User login
FDA approves daratumumab in combination with standard therapy for multiple myeloma
The Food and Drug Administration has approved daratumumab in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, for the treatment of patients with multiple myeloma who have received at least one prior therapy.
The drug was approved last year as monotherapy for patients with multiple myeloma who have received at least three prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double refractory to a proteasome inhibitor and an immunomodulatory agent.
In the POLLUX trial, median PFS had not been reached in the daratumumab plus lenalidomide and dexamethasone arm and was 18.4 months among patients getting lenalidomide and dexamethasone alone (HR=0.37; 95% CI: 0.27, 0.52; P less than.0001).
In the CASTOR trial, which compared the combination of daratumumab, bortezomib, and dexamethasone with bortezomib and dexamethasone, the estimated median PFS had not been reached in the daratumumab arm and was 7.2 months in the control arm (hazard ratio, 0.39; 95% confidence interval, 0.28-0.53; P less than .0001).
Updated results for both trials will be presented at the upcoming annual meeting of the American Society of Hematology (abstract #1150, abstract #1151).
The most frequently reported adverse reactions in POLLUX were infusion reactions, diarrhea, nausea, fatigue, pyrexia, upper respiratory tract infection, muscle spasm, cough, and dyspnea. The most frequently reported adverse reactions in CASTOR were infusion reactions, diarrhea, peripheral edema, upper respiratory tract infection, peripheral sensory neuropathy, cough, and dyspnea.
The recommended dose of daratumumab is 16 mg/kg IV (calculated on actual body weight), the FDA said.
Full prescribing information is available here.
The Food and Drug Administration has approved daratumumab in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, for the treatment of patients with multiple myeloma who have received at least one prior therapy.
The drug was approved last year as monotherapy for patients with multiple myeloma who have received at least three prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double refractory to a proteasome inhibitor and an immunomodulatory agent.
In the POLLUX trial, median PFS had not been reached in the daratumumab plus lenalidomide and dexamethasone arm and was 18.4 months among patients getting lenalidomide and dexamethasone alone (HR=0.37; 95% CI: 0.27, 0.52; P less than.0001).
In the CASTOR trial, which compared the combination of daratumumab, bortezomib, and dexamethasone with bortezomib and dexamethasone, the estimated median PFS had not been reached in the daratumumab arm and was 7.2 months in the control arm (hazard ratio, 0.39; 95% confidence interval, 0.28-0.53; P less than .0001).
Updated results for both trials will be presented at the upcoming annual meeting of the American Society of Hematology (abstract #1150, abstract #1151).
The most frequently reported adverse reactions in POLLUX were infusion reactions, diarrhea, nausea, fatigue, pyrexia, upper respiratory tract infection, muscle spasm, cough, and dyspnea. The most frequently reported adverse reactions in CASTOR were infusion reactions, diarrhea, peripheral edema, upper respiratory tract infection, peripheral sensory neuropathy, cough, and dyspnea.
The recommended dose of daratumumab is 16 mg/kg IV (calculated on actual body weight), the FDA said.
Full prescribing information is available here.
The Food and Drug Administration has approved daratumumab in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, for the treatment of patients with multiple myeloma who have received at least one prior therapy.
The drug was approved last year as monotherapy for patients with multiple myeloma who have received at least three prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double refractory to a proteasome inhibitor and an immunomodulatory agent.
In the POLLUX trial, median PFS had not been reached in the daratumumab plus lenalidomide and dexamethasone arm and was 18.4 months among patients getting lenalidomide and dexamethasone alone (HR=0.37; 95% CI: 0.27, 0.52; P less than.0001).
In the CASTOR trial, which compared the combination of daratumumab, bortezomib, and dexamethasone with bortezomib and dexamethasone, the estimated median PFS had not been reached in the daratumumab arm and was 7.2 months in the control arm (hazard ratio, 0.39; 95% confidence interval, 0.28-0.53; P less than .0001).
Updated results for both trials will be presented at the upcoming annual meeting of the American Society of Hematology (abstract #1150, abstract #1151).
The most frequently reported adverse reactions in POLLUX were infusion reactions, diarrhea, nausea, fatigue, pyrexia, upper respiratory tract infection, muscle spasm, cough, and dyspnea. The most frequently reported adverse reactions in CASTOR were infusion reactions, diarrhea, peripheral edema, upper respiratory tract infection, peripheral sensory neuropathy, cough, and dyspnea.
The recommended dose of daratumumab is 16 mg/kg IV (calculated on actual body weight), the FDA said.
Full prescribing information is available here.
Novel CLL drugs could greatly increase costs

New research suggests the increasing use of oral targeted therapies for chronic lymphocytic leukemia (CLL) could raise US treatment costs for the disease by almost 600%.
Investigators modeled the evolving management of CLL from 2011 to 2025 and found that increasing use of the oral targeted therapies ibrutinib and idelalisib could greatly increase costs for both patients and payers.
The team detailed these findings in the Journal of Clinical Oncology.
“The rising cost of cancer care is a serious concern,” said study author Jagpreet Chhatwal, PhD, of Massachusetts General Hospital in Boston.
“The average cost of annual cancer treatment, which was below $10,000 per patient before 2000, has now increased to more than $100,000. Such increasing trends can limit access to new therapies, potentially undermining their clinical effectiveness. These new drugs are highly effective, but their high costs motivated us to project their changing economic burden and affordability.”
Dr Chhatwal and his colleagues noted that ibrutinib and idelalisib each cost around $130,000 per year, and treatment with these drugs may be continued indefinitely.
So the team set out to determine the potential financial impact of the drugs on payers’ budgets, as well as on Medicare-enrolled patients, who represent the majority of CLL patients in the US.
The investigators developed a model to simulate the evolving management of CLL from 2011 to 2025.
In one scenario, chemoimmunotherapy was the standard of care before 2014, while oral targeted therapies were used for patients with del(17p) and relapsed CLL from 2014 onward and for first-line treatment of CLL from 2016 onward.
The team also modeled a scenario in which chemoimmunotherapy was the standard of care throughout the entire time period and compared the costs between these scenarios.
The model projects that:
- Per-patient lifetime costs for CLL treatment will increase from $147,000 to $604,000 from 2016 onward
- The total out-of-pocket costs for Medicare patients will increase from $9200 to $57,000 for patients initiating treatment from 2016 onward
- The total annual cost of CLL management in the US will rise from $0.74 billion in 2011 to $5.13 billion in 2025, an increase of 590%.
“Such substantial increases in the cost are mainly driven by high drug prices, prolonged treatment duration, and the increase in the number of patients living with CLL,” said study author Qiushi Chen, PhD, of Massachusetts General Hospital.
The investigators also noted that the standard measure used to determine the cost-effectiveness of a medical intervention is whether it costs less than $100,000 for each additional year of life gained. The projected cost-effectiveness ratio of oral targeted therapy in CLL is $189,000 for each year gained.
“At the current average wholesale prices, oral targeted therapies for CLL are not cost-effective, and prices would need to drop by 50% to 70% to become cost-effective,” Dr Chhatwal said.
“We are not recommending that clinicians choose less effective CLL management strategies that do not include oral targeted therapies,” said study author Nitin Jain, MD, of the University of Texas MD Anderson Cancer Center in Houston.
“Instead, we propose that the prices of these drugs need to be reduced to make the treatment cost-effective and more affordable, something we hope may happen with all cancer drugs. We also believe more research is needed to explore whether we can discontinue targeted treatment of patients who have responded well without risking worsening of their health.” ![]()
New research suggests the increasing use of oral targeted therapies for chronic lymphocytic leukemia (CLL) could raise US treatment costs for the disease by almost 600%.
Investigators modeled the evolving management of CLL from 2011 to 2025 and found that increasing use of the oral targeted therapies ibrutinib and idelalisib could greatly increase costs for both patients and payers.
The team detailed these findings in the Journal of Clinical Oncology.
“The rising cost of cancer care is a serious concern,” said study author Jagpreet Chhatwal, PhD, of Massachusetts General Hospital in Boston.
“The average cost of annual cancer treatment, which was below $10,000 per patient before 2000, has now increased to more than $100,000. Such increasing trends can limit access to new therapies, potentially undermining their clinical effectiveness. These new drugs are highly effective, but their high costs motivated us to project their changing economic burden and affordability.”
Dr Chhatwal and his colleagues noted that ibrutinib and idelalisib each cost around $130,000 per year, and treatment with these drugs may be continued indefinitely.
So the team set out to determine the potential financial impact of the drugs on payers’ budgets, as well as on Medicare-enrolled patients, who represent the majority of CLL patients in the US.
The investigators developed a model to simulate the evolving management of CLL from 2011 to 2025.
In one scenario, chemoimmunotherapy was the standard of care before 2014, while oral targeted therapies were used for patients with del(17p) and relapsed CLL from 2014 onward and for first-line treatment of CLL from 2016 onward.
The team also modeled a scenario in which chemoimmunotherapy was the standard of care throughout the entire time period and compared the costs between these scenarios.
The model projects that:
- Per-patient lifetime costs for CLL treatment will increase from $147,000 to $604,000 from 2016 onward
- The total out-of-pocket costs for Medicare patients will increase from $9200 to $57,000 for patients initiating treatment from 2016 onward
- The total annual cost of CLL management in the US will rise from $0.74 billion in 2011 to $5.13 billion in 2025, an increase of 590%.
“Such substantial increases in the cost are mainly driven by high drug prices, prolonged treatment duration, and the increase in the number of patients living with CLL,” said study author Qiushi Chen, PhD, of Massachusetts General Hospital.
The investigators also noted that the standard measure used to determine the cost-effectiveness of a medical intervention is whether it costs less than $100,000 for each additional year of life gained. The projected cost-effectiveness ratio of oral targeted therapy in CLL is $189,000 for each year gained.
“At the current average wholesale prices, oral targeted therapies for CLL are not cost-effective, and prices would need to drop by 50% to 70% to become cost-effective,” Dr Chhatwal said.
“We are not recommending that clinicians choose less effective CLL management strategies that do not include oral targeted therapies,” said study author Nitin Jain, MD, of the University of Texas MD Anderson Cancer Center in Houston.
“Instead, we propose that the prices of these drugs need to be reduced to make the treatment cost-effective and more affordable, something we hope may happen with all cancer drugs. We also believe more research is needed to explore whether we can discontinue targeted treatment of patients who have responded well without risking worsening of their health.” ![]()
New research suggests the increasing use of oral targeted therapies for chronic lymphocytic leukemia (CLL) could raise US treatment costs for the disease by almost 600%.
Investigators modeled the evolving management of CLL from 2011 to 2025 and found that increasing use of the oral targeted therapies ibrutinib and idelalisib could greatly increase costs for both patients and payers.
The team detailed these findings in the Journal of Clinical Oncology.
“The rising cost of cancer care is a serious concern,” said study author Jagpreet Chhatwal, PhD, of Massachusetts General Hospital in Boston.
“The average cost of annual cancer treatment, which was below $10,000 per patient before 2000, has now increased to more than $100,000. Such increasing trends can limit access to new therapies, potentially undermining their clinical effectiveness. These new drugs are highly effective, but their high costs motivated us to project their changing economic burden and affordability.”
Dr Chhatwal and his colleagues noted that ibrutinib and idelalisib each cost around $130,000 per year, and treatment with these drugs may be continued indefinitely.
So the team set out to determine the potential financial impact of the drugs on payers’ budgets, as well as on Medicare-enrolled patients, who represent the majority of CLL patients in the US.
The investigators developed a model to simulate the evolving management of CLL from 2011 to 2025.
In one scenario, chemoimmunotherapy was the standard of care before 2014, while oral targeted therapies were used for patients with del(17p) and relapsed CLL from 2014 onward and for first-line treatment of CLL from 2016 onward.
The team also modeled a scenario in which chemoimmunotherapy was the standard of care throughout the entire time period and compared the costs between these scenarios.
The model projects that:
- Per-patient lifetime costs for CLL treatment will increase from $147,000 to $604,000 from 2016 onward
- The total out-of-pocket costs for Medicare patients will increase from $9200 to $57,000 for patients initiating treatment from 2016 onward
- The total annual cost of CLL management in the US will rise from $0.74 billion in 2011 to $5.13 billion in 2025, an increase of 590%.
“Such substantial increases in the cost are mainly driven by high drug prices, prolonged treatment duration, and the increase in the number of patients living with CLL,” said study author Qiushi Chen, PhD, of Massachusetts General Hospital.
The investigators also noted that the standard measure used to determine the cost-effectiveness of a medical intervention is whether it costs less than $100,000 for each additional year of life gained. The projected cost-effectiveness ratio of oral targeted therapy in CLL is $189,000 for each year gained.
“At the current average wholesale prices, oral targeted therapies for CLL are not cost-effective, and prices would need to drop by 50% to 70% to become cost-effective,” Dr Chhatwal said.
“We are not recommending that clinicians choose less effective CLL management strategies that do not include oral targeted therapies,” said study author Nitin Jain, MD, of the University of Texas MD Anderson Cancer Center in Houston.
“Instead, we propose that the prices of these drugs need to be reduced to make the treatment cost-effective and more affordable, something we hope may happen with all cancer drugs. We also believe more research is needed to explore whether we can discontinue targeted treatment of patients who have responded well without risking worsening of their health.” ![]()
Central nervous system manifestations of multiple myeloma: risk and prognostic considerations
Multiple myeloma accounts for about 1% of all cancers and for 10% of hematologic malignancies in the United States. This report describes the cases of 2 patients with multiple myeloma who developed CNS involvement after autologous stem cell transplant in the context of extramedullary disease.
Click on the PDF icon at the top of this introduction to read the full article.
Multiple myeloma accounts for about 1% of all cancers and for 10% of hematologic malignancies in the United States. This report describes the cases of 2 patients with multiple myeloma who developed CNS involvement after autologous stem cell transplant in the context of extramedullary disease.
Click on the PDF icon at the top of this introduction to read the full article.
Multiple myeloma accounts for about 1% of all cancers and for 10% of hematologic malignancies in the United States. This report describes the cases of 2 patients with multiple myeloma who developed CNS involvement after autologous stem cell transplant in the context of extramedullary disease.
Click on the PDF icon at the top of this introduction to read the full article.
Large-scale tumor profiling deemed feasible, but challenges remain

Photo courtesy of the
National Institute of
General Medical Sciences
New research suggests large-scale genomic profiling is technically feasible in a broad population of cancer patients.
However, the study also revealed challenges and barriers to widespread implementation of precision medicine, according to researchers.
Specifically, half of the patients studied did not receive results of genomic profiling due to insufficient samples or sequencing failure.
Most patients who did receive results did not see a change in their care.
However, genomic profiling provided an accurate diagnosis and changed treatment for a handful of the patients studied.
Lynette M. Sholl, MD, of Brigham and Women’s Hospital in Boston, Massachusetts, and her colleagues reported these findings in JCI Insight.
The report contains data on pediatric and adult patients with a range of malignancies.
Patient samples were analyzed using OncoPanel. This platform sequences hundreds of known cancer-related genes to look for alterations that drive tumors and might be helpful in guiding treatment choice or enrolling the patient in an appropriate clinical trial.
This study began with 7397 patients, but many of these individuals did not have specimens adequate for sequencing. This left 3892 patients (53%) to undergo genomic profiling, but sequencing failed in 165 (4%) of them. So sequencing was successful in 3727 patients, or 50% of the overall population.
Of the 3727 patients in whom sequencing was successful, 73% had at least 1 genetic alteration that was considered “clinically actionable or informative” by the researchers.
This included 54% of patients with alterations that might be used to inform diagnosis or recommend enrollment in a clinical trial. It also included 19% of patients who had an alteration that “would inform standard-of-care therapeutic decision-making,” according to the researchers.
The team provided several examples of how genomic testing clarified or changed a patient’s diagnosis, which, in turn, altered treatment and prognosis.
One example was a patient who was originally diagnosed with peripheral T-cell lymphoma, which was later revised to myeloid sarcoma. Sequencing results suggested the patient actually had FIP1L1-PDGFRA-driven acute myeloid leukemia, which predicted responsiveness to imatinib.
The patient was treated with imatinib and experienced a “dramatic and sustained clinical response.” He then proceeded to allogeneic transplant and had no evidence of disease at 1 year of follow-up.
The researchers concluded that genomic sequencing results may alter the management of cancer patients in some cases, but certain barriers must be overcome to enable precision cancer medicine on a large scale. ![]()
Photo courtesy of the
National Institute of
General Medical Sciences
New research suggests large-scale genomic profiling is technically feasible in a broad population of cancer patients.
However, the study also revealed challenges and barriers to widespread implementation of precision medicine, according to researchers.
Specifically, half of the patients studied did not receive results of genomic profiling due to insufficient samples or sequencing failure.
Most patients who did receive results did not see a change in their care.
However, genomic profiling provided an accurate diagnosis and changed treatment for a handful of the patients studied.
Lynette M. Sholl, MD, of Brigham and Women’s Hospital in Boston, Massachusetts, and her colleagues reported these findings in JCI Insight.
The report contains data on pediatric and adult patients with a range of malignancies.
Patient samples were analyzed using OncoPanel. This platform sequences hundreds of known cancer-related genes to look for alterations that drive tumors and might be helpful in guiding treatment choice or enrolling the patient in an appropriate clinical trial.
This study began with 7397 patients, but many of these individuals did not have specimens adequate for sequencing. This left 3892 patients (53%) to undergo genomic profiling, but sequencing failed in 165 (4%) of them. So sequencing was successful in 3727 patients, or 50% of the overall population.
Of the 3727 patients in whom sequencing was successful, 73% had at least 1 genetic alteration that was considered “clinically actionable or informative” by the researchers.
This included 54% of patients with alterations that might be used to inform diagnosis or recommend enrollment in a clinical trial. It also included 19% of patients who had an alteration that “would inform standard-of-care therapeutic decision-making,” according to the researchers.
The team provided several examples of how genomic testing clarified or changed a patient’s diagnosis, which, in turn, altered treatment and prognosis.
One example was a patient who was originally diagnosed with peripheral T-cell lymphoma, which was later revised to myeloid sarcoma. Sequencing results suggested the patient actually had FIP1L1-PDGFRA-driven acute myeloid leukemia, which predicted responsiveness to imatinib.
The patient was treated with imatinib and experienced a “dramatic and sustained clinical response.” He then proceeded to allogeneic transplant and had no evidence of disease at 1 year of follow-up.
The researchers concluded that genomic sequencing results may alter the management of cancer patients in some cases, but certain barriers must be overcome to enable precision cancer medicine on a large scale. ![]()
Photo courtesy of the
National Institute of
General Medical Sciences
New research suggests large-scale genomic profiling is technically feasible in a broad population of cancer patients.
However, the study also revealed challenges and barriers to widespread implementation of precision medicine, according to researchers.
Specifically, half of the patients studied did not receive results of genomic profiling due to insufficient samples or sequencing failure.
Most patients who did receive results did not see a change in their care.
However, genomic profiling provided an accurate diagnosis and changed treatment for a handful of the patients studied.
Lynette M. Sholl, MD, of Brigham and Women’s Hospital in Boston, Massachusetts, and her colleagues reported these findings in JCI Insight.
The report contains data on pediatric and adult patients with a range of malignancies.
Patient samples were analyzed using OncoPanel. This platform sequences hundreds of known cancer-related genes to look for alterations that drive tumors and might be helpful in guiding treatment choice or enrolling the patient in an appropriate clinical trial.
This study began with 7397 patients, but many of these individuals did not have specimens adequate for sequencing. This left 3892 patients (53%) to undergo genomic profiling, but sequencing failed in 165 (4%) of them. So sequencing was successful in 3727 patients, or 50% of the overall population.
Of the 3727 patients in whom sequencing was successful, 73% had at least 1 genetic alteration that was considered “clinically actionable or informative” by the researchers.
This included 54% of patients with alterations that might be used to inform diagnosis or recommend enrollment in a clinical trial. It also included 19% of patients who had an alteration that “would inform standard-of-care therapeutic decision-making,” according to the researchers.
The team provided several examples of how genomic testing clarified or changed a patient’s diagnosis, which, in turn, altered treatment and prognosis.
One example was a patient who was originally diagnosed with peripheral T-cell lymphoma, which was later revised to myeloid sarcoma. Sequencing results suggested the patient actually had FIP1L1-PDGFRA-driven acute myeloid leukemia, which predicted responsiveness to imatinib.
The patient was treated with imatinib and experienced a “dramatic and sustained clinical response.” He then proceeded to allogeneic transplant and had no evidence of disease at 1 year of follow-up.
The researchers concluded that genomic sequencing results may alter the management of cancer patients in some cases, but certain barriers must be overcome to enable precision cancer medicine on a large scale. ![]()
Hashimoto’s Thyroiditis and Lymphoma
A “heightened index of suspicion” is called for when a patient with Hashimoto’s thyroiditis (HT) presents with an enlarging neck mass, say researchers from Tan Tock Seng Hospital, Singapore, in a case report. According to the researchers, because the complication of thyroid lymphoma is rare, physicians commonly forgotten it. But primary thyroid lymphomas (PTLs) have a 60-fold risk in patients with HT.
Related: Study Points to Risk Factors for Lymphoma
Hashimoto’s thyroiditis typically is treated successfully with thyroxine. The study patient, however, began to lose weight and developed a mass in her neck that was diagnosed as diffuse large B-cell lymphoma. Previously, research suggested that having HT for ≥ 20 years increased the risk of thyroid lymphoma, but small studies have found that the interval between diagnosis of HT and diagnosis of thyroid lymphoma might be shorter—4 to 9 years, the researchers note. They cite another study that found the median interval was 18 months, as with their patient. Symptoms usually last from a few days to 36 months before diagnosis; in the study patient, symptoms of compression occurred over 2 to 3 weeks.
That shorter time frame may indicate that ultrasonography surveillance should be started early and done periodically, the researchers say, to detect lymphoma development as soon as possible. Radiologic imaging is helpful but “only serves as an adjunct to the diagnosis.” Histologic diagnosis is still needed for definitive diagnosis.
Timely diagnosis and early treatment mean the prognosis can be good for PTL, with relatively high survival rates after chemotherapy and radiotherapy. In this case, the patient underwent 6 cycles of chemotherapy with adjuvant radiotherapy. She then was maintained on thyroxine 75 µg daily. She remains euthyroid and disease free 1 year after completing her cancer treatment.
Source:
Chiang B, Cheng S, Seow CJ. BMJ Case Rep. 2016;pii:bcr2016217568.
doi: 10.1136/bcr-2016-217568.
A “heightened index of suspicion” is called for when a patient with Hashimoto’s thyroiditis (HT) presents with an enlarging neck mass, say researchers from Tan Tock Seng Hospital, Singapore, in a case report. According to the researchers, because the complication of thyroid lymphoma is rare, physicians commonly forgotten it. But primary thyroid lymphomas (PTLs) have a 60-fold risk in patients with HT.
Related: Study Points to Risk Factors for Lymphoma
Hashimoto’s thyroiditis typically is treated successfully with thyroxine. The study patient, however, began to lose weight and developed a mass in her neck that was diagnosed as diffuse large B-cell lymphoma. Previously, research suggested that having HT for ≥ 20 years increased the risk of thyroid lymphoma, but small studies have found that the interval between diagnosis of HT and diagnosis of thyroid lymphoma might be shorter—4 to 9 years, the researchers note. They cite another study that found the median interval was 18 months, as with their patient. Symptoms usually last from a few days to 36 months before diagnosis; in the study patient, symptoms of compression occurred over 2 to 3 weeks.
That shorter time frame may indicate that ultrasonography surveillance should be started early and done periodically, the researchers say, to detect lymphoma development as soon as possible. Radiologic imaging is helpful but “only serves as an adjunct to the diagnosis.” Histologic diagnosis is still needed for definitive diagnosis.
Timely diagnosis and early treatment mean the prognosis can be good for PTL, with relatively high survival rates after chemotherapy and radiotherapy. In this case, the patient underwent 6 cycles of chemotherapy with adjuvant radiotherapy. She then was maintained on thyroxine 75 µg daily. She remains euthyroid and disease free 1 year after completing her cancer treatment.
Source:
Chiang B, Cheng S, Seow CJ. BMJ Case Rep. 2016;pii:bcr2016217568.
doi: 10.1136/bcr-2016-217568.
A “heightened index of suspicion” is called for when a patient with Hashimoto’s thyroiditis (HT) presents with an enlarging neck mass, say researchers from Tan Tock Seng Hospital, Singapore, in a case report. According to the researchers, because the complication of thyroid lymphoma is rare, physicians commonly forgotten it. But primary thyroid lymphomas (PTLs) have a 60-fold risk in patients with HT.
Related: Study Points to Risk Factors for Lymphoma
Hashimoto’s thyroiditis typically is treated successfully with thyroxine. The study patient, however, began to lose weight and developed a mass in her neck that was diagnosed as diffuse large B-cell lymphoma. Previously, research suggested that having HT for ≥ 20 years increased the risk of thyroid lymphoma, but small studies have found that the interval between diagnosis of HT and diagnosis of thyroid lymphoma might be shorter—4 to 9 years, the researchers note. They cite another study that found the median interval was 18 months, as with their patient. Symptoms usually last from a few days to 36 months before diagnosis; in the study patient, symptoms of compression occurred over 2 to 3 weeks.
That shorter time frame may indicate that ultrasonography surveillance should be started early and done periodically, the researchers say, to detect lymphoma development as soon as possible. Radiologic imaging is helpful but “only serves as an adjunct to the diagnosis.” Histologic diagnosis is still needed for definitive diagnosis.
Timely diagnosis and early treatment mean the prognosis can be good for PTL, with relatively high survival rates after chemotherapy and radiotherapy. In this case, the patient underwent 6 cycles of chemotherapy with adjuvant radiotherapy. She then was maintained on thyroxine 75 µg daily. She remains euthyroid and disease free 1 year after completing her cancer treatment.
Source:
Chiang B, Cheng S, Seow CJ. BMJ Case Rep. 2016;pii:bcr2016217568.
doi: 10.1136/bcr-2016-217568.
How EBV drives lymphomagenesis

Image by Ed Uthman
Results of research published in eLIFE appear to explain how Epstein-Barr virus (EBV) controls a pair of genes to drive lymphomagenesis.
Researchers set out to determine how EBV controls MYC, which is known to drive lymphoma development when activated, and BCL2L11, a gene that normally triggers apoptosis to prevent lymphoma but can be silenced by EBV.
The team discovered that EBV controls MYC and BCL2L11 by hijacking enhancer regions of DNA, which are situated far away from the genes.
These enhancers act as “control centers” and are able to contact and control genes from long distances by the looping out of the intervening stretches of DNA.
The researchers found that EBV activates MYC by increasing contacts between a specific set of enhancers and the gene.
The team said an Epstein-Barr nuclear antigen, EBNA2, activates multiple MYC enhancers and reconfigures the MYC locus to increase upstream enhancer-promoter interactions and decrease downstream interactions.
They noted that EBNA2 recruits the BRG1 ATPase of the SWI/SNF remodeller to MYC enhancers, and BRG1 is required for enhancer-promoter interactions in EBV-infected cells.
The researchers also discovered new enhancers that control BCL2L11. In this case, though, EBV stops these control centers from contacting the gene.
Specifically, the team found a hematopoietic enhancer hub that is inactivated by the Epstein-Barr nuclear antigens EBNA3A and EBNA3C through recruitment of the H3K27 methyltransferase EZH2.
Therefore, the researchers set out to determine if an EZH1/2 inhibitor, UNC1999, could reverse this effect. They found that UNC1999 did reverse enhancer inactivation, upregulated BCL2L11, and induced apoptosis in EBV-positive Burkitt lymphoma cells.
“This is a key step towards uncovering how this common virus, which affects thousands of people every year, causes blood cancer,” said study author Michelle West, PhD, of the University of Sussex in Brighton, UK.
“It is now important to carry out further studies to determine how the Epstein-Barr virus controls other genes that are associated with lymphoma. This will tell us more about how the virus drives lymphoma development and will help to identify new ways of targeting Epstein-Barr virus-infected cancer cells with specific drugs.” ![]()
Image by Ed Uthman
Results of research published in eLIFE appear to explain how Epstein-Barr virus (EBV) controls a pair of genes to drive lymphomagenesis.
Researchers set out to determine how EBV controls MYC, which is known to drive lymphoma development when activated, and BCL2L11, a gene that normally triggers apoptosis to prevent lymphoma but can be silenced by EBV.
The team discovered that EBV controls MYC and BCL2L11 by hijacking enhancer regions of DNA, which are situated far away from the genes.
These enhancers act as “control centers” and are able to contact and control genes from long distances by the looping out of the intervening stretches of DNA.
The researchers found that EBV activates MYC by increasing contacts between a specific set of enhancers and the gene.
The team said an Epstein-Barr nuclear antigen, EBNA2, activates multiple MYC enhancers and reconfigures the MYC locus to increase upstream enhancer-promoter interactions and decrease downstream interactions.
They noted that EBNA2 recruits the BRG1 ATPase of the SWI/SNF remodeller to MYC enhancers, and BRG1 is required for enhancer-promoter interactions in EBV-infected cells.
The researchers also discovered new enhancers that control BCL2L11. In this case, though, EBV stops these control centers from contacting the gene.
Specifically, the team found a hematopoietic enhancer hub that is inactivated by the Epstein-Barr nuclear antigens EBNA3A and EBNA3C through recruitment of the H3K27 methyltransferase EZH2.
Therefore, the researchers set out to determine if an EZH1/2 inhibitor, UNC1999, could reverse this effect. They found that UNC1999 did reverse enhancer inactivation, upregulated BCL2L11, and induced apoptosis in EBV-positive Burkitt lymphoma cells.
“This is a key step towards uncovering how this common virus, which affects thousands of people every year, causes blood cancer,” said study author Michelle West, PhD, of the University of Sussex in Brighton, UK.
“It is now important to carry out further studies to determine how the Epstein-Barr virus controls other genes that are associated with lymphoma. This will tell us more about how the virus drives lymphoma development and will help to identify new ways of targeting Epstein-Barr virus-infected cancer cells with specific drugs.” ![]()
Image by Ed Uthman
Results of research published in eLIFE appear to explain how Epstein-Barr virus (EBV) controls a pair of genes to drive lymphomagenesis.
Researchers set out to determine how EBV controls MYC, which is known to drive lymphoma development when activated, and BCL2L11, a gene that normally triggers apoptosis to prevent lymphoma but can be silenced by EBV.
The team discovered that EBV controls MYC and BCL2L11 by hijacking enhancer regions of DNA, which are situated far away from the genes.
These enhancers act as “control centers” and are able to contact and control genes from long distances by the looping out of the intervening stretches of DNA.
The researchers found that EBV activates MYC by increasing contacts between a specific set of enhancers and the gene.
The team said an Epstein-Barr nuclear antigen, EBNA2, activates multiple MYC enhancers and reconfigures the MYC locus to increase upstream enhancer-promoter interactions and decrease downstream interactions.
They noted that EBNA2 recruits the BRG1 ATPase of the SWI/SNF remodeller to MYC enhancers, and BRG1 is required for enhancer-promoter interactions in EBV-infected cells.
The researchers also discovered new enhancers that control BCL2L11. In this case, though, EBV stops these control centers from contacting the gene.
Specifically, the team found a hematopoietic enhancer hub that is inactivated by the Epstein-Barr nuclear antigens EBNA3A and EBNA3C through recruitment of the H3K27 methyltransferase EZH2.
Therefore, the researchers set out to determine if an EZH1/2 inhibitor, UNC1999, could reverse this effect. They found that UNC1999 did reverse enhancer inactivation, upregulated BCL2L11, and induced apoptosis in EBV-positive Burkitt lymphoma cells.
“This is a key step towards uncovering how this common virus, which affects thousands of people every year, causes blood cancer,” said study author Michelle West, PhD, of the University of Sussex in Brighton, UK.
“It is now important to carry out further studies to determine how the Epstein-Barr virus controls other genes that are associated with lymphoma. This will tell us more about how the virus drives lymphoma development and will help to identify new ways of targeting Epstein-Barr virus-infected cancer cells with specific drugs.” ![]()
Drug dubbed ‘breakthrough’ for CTCL subtypes

Photo from Business Wire
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to brentuximab vedotin (Adcetris) as a treatment for 2 subtypes of cutaneous T-cell lymphoma (CTCL).
The drug now has this designation for the treatment of patients with CD30-expressing mycosis fungoides (MF) and patients with primary cutaneous anaplastic large-cell lymphoma (pcALCL) who require systemic therapy and have received 1 prior systemic therapy.
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Brentuximab vedotin in CTCL
Brentuximab vedotin is an antibody-drug conjugate directed to CD30, which is expressed on skin lesions in approximately 50% of patients with CTCL. The drug is being developed by Seattle Genetics and Takeda Pharmaceutical Company Limited.
Brentuximab vedotin has orphan drug designation from the FDA for the treatment of MF. The drug also received orphan drug designation from the European Commission for CTCL, including subtypes pcALCL and MF.
Brentuximab vedotin has been evaluated in CD30-expressing CTCL in investigator- and corporate-sponsored clinical trials, including the phase 3 ALCANZA trial.
This trial was designed to compare single-agent brentuximab vedotin to investigator’s choice of standard therapies—methotrexate or bexarotene—in patients with CD30-expressing CTCL, including those with pcALCL or MF.
The trial has enrolled 131 patients at 50 sites globally. Patients with pcALCL must have received at least 1 prior systemic or radiation therapy, and patients with MF must have received at least 1 prior systemic therapy.
The study’s primary endpoint is objective response lasting at least 4 months (ORR4), as assessed by Global Response Score, in the brentuximab vedotin arm compared to the control arm. Key secondary endpoints are complete response rate, progression-free survival, and reduction in the burden of symptoms during treatment.
Topline results of the trial were announced in August. The data showed a significant improvement in the ORR4 for the brentuximab vedotin arm compared to the control arm. The ORR4 was 56.3% and 12.5%, respectively (P<0.0001).
The key secondary endpoints were all statistically significant in favor of the brentuximab vedotin arm. And investigators said the safety profile of brentuximab vedotin was generally consistent with the existing prescribing information.
An abstract detailing results of the ALCANZA trial was accepted for oral presentation at the upcoming ASH Annual Meeting (abstract 182). ![]()
Photo from Business Wire
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to brentuximab vedotin (Adcetris) as a treatment for 2 subtypes of cutaneous T-cell lymphoma (CTCL).
The drug now has this designation for the treatment of patients with CD30-expressing mycosis fungoides (MF) and patients with primary cutaneous anaplastic large-cell lymphoma (pcALCL) who require systemic therapy and have received 1 prior systemic therapy.
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Brentuximab vedotin in CTCL
Brentuximab vedotin is an antibody-drug conjugate directed to CD30, which is expressed on skin lesions in approximately 50% of patients with CTCL. The drug is being developed by Seattle Genetics and Takeda Pharmaceutical Company Limited.
Brentuximab vedotin has orphan drug designation from the FDA for the treatment of MF. The drug also received orphan drug designation from the European Commission for CTCL, including subtypes pcALCL and MF.
Brentuximab vedotin has been evaluated in CD30-expressing CTCL in investigator- and corporate-sponsored clinical trials, including the phase 3 ALCANZA trial.
This trial was designed to compare single-agent brentuximab vedotin to investigator’s choice of standard therapies—methotrexate or bexarotene—in patients with CD30-expressing CTCL, including those with pcALCL or MF.
The trial has enrolled 131 patients at 50 sites globally. Patients with pcALCL must have received at least 1 prior systemic or radiation therapy, and patients with MF must have received at least 1 prior systemic therapy.
The study’s primary endpoint is objective response lasting at least 4 months (ORR4), as assessed by Global Response Score, in the brentuximab vedotin arm compared to the control arm. Key secondary endpoints are complete response rate, progression-free survival, and reduction in the burden of symptoms during treatment.
Topline results of the trial were announced in August. The data showed a significant improvement in the ORR4 for the brentuximab vedotin arm compared to the control arm. The ORR4 was 56.3% and 12.5%, respectively (P<0.0001).
The key secondary endpoints were all statistically significant in favor of the brentuximab vedotin arm. And investigators said the safety profile of brentuximab vedotin was generally consistent with the existing prescribing information.
An abstract detailing results of the ALCANZA trial was accepted for oral presentation at the upcoming ASH Annual Meeting (abstract 182). ![]()
Photo from Business Wire
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to brentuximab vedotin (Adcetris) as a treatment for 2 subtypes of cutaneous T-cell lymphoma (CTCL).
The drug now has this designation for the treatment of patients with CD30-expressing mycosis fungoides (MF) and patients with primary cutaneous anaplastic large-cell lymphoma (pcALCL) who require systemic therapy and have received 1 prior systemic therapy.
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Brentuximab vedotin in CTCL
Brentuximab vedotin is an antibody-drug conjugate directed to CD30, which is expressed on skin lesions in approximately 50% of patients with CTCL. The drug is being developed by Seattle Genetics and Takeda Pharmaceutical Company Limited.
Brentuximab vedotin has orphan drug designation from the FDA for the treatment of MF. The drug also received orphan drug designation from the European Commission for CTCL, including subtypes pcALCL and MF.
Brentuximab vedotin has been evaluated in CD30-expressing CTCL in investigator- and corporate-sponsored clinical trials, including the phase 3 ALCANZA trial.
This trial was designed to compare single-agent brentuximab vedotin to investigator’s choice of standard therapies—methotrexate or bexarotene—in patients with CD30-expressing CTCL, including those with pcALCL or MF.
The trial has enrolled 131 patients at 50 sites globally. Patients with pcALCL must have received at least 1 prior systemic or radiation therapy, and patients with MF must have received at least 1 prior systemic therapy.
The study’s primary endpoint is objective response lasting at least 4 months (ORR4), as assessed by Global Response Score, in the brentuximab vedotin arm compared to the control arm. Key secondary endpoints are complete response rate, progression-free survival, and reduction in the burden of symptoms during treatment.
Topline results of the trial were announced in August. The data showed a significant improvement in the ORR4 for the brentuximab vedotin arm compared to the control arm. The ORR4 was 56.3% and 12.5%, respectively (P<0.0001).
The key secondary endpoints were all statistically significant in favor of the brentuximab vedotin arm. And investigators said the safety profile of brentuximab vedotin was generally consistent with the existing prescribing information.
An abstract detailing results of the ALCANZA trial was accepted for oral presentation at the upcoming ASH Annual Meeting (abstract 182). ![]()
NCCN: Deliver vincristine by mini IV drip bag
Always dilute chemotherapy agent vincristine and administer it by mini IV-drip bag, instead of syringe, urges the National Comprehensive Cancer Network in a new campaign.
The goal of “Just Bag It” is to prevent a rare but uniformly fatal medical error – administering vincristine to the spinal fluid. When syringes are side by side – one with vincristine for IV push, another with a chemotherapeutic agent meant for push into the spinal fluid – it is just too easy to make a mistake. When administered intrathecally, vincristine causes ascending paralysis, neurological defects, and eventually death.
Despite all the warning labels and checks, “this still happens,” Marc Stewart, MD, cochair of the National Comprehensive Cancer Network (NCCN) Best Practices Committee, as well as medical director of the Seattle Cancer Care Alliance and professor of medicine at the University of Washington, said at a press conference.

Mini IV-drip bag administration will make it “virtually impossible. No physician would hook the bag up to a needle in someone’s spine” and even if they did, there wouldn’t be enough pressure in the bag to push vincristine in, he said.
The group has encouraged drip-bag delivery of vincristine for years, but only about half of hospitals have adopted the policy. The mistake happens so rarely – about 125 cases since the 1960s – “that the motivation for change is just not there.” Until somebody like NCCN calls it out in a high-profile campaign, “it’s not high on the radar screen,” Dr. Stewart said. It should be a relatively easy fix because bagging vincristine is not more costly. In general, the cost difference versus syringe “is going to be pennies,” he said.
“We challenge all medical centers, hospitals, and oncology practices around the nation and the world to implement this medication safety policy so this error never occurs again,” NCCN Chief Executive Officer Robert Carlson, MD, said in a press release. A medical oncologist, he witnessed the death of a 21-year-old patient after an intrathecal vincristine injection in 2005.
“Some health care providers may associate the use of an IV bag with a heightened risk of extravasation, but research shows that the risk of extravasation is extremely low (less than 0.05%) regardless of how vincristine is administered,” the press release noted.
Vincristine is widely used in treating patients with leukemia or lymphoma.
The safety of intravenous administration of vincristine has been a long-standing concern for anyone who participates in the management of patients with hematologic malignancies. As we all know, accidental intrathecal administration of vincristine is uniformly fatal.

At many centers, including ours, policies related to intravenous infusion of vesicants via a peripheral line have made the implementation of the safety recommendations difficult. It is not surprising that only 50% of hospitals surveyed by NCCN have fully implemented the mini-bag recommendation given the concern for extravasation. However, the newest ONS guidelines for vesicant administration allow for short-term infusions via a peripheral line. For our center, this support has been instrumental in allowing us to move to a practice with the recommended mini-bags. The NCCN “Just Bag It” campaign will likely help to move institutions such as ours to be in compliance with this important safety initiative.
Donna Capozzi, PharmD, is associate director of ambulatory services in the department of pharmacy at the Hospital of the University of Pennsylvania Perelman Center for Advanced Medicine in Philadelphia. She is on the editorial advisory board of Hematology News, a publication of this news company.
The safety of intravenous administration of vincristine has been a long-standing concern for anyone who participates in the management of patients with hematologic malignancies. As we all know, accidental intrathecal administration of vincristine is uniformly fatal.
At many centers, including ours, policies related to intravenous infusion of vesicants via a peripheral line have made the implementation of the safety recommendations difficult. It is not surprising that only 50% of hospitals surveyed by NCCN have fully implemented the mini-bag recommendation given the concern for extravasation. However, the newest ONS guidelines for vesicant administration allow for short-term infusions via a peripheral line. For our center, this support has been instrumental in allowing us to move to a practice with the recommended mini-bags. The NCCN “Just Bag It” campaign will likely help to move institutions such as ours to be in compliance with this important safety initiative.
Donna Capozzi, PharmD, is associate director of ambulatory services in the department of pharmacy at the Hospital of the University of Pennsylvania Perelman Center for Advanced Medicine in Philadelphia. She is on the editorial advisory board of Hematology News, a publication of this news company.
The safety of intravenous administration of vincristine has been a long-standing concern for anyone who participates in the management of patients with hematologic malignancies. As we all know, accidental intrathecal administration of vincristine is uniformly fatal.
At many centers, including ours, policies related to intravenous infusion of vesicants via a peripheral line have made the implementation of the safety recommendations difficult. It is not surprising that only 50% of hospitals surveyed by NCCN have fully implemented the mini-bag recommendation given the concern for extravasation. However, the newest ONS guidelines for vesicant administration allow for short-term infusions via a peripheral line. For our center, this support has been instrumental in allowing us to move to a practice with the recommended mini-bags. The NCCN “Just Bag It” campaign will likely help to move institutions such as ours to be in compliance with this important safety initiative.
Donna Capozzi, PharmD, is associate director of ambulatory services in the department of pharmacy at the Hospital of the University of Pennsylvania Perelman Center for Advanced Medicine in Philadelphia. She is on the editorial advisory board of Hematology News, a publication of this news company.
Always dilute chemotherapy agent vincristine and administer it by mini IV-drip bag, instead of syringe, urges the National Comprehensive Cancer Network in a new campaign.
The goal of “Just Bag It” is to prevent a rare but uniformly fatal medical error – administering vincristine to the spinal fluid. When syringes are side by side – one with vincristine for IV push, another with a chemotherapeutic agent meant for push into the spinal fluid – it is just too easy to make a mistake. When administered intrathecally, vincristine causes ascending paralysis, neurological defects, and eventually death.
Despite all the warning labels and checks, “this still happens,” Marc Stewart, MD, cochair of the National Comprehensive Cancer Network (NCCN) Best Practices Committee, as well as medical director of the Seattle Cancer Care Alliance and professor of medicine at the University of Washington, said at a press conference.
Mini IV-drip bag administration will make it “virtually impossible. No physician would hook the bag up to a needle in someone’s spine” and even if they did, there wouldn’t be enough pressure in the bag to push vincristine in, he said.
The group has encouraged drip-bag delivery of vincristine for years, but only about half of hospitals have adopted the policy. The mistake happens so rarely – about 125 cases since the 1960s – “that the motivation for change is just not there.” Until somebody like NCCN calls it out in a high-profile campaign, “it’s not high on the radar screen,” Dr. Stewart said. It should be a relatively easy fix because bagging vincristine is not more costly. In general, the cost difference versus syringe “is going to be pennies,” he said.
“We challenge all medical centers, hospitals, and oncology practices around the nation and the world to implement this medication safety policy so this error never occurs again,” NCCN Chief Executive Officer Robert Carlson, MD, said in a press release. A medical oncologist, he witnessed the death of a 21-year-old patient after an intrathecal vincristine injection in 2005.
“Some health care providers may associate the use of an IV bag with a heightened risk of extravasation, but research shows that the risk of extravasation is extremely low (less than 0.05%) regardless of how vincristine is administered,” the press release noted.
Vincristine is widely used in treating patients with leukemia or lymphoma.
Always dilute chemotherapy agent vincristine and administer it by mini IV-drip bag, instead of syringe, urges the National Comprehensive Cancer Network in a new campaign.
The goal of “Just Bag It” is to prevent a rare but uniformly fatal medical error – administering vincristine to the spinal fluid. When syringes are side by side – one with vincristine for IV push, another with a chemotherapeutic agent meant for push into the spinal fluid – it is just too easy to make a mistake. When administered intrathecally, vincristine causes ascending paralysis, neurological defects, and eventually death.
Despite all the warning labels and checks, “this still happens,” Marc Stewart, MD, cochair of the National Comprehensive Cancer Network (NCCN) Best Practices Committee, as well as medical director of the Seattle Cancer Care Alliance and professor of medicine at the University of Washington, said at a press conference.
Mini IV-drip bag administration will make it “virtually impossible. No physician would hook the bag up to a needle in someone’s spine” and even if they did, there wouldn’t be enough pressure in the bag to push vincristine in, he said.
The group has encouraged drip-bag delivery of vincristine for years, but only about half of hospitals have adopted the policy. The mistake happens so rarely – about 125 cases since the 1960s – “that the motivation for change is just not there.” Until somebody like NCCN calls it out in a high-profile campaign, “it’s not high on the radar screen,” Dr. Stewart said. It should be a relatively easy fix because bagging vincristine is not more costly. In general, the cost difference versus syringe “is going to be pennies,” he said.
“We challenge all medical centers, hospitals, and oncology practices around the nation and the world to implement this medication safety policy so this error never occurs again,” NCCN Chief Executive Officer Robert Carlson, MD, said in a press release. A medical oncologist, he witnessed the death of a 21-year-old patient after an intrathecal vincristine injection in 2005.
“Some health care providers may associate the use of an IV bag with a heightened risk of extravasation, but research shows that the risk of extravasation is extremely low (less than 0.05%) regardless of how vincristine is administered,” the press release noted.
Vincristine is widely used in treating patients with leukemia or lymphoma.
CHMP recommends expanding use of drug in CLL

Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for ofatumumab (Arzerra®).
The CHMP is recommending the drug be approved for use in combination with fludarabine and cyclophosphamide to treat adults with relapsed chronic lymphocytic leukemia (CLL).
The CHMP’s recommendation will be reviewed by the European Commission.
A final decision is expected in the coming months.
Ofatumumab is a monoclonal antibody designed to target CD20. The drug is marketed under a collaboration agreement between Genmab and Novartis.
The European Commission has already approved ofatumumab for the following indications:
- As a single-agent to treat CLL patients who are refractory to fludarabine and alemtuzumab
- For use in combination with chlorambucil or bendamustine in CLL patients who
have not received prior therapy and are not eligible for
fludarabine-based therapy.
COMPLEMENT 2 trial
The CHMP’s recommendation to approve ofatumumab in combination with fludarabine and cyclophosphamide was based on results from the phase 3 COMPLEMENT 2 study. Novartis reported top-line results from this study last April.
The trial enrolled 365 patients with relapsed CLL. The patients were randomized 1:1 to receive up to 6 cycles of ofatumumab in combination with fludarabine and cyclophosphamide or up to 6 cycles of fludarabine and cyclophosphamide alone.
The primary endpoint was progression-free survival, as assessed by an independent review committee.
The median progression-free survival was 28.9 months for patients receiving ofatumumab plus fludarabine and cyclophosphamide, compared to 18.8 months for patients receiving fludarabine and cyclophosphamide alone (hazard ratio=0.67, P=0.0032).
Novartis said the safety profile observed in this study was consistent with other trials of ofatumumab, and no new safety signals were observed. ![]()
Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for ofatumumab (Arzerra®).
The CHMP is recommending the drug be approved for use in combination with fludarabine and cyclophosphamide to treat adults with relapsed chronic lymphocytic leukemia (CLL).
The CHMP’s recommendation will be reviewed by the European Commission.
A final decision is expected in the coming months.
Ofatumumab is a monoclonal antibody designed to target CD20. The drug is marketed under a collaboration agreement between Genmab and Novartis.
The European Commission has already approved ofatumumab for the following indications:
- As a single-agent to treat CLL patients who are refractory to fludarabine and alemtuzumab
- For use in combination with chlorambucil or bendamustine in CLL patients who
have not received prior therapy and are not eligible for
fludarabine-based therapy.
COMPLEMENT 2 trial
The CHMP’s recommendation to approve ofatumumab in combination with fludarabine and cyclophosphamide was based on results from the phase 3 COMPLEMENT 2 study. Novartis reported top-line results from this study last April.
The trial enrolled 365 patients with relapsed CLL. The patients were randomized 1:1 to receive up to 6 cycles of ofatumumab in combination with fludarabine and cyclophosphamide or up to 6 cycles of fludarabine and cyclophosphamide alone.
The primary endpoint was progression-free survival, as assessed by an independent review committee.
The median progression-free survival was 28.9 months for patients receiving ofatumumab plus fludarabine and cyclophosphamide, compared to 18.8 months for patients receiving fludarabine and cyclophosphamide alone (hazard ratio=0.67, P=0.0032).
Novartis said the safety profile observed in this study was consistent with other trials of ofatumumab, and no new safety signals were observed. ![]()
Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for ofatumumab (Arzerra®).
The CHMP is recommending the drug be approved for use in combination with fludarabine and cyclophosphamide to treat adults with relapsed chronic lymphocytic leukemia (CLL).
The CHMP’s recommendation will be reviewed by the European Commission.
A final decision is expected in the coming months.
Ofatumumab is a monoclonal antibody designed to target CD20. The drug is marketed under a collaboration agreement between Genmab and Novartis.
The European Commission has already approved ofatumumab for the following indications:
- As a single-agent to treat CLL patients who are refractory to fludarabine and alemtuzumab
- For use in combination with chlorambucil or bendamustine in CLL patients who
have not received prior therapy and are not eligible for
fludarabine-based therapy.
COMPLEMENT 2 trial
The CHMP’s recommendation to approve ofatumumab in combination with fludarabine and cyclophosphamide was based on results from the phase 3 COMPLEMENT 2 study. Novartis reported top-line results from this study last April.
The trial enrolled 365 patients with relapsed CLL. The patients were randomized 1:1 to receive up to 6 cycles of ofatumumab in combination with fludarabine and cyclophosphamide or up to 6 cycles of fludarabine and cyclophosphamide alone.
The primary endpoint was progression-free survival, as assessed by an independent review committee.
The median progression-free survival was 28.9 months for patients receiving ofatumumab plus fludarabine and cyclophosphamide, compared to 18.8 months for patients receiving fludarabine and cyclophosphamide alone (hazard ratio=0.67, P=0.0032).
Novartis said the safety profile observed in this study was consistent with other trials of ofatumumab, and no new safety signals were observed. ![]()
NCCN issues challenge to ‘bag’ vincristine
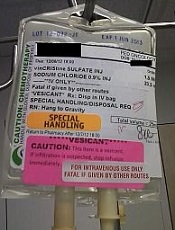
Photo courtesy of ISMP
PHILADELPHIA—To ensure proper administration of vincristine, the National Comprehensive Cancer Network (NCCN) has issued a challenge to hospitals, medical centers, and oncology practices as part of its “Just Bag It!” campaign.
Vincristine—the “O” for Oncovin in the CHOP regimen—is widely used to treat patients with leukemia or lymphoma.
It is considered an important chemotherapeutic agent. However, if administered incorrectly, vincristine is uniformly fatal, usually within a week, according to the NCCN.
Vincristine is highly neurotoxic and should always be administered intravenously. If it is mistakenly given intrathecally along with other chemotherapy drugs, it causes ascending paralysis, neurological defects, and death.
Therefore, the NCCN recommends always diluting and administering vincristine in a mini IV-drip bag, never through a syringe.
This precaution decreases the chances of improper dosage and makes it impossible to accidentally administer vincristine into the spinal fluid.
The NCCN initiated the safe-handling campaign in response to the death 11 years ago of a 21-year-old patient who received vincristine incorrectly administered into his spinal fluid. He was referred to Robert W. Carlson, MD, NCCN’s chief executive officer, who, at the time, was at Stanford Hospital, not the hospital where the error occurred.
The patient, Christopher Wibeto, had a “likely curable” non-Hodgkin lymphoma and died 4 days later.
“When I first met Christopher, he was doing well,” Dr Carlson said. “He was a delightful young gentleman, very articulate. He was funny. Even in the ICU, he had me chuckling and laughing at what he was saying.”
“But we knew that the medical error would almost certainly lead to his death. Shortly thereafter, I met his parents, Debra and Robin, . . . and had to tell them what the consequences of that medical error were likely to be. And they joined me in Christopher’s room while we talked with him about what the consequences of that medical error were likely to be.”
Making the situation even more painful, Dr Carlson, at that time, was the father of a young son who is now almost the age Christopher was then.
Dr Carlson said he realized that “we needed to come up with systems to assure that this did not happen, not today or tomorrow or ever again.”
Motivated by the tragedy, Dr Carlson spearheaded a national effort to address this mistake when he joined NCCN as CEO, enlisting the help of NCCN’s Best Practices Committee.
The NCCN developed and issued guidelines, and all 27 member institutions have adopted policies in line with the guidelines.
The Institute for Safe Medication Practices (ISMP) has undertaken efforts over more than a decade to implement procedures for safe vincristine administration.
ISMP conducted surveys and follow-up self-assessments regarding use of IV bags for vincristine at oncology practice sites. They found that only about half the institutions surveyed dilute IV vincristine for administration in a small-volume bag.
Some practitioners associate the use of an IV bag with an increased risk of extravasation (when the chemotherapy agent leaks into the tissue surrounding the administration site). Research shows, however, that the risk of extravasation is extremely low (less than 0.5%), regardless of how vincristine is administered.
And cost is not an issue when implementing the mini-bag policy, according to the president of ISMP, Michael R. Cohen, RPh.
“It cost a few pennies more,” he said. “And I mean pennies. I think probably what is an issue is just the age-old habit of putting vincristine in a syringe and being able to change that habit.”
Since the introduction of vincristine use in the 1960s, 125 documented cases of accidental death in the US and abroad have been reported. While the error is relatively rare, it is preventable and unique in its level of mortality.
“It’s hard to understand why this idea of ‘Just Bag It’ hasn’t permeated healthcare at this point,” Cohen said. “Because it is a sure-fire way to prevent this type of error.”
The ISMP, the Joint Commission, the World Health Organization, and the Oncology Nursing Society also recommend the bag-it policy. ![]()
Photo courtesy of ISMP
PHILADELPHIA—To ensure proper administration of vincristine, the National Comprehensive Cancer Network (NCCN) has issued a challenge to hospitals, medical centers, and oncology practices as part of its “Just Bag It!” campaign.
Vincristine—the “O” for Oncovin in the CHOP regimen—is widely used to treat patients with leukemia or lymphoma.
It is considered an important chemotherapeutic agent. However, if administered incorrectly, vincristine is uniformly fatal, usually within a week, according to the NCCN.
Vincristine is highly neurotoxic and should always be administered intravenously. If it is mistakenly given intrathecally along with other chemotherapy drugs, it causes ascending paralysis, neurological defects, and death.
Therefore, the NCCN recommends always diluting and administering vincristine in a mini IV-drip bag, never through a syringe.
This precaution decreases the chances of improper dosage and makes it impossible to accidentally administer vincristine into the spinal fluid.
The NCCN initiated the safe-handling campaign in response to the death 11 years ago of a 21-year-old patient who received vincristine incorrectly administered into his spinal fluid. He was referred to Robert W. Carlson, MD, NCCN’s chief executive officer, who, at the time, was at Stanford Hospital, not the hospital where the error occurred.
The patient, Christopher Wibeto, had a “likely curable” non-Hodgkin lymphoma and died 4 days later.
“When I first met Christopher, he was doing well,” Dr Carlson said. “He was a delightful young gentleman, very articulate. He was funny. Even in the ICU, he had me chuckling and laughing at what he was saying.”
“But we knew that the medical error would almost certainly lead to his death. Shortly thereafter, I met his parents, Debra and Robin, . . . and had to tell them what the consequences of that medical error were likely to be. And they joined me in Christopher’s room while we talked with him about what the consequences of that medical error were likely to be.”
Making the situation even more painful, Dr Carlson, at that time, was the father of a young son who is now almost the age Christopher was then.
Dr Carlson said he realized that “we needed to come up with systems to assure that this did not happen, not today or tomorrow or ever again.”
Motivated by the tragedy, Dr Carlson spearheaded a national effort to address this mistake when he joined NCCN as CEO, enlisting the help of NCCN’s Best Practices Committee.
The NCCN developed and issued guidelines, and all 27 member institutions have adopted policies in line with the guidelines.
The Institute for Safe Medication Practices (ISMP) has undertaken efforts over more than a decade to implement procedures for safe vincristine administration.
ISMP conducted surveys and follow-up self-assessments regarding use of IV bags for vincristine at oncology practice sites. They found that only about half the institutions surveyed dilute IV vincristine for administration in a small-volume bag.
Some practitioners associate the use of an IV bag with an increased risk of extravasation (when the chemotherapy agent leaks into the tissue surrounding the administration site). Research shows, however, that the risk of extravasation is extremely low (less than 0.5%), regardless of how vincristine is administered.
And cost is not an issue when implementing the mini-bag policy, according to the president of ISMP, Michael R. Cohen, RPh.
“It cost a few pennies more,” he said. “And I mean pennies. I think probably what is an issue is just the age-old habit of putting vincristine in a syringe and being able to change that habit.”
Since the introduction of vincristine use in the 1960s, 125 documented cases of accidental death in the US and abroad have been reported. While the error is relatively rare, it is preventable and unique in its level of mortality.
“It’s hard to understand why this idea of ‘Just Bag It’ hasn’t permeated healthcare at this point,” Cohen said. “Because it is a sure-fire way to prevent this type of error.”
The ISMP, the Joint Commission, the World Health Organization, and the Oncology Nursing Society also recommend the bag-it policy. ![]()
Photo courtesy of ISMP
PHILADELPHIA—To ensure proper administration of vincristine, the National Comprehensive Cancer Network (NCCN) has issued a challenge to hospitals, medical centers, and oncology practices as part of its “Just Bag It!” campaign.
Vincristine—the “O” for Oncovin in the CHOP regimen—is widely used to treat patients with leukemia or lymphoma.
It is considered an important chemotherapeutic agent. However, if administered incorrectly, vincristine is uniformly fatal, usually within a week, according to the NCCN.
Vincristine is highly neurotoxic and should always be administered intravenously. If it is mistakenly given intrathecally along with other chemotherapy drugs, it causes ascending paralysis, neurological defects, and death.
Therefore, the NCCN recommends always diluting and administering vincristine in a mini IV-drip bag, never through a syringe.
This precaution decreases the chances of improper dosage and makes it impossible to accidentally administer vincristine into the spinal fluid.
The NCCN initiated the safe-handling campaign in response to the death 11 years ago of a 21-year-old patient who received vincristine incorrectly administered into his spinal fluid. He was referred to Robert W. Carlson, MD, NCCN’s chief executive officer, who, at the time, was at Stanford Hospital, not the hospital where the error occurred.
The patient, Christopher Wibeto, had a “likely curable” non-Hodgkin lymphoma and died 4 days later.
“When I first met Christopher, he was doing well,” Dr Carlson said. “He was a delightful young gentleman, very articulate. He was funny. Even in the ICU, he had me chuckling and laughing at what he was saying.”
“But we knew that the medical error would almost certainly lead to his death. Shortly thereafter, I met his parents, Debra and Robin, . . . and had to tell them what the consequences of that medical error were likely to be. And they joined me in Christopher’s room while we talked with him about what the consequences of that medical error were likely to be.”
Making the situation even more painful, Dr Carlson, at that time, was the father of a young son who is now almost the age Christopher was then.
Dr Carlson said he realized that “we needed to come up with systems to assure that this did not happen, not today or tomorrow or ever again.”
Motivated by the tragedy, Dr Carlson spearheaded a national effort to address this mistake when he joined NCCN as CEO, enlisting the help of NCCN’s Best Practices Committee.
The NCCN developed and issued guidelines, and all 27 member institutions have adopted policies in line with the guidelines.
The Institute for Safe Medication Practices (ISMP) has undertaken efforts over more than a decade to implement procedures for safe vincristine administration.
ISMP conducted surveys and follow-up self-assessments regarding use of IV bags for vincristine at oncology practice sites. They found that only about half the institutions surveyed dilute IV vincristine for administration in a small-volume bag.
Some practitioners associate the use of an IV bag with an increased risk of extravasation (when the chemotherapy agent leaks into the tissue surrounding the administration site). Research shows, however, that the risk of extravasation is extremely low (less than 0.5%), regardless of how vincristine is administered.
And cost is not an issue when implementing the mini-bag policy, according to the president of ISMP, Michael R. Cohen, RPh.
“It cost a few pennies more,” he said. “And I mean pennies. I think probably what is an issue is just the age-old habit of putting vincristine in a syringe and being able to change that habit.”
Since the introduction of vincristine use in the 1960s, 125 documented cases of accidental death in the US and abroad have been reported. While the error is relatively rare, it is preventable and unique in its level of mortality.
“It’s hard to understand why this idea of ‘Just Bag It’ hasn’t permeated healthcare at this point,” Cohen said. “Because it is a sure-fire way to prevent this type of error.”
The ISMP, the Joint Commission, the World Health Organization, and the Oncology Nursing Society also recommend the bag-it policy. ![]()