User login
For MD-IQ use only
Implications of Thyroid Disease in Hospitalized Patients With Hidradenitis Suppurativa
Implications of Thyroid Disease in Hospitalized Patients With Hidradenitis Suppurativa
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition characterized by painful recurrent abscesses. Several autoimmune and endocrine diseases are associated with HS, including inflammatory bowel disease and diabetes mellitus (DM).1 Notably, the association between HS and thyroid disorders is poorly characterized,2 and there are no known nationwide studies exploring this potential association in the hospital setting. In this cross-sectional matched cohort study, we aimed to characterize HS patients with comorbid thyroid disorders as well as to explore whether thyroid disease is associated with comorbidities and hospital outcome measures in these patients.
The 2019 National Inpatient Sample (NIS) was weighted in accordance with NIS-assigned weight variables and queried for HS, hypothyroidism, and hyperthyroidism cases using International Classification of Diseases, Tenth Revision, codes L73.2, E03, and E05, respectively. Propensity score matching based on age and sex was performed using a nearest-neighbor method in the MatchIt statistical R package. Patient demographics, comorbidities, and outcome variables were collected. Univariable analysis of HS patients with thyroid disease vs those without thyroid disease vs controls without HS were performed using X2 and t-test functions in SPSS statistical software (IBM). A series of multivariate analyses were performed using SPSS logistic and linear regression models to examine the effect of thyroid disease on hospital outcome measures and comorbidities in HS patients, with statistical significance set at P=.05.
A total of 1720 HS patients with comorbid thyroid disease (hyperthyroidism/hypothyroidism), 23,785 HS patients without thyroid disease, and 25,497 age- and sex-matched controls were included in the analysis. On average, HS patients with comorbid thyroid disease were older than HS patients without thyroid disease and controls (49.36 years vs 42.17 years vs 42.66 years [P<.001]), more likely to be female (75.58% vs 58.67% vs 59.81% [P<.001]), more likely to be in the highest income quartile (17.52% vs 12.18% vs 8.14% [P<.001]), and more likely to be Medicare insured (39.07% vs 27.47% vs 18.02% [P<.001])(eTable).
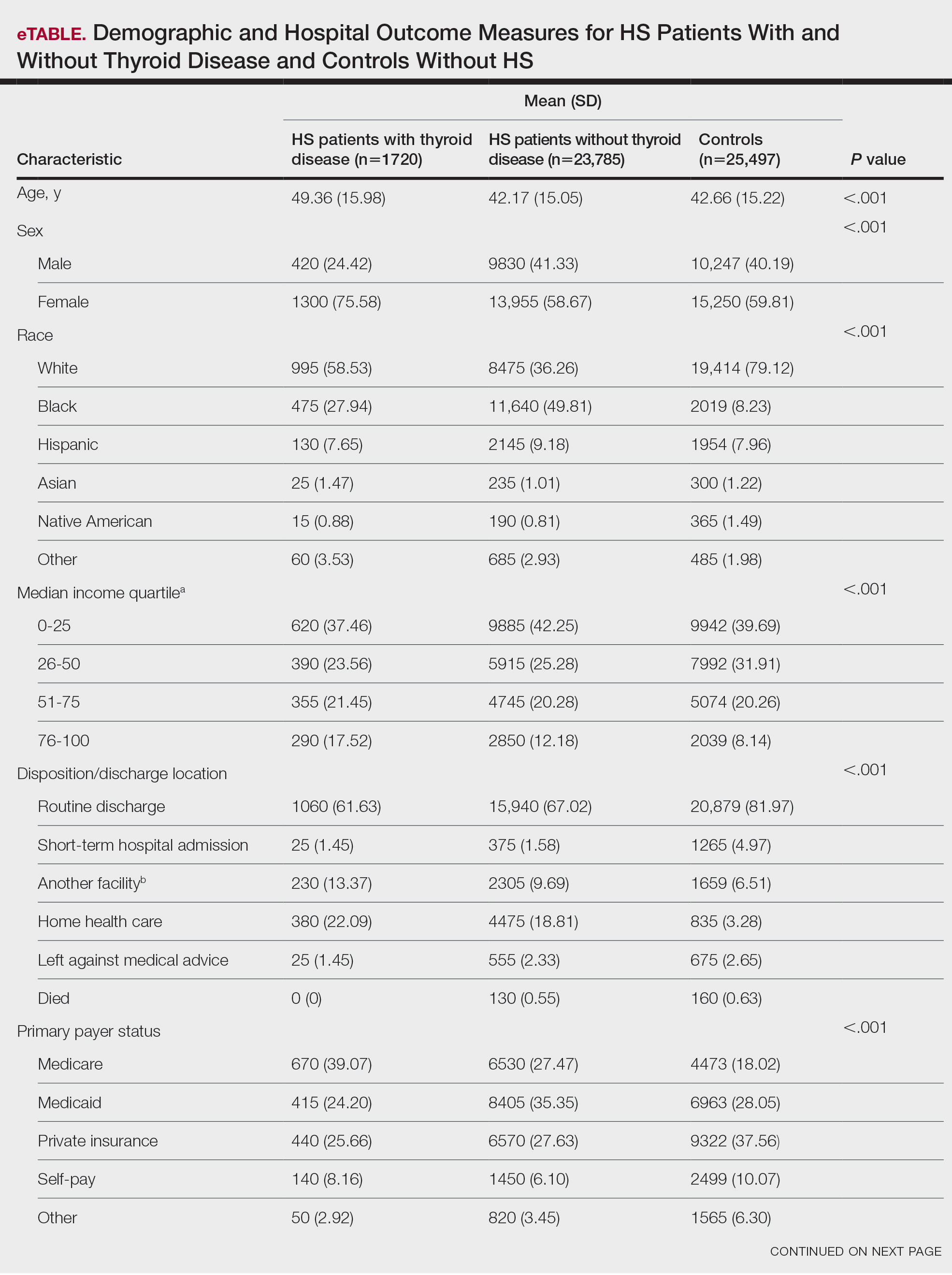
On univariate analysis of hospital outcome measures, HS patients with comorbid thyroid disease had the highest frequency of extreme likelihood of dying compared with HS patients without thyroid disease and with controls (6.40% vs 5.38% vs 2.47% [P<.001]), the highest mean number of diagnoses (18.31 vs 14.14 vs 8.57 [P<.001]), and the longest mean length of hospital stay (6.03 days vs 5.94 days vs 3.73 days [P<.001]). On univariate analysis of comorbidities, HS patients with thyroid disease had the highest incidence of the following comorbidities compared with HS patients without thyroid disease and controls: hypertension (34.01% vs 28.55% vs 22.39% [P<.001]), DM (48.26% vs 35.63% vs 18.05% [P<.001]), obesity (46.80% vs 39.65% vs 11.70% [P<.001]), and acute kidney injury (AKI)(21.80% vs 13.10% vs 6.33% [P<.001])(eTable).
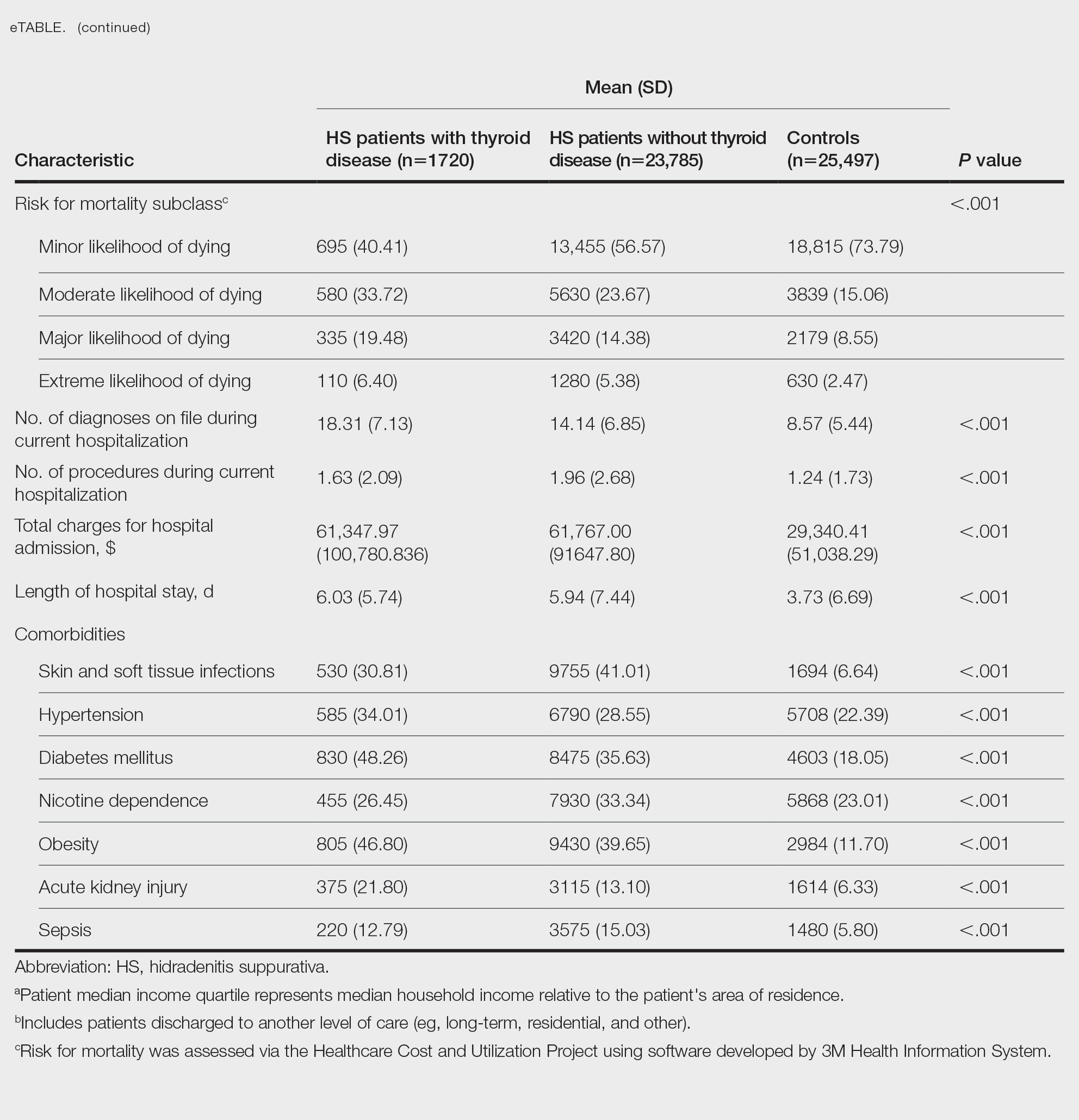
A multivariate analysis adjusting for multiple potential confounders including age, sex, race, median income quartile, disposition/discharge location, and primary payer was performed for hospital outcome measures and comorbidities. There were no significant differences in hospital outcome measures between HS patients with comorbid thyroid disease vs those without thyroid disease (P>.05)(Table 1). Thyroid disease was associated with increased odds of comorbid DM (odds ratio [OR], 1.242 [95% CI, 1.113-1.386]), obesity (OR, 1.173 [95% CI, 1.057-1.302]), and AKI (OR, 1.623 [95% CI, 1.423-1.851]) and decreased odds of comorbid nicotine dependence (OR, 0.609 [95% CI, 0.540-0.687]), skin and soft tissue infections (OR, 0.712 [95% CI, 0.637-0.797]), and sepsis (OR, 0.836 [95% CI, 0.717-0.973]) in HS patients (Table 2).
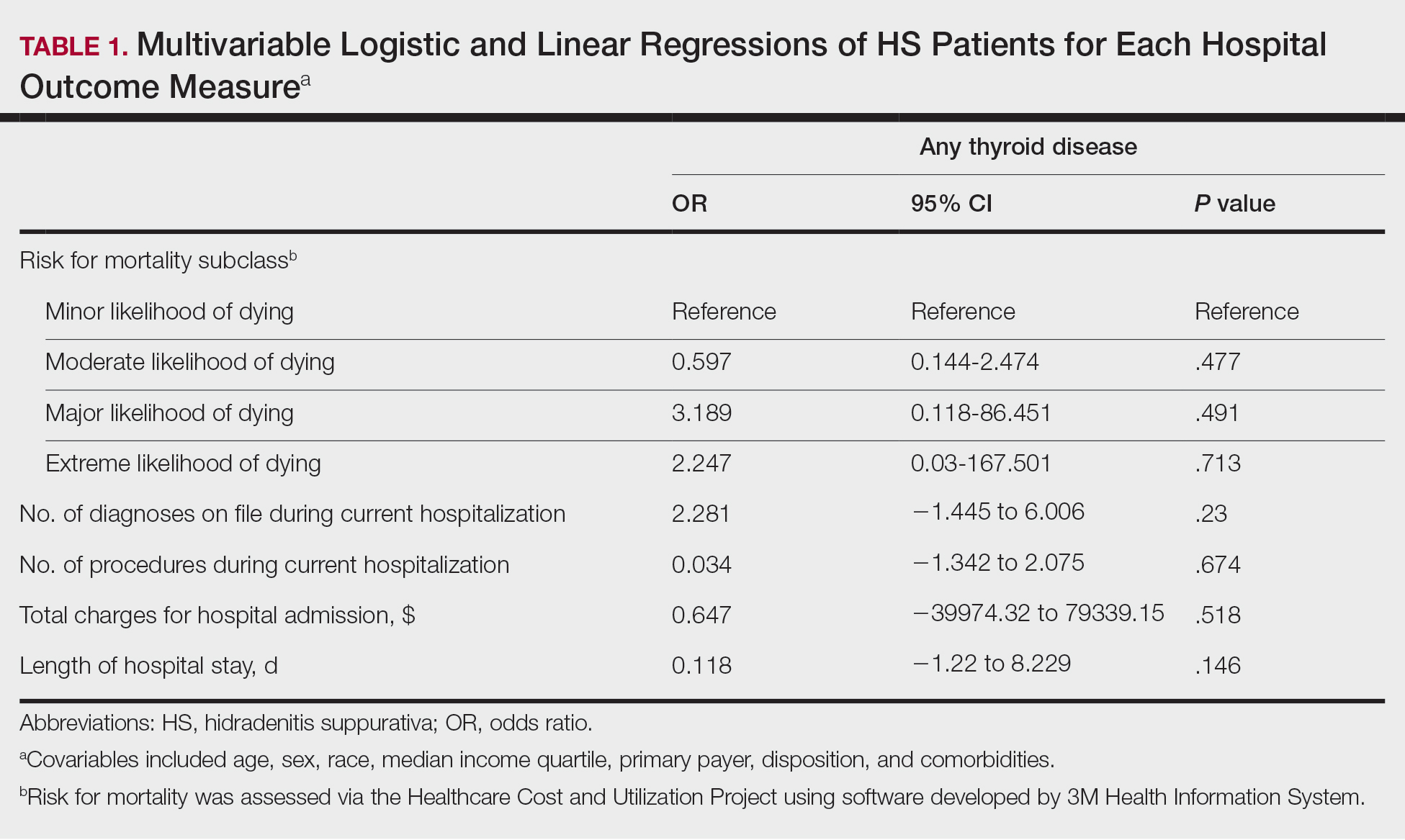

We found that HS patients with thyroid disease had increased odds of comorbid obesity, DM, and AKI compared with HS patients without thyroid disease when adjusting for potential confounders on multivariate analysis. A 2019 nationwide cross-sectional study of 18,224 patients with thyroid disease and 72,896 controls in Taiwan showed a higher prevalence of obesity (1.26% vs 0.57% [P<.0001]) and a higher hazard ratio (HR) of type 2 DM (HR, 1.23 [95% CI, 1.16-1.31]) in the thyroid disease group vs the controls.3 In a 2024 claims-based national cohort study of 4,152,830 patients with 2 or more consecutive thyroid-stimulating hormone measurements in the United States, patients with hypothyroidism and hyperthyroidism had a higher incidence risk for kidney dysfunction vs patients with euthyroidism (HRs, 1.37 [95% CI, 1.34–1.40] and 1.42 [95% CI, 1.39-1.45]).4 In addition, patients with and without DM and thyroid disease had increased risk for kidney disease compared to patients with and without DM and euthyroidism (hypothyroidism: HRs, 1.17 [95% CI, 1.13-1.22] and 1.52 [95% CI, 1.49-1.56]; hyperthyroidism: HRs, 1.34 [95% CI, 1.29-1.38] and 1.36 [95% CI, 1.33-1.39]). Furthermore, patients with and without obesity and thyroid disease had increased risk for kidney disease compared to patients with and without obesity and with euthyroidism (hypothyroidism: HRs, 1.40 [95% CI, 1.36-1.45] and 1.26 [95% CI, 1.21-1.32]; hyperthyroidism: HRs, 1.34 [95% CI, 1.30-1.39] and 1.35 [95% CI, 1.30-1.40]).4 However, these studies did not focus on HS patients.5
Hidradenitis suppurativa has a major comorbidity burden, including obesity, DM, and kidney disease.5 Our findings suggest a potential additive risk for these conditions in HS patients with comorbid thyroid disease; therefore, heightened surveillance for obesity, DM, and AKI in this population is encouraged. Prospective and retrospective studies in HS patients assessing the risk for each comorbidity while controlling for the others may help to better characterize these relationships.
Using multivariate analysis, we found that HS patients with comorbid thyroid disease had no significant differences in hospital outcome measures compared with HS patients without thyroid disease despite significant differences on univariate analysis (P<.05). Similarly, in a 2018 cross-sectional study of 430 HS patients and 20,780 controls in Denmark, the HS group had 10% lower thyroid-stimulating hormone levels vs the control group, but this did not significantly affect HS severity and thyroid function on multivariate analysis.6 In a 2020 cross-sectional analysis of 290 Greek HS patients, thyroid disease was associated with higher HS severity using Hurley classification (OR, 1.19 [95% CI, 1.03-1.51]) and International Hidradenitis Suppurativa Severity Score System 4 classification (OR, 1.29 [95% CI, 1.13-1.62]); however, this analysis was univariate and did not account for confounders.7 Taken together, our study and previous research suggest that thyroid disease is not an independent prognostic indicator for hospital outcome measures in HS patients when cofounders are considered and therefore may not warrant extra caution when treating hospitalized HS patients.
Nicotine dependence was an important potential confounder with regard to the effects of comorbid thyroid disease on outcomes of HS patients in our study. While we found that the prevalence of nicotine dependence was higher in HS patients vs matched controls, HS patients with comorbid thyroid disease had a lower prevalence of nicotine dependence than HS patients without thyroid disease. Furthermore, thyroid disease was associated with decreased odds of nicotine dependence in HS patients when adjusting for confounders. Previous studies have shown an association between cigarette smoking and HS. Smoking also may affect thyroid function via thiocyanate, sympathetic activation, or immunologic disturbances. Smoking may have both prothyroid and antithyroid effects.6 In a 2023 cross-sectional study of 108 HS patients and 52 age- and sex-matched controls in Germany, HS patients had higher thyroid antibody (TRAb) levels compared with controls (median TRAb level, 15.4 vs 14.2 [P=.026]), with even greater increases in TRAb in HS patients who were smokers or former smokers vs never smokers (median TRAb level, 1.18 vs 1.08 [P=.042]).2
There was a lower frequency of thyroid disease in our HS cohort compared with our matched controls cohort. While there are conflicting reports on the association between HS and thyroid disease in the literature, 2 recent meta-analyses of 5 and 6 case-control studies, respectively, found an association between HS and thyroid disease (OR, 1.36 [95% CI, 1.13-1.64] and 1.88 [95% CI, 1.25-2.81]).1,8 Notably, these studies were either claims or survey based, included outpatients, or were unspecified. One potential explanation for the difference in our findings vs those of other studies could be underdiagnosis of thyroid disease in hospitalized HS patients. We found that HS patients were most frequently Medicaid or Medicare insured compared to controls, who most frequently were privately insured. Increased availability and ease of access to outpatient medical care through private health insurance may be a possible contributor to the higher frequency of diagnosed thyroid disease in control patients in our study; therefore, awareness of potential underdiagnosis of thyroid disease in hospitalized HS patients is recommended.
Limitations of our study included those inherent to the NIS database, including potential miscoding and lack of data on pharmacologic treatments. Outcome measures assessed were limited by inclusion of both primary and secondary diagnoses of HS and thyroid disease in our cohort and may have been affected by other conditions. As with any observational study, there was a possibility of unidentified confounders unaccounted for in our study.
In conclusion, in this national inpatient-matched cohort study, thyroid disease was associated with increased odds of obesity, DM, and AKI in HS inpatients but was not an independent risk factor for worse hospital outcome measures. Therefore, while increased surveillance of associated comorbidities is appropriate, thyroid disease may not be a cause for increased concern for dermatologists treating hospitalized HS patients. Prospective studies are necessary to better characterize these findings.
- Phan K, Huo YR, Charlton O, et al. Hidradenitis suppurativa and thyroid disease: systematic review and meta-analysis. J Cutan Med Surg. 2020;24:23-27. doi:10.1177/1203475419874411
- Abu Rached N, Dietrich JW, Ocker L, et al. Primary thyroid dysfunction is prevalent in hidradenitis suppurativa and marked by a signature of hypothyroid Graves’ disease: a case-control study. J Clin Med. 2023;12:7490. doi:10.3390/jcm12237490
- Chen RH, Chen HY, Man KM, et al. Thyroid diseases increased the risk of type 2 diabetes mellitus: a nation-wide cohort study. Medicine (Baltimore). 2019;98:E15631. doi:10.1097/md.0000000000015631
- You AS, Kalantar-Zadeh K, Brent GA, et al. Impact of thyroid status on incident kidney dysfunction and chronic kidney disease progression in a nationally representative cohort. Mayo Clin Proc. 2024;99:39-56. doi:10.1016/j.mayocp.2023.08.028
- Almuhanna N, Tobe SW, Alhusayen R. Risk of chronic kidney disease in hospitalized patients with hidradenitis suppurativa. Dermatology. 2023;239:912-918. doi:10.1159/000531960
- Miller IM, Vinding G, Sorensen HA, et al. Thyroid function in hidradenitis suppurativa: a population]based cross]sectional study from Denmark. Clin Exp Dermatol. 2018;43:899-905. doi:10.1111/ced.13606
- Liakou AI, Kontochristopoulos G, Marnelakis I, et al. Thyroid disease and active smoking may be associated with more severe hidradenitis suppurativa: data from a prospective cross sectional single-center study. Dermatology. 2021;237:125-130. doi:10.1159/000508528
- Acharya P, Mathur M. Thyroid disorders in patients with hidradenitis suppurativa: a systematic review and meta-analysis. J Am Acad Dermatol. 2020;82:491-493. doi:10.1016/j.jaad.2019.07.025
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition characterized by painful recurrent abscesses. Several autoimmune and endocrine diseases are associated with HS, including inflammatory bowel disease and diabetes mellitus (DM).1 Notably, the association between HS and thyroid disorders is poorly characterized,2 and there are no known nationwide studies exploring this potential association in the hospital setting. In this cross-sectional matched cohort study, we aimed to characterize HS patients with comorbid thyroid disorders as well as to explore whether thyroid disease is associated with comorbidities and hospital outcome measures in these patients.
The 2019 National Inpatient Sample (NIS) was weighted in accordance with NIS-assigned weight variables and queried for HS, hypothyroidism, and hyperthyroidism cases using International Classification of Diseases, Tenth Revision, codes L73.2, E03, and E05, respectively. Propensity score matching based on age and sex was performed using a nearest-neighbor method in the MatchIt statistical R package. Patient demographics, comorbidities, and outcome variables were collected. Univariable analysis of HS patients with thyroid disease vs those without thyroid disease vs controls without HS were performed using X2 and t-test functions in SPSS statistical software (IBM). A series of multivariate analyses were performed using SPSS logistic and linear regression models to examine the effect of thyroid disease on hospital outcome measures and comorbidities in HS patients, with statistical significance set at P=.05.
A total of 1720 HS patients with comorbid thyroid disease (hyperthyroidism/hypothyroidism), 23,785 HS patients without thyroid disease, and 25,497 age- and sex-matched controls were included in the analysis. On average, HS patients with comorbid thyroid disease were older than HS patients without thyroid disease and controls (49.36 years vs 42.17 years vs 42.66 years [P<.001]), more likely to be female (75.58% vs 58.67% vs 59.81% [P<.001]), more likely to be in the highest income quartile (17.52% vs 12.18% vs 8.14% [P<.001]), and more likely to be Medicare insured (39.07% vs 27.47% vs 18.02% [P<.001])(eTable).
On univariate analysis of hospital outcome measures, HS patients with comorbid thyroid disease had the highest frequency of extreme likelihood of dying compared with HS patients without thyroid disease and with controls (6.40% vs 5.38% vs 2.47% [P<.001]), the highest mean number of diagnoses (18.31 vs 14.14 vs 8.57 [P<.001]), and the longest mean length of hospital stay (6.03 days vs 5.94 days vs 3.73 days [P<.001]). On univariate analysis of comorbidities, HS patients with thyroid disease had the highest incidence of the following comorbidities compared with HS patients without thyroid disease and controls: hypertension (34.01% vs 28.55% vs 22.39% [P<.001]), DM (48.26% vs 35.63% vs 18.05% [P<.001]), obesity (46.80% vs 39.65% vs 11.70% [P<.001]), and acute kidney injury (AKI)(21.80% vs 13.10% vs 6.33% [P<.001])(eTable).
A multivariate analysis adjusting for multiple potential confounders including age, sex, race, median income quartile, disposition/discharge location, and primary payer was performed for hospital outcome measures and comorbidities. There were no significant differences in hospital outcome measures between HS patients with comorbid thyroid disease vs those without thyroid disease (P>.05)(Table 1). Thyroid disease was associated with increased odds of comorbid DM (odds ratio [OR], 1.242 [95% CI, 1.113-1.386]), obesity (OR, 1.173 [95% CI, 1.057-1.302]), and AKI (OR, 1.623 [95% CI, 1.423-1.851]) and decreased odds of comorbid nicotine dependence (OR, 0.609 [95% CI, 0.540-0.687]), skin and soft tissue infections (OR, 0.712 [95% CI, 0.637-0.797]), and sepsis (OR, 0.836 [95% CI, 0.717-0.973]) in HS patients (Table 2).
We found that HS patients with thyroid disease had increased odds of comorbid obesity, DM, and AKI compared with HS patients without thyroid disease when adjusting for potential confounders on multivariate analysis. A 2019 nationwide cross-sectional study of 18,224 patients with thyroid disease and 72,896 controls in Taiwan showed a higher prevalence of obesity (1.26% vs 0.57% [P<.0001]) and a higher hazard ratio (HR) of type 2 DM (HR, 1.23 [95% CI, 1.16-1.31]) in the thyroid disease group vs the controls.3 In a 2024 claims-based national cohort study of 4,152,830 patients with 2 or more consecutive thyroid-stimulating hormone measurements in the United States, patients with hypothyroidism and hyperthyroidism had a higher incidence risk for kidney dysfunction vs patients with euthyroidism (HRs, 1.37 [95% CI, 1.34–1.40] and 1.42 [95% CI, 1.39-1.45]).4 In addition, patients with and without DM and thyroid disease had increased risk for kidney disease compared to patients with and without DM and euthyroidism (hypothyroidism: HRs, 1.17 [95% CI, 1.13-1.22] and 1.52 [95% CI, 1.49-1.56]; hyperthyroidism: HRs, 1.34 [95% CI, 1.29-1.38] and 1.36 [95% CI, 1.33-1.39]). Furthermore, patients with and without obesity and thyroid disease had increased risk for kidney disease compared to patients with and without obesity and with euthyroidism (hypothyroidism: HRs, 1.40 [95% CI, 1.36-1.45] and 1.26 [95% CI, 1.21-1.32]; hyperthyroidism: HRs, 1.34 [95% CI, 1.30-1.39] and 1.35 [95% CI, 1.30-1.40]).4 However, these studies did not focus on HS patients.5
Hidradenitis suppurativa has a major comorbidity burden, including obesity, DM, and kidney disease.5 Our findings suggest a potential additive risk for these conditions in HS patients with comorbid thyroid disease; therefore, heightened surveillance for obesity, DM, and AKI in this population is encouraged. Prospective and retrospective studies in HS patients assessing the risk for each comorbidity while controlling for the others may help to better characterize these relationships.
Using multivariate analysis, we found that HS patients with comorbid thyroid disease had no significant differences in hospital outcome measures compared with HS patients without thyroid disease despite significant differences on univariate analysis (P<.05). Similarly, in a 2018 cross-sectional study of 430 HS patients and 20,780 controls in Denmark, the HS group had 10% lower thyroid-stimulating hormone levels vs the control group, but this did not significantly affect HS severity and thyroid function on multivariate analysis.6 In a 2020 cross-sectional analysis of 290 Greek HS patients, thyroid disease was associated with higher HS severity using Hurley classification (OR, 1.19 [95% CI, 1.03-1.51]) and International Hidradenitis Suppurativa Severity Score System 4 classification (OR, 1.29 [95% CI, 1.13-1.62]); however, this analysis was univariate and did not account for confounders.7 Taken together, our study and previous research suggest that thyroid disease is not an independent prognostic indicator for hospital outcome measures in HS patients when cofounders are considered and therefore may not warrant extra caution when treating hospitalized HS patients.
Nicotine dependence was an important potential confounder with regard to the effects of comorbid thyroid disease on outcomes of HS patients in our study. While we found that the prevalence of nicotine dependence was higher in HS patients vs matched controls, HS patients with comorbid thyroid disease had a lower prevalence of nicotine dependence than HS patients without thyroid disease. Furthermore, thyroid disease was associated with decreased odds of nicotine dependence in HS patients when adjusting for confounders. Previous studies have shown an association between cigarette smoking and HS. Smoking also may affect thyroid function via thiocyanate, sympathetic activation, or immunologic disturbances. Smoking may have both prothyroid and antithyroid effects.6 In a 2023 cross-sectional study of 108 HS patients and 52 age- and sex-matched controls in Germany, HS patients had higher thyroid antibody (TRAb) levels compared with controls (median TRAb level, 15.4 vs 14.2 [P=.026]), with even greater increases in TRAb in HS patients who were smokers or former smokers vs never smokers (median TRAb level, 1.18 vs 1.08 [P=.042]).2
There was a lower frequency of thyroid disease in our HS cohort compared with our matched controls cohort. While there are conflicting reports on the association between HS and thyroid disease in the literature, 2 recent meta-analyses of 5 and 6 case-control studies, respectively, found an association between HS and thyroid disease (OR, 1.36 [95% CI, 1.13-1.64] and 1.88 [95% CI, 1.25-2.81]).1,8 Notably, these studies were either claims or survey based, included outpatients, or were unspecified. One potential explanation for the difference in our findings vs those of other studies could be underdiagnosis of thyroid disease in hospitalized HS patients. We found that HS patients were most frequently Medicaid or Medicare insured compared to controls, who most frequently were privately insured. Increased availability and ease of access to outpatient medical care through private health insurance may be a possible contributor to the higher frequency of diagnosed thyroid disease in control patients in our study; therefore, awareness of potential underdiagnosis of thyroid disease in hospitalized HS patients is recommended.
Limitations of our study included those inherent to the NIS database, including potential miscoding and lack of data on pharmacologic treatments. Outcome measures assessed were limited by inclusion of both primary and secondary diagnoses of HS and thyroid disease in our cohort and may have been affected by other conditions. As with any observational study, there was a possibility of unidentified confounders unaccounted for in our study.
In conclusion, in this national inpatient-matched cohort study, thyroid disease was associated with increased odds of obesity, DM, and AKI in HS inpatients but was not an independent risk factor for worse hospital outcome measures. Therefore, while increased surveillance of associated comorbidities is appropriate, thyroid disease may not be a cause for increased concern for dermatologists treating hospitalized HS patients. Prospective studies are necessary to better characterize these findings.
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition characterized by painful recurrent abscesses. Several autoimmune and endocrine diseases are associated with HS, including inflammatory bowel disease and diabetes mellitus (DM).1 Notably, the association between HS and thyroid disorders is poorly characterized,2 and there are no known nationwide studies exploring this potential association in the hospital setting. In this cross-sectional matched cohort study, we aimed to characterize HS patients with comorbid thyroid disorders as well as to explore whether thyroid disease is associated with comorbidities and hospital outcome measures in these patients.
The 2019 National Inpatient Sample (NIS) was weighted in accordance with NIS-assigned weight variables and queried for HS, hypothyroidism, and hyperthyroidism cases using International Classification of Diseases, Tenth Revision, codes L73.2, E03, and E05, respectively. Propensity score matching based on age and sex was performed using a nearest-neighbor method in the MatchIt statistical R package. Patient demographics, comorbidities, and outcome variables were collected. Univariable analysis of HS patients with thyroid disease vs those without thyroid disease vs controls without HS were performed using X2 and t-test functions in SPSS statistical software (IBM). A series of multivariate analyses were performed using SPSS logistic and linear regression models to examine the effect of thyroid disease on hospital outcome measures and comorbidities in HS patients, with statistical significance set at P=.05.
A total of 1720 HS patients with comorbid thyroid disease (hyperthyroidism/hypothyroidism), 23,785 HS patients without thyroid disease, and 25,497 age- and sex-matched controls were included in the analysis. On average, HS patients with comorbid thyroid disease were older than HS patients without thyroid disease and controls (49.36 years vs 42.17 years vs 42.66 years [P<.001]), more likely to be female (75.58% vs 58.67% vs 59.81% [P<.001]), more likely to be in the highest income quartile (17.52% vs 12.18% vs 8.14% [P<.001]), and more likely to be Medicare insured (39.07% vs 27.47% vs 18.02% [P<.001])(eTable).
On univariate analysis of hospital outcome measures, HS patients with comorbid thyroid disease had the highest frequency of extreme likelihood of dying compared with HS patients without thyroid disease and with controls (6.40% vs 5.38% vs 2.47% [P<.001]), the highest mean number of diagnoses (18.31 vs 14.14 vs 8.57 [P<.001]), and the longest mean length of hospital stay (6.03 days vs 5.94 days vs 3.73 days [P<.001]). On univariate analysis of comorbidities, HS patients with thyroid disease had the highest incidence of the following comorbidities compared with HS patients without thyroid disease and controls: hypertension (34.01% vs 28.55% vs 22.39% [P<.001]), DM (48.26% vs 35.63% vs 18.05% [P<.001]), obesity (46.80% vs 39.65% vs 11.70% [P<.001]), and acute kidney injury (AKI)(21.80% vs 13.10% vs 6.33% [P<.001])(eTable).
A multivariate analysis adjusting for multiple potential confounders including age, sex, race, median income quartile, disposition/discharge location, and primary payer was performed for hospital outcome measures and comorbidities. There were no significant differences in hospital outcome measures between HS patients with comorbid thyroid disease vs those without thyroid disease (P>.05)(Table 1). Thyroid disease was associated with increased odds of comorbid DM (odds ratio [OR], 1.242 [95% CI, 1.113-1.386]), obesity (OR, 1.173 [95% CI, 1.057-1.302]), and AKI (OR, 1.623 [95% CI, 1.423-1.851]) and decreased odds of comorbid nicotine dependence (OR, 0.609 [95% CI, 0.540-0.687]), skin and soft tissue infections (OR, 0.712 [95% CI, 0.637-0.797]), and sepsis (OR, 0.836 [95% CI, 0.717-0.973]) in HS patients (Table 2).
We found that HS patients with thyroid disease had increased odds of comorbid obesity, DM, and AKI compared with HS patients without thyroid disease when adjusting for potential confounders on multivariate analysis. A 2019 nationwide cross-sectional study of 18,224 patients with thyroid disease and 72,896 controls in Taiwan showed a higher prevalence of obesity (1.26% vs 0.57% [P<.0001]) and a higher hazard ratio (HR) of type 2 DM (HR, 1.23 [95% CI, 1.16-1.31]) in the thyroid disease group vs the controls.3 In a 2024 claims-based national cohort study of 4,152,830 patients with 2 or more consecutive thyroid-stimulating hormone measurements in the United States, patients with hypothyroidism and hyperthyroidism had a higher incidence risk for kidney dysfunction vs patients with euthyroidism (HRs, 1.37 [95% CI, 1.34–1.40] and 1.42 [95% CI, 1.39-1.45]).4 In addition, patients with and without DM and thyroid disease had increased risk for kidney disease compared to patients with and without DM and euthyroidism (hypothyroidism: HRs, 1.17 [95% CI, 1.13-1.22] and 1.52 [95% CI, 1.49-1.56]; hyperthyroidism: HRs, 1.34 [95% CI, 1.29-1.38] and 1.36 [95% CI, 1.33-1.39]). Furthermore, patients with and without obesity and thyroid disease had increased risk for kidney disease compared to patients with and without obesity and with euthyroidism (hypothyroidism: HRs, 1.40 [95% CI, 1.36-1.45] and 1.26 [95% CI, 1.21-1.32]; hyperthyroidism: HRs, 1.34 [95% CI, 1.30-1.39] and 1.35 [95% CI, 1.30-1.40]).4 However, these studies did not focus on HS patients.5
Hidradenitis suppurativa has a major comorbidity burden, including obesity, DM, and kidney disease.5 Our findings suggest a potential additive risk for these conditions in HS patients with comorbid thyroid disease; therefore, heightened surveillance for obesity, DM, and AKI in this population is encouraged. Prospective and retrospective studies in HS patients assessing the risk for each comorbidity while controlling for the others may help to better characterize these relationships.
Using multivariate analysis, we found that HS patients with comorbid thyroid disease had no significant differences in hospital outcome measures compared with HS patients without thyroid disease despite significant differences on univariate analysis (P<.05). Similarly, in a 2018 cross-sectional study of 430 HS patients and 20,780 controls in Denmark, the HS group had 10% lower thyroid-stimulating hormone levels vs the control group, but this did not significantly affect HS severity and thyroid function on multivariate analysis.6 In a 2020 cross-sectional analysis of 290 Greek HS patients, thyroid disease was associated with higher HS severity using Hurley classification (OR, 1.19 [95% CI, 1.03-1.51]) and International Hidradenitis Suppurativa Severity Score System 4 classification (OR, 1.29 [95% CI, 1.13-1.62]); however, this analysis was univariate and did not account for confounders.7 Taken together, our study and previous research suggest that thyroid disease is not an independent prognostic indicator for hospital outcome measures in HS patients when cofounders are considered and therefore may not warrant extra caution when treating hospitalized HS patients.
Nicotine dependence was an important potential confounder with regard to the effects of comorbid thyroid disease on outcomes of HS patients in our study. While we found that the prevalence of nicotine dependence was higher in HS patients vs matched controls, HS patients with comorbid thyroid disease had a lower prevalence of nicotine dependence than HS patients without thyroid disease. Furthermore, thyroid disease was associated with decreased odds of nicotine dependence in HS patients when adjusting for confounders. Previous studies have shown an association between cigarette smoking and HS. Smoking also may affect thyroid function via thiocyanate, sympathetic activation, or immunologic disturbances. Smoking may have both prothyroid and antithyroid effects.6 In a 2023 cross-sectional study of 108 HS patients and 52 age- and sex-matched controls in Germany, HS patients had higher thyroid antibody (TRAb) levels compared with controls (median TRAb level, 15.4 vs 14.2 [P=.026]), with even greater increases in TRAb in HS patients who were smokers or former smokers vs never smokers (median TRAb level, 1.18 vs 1.08 [P=.042]).2
There was a lower frequency of thyroid disease in our HS cohort compared with our matched controls cohort. While there are conflicting reports on the association between HS and thyroid disease in the literature, 2 recent meta-analyses of 5 and 6 case-control studies, respectively, found an association between HS and thyroid disease (OR, 1.36 [95% CI, 1.13-1.64] and 1.88 [95% CI, 1.25-2.81]).1,8 Notably, these studies were either claims or survey based, included outpatients, or were unspecified. One potential explanation for the difference in our findings vs those of other studies could be underdiagnosis of thyroid disease in hospitalized HS patients. We found that HS patients were most frequently Medicaid or Medicare insured compared to controls, who most frequently were privately insured. Increased availability and ease of access to outpatient medical care through private health insurance may be a possible contributor to the higher frequency of diagnosed thyroid disease in control patients in our study; therefore, awareness of potential underdiagnosis of thyroid disease in hospitalized HS patients is recommended.
Limitations of our study included those inherent to the NIS database, including potential miscoding and lack of data on pharmacologic treatments. Outcome measures assessed were limited by inclusion of both primary and secondary diagnoses of HS and thyroid disease in our cohort and may have been affected by other conditions. As with any observational study, there was a possibility of unidentified confounders unaccounted for in our study.
In conclusion, in this national inpatient-matched cohort study, thyroid disease was associated with increased odds of obesity, DM, and AKI in HS inpatients but was not an independent risk factor for worse hospital outcome measures. Therefore, while increased surveillance of associated comorbidities is appropriate, thyroid disease may not be a cause for increased concern for dermatologists treating hospitalized HS patients. Prospective studies are necessary to better characterize these findings.
- Phan K, Huo YR, Charlton O, et al. Hidradenitis suppurativa and thyroid disease: systematic review and meta-analysis. J Cutan Med Surg. 2020;24:23-27. doi:10.1177/1203475419874411
- Abu Rached N, Dietrich JW, Ocker L, et al. Primary thyroid dysfunction is prevalent in hidradenitis suppurativa and marked by a signature of hypothyroid Graves’ disease: a case-control study. J Clin Med. 2023;12:7490. doi:10.3390/jcm12237490
- Chen RH, Chen HY, Man KM, et al. Thyroid diseases increased the risk of type 2 diabetes mellitus: a nation-wide cohort study. Medicine (Baltimore). 2019;98:E15631. doi:10.1097/md.0000000000015631
- You AS, Kalantar-Zadeh K, Brent GA, et al. Impact of thyroid status on incident kidney dysfunction and chronic kidney disease progression in a nationally representative cohort. Mayo Clin Proc. 2024;99:39-56. doi:10.1016/j.mayocp.2023.08.028
- Almuhanna N, Tobe SW, Alhusayen R. Risk of chronic kidney disease in hospitalized patients with hidradenitis suppurativa. Dermatology. 2023;239:912-918. doi:10.1159/000531960
- Miller IM, Vinding G, Sorensen HA, et al. Thyroid function in hidradenitis suppurativa: a population]based cross]sectional study from Denmark. Clin Exp Dermatol. 2018;43:899-905. doi:10.1111/ced.13606
- Liakou AI, Kontochristopoulos G, Marnelakis I, et al. Thyroid disease and active smoking may be associated with more severe hidradenitis suppurativa: data from a prospective cross sectional single-center study. Dermatology. 2021;237:125-130. doi:10.1159/000508528
- Acharya P, Mathur M. Thyroid disorders in patients with hidradenitis suppurativa: a systematic review and meta-analysis. J Am Acad Dermatol. 2020;82:491-493. doi:10.1016/j.jaad.2019.07.025
- Phan K, Huo YR, Charlton O, et al. Hidradenitis suppurativa and thyroid disease: systematic review and meta-analysis. J Cutan Med Surg. 2020;24:23-27. doi:10.1177/1203475419874411
- Abu Rached N, Dietrich JW, Ocker L, et al. Primary thyroid dysfunction is prevalent in hidradenitis suppurativa and marked by a signature of hypothyroid Graves’ disease: a case-control study. J Clin Med. 2023;12:7490. doi:10.3390/jcm12237490
- Chen RH, Chen HY, Man KM, et al. Thyroid diseases increased the risk of type 2 diabetes mellitus: a nation-wide cohort study. Medicine (Baltimore). 2019;98:E15631. doi:10.1097/md.0000000000015631
- You AS, Kalantar-Zadeh K, Brent GA, et al. Impact of thyroid status on incident kidney dysfunction and chronic kidney disease progression in a nationally representative cohort. Mayo Clin Proc. 2024;99:39-56. doi:10.1016/j.mayocp.2023.08.028
- Almuhanna N, Tobe SW, Alhusayen R. Risk of chronic kidney disease in hospitalized patients with hidradenitis suppurativa. Dermatology. 2023;239:912-918. doi:10.1159/000531960
- Miller IM, Vinding G, Sorensen HA, et al. Thyroid function in hidradenitis suppurativa: a population]based cross]sectional study from Denmark. Clin Exp Dermatol. 2018;43:899-905. doi:10.1111/ced.13606
- Liakou AI, Kontochristopoulos G, Marnelakis I, et al. Thyroid disease and active smoking may be associated with more severe hidradenitis suppurativa: data from a prospective cross sectional single-center study. Dermatology. 2021;237:125-130. doi:10.1159/000508528
- Acharya P, Mathur M. Thyroid disorders in patients with hidradenitis suppurativa: a systematic review and meta-analysis. J Am Acad Dermatol. 2020;82:491-493. doi:10.1016/j.jaad.2019.07.025
Implications of Thyroid Disease in Hospitalized Patients With Hidradenitis Suppurativa
Implications of Thyroid Disease in Hospitalized Patients With Hidradenitis Suppurativa
PRACTICE
- Hidradenitis suppurativa (HS) is associated with autoimmune and endocrine conditions, but the association between HS and thyroid disorders is poorly characterized.
Not as Bland as You May Think: Celery (Apium graveolens) Commonly Induces Phytophotodermatitis
Not as Bland as You May Think: Celery (Apium graveolens) Commonly Induces Phytophotodermatitis
Celery (Apium graveolens)—that lowly vegetable that often languishes in the refrigerator crisper and apparently supplies fewer calories than are required to consume it—contains a myriad of photosensitizing chemicals known as furocoumarins and psoralens that can cause phytophotodermatitis (PPD) when handled prior to exposure to UV light.1 Individuals who are most likely to develop PPD caused by repeated contact with celery include food industry workers (eg, grocery store workers, farmers) who pick, handle, or prepare celery for consumption. While eating celery as part of a standard diet is highly unlikely to cause PPD, celery infected with Sclerotinia sclerotiorum (known as pink rot) causes more severe generalized sun sensitivity due to an increased amount of furocoumarins produced in response to the fungus.2 Contact with celery also can induce cutaneous manifestations unrelated to sun exposure in some individuals, including urticaria, allergic contact dermatitis, and anaphylaxis.3 In this article, we provide an overview of the life cycle and origin of celery as well as its irritant and allergic properties. We also describe cutaneous rashes associated with PPD caused by exposure to celery and highlight treatment options.
Morphology and Distribution
The Apiaceae family features aromatic flowering plants that comprise more than 3500 species, including many economically important vegetables, herbs, and spices.4 It also includes many alkaloid-containing species that are known to be poisonous to humans, such as poison hemlock (Conium maculatum) and water hemlock (Cicuta maculate). Most Apiaceae plants that are consumed by humans originate from the Mediterranean region.5 While known for their diversity of flavor and aroma, most of the plants from this family have low caloric value and provide minimal amounts of energy.
Members of the Apiaceae family have flowers that create a classic umbel shape mimicking the appearance of an upside-down umbrella (thus the former name for this family, Umbelliferae). The pedicles—the small stems attached to the base of each flower—spread from a common center to form the umbel.5 The Apiaceae family also includes the greatest number of plants that cause PPD due to their high concentration of furocoumarins, which deter fungus from harming the plants.6
A biennial plant, celery completes its life cycle in 2 years. During the first season, the stems, roots, and leaves sprout; in the second and final year, the flowers, fruits, and seeds proliferate, followed by decomposition. Apium graveolens approaches heights of 2 to 3 ft, growing upright and displaying grooved stems. Each stem terminates in a basal rosette of leaves. The second season brings white flower blooms in terminal or axillary umbels.7
Celery originated in the temperate Mediterranean regions of Europe, but farmers now cultivate it globally.8 It grows best in rich moist soil with full exposure to sunlight. Plants multiply their numbers through self-seeding. Celery commonly is found in suburban and rural homes, both in refrigerators for consumption as well as in medicine cabinets in capsule form for the treatment of arthritis.4
Irritant and Allergenic Properties
Despite the potential health benefits of celery, the Apiaceae family, which includes hogweed, dill, and fennel, prevails as the most common culprit for phytotoxic reactions. The Rutaceae family, including citrus plants and rue, remains runner-up for causes of PPD.9 Phytophotodermatitis is not an immunologic reaction, making anyone susceptible to formation of the cutaneous lesions when exposed to UV light after handling celery. Pruritis rarely occurs, unlike in allergic phytodermatitis.10 Upon photoexcitation from exposure to UVA light, individual psoralen molecules covalently bind to pyrimidine bases, causing interstrand cross-linking that prevents DNA replication and triggering a cascade leading to apoptosis of the cell. Apoptosis induces cell membrane edema, which manifests as cutaneous vesicles and bullae on the skin.10 Regardless of plant species, PPD reactions have similar appearance.
Celery roots contain the greatest concentration of psoralens, making it the most likely part of the plant to induce PPD.6 Phytophotodermatitis caused by celery can occur at any time of the year, but most eruptions occur during the summer months due to increased sunlight exposure and intensity. Among 320 randomly selected Michigan celery harvesters, 163 (51%) displayed evidence of vesicular and bullous dermatitis on the fingers, hands, and forearms.11 In this study, celery infected with pink rot fungus induced an erythematous eruption with vesicles and bullae within 48 hours of contact after just 30 seconds of summer sunlight exposure; however, eruptions are not limited to summer months, as the cutaneous presentation depends solely on exposure to UVA light, which can occur year-round.
Use of tanning beds is a major risk factor for PPD.12 Tanning beds utilize fluorescent bulbs that primarily emit UVA light, with UVB light emitted to a lesser degree. The UVA radiation produced by tanning beds is more than 3 times as intense as natural sunlight.12 Among grocery store employees, the combination of these 2 risk factors—regular contact with celery and tanning bed use—resulted in a prevalence ratio for PPD more than 40 times greater than that of individuals with neither risk factor.13
Cutaneous Manifestations of PPD
Phytophotodermatitis is a nonimmunologic dermatitis that forms via the interaction between UV light exposure and the photosensitizing chemicals inherent to some plant species. Development of PPD following contact with celery may be caused by the photoactive substances in celery, including the psoralens 8-methoxypsoralen and 5-methoxypsoralen.14 The psoralens must become activated by UV light with wavelengths between 320 nm and 400 nm (UVA) to initiate biologic effects.15
Once chemically activated, the photoactive mediators cause an erythematous and edematous sunburnlike reaction. Current hypotheses state that psoralen plus UVA generates reactive oxygen species, which damage the DNA within cells and alter receptors on cell membranes within the epidermis.14 The cutaneous eruption usually appears between 12 and 36 hours after sun exposure. Although they generally are not pruritic, the eruptions may induce pain. Within 7 to 10 days following development of the rash, hyperpigmentation occurs in the affected area and often persists for months to years.16 Ingestion of large amounts of celery has been cited to cause generalized phototoxic reactions; however, PPD rarely arises solely after ingestion, unless excessive amounts are consumed with concomitant exposure to psoralen plus UVA or tanning beds.17 In these cases, patients develop diffuse redness with superficial scaling, pain, and blistering if severe.
Treatment of PPD
Prevention remains the best form of treatment for PPD caused by exposure to celery. Postcontact management includes washing the affected area with soap and water and changing clothes promptly. Topical corticosteroids have mild utility in treatment of PPD.18 Oral steroid tapers, which reduce acute inflammation, also are an option for treatment. Alternatively, intramuscular triamcinolone acetonide 1 mg/kg mixed with budesonide 0.1 mg/kg is an option and is associated with a reduced risk for adverse effects compared to oral steroids. The resulting hyperpigmentation develops 1 to 2 weeks postepithelialization.19 Hyperpigmentation often fades slowly over several months in lighter-skinned individuals but may last for years or indefinitely in darker-skinned patients.
Final Thoughts
Dermatologists should be knowledgeable about the various plant culprits that can induce PPD. Understanding the mechanism and pathophysiology can help guide both therapeutic interventions and preventive counseling. Understanding that even readily available vegetables such as celery can induce cutaneous eruptions should put PPD in the differential diagnosis more commonly when unspecified dermatitides are present.
- Walansky A. Study finally confirms eating celery burns more calories than it contains. Food & Wine. June 22, 2017. Accessed January 17, 2025. https://www.foodandwine.com/news/study-finally-confirms-eating-celery-burns-more-caloriesit-contains
- Puig L. Enhancement of PUVA phototoxic effects following celery ingestion: cool broth also can burn. Arch Dermatol. 1994;130:809-810. doi:10.1001/archderm.130.6.809
- Perez-Pimiento AJ, Moneo I, Santaolalla M, et al. Anaphylactic reaction to young garlic. Allergy. 1999;54:626-629.
- The Editors of Encyclopaedia Britannica. Apiaceae. Britannica. Updated November 25, 2024. Accessed January 17, 2025. https://www.britannica.com/plant/Apiaceae
- Smith R. Celery. In: Geoffriau E, Simon PW, eds. Carrots and Related Apiaceae Crops. 2nd ed. CABI; 2021:272-282.
- Dijkstra JWE, Chang L. Severe phototoxic burn following celery ingestion. Arch Dermatol. 1992;128:1277.
- Tobyn G, Denham A, Whitelegg M. Apium graveolens, wild celery. The Western Herbal Tradition: 2000 years of Medicinal Plant Knowledge. Elsevier. 2011:79-89. doi:10.1016/b978-0-443-10344-5.00014-8
- Rademaker M. Celery. DermNet. Accessed January 17, 2025. https://dermnetnz.org/topics/celery
- Sasseville D. Clinical patterns of phytophotodermatitis. Dermatol Clin. 2009;27:299-308.
- Jin Goon AT, Goh CL. Plant dermatitis: Asian perspective. Indian J Dermatol. 2011;56:707-710. doi:10.4103/0019-5154.91833
- Birmingham DJ, Key MM, Tublich GE. Phototoxic bullae among celery harvesters. Arch Dermatol. 1961;83:73-87.
- Robb-Nicholson C. By the way, doctor: is a tanning bed safer than sunlight? Harvard Health Publishing. Harvard Medical School. September 1, 2009. Accessed January 17, 2025. https://www.health.harvard.edu/staying-healthy/is-a-tanning-bed-saferthan-sunlight
- Vester L, Thyssen JP, Menne T, et al. Consequences of occupational food-related hand dermatoses with a focus on protein contact dermatitis. Contact Dermatitis. 2012;67:328-333.
- Ling TC, Clayton TH, Crawley J, et al. British Association of Dermatologists and British Photodermatology Group guidelines for the safe and effective use of psoralen-ultraviolet A therapy 2015. Br J Dermatol. 2016;174:24-55.
- Laskin JD. Cellular and molecular mechanisms in photochemical sensitization: studies on the mechanism of action of psoralens. Food Chem Toxicol. 1994;32:119-127. doi:10.1016/0278-6915(94)90172-4
- Elmets CA. Photosensitivity disorders (photodermatoses): clinical manifestations, diagnosis, and treatment. UpToDate. Updated February 23, 2023. Accessed January 17, 2025. https://www.uptodate.com/contents/photosensitivity-disorders-photodermatoses-clinical-manifestations-diagnosis-and-treatment
- Boffa, MJ, Gilmour E, Ead RD. Celery soup causing severe phototoxity during PUVA therapy. Br J Dermatol. 1996;135:334. doi:10.1111/j.1365-2133.1996.tb01182.x
- Sarhane KA, Ibrahim A, Fagan SP, et al. Phytophotodermatitis. Eplasty. 2013;13:ic57.
- McGovern TW. Dermatoses due to plants. In: Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. Mosby; 2018:286-303.
Celery (Apium graveolens)—that lowly vegetable that often languishes in the refrigerator crisper and apparently supplies fewer calories than are required to consume it—contains a myriad of photosensitizing chemicals known as furocoumarins and psoralens that can cause phytophotodermatitis (PPD) when handled prior to exposure to UV light.1 Individuals who are most likely to develop PPD caused by repeated contact with celery include food industry workers (eg, grocery store workers, farmers) who pick, handle, or prepare celery for consumption. While eating celery as part of a standard diet is highly unlikely to cause PPD, celery infected with Sclerotinia sclerotiorum (known as pink rot) causes more severe generalized sun sensitivity due to an increased amount of furocoumarins produced in response to the fungus.2 Contact with celery also can induce cutaneous manifestations unrelated to sun exposure in some individuals, including urticaria, allergic contact dermatitis, and anaphylaxis.3 In this article, we provide an overview of the life cycle and origin of celery as well as its irritant and allergic properties. We also describe cutaneous rashes associated with PPD caused by exposure to celery and highlight treatment options.
Morphology and Distribution
The Apiaceae family features aromatic flowering plants that comprise more than 3500 species, including many economically important vegetables, herbs, and spices.4 It also includes many alkaloid-containing species that are known to be poisonous to humans, such as poison hemlock (Conium maculatum) and water hemlock (Cicuta maculate). Most Apiaceae plants that are consumed by humans originate from the Mediterranean region.5 While known for their diversity of flavor and aroma, most of the plants from this family have low caloric value and provide minimal amounts of energy.
Members of the Apiaceae family have flowers that create a classic umbel shape mimicking the appearance of an upside-down umbrella (thus the former name for this family, Umbelliferae). The pedicles—the small stems attached to the base of each flower—spread from a common center to form the umbel.5 The Apiaceae family also includes the greatest number of plants that cause PPD due to their high concentration of furocoumarins, which deter fungus from harming the plants.6
A biennial plant, celery completes its life cycle in 2 years. During the first season, the stems, roots, and leaves sprout; in the second and final year, the flowers, fruits, and seeds proliferate, followed by decomposition. Apium graveolens approaches heights of 2 to 3 ft, growing upright and displaying grooved stems. Each stem terminates in a basal rosette of leaves. The second season brings white flower blooms in terminal or axillary umbels.7
Celery originated in the temperate Mediterranean regions of Europe, but farmers now cultivate it globally.8 It grows best in rich moist soil with full exposure to sunlight. Plants multiply their numbers through self-seeding. Celery commonly is found in suburban and rural homes, both in refrigerators for consumption as well as in medicine cabinets in capsule form for the treatment of arthritis.4
Irritant and Allergenic Properties
Despite the potential health benefits of celery, the Apiaceae family, which includes hogweed, dill, and fennel, prevails as the most common culprit for phytotoxic reactions. The Rutaceae family, including citrus plants and rue, remains runner-up for causes of PPD.9 Phytophotodermatitis is not an immunologic reaction, making anyone susceptible to formation of the cutaneous lesions when exposed to UV light after handling celery. Pruritis rarely occurs, unlike in allergic phytodermatitis.10 Upon photoexcitation from exposure to UVA light, individual psoralen molecules covalently bind to pyrimidine bases, causing interstrand cross-linking that prevents DNA replication and triggering a cascade leading to apoptosis of the cell. Apoptosis induces cell membrane edema, which manifests as cutaneous vesicles and bullae on the skin.10 Regardless of plant species, PPD reactions have similar appearance.
Celery roots contain the greatest concentration of psoralens, making it the most likely part of the plant to induce PPD.6 Phytophotodermatitis caused by celery can occur at any time of the year, but most eruptions occur during the summer months due to increased sunlight exposure and intensity. Among 320 randomly selected Michigan celery harvesters, 163 (51%) displayed evidence of vesicular and bullous dermatitis on the fingers, hands, and forearms.11 In this study, celery infected with pink rot fungus induced an erythematous eruption with vesicles and bullae within 48 hours of contact after just 30 seconds of summer sunlight exposure; however, eruptions are not limited to summer months, as the cutaneous presentation depends solely on exposure to UVA light, which can occur year-round.
Use of tanning beds is a major risk factor for PPD.12 Tanning beds utilize fluorescent bulbs that primarily emit UVA light, with UVB light emitted to a lesser degree. The UVA radiation produced by tanning beds is more than 3 times as intense as natural sunlight.12 Among grocery store employees, the combination of these 2 risk factors—regular contact with celery and tanning bed use—resulted in a prevalence ratio for PPD more than 40 times greater than that of individuals with neither risk factor.13
Cutaneous Manifestations of PPD
Phytophotodermatitis is a nonimmunologic dermatitis that forms via the interaction between UV light exposure and the photosensitizing chemicals inherent to some plant species. Development of PPD following contact with celery may be caused by the photoactive substances in celery, including the psoralens 8-methoxypsoralen and 5-methoxypsoralen.14 The psoralens must become activated by UV light with wavelengths between 320 nm and 400 nm (UVA) to initiate biologic effects.15
Once chemically activated, the photoactive mediators cause an erythematous and edematous sunburnlike reaction. Current hypotheses state that psoralen plus UVA generates reactive oxygen species, which damage the DNA within cells and alter receptors on cell membranes within the epidermis.14 The cutaneous eruption usually appears between 12 and 36 hours after sun exposure. Although they generally are not pruritic, the eruptions may induce pain. Within 7 to 10 days following development of the rash, hyperpigmentation occurs in the affected area and often persists for months to years.16 Ingestion of large amounts of celery has been cited to cause generalized phototoxic reactions; however, PPD rarely arises solely after ingestion, unless excessive amounts are consumed with concomitant exposure to psoralen plus UVA or tanning beds.17 In these cases, patients develop diffuse redness with superficial scaling, pain, and blistering if severe.
Treatment of PPD
Prevention remains the best form of treatment for PPD caused by exposure to celery. Postcontact management includes washing the affected area with soap and water and changing clothes promptly. Topical corticosteroids have mild utility in treatment of PPD.18 Oral steroid tapers, which reduce acute inflammation, also are an option for treatment. Alternatively, intramuscular triamcinolone acetonide 1 mg/kg mixed with budesonide 0.1 mg/kg is an option and is associated with a reduced risk for adverse effects compared to oral steroids. The resulting hyperpigmentation develops 1 to 2 weeks postepithelialization.19 Hyperpigmentation often fades slowly over several months in lighter-skinned individuals but may last for years or indefinitely in darker-skinned patients.
Final Thoughts
Dermatologists should be knowledgeable about the various plant culprits that can induce PPD. Understanding the mechanism and pathophysiology can help guide both therapeutic interventions and preventive counseling. Understanding that even readily available vegetables such as celery can induce cutaneous eruptions should put PPD in the differential diagnosis more commonly when unspecified dermatitides are present.
Celery (Apium graveolens)—that lowly vegetable that often languishes in the refrigerator crisper and apparently supplies fewer calories than are required to consume it—contains a myriad of photosensitizing chemicals known as furocoumarins and psoralens that can cause phytophotodermatitis (PPD) when handled prior to exposure to UV light.1 Individuals who are most likely to develop PPD caused by repeated contact with celery include food industry workers (eg, grocery store workers, farmers) who pick, handle, or prepare celery for consumption. While eating celery as part of a standard diet is highly unlikely to cause PPD, celery infected with Sclerotinia sclerotiorum (known as pink rot) causes more severe generalized sun sensitivity due to an increased amount of furocoumarins produced in response to the fungus.2 Contact with celery also can induce cutaneous manifestations unrelated to sun exposure in some individuals, including urticaria, allergic contact dermatitis, and anaphylaxis.3 In this article, we provide an overview of the life cycle and origin of celery as well as its irritant and allergic properties. We also describe cutaneous rashes associated with PPD caused by exposure to celery and highlight treatment options.
Morphology and Distribution
The Apiaceae family features aromatic flowering plants that comprise more than 3500 species, including many economically important vegetables, herbs, and spices.4 It also includes many alkaloid-containing species that are known to be poisonous to humans, such as poison hemlock (Conium maculatum) and water hemlock (Cicuta maculate). Most Apiaceae plants that are consumed by humans originate from the Mediterranean region.5 While known for their diversity of flavor and aroma, most of the plants from this family have low caloric value and provide minimal amounts of energy.
Members of the Apiaceae family have flowers that create a classic umbel shape mimicking the appearance of an upside-down umbrella (thus the former name for this family, Umbelliferae). The pedicles—the small stems attached to the base of each flower—spread from a common center to form the umbel.5 The Apiaceae family also includes the greatest number of plants that cause PPD due to their high concentration of furocoumarins, which deter fungus from harming the plants.6
A biennial plant, celery completes its life cycle in 2 years. During the first season, the stems, roots, and leaves sprout; in the second and final year, the flowers, fruits, and seeds proliferate, followed by decomposition. Apium graveolens approaches heights of 2 to 3 ft, growing upright and displaying grooved stems. Each stem terminates in a basal rosette of leaves. The second season brings white flower blooms in terminal or axillary umbels.7
Celery originated in the temperate Mediterranean regions of Europe, but farmers now cultivate it globally.8 It grows best in rich moist soil with full exposure to sunlight. Plants multiply their numbers through self-seeding. Celery commonly is found in suburban and rural homes, both in refrigerators for consumption as well as in medicine cabinets in capsule form for the treatment of arthritis.4
Irritant and Allergenic Properties
Despite the potential health benefits of celery, the Apiaceae family, which includes hogweed, dill, and fennel, prevails as the most common culprit for phytotoxic reactions. The Rutaceae family, including citrus plants and rue, remains runner-up for causes of PPD.9 Phytophotodermatitis is not an immunologic reaction, making anyone susceptible to formation of the cutaneous lesions when exposed to UV light after handling celery. Pruritis rarely occurs, unlike in allergic phytodermatitis.10 Upon photoexcitation from exposure to UVA light, individual psoralen molecules covalently bind to pyrimidine bases, causing interstrand cross-linking that prevents DNA replication and triggering a cascade leading to apoptosis of the cell. Apoptosis induces cell membrane edema, which manifests as cutaneous vesicles and bullae on the skin.10 Regardless of plant species, PPD reactions have similar appearance.
Celery roots contain the greatest concentration of psoralens, making it the most likely part of the plant to induce PPD.6 Phytophotodermatitis caused by celery can occur at any time of the year, but most eruptions occur during the summer months due to increased sunlight exposure and intensity. Among 320 randomly selected Michigan celery harvesters, 163 (51%) displayed evidence of vesicular and bullous dermatitis on the fingers, hands, and forearms.11 In this study, celery infected with pink rot fungus induced an erythematous eruption with vesicles and bullae within 48 hours of contact after just 30 seconds of summer sunlight exposure; however, eruptions are not limited to summer months, as the cutaneous presentation depends solely on exposure to UVA light, which can occur year-round.
Use of tanning beds is a major risk factor for PPD.12 Tanning beds utilize fluorescent bulbs that primarily emit UVA light, with UVB light emitted to a lesser degree. The UVA radiation produced by tanning beds is more than 3 times as intense as natural sunlight.12 Among grocery store employees, the combination of these 2 risk factors—regular contact with celery and tanning bed use—resulted in a prevalence ratio for PPD more than 40 times greater than that of individuals with neither risk factor.13
Cutaneous Manifestations of PPD
Phytophotodermatitis is a nonimmunologic dermatitis that forms via the interaction between UV light exposure and the photosensitizing chemicals inherent to some plant species. Development of PPD following contact with celery may be caused by the photoactive substances in celery, including the psoralens 8-methoxypsoralen and 5-methoxypsoralen.14 The psoralens must become activated by UV light with wavelengths between 320 nm and 400 nm (UVA) to initiate biologic effects.15
Once chemically activated, the photoactive mediators cause an erythematous and edematous sunburnlike reaction. Current hypotheses state that psoralen plus UVA generates reactive oxygen species, which damage the DNA within cells and alter receptors on cell membranes within the epidermis.14 The cutaneous eruption usually appears between 12 and 36 hours after sun exposure. Although they generally are not pruritic, the eruptions may induce pain. Within 7 to 10 days following development of the rash, hyperpigmentation occurs in the affected area and often persists for months to years.16 Ingestion of large amounts of celery has been cited to cause generalized phototoxic reactions; however, PPD rarely arises solely after ingestion, unless excessive amounts are consumed with concomitant exposure to psoralen plus UVA or tanning beds.17 In these cases, patients develop diffuse redness with superficial scaling, pain, and blistering if severe.
Treatment of PPD
Prevention remains the best form of treatment for PPD caused by exposure to celery. Postcontact management includes washing the affected area with soap and water and changing clothes promptly. Topical corticosteroids have mild utility in treatment of PPD.18 Oral steroid tapers, which reduce acute inflammation, also are an option for treatment. Alternatively, intramuscular triamcinolone acetonide 1 mg/kg mixed with budesonide 0.1 mg/kg is an option and is associated with a reduced risk for adverse effects compared to oral steroids. The resulting hyperpigmentation develops 1 to 2 weeks postepithelialization.19 Hyperpigmentation often fades slowly over several months in lighter-skinned individuals but may last for years or indefinitely in darker-skinned patients.
Final Thoughts
Dermatologists should be knowledgeable about the various plant culprits that can induce PPD. Understanding the mechanism and pathophysiology can help guide both therapeutic interventions and preventive counseling. Understanding that even readily available vegetables such as celery can induce cutaneous eruptions should put PPD in the differential diagnosis more commonly when unspecified dermatitides are present.
- Walansky A. Study finally confirms eating celery burns more calories than it contains. Food & Wine. June 22, 2017. Accessed January 17, 2025. https://www.foodandwine.com/news/study-finally-confirms-eating-celery-burns-more-caloriesit-contains
- Puig L. Enhancement of PUVA phototoxic effects following celery ingestion: cool broth also can burn. Arch Dermatol. 1994;130:809-810. doi:10.1001/archderm.130.6.809
- Perez-Pimiento AJ, Moneo I, Santaolalla M, et al. Anaphylactic reaction to young garlic. Allergy. 1999;54:626-629.
- The Editors of Encyclopaedia Britannica. Apiaceae. Britannica. Updated November 25, 2024. Accessed January 17, 2025. https://www.britannica.com/plant/Apiaceae
- Smith R. Celery. In: Geoffriau E, Simon PW, eds. Carrots and Related Apiaceae Crops. 2nd ed. CABI; 2021:272-282.
- Dijkstra JWE, Chang L. Severe phototoxic burn following celery ingestion. Arch Dermatol. 1992;128:1277.
- Tobyn G, Denham A, Whitelegg M. Apium graveolens, wild celery. The Western Herbal Tradition: 2000 years of Medicinal Plant Knowledge. Elsevier. 2011:79-89. doi:10.1016/b978-0-443-10344-5.00014-8
- Rademaker M. Celery. DermNet. Accessed January 17, 2025. https://dermnetnz.org/topics/celery
- Sasseville D. Clinical patterns of phytophotodermatitis. Dermatol Clin. 2009;27:299-308.
- Jin Goon AT, Goh CL. Plant dermatitis: Asian perspective. Indian J Dermatol. 2011;56:707-710. doi:10.4103/0019-5154.91833
- Birmingham DJ, Key MM, Tublich GE. Phototoxic bullae among celery harvesters. Arch Dermatol. 1961;83:73-87.
- Robb-Nicholson C. By the way, doctor: is a tanning bed safer than sunlight? Harvard Health Publishing. Harvard Medical School. September 1, 2009. Accessed January 17, 2025. https://www.health.harvard.edu/staying-healthy/is-a-tanning-bed-saferthan-sunlight
- Vester L, Thyssen JP, Menne T, et al. Consequences of occupational food-related hand dermatoses with a focus on protein contact dermatitis. Contact Dermatitis. 2012;67:328-333.
- Ling TC, Clayton TH, Crawley J, et al. British Association of Dermatologists and British Photodermatology Group guidelines for the safe and effective use of psoralen-ultraviolet A therapy 2015. Br J Dermatol. 2016;174:24-55.
- Laskin JD. Cellular and molecular mechanisms in photochemical sensitization: studies on the mechanism of action of psoralens. Food Chem Toxicol. 1994;32:119-127. doi:10.1016/0278-6915(94)90172-4
- Elmets CA. Photosensitivity disorders (photodermatoses): clinical manifestations, diagnosis, and treatment. UpToDate. Updated February 23, 2023. Accessed January 17, 2025. https://www.uptodate.com/contents/photosensitivity-disorders-photodermatoses-clinical-manifestations-diagnosis-and-treatment
- Boffa, MJ, Gilmour E, Ead RD. Celery soup causing severe phototoxity during PUVA therapy. Br J Dermatol. 1996;135:334. doi:10.1111/j.1365-2133.1996.tb01182.x
- Sarhane KA, Ibrahim A, Fagan SP, et al. Phytophotodermatitis. Eplasty. 2013;13:ic57.
- McGovern TW. Dermatoses due to plants. In: Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. Mosby; 2018:286-303.
- Walansky A. Study finally confirms eating celery burns more calories than it contains. Food & Wine. June 22, 2017. Accessed January 17, 2025. https://www.foodandwine.com/news/study-finally-confirms-eating-celery-burns-more-caloriesit-contains
- Puig L. Enhancement of PUVA phototoxic effects following celery ingestion: cool broth also can burn. Arch Dermatol. 1994;130:809-810. doi:10.1001/archderm.130.6.809
- Perez-Pimiento AJ, Moneo I, Santaolalla M, et al. Anaphylactic reaction to young garlic. Allergy. 1999;54:626-629.
- The Editors of Encyclopaedia Britannica. Apiaceae. Britannica. Updated November 25, 2024. Accessed January 17, 2025. https://www.britannica.com/plant/Apiaceae
- Smith R. Celery. In: Geoffriau E, Simon PW, eds. Carrots and Related Apiaceae Crops. 2nd ed. CABI; 2021:272-282.
- Dijkstra JWE, Chang L. Severe phototoxic burn following celery ingestion. Arch Dermatol. 1992;128:1277.
- Tobyn G, Denham A, Whitelegg M. Apium graveolens, wild celery. The Western Herbal Tradition: 2000 years of Medicinal Plant Knowledge. Elsevier. 2011:79-89. doi:10.1016/b978-0-443-10344-5.00014-8
- Rademaker M. Celery. DermNet. Accessed January 17, 2025. https://dermnetnz.org/topics/celery
- Sasseville D. Clinical patterns of phytophotodermatitis. Dermatol Clin. 2009;27:299-308.
- Jin Goon AT, Goh CL. Plant dermatitis: Asian perspective. Indian J Dermatol. 2011;56:707-710. doi:10.4103/0019-5154.91833
- Birmingham DJ, Key MM, Tublich GE. Phototoxic bullae among celery harvesters. Arch Dermatol. 1961;83:73-87.
- Robb-Nicholson C. By the way, doctor: is a tanning bed safer than sunlight? Harvard Health Publishing. Harvard Medical School. September 1, 2009. Accessed January 17, 2025. https://www.health.harvard.edu/staying-healthy/is-a-tanning-bed-saferthan-sunlight
- Vester L, Thyssen JP, Menne T, et al. Consequences of occupational food-related hand dermatoses with a focus on protein contact dermatitis. Contact Dermatitis. 2012;67:328-333.
- Ling TC, Clayton TH, Crawley J, et al. British Association of Dermatologists and British Photodermatology Group guidelines for the safe and effective use of psoralen-ultraviolet A therapy 2015. Br J Dermatol. 2016;174:24-55.
- Laskin JD. Cellular and molecular mechanisms in photochemical sensitization: studies on the mechanism of action of psoralens. Food Chem Toxicol. 1994;32:119-127. doi:10.1016/0278-6915(94)90172-4
- Elmets CA. Photosensitivity disorders (photodermatoses): clinical manifestations, diagnosis, and treatment. UpToDate. Updated February 23, 2023. Accessed January 17, 2025. https://www.uptodate.com/contents/photosensitivity-disorders-photodermatoses-clinical-manifestations-diagnosis-and-treatment
- Boffa, MJ, Gilmour E, Ead RD. Celery soup causing severe phototoxity during PUVA therapy. Br J Dermatol. 1996;135:334. doi:10.1111/j.1365-2133.1996.tb01182.x
- Sarhane KA, Ibrahim A, Fagan SP, et al. Phytophotodermatitis. Eplasty. 2013;13:ic57.
- McGovern TW. Dermatoses due to plants. In: Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. Mosby; 2018:286-303.
Not as Bland as You May Think: Celery (Apium graveolens) Commonly Induces Phytophotodermatitis
Not as Bland as You May Think: Celery (Apium graveolens) Commonly Induces Phytophotodermatitis
PRACTICE POINTS
- Clinicians should consider phytophotodermatitis (PPD) in the differential diagnosis for erythematous eruptions with bullae and vesicles manifesting in sun-exposed distributions.
- A clinical history that includes the patient’s occupation, diet, and history of treatment with psoralen plus UVA and use of tanning beds may help diagnose PPD.
- It is important to educate patients who regularly handle celery and other plants containing furocoumarins and psoralens on how to prevent PPD and utilize effective photoprotection.
A Threat to Scientific Progress
The United States has long been recognized as a global leader in biomedical research and scientific discovery, with federal research and development (R&D) funding serving as the bedrock of national innovation. Substantial federal investment in biomedical research has stemmed from a recognition of its importance in fueling critical discoveries that improve patient care and the health of our communities.
In the United States, academic institutions play a key role in conducting research in the national interest and collaborating with industry, with most of the federal research funding distributed by the National Institutes of Health, National Science Foundation, and other agencies awarded to university-based academic investigators. In a 2014 report, the National Academies of Sciences, Engineering and Medicine identified three pillars of a highly productive research system: a talented and interconnected workforce, adequate and dependable resources, and world-class basic research in all major areas of science.

A series of recent, short-sighted federal policy decisions threaten the future of scientific discovery by eroding these pillars. Decisions to freeze previously awarded federal grant funding, delay grant review panels, fire federal scientists, and propose crippling cuts to indirect cost rates (among others) have sent shock waves through the research community and already have led some prominent research institutions to cut staff and divert resources away from groundbreaking research. While the acute effects of these changes are just beginning to be felt, it is the long-term effects of these decisions on future medical and scientific discovery that will be most devastating to society.
In our April issue, we highlight important research advancements in inflammatory bowel disease presented at February’s Congress of the European Crohn’s and Colitis Organisation (ECCO) in Berlin. In this month’s Member Spotlight, Abigail Meyers, MPAS, PA-C, outlines her impactful work as a member of AGA’s newly formed Nurse Practitioner and Physician Assistant Task Force and shares how her personal journey as a patient with inflammatory bowel disease allows her to be a more powerful advocate for important issues impacting other patients with this condition.
Megan A. Adams, MD, JD, MSc
Editor in Chief
The United States has long been recognized as a global leader in biomedical research and scientific discovery, with federal research and development (R&D) funding serving as the bedrock of national innovation. Substantial federal investment in biomedical research has stemmed from a recognition of its importance in fueling critical discoveries that improve patient care and the health of our communities.
In the United States, academic institutions play a key role in conducting research in the national interest and collaborating with industry, with most of the federal research funding distributed by the National Institutes of Health, National Science Foundation, and other agencies awarded to university-based academic investigators. In a 2014 report, the National Academies of Sciences, Engineering and Medicine identified three pillars of a highly productive research system: a talented and interconnected workforce, adequate and dependable resources, and world-class basic research in all major areas of science.
A series of recent, short-sighted federal policy decisions threaten the future of scientific discovery by eroding these pillars. Decisions to freeze previously awarded federal grant funding, delay grant review panels, fire federal scientists, and propose crippling cuts to indirect cost rates (among others) have sent shock waves through the research community and already have led some prominent research institutions to cut staff and divert resources away from groundbreaking research. While the acute effects of these changes are just beginning to be felt, it is the long-term effects of these decisions on future medical and scientific discovery that will be most devastating to society.
In our April issue, we highlight important research advancements in inflammatory bowel disease presented at February’s Congress of the European Crohn’s and Colitis Organisation (ECCO) in Berlin. In this month’s Member Spotlight, Abigail Meyers, MPAS, PA-C, outlines her impactful work as a member of AGA’s newly formed Nurse Practitioner and Physician Assistant Task Force and shares how her personal journey as a patient with inflammatory bowel disease allows her to be a more powerful advocate for important issues impacting other patients with this condition.
Megan A. Adams, MD, JD, MSc
Editor in Chief
The United States has long been recognized as a global leader in biomedical research and scientific discovery, with federal research and development (R&D) funding serving as the bedrock of national innovation. Substantial federal investment in biomedical research has stemmed from a recognition of its importance in fueling critical discoveries that improve patient care and the health of our communities.
In the United States, academic institutions play a key role in conducting research in the national interest and collaborating with industry, with most of the federal research funding distributed by the National Institutes of Health, National Science Foundation, and other agencies awarded to university-based academic investigators. In a 2014 report, the National Academies of Sciences, Engineering and Medicine identified three pillars of a highly productive research system: a talented and interconnected workforce, adequate and dependable resources, and world-class basic research in all major areas of science.
A series of recent, short-sighted federal policy decisions threaten the future of scientific discovery by eroding these pillars. Decisions to freeze previously awarded federal grant funding, delay grant review panels, fire federal scientists, and propose crippling cuts to indirect cost rates (among others) have sent shock waves through the research community and already have led some prominent research institutions to cut staff and divert resources away from groundbreaking research. While the acute effects of these changes are just beginning to be felt, it is the long-term effects of these decisions on future medical and scientific discovery that will be most devastating to society.
In our April issue, we highlight important research advancements in inflammatory bowel disease presented at February’s Congress of the European Crohn’s and Colitis Organisation (ECCO) in Berlin. In this month’s Member Spotlight, Abigail Meyers, MPAS, PA-C, outlines her impactful work as a member of AGA’s newly formed Nurse Practitioner and Physician Assistant Task Force and shares how her personal journey as a patient with inflammatory bowel disease allows her to be a more powerful advocate for important issues impacting other patients with this condition.
Megan A. Adams, MD, JD, MSc
Editor in Chief
A Painful Flesh-Colored Papule on the Shoulder
A Painful Flesh-Colored Papule on the Shoulder
The Diagnosis: Leiomyoma
Histopathology revealed a dermal mesenchymal tumor composed of fascicles of bland spindle cells with tapered nuclei, perinuclear vacuoles, eosinophilic cytoplasm, and low cellularity (Figure 1). Immunohistochemical studies of the cells stained strongly positive for smooth muscle actin and desmin, consistent with a smooth muscle neoplasm (Figure 2). Fumarate hydratase (FH) staining revealed loss of expression in tumor cells, consistent with FH deficiency (Figure 3). A diagnosis of cutaneous leiomyoma was made, and although the clinical and histologic findings suggested hereditary leiomyomatosis and renal cell cancer (HLRCC), genetic testing was negative for an FH gene mutation. This negative result indicated that HLRCC was unlikely despite the initial concerns based on the findings.
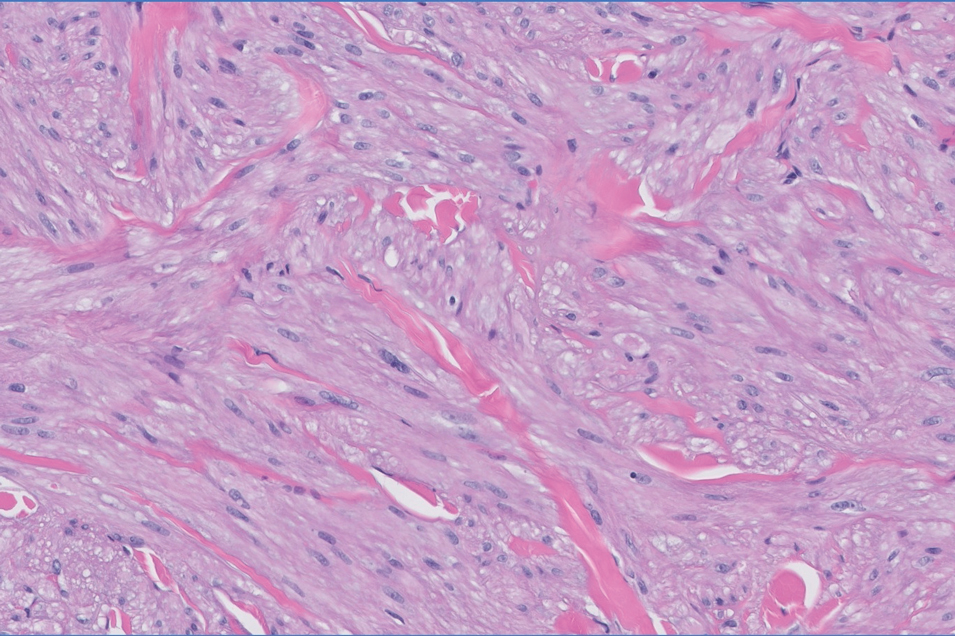
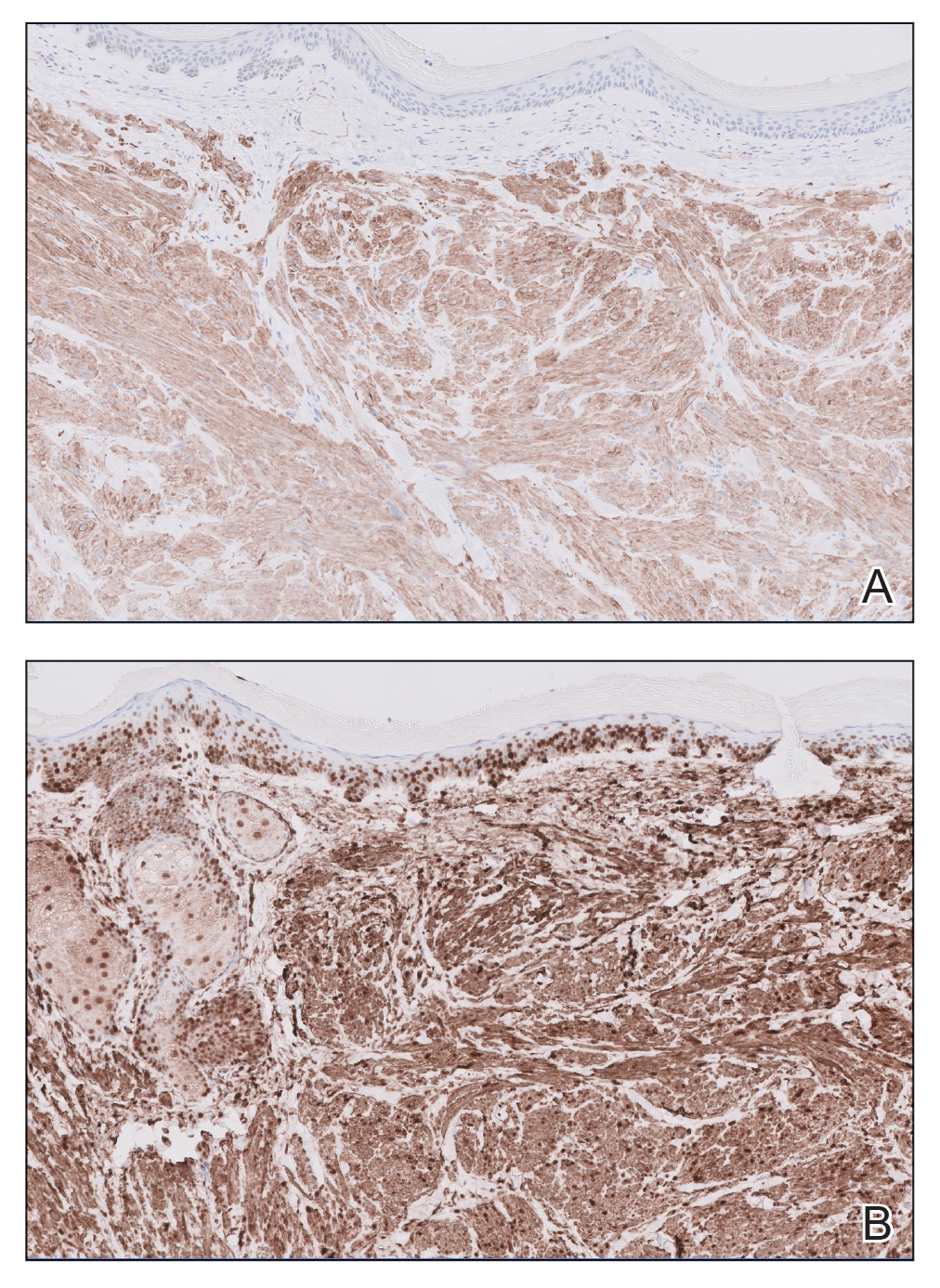

Leiomyomas are benign neoplasms that are challenging to diagnose based on the clinical picture alone. Leiomyomas most commonly are found in the genitourinary and gastrointestinal systems, with cutaneous manifestation being the second most common presentation.1 These benign smooth muscle tumors manifest as tender, firm, flesh-colored, pink or reddish-brown nodules that are subcategorized based on the derivation of the smooth muscle within the tumor.2 Angioleiomyomas, the most common type, arise from the tunica media of blood vessels, whereas piloleiomyomas and genital leiomyomas arise from the arrector pili musculature of the hair follicle and the smooth muscle found in the scrotum, labia, or nipple.2 Rare cases of cutaneous leiomyosarcomas and angioleiomyosarcomas have been reported in the literature.3,4 Solitary leiomyomas tend to develop on the lower extremities, whereas multiple lesions frequently manifest on the extensor surfaces of extremities and the trunk. Lesions often are painful, either spontaneously or in association with applied pressure, emotional stress, or exposure to cold temperatures.2
Although leiomyomas themselves are benign, patients with multiple cutaneous leiomyomas may have an underlying genetic mutation that increases their risk of developing HLRCC, an autosomal-dominant syndrome.5 Referral should be considered for individuals with a personal history of or a first-degree relative with cutaneous leiomyomas or renal cell carcinoma (RCC) with histology typical of hereditary leiomyomatosis and RCC, as recommended by the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors.6 In this case, the decision to refer the patient for genetic testing was based on her family history, specifically her paternal uncle having multiple similar lesions, which, while not a first-degree relative, still raised concerns about potential hereditary risks and warranted further evaluation. A germline mutation in the FH gene, which encodes an enzyme that converts fumarate to malate in the Krebs cycle and plays a role in tumor suppression, is the cause of HLRCC.2,7 When part of this genetic condition, cutaneous leiomyomas tend to occur around 25 years of age (range, 10-50 years).2 A diagnosis of HLRCC should be strongly considered if a patient displays multiple cutaneous leiomyomas with at least 1 histologically confirmed lesion or at least 2 of the following: solitary cutaneous leiomyoma with family history of HLRCC, onset of severely symptomatic uterine fibroids before age 40 years, type II papillary or collecting duct renal cell cancer before age 40 years, or a first-degree family member who meets 1 of these criteria.5,8
Diagnosis of cutaneous leiomyoma may be accomplished by microscopic examination of a tissue sample; however, further diagnostic workup is warranted due to the strong correlation with HLRCC.2 A definitive diagnosis of HLRCC is confirmed with a germline mutation in the FH gene, and genetic screening should be offered to patients before renal cancer surveillance to avoid unwarranted investigations.8 Timely clinical diagnosis enables early genetic testing and enhanced outcomes for patients with confirmed HLRCC who may need a multidisciplinary approach of dermatologists, gynecologists, and urologic oncologists.5,8
Cutaneous leiomyomas can be excised, and this typically is the gold standard of care for small and localized lesions, although the use of cryosurgery and carbon dioxide lasers has been reported as well.2,9,10 For more widespread lesions or for patients who are not appropriate candidates for surgery, pharmacologic therapies (α-blockers, calcium channel blockers, nitroglycerin), intralesional corticosteroids, and/or botulinum toxin injections can be utilized.2,11
The acronym BLEND AN EGG encompasses the clinical differential diagnosis for painful skin tumors: blue rubber bleb nevus, leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomangioma, and granular cell tumor. Blue rubber bleb nevi are deep blue in color, and angiolipomas sit under the skin and present as subcutaneous swellings. Dermatofibromas and neurofibromas also are included in the differential.12 Dermatofibromas are firm solitary lesions that have a pathognomonic pinch sign. Neurofibromas are soft and rubbery, have a buttonhole sign, and stain positively for S-100 protein and SOX-10 but negatively for actin and desmin.12
- Malhotra P, Walia H, Singh A, et al. Leiomyoma cutis: a clinicopathological series of 37 cases. Indian J Dermatol. 2010;55:337-341.
- Bernett CN, Mammino JJ. Cutaneous leiomyomas. In: StatPearls. StatPearls Publishing; 2023.
- Chayed Z, Kristensen LK, Ousager LB, et al. Hereditary leiomyomatosis and renal cell carcinoma: a case series and literature review. Orphanet J Rare Dis. 2021;16:34. doi:10.1186/s13023-020-01653-9
- Perkins J, Scarbrough C, Sammons D, et al. Reed syndrome: an atypical presentation of a rare disease. Dermatol Online J. 2014;21: 13030/qt5k35r5pn.
- Schmidt LS, Linehan WM. Hereditary leiomyomatosis and renal cell carcinoma. Int J Nephrol Renovasc Dis. 2014;7:253-260. doi:10.2147 /IJNRD.S42097
- Hampel H, Bennett RL, Buchanan A, et al. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med. 2015;17:70-87. doi:10.1038/gim.2014.147
- Alam NA, Barclay E, Rowan AJ, et al. Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed tumor syndrome. Arch Dermatol. 2005;141:199-206. doi:10.1001 /archderm.141.2.199
- Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637-644. doi:10.1007/s10689-014-9735-2
- Uyar B, Acar EM, Subas¸ıog˘lu A. Treatment of three hereditary leiomyomatosis patients with cryotherapy. Dermatol Ther. 2020;33:e13226. doi:10.1111/dth.13226
- Christenson LJ, Smith K, Arpey CJ. Treatment of multiple cutaneous leiomyomas with CO2 laser ablation. Dermatol Surg. 2000;26:319-322. doi:10.1046/j.1524-4725.2000.99250.x
- Onder M, Adis¸en E. A new indication of botulinum toxin: leiomyoma- related pain. J Am Acad Dermatol. 2009;60:325-328. doi:10.1016 /j.jaad.2008.05.044
- Clarey DD, Lauer SR, Adams JL. Painful papules on the arms. Cutis. 2020;106:232-249. doi:10.12788/cutis.0109
The Diagnosis: Leiomyoma
Histopathology revealed a dermal mesenchymal tumor composed of fascicles of bland spindle cells with tapered nuclei, perinuclear vacuoles, eosinophilic cytoplasm, and low cellularity (Figure 1). Immunohistochemical studies of the cells stained strongly positive for smooth muscle actin and desmin, consistent with a smooth muscle neoplasm (Figure 2). Fumarate hydratase (FH) staining revealed loss of expression in tumor cells, consistent with FH deficiency (Figure 3). A diagnosis of cutaneous leiomyoma was made, and although the clinical and histologic findings suggested hereditary leiomyomatosis and renal cell cancer (HLRCC), genetic testing was negative for an FH gene mutation. This negative result indicated that HLRCC was unlikely despite the initial concerns based on the findings.
Leiomyomas are benign neoplasms that are challenging to diagnose based on the clinical picture alone. Leiomyomas most commonly are found in the genitourinary and gastrointestinal systems, with cutaneous manifestation being the second most common presentation.1 These benign smooth muscle tumors manifest as tender, firm, flesh-colored, pink or reddish-brown nodules that are subcategorized based on the derivation of the smooth muscle within the tumor.2 Angioleiomyomas, the most common type, arise from the tunica media of blood vessels, whereas piloleiomyomas and genital leiomyomas arise from the arrector pili musculature of the hair follicle and the smooth muscle found in the scrotum, labia, or nipple.2 Rare cases of cutaneous leiomyosarcomas and angioleiomyosarcomas have been reported in the literature.3,4 Solitary leiomyomas tend to develop on the lower extremities, whereas multiple lesions frequently manifest on the extensor surfaces of extremities and the trunk. Lesions often are painful, either spontaneously or in association with applied pressure, emotional stress, or exposure to cold temperatures.2
Although leiomyomas themselves are benign, patients with multiple cutaneous leiomyomas may have an underlying genetic mutation that increases their risk of developing HLRCC, an autosomal-dominant syndrome.5 Referral should be considered for individuals with a personal history of or a first-degree relative with cutaneous leiomyomas or renal cell carcinoma (RCC) with histology typical of hereditary leiomyomatosis and RCC, as recommended by the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors.6 In this case, the decision to refer the patient for genetic testing was based on her family history, specifically her paternal uncle having multiple similar lesions, which, while not a first-degree relative, still raised concerns about potential hereditary risks and warranted further evaluation. A germline mutation in the FH gene, which encodes an enzyme that converts fumarate to malate in the Krebs cycle and plays a role in tumor suppression, is the cause of HLRCC.2,7 When part of this genetic condition, cutaneous leiomyomas tend to occur around 25 years of age (range, 10-50 years).2 A diagnosis of HLRCC should be strongly considered if a patient displays multiple cutaneous leiomyomas with at least 1 histologically confirmed lesion or at least 2 of the following: solitary cutaneous leiomyoma with family history of HLRCC, onset of severely symptomatic uterine fibroids before age 40 years, type II papillary or collecting duct renal cell cancer before age 40 years, or a first-degree family member who meets 1 of these criteria.5,8
Diagnosis of cutaneous leiomyoma may be accomplished by microscopic examination of a tissue sample; however, further diagnostic workup is warranted due to the strong correlation with HLRCC.2 A definitive diagnosis of HLRCC is confirmed with a germline mutation in the FH gene, and genetic screening should be offered to patients before renal cancer surveillance to avoid unwarranted investigations.8 Timely clinical diagnosis enables early genetic testing and enhanced outcomes for patients with confirmed HLRCC who may need a multidisciplinary approach of dermatologists, gynecologists, and urologic oncologists.5,8
Cutaneous leiomyomas can be excised, and this typically is the gold standard of care for small and localized lesions, although the use of cryosurgery and carbon dioxide lasers has been reported as well.2,9,10 For more widespread lesions or for patients who are not appropriate candidates for surgery, pharmacologic therapies (α-blockers, calcium channel blockers, nitroglycerin), intralesional corticosteroids, and/or botulinum toxin injections can be utilized.2,11
The acronym BLEND AN EGG encompasses the clinical differential diagnosis for painful skin tumors: blue rubber bleb nevus, leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomangioma, and granular cell tumor. Blue rubber bleb nevi are deep blue in color, and angiolipomas sit under the skin and present as subcutaneous swellings. Dermatofibromas and neurofibromas also are included in the differential.12 Dermatofibromas are firm solitary lesions that have a pathognomonic pinch sign. Neurofibromas are soft and rubbery, have a buttonhole sign, and stain positively for S-100 protein and SOX-10 but negatively for actin and desmin.12
The Diagnosis: Leiomyoma
Histopathology revealed a dermal mesenchymal tumor composed of fascicles of bland spindle cells with tapered nuclei, perinuclear vacuoles, eosinophilic cytoplasm, and low cellularity (Figure 1). Immunohistochemical studies of the cells stained strongly positive for smooth muscle actin and desmin, consistent with a smooth muscle neoplasm (Figure 2). Fumarate hydratase (FH) staining revealed loss of expression in tumor cells, consistent with FH deficiency (Figure 3). A diagnosis of cutaneous leiomyoma was made, and although the clinical and histologic findings suggested hereditary leiomyomatosis and renal cell cancer (HLRCC), genetic testing was negative for an FH gene mutation. This negative result indicated that HLRCC was unlikely despite the initial concerns based on the findings.
Leiomyomas are benign neoplasms that are challenging to diagnose based on the clinical picture alone. Leiomyomas most commonly are found in the genitourinary and gastrointestinal systems, with cutaneous manifestation being the second most common presentation.1 These benign smooth muscle tumors manifest as tender, firm, flesh-colored, pink or reddish-brown nodules that are subcategorized based on the derivation of the smooth muscle within the tumor.2 Angioleiomyomas, the most common type, arise from the tunica media of blood vessels, whereas piloleiomyomas and genital leiomyomas arise from the arrector pili musculature of the hair follicle and the smooth muscle found in the scrotum, labia, or nipple.2 Rare cases of cutaneous leiomyosarcomas and angioleiomyosarcomas have been reported in the literature.3,4 Solitary leiomyomas tend to develop on the lower extremities, whereas multiple lesions frequently manifest on the extensor surfaces of extremities and the trunk. Lesions often are painful, either spontaneously or in association with applied pressure, emotional stress, or exposure to cold temperatures.2
Although leiomyomas themselves are benign, patients with multiple cutaneous leiomyomas may have an underlying genetic mutation that increases their risk of developing HLRCC, an autosomal-dominant syndrome.5 Referral should be considered for individuals with a personal history of or a first-degree relative with cutaneous leiomyomas or renal cell carcinoma (RCC) with histology typical of hereditary leiomyomatosis and RCC, as recommended by the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors.6 In this case, the decision to refer the patient for genetic testing was based on her family history, specifically her paternal uncle having multiple similar lesions, which, while not a first-degree relative, still raised concerns about potential hereditary risks and warranted further evaluation. A germline mutation in the FH gene, which encodes an enzyme that converts fumarate to malate in the Krebs cycle and plays a role in tumor suppression, is the cause of HLRCC.2,7 When part of this genetic condition, cutaneous leiomyomas tend to occur around 25 years of age (range, 10-50 years).2 A diagnosis of HLRCC should be strongly considered if a patient displays multiple cutaneous leiomyomas with at least 1 histologically confirmed lesion or at least 2 of the following: solitary cutaneous leiomyoma with family history of HLRCC, onset of severely symptomatic uterine fibroids before age 40 years, type II papillary or collecting duct renal cell cancer before age 40 years, or a first-degree family member who meets 1 of these criteria.5,8
Diagnosis of cutaneous leiomyoma may be accomplished by microscopic examination of a tissue sample; however, further diagnostic workup is warranted due to the strong correlation with HLRCC.2 A definitive diagnosis of HLRCC is confirmed with a germline mutation in the FH gene, and genetic screening should be offered to patients before renal cancer surveillance to avoid unwarranted investigations.8 Timely clinical diagnosis enables early genetic testing and enhanced outcomes for patients with confirmed HLRCC who may need a multidisciplinary approach of dermatologists, gynecologists, and urologic oncologists.5,8
Cutaneous leiomyomas can be excised, and this typically is the gold standard of care for small and localized lesions, although the use of cryosurgery and carbon dioxide lasers has been reported as well.2,9,10 For more widespread lesions or for patients who are not appropriate candidates for surgery, pharmacologic therapies (α-blockers, calcium channel blockers, nitroglycerin), intralesional corticosteroids, and/or botulinum toxin injections can be utilized.2,11
The acronym BLEND AN EGG encompasses the clinical differential diagnosis for painful skin tumors: blue rubber bleb nevus, leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomangioma, and granular cell tumor. Blue rubber bleb nevi are deep blue in color, and angiolipomas sit under the skin and present as subcutaneous swellings. Dermatofibromas and neurofibromas also are included in the differential.12 Dermatofibromas are firm solitary lesions that have a pathognomonic pinch sign. Neurofibromas are soft and rubbery, have a buttonhole sign, and stain positively for S-100 protein and SOX-10 but negatively for actin and desmin.12
- Malhotra P, Walia H, Singh A, et al. Leiomyoma cutis: a clinicopathological series of 37 cases. Indian J Dermatol. 2010;55:337-341.
- Bernett CN, Mammino JJ. Cutaneous leiomyomas. In: StatPearls. StatPearls Publishing; 2023.
- Chayed Z, Kristensen LK, Ousager LB, et al. Hereditary leiomyomatosis and renal cell carcinoma: a case series and literature review. Orphanet J Rare Dis. 2021;16:34. doi:10.1186/s13023-020-01653-9
- Perkins J, Scarbrough C, Sammons D, et al. Reed syndrome: an atypical presentation of a rare disease. Dermatol Online J. 2014;21: 13030/qt5k35r5pn.
- Schmidt LS, Linehan WM. Hereditary leiomyomatosis and renal cell carcinoma. Int J Nephrol Renovasc Dis. 2014;7:253-260. doi:10.2147 /IJNRD.S42097
- Hampel H, Bennett RL, Buchanan A, et al. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med. 2015;17:70-87. doi:10.1038/gim.2014.147
- Alam NA, Barclay E, Rowan AJ, et al. Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed tumor syndrome. Arch Dermatol. 2005;141:199-206. doi:10.1001 /archderm.141.2.199
- Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637-644. doi:10.1007/s10689-014-9735-2
- Uyar B, Acar EM, Subas¸ıog˘lu A. Treatment of three hereditary leiomyomatosis patients with cryotherapy. Dermatol Ther. 2020;33:e13226. doi:10.1111/dth.13226
- Christenson LJ, Smith K, Arpey CJ. Treatment of multiple cutaneous leiomyomas with CO2 laser ablation. Dermatol Surg. 2000;26:319-322. doi:10.1046/j.1524-4725.2000.99250.x
- Onder M, Adis¸en E. A new indication of botulinum toxin: leiomyoma- related pain. J Am Acad Dermatol. 2009;60:325-328. doi:10.1016 /j.jaad.2008.05.044
- Clarey DD, Lauer SR, Adams JL. Painful papules on the arms. Cutis. 2020;106:232-249. doi:10.12788/cutis.0109
- Malhotra P, Walia H, Singh A, et al. Leiomyoma cutis: a clinicopathological series of 37 cases. Indian J Dermatol. 2010;55:337-341.
- Bernett CN, Mammino JJ. Cutaneous leiomyomas. In: StatPearls. StatPearls Publishing; 2023.
- Chayed Z, Kristensen LK, Ousager LB, et al. Hereditary leiomyomatosis and renal cell carcinoma: a case series and literature review. Orphanet J Rare Dis. 2021;16:34. doi:10.1186/s13023-020-01653-9
- Perkins J, Scarbrough C, Sammons D, et al. Reed syndrome: an atypical presentation of a rare disease. Dermatol Online J. 2014;21: 13030/qt5k35r5pn.
- Schmidt LS, Linehan WM. Hereditary leiomyomatosis and renal cell carcinoma. Int J Nephrol Renovasc Dis. 2014;7:253-260. doi:10.2147 /IJNRD.S42097
- Hampel H, Bennett RL, Buchanan A, et al. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med. 2015;17:70-87. doi:10.1038/gim.2014.147
- Alam NA, Barclay E, Rowan AJ, et al. Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed tumor syndrome. Arch Dermatol. 2005;141:199-206. doi:10.1001 /archderm.141.2.199
- Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637-644. doi:10.1007/s10689-014-9735-2
- Uyar B, Acar EM, Subas¸ıog˘lu A. Treatment of three hereditary leiomyomatosis patients with cryotherapy. Dermatol Ther. 2020;33:e13226. doi:10.1111/dth.13226
- Christenson LJ, Smith K, Arpey CJ. Treatment of multiple cutaneous leiomyomas with CO2 laser ablation. Dermatol Surg. 2000;26:319-322. doi:10.1046/j.1524-4725.2000.99250.x
- Onder M, Adis¸en E. A new indication of botulinum toxin: leiomyoma- related pain. J Am Acad Dermatol. 2009;60:325-328. doi:10.1016 /j.jaad.2008.05.044
- Clarey DD, Lauer SR, Adams JL. Painful papules on the arms. Cutis. 2020;106:232-249. doi:10.12788/cutis.0109
A Painful Flesh-Colored Papule on the Shoulder
A Painful Flesh-Colored Papule on the Shoulder
A 65-year-old woman with a history of metabolic syndrome presented to the family medicine clinic for evaluation of a papule on the right shoulder that had started small and increased in size over the past 3 years. Physical examination revealed a 1.0×0.8×0.1-cm, smooth, flesh-colored to light brown papule on the right shoulder that was notably tender to palpation. The patient reported that her paternal uncle had multiple skin lesions of similar morphology dispersed on the bilateral upper extremities. A shave biopsy of the lesion was performed.
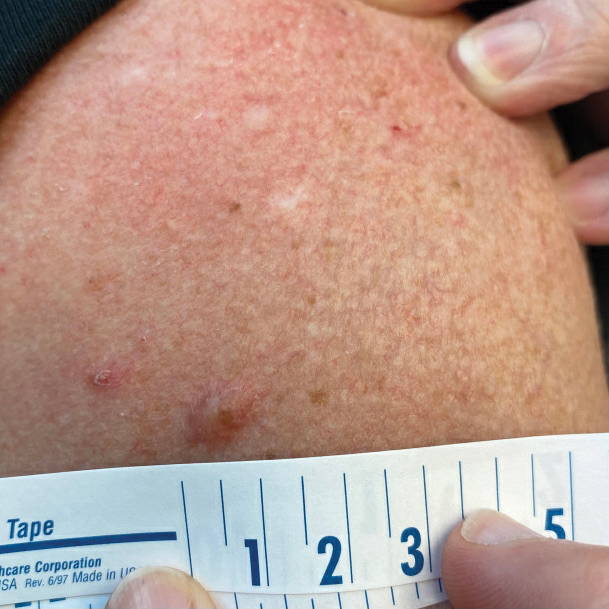
Erythematous Annular Scaly Plaques on the Upper Chest
Erythematous Annular Scaly Plaques on the Upper Chest
THE DIAGNOSIS: Tinea Corporis
Due to the scaly and acute nature of the rash, a potassium hydroxide (KOH) preparation was performed, and hyphal elements were floridly present. After further questioning, the patient reported finding a stray kitten a few weeks before the onset of the eruption and shared a picture of it lying on her chest in the area corresponding with the main distribution of the rash (Figure). Based on the patient’s personal history and the positive KOH preparation, a diagnosis of tinea corporis was made. She was immediately started on fluconazole 300 mg once weekly for 4 weeks and naftifine gel 1%, which she used for 6 to 8 weeks with complete resolution of the eruption.

Tinea corporis is a dermatophyte infection that typically affects exposed areas of the skin such as the chest, arms, and legs. Spread via human-to-human contact, Trichophyton rubrum is the most common cause worldwide. The second most common is Trichophyton mentagrophytes, which is spread through animal-to-human contact.1,2
Symptoms of tinea corporis usually appear 1 to 3 weeks after exposure and manifest as itchy scaly papules that spread outward, forming annular, circinate, and petaloid erythematous plaques with central clearing. The condition most commonly is diagnosed through the examination of scale from the affected area using a KOH preparation, which will reveal hyphae when positive.2-4 Cultures are the gold standard for identifying dermatophyte species,5 but results can take several weeks. Biopsy also can confirm the diagnosis by showing the presence of hyphae in the stratum corneum, which can be highlighted using periodic acid–Schiff or silver stains.3
Topical antifungals are the first-line treatment for cutaneous dermatophyte infections.3-5 The most effective topical therapies are allylamines and azoles, which work by inhibiting the growth of the fungus. Allylamines are more effective than azoles due to their fungicidal properties and ability to penetrate the skin more effectively.6,7 Topical medications should be applied at least 2 cm beyond the infected area for 2 to 4 weeks or until the infection has cleared.3 Systemic antifungals may be necessary in more complicated cases.
It is important to consider a broad differential and take into consideration the distribution of the plaques, the patient’s history, and other clinical features when differentiating tinea corporis from other conditions. Erythema annulare centrifugum more often presents as nonpruritic annular plaques with a trailing scale instead of a leading scale seen in tinea corporis. Biopsy exhibits a dense, perivascular, lymphocytic infiltrate in superficial vessels, resembling a coat sleeve.3,8 Pemphigus foliaceous can manifest with painful crusted scaly plaques and vesicles in a seborrheic distribution. Biopsy reveals subcorneal acantholytic vesicles and can be confirmed on direct immunofluorescence.3,8 Subacute cutaneous lupus erythematosus presents with annular plaques that often are symmetric and most prominent in sun-exposed areas, sparing the face.3,9,10 It can be associated with other autoimmune conditions as well as medications such as thiazides, terbinafine, and calcium channel blockers. Additionally, 76% to 90% of patients are Ro/SSA antibody positive.3 Biopsy often demonstrates follicular plugging, perivascular and periadnexal lymphocytic infiltrates, and mucin.3,10 Lastly, pityriasis rosea typically begins with a herald patch, followed by a widespread rash that often appears in a Christmas tree distribution.3
- Havlickova B, Czaika VA, Friedrich M. Epidemiological trends in skin mycoses worldwide. Mycoses. 2008;51 (suppl 4):2-15. doi: 10.1111 /j.1439-0507.2008.01606.x
- Yee G, Al Aboud AM. Tinea corporis. 2022 Aug 8. In: StatPearls [Internet]. StatPearls Publishing; 2023
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Elsevier; 2018.
- Diseases resulting from fungal and yeast. In: James WD, Berger TG, Elston DM, et al, eds. Andrews’ Diseases of The Skin: Clinical Dermatology. 12th ed. Elsevier; 2016: 289-290.
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9:2020-5-6 . doi:10.7573/dic.2020-5-6
- El-Gohary M, van Zuuren EJ, Fedorowicz Z, et al. Topical antifungal treatments for tinea cruris and tinea corporis. Cochrane Database Syst Rev. 2014;2014:CD009992. doi:10.1002/14651858 .CD009992.pub2
- Wolverton SE. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2018.
- Burgdorf W. Erythema annulare centrifugum and other figurate erythemas. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. McGraw-Hill; 2008: 366-368.
- Modi GM, Maender JL, Coleman N, et al. Tinea corporis masquerading as subacute cutaneous lupus erythematosus. Dermatol Online J. 2008;14:8.
- Stavropoulos PG, Goules AV, Avgerinou G, et al. Pathogenesis of subacute cutaneous lupus erythematosus. J Eur Acad Dermatol Venereol. 2008;22:1281.
THE DIAGNOSIS: Tinea Corporis
Due to the scaly and acute nature of the rash, a potassium hydroxide (KOH) preparation was performed, and hyphal elements were floridly present. After further questioning, the patient reported finding a stray kitten a few weeks before the onset of the eruption and shared a picture of it lying on her chest in the area corresponding with the main distribution of the rash (Figure). Based on the patient’s personal history and the positive KOH preparation, a diagnosis of tinea corporis was made. She was immediately started on fluconazole 300 mg once weekly for 4 weeks and naftifine gel 1%, which she used for 6 to 8 weeks with complete resolution of the eruption.
Tinea corporis is a dermatophyte infection that typically affects exposed areas of the skin such as the chest, arms, and legs. Spread via human-to-human contact, Trichophyton rubrum is the most common cause worldwide. The second most common is Trichophyton mentagrophytes, which is spread through animal-to-human contact.1,2
Symptoms of tinea corporis usually appear 1 to 3 weeks after exposure and manifest as itchy scaly papules that spread outward, forming annular, circinate, and petaloid erythematous plaques with central clearing. The condition most commonly is diagnosed through the examination of scale from the affected area using a KOH preparation, which will reveal hyphae when positive.2-4 Cultures are the gold standard for identifying dermatophyte species,5 but results can take several weeks. Biopsy also can confirm the diagnosis by showing the presence of hyphae in the stratum corneum, which can be highlighted using periodic acid–Schiff or silver stains.3
Topical antifungals are the first-line treatment for cutaneous dermatophyte infections.3-5 The most effective topical therapies are allylamines and azoles, which work by inhibiting the growth of the fungus. Allylamines are more effective than azoles due to their fungicidal properties and ability to penetrate the skin more effectively.6,7 Topical medications should be applied at least 2 cm beyond the infected area for 2 to 4 weeks or until the infection has cleared.3 Systemic antifungals may be necessary in more complicated cases.
It is important to consider a broad differential and take into consideration the distribution of the plaques, the patient’s history, and other clinical features when differentiating tinea corporis from other conditions. Erythema annulare centrifugum more often presents as nonpruritic annular plaques with a trailing scale instead of a leading scale seen in tinea corporis. Biopsy exhibits a dense, perivascular, lymphocytic infiltrate in superficial vessels, resembling a coat sleeve.3,8 Pemphigus foliaceous can manifest with painful crusted scaly plaques and vesicles in a seborrheic distribution. Biopsy reveals subcorneal acantholytic vesicles and can be confirmed on direct immunofluorescence.3,8 Subacute cutaneous lupus erythematosus presents with annular plaques that often are symmetric and most prominent in sun-exposed areas, sparing the face.3,9,10 It can be associated with other autoimmune conditions as well as medications such as thiazides, terbinafine, and calcium channel blockers. Additionally, 76% to 90% of patients are Ro/SSA antibody positive.3 Biopsy often demonstrates follicular plugging, perivascular and periadnexal lymphocytic infiltrates, and mucin.3,10 Lastly, pityriasis rosea typically begins with a herald patch, followed by a widespread rash that often appears in a Christmas tree distribution.3
THE DIAGNOSIS: Tinea Corporis
Due to the scaly and acute nature of the rash, a potassium hydroxide (KOH) preparation was performed, and hyphal elements were floridly present. After further questioning, the patient reported finding a stray kitten a few weeks before the onset of the eruption and shared a picture of it lying on her chest in the area corresponding with the main distribution of the rash (Figure). Based on the patient’s personal history and the positive KOH preparation, a diagnosis of tinea corporis was made. She was immediately started on fluconazole 300 mg once weekly for 4 weeks and naftifine gel 1%, which she used for 6 to 8 weeks with complete resolution of the eruption.
Tinea corporis is a dermatophyte infection that typically affects exposed areas of the skin such as the chest, arms, and legs. Spread via human-to-human contact, Trichophyton rubrum is the most common cause worldwide. The second most common is Trichophyton mentagrophytes, which is spread through animal-to-human contact.1,2
Symptoms of tinea corporis usually appear 1 to 3 weeks after exposure and manifest as itchy scaly papules that spread outward, forming annular, circinate, and petaloid erythematous plaques with central clearing. The condition most commonly is diagnosed through the examination of scale from the affected area using a KOH preparation, which will reveal hyphae when positive.2-4 Cultures are the gold standard for identifying dermatophyte species,5 but results can take several weeks. Biopsy also can confirm the diagnosis by showing the presence of hyphae in the stratum corneum, which can be highlighted using periodic acid–Schiff or silver stains.3
Topical antifungals are the first-line treatment for cutaneous dermatophyte infections.3-5 The most effective topical therapies are allylamines and azoles, which work by inhibiting the growth of the fungus. Allylamines are more effective than azoles due to their fungicidal properties and ability to penetrate the skin more effectively.6,7 Topical medications should be applied at least 2 cm beyond the infected area for 2 to 4 weeks or until the infection has cleared.3 Systemic antifungals may be necessary in more complicated cases.
It is important to consider a broad differential and take into consideration the distribution of the plaques, the patient’s history, and other clinical features when differentiating tinea corporis from other conditions. Erythema annulare centrifugum more often presents as nonpruritic annular plaques with a trailing scale instead of a leading scale seen in tinea corporis. Biopsy exhibits a dense, perivascular, lymphocytic infiltrate in superficial vessels, resembling a coat sleeve.3,8 Pemphigus foliaceous can manifest with painful crusted scaly plaques and vesicles in a seborrheic distribution. Biopsy reveals subcorneal acantholytic vesicles and can be confirmed on direct immunofluorescence.3,8 Subacute cutaneous lupus erythematosus presents with annular plaques that often are symmetric and most prominent in sun-exposed areas, sparing the face.3,9,10 It can be associated with other autoimmune conditions as well as medications such as thiazides, terbinafine, and calcium channel blockers. Additionally, 76% to 90% of patients are Ro/SSA antibody positive.3 Biopsy often demonstrates follicular plugging, perivascular and periadnexal lymphocytic infiltrates, and mucin.3,10 Lastly, pityriasis rosea typically begins with a herald patch, followed by a widespread rash that often appears in a Christmas tree distribution.3
- Havlickova B, Czaika VA, Friedrich M. Epidemiological trends in skin mycoses worldwide. Mycoses. 2008;51 (suppl 4):2-15. doi: 10.1111 /j.1439-0507.2008.01606.x
- Yee G, Al Aboud AM. Tinea corporis. 2022 Aug 8. In: StatPearls [Internet]. StatPearls Publishing; 2023
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Elsevier; 2018.
- Diseases resulting from fungal and yeast. In: James WD, Berger TG, Elston DM, et al, eds. Andrews’ Diseases of The Skin: Clinical Dermatology. 12th ed. Elsevier; 2016: 289-290.
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9:2020-5-6 . doi:10.7573/dic.2020-5-6
- El-Gohary M, van Zuuren EJ, Fedorowicz Z, et al. Topical antifungal treatments for tinea cruris and tinea corporis. Cochrane Database Syst Rev. 2014;2014:CD009992. doi:10.1002/14651858 .CD009992.pub2
- Wolverton SE. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2018.
- Burgdorf W. Erythema annulare centrifugum and other figurate erythemas. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. McGraw-Hill; 2008: 366-368.
- Modi GM, Maender JL, Coleman N, et al. Tinea corporis masquerading as subacute cutaneous lupus erythematosus. Dermatol Online J. 2008;14:8.
- Stavropoulos PG, Goules AV, Avgerinou G, et al. Pathogenesis of subacute cutaneous lupus erythematosus. J Eur Acad Dermatol Venereol. 2008;22:1281.
- Havlickova B, Czaika VA, Friedrich M. Epidemiological trends in skin mycoses worldwide. Mycoses. 2008;51 (suppl 4):2-15. doi: 10.1111 /j.1439-0507.2008.01606.x
- Yee G, Al Aboud AM. Tinea corporis. 2022 Aug 8. In: StatPearls [Internet]. StatPearls Publishing; 2023
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Elsevier; 2018.
- Diseases resulting from fungal and yeast. In: James WD, Berger TG, Elston DM, et al, eds. Andrews’ Diseases of The Skin: Clinical Dermatology. 12th ed. Elsevier; 2016: 289-290.
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9:2020-5-6 . doi:10.7573/dic.2020-5-6
- El-Gohary M, van Zuuren EJ, Fedorowicz Z, et al. Topical antifungal treatments for tinea cruris and tinea corporis. Cochrane Database Syst Rev. 2014;2014:CD009992. doi:10.1002/14651858 .CD009992.pub2
- Wolverton SE. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2018.
- Burgdorf W. Erythema annulare centrifugum and other figurate erythemas. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. McGraw-Hill; 2008: 366-368.
- Modi GM, Maender JL, Coleman N, et al. Tinea corporis masquerading as subacute cutaneous lupus erythematosus. Dermatol Online J. 2008;14:8.
- Stavropoulos PG, Goules AV, Avgerinou G, et al. Pathogenesis of subacute cutaneous lupus erythematosus. J Eur Acad Dermatol Venereol. 2008;22:1281.
Erythematous Annular Scaly Plaques on the Upper Chest
Erythematous Annular Scaly Plaques on the Upper Chest
A 60-year-old woman with a history of keratinocyte carcinomas, hypertension, diabetes mellitus, and anxiety presented to the dermatology department with a widespread rash of more than 2 weeks’ duration. The patient had tried 1 to 2 days of self-treatment with triamcinolone cream that she had previously been prescribed for an unknown dermatitis and zinc oxide cream, which caused considerable inflammation of the rash and prompted her to discontinue use. She could not recall any recent use of new personal care products or medications or eating any new foods. She also denied any recent yard work, known arthropod bites, illnesses, prolonged sun exposure, or constitutional symptoms. Her medications included metformin, hydrochlorothiazide, losartan, and sertraline. She also reported taking daily supplements of vitamins D, K, and C as well as acetaminophen and ibuprofen as needed. Physical examination revealed several 2- to 4-cm, erythematous, annular, circinate, petaloid plaques with scale mostly on photodistributed areas of the central anterior chest, neck, lower cheeks, and chin as well as a few scattered lesions with similar morphology on the arms, lower abdomen, left buttock, and back.
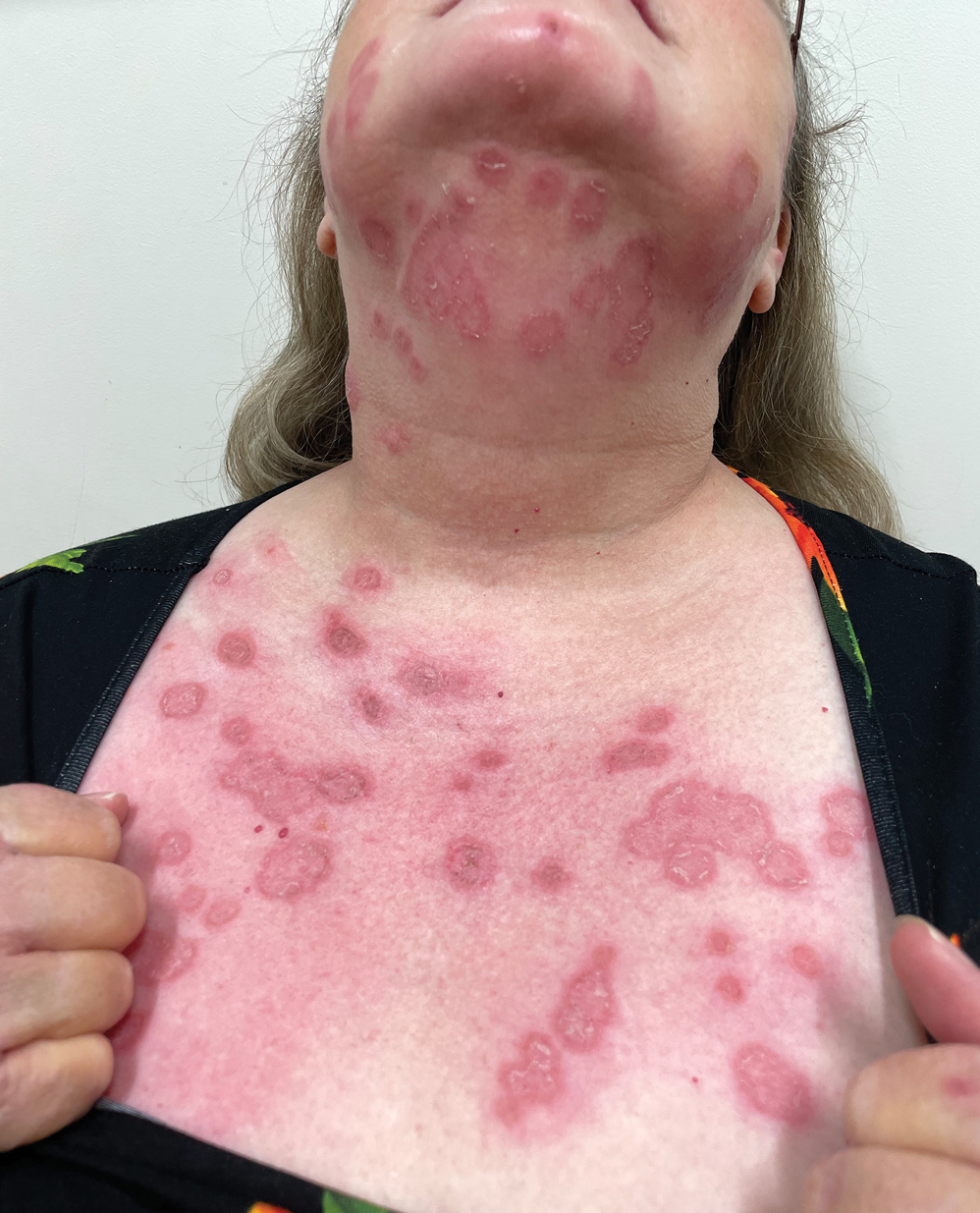

A Candida Glabrata-Associated Prosthetic Joint Infection: Case Report and Literature Review
A Candida Glabrata-Associated Prosthetic Joint Infection: Case Report and Literature Review
Prosthetic joint infection (PJI) occurs in about 1% to 2% of joint replacements. 1 Risk factors include immunosuppression, diabetes, chronic illnesses, and prolonged operative time.2 Bacterial infections constitute most of these infections, while fungal pathogens account for about 1%. Candida (C.) species, predominantly C. albicans, are responsible for most PJIs.1,3 In contrast, C. glabrata is a rare cause of fungal PJI, with only 18 PJI cases currently reported in the literature.4 C. glabrata PJI occurs more frequently among immunosuppressed patients and is associated with a higher treatment failure rate despite antifungal therapy.5 Treatment of fungal PJI is often complicated, involving multiple surgical debridements, prolonged antifungal therapy, and in some cases, prosthesis removal.6 However, given the rarity of C. glabrata as a PJI pathogen, no standardized treatment guidelines exist, leading to potential delays in diagnosis and tailored treatment.7,8
CASE PRESENTATION
A male Vietnam veteran aged 75 years presented to the emergency department in July 2023 with a fluid collection over his left hip surgical incision site. The patient had a complex medical history that included chronic kidney disease, well-controlled type 2 diabetes, hypertension, and osteoarthritis. His history was further complicated by nonalcoholic steatohepatitis with hepatocellular carcinoma that was treated with transarterial radioembolization and yttrium-90. The patient had undergone a left total hip arthroplasty in 1996 and subsequent open reduction and internal fixation about 9 months prior to his presentation. The patient reported the fluid had been present for about 6 weeks, while he received outpatient monitoring by the orthopedic surgery service. He sought emergency care after noting a moderate amount of purulent discharge on his clothing originating from his hip. In the week prior to admission, the patient observed progressive erythema, warmth, and tenderness over the incision site. Despite these symptoms, the patient remained ambulatory and able to walk long distances with the use of an assistive device.
Upon presentation, the patient was afebrile and normotensive. Laboratory testing revealed an elevated erythrocyte sedimentation rate of 77 mm/h (reference range, 0-20 mm/h) and a C-reactive protein of 9.8 mg/L (reference range, 0-2.5 mg/L), suggesting an underlying infectious process. A physical examination revealed a well-healed incision over the left hip with a poorly defined area of fluctuance and evidence of wound dehiscence. The left lower extremity was swollen with 2+ pitting edema, but tenderness was localized to the incision site. Magnetic resonance imaging of the left hip revealed a multiloculated fluid collection abutting the left greater trochanter with extension to the skin surface and inferior extension along the entire length of the surgical fixation hardware (Figure).
Upon admission, orthopedic surgery performed a bedside aspiration of the fluid collection. Samples were sent for analysis, including cell count and bacterial and fungal cultures. Initial blood cultures were sterile. Due to concerns for a bacterial infection, the patient was started on empiric intravenous (IV) ceftriaxone 2 g/day and IV vancomycin 1250 mg/day. Synovial fluid analysis revealed an elevated white blood cell count of 45,000/ìL, but bacterial cultures were negative. Five days after admission, the fungal culture from the left hip wound was notable for presence of C. glabrata, prompting an infectious diseases (ID) consultation. IV micafungin 100 mg/day was initiated as empiric antifungal therapy.
ID and orthopedic surgery teams determined that a combined medical and surgical approach would be best suited for infection control. They proposed 2 main approaches: complete hardware replacement with washout, which carried a higher morbidity risk but a better chance of infection resolution, or partial hardware replacement with washout, which was associated with a lower morbidity risk but a higher risk of infection persistence and recurrence. This decision was particularly challenging for the patient, who prioritized maintaining his functional status, including his ability to continue dancing for pleasure. The patient opted for a more conservative approach, electing to proceed with antifungal therapy and debridement while retaining the prosthetic joint.
After 11 days of hospitalization, the patient was discharged with a peripherally inserted central catheter for long-term antifungal infusions of micafungin 150 mg/day at home. Fungal sensitivity test results several days after discharge confirmed susceptibility to micafungin.
About 2 weeks after discharge, the patient underwent debridement and implant retention (DAIR). Wound cultures were positive for C. glabrata, Enterococcus faecalis, Staphylococcus epidermidis, and Corynebacterium tuberculostearicum. Based on susceptibilities, he completed a 2-month course of IV micafungin 150 mg daily and daptomycin 750 mg daily, followed by an oral suppressive regimen consisting of doxycycline 100 mg twice daily, amoxicillin-clavulanate 2 g twice daily, and fluconazole initially 800 mg daily adjusted to 400 mg daily. The patient continued wound management with twice-daily dressing changes.
Nine months after DAIR, the patient remained on suppressive antifungal and antibacterial therapy. He continued to experience serous drainage from the wound, which greatly affected his quality of life. After discussion with his family and the orthopedic surgery team, he agreed to proceed with a 2-staged revision arthroplasty involving prosthetic explant and antibiotic spacer placement. However, the surgery was postponed due to findings of anemia (hemoglobin, 8.9 g/dL) and thrombocytopenia (platelet count, 73 x 103/λL). At the time of this report, the patient was being monitored closely with his multidisciplinary care team for the planned orthopedic procedure.
DISCUSSION
PJI is the most common cause of primary hip arthroplasty failure; however, fungal species only make up about 1% of PJIs.3,9-11 Patients are typically immunocompromised, undergoing antineoplastic therapies for malignancy, or have other comorbid conditions such as diabetes.12,13 C. glabrata presents a unique diagnostic and therapeutic challenge as it is not only rare but also notorious for its resistance to common antifungal agents. C. glabrata is known to develop multidrug resistance through the rapid accumulation of genomic mutations.14 Its propensity towards forming protective biofilm also arms it with intrinsic resistance to agents like fluconazole.15 Furthermore, based on a review of the available reports in the literature, C. glabrata PJIs are often insidious and present with symptoms closely mimicking those of bacterial PJIs, as it did in the patient in this case.16
Synovial fluid analysis, fungal cultures, and sensitivity testing are paramount for ensuring proper diagnosis for fungal PJI. The patient in this case was empirically treated with micafungin based on recommendations from the ID team. When the sensitivities results were reviewed, the same antifungal therapy was continued. Echinocandins have a favorable toxicity profile in long-term use, as well as efficacy against biofilm-producing organisms like C. glabrata.17,18
While there are a few cases citing DAIR as a feasible surgical strategy for treating fungal PJI, more recent studies have reported greater success with a 2-staged revision arthroplasty involving some combination of debridement, placement of antibiotic-loaded bone cement spacers, and partial or total exchange of the infected prosthetic joint.4,19-23 In this case, complete hardware replacement would have offered the patient the most favorable outlook for eliminating this fungal infection. However, given the patient’s advanced age, significant underlying comorbidities, and functional status, medical management with antifungal therapy and DAIR was favored.
Based on the discussion from the 6-month follow-up visit, the patient was experiencing progressive and persistent wound drainage and frequent dressing changes, highlighting the limitations of medical management for PJI in the setting of retained prosthesis. If the patient ultimately proceeds with a more invasive surgical intervention, another important consideration will be the likelihood of fungal PJI recurrence. At present, fungal PJI recurrence rates following antifungal and surgical treatment have been reported to range between 0% to 50%, which is too imprecise to be considered clinically useful.22-24
Given the ambiguity surrounding management guidelines and limited treatment options, it is crucial to emphasize the timeline of this patient’s clinical presentation and subsequent course of treatment. Upon presentation to the ED in late July, fungal PJI was considered less likely. Initial blood cultures from presentation were negative, which is common with PJIs. It was not until 5 days later that the left hip wound culture showed moderate growth of C. glabrata. Identifying a PJI is clinically challenging due to the lack of standardized diagnostic criteria. However, timely identification and diagnosis of fungal PJI with appropriate antifungal therapy, in patients with limited curative options due to comorbidities, can significantly improve quality of life and overall outcomes.25 Routine fungal and mycobacterial cultures are not currently recommended in PJI guidelines, but this case illustrates it is imperative in immunocompromised hosts.26
This case and the current paucity of similar cases in the literature stress the importance of clinicians publishing their experience in the management of fungal PJI. We strongly recommend that clinicians approach each suspected PJI with careful consideration of the patient’s unique risk factors, comorbidities, and goals of care, when deciding on a curative vs suppressive approach to therapy.
CONCLUSIONS
This case report highlights the importance of considering fungal pathogens for PJIs, especially in high-risk patients, the value of obtaining fungal cultures, the necessity of a multidisciplinary approach, the role of antifungal susceptibility testing, and consideration for the feasibility of a surgical intervention. It underscores the challenges in diagnosis and treatment of C. glabrata-associated PJI, emphasizing the importance of clinician experience-sharing in developing evidence-based management strategies. As the understanding of fungal PJI evolves, continued research and clinical data collection remain crucial for improving patient outcomes in the management of these complex cases.
- Osmon DR, Berbari EF, Berendt AR, et al. Executive summary: diagnosis and management of prosthetic joint infection: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2013;56(1):1-10. doi:10.1093/cid/cis966
- Eka A, Chen AF. Patient-related medical risk factors for periprosthetic joint infection of the hip and knee. Ann Transl Med. 2015;3(16):233. doi:10.3978/j.issn.2305-5839.2015.09.26
- Darouiche RO, Hamill RJ, Musher DM, Young EJ, Harris RL. Periprosthetic candidal infections following arthroplasty. Rev Infect Dis. 1989;11(1):89-96. doi:10.1093/clinids/11.1.89
- Koutserimpas C, Zervakis SG, Maraki S, et al. Non-albicans Candida prosthetic joint infections: a systematic review of treatment. World J Clin Cases. 2019;7(12):1430- 1443. doi:10.12998/wjcc.v7.i12.1430
- Fidel PL Jr, Vazquez JA, Sobel JD. Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin Microbiol Rev. 1999;12(1):80-96. doi:10.1128/CMR.12.1.80
- Aboltins C, Daffy J, Choong P, Stanley P. Current concepts in the management of prosthetic joint infection. Intern Med J. 2014;44(9):834-840. doi:10.1111/imj.12510
- Lee YR, Kim HJ, Lee EJ, Sohn JW, Kim MJ, Yoon YK. Prosthetic joint infections caused by candida species: a systematic review and a case series. Mycopathologia. 2019;184(1):23-33. doi:10.1007/s11046-018-0286-1
- Herndon CL, Rowe TM, Metcalf RW, et al. Treatment outcomes of fungal periprosthetic joint infection. J Arthroplasty. 2023;38(11):2436-2440.e1. doi:10.1016/j.arth.2023.05.009
- Delaunay C, Hamadouche M, Girard J, Duhamel A; SoFCOT. What are the causes for failures of primary hip arthroplasties in France? Clin Orthop Relat Res. 2013;471(12): 3863-3869. doi:10.1007/s11999-013-2935-5
- Bozic KJ, Kurtz SM, Lau E, Ong K, Vail TP, Berry DJ. The epidemiology of revision total hip arthroplasty in the United States. J Bone Joint Surg Am. 2009;91(1): 128-133. doi:10.2106/JBJS.H.00155
- Furnes O, Lie SA, Espehaug B, Vollset SE, Engesaeter LB, Havelin LI. Hip disease and the prognosis of total hip replacements. A review of 53,698 primary total hip replacements reported to the Norwegian Arthroplasty Register 1987-99. J Bone Joint Surg Br. 2001;83(4):579-586. doi:10.1302/0301-620x.83b4.11223
- Gonzalez MR, Bedi ADS, Karczewski D, Lozano-Calderon SA. Treatment and outcomes of fungal prosthetic joint infections: a systematic review of 225 cases. J Arthroplasty. 2023;38(11):2464-2471.e1. doi:10.1016/j.arth.2023.05.003
- Gonzalez MR, Pretell-Mazzini J, Lozano-Calderon SA. Risk factors and management of prosthetic joint infections in megaprostheses-a review of the literature. Antibiotics (Basel). 2023;13(1):25. doi:10.3390/antibiotics13010025
- Biswas C, Chen SC, Halliday C, et al. Identification of genetic markers of resistance to echinocandins, azoles and 5-fluorocytosine in Candida glabrata by next-generation sequencing: a feasibility study. Clin Microbiol Infect. 2017;23(9):676.e7-676.e10. doi:10.1016/j.cmi.2017.03.014
- Hassan Y, Chew SY, Than LTL. Candida glabrata: pathogenicity and resistance mechanisms for adaptation and survival. J Fungi (Basel). 2021;7(8):667. doi:10.3390/jof7080667
- Aboltins C, Daffy J, Choong P, Stanley P. Current concepts in the management of prosthetic joint infection. Intern Med J. 2014;44(9):834-840. doi:10.1111/imj.12510
- Pierce CG, Uppuluri P, Tristan AR, et al. A simple and reproducible 96-well plate-based method for the formation of fungal biofilms and its application to antifungal susceptibility testing. Nat Protoc. 2008;3(9):1494-1500. doi:10.1038/nport.2008.141
- Koutserimpas C, Samonis G, Velivassakis E, Iliopoulou- Kosmadaki S, Kontakis G, Kofteridis DP. Candida glabrata prosthetic joint infection, successfully treated with anidulafungin: a case report and review of the literature. Mycoses. 2018;61(4):266-269. doi:10.1111/myc.12736
- Brooks DH, Pupparo F. Successful salvage of a primary total knee arthroplasty infected with Candida parapsilosis. J Arthroplasty. 1998;13(6):707-712. doi:10.1016/s0883-5403(98)80017-x
- Merrer J, Dupont B, Nieszkowska A, De Jonghe B, Outin H. Candida albicans prosthetic arthritis treated with fluconazole alone. J Infect. 2001;42(3):208-209. doi:10.1053/jinf.2001.0819
- Koutserimpas C, Naoum S, Alpantaki K, et al. Fungal prosthetic joint infection in revised knee arthroplasty: an orthopaedic surgeon’s nightmare. Diagnostics (Basel). 2022;12(7):1606. doi:10.3390/diagnostics12071606
- Gao Z, Li X, Du Y, Peng Y, Wu W, Zhou Y. Success rate of fungal peri-prosthetic joint infection treated by 2-stage revision and potential risk factors of treatment failure: a retrospective study. Med Sci Monit. 2018;24:5549-5557. doi:10.12659/MSM.909168
- Hwang BH, Yoon JY, Nam CH, et al. Fungal periprosthetic joint infection after primary total knee replacement. J Bone Joint Surg Br. 2012;94(5):656-659. doi:10.1302/0301-620X.94B5.28125
- Ueng SW, Lee CY, Hu CC, Hsieh PH, Chang Y. What is the success of treatment of hip and knee candidal periprosthetic joint infection? Clin Orthop Relat Res. 2013;471(9):3002-3009. doi:10.1007/s11999-013-3007-6
- Nodzo, Scott R. MD; Bauer, Thomas MD, PhD; Pottinger, et al. Conventional diagnostic challenges in periprosthetic joint infection. J Am Acad Orthop Surg. 2015;23 Suppl:S18-S25. doi:10.5435/JAAOS-D-14-00385
- American Academy of Orthopaedic Surgeons. Diagnosis and prevention of periprosthetic joint infections. March 11, 2019. Accessed February 5, 2025. https://www.aaos.org/pjicpg
Prosthetic joint infection (PJI) occurs in about 1% to 2% of joint replacements. 1 Risk factors include immunosuppression, diabetes, chronic illnesses, and prolonged operative time.2 Bacterial infections constitute most of these infections, while fungal pathogens account for about 1%. Candida (C.) species, predominantly C. albicans, are responsible for most PJIs.1,3 In contrast, C. glabrata is a rare cause of fungal PJI, with only 18 PJI cases currently reported in the literature.4 C. glabrata PJI occurs more frequently among immunosuppressed patients and is associated with a higher treatment failure rate despite antifungal therapy.5 Treatment of fungal PJI is often complicated, involving multiple surgical debridements, prolonged antifungal therapy, and in some cases, prosthesis removal.6 However, given the rarity of C. glabrata as a PJI pathogen, no standardized treatment guidelines exist, leading to potential delays in diagnosis and tailored treatment.7,8
CASE PRESENTATION
A male Vietnam veteran aged 75 years presented to the emergency department in July 2023 with a fluid collection over his left hip surgical incision site. The patient had a complex medical history that included chronic kidney disease, well-controlled type 2 diabetes, hypertension, and osteoarthritis. His history was further complicated by nonalcoholic steatohepatitis with hepatocellular carcinoma that was treated with transarterial radioembolization and yttrium-90. The patient had undergone a left total hip arthroplasty in 1996 and subsequent open reduction and internal fixation about 9 months prior to his presentation. The patient reported the fluid had been present for about 6 weeks, while he received outpatient monitoring by the orthopedic surgery service. He sought emergency care after noting a moderate amount of purulent discharge on his clothing originating from his hip. In the week prior to admission, the patient observed progressive erythema, warmth, and tenderness over the incision site. Despite these symptoms, the patient remained ambulatory and able to walk long distances with the use of an assistive device.
Upon presentation, the patient was afebrile and normotensive. Laboratory testing revealed an elevated erythrocyte sedimentation rate of 77 mm/h (reference range, 0-20 mm/h) and a C-reactive protein of 9.8 mg/L (reference range, 0-2.5 mg/L), suggesting an underlying infectious process. A physical examination revealed a well-healed incision over the left hip with a poorly defined area of fluctuance and evidence of wound dehiscence. The left lower extremity was swollen with 2+ pitting edema, but tenderness was localized to the incision site. Magnetic resonance imaging of the left hip revealed a multiloculated fluid collection abutting the left greater trochanter with extension to the skin surface and inferior extension along the entire length of the surgical fixation hardware (Figure).
Upon admission, orthopedic surgery performed a bedside aspiration of the fluid collection. Samples were sent for analysis, including cell count and bacterial and fungal cultures. Initial blood cultures were sterile. Due to concerns for a bacterial infection, the patient was started on empiric intravenous (IV) ceftriaxone 2 g/day and IV vancomycin 1250 mg/day. Synovial fluid analysis revealed an elevated white blood cell count of 45,000/ìL, but bacterial cultures were negative. Five days after admission, the fungal culture from the left hip wound was notable for presence of C. glabrata, prompting an infectious diseases (ID) consultation. IV micafungin 100 mg/day was initiated as empiric antifungal therapy.
ID and orthopedic surgery teams determined that a combined medical and surgical approach would be best suited for infection control. They proposed 2 main approaches: complete hardware replacement with washout, which carried a higher morbidity risk but a better chance of infection resolution, or partial hardware replacement with washout, which was associated with a lower morbidity risk but a higher risk of infection persistence and recurrence. This decision was particularly challenging for the patient, who prioritized maintaining his functional status, including his ability to continue dancing for pleasure. The patient opted for a more conservative approach, electing to proceed with antifungal therapy and debridement while retaining the prosthetic joint.
After 11 days of hospitalization, the patient was discharged with a peripherally inserted central catheter for long-term antifungal infusions of micafungin 150 mg/day at home. Fungal sensitivity test results several days after discharge confirmed susceptibility to micafungin.
About 2 weeks after discharge, the patient underwent debridement and implant retention (DAIR). Wound cultures were positive for C. glabrata, Enterococcus faecalis, Staphylococcus epidermidis, and Corynebacterium tuberculostearicum. Based on susceptibilities, he completed a 2-month course of IV micafungin 150 mg daily and daptomycin 750 mg daily, followed by an oral suppressive regimen consisting of doxycycline 100 mg twice daily, amoxicillin-clavulanate 2 g twice daily, and fluconazole initially 800 mg daily adjusted to 400 mg daily. The patient continued wound management with twice-daily dressing changes.
Nine months after DAIR, the patient remained on suppressive antifungal and antibacterial therapy. He continued to experience serous drainage from the wound, which greatly affected his quality of life. After discussion with his family and the orthopedic surgery team, he agreed to proceed with a 2-staged revision arthroplasty involving prosthetic explant and antibiotic spacer placement. However, the surgery was postponed due to findings of anemia (hemoglobin, 8.9 g/dL) and thrombocytopenia (platelet count, 73 x 103/λL). At the time of this report, the patient was being monitored closely with his multidisciplinary care team for the planned orthopedic procedure.
DISCUSSION
PJI is the most common cause of primary hip arthroplasty failure; however, fungal species only make up about 1% of PJIs.3,9-11 Patients are typically immunocompromised, undergoing antineoplastic therapies for malignancy, or have other comorbid conditions such as diabetes.12,13 C. glabrata presents a unique diagnostic and therapeutic challenge as it is not only rare but also notorious for its resistance to common antifungal agents. C. glabrata is known to develop multidrug resistance through the rapid accumulation of genomic mutations.14 Its propensity towards forming protective biofilm also arms it with intrinsic resistance to agents like fluconazole.15 Furthermore, based on a review of the available reports in the literature, C. glabrata PJIs are often insidious and present with symptoms closely mimicking those of bacterial PJIs, as it did in the patient in this case.16
Synovial fluid analysis, fungal cultures, and sensitivity testing are paramount for ensuring proper diagnosis for fungal PJI. The patient in this case was empirically treated with micafungin based on recommendations from the ID team. When the sensitivities results were reviewed, the same antifungal therapy was continued. Echinocandins have a favorable toxicity profile in long-term use, as well as efficacy against biofilm-producing organisms like C. glabrata.17,18
While there are a few cases citing DAIR as a feasible surgical strategy for treating fungal PJI, more recent studies have reported greater success with a 2-staged revision arthroplasty involving some combination of debridement, placement of antibiotic-loaded bone cement spacers, and partial or total exchange of the infected prosthetic joint.4,19-23 In this case, complete hardware replacement would have offered the patient the most favorable outlook for eliminating this fungal infection. However, given the patient’s advanced age, significant underlying comorbidities, and functional status, medical management with antifungal therapy and DAIR was favored.
Based on the discussion from the 6-month follow-up visit, the patient was experiencing progressive and persistent wound drainage and frequent dressing changes, highlighting the limitations of medical management for PJI in the setting of retained prosthesis. If the patient ultimately proceeds with a more invasive surgical intervention, another important consideration will be the likelihood of fungal PJI recurrence. At present, fungal PJI recurrence rates following antifungal and surgical treatment have been reported to range between 0% to 50%, which is too imprecise to be considered clinically useful.22-24
Given the ambiguity surrounding management guidelines and limited treatment options, it is crucial to emphasize the timeline of this patient’s clinical presentation and subsequent course of treatment. Upon presentation to the ED in late July, fungal PJI was considered less likely. Initial blood cultures from presentation were negative, which is common with PJIs. It was not until 5 days later that the left hip wound culture showed moderate growth of C. glabrata. Identifying a PJI is clinically challenging due to the lack of standardized diagnostic criteria. However, timely identification and diagnosis of fungal PJI with appropriate antifungal therapy, in patients with limited curative options due to comorbidities, can significantly improve quality of life and overall outcomes.25 Routine fungal and mycobacterial cultures are not currently recommended in PJI guidelines, but this case illustrates it is imperative in immunocompromised hosts.26
This case and the current paucity of similar cases in the literature stress the importance of clinicians publishing their experience in the management of fungal PJI. We strongly recommend that clinicians approach each suspected PJI with careful consideration of the patient’s unique risk factors, comorbidities, and goals of care, when deciding on a curative vs suppressive approach to therapy.
CONCLUSIONS
This case report highlights the importance of considering fungal pathogens for PJIs, especially in high-risk patients, the value of obtaining fungal cultures, the necessity of a multidisciplinary approach, the role of antifungal susceptibility testing, and consideration for the feasibility of a surgical intervention. It underscores the challenges in diagnosis and treatment of C. glabrata-associated PJI, emphasizing the importance of clinician experience-sharing in developing evidence-based management strategies. As the understanding of fungal PJI evolves, continued research and clinical data collection remain crucial for improving patient outcomes in the management of these complex cases.
Prosthetic joint infection (PJI) occurs in about 1% to 2% of joint replacements. 1 Risk factors include immunosuppression, diabetes, chronic illnesses, and prolonged operative time.2 Bacterial infections constitute most of these infections, while fungal pathogens account for about 1%. Candida (C.) species, predominantly C. albicans, are responsible for most PJIs.1,3 In contrast, C. glabrata is a rare cause of fungal PJI, with only 18 PJI cases currently reported in the literature.4 C. glabrata PJI occurs more frequently among immunosuppressed patients and is associated with a higher treatment failure rate despite antifungal therapy.5 Treatment of fungal PJI is often complicated, involving multiple surgical debridements, prolonged antifungal therapy, and in some cases, prosthesis removal.6 However, given the rarity of C. glabrata as a PJI pathogen, no standardized treatment guidelines exist, leading to potential delays in diagnosis and tailored treatment.7,8
CASE PRESENTATION
A male Vietnam veteran aged 75 years presented to the emergency department in July 2023 with a fluid collection over his left hip surgical incision site. The patient had a complex medical history that included chronic kidney disease, well-controlled type 2 diabetes, hypertension, and osteoarthritis. His history was further complicated by nonalcoholic steatohepatitis with hepatocellular carcinoma that was treated with transarterial radioembolization and yttrium-90. The patient had undergone a left total hip arthroplasty in 1996 and subsequent open reduction and internal fixation about 9 months prior to his presentation. The patient reported the fluid had been present for about 6 weeks, while he received outpatient monitoring by the orthopedic surgery service. He sought emergency care after noting a moderate amount of purulent discharge on his clothing originating from his hip. In the week prior to admission, the patient observed progressive erythema, warmth, and tenderness over the incision site. Despite these symptoms, the patient remained ambulatory and able to walk long distances with the use of an assistive device.
Upon presentation, the patient was afebrile and normotensive. Laboratory testing revealed an elevated erythrocyte sedimentation rate of 77 mm/h (reference range, 0-20 mm/h) and a C-reactive protein of 9.8 mg/L (reference range, 0-2.5 mg/L), suggesting an underlying infectious process. A physical examination revealed a well-healed incision over the left hip with a poorly defined area of fluctuance and evidence of wound dehiscence. The left lower extremity was swollen with 2+ pitting edema, but tenderness was localized to the incision site. Magnetic resonance imaging of the left hip revealed a multiloculated fluid collection abutting the left greater trochanter with extension to the skin surface and inferior extension along the entire length of the surgical fixation hardware (Figure).
Upon admission, orthopedic surgery performed a bedside aspiration of the fluid collection. Samples were sent for analysis, including cell count and bacterial and fungal cultures. Initial blood cultures were sterile. Due to concerns for a bacterial infection, the patient was started on empiric intravenous (IV) ceftriaxone 2 g/day and IV vancomycin 1250 mg/day. Synovial fluid analysis revealed an elevated white blood cell count of 45,000/ìL, but bacterial cultures were negative. Five days after admission, the fungal culture from the left hip wound was notable for presence of C. glabrata, prompting an infectious diseases (ID) consultation. IV micafungin 100 mg/day was initiated as empiric antifungal therapy.
ID and orthopedic surgery teams determined that a combined medical and surgical approach would be best suited for infection control. They proposed 2 main approaches: complete hardware replacement with washout, which carried a higher morbidity risk but a better chance of infection resolution, or partial hardware replacement with washout, which was associated with a lower morbidity risk but a higher risk of infection persistence and recurrence. This decision was particularly challenging for the patient, who prioritized maintaining his functional status, including his ability to continue dancing for pleasure. The patient opted for a more conservative approach, electing to proceed with antifungal therapy and debridement while retaining the prosthetic joint.
After 11 days of hospitalization, the patient was discharged with a peripherally inserted central catheter for long-term antifungal infusions of micafungin 150 mg/day at home. Fungal sensitivity test results several days after discharge confirmed susceptibility to micafungin.
About 2 weeks after discharge, the patient underwent debridement and implant retention (DAIR). Wound cultures were positive for C. glabrata, Enterococcus faecalis, Staphylococcus epidermidis, and Corynebacterium tuberculostearicum. Based on susceptibilities, he completed a 2-month course of IV micafungin 150 mg daily and daptomycin 750 mg daily, followed by an oral suppressive regimen consisting of doxycycline 100 mg twice daily, amoxicillin-clavulanate 2 g twice daily, and fluconazole initially 800 mg daily adjusted to 400 mg daily. The patient continued wound management with twice-daily dressing changes.
Nine months after DAIR, the patient remained on suppressive antifungal and antibacterial therapy. He continued to experience serous drainage from the wound, which greatly affected his quality of life. After discussion with his family and the orthopedic surgery team, he agreed to proceed with a 2-staged revision arthroplasty involving prosthetic explant and antibiotic spacer placement. However, the surgery was postponed due to findings of anemia (hemoglobin, 8.9 g/dL) and thrombocytopenia (platelet count, 73 x 103/λL). At the time of this report, the patient was being monitored closely with his multidisciplinary care team for the planned orthopedic procedure.
DISCUSSION
PJI is the most common cause of primary hip arthroplasty failure; however, fungal species only make up about 1% of PJIs.3,9-11 Patients are typically immunocompromised, undergoing antineoplastic therapies for malignancy, or have other comorbid conditions such as diabetes.12,13 C. glabrata presents a unique diagnostic and therapeutic challenge as it is not only rare but also notorious for its resistance to common antifungal agents. C. glabrata is known to develop multidrug resistance through the rapid accumulation of genomic mutations.14 Its propensity towards forming protective biofilm also arms it with intrinsic resistance to agents like fluconazole.15 Furthermore, based on a review of the available reports in the literature, C. glabrata PJIs are often insidious and present with symptoms closely mimicking those of bacterial PJIs, as it did in the patient in this case.16
Synovial fluid analysis, fungal cultures, and sensitivity testing are paramount for ensuring proper diagnosis for fungal PJI. The patient in this case was empirically treated with micafungin based on recommendations from the ID team. When the sensitivities results were reviewed, the same antifungal therapy was continued. Echinocandins have a favorable toxicity profile in long-term use, as well as efficacy against biofilm-producing organisms like C. glabrata.17,18
While there are a few cases citing DAIR as a feasible surgical strategy for treating fungal PJI, more recent studies have reported greater success with a 2-staged revision arthroplasty involving some combination of debridement, placement of antibiotic-loaded bone cement spacers, and partial or total exchange of the infected prosthetic joint.4,19-23 In this case, complete hardware replacement would have offered the patient the most favorable outlook for eliminating this fungal infection. However, given the patient’s advanced age, significant underlying comorbidities, and functional status, medical management with antifungal therapy and DAIR was favored.
Based on the discussion from the 6-month follow-up visit, the patient was experiencing progressive and persistent wound drainage and frequent dressing changes, highlighting the limitations of medical management for PJI in the setting of retained prosthesis. If the patient ultimately proceeds with a more invasive surgical intervention, another important consideration will be the likelihood of fungal PJI recurrence. At present, fungal PJI recurrence rates following antifungal and surgical treatment have been reported to range between 0% to 50%, which is too imprecise to be considered clinically useful.22-24
Given the ambiguity surrounding management guidelines and limited treatment options, it is crucial to emphasize the timeline of this patient’s clinical presentation and subsequent course of treatment. Upon presentation to the ED in late July, fungal PJI was considered less likely. Initial blood cultures from presentation were negative, which is common with PJIs. It was not until 5 days later that the left hip wound culture showed moderate growth of C. glabrata. Identifying a PJI is clinically challenging due to the lack of standardized diagnostic criteria. However, timely identification and diagnosis of fungal PJI with appropriate antifungal therapy, in patients with limited curative options due to comorbidities, can significantly improve quality of life and overall outcomes.25 Routine fungal and mycobacterial cultures are not currently recommended in PJI guidelines, but this case illustrates it is imperative in immunocompromised hosts.26
This case and the current paucity of similar cases in the literature stress the importance of clinicians publishing their experience in the management of fungal PJI. We strongly recommend that clinicians approach each suspected PJI with careful consideration of the patient’s unique risk factors, comorbidities, and goals of care, when deciding on a curative vs suppressive approach to therapy.
CONCLUSIONS
This case report highlights the importance of considering fungal pathogens for PJIs, especially in high-risk patients, the value of obtaining fungal cultures, the necessity of a multidisciplinary approach, the role of antifungal susceptibility testing, and consideration for the feasibility of a surgical intervention. It underscores the challenges in diagnosis and treatment of C. glabrata-associated PJI, emphasizing the importance of clinician experience-sharing in developing evidence-based management strategies. As the understanding of fungal PJI evolves, continued research and clinical data collection remain crucial for improving patient outcomes in the management of these complex cases.
- Osmon DR, Berbari EF, Berendt AR, et al. Executive summary: diagnosis and management of prosthetic joint infection: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2013;56(1):1-10. doi:10.1093/cid/cis966
- Eka A, Chen AF. Patient-related medical risk factors for periprosthetic joint infection of the hip and knee. Ann Transl Med. 2015;3(16):233. doi:10.3978/j.issn.2305-5839.2015.09.26
- Darouiche RO, Hamill RJ, Musher DM, Young EJ, Harris RL. Periprosthetic candidal infections following arthroplasty. Rev Infect Dis. 1989;11(1):89-96. doi:10.1093/clinids/11.1.89
- Koutserimpas C, Zervakis SG, Maraki S, et al. Non-albicans Candida prosthetic joint infections: a systematic review of treatment. World J Clin Cases. 2019;7(12):1430- 1443. doi:10.12998/wjcc.v7.i12.1430
- Fidel PL Jr, Vazquez JA, Sobel JD. Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin Microbiol Rev. 1999;12(1):80-96. doi:10.1128/CMR.12.1.80
- Aboltins C, Daffy J, Choong P, Stanley P. Current concepts in the management of prosthetic joint infection. Intern Med J. 2014;44(9):834-840. doi:10.1111/imj.12510
- Lee YR, Kim HJ, Lee EJ, Sohn JW, Kim MJ, Yoon YK. Prosthetic joint infections caused by candida species: a systematic review and a case series. Mycopathologia. 2019;184(1):23-33. doi:10.1007/s11046-018-0286-1
- Herndon CL, Rowe TM, Metcalf RW, et al. Treatment outcomes of fungal periprosthetic joint infection. J Arthroplasty. 2023;38(11):2436-2440.e1. doi:10.1016/j.arth.2023.05.009
- Delaunay C, Hamadouche M, Girard J, Duhamel A; SoFCOT. What are the causes for failures of primary hip arthroplasties in France? Clin Orthop Relat Res. 2013;471(12): 3863-3869. doi:10.1007/s11999-013-2935-5
- Bozic KJ, Kurtz SM, Lau E, Ong K, Vail TP, Berry DJ. The epidemiology of revision total hip arthroplasty in the United States. J Bone Joint Surg Am. 2009;91(1): 128-133. doi:10.2106/JBJS.H.00155
- Furnes O, Lie SA, Espehaug B, Vollset SE, Engesaeter LB, Havelin LI. Hip disease and the prognosis of total hip replacements. A review of 53,698 primary total hip replacements reported to the Norwegian Arthroplasty Register 1987-99. J Bone Joint Surg Br. 2001;83(4):579-586. doi:10.1302/0301-620x.83b4.11223
- Gonzalez MR, Bedi ADS, Karczewski D, Lozano-Calderon SA. Treatment and outcomes of fungal prosthetic joint infections: a systematic review of 225 cases. J Arthroplasty. 2023;38(11):2464-2471.e1. doi:10.1016/j.arth.2023.05.003
- Gonzalez MR, Pretell-Mazzini J, Lozano-Calderon SA. Risk factors and management of prosthetic joint infections in megaprostheses-a review of the literature. Antibiotics (Basel). 2023;13(1):25. doi:10.3390/antibiotics13010025
- Biswas C, Chen SC, Halliday C, et al. Identification of genetic markers of resistance to echinocandins, azoles and 5-fluorocytosine in Candida glabrata by next-generation sequencing: a feasibility study. Clin Microbiol Infect. 2017;23(9):676.e7-676.e10. doi:10.1016/j.cmi.2017.03.014
- Hassan Y, Chew SY, Than LTL. Candida glabrata: pathogenicity and resistance mechanisms for adaptation and survival. J Fungi (Basel). 2021;7(8):667. doi:10.3390/jof7080667
- Aboltins C, Daffy J, Choong P, Stanley P. Current concepts in the management of prosthetic joint infection. Intern Med J. 2014;44(9):834-840. doi:10.1111/imj.12510
- Pierce CG, Uppuluri P, Tristan AR, et al. A simple and reproducible 96-well plate-based method for the formation of fungal biofilms and its application to antifungal susceptibility testing. Nat Protoc. 2008;3(9):1494-1500. doi:10.1038/nport.2008.141
- Koutserimpas C, Samonis G, Velivassakis E, Iliopoulou- Kosmadaki S, Kontakis G, Kofteridis DP. Candida glabrata prosthetic joint infection, successfully treated with anidulafungin: a case report and review of the literature. Mycoses. 2018;61(4):266-269. doi:10.1111/myc.12736
- Brooks DH, Pupparo F. Successful salvage of a primary total knee arthroplasty infected with Candida parapsilosis. J Arthroplasty. 1998;13(6):707-712. doi:10.1016/s0883-5403(98)80017-x
- Merrer J, Dupont B, Nieszkowska A, De Jonghe B, Outin H. Candida albicans prosthetic arthritis treated with fluconazole alone. J Infect. 2001;42(3):208-209. doi:10.1053/jinf.2001.0819
- Koutserimpas C, Naoum S, Alpantaki K, et al. Fungal prosthetic joint infection in revised knee arthroplasty: an orthopaedic surgeon’s nightmare. Diagnostics (Basel). 2022;12(7):1606. doi:10.3390/diagnostics12071606
- Gao Z, Li X, Du Y, Peng Y, Wu W, Zhou Y. Success rate of fungal peri-prosthetic joint infection treated by 2-stage revision and potential risk factors of treatment failure: a retrospective study. Med Sci Monit. 2018;24:5549-5557. doi:10.12659/MSM.909168
- Hwang BH, Yoon JY, Nam CH, et al. Fungal periprosthetic joint infection after primary total knee replacement. J Bone Joint Surg Br. 2012;94(5):656-659. doi:10.1302/0301-620X.94B5.28125
- Ueng SW, Lee CY, Hu CC, Hsieh PH, Chang Y. What is the success of treatment of hip and knee candidal periprosthetic joint infection? Clin Orthop Relat Res. 2013;471(9):3002-3009. doi:10.1007/s11999-013-3007-6
- Nodzo, Scott R. MD; Bauer, Thomas MD, PhD; Pottinger, et al. Conventional diagnostic challenges in periprosthetic joint infection. J Am Acad Orthop Surg. 2015;23 Suppl:S18-S25. doi:10.5435/JAAOS-D-14-00385
- American Academy of Orthopaedic Surgeons. Diagnosis and prevention of periprosthetic joint infections. March 11, 2019. Accessed February 5, 2025. https://www.aaos.org/pjicpg
- Osmon DR, Berbari EF, Berendt AR, et al. Executive summary: diagnosis and management of prosthetic joint infection: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2013;56(1):1-10. doi:10.1093/cid/cis966
- Eka A, Chen AF. Patient-related medical risk factors for periprosthetic joint infection of the hip and knee. Ann Transl Med. 2015;3(16):233. doi:10.3978/j.issn.2305-5839.2015.09.26
- Darouiche RO, Hamill RJ, Musher DM, Young EJ, Harris RL. Periprosthetic candidal infections following arthroplasty. Rev Infect Dis. 1989;11(1):89-96. doi:10.1093/clinids/11.1.89
- Koutserimpas C, Zervakis SG, Maraki S, et al. Non-albicans Candida prosthetic joint infections: a systematic review of treatment. World J Clin Cases. 2019;7(12):1430- 1443. doi:10.12998/wjcc.v7.i12.1430
- Fidel PL Jr, Vazquez JA, Sobel JD. Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin Microbiol Rev. 1999;12(1):80-96. doi:10.1128/CMR.12.1.80
- Aboltins C, Daffy J, Choong P, Stanley P. Current concepts in the management of prosthetic joint infection. Intern Med J. 2014;44(9):834-840. doi:10.1111/imj.12510
- Lee YR, Kim HJ, Lee EJ, Sohn JW, Kim MJ, Yoon YK. Prosthetic joint infections caused by candida species: a systematic review and a case series. Mycopathologia. 2019;184(1):23-33. doi:10.1007/s11046-018-0286-1
- Herndon CL, Rowe TM, Metcalf RW, et al. Treatment outcomes of fungal periprosthetic joint infection. J Arthroplasty. 2023;38(11):2436-2440.e1. doi:10.1016/j.arth.2023.05.009
- Delaunay C, Hamadouche M, Girard J, Duhamel A; SoFCOT. What are the causes for failures of primary hip arthroplasties in France? Clin Orthop Relat Res. 2013;471(12): 3863-3869. doi:10.1007/s11999-013-2935-5
- Bozic KJ, Kurtz SM, Lau E, Ong K, Vail TP, Berry DJ. The epidemiology of revision total hip arthroplasty in the United States. J Bone Joint Surg Am. 2009;91(1): 128-133. doi:10.2106/JBJS.H.00155
- Furnes O, Lie SA, Espehaug B, Vollset SE, Engesaeter LB, Havelin LI. Hip disease and the prognosis of total hip replacements. A review of 53,698 primary total hip replacements reported to the Norwegian Arthroplasty Register 1987-99. J Bone Joint Surg Br. 2001;83(4):579-586. doi:10.1302/0301-620x.83b4.11223
- Gonzalez MR, Bedi ADS, Karczewski D, Lozano-Calderon SA. Treatment and outcomes of fungal prosthetic joint infections: a systematic review of 225 cases. J Arthroplasty. 2023;38(11):2464-2471.e1. doi:10.1016/j.arth.2023.05.003
- Gonzalez MR, Pretell-Mazzini J, Lozano-Calderon SA. Risk factors and management of prosthetic joint infections in megaprostheses-a review of the literature. Antibiotics (Basel). 2023;13(1):25. doi:10.3390/antibiotics13010025
- Biswas C, Chen SC, Halliday C, et al. Identification of genetic markers of resistance to echinocandins, azoles and 5-fluorocytosine in Candida glabrata by next-generation sequencing: a feasibility study. Clin Microbiol Infect. 2017;23(9):676.e7-676.e10. doi:10.1016/j.cmi.2017.03.014
- Hassan Y, Chew SY, Than LTL. Candida glabrata: pathogenicity and resistance mechanisms for adaptation and survival. J Fungi (Basel). 2021;7(8):667. doi:10.3390/jof7080667
- Aboltins C, Daffy J, Choong P, Stanley P. Current concepts in the management of prosthetic joint infection. Intern Med J. 2014;44(9):834-840. doi:10.1111/imj.12510
- Pierce CG, Uppuluri P, Tristan AR, et al. A simple and reproducible 96-well plate-based method for the formation of fungal biofilms and its application to antifungal susceptibility testing. Nat Protoc. 2008;3(9):1494-1500. doi:10.1038/nport.2008.141
- Koutserimpas C, Samonis G, Velivassakis E, Iliopoulou- Kosmadaki S, Kontakis G, Kofteridis DP. Candida glabrata prosthetic joint infection, successfully treated with anidulafungin: a case report and review of the literature. Mycoses. 2018;61(4):266-269. doi:10.1111/myc.12736
- Brooks DH, Pupparo F. Successful salvage of a primary total knee arthroplasty infected with Candida parapsilosis. J Arthroplasty. 1998;13(6):707-712. doi:10.1016/s0883-5403(98)80017-x
- Merrer J, Dupont B, Nieszkowska A, De Jonghe B, Outin H. Candida albicans prosthetic arthritis treated with fluconazole alone. J Infect. 2001;42(3):208-209. doi:10.1053/jinf.2001.0819
- Koutserimpas C, Naoum S, Alpantaki K, et al. Fungal prosthetic joint infection in revised knee arthroplasty: an orthopaedic surgeon’s nightmare. Diagnostics (Basel). 2022;12(7):1606. doi:10.3390/diagnostics12071606
- Gao Z, Li X, Du Y, Peng Y, Wu W, Zhou Y. Success rate of fungal peri-prosthetic joint infection treated by 2-stage revision and potential risk factors of treatment failure: a retrospective study. Med Sci Monit. 2018;24:5549-5557. doi:10.12659/MSM.909168
- Hwang BH, Yoon JY, Nam CH, et al. Fungal periprosthetic joint infection after primary total knee replacement. J Bone Joint Surg Br. 2012;94(5):656-659. doi:10.1302/0301-620X.94B5.28125
- Ueng SW, Lee CY, Hu CC, Hsieh PH, Chang Y. What is the success of treatment of hip and knee candidal periprosthetic joint infection? Clin Orthop Relat Res. 2013;471(9):3002-3009. doi:10.1007/s11999-013-3007-6
- Nodzo, Scott R. MD; Bauer, Thomas MD, PhD; Pottinger, et al. Conventional diagnostic challenges in periprosthetic joint infection. J Am Acad Orthop Surg. 2015;23 Suppl:S18-S25. doi:10.5435/JAAOS-D-14-00385
- American Academy of Orthopaedic Surgeons. Diagnosis and prevention of periprosthetic joint infections. March 11, 2019. Accessed February 5, 2025. https://www.aaos.org/pjicpg
A Candida Glabrata-Associated Prosthetic Joint Infection: Case Report and Literature Review
A Candida Glabrata-Associated Prosthetic Joint Infection: Case Report and Literature Review
Hearing Patient Stories: Use of Medical Humanities on a Large-Scale, Virtual Platform to Improve Clinician Engagement
Hearing Patient Stories: Use of Medical Humanities on a Large-Scale, Virtual Platform to Improve Clinician Engagement
The COVID-19 pandemic presented stressors for patients and health care professionals alike, and the prevalence of health care practitioner burnout and dissatisfaction has risen dramatically.1,2 This, in combination with an increasingly virtual interface between patients and care teams, has the potential to lead to increased depersonalization, anxiety, distress, and diminished overall well-being among clinicians.1,3 Within the Veterans Health Administration (VHA), women’s health primary care practitioners (PCPs) are specially trained clinicians thatprovide comprehensive care to women veterans. Data suggest that women’s health PCPs may experience higher rates of burnout and attrition (14% per year) compared to general PCPs in VHA.4 Burnout among PCPs, especially those working at VHA, is well known and likely related to poor interdisciplinary team structure, limited administrative time, high patient complexity, and isolation from additional resources (eg, rural settings).4-7 Increased clinician burnout is associated with poorer quality of care and worsening quality of the doctor-patient relationship.8
The medical humanities can act as a countermeasure to clinician burnout.9,10 Studies have demonstrated that physicians who participate in the medical humanities are more empathic and experience less burnout.11,12 Engaging with patient stories through listening and writing has been a source of fulfillment for clinicians.13 Despite the benefits of narrative medicine, programs are often limited in scope in small face-to-face group settings during elective time or outside work hours.14 The COVID-19 pandemic presented significant challenges to implementing such programming. The VHA is a large health care system with many rural locations, which further limits the availability of traditional small-group and face-to-face trainings. Few studies describe large-scale medical humanities training in virtual learning environments.
NARRATIVE MEDICINE EVENT
To improve satisfaction and engagement among PCPs who care for women veterans, we developed, implemented, and evaluated a large-scale, virtual, interprofessional narrative medicine event aimed at achieving the following: (1) gain a deeper appreciation of the impact of deployments on women veterans; (2) describe the social and emotional challenges faced by women veterans returning from deployment (reintegration); (3) identify strategies to support veterans during reintegration; (4) apply narrative medicine techniques on a large-scale, virtual platform; and (5) assess clinician engagement and satisfaction following participation. We hypothesized that clinician satisfaction and appreciation would improve with a better understanding of the unique complexities of deployment and reintegration faced by women veterans. Utilizing a novel, humanities-based intervention would lead to strong engagement and interaction from participants.
Setting
A 3-hour virtual session was conducted on November 15, 2022, for an interdisciplinary audience. This included physicians and trainees in medicine and behavioral health, nurse practitioners, social workers, dieticians, nurses, and clinical support staff. The training was advertised via emails through established mailing lists and newsletters, reaching a large interdisciplinary VHA audience 90 days prior to the event. This allowed potential participants to dedicate time to attend the session. The training was open to all VHA employees, with no inclusion or exclusion criteria for either the training or the evaluation. The training was delivered within existing space utilized for continuing medical education in women’s health.
For the session, the 93-minute documentary Journey to Normal (jtninc.org) was chosen because it focused on the impact of deployment on women veterans and their experiences when returning home. The film follows the stories of several women veterans through combat and reintegration. The screening was split into 2 segments given the emotional impact and length of the documentary.
A facilitator opened the session by reading a series of reflective prompts centered on women veteran deployment, reintegration, and the stressors surrounding these transitions. The initial prompt served to familiarize participants with the session’s interactive components. Additional prompts were interspersed and discussed in real time and were chosen to mirror the major themes of the documentary: the emotional and psychological impact of deployment and reintegration for women veterans. Short responses and word cloud generation were used and debriefed synchronously to encourage ongoing engagement. Participants responded to prompts through anonymous polling and the chat function of the virtual platform.
During intermission, we introduced My Life, My Story (MLMS). MLMS is a VHA initiative started in 2013 that, with the veteran’s permission, shares a piece of a veteran’s life story with their health care practitioner in their medical chart.15 Evaluation of MLMS has demonstrated positive impacts on assessments of patient-clinician connection.16 The MLMS goal to improve patient-centered care competencies by learning stories of veterans aligned with the overarching goals of this program. Following the film, participants were given 10 minutes to respond to a final reflective prompt. The session ended with a review of existing VHA resources to support returning veterans, followed by a question-and-answer session conducted via chat.
We used the Brightcove virtual platform to stream this program, which facilitated significant interaction between participants and facilitators, as well as between participants themselves. In addition to posing questions to the session leaders, participants could directly respond to each other’s comments within the chat function and also upvote/downvote or emphasize others’ comments.
Evaluation
The evaluation schema was 2-fold. Because this session was presented as a part of the national VA Women’s Health webinar series, a standard evaluation was dictated by the VHA Employee Education System. This survey was electronically disseminated and included questions on occupational category and overall satisfaction, plus 9 standard evaluation questions and 4 program-specific questions tied to the workshop objectives. The standard evaluation questions assessed participant satisfaction with the training, satisfaction with the training environment, and appropriateness of the content. The programspecific questions asked the participants whether the session met the stated learning objectives. All questions used a 5-point Likert scale (1, strongly disagree; 5, strongly agree). Descriptive statistics were used for analysis. Individual chat messages and spontaneous replies were analyzed as a surrogate measures of audience engagement. A qualitative analysis of participants’ final reflections to assess for attitudes related to patient care, empathy, and burnout following participation in this curriculum is forthcoming.
A total of 876 participants attended the virtual setting and 525 (59.9%) completed the immediate postevaluation survey. Respondents represented a variety of disciplines, including 179 nurses (34.1%), 100 social workers (19.0%), 65 physicians (12.4%), and 10 physician assistants (1.9%), with < 10% comprising counselors, dentists, dietitians, pharmacists, physical therapists, and psychologists. Nearly all participants reported satisfaction with the learning activity, would recommend it to others, and felt it advanced their knowledge, attitudes, and skills to better contribute to their VHA interprofessional team for patient care (Table 1). Similarly, participants reported a highlevel of agreement that the program satisfied the session-specific objectives. In response to an open-ended question on the standard VA evaluation regarding overall perceptions of the training, free-text responses included such statements as, “I think this should be mandatory training for all VA [clinicians]”; and “This webinar [opened] my mind to the various struggles women veterans may encounter when [they] return to civilian life and [increased] my understanding of how I could support.”

More than 1700 individual chat messages and > 80 spontaneous replies between participants were recorded during the interactive session (Table 2). Spontaneous quotes written in the chat included: “This is the best film representing the female veteran I have ever seen;” “Powerful and perspective changing;” “Thank you for sharing this incredible film;” and “I needed this to remind me to focus on woman veterans. Although our female veteran population is small it will remind me daily of their dedication, recognizing that there are so many facets of making the ultimate sacrifice.” Several participants said such programming should be a mandatory component of VA new employee orientation.

DISCUSSION
Clinician burnout diminishes empathetic patient-physician engagement. Patients’ stories are a known, powerful way to evoke empathy. This session provides one of the first examples of a straightforward approach to delivering a medical humanities intervention to a large audience via virtual platform. As measured by its high engagement, participant satisfaction, and narrative evaluations, this model was successful in evoking empathy and reinforcing the core VHA values for patient care: integrity, commitment, advocacy, respect, and excellence.
Rates of burnout and disengagement among PCPs are high and increased during the COVID-19 pandemic.2 This curriculum used a synchronous, narrative-based approach during work hours to address burnout. Lack of empathy is a cause and consequence of burnout and disengagement. Narrative approaches, especially those evoking patients’ stories can evoke empathy and help counteract such burnout. This curriculum demonstrates one of the first large-scale, narrative-based, virtual-platform approaches to utilizing patients’ stories for positive clinician impact, as evidenced by the extensive participation, engagement, and satisfaction of participants.
Individuals interested in implementing a similar program should consider common barriers, including time constraints, advertising, and clinician buy-in. Several key factors led to the successful implementation of this program. First, partnering with established educational efforts related to improving care for veterans provided time to implement the program and establish mechanisms for advertising. The VHA is a mission-driven organization; directly tying this intervention to the mission likely contributed to participant buy-in and programmatic success. Further, by partnering with established educational efforts, this session was conducted during business hours, allowing for widespread participation.
A diverse group of VHA clinicians were actively engaged throughout the session. Chat data demonstrated not only numerous responses to directed prompts, but also a larger extemporaneous conversation among participants. Additionally, it is clear participants were deeply engaged with the material. The quality of participant responses demonstrates the impact of narrative stories and included a new respect for our shared patients, a sense of humbleness as it relates to the women veteran experience, and a sense of pride in both the VHA mission and their roles as a part of the organization.
This session did not end with traditional take-home skills or reference handout resources typical of continuing education. This was intentional; the intended take-home message was the evoked emotional response and resultant perspective shift. The impact of this session on patient care will be examined in a forthcoming qualitative analysis of participants written reflections.
Limitations
Some participants noted that the chat could be distracting from the film. Others described that virtually attending the session allowed increased opportunity for interruption by ongoing patient care responsibilities, resulting in diverted attention. Many participants were granted protected time to attend this continuing education session; however, this was not always the case. Additionally, this evaluation is limited, as 40% of participants elected to not complete the postevent survey. The individuals who choose to respond may have been more engaged with the content or felt more strongly about the impact of the session. However, the volume of chat engagement during the session suggests strong participant involvement. The analysis was also limited by an electronic survey which did not allow more granular assessment of the data.
This session also raised an ethical consideration. The film evoked very strong emotional responses which, for some, were challenging to attend to personally in a large-scale virtual environment. Established clinician resources were highlighted during the session that were available for any participant who needed additional support. Participants were also encouraged to step away and process their emotions, if needed. Future interactions of this session might consider improved interparticipant chat management and upfront warnings about the emotional impact of the film accompanied by proactive dissemination of resources for participant support. One example of such resources includes breakout rooms facilitated by trained counselors. Prompts might also be adjusted to allow for more guided interparticipant engagement; facilitation can be brief as participants’ responses often carry the conversation.
CONCLUSIONS
This study shows that a large-scale, virtual medical humanities intervention is not only possible but well received, as evidenced by both quantity and quality of participant responses and engagement. The narrative approach of hearing patients’ stories, as portrayed in Journey to Normal, was found to be satisfying and appreciated by participants. Such an intervention has the potential to evoke empathy and help counteract burnout and disengagement among clinicians. This study directly aligned to the greater mission of the VHA: to improve quality medical care for all veterans, including women veterans, a subset population that is often overlooked. Organizations beyond the VHA may wish to leverage virtual learning as a mechanism to offer medical humanities to a wider audience. To optimize success, future programs should be tied to organizational missions, highlight patient voices and stories, and utilize platforms that allow for participant interactivity. Through virtual platforms, the medical humanities can reach a broader audience without detracting from its impact.
- Van Wert MJ, Gandhi S, Gupta I, et al. Healthcare worker mental health after the initial peak of the COVID- 19 pandemic: a US medical center cross-sectional survey. J Gen Intern Med. 2022;37(5):1169-1176. doi:10.1007/s11606-021-07251-0
- Centers for Disease Control and Prevention. Vital Signs. Health workers face a mental health crisis: workers report harassment, burnout, and poor mental health; supportive workplaces can help. Updated October 24, 2023. Accessed February 18, 2025. https://www.cdc.gov/vitalsigns/health-worker-mental-health/index.html
- Holmgren AJ, Downing NL, Tang M, Sharp C, Longhurst C, Huckman RS. Assessing the impact of the COVID-19 pandemic on clinician ambulatory electronic health record use. J Am Med Inform Assoc. 2022;29(3):453-460. doi:10.1093/jamia/ocab268
- Apaydin EA, Mohr DC, Hamilton AB, Rose DE, Haskell S, Yano EM. Differences in burnout and intent to leave between women’s health and general primary care providers in the Veterans Health Administration. J Gen Intern Med. 2022;37(10):2382-2389. doi:10.1007/s11606-021-07133-5
- Willard-Grace R, Knox M, Huang B, Hammer H, Kivlahan C, Grumbach K. Burnout and health care workforce turnover. Ann Fam Med. 2019;17(1):36-41. doi:10.1370/afm.2338
- Rinne ST, Mohr DC, Swamy L, Blok AC, Wong ES, Charns MP. National burnout trends among physicians working in the department of veterans affairs. J Gen Intern Med. 2020;35(5):1382-1388. doi:10.1007/s11606-019-05582-7
- Spinelli WM, Fernstrom KM, Galos DL, Britt HR. Extending our understanding of burnout and its associated factors: providers and staff in primary care clinics. Eval Health Prof. 2016;39(3):282-298. doi:10.1177/0163278716637900
- Abraham CM, Zheng K, Poghosyan L. Predictors and outcomes of burnout among primary care providers in the United States: a systematic review. Med Care Res Rev. 2020;77(5):387-401. doi:10.1177/1077558719888427
- Charon R, Williams P. Introduction: the humanities and medical education. Acad Med. 1995;70(9):758-760.
- Winkel AF, Yingling S, Jones A-A, Nicholson J. Reflection as a learning tool in graduate medical education: a systematic review. J Grad Med Educ. 2017;9(4):430-439. doi:10.4300/JGME-D-16-00500.1
- Charon R. The patient-physician relationship. Narrative medicine: a model for empathy, reflection, profession, and trust. JAMA. 2001;286(15):1897-1902. doi:10.1001/jama.286.15.1897
- DasGupta S, Charon R. Personal illness narratives: using reflective writing to teach empathy. Acad Med. 2004; 79(4):351-356. doi:10.1097/00001888-200404000-00013
- Liao JM, Secemsky BJ. The value of narrative medical writing in internal medicine residency. J Gen Intern Med. 2015;30(11):1707-1710. doi:10.1007/s11606-015-3460-x
- Branch WT, Kern D, Haidet P, et al. The patient-physician relationship. Teaching the human dimensions of care in clinical settings. JAMA. 2001;286(9):1067-1074. doi:10.1001/jama.286.9.1067
- Roberts TJ, Ringler T, Krahn D, Ahearn E. The my life, my story program: sustained impact of veterans’ personal narratives on healthcare providers 5 years after implementation. Health Commun. 2021;36(7):829-836. doi:10.1080/10410236.2020.1719316
- Lam JA, Feingold-Link M, Noguchi J, et al. My life, my story: integrating a life story narrative component into medical student curricula. MedEdPORTAL. 2022;18:11211. doi:10.15766/mep_2374-8265.11211
The COVID-19 pandemic presented stressors for patients and health care professionals alike, and the prevalence of health care practitioner burnout and dissatisfaction has risen dramatically.1,2 This, in combination with an increasingly virtual interface between patients and care teams, has the potential to lead to increased depersonalization, anxiety, distress, and diminished overall well-being among clinicians.1,3 Within the Veterans Health Administration (VHA), women’s health primary care practitioners (PCPs) are specially trained clinicians thatprovide comprehensive care to women veterans. Data suggest that women’s health PCPs may experience higher rates of burnout and attrition (14% per year) compared to general PCPs in VHA.4 Burnout among PCPs, especially those working at VHA, is well known and likely related to poor interdisciplinary team structure, limited administrative time, high patient complexity, and isolation from additional resources (eg, rural settings).4-7 Increased clinician burnout is associated with poorer quality of care and worsening quality of the doctor-patient relationship.8
The medical humanities can act as a countermeasure to clinician burnout.9,10 Studies have demonstrated that physicians who participate in the medical humanities are more empathic and experience less burnout.11,12 Engaging with patient stories through listening and writing has been a source of fulfillment for clinicians.13 Despite the benefits of narrative medicine, programs are often limited in scope in small face-to-face group settings during elective time or outside work hours.14 The COVID-19 pandemic presented significant challenges to implementing such programming. The VHA is a large health care system with many rural locations, which further limits the availability of traditional small-group and face-to-face trainings. Few studies describe large-scale medical humanities training in virtual learning environments.
NARRATIVE MEDICINE EVENT
To improve satisfaction and engagement among PCPs who care for women veterans, we developed, implemented, and evaluated a large-scale, virtual, interprofessional narrative medicine event aimed at achieving the following: (1) gain a deeper appreciation of the impact of deployments on women veterans; (2) describe the social and emotional challenges faced by women veterans returning from deployment (reintegration); (3) identify strategies to support veterans during reintegration; (4) apply narrative medicine techniques on a large-scale, virtual platform; and (5) assess clinician engagement and satisfaction following participation. We hypothesized that clinician satisfaction and appreciation would improve with a better understanding of the unique complexities of deployment and reintegration faced by women veterans. Utilizing a novel, humanities-based intervention would lead to strong engagement and interaction from participants.
Setting
A 3-hour virtual session was conducted on November 15, 2022, for an interdisciplinary audience. This included physicians and trainees in medicine and behavioral health, nurse practitioners, social workers, dieticians, nurses, and clinical support staff. The training was advertised via emails through established mailing lists and newsletters, reaching a large interdisciplinary VHA audience 90 days prior to the event. This allowed potential participants to dedicate time to attend the session. The training was open to all VHA employees, with no inclusion or exclusion criteria for either the training or the evaluation. The training was delivered within existing space utilized for continuing medical education in women’s health.
For the session, the 93-minute documentary Journey to Normal (jtninc.org) was chosen because it focused on the impact of deployment on women veterans and their experiences when returning home. The film follows the stories of several women veterans through combat and reintegration. The screening was split into 2 segments given the emotional impact and length of the documentary.
A facilitator opened the session by reading a series of reflective prompts centered on women veteran deployment, reintegration, and the stressors surrounding these transitions. The initial prompt served to familiarize participants with the session’s interactive components. Additional prompts were interspersed and discussed in real time and were chosen to mirror the major themes of the documentary: the emotional and psychological impact of deployment and reintegration for women veterans. Short responses and word cloud generation were used and debriefed synchronously to encourage ongoing engagement. Participants responded to prompts through anonymous polling and the chat function of the virtual platform.
During intermission, we introduced My Life, My Story (MLMS). MLMS is a VHA initiative started in 2013 that, with the veteran’s permission, shares a piece of a veteran’s life story with their health care practitioner in their medical chart.15 Evaluation of MLMS has demonstrated positive impacts on assessments of patient-clinician connection.16 The MLMS goal to improve patient-centered care competencies by learning stories of veterans aligned with the overarching goals of this program. Following the film, participants were given 10 minutes to respond to a final reflective prompt. The session ended with a review of existing VHA resources to support returning veterans, followed by a question-and-answer session conducted via chat.
We used the Brightcove virtual platform to stream this program, which facilitated significant interaction between participants and facilitators, as well as between participants themselves. In addition to posing questions to the session leaders, participants could directly respond to each other’s comments within the chat function and also upvote/downvote or emphasize others’ comments.
Evaluation
The evaluation schema was 2-fold. Because this session was presented as a part of the national VA Women’s Health webinar series, a standard evaluation was dictated by the VHA Employee Education System. This survey was electronically disseminated and included questions on occupational category and overall satisfaction, plus 9 standard evaluation questions and 4 program-specific questions tied to the workshop objectives. The standard evaluation questions assessed participant satisfaction with the training, satisfaction with the training environment, and appropriateness of the content. The programspecific questions asked the participants whether the session met the stated learning objectives. All questions used a 5-point Likert scale (1, strongly disagree; 5, strongly agree). Descriptive statistics were used for analysis. Individual chat messages and spontaneous replies were analyzed as a surrogate measures of audience engagement. A qualitative analysis of participants’ final reflections to assess for attitudes related to patient care, empathy, and burnout following participation in this curriculum is forthcoming.
A total of 876 participants attended the virtual setting and 525 (59.9%) completed the immediate postevaluation survey. Respondents represented a variety of disciplines, including 179 nurses (34.1%), 100 social workers (19.0%), 65 physicians (12.4%), and 10 physician assistants (1.9%), with < 10% comprising counselors, dentists, dietitians, pharmacists, physical therapists, and psychologists. Nearly all participants reported satisfaction with the learning activity, would recommend it to others, and felt it advanced their knowledge, attitudes, and skills to better contribute to their VHA interprofessional team for patient care (Table 1). Similarly, participants reported a highlevel of agreement that the program satisfied the session-specific objectives. In response to an open-ended question on the standard VA evaluation regarding overall perceptions of the training, free-text responses included such statements as, “I think this should be mandatory training for all VA [clinicians]”; and “This webinar [opened] my mind to the various struggles women veterans may encounter when [they] return to civilian life and [increased] my understanding of how I could support.”
More than 1700 individual chat messages and > 80 spontaneous replies between participants were recorded during the interactive session (Table 2). Spontaneous quotes written in the chat included: “This is the best film representing the female veteran I have ever seen;” “Powerful and perspective changing;” “Thank you for sharing this incredible film;” and “I needed this to remind me to focus on woman veterans. Although our female veteran population is small it will remind me daily of their dedication, recognizing that there are so many facets of making the ultimate sacrifice.” Several participants said such programming should be a mandatory component of VA new employee orientation.
DISCUSSION
Clinician burnout diminishes empathetic patient-physician engagement. Patients’ stories are a known, powerful way to evoke empathy. This session provides one of the first examples of a straightforward approach to delivering a medical humanities intervention to a large audience via virtual platform. As measured by its high engagement, participant satisfaction, and narrative evaluations, this model was successful in evoking empathy and reinforcing the core VHA values for patient care: integrity, commitment, advocacy, respect, and excellence.
Rates of burnout and disengagement among PCPs are high and increased during the COVID-19 pandemic.2 This curriculum used a synchronous, narrative-based approach during work hours to address burnout. Lack of empathy is a cause and consequence of burnout and disengagement. Narrative approaches, especially those evoking patients’ stories can evoke empathy and help counteract such burnout. This curriculum demonstrates one of the first large-scale, narrative-based, virtual-platform approaches to utilizing patients’ stories for positive clinician impact, as evidenced by the extensive participation, engagement, and satisfaction of participants.
Individuals interested in implementing a similar program should consider common barriers, including time constraints, advertising, and clinician buy-in. Several key factors led to the successful implementation of this program. First, partnering with established educational efforts related to improving care for veterans provided time to implement the program and establish mechanisms for advertising. The VHA is a mission-driven organization; directly tying this intervention to the mission likely contributed to participant buy-in and programmatic success. Further, by partnering with established educational efforts, this session was conducted during business hours, allowing for widespread participation.
A diverse group of VHA clinicians were actively engaged throughout the session. Chat data demonstrated not only numerous responses to directed prompts, but also a larger extemporaneous conversation among participants. Additionally, it is clear participants were deeply engaged with the material. The quality of participant responses demonstrates the impact of narrative stories and included a new respect for our shared patients, a sense of humbleness as it relates to the women veteran experience, and a sense of pride in both the VHA mission and their roles as a part of the organization.
This session did not end with traditional take-home skills or reference handout resources typical of continuing education. This was intentional; the intended take-home message was the evoked emotional response and resultant perspective shift. The impact of this session on patient care will be examined in a forthcoming qualitative analysis of participants written reflections.
Limitations
Some participants noted that the chat could be distracting from the film. Others described that virtually attending the session allowed increased opportunity for interruption by ongoing patient care responsibilities, resulting in diverted attention. Many participants were granted protected time to attend this continuing education session; however, this was not always the case. Additionally, this evaluation is limited, as 40% of participants elected to not complete the postevent survey. The individuals who choose to respond may have been more engaged with the content or felt more strongly about the impact of the session. However, the volume of chat engagement during the session suggests strong participant involvement. The analysis was also limited by an electronic survey which did not allow more granular assessment of the data.
This session also raised an ethical consideration. The film evoked very strong emotional responses which, for some, were challenging to attend to personally in a large-scale virtual environment. Established clinician resources were highlighted during the session that were available for any participant who needed additional support. Participants were also encouraged to step away and process their emotions, if needed. Future interactions of this session might consider improved interparticipant chat management and upfront warnings about the emotional impact of the film accompanied by proactive dissemination of resources for participant support. One example of such resources includes breakout rooms facilitated by trained counselors. Prompts might also be adjusted to allow for more guided interparticipant engagement; facilitation can be brief as participants’ responses often carry the conversation.
CONCLUSIONS
This study shows that a large-scale, virtual medical humanities intervention is not only possible but well received, as evidenced by both quantity and quality of participant responses and engagement. The narrative approach of hearing patients’ stories, as portrayed in Journey to Normal, was found to be satisfying and appreciated by participants. Such an intervention has the potential to evoke empathy and help counteract burnout and disengagement among clinicians. This study directly aligned to the greater mission of the VHA: to improve quality medical care for all veterans, including women veterans, a subset population that is often overlooked. Organizations beyond the VHA may wish to leverage virtual learning as a mechanism to offer medical humanities to a wider audience. To optimize success, future programs should be tied to organizational missions, highlight patient voices and stories, and utilize platforms that allow for participant interactivity. Through virtual platforms, the medical humanities can reach a broader audience without detracting from its impact.
The COVID-19 pandemic presented stressors for patients and health care professionals alike, and the prevalence of health care practitioner burnout and dissatisfaction has risen dramatically.1,2 This, in combination with an increasingly virtual interface between patients and care teams, has the potential to lead to increased depersonalization, anxiety, distress, and diminished overall well-being among clinicians.1,3 Within the Veterans Health Administration (VHA), women’s health primary care practitioners (PCPs) are specially trained clinicians thatprovide comprehensive care to women veterans. Data suggest that women’s health PCPs may experience higher rates of burnout and attrition (14% per year) compared to general PCPs in VHA.4 Burnout among PCPs, especially those working at VHA, is well known and likely related to poor interdisciplinary team structure, limited administrative time, high patient complexity, and isolation from additional resources (eg, rural settings).4-7 Increased clinician burnout is associated with poorer quality of care and worsening quality of the doctor-patient relationship.8
The medical humanities can act as a countermeasure to clinician burnout.9,10 Studies have demonstrated that physicians who participate in the medical humanities are more empathic and experience less burnout.11,12 Engaging with patient stories through listening and writing has been a source of fulfillment for clinicians.13 Despite the benefits of narrative medicine, programs are often limited in scope in small face-to-face group settings during elective time or outside work hours.14 The COVID-19 pandemic presented significant challenges to implementing such programming. The VHA is a large health care system with many rural locations, which further limits the availability of traditional small-group and face-to-face trainings. Few studies describe large-scale medical humanities training in virtual learning environments.
NARRATIVE MEDICINE EVENT
To improve satisfaction and engagement among PCPs who care for women veterans, we developed, implemented, and evaluated a large-scale, virtual, interprofessional narrative medicine event aimed at achieving the following: (1) gain a deeper appreciation of the impact of deployments on women veterans; (2) describe the social and emotional challenges faced by women veterans returning from deployment (reintegration); (3) identify strategies to support veterans during reintegration; (4) apply narrative medicine techniques on a large-scale, virtual platform; and (5) assess clinician engagement and satisfaction following participation. We hypothesized that clinician satisfaction and appreciation would improve with a better understanding of the unique complexities of deployment and reintegration faced by women veterans. Utilizing a novel, humanities-based intervention would lead to strong engagement and interaction from participants.
Setting
A 3-hour virtual session was conducted on November 15, 2022, for an interdisciplinary audience. This included physicians and trainees in medicine and behavioral health, nurse practitioners, social workers, dieticians, nurses, and clinical support staff. The training was advertised via emails through established mailing lists and newsletters, reaching a large interdisciplinary VHA audience 90 days prior to the event. This allowed potential participants to dedicate time to attend the session. The training was open to all VHA employees, with no inclusion or exclusion criteria for either the training or the evaluation. The training was delivered within existing space utilized for continuing medical education in women’s health.
For the session, the 93-minute documentary Journey to Normal (jtninc.org) was chosen because it focused on the impact of deployment on women veterans and their experiences when returning home. The film follows the stories of several women veterans through combat and reintegration. The screening was split into 2 segments given the emotional impact and length of the documentary.
A facilitator opened the session by reading a series of reflective prompts centered on women veteran deployment, reintegration, and the stressors surrounding these transitions. The initial prompt served to familiarize participants with the session’s interactive components. Additional prompts were interspersed and discussed in real time and were chosen to mirror the major themes of the documentary: the emotional and psychological impact of deployment and reintegration for women veterans. Short responses and word cloud generation were used and debriefed synchronously to encourage ongoing engagement. Participants responded to prompts through anonymous polling and the chat function of the virtual platform.
During intermission, we introduced My Life, My Story (MLMS). MLMS is a VHA initiative started in 2013 that, with the veteran’s permission, shares a piece of a veteran’s life story with their health care practitioner in their medical chart.15 Evaluation of MLMS has demonstrated positive impacts on assessments of patient-clinician connection.16 The MLMS goal to improve patient-centered care competencies by learning stories of veterans aligned with the overarching goals of this program. Following the film, participants were given 10 minutes to respond to a final reflective prompt. The session ended with a review of existing VHA resources to support returning veterans, followed by a question-and-answer session conducted via chat.
We used the Brightcove virtual platform to stream this program, which facilitated significant interaction between participants and facilitators, as well as between participants themselves. In addition to posing questions to the session leaders, participants could directly respond to each other’s comments within the chat function and also upvote/downvote or emphasize others’ comments.
Evaluation
The evaluation schema was 2-fold. Because this session was presented as a part of the national VA Women’s Health webinar series, a standard evaluation was dictated by the VHA Employee Education System. This survey was electronically disseminated and included questions on occupational category and overall satisfaction, plus 9 standard evaluation questions and 4 program-specific questions tied to the workshop objectives. The standard evaluation questions assessed participant satisfaction with the training, satisfaction with the training environment, and appropriateness of the content. The programspecific questions asked the participants whether the session met the stated learning objectives. All questions used a 5-point Likert scale (1, strongly disagree; 5, strongly agree). Descriptive statistics were used for analysis. Individual chat messages and spontaneous replies were analyzed as a surrogate measures of audience engagement. A qualitative analysis of participants’ final reflections to assess for attitudes related to patient care, empathy, and burnout following participation in this curriculum is forthcoming.
A total of 876 participants attended the virtual setting and 525 (59.9%) completed the immediate postevaluation survey. Respondents represented a variety of disciplines, including 179 nurses (34.1%), 100 social workers (19.0%), 65 physicians (12.4%), and 10 physician assistants (1.9%), with < 10% comprising counselors, dentists, dietitians, pharmacists, physical therapists, and psychologists. Nearly all participants reported satisfaction with the learning activity, would recommend it to others, and felt it advanced their knowledge, attitudes, and skills to better contribute to their VHA interprofessional team for patient care (Table 1). Similarly, participants reported a highlevel of agreement that the program satisfied the session-specific objectives. In response to an open-ended question on the standard VA evaluation regarding overall perceptions of the training, free-text responses included such statements as, “I think this should be mandatory training for all VA [clinicians]”; and “This webinar [opened] my mind to the various struggles women veterans may encounter when [they] return to civilian life and [increased] my understanding of how I could support.”
More than 1700 individual chat messages and > 80 spontaneous replies between participants were recorded during the interactive session (Table 2). Spontaneous quotes written in the chat included: “This is the best film representing the female veteran I have ever seen;” “Powerful and perspective changing;” “Thank you for sharing this incredible film;” and “I needed this to remind me to focus on woman veterans. Although our female veteran population is small it will remind me daily of their dedication, recognizing that there are so many facets of making the ultimate sacrifice.” Several participants said such programming should be a mandatory component of VA new employee orientation.
DISCUSSION
Clinician burnout diminishes empathetic patient-physician engagement. Patients’ stories are a known, powerful way to evoke empathy. This session provides one of the first examples of a straightforward approach to delivering a medical humanities intervention to a large audience via virtual platform. As measured by its high engagement, participant satisfaction, and narrative evaluations, this model was successful in evoking empathy and reinforcing the core VHA values for patient care: integrity, commitment, advocacy, respect, and excellence.
Rates of burnout and disengagement among PCPs are high and increased during the COVID-19 pandemic.2 This curriculum used a synchronous, narrative-based approach during work hours to address burnout. Lack of empathy is a cause and consequence of burnout and disengagement. Narrative approaches, especially those evoking patients’ stories can evoke empathy and help counteract such burnout. This curriculum demonstrates one of the first large-scale, narrative-based, virtual-platform approaches to utilizing patients’ stories for positive clinician impact, as evidenced by the extensive participation, engagement, and satisfaction of participants.
Individuals interested in implementing a similar program should consider common barriers, including time constraints, advertising, and clinician buy-in. Several key factors led to the successful implementation of this program. First, partnering with established educational efforts related to improving care for veterans provided time to implement the program and establish mechanisms for advertising. The VHA is a mission-driven organization; directly tying this intervention to the mission likely contributed to participant buy-in and programmatic success. Further, by partnering with established educational efforts, this session was conducted during business hours, allowing for widespread participation.
A diverse group of VHA clinicians were actively engaged throughout the session. Chat data demonstrated not only numerous responses to directed prompts, but also a larger extemporaneous conversation among participants. Additionally, it is clear participants were deeply engaged with the material. The quality of participant responses demonstrates the impact of narrative stories and included a new respect for our shared patients, a sense of humbleness as it relates to the women veteran experience, and a sense of pride in both the VHA mission and their roles as a part of the organization.
This session did not end with traditional take-home skills or reference handout resources typical of continuing education. This was intentional; the intended take-home message was the evoked emotional response and resultant perspective shift. The impact of this session on patient care will be examined in a forthcoming qualitative analysis of participants written reflections.
Limitations
Some participants noted that the chat could be distracting from the film. Others described that virtually attending the session allowed increased opportunity for interruption by ongoing patient care responsibilities, resulting in diverted attention. Many participants were granted protected time to attend this continuing education session; however, this was not always the case. Additionally, this evaluation is limited, as 40% of participants elected to not complete the postevent survey. The individuals who choose to respond may have been more engaged with the content or felt more strongly about the impact of the session. However, the volume of chat engagement during the session suggests strong participant involvement. The analysis was also limited by an electronic survey which did not allow more granular assessment of the data.
This session also raised an ethical consideration. The film evoked very strong emotional responses which, for some, were challenging to attend to personally in a large-scale virtual environment. Established clinician resources were highlighted during the session that were available for any participant who needed additional support. Participants were also encouraged to step away and process their emotions, if needed. Future interactions of this session might consider improved interparticipant chat management and upfront warnings about the emotional impact of the film accompanied by proactive dissemination of resources for participant support. One example of such resources includes breakout rooms facilitated by trained counselors. Prompts might also be adjusted to allow for more guided interparticipant engagement; facilitation can be brief as participants’ responses often carry the conversation.
CONCLUSIONS
This study shows that a large-scale, virtual medical humanities intervention is not only possible but well received, as evidenced by both quantity and quality of participant responses and engagement. The narrative approach of hearing patients’ stories, as portrayed in Journey to Normal, was found to be satisfying and appreciated by participants. Such an intervention has the potential to evoke empathy and help counteract burnout and disengagement among clinicians. This study directly aligned to the greater mission of the VHA: to improve quality medical care for all veterans, including women veterans, a subset population that is often overlooked. Organizations beyond the VHA may wish to leverage virtual learning as a mechanism to offer medical humanities to a wider audience. To optimize success, future programs should be tied to organizational missions, highlight patient voices and stories, and utilize platforms that allow for participant interactivity. Through virtual platforms, the medical humanities can reach a broader audience without detracting from its impact.
- Van Wert MJ, Gandhi S, Gupta I, et al. Healthcare worker mental health after the initial peak of the COVID- 19 pandemic: a US medical center cross-sectional survey. J Gen Intern Med. 2022;37(5):1169-1176. doi:10.1007/s11606-021-07251-0
- Centers for Disease Control and Prevention. Vital Signs. Health workers face a mental health crisis: workers report harassment, burnout, and poor mental health; supportive workplaces can help. Updated October 24, 2023. Accessed February 18, 2025. https://www.cdc.gov/vitalsigns/health-worker-mental-health/index.html
- Holmgren AJ, Downing NL, Tang M, Sharp C, Longhurst C, Huckman RS. Assessing the impact of the COVID-19 pandemic on clinician ambulatory electronic health record use. J Am Med Inform Assoc. 2022;29(3):453-460. doi:10.1093/jamia/ocab268
- Apaydin EA, Mohr DC, Hamilton AB, Rose DE, Haskell S, Yano EM. Differences in burnout and intent to leave between women’s health and general primary care providers in the Veterans Health Administration. J Gen Intern Med. 2022;37(10):2382-2389. doi:10.1007/s11606-021-07133-5
- Willard-Grace R, Knox M, Huang B, Hammer H, Kivlahan C, Grumbach K. Burnout and health care workforce turnover. Ann Fam Med. 2019;17(1):36-41. doi:10.1370/afm.2338
- Rinne ST, Mohr DC, Swamy L, Blok AC, Wong ES, Charns MP. National burnout trends among physicians working in the department of veterans affairs. J Gen Intern Med. 2020;35(5):1382-1388. doi:10.1007/s11606-019-05582-7
- Spinelli WM, Fernstrom KM, Galos DL, Britt HR. Extending our understanding of burnout and its associated factors: providers and staff in primary care clinics. Eval Health Prof. 2016;39(3):282-298. doi:10.1177/0163278716637900
- Abraham CM, Zheng K, Poghosyan L. Predictors and outcomes of burnout among primary care providers in the United States: a systematic review. Med Care Res Rev. 2020;77(5):387-401. doi:10.1177/1077558719888427
- Charon R, Williams P. Introduction: the humanities and medical education. Acad Med. 1995;70(9):758-760.
- Winkel AF, Yingling S, Jones A-A, Nicholson J. Reflection as a learning tool in graduate medical education: a systematic review. J Grad Med Educ. 2017;9(4):430-439. doi:10.4300/JGME-D-16-00500.1
- Charon R. The patient-physician relationship. Narrative medicine: a model for empathy, reflection, profession, and trust. JAMA. 2001;286(15):1897-1902. doi:10.1001/jama.286.15.1897
- DasGupta S, Charon R. Personal illness narratives: using reflective writing to teach empathy. Acad Med. 2004; 79(4):351-356. doi:10.1097/00001888-200404000-00013
- Liao JM, Secemsky BJ. The value of narrative medical writing in internal medicine residency. J Gen Intern Med. 2015;30(11):1707-1710. doi:10.1007/s11606-015-3460-x
- Branch WT, Kern D, Haidet P, et al. The patient-physician relationship. Teaching the human dimensions of care in clinical settings. JAMA. 2001;286(9):1067-1074. doi:10.1001/jama.286.9.1067
- Roberts TJ, Ringler T, Krahn D, Ahearn E. The my life, my story program: sustained impact of veterans’ personal narratives on healthcare providers 5 years after implementation. Health Commun. 2021;36(7):829-836. doi:10.1080/10410236.2020.1719316
- Lam JA, Feingold-Link M, Noguchi J, et al. My life, my story: integrating a life story narrative component into medical student curricula. MedEdPORTAL. 2022;18:11211. doi:10.15766/mep_2374-8265.11211
- Van Wert MJ, Gandhi S, Gupta I, et al. Healthcare worker mental health after the initial peak of the COVID- 19 pandemic: a US medical center cross-sectional survey. J Gen Intern Med. 2022;37(5):1169-1176. doi:10.1007/s11606-021-07251-0
- Centers for Disease Control and Prevention. Vital Signs. Health workers face a mental health crisis: workers report harassment, burnout, and poor mental health; supportive workplaces can help. Updated October 24, 2023. Accessed February 18, 2025. https://www.cdc.gov/vitalsigns/health-worker-mental-health/index.html
- Holmgren AJ, Downing NL, Tang M, Sharp C, Longhurst C, Huckman RS. Assessing the impact of the COVID-19 pandemic on clinician ambulatory electronic health record use. J Am Med Inform Assoc. 2022;29(3):453-460. doi:10.1093/jamia/ocab268
- Apaydin EA, Mohr DC, Hamilton AB, Rose DE, Haskell S, Yano EM. Differences in burnout and intent to leave between women’s health and general primary care providers in the Veterans Health Administration. J Gen Intern Med. 2022;37(10):2382-2389. doi:10.1007/s11606-021-07133-5
- Willard-Grace R, Knox M, Huang B, Hammer H, Kivlahan C, Grumbach K. Burnout and health care workforce turnover. Ann Fam Med. 2019;17(1):36-41. doi:10.1370/afm.2338
- Rinne ST, Mohr DC, Swamy L, Blok AC, Wong ES, Charns MP. National burnout trends among physicians working in the department of veterans affairs. J Gen Intern Med. 2020;35(5):1382-1388. doi:10.1007/s11606-019-05582-7
- Spinelli WM, Fernstrom KM, Galos DL, Britt HR. Extending our understanding of burnout and its associated factors: providers and staff in primary care clinics. Eval Health Prof. 2016;39(3):282-298. doi:10.1177/0163278716637900
- Abraham CM, Zheng K, Poghosyan L. Predictors and outcomes of burnout among primary care providers in the United States: a systematic review. Med Care Res Rev. 2020;77(5):387-401. doi:10.1177/1077558719888427
- Charon R, Williams P. Introduction: the humanities and medical education. Acad Med. 1995;70(9):758-760.
- Winkel AF, Yingling S, Jones A-A, Nicholson J. Reflection as a learning tool in graduate medical education: a systematic review. J Grad Med Educ. 2017;9(4):430-439. doi:10.4300/JGME-D-16-00500.1
- Charon R. The patient-physician relationship. Narrative medicine: a model for empathy, reflection, profession, and trust. JAMA. 2001;286(15):1897-1902. doi:10.1001/jama.286.15.1897
- DasGupta S, Charon R. Personal illness narratives: using reflective writing to teach empathy. Acad Med. 2004; 79(4):351-356. doi:10.1097/00001888-200404000-00013
- Liao JM, Secemsky BJ. The value of narrative medical writing in internal medicine residency. J Gen Intern Med. 2015;30(11):1707-1710. doi:10.1007/s11606-015-3460-x
- Branch WT, Kern D, Haidet P, et al. The patient-physician relationship. Teaching the human dimensions of care in clinical settings. JAMA. 2001;286(9):1067-1074. doi:10.1001/jama.286.9.1067
- Roberts TJ, Ringler T, Krahn D, Ahearn E. The my life, my story program: sustained impact of veterans’ personal narratives on healthcare providers 5 years after implementation. Health Commun. 2021;36(7):829-836. doi:10.1080/10410236.2020.1719316
- Lam JA, Feingold-Link M, Noguchi J, et al. My life, my story: integrating a life story narrative component into medical student curricula. MedEdPORTAL. 2022;18:11211. doi:10.15766/mep_2374-8265.11211
Hearing Patient Stories: Use of Medical Humanities on a Large-Scale, Virtual Platform to Improve Clinician Engagement
Hearing Patient Stories: Use of Medical Humanities on a Large-Scale, Virtual Platform to Improve Clinician Engagement

Where Are All the Nurses? Data Show That Some States Have a Far Higher Number of Nurses Per Capita Than Others
During their 12-hour shifts, registered nurses (RNs) in Arizona and Arkansas perform many of the same tasks as RNs in Wisconsin and Wyoming: Assessing patients, monitoring vital signs, administering medications, and charting records to provide the best patient care. The work might be similar, but there are vast differences in the number of RNs in each state.
In states like Idaho, Utah, and Nevada, which have the lowest number of nurses per capita, there are as few as 7 nurses per 1000 residents, compared with South Dakota and the District of Columbia, which have double the number of nurses than underserved states — giving them the highest number of nurses per capita.
Even states with the largest number of nurses per capita are not immune to the nursing shortage. The National Bureau of Labor Statistics estimates that there will be 195,400 job openings for RNs from 2021 to 2031.
So, what makes it easier for some states to recruit and retain RNs than others?
States With the Highest Number of Nurses Per Capita
South Dakota
RNs per 1000 residents: 15.79
Average wage: $67,030 or $32.23 per hour
Average rent in Sioux Falls: $1192 per month
The Midwestern state has more miles of shoreline than Florida, herds of wild buffalo, the highest summit east of the Rockies, and more nurses per capita than all other states . Healthcare is one of the major industries in the Mount Rushmore State.
Haifa Abou Samra, dean and professor at the University of South Dakota School of Health Sciences, Vermillion, isn’t surprised that RNs want to call the state home.
“South Dakota is a nice place to live,” Samra said. “[The] schools are wonderful. If people are growing families, there is support; neighbors support their neighbors, and it’s a relatively safe community.”
South Dakota has 19 approved nursing education programs that graduated 878 RNs in 2022. Scholarships and student loan forgiveness programs have helped attract qualified RNs, and collaborations between education and industry have been instrumental in addressing the nursing shortage, Samra told this news organization.
Even though RNs earn less than the median wage ($87,070 per year/41.38 per hour), South Dakota has a low cost of living and no individual income tax, which helps stretch those earnings.
District of Columbia
RNs per 1000 residents: 15.39
Average wage: $105,220 or $50.59 per hour
Average rent in Washington, DC: $2485 per month
After a shift at some of the top-ranking hospitals in the nation, RNs working in the compact capital region can explore museums, monuments, and cultural sites; walk along the banks of the Potomac River; or grab a bite at award-winning restaurants.
Washington, a top-ranking metro area because of its growth, high wages, and access to economic opportunities, is also home to several top-tier hospitals and some of the best healthcare in the nation, and RNs who want to pursue continuing education have access to top-tier universities.
Nurses in Washington, DC, might make some of the highest wages in the nation, but the region also has the second-highest cost of living in the United States, with average rents topping $2400 per month and an average home price of $594,337.
North Dakota
RNs per 1000 residents: 12.99
Average wage: $74,930 or $36.03 per hour
Average rent in Fargo: $1051 per month
North Dakota projects a 10.4% increase in employment for RNs, which is higher than the national average, and the state has implemented several strategies to address chronic nursing shortages. The Nurse Staffing Clearinghouse connects nursing school graduates with local employers and created a statewide nursing staffing pool for in-state recruitment of travel nurses.
But it’s not just plentiful job opportunities and a low cost of living that attract nurses to the Peace Garden State. The state and its largest cities, Bismarck and Fargo, hold several “best of” accolades, including nods for the safest places to live and among the Best Places to Raise a Family, giving it high marks for quality of life.
Sure, the winters are cold, but the outdoor recreation can’t be beaten. RNs can bundle up and see the bighorn sheep in the Badlands at Theodore Roosevelt National Park or explore expansive terrain for skiing, snowboarding, and snowmobile trails.
States With the Lowest Number of Nurses Per Capita
Nevada
RNs per 1000 residents: 7.92
Average wage: $96,201 or $46.25 per hour
Average rent in Las Vegas: $1478 per month
Despite a projected 23% job growth for RNs between 2020 and 2030, the state has struggled to fill open positions. It might be the higher-than-average cost of living (9.7% higher than the US average) or higher-than-average crime rates that make RNs reluctant to gamble on a job in the Silver State. But there are some big wins for nurses in the state.
Salaries are higher than the national average, there is no state income tax, and some of the lowest property taxes in the nation. Thanks to new legislation, RNs with student loan debt won’t have to bet on black at the casino to make their payments. The Health Equity and Loan Assistance Program is a new initiative that offers up to $120,000 in loan repayment assistance to providers, including RNs, who commit to working in underserved and rural areas across the state for 5 years.
The state also has incredible attractions, from the neon lights and over-the-top architecture in Las Vegas to iconic red rock canyons, stunning state parks, and landmarks like Hoover Dam and Lake Tahoe.
Utah
RNs per 1000 residents: 7.05
Average wage: $79,790 or $38.36 per hour
Average rent in Salt Lake City: $1611 per month
Healthcare is one of the biggest employers in Utah, and nurses are the most in-demand healthcare workers in the state. But below-average wages and a cost of living that is a whopping 28% higher than the national average could be some reasons that the Beehive State is struggling to attract nurses.
A high number of job vacancies mean higher patient-to-nurse ratios, creating additional stress for a workforce prone to burnout. Much of the state is rural, public transportation is inadequate, and poor air quality causes frequent haze and smog.
The challenges are offset by some big benefits: Utah has been ranked as the “best state” thanks to the strong economy, infrastructure, and quality education — and it doesn’t hurt that Utah is home to myriad outdoor recreation opportunities and the stunning scenery at landmarks like Bryce Canyon and Arches National Park.
Moreover, Utah is hustling to boost its RN workforce. The University of Utah, Salt Lake City, has increased enrollment by 25% and hired additional faculty to help boost the nursing workforce — and those who work in hospitals and health clinics across the state benefit from a flat 4.55% individual income tax rate.
Idaho
RNs per 1000 residents: 7.02
Average wage: $80,130 or $38.53 per hour
Average rent in Boise: $1646 per month
Although the nursing workforce in Idaho has increased, it still ranks as the lowest in the nation. Teresa Stanfill, DNP, RN, executive director for the Idaho Center for Nursing, said that the number of new nurses is too low to replace the number of retiring nurses.
The state introduced loan repayment programs that award up to $25,000 to cover student loan debt, and hospitals and health systems often offer sign-on bonuses and relocation packages to attract RNs. But long winters, an isolated location, and limited cultural options can make it harder to attract nurses to the state.
It’s easier to recruit RNs to suburban areas like Boise, Meridian, and Nampa, but rural parts of the state struggle, Stanfill added. The nursing shortage is among the reasons that 11 hospitals and emergency departments closed in 2024, and healthcare organizations slashed services across the state.
Idaho has a lot to offer RNs, from small-town charm, reasonable cost of living, and gorgeous landscapes that make it one of the top 10 fastest-growing states in the nation. Collaboration between industry leaders and nursing programs is focused on finding creative solutions to boost the nursing workforce in Idaho.
A version of this article first appeared on Medscape.com.
During their 12-hour shifts, registered nurses (RNs) in Arizona and Arkansas perform many of the same tasks as RNs in Wisconsin and Wyoming: Assessing patients, monitoring vital signs, administering medications, and charting records to provide the best patient care. The work might be similar, but there are vast differences in the number of RNs in each state.
In states like Idaho, Utah, and Nevada, which have the lowest number of nurses per capita, there are as few as 7 nurses per 1000 residents, compared with South Dakota and the District of Columbia, which have double the number of nurses than underserved states — giving them the highest number of nurses per capita.
Even states with the largest number of nurses per capita are not immune to the nursing shortage. The National Bureau of Labor Statistics estimates that there will be 195,400 job openings for RNs from 2021 to 2031.
So, what makes it easier for some states to recruit and retain RNs than others?
States With the Highest Number of Nurses Per Capita
South Dakota
RNs per 1000 residents: 15.79
Average wage: $67,030 or $32.23 per hour
Average rent in Sioux Falls: $1192 per month
The Midwestern state has more miles of shoreline than Florida, herds of wild buffalo, the highest summit east of the Rockies, and more nurses per capita than all other states . Healthcare is one of the major industries in the Mount Rushmore State.
Haifa Abou Samra, dean and professor at the University of South Dakota School of Health Sciences, Vermillion, isn’t surprised that RNs want to call the state home.
“South Dakota is a nice place to live,” Samra said. “[The] schools are wonderful. If people are growing families, there is support; neighbors support their neighbors, and it’s a relatively safe community.”
South Dakota has 19 approved nursing education programs that graduated 878 RNs in 2022. Scholarships and student loan forgiveness programs have helped attract qualified RNs, and collaborations between education and industry have been instrumental in addressing the nursing shortage, Samra told this news organization.
Even though RNs earn less than the median wage ($87,070 per year/41.38 per hour), South Dakota has a low cost of living and no individual income tax, which helps stretch those earnings.
District of Columbia
RNs per 1000 residents: 15.39
Average wage: $105,220 or $50.59 per hour
Average rent in Washington, DC: $2485 per month
After a shift at some of the top-ranking hospitals in the nation, RNs working in the compact capital region can explore museums, monuments, and cultural sites; walk along the banks of the Potomac River; or grab a bite at award-winning restaurants.
Washington, a top-ranking metro area because of its growth, high wages, and access to economic opportunities, is also home to several top-tier hospitals and some of the best healthcare in the nation, and RNs who want to pursue continuing education have access to top-tier universities.
Nurses in Washington, DC, might make some of the highest wages in the nation, but the region also has the second-highest cost of living in the United States, with average rents topping $2400 per month and an average home price of $594,337.
North Dakota
RNs per 1000 residents: 12.99
Average wage: $74,930 or $36.03 per hour
Average rent in Fargo: $1051 per month
North Dakota projects a 10.4% increase in employment for RNs, which is higher than the national average, and the state has implemented several strategies to address chronic nursing shortages. The Nurse Staffing Clearinghouse connects nursing school graduates with local employers and created a statewide nursing staffing pool for in-state recruitment of travel nurses.
But it’s not just plentiful job opportunities and a low cost of living that attract nurses to the Peace Garden State. The state and its largest cities, Bismarck and Fargo, hold several “best of” accolades, including nods for the safest places to live and among the Best Places to Raise a Family, giving it high marks for quality of life.
Sure, the winters are cold, but the outdoor recreation can’t be beaten. RNs can bundle up and see the bighorn sheep in the Badlands at Theodore Roosevelt National Park or explore expansive terrain for skiing, snowboarding, and snowmobile trails.
States With the Lowest Number of Nurses Per Capita
Nevada
RNs per 1000 residents: 7.92
Average wage: $96,201 or $46.25 per hour
Average rent in Las Vegas: $1478 per month
Despite a projected 23% job growth for RNs between 2020 and 2030, the state has struggled to fill open positions. It might be the higher-than-average cost of living (9.7% higher than the US average) or higher-than-average crime rates that make RNs reluctant to gamble on a job in the Silver State. But there are some big wins for nurses in the state.
Salaries are higher than the national average, there is no state income tax, and some of the lowest property taxes in the nation. Thanks to new legislation, RNs with student loan debt won’t have to bet on black at the casino to make their payments. The Health Equity and Loan Assistance Program is a new initiative that offers up to $120,000 in loan repayment assistance to providers, including RNs, who commit to working in underserved and rural areas across the state for 5 years.
The state also has incredible attractions, from the neon lights and over-the-top architecture in Las Vegas to iconic red rock canyons, stunning state parks, and landmarks like Hoover Dam and Lake Tahoe.
Utah
RNs per 1000 residents: 7.05
Average wage: $79,790 or $38.36 per hour
Average rent in Salt Lake City: $1611 per month
Healthcare is one of the biggest employers in Utah, and nurses are the most in-demand healthcare workers in the state. But below-average wages and a cost of living that is a whopping 28% higher than the national average could be some reasons that the Beehive State is struggling to attract nurses.
A high number of job vacancies mean higher patient-to-nurse ratios, creating additional stress for a workforce prone to burnout. Much of the state is rural, public transportation is inadequate, and poor air quality causes frequent haze and smog.
The challenges are offset by some big benefits: Utah has been ranked as the “best state” thanks to the strong economy, infrastructure, and quality education — and it doesn’t hurt that Utah is home to myriad outdoor recreation opportunities and the stunning scenery at landmarks like Bryce Canyon and Arches National Park.
Moreover, Utah is hustling to boost its RN workforce. The University of Utah, Salt Lake City, has increased enrollment by 25% and hired additional faculty to help boost the nursing workforce — and those who work in hospitals and health clinics across the state benefit from a flat 4.55% individual income tax rate.
Idaho
RNs per 1000 residents: 7.02
Average wage: $80,130 or $38.53 per hour
Average rent in Boise: $1646 per month
Although the nursing workforce in Idaho has increased, it still ranks as the lowest in the nation. Teresa Stanfill, DNP, RN, executive director for the Idaho Center for Nursing, said that the number of new nurses is too low to replace the number of retiring nurses.
The state introduced loan repayment programs that award up to $25,000 to cover student loan debt, and hospitals and health systems often offer sign-on bonuses and relocation packages to attract RNs. But long winters, an isolated location, and limited cultural options can make it harder to attract nurses to the state.
It’s easier to recruit RNs to suburban areas like Boise, Meridian, and Nampa, but rural parts of the state struggle, Stanfill added. The nursing shortage is among the reasons that 11 hospitals and emergency departments closed in 2024, and healthcare organizations slashed services across the state.
Idaho has a lot to offer RNs, from small-town charm, reasonable cost of living, and gorgeous landscapes that make it one of the top 10 fastest-growing states in the nation. Collaboration between industry leaders and nursing programs is focused on finding creative solutions to boost the nursing workforce in Idaho.
A version of this article first appeared on Medscape.com.
During their 12-hour shifts, registered nurses (RNs) in Arizona and Arkansas perform many of the same tasks as RNs in Wisconsin and Wyoming: Assessing patients, monitoring vital signs, administering medications, and charting records to provide the best patient care. The work might be similar, but there are vast differences in the number of RNs in each state.
In states like Idaho, Utah, and Nevada, which have the lowest number of nurses per capita, there are as few as 7 nurses per 1000 residents, compared with South Dakota and the District of Columbia, which have double the number of nurses than underserved states — giving them the highest number of nurses per capita.
Even states with the largest number of nurses per capita are not immune to the nursing shortage. The National Bureau of Labor Statistics estimates that there will be 195,400 job openings for RNs from 2021 to 2031.
So, what makes it easier for some states to recruit and retain RNs than others?
States With the Highest Number of Nurses Per Capita
South Dakota
RNs per 1000 residents: 15.79
Average wage: $67,030 or $32.23 per hour
Average rent in Sioux Falls: $1192 per month
The Midwestern state has more miles of shoreline than Florida, herds of wild buffalo, the highest summit east of the Rockies, and more nurses per capita than all other states . Healthcare is one of the major industries in the Mount Rushmore State.
Haifa Abou Samra, dean and professor at the University of South Dakota School of Health Sciences, Vermillion, isn’t surprised that RNs want to call the state home.
“South Dakota is a nice place to live,” Samra said. “[The] schools are wonderful. If people are growing families, there is support; neighbors support their neighbors, and it’s a relatively safe community.”
South Dakota has 19 approved nursing education programs that graduated 878 RNs in 2022. Scholarships and student loan forgiveness programs have helped attract qualified RNs, and collaborations between education and industry have been instrumental in addressing the nursing shortage, Samra told this news organization.
Even though RNs earn less than the median wage ($87,070 per year/41.38 per hour), South Dakota has a low cost of living and no individual income tax, which helps stretch those earnings.
District of Columbia
RNs per 1000 residents: 15.39
Average wage: $105,220 or $50.59 per hour
Average rent in Washington, DC: $2485 per month
After a shift at some of the top-ranking hospitals in the nation, RNs working in the compact capital region can explore museums, monuments, and cultural sites; walk along the banks of the Potomac River; or grab a bite at award-winning restaurants.
Washington, a top-ranking metro area because of its growth, high wages, and access to economic opportunities, is also home to several top-tier hospitals and some of the best healthcare in the nation, and RNs who want to pursue continuing education have access to top-tier universities.
Nurses in Washington, DC, might make some of the highest wages in the nation, but the region also has the second-highest cost of living in the United States, with average rents topping $2400 per month and an average home price of $594,337.
North Dakota
RNs per 1000 residents: 12.99
Average wage: $74,930 or $36.03 per hour
Average rent in Fargo: $1051 per month
North Dakota projects a 10.4% increase in employment for RNs, which is higher than the national average, and the state has implemented several strategies to address chronic nursing shortages. The Nurse Staffing Clearinghouse connects nursing school graduates with local employers and created a statewide nursing staffing pool for in-state recruitment of travel nurses.
But it’s not just plentiful job opportunities and a low cost of living that attract nurses to the Peace Garden State. The state and its largest cities, Bismarck and Fargo, hold several “best of” accolades, including nods for the safest places to live and among the Best Places to Raise a Family, giving it high marks for quality of life.
Sure, the winters are cold, but the outdoor recreation can’t be beaten. RNs can bundle up and see the bighorn sheep in the Badlands at Theodore Roosevelt National Park or explore expansive terrain for skiing, snowboarding, and snowmobile trails.
States With the Lowest Number of Nurses Per Capita
Nevada
RNs per 1000 residents: 7.92
Average wage: $96,201 or $46.25 per hour
Average rent in Las Vegas: $1478 per month
Despite a projected 23% job growth for RNs between 2020 and 2030, the state has struggled to fill open positions. It might be the higher-than-average cost of living (9.7% higher than the US average) or higher-than-average crime rates that make RNs reluctant to gamble on a job in the Silver State. But there are some big wins for nurses in the state.
Salaries are higher than the national average, there is no state income tax, and some of the lowest property taxes in the nation. Thanks to new legislation, RNs with student loan debt won’t have to bet on black at the casino to make their payments. The Health Equity and Loan Assistance Program is a new initiative that offers up to $120,000 in loan repayment assistance to providers, including RNs, who commit to working in underserved and rural areas across the state for 5 years.
The state also has incredible attractions, from the neon lights and over-the-top architecture in Las Vegas to iconic red rock canyons, stunning state parks, and landmarks like Hoover Dam and Lake Tahoe.
Utah
RNs per 1000 residents: 7.05
Average wage: $79,790 or $38.36 per hour
Average rent in Salt Lake City: $1611 per month
Healthcare is one of the biggest employers in Utah, and nurses are the most in-demand healthcare workers in the state. But below-average wages and a cost of living that is a whopping 28% higher than the national average could be some reasons that the Beehive State is struggling to attract nurses.
A high number of job vacancies mean higher patient-to-nurse ratios, creating additional stress for a workforce prone to burnout. Much of the state is rural, public transportation is inadequate, and poor air quality causes frequent haze and smog.
The challenges are offset by some big benefits: Utah has been ranked as the “best state” thanks to the strong economy, infrastructure, and quality education — and it doesn’t hurt that Utah is home to myriad outdoor recreation opportunities and the stunning scenery at landmarks like Bryce Canyon and Arches National Park.
Moreover, Utah is hustling to boost its RN workforce. The University of Utah, Salt Lake City, has increased enrollment by 25% and hired additional faculty to help boost the nursing workforce — and those who work in hospitals and health clinics across the state benefit from a flat 4.55% individual income tax rate.
Idaho
RNs per 1000 residents: 7.02
Average wage: $80,130 or $38.53 per hour
Average rent in Boise: $1646 per month
Although the nursing workforce in Idaho has increased, it still ranks as the lowest in the nation. Teresa Stanfill, DNP, RN, executive director for the Idaho Center for Nursing, said that the number of new nurses is too low to replace the number of retiring nurses.
The state introduced loan repayment programs that award up to $25,000 to cover student loan debt, and hospitals and health systems often offer sign-on bonuses and relocation packages to attract RNs. But long winters, an isolated location, and limited cultural options can make it harder to attract nurses to the state.
It’s easier to recruit RNs to suburban areas like Boise, Meridian, and Nampa, but rural parts of the state struggle, Stanfill added. The nursing shortage is among the reasons that 11 hospitals and emergency departments closed in 2024, and healthcare organizations slashed services across the state.
Idaho has a lot to offer RNs, from small-town charm, reasonable cost of living, and gorgeous landscapes that make it one of the top 10 fastest-growing states in the nation. Collaboration between industry leaders and nursing programs is focused on finding creative solutions to boost the nursing workforce in Idaho.
A version of this article first appeared on Medscape.com.
A Systematic Review of Dermatologic Findings in Adults With Hemophagocytic Lymphohistiocytosis
A Systematic Review of Dermatologic Findings in Adults With Hemophagocytic Lymphohistiocytosis
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening immunologic phenomenon characterized by a systemic inflammatory response syndrome—like clinical picture with additional features, including hepatosplenomegaly, hyperferritinemia, and increased natural killer cell activity. Clinical manifestations of HLH often are nonspecific, making HLH diagnosis challenging. High persistent fever is a key feature of HLH; patients also may report gastrointestinal distress, lethargy, and/or widespread rash.1
Hemophagocytic lymphohistiocytosis is believed to stem from inherited defects in several genes, such as perforin (PRF1), as well as immune dysregulation due to infections, rheumatologic diseases, hematologic malignancies, or drug reactions.2 The primary mechanism of HLH is hypothesized to be driven by aberrant immune activation, interferon gamma released from CD8+ T cells, and uncontrolled phagocytosis by activated macrophages. The cytokine cascade results in tissue injury and multiorgan dysfunction.3,4
Although HLH historically has been categorized as primary (familial) or secondary (acquired), the most recent guidelines suggest the etiology is not always binary.3,5 That said, the concept of secondary causes is useful in understanding risk factors for developing HLH. Both forms of the disease are thought to be elicited by a trigger (eg, infection), even when inherited genetic mutations exist.6 The primary form commonly affects the pediatric population,4,6-8 whereas the secondary form is more common in adults.7
Several sets of diagnostic criteria for HLH have been developed, the most well-known being the HLH-2004 criteria.1,3 The HLH-2009 modified criteria were developed after further evidence provided a refined sense of how the HLH-2004 criteria should be stratified.9 Finally, Fardet et al10 presented the HScore as an estimation of likelihood of diagnosis of HLH. These sets of HLH criteria include clinical and laboratory features that demonstrate inflammation, natrual killer cell activity, hemophagocytosis, end-organ damage, and cell lineage effects. The HScore differs from the other sets of HLH criteria in that it is designed to estimate an individual patient’s risk of having reactive hemophagocytic syndrome, which likely is equivalent to secondary HLH, although the authors do not use this exact terminology.10
While these criteria provide a framework for diagnosing HLH, they may fail to distinguish between HLH disease and HLH disease mimics, a concept described by the North American Consortium for Histiocytosis that may impact the success of immunosuppressive treatment.3 Individuals with HLH syndrome meet the aforementioned diagnostic criteria; HLH syndrome is further divided into HLH disease and HLH disease mimics (Figure 1). The “disease” label describes the traditional concept of HLH, driven by aberrant immune overactivation, in which patients benefit from immunosuppression. In contrast, HLH mimics include a subset of patients who meet the HLH criteria but are unlikely to benefit from immunosuppression because the primary mechanism driving their condition is not owed to immune overactivation, as is the case with HLH disease. Examples of HLH mimics include certain infections, such as Epstein-Barr virus (EBV), that may demonstrate clinical findings consistent with HLH but would not benefit from immunosuppression. Ironically, infections (including EBV) also are known triggers of HLH disease, making this concept difficult to understand and adopt. In this study, we refer to HLH disease simply as HLH.
Although cutaneous manifestations of HLH are not included in the diagnostic criteria, skin findings are common and may coincide with the severity and progression of the disease.11 Despite the fact that HLH can manifest with rash,1 comprehensive reviews of reported cutaneous findings in adult HLH are lacking. Thus, the goal of this study was to provide an organized characterization of reported cutaneous findings in adults with HLH and context for how the dermatologic examination may support the diagnosis or uncover the underlying etiology of this condition.
Methods
A search of PubMed articles indexed for MEDLINE using the phrase (cutaneous OR dermatologic OR skin) findings) AND hemophagocytic lymphohistiocytosis performed on September 20, 2023, yielded 423 results (Figure 2). Filters to exclude non–English language publications and pediatric populations were applied, resulting in 161 articles. Other exclusion criteria included the absence of a description of dermatologic findings. Seventy-five articles remained after screening titles and abstracts, and full-text review yielded 55 articles that were deemed appropriate for inclusion in the study. Subsequent reference searches and use of the online resource Litmaps revealed 45 additional publications that underwent full-text screening; of these articles, 5 were included in the final review.
Results
Sixty studies were included in this systematic review.5,7,11-68 The reported prevalence of skin findings among patients with HLH from the included retrospective studies ranged from 15% to 85%.12-15 Several literature reviews reported similarly varied prevalence among adult patients with HLH.7,16 Fardet et al14 categorized cutaneous manifestations of HLH into 3 types: direct manifestations of HLH not explained by systemic features (eg, generalized maculopapular eruption), indirect manifestations of HLH that are explained by systemic features of the disease (eg, purpura due to HLH-induced coagulopathy), and findings specific to the underlying etiology of HLH (eg, malar rash seen in systemic lupus erythematosus [SLE]–associated HLH). This categorization served as the outline for the results below, providing an organized review of cutaneous findings and context for how they may support the diagnosis or uncover the underlying etiology of HLH.
Category I: Direct Manifestations of HLH
Several articles reported cutaneous findings that seemed to be the direct result of HLH and not attributed to an underlying trigger or sequalae of HLH.11,14,16-31 The most common descriptions were a generalized, morbilliform, or nonspecific eruption that encompasses large areas of the skin, commonly the trunk and extremities, sometimes extending to the face and scalp.14,16-23,25,31,32 There were variations in secondary features such as pruritus and tenderness; some studies also described violaceous discoloration in addition to erythema.16,23
Other skin findings thought to be a direct result of HLH were described in detail by Zerah and DeWitt11 in their retrospective study, including pyoderma gangrenosum, panniculitis, Stevens-Johnson syndrome, atypical targetoid lesions, and bullous eruptions. The authors also analyzed dermatopathologic data that ultimately revealed that pathologic analysis was largely inconsistent and nondescript.11 There was a single case report of purpura fulminans arising alongside signs and symptoms of HLH,26 and several case reports described Sweet syndrome developing around the same time as HLH.27-29 Lastly, Collins et al30 described a case of HLH manifesting with violaceous ulcerating papules and nodules scattered across the legs, abdomen, and arms. Biopsy of this patient’s lesions showed a diffuse dermal infiltrate of histiocytes and hemophagocytosis.
Category II: Secondary Complications and Sequelae of HLH
This was the smallest group among the 3 categories, comprising a few case reports and retrospective cohort studies primarily reporting jaundice/icterus and hemorrhagic lesions such as purpura, petechiae, and scleral hemorrhage.11,21,23,33-35 Several literature reviews described these conditions as nonspecific findings in HLH.16,20 The cause of jaundice in HLH likely can be attributed to its characteristic hepatic dysfunction, whereas hemorrhagic lesions likely are the result of both hepatic and bone marrow dysfunction resulting in coagulopathy.
Category III: Manifestations of Underlying Etiology or Triggers of HLH
Infectious—Infection is known to be one of the most common triggers of HLH, with several retrospective studies reporting infectious triggers in approximately 20% of cases.13,15 Although many pathogens have been implicated, only a few of these infection-induced HLH reports described cutaneous findings, which included a case of varicella zoster virus, Escherichia coli necrotizing fasciitis, leprosy, EBV reactivation, parvovirus B19, and both focal and disseminated herpes simplex virus 2.36-42 Most of these patients presented with classic findings of each disease. The case of varicella zoster virus exhibited pruritic erythematous papules on the face, trunk, and limbs.36 The necrotizing fasciitis case presented with tender erythematous swelling of the lower extremity.37 The patient with leprosy exhibited leonine facies and numerous erythematous nodules, plaques, and superficial ulcerating plaques over the trunk and limbs with palmoplantar involvement,39 and both cases of herpes simplex virus 2 reported small bullae either diffusely over the face, trunk, and extremities or over the genitalia.38,40 Interestingly, the cases of parvovirus B19 and EBV reactivation both exhibited polyarteritis nodosa and occurred in patients with underlying autoimmune conditions, raising the question of whether these cases of HLH had a single trigger or were the result of the overall immunologic dysregulation induced by both infection and autoimmunity.41,42
Rheumatologic—Several articles reported dermatologic findings associated with macrophage activation syndrome, a term that often is used to describe HLH associated with autoimmune conditions. Cases of HLH in adult-onset Still disease, dermatomyositis, polyarteritis nodosa, and SLE described skin findings characteristic of the underlying rheumatologic disease, sometimes with acutely worse dermatologic findings at the time of HLH presentation.35,41-48 With regard to SLE, the acute manifestation of classic findings of the disease with HLH has sometimes been described as acute lupus hemophagocytic syndrome (HPS).48 Lambotte at al48 described common findings of acute lupus hemophagocytic syndrome in their retrospective study as malar rash, weight loss, polyarthralgia, and nephritis in addition to classic HLH findings including fever, lymphadenopathy, and hepatosplenomegaly. Many other rheumatologic conditions have been associated with HLH, including rheumatoid arthritis, mixed connective tissue disease, systemic sclerosis, and Sjögren disease. All these conditions can have dermatologic manifestations; however, no descriptions of dermatologic findings in cases of HLH associated with these diseases were found.13
Malignancy—Several cases of malignancy-induced HLH described cutaneous findings, the majority being cutaneous lymphomas, namely subcutaneous panniculitis-like T-cell lymphoma (SPTCL). Other less commonly reported malignancies in this group included Kaposi sarcoma, intravascular lymphoma, Sézary syndrome, mycosis fungoides, cutaneous diffuse large B-cell lymphoma, and several subtypes of primary cutaneous T-cell lymphoma.2,32,49-60 The most common description of SPTCL included multiple scattered plaques and subcutaneous nodules, some associated with tenderness, induration, drainage, or hemorrhagic features.32,50,52,55,57,60 Cases of mycosis fungoides and Sézary syndrome presented with variations in size and distribution of erythroderma with associated lymphadenopathy.2 A unique case of HLH developing in a patient with intravascular lymphoma described an eruption of multiple telangiectasias and petechial hemorrhages on the trunk,58 while one case associated with primary cutaneous anaplastic large cell lymphoma presented with a rapidly enlarging tumor with central ulceration and eschar.59
Drug Induced—Interestingly, most of the drug-induced cases of HLH identified in our search were secondary to biologic therapies used in the treatment of metastatic melanoma, specifically the immune checkpoint inhibitors (ICIs), which have been reported to have an association with HLH in prior literature reviews.61-65 Choi et al66 described an interesting case of ICI-induced HLH presenting with a concurrent severe lichenoid drug eruption that progressed from a pruritic truncal rash to mucocutaneous bullae, erosions, and desquamation resembling a Stevens-Johnson syndrome–type picture. This patient had treatment-refractory, HIV-negative Kaposi sarcoma, where the underlying immunologic dysregulation may explain the more severe cutaneous presentation not observed in other reported cases of ICI-induced HLH.
Yang et al’s67 review of 23 cases with concurrent diagnoses of HLH and DIHS found that 61% (14/23) of cases were diagnosed initially as DIHS before failing treatment and receiving a diagnosis of HLH several weeks later. Additionally, the authors found that several cases met criteria for one diagnosis while clinically presenting strongly for the other.67 This overlap in clinical presentation also was demonstrated in Zerah and DeWitt’s11 retrospective study regarding cutaneous findings in HLH, in which several of the morbilliform eruptions thought to be contributed to HLH ultimately were decided to be drug reactions.
Comment
Regarding direct (or primary) cutaneous findings in HLH (category I), there seem to be 2 groups of features associated with the onset of HLH that are not related to its characteristic hepatic dysfunction (category II) nor its underlying triggers (category III): a nonspecific, generalized, erythematous eruption; and dermatologic conditions separate from HLH itself (eg, Sweet syndrome, pyoderma gangrenosum). Whether the latter group truly is a direct manifestation of HLH is difficult to discern with the evidence available. Nevertheless, we can conclude that there is some type of association between these dermatologic diseases and HLH, and this association can serve as both a diagnostic tool for clinicians and a point of interest for further clinical research.
The relatively low number of articles identified through our systematic review that specifically reported secondary findings, such as jaundice or coagulopathy-associated hemorrhagic lesions, may lead one to believe that these are not common findings in HLH; however, it is possible that these are not regularly reported in the literature simply because these findings are nonspecific and can be considered expected results of the characteristic organ dysfunction in HLH.
As suspected, the skin findings in category III were the most broad given the variety of underlying etiologies that have been associated with HLH. Like the other 2 categories, these skin findings generally are nonspecific to HLH; however, the ones in category III are specific to underlying etiology of HLH and may aid in identifying and treating the underlying cause of a patient’s HLH when indicated.
Most of the rheumatologic diseases seem to have been known at the time of HLH development and diagnosis, which may highlight the importance of considering a diagnosis of HLH early on in patients with known autoimmune disease and systemic signs of illness or acutely worsening signs and symptoms of their underlying autoimmune disease.
Interestingly, several cases of malignancy-associated HLH reported signs and symptoms of HLH at initial presentation of the malignant disease.32,50,59 This situation seems to be somewhat common, as Go and Wester’s68 systematic analysis of 156 patients with SPTCL found HLH was the presenting feature in 37% of patients included in their study. This may call attention to the importance of considering cutaneous lymphomas as the cause of skin lesions in patients with signs and symptoms of HLH, where it may be easy to assume that skin findings are a result of their systemic disease.
In highlighting cases of HLH related to medication use, we found it pertinent to include and discuss the complex relationship between drug-induced hypersensitivity syndrome (DIHS [formerly known as drug rash with eosinophilia and systemic symptoms [DRESS] syndrome) and HLH. The results of this study suggest that DIHS may have considerable clinical overlap with HLH11 and may even lead to development of HLH,67 creating difficulty in distinguishing between these conditions where there may be similar findings, such as cutaneous eruptions, fever, and hepatic or other internal organ involvement. We agree with Yang et al67 that there can be large overlap in symptomology between these two conditions and that more investigation is necessary to explore the relationship between them.
Conclusion
Diagnosis of HLH in adults continues to be challenging, with several diagnostic tools but no true gold standard. In addition to the nonspecific symptomology, there is a myriad of cutaneous findings that can be present in adults with HLH (eTable), all of which are also nonspecific. Even so, awareness of which dermatologic findings have been associated with HLH may provide a cue to consider HLH in the systemically ill patient with a notable dermatologic examination. Furthermore, there are several avenues for further investigation that can be drawn, including further dermatologic analysis among nonspecific eruptions attributed to HLH, clinical and pathologic differentiation between DIHS/DRESS and HLH, and correlation between severity of skin manifestations and severity of HLH disease.

Limitations of this study included a lack of clarity in diagnosis of HLH in patients described in the included articles, as some reports use variable terminology (HLH vs hemophagocytic syndrome vs macrophage activation syndrome, etc), and it is impossible to know if all authors used the same diagnostic criteria—or any validated diagnostic criteria—unless specifically stated. Additionally, including case reports in our study limited the amount and quality of information described in each report. Despite its limitations, this systematic review outlines the cutaneous manifestations associated with HLH. These data will promote clinical awareness of this complex condition and allow for consideration of HLH in patients meeting criteria for HLH syndrome. More studies ultimately are needed to differentiate HLH from its mimics.
- Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124-131. doi:10.1002/pbc.21039
- Blom A, Beylot-Barry M, D’Incan M, et al. Lymphoma-associated hemophagocytic syndrome (LAHS) in advanced-stage mycosis fungoides/ Sézary syndrome cutaneous T-cell lymphoma. J Am Acad Dermatol. 2011;65:404-410. doi:10.1016/j.jaad.2010.05.029
- Jordan MB, Allen CE, Greenberg J, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer. 2019;66:e27929. doi:10.1002/pbc.27929
- Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: an update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. 2020;34:101515. doi:10.1016/j.berh.2020.101515
- Tomasini D, Berti E. Subcutaneous panniculitis-like T-cell lymphoma. G Ital Dermatol Venereol. 2013;148:395-411.
- Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672-2681. doi:10.1182/blood-2016-01-690636
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, et al. Adult haemophagocytic syndrome. Lancet. 2014;383:1503-1516. doi:10.1016/s0140-6736(13)61048-x
- Sieni E, Cetica V, Piccin A, et al. Familial hemophagocytic lymphohistiocytosis may present during adulthood: clinical and genetic features of a small series. PLoS One. 2012;7:e44649. doi:10.1371/journal.pone.0044649
- Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology. 2009:127-131. doi:10.1182 /asheducation-2009.1.127
- Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66:2613-2620. doi:10.1002/art.38690
- Zerah ML, DeWitt CA. Cutaneous findings in hemophagocytic lymphohistiocytosis. Dermatology. 2015;230:234-243. doi:10.1159/000368552
- Fardet L, Galicier L, Vignon-Pennamen MD, et al. Frequency, clinical features and prognosis of cutaneous manifestations in adult patients with reactive haemophagocytic syndrome. Br J Dermatol. 2010;162:547-553. doi:10.1111/j.1365-2133.2009.09549.x
- Dhote R, Simon J, Papo T, et al. Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum. 2003;49:633-639. doi:10.1002/art.11368
- Li J, Wang Q, Zheng W, et al. Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine (Baltimore). 2014;93:100-105. doi:10.1097/md.0000000000000022
- Tudesq JJ, Valade S, Galicier L, et al. Diagnostic strategy for trigger identification in severe reactive hemophagocytic lymphohistiocytosis: a diagnostic accuracy study. Hematol Oncol. 2021;39:114-122. doi:10.1002 /hon.2819
- Sakai H, Otsubo S, Miura T, et al. Hemophagocytic syndrome presenting with a facial erythema in a patient with systemic lupus erythematosus. J Am Acad Dermatol. 2007;57(5 Suppl):S111-S114. doi:10.1016/j .jaad.2006.11.024
- Chung SM, Song JY, Kim W, et al. Dengue-associated hemophagocytic lymphohistiocytosis in an adult: a case report and literature review. Medicine (Baltimore). 2017;96:e6159. doi:10.1097/md.0000000000006159
- Esmaili H, Rahmani O, Fouladi RF. Hemophagocytic syndrome in patients with unexplained cytopenia: report of 15 cases. Turk Patoloji Derg. 2013;29:15-18. doi:10.5146/tjpath.2013.01142
- Jiwnani S, Karimundackal G, Kulkarni A, et al. Hemophagocytic syndrome complicating lung resection. Asian Cardiovasc Thorac Ann. 2012;20:341-343. doi:10.1177/0218492311435686
- Lee WJ, Lee DW, Kim CH, et al. Dermatopathic lymphadenitis with generalized erythroderma in a patient with Epstein-Barr virusassociated hemophagocytic lymphohistiocytosis. Am J Dermatopathol. 2010;32:357-361. doi:10.1097/DAD.0b013e3181b2a50f
- Lovisari F, Terzi V, Lippi MG, et al. Hemophagocytic lymphohistiocytosis complicated by multiorgan failure: a case report. Medicine (Baltimore). 2017;96:e9198. doi:10.1097/md.0000000000009198
- Miechowiecki J, Stainer W, Wallner G, et al. Severe complication during remission of Crohn’s disease: hemophagocytic lymphohistiocytosis due to acute cytomegalovirus infection. Z Gastroenterol. 2018;56:259-263. doi:10.1055/s-0043-123999
- Ochoa S, Cheng K, Fleury CM, et al. A 28-year-old woman with fever, rash, and pancytopenia. Allergy Asthma Proc. 2017;38:322-327. doi:10.2500/aap.2017.38.4042
- Tokoro S, Namiki T, Miura K, et al. Chronic active Epstein-Barr virus infection with cutaneous lymphoproliferation: haemophagocytosis in the skin and haemophagocytic syndrome. J Eur Acad Dermatol Venereol. 2018;32:e116-e117. doi:10.1111/jdv.14640
- Tzeng HE, Teng CL, Yang Y, et al. Occult subcutaneous panniculitislike T-cell lymphoma with initial presentations of cellulitis-like skin lesion and fulminant hemophagocytosis. J Formos Med Assoc. 2007;106 (2 Suppl):S55-S59. doi:10.1016/s0929-6646(09)60354-5
- Honjo O, Kubo T, Sugaya F, et al. Severe cytokine release syndrome resulting in purpura fulminans despite successful response to nivolumab therapy in a patient with pleomorphic carcinoma of the lung: a case report. J Immunother Cancer. 2019;7:97. doi:10.1186/s40425- 019-0582-4
- Kao RL, Jacobsen AA, Billington CJ Jr, et al. A case of VEXAS syndrome associated with EBV-associated hemophagocytic lymphohistiocytosis. Blood Cells Mol Dis. 2022;93:102636. doi:10.1016/j .bcmd.2021.102636
- Koga T, Takano K, Horai Y, et al. Sweet’s syndrome complicated by Kikuchi’s disease and hemophagocytic syndrome which presented with retinoic acid-inducible gene-I in both the skin epidermal basal layer and the cervical lymph nodes. Intern Med. 2013;52:1839-1843. doi:10.2169 /internalmedicine.52.9542
- Lin WL, Lin WC, Chiu CS, et al. Paraneoplastic Sweet’s syndrome in a patient with hemophagocytic syndrome. Int J Dermatol. 2008;3:305-307.
- Collins MK, Ho J, Akilov OE. Case 52. A unique presentation of hemophagocytic lymphohistiocytosis with ulcerating papulonodules. In: Akilov OE, ed. Cutaneous Lymphomas: Unusual Cases 3. Springer International Publishing; 2021:126-127.
- Chakrapani A, Avery A, Warnke R. Primary cutaneous gamma delta T-cell lymphoma with brain involvement and hemophagocytic syndrome. Am J Dermatopathol. 2013;35:270-272. doi:10.1097 /DAD.0b013e3182624e98
- Sullivan C, Loghmani A, Thomas K, et al. Hemophagocytic lymphohistiocytosis as the initial presentation of subcutaneous panniculitis-like T-cell lymphoma: a rare case responding to cyclosporine A and steroids. J Investig Med High Impact Case Rep. 2020;8:2324709620981531. doi:10.1177/2324709620981531
- Darmawan G, Salido EO, Concepcion ML, et al. Hemophagocytic lymphohistiocytosis: “a dreadful mimic.” Int J Rheum Dis. 2015; 18:810-812. doi:10.1111/1756-185x.12506
- Maus MV, Leick MB, Cornejo KM, et al. Case 35-2019: a 66-year-old man with pancytopenia and rash. N Engl J Med. 2019;381:1951-1960. doi:10.1056/NEJMcpc1909627
- Chamseddin B, Marks E, Dominguez A, et al. Refractory macrophage activation syndrome in the setting of adult-onset Still disease with hemophagocytic lymphohistiocytosis detected on skin biopsy treated with canakinumab and tacrolimus. J Cutan Pathol. 2019;46:528-531. doi:10.1111/cup.13466
- Bérar A, Ardois S, Walter-Moraux P, et al. Primary varicella-zoster virus infection of the immunocompromised associated with acute pancreatitis and hemophagocytic lymphohistiocytosis: a case report. Medicine (Baltimore). 2021;100:e25351. doi:10.1097 /md.0000000000025351
- Chang CC, Hsiao PJ, Chiu CC, et al. Catastrophic hemophagocytic lymphohistiocytosis in a young man with nephrotic syndrome. Clin Chim Acta. 2015;439:168-171. doi:10.1016/j.cca.2014.10.025
- Kurosawa S, Sekiya N, Fukushima K, et al. Unusual manifestation of disseminated herpes simplex virus type 2 infection associated with pharyngotonsilitis, esophagitis, and hemophagocytic lymphohisitocytosis without genital involvement. BMC Infect Dis. 2019;19:65. doi:10.1186/s12879-019-3721-0
- Saidi W, Gammoudi R, Korbi M, et al. Hemophagocytic lymphohistiocytosis: an unusual complication of leprosy. Int J Dermatol. 2015;54: 1054-1059. doi:10.1111/ijd.12792
- Yamaguchi K, Yamamoto A, Hisano M, et al. Herpes simplex virus 2-associated hemophagocytic lymphohistiocytosis in a pregnant patient. Obstet Gynecol. 2005;105(5 Pt 2):1241-1244. doi:10.1097 /01.AOG.0000157757.54948.9b
- Hayakawa I, Shirasaki F, Ikeda H, et al. Reactive hemophagocytic syndrome in a patient with polyarteritis nodosa associated with Epstein- Barr virus reactivation. Rheumatol Int. 2006;26:573-576. doi:10.1007 /s00296-005-0024-0
- Jeong JY, Park JY, Ham JY, et al. Molecular evidence of parvovirus B19 in the cutaneous polyarteritis nodosa tissue from a patient with parvovirus-associated hemophagocytic syndrome: case report. Medicine (Baltimore). 2020;99:e22079. doi:10.1097 /md.0000000000022079
- Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478. doi:10.2169 /internalmedicine.1121-18
- Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune- associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246. doi:10 .1080/03009742.2019.1653493
- Jung SY. Hemophagocytic syndrome diagnosed by liver biopsy in a female patient with systemic lupus erythematosus. J Clin Rheumatol. 2013;19:449-451. doi:10.1097/rhu.0000000000000040
- Kerl K, Wolf IH, Cerroni L, et al. Hemophagocytosis in cutaneous autoimmune disease. Am J Dermatopathol. 2015;37:539-543. doi:10.1097 /dad.0000000000000166
- Komiya Y, Saito T, Mizoguchi F, et al. Hemophagocytic syndrome complicated with dermatomyositis controlled successfully with infliximab and conventional therapies. Intern Med. 2017;56:3237-3241. doi:10.2169 /internalmedicine.7966-16
- Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and long-term outcome of 15 episodes of systemic lupus erythematosusassociated hemophagocytic syndrome. Medicine (Baltimore). 2006;85: 169-182. doi:10.1097/01.md.0000224708.62510.d1
- Guitart J, Mangold AR, Martinez-Escala ME, et al. Clinical and pathological characteristics and outcomes among patients with subcutaneous panniculitis-like T-cell lymphoma and related adipotropic lymphoproliferative disorders. JAMA Dermatol. 2022;158:1167-1174. doi:10.1001/jamadermatol.2022.3347
- Hung GD, Chen YH, Chen DY, et al. Subcutaneous panniculitis-like T-cell lymphoma presenting with hemophagocytic lymphohistiocytosis and skin lesions with characteristic high-resolution ultrasonographic findings. Clin Rheumatol. 2007;26:775-778. doi:10.1007/s10067 -005-0193-y
- Jamil A, Nadzri N, Harun N, et al. Primary cutaneous diffuse large B-cell lymphoma leg type presenting with hemophagocytic syndrome. J Am Acad Dermatol. 2012;67:e222-3. doi:10.1016/j.jaad.2012.04.021
- LeBlanc RE, Lansigan F. Unraveling subcutaneous panniculitis-like T-cell lymphoma: an association between subcutaneous panniculitislike T-cell lymphoma, autoimmune lymphoproliferative syndrome, and familial hemophagocytic lymphohistiocytosis. J Cutan Pathol. 2021;48:572-577. doi:10.1111/cup.13863
- Lee DE, Martinez-Escala ME, Serrano LM, et al. Hemophagocytic lymphohistiocytosis in cutaneous T-cell lymphoma. JAMA Dermatol. 2018;154:828-831. doi:10.1001/jamadermatol.2018.1264
- Maejima H, Tanei R, Morioka T, et al. Haemophagocytosis-related intravascular large B-cell lymphoma associated with skin eruption. Acta Derm Venereol. 2011;91:339-340. doi:10.2340/00015555-0981
- Mody A, Cherry D, Georgescu G, et al. A rare case of subcutaneous panniculitis-like T cell lymphoma with hemophagocytic lymphohistiocytosis mimicking cellulitis. Am J Case Rep. 2021;22:E927142. doi:10.12659/ajcr.927142
- Park YJ, Bae HJ, Chang JY, et al. Development of Kaposi sarcoma and hemophagocytic lymphohistiocytosis associated with human herpesvirus 8 in a renal transplant recipient. Korean J Intern Med. 2017;4:750-752.
- Phatak S, Gupta L, Aggarwal A. A young woman with panniculitis and cytopenia who later developed coagulopathy. J Assoc Physicians India. 2016;64:65-67.
- Pongpairoj K, Rerknimitr P, Wititsuwannakul J, et al. Eruptive telangiectasia in a patient with fever and haemophagocytic syndrome. Clin Exp Dermatol. 2016;41:696-698. doi:10.1111/ced.12859
- Shimizu Y, Tanae K, Takahashi N, et al. Primary cutaneous anaplastic large-cell lymphoma presenting with hemophagocytic syndrome: a case report and review of the literature. Leuk Res. 2010;34:263-266. doi:10.1016/j.leukres.2009.07.001
- Sirka CS, Pradhan S, Patra S, et al. Hemophagocytic lymphohistiocytosis: a rare, potentially fatal complication in subcutaneous panniculitis like T cell lymphoma. Indian J Dermatol Venereol Leprol. 2019;5:481-485.
- Chin CK, Hall S, Green C, et al. Secondary haemophagocytic lymphohistiocytosis due to checkpoint inhibitor therapy. Eur J Cancer. 2019;115: 84-87. doi:10.1016/j.ejca.2019.04.026
- Dudda M, Mann C, Heinz J, et al. Hemophagocytic lymphohistiocytosis of a melanoma patient under BRAF/MEK-inhibitor therapy following anti-PD1 inhibitor treatment: a case report and review to the literature. Melanoma Res. 2021;31:81-84. doi:10.1097 /cmr.0000000000000703
- Mizuta H, Nakano E, Takahashi A, et al. Hemophagocytic lymphohistiocytosis with advanced malignant melanoma accompanied by ipilimumab and nivolumab: a case report and literature review. Dermatol Ther. 2020;33:e13321. doi:10.1111/dth.13321
- Satzger I, Ivanyi P, Länger F, et al. Treatment-related hemophagocytic lymphohistiocytosis secondary to checkpoint inhibition with nivolumab plus ipilimumab. Eur J Cancer. 2018;93:150-153. doi:10.1016/j.ejca.2018.01.063
- Michot JM, Lazarovici J, Tieu A, et al. Haematological immune-related adverse events with immune checkpoint inhibitors, how to manage? Eur J Cancer. 2019;122:72-90. doi:10.1016/J.EJCA.2019.07.014
- Choi S, Zhou M, Bahrani E, et al. Rare and fatal complication of immune checkpoint inhibition: a case report of haemophagocytic lymphohistiocytosis with severe lichenoid dermatitis. Br J Haematol. 2021;193:e44-e47. doi:10.1111/BJH.17442
- Yang JJ, Lei DK, Ravi V, et al. Overlap between hemophagocytic lymphohistiocytosis and drug reaction and eosinophilia with systemic symptoms: a review. Int J Dermatol. 2021;60:925-932. doi:10.1111 /ijd.15196
- Go RS, Wester SM. Immunophenotypic and molecular features, clinical outcomes, treatments, and prognostic factors associated with subcutaneous panniculitis-like T-cell lymphoma: a systematic analysis of 156 patients reported in the literature. Cancer. 2004;101:1404-1413. doi:10.1002/cncr.20502
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening immunologic phenomenon characterized by a systemic inflammatory response syndrome—like clinical picture with additional features, including hepatosplenomegaly, hyperferritinemia, and increased natural killer cell activity. Clinical manifestations of HLH often are nonspecific, making HLH diagnosis challenging. High persistent fever is a key feature of HLH; patients also may report gastrointestinal distress, lethargy, and/or widespread rash.1
Hemophagocytic lymphohistiocytosis is believed to stem from inherited defects in several genes, such as perforin (PRF1), as well as immune dysregulation due to infections, rheumatologic diseases, hematologic malignancies, or drug reactions.2 The primary mechanism of HLH is hypothesized to be driven by aberrant immune activation, interferon gamma released from CD8+ T cells, and uncontrolled phagocytosis by activated macrophages. The cytokine cascade results in tissue injury and multiorgan dysfunction.3,4
Although HLH historically has been categorized as primary (familial) or secondary (acquired), the most recent guidelines suggest the etiology is not always binary.3,5 That said, the concept of secondary causes is useful in understanding risk factors for developing HLH. Both forms of the disease are thought to be elicited by a trigger (eg, infection), even when inherited genetic mutations exist.6 The primary form commonly affects the pediatric population,4,6-8 whereas the secondary form is more common in adults.7
Several sets of diagnostic criteria for HLH have been developed, the most well-known being the HLH-2004 criteria.1,3 The HLH-2009 modified criteria were developed after further evidence provided a refined sense of how the HLH-2004 criteria should be stratified.9 Finally, Fardet et al10 presented the HScore as an estimation of likelihood of diagnosis of HLH. These sets of HLH criteria include clinical and laboratory features that demonstrate inflammation, natrual killer cell activity, hemophagocytosis, end-organ damage, and cell lineage effects. The HScore differs from the other sets of HLH criteria in that it is designed to estimate an individual patient’s risk of having reactive hemophagocytic syndrome, which likely is equivalent to secondary HLH, although the authors do not use this exact terminology.10
While these criteria provide a framework for diagnosing HLH, they may fail to distinguish between HLH disease and HLH disease mimics, a concept described by the North American Consortium for Histiocytosis that may impact the success of immunosuppressive treatment.3 Individuals with HLH syndrome meet the aforementioned diagnostic criteria; HLH syndrome is further divided into HLH disease and HLH disease mimics (Figure 1). The “disease” label describes the traditional concept of HLH, driven by aberrant immune overactivation, in which patients benefit from immunosuppression. In contrast, HLH mimics include a subset of patients who meet the HLH criteria but are unlikely to benefit from immunosuppression because the primary mechanism driving their condition is not owed to immune overactivation, as is the case with HLH disease. Examples of HLH mimics include certain infections, such as Epstein-Barr virus (EBV), that may demonstrate clinical findings consistent with HLH but would not benefit from immunosuppression. Ironically, infections (including EBV) also are known triggers of HLH disease, making this concept difficult to understand and adopt. In this study, we refer to HLH disease simply as HLH.
Although cutaneous manifestations of HLH are not included in the diagnostic criteria, skin findings are common and may coincide with the severity and progression of the disease.11 Despite the fact that HLH can manifest with rash,1 comprehensive reviews of reported cutaneous findings in adult HLH are lacking. Thus, the goal of this study was to provide an organized characterization of reported cutaneous findings in adults with HLH and context for how the dermatologic examination may support the diagnosis or uncover the underlying etiology of this condition.
Methods
A search of PubMed articles indexed for MEDLINE using the phrase (cutaneous OR dermatologic OR skin) findings) AND hemophagocytic lymphohistiocytosis performed on September 20, 2023, yielded 423 results (Figure 2). Filters to exclude non–English language publications and pediatric populations were applied, resulting in 161 articles. Other exclusion criteria included the absence of a description of dermatologic findings. Seventy-five articles remained after screening titles and abstracts, and full-text review yielded 55 articles that were deemed appropriate for inclusion in the study. Subsequent reference searches and use of the online resource Litmaps revealed 45 additional publications that underwent full-text screening; of these articles, 5 were included in the final review.
Results
Sixty studies were included in this systematic review.5,7,11-68 The reported prevalence of skin findings among patients with HLH from the included retrospective studies ranged from 15% to 85%.12-15 Several literature reviews reported similarly varied prevalence among adult patients with HLH.7,16 Fardet et al14 categorized cutaneous manifestations of HLH into 3 types: direct manifestations of HLH not explained by systemic features (eg, generalized maculopapular eruption), indirect manifestations of HLH that are explained by systemic features of the disease (eg, purpura due to HLH-induced coagulopathy), and findings specific to the underlying etiology of HLH (eg, malar rash seen in systemic lupus erythematosus [SLE]–associated HLH). This categorization served as the outline for the results below, providing an organized review of cutaneous findings and context for how they may support the diagnosis or uncover the underlying etiology of HLH.
Category I: Direct Manifestations of HLH
Several articles reported cutaneous findings that seemed to be the direct result of HLH and not attributed to an underlying trigger or sequalae of HLH.11,14,16-31 The most common descriptions were a generalized, morbilliform, or nonspecific eruption that encompasses large areas of the skin, commonly the trunk and extremities, sometimes extending to the face and scalp.14,16-23,25,31,32 There were variations in secondary features such as pruritus and tenderness; some studies also described violaceous discoloration in addition to erythema.16,23
Other skin findings thought to be a direct result of HLH were described in detail by Zerah and DeWitt11 in their retrospective study, including pyoderma gangrenosum, panniculitis, Stevens-Johnson syndrome, atypical targetoid lesions, and bullous eruptions. The authors also analyzed dermatopathologic data that ultimately revealed that pathologic analysis was largely inconsistent and nondescript.11 There was a single case report of purpura fulminans arising alongside signs and symptoms of HLH,26 and several case reports described Sweet syndrome developing around the same time as HLH.27-29 Lastly, Collins et al30 described a case of HLH manifesting with violaceous ulcerating papules and nodules scattered across the legs, abdomen, and arms. Biopsy of this patient’s lesions showed a diffuse dermal infiltrate of histiocytes and hemophagocytosis.
Category II: Secondary Complications and Sequelae of HLH
This was the smallest group among the 3 categories, comprising a few case reports and retrospective cohort studies primarily reporting jaundice/icterus and hemorrhagic lesions such as purpura, petechiae, and scleral hemorrhage.11,21,23,33-35 Several literature reviews described these conditions as nonspecific findings in HLH.16,20 The cause of jaundice in HLH likely can be attributed to its characteristic hepatic dysfunction, whereas hemorrhagic lesions likely are the result of both hepatic and bone marrow dysfunction resulting in coagulopathy.
Category III: Manifestations of Underlying Etiology or Triggers of HLH
Infectious—Infection is known to be one of the most common triggers of HLH, with several retrospective studies reporting infectious triggers in approximately 20% of cases.13,15 Although many pathogens have been implicated, only a few of these infection-induced HLH reports described cutaneous findings, which included a case of varicella zoster virus, Escherichia coli necrotizing fasciitis, leprosy, EBV reactivation, parvovirus B19, and both focal and disseminated herpes simplex virus 2.36-42 Most of these patients presented with classic findings of each disease. The case of varicella zoster virus exhibited pruritic erythematous papules on the face, trunk, and limbs.36 The necrotizing fasciitis case presented with tender erythematous swelling of the lower extremity.37 The patient with leprosy exhibited leonine facies and numerous erythematous nodules, plaques, and superficial ulcerating plaques over the trunk and limbs with palmoplantar involvement,39 and both cases of herpes simplex virus 2 reported small bullae either diffusely over the face, trunk, and extremities or over the genitalia.38,40 Interestingly, the cases of parvovirus B19 and EBV reactivation both exhibited polyarteritis nodosa and occurred in patients with underlying autoimmune conditions, raising the question of whether these cases of HLH had a single trigger or were the result of the overall immunologic dysregulation induced by both infection and autoimmunity.41,42
Rheumatologic—Several articles reported dermatologic findings associated with macrophage activation syndrome, a term that often is used to describe HLH associated with autoimmune conditions. Cases of HLH in adult-onset Still disease, dermatomyositis, polyarteritis nodosa, and SLE described skin findings characteristic of the underlying rheumatologic disease, sometimes with acutely worse dermatologic findings at the time of HLH presentation.35,41-48 With regard to SLE, the acute manifestation of classic findings of the disease with HLH has sometimes been described as acute lupus hemophagocytic syndrome (HPS).48 Lambotte at al48 described common findings of acute lupus hemophagocytic syndrome in their retrospective study as malar rash, weight loss, polyarthralgia, and nephritis in addition to classic HLH findings including fever, lymphadenopathy, and hepatosplenomegaly. Many other rheumatologic conditions have been associated with HLH, including rheumatoid arthritis, mixed connective tissue disease, systemic sclerosis, and Sjögren disease. All these conditions can have dermatologic manifestations; however, no descriptions of dermatologic findings in cases of HLH associated with these diseases were found.13
Malignancy—Several cases of malignancy-induced HLH described cutaneous findings, the majority being cutaneous lymphomas, namely subcutaneous panniculitis-like T-cell lymphoma (SPTCL). Other less commonly reported malignancies in this group included Kaposi sarcoma, intravascular lymphoma, Sézary syndrome, mycosis fungoides, cutaneous diffuse large B-cell lymphoma, and several subtypes of primary cutaneous T-cell lymphoma.2,32,49-60 The most common description of SPTCL included multiple scattered plaques and subcutaneous nodules, some associated with tenderness, induration, drainage, or hemorrhagic features.32,50,52,55,57,60 Cases of mycosis fungoides and Sézary syndrome presented with variations in size and distribution of erythroderma with associated lymphadenopathy.2 A unique case of HLH developing in a patient with intravascular lymphoma described an eruption of multiple telangiectasias and petechial hemorrhages on the trunk,58 while one case associated with primary cutaneous anaplastic large cell lymphoma presented with a rapidly enlarging tumor with central ulceration and eschar.59
Drug Induced—Interestingly, most of the drug-induced cases of HLH identified in our search were secondary to biologic therapies used in the treatment of metastatic melanoma, specifically the immune checkpoint inhibitors (ICIs), which have been reported to have an association with HLH in prior literature reviews.61-65 Choi et al66 described an interesting case of ICI-induced HLH presenting with a concurrent severe lichenoid drug eruption that progressed from a pruritic truncal rash to mucocutaneous bullae, erosions, and desquamation resembling a Stevens-Johnson syndrome–type picture. This patient had treatment-refractory, HIV-negative Kaposi sarcoma, where the underlying immunologic dysregulation may explain the more severe cutaneous presentation not observed in other reported cases of ICI-induced HLH.
Yang et al’s67 review of 23 cases with concurrent diagnoses of HLH and DIHS found that 61% (14/23) of cases were diagnosed initially as DIHS before failing treatment and receiving a diagnosis of HLH several weeks later. Additionally, the authors found that several cases met criteria for one diagnosis while clinically presenting strongly for the other.67 This overlap in clinical presentation also was demonstrated in Zerah and DeWitt’s11 retrospective study regarding cutaneous findings in HLH, in which several of the morbilliform eruptions thought to be contributed to HLH ultimately were decided to be drug reactions.
Comment
Regarding direct (or primary) cutaneous findings in HLH (category I), there seem to be 2 groups of features associated with the onset of HLH that are not related to its characteristic hepatic dysfunction (category II) nor its underlying triggers (category III): a nonspecific, generalized, erythematous eruption; and dermatologic conditions separate from HLH itself (eg, Sweet syndrome, pyoderma gangrenosum). Whether the latter group truly is a direct manifestation of HLH is difficult to discern with the evidence available. Nevertheless, we can conclude that there is some type of association between these dermatologic diseases and HLH, and this association can serve as both a diagnostic tool for clinicians and a point of interest for further clinical research.
The relatively low number of articles identified through our systematic review that specifically reported secondary findings, such as jaundice or coagulopathy-associated hemorrhagic lesions, may lead one to believe that these are not common findings in HLH; however, it is possible that these are not regularly reported in the literature simply because these findings are nonspecific and can be considered expected results of the characteristic organ dysfunction in HLH.
As suspected, the skin findings in category III were the most broad given the variety of underlying etiologies that have been associated with HLH. Like the other 2 categories, these skin findings generally are nonspecific to HLH; however, the ones in category III are specific to underlying etiology of HLH and may aid in identifying and treating the underlying cause of a patient’s HLH when indicated.
Most of the rheumatologic diseases seem to have been known at the time of HLH development and diagnosis, which may highlight the importance of considering a diagnosis of HLH early on in patients with known autoimmune disease and systemic signs of illness or acutely worsening signs and symptoms of their underlying autoimmune disease.
Interestingly, several cases of malignancy-associated HLH reported signs and symptoms of HLH at initial presentation of the malignant disease.32,50,59 This situation seems to be somewhat common, as Go and Wester’s68 systematic analysis of 156 patients with SPTCL found HLH was the presenting feature in 37% of patients included in their study. This may call attention to the importance of considering cutaneous lymphomas as the cause of skin lesions in patients with signs and symptoms of HLH, where it may be easy to assume that skin findings are a result of their systemic disease.
In highlighting cases of HLH related to medication use, we found it pertinent to include and discuss the complex relationship between drug-induced hypersensitivity syndrome (DIHS [formerly known as drug rash with eosinophilia and systemic symptoms [DRESS] syndrome) and HLH. The results of this study suggest that DIHS may have considerable clinical overlap with HLH11 and may even lead to development of HLH,67 creating difficulty in distinguishing between these conditions where there may be similar findings, such as cutaneous eruptions, fever, and hepatic or other internal organ involvement. We agree with Yang et al67 that there can be large overlap in symptomology between these two conditions and that more investigation is necessary to explore the relationship between them.
Conclusion
Diagnosis of HLH in adults continues to be challenging, with several diagnostic tools but no true gold standard. In addition to the nonspecific symptomology, there is a myriad of cutaneous findings that can be present in adults with HLH (eTable), all of which are also nonspecific. Even so, awareness of which dermatologic findings have been associated with HLH may provide a cue to consider HLH in the systemically ill patient with a notable dermatologic examination. Furthermore, there are several avenues for further investigation that can be drawn, including further dermatologic analysis among nonspecific eruptions attributed to HLH, clinical and pathologic differentiation between DIHS/DRESS and HLH, and correlation between severity of skin manifestations and severity of HLH disease.
Limitations of this study included a lack of clarity in diagnosis of HLH in patients described in the included articles, as some reports use variable terminology (HLH vs hemophagocytic syndrome vs macrophage activation syndrome, etc), and it is impossible to know if all authors used the same diagnostic criteria—or any validated diagnostic criteria—unless specifically stated. Additionally, including case reports in our study limited the amount and quality of information described in each report. Despite its limitations, this systematic review outlines the cutaneous manifestations associated with HLH. These data will promote clinical awareness of this complex condition and allow for consideration of HLH in patients meeting criteria for HLH syndrome. More studies ultimately are needed to differentiate HLH from its mimics.
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening immunologic phenomenon characterized by a systemic inflammatory response syndrome—like clinical picture with additional features, including hepatosplenomegaly, hyperferritinemia, and increased natural killer cell activity. Clinical manifestations of HLH often are nonspecific, making HLH diagnosis challenging. High persistent fever is a key feature of HLH; patients also may report gastrointestinal distress, lethargy, and/or widespread rash.1
Hemophagocytic lymphohistiocytosis is believed to stem from inherited defects in several genes, such as perforin (PRF1), as well as immune dysregulation due to infections, rheumatologic diseases, hematologic malignancies, or drug reactions.2 The primary mechanism of HLH is hypothesized to be driven by aberrant immune activation, interferon gamma released from CD8+ T cells, and uncontrolled phagocytosis by activated macrophages. The cytokine cascade results in tissue injury and multiorgan dysfunction.3,4
Although HLH historically has been categorized as primary (familial) or secondary (acquired), the most recent guidelines suggest the etiology is not always binary.3,5 That said, the concept of secondary causes is useful in understanding risk factors for developing HLH. Both forms of the disease are thought to be elicited by a trigger (eg, infection), even when inherited genetic mutations exist.6 The primary form commonly affects the pediatric population,4,6-8 whereas the secondary form is more common in adults.7
Several sets of diagnostic criteria for HLH have been developed, the most well-known being the HLH-2004 criteria.1,3 The HLH-2009 modified criteria were developed after further evidence provided a refined sense of how the HLH-2004 criteria should be stratified.9 Finally, Fardet et al10 presented the HScore as an estimation of likelihood of diagnosis of HLH. These sets of HLH criteria include clinical and laboratory features that demonstrate inflammation, natrual killer cell activity, hemophagocytosis, end-organ damage, and cell lineage effects. The HScore differs from the other sets of HLH criteria in that it is designed to estimate an individual patient’s risk of having reactive hemophagocytic syndrome, which likely is equivalent to secondary HLH, although the authors do not use this exact terminology.10
While these criteria provide a framework for diagnosing HLH, they may fail to distinguish between HLH disease and HLH disease mimics, a concept described by the North American Consortium for Histiocytosis that may impact the success of immunosuppressive treatment.3 Individuals with HLH syndrome meet the aforementioned diagnostic criteria; HLH syndrome is further divided into HLH disease and HLH disease mimics (Figure 1). The “disease” label describes the traditional concept of HLH, driven by aberrant immune overactivation, in which patients benefit from immunosuppression. In contrast, HLH mimics include a subset of patients who meet the HLH criteria but are unlikely to benefit from immunosuppression because the primary mechanism driving their condition is not owed to immune overactivation, as is the case with HLH disease. Examples of HLH mimics include certain infections, such as Epstein-Barr virus (EBV), that may demonstrate clinical findings consistent with HLH but would not benefit from immunosuppression. Ironically, infections (including EBV) also are known triggers of HLH disease, making this concept difficult to understand and adopt. In this study, we refer to HLH disease simply as HLH.
Although cutaneous manifestations of HLH are not included in the diagnostic criteria, skin findings are common and may coincide with the severity and progression of the disease.11 Despite the fact that HLH can manifest with rash,1 comprehensive reviews of reported cutaneous findings in adult HLH are lacking. Thus, the goal of this study was to provide an organized characterization of reported cutaneous findings in adults with HLH and context for how the dermatologic examination may support the diagnosis or uncover the underlying etiology of this condition.
Methods
A search of PubMed articles indexed for MEDLINE using the phrase (cutaneous OR dermatologic OR skin) findings) AND hemophagocytic lymphohistiocytosis performed on September 20, 2023, yielded 423 results (Figure 2). Filters to exclude non–English language publications and pediatric populations were applied, resulting in 161 articles. Other exclusion criteria included the absence of a description of dermatologic findings. Seventy-five articles remained after screening titles and abstracts, and full-text review yielded 55 articles that were deemed appropriate for inclusion in the study. Subsequent reference searches and use of the online resource Litmaps revealed 45 additional publications that underwent full-text screening; of these articles, 5 were included in the final review.
Results
Sixty studies were included in this systematic review.5,7,11-68 The reported prevalence of skin findings among patients with HLH from the included retrospective studies ranged from 15% to 85%.12-15 Several literature reviews reported similarly varied prevalence among adult patients with HLH.7,16 Fardet et al14 categorized cutaneous manifestations of HLH into 3 types: direct manifestations of HLH not explained by systemic features (eg, generalized maculopapular eruption), indirect manifestations of HLH that are explained by systemic features of the disease (eg, purpura due to HLH-induced coagulopathy), and findings specific to the underlying etiology of HLH (eg, malar rash seen in systemic lupus erythematosus [SLE]–associated HLH). This categorization served as the outline for the results below, providing an organized review of cutaneous findings and context for how they may support the diagnosis or uncover the underlying etiology of HLH.
Category I: Direct Manifestations of HLH
Several articles reported cutaneous findings that seemed to be the direct result of HLH and not attributed to an underlying trigger or sequalae of HLH.11,14,16-31 The most common descriptions were a generalized, morbilliform, or nonspecific eruption that encompasses large areas of the skin, commonly the trunk and extremities, sometimes extending to the face and scalp.14,16-23,25,31,32 There were variations in secondary features such as pruritus and tenderness; some studies also described violaceous discoloration in addition to erythema.16,23
Other skin findings thought to be a direct result of HLH were described in detail by Zerah and DeWitt11 in their retrospective study, including pyoderma gangrenosum, panniculitis, Stevens-Johnson syndrome, atypical targetoid lesions, and bullous eruptions. The authors also analyzed dermatopathologic data that ultimately revealed that pathologic analysis was largely inconsistent and nondescript.11 There was a single case report of purpura fulminans arising alongside signs and symptoms of HLH,26 and several case reports described Sweet syndrome developing around the same time as HLH.27-29 Lastly, Collins et al30 described a case of HLH manifesting with violaceous ulcerating papules and nodules scattered across the legs, abdomen, and arms. Biopsy of this patient’s lesions showed a diffuse dermal infiltrate of histiocytes and hemophagocytosis.
Category II: Secondary Complications and Sequelae of HLH
This was the smallest group among the 3 categories, comprising a few case reports and retrospective cohort studies primarily reporting jaundice/icterus and hemorrhagic lesions such as purpura, petechiae, and scleral hemorrhage.11,21,23,33-35 Several literature reviews described these conditions as nonspecific findings in HLH.16,20 The cause of jaundice in HLH likely can be attributed to its characteristic hepatic dysfunction, whereas hemorrhagic lesions likely are the result of both hepatic and bone marrow dysfunction resulting in coagulopathy.
Category III: Manifestations of Underlying Etiology or Triggers of HLH
Infectious—Infection is known to be one of the most common triggers of HLH, with several retrospective studies reporting infectious triggers in approximately 20% of cases.13,15 Although many pathogens have been implicated, only a few of these infection-induced HLH reports described cutaneous findings, which included a case of varicella zoster virus, Escherichia coli necrotizing fasciitis, leprosy, EBV reactivation, parvovirus B19, and both focal and disseminated herpes simplex virus 2.36-42 Most of these patients presented with classic findings of each disease. The case of varicella zoster virus exhibited pruritic erythematous papules on the face, trunk, and limbs.36 The necrotizing fasciitis case presented with tender erythematous swelling of the lower extremity.37 The patient with leprosy exhibited leonine facies and numerous erythematous nodules, plaques, and superficial ulcerating plaques over the trunk and limbs with palmoplantar involvement,39 and both cases of herpes simplex virus 2 reported small bullae either diffusely over the face, trunk, and extremities or over the genitalia.38,40 Interestingly, the cases of parvovirus B19 and EBV reactivation both exhibited polyarteritis nodosa and occurred in patients with underlying autoimmune conditions, raising the question of whether these cases of HLH had a single trigger or were the result of the overall immunologic dysregulation induced by both infection and autoimmunity.41,42
Rheumatologic—Several articles reported dermatologic findings associated with macrophage activation syndrome, a term that often is used to describe HLH associated with autoimmune conditions. Cases of HLH in adult-onset Still disease, dermatomyositis, polyarteritis nodosa, and SLE described skin findings characteristic of the underlying rheumatologic disease, sometimes with acutely worse dermatologic findings at the time of HLH presentation.35,41-48 With regard to SLE, the acute manifestation of classic findings of the disease with HLH has sometimes been described as acute lupus hemophagocytic syndrome (HPS).48 Lambotte at al48 described common findings of acute lupus hemophagocytic syndrome in their retrospective study as malar rash, weight loss, polyarthralgia, and nephritis in addition to classic HLH findings including fever, lymphadenopathy, and hepatosplenomegaly. Many other rheumatologic conditions have been associated with HLH, including rheumatoid arthritis, mixed connective tissue disease, systemic sclerosis, and Sjögren disease. All these conditions can have dermatologic manifestations; however, no descriptions of dermatologic findings in cases of HLH associated with these diseases were found.13
Malignancy—Several cases of malignancy-induced HLH described cutaneous findings, the majority being cutaneous lymphomas, namely subcutaneous panniculitis-like T-cell lymphoma (SPTCL). Other less commonly reported malignancies in this group included Kaposi sarcoma, intravascular lymphoma, Sézary syndrome, mycosis fungoides, cutaneous diffuse large B-cell lymphoma, and several subtypes of primary cutaneous T-cell lymphoma.2,32,49-60 The most common description of SPTCL included multiple scattered plaques and subcutaneous nodules, some associated with tenderness, induration, drainage, or hemorrhagic features.32,50,52,55,57,60 Cases of mycosis fungoides and Sézary syndrome presented with variations in size and distribution of erythroderma with associated lymphadenopathy.2 A unique case of HLH developing in a patient with intravascular lymphoma described an eruption of multiple telangiectasias and petechial hemorrhages on the trunk,58 while one case associated with primary cutaneous anaplastic large cell lymphoma presented with a rapidly enlarging tumor with central ulceration and eschar.59
Drug Induced—Interestingly, most of the drug-induced cases of HLH identified in our search were secondary to biologic therapies used in the treatment of metastatic melanoma, specifically the immune checkpoint inhibitors (ICIs), which have been reported to have an association with HLH in prior literature reviews.61-65 Choi et al66 described an interesting case of ICI-induced HLH presenting with a concurrent severe lichenoid drug eruption that progressed from a pruritic truncal rash to mucocutaneous bullae, erosions, and desquamation resembling a Stevens-Johnson syndrome–type picture. This patient had treatment-refractory, HIV-negative Kaposi sarcoma, where the underlying immunologic dysregulation may explain the more severe cutaneous presentation not observed in other reported cases of ICI-induced HLH.
Yang et al’s67 review of 23 cases with concurrent diagnoses of HLH and DIHS found that 61% (14/23) of cases were diagnosed initially as DIHS before failing treatment and receiving a diagnosis of HLH several weeks later. Additionally, the authors found that several cases met criteria for one diagnosis while clinically presenting strongly for the other.67 This overlap in clinical presentation also was demonstrated in Zerah and DeWitt’s11 retrospective study regarding cutaneous findings in HLH, in which several of the morbilliform eruptions thought to be contributed to HLH ultimately were decided to be drug reactions.
Comment
Regarding direct (or primary) cutaneous findings in HLH (category I), there seem to be 2 groups of features associated with the onset of HLH that are not related to its characteristic hepatic dysfunction (category II) nor its underlying triggers (category III): a nonspecific, generalized, erythematous eruption; and dermatologic conditions separate from HLH itself (eg, Sweet syndrome, pyoderma gangrenosum). Whether the latter group truly is a direct manifestation of HLH is difficult to discern with the evidence available. Nevertheless, we can conclude that there is some type of association between these dermatologic diseases and HLH, and this association can serve as both a diagnostic tool for clinicians and a point of interest for further clinical research.
The relatively low number of articles identified through our systematic review that specifically reported secondary findings, such as jaundice or coagulopathy-associated hemorrhagic lesions, may lead one to believe that these are not common findings in HLH; however, it is possible that these are not regularly reported in the literature simply because these findings are nonspecific and can be considered expected results of the characteristic organ dysfunction in HLH.
As suspected, the skin findings in category III were the most broad given the variety of underlying etiologies that have been associated with HLH. Like the other 2 categories, these skin findings generally are nonspecific to HLH; however, the ones in category III are specific to underlying etiology of HLH and may aid in identifying and treating the underlying cause of a patient’s HLH when indicated.
Most of the rheumatologic diseases seem to have been known at the time of HLH development and diagnosis, which may highlight the importance of considering a diagnosis of HLH early on in patients with known autoimmune disease and systemic signs of illness or acutely worsening signs and symptoms of their underlying autoimmune disease.
Interestingly, several cases of malignancy-associated HLH reported signs and symptoms of HLH at initial presentation of the malignant disease.32,50,59 This situation seems to be somewhat common, as Go and Wester’s68 systematic analysis of 156 patients with SPTCL found HLH was the presenting feature in 37% of patients included in their study. This may call attention to the importance of considering cutaneous lymphomas as the cause of skin lesions in patients with signs and symptoms of HLH, where it may be easy to assume that skin findings are a result of their systemic disease.
In highlighting cases of HLH related to medication use, we found it pertinent to include and discuss the complex relationship between drug-induced hypersensitivity syndrome (DIHS [formerly known as drug rash with eosinophilia and systemic symptoms [DRESS] syndrome) and HLH. The results of this study suggest that DIHS may have considerable clinical overlap with HLH11 and may even lead to development of HLH,67 creating difficulty in distinguishing between these conditions where there may be similar findings, such as cutaneous eruptions, fever, and hepatic or other internal organ involvement. We agree with Yang et al67 that there can be large overlap in symptomology between these two conditions and that more investigation is necessary to explore the relationship between them.
Conclusion
Diagnosis of HLH in adults continues to be challenging, with several diagnostic tools but no true gold standard. In addition to the nonspecific symptomology, there is a myriad of cutaneous findings that can be present in adults with HLH (eTable), all of which are also nonspecific. Even so, awareness of which dermatologic findings have been associated with HLH may provide a cue to consider HLH in the systemically ill patient with a notable dermatologic examination. Furthermore, there are several avenues for further investigation that can be drawn, including further dermatologic analysis among nonspecific eruptions attributed to HLH, clinical and pathologic differentiation between DIHS/DRESS and HLH, and correlation between severity of skin manifestations and severity of HLH disease.
Limitations of this study included a lack of clarity in diagnosis of HLH in patients described in the included articles, as some reports use variable terminology (HLH vs hemophagocytic syndrome vs macrophage activation syndrome, etc), and it is impossible to know if all authors used the same diagnostic criteria—or any validated diagnostic criteria—unless specifically stated. Additionally, including case reports in our study limited the amount and quality of information described in each report. Despite its limitations, this systematic review outlines the cutaneous manifestations associated with HLH. These data will promote clinical awareness of this complex condition and allow for consideration of HLH in patients meeting criteria for HLH syndrome. More studies ultimately are needed to differentiate HLH from its mimics.
- Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124-131. doi:10.1002/pbc.21039
- Blom A, Beylot-Barry M, D’Incan M, et al. Lymphoma-associated hemophagocytic syndrome (LAHS) in advanced-stage mycosis fungoides/ Sézary syndrome cutaneous T-cell lymphoma. J Am Acad Dermatol. 2011;65:404-410. doi:10.1016/j.jaad.2010.05.029
- Jordan MB, Allen CE, Greenberg J, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer. 2019;66:e27929. doi:10.1002/pbc.27929
- Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: an update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. 2020;34:101515. doi:10.1016/j.berh.2020.101515
- Tomasini D, Berti E. Subcutaneous panniculitis-like T-cell lymphoma. G Ital Dermatol Venereol. 2013;148:395-411.
- Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672-2681. doi:10.1182/blood-2016-01-690636
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, et al. Adult haemophagocytic syndrome. Lancet. 2014;383:1503-1516. doi:10.1016/s0140-6736(13)61048-x
- Sieni E, Cetica V, Piccin A, et al. Familial hemophagocytic lymphohistiocytosis may present during adulthood: clinical and genetic features of a small series. PLoS One. 2012;7:e44649. doi:10.1371/journal.pone.0044649
- Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology. 2009:127-131. doi:10.1182 /asheducation-2009.1.127
- Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66:2613-2620. doi:10.1002/art.38690
- Zerah ML, DeWitt CA. Cutaneous findings in hemophagocytic lymphohistiocytosis. Dermatology. 2015;230:234-243. doi:10.1159/000368552
- Fardet L, Galicier L, Vignon-Pennamen MD, et al. Frequency, clinical features and prognosis of cutaneous manifestations in adult patients with reactive haemophagocytic syndrome. Br J Dermatol. 2010;162:547-553. doi:10.1111/j.1365-2133.2009.09549.x
- Dhote R, Simon J, Papo T, et al. Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum. 2003;49:633-639. doi:10.1002/art.11368
- Li J, Wang Q, Zheng W, et al. Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine (Baltimore). 2014;93:100-105. doi:10.1097/md.0000000000000022
- Tudesq JJ, Valade S, Galicier L, et al. Diagnostic strategy for trigger identification in severe reactive hemophagocytic lymphohistiocytosis: a diagnostic accuracy study. Hematol Oncol. 2021;39:114-122. doi:10.1002 /hon.2819
- Sakai H, Otsubo S, Miura T, et al. Hemophagocytic syndrome presenting with a facial erythema in a patient with systemic lupus erythematosus. J Am Acad Dermatol. 2007;57(5 Suppl):S111-S114. doi:10.1016/j .jaad.2006.11.024
- Chung SM, Song JY, Kim W, et al. Dengue-associated hemophagocytic lymphohistiocytosis in an adult: a case report and literature review. Medicine (Baltimore). 2017;96:e6159. doi:10.1097/md.0000000000006159
- Esmaili H, Rahmani O, Fouladi RF. Hemophagocytic syndrome in patients with unexplained cytopenia: report of 15 cases. Turk Patoloji Derg. 2013;29:15-18. doi:10.5146/tjpath.2013.01142
- Jiwnani S, Karimundackal G, Kulkarni A, et al. Hemophagocytic syndrome complicating lung resection. Asian Cardiovasc Thorac Ann. 2012;20:341-343. doi:10.1177/0218492311435686
- Lee WJ, Lee DW, Kim CH, et al. Dermatopathic lymphadenitis with generalized erythroderma in a patient with Epstein-Barr virusassociated hemophagocytic lymphohistiocytosis. Am J Dermatopathol. 2010;32:357-361. doi:10.1097/DAD.0b013e3181b2a50f
- Lovisari F, Terzi V, Lippi MG, et al. Hemophagocytic lymphohistiocytosis complicated by multiorgan failure: a case report. Medicine (Baltimore). 2017;96:e9198. doi:10.1097/md.0000000000009198
- Miechowiecki J, Stainer W, Wallner G, et al. Severe complication during remission of Crohn’s disease: hemophagocytic lymphohistiocytosis due to acute cytomegalovirus infection. Z Gastroenterol. 2018;56:259-263. doi:10.1055/s-0043-123999
- Ochoa S, Cheng K, Fleury CM, et al. A 28-year-old woman with fever, rash, and pancytopenia. Allergy Asthma Proc. 2017;38:322-327. doi:10.2500/aap.2017.38.4042
- Tokoro S, Namiki T, Miura K, et al. Chronic active Epstein-Barr virus infection with cutaneous lymphoproliferation: haemophagocytosis in the skin and haemophagocytic syndrome. J Eur Acad Dermatol Venereol. 2018;32:e116-e117. doi:10.1111/jdv.14640
- Tzeng HE, Teng CL, Yang Y, et al. Occult subcutaneous panniculitislike T-cell lymphoma with initial presentations of cellulitis-like skin lesion and fulminant hemophagocytosis. J Formos Med Assoc. 2007;106 (2 Suppl):S55-S59. doi:10.1016/s0929-6646(09)60354-5
- Honjo O, Kubo T, Sugaya F, et al. Severe cytokine release syndrome resulting in purpura fulminans despite successful response to nivolumab therapy in a patient with pleomorphic carcinoma of the lung: a case report. J Immunother Cancer. 2019;7:97. doi:10.1186/s40425- 019-0582-4
- Kao RL, Jacobsen AA, Billington CJ Jr, et al. A case of VEXAS syndrome associated with EBV-associated hemophagocytic lymphohistiocytosis. Blood Cells Mol Dis. 2022;93:102636. doi:10.1016/j .bcmd.2021.102636
- Koga T, Takano K, Horai Y, et al. Sweet’s syndrome complicated by Kikuchi’s disease and hemophagocytic syndrome which presented with retinoic acid-inducible gene-I in both the skin epidermal basal layer and the cervical lymph nodes. Intern Med. 2013;52:1839-1843. doi:10.2169 /internalmedicine.52.9542
- Lin WL, Lin WC, Chiu CS, et al. Paraneoplastic Sweet’s syndrome in a patient with hemophagocytic syndrome. Int J Dermatol. 2008;3:305-307.
- Collins MK, Ho J, Akilov OE. Case 52. A unique presentation of hemophagocytic lymphohistiocytosis with ulcerating papulonodules. In: Akilov OE, ed. Cutaneous Lymphomas: Unusual Cases 3. Springer International Publishing; 2021:126-127.
- Chakrapani A, Avery A, Warnke R. Primary cutaneous gamma delta T-cell lymphoma with brain involvement and hemophagocytic syndrome. Am J Dermatopathol. 2013;35:270-272. doi:10.1097 /DAD.0b013e3182624e98
- Sullivan C, Loghmani A, Thomas K, et al. Hemophagocytic lymphohistiocytosis as the initial presentation of subcutaneous panniculitis-like T-cell lymphoma: a rare case responding to cyclosporine A and steroids. J Investig Med High Impact Case Rep. 2020;8:2324709620981531. doi:10.1177/2324709620981531
- Darmawan G, Salido EO, Concepcion ML, et al. Hemophagocytic lymphohistiocytosis: “a dreadful mimic.” Int J Rheum Dis. 2015; 18:810-812. doi:10.1111/1756-185x.12506
- Maus MV, Leick MB, Cornejo KM, et al. Case 35-2019: a 66-year-old man with pancytopenia and rash. N Engl J Med. 2019;381:1951-1960. doi:10.1056/NEJMcpc1909627
- Chamseddin B, Marks E, Dominguez A, et al. Refractory macrophage activation syndrome in the setting of adult-onset Still disease with hemophagocytic lymphohistiocytosis detected on skin biopsy treated with canakinumab and tacrolimus. J Cutan Pathol. 2019;46:528-531. doi:10.1111/cup.13466
- Bérar A, Ardois S, Walter-Moraux P, et al. Primary varicella-zoster virus infection of the immunocompromised associated with acute pancreatitis and hemophagocytic lymphohistiocytosis: a case report. Medicine (Baltimore). 2021;100:e25351. doi:10.1097 /md.0000000000025351
- Chang CC, Hsiao PJ, Chiu CC, et al. Catastrophic hemophagocytic lymphohistiocytosis in a young man with nephrotic syndrome. Clin Chim Acta. 2015;439:168-171. doi:10.1016/j.cca.2014.10.025
- Kurosawa S, Sekiya N, Fukushima K, et al. Unusual manifestation of disseminated herpes simplex virus type 2 infection associated with pharyngotonsilitis, esophagitis, and hemophagocytic lymphohisitocytosis without genital involvement. BMC Infect Dis. 2019;19:65. doi:10.1186/s12879-019-3721-0
- Saidi W, Gammoudi R, Korbi M, et al. Hemophagocytic lymphohistiocytosis: an unusual complication of leprosy. Int J Dermatol. 2015;54: 1054-1059. doi:10.1111/ijd.12792
- Yamaguchi K, Yamamoto A, Hisano M, et al. Herpes simplex virus 2-associated hemophagocytic lymphohistiocytosis in a pregnant patient. Obstet Gynecol. 2005;105(5 Pt 2):1241-1244. doi:10.1097 /01.AOG.0000157757.54948.9b
- Hayakawa I, Shirasaki F, Ikeda H, et al. Reactive hemophagocytic syndrome in a patient with polyarteritis nodosa associated with Epstein- Barr virus reactivation. Rheumatol Int. 2006;26:573-576. doi:10.1007 /s00296-005-0024-0
- Jeong JY, Park JY, Ham JY, et al. Molecular evidence of parvovirus B19 in the cutaneous polyarteritis nodosa tissue from a patient with parvovirus-associated hemophagocytic syndrome: case report. Medicine (Baltimore). 2020;99:e22079. doi:10.1097 /md.0000000000022079
- Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478. doi:10.2169 /internalmedicine.1121-18
- Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune- associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246. doi:10 .1080/03009742.2019.1653493
- Jung SY. Hemophagocytic syndrome diagnosed by liver biopsy in a female patient with systemic lupus erythematosus. J Clin Rheumatol. 2013;19:449-451. doi:10.1097/rhu.0000000000000040
- Kerl K, Wolf IH, Cerroni L, et al. Hemophagocytosis in cutaneous autoimmune disease. Am J Dermatopathol. 2015;37:539-543. doi:10.1097 /dad.0000000000000166
- Komiya Y, Saito T, Mizoguchi F, et al. Hemophagocytic syndrome complicated with dermatomyositis controlled successfully with infliximab and conventional therapies. Intern Med. 2017;56:3237-3241. doi:10.2169 /internalmedicine.7966-16
- Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and long-term outcome of 15 episodes of systemic lupus erythematosusassociated hemophagocytic syndrome. Medicine (Baltimore). 2006;85: 169-182. doi:10.1097/01.md.0000224708.62510.d1
- Guitart J, Mangold AR, Martinez-Escala ME, et al. Clinical and pathological characteristics and outcomes among patients with subcutaneous panniculitis-like T-cell lymphoma and related adipotropic lymphoproliferative disorders. JAMA Dermatol. 2022;158:1167-1174. doi:10.1001/jamadermatol.2022.3347
- Hung GD, Chen YH, Chen DY, et al. Subcutaneous panniculitis-like T-cell lymphoma presenting with hemophagocytic lymphohistiocytosis and skin lesions with characteristic high-resolution ultrasonographic findings. Clin Rheumatol. 2007;26:775-778. doi:10.1007/s10067 -005-0193-y
- Jamil A, Nadzri N, Harun N, et al. Primary cutaneous diffuse large B-cell lymphoma leg type presenting with hemophagocytic syndrome. J Am Acad Dermatol. 2012;67:e222-3. doi:10.1016/j.jaad.2012.04.021
- LeBlanc RE, Lansigan F. Unraveling subcutaneous panniculitis-like T-cell lymphoma: an association between subcutaneous panniculitislike T-cell lymphoma, autoimmune lymphoproliferative syndrome, and familial hemophagocytic lymphohistiocytosis. J Cutan Pathol. 2021;48:572-577. doi:10.1111/cup.13863
- Lee DE, Martinez-Escala ME, Serrano LM, et al. Hemophagocytic lymphohistiocytosis in cutaneous T-cell lymphoma. JAMA Dermatol. 2018;154:828-831. doi:10.1001/jamadermatol.2018.1264
- Maejima H, Tanei R, Morioka T, et al. Haemophagocytosis-related intravascular large B-cell lymphoma associated with skin eruption. Acta Derm Venereol. 2011;91:339-340. doi:10.2340/00015555-0981
- Mody A, Cherry D, Georgescu G, et al. A rare case of subcutaneous panniculitis-like T cell lymphoma with hemophagocytic lymphohistiocytosis mimicking cellulitis. Am J Case Rep. 2021;22:E927142. doi:10.12659/ajcr.927142
- Park YJ, Bae HJ, Chang JY, et al. Development of Kaposi sarcoma and hemophagocytic lymphohistiocytosis associated with human herpesvirus 8 in a renal transplant recipient. Korean J Intern Med. 2017;4:750-752.
- Phatak S, Gupta L, Aggarwal A. A young woman with panniculitis and cytopenia who later developed coagulopathy. J Assoc Physicians India. 2016;64:65-67.
- Pongpairoj K, Rerknimitr P, Wititsuwannakul J, et al. Eruptive telangiectasia in a patient with fever and haemophagocytic syndrome. Clin Exp Dermatol. 2016;41:696-698. doi:10.1111/ced.12859
- Shimizu Y, Tanae K, Takahashi N, et al. Primary cutaneous anaplastic large-cell lymphoma presenting with hemophagocytic syndrome: a case report and review of the literature. Leuk Res. 2010;34:263-266. doi:10.1016/j.leukres.2009.07.001
- Sirka CS, Pradhan S, Patra S, et al. Hemophagocytic lymphohistiocytosis: a rare, potentially fatal complication in subcutaneous panniculitis like T cell lymphoma. Indian J Dermatol Venereol Leprol. 2019;5:481-485.
- Chin CK, Hall S, Green C, et al. Secondary haemophagocytic lymphohistiocytosis due to checkpoint inhibitor therapy. Eur J Cancer. 2019;115: 84-87. doi:10.1016/j.ejca.2019.04.026
- Dudda M, Mann C, Heinz J, et al. Hemophagocytic lymphohistiocytosis of a melanoma patient under BRAF/MEK-inhibitor therapy following anti-PD1 inhibitor treatment: a case report and review to the literature. Melanoma Res. 2021;31:81-84. doi:10.1097 /cmr.0000000000000703
- Mizuta H, Nakano E, Takahashi A, et al. Hemophagocytic lymphohistiocytosis with advanced malignant melanoma accompanied by ipilimumab and nivolumab: a case report and literature review. Dermatol Ther. 2020;33:e13321. doi:10.1111/dth.13321
- Satzger I, Ivanyi P, Länger F, et al. Treatment-related hemophagocytic lymphohistiocytosis secondary to checkpoint inhibition with nivolumab plus ipilimumab. Eur J Cancer. 2018;93:150-153. doi:10.1016/j.ejca.2018.01.063
- Michot JM, Lazarovici J, Tieu A, et al. Haematological immune-related adverse events with immune checkpoint inhibitors, how to manage? Eur J Cancer. 2019;122:72-90. doi:10.1016/J.EJCA.2019.07.014
- Choi S, Zhou M, Bahrani E, et al. Rare and fatal complication of immune checkpoint inhibition: a case report of haemophagocytic lymphohistiocytosis with severe lichenoid dermatitis. Br J Haematol. 2021;193:e44-e47. doi:10.1111/BJH.17442
- Yang JJ, Lei DK, Ravi V, et al. Overlap between hemophagocytic lymphohistiocytosis and drug reaction and eosinophilia with systemic symptoms: a review. Int J Dermatol. 2021;60:925-932. doi:10.1111 /ijd.15196
- Go RS, Wester SM. Immunophenotypic and molecular features, clinical outcomes, treatments, and prognostic factors associated with subcutaneous panniculitis-like T-cell lymphoma: a systematic analysis of 156 patients reported in the literature. Cancer. 2004;101:1404-1413. doi:10.1002/cncr.20502
- Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124-131. doi:10.1002/pbc.21039
- Blom A, Beylot-Barry M, D’Incan M, et al. Lymphoma-associated hemophagocytic syndrome (LAHS) in advanced-stage mycosis fungoides/ Sézary syndrome cutaneous T-cell lymphoma. J Am Acad Dermatol. 2011;65:404-410. doi:10.1016/j.jaad.2010.05.029
- Jordan MB, Allen CE, Greenberg J, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer. 2019;66:e27929. doi:10.1002/pbc.27929
- Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: an update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. 2020;34:101515. doi:10.1016/j.berh.2020.101515
- Tomasini D, Berti E. Subcutaneous panniculitis-like T-cell lymphoma. G Ital Dermatol Venereol. 2013;148:395-411.
- Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672-2681. doi:10.1182/blood-2016-01-690636
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, et al. Adult haemophagocytic syndrome. Lancet. 2014;383:1503-1516. doi:10.1016/s0140-6736(13)61048-x
- Sieni E, Cetica V, Piccin A, et al. Familial hemophagocytic lymphohistiocytosis may present during adulthood: clinical and genetic features of a small series. PLoS One. 2012;7:e44649. doi:10.1371/journal.pone.0044649
- Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology. 2009:127-131. doi:10.1182 /asheducation-2009.1.127
- Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66:2613-2620. doi:10.1002/art.38690
- Zerah ML, DeWitt CA. Cutaneous findings in hemophagocytic lymphohistiocytosis. Dermatology. 2015;230:234-243. doi:10.1159/000368552
- Fardet L, Galicier L, Vignon-Pennamen MD, et al. Frequency, clinical features and prognosis of cutaneous manifestations in adult patients with reactive haemophagocytic syndrome. Br J Dermatol. 2010;162:547-553. doi:10.1111/j.1365-2133.2009.09549.x
- Dhote R, Simon J, Papo T, et al. Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum. 2003;49:633-639. doi:10.1002/art.11368
- Li J, Wang Q, Zheng W, et al. Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine (Baltimore). 2014;93:100-105. doi:10.1097/md.0000000000000022
- Tudesq JJ, Valade S, Galicier L, et al. Diagnostic strategy for trigger identification in severe reactive hemophagocytic lymphohistiocytosis: a diagnostic accuracy study. Hematol Oncol. 2021;39:114-122. doi:10.1002 /hon.2819
- Sakai H, Otsubo S, Miura T, et al. Hemophagocytic syndrome presenting with a facial erythema in a patient with systemic lupus erythematosus. J Am Acad Dermatol. 2007;57(5 Suppl):S111-S114. doi:10.1016/j .jaad.2006.11.024
- Chung SM, Song JY, Kim W, et al. Dengue-associated hemophagocytic lymphohistiocytosis in an adult: a case report and literature review. Medicine (Baltimore). 2017;96:e6159. doi:10.1097/md.0000000000006159
- Esmaili H, Rahmani O, Fouladi RF. Hemophagocytic syndrome in patients with unexplained cytopenia: report of 15 cases. Turk Patoloji Derg. 2013;29:15-18. doi:10.5146/tjpath.2013.01142
- Jiwnani S, Karimundackal G, Kulkarni A, et al. Hemophagocytic syndrome complicating lung resection. Asian Cardiovasc Thorac Ann. 2012;20:341-343. doi:10.1177/0218492311435686
- Lee WJ, Lee DW, Kim CH, et al. Dermatopathic lymphadenitis with generalized erythroderma in a patient with Epstein-Barr virusassociated hemophagocytic lymphohistiocytosis. Am J Dermatopathol. 2010;32:357-361. doi:10.1097/DAD.0b013e3181b2a50f
- Lovisari F, Terzi V, Lippi MG, et al. Hemophagocytic lymphohistiocytosis complicated by multiorgan failure: a case report. Medicine (Baltimore). 2017;96:e9198. doi:10.1097/md.0000000000009198
- Miechowiecki J, Stainer W, Wallner G, et al. Severe complication during remission of Crohn’s disease: hemophagocytic lymphohistiocytosis due to acute cytomegalovirus infection. Z Gastroenterol. 2018;56:259-263. doi:10.1055/s-0043-123999
- Ochoa S, Cheng K, Fleury CM, et al. A 28-year-old woman with fever, rash, and pancytopenia. Allergy Asthma Proc. 2017;38:322-327. doi:10.2500/aap.2017.38.4042
- Tokoro S, Namiki T, Miura K, et al. Chronic active Epstein-Barr virus infection with cutaneous lymphoproliferation: haemophagocytosis in the skin and haemophagocytic syndrome. J Eur Acad Dermatol Venereol. 2018;32:e116-e117. doi:10.1111/jdv.14640
- Tzeng HE, Teng CL, Yang Y, et al. Occult subcutaneous panniculitislike T-cell lymphoma with initial presentations of cellulitis-like skin lesion and fulminant hemophagocytosis. J Formos Med Assoc. 2007;106 (2 Suppl):S55-S59. doi:10.1016/s0929-6646(09)60354-5
- Honjo O, Kubo T, Sugaya F, et al. Severe cytokine release syndrome resulting in purpura fulminans despite successful response to nivolumab therapy in a patient with pleomorphic carcinoma of the lung: a case report. J Immunother Cancer. 2019;7:97. doi:10.1186/s40425- 019-0582-4
- Kao RL, Jacobsen AA, Billington CJ Jr, et al. A case of VEXAS syndrome associated with EBV-associated hemophagocytic lymphohistiocytosis. Blood Cells Mol Dis. 2022;93:102636. doi:10.1016/j .bcmd.2021.102636
- Koga T, Takano K, Horai Y, et al. Sweet’s syndrome complicated by Kikuchi’s disease and hemophagocytic syndrome which presented with retinoic acid-inducible gene-I in both the skin epidermal basal layer and the cervical lymph nodes. Intern Med. 2013;52:1839-1843. doi:10.2169 /internalmedicine.52.9542
- Lin WL, Lin WC, Chiu CS, et al. Paraneoplastic Sweet’s syndrome in a patient with hemophagocytic syndrome. Int J Dermatol. 2008;3:305-307.
- Collins MK, Ho J, Akilov OE. Case 52. A unique presentation of hemophagocytic lymphohistiocytosis with ulcerating papulonodules. In: Akilov OE, ed. Cutaneous Lymphomas: Unusual Cases 3. Springer International Publishing; 2021:126-127.
- Chakrapani A, Avery A, Warnke R. Primary cutaneous gamma delta T-cell lymphoma with brain involvement and hemophagocytic syndrome. Am J Dermatopathol. 2013;35:270-272. doi:10.1097 /DAD.0b013e3182624e98
- Sullivan C, Loghmani A, Thomas K, et al. Hemophagocytic lymphohistiocytosis as the initial presentation of subcutaneous panniculitis-like T-cell lymphoma: a rare case responding to cyclosporine A and steroids. J Investig Med High Impact Case Rep. 2020;8:2324709620981531. doi:10.1177/2324709620981531
- Darmawan G, Salido EO, Concepcion ML, et al. Hemophagocytic lymphohistiocytosis: “a dreadful mimic.” Int J Rheum Dis. 2015; 18:810-812. doi:10.1111/1756-185x.12506
- Maus MV, Leick MB, Cornejo KM, et al. Case 35-2019: a 66-year-old man with pancytopenia and rash. N Engl J Med. 2019;381:1951-1960. doi:10.1056/NEJMcpc1909627
- Chamseddin B, Marks E, Dominguez A, et al. Refractory macrophage activation syndrome in the setting of adult-onset Still disease with hemophagocytic lymphohistiocytosis detected on skin biopsy treated with canakinumab and tacrolimus. J Cutan Pathol. 2019;46:528-531. doi:10.1111/cup.13466
- Bérar A, Ardois S, Walter-Moraux P, et al. Primary varicella-zoster virus infection of the immunocompromised associated with acute pancreatitis and hemophagocytic lymphohistiocytosis: a case report. Medicine (Baltimore). 2021;100:e25351. doi:10.1097 /md.0000000000025351
- Chang CC, Hsiao PJ, Chiu CC, et al. Catastrophic hemophagocytic lymphohistiocytosis in a young man with nephrotic syndrome. Clin Chim Acta. 2015;439:168-171. doi:10.1016/j.cca.2014.10.025
- Kurosawa S, Sekiya N, Fukushima K, et al. Unusual manifestation of disseminated herpes simplex virus type 2 infection associated with pharyngotonsilitis, esophagitis, and hemophagocytic lymphohisitocytosis without genital involvement. BMC Infect Dis. 2019;19:65. doi:10.1186/s12879-019-3721-0
- Saidi W, Gammoudi R, Korbi M, et al. Hemophagocytic lymphohistiocytosis: an unusual complication of leprosy. Int J Dermatol. 2015;54: 1054-1059. doi:10.1111/ijd.12792
- Yamaguchi K, Yamamoto A, Hisano M, et al. Herpes simplex virus 2-associated hemophagocytic lymphohistiocytosis in a pregnant patient. Obstet Gynecol. 2005;105(5 Pt 2):1241-1244. doi:10.1097 /01.AOG.0000157757.54948.9b
- Hayakawa I, Shirasaki F, Ikeda H, et al. Reactive hemophagocytic syndrome in a patient with polyarteritis nodosa associated with Epstein- Barr virus reactivation. Rheumatol Int. 2006;26:573-576. doi:10.1007 /s00296-005-0024-0
- Jeong JY, Park JY, Ham JY, et al. Molecular evidence of parvovirus B19 in the cutaneous polyarteritis nodosa tissue from a patient with parvovirus-associated hemophagocytic syndrome: case report. Medicine (Baltimore). 2020;99:e22079. doi:10.1097 /md.0000000000022079
- Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478. doi:10.2169 /internalmedicine.1121-18
- Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune- associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246. doi:10 .1080/03009742.2019.1653493
- Jung SY. Hemophagocytic syndrome diagnosed by liver biopsy in a female patient with systemic lupus erythematosus. J Clin Rheumatol. 2013;19:449-451. doi:10.1097/rhu.0000000000000040
- Kerl K, Wolf IH, Cerroni L, et al. Hemophagocytosis in cutaneous autoimmune disease. Am J Dermatopathol. 2015;37:539-543. doi:10.1097 /dad.0000000000000166
- Komiya Y, Saito T, Mizoguchi F, et al. Hemophagocytic syndrome complicated with dermatomyositis controlled successfully with infliximab and conventional therapies. Intern Med. 2017;56:3237-3241. doi:10.2169 /internalmedicine.7966-16
- Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and long-term outcome of 15 episodes of systemic lupus erythematosusassociated hemophagocytic syndrome. Medicine (Baltimore). 2006;85: 169-182. doi:10.1097/01.md.0000224708.62510.d1
- Guitart J, Mangold AR, Martinez-Escala ME, et al. Clinical and pathological characteristics and outcomes among patients with subcutaneous panniculitis-like T-cell lymphoma and related adipotropic lymphoproliferative disorders. JAMA Dermatol. 2022;158:1167-1174. doi:10.1001/jamadermatol.2022.3347
- Hung GD, Chen YH, Chen DY, et al. Subcutaneous panniculitis-like T-cell lymphoma presenting with hemophagocytic lymphohistiocytosis and skin lesions with characteristic high-resolution ultrasonographic findings. Clin Rheumatol. 2007;26:775-778. doi:10.1007/s10067 -005-0193-y
- Jamil A, Nadzri N, Harun N, et al. Primary cutaneous diffuse large B-cell lymphoma leg type presenting with hemophagocytic syndrome. J Am Acad Dermatol. 2012;67:e222-3. doi:10.1016/j.jaad.2012.04.021
- LeBlanc RE, Lansigan F. Unraveling subcutaneous panniculitis-like T-cell lymphoma: an association between subcutaneous panniculitislike T-cell lymphoma, autoimmune lymphoproliferative syndrome, and familial hemophagocytic lymphohistiocytosis. J Cutan Pathol. 2021;48:572-577. doi:10.1111/cup.13863
- Lee DE, Martinez-Escala ME, Serrano LM, et al. Hemophagocytic lymphohistiocytosis in cutaneous T-cell lymphoma. JAMA Dermatol. 2018;154:828-831. doi:10.1001/jamadermatol.2018.1264
- Maejima H, Tanei R, Morioka T, et al. Haemophagocytosis-related intravascular large B-cell lymphoma associated with skin eruption. Acta Derm Venereol. 2011;91:339-340. doi:10.2340/00015555-0981
- Mody A, Cherry D, Georgescu G, et al. A rare case of subcutaneous panniculitis-like T cell lymphoma with hemophagocytic lymphohistiocytosis mimicking cellulitis. Am J Case Rep. 2021;22:E927142. doi:10.12659/ajcr.927142
- Park YJ, Bae HJ, Chang JY, et al. Development of Kaposi sarcoma and hemophagocytic lymphohistiocytosis associated with human herpesvirus 8 in a renal transplant recipient. Korean J Intern Med. 2017;4:750-752.
- Phatak S, Gupta L, Aggarwal A. A young woman with panniculitis and cytopenia who later developed coagulopathy. J Assoc Physicians India. 2016;64:65-67.
- Pongpairoj K, Rerknimitr P, Wititsuwannakul J, et al. Eruptive telangiectasia in a patient with fever and haemophagocytic syndrome. Clin Exp Dermatol. 2016;41:696-698. doi:10.1111/ced.12859
- Shimizu Y, Tanae K, Takahashi N, et al. Primary cutaneous anaplastic large-cell lymphoma presenting with hemophagocytic syndrome: a case report and review of the literature. Leuk Res. 2010;34:263-266. doi:10.1016/j.leukres.2009.07.001
- Sirka CS, Pradhan S, Patra S, et al. Hemophagocytic lymphohistiocytosis: a rare, potentially fatal complication in subcutaneous panniculitis like T cell lymphoma. Indian J Dermatol Venereol Leprol. 2019;5:481-485.
- Chin CK, Hall S, Green C, et al. Secondary haemophagocytic lymphohistiocytosis due to checkpoint inhibitor therapy. Eur J Cancer. 2019;115: 84-87. doi:10.1016/j.ejca.2019.04.026
- Dudda M, Mann C, Heinz J, et al. Hemophagocytic lymphohistiocytosis of a melanoma patient under BRAF/MEK-inhibitor therapy following anti-PD1 inhibitor treatment: a case report and review to the literature. Melanoma Res. 2021;31:81-84. doi:10.1097 /cmr.0000000000000703
- Mizuta H, Nakano E, Takahashi A, et al. Hemophagocytic lymphohistiocytosis with advanced malignant melanoma accompanied by ipilimumab and nivolumab: a case report and literature review. Dermatol Ther. 2020;33:e13321. doi:10.1111/dth.13321
- Satzger I, Ivanyi P, Länger F, et al. Treatment-related hemophagocytic lymphohistiocytosis secondary to checkpoint inhibition with nivolumab plus ipilimumab. Eur J Cancer. 2018;93:150-153. doi:10.1016/j.ejca.2018.01.063
- Michot JM, Lazarovici J, Tieu A, et al. Haematological immune-related adverse events with immune checkpoint inhibitors, how to manage? Eur J Cancer. 2019;122:72-90. doi:10.1016/J.EJCA.2019.07.014
- Choi S, Zhou M, Bahrani E, et al. Rare and fatal complication of immune checkpoint inhibition: a case report of haemophagocytic lymphohistiocytosis with severe lichenoid dermatitis. Br J Haematol. 2021;193:e44-e47. doi:10.1111/BJH.17442
- Yang JJ, Lei DK, Ravi V, et al. Overlap between hemophagocytic lymphohistiocytosis and drug reaction and eosinophilia with systemic symptoms: a review. Int J Dermatol. 2021;60:925-932. doi:10.1111 /ijd.15196
- Go RS, Wester SM. Immunophenotypic and molecular features, clinical outcomes, treatments, and prognostic factors associated with subcutaneous panniculitis-like T-cell lymphoma: a systematic analysis of 156 patients reported in the literature. Cancer. 2004;101:1404-1413. doi:10.1002/cncr.20502
A Systematic Review of Dermatologic Findings in Adults With Hemophagocytic Lymphohistiocytosis
A Systematic Review of Dermatologic Findings in Adults With Hemophagocytic Lymphohistiocytosis
PRACTICE POINTS
- Hemophagocytic lymphohistiocytosis (HLH) is a complex, life-threatening immunologic condition that is associated with various diagnostic tools.
- Physicians who care for patients with HLH should know that skin findings are not uncommon but are largely nonspecific and can be a direct result of HLH itself, systemic complications, or the underlying etiology of the condition.
- There is a myriad of cutaneous findings that can manifest in adult patients with HLH. Awareness of HLH-associated dermatologic conditions and available diagnostic tools among multidisciplinary teams will aid in diagnosis.
A Veteran Presenting With Symptomatic Postprandial Episodes
A Veteran Presenting With Symptomatic Postprandial Episodes
Idiopathic postprandial syndrome (IPP), initially termed reactive hypoglycemia, presents with hypoglycemic-like symptoms in the absence of biochemical hypoglycemia and remains a diagnosis of exclusion. Its pathophysiology is poorly understood. The diagnosis requires thorough evaluation of cardiac, metabolic, neurologic, and gastrointestinal causes, as well as Whipple triad criteria. Dietary modifications, including reduced carbohydrate intake, increased protein and fiber, and frequent small meals, remain the cornerstone of IPP management. Continuous glucose monitoring (CGM) may be a useful adjunct in correlating symptoms with glucose trends, but its role is still evolving.
In the evaluation of patients with symptoms suggestive of hypoglycemia (Figure 1), patients should first be assessed for Whipple triad: symptoms consistent with hypoglycemia, blood glucose level < 55 mg/dL, and reversal of symptoms with glucose.1 Patients who meet Whipple triad criteria should be investigated to identify further etiologies of hypoglycemia. They may include insulinoma, medication-induced (insulin, sulfonylurea, meglitinide, or β blocker use), postbariatric surgery complications, noninsulinoma pancreatogenous hypoglycemia syndrome, ackee fruit consumption, or familial conditions.2 The presence of hypoglycemic symptoms in the postprandial or fasting state can provide valuable insights into underlying etiology.
Patients who do not meet Whipple triad criteria, but exhibit postprandial symptoms consistent with hypoglycemia, as in this case, present a diagnostic dilemma. IPP is defined as hypoglycemic symptoms occuring after carbohydrate ingestion without biochemical hypoglycemia. Initially termed reactive hypoglycemia, it was renamed in 1981to reflect the absence of low blood glucose levels.3
The understanding of this diagnosis has not significantly progressed since the 1980s. Its prevalence, incidence, risk factors, and societal burden remain unclear. IPP is a challenging diagnosis due to nonspecific symptoms that overlap with a myriad of conditions. These symptoms may include adrenergic symptoms such as diaphoresis, tremulousness, palpitations, anxiety, and hunger. Potentially severe neuroglycopenic symptoms, including weakness, dizziness, behavior changes, confusion, and coma, are not typically observed.4 Given that objective criteria are not well established, IPP remains a diagnosis of exclusion. It is imperative to rule out alternative etiologies, particularly cardiac, gastrointestinal, and neurologic causes.
CASE PRESENTATION
A male aged 41 years presented to primary care for evaluation of acute on chronic symptomatic postprandial episodes. He reported a history of symptomatic sinus bradycardia in the setting of sick sinus syndrome following dual-chamber pacemaker placement, posttraumatic stress disorder, and gastroesophageal reflux disease. He was a retired Navy sailor without any known occupational exposures who worked in the real estate industry. The patient reported feeling lightheaded, tremulous, and anxious most afternoons after lunch for several years. He also reported that meals heavy in carbohydrates exacerbated his symptoms, whereas skipping meals or lying down alleviated his symptoms. The patient also reported concomitant arm numbness, shortness of breath, palpitations, and nausea during these episodes. Review of systems was otherwise negative, including no weight changes, fever, chills, night sweats, chest pain, or syncope.
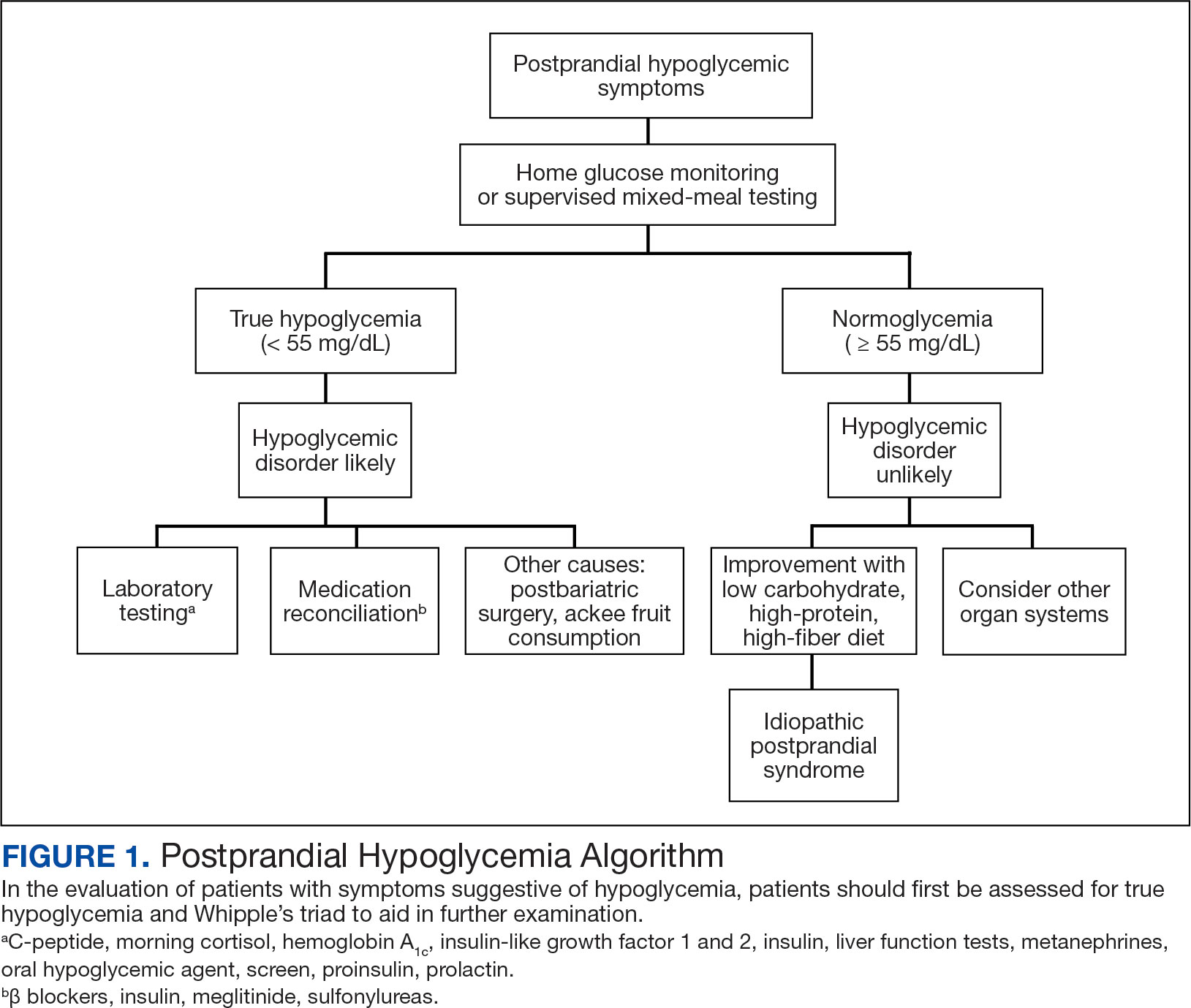
The patient’s medications included ferrous sulfate 325 mg once every other day, bupropion 200 mg once daily, metoprolol succinate 25 mg once daily, and as-needed lorazepam 1 mg once daily. The patient reported no current substance use but reported previous tobacco use 3 years prior (maximum 1 pack/week) and alcohol use 5 years prior (750 ml/day for 15 years). The patient did not exercise and typically ate oatmeal for breakfast, a sandwich or salad for lunch, and taquitos or salad for dinner, with snacks throughout the day. Notable family history included a maternal grandmother with colon cancer. The patient’s vital signs included a 36.8 °C temperature, heart rate 87 beats/min, 118/71 mm Hg blood pressure, oxygen saturation 98% on room air, 125.2 kg weight, and 38.5 body mass index. There were no orthostatic vital sign changes. A physical examination demonstrated obesity with an unremarkable cardiopulmonary and volume examination.
Additional testing included Gallium-68 dototate positron emission tomography/computed tomography, brain magnetic resonance imaging, echocardiogram, electromyogram, exercise tolerance test, Holter monitoring, invasive cardiopulmonary exercise testing, pacemaker interrogation, pulmonary function testing, stress echocardiogram, tilt table test, and venogram computed tomography of the chest, but the results were unremarkable (Appendix). His afternoon nonfasting glucose level was 138 mg/dL with a concurrent hemoglobin A1c of 5.2%. The patient had a fasting C-peptide level of 3.7 ng/mL (reference range 0.5-2.0 ng/mL), fasting insulin level 19.1 mIU/L (reference range < 25 mIU/L), and a fasting glucose level of 93 mg/dL (reference range 70-99 mg/dL). The patient’s urine 5-HIAA, plasma metanephrines, urine metanephrines, insulin-like growth factor 1, prolactin, corticotropin, fasting cortisol, and thyrotropin yielded results within reference ranges (Table). The veteran was prescribed a CGM, which demonstrated normal glucose levels (≥ 55 mg/dL) during symptomatic episodes (Figure 2).
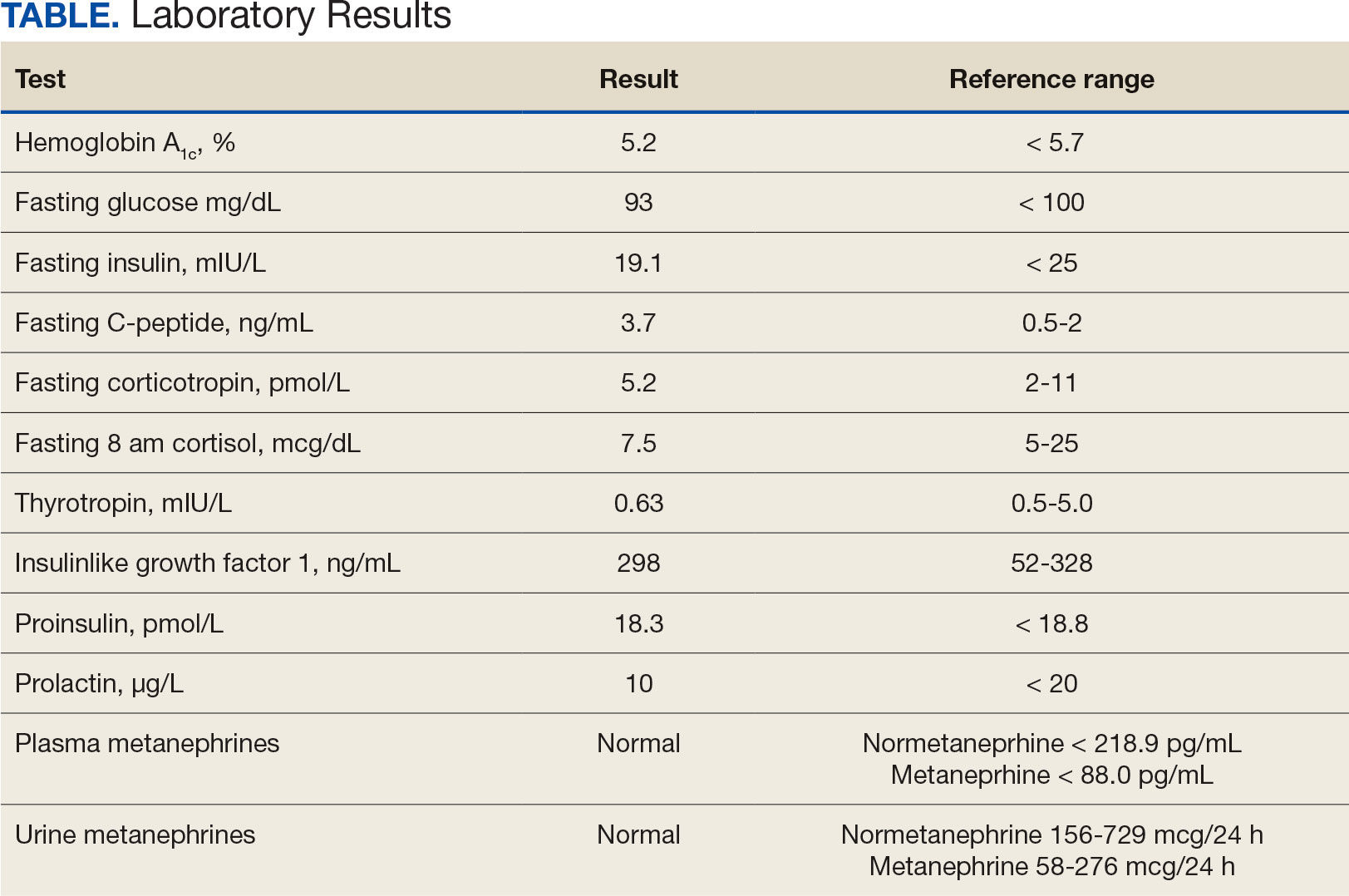
The patient was diagnosed with IPP given normoglycemia, exclusion of alternative diagnoses, and symptomatic improvement with dietary changes. He was referred to a nutritionist for a high-protein, high-fiber, and low-carbohydrate diet.
DISCUSSION
Seemingly simple diagnostic tools can lead to diagnostic pitfalls. Home glucose monitoring with the use of a standard glucometer during an episode is the typical first step in identifying hypoglycemia, as it is both pragmatic and accurate, with a mean absolute relative difference (MARD) of about 10% in hypoglycemic ranges.5 While the advent of CGM provides real-time data and can reveal clinically relevant fluctuations, it reveals mild hypoglycemia (54 to 70 mg/dL) of no clinical significance in a large proportion of individuals.
Additionally, CGM is less accurate than glucometers with a MARD of about 20% in hypoglycemia ranges.6 CGM technology, however, is rapidly evolving and undergoing further investigation for hypoglycemia detection. Therefore, CGM may be considered in select patients as prospective study results are established; the newest CGMs have MARDs very similar to fingerstick blood glucose data.7,8 In the patient described in this case, CGM helped corroborate the diagnosis, given that symptomatic episodes correlated with lower glucose levels. Provocative testing with oral glucose tolerance testing can frequently result in false positive hypoglycemic readings and is not recommended.9 Supervised mixed meal testing can also be used, which entails monitoring after consuming a mixed macronutrient meal. The test concludes after hypoglycemic symptoms develop or 5 hours elapse, whichever occurs first.1
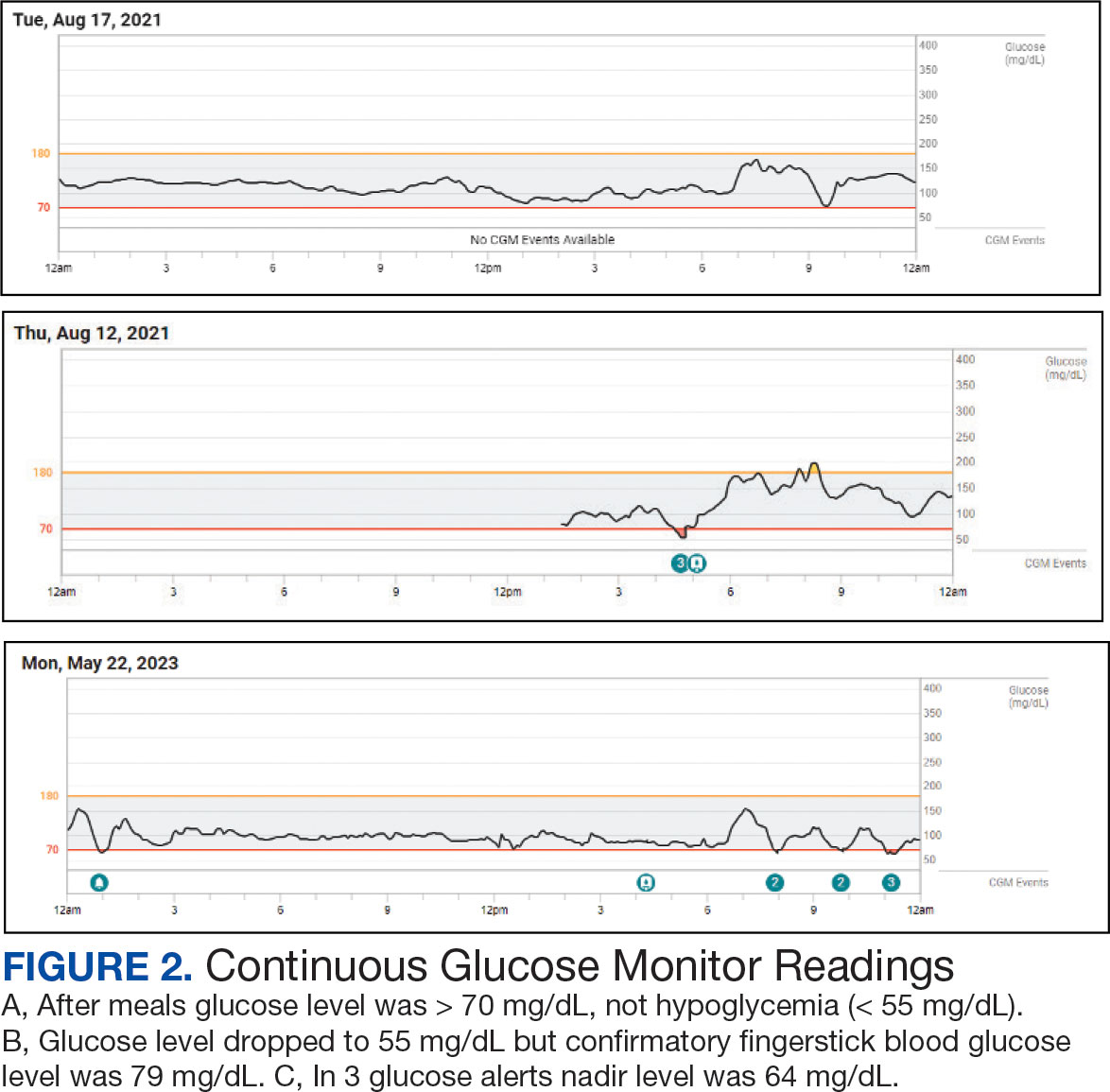
The pathophysiology of IPP is poorly understood. Proposed mechanisms include increased insulin sensitivity, increased adrenergic sensitivity, impaired glucagon regulation, emotional distress, insulin resistance, and increased glucagon-like peptide-1 production.10-13 Research suggests this may occur as pancreatic β cells fail in early type 2 diabetes mellitus, with diminished first-phase insulin release leading to an initial exuberant rise in blood glucose, an overshooting of the second phase of insulin secretion, and the feeling of the postprandial blood glucose falling, even though the final glucose level achieved is not truly low.13 There are contradictory studies in the literature demonstrating no association between insulin resistance and hypoglycemic symptoms.14 In 2022, Kosuda and colleagues looked at homeostatic model assessment for insulin resistance in patients with postprandial syndrome. They found that the patients were slightly insulin resistant but had normal or exaggerated insulin secretory capacity compared to an oral glucose load, whereas glucagon levels were robustly suppressed by a glucose load. The observed hormonal responses may result in the glycemic patterns and symptoms observed; further study is warranted to elucidate the mechanism.15
Dietary modification is the cornerstone treatment for postprandial syndrome, including reduced carbohydrate intake, increased protein and fiber intake, and more frequent and smaller meals. There is also evidence that a Mediterranean diet may be beneficial for managing hypoglycemic symptoms.16 Furthermore, α-glucosidase inhibitors, whose mechanism of action delays the digestion of carbohydrates, have demonstrated promise. This medication class has demonstrated significance in raising postprandial glucose levels and alleviating hypoglycemic symptoms in patients with true postprandial hypoglycemia.17
CONCLUSIONS
IPP is a benign diagnosis encompassing hypoglycemic symptoms without biochemical hypoglycemia. It is not a true hypoglycemic disorder. IPP is challenging to diagnose, given that it is an interpretation of exclusion, supported by symptom improvement with dietary changes (ie, reduced carbohydrate intake, increased protein and fiber intake, and more frequent and smaller meals). Supervised mixed meal testing or CGM can be used to assist with diagnosis. Even though CGM is undergoing further study in this patient population, it corroborated the diagnosis in the patient described in this case.
For hypoglycemic symptoms, physicians should first assess for evidence of Whipple triad to evaluate for true biochemical hypoglycemia. For true hypoglycemia (< 55 mg/dL), physicians may conduct an examination in conjunction with an endocrinologist. For normoglycemia (≥ 55 mg/dL), physicians should first exclude alternative etiologies (including cardiac and neurologic), and subsequently consider IPP.
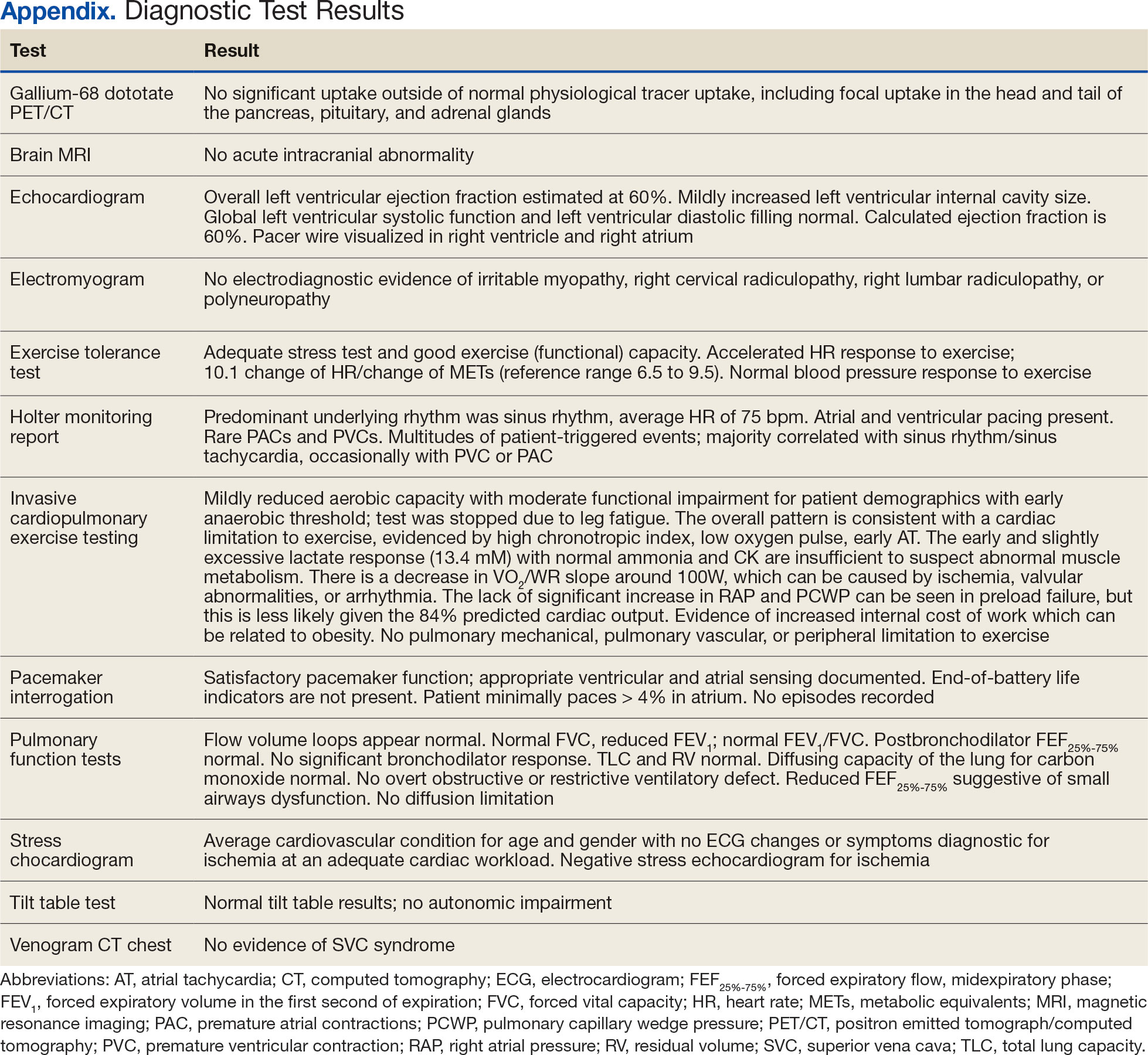
Bansal N, Weinstock RS. Non-Diabetic Hypoglycemia. In: Feingold KR, Anawalt B, Blackman MR, et al, eds. Endotext. MDText.com, Inc.; 2000.
Service FJ. Hypoglycemic disorders. New Engl J Med. 1995;332(17):1144-1152.doi:10.1056/NEJM199504273321707
Charles MA, Hofeldt F, Shackelford A, et al. Comparison of oral glucose tolerance tests and mixed meals in patients with apparent idiopathic postabsorptive hypoglycemia: absence of hypoglycemia after meals. Diabetes. 1981;30(6):465-470.
Douillard C, Jannin A, Vantyghem MC. Rare causes of hypoglycemia in adults. Ann Endocrinol (Paris). 2020;81(2-3):110-117. doi:10.1016/j.ando.2020.04.003
Ekhlaspour L, Mondesir D, Lautsch N, et al. Comparative accuracy of 17 point-of-care glucose meters. J Diabetes Sci Technol. 2017;11(3):558-566. doi:10.1177/1932296816672237
Alitta Q, Grino M, Adjemout L, Langar A, Retornaz F, Oliver C. Overestimation of hypoglycemia diagnosis by FreeStyle Libre continuous glucose monitoring in long-term care home residents with diabetes. J Diabetes Sci Technol. 2018;12(3):727-728. doi:10.1177/1932296817747887
Mongraw-Chaffin M, Beavers DP, McClain DA. Hypoglycemic symptoms in the absence of diabetes: pilot evidence of clinical hypoglycemia in young women. J Clin Transl Endocrinol. 2019;18:100202. doi:10.1016/j.jcte.2019.100202
Shah VN, DuBose SN, Li Z, et al. Continuous glucose monitoring profiles in healthy nondiabetic participants: a multicenter prospective study. J Clin Endocrinol Metab. 2019;104(10):4356-4364. doi:10.1210/jc.2018-02763
Cryer PE, Axelrod L, Grossman AB, et al. Evaluation and management of adult hypoglycemic disorders: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2009;94(3):709-728. doi:10.1210/jc.2008-1410
Galati SJ, Rayfield EJ. Approach to the patient with postprandial hypoglycemia. Endocr Pract. 2014;20(4):331-340. doi:10.4158/EP13132.RA
Altuntas Y. Postprandial reactive hypoglycemia. Sisli Etfal Hastan Tip Bul. 2019;53(3):215-220.doi:10.14744/SEMB.2019.59455
HARRIS S. HYPERINSULINISM AND DYSINSULINISM. JAMA. 1924;83(10):729-733.doi:10.1001/jama.1924.02660100003002
Harris S. HYPERINSULINISM AND DYSINSULINISM (INSULOGENIC HYPOGLYCBMIA). Endocrinology. 1932;16(1):29-42. doi:10.1210/endo-16-1-29
Hall M, Walicka M, Panczyk M, Traczyk I. Metabolic parameters in patients with suspected reactive hypoglycemia. J Pers Med. 2021;11(4):276. doi:10.3390/jpm11040276
Kosuda M, Watanabe K, Koike M, et al. Glucagon response to glucose challenge in patients with idiopathic postprandial syndrome. J Nippon Med Sch. 2022;89(1):102-107. doi:10.1272/jnms.JNMS.2022_89-205
Hall M, Walicka M, Panczyk M, Traczyk I. Assessing long-term impact of dietary interventions on occurrence of symptoms consistent with hypoglycemia in patients without diabetes: a one-year follow-up study. Nutrients. 2022;14(3):497. doi:10.3390/nu14030497
Ozgen AG, Hamulu F, Bayraktar F, et al. Long-term treatment with acarbose for the treatment of reactive hypoglycemia. Eat Weight Disord. 1998;3(3):136-140. doi:10.1007/BF03340001
Idiopathic postprandial syndrome (IPP), initially termed reactive hypoglycemia, presents with hypoglycemic-like symptoms in the absence of biochemical hypoglycemia and remains a diagnosis of exclusion. Its pathophysiology is poorly understood. The diagnosis requires thorough evaluation of cardiac, metabolic, neurologic, and gastrointestinal causes, as well as Whipple triad criteria. Dietary modifications, including reduced carbohydrate intake, increased protein and fiber, and frequent small meals, remain the cornerstone of IPP management. Continuous glucose monitoring (CGM) may be a useful adjunct in correlating symptoms with glucose trends, but its role is still evolving.
In the evaluation of patients with symptoms suggestive of hypoglycemia (Figure 1), patients should first be assessed for Whipple triad: symptoms consistent with hypoglycemia, blood glucose level < 55 mg/dL, and reversal of symptoms with glucose.1 Patients who meet Whipple triad criteria should be investigated to identify further etiologies of hypoglycemia. They may include insulinoma, medication-induced (insulin, sulfonylurea, meglitinide, or β blocker use), postbariatric surgery complications, noninsulinoma pancreatogenous hypoglycemia syndrome, ackee fruit consumption, or familial conditions.2 The presence of hypoglycemic symptoms in the postprandial or fasting state can provide valuable insights into underlying etiology.
Patients who do not meet Whipple triad criteria, but exhibit postprandial symptoms consistent with hypoglycemia, as in this case, present a diagnostic dilemma. IPP is defined as hypoglycemic symptoms occuring after carbohydrate ingestion without biochemical hypoglycemia. Initially termed reactive hypoglycemia, it was renamed in 1981to reflect the absence of low blood glucose levels.3
The understanding of this diagnosis has not significantly progressed since the 1980s. Its prevalence, incidence, risk factors, and societal burden remain unclear. IPP is a challenging diagnosis due to nonspecific symptoms that overlap with a myriad of conditions. These symptoms may include adrenergic symptoms such as diaphoresis, tremulousness, palpitations, anxiety, and hunger. Potentially severe neuroglycopenic symptoms, including weakness, dizziness, behavior changes, confusion, and coma, are not typically observed.4 Given that objective criteria are not well established, IPP remains a diagnosis of exclusion. It is imperative to rule out alternative etiologies, particularly cardiac, gastrointestinal, and neurologic causes.
CASE PRESENTATION
A male aged 41 years presented to primary care for evaluation of acute on chronic symptomatic postprandial episodes. He reported a history of symptomatic sinus bradycardia in the setting of sick sinus syndrome following dual-chamber pacemaker placement, posttraumatic stress disorder, and gastroesophageal reflux disease. He was a retired Navy sailor without any known occupational exposures who worked in the real estate industry. The patient reported feeling lightheaded, tremulous, and anxious most afternoons after lunch for several years. He also reported that meals heavy in carbohydrates exacerbated his symptoms, whereas skipping meals or lying down alleviated his symptoms. The patient also reported concomitant arm numbness, shortness of breath, palpitations, and nausea during these episodes. Review of systems was otherwise negative, including no weight changes, fever, chills, night sweats, chest pain, or syncope.
The patient’s medications included ferrous sulfate 325 mg once every other day, bupropion 200 mg once daily, metoprolol succinate 25 mg once daily, and as-needed lorazepam 1 mg once daily. The patient reported no current substance use but reported previous tobacco use 3 years prior (maximum 1 pack/week) and alcohol use 5 years prior (750 ml/day for 15 years). The patient did not exercise and typically ate oatmeal for breakfast, a sandwich or salad for lunch, and taquitos or salad for dinner, with snacks throughout the day. Notable family history included a maternal grandmother with colon cancer. The patient’s vital signs included a 36.8 °C temperature, heart rate 87 beats/min, 118/71 mm Hg blood pressure, oxygen saturation 98% on room air, 125.2 kg weight, and 38.5 body mass index. There were no orthostatic vital sign changes. A physical examination demonstrated obesity with an unremarkable cardiopulmonary and volume examination.
Additional testing included Gallium-68 dototate positron emission tomography/computed tomography, brain magnetic resonance imaging, echocardiogram, electromyogram, exercise tolerance test, Holter monitoring, invasive cardiopulmonary exercise testing, pacemaker interrogation, pulmonary function testing, stress echocardiogram, tilt table test, and venogram computed tomography of the chest, but the results were unremarkable (Appendix). His afternoon nonfasting glucose level was 138 mg/dL with a concurrent hemoglobin A1c of 5.2%. The patient had a fasting C-peptide level of 3.7 ng/mL (reference range 0.5-2.0 ng/mL), fasting insulin level 19.1 mIU/L (reference range < 25 mIU/L), and a fasting glucose level of 93 mg/dL (reference range 70-99 mg/dL). The patient’s urine 5-HIAA, plasma metanephrines, urine metanephrines, insulin-like growth factor 1, prolactin, corticotropin, fasting cortisol, and thyrotropin yielded results within reference ranges (Table). The veteran was prescribed a CGM, which demonstrated normal glucose levels (≥ 55 mg/dL) during symptomatic episodes (Figure 2).
The patient was diagnosed with IPP given normoglycemia, exclusion of alternative diagnoses, and symptomatic improvement with dietary changes. He was referred to a nutritionist for a high-protein, high-fiber, and low-carbohydrate diet.
DISCUSSION
Seemingly simple diagnostic tools can lead to diagnostic pitfalls. Home glucose monitoring with the use of a standard glucometer during an episode is the typical first step in identifying hypoglycemia, as it is both pragmatic and accurate, with a mean absolute relative difference (MARD) of about 10% in hypoglycemic ranges.5 While the advent of CGM provides real-time data and can reveal clinically relevant fluctuations, it reveals mild hypoglycemia (54 to 70 mg/dL) of no clinical significance in a large proportion of individuals.
Additionally, CGM is less accurate than glucometers with a MARD of about 20% in hypoglycemia ranges.6 CGM technology, however, is rapidly evolving and undergoing further investigation for hypoglycemia detection. Therefore, CGM may be considered in select patients as prospective study results are established; the newest CGMs have MARDs very similar to fingerstick blood glucose data.7,8 In the patient described in this case, CGM helped corroborate the diagnosis, given that symptomatic episodes correlated with lower glucose levels. Provocative testing with oral glucose tolerance testing can frequently result in false positive hypoglycemic readings and is not recommended.9 Supervised mixed meal testing can also be used, which entails monitoring after consuming a mixed macronutrient meal. The test concludes after hypoglycemic symptoms develop or 5 hours elapse, whichever occurs first.1
The pathophysiology of IPP is poorly understood. Proposed mechanisms include increased insulin sensitivity, increased adrenergic sensitivity, impaired glucagon regulation, emotional distress, insulin resistance, and increased glucagon-like peptide-1 production.10-13 Research suggests this may occur as pancreatic β cells fail in early type 2 diabetes mellitus, with diminished first-phase insulin release leading to an initial exuberant rise in blood glucose, an overshooting of the second phase of insulin secretion, and the feeling of the postprandial blood glucose falling, even though the final glucose level achieved is not truly low.13 There are contradictory studies in the literature demonstrating no association between insulin resistance and hypoglycemic symptoms.14 In 2022, Kosuda and colleagues looked at homeostatic model assessment for insulin resistance in patients with postprandial syndrome. They found that the patients were slightly insulin resistant but had normal or exaggerated insulin secretory capacity compared to an oral glucose load, whereas glucagon levels were robustly suppressed by a glucose load. The observed hormonal responses may result in the glycemic patterns and symptoms observed; further study is warranted to elucidate the mechanism.15
Dietary modification is the cornerstone treatment for postprandial syndrome, including reduced carbohydrate intake, increased protein and fiber intake, and more frequent and smaller meals. There is also evidence that a Mediterranean diet may be beneficial for managing hypoglycemic symptoms.16 Furthermore, α-glucosidase inhibitors, whose mechanism of action delays the digestion of carbohydrates, have demonstrated promise. This medication class has demonstrated significance in raising postprandial glucose levels and alleviating hypoglycemic symptoms in patients with true postprandial hypoglycemia.17
CONCLUSIONS
IPP is a benign diagnosis encompassing hypoglycemic symptoms without biochemical hypoglycemia. It is not a true hypoglycemic disorder. IPP is challenging to diagnose, given that it is an interpretation of exclusion, supported by symptom improvement with dietary changes (ie, reduced carbohydrate intake, increased protein and fiber intake, and more frequent and smaller meals). Supervised mixed meal testing or CGM can be used to assist with diagnosis. Even though CGM is undergoing further study in this patient population, it corroborated the diagnosis in the patient described in this case.
For hypoglycemic symptoms, physicians should first assess for evidence of Whipple triad to evaluate for true biochemical hypoglycemia. For true hypoglycemia (< 55 mg/dL), physicians may conduct an examination in conjunction with an endocrinologist. For normoglycemia (≥ 55 mg/dL), physicians should first exclude alternative etiologies (including cardiac and neurologic), and subsequently consider IPP.
Idiopathic postprandial syndrome (IPP), initially termed reactive hypoglycemia, presents with hypoglycemic-like symptoms in the absence of biochemical hypoglycemia and remains a diagnosis of exclusion. Its pathophysiology is poorly understood. The diagnosis requires thorough evaluation of cardiac, metabolic, neurologic, and gastrointestinal causes, as well as Whipple triad criteria. Dietary modifications, including reduced carbohydrate intake, increased protein and fiber, and frequent small meals, remain the cornerstone of IPP management. Continuous glucose monitoring (CGM) may be a useful adjunct in correlating symptoms with glucose trends, but its role is still evolving.
In the evaluation of patients with symptoms suggestive of hypoglycemia (Figure 1), patients should first be assessed for Whipple triad: symptoms consistent with hypoglycemia, blood glucose level < 55 mg/dL, and reversal of symptoms with glucose.1 Patients who meet Whipple triad criteria should be investigated to identify further etiologies of hypoglycemia. They may include insulinoma, medication-induced (insulin, sulfonylurea, meglitinide, or β blocker use), postbariatric surgery complications, noninsulinoma pancreatogenous hypoglycemia syndrome, ackee fruit consumption, or familial conditions.2 The presence of hypoglycemic symptoms in the postprandial or fasting state can provide valuable insights into underlying etiology.
Patients who do not meet Whipple triad criteria, but exhibit postprandial symptoms consistent with hypoglycemia, as in this case, present a diagnostic dilemma. IPP is defined as hypoglycemic symptoms occuring after carbohydrate ingestion without biochemical hypoglycemia. Initially termed reactive hypoglycemia, it was renamed in 1981to reflect the absence of low blood glucose levels.3
The understanding of this diagnosis has not significantly progressed since the 1980s. Its prevalence, incidence, risk factors, and societal burden remain unclear. IPP is a challenging diagnosis due to nonspecific symptoms that overlap with a myriad of conditions. These symptoms may include adrenergic symptoms such as diaphoresis, tremulousness, palpitations, anxiety, and hunger. Potentially severe neuroglycopenic symptoms, including weakness, dizziness, behavior changes, confusion, and coma, are not typically observed.4 Given that objective criteria are not well established, IPP remains a diagnosis of exclusion. It is imperative to rule out alternative etiologies, particularly cardiac, gastrointestinal, and neurologic causes.
CASE PRESENTATION
A male aged 41 years presented to primary care for evaluation of acute on chronic symptomatic postprandial episodes. He reported a history of symptomatic sinus bradycardia in the setting of sick sinus syndrome following dual-chamber pacemaker placement, posttraumatic stress disorder, and gastroesophageal reflux disease. He was a retired Navy sailor without any known occupational exposures who worked in the real estate industry. The patient reported feeling lightheaded, tremulous, and anxious most afternoons after lunch for several years. He also reported that meals heavy in carbohydrates exacerbated his symptoms, whereas skipping meals or lying down alleviated his symptoms. The patient also reported concomitant arm numbness, shortness of breath, palpitations, and nausea during these episodes. Review of systems was otherwise negative, including no weight changes, fever, chills, night sweats, chest pain, or syncope.
The patient’s medications included ferrous sulfate 325 mg once every other day, bupropion 200 mg once daily, metoprolol succinate 25 mg once daily, and as-needed lorazepam 1 mg once daily. The patient reported no current substance use but reported previous tobacco use 3 years prior (maximum 1 pack/week) and alcohol use 5 years prior (750 ml/day for 15 years). The patient did not exercise and typically ate oatmeal for breakfast, a sandwich or salad for lunch, and taquitos or salad for dinner, with snacks throughout the day. Notable family history included a maternal grandmother with colon cancer. The patient’s vital signs included a 36.8 °C temperature, heart rate 87 beats/min, 118/71 mm Hg blood pressure, oxygen saturation 98% on room air, 125.2 kg weight, and 38.5 body mass index. There were no orthostatic vital sign changes. A physical examination demonstrated obesity with an unremarkable cardiopulmonary and volume examination.
Additional testing included Gallium-68 dototate positron emission tomography/computed tomography, brain magnetic resonance imaging, echocardiogram, electromyogram, exercise tolerance test, Holter monitoring, invasive cardiopulmonary exercise testing, pacemaker interrogation, pulmonary function testing, stress echocardiogram, tilt table test, and venogram computed tomography of the chest, but the results were unremarkable (Appendix). His afternoon nonfasting glucose level was 138 mg/dL with a concurrent hemoglobin A1c of 5.2%. The patient had a fasting C-peptide level of 3.7 ng/mL (reference range 0.5-2.0 ng/mL), fasting insulin level 19.1 mIU/L (reference range < 25 mIU/L), and a fasting glucose level of 93 mg/dL (reference range 70-99 mg/dL). The patient’s urine 5-HIAA, plasma metanephrines, urine metanephrines, insulin-like growth factor 1, prolactin, corticotropin, fasting cortisol, and thyrotropin yielded results within reference ranges (Table). The veteran was prescribed a CGM, which demonstrated normal glucose levels (≥ 55 mg/dL) during symptomatic episodes (Figure 2).
The patient was diagnosed with IPP given normoglycemia, exclusion of alternative diagnoses, and symptomatic improvement with dietary changes. He was referred to a nutritionist for a high-protein, high-fiber, and low-carbohydrate diet.
DISCUSSION
Seemingly simple diagnostic tools can lead to diagnostic pitfalls. Home glucose monitoring with the use of a standard glucometer during an episode is the typical first step in identifying hypoglycemia, as it is both pragmatic and accurate, with a mean absolute relative difference (MARD) of about 10% in hypoglycemic ranges.5 While the advent of CGM provides real-time data and can reveal clinically relevant fluctuations, it reveals mild hypoglycemia (54 to 70 mg/dL) of no clinical significance in a large proportion of individuals.
Additionally, CGM is less accurate than glucometers with a MARD of about 20% in hypoglycemia ranges.6 CGM technology, however, is rapidly evolving and undergoing further investigation for hypoglycemia detection. Therefore, CGM may be considered in select patients as prospective study results are established; the newest CGMs have MARDs very similar to fingerstick blood glucose data.7,8 In the patient described in this case, CGM helped corroborate the diagnosis, given that symptomatic episodes correlated with lower glucose levels. Provocative testing with oral glucose tolerance testing can frequently result in false positive hypoglycemic readings and is not recommended.9 Supervised mixed meal testing can also be used, which entails monitoring after consuming a mixed macronutrient meal. The test concludes after hypoglycemic symptoms develop or 5 hours elapse, whichever occurs first.1
The pathophysiology of IPP is poorly understood. Proposed mechanisms include increased insulin sensitivity, increased adrenergic sensitivity, impaired glucagon regulation, emotional distress, insulin resistance, and increased glucagon-like peptide-1 production.10-13 Research suggests this may occur as pancreatic β cells fail in early type 2 diabetes mellitus, with diminished first-phase insulin release leading to an initial exuberant rise in blood glucose, an overshooting of the second phase of insulin secretion, and the feeling of the postprandial blood glucose falling, even though the final glucose level achieved is not truly low.13 There are contradictory studies in the literature demonstrating no association between insulin resistance and hypoglycemic symptoms.14 In 2022, Kosuda and colleagues looked at homeostatic model assessment for insulin resistance in patients with postprandial syndrome. They found that the patients were slightly insulin resistant but had normal or exaggerated insulin secretory capacity compared to an oral glucose load, whereas glucagon levels were robustly suppressed by a glucose load. The observed hormonal responses may result in the glycemic patterns and symptoms observed; further study is warranted to elucidate the mechanism.15
Dietary modification is the cornerstone treatment for postprandial syndrome, including reduced carbohydrate intake, increased protein and fiber intake, and more frequent and smaller meals. There is also evidence that a Mediterranean diet may be beneficial for managing hypoglycemic symptoms.16 Furthermore, α-glucosidase inhibitors, whose mechanism of action delays the digestion of carbohydrates, have demonstrated promise. This medication class has demonstrated significance in raising postprandial glucose levels and alleviating hypoglycemic symptoms in patients with true postprandial hypoglycemia.17
CONCLUSIONS
IPP is a benign diagnosis encompassing hypoglycemic symptoms without biochemical hypoglycemia. It is not a true hypoglycemic disorder. IPP is challenging to diagnose, given that it is an interpretation of exclusion, supported by symptom improvement with dietary changes (ie, reduced carbohydrate intake, increased protein and fiber intake, and more frequent and smaller meals). Supervised mixed meal testing or CGM can be used to assist with diagnosis. Even though CGM is undergoing further study in this patient population, it corroborated the diagnosis in the patient described in this case.
For hypoglycemic symptoms, physicians should first assess for evidence of Whipple triad to evaluate for true biochemical hypoglycemia. For true hypoglycemia (< 55 mg/dL), physicians may conduct an examination in conjunction with an endocrinologist. For normoglycemia (≥ 55 mg/dL), physicians should first exclude alternative etiologies (including cardiac and neurologic), and subsequently consider IPP.
Bansal N, Weinstock RS. Non-Diabetic Hypoglycemia. In: Feingold KR, Anawalt B, Blackman MR, et al, eds. Endotext. MDText.com, Inc.; 2000.
Service FJ. Hypoglycemic disorders. New Engl J Med. 1995;332(17):1144-1152.doi:10.1056/NEJM199504273321707
Charles MA, Hofeldt F, Shackelford A, et al. Comparison of oral glucose tolerance tests and mixed meals in patients with apparent idiopathic postabsorptive hypoglycemia: absence of hypoglycemia after meals. Diabetes. 1981;30(6):465-470.
Douillard C, Jannin A, Vantyghem MC. Rare causes of hypoglycemia in adults. Ann Endocrinol (Paris). 2020;81(2-3):110-117. doi:10.1016/j.ando.2020.04.003
Ekhlaspour L, Mondesir D, Lautsch N, et al. Comparative accuracy of 17 point-of-care glucose meters. J Diabetes Sci Technol. 2017;11(3):558-566. doi:10.1177/1932296816672237
Alitta Q, Grino M, Adjemout L, Langar A, Retornaz F, Oliver C. Overestimation of hypoglycemia diagnosis by FreeStyle Libre continuous glucose monitoring in long-term care home residents with diabetes. J Diabetes Sci Technol. 2018;12(3):727-728. doi:10.1177/1932296817747887
Mongraw-Chaffin M, Beavers DP, McClain DA. Hypoglycemic symptoms in the absence of diabetes: pilot evidence of clinical hypoglycemia in young women. J Clin Transl Endocrinol. 2019;18:100202. doi:10.1016/j.jcte.2019.100202
Shah VN, DuBose SN, Li Z, et al. Continuous glucose monitoring profiles in healthy nondiabetic participants: a multicenter prospective study. J Clin Endocrinol Metab. 2019;104(10):4356-4364. doi:10.1210/jc.2018-02763
Cryer PE, Axelrod L, Grossman AB, et al. Evaluation and management of adult hypoglycemic disorders: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2009;94(3):709-728. doi:10.1210/jc.2008-1410
Galati SJ, Rayfield EJ. Approach to the patient with postprandial hypoglycemia. Endocr Pract. 2014;20(4):331-340. doi:10.4158/EP13132.RA
Altuntas Y. Postprandial reactive hypoglycemia. Sisli Etfal Hastan Tip Bul. 2019;53(3):215-220.doi:10.14744/SEMB.2019.59455
HARRIS S. HYPERINSULINISM AND DYSINSULINISM. JAMA. 1924;83(10):729-733.doi:10.1001/jama.1924.02660100003002
Harris S. HYPERINSULINISM AND DYSINSULINISM (INSULOGENIC HYPOGLYCBMIA). Endocrinology. 1932;16(1):29-42. doi:10.1210/endo-16-1-29
Hall M, Walicka M, Panczyk M, Traczyk I. Metabolic parameters in patients with suspected reactive hypoglycemia. J Pers Med. 2021;11(4):276. doi:10.3390/jpm11040276
Kosuda M, Watanabe K, Koike M, et al. Glucagon response to glucose challenge in patients with idiopathic postprandial syndrome. J Nippon Med Sch. 2022;89(1):102-107. doi:10.1272/jnms.JNMS.2022_89-205
Hall M, Walicka M, Panczyk M, Traczyk I. Assessing long-term impact of dietary interventions on occurrence of symptoms consistent with hypoglycemia in patients without diabetes: a one-year follow-up study. Nutrients. 2022;14(3):497. doi:10.3390/nu14030497
Ozgen AG, Hamulu F, Bayraktar F, et al. Long-term treatment with acarbose for the treatment of reactive hypoglycemia. Eat Weight Disord. 1998;3(3):136-140. doi:10.1007/BF03340001
Bansal N, Weinstock RS. Non-Diabetic Hypoglycemia. In: Feingold KR, Anawalt B, Blackman MR, et al, eds. Endotext. MDText.com, Inc.; 2000.
Service FJ. Hypoglycemic disorders. New Engl J Med. 1995;332(17):1144-1152.doi:10.1056/NEJM199504273321707
Charles MA, Hofeldt F, Shackelford A, et al. Comparison of oral glucose tolerance tests and mixed meals in patients with apparent idiopathic postabsorptive hypoglycemia: absence of hypoglycemia after meals. Diabetes. 1981;30(6):465-470.
Douillard C, Jannin A, Vantyghem MC. Rare causes of hypoglycemia in adults. Ann Endocrinol (Paris). 2020;81(2-3):110-117. doi:10.1016/j.ando.2020.04.003
Ekhlaspour L, Mondesir D, Lautsch N, et al. Comparative accuracy of 17 point-of-care glucose meters. J Diabetes Sci Technol. 2017;11(3):558-566. doi:10.1177/1932296816672237
Alitta Q, Grino M, Adjemout L, Langar A, Retornaz F, Oliver C. Overestimation of hypoglycemia diagnosis by FreeStyle Libre continuous glucose monitoring in long-term care home residents with diabetes. J Diabetes Sci Technol. 2018;12(3):727-728. doi:10.1177/1932296817747887
Mongraw-Chaffin M, Beavers DP, McClain DA. Hypoglycemic symptoms in the absence of diabetes: pilot evidence of clinical hypoglycemia in young women. J Clin Transl Endocrinol. 2019;18:100202. doi:10.1016/j.jcte.2019.100202
Shah VN, DuBose SN, Li Z, et al. Continuous glucose monitoring profiles in healthy nondiabetic participants: a multicenter prospective study. J Clin Endocrinol Metab. 2019;104(10):4356-4364. doi:10.1210/jc.2018-02763
Cryer PE, Axelrod L, Grossman AB, et al. Evaluation and management of adult hypoglycemic disorders: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2009;94(3):709-728. doi:10.1210/jc.2008-1410
Galati SJ, Rayfield EJ. Approach to the patient with postprandial hypoglycemia. Endocr Pract. 2014;20(4):331-340. doi:10.4158/EP13132.RA
Altuntas Y. Postprandial reactive hypoglycemia. Sisli Etfal Hastan Tip Bul. 2019;53(3):215-220.doi:10.14744/SEMB.2019.59455
HARRIS S. HYPERINSULINISM AND DYSINSULINISM. JAMA. 1924;83(10):729-733.doi:10.1001/jama.1924.02660100003002
Harris S. HYPERINSULINISM AND DYSINSULINISM (INSULOGENIC HYPOGLYCBMIA). Endocrinology. 1932;16(1):29-42. doi:10.1210/endo-16-1-29
Hall M, Walicka M, Panczyk M, Traczyk I. Metabolic parameters in patients with suspected reactive hypoglycemia. J Pers Med. 2021;11(4):276. doi:10.3390/jpm11040276
Kosuda M, Watanabe K, Koike M, et al. Glucagon response to glucose challenge in patients with idiopathic postprandial syndrome. J Nippon Med Sch. 2022;89(1):102-107. doi:10.1272/jnms.JNMS.2022_89-205
Hall M, Walicka M, Panczyk M, Traczyk I. Assessing long-term impact of dietary interventions on occurrence of symptoms consistent with hypoglycemia in patients without diabetes: a one-year follow-up study. Nutrients. 2022;14(3):497. doi:10.3390/nu14030497
Ozgen AG, Hamulu F, Bayraktar F, et al. Long-term treatment with acarbose for the treatment of reactive hypoglycemia. Eat Weight Disord. 1998;3(3):136-140. doi:10.1007/BF03340001
A Veteran Presenting With Symptomatic Postprandial Episodes
A Veteran Presenting With Symptomatic Postprandial Episodes