcinolone resulted in slight improvement of the lesion. A therapeutic trial of targeted pulsed dye laser treatment was ineffective. The lesion gradually increased in size over the next year with no therapy, and a repeat biopsy revealed a lichenoid lymphohistiocytic infiltrate with abundant extravasated RBCs consistent with persistent PPD. A granulomatous infiltrate was not evident in the superficial shave biopsy specimen.
Patient 3
A 75-year-old white woman presented with scattered, speckled, cayenne pepper–like, red-brown macules on the legs. Two years prior to presentation, a few scattered symmetrical macules appeared on the dorsal aspects of the feet, which gradually increased in number to form larger confluent patches that spread to the lower legs. The patient denied itching or burning but reported that the patches became painful when scratched and were aggravated by sun exposure. Her medical history was remarkable for asthma, chronic renal insufficiency, coronary artery disease, Barrett esophagus, obstructive sleep apnea, hypothyroidism, renal calculi, type 2 diabetes mellitus, and hyperlipidemia. Her current medications included carvedilol, valsartan, levothyroxine, aspirin, clopidogrel, furosemide, nitrofurantoin, temazepam, insulin, ezetimibe-simvastatin, and lansoprazole. Computed tomography of the chest revealed no signs of sarcoidosis. Pulmonary function tests revealed moderate obstructive lung disease. An ophthalmology examination showed no evidence of sarcoidosis. Laboratory results revealed an elevated glucose, blood urea nitrogen, creatinine, and triglyceride levels, as well as low hematocrit and vitamin D levels. Urinalysis, thyroid-stimulating hormone (thyrotropin) test, liver function tests, angiotensin-converting enzyme test, hepatitis B surface antigen, and IFN-g release assay were normal.
Histologic examination of a punch biopsy specimen revealed an inflammatory infiltrate confined to the papillary dermis. This infiltrate was comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated RBC extravasation and intimately associated granulomas (Figure, F). Additional inflammation in the deeper aspects of the dermis was not identified. Periodic acid–Schiff, AFB, and Fite stains were negative for organisms. Polarization was negative for refractile foreign material.
|
|
|
| ||
|
|
|
| ||
| A 3-cm asymptomatic light brown patch with a nonblanching violaceous center on the right posterior thigh that was studded with pinpoint yellow papules (A). Lichenoid lymphohistiocytic infiltrate in the papillary dermis with marked red blood cell extravasation (B)(H&E, original magnification ×20). Superficial and deep perivascular mononuclear inflammatory infiltrate intermixed with numerous small granulomas comprised of epithelioid histiocytes in the mid and deep dermis (C)(H&E, original magnification ×20). Perivascular and interstitial lymphohistiocytic infiltrate with abundant extravasated red blood cells in the papillary dermis (D)(H&E, original magnification ×10). Perivascular lymphohistiocytic inflammation with epithelioid granulomas in the mid dermis (E)(H&E, original magnification ×20). Lymphohistiocytic inflammation in the papillary dermis comprised of an admixture of lymphocytes and histiocytes in a perivascular distribution with associated red blood cell extravasation and intimately associated granulomas (F)(H&E, original magnification ×20). | ||||
The patient was treated with topical steroids and minocycline 50 mg twice daily without improvement. The lesions improved after the patient underwent treatment with oral corticosteroids for pulmonary disease.
Comment
Pigmented purpuric dermatoses comprise a spectrum of clinical and pathologic conditions.1,2 Granulomatous PPD is a much less common variant, characterized by a granulomatous infiltrate admixed with PPD. We report 3 additional cases and review the literature on this rare and interesting variant of PPD.
We noted several unifying clinicopathologic features among our patients and those previously reported in the literature (Table).3-7 Including our cases, our review yielded 13 GPPD patients ranging in age from 9 to 75 years, with a mean age of 49.1 years. Two of our patients—patients 1 and 3—were the youngest and oldest patients, respectively, among the cases we reviewed. The majority of the cases we reviewed included patients of East Asian descent (6 Taiwanese; 2 Japanese; 1 Korean) as well as 4 white patients. No distinctive gender predilection was apparent, as our review included 8 females and 5 males.
Our review revealed that GPPD lesions typically involve the lower extremities and usually are asymptomatic, with the exception of occasional pruritus. Additional lesions have been reported on the dorsal aspect of the hands, and 1 case noted exclusive involvement of the wrist.6 Lesions of GPPD can range in their clinical appearance. Three of 13 patients presented with purpuric papules and 2 had brown pigmentation with hemorrhagic papules3,4,6; the remaining 8 patients had erythematous or brown macules, papules, or plaques.5-7 The most commonly associated disease condition was hyperlipidemia, which was reported in 7 of 13 cases.5-7 Additional reported comorbidities included meningioma, renal calculi, obesity, hypertension, diabetes mellitus, chronic obstructive pulmonary disease, hepatitis C virus, ulcerative colitis, thrombocytopenia, and hyperuricemia. Reported serologic abnormalities included a rare positive antinuclear antibody, rheumatoid factor, and cryoglobulins.3,6 Therapeutic efficacy in the management of GPPD has not been well described; however, for the rare cases in which therapies were described, they were largely unsuccessful, with 1 patient exhibiting spontaneous improvement.3,4
Granulomatous PPD also appears to exhibit a range of histologic findings. All cases of GPPD shared fundamental components, such as a brisk perivascular infiltrate accompanied by RBC extravasation with variable hemosiderin-laden macrophages and a granulomatous infiltrate. All of the reports we reviewed described an intimate association between these components, with the granulomas being essentially superimposed on typical PPD. As for other types of PPD, obvious vasculitis characterized by a vasculocentric inflammatory infiltrate and evidence of vascular injury, such as fibrinoid necrosis of the vessel wall, has not been described in GPPD.3-7 Finally, histologic changes suggestive of a relationship with cutaneous T-cell lymphoma, cytologic atypia, and epidermotropism have been described for some forms of PPD but have not been described for GPPD.3-8
Our case reports expand the histologic spectrum of GPPD. Although patient 3 exhibited a relatively intimate association of granulomas and PPD, 2 of our cases (patients 1 and 2) demonstrated a granulomatous infiltrate in the mid to deep dermis, which is separate from the more superficially situated lichenoid lymphohistiocytic infiltrate, RBC extravasation, and hemosiderin-laden macrophages noted in the papillary dermis. Considered along with the absence of an obvious clinicopathologic explanation for the granulomatous infiltrates (eg, polarizable material, infectious organisms, systemic disease), these 2 cases (patients 1 and 2) suggest a composite form of PPD that combines the lichenoid pattern of PPD of Gougerot and Blum with a deep granulomatous component of GPPD. The importance of this distinction lies in the potential for physicians to overlook this potentially informative histologic pattern if only a superficial biopsy is performed. The clinical relevance is unclear; however, in our experience, it has been challenging to treat this relatively small subset of patients who have exhibited a limited response to treatment with topical steroids, intralesional steroids, pulsed dye laser, and vitamin supplementation.
The cause of the granulomatous infiltrate in GPPD is poorly understood. Seven of 13 cases included in our review occurred in patients with a history of hyperlipidemia.5-7 Some have postulated that the constellation of findings of GPPD in hyperlipidemic patients reflects an underlying vascular injury process induced by lipid deposition in the endothelial cells with subsequent RBC extravasation and a secondary granulomatous response to the lipid deposits.6,7 However, given the occurrence of GPPD in patients without hyperlipidemia, other mechanisms also must be considered in the pathogenesis of GPPD, including a reaction to medications, systemic diseases, and infectious etiologies (eg, hepatitis B virus).4,6 As additional cases are described in the literature, other unifying clinical etiologies for this histopathologic reaction pattern may emerge.
Conclusion
Granulomatous PPD may comprise an underrecognized variant of PPD in cases when only a superficial biopsy is evaluated. Clinicians and pathologists should be aware of this variant, and in refractory cases of PPD, deeper sampling may be warranted to identify granulomas.
1. Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
2. Piette WW. Purpura: mechanisms and differential diagnosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby; 2008:321-330.
3. Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551-555.
4. Kerns MJ, Mallatt BD, Shamma HN. Granulomatous pigmented purpura: an unusual histological variant. Am J Dermatopathol. 2009;31:77-80.
5. Wong WR, Kuo TT, Chen MJ, et al. Granulomatous variant of chronic pigmented purpuric dermatosis: report of two cases. Br J Dermatol. 2001;145:162-164.
6. Lin WL, Kou TT, Shih PY, et al. Granulomatous variant of chronic pigmented purpuric dermatoses: report of four new cases and an association with hyperlipidaemia [published online ahead of print May 29, 2007]. Clin Exp Dermatol. 2007;32:513-515.
7. Lee SH, Kwon JE, Lee KG, et al. Granulomatous variant of chronic pigmented purpuric dermatosis associated with hyperlipidemia. J Eur Acad Dermatol Venereol. 2010;24:1243-1245.
8. Toro JR, Sander CA, LeBoit PE. Persistent pigmented dermatoses and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
1. Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
2. Piette WW. Purpura: mechanisms and differential diagnosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby; 2008:321-330.
3. Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551-555.
4. Kerns MJ, Mallatt BD, Shamma HN. Granulomatous pigmented purpura: an unusual histological variant. Am J Dermatopathol. 2009;31:77-80.
5. Wong WR, Kuo TT, Chen MJ, et al. Granulomatous variant of chronic pigmented purpuric dermatosis: report of two cases. Br J Dermatol. 2001;145:162-164.
6. Lin WL, Kou TT, Shih PY, et al. Granulomatous variant of chronic pigmented purpuric dermatoses: report of four new cases and an association with hyperlipidaemia [published online ahead of print May 29, 2007]. Clin Exp Dermatol. 2007;32:513-515.
7. Lee SH, Kwon JE, Lee KG, et al. Granulomatous variant of chronic pigmented purpuric dermatosis associated with hyperlipidemia. J Eur Acad Dermatol Venereol. 2010;24:1243-1245.
8. Toro JR, Sander CA, LeBoit PE. Persistent pigmented dermatoses and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
Practice Points
- Consider a punch biopsy when sampling suspected inflammatory dermatoses, such as pigmented purpuric dermatosis, to allow deeper sampling.
- Provide all clinical details to the dermatopathologist to assist with clinicopathologic correlation and diagnostic accuracy.
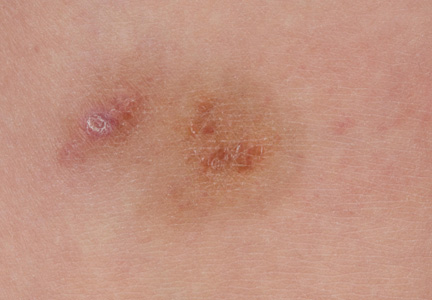
HATS Syndrome: Hemimaxillary Enlargement, Asymmetry of the Face, Tooth Abnormalities, and Skin Findings
Case Report
A 14-year-old adolescent boy presented to the dermatology clinic at our institution for evaluation of a hyperpigmented hairy patch on the right side of the face that had been present since birth. The patient reported the lesion originally had involved the right cheek, neck, and back but had gradually expanded to include the right side of the upper lip and oral mucosa. His medical history was remarkable for acne, which was currently being managed with topical treatments. There was no family history of similar conditions. There were no mental or developmental deformities since birth.
Physical examination revealed a hyperpigmented patch with hypertrichosis on the right side of the body involving the back, neck, and cheek (Figure 1), as well as hyperpigmentation involving the right side of the upper lip and oral mucosa (Figure 2A). Slight facial asymmetry also was noted. Dental examination revealed irregular spacing and decreased growth of the teeth on the right side of the mouth (Figure 2B).
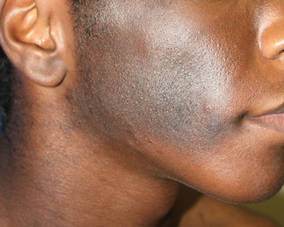
| 
|
Figure 2. Some hyperpigmentation involving the oral mucosa on the right side (A) and dental abnormalities (B). | |
A biopsy of the hyperpigmented patch on the back revealed mild regular acanthosis, basal hypermelanosis, slight papillomatosis, and hair structures within the dermis with features that were consistent with a Becker nevus. A dental radiograph demonstrated hyperplasia of the right maxillary alveolus and basal bone area with 2 missing permanent teeth (fourth and fifth premolars)(Figure 3). Computed axial tomography revealed enlargement of the maxillary bone on the right side.
The constellation of clinical, histopathologic, and radiologic findings was consistent with a diagnosis of hemimaxillary enlargement, asymmetry of the face, tooth abnormalities, and skin findings (HATS syndrome). The treatment plan involved surgical modification of the maxillary bone to correct the hyperplasia on the affected side and implanting 2 artificial premolars. Additionally, laser therapy using a Q-switched ruby laser, frequency-doubled Nd:YAG, 1550-nm erbium-doped fiber laser, or 755-nm alexandrite laser was considered to treat the hyperpigmentation associ-ated with the Becker nevus.
Comment
HATS syndrome is a rare, local developmental defect involving the first and second branchial arches. It generally is detected at birth or in early childhood and is associated with unilateral abnormalities of the bones, teeth, gums, and skin. It is more common in boys than girls (1.8:1.0 ratio), with an age range of 2 to 28 years; there is a peak in the first decade of life.1 It was first described by Miles et al2 in 1987 in a case of congenital mild facial asymmetry, unilateral enlargement of the maxillary gingiva and alveolar bone, hypoplastic teeth, and hypertrichosis in the affected area. The investigators at that time suggested the term hemimaxillofacial dysplasia (HD). In 1990, Danforth et al3 reported 8 additional cases with similar features but without known skin changes; they proposed the term segmental odontomaxillary dysplasia (SOD). In 1996, Desalvo et al4 reported a case of SOD involving a 7-year-old girl with an area of hypopigmentation of the lip on the affected side, and Packota et al5 described the radiographic features of 12 cases of SOD. In subsequent years, other cases of HD or SOD were reported in the literature.1,6-16 In 2004, Welsch and Stein17 reported 1 patient with a Becker nevus of the skin and recommended the acronym HATS. Armstrong et al18 reported 2 cases of SOD with new histopathologic findings of the teeth (eg, fibrous enlargement of the pulps, an irregular pulp-dentin interface displaying many pseudoinclusions, pulp stones). In 2008, Porwal et al19 reported a case of HD in which maxillary hypoplasia rather than hyperplasia was noted, which emphasized the variability of the maxillary dysplasia. Koenig et al20 reported a case of SOD with facial hypertrichosis, commissural lip clefting, and hyperlinear palms. Bhatia et al21 reported another case of SOD with a new finding of unilateral ectopic eyelashes.
The etiology remains unknown, but theories include an alteration that occurs in utero or in in-fancy; the possibility of a systemic or endocrine aberration; a postzygotic mutation resulting in genotypic and phenotypic mosaicism of bone and skin, similar to McCune-Albright syndrome; and viral or bacterial infection along the branches of the maxillary division of the trigeminal nerve.1,15 Bone defects include unilateral enlargement of the maxillary alveolar process and thickening of the vertically oriented trabeculae, which is detected radiographically. A reduction in size of the maxillary sinus and nasal airway was reported in about one-half of cases1 and can be detected easily by computed tomography scanning. Missing permanent premolar teeth, tooth shape abnormalities, delayed eruption of teeth, abnormal spacing of teeth, hypoplastic teeth, enlarged teeth, and gingival thickening also are common oral findings.1 The skin manifestations of HATS syndrome are not static but progress well into adolescence15 and can include facial asymmetry, hypertrichosis, Becker nevus, hairy nevus, lip hypopigmentation, discontinuity of the vermilion border, depression of the cheek, and facial erythema.17
The differential diagnosis includes hemifacial hyperplasia, monostotic fibrous dysplasia, and regional odontodysplasia.1 Little information is available concerning the treatment of patients with this condition.15 The reported treatment modalities include combined surgical and orthodontic treatment of unerupted teeth (premolar/canine), prosthodontic treatment, gingivoplasty, recontouring osteotomy for severe facial asymmetry, and reconstructive jaw surgery.1,6,11,15 Successful treatment of Becker nevi with the Q-switched ruby laser, erbium:YAG laser, and 755-nm alexandrite laser have been reported.22-24
Conclusion
There is a need for continued reporting of cases of HATS syndrome in addition to long-term follow-up to document the natural history of the condition and to establish the appropriate treatment.
1. Friedlander-Barenboim S, Kamburoglu K, Kaffe I. Clinical and radiological presentation of hemimaxillofacial dysplasia/segmental odontomaxillary dysplasia: critical analysis and report of a case. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;113:268-273.
2. Miles DA, Lovas JL, Cohen MM. Hemimaxillofacial dysplasia: a newly recognized disorder of facial asymmetry, hypertrichosis of the facial skin, unilateral enlargement of the maxilla, and hypoplastic teeth in two patients. Oral Surg Oral Med Oral Pathol. 1987;64:445-448.
3. Danforth RA, Melrose RJ, Abrams AM, et al. Segmental odontomaxillary dysplasia: report of eight cases and comparison with hemimaxillofacial dysplasia. Oral Surg Oral Med Oral Pathol. 1990;70:81-85.
4. Desalvo MS, Copete MA, Riesenberger RE, et al. Segmental odontomaxillary dysplasia (hemimaxillofacial dysplasia): case report. Pediatr Dent. 1996;18:154-156.
5. Packota GV, Pharoah MJ, Petrikowski CG. Radiographic features of segmental odontomaxillary dysplasia: a study of 12 cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;82:577-584.
6. Paticoff K, Marion RW, Shprintzen RJ, et al. Hemimaxillofacial dysplasia: a report of two new cases and further delineation of the disorder. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;83:484-488.
7. Jones AC, Ford MJ. Simultaneous occurrence of segmental odontomaxillary dysplasia and Becker’s nevus. J Oral Maxillofac Surg. 1990;57:1251-1254.
8. Prusack N, Pringle G, Scotti V, et al. Segmental odontomaxillary dysplasia: a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:483-488.
9. Becktor KB, Reibel J, Vedel B, et al. Segmental odontomaxillary dysplasia: clinical, radiological and histological aspects of four cases. Oral Dis. 2002;8:106-110.
10. Velez I, Vedrenne D, Cralle P, et al. Segmental odontomaxillary dysplasia: report of two cases and review of the literature. Todays FDA. 2002;14:20-21.
11. Drake DL. Segmental odontomaxillary dysplasia: an unusual orthodontic challenge. Am J Orthod Dentofac Orthop. 2003;123:84-86.
12. Gavalda C. Segmental odontomaxillary dysplasia. Med Oral. 2004;9:181.
13. Ozpinar B, Gokce B, Uzel G, et al. Prosthetic rehabilitation of segmental odontomaxillary dysplasia: seven-year follow-up. Cleft Palate Craniofac J. 2009;46:103-107.
14. Kuklani RM, Nair MK. Segmental odontomaxillary dysplasia: review of the literature and case report [published online ahead of print December 14, 2010]. Int J Dent. 2010;2010:837283.
15. Minett CP, Daley TD. Hemimaxillofacial dysplasia (segmental odontomaxillary dysplasia): case study with 11 years of follow-up from primary to adult dentition. J Oral Maxillofac Surg. 2012;70:1183-1191.
16. Whitt JC, Rokos JW, Dunlap CL, et al. Segmental odontomaxillary dysplasia: report of a series of 5 cases with long-term follow-up. Oral Surg Oral Med Oral Path Oral Radiol Endod. 2011;112:E29-E47.
17. Welsch MJ, Stein SL. A syndrome of hemimaxillary enlargement, asymmetry of the face, tooth abnormalities, and skin findings (HATS). Pediatr Dermatol. 2004;21:448-451.
18. Armstrong C, Napier SS, Boyd RC, et al. Histopathology of teeth in segmental odontomaxillary dysplasia: new findings. J Oral Pathol Med. 2004;33:246-248.
19. Porwal R, Ousterhout DK, Hoffman WY, et al. Hemimaxillofacial dysplasia: a variable presentation. J Craniofac Surg. 2008;19:1554-1557.
20. Koenig LJ, Lynch DP, Yancey KB. Segmental odontomaxillary dysplasia presenting with facial hypertrichosis, commissural lip clefting, and hyperlinear palm. Pediatr Dermatol. 2008;25:491-492.
21. Bhatia SK, Drage N, Cronin AJ, et al. Case report: segmental odontomaxillary dysplasia—a rare disorder. Eur Arch Paediatr Dent. 2008;9:245-248.
22. Raulin C, Schönermark MP, Greve B, et al. Q-switched ruby laser treatment of tattoos and benign pigmented skin lesions: a critical review. Ann Plast Surg. 1998;41:555-565.
23. Trelles MA, Allones I, Moreno-Arias GA, et al. Becker’s naevus: a comparative study between erbium: YAG and Q-switched neodymium:YAG; clinical and histopathological findings. Br J Dermatol. 2005;152:308-313.
24. Choi JE, Kim JW, Seo SH, et al. Treatment of Becker’s nevi with a long-pulse alexandrite laser. Dermatol Surg. 2009;35:1105-1108.
Case Report
A 14-year-old adolescent boy presented to the dermatology clinic at our institution for evaluation of a hyperpigmented hairy patch on the right side of the face that had been present since birth. The patient reported the lesion originally had involved the right cheek, neck, and back but had gradually expanded to include the right side of the upper lip and oral mucosa. His medical history was remarkable for acne, which was currently being managed with topical treatments. There was no family history of similar conditions. There were no mental or developmental deformities since birth.
Physical examination revealed a hyperpigmented patch with hypertrichosis on the right side of the body involving the back, neck, and cheek (Figure 1), as well as hyperpigmentation involving the right side of the upper lip and oral mucosa (Figure 2A). Slight facial asymmetry also was noted. Dental examination revealed irregular spacing and decreased growth of the teeth on the right side of the mouth (Figure 2B).
|
|
|
Figure 2. Some hyperpigmentation involving the oral mucosa on the right side (A) and dental abnormalities (B). | |
A biopsy of the hyperpigmented patch on the back revealed mild regular acanthosis, basal hypermelanosis, slight papillomatosis, and hair structures within the dermis with features that were consistent with a Becker nevus. A dental radiograph demonstrated hyperplasia of the right maxillary alveolus and basal bone area with 2 missing permanent teeth (fourth and fifth premolars)(Figure 3). Computed axial tomography revealed enlargement of the maxillary bone on the right side.
The constellation of clinical, histopathologic, and radiologic findings was consistent with a diagnosis of hemimaxillary enlargement, asymmetry of the face, tooth abnormalities, and skin findings (HATS syndrome). The treatment plan involved surgical modification of the maxillary bone to correct the hyperplasia on the affected side and implanting 2 artificial premolars. Additionally, laser therapy using a Q-switched ruby laser, frequency-doubled Nd:YAG, 1550-nm erbium-doped fiber laser, or 755-nm alexandrite laser was considered to treat the hyperpigmentation associ-ated with the Becker nevus.
Comment
HATS syndrome is a rare, local developmental defect involving the first and second branchial arches. It generally is detected at birth or in early childhood and is associated with unilateral abnormalities of the bones, teeth, gums, and skin. It is more common in boys than girls (1.8:1.0 ratio), with an age range of 2 to 28 years; there is a peak in the first decade of life.1 It was first described by Miles et al2 in 1987 in a case of congenital mild facial asymmetry, unilateral enlargement of the maxillary gingiva and alveolar bone, hypoplastic teeth, and hypertrichosis in the affected area. The investigators at that time suggested the term hemimaxillofacial dysplasia (HD). In 1990, Danforth et al3 reported 8 additional cases with similar features but without known skin changes; they proposed the term segmental odontomaxillary dysplasia (SOD). In 1996, Desalvo et al4 reported a case of SOD involving a 7-year-old girl with an area of hypopigmentation of the lip on the affected side, and Packota et al5 described the radiographic features of 12 cases of SOD. In subsequent years, other cases of HD or SOD were reported in the literature.1,6-16 In 2004, Welsch and Stein17 reported 1 patient with a Becker nevus of the skin and recommended the acronym HATS. Armstrong et al18 reported 2 cases of SOD with new histopathologic findings of the teeth (eg, fibrous enlargement of the pulps, an irregular pulp-dentin interface displaying many pseudoinclusions, pulp stones). In 2008, Porwal et al19 reported a case of HD in which maxillary hypoplasia rather than hyperplasia was noted, which emphasized the variability of the maxillary dysplasia. Koenig et al20 reported a case of SOD with facial hypertrichosis, commissural lip clefting, and hyperlinear palms. Bhatia et al21 reported another case of SOD with a new finding of unilateral ectopic eyelashes.
The etiology remains unknown, but theories include an alteration that occurs in utero or in in-fancy; the possibility of a systemic or endocrine aberration; a postzygotic mutation resulting in genotypic and phenotypic mosaicism of bone and skin, similar to McCune-Albright syndrome; and viral or bacterial infection along the branches of the maxillary division of the trigeminal nerve.1,15 Bone defects include unilateral enlargement of the maxillary alveolar process and thickening of the vertically oriented trabeculae, which is detected radiographically. A reduction in size of the maxillary sinus and nasal airway was reported in about one-half of cases1 and can be detected easily by computed tomography scanning. Missing permanent premolar teeth, tooth shape abnormalities, delayed eruption of teeth, abnormal spacing of teeth, hypoplastic teeth, enlarged teeth, and gingival thickening also are common oral findings.1 The skin manifestations of HATS syndrome are not static but progress well into adolescence15 and can include facial asymmetry, hypertrichosis, Becker nevus, hairy nevus, lip hypopigmentation, discontinuity of the vermilion border, depression of the cheek, and facial erythema.17
The differential diagnosis includes hemifacial hyperplasia, monostotic fibrous dysplasia, and regional odontodysplasia.1 Little information is available concerning the treatment of patients with this condition.15 The reported treatment modalities include combined surgical and orthodontic treatment of unerupted teeth (premolar/canine), prosthodontic treatment, gingivoplasty, recontouring osteotomy for severe facial asymmetry, and reconstructive jaw surgery.1,6,11,15 Successful treatment of Becker nevi with the Q-switched ruby laser, erbium:YAG laser, and 755-nm alexandrite laser have been reported.22-24
Conclusion
There is a need for continued reporting of cases of HATS syndrome in addition to long-term follow-up to document the natural history of the condition and to establish the appropriate treatment.
Case Report
A 14-year-old adolescent boy presented to the dermatology clinic at our institution for evaluation of a hyperpigmented hairy patch on the right side of the face that had been present since birth. The patient reported the lesion originally had involved the right cheek, neck, and back but had gradually expanded to include the right side of the upper lip and oral mucosa. His medical history was remarkable for acne, which was currently being managed with topical treatments. There was no family history of similar conditions. There were no mental or developmental deformities since birth.
Physical examination revealed a hyperpigmented patch with hypertrichosis on the right side of the body involving the back, neck, and cheek (Figure 1), as well as hyperpigmentation involving the right side of the upper lip and oral mucosa (Figure 2A). Slight facial asymmetry also was noted. Dental examination revealed irregular spacing and decreased growth of the teeth on the right side of the mouth (Figure 2B).
|
|
|
Figure 2. Some hyperpigmentation involving the oral mucosa on the right side (A) and dental abnormalities (B). | |
A biopsy of the hyperpigmented patch on the back revealed mild regular acanthosis, basal hypermelanosis, slight papillomatosis, and hair structures within the dermis with features that were consistent with a Becker nevus. A dental radiograph demonstrated hyperplasia of the right maxillary alveolus and basal bone area with 2 missing permanent teeth (fourth and fifth premolars)(Figure 3). Computed axial tomography revealed enlargement of the maxillary bone on the right side.
The constellation of clinical, histopathologic, and radiologic findings was consistent with a diagnosis of hemimaxillary enlargement, asymmetry of the face, tooth abnormalities, and skin findings (HATS syndrome). The treatment plan involved surgical modification of the maxillary bone to correct the hyperplasia on the affected side and implanting 2 artificial premolars. Additionally, laser therapy using a Q-switched ruby laser, frequency-doubled Nd:YAG, 1550-nm erbium-doped fiber laser, or 755-nm alexandrite laser was considered to treat the hyperpigmentation associ-ated with the Becker nevus.
Comment
HATS syndrome is a rare, local developmental defect involving the first and second branchial arches. It generally is detected at birth or in early childhood and is associated with unilateral abnormalities of the bones, teeth, gums, and skin. It is more common in boys than girls (1.8:1.0 ratio), with an age range of 2 to 28 years; there is a peak in the first decade of life.1 It was first described by Miles et al2 in 1987 in a case of congenital mild facial asymmetry, unilateral enlargement of the maxillary gingiva and alveolar bone, hypoplastic teeth, and hypertrichosis in the affected area. The investigators at that time suggested the term hemimaxillofacial dysplasia (HD). In 1990, Danforth et al3 reported 8 additional cases with similar features but without known skin changes; they proposed the term segmental odontomaxillary dysplasia (SOD). In 1996, Desalvo et al4 reported a case of SOD involving a 7-year-old girl with an area of hypopigmentation of the lip on the affected side, and Packota et al5 described the radiographic features of 12 cases of SOD. In subsequent years, other cases of HD or SOD were reported in the literature.1,6-16 In 2004, Welsch and Stein17 reported 1 patient with a Becker nevus of the skin and recommended the acronym HATS. Armstrong et al18 reported 2 cases of SOD with new histopathologic findings of the teeth (eg, fibrous enlargement of the pulps, an irregular pulp-dentin interface displaying many pseudoinclusions, pulp stones). In 2008, Porwal et al19 reported a case of HD in which maxillary hypoplasia rather than hyperplasia was noted, which emphasized the variability of the maxillary dysplasia. Koenig et al20 reported a case of SOD with facial hypertrichosis, commissural lip clefting, and hyperlinear palms. Bhatia et al21 reported another case of SOD with a new finding of unilateral ectopic eyelashes.
The etiology remains unknown, but theories include an alteration that occurs in utero or in in-fancy; the possibility of a systemic or endocrine aberration; a postzygotic mutation resulting in genotypic and phenotypic mosaicism of bone and skin, similar to McCune-Albright syndrome; and viral or bacterial infection along the branches of the maxillary division of the trigeminal nerve.1,15 Bone defects include unilateral enlargement of the maxillary alveolar process and thickening of the vertically oriented trabeculae, which is detected radiographically. A reduction in size of the maxillary sinus and nasal airway was reported in about one-half of cases1 and can be detected easily by computed tomography scanning. Missing permanent premolar teeth, tooth shape abnormalities, delayed eruption of teeth, abnormal spacing of teeth, hypoplastic teeth, enlarged teeth, and gingival thickening also are common oral findings.1 The skin manifestations of HATS syndrome are not static but progress well into adolescence15 and can include facial asymmetry, hypertrichosis, Becker nevus, hairy nevus, lip hypopigmentation, discontinuity of the vermilion border, depression of the cheek, and facial erythema.17
The differential diagnosis includes hemifacial hyperplasia, monostotic fibrous dysplasia, and regional odontodysplasia.1 Little information is available concerning the treatment of patients with this condition.15 The reported treatment modalities include combined surgical and orthodontic treatment of unerupted teeth (premolar/canine), prosthodontic treatment, gingivoplasty, recontouring osteotomy for severe facial asymmetry, and reconstructive jaw surgery.1,6,11,15 Successful treatment of Becker nevi with the Q-switched ruby laser, erbium:YAG laser, and 755-nm alexandrite laser have been reported.22-24
Conclusion
There is a need for continued reporting of cases of HATS syndrome in addition to long-term follow-up to document the natural history of the condition and to establish the appropriate treatment.
1. Friedlander-Barenboim S, Kamburoglu K, Kaffe I. Clinical and radiological presentation of hemimaxillofacial dysplasia/segmental odontomaxillary dysplasia: critical analysis and report of a case. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;113:268-273.
2. Miles DA, Lovas JL, Cohen MM. Hemimaxillofacial dysplasia: a newly recognized disorder of facial asymmetry, hypertrichosis of the facial skin, unilateral enlargement of the maxilla, and hypoplastic teeth in two patients. Oral Surg Oral Med Oral Pathol. 1987;64:445-448.
3. Danforth RA, Melrose RJ, Abrams AM, et al. Segmental odontomaxillary dysplasia: report of eight cases and comparison with hemimaxillofacial dysplasia. Oral Surg Oral Med Oral Pathol. 1990;70:81-85.
4. Desalvo MS, Copete MA, Riesenberger RE, et al. Segmental odontomaxillary dysplasia (hemimaxillofacial dysplasia): case report. Pediatr Dent. 1996;18:154-156.
5. Packota GV, Pharoah MJ, Petrikowski CG. Radiographic features of segmental odontomaxillary dysplasia: a study of 12 cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;82:577-584.
6. Paticoff K, Marion RW, Shprintzen RJ, et al. Hemimaxillofacial dysplasia: a report of two new cases and further delineation of the disorder. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;83:484-488.
7. Jones AC, Ford MJ. Simultaneous occurrence of segmental odontomaxillary dysplasia and Becker’s nevus. J Oral Maxillofac Surg. 1990;57:1251-1254.
8. Prusack N, Pringle G, Scotti V, et al. Segmental odontomaxillary dysplasia: a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:483-488.
9. Becktor KB, Reibel J, Vedel B, et al. Segmental odontomaxillary dysplasia: clinical, radiological and histological aspects of four cases. Oral Dis. 2002;8:106-110.
10. Velez I, Vedrenne D, Cralle P, et al. Segmental odontomaxillary dysplasia: report of two cases and review of the literature. Todays FDA. 2002;14:20-21.
11. Drake DL. Segmental odontomaxillary dysplasia: an unusual orthodontic challenge. Am J Orthod Dentofac Orthop. 2003;123:84-86.
12. Gavalda C. Segmental odontomaxillary dysplasia. Med Oral. 2004;9:181.
13. Ozpinar B, Gokce B, Uzel G, et al. Prosthetic rehabilitation of segmental odontomaxillary dysplasia: seven-year follow-up. Cleft Palate Craniofac J. 2009;46:103-107.
14. Kuklani RM, Nair MK. Segmental odontomaxillary dysplasia: review of the literature and case report [published online ahead of print December 14, 2010]. Int J Dent. 2010;2010:837283.
15. Minett CP, Daley TD. Hemimaxillofacial dysplasia (segmental odontomaxillary dysplasia): case study with 11 years of follow-up from primary to adult dentition. J Oral Maxillofac Surg. 2012;70:1183-1191.
16. Whitt JC, Rokos JW, Dunlap CL, et al. Segmental odontomaxillary dysplasia: report of a series of 5 cases with long-term follow-up. Oral Surg Oral Med Oral Path Oral Radiol Endod. 2011;112:E29-E47.
17. Welsch MJ, Stein SL. A syndrome of hemimaxillary enlargement, asymmetry of the face, tooth abnormalities, and skin findings (HATS). Pediatr Dermatol. 2004;21:448-451.
18. Armstrong C, Napier SS, Boyd RC, et al. Histopathology of teeth in segmental odontomaxillary dysplasia: new findings. J Oral Pathol Med. 2004;33:246-248.
19. Porwal R, Ousterhout DK, Hoffman WY, et al. Hemimaxillofacial dysplasia: a variable presentation. J Craniofac Surg. 2008;19:1554-1557.
20. Koenig LJ, Lynch DP, Yancey KB. Segmental odontomaxillary dysplasia presenting with facial hypertrichosis, commissural lip clefting, and hyperlinear palm. Pediatr Dermatol. 2008;25:491-492.
21. Bhatia SK, Drage N, Cronin AJ, et al. Case report: segmental odontomaxillary dysplasia—a rare disorder. Eur Arch Paediatr Dent. 2008;9:245-248.
22. Raulin C, Schönermark MP, Greve B, et al. Q-switched ruby laser treatment of tattoos and benign pigmented skin lesions: a critical review. Ann Plast Surg. 1998;41:555-565.
23. Trelles MA, Allones I, Moreno-Arias GA, et al. Becker’s naevus: a comparative study between erbium: YAG and Q-switched neodymium:YAG; clinical and histopathological findings. Br J Dermatol. 2005;152:308-313.
24. Choi JE, Kim JW, Seo SH, et al. Treatment of Becker’s nevi with a long-pulse alexandrite laser. Dermatol Surg. 2009;35:1105-1108.
1. Friedlander-Barenboim S, Kamburoglu K, Kaffe I. Clinical and radiological presentation of hemimaxillofacial dysplasia/segmental odontomaxillary dysplasia: critical analysis and report of a case. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;113:268-273.
2. Miles DA, Lovas JL, Cohen MM. Hemimaxillofacial dysplasia: a newly recognized disorder of facial asymmetry, hypertrichosis of the facial skin, unilateral enlargement of the maxilla, and hypoplastic teeth in two patients. Oral Surg Oral Med Oral Pathol. 1987;64:445-448.
3. Danforth RA, Melrose RJ, Abrams AM, et al. Segmental odontomaxillary dysplasia: report of eight cases and comparison with hemimaxillofacial dysplasia. Oral Surg Oral Med Oral Pathol. 1990;70:81-85.
4. Desalvo MS, Copete MA, Riesenberger RE, et al. Segmental odontomaxillary dysplasia (hemimaxillofacial dysplasia): case report. Pediatr Dent. 1996;18:154-156.
5. Packota GV, Pharoah MJ, Petrikowski CG. Radiographic features of segmental odontomaxillary dysplasia: a study of 12 cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;82:577-584.
6. Paticoff K, Marion RW, Shprintzen RJ, et al. Hemimaxillofacial dysplasia: a report of two new cases and further delineation of the disorder. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;83:484-488.
7. Jones AC, Ford MJ. Simultaneous occurrence of segmental odontomaxillary dysplasia and Becker’s nevus. J Oral Maxillofac Surg. 1990;57:1251-1254.
8. Prusack N, Pringle G, Scotti V, et al. Segmental odontomaxillary dysplasia: a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:483-488.
9. Becktor KB, Reibel J, Vedel B, et al. Segmental odontomaxillary dysplasia: clinical, radiological and histological aspects of four cases. Oral Dis. 2002;8:106-110.
10. Velez I, Vedrenne D, Cralle P, et al. Segmental odontomaxillary dysplasia: report of two cases and review of the literature. Todays FDA. 2002;14:20-21.
11. Drake DL. Segmental odontomaxillary dysplasia: an unusual orthodontic challenge. Am J Orthod Dentofac Orthop. 2003;123:84-86.
12. Gavalda C. Segmental odontomaxillary dysplasia. Med Oral. 2004;9:181.
13. Ozpinar B, Gokce B, Uzel G, et al. Prosthetic rehabilitation of segmental odontomaxillary dysplasia: seven-year follow-up. Cleft Palate Craniofac J. 2009;46:103-107.
14. Kuklani RM, Nair MK. Segmental odontomaxillary dysplasia: review of the literature and case report [published online ahead of print December 14, 2010]. Int J Dent. 2010;2010:837283.
15. Minett CP, Daley TD. Hemimaxillofacial dysplasia (segmental odontomaxillary dysplasia): case study with 11 years of follow-up from primary to adult dentition. J Oral Maxillofac Surg. 2012;70:1183-1191.
16. Whitt JC, Rokos JW, Dunlap CL, et al. Segmental odontomaxillary dysplasia: report of a series of 5 cases with long-term follow-up. Oral Surg Oral Med Oral Path Oral Radiol Endod. 2011;112:E29-E47.
17. Welsch MJ, Stein SL. A syndrome of hemimaxillary enlargement, asymmetry of the face, tooth abnormalities, and skin findings (HATS). Pediatr Dermatol. 2004;21:448-451.
18. Armstrong C, Napier SS, Boyd RC, et al. Histopathology of teeth in segmental odontomaxillary dysplasia: new findings. J Oral Pathol Med. 2004;33:246-248.
19. Porwal R, Ousterhout DK, Hoffman WY, et al. Hemimaxillofacial dysplasia: a variable presentation. J Craniofac Surg. 2008;19:1554-1557.
20. Koenig LJ, Lynch DP, Yancey KB. Segmental odontomaxillary dysplasia presenting with facial hypertrichosis, commissural lip clefting, and hyperlinear palm. Pediatr Dermatol. 2008;25:491-492.
21. Bhatia SK, Drage N, Cronin AJ, et al. Case report: segmental odontomaxillary dysplasia—a rare disorder. Eur Arch Paediatr Dent. 2008;9:245-248.
22. Raulin C, Schönermark MP, Greve B, et al. Q-switched ruby laser treatment of tattoos and benign pigmented skin lesions: a critical review. Ann Plast Surg. 1998;41:555-565.
23. Trelles MA, Allones I, Moreno-Arias GA, et al. Becker’s naevus: a comparative study between erbium: YAG and Q-switched neodymium:YAG; clinical and histopathological findings. Br J Dermatol. 2005;152:308-313.
24. Choi JE, Kim JW, Seo SH, et al. Treatment of Becker’s nevi with a long-pulse alexandrite laser. Dermatol Surg. 2009;35:1105-1108.
Practice Points
- Appropriate management and treatment of hemimaxillary enlargement, asymmetry of the face, tooth abnormalities, and skin findings (HATS syndrome) requires a multidisciplinary team including a dermatologist, dentist, radiologist, and orthopedic surgeon.
- Becker nevus is among the most common cutaneous manifestations of HATS syndrome and can be treated effectively with the Q-switched laser or the erbium:YAG laser.
Interventions for the Treatment of Stretch Marks: A Systematic Review
Stretch marks (striae cutis distensae) are a common disfiguring skin condition characterized by linear bands of atrophic-appearing skin.1 The prevalence of stretch marks associated with pregnancy ranges from 50% to 90%.2 Although stretch marks do not pose a health risk, they often cause burning, itching, and emotional distress, and they can have a deep psychological impact on patients, particularly in young healthy women who are commonly affected by this condition.3
The cause of stretch marks currently is unknown, but they are known to develop in a variety of physiological and pathological states (eg, pregnancy, adolescent growth spurts, obesity, large weight gain, Cushing syndrome, Marfan syndrome, diabetes mellitus, long-term systemic or topical steroid use).2-5 Clinically, newly formed stretch marks present as pink or purple linear lesions without substantial depression of the skin (striae rubra). Over time, the lesions lose their pigmentation, becoming depressed, atrophic, and white (striae alba).2,3,6 The most commonly affected sites are the breasts, upper arms, abdomen, buttocks, and thighs.3,4
Regardless of the etiology, the same histologic changes can be noted in the epidermis of all stretch marks, such as atrophy and loss of rete ridges, with features that are similar to scarring.3 Additionally, reorganization and diminution of the elastic fiber network of skin can be observed.7
A variety of treatment strategies are available for stretch marks, including topical preparations (eg, tretinoin, glycolic acid) and lasers.4 With current methods, no consistently effective therapies have been established yet. In this article, we present the results of a systematic review of the literature to address the effectiveness and safety of the available treatment options for stretch marks.
Methods
A literature search for randomized controlled trials (RCTs) related to the treatment of stretch marks was conducted on March 13, 2013, using the Cochrane Central Register of Controlled Trials, PubMed (from 1966), Embase (from 1974), Chinese Biomedical Literature Database (from 1978), China National Knowledge Infrastructure (from 1994), Chinese Science Journals Database (from 1989), and Wanfang Data (from 1995). Search terms included stretch marks, stretch mark, striae atrophicae, striae distensae, striae gravidarum, striae rubra, striae alba, lineae albicantes, striae, kikkisa, and random*.
We attempted to contact the original investigators of the 25 articles assessed for eligibility by e-mail to identify the randomization and answer other methodology questions to ensure that the studies included in the analysis were RCTs. Each of the 8 RCTs selected for inclusion was assessed independently by 2 investigators (L.L. and H.M.), and data extraction also was performed independently. Any differences in opinion were resolved by discussion. The risk of biases were assessed and 5 domains—random sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, incomplete outcome data—were judged for each study included in the analysis using the Cochrane Collaboration’s domain-based evaluation tool as described in the Cochrane Handbook for Systematic Reviews of Interventions.8 Publication bias was not assessed due to insufficient data.
Studies ultimately were classified into 3 categories based on the risk of bias: (1) low risk of bias or low risk of bias for all key domains; (2) unclear risk of bias or unclear risk of bias for 1 or more key domains; and (3) high risk of bias or high risk of 1 or more key domains.
Results
Search Results
Figure 1 presents the literature search results. Of 300 total search results, 8 RCTs were selected for assessment,9-16 which included a total of 240 patients (Table). The investigators of all 8 reports were contacted, but only 2 responses were received.11,14 The full text of one article could not be obtained; therefore, we could not confirm that it was a true RCT and excluded it.17
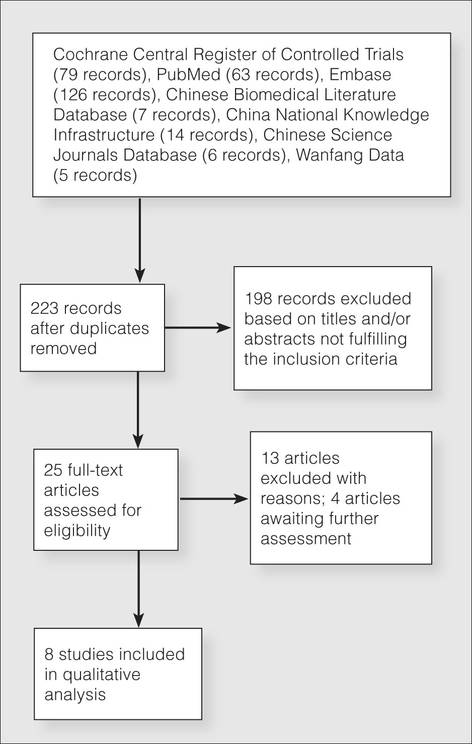
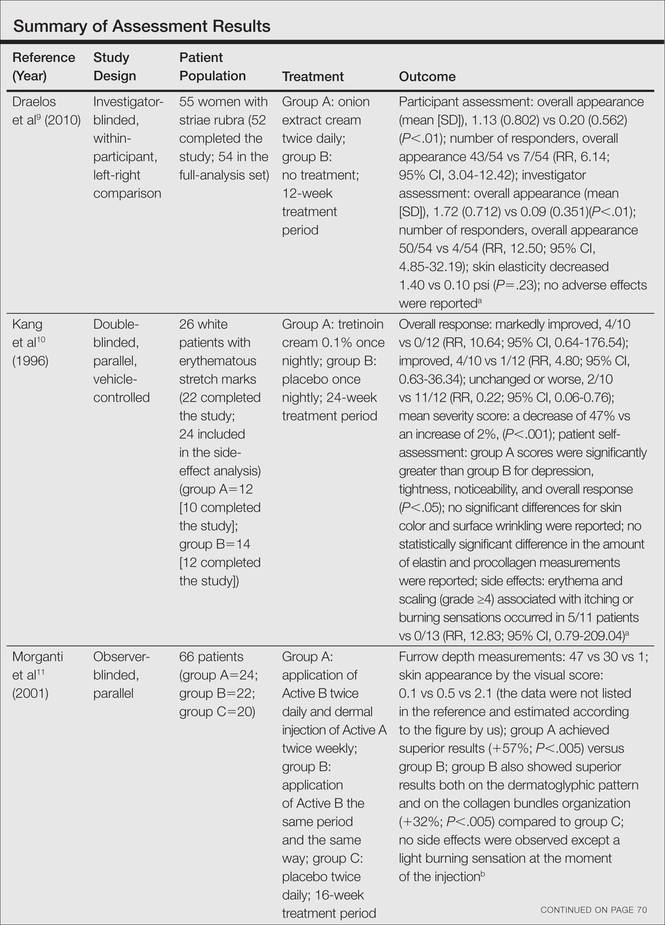
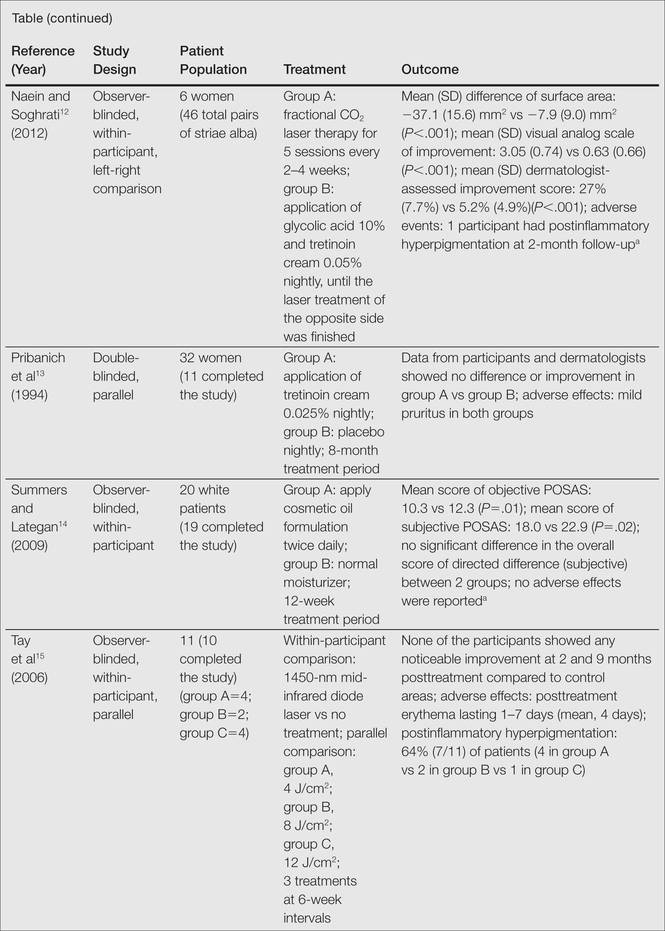
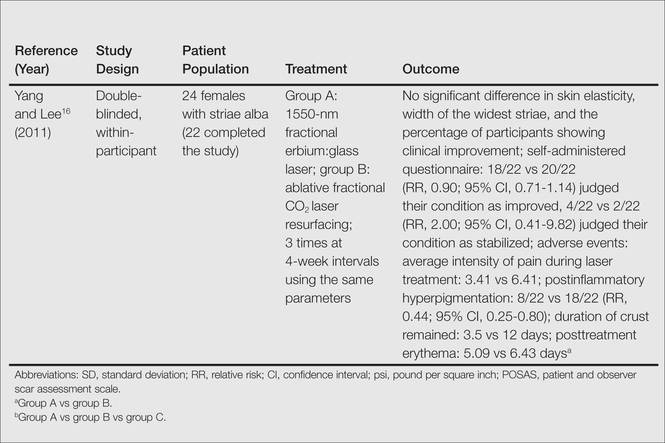
Risk of Bias
The risk of bias in methodology was evaluated for all 8 RCTs and the judgments were given for each domain (Figure 2). All the included studies claimed to be RCTs, but only 37.5% (3/8) of them used adequate randomizations, which were from a including computer-generated code,10 a table of randomized numbers,13 or the Microsoft Excel RND function (from the author by e-mail).14 The randomization methods in the other 5 studies were unclear. Allocation concealment was adequate in 1 trial13 but was unclear in the others. Three trials were double-blinded with the participants and outcome assessors blinded10,13,16; in 2 of these studies investigators also were blinded.10,13 There were 5 single-blinded trials; in 3 of these trials the outcome assessors were blinded12,14,15 and 1 was investigator-blinded.9 The other study was stated to be single-blinded but with no further detail.11 Due to the nature of the experimental design in 2 of the trials12,15 (ie, effects of laser therapy compared to topical treatment or no therapy), participants could not be blinded to treatment types; however, participants were blinded in 1 trial that compared different types of lasers.16 Investigators from all studies reported participants who did not complete the trial or were lost to follow-up, ranging from 0% to 65.6%. Two trials reported no loss of follow-up.11,12 Most trials had losses less than 20% except Pribanich et al13 who reported a loss of 65.6% of participants. One trial included a full analysis set,9 and none of the studies included an intention-to-treat analysis.

The overall risk of bias was assessed for each study and none could be categorized as low risk. Six studies had 1 or more domains assessed as high risk of bias and were classified as high risk of bias.9,11-15 The remaining 2 studies without high-risk domains had one or more domains assessed as unclear10,16 and were therefore considered to be at unclear risk of bias overall.
Effects of Treatments
Among the 8 studies we assessed, there were different treatments, methods of comparison, product concentrations, and times of application. The methods for assessing outcomes (eg, the size and severity of stretch marks) also were varied. Therefore, it is difficult to perform a meta-analysis of the data, and all the evidence was from individual studies. A summary of the results is presented in the Table.
All of the studies we evaluated assessed clinical improvement. Three studies reported the effects of topical tretinoin on stretch marks.10,12,13 A small parallel study with unclear risk of bias indicated that white participants with erythematous stretch marks seemed to have a better response to treatment with tretinoin cream 0.1% for 24 weeks versus placebo.10 However, there was no significant difference between tretinoin cream 0.025% and placebo for patients with abdominal striae in another trial.13 The latter trial was performed with low risk of bias in methodology, but the dropout rate was high (65.6%), with only 11 of 32 participants completing the trial. It is likely that the small number of patients makes the power too low to detect significant differences between tretinoin cream 0.025% and placebo if such a difference indeed existed.13 Because the outcomes in these 2 trials were assessed in different ways, it is difficult to perform a meta-analysis on the data. More adverse effects, mainly erythema and scaling associated with itching or burning sensations, were reported with the higher concentration (0.05%) of tretinoin.10 Another study at a high risk of bias found that the combined use of tretinoin cream 0.05% and glycolic acid 10% was not as effective as fractional CO2 laser therapy in improving the appearance of striae alba.12 There also were 3 studies comparing the effects of laser therapy with another treatment or no treatment.12,15,16 Two within-participant comparison studies with unclear or high risk of bias compared CO2 fractional laser therapy with other active treatment methods in female participants with striae alba.12,16 No difference between the fractional CO2 laser and the 1550-nm nonablative fractional erbium:glass laser was reported,16 but the fractional CO2 laser may be more effective than the topical therapy.12 A small study (11 participants) at high risk of bias reported negative results for the 1450-nm mid-infrared diode laser compared to no treatment.15 Data on the adverse effects of laser therapy were available from these studies. Postinflammatory hyperpigmentation was found in all the 3 studies12,15,16 and posttreatment erythema was mentioned in 2 studies.15,16 Based on the individual studies, treatment with a cosmetic oil formulation was more effective than a moisturizer in improving clinical presentation of stretch marks in white patients.14 Women with striae rubra showed better response to treatment with onion extract cream versus no treatment.9 Limited data from 1 study showed that combined use of Active B (sodium ascorbyl phosphate, 3-aminopropyl-L-ascorbyl phosphate, carboxybetaglucan, hyaluronic acid) and Active A (hyaluronic acid, sodium salt 2 mg, sodium carboxymethyl betaglucan 0.1 mg, ascorbic acid 0.5 mg, arginine 1 mg, sodium chloride 9 mg, sterile water) might be more effective than the use of Active B or placebo.11 These 3 studies are at high risk of bias and no obvious adverse effects were reported.9,11,14
Comment
In the 8 trials included in our assessment, 5 used a within-participant design in which 2 different treatments were randomly administered to the left and right sides of the body, respectively.9,12,14-16 Because the comparison of treatments was made based on results in the same patient versus 2 different treatment groups, the results may be more accurate. In the studies we reviewed, only 3 were placebo-controlled, which may only provide limited evidence on the comparative efficacy of the treatments used in these studies.10,11,13 Most treatments were evaluated in single studies, and most studies had a small number of participants (range, 6–66 participants). A considerable number of the total participants withdrew from their respective studies or were lost during follow-up. In some cases, no reason was given,13 but in the others, it was because of an obvious side effect16 or noncompliance.14 Overall, the methodology quality was low, especially the methods of randomization and allocation concealment. Unsuccessful attempts to contact the original investigators made it difficult to make accurate assessments of the risk of bias in most of the studies included in our assessment. No study met all the risk of bias criteria, and none were classified as having a low risk of bias.
The impact of industry sponsorship on the direction and completeness of the results of the studies we reviewed is unclear. One study was funded by a grant from the manufacturer of the study product,14 and the medication used in another study was supplied by the manufacturer.13 Another study was supported in part by a company that had no part in the conduct, analysis, or reporting of the study.10 In one instance, the authors were employees of the manufacturer of the study product.9 The remaining studies made no declaration.11,12,15,16
Thus the evidence from this review was insufficient to provide clear guidelines for practice. Because the results were based on a small number of patients and were of high or unclear risk of bias, caution must be taken when comparing the efficacy of the treatments administered in these studies; however, given the negligible reported side effects, tretinoin cream 0.1%, a cosmetic oil formulation, onion extract cream, or the combined use of Active A and Active B could reasonably be considered for the treatment of stretch marks. Laser therapies such as the fractional CO2 laser or the 1550-nm fractional erbium:glass laser may be another effective choice.
Conclusion
In future investigations of stretch mark treatments, more high-quality, placebo-controlled trials are needed. One important issue is the varied outcome assessment among different studies, which makes the evaluation and pooling of different studies difficult. Therefore, future RCTs should measure clinical features with a uniform score system such as the visual analog scale or the patient and observer scar assessment scale and try to avoid individual made-up system to assess the outcome. Furthermore, quality-of-life assessment was not included in any of the reports we evaluated; rather all 8 studies focused on changes in the appearance of stretch marks only. Given the deep psychological impact that stretch marks can have on patients, measures for quality-of-life assessment, such as the dermatology life quality index, should be incorporated into future study designs to improve the relevance of the trial and allow comparisons among studies using different interventions.
1. Viennet C, Bride J, Cohen-Letessier A, et al. Mechanical behavior of fibroblasts included in collagen lattices. J Soc Biol. 2001;195:427-430.
2. Chang AL, Agredano YZ, Kimball AB. Risk factors associated with striae gravidarum. J Am Acad Dermatol. 2004;51:881-885.
3. Salter SA, Kimball AB. Striae gravidarum. Clin Dermatol. 2006;24:97-100.
4. Elsaie ML, Baumann LS, Elsaaiee LT. Striae distensae (stretch marks) and different modalities of therapy: an update. Dermatol Surg. 2009;35:563-573.
5. Singh G, Kumar LP. Striae distensae. Indian J Dermatol Venereol Leprol. 2005;71:370-372.
6. Jiménez GP, Flores F, Berman B, et al. Treatment of striae rubra and striae alba with the 585-nm pulsed-dye laser. Dermatol Surg. 2003;29:362-365.
7. Watson RE, Parry EJ, Humphries JD, et al. Fibrillin microfibrils are reduced in skin exhibiting striae distensae. Br J Dermatol. 1998;138:931-937.
8. Higgins JPT, Green S, eds. Cochrane Handbook for Systematic Reviews of Interventions. Oxford, United Kingdom: The Cochrane Collaboration; 2011. http://handbook.cochrane.org/. Accessed July 8, 2014.
9. Draelos ZD, Gold MH, Kaur M, et al. Evaluation of an onion extract, Centella asiatica, and hyaluronic acid cream in the appearance of striae rubra. Skinmed. 2010;8:80-86.
10. Kang S, Kim KJ, Griffiths CE, et al. Topical tretinoin (retinoic acid) improves early stretch marks. Arch Dermatol. 1996;132:519-526.
11. Morganti P, Palombo P, Fabrizi G, et al. Biweekly in-office injectable treatment of striae distensae vs a long-term daily use of topical vitamin C. J Appl Cosmetol. 2001;19:107-112.
12. Naein FF, Soghrati M. Fractional CO2 laser as an effective modality in treatment of striae alba in skin types III and IV. J Res Med Sci. 2012;17:928-933.
13. Pribanich S, Simpson FG, Held B, et al. Low-dose tretinoin does not improve striae distensae: a double-blind, placebo-controlled study. Cutis. 1994;54:121-124.
14. Summers B, Lategan M. The effect of a topically-applied cosmetic oil formulation on striae distensae. SA Fam Pract. 2009;51:332-336.
15. Tay YK, Kwok C, Tan E. Non-ablative 1,450-nm diode laser treatment of striae distensae. Lasers Surg Med. 2006;38:196-199.
16. Yang YJ, Lee GY. Treatment of striae distensae with nonablative fractional laser versus ablative CO2 fractional laser: a randomized controlled trial. Ann Dermatol. 2011;23:481-489.
17. Joshi J, Donga SB, Pandya MA. A comparative study of Savarnakara Ghrita and Savarnakara Cream in the management of Kikkisa w.s.r. to Striae Gravidarum. Ayu. 2008;29:260-265.
Stretch marks (striae cutis distensae) are a common disfiguring skin condition characterized by linear bands of atrophic-appearing skin.1 The prevalence of stretch marks associated with pregnancy ranges from 50% to 90%.2 Although stretch marks do not pose a health risk, they often cause burning, itching, and emotional distress, and they can have a deep psychological impact on patients, particularly in young healthy women who are commonly affected by this condition.3
The cause of stretch marks currently is unknown, but they are known to develop in a variety of physiological and pathological states (eg, pregnancy, adolescent growth spurts, obesity, large weight gain, Cushing syndrome, Marfan syndrome, diabetes mellitus, long-term systemic or topical steroid use).2-5 Clinically, newly formed stretch marks present as pink or purple linear lesions without substantial depression of the skin (striae rubra). Over time, the lesions lose their pigmentation, becoming depressed, atrophic, and white (striae alba).2,3,6 The most commonly affected sites are the breasts, upper arms, abdomen, buttocks, and thighs.3,4
Regardless of the etiology, the same histologic changes can be noted in the epidermis of all stretch marks, such as atrophy and loss of rete ridges, with features that are similar to scarring.3 Additionally, reorganization and diminution of the elastic fiber network of skin can be observed.7
A variety of treatment strategies are available for stretch marks, including topical preparations (eg, tretinoin, glycolic acid) and lasers.4 With current methods, no consistently effective therapies have been established yet. In this article, we present the results of a systematic review of the literature to address the effectiveness and safety of the available treatment options for stretch marks.
Methods
A literature search for randomized controlled trials (RCTs) related to the treatment of stretch marks was conducted on March 13, 2013, using the Cochrane Central Register of Controlled Trials, PubMed (from 1966), Embase (from 1974), Chinese Biomedical Literature Database (from 1978), China National Knowledge Infrastructure (from 1994), Chinese Science Journals Database (from 1989), and Wanfang Data (from 1995). Search terms included stretch marks, stretch mark, striae atrophicae, striae distensae, striae gravidarum, striae rubra, striae alba, lineae albicantes, striae, kikkisa, and random*.
We attempted to contact the original investigators of the 25 articles assessed for eligibility by e-mail to identify the randomization and answer other methodology questions to ensure that the studies included in the analysis were RCTs. Each of the 8 RCTs selected for inclusion was assessed independently by 2 investigators (L.L. and H.M.), and data extraction also was performed independently. Any differences in opinion were resolved by discussion. The risk of biases were assessed and 5 domains—random sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, incomplete outcome data—were judged for each study included in the analysis using the Cochrane Collaboration’s domain-based evaluation tool as described in the Cochrane Handbook for Systematic Reviews of Interventions.8 Publication bias was not assessed due to insufficient data.
Studies ultimately were classified into 3 categories based on the risk of bias: (1) low risk of bias or low risk of bias for all key domains; (2) unclear risk of bias or unclear risk of bias for 1 or more key domains; and (3) high risk of bias or high risk of 1 or more key domains.
Results
Search Results
Figure 1 presents the literature search results. Of 300 total search results, 8 RCTs were selected for assessment,9-16 which included a total of 240 patients (Table). The investigators of all 8 reports were contacted, but only 2 responses were received.11,14 The full text of one article could not be obtained; therefore, we could not confirm that it was a true RCT and excluded it.17
Risk of Bias
The risk of bias in methodology was evaluated for all 8 RCTs and the judgments were given for each domain (Figure 2). All the included studies claimed to be RCTs, but only 37.5% (3/8) of them used adequate randomizations, which were from a including computer-generated code,10 a table of randomized numbers,13 or the Microsoft Excel RND function (from the author by e-mail).14 The randomization methods in the other 5 studies were unclear. Allocation concealment was adequate in 1 trial13 but was unclear in the others. Three trials were double-blinded with the participants and outcome assessors blinded10,13,16; in 2 of these studies investigators also were blinded.10,13 There were 5 single-blinded trials; in 3 of these trials the outcome assessors were blinded12,14,15 and 1 was investigator-blinded.9 The other study was stated to be single-blinded but with no further detail.11 Due to the nature of the experimental design in 2 of the trials12,15 (ie, effects of laser therapy compared to topical treatment or no therapy), participants could not be blinded to treatment types; however, participants were blinded in 1 trial that compared different types of lasers.16 Investigators from all studies reported participants who did not complete the trial or were lost to follow-up, ranging from 0% to 65.6%. Two trials reported no loss of follow-up.11,12 Most trials had losses less than 20% except Pribanich et al13 who reported a loss of 65.6% of participants. One trial included a full analysis set,9 and none of the studies included an intention-to-treat analysis.
The overall risk of bias was assessed for each study and none could be categorized as low risk. Six studies had 1 or more domains assessed as high risk of bias and were classified as high risk of bias.9,11-15 The remaining 2 studies without high-risk domains had one or more domains assessed as unclear10,16 and were therefore considered to be at unclear risk of bias overall.
Effects of Treatments
Among the 8 studies we assessed, there were different treatments, methods of comparison, product concentrations, and times of application. The methods for assessing outcomes (eg, the size and severity of stretch marks) also were varied. Therefore, it is difficult to perform a meta-analysis of the data, and all the evidence was from individual studies. A summary of the results is presented in the Table.
All of the studies we evaluated assessed clinical improvement. Three studies reported the effects of topical tretinoin on stretch marks.10,12,13 A small parallel study with unclear risk of bias indicated that white participants with erythematous stretch marks seemed to have a better response to treatment with tretinoin cream 0.1% for 24 weeks versus placebo.10 However, there was no significant difference between tretinoin cream 0.025% and placebo for patients with abdominal striae in another trial.13 The latter trial was performed with low risk of bias in methodology, but the dropout rate was high (65.6%), with only 11 of 32 participants completing the trial. It is likely that the small number of patients makes the power too low to detect significant differences between tretinoin cream 0.025% and placebo if such a difference indeed existed.13 Because the outcomes in these 2 trials were assessed in different ways, it is difficult to perform a meta-analysis on the data. More adverse effects, mainly erythema and scaling associated with itching or burning sensations, were reported with the higher concentration (0.05%) of tretinoin.10 Another study at a high risk of bias found that the combined use of tretinoin cream 0.05% and glycolic acid 10% was not as effective as fractional CO2 laser therapy in improving the appearance of striae alba.12 There also were 3 studies comparing the effects of laser therapy with another treatment or no treatment.12,15,16 Two within-participant comparison studies with unclear or high risk of bias compared CO2 fractional laser therapy with other active treatment methods in female participants with striae alba.12,16 No difference between the fractional CO2 laser and the 1550-nm nonablative fractional erbium:glass laser was reported,16 but the fractional CO2 laser may be more effective than the topical therapy.12 A small study (11 participants) at high risk of bias reported negative results for the 1450-nm mid-infrared diode laser compared to no treatment.15 Data on the adverse effects of laser therapy were available from these studies. Postinflammatory hyperpigmentation was found in all the 3 studies12,15,16 and posttreatment erythema was mentioned in 2 studies.15,16 Based on the individual studies, treatment with a cosmetic oil formulation was more effective than a moisturizer in improving clinical presentation of stretch marks in white patients.14 Women with striae rubra showed better response to treatment with onion extract cream versus no treatment.9 Limited data from 1 study showed that combined use of Active B (sodium ascorbyl phosphate, 3-aminopropyl-L-ascorbyl phosphate, carboxybetaglucan, hyaluronic acid) and Active A (hyaluronic acid, sodium salt 2 mg, sodium carboxymethyl betaglucan 0.1 mg, ascorbic acid 0.5 mg, arginine 1 mg, sodium chloride 9 mg, sterile water) might be more effective than the use of Active B or placebo.11 These 3 studies are at high risk of bias and no obvious adverse effects were reported.9,11,14
Comment
In the 8 trials included in our assessment, 5 used a within-participant design in which 2 different treatments were randomly administered to the left and right sides of the body, respectively.9,12,14-16 Because the comparison of treatments was made based on results in the same patient versus 2 different treatment groups, the results may be more accurate. In the studies we reviewed, only 3 were placebo-controlled, which may only provide limited evidence on the comparative efficacy of the treatments used in these studies.10,11,13 Most treatments were evaluated in single studies, and most studies had a small number of participants (range, 6–66 participants). A considerable number of the total participants withdrew from their respective studies or were lost during follow-up. In some cases, no reason was given,13 but in the others, it was because of an obvious side effect16 or noncompliance.14 Overall, the methodology quality was low, especially the methods of randomization and allocation concealment. Unsuccessful attempts to contact the original investigators made it difficult to make accurate assessments of the risk of bias in most of the studies included in our assessment. No study met all the risk of bias criteria, and none were classified as having a low risk of bias.
The impact of industry sponsorship on the direction and completeness of the results of the studies we reviewed is unclear. One study was funded by a grant from the manufacturer of the study product,14 and the medication used in another study was supplied by the manufacturer.13 Another study was supported in part by a company that had no part in the conduct, analysis, or reporting of the study.10 In one instance, the authors were employees of the manufacturer of the study product.9 The remaining studies made no declaration.11,12,15,16
Thus the evidence from this review was insufficient to provide clear guidelines for practice. Because the results were based on a small number of patients and were of high or unclear risk of bias, caution must be taken when comparing the efficacy of the treatments administered in these studies; however, given the negligible reported side effects, tretinoin cream 0.1%, a cosmetic oil formulation, onion extract cream, or the combined use of Active A and Active B could reasonably be considered for the treatment of stretch marks. Laser therapies such as the fractional CO2 laser or the 1550-nm fractional erbium:glass laser may be another effective choice.
Conclusion
In future investigations of stretch mark treatments, more high-quality, placebo-controlled trials are needed. One important issue is the varied outcome assessment among different studies, which makes the evaluation and pooling of different studies difficult. Therefore, future RCTs should measure clinical features with a uniform score system such as the visual analog scale or the patient and observer scar assessment scale and try to avoid individual made-up system to assess the outcome. Furthermore, quality-of-life assessment was not included in any of the reports we evaluated; rather all 8 studies focused on changes in the appearance of stretch marks only. Given the deep psychological impact that stretch marks can have on patients, measures for quality-of-life assessment, such as the dermatology life quality index, should be incorporated into future study designs to improve the relevance of the trial and allow comparisons among studies using different interventions.
Stretch marks (striae cutis distensae) are a common disfiguring skin condition characterized by linear bands of atrophic-appearing skin.1 The prevalence of stretch marks associated with pregnancy ranges from 50% to 90%.2 Although stretch marks do not pose a health risk, they often cause burning, itching, and emotional distress, and they can have a deep psychological impact on patients, particularly in young healthy women who are commonly affected by this condition.3
The cause of stretch marks currently is unknown, but they are known to develop in a variety of physiological and pathological states (eg, pregnancy, adolescent growth spurts, obesity, large weight gain, Cushing syndrome, Marfan syndrome, diabetes mellitus, long-term systemic or topical steroid use).2-5 Clinically, newly formed stretch marks present as pink or purple linear lesions without substantial depression of the skin (striae rubra). Over time, the lesions lose their pigmentation, becoming depressed, atrophic, and white (striae alba).2,3,6 The most commonly affected sites are the breasts, upper arms, abdomen, buttocks, and thighs.3,4
Regardless of the etiology, the same histologic changes can be noted in the epidermis of all stretch marks, such as atrophy and loss of rete ridges, with features that are similar to scarring.3 Additionally, reorganization and diminution of the elastic fiber network of skin can be observed.7
A variety of treatment strategies are available for stretch marks, including topical preparations (eg, tretinoin, glycolic acid) and lasers.4 With current methods, no consistently effective therapies have been established yet. In this article, we present the results of a systematic review of the literature to address the effectiveness and safety of the available treatment options for stretch marks.
Methods
A literature search for randomized controlled trials (RCTs) related to the treatment of stretch marks was conducted on March 13, 2013, using the Cochrane Central Register of Controlled Trials, PubMed (from 1966), Embase (from 1974), Chinese Biomedical Literature Database (from 1978), China National Knowledge Infrastructure (from 1994), Chinese Science Journals Database (from 1989), and Wanfang Data (from 1995). Search terms included stretch marks, stretch mark, striae atrophicae, striae distensae, striae gravidarum, striae rubra, striae alba, lineae albicantes, striae, kikkisa, and random*.
We attempted to contact the original investigators of the 25 articles assessed for eligibility by e-mail to identify the randomization and answer other methodology questions to ensure that the studies included in the analysis were RCTs. Each of the 8 RCTs selected for inclusion was assessed independently by 2 investigators (L.L. and H.M.), and data extraction also was performed independently. Any differences in opinion were resolved by discussion. The risk of biases were assessed and 5 domains—random sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, incomplete outcome data—were judged for each study included in the analysis using the Cochrane Collaboration’s domain-based evaluation tool as described in the Cochrane Handbook for Systematic Reviews of Interventions.8 Publication bias was not assessed due to insufficient data.
Studies ultimately were classified into 3 categories based on the risk of bias: (1) low risk of bias or low risk of bias for all key domains; (2) unclear risk of bias or unclear risk of bias for 1 or more key domains; and (3) high risk of bias or high risk of 1 or more key domains.
Results
Search Results
Figure 1 presents the literature search results. Of 300 total search results, 8 RCTs were selected for assessment,9-16 which included a total of 240 patients (Table). The investigators of all 8 reports were contacted, but only 2 responses were received.11,14 The full text of one article could not be obtained; therefore, we could not confirm that it was a true RCT and excluded it.17
Risk of Bias
The risk of bias in methodology was evaluated for all 8 RCTs and the judgments were given for each domain (Figure 2). All the included studies claimed to be RCTs, but only 37.5% (3/8) of them used adequate randomizations, which were from a including computer-generated code,10 a table of randomized numbers,13 or the Microsoft Excel RND function (from the author by e-mail).14 The randomization methods in the other 5 studies were unclear. Allocation concealment was adequate in 1 trial13 but was unclear in the others. Three trials were double-blinded with the participants and outcome assessors blinded10,13,16; in 2 of these studies investigators also were blinded.10,13 There were 5 single-blinded trials; in 3 of these trials the outcome assessors were blinded12,14,15 and 1 was investigator-blinded.9 The other study was stated to be single-blinded but with no further detail.11 Due to the nature of the experimental design in 2 of the trials12,15 (ie, effects of laser therapy compared to topical treatment or no therapy), participants could not be blinded to treatment types; however, participants were blinded in 1 trial that compared different types of lasers.16 Investigators from all studies reported participants who did not complete the trial or were lost to follow-up, ranging from 0% to 65.6%. Two trials reported no loss of follow-up.11,12 Most trials had losses less than 20% except Pribanich et al13 who reported a loss of 65.6% of participants. One trial included a full analysis set,9 and none of the studies included an intention-to-treat analysis.
The overall risk of bias was assessed for each study and none could be categorized as low risk. Six studies had 1 or more domains assessed as high risk of bias and were classified as high risk of bias.9,11-15 The remaining 2 studies without high-risk domains had one or more domains assessed as unclear10,16 and were therefore considered to be at unclear risk of bias overall.
Effects of Treatments
Among the 8 studies we assessed, there were different treatments, methods of comparison, product concentrations, and times of application. The methods for assessing outcomes (eg, the size and severity of stretch marks) also were varied. Therefore, it is difficult to perform a meta-analysis of the data, and all the evidence was from individual studies. A summary of the results is presented in the Table.
All of the studies we evaluated assessed clinical improvement. Three studies reported the effects of topical tretinoin on stretch marks.10,12,13 A small parallel study with unclear risk of bias indicated that white participants with erythematous stretch marks seemed to have a better response to treatment with tretinoin cream 0.1% for 24 weeks versus placebo.10 However, there was no significant difference between tretinoin cream 0.025% and placebo for patients with abdominal striae in another trial.13 The latter trial was performed with low risk of bias in methodology, but the dropout rate was high (65.6%), with only 11 of 32 participants completing the trial. It is likely that the small number of patients makes the power too low to detect significant differences between tretinoin cream 0.025% and placebo if such a difference indeed existed.13 Because the outcomes in these 2 trials were assessed in different ways, it is difficult to perform a meta-analysis on the data. More adverse effects, mainly erythema and scaling associated with itching or burning sensations, were reported with the higher concentration (0.05%) of tretinoin.10 Another study at a high risk of bias found that the combined use of tretinoin cream 0.05% and glycolic acid 10% was not as effective as fractional CO2 laser therapy in improving the appearance of striae alba.12 There also were 3 studies comparing the effects of laser therapy with another treatment or no treatment.12,15,16 Two within-participant comparison studies with unclear or high risk of bias compared CO2 fractional laser therapy with other active treatment methods in female participants with striae alba.12,16 No difference between the fractional CO2 laser and the 1550-nm nonablative fractional erbium:glass laser was reported,16 but the fractional CO2 laser may be more effective than the topical therapy.12 A small study (11 participants) at high risk of bias reported negative results for the 1450-nm mid-infrared diode laser compared to no treatment.15 Data on the adverse effects of laser therapy were available from these studies. Postinflammatory hyperpigmentation was found in all the 3 studies12,15,16 and posttreatment erythema was mentioned in 2 studies.15,16 Based on the individual studies, treatment with a cosmetic oil formulation was more effective than a moisturizer in improving clinical presentation of stretch marks in white patients.14 Women with striae rubra showed better response to treatment with onion extract cream versus no treatment.9 Limited data from 1 study showed that combined use of Active B (sodium ascorbyl phosphate, 3-aminopropyl-L-ascorbyl phosphate, carboxybetaglucan, hyaluronic acid) and Active A (hyaluronic acid, sodium salt 2 mg, sodium carboxymethyl betaglucan 0.1 mg, ascorbic acid 0.5 mg, arginine 1 mg, sodium chloride 9 mg, sterile water) might be more effective than the use of Active B or placebo.11 These 3 studies are at high risk of bias and no obvious adverse effects were reported.9,11,14
Comment
In the 8 trials included in our assessment, 5 used a within-participant design in which 2 different treatments were randomly administered to the left and right sides of the body, respectively.9,12,14-16 Because the comparison of treatments was made based on results in the same patient versus 2 different treatment groups, the results may be more accurate. In the studies we reviewed, only 3 were placebo-controlled, which may only provide limited evidence on the comparative efficacy of the treatments used in these studies.10,11,13 Most treatments were evaluated in single studies, and most studies had a small number of participants (range, 6–66 participants). A considerable number of the total participants withdrew from their respective studies or were lost during follow-up. In some cases, no reason was given,13 but in the others, it was because of an obvious side effect16 or noncompliance.14 Overall, the methodology quality was low, especially the methods of randomization and allocation concealment. Unsuccessful attempts to contact the original investigators made it difficult to make accurate assessments of the risk of bias in most of the studies included in our assessment. No study met all the risk of bias criteria, and none were classified as having a low risk of bias.
The impact of industry sponsorship on the direction and completeness of the results of the studies we reviewed is unclear. One study was funded by a grant from the manufacturer of the study product,14 and the medication used in another study was supplied by the manufacturer.13 Another study was supported in part by a company that had no part in the conduct, analysis, or reporting of the study.10 In one instance, the authors were employees of the manufacturer of the study product.9 The remaining studies made no declaration.11,12,15,16
Thus the evidence from this review was insufficient to provide clear guidelines for practice. Because the results were based on a small number of patients and were of high or unclear risk of bias, caution must be taken when comparing the efficacy of the treatments administered in these studies; however, given the negligible reported side effects, tretinoin cream 0.1%, a cosmetic oil formulation, onion extract cream, or the combined use of Active A and Active B could reasonably be considered for the treatment of stretch marks. Laser therapies such as the fractional CO2 laser or the 1550-nm fractional erbium:glass laser may be another effective choice.
Conclusion
In future investigations of stretch mark treatments, more high-quality, placebo-controlled trials are needed. One important issue is the varied outcome assessment among different studies, which makes the evaluation and pooling of different studies difficult. Therefore, future RCTs should measure clinical features with a uniform score system such as the visual analog scale or the patient and observer scar assessment scale and try to avoid individual made-up system to assess the outcome. Furthermore, quality-of-life assessment was not included in any of the reports we evaluated; rather all 8 studies focused on changes in the appearance of stretch marks only. Given the deep psychological impact that stretch marks can have on patients, measures for quality-of-life assessment, such as the dermatology life quality index, should be incorporated into future study designs to improve the relevance of the trial and allow comparisons among studies using different interventions.
1. Viennet C, Bride J, Cohen-Letessier A, et al. Mechanical behavior of fibroblasts included in collagen lattices. J Soc Biol. 2001;195:427-430.
2. Chang AL, Agredano YZ, Kimball AB. Risk factors associated with striae gravidarum. J Am Acad Dermatol. 2004;51:881-885.
3. Salter SA, Kimball AB. Striae gravidarum. Clin Dermatol. 2006;24:97-100.
4. Elsaie ML, Baumann LS, Elsaaiee LT. Striae distensae (stretch marks) and different modalities of therapy: an update. Dermatol Surg. 2009;35:563-573.
5. Singh G, Kumar LP. Striae distensae. Indian J Dermatol Venereol Leprol. 2005;71:370-372.
6. Jiménez GP, Flores F, Berman B, et al. Treatment of striae rubra and striae alba with the 585-nm pulsed-dye laser. Dermatol Surg. 2003;29:362-365.
7. Watson RE, Parry EJ, Humphries JD, et al. Fibrillin microfibrils are reduced in skin exhibiting striae distensae. Br J Dermatol. 1998;138:931-937.
8. Higgins JPT, Green S, eds. Cochrane Handbook for Systematic Reviews of Interventions. Oxford, United Kingdom: The Cochrane Collaboration; 2011. http://handbook.cochrane.org/. Accessed July 8, 2014.
9. Draelos ZD, Gold MH, Kaur M, et al. Evaluation of an onion extract, Centella asiatica, and hyaluronic acid cream in the appearance of striae rubra. Skinmed. 2010;8:80-86.
10. Kang S, Kim KJ, Griffiths CE, et al. Topical tretinoin (retinoic acid) improves early stretch marks. Arch Dermatol. 1996;132:519-526.
11. Morganti P, Palombo P, Fabrizi G, et al. Biweekly in-office injectable treatment of striae distensae vs a long-term daily use of topical vitamin C. J Appl Cosmetol. 2001;19:107-112.
12. Naein FF, Soghrati M. Fractional CO2 laser as an effective modality in treatment of striae alba in skin types III and IV. J Res Med Sci. 2012;17:928-933.
13. Pribanich S, Simpson FG, Held B, et al. Low-dose tretinoin does not improve striae distensae: a double-blind, placebo-controlled study. Cutis. 1994;54:121-124.
14. Summers B, Lategan M. The effect of a topically-applied cosmetic oil formulation on striae distensae. SA Fam Pract. 2009;51:332-336.
15. Tay YK, Kwok C, Tan E. Non-ablative 1,450-nm diode laser treatment of striae distensae. Lasers Surg Med. 2006;38:196-199.
16. Yang YJ, Lee GY. Treatment of striae distensae with nonablative fractional laser versus ablative CO2 fractional laser: a randomized controlled trial. Ann Dermatol. 2011;23:481-489.
17. Joshi J, Donga SB, Pandya MA. A comparative study of Savarnakara Ghrita and Savarnakara Cream in the management of Kikkisa w.s.r. to Striae Gravidarum. Ayu. 2008;29:260-265.
1. Viennet C, Bride J, Cohen-Letessier A, et al. Mechanical behavior of fibroblasts included in collagen lattices. J Soc Biol. 2001;195:427-430.
2. Chang AL, Agredano YZ, Kimball AB. Risk factors associated with striae gravidarum. J Am Acad Dermatol. 2004;51:881-885.
3. Salter SA, Kimball AB. Striae gravidarum. Clin Dermatol. 2006;24:97-100.
4. Elsaie ML, Baumann LS, Elsaaiee LT. Striae distensae (stretch marks) and different modalities of therapy: an update. Dermatol Surg. 2009;35:563-573.
5. Singh G, Kumar LP. Striae distensae. Indian J Dermatol Venereol Leprol. 2005;71:370-372.
6. Jiménez GP, Flores F, Berman B, et al. Treatment of striae rubra and striae alba with the 585-nm pulsed-dye laser. Dermatol Surg. 2003;29:362-365.
7. Watson RE, Parry EJ, Humphries JD, et al. Fibrillin microfibrils are reduced in skin exhibiting striae distensae. Br J Dermatol. 1998;138:931-937.
8. Higgins JPT, Green S, eds. Cochrane Handbook for Systematic Reviews of Interventions. Oxford, United Kingdom: The Cochrane Collaboration; 2011. http://handbook.cochrane.org/. Accessed July 8, 2014.
9. Draelos ZD, Gold MH, Kaur M, et al. Evaluation of an onion extract, Centella asiatica, and hyaluronic acid cream in the appearance of striae rubra. Skinmed. 2010;8:80-86.
10. Kang S, Kim KJ, Griffiths CE, et al. Topical tretinoin (retinoic acid) improves early stretch marks. Arch Dermatol. 1996;132:519-526.
11. Morganti P, Palombo P, Fabrizi G, et al. Biweekly in-office injectable treatment of striae distensae vs a long-term daily use of topical vitamin C. J Appl Cosmetol. 2001;19:107-112.
12. Naein FF, Soghrati M. Fractional CO2 laser as an effective modality in treatment of striae alba in skin types III and IV. J Res Med Sci. 2012;17:928-933.
13. Pribanich S, Simpson FG, Held B, et al. Low-dose tretinoin does not improve striae distensae: a double-blind, placebo-controlled study. Cutis. 1994;54:121-124.
14. Summers B, Lategan M. The effect of a topically-applied cosmetic oil formulation on striae distensae. SA Fam Pract. 2009;51:332-336.
15. Tay YK, Kwok C, Tan E. Non-ablative 1,450-nm diode laser treatment of striae distensae. Lasers Surg Med. 2006;38:196-199.
16. Yang YJ, Lee GY. Treatment of striae distensae with nonablative fractional laser versus ablative CO2 fractional laser: a randomized controlled trial. Ann Dermatol. 2011;23:481-489.
17. Joshi J, Donga SB, Pandya MA. A comparative study of Savarnakara Ghrita and Savarnakara Cream in the management of Kikkisa w.s.r. to Striae Gravidarum. Ayu. 2008;29:260-265.
Practice Points
- Given the negligible reported side effects, tretinoin cream 0.1%, a cosmetic oil formulation, onion extract cream, or the combined use of Active A and Active B could be considered for the treatment of stretch marks, though the evidence is insufficient.
- High-quality, randomized, placebo-controlled trials are needed in the future.