User login
Treatment for hepatitis C reduces risk of Parkinson’s disease
, according to a cohort study published online June 5 in JAMA Neurology. The results provide evidence that hepatitis C virus is a risk factor for Parkinson’s disease.
In the past several years, epidemiologic studies have suggested an association between hepatitis C virus infection and Parkinson’s disease. A study published in 2017, however, found no association between the two. In addition, these investigations did not consider antiviral therapy as a potential modifying factor.
Wey-Yil Lin, MD, a neurologist at Landseed International Hospital in Taoyuan, Taiwan, and colleagues examined claims data from the Taiwan National Health Insurance Research Database to identify the risk of incident Parkinson’s disease in patients with hepatitis C virus infection who received antiviral treatment, compared with those who did not receive treatment.
The investigators selected all patients with a new diagnosis of hepatitis C virus infection with or without hepatitis from January 1, 2003, to December 31, 2013. They excluded patients who were aged 20 years or younger; had Parkinson’s disease, dementia, or stroke; or had had major hepatic diseases on the index date. To ensure that treated patients had had an effective course of therapy, the researchers excluded patients who were lost to follow-up within 6 months of the index date, received antiviral therapy for fewer than 16 weeks, or developed Parkinson’s disease within 6 months of the index date.
The primary outcome was incident Parkinson’s disease. Dr. Lin and colleagues excluded participants with a diagnosis of stroke and dementia before the index date to reduce the possibility of enrolling participants with secondary and atypical parkinsonism.
To minimize the potential selection bias to which observational studies are subject, the investigators performed propensity score matching with sex, age, comorbidities, and medication as covariates. This method was intended to create treated and untreated cohorts with comparable characteristics.
Dr. Lin and colleagues included 188,152 patients in their analysis. After matching, each group included 39,936 participants. In the group that received antiviral treatment, 45.0% of participants were female, and mean age was 52.8 years. In the untreated group, 44.4% of participants were female, and mean age was 52.5 years.
The incidence density of Parkinson’s disease per 1,000 person-years was 1.00 in the treated group and 1.39 in the untreated group. The difference in risk of Parkinson’s disease between the treated and untreated groups was statistically significant at year 5 of follow-up (hazard ratio [HR], 0.75) and at the end of the cohort (HR, 0.71). The risk did not differ significantly at year 1 and year 3, however. A subgroup analysis found a greater benefit of antiviral therapy among patients who concurrently used dihydropyridine calcium channel blockers.
“To our knowledge, this is the first cohort study to investigate the association between antiviral therapy and risk of Parkinson’s disease in patients with chronic hepatitis C viral infection,” said Dr. Lin and colleagues. Although it is possible that interferon-based antiviral therapy directly protected against the development of Parkinson’s disease, the short time of exposure to the antiviral agent “makes protecting against Parkinson’s disease development in 5 years less likely,” they added.
Among the study limitations that the authors acknowledged was the lack of data about hepatic function profile, serum virologic response, viral genotype, and hepatitis C virus RNA-level. The database that the investigators used also lacked data about behavioral factors (e.g., smoking status, coffee consumption, and alcohol consumption) that may have affected the incidence of Parkinson’s disease in the cohort. Investigations with longer follow-up periods will be needed to provide clearer information, they concluded.
The authors reported no conflicts of interest. The study was funded by grants from Chang Gung Medical Research Fund and from Chang Gung Memorial Hospital.
SOURCE: Lin W-Y et al. JAMA Neurol. 2019 Jun 5. doi: 10.1001/jamaneurol.2019.1368.
The findings of Lin et al. suggest a potentially modifiable hepatologic risk factor for Parkinson’s disease, Adolfo Ramirez-Zamora, MD, associate professor of neurology; Christopher W. Hess, MD, assistant professor of neurology; and David R. Nelson, MD, senior vice president for health affairs, all at the University of Florida in Gainesville, wrote in an accompanying editorial. Hepatitis C virus infection might enter the brain through the microvasculature and might induce microglial and macrophage-related inflammatory changes (JAMA Neurol. 2019 June 5. doi: 10.1001/jamaneurol.2019.1377).
Lin et al. estimated high diagnostic accuracy for Parkinson’s disease in their study. Nevertheless, clinical, neuroimaging, and pathological confirmation was unavailable, which is a limitation of their investigation, said Dr. Ramirez-Zamora and colleagues. “The diagnosis of Parkinson’s disease in early stages can be challenging, as other related conditions can mimic Parkinson’s disease, including cirrhosis-related parkinsonism. Moreover, using record-linkage systems excludes patients who did not seek medical advice or those who were misdiagnosed by symptoms alone, which may also underestimate the prevalence of Parkinson’s disease. Using population-based studies would be a more accurate method.”
Because interferon, which was the antiviral therapy used in this study, greatly affects the immune system and has a modest rate of eradicating viral hepatitis C infection, future research should examine the association between Parkinson’s disease and patients who cleared the virus, as well as patients who did not, said Dr. Ramirez-Zamora and colleagues. Such research could shed light on potential mechanisms of treatment response. Lin et al. did not examine the newer direct-acting antiviral therapies for hepatitis C virus infection, which cure more than 90% of patients. Nor did they analyze other well established lifestyle and demographic risk factors for developing the disease. In addition, “the authors could not generalize the results to those aged 75 years or older because of the substantially smaller number of patients in this age group,” said Dr. Ramirez-Zamora and colleagues.
Still, “identification of potentially treatable Parkinson’s disease risk factors presents a unique opportunity for treatment. Additional studies with detailed viral analysis and exposure are needed, including in other geographic and ethnic distributions,” they concluded.
The findings of Lin et al. suggest a potentially modifiable hepatologic risk factor for Parkinson’s disease, Adolfo Ramirez-Zamora, MD, associate professor of neurology; Christopher W. Hess, MD, assistant professor of neurology; and David R. Nelson, MD, senior vice president for health affairs, all at the University of Florida in Gainesville, wrote in an accompanying editorial. Hepatitis C virus infection might enter the brain through the microvasculature and might induce microglial and macrophage-related inflammatory changes (JAMA Neurol. 2019 June 5. doi: 10.1001/jamaneurol.2019.1377).
Lin et al. estimated high diagnostic accuracy for Parkinson’s disease in their study. Nevertheless, clinical, neuroimaging, and pathological confirmation was unavailable, which is a limitation of their investigation, said Dr. Ramirez-Zamora and colleagues. “The diagnosis of Parkinson’s disease in early stages can be challenging, as other related conditions can mimic Parkinson’s disease, including cirrhosis-related parkinsonism. Moreover, using record-linkage systems excludes patients who did not seek medical advice or those who were misdiagnosed by symptoms alone, which may also underestimate the prevalence of Parkinson’s disease. Using population-based studies would be a more accurate method.”
Because interferon, which was the antiviral therapy used in this study, greatly affects the immune system and has a modest rate of eradicating viral hepatitis C infection, future research should examine the association between Parkinson’s disease and patients who cleared the virus, as well as patients who did not, said Dr. Ramirez-Zamora and colleagues. Such research could shed light on potential mechanisms of treatment response. Lin et al. did not examine the newer direct-acting antiviral therapies for hepatitis C virus infection, which cure more than 90% of patients. Nor did they analyze other well established lifestyle and demographic risk factors for developing the disease. In addition, “the authors could not generalize the results to those aged 75 years or older because of the substantially smaller number of patients in this age group,” said Dr. Ramirez-Zamora and colleagues.
Still, “identification of potentially treatable Parkinson’s disease risk factors presents a unique opportunity for treatment. Additional studies with detailed viral analysis and exposure are needed, including in other geographic and ethnic distributions,” they concluded.
The findings of Lin et al. suggest a potentially modifiable hepatologic risk factor for Parkinson’s disease, Adolfo Ramirez-Zamora, MD, associate professor of neurology; Christopher W. Hess, MD, assistant professor of neurology; and David R. Nelson, MD, senior vice president for health affairs, all at the University of Florida in Gainesville, wrote in an accompanying editorial. Hepatitis C virus infection might enter the brain through the microvasculature and might induce microglial and macrophage-related inflammatory changes (JAMA Neurol. 2019 June 5. doi: 10.1001/jamaneurol.2019.1377).
Lin et al. estimated high diagnostic accuracy for Parkinson’s disease in their study. Nevertheless, clinical, neuroimaging, and pathological confirmation was unavailable, which is a limitation of their investigation, said Dr. Ramirez-Zamora and colleagues. “The diagnosis of Parkinson’s disease in early stages can be challenging, as other related conditions can mimic Parkinson’s disease, including cirrhosis-related parkinsonism. Moreover, using record-linkage systems excludes patients who did not seek medical advice or those who were misdiagnosed by symptoms alone, which may also underestimate the prevalence of Parkinson’s disease. Using population-based studies would be a more accurate method.”
Because interferon, which was the antiviral therapy used in this study, greatly affects the immune system and has a modest rate of eradicating viral hepatitis C infection, future research should examine the association between Parkinson’s disease and patients who cleared the virus, as well as patients who did not, said Dr. Ramirez-Zamora and colleagues. Such research could shed light on potential mechanisms of treatment response. Lin et al. did not examine the newer direct-acting antiviral therapies for hepatitis C virus infection, which cure more than 90% of patients. Nor did they analyze other well established lifestyle and demographic risk factors for developing the disease. In addition, “the authors could not generalize the results to those aged 75 years or older because of the substantially smaller number of patients in this age group,” said Dr. Ramirez-Zamora and colleagues.
Still, “identification of potentially treatable Parkinson’s disease risk factors presents a unique opportunity for treatment. Additional studies with detailed viral analysis and exposure are needed, including in other geographic and ethnic distributions,” they concluded.
, according to a cohort study published online June 5 in JAMA Neurology. The results provide evidence that hepatitis C virus is a risk factor for Parkinson’s disease.
In the past several years, epidemiologic studies have suggested an association between hepatitis C virus infection and Parkinson’s disease. A study published in 2017, however, found no association between the two. In addition, these investigations did not consider antiviral therapy as a potential modifying factor.
Wey-Yil Lin, MD, a neurologist at Landseed International Hospital in Taoyuan, Taiwan, and colleagues examined claims data from the Taiwan National Health Insurance Research Database to identify the risk of incident Parkinson’s disease in patients with hepatitis C virus infection who received antiviral treatment, compared with those who did not receive treatment.
The investigators selected all patients with a new diagnosis of hepatitis C virus infection with or without hepatitis from January 1, 2003, to December 31, 2013. They excluded patients who were aged 20 years or younger; had Parkinson’s disease, dementia, or stroke; or had had major hepatic diseases on the index date. To ensure that treated patients had had an effective course of therapy, the researchers excluded patients who were lost to follow-up within 6 months of the index date, received antiviral therapy for fewer than 16 weeks, or developed Parkinson’s disease within 6 months of the index date.
The primary outcome was incident Parkinson’s disease. Dr. Lin and colleagues excluded participants with a diagnosis of stroke and dementia before the index date to reduce the possibility of enrolling participants with secondary and atypical parkinsonism.
To minimize the potential selection bias to which observational studies are subject, the investigators performed propensity score matching with sex, age, comorbidities, and medication as covariates. This method was intended to create treated and untreated cohorts with comparable characteristics.
Dr. Lin and colleagues included 188,152 patients in their analysis. After matching, each group included 39,936 participants. In the group that received antiviral treatment, 45.0% of participants were female, and mean age was 52.8 years. In the untreated group, 44.4% of participants were female, and mean age was 52.5 years.
The incidence density of Parkinson’s disease per 1,000 person-years was 1.00 in the treated group and 1.39 in the untreated group. The difference in risk of Parkinson’s disease between the treated and untreated groups was statistically significant at year 5 of follow-up (hazard ratio [HR], 0.75) and at the end of the cohort (HR, 0.71). The risk did not differ significantly at year 1 and year 3, however. A subgroup analysis found a greater benefit of antiviral therapy among patients who concurrently used dihydropyridine calcium channel blockers.
“To our knowledge, this is the first cohort study to investigate the association between antiviral therapy and risk of Parkinson’s disease in patients with chronic hepatitis C viral infection,” said Dr. Lin and colleagues. Although it is possible that interferon-based antiviral therapy directly protected against the development of Parkinson’s disease, the short time of exposure to the antiviral agent “makes protecting against Parkinson’s disease development in 5 years less likely,” they added.
Among the study limitations that the authors acknowledged was the lack of data about hepatic function profile, serum virologic response, viral genotype, and hepatitis C virus RNA-level. The database that the investigators used also lacked data about behavioral factors (e.g., smoking status, coffee consumption, and alcohol consumption) that may have affected the incidence of Parkinson’s disease in the cohort. Investigations with longer follow-up periods will be needed to provide clearer information, they concluded.
The authors reported no conflicts of interest. The study was funded by grants from Chang Gung Medical Research Fund and from Chang Gung Memorial Hospital.
SOURCE: Lin W-Y et al. JAMA Neurol. 2019 Jun 5. doi: 10.1001/jamaneurol.2019.1368.
, according to a cohort study published online June 5 in JAMA Neurology. The results provide evidence that hepatitis C virus is a risk factor for Parkinson’s disease.
In the past several years, epidemiologic studies have suggested an association between hepatitis C virus infection and Parkinson’s disease. A study published in 2017, however, found no association between the two. In addition, these investigations did not consider antiviral therapy as a potential modifying factor.
Wey-Yil Lin, MD, a neurologist at Landseed International Hospital in Taoyuan, Taiwan, and colleagues examined claims data from the Taiwan National Health Insurance Research Database to identify the risk of incident Parkinson’s disease in patients with hepatitis C virus infection who received antiviral treatment, compared with those who did not receive treatment.
The investigators selected all patients with a new diagnosis of hepatitis C virus infection with or without hepatitis from January 1, 2003, to December 31, 2013. They excluded patients who were aged 20 years or younger; had Parkinson’s disease, dementia, or stroke; or had had major hepatic diseases on the index date. To ensure that treated patients had had an effective course of therapy, the researchers excluded patients who were lost to follow-up within 6 months of the index date, received antiviral therapy for fewer than 16 weeks, or developed Parkinson’s disease within 6 months of the index date.
The primary outcome was incident Parkinson’s disease. Dr. Lin and colleagues excluded participants with a diagnosis of stroke and dementia before the index date to reduce the possibility of enrolling participants with secondary and atypical parkinsonism.
To minimize the potential selection bias to which observational studies are subject, the investigators performed propensity score matching with sex, age, comorbidities, and medication as covariates. This method was intended to create treated and untreated cohorts with comparable characteristics.
Dr. Lin and colleagues included 188,152 patients in their analysis. After matching, each group included 39,936 participants. In the group that received antiviral treatment, 45.0% of participants were female, and mean age was 52.8 years. In the untreated group, 44.4% of participants were female, and mean age was 52.5 years.
The incidence density of Parkinson’s disease per 1,000 person-years was 1.00 in the treated group and 1.39 in the untreated group. The difference in risk of Parkinson’s disease between the treated and untreated groups was statistically significant at year 5 of follow-up (hazard ratio [HR], 0.75) and at the end of the cohort (HR, 0.71). The risk did not differ significantly at year 1 and year 3, however. A subgroup analysis found a greater benefit of antiviral therapy among patients who concurrently used dihydropyridine calcium channel blockers.
“To our knowledge, this is the first cohort study to investigate the association between antiviral therapy and risk of Parkinson’s disease in patients with chronic hepatitis C viral infection,” said Dr. Lin and colleagues. Although it is possible that interferon-based antiviral therapy directly protected against the development of Parkinson’s disease, the short time of exposure to the antiviral agent “makes protecting against Parkinson’s disease development in 5 years less likely,” they added.
Among the study limitations that the authors acknowledged was the lack of data about hepatic function profile, serum virologic response, viral genotype, and hepatitis C virus RNA-level. The database that the investigators used also lacked data about behavioral factors (e.g., smoking status, coffee consumption, and alcohol consumption) that may have affected the incidence of Parkinson’s disease in the cohort. Investigations with longer follow-up periods will be needed to provide clearer information, they concluded.
The authors reported no conflicts of interest. The study was funded by grants from Chang Gung Medical Research Fund and from Chang Gung Memorial Hospital.
SOURCE: Lin W-Y et al. JAMA Neurol. 2019 Jun 5. doi: 10.1001/jamaneurol.2019.1368.
FROM JAMA NEUROLOGY
Stewart Tepper: Emgality approval ‘very exciting’
The drug, a humanized monoclonal antibody that binds to calcitonin gene-related peptide (CGRP), is administered by self-injection in 300-mg doses.

Galcanezumab is the first medication for episodic cluster headache that reduces the frequency of attacks, the agency said in an announcement.
Cluster headache can be more intense than migraine. The pain is unilateral and occurs in the orbital, supraorbital, or temporal regions. It reaches its peak intensity within 5-10 minutes and generally lasts for 30-90 minutes. Symptoms include a burning sensation, conjunctival injection, rhinorrhea, and photosensitivity. Patients often have one to three of these headaches per day, and the headaches appear to be linked to the circadian rhythm. An episodic cluster cycle can last for weeks to months of daily or near daily attacks.
A study presented at the recent meeting of the American Academy of Neurology provided evidence of the drug’s efficacy in cluster headache. In this trial, researchers randomized 106 patients with episodic cluster headache to galcanezumab or placebo. The baseline cluster headache frequency was 17.3 attacks per week, and galcanezumab reduced this frequency to 9.1 attacks per week, compared with 12.1 attacks per week with placebo. The most common side effect reported in this and other clinical trials was injection-site reactions.
Galcanezumab entails a risk of hypersensitivity reactions, according to the FDA. These reactions may occur several days after administration and may be prolonged. “If a serious hypersensitivity reaction occurs, treatment should be discontinued,” the agency said.
“It’s a very exciting day. There had never been a drug approved for prevention of cluster headache,” said Stewart J. Tepper, MD, professor of neurology at the Geisel School of Medicine at Dartmouth and director of the Dartmouth Headache Center, Dartmouth-Hitchcock Medical Center, Lebanon, N.H.

It is difficult to achieve therapeutic concentrations of current preventive medications that do not have FDA approval for this indication, such as verapamil, lithium, or antiepileptic drugs. Galcanezumab, in contrast, works quickly. It is important to note that the approval was for preventive treatment of episodic cluster headache, not for prevention of chronic cluster headache, and not for acute treatment, Dr. Tepper said.
“It’s important to get optimal therapy for cluster headache. It is one of the most disabling, terrible disorders on Earth,” Dr. Tepper said. “The importance [of this approval] cannot be overestimated.”
When asked for comment, Alan M. Rapoport, MD, clinical professor of neurology at the University of California, Los Angeles, said “If this monoclonal antibody to the CGRP ligand works as well in real life as in the trial, it will be an important advance in the treatment of cluster headache.”
Prior to the approval of galcanezumab, noninvasive vagal nerve stimulation was approved in November 2018 for adjunctive use in the preventive treatment of cluster headache in adults.
The FDA granted the application for galcanezumab using a Priority Review and Breakthrough Therapy designation. The agency approved galcanezumab for the preventive treatment of migraine in adults in September 2018. The drug appears to have a similar safety profile in both patient populations. Eli Lilly, which is based in Indianapolis, Indiana, manufactures the drug.
This article was updated June 5, 2019.
The drug, a humanized monoclonal antibody that binds to calcitonin gene-related peptide (CGRP), is administered by self-injection in 300-mg doses.
Galcanezumab is the first medication for episodic cluster headache that reduces the frequency of attacks, the agency said in an announcement.
Cluster headache can be more intense than migraine. The pain is unilateral and occurs in the orbital, supraorbital, or temporal regions. It reaches its peak intensity within 5-10 minutes and generally lasts for 30-90 minutes. Symptoms include a burning sensation, conjunctival injection, rhinorrhea, and photosensitivity. Patients often have one to three of these headaches per day, and the headaches appear to be linked to the circadian rhythm. An episodic cluster cycle can last for weeks to months of daily or near daily attacks.
A study presented at the recent meeting of the American Academy of Neurology provided evidence of the drug’s efficacy in cluster headache. In this trial, researchers randomized 106 patients with episodic cluster headache to galcanezumab or placebo. The baseline cluster headache frequency was 17.3 attacks per week, and galcanezumab reduced this frequency to 9.1 attacks per week, compared with 12.1 attacks per week with placebo. The most common side effect reported in this and other clinical trials was injection-site reactions.
Galcanezumab entails a risk of hypersensitivity reactions, according to the FDA. These reactions may occur several days after administration and may be prolonged. “If a serious hypersensitivity reaction occurs, treatment should be discontinued,” the agency said.
“It’s a very exciting day. There had never been a drug approved for prevention of cluster headache,” said Stewart J. Tepper, MD, professor of neurology at the Geisel School of Medicine at Dartmouth and director of the Dartmouth Headache Center, Dartmouth-Hitchcock Medical Center, Lebanon, N.H.
It is difficult to achieve therapeutic concentrations of current preventive medications that do not have FDA approval for this indication, such as verapamil, lithium, or antiepileptic drugs. Galcanezumab, in contrast, works quickly. It is important to note that the approval was for preventive treatment of episodic cluster headache, not for prevention of chronic cluster headache, and not for acute treatment, Dr. Tepper said.
“It’s important to get optimal therapy for cluster headache. It is one of the most disabling, terrible disorders on Earth,” Dr. Tepper said. “The importance [of this approval] cannot be overestimated.”
When asked for comment, Alan M. Rapoport, MD, clinical professor of neurology at the University of California, Los Angeles, said “If this monoclonal antibody to the CGRP ligand works as well in real life as in the trial, it will be an important advance in the treatment of cluster headache.”
Prior to the approval of galcanezumab, noninvasive vagal nerve stimulation was approved in November 2018 for adjunctive use in the preventive treatment of cluster headache in adults.
The FDA granted the application for galcanezumab using a Priority Review and Breakthrough Therapy designation. The agency approved galcanezumab for the preventive treatment of migraine in adults in September 2018. The drug appears to have a similar safety profile in both patient populations. Eli Lilly, which is based in Indianapolis, Indiana, manufactures the drug.
This article was updated June 5, 2019.
The drug, a humanized monoclonal antibody that binds to calcitonin gene-related peptide (CGRP), is administered by self-injection in 300-mg doses.
Galcanezumab is the first medication for episodic cluster headache that reduces the frequency of attacks, the agency said in an announcement.
Cluster headache can be more intense than migraine. The pain is unilateral and occurs in the orbital, supraorbital, or temporal regions. It reaches its peak intensity within 5-10 minutes and generally lasts for 30-90 minutes. Symptoms include a burning sensation, conjunctival injection, rhinorrhea, and photosensitivity. Patients often have one to three of these headaches per day, and the headaches appear to be linked to the circadian rhythm. An episodic cluster cycle can last for weeks to months of daily or near daily attacks.
A study presented at the recent meeting of the American Academy of Neurology provided evidence of the drug’s efficacy in cluster headache. In this trial, researchers randomized 106 patients with episodic cluster headache to galcanezumab or placebo. The baseline cluster headache frequency was 17.3 attacks per week, and galcanezumab reduced this frequency to 9.1 attacks per week, compared with 12.1 attacks per week with placebo. The most common side effect reported in this and other clinical trials was injection-site reactions.
Galcanezumab entails a risk of hypersensitivity reactions, according to the FDA. These reactions may occur several days after administration and may be prolonged. “If a serious hypersensitivity reaction occurs, treatment should be discontinued,” the agency said.
“It’s a very exciting day. There had never been a drug approved for prevention of cluster headache,” said Stewart J. Tepper, MD, professor of neurology at the Geisel School of Medicine at Dartmouth and director of the Dartmouth Headache Center, Dartmouth-Hitchcock Medical Center, Lebanon, N.H.
It is difficult to achieve therapeutic concentrations of current preventive medications that do not have FDA approval for this indication, such as verapamil, lithium, or antiepileptic drugs. Galcanezumab, in contrast, works quickly. It is important to note that the approval was for preventive treatment of episodic cluster headache, not for prevention of chronic cluster headache, and not for acute treatment, Dr. Tepper said.
“It’s important to get optimal therapy for cluster headache. It is one of the most disabling, terrible disorders on Earth,” Dr. Tepper said. “The importance [of this approval] cannot be overestimated.”
When asked for comment, Alan M. Rapoport, MD, clinical professor of neurology at the University of California, Los Angeles, said “If this monoclonal antibody to the CGRP ligand works as well in real life as in the trial, it will be an important advance in the treatment of cluster headache.”
Prior to the approval of galcanezumab, noninvasive vagal nerve stimulation was approved in November 2018 for adjunctive use in the preventive treatment of cluster headache in adults.
The FDA granted the application for galcanezumab using a Priority Review and Breakthrough Therapy designation. The agency approved galcanezumab for the preventive treatment of migraine in adults in September 2018. The drug appears to have a similar safety profile in both patient populations. Eli Lilly, which is based in Indianapolis, Indiana, manufactures the drug.
This article was updated June 5, 2019.
DOACs surpass warfarin in low-weight AFib patients
SAN FRANCISCO – The direct-acting anticoagulants, as a class, were more effective and at least as safe as warfarin in low-weight and very-low-weight patients with atrial fibrillation in an adjusted analysis of real-world outcomes data from more than 21,000 Korean patients.

The analysis also showed that the direct-acting oral anticoagulants (DOACs) had the best safety and efficacy on low-weight patients when used at the labeled dosages, with blunted efficacy and safety at dosages that either exceeded or fell short of labeled levels, So-Ryoung Lee, MD, said at the annual scientific sessions of the Heart Rhythm Society.
The overall superiority of DOACs by both efficacy and safety also generally extended to the subgroup of very-low-weight patients, those with weights of less than 50 kg. In this subgroup, which was 28% of the total population studied, the composite adverse event outcome occurred 33% less often among patients treated with a DOAC relative to patients treated with warfarin, a statistically significant difference, said Dr. Lee, a cardiologist at Seoul (South Korea) National University Hospital. Among all patients with weights of 60 kg (132 pounds) or less, the composite outcome occurred 34% less often in the DOAC-treated patients relative to the warfarin-treated patients, also a statistically significant difference.
Dr. Lee and colleagues used a Korean National Health Insurance database that included information on more than 600,000 adults with atrial fibrillation (AFib) as of January 2013. The researchers whittled this down to 21,678 patients who began for the first time treatment with an oral anticoagulant starting during or after January 2014; had no history of a stroke, intracranial hemorrhage, or gastrointestinal bleed; and weighed no more than 60 kg. This cohort included 7,575 (35%) who received warfarin treatment, and 14,103 (65%) who received a DOAC. Within the DOAC-treated group, 42% received rivaroxaban (Xarelto), 26% dabigatran (Pradaxa), 24% apixaban (Eliquis), and 8% edoxaban (Savaysa).
To account for baseline differences in demographics and comorbidities between the patients treated with a DOAC and those who received warfarin, Dr. Lee and her associates did propensity score adjustment, which resulted in similar cohorts of 6,692 patients treated with warfarin and 12,810 patients treated with a DOAC. The average age of these patients was 73 years, a third were men, and the average body mass index was just over 22 kg/m2.
The events that the researchers tallied during follow-up through December 2016 included rates of all-cause death, ischemic stroke, intracranial hemorrhage, hospitalization for GI bleeding, hospitalization for major bleeding, and the composite of these five outcomes.
In the propensity-score adjusted full cohort of all patients who weighed 60 kg or less, the rate of each of these five outcomes, as well as the composite outcome, occurred statistically significantly less often among the DOAC-treated patients than in those on warfarin. The reductions ranged from a 41% lower incidence of ischemic stroke on DOAC treatment compared with warfarin treatment, to an 18% reduced rate of hospitalization for a GI bleed, compared with warfarin-treated patients. In the subgroup of patients who weighed less than 50 kg (110 pounds), the reductions ranged from a 41% cut in ischemic stroke on a DOAC compared with warfarin to a 24% relative reduction in the rate of hospitalization for a major bleed, a difference that just reached statistical significance. The outcome of hospitalization for a GI bleed showed no significant between-group difference among very-low-weight patients, but the rates of intracranial hemorrhage and all-cause death also showed statistically significant lower rates among DOAC-treated patients.
Nearly two-thirds of the patients on a DOAC received the label-appropriate dose of the drug, but 31% received a dosage that was below the labeled level while 4% received a dosage above the labeled level. An analysis that divided the NOAC patients by the appropriateness of their treatment dosages showed that patients on the correct dosages fared best. For example, in the total cohort of patients who weighed 60 kg or less, those on the correct DOAC dosage had a 9.1% rate of the combined endpoint. Patients on a low DOAC dosage did about as well as did the patients on warfarin, with a combined event rate of 11.6% in each of these subgroups. The worst outcomes occurred among the small number of patients on an inappropriately-high DOAC dosage, with a combined event rate of 15.4%. The researchers found a similar pattern among patients who weighed less than 50 kg.
Dr. Lee had no disclosures.
SOURCE: Lee SR et al. HRS 2019, Abstract S-AB30-05.
SAN FRANCISCO – The direct-acting anticoagulants, as a class, were more effective and at least as safe as warfarin in low-weight and very-low-weight patients with atrial fibrillation in an adjusted analysis of real-world outcomes data from more than 21,000 Korean patients.
The analysis also showed that the direct-acting oral anticoagulants (DOACs) had the best safety and efficacy on low-weight patients when used at the labeled dosages, with blunted efficacy and safety at dosages that either exceeded or fell short of labeled levels, So-Ryoung Lee, MD, said at the annual scientific sessions of the Heart Rhythm Society.
The overall superiority of DOACs by both efficacy and safety also generally extended to the subgroup of very-low-weight patients, those with weights of less than 50 kg. In this subgroup, which was 28% of the total population studied, the composite adverse event outcome occurred 33% less often among patients treated with a DOAC relative to patients treated with warfarin, a statistically significant difference, said Dr. Lee, a cardiologist at Seoul (South Korea) National University Hospital. Among all patients with weights of 60 kg (132 pounds) or less, the composite outcome occurred 34% less often in the DOAC-treated patients relative to the warfarin-treated patients, also a statistically significant difference.
Dr. Lee and colleagues used a Korean National Health Insurance database that included information on more than 600,000 adults with atrial fibrillation (AFib) as of January 2013. The researchers whittled this down to 21,678 patients who began for the first time treatment with an oral anticoagulant starting during or after January 2014; had no history of a stroke, intracranial hemorrhage, or gastrointestinal bleed; and weighed no more than 60 kg. This cohort included 7,575 (35%) who received warfarin treatment, and 14,103 (65%) who received a DOAC. Within the DOAC-treated group, 42% received rivaroxaban (Xarelto), 26% dabigatran (Pradaxa), 24% apixaban (Eliquis), and 8% edoxaban (Savaysa).
To account for baseline differences in demographics and comorbidities between the patients treated with a DOAC and those who received warfarin, Dr. Lee and her associates did propensity score adjustment, which resulted in similar cohorts of 6,692 patients treated with warfarin and 12,810 patients treated with a DOAC. The average age of these patients was 73 years, a third were men, and the average body mass index was just over 22 kg/m2.
The events that the researchers tallied during follow-up through December 2016 included rates of all-cause death, ischemic stroke, intracranial hemorrhage, hospitalization for GI bleeding, hospitalization for major bleeding, and the composite of these five outcomes.
In the propensity-score adjusted full cohort of all patients who weighed 60 kg or less, the rate of each of these five outcomes, as well as the composite outcome, occurred statistically significantly less often among the DOAC-treated patients than in those on warfarin. The reductions ranged from a 41% lower incidence of ischemic stroke on DOAC treatment compared with warfarin treatment, to an 18% reduced rate of hospitalization for a GI bleed, compared with warfarin-treated patients. In the subgroup of patients who weighed less than 50 kg (110 pounds), the reductions ranged from a 41% cut in ischemic stroke on a DOAC compared with warfarin to a 24% relative reduction in the rate of hospitalization for a major bleed, a difference that just reached statistical significance. The outcome of hospitalization for a GI bleed showed no significant between-group difference among very-low-weight patients, but the rates of intracranial hemorrhage and all-cause death also showed statistically significant lower rates among DOAC-treated patients.
Nearly two-thirds of the patients on a DOAC received the label-appropriate dose of the drug, but 31% received a dosage that was below the labeled level while 4% received a dosage above the labeled level. An analysis that divided the NOAC patients by the appropriateness of their treatment dosages showed that patients on the correct dosages fared best. For example, in the total cohort of patients who weighed 60 kg or less, those on the correct DOAC dosage had a 9.1% rate of the combined endpoint. Patients on a low DOAC dosage did about as well as did the patients on warfarin, with a combined event rate of 11.6% in each of these subgroups. The worst outcomes occurred among the small number of patients on an inappropriately-high DOAC dosage, with a combined event rate of 15.4%. The researchers found a similar pattern among patients who weighed less than 50 kg.
Dr. Lee had no disclosures.
SOURCE: Lee SR et al. HRS 2019, Abstract S-AB30-05.
SAN FRANCISCO – The direct-acting anticoagulants, as a class, were more effective and at least as safe as warfarin in low-weight and very-low-weight patients with atrial fibrillation in an adjusted analysis of real-world outcomes data from more than 21,000 Korean patients.
The analysis also showed that the direct-acting oral anticoagulants (DOACs) had the best safety and efficacy on low-weight patients when used at the labeled dosages, with blunted efficacy and safety at dosages that either exceeded or fell short of labeled levels, So-Ryoung Lee, MD, said at the annual scientific sessions of the Heart Rhythm Society.
The overall superiority of DOACs by both efficacy and safety also generally extended to the subgroup of very-low-weight patients, those with weights of less than 50 kg. In this subgroup, which was 28% of the total population studied, the composite adverse event outcome occurred 33% less often among patients treated with a DOAC relative to patients treated with warfarin, a statistically significant difference, said Dr. Lee, a cardiologist at Seoul (South Korea) National University Hospital. Among all patients with weights of 60 kg (132 pounds) or less, the composite outcome occurred 34% less often in the DOAC-treated patients relative to the warfarin-treated patients, also a statistically significant difference.
Dr. Lee and colleagues used a Korean National Health Insurance database that included information on more than 600,000 adults with atrial fibrillation (AFib) as of January 2013. The researchers whittled this down to 21,678 patients who began for the first time treatment with an oral anticoagulant starting during or after January 2014; had no history of a stroke, intracranial hemorrhage, or gastrointestinal bleed; and weighed no more than 60 kg. This cohort included 7,575 (35%) who received warfarin treatment, and 14,103 (65%) who received a DOAC. Within the DOAC-treated group, 42% received rivaroxaban (Xarelto), 26% dabigatran (Pradaxa), 24% apixaban (Eliquis), and 8% edoxaban (Savaysa).
To account for baseline differences in demographics and comorbidities between the patients treated with a DOAC and those who received warfarin, Dr. Lee and her associates did propensity score adjustment, which resulted in similar cohorts of 6,692 patients treated with warfarin and 12,810 patients treated with a DOAC. The average age of these patients was 73 years, a third were men, and the average body mass index was just over 22 kg/m2.
The events that the researchers tallied during follow-up through December 2016 included rates of all-cause death, ischemic stroke, intracranial hemorrhage, hospitalization for GI bleeding, hospitalization for major bleeding, and the composite of these five outcomes.
In the propensity-score adjusted full cohort of all patients who weighed 60 kg or less, the rate of each of these five outcomes, as well as the composite outcome, occurred statistically significantly less often among the DOAC-treated patients than in those on warfarin. The reductions ranged from a 41% lower incidence of ischemic stroke on DOAC treatment compared with warfarin treatment, to an 18% reduced rate of hospitalization for a GI bleed, compared with warfarin-treated patients. In the subgroup of patients who weighed less than 50 kg (110 pounds), the reductions ranged from a 41% cut in ischemic stroke on a DOAC compared with warfarin to a 24% relative reduction in the rate of hospitalization for a major bleed, a difference that just reached statistical significance. The outcome of hospitalization for a GI bleed showed no significant between-group difference among very-low-weight patients, but the rates of intracranial hemorrhage and all-cause death also showed statistically significant lower rates among DOAC-treated patients.
Nearly two-thirds of the patients on a DOAC received the label-appropriate dose of the drug, but 31% received a dosage that was below the labeled level while 4% received a dosage above the labeled level. An analysis that divided the NOAC patients by the appropriateness of their treatment dosages showed that patients on the correct dosages fared best. For example, in the total cohort of patients who weighed 60 kg or less, those on the correct DOAC dosage had a 9.1% rate of the combined endpoint. Patients on a low DOAC dosage did about as well as did the patients on warfarin, with a combined event rate of 11.6% in each of these subgroups. The worst outcomes occurred among the small number of patients on an inappropriately-high DOAC dosage, with a combined event rate of 15.4%. The researchers found a similar pattern among patients who weighed less than 50 kg.
Dr. Lee had no disclosures.
SOURCE: Lee SR et al. HRS 2019, Abstract S-AB30-05.
REPORTING FROM HEART RHYTHM 2019
Restless legs syndrome in MS linked to cognitive impairment
SEATTLE – The results suggest that sleep dysfunction exacerbated by RLS could affect cognition in patients with MS, study lead author Katie L. Cederberg, CPT, a doctoral student in the department of physical therapy at the University of Alabama at Birmingham, said in an interview. She spoke at the annual meeting of the Consortium of Multiple Sclerosis Centers, where she presented the findings.
“RLS severity did predict cognitive impairment,” she said. However, she added, “this is just a snapshot, and we need to do more research.”
Sleep problems, including RLS, are more common in patients with MS than in the general population. “Current research suggests that anywhere from 19% to 67% of individuals with MS experience some sort of sleep difficulty, with rates as high as 80% in some samples,” a 2015 report noted.
As for RLS, a 2018 systematic review and meta-analysis found that “pooled RLS prevalence among MS patients of various ethnicities was 26%, and prevalence was lower in Asia (20%) than outside Asia (27%). Prevalence was higher among cross-sectional studies (30%) than among case-control studies (23%). RLS prevalence was higher among female than among male MS patients (26% vs. 17%), and it was higher among MS patients than among healthy controls (odds ratio, 3.96, 95% confidence interval, 3.29-4.77, P less than .001) (Sleep Med. 2018 Oct;50:97-104).
Ms. Cederberg said the frequency of RLS in patients with MS spurred her and colleagues to explore whether it may affect cognitive function.
For their study, the researchers surveyed 275 patients with MS (mean age = 60, 81% female, 33% employed, 95% white, 66% with relapsing-remitting MS). Of the 275, 75 appeared to have RLS. These patients were similar to the non-RLS patients in multiple areas, but they diverged in scores on the brief Multiple Sclerosis Neuropsychological Questionnaire, which measures self-perception of cognition.
Those with both MS and RLS scored 21.9 (± 11.7) on the test, while those with MS scored 18.0 (± 11.0), P = 0.023.
Analyses linked greater RLS severity to worse self-perceived cognitive impairment and sleep quality. “The diagnosis and treatment of RLS symptoms and other effectors of sleep quality could improve cognitive consequences of MS,” the authors concluded.
The National MS Society funded the study. The study authors reported no relevant disclosures.
SEATTLE – The results suggest that sleep dysfunction exacerbated by RLS could affect cognition in patients with MS, study lead author Katie L. Cederberg, CPT, a doctoral student in the department of physical therapy at the University of Alabama at Birmingham, said in an interview. She spoke at the annual meeting of the Consortium of Multiple Sclerosis Centers, where she presented the findings.
“RLS severity did predict cognitive impairment,” she said. However, she added, “this is just a snapshot, and we need to do more research.”
Sleep problems, including RLS, are more common in patients with MS than in the general population. “Current research suggests that anywhere from 19% to 67% of individuals with MS experience some sort of sleep difficulty, with rates as high as 80% in some samples,” a 2015 report noted.
As for RLS, a 2018 systematic review and meta-analysis found that “pooled RLS prevalence among MS patients of various ethnicities was 26%, and prevalence was lower in Asia (20%) than outside Asia (27%). Prevalence was higher among cross-sectional studies (30%) than among case-control studies (23%). RLS prevalence was higher among female than among male MS patients (26% vs. 17%), and it was higher among MS patients than among healthy controls (odds ratio, 3.96, 95% confidence interval, 3.29-4.77, P less than .001) (Sleep Med. 2018 Oct;50:97-104).
Ms. Cederberg said the frequency of RLS in patients with MS spurred her and colleagues to explore whether it may affect cognitive function.
For their study, the researchers surveyed 275 patients with MS (mean age = 60, 81% female, 33% employed, 95% white, 66% with relapsing-remitting MS). Of the 275, 75 appeared to have RLS. These patients were similar to the non-RLS patients in multiple areas, but they diverged in scores on the brief Multiple Sclerosis Neuropsychological Questionnaire, which measures self-perception of cognition.
Those with both MS and RLS scored 21.9 (± 11.7) on the test, while those with MS scored 18.0 (± 11.0), P = 0.023.
Analyses linked greater RLS severity to worse self-perceived cognitive impairment and sleep quality. “The diagnosis and treatment of RLS symptoms and other effectors of sleep quality could improve cognitive consequences of MS,” the authors concluded.
The National MS Society funded the study. The study authors reported no relevant disclosures.
SEATTLE – The results suggest that sleep dysfunction exacerbated by RLS could affect cognition in patients with MS, study lead author Katie L. Cederberg, CPT, a doctoral student in the department of physical therapy at the University of Alabama at Birmingham, said in an interview. She spoke at the annual meeting of the Consortium of Multiple Sclerosis Centers, where she presented the findings.
“RLS severity did predict cognitive impairment,” she said. However, she added, “this is just a snapshot, and we need to do more research.”
Sleep problems, including RLS, are more common in patients with MS than in the general population. “Current research suggests that anywhere from 19% to 67% of individuals with MS experience some sort of sleep difficulty, with rates as high as 80% in some samples,” a 2015 report noted.
As for RLS, a 2018 systematic review and meta-analysis found that “pooled RLS prevalence among MS patients of various ethnicities was 26%, and prevalence was lower in Asia (20%) than outside Asia (27%). Prevalence was higher among cross-sectional studies (30%) than among case-control studies (23%). RLS prevalence was higher among female than among male MS patients (26% vs. 17%), and it was higher among MS patients than among healthy controls (odds ratio, 3.96, 95% confidence interval, 3.29-4.77, P less than .001) (Sleep Med. 2018 Oct;50:97-104).
Ms. Cederberg said the frequency of RLS in patients with MS spurred her and colleagues to explore whether it may affect cognitive function.
For their study, the researchers surveyed 275 patients with MS (mean age = 60, 81% female, 33% employed, 95% white, 66% with relapsing-remitting MS). Of the 275, 75 appeared to have RLS. These patients were similar to the non-RLS patients in multiple areas, but they diverged in scores on the brief Multiple Sclerosis Neuropsychological Questionnaire, which measures self-perception of cognition.
Those with both MS and RLS scored 21.9 (± 11.7) on the test, while those with MS scored 18.0 (± 11.0), P = 0.023.
Analyses linked greater RLS severity to worse self-perceived cognitive impairment and sleep quality. “The diagnosis and treatment of RLS symptoms and other effectors of sleep quality could improve cognitive consequences of MS,” the authors concluded.
The National MS Society funded the study. The study authors reported no relevant disclosures.
REPORTING FROM CMSC 2019
Developing new measurements for better MS outcomes
SEATTLE – , according to Jared Srinivasan.
Mr. Srinivasan, a research coordinator at South Shore Neurologic Associates in Patchogue, N.Y., sat down at the annual meeting of the Consortium of Multiple Sclerosis Centers for a video interview summarizing his work on new measurement tools for assessing disease status in MS patients with Mark Gudesblatt, MD, and other colleagues at South Shore Neurologic Associates.
“We are trying to find better ways of measuring disease status, rather than the EDSS [Expanded Disability Status Scale] ... It is not as sensitive as some other measures can be,” Mr. Srinivasan said. “We are trying to shed light on some new tools regarding objectively measuring cognition, manual dexterity, gait, and ocular coherence tomography.”
The overall goal, he said, “is to use a combination of these granular outcome measures to create a bigger picture of a patient’s disease so we can better treat them.”
One of the tools is called Neurotrax, which measures cognition in multiple dimensions (e.g., attention, information processing, motor skills, verbal functioning). With this and other new tools for manual dexterity and its cognitive aspects, as well as other dimensions of MS, the researchers are trying capture a fuller picture of MS in individual patients.
“The end goal of this is that if we can show that MS is such a complex disease that the current tools we are using do not quite capture the full nuances and granularity in it, then we can move toward using better measures that will capture that, which will move patient care forward.”
Mr. Srinivasan had nothing to disclose.
SEATTLE – , according to Jared Srinivasan.
Mr. Srinivasan, a research coordinator at South Shore Neurologic Associates in Patchogue, N.Y., sat down at the annual meeting of the Consortium of Multiple Sclerosis Centers for a video interview summarizing his work on new measurement tools for assessing disease status in MS patients with Mark Gudesblatt, MD, and other colleagues at South Shore Neurologic Associates.
“We are trying to find better ways of measuring disease status, rather than the EDSS [Expanded Disability Status Scale] ... It is not as sensitive as some other measures can be,” Mr. Srinivasan said. “We are trying to shed light on some new tools regarding objectively measuring cognition, manual dexterity, gait, and ocular coherence tomography.”
The overall goal, he said, “is to use a combination of these granular outcome measures to create a bigger picture of a patient’s disease so we can better treat them.”
One of the tools is called Neurotrax, which measures cognition in multiple dimensions (e.g., attention, information processing, motor skills, verbal functioning). With this and other new tools for manual dexterity and its cognitive aspects, as well as other dimensions of MS, the researchers are trying capture a fuller picture of MS in individual patients.
“The end goal of this is that if we can show that MS is such a complex disease that the current tools we are using do not quite capture the full nuances and granularity in it, then we can move toward using better measures that will capture that, which will move patient care forward.”
Mr. Srinivasan had nothing to disclose.
SEATTLE – , according to Jared Srinivasan.
Mr. Srinivasan, a research coordinator at South Shore Neurologic Associates in Patchogue, N.Y., sat down at the annual meeting of the Consortium of Multiple Sclerosis Centers for a video interview summarizing his work on new measurement tools for assessing disease status in MS patients with Mark Gudesblatt, MD, and other colleagues at South Shore Neurologic Associates.
“We are trying to find better ways of measuring disease status, rather than the EDSS [Expanded Disability Status Scale] ... It is not as sensitive as some other measures can be,” Mr. Srinivasan said. “We are trying to shed light on some new tools regarding objectively measuring cognition, manual dexterity, gait, and ocular coherence tomography.”
The overall goal, he said, “is to use a combination of these granular outcome measures to create a bigger picture of a patient’s disease so we can better treat them.”
One of the tools is called Neurotrax, which measures cognition in multiple dimensions (e.g., attention, information processing, motor skills, verbal functioning). With this and other new tools for manual dexterity and its cognitive aspects, as well as other dimensions of MS, the researchers are trying capture a fuller picture of MS in individual patients.
“The end goal of this is that if we can show that MS is such a complex disease that the current tools we are using do not quite capture the full nuances and granularity in it, then we can move toward using better measures that will capture that, which will move patient care forward.”
Mr. Srinivasan had nothing to disclose.
EXPERT ANALYSIS FROM CMSC 2019

Mindfulness meditation may boost cognition in MS
SEATTLE – a new report suggests.

“The present study demonstrated significant improvement in processing speed, a core area of impairment in individuals with MS, following 4 weeks of mindfulness meditation,” said lead author Heena R. Manglani, a graduate student at the Ohio State University, Columbus. She spoke in an interview and in a presentation about the study findings at the annual meeting of the Consortium of Multiple Sclerosis Centers.
An estimated 43%-70% of people with MS experience cognitive decline. This decline “has a sophisticated neuroanatomic and pathophysiologic background and disturbs such vital cognitive domains as speed of information processing, memory, attention, executive functions, and visual perceptual function,” reported the authors of a 2017 review (Rev Neurosci. 2017 Nov 27;28[8]:845-860).
For the new study, researchers tested two strategies for cognitive enhancement in patients with MS. All study participants were aged 31-59 years and relapse free within the previous 30 days; most had relapsing remitting MS, and most did not show signs of cognitive decline.
The researchers assigned 20 patients to a 4-week adaptive computerized cognitive training program and 20 patients to a 4-week mindfulness meditation training program. Another 21 patients were assigned to a control group.
The adaptive training program relied on computerized games designed to boost processing speed, attention, and working memory. The mindfulness training focused on components such as awareness of breathing and of bodily sensations.
Researchers found that “the magnitude of cognitive gain from pre- to post training was greatest in participants in the mindfulness group, who did better than participants in either of the other two groups,” Ms. Manglani said.
Compared with the adaptive cognitive training and the control group, she said, the mindfulness meditation group showed statistically significant improvement in processing speed per scores on the Symbol Digit Modalities Test, which rose from 52.2 before training to 58.4 post training.
The interventions did not appear to have any effect on Paced Auditory Serial Addition scores, which measure working memory.
The findings suggest that “less than 20 hours of mindfulness may be effective in significantly improving processing speed in MS,” Ms. Manglani said. “It is much shorter than a typically delivered program. We hypothesize that you are training attention with mindfulness training. Attention has a lot of overlap with processing speed.”
Ms. Manglani noted that this was a pilot study, and she acknowledged that fairly few participants – only five or six in each group – showed signs of cognitive decline. The study also did not explore whether cognitive improvements translated to real-life changes in cognition.
“This effect needs to be replicated in a larger sample,” Ms. Manglani said, “and future studies are needed to establish the lasting effects of such training and how improvements in cognitive function may generalize to greater engagement in vocational and leisure activities and higher quality of life.”
The study was funded by the National Multiple Sclerosis Society and the National Institutes of Health. The authors reported no relevant disclosures except for one coauthor who received honoraria from Sanofi Genzyme and funding from the National Multiple Sclerosis Society and the NIH.
SEATTLE – a new report suggests.
“The present study demonstrated significant improvement in processing speed, a core area of impairment in individuals with MS, following 4 weeks of mindfulness meditation,” said lead author Heena R. Manglani, a graduate student at the Ohio State University, Columbus. She spoke in an interview and in a presentation about the study findings at the annual meeting of the Consortium of Multiple Sclerosis Centers.
An estimated 43%-70% of people with MS experience cognitive decline. This decline “has a sophisticated neuroanatomic and pathophysiologic background and disturbs such vital cognitive domains as speed of information processing, memory, attention, executive functions, and visual perceptual function,” reported the authors of a 2017 review (Rev Neurosci. 2017 Nov 27;28[8]:845-860).
For the new study, researchers tested two strategies for cognitive enhancement in patients with MS. All study participants were aged 31-59 years and relapse free within the previous 30 days; most had relapsing remitting MS, and most did not show signs of cognitive decline.
The researchers assigned 20 patients to a 4-week adaptive computerized cognitive training program and 20 patients to a 4-week mindfulness meditation training program. Another 21 patients were assigned to a control group.
The adaptive training program relied on computerized games designed to boost processing speed, attention, and working memory. The mindfulness training focused on components such as awareness of breathing and of bodily sensations.
Researchers found that “the magnitude of cognitive gain from pre- to post training was greatest in participants in the mindfulness group, who did better than participants in either of the other two groups,” Ms. Manglani said.
Compared with the adaptive cognitive training and the control group, she said, the mindfulness meditation group showed statistically significant improvement in processing speed per scores on the Symbol Digit Modalities Test, which rose from 52.2 before training to 58.4 post training.
The interventions did not appear to have any effect on Paced Auditory Serial Addition scores, which measure working memory.
The findings suggest that “less than 20 hours of mindfulness may be effective in significantly improving processing speed in MS,” Ms. Manglani said. “It is much shorter than a typically delivered program. We hypothesize that you are training attention with mindfulness training. Attention has a lot of overlap with processing speed.”
Ms. Manglani noted that this was a pilot study, and she acknowledged that fairly few participants – only five or six in each group – showed signs of cognitive decline. The study also did not explore whether cognitive improvements translated to real-life changes in cognition.
“This effect needs to be replicated in a larger sample,” Ms. Manglani said, “and future studies are needed to establish the lasting effects of such training and how improvements in cognitive function may generalize to greater engagement in vocational and leisure activities and higher quality of life.”
The study was funded by the National Multiple Sclerosis Society and the National Institutes of Health. The authors reported no relevant disclosures except for one coauthor who received honoraria from Sanofi Genzyme and funding from the National Multiple Sclerosis Society and the NIH.
SEATTLE – a new report suggests.
“The present study demonstrated significant improvement in processing speed, a core area of impairment in individuals with MS, following 4 weeks of mindfulness meditation,” said lead author Heena R. Manglani, a graduate student at the Ohio State University, Columbus. She spoke in an interview and in a presentation about the study findings at the annual meeting of the Consortium of Multiple Sclerosis Centers.
An estimated 43%-70% of people with MS experience cognitive decline. This decline “has a sophisticated neuroanatomic and pathophysiologic background and disturbs such vital cognitive domains as speed of information processing, memory, attention, executive functions, and visual perceptual function,” reported the authors of a 2017 review (Rev Neurosci. 2017 Nov 27;28[8]:845-860).
For the new study, researchers tested two strategies for cognitive enhancement in patients with MS. All study participants were aged 31-59 years and relapse free within the previous 30 days; most had relapsing remitting MS, and most did not show signs of cognitive decline.
The researchers assigned 20 patients to a 4-week adaptive computerized cognitive training program and 20 patients to a 4-week mindfulness meditation training program. Another 21 patients were assigned to a control group.
The adaptive training program relied on computerized games designed to boost processing speed, attention, and working memory. The mindfulness training focused on components such as awareness of breathing and of bodily sensations.
Researchers found that “the magnitude of cognitive gain from pre- to post training was greatest in participants in the mindfulness group, who did better than participants in either of the other two groups,” Ms. Manglani said.
Compared with the adaptive cognitive training and the control group, she said, the mindfulness meditation group showed statistically significant improvement in processing speed per scores on the Symbol Digit Modalities Test, which rose from 52.2 before training to 58.4 post training.
The interventions did not appear to have any effect on Paced Auditory Serial Addition scores, which measure working memory.
The findings suggest that “less than 20 hours of mindfulness may be effective in significantly improving processing speed in MS,” Ms. Manglani said. “It is much shorter than a typically delivered program. We hypothesize that you are training attention with mindfulness training. Attention has a lot of overlap with processing speed.”
Ms. Manglani noted that this was a pilot study, and she acknowledged that fairly few participants – only five or six in each group – showed signs of cognitive decline. The study also did not explore whether cognitive improvements translated to real-life changes in cognition.
“This effect needs to be replicated in a larger sample,” Ms. Manglani said, “and future studies are needed to establish the lasting effects of such training and how improvements in cognitive function may generalize to greater engagement in vocational and leisure activities and higher quality of life.”
The study was funded by the National Multiple Sclerosis Society and the National Institutes of Health. The authors reported no relevant disclosures except for one coauthor who received honoraria from Sanofi Genzyme and funding from the National Multiple Sclerosis Society and the NIH.
REPORTING FROM CMSC 2019
Consider patients’ perceptions of tardive dyskinesia
SAN FRANCISCO – Stanley N. Caroff, MD, said at the annual meeting of the American Psychiatric Association.
“You really need to ask the patient a lot of questions – and the family and the caregivers – about how much tardive dyskinesia affects their lives,” he said.
Those were some of the early results of RE-KINECT, an ongoing study of patients with schizophrenia and schizoaffective disorder who were being treated with antipsychotic agents.
TD occurs in more than 25% of patients in outpatient practices who are exposed to dopamine receptor blockers. Symptoms can include involuntary movements of the tongue, hands, and feet; facial distortions; rapid eye blinking; and difficulty speaking. In some cases, the side effects resolve after patients stop taking the medications.
In this video, Dr. Caroff discussed the studies’ findings and their implications for everyday clinical practice. He also presented some of the early RE-KINECT findings in a poster at the meeting.
Dr. Caroff is professor of psychiatry at the University of Pennsylvania, Philadelphia. He also is affiliated with the Michael J. Crescenz VA Medical Center in Philadelphia. He disclosed working as a consultant for and receiving research funding from Neurocrine Biosciences. He also is a consultant for DisperSol Technologies, Osmotica Pharmaceuticals, Teva Pharmaceutical.
SAN FRANCISCO – Stanley N. Caroff, MD, said at the annual meeting of the American Psychiatric Association.
“You really need to ask the patient a lot of questions – and the family and the caregivers – about how much tardive dyskinesia affects their lives,” he said.
Those were some of the early results of RE-KINECT, an ongoing study of patients with schizophrenia and schizoaffective disorder who were being treated with antipsychotic agents.
TD occurs in more than 25% of patients in outpatient practices who are exposed to dopamine receptor blockers. Symptoms can include involuntary movements of the tongue, hands, and feet; facial distortions; rapid eye blinking; and difficulty speaking. In some cases, the side effects resolve after patients stop taking the medications.
In this video, Dr. Caroff discussed the studies’ findings and their implications for everyday clinical practice. He also presented some of the early RE-KINECT findings in a poster at the meeting.
Dr. Caroff is professor of psychiatry at the University of Pennsylvania, Philadelphia. He also is affiliated with the Michael J. Crescenz VA Medical Center in Philadelphia. He disclosed working as a consultant for and receiving research funding from Neurocrine Biosciences. He also is a consultant for DisperSol Technologies, Osmotica Pharmaceuticals, Teva Pharmaceutical.
SAN FRANCISCO – Stanley N. Caroff, MD, said at the annual meeting of the American Psychiatric Association.
“You really need to ask the patient a lot of questions – and the family and the caregivers – about how much tardive dyskinesia affects their lives,” he said.
Those were some of the early results of RE-KINECT, an ongoing study of patients with schizophrenia and schizoaffective disorder who were being treated with antipsychotic agents.
TD occurs in more than 25% of patients in outpatient practices who are exposed to dopamine receptor blockers. Symptoms can include involuntary movements of the tongue, hands, and feet; facial distortions; rapid eye blinking; and difficulty speaking. In some cases, the side effects resolve after patients stop taking the medications.
In this video, Dr. Caroff discussed the studies’ findings and their implications for everyday clinical practice. He also presented some of the early RE-KINECT findings in a poster at the meeting.
Dr. Caroff is professor of psychiatry at the University of Pennsylvania, Philadelphia. He also is affiliated with the Michael J. Crescenz VA Medical Center in Philadelphia. He disclosed working as a consultant for and receiving research funding from Neurocrine Biosciences. He also is a consultant for DisperSol Technologies, Osmotica Pharmaceuticals, Teva Pharmaceutical.
REPORTING FROM APA 2019

Meta-analysis finds no link between PPI use and risk of dementia

The finding runs counter to recent studies, including a large pharmacoepidemiological claims data analysis from Germany, that propose an association between proton pump inhibitor (PPI) use and the development of dementia (JAMA Neurol. 2016;73[4]:410-6). “The issue with these studies is that they’re based on retrospective claims data and pharmacoepidemiological studies and insurance databases that don’t really give you a good causality basis,” lead study author Saad Alrajhi, MD, said in an interview at the annual Digestive Disease Week.
In an effort to better characterize the association between PPI exposure and dementia, Dr. Alrajhi, a gastroenterology fellow at McGill University, Montreal, and colleagues conducted a meta-analysis of all fully published randomized clinical trials or observational studies comparing use of PPIs and occurrence of dementia. The researchers queried Embase, MEDLINE, and ISI Web of Knowledge for relevant studies that were published from 1995 through September 2018. Next, they assessed the quality of the studies by using the Cochrane risk assessment tool for RCTs or the Newcastle-Ottawa Scale for observational studies.
As the primary outcome, the researchers compared dementia incidence after PPI exposure (experimental group) versus no PPI exposure (control group). Development of Alzheimer’s dementia was a secondary outcome. Sensitivity analyses consisted of excluding one study at a time, and assessing results among studies of highest qualities. Subgroup analyses included stratifying patients by age. To report odds ratios, Dr. Alrajhi and colleagues used fixed or random effects models based on the absence or presence of heterogeneity.
Of 549 studies assessed, 5 met the criteria for inclusion in the final analysis: 3 case-control studies and 2 cohort studies, with a total of 472,933 patients. All of the studies scored 8 or 9 on the Newcastle-Ottawa scale, indicating high quality. Significant heterogeneity was noted for all analyses. The researchers found that the incidence of dementia was not significantly increased among patients in the PPI-exposed group (odd ratio, 1.08 (95% confidence interval, 0.97-1.20; P = .18). Sensitivity analyses confirmed the robustness of the results. Subgroup analysis showed no between-group differences among studies that included a minimum age above 65 years (three studies) or less than age 65 (two studies). PPI exposure was not associated with the development of Alzheimer’s dementia (two studies) (OR, 1.32 (95% CI, 0.80-2.17; P = .27).
“In the absence of randomized trial evidence, a PPI prescribing approach based on appropriate utilization of guideline-based prescription should be done without the extra fear of the association of dementia,” Dr. Alrajhi said.
The researchers reported having no financial disclosures.
The finding runs counter to recent studies, including a large pharmacoepidemiological claims data analysis from Germany, that propose an association between proton pump inhibitor (PPI) use and the development of dementia (JAMA Neurol. 2016;73[4]:410-6). “The issue with these studies is that they’re based on retrospective claims data and pharmacoepidemiological studies and insurance databases that don’t really give you a good causality basis,” lead study author Saad Alrajhi, MD, said in an interview at the annual Digestive Disease Week.
In an effort to better characterize the association between PPI exposure and dementia, Dr. Alrajhi, a gastroenterology fellow at McGill University, Montreal, and colleagues conducted a meta-analysis of all fully published randomized clinical trials or observational studies comparing use of PPIs and occurrence of dementia. The researchers queried Embase, MEDLINE, and ISI Web of Knowledge for relevant studies that were published from 1995 through September 2018. Next, they assessed the quality of the studies by using the Cochrane risk assessment tool for RCTs or the Newcastle-Ottawa Scale for observational studies.
As the primary outcome, the researchers compared dementia incidence after PPI exposure (experimental group) versus no PPI exposure (control group). Development of Alzheimer’s dementia was a secondary outcome. Sensitivity analyses consisted of excluding one study at a time, and assessing results among studies of highest qualities. Subgroup analyses included stratifying patients by age. To report odds ratios, Dr. Alrajhi and colleagues used fixed or random effects models based on the absence or presence of heterogeneity.
Of 549 studies assessed, 5 met the criteria for inclusion in the final analysis: 3 case-control studies and 2 cohort studies, with a total of 472,933 patients. All of the studies scored 8 or 9 on the Newcastle-Ottawa scale, indicating high quality. Significant heterogeneity was noted for all analyses. The researchers found that the incidence of dementia was not significantly increased among patients in the PPI-exposed group (odd ratio, 1.08 (95% confidence interval, 0.97-1.20; P = .18). Sensitivity analyses confirmed the robustness of the results. Subgroup analysis showed no between-group differences among studies that included a minimum age above 65 years (three studies) or less than age 65 (two studies). PPI exposure was not associated with the development of Alzheimer’s dementia (two studies) (OR, 1.32 (95% CI, 0.80-2.17; P = .27).
“In the absence of randomized trial evidence, a PPI prescribing approach based on appropriate utilization of guideline-based prescription should be done without the extra fear of the association of dementia,” Dr. Alrajhi said.
The researchers reported having no financial disclosures.
The finding runs counter to recent studies, including a large pharmacoepidemiological claims data analysis from Germany, that propose an association between proton pump inhibitor (PPI) use and the development of dementia (JAMA Neurol. 2016;73[4]:410-6). “The issue with these studies is that they’re based on retrospective claims data and pharmacoepidemiological studies and insurance databases that don’t really give you a good causality basis,” lead study author Saad Alrajhi, MD, said in an interview at the annual Digestive Disease Week.
In an effort to better characterize the association between PPI exposure and dementia, Dr. Alrajhi, a gastroenterology fellow at McGill University, Montreal, and colleagues conducted a meta-analysis of all fully published randomized clinical trials or observational studies comparing use of PPIs and occurrence of dementia. The researchers queried Embase, MEDLINE, and ISI Web of Knowledge for relevant studies that were published from 1995 through September 2018. Next, they assessed the quality of the studies by using the Cochrane risk assessment tool for RCTs or the Newcastle-Ottawa Scale for observational studies.
As the primary outcome, the researchers compared dementia incidence after PPI exposure (experimental group) versus no PPI exposure (control group). Development of Alzheimer’s dementia was a secondary outcome. Sensitivity analyses consisted of excluding one study at a time, and assessing results among studies of highest qualities. Subgroup analyses included stratifying patients by age. To report odds ratios, Dr. Alrajhi and colleagues used fixed or random effects models based on the absence or presence of heterogeneity.
Of 549 studies assessed, 5 met the criteria for inclusion in the final analysis: 3 case-control studies and 2 cohort studies, with a total of 472,933 patients. All of the studies scored 8 or 9 on the Newcastle-Ottawa scale, indicating high quality. Significant heterogeneity was noted for all analyses. The researchers found that the incidence of dementia was not significantly increased among patients in the PPI-exposed group (odd ratio, 1.08 (95% confidence interval, 0.97-1.20; P = .18). Sensitivity analyses confirmed the robustness of the results. Subgroup analysis showed no between-group differences among studies that included a minimum age above 65 years (three studies) or less than age 65 (two studies). PPI exposure was not associated with the development of Alzheimer’s dementia (two studies) (OR, 1.32 (95% CI, 0.80-2.17; P = .27).
“In the absence of randomized trial evidence, a PPI prescribing approach based on appropriate utilization of guideline-based prescription should be done without the extra fear of the association of dementia,” Dr. Alrajhi said.
The researchers reported having no financial disclosures.
REPORTING FROM DDW 2019
A sleeping beast: Obstructive sleep apnea and stroke
Obstructive sleep apnea (OSA) is an independent risk factor for ischemic stroke and may also, infrequently, be a consequence of stroke. It is significantly underdiagnosed in the general population and is highly prevalent in patients who have had a stroke. Many patients likely had their stroke because of this chronic untreated condition.
This review focuses on OSA and its prevalence, consequences, and treatment in patients after a stroke.
DEFINING AND QUANTIFYING OSA
OSA is the most common type of sleep-disordered breathing.1,2 It involves repeated narrowing or complete collapse of the upper airway despite ongoing respiratory effort.3,4 Apneic episodes are terminated by arousals from hypoxemia or efforts to breathe.5 In contrast, central sleep apnea is characterized by a patent airway but lack of airflow due to absent respiratory effort.5
In OSA, the number of episodes of apnea (absent airflow) and hypopnea (reduced airflow) are added together and divided by hours of sleep to calculate the apnea-hypopnea index (AHI). OSA is diagnosed by either of the following3,4:
- AHI of 5 or higher, with clinical symptoms related to OSA (described below)
- AHI of 15 or higher, regardless of symptoms.
The AHI also defines OSA severity, as follows3:
- Mild: AHI 5 to 15
- Moderate: AHI 15 to 30
- Severe: AHI greater than 30.
Diagnostic criteria (eg, definition of hypopnea, testing methods, and AHI thresholds) have varied over time, an important consideration when reviewing the literature.
OSA IS MORE COMMON THAN EXPECTED AFTER STROKE
In the most methodologically sound and generalizable study of this topic to date, the Wisconsin Sleep Cohort Study6 reported in 2013 that about 14% of men and 5% of women ages 30 to 70 have an AHI greater than 5 (using 4% desaturation to score hypopneic episodes) with daytime sleepiness. Other studies suggest that 80% to 90% of people with OSA are undiagnosed and untreated.1,7
The prevalence of OSA in patients who have had a stroke is much higher, ranging from 30% to 96% depending on the study methods and population.1,8–12 A 2010 meta-analysis11 of 29 studies reported that 72% of patients who had a stroke had an AHI greater than 5, and 29% had severe OSA. In this analysis, 7% of those with sleep-disordered breathing had central sleep apnea; still, these data indicate that the prevalence of OSA in these patients is about 5 times higher than in the general population.
RISK FACTORS MAY DIFFER IN STROKE POPULATION
Several risk factors for OSA have been identified.
Obesity is one of the strongest risk factors, with increasing body mass index (BMI) associated with increased OSA prevalence.4,6,13 However, obesity appears to be a less significant risk factor in patients who have had a stroke than in the general population. In the 2010 meta-analysis11 of OSA after stroke, the average BMI was only 26.4 kg/m2 (with obesity defined as a BMI > 30.0 kg/m2), and increasing BMI was not associated with increasing AHI.
Male sex and advanced age are also OSA risk factors.4,5 They remain significant in patients after a stroke; about 65% of poststroke patients who have OSA are men, and the older the patient, the more likely the AHI is greater than 10.11
Ethnicity and genetics may also play important roles in OSA risk, with roughly 25% of OSA prevalence estimated to have a genetic basis.14,15 Some risk factors for OSA such as craniofacial shape, upper airway anatomy, upper airway muscle dysfunction, increased respiratory chemosensitivity, and poor arousal threshold during sleep are likely determined by genetics and ethnicity.14,15 Compared with people of European origin, Asians have a similar prevalence of OSA, but at a much lower average BMI, suggesting that other factors are significant.14 Possible genetically determined anatomic risk factors have not been specifically studied in the poststroke population, but it can be assumed they remain relevant.
Several studies have tried to find an association between OSA and type, location, etiology, or pattern of stroke.10,11,16–19 Although some suggest links between cardioembolic stroke and OSA,16,20 or thrombolysis and OSA,10 most have found no association between OSA and stroke features.11,12,21,22
HOW DOES OSA INCREASE STROKE RISK?
Untreated severe OSA is associated with increased cardiovascular mortality,21,22 and OSA is an independent risk factor for incident stroke.23 A number of mechanisms may explain these relationships.
Intermittent hypoxemia and recurrent sympathetic arousals resulting from OSA are thought to lead to many of the comorbid conditions with which it is associated: hypertension, coronary artery disease, heart failure, arrhythmias, pulmonary hypertension, and stroke. Repetitive decreases in ventilation lead to oxygen desaturations that result in cycles of increased sympathetic outflow and eventual sustained nocturnal hypertension and daytime chronic hypertension.1,5,9,13 Also implicated are various changes in vasodilator and vasoconstrictor substances due to endothelial dysfunction and inflammation, which are thought to play a role in the atherogenic and prothrombotic states induced by OSA.1,5,13
Cerebral circulation is altered primarily by the changes in partial pressure of carbon dioxide (Pco2). During apnea, the Pco2 rises, causing vasodilation and increased blood flow. After the apnea resolves, there is hyperpnea with resultant decreased Pco2, and vasoconstriction. In a patient who already has vascular disease, the enhanced vasoconstriction could lead to ischemia.1,5
Changes in intrathoracic pressure result in distortion of cardiac architecture. When the patient tries to breathe against an occluded airway, the intrathoracic pressure becomes more and more negative, increasing preload and afterload. When this happens repeatedly every night for years, it leads to remodeling of the heart such as left and right ventricular hypertrophy, with reduced stroke volume, myocardial ischemia, and increased risk of arrhythmia.1,5,13
Untreated OSA is believed to predispose patients to develop atrial fibrillation through sympathetic overactivity, vascular inflammation, heart rate variability, and cardiac remodeling.24 As atrial fibrillation is a major risk factor for stroke, particularly cardioembolic stroke, it may be another pathway of increased stroke risk in OSA.16,20,25
CLINICAL MANIFESTATIONS OF OSA NOT OBVIOUS AFTER STROKE
OSA typically causes both daytime symptoms (excessive sleepiness, poor concentration, morning headache, depressive symptoms) and nighttime signs and symptoms (snoring, choking, gasping, night sweats, insomnia, nocturia, witnessed episodes of apnea).3,4,26 Unfortunately, because these are nonspecific, OSA is often underdiagnosed.4,26
Identifying OSA after a stroke may be a particular challenge, as patients often do not report classic symptoms, and the typical picture of OSA may have less predictive validity in these patients.1,27,28 Within the first 24 hours after a stroke, hypersomnia, snoring history, and age are not predictive of OSA.1 Patients found to have OSA after a stroke frequently do not have the traditional symptoms (sleepiness, snoring) seen in usual OSA patients. And they have higher rates of OSA at a younger age than the usual OSA patients, so age is not a predictive risk factor. In addition, daytime sleepiness and obesity are often absent or less prominent.1,9,27,28 Finally, typical OSA signs and symptoms may be attributed to the stroke itself or to comorbidities affecting the patient, lowering suspicion for OSA.
OSA MAY HINDER STROKE RECOVERY, WORSEN OUTCOMES
OSA, particularly when moderate to severe, is linked to pathophysiologic changes that can hinder recovery from a stroke.
Intermittent hypoxemia during sleep can worsen vascular damage of at-risk tissue: nocturnal hypoxemia correlates with white matter hyperintensities on magnetic resonance imaging, a marker of ischemic demyelination.29 Oxidative stress and release of inflammatory mediators associated with intermittent hypoxemia may impair vascular blood flow to brain tissue attempting to repair itself.30 In addition, sympathetic overactivity and Pco2 fluctuations associated with OSA may impede cerebral circulation.
Taken together, such ongoing nocturnal insults can lead to clinical consequences during this vulnerable period.
A 1996 study31 of patients recovering from a stroke found that an oxygen desaturation index (number of times that the blood oxygen level drops below a certain threshold, as measured by overnight oximetry) of more than 10 per hour was associated with worse functional recovery at discharge and at 3 and 12 months after discharge. This study also noted an association between time spent with oxygen saturations below 90% and the rate of death at 1 year.
A 2003 study32 reported that patients with an AHI greater than 10 by polysomnography spent an average of 13 days longer on the rehabilitation service and had worse functional and cognitive status on discharge, even after controlling for multiple confounders. Several subsequent studies have confirmed these and similar findings.8,33,34
OSA has also been linked to depression,35 which is common after stroke and may worsen outcomes.36 The interaction between OSA, depression, and poststroke outcomes warrants further study.
In the general population, OSA has been independently associated with increased risk of stroke or death from any cause.21,22,37 These associations have also been reported in the poststroke population: a 2014 meta-analysis found that OSA increased the risk of a repeat stroke (relative risk [RR] 1.8, 95% confidence interval [CI] 1.2–2.6) and all-cause mortality (RR 1.69, 95% CI 1.4–2.1).38
TESTING FOR OSA AFTER STROKE
Because of the high prevalence of OSA in patients who have had a stroke and the potential for worse outcomes associated with untreated OSA, there should be a low threshold for evaluating for OSA soon after stroke. Objective testing is required to qualify for therapy, and the gold standard for diagnosis of OSA is formal polysomnography conducted in a sleep laboratory.2–4 Unfortunately, polysomnography may be unacceptable to some patients, is costly, and is resource-intensive, particularly in an inpatient or rehabilitation setting.28 Ideally, to optimize testing efficiency, patients should be screened for the likelihood of OSA before polysomnography is ordered.
Questionnaires can help determine the need for further testing
Questionnaires developed to assess OSA risk39 include the following:
The Berlin questionnaire, developed in 1999, has 10 questions assessing daytime and nighttime signs and symptoms and presence of hypertension.
The STOP questionnaire, developed in 2008, assesses snoring, tiredness, observed apneic episodes, and elevated blood pressure.
The STOP-BANG questionnaire, published in 2010, includes the STOP questions plus BMI over 35 kg/m2, age over 50, neck circumference over 41 cm, and male gender.
A 2017 meta-analysis39 of 108 studies with nearly 50,000 people found that the STOP-BANG questionnaire performed best with regard to sensitivity and diagnostic odds ratio, but with poor specificity.
These screening tools and modified versions of them have also been evaluated in patients who have had a stroke.
In 2015, Boulos et al28 found that the STOP-BAG (a version of STOP-BANG that excludes neck circumference) and the 4-variable (4V) questionnaire (sex, BMI, blood pressure, snoring) had moderate predictive value for OSA within 6 months after sroke.
In 2016, Katzan et al40 found that the STOP-BAG2 (STOP-BAG criteria plus continuous variables for BMI and age) had a high sensitivity for polysomnographically diagnosed OSA within the first year after a stroke. The specificity was significantly better than the STOP-BANG or the STOP-BAG questionnaire, although it remained suboptimal at 60.5%.
In 2017, Sico et al41 developed and assessed the SLEEP Inventory (sex, left heart failure, Epworth Sleepiness Scale, enlarged neck, weight in pounds, insulin resistance or diabetes, and National Institutes of Health Stroke Scale) and found that it outperformed the Berlin and STOP-BANG questionnaires in the poststroke setting. The SLEEP Inventory had the best specificity and negative predictive value, and a slightly better ability to correctly classify patients as having OSA or not, classifying 80% of patients correctly.
These newer screening tools (eg, STOP-BAG, STOP-BAG2, SLEEP) can be used to identify with reasonable accuracy which patients need definitive testing after stroke.
Pulse oximetry is another possible screening tool
Overnight pulse oximetry may also help screen for sleep apnea and stratify risk after a stroke. A 2012 study42 of overnight oximetry to screen patients before surgery found that the oxygen desaturation index was significantly associated with the AHI measured by polysomnography. However, oximetry testing cannot distinguish between OSA and central sleep apnea, so it is insufficient to diagnose OSA or qualify patients for therapy. Further study is needed to examine the ability of overnight pulse oximetry to screen or to stratify risk for OSA after stroke.
Polysomnography vs home testing
Polysomnography is the gold standard for diagnosing OSA. Benefits include technical support and trouble-shooting, determining relationships between OSA, body position, and sleep stage, and the ability to intervene with treatment.2 However, polysomnography can be cumbersome, costly, and resource-intensive.
A home sleep apnea test, ie, an unattended, limited-channel sleep study, may be an acceptable alternative.2–4,43,44 Home testing does not require a sleep technologist to be present during testing, uses fewer sensors, and is less expensive than overnight polysomnography, but its utility can be limited: it fails to accurately discriminate between episodes of OSA and central sleep apnea, there is potential for false-negative results, and it can underestimate sleep apnea burden because it does not measure sleep.2
Institutional resources and logistics may influence the choice of diagnostic modality. No data exist on outcomes from different diagnostic testing methods in poststroke patients. Further research is needed.
POSITIVE AIRWAY PRESSURE THERAPY: BENEFITS, CHALLENGES, ALTERNATIVES
The first-line treatment for OSA is positive airway pressure (PAP).3 For most patients, this is continuous PAP (CPAP) or autoadjusting PAP (APAP). In some instances, particularly for those who cannot tolerate CPAP or who have comorbid hypoventilation, bilevel PAP (BPAP) may be indicated. More advanced PAP therapies are unlikely to be used after stroke.
PAP therapy is associated with reduced daytime sleepiness, improved mood, normalization of sleep architecture, improved systemic and pulmonary artery blood pressure, reduced rates of atrial fibrillation after ablation, and improved insulin sensitivity.45–49 Whether it reduces the risk of cardiovascular events, including stroke, remains controversial; most data suggest that it does not.50,51 However, when adherence to PAP therapy is considered rather than intention to treat, treatment has been found to lead to improved cardiovascular outcomes.52
Mixed evidence of benefits after stroke
Observational studies provide evidence that CPAP may help patients with OSA after stroke, although results are mixed.53–58 The studies ranged in size from 14 to 105 patients, enrolled patients with mostly moderate to severe OSA, and followed patients from 10 days to 7 years. Adherence to therapy was generally good in the short term (50%–70%), but only 15% to 30% of patients remained adherent at 5 to 7 years. Variable outcomes were reported, with some studies finding improved symptoms in the near term and mixed evidence of cardiovascular benefit in the longer ones. However, as these studies lacked randomization, drawing definitive conclusions on CPAP efficacy is difficult.

Patients were enrolled in the index admission or when starting a rehabilitation service—generally 2 to 3 weeks after their stroke. No clear association was found between the timing of initiating PAP therapy and outcomes. All patients had ischemic strokes, but few details were provided regarding stroke location, size, and severity. Exclusion criteria included severe underlying cardiopulmonary disease, confusion, severe stroke with marked impairment, and inability to cooperate. Almost all patients had moderate to severe OSA, and patients with central sleep apnea were excluded.
The major outcomes examined were drop-out rates, PAP adherence, and neurologic improvement based on neurologic functional scales (National Institutes of Health Stroke Scale and Canadian Neurologic Scale). As expected, dropout rates were higher in patients randomized to CPAP (OR 1.83, 95% CI 1.05–3.21, P = .03), although overall adherence was better than anticipated, with mean CPAP use across trials of 4.5 hours per night (95% CI 3.97–5.08) and with about 50% to 60% of patients adhering to therapy for at least 4 hours nightly.
Improvement in neurologic outcomes favored CPAP (standard mean difference 0.54, 95% CI 0.026–1.05), although considerable heterogeneity was seen. Improved sleepiness outcomes were inconsistent. Major cardiovascular outcomes were reported in only 2 studies (using the same data set) and showed delayed time to the next cardiovascular event for those treated with CPAP but no difference in cardiovascular event-free survival.
PAP poses more challenges after stroke
The primary limitation to PAP therapy is poor acceptance and adherence to therapy.59 High rates of refusal of therapy and difficulty complying with treatment have been noted in the poststroke population, although recent studies have reported better adherence rates. How rates of adherence play out in real-world settings, outside of the controlled environment of a research study, has yet to be determined.
In general, CPAP adherence is affected by claustrophobia, difficulty tolerating a mask, problems with pressure intolerance, irritating air leaks, nasal congestion, and naso-oral dryness. Many such barriers can be overcome with use of a properly fitted mask, an appropriate pressure setting, heated humidification, nasal sprays (eg, saline, inhaled steroids), and education, encouragement, and reassurance.
After a stroke, additional obstacles may impede the ability to use PAP therapy.68 Facial paresis (hemi- or bifacial) may make fitting of the mask problematic. Paralysis or weakness of the extremities may limit the ability to adjust or remove a mask. Aphasia can impair communication and understanding of the need to use PAP therapy, and upper-airway problems related to stroke, including dysphagia, may lead to pressure intolerance or risk of aspiration. Finally, a lack of perceived benefit, particularly if the patient does not have daytime sleepiness, may limit motivation.
Consider alternatives
For patients unlikely to succeed with PAP therapy, there are alternatives. Surgery and oral appliances are not usually realistic options in the setting of recent stroke, but positional therapy, including the use of body positioners to prevent supine sleep, as well as elevating the head of the bed, may be of some benefit.69,70 A nasopharyngeal airway stenting device (nasal trumpet) may also be tolerated by some patients.

A proposed algorithm for screening, diagnosing, and treating OSA in patients after stroke is presented in Figure 1.
- Selim B, Roux FJ. Stroke and sleep disorders. Sleep Med Clin 2012; 7(4):597–607. doi:10.1016/j.jsmc.2012.08.007
- Kapur VK, Auckley DH, Chowdhuri S, et al. Clinical practice guideline for diagnostic testing for adult obstructive sleep apnea: an American Academy of Sleep Medicine Clinical Practice Guideline. J Clin Sleep Med 2017; 13(3):479–504. doi:10.5664/jcsm.6506
- Epstein LJ, Kristo D, Strollo PJ Jr, et al; Adult Obstructive Sleep Apnea Task Force of the American Academy of Sleep Medicine. Clinical guideline for the evaluation, management, and long-term care of obstructive sleep apnea in adults. J Clin Sleep Med 2009; 5(3):263–276. pmid:19960649
- Patil SP, Schneider H, Schwartz AR, Smith PL. Adult obstructive sleep apnea: pathophysiology and diagnosis. Chest 2007; 132(1):325–337. doi:10.1378/chest.07-0040
- Dempsey JA, Veasey SC, Morgan BJ, O’Donnell CP. Pathophysiology of sleep apnea. Physiol Rev 2010; 90(1):47–112. doi:10.1152/physrev.00043.2008
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177(9):1006–1014. doi:10.1093/aje/kws342
- Redline S, Sotres-Alvarez D, Loredo J, et al. Sleep-disordered breathing in Hispanic/Latino individuals of diverse backgrounds. The Hispanic Community Health Study/Study of Latinos. Am J Resp Crit Care Med 2014; 189(3):335–344. doi:10.1164/rccm.201309-1735OC
- Aaronson JA, van Bennekom CA, Hofman WF, et al. Obstructive sleep apnea is related to impaired cognitive and functional status after stroke. Sleep 2015; 38(9):1431–1437. doi:10.5665/sleep.4984
- Sharma S, Culebras A. Sleep apnoea and stroke. Stroke Vasc Neurol 2016; 1(4):185–191. doi:10.1136/svn-2016-000038
- Huhtakangas JK, Huhtakangas J, Bloigu R, Saaresranta T. Prevalence of sleep apnea at the acute phase of ischemic stroke with or without thrombolysis. Sleep Med 2017; 40:40–46. doi:10.1016/j.sleep.2017.08.018
- Johnson KG, Johnson DC. Frequency of sleep apnea in stroke and TIA patients: a meta-analysis. J Clin Sleep Med 2010; 6(2):131–137. pmid:20411688
- Iranzo A, Santamaria J, Berenguer J, Sanchez M, Chamorro A. Prevalence and clinical importance of sleep apnea in the first night after cerebral infarction. Neurology 2002; 58:911–916. pmid:11914407
- Javaheri S, Barbe F, Campos-Rodriguez F, et al. Sleep apnea: types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol 2017; 69(7):841–858. doi:10.1016/j.jacc.2016.11.069
- Dudley KA, Patel SR. Disparities and genetic risk factors in obstructive sleep apnea. Sleep Med 2016; 18:96–102. doi:10.1016/j.sleep.2015.01.015
- Redline S, Tishler PV. The genetics of sleep apnea. Sleep Med Rev 2000; 4(6):583–602. doi:10.1053/smrv.2000.0120
- Lipford MC, Flemming KD, Calvin AD, et al. Associations between cardioembolic stroke and obstructive sleep apnea. Sleep 2015; 38(11):1699–1705. doi:10.5665/sleep.5146
- Wang Y, Wang Y, Chen J, Yi X, Dong S, Cao L. Stroke patterns, topography, and etiology in patients with obstructive sleep apnea hypopnea syndrome. Int J Clin Exp Med 2017; 10(4):7137–7143.
- Fisse AL, Kemmling A, Teuber A, et al. The association of lesion location and sleep related breathing disorder in patients with acute ischemic stroke. PLoS One 2017; 12(1):e0171243. doi:10.1371/journal.pone.0171243
- Brown DL, Mowla A, McDermott M, et al. Ischemic stroke subtype and presence of sleep-disordered breathing: the BASIC sleep apnea study. J Stroke Cerebrovasc Dis 2015; 24(2):388–393. doi:10.1016/j.jstrokecerebrovasdis.2014.09.007
- Poli M, Philip P, Taillard J, et al. Atrial fibrillation as a major cause of stroke in apneic patients: a prospective study. Sleep Med 2017; 30:251–254. doi:10.1016/j.sleep.2015.07.031
- Young T, Finn L, Peppard PE, et al. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin Sleep Cohort. Sleep 2008; 31(8):1071–1078. pmid:18714778
- Molnar MZ, Mucsi I, Novak M, et al. Association of incident obstructive sleep apnoea with outcomes in a large cohort of US veterans. Thorax 2015; 70(9):888–895. doi:10.1136/thoraxjnl-2015-206970
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the sleep heart health study. Am J Respir Crit Care Med 2010; 182(2):269–277. doi:10.1164/rccm.200911-1746OC
- Marulanda-Londono E, Chaturvedi S. The interplay between obstructive sleep apnea and atrial fibrillation. Fron Neurol 2017; 8:668. doi:10.3389/fneur.2017.00668
- Szymanski FM, Filipiak KJ, Platek AE, Hrynkiewicz-Szymanska A, Karpinski G, Opolski G. Assessment of CHADS2 and CHA 2DS 2-VASc scores in obstructive sleep apnea patients with atrial fibrillation. Sleep Breath 2015; 19(2):531–537. doi:10.1007/s11325-014-1042-5
- Stansbury RC, Strollo PJ. Clinical manifestations of sleep apnea. J Thoracic Dis 2015; 7(9):E298–E310. doi:10.3978/j.issn.2072-1439.2015.09.13
- Chan W, Coutts SB, Hanly P. Sleep apnea in patients with transient ischemic attack and minor stroke: opportunity for risk reduction of recurrent stroke? Stroke 2010; 41(12):2973–2975. doi:10.1161/STROKEAHA.110.596759
- Boulos MI, Wan A, Im J, et al. Identifying obstructive sleep apnea after stroke/TIA: evaluating four simple screening tools. Sleep Med 2016; 21:133–139. doi:10.1016/j.sleep.2015.12.013
- Patel SK, Hanly PJ, Smith EE, Chan W, Coutts SB. Nocturnal hypoxemia is associated with white matter hyperintensities in patients with a minor stroke or transient ischemic attack. J Clin Sleep Med 2015; 11(12):1417–1424. doi:10.5664/jcsm.5278
- McCarty MF, DiNicolantonio JJ, O’Keefe JH. NADPH oxidase, uncoupled endothelial nitric oxide synthase, and NF-KappaB are key mediators of the pathogenic impact of obstructive sleep apnea—therapeutic implications. J Integr Cardiol 2016; 2(5):367–374. doi:10.15761/JIC.1000177
- Good DC, Henkle JQ, Gelber D, Welsh J, Verhulst S. Sleep-disordered breathing and poor functional outcome after stroke. Stroke 1996; 27(2):252–259. pmid:8571419
- Kaneko Y, Hajek VE, Zivanovic V, Raboud J, Bradley TD. Relationship of sleep apnea to functional capacity and length of hospitalization following stroke. Sleep 2003; 26(3):293–297. pmid:12749548
- Yan-fang S, Yu-ping W. Sleep-disordered breathing: impact on functional outcome of ischemic stroke patients. Sleep Med 2009; 10(7):717–719. doi:10.1016/j.sleep.2008.08.006
- Kumar R, Suri JC, Manocha R. Study of association of severity of sleep disordered breathing and functional outcome in stroke patients. Sleep Med 2017; 34:50–56. doi:10.1016/j.sleep.2017.02.025
- Kerner NA, Roose SP. Obstructive sleep apnea is linked to depression and cognitive impairment: evidence and potential mechanisms. Am J Geriatr Psychiatry 2016; 24(6):496–508. doi:10.1016/j.jagp.2016.01.134
- Bartoli F, Lillia N, Lax A, et al. Depression after stroke and risk of mortality: a systematic review and meta-analysis. Stroke Res Treat 2013; 2013:862978. doi:10.1155/2013/862978
- Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 2005; 353(19):2034–2041. doi:10.1056/NEJMoa043104
- Xie W, Zheng F, Song X. Obstructive sleep apnea and serious adverse outcomes in patients with cardiovascular or cerebrovascular disease: a PRISMA-compliant systematic review and meta-analysis. Medicine (Baltimore) 2014; 93(29):e336. doi:10.1097/MD.0000000000000336
- Chiu HY, Chen PY, Chuang LP, et al. Diagnostic accuracy of the Berlin questionnaire, STOP-BANG, STOP, and Epworth sleepiness scale in detecting obstructive sleep apnea: a bivariate meta-analysis. Sleep Med Rev 2017; 36:57–70. doi:10.1016/j.smrv.2016.10.004
- Katzan IL, Thompson NR, Uchino K, Foldvary-Schaefer N. A screening tool for obstructive sleep apnea in cerebrovascular patients. Sleep Med 2016; 21:70–76. doi:10.1016/j.sleep.2016.02.001
- Sico JJ, Yaggi HK, Ofner S, et al. Development, validation, and assessment of an ischemic stroke or transient ischemic attack-specific prediction tool for obstructive sleep apnea. J Stroke Cerebrovasc Dis 2017; 26(8):1745–1754. doi:10.1016/j.jstrokecerebrovasdis.2017.03.042
- Chung F, Liao P, Elsaid H, Islam S, Shapiro CM, Sun Y. Oxygen desaturation index from nocturnal oximetry: a sensitive and specific tool to detect sleep-disordered breathing in surgical patients. Anesth Analg 2012; 114(5):993–1000. doi:10.1213/ANE.0b013e318248f4f5
- Boulos MI, Elias S, Wan A, et al. Unattended hospital and home sleep apnea testing following cerebrovascular events. J Stroke Cerebrovasc Dis 2017; 26(1):143–149. doi:10.1016/j.jstrokecerebrovasdis.2016.09.001
- Saletu MT, Kotzian ST, Schwarzinger A, Haider S, Spatt J, Saletu B. Home sleep apnea testing is a feasible and accurate method to diagnose obstructive sleep apnea in stroke patients during in-hospital rehabilitation. J Clin Sleep Med 2018; 14(9):1495–1501. doi:10.5664/jcsm.7322
- Giles TL, Lasserson TJ, Smith BH, White J, Wright J, Cates CJ. Continuous positive airways pressure for obstructive sleep apnoea in adults. Cochrane Database Syst Rev 2006; (3):CD001106. doi:10.1002/14651858.CD001106.pub3
- Fatureto-Borges F, Lorenzi-Filho G, Drager LF. Effectiveness of continuous positive airway pressure in lowering blood pressure in patients with obstructive sleep apnea: a critical review of the literature. Integr Blood Press Control 2016; 9:43–47. doi:10.2147/IBPC.S70402
- Imran TF, Gharzipura M, Liu S, et al. Effect of continuous positive airway pressure treatment on pulmonary artery pressure in patients with isolated obstructive sleep apnea: a meta-analysis. Heart Fail Rev 2016; 21(5):591–598. doi:10.1007/s10741-016-9548-5
- Deng F, Raza A, Guo J. Treating obstructive sleep apnea with continuous positive airway pressure reduces risk of recurrent atrial fibrillation after catheter ablation: a meta-analysis. Sleep Med 2018; 46:5–11. doi:10.1016/j.sleep.2018.02.013
- Seetho IW, Wilding JPH. Sleep-disordered breathing, type 2 diabetes, and the metabolic syndrome. Chronic Resp Dis 2014; 11(4):257–275. doi:10.1177/1479972314552806
- Kim Y, Koo YS, Lee HY, Lee SY. Can continuous positive airway pressure reduce the risk of stroke in obstructive sleep apnea patients? A systematic review and meta-analysis. PloS ONE 2016; 11(1):e0146317. doi:10.1371/journal.pone.0146317
- Yu J, Zhou Z, McEvoy RD, et al. Association of positive airway pressure with cardiovascular events and death in adults with sleep apnea: a systematic review and meta-analysis. JAMA 2017; 318(2):156–166. doi:10.1001/jama.2017.7967
- Peker Y, Glantz H, Eulenburg C, Wegscheider K, Herlitz J, Thunström E. Effect of positive airway pressure on cardiovascular outcomes in coronary artery disease patients with nonsleepy obstructive sleep apnea. The RICCADSA randomized controlled trial. Am J Respir Crit Care Med 2016; 194(5):613–620. doi:10.1164/rccm.201601-0088OC
- Martinez-Garcia MA, Soler-Cataluna JJ, Ejarque-Martinez L, et al. Continuous positive airway pressure treatment reduces mortality in patients with ischemic stroke and obstructive sleep apnea: a 5-year follow-up study. Am J Respir Crit Care Med 2009; 180(1):36–41. doi:10.1164/rccm.200808-1341OC
- Broadley SA, Jorgensen L, Cheek A, et al. Early investigation and treatment of obstructive sleep apnoea after acute stroke. J Clin Neurosci 2007; 14(4):328–333. doi:10.1016/j.jocn.2006.01.017
- Wessendorf TE, Wang YM, Thilmann AF, Sorgenfrei U, Konietzko N, Teschler H. Treatment of obstructive sleep apnoea with nasal continuous positive airway pressure in stroke. Eur Respir J 2001; 18(4):623–629. pmid:11716165
- Bassetti CL, Milanova M, Gugger M. Sleep-disordered breathing and acute ischemic stroke: diagnosis, risk factors, treatment, evolution, and long-term clinical outcome. Stroke 2006; 37(4):967–972. doi:10.1161/01.STR.0000208215.49243.c3
- Palombini L, Guilleminault C. Stroke and treatment with nasal CPAP. Eur J Neurol 2006; 13(2):198–200. doi:10.1111/j.1468-1331.2006.01169.x
- Martínez-García MA, Campos-Rodríguez F, Soler-Cataluña JJ, Catalán-Serra P, Román-Sánchez P, Montserrat JM. Increased incidence of nonfatal cardiovascular events in stroke patients with sleep apnoea: effect of CPAP treatment. Eur Respir J 2012; 39(4):906–912. doi:10.1183/09031936.00011311
- Brill AK, Horvath T, Seiler A, et al. CPAP as treatment of sleep apnea after stroke: a meta-analysis of randomized trials. Neurology 2018; 90(14):e1222–e1230. doi:10.1212/WNL.0000000000005262
- Hsu C, Vennelle M, Li H, Engleman HM, Dennis MS, Douglas NJ. Sleep-disordered breathing after stroke: a randomised controlled trial of continuous positive airway pressure. J Neurol Neurosurg Psychiatry 2006; 77(10):1143–1149. doi:10.1136/jnnp.2005.086686
- Parra O, Sanchez-Armengol A, Bonnin M, et al. Early treatment of obstructive apnoea and stroke outcome: a randomised controlled trial. Eur Resp J 2011; 37(5):1128–1136. doi:10.1183/09031936.00034410
- Ryan CM, Bayley M, Green R, Murray BJ, Bradley TD. Influence of continuous positive airway pressure on outcomes of rehabilitation in stroke patients with obstructive sleep apnea. Stroke 2011; 42(4):1062–1067. doi:10.1161/STROKEAHA.110.597468
- Bravata DM, Concato J, Fried T, et al. Continuous positive airway pressure: evaluation of a novel therapy for patients with acute ischemic stroke. Sleep 2011; 34(9):1271–1277. doi:10.5665/SLEEP.1254
- Parra O, Sanchez-Armengol A, Capote F, et al. Efficacy of continuous positive airway pressure treatment on 5-year survival in patients with ischaemic stroke and obstructive sleep apnea: a randomized controlled trial. J Sleep Res 2015; 24(1):47–53. doi:10.1111/jsr.12181
- Khot SP, Davis AP, Crane DA, et al. Effect of continuous positive airway pressure on stroke rehabilitation: a pilot randomized sham-controlled trial. J Clin Sleep Med 2016; 12(7):1019–1026. doi:10.5664/jcsm.5940
- Aaronson JA, Hofman WF, van Bennekom CA, et al. Effects of continuous positive airway pressure on cognitive and functional outcome of stroke patients with obstructive sleep apnea: a randomized controlled trial. J Clin Sleep Med 2016; 12(4):533–541. doi:10.5664/jcsm.5684
- Gupta A, Shukla G, Afsar M, et al. Role of positive airway pressure therapy for obstructive sleep apnea in patients with stroke: a randomized controlled trial. J Clin Sleep Med 2018; 14(4):511–521. doi:10.5664/jcsm.7034
- Mello-Fujita L, Kim LJ, Palombini Lde O, et al. Treatment of obstructive sleep apnea syndrome associated with stroke. Sleep Med 2015; 16(6):691–696. doi:10.1016/j.sleep.2014.12.017
- Svatikova A, Chervin RD, Wing JJ, Sanchez BN, Migda EM, Brown DL. Positional therapy in ischemic stroke patients with obstructive sleep apnea. Sleep Med 2011; 12(3):262–266. doi:10.1016/j.sleep.2010.12.008
- Souza FJ, Genta PR, de Souza Filho AJ, Wellman A, Lorenzi-Filho G. The influence of head-of-bed elevation in patients with obstructive sleep apnea. Sleep Breath 2017; 21(4):815–820. doi:10.1007/s11325-017-1524-3
Obstructive sleep apnea (OSA) is an independent risk factor for ischemic stroke and may also, infrequently, be a consequence of stroke. It is significantly underdiagnosed in the general population and is highly prevalent in patients who have had a stroke. Many patients likely had their stroke because of this chronic untreated condition.
This review focuses on OSA and its prevalence, consequences, and treatment in patients after a stroke.
DEFINING AND QUANTIFYING OSA
OSA is the most common type of sleep-disordered breathing.1,2 It involves repeated narrowing or complete collapse of the upper airway despite ongoing respiratory effort.3,4 Apneic episodes are terminated by arousals from hypoxemia or efforts to breathe.5 In contrast, central sleep apnea is characterized by a patent airway but lack of airflow due to absent respiratory effort.5
In OSA, the number of episodes of apnea (absent airflow) and hypopnea (reduced airflow) are added together and divided by hours of sleep to calculate the apnea-hypopnea index (AHI). OSA is diagnosed by either of the following3,4:
- AHI of 5 or higher, with clinical symptoms related to OSA (described below)
- AHI of 15 or higher, regardless of symptoms.
The AHI also defines OSA severity, as follows3:
- Mild: AHI 5 to 15
- Moderate: AHI 15 to 30
- Severe: AHI greater than 30.
Diagnostic criteria (eg, definition of hypopnea, testing methods, and AHI thresholds) have varied over time, an important consideration when reviewing the literature.
OSA IS MORE COMMON THAN EXPECTED AFTER STROKE
In the most methodologically sound and generalizable study of this topic to date, the Wisconsin Sleep Cohort Study6 reported in 2013 that about 14% of men and 5% of women ages 30 to 70 have an AHI greater than 5 (using 4% desaturation to score hypopneic episodes) with daytime sleepiness. Other studies suggest that 80% to 90% of people with OSA are undiagnosed and untreated.1,7
The prevalence of OSA in patients who have had a stroke is much higher, ranging from 30% to 96% depending on the study methods and population.1,8–12 A 2010 meta-analysis11 of 29 studies reported that 72% of patients who had a stroke had an AHI greater than 5, and 29% had severe OSA. In this analysis, 7% of those with sleep-disordered breathing had central sleep apnea; still, these data indicate that the prevalence of OSA in these patients is about 5 times higher than in the general population.
RISK FACTORS MAY DIFFER IN STROKE POPULATION
Several risk factors for OSA have been identified.
Obesity is one of the strongest risk factors, with increasing body mass index (BMI) associated with increased OSA prevalence.4,6,13 However, obesity appears to be a less significant risk factor in patients who have had a stroke than in the general population. In the 2010 meta-analysis11 of OSA after stroke, the average BMI was only 26.4 kg/m2 (with obesity defined as a BMI > 30.0 kg/m2), and increasing BMI was not associated with increasing AHI.
Male sex and advanced age are also OSA risk factors.4,5 They remain significant in patients after a stroke; about 65% of poststroke patients who have OSA are men, and the older the patient, the more likely the AHI is greater than 10.11
Ethnicity and genetics may also play important roles in OSA risk, with roughly 25% of OSA prevalence estimated to have a genetic basis.14,15 Some risk factors for OSA such as craniofacial shape, upper airway anatomy, upper airway muscle dysfunction, increased respiratory chemosensitivity, and poor arousal threshold during sleep are likely determined by genetics and ethnicity.14,15 Compared with people of European origin, Asians have a similar prevalence of OSA, but at a much lower average BMI, suggesting that other factors are significant.14 Possible genetically determined anatomic risk factors have not been specifically studied in the poststroke population, but it can be assumed they remain relevant.
Several studies have tried to find an association between OSA and type, location, etiology, or pattern of stroke.10,11,16–19 Although some suggest links between cardioembolic stroke and OSA,16,20 or thrombolysis and OSA,10 most have found no association between OSA and stroke features.11,12,21,22
HOW DOES OSA INCREASE STROKE RISK?
Untreated severe OSA is associated with increased cardiovascular mortality,21,22 and OSA is an independent risk factor for incident stroke.23 A number of mechanisms may explain these relationships.
Intermittent hypoxemia and recurrent sympathetic arousals resulting from OSA are thought to lead to many of the comorbid conditions with which it is associated: hypertension, coronary artery disease, heart failure, arrhythmias, pulmonary hypertension, and stroke. Repetitive decreases in ventilation lead to oxygen desaturations that result in cycles of increased sympathetic outflow and eventual sustained nocturnal hypertension and daytime chronic hypertension.1,5,9,13 Also implicated are various changes in vasodilator and vasoconstrictor substances due to endothelial dysfunction and inflammation, which are thought to play a role in the atherogenic and prothrombotic states induced by OSA.1,5,13
Cerebral circulation is altered primarily by the changes in partial pressure of carbon dioxide (Pco2). During apnea, the Pco2 rises, causing vasodilation and increased blood flow. After the apnea resolves, there is hyperpnea with resultant decreased Pco2, and vasoconstriction. In a patient who already has vascular disease, the enhanced vasoconstriction could lead to ischemia.1,5
Changes in intrathoracic pressure result in distortion of cardiac architecture. When the patient tries to breathe against an occluded airway, the intrathoracic pressure becomes more and more negative, increasing preload and afterload. When this happens repeatedly every night for years, it leads to remodeling of the heart such as left and right ventricular hypertrophy, with reduced stroke volume, myocardial ischemia, and increased risk of arrhythmia.1,5,13
Untreated OSA is believed to predispose patients to develop atrial fibrillation through sympathetic overactivity, vascular inflammation, heart rate variability, and cardiac remodeling.24 As atrial fibrillation is a major risk factor for stroke, particularly cardioembolic stroke, it may be another pathway of increased stroke risk in OSA.16,20,25
CLINICAL MANIFESTATIONS OF OSA NOT OBVIOUS AFTER STROKE
OSA typically causes both daytime symptoms (excessive sleepiness, poor concentration, morning headache, depressive symptoms) and nighttime signs and symptoms (snoring, choking, gasping, night sweats, insomnia, nocturia, witnessed episodes of apnea).3,4,26 Unfortunately, because these are nonspecific, OSA is often underdiagnosed.4,26
Identifying OSA after a stroke may be a particular challenge, as patients often do not report classic symptoms, and the typical picture of OSA may have less predictive validity in these patients.1,27,28 Within the first 24 hours after a stroke, hypersomnia, snoring history, and age are not predictive of OSA.1 Patients found to have OSA after a stroke frequently do not have the traditional symptoms (sleepiness, snoring) seen in usual OSA patients. And they have higher rates of OSA at a younger age than the usual OSA patients, so age is not a predictive risk factor. In addition, daytime sleepiness and obesity are often absent or less prominent.1,9,27,28 Finally, typical OSA signs and symptoms may be attributed to the stroke itself or to comorbidities affecting the patient, lowering suspicion for OSA.
OSA MAY HINDER STROKE RECOVERY, WORSEN OUTCOMES
OSA, particularly when moderate to severe, is linked to pathophysiologic changes that can hinder recovery from a stroke.
Intermittent hypoxemia during sleep can worsen vascular damage of at-risk tissue: nocturnal hypoxemia correlates with white matter hyperintensities on magnetic resonance imaging, a marker of ischemic demyelination.29 Oxidative stress and release of inflammatory mediators associated with intermittent hypoxemia may impair vascular blood flow to brain tissue attempting to repair itself.30 In addition, sympathetic overactivity and Pco2 fluctuations associated with OSA may impede cerebral circulation.
Taken together, such ongoing nocturnal insults can lead to clinical consequences during this vulnerable period.
A 1996 study31 of patients recovering from a stroke found that an oxygen desaturation index (number of times that the blood oxygen level drops below a certain threshold, as measured by overnight oximetry) of more than 10 per hour was associated with worse functional recovery at discharge and at 3 and 12 months after discharge. This study also noted an association between time spent with oxygen saturations below 90% and the rate of death at 1 year.
A 2003 study32 reported that patients with an AHI greater than 10 by polysomnography spent an average of 13 days longer on the rehabilitation service and had worse functional and cognitive status on discharge, even after controlling for multiple confounders. Several subsequent studies have confirmed these and similar findings.8,33,34
OSA has also been linked to depression,35 which is common after stroke and may worsen outcomes.36 The interaction between OSA, depression, and poststroke outcomes warrants further study.
In the general population, OSA has been independently associated with increased risk of stroke or death from any cause.21,22,37 These associations have also been reported in the poststroke population: a 2014 meta-analysis found that OSA increased the risk of a repeat stroke (relative risk [RR] 1.8, 95% confidence interval [CI] 1.2–2.6) and all-cause mortality (RR 1.69, 95% CI 1.4–2.1).38
TESTING FOR OSA AFTER STROKE
Because of the high prevalence of OSA in patients who have had a stroke and the potential for worse outcomes associated with untreated OSA, there should be a low threshold for evaluating for OSA soon after stroke. Objective testing is required to qualify for therapy, and the gold standard for diagnosis of OSA is formal polysomnography conducted in a sleep laboratory.2–4 Unfortunately, polysomnography may be unacceptable to some patients, is costly, and is resource-intensive, particularly in an inpatient or rehabilitation setting.28 Ideally, to optimize testing efficiency, patients should be screened for the likelihood of OSA before polysomnography is ordered.
Questionnaires can help determine the need for further testing
Questionnaires developed to assess OSA risk39 include the following:
The Berlin questionnaire, developed in 1999, has 10 questions assessing daytime and nighttime signs and symptoms and presence of hypertension.
The STOP questionnaire, developed in 2008, assesses snoring, tiredness, observed apneic episodes, and elevated blood pressure.
The STOP-BANG questionnaire, published in 2010, includes the STOP questions plus BMI over 35 kg/m2, age over 50, neck circumference over 41 cm, and male gender.
A 2017 meta-analysis39 of 108 studies with nearly 50,000 people found that the STOP-BANG questionnaire performed best with regard to sensitivity and diagnostic odds ratio, but with poor specificity.
These screening tools and modified versions of them have also been evaluated in patients who have had a stroke.
In 2015, Boulos et al28 found that the STOP-BAG (a version of STOP-BANG that excludes neck circumference) and the 4-variable (4V) questionnaire (sex, BMI, blood pressure, snoring) had moderate predictive value for OSA within 6 months after sroke.
In 2016, Katzan et al40 found that the STOP-BAG2 (STOP-BAG criteria plus continuous variables for BMI and age) had a high sensitivity for polysomnographically diagnosed OSA within the first year after a stroke. The specificity was significantly better than the STOP-BANG or the STOP-BAG questionnaire, although it remained suboptimal at 60.5%.
In 2017, Sico et al41 developed and assessed the SLEEP Inventory (sex, left heart failure, Epworth Sleepiness Scale, enlarged neck, weight in pounds, insulin resistance or diabetes, and National Institutes of Health Stroke Scale) and found that it outperformed the Berlin and STOP-BANG questionnaires in the poststroke setting. The SLEEP Inventory had the best specificity and negative predictive value, and a slightly better ability to correctly classify patients as having OSA or not, classifying 80% of patients correctly.
These newer screening tools (eg, STOP-BAG, STOP-BAG2, SLEEP) can be used to identify with reasonable accuracy which patients need definitive testing after stroke.
Pulse oximetry is another possible screening tool
Overnight pulse oximetry may also help screen for sleep apnea and stratify risk after a stroke. A 2012 study42 of overnight oximetry to screen patients before surgery found that the oxygen desaturation index was significantly associated with the AHI measured by polysomnography. However, oximetry testing cannot distinguish between OSA and central sleep apnea, so it is insufficient to diagnose OSA or qualify patients for therapy. Further study is needed to examine the ability of overnight pulse oximetry to screen or to stratify risk for OSA after stroke.
Polysomnography vs home testing
Polysomnography is the gold standard for diagnosing OSA. Benefits include technical support and trouble-shooting, determining relationships between OSA, body position, and sleep stage, and the ability to intervene with treatment.2 However, polysomnography can be cumbersome, costly, and resource-intensive.
A home sleep apnea test, ie, an unattended, limited-channel sleep study, may be an acceptable alternative.2–4,43,44 Home testing does not require a sleep technologist to be present during testing, uses fewer sensors, and is less expensive than overnight polysomnography, but its utility can be limited: it fails to accurately discriminate between episodes of OSA and central sleep apnea, there is potential for false-negative results, and it can underestimate sleep apnea burden because it does not measure sleep.2
Institutional resources and logistics may influence the choice of diagnostic modality. No data exist on outcomes from different diagnostic testing methods in poststroke patients. Further research is needed.
POSITIVE AIRWAY PRESSURE THERAPY: BENEFITS, CHALLENGES, ALTERNATIVES
The first-line treatment for OSA is positive airway pressure (PAP).3 For most patients, this is continuous PAP (CPAP) or autoadjusting PAP (APAP). In some instances, particularly for those who cannot tolerate CPAP or who have comorbid hypoventilation, bilevel PAP (BPAP) may be indicated. More advanced PAP therapies are unlikely to be used after stroke.
PAP therapy is associated with reduced daytime sleepiness, improved mood, normalization of sleep architecture, improved systemic and pulmonary artery blood pressure, reduced rates of atrial fibrillation after ablation, and improved insulin sensitivity.45–49 Whether it reduces the risk of cardiovascular events, including stroke, remains controversial; most data suggest that it does not.50,51 However, when adherence to PAP therapy is considered rather than intention to treat, treatment has been found to lead to improved cardiovascular outcomes.52
Mixed evidence of benefits after stroke
Observational studies provide evidence that CPAP may help patients with OSA after stroke, although results are mixed.53–58 The studies ranged in size from 14 to 105 patients, enrolled patients with mostly moderate to severe OSA, and followed patients from 10 days to 7 years. Adherence to therapy was generally good in the short term (50%–70%), but only 15% to 30% of patients remained adherent at 5 to 7 years. Variable outcomes were reported, with some studies finding improved symptoms in the near term and mixed evidence of cardiovascular benefit in the longer ones. However, as these studies lacked randomization, drawing definitive conclusions on CPAP efficacy is difficult.
Patients were enrolled in the index admission or when starting a rehabilitation service—generally 2 to 3 weeks after their stroke. No clear association was found between the timing of initiating PAP therapy and outcomes. All patients had ischemic strokes, but few details were provided regarding stroke location, size, and severity. Exclusion criteria included severe underlying cardiopulmonary disease, confusion, severe stroke with marked impairment, and inability to cooperate. Almost all patients had moderate to severe OSA, and patients with central sleep apnea were excluded.
The major outcomes examined were drop-out rates, PAP adherence, and neurologic improvement based on neurologic functional scales (National Institutes of Health Stroke Scale and Canadian Neurologic Scale). As expected, dropout rates were higher in patients randomized to CPAP (OR 1.83, 95% CI 1.05–3.21, P = .03), although overall adherence was better than anticipated, with mean CPAP use across trials of 4.5 hours per night (95% CI 3.97–5.08) and with about 50% to 60% of patients adhering to therapy for at least 4 hours nightly.
Improvement in neurologic outcomes favored CPAP (standard mean difference 0.54, 95% CI 0.026–1.05), although considerable heterogeneity was seen. Improved sleepiness outcomes were inconsistent. Major cardiovascular outcomes were reported in only 2 studies (using the same data set) and showed delayed time to the next cardiovascular event for those treated with CPAP but no difference in cardiovascular event-free survival.
PAP poses more challenges after stroke
The primary limitation to PAP therapy is poor acceptance and adherence to therapy.59 High rates of refusal of therapy and difficulty complying with treatment have been noted in the poststroke population, although recent studies have reported better adherence rates. How rates of adherence play out in real-world settings, outside of the controlled environment of a research study, has yet to be determined.
In general, CPAP adherence is affected by claustrophobia, difficulty tolerating a mask, problems with pressure intolerance, irritating air leaks, nasal congestion, and naso-oral dryness. Many such barriers can be overcome with use of a properly fitted mask, an appropriate pressure setting, heated humidification, nasal sprays (eg, saline, inhaled steroids), and education, encouragement, and reassurance.
After a stroke, additional obstacles may impede the ability to use PAP therapy.68 Facial paresis (hemi- or bifacial) may make fitting of the mask problematic. Paralysis or weakness of the extremities may limit the ability to adjust or remove a mask. Aphasia can impair communication and understanding of the need to use PAP therapy, and upper-airway problems related to stroke, including dysphagia, may lead to pressure intolerance or risk of aspiration. Finally, a lack of perceived benefit, particularly if the patient does not have daytime sleepiness, may limit motivation.
Consider alternatives
For patients unlikely to succeed with PAP therapy, there are alternatives. Surgery and oral appliances are not usually realistic options in the setting of recent stroke, but positional therapy, including the use of body positioners to prevent supine sleep, as well as elevating the head of the bed, may be of some benefit.69,70 A nasopharyngeal airway stenting device (nasal trumpet) may also be tolerated by some patients.
A proposed algorithm for screening, diagnosing, and treating OSA in patients after stroke is presented in Figure 1.
Obstructive sleep apnea (OSA) is an independent risk factor for ischemic stroke and may also, infrequently, be a consequence of stroke. It is significantly underdiagnosed in the general population and is highly prevalent in patients who have had a stroke. Many patients likely had their stroke because of this chronic untreated condition.
This review focuses on OSA and its prevalence, consequences, and treatment in patients after a stroke.
DEFINING AND QUANTIFYING OSA
OSA is the most common type of sleep-disordered breathing.1,2 It involves repeated narrowing or complete collapse of the upper airway despite ongoing respiratory effort.3,4 Apneic episodes are terminated by arousals from hypoxemia or efforts to breathe.5 In contrast, central sleep apnea is characterized by a patent airway but lack of airflow due to absent respiratory effort.5
In OSA, the number of episodes of apnea (absent airflow) and hypopnea (reduced airflow) are added together and divided by hours of sleep to calculate the apnea-hypopnea index (AHI). OSA is diagnosed by either of the following3,4:
- AHI of 5 or higher, with clinical symptoms related to OSA (described below)
- AHI of 15 or higher, regardless of symptoms.
The AHI also defines OSA severity, as follows3:
- Mild: AHI 5 to 15
- Moderate: AHI 15 to 30
- Severe: AHI greater than 30.
Diagnostic criteria (eg, definition of hypopnea, testing methods, and AHI thresholds) have varied over time, an important consideration when reviewing the literature.
OSA IS MORE COMMON THAN EXPECTED AFTER STROKE
In the most methodologically sound and generalizable study of this topic to date, the Wisconsin Sleep Cohort Study6 reported in 2013 that about 14% of men and 5% of women ages 30 to 70 have an AHI greater than 5 (using 4% desaturation to score hypopneic episodes) with daytime sleepiness. Other studies suggest that 80% to 90% of people with OSA are undiagnosed and untreated.1,7
The prevalence of OSA in patients who have had a stroke is much higher, ranging from 30% to 96% depending on the study methods and population.1,8–12 A 2010 meta-analysis11 of 29 studies reported that 72% of patients who had a stroke had an AHI greater than 5, and 29% had severe OSA. In this analysis, 7% of those with sleep-disordered breathing had central sleep apnea; still, these data indicate that the prevalence of OSA in these patients is about 5 times higher than in the general population.
RISK FACTORS MAY DIFFER IN STROKE POPULATION
Several risk factors for OSA have been identified.
Obesity is one of the strongest risk factors, with increasing body mass index (BMI) associated with increased OSA prevalence.4,6,13 However, obesity appears to be a less significant risk factor in patients who have had a stroke than in the general population. In the 2010 meta-analysis11 of OSA after stroke, the average BMI was only 26.4 kg/m2 (with obesity defined as a BMI > 30.0 kg/m2), and increasing BMI was not associated with increasing AHI.
Male sex and advanced age are also OSA risk factors.4,5 They remain significant in patients after a stroke; about 65% of poststroke patients who have OSA are men, and the older the patient, the more likely the AHI is greater than 10.11
Ethnicity and genetics may also play important roles in OSA risk, with roughly 25% of OSA prevalence estimated to have a genetic basis.14,15 Some risk factors for OSA such as craniofacial shape, upper airway anatomy, upper airway muscle dysfunction, increased respiratory chemosensitivity, and poor arousal threshold during sleep are likely determined by genetics and ethnicity.14,15 Compared with people of European origin, Asians have a similar prevalence of OSA, but at a much lower average BMI, suggesting that other factors are significant.14 Possible genetically determined anatomic risk factors have not been specifically studied in the poststroke population, but it can be assumed they remain relevant.
Several studies have tried to find an association between OSA and type, location, etiology, or pattern of stroke.10,11,16–19 Although some suggest links between cardioembolic stroke and OSA,16,20 or thrombolysis and OSA,10 most have found no association between OSA and stroke features.11,12,21,22
HOW DOES OSA INCREASE STROKE RISK?
Untreated severe OSA is associated with increased cardiovascular mortality,21,22 and OSA is an independent risk factor for incident stroke.23 A number of mechanisms may explain these relationships.
Intermittent hypoxemia and recurrent sympathetic arousals resulting from OSA are thought to lead to many of the comorbid conditions with which it is associated: hypertension, coronary artery disease, heart failure, arrhythmias, pulmonary hypertension, and stroke. Repetitive decreases in ventilation lead to oxygen desaturations that result in cycles of increased sympathetic outflow and eventual sustained nocturnal hypertension and daytime chronic hypertension.1,5,9,13 Also implicated are various changes in vasodilator and vasoconstrictor substances due to endothelial dysfunction and inflammation, which are thought to play a role in the atherogenic and prothrombotic states induced by OSA.1,5,13
Cerebral circulation is altered primarily by the changes in partial pressure of carbon dioxide (Pco2). During apnea, the Pco2 rises, causing vasodilation and increased blood flow. After the apnea resolves, there is hyperpnea with resultant decreased Pco2, and vasoconstriction. In a patient who already has vascular disease, the enhanced vasoconstriction could lead to ischemia.1,5
Changes in intrathoracic pressure result in distortion of cardiac architecture. When the patient tries to breathe against an occluded airway, the intrathoracic pressure becomes more and more negative, increasing preload and afterload. When this happens repeatedly every night for years, it leads to remodeling of the heart such as left and right ventricular hypertrophy, with reduced stroke volume, myocardial ischemia, and increased risk of arrhythmia.1,5,13
Untreated OSA is believed to predispose patients to develop atrial fibrillation through sympathetic overactivity, vascular inflammation, heart rate variability, and cardiac remodeling.24 As atrial fibrillation is a major risk factor for stroke, particularly cardioembolic stroke, it may be another pathway of increased stroke risk in OSA.16,20,25
CLINICAL MANIFESTATIONS OF OSA NOT OBVIOUS AFTER STROKE
OSA typically causes both daytime symptoms (excessive sleepiness, poor concentration, morning headache, depressive symptoms) and nighttime signs and symptoms (snoring, choking, gasping, night sweats, insomnia, nocturia, witnessed episodes of apnea).3,4,26 Unfortunately, because these are nonspecific, OSA is often underdiagnosed.4,26
Identifying OSA after a stroke may be a particular challenge, as patients often do not report classic symptoms, and the typical picture of OSA may have less predictive validity in these patients.1,27,28 Within the first 24 hours after a stroke, hypersomnia, snoring history, and age are not predictive of OSA.1 Patients found to have OSA after a stroke frequently do not have the traditional symptoms (sleepiness, snoring) seen in usual OSA patients. And they have higher rates of OSA at a younger age than the usual OSA patients, so age is not a predictive risk factor. In addition, daytime sleepiness and obesity are often absent or less prominent.1,9,27,28 Finally, typical OSA signs and symptoms may be attributed to the stroke itself or to comorbidities affecting the patient, lowering suspicion for OSA.
OSA MAY HINDER STROKE RECOVERY, WORSEN OUTCOMES
OSA, particularly when moderate to severe, is linked to pathophysiologic changes that can hinder recovery from a stroke.
Intermittent hypoxemia during sleep can worsen vascular damage of at-risk tissue: nocturnal hypoxemia correlates with white matter hyperintensities on magnetic resonance imaging, a marker of ischemic demyelination.29 Oxidative stress and release of inflammatory mediators associated with intermittent hypoxemia may impair vascular blood flow to brain tissue attempting to repair itself.30 In addition, sympathetic overactivity and Pco2 fluctuations associated with OSA may impede cerebral circulation.
Taken together, such ongoing nocturnal insults can lead to clinical consequences during this vulnerable period.
A 1996 study31 of patients recovering from a stroke found that an oxygen desaturation index (number of times that the blood oxygen level drops below a certain threshold, as measured by overnight oximetry) of more than 10 per hour was associated with worse functional recovery at discharge and at 3 and 12 months after discharge. This study also noted an association between time spent with oxygen saturations below 90% and the rate of death at 1 year.
A 2003 study32 reported that patients with an AHI greater than 10 by polysomnography spent an average of 13 days longer on the rehabilitation service and had worse functional and cognitive status on discharge, even after controlling for multiple confounders. Several subsequent studies have confirmed these and similar findings.8,33,34
OSA has also been linked to depression,35 which is common after stroke and may worsen outcomes.36 The interaction between OSA, depression, and poststroke outcomes warrants further study.
In the general population, OSA has been independently associated with increased risk of stroke or death from any cause.21,22,37 These associations have also been reported in the poststroke population: a 2014 meta-analysis found that OSA increased the risk of a repeat stroke (relative risk [RR] 1.8, 95% confidence interval [CI] 1.2–2.6) and all-cause mortality (RR 1.69, 95% CI 1.4–2.1).38
TESTING FOR OSA AFTER STROKE
Because of the high prevalence of OSA in patients who have had a stroke and the potential for worse outcomes associated with untreated OSA, there should be a low threshold for evaluating for OSA soon after stroke. Objective testing is required to qualify for therapy, and the gold standard for diagnosis of OSA is formal polysomnography conducted in a sleep laboratory.2–4 Unfortunately, polysomnography may be unacceptable to some patients, is costly, and is resource-intensive, particularly in an inpatient or rehabilitation setting.28 Ideally, to optimize testing efficiency, patients should be screened for the likelihood of OSA before polysomnography is ordered.
Questionnaires can help determine the need for further testing
Questionnaires developed to assess OSA risk39 include the following:
The Berlin questionnaire, developed in 1999, has 10 questions assessing daytime and nighttime signs and symptoms and presence of hypertension.
The STOP questionnaire, developed in 2008, assesses snoring, tiredness, observed apneic episodes, and elevated blood pressure.
The STOP-BANG questionnaire, published in 2010, includes the STOP questions plus BMI over 35 kg/m2, age over 50, neck circumference over 41 cm, and male gender.
A 2017 meta-analysis39 of 108 studies with nearly 50,000 people found that the STOP-BANG questionnaire performed best with regard to sensitivity and diagnostic odds ratio, but with poor specificity.
These screening tools and modified versions of them have also been evaluated in patients who have had a stroke.
In 2015, Boulos et al28 found that the STOP-BAG (a version of STOP-BANG that excludes neck circumference) and the 4-variable (4V) questionnaire (sex, BMI, blood pressure, snoring) had moderate predictive value for OSA within 6 months after sroke.
In 2016, Katzan et al40 found that the STOP-BAG2 (STOP-BAG criteria plus continuous variables for BMI and age) had a high sensitivity for polysomnographically diagnosed OSA within the first year after a stroke. The specificity was significantly better than the STOP-BANG or the STOP-BAG questionnaire, although it remained suboptimal at 60.5%.
In 2017, Sico et al41 developed and assessed the SLEEP Inventory (sex, left heart failure, Epworth Sleepiness Scale, enlarged neck, weight in pounds, insulin resistance or diabetes, and National Institutes of Health Stroke Scale) and found that it outperformed the Berlin and STOP-BANG questionnaires in the poststroke setting. The SLEEP Inventory had the best specificity and negative predictive value, and a slightly better ability to correctly classify patients as having OSA or not, classifying 80% of patients correctly.
These newer screening tools (eg, STOP-BAG, STOP-BAG2, SLEEP) can be used to identify with reasonable accuracy which patients need definitive testing after stroke.
Pulse oximetry is another possible screening tool
Overnight pulse oximetry may also help screen for sleep apnea and stratify risk after a stroke. A 2012 study42 of overnight oximetry to screen patients before surgery found that the oxygen desaturation index was significantly associated with the AHI measured by polysomnography. However, oximetry testing cannot distinguish between OSA and central sleep apnea, so it is insufficient to diagnose OSA or qualify patients for therapy. Further study is needed to examine the ability of overnight pulse oximetry to screen or to stratify risk for OSA after stroke.
Polysomnography vs home testing
Polysomnography is the gold standard for diagnosing OSA. Benefits include technical support and trouble-shooting, determining relationships between OSA, body position, and sleep stage, and the ability to intervene with treatment.2 However, polysomnography can be cumbersome, costly, and resource-intensive.
A home sleep apnea test, ie, an unattended, limited-channel sleep study, may be an acceptable alternative.2–4,43,44 Home testing does not require a sleep technologist to be present during testing, uses fewer sensors, and is less expensive than overnight polysomnography, but its utility can be limited: it fails to accurately discriminate between episodes of OSA and central sleep apnea, there is potential for false-negative results, and it can underestimate sleep apnea burden because it does not measure sleep.2
Institutional resources and logistics may influence the choice of diagnostic modality. No data exist on outcomes from different diagnostic testing methods in poststroke patients. Further research is needed.
POSITIVE AIRWAY PRESSURE THERAPY: BENEFITS, CHALLENGES, ALTERNATIVES
The first-line treatment for OSA is positive airway pressure (PAP).3 For most patients, this is continuous PAP (CPAP) or autoadjusting PAP (APAP). In some instances, particularly for those who cannot tolerate CPAP or who have comorbid hypoventilation, bilevel PAP (BPAP) may be indicated. More advanced PAP therapies are unlikely to be used after stroke.
PAP therapy is associated with reduced daytime sleepiness, improved mood, normalization of sleep architecture, improved systemic and pulmonary artery blood pressure, reduced rates of atrial fibrillation after ablation, and improved insulin sensitivity.45–49 Whether it reduces the risk of cardiovascular events, including stroke, remains controversial; most data suggest that it does not.50,51 However, when adherence to PAP therapy is considered rather than intention to treat, treatment has been found to lead to improved cardiovascular outcomes.52
Mixed evidence of benefits after stroke
Observational studies provide evidence that CPAP may help patients with OSA after stroke, although results are mixed.53–58 The studies ranged in size from 14 to 105 patients, enrolled patients with mostly moderate to severe OSA, and followed patients from 10 days to 7 years. Adherence to therapy was generally good in the short term (50%–70%), but only 15% to 30% of patients remained adherent at 5 to 7 years. Variable outcomes were reported, with some studies finding improved symptoms in the near term and mixed evidence of cardiovascular benefit in the longer ones. However, as these studies lacked randomization, drawing definitive conclusions on CPAP efficacy is difficult.
Patients were enrolled in the index admission or when starting a rehabilitation service—generally 2 to 3 weeks after their stroke. No clear association was found between the timing of initiating PAP therapy and outcomes. All patients had ischemic strokes, but few details were provided regarding stroke location, size, and severity. Exclusion criteria included severe underlying cardiopulmonary disease, confusion, severe stroke with marked impairment, and inability to cooperate. Almost all patients had moderate to severe OSA, and patients with central sleep apnea were excluded.
The major outcomes examined were drop-out rates, PAP adherence, and neurologic improvement based on neurologic functional scales (National Institutes of Health Stroke Scale and Canadian Neurologic Scale). As expected, dropout rates were higher in patients randomized to CPAP (OR 1.83, 95% CI 1.05–3.21, P = .03), although overall adherence was better than anticipated, with mean CPAP use across trials of 4.5 hours per night (95% CI 3.97–5.08) and with about 50% to 60% of patients adhering to therapy for at least 4 hours nightly.
Improvement in neurologic outcomes favored CPAP (standard mean difference 0.54, 95% CI 0.026–1.05), although considerable heterogeneity was seen. Improved sleepiness outcomes were inconsistent. Major cardiovascular outcomes were reported in only 2 studies (using the same data set) and showed delayed time to the next cardiovascular event for those treated with CPAP but no difference in cardiovascular event-free survival.
PAP poses more challenges after stroke
The primary limitation to PAP therapy is poor acceptance and adherence to therapy.59 High rates of refusal of therapy and difficulty complying with treatment have been noted in the poststroke population, although recent studies have reported better adherence rates. How rates of adherence play out in real-world settings, outside of the controlled environment of a research study, has yet to be determined.
In general, CPAP adherence is affected by claustrophobia, difficulty tolerating a mask, problems with pressure intolerance, irritating air leaks, nasal congestion, and naso-oral dryness. Many such barriers can be overcome with use of a properly fitted mask, an appropriate pressure setting, heated humidification, nasal sprays (eg, saline, inhaled steroids), and education, encouragement, and reassurance.
After a stroke, additional obstacles may impede the ability to use PAP therapy.68 Facial paresis (hemi- or bifacial) may make fitting of the mask problematic. Paralysis or weakness of the extremities may limit the ability to adjust or remove a mask. Aphasia can impair communication and understanding of the need to use PAP therapy, and upper-airway problems related to stroke, including dysphagia, may lead to pressure intolerance or risk of aspiration. Finally, a lack of perceived benefit, particularly if the patient does not have daytime sleepiness, may limit motivation.
Consider alternatives
For patients unlikely to succeed with PAP therapy, there are alternatives. Surgery and oral appliances are not usually realistic options in the setting of recent stroke, but positional therapy, including the use of body positioners to prevent supine sleep, as well as elevating the head of the bed, may be of some benefit.69,70 A nasopharyngeal airway stenting device (nasal trumpet) may also be tolerated by some patients.
A proposed algorithm for screening, diagnosing, and treating OSA in patients after stroke is presented in Figure 1.
- Selim B, Roux FJ. Stroke and sleep disorders. Sleep Med Clin 2012; 7(4):597–607. doi:10.1016/j.jsmc.2012.08.007
- Kapur VK, Auckley DH, Chowdhuri S, et al. Clinical practice guideline for diagnostic testing for adult obstructive sleep apnea: an American Academy of Sleep Medicine Clinical Practice Guideline. J Clin Sleep Med 2017; 13(3):479–504. doi:10.5664/jcsm.6506
- Epstein LJ, Kristo D, Strollo PJ Jr, et al; Adult Obstructive Sleep Apnea Task Force of the American Academy of Sleep Medicine. Clinical guideline for the evaluation, management, and long-term care of obstructive sleep apnea in adults. J Clin Sleep Med 2009; 5(3):263–276. pmid:19960649
- Patil SP, Schneider H, Schwartz AR, Smith PL. Adult obstructive sleep apnea: pathophysiology and diagnosis. Chest 2007; 132(1):325–337. doi:10.1378/chest.07-0040
- Dempsey JA, Veasey SC, Morgan BJ, O’Donnell CP. Pathophysiology of sleep apnea. Physiol Rev 2010; 90(1):47–112. doi:10.1152/physrev.00043.2008
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177(9):1006–1014. doi:10.1093/aje/kws342
- Redline S, Sotres-Alvarez D, Loredo J, et al. Sleep-disordered breathing in Hispanic/Latino individuals of diverse backgrounds. The Hispanic Community Health Study/Study of Latinos. Am J Resp Crit Care Med 2014; 189(3):335–344. doi:10.1164/rccm.201309-1735OC
- Aaronson JA, van Bennekom CA, Hofman WF, et al. Obstructive sleep apnea is related to impaired cognitive and functional status after stroke. Sleep 2015; 38(9):1431–1437. doi:10.5665/sleep.4984
- Sharma S, Culebras A. Sleep apnoea and stroke. Stroke Vasc Neurol 2016; 1(4):185–191. doi:10.1136/svn-2016-000038
- Huhtakangas JK, Huhtakangas J, Bloigu R, Saaresranta T. Prevalence of sleep apnea at the acute phase of ischemic stroke with or without thrombolysis. Sleep Med 2017; 40:40–46. doi:10.1016/j.sleep.2017.08.018
- Johnson KG, Johnson DC. Frequency of sleep apnea in stroke and TIA patients: a meta-analysis. J Clin Sleep Med 2010; 6(2):131–137. pmid:20411688
- Iranzo A, Santamaria J, Berenguer J, Sanchez M, Chamorro A. Prevalence and clinical importance of sleep apnea in the first night after cerebral infarction. Neurology 2002; 58:911–916. pmid:11914407
- Javaheri S, Barbe F, Campos-Rodriguez F, et al. Sleep apnea: types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol 2017; 69(7):841–858. doi:10.1016/j.jacc.2016.11.069
- Dudley KA, Patel SR. Disparities and genetic risk factors in obstructive sleep apnea. Sleep Med 2016; 18:96–102. doi:10.1016/j.sleep.2015.01.015
- Redline S, Tishler PV. The genetics of sleep apnea. Sleep Med Rev 2000; 4(6):583–602. doi:10.1053/smrv.2000.0120
- Lipford MC, Flemming KD, Calvin AD, et al. Associations between cardioembolic stroke and obstructive sleep apnea. Sleep 2015; 38(11):1699–1705. doi:10.5665/sleep.5146
- Wang Y, Wang Y, Chen J, Yi X, Dong S, Cao L. Stroke patterns, topography, and etiology in patients with obstructive sleep apnea hypopnea syndrome. Int J Clin Exp Med 2017; 10(4):7137–7143.
- Fisse AL, Kemmling A, Teuber A, et al. The association of lesion location and sleep related breathing disorder in patients with acute ischemic stroke. PLoS One 2017; 12(1):e0171243. doi:10.1371/journal.pone.0171243
- Brown DL, Mowla A, McDermott M, et al. Ischemic stroke subtype and presence of sleep-disordered breathing: the BASIC sleep apnea study. J Stroke Cerebrovasc Dis 2015; 24(2):388–393. doi:10.1016/j.jstrokecerebrovasdis.2014.09.007
- Poli M, Philip P, Taillard J, et al. Atrial fibrillation as a major cause of stroke in apneic patients: a prospective study. Sleep Med 2017; 30:251–254. doi:10.1016/j.sleep.2015.07.031
- Young T, Finn L, Peppard PE, et al. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin Sleep Cohort. Sleep 2008; 31(8):1071–1078. pmid:18714778
- Molnar MZ, Mucsi I, Novak M, et al. Association of incident obstructive sleep apnoea with outcomes in a large cohort of US veterans. Thorax 2015; 70(9):888–895. doi:10.1136/thoraxjnl-2015-206970
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the sleep heart health study. Am J Respir Crit Care Med 2010; 182(2):269–277. doi:10.1164/rccm.200911-1746OC
- Marulanda-Londono E, Chaturvedi S. The interplay between obstructive sleep apnea and atrial fibrillation. Fron Neurol 2017; 8:668. doi:10.3389/fneur.2017.00668
- Szymanski FM, Filipiak KJ, Platek AE, Hrynkiewicz-Szymanska A, Karpinski G, Opolski G. Assessment of CHADS2 and CHA 2DS 2-VASc scores in obstructive sleep apnea patients with atrial fibrillation. Sleep Breath 2015; 19(2):531–537. doi:10.1007/s11325-014-1042-5
- Stansbury RC, Strollo PJ. Clinical manifestations of sleep apnea. J Thoracic Dis 2015; 7(9):E298–E310. doi:10.3978/j.issn.2072-1439.2015.09.13
- Chan W, Coutts SB, Hanly P. Sleep apnea in patients with transient ischemic attack and minor stroke: opportunity for risk reduction of recurrent stroke? Stroke 2010; 41(12):2973–2975. doi:10.1161/STROKEAHA.110.596759
- Boulos MI, Wan A, Im J, et al. Identifying obstructive sleep apnea after stroke/TIA: evaluating four simple screening tools. Sleep Med 2016; 21:133–139. doi:10.1016/j.sleep.2015.12.013
- Patel SK, Hanly PJ, Smith EE, Chan W, Coutts SB. Nocturnal hypoxemia is associated with white matter hyperintensities in patients with a minor stroke or transient ischemic attack. J Clin Sleep Med 2015; 11(12):1417–1424. doi:10.5664/jcsm.5278
- McCarty MF, DiNicolantonio JJ, O’Keefe JH. NADPH oxidase, uncoupled endothelial nitric oxide synthase, and NF-KappaB are key mediators of the pathogenic impact of obstructive sleep apnea—therapeutic implications. J Integr Cardiol 2016; 2(5):367–374. doi:10.15761/JIC.1000177
- Good DC, Henkle JQ, Gelber D, Welsh J, Verhulst S. Sleep-disordered breathing and poor functional outcome after stroke. Stroke 1996; 27(2):252–259. pmid:8571419
- Kaneko Y, Hajek VE, Zivanovic V, Raboud J, Bradley TD. Relationship of sleep apnea to functional capacity and length of hospitalization following stroke. Sleep 2003; 26(3):293–297. pmid:12749548
- Yan-fang S, Yu-ping W. Sleep-disordered breathing: impact on functional outcome of ischemic stroke patients. Sleep Med 2009; 10(7):717–719. doi:10.1016/j.sleep.2008.08.006
- Kumar R, Suri JC, Manocha R. Study of association of severity of sleep disordered breathing and functional outcome in stroke patients. Sleep Med 2017; 34:50–56. doi:10.1016/j.sleep.2017.02.025
- Kerner NA, Roose SP. Obstructive sleep apnea is linked to depression and cognitive impairment: evidence and potential mechanisms. Am J Geriatr Psychiatry 2016; 24(6):496–508. doi:10.1016/j.jagp.2016.01.134
- Bartoli F, Lillia N, Lax A, et al. Depression after stroke and risk of mortality: a systematic review and meta-analysis. Stroke Res Treat 2013; 2013:862978. doi:10.1155/2013/862978
- Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 2005; 353(19):2034–2041. doi:10.1056/NEJMoa043104
- Xie W, Zheng F, Song X. Obstructive sleep apnea and serious adverse outcomes in patients with cardiovascular or cerebrovascular disease: a PRISMA-compliant systematic review and meta-analysis. Medicine (Baltimore) 2014; 93(29):e336. doi:10.1097/MD.0000000000000336
- Chiu HY, Chen PY, Chuang LP, et al. Diagnostic accuracy of the Berlin questionnaire, STOP-BANG, STOP, and Epworth sleepiness scale in detecting obstructive sleep apnea: a bivariate meta-analysis. Sleep Med Rev 2017; 36:57–70. doi:10.1016/j.smrv.2016.10.004
- Katzan IL, Thompson NR, Uchino K, Foldvary-Schaefer N. A screening tool for obstructive sleep apnea in cerebrovascular patients. Sleep Med 2016; 21:70–76. doi:10.1016/j.sleep.2016.02.001
- Sico JJ, Yaggi HK, Ofner S, et al. Development, validation, and assessment of an ischemic stroke or transient ischemic attack-specific prediction tool for obstructive sleep apnea. J Stroke Cerebrovasc Dis 2017; 26(8):1745–1754. doi:10.1016/j.jstrokecerebrovasdis.2017.03.042
- Chung F, Liao P, Elsaid H, Islam S, Shapiro CM, Sun Y. Oxygen desaturation index from nocturnal oximetry: a sensitive and specific tool to detect sleep-disordered breathing in surgical patients. Anesth Analg 2012; 114(5):993–1000. doi:10.1213/ANE.0b013e318248f4f5
- Boulos MI, Elias S, Wan A, et al. Unattended hospital and home sleep apnea testing following cerebrovascular events. J Stroke Cerebrovasc Dis 2017; 26(1):143–149. doi:10.1016/j.jstrokecerebrovasdis.2016.09.001
- Saletu MT, Kotzian ST, Schwarzinger A, Haider S, Spatt J, Saletu B. Home sleep apnea testing is a feasible and accurate method to diagnose obstructive sleep apnea in stroke patients during in-hospital rehabilitation. J Clin Sleep Med 2018; 14(9):1495–1501. doi:10.5664/jcsm.7322
- Giles TL, Lasserson TJ, Smith BH, White J, Wright J, Cates CJ. Continuous positive airways pressure for obstructive sleep apnoea in adults. Cochrane Database Syst Rev 2006; (3):CD001106. doi:10.1002/14651858.CD001106.pub3
- Fatureto-Borges F, Lorenzi-Filho G, Drager LF. Effectiveness of continuous positive airway pressure in lowering blood pressure in patients with obstructive sleep apnea: a critical review of the literature. Integr Blood Press Control 2016; 9:43–47. doi:10.2147/IBPC.S70402
- Imran TF, Gharzipura M, Liu S, et al. Effect of continuous positive airway pressure treatment on pulmonary artery pressure in patients with isolated obstructive sleep apnea: a meta-analysis. Heart Fail Rev 2016; 21(5):591–598. doi:10.1007/s10741-016-9548-5
- Deng F, Raza A, Guo J. Treating obstructive sleep apnea with continuous positive airway pressure reduces risk of recurrent atrial fibrillation after catheter ablation: a meta-analysis. Sleep Med 2018; 46:5–11. doi:10.1016/j.sleep.2018.02.013
- Seetho IW, Wilding JPH. Sleep-disordered breathing, type 2 diabetes, and the metabolic syndrome. Chronic Resp Dis 2014; 11(4):257–275. doi:10.1177/1479972314552806
- Kim Y, Koo YS, Lee HY, Lee SY. Can continuous positive airway pressure reduce the risk of stroke in obstructive sleep apnea patients? A systematic review and meta-analysis. PloS ONE 2016; 11(1):e0146317. doi:10.1371/journal.pone.0146317
- Yu J, Zhou Z, McEvoy RD, et al. Association of positive airway pressure with cardiovascular events and death in adults with sleep apnea: a systematic review and meta-analysis. JAMA 2017; 318(2):156–166. doi:10.1001/jama.2017.7967
- Peker Y, Glantz H, Eulenburg C, Wegscheider K, Herlitz J, Thunström E. Effect of positive airway pressure on cardiovascular outcomes in coronary artery disease patients with nonsleepy obstructive sleep apnea. The RICCADSA randomized controlled trial. Am J Respir Crit Care Med 2016; 194(5):613–620. doi:10.1164/rccm.201601-0088OC
- Martinez-Garcia MA, Soler-Cataluna JJ, Ejarque-Martinez L, et al. Continuous positive airway pressure treatment reduces mortality in patients with ischemic stroke and obstructive sleep apnea: a 5-year follow-up study. Am J Respir Crit Care Med 2009; 180(1):36–41. doi:10.1164/rccm.200808-1341OC
- Broadley SA, Jorgensen L, Cheek A, et al. Early investigation and treatment of obstructive sleep apnoea after acute stroke. J Clin Neurosci 2007; 14(4):328–333. doi:10.1016/j.jocn.2006.01.017
- Wessendorf TE, Wang YM, Thilmann AF, Sorgenfrei U, Konietzko N, Teschler H. Treatment of obstructive sleep apnoea with nasal continuous positive airway pressure in stroke. Eur Respir J 2001; 18(4):623–629. pmid:11716165
- Bassetti CL, Milanova M, Gugger M. Sleep-disordered breathing and acute ischemic stroke: diagnosis, risk factors, treatment, evolution, and long-term clinical outcome. Stroke 2006; 37(4):967–972. doi:10.1161/01.STR.0000208215.49243.c3
- Palombini L, Guilleminault C. Stroke and treatment with nasal CPAP. Eur J Neurol 2006; 13(2):198–200. doi:10.1111/j.1468-1331.2006.01169.x
- Martínez-García MA, Campos-Rodríguez F, Soler-Cataluña JJ, Catalán-Serra P, Román-Sánchez P, Montserrat JM. Increased incidence of nonfatal cardiovascular events in stroke patients with sleep apnoea: effect of CPAP treatment. Eur Respir J 2012; 39(4):906–912. doi:10.1183/09031936.00011311
- Brill AK, Horvath T, Seiler A, et al. CPAP as treatment of sleep apnea after stroke: a meta-analysis of randomized trials. Neurology 2018; 90(14):e1222–e1230. doi:10.1212/WNL.0000000000005262
- Hsu C, Vennelle M, Li H, Engleman HM, Dennis MS, Douglas NJ. Sleep-disordered breathing after stroke: a randomised controlled trial of continuous positive airway pressure. J Neurol Neurosurg Psychiatry 2006; 77(10):1143–1149. doi:10.1136/jnnp.2005.086686
- Parra O, Sanchez-Armengol A, Bonnin M, et al. Early treatment of obstructive apnoea and stroke outcome: a randomised controlled trial. Eur Resp J 2011; 37(5):1128–1136. doi:10.1183/09031936.00034410
- Ryan CM, Bayley M, Green R, Murray BJ, Bradley TD. Influence of continuous positive airway pressure on outcomes of rehabilitation in stroke patients with obstructive sleep apnea. Stroke 2011; 42(4):1062–1067. doi:10.1161/STROKEAHA.110.597468
- Bravata DM, Concato J, Fried T, et al. Continuous positive airway pressure: evaluation of a novel therapy for patients with acute ischemic stroke. Sleep 2011; 34(9):1271–1277. doi:10.5665/SLEEP.1254
- Parra O, Sanchez-Armengol A, Capote F, et al. Efficacy of continuous positive airway pressure treatment on 5-year survival in patients with ischaemic stroke and obstructive sleep apnea: a randomized controlled trial. J Sleep Res 2015; 24(1):47–53. doi:10.1111/jsr.12181
- Khot SP, Davis AP, Crane DA, et al. Effect of continuous positive airway pressure on stroke rehabilitation: a pilot randomized sham-controlled trial. J Clin Sleep Med 2016; 12(7):1019–1026. doi:10.5664/jcsm.5940
- Aaronson JA, Hofman WF, van Bennekom CA, et al. Effects of continuous positive airway pressure on cognitive and functional outcome of stroke patients with obstructive sleep apnea: a randomized controlled trial. J Clin Sleep Med 2016; 12(4):533–541. doi:10.5664/jcsm.5684
- Gupta A, Shukla G, Afsar M, et al. Role of positive airway pressure therapy for obstructive sleep apnea in patients with stroke: a randomized controlled trial. J Clin Sleep Med 2018; 14(4):511–521. doi:10.5664/jcsm.7034
- Mello-Fujita L, Kim LJ, Palombini Lde O, et al. Treatment of obstructive sleep apnea syndrome associated with stroke. Sleep Med 2015; 16(6):691–696. doi:10.1016/j.sleep.2014.12.017
- Svatikova A, Chervin RD, Wing JJ, Sanchez BN, Migda EM, Brown DL. Positional therapy in ischemic stroke patients with obstructive sleep apnea. Sleep Med 2011; 12(3):262–266. doi:10.1016/j.sleep.2010.12.008
- Souza FJ, Genta PR, de Souza Filho AJ, Wellman A, Lorenzi-Filho G. The influence of head-of-bed elevation in patients with obstructive sleep apnea. Sleep Breath 2017; 21(4):815–820. doi:10.1007/s11325-017-1524-3
- Selim B, Roux FJ. Stroke and sleep disorders. Sleep Med Clin 2012; 7(4):597–607. doi:10.1016/j.jsmc.2012.08.007
- Kapur VK, Auckley DH, Chowdhuri S, et al. Clinical practice guideline for diagnostic testing for adult obstructive sleep apnea: an American Academy of Sleep Medicine Clinical Practice Guideline. J Clin Sleep Med 2017; 13(3):479–504. doi:10.5664/jcsm.6506
- Epstein LJ, Kristo D, Strollo PJ Jr, et al; Adult Obstructive Sleep Apnea Task Force of the American Academy of Sleep Medicine. Clinical guideline for the evaluation, management, and long-term care of obstructive sleep apnea in adults. J Clin Sleep Med 2009; 5(3):263–276. pmid:19960649
- Patil SP, Schneider H, Schwartz AR, Smith PL. Adult obstructive sleep apnea: pathophysiology and diagnosis. Chest 2007; 132(1):325–337. doi:10.1378/chest.07-0040
- Dempsey JA, Veasey SC, Morgan BJ, O’Donnell CP. Pathophysiology of sleep apnea. Physiol Rev 2010; 90(1):47–112. doi:10.1152/physrev.00043.2008
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177(9):1006–1014. doi:10.1093/aje/kws342
- Redline S, Sotres-Alvarez D, Loredo J, et al. Sleep-disordered breathing in Hispanic/Latino individuals of diverse backgrounds. The Hispanic Community Health Study/Study of Latinos. Am J Resp Crit Care Med 2014; 189(3):335–344. doi:10.1164/rccm.201309-1735OC
- Aaronson JA, van Bennekom CA, Hofman WF, et al. Obstructive sleep apnea is related to impaired cognitive and functional status after stroke. Sleep 2015; 38(9):1431–1437. doi:10.5665/sleep.4984
- Sharma S, Culebras A. Sleep apnoea and stroke. Stroke Vasc Neurol 2016; 1(4):185–191. doi:10.1136/svn-2016-000038
- Huhtakangas JK, Huhtakangas J, Bloigu R, Saaresranta T. Prevalence of sleep apnea at the acute phase of ischemic stroke with or without thrombolysis. Sleep Med 2017; 40:40–46. doi:10.1016/j.sleep.2017.08.018
- Johnson KG, Johnson DC. Frequency of sleep apnea in stroke and TIA patients: a meta-analysis. J Clin Sleep Med 2010; 6(2):131–137. pmid:20411688
- Iranzo A, Santamaria J, Berenguer J, Sanchez M, Chamorro A. Prevalence and clinical importance of sleep apnea in the first night after cerebral infarction. Neurology 2002; 58:911–916. pmid:11914407
- Javaheri S, Barbe F, Campos-Rodriguez F, et al. Sleep apnea: types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol 2017; 69(7):841–858. doi:10.1016/j.jacc.2016.11.069
- Dudley KA, Patel SR. Disparities and genetic risk factors in obstructive sleep apnea. Sleep Med 2016; 18:96–102. doi:10.1016/j.sleep.2015.01.015
- Redline S, Tishler PV. The genetics of sleep apnea. Sleep Med Rev 2000; 4(6):583–602. doi:10.1053/smrv.2000.0120
- Lipford MC, Flemming KD, Calvin AD, et al. Associations between cardioembolic stroke and obstructive sleep apnea. Sleep 2015; 38(11):1699–1705. doi:10.5665/sleep.5146
- Wang Y, Wang Y, Chen J, Yi X, Dong S, Cao L. Stroke patterns, topography, and etiology in patients with obstructive sleep apnea hypopnea syndrome. Int J Clin Exp Med 2017; 10(4):7137–7143.
- Fisse AL, Kemmling A, Teuber A, et al. The association of lesion location and sleep related breathing disorder in patients with acute ischemic stroke. PLoS One 2017; 12(1):e0171243. doi:10.1371/journal.pone.0171243
- Brown DL, Mowla A, McDermott M, et al. Ischemic stroke subtype and presence of sleep-disordered breathing: the BASIC sleep apnea study. J Stroke Cerebrovasc Dis 2015; 24(2):388–393. doi:10.1016/j.jstrokecerebrovasdis.2014.09.007
- Poli M, Philip P, Taillard J, et al. Atrial fibrillation as a major cause of stroke in apneic patients: a prospective study. Sleep Med 2017; 30:251–254. doi:10.1016/j.sleep.2015.07.031
- Young T, Finn L, Peppard PE, et al. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin Sleep Cohort. Sleep 2008; 31(8):1071–1078. pmid:18714778
- Molnar MZ, Mucsi I, Novak M, et al. Association of incident obstructive sleep apnoea with outcomes in a large cohort of US veterans. Thorax 2015; 70(9):888–895. doi:10.1136/thoraxjnl-2015-206970
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the sleep heart health study. Am J Respir Crit Care Med 2010; 182(2):269–277. doi:10.1164/rccm.200911-1746OC
- Marulanda-Londono E, Chaturvedi S. The interplay between obstructive sleep apnea and atrial fibrillation. Fron Neurol 2017; 8:668. doi:10.3389/fneur.2017.00668
- Szymanski FM, Filipiak KJ, Platek AE, Hrynkiewicz-Szymanska A, Karpinski G, Opolski G. Assessment of CHADS2 and CHA 2DS 2-VASc scores in obstructive sleep apnea patients with atrial fibrillation. Sleep Breath 2015; 19(2):531–537. doi:10.1007/s11325-014-1042-5
- Stansbury RC, Strollo PJ. Clinical manifestations of sleep apnea. J Thoracic Dis 2015; 7(9):E298–E310. doi:10.3978/j.issn.2072-1439.2015.09.13
- Chan W, Coutts SB, Hanly P. Sleep apnea in patients with transient ischemic attack and minor stroke: opportunity for risk reduction of recurrent stroke? Stroke 2010; 41(12):2973–2975. doi:10.1161/STROKEAHA.110.596759
- Boulos MI, Wan A, Im J, et al. Identifying obstructive sleep apnea after stroke/TIA: evaluating four simple screening tools. Sleep Med 2016; 21:133–139. doi:10.1016/j.sleep.2015.12.013
- Patel SK, Hanly PJ, Smith EE, Chan W, Coutts SB. Nocturnal hypoxemia is associated with white matter hyperintensities in patients with a minor stroke or transient ischemic attack. J Clin Sleep Med 2015; 11(12):1417–1424. doi:10.5664/jcsm.5278
- McCarty MF, DiNicolantonio JJ, O’Keefe JH. NADPH oxidase, uncoupled endothelial nitric oxide synthase, and NF-KappaB are key mediators of the pathogenic impact of obstructive sleep apnea—therapeutic implications. J Integr Cardiol 2016; 2(5):367–374. doi:10.15761/JIC.1000177
- Good DC, Henkle JQ, Gelber D, Welsh J, Verhulst S. Sleep-disordered breathing and poor functional outcome after stroke. Stroke 1996; 27(2):252–259. pmid:8571419
- Kaneko Y, Hajek VE, Zivanovic V, Raboud J, Bradley TD. Relationship of sleep apnea to functional capacity and length of hospitalization following stroke. Sleep 2003; 26(3):293–297. pmid:12749548
- Yan-fang S, Yu-ping W. Sleep-disordered breathing: impact on functional outcome of ischemic stroke patients. Sleep Med 2009; 10(7):717–719. doi:10.1016/j.sleep.2008.08.006
- Kumar R, Suri JC, Manocha R. Study of association of severity of sleep disordered breathing and functional outcome in stroke patients. Sleep Med 2017; 34:50–56. doi:10.1016/j.sleep.2017.02.025
- Kerner NA, Roose SP. Obstructive sleep apnea is linked to depression and cognitive impairment: evidence and potential mechanisms. Am J Geriatr Psychiatry 2016; 24(6):496–508. doi:10.1016/j.jagp.2016.01.134
- Bartoli F, Lillia N, Lax A, et al. Depression after stroke and risk of mortality: a systematic review and meta-analysis. Stroke Res Treat 2013; 2013:862978. doi:10.1155/2013/862978
- Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 2005; 353(19):2034–2041. doi:10.1056/NEJMoa043104
- Xie W, Zheng F, Song X. Obstructive sleep apnea and serious adverse outcomes in patients with cardiovascular or cerebrovascular disease: a PRISMA-compliant systematic review and meta-analysis. Medicine (Baltimore) 2014; 93(29):e336. doi:10.1097/MD.0000000000000336
- Chiu HY, Chen PY, Chuang LP, et al. Diagnostic accuracy of the Berlin questionnaire, STOP-BANG, STOP, and Epworth sleepiness scale in detecting obstructive sleep apnea: a bivariate meta-analysis. Sleep Med Rev 2017; 36:57–70. doi:10.1016/j.smrv.2016.10.004
- Katzan IL, Thompson NR, Uchino K, Foldvary-Schaefer N. A screening tool for obstructive sleep apnea in cerebrovascular patients. Sleep Med 2016; 21:70–76. doi:10.1016/j.sleep.2016.02.001
- Sico JJ, Yaggi HK, Ofner S, et al. Development, validation, and assessment of an ischemic stroke or transient ischemic attack-specific prediction tool for obstructive sleep apnea. J Stroke Cerebrovasc Dis 2017; 26(8):1745–1754. doi:10.1016/j.jstrokecerebrovasdis.2017.03.042
- Chung F, Liao P, Elsaid H, Islam S, Shapiro CM, Sun Y. Oxygen desaturation index from nocturnal oximetry: a sensitive and specific tool to detect sleep-disordered breathing in surgical patients. Anesth Analg 2012; 114(5):993–1000. doi:10.1213/ANE.0b013e318248f4f5
- Boulos MI, Elias S, Wan A, et al. Unattended hospital and home sleep apnea testing following cerebrovascular events. J Stroke Cerebrovasc Dis 2017; 26(1):143–149. doi:10.1016/j.jstrokecerebrovasdis.2016.09.001
- Saletu MT, Kotzian ST, Schwarzinger A, Haider S, Spatt J, Saletu B. Home sleep apnea testing is a feasible and accurate method to diagnose obstructive sleep apnea in stroke patients during in-hospital rehabilitation. J Clin Sleep Med 2018; 14(9):1495–1501. doi:10.5664/jcsm.7322
- Giles TL, Lasserson TJ, Smith BH, White J, Wright J, Cates CJ. Continuous positive airways pressure for obstructive sleep apnoea in adults. Cochrane Database Syst Rev 2006; (3):CD001106. doi:10.1002/14651858.CD001106.pub3
- Fatureto-Borges F, Lorenzi-Filho G, Drager LF. Effectiveness of continuous positive airway pressure in lowering blood pressure in patients with obstructive sleep apnea: a critical review of the literature. Integr Blood Press Control 2016; 9:43–47. doi:10.2147/IBPC.S70402
- Imran TF, Gharzipura M, Liu S, et al. Effect of continuous positive airway pressure treatment on pulmonary artery pressure in patients with isolated obstructive sleep apnea: a meta-analysis. Heart Fail Rev 2016; 21(5):591–598. doi:10.1007/s10741-016-9548-5
- Deng F, Raza A, Guo J. Treating obstructive sleep apnea with continuous positive airway pressure reduces risk of recurrent atrial fibrillation after catheter ablation: a meta-analysis. Sleep Med 2018; 46:5–11. doi:10.1016/j.sleep.2018.02.013
- Seetho IW, Wilding JPH. Sleep-disordered breathing, type 2 diabetes, and the metabolic syndrome. Chronic Resp Dis 2014; 11(4):257–275. doi:10.1177/1479972314552806
- Kim Y, Koo YS, Lee HY, Lee SY. Can continuous positive airway pressure reduce the risk of stroke in obstructive sleep apnea patients? A systematic review and meta-analysis. PloS ONE 2016; 11(1):e0146317. doi:10.1371/journal.pone.0146317
- Yu J, Zhou Z, McEvoy RD, et al. Association of positive airway pressure with cardiovascular events and death in adults with sleep apnea: a systematic review and meta-analysis. JAMA 2017; 318(2):156–166. doi:10.1001/jama.2017.7967
- Peker Y, Glantz H, Eulenburg C, Wegscheider K, Herlitz J, Thunström E. Effect of positive airway pressure on cardiovascular outcomes in coronary artery disease patients with nonsleepy obstructive sleep apnea. The RICCADSA randomized controlled trial. Am J Respir Crit Care Med 2016; 194(5):613–620. doi:10.1164/rccm.201601-0088OC
- Martinez-Garcia MA, Soler-Cataluna JJ, Ejarque-Martinez L, et al. Continuous positive airway pressure treatment reduces mortality in patients with ischemic stroke and obstructive sleep apnea: a 5-year follow-up study. Am J Respir Crit Care Med 2009; 180(1):36–41. doi:10.1164/rccm.200808-1341OC
- Broadley SA, Jorgensen L, Cheek A, et al. Early investigation and treatment of obstructive sleep apnoea after acute stroke. J Clin Neurosci 2007; 14(4):328–333. doi:10.1016/j.jocn.2006.01.017
- Wessendorf TE, Wang YM, Thilmann AF, Sorgenfrei U, Konietzko N, Teschler H. Treatment of obstructive sleep apnoea with nasal continuous positive airway pressure in stroke. Eur Respir J 2001; 18(4):623–629. pmid:11716165
- Bassetti CL, Milanova M, Gugger M. Sleep-disordered breathing and acute ischemic stroke: diagnosis, risk factors, treatment, evolution, and long-term clinical outcome. Stroke 2006; 37(4):967–972. doi:10.1161/01.STR.0000208215.49243.c3
- Palombini L, Guilleminault C. Stroke and treatment with nasal CPAP. Eur J Neurol 2006; 13(2):198–200. doi:10.1111/j.1468-1331.2006.01169.x
- Martínez-García MA, Campos-Rodríguez F, Soler-Cataluña JJ, Catalán-Serra P, Román-Sánchez P, Montserrat JM. Increased incidence of nonfatal cardiovascular events in stroke patients with sleep apnoea: effect of CPAP treatment. Eur Respir J 2012; 39(4):906–912. doi:10.1183/09031936.00011311
- Brill AK, Horvath T, Seiler A, et al. CPAP as treatment of sleep apnea after stroke: a meta-analysis of randomized trials. Neurology 2018; 90(14):e1222–e1230. doi:10.1212/WNL.0000000000005262
- Hsu C, Vennelle M, Li H, Engleman HM, Dennis MS, Douglas NJ. Sleep-disordered breathing after stroke: a randomised controlled trial of continuous positive airway pressure. J Neurol Neurosurg Psychiatry 2006; 77(10):1143–1149. doi:10.1136/jnnp.2005.086686
- Parra O, Sanchez-Armengol A, Bonnin M, et al. Early treatment of obstructive apnoea and stroke outcome: a randomised controlled trial. Eur Resp J 2011; 37(5):1128–1136. doi:10.1183/09031936.00034410
- Ryan CM, Bayley M, Green R, Murray BJ, Bradley TD. Influence of continuous positive airway pressure on outcomes of rehabilitation in stroke patients with obstructive sleep apnea. Stroke 2011; 42(4):1062–1067. doi:10.1161/STROKEAHA.110.597468
- Bravata DM, Concato J, Fried T, et al. Continuous positive airway pressure: evaluation of a novel therapy for patients with acute ischemic stroke. Sleep 2011; 34(9):1271–1277. doi:10.5665/SLEEP.1254
- Parra O, Sanchez-Armengol A, Capote F, et al. Efficacy of continuous positive airway pressure treatment on 5-year survival in patients with ischaemic stroke and obstructive sleep apnea: a randomized controlled trial. J Sleep Res 2015; 24(1):47–53. doi:10.1111/jsr.12181
- Khot SP, Davis AP, Crane DA, et al. Effect of continuous positive airway pressure on stroke rehabilitation: a pilot randomized sham-controlled trial. J Clin Sleep Med 2016; 12(7):1019–1026. doi:10.5664/jcsm.5940
- Aaronson JA, Hofman WF, van Bennekom CA, et al. Effects of continuous positive airway pressure on cognitive and functional outcome of stroke patients with obstructive sleep apnea: a randomized controlled trial. J Clin Sleep Med 2016; 12(4):533–541. doi:10.5664/jcsm.5684
- Gupta A, Shukla G, Afsar M, et al. Role of positive airway pressure therapy for obstructive sleep apnea in patients with stroke: a randomized controlled trial. J Clin Sleep Med 2018; 14(4):511–521. doi:10.5664/jcsm.7034
- Mello-Fujita L, Kim LJ, Palombini Lde O, et al. Treatment of obstructive sleep apnea syndrome associated with stroke. Sleep Med 2015; 16(6):691–696. doi:10.1016/j.sleep.2014.12.017
- Svatikova A, Chervin RD, Wing JJ, Sanchez BN, Migda EM, Brown DL. Positional therapy in ischemic stroke patients with obstructive sleep apnea. Sleep Med 2011; 12(3):262–266. doi:10.1016/j.sleep.2010.12.008
- Souza FJ, Genta PR, de Souza Filho AJ, Wellman A, Lorenzi-Filho G. The influence of head-of-bed elevation in patients with obstructive sleep apnea. Sleep Breath 2017; 21(4):815–820. doi:10.1007/s11325-017-1524-3
KEY POINTS
- A low threshold for evaluating for OSA after a stroke is warranted: the prevalence is high in this population, and risk factors for OSA and its typical clinical picture may not be present.
- Questionnaires can help screen for the likelihood of OSA and the need for more definitive assessment with polysomnography or home sleep apnea testing, tests that pose additional challenges after stroke.
- Positive airway pressure (PAP) therapy remains the first-line treatment for OSA after stroke; it may improve recovery and reduce long-term sequelae of untreated OSA.
- Acceptance of and adherence to PAP therapy can be especially problematic in this population, and alternatives should be considered if needed.
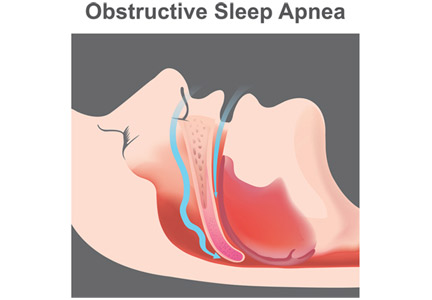
A 69-year-old woman with double vision and lower-extremity weakness
A 69-year-old woman was admitted to the hospital with double vision, weakness in the lower extremities, sensory loss, pain, and falls. Her symptoms started with sudden onset of horizontal diplopia 6 weeks before, followed by gradually worsening lower-extremity weakness, as well as ataxia and patchy and bilateral radicular burning leg pain more pronounced on the right. Her medical history included narcolepsy, obstructive sleep apnea, hypertension, hyperlipidemia, and bilateral knee replacements for osteoarthritis.
Neurologic examination showed inability to abduct the right eye, bilateral hip flexion weakness, decreased pinprick response, decreased proprioception, and diminished muscle stretch reflexes in the lower extremities. Magnetic resonance imaging (MRI) of the brain without contrast and magnetic resonance angiography of the brain and carotid arteries showed no evidence of acute stroke. No abnormalities were noted on electrocardiography and echocardiography.
A diagnosis of idiopathic peripheral neuropathy was made, and outpatient physical therapy was recommended. Over the subsequent 2 weeks, her condition declined to the point where she needed a walker. She continued to have worsening leg weakness with falls, prompting hospital readmission.
INITIAL EVALUATION
In addition to her diplopia and weakness, she said she had lost 15 pounds since the onset of symptoms and had experienced symptoms suggesting urinary retention.
Physical examination
Her temperature was 37°C (98.6°F), heart rate 79 beats per minute, blood pressure 117/86 mm Hg, respiratory rate 14 breaths per minute, and oxygen saturation 98% on room air. Examination of the head, neck, heart, lung, abdomen, lymph nodes, and extremities yielded nothing remarkable except for chronic venous changes in the lower extremities.
The neurologic examination showed incomplete lateral gaze bilaterally (cranial nerve VI dysfunction). Strength in the upper extremities was normal. In the legs, the Medical Research Council scale score for proximal muscle strength was 2 to 3 out of 5, and for distal muscles 3 to 4 out of 5, with the right side worse than the left and flexors and extensors affected equally. Muscle stretch reflexes were absent in both lower extremities and the left upper extremity, but intact in the right upper extremity. No abnormal corticospinal tract reflexes were elicited.
Sensory testing revealed diminished pin-prick perception in a length-dependent fashion in the lower extremities, reduced 50% compared with the hands. Gait could not be assessed due to weakness.
Initial laboratory testing
Results of initial laboratory tests—complete blood cell count, complete metabolic panel, erythrocyte sedimentation rate, C-reactive protein, thyroid-stimulating hormone, and hemoglobin A1c—were unremarkable.
FURTHER EVALUATION AND DIFFERENTIAL DIAGNOSIS
1. Which of the following is the most likely diagnosis at this point?
- Cerebral infarction
- Guillain-Barré syndrome
- Progressive polyneuropathy
- Transverse myelitis
- Polyradiculopathy
In the absence of definitive diagnostic tests, all of the above options were considered in the differential diagnosis for this patient.
Cerebral infarction
Although acute-onset diplopia can be explained by brainstem stroke involving cranial nerve nuclei or their projections, the onset of diplopia with progressive bilateral lower-extremity weakness makes stroke unlikely. Flaccid paralysis, areflexia of the lower extremities, and sensory involvement can also be caused by acute anterior spinal artery occlusion leading to spinal cord infarction; however, the deficits are usually maximal at onset.
Guillain-Barré syndrome
The combination of acute-subacute progressive ascending weakness, sensory involvement, and diminished or absent reflexes is typical of Guillain-Barré syndrome. Cranial nerve involvement can overlap with the more typical features of the syndrome. However, most patients reach the nadir of their disease by 4 weeks after initial symptom onset, even without treatment.1 This patient’s condition continued to worsen over 8 weeks. In addition, the asymmetric lower-extremity weakness and sparing of the arms are atypical for Guillain-Barré syndrome.
Given the progression of symptoms, chronic inflammatory demyelinating polyneuropathy is also a consideration, typically presenting as a relapsing or progressive neuropathy in proximal and distal muscles and worsening over at least an 8-week period.2
The initial workup for Guillain-Barré syndrome or chronic inflammatory demyelinating polyneuropathy includes lumbar puncture to assess for albuminocytologic dissociation (elevated protein with normal white blood cell count) in cerebrospinal fluid (CSF), and electromyography (EMG) to assess for neurophysiologic evidence of peripheral nerve demyelination. In Miller-Fisher syndrome, a rare variant of Guillain-Barré syndrome characterized by ataxia, ophthalmoparesis, and areflexia, serum ganglioside antibodies to GQ1b are found in over 90% of patients.3,4 Although MRI of the spine is not necessary to diagnose Guillain-Barré syndrome, it is often done to exclude other causes of lower-extremity weakness such as spinal cord or cauda equina compression that would require urgent neurosurgical consultation. MRI can support the diagnosis of Guillain-Barré syndrome when it reveals enhancement of the spinal nerve roots or cauda equina.
Other polyneuropathies
Polyneuropathy is caused by a variety of diseases that affect the function of peripheral motor, sensory, or autonomic nerves. The differential diagnosis is broad and involves inflammatory diseases (including autoimmune and paraneoplastic causes), hereditary disorders, infection, toxicity, and ischemic and nutritional deficiencies.5 Polyneuropathy can present in a distal-predominant, generalized, or asymmetric pattern involving individual nerve trunks termed “mononeuropathy multiplex,” as in our patient’s presentation. The initial workup includes EMG and a battery of serologic tests. In cases of severe and progressive polyneuropathy, nerve biopsy can assess for the presence of vasculitis, amyloidosis, and paraprotein deposition.
Transverse myelitis
Transverse myelitis is an inflammatory myelopathy that usually presents with acute or subacute weakness of the upper extremities or lower extremities, or both, corresponding to the level of the lesion, hyperreflexia, bladder and bowel dysfunction, spinal level of sensory loss, and autonomic involvement.6 The differential diagnosis of acute myelopathy includes:
- Infection (eg, herpes simplex virus, West Nile virus, Lyme disease, Mycoplasma pneumoniae, human immunodeficiency virus)
- Systemic inflammatory disease (systemic lupus erythematosus, sarcoidosis, Sjögren syndrome, scleroderma, paraneoplastic syndrome)
- Central nervous system demyelinating disease (acute disseminated encephalomyelitis, multiple sclerosis, neuromyelitis optica)
- Vascular malformation (dural arteriovenous fistula)
- Compression due to tumor, bleeding, disc herniation, infection, or abscess.
The workup involves laboratory tests to exclude systemic inflammatory and infectious causes, as well as MRI of the spine with and without contrast to identify a causative lesion. Lumbar puncture and CSF analysis may show pleocytosis, elevated protein concentration, and increased intrathecal immunoglobulin G (IgG) index.7
Although our patient’s presentation with subacute lower-extremity weakness, sensory changes, and bladder dysfunction were consistent with transverse myelitis, her cranial nerve abnormalities would be atypical for it.
Polyradiculopathy
Polyradiculopathy has many possible causes. In the United States, the most common causes are lumbar spondylosis, lumbar canal stenosis, and diabetic polyradiculoneuropathy.
When multiple spinal segments are affected, leptomeningeal disease involving the arachnoid and pia mater should be considered. Causes include malignant invasion, inflammatory cell accumulation, and protein deposition, leading to patchy but widespread dysfunction of spinal nerve roots and cranial nerves. Specific causes are myriad and include carcinomatous meningitis,8 syphilis, tuberculosis, sarcoidosis, and paraproteinemias. CSF and MRI changes are often nonspecific, leading to the need for meningeal biopsy for diagnosis.
CASE CONTINUED
During her hospitalization, our patient developed acute right upper and lower facial weakness consistent with peripheral facial mononeuropathy. Bilateral lower-extremity weakness progressed to disabling paraparesis.

She underwent lumbar puncture and CSF analysis (Table 1). The most notable findings were significant pleocytosis (72% lymphocytic predominance), protein elevation, and elevated IgG index (indicative of elevated intrathecal immunoglobulin synthesis in the central nervous system). Viral, bacterial, and fungal studies were negative. Guillain-Barré syndrome, other polyneuropathies, and spinal cord infarction would not be expected with these CSF features.
Surface EMG demonstrated normal sensory responses, and needle EMG showed chronic and active motor axon loss in the L3 and S1 root distributions, suggesting polyradiculopathy without polyneuropathy. These findings would not be expected in typical acute transverse myelitis but could be seen with spinal cord infarction.
 in axial T1 sequence (top) and sagittal T1 sequence (bottom).")
MRI of the entire spine with and without contrast showed cauda equina nerve root thickening and enhancement, especially involving the L5 and S1 roots (Figure 1). The spinal cord appeared normal. These findings further supported polyradiculopathy and a leptomeningeal process.
Further evaluation included chest radiography, erythrocyte sedimentation rate, C-reactive protein, hemoglobin A1c, human immunodeficiency virus testing, antinuclear antibody, antineutrophil cytoplasmic antibody, extractable nuclear antibody, GQ1b antibody, serum and CSF paraneoplastic panels, levels of vitamin B1, B12, and B6, copper, and ceruloplasmin, and a screen for heavy metals. All results were within normal ranges.
ESTABLISHING THE DIAGNOSIS
Serum monoclonal protein analysis with immunofixation revealed IgM kappa monoclonal gammopathy with an IgM level of 1,570 (reference range 53–334 mg/dL) and M-spike 0.75 (0.00 mg/dL), serum free kappa light chains 61.1 (3.30–19.40 mg/L), lambda 9.3 (5.7–26.3 mg/L), and kappa-lambda ratio 6.57 (0.26–1.65).
2. Which is the best next step in this patient’s neurologic evaluation?
- Test CSF angiotensin-converting enzyme level
- CSF cytology
- Meningeal biopsy
- Peripheral nerve biopsy
Given the high suspicion for malignancy, CSF cytology was performed and showed increased numbers of mononuclear chronic inflammatory cells, including a mixture of lymphocytes and monocytes, favoring a reactive lymphoid pleocytosis. Flow cytometry indicated the presence of a monoclonal, CD5- and CD10- negative, B-cell lymphoproliferative disorder. The immunophenotypic findings were not specific for a single diagnosis. The differential diagnosis included marginal zone lymphoma and lymphoplasmacytic lymphoma.
3. Given the presence of serum IgM monoclonal gammopathy in this patient, which is the most likely diagnosis?
- Neurosarcoidosis
- Multiple myeloma
- Waldenström macroglobulinemia
- Carcinomatous meningitis
Study of bone marrow biopsy demonstrated limited bone marrow involvement (1%) by a lymphoproliferative disorder with plasmacytoid features, and DNA testing detected an MYD88 L265P mutation, reported to be present in 90% of patients with Waldenström macroglobulinemia.9 This finding confirmed the diagnosis of Waldenström macroglobulinemia with central nervous system involvement. Our patient began therapy with rituximab and methotrexate, which resulted in some improvement in strength, gait, and vision.
WALDENSTRÖM MACROGLOBULINEMIA AND BING-NEEL SYNDROME
Waldenström macroglobulinemia is a lymphoplasmacytic lymphoma associated with a monoclonal IgM protein.10 It is considered a paraproteinemic disorder, similar to multiple myeloma. The presenting symptoms and complications are related to direct tumor infiltration, hyperviscosity syndrome, and deposition of IgM in various tissues.11,12
Waldenström macroglobulinemia is usually indolent, and treatment is reserved for patients with symptoms.13,14 It includes rituximab, usually in combination with chemotherapy or other targeted agents.15,16
Paraneoplastic antibody-mediated polyneuropathy may occur in these patients. However, the pattern is usually symmetrical clinically, with demyelination on EMG, and is not associated with cranial nerve or meningeal involvement. Management with plasmapheresis, corticosteroids, and intravenous immunoglobulin has not been shown to be effective.17
Involvement of the central nervous system as a complication of Waldenström macroglobulinemia has been described as Bing-Neel syndrome. It can present as diffuse malignant cell infiltration of the leptomeningeal space, white matter, or spinal cord, or in a tumoral form presenting as intraparenchymal masses or nodular lesions. The distinction between the tumoral and diffuse forms is based primarily on imaging findings.18
In a report of 44 patients with Bing-Neel syndrome, 36% presented with the disorder as the initial manifestation of Waldenström macroglobulinemia.18 The primary presenting symptoms were imbalance and gait difficulty (48%) and cranial nerve involvement (36%), which presented as predominantly facial or oculomotor nerve palsy. Cauda equina syndrome with motor involvement (seen in our patient) occurred in 14% of patients. Other presenting symptoms included cognitive impairment, sensory deficits, headache, dysarthria, aphasia, and seizures.
LEARNING POINTS
The differential diagnosis for patients presenting with multifocal neurologic symptoms can be broad, and a systematic approach to the diagnosis is necessary. Localizing the lesion is important in determining the diagnosis for patients presenting with neurologic symptoms. The process of localization begins with taking the history, is further refined during the examination, and is confirmed with diagnostic studies. Atypical presentations of relatively common neurologic diseases such as Guillain-Barré syndrome, transverse myelitis, and peripheral polyneuropathy do occur, but uncommon diagnoses need to be considered when support for the initial diagnosis is lacking.
- Fokke C, van den Berg B, Drenthen J, Walgaard C, van Doorn PA, Jacobs BC. Diagnosis of Guillain-Barre syndrome and validation of Brighton criteria. Brain 2014; 137(Pt 1):33–43. doi:10.1093/brain/awt285
- Mathey EK, Park SB, Hughes RA, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry 2015; 86(9):973–985. doi:10.1136/jnnp-2014-309697
- Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Serum anti-GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain-Barré syndrome: clinical and immunohistochemical studies. Neurology 1993; 43(10):1911–1917. pmid:8413947
- Teener J. Miller Fisher’s syndrome. Semin Neurol 2012; 32(5):512–516. doi:10.1055/s-0033-1334470
- Watson JC, Dyck PJ. Peripheral neuropathy: a practical approach to diagnosis and symptom management. Mayo Clin Proc 2015; 90(7):940–951. doi:10.1016/j.mayocp.2015.05.004
- Greenberg BM. Treatment of acute transverse myelitis and its early complications. Continuum (Minneap Minn) 2011; 17(4):733–743. doi:10.1212/01.CON.0000403792.36161.f5
- West TW. Transverse myelitis—a review of the presentation, diagnosis, and initial management. Discov Med 2013; 16(88):167–177. pmid:24099672
- Le Rhun E, Taillibert S, Chamberlain MC. Carcinomatous meningitis: leptomeningeal metastases in solid tumors. Surg Neurol Int 2013; 4(suppl 4):S265–S288. doi:10.4103/2152-7806.111304
- Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med 2012; 367(9):826–833. doi:10.1056/NEJMoa1200710
- Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol 2003; 30(2):110–115. doi:10.1053/sonc.2003.50082
- Björkholm M, Johansson E, Papamichael D, et al. Patterns of clinical presentation, treatment, and outcome in patients with Waldenstrom’s macroglobulinemia: a two-institution study. Semin Oncol 2003; 30(2):226–230. doi:10.1053/sonc.2003.50054
- Rison RA, Beydoun SR. Paraproteinemic neuropathy: a practical review. BMC Neurol 2016; 16:13. doi:10.1186/s12883-016-0532-4
- Kyle RA, Benson J, Larson D, et al. IgM monoclonal gammopathy of undetermined significance and smoldering Waldenström’s macroglobulinemia. Clin Lymphoma Myeloma 2009; 9(1):17–18. doi:10.3816/CLM.2009.n.002
- Kyle RA, Benson JT, Larson DR, et al. Progression in smoldering Waldenstrom macroglobulinemia: long-term results. Blood 2012; 119(19):4462–4466. doi:10.1182/blood-2011-10-384768
- Leblond V, Kastritis E, Advani R, et al. Treatment recommendations from the Eighth International Workshop on Waldenström’s macroglobulinemia. Blood 2016; 128(10):1321–1328. doi:10.1182/blood-2016-04-711234
- Kapoor P, Ansell SM, Fonseca R, et al. Diagnosis and management of Waldenström macroglobulinemia: Mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines 2016. JAMA Oncol 2017; 3(9):1257–1265. doi:10.1001/jamaoncol.2016.5763
- D’Sa S, Kersten MJ, Castillo JJ, et al. Investigation and management of IgM and Waldenström-associated peripheral neuropathies: recommendations from the IWWM-8 consensus panel. Br J Haematol 2017; 176(5):728–742. doi:10.1111/bjh.14492
- Simon L, Fitsiori A, Lemal R, et al. Bing-Neel syndrome, a rare complication of Waldenström macroglobulinemia: analysis of 44 cases and review of the literature. A study on behalf of the French Innovative Leukemia Organization (FILO). Haematologica 2015; 100(12):1587–1594. doi:10.3324/haematol.2015.133744
A 69-year-old woman was admitted to the hospital with double vision, weakness in the lower extremities, sensory loss, pain, and falls. Her symptoms started with sudden onset of horizontal diplopia 6 weeks before, followed by gradually worsening lower-extremity weakness, as well as ataxia and patchy and bilateral radicular burning leg pain more pronounced on the right. Her medical history included narcolepsy, obstructive sleep apnea, hypertension, hyperlipidemia, and bilateral knee replacements for osteoarthritis.
Neurologic examination showed inability to abduct the right eye, bilateral hip flexion weakness, decreased pinprick response, decreased proprioception, and diminished muscle stretch reflexes in the lower extremities. Magnetic resonance imaging (MRI) of the brain without contrast and magnetic resonance angiography of the brain and carotid arteries showed no evidence of acute stroke. No abnormalities were noted on electrocardiography and echocardiography.
A diagnosis of idiopathic peripheral neuropathy was made, and outpatient physical therapy was recommended. Over the subsequent 2 weeks, her condition declined to the point where she needed a walker. She continued to have worsening leg weakness with falls, prompting hospital readmission.
INITIAL EVALUATION
In addition to her diplopia and weakness, she said she had lost 15 pounds since the onset of symptoms and had experienced symptoms suggesting urinary retention.
Physical examination
Her temperature was 37°C (98.6°F), heart rate 79 beats per minute, blood pressure 117/86 mm Hg, respiratory rate 14 breaths per minute, and oxygen saturation 98% on room air. Examination of the head, neck, heart, lung, abdomen, lymph nodes, and extremities yielded nothing remarkable except for chronic venous changes in the lower extremities.
The neurologic examination showed incomplete lateral gaze bilaterally (cranial nerve VI dysfunction). Strength in the upper extremities was normal. In the legs, the Medical Research Council scale score for proximal muscle strength was 2 to 3 out of 5, and for distal muscles 3 to 4 out of 5, with the right side worse than the left and flexors and extensors affected equally. Muscle stretch reflexes were absent in both lower extremities and the left upper extremity, but intact in the right upper extremity. No abnormal corticospinal tract reflexes were elicited.
Sensory testing revealed diminished pin-prick perception in a length-dependent fashion in the lower extremities, reduced 50% compared with the hands. Gait could not be assessed due to weakness.
Initial laboratory testing
Results of initial laboratory tests—complete blood cell count, complete metabolic panel, erythrocyte sedimentation rate, C-reactive protein, thyroid-stimulating hormone, and hemoglobin A1c—were unremarkable.
FURTHER EVALUATION AND DIFFERENTIAL DIAGNOSIS
1. Which of the following is the most likely diagnosis at this point?
- Cerebral infarction
- Guillain-Barré syndrome
- Progressive polyneuropathy
- Transverse myelitis
- Polyradiculopathy
In the absence of definitive diagnostic tests, all of the above options were considered in the differential diagnosis for this patient.
Cerebral infarction
Although acute-onset diplopia can be explained by brainstem stroke involving cranial nerve nuclei or their projections, the onset of diplopia with progressive bilateral lower-extremity weakness makes stroke unlikely. Flaccid paralysis, areflexia of the lower extremities, and sensory involvement can also be caused by acute anterior spinal artery occlusion leading to spinal cord infarction; however, the deficits are usually maximal at onset.
Guillain-Barré syndrome
The combination of acute-subacute progressive ascending weakness, sensory involvement, and diminished or absent reflexes is typical of Guillain-Barré syndrome. Cranial nerve involvement can overlap with the more typical features of the syndrome. However, most patients reach the nadir of their disease by 4 weeks after initial symptom onset, even without treatment.1 This patient’s condition continued to worsen over 8 weeks. In addition, the asymmetric lower-extremity weakness and sparing of the arms are atypical for Guillain-Barré syndrome.
Given the progression of symptoms, chronic inflammatory demyelinating polyneuropathy is also a consideration, typically presenting as a relapsing or progressive neuropathy in proximal and distal muscles and worsening over at least an 8-week period.2
The initial workup for Guillain-Barré syndrome or chronic inflammatory demyelinating polyneuropathy includes lumbar puncture to assess for albuminocytologic dissociation (elevated protein with normal white blood cell count) in cerebrospinal fluid (CSF), and electromyography (EMG) to assess for neurophysiologic evidence of peripheral nerve demyelination. In Miller-Fisher syndrome, a rare variant of Guillain-Barré syndrome characterized by ataxia, ophthalmoparesis, and areflexia, serum ganglioside antibodies to GQ1b are found in over 90% of patients.3,4 Although MRI of the spine is not necessary to diagnose Guillain-Barré syndrome, it is often done to exclude other causes of lower-extremity weakness such as spinal cord or cauda equina compression that would require urgent neurosurgical consultation. MRI can support the diagnosis of Guillain-Barré syndrome when it reveals enhancement of the spinal nerve roots or cauda equina.
Other polyneuropathies
Polyneuropathy is caused by a variety of diseases that affect the function of peripheral motor, sensory, or autonomic nerves. The differential diagnosis is broad and involves inflammatory diseases (including autoimmune and paraneoplastic causes), hereditary disorders, infection, toxicity, and ischemic and nutritional deficiencies.5 Polyneuropathy can present in a distal-predominant, generalized, or asymmetric pattern involving individual nerve trunks termed “mononeuropathy multiplex,” as in our patient’s presentation. The initial workup includes EMG and a battery of serologic tests. In cases of severe and progressive polyneuropathy, nerve biopsy can assess for the presence of vasculitis, amyloidosis, and paraprotein deposition.
Transverse myelitis
Transverse myelitis is an inflammatory myelopathy that usually presents with acute or subacute weakness of the upper extremities or lower extremities, or both, corresponding to the level of the lesion, hyperreflexia, bladder and bowel dysfunction, spinal level of sensory loss, and autonomic involvement.6 The differential diagnosis of acute myelopathy includes:
- Infection (eg, herpes simplex virus, West Nile virus, Lyme disease, Mycoplasma pneumoniae, human immunodeficiency virus)
- Systemic inflammatory disease (systemic lupus erythematosus, sarcoidosis, Sjögren syndrome, scleroderma, paraneoplastic syndrome)
- Central nervous system demyelinating disease (acute disseminated encephalomyelitis, multiple sclerosis, neuromyelitis optica)
- Vascular malformation (dural arteriovenous fistula)
- Compression due to tumor, bleeding, disc herniation, infection, or abscess.
The workup involves laboratory tests to exclude systemic inflammatory and infectious causes, as well as MRI of the spine with and without contrast to identify a causative lesion. Lumbar puncture and CSF analysis may show pleocytosis, elevated protein concentration, and increased intrathecal immunoglobulin G (IgG) index.7
Although our patient’s presentation with subacute lower-extremity weakness, sensory changes, and bladder dysfunction were consistent with transverse myelitis, her cranial nerve abnormalities would be atypical for it.
Polyradiculopathy
Polyradiculopathy has many possible causes. In the United States, the most common causes are lumbar spondylosis, lumbar canal stenosis, and diabetic polyradiculoneuropathy.
When multiple spinal segments are affected, leptomeningeal disease involving the arachnoid and pia mater should be considered. Causes include malignant invasion, inflammatory cell accumulation, and protein deposition, leading to patchy but widespread dysfunction of spinal nerve roots and cranial nerves. Specific causes are myriad and include carcinomatous meningitis,8 syphilis, tuberculosis, sarcoidosis, and paraproteinemias. CSF and MRI changes are often nonspecific, leading to the need for meningeal biopsy for diagnosis.
CASE CONTINUED
During her hospitalization, our patient developed acute right upper and lower facial weakness consistent with peripheral facial mononeuropathy. Bilateral lower-extremity weakness progressed to disabling paraparesis.
She underwent lumbar puncture and CSF analysis (Table 1). The most notable findings were significant pleocytosis (72% lymphocytic predominance), protein elevation, and elevated IgG index (indicative of elevated intrathecal immunoglobulin synthesis in the central nervous system). Viral, bacterial, and fungal studies were negative. Guillain-Barré syndrome, other polyneuropathies, and spinal cord infarction would not be expected with these CSF features.
Surface EMG demonstrated normal sensory responses, and needle EMG showed chronic and active motor axon loss in the L3 and S1 root distributions, suggesting polyradiculopathy without polyneuropathy. These findings would not be expected in typical acute transverse myelitis but could be seen with spinal cord infarction.
MRI of the entire spine with and without contrast showed cauda equina nerve root thickening and enhancement, especially involving the L5 and S1 roots (Figure 1). The spinal cord appeared normal. These findings further supported polyradiculopathy and a leptomeningeal process.
Further evaluation included chest radiography, erythrocyte sedimentation rate, C-reactive protein, hemoglobin A1c, human immunodeficiency virus testing, antinuclear antibody, antineutrophil cytoplasmic antibody, extractable nuclear antibody, GQ1b antibody, serum and CSF paraneoplastic panels, levels of vitamin B1, B12, and B6, copper, and ceruloplasmin, and a screen for heavy metals. All results were within normal ranges.
ESTABLISHING THE DIAGNOSIS
Serum monoclonal protein analysis with immunofixation revealed IgM kappa monoclonal gammopathy with an IgM level of 1,570 (reference range 53–334 mg/dL) and M-spike 0.75 (0.00 mg/dL), serum free kappa light chains 61.1 (3.30–19.40 mg/L), lambda 9.3 (5.7–26.3 mg/L), and kappa-lambda ratio 6.57 (0.26–1.65).
2. Which is the best next step in this patient’s neurologic evaluation?
- Test CSF angiotensin-converting enzyme level
- CSF cytology
- Meningeal biopsy
- Peripheral nerve biopsy
Given the high suspicion for malignancy, CSF cytology was performed and showed increased numbers of mononuclear chronic inflammatory cells, including a mixture of lymphocytes and monocytes, favoring a reactive lymphoid pleocytosis. Flow cytometry indicated the presence of a monoclonal, CD5- and CD10- negative, B-cell lymphoproliferative disorder. The immunophenotypic findings were not specific for a single diagnosis. The differential diagnosis included marginal zone lymphoma and lymphoplasmacytic lymphoma.
3. Given the presence of serum IgM monoclonal gammopathy in this patient, which is the most likely diagnosis?
- Neurosarcoidosis
- Multiple myeloma
- Waldenström macroglobulinemia
- Carcinomatous meningitis
Study of bone marrow biopsy demonstrated limited bone marrow involvement (1%) by a lymphoproliferative disorder with plasmacytoid features, and DNA testing detected an MYD88 L265P mutation, reported to be present in 90% of patients with Waldenström macroglobulinemia.9 This finding confirmed the diagnosis of Waldenström macroglobulinemia with central nervous system involvement. Our patient began therapy with rituximab and methotrexate, which resulted in some improvement in strength, gait, and vision.
WALDENSTRÖM MACROGLOBULINEMIA AND BING-NEEL SYNDROME
Waldenström macroglobulinemia is a lymphoplasmacytic lymphoma associated with a monoclonal IgM protein.10 It is considered a paraproteinemic disorder, similar to multiple myeloma. The presenting symptoms and complications are related to direct tumor infiltration, hyperviscosity syndrome, and deposition of IgM in various tissues.11,12
Waldenström macroglobulinemia is usually indolent, and treatment is reserved for patients with symptoms.13,14 It includes rituximab, usually in combination with chemotherapy or other targeted agents.15,16
Paraneoplastic antibody-mediated polyneuropathy may occur in these patients. However, the pattern is usually symmetrical clinically, with demyelination on EMG, and is not associated with cranial nerve or meningeal involvement. Management with plasmapheresis, corticosteroids, and intravenous immunoglobulin has not been shown to be effective.17
Involvement of the central nervous system as a complication of Waldenström macroglobulinemia has been described as Bing-Neel syndrome. It can present as diffuse malignant cell infiltration of the leptomeningeal space, white matter, or spinal cord, or in a tumoral form presenting as intraparenchymal masses or nodular lesions. The distinction between the tumoral and diffuse forms is based primarily on imaging findings.18
In a report of 44 patients with Bing-Neel syndrome, 36% presented with the disorder as the initial manifestation of Waldenström macroglobulinemia.18 The primary presenting symptoms were imbalance and gait difficulty (48%) and cranial nerve involvement (36%), which presented as predominantly facial or oculomotor nerve palsy. Cauda equina syndrome with motor involvement (seen in our patient) occurred in 14% of patients. Other presenting symptoms included cognitive impairment, sensory deficits, headache, dysarthria, aphasia, and seizures.
LEARNING POINTS
The differential diagnosis for patients presenting with multifocal neurologic symptoms can be broad, and a systematic approach to the diagnosis is necessary. Localizing the lesion is important in determining the diagnosis for patients presenting with neurologic symptoms. The process of localization begins with taking the history, is further refined during the examination, and is confirmed with diagnostic studies. Atypical presentations of relatively common neurologic diseases such as Guillain-Barré syndrome, transverse myelitis, and peripheral polyneuropathy do occur, but uncommon diagnoses need to be considered when support for the initial diagnosis is lacking.
A 69-year-old woman was admitted to the hospital with double vision, weakness in the lower extremities, sensory loss, pain, and falls. Her symptoms started with sudden onset of horizontal diplopia 6 weeks before, followed by gradually worsening lower-extremity weakness, as well as ataxia and patchy and bilateral radicular burning leg pain more pronounced on the right. Her medical history included narcolepsy, obstructive sleep apnea, hypertension, hyperlipidemia, and bilateral knee replacements for osteoarthritis.
Neurologic examination showed inability to abduct the right eye, bilateral hip flexion weakness, decreased pinprick response, decreased proprioception, and diminished muscle stretch reflexes in the lower extremities. Magnetic resonance imaging (MRI) of the brain without contrast and magnetic resonance angiography of the brain and carotid arteries showed no evidence of acute stroke. No abnormalities were noted on electrocardiography and echocardiography.
A diagnosis of idiopathic peripheral neuropathy was made, and outpatient physical therapy was recommended. Over the subsequent 2 weeks, her condition declined to the point where she needed a walker. She continued to have worsening leg weakness with falls, prompting hospital readmission.
INITIAL EVALUATION
In addition to her diplopia and weakness, she said she had lost 15 pounds since the onset of symptoms and had experienced symptoms suggesting urinary retention.
Physical examination
Her temperature was 37°C (98.6°F), heart rate 79 beats per minute, blood pressure 117/86 mm Hg, respiratory rate 14 breaths per minute, and oxygen saturation 98% on room air. Examination of the head, neck, heart, lung, abdomen, lymph nodes, and extremities yielded nothing remarkable except for chronic venous changes in the lower extremities.
The neurologic examination showed incomplete lateral gaze bilaterally (cranial nerve VI dysfunction). Strength in the upper extremities was normal. In the legs, the Medical Research Council scale score for proximal muscle strength was 2 to 3 out of 5, and for distal muscles 3 to 4 out of 5, with the right side worse than the left and flexors and extensors affected equally. Muscle stretch reflexes were absent in both lower extremities and the left upper extremity, but intact in the right upper extremity. No abnormal corticospinal tract reflexes were elicited.
Sensory testing revealed diminished pin-prick perception in a length-dependent fashion in the lower extremities, reduced 50% compared with the hands. Gait could not be assessed due to weakness.
Initial laboratory testing
Results of initial laboratory tests—complete blood cell count, complete metabolic panel, erythrocyte sedimentation rate, C-reactive protein, thyroid-stimulating hormone, and hemoglobin A1c—were unremarkable.
FURTHER EVALUATION AND DIFFERENTIAL DIAGNOSIS
1. Which of the following is the most likely diagnosis at this point?
- Cerebral infarction
- Guillain-Barré syndrome
- Progressive polyneuropathy
- Transverse myelitis
- Polyradiculopathy
In the absence of definitive diagnostic tests, all of the above options were considered in the differential diagnosis for this patient.
Cerebral infarction
Although acute-onset diplopia can be explained by brainstem stroke involving cranial nerve nuclei or their projections, the onset of diplopia with progressive bilateral lower-extremity weakness makes stroke unlikely. Flaccid paralysis, areflexia of the lower extremities, and sensory involvement can also be caused by acute anterior spinal artery occlusion leading to spinal cord infarction; however, the deficits are usually maximal at onset.
Guillain-Barré syndrome
The combination of acute-subacute progressive ascending weakness, sensory involvement, and diminished or absent reflexes is typical of Guillain-Barré syndrome. Cranial nerve involvement can overlap with the more typical features of the syndrome. However, most patients reach the nadir of their disease by 4 weeks after initial symptom onset, even without treatment.1 This patient’s condition continued to worsen over 8 weeks. In addition, the asymmetric lower-extremity weakness and sparing of the arms are atypical for Guillain-Barré syndrome.
Given the progression of symptoms, chronic inflammatory demyelinating polyneuropathy is also a consideration, typically presenting as a relapsing or progressive neuropathy in proximal and distal muscles and worsening over at least an 8-week period.2
The initial workup for Guillain-Barré syndrome or chronic inflammatory demyelinating polyneuropathy includes lumbar puncture to assess for albuminocytologic dissociation (elevated protein with normal white blood cell count) in cerebrospinal fluid (CSF), and electromyography (EMG) to assess for neurophysiologic evidence of peripheral nerve demyelination. In Miller-Fisher syndrome, a rare variant of Guillain-Barré syndrome characterized by ataxia, ophthalmoparesis, and areflexia, serum ganglioside antibodies to GQ1b are found in over 90% of patients.3,4 Although MRI of the spine is not necessary to diagnose Guillain-Barré syndrome, it is often done to exclude other causes of lower-extremity weakness such as spinal cord or cauda equina compression that would require urgent neurosurgical consultation. MRI can support the diagnosis of Guillain-Barré syndrome when it reveals enhancement of the spinal nerve roots or cauda equina.
Other polyneuropathies
Polyneuropathy is caused by a variety of diseases that affect the function of peripheral motor, sensory, or autonomic nerves. The differential diagnosis is broad and involves inflammatory diseases (including autoimmune and paraneoplastic causes), hereditary disorders, infection, toxicity, and ischemic and nutritional deficiencies.5 Polyneuropathy can present in a distal-predominant, generalized, or asymmetric pattern involving individual nerve trunks termed “mononeuropathy multiplex,” as in our patient’s presentation. The initial workup includes EMG and a battery of serologic tests. In cases of severe and progressive polyneuropathy, nerve biopsy can assess for the presence of vasculitis, amyloidosis, and paraprotein deposition.
Transverse myelitis
Transverse myelitis is an inflammatory myelopathy that usually presents with acute or subacute weakness of the upper extremities or lower extremities, or both, corresponding to the level of the lesion, hyperreflexia, bladder and bowel dysfunction, spinal level of sensory loss, and autonomic involvement.6 The differential diagnosis of acute myelopathy includes:
- Infection (eg, herpes simplex virus, West Nile virus, Lyme disease, Mycoplasma pneumoniae, human immunodeficiency virus)
- Systemic inflammatory disease (systemic lupus erythematosus, sarcoidosis, Sjögren syndrome, scleroderma, paraneoplastic syndrome)
- Central nervous system demyelinating disease (acute disseminated encephalomyelitis, multiple sclerosis, neuromyelitis optica)
- Vascular malformation (dural arteriovenous fistula)
- Compression due to tumor, bleeding, disc herniation, infection, or abscess.
The workup involves laboratory tests to exclude systemic inflammatory and infectious causes, as well as MRI of the spine with and without contrast to identify a causative lesion. Lumbar puncture and CSF analysis may show pleocytosis, elevated protein concentration, and increased intrathecal immunoglobulin G (IgG) index.7
Although our patient’s presentation with subacute lower-extremity weakness, sensory changes, and bladder dysfunction were consistent with transverse myelitis, her cranial nerve abnormalities would be atypical for it.
Polyradiculopathy
Polyradiculopathy has many possible causes. In the United States, the most common causes are lumbar spondylosis, lumbar canal stenosis, and diabetic polyradiculoneuropathy.
When multiple spinal segments are affected, leptomeningeal disease involving the arachnoid and pia mater should be considered. Causes include malignant invasion, inflammatory cell accumulation, and protein deposition, leading to patchy but widespread dysfunction of spinal nerve roots and cranial nerves. Specific causes are myriad and include carcinomatous meningitis,8 syphilis, tuberculosis, sarcoidosis, and paraproteinemias. CSF and MRI changes are often nonspecific, leading to the need for meningeal biopsy for diagnosis.
CASE CONTINUED
During her hospitalization, our patient developed acute right upper and lower facial weakness consistent with peripheral facial mononeuropathy. Bilateral lower-extremity weakness progressed to disabling paraparesis.
She underwent lumbar puncture and CSF analysis (Table 1). The most notable findings were significant pleocytosis (72% lymphocytic predominance), protein elevation, and elevated IgG index (indicative of elevated intrathecal immunoglobulin synthesis in the central nervous system). Viral, bacterial, and fungal studies were negative. Guillain-Barré syndrome, other polyneuropathies, and spinal cord infarction would not be expected with these CSF features.
Surface EMG demonstrated normal sensory responses, and needle EMG showed chronic and active motor axon loss in the L3 and S1 root distributions, suggesting polyradiculopathy without polyneuropathy. These findings would not be expected in typical acute transverse myelitis but could be seen with spinal cord infarction.
MRI of the entire spine with and without contrast showed cauda equina nerve root thickening and enhancement, especially involving the L5 and S1 roots (Figure 1). The spinal cord appeared normal. These findings further supported polyradiculopathy and a leptomeningeal process.
Further evaluation included chest radiography, erythrocyte sedimentation rate, C-reactive protein, hemoglobin A1c, human immunodeficiency virus testing, antinuclear antibody, antineutrophil cytoplasmic antibody, extractable nuclear antibody, GQ1b antibody, serum and CSF paraneoplastic panels, levels of vitamin B1, B12, and B6, copper, and ceruloplasmin, and a screen for heavy metals. All results were within normal ranges.
ESTABLISHING THE DIAGNOSIS
Serum monoclonal protein analysis with immunofixation revealed IgM kappa monoclonal gammopathy with an IgM level of 1,570 (reference range 53–334 mg/dL) and M-spike 0.75 (0.00 mg/dL), serum free kappa light chains 61.1 (3.30–19.40 mg/L), lambda 9.3 (5.7–26.3 mg/L), and kappa-lambda ratio 6.57 (0.26–1.65).
2. Which is the best next step in this patient’s neurologic evaluation?
- Test CSF angiotensin-converting enzyme level
- CSF cytology
- Meningeal biopsy
- Peripheral nerve biopsy
Given the high suspicion for malignancy, CSF cytology was performed and showed increased numbers of mononuclear chronic inflammatory cells, including a mixture of lymphocytes and monocytes, favoring a reactive lymphoid pleocytosis. Flow cytometry indicated the presence of a monoclonal, CD5- and CD10- negative, B-cell lymphoproliferative disorder. The immunophenotypic findings were not specific for a single diagnosis. The differential diagnosis included marginal zone lymphoma and lymphoplasmacytic lymphoma.
3. Given the presence of serum IgM monoclonal gammopathy in this patient, which is the most likely diagnosis?
- Neurosarcoidosis
- Multiple myeloma
- Waldenström macroglobulinemia
- Carcinomatous meningitis
Study of bone marrow biopsy demonstrated limited bone marrow involvement (1%) by a lymphoproliferative disorder with plasmacytoid features, and DNA testing detected an MYD88 L265P mutation, reported to be present in 90% of patients with Waldenström macroglobulinemia.9 This finding confirmed the diagnosis of Waldenström macroglobulinemia with central nervous system involvement. Our patient began therapy with rituximab and methotrexate, which resulted in some improvement in strength, gait, and vision.
WALDENSTRÖM MACROGLOBULINEMIA AND BING-NEEL SYNDROME
Waldenström macroglobulinemia is a lymphoplasmacytic lymphoma associated with a monoclonal IgM protein.10 It is considered a paraproteinemic disorder, similar to multiple myeloma. The presenting symptoms and complications are related to direct tumor infiltration, hyperviscosity syndrome, and deposition of IgM in various tissues.11,12
Waldenström macroglobulinemia is usually indolent, and treatment is reserved for patients with symptoms.13,14 It includes rituximab, usually in combination with chemotherapy or other targeted agents.15,16
Paraneoplastic antibody-mediated polyneuropathy may occur in these patients. However, the pattern is usually symmetrical clinically, with demyelination on EMG, and is not associated with cranial nerve or meningeal involvement. Management with plasmapheresis, corticosteroids, and intravenous immunoglobulin has not been shown to be effective.17
Involvement of the central nervous system as a complication of Waldenström macroglobulinemia has been described as Bing-Neel syndrome. It can present as diffuse malignant cell infiltration of the leptomeningeal space, white matter, or spinal cord, or in a tumoral form presenting as intraparenchymal masses or nodular lesions. The distinction between the tumoral and diffuse forms is based primarily on imaging findings.18
In a report of 44 patients with Bing-Neel syndrome, 36% presented with the disorder as the initial manifestation of Waldenström macroglobulinemia.18 The primary presenting symptoms were imbalance and gait difficulty (48%) and cranial nerve involvement (36%), which presented as predominantly facial or oculomotor nerve palsy. Cauda equina syndrome with motor involvement (seen in our patient) occurred in 14% of patients. Other presenting symptoms included cognitive impairment, sensory deficits, headache, dysarthria, aphasia, and seizures.
LEARNING POINTS
The differential diagnosis for patients presenting with multifocal neurologic symptoms can be broad, and a systematic approach to the diagnosis is necessary. Localizing the lesion is important in determining the diagnosis for patients presenting with neurologic symptoms. The process of localization begins with taking the history, is further refined during the examination, and is confirmed with diagnostic studies. Atypical presentations of relatively common neurologic diseases such as Guillain-Barré syndrome, transverse myelitis, and peripheral polyneuropathy do occur, but uncommon diagnoses need to be considered when support for the initial diagnosis is lacking.
- Fokke C, van den Berg B, Drenthen J, Walgaard C, van Doorn PA, Jacobs BC. Diagnosis of Guillain-Barre syndrome and validation of Brighton criteria. Brain 2014; 137(Pt 1):33–43. doi:10.1093/brain/awt285
- Mathey EK, Park SB, Hughes RA, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry 2015; 86(9):973–985. doi:10.1136/jnnp-2014-309697
- Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Serum anti-GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain-Barré syndrome: clinical and immunohistochemical studies. Neurology 1993; 43(10):1911–1917. pmid:8413947
- Teener J. Miller Fisher’s syndrome. Semin Neurol 2012; 32(5):512–516. doi:10.1055/s-0033-1334470
- Watson JC, Dyck PJ. Peripheral neuropathy: a practical approach to diagnosis and symptom management. Mayo Clin Proc 2015; 90(7):940–951. doi:10.1016/j.mayocp.2015.05.004
- Greenberg BM. Treatment of acute transverse myelitis and its early complications. Continuum (Minneap Minn) 2011; 17(4):733–743. doi:10.1212/01.CON.0000403792.36161.f5
- West TW. Transverse myelitis—a review of the presentation, diagnosis, and initial management. Discov Med 2013; 16(88):167–177. pmid:24099672
- Le Rhun E, Taillibert S, Chamberlain MC. Carcinomatous meningitis: leptomeningeal metastases in solid tumors. Surg Neurol Int 2013; 4(suppl 4):S265–S288. doi:10.4103/2152-7806.111304
- Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med 2012; 367(9):826–833. doi:10.1056/NEJMoa1200710
- Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol 2003; 30(2):110–115. doi:10.1053/sonc.2003.50082
- Björkholm M, Johansson E, Papamichael D, et al. Patterns of clinical presentation, treatment, and outcome in patients with Waldenstrom’s macroglobulinemia: a two-institution study. Semin Oncol 2003; 30(2):226–230. doi:10.1053/sonc.2003.50054
- Rison RA, Beydoun SR. Paraproteinemic neuropathy: a practical review. BMC Neurol 2016; 16:13. doi:10.1186/s12883-016-0532-4
- Kyle RA, Benson J, Larson D, et al. IgM monoclonal gammopathy of undetermined significance and smoldering Waldenström’s macroglobulinemia. Clin Lymphoma Myeloma 2009; 9(1):17–18. doi:10.3816/CLM.2009.n.002
- Kyle RA, Benson JT, Larson DR, et al. Progression in smoldering Waldenstrom macroglobulinemia: long-term results. Blood 2012; 119(19):4462–4466. doi:10.1182/blood-2011-10-384768
- Leblond V, Kastritis E, Advani R, et al. Treatment recommendations from the Eighth International Workshop on Waldenström’s macroglobulinemia. Blood 2016; 128(10):1321–1328. doi:10.1182/blood-2016-04-711234
- Kapoor P, Ansell SM, Fonseca R, et al. Diagnosis and management of Waldenström macroglobulinemia: Mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines 2016. JAMA Oncol 2017; 3(9):1257–1265. doi:10.1001/jamaoncol.2016.5763
- D’Sa S, Kersten MJ, Castillo JJ, et al. Investigation and management of IgM and Waldenström-associated peripheral neuropathies: recommendations from the IWWM-8 consensus panel. Br J Haematol 2017; 176(5):728–742. doi:10.1111/bjh.14492
- Simon L, Fitsiori A, Lemal R, et al. Bing-Neel syndrome, a rare complication of Waldenström macroglobulinemia: analysis of 44 cases and review of the literature. A study on behalf of the French Innovative Leukemia Organization (FILO). Haematologica 2015; 100(12):1587–1594. doi:10.3324/haematol.2015.133744
- Fokke C, van den Berg B, Drenthen J, Walgaard C, van Doorn PA, Jacobs BC. Diagnosis of Guillain-Barre syndrome and validation of Brighton criteria. Brain 2014; 137(Pt 1):33–43. doi:10.1093/brain/awt285
- Mathey EK, Park SB, Hughes RA, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry 2015; 86(9):973–985. doi:10.1136/jnnp-2014-309697
- Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Serum anti-GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain-Barré syndrome: clinical and immunohistochemical studies. Neurology 1993; 43(10):1911–1917. pmid:8413947
- Teener J. Miller Fisher’s syndrome. Semin Neurol 2012; 32(5):512–516. doi:10.1055/s-0033-1334470
- Watson JC, Dyck PJ. Peripheral neuropathy: a practical approach to diagnosis and symptom management. Mayo Clin Proc 2015; 90(7):940–951. doi:10.1016/j.mayocp.2015.05.004
- Greenberg BM. Treatment of acute transverse myelitis and its early complications. Continuum (Minneap Minn) 2011; 17(4):733–743. doi:10.1212/01.CON.0000403792.36161.f5
- West TW. Transverse myelitis—a review of the presentation, diagnosis, and initial management. Discov Med 2013; 16(88):167–177. pmid:24099672
- Le Rhun E, Taillibert S, Chamberlain MC. Carcinomatous meningitis: leptomeningeal metastases in solid tumors. Surg Neurol Int 2013; 4(suppl 4):S265–S288. doi:10.4103/2152-7806.111304
- Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med 2012; 367(9):826–833. doi:10.1056/NEJMoa1200710
- Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol 2003; 30(2):110–115. doi:10.1053/sonc.2003.50082
- Björkholm M, Johansson E, Papamichael D, et al. Patterns of clinical presentation, treatment, and outcome in patients with Waldenstrom’s macroglobulinemia: a two-institution study. Semin Oncol 2003; 30(2):226–230. doi:10.1053/sonc.2003.50054
- Rison RA, Beydoun SR. Paraproteinemic neuropathy: a practical review. BMC Neurol 2016; 16:13. doi:10.1186/s12883-016-0532-4
- Kyle RA, Benson J, Larson D, et al. IgM monoclonal gammopathy of undetermined significance and smoldering Waldenström’s macroglobulinemia. Clin Lymphoma Myeloma 2009; 9(1):17–18. doi:10.3816/CLM.2009.n.002
- Kyle RA, Benson JT, Larson DR, et al. Progression in smoldering Waldenstrom macroglobulinemia: long-term results. Blood 2012; 119(19):4462–4466. doi:10.1182/blood-2011-10-384768
- Leblond V, Kastritis E, Advani R, et al. Treatment recommendations from the Eighth International Workshop on Waldenström’s macroglobulinemia. Blood 2016; 128(10):1321–1328. doi:10.1182/blood-2016-04-711234
- Kapoor P, Ansell SM, Fonseca R, et al. Diagnosis and management of Waldenström macroglobulinemia: Mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines 2016. JAMA Oncol 2017; 3(9):1257–1265. doi:10.1001/jamaoncol.2016.5763
- D’Sa S, Kersten MJ, Castillo JJ, et al. Investigation and management of IgM and Waldenström-associated peripheral neuropathies: recommendations from the IWWM-8 consensus panel. Br J Haematol 2017; 176(5):728–742. doi:10.1111/bjh.14492
- Simon L, Fitsiori A, Lemal R, et al. Bing-Neel syndrome, a rare complication of Waldenström macroglobulinemia: analysis of 44 cases and review of the literature. A study on behalf of the French Innovative Leukemia Organization (FILO). Haematologica 2015; 100(12):1587–1594. doi:10.3324/haematol.2015.133744