User login
Len plus anti-CD19 Mab MOR208 active against advanced DLBCL
LUGANO, SWITZERLAND – Combining lenalidomide (Revlimid) with an anti-CD19 monoclonal antibody labeled MOR208 showed promising activity in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who were ineligible for stem cell transplant and had poor prognosis, early interim results from a clinical study indicate.
Among 34 patients evaluable for response, the preliminary objective response rate (ORR) was 56%, including complete responses in 32% of patients, reported Gilles Salles, MD, PhD, of the University of Lyon, France.

MOR208 is a humanized anti-CD19 monoclonal antibody with the Fc-antibody region enhanced to improve cytotoxicity. Its mechanisms of action include natural killer cell–mediated antibody-dependent cell-mediated cytotoxicity, antibody-dependent cellular phagocytosis, and direct cytotoxicity.
In a preclinical study, a combination of MOR208 and lenalidomide showed synergistic antileukemic and antilymphoma activity both in vivo and in vitro, Dr. Salles said.
In addition, both lenalidomide and MOR208 have shown significant activity against relapsed, refractory B-cell non-Hodgkin lymphomas.
In an ongoing phase II, open-label study, Dr. Salles and his colleagues are enrolling transplant-ineligible patients 18 years and older with relapsed/refractory DLBCL, Eastern Cooperative Oncology Group status 0-2, and adequate organ function who had disease progression after 1-3 prior lines of therapy.
Patients with primary refractory DLBCL, double-hit or triple-hit DLBCL (i.e., mutations in Myc, BCL2, and/or BCL6), other NHL histological subtypes, or central nervous system lymphoma involvement are excluded.
Patients receive MOR208 12 mg/kg intravenously on days 1, 8, 15, and 22 for cycles 1-3 and on days 1 and 15 of cycles 4-12. Lenalidomide 25 mg orally is delivered on days 1-21 of each cycle. Patients who have stable disease or better at the end of 12 cycles can be maintained on MOR208 at the same dose on days 1 and 15.
As of the data cutoff on March 6, 2017, 44 patients had been enrolled, and 34 were evaluable for response. The median patient age was 73 years (range, 47-82 years).
At the time of the data presentation, ORR, the primary endpoint, was 56%, consisting of 32% complete responses (11 patients), 24% partial responses (8), 12% stable disease (4), and 32% of patients who either had disease progression or had not yet had a postbaseline response assessment.
The median time to response was 1.8 months, with a median time to complete response of 3.4 months. Of 19 responders, 16 continue to have a response, including 10 of 11 patients with complete responses.
The most common grade 3 or 4 hematologic toxicities were neutropenia, anemia, and thrombocytopenia. Nonhematologic toxicities of any grade included rashes in 20% of patients, pyrexia in 16%, diarrhea in 16%, asthenia in 14%, and pneumonia, bronchitis, and nausea in 11% each.
There were no reported infusion-related reactions with the antibody. In all, 27% of patients required a lenalidomide dose reduction – to 20 mg/day in 20% of patients and to 15 mg/day in 7%.
Study accrual, follow-up of patients on therapy, investigations of cell origin, and subgroup analyses are ongoing.
MorphoSys is sponsoring the study. Dr. Salles has received honoraria from Amgen, BMS, Celgene, Gilead, Janssen, Roche/Genentech, and Servier and is an advisor/consultant to many of the same companies.
LUGANO, SWITZERLAND – Combining lenalidomide (Revlimid) with an anti-CD19 monoclonal antibody labeled MOR208 showed promising activity in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who were ineligible for stem cell transplant and had poor prognosis, early interim results from a clinical study indicate.
Among 34 patients evaluable for response, the preliminary objective response rate (ORR) was 56%, including complete responses in 32% of patients, reported Gilles Salles, MD, PhD, of the University of Lyon, France.
MOR208 is a humanized anti-CD19 monoclonal antibody with the Fc-antibody region enhanced to improve cytotoxicity. Its mechanisms of action include natural killer cell–mediated antibody-dependent cell-mediated cytotoxicity, antibody-dependent cellular phagocytosis, and direct cytotoxicity.
In a preclinical study, a combination of MOR208 and lenalidomide showed synergistic antileukemic and antilymphoma activity both in vivo and in vitro, Dr. Salles said.
In addition, both lenalidomide and MOR208 have shown significant activity against relapsed, refractory B-cell non-Hodgkin lymphomas.
In an ongoing phase II, open-label study, Dr. Salles and his colleagues are enrolling transplant-ineligible patients 18 years and older with relapsed/refractory DLBCL, Eastern Cooperative Oncology Group status 0-2, and adequate organ function who had disease progression after 1-3 prior lines of therapy.
Patients with primary refractory DLBCL, double-hit or triple-hit DLBCL (i.e., mutations in Myc, BCL2, and/or BCL6), other NHL histological subtypes, or central nervous system lymphoma involvement are excluded.
Patients receive MOR208 12 mg/kg intravenously on days 1, 8, 15, and 22 for cycles 1-3 and on days 1 and 15 of cycles 4-12. Lenalidomide 25 mg orally is delivered on days 1-21 of each cycle. Patients who have stable disease or better at the end of 12 cycles can be maintained on MOR208 at the same dose on days 1 and 15.
As of the data cutoff on March 6, 2017, 44 patients had been enrolled, and 34 were evaluable for response. The median patient age was 73 years (range, 47-82 years).
At the time of the data presentation, ORR, the primary endpoint, was 56%, consisting of 32% complete responses (11 patients), 24% partial responses (8), 12% stable disease (4), and 32% of patients who either had disease progression or had not yet had a postbaseline response assessment.
The median time to response was 1.8 months, with a median time to complete response of 3.4 months. Of 19 responders, 16 continue to have a response, including 10 of 11 patients with complete responses.
The most common grade 3 or 4 hematologic toxicities were neutropenia, anemia, and thrombocytopenia. Nonhematologic toxicities of any grade included rashes in 20% of patients, pyrexia in 16%, diarrhea in 16%, asthenia in 14%, and pneumonia, bronchitis, and nausea in 11% each.
There were no reported infusion-related reactions with the antibody. In all, 27% of patients required a lenalidomide dose reduction – to 20 mg/day in 20% of patients and to 15 mg/day in 7%.
Study accrual, follow-up of patients on therapy, investigations of cell origin, and subgroup analyses are ongoing.
MorphoSys is sponsoring the study. Dr. Salles has received honoraria from Amgen, BMS, Celgene, Gilead, Janssen, Roche/Genentech, and Servier and is an advisor/consultant to many of the same companies.
LUGANO, SWITZERLAND – Combining lenalidomide (Revlimid) with an anti-CD19 monoclonal antibody labeled MOR208 showed promising activity in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who were ineligible for stem cell transplant and had poor prognosis, early interim results from a clinical study indicate.
Among 34 patients evaluable for response, the preliminary objective response rate (ORR) was 56%, including complete responses in 32% of patients, reported Gilles Salles, MD, PhD, of the University of Lyon, France.
MOR208 is a humanized anti-CD19 monoclonal antibody with the Fc-antibody region enhanced to improve cytotoxicity. Its mechanisms of action include natural killer cell–mediated antibody-dependent cell-mediated cytotoxicity, antibody-dependent cellular phagocytosis, and direct cytotoxicity.
In a preclinical study, a combination of MOR208 and lenalidomide showed synergistic antileukemic and antilymphoma activity both in vivo and in vitro, Dr. Salles said.
In addition, both lenalidomide and MOR208 have shown significant activity against relapsed, refractory B-cell non-Hodgkin lymphomas.
In an ongoing phase II, open-label study, Dr. Salles and his colleagues are enrolling transplant-ineligible patients 18 years and older with relapsed/refractory DLBCL, Eastern Cooperative Oncology Group status 0-2, and adequate organ function who had disease progression after 1-3 prior lines of therapy.
Patients with primary refractory DLBCL, double-hit or triple-hit DLBCL (i.e., mutations in Myc, BCL2, and/or BCL6), other NHL histological subtypes, or central nervous system lymphoma involvement are excluded.
Patients receive MOR208 12 mg/kg intravenously on days 1, 8, 15, and 22 for cycles 1-3 and on days 1 and 15 of cycles 4-12. Lenalidomide 25 mg orally is delivered on days 1-21 of each cycle. Patients who have stable disease or better at the end of 12 cycles can be maintained on MOR208 at the same dose on days 1 and 15.
As of the data cutoff on March 6, 2017, 44 patients had been enrolled, and 34 were evaluable for response. The median patient age was 73 years (range, 47-82 years).
At the time of the data presentation, ORR, the primary endpoint, was 56%, consisting of 32% complete responses (11 patients), 24% partial responses (8), 12% stable disease (4), and 32% of patients who either had disease progression or had not yet had a postbaseline response assessment.
The median time to response was 1.8 months, with a median time to complete response of 3.4 months. Of 19 responders, 16 continue to have a response, including 10 of 11 patients with complete responses.
The most common grade 3 or 4 hematologic toxicities were neutropenia, anemia, and thrombocytopenia. Nonhematologic toxicities of any grade included rashes in 20% of patients, pyrexia in 16%, diarrhea in 16%, asthenia in 14%, and pneumonia, bronchitis, and nausea in 11% each.
There were no reported infusion-related reactions with the antibody. In all, 27% of patients required a lenalidomide dose reduction – to 20 mg/day in 20% of patients and to 15 mg/day in 7%.
Study accrual, follow-up of patients on therapy, investigations of cell origin, and subgroup analyses are ongoing.
MorphoSys is sponsoring the study. Dr. Salles has received honoraria from Amgen, BMS, Celgene, Gilead, Janssen, Roche/Genentech, and Servier and is an advisor/consultant to many of the same companies.
AT 14-ICML
Key clinical point: A combination of the anti-CD19 monoclonal antibody MOR208 and the immunomodulator lenalidomide has shown good activity against relapsed/refractory diffuse large B-cell lymphoma.
Major finding: The preliminary objective response rate was 56%, including 32% complete responses.
Data source: An ongoing open-label phase II study with 44 patients out of a planned 80 enrolled.
Disclosures: MorphoSys is sponsoring the study. Dr. Salles has received honoraria from Amgen, BMS, Celgene, Gilead, Janssen, Roche/Genentech, and Servier and is an advisor or consultant to many of the same companies.
Hitting BTK, PI3K pays off in B-cell malignancies
LUGANO, SWITZERLAND – A combination of ibrutinib and umbralisib, an investigational inhibitor of phosphatidylinostiol 3-kinase (PI3K), induced high response rates in patients with relapsed/refractory B-cell malignancies, with no dose-limiting toxicities, based on updated early efficacy results from a phase I/IB dose-escalation study.
One-year progression-free survival (PFS) was 88% for patients with chronic lymphocytic leukemia (CLL), and 1-year overall survival (OS) was 94%, reported Matthew S. Davids, MD, MMSc, of the Dana-Farber Cancer Institute in Boston.

Single-agent ibrutinib (Imbruvica), an inhibitor of Bruton’s tyrosine kinase, is effective in patients with high-risk CLL or MCL, but the depth and durability of response are limited, he said. Umbralisib (TGR-1202) is a second-generation PI3K inhibitor with a high degree of specificity for the delta isoform of the kinase. It was designed to have a better safety profile than the first-in-class agent idelalisib (Zydelig).
“We hypothesized that inhibiting multiple BCR [B-cell receptor] pathways with kinase inhibitors may both deepen and prolong response and potentially overcome resistance mutations,” he said at the International Conference on Malignant Lymphoma.
In an ongoing, investigator-initiated phase I/IB trial, Dr. Davids and his colleagues enrolled 14 patients with MCL and 18 with CLL into parallel dose-escalation arms. Data were insufficient for the preliminary efficacy analysis.
Among patients with CLL, the objective response rate was 94% (16 of 17 patients). Of the 17 patients, 15 had a partial response or a partial response with lymphocytosis. One patient had a complete response, and three had radiographic complete responses, but these were not included in the objective response rate.
All three patients who had prior exposure to a PI3K inhibitor had responses, as did one of two patients with prior ibrutinib exposure.
For the patients with MCL, the objective response rate was 79% (11 of 14 patients); 10 had a partial response and 1 had a complete response. One other patient with a radiographic complete response was not included in the objective response rate.
Median follow-up among survivors was 14 months. As noted, the 1-year PFS and OS for patients with CLL were 88% and 94%, and the median PFS and OS for patients with MCL were 8.4 and 11.6 months.
One patient with CLL and five with MCL died of disease progression. A sixth patient with MCL did not have an adequate response to ibrutinib/umbralisib and died of toxicities related to the next line of therapy.
The safety analysis showed no dose-limiting toxicities, and the maximum tolerated dose was not identified with umbralisib at doses of 400 mg, 600 mg, or 800 mg daily in patients with either CLL or MCL.
The most common hematologic adverse events were grade 3/4 neutropenia in approximately 37% of patients in each arm, thrombocytopenia in 11% of CLL patients and 36% of MCL patients, and anemia in 15% and 29%, respectively.
The MCL arm of the study is still accruing patients, and correlative studies are in progress, Dr. Davids said.
The study is supported by TG Therapeutics, BCRP/LLS TAP, and grants from ASCO and the National Institutes of Health. Dr. Davids disclosed honoraria from Janssen and research funding to his institution from Phamarcyclics.
LUGANO, SWITZERLAND – A combination of ibrutinib and umbralisib, an investigational inhibitor of phosphatidylinostiol 3-kinase (PI3K), induced high response rates in patients with relapsed/refractory B-cell malignancies, with no dose-limiting toxicities, based on updated early efficacy results from a phase I/IB dose-escalation study.
One-year progression-free survival (PFS) was 88% for patients with chronic lymphocytic leukemia (CLL), and 1-year overall survival (OS) was 94%, reported Matthew S. Davids, MD, MMSc, of the Dana-Farber Cancer Institute in Boston.
Single-agent ibrutinib (Imbruvica), an inhibitor of Bruton’s tyrosine kinase, is effective in patients with high-risk CLL or MCL, but the depth and durability of response are limited, he said. Umbralisib (TGR-1202) is a second-generation PI3K inhibitor with a high degree of specificity for the delta isoform of the kinase. It was designed to have a better safety profile than the first-in-class agent idelalisib (Zydelig).
“We hypothesized that inhibiting multiple BCR [B-cell receptor] pathways with kinase inhibitors may both deepen and prolong response and potentially overcome resistance mutations,” he said at the International Conference on Malignant Lymphoma.
In an ongoing, investigator-initiated phase I/IB trial, Dr. Davids and his colleagues enrolled 14 patients with MCL and 18 with CLL into parallel dose-escalation arms. Data were insufficient for the preliminary efficacy analysis.
Among patients with CLL, the objective response rate was 94% (16 of 17 patients). Of the 17 patients, 15 had a partial response or a partial response with lymphocytosis. One patient had a complete response, and three had radiographic complete responses, but these were not included in the objective response rate.
All three patients who had prior exposure to a PI3K inhibitor had responses, as did one of two patients with prior ibrutinib exposure.
For the patients with MCL, the objective response rate was 79% (11 of 14 patients); 10 had a partial response and 1 had a complete response. One other patient with a radiographic complete response was not included in the objective response rate.
Median follow-up among survivors was 14 months. As noted, the 1-year PFS and OS for patients with CLL were 88% and 94%, and the median PFS and OS for patients with MCL were 8.4 and 11.6 months.
One patient with CLL and five with MCL died of disease progression. A sixth patient with MCL did not have an adequate response to ibrutinib/umbralisib and died of toxicities related to the next line of therapy.
The safety analysis showed no dose-limiting toxicities, and the maximum tolerated dose was not identified with umbralisib at doses of 400 mg, 600 mg, or 800 mg daily in patients with either CLL or MCL.
The most common hematologic adverse events were grade 3/4 neutropenia in approximately 37% of patients in each arm, thrombocytopenia in 11% of CLL patients and 36% of MCL patients, and anemia in 15% and 29%, respectively.
The MCL arm of the study is still accruing patients, and correlative studies are in progress, Dr. Davids said.
The study is supported by TG Therapeutics, BCRP/LLS TAP, and grants from ASCO and the National Institutes of Health. Dr. Davids disclosed honoraria from Janssen and research funding to his institution from Phamarcyclics.
LUGANO, SWITZERLAND – A combination of ibrutinib and umbralisib, an investigational inhibitor of phosphatidylinostiol 3-kinase (PI3K), induced high response rates in patients with relapsed/refractory B-cell malignancies, with no dose-limiting toxicities, based on updated early efficacy results from a phase I/IB dose-escalation study.
One-year progression-free survival (PFS) was 88% for patients with chronic lymphocytic leukemia (CLL), and 1-year overall survival (OS) was 94%, reported Matthew S. Davids, MD, MMSc, of the Dana-Farber Cancer Institute in Boston.
Single-agent ibrutinib (Imbruvica), an inhibitor of Bruton’s tyrosine kinase, is effective in patients with high-risk CLL or MCL, but the depth and durability of response are limited, he said. Umbralisib (TGR-1202) is a second-generation PI3K inhibitor with a high degree of specificity for the delta isoform of the kinase. It was designed to have a better safety profile than the first-in-class agent idelalisib (Zydelig).
“We hypothesized that inhibiting multiple BCR [B-cell receptor] pathways with kinase inhibitors may both deepen and prolong response and potentially overcome resistance mutations,” he said at the International Conference on Malignant Lymphoma.
In an ongoing, investigator-initiated phase I/IB trial, Dr. Davids and his colleagues enrolled 14 patients with MCL and 18 with CLL into parallel dose-escalation arms. Data were insufficient for the preliminary efficacy analysis.
Among patients with CLL, the objective response rate was 94% (16 of 17 patients). Of the 17 patients, 15 had a partial response or a partial response with lymphocytosis. One patient had a complete response, and three had radiographic complete responses, but these were not included in the objective response rate.
All three patients who had prior exposure to a PI3K inhibitor had responses, as did one of two patients with prior ibrutinib exposure.
For the patients with MCL, the objective response rate was 79% (11 of 14 patients); 10 had a partial response and 1 had a complete response. One other patient with a radiographic complete response was not included in the objective response rate.
Median follow-up among survivors was 14 months. As noted, the 1-year PFS and OS for patients with CLL were 88% and 94%, and the median PFS and OS for patients with MCL were 8.4 and 11.6 months.
One patient with CLL and five with MCL died of disease progression. A sixth patient with MCL did not have an adequate response to ibrutinib/umbralisib and died of toxicities related to the next line of therapy.
The safety analysis showed no dose-limiting toxicities, and the maximum tolerated dose was not identified with umbralisib at doses of 400 mg, 600 mg, or 800 mg daily in patients with either CLL or MCL.
The most common hematologic adverse events were grade 3/4 neutropenia in approximately 37% of patients in each arm, thrombocytopenia in 11% of CLL patients and 36% of MCL patients, and anemia in 15% and 29%, respectively.
The MCL arm of the study is still accruing patients, and correlative studies are in progress, Dr. Davids said.
The study is supported by TG Therapeutics, BCRP/LLS TAP, and grants from ASCO and the National Institutes of Health. Dr. Davids disclosed honoraria from Janssen and research funding to his institution from Phamarcyclics.
AT 14-ICML
Key clinical point:
Major finding: The objective response rate to the combination was 94% in 18 patients with chronic lymphocytic leukemia and 79% in 14 patients with mantle cell lymphoma.
Data source: A phase I/IB dose-escalation study.
Disclosures: The study is supported by TG Therapeutics, BCRP/LLS TAP, and grants from ASCO and the National Institutes of Health. Dr. Davids disclosed honoraria from Janssen and research funding to his institution from Phamarcyclics.
Biosimilar rituximab approved in Europe

The European Commission (EC) has approved the Sandoz biosimilar rituximab (Rixathon®) for use in the European Economic Area.
Rixathon is approved for all indications of the reference medicine, MabThera®, including follicular lymphoma, diffuse large B-cell lymphoma, chronic lymphocytic leukemia, and immunologic diseases such as rheumatoid arthritis, granulomatosis with polyangiitis, and microscopic polyangiitis.
This approval allows Rixathon to be marketed in the member states of the European Union and Iceland, Liechtenstein, and Norway, members of the European Free Trade Association.
The approval “represents a big win for patients in Europe with blood cancers or immunological diseases,” according to Carol Lynch, global head of Biopharmaceuticals at Sandoz.
“Rixathon will be one of the 5 major launches we plan in the next 4 years,” she said.
Earlier in the year, the European Medicines Agency’s Committee for Medicinal Products for Human Use had recommended marketing authorization for Rixathon.
The EC based its approval on a comprehensive development program generating analytical, preclinical, and clinical data. Clinical studies included ASSIST-RA and ASSIST-FL.
ASSIST-RA demonstrated that the biosimilar product has equivalent pharmacokinetic and pharmacodynamic profiles to the reference medicine, with no clinically meaningful differences in safety, tolerability, or immunogenicity in patients with rheumatoid arthritis.
ASSIST-FL was a phase 3 study confirming efficacy and safety. The study met its primary endpoint of equivalence in overall response rate between the biosimilar product and the reference medicine after 6 months.
ASSIST-FL also confirmed the comparable safety profiles of the 2 medicines.
Sandoz is a division of the Swiss pharmaceutical company Novartis. MabThera is a registered trademark of F. Hoffmann-La-Roche AG.
Another Sandoz biosimilar rituximab has been approved in the EU as Riximyo® under a duplicate marketing authorization. ![]()
The European Commission (EC) has approved the Sandoz biosimilar rituximab (Rixathon®) for use in the European Economic Area.
Rixathon is approved for all indications of the reference medicine, MabThera®, including follicular lymphoma, diffuse large B-cell lymphoma, chronic lymphocytic leukemia, and immunologic diseases such as rheumatoid arthritis, granulomatosis with polyangiitis, and microscopic polyangiitis.
This approval allows Rixathon to be marketed in the member states of the European Union and Iceland, Liechtenstein, and Norway, members of the European Free Trade Association.
The approval “represents a big win for patients in Europe with blood cancers or immunological diseases,” according to Carol Lynch, global head of Biopharmaceuticals at Sandoz.
“Rixathon will be one of the 5 major launches we plan in the next 4 years,” she said.
Earlier in the year, the European Medicines Agency’s Committee for Medicinal Products for Human Use had recommended marketing authorization for Rixathon.
The EC based its approval on a comprehensive development program generating analytical, preclinical, and clinical data. Clinical studies included ASSIST-RA and ASSIST-FL.
ASSIST-RA demonstrated that the biosimilar product has equivalent pharmacokinetic and pharmacodynamic profiles to the reference medicine, with no clinically meaningful differences in safety, tolerability, or immunogenicity in patients with rheumatoid arthritis.
ASSIST-FL was a phase 3 study confirming efficacy and safety. The study met its primary endpoint of equivalence in overall response rate between the biosimilar product and the reference medicine after 6 months.
ASSIST-FL also confirmed the comparable safety profiles of the 2 medicines.
Sandoz is a division of the Swiss pharmaceutical company Novartis. MabThera is a registered trademark of F. Hoffmann-La-Roche AG.
Another Sandoz biosimilar rituximab has been approved in the EU as Riximyo® under a duplicate marketing authorization. ![]()
The European Commission (EC) has approved the Sandoz biosimilar rituximab (Rixathon®) for use in the European Economic Area.
Rixathon is approved for all indications of the reference medicine, MabThera®, including follicular lymphoma, diffuse large B-cell lymphoma, chronic lymphocytic leukemia, and immunologic diseases such as rheumatoid arthritis, granulomatosis with polyangiitis, and microscopic polyangiitis.
This approval allows Rixathon to be marketed in the member states of the European Union and Iceland, Liechtenstein, and Norway, members of the European Free Trade Association.
The approval “represents a big win for patients in Europe with blood cancers or immunological diseases,” according to Carol Lynch, global head of Biopharmaceuticals at Sandoz.
“Rixathon will be one of the 5 major launches we plan in the next 4 years,” she said.
Earlier in the year, the European Medicines Agency’s Committee for Medicinal Products for Human Use had recommended marketing authorization for Rixathon.
The EC based its approval on a comprehensive development program generating analytical, preclinical, and clinical data. Clinical studies included ASSIST-RA and ASSIST-FL.
ASSIST-RA demonstrated that the biosimilar product has equivalent pharmacokinetic and pharmacodynamic profiles to the reference medicine, with no clinically meaningful differences in safety, tolerability, or immunogenicity in patients with rheumatoid arthritis.
ASSIST-FL was a phase 3 study confirming efficacy and safety. The study met its primary endpoint of equivalence in overall response rate between the biosimilar product and the reference medicine after 6 months.
ASSIST-FL also confirmed the comparable safety profiles of the 2 medicines.
Sandoz is a division of the Swiss pharmaceutical company Novartis. MabThera is a registered trademark of F. Hoffmann-La-Roche AG.
Another Sandoz biosimilar rituximab has been approved in the EU as Riximyo® under a duplicate marketing authorization. ![]()
CAR T cells plus ibrutinib induce CLL remissions

CHICAGO—Chimeric antigen receptor (CAR) T cells combined with ibrutinib enhance T-cell function and can induce complete remission (CR) in patients with chronic lymphocytic leukemia (CLL), researchers report.
Many CLL patients receive ibrutinib treatment, which is well tolerated, but few patients achieve CR.
Immunotherapy with anti-CD19 CAR T cells has induced CR in 25% - 45% of patients with CLL, Saar Gill, MD, of the University of Pennsylvania in Philadelphia, told Hematology Times, and these CRs tend to be durable.
So investigators conducted a pilot trial in 10 patients to test whether combining anti-CD19 CAR T cells with ibrutinib would enhance the CR rate.
Dr Gill reported the findings of the pilot trial at the ASCO 2017 Annual Meeting (abstract 7509).
The patients must have failed at least 1 regimen before ibrutinib, unless they had del(17)(p13.1) or a TP53 mutation.
T cells were lentivirally transduced to express a CAR that included humanized anti-CD19.
Patients were lymphodepleted 1 week before infusion, and ibrutinib was continued throughout the trial.
After a median follow-up of 6 months, 8 of the 9 evaluable patients show absence of CLL in the bone marrow by flow cytometry or minimal residual disease (MRD) negative, and all remain in marrow CR at last follow-up, Dr Gill said.
Radiologic responses are less clear-cut and may require longer follow-up.
“All but 1 patient achieved MRD with deep sequencing. We have deep response in the bone marrow,” Dr Gill said. He also noted that the treatment was well tolerated.
Cytokine release syndrome (CRS) developed in 9 patients: grade 1 in 2 patients, grade 2 in 6 patients, and grade 3 in 1 patient. One patient developed grade 4 tumor lysis syndrome. Treatment of CRS with the IL-6 receptor antagonist tocilizumab was not required.
There was modest residual splenomegaly in 3 of 5 patients, and adenopathy resolved in 4 of 6 patients, with progression in 1 patient.
Ibrutinib reduced CRS apparently by blocking cytokine production by T cells, said Dr Gill, adding, “The combination led to improved efficacy without increased toxicity.”
Ibrutinib may make CAR T-cell therapy more feasible.
Patients who receive ibrutinib for 6 months have a better T-cell response.
“This opens up future discussions of bringing CAR T-cell therapy earlier into CLL treatment,” said Dr Gill.
He envisions patients receiving ibrutinib for 6 months, which would allow time to manufacture T cells, and then have a T-cell infusion.
“Once patients achieve MRD, then we can discuss the possibility of stopping ibrutinib therapy,” he said.
“Most patients remain on ibrutinib, but longer follow-up may show whether remissions are sustained off ibrutinib.”
The researchers have ongoing plans to treat 25 patients with CTL19 plus ibrutinib in a continuation of this trial.
Dr Gill said longer follow-up will reveal the durability of these results “and could support evaluation of a first-line combination approach in an attempt to obviate the need for chronic therapy.” ![]()
CHICAGO—Chimeric antigen receptor (CAR) T cells combined with ibrutinib enhance T-cell function and can induce complete remission (CR) in patients with chronic lymphocytic leukemia (CLL), researchers report.
Many CLL patients receive ibrutinib treatment, which is well tolerated, but few patients achieve CR.
Immunotherapy with anti-CD19 CAR T cells has induced CR in 25% - 45% of patients with CLL, Saar Gill, MD, of the University of Pennsylvania in Philadelphia, told Hematology Times, and these CRs tend to be durable.
So investigators conducted a pilot trial in 10 patients to test whether combining anti-CD19 CAR T cells with ibrutinib would enhance the CR rate.
Dr Gill reported the findings of the pilot trial at the ASCO 2017 Annual Meeting (abstract 7509).
The patients must have failed at least 1 regimen before ibrutinib, unless they had del(17)(p13.1) or a TP53 mutation.
T cells were lentivirally transduced to express a CAR that included humanized anti-CD19.
Patients were lymphodepleted 1 week before infusion, and ibrutinib was continued throughout the trial.
After a median follow-up of 6 months, 8 of the 9 evaluable patients show absence of CLL in the bone marrow by flow cytometry or minimal residual disease (MRD) negative, and all remain in marrow CR at last follow-up, Dr Gill said.
Radiologic responses are less clear-cut and may require longer follow-up.
“All but 1 patient achieved MRD with deep sequencing. We have deep response in the bone marrow,” Dr Gill said. He also noted that the treatment was well tolerated.
Cytokine release syndrome (CRS) developed in 9 patients: grade 1 in 2 patients, grade 2 in 6 patients, and grade 3 in 1 patient. One patient developed grade 4 tumor lysis syndrome. Treatment of CRS with the IL-6 receptor antagonist tocilizumab was not required.
There was modest residual splenomegaly in 3 of 5 patients, and adenopathy resolved in 4 of 6 patients, with progression in 1 patient.
Ibrutinib reduced CRS apparently by blocking cytokine production by T cells, said Dr Gill, adding, “The combination led to improved efficacy without increased toxicity.”
Ibrutinib may make CAR T-cell therapy more feasible.
Patients who receive ibrutinib for 6 months have a better T-cell response.
“This opens up future discussions of bringing CAR T-cell therapy earlier into CLL treatment,” said Dr Gill.
He envisions patients receiving ibrutinib for 6 months, which would allow time to manufacture T cells, and then have a T-cell infusion.
“Once patients achieve MRD, then we can discuss the possibility of stopping ibrutinib therapy,” he said.
“Most patients remain on ibrutinib, but longer follow-up may show whether remissions are sustained off ibrutinib.”
The researchers have ongoing plans to treat 25 patients with CTL19 plus ibrutinib in a continuation of this trial.
Dr Gill said longer follow-up will reveal the durability of these results “and could support evaluation of a first-line combination approach in an attempt to obviate the need for chronic therapy.” ![]()
CHICAGO—Chimeric antigen receptor (CAR) T cells combined with ibrutinib enhance T-cell function and can induce complete remission (CR) in patients with chronic lymphocytic leukemia (CLL), researchers report.
Many CLL patients receive ibrutinib treatment, which is well tolerated, but few patients achieve CR.
Immunotherapy with anti-CD19 CAR T cells has induced CR in 25% - 45% of patients with CLL, Saar Gill, MD, of the University of Pennsylvania in Philadelphia, told Hematology Times, and these CRs tend to be durable.
So investigators conducted a pilot trial in 10 patients to test whether combining anti-CD19 CAR T cells with ibrutinib would enhance the CR rate.
Dr Gill reported the findings of the pilot trial at the ASCO 2017 Annual Meeting (abstract 7509).
The patients must have failed at least 1 regimen before ibrutinib, unless they had del(17)(p13.1) or a TP53 mutation.
T cells were lentivirally transduced to express a CAR that included humanized anti-CD19.
Patients were lymphodepleted 1 week before infusion, and ibrutinib was continued throughout the trial.
After a median follow-up of 6 months, 8 of the 9 evaluable patients show absence of CLL in the bone marrow by flow cytometry or minimal residual disease (MRD) negative, and all remain in marrow CR at last follow-up, Dr Gill said.
Radiologic responses are less clear-cut and may require longer follow-up.
“All but 1 patient achieved MRD with deep sequencing. We have deep response in the bone marrow,” Dr Gill said. He also noted that the treatment was well tolerated.
Cytokine release syndrome (CRS) developed in 9 patients: grade 1 in 2 patients, grade 2 in 6 patients, and grade 3 in 1 patient. One patient developed grade 4 tumor lysis syndrome. Treatment of CRS with the IL-6 receptor antagonist tocilizumab was not required.
There was modest residual splenomegaly in 3 of 5 patients, and adenopathy resolved in 4 of 6 patients, with progression in 1 patient.
Ibrutinib reduced CRS apparently by blocking cytokine production by T cells, said Dr Gill, adding, “The combination led to improved efficacy without increased toxicity.”
Ibrutinib may make CAR T-cell therapy more feasible.
Patients who receive ibrutinib for 6 months have a better T-cell response.
“This opens up future discussions of bringing CAR T-cell therapy earlier into CLL treatment,” said Dr Gill.
He envisions patients receiving ibrutinib for 6 months, which would allow time to manufacture T cells, and then have a T-cell infusion.
“Once patients achieve MRD, then we can discuss the possibility of stopping ibrutinib therapy,” he said.
“Most patients remain on ibrutinib, but longer follow-up may show whether remissions are sustained off ibrutinib.”
The researchers have ongoing plans to treat 25 patients with CTL19 plus ibrutinib in a continuation of this trial.
Dr Gill said longer follow-up will reveal the durability of these results “and could support evaluation of a first-line combination approach in an attempt to obviate the need for chronic therapy.” ![]()
Two SNPs linked to survival in R-CHOP–treated DLBCL
Two variations of the BCL2 gene are linked with the survival prospects of patients with diffuse large B-cell lymphoma (DLBCL) who are treated with the R-CHOP regimen, based on a study published in Haematologica.
In the population-based, case-control study of patients with non-Hodgkin’s lymphoma across the British Columbia province, Morteza Bashash, PhD, of the Dalla Lana School of Public Health, Toronto, and researchers at the British Columbia Cancer Agency analyzed 217 germline DLBCL samples, excluding those with primary mediastinal large B-cell lymphoma, specifically looking at nine single nucleotide polymorphisms (SNPs).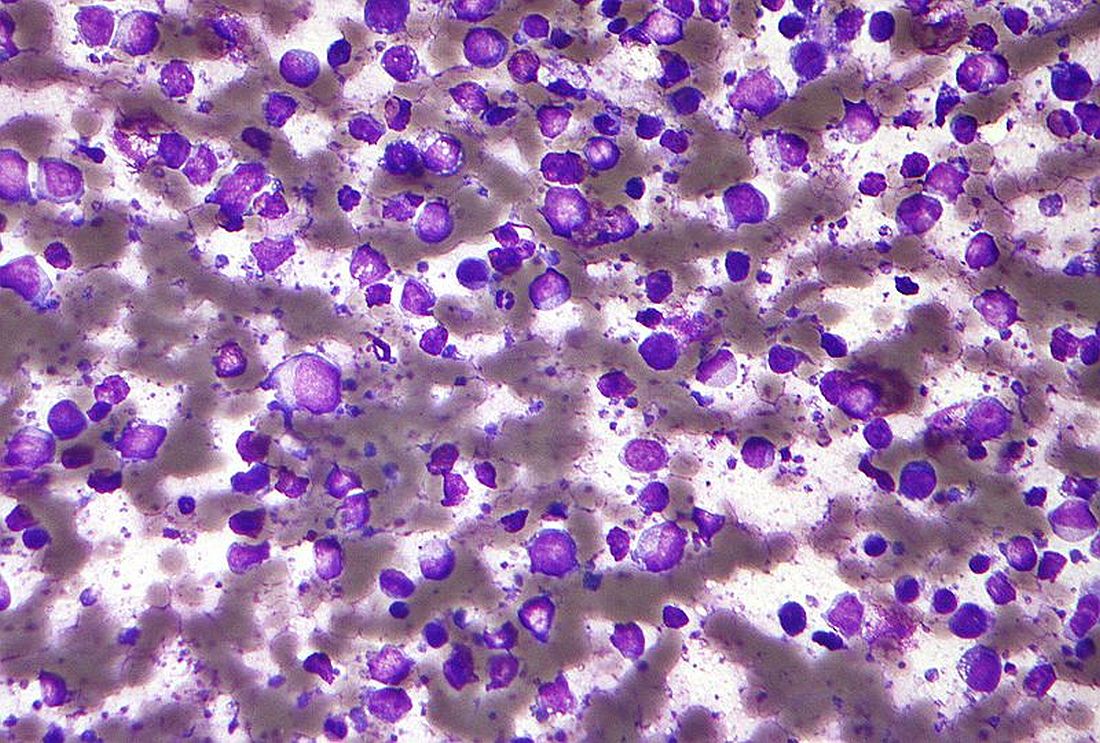
Patients receiving R-CHOP who had the AA genotype at rs7226979 had a risk of death that was four times higher than that of those with a G allele, researchers said (P less than .01). The same pattern was seen for PFS, with AA carriers having twice the risk of an event, compared with the other genotypes (P less than .05).
For those with rs4456611, patients with the GG genotype had a risk of death that was 3 times greater than that of those with an A allele (P less than .01), but there was no association with PFS for that SNP.
In an analysis of an independent cohort, only the associations that were seen with rs7226979 – and not those with rs4456611 – were able to be replicated.
The researchers noted that, while most predictive markers that are used to guide clinical treatment are drawn from actual tumor material, host-related factors could also be important.
“Compared to genetic analysis of the tumor, the patient’s constitutional genetic profile is relatively easy to obtain and can be assessed before treatment is started,” they wrote. “Our result has the potential to be useful as a complementary tool to predict the outcome of patients treated with R-CHOP and enhance clinical decision-making after confirmation by further studies.”
Two variations of the BCL2 gene are linked with the survival prospects of patients with diffuse large B-cell lymphoma (DLBCL) who are treated with the R-CHOP regimen, based on a study published in Haematologica.
In the population-based, case-control study of patients with non-Hodgkin’s lymphoma across the British Columbia province, Morteza Bashash, PhD, of the Dalla Lana School of Public Health, Toronto, and researchers at the British Columbia Cancer Agency analyzed 217 germline DLBCL samples, excluding those with primary mediastinal large B-cell lymphoma, specifically looking at nine single nucleotide polymorphisms (SNPs).
Patients receiving R-CHOP who had the AA genotype at rs7226979 had a risk of death that was four times higher than that of those with a G allele, researchers said (P less than .01). The same pattern was seen for PFS, with AA carriers having twice the risk of an event, compared with the other genotypes (P less than .05).
For those with rs4456611, patients with the GG genotype had a risk of death that was 3 times greater than that of those with an A allele (P less than .01), but there was no association with PFS for that SNP.
In an analysis of an independent cohort, only the associations that were seen with rs7226979 – and not those with rs4456611 – were able to be replicated.
The researchers noted that, while most predictive markers that are used to guide clinical treatment are drawn from actual tumor material, host-related factors could also be important.
“Compared to genetic analysis of the tumor, the patient’s constitutional genetic profile is relatively easy to obtain and can be assessed before treatment is started,” they wrote. “Our result has the potential to be useful as a complementary tool to predict the outcome of patients treated with R-CHOP and enhance clinical decision-making after confirmation by further studies.”
Two variations of the BCL2 gene are linked with the survival prospects of patients with diffuse large B-cell lymphoma (DLBCL) who are treated with the R-CHOP regimen, based on a study published in Haematologica.
In the population-based, case-control study of patients with non-Hodgkin’s lymphoma across the British Columbia province, Morteza Bashash, PhD, of the Dalla Lana School of Public Health, Toronto, and researchers at the British Columbia Cancer Agency analyzed 217 germline DLBCL samples, excluding those with primary mediastinal large B-cell lymphoma, specifically looking at nine single nucleotide polymorphisms (SNPs).
Patients receiving R-CHOP who had the AA genotype at rs7226979 had a risk of death that was four times higher than that of those with a G allele, researchers said (P less than .01). The same pattern was seen for PFS, with AA carriers having twice the risk of an event, compared with the other genotypes (P less than .05).
For those with rs4456611, patients with the GG genotype had a risk of death that was 3 times greater than that of those with an A allele (P less than .01), but there was no association with PFS for that SNP.
In an analysis of an independent cohort, only the associations that were seen with rs7226979 – and not those with rs4456611 – were able to be replicated.
The researchers noted that, while most predictive markers that are used to guide clinical treatment are drawn from actual tumor material, host-related factors could also be important.
“Compared to genetic analysis of the tumor, the patient’s constitutional genetic profile is relatively easy to obtain and can be assessed before treatment is started,” they wrote. “Our result has the potential to be useful as a complementary tool to predict the outcome of patients treated with R-CHOP and enhance clinical decision-making after confirmation by further studies.”
FROM HAEMATOLOGICA
Key clinical point: Two SNPs were found to be linked to survival prospects in patients with diffuse large B-cell lymphoma who receive primary R-CHOP therapy.
Major finding: For the rs7226979 SNP, those with the AA genotype had a four times higher risk of death than those with a G allele.
Data source: A population-based, case-control study of patients with non-Hodgkin’s lymphoma in British Columbia, with DNA samples analyzed for 9nine SNPs among the DLBCL patients, excluding those with primary mediastinal large B-cell lymphoma.
Disclosures: Some of the study authors reported institutional research funding from Roche; honoraria from Roche/Genentech, Janssen Pharmaceuticals and Celgene; and/or consultant or advisory roles with Roche/Genentech, Janssen Pharmaceuticals, Celgene, and NanoString Technologies.
Novel CAR T cells drive high objective response rate in multiple myeloma
CHICAGO – CARs just keep getting better: In an early clinical trial, a chimeric antigen receptor (CAR) T-cell construct targeting B-cell maturation protein induced clinical remissions in 33 of 35 patients with relapsed/refractory multiple myeloma who were treated in an early clinical trial.
“In our current trials we have observed revolutionary, quick, and durable remissions in patients with multiple myeloma,” said Wanhong Zhao, MD, of the Second Affiliated Hospital of Xi’an (China) Jiaotong University.

“I think what you’re seeing here is the expansion of immunotherapy to cancers that really are refractory to chemotherapy and how immunotherapy is now providing hope to a lot of patients with cancers that were not really responding to our standard chemotherapies,” commented ASCO expert Michael S. Sabel, MD, of the University of Michigan, Ann Arbor. “What I also think is really fascinating about this and similar forms of research is that you are now seeing the merger of immunotherapy with personalized medicine.”
Current CAR T-cell technologies targeting CD19 or a similar antigen have shown efficacy against acute lymphoblastic leukemia and some forms of lymphoma, but it has been difficult to identify a suitable target in multiple myeloma.
B-cell maturation antigen (BCMA) was first described in myeloma in 2004 as a mechanism for the growth and survival of malignant plasma cells.
Several research groups are currently investigating CAR T cells or monoclonal antibodies targeted to BCMA.
In the study by Dr. Zhao and his colleagues, 19 patients had been followed for more than 4 months before the data cutoff in January 2017. Four months is the minimum established by the International Myeloma Working Group for efficacy assessment.
Of the 19 patients, 14 had achieved a stringent complete response (sCR), 4 had very good partial responses, and 1 had a partial response, for an objective response rate of 100%.
No patients who achieved an sCR have had relapses, and all five patients who have been in follow-up for more than a year have maintained their sCRs and are free of minimal residual disease, Dr. Zhao reported.
One patient with a very good partial response had disease progression, with recurrence of an extramedullary lesion that had previously disappeared.
The most common adverse event was cytokine release syndrome, which occurred in 85% of patients, but the condition was transient and manageable in a majority, Dr. Zhao said.
Two patients developed grade 3 cytokine release syndrome and were treated with tocilizumab (Actemra).
The investigators plan to enroll a total of 100 patients from participating hospitals in China and are planning a U.S. trial for launch in early 2018.
The investigators hope to look at BCMA CAR-T cell therapy in the frontline for patients with newly diagnosed multiple myeloma.
The study was funded by Legend Biotech. Coauthor Fran (Xiaohu) Fan, MD, PhD, is employed by the company. Dr. Zhao did not report disclosures. Dr. Sabel had no disclosures relevant to the study.
CHICAGO – CARs just keep getting better: In an early clinical trial, a chimeric antigen receptor (CAR) T-cell construct targeting B-cell maturation protein induced clinical remissions in 33 of 35 patients with relapsed/refractory multiple myeloma who were treated in an early clinical trial.
“In our current trials we have observed revolutionary, quick, and durable remissions in patients with multiple myeloma,” said Wanhong Zhao, MD, of the Second Affiliated Hospital of Xi’an (China) Jiaotong University.
“I think what you’re seeing here is the expansion of immunotherapy to cancers that really are refractory to chemotherapy and how immunotherapy is now providing hope to a lot of patients with cancers that were not really responding to our standard chemotherapies,” commented ASCO expert Michael S. Sabel, MD, of the University of Michigan, Ann Arbor. “What I also think is really fascinating about this and similar forms of research is that you are now seeing the merger of immunotherapy with personalized medicine.”
Current CAR T-cell technologies targeting CD19 or a similar antigen have shown efficacy against acute lymphoblastic leukemia and some forms of lymphoma, but it has been difficult to identify a suitable target in multiple myeloma.
B-cell maturation antigen (BCMA) was first described in myeloma in 2004 as a mechanism for the growth and survival of malignant plasma cells.
Several research groups are currently investigating CAR T cells or monoclonal antibodies targeted to BCMA.
In the study by Dr. Zhao and his colleagues, 19 patients had been followed for more than 4 months before the data cutoff in January 2017. Four months is the minimum established by the International Myeloma Working Group for efficacy assessment.
Of the 19 patients, 14 had achieved a stringent complete response (sCR), 4 had very good partial responses, and 1 had a partial response, for an objective response rate of 100%.
No patients who achieved an sCR have had relapses, and all five patients who have been in follow-up for more than a year have maintained their sCRs and are free of minimal residual disease, Dr. Zhao reported.
One patient with a very good partial response had disease progression, with recurrence of an extramedullary lesion that had previously disappeared.
The most common adverse event was cytokine release syndrome, which occurred in 85% of patients, but the condition was transient and manageable in a majority, Dr. Zhao said.
Two patients developed grade 3 cytokine release syndrome and were treated with tocilizumab (Actemra).
The investigators plan to enroll a total of 100 patients from participating hospitals in China and are planning a U.S. trial for launch in early 2018.
The investigators hope to look at BCMA CAR-T cell therapy in the frontline for patients with newly diagnosed multiple myeloma.
The study was funded by Legend Biotech. Coauthor Fran (Xiaohu) Fan, MD, PhD, is employed by the company. Dr. Zhao did not report disclosures. Dr. Sabel had no disclosures relevant to the study.
CHICAGO – CARs just keep getting better: In an early clinical trial, a chimeric antigen receptor (CAR) T-cell construct targeting B-cell maturation protein induced clinical remissions in 33 of 35 patients with relapsed/refractory multiple myeloma who were treated in an early clinical trial.
“In our current trials we have observed revolutionary, quick, and durable remissions in patients with multiple myeloma,” said Wanhong Zhao, MD, of the Second Affiliated Hospital of Xi’an (China) Jiaotong University.
“I think what you’re seeing here is the expansion of immunotherapy to cancers that really are refractory to chemotherapy and how immunotherapy is now providing hope to a lot of patients with cancers that were not really responding to our standard chemotherapies,” commented ASCO expert Michael S. Sabel, MD, of the University of Michigan, Ann Arbor. “What I also think is really fascinating about this and similar forms of research is that you are now seeing the merger of immunotherapy with personalized medicine.”
Current CAR T-cell technologies targeting CD19 or a similar antigen have shown efficacy against acute lymphoblastic leukemia and some forms of lymphoma, but it has been difficult to identify a suitable target in multiple myeloma.
B-cell maturation antigen (BCMA) was first described in myeloma in 2004 as a mechanism for the growth and survival of malignant plasma cells.
Several research groups are currently investigating CAR T cells or monoclonal antibodies targeted to BCMA.
In the study by Dr. Zhao and his colleagues, 19 patients had been followed for more than 4 months before the data cutoff in January 2017. Four months is the minimum established by the International Myeloma Working Group for efficacy assessment.
Of the 19 patients, 14 had achieved a stringent complete response (sCR), 4 had very good partial responses, and 1 had a partial response, for an objective response rate of 100%.
No patients who achieved an sCR have had relapses, and all five patients who have been in follow-up for more than a year have maintained their sCRs and are free of minimal residual disease, Dr. Zhao reported.
One patient with a very good partial response had disease progression, with recurrence of an extramedullary lesion that had previously disappeared.
The most common adverse event was cytokine release syndrome, which occurred in 85% of patients, but the condition was transient and manageable in a majority, Dr. Zhao said.
Two patients developed grade 3 cytokine release syndrome and were treated with tocilizumab (Actemra).
The investigators plan to enroll a total of 100 patients from participating hospitals in China and are planning a U.S. trial for launch in early 2018.
The investigators hope to look at BCMA CAR-T cell therapy in the frontline for patients with newly diagnosed multiple myeloma.
The study was funded by Legend Biotech. Coauthor Fran (Xiaohu) Fan, MD, PhD, is employed by the company. Dr. Zhao did not report disclosures. Dr. Sabel had no disclosures relevant to the study.
AT THE 2017 ASCO ANNUAL MEETING
Key clinical point: All of 19 patients treated with the CAR T-cell construct targeting B-cell maturation antigen had an objective response.
Major finding: Of 35 patients with relapsed/refractory multiple myeloma treated with BCMA, 33 had remissions.
Data source: A prospective single-arm study of 35 patients, with enrollment planned for 100.
Disclosures: The study was funded by Legend Biotech. Coauthor Fran (Xiaohu) Fan, MD, PhD, is employed by the company. Dr. Zhao did not report disclosures. Dr. Sabel had no disclosures relevant to the study.
Addition of ublituximab to ibrutinib improves response in r/r CLL

Ibrutinib, the Bruton’s tyrosine kinase (BTK) inhibitor, has transformed the treatment landscape for patients with relapsed or refractory (r/r) chronic lymphocytic leukemia (CLL).
Yet for patients with high-risk molecular features, such as 11q deletion, 17p deletion, or TP53 mutation, relapse remains problematic.
Investigators evaluated whether the addition of ublituximab to ibrutinib would improve the outcome of patients with genetically high-risk CLL in the GENUINE (UTX-IB-301) phase 3 study.
Jeff P. Sharman, MD, of Willamette Valley Cancer Institute and Research Center in Springfield, Oregon, reported the results at the 2017 ASCO Annual Meeting (abstract 7504).*
Ublituximab is a glycoengineered, anti-CD20 type 1 monoclonal antibody that maintains complement-dependent cytotoxicity and enhances antibody-dependent cell-mediated cytotoxicity. In a phase 2 study in combination with ibrutinib, it achieved an ORR of approximately 88%.
Protocol design
Originally, the study had co-primary endpoints of overall response rate (ORR) and progression-free survival (PFS). To adequately power for both endpoints, the target enrollment was 330 patients.
Dr Sharman explained that after 22 months of open enrollment, the trial sponsor determined that the original enrollment goal could not be met in a timely manner and elected to redesign the protocol.
In the modified protocol, ORR became the primary response rate and PFS a secondary endpoint. This allowed for a reduced target enrollment of 120. However, the study was no longer powered to detect a change in PFS.
Investigators stratified the patients by lines of prior therapy and then randomized them to receive ibrutinib or ublituximab plus ibrutinib.
The ibrutinib dose was 420 mg daily in both arms. Ublituximab dose was 900 mg on days 1, 8, and 15 of cycle 1, day 1 of cycles 2 through 6 and every third cycle thereafter.
The primary endpoint was ORR as assessed by Independent Central Review (IRC) using the iwCLL 2008 criteria.
Secondary endpoints included PFS, the complete response (CR) rate and depth of response (minimal residual disease [MRD] negativity), and safety.
The investigators assessed patients for response on weeks 8, 16, 24, and every 12 weeks thereafter.
The primary endpoint was evaluated when all enrolled patients had at least 2 efficacy evaluations.
The median follow-up was 11.4 months.
Patient characteristics
Patients with relapsed or refractory high-risk CLL had their disease centrally confirmed for the presence of deletion 17p, deletion 11q, and/or TP53 mutation.
They had measurable disease, ECOG performance status of 2 or less, no history of transformation of CLL, and no prior BTK inhibitor therapy.
The investigators randomized 126 patients, and 117 received any dose of therapy.
“The dropout was because in part ibrutinib was via commercial supply and not every patient could get access,” Dr Sharman noted.
Fifty-nine patients were treated in the combination arm and 58 in the monotherapy arm.
All patients had at least one of the specified mutations, which were relatively balanced between the 2 arms.
Patients were a mean age of 67 (range, 43 – 87), had a median of 3 prior therapies (range, 1 – 8), and more than 70% were male.
Patient characteristics were similar in each arm except for bulky disease, with 45% in the combination arm having bulky disease of 5 cm or more at baseline, compared with 26% in the monotherapy arm.
Twenty percent of the patients were considered refractory to rituximab.
Safety
Infusion reactions occurred in 54% of patients in the combination arm and 5% had grade 3/4 reactions. None occurred in the ibrutinib arm, since the latter is an orally bioavailable drug.
Other adverse events of all grades occurring in 10% of patients or more for the combination and monotherapy arms, respectively, were: diarrhea (42% and 40%), fatigue (27% and 33%), insomnia (24% and 10%), nausea (22% and 21%), headache (20% and 28%), arthralgia (19% and 17%), cough (19% and 24%), abdominal pain (15% and 9%), stomatitis (15% and 9%), upper respiratory infection (15% and 12%), dizziness (15% and 22%), contusion (15% and 29%), anemia (14% and 17%), and peripheral edema (10% and 21%).
Neutropenia was higher in the experimental arm, 22% any grade, compared with 12% in the ibrutinib arm, although grade 3 or higher neutropenia was similar in the 2 arms. Other laboratory abnormalities were similar between the arms.
Efficacy
The best ORR in the combination arm was 78%, with 7% achieving CR compared with 45% in the monotherapy arm with no CRs (P<0.001).
Nineteen percent of the combination arm achieved MRD negativity in peripheral blood compared with 2% of the monotherapy arm (P<0.01).
The reduction in lymph node size was similar between the arms.
In contrast, lymphocytosis was very different between the arms.
“As has been reported multiple times with targeted B-cell receptor signaling inhibitors,” Dr Sharman said, “patients treated with ibrutinib experienced rapid increase in their lymphocytes, returning approximately to baseline by 3 months and decreasing thereafter.”
“By contrast,” he continued, “those patients treated with the additional antibody had much more rapid resolution of their lymphocytosis. This was true whether patients were considered rituximab refractory or not.”
The investigators performed an additional analysis of ORR, this time including patients who achieved partial response with lymphocytosis (PR-L). These patients were not included in the primary endpoint because the iwCLL 2008 criteria had not yet been updated to include PR-L.
The best overall response including active PR-L patients was 83% in the experimental arm and 59% in the ibrutinib monotherapy arm (P<0.01).
PFS showed a trend toward improvement in the patients treated with the combination, with a hazard ratio of 0.559, which was not of statistical significance at the time of analysis.
The investigators concluded that the study met its primary endpoint, with a greater response rate and a greater depth of response than ibrutinib alone.
And the addition of ublituximab did not alter the safety profile of ibrutinib monotherapy.
TG Therapeutics, Inc, funded the study. ![]()
*Data in the abstract differ from the meeting presentation.
Ibrutinib, the Bruton’s tyrosine kinase (BTK) inhibitor, has transformed the treatment landscape for patients with relapsed or refractory (r/r) chronic lymphocytic leukemia (CLL).
Yet for patients with high-risk molecular features, such as 11q deletion, 17p deletion, or TP53 mutation, relapse remains problematic.
Investigators evaluated whether the addition of ublituximab to ibrutinib would improve the outcome of patients with genetically high-risk CLL in the GENUINE (UTX-IB-301) phase 3 study.
Jeff P. Sharman, MD, of Willamette Valley Cancer Institute and Research Center in Springfield, Oregon, reported the results at the 2017 ASCO Annual Meeting (abstract 7504).*
Ublituximab is a glycoengineered, anti-CD20 type 1 monoclonal antibody that maintains complement-dependent cytotoxicity and enhances antibody-dependent cell-mediated cytotoxicity. In a phase 2 study in combination with ibrutinib, it achieved an ORR of approximately 88%.
Protocol design
Originally, the study had co-primary endpoints of overall response rate (ORR) and progression-free survival (PFS). To adequately power for both endpoints, the target enrollment was 330 patients.
Dr Sharman explained that after 22 months of open enrollment, the trial sponsor determined that the original enrollment goal could not be met in a timely manner and elected to redesign the protocol.
In the modified protocol, ORR became the primary response rate and PFS a secondary endpoint. This allowed for a reduced target enrollment of 120. However, the study was no longer powered to detect a change in PFS.
Investigators stratified the patients by lines of prior therapy and then randomized them to receive ibrutinib or ublituximab plus ibrutinib.
The ibrutinib dose was 420 mg daily in both arms. Ublituximab dose was 900 mg on days 1, 8, and 15 of cycle 1, day 1 of cycles 2 through 6 and every third cycle thereafter.
The primary endpoint was ORR as assessed by Independent Central Review (IRC) using the iwCLL 2008 criteria.
Secondary endpoints included PFS, the complete response (CR) rate and depth of response (minimal residual disease [MRD] negativity), and safety.
The investigators assessed patients for response on weeks 8, 16, 24, and every 12 weeks thereafter.
The primary endpoint was evaluated when all enrolled patients had at least 2 efficacy evaluations.
The median follow-up was 11.4 months.
Patient characteristics
Patients with relapsed or refractory high-risk CLL had their disease centrally confirmed for the presence of deletion 17p, deletion 11q, and/or TP53 mutation.
They had measurable disease, ECOG performance status of 2 or less, no history of transformation of CLL, and no prior BTK inhibitor therapy.
The investigators randomized 126 patients, and 117 received any dose of therapy.
“The dropout was because in part ibrutinib was via commercial supply and not every patient could get access,” Dr Sharman noted.
Fifty-nine patients were treated in the combination arm and 58 in the monotherapy arm.
All patients had at least one of the specified mutations, which were relatively balanced between the 2 arms.
Patients were a mean age of 67 (range, 43 – 87), had a median of 3 prior therapies (range, 1 – 8), and more than 70% were male.
Patient characteristics were similar in each arm except for bulky disease, with 45% in the combination arm having bulky disease of 5 cm or more at baseline, compared with 26% in the monotherapy arm.
Twenty percent of the patients were considered refractory to rituximab.
Safety
Infusion reactions occurred in 54% of patients in the combination arm and 5% had grade 3/4 reactions. None occurred in the ibrutinib arm, since the latter is an orally bioavailable drug.
Other adverse events of all grades occurring in 10% of patients or more for the combination and monotherapy arms, respectively, were: diarrhea (42% and 40%), fatigue (27% and 33%), insomnia (24% and 10%), nausea (22% and 21%), headache (20% and 28%), arthralgia (19% and 17%), cough (19% and 24%), abdominal pain (15% and 9%), stomatitis (15% and 9%), upper respiratory infection (15% and 12%), dizziness (15% and 22%), contusion (15% and 29%), anemia (14% and 17%), and peripheral edema (10% and 21%).
Neutropenia was higher in the experimental arm, 22% any grade, compared with 12% in the ibrutinib arm, although grade 3 or higher neutropenia was similar in the 2 arms. Other laboratory abnormalities were similar between the arms.
Efficacy
The best ORR in the combination arm was 78%, with 7% achieving CR compared with 45% in the monotherapy arm with no CRs (P<0.001).
Nineteen percent of the combination arm achieved MRD negativity in peripheral blood compared with 2% of the monotherapy arm (P<0.01).
The reduction in lymph node size was similar between the arms.
In contrast, lymphocytosis was very different between the arms.
“As has been reported multiple times with targeted B-cell receptor signaling inhibitors,” Dr Sharman said, “patients treated with ibrutinib experienced rapid increase in their lymphocytes, returning approximately to baseline by 3 months and decreasing thereafter.”
“By contrast,” he continued, “those patients treated with the additional antibody had much more rapid resolution of their lymphocytosis. This was true whether patients were considered rituximab refractory or not.”
The investigators performed an additional analysis of ORR, this time including patients who achieved partial response with lymphocytosis (PR-L). These patients were not included in the primary endpoint because the iwCLL 2008 criteria had not yet been updated to include PR-L.
The best overall response including active PR-L patients was 83% in the experimental arm and 59% in the ibrutinib monotherapy arm (P<0.01).
PFS showed a trend toward improvement in the patients treated with the combination, with a hazard ratio of 0.559, which was not of statistical significance at the time of analysis.
The investigators concluded that the study met its primary endpoint, with a greater response rate and a greater depth of response than ibrutinib alone.
And the addition of ublituximab did not alter the safety profile of ibrutinib monotherapy.
TG Therapeutics, Inc, funded the study. ![]()
*Data in the abstract differ from the meeting presentation.
Ibrutinib, the Bruton’s tyrosine kinase (BTK) inhibitor, has transformed the treatment landscape for patients with relapsed or refractory (r/r) chronic lymphocytic leukemia (CLL).
Yet for patients with high-risk molecular features, such as 11q deletion, 17p deletion, or TP53 mutation, relapse remains problematic.
Investigators evaluated whether the addition of ublituximab to ibrutinib would improve the outcome of patients with genetically high-risk CLL in the GENUINE (UTX-IB-301) phase 3 study.
Jeff P. Sharman, MD, of Willamette Valley Cancer Institute and Research Center in Springfield, Oregon, reported the results at the 2017 ASCO Annual Meeting (abstract 7504).*
Ublituximab is a glycoengineered, anti-CD20 type 1 monoclonal antibody that maintains complement-dependent cytotoxicity and enhances antibody-dependent cell-mediated cytotoxicity. In a phase 2 study in combination with ibrutinib, it achieved an ORR of approximately 88%.
Protocol design
Originally, the study had co-primary endpoints of overall response rate (ORR) and progression-free survival (PFS). To adequately power for both endpoints, the target enrollment was 330 patients.
Dr Sharman explained that after 22 months of open enrollment, the trial sponsor determined that the original enrollment goal could not be met in a timely manner and elected to redesign the protocol.
In the modified protocol, ORR became the primary response rate and PFS a secondary endpoint. This allowed for a reduced target enrollment of 120. However, the study was no longer powered to detect a change in PFS.
Investigators stratified the patients by lines of prior therapy and then randomized them to receive ibrutinib or ublituximab plus ibrutinib.
The ibrutinib dose was 420 mg daily in both arms. Ublituximab dose was 900 mg on days 1, 8, and 15 of cycle 1, day 1 of cycles 2 through 6 and every third cycle thereafter.
The primary endpoint was ORR as assessed by Independent Central Review (IRC) using the iwCLL 2008 criteria.
Secondary endpoints included PFS, the complete response (CR) rate and depth of response (minimal residual disease [MRD] negativity), and safety.
The investigators assessed patients for response on weeks 8, 16, 24, and every 12 weeks thereafter.
The primary endpoint was evaluated when all enrolled patients had at least 2 efficacy evaluations.
The median follow-up was 11.4 months.
Patient characteristics
Patients with relapsed or refractory high-risk CLL had their disease centrally confirmed for the presence of deletion 17p, deletion 11q, and/or TP53 mutation.
They had measurable disease, ECOG performance status of 2 or less, no history of transformation of CLL, and no prior BTK inhibitor therapy.
The investigators randomized 126 patients, and 117 received any dose of therapy.
“The dropout was because in part ibrutinib was via commercial supply and not every patient could get access,” Dr Sharman noted.
Fifty-nine patients were treated in the combination arm and 58 in the monotherapy arm.
All patients had at least one of the specified mutations, which were relatively balanced between the 2 arms.
Patients were a mean age of 67 (range, 43 – 87), had a median of 3 prior therapies (range, 1 – 8), and more than 70% were male.
Patient characteristics were similar in each arm except for bulky disease, with 45% in the combination arm having bulky disease of 5 cm or more at baseline, compared with 26% in the monotherapy arm.
Twenty percent of the patients were considered refractory to rituximab.
Safety
Infusion reactions occurred in 54% of patients in the combination arm and 5% had grade 3/4 reactions. None occurred in the ibrutinib arm, since the latter is an orally bioavailable drug.
Other adverse events of all grades occurring in 10% of patients or more for the combination and monotherapy arms, respectively, were: diarrhea (42% and 40%), fatigue (27% and 33%), insomnia (24% and 10%), nausea (22% and 21%), headache (20% and 28%), arthralgia (19% and 17%), cough (19% and 24%), abdominal pain (15% and 9%), stomatitis (15% and 9%), upper respiratory infection (15% and 12%), dizziness (15% and 22%), contusion (15% and 29%), anemia (14% and 17%), and peripheral edema (10% and 21%).
Neutropenia was higher in the experimental arm, 22% any grade, compared with 12% in the ibrutinib arm, although grade 3 or higher neutropenia was similar in the 2 arms. Other laboratory abnormalities were similar between the arms.
Efficacy
The best ORR in the combination arm was 78%, with 7% achieving CR compared with 45% in the monotherapy arm with no CRs (P<0.001).
Nineteen percent of the combination arm achieved MRD negativity in peripheral blood compared with 2% of the monotherapy arm (P<0.01).
The reduction in lymph node size was similar between the arms.
In contrast, lymphocytosis was very different between the arms.
“As has been reported multiple times with targeted B-cell receptor signaling inhibitors,” Dr Sharman said, “patients treated with ibrutinib experienced rapid increase in their lymphocytes, returning approximately to baseline by 3 months and decreasing thereafter.”
“By contrast,” he continued, “those patients treated with the additional antibody had much more rapid resolution of their lymphocytosis. This was true whether patients were considered rituximab refractory or not.”
The investigators performed an additional analysis of ORR, this time including patients who achieved partial response with lymphocytosis (PR-L). These patients were not included in the primary endpoint because the iwCLL 2008 criteria had not yet been updated to include PR-L.
The best overall response including active PR-L patients was 83% in the experimental arm and 59% in the ibrutinib monotherapy arm (P<0.01).
PFS showed a trend toward improvement in the patients treated with the combination, with a hazard ratio of 0.559, which was not of statistical significance at the time of analysis.
The investigators concluded that the study met its primary endpoint, with a greater response rate and a greater depth of response than ibrutinib alone.
And the addition of ublituximab did not alter the safety profile of ibrutinib monotherapy.
TG Therapeutics, Inc, funded the study. ![]()
*Data in the abstract differ from the meeting presentation.
Severe health conditions decrease among childhood cancer survivors

CHICAGO—The 15-year cumulative incidence of severe health conditions for survivors of childhood cancer has decreased over the past 30 years, from 12.7% for those diagnosed in the 1970s to 10.1% and 8.9% for those diagnosed in the 1980s and 1990s, respectively. And the decreases were greatest for patients with Wilms’ tumor and Hodgkin lymphoma (HL), followed by patients with astrocytoma, non-Hodgkin lymphoma (NHL), and acute lymphoblastic leukemia (ALL).
Investigators of the Childhood Cancer Survivor Study (CCSS) undertook a retrospective cohort analysis of children aged 0 – 14 years diagnosed with cancer between 1970 and 1999. Their goal was to determine whether cancer therapy modifications have maintained cure rates while decreasing the risk of late effects of therapy.
Todd M. Gibson, PhD, of St Jude Children’s Research Hospital in Memphis, Tennessee, presented the findings at the 2017 annual meeting of the American Society for Clinical Oncology (ASCO) as abstract LBA10500.
Researchers analyzed data from 23,600 childhood cancer survivors in the CCSS who were alive 5 years after diagnosis. The patients had leukemia, lymphoma, CNS malignancies, Wilms tumor, neuroblastoma, or soft-tissue/bone sarcoma.
Dr Gibson noted that while 83% of children with a malignancy achieve a 5-year survival, more than half develop at least one severe, disabling, life-threatening health condition by age 50.
The survivors were a median age at last follow-up of 28 years (range, 5-63) and the median time since diagnosis was 21 years (range, 5-43).
The investigators found significant decreases in severe health conditions in 6 diagnostic groups:
- Wilms tumor, decreased from 13% to 5% (P<0.0001)
- HL, decreased from 18% to 11% (P<0.0001)
- Astrocytoma, decreased from 15% to 9% (P=0.004)
- NHL, decreased from 10% to 6% (P=0.04)
- ALL, decreased from 9% to 7% (P=0.002)
- Ewings sarcoma, decreased from 19% to 10% (P=0.01)
They found no reductions in subsequent severe health conditions among survivors of neuroblastoma, acute myeloid leukemia (AML), soft tissue sarcoma, or osteosarcoma.
The investigators believe the decreases were driven mainly by a reduced incidence of endocrine conditions, subsequent malignant neoplasms, gastrointestinal and neurological conditions, but not cardiac or pulmonary conditions.
They also analyzed the reduction in treatment intensities by decade for different diseases and found they correlated with the reduced incidence of serious chronic health conditions by 15 years after diagnosis.
The National Institutes of Health funded the study.
CHICAGO—The 15-year cumulative incidence of severe health conditions for survivors of childhood cancer has decreased over the past 30 years, from 12.7% for those diagnosed in the 1970s to 10.1% and 8.9% for those diagnosed in the 1980s and 1990s, respectively. And the decreases were greatest for patients with Wilms’ tumor and Hodgkin lymphoma (HL), followed by patients with astrocytoma, non-Hodgkin lymphoma (NHL), and acute lymphoblastic leukemia (ALL).
Investigators of the Childhood Cancer Survivor Study (CCSS) undertook a retrospective cohort analysis of children aged 0 – 14 years diagnosed with cancer between 1970 and 1999. Their goal was to determine whether cancer therapy modifications have maintained cure rates while decreasing the risk of late effects of therapy.
Todd M. Gibson, PhD, of St Jude Children’s Research Hospital in Memphis, Tennessee, presented the findings at the 2017 annual meeting of the American Society for Clinical Oncology (ASCO) as abstract LBA10500.
Researchers analyzed data from 23,600 childhood cancer survivors in the CCSS who were alive 5 years after diagnosis. The patients had leukemia, lymphoma, CNS malignancies, Wilms tumor, neuroblastoma, or soft-tissue/bone sarcoma.
Dr Gibson noted that while 83% of children with a malignancy achieve a 5-year survival, more than half develop at least one severe, disabling, life-threatening health condition by age 50.
The survivors were a median age at last follow-up of 28 years (range, 5-63) and the median time since diagnosis was 21 years (range, 5-43).
The investigators found significant decreases in severe health conditions in 6 diagnostic groups:
- Wilms tumor, decreased from 13% to 5% (P<0.0001)
- HL, decreased from 18% to 11% (P<0.0001)
- Astrocytoma, decreased from 15% to 9% (P=0.004)
- NHL, decreased from 10% to 6% (P=0.04)
- ALL, decreased from 9% to 7% (P=0.002)
- Ewings sarcoma, decreased from 19% to 10% (P=0.01)
They found no reductions in subsequent severe health conditions among survivors of neuroblastoma, acute myeloid leukemia (AML), soft tissue sarcoma, or osteosarcoma.
The investigators believe the decreases were driven mainly by a reduced incidence of endocrine conditions, subsequent malignant neoplasms, gastrointestinal and neurological conditions, but not cardiac or pulmonary conditions.
They also analyzed the reduction in treatment intensities by decade for different diseases and found they correlated with the reduced incidence of serious chronic health conditions by 15 years after diagnosis.
The National Institutes of Health funded the study.
CHICAGO—The 15-year cumulative incidence of severe health conditions for survivors of childhood cancer has decreased over the past 30 years, from 12.7% for those diagnosed in the 1970s to 10.1% and 8.9% for those diagnosed in the 1980s and 1990s, respectively. And the decreases were greatest for patients with Wilms’ tumor and Hodgkin lymphoma (HL), followed by patients with astrocytoma, non-Hodgkin lymphoma (NHL), and acute lymphoblastic leukemia (ALL).
Investigators of the Childhood Cancer Survivor Study (CCSS) undertook a retrospective cohort analysis of children aged 0 – 14 years diagnosed with cancer between 1970 and 1999. Their goal was to determine whether cancer therapy modifications have maintained cure rates while decreasing the risk of late effects of therapy.
Todd M. Gibson, PhD, of St Jude Children’s Research Hospital in Memphis, Tennessee, presented the findings at the 2017 annual meeting of the American Society for Clinical Oncology (ASCO) as abstract LBA10500.
Researchers analyzed data from 23,600 childhood cancer survivors in the CCSS who were alive 5 years after diagnosis. The patients had leukemia, lymphoma, CNS malignancies, Wilms tumor, neuroblastoma, or soft-tissue/bone sarcoma.
Dr Gibson noted that while 83% of children with a malignancy achieve a 5-year survival, more than half develop at least one severe, disabling, life-threatening health condition by age 50.
The survivors were a median age at last follow-up of 28 years (range, 5-63) and the median time since diagnosis was 21 years (range, 5-43).
The investigators found significant decreases in severe health conditions in 6 diagnostic groups:
- Wilms tumor, decreased from 13% to 5% (P<0.0001)
- HL, decreased from 18% to 11% (P<0.0001)
- Astrocytoma, decreased from 15% to 9% (P=0.004)
- NHL, decreased from 10% to 6% (P=0.04)
- ALL, decreased from 9% to 7% (P=0.002)
- Ewings sarcoma, decreased from 19% to 10% (P=0.01)
They found no reductions in subsequent severe health conditions among survivors of neuroblastoma, acute myeloid leukemia (AML), soft tissue sarcoma, or osteosarcoma.
The investigators believe the decreases were driven mainly by a reduced incidence of endocrine conditions, subsequent malignant neoplasms, gastrointestinal and neurological conditions, but not cardiac or pulmonary conditions.
They also analyzed the reduction in treatment intensities by decade for different diseases and found they correlated with the reduced incidence of serious chronic health conditions by 15 years after diagnosis.
The National Institutes of Health funded the study.
Differences emerge in new guidelines for managing FN in kids

A multidisciplinary, international panel of experts has updated earlier clinical practice guidelines on managing fever and neutropenia (FN) in children with cancer and in those undergoing hematopoietic stem cell transplantation (HSCT). And while most of the recommendations remained unchanged from the 2012 guidelines, a few key differences emerged. The changes included addition of a 4th generation cephalosporin for empirical antifungal therapy and refinements in risk stratification for invasive fungal disease (IFD), among others.
The new guidelines were published by The International Pediatric Fever and Neutropenia Guideline Panel in the Journal of Clinical Oncology.
The recommendations were organized into 3 major sections: initial presentation, ongoing management, and empirical antifungal therapy. The guidelines panel followed procedures previously validated for creating evidence-based guidelines and used the Appraisal of Guidelines for Research & Evaluation II instrument as a framework.
For the initial presentation of FN, the panel increased the quality of evidence from low to moderate in the recommendation to obtain peripheral blood cultures concurrent with central venous catheter cultures.
In the treatment of FN, the panel added a 4th-generation cephalosporin as empirical therapy in high-risk FN.
The panel refined the IFD risk factors and decreased the quality of evidence from moderate to low. Children with acute myeloid leukemia (AML), high-risk acute lymphoblastic leukemia (ALL), relapsed acute leukemia, those undergoing allogeneic HSCT, those with prolonged neutropenia, and those receiving high-dose corticosteroids are at high risk of IFD. All others should be categorized as IFD low risk.
The panel suggested serum galactomannan not be used to guide empirical antifungal management for prolonged FN lasting 96 hours or more in high-risk IFD patients. GM does not rule out non-Aspergillus molds, and therefore high negative values provide less useful predictions. Previously, the use of galactomannan was a weak recommendation.
The panel added a new recommendation against using fungal polymerase chain reaction (PCR) testing in blood. They explained PCR testing provides poor positive predictive values and negative predictive values are not sufficiently high to be clinically useful. Also, PCR testing is not yet standardized.
Another new recommendation is the addition of imaging of the abdomen in patients without localizing signs or symptoms. Even though the ideal imaging modality is not known, ultrasound is readily available, not associated with radiation exposure, and usually does not require sedation. For these reasons, the panel said it is preferable to computed tomography or magnetic resonance imaging.
The panel also changed a previously weak recommendation to administer empirical therapy for IFD low-risk patients with prolonged FN to a weak recommendation against administering therapy for these patients.
The panel's recommendations and their rationale can be found in the JCO article.
The guidelines update was supported by meeting grants from the Canadian Institutes of Health Research and the Garron Comprehensive Cancer Centre. ![]()
A multidisciplinary, international panel of experts has updated earlier clinical practice guidelines on managing fever and neutropenia (FN) in children with cancer and in those undergoing hematopoietic stem cell transplantation (HSCT). And while most of the recommendations remained unchanged from the 2012 guidelines, a few key differences emerged. The changes included addition of a 4th generation cephalosporin for empirical antifungal therapy and refinements in risk stratification for invasive fungal disease (IFD), among others.
The new guidelines were published by The International Pediatric Fever and Neutropenia Guideline Panel in the Journal of Clinical Oncology.
The recommendations were organized into 3 major sections: initial presentation, ongoing management, and empirical antifungal therapy. The guidelines panel followed procedures previously validated for creating evidence-based guidelines and used the Appraisal of Guidelines for Research & Evaluation II instrument as a framework.
For the initial presentation of FN, the panel increased the quality of evidence from low to moderate in the recommendation to obtain peripheral blood cultures concurrent with central venous catheter cultures.
In the treatment of FN, the panel added a 4th-generation cephalosporin as empirical therapy in high-risk FN.
The panel refined the IFD risk factors and decreased the quality of evidence from moderate to low. Children with acute myeloid leukemia (AML), high-risk acute lymphoblastic leukemia (ALL), relapsed acute leukemia, those undergoing allogeneic HSCT, those with prolonged neutropenia, and those receiving high-dose corticosteroids are at high risk of IFD. All others should be categorized as IFD low risk.
The panel suggested serum galactomannan not be used to guide empirical antifungal management for prolonged FN lasting 96 hours or more in high-risk IFD patients. GM does not rule out non-Aspergillus molds, and therefore high negative values provide less useful predictions. Previously, the use of galactomannan was a weak recommendation.
The panel added a new recommendation against using fungal polymerase chain reaction (PCR) testing in blood. They explained PCR testing provides poor positive predictive values and negative predictive values are not sufficiently high to be clinically useful. Also, PCR testing is not yet standardized.
Another new recommendation is the addition of imaging of the abdomen in patients without localizing signs or symptoms. Even though the ideal imaging modality is not known, ultrasound is readily available, not associated with radiation exposure, and usually does not require sedation. For these reasons, the panel said it is preferable to computed tomography or magnetic resonance imaging.
The panel also changed a previously weak recommendation to administer empirical therapy for IFD low-risk patients with prolonged FN to a weak recommendation against administering therapy for these patients.
The panel's recommendations and their rationale can be found in the JCO article.
The guidelines update was supported by meeting grants from the Canadian Institutes of Health Research and the Garron Comprehensive Cancer Centre. ![]()
A multidisciplinary, international panel of experts has updated earlier clinical practice guidelines on managing fever and neutropenia (FN) in children with cancer and in those undergoing hematopoietic stem cell transplantation (HSCT). And while most of the recommendations remained unchanged from the 2012 guidelines, a few key differences emerged. The changes included addition of a 4th generation cephalosporin for empirical antifungal therapy and refinements in risk stratification for invasive fungal disease (IFD), among others.
The new guidelines were published by The International Pediatric Fever and Neutropenia Guideline Panel in the Journal of Clinical Oncology.
The recommendations were organized into 3 major sections: initial presentation, ongoing management, and empirical antifungal therapy. The guidelines panel followed procedures previously validated for creating evidence-based guidelines and used the Appraisal of Guidelines for Research & Evaluation II instrument as a framework.
For the initial presentation of FN, the panel increased the quality of evidence from low to moderate in the recommendation to obtain peripheral blood cultures concurrent with central venous catheter cultures.
In the treatment of FN, the panel added a 4th-generation cephalosporin as empirical therapy in high-risk FN.
The panel refined the IFD risk factors and decreased the quality of evidence from moderate to low. Children with acute myeloid leukemia (AML), high-risk acute lymphoblastic leukemia (ALL), relapsed acute leukemia, those undergoing allogeneic HSCT, those with prolonged neutropenia, and those receiving high-dose corticosteroids are at high risk of IFD. All others should be categorized as IFD low risk.
The panel suggested serum galactomannan not be used to guide empirical antifungal management for prolonged FN lasting 96 hours or more in high-risk IFD patients. GM does not rule out non-Aspergillus molds, and therefore high negative values provide less useful predictions. Previously, the use of galactomannan was a weak recommendation.
The panel added a new recommendation against using fungal polymerase chain reaction (PCR) testing in blood. They explained PCR testing provides poor positive predictive values and negative predictive values are not sufficiently high to be clinically useful. Also, PCR testing is not yet standardized.
Another new recommendation is the addition of imaging of the abdomen in patients without localizing signs or symptoms. Even though the ideal imaging modality is not known, ultrasound is readily available, not associated with radiation exposure, and usually does not require sedation. For these reasons, the panel said it is preferable to computed tomography or magnetic resonance imaging.
The panel also changed a previously weak recommendation to administer empirical therapy for IFD low-risk patients with prolonged FN to a weak recommendation against administering therapy for these patients.
The panel's recommendations and their rationale can be found in the JCO article.
The guidelines update was supported by meeting grants from the Canadian Institutes of Health Research and the Garron Comprehensive Cancer Centre. ![]()
BLA for CAR T-cell therapy granted priority review

The US Food and Drug Administration (FDA) has accepted for priority review the biologics license application (BLA) for axicabtagene ciloleucel (formerly KTE-C19), a chimeric antigen receptor (CAR) T-cell therapy.
Kite Pharma, Inc. is seeking approval for axicabtagene ciloleucel as a treatment for patients with relapsed or refractory, aggressive non-Hodgkin lymphoma (NHL) who are ineligible for autologous stem cell transplant.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The FDA has set a review deadline of November 29, 2017, for the axicabtagene ciloleucel BLA.
Axicabtagene ciloleucel also has breakthrough therapy designation from the FDA as a treatment for diffuse large B-cell lymphoma, transformed follicular lymphoma, and primary mediastinal B-cell lymphoma.
ZUMA-1 trial
The BLA for axicabtagene ciloleucel is supported by data from the phase 2 ZUMA-1 trial, which enrolled 111 patients with relapsed/refractory B-cell NHL.
After a single infusion of axicabtagene ciloleucel, the objective response rate was 82%. At a median follow-up of 8.7 months, 44% of patients were still in response, which included 39% of patients in complete response.
The most common grade 3 or higher adverse events were anemia (43%), neutropenia (39%), decreased neutrophil count (32%), febrile neutropenia (31%), decreased white blood cell count (29%), thrombocytopenia (24%), encephalopathy (21%), and decreased lymphocyte count (20%).
There were 3 deaths during the trial that were not due to disease progression. Two of these deaths were deemed related to axicabtagene ciloleucel. ![]()
The US Food and Drug Administration (FDA) has accepted for priority review the biologics license application (BLA) for axicabtagene ciloleucel (formerly KTE-C19), a chimeric antigen receptor (CAR) T-cell therapy.
Kite Pharma, Inc. is seeking approval for axicabtagene ciloleucel as a treatment for patients with relapsed or refractory, aggressive non-Hodgkin lymphoma (NHL) who are ineligible for autologous stem cell transplant.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The FDA has set a review deadline of November 29, 2017, for the axicabtagene ciloleucel BLA.
Axicabtagene ciloleucel also has breakthrough therapy designation from the FDA as a treatment for diffuse large B-cell lymphoma, transformed follicular lymphoma, and primary mediastinal B-cell lymphoma.
ZUMA-1 trial
The BLA for axicabtagene ciloleucel is supported by data from the phase 2 ZUMA-1 trial, which enrolled 111 patients with relapsed/refractory B-cell NHL.
After a single infusion of axicabtagene ciloleucel, the objective response rate was 82%. At a median follow-up of 8.7 months, 44% of patients were still in response, which included 39% of patients in complete response.
The most common grade 3 or higher adverse events were anemia (43%), neutropenia (39%), decreased neutrophil count (32%), febrile neutropenia (31%), decreased white blood cell count (29%), thrombocytopenia (24%), encephalopathy (21%), and decreased lymphocyte count (20%).
There were 3 deaths during the trial that were not due to disease progression. Two of these deaths were deemed related to axicabtagene ciloleucel. ![]()
The US Food and Drug Administration (FDA) has accepted for priority review the biologics license application (BLA) for axicabtagene ciloleucel (formerly KTE-C19), a chimeric antigen receptor (CAR) T-cell therapy.
Kite Pharma, Inc. is seeking approval for axicabtagene ciloleucel as a treatment for patients with relapsed or refractory, aggressive non-Hodgkin lymphoma (NHL) who are ineligible for autologous stem cell transplant.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The FDA has set a review deadline of November 29, 2017, for the axicabtagene ciloleucel BLA.
Axicabtagene ciloleucel also has breakthrough therapy designation from the FDA as a treatment for diffuse large B-cell lymphoma, transformed follicular lymphoma, and primary mediastinal B-cell lymphoma.
ZUMA-1 trial
The BLA for axicabtagene ciloleucel is supported by data from the phase 2 ZUMA-1 trial, which enrolled 111 patients with relapsed/refractory B-cell NHL.
After a single infusion of axicabtagene ciloleucel, the objective response rate was 82%. At a median follow-up of 8.7 months, 44% of patients were still in response, which included 39% of patients in complete response.
The most common grade 3 or higher adverse events were anemia (43%), neutropenia (39%), decreased neutrophil count (32%), febrile neutropenia (31%), decreased white blood cell count (29%), thrombocytopenia (24%), encephalopathy (21%), and decreased lymphocyte count (20%).
There were 3 deaths during the trial that were not due to disease progression. Two of these deaths were deemed related to axicabtagene ciloleucel. ![]()