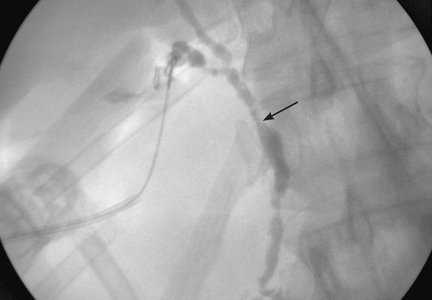
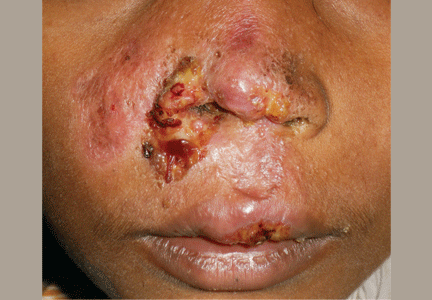
There was an error in the caption for Figure 2 in: Villa-Forte A. Giant cell arteritis: Suspect it, treat it promptly. Cleve Clin J Med 2011; 78:265–270. The image was of digital subtraction angiography, not magnetic resonance angiography. The caption has been corrected in the online version of the article.
There was an error in the caption for Figure 2 in: Villa-Forte A. Giant cell arteritis: Suspect it, treat it promptly. Cleve Clin J Med 2011; 78:265–270. The image was of digital subtraction angiography, not magnetic resonance angiography. The caption has been corrected in the online version of the article.
There was an error in the caption for Figure 2 in: Villa-Forte A. Giant cell arteritis: Suspect it, treat it promptly. Cleve Clin J Med 2011; 78:265–270. The image was of digital subtraction angiography, not magnetic resonance angiography. The caption has been corrected in the online version of the article.
A 52-year-old woman is referred to our dermatology clinic by her primary care physician for swelling and redness of seven old scars. The swelling began 3 months ago.
She is on no regular medications. She has never smoked. She underwent liposuction 7 years ago and appendectomy at age 15.
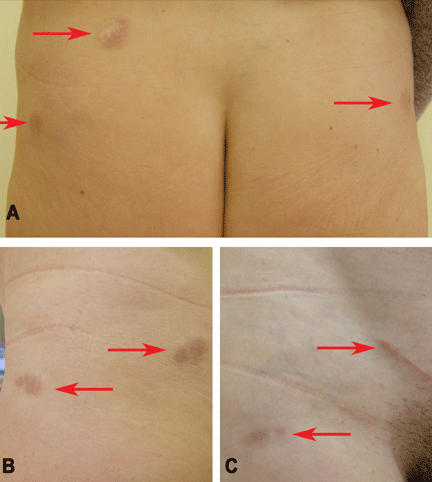
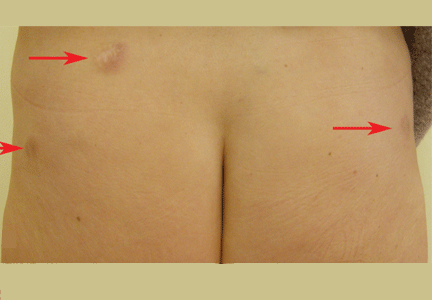
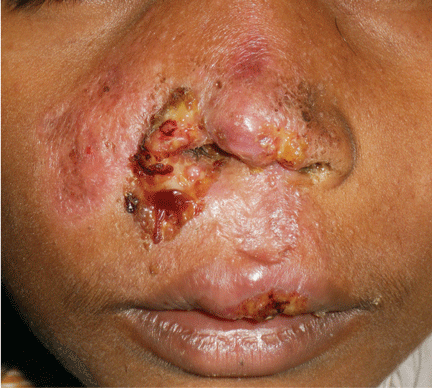
Figure 1. Indurated and painful brownish-red plaques localized on old scars: (A) lower trunk, (B) left side, (C) right side.
Physical examination reveals indurated and painful brownish-red plaques on the thighs and lower trunk, extending beyond the borders of six scars from the liposuction surgery and one scar from the appendectomy (Figure 1). She reports no dyspnea, night sweats, weight loss, fever, or other constitutional symptoms, but she has a dry cough that began 2 months after the onset of the skin symptoms. Her primary care physician has managed the cough symptomatically.
Laboratory testing shows mild leukopenia, with a white blood cell count of 3.0 × 109/L (reference range 4.2–9.0). Other routine laboratory values are normal, including antinuclear antibody, extractable nuclear antibody, anti-double-stranded DNA, rheumatoid factor, urinalysis, erythrocyte sedimentation rate, C-reactive protein, serum calcium concentration, and liver and renal function tests.
Q: What is the next most appropriate diagnostic procedure?
Skin biopsy
High-resolution computed tomography (CT) of the chest
QuantiFERON-TB Gold test
Ventilatory function tests
Serum angiotensin-converting enzyme (ACE) level
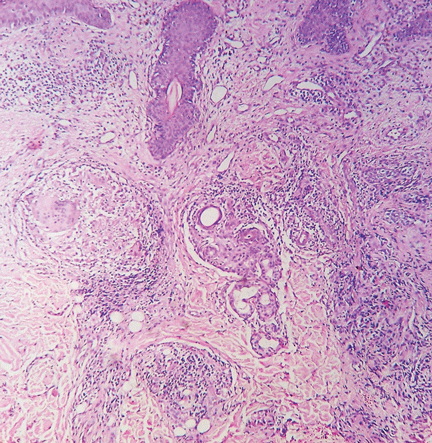
A: All are appropriate at this point. In this case, skin biopsy and high-resolution CT were performed. Histopathologic examination of one of the scars showed multiple well-demarcated, large, noncaseating epitheloid granulomas with histiocytes and multinucleated giant cells. High-resolution CT confirmed bilateral hilar and mediastinal lymphadenopathy and revealed micronodular densities with a bronchovascular and subpleural distribution.
An interferon-gamma-release assay for tuberculosis—QuantiFERON-TB Gold (Cellestis, Carnegie, Australia)—was negative. Ventilatory function tests showed a normal pattern, while the serum ACE level, electrocardiography, and an eye examination revealed no pathologic findings.
Q: What is the diagnosis?
Keloids
Scar sarcoidosis
Paraneoplastic sign
Dermatofibrosarcoma protuberans
Tubercolosis
A: Based on the data outlined above, we made the diagnosis of scar sarcoidosis with involvement of hilar and mediastinal lymph nodes. The patient began systemic treatment with oral prednisone 1 mg/kg/day for 6 weeks, which was then gradually withdrawn, until the skin and hilar lesions resolved completely.
SCAR SARCOIDOSIS
Sarcoidosis is a multisystem disorder of unknown cause characterized by the formation of noncaseating granulomas in the affected organs. Patients may present with symptoms related to the specific organ affected, but they may have no symptoms or only general symptoms such as fever or general malaise.
The skin is involved in 25% of cases and presents so many polymorphous manifestations that sarcoidosis has become known as one of the “great imitators” in dermatology.1,2
Although sarcoidosis on liposuction scars has not been reported previously, the reactivation of old scars is well known on sites of previous injections, tattoos, herpes zoster, and burns.2,3
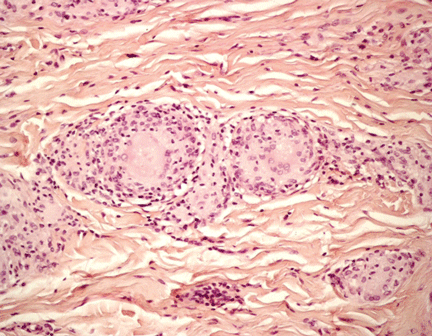
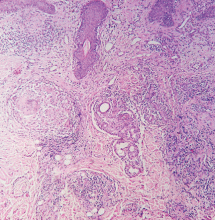
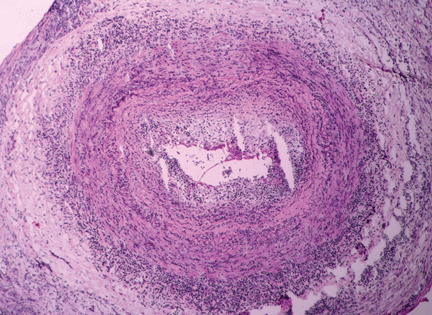
Figure 2. Noncaseating granuloma seen on skin biopsy study.
The finding of granuloma is not specific for sarcoidosis (Figure 2). The histologic differential diagnosis of sarcoidosis includes tuberculosis, atypical mycobacteriosis, fungal infection, reaction to a foreign body, rheumatoid nodules, leishmaniasis, Crohn disease, and necrobiosis lipoidica diabeticorum.
The diagnosis of scar sarcoidosis is confirmed only by excluding other conditions via a comprehensive evaluation of clinical manifestations, histology, history, and radiologic and laboratory findings.
It has been suggested that the most satisfying therapy for the patient and physician in sarcoidosis is no treatment at all,4 and in fact sarcoidosis often remits spontaneously. Currently, the choice of treatment depends on the degree of systemic involvement, and the oral corticosteroid prednisone remains the first-line treatment. If the condition does not respond, the use of other systemic agents has been reported, but their effectiveness has not been evaluated in controlled clinical trials.
Recurrence is common after the suspension of treatment; therefore, treatment may need to be continued for several years, with frequent checkups.
Skin lesions are a visible clue to the diagnosis. Reactivation of old scars may be the single manifestation of cutaneous sarcoidosis, but it may also precede or accompany systemic involvement, often representing the main sign of an exacerbation or a relapse of systemic sarcoidosis, as in our patient.5
Tchernev G. Cutaneous sarcoidosis: the “great imitator”: etiopathogenesis, morphology, differential diagnosis, and clinical management. Am J Clin Dermatol2006; 7:375–382.
Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol2007; 25:276–287.
Baughman RP, Lower EE, du Bois RM. Sarcoidosis. Lancet2003; 361:1111–1118.
Sorabjee JS, Garje R. Reactivation of old scars: inevitably sarcoid. Postgrad Med J2005; 81:60–61.
Giulia Rech, MD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Riccardo Balestri, MD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Federico Bardazzi, MD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Bianca Maria Piraccini, MD, PhD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Annalisa Patrizi, MD, PhD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Giulia Rech, MD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Riccardo Balestri, MD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Federico Bardazzi, MD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Bianca Maria Piraccini, MD, PhD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Annalisa Patrizi, MD, PhD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Giulia Rech, MD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Riccardo Balestri, MD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Federico Bardazzi, MD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Bianca Maria Piraccini, MD, PhD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Annalisa Patrizi, MD, PhD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
A 52-year-old woman is referred to our dermatology clinic by her primary care physician for swelling and redness of seven old scars. The swelling began 3 months ago.
She is on no regular medications. She has never smoked. She underwent liposuction 7 years ago and appendectomy at age 15.
Figure 1. Indurated and painful brownish-red plaques localized on old scars: (A) lower trunk, (B) left side, (C) right side.
Physical examination reveals indurated and painful brownish-red plaques on the thighs and lower trunk, extending beyond the borders of six scars from the liposuction surgery and one scar from the appendectomy (Figure 1). She reports no dyspnea, night sweats, weight loss, fever, or other constitutional symptoms, but she has a dry cough that began 2 months after the onset of the skin symptoms. Her primary care physician has managed the cough symptomatically.
Laboratory testing shows mild leukopenia, with a white blood cell count of 3.0 × 109/L (reference range 4.2–9.0). Other routine laboratory values are normal, including antinuclear antibody, extractable nuclear antibody, anti-double-stranded DNA, rheumatoid factor, urinalysis, erythrocyte sedimentation rate, C-reactive protein, serum calcium concentration, and liver and renal function tests.
Q: What is the next most appropriate diagnostic procedure?
Skin biopsy
High-resolution computed tomography (CT) of the chest
QuantiFERON-TB Gold test
Ventilatory function tests
Serum angiotensin-converting enzyme (ACE) level
A: All are appropriate at this point. In this case, skin biopsy and high-resolution CT were performed. Histopathologic examination of one of the scars showed multiple well-demarcated, large, noncaseating epitheloid granulomas with histiocytes and multinucleated giant cells. High-resolution CT confirmed bilateral hilar and mediastinal lymphadenopathy and revealed micronodular densities with a bronchovascular and subpleural distribution.
An interferon-gamma-release assay for tuberculosis—QuantiFERON-TB Gold (Cellestis, Carnegie, Australia)—was negative. Ventilatory function tests showed a normal pattern, while the serum ACE level, electrocardiography, and an eye examination revealed no pathologic findings.
Q: What is the diagnosis?
Keloids
Scar sarcoidosis
Paraneoplastic sign
Dermatofibrosarcoma protuberans
Tubercolosis
A: Based on the data outlined above, we made the diagnosis of scar sarcoidosis with involvement of hilar and mediastinal lymph nodes. The patient began systemic treatment with oral prednisone 1 mg/kg/day for 6 weeks, which was then gradually withdrawn, until the skin and hilar lesions resolved completely.
SCAR SARCOIDOSIS
Sarcoidosis is a multisystem disorder of unknown cause characterized by the formation of noncaseating granulomas in the affected organs. Patients may present with symptoms related to the specific organ affected, but they may have no symptoms or only general symptoms such as fever or general malaise.
The skin is involved in 25% of cases and presents so many polymorphous manifestations that sarcoidosis has become known as one of the “great imitators” in dermatology.1,2
Although sarcoidosis on liposuction scars has not been reported previously, the reactivation of old scars is well known on sites of previous injections, tattoos, herpes zoster, and burns.2,3
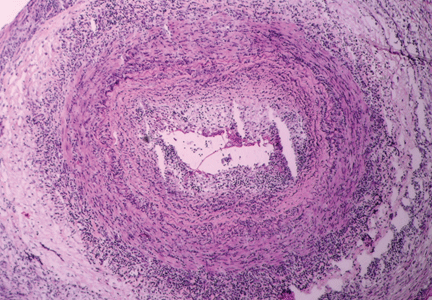
Figure 2. Noncaseating granuloma seen on skin biopsy study.
The finding of granuloma is not specific for sarcoidosis (Figure 2). The histologic differential diagnosis of sarcoidosis includes tuberculosis, atypical mycobacteriosis, fungal infection, reaction to a foreign body, rheumatoid nodules, leishmaniasis, Crohn disease, and necrobiosis lipoidica diabeticorum.
The diagnosis of scar sarcoidosis is confirmed only by excluding other conditions via a comprehensive evaluation of clinical manifestations, histology, history, and radiologic and laboratory findings.
It has been suggested that the most satisfying therapy for the patient and physician in sarcoidosis is no treatment at all,4 and in fact sarcoidosis often remits spontaneously. Currently, the choice of treatment depends on the degree of systemic involvement, and the oral corticosteroid prednisone remains the first-line treatment. If the condition does not respond, the use of other systemic agents has been reported, but their effectiveness has not been evaluated in controlled clinical trials.
Recurrence is common after the suspension of treatment; therefore, treatment may need to be continued for several years, with frequent checkups.
Skin lesions are a visible clue to the diagnosis. Reactivation of old scars may be the single manifestation of cutaneous sarcoidosis, but it may also precede or accompany systemic involvement, often representing the main sign of an exacerbation or a relapse of systemic sarcoidosis, as in our patient.5
A 52-year-old woman is referred to our dermatology clinic by her primary care physician for swelling and redness of seven old scars. The swelling began 3 months ago.
She is on no regular medications. She has never smoked. She underwent liposuction 7 years ago and appendectomy at age 15.
Figure 1. Indurated and painful brownish-red plaques localized on old scars: (A) lower trunk, (B) left side, (C) right side.
Physical examination reveals indurated and painful brownish-red plaques on the thighs and lower trunk, extending beyond the borders of six scars from the liposuction surgery and one scar from the appendectomy (Figure 1). She reports no dyspnea, night sweats, weight loss, fever, or other constitutional symptoms, but she has a dry cough that began 2 months after the onset of the skin symptoms. Her primary care physician has managed the cough symptomatically.
Laboratory testing shows mild leukopenia, with a white blood cell count of 3.0 × 109/L (reference range 4.2–9.0). Other routine laboratory values are normal, including antinuclear antibody, extractable nuclear antibody, anti-double-stranded DNA, rheumatoid factor, urinalysis, erythrocyte sedimentation rate, C-reactive protein, serum calcium concentration, and liver and renal function tests.
Q: What is the next most appropriate diagnostic procedure?
Skin biopsy
High-resolution computed tomography (CT) of the chest
QuantiFERON-TB Gold test
Ventilatory function tests
Serum angiotensin-converting enzyme (ACE) level
A: All are appropriate at this point. In this case, skin biopsy and high-resolution CT were performed. Histopathologic examination of one of the scars showed multiple well-demarcated, large, noncaseating epitheloid granulomas with histiocytes and multinucleated giant cells. High-resolution CT confirmed bilateral hilar and mediastinal lymphadenopathy and revealed micronodular densities with a bronchovascular and subpleural distribution.
An interferon-gamma-release assay for tuberculosis—QuantiFERON-TB Gold (Cellestis, Carnegie, Australia)—was negative. Ventilatory function tests showed a normal pattern, while the serum ACE level, electrocardiography, and an eye examination revealed no pathologic findings.
Q: What is the diagnosis?
Keloids
Scar sarcoidosis
Paraneoplastic sign
Dermatofibrosarcoma protuberans
Tubercolosis
A: Based on the data outlined above, we made the diagnosis of scar sarcoidosis with involvement of hilar and mediastinal lymph nodes. The patient began systemic treatment with oral prednisone 1 mg/kg/day for 6 weeks, which was then gradually withdrawn, until the skin and hilar lesions resolved completely.
SCAR SARCOIDOSIS
Sarcoidosis is a multisystem disorder of unknown cause characterized by the formation of noncaseating granulomas in the affected organs. Patients may present with symptoms related to the specific organ affected, but they may have no symptoms or only general symptoms such as fever or general malaise.
The skin is involved in 25% of cases and presents so many polymorphous manifestations that sarcoidosis has become known as one of the “great imitators” in dermatology.1,2
Although sarcoidosis on liposuction scars has not been reported previously, the reactivation of old scars is well known on sites of previous injections, tattoos, herpes zoster, and burns.2,3
Figure 2. Noncaseating granuloma seen on skin biopsy study.
The finding of granuloma is not specific for sarcoidosis (Figure 2). The histologic differential diagnosis of sarcoidosis includes tuberculosis, atypical mycobacteriosis, fungal infection, reaction to a foreign body, rheumatoid nodules, leishmaniasis, Crohn disease, and necrobiosis lipoidica diabeticorum.
The diagnosis of scar sarcoidosis is confirmed only by excluding other conditions via a comprehensive evaluation of clinical manifestations, histology, history, and radiologic and laboratory findings.
It has been suggested that the most satisfying therapy for the patient and physician in sarcoidosis is no treatment at all,4 and in fact sarcoidosis often remits spontaneously. Currently, the choice of treatment depends on the degree of systemic involvement, and the oral corticosteroid prednisone remains the first-line treatment. If the condition does not respond, the use of other systemic agents has been reported, but their effectiveness has not been evaluated in controlled clinical trials.
Recurrence is common after the suspension of treatment; therefore, treatment may need to be continued for several years, with frequent checkups.
Skin lesions are a visible clue to the diagnosis. Reactivation of old scars may be the single manifestation of cutaneous sarcoidosis, but it may also precede or accompany systemic involvement, often representing the main sign of an exacerbation or a relapse of systemic sarcoidosis, as in our patient.5
Tchernev G. Cutaneous sarcoidosis: the “great imitator”: etiopathogenesis, morphology, differential diagnosis, and clinical management. Am J Clin Dermatol2006; 7:375–382.
Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol2007; 25:276–287.
Baughman RP, Lower EE, du Bois RM. Sarcoidosis. Lancet2003; 361:1111–1118.
Sorabjee JS, Garje R. Reactivation of old scars: inevitably sarcoid. Postgrad Med J2005; 81:60–61.
Tchernev G. Cutaneous sarcoidosis: the “great imitator”: etiopathogenesis, morphology, differential diagnosis, and clinical management. Am J Clin Dermatol2006; 7:375–382.
Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol2007; 25:276–287.
Baughman RP, Lower EE, du Bois RM. Sarcoidosis. Lancet2003; 361:1111–1118.
Sorabjee JS, Garje R. Reactivation of old scars: inevitably sarcoid. Postgrad Med J2005; 81:60–61.
Although diagnosis of a bee sting allergy is often straightforward, it’s important to go through the history. Ask when the child was stung, what type of reaction they had, and how soon after the sting they experienced symptoms.
A large local reaction can be impressive in size, but it may not be as serious as the child who presents with systemic symptoms such as hives or difficulty breathing.
Immediately direct a child experiencing acute anaphylaxis to emergency care. Acute effects will be seen right away, generally within 15-30 minutes. The parents of a child with a known sensitivity to bee stings, in particular, will know to head to the emergency department right away, especially after self-administration of epinephrine by an autoinjector.
It is more likely that a patient will come to you with a less severe reaction or for advice on how to manage their potential allergy. In general, local reactions are no larger than 10 cm, and you can treat the area with ice or cold compresses in your office. Typical local reactions are a little bump, a local hive, or an indurated area of swelling that is warm or hot.
Take photos of the allergic reaction. This can be very helpful if you later refer the child to a specialist. It helps to immediately see the size and location of the reaction.
Check to see if the stinger is still in place when a flustered child (or parent) comes in right after a bee sting. Although most people remove it immediately, some patients come in with the stinger still in their skin. You want to scrape or brush across the skin with a credit card or coin to remove the stinger. The removal technique is important because honey bees can leave both their stinger and venom sac behind as a last defense. If you just try to pull out the stinger, unintentional squeezing of the venom sac can mean more venom gets injected into the allergic child.
Consider referral to a pediatric allergy specialist if a child has a history of adverse or severe reactions to bee stings. The risk of future severe reactions, including anaphylaxis, will be elevated in a patient who has already spent any time in the emergency department, for example. When you refer, include a list of any local or systemic symptoms and any medications the child is taking.
Each subsequent exposure to bee venom increases the risk of a more severe reaction. One question I always get is: "I’ve been stung 15 times before. How come this time I developed an anaphylactic reaction?" I explain that a person needs to be stung only once before the body can develop an allergy, and any exposure after that may trigger a serious or life-threatening reaction.
You can perform allergy testing in your primary care office, but the question is what to do with the results. Such testing prior to referral does not tend to help us a lot. We often perform a more comprehensive evaluation. For example, as a general rule I order IgE protein-specific tests for the five common flying insect venoms, because most children cannot tell if a wasp, hornet, or bee stung them.
The good news is that if an individual meets criteria and is treated with immunotherapy or allergy shots, he or she has a success rate of about 98%. Even so, I recommend that a child with a history of bee sting adverse reactions carry an autoinjectable epinephrine device and practice bee avoidance measures.
You can educate children in the primary care setting how to stay away from bees. Tell them not to play in or around woods, for example. Make sure the child knows not to provoke or aggravate any bees they encounter, and that bees are attracted by bright-colored clothing, perfume, and cologne. I also tell patients to avoid drinking cans of soda outdoors. Bees attracted to the sweet soda will fly into these cans and, unfortunately, it is not uncommon for people to be very surprised and get stung in the mouth, on the tongue, or on the lips this way.
Dr. Doshi is director of pediatric allergy and immunology at Beaumont Hospital, Royal Oak, Mich.
Although diagnosis of a bee sting allergy is often straightforward, it’s important to go through the history. Ask when the child was stung, what type of reaction they had, and how soon after the sting they experienced symptoms.
A large local reaction can be impressive in size, but it may not be as serious as the child who presents with systemic symptoms such as hives or difficulty breathing.
Immediately direct a child experiencing acute anaphylaxis to emergency care. Acute effects will be seen right away, generally within 15-30 minutes. The parents of a child with a known sensitivity to bee stings, in particular, will know to head to the emergency department right away, especially after self-administration of epinephrine by an autoinjector.
It is more likely that a patient will come to you with a less severe reaction or for advice on how to manage their potential allergy. In general, local reactions are no larger than 10 cm, and you can treat the area with ice or cold compresses in your office. Typical local reactions are a little bump, a local hive, or an indurated area of swelling that is warm or hot.
Take photos of the allergic reaction. This can be very helpful if you later refer the child to a specialist. It helps to immediately see the size and location of the reaction.
Check to see if the stinger is still in place when a flustered child (or parent) comes in right after a bee sting. Although most people remove it immediately, some patients come in with the stinger still in their skin. You want to scrape or brush across the skin with a credit card or coin to remove the stinger. The removal technique is important because honey bees can leave both their stinger and venom sac behind as a last defense. If you just try to pull out the stinger, unintentional squeezing of the venom sac can mean more venom gets injected into the allergic child.
Consider referral to a pediatric allergy specialist if a child has a history of adverse or severe reactions to bee stings. The risk of future severe reactions, including anaphylaxis, will be elevated in a patient who has already spent any time in the emergency department, for example. When you refer, include a list of any local or systemic symptoms and any medications the child is taking.
Each subsequent exposure to bee venom increases the risk of a more severe reaction. One question I always get is: "I’ve been stung 15 times before. How come this time I developed an anaphylactic reaction?" I explain that a person needs to be stung only once before the body can develop an allergy, and any exposure after that may trigger a serious or life-threatening reaction.
You can perform allergy testing in your primary care office, but the question is what to do with the results. Such testing prior to referral does not tend to help us a lot. We often perform a more comprehensive evaluation. For example, as a general rule I order IgE protein-specific tests for the five common flying insect venoms, because most children cannot tell if a wasp, hornet, or bee stung them.
The good news is that if an individual meets criteria and is treated with immunotherapy or allergy shots, he or she has a success rate of about 98%. Even so, I recommend that a child with a history of bee sting adverse reactions carry an autoinjectable epinephrine device and practice bee avoidance measures.
You can educate children in the primary care setting how to stay away from bees. Tell them not to play in or around woods, for example. Make sure the child knows not to provoke or aggravate any bees they encounter, and that bees are attracted by bright-colored clothing, perfume, and cologne. I also tell patients to avoid drinking cans of soda outdoors. Bees attracted to the sweet soda will fly into these cans and, unfortunately, it is not uncommon for people to be very surprised and get stung in the mouth, on the tongue, or on the lips this way.
Dr. Doshi is director of pediatric allergy and immunology at Beaumont Hospital, Royal Oak, Mich.
Although diagnosis of a bee sting allergy is often straightforward, it’s important to go through the history. Ask when the child was stung, what type of reaction they had, and how soon after the sting they experienced symptoms.
A large local reaction can be impressive in size, but it may not be as serious as the child who presents with systemic symptoms such as hives or difficulty breathing.
Immediately direct a child experiencing acute anaphylaxis to emergency care. Acute effects will be seen right away, generally within 15-30 minutes. The parents of a child with a known sensitivity to bee stings, in particular, will know to head to the emergency department right away, especially after self-administration of epinephrine by an autoinjector.
It is more likely that a patient will come to you with a less severe reaction or for advice on how to manage their potential allergy. In general, local reactions are no larger than 10 cm, and you can treat the area with ice or cold compresses in your office. Typical local reactions are a little bump, a local hive, or an indurated area of swelling that is warm or hot.
Take photos of the allergic reaction. This can be very helpful if you later refer the child to a specialist. It helps to immediately see the size and location of the reaction.
Check to see if the stinger is still in place when a flustered child (or parent) comes in right after a bee sting. Although most people remove it immediately, some patients come in with the stinger still in their skin. You want to scrape or brush across the skin with a credit card or coin to remove the stinger. The removal technique is important because honey bees can leave both their stinger and venom sac behind as a last defense. If you just try to pull out the stinger, unintentional squeezing of the venom sac can mean more venom gets injected into the allergic child.
Consider referral to a pediatric allergy specialist if a child has a history of adverse or severe reactions to bee stings. The risk of future severe reactions, including anaphylaxis, will be elevated in a patient who has already spent any time in the emergency department, for example. When you refer, include a list of any local or systemic symptoms and any medications the child is taking.
Each subsequent exposure to bee venom increases the risk of a more severe reaction. One question I always get is: "I’ve been stung 15 times before. How come this time I developed an anaphylactic reaction?" I explain that a person needs to be stung only once before the body can develop an allergy, and any exposure after that may trigger a serious or life-threatening reaction.
You can perform allergy testing in your primary care office, but the question is what to do with the results. Such testing prior to referral does not tend to help us a lot. We often perform a more comprehensive evaluation. For example, as a general rule I order IgE protein-specific tests for the five common flying insect venoms, because most children cannot tell if a wasp, hornet, or bee stung them.
The good news is that if an individual meets criteria and is treated with immunotherapy or allergy shots, he or she has a success rate of about 98%. Even so, I recommend that a child with a history of bee sting adverse reactions carry an autoinjectable epinephrine device and practice bee avoidance measures.
You can educate children in the primary care setting how to stay away from bees. Tell them not to play in or around woods, for example. Make sure the child knows not to provoke or aggravate any bees they encounter, and that bees are attracted by bright-colored clothing, perfume, and cologne. I also tell patients to avoid drinking cans of soda outdoors. Bees attracted to the sweet soda will fly into these cans and, unfortunately, it is not uncommon for people to be very surprised and get stung in the mouth, on the tongue, or on the lips this way.
Dr. Doshi is director of pediatric allergy and immunology at Beaumont Hospital, Royal Oak, Mich.
A 49-year-old man has had ulcerative colitis for more than 30 years. It is well controlled with sulfasalazine (Azulfidine). Now, he has come to see his primary care physician because for the past 3 months he has had mild, intermittent pain in his right upper abdominal quadrant.
His physical examination is normal. Routine laboratory testing shows the following:
Hemoglobin 14.2 g/dL (reference range 13.5–17.5)
White blood cell count 6.7 × 109/L (3.5–10.5)
Platelet count 279 × 109/L (150–450)
Alkaline phosphatase 387 U/L (45–115)
Total bilirubin 0.9 mg/dL (0.1–1.0)
Aspartate aminotransferase (AST) 35 U/L (35–48)
Alanine aminotransferase (ALT) 30 U/L (7–55).
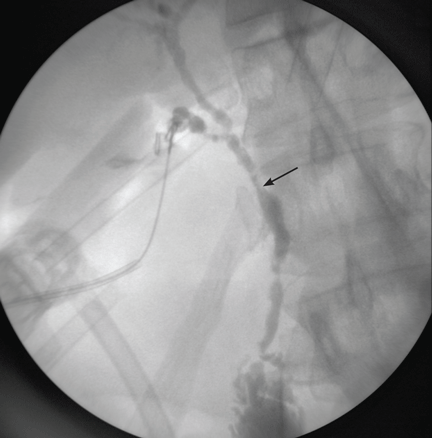
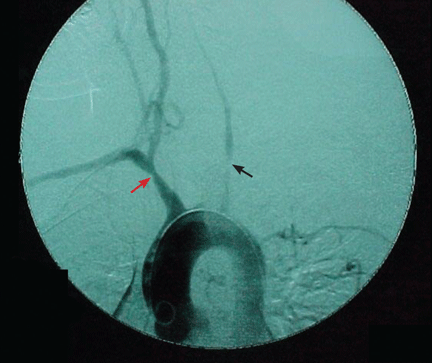
Figure 1. Intraoperative cholangiography demonstrates annular, multifocal stricturing and beading of the extrahepatic biliary system (arrow).
His physician is concerned about his elevated alkaline phosphatase level, which can be a sign of cholestatic liver disease (ie, involving blockage of the flow of bile). He sends him for ultrasonography, which reveals mild thickening of the gallbladder wall. The patient is referred to a general surgeon, who decides to remove the gallbladder. The procedure goes well, but when contrast dye is injected into the biliary system during cholangiography, the image is markedly abnormal (Figure 1). The patient is referred to Mayo Clinic for further evaluation.
WHAT IS THE DIAGNOSIS?
1.Based on this information, which of the following is the most likely diagnosis?
Autoimmune hepatitis
Primary sclerosing cholangitis
Primary biliary cirrhosis
Idiopathic adulthood ductopenia
Primary sclerosing cholangitis
The most likely diagnosis is primary sclerosing cholangitis, a chronic cholestatic liver disease characterized by diffuse inflammatory destruction of intrahepatic and extrahepatic bile ducts, resulting in fibrosis, cirrhosis, and liver failure. Its cause is unknown, but it is likely the result of acquired exposures interacting with predisposing host factors. Current diagnostic criteria include:
Characteristic cholangiographic abnormalities of the biliary tree
Compatible clinical and biochemical findings (typically cholestasis with elevated alkaline phosphatase levels for at least 6 months)
Exclusion of causes of secondary sclerosing cholangitis: secondary sclerosing cholangitis is characterized by a similar multifocal biliary stricturing process, but with an identifiable cause such as long-term biliary obstruction, surgical biliary trauma, or recurrent pancreatitis.1
At presentation, the most common liver enzyme abnormality is an elevated alkaline phosphatase level, often three or four times the normal level.2 In contrast, aminotransferase levels are only modestly elevated, less than three times the upper limit of normal.3 At the time of diagnosis, serum bilirubin levels are normal in 60% of patients.4
Two large epidemiologic studies (one from Olmsted County, MN,5 the other from Swansea, Wales, UK6) estimated the age-adjusted incidence of primary sclerosing cholangitis to be 0.9 per 100,000 individuals. The median age of the patients at onset was in the 30s or 40s, and most were men. At 10 years, an estimated 65% were still alive and had not undergone liver transplantation—a significantly lower percentage than in age- and sex-matched populations.
It is estimated that more than 70% of patients with primary sclerosing cholangitis also have inflammatory bowel disease.5 In fact, the most common presentation of primary sclerosing cholangitis is asymptomatic inflammatory bowel disease and persistently elevated alkaline phosphatase—usually first noted on routine biochemical screening, as in our patient.
Imaging of the biliary tree is essential for the diagnosis of primary sclerosing cholangitis. Typical findings on cholangiography include multifocal stricturing and beading, usually involving both the intrahepatic and the extrahepatic biliary systems, as in our patient (Figure 1). Endoscopic retrograde cholangiopancreatography (ERCP) is considered the gold standard imaging test, but recent studies have shown that magnetic resonance cholangiopancreatography (MRCP) is an acceptable noninvasive substitute,7 and it may cost less per diagnosis.8
Liver biopsy alone is generally nondiagnostic because the histologic changes are quite variable in different segments of the same liver. The classic “onion-skin fibrosis” of primary sclerosing cholangitis is seen in fewer than 10% of biopsy specimens.9
Autoimmune hepatitis
Autoimmune hepatitis is chronic and is characterized by circulating autoantibodies and high serum globulin concentrations.10 Its presentation is heterogeneous, varying from no symptoms to nonspecific symptoms of malaise, fatigue, abdominal pain, itching, and arthralgia. Generally, elevations in aminotransferases are much more prominent than abnormalities in bilirubin and alkaline phosphatase levels10—unlike the pattern in our patient.
Primary biliary cirrhosis
Primary biliary cirrhosis is diagnosed if the patient has at least two of these three clinical criteria:
Biochemical evidence of cholestasis, with elevation of alkaline phosphatase for at least 6 months
Antimitochondrial antibody
Histologic evidence of nonsuppurative cholangitis and destruction of small or medium-sized bile ducts.11
In patients who lack antimitochondrial antibody, liver biopsy is necessary to establish the diagnosis. Given that primary biliary cirrhosis involves only small and medium-sized bile ducts, cholangiography is usually normal unless the patient has advanced cirrhosis.
Idiopathic adulthood ductopenia
Idiopathic adulthood ductopenia is a rare condition of unknown cause that involves the progressive destruction of segments of the small bile ducts inside the liver (“small-duct” biliary disease).12 Laboratory findings reveal a cholestatic pattern of liver injury, but biopsy samples show no features diagnostic or suggestive of another biliary disease; cholangiography is typically normal.12,13
ASSOCIATION WITH INFLAMMATORY BOWEL DISEASE
2. Which statement best characterizes inflammatory bowel disease associated with primary sclerosing cholangitis?
Crohn disease of the small bowel is the most common form
Liver disease often precedes the bowel disease
Treating the underlying bowel disease improves the long-term prognosis for the liver condition
Patients with primary sclerosing cholangitis and chronic ulcerative colitis are at higher risk of colonic dysplasia than patients with chronic ulcerative colitis alone
From 70% to 80% of patients with primary sclerosing cholangitis also have inflammatory bowel disease, usually chronic ulcerative colitis.14,15 Conversely, 2.4% to 4% of patients with ulcerative colitis and 1.4% to 3.4% of patients with Crohn disease have primary sclerosing cholangitis.1
Typically, the diagnosis of inflammatory bowel disease is made 8 to 10 years before the diagnosis of liver disease, although cases have also been reported to occur years after the diagnosis of cholangitis.15,16
No association between the severity of bowel disease and liver disease has been reported, and treating the inflammatory bowel disease does not alter the natural history of primary sclerosing cholangitis. Particularly, proctocolectomy, the most aggressive treatment for chronic ulcerative colitis, appears to have no effect on the course of the cholangitis.17
In patients with both primary sclerosing cholangitis and chronic ulcerative colitis, the risk of colonic dysplasia is higher than in patients with chronic ulcerative colitis alone.18 Recent studies have predicted that the risk of colorectal carcinoma in patients with primary sclerosing cholangitis and inflammatory bowel disease is as high as 25% after 10 years.19,20 Therefore, annual colonoscopy with surveillance biopsy is recommended in patients with both primary sclerosing cholangitis and chronic ulcerative colitis, since screening and early detection improve survival rates.15
TREATMENT AND PROGNOSIS
After being diagnosed with primary sclerosing cholangitis, the patient inquires about ongoing medical therapy and long-term prognosis.
3.Which is the only life-prolonging therapy for primary sclerosing cholangitis?
Methotrexate (Trexall)
Ursodeoxycholic acid (UDCA) (Actigall) at a standard dosage (13–15 mg/kg/day)
UDCA at a high dosage (20–30 mg/kg/day)
Liver transplantation
Drug therapy has not been shown to improve the prognosis of primary sclerosing cholangitis.
In randomized placebo-controlled trials, penicillamine (Depen), colchicine (Colcrys), methotrexate, and UDCA (13–15 mg/kg per day) failed to show efficacy.21–23
In pilot studies, high-dose UDCA (20 to 30 mg/kg/day) initially appeared to bring an improvement in survival probability, with trends toward histologic improvement,24,25 but larger randomized placebo-controlled trials found no improvement in symptoms, quality of life, survival rates, or risk of cholangiocarcinoma with high-dose UDCA.26,27 In fact, in 5 years of follow-up, patients on high-dose UDCA had a risk of death or transplantation two times higher than with placebo.27 One study indicated UDCA may decrease the incidence of colonic dysplasia in patients with primary sclerosing cholangitis and chronic ulcerative colitis.28 However, more prospective studies are required to better define the routine use of UDCA as a prophylactic agent.
Liver transplantation remains the most effective treatment for primary sclerosing cholangitis, and it improves the rate of survival.29 Nevertheless, about 20% of patients who undergo transplantation have a recurrence of cholangitis, and it may recur earlier after living-donor liver transplantation, particularly when the graft is from a biologically related donor.30 Proposed risk factors for recurrence include inflammatory bowel disease, prolonged ischemia time, the number of cellular rejection events, prior biliary surgery, cytomegalovirus infection, and lymphocytotoxic cross-match.31
4.In addition to cirrhosis and cholangitis, which of the following is a potential long-term complication of primary sclerosing cholangitis?
Colon cancer
Cholangiocarcinoma
Osteoporosis
Fat-soluble vitamin deficiency
All of the above
All are potential long-term complications.
Colon cancer. Concomitant chronic ulcerative colitis puts the patient at a higher risk of colonic dysplasia compared with patients with chronic ulcerative colitis alone.18 According to recent studies of patients with primary sclerosing cholangitis and inflammatory bowel disease, 19,20 the risk of colorectal carcinoma after 10 years of disease is as high as 25%.
Cholangiocarcinoma. Primary sclerosing cholangitis is considered a risk factor for cholangiocarcinoma, with an estimated 10-year cumulative incidence of 7% to 9%.1,20 In a retrospective study of 30 patients,32 the median survival was 5 months from the time of diagnosis of cholangiocarcinoma; at the time of diagnosis approximately 19 patients (63%) had metastatic disease.
At present, early detection of cholangiocarcinoma is hampered by the low sensitivity and specificity of standard diagnostic approaches. Carbohydrate antigen 19-9 has been used as a marker, but it has questionable accuracy, since elevations of this antigen can also be a result of pancreatic malignancy and bacterial cholangitis. However, cholangiocarcinoma should be suspected when patients present with progressive jaundice, weight loss, abdominal discomfort, and a sudden rise in carbohydrate antigen 19-9.
Conventional ultrasonography and computed tomography (CT) have poor sensitivity for detecting this malignancy. ERCP with biliary brushings should be considered when evaluating for biliary malignancy. New diagnostic methods such as digitized image analysis and fluorescence in situ hybridization on biliary brushings offer promise to evaluate bile duct lesions for cellular aneuploidy and chromosomal aberrations, which may improve the detection of cholangiocarcinoma.33 A recent large-scale study of nearly 500 patients showed that fluorescence in situ hybridization had a higher sensitivity (42.9%) than routine cytology (20.1%) with identical specificity (99.6%) for malignancy.34
Metabolic bone disease, usually osteoporosis rather than osteomalacia, is relatively common and is an important complication of primary sclerosing cholangitis.35 Patients with osteoporosis should be treated with vitamin D and calcium supplementation. Bisphosphonates have been used with varying results in primary biliary cirrhosis36 and can be considered in patients with advanced osteoporosis.
Fat-soluble vitamin deficiency is relatively common in primary sclerosing cholangitis, particularly as it progresses to advanced liver disease. Up to 40% of patients have vitamin A deficiency, 14% have vitamin D deficiency, and 2% have vitamin E deficiency.37 Patients can undergo simple oral replacement therapy.
A stone is removed, fever develops
Three years after the diagnosis of primary sclerosing cholangitis, the patient develops mild hyperbilirubinemia and undergoes ERCP at his local hospital. A stone is found obstructing the common bile duct and is successfully extracted.
Twenty-four hours after this procedure, he develops severe right-upper-quadrant pain and fever. He is seen at his local emergency department and blood cultures are drawn. He is started on antibiotics and is transferred to Mayo Clinic for further management.
5.In addition to continuing a broad-spectrum antibiotic, which would be the next best step for this patient?
ERCP
MRCP
Abdominal ultrasonography
Abdominal CT
The patient’s clinical presentation is consistent with acute bacterial cholangitis. The classic Charcot triad of fever, right-upper-quadrant pain, and jaundice occurs in only 50% to 75% of patients with acute cholangitis.38 In addition to receiving a broad-spectrum antibiotic, patients with bacterial cholangitis require emergency endoscopic evaluation—ERCP—to find and remove stones from the bile ducts and, if necessary, to dilate the biliary strictures to allow adequate drainage.
In our experience, more than 10% of patients with primary sclerosing cholangitis who undergo ERCP develop complications requiring hospitalization.39 The procedure generally takes longer to perform and the incidence of cholangitis is higher, despite routine antibiotic prophylaxis, in patients with primary sclerosing cholangitis than in those without it. However, the overall risk of pancreatitis, perforation, and bleeding was similar in patients with or without sclerosing cholangitis.39
MRCP is a promising noninvasive substitute for ERCP in establishing the diagnosis of primary sclerosing cholangitis.7,8 Unfortunately, as with other noninvasive imaging studies such as abdominal ultrasonography and CT, MRCP does not allow for therapeutic biliary decompression.
The patient undergoes ERCP with stenting
The patient’s acute cholangitis is thought to be a complication of his recent ERCP procedure. He undergoes emergency ERCP with balloon dilation and placement of a temporary left hepatic stent. His fever improves and he is discharged 48 hours later. He completes a 14-day course of antibiotics for Enterococcus faecalis bacteremia. Six weeks later, he undergoes ERCP yet again to remove the stent and tolerates the procedure well without complications.
TAKE-HOME POINTS
Primary sclerosing cholangitis is a progressive cholestatic liver disease of unknown etiology that primarily affects men during the fourth decade of life.
This condition is strongly associated with inflammatory bowel disease, particularly with ulcerative colitis.
Cholangiocarcinoma and colon cancer are dreaded complications.
Liver transplantation is the only life-extending therapy for primary sclerosing cholangitis; however, the condition can recur in the allograft.
References
Chapman R, Fevery J, Kalloo A, et al; American Association for the Study of Liver Diseases. Diagnosis and management of primary sclerosing cholangitis. Hepatology2010; 51:660–678.
Silveira MG, Lindor KD. Clinical features and management of primary sclerosing cholangitis. World J Gastroenterol2008; 14:3338–3349.
Lee YM, Kaplan MM. Primary sclerosing cholangitis. N Engl J Med1995; 332:924–933.
Bambha K, Kim WR, Talwalkar J, et al. Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterology2003; 125:1364–1369.
Kingham JG, Kochar N, Gravenor MB. Incidence, clinical patterns, and outcomes of primary sclerosing cholangitis in South Wales, United Kingdom. Gastroenterology2004; 126:1929–1930.
Berstad AE, Aabakken L, Smith HJ, Aasen S, Boberg KM, Schrumpf E. Diagnostic accuracy of magnetic resonance and endoscopic retrograde cholangiography in primary sclerosing cholangitis. Clin Gastroenterol Hepatol2006; 4:514–520.
Talwalkar JA, Angulo P, Johnson CD, Petersen BT, Lindor KD. Cost-minimization analysis of MRC versus ERCP for the diagnosis of primary sclerosing cholangitis. Hepatology2004; 40:39–45.
Ludwig J, Barham SS, LaRusso NF, Elveback LR, Wiesner RH, McCall JT. Morphologic features of chronic hepatitis associated with primary sclerosing cholangitis and chronic ulcerative colitis. Hepatology1981; 1:632–640.
Krawitt EL. Autoimmune hepatitis. N Engl J Med2006; 354:54–66.
Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ; American Association for Study of Liver Diseases. Primary biliary cirrhosis. Hepatology2009; 50:291–308.
Ludwig J, Wiesner RH, LaRusso NF. Idiopathic adulthood ductopenia. A cause of chronic cholestatic liver disease and biliary cirrhosis. J Hepatol1988; 7:193–199.
Ludwig J. Idiopathic adulthood ductopenia: an update. Mayo Clin Proc1998; 73:285–291.
Fausa O, Schrumpf E, Elgjo K. Relationship of inflammatory bowel disease and primary sclerosing cholangitis. Semin Liver Dis1991; 11:31–39.
Loftus EV, Aguilar HI, Sandborn WJ, et al. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis following orthotopic liver transplantation. Hepatology1998; 27:685–690.
Loftus EV, Sandborn WJ, Tremaine WJ, et al. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis. Gastroenterology1996; 110:432–440.
Cangemi JR, Wiesner RH, Beaver SJ, et al. Effect of proctocolectomy for chronic ulcerative colitis on the natural history of primary sclerosing cholangitis. Gastroenterology1989; 96:790–794.
Broomé U, Löfberg R, Veress B, Eriksson LS. Primary sclerosing cholangitis and ulcerative colitis: evidence for increased neoplastic potential. Hepatology1995; 22:1404–1408.
Kornfeld D, Ekbom A, Ihre T. Is there an excess risk for colorectal cancer in patients with ulcerative colitis and concomitant primary sclerosing cholangitis? A population based study. Gut1997; 41:522–525.
Claessen MM, Vleggaar FP, Tytgat KM, Siersema PD, van Buuren HR. High lifetime risk of cancer in primary sclerosing cholangitis. J Hepatol2009; 50:158–164.
Lindor KD. Ursodiol for primary sclerosing cholangitis. Mayo Primary Sclerosing Cholangitis-Ursodeoxycholic Acid Study Group. N Engl J Med1997; 336:691–695.
Olsson R, Broomé U, Danielsson A, et al. Colchicine treatment of primary sclerosing cholangitis. Gastroenterology1995; 108:1199–1203.
LaRusso NF, Wiesner RH, Ludwig J, MacCarty RL, Beaver SJ, Zinsmeister AR. Prospective trial of penicillamine in primary sclerosing cholangitis. Gastroenterology1988; 95:1036–1042.
Mitchell SA, Bansi DS, Hunt N, Von Bergmann K, Fleming KA, Chapman RW. A preliminary trial of high-dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology2001; 121:900–907.
Cullen SN, Rust C, Fleming K, Edwards C, Beuers U, Chapman RW. High dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis is safe and effective. J Hepatol2008; 48:792–800.
Olsson R, Boberg KM, de Muckadell OS, et al. High-dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5-year multicenter, randomized, controlled study. Gastroenterology2005; 129:1464–1472.
Lindor KD, Kowdley KV, Luketic VA, et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology2009; 50:808–814.
Tung BY, Emond MJ, Haggitt RC, et al. Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann Intern Med2001; 134:89–95.
Wiesner RH, Porayko MK, Hay JE, et al. Liver transplantation for primary sclerosing cholangitis: impact of risk factors on outcome. Liver Transpl Surg1996; 2(suppl 1):99–108..
Tamura S, Sugawara Y, Kaneko J, Matsui Y, Togashi J, Makuuchi M. Recurrence of primary sclerosing cholangitis after living donor liver transplantation. Liver Int2007; 27:86–94.
Gautam M, Cheruvattath R, Balan V. Recurrence of autoimmune liver disease after liver transplantation: a systematic review. Liver Transpl2006; 12:1813–1824.
Fritcher EG, Kipp BR, Halling KC, et al. A multivariable model using advanced cytologic methods for the evaluation of indeterminate pancreatobiliary strictures. Gastroenterology2009; 136:2180–2186.
Hay JE, Lindor KD, Wiesner RH, Dickson ER, Krom RA, LaRusso NF. The metabolic bone disease of primary sclerosing cholangitis. Hepatology1991; 14:257–261.
Guañabens N, Parés A, Ros I, et al. Alendronate is more effective than etidronate for increasing bone mass in osteopenic patients with primary biliary cirrhosis. Am J Gastroenterol2003; 98:2268–2274.
Douglas L. Nguyen, MD Resident Physician, Department of Internal Medicine, Mayo Clinic College of Medicine, Rochester, MN
Konstantinos N. Lazaridis, MD Associate Professor of Medicine, Center for Basic Research in Digestive Diseases, Division of Gastroenterology and Hepatology, Mayo Clinic College of Medicine, Rochester, MN
Address: Konstantinos N. Lazaridis, MD, Division of Gastroenterology and Hepatology, Mayo Clinic College of Medicine, 200 First Street SW, Rochester, MN 55905; e-mail lazaridis.konstantinos@mayo.edu
Douglas L. Nguyen, MD Resident Physician, Department of Internal Medicine, Mayo Clinic College of Medicine, Rochester, MN
Konstantinos N. Lazaridis, MD Associate Professor of Medicine, Center for Basic Research in Digestive Diseases, Division of Gastroenterology and Hepatology, Mayo Clinic College of Medicine, Rochester, MN
Address: Konstantinos N. Lazaridis, MD, Division of Gastroenterology and Hepatology, Mayo Clinic College of Medicine, 200 First Street SW, Rochester, MN 55905; e-mail lazaridis.konstantinos@mayo.edu
Author and Disclosure Information
Douglas L. Nguyen, MD Resident Physician, Department of Internal Medicine, Mayo Clinic College of Medicine, Rochester, MN
Konstantinos N. Lazaridis, MD Associate Professor of Medicine, Center for Basic Research in Digestive Diseases, Division of Gastroenterology and Hepatology, Mayo Clinic College of Medicine, Rochester, MN
Address: Konstantinos N. Lazaridis, MD, Division of Gastroenterology and Hepatology, Mayo Clinic College of Medicine, 200 First Street SW, Rochester, MN 55905; e-mail lazaridis.konstantinos@mayo.edu
A 49-year-old man has had ulcerative colitis for more than 30 years. It is well controlled with sulfasalazine (Azulfidine). Now, he has come to see his primary care physician because for the past 3 months he has had mild, intermittent pain in his right upper abdominal quadrant.
His physical examination is normal. Routine laboratory testing shows the following:
Hemoglobin 14.2 g/dL (reference range 13.5–17.5)
White blood cell count 6.7 × 109/L (3.5–10.5)
Platelet count 279 × 109/L (150–450)
Alkaline phosphatase 387 U/L (45–115)
Total bilirubin 0.9 mg/dL (0.1–1.0)
Aspartate aminotransferase (AST) 35 U/L (35–48)
Alanine aminotransferase (ALT) 30 U/L (7–55).
Figure 1. Intraoperative cholangiography demonstrates annular, multifocal stricturing and beading of the extrahepatic biliary system (arrow).
His physician is concerned about his elevated alkaline phosphatase level, which can be a sign of cholestatic liver disease (ie, involving blockage of the flow of bile). He sends him for ultrasonography, which reveals mild thickening of the gallbladder wall. The patient is referred to a general surgeon, who decides to remove the gallbladder. The procedure goes well, but when contrast dye is injected into the biliary system during cholangiography, the image is markedly abnormal (Figure 1). The patient is referred to Mayo Clinic for further evaluation.
WHAT IS THE DIAGNOSIS?
1.Based on this information, which of the following is the most likely diagnosis?
Autoimmune hepatitis
Primary sclerosing cholangitis
Primary biliary cirrhosis
Idiopathic adulthood ductopenia
Primary sclerosing cholangitis
The most likely diagnosis is primary sclerosing cholangitis, a chronic cholestatic liver disease characterized by diffuse inflammatory destruction of intrahepatic and extrahepatic bile ducts, resulting in fibrosis, cirrhosis, and liver failure. Its cause is unknown, but it is likely the result of acquired exposures interacting with predisposing host factors. Current diagnostic criteria include:
Characteristic cholangiographic abnormalities of the biliary tree
Compatible clinical and biochemical findings (typically cholestasis with elevated alkaline phosphatase levels for at least 6 months)
Exclusion of causes of secondary sclerosing cholangitis: secondary sclerosing cholangitis is characterized by a similar multifocal biliary stricturing process, but with an identifiable cause such as long-term biliary obstruction, surgical biliary trauma, or recurrent pancreatitis.1
At presentation, the most common liver enzyme abnormality is an elevated alkaline phosphatase level, often three or four times the normal level.2 In contrast, aminotransferase levels are only modestly elevated, less than three times the upper limit of normal.3 At the time of diagnosis, serum bilirubin levels are normal in 60% of patients.4
Two large epidemiologic studies (one from Olmsted County, MN,5 the other from Swansea, Wales, UK6) estimated the age-adjusted incidence of primary sclerosing cholangitis to be 0.9 per 100,000 individuals. The median age of the patients at onset was in the 30s or 40s, and most were men. At 10 years, an estimated 65% were still alive and had not undergone liver transplantation—a significantly lower percentage than in age- and sex-matched populations.
It is estimated that more than 70% of patients with primary sclerosing cholangitis also have inflammatory bowel disease.5 In fact, the most common presentation of primary sclerosing cholangitis is asymptomatic inflammatory bowel disease and persistently elevated alkaline phosphatase—usually first noted on routine biochemical screening, as in our patient.
Imaging of the biliary tree is essential for the diagnosis of primary sclerosing cholangitis. Typical findings on cholangiography include multifocal stricturing and beading, usually involving both the intrahepatic and the extrahepatic biliary systems, as in our patient (Figure 1). Endoscopic retrograde cholangiopancreatography (ERCP) is considered the gold standard imaging test, but recent studies have shown that magnetic resonance cholangiopancreatography (MRCP) is an acceptable noninvasive substitute,7 and it may cost less per diagnosis.8
Liver biopsy alone is generally nondiagnostic because the histologic changes are quite variable in different segments of the same liver. The classic “onion-skin fibrosis” of primary sclerosing cholangitis is seen in fewer than 10% of biopsy specimens.9
Autoimmune hepatitis
Autoimmune hepatitis is chronic and is characterized by circulating autoantibodies and high serum globulin concentrations.10 Its presentation is heterogeneous, varying from no symptoms to nonspecific symptoms of malaise, fatigue, abdominal pain, itching, and arthralgia. Generally, elevations in aminotransferases are much more prominent than abnormalities in bilirubin and alkaline phosphatase levels10—unlike the pattern in our patient.
Primary biliary cirrhosis
Primary biliary cirrhosis is diagnosed if the patient has at least two of these three clinical criteria:
Biochemical evidence of cholestasis, with elevation of alkaline phosphatase for at least 6 months
Antimitochondrial antibody
Histologic evidence of nonsuppurative cholangitis and destruction of small or medium-sized bile ducts.11
In patients who lack antimitochondrial antibody, liver biopsy is necessary to establish the diagnosis. Given that primary biliary cirrhosis involves only small and medium-sized bile ducts, cholangiography is usually normal unless the patient has advanced cirrhosis.
Idiopathic adulthood ductopenia
Idiopathic adulthood ductopenia is a rare condition of unknown cause that involves the progressive destruction of segments of the small bile ducts inside the liver (“small-duct” biliary disease).12 Laboratory findings reveal a cholestatic pattern of liver injury, but biopsy samples show no features diagnostic or suggestive of another biliary disease; cholangiography is typically normal.12,13
ASSOCIATION WITH INFLAMMATORY BOWEL DISEASE
2. Which statement best characterizes inflammatory bowel disease associated with primary sclerosing cholangitis?
Crohn disease of the small bowel is the most common form
Liver disease often precedes the bowel disease
Treating the underlying bowel disease improves the long-term prognosis for the liver condition
Patients with primary sclerosing cholangitis and chronic ulcerative colitis are at higher risk of colonic dysplasia than patients with chronic ulcerative colitis alone
From 70% to 80% of patients with primary sclerosing cholangitis also have inflammatory bowel disease, usually chronic ulcerative colitis.14,15 Conversely, 2.4% to 4% of patients with ulcerative colitis and 1.4% to 3.4% of patients with Crohn disease have primary sclerosing cholangitis.1
Typically, the diagnosis of inflammatory bowel disease is made 8 to 10 years before the diagnosis of liver disease, although cases have also been reported to occur years after the diagnosis of cholangitis.15,16
No association between the severity of bowel disease and liver disease has been reported, and treating the inflammatory bowel disease does not alter the natural history of primary sclerosing cholangitis. Particularly, proctocolectomy, the most aggressive treatment for chronic ulcerative colitis, appears to have no effect on the course of the cholangitis.17
In patients with both primary sclerosing cholangitis and chronic ulcerative colitis, the risk of colonic dysplasia is higher than in patients with chronic ulcerative colitis alone.18 Recent studies have predicted that the risk of colorectal carcinoma in patients with primary sclerosing cholangitis and inflammatory bowel disease is as high as 25% after 10 years.19,20 Therefore, annual colonoscopy with surveillance biopsy is recommended in patients with both primary sclerosing cholangitis and chronic ulcerative colitis, since screening and early detection improve survival rates.15
TREATMENT AND PROGNOSIS
After being diagnosed with primary sclerosing cholangitis, the patient inquires about ongoing medical therapy and long-term prognosis.
3.Which is the only life-prolonging therapy for primary sclerosing cholangitis?
Methotrexate (Trexall)
Ursodeoxycholic acid (UDCA) (Actigall) at a standard dosage (13–15 mg/kg/day)
UDCA at a high dosage (20–30 mg/kg/day)
Liver transplantation
Drug therapy has not been shown to improve the prognosis of primary sclerosing cholangitis.
In randomized placebo-controlled trials, penicillamine (Depen), colchicine (Colcrys), methotrexate, and UDCA (13–15 mg/kg per day) failed to show efficacy.21–23
In pilot studies, high-dose UDCA (20 to 30 mg/kg/day) initially appeared to bring an improvement in survival probability, with trends toward histologic improvement,24,25 but larger randomized placebo-controlled trials found no improvement in symptoms, quality of life, survival rates, or risk of cholangiocarcinoma with high-dose UDCA.26,27 In fact, in 5 years of follow-up, patients on high-dose UDCA had a risk of death or transplantation two times higher than with placebo.27 One study indicated UDCA may decrease the incidence of colonic dysplasia in patients with primary sclerosing cholangitis and chronic ulcerative colitis.28 However, more prospective studies are required to better define the routine use of UDCA as a prophylactic agent.
Liver transplantation remains the most effective treatment for primary sclerosing cholangitis, and it improves the rate of survival.29 Nevertheless, about 20% of patients who undergo transplantation have a recurrence of cholangitis, and it may recur earlier after living-donor liver transplantation, particularly when the graft is from a biologically related donor.30 Proposed risk factors for recurrence include inflammatory bowel disease, prolonged ischemia time, the number of cellular rejection events, prior biliary surgery, cytomegalovirus infection, and lymphocytotoxic cross-match.31
4.In addition to cirrhosis and cholangitis, which of the following is a potential long-term complication of primary sclerosing cholangitis?
Colon cancer
Cholangiocarcinoma
Osteoporosis
Fat-soluble vitamin deficiency
All of the above
All are potential long-term complications.
Colon cancer. Concomitant chronic ulcerative colitis puts the patient at a higher risk of colonic dysplasia compared with patients with chronic ulcerative colitis alone.18 According to recent studies of patients with primary sclerosing cholangitis and inflammatory bowel disease, 19,20 the risk of colorectal carcinoma after 10 years of disease is as high as 25%.
Cholangiocarcinoma. Primary sclerosing cholangitis is considered a risk factor for cholangiocarcinoma, with an estimated 10-year cumulative incidence of 7% to 9%.1,20 In a retrospective study of 30 patients,32 the median survival was 5 months from the time of diagnosis of cholangiocarcinoma; at the time of diagnosis approximately 19 patients (63%) had metastatic disease.
At present, early detection of cholangiocarcinoma is hampered by the low sensitivity and specificity of standard diagnostic approaches. Carbohydrate antigen 19-9 has been used as a marker, but it has questionable accuracy, since elevations of this antigen can also be a result of pancreatic malignancy and bacterial cholangitis. However, cholangiocarcinoma should be suspected when patients present with progressive jaundice, weight loss, abdominal discomfort, and a sudden rise in carbohydrate antigen 19-9.
Conventional ultrasonography and computed tomography (CT) have poor sensitivity for detecting this malignancy. ERCP with biliary brushings should be considered when evaluating for biliary malignancy. New diagnostic methods such as digitized image analysis and fluorescence in situ hybridization on biliary brushings offer promise to evaluate bile duct lesions for cellular aneuploidy and chromosomal aberrations, which may improve the detection of cholangiocarcinoma.33 A recent large-scale study of nearly 500 patients showed that fluorescence in situ hybridization had a higher sensitivity (42.9%) than routine cytology (20.1%) with identical specificity (99.6%) for malignancy.34
Metabolic bone disease, usually osteoporosis rather than osteomalacia, is relatively common and is an important complication of primary sclerosing cholangitis.35 Patients with osteoporosis should be treated with vitamin D and calcium supplementation. Bisphosphonates have been used with varying results in primary biliary cirrhosis36 and can be considered in patients with advanced osteoporosis.
Fat-soluble vitamin deficiency is relatively common in primary sclerosing cholangitis, particularly as it progresses to advanced liver disease. Up to 40% of patients have vitamin A deficiency, 14% have vitamin D deficiency, and 2% have vitamin E deficiency.37 Patients can undergo simple oral replacement therapy.
A stone is removed, fever develops
Three years after the diagnosis of primary sclerosing cholangitis, the patient develops mild hyperbilirubinemia and undergoes ERCP at his local hospital. A stone is found obstructing the common bile duct and is successfully extracted.
Twenty-four hours after this procedure, he develops severe right-upper-quadrant pain and fever. He is seen at his local emergency department and blood cultures are drawn. He is started on antibiotics and is transferred to Mayo Clinic for further management.
5.In addition to continuing a broad-spectrum antibiotic, which would be the next best step for this patient?
ERCP
MRCP
Abdominal ultrasonography
Abdominal CT
The patient’s clinical presentation is consistent with acute bacterial cholangitis. The classic Charcot triad of fever, right-upper-quadrant pain, and jaundice occurs in only 50% to 75% of patients with acute cholangitis.38 In addition to receiving a broad-spectrum antibiotic, patients with bacterial cholangitis require emergency endoscopic evaluation—ERCP—to find and remove stones from the bile ducts and, if necessary, to dilate the biliary strictures to allow adequate drainage.
In our experience, more than 10% of patients with primary sclerosing cholangitis who undergo ERCP develop complications requiring hospitalization.39 The procedure generally takes longer to perform and the incidence of cholangitis is higher, despite routine antibiotic prophylaxis, in patients with primary sclerosing cholangitis than in those without it. However, the overall risk of pancreatitis, perforation, and bleeding was similar in patients with or without sclerosing cholangitis.39
MRCP is a promising noninvasive substitute for ERCP in establishing the diagnosis of primary sclerosing cholangitis.7,8 Unfortunately, as with other noninvasive imaging studies such as abdominal ultrasonography and CT, MRCP does not allow for therapeutic biliary decompression.
The patient undergoes ERCP with stenting
The patient’s acute cholangitis is thought to be a complication of his recent ERCP procedure. He undergoes emergency ERCP with balloon dilation and placement of a temporary left hepatic stent. His fever improves and he is discharged 48 hours later. He completes a 14-day course of antibiotics for Enterococcus faecalis bacteremia. Six weeks later, he undergoes ERCP yet again to remove the stent and tolerates the procedure well without complications.
TAKE-HOME POINTS
Primary sclerosing cholangitis is a progressive cholestatic liver disease of unknown etiology that primarily affects men during the fourth decade of life.
This condition is strongly associated with inflammatory bowel disease, particularly with ulcerative colitis.
Cholangiocarcinoma and colon cancer are dreaded complications.
Liver transplantation is the only life-extending therapy for primary sclerosing cholangitis; however, the condition can recur in the allograft.
A 49-year-old man has had ulcerative colitis for more than 30 years. It is well controlled with sulfasalazine (Azulfidine). Now, he has come to see his primary care physician because for the past 3 months he has had mild, intermittent pain in his right upper abdominal quadrant.
His physical examination is normal. Routine laboratory testing shows the following:
Hemoglobin 14.2 g/dL (reference range 13.5–17.5)
White blood cell count 6.7 × 109/L (3.5–10.5)
Platelet count 279 × 109/L (150–450)
Alkaline phosphatase 387 U/L (45–115)
Total bilirubin 0.9 mg/dL (0.1–1.0)
Aspartate aminotransferase (AST) 35 U/L (35–48)
Alanine aminotransferase (ALT) 30 U/L (7–55).
Figure 1. Intraoperative cholangiography demonstrates annular, multifocal stricturing and beading of the extrahepatic biliary system (arrow).
His physician is concerned about his elevated alkaline phosphatase level, which can be a sign of cholestatic liver disease (ie, involving blockage of the flow of bile). He sends him for ultrasonography, which reveals mild thickening of the gallbladder wall. The patient is referred to a general surgeon, who decides to remove the gallbladder. The procedure goes well, but when contrast dye is injected into the biliary system during cholangiography, the image is markedly abnormal (Figure 1). The patient is referred to Mayo Clinic for further evaluation.
WHAT IS THE DIAGNOSIS?
1.Based on this information, which of the following is the most likely diagnosis?
Autoimmune hepatitis
Primary sclerosing cholangitis
Primary biliary cirrhosis
Idiopathic adulthood ductopenia
Primary sclerosing cholangitis
The most likely diagnosis is primary sclerosing cholangitis, a chronic cholestatic liver disease characterized by diffuse inflammatory destruction of intrahepatic and extrahepatic bile ducts, resulting in fibrosis, cirrhosis, and liver failure. Its cause is unknown, but it is likely the result of acquired exposures interacting with predisposing host factors. Current diagnostic criteria include:
Characteristic cholangiographic abnormalities of the biliary tree
Compatible clinical and biochemical findings (typically cholestasis with elevated alkaline phosphatase levels for at least 6 months)
Exclusion of causes of secondary sclerosing cholangitis: secondary sclerosing cholangitis is characterized by a similar multifocal biliary stricturing process, but with an identifiable cause such as long-term biliary obstruction, surgical biliary trauma, or recurrent pancreatitis.1
At presentation, the most common liver enzyme abnormality is an elevated alkaline phosphatase level, often three or four times the normal level.2 In contrast, aminotransferase levels are only modestly elevated, less than three times the upper limit of normal.3 At the time of diagnosis, serum bilirubin levels are normal in 60% of patients.4
Two large epidemiologic studies (one from Olmsted County, MN,5 the other from Swansea, Wales, UK6) estimated the age-adjusted incidence of primary sclerosing cholangitis to be 0.9 per 100,000 individuals. The median age of the patients at onset was in the 30s or 40s, and most were men. At 10 years, an estimated 65% were still alive and had not undergone liver transplantation—a significantly lower percentage than in age- and sex-matched populations.
It is estimated that more than 70% of patients with primary sclerosing cholangitis also have inflammatory bowel disease.5 In fact, the most common presentation of primary sclerosing cholangitis is asymptomatic inflammatory bowel disease and persistently elevated alkaline phosphatase—usually first noted on routine biochemical screening, as in our patient.
Imaging of the biliary tree is essential for the diagnosis of primary sclerosing cholangitis. Typical findings on cholangiography include multifocal stricturing and beading, usually involving both the intrahepatic and the extrahepatic biliary systems, as in our patient (Figure 1). Endoscopic retrograde cholangiopancreatography (ERCP) is considered the gold standard imaging test, but recent studies have shown that magnetic resonance cholangiopancreatography (MRCP) is an acceptable noninvasive substitute,7 and it may cost less per diagnosis.8
Liver biopsy alone is generally nondiagnostic because the histologic changes are quite variable in different segments of the same liver. The classic “onion-skin fibrosis” of primary sclerosing cholangitis is seen in fewer than 10% of biopsy specimens.9
Autoimmune hepatitis
Autoimmune hepatitis is chronic and is characterized by circulating autoantibodies and high serum globulin concentrations.10 Its presentation is heterogeneous, varying from no symptoms to nonspecific symptoms of malaise, fatigue, abdominal pain, itching, and arthralgia. Generally, elevations in aminotransferases are much more prominent than abnormalities in bilirubin and alkaline phosphatase levels10—unlike the pattern in our patient.
Primary biliary cirrhosis
Primary biliary cirrhosis is diagnosed if the patient has at least two of these three clinical criteria:
Biochemical evidence of cholestasis, with elevation of alkaline phosphatase for at least 6 months
Antimitochondrial antibody
Histologic evidence of nonsuppurative cholangitis and destruction of small or medium-sized bile ducts.11
In patients who lack antimitochondrial antibody, liver biopsy is necessary to establish the diagnosis. Given that primary biliary cirrhosis involves only small and medium-sized bile ducts, cholangiography is usually normal unless the patient has advanced cirrhosis.
Idiopathic adulthood ductopenia
Idiopathic adulthood ductopenia is a rare condition of unknown cause that involves the progressive destruction of segments of the small bile ducts inside the liver (“small-duct” biliary disease).12 Laboratory findings reveal a cholestatic pattern of liver injury, but biopsy samples show no features diagnostic or suggestive of another biliary disease; cholangiography is typically normal.12,13
ASSOCIATION WITH INFLAMMATORY BOWEL DISEASE
2. Which statement best characterizes inflammatory bowel disease associated with primary sclerosing cholangitis?
Crohn disease of the small bowel is the most common form
Liver disease often precedes the bowel disease
Treating the underlying bowel disease improves the long-term prognosis for the liver condition
Patients with primary sclerosing cholangitis and chronic ulcerative colitis are at higher risk of colonic dysplasia than patients with chronic ulcerative colitis alone
From 70% to 80% of patients with primary sclerosing cholangitis also have inflammatory bowel disease, usually chronic ulcerative colitis.14,15 Conversely, 2.4% to 4% of patients with ulcerative colitis and 1.4% to 3.4% of patients with Crohn disease have primary sclerosing cholangitis.1
Typically, the diagnosis of inflammatory bowel disease is made 8 to 10 years before the diagnosis of liver disease, although cases have also been reported to occur years after the diagnosis of cholangitis.15,16
No association between the severity of bowel disease and liver disease has been reported, and treating the inflammatory bowel disease does not alter the natural history of primary sclerosing cholangitis. Particularly, proctocolectomy, the most aggressive treatment for chronic ulcerative colitis, appears to have no effect on the course of the cholangitis.17
In patients with both primary sclerosing cholangitis and chronic ulcerative colitis, the risk of colonic dysplasia is higher than in patients with chronic ulcerative colitis alone.18 Recent studies have predicted that the risk of colorectal carcinoma in patients with primary sclerosing cholangitis and inflammatory bowel disease is as high as 25% after 10 years.19,20 Therefore, annual colonoscopy with surveillance biopsy is recommended in patients with both primary sclerosing cholangitis and chronic ulcerative colitis, since screening and early detection improve survival rates.15
TREATMENT AND PROGNOSIS
After being diagnosed with primary sclerosing cholangitis, the patient inquires about ongoing medical therapy and long-term prognosis.
3.Which is the only life-prolonging therapy for primary sclerosing cholangitis?
Methotrexate (Trexall)
Ursodeoxycholic acid (UDCA) (Actigall) at a standard dosage (13–15 mg/kg/day)
UDCA at a high dosage (20–30 mg/kg/day)
Liver transplantation
Drug therapy has not been shown to improve the prognosis of primary sclerosing cholangitis.
In randomized placebo-controlled trials, penicillamine (Depen), colchicine (Colcrys), methotrexate, and UDCA (13–15 mg/kg per day) failed to show efficacy.21–23
In pilot studies, high-dose UDCA (20 to 30 mg/kg/day) initially appeared to bring an improvement in survival probability, with trends toward histologic improvement,24,25 but larger randomized placebo-controlled trials found no improvement in symptoms, quality of life, survival rates, or risk of cholangiocarcinoma with high-dose UDCA.26,27 In fact, in 5 years of follow-up, patients on high-dose UDCA had a risk of death or transplantation two times higher than with placebo.27 One study indicated UDCA may decrease the incidence of colonic dysplasia in patients with primary sclerosing cholangitis and chronic ulcerative colitis.28 However, more prospective studies are required to better define the routine use of UDCA as a prophylactic agent.
Liver transplantation remains the most effective treatment for primary sclerosing cholangitis, and it improves the rate of survival.29 Nevertheless, about 20% of patients who undergo transplantation have a recurrence of cholangitis, and it may recur earlier after living-donor liver transplantation, particularly when the graft is from a biologically related donor.30 Proposed risk factors for recurrence include inflammatory bowel disease, prolonged ischemia time, the number of cellular rejection events, prior biliary surgery, cytomegalovirus infection, and lymphocytotoxic cross-match.31
4.In addition to cirrhosis and cholangitis, which of the following is a potential long-term complication of primary sclerosing cholangitis?
Colon cancer
Cholangiocarcinoma
Osteoporosis
Fat-soluble vitamin deficiency
All of the above
All are potential long-term complications.
Colon cancer. Concomitant chronic ulcerative colitis puts the patient at a higher risk of colonic dysplasia compared with patients with chronic ulcerative colitis alone.18 According to recent studies of patients with primary sclerosing cholangitis and inflammatory bowel disease, 19,20 the risk of colorectal carcinoma after 10 years of disease is as high as 25%.
Cholangiocarcinoma. Primary sclerosing cholangitis is considered a risk factor for cholangiocarcinoma, with an estimated 10-year cumulative incidence of 7% to 9%.1,20 In a retrospective study of 30 patients,32 the median survival was 5 months from the time of diagnosis of cholangiocarcinoma; at the time of diagnosis approximately 19 patients (63%) had metastatic disease.
At present, early detection of cholangiocarcinoma is hampered by the low sensitivity and specificity of standard diagnostic approaches. Carbohydrate antigen 19-9 has been used as a marker, but it has questionable accuracy, since elevations of this antigen can also be a result of pancreatic malignancy and bacterial cholangitis. However, cholangiocarcinoma should be suspected when patients present with progressive jaundice, weight loss, abdominal discomfort, and a sudden rise in carbohydrate antigen 19-9.
Conventional ultrasonography and computed tomography (CT) have poor sensitivity for detecting this malignancy. ERCP with biliary brushings should be considered when evaluating for biliary malignancy. New diagnostic methods such as digitized image analysis and fluorescence in situ hybridization on biliary brushings offer promise to evaluate bile duct lesions for cellular aneuploidy and chromosomal aberrations, which may improve the detection of cholangiocarcinoma.33 A recent large-scale study of nearly 500 patients showed that fluorescence in situ hybridization had a higher sensitivity (42.9%) than routine cytology (20.1%) with identical specificity (99.6%) for malignancy.34
Metabolic bone disease, usually osteoporosis rather than osteomalacia, is relatively common and is an important complication of primary sclerosing cholangitis.35 Patients with osteoporosis should be treated with vitamin D and calcium supplementation. Bisphosphonates have been used with varying results in primary biliary cirrhosis36 and can be considered in patients with advanced osteoporosis.
Fat-soluble vitamin deficiency is relatively common in primary sclerosing cholangitis, particularly as it progresses to advanced liver disease. Up to 40% of patients have vitamin A deficiency, 14% have vitamin D deficiency, and 2% have vitamin E deficiency.37 Patients can undergo simple oral replacement therapy.
A stone is removed, fever develops
Three years after the diagnosis of primary sclerosing cholangitis, the patient develops mild hyperbilirubinemia and undergoes ERCP at his local hospital. A stone is found obstructing the common bile duct and is successfully extracted.
Twenty-four hours after this procedure, he develops severe right-upper-quadrant pain and fever. He is seen at his local emergency department and blood cultures are drawn. He is started on antibiotics and is transferred to Mayo Clinic for further management.
5.In addition to continuing a broad-spectrum antibiotic, which would be the next best step for this patient?
ERCP
MRCP
Abdominal ultrasonography
Abdominal CT
The patient’s clinical presentation is consistent with acute bacterial cholangitis. The classic Charcot triad of fever, right-upper-quadrant pain, and jaundice occurs in only 50% to 75% of patients with acute cholangitis.38 In addition to receiving a broad-spectrum antibiotic, patients with bacterial cholangitis require emergency endoscopic evaluation—ERCP—to find and remove stones from the bile ducts and, if necessary, to dilate the biliary strictures to allow adequate drainage.
In our experience, more than 10% of patients with primary sclerosing cholangitis who undergo ERCP develop complications requiring hospitalization.39 The procedure generally takes longer to perform and the incidence of cholangitis is higher, despite routine antibiotic prophylaxis, in patients with primary sclerosing cholangitis than in those without it. However, the overall risk of pancreatitis, perforation, and bleeding was similar in patients with or without sclerosing cholangitis.39
MRCP is a promising noninvasive substitute for ERCP in establishing the diagnosis of primary sclerosing cholangitis.7,8 Unfortunately, as with other noninvasive imaging studies such as abdominal ultrasonography and CT, MRCP does not allow for therapeutic biliary decompression.
The patient undergoes ERCP with stenting
The patient’s acute cholangitis is thought to be a complication of his recent ERCP procedure. He undergoes emergency ERCP with balloon dilation and placement of a temporary left hepatic stent. His fever improves and he is discharged 48 hours later. He completes a 14-day course of antibiotics for Enterococcus faecalis bacteremia. Six weeks later, he undergoes ERCP yet again to remove the stent and tolerates the procedure well without complications.
TAKE-HOME POINTS
Primary sclerosing cholangitis is a progressive cholestatic liver disease of unknown etiology that primarily affects men during the fourth decade of life.
This condition is strongly associated with inflammatory bowel disease, particularly with ulcerative colitis.
Cholangiocarcinoma and colon cancer are dreaded complications.
Liver transplantation is the only life-extending therapy for primary sclerosing cholangitis; however, the condition can recur in the allograft.
References
Chapman R, Fevery J, Kalloo A, et al; American Association for the Study of Liver Diseases. Diagnosis and management of primary sclerosing cholangitis. Hepatology2010; 51:660–678.
Silveira MG, Lindor KD. Clinical features and management of primary sclerosing cholangitis. World J Gastroenterol2008; 14:3338–3349.
Lee YM, Kaplan MM. Primary sclerosing cholangitis. N Engl J Med1995; 332:924–933.
Bambha K, Kim WR, Talwalkar J, et al. Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterology2003; 125:1364–1369.
Kingham JG, Kochar N, Gravenor MB. Incidence, clinical patterns, and outcomes of primary sclerosing cholangitis in South Wales, United Kingdom. Gastroenterology2004; 126:1929–1930.
Berstad AE, Aabakken L, Smith HJ, Aasen S, Boberg KM, Schrumpf E. Diagnostic accuracy of magnetic resonance and endoscopic retrograde cholangiography in primary sclerosing cholangitis. Clin Gastroenterol Hepatol2006; 4:514–520.
Talwalkar JA, Angulo P, Johnson CD, Petersen BT, Lindor KD. Cost-minimization analysis of MRC versus ERCP for the diagnosis of primary sclerosing cholangitis. Hepatology2004; 40:39–45.
Ludwig J, Barham SS, LaRusso NF, Elveback LR, Wiesner RH, McCall JT. Morphologic features of chronic hepatitis associated with primary sclerosing cholangitis and chronic ulcerative colitis. Hepatology1981; 1:632–640.
Krawitt EL. Autoimmune hepatitis. N Engl J Med2006; 354:54–66.
Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ; American Association for Study of Liver Diseases. Primary biliary cirrhosis. Hepatology2009; 50:291–308.
Ludwig J, Wiesner RH, LaRusso NF. Idiopathic adulthood ductopenia. A cause of chronic cholestatic liver disease and biliary cirrhosis. J Hepatol1988; 7:193–199.
Ludwig J. Idiopathic adulthood ductopenia: an update. Mayo Clin Proc1998; 73:285–291.
Fausa O, Schrumpf E, Elgjo K. Relationship of inflammatory bowel disease and primary sclerosing cholangitis. Semin Liver Dis1991; 11:31–39.
Loftus EV, Aguilar HI, Sandborn WJ, et al. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis following orthotopic liver transplantation. Hepatology1998; 27:685–690.
Loftus EV, Sandborn WJ, Tremaine WJ, et al. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis. Gastroenterology1996; 110:432–440.
Cangemi JR, Wiesner RH, Beaver SJ, et al. Effect of proctocolectomy for chronic ulcerative colitis on the natural history of primary sclerosing cholangitis. Gastroenterology1989; 96:790–794.
Broomé U, Löfberg R, Veress B, Eriksson LS. Primary sclerosing cholangitis and ulcerative colitis: evidence for increased neoplastic potential. Hepatology1995; 22:1404–1408.
Kornfeld D, Ekbom A, Ihre T. Is there an excess risk for colorectal cancer in patients with ulcerative colitis and concomitant primary sclerosing cholangitis? A population based study. Gut1997; 41:522–525.
Claessen MM, Vleggaar FP, Tytgat KM, Siersema PD, van Buuren HR. High lifetime risk of cancer in primary sclerosing cholangitis. J Hepatol2009; 50:158–164.
Lindor KD. Ursodiol for primary sclerosing cholangitis. Mayo Primary Sclerosing Cholangitis-Ursodeoxycholic Acid Study Group. N Engl J Med1997; 336:691–695.
Olsson R, Broomé U, Danielsson A, et al. Colchicine treatment of primary sclerosing cholangitis. Gastroenterology1995; 108:1199–1203.
LaRusso NF, Wiesner RH, Ludwig J, MacCarty RL, Beaver SJ, Zinsmeister AR. Prospective trial of penicillamine in primary sclerosing cholangitis. Gastroenterology1988; 95:1036–1042.
Mitchell SA, Bansi DS, Hunt N, Von Bergmann K, Fleming KA, Chapman RW. A preliminary trial of high-dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology2001; 121:900–907.
Cullen SN, Rust C, Fleming K, Edwards C, Beuers U, Chapman RW. High dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis is safe and effective. J Hepatol2008; 48:792–800.
Olsson R, Boberg KM, de Muckadell OS, et al. High-dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5-year multicenter, randomized, controlled study. Gastroenterology2005; 129:1464–1472.
Lindor KD, Kowdley KV, Luketic VA, et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology2009; 50:808–814.
Tung BY, Emond MJ, Haggitt RC, et al. Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann Intern Med2001; 134:89–95.
Wiesner RH, Porayko MK, Hay JE, et al. Liver transplantation for primary sclerosing cholangitis: impact of risk factors on outcome. Liver Transpl Surg1996; 2(suppl 1):99–108..
Tamura S, Sugawara Y, Kaneko J, Matsui Y, Togashi J, Makuuchi M. Recurrence of primary sclerosing cholangitis after living donor liver transplantation. Liver Int2007; 27:86–94.
Gautam M, Cheruvattath R, Balan V. Recurrence of autoimmune liver disease after liver transplantation: a systematic review. Liver Transpl2006; 12:1813–1824.
Fritcher EG, Kipp BR, Halling KC, et al. A multivariable model using advanced cytologic methods for the evaluation of indeterminate pancreatobiliary strictures. Gastroenterology2009; 136:2180–2186.
Hay JE, Lindor KD, Wiesner RH, Dickson ER, Krom RA, LaRusso NF. The metabolic bone disease of primary sclerosing cholangitis. Hepatology1991; 14:257–261.
Guañabens N, Parés A, Ros I, et al. Alendronate is more effective than etidronate for increasing bone mass in osteopenic patients with primary biliary cirrhosis. Am J Gastroenterol2003; 98:2268–2274.
Saik RP, Greenburg AG, Farris JM, Peskin GW. Spectrum of cholangitis. Am J Surg1975; 130:143–150.
Bangarulingam SY, Gossard AA, Petersen BT, Ott BJ, Lindor KD. Complications of endoscopic retrograde cholangiopancreatography in primary sclerosing cholangitis. Am J Gastroenterol2009; 104:855–860.
References
Chapman R, Fevery J, Kalloo A, et al; American Association for the Study of Liver Diseases. Diagnosis and management of primary sclerosing cholangitis. Hepatology2010; 51:660–678.
Silveira MG, Lindor KD. Clinical features and management of primary sclerosing cholangitis. World J Gastroenterol2008; 14:3338–3349.
Lee YM, Kaplan MM. Primary sclerosing cholangitis. N Engl J Med1995; 332:924–933.
Bambha K, Kim WR, Talwalkar J, et al. Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterology2003; 125:1364–1369.
Kingham JG, Kochar N, Gravenor MB. Incidence, clinical patterns, and outcomes of primary sclerosing cholangitis in South Wales, United Kingdom. Gastroenterology2004; 126:1929–1930.
Berstad AE, Aabakken L, Smith HJ, Aasen S, Boberg KM, Schrumpf E. Diagnostic accuracy of magnetic resonance and endoscopic retrograde cholangiography in primary sclerosing cholangitis. Clin Gastroenterol Hepatol2006; 4:514–520.
Talwalkar JA, Angulo P, Johnson CD, Petersen BT, Lindor KD. Cost-minimization analysis of MRC versus ERCP for the diagnosis of primary sclerosing cholangitis. Hepatology2004; 40:39–45.
Ludwig J, Barham SS, LaRusso NF, Elveback LR, Wiesner RH, McCall JT. Morphologic features of chronic hepatitis associated with primary sclerosing cholangitis and chronic ulcerative colitis. Hepatology1981; 1:632–640.
Krawitt EL. Autoimmune hepatitis. N Engl J Med2006; 354:54–66.
Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ; American Association for Study of Liver Diseases. Primary biliary cirrhosis. Hepatology2009; 50:291–308.
Ludwig J, Wiesner RH, LaRusso NF. Idiopathic adulthood ductopenia. A cause of chronic cholestatic liver disease and biliary cirrhosis. J Hepatol1988; 7:193–199.
Ludwig J. Idiopathic adulthood ductopenia: an update. Mayo Clin Proc1998; 73:285–291.
Fausa O, Schrumpf E, Elgjo K. Relationship of inflammatory bowel disease and primary sclerosing cholangitis. Semin Liver Dis1991; 11:31–39.
Loftus EV, Aguilar HI, Sandborn WJ, et al. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis following orthotopic liver transplantation. Hepatology1998; 27:685–690.
Loftus EV, Sandborn WJ, Tremaine WJ, et al. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis. Gastroenterology1996; 110:432–440.
Cangemi JR, Wiesner RH, Beaver SJ, et al. Effect of proctocolectomy for chronic ulcerative colitis on the natural history of primary sclerosing cholangitis. Gastroenterology1989; 96:790–794.
Broomé U, Löfberg R, Veress B, Eriksson LS. Primary sclerosing cholangitis and ulcerative colitis: evidence for increased neoplastic potential. Hepatology1995; 22:1404–1408.
Kornfeld D, Ekbom A, Ihre T. Is there an excess risk for colorectal cancer in patients with ulcerative colitis and concomitant primary sclerosing cholangitis? A population based study. Gut1997; 41:522–525.
Claessen MM, Vleggaar FP, Tytgat KM, Siersema PD, van Buuren HR. High lifetime risk of cancer in primary sclerosing cholangitis. J Hepatol2009; 50:158–164.
Lindor KD. Ursodiol for primary sclerosing cholangitis. Mayo Primary Sclerosing Cholangitis-Ursodeoxycholic Acid Study Group. N Engl J Med1997; 336:691–695.
Olsson R, Broomé U, Danielsson A, et al. Colchicine treatment of primary sclerosing cholangitis. Gastroenterology1995; 108:1199–1203.
LaRusso NF, Wiesner RH, Ludwig J, MacCarty RL, Beaver SJ, Zinsmeister AR. Prospective trial of penicillamine in primary sclerosing cholangitis. Gastroenterology1988; 95:1036–1042.
Mitchell SA, Bansi DS, Hunt N, Von Bergmann K, Fleming KA, Chapman RW. A preliminary trial of high-dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology2001; 121:900–907.
Cullen SN, Rust C, Fleming K, Edwards C, Beuers U, Chapman RW. High dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis is safe and effective. J Hepatol2008; 48:792–800.
Olsson R, Boberg KM, de Muckadell OS, et al. High-dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5-year multicenter, randomized, controlled study. Gastroenterology2005; 129:1464–1472.
Lindor KD, Kowdley KV, Luketic VA, et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology2009; 50:808–814.
Tung BY, Emond MJ, Haggitt RC, et al. Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann Intern Med2001; 134:89–95.
Wiesner RH, Porayko MK, Hay JE, et al. Liver transplantation for primary sclerosing cholangitis: impact of risk factors on outcome. Liver Transpl Surg1996; 2(suppl 1):99–108..
Tamura S, Sugawara Y, Kaneko J, Matsui Y, Togashi J, Makuuchi M. Recurrence of primary sclerosing cholangitis after living donor liver transplantation. Liver Int2007; 27:86–94.
Gautam M, Cheruvattath R, Balan V. Recurrence of autoimmune liver disease after liver transplantation: a systematic review. Liver Transpl2006; 12:1813–1824.
Fritcher EG, Kipp BR, Halling KC, et al. A multivariable model using advanced cytologic methods for the evaluation of indeterminate pancreatobiliary strictures. Gastroenterology2009; 136:2180–2186.
Hay JE, Lindor KD, Wiesner RH, Dickson ER, Krom RA, LaRusso NF. The metabolic bone disease of primary sclerosing cholangitis. Hepatology1991; 14:257–261.
Guañabens N, Parés A, Ros I, et al. Alendronate is more effective than etidronate for increasing bone mass in osteopenic patients with primary biliary cirrhosis. Am J Gastroenterol2003; 98:2268–2274.
A 12-year-old boy presents with painless swelling and ulceration on and around his nose that has progressed gradually over the last 6 months. The lesion has increased in size despite treatment with topical neomycin and oral erythromycin. He has no systemic symptoms.
Figure 1. Ulcerated plaque with destruction of the right nasal wing.
On examination (Figure 1), we note an indurated, nontender plaque with scarring at places on his right cheek, nose, and the vermilion border of the lip. In addition, there are two purulent ulcerations on the nose partly destroying the right nasal wing. The upper lip is also infiltrated, studded with a solitary ulceration. There is no regional lymphadenopathy. An examination of systems is normal.
Cutaneous tuberculosis occurs in many forms, and lupus vulgaris is one of the most common.1 Lupus vulgaris usually arises as a result of hematogenous spread from an endogenous source. It may also arise from exogenous inoculation or as a complication of vaccination with bacille Calmette-Guérin.2
Several morphologic variants have been described.1,2 One form is characterized by plaques, often studded with psoriasiform scales. Large plaques may show irregular areas of scarring with islands of active lupus tissue and a thickened and hyperkeratotic margin. Ulcerative and mutilating variants of lupus vulgaris are characterized by scarring, ulceration, crusts over areas of necrosis, and destruction of the deep tissues and cartilage, resulting in deformities. The vegetative form produces marked infiltration, ulceration, and necrosis, with minimal scarring. Mucous membranes and cartilages are often destroyed. Tumor-like hypertrophic lesions and multiple papular and nodular lesions may also be seen. Nasal lesions may start as nodules, which may bleed and then ulcerate, sometimes resulting in cartilage destruction.
CLINICAL FEATURES AND LABORATORY WORKUP CLINCHED THE DIAGNOSIS
A number of factors helped to confirm the diagnosis in this patient:
A strongly positive Mantoux test (22-mm induration at 48 hours)
Acid-fast bacilli on Ziehl-Neelsen staining of the smear taken from the purulent ulceration
Isolation of Mycobacterium tuberculosis from the purulent exudates via culture in Lowenstein-Jensen medium
Figure 2. Epithelioid cell granuloma and giant cells (hematoxylin and eosin, × 100).
A suggestive histopathologic picture (Figure 2)
The features on presentation
A significant clinical improvement within 2 months of starting antituberculosis therapy.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis includes all the conditions in the question above. However, the absence of respiratory and renal involvement helps rule out Wegener granulomatosis; the absence of impaired sensation and nerve thickening helps rule out Hansen disease; and the absence of a nasal septal defect helps rule out Wegener granulomatosis, midline lethal granuloma, and Hansen disease.
On the other hand, the lupoid form of cutaneous leishmaniasis usually presents as an erythematous, infiltrated plaque that often closely resembles lupus vulgaris, but these lesions are usually less destructive than lupus vulgaris. However, the laboratory workup including the microbiological and histopathologic examination clearly excluded the other potential diagnoses in this patient.
TREATMENT
Lupus vulgaris is treated with standard antituberculosis therapy.3 The first phase of a fourdrug regimen is given for 2 months—isoniazid, rifampin (Rifadin), pyrazinamide, and ethambutol (Myambutol). The second phase consists of isoniazid and rifampin for 4 months.3
Early recognition and confirmation of the diagnosis followed by treatment are of immense importance for preventing permanent disfigurement.
References
Freitag DS, Chin R. Facial granulomas with nasal destruction. Chest1988; 93:422–423.
Sudip Kumar Ghosh, MD, DNB Assistant Professor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Debarbrata Bandyopadhyay, MD Professor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Loknath Ghoshal, MD Resident Medical Officer-cum-Clinical Tutor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Address: Sudip Kumar Ghosh, MD, DNB, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, 1, Khudiram Bose Sarani, 700004 Kolkata, India; e-mail dr_skghosh@yahoo.co.in
Sudip Kumar Ghosh, MD, DNB Assistant Professor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Debarbrata Bandyopadhyay, MD Professor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Loknath Ghoshal, MD Resident Medical Officer-cum-Clinical Tutor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Address: Sudip Kumar Ghosh, MD, DNB, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, 1, Khudiram Bose Sarani, 700004 Kolkata, India; e-mail dr_skghosh@yahoo.co.in
Author and Disclosure Information
Sudip Kumar Ghosh, MD, DNB Assistant Professor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Debarbrata Bandyopadhyay, MD Professor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Loknath Ghoshal, MD Resident Medical Officer-cum-Clinical Tutor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Address: Sudip Kumar Ghosh, MD, DNB, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, 1, Khudiram Bose Sarani, 700004 Kolkata, India; e-mail dr_skghosh@yahoo.co.in
A 12-year-old boy presents with painless swelling and ulceration on and around his nose that has progressed gradually over the last 6 months. The lesion has increased in size despite treatment with topical neomycin and oral erythromycin. He has no systemic symptoms.
Figure 1. Ulcerated plaque with destruction of the right nasal wing.
On examination (Figure 1), we note an indurated, nontender plaque with scarring at places on his right cheek, nose, and the vermilion border of the lip. In addition, there are two purulent ulcerations on the nose partly destroying the right nasal wing. The upper lip is also infiltrated, studded with a solitary ulceration. There is no regional lymphadenopathy. An examination of systems is normal.
Cutaneous tuberculosis occurs in many forms, and lupus vulgaris is one of the most common.1 Lupus vulgaris usually arises as a result of hematogenous spread from an endogenous source. It may also arise from exogenous inoculation or as a complication of vaccination with bacille Calmette-Guérin.2
Several morphologic variants have been described.1,2 One form is characterized by plaques, often studded with psoriasiform scales. Large plaques may show irregular areas of scarring with islands of active lupus tissue and a thickened and hyperkeratotic margin. Ulcerative and mutilating variants of lupus vulgaris are characterized by scarring, ulceration, crusts over areas of necrosis, and destruction of the deep tissues and cartilage, resulting in deformities. The vegetative form produces marked infiltration, ulceration, and necrosis, with minimal scarring. Mucous membranes and cartilages are often destroyed. Tumor-like hypertrophic lesions and multiple papular and nodular lesions may also be seen. Nasal lesions may start as nodules, which may bleed and then ulcerate, sometimes resulting in cartilage destruction.
CLINICAL FEATURES AND LABORATORY WORKUP CLINCHED THE DIAGNOSIS
A number of factors helped to confirm the diagnosis in this patient:
A strongly positive Mantoux test (22-mm induration at 48 hours)
Acid-fast bacilli on Ziehl-Neelsen staining of the smear taken from the purulent ulceration
Isolation of Mycobacterium tuberculosis from the purulent exudates via culture in Lowenstein-Jensen medium
Figure 2. Epithelioid cell granuloma and giant cells (hematoxylin and eosin, × 100).
A suggestive histopathologic picture (Figure 2)
The features on presentation
A significant clinical improvement within 2 months of starting antituberculosis therapy.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis includes all the conditions in the question above. However, the absence of respiratory and renal involvement helps rule out Wegener granulomatosis; the absence of impaired sensation and nerve thickening helps rule out Hansen disease; and the absence of a nasal septal defect helps rule out Wegener granulomatosis, midline lethal granuloma, and Hansen disease.
On the other hand, the lupoid form of cutaneous leishmaniasis usually presents as an erythematous, infiltrated plaque that often closely resembles lupus vulgaris, but these lesions are usually less destructive than lupus vulgaris. However, the laboratory workup including the microbiological and histopathologic examination clearly excluded the other potential diagnoses in this patient.
TREATMENT
Lupus vulgaris is treated with standard antituberculosis therapy.3 The first phase of a fourdrug regimen is given for 2 months—isoniazid, rifampin (Rifadin), pyrazinamide, and ethambutol (Myambutol). The second phase consists of isoniazid and rifampin for 4 months.3
Early recognition and confirmation of the diagnosis followed by treatment are of immense importance for preventing permanent disfigurement.
A 12-year-old boy presents with painless swelling and ulceration on and around his nose that has progressed gradually over the last 6 months. The lesion has increased in size despite treatment with topical neomycin and oral erythromycin. He has no systemic symptoms.
Figure 1. Ulcerated plaque with destruction of the right nasal wing.
On examination (Figure 1), we note an indurated, nontender plaque with scarring at places on his right cheek, nose, and the vermilion border of the lip. In addition, there are two purulent ulcerations on the nose partly destroying the right nasal wing. The upper lip is also infiltrated, studded with a solitary ulceration. There is no regional lymphadenopathy. An examination of systems is normal.
Cutaneous tuberculosis occurs in many forms, and lupus vulgaris is one of the most common.1 Lupus vulgaris usually arises as a result of hematogenous spread from an endogenous source. It may also arise from exogenous inoculation or as a complication of vaccination with bacille Calmette-Guérin.2
Several morphologic variants have been described.1,2 One form is characterized by plaques, often studded with psoriasiform scales. Large plaques may show irregular areas of scarring with islands of active lupus tissue and a thickened and hyperkeratotic margin. Ulcerative and mutilating variants of lupus vulgaris are characterized by scarring, ulceration, crusts over areas of necrosis, and destruction of the deep tissues and cartilage, resulting in deformities. The vegetative form produces marked infiltration, ulceration, and necrosis, with minimal scarring. Mucous membranes and cartilages are often destroyed. Tumor-like hypertrophic lesions and multiple papular and nodular lesions may also be seen. Nasal lesions may start as nodules, which may bleed and then ulcerate, sometimes resulting in cartilage destruction.
CLINICAL FEATURES AND LABORATORY WORKUP CLINCHED THE DIAGNOSIS
A number of factors helped to confirm the diagnosis in this patient:
A strongly positive Mantoux test (22-mm induration at 48 hours)
Acid-fast bacilli on Ziehl-Neelsen staining of the smear taken from the purulent ulceration
Isolation of Mycobacterium tuberculosis from the purulent exudates via culture in Lowenstein-Jensen medium
Figure 2. Epithelioid cell granuloma and giant cells (hematoxylin and eosin, × 100).
A suggestive histopathologic picture (Figure 2)
The features on presentation
A significant clinical improvement within 2 months of starting antituberculosis therapy.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis includes all the conditions in the question above. However, the absence of respiratory and renal involvement helps rule out Wegener granulomatosis; the absence of impaired sensation and nerve thickening helps rule out Hansen disease; and the absence of a nasal septal defect helps rule out Wegener granulomatosis, midline lethal granuloma, and Hansen disease.
On the other hand, the lupoid form of cutaneous leishmaniasis usually presents as an erythematous, infiltrated plaque that often closely resembles lupus vulgaris, but these lesions are usually less destructive than lupus vulgaris. However, the laboratory workup including the microbiological and histopathologic examination clearly excluded the other potential diagnoses in this patient.
TREATMENT
Lupus vulgaris is treated with standard antituberculosis therapy.3 The first phase of a fourdrug regimen is given for 2 months—isoniazid, rifampin (Rifadin), pyrazinamide, and ethambutol (Myambutol). The second phase consists of isoniazid and rifampin for 4 months.3
Early recognition and confirmation of the diagnosis followed by treatment are of immense importance for preventing permanent disfigurement.
References
Freitag DS, Chin R. Facial granulomas with nasal destruction. Chest1988; 93:422–423.
Giant cell arteritis is the most common primary systemic vasculitis. The disease occurs almost exclusively in people over age 50, with an annual incidence of 15 to 25 per 100,000.1 Incidence rates vary significantly depending on ethnicity. The highest rates are in whites, particularly those of North European descent.2 Incidence rates progressively increase after age 50. The disease is more prevalent in women. Its cause is unknown; both genetic and environmental factors are thought to play a role.
INFLAMED ARTERIES
Giant cell arteritis is characterized by a granulomatous inflammatory infiltrate affecting large and medium-size arteries. Not all vessels are equally affected: the most susceptible are the cranial arteries, the aorta, and the aorta’s primary branches, particularly those in the upper extremities.
The disease is usually associated with an intense acute-phase response. Vessel wall inflammation results in intimal hyperplasia, luminal occlusion, and tissue ischemia. Typical histologic features include a mononuclear inflammatory infiltrate primarily composed of CD4+ T cells and activated macrophages. Multinucleated giant cells are seen in only about 50% of positive biopsies; therefore, their presence is not essential for the diagnosis.3
FOUR MAIN PHENOTYPES
Some of the possible symptoms of giant cell arteritis readily point to the correct diagnosis, eg, those due to cranial artery involvement, such as temporal headache, claudication of masticatory muscles, and visual changes. However, the clinical presentation can be quite varied.
There are four predominant clinical phenotypes, which may be present at the onset of disease or appear later as the disease progresses. Although they will be described separately in this review, these clinical presentations often overlap.
Cranial arteritis
Cranial arteritis is the clinical presentation most readily associated with giant cell arteritis. Clinical features result from involvement of branches of the external or internal carotid artery.
Headache, the most frequent symptom, is typically but not exclusively localized to the temporal areas.
Visual loss is due to involvement of the branches of the ophthalmic or posterior ciliary arteries, resulting in ischemia of the optic nerve or its tracts. It occurs in up to 20% of patients.4,5
Other symptoms and complications from cranial arteritis include tenderness of the scalp and temporal areas, claudication of the tongue or jaw muscles, stroke, and more rarely, tongue infarction.
Polymyalgia rheumatica
Polymyalgia rheumatica is a clinical syndrome that can occur by itself or in conjunction with giant cell arteritis. It may occur independently of giant cell arteritis, but also occurs in about 40% of patients with giant cell arteritis. It may precede, develop simultaneously with, or develop later during the course of the giant cell arteritis.6,7 It is a common clinical manifestation in relapses of giant cell arteritis, even in those who did not have symptoms of polymyalgia rheumatica at the time giant cell arteritis was diagnosed.
Polymyalgia rheumatica is characterized by aching of the shoulder and hip girdle, with morning stiffness. Fatigue and malaise are often present and may be severe. Some patients with polymyalgia rheumatica may also present with peripheral joint synovitis, which may be mistakenly diagnosed as rheumatoid arthritis.8 Muscle weakness and elevated muscle enzymes are not associated with polymyalgia rheumatica.
Polymyalgia rheumatica is a clinical diagnosis. Approximately 80% of patients with polymyalgia rheumatica have an elevated erythrocyte sedimentation rate or an elevated C-reactive protein level.9 When it occurs in the absence of giant cell arteritis, it is treated differently, with less intense doses of corticosteroids. All patients with polymyalgia rheumatica should be routinely questioned regarding symptoms of giant cell arteritis.
Nonspecific systemic inflammatory disease
Some patients with giant cell arteritis may present with a nonspecific systemic inflammatory disease characterized by some combination of fever, night sweats, fatigue, malaise, and weight loss. In these patients, the diagnosis may be delayed by the lack of localizing symptoms.
Laboratory tests typically show anemia, leukocytosis, and thrombocytosis. The erythrocyte sedimentation rate and the C-reactive protein level are usually very high.
Giant cell arteritis should be in the differential diagnosis when these signs and symptoms are found in patients over age 50.
Large-vessel vasculitis
Although thoracic aortic aneurysm and dissection have been described as late complications of giant cell arteritis, large-vessel vasculitis may precede or occur concomitantly with cranial arteritis early in the disease.10,11
Population-based surveys have shown that large-vessel vasculitis is extremely frequent in patients with giant cell arteritis. In a postmortem study of 11 patients with giant cell arteritis, all of them had evidence of arteritis involving the subclavian artery, the carotid artery, and the aorta.12
Patients may have no symptoms or may present with symptoms or signs of tissue ischemia such as claudication of the extremities, carotid artery tenderness, decreased or absent pulses, and large-vessel bruits on physical examination.
CONSIDER THE DIAGNOSIS IN OLDER PATIENTS
Giant cell arteritis should always be considered in patients over age 50 who have any of the clinical features described above. It is therefore very important to be familiar with its symptoms and signs.
A complete and detailed history and a detailed but focused physical examination that includes a comprehensive vascular examination are the first and most important steps in establishing the diagnosis. The vascular examination includes measuring the blood pressure in all four extremities, palpating the peripheral pulses, listening for bruits, and palpating the temporal arteries.
Temporal artery biopsy: The gold standard
Confirming the diagnosis of giant cell arteritis requires histologic findings of inflammation in the temporal artery. Superficial temporal artery biopsy is recommended for diagnostic confirmation in patients who have cranial symptoms and other signs suggesting the disease.
The biopsy should be performed on the same side as the symptoms or abnormal findings on examination. Performing a biopsy in both temporal arteries may increase the diagnostic yield but may not need to be done routinely.13
Although some experts recommend temporal artery biopsy in all patients in whom giant cell arteritis is suspected, biopsy has a lower diagnostic yield in patients who have no cranial symptoms. Interestingly, 5% to 15% of temporal artery biopsies performed in patients who had isolated polymyalgia rheumatica were found to be positive.14,15 Patients with polymyalgia rheumatica and no clinical symptoms to suggest giant cell arteritis generally are not biopsied.
The segmental nature of the inflammation involving the temporal artery in giant cell arteritis may result in negative biopsy results in patients with giant cell arteritis. A temporal artery biopsy length of 5 mm or less has a very low (8%) rate of positive results, whereas a length longer than 20 mm exceeds a 50% rate of positive results. Although the optimal length of a temporal artery specimen is still debated, a longer biopsy specimen should be obtained to increase the chance of arterial specimens showing inflammatory changes.16,17
Figure 1. Temporal arteritis with intense inflammatory infiltrate within the arterial wall causing intimal thickening with nearly complete occlusion of the arterial lumen (hematoxylin and eosin, × 90).
Typical findings in an inflamed temporal artery (Figure 1) include a lymphocytic infiltrate with activated macrophages and multinucleated giant cells (in 50% of cases). Typical panarteritis is not always seen, and infiltrates limited to the adventitia may be the only histologic finding in some patients.18
Laboratory studies: Acute-phase reactants may be elevated
High levels of acute-phase reactants should increase one’s suspicion of giant cell arteritis. Elevations in the erythrocyte sedimentation rate and C-reactive protein and interleukin 6 levels reflect the inflammatory process in this disease.19 However, not all patients with giant cell arteritis have a high sedimentation rate, and as many as 20% of patients with biopsy-proven giant cell arteritis have a normal sedimentation rate before therapy.20 Therefore, a normal sedimentation rate does not exclude the diagnosis of giant cell arteritis and should not delay its diagnosis and treatment.
As a result of systemic inflammation, the patient may also present with normochromic normocytic anemia, leukocytosis, and thrombocytosis.
Imaging studies are controversial
Imaging studies are potentially useful diagnostic tools in large-vessel vasculitis but are still the subject of significant controversy.
Ultrasonography of the temporal artery has been a controversial subject in many studies.21,22 Color duplex ultrasonography of the temporal artery has been reported to be helpful in the diagnosis of giant cell arteritis (showing a “halo” around the arterial lumen), but further studies are needed to establish its clinical utility.
At this time, temporal artery biopsy remains the gold standard diagnostic test for giant cell arteritis, and ultrasonography is neither a substitute for biopsy nor a screening test for this disease.23 Some have suggested, however, that ultrasonography may help to identify the best site for biopsy of the temporal artery in some patients.
Arteriography is an accurate technique for evaluating the vessel lumen and allows for measuring central blood pressure and performing vascular interventions. However, because of potential complications, it has been largely replaced by noninvasive angiographic imaging to delineate vascular anatomy.
Figure 2. Digital subtraction angiography shows occlusion of the left subclavian artery and the left common carotid artery (black arrow), brachiocephalic dilatation, and post-dilatation stenosis (red arrow).Magnetic resonance angiography and computed angiography. These two noninvasive imaging tests have been used in the diagnosis and serial monitoring of patients with large-vessel involvement from giant cell arteritis (Figure 2). In addition to measuring lumen dimensions, magnetic resonance angiography (edema-weighted images) may also give information on vessel-wall signal intensity that may reflect inflammation. This information may be helpful in serial monitoring of patients with established large-vessel involvement, but it should be interpreted with great caution as it does not always correlate with active inflammation or with new structural changes in the vessel.24,25
TREATMENT
Glucocorticoid therapy remains the standard of care
Once the diagnosis of giant cell arteritis is established, glucocorticoid treatment should be started. Glucocorticoids are the standard therapy, and they usually bring about a prompt clinical response. Although never evaluated in placebo-controlled trials, these drugs have been shown to prevent progression of visual loss in a retrospective study.26
In patients with visual symptoms or imminent visual loss, therapy should be started promptly once suspicion of giant cell arteritis is raised; ie, one should not wait until the diagnosis is confirmed by biopsy.
Ideally, a glucocorticoid should be started after a temporal artery biopsy is done, but treatment should not be delayed, as it rapidly suppresses the inflammatory response and may prevent complications from tissue ischemia, such as blindness. Visual loss is usually irreversible.
There is still a role for obtaining a temporal artery biopsy up to several weeks after glucocorticoid therapy is started, as the pathological abnormalities of arteritis do not rapidly resolve.27
Glucocorticoid therapy is highly effective in inducing disease remission in patients with giant cell arteritis. Nearly all patients respond to 1 mg/kg (40–60 mg) per day of prednisone or its equivalent.
The initial dosing is usually maintained for 4 weeks and then decreased slowly. The duration of therapy varies; most patients remain on therapy for at least 1 year, and some cannot stop it completely without recurrence of symptoms.
If a patient is about to lose his or her vision or has lost all or some vision in one eye, a higher initial dose of a glucocorticoid is usually used (ie, a pulse of 500 or 1,000 mg of intravenous methylprednisolone) and may be beneficial.28
Although a rapid clinical response to therapy is usually seen within 48 hours, some patients may have a more delayed clinical improvement.
Alternate-day therapy was compared with daily therapy and was found to be less effective, and as a result it is not recommended.29
Glucocorticoid therapy can cause significant toxicity in patients with giant cell arteritis, as they commonly must take these drugs for long periods. The rate of relapse in those who discontinue therapy is quite high—as high as 77% within 12 months.30
Given the concern about glucocorticoid toxicity, several studies have evaluated alternative strategies and other immunosuppressive drugs. However, no study has concluded that other medications are effective in the treatment of giant cell arteritis.
Mazlumzadeh et al31 evaluated the initial use of intravenous pulse methylprednisolone therapy (15 mg/kg ideal body weight on 3 consecutive days) in an attempt to decrease the glucocorticoid requirement. Although the group receiving this therapy had a lower relapse rate than in the placebo group, and their cumulative dose of glucocorticoid was lower (all patients also received oral prednisone), there was no reduction in the rate of glucocorticoid-associated toxicity.31 Care must be taken to prevent and monitor for corticosteroid complications such as osteoporosis, glaucoma, diabetes mellitus, and hypertension.
Methotrexate: Mixed results in clinical trials
Methotrexate has been evaluated in three prospective randomized trials,30,32,33 with mixed results.
Spiera et al32 enrolled 21 patients in a double-blind placebo-controlled trial: 12 patients received low-dose methotrexate (7.5 mg/week) and 9 received placebo. In addition, all 21 received a glucocorticoid. There was no significant difference between the methotrexate- and placebo-treated patients in the cumulative dose of glucocorticoid, duration of glucocorticoid therapy, time to taper off the glucocorticoid to less than 10 mg of prednisone per day, or glucocorticoidrelated adverse effects.
Jover et al,33 in another double-blind placebo-controlled trial, studied 42 patients with giant cell arteritis, half of whom were randomized to receive methotrexate 10 mg/week, while the other half received placebo. All patients received prednisone. Patients in the methotrexate group had fewer relapses and a 25% lower cumulative dose of prednisone during follow-up. However, the incidence of adverse events was similar in both groups. Methotrexate was discontinued in 3 patients who developed drug-related adverse events.
Hoffman et al30 randomized 98 patients to receive either methotrexate (up to 15 mg/week) or placebo in a double-blind fashion. All patients also received prednisone at an initial dose of 1 mg/kg/day (up to 60 mg/day). At completion of the study, no differences between the groups were noted in the rates of relapse or treatment-related morbidity or in the cumulative dose of glucocorticoid. However, treatment with methotrexate appeared to lower the rate of recurrence of isolated polymyalgia rheumatica in a small number of patients.30
Comment. Differences in the results of these trials may be attributed to several factors, including different definitions of relapses and different glucocorticoid doses and tapering regimens.
A meta-analysis of these three trials34 showed a reduction in the risk of relapse: 4 patients would have to be treated to prevent one first relapse, 5 would have to be treated to prevent one second relapse, and 11 would need to be treated to prevent one first relapse of cranial symptoms in the first 48 weeks. However, the main goal of methotrexate therapy is to decrease the frequency of adverse events from glucocorticoids, and this meta-analysis found no difference in rates of glucocorticoid-related adverse events in patients treated with methotrexate.
The study raises the question of whether methotrexate should be further evaluated in in different patient populations and at higher doses.34
Infliximab is not recommended
In a prospective study, patients with giant cell arteritis were randomly assigned to receive either infliximab (Remicade) 5 mg/kg every 8 weeks or placebo, in addition to standard glucocorticoid therapy. The study showed no significant difference in the relapse rate and a higher rate of infection in the infliximab group (71%) than in the placebo group (56%). Given the lack of any benefit observed in this study, infliximab is not recommended in the treatment of patients with giant cell arteritis.35
Aspirin is recommended
Daily low-dose aspirin therapy has been shown in several studies to be effective in preventing ischemic complications of giant cell arteritis, including stroke and visual loss. It is currently recommended that all patients with giant cell arteritis without a major contraindication take aspirin 81 mg daily.36–38
References
Salvarani C, Gabriel SE, O’Fallon WM, Hunder GG. The incidence of giant cell arteritis in Olmsted County, Minnesota: apparent fluctuations in a cyclic pattern. Ann Intern Med1995; 123:192–194.
Baldursson O, Steinsson K, Björnsson J, Lie JT. Giant cell arteritis in Iceland. An epidemiologic and histopathologic analysis. Arthritis Rheum1994; 37:1007–1012.
Weyand CM, Goronzy JJ. Medium- and large-vessel vasculitis. N Engl J Med2003; 349:160–169.
Salvarani C, Cimino L, Macchioni P, et al. Risk factors for visual loss in an Italian population-based cohort of patients with giant cell arteritis. Arthritis Rheum2005; 53:293–297.
Bahlas S, Ramos-Remus C, Davis P. Clinical outcome of 149 patients with polymyalgia rheumatica and giant cell arteritis. J Rheumatol1998; 25:99–104.
Gonzalez-Gay MA, Barros S, Lopez-Diaz MJ, Garcia-Porrua C, Sanchez-Andrade A, Llorca J. Giant cell arteritis: disease patterns of clinical presentation in a series of 240 patients. Medicine (Baltimore)2005; 84:269–276.
Salvarani C, Cantini F, Macchioni P, et al. Distal musculoskeletal manifestations in polymyalgia rheumatica: a prospective followup study. Arthritis Rheum1998; 41:1221–1226.
Salvarani C, Cantini F, Boiardi L, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med2002; 347:261–271.
Lie JT. Aortic and extracranial large vessel giant cell arteritis: a review of 72 cases with histopathologic documentation. Semin Arthritis Rheum1995; 24:422–431.
Evans JM, O’Fallon WM, Hunder GG. Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis. A population-based study. Ann Intern Med1995; 122:502–507.
Ostberg G. An arteritis with special reference to polymyalgia arteritica. Acta Pathol Microbiol Scand Suppl1973; 237(suppl 237):1–59.
Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol1999; 128:211–215.
González-Gay MA, Garcia-Porrua C, Rivas MJ, Rodriguez-Ledo P, Llorca J. Epidemiology of biopsy proven giant cell arteritis in northwestern Spain: trend over an 18 year period. Ann Rheum Dis2001; 60:367–371.
Rodriguez-Valverde V, Sarabia JM, González-Gay MA, et al. Risk factors and predictive models of giant cell arteritis in polymyalgia rheumatica. Am J Med1997; 102:331–336.
Mahr A, Saba M, Kambouchner M, et al. Temporal artery biopsy for diagnosing giant cell arteritis: the longer, the better?Ann Rheum Dis2006; 65:826–828.
Breuer GS, Nesher R, Nesher G. Effect of biopsy length on the rate of positive temporal artery biopsies. Clin Exp Rheumatol2009; 27(1 suppl 52):S10–S13.
Weyand CM, Goronzy JJ. Giant-cell arteritis and polymyalgia rheumatica. Ann Intern Med2003; 139:505–515.
Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurence in a population-based study. Arthritis Rheum2001; 45:140–145.
Schmidt WA, Kraft HE, Vorpahl K, Völker L, Gromnica-Ihle EJ. Color duplex ultrasonography in the diagnosis of temporal arteritis. N Engl J Med1997; 337:1336–1342.
Karassa FB, Matsagas MI, Schmidt WA, Ioannidis JP. Meta-analysis: test performance of ultrasonography for giant-cell arteritis. Ann Intern Med2005; 142:359–369.
Maldini C, Dépinay-Dhellemmes C, Tra TT, et al. Limited value of temporal artery ultrasonography examinations for diagnosis of giant cell arteritis: analysis of 77 subjects. J Rheumatol2010; Epub ahead of print.
Both M, Ahmadi-Simab K, Reuter M, et al. MRI and FDG-PET in the assessment of inflammatory aortic arch syndrome in complicated courses of giant cell arteritis. Ann Rheum Dis2008; 67:1030–1033.
Tso E, Flamm SD, White RD, Schvartzman PR, Mascha E, Hoffman GS. Takayasu arteritis: utility and limitations of magnetic resonance imaging in diagnosis and treatment. Arthritis Rheum2002; 46:1634–1642.
Birkhead NC, Wagener HP, Shick RM. Treatment of temporal arteritis with adrenal corticosteroids; results in fifty-five cases in which lesion was proved at biopsy. J Am Med Assoc1957; 163:821–827.
Ray-Chaudhuri N, Kiné DA, Tijani SO, et al. Effect of prior steroid treatment on temporal artery biopsy findings in giant cell arteritis. Br J Ophthalmol2002; 86:530–532.
Chan CC, Paine M, O’Day J. Steroid management in giant cell arteritis. Br J Ophthalmol2001; 85:1061–1064.
Hunder GG, Sheps SG, Allen GL, Joyce JW. Daily and alternate-day corticosteroid regimens in treatment of giant cell arteritis: comparison in a prospective study. Ann Intern Med1975; 82:613–618.
Hoffman GS, Cid MC, Hellmann DB, et al; International Network for the Study of Systemic Vasculitides. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum2002; 46:1309–1318.
Mazlumzadeh M, Hunder GG, Easley KA, et al. Treatment of giant cell arteritis using induction therapy with high-dose glucocorticoids: a double-blind, placebo-controlled, randomized prospective clinical trial. Arthritis Rheum2006; 54:3310–3318.
Spiera RF, Mitnick HJ, Kupersmith M, et al. A prospective, doubleblind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA). Clin Exp Rheumatol2001; 19:495–501.
Jover JA, Hernández-García C, Morado IC, Vargas E, Bañares A, Fernández-Gutiérrez B. Combined treatment of giant-cell arteritis with methotrexate and prednisone. a randomized, double-blind, placebo-controlled trial. Ann Intern Med2001; 134:106–114.
Mahr AD, Jover JA, Spiera RF, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum2007; 56:2789–2797.
Hoffman GS, Cid MC, Rendt-Zagar KE, et al; Infliximab-GCA Study Group. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med2007; 146:621–630.
Weyand CM, Kaiser M, Yang H, Younge B, Goronzy JJ. Therapeutic effects of acetylsalicylic acid in giant cell arteritis. Arthritis Rheum2002; 46:457–466.
Nesher G, Berkun Y, Mates M, Baras M, Rubinow A, Sonnenblick M. Low-dose aspirin and prevention of cranial ischemic complications in giant cell arteritis. Arthritis Rheum2004; 50:1332–1337.
Lee MS, Smith SD, Galor A, Hoffman GS. Antiplatelet and anticoagulant therapy in patients with giant cell arteritis. Arthritis Rheum2006; 54:3306–3309.
Giant cell arteritis is the most common primary systemic vasculitis. The disease occurs almost exclusively in people over age 50, with an annual incidence of 15 to 25 per 100,000.1 Incidence rates vary significantly depending on ethnicity. The highest rates are in whites, particularly those of North European descent.2 Incidence rates progressively increase after age 50. The disease is more prevalent in women. Its cause is unknown; both genetic and environmental factors are thought to play a role.
INFLAMED ARTERIES
Giant cell arteritis is characterized by a granulomatous inflammatory infiltrate affecting large and medium-size arteries. Not all vessels are equally affected: the most susceptible are the cranial arteries, the aorta, and the aorta’s primary branches, particularly those in the upper extremities.
The disease is usually associated with an intense acute-phase response. Vessel wall inflammation results in intimal hyperplasia, luminal occlusion, and tissue ischemia. Typical histologic features include a mononuclear inflammatory infiltrate primarily composed of CD4+ T cells and activated macrophages. Multinucleated giant cells are seen in only about 50% of positive biopsies; therefore, their presence is not essential for the diagnosis.3
FOUR MAIN PHENOTYPES
Some of the possible symptoms of giant cell arteritis readily point to the correct diagnosis, eg, those due to cranial artery involvement, such as temporal headache, claudication of masticatory muscles, and visual changes. However, the clinical presentation can be quite varied.
There are four predominant clinical phenotypes, which may be present at the onset of disease or appear later as the disease progresses. Although they will be described separately in this review, these clinical presentations often overlap.
Cranial arteritis
Cranial arteritis is the clinical presentation most readily associated with giant cell arteritis. Clinical features result from involvement of branches of the external or internal carotid artery.
Headache, the most frequent symptom, is typically but not exclusively localized to the temporal areas.
Visual loss is due to involvement of the branches of the ophthalmic or posterior ciliary arteries, resulting in ischemia of the optic nerve or its tracts. It occurs in up to 20% of patients.4,5
Other symptoms and complications from cranial arteritis include tenderness of the scalp and temporal areas, claudication of the tongue or jaw muscles, stroke, and more rarely, tongue infarction.
Polymyalgia rheumatica
Polymyalgia rheumatica is a clinical syndrome that can occur by itself or in conjunction with giant cell arteritis. It may occur independently of giant cell arteritis, but also occurs in about 40% of patients with giant cell arteritis. It may precede, develop simultaneously with, or develop later during the course of the giant cell arteritis.6,7 It is a common clinical manifestation in relapses of giant cell arteritis, even in those who did not have symptoms of polymyalgia rheumatica at the time giant cell arteritis was diagnosed.
Polymyalgia rheumatica is characterized by aching of the shoulder and hip girdle, with morning stiffness. Fatigue and malaise are often present and may be severe. Some patients with polymyalgia rheumatica may also present with peripheral joint synovitis, which may be mistakenly diagnosed as rheumatoid arthritis.8 Muscle weakness and elevated muscle enzymes are not associated with polymyalgia rheumatica.
Polymyalgia rheumatica is a clinical diagnosis. Approximately 80% of patients with polymyalgia rheumatica have an elevated erythrocyte sedimentation rate or an elevated C-reactive protein level.9 When it occurs in the absence of giant cell arteritis, it is treated differently, with less intense doses of corticosteroids. All patients with polymyalgia rheumatica should be routinely questioned regarding symptoms of giant cell arteritis.
Nonspecific systemic inflammatory disease
Some patients with giant cell arteritis may present with a nonspecific systemic inflammatory disease characterized by some combination of fever, night sweats, fatigue, malaise, and weight loss. In these patients, the diagnosis may be delayed by the lack of localizing symptoms.
Laboratory tests typically show anemia, leukocytosis, and thrombocytosis. The erythrocyte sedimentation rate and the C-reactive protein level are usually very high.
Giant cell arteritis should be in the differential diagnosis when these signs and symptoms are found in patients over age 50.
Large-vessel vasculitis
Although thoracic aortic aneurysm and dissection have been described as late complications of giant cell arteritis, large-vessel vasculitis may precede or occur concomitantly with cranial arteritis early in the disease.10,11
Population-based surveys have shown that large-vessel vasculitis is extremely frequent in patients with giant cell arteritis. In a postmortem study of 11 patients with giant cell arteritis, all of them had evidence of arteritis involving the subclavian artery, the carotid artery, and the aorta.12
Patients may have no symptoms or may present with symptoms or signs of tissue ischemia such as claudication of the extremities, carotid artery tenderness, decreased or absent pulses, and large-vessel bruits on physical examination.
CONSIDER THE DIAGNOSIS IN OLDER PATIENTS
Giant cell arteritis should always be considered in patients over age 50 who have any of the clinical features described above. It is therefore very important to be familiar with its symptoms and signs.
A complete and detailed history and a detailed but focused physical examination that includes a comprehensive vascular examination are the first and most important steps in establishing the diagnosis. The vascular examination includes measuring the blood pressure in all four extremities, palpating the peripheral pulses, listening for bruits, and palpating the temporal arteries.
Temporal artery biopsy: The gold standard
Confirming the diagnosis of giant cell arteritis requires histologic findings of inflammation in the temporal artery. Superficial temporal artery biopsy is recommended for diagnostic confirmation in patients who have cranial symptoms and other signs suggesting the disease.
The biopsy should be performed on the same side as the symptoms or abnormal findings on examination. Performing a biopsy in both temporal arteries may increase the diagnostic yield but may not need to be done routinely.13
Although some experts recommend temporal artery biopsy in all patients in whom giant cell arteritis is suspected, biopsy has a lower diagnostic yield in patients who have no cranial symptoms. Interestingly, 5% to 15% of temporal artery biopsies performed in patients who had isolated polymyalgia rheumatica were found to be positive.14,15 Patients with polymyalgia rheumatica and no clinical symptoms to suggest giant cell arteritis generally are not biopsied.
The segmental nature of the inflammation involving the temporal artery in giant cell arteritis may result in negative biopsy results in patients with giant cell arteritis. A temporal artery biopsy length of 5 mm or less has a very low (8%) rate of positive results, whereas a length longer than 20 mm exceeds a 50% rate of positive results. Although the optimal length of a temporal artery specimen is still debated, a longer biopsy specimen should be obtained to increase the chance of arterial specimens showing inflammatory changes.16,17
Figure 1. Temporal arteritis with intense inflammatory infiltrate within the arterial wall causing intimal thickening with nearly complete occlusion of the arterial lumen (hematoxylin and eosin, × 90).
Typical findings in an inflamed temporal artery (Figure 1) include a lymphocytic infiltrate with activated macrophages and multinucleated giant cells (in 50% of cases). Typical panarteritis is not always seen, and infiltrates limited to the adventitia may be the only histologic finding in some patients.18
Laboratory studies: Acute-phase reactants may be elevated
High levels of acute-phase reactants should increase one’s suspicion of giant cell arteritis. Elevations in the erythrocyte sedimentation rate and C-reactive protein and interleukin 6 levels reflect the inflammatory process in this disease.19 However, not all patients with giant cell arteritis have a high sedimentation rate, and as many as 20% of patients with biopsy-proven giant cell arteritis have a normal sedimentation rate before therapy.20 Therefore, a normal sedimentation rate does not exclude the diagnosis of giant cell arteritis and should not delay its diagnosis and treatment.
As a result of systemic inflammation, the patient may also present with normochromic normocytic anemia, leukocytosis, and thrombocytosis.
Imaging studies are controversial
Imaging studies are potentially useful diagnostic tools in large-vessel vasculitis but are still the subject of significant controversy.
Ultrasonography of the temporal artery has been a controversial subject in many studies.21,22 Color duplex ultrasonography of the temporal artery has been reported to be helpful in the diagnosis of giant cell arteritis (showing a “halo” around the arterial lumen), but further studies are needed to establish its clinical utility.
At this time, temporal artery biopsy remains the gold standard diagnostic test for giant cell arteritis, and ultrasonography is neither a substitute for biopsy nor a screening test for this disease.23 Some have suggested, however, that ultrasonography may help to identify the best site for biopsy of the temporal artery in some patients.
Arteriography is an accurate technique for evaluating the vessel lumen and allows for measuring central blood pressure and performing vascular interventions. However, because of potential complications, it has been largely replaced by noninvasive angiographic imaging to delineate vascular anatomy.
Figure 2. Digital subtraction angiography shows occlusion of the left subclavian artery and the left common carotid artery (black arrow), brachiocephalic dilatation, and post-dilatation stenosis (red arrow).Magnetic resonance angiography and computed angiography. These two noninvasive imaging tests have been used in the diagnosis and serial monitoring of patients with large-vessel involvement from giant cell arteritis (Figure 2). In addition to measuring lumen dimensions, magnetic resonance angiography (edema-weighted images) may also give information on vessel-wall signal intensity that may reflect inflammation. This information may be helpful in serial monitoring of patients with established large-vessel involvement, but it should be interpreted with great caution as it does not always correlate with active inflammation or with new structural changes in the vessel.24,25
TREATMENT
Glucocorticoid therapy remains the standard of care
Once the diagnosis of giant cell arteritis is established, glucocorticoid treatment should be started. Glucocorticoids are the standard therapy, and they usually bring about a prompt clinical response. Although never evaluated in placebo-controlled trials, these drugs have been shown to prevent progression of visual loss in a retrospective study.26
In patients with visual symptoms or imminent visual loss, therapy should be started promptly once suspicion of giant cell arteritis is raised; ie, one should not wait until the diagnosis is confirmed by biopsy.
Ideally, a glucocorticoid should be started after a temporal artery biopsy is done, but treatment should not be delayed, as it rapidly suppresses the inflammatory response and may prevent complications from tissue ischemia, such as blindness. Visual loss is usually irreversible.
There is still a role for obtaining a temporal artery biopsy up to several weeks after glucocorticoid therapy is started, as the pathological abnormalities of arteritis do not rapidly resolve.27
Glucocorticoid therapy is highly effective in inducing disease remission in patients with giant cell arteritis. Nearly all patients respond to 1 mg/kg (40–60 mg) per day of prednisone or its equivalent.
The initial dosing is usually maintained for 4 weeks and then decreased slowly. The duration of therapy varies; most patients remain on therapy for at least 1 year, and some cannot stop it completely without recurrence of symptoms.
If a patient is about to lose his or her vision or has lost all or some vision in one eye, a higher initial dose of a glucocorticoid is usually used (ie, a pulse of 500 or 1,000 mg of intravenous methylprednisolone) and may be beneficial.28
Although a rapid clinical response to therapy is usually seen within 48 hours, some patients may have a more delayed clinical improvement.
Alternate-day therapy was compared with daily therapy and was found to be less effective, and as a result it is not recommended.29
Glucocorticoid therapy can cause significant toxicity in patients with giant cell arteritis, as they commonly must take these drugs for long periods. The rate of relapse in those who discontinue therapy is quite high—as high as 77% within 12 months.30
Given the concern about glucocorticoid toxicity, several studies have evaluated alternative strategies and other immunosuppressive drugs. However, no study has concluded that other medications are effective in the treatment of giant cell arteritis.
Mazlumzadeh et al31 evaluated the initial use of intravenous pulse methylprednisolone therapy (15 mg/kg ideal body weight on 3 consecutive days) in an attempt to decrease the glucocorticoid requirement. Although the group receiving this therapy had a lower relapse rate than in the placebo group, and their cumulative dose of glucocorticoid was lower (all patients also received oral prednisone), there was no reduction in the rate of glucocorticoid-associated toxicity.31 Care must be taken to prevent and monitor for corticosteroid complications such as osteoporosis, glaucoma, diabetes mellitus, and hypertension.
Methotrexate: Mixed results in clinical trials
Methotrexate has been evaluated in three prospective randomized trials,30,32,33 with mixed results.
Spiera et al32 enrolled 21 patients in a double-blind placebo-controlled trial: 12 patients received low-dose methotrexate (7.5 mg/week) and 9 received placebo. In addition, all 21 received a glucocorticoid. There was no significant difference between the methotrexate- and placebo-treated patients in the cumulative dose of glucocorticoid, duration of glucocorticoid therapy, time to taper off the glucocorticoid to less than 10 mg of prednisone per day, or glucocorticoidrelated adverse effects.
Jover et al,33 in another double-blind placebo-controlled trial, studied 42 patients with giant cell arteritis, half of whom were randomized to receive methotrexate 10 mg/week, while the other half received placebo. All patients received prednisone. Patients in the methotrexate group had fewer relapses and a 25% lower cumulative dose of prednisone during follow-up. However, the incidence of adverse events was similar in both groups. Methotrexate was discontinued in 3 patients who developed drug-related adverse events.
Hoffman et al30 randomized 98 patients to receive either methotrexate (up to 15 mg/week) or placebo in a double-blind fashion. All patients also received prednisone at an initial dose of 1 mg/kg/day (up to 60 mg/day). At completion of the study, no differences between the groups were noted in the rates of relapse or treatment-related morbidity or in the cumulative dose of glucocorticoid. However, treatment with methotrexate appeared to lower the rate of recurrence of isolated polymyalgia rheumatica in a small number of patients.30
Comment. Differences in the results of these trials may be attributed to several factors, including different definitions of relapses and different glucocorticoid doses and tapering regimens.
A meta-analysis of these three trials34 showed a reduction in the risk of relapse: 4 patients would have to be treated to prevent one first relapse, 5 would have to be treated to prevent one second relapse, and 11 would need to be treated to prevent one first relapse of cranial symptoms in the first 48 weeks. However, the main goal of methotrexate therapy is to decrease the frequency of adverse events from glucocorticoids, and this meta-analysis found no difference in rates of glucocorticoid-related adverse events in patients treated with methotrexate.
The study raises the question of whether methotrexate should be further evaluated in in different patient populations and at higher doses.34
Infliximab is not recommended
In a prospective study, patients with giant cell arteritis were randomly assigned to receive either infliximab (Remicade) 5 mg/kg every 8 weeks or placebo, in addition to standard glucocorticoid therapy. The study showed no significant difference in the relapse rate and a higher rate of infection in the infliximab group (71%) than in the placebo group (56%). Given the lack of any benefit observed in this study, infliximab is not recommended in the treatment of patients with giant cell arteritis.35
Aspirin is recommended
Daily low-dose aspirin therapy has been shown in several studies to be effective in preventing ischemic complications of giant cell arteritis, including stroke and visual loss. It is currently recommended that all patients with giant cell arteritis without a major contraindication take aspirin 81 mg daily.36–38
Giant cell arteritis is the most common primary systemic vasculitis. The disease occurs almost exclusively in people over age 50, with an annual incidence of 15 to 25 per 100,000.1 Incidence rates vary significantly depending on ethnicity. The highest rates are in whites, particularly those of North European descent.2 Incidence rates progressively increase after age 50. The disease is more prevalent in women. Its cause is unknown; both genetic and environmental factors are thought to play a role.
INFLAMED ARTERIES
Giant cell arteritis is characterized by a granulomatous inflammatory infiltrate affecting large and medium-size arteries. Not all vessels are equally affected: the most susceptible are the cranial arteries, the aorta, and the aorta’s primary branches, particularly those in the upper extremities.
The disease is usually associated with an intense acute-phase response. Vessel wall inflammation results in intimal hyperplasia, luminal occlusion, and tissue ischemia. Typical histologic features include a mononuclear inflammatory infiltrate primarily composed of CD4+ T cells and activated macrophages. Multinucleated giant cells are seen in only about 50% of positive biopsies; therefore, their presence is not essential for the diagnosis.3
FOUR MAIN PHENOTYPES
Some of the possible symptoms of giant cell arteritis readily point to the correct diagnosis, eg, those due to cranial artery involvement, such as temporal headache, claudication of masticatory muscles, and visual changes. However, the clinical presentation can be quite varied.
There are four predominant clinical phenotypes, which may be present at the onset of disease or appear later as the disease progresses. Although they will be described separately in this review, these clinical presentations often overlap.
Cranial arteritis
Cranial arteritis is the clinical presentation most readily associated with giant cell arteritis. Clinical features result from involvement of branches of the external or internal carotid artery.
Headache, the most frequent symptom, is typically but not exclusively localized to the temporal areas.
Visual loss is due to involvement of the branches of the ophthalmic or posterior ciliary arteries, resulting in ischemia of the optic nerve or its tracts. It occurs in up to 20% of patients.4,5
Other symptoms and complications from cranial arteritis include tenderness of the scalp and temporal areas, claudication of the tongue or jaw muscles, stroke, and more rarely, tongue infarction.
Polymyalgia rheumatica
Polymyalgia rheumatica is a clinical syndrome that can occur by itself or in conjunction with giant cell arteritis. It may occur independently of giant cell arteritis, but also occurs in about 40% of patients with giant cell arteritis. It may precede, develop simultaneously with, or develop later during the course of the giant cell arteritis.6,7 It is a common clinical manifestation in relapses of giant cell arteritis, even in those who did not have symptoms of polymyalgia rheumatica at the time giant cell arteritis was diagnosed.
Polymyalgia rheumatica is characterized by aching of the shoulder and hip girdle, with morning stiffness. Fatigue and malaise are often present and may be severe. Some patients with polymyalgia rheumatica may also present with peripheral joint synovitis, which may be mistakenly diagnosed as rheumatoid arthritis.8 Muscle weakness and elevated muscle enzymes are not associated with polymyalgia rheumatica.
Polymyalgia rheumatica is a clinical diagnosis. Approximately 80% of patients with polymyalgia rheumatica have an elevated erythrocyte sedimentation rate or an elevated C-reactive protein level.9 When it occurs in the absence of giant cell arteritis, it is treated differently, with less intense doses of corticosteroids. All patients with polymyalgia rheumatica should be routinely questioned regarding symptoms of giant cell arteritis.
Nonspecific systemic inflammatory disease
Some patients with giant cell arteritis may present with a nonspecific systemic inflammatory disease characterized by some combination of fever, night sweats, fatigue, malaise, and weight loss. In these patients, the diagnosis may be delayed by the lack of localizing symptoms.
Laboratory tests typically show anemia, leukocytosis, and thrombocytosis. The erythrocyte sedimentation rate and the C-reactive protein level are usually very high.
Giant cell arteritis should be in the differential diagnosis when these signs and symptoms are found in patients over age 50.
Large-vessel vasculitis
Although thoracic aortic aneurysm and dissection have been described as late complications of giant cell arteritis, large-vessel vasculitis may precede or occur concomitantly with cranial arteritis early in the disease.10,11
Population-based surveys have shown that large-vessel vasculitis is extremely frequent in patients with giant cell arteritis. In a postmortem study of 11 patients with giant cell arteritis, all of them had evidence of arteritis involving the subclavian artery, the carotid artery, and the aorta.12
Patients may have no symptoms or may present with symptoms or signs of tissue ischemia such as claudication of the extremities, carotid artery tenderness, decreased or absent pulses, and large-vessel bruits on physical examination.
CONSIDER THE DIAGNOSIS IN OLDER PATIENTS
Giant cell arteritis should always be considered in patients over age 50 who have any of the clinical features described above. It is therefore very important to be familiar with its symptoms and signs.
A complete and detailed history and a detailed but focused physical examination that includes a comprehensive vascular examination are the first and most important steps in establishing the diagnosis. The vascular examination includes measuring the blood pressure in all four extremities, palpating the peripheral pulses, listening for bruits, and palpating the temporal arteries.
Temporal artery biopsy: The gold standard
Confirming the diagnosis of giant cell arteritis requires histologic findings of inflammation in the temporal artery. Superficial temporal artery biopsy is recommended for diagnostic confirmation in patients who have cranial symptoms and other signs suggesting the disease.
The biopsy should be performed on the same side as the symptoms or abnormal findings on examination. Performing a biopsy in both temporal arteries may increase the diagnostic yield but may not need to be done routinely.13
Although some experts recommend temporal artery biopsy in all patients in whom giant cell arteritis is suspected, biopsy has a lower diagnostic yield in patients who have no cranial symptoms. Interestingly, 5% to 15% of temporal artery biopsies performed in patients who had isolated polymyalgia rheumatica were found to be positive.14,15 Patients with polymyalgia rheumatica and no clinical symptoms to suggest giant cell arteritis generally are not biopsied.
The segmental nature of the inflammation involving the temporal artery in giant cell arteritis may result in negative biopsy results in patients with giant cell arteritis. A temporal artery biopsy length of 5 mm or less has a very low (8%) rate of positive results, whereas a length longer than 20 mm exceeds a 50% rate of positive results. Although the optimal length of a temporal artery specimen is still debated, a longer biopsy specimen should be obtained to increase the chance of arterial specimens showing inflammatory changes.16,17
Figure 1. Temporal arteritis with intense inflammatory infiltrate within the arterial wall causing intimal thickening with nearly complete occlusion of the arterial lumen (hematoxylin and eosin, × 90).
Typical findings in an inflamed temporal artery (Figure 1) include a lymphocytic infiltrate with activated macrophages and multinucleated giant cells (in 50% of cases). Typical panarteritis is not always seen, and infiltrates limited to the adventitia may be the only histologic finding in some patients.18
Laboratory studies: Acute-phase reactants may be elevated
High levels of acute-phase reactants should increase one’s suspicion of giant cell arteritis. Elevations in the erythrocyte sedimentation rate and C-reactive protein and interleukin 6 levels reflect the inflammatory process in this disease.19 However, not all patients with giant cell arteritis have a high sedimentation rate, and as many as 20% of patients with biopsy-proven giant cell arteritis have a normal sedimentation rate before therapy.20 Therefore, a normal sedimentation rate does not exclude the diagnosis of giant cell arteritis and should not delay its diagnosis and treatment.
As a result of systemic inflammation, the patient may also present with normochromic normocytic anemia, leukocytosis, and thrombocytosis.
Imaging studies are controversial
Imaging studies are potentially useful diagnostic tools in large-vessel vasculitis but are still the subject of significant controversy.
Ultrasonography of the temporal artery has been a controversial subject in many studies.21,22 Color duplex ultrasonography of the temporal artery has been reported to be helpful in the diagnosis of giant cell arteritis (showing a “halo” around the arterial lumen), but further studies are needed to establish its clinical utility.
At this time, temporal artery biopsy remains the gold standard diagnostic test for giant cell arteritis, and ultrasonography is neither a substitute for biopsy nor a screening test for this disease.23 Some have suggested, however, that ultrasonography may help to identify the best site for biopsy of the temporal artery in some patients.
Arteriography is an accurate technique for evaluating the vessel lumen and allows for measuring central blood pressure and performing vascular interventions. However, because of potential complications, it has been largely replaced by noninvasive angiographic imaging to delineate vascular anatomy.
Figure 2. Digital subtraction angiography shows occlusion of the left subclavian artery and the left common carotid artery (black arrow), brachiocephalic dilatation, and post-dilatation stenosis (red arrow).Magnetic resonance angiography and computed angiography. These two noninvasive imaging tests have been used in the diagnosis and serial monitoring of patients with large-vessel involvement from giant cell arteritis (Figure 2). In addition to measuring lumen dimensions, magnetic resonance angiography (edema-weighted images) may also give information on vessel-wall signal intensity that may reflect inflammation. This information may be helpful in serial monitoring of patients with established large-vessel involvement, but it should be interpreted with great caution as it does not always correlate with active inflammation or with new structural changes in the vessel.24,25
TREATMENT
Glucocorticoid therapy remains the standard of care
Once the diagnosis of giant cell arteritis is established, glucocorticoid treatment should be started. Glucocorticoids are the standard therapy, and they usually bring about a prompt clinical response. Although never evaluated in placebo-controlled trials, these drugs have been shown to prevent progression of visual loss in a retrospective study.26
In patients with visual symptoms or imminent visual loss, therapy should be started promptly once suspicion of giant cell arteritis is raised; ie, one should not wait until the diagnosis is confirmed by biopsy.
Ideally, a glucocorticoid should be started after a temporal artery biopsy is done, but treatment should not be delayed, as it rapidly suppresses the inflammatory response and may prevent complications from tissue ischemia, such as blindness. Visual loss is usually irreversible.
There is still a role for obtaining a temporal artery biopsy up to several weeks after glucocorticoid therapy is started, as the pathological abnormalities of arteritis do not rapidly resolve.27
Glucocorticoid therapy is highly effective in inducing disease remission in patients with giant cell arteritis. Nearly all patients respond to 1 mg/kg (40–60 mg) per day of prednisone or its equivalent.
The initial dosing is usually maintained for 4 weeks and then decreased slowly. The duration of therapy varies; most patients remain on therapy for at least 1 year, and some cannot stop it completely without recurrence of symptoms.
If a patient is about to lose his or her vision or has lost all or some vision in one eye, a higher initial dose of a glucocorticoid is usually used (ie, a pulse of 500 or 1,000 mg of intravenous methylprednisolone) and may be beneficial.28
Although a rapid clinical response to therapy is usually seen within 48 hours, some patients may have a more delayed clinical improvement.
Alternate-day therapy was compared with daily therapy and was found to be less effective, and as a result it is not recommended.29
Glucocorticoid therapy can cause significant toxicity in patients with giant cell arteritis, as they commonly must take these drugs for long periods. The rate of relapse in those who discontinue therapy is quite high—as high as 77% within 12 months.30
Given the concern about glucocorticoid toxicity, several studies have evaluated alternative strategies and other immunosuppressive drugs. However, no study has concluded that other medications are effective in the treatment of giant cell arteritis.
Mazlumzadeh et al31 evaluated the initial use of intravenous pulse methylprednisolone therapy (15 mg/kg ideal body weight on 3 consecutive days) in an attempt to decrease the glucocorticoid requirement. Although the group receiving this therapy had a lower relapse rate than in the placebo group, and their cumulative dose of glucocorticoid was lower (all patients also received oral prednisone), there was no reduction in the rate of glucocorticoid-associated toxicity.31 Care must be taken to prevent and monitor for corticosteroid complications such as osteoporosis, glaucoma, diabetes mellitus, and hypertension.
Methotrexate: Mixed results in clinical trials
Methotrexate has been evaluated in three prospective randomized trials,30,32,33 with mixed results.
Spiera et al32 enrolled 21 patients in a double-blind placebo-controlled trial: 12 patients received low-dose methotrexate (7.5 mg/week) and 9 received placebo. In addition, all 21 received a glucocorticoid. There was no significant difference between the methotrexate- and placebo-treated patients in the cumulative dose of glucocorticoid, duration of glucocorticoid therapy, time to taper off the glucocorticoid to less than 10 mg of prednisone per day, or glucocorticoidrelated adverse effects.
Jover et al,33 in another double-blind placebo-controlled trial, studied 42 patients with giant cell arteritis, half of whom were randomized to receive methotrexate 10 mg/week, while the other half received placebo. All patients received prednisone. Patients in the methotrexate group had fewer relapses and a 25% lower cumulative dose of prednisone during follow-up. However, the incidence of adverse events was similar in both groups. Methotrexate was discontinued in 3 patients who developed drug-related adverse events.
Hoffman et al30 randomized 98 patients to receive either methotrexate (up to 15 mg/week) or placebo in a double-blind fashion. All patients also received prednisone at an initial dose of 1 mg/kg/day (up to 60 mg/day). At completion of the study, no differences between the groups were noted in the rates of relapse or treatment-related morbidity or in the cumulative dose of glucocorticoid. However, treatment with methotrexate appeared to lower the rate of recurrence of isolated polymyalgia rheumatica in a small number of patients.30
Comment. Differences in the results of these trials may be attributed to several factors, including different definitions of relapses and different glucocorticoid doses and tapering regimens.
A meta-analysis of these three trials34 showed a reduction in the risk of relapse: 4 patients would have to be treated to prevent one first relapse, 5 would have to be treated to prevent one second relapse, and 11 would need to be treated to prevent one first relapse of cranial symptoms in the first 48 weeks. However, the main goal of methotrexate therapy is to decrease the frequency of adverse events from glucocorticoids, and this meta-analysis found no difference in rates of glucocorticoid-related adverse events in patients treated with methotrexate.
The study raises the question of whether methotrexate should be further evaluated in in different patient populations and at higher doses.34
Infliximab is not recommended
In a prospective study, patients with giant cell arteritis were randomly assigned to receive either infliximab (Remicade) 5 mg/kg every 8 weeks or placebo, in addition to standard glucocorticoid therapy. The study showed no significant difference in the relapse rate and a higher rate of infection in the infliximab group (71%) than in the placebo group (56%). Given the lack of any benefit observed in this study, infliximab is not recommended in the treatment of patients with giant cell arteritis.35
Aspirin is recommended
Daily low-dose aspirin therapy has been shown in several studies to be effective in preventing ischemic complications of giant cell arteritis, including stroke and visual loss. It is currently recommended that all patients with giant cell arteritis without a major contraindication take aspirin 81 mg daily.36–38
References
Salvarani C, Gabriel SE, O’Fallon WM, Hunder GG. The incidence of giant cell arteritis in Olmsted County, Minnesota: apparent fluctuations in a cyclic pattern. Ann Intern Med1995; 123:192–194.
Baldursson O, Steinsson K, Björnsson J, Lie JT. Giant cell arteritis in Iceland. An epidemiologic and histopathologic analysis. Arthritis Rheum1994; 37:1007–1012.
Weyand CM, Goronzy JJ. Medium- and large-vessel vasculitis. N Engl J Med2003; 349:160–169.
Salvarani C, Cimino L, Macchioni P, et al. Risk factors for visual loss in an Italian population-based cohort of patients with giant cell arteritis. Arthritis Rheum2005; 53:293–297.
Bahlas S, Ramos-Remus C, Davis P. Clinical outcome of 149 patients with polymyalgia rheumatica and giant cell arteritis. J Rheumatol1998; 25:99–104.
Gonzalez-Gay MA, Barros S, Lopez-Diaz MJ, Garcia-Porrua C, Sanchez-Andrade A, Llorca J. Giant cell arteritis: disease patterns of clinical presentation in a series of 240 patients. Medicine (Baltimore)2005; 84:269–276.
Salvarani C, Cantini F, Macchioni P, et al. Distal musculoskeletal manifestations in polymyalgia rheumatica: a prospective followup study. Arthritis Rheum1998; 41:1221–1226.
Salvarani C, Cantini F, Boiardi L, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med2002; 347:261–271.
Lie JT. Aortic and extracranial large vessel giant cell arteritis: a review of 72 cases with histopathologic documentation. Semin Arthritis Rheum1995; 24:422–431.
Evans JM, O’Fallon WM, Hunder GG. Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis. A population-based study. Ann Intern Med1995; 122:502–507.
Ostberg G. An arteritis with special reference to polymyalgia arteritica. Acta Pathol Microbiol Scand Suppl1973; 237(suppl 237):1–59.
Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol1999; 128:211–215.
González-Gay MA, Garcia-Porrua C, Rivas MJ, Rodriguez-Ledo P, Llorca J. Epidemiology of biopsy proven giant cell arteritis in northwestern Spain: trend over an 18 year period. Ann Rheum Dis2001; 60:367–371.
Rodriguez-Valverde V, Sarabia JM, González-Gay MA, et al. Risk factors and predictive models of giant cell arteritis in polymyalgia rheumatica. Am J Med1997; 102:331–336.
Mahr A, Saba M, Kambouchner M, et al. Temporal artery biopsy for diagnosing giant cell arteritis: the longer, the better?Ann Rheum Dis2006; 65:826–828.
Breuer GS, Nesher R, Nesher G. Effect of biopsy length on the rate of positive temporal artery biopsies. Clin Exp Rheumatol2009; 27(1 suppl 52):S10–S13.
Weyand CM, Goronzy JJ. Giant-cell arteritis and polymyalgia rheumatica. Ann Intern Med2003; 139:505–515.
Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurence in a population-based study. Arthritis Rheum2001; 45:140–145.
Schmidt WA, Kraft HE, Vorpahl K, Völker L, Gromnica-Ihle EJ. Color duplex ultrasonography in the diagnosis of temporal arteritis. N Engl J Med1997; 337:1336–1342.
Karassa FB, Matsagas MI, Schmidt WA, Ioannidis JP. Meta-analysis: test performance of ultrasonography for giant-cell arteritis. Ann Intern Med2005; 142:359–369.
Maldini C, Dépinay-Dhellemmes C, Tra TT, et al. Limited value of temporal artery ultrasonography examinations for diagnosis of giant cell arteritis: analysis of 77 subjects. J Rheumatol2010; Epub ahead of print.
Both M, Ahmadi-Simab K, Reuter M, et al. MRI and FDG-PET in the assessment of inflammatory aortic arch syndrome in complicated courses of giant cell arteritis. Ann Rheum Dis2008; 67:1030–1033.
Tso E, Flamm SD, White RD, Schvartzman PR, Mascha E, Hoffman GS. Takayasu arteritis: utility and limitations of magnetic resonance imaging in diagnosis and treatment. Arthritis Rheum2002; 46:1634–1642.
Birkhead NC, Wagener HP, Shick RM. Treatment of temporal arteritis with adrenal corticosteroids; results in fifty-five cases in which lesion was proved at biopsy. J Am Med Assoc1957; 163:821–827.
Ray-Chaudhuri N, Kiné DA, Tijani SO, et al. Effect of prior steroid treatment on temporal artery biopsy findings in giant cell arteritis. Br J Ophthalmol2002; 86:530–532.
Chan CC, Paine M, O’Day J. Steroid management in giant cell arteritis. Br J Ophthalmol2001; 85:1061–1064.
Hunder GG, Sheps SG, Allen GL, Joyce JW. Daily and alternate-day corticosteroid regimens in treatment of giant cell arteritis: comparison in a prospective study. Ann Intern Med1975; 82:613–618.
Hoffman GS, Cid MC, Hellmann DB, et al; International Network for the Study of Systemic Vasculitides. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum2002; 46:1309–1318.
Mazlumzadeh M, Hunder GG, Easley KA, et al. Treatment of giant cell arteritis using induction therapy with high-dose glucocorticoids: a double-blind, placebo-controlled, randomized prospective clinical trial. Arthritis Rheum2006; 54:3310–3318.
Spiera RF, Mitnick HJ, Kupersmith M, et al. A prospective, doubleblind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA). Clin Exp Rheumatol2001; 19:495–501.
Jover JA, Hernández-García C, Morado IC, Vargas E, Bañares A, Fernández-Gutiérrez B. Combined treatment of giant-cell arteritis with methotrexate and prednisone. a randomized, double-blind, placebo-controlled trial. Ann Intern Med2001; 134:106–114.
Mahr AD, Jover JA, Spiera RF, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum2007; 56:2789–2797.
Hoffman GS, Cid MC, Rendt-Zagar KE, et al; Infliximab-GCA Study Group. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med2007; 146:621–630.
Weyand CM, Kaiser M, Yang H, Younge B, Goronzy JJ. Therapeutic effects of acetylsalicylic acid in giant cell arteritis. Arthritis Rheum2002; 46:457–466.
Nesher G, Berkun Y, Mates M, Baras M, Rubinow A, Sonnenblick M. Low-dose aspirin and prevention of cranial ischemic complications in giant cell arteritis. Arthritis Rheum2004; 50:1332–1337.
Lee MS, Smith SD, Galor A, Hoffman GS. Antiplatelet and anticoagulant therapy in patients with giant cell arteritis. Arthritis Rheum2006; 54:3306–3309.
References
Salvarani C, Gabriel SE, O’Fallon WM, Hunder GG. The incidence of giant cell arteritis in Olmsted County, Minnesota: apparent fluctuations in a cyclic pattern. Ann Intern Med1995; 123:192–194.
Baldursson O, Steinsson K, Björnsson J, Lie JT. Giant cell arteritis in Iceland. An epidemiologic and histopathologic analysis. Arthritis Rheum1994; 37:1007–1012.
Weyand CM, Goronzy JJ. Medium- and large-vessel vasculitis. N Engl J Med2003; 349:160–169.
Salvarani C, Cimino L, Macchioni P, et al. Risk factors for visual loss in an Italian population-based cohort of patients with giant cell arteritis. Arthritis Rheum2005; 53:293–297.
Bahlas S, Ramos-Remus C, Davis P. Clinical outcome of 149 patients with polymyalgia rheumatica and giant cell arteritis. J Rheumatol1998; 25:99–104.
Gonzalez-Gay MA, Barros S, Lopez-Diaz MJ, Garcia-Porrua C, Sanchez-Andrade A, Llorca J. Giant cell arteritis: disease patterns of clinical presentation in a series of 240 patients. Medicine (Baltimore)2005; 84:269–276.
Salvarani C, Cantini F, Macchioni P, et al. Distal musculoskeletal manifestations in polymyalgia rheumatica: a prospective followup study. Arthritis Rheum1998; 41:1221–1226.
Salvarani C, Cantini F, Boiardi L, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med2002; 347:261–271.
Lie JT. Aortic and extracranial large vessel giant cell arteritis: a review of 72 cases with histopathologic documentation. Semin Arthritis Rheum1995; 24:422–431.
Evans JM, O’Fallon WM, Hunder GG. Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis. A population-based study. Ann Intern Med1995; 122:502–507.
Ostberg G. An arteritis with special reference to polymyalgia arteritica. Acta Pathol Microbiol Scand Suppl1973; 237(suppl 237):1–59.
Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol1999; 128:211–215.
González-Gay MA, Garcia-Porrua C, Rivas MJ, Rodriguez-Ledo P, Llorca J. Epidemiology of biopsy proven giant cell arteritis in northwestern Spain: trend over an 18 year period. Ann Rheum Dis2001; 60:367–371.
Rodriguez-Valverde V, Sarabia JM, González-Gay MA, et al. Risk factors and predictive models of giant cell arteritis in polymyalgia rheumatica. Am J Med1997; 102:331–336.
Mahr A, Saba M, Kambouchner M, et al. Temporal artery biopsy for diagnosing giant cell arteritis: the longer, the better?Ann Rheum Dis2006; 65:826–828.
Breuer GS, Nesher R, Nesher G. Effect of biopsy length on the rate of positive temporal artery biopsies. Clin Exp Rheumatol2009; 27(1 suppl 52):S10–S13.
Weyand CM, Goronzy JJ. Giant-cell arteritis and polymyalgia rheumatica. Ann Intern Med2003; 139:505–515.
Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurence in a population-based study. Arthritis Rheum2001; 45:140–145.
Schmidt WA, Kraft HE, Vorpahl K, Völker L, Gromnica-Ihle EJ. Color duplex ultrasonography in the diagnosis of temporal arteritis. N Engl J Med1997; 337:1336–1342.
Karassa FB, Matsagas MI, Schmidt WA, Ioannidis JP. Meta-analysis: test performance of ultrasonography for giant-cell arteritis. Ann Intern Med2005; 142:359–369.
Maldini C, Dépinay-Dhellemmes C, Tra TT, et al. Limited value of temporal artery ultrasonography examinations for diagnosis of giant cell arteritis: analysis of 77 subjects. J Rheumatol2010; Epub ahead of print.
Both M, Ahmadi-Simab K, Reuter M, et al. MRI and FDG-PET in the assessment of inflammatory aortic arch syndrome in complicated courses of giant cell arteritis. Ann Rheum Dis2008; 67:1030–1033.
Tso E, Flamm SD, White RD, Schvartzman PR, Mascha E, Hoffman GS. Takayasu arteritis: utility and limitations of magnetic resonance imaging in diagnosis and treatment. Arthritis Rheum2002; 46:1634–1642.
Birkhead NC, Wagener HP, Shick RM. Treatment of temporal arteritis with adrenal corticosteroids; results in fifty-five cases in which lesion was proved at biopsy. J Am Med Assoc1957; 163:821–827.
Ray-Chaudhuri N, Kiné DA, Tijani SO, et al. Effect of prior steroid treatment on temporal artery biopsy findings in giant cell arteritis. Br J Ophthalmol2002; 86:530–532.
Chan CC, Paine M, O’Day J. Steroid management in giant cell arteritis. Br J Ophthalmol2001; 85:1061–1064.
Hunder GG, Sheps SG, Allen GL, Joyce JW. Daily and alternate-day corticosteroid regimens in treatment of giant cell arteritis: comparison in a prospective study. Ann Intern Med1975; 82:613–618.
Hoffman GS, Cid MC, Hellmann DB, et al; International Network for the Study of Systemic Vasculitides. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum2002; 46:1309–1318.
Mazlumzadeh M, Hunder GG, Easley KA, et al. Treatment of giant cell arteritis using induction therapy with high-dose glucocorticoids: a double-blind, placebo-controlled, randomized prospective clinical trial. Arthritis Rheum2006; 54:3310–3318.
Spiera RF, Mitnick HJ, Kupersmith M, et al. A prospective, doubleblind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA). Clin Exp Rheumatol2001; 19:495–501.
Jover JA, Hernández-García C, Morado IC, Vargas E, Bañares A, Fernández-Gutiérrez B. Combined treatment of giant-cell arteritis with methotrexate and prednisone. a randomized, double-blind, placebo-controlled trial. Ann Intern Med2001; 134:106–114.
Mahr AD, Jover JA, Spiera RF, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum2007; 56:2789–2797.
Hoffman GS, Cid MC, Rendt-Zagar KE, et al; Infliximab-GCA Study Group. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med2007; 146:621–630.
Weyand CM, Kaiser M, Yang H, Younge B, Goronzy JJ. Therapeutic effects of acetylsalicylic acid in giant cell arteritis. Arthritis Rheum2002; 46:457–466.
Nesher G, Berkun Y, Mates M, Baras M, Rubinow A, Sonnenblick M. Low-dose aspirin and prevention of cranial ischemic complications in giant cell arteritis. Arthritis Rheum2004; 50:1332–1337.
Lee MS, Smith SD, Galor A, Hoffman GS. Antiplatelet and anticoagulant therapy in patients with giant cell arteritis. Arthritis Rheum2006; 54:3306–3309.
Giant cell arteritis is often associated with an intense acute-phase response and cranial symptoms.
Large-vessel involvement is commonly present and may result in serious complications such as visual loss, stroke, limb claudication, and aortic aneurysm.
The diagnosis is usually confirmed by an abnormal temporal artery biopsy.
Symptoms of giant cell arteritis usually respond rapidly and completely to glucocorticoid therapy, still the mainstay of treatment. Most patients need prolonged therapy.
Several studies have evaluated alternative drugs in an attempt to avoid toxicity from long-term use of glucocorticoids. Results have been mixed, and further study is needed.
Figure 1.
A chronic cough (ie, a cough lasting more than 8 weeks1) has many possible causes. Physicians should use a structured diagnostic approach based on observing the clinical picture, trying therapy for the likely cause, obtaining targeted investigations, and referring to a specialist when needed (Figure 1).
To begin, obtain a clinical history, perform a physical examination, and order a chest radiograph.
In the history, look for exposure to environmental irritants such as tobacco smoke, allergens, or dust, or medications such as angiotensin-converting enzyme (ACE) inhibitors or oxymetazoline (Afrin). If a potential irritant is present, it should be avoided or stopped immediately.1–3 If the cough improves partially or fully when exposure to the irritant is stopped, this supports a diagnosis of chronic bronchitis or, in the case of ACE inhibitors, ACE-inhibitor-induced cough. The character of the cough (eg, paroxysmal, loose, dry, or productive1) has not been shown to be diagnostically useful or specific.
If the chest radiograph is abnormal, then the diagnostic inquiry should be guided by the abnormality. Abnormalities that cause cough include bronchogenic carcinoma, sarcoidosis, and bronchiectasis. If the radiograph is normal, then upper airway cough syndrome, asthma, gastroesophageal reflux disease (GERD), chronic bronchitis, or nonasthmatic eosinophilic bronchitis is more likely.
COMMON CAUSES OF CHRONIC COUGH
The most common causes of chronic cough, accounting for 95% of cases, are chronic bronchitis due to environmental irritants, upper airway cough syndrome, GERD, asthma, nonasthmatic eosinophilic bronchitis, and bronchiectasis (Table 1).1–8
Chronic bronchitis
As noted above, a history of exposure to an irritant suggests this diagnosis.
Upper airway cough syndrome
Upper airway cough syndrome (formerly known as postnasal drip) is due to chronic upper respiratory tract irritation and hypersensitivity of cough receptors.3,4 Sources of irritation vary and include sinusitis and any form of rhinitis: allergic and nonallergic, postinfectious, environmental irritant-induced, vasomotor, and drug-induced.
Patients complain of postnasal drip or frequent clearing of the throat. On physical examination one can see mucus in the oropharnyx or a cobblestone appearance. However, these symptoms and signs are not specific and may be absent.
A therapeutic trial is warranted, but be aware that different rhinitides respond to specific treatments:
Histamine-mediated or allergic rhinitis will respond to allergen avoidance, new-generation antihistamines such as loratadine (Claritin), mast cell stabilizers such as cromolyn (Intal), and intranasal glucocorticoids such as fluticasone (Flovent).4,5
Nonhistamine-mediated rhinitides (the common cold and perennial nonallergic rhinitis) respond to older-generation antihistamines such as diphenhydramine (Benadryl) and decongestant combinations. If these cannot be used, intranasal glucocorticoids and ipratropium (Atrovent) are alternatives.
Vasomotor rhinitis will respond to intranasal ipratropium 0.3% for 3 weeks and then as required.
Postinfective rhinitis, ie, a cough that began as severe bronchitis, would warrant an antihistamine-decongestant combination.
With adequate treatment, the cough should improve after 1 to 2 weeks; if rhinosinus symptoms persist, consider bacterial sinusitis and obtain radiographs of the sinuses. If imaging shows mucosal thickening (> 5 mm) or an air-fluid level, treat with decongestants and antibiotics for 3 weeks.1,4,5
Gastroesophageal reflux disease
GERD is another common cause of cough, and the most difficult to exclude.5 Look for a history of reflux or heartburn and positional coughing, and have a low threshold for beginning empiric therapy. Indeed, according to the 2006 American College of Chest Physicians Cough Guideline Committee,5,6 should a patient arrive in your clinic with a chronic cough and a normal chest radiograph who does not smoke and is not on an ACE inhibitor, then you should start empiric reflux therapy. Begin with lifestyle changes, acid suppression, and prokinetics. The cough may take 1 to 2 months before it begins to improve, and even longer to resolve.
The gold standard for diagnosis is 24-hour pH and impedance monitoring with patient self-reporting of symptoms. However, this test is not available everywhere, and there is no consensus on how to interpret the results.1,5,6 If you strongly suspect the patient has GERD-related cough but it fails to improve with intense medical management, then refer to a specialist, as antireflux surgery may be required.
Cough-variant asthma
Cough is the only symptom of asthma in cough-variant asthma, in which the usual features of dyspnea and wheezing are absent.7 A methacholine challenge shows bronchial hyperresponsiveness, and asthma therapy resolves the cough.
Nonasthmatic eosinophilic bronchitis
It is important to distinguish asthma from nonasthmatic eosinophilic bronchitis,7,8 an underdiagnosed condition. Both conditions respond equally well to treatment with inhaled or oral steroids. However, patients who have nonasthmatic eosinophilic bronchitis have normal results on spirometry and the methacholine challenge test. The diagnosis of nonasthmatic eosinophilic bronchitis is made if more than 3% of the nonsquamous cells in an induced sputum sample are eosinophils.
UNCOMMON CAUSES OF COUGH
The remaining 5% of cases of cough are caused by conditions that include bronchogenic carcinoma, chronic interstitial pneumonia, sarcoidosis, left ventricular dysfunction, use of ACE inhibitors, neurosensory cough, dynamic airway collapse, aspiration due to pharyngeal dysfunction, and psychogenic causes.1
MULTIPLE CAUSES
Therapeutic trials will support the diagnosis. If more than one cause is suggested, start treatment in the order in which the abnormalities are discovered. If treatment is only partially successful, then pursue further causes and add to the existing treatment without stopping it.
Cough may have more than one cause, but in up to 98% of patients it can be successfully treated.
IMPORTANT POINTS
Multiple causes of chronic cough can coexist.
Therapeutic trials are part of the workup.
Do not stop therapy if it is only partially successful: add to existing therapies
Start the investigation with the most likely cause.
Treatment is 84% to 98% successful.
References
Irwin RS, Madison JM. The diagnosis and treatment of cough. N Engl J Med2000; 343:1715–1721.
Vegter S, de Jong-van den Berg LT. Misdiagnosis and mistreatment of a common side-effect—angiotensin-converting enzyme inhibitor-induced cough. Br J Clin Pharmacol2010; 69:200–203.
Irwin RS, Baumann MH, Bolser DC, et al; American College of Chest Physicians (ACCP). Diagnosis and management of cough executive summary: ACCP evidence-based clinical practice guidelines. Chest2006; 129(suppl):1S–23S.
Pratter MR. Chronic upper airway cough syndrome secondary to rhinosinus diseases (previously referred to as postnasal drip syndrome): ACCP evidence-based clinical practice guidelines. Chest2006; 129(suppl):63S–71S.
Irwin RS. Chronic cough due to gastroesophageal reflux disease: ACCP evidence-based clinical practice guidelines. Chest2006; 129(suppl):80S–94S.
Figure 1.
A chronic cough (ie, a cough lasting more than 8 weeks1) has many possible causes. Physicians should use a structured diagnostic approach based on observing the clinical picture, trying therapy for the likely cause, obtaining targeted investigations, and referring to a specialist when needed (Figure 1).
To begin, obtain a clinical history, perform a physical examination, and order a chest radiograph.
In the history, look for exposure to environmental irritants such as tobacco smoke, allergens, or dust, or medications such as angiotensin-converting enzyme (ACE) inhibitors or oxymetazoline (Afrin). If a potential irritant is present, it should be avoided or stopped immediately.1–3 If the cough improves partially or fully when exposure to the irritant is stopped, this supports a diagnosis of chronic bronchitis or, in the case of ACE inhibitors, ACE-inhibitor-induced cough. The character of the cough (eg, paroxysmal, loose, dry, or productive1) has not been shown to be diagnostically useful or specific.
If the chest radiograph is abnormal, then the diagnostic inquiry should be guided by the abnormality. Abnormalities that cause cough include bronchogenic carcinoma, sarcoidosis, and bronchiectasis. If the radiograph is normal, then upper airway cough syndrome, asthma, gastroesophageal reflux disease (GERD), chronic bronchitis, or nonasthmatic eosinophilic bronchitis is more likely.
COMMON CAUSES OF CHRONIC COUGH
The most common causes of chronic cough, accounting for 95% of cases, are chronic bronchitis due to environmental irritants, upper airway cough syndrome, GERD, asthma, nonasthmatic eosinophilic bronchitis, and bronchiectasis (Table 1).1–8
Chronic bronchitis
As noted above, a history of exposure to an irritant suggests this diagnosis.
Upper airway cough syndrome
Upper airway cough syndrome (formerly known as postnasal drip) is due to chronic upper respiratory tract irritation and hypersensitivity of cough receptors.3,4 Sources of irritation vary and include sinusitis and any form of rhinitis: allergic and nonallergic, postinfectious, environmental irritant-induced, vasomotor, and drug-induced.
Patients complain of postnasal drip or frequent clearing of the throat. On physical examination one can see mucus in the oropharnyx or a cobblestone appearance. However, these symptoms and signs are not specific and may be absent.
A therapeutic trial is warranted, but be aware that different rhinitides respond to specific treatments:
Histamine-mediated or allergic rhinitis will respond to allergen avoidance, new-generation antihistamines such as loratadine (Claritin), mast cell stabilizers such as cromolyn (Intal), and intranasal glucocorticoids such as fluticasone (Flovent).4,5
Nonhistamine-mediated rhinitides (the common cold and perennial nonallergic rhinitis) respond to older-generation antihistamines such as diphenhydramine (Benadryl) and decongestant combinations. If these cannot be used, intranasal glucocorticoids and ipratropium (Atrovent) are alternatives.
Vasomotor rhinitis will respond to intranasal ipratropium 0.3% for 3 weeks and then as required.
Postinfective rhinitis, ie, a cough that began as severe bronchitis, would warrant an antihistamine-decongestant combination.
With adequate treatment, the cough should improve after 1 to 2 weeks; if rhinosinus symptoms persist, consider bacterial sinusitis and obtain radiographs of the sinuses. If imaging shows mucosal thickening (> 5 mm) or an air-fluid level, treat with decongestants and antibiotics for 3 weeks.1,4,5
Gastroesophageal reflux disease
GERD is another common cause of cough, and the most difficult to exclude.5 Look for a history of reflux or heartburn and positional coughing, and have a low threshold for beginning empiric therapy. Indeed, according to the 2006 American College of Chest Physicians Cough Guideline Committee,5,6 should a patient arrive in your clinic with a chronic cough and a normal chest radiograph who does not smoke and is not on an ACE inhibitor, then you should start empiric reflux therapy. Begin with lifestyle changes, acid suppression, and prokinetics. The cough may take 1 to 2 months before it begins to improve, and even longer to resolve.
The gold standard for diagnosis is 24-hour pH and impedance monitoring with patient self-reporting of symptoms. However, this test is not available everywhere, and there is no consensus on how to interpret the results.1,5,6 If you strongly suspect the patient has GERD-related cough but it fails to improve with intense medical management, then refer to a specialist, as antireflux surgery may be required.
Cough-variant asthma
Cough is the only symptom of asthma in cough-variant asthma, in which the usual features of dyspnea and wheezing are absent.7 A methacholine challenge shows bronchial hyperresponsiveness, and asthma therapy resolves the cough.
Nonasthmatic eosinophilic bronchitis
It is important to distinguish asthma from nonasthmatic eosinophilic bronchitis,7,8 an underdiagnosed condition. Both conditions respond equally well to treatment with inhaled or oral steroids. However, patients who have nonasthmatic eosinophilic bronchitis have normal results on spirometry and the methacholine challenge test. The diagnosis of nonasthmatic eosinophilic bronchitis is made if more than 3% of the nonsquamous cells in an induced sputum sample are eosinophils.
UNCOMMON CAUSES OF COUGH
The remaining 5% of cases of cough are caused by conditions that include bronchogenic carcinoma, chronic interstitial pneumonia, sarcoidosis, left ventricular dysfunction, use of ACE inhibitors, neurosensory cough, dynamic airway collapse, aspiration due to pharyngeal dysfunction, and psychogenic causes.1
MULTIPLE CAUSES
Therapeutic trials will support the diagnosis. If more than one cause is suggested, start treatment in the order in which the abnormalities are discovered. If treatment is only partially successful, then pursue further causes and add to the existing treatment without stopping it.
Cough may have more than one cause, but in up to 98% of patients it can be successfully treated.
IMPORTANT POINTS
Multiple causes of chronic cough can coexist.
Therapeutic trials are part of the workup.
Do not stop therapy if it is only partially successful: add to existing therapies
Start the investigation with the most likely cause.
Treatment is 84% to 98% successful.
Figure 1.
A chronic cough (ie, a cough lasting more than 8 weeks1) has many possible causes. Physicians should use a structured diagnostic approach based on observing the clinical picture, trying therapy for the likely cause, obtaining targeted investigations, and referring to a specialist when needed (Figure 1).
To begin, obtain a clinical history, perform a physical examination, and order a chest radiograph.
In the history, look for exposure to environmental irritants such as tobacco smoke, allergens, or dust, or medications such as angiotensin-converting enzyme (ACE) inhibitors or oxymetazoline (Afrin). If a potential irritant is present, it should be avoided or stopped immediately.1–3 If the cough improves partially or fully when exposure to the irritant is stopped, this supports a diagnosis of chronic bronchitis or, in the case of ACE inhibitors, ACE-inhibitor-induced cough. The character of the cough (eg, paroxysmal, loose, dry, or productive1) has not been shown to be diagnostically useful or specific.
If the chest radiograph is abnormal, then the diagnostic inquiry should be guided by the abnormality. Abnormalities that cause cough include bronchogenic carcinoma, sarcoidosis, and bronchiectasis. If the radiograph is normal, then upper airway cough syndrome, asthma, gastroesophageal reflux disease (GERD), chronic bronchitis, or nonasthmatic eosinophilic bronchitis is more likely.
COMMON CAUSES OF CHRONIC COUGH
The most common causes of chronic cough, accounting for 95% of cases, are chronic bronchitis due to environmental irritants, upper airway cough syndrome, GERD, asthma, nonasthmatic eosinophilic bronchitis, and bronchiectasis (Table 1).1–8
Chronic bronchitis
As noted above, a history of exposure to an irritant suggests this diagnosis.
Upper airway cough syndrome
Upper airway cough syndrome (formerly known as postnasal drip) is due to chronic upper respiratory tract irritation and hypersensitivity of cough receptors.3,4 Sources of irritation vary and include sinusitis and any form of rhinitis: allergic and nonallergic, postinfectious, environmental irritant-induced, vasomotor, and drug-induced.
Patients complain of postnasal drip or frequent clearing of the throat. On physical examination one can see mucus in the oropharnyx or a cobblestone appearance. However, these symptoms and signs are not specific and may be absent.
A therapeutic trial is warranted, but be aware that different rhinitides respond to specific treatments:
Histamine-mediated or allergic rhinitis will respond to allergen avoidance, new-generation antihistamines such as loratadine (Claritin), mast cell stabilizers such as cromolyn (Intal), and intranasal glucocorticoids such as fluticasone (Flovent).4,5
Nonhistamine-mediated rhinitides (the common cold and perennial nonallergic rhinitis) respond to older-generation antihistamines such as diphenhydramine (Benadryl) and decongestant combinations. If these cannot be used, intranasal glucocorticoids and ipratropium (Atrovent) are alternatives.
Vasomotor rhinitis will respond to intranasal ipratropium 0.3% for 3 weeks and then as required.
Postinfective rhinitis, ie, a cough that began as severe bronchitis, would warrant an antihistamine-decongestant combination.
With adequate treatment, the cough should improve after 1 to 2 weeks; if rhinosinus symptoms persist, consider bacterial sinusitis and obtain radiographs of the sinuses. If imaging shows mucosal thickening (> 5 mm) or an air-fluid level, treat with decongestants and antibiotics for 3 weeks.1,4,5
Gastroesophageal reflux disease
GERD is another common cause of cough, and the most difficult to exclude.5 Look for a history of reflux or heartburn and positional coughing, and have a low threshold for beginning empiric therapy. Indeed, according to the 2006 American College of Chest Physicians Cough Guideline Committee,5,6 should a patient arrive in your clinic with a chronic cough and a normal chest radiograph who does not smoke and is not on an ACE inhibitor, then you should start empiric reflux therapy. Begin with lifestyle changes, acid suppression, and prokinetics. The cough may take 1 to 2 months before it begins to improve, and even longer to resolve.
The gold standard for diagnosis is 24-hour pH and impedance monitoring with patient self-reporting of symptoms. However, this test is not available everywhere, and there is no consensus on how to interpret the results.1,5,6 If you strongly suspect the patient has GERD-related cough but it fails to improve with intense medical management, then refer to a specialist, as antireflux surgery may be required.
Cough-variant asthma
Cough is the only symptom of asthma in cough-variant asthma, in which the usual features of dyspnea and wheezing are absent.7 A methacholine challenge shows bronchial hyperresponsiveness, and asthma therapy resolves the cough.
Nonasthmatic eosinophilic bronchitis
It is important to distinguish asthma from nonasthmatic eosinophilic bronchitis,7,8 an underdiagnosed condition. Both conditions respond equally well to treatment with inhaled or oral steroids. However, patients who have nonasthmatic eosinophilic bronchitis have normal results on spirometry and the methacholine challenge test. The diagnosis of nonasthmatic eosinophilic bronchitis is made if more than 3% of the nonsquamous cells in an induced sputum sample are eosinophils.
UNCOMMON CAUSES OF COUGH
The remaining 5% of cases of cough are caused by conditions that include bronchogenic carcinoma, chronic interstitial pneumonia, sarcoidosis, left ventricular dysfunction, use of ACE inhibitors, neurosensory cough, dynamic airway collapse, aspiration due to pharyngeal dysfunction, and psychogenic causes.1
MULTIPLE CAUSES
Therapeutic trials will support the diagnosis. If more than one cause is suggested, start treatment in the order in which the abnormalities are discovered. If treatment is only partially successful, then pursue further causes and add to the existing treatment without stopping it.
Cough may have more than one cause, but in up to 98% of patients it can be successfully treated.
IMPORTANT POINTS
Multiple causes of chronic cough can coexist.
Therapeutic trials are part of the workup.
Do not stop therapy if it is only partially successful: add to existing therapies
Start the investigation with the most likely cause.
Treatment is 84% to 98% successful.
References
Irwin RS, Madison JM. The diagnosis and treatment of cough. N Engl J Med2000; 343:1715–1721.
Vegter S, de Jong-van den Berg LT. Misdiagnosis and mistreatment of a common side-effect—angiotensin-converting enzyme inhibitor-induced cough. Br J Clin Pharmacol2010; 69:200–203.
Irwin RS, Baumann MH, Bolser DC, et al; American College of Chest Physicians (ACCP). Diagnosis and management of cough executive summary: ACCP evidence-based clinical practice guidelines. Chest2006; 129(suppl):1S–23S.
Pratter MR. Chronic upper airway cough syndrome secondary to rhinosinus diseases (previously referred to as postnasal drip syndrome): ACCP evidence-based clinical practice guidelines. Chest2006; 129(suppl):63S–71S.
Irwin RS. Chronic cough due to gastroesophageal reflux disease: ACCP evidence-based clinical practice guidelines. Chest2006; 129(suppl):80S–94S.
Dicpinigaitis PV. Chronic cough due to asthma: ACCP evidence-based clinical practice guidelines. Chest2006; 129(suppl):75S–79S.
Brightling CE. Chronic cough due to nonasthmatic eosinophilic bronchitis: ACCP evidence-based clinical practice guidelines. Chest2006; 129(suppl):116S–121S.
References
Irwin RS, Madison JM. The diagnosis and treatment of cough. N Engl J Med2000; 343:1715–1721.
Vegter S, de Jong-van den Berg LT. Misdiagnosis and mistreatment of a common side-effect—angiotensin-converting enzyme inhibitor-induced cough. Br J Clin Pharmacol2010; 69:200–203.
Irwin RS, Baumann MH, Bolser DC, et al; American College of Chest Physicians (ACCP). Diagnosis and management of cough executive summary: ACCP evidence-based clinical practice guidelines. Chest2006; 129(suppl):1S–23S.
Pratter MR. Chronic upper airway cough syndrome secondary to rhinosinus diseases (previously referred to as postnasal drip syndrome): ACCP evidence-based clinical practice guidelines. Chest2006; 129(suppl):63S–71S.
Irwin RS. Chronic cough due to gastroesophageal reflux disease: ACCP evidence-based clinical practice guidelines. Chest2006; 129(suppl):80S–94S.
You have a 54-year-old black patient with gout, diabetes mellitus, hypertension, and chronic kidney disease (CKD). He has an acute gout flare involving his right knee. In the past year he has had four attacks of gout in the ankles and knees, which you treated with intra-articular glucocorticoid injections. He has been on allopurinol (Zyloprim) 200 mg daily, but his last serum urate level was 9.4 mg/dL (reference range 3.0–8.0). His creatinine clearance is 45 mL/minute (reference range 85–125).
In view of his kidney disease, you are concerned about increasing his dose of allopurinol, but also about the need to treat his frequent attacks. How should you manage this patient?
GOUT IS CHALLENGING TO TREAT IN PATIENTS WITH KIDNEY DISEASE
A major challenge in treating patients with gout is to avoid therapeutic interactions with common comorbidities, including hypertension, insulin resistance, coronary artery disease, heart failure, and especially CKD.1
In this paper, we discuss approaches to and controversies in the management of gout and hyperuricemia in patients with CKD. Unfortunately, the evidence from clinical trials to guide treatment decisions is limited; therefore, decisions must often be based on experience and pathophysiologic principles.
GENERAL GOALS OF GOUT THERAPY
Depending on the patient and the stage of the disease, the goals in treating patients with gout are to:
Terminate acute attacks as promptly and safely as possible
Prevent recurrences of acute gout attacks
Prevent or reverse complications resulting from deposition of monosodium urate in the joints, in the kidneys, or at other sites.
These goals are more difficult to achieve in patients with CKD because of the potential complications from many of the available drugs.
TERMINATING ACUTE GOUT FLARES
In patients with acute gout, treatment is aimed at quickly resolving pain and inflammation.
Several types of drugs can terminate acute gout flares. The choice in most situations is colchicine (Colcrys); a nonsteroidal anti-inflammatory drug (NSAID); a corticosteroid; or corticotropin (ACTH).
However, in patients with CKD, there are concerns about using colchicine or NSAIDs, and corticotropin is very expensive; thus, corticosteroids are often used.
Colchicine’s clearance is reduced in CKD
Colchicine is somewhat effective in treating acute gout attacks and probably more effective in preventing attacks.
Due to concerns about inappropriate dosing and reported deaths,2 the intravenous formulation is not available in many countries, including the United States.
After oral administration, colchicine is rapidly absorbed, with a bioavailability of up to 50%. It undergoes metabolism by the liver, and its metabolites are excreted by renal and biliary-intestinal routes. Up to 20% of the active drug is excreted by the kidneys.3
Colchicine’s clearance is significantly reduced in patients with renal or hepatic insufficiency, and the drug may accumulate in cells, with resultant toxicity.4 Colchicine-induced toxicity has been observed when the drug was used for acute treatment, as well as for chronic prophylaxis of gout in patients with CKD; thus, alternative agents for treating acute attacks should be considered.5,6 With prolonged use, reversible colchicine-induced axonal neuropathy, neutropenia, and vacuolar myopathy can develop in patients with CKD.7
In a trial in patients with normal renal function, nearly 100% who received an initial dose of 1 mg followed by 0.5 mg every 2 hours developed diarrhea at a median time of 24 hours.8 Emesis may also occur.
A lower dose of 1.8 mg (two 0.6-mg pills followed by one pill an hour later) was well tolerated but only moderately effective in treating acute gout, causing at least a 50% reduction in pain at 24 hours in only 38% of patients.9 This study does not clarify the dosage to use to completely resolve attacks. Using additional colchicine likely will increase the response rate, but will also increase side effects. Patients with CKD were not included.
Some patients, as shown in the above trial, can abort attacks by taking only one or two colchicine tablets when they feel the first “twinge” of an attack. This approach is likely to be safe in CKD, but it may be of value to only a few patients.
Nonsteroidal anti-inflammatory drugs can worsen chronic kidney disease
NSAIDs in high doses can effectively treat the pain and inflammation of acute gout. Indomethacin (Indocin) 50 mg three times daily has been standard NSAID therapy.
Other nonselective NSAIDs and NSAIDs that selectively inhibit cyclooxygenase 2 (COX-2) are effective, but all can cause acute renal toxicity or worsen CKD.10 Renal side effects include salt and water retention, acute tubular necrosis, acute interstitial nephritis, proteinuria, hypertension, hyperkalemia, and chronic renal injury.11
Even short-term use of high-dose NSAIDs should generally be avoided in patients with preexisting CKD, for whom there is no established safe threshold dose. When NSAIDs (including selective COX-2 inhibitors) are used, renal function should be monitored closely and the duration limited as much as possible.
Corticosteroids are often used to treat acute attacks
Due to the concerns about NSAIDs or colchicine to treat acute gout attacks in patients with CKD, corticosteroids are often used in this setting.
Intra-articular steroid injections are useful in treating acute gout limited to a single joint or bursa.12 However, one should first make sure that the joint is not infected: septic arthritis should ideally be excluded by arthrocentesis, particularly in immunosuppressed patients13 or those with end-stage renal disease, who are predisposed to bacteremia.
Oral, intramuscular, or intravenous steroids can provide complete relief from acute gout, although high doses (eg, prednisone 30–60 mg/day or the equivalent) are often needed. Common errors resulting in inefficacy include using too low a dose or not treating for a sufficient time before tapering or stopping. Groff and colleagues14 described 13 patients who received oral or intravenous steroids for acute gout. Nine patients received an initial single dose of prednisone ranging from 20 to 50 mg, with tapering over a mean of 10 days. Twelve of the 13 patients had improvement within 48 hours, and the signs and symptoms of acute gout resolved completely within 7 to 10 days.
We often give prednisone 40 mg daily until a day after the acute attack resolves and then taper over another 7 to 10 days. There are no data to guide steroid dosing in an evidence-based way, but we believe too short a course of therapy may result in return of symptoms.
Corticotropin and other agents: Effective but costly
Corticotropin shares the same indications as systemic corticosteroids, being used to treat flares when NSAIDs, intra-articular steroids, and colchicine are contraindicated. However, corticotropin is far more expensive than generic corticosteroids, costing nearly $2,000 for a single 80-IU dose, which may need to be repeated.
Corticotropin is available for subcutaneous or intramuscular injection. A single intramuscular injection of corticotropin gel (H.P. Acthar, 25–80 IU) may terminate an acute gout attack.15 However, many patients need another injection after 24 to 72 hours, which would require another visit to the physician. This treatment has been touted by some as being more effective than corticosteroid therapy, possibly because of a unique peripheral mechanism of action in addition to stimulating cortisol release.16
We rarely use corticotropin, in view of its cost as well as concerns about excessive sodium and water retention due to the release of multiple hormones from the adrenal gland. This may be especially deleterious in patients with CKD or congestive heart failure.17
Parenteral anti-tumor necrosis factor agents or interleukin 1 antagonists can be dramatically effective but are also expensive.18,19 For example, anakinra (Kineret) 100 mg costs about $73, and multiple daily doses may be necessary.
Under unique conditions in which they can be safely used (eg, patients with CKD, diabetes mellitus, liver disease), they may be cost-effective if they can shorten the stay of a hospitalized patient with acute gout.
PROPHYLACTIC ANTI-INFLAMMATORY THERAPY FOR PATIENTS WITH GOUT
Between attacks, the goal is to prevent new attacks through prophylactic management, which may include anti-inflammatory and hypouricemic therapy along with dietary instruction (such as avoiding excessive beer, liquor, and fructose ingestion).
Colchicine can be used as prophylaxis, with caution and monitoring
Although colchicine is not 100% effective, it markedly reduces the flare rate when started in low doses at the time hypouricemic therapy is initiated.20,21 (Hypouricemic therapy is discussed below.) We generally try to continue this prophylactic therapy, if the patient tolerates it, for at least 6 months—longer if tophi are still present or if attacks continue to occur.
If renal function is intact, colchicine can be prescribed at a dosage of 0.6 mg orally once or twice daily.21 In CKD, since the clearance of colchicine is reduced,4 the dosage should be reduced. Patients on colchicine for prophylaxis must be carefully monitored if the glomerular filtration rate is less than 50 mL/minute, or colchicine should be avoided altogether.6 Laboratory testing for colchicine levels is not routinely available and may be of limited value in predicting adverse effects; thus, recommendations about dose adjustments in CKD are empiric.
Wallace et al22 recommended a dose of 0.6 mg once daily if the creatinine clearance is 35 to 49 mL/minute and 0.6 mg every 2 to 3 days if it is 10 to 34 mL/minute, but there are no published long-term safety or efficacy data validating these reasonable (based on available information) dosing regimens.
Even with dose adjustment, caution is needed. Low-dose daily colchicine may be associated with reversible neuromyopathy and bone marrow suppression.7,23 Patients with neuromyopathy may complain of myalgias, proximal muscle weakness, and numbness and may have areflexia and decreased sensation. Laboratory findings include elevated creatine kinase and aminotransferase levels. We regularly check for leukopenia or elevated creatine kinase and aspartate aminotransferase levels in patients with CKD who are receiving colchicine in any dose.
Prolonged colchicine therapy should probably be avoided in patients on hemodialysis, as this drug is not removed by dialysis or by exchange transfusion, and the risk of toxicity under these circumstances may be high.22 When there is no viable alternative and the drug is given, patients should be closely monitored for signs of toxicity.
Concurrent (even short-term) treatment with most macrolide antibiotics, particularly clarithromycin (Biaxin), most statin drugs, ketoconazole (Nizoral), cyclosporine, and likely other drugs predisposes to colchicine toxicity by altering its distribution and elimination, and can in rare cases cause morbidity or death.24–26
NSAIDs are not optimal as prophylaxis in patients with chronic kidney disease
Little information has been published about using NSAIDs chronically to prevent flares, but they are not the optimal drugs to use in patients with CKD, as discussed above. In patients with end-stage renal disease, there are also concerns about NSAID-induced gastric and intestinal bleeding.
Low-dose steroids may not be effective as prophylaxis
Lower doses of steroids may not be effective as prophylaxis against gout flares, consistent with the common observation that gout flares still occur in organ transplant recipients who are taking maintenance doses of prednisone.13
PREVENTING FLARES BY LOWERING SERUM URATE LEVELS
If tophi are present, if radiography shows evidence of damage, if attacks are frequent or disabling, or if there are relative contraindications to the drugs that would be needed to treat acute attacks, then hypouricemic therapy should be strongly considered to reduce the burden of urate in the body, resorb tophi, and ultimately reduce the frequency of gout flares.20
Although intermittent therapy for attacks or prolonged prophylactic use of colchicine may prevent recurrent episodes of gouty arthritis and may be reasonable for many patients, this approach does not prevent continued urate deposition, with the potential development of bony erosions, tophaceous deposits, and chronic arthritis.
The definitive therapy for gouty arthritis is to deplete the periarticular deposits of urate by maintaining a low serum urate level. Urate-lowering therapy, when indicated, is almost always lifelong.
Four strategies for lowering serum urate
The serum urate concentration can be lowered in four ways:
Increasing renal uric acid excretion
Altering the diet
Decreasing urate synthesis
Converting urate to a more soluble metabolite.
Increasing uric acid excretion is rarely effective if renal function is impaired
Probenecid, sulfinpyrazone (Anturane), and losartan (Cozaar) modestly increase uric acid secretion and reduce serum urate levels, but they are rarely effective if the creatinine clearance rate is less than 60 mL/minute, and they require significant fluid intake for maximal efficacy.
Uricosuric drugs probably should be avoided in patients who excrete more than 1,000 mg of uric acid per day on a normal diet, since urinary uric acid stones may form. In practice, however, patients are given losartan to treat hypertension without attention to uric acid excretion.
More-potent urocosuric drugs are being tested in clinical trials.
Altering the diet: Traditional advice confirmed
The Health Professionals Follow-up Study27,28 prospectively examined the relation between diet and gout over 12 years in 47,150 men. The study confirmed some long-standing beliefs, such as that consuming meat, seafood, beer, and liquor increases the risk. Other risk factors were consumption of sugar-sweetened soft drinks and fructose, adiposity, weight gain, hypertension, and diuretic use. On the other hand, protein, wine, and purine-rich vegetables were not associated with gout flares. Low-fat dairy products may have a protective effect. Weight loss was found to be protective.
Low-purine diets are not very palatable, are difficult to adhere to, and are at best only minimally effective, lowering serum urate by 1 to 2 mg/dL. Low-protein diets designed to slow progression of CKD will likely also have only a slight effect on serum urate. Dietary change alone is not likely to dramatically lower serum urate levels.
Metabolizing urate with exogenous uricase
Rasburicase (Elitek) effectively converts urate to allantoin, which is more soluble, but rasburicase is fraught with allergic reactions and cannot be used as chronic therapy.
A pegylated intravenous uricase29 has just been approved by the US Food and Drug Administration (FDA); the retail cost is not yet known. It is dramatically effective in those patients able to use it chronically, but it has not been fully evaluated in patients with CKD.
Decreasing urate synthesis with allopurinol
Allopurinol acts by competitively inhibiting xanthine oxidase, the enzyme that converts hypoxanthine to xanthine and xanthine to uric acid. The drug, a structural analogue of hypoxanthine, is converted by xanthine oxidase to oxypurinol, which is an even more effective inhibitor of xanthine oxidase than allopurinol.
Allopurinol is metabolized in the liver and has a half-life of 1 to 3 hours, but oxypurinol, which is excreted in the urine, has a half-life of 12 to 17 hours. Because of these pharmacokinetic properties, allopurinol can usually be given once daily, and the dosage required to reduce serum urate levels should in theory be lower in patients with lower glomerular filtration rates.
Allopurinol (100- and 300-mg tablets) is approved by the FDA in doses of up to 800 mg/day to treat hyperuricemia in patients with gout,30 while guidelines from the British Society of Rheumatology advocate a maximum dose of 900 mg/day.31 These maximum doses are based on the limited amount of data with higher doses, not on documented toxicity.
Practice survey data in the United States indicate that most physicians prescribe no greater than 300 mg daily, although this dosage is likely to reduce the serum urate to less than 6 mg/dL—the goal level—in fewer than 50% of patients.20,32 Patients with normal renal function occasionally require more than 1,000 mg daily to reduce the serum urate level to less than 6 mg/dL.
How low should the serum urate level be?
Ideally, therapy should keep the serum urate level significantly below 6.7 mg/dL, the approximate saturation point of urate in physiologic fluids.
Lowering the serum urate level from 10 mg/dL to 7 mg/dL may seem encouraging, and the urate level may be in the laboratory “normal” range; however, urate may continue to precipitate in tissues if the concentration is greater than 6.7 mg/dL. A target of 6 mg/dL, used in clinical studies, is far enough below the saturation level to provide some margin for fluctuations in serum levels. A serum level of 6.0 mg/dL has thus been arbitrarily proposed as a reasonable therapeutic target.
The lower the serum urate level achieved during hypouricemic therapy, the faster the reduction in tophaceous deposits. With adequate urate lowering, tophi can be visibly reduced in less than a year of hypouricemic therapy.33,34
We have as yet no convincing evidence that lowering the serum urate level to less than 6.0 mg/dL is harmful, despite theoretical concerns that urate is a beneficial circulating antioxidant and epidemiologic observations that urate levels have been inversely correlated with progression of Parkinson disease.
Start low, go slow to avoid a flare
Rapid reduction of the serum urate level in a patient with chronic hyperuricemia and gout is likely to induce an acute flare.20 We have traditionally used a “start low and increase slowly” approach to escalating hypouricemic therapy in hopes of reducing the likelihood of causing a gout flare.
Without anti-inflammatory prophylaxis, acute flares associated with urate-lowering are extremely likely. In a 28-week trial of allopurinol, febuxostat, and placebo by Schumacher et al,33 during the first 8 weeks, when prophylaxis against gout flare was provided with either colchicine 0.6 mg once daily or naproxen (Naprosyn) 250 mg twice daily, the proportion of patients requiring treatment of gout flares was still 23% to 46%. When prophylaxis was stopped, the flare rate increased further.33
The more we acutely lower serum urate levels, the more likely flares are to occur. In the study by Schumacher et al,33 the percentage of patients needing treatment for gout flares during the first 8 weeks of the study, despite gout flare prophylaxis, was related to the percent reduction in serum urate by week 28 of the trial (Table 2).
IS IT NECESSARY TO ADJUST THE ALLOPURINOL DOSE IN CHRONIC KIDNEY DISEASE?
In 1984, Hande et al35 proposed that allopurinol doses be lower in patients with renal insufficiency, with a dosage scale based on creatinine clearance.
Their thoughtful proposal was based on data from six of their own patients and 72 others with severe allopurinol toxicity, mainly allopurinol hypersensitivity syndrome, reported in the literature.
Perez-Ruiz et al36 noted that patients who had experienced adverse effects from allopurinol in their series were likely to have had received “higher” doses of allopurinol, if the dosage was corrected for reduced oxypurinol elimination based on their estimated creatinine clearance.
However, most of these reactions occurred soon after initiating therapy, a temporal pattern more typical of non-dose-dependent allergic reactions. Additionally, allopurinol hypersensitivity has been linked to T-cell-mediated immune reactions to oxypurinol,37 a mechanism not likely linked to drug levels.
Arguments against dose adjustment
Despite the compelling information that allopurinol reactions are more common in CKD, adjusting the dosage of allopurinol has not been clearly shown to reduce the frequency of these reactions.
In a small retrospective analysis, Vázquez-Mellado et al38 reported that adjusting the allopurinol dosage according to creatinine clearance did not decrease the incidence of allopurinol hypersensitivity.
In a study in 250 patients, Dalbeth et al39 showed that the overall incidence of hypersensitivity reaction was 1.6%, and the incidence of allergic reactions did not decrease when allopurinol was given according to the dosing guidelines proposed by Hande et al.35 However, it is worth noting that, of the patients who received the recommended lower doses, only 19% achieved the target serum urate level of 6 mg/dL.39
Silverberg et al40 found that of 15 patients who developed hypersensitivity reactions to allopurinol, 10 had received doses that were low or appropriate according to the guidelines of Hande et al.35
More recently, Stamp et al41 found that gradually increasing the allopurinol dose above the proposed creatinine clearance-based dose was safe and effective. Thirty-one (89%) of the 35 patients who completed the study achieved the target serum urate level of 6 mg/dL, while only 3 of 45 who started the study developed rashes, which were not serious.
The small number of patients in these studies limits any strong conclusion, but at present there is no interventional study showing that allopurinol dosing adjustment based on glomerular filtration rate is effective or safer than dosing based on the serum urate level.
Our view on allopurinol dosing adjustment
We believe the initial observations of Hande et al35 and the subsequent meticulous data from Perez-Ruiz et al36 suggest a relationship between CKD and the occurrence of severe allopurinol reactions. However, these observations do not prove that dose adjustment will prevent these reactions.
In patients with normal kidney function, the FDA30 and the European League Against Rheumatism (EULAR)42 recommend slow upward titration, starting with 100 to 200 mg/day, which we agree should decrease the frequency of acute gout flares. The dose is increased by increments of 100 mg/day at intervals of 1 week (FDA recommendation) or 2 to 4 weeks (EULAR recommendation) until the serum urate level is lower than 6 mg/dL.
We believe the optimal approach to allopurinol dosing in patients with CKD remains uncertain. We generally escalate the dose slowly, with ongoing frequent laboratory and clinical monitoring, and we do not limit the maximal dose as suggested by Hande et al.35
An alternative strategy is to use the newer, far more expensive xanthine oxidase inhibitor febuxostat in patients with CKD, since it is not excreted by the kidney. We usually first try escalating doses of allopurinol.
FEBUXOSTAT, AN ALTERNATIVE TO ALLOPURINOL
Febuxostat is an oral nonpurine inhibitor of xanthine oxidase.43 Approved by the FDA in 2009, it is available in 40- and 80-mg tablets.
Unlike allopurinol, febuxostat is metabolized primarily by hepatic glucuronide formation and oxidation and then excreted in stool and urine,44 making it in theory an attractive agent in patients with renal insufficiency, bypassing the controversial dose-adjustment issue with allopurinol.
In the Febuxostat Versus Allopurinol Controlled Trial (FACT),20 a 52-week randomized, double-blind study in hyperuricemic patients with gout, serum urate levels were reduced to less than 6.0 mg/dL in over 50% of patients receiving febuxostat 80 mg or 120 mg once daily, while only 21% of patients receiving 300 mg of allopurinol achieved this goal. This does not imply that allopurinol at higher doses, as should be used in clinical practice,45 would not be equally effective. Patients with CKD were not included in this trial.
In the study by Schumacher et al,33 febuxostat 80, 120, or 240 mg once daily reduced serum urate. A small subset (35 patients) had mild to moderate renal insufficiency (serum creatinine 1.5–2 mg/dL).33 The number of patients with renal insufficiency who achieved the primary end point of a serum urate level lower than 6 mg/dL was 4 (44%) of 9 in the febuxostat 80-mg group, 5 (46%) of 11 in the 120-mg group, and 3 (60%) of 5 in the 240-mg group, while none of the 10 patients in the dose-adjusted allopurinol group achieved the primary end point (P < .05). Of note, 41% of the patients with normal renal function who received allopurinol achieved the primary end point.33 As proposed above, if the allopurinol dose had been slowly increased in the patients with renal insufficiency, it might have been equally effective.
Febuxostat has not been thoroughly evaluated in patients with severe CKD or in patients on hemodialysis.
A presumed niche indication of febuxostat is in patients allergic to allopurinol, since the drugs are not similar in chemical structure. However, at present, experience with this use is limited. Allopurinol-allergic patients were excluded from the clinical trials; thus, if there is any allergic overlap, it would not likely have been recognized in those studies. The FDA has received reports of patients who were allergic to allopurinol also having reactions to febuxostat, and it is currently evaluating these reports (personal communication).
Concern was raised over cardiovascular adverse events in patients treated with febuxostat during clinical trials. In the FACT trial, two patients died of cardiac causes.20 In the study by Schumacher et al,33 11 of 670 patients experienced cardiac adverse events in the febuxostat group vs 3 of 268 in the allopurinol group. Events included atrial fibrillation, chest pain, coronary artery disease, and myocardial infarction. However, this difference was not statistically significant.
Febuxostat costs much more than allopurinol. Currently, patients pay $153.88 for 1 month of febuxostat 40 or 80 mg from Cleveland Clinic pharmacy; 1 month of allopurinol costs $17.45 (300 mg) or $14.00 (100 mg). We believe febuxostat should be reserved for patients with documented intolerance to allopurinol in effective doses.
Monitoring serum urate levels is important in all patients on hypouricemic therapy so that dosage adjustments can be made until the target serum urate concentration is reached. In patients failing to meet target serum urate levels, patient adherence with the prescribed dosing should be specifically addressed because as many as 50% of patients do not adhere to their prescribed regimen.
DOES URATE-LOWERING THERAPY HAVE BENEFITS BEYOND GOUT?
Despite experimental animal data and a strong epidemiologic association between hyperuri-cemia and hypertension,46 metabolic syndrome, and rates of cardiovascular and all-cause mortality,47 the evidence from interventional trials so far does not support the routine use of hypo-uricemic therapy to prevent these outcomes.
Similarly, hyperuricemia has long been associated with renal disease, and there has been debate as to whether hyperuricemia is a result of kidney dysfunction or a contributing factor.46,48–51 A few studies have documented improvement of renal function after initiation of hypouricemic therapy.52 However, treating asymptomatic hyperuricemia to preserve kidney function remains controversial.
A recent study indicates that lowering the serum urate level with allopurinol can lower the blood pressure in hyperuricemic adolescents who have newly diagnosed primary hypertension.53 This does not indicate, however, that initiating hypouricemic therapy in patients with preexisting, long-standing hypertension will be successful.
RECOMMENDED FOR OUR PATIENT
As for our diabetic patient with an acute gout flare and creatinine clearance rate of 45 mL/minute, we would recommend:
Aspirating the knee, sending the fluid for bacterial culture, and then treating it with a local glucocorticoid injection
Starting colchicine 0.6 mg every day, with frequent monitoring for signs of toxicity (muscle pain, weakness, leukopenia, and elevations of creatine kinase and aspartate aminotransferase)
Increasing his allopurinol dose by 100 mg every 2 to 4 weeks until the target serum urate level of less than 6.0 mg/dL is reached
If he cannot tolerate allopurinol or if the target serum urate level is not achieved despite adequate doses of allopurinol (about 800 mg), we would switch to febuxostat 40 mg and increase the dose as needed to achieve the desired urate level.
References
Vázquez-Mellado J, García CG, Vázquez SG, et al. Metabolic syndrome and ischemic heart disease in gout. J Clin Rheumatol2004; 10:105–109.
Bonnel RA, Villalba ML, Karwoski CB, Beitz J. Deaths associated with inappropriate intravenous colchicine administration. J Emerg Med2002; 22:385–387.
Achtert G, Scherrmann JM, Christen MO. Pharmacokinetics/bioavailability of colchicine in healthy male volunteers. Eur J Drug Metab Pharmacokinet1989; 14:317–322.
Ben-Chetrit E, Scherrmann JM, Zylber-Katz E, Levy M. Colchicine disposition in patients with familial Mediterranean fever with renal impairment. J Rheumatol1994; 21:710–713.
Putterman C, Ben-Chetrit E, Caraco Y, Levy M. Colchicine intoxication: clinical pharmacology, risk factors, features, and management. Semin Arthritis Rheum1991; 21:143–155.
Aronoff G, Brater DC, Schrier R, Bennett WM. Use of drugs in patients with renal insufficiency. Workshop report. Blood Purif1994; 12:14–19.
Kuncl RW, Duncan G, Watson D, Alderson K, Rogawski MA, Peper M. Colchicine myopathy and neuropathy. N Engl J Med1987; 316:1562–1568.
Ahern MJ, Reid C, Gordon TP, McCredie M, Brooks PM, Jones M. Does colchicine work? The results of the first controlled study in acute gout. Aust N Z J Med1987; 17:301–304.
Terkeltaub R, Furst D, Bennett K, Kook K, Crockett RS, Davis WM. High versus low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum2010, 62:1060–1068.
Whelton A. Nephrotoxicity of nonsteroidal anti-inflammatory drugs: physiologic foundations and clinical implications. Am J Med1999; 106:13S–24S.
Wali RK, Henrich WL. Recent developments in toxic nephropathy. Curr Opin Nephrol Hypertens2002; 11:155–163.
Fernández C, Noguera R, González JA, Pascual E. Treatment of acute attacks of gout with a small dose of intraarticular triamcinolone acetonide. J Rheumatol1999; 26:2285–2286.
Clive DM. Renal transplant-associated hyperuricemia and gout. J Am Soc Nephrol2000; 11:974–979.
Groff GD, Franck WA, Raddatz DA. Systemic steroid therapy for acute gout: a clinical trial and review of the literature. Semin Arthritis Rheum1990; 19:329–336.
Ritter J, Kerr LD, Valeriano-Marcet J, Spiera H. ACTH revisited: effective treatment for acute crystal induced synovitis in patients with multiple medical problems. J Rheumatol1994; 21:696–699.
Getting SJ, Christian HC, Flower RJ, Perretti M. Activation of melanocortin type 3 receptor as a molecular mechanism for adrenocorticotropic hormone efficacy in gouty arthritis. Arthritis Rheum2002; 46:2765–2775.
Connell JM, Whitworth JA, Davies DL, Lever AF, Richards AM, Fraser R. Effects of ACTH and cortisol administration on blood pressure, electrolyte metabolism, atrial natriuretic peptide and renal function in normal man. J Hypertens1987; 5:425–433.
Tausche AK, Richter K, Grässler A, Hänsel S, Roch B, Schröder HE. Severe gouty arthritis refractory to anti-inflammatory drugs: treatment with anti-tumour necrosis factor alpha as a new therapeutic option. Ann Rheum Dis2004; 63:1351–1352.
So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther2007; 9:R28.
Becker MA, Schumacher HR, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med2005; 353:2450–2461.
Borstad GC, Bryant LR, Abel MP, Scroggie DA, Harris MD, Alloway JA. Colchicine for prophylaxis of acute flares when initiating allopurinol for chronic gouty arthritis. J Rheumatol2004; 31:2429–2432.
Wallace SL, Singer JZ, Duncan GJ, Wigley FM, Kuncl RW. Renal function predicts colchicine toxicity: guidelines for the prophylactic use of colchicine in gout. J Rheumatol1991; 18:264–269.
Wilbur K, Makowsky M. Colchicine myotoxicity: case reports and literature review. Pharmacotherapy2004; 24:1784–1792.
Hung IF, Wu AK, Cheng VC, et al. Fatal interaction between clarithromycin and colchicine in patients with renal insufficiency: a retrospective study. Clin Infect Dis2005; 41:291–300.
Alayli G, Cengiz K, Cantürk F, Durmus D, Akyol Y, Menekse EB. Acute myopathy in a patient with concomitant use of pravastatin and colchicine. Ann Pharmacother2005; 39:1358–1361.
Ducloux D, Schuller V, Bresson-Vautrin C, Chalopin JM. Colchicine myopathy in renal transplant recipients on cyclosporin. Nephrol Dial Transplant1997; 12:2389–2392.
Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med2004; 350:1093–1103.
Choi HK, Curhan G. Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. BMJ2008; 336:309–312.
Sundy JS, Becker MA, Baraf HS, et al; Pegloticase Phase 2 Study Investigators. Reduction of plasma urate levels following treatment with multiple doses of pegloticase (polyethylene glycol-conjugated uricase) in patients with treatment-failure gout: results of a phase II randomized study. Arthritis Rheum2008; 58:2882–2891.
Jordan KM, Cameron JS, Snaith M, et al; British Society for Rheumatology and British Health Professionals in Rheumatology Standards, Guidelines and Audit Working Group (SGAWG). British Society for Rheumatology and British Health Professionals in Rheumatology guideline for the management of gout. Rheumatology (Oxford)2007; 46:1372–1374.
Perez-Ruiz F, Alonso-Ruiz A, Calabozo M, Herrero-Beites A, García-Erauskin G, Ruiz-Lucea E. Efficacy of allopurinol and benzbromarone for the control of hyperuricaemia. A pathogenic approach to the treatment of primary chronic gout. Ann Rheum Dis1998; 57:545–549.
Schumacher HR, Becker MA, Wortmann RL, et al. Effects of febuxostat versus allopurinol and placebo in reducing serum urate in subjects with hyperuricemia and gout: a 28-week, phase III, randomized, double-blind, parallel-group trial. Arthritis Rheum2008; 59:1540–1548.
Perez-Ruiz F, Calabozo M, Pijoan JI, Herrero-Beites AM, Ruibal A. Effect of urate-lowering therapy on the velocity of size reduction of tophi in chronic gout. Arthritis Rheum2002; 47:356–360.
Hande KR, Noone RM, Stone WJ. Severe allopurinol toxicity. Description and guidelines for prevention in patients with renal insufficiency. Am J Med1984; 76:47–56.
Perez-Ruiz F, Hernando I, Villar I, Nolla JM. Correction of allopurinol dosing should be based on clearance of creatinine, but not plasma creatinine levels: another insight to allopurinol-related toxicity. J Clin Rheumatol2005; 11:129–133.
Hung SI, Chung WH, Liou LB, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A2005; 102:4134–4139.
Vázquez-Mellado J, Morales EM, Pacheco-Tena C, Burgos-Vargas R. Relation between adverse events associated with allopurinol and renal function in patients with gout. Ann Rheum Dis2001; 60:981–983.
Dalbeth N, Kumar S, Stamp L, Gow P. Dose adjustment of allopurinol according to creatinine clearance does not provide adequate control of hyperuricemia in patients with gout. J Rheumatol2006; 33:1646–1650.
Silverberg MS, Mallela R, Lesse AJ, Bonner MR, Baer AN, Li C. Allopurinol hypersensitivity reactions: a case-control study of the role of renal dosing (abstract). Arthritis Rheum2009; 60(suppl 10):1106.
Stamp LK, O'Donnell JL, Zhang M, et al. Using allopurinol above the dose based on creatinine clearance is effective and safe in chronic gout, including in those with renal impairment. Arthritis Rhem2010; doi:10.1002/art.30119. E-pub ahead of print. Accessed 10/29/2010.
Zhang W, Doherty M, Bardin T, et al; EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis2006; 65:1312–1324.
Okamoto K, Eger BT, Nishino T, Kondo S, Pai EF, Nishino T. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J Biol Chem2003; 278:1848–1855.
Khosravan R, Grabowski BA, Wu JT, Joseph-Ridge N, Vernillet L. Pharmacokinetics, pharmacodynamics and safety of febuxostat, a non-purine selective inhibitor of xanthine oxidase, in a dose escalation study in healthy subjects. Clin Pharmacokinet2006; 45:821–841.
Reinders MK, Haagsma C, Jansen TL, et al. A randomised controlled trial on the efficacy and tolerability with dose escalation of allopurinol 300–600 mg/day versus benzbromarone 100–200 mg/day in patients with gout. Ann Rheum Dis2009; 68:892–897.
Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension2001; 38:1101–1106.
Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med2008; 359:1811–1821.
Beck LH. Requiem for gouty nephropathy. Kidney Int1986; 30:280–287.
Tomita M, Mizuno S, Yamanaka H, et al. Does hyperuricemia affect mortality? A prospective cohort study of Japanese male workers. J Epidemiol2000; 10:403–409.
Kang DH, Nakagawa T. Uric acid and chronic renal disease: possible implication of hyperuricemia on progression of renal disease. Semin Nephrol2005; 25:43–49.
Iseki K, Oshiro S, Tozawa M, Iseki C, Ikemiya Y, Takishita S. Significance of hyperuricemia on the early detection of renal failure in a cohort of screened subjects. Hypertens Res2001; 24:691–697.
Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med1987; 82:421–426.
Siu YP, Leung KT, Tong MK, Kwan TH. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis2006; 47:51–59.
Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA2008; 300:924–932.
Hossam El-Zawawy, MD, MS Assistant Professor, Charles E. Schmidt College of Medicine, Florida Atlantic University, Boca Raton; Associate Staff, Department of Rheumatic and Immunologic Diseases, Cleveland Clinic, Weston, FL
Brian F. Mandell, MD, PhD Chairman, Department of Medicine, Education Institute, Cleveland Clinic; Professor of Medicine, Lerner College of Medicine of Case Western Reserve University, Cleveland, OH; Department of Rheumatic and Immunologic Diseases, Cleveland Clinic; and Editor-in-Chief, Cleveland Clinic Journal of Medicine
Address: Brian F. Mandell, MD, PhD, Education Institute, NA10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail mandelb@ccf.org
Dr. Mandell has disclosed that he has been a consultant for Takeda Pharmaceutical Company and has conducted a clinical trial for Savient Pharmaceuticals.
Hossam El-Zawawy, MD, MS Assistant Professor, Charles E. Schmidt College of Medicine, Florida Atlantic University, Boca Raton; Associate Staff, Department of Rheumatic and Immunologic Diseases, Cleveland Clinic, Weston, FL
Brian F. Mandell, MD, PhD Chairman, Department of Medicine, Education Institute, Cleveland Clinic; Professor of Medicine, Lerner College of Medicine of Case Western Reserve University, Cleveland, OH; Department of Rheumatic and Immunologic Diseases, Cleveland Clinic; and Editor-in-Chief, Cleveland Clinic Journal of Medicine
Address: Brian F. Mandell, MD, PhD, Education Institute, NA10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail mandelb@ccf.org
Dr. Mandell has disclosed that he has been a consultant for Takeda Pharmaceutical Company and has conducted a clinical trial for Savient Pharmaceuticals.
Author and Disclosure Information
Hossam El-Zawawy, MD, MS Assistant Professor, Charles E. Schmidt College of Medicine, Florida Atlantic University, Boca Raton; Associate Staff, Department of Rheumatic and Immunologic Diseases, Cleveland Clinic, Weston, FL
Brian F. Mandell, MD, PhD Chairman, Department of Medicine, Education Institute, Cleveland Clinic; Professor of Medicine, Lerner College of Medicine of Case Western Reserve University, Cleveland, OH; Department of Rheumatic and Immunologic Diseases, Cleveland Clinic; and Editor-in-Chief, Cleveland Clinic Journal of Medicine
Address: Brian F. Mandell, MD, PhD, Education Institute, NA10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail mandelb@ccf.org
Dr. Mandell has disclosed that he has been a consultant for Takeda Pharmaceutical Company and has conducted a clinical trial for Savient Pharmaceuticals.
You have a 54-year-old black patient with gout, diabetes mellitus, hypertension, and chronic kidney disease (CKD). He has an acute gout flare involving his right knee. In the past year he has had four attacks of gout in the ankles and knees, which you treated with intra-articular glucocorticoid injections. He has been on allopurinol (Zyloprim) 200 mg daily, but his last serum urate level was 9.4 mg/dL (reference range 3.0–8.0). His creatinine clearance is 45 mL/minute (reference range 85–125).
In view of his kidney disease, you are concerned about increasing his dose of allopurinol, but also about the need to treat his frequent attacks. How should you manage this patient?
GOUT IS CHALLENGING TO TREAT IN PATIENTS WITH KIDNEY DISEASE
A major challenge in treating patients with gout is to avoid therapeutic interactions with common comorbidities, including hypertension, insulin resistance, coronary artery disease, heart failure, and especially CKD.1
In this paper, we discuss approaches to and controversies in the management of gout and hyperuricemia in patients with CKD. Unfortunately, the evidence from clinical trials to guide treatment decisions is limited; therefore, decisions must often be based on experience and pathophysiologic principles.
GENERAL GOALS OF GOUT THERAPY
Depending on the patient and the stage of the disease, the goals in treating patients with gout are to:
Terminate acute attacks as promptly and safely as possible
Prevent recurrences of acute gout attacks
Prevent or reverse complications resulting from deposition of monosodium urate in the joints, in the kidneys, or at other sites.
These goals are more difficult to achieve in patients with CKD because of the potential complications from many of the available drugs.
TERMINATING ACUTE GOUT FLARES
In patients with acute gout, treatment is aimed at quickly resolving pain and inflammation.
Several types of drugs can terminate acute gout flares. The choice in most situations is colchicine (Colcrys); a nonsteroidal anti-inflammatory drug (NSAID); a corticosteroid; or corticotropin (ACTH).
However, in patients with CKD, there are concerns about using colchicine or NSAIDs, and corticotropin is very expensive; thus, corticosteroids are often used.
Colchicine’s clearance is reduced in CKD
Colchicine is somewhat effective in treating acute gout attacks and probably more effective in preventing attacks.
Due to concerns about inappropriate dosing and reported deaths,2 the intravenous formulation is not available in many countries, including the United States.
After oral administration, colchicine is rapidly absorbed, with a bioavailability of up to 50%. It undergoes metabolism by the liver, and its metabolites are excreted by renal and biliary-intestinal routes. Up to 20% of the active drug is excreted by the kidneys.3
Colchicine’s clearance is significantly reduced in patients with renal or hepatic insufficiency, and the drug may accumulate in cells, with resultant toxicity.4 Colchicine-induced toxicity has been observed when the drug was used for acute treatment, as well as for chronic prophylaxis of gout in patients with CKD; thus, alternative agents for treating acute attacks should be considered.5,6 With prolonged use, reversible colchicine-induced axonal neuropathy, neutropenia, and vacuolar myopathy can develop in patients with CKD.7
In a trial in patients with normal renal function, nearly 100% who received an initial dose of 1 mg followed by 0.5 mg every 2 hours developed diarrhea at a median time of 24 hours.8 Emesis may also occur.
A lower dose of 1.8 mg (two 0.6-mg pills followed by one pill an hour later) was well tolerated but only moderately effective in treating acute gout, causing at least a 50% reduction in pain at 24 hours in only 38% of patients.9 This study does not clarify the dosage to use to completely resolve attacks. Using additional colchicine likely will increase the response rate, but will also increase side effects. Patients with CKD were not included.
Some patients, as shown in the above trial, can abort attacks by taking only one or two colchicine tablets when they feel the first “twinge” of an attack. This approach is likely to be safe in CKD, but it may be of value to only a few patients.
Nonsteroidal anti-inflammatory drugs can worsen chronic kidney disease
NSAIDs in high doses can effectively treat the pain and inflammation of acute gout. Indomethacin (Indocin) 50 mg three times daily has been standard NSAID therapy.
Other nonselective NSAIDs and NSAIDs that selectively inhibit cyclooxygenase 2 (COX-2) are effective, but all can cause acute renal toxicity or worsen CKD.10 Renal side effects include salt and water retention, acute tubular necrosis, acute interstitial nephritis, proteinuria, hypertension, hyperkalemia, and chronic renal injury.11
Even short-term use of high-dose NSAIDs should generally be avoided in patients with preexisting CKD, for whom there is no established safe threshold dose. When NSAIDs (including selective COX-2 inhibitors) are used, renal function should be monitored closely and the duration limited as much as possible.
Corticosteroids are often used to treat acute attacks
Due to the concerns about NSAIDs or colchicine to treat acute gout attacks in patients with CKD, corticosteroids are often used in this setting.
Intra-articular steroid injections are useful in treating acute gout limited to a single joint or bursa.12 However, one should first make sure that the joint is not infected: septic arthritis should ideally be excluded by arthrocentesis, particularly in immunosuppressed patients13 or those with end-stage renal disease, who are predisposed to bacteremia.
Oral, intramuscular, or intravenous steroids can provide complete relief from acute gout, although high doses (eg, prednisone 30–60 mg/day or the equivalent) are often needed. Common errors resulting in inefficacy include using too low a dose or not treating for a sufficient time before tapering or stopping. Groff and colleagues14 described 13 patients who received oral or intravenous steroids for acute gout. Nine patients received an initial single dose of prednisone ranging from 20 to 50 mg, with tapering over a mean of 10 days. Twelve of the 13 patients had improvement within 48 hours, and the signs and symptoms of acute gout resolved completely within 7 to 10 days.
We often give prednisone 40 mg daily until a day after the acute attack resolves and then taper over another 7 to 10 days. There are no data to guide steroid dosing in an evidence-based way, but we believe too short a course of therapy may result in return of symptoms.
Corticotropin and other agents: Effective but costly
Corticotropin shares the same indications as systemic corticosteroids, being used to treat flares when NSAIDs, intra-articular steroids, and colchicine are contraindicated. However, corticotropin is far more expensive than generic corticosteroids, costing nearly $2,000 for a single 80-IU dose, which may need to be repeated.
Corticotropin is available for subcutaneous or intramuscular injection. A single intramuscular injection of corticotropin gel (H.P. Acthar, 25–80 IU) may terminate an acute gout attack.15 However, many patients need another injection after 24 to 72 hours, which would require another visit to the physician. This treatment has been touted by some as being more effective than corticosteroid therapy, possibly because of a unique peripheral mechanism of action in addition to stimulating cortisol release.16
We rarely use corticotropin, in view of its cost as well as concerns about excessive sodium and water retention due to the release of multiple hormones from the adrenal gland. This may be especially deleterious in patients with CKD or congestive heart failure.17
Parenteral anti-tumor necrosis factor agents or interleukin 1 antagonists can be dramatically effective but are also expensive.18,19 For example, anakinra (Kineret) 100 mg costs about $73, and multiple daily doses may be necessary.
Under unique conditions in which they can be safely used (eg, patients with CKD, diabetes mellitus, liver disease), they may be cost-effective if they can shorten the stay of a hospitalized patient with acute gout.
PROPHYLACTIC ANTI-INFLAMMATORY THERAPY FOR PATIENTS WITH GOUT
Between attacks, the goal is to prevent new attacks through prophylactic management, which may include anti-inflammatory and hypouricemic therapy along with dietary instruction (such as avoiding excessive beer, liquor, and fructose ingestion).
Colchicine can be used as prophylaxis, with caution and monitoring
Although colchicine is not 100% effective, it markedly reduces the flare rate when started in low doses at the time hypouricemic therapy is initiated.20,21 (Hypouricemic therapy is discussed below.) We generally try to continue this prophylactic therapy, if the patient tolerates it, for at least 6 months—longer if tophi are still present or if attacks continue to occur.
If renal function is intact, colchicine can be prescribed at a dosage of 0.6 mg orally once or twice daily.21 In CKD, since the clearance of colchicine is reduced,4 the dosage should be reduced. Patients on colchicine for prophylaxis must be carefully monitored if the glomerular filtration rate is less than 50 mL/minute, or colchicine should be avoided altogether.6 Laboratory testing for colchicine levels is not routinely available and may be of limited value in predicting adverse effects; thus, recommendations about dose adjustments in CKD are empiric.
Wallace et al22 recommended a dose of 0.6 mg once daily if the creatinine clearance is 35 to 49 mL/minute and 0.6 mg every 2 to 3 days if it is 10 to 34 mL/minute, but there are no published long-term safety or efficacy data validating these reasonable (based on available information) dosing regimens.
Even with dose adjustment, caution is needed. Low-dose daily colchicine may be associated with reversible neuromyopathy and bone marrow suppression.7,23 Patients with neuromyopathy may complain of myalgias, proximal muscle weakness, and numbness and may have areflexia and decreased sensation. Laboratory findings include elevated creatine kinase and aminotransferase levels. We regularly check for leukopenia or elevated creatine kinase and aspartate aminotransferase levels in patients with CKD who are receiving colchicine in any dose.
Prolonged colchicine therapy should probably be avoided in patients on hemodialysis, as this drug is not removed by dialysis or by exchange transfusion, and the risk of toxicity under these circumstances may be high.22 When there is no viable alternative and the drug is given, patients should be closely monitored for signs of toxicity.
Concurrent (even short-term) treatment with most macrolide antibiotics, particularly clarithromycin (Biaxin), most statin drugs, ketoconazole (Nizoral), cyclosporine, and likely other drugs predisposes to colchicine toxicity by altering its distribution and elimination, and can in rare cases cause morbidity or death.24–26
NSAIDs are not optimal as prophylaxis in patients with chronic kidney disease
Little information has been published about using NSAIDs chronically to prevent flares, but they are not the optimal drugs to use in patients with CKD, as discussed above. In patients with end-stage renal disease, there are also concerns about NSAID-induced gastric and intestinal bleeding.
Low-dose steroids may not be effective as prophylaxis
Lower doses of steroids may not be effective as prophylaxis against gout flares, consistent with the common observation that gout flares still occur in organ transplant recipients who are taking maintenance doses of prednisone.13
PREVENTING FLARES BY LOWERING SERUM URATE LEVELS
If tophi are present, if radiography shows evidence of damage, if attacks are frequent or disabling, or if there are relative contraindications to the drugs that would be needed to treat acute attacks, then hypouricemic therapy should be strongly considered to reduce the burden of urate in the body, resorb tophi, and ultimately reduce the frequency of gout flares.20
Although intermittent therapy for attacks or prolonged prophylactic use of colchicine may prevent recurrent episodes of gouty arthritis and may be reasonable for many patients, this approach does not prevent continued urate deposition, with the potential development of bony erosions, tophaceous deposits, and chronic arthritis.
The definitive therapy for gouty arthritis is to deplete the periarticular deposits of urate by maintaining a low serum urate level. Urate-lowering therapy, when indicated, is almost always lifelong.
Four strategies for lowering serum urate
The serum urate concentration can be lowered in four ways:
Increasing renal uric acid excretion
Altering the diet
Decreasing urate synthesis
Converting urate to a more soluble metabolite.
Increasing uric acid excretion is rarely effective if renal function is impaired
Probenecid, sulfinpyrazone (Anturane), and losartan (Cozaar) modestly increase uric acid secretion and reduce serum urate levels, but they are rarely effective if the creatinine clearance rate is less than 60 mL/minute, and they require significant fluid intake for maximal efficacy.
Uricosuric drugs probably should be avoided in patients who excrete more than 1,000 mg of uric acid per day on a normal diet, since urinary uric acid stones may form. In practice, however, patients are given losartan to treat hypertension without attention to uric acid excretion.
More-potent urocosuric drugs are being tested in clinical trials.
Altering the diet: Traditional advice confirmed
The Health Professionals Follow-up Study27,28 prospectively examined the relation between diet and gout over 12 years in 47,150 men. The study confirmed some long-standing beliefs, such as that consuming meat, seafood, beer, and liquor increases the risk. Other risk factors were consumption of sugar-sweetened soft drinks and fructose, adiposity, weight gain, hypertension, and diuretic use. On the other hand, protein, wine, and purine-rich vegetables were not associated with gout flares. Low-fat dairy products may have a protective effect. Weight loss was found to be protective.
Low-purine diets are not very palatable, are difficult to adhere to, and are at best only minimally effective, lowering serum urate by 1 to 2 mg/dL. Low-protein diets designed to slow progression of CKD will likely also have only a slight effect on serum urate. Dietary change alone is not likely to dramatically lower serum urate levels.
Metabolizing urate with exogenous uricase
Rasburicase (Elitek) effectively converts urate to allantoin, which is more soluble, but rasburicase is fraught with allergic reactions and cannot be used as chronic therapy.
A pegylated intravenous uricase29 has just been approved by the US Food and Drug Administration (FDA); the retail cost is not yet known. It is dramatically effective in those patients able to use it chronically, but it has not been fully evaluated in patients with CKD.
Decreasing urate synthesis with allopurinol
Allopurinol acts by competitively inhibiting xanthine oxidase, the enzyme that converts hypoxanthine to xanthine and xanthine to uric acid. The drug, a structural analogue of hypoxanthine, is converted by xanthine oxidase to oxypurinol, which is an even more effective inhibitor of xanthine oxidase than allopurinol.
Allopurinol is metabolized in the liver and has a half-life of 1 to 3 hours, but oxypurinol, which is excreted in the urine, has a half-life of 12 to 17 hours. Because of these pharmacokinetic properties, allopurinol can usually be given once daily, and the dosage required to reduce serum urate levels should in theory be lower in patients with lower glomerular filtration rates.
Allopurinol (100- and 300-mg tablets) is approved by the FDA in doses of up to 800 mg/day to treat hyperuricemia in patients with gout,30 while guidelines from the British Society of Rheumatology advocate a maximum dose of 900 mg/day.31 These maximum doses are based on the limited amount of data with higher doses, not on documented toxicity.
Practice survey data in the United States indicate that most physicians prescribe no greater than 300 mg daily, although this dosage is likely to reduce the serum urate to less than 6 mg/dL—the goal level—in fewer than 50% of patients.20,32 Patients with normal renal function occasionally require more than 1,000 mg daily to reduce the serum urate level to less than 6 mg/dL.
How low should the serum urate level be?
Ideally, therapy should keep the serum urate level significantly below 6.7 mg/dL, the approximate saturation point of urate in physiologic fluids.
Lowering the serum urate level from 10 mg/dL to 7 mg/dL may seem encouraging, and the urate level may be in the laboratory “normal” range; however, urate may continue to precipitate in tissues if the concentration is greater than 6.7 mg/dL. A target of 6 mg/dL, used in clinical studies, is far enough below the saturation level to provide some margin for fluctuations in serum levels. A serum level of 6.0 mg/dL has thus been arbitrarily proposed as a reasonable therapeutic target.
The lower the serum urate level achieved during hypouricemic therapy, the faster the reduction in tophaceous deposits. With adequate urate lowering, tophi can be visibly reduced in less than a year of hypouricemic therapy.33,34
We have as yet no convincing evidence that lowering the serum urate level to less than 6.0 mg/dL is harmful, despite theoretical concerns that urate is a beneficial circulating antioxidant and epidemiologic observations that urate levels have been inversely correlated with progression of Parkinson disease.
Start low, go slow to avoid a flare
Rapid reduction of the serum urate level in a patient with chronic hyperuricemia and gout is likely to induce an acute flare.20 We have traditionally used a “start low and increase slowly” approach to escalating hypouricemic therapy in hopes of reducing the likelihood of causing a gout flare.
Without anti-inflammatory prophylaxis, acute flares associated with urate-lowering are extremely likely. In a 28-week trial of allopurinol, febuxostat, and placebo by Schumacher et al,33 during the first 8 weeks, when prophylaxis against gout flare was provided with either colchicine 0.6 mg once daily or naproxen (Naprosyn) 250 mg twice daily, the proportion of patients requiring treatment of gout flares was still 23% to 46%. When prophylaxis was stopped, the flare rate increased further.33
The more we acutely lower serum urate levels, the more likely flares are to occur. In the study by Schumacher et al,33 the percentage of patients needing treatment for gout flares during the first 8 weeks of the study, despite gout flare prophylaxis, was related to the percent reduction in serum urate by week 28 of the trial (Table 2).
IS IT NECESSARY TO ADJUST THE ALLOPURINOL DOSE IN CHRONIC KIDNEY DISEASE?
In 1984, Hande et al35 proposed that allopurinol doses be lower in patients with renal insufficiency, with a dosage scale based on creatinine clearance.
Their thoughtful proposal was based on data from six of their own patients and 72 others with severe allopurinol toxicity, mainly allopurinol hypersensitivity syndrome, reported in the literature.
Perez-Ruiz et al36 noted that patients who had experienced adverse effects from allopurinol in their series were likely to have had received “higher” doses of allopurinol, if the dosage was corrected for reduced oxypurinol elimination based on their estimated creatinine clearance.
However, most of these reactions occurred soon after initiating therapy, a temporal pattern more typical of non-dose-dependent allergic reactions. Additionally, allopurinol hypersensitivity has been linked to T-cell-mediated immune reactions to oxypurinol,37 a mechanism not likely linked to drug levels.
Arguments against dose adjustment
Despite the compelling information that allopurinol reactions are more common in CKD, adjusting the dosage of allopurinol has not been clearly shown to reduce the frequency of these reactions.
In a small retrospective analysis, Vázquez-Mellado et al38 reported that adjusting the allopurinol dosage according to creatinine clearance did not decrease the incidence of allopurinol hypersensitivity.
In a study in 250 patients, Dalbeth et al39 showed that the overall incidence of hypersensitivity reaction was 1.6%, and the incidence of allergic reactions did not decrease when allopurinol was given according to the dosing guidelines proposed by Hande et al.35 However, it is worth noting that, of the patients who received the recommended lower doses, only 19% achieved the target serum urate level of 6 mg/dL.39
Silverberg et al40 found that of 15 patients who developed hypersensitivity reactions to allopurinol, 10 had received doses that were low or appropriate according to the guidelines of Hande et al.35
More recently, Stamp et al41 found that gradually increasing the allopurinol dose above the proposed creatinine clearance-based dose was safe and effective. Thirty-one (89%) of the 35 patients who completed the study achieved the target serum urate level of 6 mg/dL, while only 3 of 45 who started the study developed rashes, which were not serious.
The small number of patients in these studies limits any strong conclusion, but at present there is no interventional study showing that allopurinol dosing adjustment based on glomerular filtration rate is effective or safer than dosing based on the serum urate level.
Our view on allopurinol dosing adjustment
We believe the initial observations of Hande et al35 and the subsequent meticulous data from Perez-Ruiz et al36 suggest a relationship between CKD and the occurrence of severe allopurinol reactions. However, these observations do not prove that dose adjustment will prevent these reactions.
In patients with normal kidney function, the FDA30 and the European League Against Rheumatism (EULAR)42 recommend slow upward titration, starting with 100 to 200 mg/day, which we agree should decrease the frequency of acute gout flares. The dose is increased by increments of 100 mg/day at intervals of 1 week (FDA recommendation) or 2 to 4 weeks (EULAR recommendation) until the serum urate level is lower than 6 mg/dL.
We believe the optimal approach to allopurinol dosing in patients with CKD remains uncertain. We generally escalate the dose slowly, with ongoing frequent laboratory and clinical monitoring, and we do not limit the maximal dose as suggested by Hande et al.35
An alternative strategy is to use the newer, far more expensive xanthine oxidase inhibitor febuxostat in patients with CKD, since it is not excreted by the kidney. We usually first try escalating doses of allopurinol.
FEBUXOSTAT, AN ALTERNATIVE TO ALLOPURINOL
Febuxostat is an oral nonpurine inhibitor of xanthine oxidase.43 Approved by the FDA in 2009, it is available in 40- and 80-mg tablets.
Unlike allopurinol, febuxostat is metabolized primarily by hepatic glucuronide formation and oxidation and then excreted in stool and urine,44 making it in theory an attractive agent in patients with renal insufficiency, bypassing the controversial dose-adjustment issue with allopurinol.
In the Febuxostat Versus Allopurinol Controlled Trial (FACT),20 a 52-week randomized, double-blind study in hyperuricemic patients with gout, serum urate levels were reduced to less than 6.0 mg/dL in over 50% of patients receiving febuxostat 80 mg or 120 mg once daily, while only 21% of patients receiving 300 mg of allopurinol achieved this goal. This does not imply that allopurinol at higher doses, as should be used in clinical practice,45 would not be equally effective. Patients with CKD were not included in this trial.
In the study by Schumacher et al,33 febuxostat 80, 120, or 240 mg once daily reduced serum urate. A small subset (35 patients) had mild to moderate renal insufficiency (serum creatinine 1.5–2 mg/dL).33 The number of patients with renal insufficiency who achieved the primary end point of a serum urate level lower than 6 mg/dL was 4 (44%) of 9 in the febuxostat 80-mg group, 5 (46%) of 11 in the 120-mg group, and 3 (60%) of 5 in the 240-mg group, while none of the 10 patients in the dose-adjusted allopurinol group achieved the primary end point (P < .05). Of note, 41% of the patients with normal renal function who received allopurinol achieved the primary end point.33 As proposed above, if the allopurinol dose had been slowly increased in the patients with renal insufficiency, it might have been equally effective.
Febuxostat has not been thoroughly evaluated in patients with severe CKD or in patients on hemodialysis.
A presumed niche indication of febuxostat is in patients allergic to allopurinol, since the drugs are not similar in chemical structure. However, at present, experience with this use is limited. Allopurinol-allergic patients were excluded from the clinical trials; thus, if there is any allergic overlap, it would not likely have been recognized in those studies. The FDA has received reports of patients who were allergic to allopurinol also having reactions to febuxostat, and it is currently evaluating these reports (personal communication).
Concern was raised over cardiovascular adverse events in patients treated with febuxostat during clinical trials. In the FACT trial, two patients died of cardiac causes.20 In the study by Schumacher et al,33 11 of 670 patients experienced cardiac adverse events in the febuxostat group vs 3 of 268 in the allopurinol group. Events included atrial fibrillation, chest pain, coronary artery disease, and myocardial infarction. However, this difference was not statistically significant.
Febuxostat costs much more than allopurinol. Currently, patients pay $153.88 for 1 month of febuxostat 40 or 80 mg from Cleveland Clinic pharmacy; 1 month of allopurinol costs $17.45 (300 mg) or $14.00 (100 mg). We believe febuxostat should be reserved for patients with documented intolerance to allopurinol in effective doses.
Monitoring serum urate levels is important in all patients on hypouricemic therapy so that dosage adjustments can be made until the target serum urate concentration is reached. In patients failing to meet target serum urate levels, patient adherence with the prescribed dosing should be specifically addressed because as many as 50% of patients do not adhere to their prescribed regimen.
DOES URATE-LOWERING THERAPY HAVE BENEFITS BEYOND GOUT?
Despite experimental animal data and a strong epidemiologic association between hyperuri-cemia and hypertension,46 metabolic syndrome, and rates of cardiovascular and all-cause mortality,47 the evidence from interventional trials so far does not support the routine use of hypo-uricemic therapy to prevent these outcomes.
Similarly, hyperuricemia has long been associated with renal disease, and there has been debate as to whether hyperuricemia is a result of kidney dysfunction or a contributing factor.46,48–51 A few studies have documented improvement of renal function after initiation of hypouricemic therapy.52 However, treating asymptomatic hyperuricemia to preserve kidney function remains controversial.
A recent study indicates that lowering the serum urate level with allopurinol can lower the blood pressure in hyperuricemic adolescents who have newly diagnosed primary hypertension.53 This does not indicate, however, that initiating hypouricemic therapy in patients with preexisting, long-standing hypertension will be successful.
RECOMMENDED FOR OUR PATIENT
As for our diabetic patient with an acute gout flare and creatinine clearance rate of 45 mL/minute, we would recommend:
Aspirating the knee, sending the fluid for bacterial culture, and then treating it with a local glucocorticoid injection
Starting colchicine 0.6 mg every day, with frequent monitoring for signs of toxicity (muscle pain, weakness, leukopenia, and elevations of creatine kinase and aspartate aminotransferase)
Increasing his allopurinol dose by 100 mg every 2 to 4 weeks until the target serum urate level of less than 6.0 mg/dL is reached
If he cannot tolerate allopurinol or if the target serum urate level is not achieved despite adequate doses of allopurinol (about 800 mg), we would switch to febuxostat 40 mg and increase the dose as needed to achieve the desired urate level.
You have a 54-year-old black patient with gout, diabetes mellitus, hypertension, and chronic kidney disease (CKD). He has an acute gout flare involving his right knee. In the past year he has had four attacks of gout in the ankles and knees, which you treated with intra-articular glucocorticoid injections. He has been on allopurinol (Zyloprim) 200 mg daily, but his last serum urate level was 9.4 mg/dL (reference range 3.0–8.0). His creatinine clearance is 45 mL/minute (reference range 85–125).
In view of his kidney disease, you are concerned about increasing his dose of allopurinol, but also about the need to treat his frequent attacks. How should you manage this patient?
GOUT IS CHALLENGING TO TREAT IN PATIENTS WITH KIDNEY DISEASE
A major challenge in treating patients with gout is to avoid therapeutic interactions with common comorbidities, including hypertension, insulin resistance, coronary artery disease, heart failure, and especially CKD.1
In this paper, we discuss approaches to and controversies in the management of gout and hyperuricemia in patients with CKD. Unfortunately, the evidence from clinical trials to guide treatment decisions is limited; therefore, decisions must often be based on experience and pathophysiologic principles.
GENERAL GOALS OF GOUT THERAPY
Depending on the patient and the stage of the disease, the goals in treating patients with gout are to:
Terminate acute attacks as promptly and safely as possible
Prevent recurrences of acute gout attacks
Prevent or reverse complications resulting from deposition of monosodium urate in the joints, in the kidneys, or at other sites.
These goals are more difficult to achieve in patients with CKD because of the potential complications from many of the available drugs.
TERMINATING ACUTE GOUT FLARES
In patients with acute gout, treatment is aimed at quickly resolving pain and inflammation.
Several types of drugs can terminate acute gout flares. The choice in most situations is colchicine (Colcrys); a nonsteroidal anti-inflammatory drug (NSAID); a corticosteroid; or corticotropin (ACTH).
However, in patients with CKD, there are concerns about using colchicine or NSAIDs, and corticotropin is very expensive; thus, corticosteroids are often used.
Colchicine’s clearance is reduced in CKD
Colchicine is somewhat effective in treating acute gout attacks and probably more effective in preventing attacks.
Due to concerns about inappropriate dosing and reported deaths,2 the intravenous formulation is not available in many countries, including the United States.
After oral administration, colchicine is rapidly absorbed, with a bioavailability of up to 50%. It undergoes metabolism by the liver, and its metabolites are excreted by renal and biliary-intestinal routes. Up to 20% of the active drug is excreted by the kidneys.3
Colchicine’s clearance is significantly reduced in patients with renal or hepatic insufficiency, and the drug may accumulate in cells, with resultant toxicity.4 Colchicine-induced toxicity has been observed when the drug was used for acute treatment, as well as for chronic prophylaxis of gout in patients with CKD; thus, alternative agents for treating acute attacks should be considered.5,6 With prolonged use, reversible colchicine-induced axonal neuropathy, neutropenia, and vacuolar myopathy can develop in patients with CKD.7
In a trial in patients with normal renal function, nearly 100% who received an initial dose of 1 mg followed by 0.5 mg every 2 hours developed diarrhea at a median time of 24 hours.8 Emesis may also occur.
A lower dose of 1.8 mg (two 0.6-mg pills followed by one pill an hour later) was well tolerated but only moderately effective in treating acute gout, causing at least a 50% reduction in pain at 24 hours in only 38% of patients.9 This study does not clarify the dosage to use to completely resolve attacks. Using additional colchicine likely will increase the response rate, but will also increase side effects. Patients with CKD were not included.
Some patients, as shown in the above trial, can abort attacks by taking only one or two colchicine tablets when they feel the first “twinge” of an attack. This approach is likely to be safe in CKD, but it may be of value to only a few patients.
Nonsteroidal anti-inflammatory drugs can worsen chronic kidney disease
NSAIDs in high doses can effectively treat the pain and inflammation of acute gout. Indomethacin (Indocin) 50 mg three times daily has been standard NSAID therapy.
Other nonselective NSAIDs and NSAIDs that selectively inhibit cyclooxygenase 2 (COX-2) are effective, but all can cause acute renal toxicity or worsen CKD.10 Renal side effects include salt and water retention, acute tubular necrosis, acute interstitial nephritis, proteinuria, hypertension, hyperkalemia, and chronic renal injury.11
Even short-term use of high-dose NSAIDs should generally be avoided in patients with preexisting CKD, for whom there is no established safe threshold dose. When NSAIDs (including selective COX-2 inhibitors) are used, renal function should be monitored closely and the duration limited as much as possible.
Corticosteroids are often used to treat acute attacks
Due to the concerns about NSAIDs or colchicine to treat acute gout attacks in patients with CKD, corticosteroids are often used in this setting.
Intra-articular steroid injections are useful in treating acute gout limited to a single joint or bursa.12 However, one should first make sure that the joint is not infected: septic arthritis should ideally be excluded by arthrocentesis, particularly in immunosuppressed patients13 or those with end-stage renal disease, who are predisposed to bacteremia.
Oral, intramuscular, or intravenous steroids can provide complete relief from acute gout, although high doses (eg, prednisone 30–60 mg/day or the equivalent) are often needed. Common errors resulting in inefficacy include using too low a dose or not treating for a sufficient time before tapering or stopping. Groff and colleagues14 described 13 patients who received oral or intravenous steroids for acute gout. Nine patients received an initial single dose of prednisone ranging from 20 to 50 mg, with tapering over a mean of 10 days. Twelve of the 13 patients had improvement within 48 hours, and the signs and symptoms of acute gout resolved completely within 7 to 10 days.
We often give prednisone 40 mg daily until a day after the acute attack resolves and then taper over another 7 to 10 days. There are no data to guide steroid dosing in an evidence-based way, but we believe too short a course of therapy may result in return of symptoms.
Corticotropin and other agents: Effective but costly
Corticotropin shares the same indications as systemic corticosteroids, being used to treat flares when NSAIDs, intra-articular steroids, and colchicine are contraindicated. However, corticotropin is far more expensive than generic corticosteroids, costing nearly $2,000 for a single 80-IU dose, which may need to be repeated.
Corticotropin is available for subcutaneous or intramuscular injection. A single intramuscular injection of corticotropin gel (H.P. Acthar, 25–80 IU) may terminate an acute gout attack.15 However, many patients need another injection after 24 to 72 hours, which would require another visit to the physician. This treatment has been touted by some as being more effective than corticosteroid therapy, possibly because of a unique peripheral mechanism of action in addition to stimulating cortisol release.16
We rarely use corticotropin, in view of its cost as well as concerns about excessive sodium and water retention due to the release of multiple hormones from the adrenal gland. This may be especially deleterious in patients with CKD or congestive heart failure.17
Parenteral anti-tumor necrosis factor agents or interleukin 1 antagonists can be dramatically effective but are also expensive.18,19 For example, anakinra (Kineret) 100 mg costs about $73, and multiple daily doses may be necessary.
Under unique conditions in which they can be safely used (eg, patients with CKD, diabetes mellitus, liver disease), they may be cost-effective if they can shorten the stay of a hospitalized patient with acute gout.
PROPHYLACTIC ANTI-INFLAMMATORY THERAPY FOR PATIENTS WITH GOUT
Between attacks, the goal is to prevent new attacks through prophylactic management, which may include anti-inflammatory and hypouricemic therapy along with dietary instruction (such as avoiding excessive beer, liquor, and fructose ingestion).
Colchicine can be used as prophylaxis, with caution and monitoring
Although colchicine is not 100% effective, it markedly reduces the flare rate when started in low doses at the time hypouricemic therapy is initiated.20,21 (Hypouricemic therapy is discussed below.) We generally try to continue this prophylactic therapy, if the patient tolerates it, for at least 6 months—longer if tophi are still present or if attacks continue to occur.
If renal function is intact, colchicine can be prescribed at a dosage of 0.6 mg orally once or twice daily.21 In CKD, since the clearance of colchicine is reduced,4 the dosage should be reduced. Patients on colchicine for prophylaxis must be carefully monitored if the glomerular filtration rate is less than 50 mL/minute, or colchicine should be avoided altogether.6 Laboratory testing for colchicine levels is not routinely available and may be of limited value in predicting adverse effects; thus, recommendations about dose adjustments in CKD are empiric.
Wallace et al22 recommended a dose of 0.6 mg once daily if the creatinine clearance is 35 to 49 mL/minute and 0.6 mg every 2 to 3 days if it is 10 to 34 mL/minute, but there are no published long-term safety or efficacy data validating these reasonable (based on available information) dosing regimens.
Even with dose adjustment, caution is needed. Low-dose daily colchicine may be associated with reversible neuromyopathy and bone marrow suppression.7,23 Patients with neuromyopathy may complain of myalgias, proximal muscle weakness, and numbness and may have areflexia and decreased sensation. Laboratory findings include elevated creatine kinase and aminotransferase levels. We regularly check for leukopenia or elevated creatine kinase and aspartate aminotransferase levels in patients with CKD who are receiving colchicine in any dose.
Prolonged colchicine therapy should probably be avoided in patients on hemodialysis, as this drug is not removed by dialysis or by exchange transfusion, and the risk of toxicity under these circumstances may be high.22 When there is no viable alternative and the drug is given, patients should be closely monitored for signs of toxicity.
Concurrent (even short-term) treatment with most macrolide antibiotics, particularly clarithromycin (Biaxin), most statin drugs, ketoconazole (Nizoral), cyclosporine, and likely other drugs predisposes to colchicine toxicity by altering its distribution and elimination, and can in rare cases cause morbidity or death.24–26
NSAIDs are not optimal as prophylaxis in patients with chronic kidney disease
Little information has been published about using NSAIDs chronically to prevent flares, but they are not the optimal drugs to use in patients with CKD, as discussed above. In patients with end-stage renal disease, there are also concerns about NSAID-induced gastric and intestinal bleeding.
Low-dose steroids may not be effective as prophylaxis
Lower doses of steroids may not be effective as prophylaxis against gout flares, consistent with the common observation that gout flares still occur in organ transplant recipients who are taking maintenance doses of prednisone.13
PREVENTING FLARES BY LOWERING SERUM URATE LEVELS
If tophi are present, if radiography shows evidence of damage, if attacks are frequent or disabling, or if there are relative contraindications to the drugs that would be needed to treat acute attacks, then hypouricemic therapy should be strongly considered to reduce the burden of urate in the body, resorb tophi, and ultimately reduce the frequency of gout flares.20
Although intermittent therapy for attacks or prolonged prophylactic use of colchicine may prevent recurrent episodes of gouty arthritis and may be reasonable for many patients, this approach does not prevent continued urate deposition, with the potential development of bony erosions, tophaceous deposits, and chronic arthritis.
The definitive therapy for gouty arthritis is to deplete the periarticular deposits of urate by maintaining a low serum urate level. Urate-lowering therapy, when indicated, is almost always lifelong.
Four strategies for lowering serum urate
The serum urate concentration can be lowered in four ways:
Increasing renal uric acid excretion
Altering the diet
Decreasing urate synthesis
Converting urate to a more soluble metabolite.
Increasing uric acid excretion is rarely effective if renal function is impaired
Probenecid, sulfinpyrazone (Anturane), and losartan (Cozaar) modestly increase uric acid secretion and reduce serum urate levels, but they are rarely effective if the creatinine clearance rate is less than 60 mL/minute, and they require significant fluid intake for maximal efficacy.
Uricosuric drugs probably should be avoided in patients who excrete more than 1,000 mg of uric acid per day on a normal diet, since urinary uric acid stones may form. In practice, however, patients are given losartan to treat hypertension without attention to uric acid excretion.
More-potent urocosuric drugs are being tested in clinical trials.
Altering the diet: Traditional advice confirmed
The Health Professionals Follow-up Study27,28 prospectively examined the relation between diet and gout over 12 years in 47,150 men. The study confirmed some long-standing beliefs, such as that consuming meat, seafood, beer, and liquor increases the risk. Other risk factors were consumption of sugar-sweetened soft drinks and fructose, adiposity, weight gain, hypertension, and diuretic use. On the other hand, protein, wine, and purine-rich vegetables were not associated with gout flares. Low-fat dairy products may have a protective effect. Weight loss was found to be protective.
Low-purine diets are not very palatable, are difficult to adhere to, and are at best only minimally effective, lowering serum urate by 1 to 2 mg/dL. Low-protein diets designed to slow progression of CKD will likely also have only a slight effect on serum urate. Dietary change alone is not likely to dramatically lower serum urate levels.
Metabolizing urate with exogenous uricase
Rasburicase (Elitek) effectively converts urate to allantoin, which is more soluble, but rasburicase is fraught with allergic reactions and cannot be used as chronic therapy.
A pegylated intravenous uricase29 has just been approved by the US Food and Drug Administration (FDA); the retail cost is not yet known. It is dramatically effective in those patients able to use it chronically, but it has not been fully evaluated in patients with CKD.
Decreasing urate synthesis with allopurinol
Allopurinol acts by competitively inhibiting xanthine oxidase, the enzyme that converts hypoxanthine to xanthine and xanthine to uric acid. The drug, a structural analogue of hypoxanthine, is converted by xanthine oxidase to oxypurinol, which is an even more effective inhibitor of xanthine oxidase than allopurinol.
Allopurinol is metabolized in the liver and has a half-life of 1 to 3 hours, but oxypurinol, which is excreted in the urine, has a half-life of 12 to 17 hours. Because of these pharmacokinetic properties, allopurinol can usually be given once daily, and the dosage required to reduce serum urate levels should in theory be lower in patients with lower glomerular filtration rates.
Allopurinol (100- and 300-mg tablets) is approved by the FDA in doses of up to 800 mg/day to treat hyperuricemia in patients with gout,30 while guidelines from the British Society of Rheumatology advocate a maximum dose of 900 mg/day.31 These maximum doses are based on the limited amount of data with higher doses, not on documented toxicity.
Practice survey data in the United States indicate that most physicians prescribe no greater than 300 mg daily, although this dosage is likely to reduce the serum urate to less than 6 mg/dL—the goal level—in fewer than 50% of patients.20,32 Patients with normal renal function occasionally require more than 1,000 mg daily to reduce the serum urate level to less than 6 mg/dL.
How low should the serum urate level be?
Ideally, therapy should keep the serum urate level significantly below 6.7 mg/dL, the approximate saturation point of urate in physiologic fluids.
Lowering the serum urate level from 10 mg/dL to 7 mg/dL may seem encouraging, and the urate level may be in the laboratory “normal” range; however, urate may continue to precipitate in tissues if the concentration is greater than 6.7 mg/dL. A target of 6 mg/dL, used in clinical studies, is far enough below the saturation level to provide some margin for fluctuations in serum levels. A serum level of 6.0 mg/dL has thus been arbitrarily proposed as a reasonable therapeutic target.
The lower the serum urate level achieved during hypouricemic therapy, the faster the reduction in tophaceous deposits. With adequate urate lowering, tophi can be visibly reduced in less than a year of hypouricemic therapy.33,34
We have as yet no convincing evidence that lowering the serum urate level to less than 6.0 mg/dL is harmful, despite theoretical concerns that urate is a beneficial circulating antioxidant and epidemiologic observations that urate levels have been inversely correlated with progression of Parkinson disease.
Start low, go slow to avoid a flare
Rapid reduction of the serum urate level in a patient with chronic hyperuricemia and gout is likely to induce an acute flare.20 We have traditionally used a “start low and increase slowly” approach to escalating hypouricemic therapy in hopes of reducing the likelihood of causing a gout flare.
Without anti-inflammatory prophylaxis, acute flares associated with urate-lowering are extremely likely. In a 28-week trial of allopurinol, febuxostat, and placebo by Schumacher et al,33 during the first 8 weeks, when prophylaxis against gout flare was provided with either colchicine 0.6 mg once daily or naproxen (Naprosyn) 250 mg twice daily, the proportion of patients requiring treatment of gout flares was still 23% to 46%. When prophylaxis was stopped, the flare rate increased further.33
The more we acutely lower serum urate levels, the more likely flares are to occur. In the study by Schumacher et al,33 the percentage of patients needing treatment for gout flares during the first 8 weeks of the study, despite gout flare prophylaxis, was related to the percent reduction in serum urate by week 28 of the trial (Table 2).
IS IT NECESSARY TO ADJUST THE ALLOPURINOL DOSE IN CHRONIC KIDNEY DISEASE?
In 1984, Hande et al35 proposed that allopurinol doses be lower in patients with renal insufficiency, with a dosage scale based on creatinine clearance.
Their thoughtful proposal was based on data from six of their own patients and 72 others with severe allopurinol toxicity, mainly allopurinol hypersensitivity syndrome, reported in the literature.
Perez-Ruiz et al36 noted that patients who had experienced adverse effects from allopurinol in their series were likely to have had received “higher” doses of allopurinol, if the dosage was corrected for reduced oxypurinol elimination based on their estimated creatinine clearance.
However, most of these reactions occurred soon after initiating therapy, a temporal pattern more typical of non-dose-dependent allergic reactions. Additionally, allopurinol hypersensitivity has been linked to T-cell-mediated immune reactions to oxypurinol,37 a mechanism not likely linked to drug levels.
Arguments against dose adjustment
Despite the compelling information that allopurinol reactions are more common in CKD, adjusting the dosage of allopurinol has not been clearly shown to reduce the frequency of these reactions.
In a small retrospective analysis, Vázquez-Mellado et al38 reported that adjusting the allopurinol dosage according to creatinine clearance did not decrease the incidence of allopurinol hypersensitivity.
In a study in 250 patients, Dalbeth et al39 showed that the overall incidence of hypersensitivity reaction was 1.6%, and the incidence of allergic reactions did not decrease when allopurinol was given according to the dosing guidelines proposed by Hande et al.35 However, it is worth noting that, of the patients who received the recommended lower doses, only 19% achieved the target serum urate level of 6 mg/dL.39
Silverberg et al40 found that of 15 patients who developed hypersensitivity reactions to allopurinol, 10 had received doses that were low or appropriate according to the guidelines of Hande et al.35
More recently, Stamp et al41 found that gradually increasing the allopurinol dose above the proposed creatinine clearance-based dose was safe and effective. Thirty-one (89%) of the 35 patients who completed the study achieved the target serum urate level of 6 mg/dL, while only 3 of 45 who started the study developed rashes, which were not serious.
The small number of patients in these studies limits any strong conclusion, but at present there is no interventional study showing that allopurinol dosing adjustment based on glomerular filtration rate is effective or safer than dosing based on the serum urate level.
Our view on allopurinol dosing adjustment
We believe the initial observations of Hande et al35 and the subsequent meticulous data from Perez-Ruiz et al36 suggest a relationship between CKD and the occurrence of severe allopurinol reactions. However, these observations do not prove that dose adjustment will prevent these reactions.
In patients with normal kidney function, the FDA30 and the European League Against Rheumatism (EULAR)42 recommend slow upward titration, starting with 100 to 200 mg/day, which we agree should decrease the frequency of acute gout flares. The dose is increased by increments of 100 mg/day at intervals of 1 week (FDA recommendation) or 2 to 4 weeks (EULAR recommendation) until the serum urate level is lower than 6 mg/dL.
We believe the optimal approach to allopurinol dosing in patients with CKD remains uncertain. We generally escalate the dose slowly, with ongoing frequent laboratory and clinical monitoring, and we do not limit the maximal dose as suggested by Hande et al.35
An alternative strategy is to use the newer, far more expensive xanthine oxidase inhibitor febuxostat in patients with CKD, since it is not excreted by the kidney. We usually first try escalating doses of allopurinol.
FEBUXOSTAT, AN ALTERNATIVE TO ALLOPURINOL
Febuxostat is an oral nonpurine inhibitor of xanthine oxidase.43 Approved by the FDA in 2009, it is available in 40- and 80-mg tablets.
Unlike allopurinol, febuxostat is metabolized primarily by hepatic glucuronide formation and oxidation and then excreted in stool and urine,44 making it in theory an attractive agent in patients with renal insufficiency, bypassing the controversial dose-adjustment issue with allopurinol.
In the Febuxostat Versus Allopurinol Controlled Trial (FACT),20 a 52-week randomized, double-blind study in hyperuricemic patients with gout, serum urate levels were reduced to less than 6.0 mg/dL in over 50% of patients receiving febuxostat 80 mg or 120 mg once daily, while only 21% of patients receiving 300 mg of allopurinol achieved this goal. This does not imply that allopurinol at higher doses, as should be used in clinical practice,45 would not be equally effective. Patients with CKD were not included in this trial.
In the study by Schumacher et al,33 febuxostat 80, 120, or 240 mg once daily reduced serum urate. A small subset (35 patients) had mild to moderate renal insufficiency (serum creatinine 1.5–2 mg/dL).33 The number of patients with renal insufficiency who achieved the primary end point of a serum urate level lower than 6 mg/dL was 4 (44%) of 9 in the febuxostat 80-mg group, 5 (46%) of 11 in the 120-mg group, and 3 (60%) of 5 in the 240-mg group, while none of the 10 patients in the dose-adjusted allopurinol group achieved the primary end point (P < .05). Of note, 41% of the patients with normal renal function who received allopurinol achieved the primary end point.33 As proposed above, if the allopurinol dose had been slowly increased in the patients with renal insufficiency, it might have been equally effective.
Febuxostat has not been thoroughly evaluated in patients with severe CKD or in patients on hemodialysis.
A presumed niche indication of febuxostat is in patients allergic to allopurinol, since the drugs are not similar in chemical structure. However, at present, experience with this use is limited. Allopurinol-allergic patients were excluded from the clinical trials; thus, if there is any allergic overlap, it would not likely have been recognized in those studies. The FDA has received reports of patients who were allergic to allopurinol also having reactions to febuxostat, and it is currently evaluating these reports (personal communication).
Concern was raised over cardiovascular adverse events in patients treated with febuxostat during clinical trials. In the FACT trial, two patients died of cardiac causes.20 In the study by Schumacher et al,33 11 of 670 patients experienced cardiac adverse events in the febuxostat group vs 3 of 268 in the allopurinol group. Events included atrial fibrillation, chest pain, coronary artery disease, and myocardial infarction. However, this difference was not statistically significant.
Febuxostat costs much more than allopurinol. Currently, patients pay $153.88 for 1 month of febuxostat 40 or 80 mg from Cleveland Clinic pharmacy; 1 month of allopurinol costs $17.45 (300 mg) or $14.00 (100 mg). We believe febuxostat should be reserved for patients with documented intolerance to allopurinol in effective doses.
Monitoring serum urate levels is important in all patients on hypouricemic therapy so that dosage adjustments can be made until the target serum urate concentration is reached. In patients failing to meet target serum urate levels, patient adherence with the prescribed dosing should be specifically addressed because as many as 50% of patients do not adhere to their prescribed regimen.
DOES URATE-LOWERING THERAPY HAVE BENEFITS BEYOND GOUT?
Despite experimental animal data and a strong epidemiologic association between hyperuri-cemia and hypertension,46 metabolic syndrome, and rates of cardiovascular and all-cause mortality,47 the evidence from interventional trials so far does not support the routine use of hypo-uricemic therapy to prevent these outcomes.
Similarly, hyperuricemia has long been associated with renal disease, and there has been debate as to whether hyperuricemia is a result of kidney dysfunction or a contributing factor.46,48–51 A few studies have documented improvement of renal function after initiation of hypouricemic therapy.52 However, treating asymptomatic hyperuricemia to preserve kidney function remains controversial.
A recent study indicates that lowering the serum urate level with allopurinol can lower the blood pressure in hyperuricemic adolescents who have newly diagnosed primary hypertension.53 This does not indicate, however, that initiating hypouricemic therapy in patients with preexisting, long-standing hypertension will be successful.
RECOMMENDED FOR OUR PATIENT
As for our diabetic patient with an acute gout flare and creatinine clearance rate of 45 mL/minute, we would recommend:
Aspirating the knee, sending the fluid for bacterial culture, and then treating it with a local glucocorticoid injection
Starting colchicine 0.6 mg every day, with frequent monitoring for signs of toxicity (muscle pain, weakness, leukopenia, and elevations of creatine kinase and aspartate aminotransferase)
Increasing his allopurinol dose by 100 mg every 2 to 4 weeks until the target serum urate level of less than 6.0 mg/dL is reached
If he cannot tolerate allopurinol or if the target serum urate level is not achieved despite adequate doses of allopurinol (about 800 mg), we would switch to febuxostat 40 mg and increase the dose as needed to achieve the desired urate level.
References
Vázquez-Mellado J, García CG, Vázquez SG, et al. Metabolic syndrome and ischemic heart disease in gout. J Clin Rheumatol2004; 10:105–109.
Bonnel RA, Villalba ML, Karwoski CB, Beitz J. Deaths associated with inappropriate intravenous colchicine administration. J Emerg Med2002; 22:385–387.
Achtert G, Scherrmann JM, Christen MO. Pharmacokinetics/bioavailability of colchicine in healthy male volunteers. Eur J Drug Metab Pharmacokinet1989; 14:317–322.
Ben-Chetrit E, Scherrmann JM, Zylber-Katz E, Levy M. Colchicine disposition in patients with familial Mediterranean fever with renal impairment. J Rheumatol1994; 21:710–713.
Putterman C, Ben-Chetrit E, Caraco Y, Levy M. Colchicine intoxication: clinical pharmacology, risk factors, features, and management. Semin Arthritis Rheum1991; 21:143–155.
Aronoff G, Brater DC, Schrier R, Bennett WM. Use of drugs in patients with renal insufficiency. Workshop report. Blood Purif1994; 12:14–19.
Kuncl RW, Duncan G, Watson D, Alderson K, Rogawski MA, Peper M. Colchicine myopathy and neuropathy. N Engl J Med1987; 316:1562–1568.
Ahern MJ, Reid C, Gordon TP, McCredie M, Brooks PM, Jones M. Does colchicine work? The results of the first controlled study in acute gout. Aust N Z J Med1987; 17:301–304.
Terkeltaub R, Furst D, Bennett K, Kook K, Crockett RS, Davis WM. High versus low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum2010, 62:1060–1068.
Whelton A. Nephrotoxicity of nonsteroidal anti-inflammatory drugs: physiologic foundations and clinical implications. Am J Med1999; 106:13S–24S.
Wali RK, Henrich WL. Recent developments in toxic nephropathy. Curr Opin Nephrol Hypertens2002; 11:155–163.
Fernández C, Noguera R, González JA, Pascual E. Treatment of acute attacks of gout with a small dose of intraarticular triamcinolone acetonide. J Rheumatol1999; 26:2285–2286.
Clive DM. Renal transplant-associated hyperuricemia and gout. J Am Soc Nephrol2000; 11:974–979.
Groff GD, Franck WA, Raddatz DA. Systemic steroid therapy for acute gout: a clinical trial and review of the literature. Semin Arthritis Rheum1990; 19:329–336.
Ritter J, Kerr LD, Valeriano-Marcet J, Spiera H. ACTH revisited: effective treatment for acute crystal induced synovitis in patients with multiple medical problems. J Rheumatol1994; 21:696–699.
Getting SJ, Christian HC, Flower RJ, Perretti M. Activation of melanocortin type 3 receptor as a molecular mechanism for adrenocorticotropic hormone efficacy in gouty arthritis. Arthritis Rheum2002; 46:2765–2775.
Connell JM, Whitworth JA, Davies DL, Lever AF, Richards AM, Fraser R. Effects of ACTH and cortisol administration on blood pressure, electrolyte metabolism, atrial natriuretic peptide and renal function in normal man. J Hypertens1987; 5:425–433.
Tausche AK, Richter K, Grässler A, Hänsel S, Roch B, Schröder HE. Severe gouty arthritis refractory to anti-inflammatory drugs: treatment with anti-tumour necrosis factor alpha as a new therapeutic option. Ann Rheum Dis2004; 63:1351–1352.
So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther2007; 9:R28.
Becker MA, Schumacher HR, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med2005; 353:2450–2461.
Borstad GC, Bryant LR, Abel MP, Scroggie DA, Harris MD, Alloway JA. Colchicine for prophylaxis of acute flares when initiating allopurinol for chronic gouty arthritis. J Rheumatol2004; 31:2429–2432.
Wallace SL, Singer JZ, Duncan GJ, Wigley FM, Kuncl RW. Renal function predicts colchicine toxicity: guidelines for the prophylactic use of colchicine in gout. J Rheumatol1991; 18:264–269.
Wilbur K, Makowsky M. Colchicine myotoxicity: case reports and literature review. Pharmacotherapy2004; 24:1784–1792.
Hung IF, Wu AK, Cheng VC, et al. Fatal interaction between clarithromycin and colchicine in patients with renal insufficiency: a retrospective study. Clin Infect Dis2005; 41:291–300.
Alayli G, Cengiz K, Cantürk F, Durmus D, Akyol Y, Menekse EB. Acute myopathy in a patient with concomitant use of pravastatin and colchicine. Ann Pharmacother2005; 39:1358–1361.
Ducloux D, Schuller V, Bresson-Vautrin C, Chalopin JM. Colchicine myopathy in renal transplant recipients on cyclosporin. Nephrol Dial Transplant1997; 12:2389–2392.
Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med2004; 350:1093–1103.
Choi HK, Curhan G. Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. BMJ2008; 336:309–312.
Sundy JS, Becker MA, Baraf HS, et al; Pegloticase Phase 2 Study Investigators. Reduction of plasma urate levels following treatment with multiple doses of pegloticase (polyethylene glycol-conjugated uricase) in patients with treatment-failure gout: results of a phase II randomized study. Arthritis Rheum2008; 58:2882–2891.
Jordan KM, Cameron JS, Snaith M, et al; British Society for Rheumatology and British Health Professionals in Rheumatology Standards, Guidelines and Audit Working Group (SGAWG). British Society for Rheumatology and British Health Professionals in Rheumatology guideline for the management of gout. Rheumatology (Oxford)2007; 46:1372–1374.
Perez-Ruiz F, Alonso-Ruiz A, Calabozo M, Herrero-Beites A, García-Erauskin G, Ruiz-Lucea E. Efficacy of allopurinol and benzbromarone for the control of hyperuricaemia. A pathogenic approach to the treatment of primary chronic gout. Ann Rheum Dis1998; 57:545–549.
Schumacher HR, Becker MA, Wortmann RL, et al. Effects of febuxostat versus allopurinol and placebo in reducing serum urate in subjects with hyperuricemia and gout: a 28-week, phase III, randomized, double-blind, parallel-group trial. Arthritis Rheum2008; 59:1540–1548.
Perez-Ruiz F, Calabozo M, Pijoan JI, Herrero-Beites AM, Ruibal A. Effect of urate-lowering therapy on the velocity of size reduction of tophi in chronic gout. Arthritis Rheum2002; 47:356–360.
Hande KR, Noone RM, Stone WJ. Severe allopurinol toxicity. Description and guidelines for prevention in patients with renal insufficiency. Am J Med1984; 76:47–56.
Perez-Ruiz F, Hernando I, Villar I, Nolla JM. Correction of allopurinol dosing should be based on clearance of creatinine, but not plasma creatinine levels: another insight to allopurinol-related toxicity. J Clin Rheumatol2005; 11:129–133.
Hung SI, Chung WH, Liou LB, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A2005; 102:4134–4139.
Vázquez-Mellado J, Morales EM, Pacheco-Tena C, Burgos-Vargas R. Relation between adverse events associated with allopurinol and renal function in patients with gout. Ann Rheum Dis2001; 60:981–983.
Dalbeth N, Kumar S, Stamp L, Gow P. Dose adjustment of allopurinol according to creatinine clearance does not provide adequate control of hyperuricemia in patients with gout. J Rheumatol2006; 33:1646–1650.
Silverberg MS, Mallela R, Lesse AJ, Bonner MR, Baer AN, Li C. Allopurinol hypersensitivity reactions: a case-control study of the role of renal dosing (abstract). Arthritis Rheum2009; 60(suppl 10):1106.
Stamp LK, O'Donnell JL, Zhang M, et al. Using allopurinol above the dose based on creatinine clearance is effective and safe in chronic gout, including in those with renal impairment. Arthritis Rhem2010; doi:10.1002/art.30119. E-pub ahead of print. Accessed 10/29/2010.
Zhang W, Doherty M, Bardin T, et al; EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis2006; 65:1312–1324.
Okamoto K, Eger BT, Nishino T, Kondo S, Pai EF, Nishino T. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J Biol Chem2003; 278:1848–1855.
Khosravan R, Grabowski BA, Wu JT, Joseph-Ridge N, Vernillet L. Pharmacokinetics, pharmacodynamics and safety of febuxostat, a non-purine selective inhibitor of xanthine oxidase, in a dose escalation study in healthy subjects. Clin Pharmacokinet2006; 45:821–841.
Reinders MK, Haagsma C, Jansen TL, et al. A randomised controlled trial on the efficacy and tolerability with dose escalation of allopurinol 300–600 mg/day versus benzbromarone 100–200 mg/day in patients with gout. Ann Rheum Dis2009; 68:892–897.
Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension2001; 38:1101–1106.
Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med2008; 359:1811–1821.
Beck LH. Requiem for gouty nephropathy. Kidney Int1986; 30:280–287.
Tomita M, Mizuno S, Yamanaka H, et al. Does hyperuricemia affect mortality? A prospective cohort study of Japanese male workers. J Epidemiol2000; 10:403–409.
Kang DH, Nakagawa T. Uric acid and chronic renal disease: possible implication of hyperuricemia on progression of renal disease. Semin Nephrol2005; 25:43–49.
Iseki K, Oshiro S, Tozawa M, Iseki C, Ikemiya Y, Takishita S. Significance of hyperuricemia on the early detection of renal failure in a cohort of screened subjects. Hypertens Res2001; 24:691–697.
Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med1987; 82:421–426.
Siu YP, Leung KT, Tong MK, Kwan TH. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis2006; 47:51–59.
Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA2008; 300:924–932.
References
Vázquez-Mellado J, García CG, Vázquez SG, et al. Metabolic syndrome and ischemic heart disease in gout. J Clin Rheumatol2004; 10:105–109.
Bonnel RA, Villalba ML, Karwoski CB, Beitz J. Deaths associated with inappropriate intravenous colchicine administration. J Emerg Med2002; 22:385–387.
Achtert G, Scherrmann JM, Christen MO. Pharmacokinetics/bioavailability of colchicine in healthy male volunteers. Eur J Drug Metab Pharmacokinet1989; 14:317–322.
Ben-Chetrit E, Scherrmann JM, Zylber-Katz E, Levy M. Colchicine disposition in patients with familial Mediterranean fever with renal impairment. J Rheumatol1994; 21:710–713.
Putterman C, Ben-Chetrit E, Caraco Y, Levy M. Colchicine intoxication: clinical pharmacology, risk factors, features, and management. Semin Arthritis Rheum1991; 21:143–155.
Aronoff G, Brater DC, Schrier R, Bennett WM. Use of drugs in patients with renal insufficiency. Workshop report. Blood Purif1994; 12:14–19.
Kuncl RW, Duncan G, Watson D, Alderson K, Rogawski MA, Peper M. Colchicine myopathy and neuropathy. N Engl J Med1987; 316:1562–1568.
Ahern MJ, Reid C, Gordon TP, McCredie M, Brooks PM, Jones M. Does colchicine work? The results of the first controlled study in acute gout. Aust N Z J Med1987; 17:301–304.
Terkeltaub R, Furst D, Bennett K, Kook K, Crockett RS, Davis WM. High versus low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum2010, 62:1060–1068.
Whelton A. Nephrotoxicity of nonsteroidal anti-inflammatory drugs: physiologic foundations and clinical implications. Am J Med1999; 106:13S–24S.
Wali RK, Henrich WL. Recent developments in toxic nephropathy. Curr Opin Nephrol Hypertens2002; 11:155–163.
Fernández C, Noguera R, González JA, Pascual E. Treatment of acute attacks of gout with a small dose of intraarticular triamcinolone acetonide. J Rheumatol1999; 26:2285–2286.
Clive DM. Renal transplant-associated hyperuricemia and gout. J Am Soc Nephrol2000; 11:974–979.
Groff GD, Franck WA, Raddatz DA. Systemic steroid therapy for acute gout: a clinical trial and review of the literature. Semin Arthritis Rheum1990; 19:329–336.
Ritter J, Kerr LD, Valeriano-Marcet J, Spiera H. ACTH revisited: effective treatment for acute crystal induced synovitis in patients with multiple medical problems. J Rheumatol1994; 21:696–699.
Getting SJ, Christian HC, Flower RJ, Perretti M. Activation of melanocortin type 3 receptor as a molecular mechanism for adrenocorticotropic hormone efficacy in gouty arthritis. Arthritis Rheum2002; 46:2765–2775.
Connell JM, Whitworth JA, Davies DL, Lever AF, Richards AM, Fraser R. Effects of ACTH and cortisol administration on blood pressure, electrolyte metabolism, atrial natriuretic peptide and renal function in normal man. J Hypertens1987; 5:425–433.
Tausche AK, Richter K, Grässler A, Hänsel S, Roch B, Schröder HE. Severe gouty arthritis refractory to anti-inflammatory drugs: treatment with anti-tumour necrosis factor alpha as a new therapeutic option. Ann Rheum Dis2004; 63:1351–1352.
So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther2007; 9:R28.
Becker MA, Schumacher HR, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med2005; 353:2450–2461.
Borstad GC, Bryant LR, Abel MP, Scroggie DA, Harris MD, Alloway JA. Colchicine for prophylaxis of acute flares when initiating allopurinol for chronic gouty arthritis. J Rheumatol2004; 31:2429–2432.
Wallace SL, Singer JZ, Duncan GJ, Wigley FM, Kuncl RW. Renal function predicts colchicine toxicity: guidelines for the prophylactic use of colchicine in gout. J Rheumatol1991; 18:264–269.
Wilbur K, Makowsky M. Colchicine myotoxicity: case reports and literature review. Pharmacotherapy2004; 24:1784–1792.
Hung IF, Wu AK, Cheng VC, et al. Fatal interaction between clarithromycin and colchicine in patients with renal insufficiency: a retrospective study. Clin Infect Dis2005; 41:291–300.
Alayli G, Cengiz K, Cantürk F, Durmus D, Akyol Y, Menekse EB. Acute myopathy in a patient with concomitant use of pravastatin and colchicine. Ann Pharmacother2005; 39:1358–1361.
Ducloux D, Schuller V, Bresson-Vautrin C, Chalopin JM. Colchicine myopathy in renal transplant recipients on cyclosporin. Nephrol Dial Transplant1997; 12:2389–2392.
Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med2004; 350:1093–1103.
Choi HK, Curhan G. Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. BMJ2008; 336:309–312.
Sundy JS, Becker MA, Baraf HS, et al; Pegloticase Phase 2 Study Investigators. Reduction of plasma urate levels following treatment with multiple doses of pegloticase (polyethylene glycol-conjugated uricase) in patients with treatment-failure gout: results of a phase II randomized study. Arthritis Rheum2008; 58:2882–2891.
Jordan KM, Cameron JS, Snaith M, et al; British Society for Rheumatology and British Health Professionals in Rheumatology Standards, Guidelines and Audit Working Group (SGAWG). British Society for Rheumatology and British Health Professionals in Rheumatology guideline for the management of gout. Rheumatology (Oxford)2007; 46:1372–1374.
Perez-Ruiz F, Alonso-Ruiz A, Calabozo M, Herrero-Beites A, García-Erauskin G, Ruiz-Lucea E. Efficacy of allopurinol and benzbromarone for the control of hyperuricaemia. A pathogenic approach to the treatment of primary chronic gout. Ann Rheum Dis1998; 57:545–549.
Schumacher HR, Becker MA, Wortmann RL, et al. Effects of febuxostat versus allopurinol and placebo in reducing serum urate in subjects with hyperuricemia and gout: a 28-week, phase III, randomized, double-blind, parallel-group trial. Arthritis Rheum2008; 59:1540–1548.
Perez-Ruiz F, Calabozo M, Pijoan JI, Herrero-Beites AM, Ruibal A. Effect of urate-lowering therapy on the velocity of size reduction of tophi in chronic gout. Arthritis Rheum2002; 47:356–360.
Hande KR, Noone RM, Stone WJ. Severe allopurinol toxicity. Description and guidelines for prevention in patients with renal insufficiency. Am J Med1984; 76:47–56.
Perez-Ruiz F, Hernando I, Villar I, Nolla JM. Correction of allopurinol dosing should be based on clearance of creatinine, but not plasma creatinine levels: another insight to allopurinol-related toxicity. J Clin Rheumatol2005; 11:129–133.
Hung SI, Chung WH, Liou LB, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A2005; 102:4134–4139.
Vázquez-Mellado J, Morales EM, Pacheco-Tena C, Burgos-Vargas R. Relation between adverse events associated with allopurinol and renal function in patients with gout. Ann Rheum Dis2001; 60:981–983.
Dalbeth N, Kumar S, Stamp L, Gow P. Dose adjustment of allopurinol according to creatinine clearance does not provide adequate control of hyperuricemia in patients with gout. J Rheumatol2006; 33:1646–1650.
Silverberg MS, Mallela R, Lesse AJ, Bonner MR, Baer AN, Li C. Allopurinol hypersensitivity reactions: a case-control study of the role of renal dosing (abstract). Arthritis Rheum2009; 60(suppl 10):1106.
Stamp LK, O'Donnell JL, Zhang M, et al. Using allopurinol above the dose based on creatinine clearance is effective and safe in chronic gout, including in those with renal impairment. Arthritis Rhem2010; doi:10.1002/art.30119. E-pub ahead of print. Accessed 10/29/2010.
Zhang W, Doherty M, Bardin T, et al; EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis2006; 65:1312–1324.
Okamoto K, Eger BT, Nishino T, Kondo S, Pai EF, Nishino T. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J Biol Chem2003; 278:1848–1855.
Khosravan R, Grabowski BA, Wu JT, Joseph-Ridge N, Vernillet L. Pharmacokinetics, pharmacodynamics and safety of febuxostat, a non-purine selective inhibitor of xanthine oxidase, in a dose escalation study in healthy subjects. Clin Pharmacokinet2006; 45:821–841.
Reinders MK, Haagsma C, Jansen TL, et al. A randomised controlled trial on the efficacy and tolerability with dose escalation of allopurinol 300–600 mg/day versus benzbromarone 100–200 mg/day in patients with gout. Ann Rheum Dis2009; 68:892–897.
Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension2001; 38:1101–1106.
Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med2008; 359:1811–1821.
Beck LH. Requiem for gouty nephropathy. Kidney Int1986; 30:280–287.
Tomita M, Mizuno S, Yamanaka H, et al. Does hyperuricemia affect mortality? A prospective cohort study of Japanese male workers. J Epidemiol2000; 10:403–409.
Kang DH, Nakagawa T. Uric acid and chronic renal disease: possible implication of hyperuricemia on progression of renal disease. Semin Nephrol2005; 25:43–49.
Iseki K, Oshiro S, Tozawa M, Iseki C, Ikemiya Y, Takishita S. Significance of hyperuricemia on the early detection of renal failure in a cohort of screened subjects. Hypertens Res2001; 24:691–697.
Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med1987; 82:421–426.
Siu YP, Leung KT, Tong MK, Kwan TH. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis2006; 47:51–59.
Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA2008; 300:924–932.
Owing to concerns about using colchicine and nonsteroidal anti-inflammatory drugs (NSAIDs) in patients with CKD, glucocorticoids (local injections or systemic therapy) are often used to treat acute attacks. Corticotropin (Acthar), anti-tumor necrosis factor agents, and interleukin 1 antagonists are effective but expensive.
Colchicine can be used in low doses as prophylaxis, with caution and appropriate monitoring. NSAIDs should be avoided, and glucocorticoids may not be effective for this purpose.
Whether the dosage of allopurinol should be lower in patients with CKD remains controversial. We start with a low dose and slowly increase it, with a goal serum urate level of less than 6.0 mg/dL.
Febuxostat (Uloric), like allopurinol, is a xanthine oxidase inhibitor, but the elimination of the active drug is not by the kidney. Nevertheless, we try allopurinol in escalating doses first, due to major cost differences.