User login
EMA recommends safety measures for idelalisib

Photo courtesy of
Gilead Sciences, Inc.
In light of recently reported safety concerns, the European Medicines Agency’s (EMA) Pharmacovigilance Risk Assessment Committee (PRAC) has issued provisional advice for doctors and patients using idelalisib (Zydelig).
The EMA recently began reviewing the safety of idelalisib because of serious adverse events, most of which were infection-related, that were reported in 3 clinical trials investigating idelalisib in combination with other drugs.
These trials have since been stopped.
Now, the PRAC is making provisional recommendations to ensure idelalisib is used as safely as possible outside of clinical trials.
These recommendations will be forwarded to the European Commission, which will issue a provisional legally binding decision applicable in all European Union (EU) member states.
In the EU, idelalisib is approved as monotherapy for adults with follicular lymphoma that is refractory to 2 prior lines of treatment.
Idelalisib is also approved for use in combination with rituximab to treat adults with chronic lymphocytic leukemia (CLL) who have received at least 1 prior therapy or as first-line treatment in the presence of 17p deletion or TP53 mutation in CLL patients considered unsuitable for chemo-immunotherapy.
Recommendations
The PRAC is recommending that previously untreated CLL patients with 17p deletion or TP53 mutation do not begin treatment with idelalisib. In addition, doctors should re-evaluate each patient taking idelalisib as first-line treatment for CLL and only continue treatment if the benefits outweigh the risks.
Idelalisib can continue to be used in combination, only with rituximab, in CLL patients who have received at least 1 prior therapy and as monotherapy in patients with follicular lymphoma that is refractory to 2 lines of treatment.
Patients with any evidence of ongoing systemic infection should not be started on treatment with idelalisib.
And patients should be informed about the risk of serious infections with idelalisib.
All patients who do receive idelalisib should take antibiotics to prevent Pneumocystis jirovecii pneumonia, and they should be monitored for respiratory signs and symptoms. Regular clinical and laboratory monitoring for cytomegalovirus infection is also recommended.
Patients should have regular blood tests to detect neutropenia. In case a patient has moderate or severe neutropenia, treatment with idelalisib may have to be interrupted, in line with the updated summary of product characteristics.
The EMA said further details on these provisional measures will be provided in writing to healthcare professionals, and the product information will be updated accordingly.
Likewise, further information on the review of idelalisib will be provided as necessary and once the review is finished.
About the review
The EMA’s review of idelalisib started after a higher rate of serious adverse events, including deaths, was seen in 3 clinical trials evaluating the addition of idelalisib to standard therapy in first-line CLL and relapsed indolent non-Hodgkin lymphoma (NHL).
Most of the deaths were related to infections such as Pneumocystis jirovecii pneumonia and cytomegalovirus infection. Other excess deaths were related mainly to respiratory events.
The NHL studies (NCT01732926 and NCT01732913) included patients with disease characteristics different from those covered by the currently approved indication for idelalisib and investigated combinations of drugs that are not currently approved in the EU—idelalisib plus rituximab for NHL and idelalisib plus bendamustine and rituximab for NHL.
The CLL trial (NCT01980888) involved patients who had not received previous treatment, some of whom had the 17p deletion or TP53 mutation. However, the trial also investigated a combination of drugs not currently approved in the EU—idelalisib plus bendamustine and rituximab.
At present, the PRAC is conducting the review. Once the PRAC is finished, its recommendations will be forwarded to the EMA’s Committee for Medicinal Products for Human Use, which will adopt a final opinion.
The European Commission will take this opinion into account and adopt a legally binding decision that is applicable in all EU member states. ![]()
Photo courtesy of
Gilead Sciences, Inc.
In light of recently reported safety concerns, the European Medicines Agency’s (EMA) Pharmacovigilance Risk Assessment Committee (PRAC) has issued provisional advice for doctors and patients using idelalisib (Zydelig).
The EMA recently began reviewing the safety of idelalisib because of serious adverse events, most of which were infection-related, that were reported in 3 clinical trials investigating idelalisib in combination with other drugs.
These trials have since been stopped.
Now, the PRAC is making provisional recommendations to ensure idelalisib is used as safely as possible outside of clinical trials.
These recommendations will be forwarded to the European Commission, which will issue a provisional legally binding decision applicable in all European Union (EU) member states.
In the EU, idelalisib is approved as monotherapy for adults with follicular lymphoma that is refractory to 2 prior lines of treatment.
Idelalisib is also approved for use in combination with rituximab to treat adults with chronic lymphocytic leukemia (CLL) who have received at least 1 prior therapy or as first-line treatment in the presence of 17p deletion or TP53 mutation in CLL patients considered unsuitable for chemo-immunotherapy.
Recommendations
The PRAC is recommending that previously untreated CLL patients with 17p deletion or TP53 mutation do not begin treatment with idelalisib. In addition, doctors should re-evaluate each patient taking idelalisib as first-line treatment for CLL and only continue treatment if the benefits outweigh the risks.
Idelalisib can continue to be used in combination, only with rituximab, in CLL patients who have received at least 1 prior therapy and as monotherapy in patients with follicular lymphoma that is refractory to 2 lines of treatment.
Patients with any evidence of ongoing systemic infection should not be started on treatment with idelalisib.
And patients should be informed about the risk of serious infections with idelalisib.
All patients who do receive idelalisib should take antibiotics to prevent Pneumocystis jirovecii pneumonia, and they should be monitored for respiratory signs and symptoms. Regular clinical and laboratory monitoring for cytomegalovirus infection is also recommended.
Patients should have regular blood tests to detect neutropenia. In case a patient has moderate or severe neutropenia, treatment with idelalisib may have to be interrupted, in line with the updated summary of product characteristics.
The EMA said further details on these provisional measures will be provided in writing to healthcare professionals, and the product information will be updated accordingly.
Likewise, further information on the review of idelalisib will be provided as necessary and once the review is finished.
About the review
The EMA’s review of idelalisib started after a higher rate of serious adverse events, including deaths, was seen in 3 clinical trials evaluating the addition of idelalisib to standard therapy in first-line CLL and relapsed indolent non-Hodgkin lymphoma (NHL).
Most of the deaths were related to infections such as Pneumocystis jirovecii pneumonia and cytomegalovirus infection. Other excess deaths were related mainly to respiratory events.
The NHL studies (NCT01732926 and NCT01732913) included patients with disease characteristics different from those covered by the currently approved indication for idelalisib and investigated combinations of drugs that are not currently approved in the EU—idelalisib plus rituximab for NHL and idelalisib plus bendamustine and rituximab for NHL.
The CLL trial (NCT01980888) involved patients who had not received previous treatment, some of whom had the 17p deletion or TP53 mutation. However, the trial also investigated a combination of drugs not currently approved in the EU—idelalisib plus bendamustine and rituximab.
At present, the PRAC is conducting the review. Once the PRAC is finished, its recommendations will be forwarded to the EMA’s Committee for Medicinal Products for Human Use, which will adopt a final opinion.
The European Commission will take this opinion into account and adopt a legally binding decision that is applicable in all EU member states. ![]()
Photo courtesy of
Gilead Sciences, Inc.
In light of recently reported safety concerns, the European Medicines Agency’s (EMA) Pharmacovigilance Risk Assessment Committee (PRAC) has issued provisional advice for doctors and patients using idelalisib (Zydelig).
The EMA recently began reviewing the safety of idelalisib because of serious adverse events, most of which were infection-related, that were reported in 3 clinical trials investigating idelalisib in combination with other drugs.
These trials have since been stopped.
Now, the PRAC is making provisional recommendations to ensure idelalisib is used as safely as possible outside of clinical trials.
These recommendations will be forwarded to the European Commission, which will issue a provisional legally binding decision applicable in all European Union (EU) member states.
In the EU, idelalisib is approved as monotherapy for adults with follicular lymphoma that is refractory to 2 prior lines of treatment.
Idelalisib is also approved for use in combination with rituximab to treat adults with chronic lymphocytic leukemia (CLL) who have received at least 1 prior therapy or as first-line treatment in the presence of 17p deletion or TP53 mutation in CLL patients considered unsuitable for chemo-immunotherapy.
Recommendations
The PRAC is recommending that previously untreated CLL patients with 17p deletion or TP53 mutation do not begin treatment with idelalisib. In addition, doctors should re-evaluate each patient taking idelalisib as first-line treatment for CLL and only continue treatment if the benefits outweigh the risks.
Idelalisib can continue to be used in combination, only with rituximab, in CLL patients who have received at least 1 prior therapy and as monotherapy in patients with follicular lymphoma that is refractory to 2 lines of treatment.
Patients with any evidence of ongoing systemic infection should not be started on treatment with idelalisib.
And patients should be informed about the risk of serious infections with idelalisib.
All patients who do receive idelalisib should take antibiotics to prevent Pneumocystis jirovecii pneumonia, and they should be monitored for respiratory signs and symptoms. Regular clinical and laboratory monitoring for cytomegalovirus infection is also recommended.
Patients should have regular blood tests to detect neutropenia. In case a patient has moderate or severe neutropenia, treatment with idelalisib may have to be interrupted, in line with the updated summary of product characteristics.
The EMA said further details on these provisional measures will be provided in writing to healthcare professionals, and the product information will be updated accordingly.
Likewise, further information on the review of idelalisib will be provided as necessary and once the review is finished.
About the review
The EMA’s review of idelalisib started after a higher rate of serious adverse events, including deaths, was seen in 3 clinical trials evaluating the addition of idelalisib to standard therapy in first-line CLL and relapsed indolent non-Hodgkin lymphoma (NHL).
Most of the deaths were related to infections such as Pneumocystis jirovecii pneumonia and cytomegalovirus infection. Other excess deaths were related mainly to respiratory events.
The NHL studies (NCT01732926 and NCT01732913) included patients with disease characteristics different from those covered by the currently approved indication for idelalisib and investigated combinations of drugs that are not currently approved in the EU—idelalisib plus rituximab for NHL and idelalisib plus bendamustine and rituximab for NHL.
The CLL trial (NCT01980888) involved patients who had not received previous treatment, some of whom had the 17p deletion or TP53 mutation. However, the trial also investigated a combination of drugs not currently approved in the EU—idelalisib plus bendamustine and rituximab.
At present, the PRAC is conducting the review. Once the PRAC is finished, its recommendations will be forwarded to the EMA’s Committee for Medicinal Products for Human Use, which will adopt a final opinion.
The European Commission will take this opinion into account and adopt a legally binding decision that is applicable in all EU member states. ![]()
Financial burdens reduce QOL for cancer survivors

receiving treatment
Photo by Rhoda Baer
An analysis of nearly 20 million cancer survivors showed that almost 29% had financial burdens as a result of their cancer diagnosis and/or treatment.
In other words, they borrowed money, declared bankruptcy, worried about paying large medical bills, were unable to cover the cost of medical visits, or made other financial sacrifices.
Furthermore, such hardships could have lasting effects on a cancer survivor’s quality of life (QOL).
Hrishikesh Kale and Norman Carroll, PhD, both of Virginia Commonwealth University School of Pharmacy in Richmond, reported these findings in Cancer.
The pair analyzed 2011 Medical Expenditure Panel Survey data on 19.6 million cancer survivors, assessing financial burden and QOL.
Subjects were considered to have financial burden if they reported 1 of the following problems: borrowed money/declared bankruptcy, worried about paying large medical bills, unable to cover the cost of medical care visits, or other financial sacrifices.
Nearly 29% of the cancer survivors reported at least 1 financial problem resulting from cancer diagnosis, treatment, or lasting effects of that treatment.
Of all the cancer survivors in the analysis, 20.9% worried about paying large medical bills, 11.5% were unable to cover the cost of medical care visits, 7.6% reported borrowing money or going into debt, 1.4% declared bankruptcy, and 8.6% reported other financial sacrifices.
Cancer survivors who faced such financial difficulties had lower physical and mental health-related QOL, higher risk for depressed mood and psychological distress, and were more likely to worry about cancer recurrence, when compared with cancer survivors who did not face financial problems.
In addition, as the number of financial problems reported by cancer survivors increased, their QOL continued to decrease. And their risk for depressed mood, psychological distress, and worries about cancer recurrence continued to increase.
“Our results suggest that policies and practices that minimize cancer patients’ out-of-pocket costs can improve survivors’ health-related quality of life and psychological health,” Dr Carroll said.
“Reducing the financial burden of cancer care requires integrated efforts, and the study findings are useful for survivorship care programs, oncologists, payers, pharmaceutical companies, and patients and their family members.” ![]()
receiving treatment
Photo by Rhoda Baer
An analysis of nearly 20 million cancer survivors showed that almost 29% had financial burdens as a result of their cancer diagnosis and/or treatment.
In other words, they borrowed money, declared bankruptcy, worried about paying large medical bills, were unable to cover the cost of medical visits, or made other financial sacrifices.
Furthermore, such hardships could have lasting effects on a cancer survivor’s quality of life (QOL).
Hrishikesh Kale and Norman Carroll, PhD, both of Virginia Commonwealth University School of Pharmacy in Richmond, reported these findings in Cancer.
The pair analyzed 2011 Medical Expenditure Panel Survey data on 19.6 million cancer survivors, assessing financial burden and QOL.
Subjects were considered to have financial burden if they reported 1 of the following problems: borrowed money/declared bankruptcy, worried about paying large medical bills, unable to cover the cost of medical care visits, or other financial sacrifices.
Nearly 29% of the cancer survivors reported at least 1 financial problem resulting from cancer diagnosis, treatment, or lasting effects of that treatment.
Of all the cancer survivors in the analysis, 20.9% worried about paying large medical bills, 11.5% were unable to cover the cost of medical care visits, 7.6% reported borrowing money or going into debt, 1.4% declared bankruptcy, and 8.6% reported other financial sacrifices.
Cancer survivors who faced such financial difficulties had lower physical and mental health-related QOL, higher risk for depressed mood and psychological distress, and were more likely to worry about cancer recurrence, when compared with cancer survivors who did not face financial problems.
In addition, as the number of financial problems reported by cancer survivors increased, their QOL continued to decrease. And their risk for depressed mood, psychological distress, and worries about cancer recurrence continued to increase.
“Our results suggest that policies and practices that minimize cancer patients’ out-of-pocket costs can improve survivors’ health-related quality of life and psychological health,” Dr Carroll said.
“Reducing the financial burden of cancer care requires integrated efforts, and the study findings are useful for survivorship care programs, oncologists, payers, pharmaceutical companies, and patients and their family members.” ![]()
receiving treatment
Photo by Rhoda Baer
An analysis of nearly 20 million cancer survivors showed that almost 29% had financial burdens as a result of their cancer diagnosis and/or treatment.
In other words, they borrowed money, declared bankruptcy, worried about paying large medical bills, were unable to cover the cost of medical visits, or made other financial sacrifices.
Furthermore, such hardships could have lasting effects on a cancer survivor’s quality of life (QOL).
Hrishikesh Kale and Norman Carroll, PhD, both of Virginia Commonwealth University School of Pharmacy in Richmond, reported these findings in Cancer.
The pair analyzed 2011 Medical Expenditure Panel Survey data on 19.6 million cancer survivors, assessing financial burden and QOL.
Subjects were considered to have financial burden if they reported 1 of the following problems: borrowed money/declared bankruptcy, worried about paying large medical bills, unable to cover the cost of medical care visits, or other financial sacrifices.
Nearly 29% of the cancer survivors reported at least 1 financial problem resulting from cancer diagnosis, treatment, or lasting effects of that treatment.
Of all the cancer survivors in the analysis, 20.9% worried about paying large medical bills, 11.5% were unable to cover the cost of medical care visits, 7.6% reported borrowing money or going into debt, 1.4% declared bankruptcy, and 8.6% reported other financial sacrifices.
Cancer survivors who faced such financial difficulties had lower physical and mental health-related QOL, higher risk for depressed mood and psychological distress, and were more likely to worry about cancer recurrence, when compared with cancer survivors who did not face financial problems.
In addition, as the number of financial problems reported by cancer survivors increased, their QOL continued to decrease. And their risk for depressed mood, psychological distress, and worries about cancer recurrence continued to increase.
“Our results suggest that policies and practices that minimize cancer patients’ out-of-pocket costs can improve survivors’ health-related quality of life and psychological health,” Dr Carroll said.
“Reducing the financial burden of cancer care requires integrated efforts, and the study findings are useful for survivorship care programs, oncologists, payers, pharmaceutical companies, and patients and their family members.” ![]()
FDA approves propylene glycol–free melphalan for multiple myeloma
The Food and Drug Administration has approved a new propylene glycol–free formulation of melphalan hydrochloride as a high-dose conditioning treatment for autologous stem cell transplantation, and as a palliative therapy for multiple myeloma.
Evomela (Spectrum Pharmaceuticals) is intended to be reconstituted with normal saline at the time of intravenous administration. The solution is stable for 4 hours using Captisol, a proprietary agent containing modified cyclodextrin. It is also stable for 1 hour after reconstitution.
Extended stability without the need for propylene glycol is its major advantage over the other formulations of melphalan, a chemotherapy agent originally approved in 1964, according to a statement by Spectrum.
Evomela was approved on the basis of a phase IIa pharmacokinetic trial comprising 24 patients undergoing autologous stem cell transplantation. Evomela was bioequivalent with Alkeran, with a 10% higher maximum plasma concentration and area under the plasma concentration-time curve (Bone Marrow Transplant. 2014 Aug;49[8]:1042-5).
According to that, and other studies, adverse reactions included decreased neutrophil count (100%), decreased white blood cell count (100%), decreased lymphocyte count (98%), decreased platelet count (98%), diarrhea (93%), nausea (90%), fatigue (77%), hypokalemia (74%), anemia (66%), and vomiting (64%). About 2% of patients have experienced hypersensitivity reactions.
For palliative treatment, the recommended does is 16 mg/m2 infused over 15-20 minutes at 2-week intervals for four doses, and then, after adequate recovery from toxicity, at 4-week intervals.
For conditioning treatment, the recommended does is 100 mg/m2 per day infused over 30 minutes for 2 consecutive days before stem cell transplant.
The Food and Drug Administration has approved a new propylene glycol–free formulation of melphalan hydrochloride as a high-dose conditioning treatment for autologous stem cell transplantation, and as a palliative therapy for multiple myeloma.
Evomela (Spectrum Pharmaceuticals) is intended to be reconstituted with normal saline at the time of intravenous administration. The solution is stable for 4 hours using Captisol, a proprietary agent containing modified cyclodextrin. It is also stable for 1 hour after reconstitution.
Extended stability without the need for propylene glycol is its major advantage over the other formulations of melphalan, a chemotherapy agent originally approved in 1964, according to a statement by Spectrum.
Evomela was approved on the basis of a phase IIa pharmacokinetic trial comprising 24 patients undergoing autologous stem cell transplantation. Evomela was bioequivalent with Alkeran, with a 10% higher maximum plasma concentration and area under the plasma concentration-time curve (Bone Marrow Transplant. 2014 Aug;49[8]:1042-5).
According to that, and other studies, adverse reactions included decreased neutrophil count (100%), decreased white blood cell count (100%), decreased lymphocyte count (98%), decreased platelet count (98%), diarrhea (93%), nausea (90%), fatigue (77%), hypokalemia (74%), anemia (66%), and vomiting (64%). About 2% of patients have experienced hypersensitivity reactions.
For palliative treatment, the recommended does is 16 mg/m2 infused over 15-20 minutes at 2-week intervals for four doses, and then, after adequate recovery from toxicity, at 4-week intervals.
For conditioning treatment, the recommended does is 100 mg/m2 per day infused over 30 minutes for 2 consecutive days before stem cell transplant.
The Food and Drug Administration has approved a new propylene glycol–free formulation of melphalan hydrochloride as a high-dose conditioning treatment for autologous stem cell transplantation, and as a palliative therapy for multiple myeloma.
Evomela (Spectrum Pharmaceuticals) is intended to be reconstituted with normal saline at the time of intravenous administration. The solution is stable for 4 hours using Captisol, a proprietary agent containing modified cyclodextrin. It is also stable for 1 hour after reconstitution.
Extended stability without the need for propylene glycol is its major advantage over the other formulations of melphalan, a chemotherapy agent originally approved in 1964, according to a statement by Spectrum.
Evomela was approved on the basis of a phase IIa pharmacokinetic trial comprising 24 patients undergoing autologous stem cell transplantation. Evomela was bioequivalent with Alkeran, with a 10% higher maximum plasma concentration and area under the plasma concentration-time curve (Bone Marrow Transplant. 2014 Aug;49[8]:1042-5).
According to that, and other studies, adverse reactions included decreased neutrophil count (100%), decreased white blood cell count (100%), decreased lymphocyte count (98%), decreased platelet count (98%), diarrhea (93%), nausea (90%), fatigue (77%), hypokalemia (74%), anemia (66%), and vomiting (64%). About 2% of patients have experienced hypersensitivity reactions.
For palliative treatment, the recommended does is 16 mg/m2 infused over 15-20 minutes at 2-week intervals for four doses, and then, after adequate recovery from toxicity, at 4-week intervals.
For conditioning treatment, the recommended does is 100 mg/m2 per day infused over 30 minutes for 2 consecutive days before stem cell transplant.
Idelalisib use halted in six combo therapy trials, FDA announces
An increased rate of adverse events, including deaths, have been reported in clinical trials with idelalisib (Zydelig) in combination with other cancer medicines, the U.S. Food and Drug Administration announced.
Gilead Sciences, Inc. has confirmed that they are stopping six clinical trials in patients with chronic lymphocytic leukemia, small lymphocytic lymphoma and indolent non-Hodgkin lymphomas. The FDA is reviewing the findings of the clinical trials and will communicate new information as necessary, according to the FDA press release.
![]()
Idelalisib is not approved for previously untreated chronic lymphocytic leukemia. It is approved by the FDA for the treatment of:
• Relapsed chronic lymphocytic leukemia, in combination with rituximab, in patients for whom rituximab alone would be considered appropriate therapy due to other co-morbidities.
• Relapsed follicular B-cell non-Hodgkin lymphoma in patients who have received at least two prior systemic therapies.
• Relapsed small lymphocytic lymphoma in patients who have received at least two prior systemic therapies.
Adverse events involving idelalisib should be reported to the FDA MedWatch program, the release advised.
On Twitter @maryjodales
An increased rate of adverse events, including deaths, have been reported in clinical trials with idelalisib (Zydelig) in combination with other cancer medicines, the U.S. Food and Drug Administration announced.
Gilead Sciences, Inc. has confirmed that they are stopping six clinical trials in patients with chronic lymphocytic leukemia, small lymphocytic lymphoma and indolent non-Hodgkin lymphomas. The FDA is reviewing the findings of the clinical trials and will communicate new information as necessary, according to the FDA press release.
![]()
Idelalisib is not approved for previously untreated chronic lymphocytic leukemia. It is approved by the FDA for the treatment of:
• Relapsed chronic lymphocytic leukemia, in combination with rituximab, in patients for whom rituximab alone would be considered appropriate therapy due to other co-morbidities.
• Relapsed follicular B-cell non-Hodgkin lymphoma in patients who have received at least two prior systemic therapies.
• Relapsed small lymphocytic lymphoma in patients who have received at least two prior systemic therapies.
Adverse events involving idelalisib should be reported to the FDA MedWatch program, the release advised.
On Twitter @maryjodales
An increased rate of adverse events, including deaths, have been reported in clinical trials with idelalisib (Zydelig) in combination with other cancer medicines, the U.S. Food and Drug Administration announced.
Gilead Sciences, Inc. has confirmed that they are stopping six clinical trials in patients with chronic lymphocytic leukemia, small lymphocytic lymphoma and indolent non-Hodgkin lymphomas. The FDA is reviewing the findings of the clinical trials and will communicate new information as necessary, according to the FDA press release.
![]()
Idelalisib is not approved for previously untreated chronic lymphocytic leukemia. It is approved by the FDA for the treatment of:
• Relapsed chronic lymphocytic leukemia, in combination with rituximab, in patients for whom rituximab alone would be considered appropriate therapy due to other co-morbidities.
• Relapsed follicular B-cell non-Hodgkin lymphoma in patients who have received at least two prior systemic therapies.
• Relapsed small lymphocytic lymphoma in patients who have received at least two prior systemic therapies.
Adverse events involving idelalisib should be reported to the FDA MedWatch program, the release advised.
On Twitter @maryjodales
Idelalisib trials stopped due to AEs
Photo courtesy of
Gilead Sciences
The US Food and Drug Administration (FDA) has reported that Gilead Sciences, Inc., is stopping 6 clinical trials of idelalisib (Zydelig) due to adverse events (AEs) observed in patients receiving idelalisib in combination with other drugs.
The AEs, which include deaths, were mostly related to infections.
The trials include patients with chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma, and indolent non-Hodgkin lymphomas.
The FDA said it is reviewing the findings of these trials and will communicate new information as necessary.
A few days ago, the European Medicines Agency (EMA) announced its decision to review the safety of idelalisib due to the aforementioned AEs. The EMA said it is reviewing data from 3 idelalisib trials.
While this review is underway, the EMA advised that patients starting or already on treatment with idelalisib be carefully monitored for signs of infection. If the drug is well tolerated, treatment should not be stopped.
The FDA has not made any recommendations about treatment with idelalisib.
About idelalisib
Idelalisib is currently approved by the FDA for use in combination with rituximab to treat patients with relapsed CLL who cannot receive rituximab alone.
Idelalisib also has accelerated approval from the FDA to treat patients with relapsed follicular lymphoma who have received at least 2 prior systemic therapies and patients with relapsed small lymphocytic lymphoma who have received at least 2 prior systemic therapies.
In the European Union, idelalisib is approved for use in combination with rituximab to treat adults with CLL who have received at least 1 prior therapy or as first-line treatment in the presence of 17p deletion or TP53 mutation in CLL patients deemed unsuitable for chemo-immunotherapy.
Idelalisib is also approved in the European Union as monotherapy for adults with follicular lymphoma that is refractory to 2 prior lines of treatment. ![]()
Photo courtesy of
Gilead Sciences
The US Food and Drug Administration (FDA) has reported that Gilead Sciences, Inc., is stopping 6 clinical trials of idelalisib (Zydelig) due to adverse events (AEs) observed in patients receiving idelalisib in combination with other drugs.
The AEs, which include deaths, were mostly related to infections.
The trials include patients with chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma, and indolent non-Hodgkin lymphomas.
The FDA said it is reviewing the findings of these trials and will communicate new information as necessary.
A few days ago, the European Medicines Agency (EMA) announced its decision to review the safety of idelalisib due to the aforementioned AEs. The EMA said it is reviewing data from 3 idelalisib trials.
While this review is underway, the EMA advised that patients starting or already on treatment with idelalisib be carefully monitored for signs of infection. If the drug is well tolerated, treatment should not be stopped.
The FDA has not made any recommendations about treatment with idelalisib.
About idelalisib
Idelalisib is currently approved by the FDA for use in combination with rituximab to treat patients with relapsed CLL who cannot receive rituximab alone.
Idelalisib also has accelerated approval from the FDA to treat patients with relapsed follicular lymphoma who have received at least 2 prior systemic therapies and patients with relapsed small lymphocytic lymphoma who have received at least 2 prior systemic therapies.
In the European Union, idelalisib is approved for use in combination with rituximab to treat adults with CLL who have received at least 1 prior therapy or as first-line treatment in the presence of 17p deletion or TP53 mutation in CLL patients deemed unsuitable for chemo-immunotherapy.
Idelalisib is also approved in the European Union as monotherapy for adults with follicular lymphoma that is refractory to 2 prior lines of treatment. ![]()
Photo courtesy of
Gilead Sciences
The US Food and Drug Administration (FDA) has reported that Gilead Sciences, Inc., is stopping 6 clinical trials of idelalisib (Zydelig) due to adverse events (AEs) observed in patients receiving idelalisib in combination with other drugs.
The AEs, which include deaths, were mostly related to infections.
The trials include patients with chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma, and indolent non-Hodgkin lymphomas.
The FDA said it is reviewing the findings of these trials and will communicate new information as necessary.
A few days ago, the European Medicines Agency (EMA) announced its decision to review the safety of idelalisib due to the aforementioned AEs. The EMA said it is reviewing data from 3 idelalisib trials.
While this review is underway, the EMA advised that patients starting or already on treatment with idelalisib be carefully monitored for signs of infection. If the drug is well tolerated, treatment should not be stopped.
The FDA has not made any recommendations about treatment with idelalisib.
About idelalisib
Idelalisib is currently approved by the FDA for use in combination with rituximab to treat patients with relapsed CLL who cannot receive rituximab alone.
Idelalisib also has accelerated approval from the FDA to treat patients with relapsed follicular lymphoma who have received at least 2 prior systemic therapies and patients with relapsed small lymphocytic lymphoma who have received at least 2 prior systemic therapies.
In the European Union, idelalisib is approved for use in combination with rituximab to treat adults with CLL who have received at least 1 prior therapy or as first-line treatment in the presence of 17p deletion or TP53 mutation in CLL patients deemed unsuitable for chemo-immunotherapy.
Idelalisib is also approved in the European Union as monotherapy for adults with follicular lymphoma that is refractory to 2 prior lines of treatment. ![]()
Drug granted orphan designation for DLBCL

The US Food and Drug Administration (FDA) has granted orphan designation to the oncology drug candidate PNT2258 for the treatment of diffuse large B-cell lymphoma (DLBCL).
The FDA grants orphan designation to drugs intended to treat conditions affecting fewer than 200,000 patients in the US.
The designation provides the drug’s sponsor with various development incentives, including opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, and 7 years of US market exclusivity if the drug is approved.
About PNT2258
PNT2258 is designed to target cancers that overexpress BCL2, and BCL2 overexpression is thought to be a key driver of DLBCL.
PNT2258 consists of a single-stranded, 24-base DNAi oligonucleotide known as PNT100 that is encapsulated in lipid nanoparticles (LNPs).
The DNAi technology platform is based on a discovery that single-stranded DNA oligonucleotides can interact with genomic DNA to interfere with oncogenes. PNT100 DNAi is designed to target a genetic regulatory region associated with BCL2.
The LNPs are designed to provide enhanced serum stability and optimized pharmacokinetic properties to facilitate broad systemic distribution after intravenous infusion. Within the acidic environment found in tumors, the LNPs become positively charged and therefore more amenable to cellular uptake and cytoplasmic release of their payloads.
Trials of PNT2258
PNT2258 is being developed by ProNAi Therapeutics, Inc. The company has completed 2 trials of PNT2258 to date—a phase 1 trial of patients with solid tumors and a phase 2 trial of patients with non-Hodgkin lymphoma.
The phase 1 trial enrolled 22 patients with relapsed or refractory solid tumor malignancies. Results were published in Cancer Chemotherapy and Pharmacology in February 2014.
PNT2258 was deemed well-tolerated in this trial. There was no evidence of a systemic immune response to the LNPs or PNT100. There were no significant changes in immune-stimulatory cytokines or clinical signs of anaphylaxis following PNT2258 administration.
The phase 2 trial enrolled 13 patients with relapsed or refractory non-Hodgkin lymphoma. Results were presented at ASH 2014 (abstract 1716).
Six patients responded to PNT2258—4 with complete responses and 2 with partial responses. Five patients had stable disease, and 2 progressed. All 4 of the DLBCL patients in this trial responded—3 with complete responses and 1 with a partial response.
Adverse events reported in this trial include nausea (n=11), pain (n=9), chills (n=7), diarrhea (n=7), vomiting (n=7), fatigue (n=6), fever (n=6), headache (n=6), dyspnea (n=5), generalized aching (n=4), anorexia (n=4), back pain (n=4), sensory neuropathy (n=4), hypophosphatemia (n=4), anemia (n=3), hypokalemia (n=3), hyperuricemia (n=2), neutropenia (n=2), thrombocytopenia (n=4), and elevated AST/ALT (n=1).
ProNAi Therapeutics is now enrolling patients in “Wolverine,” a phase 2 trial evaluating PNT2258 in patients with relapsed or refractory DLBCL, and in “Brighton,” a phase 2 trial evaluating PNT2258 for the treatment of Richter’s transformation. ![]()
The US Food and Drug Administration (FDA) has granted orphan designation to the oncology drug candidate PNT2258 for the treatment of diffuse large B-cell lymphoma (DLBCL).
The FDA grants orphan designation to drugs intended to treat conditions affecting fewer than 200,000 patients in the US.
The designation provides the drug’s sponsor with various development incentives, including opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, and 7 years of US market exclusivity if the drug is approved.
About PNT2258
PNT2258 is designed to target cancers that overexpress BCL2, and BCL2 overexpression is thought to be a key driver of DLBCL.
PNT2258 consists of a single-stranded, 24-base DNAi oligonucleotide known as PNT100 that is encapsulated in lipid nanoparticles (LNPs).
The DNAi technology platform is based on a discovery that single-stranded DNA oligonucleotides can interact with genomic DNA to interfere with oncogenes. PNT100 DNAi is designed to target a genetic regulatory region associated with BCL2.
The LNPs are designed to provide enhanced serum stability and optimized pharmacokinetic properties to facilitate broad systemic distribution after intravenous infusion. Within the acidic environment found in tumors, the LNPs become positively charged and therefore more amenable to cellular uptake and cytoplasmic release of their payloads.
Trials of PNT2258
PNT2258 is being developed by ProNAi Therapeutics, Inc. The company has completed 2 trials of PNT2258 to date—a phase 1 trial of patients with solid tumors and a phase 2 trial of patients with non-Hodgkin lymphoma.
The phase 1 trial enrolled 22 patients with relapsed or refractory solid tumor malignancies. Results were published in Cancer Chemotherapy and Pharmacology in February 2014.
PNT2258 was deemed well-tolerated in this trial. There was no evidence of a systemic immune response to the LNPs or PNT100. There were no significant changes in immune-stimulatory cytokines or clinical signs of anaphylaxis following PNT2258 administration.
The phase 2 trial enrolled 13 patients with relapsed or refractory non-Hodgkin lymphoma. Results were presented at ASH 2014 (abstract 1716).
Six patients responded to PNT2258—4 with complete responses and 2 with partial responses. Five patients had stable disease, and 2 progressed. All 4 of the DLBCL patients in this trial responded—3 with complete responses and 1 with a partial response.
Adverse events reported in this trial include nausea (n=11), pain (n=9), chills (n=7), diarrhea (n=7), vomiting (n=7), fatigue (n=6), fever (n=6), headache (n=6), dyspnea (n=5), generalized aching (n=4), anorexia (n=4), back pain (n=4), sensory neuropathy (n=4), hypophosphatemia (n=4), anemia (n=3), hypokalemia (n=3), hyperuricemia (n=2), neutropenia (n=2), thrombocytopenia (n=4), and elevated AST/ALT (n=1).
ProNAi Therapeutics is now enrolling patients in “Wolverine,” a phase 2 trial evaluating PNT2258 in patients with relapsed or refractory DLBCL, and in “Brighton,” a phase 2 trial evaluating PNT2258 for the treatment of Richter’s transformation. ![]()
The US Food and Drug Administration (FDA) has granted orphan designation to the oncology drug candidate PNT2258 for the treatment of diffuse large B-cell lymphoma (DLBCL).
The FDA grants orphan designation to drugs intended to treat conditions affecting fewer than 200,000 patients in the US.
The designation provides the drug’s sponsor with various development incentives, including opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, and 7 years of US market exclusivity if the drug is approved.
About PNT2258
PNT2258 is designed to target cancers that overexpress BCL2, and BCL2 overexpression is thought to be a key driver of DLBCL.
PNT2258 consists of a single-stranded, 24-base DNAi oligonucleotide known as PNT100 that is encapsulated in lipid nanoparticles (LNPs).
The DNAi technology platform is based on a discovery that single-stranded DNA oligonucleotides can interact with genomic DNA to interfere with oncogenes. PNT100 DNAi is designed to target a genetic regulatory region associated with BCL2.
The LNPs are designed to provide enhanced serum stability and optimized pharmacokinetic properties to facilitate broad systemic distribution after intravenous infusion. Within the acidic environment found in tumors, the LNPs become positively charged and therefore more amenable to cellular uptake and cytoplasmic release of their payloads.
Trials of PNT2258
PNT2258 is being developed by ProNAi Therapeutics, Inc. The company has completed 2 trials of PNT2258 to date—a phase 1 trial of patients with solid tumors and a phase 2 trial of patients with non-Hodgkin lymphoma.
The phase 1 trial enrolled 22 patients with relapsed or refractory solid tumor malignancies. Results were published in Cancer Chemotherapy and Pharmacology in February 2014.
PNT2258 was deemed well-tolerated in this trial. There was no evidence of a systemic immune response to the LNPs or PNT100. There were no significant changes in immune-stimulatory cytokines or clinical signs of anaphylaxis following PNT2258 administration.
The phase 2 trial enrolled 13 patients with relapsed or refractory non-Hodgkin lymphoma. Results were presented at ASH 2014 (abstract 1716).
Six patients responded to PNT2258—4 with complete responses and 2 with partial responses. Five patients had stable disease, and 2 progressed. All 4 of the DLBCL patients in this trial responded—3 with complete responses and 1 with a partial response.
Adverse events reported in this trial include nausea (n=11), pain (n=9), chills (n=7), diarrhea (n=7), vomiting (n=7), fatigue (n=6), fever (n=6), headache (n=6), dyspnea (n=5), generalized aching (n=4), anorexia (n=4), back pain (n=4), sensory neuropathy (n=4), hypophosphatemia (n=4), anemia (n=3), hypokalemia (n=3), hyperuricemia (n=2), neutropenia (n=2), thrombocytopenia (n=4), and elevated AST/ALT (n=1).
ProNAi Therapeutics is now enrolling patients in “Wolverine,” a phase 2 trial evaluating PNT2258 in patients with relapsed or refractory DLBCL, and in “Brighton,” a phase 2 trial evaluating PNT2258 for the treatment of Richter’s transformation. ![]()
Temsirolimus results in good but short-duration responses in primary CNS lymphoma
Single-agent therapy with temsirolimus was active in patients with relapsed/refractory primary central nervous system lymphoma, but most of the responses were short lived, results of a phase II trial show.
Among 37 patients with primary CNS lymphoma (PCNSL) for whom firstline therapy had failed, there were five complete responses (CR), three CR unconfirmed, and 12 partial responses (PR), for an overall response rate (ORR) of 54%, reported Dr. Agnieszka Korfel from Charité University Medicine Berlin (Germany) and colleagues.
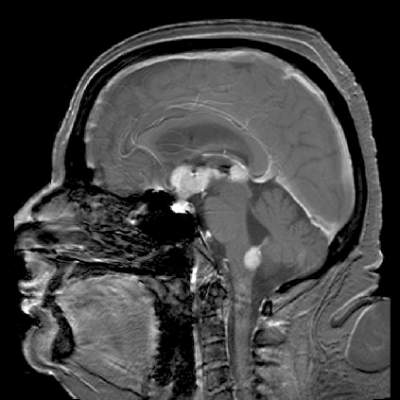
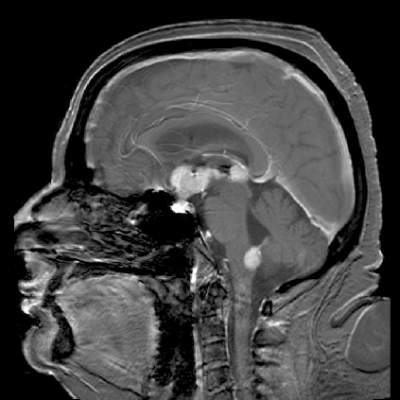
The median progression-free survival (PFS), however, was just 2.1 months, although 1 patient had PFS of 15.8 months duration, and another had a response lasting for more than 44 months, the investigators noted in a study published online in the Journal of Clinical Oncology (doi: 10.1200/JCO.2015.64.9897).
The rationale for trying temsirolimus (Torisel), an inhibitor of the mammalian target of rapamycin (mTOR), came from studies showing the drug’s efficacy against relapsed/refractory mantle-cell lymphoma and against other, more aggressive forms of non-Hodgkin lymphoma. Patients with relapsed/refractory aggressive lymphomas tolerate temsirolimus relatively well, and the drug has the ability to penetrate brain tumor tissue, the authors noted.
They enrolled 37 patients with a median age of 70 years and a median time since their last treatment of 3.9 months into an open-label trial. The patients were all immuncompetent with histologically confirmed primary central nervous system lymphoma for whom high-dose methotrexate-based chemotherapy had failed and for whom high-dose chemotherapy with autologous stem cell transplant had either failed or was not an option.
The first six patients were treated with temsirolimus 25 mg intravenously once weekly, and the remaining 31 were treated with 75 mg IV once weekly until disease progression, intolerable toxicity, patient or physician decision to terminate, or death.
As noted before, ORR, the primary endpoint, was 54%. Median overall survival (OS), a secondary endpoint, was 3.7 months, and 1-year and 2-year OS were 19% and 16.2%, respectively.
The most frequently occurring toxicities include hyperglycemia, myelosuppression, pneumonias and other infections, and fatigue. A total of 28 severe adverse events occurred in 21 patients, including infectious episodes, hospitalizations because of disease progression, deep-vein thromboses, hyperglycemia, and one case each of seizures, grade 4 thrombocytopenia, drug fever, hyponatremia, renal insufficiency, and atrial fibrillation.
“Although most responses were short lived, some patients achieved long-term control. Thus, further evaluation in combination with other drugs seems reasonable. However, one has to be aware of the risk of hematotoxicity and infections necessitating primary antibiotic prophylaxis. Definition of biomarkers allowing identification of potential responders and those who are at particular risk for toxicity would be highly desirable,” the investigators concluded.
Single-agent therapy with temsirolimus was active in patients with relapsed/refractory primary central nervous system lymphoma, but most of the responses were short lived, results of a phase II trial show.
Among 37 patients with primary CNS lymphoma (PCNSL) for whom firstline therapy had failed, there were five complete responses (CR), three CR unconfirmed, and 12 partial responses (PR), for an overall response rate (ORR) of 54%, reported Dr. Agnieszka Korfel from Charité University Medicine Berlin (Germany) and colleagues.
The median progression-free survival (PFS), however, was just 2.1 months, although 1 patient had PFS of 15.8 months duration, and another had a response lasting for more than 44 months, the investigators noted in a study published online in the Journal of Clinical Oncology (doi: 10.1200/JCO.2015.64.9897).
The rationale for trying temsirolimus (Torisel), an inhibitor of the mammalian target of rapamycin (mTOR), came from studies showing the drug’s efficacy against relapsed/refractory mantle-cell lymphoma and against other, more aggressive forms of non-Hodgkin lymphoma. Patients with relapsed/refractory aggressive lymphomas tolerate temsirolimus relatively well, and the drug has the ability to penetrate brain tumor tissue, the authors noted.
They enrolled 37 patients with a median age of 70 years and a median time since their last treatment of 3.9 months into an open-label trial. The patients were all immuncompetent with histologically confirmed primary central nervous system lymphoma for whom high-dose methotrexate-based chemotherapy had failed and for whom high-dose chemotherapy with autologous stem cell transplant had either failed or was not an option.
The first six patients were treated with temsirolimus 25 mg intravenously once weekly, and the remaining 31 were treated with 75 mg IV once weekly until disease progression, intolerable toxicity, patient or physician decision to terminate, or death.
As noted before, ORR, the primary endpoint, was 54%. Median overall survival (OS), a secondary endpoint, was 3.7 months, and 1-year and 2-year OS were 19% and 16.2%, respectively.
The most frequently occurring toxicities include hyperglycemia, myelosuppression, pneumonias and other infections, and fatigue. A total of 28 severe adverse events occurred in 21 patients, including infectious episodes, hospitalizations because of disease progression, deep-vein thromboses, hyperglycemia, and one case each of seizures, grade 4 thrombocytopenia, drug fever, hyponatremia, renal insufficiency, and atrial fibrillation.
“Although most responses were short lived, some patients achieved long-term control. Thus, further evaluation in combination with other drugs seems reasonable. However, one has to be aware of the risk of hematotoxicity and infections necessitating primary antibiotic prophylaxis. Definition of biomarkers allowing identification of potential responders and those who are at particular risk for toxicity would be highly desirable,” the investigators concluded.
Single-agent therapy with temsirolimus was active in patients with relapsed/refractory primary central nervous system lymphoma, but most of the responses were short lived, results of a phase II trial show.
Among 37 patients with primary CNS lymphoma (PCNSL) for whom firstline therapy had failed, there were five complete responses (CR), three CR unconfirmed, and 12 partial responses (PR), for an overall response rate (ORR) of 54%, reported Dr. Agnieszka Korfel from Charité University Medicine Berlin (Germany) and colleagues.
The median progression-free survival (PFS), however, was just 2.1 months, although 1 patient had PFS of 15.8 months duration, and another had a response lasting for more than 44 months, the investigators noted in a study published online in the Journal of Clinical Oncology (doi: 10.1200/JCO.2015.64.9897).
The rationale for trying temsirolimus (Torisel), an inhibitor of the mammalian target of rapamycin (mTOR), came from studies showing the drug’s efficacy against relapsed/refractory mantle-cell lymphoma and against other, more aggressive forms of non-Hodgkin lymphoma. Patients with relapsed/refractory aggressive lymphomas tolerate temsirolimus relatively well, and the drug has the ability to penetrate brain tumor tissue, the authors noted.
They enrolled 37 patients with a median age of 70 years and a median time since their last treatment of 3.9 months into an open-label trial. The patients were all immuncompetent with histologically confirmed primary central nervous system lymphoma for whom high-dose methotrexate-based chemotherapy had failed and for whom high-dose chemotherapy with autologous stem cell transplant had either failed or was not an option.
The first six patients were treated with temsirolimus 25 mg intravenously once weekly, and the remaining 31 were treated with 75 mg IV once weekly until disease progression, intolerable toxicity, patient or physician decision to terminate, or death.
As noted before, ORR, the primary endpoint, was 54%. Median overall survival (OS), a secondary endpoint, was 3.7 months, and 1-year and 2-year OS were 19% and 16.2%, respectively.
The most frequently occurring toxicities include hyperglycemia, myelosuppression, pneumonias and other infections, and fatigue. A total of 28 severe adverse events occurred in 21 patients, including infectious episodes, hospitalizations because of disease progression, deep-vein thromboses, hyperglycemia, and one case each of seizures, grade 4 thrombocytopenia, drug fever, hyponatremia, renal insufficiency, and atrial fibrillation.
“Although most responses were short lived, some patients achieved long-term control. Thus, further evaluation in combination with other drugs seems reasonable. However, one has to be aware of the risk of hematotoxicity and infections necessitating primary antibiotic prophylaxis. Definition of biomarkers allowing identification of potential responders and those who are at particular risk for toxicity would be highly desirable,” the investigators concluded.
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Relapsed/refractory primary CNS lymphoma has a poor prognosis and no standard treatment option.
Major finding: The overall response rate to once-weekly temsirolimus was 54%; most responses were short lived.
Data source: Open-label phase 2 study in 37 adults with relapsed/refractory primary CNS lymphoma.
Disclosures: The study was supported by Pfizer Germany. Dr. Korfel and several colleagues disclosed research support from or consulting/advising for the company.
IMWG issues renal impairment recommendations for myeloma patients
The International Myeloma Working Group has issued new recommendations for the diagnosis and management of multiple myeloma–related renal impairment. Depending on whether the condition is defined as elevated serum creatinine or decreased estimated glomerular filtration rate (eGFR), an estimated 20%-50% of patients with multiple myeloma have renal impairment at the time of diagnosis.
The guidelines recommend that all patients with multiple myeloma (MM) at diagnosis and at disease assessment should be tested for serum creatinine, eGFR, electrolytes, and serum free light chain, if available. Additionally, they recommend that all patients have urine electrophoresis of a sample from a 24-hour urine collection. All of the above are grade A recommendations (J Clin Oncol. 2016 Mar 14. doi: 10.1200/JCO.2015.65.0044).
Patients with nonselective proteinuria or significant albuminuria should undergo renal biopsy to determine the cause of the underlying impairment, the IMWG says (grade B recommendation).
For evaluation of eGFR in patients with stabilized serum creatinine, the IMWG favors the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation, but also acknowledges that eGFR can be assessed with the Modification of Diet in Renal Disease (MDRD) formula (grade A).
“CKD-EPI seems to more accurately reflect GFR than does MDRD, mostly in higher levels of GFR,” the IMWG wrote.
Because the reversibility of renal dysfunction can affect treatment choice, the recommendations noted that for patients on dialysis, achieving independence from dialysis is “strong indication of improvement. For all other patients, IMWG criteria for renal response to therapy are recommended (grade B).
Management
“Acute renal impairment is a myeloma emergency. Diagnosis should be established as fast as possible, and antimyeloma therapy should be started immediately after confirmation of diagnosis to rapidly restore renal function,” working group members wrote.
Supportive care with increased hydration – at least 3 liters per day – is “mandatory” for all with suspected MM-related renal impairment, they add.
The recommendations also noted that antimyeloma therapy should be initiated immediately to reduce the load of toxic serum free light chains, which can help to improve renal function.
“Bortezomib [Velcade]-based regimens remain the cornerstone of the management of myeloma-related renal impairment (grade A). High-dose dexamethasone should be administered at least for the first month of therapy (grade B),” the working group members wrote.
Lenalidomide (Revlimid) can be given, but because it is excreted through the kidneys, the dose must be adjusted according to the degree of renal impairment. In contrast, thalidomide is not excreted and does not require dose modification in this population.
Patients who are eligible for autologous stem cell transplant could receive bortezomib in a three-drug regimen with thalidomide and dexamethasone, or in combination with a conventional chemotherapeutic agent, either doxorubicin or cyclophosphamide. Patients who are not eligible for transplant can be treated with bortezomib, melphalan, and prednisone, the recommendations said, but add that there are no data on the use of this regimen in patients who are on dialysis.
Regarding newer proteasome inhibitors, the guidelines note that carfilzomib (Kyprolis) can be given safely to patients with creatinine clearance above 15 mL/min, and that the recently approved oral agent, ixazomib (Ninlaro), with lenalidomide and dexamethasone can be administered to patients with clearance rates above 30 mL/min (grade A).
The International Myeloma Working Group has issued new recommendations for the diagnosis and management of multiple myeloma–related renal impairment. Depending on whether the condition is defined as elevated serum creatinine or decreased estimated glomerular filtration rate (eGFR), an estimated 20%-50% of patients with multiple myeloma have renal impairment at the time of diagnosis.
The guidelines recommend that all patients with multiple myeloma (MM) at diagnosis and at disease assessment should be tested for serum creatinine, eGFR, electrolytes, and serum free light chain, if available. Additionally, they recommend that all patients have urine electrophoresis of a sample from a 24-hour urine collection. All of the above are grade A recommendations (J Clin Oncol. 2016 Mar 14. doi: 10.1200/JCO.2015.65.0044).
Patients with nonselective proteinuria or significant albuminuria should undergo renal biopsy to determine the cause of the underlying impairment, the IMWG says (grade B recommendation).
For evaluation of eGFR in patients with stabilized serum creatinine, the IMWG favors the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation, but also acknowledges that eGFR can be assessed with the Modification of Diet in Renal Disease (MDRD) formula (grade A).
“CKD-EPI seems to more accurately reflect GFR than does MDRD, mostly in higher levels of GFR,” the IMWG wrote.
Because the reversibility of renal dysfunction can affect treatment choice, the recommendations noted that for patients on dialysis, achieving independence from dialysis is “strong indication of improvement. For all other patients, IMWG criteria for renal response to therapy are recommended (grade B).
Management
“Acute renal impairment is a myeloma emergency. Diagnosis should be established as fast as possible, and antimyeloma therapy should be started immediately after confirmation of diagnosis to rapidly restore renal function,” working group members wrote.
Supportive care with increased hydration – at least 3 liters per day – is “mandatory” for all with suspected MM-related renal impairment, they add.
The recommendations also noted that antimyeloma therapy should be initiated immediately to reduce the load of toxic serum free light chains, which can help to improve renal function.
“Bortezomib [Velcade]-based regimens remain the cornerstone of the management of myeloma-related renal impairment (grade A). High-dose dexamethasone should be administered at least for the first month of therapy (grade B),” the working group members wrote.
Lenalidomide (Revlimid) can be given, but because it is excreted through the kidneys, the dose must be adjusted according to the degree of renal impairment. In contrast, thalidomide is not excreted and does not require dose modification in this population.
Patients who are eligible for autologous stem cell transplant could receive bortezomib in a three-drug regimen with thalidomide and dexamethasone, or in combination with a conventional chemotherapeutic agent, either doxorubicin or cyclophosphamide. Patients who are not eligible for transplant can be treated with bortezomib, melphalan, and prednisone, the recommendations said, but add that there are no data on the use of this regimen in patients who are on dialysis.
Regarding newer proteasome inhibitors, the guidelines note that carfilzomib (Kyprolis) can be given safely to patients with creatinine clearance above 15 mL/min, and that the recently approved oral agent, ixazomib (Ninlaro), with lenalidomide and dexamethasone can be administered to patients with clearance rates above 30 mL/min (grade A).
The International Myeloma Working Group has issued new recommendations for the diagnosis and management of multiple myeloma–related renal impairment. Depending on whether the condition is defined as elevated serum creatinine or decreased estimated glomerular filtration rate (eGFR), an estimated 20%-50% of patients with multiple myeloma have renal impairment at the time of diagnosis.
The guidelines recommend that all patients with multiple myeloma (MM) at diagnosis and at disease assessment should be tested for serum creatinine, eGFR, electrolytes, and serum free light chain, if available. Additionally, they recommend that all patients have urine electrophoresis of a sample from a 24-hour urine collection. All of the above are grade A recommendations (J Clin Oncol. 2016 Mar 14. doi: 10.1200/JCO.2015.65.0044).
Patients with nonselective proteinuria or significant albuminuria should undergo renal biopsy to determine the cause of the underlying impairment, the IMWG says (grade B recommendation).
For evaluation of eGFR in patients with stabilized serum creatinine, the IMWG favors the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation, but also acknowledges that eGFR can be assessed with the Modification of Diet in Renal Disease (MDRD) formula (grade A).
“CKD-EPI seems to more accurately reflect GFR than does MDRD, mostly in higher levels of GFR,” the IMWG wrote.
Because the reversibility of renal dysfunction can affect treatment choice, the recommendations noted that for patients on dialysis, achieving independence from dialysis is “strong indication of improvement. For all other patients, IMWG criteria for renal response to therapy are recommended (grade B).
Management
“Acute renal impairment is a myeloma emergency. Diagnosis should be established as fast as possible, and antimyeloma therapy should be started immediately after confirmation of diagnosis to rapidly restore renal function,” working group members wrote.
Supportive care with increased hydration – at least 3 liters per day – is “mandatory” for all with suspected MM-related renal impairment, they add.
The recommendations also noted that antimyeloma therapy should be initiated immediately to reduce the load of toxic serum free light chains, which can help to improve renal function.
“Bortezomib [Velcade]-based regimens remain the cornerstone of the management of myeloma-related renal impairment (grade A). High-dose dexamethasone should be administered at least for the first month of therapy (grade B),” the working group members wrote.
Lenalidomide (Revlimid) can be given, but because it is excreted through the kidneys, the dose must be adjusted according to the degree of renal impairment. In contrast, thalidomide is not excreted and does not require dose modification in this population.
Patients who are eligible for autologous stem cell transplant could receive bortezomib in a three-drug regimen with thalidomide and dexamethasone, or in combination with a conventional chemotherapeutic agent, either doxorubicin or cyclophosphamide. Patients who are not eligible for transplant can be treated with bortezomib, melphalan, and prednisone, the recommendations said, but add that there are no data on the use of this regimen in patients who are on dialysis.
Regarding newer proteasome inhibitors, the guidelines note that carfilzomib (Kyprolis) can be given safely to patients with creatinine clearance above 15 mL/min, and that the recently approved oral agent, ixazomib (Ninlaro), with lenalidomide and dexamethasone can be administered to patients with clearance rates above 30 mL/min (grade A).
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: All patients diagnosed with multiple myeloma should be evaluated for renal impairment.
Major finding: Bortezomib-based regimens are the standard of care for patients with multiple myeloma.
Data source: Evidence-based clinical recommendations.
Disclosures: Many coauthors disclosed multiple relationships with companies that make antimyeloma therapies and other medications.
Lenalidomide, thalidomide in melphalan regimen yields similar progression-free survival
Swapping lenalidomide for thalidomide in a standard regimen for transplant-ineligible patients with untreated multiple myeloma did not improve efficacy, but the toxicity profile may favor the use of lenalidomide in a maintenance regimen, results of a randomized trial suggest.
Among patients with previously untreated multiple myeloma who were not eligible for autologous stem cell transplant, neither median progression-free survival (PFS) nor overall survival (OS) were significantly different for patients treated with either melphalan, prednisone, and thalidomide (MPT-T) followed by thalidomide maintenance, or with the same regimen with lenalidomide (Revlimid) substituted for thalidomide (MPR-R), reported Dr. Sonja Zweegman of Vrije University Medical Center in Amsterdam.
“MPR-R has no advantage over MPT-T with respect to response rate, PFS, and OS. However, the use of thalidomide as maintenance therapy was associated with a high rate of clinically significant neuropathy and is therefore not preferred for maintenance strategies,” they wrote (Blood 2016;127[9];1109-16).
The investigators randomly assigned 637 transplant-ineligible patients with newly-diagnosed multiple myeloma to receive nine 4-week cycles of either MPT-T (318 patients) or MPR-R (319 patients). At 36 months’ median follow-up, median PFS, the primary endpoint, was 20 months for patients treated with MPT-T, compared with 23 months for those treated with MPR-R. This translated into a hazard ratio (HR) of 0.87, P = .12).
The overall response rates were 81% for MPT-T and 84% for MPR-R. Very good partial responses or better were seen in 47% and 45%, of patients, respectively. The complete response rate with MPT-T was 10%, compared with 13% for MPR-R. Median time to response and time to maximum response were similar between the arms.
OS at 2, 3, and 4 years in the MPT-T and MPR-R arms was 73% vs. 84%, 64% vs 69%, and 52% vs. 56%, respectively. These differences were not statistically significant.
The proportion of patients with one or more grade 3 or 4 adverse events was 81% with thalidomide and 86% with lenalidomide.
The investigators noted, however, that there was a high rate of discontinuation during induction therapy in each arm, with 49% of those starting on MPT-T and 41% of those starting on MPR-R halting therapy. Most of the patients who discontinued were older than age 75. Early treatment deaths (within three cycles) occurred in 13 patients on MPT-T, and 8 on MPR-R.
Among patients who started on maintenance therapy, significantly more patients on thalidomide had to discontinue thalidomide than did patients who started on lenalidomide maintenance (60% vs, 17%, P = .017).
The primary reason for the higher rate of discontinuation of MPT-T maintenance was neuropathy, which occurred in 87% of the discontinuations in this study arm, compared with 3% of those in the lenalidomide arm. Neuropathy of at least grade 3 was 16% in the MPT-T arm vs. 2% in MPR-R, resulting in a significantly shorter duration of maintenance therapy (5 vs. 17 months in MPR-R), irrespective of age.
Hematologic toxicities were higher in the MPR-R group, especially grades 3 and 4 neutropenia (64% vs 27%), but this did not translate into a higher clinical infection rate, and the toxicities were manageable in older patients, the investigators reported.
Swapping lenalidomide for thalidomide in a standard regimen for transplant-ineligible patients with untreated multiple myeloma did not improve efficacy, but the toxicity profile may favor the use of lenalidomide in a maintenance regimen, results of a randomized trial suggest.
Among patients with previously untreated multiple myeloma who were not eligible for autologous stem cell transplant, neither median progression-free survival (PFS) nor overall survival (OS) were significantly different for patients treated with either melphalan, prednisone, and thalidomide (MPT-T) followed by thalidomide maintenance, or with the same regimen with lenalidomide (Revlimid) substituted for thalidomide (MPR-R), reported Dr. Sonja Zweegman of Vrije University Medical Center in Amsterdam.
“MPR-R has no advantage over MPT-T with respect to response rate, PFS, and OS. However, the use of thalidomide as maintenance therapy was associated with a high rate of clinically significant neuropathy and is therefore not preferred for maintenance strategies,” they wrote (Blood 2016;127[9];1109-16).
The investigators randomly assigned 637 transplant-ineligible patients with newly-diagnosed multiple myeloma to receive nine 4-week cycles of either MPT-T (318 patients) or MPR-R (319 patients). At 36 months’ median follow-up, median PFS, the primary endpoint, was 20 months for patients treated with MPT-T, compared with 23 months for those treated with MPR-R. This translated into a hazard ratio (HR) of 0.87, P = .12).
The overall response rates were 81% for MPT-T and 84% for MPR-R. Very good partial responses or better were seen in 47% and 45%, of patients, respectively. The complete response rate with MPT-T was 10%, compared with 13% for MPR-R. Median time to response and time to maximum response were similar between the arms.
OS at 2, 3, and 4 years in the MPT-T and MPR-R arms was 73% vs. 84%, 64% vs 69%, and 52% vs. 56%, respectively. These differences were not statistically significant.
The proportion of patients with one or more grade 3 or 4 adverse events was 81% with thalidomide and 86% with lenalidomide.
The investigators noted, however, that there was a high rate of discontinuation during induction therapy in each arm, with 49% of those starting on MPT-T and 41% of those starting on MPR-R halting therapy. Most of the patients who discontinued were older than age 75. Early treatment deaths (within three cycles) occurred in 13 patients on MPT-T, and 8 on MPR-R.
Among patients who started on maintenance therapy, significantly more patients on thalidomide had to discontinue thalidomide than did patients who started on lenalidomide maintenance (60% vs, 17%, P = .017).
The primary reason for the higher rate of discontinuation of MPT-T maintenance was neuropathy, which occurred in 87% of the discontinuations in this study arm, compared with 3% of those in the lenalidomide arm. Neuropathy of at least grade 3 was 16% in the MPT-T arm vs. 2% in MPR-R, resulting in a significantly shorter duration of maintenance therapy (5 vs. 17 months in MPR-R), irrespective of age.
Hematologic toxicities were higher in the MPR-R group, especially grades 3 and 4 neutropenia (64% vs 27%), but this did not translate into a higher clinical infection rate, and the toxicities were manageable in older patients, the investigators reported.
Swapping lenalidomide for thalidomide in a standard regimen for transplant-ineligible patients with untreated multiple myeloma did not improve efficacy, but the toxicity profile may favor the use of lenalidomide in a maintenance regimen, results of a randomized trial suggest.
Among patients with previously untreated multiple myeloma who were not eligible for autologous stem cell transplant, neither median progression-free survival (PFS) nor overall survival (OS) were significantly different for patients treated with either melphalan, prednisone, and thalidomide (MPT-T) followed by thalidomide maintenance, or with the same regimen with lenalidomide (Revlimid) substituted for thalidomide (MPR-R), reported Dr. Sonja Zweegman of Vrije University Medical Center in Amsterdam.
“MPR-R has no advantage over MPT-T with respect to response rate, PFS, and OS. However, the use of thalidomide as maintenance therapy was associated with a high rate of clinically significant neuropathy and is therefore not preferred for maintenance strategies,” they wrote (Blood 2016;127[9];1109-16).
The investigators randomly assigned 637 transplant-ineligible patients with newly-diagnosed multiple myeloma to receive nine 4-week cycles of either MPT-T (318 patients) or MPR-R (319 patients). At 36 months’ median follow-up, median PFS, the primary endpoint, was 20 months for patients treated with MPT-T, compared with 23 months for those treated with MPR-R. This translated into a hazard ratio (HR) of 0.87, P = .12).
The overall response rates were 81% for MPT-T and 84% for MPR-R. Very good partial responses or better were seen in 47% and 45%, of patients, respectively. The complete response rate with MPT-T was 10%, compared with 13% for MPR-R. Median time to response and time to maximum response were similar between the arms.
OS at 2, 3, and 4 years in the MPT-T and MPR-R arms was 73% vs. 84%, 64% vs 69%, and 52% vs. 56%, respectively. These differences were not statistically significant.
The proportion of patients with one or more grade 3 or 4 adverse events was 81% with thalidomide and 86% with lenalidomide.
The investigators noted, however, that there was a high rate of discontinuation during induction therapy in each arm, with 49% of those starting on MPT-T and 41% of those starting on MPR-R halting therapy. Most of the patients who discontinued were older than age 75. Early treatment deaths (within three cycles) occurred in 13 patients on MPT-T, and 8 on MPR-R.
Among patients who started on maintenance therapy, significantly more patients on thalidomide had to discontinue thalidomide than did patients who started on lenalidomide maintenance (60% vs, 17%, P = .017).
The primary reason for the higher rate of discontinuation of MPT-T maintenance was neuropathy, which occurred in 87% of the discontinuations in this study arm, compared with 3% of those in the lenalidomide arm. Neuropathy of at least grade 3 was 16% in the MPT-T arm vs. 2% in MPR-R, resulting in a significantly shorter duration of maintenance therapy (5 vs. 17 months in MPR-R), irrespective of age.
Hematologic toxicities were higher in the MPR-R group, especially grades 3 and 4 neutropenia (64% vs 27%), but this did not translate into a higher clinical infection rate, and the toxicities were manageable in older patients, the investigators reported.
FROM BLOOD
Key clinical point: Lenalidomide or thalidomide added to a melphalan backbone regimen yielded similar outcomes in transplant-ineligible patients with newly diagnosed multiple myeloma.
Major finding: At 36 months median follow-up, median PFS was 20 months for patients treated with thalidomide, compared with 23 months for those treated with lenalidomide.
Data source: Randomized controlled trial of 637 patients with multiple myeloma who were not candidates for autologous stem cell transplantation.
Disclosures: The Dutch Cancer Society, Norwegian Cancer Society, and Celgene supported the study. Dr. Zweegman and other colleagues reported financial relationships with Celgene.
AEs prompt EMA review of idelalisib
Photo courtesy of
Gilead Sciences, Inc.
The European Medicines Agency (EMA) is reviewing the safety of idelalisib (Zydelig), a drug approved to treat chronic lymphocytic leukemia (CLL) and follicular lymphoma in the European Union (EU).
The European Commission (EC) requested the review because of serious adverse events (AEs), including deaths, reported in 3 clinical trials investigating idelalisib in combination with other drugs.
The AEs were mostly infection-related.
The EMA is reviewing data from these studies to assess whether the findings have any consequences for the authorized uses of idelalisib.
In the meantime, the EMA advises that patients starting or already on treatment with idelalisib be carefully monitored for signs of infections. If the drug is well tolerated, treatment should not be stopped.
The EMA is considering whether any other immediate measures are necessary during the review period. The agency said it will communicate further and keep doctors and patients informed as appropriate.
About idelalisib
In the EU, idelalisib is approved for use in combination with rituximab to treat adults with CLL who have received at least 1 prior therapy or as first-line treatment in the presence of 17p deletion or TP53 mutation in CLL patients unsuitable for chemo-immunotherapy.
Idelalisib is also approved as monotherapy for adults with follicular lymphoma that is refractory to 2 prior lines of treatment.
About the trials
The trials in which patients have experienced serious AEs involve patients with CLL and indolent non-Hodgkin lymphoma (NHL).
In one trial (NCT01732926), researchers are evaluating idelalisib in combination with bendamustine and rituximab for previously treated indolent NHL.
In another (NCT01732913), researchers are testing idelalisib in combination with rituximab for previously treated indolent NHL.
And in the third (NCT01980888), researchers are evaluating idelalisib in combination with bendamustine and rituximab in patients with previously untreated CLL.
The EMA noted that these studies are investigating combinations of drugs that are currently not approved in the EU and include patients with disease characteristics different from those covered by the approved indications for idelalisib.
About the review
The EMA has begun the review of idelalisib at the request of the EC, under Article 20 of Directive 2001/83/EC.
The review is being carried out by the EMA’s Pharmacovigilance Risk Assessment Committee, the committee responsible for the evaluation of safety issues for human medicines, which will make a set of recommendations.
Those recommendations will then be forwarded to the Committee for Medicinal Products for Human Use, which is responsible for questions concerning medicines for human use and will adopt a final opinion on the safety of idelalisib.
The final stage of the review procedure is the EC’s adoption of a legally binding decision that is applicable in all EU member states. ![]()
Photo courtesy of
Gilead Sciences, Inc.
The European Medicines Agency (EMA) is reviewing the safety of idelalisib (Zydelig), a drug approved to treat chronic lymphocytic leukemia (CLL) and follicular lymphoma in the European Union (EU).
The European Commission (EC) requested the review because of serious adverse events (AEs), including deaths, reported in 3 clinical trials investigating idelalisib in combination with other drugs.
The AEs were mostly infection-related.
The EMA is reviewing data from these studies to assess whether the findings have any consequences for the authorized uses of idelalisib.
In the meantime, the EMA advises that patients starting or already on treatment with idelalisib be carefully monitored for signs of infections. If the drug is well tolerated, treatment should not be stopped.
The EMA is considering whether any other immediate measures are necessary during the review period. The agency said it will communicate further and keep doctors and patients informed as appropriate.
About idelalisib
In the EU, idelalisib is approved for use in combination with rituximab to treat adults with CLL who have received at least 1 prior therapy or as first-line treatment in the presence of 17p deletion or TP53 mutation in CLL patients unsuitable for chemo-immunotherapy.
Idelalisib is also approved as monotherapy for adults with follicular lymphoma that is refractory to 2 prior lines of treatment.
About the trials
The trials in which patients have experienced serious AEs involve patients with CLL and indolent non-Hodgkin lymphoma (NHL).
In one trial (NCT01732926), researchers are evaluating idelalisib in combination with bendamustine and rituximab for previously treated indolent NHL.
In another (NCT01732913), researchers are testing idelalisib in combination with rituximab for previously treated indolent NHL.
And in the third (NCT01980888), researchers are evaluating idelalisib in combination with bendamustine and rituximab in patients with previously untreated CLL.
The EMA noted that these studies are investigating combinations of drugs that are currently not approved in the EU and include patients with disease characteristics different from those covered by the approved indications for idelalisib.
About the review
The EMA has begun the review of idelalisib at the request of the EC, under Article 20 of Directive 2001/83/EC.
The review is being carried out by the EMA’s Pharmacovigilance Risk Assessment Committee, the committee responsible for the evaluation of safety issues for human medicines, which will make a set of recommendations.
Those recommendations will then be forwarded to the Committee for Medicinal Products for Human Use, which is responsible for questions concerning medicines for human use and will adopt a final opinion on the safety of idelalisib.
The final stage of the review procedure is the EC’s adoption of a legally binding decision that is applicable in all EU member states. ![]()
Photo courtesy of
Gilead Sciences, Inc.
The European Medicines Agency (EMA) is reviewing the safety of idelalisib (Zydelig), a drug approved to treat chronic lymphocytic leukemia (CLL) and follicular lymphoma in the European Union (EU).
The European Commission (EC) requested the review because of serious adverse events (AEs), including deaths, reported in 3 clinical trials investigating idelalisib in combination with other drugs.
The AEs were mostly infection-related.
The EMA is reviewing data from these studies to assess whether the findings have any consequences for the authorized uses of idelalisib.
In the meantime, the EMA advises that patients starting or already on treatment with idelalisib be carefully monitored for signs of infections. If the drug is well tolerated, treatment should not be stopped.
The EMA is considering whether any other immediate measures are necessary during the review period. The agency said it will communicate further and keep doctors and patients informed as appropriate.
About idelalisib
In the EU, idelalisib is approved for use in combination with rituximab to treat adults with CLL who have received at least 1 prior therapy or as first-line treatment in the presence of 17p deletion or TP53 mutation in CLL patients unsuitable for chemo-immunotherapy.
Idelalisib is also approved as monotherapy for adults with follicular lymphoma that is refractory to 2 prior lines of treatment.
About the trials
The trials in which patients have experienced serious AEs involve patients with CLL and indolent non-Hodgkin lymphoma (NHL).
In one trial (NCT01732926), researchers are evaluating idelalisib in combination with bendamustine and rituximab for previously treated indolent NHL.
In another (NCT01732913), researchers are testing idelalisib in combination with rituximab for previously treated indolent NHL.
And in the third (NCT01980888), researchers are evaluating idelalisib in combination with bendamustine and rituximab in patients with previously untreated CLL.
The EMA noted that these studies are investigating combinations of drugs that are currently not approved in the EU and include patients with disease characteristics different from those covered by the approved indications for idelalisib.
About the review
The EMA has begun the review of idelalisib at the request of the EC, under Article 20 of Directive 2001/83/EC.
The review is being carried out by the EMA’s Pharmacovigilance Risk Assessment Committee, the committee responsible for the evaluation of safety issues for human medicines, which will make a set of recommendations.
Those recommendations will then be forwarded to the Committee for Medicinal Products for Human Use, which is responsible for questions concerning medicines for human use and will adopt a final opinion on the safety of idelalisib.
The final stage of the review procedure is the EC’s adoption of a legally binding decision that is applicable in all EU member states. ![]()