User login
Antioxidants plus sunscreens may be two-punch knockout for melasma
ORLANDO – A two-step regimen of high-potency topical antioxidants followed by a mineral-based sunscreen may help repair light-induced skin damage and protect against new damage in patients with melasma.
Recent studies suggest that the antioxidants tamp down inflammatory cytokines and damage of oxidative stress, Maria Ivonne Arellano-Mendoza, MD, said during a special focus session on Latino skin held at the annual meeting of the American Academy of Dermatology.
“Many compounds are being studied for this purpose,” said Dr. Arellano-Mendoza, head of the dermatology department of the General Hospital of Mexico, Mexico City. “One combination is vitamin C, vitamin E, ubiquinone, and grape-seed extract. This effectively prevented infrared-A radiation–induced matrix metalloproteinase-1 messenger RNA expression in human skin. This combination can be found now in some sunscreens and daily care products.”
Another effective combination seems to be a mixture of ferulic acid, tocopherol, and vitamin C, she said.
The two-step process of regularly using a topical product with antioxidants before applying a sunscreen is all there is for now, she added, because so far it’s been impossible to combine the agents in a single product.
“The challenge will be how to create a product that stays on the surface of the skin to protect it from light, while liberating the antioxidants to penetrate the skin,” she commented.
The antioxidants’ benefits, however, will be obliterated by the continued effects of some light wavelengths that aggravate melasma unless they are used in sequence with a light-scattering sunscreen.
Sunscreens are critical components of a melasma treatment regimen, Dr. Arellano-Mendoza said. “Sunscreens are a cornerstone of treatment. We clearly tell our patients that sunscreens have to be used every day, forever, and if they are not used properly, they will have no improvement.”
Patients with hyperpigmentation disorders are susceptible to longer wavelengths that aren’t covered by chemically derived sunscreens. Longer wavelengths, including infrared light and visible light, have been shown to increase expression of matrix metalloproteinase (MMP) -1 and -9, decrease expression of type 1 procollagen, and can induce macrophage infiltration. These wavelengths also increase reactive oxygen species and proinflammatory cytokines in vitro, Dr. Arellano-Mendoza noted.
Visible light can cause erythema, transient and long-lasting hyperpigmentation, thermal damage, free radical production, and premature photoaging. It also can stimulate the production of reactive oxygen species that can damage DNA.
Mineral-based, inorganic sunscreens, however – like those with titanium dioxide, zinc oxide, and iron oxide – scatter all wavelengths.
“These micronized forms of metal oxides not only scatter and reflect light, they also absorb ultraviolet radiation. The compounds aren’t new,” she said. In 1991, Dr. Elaine Kaye of Harvard University, Boston, and associates described (Arch Dermatol. 1991;127:351-5) opaque physical sunscreens that were useful blockers of visible light and found that transmittance of light can be lowered by adding iron oxide, Dr. Arellano-Mendoza pointed out.
The inorganic sunscreens have never been widely adopted because they are highly pigmented with white or, in the case of iron oxide, with red. “Not many people accepted [the iron-containing compounds] because of the redness, but now different shades are going to be hitting the market soon,” and the hope is that consumers will find them more appealing, she said.
This is good news, as the data emerging around iron oxide are intriguingly positive. A 2015 study showed that a sunscreen with iron oxides prevented melasma relapse during the summer months. Patients were randomized to the same ultraviolet filter topical sunscreen, but for one group, micronized iron oxide was added to it. After 6 months, the median melasma area severity index score was significantly better in the group using the iron oxide compound (J Am Acad Dermatol. 2015 Jan;72[1]:189-90.e1).
Support for the two-step regimen appeared in 2014, when a small study randomized 30 healthy volunteers to an SPF 30 sunscreen or the same sunscreen supplemented with an antioxidant cocktail of grape seed extract, vitamin E, ubiquinone, and vitamin C. The endpoint was MMP-1 upregulation after exposure to infrared-A light. Skin treated with the combination regimen showed significantly lower MMP-1 activation, leading the authors to conclude that the combination of topical antioxidants conferred protection against the irradiation (Photochem Photobiol. 2015 Jan-Feb;91[1]:248-50).
Those same authors published a companion article (Photodermatol Photoimmunol Photomed. 2014;30:167-74) suggesting that another antioxidant mixture (ferulic acid, tocopherol, and vitamin C) was similarly effective.
Using a combination of antioxidants is important, Dr. Arellano-Mendoza said, because different antioxidants work differently. Some (catalase, glutathione peroxidase, and superoxide dismutase) are enzymatic, catalyzing reactions that convert free radicals to oxygen and water. Others terminate free radicals by preventing the propagation of oxidative chain reactions (vitamins A and C, flavonoids, uric acid, bilirubin, albumin, and members of the thiol group). A third group consists of metal-binding proteins that sequester free iron or copper to prevent free radical production (ferritin, transferrin, lactoferrin, and ceruloplasmin).
“I think we can now consider antioxidants a part of the tools we use in treating some pigmentary disorders as melasma,” she said. “We need to choose the compounds carefully, and we definitely need more in vivo research, but the findings are very encouraging.”
Dr. Arellano-Mendoza had no relevant financial disclosures.
This article was updated 3/20/17.
msullivan@frontlinemedcom.com
On Twitter @Alz_Gal
ORLANDO – A two-step regimen of high-potency topical antioxidants followed by a mineral-based sunscreen may help repair light-induced skin damage and protect against new damage in patients with melasma.
Recent studies suggest that the antioxidants tamp down inflammatory cytokines and damage of oxidative stress, Maria Ivonne Arellano-Mendoza, MD, said during a special focus session on Latino skin held at the annual meeting of the American Academy of Dermatology.
“Many compounds are being studied for this purpose,” said Dr. Arellano-Mendoza, head of the dermatology department of the General Hospital of Mexico, Mexico City. “One combination is vitamin C, vitamin E, ubiquinone, and grape-seed extract. This effectively prevented infrared-A radiation–induced matrix metalloproteinase-1 messenger RNA expression in human skin. This combination can be found now in some sunscreens and daily care products.”
Another effective combination seems to be a mixture of ferulic acid, tocopherol, and vitamin C, she said.
The two-step process of regularly using a topical product with antioxidants before applying a sunscreen is all there is for now, she added, because so far it’s been impossible to combine the agents in a single product.
“The challenge will be how to create a product that stays on the surface of the skin to protect it from light, while liberating the antioxidants to penetrate the skin,” she commented.
The antioxidants’ benefits, however, will be obliterated by the continued effects of some light wavelengths that aggravate melasma unless they are used in sequence with a light-scattering sunscreen.
Sunscreens are critical components of a melasma treatment regimen, Dr. Arellano-Mendoza said. “Sunscreens are a cornerstone of treatment. We clearly tell our patients that sunscreens have to be used every day, forever, and if they are not used properly, they will have no improvement.”
Patients with hyperpigmentation disorders are susceptible to longer wavelengths that aren’t covered by chemically derived sunscreens. Longer wavelengths, including infrared light and visible light, have been shown to increase expression of matrix metalloproteinase (MMP) -1 and -9, decrease expression of type 1 procollagen, and can induce macrophage infiltration. These wavelengths also increase reactive oxygen species and proinflammatory cytokines in vitro, Dr. Arellano-Mendoza noted.
Visible light can cause erythema, transient and long-lasting hyperpigmentation, thermal damage, free radical production, and premature photoaging. It also can stimulate the production of reactive oxygen species that can damage DNA.
Mineral-based, inorganic sunscreens, however – like those with titanium dioxide, zinc oxide, and iron oxide – scatter all wavelengths.
“These micronized forms of metal oxides not only scatter and reflect light, they also absorb ultraviolet radiation. The compounds aren’t new,” she said. In 1991, Dr. Elaine Kaye of Harvard University, Boston, and associates described (Arch Dermatol. 1991;127:351-5) opaque physical sunscreens that were useful blockers of visible light and found that transmittance of light can be lowered by adding iron oxide, Dr. Arellano-Mendoza pointed out.
The inorganic sunscreens have never been widely adopted because they are highly pigmented with white or, in the case of iron oxide, with red. “Not many people accepted [the iron-containing compounds] because of the redness, but now different shades are going to be hitting the market soon,” and the hope is that consumers will find them more appealing, she said.
This is good news, as the data emerging around iron oxide are intriguingly positive. A 2015 study showed that a sunscreen with iron oxides prevented melasma relapse during the summer months. Patients were randomized to the same ultraviolet filter topical sunscreen, but for one group, micronized iron oxide was added to it. After 6 months, the median melasma area severity index score was significantly better in the group using the iron oxide compound (J Am Acad Dermatol. 2015 Jan;72[1]:189-90.e1).
Support for the two-step regimen appeared in 2014, when a small study randomized 30 healthy volunteers to an SPF 30 sunscreen or the same sunscreen supplemented with an antioxidant cocktail of grape seed extract, vitamin E, ubiquinone, and vitamin C. The endpoint was MMP-1 upregulation after exposure to infrared-A light. Skin treated with the combination regimen showed significantly lower MMP-1 activation, leading the authors to conclude that the combination of topical antioxidants conferred protection against the irradiation (Photochem Photobiol. 2015 Jan-Feb;91[1]:248-50).
Those same authors published a companion article (Photodermatol Photoimmunol Photomed. 2014;30:167-74) suggesting that another antioxidant mixture (ferulic acid, tocopherol, and vitamin C) was similarly effective.
Using a combination of antioxidants is important, Dr. Arellano-Mendoza said, because different antioxidants work differently. Some (catalase, glutathione peroxidase, and superoxide dismutase) are enzymatic, catalyzing reactions that convert free radicals to oxygen and water. Others terminate free radicals by preventing the propagation of oxidative chain reactions (vitamins A and C, flavonoids, uric acid, bilirubin, albumin, and members of the thiol group). A third group consists of metal-binding proteins that sequester free iron or copper to prevent free radical production (ferritin, transferrin, lactoferrin, and ceruloplasmin).
“I think we can now consider antioxidants a part of the tools we use in treating some pigmentary disorders as melasma,” she said. “We need to choose the compounds carefully, and we definitely need more in vivo research, but the findings are very encouraging.”
Dr. Arellano-Mendoza had no relevant financial disclosures.
This article was updated 3/20/17.
msullivan@frontlinemedcom.com
On Twitter @Alz_Gal
ORLANDO – A two-step regimen of high-potency topical antioxidants followed by a mineral-based sunscreen may help repair light-induced skin damage and protect against new damage in patients with melasma.
Recent studies suggest that the antioxidants tamp down inflammatory cytokines and damage of oxidative stress, Maria Ivonne Arellano-Mendoza, MD, said during a special focus session on Latino skin held at the annual meeting of the American Academy of Dermatology.
“Many compounds are being studied for this purpose,” said Dr. Arellano-Mendoza, head of the dermatology department of the General Hospital of Mexico, Mexico City. “One combination is vitamin C, vitamin E, ubiquinone, and grape-seed extract. This effectively prevented infrared-A radiation–induced matrix metalloproteinase-1 messenger RNA expression in human skin. This combination can be found now in some sunscreens and daily care products.”
Another effective combination seems to be a mixture of ferulic acid, tocopherol, and vitamin C, she said.
The two-step process of regularly using a topical product with antioxidants before applying a sunscreen is all there is for now, she added, because so far it’s been impossible to combine the agents in a single product.
“The challenge will be how to create a product that stays on the surface of the skin to protect it from light, while liberating the antioxidants to penetrate the skin,” she commented.
The antioxidants’ benefits, however, will be obliterated by the continued effects of some light wavelengths that aggravate melasma unless they are used in sequence with a light-scattering sunscreen.
Sunscreens are critical components of a melasma treatment regimen, Dr. Arellano-Mendoza said. “Sunscreens are a cornerstone of treatment. We clearly tell our patients that sunscreens have to be used every day, forever, and if they are not used properly, they will have no improvement.”
Patients with hyperpigmentation disorders are susceptible to longer wavelengths that aren’t covered by chemically derived sunscreens. Longer wavelengths, including infrared light and visible light, have been shown to increase expression of matrix metalloproteinase (MMP) -1 and -9, decrease expression of type 1 procollagen, and can induce macrophage infiltration. These wavelengths also increase reactive oxygen species and proinflammatory cytokines in vitro, Dr. Arellano-Mendoza noted.
Visible light can cause erythema, transient and long-lasting hyperpigmentation, thermal damage, free radical production, and premature photoaging. It also can stimulate the production of reactive oxygen species that can damage DNA.
Mineral-based, inorganic sunscreens, however – like those with titanium dioxide, zinc oxide, and iron oxide – scatter all wavelengths.
“These micronized forms of metal oxides not only scatter and reflect light, they also absorb ultraviolet radiation. The compounds aren’t new,” she said. In 1991, Dr. Elaine Kaye of Harvard University, Boston, and associates described (Arch Dermatol. 1991;127:351-5) opaque physical sunscreens that were useful blockers of visible light and found that transmittance of light can be lowered by adding iron oxide, Dr. Arellano-Mendoza pointed out.
The inorganic sunscreens have never been widely adopted because they are highly pigmented with white or, in the case of iron oxide, with red. “Not many people accepted [the iron-containing compounds] because of the redness, but now different shades are going to be hitting the market soon,” and the hope is that consumers will find them more appealing, she said.
This is good news, as the data emerging around iron oxide are intriguingly positive. A 2015 study showed that a sunscreen with iron oxides prevented melasma relapse during the summer months. Patients were randomized to the same ultraviolet filter topical sunscreen, but for one group, micronized iron oxide was added to it. After 6 months, the median melasma area severity index score was significantly better in the group using the iron oxide compound (J Am Acad Dermatol. 2015 Jan;72[1]:189-90.e1).
Support for the two-step regimen appeared in 2014, when a small study randomized 30 healthy volunteers to an SPF 30 sunscreen or the same sunscreen supplemented with an antioxidant cocktail of grape seed extract, vitamin E, ubiquinone, and vitamin C. The endpoint was MMP-1 upregulation after exposure to infrared-A light. Skin treated with the combination regimen showed significantly lower MMP-1 activation, leading the authors to conclude that the combination of topical antioxidants conferred protection against the irradiation (Photochem Photobiol. 2015 Jan-Feb;91[1]:248-50).
Those same authors published a companion article (Photodermatol Photoimmunol Photomed. 2014;30:167-74) suggesting that another antioxidant mixture (ferulic acid, tocopherol, and vitamin C) was similarly effective.
Using a combination of antioxidants is important, Dr. Arellano-Mendoza said, because different antioxidants work differently. Some (catalase, glutathione peroxidase, and superoxide dismutase) are enzymatic, catalyzing reactions that convert free radicals to oxygen and water. Others terminate free radicals by preventing the propagation of oxidative chain reactions (vitamins A and C, flavonoids, uric acid, bilirubin, albumin, and members of the thiol group). A third group consists of metal-binding proteins that sequester free iron or copper to prevent free radical production (ferritin, transferrin, lactoferrin, and ceruloplasmin).
“I think we can now consider antioxidants a part of the tools we use in treating some pigmentary disorders as melasma,” she said. “We need to choose the compounds carefully, and we definitely need more in vivo research, but the findings are very encouraging.”
Dr. Arellano-Mendoza had no relevant financial disclosures.
This article was updated 3/20/17.
msullivan@frontlinemedcom.com
On Twitter @Alz_Gal
EXPERT ANALYSIS FROM AAD 2017
EXPERT ANALYSIS FROM AAD 2017
Bluish Gray Hyperpigmentation on the Face and Neck
The Diagnosis: Erythema Dyschromicum Perstans
Erythema dyschromicum perstans (EDP), also referred to as ashy dermatosis, was first described by Ramirez1 in 1957 who labeled the patients los cenicientos (the ashen ones). It preferentially affects women in the second decade of life; however, patients of all ages can be affected, with reported cases occurring in children as young as 2 years of age.2 Most patients have Fitzpatrick skin type IV, mainly Amerindian, Hispanic South Asian, and Southwest Asian; however, there are cases reported worldwide.3 A genetic predisposition is proposed, as major histocompatibility complex genes associated with HLA-DR4⁎0407 are frequent in Mexican patients with ashy dermatosis and in the Amerindian population.4
The etiology of EDP is unknown. Various contributing factors have been reported including alimentary, occupational, and climatic factors,5,6 yet none have been conclusively demonstrated. High expression of CD36 (thrombospondin receptor not found in normal skin) in spinous and granular layers, CD94 (cytotoxic cell marker) in the basal cell layer and in the inflammatory dermal infiltrate,7 and focal keratinocytic expression of intercellular adhesion molecule I (CD54) in the active lesions of EDP, as well as the absence of these findings in normal skin, suggests an immunologic role in the development of the disease.8
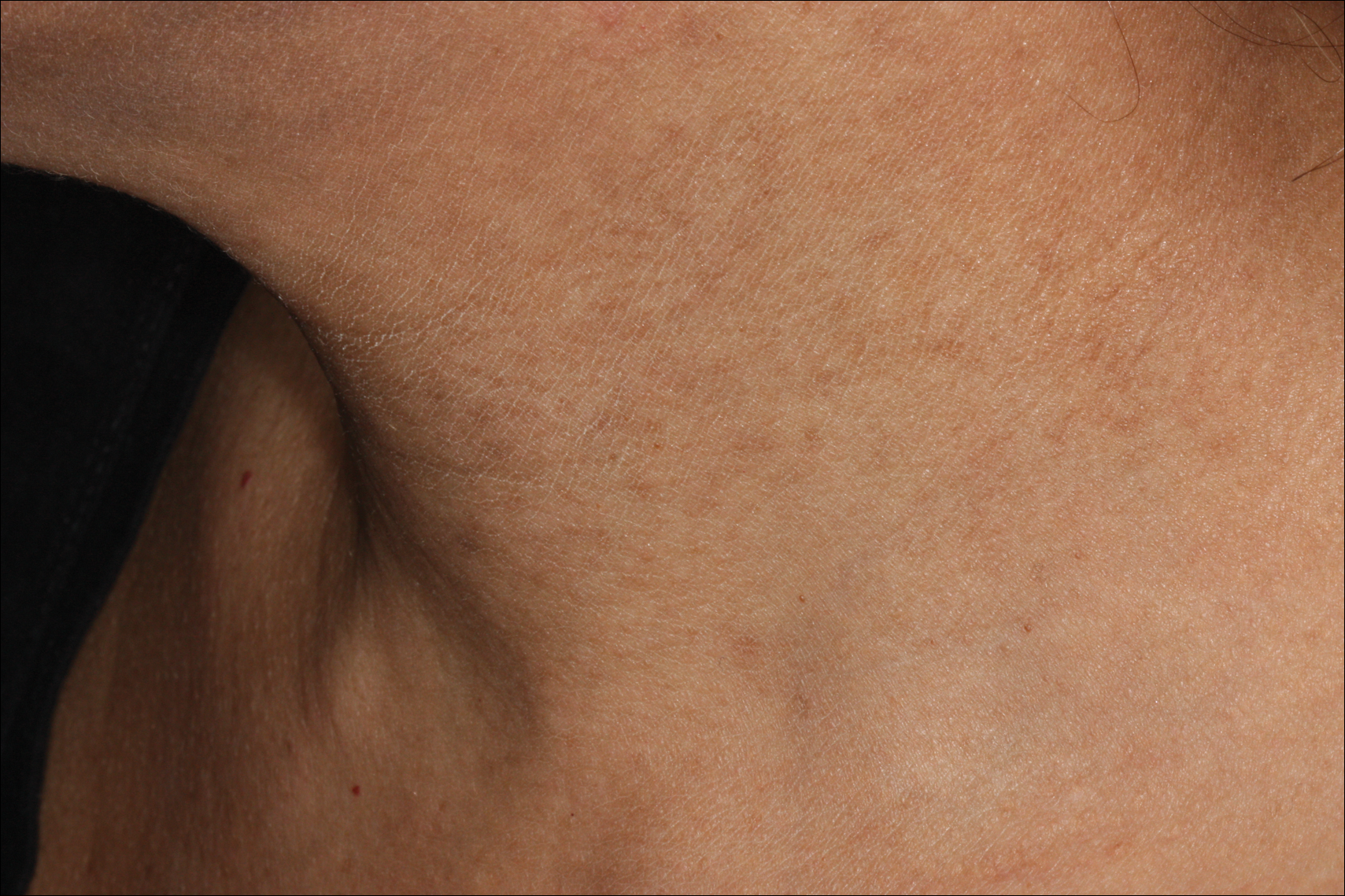
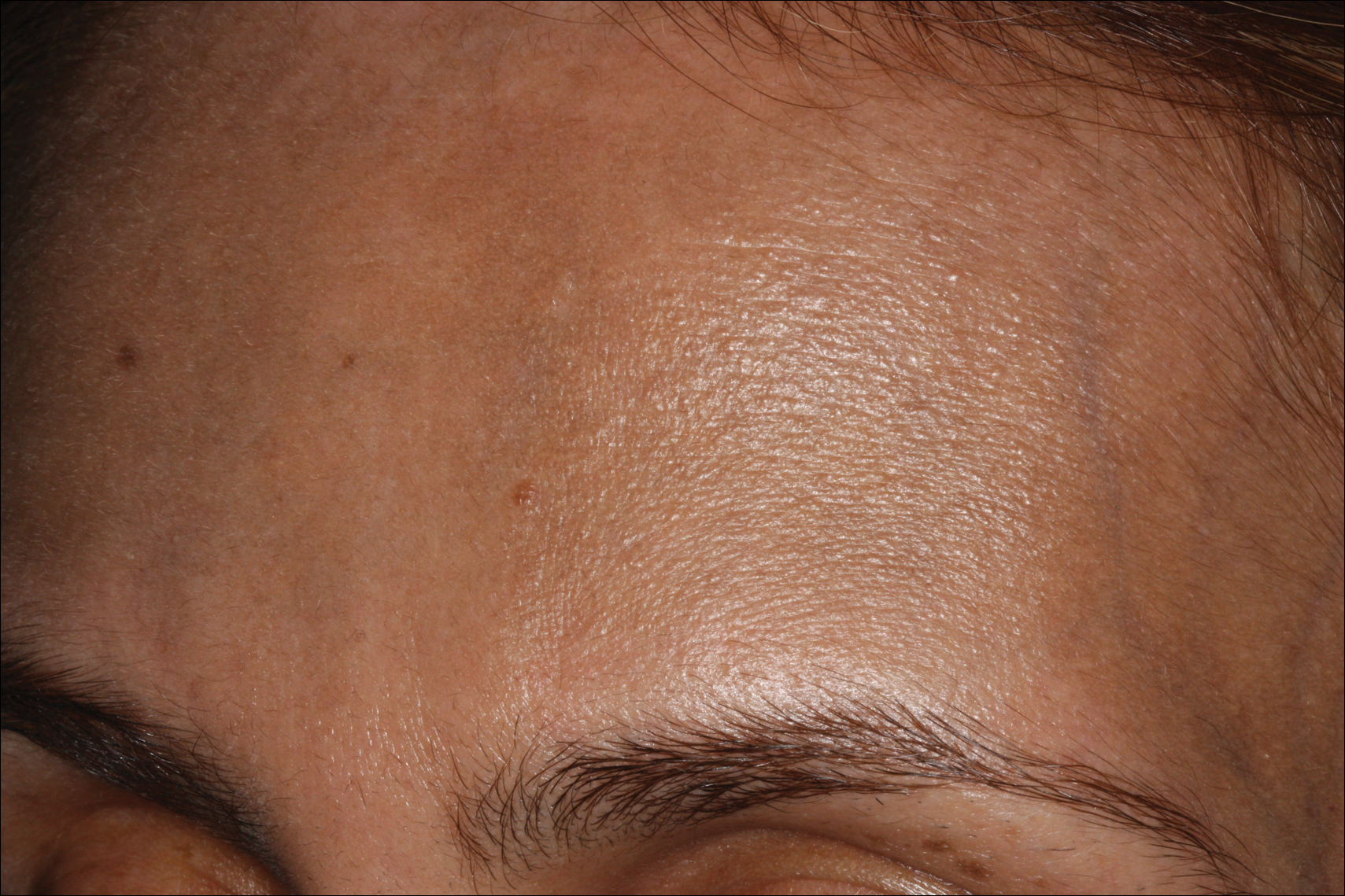
Erythema dyschromicum perstans presents clinically with blue-gray hyperpigmented macules varying in size and shape and developing symmetrically in both sun-exposed and sun-protected areas of the face, neck, trunk, arms, and sometimes the dorsal hands (Figures 1 and 2). Notable sparing of the palms, soles, scalp, and mucous membranes occurs.
Occasionally, in the early active stage of the disease, elevated erythematous borders are noted surrounding the hyperpigmented macules. Eventually a hypopigmented halo develops after a prolonged duration of disease.9 The eruption typically is chronic and asymptomatic, though some cases may be pruritic.10
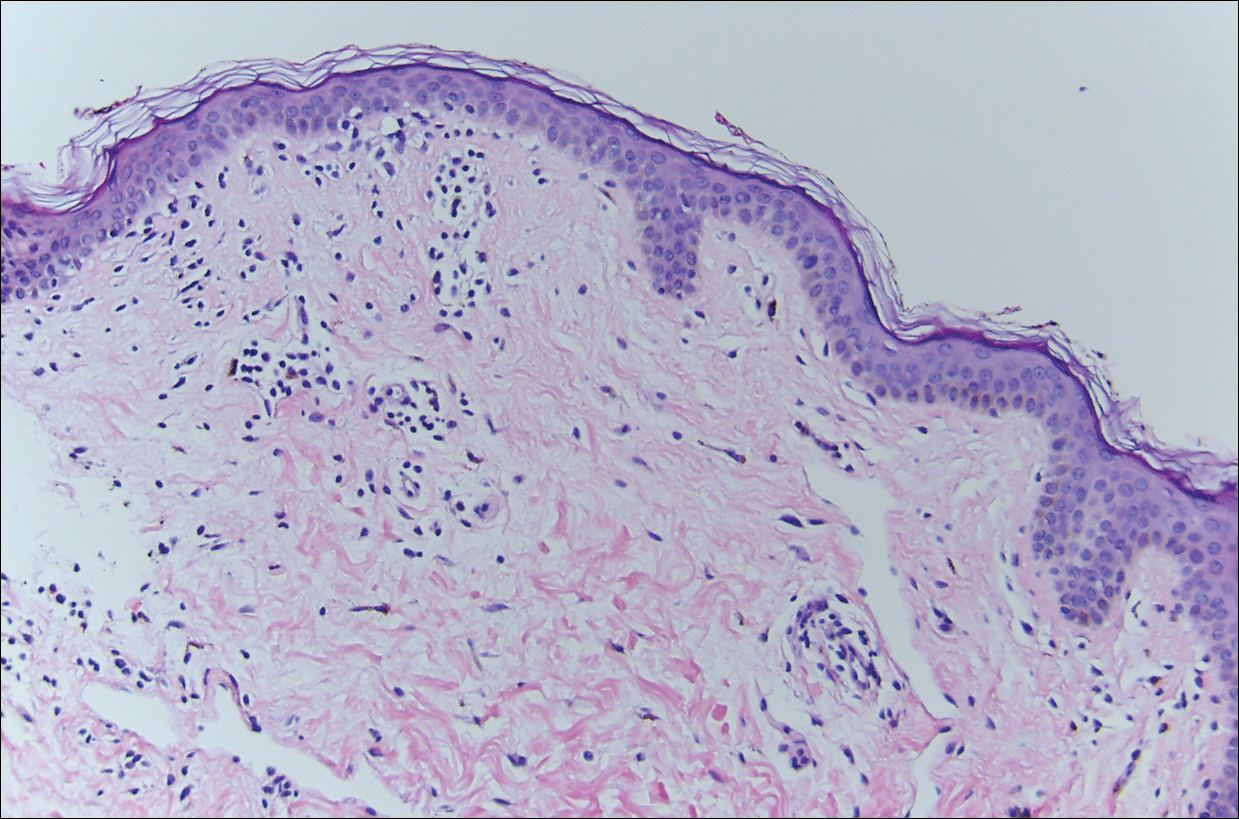
Histopathologically, the early lesions of EDP with an erythematous active border reveal lichenoid dermatitis with basal vacuolar change and occasional Civatte bodies. A mild to moderate perivascular lymphohistiocytic infiltrate admixed with melanophages can be seen in the papillary dermis (Figure 3). In older lesions, the inflammatory infiltrate is sparse, and pigment incontinence consistent with postinflammatory pigmentation is prominent, though melanophages extending deep into the reticular dermis may aid in distinguishing EDP from other causes of postinflammatory pigment alteration.7,11
Erythema dyschromicum perstans and lichen planus pigmentosus (LPP) may be indistinguishable histopathologically and may both be variants of lichen planus actinicus. Lichen planus pigmentosus often differs from EDP in that it presents with brown-black macules and patches often on the face and flexural areas. A subset of cases of LPP also may have mucous membrane involvement. The erythematous border that characterizes the active lesion of EDP is characteristically absent in LPP. In addition, pruritus often is reported with LPP. Direct immunofluorescence is not a beneficial tool in distinguishing the entities.12
Other differential diagnoses of predominantly facial hyperpigmentation include a lichenoid drug eruption; drug-induced hyperpigmentation (deposition disorder); postinflammatory hyperpigmentation following atopic dermatitis; contact dermatitis or photosensitivity reaction; early pinta; and cutaneous findings of systemic diseases manifesting with diffuse hyperpigmentation such as lupus erythematosus, dermatomyositis, hemochromatosis, and Addison disease. A detailed history including medication use, thorough clinical examination, and careful histopathologic evaluation will help distinguish these conditions.
Chrysiasis is a rare bluish to slate gray discoloration of the skin that predominantly occurs in sun-exposed areas. It is caused by chronic use of gold salts, which have been used to treat rheumatoid arthritis. UV light may contribute to induce the uptake of gold and subsequently stimulate tyrosinase activity.13 Histologic features of chrysiasis include dermal and perivascular gold deposition within the macrophages and endothelial cells as well as extracellular granules. It demonstrates an orange-red birefringence on fluorescent microscopy.14,15
Minocycline-induced hyperpigmentation is a well-recognized side effect of this drug. It is dose dependent and appears as a blue-black pigmentation that most frequently affects the shins, ankles, and arms.16 Three distinct types were documented: abnormal discoloration of the skin that has been linked to deposition of pigmented metabolites of minocycline producing blue-black pigmentation at the site of scarring or prior inflammation (type 1); blue-gray pigmentation affecting normal skin, mainly the legs (type 2); and elevated levels of melanin on the sun-exposed areas producing dirty skin syndrome (type 3).17,18
Topical and systemic corticosteroids, UV light therapy, oral dapsone, griseofulvin, retinoids, and clofazimine are reported as treatment options for ashy dermatosis, though results typically are disappointing.7
- Ramirez CO. Los cenicientos: problema clinica. In: Memoria del Primer Congresso Centroamericano de Dermatologica, December 5-8, 1957. San Salvador, El Salvador; 1957:122-130.
- Lee SJ, Chung KY. Erythema dyschromicum perstans in early childhood. J Dermatol. 1999;26:119-121.
- Homez-Chacin, Barroso C. On the etiopathogenic of the erythema dyschromicum perstans: possibility of a melanosis neurocutaneous. Dermatol Venez. 1996;4:149-151.
- Correa MC, Memije EV, Vargas-Alarcon G, et al. HLA-DR association with the genetic susceptibility to develop ashy dermatosis in Mexican Mestizo patients [published online November 20, 2006]. J Am Acad Dermatol. 2007;56:617-620.
- Jablonska S. Ingestion of ammonium nitrate as a possible cause of erythema dyschromicum perstans (ashy dermatosis). Dermatologica. 1975;150:287-291.
- Stevenson JR, Miura M. Erythema dyschromicum perstans (ashy dermatosis). Arch Dermatol. 1966;94:196-199.
- Baranda L, Torres-Alvarez B, Cortes-Franco R, et al. Involvement of cell adhesion and activation molecules in the pathogenesis of erythema dyschromicum perstans (ashy dermatitis). the effect of clofazimine therapy. Arch Dermatol. 1997;133:325-329.
- Vasquez-Ochoa LA, Isaza-Guzman DM, Orozco-Mora B, et al. Immunopathologic study of erythema dyschromicum perstans (ashy dermatosis). Int J Dermatol. 2006;45:937-941.
- Convit J, Kerdel-Vegas F, Roderiguez G. Erythema dyschromicum perstans: a hiltherto undescribed skin disease. J Invest Dermatol. 1961;36:457-462.
- Ono S, Miyachi Y, Kabashima K. Ashy dermatosis with prior pruritic and scaling skin lesions. J Dermatol. 2012;39:1103-1104.
- Sanchez NP, Pathak MA, Sato SS, et al. Circumscribed dermal melaninoses: classification, light, histochemical, and electron microscopic studies on three patients with the erythema dyschromicum perstans type. Int J Dermatol. 1982;21:25-32.
- Vega ME, Waxtein L, Arenas R, et al. Ashy dermatosis and lichen planus pigmentosus: a clinicopathologic study of 31 cases. Int J Dermatol. 1992;31:90-94.
- Ahmed SV, Sajjan R. Chrysiasis: a gold "curse!" [published online May 21, 2009]. BMJ Case Rep. 2009;2009.
- Fiscus V, Hankinson A, Alweis R. Minocycline-induced hyperpigmentation. J Community Hosp Intern Med Perspect. 2014;4. doi:10.3402/jchimp.v4.24063.
- Cox AJ, Marich KW. Gold in the dermis following gold therapy for rheumatoid arthritis. Arch Dermatol. 1973;108:655-657.
- al-Talib RK, Wright DH, Theaker JM. Orange-red birefringence of gold particles in paraffin wax embedded sections: an aid to the diagnosis of chrysiasis. Histopathology. 1994;24:176-178.
- Meyer AJ, Nahass GT. Hyperpigmented patches on the dorsa of the feet. minocycline pigmentation. Arch Dermatol. 1995;131:1447-1450.
- Bayne-Poorman M, Shubrook J. Bluish pigmentation of face and sclera. J Fam Pract. 2010;59:519-522.
The Diagnosis: Erythema Dyschromicum Perstans
Erythema dyschromicum perstans (EDP), also referred to as ashy dermatosis, was first described by Ramirez1 in 1957 who labeled the patients los cenicientos (the ashen ones). It preferentially affects women in the second decade of life; however, patients of all ages can be affected, with reported cases occurring in children as young as 2 years of age.2 Most patients have Fitzpatrick skin type IV, mainly Amerindian, Hispanic South Asian, and Southwest Asian; however, there are cases reported worldwide.3 A genetic predisposition is proposed, as major histocompatibility complex genes associated with HLA-DR4⁎0407 are frequent in Mexican patients with ashy dermatosis and in the Amerindian population.4
The etiology of EDP is unknown. Various contributing factors have been reported including alimentary, occupational, and climatic factors,5,6 yet none have been conclusively demonstrated. High expression of CD36 (thrombospondin receptor not found in normal skin) in spinous and granular layers, CD94 (cytotoxic cell marker) in the basal cell layer and in the inflammatory dermal infiltrate,7 and focal keratinocytic expression of intercellular adhesion molecule I (CD54) in the active lesions of EDP, as well as the absence of these findings in normal skin, suggests an immunologic role in the development of the disease.8
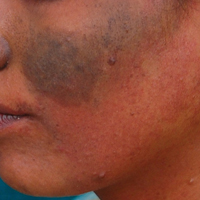
Erythema dyschromicum perstans presents clinically with blue-gray hyperpigmented macules varying in size and shape and developing symmetrically in both sun-exposed and sun-protected areas of the face, neck, trunk, arms, and sometimes the dorsal hands (Figures 1 and 2). Notable sparing of the palms, soles, scalp, and mucous membranes occurs.
Occasionally, in the early active stage of the disease, elevated erythematous borders are noted surrounding the hyperpigmented macules. Eventually a hypopigmented halo develops after a prolonged duration of disease.9 The eruption typically is chronic and asymptomatic, though some cases may be pruritic.10
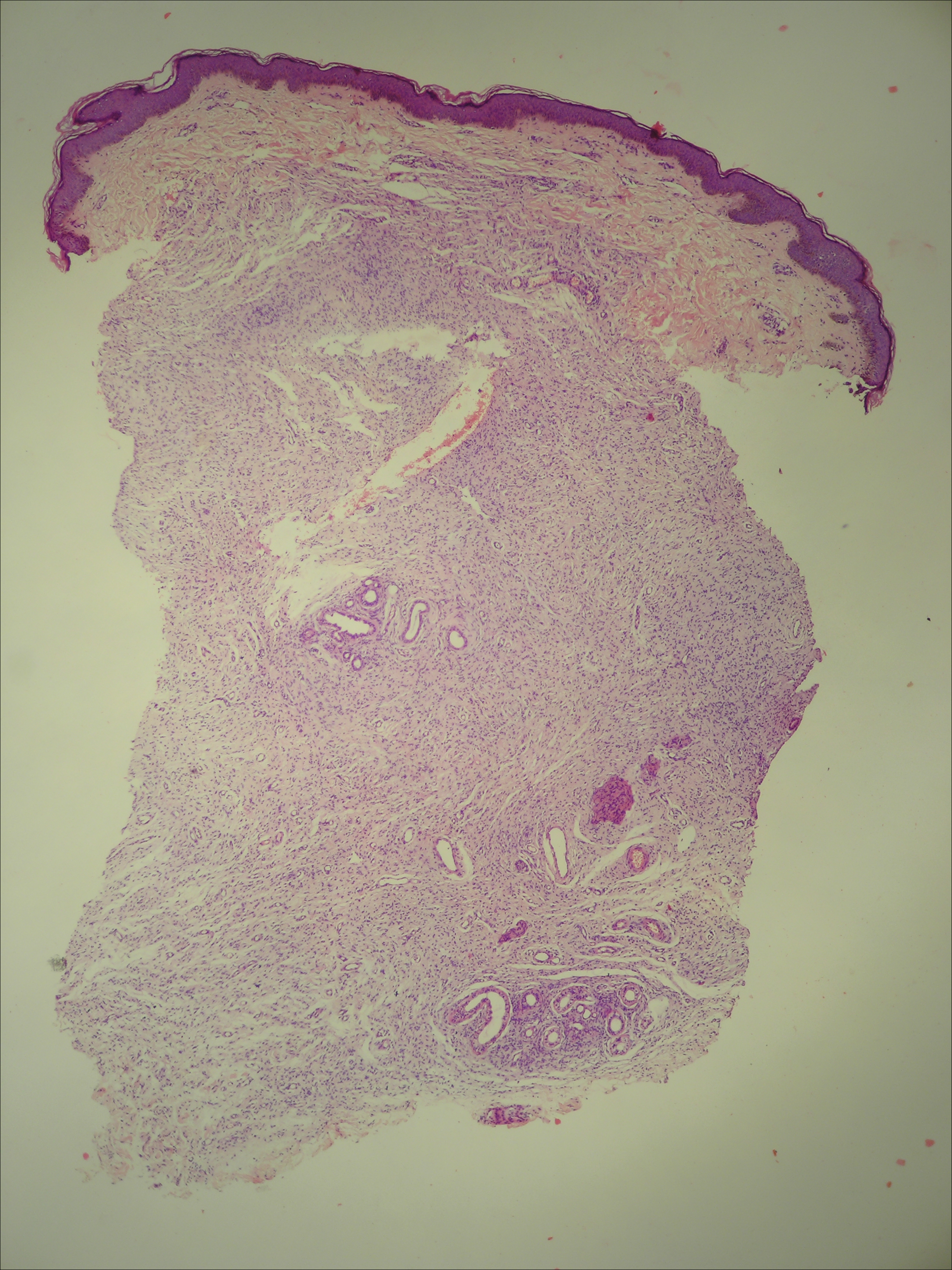
Histopathologically, the early lesions of EDP with an erythematous active border reveal lichenoid dermatitis with basal vacuolar change and occasional Civatte bodies. A mild to moderate perivascular lymphohistiocytic infiltrate admixed with melanophages can be seen in the papillary dermis (Figure 3). In older lesions, the inflammatory infiltrate is sparse, and pigment incontinence consistent with postinflammatory pigmentation is prominent, though melanophages extending deep into the reticular dermis may aid in distinguishing EDP from other causes of postinflammatory pigment alteration.7,11
Erythema dyschromicum perstans and lichen planus pigmentosus (LPP) may be indistinguishable histopathologically and may both be variants of lichen planus actinicus. Lichen planus pigmentosus often differs from EDP in that it presents with brown-black macules and patches often on the face and flexural areas. A subset of cases of LPP also may have mucous membrane involvement. The erythematous border that characterizes the active lesion of EDP is characteristically absent in LPP. In addition, pruritus often is reported with LPP. Direct immunofluorescence is not a beneficial tool in distinguishing the entities.12
Other differential diagnoses of predominantly facial hyperpigmentation include a lichenoid drug eruption; drug-induced hyperpigmentation (deposition disorder); postinflammatory hyperpigmentation following atopic dermatitis; contact dermatitis or photosensitivity reaction; early pinta; and cutaneous findings of systemic diseases manifesting with diffuse hyperpigmentation such as lupus erythematosus, dermatomyositis, hemochromatosis, and Addison disease. A detailed history including medication use, thorough clinical examination, and careful histopathologic evaluation will help distinguish these conditions.
Chrysiasis is a rare bluish to slate gray discoloration of the skin that predominantly occurs in sun-exposed areas. It is caused by chronic use of gold salts, which have been used to treat rheumatoid arthritis. UV light may contribute to induce the uptake of gold and subsequently stimulate tyrosinase activity.13 Histologic features of chrysiasis include dermal and perivascular gold deposition within the macrophages and endothelial cells as well as extracellular granules. It demonstrates an orange-red birefringence on fluorescent microscopy.14,15
Minocycline-induced hyperpigmentation is a well-recognized side effect of this drug. It is dose dependent and appears as a blue-black pigmentation that most frequently affects the shins, ankles, and arms.16 Three distinct types were documented: abnormal discoloration of the skin that has been linked to deposition of pigmented metabolites of minocycline producing blue-black pigmentation at the site of scarring or prior inflammation (type 1); blue-gray pigmentation affecting normal skin, mainly the legs (type 2); and elevated levels of melanin on the sun-exposed areas producing dirty skin syndrome (type 3).17,18
Topical and systemic corticosteroids, UV light therapy, oral dapsone, griseofulvin, retinoids, and clofazimine are reported as treatment options for ashy dermatosis, though results typically are disappointing.7
The Diagnosis: Erythema Dyschromicum Perstans
Erythema dyschromicum perstans (EDP), also referred to as ashy dermatosis, was first described by Ramirez1 in 1957 who labeled the patients los cenicientos (the ashen ones). It preferentially affects women in the second decade of life; however, patients of all ages can be affected, with reported cases occurring in children as young as 2 years of age.2 Most patients have Fitzpatrick skin type IV, mainly Amerindian, Hispanic South Asian, and Southwest Asian; however, there are cases reported worldwide.3 A genetic predisposition is proposed, as major histocompatibility complex genes associated with HLA-DR4⁎0407 are frequent in Mexican patients with ashy dermatosis and in the Amerindian population.4
The etiology of EDP is unknown. Various contributing factors have been reported including alimentary, occupational, and climatic factors,5,6 yet none have been conclusively demonstrated. High expression of CD36 (thrombospondin receptor not found in normal skin) in spinous and granular layers, CD94 (cytotoxic cell marker) in the basal cell layer and in the inflammatory dermal infiltrate,7 and focal keratinocytic expression of intercellular adhesion molecule I (CD54) in the active lesions of EDP, as well as the absence of these findings in normal skin, suggests an immunologic role in the development of the disease.8
Erythema dyschromicum perstans presents clinically with blue-gray hyperpigmented macules varying in size and shape and developing symmetrically in both sun-exposed and sun-protected areas of the face, neck, trunk, arms, and sometimes the dorsal hands (Figures 1 and 2). Notable sparing of the palms, soles, scalp, and mucous membranes occurs.
Occasionally, in the early active stage of the disease, elevated erythematous borders are noted surrounding the hyperpigmented macules. Eventually a hypopigmented halo develops after a prolonged duration of disease.9 The eruption typically is chronic and asymptomatic, though some cases may be pruritic.10
Histopathologically, the early lesions of EDP with an erythematous active border reveal lichenoid dermatitis with basal vacuolar change and occasional Civatte bodies. A mild to moderate perivascular lymphohistiocytic infiltrate admixed with melanophages can be seen in the papillary dermis (Figure 3). In older lesions, the inflammatory infiltrate is sparse, and pigment incontinence consistent with postinflammatory pigmentation is prominent, though melanophages extending deep into the reticular dermis may aid in distinguishing EDP from other causes of postinflammatory pigment alteration.7,11
Erythema dyschromicum perstans and lichen planus pigmentosus (LPP) may be indistinguishable histopathologically and may both be variants of lichen planus actinicus. Lichen planus pigmentosus often differs from EDP in that it presents with brown-black macules and patches often on the face and flexural areas. A subset of cases of LPP also may have mucous membrane involvement. The erythematous border that characterizes the active lesion of EDP is characteristically absent in LPP. In addition, pruritus often is reported with LPP. Direct immunofluorescence is not a beneficial tool in distinguishing the entities.12
Other differential diagnoses of predominantly facial hyperpigmentation include a lichenoid drug eruption; drug-induced hyperpigmentation (deposition disorder); postinflammatory hyperpigmentation following atopic dermatitis; contact dermatitis or photosensitivity reaction; early pinta; and cutaneous findings of systemic diseases manifesting with diffuse hyperpigmentation such as lupus erythematosus, dermatomyositis, hemochromatosis, and Addison disease. A detailed history including medication use, thorough clinical examination, and careful histopathologic evaluation will help distinguish these conditions.
Chrysiasis is a rare bluish to slate gray discoloration of the skin that predominantly occurs in sun-exposed areas. It is caused by chronic use of gold salts, which have been used to treat rheumatoid arthritis. UV light may contribute to induce the uptake of gold and subsequently stimulate tyrosinase activity.13 Histologic features of chrysiasis include dermal and perivascular gold deposition within the macrophages and endothelial cells as well as extracellular granules. It demonstrates an orange-red birefringence on fluorescent microscopy.14,15
Minocycline-induced hyperpigmentation is a well-recognized side effect of this drug. It is dose dependent and appears as a blue-black pigmentation that most frequently affects the shins, ankles, and arms.16 Three distinct types were documented: abnormal discoloration of the skin that has been linked to deposition of pigmented metabolites of minocycline producing blue-black pigmentation at the site of scarring or prior inflammation (type 1); blue-gray pigmentation affecting normal skin, mainly the legs (type 2); and elevated levels of melanin on the sun-exposed areas producing dirty skin syndrome (type 3).17,18
Topical and systemic corticosteroids, UV light therapy, oral dapsone, griseofulvin, retinoids, and clofazimine are reported as treatment options for ashy dermatosis, though results typically are disappointing.7
- Ramirez CO. Los cenicientos: problema clinica. In: Memoria del Primer Congresso Centroamericano de Dermatologica, December 5-8, 1957. San Salvador, El Salvador; 1957:122-130.
- Lee SJ, Chung KY. Erythema dyschromicum perstans in early childhood. J Dermatol. 1999;26:119-121.
- Homez-Chacin, Barroso C. On the etiopathogenic of the erythema dyschromicum perstans: possibility of a melanosis neurocutaneous. Dermatol Venez. 1996;4:149-151.
- Correa MC, Memije EV, Vargas-Alarcon G, et al. HLA-DR association with the genetic susceptibility to develop ashy dermatosis in Mexican Mestizo patients [published online November 20, 2006]. J Am Acad Dermatol. 2007;56:617-620.
- Jablonska S. Ingestion of ammonium nitrate as a possible cause of erythema dyschromicum perstans (ashy dermatosis). Dermatologica. 1975;150:287-291.
- Stevenson JR, Miura M. Erythema dyschromicum perstans (ashy dermatosis). Arch Dermatol. 1966;94:196-199.
- Baranda L, Torres-Alvarez B, Cortes-Franco R, et al. Involvement of cell adhesion and activation molecules in the pathogenesis of erythema dyschromicum perstans (ashy dermatitis). the effect of clofazimine therapy. Arch Dermatol. 1997;133:325-329.
- Vasquez-Ochoa LA, Isaza-Guzman DM, Orozco-Mora B, et al. Immunopathologic study of erythema dyschromicum perstans (ashy dermatosis). Int J Dermatol. 2006;45:937-941.
- Convit J, Kerdel-Vegas F, Roderiguez G. Erythema dyschromicum perstans: a hiltherto undescribed skin disease. J Invest Dermatol. 1961;36:457-462.
- Ono S, Miyachi Y, Kabashima K. Ashy dermatosis with prior pruritic and scaling skin lesions. J Dermatol. 2012;39:1103-1104.
- Sanchez NP, Pathak MA, Sato SS, et al. Circumscribed dermal melaninoses: classification, light, histochemical, and electron microscopic studies on three patients with the erythema dyschromicum perstans type. Int J Dermatol. 1982;21:25-32.
- Vega ME, Waxtein L, Arenas R, et al. Ashy dermatosis and lichen planus pigmentosus: a clinicopathologic study of 31 cases. Int J Dermatol. 1992;31:90-94.
- Ahmed SV, Sajjan R. Chrysiasis: a gold "curse!" [published online May 21, 2009]. BMJ Case Rep. 2009;2009.
- Fiscus V, Hankinson A, Alweis R. Minocycline-induced hyperpigmentation. J Community Hosp Intern Med Perspect. 2014;4. doi:10.3402/jchimp.v4.24063.
- Cox AJ, Marich KW. Gold in the dermis following gold therapy for rheumatoid arthritis. Arch Dermatol. 1973;108:655-657.
- al-Talib RK, Wright DH, Theaker JM. Orange-red birefringence of gold particles in paraffin wax embedded sections: an aid to the diagnosis of chrysiasis. Histopathology. 1994;24:176-178.
- Meyer AJ, Nahass GT. Hyperpigmented patches on the dorsa of the feet. minocycline pigmentation. Arch Dermatol. 1995;131:1447-1450.
- Bayne-Poorman M, Shubrook J. Bluish pigmentation of face and sclera. J Fam Pract. 2010;59:519-522.
- Ramirez CO. Los cenicientos: problema clinica. In: Memoria del Primer Congresso Centroamericano de Dermatologica, December 5-8, 1957. San Salvador, El Salvador; 1957:122-130.
- Lee SJ, Chung KY. Erythema dyschromicum perstans in early childhood. J Dermatol. 1999;26:119-121.
- Homez-Chacin, Barroso C. On the etiopathogenic of the erythema dyschromicum perstans: possibility of a melanosis neurocutaneous. Dermatol Venez. 1996;4:149-151.
- Correa MC, Memije EV, Vargas-Alarcon G, et al. HLA-DR association with the genetic susceptibility to develop ashy dermatosis in Mexican Mestizo patients [published online November 20, 2006]. J Am Acad Dermatol. 2007;56:617-620.
- Jablonska S. Ingestion of ammonium nitrate as a possible cause of erythema dyschromicum perstans (ashy dermatosis). Dermatologica. 1975;150:287-291.
- Stevenson JR, Miura M. Erythema dyschromicum perstans (ashy dermatosis). Arch Dermatol. 1966;94:196-199.
- Baranda L, Torres-Alvarez B, Cortes-Franco R, et al. Involvement of cell adhesion and activation molecules in the pathogenesis of erythema dyschromicum perstans (ashy dermatitis). the effect of clofazimine therapy. Arch Dermatol. 1997;133:325-329.
- Vasquez-Ochoa LA, Isaza-Guzman DM, Orozco-Mora B, et al. Immunopathologic study of erythema dyschromicum perstans (ashy dermatosis). Int J Dermatol. 2006;45:937-941.
- Convit J, Kerdel-Vegas F, Roderiguez G. Erythema dyschromicum perstans: a hiltherto undescribed skin disease. J Invest Dermatol. 1961;36:457-462.
- Ono S, Miyachi Y, Kabashima K. Ashy dermatosis with prior pruritic and scaling skin lesions. J Dermatol. 2012;39:1103-1104.
- Sanchez NP, Pathak MA, Sato SS, et al. Circumscribed dermal melaninoses: classification, light, histochemical, and electron microscopic studies on three patients with the erythema dyschromicum perstans type. Int J Dermatol. 1982;21:25-32.
- Vega ME, Waxtein L, Arenas R, et al. Ashy dermatosis and lichen planus pigmentosus: a clinicopathologic study of 31 cases. Int J Dermatol. 1992;31:90-94.
- Ahmed SV, Sajjan R. Chrysiasis: a gold "curse!" [published online May 21, 2009]. BMJ Case Rep. 2009;2009.
- Fiscus V, Hankinson A, Alweis R. Minocycline-induced hyperpigmentation. J Community Hosp Intern Med Perspect. 2014;4. doi:10.3402/jchimp.v4.24063.
- Cox AJ, Marich KW. Gold in the dermis following gold therapy for rheumatoid arthritis. Arch Dermatol. 1973;108:655-657.
- al-Talib RK, Wright DH, Theaker JM. Orange-red birefringence of gold particles in paraffin wax embedded sections: an aid to the diagnosis of chrysiasis. Histopathology. 1994;24:176-178.
- Meyer AJ, Nahass GT. Hyperpigmented patches on the dorsa of the feet. minocycline pigmentation. Arch Dermatol. 1995;131:1447-1450.
- Bayne-Poorman M, Shubrook J. Bluish pigmentation of face and sclera. J Fam Pract. 2010;59:519-522.
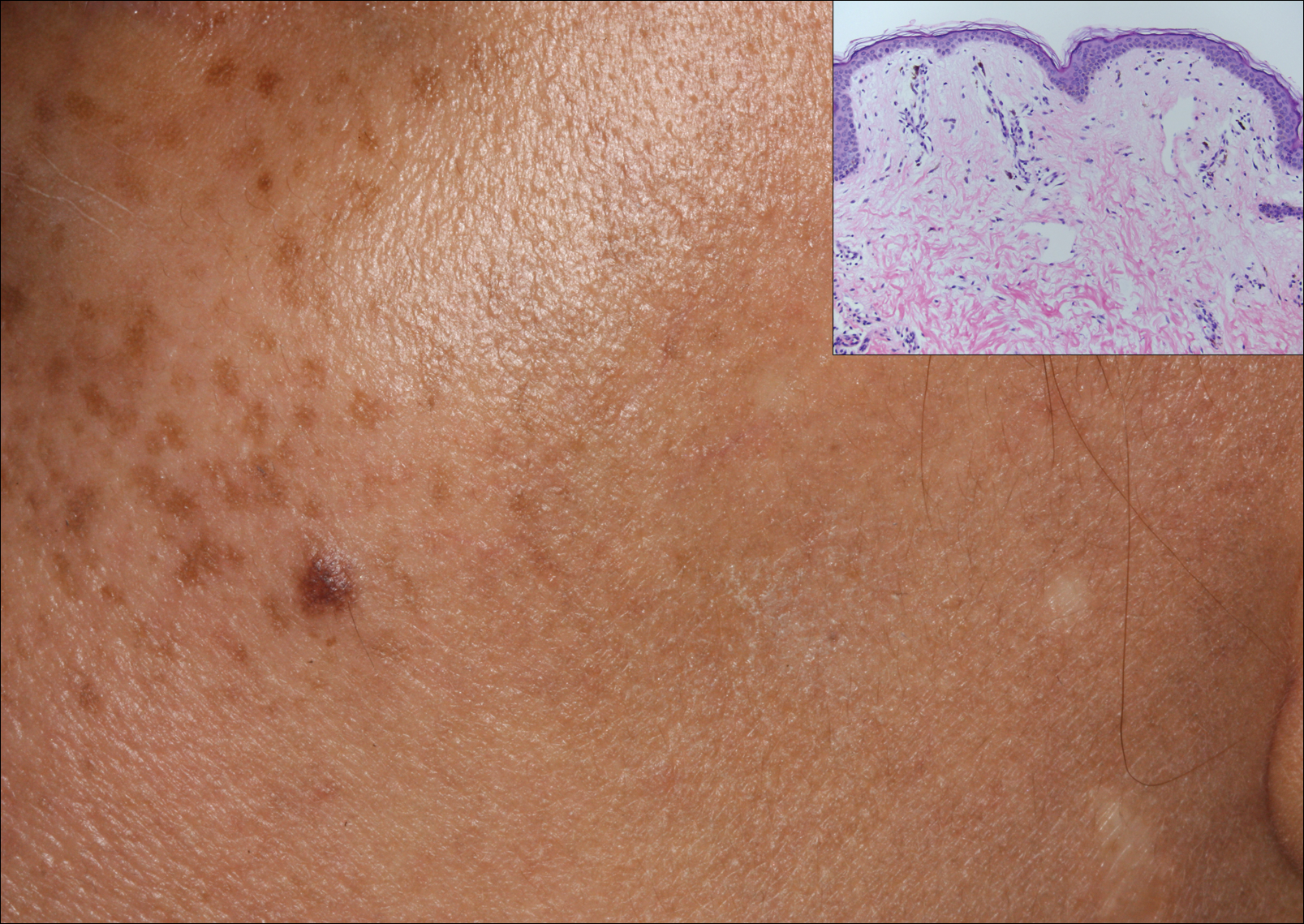
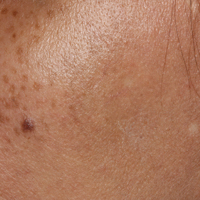
A middle-aged woman with Fitzpatrick skin type IV was evaluated for progressive hyperpigmentation of several months' duration involving the neck, jawline, both sides of the face, and forehead. The lesions were mildly pruritic. She denied contact with any new substance and there was no history of an eruption preceding the hyperpigmentation. Medical history included chronic anemia that was managed with iron supplementation. On physical examination, blue-gray nonscaly macules and patches were observed distributed symmetrically on the neck, jawline, sides of the face, and forehead. Microscopic examination of 2 shave biopsies revealed subtle vacuolar interface dermatitis with mild perivascular lymphocytic infiltrate and dermal melanophages (inset).
Phacomatosis Cesioflammea in Association With von Recklinghausen Disease (Neurofibromatosis Type I)
To the Editor:
Vascular lesions associated with melanocytic nevi were first described by Ota et al1 in 1947 and given the name phacomatosis pigmentovascularis. In 2005, Happle2 reclassified phacomatosis pigmentovascularis into 3 well-defined types: (1) phacomatosis cesioflammea: blue spots (caesius means bluish gray in Latin) and nevus flammeus; (2) phacomatosis spilorosea: nevus spilus coexisting with a pale pink telangiectatic nevus; and (3) phacomatosis cesiomarmorata: blue spots and cutis marmorata telangiectatica congenita. In 2011 Joshi et al3 described a case of a 31-year-old woman who had a port-wine stain in association with neurofibromatosis type I (NF-1). We present a case of phacomatosis cesioflammea in association with NF-1.
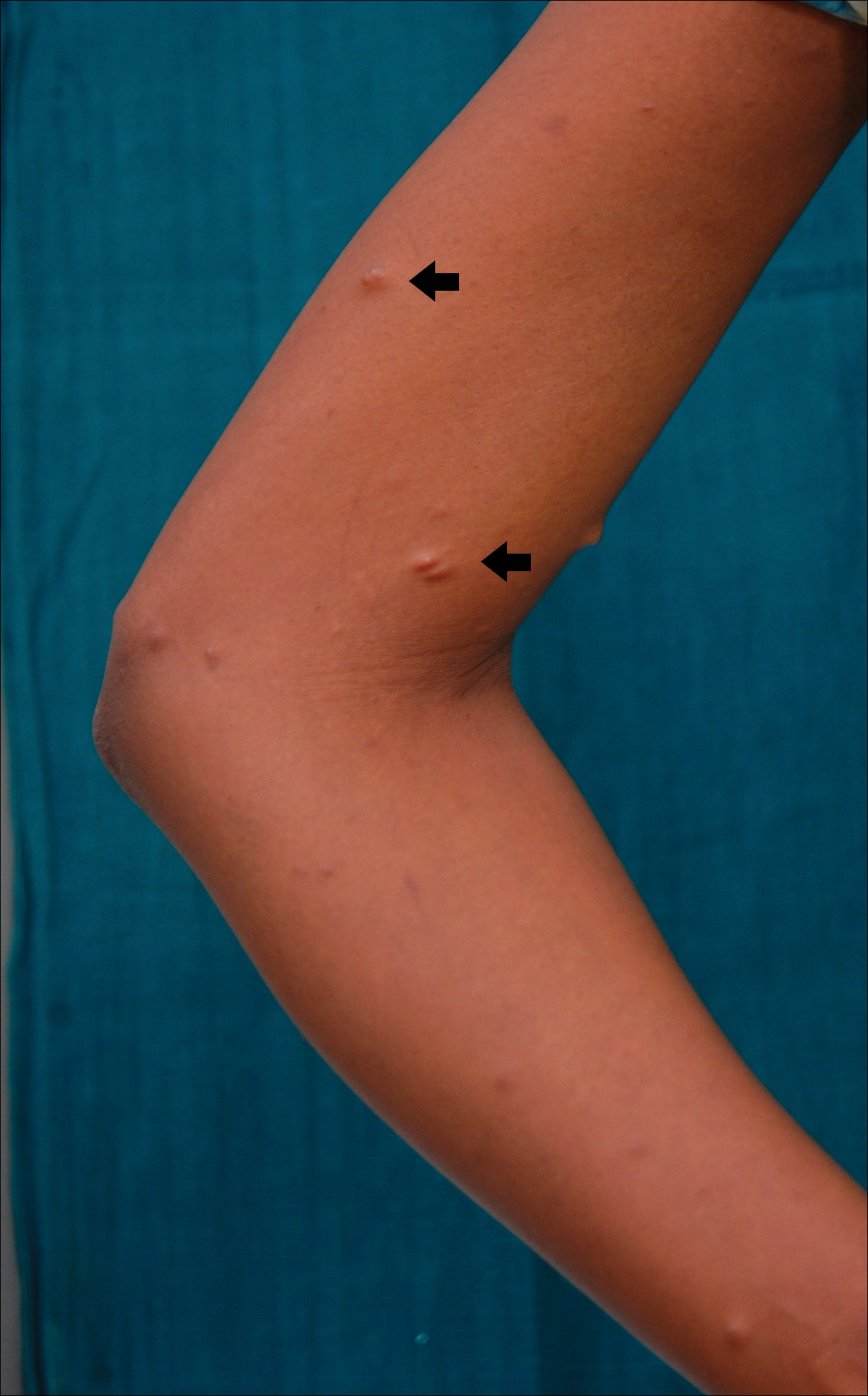
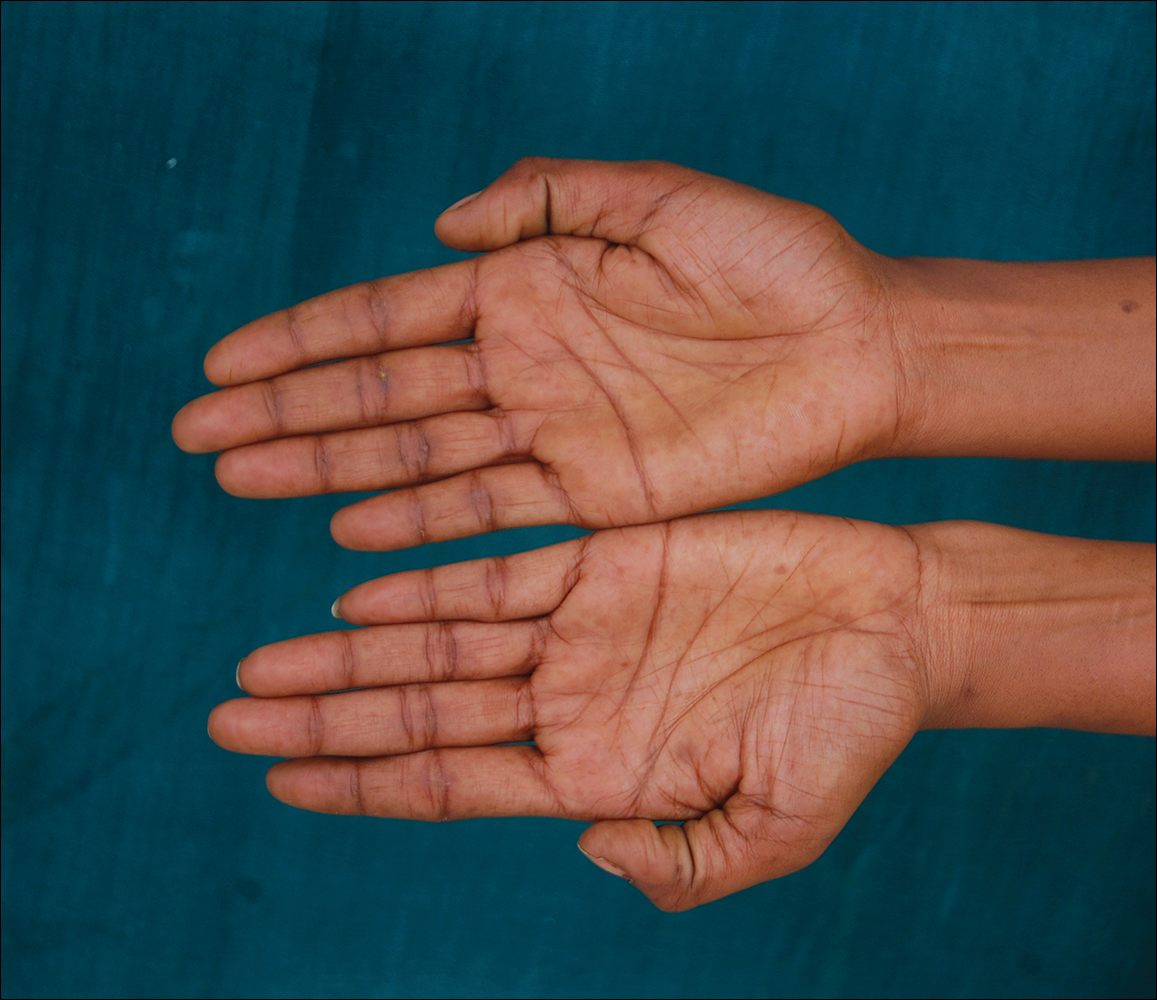
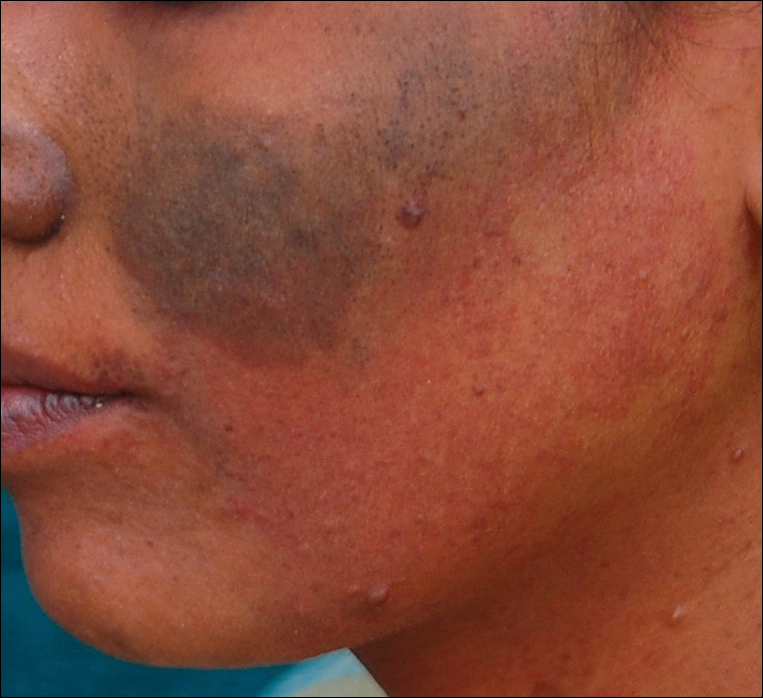
A 20-year-old woman presented to our outpatient section with a bluish black birthmark on the left side of the face since birth with the onset of multiple painless flesh-colored nodules on the trunk and arms of 1 year’s duration. She reported having occasional pruritus over the nodular lesions. Cutaneous examination showed multiple well-defined café au lait macules (0.5–3.0 cm) with regular margins. Multiple flesh-colored nodules were evident on the upper arms (Figure 1) and trunk. The nodules were firm in consistency and showed buttonholing phenomenon with some of the lesions demonstrating bag-of-worms consistency on palpation. Both palms showed multiple brownish frecklelike macules (Figure 2). A single bluish patch extended from the left ala of the nose to the sideburns. Adjoining the bluish patch was a subtle, ill-defined, nonblanchable red patch extending from the lower margin of the bluish patch to the mandibular ridge (Figure 3). Ocular examination showed melanosis bulbi of the left sclera and a few iris hamartomas (Lisch nodules) in both eyes. A biopsy of the skin nodule was obtained under local anesthesia after obtaining the patient’s informed consent; the specimen was fixed in 10% buffered formalin. A hematoxylin and eosin–stained section showed a well-circumscribed nonencapsulated tumor in the dermis composed of loosely spaced spindle cells and wavy collagenous strands (Figure 4). Routine hemogram and blood biochemistry including urinalysis were within reference range. Radiologic examination of the long bones was unremarkable. Our patient had 3 of 6 criteria defined by the National Institutes of Health for diagnosis of NF-1.4 On clinicopathological correlation we made a diagnosis of phacomatosis cesioflammea in association with NF-1. We have reassured the patient about the benign nature of vascular nevus. She was informed that the skin nodules could increase in size during pregnancy and to regularly follow-up with an eye specialist if any visual abnormalities occur.
The term phacomatosis is applied to genetically determined disorders of tissue derived from ectodermal origin (eg, skin, central nervous system, eyes) and commonly includes NF-1, tuberous sclerosis, and von Hippel-Lindau syndrome. Neurofibromatosis type I was first described by German pathologist Friedrich Daniel von Recklinghausen.5 Phacomatosis pigmentovascularis has been defined as the association of vascular nevus with a pigmentary nevus. Its pathogenesis can be explained by the twin spotting phenomenon.6 Twin spots are paired patches of mutant tissue that differ from each other and from the surrounding normal background skin. They can occur as 2 clinical types: allelic and nonallelic twin spotting. Our patient had nonallelic twin spots for 2 nevoid conditions: vascular (nevus flammeus) and pigmentary (nevus of Ota). Nevus of Ota was distributed in the V2 segment (maxillary nerve) of the fifth cranial nerve along with classical melanosis bulbi, which is considered a characteristic clinical feature of nevus of Ota (nevus cesius).7 Nevus flammeus (port-wine stain) is a vascular malformation presenting with flat lesions that persists throughout a patient’s life. The phenomenon of twin spotting, or didymosis (didymos means twin in Greek), has been proposed for co-occurrence of vascular and pigmented nevi.8 The association of NF-1 along with phacomatosis cesioflammea (a twin spot) could be explained from mosaicism of tissues derived from neuroectodermal and mesenchymal elements. Neurofibromatosis type I can occur as a mosaic disorder due to either postzygotic germ line or somatic mutations in the NF1 gene located on the proximal long arm of chromosome 17.9 Irrespective of the mutational event, a mosaic patient has a mixture of cells, some have normal copies of a particular gene and others have an abnormal copy of the same gene. Somatic mutation can lead to segmental (localized), generalized, or gonadal mosaicism. Somatic mutations occurring early during embryonic development produce generalized mosaicism, and generalized mosaics clinically appear similar to nonmosaic NF-1 cases.10,11 However, due to a lack of adequate facilities for mutation analysis and financial constraints, we were unable to confirm our case as generalized somatic mosaic for NF1 gene.
Several morphologic abnormalities have been reported with phacomatosis cesioflammea. Wu et al12 reported a single case of phacomatosis cesioflammea associated with pectus excavatum in a 9-month-old infant. Shields et al13 suggested that a thorough ocular examination on a periodic basis is essential to rule out melanoma of ocular tissues in patients with nevus flammeus and ocular melanosis.
Phacomatosis cesioflammea can occur in association with NF-1. The exact incidence of association is not known. The nevoid condition can be treated with appropriate lasers.
- Ota M, Kawamura T, Ito N. Phacomatosis pigmentovascularis (Ota). Jpn J Dermatol. 1947;52:1-3.
- Happle R. Phacomatosis pigmentovascularis revisited and reclassified. Arch Dermatol. 2005;141:385-388.
- Joshi A, Manchanda Y, Rijhwani M. Port-wine-stain with rare associations in two cases from Kuwait: phakomatosis pigmentovascularis redefined. Gulf J Dermatol Venereol. 2011;18:59-64.
- Neurofibromatosis. Conference Statement. National Institutes of Health Consensus. Arch Neurol. 1988;45:575-578.
- Gerber PA, Antal AS, Neumann NJ, et al. Neurofibromatosis. Eur J Med Res. 2009;14:102-105.
- Goyal T, Varshney A. Phacomatosis cesioflammea: first case report from India. Indian J Dermatol Venereol Leprol. 2010;76:307.
- Happle R. Didymosis cesioanemica: an unusual counterpart of phakomatosis cesioflammea. Eur J Dermatol. 2011;21:471.
- Happle R, Steijlen PM. Phacomatosis pigmentovascularis interpreted as a phenomenon of twin spots [in German]. Hautarzt. 1989;40:721-724.
- Adigun CG, Stein J. Segmental neurofibromatosis. Dermatol Online J. 2011;17:25.
- Ruggieri M, Huson SM. The clinical and diagnostic implications of mosaicism in the neurofibromatoses. Neurology. 2001;56:1433-1443.
- Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009;61:1-14.
- Wu CY, Chen PH, Chen GS. Phacomatosis cesioflammea associated with pectus excavatum. Acta Derm Venereol. 2009;89:309-310.
- Shields CL, Kligman BE, Suriano M, et al. Phacomatosis pigmentovascularis of cesioflammea type in 7 patients: combination of ocular pigmentation (melanocytosis or melanosis) and nevus flammeus with risk for melanoma. Arch Ophthalmol. 2011;129:746-750.
To the Editor:
Vascular lesions associated with melanocytic nevi were first described by Ota et al1 in 1947 and given the name phacomatosis pigmentovascularis. In 2005, Happle2 reclassified phacomatosis pigmentovascularis into 3 well-defined types: (1) phacomatosis cesioflammea: blue spots (caesius means bluish gray in Latin) and nevus flammeus; (2) phacomatosis spilorosea: nevus spilus coexisting with a pale pink telangiectatic nevus; and (3) phacomatosis cesiomarmorata: blue spots and cutis marmorata telangiectatica congenita. In 2011 Joshi et al3 described a case of a 31-year-old woman who had a port-wine stain in association with neurofibromatosis type I (NF-1). We present a case of phacomatosis cesioflammea in association with NF-1.
A 20-year-old woman presented to our outpatient section with a bluish black birthmark on the left side of the face since birth with the onset of multiple painless flesh-colored nodules on the trunk and arms of 1 year’s duration. She reported having occasional pruritus over the nodular lesions. Cutaneous examination showed multiple well-defined café au lait macules (0.5–3.0 cm) with regular margins. Multiple flesh-colored nodules were evident on the upper arms (Figure 1) and trunk. The nodules were firm in consistency and showed buttonholing phenomenon with some of the lesions demonstrating bag-of-worms consistency on palpation. Both palms showed multiple brownish frecklelike macules (Figure 2). A single bluish patch extended from the left ala of the nose to the sideburns. Adjoining the bluish patch was a subtle, ill-defined, nonblanchable red patch extending from the lower margin of the bluish patch to the mandibular ridge (Figure 3). Ocular examination showed melanosis bulbi of the left sclera and a few iris hamartomas (Lisch nodules) in both eyes. A biopsy of the skin nodule was obtained under local anesthesia after obtaining the patient’s informed consent; the specimen was fixed in 10% buffered formalin. A hematoxylin and eosin–stained section showed a well-circumscribed nonencapsulated tumor in the dermis composed of loosely spaced spindle cells and wavy collagenous strands (Figure 4). Routine hemogram and blood biochemistry including urinalysis were within reference range. Radiologic examination of the long bones was unremarkable. Our patient had 3 of 6 criteria defined by the National Institutes of Health for diagnosis of NF-1.4 On clinicopathological correlation we made a diagnosis of phacomatosis cesioflammea in association with NF-1. We have reassured the patient about the benign nature of vascular nevus. She was informed that the skin nodules could increase in size during pregnancy and to regularly follow-up with an eye specialist if any visual abnormalities occur.
The term phacomatosis is applied to genetically determined disorders of tissue derived from ectodermal origin (eg, skin, central nervous system, eyes) and commonly includes NF-1, tuberous sclerosis, and von Hippel-Lindau syndrome. Neurofibromatosis type I was first described by German pathologist Friedrich Daniel von Recklinghausen.5 Phacomatosis pigmentovascularis has been defined as the association of vascular nevus with a pigmentary nevus. Its pathogenesis can be explained by the twin spotting phenomenon.6 Twin spots are paired patches of mutant tissue that differ from each other and from the surrounding normal background skin. They can occur as 2 clinical types: allelic and nonallelic twin spotting. Our patient had nonallelic twin spots for 2 nevoid conditions: vascular (nevus flammeus) and pigmentary (nevus of Ota). Nevus of Ota was distributed in the V2 segment (maxillary nerve) of the fifth cranial nerve along with classical melanosis bulbi, which is considered a characteristic clinical feature of nevus of Ota (nevus cesius).7 Nevus flammeus (port-wine stain) is a vascular malformation presenting with flat lesions that persists throughout a patient’s life. The phenomenon of twin spotting, or didymosis (didymos means twin in Greek), has been proposed for co-occurrence of vascular and pigmented nevi.8 The association of NF-1 along with phacomatosis cesioflammea (a twin spot) could be explained from mosaicism of tissues derived from neuroectodermal and mesenchymal elements. Neurofibromatosis type I can occur as a mosaic disorder due to either postzygotic germ line or somatic mutations in the NF1 gene located on the proximal long arm of chromosome 17.9 Irrespective of the mutational event, a mosaic patient has a mixture of cells, some have normal copies of a particular gene and others have an abnormal copy of the same gene. Somatic mutation can lead to segmental (localized), generalized, or gonadal mosaicism. Somatic mutations occurring early during embryonic development produce generalized mosaicism, and generalized mosaics clinically appear similar to nonmosaic NF-1 cases.10,11 However, due to a lack of adequate facilities for mutation analysis and financial constraints, we were unable to confirm our case as generalized somatic mosaic for NF1 gene.
Several morphologic abnormalities have been reported with phacomatosis cesioflammea. Wu et al12 reported a single case of phacomatosis cesioflammea associated with pectus excavatum in a 9-month-old infant. Shields et al13 suggested that a thorough ocular examination on a periodic basis is essential to rule out melanoma of ocular tissues in patients with nevus flammeus and ocular melanosis.
Phacomatosis cesioflammea can occur in association with NF-1. The exact incidence of association is not known. The nevoid condition can be treated with appropriate lasers.
To the Editor:
Vascular lesions associated with melanocytic nevi were first described by Ota et al1 in 1947 and given the name phacomatosis pigmentovascularis. In 2005, Happle2 reclassified phacomatosis pigmentovascularis into 3 well-defined types: (1) phacomatosis cesioflammea: blue spots (caesius means bluish gray in Latin) and nevus flammeus; (2) phacomatosis spilorosea: nevus spilus coexisting with a pale pink telangiectatic nevus; and (3) phacomatosis cesiomarmorata: blue spots and cutis marmorata telangiectatica congenita. In 2011 Joshi et al3 described a case of a 31-year-old woman who had a port-wine stain in association with neurofibromatosis type I (NF-1). We present a case of phacomatosis cesioflammea in association with NF-1.
A 20-year-old woman presented to our outpatient section with a bluish black birthmark on the left side of the face since birth with the onset of multiple painless flesh-colored nodules on the trunk and arms of 1 year’s duration. She reported having occasional pruritus over the nodular lesions. Cutaneous examination showed multiple well-defined café au lait macules (0.5–3.0 cm) with regular margins. Multiple flesh-colored nodules were evident on the upper arms (Figure 1) and trunk. The nodules were firm in consistency and showed buttonholing phenomenon with some of the lesions demonstrating bag-of-worms consistency on palpation. Both palms showed multiple brownish frecklelike macules (Figure 2). A single bluish patch extended from the left ala of the nose to the sideburns. Adjoining the bluish patch was a subtle, ill-defined, nonblanchable red patch extending from the lower margin of the bluish patch to the mandibular ridge (Figure 3). Ocular examination showed melanosis bulbi of the left sclera and a few iris hamartomas (Lisch nodules) in both eyes. A biopsy of the skin nodule was obtained under local anesthesia after obtaining the patient’s informed consent; the specimen was fixed in 10% buffered formalin. A hematoxylin and eosin–stained section showed a well-circumscribed nonencapsulated tumor in the dermis composed of loosely spaced spindle cells and wavy collagenous strands (Figure 4). Routine hemogram and blood biochemistry including urinalysis were within reference range. Radiologic examination of the long bones was unremarkable. Our patient had 3 of 6 criteria defined by the National Institutes of Health for diagnosis of NF-1.4 On clinicopathological correlation we made a diagnosis of phacomatosis cesioflammea in association with NF-1. We have reassured the patient about the benign nature of vascular nevus. She was informed that the skin nodules could increase in size during pregnancy and to regularly follow-up with an eye specialist if any visual abnormalities occur.
The term phacomatosis is applied to genetically determined disorders of tissue derived from ectodermal origin (eg, skin, central nervous system, eyes) and commonly includes NF-1, tuberous sclerosis, and von Hippel-Lindau syndrome. Neurofibromatosis type I was first described by German pathologist Friedrich Daniel von Recklinghausen.5 Phacomatosis pigmentovascularis has been defined as the association of vascular nevus with a pigmentary nevus. Its pathogenesis can be explained by the twin spotting phenomenon.6 Twin spots are paired patches of mutant tissue that differ from each other and from the surrounding normal background skin. They can occur as 2 clinical types: allelic and nonallelic twin spotting. Our patient had nonallelic twin spots for 2 nevoid conditions: vascular (nevus flammeus) and pigmentary (nevus of Ota). Nevus of Ota was distributed in the V2 segment (maxillary nerve) of the fifth cranial nerve along with classical melanosis bulbi, which is considered a characteristic clinical feature of nevus of Ota (nevus cesius).7 Nevus flammeus (port-wine stain) is a vascular malformation presenting with flat lesions that persists throughout a patient’s life. The phenomenon of twin spotting, or didymosis (didymos means twin in Greek), has been proposed for co-occurrence of vascular and pigmented nevi.8 The association of NF-1 along with phacomatosis cesioflammea (a twin spot) could be explained from mosaicism of tissues derived from neuroectodermal and mesenchymal elements. Neurofibromatosis type I can occur as a mosaic disorder due to either postzygotic germ line or somatic mutations in the NF1 gene located on the proximal long arm of chromosome 17.9 Irrespective of the mutational event, a mosaic patient has a mixture of cells, some have normal copies of a particular gene and others have an abnormal copy of the same gene. Somatic mutation can lead to segmental (localized), generalized, or gonadal mosaicism. Somatic mutations occurring early during embryonic development produce generalized mosaicism, and generalized mosaics clinically appear similar to nonmosaic NF-1 cases.10,11 However, due to a lack of adequate facilities for mutation analysis and financial constraints, we were unable to confirm our case as generalized somatic mosaic for NF1 gene.
Several morphologic abnormalities have been reported with phacomatosis cesioflammea. Wu et al12 reported a single case of phacomatosis cesioflammea associated with pectus excavatum in a 9-month-old infant. Shields et al13 suggested that a thorough ocular examination on a periodic basis is essential to rule out melanoma of ocular tissues in patients with nevus flammeus and ocular melanosis.
Phacomatosis cesioflammea can occur in association with NF-1. The exact incidence of association is not known. The nevoid condition can be treated with appropriate lasers.
- Ota M, Kawamura T, Ito N. Phacomatosis pigmentovascularis (Ota). Jpn J Dermatol. 1947;52:1-3.
- Happle R. Phacomatosis pigmentovascularis revisited and reclassified. Arch Dermatol. 2005;141:385-388.
- Joshi A, Manchanda Y, Rijhwani M. Port-wine-stain with rare associations in two cases from Kuwait: phakomatosis pigmentovascularis redefined. Gulf J Dermatol Venereol. 2011;18:59-64.
- Neurofibromatosis. Conference Statement. National Institutes of Health Consensus. Arch Neurol. 1988;45:575-578.
- Gerber PA, Antal AS, Neumann NJ, et al. Neurofibromatosis. Eur J Med Res. 2009;14:102-105.
- Goyal T, Varshney A. Phacomatosis cesioflammea: first case report from India. Indian J Dermatol Venereol Leprol. 2010;76:307.
- Happle R. Didymosis cesioanemica: an unusual counterpart of phakomatosis cesioflammea. Eur J Dermatol. 2011;21:471.
- Happle R, Steijlen PM. Phacomatosis pigmentovascularis interpreted as a phenomenon of twin spots [in German]. Hautarzt. 1989;40:721-724.
- Adigun CG, Stein J. Segmental neurofibromatosis. Dermatol Online J. 2011;17:25.
- Ruggieri M, Huson SM. The clinical and diagnostic implications of mosaicism in the neurofibromatoses. Neurology. 2001;56:1433-1443.
- Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009;61:1-14.
- Wu CY, Chen PH, Chen GS. Phacomatosis cesioflammea associated with pectus excavatum. Acta Derm Venereol. 2009;89:309-310.
- Shields CL, Kligman BE, Suriano M, et al. Phacomatosis pigmentovascularis of cesioflammea type in 7 patients: combination of ocular pigmentation (melanocytosis or melanosis) and nevus flammeus with risk for melanoma. Arch Ophthalmol. 2011;129:746-750.
- Ota M, Kawamura T, Ito N. Phacomatosis pigmentovascularis (Ota). Jpn J Dermatol. 1947;52:1-3.
- Happle R. Phacomatosis pigmentovascularis revisited and reclassified. Arch Dermatol. 2005;141:385-388.
- Joshi A, Manchanda Y, Rijhwani M. Port-wine-stain with rare associations in two cases from Kuwait: phakomatosis pigmentovascularis redefined. Gulf J Dermatol Venereol. 2011;18:59-64.
- Neurofibromatosis. Conference Statement. National Institutes of Health Consensus. Arch Neurol. 1988;45:575-578.
- Gerber PA, Antal AS, Neumann NJ, et al. Neurofibromatosis. Eur J Med Res. 2009;14:102-105.
- Goyal T, Varshney A. Phacomatosis cesioflammea: first case report from India. Indian J Dermatol Venereol Leprol. 2010;76:307.
- Happle R. Didymosis cesioanemica: an unusual counterpart of phakomatosis cesioflammea. Eur J Dermatol. 2011;21:471.
- Happle R, Steijlen PM. Phacomatosis pigmentovascularis interpreted as a phenomenon of twin spots [in German]. Hautarzt. 1989;40:721-724.
- Adigun CG, Stein J. Segmental neurofibromatosis. Dermatol Online J. 2011;17:25.
- Ruggieri M, Huson SM. The clinical and diagnostic implications of mosaicism in the neurofibromatoses. Neurology. 2001;56:1433-1443.
- Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009;61:1-14.
- Wu CY, Chen PH, Chen GS. Phacomatosis cesioflammea associated with pectus excavatum. Acta Derm Venereol. 2009;89:309-310.
- Shields CL, Kligman BE, Suriano M, et al. Phacomatosis pigmentovascularis of cesioflammea type in 7 patients: combination of ocular pigmentation (melanocytosis or melanosis) and nevus flammeus with risk for melanoma. Arch Ophthalmol. 2011;129:746-750.
Practice Points
- Phacomatosis cesioflammea can be associated with neurofibromatosis type I.
- The port-wine stain component of phacomatosis cesioflammea may develop nodularity in long-standing cases.
- The Nd:YAG laser is beneficial for treating blue spots of phacomatosis cesioflammea.
Cosmetic Treatments for Skin of Color: Report From the AAD Meeting
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Vitiligo linked to moles, tanning ability, blistering sunburn
Upper extremity moles were associated with a 37% increase in the likelihood of vitiligo among white women, according to an analysis of the prospective Nurses’ Health Study.
“Women with a higher tanning ability and women who had a history of blistering sunburns in childhood were also found to have a higher risk of developing vitiligo,” Rachel Dunlap, MD, of the department of dermatology, Brown University in Providence, R.I., and her associates wrote in the Journal of Investigative Dermatology.
Vitiligo is the most common cutaneous depigmentation disorder, but associated risk factors are poorly understood, the investigators noted. They examined ties between skin pigmentation, reactions to sun exposure, and new onset vitiligo in the Nurses’ Health Study, a population-based prospective cohort study. Study participants were asked to report the number of moles on their left arms measuring at least 3 mm in diameter, their reactions to sunburn and ability to tan during childhood, and whether they had vitiligo diagnosed by a physician. A total of 51,337 women answered the question about moles, and 68,590 women answered the question about vitiligo, the investigators said (J Invest Dermatol. 2017 Feb 14. doi: 10.1016/j.jid.2017.02.004).
A total of 271 cases of vitiligo developed over 835,594 person-years. Women who reported at least one left arm mole larger than 3 mm were significantly more likely to report incident vitiligo, compared with women without moles (hazard ratio, 1.37; 95% confidence interval, 1.02-1.83), even after controlling for age, hair color, history of exposure to direct sunlight, skin tanning ability, and severity of reaction to sunburn. Developing an “average” tan or a “deep” tan after prolonged sun exposure also were significantly associated with vitiligo with hazard ratios of 2.28 (95% CI, 1.12-4.65) and 2.59 (95% CI, 1.21-5.54), respectively, “when compared to those who had minimal skin reactions or less severe burns when exposed to the sun,” the authors wrote.
A history of at least one blistering sunburn after 2 hours of sun exposure also predicted vitiligo (HR, 2.17; 95% CI, 1.15-4.10), while hair color did not.
“The benefits of good sun protection can be expanded to include potential vitiligo prevention, which may be particularly applicable to adult patients with vitiligo who are concerned about their children developing the condition,” the investigators commented. “Future studies will examine the incidence of other influencing factors, such as melanoma and melanoma associated leukoderma in this population.”
External funding sources included the National Institutes of Health and Dermatology Foundation. The investigators reported having no conflicts of interest.
Upper extremity moles were associated with a 37% increase in the likelihood of vitiligo among white women, according to an analysis of the prospective Nurses’ Health Study.
“Women with a higher tanning ability and women who had a history of blistering sunburns in childhood were also found to have a higher risk of developing vitiligo,” Rachel Dunlap, MD, of the department of dermatology, Brown University in Providence, R.I., and her associates wrote in the Journal of Investigative Dermatology.
Vitiligo is the most common cutaneous depigmentation disorder, but associated risk factors are poorly understood, the investigators noted. They examined ties between skin pigmentation, reactions to sun exposure, and new onset vitiligo in the Nurses’ Health Study, a population-based prospective cohort study. Study participants were asked to report the number of moles on their left arms measuring at least 3 mm in diameter, their reactions to sunburn and ability to tan during childhood, and whether they had vitiligo diagnosed by a physician. A total of 51,337 women answered the question about moles, and 68,590 women answered the question about vitiligo, the investigators said (J Invest Dermatol. 2017 Feb 14. doi: 10.1016/j.jid.2017.02.004).
A total of 271 cases of vitiligo developed over 835,594 person-years. Women who reported at least one left arm mole larger than 3 mm were significantly more likely to report incident vitiligo, compared with women without moles (hazard ratio, 1.37; 95% confidence interval, 1.02-1.83), even after controlling for age, hair color, history of exposure to direct sunlight, skin tanning ability, and severity of reaction to sunburn. Developing an “average” tan or a “deep” tan after prolonged sun exposure also were significantly associated with vitiligo with hazard ratios of 2.28 (95% CI, 1.12-4.65) and 2.59 (95% CI, 1.21-5.54), respectively, “when compared to those who had minimal skin reactions or less severe burns when exposed to the sun,” the authors wrote.
A history of at least one blistering sunburn after 2 hours of sun exposure also predicted vitiligo (HR, 2.17; 95% CI, 1.15-4.10), while hair color did not.
“The benefits of good sun protection can be expanded to include potential vitiligo prevention, which may be particularly applicable to adult patients with vitiligo who are concerned about their children developing the condition,” the investigators commented. “Future studies will examine the incidence of other influencing factors, such as melanoma and melanoma associated leukoderma in this population.”
External funding sources included the National Institutes of Health and Dermatology Foundation. The investigators reported having no conflicts of interest.
Upper extremity moles were associated with a 37% increase in the likelihood of vitiligo among white women, according to an analysis of the prospective Nurses’ Health Study.
“Women with a higher tanning ability and women who had a history of blistering sunburns in childhood were also found to have a higher risk of developing vitiligo,” Rachel Dunlap, MD, of the department of dermatology, Brown University in Providence, R.I., and her associates wrote in the Journal of Investigative Dermatology.
Vitiligo is the most common cutaneous depigmentation disorder, but associated risk factors are poorly understood, the investigators noted. They examined ties between skin pigmentation, reactions to sun exposure, and new onset vitiligo in the Nurses’ Health Study, a population-based prospective cohort study. Study participants were asked to report the number of moles on their left arms measuring at least 3 mm in diameter, their reactions to sunburn and ability to tan during childhood, and whether they had vitiligo diagnosed by a physician. A total of 51,337 women answered the question about moles, and 68,590 women answered the question about vitiligo, the investigators said (J Invest Dermatol. 2017 Feb 14. doi: 10.1016/j.jid.2017.02.004).
A total of 271 cases of vitiligo developed over 835,594 person-years. Women who reported at least one left arm mole larger than 3 mm were significantly more likely to report incident vitiligo, compared with women without moles (hazard ratio, 1.37; 95% confidence interval, 1.02-1.83), even after controlling for age, hair color, history of exposure to direct sunlight, skin tanning ability, and severity of reaction to sunburn. Developing an “average” tan or a “deep” tan after prolonged sun exposure also were significantly associated with vitiligo with hazard ratios of 2.28 (95% CI, 1.12-4.65) and 2.59 (95% CI, 1.21-5.54), respectively, “when compared to those who had minimal skin reactions or less severe burns when exposed to the sun,” the authors wrote.
A history of at least one blistering sunburn after 2 hours of sun exposure also predicted vitiligo (HR, 2.17; 95% CI, 1.15-4.10), while hair color did not.
“The benefits of good sun protection can be expanded to include potential vitiligo prevention, which may be particularly applicable to adult patients with vitiligo who are concerned about their children developing the condition,” the investigators commented. “Future studies will examine the incidence of other influencing factors, such as melanoma and melanoma associated leukoderma in this population.”
External funding sources included the National Institutes of Health and Dermatology Foundation. The investigators reported having no conflicts of interest.
FROM THE JOURNAL OF INVESTIGATIVE DERMATOLOGY
Key clinical point: Upper extremity moles, tanning ability, and a history of blistering sunburn were significant risk factors for vitiligo among white women.
Major finding: In the multivariate analysis, hazard ratios were 1.37 (95% confidence interval, 1.02-1.83), 2.28 (95% CI, 1.12-4.65), 2.59 (95% CI, 1.21-5.54), and 2.17 (95% CI, 1.15-4.10), respectively.
Data source: An analysis of 51,337 white women from the Nurses’ Health Study.
Disclosures: Funding sources included the National Institutes of Health and Dermatology Foundation. The investigators reported having no conflicts of interest.
Widespread Poikilodermatous Dermatomyositis Associated With Chronic Lymphocytic Leukemia
To the Editor:
Dermatomyositis represents a rare idiopathic inflammatory process presenting with cutaneous lesions and muscular weakness. It often represents a paraneoplastic syndrome. We report the case of a 62-year-old man with a history of total-body poikiloderma and a recent diagnosis of chronic lymphocytic leukemia (CLL). Despite lacking typical features of the disease, a diagnosis of dermatomyositis was made. Our patient may represent a distinct poikilodermatous variant of dermatomyositis, sharing the generalized distribution of the erythrodermic subtype.
A 62-year-old man presented with pruritic poikiloderma involving the neck, arms, legs, abdomen, chest, and back of 2 years’ duration (Figure). He also experienced dysphagia and weakness of the legs. The rash was previously treated by other dermatologists with a combination of high-potency topical steroids and topical tacrolimus 0.1% without success. His history was notable for CLL, which had been diagnosed by a dermatologist 6 months prior to the current presentation. Prior to his visit to the dermatologist, the patient had received 6 chemotherapeutic sessions with a combination of rituximab and cyclophosphamide for the treatment of CLL. The rash did not improve with chemotherapy.

Repeat biopsies of affected regions only demonstrated features of mild interface dermatitis. Direct immunofluorescence studies showed scattered colloid body fluorescence for IgM. Because of bilateral weakness of the legs, a muscle biopsy was taken, which demonstrated severe atrophy and interstitial fibrosis, with neurogenic abnormalities detected in areas of lesser atrophy via abnormal muscle fiber–type grouping. Metabolic panel showed elevated muscle enzymes in the blood: creatine kinase, 243 U/L (reference range, 10–225 U/L); serum aldolase, 16 U/L (reference range, ≤8.1 U/L); lactate dehydrogenase, 314 U/L (reference range, 60–200 U/L). An autoimmune panel was negative for Jo-1, Scl-70, U1 ribonucleoprotein, DNA, desmoglein 1 and 3, and antiacetylcholine receptor antibodies. An elevated erythrocyte sedimentation rate was measured at 16 mm/h (reference range, 0–10 mm/h). Given these findings, the lesions were confirmed as a widespread poikilodermatous variant of dermatomyositis.
The patient was placed on a daily 50-mg dose of prednisone, which produced rapid improvement in scaling and erythema. Creatine kinase and serum aldolase levels normalized and motor strength increased. After 1 week the prednisone dosage was reduced to a daily 30-mg dose, and then 20 mg a week later. The skin lesions completely resolved within 4 to 5 months and the patient is currently on a prednisone dose of 5 mg, alternating with 2.5 mg of prednisone and rituximab infusion every 2 months.
Dermatomyositis is a rare entity with an incidence of approximately 0.5 to 1 per 100,000 individuals.1 It presents with a characteristic rash composed of Gottron papules; pathognomonic flat violaceous papules on the dorsal interphalangeal joints, elbows, or knees; and a heliotrope rash, a violaceous erythema involving the eyelids. Poikiloderma frequently is reported to present in a shawl-like distribution, encompassing the shoulders, arms, and upper back.1,2 Dermatomyositis of the poikilodermatous type can present in nonphotoexposed areas and photoexposed areas. The unusual feature is the total-body involvement, which is analogous to erythroderma.3
Our case may represent a distinct poikilodermatous manifestation sharing the distribution of the erythrodermic subtype. We believe that the skin lesions may have represented a paraneoplastic event presenting prior to diagnosis with CLL. Dermatomyositis has a strong association with cancer, with patients 3 times more likely to develop internal malignancy.4 Association is strongest for non-Hodgkin lymphoma, as well as ovarian, lung, colorectal, pancreatic, and gastric cancer. When associated with malignancy, symptoms of dermatomyositis or myositis typically precede the discovery of malignancy by an average of 1.9 years.5 Dermatomyositis has been previously reported to present as a paraneoplastic manifestation of CLL.6 One case has been reported of a patient with CLL who developed leukemia cutis presenting with poikiloderma in the characteristic dermatomyositis shawl-like distribution.7 The lack of dermal infiltration with leukemic cells in our patient, however, makes a paraneoplastic etiology much more likely.
Our patient’s rash did not initially improve with treatment of CLL, but dermatomyositis associated with hematological malignancy may precede, occur simultaneously, or follow the diagnosis of malignancy.8 Additionally, symptoms of dermatomyositis do not always parallel the course of hematological malignancy outcome. However, rituximab has been used as a treatment of dermatomyositis and may have contributed some synergistic effect in combination with prednisone in our patient.9
- Dourmishev LA, Dourmishev AL, Schwartz RA. Dermatomyositis: cutaneous manifestations of its variants. Int J Dermatol. 2002;41:625-630.
- Kovacs SO, Kovacs SC. Dermatomyositis. J Am Acad Dermatol. 1998;39:899-920; quiz 921-992.
- Liu ZH, Wang XD. Acute-onset adult dermatomyositis presenting with erythroderma and diplopia. Clin Exp Dermatol. 2007;32:751-752.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Bohan A, Peter JB, Bowman RL, et al. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977;56:255-286.
- Ishida T, Aikawa K, Tamura T, et al. Chronic lymphocytic leukemia associated with nephrotic syndrome and dermatomyositis. Intern Med. 1995;34:15-17.
- Nousari HC, Kimyai-Asadi A, Huang CH, et al. T-cell chronic lymphocytic leukemia mimicking dermatomyositis. Int J Dermatol. 2000;39:144-146.
- Marie I, Guillevin L, Menard JF, et al. Hematological malignancy associated with polymyositis and dermatomyositis. Autoimmun Rev. 2012;11:615-620.
- Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601-607.
To the Editor:
Dermatomyositis represents a rare idiopathic inflammatory process presenting with cutaneous lesions and muscular weakness. It often represents a paraneoplastic syndrome. We report the case of a 62-year-old man with a history of total-body poikiloderma and a recent diagnosis of chronic lymphocytic leukemia (CLL). Despite lacking typical features of the disease, a diagnosis of dermatomyositis was made. Our patient may represent a distinct poikilodermatous variant of dermatomyositis, sharing the generalized distribution of the erythrodermic subtype.
A 62-year-old man presented with pruritic poikiloderma involving the neck, arms, legs, abdomen, chest, and back of 2 years’ duration (Figure). He also experienced dysphagia and weakness of the legs. The rash was previously treated by other dermatologists with a combination of high-potency topical steroids and topical tacrolimus 0.1% without success. His history was notable for CLL, which had been diagnosed by a dermatologist 6 months prior to the current presentation. Prior to his visit to the dermatologist, the patient had received 6 chemotherapeutic sessions with a combination of rituximab and cyclophosphamide for the treatment of CLL. The rash did not improve with chemotherapy.
Repeat biopsies of affected regions only demonstrated features of mild interface dermatitis. Direct immunofluorescence studies showed scattered colloid body fluorescence for IgM. Because of bilateral weakness of the legs, a muscle biopsy was taken, which demonstrated severe atrophy and interstitial fibrosis, with neurogenic abnormalities detected in areas of lesser atrophy via abnormal muscle fiber–type grouping. Metabolic panel showed elevated muscle enzymes in the blood: creatine kinase, 243 U/L (reference range, 10–225 U/L); serum aldolase, 16 U/L (reference range, ≤8.1 U/L); lactate dehydrogenase, 314 U/L (reference range, 60–200 U/L). An autoimmune panel was negative for Jo-1, Scl-70, U1 ribonucleoprotein, DNA, desmoglein 1 and 3, and antiacetylcholine receptor antibodies. An elevated erythrocyte sedimentation rate was measured at 16 mm/h (reference range, 0–10 mm/h). Given these findings, the lesions were confirmed as a widespread poikilodermatous variant of dermatomyositis.
The patient was placed on a daily 50-mg dose of prednisone, which produced rapid improvement in scaling and erythema. Creatine kinase and serum aldolase levels normalized and motor strength increased. After 1 week the prednisone dosage was reduced to a daily 30-mg dose, and then 20 mg a week later. The skin lesions completely resolved within 4 to 5 months and the patient is currently on a prednisone dose of 5 mg, alternating with 2.5 mg of prednisone and rituximab infusion every 2 months.
Dermatomyositis is a rare entity with an incidence of approximately 0.5 to 1 per 100,000 individuals.1 It presents with a characteristic rash composed of Gottron papules; pathognomonic flat violaceous papules on the dorsal interphalangeal joints, elbows, or knees; and a heliotrope rash, a violaceous erythema involving the eyelids. Poikiloderma frequently is reported to present in a shawl-like distribution, encompassing the shoulders, arms, and upper back.1,2 Dermatomyositis of the poikilodermatous type can present in nonphotoexposed areas and photoexposed areas. The unusual feature is the total-body involvement, which is analogous to erythroderma.3
Our case may represent a distinct poikilodermatous manifestation sharing the distribution of the erythrodermic subtype. We believe that the skin lesions may have represented a paraneoplastic event presenting prior to diagnosis with CLL. Dermatomyositis has a strong association with cancer, with patients 3 times more likely to develop internal malignancy.4 Association is strongest for non-Hodgkin lymphoma, as well as ovarian, lung, colorectal, pancreatic, and gastric cancer. When associated with malignancy, symptoms of dermatomyositis or myositis typically precede the discovery of malignancy by an average of 1.9 years.5 Dermatomyositis has been previously reported to present as a paraneoplastic manifestation of CLL.6 One case has been reported of a patient with CLL who developed leukemia cutis presenting with poikiloderma in the characteristic dermatomyositis shawl-like distribution.7 The lack of dermal infiltration with leukemic cells in our patient, however, makes a paraneoplastic etiology much more likely.
Our patient’s rash did not initially improve with treatment of CLL, but dermatomyositis associated with hematological malignancy may precede, occur simultaneously, or follow the diagnosis of malignancy.8 Additionally, symptoms of dermatomyositis do not always parallel the course of hematological malignancy outcome. However, rituximab has been used as a treatment of dermatomyositis and may have contributed some synergistic effect in combination with prednisone in our patient.9
To the Editor:
Dermatomyositis represents a rare idiopathic inflammatory process presenting with cutaneous lesions and muscular weakness. It often represents a paraneoplastic syndrome. We report the case of a 62-year-old man with a history of total-body poikiloderma and a recent diagnosis of chronic lymphocytic leukemia (CLL). Despite lacking typical features of the disease, a diagnosis of dermatomyositis was made. Our patient may represent a distinct poikilodermatous variant of dermatomyositis, sharing the generalized distribution of the erythrodermic subtype.
A 62-year-old man presented with pruritic poikiloderma involving the neck, arms, legs, abdomen, chest, and back of 2 years’ duration (Figure). He also experienced dysphagia and weakness of the legs. The rash was previously treated by other dermatologists with a combination of high-potency topical steroids and topical tacrolimus 0.1% without success. His history was notable for CLL, which had been diagnosed by a dermatologist 6 months prior to the current presentation. Prior to his visit to the dermatologist, the patient had received 6 chemotherapeutic sessions with a combination of rituximab and cyclophosphamide for the treatment of CLL. The rash did not improve with chemotherapy.
Repeat biopsies of affected regions only demonstrated features of mild interface dermatitis. Direct immunofluorescence studies showed scattered colloid body fluorescence for IgM. Because of bilateral weakness of the legs, a muscle biopsy was taken, which demonstrated severe atrophy and interstitial fibrosis, with neurogenic abnormalities detected in areas of lesser atrophy via abnormal muscle fiber–type grouping. Metabolic panel showed elevated muscle enzymes in the blood: creatine kinase, 243 U/L (reference range, 10–225 U/L); serum aldolase, 16 U/L (reference range, ≤8.1 U/L); lactate dehydrogenase, 314 U/L (reference range, 60–200 U/L). An autoimmune panel was negative for Jo-1, Scl-70, U1 ribonucleoprotein, DNA, desmoglein 1 and 3, and antiacetylcholine receptor antibodies. An elevated erythrocyte sedimentation rate was measured at 16 mm/h (reference range, 0–10 mm/h). Given these findings, the lesions were confirmed as a widespread poikilodermatous variant of dermatomyositis.
The patient was placed on a daily 50-mg dose of prednisone, which produced rapid improvement in scaling and erythema. Creatine kinase and serum aldolase levels normalized and motor strength increased. After 1 week the prednisone dosage was reduced to a daily 30-mg dose, and then 20 mg a week later. The skin lesions completely resolved within 4 to 5 months and the patient is currently on a prednisone dose of 5 mg, alternating with 2.5 mg of prednisone and rituximab infusion every 2 months.
Dermatomyositis is a rare entity with an incidence of approximately 0.5 to 1 per 100,000 individuals.1 It presents with a characteristic rash composed of Gottron papules; pathognomonic flat violaceous papules on the dorsal interphalangeal joints, elbows, or knees; and a heliotrope rash, a violaceous erythema involving the eyelids. Poikiloderma frequently is reported to present in a shawl-like distribution, encompassing the shoulders, arms, and upper back.1,2 Dermatomyositis of the poikilodermatous type can present in nonphotoexposed areas and photoexposed areas. The unusual feature is the total-body involvement, which is analogous to erythroderma.3
Our case may represent a distinct poikilodermatous manifestation sharing the distribution of the erythrodermic subtype. We believe that the skin lesions may have represented a paraneoplastic event presenting prior to diagnosis with CLL. Dermatomyositis has a strong association with cancer, with patients 3 times more likely to develop internal malignancy.4 Association is strongest for non-Hodgkin lymphoma, as well as ovarian, lung, colorectal, pancreatic, and gastric cancer. When associated with malignancy, symptoms of dermatomyositis or myositis typically precede the discovery of malignancy by an average of 1.9 years.5 Dermatomyositis has been previously reported to present as a paraneoplastic manifestation of CLL.6 One case has been reported of a patient with CLL who developed leukemia cutis presenting with poikiloderma in the characteristic dermatomyositis shawl-like distribution.7 The lack of dermal infiltration with leukemic cells in our patient, however, makes a paraneoplastic etiology much more likely.
Our patient’s rash did not initially improve with treatment of CLL, but dermatomyositis associated with hematological malignancy may precede, occur simultaneously, or follow the diagnosis of malignancy.8 Additionally, symptoms of dermatomyositis do not always parallel the course of hematological malignancy outcome. However, rituximab has been used as a treatment of dermatomyositis and may have contributed some synergistic effect in combination with prednisone in our patient.9
- Dourmishev LA, Dourmishev AL, Schwartz RA. Dermatomyositis: cutaneous manifestations of its variants. Int J Dermatol. 2002;41:625-630.
- Kovacs SO, Kovacs SC. Dermatomyositis. J Am Acad Dermatol. 1998;39:899-920; quiz 921-992.
- Liu ZH, Wang XD. Acute-onset adult dermatomyositis presenting with erythroderma and diplopia. Clin Exp Dermatol. 2007;32:751-752.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Bohan A, Peter JB, Bowman RL, et al. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977;56:255-286.
- Ishida T, Aikawa K, Tamura T, et al. Chronic lymphocytic leukemia associated with nephrotic syndrome and dermatomyositis. Intern Med. 1995;34:15-17.
- Nousari HC, Kimyai-Asadi A, Huang CH, et al. T-cell chronic lymphocytic leukemia mimicking dermatomyositis. Int J Dermatol. 2000;39:144-146.
- Marie I, Guillevin L, Menard JF, et al. Hematological malignancy associated with polymyositis and dermatomyositis. Autoimmun Rev. 2012;11:615-620.
- Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601-607.
- Dourmishev LA, Dourmishev AL, Schwartz RA. Dermatomyositis: cutaneous manifestations of its variants. Int J Dermatol. 2002;41:625-630.
- Kovacs SO, Kovacs SC. Dermatomyositis. J Am Acad Dermatol. 1998;39:899-920; quiz 921-992.
- Liu ZH, Wang XD. Acute-onset adult dermatomyositis presenting with erythroderma and diplopia. Clin Exp Dermatol. 2007;32:751-752.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Bohan A, Peter JB, Bowman RL, et al. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977;56:255-286.
- Ishida T, Aikawa K, Tamura T, et al. Chronic lymphocytic leukemia associated with nephrotic syndrome and dermatomyositis. Intern Med. 1995;34:15-17.
- Nousari HC, Kimyai-Asadi A, Huang CH, et al. T-cell chronic lymphocytic leukemia mimicking dermatomyositis. Int J Dermatol. 2000;39:144-146.
- Marie I, Guillevin L, Menard JF, et al. Hematological malignancy associated with polymyositis and dermatomyositis. Autoimmun Rev. 2012;11:615-620.
- Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601-607.
Practice Points
- Poikiloderma, even with an unusual clinical presentation, can be a useful clinical clue for the diagnosis of dermatomyositis or other collagen vascular disease.
- Dermatomyositis can be paraneoplastic and though often associated with epithelial malignancies and solid tumors can also be associated with leukemias.
Microneedling With Platelet-Rich Plasma

Hyperpigmented Papules and Plaques
The Diagnosis: Persistent Still Disease
At the time of presentation, the patient had not taken systemic medications for a year. Laboratory studies revealed leukocytosis with neutrophilia and a serum ferritin level of 5493 ng/mL (reference range, 15-200 ng/mL). Rheumatoid factor and antinuclear antibody serologies were within reference range. Microbiologic workup was negative. Lymph node and bone marrow biopsies were negative for a lymphoproliferative disorder. Skin biopsies were performed on the back and forearm. Histologic evaluation revealed orthokeratosis, slight acanthosis, and dyskeratosis confined to the upper layers of the epidermis without evidence of interface dermatitis. There was a mixed perivascular infiltrate composed of lymphocytes and neutrophils with no attendant vasculitic change (Figure).
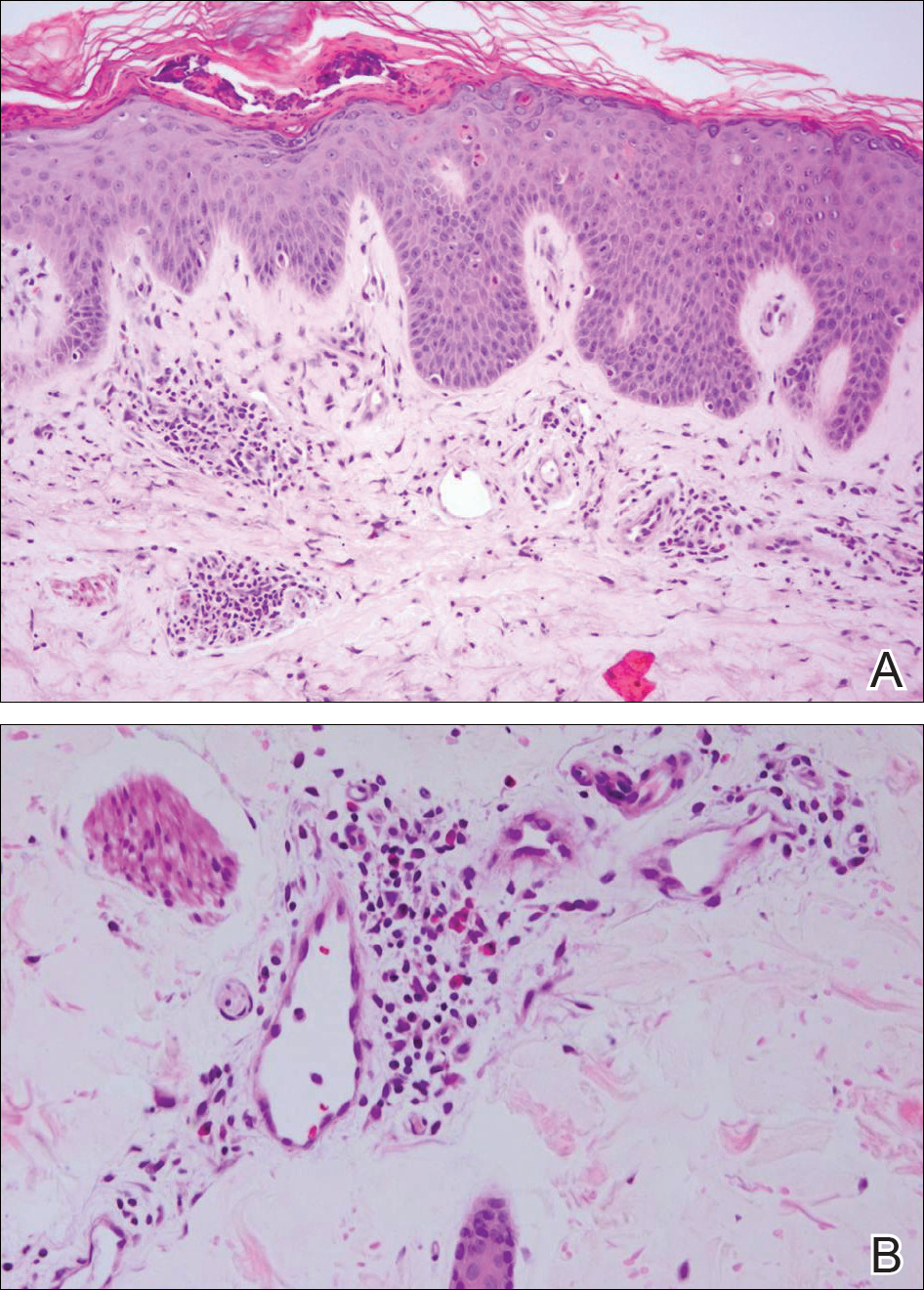
The patient was discharged on prednisone and seen for outpatient follow-up weeks later. Six weeks later, the cutaneous eruption remained unchanged. The patient was unable to start other systemic medications due to lack of insurance and ineligibility for the local patient-assistance program; he was subsequently lost to follow-up.
Adult-onset Still disease is a rare, systemic, inflammatory condition with a broad spectrum of clinical presentations.1-3 Still disease affects all age groups, and children with Still disease (<16 years) usually have a concurrent diagnosis of juvenile idiopathic arthritis (formerly known as juvenile rheumatoid arthritis).1,2,4 Still disease preferentially affects adolescents and adults aged 16 to 35 years, with more than 75% of new cases occurring in this age range.1 Worldwide, the incidence and prevalence of Still disease is disputed with no conclusive rates established.1,3
Still disease is characterized by 4 cardinal signs: high spiking fevers (temperature, ≥39°C); leukocytosis with a predominance of neutrophils (≥10,000 cells/mm3 with ≥80% neutrophils); arthralgia or arthritis; and an evanescent, nonpruritic, salmon-colored morbilliform eruption of the skin, typically on the trunk or extremities.2 Histologic evaluation of the classic Still disease eruption displays perivascular inflammation of the superficial dermis with infiltration by lymphocytes and histiocytes.3
In 1992, major and minor diagnostic criteria were established for adult-onset Still disease. For diagnosis, patients must meet 5 criteria, including 2 major criteria.5 Major criteria include arthralgia or arthritis present for more than 2 weeks, fever (temperature, >39°C) for at least 1 week, the classic Still disease morbilliform eruption (ie, salmon colored, evanescent, morbilliform), and leukocytosis with more than 80% neutrophils. Minor criteria include sore throat, lymphadenopathy and/or splenomegaly, negative rheumatoid factor and antinuclear antibody serologies, and abnormal liver function (defined as elevated transaminases).5 Although not included in the diagnostic criteria, there have been reports of elevated serum ferritin levels in patients with Still disease, a finding that potentially is useful in distinguishing between active and inactive rheumatic conditions.6,7
Several case reports have described persistent Still disease, a subtype of Still disease in which patients present with brown-red, persistent, pruritic macules, papules, and plaques that are widespread and oddly shaped.8,9 Histologically, this subtype is characterized by necrotic keratinocytes in the epidermis and dermal perivascular inflammation composed of neutrophils and lymphocytes.10 This histology differs from classic Still disease in that the latter typically does not have superficial epidermal dyskeratosis. Our case is consistent with reports of persistent Still disease.
Although the etiology of Still disease remains to be elucidated, HLA-B17, -B18, -B35, and -DR2 have been associated with the disease.3 Furthermore, helper T cell TH1, IL-2, IFN-γ, and tumor necrosis factor α have been implicated in disease pathology, enabling the use of newer targeted pharmacologic therapies. Canakinumab, an IL-1β inhibitor, has been found to improve arthritis, fever, and rash in patients with Still disease.11 These findings are particularly encouraging for patients who have not experienced improvement with traditional antirheumatic drugs, such as our patient who was not steroid responsive.3
Although a salmon-colored, evanescent, morbilliform eruption in the context of other systemic signs and symptoms readily evokes consideration of Still disease, the less common fixed cutaneous eruption seen in our case may evade accurate diagnosis. Our case aims to increase awareness of this unusual and rare subtype of the cutaneous eruption of Still disease, as a timely diagnosis may prevent potentially life-threatening sequelae including cardiopulmonary disease and respiratory failure.3,5,9
- Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still's disease [published online October 11, 2005]. Ann Rheum Dis. 2006;65:564-572.
- Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol. 2008;22:773-792.
- Bagnari V, Colina M, Ciancio G, et al. Adult-onset Still's disease. Rheumatol Int. 2010;30:855-862.
- Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007;369:767-778.
- Yamaguchi M, Ohta A, Tsunematsu, T, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992;19:424-430.
- Van Reeth C, Le Moel G, Lasne Y, et al. Serum ferritin and isoferritins are tools for diagnosis of active adult Still's disease. J Rheumatol. 1994;21:890-895.
- Novak S, Anic F, Luke-Vrbanic TS. Extremely high serum ferritin levels as a main diagnostic tool of adult-onset Still's disease. Rheumatol Int. 2012;32:1091-1094.
- Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still's disease. J Cutan Pathol. 2010;37:932-937.
- Yang CC, Lee JY, Liu MF, et al. Adult-onset Still's disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol. 2006;16:593-594.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still's disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Kontzias A, Efthimiou P. The use of canakinumab, a novel IL-1β long-acting inhibitor in refractory adult-onset Still's disease. Sem Arthritis Rheum. 2012;42:201-205.
The Diagnosis: Persistent Still Disease
At the time of presentation, the patient had not taken systemic medications for a year. Laboratory studies revealed leukocytosis with neutrophilia and a serum ferritin level of 5493 ng/mL (reference range, 15-200 ng/mL). Rheumatoid factor and antinuclear antibody serologies were within reference range. Microbiologic workup was negative. Lymph node and bone marrow biopsies were negative for a lymphoproliferative disorder. Skin biopsies were performed on the back and forearm. Histologic evaluation revealed orthokeratosis, slight acanthosis, and dyskeratosis confined to the upper layers of the epidermis without evidence of interface dermatitis. There was a mixed perivascular infiltrate composed of lymphocytes and neutrophils with no attendant vasculitic change (Figure).
The patient was discharged on prednisone and seen for outpatient follow-up weeks later. Six weeks later, the cutaneous eruption remained unchanged. The patient was unable to start other systemic medications due to lack of insurance and ineligibility for the local patient-assistance program; he was subsequently lost to follow-up.
Adult-onset Still disease is a rare, systemic, inflammatory condition with a broad spectrum of clinical presentations.1-3 Still disease affects all age groups, and children with Still disease (<16 years) usually have a concurrent diagnosis of juvenile idiopathic arthritis (formerly known as juvenile rheumatoid arthritis).1,2,4 Still disease preferentially affects adolescents and adults aged 16 to 35 years, with more than 75% of new cases occurring in this age range.1 Worldwide, the incidence and prevalence of Still disease is disputed with no conclusive rates established.1,3
Still disease is characterized by 4 cardinal signs: high spiking fevers (temperature, ≥39°C); leukocytosis with a predominance of neutrophils (≥10,000 cells/mm3 with ≥80% neutrophils); arthralgia or arthritis; and an evanescent, nonpruritic, salmon-colored morbilliform eruption of the skin, typically on the trunk or extremities.2 Histologic evaluation of the classic Still disease eruption displays perivascular inflammation of the superficial dermis with infiltration by lymphocytes and histiocytes.3
In 1992, major and minor diagnostic criteria were established for adult-onset Still disease. For diagnosis, patients must meet 5 criteria, including 2 major criteria.5 Major criteria include arthralgia or arthritis present for more than 2 weeks, fever (temperature, >39°C) for at least 1 week, the classic Still disease morbilliform eruption (ie, salmon colored, evanescent, morbilliform), and leukocytosis with more than 80% neutrophils. Minor criteria include sore throat, lymphadenopathy and/or splenomegaly, negative rheumatoid factor and antinuclear antibody serologies, and abnormal liver function (defined as elevated transaminases).5 Although not included in the diagnostic criteria, there have been reports of elevated serum ferritin levels in patients with Still disease, a finding that potentially is useful in distinguishing between active and inactive rheumatic conditions.6,7
Several case reports have described persistent Still disease, a subtype of Still disease in which patients present with brown-red, persistent, pruritic macules, papules, and plaques that are widespread and oddly shaped.8,9 Histologically, this subtype is characterized by necrotic keratinocytes in the epidermis and dermal perivascular inflammation composed of neutrophils and lymphocytes.10 This histology differs from classic Still disease in that the latter typically does not have superficial epidermal dyskeratosis. Our case is consistent with reports of persistent Still disease.
Although the etiology of Still disease remains to be elucidated, HLA-B17, -B18, -B35, and -DR2 have been associated with the disease.3 Furthermore, helper T cell TH1, IL-2, IFN-γ, and tumor necrosis factor α have been implicated in disease pathology, enabling the use of newer targeted pharmacologic therapies. Canakinumab, an IL-1β inhibitor, has been found to improve arthritis, fever, and rash in patients with Still disease.11 These findings are particularly encouraging for patients who have not experienced improvement with traditional antirheumatic drugs, such as our patient who was not steroid responsive.3
Although a salmon-colored, evanescent, morbilliform eruption in the context of other systemic signs and symptoms readily evokes consideration of Still disease, the less common fixed cutaneous eruption seen in our case may evade accurate diagnosis. Our case aims to increase awareness of this unusual and rare subtype of the cutaneous eruption of Still disease, as a timely diagnosis may prevent potentially life-threatening sequelae including cardiopulmonary disease and respiratory failure.3,5,9
The Diagnosis: Persistent Still Disease
At the time of presentation, the patient had not taken systemic medications for a year. Laboratory studies revealed leukocytosis with neutrophilia and a serum ferritin level of 5493 ng/mL (reference range, 15-200 ng/mL). Rheumatoid factor and antinuclear antibody serologies were within reference range. Microbiologic workup was negative. Lymph node and bone marrow biopsies were negative for a lymphoproliferative disorder. Skin biopsies were performed on the back and forearm. Histologic evaluation revealed orthokeratosis, slight acanthosis, and dyskeratosis confined to the upper layers of the epidermis without evidence of interface dermatitis. There was a mixed perivascular infiltrate composed of lymphocytes and neutrophils with no attendant vasculitic change (Figure).
The patient was discharged on prednisone and seen for outpatient follow-up weeks later. Six weeks later, the cutaneous eruption remained unchanged. The patient was unable to start other systemic medications due to lack of insurance and ineligibility for the local patient-assistance program; he was subsequently lost to follow-up.
Adult-onset Still disease is a rare, systemic, inflammatory condition with a broad spectrum of clinical presentations.1-3 Still disease affects all age groups, and children with Still disease (<16 years) usually have a concurrent diagnosis of juvenile idiopathic arthritis (formerly known as juvenile rheumatoid arthritis).1,2,4 Still disease preferentially affects adolescents and adults aged 16 to 35 years, with more than 75% of new cases occurring in this age range.1 Worldwide, the incidence and prevalence of Still disease is disputed with no conclusive rates established.1,3
Still disease is characterized by 4 cardinal signs: high spiking fevers (temperature, ≥39°C); leukocytosis with a predominance of neutrophils (≥10,000 cells/mm3 with ≥80% neutrophils); arthralgia or arthritis; and an evanescent, nonpruritic, salmon-colored morbilliform eruption of the skin, typically on the trunk or extremities.2 Histologic evaluation of the classic Still disease eruption displays perivascular inflammation of the superficial dermis with infiltration by lymphocytes and histiocytes.3
In 1992, major and minor diagnostic criteria were established for adult-onset Still disease. For diagnosis, patients must meet 5 criteria, including 2 major criteria.5 Major criteria include arthralgia or arthritis present for more than 2 weeks, fever (temperature, >39°C) for at least 1 week, the classic Still disease morbilliform eruption (ie, salmon colored, evanescent, morbilliform), and leukocytosis with more than 80% neutrophils. Minor criteria include sore throat, lymphadenopathy and/or splenomegaly, negative rheumatoid factor and antinuclear antibody serologies, and abnormal liver function (defined as elevated transaminases).5 Although not included in the diagnostic criteria, there have been reports of elevated serum ferritin levels in patients with Still disease, a finding that potentially is useful in distinguishing between active and inactive rheumatic conditions.6,7
Several case reports have described persistent Still disease, a subtype of Still disease in which patients present with brown-red, persistent, pruritic macules, papules, and plaques that are widespread and oddly shaped.8,9 Histologically, this subtype is characterized by necrotic keratinocytes in the epidermis and dermal perivascular inflammation composed of neutrophils and lymphocytes.10 This histology differs from classic Still disease in that the latter typically does not have superficial epidermal dyskeratosis. Our case is consistent with reports of persistent Still disease.
Although the etiology of Still disease remains to be elucidated, HLA-B17, -B18, -B35, and -DR2 have been associated with the disease.3 Furthermore, helper T cell TH1, IL-2, IFN-γ, and tumor necrosis factor α have been implicated in disease pathology, enabling the use of newer targeted pharmacologic therapies. Canakinumab, an IL-1β inhibitor, has been found to improve arthritis, fever, and rash in patients with Still disease.11 These findings are particularly encouraging for patients who have not experienced improvement with traditional antirheumatic drugs, such as our patient who was not steroid responsive.3
Although a salmon-colored, evanescent, morbilliform eruption in the context of other systemic signs and symptoms readily evokes consideration of Still disease, the less common fixed cutaneous eruption seen in our case may evade accurate diagnosis. Our case aims to increase awareness of this unusual and rare subtype of the cutaneous eruption of Still disease, as a timely diagnosis may prevent potentially life-threatening sequelae including cardiopulmonary disease and respiratory failure.3,5,9
- Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still's disease [published online October 11, 2005]. Ann Rheum Dis. 2006;65:564-572.
- Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol. 2008;22:773-792.
- Bagnari V, Colina M, Ciancio G, et al. Adult-onset Still's disease. Rheumatol Int. 2010;30:855-862.
- Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007;369:767-778.
- Yamaguchi M, Ohta A, Tsunematsu, T, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992;19:424-430.
- Van Reeth C, Le Moel G, Lasne Y, et al. Serum ferritin and isoferritins are tools for diagnosis of active adult Still's disease. J Rheumatol. 1994;21:890-895.
- Novak S, Anic F, Luke-Vrbanic TS. Extremely high serum ferritin levels as a main diagnostic tool of adult-onset Still's disease. Rheumatol Int. 2012;32:1091-1094.
- Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still's disease. J Cutan Pathol. 2010;37:932-937.
- Yang CC, Lee JY, Liu MF, et al. Adult-onset Still's disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol. 2006;16:593-594.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still's disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Kontzias A, Efthimiou P. The use of canakinumab, a novel IL-1β long-acting inhibitor in refractory adult-onset Still's disease. Sem Arthritis Rheum. 2012;42:201-205.
- Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still's disease [published online October 11, 2005]. Ann Rheum Dis. 2006;65:564-572.
- Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol. 2008;22:773-792.
- Bagnari V, Colina M, Ciancio G, et al. Adult-onset Still's disease. Rheumatol Int. 2010;30:855-862.
- Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007;369:767-778.
- Yamaguchi M, Ohta A, Tsunematsu, T, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992;19:424-430.
- Van Reeth C, Le Moel G, Lasne Y, et al. Serum ferritin and isoferritins are tools for diagnosis of active adult Still's disease. J Rheumatol. 1994;21:890-895.
- Novak S, Anic F, Luke-Vrbanic TS. Extremely high serum ferritin levels as a main diagnostic tool of adult-onset Still's disease. Rheumatol Int. 2012;32:1091-1094.
- Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still's disease. J Cutan Pathol. 2010;37:932-937.
- Yang CC, Lee JY, Liu MF, et al. Adult-onset Still's disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol. 2006;16:593-594.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still's disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Kontzias A, Efthimiou P. The use of canakinumab, a novel IL-1β long-acting inhibitor in refractory adult-onset Still's disease. Sem Arthritis Rheum. 2012;42:201-205.
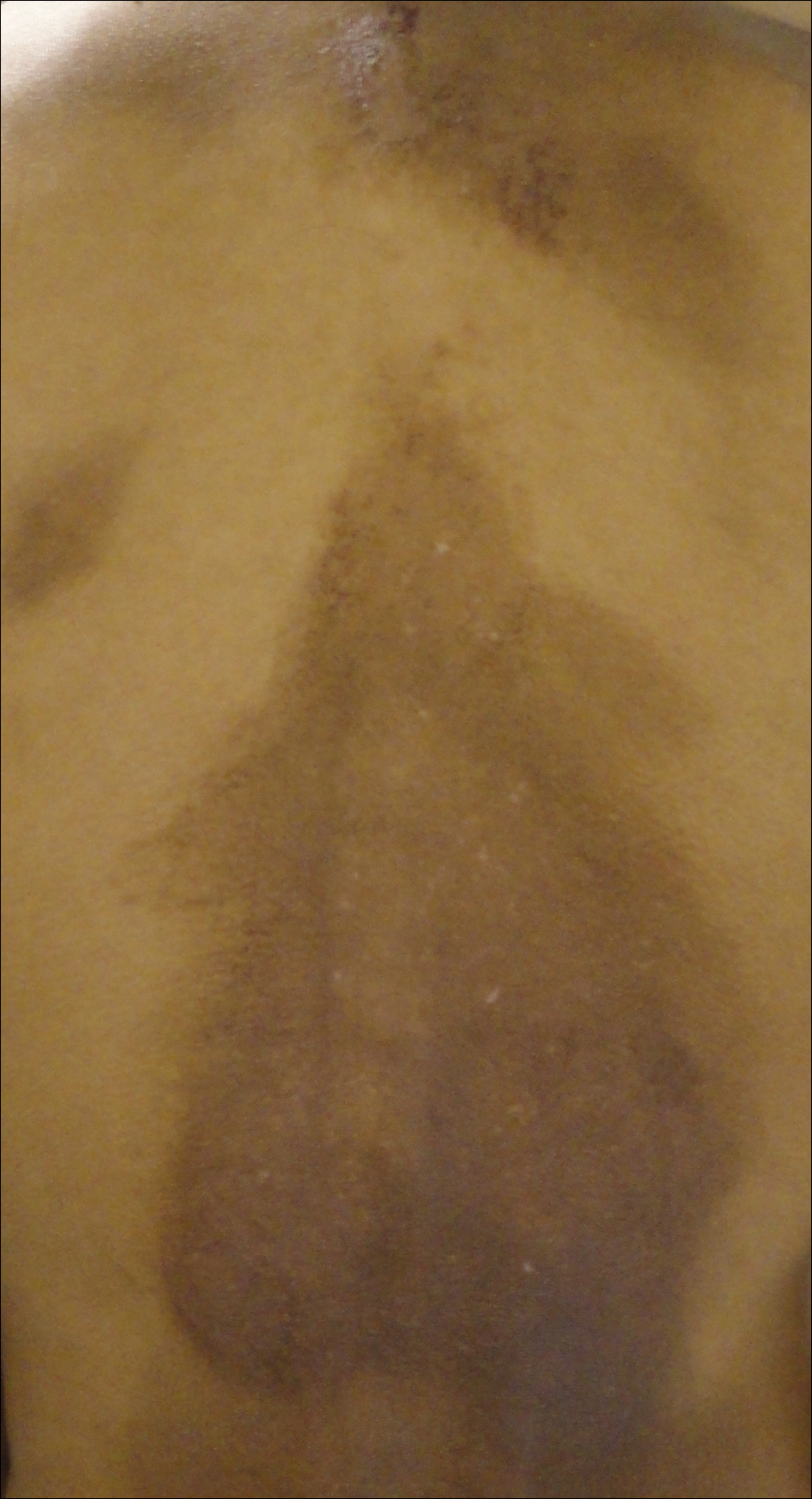
A 25-year-old Hispanic man with a history of juvenile idiopathic arthritis was admitted with a high-grade fever (temperature, >38.9°C) and diffuse nonlocalized abdominal pain of 2 days' duration. Physical examination revealed tachycardia, axillary lymphadenopathy, and hepatosplenomegaly. Cutaneous findings consisted of striking hyperpigmented patches on the chest and back, and hyperpigmented scaly lichenoid papules and plaques on the upper and lower extremities. The plaques on the lower extremities exhibited koebnerization. The patient reported that the eruption initially presented at 16 years of age as pruritic papules on the legs, which gradually spread to involve the arms, chest, and back. Prior treatments of juvenile idiopathic arthritis included prednisone, methotrexate, infliximab, and etanercept, though they were intermittent and temporary. Over time, the cutaneous eruption evolved into its current morphology and distribution, with periods of clearance observed while receiving systemic medications.
Telmisartan-Induced Lichen Planus Eruption Manifested on Vitiliginous Skin
To the Editor:
A 39-year-old man with a history of hypertension and vitiligo presented with a rapid-onset, generalized, pruritic rash covering the body of 4 weeks’ duration. He reported that the rash progressively worsened after developing mild sunburn. The patient stated that the rash was extremely pruritic with a burning sensation and was tender to touch. He was treated with betamethasone valerate cream 0.1% by an outside physician and an over-the-counter anti-itch lotion with no notable improvement. His only medication was telmisartan-hydrochlorothiazide (HCTZ) for hypertension. He denied any drug allergies.
Physical examination revealed multiple discrete and coalescent planar erythematous papules and plaques involving only the depigmented vitiliginous skin of the forehead, eyelids, and nape of the neck (Figure 1A), and confluent on the lateral aspect of the bilateral forearms (Figure 1B), dorsal aspect of the right hand, and bilateral dorsi of the feet. Wickham striae were noted on the lips (Figure 1C). A clinical diagnosis of lichen planus (LP) was made. The patient initially was prescribed halobetasol propionate ointment 0.05% twice daily. He reported notable relief of pruritus with reduction of overall symptoms and new lesion formation.
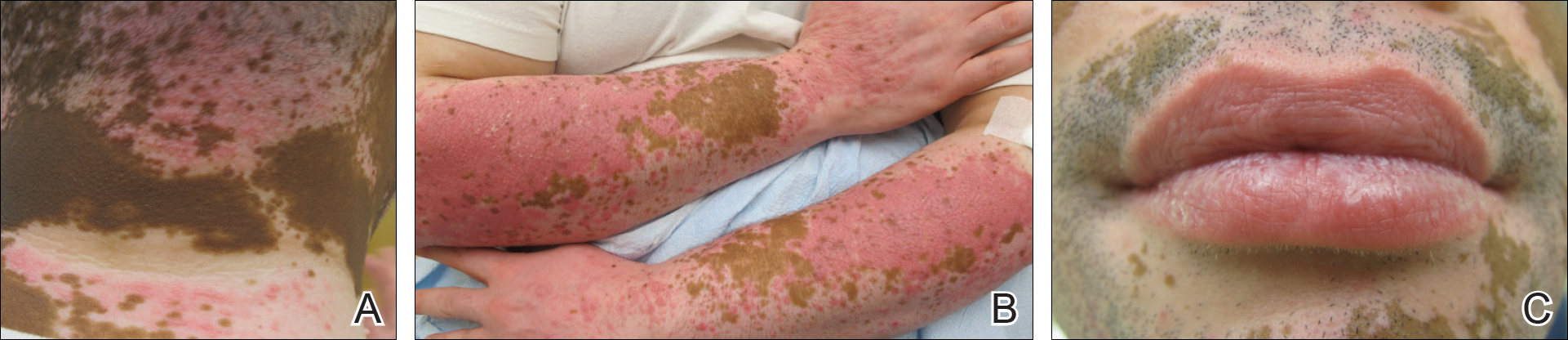
A 4-mm punch biopsy was performed on the left forearm. Histopathology revealed LP. Microscopic examination of the hematoxylin and eosin–stained specimen revealed a bandlike lymphohistiocytic infiltrate that extended across the papillary dermis, focally obscuring the dermoepidermal junction where there were vacuolar changes and colloid bodies. The epidermis showed sawtooth rete ridges, wedge-shaped foci of hypergranulosis, and compact hyperkeratosis (Figure 2).
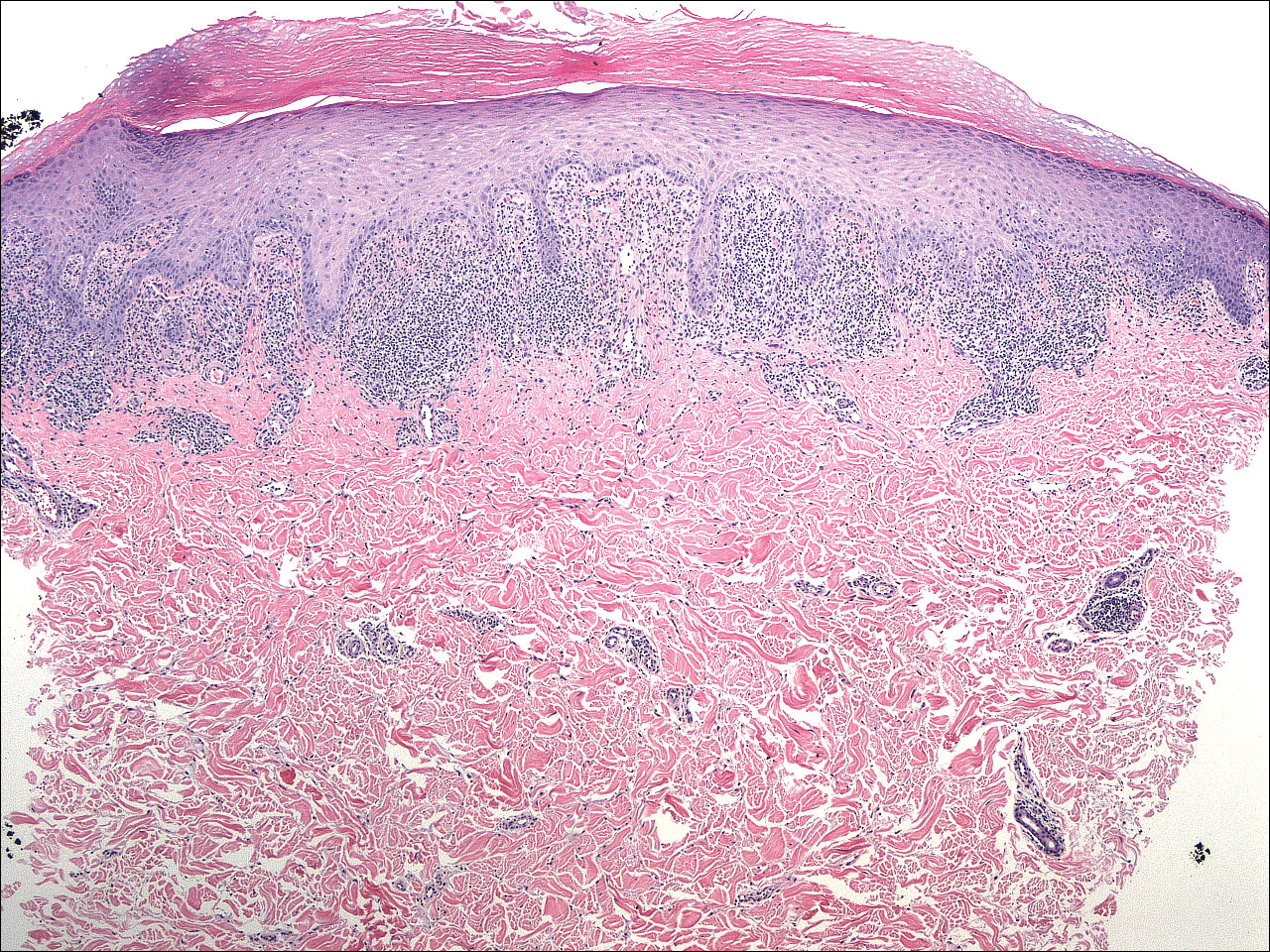
On further questioning during follow-up, the patient revealed that his hypertensive medication was changed from HCTZ, which he had been taking for the last 8 years, to the combination antihypertensive medication telmisartan-HCTZ before the onset of the skin eruption. Due to the temporal relationship between the new medication and onset of the eruption, the clinical impression was highly suspicious for drug-induced eruptive LP with Köbner phenomenon caused by the recent sunburn. Systemic workup for underlying causes of LP was negative. Laboratory tests revealed normal complete blood cell counts. The hepatitis panel included hepatitis A antibodies; hepatitis B surface, e antigen, and core antibodies; hepatitis B surface antigen and e antibodies; hepatitis C antibodies; and antinuclear antibodies, which were all negative.
The patient continued to develop new pruritic papules clinically consistent with LP. He was instructed to return to his primary care physician to change the telmisartan-HCTZ to a different class of antihypertensive medication. His medication was changed to atenolol. The patient also was instructed to continue the halobetasol propionate ointment 0.05% twice daily to the affected areas.
The patient returned for a follow-up visit 1 month later and reported notable improvement in pruritus and near-complete resolution of the LP after discontinuation of telmisartan-HCTZ. He also noted some degree of perifollicular repigmentation of the vitiliginous skin that had been unresponsive to prior therapy (Figure 3).
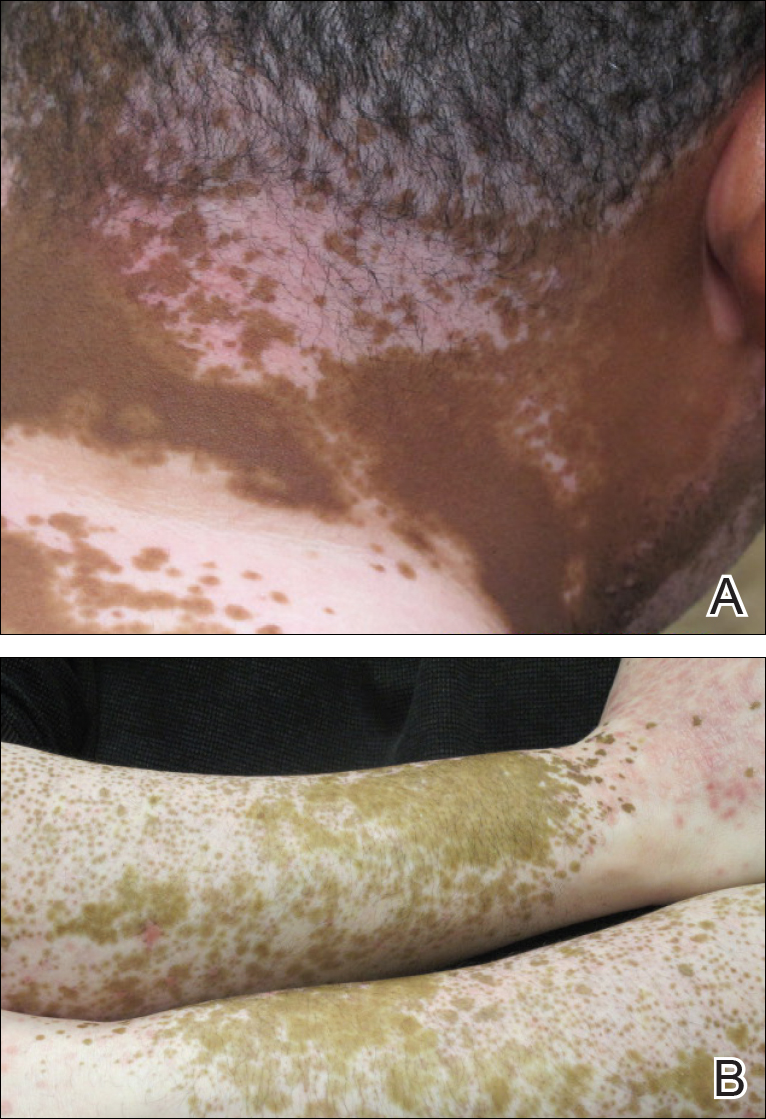
Lichen planus is a pruritic and inflammatory papulosquamous skin condition that presents as scaly, flat-topped, violaceous, polygonal-shaped papules commonly involving the flexor surface of the arms and legs, oral mucosa, scalp, nails, and genitalia. Clinically, LP can present in various forms including actinic, annular, atrophic, erosive, follicular, hypertrophic, linear, pigmented, and vesicular/bullous types. Koebnerization is common, especially in the linear form of LP. There are no specific laboratory findings or serologic markers seen in LP.
The exact cause of LP remains unknown. Clinical observations and anecdotal evidence have directed the cell-mediated immune response to insulting agents such as medications or contact allergy to metals triggering an abnormal cellular immune response. Various viral agents have been reported including hepatitis C virus, human herpesvirus, herpes simplex virus, and varicella-zoster virus.1-5 Other factors such as seasonal change and the environment may contribute to the development of LP and an increase in the incidence of LP eruption has been observed from January to July throughout the United States.6 Lichen planus also has been associated with other altered immune-related disease such as ulcerative colitis, alopecia areata, vitiligo, dermatomyositis, morphea, lichen sclerosis, and myasthenia gravis.7 Increased levels of emotional stress, particularly related to family members, often is related to the onset or aggravation of symptoms.8,9
Many drug-related LP-like and lichenoid eruptions have been reported with antihypertensive drugs, antimalarial drugs, diuretics, antidepressants, nonsteroidal anti-inflammatory drugs, antimicrobial drugs, and metals. In particular, medications such as captopril, enalapril, labetalol, propranolol, chlorothiazide, HCTZ, methyldopa, chloroquine, hydroxychloroquine, quinacrine, gold salts, penicillamine, and quinidine commonly are reported to induce lichenoid drug eruption.10
Several inflammatory papulosquamous skin conditions should be considered in the differential diagnosis before confirming the diagnosis of LP. It is important to rule out lupus erythematosus, especially if the oral mucosa and scalp are involved. In addition, erosive paraneoplastic pemphigus involving primarily the oral mucosa can resemble oral LP. Nail diseases such as psoriasis, onychomycosis, and alopecia areata should be considered as the differential diagnosis of nail disease. Genital involvement also can be seen in psoriasis and lichen sclerosus.
Treatment of LP is mainly symptomatic because of the benign nature of the disease and the high spontaneous remission rate with varying amount of time. If drugs, dental/metal implants, or underlying viral infections are the identifiable triggering factors of LP, the offending agents should be discontinued or removed. Additionally, topical or systemic treatments can be given depending on the severity of the disease, focusing mainly on symptomatic relief as well as the balance of risks and benefits associated with treatment.
Treatment options include topical and intralesional corticosteroids. Systemic medications such as oral corticosteroids and/or acitretin commonly are used in acute, severe, and disseminated cases, though treatment duration varies depending on the clinical response. Other systemic agents used to treat LP include griseofulvin, metronidazole, sulfasalazine, cyclosporine, and mycophenolate mofetil.
Phototherapy is considered an alternative therapy, especially for recalcitrant LP. UVA1 and narrowband UVB (wavelength, 311 nm) have been reported to effectively treat long-standing and therapy-resistant LP.11 In addition, a small study used the excimer laser (wavelength, 308 nm), which is well tolerated by patients, to treat focal recalcitrant oral lesions with excellent results.12 Photochemotherapy has been used with notable improvement, but the potential of carcinogenicity, especially in patients with Fitzpatrick skin types I and II, has limited its use.13
Our patient developed an unusual extensive LP eruption involving only vitiliginous skin shortly after initiation of the combined antihypertensive medication telmisartan-HCTZ, an angiotensin receptor blocker with a thiazide diuretic. Telmisartan and other angiotensin receptor blockers have not been reported to trigger LP; HCTZ is listed as one of the common drugs causing photosensitivity and LP.14,15 Although it is possible that our patient exhibited a delayed lichenoid drug eruption from the HCTZ, it is noteworthy that he did not experience a single episode of LP during his 8-year history of taking HCTZ. Instead, he developed the LP eruption shortly after the addition of telmisartan to his HCTZ antihypertensive regimen. The temporal relationship led us to direct the patient to the prescribing physician to discontinue telmisartan-HCTZ. After changing his antihypertensive medication to atenolol, the patient presented with improvement within the first month and near-complete resolution 2 months after the discontinuation of telmisartan-HCTZ.
Our patient’s LP lesions only manifested on the skin affected by vitiligo, sparing the normal-pigmented skin. Studies have demonstrated an increased ratio of CD8+ T cells to CD4+ T cells as well as increased intercellular adhesion molecule 1 at the dermal level.10,16 Both vitiligo and LP share some common histopathologic features including highly populated CD8+ T cells and intercellular adhesion molecule 1. In our case, LP was triggered on the vitiliginous skin by telmisartan. Vitiligo in combination with trauma induced by sunburn may represent the trigger that altered the cellular immune response and created the telmisartan-induced LP. As a result, the LP eruption was confined to the vitiliginous skin lesions.
Perifollicular repigmentation was observed in our patient after the LP lesions resolved; the patient’s vitiligo was unresponsive to prior treatment. The inflammatory process occurring in LP may exert and interfere in the underlying autoimmune cytotoxic effect toward the melanocytes and the melanin synthesis. It may be of interest to find out if the inflammatory response of LP has a positive influence on the effect of melanogenesis pathways or on the underlying autoimmune-related inflammatory process in vitiligo. Further studies are needed to investigate the role of immunotherapy targeting specific inflammatory pathways and the impact on the repigmentation in vitiligo.
Acknowledgment—Special thanks to Paul Chu, MD (Port Chester, New York).
- Pilli M, Zerbini A, Vescovi P, et al. Oral lichen planus pathogenesis: a role for the HCV-specific cellular immune response. Hepatology. 2002;36:1446-1452.
- De Vries HJ, van Marle J, Teunissen MB, et al. Lichen planus is associated with human herpesvirus type 7 replication and infiltration of plasmacytoid dendritic cells. Br J Dermatol. 2006;154:361-364.
- De Vries HJ, Teunissen MB, Zorgdrager F, et al. Lichen planus remission is associated with a decrease of human herpes virus type 7 protein expression in plasmacytoid dendritic cells. Arch Dermatol Res. 2007;299:213-219.
- Requena L, Kutzner H, Escalonilla P, et al. Cutaneous reactions at sites of herpes zoster scars: an expanded spectrum. Br J Dermatol. 1998;138:161-168.
- Al-Khenaizan S. Lichen planus occurring after hepatitis B vaccination: a new case. J Am Acad Dermatol. 2001;45:614-615.
- Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol. 1991;25:593-619.
- Sadr-Ashkevari S. Familial actinic lichen planus: case reports in two brothers. Arch Int Med. 2001;4:204-206.
- Manolache L, Seceleanu-Petrescu D, Benea V. Lichen planus patients and stressful events. J Eur Acad Dermatol Venereol. 2008;22:437-441.
- Mahood JM. Familial lichen planus. Arch Dermatol. 1983;119:292-294.
- Shimizu M, Higaki Y, Higaki M, et al. The role of granzyme B-expressing CD8-positive T cells in apoptosis of keratinocytes in lichen planus. Arch Dermatol Res. 1997;289:527-532.
- Bécherel PA, Bussel A, Chosidow O, et al. Extracorporeal photochemotherapy for chronic erosive lichen planus. Lancet. 1998;351:805.
- Trehan M, Taylar CR. Low-dose excimer 308-nm laser for the treatment of oral lichen planus. Arch Dermatol. 2004;140:415-420.
- Wackernagel A, Legat FJ, Hofer A, et al. Psoralen plus UVA vs. UVB-311 nm for the treatment of lichen planus. Photodermatol Photoimmunol Photomed. 2007;23:15-19.
- Fellner MJ. Lichen planus. Int J Dermatol. 1980;19:71-75.
- Moore DE. Drug-induced cutaneous photosensitivity: incidence, mechanism, prevention and management. Drug Saf. 2002;25:345-372.
- Ongenae K, Van Geel N, Naeyaert JM. Evidence for an autoimmune pathogenesis of vitiligo. Pigment Cell Res. 2003;16:90-100.
To the Editor:
A 39-year-old man with a history of hypertension and vitiligo presented with a rapid-onset, generalized, pruritic rash covering the body of 4 weeks’ duration. He reported that the rash progressively worsened after developing mild sunburn. The patient stated that the rash was extremely pruritic with a burning sensation and was tender to touch. He was treated with betamethasone valerate cream 0.1% by an outside physician and an over-the-counter anti-itch lotion with no notable improvement. His only medication was telmisartan-hydrochlorothiazide (HCTZ) for hypertension. He denied any drug allergies.
Physical examination revealed multiple discrete and coalescent planar erythematous papules and plaques involving only the depigmented vitiliginous skin of the forehead, eyelids, and nape of the neck (Figure 1A), and confluent on the lateral aspect of the bilateral forearms (Figure 1B), dorsal aspect of the right hand, and bilateral dorsi of the feet. Wickham striae were noted on the lips (Figure 1C). A clinical diagnosis of lichen planus (LP) was made. The patient initially was prescribed halobetasol propionate ointment 0.05% twice daily. He reported notable relief of pruritus with reduction of overall symptoms and new lesion formation.
A 4-mm punch biopsy was performed on the left forearm. Histopathology revealed LP. Microscopic examination of the hematoxylin and eosin–stained specimen revealed a bandlike lymphohistiocytic infiltrate that extended across the papillary dermis, focally obscuring the dermoepidermal junction where there were vacuolar changes and colloid bodies. The epidermis showed sawtooth rete ridges, wedge-shaped foci of hypergranulosis, and compact hyperkeratosis (Figure 2).
On further questioning during follow-up, the patient revealed that his hypertensive medication was changed from HCTZ, which he had been taking for the last 8 years, to the combination antihypertensive medication telmisartan-HCTZ before the onset of the skin eruption. Due to the temporal relationship between the new medication and onset of the eruption, the clinical impression was highly suspicious for drug-induced eruptive LP with Köbner phenomenon caused by the recent sunburn. Systemic workup for underlying causes of LP was negative. Laboratory tests revealed normal complete blood cell counts. The hepatitis panel included hepatitis A antibodies; hepatitis B surface, e antigen, and core antibodies; hepatitis B surface antigen and e antibodies; hepatitis C antibodies; and antinuclear antibodies, which were all negative.
The patient continued to develop new pruritic papules clinically consistent with LP. He was instructed to return to his primary care physician to change the telmisartan-HCTZ to a different class of antihypertensive medication. His medication was changed to atenolol. The patient also was instructed to continue the halobetasol propionate ointment 0.05% twice daily to the affected areas.
The patient returned for a follow-up visit 1 month later and reported notable improvement in pruritus and near-complete resolution of the LP after discontinuation of telmisartan-HCTZ. He also noted some degree of perifollicular repigmentation of the vitiliginous skin that had been unresponsive to prior therapy (Figure 3).
Lichen planus is a pruritic and inflammatory papulosquamous skin condition that presents as scaly, flat-topped, violaceous, polygonal-shaped papules commonly involving the flexor surface of the arms and legs, oral mucosa, scalp, nails, and genitalia. Clinically, LP can present in various forms including actinic, annular, atrophic, erosive, follicular, hypertrophic, linear, pigmented, and vesicular/bullous types. Koebnerization is common, especially in the linear form of LP. There are no specific laboratory findings or serologic markers seen in LP.
The exact cause of LP remains unknown. Clinical observations and anecdotal evidence have directed the cell-mediated immune response to insulting agents such as medications or contact allergy to metals triggering an abnormal cellular immune response. Various viral agents have been reported including hepatitis C virus, human herpesvirus, herpes simplex virus, and varicella-zoster virus.1-5 Other factors such as seasonal change and the environment may contribute to the development of LP and an increase in the incidence of LP eruption has been observed from January to July throughout the United States.6 Lichen planus also has been associated with other altered immune-related disease such as ulcerative colitis, alopecia areata, vitiligo, dermatomyositis, morphea, lichen sclerosis, and myasthenia gravis.7 Increased levels of emotional stress, particularly related to family members, often is related to the onset or aggravation of symptoms.8,9
Many drug-related LP-like and lichenoid eruptions have been reported with antihypertensive drugs, antimalarial drugs, diuretics, antidepressants, nonsteroidal anti-inflammatory drugs, antimicrobial drugs, and metals. In particular, medications such as captopril, enalapril, labetalol, propranolol, chlorothiazide, HCTZ, methyldopa, chloroquine, hydroxychloroquine, quinacrine, gold salts, penicillamine, and quinidine commonly are reported to induce lichenoid drug eruption.10
Several inflammatory papulosquamous skin conditions should be considered in the differential diagnosis before confirming the diagnosis of LP. It is important to rule out lupus erythematosus, especially if the oral mucosa and scalp are involved. In addition, erosive paraneoplastic pemphigus involving primarily the oral mucosa can resemble oral LP. Nail diseases such as psoriasis, onychomycosis, and alopecia areata should be considered as the differential diagnosis of nail disease. Genital involvement also can be seen in psoriasis and lichen sclerosus.
Treatment of LP is mainly symptomatic because of the benign nature of the disease and the high spontaneous remission rate with varying amount of time. If drugs, dental/metal implants, or underlying viral infections are the identifiable triggering factors of LP, the offending agents should be discontinued or removed. Additionally, topical or systemic treatments can be given depending on the severity of the disease, focusing mainly on symptomatic relief as well as the balance of risks and benefits associated with treatment.
Treatment options include topical and intralesional corticosteroids. Systemic medications such as oral corticosteroids and/or acitretin commonly are used in acute, severe, and disseminated cases, though treatment duration varies depending on the clinical response. Other systemic agents used to treat LP include griseofulvin, metronidazole, sulfasalazine, cyclosporine, and mycophenolate mofetil.
Phototherapy is considered an alternative therapy, especially for recalcitrant LP. UVA1 and narrowband UVB (wavelength, 311 nm) have been reported to effectively treat long-standing and therapy-resistant LP.11 In addition, a small study used the excimer laser (wavelength, 308 nm), which is well tolerated by patients, to treat focal recalcitrant oral lesions with excellent results.12 Photochemotherapy has been used with notable improvement, but the potential of carcinogenicity, especially in patients with Fitzpatrick skin types I and II, has limited its use.13
Our patient developed an unusual extensive LP eruption involving only vitiliginous skin shortly after initiation of the combined antihypertensive medication telmisartan-HCTZ, an angiotensin receptor blocker with a thiazide diuretic. Telmisartan and other angiotensin receptor blockers have not been reported to trigger LP; HCTZ is listed as one of the common drugs causing photosensitivity and LP.14,15 Although it is possible that our patient exhibited a delayed lichenoid drug eruption from the HCTZ, it is noteworthy that he did not experience a single episode of LP during his 8-year history of taking HCTZ. Instead, he developed the LP eruption shortly after the addition of telmisartan to his HCTZ antihypertensive regimen. The temporal relationship led us to direct the patient to the prescribing physician to discontinue telmisartan-HCTZ. After changing his antihypertensive medication to atenolol, the patient presented with improvement within the first month and near-complete resolution 2 months after the discontinuation of telmisartan-HCTZ.
Our patient’s LP lesions only manifested on the skin affected by vitiligo, sparing the normal-pigmented skin. Studies have demonstrated an increased ratio of CD8+ T cells to CD4+ T cells as well as increased intercellular adhesion molecule 1 at the dermal level.10,16 Both vitiligo and LP share some common histopathologic features including highly populated CD8+ T cells and intercellular adhesion molecule 1. In our case, LP was triggered on the vitiliginous skin by telmisartan. Vitiligo in combination with trauma induced by sunburn may represent the trigger that altered the cellular immune response and created the telmisartan-induced LP. As a result, the LP eruption was confined to the vitiliginous skin lesions.
Perifollicular repigmentation was observed in our patient after the LP lesions resolved; the patient’s vitiligo was unresponsive to prior treatment. The inflammatory process occurring in LP may exert and interfere in the underlying autoimmune cytotoxic effect toward the melanocytes and the melanin synthesis. It may be of interest to find out if the inflammatory response of LP has a positive influence on the effect of melanogenesis pathways or on the underlying autoimmune-related inflammatory process in vitiligo. Further studies are needed to investigate the role of immunotherapy targeting specific inflammatory pathways and the impact on the repigmentation in vitiligo.
Acknowledgment—Special thanks to Paul Chu, MD (Port Chester, New York).
To the Editor:
A 39-year-old man with a history of hypertension and vitiligo presented with a rapid-onset, generalized, pruritic rash covering the body of 4 weeks’ duration. He reported that the rash progressively worsened after developing mild sunburn. The patient stated that the rash was extremely pruritic with a burning sensation and was tender to touch. He was treated with betamethasone valerate cream 0.1% by an outside physician and an over-the-counter anti-itch lotion with no notable improvement. His only medication was telmisartan-hydrochlorothiazide (HCTZ) for hypertension. He denied any drug allergies.
Physical examination revealed multiple discrete and coalescent planar erythematous papules and plaques involving only the depigmented vitiliginous skin of the forehead, eyelids, and nape of the neck (Figure 1A), and confluent on the lateral aspect of the bilateral forearms (Figure 1B), dorsal aspect of the right hand, and bilateral dorsi of the feet. Wickham striae were noted on the lips (Figure 1C). A clinical diagnosis of lichen planus (LP) was made. The patient initially was prescribed halobetasol propionate ointment 0.05% twice daily. He reported notable relief of pruritus with reduction of overall symptoms and new lesion formation.
A 4-mm punch biopsy was performed on the left forearm. Histopathology revealed LP. Microscopic examination of the hematoxylin and eosin–stained specimen revealed a bandlike lymphohistiocytic infiltrate that extended across the papillary dermis, focally obscuring the dermoepidermal junction where there were vacuolar changes and colloid bodies. The epidermis showed sawtooth rete ridges, wedge-shaped foci of hypergranulosis, and compact hyperkeratosis (Figure 2).
On further questioning during follow-up, the patient revealed that his hypertensive medication was changed from HCTZ, which he had been taking for the last 8 years, to the combination antihypertensive medication telmisartan-HCTZ before the onset of the skin eruption. Due to the temporal relationship between the new medication and onset of the eruption, the clinical impression was highly suspicious for drug-induced eruptive LP with Köbner phenomenon caused by the recent sunburn. Systemic workup for underlying causes of LP was negative. Laboratory tests revealed normal complete blood cell counts. The hepatitis panel included hepatitis A antibodies; hepatitis B surface, e antigen, and core antibodies; hepatitis B surface antigen and e antibodies; hepatitis C antibodies; and antinuclear antibodies, which were all negative.
The patient continued to develop new pruritic papules clinically consistent with LP. He was instructed to return to his primary care physician to change the telmisartan-HCTZ to a different class of antihypertensive medication. His medication was changed to atenolol. The patient also was instructed to continue the halobetasol propionate ointment 0.05% twice daily to the affected areas.
The patient returned for a follow-up visit 1 month later and reported notable improvement in pruritus and near-complete resolution of the LP after discontinuation of telmisartan-HCTZ. He also noted some degree of perifollicular repigmentation of the vitiliginous skin that had been unresponsive to prior therapy (Figure 3).
Lichen planus is a pruritic and inflammatory papulosquamous skin condition that presents as scaly, flat-topped, violaceous, polygonal-shaped papules commonly involving the flexor surface of the arms and legs, oral mucosa, scalp, nails, and genitalia. Clinically, LP can present in various forms including actinic, annular, atrophic, erosive, follicular, hypertrophic, linear, pigmented, and vesicular/bullous types. Koebnerization is common, especially in the linear form of LP. There are no specific laboratory findings or serologic markers seen in LP.
The exact cause of LP remains unknown. Clinical observations and anecdotal evidence have directed the cell-mediated immune response to insulting agents such as medications or contact allergy to metals triggering an abnormal cellular immune response. Various viral agents have been reported including hepatitis C virus, human herpesvirus, herpes simplex virus, and varicella-zoster virus.1-5 Other factors such as seasonal change and the environment may contribute to the development of LP and an increase in the incidence of LP eruption has been observed from January to July throughout the United States.6 Lichen planus also has been associated with other altered immune-related disease such as ulcerative colitis, alopecia areata, vitiligo, dermatomyositis, morphea, lichen sclerosis, and myasthenia gravis.7 Increased levels of emotional stress, particularly related to family members, often is related to the onset or aggravation of symptoms.8,9
Many drug-related LP-like and lichenoid eruptions have been reported with antihypertensive drugs, antimalarial drugs, diuretics, antidepressants, nonsteroidal anti-inflammatory drugs, antimicrobial drugs, and metals. In particular, medications such as captopril, enalapril, labetalol, propranolol, chlorothiazide, HCTZ, methyldopa, chloroquine, hydroxychloroquine, quinacrine, gold salts, penicillamine, and quinidine commonly are reported to induce lichenoid drug eruption.10
Several inflammatory papulosquamous skin conditions should be considered in the differential diagnosis before confirming the diagnosis of LP. It is important to rule out lupus erythematosus, especially if the oral mucosa and scalp are involved. In addition, erosive paraneoplastic pemphigus involving primarily the oral mucosa can resemble oral LP. Nail diseases such as psoriasis, onychomycosis, and alopecia areata should be considered as the differential diagnosis of nail disease. Genital involvement also can be seen in psoriasis and lichen sclerosus.
Treatment of LP is mainly symptomatic because of the benign nature of the disease and the high spontaneous remission rate with varying amount of time. If drugs, dental/metal implants, or underlying viral infections are the identifiable triggering factors of LP, the offending agents should be discontinued or removed. Additionally, topical or systemic treatments can be given depending on the severity of the disease, focusing mainly on symptomatic relief as well as the balance of risks and benefits associated with treatment.
Treatment options include topical and intralesional corticosteroids. Systemic medications such as oral corticosteroids and/or acitretin commonly are used in acute, severe, and disseminated cases, though treatment duration varies depending on the clinical response. Other systemic agents used to treat LP include griseofulvin, metronidazole, sulfasalazine, cyclosporine, and mycophenolate mofetil.
Phototherapy is considered an alternative therapy, especially for recalcitrant LP. UVA1 and narrowband UVB (wavelength, 311 nm) have been reported to effectively treat long-standing and therapy-resistant LP.11 In addition, a small study used the excimer laser (wavelength, 308 nm), which is well tolerated by patients, to treat focal recalcitrant oral lesions with excellent results.12 Photochemotherapy has been used with notable improvement, but the potential of carcinogenicity, especially in patients with Fitzpatrick skin types I and II, has limited its use.13
Our patient developed an unusual extensive LP eruption involving only vitiliginous skin shortly after initiation of the combined antihypertensive medication telmisartan-HCTZ, an angiotensin receptor blocker with a thiazide diuretic. Telmisartan and other angiotensin receptor blockers have not been reported to trigger LP; HCTZ is listed as one of the common drugs causing photosensitivity and LP.14,15 Although it is possible that our patient exhibited a delayed lichenoid drug eruption from the HCTZ, it is noteworthy that he did not experience a single episode of LP during his 8-year history of taking HCTZ. Instead, he developed the LP eruption shortly after the addition of telmisartan to his HCTZ antihypertensive regimen. The temporal relationship led us to direct the patient to the prescribing physician to discontinue telmisartan-HCTZ. After changing his antihypertensive medication to atenolol, the patient presented with improvement within the first month and near-complete resolution 2 months after the discontinuation of telmisartan-HCTZ.
Our patient’s LP lesions only manifested on the skin affected by vitiligo, sparing the normal-pigmented skin. Studies have demonstrated an increased ratio of CD8+ T cells to CD4+ T cells as well as increased intercellular adhesion molecule 1 at the dermal level.10,16 Both vitiligo and LP share some common histopathologic features including highly populated CD8+ T cells and intercellular adhesion molecule 1. In our case, LP was triggered on the vitiliginous skin by telmisartan. Vitiligo in combination with trauma induced by sunburn may represent the trigger that altered the cellular immune response and created the telmisartan-induced LP. As a result, the LP eruption was confined to the vitiliginous skin lesions.
Perifollicular repigmentation was observed in our patient after the LP lesions resolved; the patient’s vitiligo was unresponsive to prior treatment. The inflammatory process occurring in LP may exert and interfere in the underlying autoimmune cytotoxic effect toward the melanocytes and the melanin synthesis. It may be of interest to find out if the inflammatory response of LP has a positive influence on the effect of melanogenesis pathways or on the underlying autoimmune-related inflammatory process in vitiligo. Further studies are needed to investigate the role of immunotherapy targeting specific inflammatory pathways and the impact on the repigmentation in vitiligo.
Acknowledgment—Special thanks to Paul Chu, MD (Port Chester, New York).
- Pilli M, Zerbini A, Vescovi P, et al. Oral lichen planus pathogenesis: a role for the HCV-specific cellular immune response. Hepatology. 2002;36:1446-1452.
- De Vries HJ, van Marle J, Teunissen MB, et al. Lichen planus is associated with human herpesvirus type 7 replication and infiltration of plasmacytoid dendritic cells. Br J Dermatol. 2006;154:361-364.
- De Vries HJ, Teunissen MB, Zorgdrager F, et al. Lichen planus remission is associated with a decrease of human herpes virus type 7 protein expression in plasmacytoid dendritic cells. Arch Dermatol Res. 2007;299:213-219.
- Requena L, Kutzner H, Escalonilla P, et al. Cutaneous reactions at sites of herpes zoster scars: an expanded spectrum. Br J Dermatol. 1998;138:161-168.
- Al-Khenaizan S. Lichen planus occurring after hepatitis B vaccination: a new case. J Am Acad Dermatol. 2001;45:614-615.
- Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol. 1991;25:593-619.
- Sadr-Ashkevari S. Familial actinic lichen planus: case reports in two brothers. Arch Int Med. 2001;4:204-206.
- Manolache L, Seceleanu-Petrescu D, Benea V. Lichen planus patients and stressful events. J Eur Acad Dermatol Venereol. 2008;22:437-441.
- Mahood JM. Familial lichen planus. Arch Dermatol. 1983;119:292-294.
- Shimizu M, Higaki Y, Higaki M, et al. The role of granzyme B-expressing CD8-positive T cells in apoptosis of keratinocytes in lichen planus. Arch Dermatol Res. 1997;289:527-532.
- Bécherel PA, Bussel A, Chosidow O, et al. Extracorporeal photochemotherapy for chronic erosive lichen planus. Lancet. 1998;351:805.
- Trehan M, Taylar CR. Low-dose excimer 308-nm laser for the treatment of oral lichen planus. Arch Dermatol. 2004;140:415-420.
- Wackernagel A, Legat FJ, Hofer A, et al. Psoralen plus UVA vs. UVB-311 nm for the treatment of lichen planus. Photodermatol Photoimmunol Photomed. 2007;23:15-19.
- Fellner MJ. Lichen planus. Int J Dermatol. 1980;19:71-75.
- Moore DE. Drug-induced cutaneous photosensitivity: incidence, mechanism, prevention and management. Drug Saf. 2002;25:345-372.
- Ongenae K, Van Geel N, Naeyaert JM. Evidence for an autoimmune pathogenesis of vitiligo. Pigment Cell Res. 2003;16:90-100.
- Pilli M, Zerbini A, Vescovi P, et al. Oral lichen planus pathogenesis: a role for the HCV-specific cellular immune response. Hepatology. 2002;36:1446-1452.
- De Vries HJ, van Marle J, Teunissen MB, et al. Lichen planus is associated with human herpesvirus type 7 replication and infiltration of plasmacytoid dendritic cells. Br J Dermatol. 2006;154:361-364.
- De Vries HJ, Teunissen MB, Zorgdrager F, et al. Lichen planus remission is associated with a decrease of human herpes virus type 7 protein expression in plasmacytoid dendritic cells. Arch Dermatol Res. 2007;299:213-219.
- Requena L, Kutzner H, Escalonilla P, et al. Cutaneous reactions at sites of herpes zoster scars: an expanded spectrum. Br J Dermatol. 1998;138:161-168.
- Al-Khenaizan S. Lichen planus occurring after hepatitis B vaccination: a new case. J Am Acad Dermatol. 2001;45:614-615.
- Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol. 1991;25:593-619.
- Sadr-Ashkevari S. Familial actinic lichen planus: case reports in two brothers. Arch Int Med. 2001;4:204-206.
- Manolache L, Seceleanu-Petrescu D, Benea V. Lichen planus patients and stressful events. J Eur Acad Dermatol Venereol. 2008;22:437-441.
- Mahood JM. Familial lichen planus. Arch Dermatol. 1983;119:292-294.
- Shimizu M, Higaki Y, Higaki M, et al. The role of granzyme B-expressing CD8-positive T cells in apoptosis of keratinocytes in lichen planus. Arch Dermatol Res. 1997;289:527-532.
- Bécherel PA, Bussel A, Chosidow O, et al. Extracorporeal photochemotherapy for chronic erosive lichen planus. Lancet. 1998;351:805.
- Trehan M, Taylar CR. Low-dose excimer 308-nm laser for the treatment of oral lichen planus. Arch Dermatol. 2004;140:415-420.
- Wackernagel A, Legat FJ, Hofer A, et al. Psoralen plus UVA vs. UVB-311 nm for the treatment of lichen planus. Photodermatol Photoimmunol Photomed. 2007;23:15-19.
- Fellner MJ. Lichen planus. Int J Dermatol. 1980;19:71-75.
- Moore DE. Drug-induced cutaneous photosensitivity: incidence, mechanism, prevention and management. Drug Saf. 2002;25:345-372.
- Ongenae K, Van Geel N, Naeyaert JM. Evidence for an autoimmune pathogenesis of vitiligo. Pigment Cell Res. 2003;16:90-100.
Practice Points
- Lichen planus (LP) is a T-cell–mediated autoimmune disease that affects the skin and often the mucosa, nails, and scalp.
- The etiology of LP is unknown. It can be induced by a variety of medications and may spread through the isomorphic phenomenon.
- Immune factors play a role in the development of LP, drug-induced LP, and vitiligo.
Prescribing the landmark hemangioma drug: The challenges and the benefits
For Beth Drolet, MD, a pediatric dermatologist in Wisconsin, the tremendous impact oral propranolol has had on the treatment of severe infantile hemangioma is written on the faces of children diagnosed with the condition in recent years.
“You can tell which drugs the kids were on by their age,” said Dr. Drolet, professor of dermatology and pediatrics at the Medical College of Wisconsin, Milwaukee. “If they were born before 2008, before we used this medication, those kids have had multiple surgeries and are still not looking that good. But we rarely see that in the kids born after.”

Still, it is possible for dermatologists to successfully treat their smallest patients with oral propranolol, according to Dr. Drolet and Ilona J. Frieden, MD, professor of dermatology and pediatrics at the University of California, San Francisco.
In interviews, the two pediatric dermatologists spoke about the challenges and benefits of treating hemangioma patients with oral propranolol solution, which was approved by the Food and Drug Administration in 2014 for “proliferating infantile hemangioma requiring systemic therapy.” It is the only FDA-approved systemic treatment for this indication.

The oral form of the drug was used off label to treat patients with hemangioma after a French dermatologist discovered in 2007 that it could effectively treat the condition. A topical form of propranolol is also used for hemangiomas that do not require systemic treatment.
Prior to about a decade ago, Dr. Drolet said, steroids were used to treat severe hemangiomas with limited success.
In general, infantile hemangiomas “have a natural course of gradually involuting even without treatment,” Dr. Frieden noted. But the most severe cases can produce functional impairment, scarring, and anatomic distortion.
Dr. Drolet said she considers treatment if hemangioma threatens a vital function (hearing, sight, breathing) or can lead to pain, infection, or scarring.
One challenge for dermatologists is that standard of care treatment with oral propranolol requires in-office cardiac monitoring, especially as the dose is increased over the first week or two of treatment.
“I don’t think most dermatologists are comfortable taking a heart rate and blood pressure in an infant,” said Dr. Drolet, who is director of the birthmarks and vascular anomalies section at Children’s Hospital of Wisconsin, Milwaukee. Instead, they tend to refer patients to a pediatrician or pediatric cardiologist.
Her clinic hired a cardiac nurse to train the staff in how to take heart rate and blood pressure in babies. “Partnering with cardiology was really important for us,” she commented. “We worked really closely with our pediatric cardiology team to gain that expertise for our staff to assess that. You have to be pretty comfortable with it. If you’re not, you’re going to have to find someone else.”
Another option for dermatologists, Dr. Frieden said, is to focus on heart rate alone since blood pressure in infants is difficult to measure. “It’s not FDA sanctioned, but many people seem to do that and it’s OK,” she said.
Dr. Frieden and Dr. Drolet provided the following recommendations about treating babies with oral propranolol:
• Caution parents about side effects. Cardiac side effects have been “extraordinarily rare,” Dr. Drolet said. “We have seen problems with wheezing and, very rarely, severe hypoglycemia,” which can be prevented by educating the family. While it’s uncommon for the medication alone to produce wheezing, this may occur when a respiratory infection and propranolol combine to stress the body, she noted.
In some cases, physicians prescribe albuterol for wheezing without realizing that it will interact with propranolol, she added. “One is a beta-blocker, and the other is a beta-antagonist. They completely cancel each other out.”
To prevent hypoglycemia, Dr. Frieden said she recommends that children be fed every 6 hours if they’re under 6 months old or every 8 hours if they’re over 6 months of age. And Dr. Drolet said she advises parents to stop propranolol when their infants are sick.
A major focus of an educational video provided by Dr. Drolet’s clinic is advising parents “to stop the medication if the infant is not eating regularly, vomiting, or has diarrhea. It interferes with how you respond to low blood sugar if you’re not eating,” she said. “That surprised us. Now that we’ve been teaching parents about when to call us, that’s been pretty preventable.”
Minor side effects include cold hands and feet and sleep disturbances such as sleepiness and apparent nightmares, Dr. Frieden pointed out.
• Monitor guidelines regarding safety and protocols. “Over time, we’re getting more and more expertise,” Dr. Drolet said. For example, her clinic no longer performs ECGs on babies who take the medication because research has suggested they are not needed.
• Spend time developing an education program for parents. Dr. Drolet’s clinic provides the educational video to teach parents about how oral propranolol is used. “We haven’t done that for any other drugs,” she said. “But we want to make sure we aren’t overdosing it. We’ve been very careful about our parent education to prevent that.”
Guidelines on the diagnosis and management of infantile hemangioma were published in 2015 in Pediatrics (2015 Oct;136[4]:e1060-104).
Dr. Frieden has consulted for Pierre Fabre Dermatologie, the manufacturer of the oral propranolol product, marketed as Hemangeol. Dr. Drolet has received an investigator-initiated grant from the company.
For Beth Drolet, MD, a pediatric dermatologist in Wisconsin, the tremendous impact oral propranolol has had on the treatment of severe infantile hemangioma is written on the faces of children diagnosed with the condition in recent years.
“You can tell which drugs the kids were on by their age,” said Dr. Drolet, professor of dermatology and pediatrics at the Medical College of Wisconsin, Milwaukee. “If they were born before 2008, before we used this medication, those kids have had multiple surgeries and are still not looking that good. But we rarely see that in the kids born after.”
Still, it is possible for dermatologists to successfully treat their smallest patients with oral propranolol, according to Dr. Drolet and Ilona J. Frieden, MD, professor of dermatology and pediatrics at the University of California, San Francisco.
In interviews, the two pediatric dermatologists spoke about the challenges and benefits of treating hemangioma patients with oral propranolol solution, which was approved by the Food and Drug Administration in 2014 for “proliferating infantile hemangioma requiring systemic therapy.” It is the only FDA-approved systemic treatment for this indication.
The oral form of the drug was used off label to treat patients with hemangioma after a French dermatologist discovered in 2007 that it could effectively treat the condition. A topical form of propranolol is also used for hemangiomas that do not require systemic treatment.
Prior to about a decade ago, Dr. Drolet said, steroids were used to treat severe hemangiomas with limited success.
In general, infantile hemangiomas “have a natural course of gradually involuting even without treatment,” Dr. Frieden noted. But the most severe cases can produce functional impairment, scarring, and anatomic distortion.
Dr. Drolet said she considers treatment if hemangioma threatens a vital function (hearing, sight, breathing) or can lead to pain, infection, or scarring.
One challenge for dermatologists is that standard of care treatment with oral propranolol requires in-office cardiac monitoring, especially as the dose is increased over the first week or two of treatment.
“I don’t think most dermatologists are comfortable taking a heart rate and blood pressure in an infant,” said Dr. Drolet, who is director of the birthmarks and vascular anomalies section at Children’s Hospital of Wisconsin, Milwaukee. Instead, they tend to refer patients to a pediatrician or pediatric cardiologist.
Her clinic hired a cardiac nurse to train the staff in how to take heart rate and blood pressure in babies. “Partnering with cardiology was really important for us,” she commented. “We worked really closely with our pediatric cardiology team to gain that expertise for our staff to assess that. You have to be pretty comfortable with it. If you’re not, you’re going to have to find someone else.”
Another option for dermatologists, Dr. Frieden said, is to focus on heart rate alone since blood pressure in infants is difficult to measure. “It’s not FDA sanctioned, but many people seem to do that and it’s OK,” she said.
Dr. Frieden and Dr. Drolet provided the following recommendations about treating babies with oral propranolol:
• Caution parents about side effects. Cardiac side effects have been “extraordinarily rare,” Dr. Drolet said. “We have seen problems with wheezing and, very rarely, severe hypoglycemia,” which can be prevented by educating the family. While it’s uncommon for the medication alone to produce wheezing, this may occur when a respiratory infection and propranolol combine to stress the body, she noted.
In some cases, physicians prescribe albuterol for wheezing without realizing that it will interact with propranolol, she added. “One is a beta-blocker, and the other is a beta-antagonist. They completely cancel each other out.”
To prevent hypoglycemia, Dr. Frieden said she recommends that children be fed every 6 hours if they’re under 6 months old or every 8 hours if they’re over 6 months of age. And Dr. Drolet said she advises parents to stop propranolol when their infants are sick.
A major focus of an educational video provided by Dr. Drolet’s clinic is advising parents “to stop the medication if the infant is not eating regularly, vomiting, or has diarrhea. It interferes with how you respond to low blood sugar if you’re not eating,” she said. “That surprised us. Now that we’ve been teaching parents about when to call us, that’s been pretty preventable.”
Minor side effects include cold hands and feet and sleep disturbances such as sleepiness and apparent nightmares, Dr. Frieden pointed out.
• Monitor guidelines regarding safety and protocols. “Over time, we’re getting more and more expertise,” Dr. Drolet said. For example, her clinic no longer performs ECGs on babies who take the medication because research has suggested they are not needed.
• Spend time developing an education program for parents. Dr. Drolet’s clinic provides the educational video to teach parents about how oral propranolol is used. “We haven’t done that for any other drugs,” she said. “But we want to make sure we aren’t overdosing it. We’ve been very careful about our parent education to prevent that.”
Guidelines on the diagnosis and management of infantile hemangioma were published in 2015 in Pediatrics (2015 Oct;136[4]:e1060-104).
Dr. Frieden has consulted for Pierre Fabre Dermatologie, the manufacturer of the oral propranolol product, marketed as Hemangeol. Dr. Drolet has received an investigator-initiated grant from the company.
For Beth Drolet, MD, a pediatric dermatologist in Wisconsin, the tremendous impact oral propranolol has had on the treatment of severe infantile hemangioma is written on the faces of children diagnosed with the condition in recent years.
“You can tell which drugs the kids were on by their age,” said Dr. Drolet, professor of dermatology and pediatrics at the Medical College of Wisconsin, Milwaukee. “If they were born before 2008, before we used this medication, those kids have had multiple surgeries and are still not looking that good. But we rarely see that in the kids born after.”
Still, it is possible for dermatologists to successfully treat their smallest patients with oral propranolol, according to Dr. Drolet and Ilona J. Frieden, MD, professor of dermatology and pediatrics at the University of California, San Francisco.
In interviews, the two pediatric dermatologists spoke about the challenges and benefits of treating hemangioma patients with oral propranolol solution, which was approved by the Food and Drug Administration in 2014 for “proliferating infantile hemangioma requiring systemic therapy.” It is the only FDA-approved systemic treatment for this indication.
The oral form of the drug was used off label to treat patients with hemangioma after a French dermatologist discovered in 2007 that it could effectively treat the condition. A topical form of propranolol is also used for hemangiomas that do not require systemic treatment.
Prior to about a decade ago, Dr. Drolet said, steroids were used to treat severe hemangiomas with limited success.
In general, infantile hemangiomas “have a natural course of gradually involuting even without treatment,” Dr. Frieden noted. But the most severe cases can produce functional impairment, scarring, and anatomic distortion.
Dr. Drolet said she considers treatment if hemangioma threatens a vital function (hearing, sight, breathing) or can lead to pain, infection, or scarring.
One challenge for dermatologists is that standard of care treatment with oral propranolol requires in-office cardiac monitoring, especially as the dose is increased over the first week or two of treatment.
“I don’t think most dermatologists are comfortable taking a heart rate and blood pressure in an infant,” said Dr. Drolet, who is director of the birthmarks and vascular anomalies section at Children’s Hospital of Wisconsin, Milwaukee. Instead, they tend to refer patients to a pediatrician or pediatric cardiologist.
Her clinic hired a cardiac nurse to train the staff in how to take heart rate and blood pressure in babies. “Partnering with cardiology was really important for us,” she commented. “We worked really closely with our pediatric cardiology team to gain that expertise for our staff to assess that. You have to be pretty comfortable with it. If you’re not, you’re going to have to find someone else.”
Another option for dermatologists, Dr. Frieden said, is to focus on heart rate alone since blood pressure in infants is difficult to measure. “It’s not FDA sanctioned, but many people seem to do that and it’s OK,” she said.
Dr. Frieden and Dr. Drolet provided the following recommendations about treating babies with oral propranolol:
• Caution parents about side effects. Cardiac side effects have been “extraordinarily rare,” Dr. Drolet said. “We have seen problems with wheezing and, very rarely, severe hypoglycemia,” which can be prevented by educating the family. While it’s uncommon for the medication alone to produce wheezing, this may occur when a respiratory infection and propranolol combine to stress the body, she noted.
In some cases, physicians prescribe albuterol for wheezing without realizing that it will interact with propranolol, she added. “One is a beta-blocker, and the other is a beta-antagonist. They completely cancel each other out.”
To prevent hypoglycemia, Dr. Frieden said she recommends that children be fed every 6 hours if they’re under 6 months old or every 8 hours if they’re over 6 months of age. And Dr. Drolet said she advises parents to stop propranolol when their infants are sick.
A major focus of an educational video provided by Dr. Drolet’s clinic is advising parents “to stop the medication if the infant is not eating regularly, vomiting, or has diarrhea. It interferes with how you respond to low blood sugar if you’re not eating,” she said. “That surprised us. Now that we’ve been teaching parents about when to call us, that’s been pretty preventable.”
Minor side effects include cold hands and feet and sleep disturbances such as sleepiness and apparent nightmares, Dr. Frieden pointed out.
• Monitor guidelines regarding safety and protocols. “Over time, we’re getting more and more expertise,” Dr. Drolet said. For example, her clinic no longer performs ECGs on babies who take the medication because research has suggested they are not needed.
• Spend time developing an education program for parents. Dr. Drolet’s clinic provides the educational video to teach parents about how oral propranolol is used. “We haven’t done that for any other drugs,” she said. “But we want to make sure we aren’t overdosing it. We’ve been very careful about our parent education to prevent that.”
Guidelines on the diagnosis and management of infantile hemangioma were published in 2015 in Pediatrics (2015 Oct;136[4]:e1060-104).
Dr. Frieden has consulted for Pierre Fabre Dermatologie, the manufacturer of the oral propranolol product, marketed as Hemangeol. Dr. Drolet has received an investigator-initiated grant from the company.