User login
PD-1 inhibitor granted accelerated approval for cHL

Photo courtesy of Business Wire
The US Food and Drug Administration (FDA) has granted accelerated approval for the PD-1 inhibitor nivolumab (Opdivo) to treat classical Hodgkin lymphoma (cHL).
The drug is approved to treat patients with relapsed or refractory cHL who have received an autologous hematopoietic stem cell transplant (HSCT) and post-transplant brentuximab vedotin.
Nivolumab received accelerated approval because it has not yet shown a clinical benefit in these patients. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of nivolumab for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted nivolumab breakthrough therapy designation, priority review status, and orphan drug designation.
Dosing and precautions
The recommended dose and schedule of nivolumab for cHL patients is 3 mg/kg intravenously every 2 weeks until disease progression or unacceptable toxicity.
The FDA added a new “Warning and Precaution” to the label for nivolumab, regarding complications of allogeneic HSCT after nivolumab.
Transplant-related deaths have occurred. So the FDA said healthcare professionals should follow patients closely for early evidence of transplant-related complications, such as hyperacute graft-versus-host disease (GVHD), severe acute GVHD, steroid-requiring febrile syndrome, hepatic veno-occlusive disease, and other immune-mediated adverse reactions.
The FDA has required the manufacturer of nivolumab, Bristol-Myers Squibb, to further study the safety of allogeneic HSCT after nivolumab.
Full prescribing information for the drug is available here.
Trials of nivolumab
The FDA granted nivolumab accelerated approval in cHL patients based on the results of 2 single-arm, multicenter trials—the phase 1 Checkmate 039 trial (presented at ICML last year) and the phase 2 CheckMate 205 trial (to be presented at ASCO 2016).
Efficacy
Thus far, researchers have evaluated the efficacy of nivolumab in 95 cHL patients from both trials. All of these patients previously received an autologous HSCT and post-transplant brentuximab vedotin. They received a median of 5 prior systemic regimens (range, 3 to 15).
The patients received a median of 17 doses of nivolumab (range, 3 to 48). The overall response rate was 65%, and the complete response rate was 7%.
The median time to response was 2.1 months (range, 0.7 to 5.7), and the estimated median duration of response was 8.7 months (range, 0+ to 23.1+).
Safety
Researchers evaluated the safety of nivolumab in 263 patients with relapsed or refractory cHL. Ninety-eight percent of these patients had received an autologous HSCT. The patients received a median of 10 doses of nivolumab (range, 1 to 48) at the approved dose and schedule.
The most common (≥20%) adverse events (AEs) of any grade were fatigue, upper respiratory tract infection, cough, pyrexia, and diarrhea.
Additional common (≥10%) AEs included rash, pruritus, musculoskeletal pain, nausea, vomiting, abdominal pain, headache, peripheral neuropathy, arthralgia, dyspnea, infusion-related reactions, and hypothyroidism or thyroiditis.
Serious AEs were reported in 21% of patients. The most common, reported in 1% to 3% of patients, were pneumonia, pleural effusion, pneumonitis, pyrexia, infusion-related reaction, and rash. ![]()
Photo courtesy of Business Wire
The US Food and Drug Administration (FDA) has granted accelerated approval for the PD-1 inhibitor nivolumab (Opdivo) to treat classical Hodgkin lymphoma (cHL).
The drug is approved to treat patients with relapsed or refractory cHL who have received an autologous hematopoietic stem cell transplant (HSCT) and post-transplant brentuximab vedotin.
Nivolumab received accelerated approval because it has not yet shown a clinical benefit in these patients. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of nivolumab for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted nivolumab breakthrough therapy designation, priority review status, and orphan drug designation.
Dosing and precautions
The recommended dose and schedule of nivolumab for cHL patients is 3 mg/kg intravenously every 2 weeks until disease progression or unacceptable toxicity.
The FDA added a new “Warning and Precaution” to the label for nivolumab, regarding complications of allogeneic HSCT after nivolumab.
Transplant-related deaths have occurred. So the FDA said healthcare professionals should follow patients closely for early evidence of transplant-related complications, such as hyperacute graft-versus-host disease (GVHD), severe acute GVHD, steroid-requiring febrile syndrome, hepatic veno-occlusive disease, and other immune-mediated adverse reactions.
The FDA has required the manufacturer of nivolumab, Bristol-Myers Squibb, to further study the safety of allogeneic HSCT after nivolumab.
Full prescribing information for the drug is available here.
Trials of nivolumab
The FDA granted nivolumab accelerated approval in cHL patients based on the results of 2 single-arm, multicenter trials—the phase 1 Checkmate 039 trial (presented at ICML last year) and the phase 2 CheckMate 205 trial (to be presented at ASCO 2016).
Efficacy
Thus far, researchers have evaluated the efficacy of nivolumab in 95 cHL patients from both trials. All of these patients previously received an autologous HSCT and post-transplant brentuximab vedotin. They received a median of 5 prior systemic regimens (range, 3 to 15).
The patients received a median of 17 doses of nivolumab (range, 3 to 48). The overall response rate was 65%, and the complete response rate was 7%.
The median time to response was 2.1 months (range, 0.7 to 5.7), and the estimated median duration of response was 8.7 months (range, 0+ to 23.1+).
Safety
Researchers evaluated the safety of nivolumab in 263 patients with relapsed or refractory cHL. Ninety-eight percent of these patients had received an autologous HSCT. The patients received a median of 10 doses of nivolumab (range, 1 to 48) at the approved dose and schedule.
The most common (≥20%) adverse events (AEs) of any grade were fatigue, upper respiratory tract infection, cough, pyrexia, and diarrhea.
Additional common (≥10%) AEs included rash, pruritus, musculoskeletal pain, nausea, vomiting, abdominal pain, headache, peripheral neuropathy, arthralgia, dyspnea, infusion-related reactions, and hypothyroidism or thyroiditis.
Serious AEs were reported in 21% of patients. The most common, reported in 1% to 3% of patients, were pneumonia, pleural effusion, pneumonitis, pyrexia, infusion-related reaction, and rash. ![]()
Photo courtesy of Business Wire
The US Food and Drug Administration (FDA) has granted accelerated approval for the PD-1 inhibitor nivolumab (Opdivo) to treat classical Hodgkin lymphoma (cHL).
The drug is approved to treat patients with relapsed or refractory cHL who have received an autologous hematopoietic stem cell transplant (HSCT) and post-transplant brentuximab vedotin.
Nivolumab received accelerated approval because it has not yet shown a clinical benefit in these patients. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of nivolumab for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted nivolumab breakthrough therapy designation, priority review status, and orphan drug designation.
Dosing and precautions
The recommended dose and schedule of nivolumab for cHL patients is 3 mg/kg intravenously every 2 weeks until disease progression or unacceptable toxicity.
The FDA added a new “Warning and Precaution” to the label for nivolumab, regarding complications of allogeneic HSCT after nivolumab.
Transplant-related deaths have occurred. So the FDA said healthcare professionals should follow patients closely for early evidence of transplant-related complications, such as hyperacute graft-versus-host disease (GVHD), severe acute GVHD, steroid-requiring febrile syndrome, hepatic veno-occlusive disease, and other immune-mediated adverse reactions.
The FDA has required the manufacturer of nivolumab, Bristol-Myers Squibb, to further study the safety of allogeneic HSCT after nivolumab.
Full prescribing information for the drug is available here.
Trials of nivolumab
The FDA granted nivolumab accelerated approval in cHL patients based on the results of 2 single-arm, multicenter trials—the phase 1 Checkmate 039 trial (presented at ICML last year) and the phase 2 CheckMate 205 trial (to be presented at ASCO 2016).
Efficacy
Thus far, researchers have evaluated the efficacy of nivolumab in 95 cHL patients from both trials. All of these patients previously received an autologous HSCT and post-transplant brentuximab vedotin. They received a median of 5 prior systemic regimens (range, 3 to 15).
The patients received a median of 17 doses of nivolumab (range, 3 to 48). The overall response rate was 65%, and the complete response rate was 7%.
The median time to response was 2.1 months (range, 0.7 to 5.7), and the estimated median duration of response was 8.7 months (range, 0+ to 23.1+).
Safety
Researchers evaluated the safety of nivolumab in 263 patients with relapsed or refractory cHL. Ninety-eight percent of these patients had received an autologous HSCT. The patients received a median of 10 doses of nivolumab (range, 1 to 48) at the approved dose and schedule.
The most common (≥20%) adverse events (AEs) of any grade were fatigue, upper respiratory tract infection, cough, pyrexia, and diarrhea.
Additional common (≥10%) AEs included rash, pruritus, musculoskeletal pain, nausea, vomiting, abdominal pain, headache, peripheral neuropathy, arthralgia, dyspnea, infusion-related reactions, and hypothyroidism or thyroiditis.
Serious AEs were reported in 21% of patients. The most common, reported in 1% to 3% of patients, were pneumonia, pleural effusion, pneumonitis, pyrexia, infusion-related reaction, and rash. ![]()
FDA grants accelerated approval to nivolumab for Hodgkin lymphoma
The Food and Drug Administration has granted accelerated approval to nivolumab for the treatment of patients with classical Hodgkin lymphoma (cHL) that has relapsed or progressed after autologous hematopoietic stem cell transplantation (HSCT) and posttransplantation brentuximab vedotin.
Approval was based on a 65% objective response rate in 95 patients treated with nivolumab following autologous HSCT and posttransplantation brentuximab vedotin. All patients in the single-arm, multicenter trial had relapsed or refractory cHL and were enrolled regardless of PD-L1 expression status. Patients received a median of 17 doses of nivolumab, the FDA said in a written statement.
![]()
The median time to response was 2.1 months (range, 0.7-5.7 months). The estimated median duration of response was 8.7 months.
The FDA also issued a warning for complications of allogeneic HSCT after nivolumab, reporting that transplant-related deaths have occurred. Health care professionals should follow patients closely for early evidence of transplant-related complications, such as hyperacute graft-versus-host disease (GVHD), severe acute GVHD, steroid-requiring febrile syndrome, hepatic veno-occlusive disease, and other immune-mediated adverse reactions, they said.
The most common adverse reactions in a second single-arm study used to evaluate safety (n = 263) were upper respiratory tract infection, cough, pyrexia, and diarrhea. Other immune-mediated adverse reactions, occurring in 1%-5% of patients, included rash, pneumonitis, hepatitis, hyperthyroidism, and colitis. The most common serious adverse reactions, which were reported in 1%-3% of patients, were pneumonia, pleural effusion, pneumonitis, pyrexia, infusion-related reaction, and rash.
Nivolumab is marketed as Opdivo by Bristol-Myers Squibb and has been previously approved to treat advanced renal cell carcinoma, lung cancer, and melanoma.
lnikolaides@frontlinemedcom.com
On Twitter @NikolaidesLaura
The Food and Drug Administration has granted accelerated approval to nivolumab for the treatment of patients with classical Hodgkin lymphoma (cHL) that has relapsed or progressed after autologous hematopoietic stem cell transplantation (HSCT) and posttransplantation brentuximab vedotin.
Approval was based on a 65% objective response rate in 95 patients treated with nivolumab following autologous HSCT and posttransplantation brentuximab vedotin. All patients in the single-arm, multicenter trial had relapsed or refractory cHL and were enrolled regardless of PD-L1 expression status. Patients received a median of 17 doses of nivolumab, the FDA said in a written statement.
![]()
The median time to response was 2.1 months (range, 0.7-5.7 months). The estimated median duration of response was 8.7 months.
The FDA also issued a warning for complications of allogeneic HSCT after nivolumab, reporting that transplant-related deaths have occurred. Health care professionals should follow patients closely for early evidence of transplant-related complications, such as hyperacute graft-versus-host disease (GVHD), severe acute GVHD, steroid-requiring febrile syndrome, hepatic veno-occlusive disease, and other immune-mediated adverse reactions, they said.
The most common adverse reactions in a second single-arm study used to evaluate safety (n = 263) were upper respiratory tract infection, cough, pyrexia, and diarrhea. Other immune-mediated adverse reactions, occurring in 1%-5% of patients, included rash, pneumonitis, hepatitis, hyperthyroidism, and colitis. The most common serious adverse reactions, which were reported in 1%-3% of patients, were pneumonia, pleural effusion, pneumonitis, pyrexia, infusion-related reaction, and rash.
Nivolumab is marketed as Opdivo by Bristol-Myers Squibb and has been previously approved to treat advanced renal cell carcinoma, lung cancer, and melanoma.
lnikolaides@frontlinemedcom.com
On Twitter @NikolaidesLaura
The Food and Drug Administration has granted accelerated approval to nivolumab for the treatment of patients with classical Hodgkin lymphoma (cHL) that has relapsed or progressed after autologous hematopoietic stem cell transplantation (HSCT) and posttransplantation brentuximab vedotin.
Approval was based on a 65% objective response rate in 95 patients treated with nivolumab following autologous HSCT and posttransplantation brentuximab vedotin. All patients in the single-arm, multicenter trial had relapsed or refractory cHL and were enrolled regardless of PD-L1 expression status. Patients received a median of 17 doses of nivolumab, the FDA said in a written statement.
![]()
The median time to response was 2.1 months (range, 0.7-5.7 months). The estimated median duration of response was 8.7 months.
The FDA also issued a warning for complications of allogeneic HSCT after nivolumab, reporting that transplant-related deaths have occurred. Health care professionals should follow patients closely for early evidence of transplant-related complications, such as hyperacute graft-versus-host disease (GVHD), severe acute GVHD, steroid-requiring febrile syndrome, hepatic veno-occlusive disease, and other immune-mediated adverse reactions, they said.
The most common adverse reactions in a second single-arm study used to evaluate safety (n = 263) were upper respiratory tract infection, cough, pyrexia, and diarrhea. Other immune-mediated adverse reactions, occurring in 1%-5% of patients, included rash, pneumonitis, hepatitis, hyperthyroidism, and colitis. The most common serious adverse reactions, which were reported in 1%-3% of patients, were pneumonia, pleural effusion, pneumonitis, pyrexia, infusion-related reaction, and rash.
Nivolumab is marketed as Opdivo by Bristol-Myers Squibb and has been previously approved to treat advanced renal cell carcinoma, lung cancer, and melanoma.
lnikolaides@frontlinemedcom.com
On Twitter @NikolaidesLaura
Physical activity may lower risk of some cancers

Photo by K. Johansson
Being physically active during leisure time may lower a person’s risk of certain cancers, according to a new study.
A high level of physical activity was associated with a 20% lower risk of myeloid leukemia, a 17% lower risk of myeloma, a 9% lower risk of non-Hodgkin lymphoma, and a 7% lower risk of cancer in general.
On the other hand, a high level of physical activity was also associated with a higher risk of malignant melanoma and prostate cancer.
Steven C. Moore, PhD, of the National Cancer Institute in Bethesda, Maryland, and his colleagues reported these findings in JAMA Internal Medicine.
The researchers pooled data from 12 US and European study cohorts with self-reported physical activity (1987-2004). And they analyzed associations between physical activity and 26 types of cancer.
The study included 1.4 million participants, and 186,932 cancers were identified during a median of 11 years of follow-up.
Compared with the lowest level of leisure-time physical activity (10th percentile), the highest level of activity (90th percentile) had strong inverse associations (a 20% or greater reduction in risk) for 7 cancer types:
- Myeloid leukemia (hazard ratio [HR]=0.80 [95% CI, 0.70-0.92])
- Esophageal adenocarcinoma (HR=0.58 [95% CI, 0.37-0.89])
- Liver cancer (HR=0.73 [95% CI, 0.55-0.98])
- Lung cancer (HR=0.74 [95% CI, 0.71-0.77])
- Kidney cancer (HR=0.77 [95% CI, 0.70-0.85])
- Gastric cardia (HR=0.78 [95% CI, 0.64-0.95])
- Endometrial cancer (HR=0.79 [95% CI, 0.68-0.92]).
There were moderate inverse associations (a 10% to 20% reduction in risk) between the highest level of activity and 6 cancers:
- Myeloma (HR=0.83 [95% CI, 0.72-0.95])
- Colon cancer (HR=0.84 [95% CI, 0.77-0.91])
- Head and neck cancer (HR=0.85 [95% CI, 0.78-0.93])
- Rectal cancer (HR=0.87 [95% CI, 0.80-0.95])
- Bladder cancer (HR=0.87 [95% CI, 0.82-0.92])
- Breast cancer (HR=0.90 [95% CI, 0.87-0.93]).
And there were suggestive inverse associations between the highest level of activity and 3 cancers:
- Non-Hodgkin lymphoma (HR=0.91 [95% CI, 0.83-1.00])
- Gallbladder cancer (HR=0.72 [95% CI, 0.51-1.01])
- Small intestine cancer (HR=0.78 [95% CI, 0.60-1.00]).
However, the highest level of activity was also associated with an increased risk of prostate cancer (HR=1.05 [95% CI, 1.03-1.08]) and malignant melanoma (HR=1.27 [95% CI, 1.16-1.40]).
The researchers said the main limitation of this study is that they cannot fully exclude the possibility that diet, smoking, and other factors may have affected these results. Also, the study used self-reported physical activity, which can mean errors in recall.
Still, the team said these findings support promoting physical activity as a key component of population-wide cancer prevention and control efforts. ![]()
Photo by K. Johansson
Being physically active during leisure time may lower a person’s risk of certain cancers, according to a new study.
A high level of physical activity was associated with a 20% lower risk of myeloid leukemia, a 17% lower risk of myeloma, a 9% lower risk of non-Hodgkin lymphoma, and a 7% lower risk of cancer in general.
On the other hand, a high level of physical activity was also associated with a higher risk of malignant melanoma and prostate cancer.
Steven C. Moore, PhD, of the National Cancer Institute in Bethesda, Maryland, and his colleagues reported these findings in JAMA Internal Medicine.
The researchers pooled data from 12 US and European study cohorts with self-reported physical activity (1987-2004). And they analyzed associations between physical activity and 26 types of cancer.
The study included 1.4 million participants, and 186,932 cancers were identified during a median of 11 years of follow-up.
Compared with the lowest level of leisure-time physical activity (10th percentile), the highest level of activity (90th percentile) had strong inverse associations (a 20% or greater reduction in risk) for 7 cancer types:
- Myeloid leukemia (hazard ratio [HR]=0.80 [95% CI, 0.70-0.92])
- Esophageal adenocarcinoma (HR=0.58 [95% CI, 0.37-0.89])
- Liver cancer (HR=0.73 [95% CI, 0.55-0.98])
- Lung cancer (HR=0.74 [95% CI, 0.71-0.77])
- Kidney cancer (HR=0.77 [95% CI, 0.70-0.85])
- Gastric cardia (HR=0.78 [95% CI, 0.64-0.95])
- Endometrial cancer (HR=0.79 [95% CI, 0.68-0.92]).
There were moderate inverse associations (a 10% to 20% reduction in risk) between the highest level of activity and 6 cancers:
- Myeloma (HR=0.83 [95% CI, 0.72-0.95])
- Colon cancer (HR=0.84 [95% CI, 0.77-0.91])
- Head and neck cancer (HR=0.85 [95% CI, 0.78-0.93])
- Rectal cancer (HR=0.87 [95% CI, 0.80-0.95])
- Bladder cancer (HR=0.87 [95% CI, 0.82-0.92])
- Breast cancer (HR=0.90 [95% CI, 0.87-0.93]).
And there were suggestive inverse associations between the highest level of activity and 3 cancers:
- Non-Hodgkin lymphoma (HR=0.91 [95% CI, 0.83-1.00])
- Gallbladder cancer (HR=0.72 [95% CI, 0.51-1.01])
- Small intestine cancer (HR=0.78 [95% CI, 0.60-1.00]).
However, the highest level of activity was also associated with an increased risk of prostate cancer (HR=1.05 [95% CI, 1.03-1.08]) and malignant melanoma (HR=1.27 [95% CI, 1.16-1.40]).
The researchers said the main limitation of this study is that they cannot fully exclude the possibility that diet, smoking, and other factors may have affected these results. Also, the study used self-reported physical activity, which can mean errors in recall.
Still, the team said these findings support promoting physical activity as a key component of population-wide cancer prevention and control efforts. ![]()
Photo by K. Johansson
Being physically active during leisure time may lower a person’s risk of certain cancers, according to a new study.
A high level of physical activity was associated with a 20% lower risk of myeloid leukemia, a 17% lower risk of myeloma, a 9% lower risk of non-Hodgkin lymphoma, and a 7% lower risk of cancer in general.
On the other hand, a high level of physical activity was also associated with a higher risk of malignant melanoma and prostate cancer.
Steven C. Moore, PhD, of the National Cancer Institute in Bethesda, Maryland, and his colleagues reported these findings in JAMA Internal Medicine.
The researchers pooled data from 12 US and European study cohorts with self-reported physical activity (1987-2004). And they analyzed associations between physical activity and 26 types of cancer.
The study included 1.4 million participants, and 186,932 cancers were identified during a median of 11 years of follow-up.
Compared with the lowest level of leisure-time physical activity (10th percentile), the highest level of activity (90th percentile) had strong inverse associations (a 20% or greater reduction in risk) for 7 cancer types:
- Myeloid leukemia (hazard ratio [HR]=0.80 [95% CI, 0.70-0.92])
- Esophageal adenocarcinoma (HR=0.58 [95% CI, 0.37-0.89])
- Liver cancer (HR=0.73 [95% CI, 0.55-0.98])
- Lung cancer (HR=0.74 [95% CI, 0.71-0.77])
- Kidney cancer (HR=0.77 [95% CI, 0.70-0.85])
- Gastric cardia (HR=0.78 [95% CI, 0.64-0.95])
- Endometrial cancer (HR=0.79 [95% CI, 0.68-0.92]).
There were moderate inverse associations (a 10% to 20% reduction in risk) between the highest level of activity and 6 cancers:
- Myeloma (HR=0.83 [95% CI, 0.72-0.95])
- Colon cancer (HR=0.84 [95% CI, 0.77-0.91])
- Head and neck cancer (HR=0.85 [95% CI, 0.78-0.93])
- Rectal cancer (HR=0.87 [95% CI, 0.80-0.95])
- Bladder cancer (HR=0.87 [95% CI, 0.82-0.92])
- Breast cancer (HR=0.90 [95% CI, 0.87-0.93]).
And there were suggestive inverse associations between the highest level of activity and 3 cancers:
- Non-Hodgkin lymphoma (HR=0.91 [95% CI, 0.83-1.00])
- Gallbladder cancer (HR=0.72 [95% CI, 0.51-1.01])
- Small intestine cancer (HR=0.78 [95% CI, 0.60-1.00]).
However, the highest level of activity was also associated with an increased risk of prostate cancer (HR=1.05 [95% CI, 1.03-1.08]) and malignant melanoma (HR=1.27 [95% CI, 1.16-1.40]).
The researchers said the main limitation of this study is that they cannot fully exclude the possibility that diet, smoking, and other factors may have affected these results. Also, the study used self-reported physical activity, which can mean errors in recall.
Still, the team said these findings support promoting physical activity as a key component of population-wide cancer prevention and control efforts. ![]()
FDA warns of counterfeit BiCNU
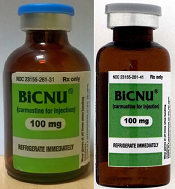
shown on the left and a
counterfeit vial on the right
Photo courtesy of the FDA
The US Food and Drug Administration (FDA) is warning healthcare professionals that a counterfeit version of BiCNU (carmustine for injection) 100 mg has been detected in some foreign countries.
The agency said there is no indication that counterfeit BiCNU has entered the legitimate US drug supply chain and no indication that any US patients have received counterfeit BiCNU.
Still, the FDA is advising that healthcare professionals inspect BiCNU vials as an added precaution to ensure the product administered to patients is authentic.
BiCNU is approved to treat brain cancers, multiple myeloma, and lymphoma. It is manufactured by Emcure Pharmaceuticals Ltd. and distributed in the US by Heritage Pharmaceuticals Inc.
Heritage previously announced that counterfeit BiCNU had been found in India, Ireland, and Israel.
How to identify counterfeit BiCNU
BiCNU is available as a vial of BiCNU and dehydrated alcohol co-packaged together.
While the NDC on the outer package of the authentic and counterfeit versions might match, the best way to distinguish a counterfeit is to look at the BiCNU vial inside the packaging. The authentic product has a blue flip top, while the counterfeit product may have a gray flip top.
The product may also be counterfeit if the vial displays the following lot numbers, batch numbers, manufacturing dates, and expiration dates.
| Product | Expiration
date |
Manufacturing
date |
Lot number | Batch number |
| BiCNU | 01/18 | 2/16 | BCEM771322 | EM/BC20161990 |
| Diluent | 01/18 | 2/16 | SBCDA224736 | EM/BCD2220 |
| BiCNU | 12/17 | 1/16 | BCEM771318 | EM/BC20151896 |
| Diluent | 12/17 | 1/16 | SBCDA224732 | EM/BCD2216 |
| BiCNU | 10/17 | 11/15 | BCEM771317 | EM/BC20151895 |
| Diluent | 10/17 | 11/15 | SBCDA224731 | EM/BCD2215 |
The FDA urges healthcare professionals to purchase drug products only from legitimate suppliers.
Healthcare professionals are encouraged to report sales solicitation of suspect drug products by calling the FDA’s Office of Criminal Investigations (OCI) at 800-551-3989, reporting via OCI’s website, or emailing DrugSupplyChainIntegrity@fda.hhs.gov.
Healthcare professionals and patients should report adverse events related to the use of any suspect medications to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
shown on the left and a
counterfeit vial on the right
Photo courtesy of the FDA
The US Food and Drug Administration (FDA) is warning healthcare professionals that a counterfeit version of BiCNU (carmustine for injection) 100 mg has been detected in some foreign countries.
The agency said there is no indication that counterfeit BiCNU has entered the legitimate US drug supply chain and no indication that any US patients have received counterfeit BiCNU.
Still, the FDA is advising that healthcare professionals inspect BiCNU vials as an added precaution to ensure the product administered to patients is authentic.
BiCNU is approved to treat brain cancers, multiple myeloma, and lymphoma. It is manufactured by Emcure Pharmaceuticals Ltd. and distributed in the US by Heritage Pharmaceuticals Inc.
Heritage previously announced that counterfeit BiCNU had been found in India, Ireland, and Israel.
How to identify counterfeit BiCNU
BiCNU is available as a vial of BiCNU and dehydrated alcohol co-packaged together.
While the NDC on the outer package of the authentic and counterfeit versions might match, the best way to distinguish a counterfeit is to look at the BiCNU vial inside the packaging. The authentic product has a blue flip top, while the counterfeit product may have a gray flip top.
The product may also be counterfeit if the vial displays the following lot numbers, batch numbers, manufacturing dates, and expiration dates.
| Product | Expiration
date |
Manufacturing
date |
Lot number | Batch number |
| BiCNU | 01/18 | 2/16 | BCEM771322 | EM/BC20161990 |
| Diluent | 01/18 | 2/16 | SBCDA224736 | EM/BCD2220 |
| BiCNU | 12/17 | 1/16 | BCEM771318 | EM/BC20151896 |
| Diluent | 12/17 | 1/16 | SBCDA224732 | EM/BCD2216 |
| BiCNU | 10/17 | 11/15 | BCEM771317 | EM/BC20151895 |
| Diluent | 10/17 | 11/15 | SBCDA224731 | EM/BCD2215 |
The FDA urges healthcare professionals to purchase drug products only from legitimate suppliers.
Healthcare professionals are encouraged to report sales solicitation of suspect drug products by calling the FDA’s Office of Criminal Investigations (OCI) at 800-551-3989, reporting via OCI’s website, or emailing DrugSupplyChainIntegrity@fda.hhs.gov.
Healthcare professionals and patients should report adverse events related to the use of any suspect medications to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
shown on the left and a
counterfeit vial on the right
Photo courtesy of the FDA
The US Food and Drug Administration (FDA) is warning healthcare professionals that a counterfeit version of BiCNU (carmustine for injection) 100 mg has been detected in some foreign countries.
The agency said there is no indication that counterfeit BiCNU has entered the legitimate US drug supply chain and no indication that any US patients have received counterfeit BiCNU.
Still, the FDA is advising that healthcare professionals inspect BiCNU vials as an added precaution to ensure the product administered to patients is authentic.
BiCNU is approved to treat brain cancers, multiple myeloma, and lymphoma. It is manufactured by Emcure Pharmaceuticals Ltd. and distributed in the US by Heritage Pharmaceuticals Inc.
Heritage previously announced that counterfeit BiCNU had been found in India, Ireland, and Israel.
How to identify counterfeit BiCNU
BiCNU is available as a vial of BiCNU and dehydrated alcohol co-packaged together.
While the NDC on the outer package of the authentic and counterfeit versions might match, the best way to distinguish a counterfeit is to look at the BiCNU vial inside the packaging. The authentic product has a blue flip top, while the counterfeit product may have a gray flip top.
The product may also be counterfeit if the vial displays the following lot numbers, batch numbers, manufacturing dates, and expiration dates.
| Product | Expiration
date |
Manufacturing
date |
Lot number | Batch number |
| BiCNU | 01/18 | 2/16 | BCEM771322 | EM/BC20161990 |
| Diluent | 01/18 | 2/16 | SBCDA224736 | EM/BCD2220 |
| BiCNU | 12/17 | 1/16 | BCEM771318 | EM/BC20151896 |
| Diluent | 12/17 | 1/16 | SBCDA224732 | EM/BCD2216 |
| BiCNU | 10/17 | 11/15 | BCEM771317 | EM/BC20151895 |
| Diluent | 10/17 | 11/15 | SBCDA224731 | EM/BCD2215 |
The FDA urges healthcare professionals to purchase drug products only from legitimate suppliers.
Healthcare professionals are encouraged to report sales solicitation of suspect drug products by calling the FDA’s Office of Criminal Investigations (OCI) at 800-551-3989, reporting via OCI’s website, or emailing DrugSupplyChainIntegrity@fda.hhs.gov.
Healthcare professionals and patients should report adverse events related to the use of any suspect medications to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
FDA issues warning about counterfeit BiCNU
A counterfeit version of BiCNU has been detected in foreign countries, the Food and Drug Administration reports.
BiCNU is approved to treat different types of brain cancer, multiple myeloma, and lymphoma (Hodgkin’s and non-Hodgkin’s), manufactured by Emcure Pharmaceuticals, and distributed by Heritage Pharmaceuticals.
There has been no counterfeit BiCNU detected in the United States, but the FDA encourages health care professionals to diligently inspect BiCNU vials before administering the drug to patients.
“While the [National Drug Code] on the outer package of the authentic and counterfeit version might match, the best way to distinguish a counterfeit is to look at the BiCNU vial inside the packaging,” the FDA reported in a written statement, which includes a list of the counterfeit lots.
To report sales solicitation of suspect drugs call the FDA’s office of criminal investigations at 800-551-3989 or e-mail DrugSupplyChainIntegrity@fda.hhs.gov. To report adverse events related to suspect medications, submit a report online at www.fda.gov/medwatch/report.htm.
On Twitter @jess_craig94
A counterfeit version of BiCNU has been detected in foreign countries, the Food and Drug Administration reports.
BiCNU is approved to treat different types of brain cancer, multiple myeloma, and lymphoma (Hodgkin’s and non-Hodgkin’s), manufactured by Emcure Pharmaceuticals, and distributed by Heritage Pharmaceuticals.
There has been no counterfeit BiCNU detected in the United States, but the FDA encourages health care professionals to diligently inspect BiCNU vials before administering the drug to patients.
“While the [National Drug Code] on the outer package of the authentic and counterfeit version might match, the best way to distinguish a counterfeit is to look at the BiCNU vial inside the packaging,” the FDA reported in a written statement, which includes a list of the counterfeit lots.
To report sales solicitation of suspect drugs call the FDA’s office of criminal investigations at 800-551-3989 or e-mail DrugSupplyChainIntegrity@fda.hhs.gov. To report adverse events related to suspect medications, submit a report online at www.fda.gov/medwatch/report.htm.
On Twitter @jess_craig94
A counterfeit version of BiCNU has been detected in foreign countries, the Food and Drug Administration reports.
BiCNU is approved to treat different types of brain cancer, multiple myeloma, and lymphoma (Hodgkin’s and non-Hodgkin’s), manufactured by Emcure Pharmaceuticals, and distributed by Heritage Pharmaceuticals.
There has been no counterfeit BiCNU detected in the United States, but the FDA encourages health care professionals to diligently inspect BiCNU vials before administering the drug to patients.
“While the [National Drug Code] on the outer package of the authentic and counterfeit version might match, the best way to distinguish a counterfeit is to look at the BiCNU vial inside the packaging,” the FDA reported in a written statement, which includes a list of the counterfeit lots.
To report sales solicitation of suspect drugs call the FDA’s office of criminal investigations at 800-551-3989 or e-mail DrugSupplyChainIntegrity@fda.hhs.gov. To report adverse events related to suspect medications, submit a report online at www.fda.gov/medwatch/report.htm.
On Twitter @jess_craig94
Health Canada approves ibrutinib for WM

Photo from Janssen Biotech
Health Canada has approved the BTK inhibitor ibrutinib (Imbruvica) as a treatment for patients with Waldenström’s macroglobulinemia (WM).
Ibrutinib was first approved in Canada in November 2014 for the treatment of patients with chronic lymphocytic leukemia (CLL), including those with 17p deletion, who have received at least one prior therapy, or for the frontline treatment of patients with CLL and 17p deletion.
In July 2015, ibrutinib was granted conditional approval for the treatment of patients with relapsed or refractory mantle cell lymphoma.
Health Canada’s approval of ibrutinib for WM was based on results of a multicenter, phase 2 study in which researchers tested the drug (given at 420 mg once daily) in 63 patients with previously treated WM.
The patients’ median age was 63 (range, 44-86), and their median number of prior therapies was 2 (range, 1-11).
Initial data showed an overall response rate of 87.3% in patients who received ibrutinib for a median of 11.7 months.
Updated results from the study were published in NEJM in April 2015. After a median treatment duration of 19.1 months, the overall response rate was 91%.
At 24 months, the estimated rate of progression-free survival was 69%, and the estimated rate of overall survival was 95%.
The most common grade 2-4 adverse events were neutropenia (22%) and thrombocytopenia (14%). Ibrutinib-related neutropenia and thrombocytopenia were reversible but required a dose reduction in 3 patients and treatment discontinuation in 4 patients.
Grade 2 or higher bleeding events occurred in 4 patients, and there were 15 infections considered possibly related to ibrutinib.
Treatment-related atrial fibrillation (AFib) occurred in 3 patients, all of whom had a prior history of paroxysmal AFib. AFib resolved when treatment was withheld, and all 3 patients were able to continue on therapy per protocol without an additional event.
Ibrutinib is co-developed by Cilag GmbH International (a member of the Janssen Pharmaceutical Companies) and Pharmacyclics LLC, an AbbVie company. Janssen Inc. markets ibrutinib as Imbruvica in Canada. ![]()
Photo from Janssen Biotech
Health Canada has approved the BTK inhibitor ibrutinib (Imbruvica) as a treatment for patients with Waldenström’s macroglobulinemia (WM).
Ibrutinib was first approved in Canada in November 2014 for the treatment of patients with chronic lymphocytic leukemia (CLL), including those with 17p deletion, who have received at least one prior therapy, or for the frontline treatment of patients with CLL and 17p deletion.
In July 2015, ibrutinib was granted conditional approval for the treatment of patients with relapsed or refractory mantle cell lymphoma.
Health Canada’s approval of ibrutinib for WM was based on results of a multicenter, phase 2 study in which researchers tested the drug (given at 420 mg once daily) in 63 patients with previously treated WM.
The patients’ median age was 63 (range, 44-86), and their median number of prior therapies was 2 (range, 1-11).
Initial data showed an overall response rate of 87.3% in patients who received ibrutinib for a median of 11.7 months.
Updated results from the study were published in NEJM in April 2015. After a median treatment duration of 19.1 months, the overall response rate was 91%.
At 24 months, the estimated rate of progression-free survival was 69%, and the estimated rate of overall survival was 95%.
The most common grade 2-4 adverse events were neutropenia (22%) and thrombocytopenia (14%). Ibrutinib-related neutropenia and thrombocytopenia were reversible but required a dose reduction in 3 patients and treatment discontinuation in 4 patients.
Grade 2 or higher bleeding events occurred in 4 patients, and there were 15 infections considered possibly related to ibrutinib.
Treatment-related atrial fibrillation (AFib) occurred in 3 patients, all of whom had a prior history of paroxysmal AFib. AFib resolved when treatment was withheld, and all 3 patients were able to continue on therapy per protocol without an additional event.
Ibrutinib is co-developed by Cilag GmbH International (a member of the Janssen Pharmaceutical Companies) and Pharmacyclics LLC, an AbbVie company. Janssen Inc. markets ibrutinib as Imbruvica in Canada. ![]()
Photo from Janssen Biotech
Health Canada has approved the BTK inhibitor ibrutinib (Imbruvica) as a treatment for patients with Waldenström’s macroglobulinemia (WM).
Ibrutinib was first approved in Canada in November 2014 for the treatment of patients with chronic lymphocytic leukemia (CLL), including those with 17p deletion, who have received at least one prior therapy, or for the frontline treatment of patients with CLL and 17p deletion.
In July 2015, ibrutinib was granted conditional approval for the treatment of patients with relapsed or refractory mantle cell lymphoma.
Health Canada’s approval of ibrutinib for WM was based on results of a multicenter, phase 2 study in which researchers tested the drug (given at 420 mg once daily) in 63 patients with previously treated WM.
The patients’ median age was 63 (range, 44-86), and their median number of prior therapies was 2 (range, 1-11).
Initial data showed an overall response rate of 87.3% in patients who received ibrutinib for a median of 11.7 months.
Updated results from the study were published in NEJM in April 2015. After a median treatment duration of 19.1 months, the overall response rate was 91%.
At 24 months, the estimated rate of progression-free survival was 69%, and the estimated rate of overall survival was 95%.
The most common grade 2-4 adverse events were neutropenia (22%) and thrombocytopenia (14%). Ibrutinib-related neutropenia and thrombocytopenia were reversible but required a dose reduction in 3 patients and treatment discontinuation in 4 patients.
Grade 2 or higher bleeding events occurred in 4 patients, and there were 15 infections considered possibly related to ibrutinib.
Treatment-related atrial fibrillation (AFib) occurred in 3 patients, all of whom had a prior history of paroxysmal AFib. AFib resolved when treatment was withheld, and all 3 patients were able to continue on therapy per protocol without an additional event.
Ibrutinib is co-developed by Cilag GmbH International (a member of the Janssen Pharmaceutical Companies) and Pharmacyclics LLC, an AbbVie company. Janssen Inc. markets ibrutinib as Imbruvica in Canada. ![]()
Tools may aid transition from pediatric to adult care

Photo courtesy of the CDC
WASHINGTON, DC—The American Society of Hematology (ASH) has created a toolkit to help hematologists aid patients who are transitioning from pediatric to adult practices.
The toolkit contains general resources for all hematologic conditions, as well as specific resources for patients with hemophilia and sickle cell disease.
It includes 2 types of forms—a transition-readiness assessment and a clinical summary.
The toolkit was presented at the American College of Physicians (ACP) Internal Medicine Meeting 2016.
“Transitioning from pediatric to adult healthcare practices is often a challenge for patients with chronic medical issues because it can be difficult to adhere to a treatment regimen or attend regular appointments without the assistance of a parent or guardian,” said ASH President Charles S. Abrams, MD, of the University of Pennsylvania in Philadelphia.
“ASH recognizes that understanding a patient’s preparedness to take control of his or her medical condition in adulthood can make a huge difference in quality of care, which is why we are pleased to join the American College of Physicians and partner societies in this important initiative.”
ASH joined more than 2 dozen groups to participate in the ACP’s Pediatric to Adult Care Transition Initiative. The goal of this initiative was to develop guidance and tools that both primary care internal medicine and subspecialty practices can use for patients who are transitioning from pediatric/adolescent practices to adult care.
An ASH Transitions Work Group, made up of society members from pediatric and adult practices, developed 3 segments of the hematology-specific toolkit:
- generic forms for patients with any hematologic condition, with an addendum that includes links to additional condition-specific guidelines and resources
- specific forms for hemophilia
- specific forms for sickle cell disease.
For each segment, there are 2 types of forms— a transition-readiness assessment and a clinical summary.
The transition-readiness assessment should be completed by the patient. It assesses the patient’s readiness for the transition to adult care by evaluating the patient’s understanding of his or her condition and ability to manage medications, appointments, insurance, and medical privacy issues.
This assessment should be used by the adult care team to assess any remaining gaps in the patient’s self-care knowledge or additional issues that should be addressed to ensure optimal care.
The clinical summary is a medical record summary to be completed by the referring provider and the patient. The summary contains essential clinical information regarding the patient’s condition that is to be included in the patient’s medical record upon transfer to the adult practice.
More information on the ACP Pediatric to Adult Care Transitions Initiative is available on the ACP website. The forms for the ASH transitions toolkit are available in the “Hematology” section of the Condition-Specific Tools page. ![]()
Photo courtesy of the CDC
WASHINGTON, DC—The American Society of Hematology (ASH) has created a toolkit to help hematologists aid patients who are transitioning from pediatric to adult practices.
The toolkit contains general resources for all hematologic conditions, as well as specific resources for patients with hemophilia and sickle cell disease.
It includes 2 types of forms—a transition-readiness assessment and a clinical summary.
The toolkit was presented at the American College of Physicians (ACP) Internal Medicine Meeting 2016.
“Transitioning from pediatric to adult healthcare practices is often a challenge for patients with chronic medical issues because it can be difficult to adhere to a treatment regimen or attend regular appointments without the assistance of a parent or guardian,” said ASH President Charles S. Abrams, MD, of the University of Pennsylvania in Philadelphia.
“ASH recognizes that understanding a patient’s preparedness to take control of his or her medical condition in adulthood can make a huge difference in quality of care, which is why we are pleased to join the American College of Physicians and partner societies in this important initiative.”
ASH joined more than 2 dozen groups to participate in the ACP’s Pediatric to Adult Care Transition Initiative. The goal of this initiative was to develop guidance and tools that both primary care internal medicine and subspecialty practices can use for patients who are transitioning from pediatric/adolescent practices to adult care.
An ASH Transitions Work Group, made up of society members from pediatric and adult practices, developed 3 segments of the hematology-specific toolkit:
- generic forms for patients with any hematologic condition, with an addendum that includes links to additional condition-specific guidelines and resources
- specific forms for hemophilia
- specific forms for sickle cell disease.
For each segment, there are 2 types of forms— a transition-readiness assessment and a clinical summary.
The transition-readiness assessment should be completed by the patient. It assesses the patient’s readiness for the transition to adult care by evaluating the patient’s understanding of his or her condition and ability to manage medications, appointments, insurance, and medical privacy issues.
This assessment should be used by the adult care team to assess any remaining gaps in the patient’s self-care knowledge or additional issues that should be addressed to ensure optimal care.
The clinical summary is a medical record summary to be completed by the referring provider and the patient. The summary contains essential clinical information regarding the patient’s condition that is to be included in the patient’s medical record upon transfer to the adult practice.
More information on the ACP Pediatric to Adult Care Transitions Initiative is available on the ACP website. The forms for the ASH transitions toolkit are available in the “Hematology” section of the Condition-Specific Tools page. ![]()
Photo courtesy of the CDC
WASHINGTON, DC—The American Society of Hematology (ASH) has created a toolkit to help hematologists aid patients who are transitioning from pediatric to adult practices.
The toolkit contains general resources for all hematologic conditions, as well as specific resources for patients with hemophilia and sickle cell disease.
It includes 2 types of forms—a transition-readiness assessment and a clinical summary.
The toolkit was presented at the American College of Physicians (ACP) Internal Medicine Meeting 2016.
“Transitioning from pediatric to adult healthcare practices is often a challenge for patients with chronic medical issues because it can be difficult to adhere to a treatment regimen or attend regular appointments without the assistance of a parent or guardian,” said ASH President Charles S. Abrams, MD, of the University of Pennsylvania in Philadelphia.
“ASH recognizes that understanding a patient’s preparedness to take control of his or her medical condition in adulthood can make a huge difference in quality of care, which is why we are pleased to join the American College of Physicians and partner societies in this important initiative.”
ASH joined more than 2 dozen groups to participate in the ACP’s Pediatric to Adult Care Transition Initiative. The goal of this initiative was to develop guidance and tools that both primary care internal medicine and subspecialty practices can use for patients who are transitioning from pediatric/adolescent practices to adult care.
An ASH Transitions Work Group, made up of society members from pediatric and adult practices, developed 3 segments of the hematology-specific toolkit:
- generic forms for patients with any hematologic condition, with an addendum that includes links to additional condition-specific guidelines and resources
- specific forms for hemophilia
- specific forms for sickle cell disease.
For each segment, there are 2 types of forms— a transition-readiness assessment and a clinical summary.
The transition-readiness assessment should be completed by the patient. It assesses the patient’s readiness for the transition to adult care by evaluating the patient’s understanding of his or her condition and ability to manage medications, appointments, insurance, and medical privacy issues.
This assessment should be used by the adult care team to assess any remaining gaps in the patient’s self-care knowledge or additional issues that should be addressed to ensure optimal care.
The clinical summary is a medical record summary to be completed by the referring provider and the patient. The summary contains essential clinical information regarding the patient’s condition that is to be included in the patient’s medical record upon transfer to the adult practice.
More information on the ACP Pediatric to Adult Care Transitions Initiative is available on the ACP website. The forms for the ASH transitions toolkit are available in the “Hematology” section of the Condition-Specific Tools page. ![]()
CAR T-cell therapy granted orphan designation

The US Food and Drug Administration (FDA) has granted orphan drug designation for the chimeric antigen receptor (CAR) T-cell therapy KTE-C19 as a treatment for several hematologic malignancies.
This includes primary mediastinal B-cell lymphoma (PMBCL), mantle cell lymphoma (MCL), follicular lymphoma (FL), acute lymphoblastic leukemia (ALL), and chronic lymphocytic leukemia (CLL).
KTE-C19 previously received orphan designation from the FDA for the treatment of diffuse large B-cell lymphoma (DLBCL).
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity.
KTE-C19 also has breakthrough therapy designation from the FDA as a treatment for DLBCL, PMBCL, and transformed FL.
About KTE-C19
KTE-C19 is an investigational therapy in which a patient’s T cells are genetically modified to express a CAR designed to target CD19. The product is being developed by Kite Pharma, Inc.
In a study published in the Journal of Clinical Oncology, researchers evaluated KTE-C19 in 15 patients with advanced B-cell malignancies.
The patients received a conditioning regimen of cyclophosphamide and fludarabine, followed 1 day later by a single infusion of KTE-C19. The researchers noted that the conditioning regimen is known to be active against B-cell malignancies and could have made a direct contribution to patient responses.
Thirteen patients were evaluable for response. The overall response rate was 92%. Eight patients achieved a complete response (CR), and 4 had a partial response (PR).
Of the 7 patients with DLBCL, 4 achieved a CR, 2 achieved a PR, and 1 had stable disease. Three of the CRs were ongoing at the time of publication, with the duration ranging from 9 months to 22 months.
Of the 4 patients with CLL, 3 had a CR, and 1 had a PR. All 3 CRs were ongoing at the time of publication, with the duration ranging from 14 months to 23 months.
Among the 2 patients with indolent lymphomas, 1 achieved a CR, and 1 had a PR. The duration of the CR was 11 months at the time of publication.
KTE-C19 elicited a number of adverse events, including fever, hypotension, delirium, and other neurologic toxicities. All but 2 patients experienced grade 3/4 adverse events.
Three patients developed unexpected neurologic abnormalities. One patient experienced aphasia and right-sided facial paresis. One patient developed aphasia, confusion, and severe, generalized myoclonus. And 1 patient had aphasia, confusion, hemifacial spasms, apraxia, and gait disturbances.
KTE-C19 is currently under investigation in a phase 2 trial of refractory DLBCL, PMBCL, and transformed FL (ZUMA-1), a phase 2 trial of relapsed/refractory MCL (ZUMA-2), a phase 1/2 trial of relapsed/refractory adult ALL (ZUMA-3), and a phase 1/2 trial of relapsed/refractory pediatric ALL (ZUMA-4).
Results from ZUMA-1 were recently presented at the 2016 AACR Annual Meeting (abstract CT135). ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation for the chimeric antigen receptor (CAR) T-cell therapy KTE-C19 as a treatment for several hematologic malignancies.
This includes primary mediastinal B-cell lymphoma (PMBCL), mantle cell lymphoma (MCL), follicular lymphoma (FL), acute lymphoblastic leukemia (ALL), and chronic lymphocytic leukemia (CLL).
KTE-C19 previously received orphan designation from the FDA for the treatment of diffuse large B-cell lymphoma (DLBCL).
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity.
KTE-C19 also has breakthrough therapy designation from the FDA as a treatment for DLBCL, PMBCL, and transformed FL.
About KTE-C19
KTE-C19 is an investigational therapy in which a patient’s T cells are genetically modified to express a CAR designed to target CD19. The product is being developed by Kite Pharma, Inc.
In a study published in the Journal of Clinical Oncology, researchers evaluated KTE-C19 in 15 patients with advanced B-cell malignancies.
The patients received a conditioning regimen of cyclophosphamide and fludarabine, followed 1 day later by a single infusion of KTE-C19. The researchers noted that the conditioning regimen is known to be active against B-cell malignancies and could have made a direct contribution to patient responses.
Thirteen patients were evaluable for response. The overall response rate was 92%. Eight patients achieved a complete response (CR), and 4 had a partial response (PR).
Of the 7 patients with DLBCL, 4 achieved a CR, 2 achieved a PR, and 1 had stable disease. Three of the CRs were ongoing at the time of publication, with the duration ranging from 9 months to 22 months.
Of the 4 patients with CLL, 3 had a CR, and 1 had a PR. All 3 CRs were ongoing at the time of publication, with the duration ranging from 14 months to 23 months.
Among the 2 patients with indolent lymphomas, 1 achieved a CR, and 1 had a PR. The duration of the CR was 11 months at the time of publication.
KTE-C19 elicited a number of adverse events, including fever, hypotension, delirium, and other neurologic toxicities. All but 2 patients experienced grade 3/4 adverse events.
Three patients developed unexpected neurologic abnormalities. One patient experienced aphasia and right-sided facial paresis. One patient developed aphasia, confusion, and severe, generalized myoclonus. And 1 patient had aphasia, confusion, hemifacial spasms, apraxia, and gait disturbances.
KTE-C19 is currently under investigation in a phase 2 trial of refractory DLBCL, PMBCL, and transformed FL (ZUMA-1), a phase 2 trial of relapsed/refractory MCL (ZUMA-2), a phase 1/2 trial of relapsed/refractory adult ALL (ZUMA-3), and a phase 1/2 trial of relapsed/refractory pediatric ALL (ZUMA-4).
Results from ZUMA-1 were recently presented at the 2016 AACR Annual Meeting (abstract CT135). ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation for the chimeric antigen receptor (CAR) T-cell therapy KTE-C19 as a treatment for several hematologic malignancies.
This includes primary mediastinal B-cell lymphoma (PMBCL), mantle cell lymphoma (MCL), follicular lymphoma (FL), acute lymphoblastic leukemia (ALL), and chronic lymphocytic leukemia (CLL).
KTE-C19 previously received orphan designation from the FDA for the treatment of diffuse large B-cell lymphoma (DLBCL).
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity.
KTE-C19 also has breakthrough therapy designation from the FDA as a treatment for DLBCL, PMBCL, and transformed FL.
About KTE-C19
KTE-C19 is an investigational therapy in which a patient’s T cells are genetically modified to express a CAR designed to target CD19. The product is being developed by Kite Pharma, Inc.
In a study published in the Journal of Clinical Oncology, researchers evaluated KTE-C19 in 15 patients with advanced B-cell malignancies.
The patients received a conditioning regimen of cyclophosphamide and fludarabine, followed 1 day later by a single infusion of KTE-C19. The researchers noted that the conditioning regimen is known to be active against B-cell malignancies and could have made a direct contribution to patient responses.
Thirteen patients were evaluable for response. The overall response rate was 92%. Eight patients achieved a complete response (CR), and 4 had a partial response (PR).
Of the 7 patients with DLBCL, 4 achieved a CR, 2 achieved a PR, and 1 had stable disease. Three of the CRs were ongoing at the time of publication, with the duration ranging from 9 months to 22 months.
Of the 4 patients with CLL, 3 had a CR, and 1 had a PR. All 3 CRs were ongoing at the time of publication, with the duration ranging from 14 months to 23 months.
Among the 2 patients with indolent lymphomas, 1 achieved a CR, and 1 had a PR. The duration of the CR was 11 months at the time of publication.
KTE-C19 elicited a number of adverse events, including fever, hypotension, delirium, and other neurologic toxicities. All but 2 patients experienced grade 3/4 adverse events.
Three patients developed unexpected neurologic abnormalities. One patient experienced aphasia and right-sided facial paresis. One patient developed aphasia, confusion, and severe, generalized myoclonus. And 1 patient had aphasia, confusion, hemifacial spasms, apraxia, and gait disturbances.
KTE-C19 is currently under investigation in a phase 2 trial of refractory DLBCL, PMBCL, and transformed FL (ZUMA-1), a phase 2 trial of relapsed/refractory MCL (ZUMA-2), a phase 1/2 trial of relapsed/refractory adult ALL (ZUMA-3), and a phase 1/2 trial of relapsed/refractory pediatric ALL (ZUMA-4).
Results from ZUMA-1 were recently presented at the 2016 AACR Annual Meeting (abstract CT135). ![]()
Pre-treatment gut bacteria may predict risk of BSI
A new study suggests the composition of a cancer patient’s intestinal microbiome before treatment may predict his risk of developing a bloodstream infection (BSI) after treatment.
Researchers analyzed fecal samples from patients with non-Hodgkin lymphoma who were set to receive an allogeneic hematopoietic stem cell transplant (allo-HSCT) with myeloablative conditioning.
The team found that patients with less diversity in their fecal samples before this treatment were more likely to develop a BSI after.
Emmanuel Montassier, MD, PhD, of Nantes University Hospital in France, and his colleagues conducted this study and reported the result in Genome Medicine.
A previous study suggested that intestinal domination—when a single bacterial taxon occupies at least 30% of the microbiota—is associated with BSIs in patients undergoing allo-HSCT. However, the role of the intestinal microbiome before treatment was not clear.
So Dr Montassier and his colleagues set out to characterize the fecal microbiome before treatment. To do this, they sequenced the bacterial DNA of fecal samples from 28 patients with non-Hodgkin lymphoma.
The team collected the samples before patients began a 5-day myeloablative conditioning regimen (high-dose carmustine, etoposide, aracytine, and melphalan), followed by allo-HSCT on the seventh day.
Eleven of these patients developed a BSI at a mean of 12 days after sample collection. Two patients (18.2%) developed Enterococcus BSI, 4 (36.4%) developed Escherichia coli BSI, and 5 (45.5%) developed other Gammaproteobacteria BSI.
The researchers said that alpha diversity in samples from these patients was significantly lower than alpha diversity from patients who did not develop a BSI, with reduced evenness (Shannon index, Monte Carlo permuted t-test two-sided P value = 0.004) and reduced richness (Observed species, Monte Carlo permuted t-test two-sided P value = 0.001)
The team also noted that, compared to patients who did not develop a BSI, those who did had decreased abundance of Barnesiellaceae, Coriobacteriaceae, Faecalibacterium, Christensenella, Dehalobacterium, Desulfovibrio, and Sutterella.
Using this information, the researchers developed a BSI risk index that could predict the likelihood of a BSI with 90% sensitivity and specificity.
“This method worked even better than we expected because we found a consistent difference between the gut bacteria in those who developed infections and those who did not,” said study author Dan Knights, PhD, of the University of Minnesota in Minneapolis.
“This research is an early demonstration that we may be able to use the bugs in our gut to predict infections and possibly develop new prognostic models in other diseases.”
Still, the researchers said these findings are based on a limited number of patients treated with the same regimen at a single clinic. So the next step for this research is to validate the findings in a much larger cohort including patients with different cancer types, different treatment types, and from multiple treatment centers.
“We still need to determine if these bacteria are playing any kind of causal role in the infections or if they are simply acting as biomarkers for some other predisposing condition in the patient,” Dr Montassier said. ![]()
A new study suggests the composition of a cancer patient’s intestinal microbiome before treatment may predict his risk of developing a bloodstream infection (BSI) after treatment.
Researchers analyzed fecal samples from patients with non-Hodgkin lymphoma who were set to receive an allogeneic hematopoietic stem cell transplant (allo-HSCT) with myeloablative conditioning.
The team found that patients with less diversity in their fecal samples before this treatment were more likely to develop a BSI after.
Emmanuel Montassier, MD, PhD, of Nantes University Hospital in France, and his colleagues conducted this study and reported the result in Genome Medicine.
A previous study suggested that intestinal domination—when a single bacterial taxon occupies at least 30% of the microbiota—is associated with BSIs in patients undergoing allo-HSCT. However, the role of the intestinal microbiome before treatment was not clear.
So Dr Montassier and his colleagues set out to characterize the fecal microbiome before treatment. To do this, they sequenced the bacterial DNA of fecal samples from 28 patients with non-Hodgkin lymphoma.
The team collected the samples before patients began a 5-day myeloablative conditioning regimen (high-dose carmustine, etoposide, aracytine, and melphalan), followed by allo-HSCT on the seventh day.
Eleven of these patients developed a BSI at a mean of 12 days after sample collection. Two patients (18.2%) developed Enterococcus BSI, 4 (36.4%) developed Escherichia coli BSI, and 5 (45.5%) developed other Gammaproteobacteria BSI.
The researchers said that alpha diversity in samples from these patients was significantly lower than alpha diversity from patients who did not develop a BSI, with reduced evenness (Shannon index, Monte Carlo permuted t-test two-sided P value = 0.004) and reduced richness (Observed species, Monte Carlo permuted t-test two-sided P value = 0.001)
The team also noted that, compared to patients who did not develop a BSI, those who did had decreased abundance of Barnesiellaceae, Coriobacteriaceae, Faecalibacterium, Christensenella, Dehalobacterium, Desulfovibrio, and Sutterella.
Using this information, the researchers developed a BSI risk index that could predict the likelihood of a BSI with 90% sensitivity and specificity.
“This method worked even better than we expected because we found a consistent difference between the gut bacteria in those who developed infections and those who did not,” said study author Dan Knights, PhD, of the University of Minnesota in Minneapolis.
“This research is an early demonstration that we may be able to use the bugs in our gut to predict infections and possibly develop new prognostic models in other diseases.”
Still, the researchers said these findings are based on a limited number of patients treated with the same regimen at a single clinic. So the next step for this research is to validate the findings in a much larger cohort including patients with different cancer types, different treatment types, and from multiple treatment centers.
“We still need to determine if these bacteria are playing any kind of causal role in the infections or if they are simply acting as biomarkers for some other predisposing condition in the patient,” Dr Montassier said. ![]()
A new study suggests the composition of a cancer patient’s intestinal microbiome before treatment may predict his risk of developing a bloodstream infection (BSI) after treatment.
Researchers analyzed fecal samples from patients with non-Hodgkin lymphoma who were set to receive an allogeneic hematopoietic stem cell transplant (allo-HSCT) with myeloablative conditioning.
The team found that patients with less diversity in their fecal samples before this treatment were more likely to develop a BSI after.
Emmanuel Montassier, MD, PhD, of Nantes University Hospital in France, and his colleagues conducted this study and reported the result in Genome Medicine.
A previous study suggested that intestinal domination—when a single bacterial taxon occupies at least 30% of the microbiota—is associated with BSIs in patients undergoing allo-HSCT. However, the role of the intestinal microbiome before treatment was not clear.
So Dr Montassier and his colleagues set out to characterize the fecal microbiome before treatment. To do this, they sequenced the bacterial DNA of fecal samples from 28 patients with non-Hodgkin lymphoma.
The team collected the samples before patients began a 5-day myeloablative conditioning regimen (high-dose carmustine, etoposide, aracytine, and melphalan), followed by allo-HSCT on the seventh day.
Eleven of these patients developed a BSI at a mean of 12 days after sample collection. Two patients (18.2%) developed Enterococcus BSI, 4 (36.4%) developed Escherichia coli BSI, and 5 (45.5%) developed other Gammaproteobacteria BSI.
The researchers said that alpha diversity in samples from these patients was significantly lower than alpha diversity from patients who did not develop a BSI, with reduced evenness (Shannon index, Monte Carlo permuted t-test two-sided P value = 0.004) and reduced richness (Observed species, Monte Carlo permuted t-test two-sided P value = 0.001)
The team also noted that, compared to patients who did not develop a BSI, those who did had decreased abundance of Barnesiellaceae, Coriobacteriaceae, Faecalibacterium, Christensenella, Dehalobacterium, Desulfovibrio, and Sutterella.
Using this information, the researchers developed a BSI risk index that could predict the likelihood of a BSI with 90% sensitivity and specificity.
“This method worked even better than we expected because we found a consistent difference between the gut bacteria in those who developed infections and those who did not,” said study author Dan Knights, PhD, of the University of Minnesota in Minneapolis.
“This research is an early demonstration that we may be able to use the bugs in our gut to predict infections and possibly develop new prognostic models in other diseases.”
Still, the researchers said these findings are based on a limited number of patients treated with the same regimen at a single clinic. So the next step for this research is to validate the findings in a much larger cohort including patients with different cancer types, different treatment types, and from multiple treatment centers.
“We still need to determine if these bacteria are playing any kind of causal role in the infections or if they are simply acting as biomarkers for some other predisposing condition in the patient,” Dr Montassier said.
Radiotherapy trial results going unpublished

woman for radiotherapy
Photo by Rhoda Baer
TURIN, ITALY—Results from many phase 3 radiotherapy trials conducted in the US are not being published on ClincalTrials.gov, according to a study presented at ESTRO 35.
Since 2007, it has been mandatory to publish the results of clinical trials carried out in the US on ClinicalTrials.gov within 12 months of trial completion.
However, an analysis of more than 800 radiotherapy trials revealed that more than 80% did not meet this requirement.
Jaime Pérez-Alija, of Hospital Plató in Barcelona, Spain, and his colleagues presented these results at ESTRO 35 as abstract PV-0087.
In 2007, the Food and Drug Administration Amendments Act (FDAAA) mandated that sponsors of most US trials begin registering and reporting basic summary results on ClinicalTrials.gov so the American public could have access to the resulting data.
The requirement covers non-phase-1 trials of drugs, medical devices, or biologics that had at least 1 US research site. Trial results are to be reported by the sponsor within a year of completing data collection.
To investigate how well this mandate has been followed by sponsors of phase 3 radiotherapy trials, Pérez-Alija and his colleagues analyzed 802 trials with a primary completion date prior to January 1, 2013.
The team found that 81.7% of these trials (n=655) did not have even summary results published on ClinicalTrials.gov.
The researchers also looked specifically at those trials that began after the 2007 act was passed and found that 76.4% of these trials (422/552) did not have results published.
When the researchers looked at publication by cancer type, they found that 78% of lymphoma trial results were unpublished.
The most-published cancer type was glioblastoma, with 62.5% of results unpublished. And the least-published cancer types were anal and testicular cancers, for which 100% of trial results were unpublished.
“These findings came as a surprise for many reasons, not least of which was that many of the trials had been funded by the US National Institutes of Health,” Pérez-Alija said.
“Interestingly, we found that company-funded trials are far better at complying with the rules than academic trials—55% and 30% respectively. However, only one-third of all the trials we studied were company trials. Since we know that clinical trials produce the best data for decision-making in modern evidenced-based medicine, it is particularly worrying that the law is being ignored on such a wide scale.”
One possible reason for non-publication, according to the researchers, is that some of the trials may have been granted a deadline extension. However, if this is the case, it is not publicly known.
“Therefore, our first problem is that we do not know with any certainty whether a trial is truly overdue,” Pérez-Alija said. “The registry says clearly that all dates must be updated if an extension has been allowed, but it seems likely that this is not happening in many cases.”
The researchers are investigating the issue further to see, for example, how many of the trials registered in ClinicalTrials.gov or in other databases are being published in medical journals.
They intend to email principal investigators to ask why the mandatory deposition of results did not take place and to enquire about the reasons for non-publication in medical journals of those trials where there is a published deposition.
“We have shown that a large number of study participants are routinely exposed to the risks of trial participation without the benefits that sharing and publishing results would have for patients in the future,” Pérez-Alija said.
“Information about what was done and what was found in these trials could be lost forever, leading to bad treatment decisions, missed opportunities for good medicine, and trials being repeated unnecessarily. This situation should not be allowed to continue.”
woman for radiotherapy
Photo by Rhoda Baer
TURIN, ITALY—Results from many phase 3 radiotherapy trials conducted in the US are not being published on ClincalTrials.gov, according to a study presented at ESTRO 35.
Since 2007, it has been mandatory to publish the results of clinical trials carried out in the US on ClinicalTrials.gov within 12 months of trial completion.
However, an analysis of more than 800 radiotherapy trials revealed that more than 80% did not meet this requirement.
Jaime Pérez-Alija, of Hospital Plató in Barcelona, Spain, and his colleagues presented these results at ESTRO 35 as abstract PV-0087.
In 2007, the Food and Drug Administration Amendments Act (FDAAA) mandated that sponsors of most US trials begin registering and reporting basic summary results on ClinicalTrials.gov so the American public could have access to the resulting data.
The requirement covers non-phase-1 trials of drugs, medical devices, or biologics that had at least 1 US research site. Trial results are to be reported by the sponsor within a year of completing data collection.
To investigate how well this mandate has been followed by sponsors of phase 3 radiotherapy trials, Pérez-Alija and his colleagues analyzed 802 trials with a primary completion date prior to January 1, 2013.
The team found that 81.7% of these trials (n=655) did not have even summary results published on ClinicalTrials.gov.
The researchers also looked specifically at those trials that began after the 2007 act was passed and found that 76.4% of these trials (422/552) did not have results published.
When the researchers looked at publication by cancer type, they found that 78% of lymphoma trial results were unpublished.
The most-published cancer type was glioblastoma, with 62.5% of results unpublished. And the least-published cancer types were anal and testicular cancers, for which 100% of trial results were unpublished.
“These findings came as a surprise for many reasons, not least of which was that many of the trials had been funded by the US National Institutes of Health,” Pérez-Alija said.
“Interestingly, we found that company-funded trials are far better at complying with the rules than academic trials—55% and 30% respectively. However, only one-third of all the trials we studied were company trials. Since we know that clinical trials produce the best data for decision-making in modern evidenced-based medicine, it is particularly worrying that the law is being ignored on such a wide scale.”
One possible reason for non-publication, according to the researchers, is that some of the trials may have been granted a deadline extension. However, if this is the case, it is not publicly known.
“Therefore, our first problem is that we do not know with any certainty whether a trial is truly overdue,” Pérez-Alija said. “The registry says clearly that all dates must be updated if an extension has been allowed, but it seems likely that this is not happening in many cases.”
The researchers are investigating the issue further to see, for example, how many of the trials registered in ClinicalTrials.gov or in other databases are being published in medical journals.
They intend to email principal investigators to ask why the mandatory deposition of results did not take place and to enquire about the reasons for non-publication in medical journals of those trials where there is a published deposition.
“We have shown that a large number of study participants are routinely exposed to the risks of trial participation without the benefits that sharing and publishing results would have for patients in the future,” Pérez-Alija said.
“Information about what was done and what was found in these trials could be lost forever, leading to bad treatment decisions, missed opportunities for good medicine, and trials being repeated unnecessarily. This situation should not be allowed to continue.”
woman for radiotherapy
Photo by Rhoda Baer
TURIN, ITALY—Results from many phase 3 radiotherapy trials conducted in the US are not being published on ClincalTrials.gov, according to a study presented at ESTRO 35.
Since 2007, it has been mandatory to publish the results of clinical trials carried out in the US on ClinicalTrials.gov within 12 months of trial completion.
However, an analysis of more than 800 radiotherapy trials revealed that more than 80% did not meet this requirement.
Jaime Pérez-Alija, of Hospital Plató in Barcelona, Spain, and his colleagues presented these results at ESTRO 35 as abstract PV-0087.
In 2007, the Food and Drug Administration Amendments Act (FDAAA) mandated that sponsors of most US trials begin registering and reporting basic summary results on ClinicalTrials.gov so the American public could have access to the resulting data.
The requirement covers non-phase-1 trials of drugs, medical devices, or biologics that had at least 1 US research site. Trial results are to be reported by the sponsor within a year of completing data collection.
To investigate how well this mandate has been followed by sponsors of phase 3 radiotherapy trials, Pérez-Alija and his colleagues analyzed 802 trials with a primary completion date prior to January 1, 2013.
The team found that 81.7% of these trials (n=655) did not have even summary results published on ClinicalTrials.gov.
The researchers also looked specifically at those trials that began after the 2007 act was passed and found that 76.4% of these trials (422/552) did not have results published.
When the researchers looked at publication by cancer type, they found that 78% of lymphoma trial results were unpublished.
The most-published cancer type was glioblastoma, with 62.5% of results unpublished. And the least-published cancer types were anal and testicular cancers, for which 100% of trial results were unpublished.
“These findings came as a surprise for many reasons, not least of which was that many of the trials had been funded by the US National Institutes of Health,” Pérez-Alija said.
“Interestingly, we found that company-funded trials are far better at complying with the rules than academic trials—55% and 30% respectively. However, only one-third of all the trials we studied were company trials. Since we know that clinical trials produce the best data for decision-making in modern evidenced-based medicine, it is particularly worrying that the law is being ignored on such a wide scale.”
One possible reason for non-publication, according to the researchers, is that some of the trials may have been granted a deadline extension. However, if this is the case, it is not publicly known.
“Therefore, our first problem is that we do not know with any certainty whether a trial is truly overdue,” Pérez-Alija said. “The registry says clearly that all dates must be updated if an extension has been allowed, but it seems likely that this is not happening in many cases.”
The researchers are investigating the issue further to see, for example, how many of the trials registered in ClinicalTrials.gov or in other databases are being published in medical journals.
They intend to email principal investigators to ask why the mandatory deposition of results did not take place and to enquire about the reasons for non-publication in medical journals of those trials where there is a published deposition.
“We have shown that a large number of study participants are routinely exposed to the risks of trial participation without the benefits that sharing and publishing results would have for patients in the future,” Pérez-Alija said.
“Information about what was done and what was found in these trials could be lost forever, leading to bad treatment decisions, missed opportunities for good medicine, and trials being repeated unnecessarily. This situation should not be allowed to continue.”