User login
Influenza Overview and Update: 2010-2011
Are antibiotics indicated for the treatment of aspiration pneumonia?
Antibiotics are indicated for primary bacterial aspiration pneumonia and secondary bacterial infection of aspiration (chemical) pneumonitis, but not for uncomplicated chemical pneumonitis.
THREE TYPES OF ‘ASPIRATION PNEUMONIA’
Aspiration pneumonia is a broad and vague term mainly used to refer to the pulmonary consequences of abnormal entry of exogenous or endogenous substances into the lower airways. It can be classified as:
- Aspiration (chemical) pneumonitis
- Primary bacterial aspiration pneumonia
- Secondary bacterial infection of chemical pneumonitis.
These three are sometimes difficult to differentiate, as their signs and symptoms can overlap.
CHEMICAL PNEUMONITIS
Aspiration of stomach contents is relatively common, even in healthy people, and usually has no clinical consequences.1 However, it has also been closely related to community-acquired and nosocomial pneumonia in some studies.2,3
Chemical pneumonitis is usually a consequence of the aspiration of a large volume (≥ 4 mL/kg) of sterile acidic (pH < 2.5) gastric contents into the lower airways (Mendelson syndrome).4,5 The clinical picture varies from asymptomatic to signs of severe dyspnea, hypoxia, cough, and low-grade fever; these signs and symptoms may develop rapidly, within minutes to hours after a witnessed or suspected episode of aspiration.2,6,7 However, they represent an inflammatory reaction to the gastric acid rather than a reaction to bacterial infection.8–10
Chemical pneumonitis affects the most dependent regions of the lungs
Chest radiography shows infiltrates in the most dependent regions of the lung. If aspiration occurs while the patient is supine, the posterior segments of the upper lobes and the apical segments of the lower lobes are most affected. The basal segments of the lower lobes are usually affected if aspiration occurs while the patient is standing or upright.1,2,11,12
Clinical course varies
The clinical course varies. In almost 60% of cases, the patient’s condition improves and the lung infiltrates resolve rapidly, within 2 to 4 days. On the other hand, in about 15% of cases, the patient’s condition deteriorates quickly, within 24 to 36 hours, and progresses to hypoxic respiratory failure and acute respiratory distress syndrome.
In the other 25% of cases, the patient’s condition may improve initially but then worsen as a secondary bacterial infection sets in. The death rate in these patients is almost three times higher than the rate in patients with uncomplicated chemical pneumonitis.11,13
Treatment of uncomplicated cases is mainly supportive
The treatment of uncomplicated chemical pneumonitis involves supportive measures such as airway clearance, oxygen supplementation, and positive pressure ventilation if needed. An obstructing foreign body may need to be removed.12,14 Corticosteroids have been tried, without success.11–13,15
Empiric antibiotic treatment is controversial
Chemical pneumonitis can be difficult to differentiate from bacterial aspiration pneumonia, and whether to give antibiotics is controversial. 16 A survey of current practices among intensivists showed that antimicrobial therapy was often given empirically for noninfectious chemical pneumonitis.17 This practice raises concerns of higher treatment costs and antibiotic resistance.16,18,19 Additionally, antibiotics do not seem to alter the clinical outcome, including radiographic resolution, duration of hospitalization, or death rate, nor do they influence the subsequent development of infection.1,11,13,20
In cases of witnessed or strongly suspected aspiration of gastric contents, antibiotics are not warranted since bacterial infection is not likely to be the cause of any signs or symptoms. 2,7,16 However, to detect secondary infection early, the patient’s respiratory status should be monitored carefully and chest radiography should be repeated.
In less clear-cut cases, ie, if it is not clear whether the patient actually has chemical pneumonitis or primary bacterial aspiration pneumonia, it is prudent to start antibiotics empirically after obtaining lower-respiratory-tract secretions for stains and cultures, and then to reassess within 48 to 72 hours. The antibiotics can be discontinued if the patient has rapid clinical and radiographic improvement and negative cultures. Those whose condition does not improve or who have positive cultures should receive a full course of antibiotics.21,22
PRIMARY BACTERIAL ASPIRATION PNEUMONIA
Primary bacterial aspiration pneumonia—ie, caused by bacteria residing in the upper airways and stomach gaining access to lower airways through aspiration in small or large amounts—is the most common form of aspiration pneumonia, although the actual episode of aspiration is seldom observed.
Signs of bacterial pneumonia
Primary bacterial aspiration pneumonia bears the hallmarks of bacterial pneumonia.12 The clinical picture is more indolent than chemical pneumonitis and includes cough, fever, and putrid sputum, mainly in patients who have clinical conditions predisposing to aspiration (eg, coma, stroke, alcoholism, poor dentition, tube feedings).1,12,20
The characteristic signs on chest radiography are infiltrates involving mainly the lung bases (the right more then the left). If untreated or inadequately treated, complications such as lung abscess, empyema, bronchiectasis, and broncopleural fistula are common.23
Are aerobic organisms replacing anaerobic ones in the community?
The causative organisms in community-acquired aspiration pneumonia are still debated despite abundant research. Older studies1,24,25 found mostly anaerobic organisms (pepto-streptococci, peptococci, Fusobacterium, Prevotela, Bacteroides) as the underlying pathogens, whereas more recent studies16,26,27 found mostly aerobic organisms (Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, Enterobacteriaceae) and failed to recover anaerobic organisms. These discrepancies may be the result of different techniques used to isolate organisms: older studies used transtracheal sampling, and transtracheal aspirates may be easily contaminated or colonized by oropharyngeal flora; more recent studies used protected specimen brushes to collect lower-airway specimens.2
In addition, the pathogenic organisms that predominate in community-acquired aspiration pneumonia, as listed above, are different from those most often found in nosocomial cases; gram-negative bacilli (Pseudomonas aeruginosa, Klebsiella pneumoniae, Escherichia coli) are most often isolated in patients with aspiration pneumonia acquired in hospitals and nursing homes.16,27,28S aureus also is an important causative organism in nosocomial cases.16,28
Knowing the causative organisms in bacterial aspiration pneumonia is important for guiding antimicrobial therapy.
Antibiotics are required for bacterial aspiration pneumonia
A course of antibiotics is required for bacterial aspiration pneumonia. While there are no definitive recommendations for the duration of treatment, 7 to 8 days is probably appropriate in uncomplicated cases (ie, no lung abscess, empyema, bronchopleural fistula).22,29 Patients who have complications may need drainage of abscesses or empyema along with a longer duration of antibiotic therapy until clinical and radiographic signs improve.
For community-acquired cases of aspiration pneumonia, a number of antibiotics have proven effective:
- Clindamycin (Cleocin) is still the agent most commonly used, although it lacks gram-negative bacterial coverage.
- Beta-lactam penicillins and newer quinolones have been used successfully.2,29–31 In addition to covering the previously mentioned bacteria, these antibiotics have the added benefit of covering anaerobic bacteria.
- Metronidazole (Flagyl) should not be used alone because it has a higher clinical failure rate.32,33
For nosocomial aspiration pneumonia, giving a broad-spectrum antibiotic empirically is warranted. Beta-lactam penicillins with extended gram-negative coverage, carbapenems, or monobactams in combination with an anti-staphylococcal drug have been advocated for nosocomial aspiration.2,22 A strategy of broad-spectrum coverage followed by narrowing or de-escalating coverage according to lower respiratory tract cultures is encouraged.21,22,34
SECONDARY BACTERIAL INFECTION OF CHEMICAL PNEUMONITIS
Nearly 25% of patients with chemical pneumonitis improve initially, then show clinical deterioration secondary to superimposed bacterial infection.13 Chest radiographs show worsening of initial infiltrates or the development of new ones. The causative organisms and treatment depend on whether the superimposed infection is community-acquired or nosocomial, as is the case in primary bacterial aspiration pneumonia.
PREVENTING ASPIRATION
Measures should be taken to prevent aspiration pneumonia and chemical pneumonitis, especially in institutionalized patients at high risk.12
Elevation of the head of the bed while feeding, dental prophylaxis, and good oral hygiene are known to reduce the incidence of these problems.35–37
A swallowing evaluation for patients with dysphagia can identify those at higher risk of aspiration. These patients may be candidates for postural adjustments, diet modification, strengthening, and other measures offered by the speech and language pathology teams to improve swallowing physiology, biomechanics, safety, and endurance.2,35
Although percutaneous endoscopic gastrostomy tubes are often placed in patients who have aspirated or who are at high risk of aspiration, they do not protect against aspiration, nor do orogastric or nasogastric tubes.38
To date, we have no evidence that prophylactic antibiotic therapy prevents bacterial aspiration pneumonia. In addition, this practice encourages the development of resistant organisms.19,39,40
- Bartlett JG, Gorbach SL. The triple threat of aspiration pneumonia. Chest 1975; 68:560–566.
- Marik PE. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med 2001; 344:665–671.
- Kikuchi R, Watabe N, Konno T, Mishina N, Sekizawa K, Sasaki H. High incidence of silent aspiration in elderly patients with community-acquired pneumonia. Am J Respir Crit Care Med 1994; 150:251–253.
- Mendelson CL. The aspiration of stomach contents into lungs during obstetric anesthesia. Am J Obstet Gynecol 1946; 52:191–205.
- Cameron JL, Caldini P, Toung JK, Zuidema GD. Aspiration pneumonia: physiologic data following experimental aspiration. Surgery 1972; 72:238–245.
- Warner MA, Warner ME, Weber JG. Clinical significance of pulmonary aspiration during the perioperative period. Anesthesiology 1993; 78:56–62.
- DePaso WJ. Aspiration pneumonia. Clin Chest Med 1991; 12:269–284.
- Folkesson HG, Matthay MA, Hébert CA, Broaddus VC. Acid aspiration-induced lung injury in rabbits is mediated by interleukin-8-dependent mechanisms. J Clin Invest 1995; 96:107–116.
- Goldman G, Welbourn R, Kobzik L, Valeri CR, Shepro D, Hechtman HB. Tumor necrosis factor-alpha mediates acid aspiration-induced systemic organ injury. Ann Surg 1990; 212:513–519.
- LeFrock JL, Clark TS, Davies B, Klainer AS. Aspiration pneumonia: a ten-year review. Am Surg 1979; 45:305–313.
- Cameron JL, Mitchell WH, Zuidema GD. Aspiration pneumonia. Clinical outcome following documented aspiration. Arch Surg 1973; 106:49–52.
- Arms RA, Dines DE, Tinstman TC. Aspiration pneumonia. Chest 1974; 65:136–139.
- Bynum LJ, Pierce AK. Pulmonary aspiration of gastric contents. Am Rev Respir Dis 1976; 114:1129–1136.
- Merchant SN, Kirtane MV, Shah KL, Karnik PP. Foreign bodies in the bronchi (a 10 year review of 132 cases). J Postgrad Med 1984; 30:219–223.
- Wolfe JE, Bone RC, Ruth WE. Effects of corticosteroids in the treatment of patients with gastric aspiration. Am J Med 1977; 63:719–722.
- Kane-Gill SL, Olsen KM, Rebuck JA, et al; Aspiration Evaluation Group of the Clinical Pharmacy and Pharmacology Section. Multicenter treatment and outcome evaluation of aspiration syndromes in critically ill patients. Ann Pharmacother 2007; 41:549–555.
- Rebuck JA, Rasmussen JR, Olsen KM. Clinical aspiration-related practice patterns in the intensive care unit: a physician survey. Crit Care Med 2001; 29:2239–2244.
- Singh N, Rogers P, Atwood CW, Wagener MM, Yu VL. Short-course empiric antibiotic therapy for patients with pulmonary infiltrates in the intensive care unit. A proposed solution for indiscriminate antibiotic prescription. Am J Respir Crit Care Med 2000; 162:505–511.
- Kollef MH, Fraser VJ. Antibiotic resistance in the intensive care unit. Ann Intern Med 2001; 134:298–314.
- Lewis RT, Burgess JH, Hampson LG. Cardiorespiratory studies in critical illness. Changes in aspiration pneumonitis. Arch Surg 1971; 103:335–340.
- Rello J. Importance of appropriate initial antibiotic therapy and de-escalation in the treatment of nosocomial pneumonia. Eur Respir Rev 2007; 16:33–39.
- American Thoracic Society. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med 2005; 171:388–416.
- Bartlett JG. Anaerobic bacterial infections of the lung and pleural space. Clin Infect Dis 1993; 16(suppl 4):S248–S255.
- Lorber B, Swenson RM. Bacteriology of aspiration pneumonia. A prospective study of community- and hospital-acquired cases. Ann Intern Med 1974; 81:329–331.
- Bartlett JG, Gorbach SL, Finegold SM. The bacteriology of aspiration pneumonia. Am J Med 1974; 56:202–207.
- Mier L, Dreyfuss D, Darchy B, et al. Is penicillin G an adequate initial treatment for aspiration pneumonia? A prospective evaluation using a protected specimen brush and quantitative cultures. Intensive Care Med 1993; 19:279–284.
- Marik PE, Careau P. The role of anaerobes in patients with ventilator-associated pneumonia and aspiration pneumonia: a prospective study. Chest 1999; 115:178–183.
- El-Solh AA, Pietrantoni C, Bhat A, et al. Microbiology of severe aspiration pneumonia in institutionalized elderly. Am J Respir Crit Care Med 2003; 167:1650–1654.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44(suppl 2):S27–S72.
- Kadowaki M, Demura Y, Mizuno S, et al. Reappraisal of clindamycin IV monotherapy for treatment of mild-to-moderate aspiration pneumonia in elderly patients. Chest 2005; 127:1276–1282.
- Ott SR, Allewelt M, Lorenz J, Reimnitz P, Lode H; German Lung Abscess Study Group. Moxifloxacin vs ampicillin/sulbactam in aspiration pneumonia and primary lung abscess. Infection 2008; 36:23–30.
- Perlino CA. Metronidazole vs clindamycin treatment of anerobic pulmonary infection. Failure of metronidazole therapy. Arch Intern Med 1981; 141:1424–1427.
- Sanders CV, Hanna BJ, Lewis AC. Metronidazole in the treatment of anaerobic infections. Am Rev Respir Dis 1979; 120:337–343.
- Alvarez-Lerma F, Alvarez B, Luque P, et al; ADANN Study Group. Empiric broad-spectrum antibiotic therapy of nosocomial pneumonia in the intensive care unit: a prospective observational study. Crit Care 2006; 10:R78.
- Johnson JL, Hirsch CS. Aspiration pneumonia. Recognizing and managing a potentially growing disorder. Postgrad Med 2003; 113:99–112.
- Scolapio JS. Methods for decreasing risk of aspiration pneumonia in critically ill patients. JPEN J Parenter Enteral Nutr 2002; 26(suppl 6):S58–S61.
- Orozco-Levi M, Torres A, Ferrer M, et al. Semirecumbent position protects from pulmonary aspiration but not completely from gastroesophageal reflux in mechanically ventilated patients. Am J Respir Crit Care Med 1995; 152:1387–1390.
- Park RH, Allison MC, Lang J, et al. Randomised comparison of percutaneous endoscopic gastrostomy and nasogastric tube feeding in patients with persisting neurological dysphagia. BMJ 1992; 304( 6839):1406–1409.
- Donskey CJ, Chowdhry TK, Hecker MT, et al. Effect of antibiotic therapy on the density of vancomycin-resistant enterococci in the stool of colonized patients. N Engl J Med 2000; 343:1925–1932.
- Mouw DR, Langlois JP, Turner LF, Neher JO. Clinical inquiries. Are antibiotics effective in preventing pneumonia for nursing home patients? J Fam Pract 2004; 53:994–996.
Antibiotics are indicated for primary bacterial aspiration pneumonia and secondary bacterial infection of aspiration (chemical) pneumonitis, but not for uncomplicated chemical pneumonitis.
THREE TYPES OF ‘ASPIRATION PNEUMONIA’
Aspiration pneumonia is a broad and vague term mainly used to refer to the pulmonary consequences of abnormal entry of exogenous or endogenous substances into the lower airways. It can be classified as:
- Aspiration (chemical) pneumonitis
- Primary bacterial aspiration pneumonia
- Secondary bacterial infection of chemical pneumonitis.
These three are sometimes difficult to differentiate, as their signs and symptoms can overlap.
CHEMICAL PNEUMONITIS
Aspiration of stomach contents is relatively common, even in healthy people, and usually has no clinical consequences.1 However, it has also been closely related to community-acquired and nosocomial pneumonia in some studies.2,3
Chemical pneumonitis is usually a consequence of the aspiration of a large volume (≥ 4 mL/kg) of sterile acidic (pH < 2.5) gastric contents into the lower airways (Mendelson syndrome).4,5 The clinical picture varies from asymptomatic to signs of severe dyspnea, hypoxia, cough, and low-grade fever; these signs and symptoms may develop rapidly, within minutes to hours after a witnessed or suspected episode of aspiration.2,6,7 However, they represent an inflammatory reaction to the gastric acid rather than a reaction to bacterial infection.8–10
Chemical pneumonitis affects the most dependent regions of the lungs
Chest radiography shows infiltrates in the most dependent regions of the lung. If aspiration occurs while the patient is supine, the posterior segments of the upper lobes and the apical segments of the lower lobes are most affected. The basal segments of the lower lobes are usually affected if aspiration occurs while the patient is standing or upright.1,2,11,12
Clinical course varies
The clinical course varies. In almost 60% of cases, the patient’s condition improves and the lung infiltrates resolve rapidly, within 2 to 4 days. On the other hand, in about 15% of cases, the patient’s condition deteriorates quickly, within 24 to 36 hours, and progresses to hypoxic respiratory failure and acute respiratory distress syndrome.
In the other 25% of cases, the patient’s condition may improve initially but then worsen as a secondary bacterial infection sets in. The death rate in these patients is almost three times higher than the rate in patients with uncomplicated chemical pneumonitis.11,13
Treatment of uncomplicated cases is mainly supportive
The treatment of uncomplicated chemical pneumonitis involves supportive measures such as airway clearance, oxygen supplementation, and positive pressure ventilation if needed. An obstructing foreign body may need to be removed.12,14 Corticosteroids have been tried, without success.11–13,15
Empiric antibiotic treatment is controversial
Chemical pneumonitis can be difficult to differentiate from bacterial aspiration pneumonia, and whether to give antibiotics is controversial. 16 A survey of current practices among intensivists showed that antimicrobial therapy was often given empirically for noninfectious chemical pneumonitis.17 This practice raises concerns of higher treatment costs and antibiotic resistance.16,18,19 Additionally, antibiotics do not seem to alter the clinical outcome, including radiographic resolution, duration of hospitalization, or death rate, nor do they influence the subsequent development of infection.1,11,13,20
In cases of witnessed or strongly suspected aspiration of gastric contents, antibiotics are not warranted since bacterial infection is not likely to be the cause of any signs or symptoms. 2,7,16 However, to detect secondary infection early, the patient’s respiratory status should be monitored carefully and chest radiography should be repeated.
In less clear-cut cases, ie, if it is not clear whether the patient actually has chemical pneumonitis or primary bacterial aspiration pneumonia, it is prudent to start antibiotics empirically after obtaining lower-respiratory-tract secretions for stains and cultures, and then to reassess within 48 to 72 hours. The antibiotics can be discontinued if the patient has rapid clinical and radiographic improvement and negative cultures. Those whose condition does not improve or who have positive cultures should receive a full course of antibiotics.21,22
PRIMARY BACTERIAL ASPIRATION PNEUMONIA
Primary bacterial aspiration pneumonia—ie, caused by bacteria residing in the upper airways and stomach gaining access to lower airways through aspiration in small or large amounts—is the most common form of aspiration pneumonia, although the actual episode of aspiration is seldom observed.
Signs of bacterial pneumonia
Primary bacterial aspiration pneumonia bears the hallmarks of bacterial pneumonia.12 The clinical picture is more indolent than chemical pneumonitis and includes cough, fever, and putrid sputum, mainly in patients who have clinical conditions predisposing to aspiration (eg, coma, stroke, alcoholism, poor dentition, tube feedings).1,12,20
The characteristic signs on chest radiography are infiltrates involving mainly the lung bases (the right more then the left). If untreated or inadequately treated, complications such as lung abscess, empyema, bronchiectasis, and broncopleural fistula are common.23
Are aerobic organisms replacing anaerobic ones in the community?
The causative organisms in community-acquired aspiration pneumonia are still debated despite abundant research. Older studies1,24,25 found mostly anaerobic organisms (pepto-streptococci, peptococci, Fusobacterium, Prevotela, Bacteroides) as the underlying pathogens, whereas more recent studies16,26,27 found mostly aerobic organisms (Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, Enterobacteriaceae) and failed to recover anaerobic organisms. These discrepancies may be the result of different techniques used to isolate organisms: older studies used transtracheal sampling, and transtracheal aspirates may be easily contaminated or colonized by oropharyngeal flora; more recent studies used protected specimen brushes to collect lower-airway specimens.2
In addition, the pathogenic organisms that predominate in community-acquired aspiration pneumonia, as listed above, are different from those most often found in nosocomial cases; gram-negative bacilli (Pseudomonas aeruginosa, Klebsiella pneumoniae, Escherichia coli) are most often isolated in patients with aspiration pneumonia acquired in hospitals and nursing homes.16,27,28S aureus also is an important causative organism in nosocomial cases.16,28
Knowing the causative organisms in bacterial aspiration pneumonia is important for guiding antimicrobial therapy.
Antibiotics are required for bacterial aspiration pneumonia
A course of antibiotics is required for bacterial aspiration pneumonia. While there are no definitive recommendations for the duration of treatment, 7 to 8 days is probably appropriate in uncomplicated cases (ie, no lung abscess, empyema, bronchopleural fistula).22,29 Patients who have complications may need drainage of abscesses or empyema along with a longer duration of antibiotic therapy until clinical and radiographic signs improve.
For community-acquired cases of aspiration pneumonia, a number of antibiotics have proven effective:
- Clindamycin (Cleocin) is still the agent most commonly used, although it lacks gram-negative bacterial coverage.
- Beta-lactam penicillins and newer quinolones have been used successfully.2,29–31 In addition to covering the previously mentioned bacteria, these antibiotics have the added benefit of covering anaerobic bacteria.
- Metronidazole (Flagyl) should not be used alone because it has a higher clinical failure rate.32,33
For nosocomial aspiration pneumonia, giving a broad-spectrum antibiotic empirically is warranted. Beta-lactam penicillins with extended gram-negative coverage, carbapenems, or monobactams in combination with an anti-staphylococcal drug have been advocated for nosocomial aspiration.2,22 A strategy of broad-spectrum coverage followed by narrowing or de-escalating coverage according to lower respiratory tract cultures is encouraged.21,22,34
SECONDARY BACTERIAL INFECTION OF CHEMICAL PNEUMONITIS
Nearly 25% of patients with chemical pneumonitis improve initially, then show clinical deterioration secondary to superimposed bacterial infection.13 Chest radiographs show worsening of initial infiltrates or the development of new ones. The causative organisms and treatment depend on whether the superimposed infection is community-acquired or nosocomial, as is the case in primary bacterial aspiration pneumonia.
PREVENTING ASPIRATION
Measures should be taken to prevent aspiration pneumonia and chemical pneumonitis, especially in institutionalized patients at high risk.12
Elevation of the head of the bed while feeding, dental prophylaxis, and good oral hygiene are known to reduce the incidence of these problems.35–37
A swallowing evaluation for patients with dysphagia can identify those at higher risk of aspiration. These patients may be candidates for postural adjustments, diet modification, strengthening, and other measures offered by the speech and language pathology teams to improve swallowing physiology, biomechanics, safety, and endurance.2,35
Although percutaneous endoscopic gastrostomy tubes are often placed in patients who have aspirated or who are at high risk of aspiration, they do not protect against aspiration, nor do orogastric or nasogastric tubes.38
To date, we have no evidence that prophylactic antibiotic therapy prevents bacterial aspiration pneumonia. In addition, this practice encourages the development of resistant organisms.19,39,40
Antibiotics are indicated for primary bacterial aspiration pneumonia and secondary bacterial infection of aspiration (chemical) pneumonitis, but not for uncomplicated chemical pneumonitis.
THREE TYPES OF ‘ASPIRATION PNEUMONIA’
Aspiration pneumonia is a broad and vague term mainly used to refer to the pulmonary consequences of abnormal entry of exogenous or endogenous substances into the lower airways. It can be classified as:
- Aspiration (chemical) pneumonitis
- Primary bacterial aspiration pneumonia
- Secondary bacterial infection of chemical pneumonitis.
These three are sometimes difficult to differentiate, as their signs and symptoms can overlap.
CHEMICAL PNEUMONITIS
Aspiration of stomach contents is relatively common, even in healthy people, and usually has no clinical consequences.1 However, it has also been closely related to community-acquired and nosocomial pneumonia in some studies.2,3
Chemical pneumonitis is usually a consequence of the aspiration of a large volume (≥ 4 mL/kg) of sterile acidic (pH < 2.5) gastric contents into the lower airways (Mendelson syndrome).4,5 The clinical picture varies from asymptomatic to signs of severe dyspnea, hypoxia, cough, and low-grade fever; these signs and symptoms may develop rapidly, within minutes to hours after a witnessed or suspected episode of aspiration.2,6,7 However, they represent an inflammatory reaction to the gastric acid rather than a reaction to bacterial infection.8–10
Chemical pneumonitis affects the most dependent regions of the lungs
Chest radiography shows infiltrates in the most dependent regions of the lung. If aspiration occurs while the patient is supine, the posterior segments of the upper lobes and the apical segments of the lower lobes are most affected. The basal segments of the lower lobes are usually affected if aspiration occurs while the patient is standing or upright.1,2,11,12
Clinical course varies
The clinical course varies. In almost 60% of cases, the patient’s condition improves and the lung infiltrates resolve rapidly, within 2 to 4 days. On the other hand, in about 15% of cases, the patient’s condition deteriorates quickly, within 24 to 36 hours, and progresses to hypoxic respiratory failure and acute respiratory distress syndrome.
In the other 25% of cases, the patient’s condition may improve initially but then worsen as a secondary bacterial infection sets in. The death rate in these patients is almost three times higher than the rate in patients with uncomplicated chemical pneumonitis.11,13
Treatment of uncomplicated cases is mainly supportive
The treatment of uncomplicated chemical pneumonitis involves supportive measures such as airway clearance, oxygen supplementation, and positive pressure ventilation if needed. An obstructing foreign body may need to be removed.12,14 Corticosteroids have been tried, without success.11–13,15
Empiric antibiotic treatment is controversial
Chemical pneumonitis can be difficult to differentiate from bacterial aspiration pneumonia, and whether to give antibiotics is controversial. 16 A survey of current practices among intensivists showed that antimicrobial therapy was often given empirically for noninfectious chemical pneumonitis.17 This practice raises concerns of higher treatment costs and antibiotic resistance.16,18,19 Additionally, antibiotics do not seem to alter the clinical outcome, including radiographic resolution, duration of hospitalization, or death rate, nor do they influence the subsequent development of infection.1,11,13,20
In cases of witnessed or strongly suspected aspiration of gastric contents, antibiotics are not warranted since bacterial infection is not likely to be the cause of any signs or symptoms. 2,7,16 However, to detect secondary infection early, the patient’s respiratory status should be monitored carefully and chest radiography should be repeated.
In less clear-cut cases, ie, if it is not clear whether the patient actually has chemical pneumonitis or primary bacterial aspiration pneumonia, it is prudent to start antibiotics empirically after obtaining lower-respiratory-tract secretions for stains and cultures, and then to reassess within 48 to 72 hours. The antibiotics can be discontinued if the patient has rapid clinical and radiographic improvement and negative cultures. Those whose condition does not improve or who have positive cultures should receive a full course of antibiotics.21,22
PRIMARY BACTERIAL ASPIRATION PNEUMONIA
Primary bacterial aspiration pneumonia—ie, caused by bacteria residing in the upper airways and stomach gaining access to lower airways through aspiration in small or large amounts—is the most common form of aspiration pneumonia, although the actual episode of aspiration is seldom observed.
Signs of bacterial pneumonia
Primary bacterial aspiration pneumonia bears the hallmarks of bacterial pneumonia.12 The clinical picture is more indolent than chemical pneumonitis and includes cough, fever, and putrid sputum, mainly in patients who have clinical conditions predisposing to aspiration (eg, coma, stroke, alcoholism, poor dentition, tube feedings).1,12,20
The characteristic signs on chest radiography are infiltrates involving mainly the lung bases (the right more then the left). If untreated or inadequately treated, complications such as lung abscess, empyema, bronchiectasis, and broncopleural fistula are common.23
Are aerobic organisms replacing anaerobic ones in the community?
The causative organisms in community-acquired aspiration pneumonia are still debated despite abundant research. Older studies1,24,25 found mostly anaerobic organisms (pepto-streptococci, peptococci, Fusobacterium, Prevotela, Bacteroides) as the underlying pathogens, whereas more recent studies16,26,27 found mostly aerobic organisms (Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, Enterobacteriaceae) and failed to recover anaerobic organisms. These discrepancies may be the result of different techniques used to isolate organisms: older studies used transtracheal sampling, and transtracheal aspirates may be easily contaminated or colonized by oropharyngeal flora; more recent studies used protected specimen brushes to collect lower-airway specimens.2
In addition, the pathogenic organisms that predominate in community-acquired aspiration pneumonia, as listed above, are different from those most often found in nosocomial cases; gram-negative bacilli (Pseudomonas aeruginosa, Klebsiella pneumoniae, Escherichia coli) are most often isolated in patients with aspiration pneumonia acquired in hospitals and nursing homes.16,27,28S aureus also is an important causative organism in nosocomial cases.16,28
Knowing the causative organisms in bacterial aspiration pneumonia is important for guiding antimicrobial therapy.
Antibiotics are required for bacterial aspiration pneumonia
A course of antibiotics is required for bacterial aspiration pneumonia. While there are no definitive recommendations for the duration of treatment, 7 to 8 days is probably appropriate in uncomplicated cases (ie, no lung abscess, empyema, bronchopleural fistula).22,29 Patients who have complications may need drainage of abscesses or empyema along with a longer duration of antibiotic therapy until clinical and radiographic signs improve.
For community-acquired cases of aspiration pneumonia, a number of antibiotics have proven effective:
- Clindamycin (Cleocin) is still the agent most commonly used, although it lacks gram-negative bacterial coverage.
- Beta-lactam penicillins and newer quinolones have been used successfully.2,29–31 In addition to covering the previously mentioned bacteria, these antibiotics have the added benefit of covering anaerobic bacteria.
- Metronidazole (Flagyl) should not be used alone because it has a higher clinical failure rate.32,33
For nosocomial aspiration pneumonia, giving a broad-spectrum antibiotic empirically is warranted. Beta-lactam penicillins with extended gram-negative coverage, carbapenems, or monobactams in combination with an anti-staphylococcal drug have been advocated for nosocomial aspiration.2,22 A strategy of broad-spectrum coverage followed by narrowing or de-escalating coverage according to lower respiratory tract cultures is encouraged.21,22,34
SECONDARY BACTERIAL INFECTION OF CHEMICAL PNEUMONITIS
Nearly 25% of patients with chemical pneumonitis improve initially, then show clinical deterioration secondary to superimposed bacterial infection.13 Chest radiographs show worsening of initial infiltrates or the development of new ones. The causative organisms and treatment depend on whether the superimposed infection is community-acquired or nosocomial, as is the case in primary bacterial aspiration pneumonia.
PREVENTING ASPIRATION
Measures should be taken to prevent aspiration pneumonia and chemical pneumonitis, especially in institutionalized patients at high risk.12
Elevation of the head of the bed while feeding, dental prophylaxis, and good oral hygiene are known to reduce the incidence of these problems.35–37
A swallowing evaluation for patients with dysphagia can identify those at higher risk of aspiration. These patients may be candidates for postural adjustments, diet modification, strengthening, and other measures offered by the speech and language pathology teams to improve swallowing physiology, biomechanics, safety, and endurance.2,35
Although percutaneous endoscopic gastrostomy tubes are often placed in patients who have aspirated or who are at high risk of aspiration, they do not protect against aspiration, nor do orogastric or nasogastric tubes.38
To date, we have no evidence that prophylactic antibiotic therapy prevents bacterial aspiration pneumonia. In addition, this practice encourages the development of resistant organisms.19,39,40
- Bartlett JG, Gorbach SL. The triple threat of aspiration pneumonia. Chest 1975; 68:560–566.
- Marik PE. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med 2001; 344:665–671.
- Kikuchi R, Watabe N, Konno T, Mishina N, Sekizawa K, Sasaki H. High incidence of silent aspiration in elderly patients with community-acquired pneumonia. Am J Respir Crit Care Med 1994; 150:251–253.
- Mendelson CL. The aspiration of stomach contents into lungs during obstetric anesthesia. Am J Obstet Gynecol 1946; 52:191–205.
- Cameron JL, Caldini P, Toung JK, Zuidema GD. Aspiration pneumonia: physiologic data following experimental aspiration. Surgery 1972; 72:238–245.
- Warner MA, Warner ME, Weber JG. Clinical significance of pulmonary aspiration during the perioperative period. Anesthesiology 1993; 78:56–62.
- DePaso WJ. Aspiration pneumonia. Clin Chest Med 1991; 12:269–284.
- Folkesson HG, Matthay MA, Hébert CA, Broaddus VC. Acid aspiration-induced lung injury in rabbits is mediated by interleukin-8-dependent mechanisms. J Clin Invest 1995; 96:107–116.
- Goldman G, Welbourn R, Kobzik L, Valeri CR, Shepro D, Hechtman HB. Tumor necrosis factor-alpha mediates acid aspiration-induced systemic organ injury. Ann Surg 1990; 212:513–519.
- LeFrock JL, Clark TS, Davies B, Klainer AS. Aspiration pneumonia: a ten-year review. Am Surg 1979; 45:305–313.
- Cameron JL, Mitchell WH, Zuidema GD. Aspiration pneumonia. Clinical outcome following documented aspiration. Arch Surg 1973; 106:49–52.
- Arms RA, Dines DE, Tinstman TC. Aspiration pneumonia. Chest 1974; 65:136–139.
- Bynum LJ, Pierce AK. Pulmonary aspiration of gastric contents. Am Rev Respir Dis 1976; 114:1129–1136.
- Merchant SN, Kirtane MV, Shah KL, Karnik PP. Foreign bodies in the bronchi (a 10 year review of 132 cases). J Postgrad Med 1984; 30:219–223.
- Wolfe JE, Bone RC, Ruth WE. Effects of corticosteroids in the treatment of patients with gastric aspiration. Am J Med 1977; 63:719–722.
- Kane-Gill SL, Olsen KM, Rebuck JA, et al; Aspiration Evaluation Group of the Clinical Pharmacy and Pharmacology Section. Multicenter treatment and outcome evaluation of aspiration syndromes in critically ill patients. Ann Pharmacother 2007; 41:549–555.
- Rebuck JA, Rasmussen JR, Olsen KM. Clinical aspiration-related practice patterns in the intensive care unit: a physician survey. Crit Care Med 2001; 29:2239–2244.
- Singh N, Rogers P, Atwood CW, Wagener MM, Yu VL. Short-course empiric antibiotic therapy for patients with pulmonary infiltrates in the intensive care unit. A proposed solution for indiscriminate antibiotic prescription. Am J Respir Crit Care Med 2000; 162:505–511.
- Kollef MH, Fraser VJ. Antibiotic resistance in the intensive care unit. Ann Intern Med 2001; 134:298–314.
- Lewis RT, Burgess JH, Hampson LG. Cardiorespiratory studies in critical illness. Changes in aspiration pneumonitis. Arch Surg 1971; 103:335–340.
- Rello J. Importance of appropriate initial antibiotic therapy and de-escalation in the treatment of nosocomial pneumonia. Eur Respir Rev 2007; 16:33–39.
- American Thoracic Society. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med 2005; 171:388–416.
- Bartlett JG. Anaerobic bacterial infections of the lung and pleural space. Clin Infect Dis 1993; 16(suppl 4):S248–S255.
- Lorber B, Swenson RM. Bacteriology of aspiration pneumonia. A prospective study of community- and hospital-acquired cases. Ann Intern Med 1974; 81:329–331.
- Bartlett JG, Gorbach SL, Finegold SM. The bacteriology of aspiration pneumonia. Am J Med 1974; 56:202–207.
- Mier L, Dreyfuss D, Darchy B, et al. Is penicillin G an adequate initial treatment for aspiration pneumonia? A prospective evaluation using a protected specimen brush and quantitative cultures. Intensive Care Med 1993; 19:279–284.
- Marik PE, Careau P. The role of anaerobes in patients with ventilator-associated pneumonia and aspiration pneumonia: a prospective study. Chest 1999; 115:178–183.
- El-Solh AA, Pietrantoni C, Bhat A, et al. Microbiology of severe aspiration pneumonia in institutionalized elderly. Am J Respir Crit Care Med 2003; 167:1650–1654.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44(suppl 2):S27–S72.
- Kadowaki M, Demura Y, Mizuno S, et al. Reappraisal of clindamycin IV monotherapy for treatment of mild-to-moderate aspiration pneumonia in elderly patients. Chest 2005; 127:1276–1282.
- Ott SR, Allewelt M, Lorenz J, Reimnitz P, Lode H; German Lung Abscess Study Group. Moxifloxacin vs ampicillin/sulbactam in aspiration pneumonia and primary lung abscess. Infection 2008; 36:23–30.
- Perlino CA. Metronidazole vs clindamycin treatment of anerobic pulmonary infection. Failure of metronidazole therapy. Arch Intern Med 1981; 141:1424–1427.
- Sanders CV, Hanna BJ, Lewis AC. Metronidazole in the treatment of anaerobic infections. Am Rev Respir Dis 1979; 120:337–343.
- Alvarez-Lerma F, Alvarez B, Luque P, et al; ADANN Study Group. Empiric broad-spectrum antibiotic therapy of nosocomial pneumonia in the intensive care unit: a prospective observational study. Crit Care 2006; 10:R78.
- Johnson JL, Hirsch CS. Aspiration pneumonia. Recognizing and managing a potentially growing disorder. Postgrad Med 2003; 113:99–112.
- Scolapio JS. Methods for decreasing risk of aspiration pneumonia in critically ill patients. JPEN J Parenter Enteral Nutr 2002; 26(suppl 6):S58–S61.
- Orozco-Levi M, Torres A, Ferrer M, et al. Semirecumbent position protects from pulmonary aspiration but not completely from gastroesophageal reflux in mechanically ventilated patients. Am J Respir Crit Care Med 1995; 152:1387–1390.
- Park RH, Allison MC, Lang J, et al. Randomised comparison of percutaneous endoscopic gastrostomy and nasogastric tube feeding in patients with persisting neurological dysphagia. BMJ 1992; 304( 6839):1406–1409.
- Donskey CJ, Chowdhry TK, Hecker MT, et al. Effect of antibiotic therapy on the density of vancomycin-resistant enterococci in the stool of colonized patients. N Engl J Med 2000; 343:1925–1932.
- Mouw DR, Langlois JP, Turner LF, Neher JO. Clinical inquiries. Are antibiotics effective in preventing pneumonia for nursing home patients? J Fam Pract 2004; 53:994–996.
- Bartlett JG, Gorbach SL. The triple threat of aspiration pneumonia. Chest 1975; 68:560–566.
- Marik PE. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med 2001; 344:665–671.
- Kikuchi R, Watabe N, Konno T, Mishina N, Sekizawa K, Sasaki H. High incidence of silent aspiration in elderly patients with community-acquired pneumonia. Am J Respir Crit Care Med 1994; 150:251–253.
- Mendelson CL. The aspiration of stomach contents into lungs during obstetric anesthesia. Am J Obstet Gynecol 1946; 52:191–205.
- Cameron JL, Caldini P, Toung JK, Zuidema GD. Aspiration pneumonia: physiologic data following experimental aspiration. Surgery 1972; 72:238–245.
- Warner MA, Warner ME, Weber JG. Clinical significance of pulmonary aspiration during the perioperative period. Anesthesiology 1993; 78:56–62.
- DePaso WJ. Aspiration pneumonia. Clin Chest Med 1991; 12:269–284.
- Folkesson HG, Matthay MA, Hébert CA, Broaddus VC. Acid aspiration-induced lung injury in rabbits is mediated by interleukin-8-dependent mechanisms. J Clin Invest 1995; 96:107–116.
- Goldman G, Welbourn R, Kobzik L, Valeri CR, Shepro D, Hechtman HB. Tumor necrosis factor-alpha mediates acid aspiration-induced systemic organ injury. Ann Surg 1990; 212:513–519.
- LeFrock JL, Clark TS, Davies B, Klainer AS. Aspiration pneumonia: a ten-year review. Am Surg 1979; 45:305–313.
- Cameron JL, Mitchell WH, Zuidema GD. Aspiration pneumonia. Clinical outcome following documented aspiration. Arch Surg 1973; 106:49–52.
- Arms RA, Dines DE, Tinstman TC. Aspiration pneumonia. Chest 1974; 65:136–139.
- Bynum LJ, Pierce AK. Pulmonary aspiration of gastric contents. Am Rev Respir Dis 1976; 114:1129–1136.
- Merchant SN, Kirtane MV, Shah KL, Karnik PP. Foreign bodies in the bronchi (a 10 year review of 132 cases). J Postgrad Med 1984; 30:219–223.
- Wolfe JE, Bone RC, Ruth WE. Effects of corticosteroids in the treatment of patients with gastric aspiration. Am J Med 1977; 63:719–722.
- Kane-Gill SL, Olsen KM, Rebuck JA, et al; Aspiration Evaluation Group of the Clinical Pharmacy and Pharmacology Section. Multicenter treatment and outcome evaluation of aspiration syndromes in critically ill patients. Ann Pharmacother 2007; 41:549–555.
- Rebuck JA, Rasmussen JR, Olsen KM. Clinical aspiration-related practice patterns in the intensive care unit: a physician survey. Crit Care Med 2001; 29:2239–2244.
- Singh N, Rogers P, Atwood CW, Wagener MM, Yu VL. Short-course empiric antibiotic therapy for patients with pulmonary infiltrates in the intensive care unit. A proposed solution for indiscriminate antibiotic prescription. Am J Respir Crit Care Med 2000; 162:505–511.
- Kollef MH, Fraser VJ. Antibiotic resistance in the intensive care unit. Ann Intern Med 2001; 134:298–314.
- Lewis RT, Burgess JH, Hampson LG. Cardiorespiratory studies in critical illness. Changes in aspiration pneumonitis. Arch Surg 1971; 103:335–340.
- Rello J. Importance of appropriate initial antibiotic therapy and de-escalation in the treatment of nosocomial pneumonia. Eur Respir Rev 2007; 16:33–39.
- American Thoracic Society. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med 2005; 171:388–416.
- Bartlett JG. Anaerobic bacterial infections of the lung and pleural space. Clin Infect Dis 1993; 16(suppl 4):S248–S255.
- Lorber B, Swenson RM. Bacteriology of aspiration pneumonia. A prospective study of community- and hospital-acquired cases. Ann Intern Med 1974; 81:329–331.
- Bartlett JG, Gorbach SL, Finegold SM. The bacteriology of aspiration pneumonia. Am J Med 1974; 56:202–207.
- Mier L, Dreyfuss D, Darchy B, et al. Is penicillin G an adequate initial treatment for aspiration pneumonia? A prospective evaluation using a protected specimen brush and quantitative cultures. Intensive Care Med 1993; 19:279–284.
- Marik PE, Careau P. The role of anaerobes in patients with ventilator-associated pneumonia and aspiration pneumonia: a prospective study. Chest 1999; 115:178–183.
- El-Solh AA, Pietrantoni C, Bhat A, et al. Microbiology of severe aspiration pneumonia in institutionalized elderly. Am J Respir Crit Care Med 2003; 167:1650–1654.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44(suppl 2):S27–S72.
- Kadowaki M, Demura Y, Mizuno S, et al. Reappraisal of clindamycin IV monotherapy for treatment of mild-to-moderate aspiration pneumonia in elderly patients. Chest 2005; 127:1276–1282.
- Ott SR, Allewelt M, Lorenz J, Reimnitz P, Lode H; German Lung Abscess Study Group. Moxifloxacin vs ampicillin/sulbactam in aspiration pneumonia and primary lung abscess. Infection 2008; 36:23–30.
- Perlino CA. Metronidazole vs clindamycin treatment of anerobic pulmonary infection. Failure of metronidazole therapy. Arch Intern Med 1981; 141:1424–1427.
- Sanders CV, Hanna BJ, Lewis AC. Metronidazole in the treatment of anaerobic infections. Am Rev Respir Dis 1979; 120:337–343.
- Alvarez-Lerma F, Alvarez B, Luque P, et al; ADANN Study Group. Empiric broad-spectrum antibiotic therapy of nosocomial pneumonia in the intensive care unit: a prospective observational study. Crit Care 2006; 10:R78.
- Johnson JL, Hirsch CS. Aspiration pneumonia. Recognizing and managing a potentially growing disorder. Postgrad Med 2003; 113:99–112.
- Scolapio JS. Methods for decreasing risk of aspiration pneumonia in critically ill patients. JPEN J Parenter Enteral Nutr 2002; 26(suppl 6):S58–S61.
- Orozco-Levi M, Torres A, Ferrer M, et al. Semirecumbent position protects from pulmonary aspiration but not completely from gastroesophageal reflux in mechanically ventilated patients. Am J Respir Crit Care Med 1995; 152:1387–1390.
- Park RH, Allison MC, Lang J, et al. Randomised comparison of percutaneous endoscopic gastrostomy and nasogastric tube feeding in patients with persisting neurological dysphagia. BMJ 1992; 304( 6839):1406–1409.
- Donskey CJ, Chowdhry TK, Hecker MT, et al. Effect of antibiotic therapy on the density of vancomycin-resistant enterococci in the stool of colonized patients. N Engl J Med 2000; 343:1925–1932.
- Mouw DR, Langlois JP, Turner LF, Neher JO. Clinical inquiries. Are antibiotics effective in preventing pneumonia for nursing home patients? J Fam Pract 2004; 53:994–996.
Should patients with mild asthma use inhaled steroids?
Yes. A number of large randomized controlled trials have shown inhaled corticosteroids to be beneficial in low doses for patients who have mild persistent asthma, and therefore these drugs are strongly recommended in this situation.1
Asthma care providers should, however, consider this “yes” in the context of asthma severity, the goals of therapy, and the benefits and risks associated with inhaled corticosteroids.
CLASSIFICATION OF ASTHMA SEVERITY
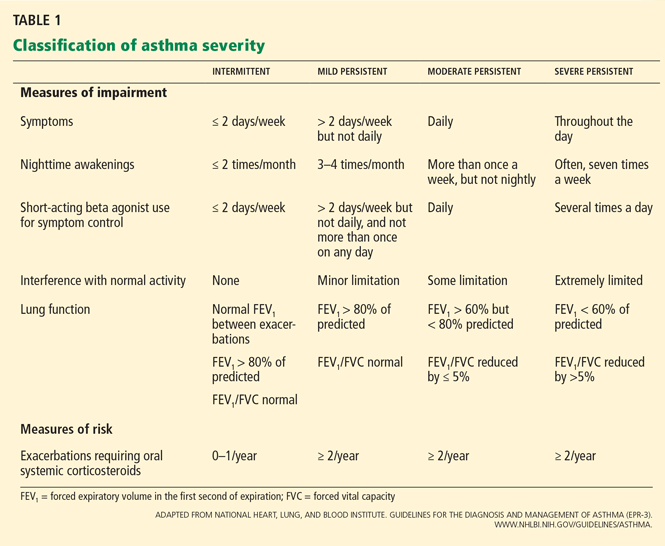
Although the studies of asthma prevalence had methodologic limitations and therefore the true prevalence of mild persistent asthma cannot be determined, it is common. Fuhlbrigge et al2 reported that most asthma patients have some form of persistent asthma. In contrast, Dusser et al3 reviewed available studies and concluded that most patients with asthma have either intermittent or mild persistent asthma.
GOALS: REDUCE IMPAIRMENT AND RISK
The goals of asthma management are to:
Reduce impairment by controlling symptoms so that normal activity levels can be maintained, by minimizing the need for short-acting bronchodilator use, and by maintaining normal pulmonary function; and to
Reduce risk by preventing progressive loss of lung function and recurrent exacerbations, and by optimizing pharmacotherapy while minimizing potential adverse effects.1
EVIDENCE OF BENEFIT

The OPTIMA trial4 (Low Dose Inhaled Budesonide and Formoterol in Mild Persistent Asthma) was a double-blind, randomized trial carried out in 198 centers in 17 countries. Compared with those randomized to receive placebo, patients who were randomized to receive an inhaled corticosteroid, ie, budesonide (Pulmicort) 100 μg twice daily, had 60% fewer severe exacerbations (relative risk [RR] 0.4, 95% confidence interval [CI] 0.27–0.59) and 48% fewer days when their asthma was poorly controlled (RR 0.52, 95% CI 0.4–0.67). Adding a long-acting beta-agonist did not change this outcome.
The START study5 (Inhaled Steroid Treatment as Regular Therapy in Early Asthma) showed that, compared with placebo, starting inhaled budesonide within the first 2 years of asthma symptoms in patients with mild persistent asthma was associated with better asthma control and less need for additional asthma medication.
The IMPACT study6 (Improving Asthma Control Trial) showed that inhaled steroids need to be taken daily, on a regular schedule, rather than intermittently as needed. Patients received either inhaled budesonide as needed, budesonide 200 μg twice daily every day, or zafirlukast (Accolate) 20 mg twice daily. Daily budesonide therapy resulted in better asthma control, less bronchial hyperresponsiveness, and less airway inflammation compared with intermittent use, zafirlukast therapy, or placebo. Daily zafirlukast and intermittent steroid treatment produced similar results for all outcomes measured.
Despite this strong evidence supporting regular use of inhaled corticosteroids in patients with mild persistent asthma, many patients choose to take them intermittently.
Suissa et al7 found, in a large observational cohort study, that fewer patients died of asthma if they were receiving low-dose inhaled corticosteroids than if they were not. The rate of death due to asthma was lower in patients who had used more inhaled corticosteroids over the previous year, and the death rate was higher in those who had discontinued inhaled corticosteroids in the previous 3 months than in those who continued using them.
STEROIDS DO NOT SLOW THE LOSS OF LUNG FUNCTION
Compared with people without asthma, asthma patients have substantially lower values of forced expiratory volume in the first second of expiration (FEV1). They also have a faster rate of functional decline: the average decrease in FEV1 in asthma patients is 38 mL per year, compared with 22 mL per year in nonasthmatic people.9
Although inhaled corticosteroids have been shown to increase lung function in asthma patients in the short term, there is little convincing evidence to suggest that they affect the rate of decline in the long term.10 In fact, airway inflammation and bronchial hyperresponsiveness return to baseline within 2 weeks after inhaled corticosteroids are discontinued.10
DO INHALED CORTICOSTEROIDS STUNT CHILDREN’S GROWTH?
The safety of long-term low-dose inhaled corticosteroids is well established in adults. However, two large randomized controlled trials found that children treated with low-dose inhaled steroids (budesonide 200–400 μg per day) grew 1 to 1.5 cm less over 3 to 5 years of treatment than children receiving placebo.11 However, this effect was primarily evident within the first year of therapy, and growth velocity was similar to that with placebo at the end of the treatment period (4 to 6 years).12
Agertoft and Pedersen13 found that taking inhaled corticosteroids long-term is unlikely to have an effect on final height. Children who took inhaled budesonide (up to an average daily dose of 500 μg) into adulthood ended up no shorter than those who did not.
Based on these and other data, inhaled corticosteroids are generally considered safe at recommended doses. However, the decision to prescribe them for long-term therapy should be based on the risks and benefits to the individual patient.1
ALTERNATIVE DRUGS FOR MILD PERSISTENT ASTHMA
Leukotriene-modifying drugs include the leukotriene receptor antagonists montelukast (Singulair) and zafirlukast and the 5-lipoxygenase inhibitor zileuton (Zyflo CR). These drugs have been associated with statistically significant improvement in FEV1 compared with placebo in patients with mild to moderate asthma, reductions in both blood and sputum eosinophils,14 and attenuation of bronchoconstriction with exercise.11
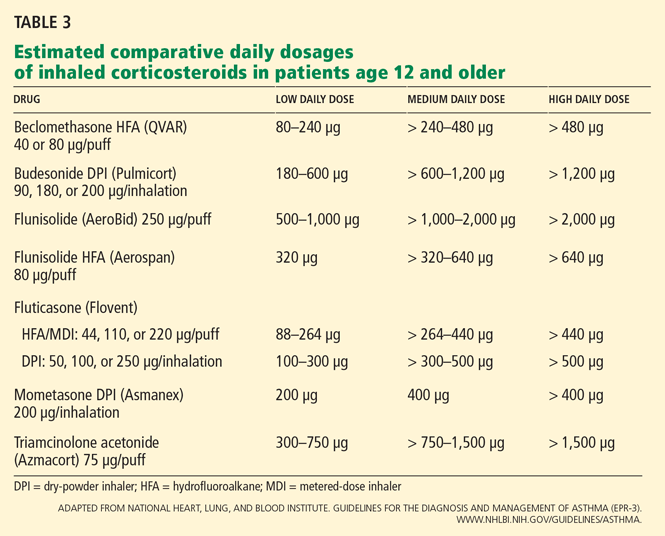
Large randomized trials comparing leukotriene modifier therapy with low-dose inhaled steroids in adults and children with mild persistent asthma have found that although outcomes improve with either therapy, the improvement is statistically superior with inhaled steroids for most asthma-control measures. 6,8 Low-dose inhaled steroid therapy in patients with mild persistent and moderate persistent asthma has been associated with superior clinical outcomes as well as greater improvement in pulmonary function than treatment with antileukotriene drugs (Table 2).8
Asthma is heterogeneous, and properly selected patients with mild persistent asthma may achieve good control with leukotrienemodifier monotherapy.15 Alternatives for patients with mild persistent asthma include the methylxanthine theophylline, but this drug is less desirable due to its narrow therapeutic index. 1 The inhaled cromones nedocromil (Tilade) and cromolyn (Intal) were other options in this patient population, but their short half-lives made them less practical, and US production has been discontinued.
THE BOTTOM LINE
Though leukotriene receptor antagonists can be effective, the daily use of inhaled corticosteroids results in higher asthma control test scores, more symptom-free days, greater pre-bronchodilator FEV1, and decreased percentage of sputum eosinophils6 in patients with mild persistent asthma, and the addition of a long-acting beta agonist does not provide additional benefit.4 Furthermore, daily use of inhaled corticosteroids in these patients has also been associated with a lower rate of asthma-related deaths and with less need for systemic corticosteroid therapy,7,8 even though inhaled corticosteroids have not yet been shown to alter the progressive loss of lung function.10
- National Heart, Lung, and Blood Institute. Guidelines for the Diagnosis and Management of Asthma (EPR-3). www.nhlbi.nih.gov/guidelines/asthma/. Accessed March 26, 2010.
- Fuhlbrigge AL, Adams RJ, Guilbert TW, et al. The burden of asthma in the United States: level and distribution are dependent on interpretation of the National Asthma Education and Prevention Program. Am J Respir Crit Care Med 2002; 166:1044–1049.
- Dusser D, Montani D, Chanez P, et al. Mild asthma: an expert review on epidemiology, clinical characteristics and treatment recommendations. Allergy 2007; 62:591–604.
- O’Byrne PM, Barnes PJ, Rodriguez-Roisin R, et al. Low dose inhaled budesonide and formoterol in mild persistent asthma: the OPTIMA randomized trial. Am J Respir Crit Care Med 2001; 164:1392–1397.
- Busse WW, Pedersen S, Pauwels RA, et al; START Investigators Group. The Inhaled Steroid Treatment As Regular Therapy in Early Asthma (START) study 5-year follow-up: effectiveness of early intervention with budesonide in mild persistent asthma. J Allergy Clin Immunol 2008; 121:1167–1174.
- Boushey HA, Sorkness CA, King TS, et al; National Heart, Lung, and Blood Institute’s Asthma Clinical Research Network. Daily versus as-needed corticosteroids for mild persistent asthma. N Engl J Med 2005; 352:1519–1528.
- Suissa S, Ernst P, Benayoun S, Baltzan M, Cai B. Low-dose inhaled corticosteroids and the prevention of death from asthma. N Engl J Med 2000; 343:332–356.
- Busse W, Wolfe J, Storms W, et al. Fluticasone propionate compared with zafirlukast in controlling persistent asthma: a randomized double-blind, placebo-controlled trial. J Fam Pract 2001; 50:595–602.
- Lange P, Parner J, Vestbo J, Schnohr P, Jensen G. A 15-year follow-up study of ventilatory function in adults with asthma. N Engl J Med 1998; 339:1194–1200.
- Fanta CH. Asthma. N Engl J Med 2009; 360:1002–1014.
- O’Byrne PM, Parameswaran K. Pharmacological management of mild or moderate persistent asthma. Lancet 2006; 368:794–803.
- The Childhood Asthma Management Program Research Group. Long-term effects of budesonide or nedocromil in children with asthma. N Engl J Med 2000; 343:1054–1063.
- Agertoft L, Pedersen S. Effect of long-term treatment with inhaled budesonide on adult height in children with asthma. N Engl J Med 2000; 343:1064–1069.
- Pizzichini E, Leff JA, Reiss TF, et al. Montelukast reduces airway eosinophilic inflammation in asthma: a randomized, controlled trial. Eur Respir J 1999; 14:12–18.
- Kraft M, Israel E, O’Connor GT. Clinical decisions. Treatment of mild persistent asthma. N Engl J Med 2007; 356:2096–2100.
Yes. A number of large randomized controlled trials have shown inhaled corticosteroids to be beneficial in low doses for patients who have mild persistent asthma, and therefore these drugs are strongly recommended in this situation.1
Asthma care providers should, however, consider this “yes” in the context of asthma severity, the goals of therapy, and the benefits and risks associated with inhaled corticosteroids.
CLASSIFICATION OF ASTHMA SEVERITY
Although the studies of asthma prevalence had methodologic limitations and therefore the true prevalence of mild persistent asthma cannot be determined, it is common. Fuhlbrigge et al2 reported that most asthma patients have some form of persistent asthma. In contrast, Dusser et al3 reviewed available studies and concluded that most patients with asthma have either intermittent or mild persistent asthma.
GOALS: REDUCE IMPAIRMENT AND RISK
The goals of asthma management are to:
Reduce impairment by controlling symptoms so that normal activity levels can be maintained, by minimizing the need for short-acting bronchodilator use, and by maintaining normal pulmonary function; and to
Reduce risk by preventing progressive loss of lung function and recurrent exacerbations, and by optimizing pharmacotherapy while minimizing potential adverse effects.1
EVIDENCE OF BENEFIT
The OPTIMA trial4 (Low Dose Inhaled Budesonide and Formoterol in Mild Persistent Asthma) was a double-blind, randomized trial carried out in 198 centers in 17 countries. Compared with those randomized to receive placebo, patients who were randomized to receive an inhaled corticosteroid, ie, budesonide (Pulmicort) 100 μg twice daily, had 60% fewer severe exacerbations (relative risk [RR] 0.4, 95% confidence interval [CI] 0.27–0.59) and 48% fewer days when their asthma was poorly controlled (RR 0.52, 95% CI 0.4–0.67). Adding a long-acting beta-agonist did not change this outcome.
The START study5 (Inhaled Steroid Treatment as Regular Therapy in Early Asthma) showed that, compared with placebo, starting inhaled budesonide within the first 2 years of asthma symptoms in patients with mild persistent asthma was associated with better asthma control and less need for additional asthma medication.
The IMPACT study6 (Improving Asthma Control Trial) showed that inhaled steroids need to be taken daily, on a regular schedule, rather than intermittently as needed. Patients received either inhaled budesonide as needed, budesonide 200 μg twice daily every day, or zafirlukast (Accolate) 20 mg twice daily. Daily budesonide therapy resulted in better asthma control, less bronchial hyperresponsiveness, and less airway inflammation compared with intermittent use, zafirlukast therapy, or placebo. Daily zafirlukast and intermittent steroid treatment produced similar results for all outcomes measured.
Despite this strong evidence supporting regular use of inhaled corticosteroids in patients with mild persistent asthma, many patients choose to take them intermittently.
Suissa et al7 found, in a large observational cohort study, that fewer patients died of asthma if they were receiving low-dose inhaled corticosteroids than if they were not. The rate of death due to asthma was lower in patients who had used more inhaled corticosteroids over the previous year, and the death rate was higher in those who had discontinued inhaled corticosteroids in the previous 3 months than in those who continued using them.
STEROIDS DO NOT SLOW THE LOSS OF LUNG FUNCTION
Compared with people without asthma, asthma patients have substantially lower values of forced expiratory volume in the first second of expiration (FEV1). They also have a faster rate of functional decline: the average decrease in FEV1 in asthma patients is 38 mL per year, compared with 22 mL per year in nonasthmatic people.9
Although inhaled corticosteroids have been shown to increase lung function in asthma patients in the short term, there is little convincing evidence to suggest that they affect the rate of decline in the long term.10 In fact, airway inflammation and bronchial hyperresponsiveness return to baseline within 2 weeks after inhaled corticosteroids are discontinued.10
DO INHALED CORTICOSTEROIDS STUNT CHILDREN’S GROWTH?
The safety of long-term low-dose inhaled corticosteroids is well established in adults. However, two large randomized controlled trials found that children treated with low-dose inhaled steroids (budesonide 200–400 μg per day) grew 1 to 1.5 cm less over 3 to 5 years of treatment than children receiving placebo.11 However, this effect was primarily evident within the first year of therapy, and growth velocity was similar to that with placebo at the end of the treatment period (4 to 6 years).12
Agertoft and Pedersen13 found that taking inhaled corticosteroids long-term is unlikely to have an effect on final height. Children who took inhaled budesonide (up to an average daily dose of 500 μg) into adulthood ended up no shorter than those who did not.
Based on these and other data, inhaled corticosteroids are generally considered safe at recommended doses. However, the decision to prescribe them for long-term therapy should be based on the risks and benefits to the individual patient.1
ALTERNATIVE DRUGS FOR MILD PERSISTENT ASTHMA
Leukotriene-modifying drugs include the leukotriene receptor antagonists montelukast (Singulair) and zafirlukast and the 5-lipoxygenase inhibitor zileuton (Zyflo CR). These drugs have been associated with statistically significant improvement in FEV1 compared with placebo in patients with mild to moderate asthma, reductions in both blood and sputum eosinophils,14 and attenuation of bronchoconstriction with exercise.11
Large randomized trials comparing leukotriene modifier therapy with low-dose inhaled steroids in adults and children with mild persistent asthma have found that although outcomes improve with either therapy, the improvement is statistically superior with inhaled steroids for most asthma-control measures. 6,8 Low-dose inhaled steroid therapy in patients with mild persistent and moderate persistent asthma has been associated with superior clinical outcomes as well as greater improvement in pulmonary function than treatment with antileukotriene drugs (Table 2).8
Asthma is heterogeneous, and properly selected patients with mild persistent asthma may achieve good control with leukotrienemodifier monotherapy.15 Alternatives for patients with mild persistent asthma include the methylxanthine theophylline, but this drug is less desirable due to its narrow therapeutic index. 1 The inhaled cromones nedocromil (Tilade) and cromolyn (Intal) were other options in this patient population, but their short half-lives made them less practical, and US production has been discontinued.
THE BOTTOM LINE
Though leukotriene receptor antagonists can be effective, the daily use of inhaled corticosteroids results in higher asthma control test scores, more symptom-free days, greater pre-bronchodilator FEV1, and decreased percentage of sputum eosinophils6 in patients with mild persistent asthma, and the addition of a long-acting beta agonist does not provide additional benefit.4 Furthermore, daily use of inhaled corticosteroids in these patients has also been associated with a lower rate of asthma-related deaths and with less need for systemic corticosteroid therapy,7,8 even though inhaled corticosteroids have not yet been shown to alter the progressive loss of lung function.10
Yes. A number of large randomized controlled trials have shown inhaled corticosteroids to be beneficial in low doses for patients who have mild persistent asthma, and therefore these drugs are strongly recommended in this situation.1
Asthma care providers should, however, consider this “yes” in the context of asthma severity, the goals of therapy, and the benefits and risks associated with inhaled corticosteroids.
CLASSIFICATION OF ASTHMA SEVERITY
Although the studies of asthma prevalence had methodologic limitations and therefore the true prevalence of mild persistent asthma cannot be determined, it is common. Fuhlbrigge et al2 reported that most asthma patients have some form of persistent asthma. In contrast, Dusser et al3 reviewed available studies and concluded that most patients with asthma have either intermittent or mild persistent asthma.
GOALS: REDUCE IMPAIRMENT AND RISK
The goals of asthma management are to:
Reduce impairment by controlling symptoms so that normal activity levels can be maintained, by minimizing the need for short-acting bronchodilator use, and by maintaining normal pulmonary function; and to
Reduce risk by preventing progressive loss of lung function and recurrent exacerbations, and by optimizing pharmacotherapy while minimizing potential adverse effects.1
EVIDENCE OF BENEFIT
The OPTIMA trial4 (Low Dose Inhaled Budesonide and Formoterol in Mild Persistent Asthma) was a double-blind, randomized trial carried out in 198 centers in 17 countries. Compared with those randomized to receive placebo, patients who were randomized to receive an inhaled corticosteroid, ie, budesonide (Pulmicort) 100 μg twice daily, had 60% fewer severe exacerbations (relative risk [RR] 0.4, 95% confidence interval [CI] 0.27–0.59) and 48% fewer days when their asthma was poorly controlled (RR 0.52, 95% CI 0.4–0.67). Adding a long-acting beta-agonist did not change this outcome.
The START study5 (Inhaled Steroid Treatment as Regular Therapy in Early Asthma) showed that, compared with placebo, starting inhaled budesonide within the first 2 years of asthma symptoms in patients with mild persistent asthma was associated with better asthma control and less need for additional asthma medication.
The IMPACT study6 (Improving Asthma Control Trial) showed that inhaled steroids need to be taken daily, on a regular schedule, rather than intermittently as needed. Patients received either inhaled budesonide as needed, budesonide 200 μg twice daily every day, or zafirlukast (Accolate) 20 mg twice daily. Daily budesonide therapy resulted in better asthma control, less bronchial hyperresponsiveness, and less airway inflammation compared with intermittent use, zafirlukast therapy, or placebo. Daily zafirlukast and intermittent steroid treatment produced similar results for all outcomes measured.
Despite this strong evidence supporting regular use of inhaled corticosteroids in patients with mild persistent asthma, many patients choose to take them intermittently.
Suissa et al7 found, in a large observational cohort study, that fewer patients died of asthma if they were receiving low-dose inhaled corticosteroids than if they were not. The rate of death due to asthma was lower in patients who had used more inhaled corticosteroids over the previous year, and the death rate was higher in those who had discontinued inhaled corticosteroids in the previous 3 months than in those who continued using them.
STEROIDS DO NOT SLOW THE LOSS OF LUNG FUNCTION
Compared with people without asthma, asthma patients have substantially lower values of forced expiratory volume in the first second of expiration (FEV1). They also have a faster rate of functional decline: the average decrease in FEV1 in asthma patients is 38 mL per year, compared with 22 mL per year in nonasthmatic people.9
Although inhaled corticosteroids have been shown to increase lung function in asthma patients in the short term, there is little convincing evidence to suggest that they affect the rate of decline in the long term.10 In fact, airway inflammation and bronchial hyperresponsiveness return to baseline within 2 weeks after inhaled corticosteroids are discontinued.10
DO INHALED CORTICOSTEROIDS STUNT CHILDREN’S GROWTH?
The safety of long-term low-dose inhaled corticosteroids is well established in adults. However, two large randomized controlled trials found that children treated with low-dose inhaled steroids (budesonide 200–400 μg per day) grew 1 to 1.5 cm less over 3 to 5 years of treatment than children receiving placebo.11 However, this effect was primarily evident within the first year of therapy, and growth velocity was similar to that with placebo at the end of the treatment period (4 to 6 years).12
Agertoft and Pedersen13 found that taking inhaled corticosteroids long-term is unlikely to have an effect on final height. Children who took inhaled budesonide (up to an average daily dose of 500 μg) into adulthood ended up no shorter than those who did not.
Based on these and other data, inhaled corticosteroids are generally considered safe at recommended doses. However, the decision to prescribe them for long-term therapy should be based on the risks and benefits to the individual patient.1
ALTERNATIVE DRUGS FOR MILD PERSISTENT ASTHMA
Leukotriene-modifying drugs include the leukotriene receptor antagonists montelukast (Singulair) and zafirlukast and the 5-lipoxygenase inhibitor zileuton (Zyflo CR). These drugs have been associated with statistically significant improvement in FEV1 compared with placebo in patients with mild to moderate asthma, reductions in both blood and sputum eosinophils,14 and attenuation of bronchoconstriction with exercise.11
Large randomized trials comparing leukotriene modifier therapy with low-dose inhaled steroids in adults and children with mild persistent asthma have found that although outcomes improve with either therapy, the improvement is statistically superior with inhaled steroids for most asthma-control measures. 6,8 Low-dose inhaled steroid therapy in patients with mild persistent and moderate persistent asthma has been associated with superior clinical outcomes as well as greater improvement in pulmonary function than treatment with antileukotriene drugs (Table 2).8
Asthma is heterogeneous, and properly selected patients with mild persistent asthma may achieve good control with leukotrienemodifier monotherapy.15 Alternatives for patients with mild persistent asthma include the methylxanthine theophylline, but this drug is less desirable due to its narrow therapeutic index. 1 The inhaled cromones nedocromil (Tilade) and cromolyn (Intal) were other options in this patient population, but their short half-lives made them less practical, and US production has been discontinued.
THE BOTTOM LINE
Though leukotriene receptor antagonists can be effective, the daily use of inhaled corticosteroids results in higher asthma control test scores, more symptom-free days, greater pre-bronchodilator FEV1, and decreased percentage of sputum eosinophils6 in patients with mild persistent asthma, and the addition of a long-acting beta agonist does not provide additional benefit.4 Furthermore, daily use of inhaled corticosteroids in these patients has also been associated with a lower rate of asthma-related deaths and with less need for systemic corticosteroid therapy,7,8 even though inhaled corticosteroids have not yet been shown to alter the progressive loss of lung function.10
- National Heart, Lung, and Blood Institute. Guidelines for the Diagnosis and Management of Asthma (EPR-3). www.nhlbi.nih.gov/guidelines/asthma/. Accessed March 26, 2010.
- Fuhlbrigge AL, Adams RJ, Guilbert TW, et al. The burden of asthma in the United States: level and distribution are dependent on interpretation of the National Asthma Education and Prevention Program. Am J Respir Crit Care Med 2002; 166:1044–1049.
- Dusser D, Montani D, Chanez P, et al. Mild asthma: an expert review on epidemiology, clinical characteristics and treatment recommendations. Allergy 2007; 62:591–604.
- O’Byrne PM, Barnes PJ, Rodriguez-Roisin R, et al. Low dose inhaled budesonide and formoterol in mild persistent asthma: the OPTIMA randomized trial. Am J Respir Crit Care Med 2001; 164:1392–1397.
- Busse WW, Pedersen S, Pauwels RA, et al; START Investigators Group. The Inhaled Steroid Treatment As Regular Therapy in Early Asthma (START) study 5-year follow-up: effectiveness of early intervention with budesonide in mild persistent asthma. J Allergy Clin Immunol 2008; 121:1167–1174.
- Boushey HA, Sorkness CA, King TS, et al; National Heart, Lung, and Blood Institute’s Asthma Clinical Research Network. Daily versus as-needed corticosteroids for mild persistent asthma. N Engl J Med 2005; 352:1519–1528.
- Suissa S, Ernst P, Benayoun S, Baltzan M, Cai B. Low-dose inhaled corticosteroids and the prevention of death from asthma. N Engl J Med 2000; 343:332–356.
- Busse W, Wolfe J, Storms W, et al. Fluticasone propionate compared with zafirlukast in controlling persistent asthma: a randomized double-blind, placebo-controlled trial. J Fam Pract 2001; 50:595–602.
- Lange P, Parner J, Vestbo J, Schnohr P, Jensen G. A 15-year follow-up study of ventilatory function in adults with asthma. N Engl J Med 1998; 339:1194–1200.
- Fanta CH. Asthma. N Engl J Med 2009; 360:1002–1014.
- O’Byrne PM, Parameswaran K. Pharmacological management of mild or moderate persistent asthma. Lancet 2006; 368:794–803.
- The Childhood Asthma Management Program Research Group. Long-term effects of budesonide or nedocromil in children with asthma. N Engl J Med 2000; 343:1054–1063.
- Agertoft L, Pedersen S. Effect of long-term treatment with inhaled budesonide on adult height in children with asthma. N Engl J Med 2000; 343:1064–1069.
- Pizzichini E, Leff JA, Reiss TF, et al. Montelukast reduces airway eosinophilic inflammation in asthma: a randomized, controlled trial. Eur Respir J 1999; 14:12–18.
- Kraft M, Israel E, O’Connor GT. Clinical decisions. Treatment of mild persistent asthma. N Engl J Med 2007; 356:2096–2100.
- National Heart, Lung, and Blood Institute. Guidelines for the Diagnosis and Management of Asthma (EPR-3). www.nhlbi.nih.gov/guidelines/asthma/. Accessed March 26, 2010.
- Fuhlbrigge AL, Adams RJ, Guilbert TW, et al. The burden of asthma in the United States: level and distribution are dependent on interpretation of the National Asthma Education and Prevention Program. Am J Respir Crit Care Med 2002; 166:1044–1049.
- Dusser D, Montani D, Chanez P, et al. Mild asthma: an expert review on epidemiology, clinical characteristics and treatment recommendations. Allergy 2007; 62:591–604.
- O’Byrne PM, Barnes PJ, Rodriguez-Roisin R, et al. Low dose inhaled budesonide and formoterol in mild persistent asthma: the OPTIMA randomized trial. Am J Respir Crit Care Med 2001; 164:1392–1397.
- Busse WW, Pedersen S, Pauwels RA, et al; START Investigators Group. The Inhaled Steroid Treatment As Regular Therapy in Early Asthma (START) study 5-year follow-up: effectiveness of early intervention with budesonide in mild persistent asthma. J Allergy Clin Immunol 2008; 121:1167–1174.
- Boushey HA, Sorkness CA, King TS, et al; National Heart, Lung, and Blood Institute’s Asthma Clinical Research Network. Daily versus as-needed corticosteroids for mild persistent asthma. N Engl J Med 2005; 352:1519–1528.
- Suissa S, Ernst P, Benayoun S, Baltzan M, Cai B. Low-dose inhaled corticosteroids and the prevention of death from asthma. N Engl J Med 2000; 343:332–356.
- Busse W, Wolfe J, Storms W, et al. Fluticasone propionate compared with zafirlukast in controlling persistent asthma: a randomized double-blind, placebo-controlled trial. J Fam Pract 2001; 50:595–602.
- Lange P, Parner J, Vestbo J, Schnohr P, Jensen G. A 15-year follow-up study of ventilatory function in adults with asthma. N Engl J Med 1998; 339:1194–1200.
- Fanta CH. Asthma. N Engl J Med 2009; 360:1002–1014.
- O’Byrne PM, Parameswaran K. Pharmacological management of mild or moderate persistent asthma. Lancet 2006; 368:794–803.
- The Childhood Asthma Management Program Research Group. Long-term effects of budesonide or nedocromil in children with asthma. N Engl J Med 2000; 343:1054–1063.
- Agertoft L, Pedersen S. Effect of long-term treatment with inhaled budesonide on adult height in children with asthma. N Engl J Med 2000; 343:1064–1069.
- Pizzichini E, Leff JA, Reiss TF, et al. Montelukast reduces airway eosinophilic inflammation in asthma: a randomized, controlled trial. Eur Respir J 1999; 14:12–18.
- Kraft M, Israel E, O’Connor GT. Clinical decisions. Treatment of mild persistent asthma. N Engl J Med 2007; 356:2096–2100.
Food allergy and eosinophilic esophagitis: Learning what to avoid
More children and even adults seem to be allergic to various foods these days than in the past. Also apparently on the rise is a linked condition, eosinophilic esophagitis.
The reason for these increases is not clear. This article confines itself to what we know about the mechanisms of food allergies and eosinophilic esophagitis, how to diagnose them, and how to treat them.
FOOD ALLERGIES ARE COMMON, AND MORE PREVALENT THAN EVER
Food allergies—abnormal immune responses to food proteins1—affect an estimated 6% to 8% of young children and 3% to 4% of adults in the United States,2,3 and their prevalence appears to be rising in developed countries. Studies in US and British children indicate that peanut allergy has doubled in the past decade. 4
Any food can provoke a reaction, but only a few foods account for most of the significant allergic reactions: cow’s milk, soy, wheat, eggs, peanuts, tree nuts, fish, and shellfish.
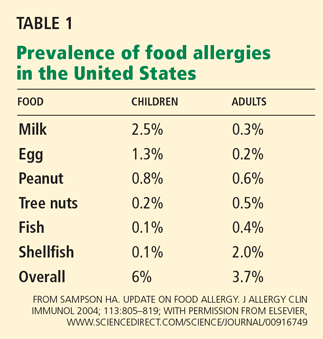
Approximately 80% of allergies to milk, egg, wheat, and soy resolve by the time the patient reaches early adolescence.6 Fewer cases resolve in children with tree nut allergies (approximately 9%) or peanut allergy (20%),7,8 and allergies to fish and shellfish often develop or persist in adulthood.
A family history of an atopic disease such as asthma, allergic rhinitis, eczema, or food allergy is a risk factor for developing a food allergy. 3 Considering that the rate of peanut allergy has doubled in children over the past 10 years, environmental factors may also play a role.3
How we tolerate foods or become allergic to them
The gut, the largest mucosal organ in the body, is exposed to large quantities of foreign proteins daily. Most protein is broken down by stomach acid and digestive enzymes into lessantigenic peptides or is bound by secretory immunoglobulin A (IgA), which prevents it from being absorbed. Further, the epithelial cells lining the gut do not allow large molecules to pass easily, having tight intracellular junctions and being covered with mucus.
For these reasons, less than 2% of the protein in food is absorbed in an allergenic form.9 The reason food allergies are more prevalent in children is most likely that children have an immature gut barrier, lower IgA levels, a higher gastric pH, and lower proteolytic enzyme levels.
When dietary proteins do cross the gut barrier, the immune system normally suppresses the allergic response. Regulatory T cells, dendritic cells, and local immune responses play critical roles in the development of tolerance. Several types of regulatory T cells, such as Tr1 cells (which secrete interleukin 10), TH3 cells (which secrete transforming growth factor beta), CD4+CD25+ regulatory T cells, gamma-delta T cells, and CD8+ suppressor cells can all contribute to suppressing allergic responses.10 Dendritic cells also help induce tolerance by stimulating CD4+ T cells to secrete transforming growth factor beta, which leads to the production of interleukin 10 and additional transforming growth factor beta.11
Factors that contribute to food allergy
Many factors may contribute to whether a person becomes tolerant to or sensitized to a specific food protein.
The dose of antigen. Tolerance can develop after either high or low doses of antigens, but by different mechanisms.
The antigen structure. Soluble antigens are less sensitizing than particulate antigens.12,13
Processing of foods. Dry-roasted peanuts are more allergenic than raw or boiled peanuts, partly because they are less soluble.13
The route of initial exposure. Sensitization to food proteins can occur directly through the gut or the skin. Alternatively, it can occur indirectly via the respiratory tract. Skin exposure may be especially sensitizing in children with atopic dermatitis.14,15
The gut flora. When mice are raised in a germ-free environment, they fail to develop normal tolerance.16 They are also more likely to become sensitized if they are treated with antibiotics or if they lack toll-like receptors that recognize bacterial lipopolysaccharides.17 Furthermore, human studies suggest that probiotics promote tolerance, especially in preventing atopic dermatitis, although the studies have had conflicting results.18–21
The gastric pH. Murine and human studies reveal that antacid medications increase the risk of food allergy.22,23
Genetic susceptibility. A child with a sibling who is allergic to peanuts is approximately 10 times more likely to be allergic to peanuts than predicted by the rate in the general population. Although no risk-conferring gene has been identified, a study of twins showed concordance for peanut allergy in 64.3% of identical twins vs 6.8% of fraternal twins.24
Three types of immune responses to food

Immunologic reactions to foods can be divided into three categories: mediated by immunoglobulin E (IgE), non-IgE-mediated, and mixed. Therefore, these disorders can present as an acute, potentially life-threatening reaction or as a chronic disease such as eosinophilic gastoenteropathy.
IgE-mediated reactions are immediate hypersensitivity responses. In most patients, an IgE-mediated mechanism can be confirmed by a positive skin test or a test for food-specific IgE in the serum. In this article, the term “food allergy” refers to an IgE-mediated reaction to a food, unless otherwise indicated.
Non-IgE-mediated reactions have a delayed onset and chronic symptoms. Commonly, they are confined to the gastrointestinal tract; examples are food-protein-induced enterocolitis, proctitis, and proctocolitis and celiac disease.3,26,27 However, other diseases such as contact dermatitis, dermatitis herpetiformis, and food-induced pulmonary hemosiderosis (Heiner syndrome) are also considered non-IgE-mediated allergies.
Mixed-reaction disorders are chronic and include the eosinophilic gastroenteropathies, ie, eosinophilic proctocolitis, eosinophilic gastroenteritis, and eosinophilic esophagitis.28 The pathophysiology of these diseases is poorly understood. Many patients have evidence of allergic sensitivities to food or to environmental allergens, or both, but whether these sensitivities have a causal role in these disorders is not clear.
Atopic dermatitis, another complicated disease process, may be associated with mixedreaction food allergy, as approximately 35% of young children with moderate to severe atopic dermatitis have food allergies.29
Diagnosis of IgE-mediated food allergies
A thorough history and physical examination are key to diagnosing an IgE-mediated food allergy.
The history should include potential culprit foods, the quantity eaten, the timing of the onset of symptoms, and related factors such as exercise, alcohol intake, or medication use. Symptoms of an IgE-mediated reaction are generally rapid in onset but may be delayed up to a few hours, while non-IgE mediated symptoms may present several hours to days later.
Food challenge. A double-blind, placebocontrolled oral food challenge is the gold standard for the diagnosis of food allergies. (The food to be tested is hidden in other food or in capsules.) However, this test poses significant risks, and other diagnostic methods are more practical for screening.
Skin-prick tests with commercially available extracts are a rapid and sensitive method of screening for allergy to several foods.
Negative skin-prick tests have an estimated negative predictive value of more than 95% and can therefore exclude IgE-mediated food allergies.
A positive test indicates the presence of IgE against a specific food allergen and suggests a clinical food allergy, although the specificity of the test is only about 50%, making a positive result difficult to interpret. Although the size of the skin-test response does not necessarily correlate with the potential severity of a reaction, a response larger than 3 mm does indicate a greater likelihood of clinical reactivity. A positive test is most helpful in confirming the diagnosis of IgE-mediated food allergy when combined with a clear history of food-induced symptoms.
The proteins in commercially based extracts of most fruits and vegetables are often labile; therefore, skin testing with fresh fruits and vegetables may be indicated.30
Immunoassays. Radioallergosorbent tests (RASTs) and fluorescent enzyme immunoassays are used to identity food-specific IgE antibodies in the serum. The commercially available tests do not use radioactivity, but the term “RAST” is still commonly used.
Immunoassays are generally less sensitive and more costly than skin-prick tests, and their results are not immediately available, unlike those of skin-prick testing. However, these in vitro tests are not affected by antihistamine use and are useful in patients with severe dermatologic conditions or severe anaphylaxis, for whom skin-prick testing would not be appropriate.

However, unlike a negative skin-prick test, an undetectable serum food-specific IgE level has a low negative predictive value, and an undetectable level may be associated with symptoms of an allergic reaction for 10% to 25% of patients.29 Therefore, if one suspects an allergic reaction but no food-specific IgE can be detected in the serum, confirming the absence of a clinical allergy must be done with a skin-prick test or with a physician-supervised oral challenge, or both.
Managing food allergy by avoiding the allergen
Food allergies are managed by strictly avoiding food allergens and by taking medications such as self-injectable epinephrine for anaphylactic symptoms.
Patients and caregivers must be educated about reading food labels, avoiding high-risk situations such as eating at buffets and other restaurants with high risk of cross-contamination, wearing a medical-alert bracelet, recognizing and managing early symptoms of an allergic reaction, and calling for emergency services if they are having an allergic reaction. Since January 2006, the US Food and Drug Administration has required food manufacturers to list common food allergens on food labels (cow’s milk, soy, wheat, egg, peanut, tree nuts, fish, and shellfish), and the labeling must use simple, easily understood terms, such as “milk” instead of “whey.” However, it is still prudent to read all ingredients listed on the label.
Experimental treatments for food allergies
Humanized monoclonal anti-IgE antibodies such as talizumab (also known as TNX-901) and omalizumab (Xolair) have been developed, but their use in food allergy has been limited. In a study in patients with peanut allergy, injections of talizumab increased the threshold for sensitivity to peanuts in most patients, but 25% of the patients did not have any improvement.32 A study of omalizumab in patients with peanut allergy was stopped after adverse reactions developed during oral peanut challenges.33
Oral immunotherapy. Recent studies suggest it may be possible to induce oral tolerance in patients with IgE-mediated food allergy. Pilot studies have shown that frequent, increasing doses of food allergens (egg, milk, and peanut) may raise the threshold at which symptoms occur.34–36 Though these studies suggest that oral immunotherapy may protect some patients against a reaction if they accidentally ingest a food they are allergic to, some patients could not reach the goal doses because allergic symptoms were provoked.
At this early stage, these strategies must be considered investigational, and more randomized, placebo-controlled studies are needed. Further studies will also be needed to assess whether oral immunotherapy induces only short-term desensitization (in which case the allergen needs to be ingested daily to prevent reactions) or sustained tolerance (in which case the antigenic protein can be ingested without symptoms despite periods of abstinence).
THE ROLE OF FOOD ALLERGY IN EOSINOPHILIC ESOPHAGITIS
Eosinophilic esophagitis has been recognized with increasing frequency in both children and adults over the past several years. Symptoms can include difficulty feeding, failure to thrive, vomiting, epigastric or chest pain, dysphagia, and food impaction.
Diagnostic criteria for eosinophilic esophagitis are37:
- Clinical symptoms of esophageal dysfunction
- At least 15 eosinophils per high-power field in at least one esophageal biopsy specimen
- No response to a proton-pump inhibitor in high doses (up to 2 mg/kg/day) for 1 to 2 months, or normal results on pH probe monitoring of the esophagus (the reason for this criterion is that patients with gastroesophageal reflux disease can also have large numbers of eosinophils in the esophagus—more than 100 per highpower field38)
- Exclusion of other causes.
Though the cause of eosinophilic esophagitis is not completely understood, atopy has been strongly implicated as a factor. More than 50% of patients with eosinophilic esophagitis also have an atopic condition (eg, atopic dermatitis, allergic rhinitis, asthma), as well as positive results on skin-prick testing or measurement of antigen-specific IgE in the serum.39–41 Also, since most patients improve with either dietary restriction or elemental diets, food sensitization appears to play a considerable role.
As with atopic conditions such as asthma, atopic dermatitis, allergic rhinitis, and food allergy, eosinophilic esophagitis has been linked with immune responses involving helper T cell 2 (TH2). Adults and children with eosinophilic esophagitis have been found to have elevated eosinophil counts and total IgE levels in peripheral blood.37 In the esophagus, patients have elevated levels of the TH2 cytokines often seen in atopic patients (eg, interleukins 4, 5, and 13) and mast cells.42,43 In mice, eosinophilic esophagitis can be induced by allergen exposure and overexpression of TH2 cytokines.44,45 Expression of eotaxin-3, a potent eosinophil chemoattractant, was noted to be higher in children with eosinophilic esophagitis than in controls.46
Of interest, some patients with eosinophilic esophagitis say their symptoms vary with the seasons, correlating with seasonal changes in esophageal eosinophil levels.47,48
Studies linking eosinophilic esophagitis and food allergy in children
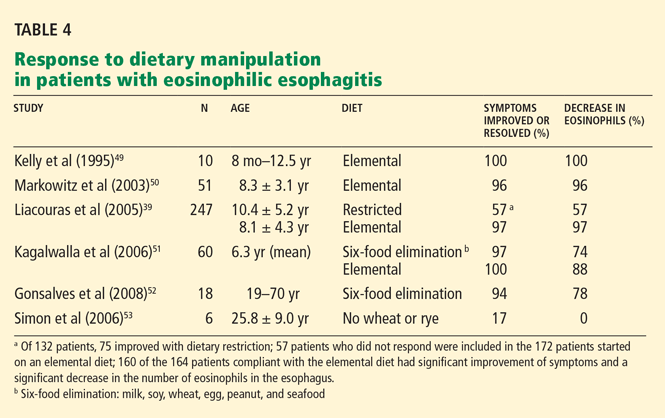
Kelly et al49 reported that 10 children with chronic symptomatic gastroesophageal reflux and eosinophilic esophagitis all had partial or complete resolution of symptoms on an elemental diet.
Markowitz et al50 found that symptoms of chronic reflux disease and eosinophilic esophagitis improved in 49 of 51 children on an elemental diet, and the number of eosinophils in the distal esophagus decreased significantly.
Liacouras et al39 reported similar findings in a 10-year experience. Of 132 children who had eosinophilic esophagitis, 75 improved with dietary restriction based on results of skin-prick and patch testing. The 57 patients who did not respond and 115 others were started on an elemental diet. Of the 164 patients who complied with the elemental diet, 160 had significant improvement of symptoms and a significant decrease in the number of eosinophils in the esophagus. Individual foods were reintroduced approximately every 5 days, and esophagogastroduodenoscopy with biopsies was performed 4 to 8 weeks after the last was reintroduced into the diet.
In a retrospective study, Kagalwalla et al51 reported that 60 children with eosinophilic esophagitis were treated with either an elemental diet or a six-food elimination diet (no milk, soy, wheat, egg, peanut, or seafood). The two groups showed similar clinical and histologic improvements.
Collectively, these studies in pediatric patients imply that food allergy is a significant factor in the pathogenesis of eosinophilic esophagitis.
Studies in adults
Fewer studies of the link between food allergy and eosinophilic esophagitis have been done in adults.
In a preliminary study, 18 adults followed the six-food elimination diet. Symptoms improved in 17 (94%), and histologic findings improved in 14 (78%).52
On the other hand, in six adult patients with eosinophilic esophagitis, Simon et al53 found that only one had improvement in symptoms after eliminating wheat and rye from the diet, and none had significant changes in the number of eosinophils in the esophagus.
In a 37-year-old man with eosinophilic esophagitis, symptoms improved after eliminating egg from his diet.54
Yamazaki et al55 measured expression of interleukin 5 and interleukin 13 in 15 adult patients with eosinophilic esophagitis. Food and aeroallergens that included milk, soy, dust mite, ragweed, and Aspergillus induced significantly more interleukin 5 production in these patients than in atopic controls, suggesting that both foods and aeroallergens may have a role in the pathogenesis of eosinophilic esophagitis in adults.
How to identify potential food triggers of eosinophilic esophagitis
Though elemental diets have been associated with a decrease in symptoms and esophageal eosinophilia, elemental formulas are expensive and unpalatable and pose a risk of nutritional deprivation. Identifying specific food allergens to eliminate from the diet in patients with eosinophilic esophagitis may be less expensive and more desirable than a very limited or elemental diet.
However, potential food triggers have been hard to identify in eosinophilic esophagitis. A recent consensus report did not recommend in vitro food allergy testing,37 owing to a lack of positive or negative predictive values for food-specific IgE level testing in eosinophilic esophagitis. Furthermore, the absence of IgE does not eliminate a food as a potential trigger, since non-IgE mechanisms may play a role.
Skin-prick testing is one of the currently validated diagnostic methods. Several studies have used skin-prick testing of foods in patients with eosinophilic esophagitis. In these studies, approximately two-thirds of patients had positive test reactions to at least one food, most often to common food allergens such as cow’s milk, egg, soy, wheat, and peanut, but also to rye, beef, and bean.37 In a recent article,56 81% of adult patients with eosinophilic esophagitis had one or more allergens identified by skin-prick testing, and 50% of the patients tested positive for one or more food allergens.
Atopy patch testing. The combination of skin-prick testing and atopy patch testing may be more effective than skin-prick testing alone in identifying potential food triggers. Atopy patch testing has been used in the diagnosis of non-IgE cell-mediated (delayed) immune responses, in which T cells may play a significant role.
Atopy patch testing is similar to patch testing for contact dermatitis. It involves placing a small quantity of food on the skin and evaluating for a local delayed reaction after a set time.
In two studies,50,57 146 children with biopsy-proven eosinophilic esophagitis had foods eliminated from the diet on the basis of positive skin-prick tests and atopy patch tests. Approximately 77% of the children had significant reduction of esophageal eosinophils in biopsy specimens (from 20 per high-power field to 1.1). The foods most commonly implicated by skin-prick testing were cow’s milk, egg, wheat, peanut, shellfish, peas, beef, fish, rye, and tomato; those identified by atopy patch testing were cow’s milk, egg, wheat, corn, beef, milk, soy, rye, chicken, oats, and potato. The combination of both types of testing had a negative predictive value of 88% to 100% for all foods except milk, while the positive predictive value was greater than 74% for the most common foods causing eosinophilic esophagitis.58
Though atopy patch testing shows some usefulness in identifying foods that may elicit non-IgE-mediated reactions, currently these tests are not validated and have been evaluated in only a small number of studies. Currently, no standardized testing materials, methods of application, or interpretation of results exist, and no studies have included a control population to validate atopy patch testing. More studies are needed to validate atopy patch testing as a reliable diagnostic tool before it can be recommended as a component of routine diagnostic evaluation in patients with eosinophilic esophagitis.
- Bruijnzeel-Koomen C, Ortolani C, Aas K, et al. Adverse reactions to food. European Academy of Allergology and Clinical Immunology Subcommittee. Allergy 1995; 50:623–635.
- Sampson HA. Update on food allergy. J Allergy Clin Immunol 2004; 113:805–819.
- Sicherer SH, Sampson HA. 9. Food allergy. J Allergy Clin Immunol 2006; 117 (suppl 2):S470–S475.
- Sicherer SH, Munoz-Furlong A, Sampson HA. Prevalence of peanut and tree nut allergy in the United States determined by means of a random digit dial telephone survey: a 5-year follow-up study. J Allergy Clin Immunol 2003; 112:1203–1207.
- American College of Allergy, Asthma, & Immunology. Food allergy: a practice parameter. Ann Allergy Asthma Immunol 2006; 96( suppl 2):S1–S68.
- Wood RA. The natural history of food allergy. Pediatrics 2003; 111:1631–1637.
- Hourihane JO, Roberts SA, Warner JO. Resolution of peanut allergy: case-control study. BMJ 1998; 316:1271–1275.
- Fleischer DM, Conover-Walker MK, Matsui EC, Wood RA. The natural history of tree nut allergy. J Allergy Clin Immunol 2005; 116:1087–1093.
- Husby S, Foged N, Host A, Svehag SE. Passage of dietary antigens into the blood of children with coeliac disease. Quantification and size distribution of absorbed antigens. Gut 1987; 28:1062–1072.
- Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens. Nat Rev Immunol 2003; 3:331–341.
- Frossard CP, Tropia L, Hauser C, Eigenmann PA. Lymphocytes in Peyer patches regulate clinical tolerance in a murine model of food allergy. J Allergy Clin Immunol 2004; 113:958–964.
- Jain SL, Barone KS, Flanagan MP, Michael JG. Activation patterns of murine B cells after oral administration of an encapsulated soluble antigen. Vaccine 1996; 14:1291–1297.
- Kopper RA, Odum NJ, Sen M, Helm RM, Stanley JS, Burks AW. Peanut protein allergens: the effect of roasting on solubility and allergenicity. Int Arch Allergy Immunol 2005; 136:16–22.
- Lack G. Epidemiologic risks for food allergy. J Allergy Clin Immunol 2008; 121:1331–1336.
- Lack G, Fox D, Northstone K, Golding J; Avon Longitudinal Study of Parents and Children Study Team. Factors associated with the development of peanut allergy in childhood. N Engl J Med 2003; 348:977–985.
- Sudo N, Sawamura S, Tanaka K, Aiba Y, Kubo C, Koga Y. The requirement of intestinal bacterial flora for the development of an IgE production system fully susceptible to oral tolerance induction. J Immunol 1997; 159:1739–1745.
- Bashir ME, Louie S, Shi HN, Nagler-Anderson C. Toll-like receptor 4 signaling by intestinal microbes influences susceptibility to food allergy. J Immunol 2004; 172:6978–6987.
- Kopp MV, Hennemuth I, Heinzmann A, Urbanek R. Randomized, double-blind, placebo-controlled trial of probiotics for primary prevention: no clinical effects of lactobacillus GG supplementation. Pediatrics 2008; 121:e850–e856.
- Kukkonen K, Savilahti E, Haahtela T, et al. Probiotics and prebiotic galacto-oligosaccharides in the prevention of allergic diseases: a randomized, double-blind, placebo-controlled trial. J Allergy Clin Immunol 2007; 119:192–198.
- Osborn DA, Sinn JK. Probiotics in infants for prevention of allergic disease and food hypersensitivity. Cochrane Database Syst Rev 2007;CD006475.
- Prescott SL, Bjorksten B. Probiotics for the prevention or treatment of allergic diseases. J Allergy Clin Immunol 2007; 120:255–262.
- Untersmayr E, Jensen-Jarolim E. The role of protein digestibility and antacids on food allergy outcomes. J Allergy Clin Immunol 2008; 121:1301–1308.
- Untersmayr E, Scholl I, Swoboda I, et al. Antacid medication inhibits digestion of dietary proteins and causes food allergy: a fish allergy model in BALB/c mice. J Allergy Clin Immunol 2003; 112:616–623.
- Sicherer SH, Furlong TJ, Maes HH, Desnick RJ, Sampson HA, Gelb BD. Genetics of peanut allergy: a twin study. J Allergy Clin Immunol 2000; 106:53–56.
- Sicherer SH, Sampson HA. Food allergy: recent advances in pathophysiology and treatment. Annu Rev Med 2009; 60:261–277.
- Sampson HA, Anderson JA. Summary and recommendations: classification of gastrointestinal manifestations due to immunologic reactions to foods in infants and young children. J Pediatr Gastroenterol Nutr 2000; 30( suppl 1):S87–S94.
- Sampson HA, Sicherer SH, Birnbaum AH. AGA technical review on the evaluation of food allergy in gastrointestinal disorders. American Gastroenterological Association. Gastroenterology 2001; 120:1026–1040.
- Spergel JM, Pawlowski NA. Food allergy. Mechanisms, diagnosis, and management in children. Pediatr Clin North Am 2002; 49:73–96.
- Sampson HA. Utility of food-specific IgE concentrations in predicting symptomatic food allergy. J Allergy Clin Immunol 2001; 107:891–896.
- Ortolani C, Ispano M, Pastorello EA, Ansaloni R, Magri GC. Comparison of results of skin prick tests (with fresh foods and commercial food extracts) and RAST in 100 patients with oral allergy syndrome. J Allergy Clin Immunol 1989; 83:683–690.
- Perry TT, Matsui EC, Kay Conover-Walker M, Wood RA. The relationship of allergen-specific IgE levels and oral food challenge outcome. J Allergy Clin Immunol 2004; 114:144–149.
- Leung DY, Sampson HA, Yunginger JW, et al; Avon Longitudinal Study of Parents and Children Study Team. Effect of anti-IgE therapy in patients with peanut allergy. N Engl J Med 2003; 348:986–993.
- Sampson HA. A phase II, randomized double-blind, parallel-group, placebo-controlled, oral food challenge trial of Xolair (omalizumab) in peanut allergy (TOPS). J Allergy Clin Immunol 2007; 119 (suppl 1):S117.
- Buchanan AD, Green TD, Jones SM, et al Egg oral immunotherapy in nonanaphylactic children with egg allergy. J Allergy Clin Immunol 2007; 119:199–205.
- Burks AW, Jones SM. Egg oral immunotherapy in non-anaphylactic children with egg allergy: follow-up. J Allergy Clin Immunol 2008; 121:270–271.
- Skripak JM, Nash SD, Rowley H, et al. A randomized, double-blind, placebo-controlled study of milk oral immunotherapy for cow's milk allergy. J Allergy Clin Immunol 2008; 122:1154–1160.
- Furuta GT, Liacouras CA, Collins MH, et al; First International Gastrointestinal Eosinophil Research Symposium (FIGERS) Subcommittees. Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology 2007; 133:1342–1363.
- Rodrigo S, Abboud G, Oh D, et al. High intraepithelial eosinophil counts in esophageal squamous epithelium are not specific for eosinophilic esophagitis in adults. Am J Gastroenterol 2008; 103:435–442.
- Liacouras CA, Spergel JM, Ruchelli E, et al. Eosinophilic esophagitis: a 10-year experience in 381 children. Clin Gastroenterol Hepatol 2005; 3:1198–1206.
- Simon D, Marti H, Heer P, Simon HU, Braathen LR, Straumann A. Eosinophilic esophagitis is frequently associated with IgE-mediated allergic airway diseases. J Allergy Clin Immunol 2005; 115:1090–1092.
- Rothenberg ME, Mishra A, Collins MH, Putnam PE. Pathogenesis and clinical features of eosinophilic esophagitis. J Allergy Clin Immunol 2001; 108:891–894.
- Gupta SK, Fitzgerald JF, Kondratyuk T, HogenEsch H. Cytokine expression in normal and inflamed esophageal mucosa: a study into the pathogenesis of allergic eosinophilic esophagitis. J Pediatr Gastroenterol Nutr 2006; 42:22–26.
- Straumann A, Bauer M, Fischer B, Blaser K, Simon HU. Idiopathic eosinophilic esophagitis is associated with a T(H)2-type allergic inflammatory response. J Allergy Clin Immunol 2001; 108:954–961.
- Mishra A, Rothenberg ME. Intratracheal IL-13 induces eosinophilic esophagitis by an IL-5, eotaxin-1, and STAT6-dependent mechanism. Gastroenterology 2003; 125:1419–1427.
- Akei HS, Mishra A, Blanchard C, Rothenberg ME. Epicutaneous antigen exposure primes for experimental eosinophilic esophagitis in mice. Gastroenterology 2005; 129:985–994.
- Blanchard C, Wang N, Stringer KF, et al. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest 2006; 116:536–547.
- Fogg MI, Ruchelli E, Spergel JM. Pollen and eosinophilic esophagitis. J Allergy Clin Immunol 2003; 112:796–797.
- Almansa C, Krishna M, Buchner AM, et al. Seasonal distribution in newly diagnosed cases of eosinophilic esophagitis in adults. Am J Gastroenterol 2009; 104:828–833.
- Kelly KJ, Lazenby AJ, Rowe PC, Yardley JH, Perman JA, Sampson HA. Eosinophilic esophagitis attributed to gastroesophageal reflux: improvement with an amino acid-based formula. Gastroenterology 1995; 109:1503–1512.
- Markowitz JE, Spergel JM, Ruchelli E, Liacouras CA. Elemental diet is an effective treatment for eosinophilic esophagitis in children and adolescents. Am J Gastroenterol 2003; 98:777–782.
- Kagalwalla AF, Sentongo TA, Ritz S, et al. Effect of six-food elimination diet on clinical and histologic outcomes in eosinophilic esophagitis. Clin Gastroenterol Hepatol 2006; 4:1097–1102.
- Gonsalves N, Yang GY, Doerfler B, et al. A prospective clinical trial of six food elimination diet and reintroduction of causative agents in adults with eosinophilic esophagitis [abstract]. Gastroenterology 2008; 134( suppl 1):A104–A105.
- Simon D, Straumann A, Wenk A, Spichtin H, Simon HU, Braathen LR. Eosinophilic esophagitis in adults—no clinical relevance of wheat and rye sensitizations. Allergy 2006; 61:1480–1483.
- Antón Remirez J, Escudero R, Caceres O, Fernandez-Benitez M. Eosinophilic esophagitis. Allergol Immunopathol (Madr) 2006; 34:79–81.
- Yamazaki K, Murray JA, Arora AS, et al. Allergen-specific in vitro cytokine production in adult patients with eosinophilic esophagitis. Dig Dis Sci 2006; 51:1934–1941.
- Penfield JD, Lang DM, Goldblum JR, Lopez R, Falk GW. The role of allergy evaluation in adults with eosinophilic esophagitis. J Clin Gastroenterol 2009(Epub ahead of print)
- Spergel JM, Andrews T, Brown-Whitehorn TF, Beausoleil JL, Liacouras CA. Treatment of eosinophilic esophagitis with specific food elimination diet directed by a combination of skin prick and patch tests. Ann Allergy Asthma Immunol 2005; 95:336–343.
- Spergel JM, Brown-Whitehorn T, Beausoleil JL, Shuker M, Liacouras CA. Predictive values for skin prick test and atopy patch test for eosinophilic esophagitis. J Allergy Clin Immunol 2007; 119:509–511.
More children and even adults seem to be allergic to various foods these days than in the past. Also apparently on the rise is a linked condition, eosinophilic esophagitis.
The reason for these increases is not clear. This article confines itself to what we know about the mechanisms of food allergies and eosinophilic esophagitis, how to diagnose them, and how to treat them.
FOOD ALLERGIES ARE COMMON, AND MORE PREVALENT THAN EVER
Food allergies—abnormal immune responses to food proteins1—affect an estimated 6% to 8% of young children and 3% to 4% of adults in the United States,2,3 and their prevalence appears to be rising in developed countries. Studies in US and British children indicate that peanut allergy has doubled in the past decade. 4
Any food can provoke a reaction, but only a few foods account for most of the significant allergic reactions: cow’s milk, soy, wheat, eggs, peanuts, tree nuts, fish, and shellfish.
Approximately 80% of allergies to milk, egg, wheat, and soy resolve by the time the patient reaches early adolescence.6 Fewer cases resolve in children with tree nut allergies (approximately 9%) or peanut allergy (20%),7,8 and allergies to fish and shellfish often develop or persist in adulthood.
A family history of an atopic disease such as asthma, allergic rhinitis, eczema, or food allergy is a risk factor for developing a food allergy. 3 Considering that the rate of peanut allergy has doubled in children over the past 10 years, environmental factors may also play a role.3
How we tolerate foods or become allergic to them
The gut, the largest mucosal organ in the body, is exposed to large quantities of foreign proteins daily. Most protein is broken down by stomach acid and digestive enzymes into lessantigenic peptides or is bound by secretory immunoglobulin A (IgA), which prevents it from being absorbed. Further, the epithelial cells lining the gut do not allow large molecules to pass easily, having tight intracellular junctions and being covered with mucus.
For these reasons, less than 2% of the protein in food is absorbed in an allergenic form.9 The reason food allergies are more prevalent in children is most likely that children have an immature gut barrier, lower IgA levels, a higher gastric pH, and lower proteolytic enzyme levels.
When dietary proteins do cross the gut barrier, the immune system normally suppresses the allergic response. Regulatory T cells, dendritic cells, and local immune responses play critical roles in the development of tolerance. Several types of regulatory T cells, such as Tr1 cells (which secrete interleukin 10), TH3 cells (which secrete transforming growth factor beta), CD4+CD25+ regulatory T cells, gamma-delta T cells, and CD8+ suppressor cells can all contribute to suppressing allergic responses.10 Dendritic cells also help induce tolerance by stimulating CD4+ T cells to secrete transforming growth factor beta, which leads to the production of interleukin 10 and additional transforming growth factor beta.11
Factors that contribute to food allergy
Many factors may contribute to whether a person becomes tolerant to or sensitized to a specific food protein.
The dose of antigen. Tolerance can develop after either high or low doses of antigens, but by different mechanisms.
The antigen structure. Soluble antigens are less sensitizing than particulate antigens.12,13
Processing of foods. Dry-roasted peanuts are more allergenic than raw or boiled peanuts, partly because they are less soluble.13
The route of initial exposure. Sensitization to food proteins can occur directly through the gut or the skin. Alternatively, it can occur indirectly via the respiratory tract. Skin exposure may be especially sensitizing in children with atopic dermatitis.14,15
The gut flora. When mice are raised in a germ-free environment, they fail to develop normal tolerance.16 They are also more likely to become sensitized if they are treated with antibiotics or if they lack toll-like receptors that recognize bacterial lipopolysaccharides.17 Furthermore, human studies suggest that probiotics promote tolerance, especially in preventing atopic dermatitis, although the studies have had conflicting results.18–21
The gastric pH. Murine and human studies reveal that antacid medications increase the risk of food allergy.22,23
Genetic susceptibility. A child with a sibling who is allergic to peanuts is approximately 10 times more likely to be allergic to peanuts than predicted by the rate in the general population. Although no risk-conferring gene has been identified, a study of twins showed concordance for peanut allergy in 64.3% of identical twins vs 6.8% of fraternal twins.24
Three types of immune responses to food
Immunologic reactions to foods can be divided into three categories: mediated by immunoglobulin E (IgE), non-IgE-mediated, and mixed. Therefore, these disorders can present as an acute, potentially life-threatening reaction or as a chronic disease such as eosinophilic gastoenteropathy.
IgE-mediated reactions are immediate hypersensitivity responses. In most patients, an IgE-mediated mechanism can be confirmed by a positive skin test or a test for food-specific IgE in the serum. In this article, the term “food allergy” refers to an IgE-mediated reaction to a food, unless otherwise indicated.
Non-IgE-mediated reactions have a delayed onset and chronic symptoms. Commonly, they are confined to the gastrointestinal tract; examples are food-protein-induced enterocolitis, proctitis, and proctocolitis and celiac disease.3,26,27 However, other diseases such as contact dermatitis, dermatitis herpetiformis, and food-induced pulmonary hemosiderosis (Heiner syndrome) are also considered non-IgE-mediated allergies.
Mixed-reaction disorders are chronic and include the eosinophilic gastroenteropathies, ie, eosinophilic proctocolitis, eosinophilic gastroenteritis, and eosinophilic esophagitis.28 The pathophysiology of these diseases is poorly understood. Many patients have evidence of allergic sensitivities to food or to environmental allergens, or both, but whether these sensitivities have a causal role in these disorders is not clear.
Atopic dermatitis, another complicated disease process, may be associated with mixedreaction food allergy, as approximately 35% of young children with moderate to severe atopic dermatitis have food allergies.29
Diagnosis of IgE-mediated food allergies
A thorough history and physical examination are key to diagnosing an IgE-mediated food allergy.
The history should include potential culprit foods, the quantity eaten, the timing of the onset of symptoms, and related factors such as exercise, alcohol intake, or medication use. Symptoms of an IgE-mediated reaction are generally rapid in onset but may be delayed up to a few hours, while non-IgE mediated symptoms may present several hours to days later.
Food challenge. A double-blind, placebocontrolled oral food challenge is the gold standard for the diagnosis of food allergies. (The food to be tested is hidden in other food or in capsules.) However, this test poses significant risks, and other diagnostic methods are more practical for screening.
Skin-prick tests with commercially available extracts are a rapid and sensitive method of screening for allergy to several foods.
Negative skin-prick tests have an estimated negative predictive value of more than 95% and can therefore exclude IgE-mediated food allergies.
A positive test indicates the presence of IgE against a specific food allergen and suggests a clinical food allergy, although the specificity of the test is only about 50%, making a positive result difficult to interpret. Although the size of the skin-test response does not necessarily correlate with the potential severity of a reaction, a response larger than 3 mm does indicate a greater likelihood of clinical reactivity. A positive test is most helpful in confirming the diagnosis of IgE-mediated food allergy when combined with a clear history of food-induced symptoms.
The proteins in commercially based extracts of most fruits and vegetables are often labile; therefore, skin testing with fresh fruits and vegetables may be indicated.30
Immunoassays. Radioallergosorbent tests (RASTs) and fluorescent enzyme immunoassays are used to identity food-specific IgE antibodies in the serum. The commercially available tests do not use radioactivity, but the term “RAST” is still commonly used.
Immunoassays are generally less sensitive and more costly than skin-prick tests, and their results are not immediately available, unlike those of skin-prick testing. However, these in vitro tests are not affected by antihistamine use and are useful in patients with severe dermatologic conditions or severe anaphylaxis, for whom skin-prick testing would not be appropriate.
However, unlike a negative skin-prick test, an undetectable serum food-specific IgE level has a low negative predictive value, and an undetectable level may be associated with symptoms of an allergic reaction for 10% to 25% of patients.29 Therefore, if one suspects an allergic reaction but no food-specific IgE can be detected in the serum, confirming the absence of a clinical allergy must be done with a skin-prick test or with a physician-supervised oral challenge, or both.
Managing food allergy by avoiding the allergen
Food allergies are managed by strictly avoiding food allergens and by taking medications such as self-injectable epinephrine for anaphylactic symptoms.
Patients and caregivers must be educated about reading food labels, avoiding high-risk situations such as eating at buffets and other restaurants with high risk of cross-contamination, wearing a medical-alert bracelet, recognizing and managing early symptoms of an allergic reaction, and calling for emergency services if they are having an allergic reaction. Since January 2006, the US Food and Drug Administration has required food manufacturers to list common food allergens on food labels (cow’s milk, soy, wheat, egg, peanut, tree nuts, fish, and shellfish), and the labeling must use simple, easily understood terms, such as “milk” instead of “whey.” However, it is still prudent to read all ingredients listed on the label.
Experimental treatments for food allergies
Humanized monoclonal anti-IgE antibodies such as talizumab (also known as TNX-901) and omalizumab (Xolair) have been developed, but their use in food allergy has been limited. In a study in patients with peanut allergy, injections of talizumab increased the threshold for sensitivity to peanuts in most patients, but 25% of the patients did not have any improvement.32 A study of omalizumab in patients with peanut allergy was stopped after adverse reactions developed during oral peanut challenges.33
Oral immunotherapy. Recent studies suggest it may be possible to induce oral tolerance in patients with IgE-mediated food allergy. Pilot studies have shown that frequent, increasing doses of food allergens (egg, milk, and peanut) may raise the threshold at which symptoms occur.34–36 Though these studies suggest that oral immunotherapy may protect some patients against a reaction if they accidentally ingest a food they are allergic to, some patients could not reach the goal doses because allergic symptoms were provoked.
At this early stage, these strategies must be considered investigational, and more randomized, placebo-controlled studies are needed. Further studies will also be needed to assess whether oral immunotherapy induces only short-term desensitization (in which case the allergen needs to be ingested daily to prevent reactions) or sustained tolerance (in which case the antigenic protein can be ingested without symptoms despite periods of abstinence).
THE ROLE OF FOOD ALLERGY IN EOSINOPHILIC ESOPHAGITIS
Eosinophilic esophagitis has been recognized with increasing frequency in both children and adults over the past several years. Symptoms can include difficulty feeding, failure to thrive, vomiting, epigastric or chest pain, dysphagia, and food impaction.
Diagnostic criteria for eosinophilic esophagitis are37:
- Clinical symptoms of esophageal dysfunction
- At least 15 eosinophils per high-power field in at least one esophageal biopsy specimen
- No response to a proton-pump inhibitor in high doses (up to 2 mg/kg/day) for 1 to 2 months, or normal results on pH probe monitoring of the esophagus (the reason for this criterion is that patients with gastroesophageal reflux disease can also have large numbers of eosinophils in the esophagus—more than 100 per highpower field38)
- Exclusion of other causes.
Though the cause of eosinophilic esophagitis is not completely understood, atopy has been strongly implicated as a factor. More than 50% of patients with eosinophilic esophagitis also have an atopic condition (eg, atopic dermatitis, allergic rhinitis, asthma), as well as positive results on skin-prick testing or measurement of antigen-specific IgE in the serum.39–41 Also, since most patients improve with either dietary restriction or elemental diets, food sensitization appears to play a considerable role.
As with atopic conditions such as asthma, atopic dermatitis, allergic rhinitis, and food allergy, eosinophilic esophagitis has been linked with immune responses involving helper T cell 2 (TH2). Adults and children with eosinophilic esophagitis have been found to have elevated eosinophil counts and total IgE levels in peripheral blood.37 In the esophagus, patients have elevated levels of the TH2 cytokines often seen in atopic patients (eg, interleukins 4, 5, and 13) and mast cells.42,43 In mice, eosinophilic esophagitis can be induced by allergen exposure and overexpression of TH2 cytokines.44,45 Expression of eotaxin-3, a potent eosinophil chemoattractant, was noted to be higher in children with eosinophilic esophagitis than in controls.46
Of interest, some patients with eosinophilic esophagitis say their symptoms vary with the seasons, correlating with seasonal changes in esophageal eosinophil levels.47,48
Studies linking eosinophilic esophagitis and food allergy in children
Kelly et al49 reported that 10 children with chronic symptomatic gastroesophageal reflux and eosinophilic esophagitis all had partial or complete resolution of symptoms on an elemental diet.
Markowitz et al50 found that symptoms of chronic reflux disease and eosinophilic esophagitis improved in 49 of 51 children on an elemental diet, and the number of eosinophils in the distal esophagus decreased significantly.
Liacouras et al39 reported similar findings in a 10-year experience. Of 132 children who had eosinophilic esophagitis, 75 improved with dietary restriction based on results of skin-prick and patch testing. The 57 patients who did not respond and 115 others were started on an elemental diet. Of the 164 patients who complied with the elemental diet, 160 had significant improvement of symptoms and a significant decrease in the number of eosinophils in the esophagus. Individual foods were reintroduced approximately every 5 days, and esophagogastroduodenoscopy with biopsies was performed 4 to 8 weeks after the last was reintroduced into the diet.
In a retrospective study, Kagalwalla et al51 reported that 60 children with eosinophilic esophagitis were treated with either an elemental diet or a six-food elimination diet (no milk, soy, wheat, egg, peanut, or seafood). The two groups showed similar clinical and histologic improvements.
Collectively, these studies in pediatric patients imply that food allergy is a significant factor in the pathogenesis of eosinophilic esophagitis.
Studies in adults
Fewer studies of the link between food allergy and eosinophilic esophagitis have been done in adults.
In a preliminary study, 18 adults followed the six-food elimination diet. Symptoms improved in 17 (94%), and histologic findings improved in 14 (78%).52
On the other hand, in six adult patients with eosinophilic esophagitis, Simon et al53 found that only one had improvement in symptoms after eliminating wheat and rye from the diet, and none had significant changes in the number of eosinophils in the esophagus.
In a 37-year-old man with eosinophilic esophagitis, symptoms improved after eliminating egg from his diet.54
Yamazaki et al55 measured expression of interleukin 5 and interleukin 13 in 15 adult patients with eosinophilic esophagitis. Food and aeroallergens that included milk, soy, dust mite, ragweed, and Aspergillus induced significantly more interleukin 5 production in these patients than in atopic controls, suggesting that both foods and aeroallergens may have a role in the pathogenesis of eosinophilic esophagitis in adults.
How to identify potential food triggers of eosinophilic esophagitis
Though elemental diets have been associated with a decrease in symptoms and esophageal eosinophilia, elemental formulas are expensive and unpalatable and pose a risk of nutritional deprivation. Identifying specific food allergens to eliminate from the diet in patients with eosinophilic esophagitis may be less expensive and more desirable than a very limited or elemental diet.
However, potential food triggers have been hard to identify in eosinophilic esophagitis. A recent consensus report did not recommend in vitro food allergy testing,37 owing to a lack of positive or negative predictive values for food-specific IgE level testing in eosinophilic esophagitis. Furthermore, the absence of IgE does not eliminate a food as a potential trigger, since non-IgE mechanisms may play a role.
Skin-prick testing is one of the currently validated diagnostic methods. Several studies have used skin-prick testing of foods in patients with eosinophilic esophagitis. In these studies, approximately two-thirds of patients had positive test reactions to at least one food, most often to common food allergens such as cow’s milk, egg, soy, wheat, and peanut, but also to rye, beef, and bean.37 In a recent article,56 81% of adult patients with eosinophilic esophagitis had one or more allergens identified by skin-prick testing, and 50% of the patients tested positive for one or more food allergens.
Atopy patch testing. The combination of skin-prick testing and atopy patch testing may be more effective than skin-prick testing alone in identifying potential food triggers. Atopy patch testing has been used in the diagnosis of non-IgE cell-mediated (delayed) immune responses, in which T cells may play a significant role.
Atopy patch testing is similar to patch testing for contact dermatitis. It involves placing a small quantity of food on the skin and evaluating for a local delayed reaction after a set time.
In two studies,50,57 146 children with biopsy-proven eosinophilic esophagitis had foods eliminated from the diet on the basis of positive skin-prick tests and atopy patch tests. Approximately 77% of the children had significant reduction of esophageal eosinophils in biopsy specimens (from 20 per high-power field to 1.1). The foods most commonly implicated by skin-prick testing were cow’s milk, egg, wheat, peanut, shellfish, peas, beef, fish, rye, and tomato; those identified by atopy patch testing were cow’s milk, egg, wheat, corn, beef, milk, soy, rye, chicken, oats, and potato. The combination of both types of testing had a negative predictive value of 88% to 100% for all foods except milk, while the positive predictive value was greater than 74% for the most common foods causing eosinophilic esophagitis.58
Though atopy patch testing shows some usefulness in identifying foods that may elicit non-IgE-mediated reactions, currently these tests are not validated and have been evaluated in only a small number of studies. Currently, no standardized testing materials, methods of application, or interpretation of results exist, and no studies have included a control population to validate atopy patch testing. More studies are needed to validate atopy patch testing as a reliable diagnostic tool before it can be recommended as a component of routine diagnostic evaluation in patients with eosinophilic esophagitis.
More children and even adults seem to be allergic to various foods these days than in the past. Also apparently on the rise is a linked condition, eosinophilic esophagitis.
The reason for these increases is not clear. This article confines itself to what we know about the mechanisms of food allergies and eosinophilic esophagitis, how to diagnose them, and how to treat them.
FOOD ALLERGIES ARE COMMON, AND MORE PREVALENT THAN EVER
Food allergies—abnormal immune responses to food proteins1—affect an estimated 6% to 8% of young children and 3% to 4% of adults in the United States,2,3 and their prevalence appears to be rising in developed countries. Studies in US and British children indicate that peanut allergy has doubled in the past decade. 4
Any food can provoke a reaction, but only a few foods account for most of the significant allergic reactions: cow’s milk, soy, wheat, eggs, peanuts, tree nuts, fish, and shellfish.
Approximately 80% of allergies to milk, egg, wheat, and soy resolve by the time the patient reaches early adolescence.6 Fewer cases resolve in children with tree nut allergies (approximately 9%) or peanut allergy (20%),7,8 and allergies to fish and shellfish often develop or persist in adulthood.
A family history of an atopic disease such as asthma, allergic rhinitis, eczema, or food allergy is a risk factor for developing a food allergy. 3 Considering that the rate of peanut allergy has doubled in children over the past 10 years, environmental factors may also play a role.3
How we tolerate foods or become allergic to them
The gut, the largest mucosal organ in the body, is exposed to large quantities of foreign proteins daily. Most protein is broken down by stomach acid and digestive enzymes into lessantigenic peptides or is bound by secretory immunoglobulin A (IgA), which prevents it from being absorbed. Further, the epithelial cells lining the gut do not allow large molecules to pass easily, having tight intracellular junctions and being covered with mucus.
For these reasons, less than 2% of the protein in food is absorbed in an allergenic form.9 The reason food allergies are more prevalent in children is most likely that children have an immature gut barrier, lower IgA levels, a higher gastric pH, and lower proteolytic enzyme levels.
When dietary proteins do cross the gut barrier, the immune system normally suppresses the allergic response. Regulatory T cells, dendritic cells, and local immune responses play critical roles in the development of tolerance. Several types of regulatory T cells, such as Tr1 cells (which secrete interleukin 10), TH3 cells (which secrete transforming growth factor beta), CD4+CD25+ regulatory T cells, gamma-delta T cells, and CD8+ suppressor cells can all contribute to suppressing allergic responses.10 Dendritic cells also help induce tolerance by stimulating CD4+ T cells to secrete transforming growth factor beta, which leads to the production of interleukin 10 and additional transforming growth factor beta.11
Factors that contribute to food allergy
Many factors may contribute to whether a person becomes tolerant to or sensitized to a specific food protein.
The dose of antigen. Tolerance can develop after either high or low doses of antigens, but by different mechanisms.
The antigen structure. Soluble antigens are less sensitizing than particulate antigens.12,13
Processing of foods. Dry-roasted peanuts are more allergenic than raw or boiled peanuts, partly because they are less soluble.13
The route of initial exposure. Sensitization to food proteins can occur directly through the gut or the skin. Alternatively, it can occur indirectly via the respiratory tract. Skin exposure may be especially sensitizing in children with atopic dermatitis.14,15
The gut flora. When mice are raised in a germ-free environment, they fail to develop normal tolerance.16 They are also more likely to become sensitized if they are treated with antibiotics or if they lack toll-like receptors that recognize bacterial lipopolysaccharides.17 Furthermore, human studies suggest that probiotics promote tolerance, especially in preventing atopic dermatitis, although the studies have had conflicting results.18–21
The gastric pH. Murine and human studies reveal that antacid medications increase the risk of food allergy.22,23
Genetic susceptibility. A child with a sibling who is allergic to peanuts is approximately 10 times more likely to be allergic to peanuts than predicted by the rate in the general population. Although no risk-conferring gene has been identified, a study of twins showed concordance for peanut allergy in 64.3% of identical twins vs 6.8% of fraternal twins.24
Three types of immune responses to food
Immunologic reactions to foods can be divided into three categories: mediated by immunoglobulin E (IgE), non-IgE-mediated, and mixed. Therefore, these disorders can present as an acute, potentially life-threatening reaction or as a chronic disease such as eosinophilic gastoenteropathy.
IgE-mediated reactions are immediate hypersensitivity responses. In most patients, an IgE-mediated mechanism can be confirmed by a positive skin test or a test for food-specific IgE in the serum. In this article, the term “food allergy” refers to an IgE-mediated reaction to a food, unless otherwise indicated.
Non-IgE-mediated reactions have a delayed onset and chronic symptoms. Commonly, they are confined to the gastrointestinal tract; examples are food-protein-induced enterocolitis, proctitis, and proctocolitis and celiac disease.3,26,27 However, other diseases such as contact dermatitis, dermatitis herpetiformis, and food-induced pulmonary hemosiderosis (Heiner syndrome) are also considered non-IgE-mediated allergies.
Mixed-reaction disorders are chronic and include the eosinophilic gastroenteropathies, ie, eosinophilic proctocolitis, eosinophilic gastroenteritis, and eosinophilic esophagitis.28 The pathophysiology of these diseases is poorly understood. Many patients have evidence of allergic sensitivities to food or to environmental allergens, or both, but whether these sensitivities have a causal role in these disorders is not clear.
Atopic dermatitis, another complicated disease process, may be associated with mixedreaction food allergy, as approximately 35% of young children with moderate to severe atopic dermatitis have food allergies.29
Diagnosis of IgE-mediated food allergies
A thorough history and physical examination are key to diagnosing an IgE-mediated food allergy.
The history should include potential culprit foods, the quantity eaten, the timing of the onset of symptoms, and related factors such as exercise, alcohol intake, or medication use. Symptoms of an IgE-mediated reaction are generally rapid in onset but may be delayed up to a few hours, while non-IgE mediated symptoms may present several hours to days later.
Food challenge. A double-blind, placebocontrolled oral food challenge is the gold standard for the diagnosis of food allergies. (The food to be tested is hidden in other food or in capsules.) However, this test poses significant risks, and other diagnostic methods are more practical for screening.
Skin-prick tests with commercially available extracts are a rapid and sensitive method of screening for allergy to several foods.
Negative skin-prick tests have an estimated negative predictive value of more than 95% and can therefore exclude IgE-mediated food allergies.
A positive test indicates the presence of IgE against a specific food allergen and suggests a clinical food allergy, although the specificity of the test is only about 50%, making a positive result difficult to interpret. Although the size of the skin-test response does not necessarily correlate with the potential severity of a reaction, a response larger than 3 mm does indicate a greater likelihood of clinical reactivity. A positive test is most helpful in confirming the diagnosis of IgE-mediated food allergy when combined with a clear history of food-induced symptoms.
The proteins in commercially based extracts of most fruits and vegetables are often labile; therefore, skin testing with fresh fruits and vegetables may be indicated.30
Immunoassays. Radioallergosorbent tests (RASTs) and fluorescent enzyme immunoassays are used to identity food-specific IgE antibodies in the serum. The commercially available tests do not use radioactivity, but the term “RAST” is still commonly used.
Immunoassays are generally less sensitive and more costly than skin-prick tests, and their results are not immediately available, unlike those of skin-prick testing. However, these in vitro tests are not affected by antihistamine use and are useful in patients with severe dermatologic conditions or severe anaphylaxis, for whom skin-prick testing would not be appropriate.
However, unlike a negative skin-prick test, an undetectable serum food-specific IgE level has a low negative predictive value, and an undetectable level may be associated with symptoms of an allergic reaction for 10% to 25% of patients.29 Therefore, if one suspects an allergic reaction but no food-specific IgE can be detected in the serum, confirming the absence of a clinical allergy must be done with a skin-prick test or with a physician-supervised oral challenge, or both.
Managing food allergy by avoiding the allergen
Food allergies are managed by strictly avoiding food allergens and by taking medications such as self-injectable epinephrine for anaphylactic symptoms.
Patients and caregivers must be educated about reading food labels, avoiding high-risk situations such as eating at buffets and other restaurants with high risk of cross-contamination, wearing a medical-alert bracelet, recognizing and managing early symptoms of an allergic reaction, and calling for emergency services if they are having an allergic reaction. Since January 2006, the US Food and Drug Administration has required food manufacturers to list common food allergens on food labels (cow’s milk, soy, wheat, egg, peanut, tree nuts, fish, and shellfish), and the labeling must use simple, easily understood terms, such as “milk” instead of “whey.” However, it is still prudent to read all ingredients listed on the label.
Experimental treatments for food allergies
Humanized monoclonal anti-IgE antibodies such as talizumab (also known as TNX-901) and omalizumab (Xolair) have been developed, but their use in food allergy has been limited. In a study in patients with peanut allergy, injections of talizumab increased the threshold for sensitivity to peanuts in most patients, but 25% of the patients did not have any improvement.32 A study of omalizumab in patients with peanut allergy was stopped after adverse reactions developed during oral peanut challenges.33
Oral immunotherapy. Recent studies suggest it may be possible to induce oral tolerance in patients with IgE-mediated food allergy. Pilot studies have shown that frequent, increasing doses of food allergens (egg, milk, and peanut) may raise the threshold at which symptoms occur.34–36 Though these studies suggest that oral immunotherapy may protect some patients against a reaction if they accidentally ingest a food they are allergic to, some patients could not reach the goal doses because allergic symptoms were provoked.
At this early stage, these strategies must be considered investigational, and more randomized, placebo-controlled studies are needed. Further studies will also be needed to assess whether oral immunotherapy induces only short-term desensitization (in which case the allergen needs to be ingested daily to prevent reactions) or sustained tolerance (in which case the antigenic protein can be ingested without symptoms despite periods of abstinence).
THE ROLE OF FOOD ALLERGY IN EOSINOPHILIC ESOPHAGITIS
Eosinophilic esophagitis has been recognized with increasing frequency in both children and adults over the past several years. Symptoms can include difficulty feeding, failure to thrive, vomiting, epigastric or chest pain, dysphagia, and food impaction.
Diagnostic criteria for eosinophilic esophagitis are37:
- Clinical symptoms of esophageal dysfunction
- At least 15 eosinophils per high-power field in at least one esophageal biopsy specimen
- No response to a proton-pump inhibitor in high doses (up to 2 mg/kg/day) for 1 to 2 months, or normal results on pH probe monitoring of the esophagus (the reason for this criterion is that patients with gastroesophageal reflux disease can also have large numbers of eosinophils in the esophagus—more than 100 per highpower field38)
- Exclusion of other causes.
Though the cause of eosinophilic esophagitis is not completely understood, atopy has been strongly implicated as a factor. More than 50% of patients with eosinophilic esophagitis also have an atopic condition (eg, atopic dermatitis, allergic rhinitis, asthma), as well as positive results on skin-prick testing or measurement of antigen-specific IgE in the serum.39–41 Also, since most patients improve with either dietary restriction or elemental diets, food sensitization appears to play a considerable role.
As with atopic conditions such as asthma, atopic dermatitis, allergic rhinitis, and food allergy, eosinophilic esophagitis has been linked with immune responses involving helper T cell 2 (TH2). Adults and children with eosinophilic esophagitis have been found to have elevated eosinophil counts and total IgE levels in peripheral blood.37 In the esophagus, patients have elevated levels of the TH2 cytokines often seen in atopic patients (eg, interleukins 4, 5, and 13) and mast cells.42,43 In mice, eosinophilic esophagitis can be induced by allergen exposure and overexpression of TH2 cytokines.44,45 Expression of eotaxin-3, a potent eosinophil chemoattractant, was noted to be higher in children with eosinophilic esophagitis than in controls.46
Of interest, some patients with eosinophilic esophagitis say their symptoms vary with the seasons, correlating with seasonal changes in esophageal eosinophil levels.47,48
Studies linking eosinophilic esophagitis and food allergy in children
Kelly et al49 reported that 10 children with chronic symptomatic gastroesophageal reflux and eosinophilic esophagitis all had partial or complete resolution of symptoms on an elemental diet.
Markowitz et al50 found that symptoms of chronic reflux disease and eosinophilic esophagitis improved in 49 of 51 children on an elemental diet, and the number of eosinophils in the distal esophagus decreased significantly.
Liacouras et al39 reported similar findings in a 10-year experience. Of 132 children who had eosinophilic esophagitis, 75 improved with dietary restriction based on results of skin-prick and patch testing. The 57 patients who did not respond and 115 others were started on an elemental diet. Of the 164 patients who complied with the elemental diet, 160 had significant improvement of symptoms and a significant decrease in the number of eosinophils in the esophagus. Individual foods were reintroduced approximately every 5 days, and esophagogastroduodenoscopy with biopsies was performed 4 to 8 weeks after the last was reintroduced into the diet.
In a retrospective study, Kagalwalla et al51 reported that 60 children with eosinophilic esophagitis were treated with either an elemental diet or a six-food elimination diet (no milk, soy, wheat, egg, peanut, or seafood). The two groups showed similar clinical and histologic improvements.
Collectively, these studies in pediatric patients imply that food allergy is a significant factor in the pathogenesis of eosinophilic esophagitis.
Studies in adults
Fewer studies of the link between food allergy and eosinophilic esophagitis have been done in adults.
In a preliminary study, 18 adults followed the six-food elimination diet. Symptoms improved in 17 (94%), and histologic findings improved in 14 (78%).52
On the other hand, in six adult patients with eosinophilic esophagitis, Simon et al53 found that only one had improvement in symptoms after eliminating wheat and rye from the diet, and none had significant changes in the number of eosinophils in the esophagus.
In a 37-year-old man with eosinophilic esophagitis, symptoms improved after eliminating egg from his diet.54
Yamazaki et al55 measured expression of interleukin 5 and interleukin 13 in 15 adult patients with eosinophilic esophagitis. Food and aeroallergens that included milk, soy, dust mite, ragweed, and Aspergillus induced significantly more interleukin 5 production in these patients than in atopic controls, suggesting that both foods and aeroallergens may have a role in the pathogenesis of eosinophilic esophagitis in adults.
How to identify potential food triggers of eosinophilic esophagitis
Though elemental diets have been associated with a decrease in symptoms and esophageal eosinophilia, elemental formulas are expensive and unpalatable and pose a risk of nutritional deprivation. Identifying specific food allergens to eliminate from the diet in patients with eosinophilic esophagitis may be less expensive and more desirable than a very limited or elemental diet.
However, potential food triggers have been hard to identify in eosinophilic esophagitis. A recent consensus report did not recommend in vitro food allergy testing,37 owing to a lack of positive or negative predictive values for food-specific IgE level testing in eosinophilic esophagitis. Furthermore, the absence of IgE does not eliminate a food as a potential trigger, since non-IgE mechanisms may play a role.
Skin-prick testing is one of the currently validated diagnostic methods. Several studies have used skin-prick testing of foods in patients with eosinophilic esophagitis. In these studies, approximately two-thirds of patients had positive test reactions to at least one food, most often to common food allergens such as cow’s milk, egg, soy, wheat, and peanut, but also to rye, beef, and bean.37 In a recent article,56 81% of adult patients with eosinophilic esophagitis had one or more allergens identified by skin-prick testing, and 50% of the patients tested positive for one or more food allergens.
Atopy patch testing. The combination of skin-prick testing and atopy patch testing may be more effective than skin-prick testing alone in identifying potential food triggers. Atopy patch testing has been used in the diagnosis of non-IgE cell-mediated (delayed) immune responses, in which T cells may play a significant role.
Atopy patch testing is similar to patch testing for contact dermatitis. It involves placing a small quantity of food on the skin and evaluating for a local delayed reaction after a set time.
In two studies,50,57 146 children with biopsy-proven eosinophilic esophagitis had foods eliminated from the diet on the basis of positive skin-prick tests and atopy patch tests. Approximately 77% of the children had significant reduction of esophageal eosinophils in biopsy specimens (from 20 per high-power field to 1.1). The foods most commonly implicated by skin-prick testing were cow’s milk, egg, wheat, peanut, shellfish, peas, beef, fish, rye, and tomato; those identified by atopy patch testing were cow’s milk, egg, wheat, corn, beef, milk, soy, rye, chicken, oats, and potato. The combination of both types of testing had a negative predictive value of 88% to 100% for all foods except milk, while the positive predictive value was greater than 74% for the most common foods causing eosinophilic esophagitis.58
Though atopy patch testing shows some usefulness in identifying foods that may elicit non-IgE-mediated reactions, currently these tests are not validated and have been evaluated in only a small number of studies. Currently, no standardized testing materials, methods of application, or interpretation of results exist, and no studies have included a control population to validate atopy patch testing. More studies are needed to validate atopy patch testing as a reliable diagnostic tool before it can be recommended as a component of routine diagnostic evaluation in patients with eosinophilic esophagitis.
- Bruijnzeel-Koomen C, Ortolani C, Aas K, et al. Adverse reactions to food. European Academy of Allergology and Clinical Immunology Subcommittee. Allergy 1995; 50:623–635.
- Sampson HA. Update on food allergy. J Allergy Clin Immunol 2004; 113:805–819.
- Sicherer SH, Sampson HA. 9. Food allergy. J Allergy Clin Immunol 2006; 117 (suppl 2):S470–S475.
- Sicherer SH, Munoz-Furlong A, Sampson HA. Prevalence of peanut and tree nut allergy in the United States determined by means of a random digit dial telephone survey: a 5-year follow-up study. J Allergy Clin Immunol 2003; 112:1203–1207.
- American College of Allergy, Asthma, & Immunology. Food allergy: a practice parameter. Ann Allergy Asthma Immunol 2006; 96( suppl 2):S1–S68.
- Wood RA. The natural history of food allergy. Pediatrics 2003; 111:1631–1637.
- Hourihane JO, Roberts SA, Warner JO. Resolution of peanut allergy: case-control study. BMJ 1998; 316:1271–1275.
- Fleischer DM, Conover-Walker MK, Matsui EC, Wood RA. The natural history of tree nut allergy. J Allergy Clin Immunol 2005; 116:1087–1093.
- Husby S, Foged N, Host A, Svehag SE. Passage of dietary antigens into the blood of children with coeliac disease. Quantification and size distribution of absorbed antigens. Gut 1987; 28:1062–1072.
- Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens. Nat Rev Immunol 2003; 3:331–341.
- Frossard CP, Tropia L, Hauser C, Eigenmann PA. Lymphocytes in Peyer patches regulate clinical tolerance in a murine model of food allergy. J Allergy Clin Immunol 2004; 113:958–964.
- Jain SL, Barone KS, Flanagan MP, Michael JG. Activation patterns of murine B cells after oral administration of an encapsulated soluble antigen. Vaccine 1996; 14:1291–1297.
- Kopper RA, Odum NJ, Sen M, Helm RM, Stanley JS, Burks AW. Peanut protein allergens: the effect of roasting on solubility and allergenicity. Int Arch Allergy Immunol 2005; 136:16–22.
- Lack G. Epidemiologic risks for food allergy. J Allergy Clin Immunol 2008; 121:1331–1336.
- Lack G, Fox D, Northstone K, Golding J; Avon Longitudinal Study of Parents and Children Study Team. Factors associated with the development of peanut allergy in childhood. N Engl J Med 2003; 348:977–985.
- Sudo N, Sawamura S, Tanaka K, Aiba Y, Kubo C, Koga Y. The requirement of intestinal bacterial flora for the development of an IgE production system fully susceptible to oral tolerance induction. J Immunol 1997; 159:1739–1745.
- Bashir ME, Louie S, Shi HN, Nagler-Anderson C. Toll-like receptor 4 signaling by intestinal microbes influences susceptibility to food allergy. J Immunol 2004; 172:6978–6987.
- Kopp MV, Hennemuth I, Heinzmann A, Urbanek R. Randomized, double-blind, placebo-controlled trial of probiotics for primary prevention: no clinical effects of lactobacillus GG supplementation. Pediatrics 2008; 121:e850–e856.
- Kukkonen K, Savilahti E, Haahtela T, et al. Probiotics and prebiotic galacto-oligosaccharides in the prevention of allergic diseases: a randomized, double-blind, placebo-controlled trial. J Allergy Clin Immunol 2007; 119:192–198.
- Osborn DA, Sinn JK. Probiotics in infants for prevention of allergic disease and food hypersensitivity. Cochrane Database Syst Rev 2007;CD006475.
- Prescott SL, Bjorksten B. Probiotics for the prevention or treatment of allergic diseases. J Allergy Clin Immunol 2007; 120:255–262.
- Untersmayr E, Jensen-Jarolim E. The role of protein digestibility and antacids on food allergy outcomes. J Allergy Clin Immunol 2008; 121:1301–1308.
- Untersmayr E, Scholl I, Swoboda I, et al. Antacid medication inhibits digestion of dietary proteins and causes food allergy: a fish allergy model in BALB/c mice. J Allergy Clin Immunol 2003; 112:616–623.
- Sicherer SH, Furlong TJ, Maes HH, Desnick RJ, Sampson HA, Gelb BD. Genetics of peanut allergy: a twin study. J Allergy Clin Immunol 2000; 106:53–56.
- Sicherer SH, Sampson HA. Food allergy: recent advances in pathophysiology and treatment. Annu Rev Med 2009; 60:261–277.
- Sampson HA, Anderson JA. Summary and recommendations: classification of gastrointestinal manifestations due to immunologic reactions to foods in infants and young children. J Pediatr Gastroenterol Nutr 2000; 30( suppl 1):S87–S94.
- Sampson HA, Sicherer SH, Birnbaum AH. AGA technical review on the evaluation of food allergy in gastrointestinal disorders. American Gastroenterological Association. Gastroenterology 2001; 120:1026–1040.
- Spergel JM, Pawlowski NA. Food allergy. Mechanisms, diagnosis, and management in children. Pediatr Clin North Am 2002; 49:73–96.
- Sampson HA. Utility of food-specific IgE concentrations in predicting symptomatic food allergy. J Allergy Clin Immunol 2001; 107:891–896.
- Ortolani C, Ispano M, Pastorello EA, Ansaloni R, Magri GC. Comparison of results of skin prick tests (with fresh foods and commercial food extracts) and RAST in 100 patients with oral allergy syndrome. J Allergy Clin Immunol 1989; 83:683–690.
- Perry TT, Matsui EC, Kay Conover-Walker M, Wood RA. The relationship of allergen-specific IgE levels and oral food challenge outcome. J Allergy Clin Immunol 2004; 114:144–149.
- Leung DY, Sampson HA, Yunginger JW, et al; Avon Longitudinal Study of Parents and Children Study Team. Effect of anti-IgE therapy in patients with peanut allergy. N Engl J Med 2003; 348:986–993.
- Sampson HA. A phase II, randomized double-blind, parallel-group, placebo-controlled, oral food challenge trial of Xolair (omalizumab) in peanut allergy (TOPS). J Allergy Clin Immunol 2007; 119 (suppl 1):S117.
- Buchanan AD, Green TD, Jones SM, et al Egg oral immunotherapy in nonanaphylactic children with egg allergy. J Allergy Clin Immunol 2007; 119:199–205.
- Burks AW, Jones SM. Egg oral immunotherapy in non-anaphylactic children with egg allergy: follow-up. J Allergy Clin Immunol 2008; 121:270–271.
- Skripak JM, Nash SD, Rowley H, et al. A randomized, double-blind, placebo-controlled study of milk oral immunotherapy for cow's milk allergy. J Allergy Clin Immunol 2008; 122:1154–1160.
- Furuta GT, Liacouras CA, Collins MH, et al; First International Gastrointestinal Eosinophil Research Symposium (FIGERS) Subcommittees. Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology 2007; 133:1342–1363.
- Rodrigo S, Abboud G, Oh D, et al. High intraepithelial eosinophil counts in esophageal squamous epithelium are not specific for eosinophilic esophagitis in adults. Am J Gastroenterol 2008; 103:435–442.
- Liacouras CA, Spergel JM, Ruchelli E, et al. Eosinophilic esophagitis: a 10-year experience in 381 children. Clin Gastroenterol Hepatol 2005; 3:1198–1206.
- Simon D, Marti H, Heer P, Simon HU, Braathen LR, Straumann A. Eosinophilic esophagitis is frequently associated with IgE-mediated allergic airway diseases. J Allergy Clin Immunol 2005; 115:1090–1092.
- Rothenberg ME, Mishra A, Collins MH, Putnam PE. Pathogenesis and clinical features of eosinophilic esophagitis. J Allergy Clin Immunol 2001; 108:891–894.
- Gupta SK, Fitzgerald JF, Kondratyuk T, HogenEsch H. Cytokine expression in normal and inflamed esophageal mucosa: a study into the pathogenesis of allergic eosinophilic esophagitis. J Pediatr Gastroenterol Nutr 2006; 42:22–26.
- Straumann A, Bauer M, Fischer B, Blaser K, Simon HU. Idiopathic eosinophilic esophagitis is associated with a T(H)2-type allergic inflammatory response. J Allergy Clin Immunol 2001; 108:954–961.
- Mishra A, Rothenberg ME. Intratracheal IL-13 induces eosinophilic esophagitis by an IL-5, eotaxin-1, and STAT6-dependent mechanism. Gastroenterology 2003; 125:1419–1427.
- Akei HS, Mishra A, Blanchard C, Rothenberg ME. Epicutaneous antigen exposure primes for experimental eosinophilic esophagitis in mice. Gastroenterology 2005; 129:985–994.
- Blanchard C, Wang N, Stringer KF, et al. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest 2006; 116:536–547.
- Fogg MI, Ruchelli E, Spergel JM. Pollen and eosinophilic esophagitis. J Allergy Clin Immunol 2003; 112:796–797.
- Almansa C, Krishna M, Buchner AM, et al. Seasonal distribution in newly diagnosed cases of eosinophilic esophagitis in adults. Am J Gastroenterol 2009; 104:828–833.
- Kelly KJ, Lazenby AJ, Rowe PC, Yardley JH, Perman JA, Sampson HA. Eosinophilic esophagitis attributed to gastroesophageal reflux: improvement with an amino acid-based formula. Gastroenterology 1995; 109:1503–1512.
- Markowitz JE, Spergel JM, Ruchelli E, Liacouras CA. Elemental diet is an effective treatment for eosinophilic esophagitis in children and adolescents. Am J Gastroenterol 2003; 98:777–782.
- Kagalwalla AF, Sentongo TA, Ritz S, et al. Effect of six-food elimination diet on clinical and histologic outcomes in eosinophilic esophagitis. Clin Gastroenterol Hepatol 2006; 4:1097–1102.
- Gonsalves N, Yang GY, Doerfler B, et al. A prospective clinical trial of six food elimination diet and reintroduction of causative agents in adults with eosinophilic esophagitis [abstract]. Gastroenterology 2008; 134( suppl 1):A104–A105.
- Simon D, Straumann A, Wenk A, Spichtin H, Simon HU, Braathen LR. Eosinophilic esophagitis in adults—no clinical relevance of wheat and rye sensitizations. Allergy 2006; 61:1480–1483.
- Antón Remirez J, Escudero R, Caceres O, Fernandez-Benitez M. Eosinophilic esophagitis. Allergol Immunopathol (Madr) 2006; 34:79–81.
- Yamazaki K, Murray JA, Arora AS, et al. Allergen-specific in vitro cytokine production in adult patients with eosinophilic esophagitis. Dig Dis Sci 2006; 51:1934–1941.
- Penfield JD, Lang DM, Goldblum JR, Lopez R, Falk GW. The role of allergy evaluation in adults with eosinophilic esophagitis. J Clin Gastroenterol 2009(Epub ahead of print)
- Spergel JM, Andrews T, Brown-Whitehorn TF, Beausoleil JL, Liacouras CA. Treatment of eosinophilic esophagitis with specific food elimination diet directed by a combination of skin prick and patch tests. Ann Allergy Asthma Immunol 2005; 95:336–343.
- Spergel JM, Brown-Whitehorn T, Beausoleil JL, Shuker M, Liacouras CA. Predictive values for skin prick test and atopy patch test for eosinophilic esophagitis. J Allergy Clin Immunol 2007; 119:509–511.
- Bruijnzeel-Koomen C, Ortolani C, Aas K, et al. Adverse reactions to food. European Academy of Allergology and Clinical Immunology Subcommittee. Allergy 1995; 50:623–635.
- Sampson HA. Update on food allergy. J Allergy Clin Immunol 2004; 113:805–819.
- Sicherer SH, Sampson HA. 9. Food allergy. J Allergy Clin Immunol 2006; 117 (suppl 2):S470–S475.
- Sicherer SH, Munoz-Furlong A, Sampson HA. Prevalence of peanut and tree nut allergy in the United States determined by means of a random digit dial telephone survey: a 5-year follow-up study. J Allergy Clin Immunol 2003; 112:1203–1207.
- American College of Allergy, Asthma, & Immunology. Food allergy: a practice parameter. Ann Allergy Asthma Immunol 2006; 96( suppl 2):S1–S68.
- Wood RA. The natural history of food allergy. Pediatrics 2003; 111:1631–1637.
- Hourihane JO, Roberts SA, Warner JO. Resolution of peanut allergy: case-control study. BMJ 1998; 316:1271–1275.
- Fleischer DM, Conover-Walker MK, Matsui EC, Wood RA. The natural history of tree nut allergy. J Allergy Clin Immunol 2005; 116:1087–1093.
- Husby S, Foged N, Host A, Svehag SE. Passage of dietary antigens into the blood of children with coeliac disease. Quantification and size distribution of absorbed antigens. Gut 1987; 28:1062–1072.
- Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens. Nat Rev Immunol 2003; 3:331–341.
- Frossard CP, Tropia L, Hauser C, Eigenmann PA. Lymphocytes in Peyer patches regulate clinical tolerance in a murine model of food allergy. J Allergy Clin Immunol 2004; 113:958–964.
- Jain SL, Barone KS, Flanagan MP, Michael JG. Activation patterns of murine B cells after oral administration of an encapsulated soluble antigen. Vaccine 1996; 14:1291–1297.
- Kopper RA, Odum NJ, Sen M, Helm RM, Stanley JS, Burks AW. Peanut protein allergens: the effect of roasting on solubility and allergenicity. Int Arch Allergy Immunol 2005; 136:16–22.
- Lack G. Epidemiologic risks for food allergy. J Allergy Clin Immunol 2008; 121:1331–1336.
- Lack G, Fox D, Northstone K, Golding J; Avon Longitudinal Study of Parents and Children Study Team. Factors associated with the development of peanut allergy in childhood. N Engl J Med 2003; 348:977–985.
- Sudo N, Sawamura S, Tanaka K, Aiba Y, Kubo C, Koga Y. The requirement of intestinal bacterial flora for the development of an IgE production system fully susceptible to oral tolerance induction. J Immunol 1997; 159:1739–1745.
- Bashir ME, Louie S, Shi HN, Nagler-Anderson C. Toll-like receptor 4 signaling by intestinal microbes influences susceptibility to food allergy. J Immunol 2004; 172:6978–6987.
- Kopp MV, Hennemuth I, Heinzmann A, Urbanek R. Randomized, double-blind, placebo-controlled trial of probiotics for primary prevention: no clinical effects of lactobacillus GG supplementation. Pediatrics 2008; 121:e850–e856.
- Kukkonen K, Savilahti E, Haahtela T, et al. Probiotics and prebiotic galacto-oligosaccharides in the prevention of allergic diseases: a randomized, double-blind, placebo-controlled trial. J Allergy Clin Immunol 2007; 119:192–198.
- Osborn DA, Sinn JK. Probiotics in infants for prevention of allergic disease and food hypersensitivity. Cochrane Database Syst Rev 2007;CD006475.
- Prescott SL, Bjorksten B. Probiotics for the prevention or treatment of allergic diseases. J Allergy Clin Immunol 2007; 120:255–262.
- Untersmayr E, Jensen-Jarolim E. The role of protein digestibility and antacids on food allergy outcomes. J Allergy Clin Immunol 2008; 121:1301–1308.
- Untersmayr E, Scholl I, Swoboda I, et al. Antacid medication inhibits digestion of dietary proteins and causes food allergy: a fish allergy model in BALB/c mice. J Allergy Clin Immunol 2003; 112:616–623.
- Sicherer SH, Furlong TJ, Maes HH, Desnick RJ, Sampson HA, Gelb BD. Genetics of peanut allergy: a twin study. J Allergy Clin Immunol 2000; 106:53–56.
- Sicherer SH, Sampson HA. Food allergy: recent advances in pathophysiology and treatment. Annu Rev Med 2009; 60:261–277.
- Sampson HA, Anderson JA. Summary and recommendations: classification of gastrointestinal manifestations due to immunologic reactions to foods in infants and young children. J Pediatr Gastroenterol Nutr 2000; 30( suppl 1):S87–S94.
- Sampson HA, Sicherer SH, Birnbaum AH. AGA technical review on the evaluation of food allergy in gastrointestinal disorders. American Gastroenterological Association. Gastroenterology 2001; 120:1026–1040.
- Spergel JM, Pawlowski NA. Food allergy. Mechanisms, diagnosis, and management in children. Pediatr Clin North Am 2002; 49:73–96.
- Sampson HA. Utility of food-specific IgE concentrations in predicting symptomatic food allergy. J Allergy Clin Immunol 2001; 107:891–896.
- Ortolani C, Ispano M, Pastorello EA, Ansaloni R, Magri GC. Comparison of results of skin prick tests (with fresh foods and commercial food extracts) and RAST in 100 patients with oral allergy syndrome. J Allergy Clin Immunol 1989; 83:683–690.
- Perry TT, Matsui EC, Kay Conover-Walker M, Wood RA. The relationship of allergen-specific IgE levels and oral food challenge outcome. J Allergy Clin Immunol 2004; 114:144–149.
- Leung DY, Sampson HA, Yunginger JW, et al; Avon Longitudinal Study of Parents and Children Study Team. Effect of anti-IgE therapy in patients with peanut allergy. N Engl J Med 2003; 348:986–993.
- Sampson HA. A phase II, randomized double-blind, parallel-group, placebo-controlled, oral food challenge trial of Xolair (omalizumab) in peanut allergy (TOPS). J Allergy Clin Immunol 2007; 119 (suppl 1):S117.
- Buchanan AD, Green TD, Jones SM, et al Egg oral immunotherapy in nonanaphylactic children with egg allergy. J Allergy Clin Immunol 2007; 119:199–205.
- Burks AW, Jones SM. Egg oral immunotherapy in non-anaphylactic children with egg allergy: follow-up. J Allergy Clin Immunol 2008; 121:270–271.
- Skripak JM, Nash SD, Rowley H, et al. A randomized, double-blind, placebo-controlled study of milk oral immunotherapy for cow's milk allergy. J Allergy Clin Immunol 2008; 122:1154–1160.
- Furuta GT, Liacouras CA, Collins MH, et al; First International Gastrointestinal Eosinophil Research Symposium (FIGERS) Subcommittees. Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology 2007; 133:1342–1363.
- Rodrigo S, Abboud G, Oh D, et al. High intraepithelial eosinophil counts in esophageal squamous epithelium are not specific for eosinophilic esophagitis in adults. Am J Gastroenterol 2008; 103:435–442.
- Liacouras CA, Spergel JM, Ruchelli E, et al. Eosinophilic esophagitis: a 10-year experience in 381 children. Clin Gastroenterol Hepatol 2005; 3:1198–1206.
- Simon D, Marti H, Heer P, Simon HU, Braathen LR, Straumann A. Eosinophilic esophagitis is frequently associated with IgE-mediated allergic airway diseases. J Allergy Clin Immunol 2005; 115:1090–1092.
- Rothenberg ME, Mishra A, Collins MH, Putnam PE. Pathogenesis and clinical features of eosinophilic esophagitis. J Allergy Clin Immunol 2001; 108:891–894.
- Gupta SK, Fitzgerald JF, Kondratyuk T, HogenEsch H. Cytokine expression in normal and inflamed esophageal mucosa: a study into the pathogenesis of allergic eosinophilic esophagitis. J Pediatr Gastroenterol Nutr 2006; 42:22–26.
- Straumann A, Bauer M, Fischer B, Blaser K, Simon HU. Idiopathic eosinophilic esophagitis is associated with a T(H)2-type allergic inflammatory response. J Allergy Clin Immunol 2001; 108:954–961.
- Mishra A, Rothenberg ME. Intratracheal IL-13 induces eosinophilic esophagitis by an IL-5, eotaxin-1, and STAT6-dependent mechanism. Gastroenterology 2003; 125:1419–1427.
- Akei HS, Mishra A, Blanchard C, Rothenberg ME. Epicutaneous antigen exposure primes for experimental eosinophilic esophagitis in mice. Gastroenterology 2005; 129:985–994.
- Blanchard C, Wang N, Stringer KF, et al. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest 2006; 116:536–547.
- Fogg MI, Ruchelli E, Spergel JM. Pollen and eosinophilic esophagitis. J Allergy Clin Immunol 2003; 112:796–797.
- Almansa C, Krishna M, Buchner AM, et al. Seasonal distribution in newly diagnosed cases of eosinophilic esophagitis in adults. Am J Gastroenterol 2009; 104:828–833.
- Kelly KJ, Lazenby AJ, Rowe PC, Yardley JH, Perman JA, Sampson HA. Eosinophilic esophagitis attributed to gastroesophageal reflux: improvement with an amino acid-based formula. Gastroenterology 1995; 109:1503–1512.
- Markowitz JE, Spergel JM, Ruchelli E, Liacouras CA. Elemental diet is an effective treatment for eosinophilic esophagitis in children and adolescents. Am J Gastroenterol 2003; 98:777–782.
- Kagalwalla AF, Sentongo TA, Ritz S, et al. Effect of six-food elimination diet on clinical and histologic outcomes in eosinophilic esophagitis. Clin Gastroenterol Hepatol 2006; 4:1097–1102.
- Gonsalves N, Yang GY, Doerfler B, et al. A prospective clinical trial of six food elimination diet and reintroduction of causative agents in adults with eosinophilic esophagitis [abstract]. Gastroenterology 2008; 134( suppl 1):A104–A105.
- Simon D, Straumann A, Wenk A, Spichtin H, Simon HU, Braathen LR. Eosinophilic esophagitis in adults—no clinical relevance of wheat and rye sensitizations. Allergy 2006; 61:1480–1483.
- Antón Remirez J, Escudero R, Caceres O, Fernandez-Benitez M. Eosinophilic esophagitis. Allergol Immunopathol (Madr) 2006; 34:79–81.
- Yamazaki K, Murray JA, Arora AS, et al. Allergen-specific in vitro cytokine production in adult patients with eosinophilic esophagitis. Dig Dis Sci 2006; 51:1934–1941.
- Penfield JD, Lang DM, Goldblum JR, Lopez R, Falk GW. The role of allergy evaluation in adults with eosinophilic esophagitis. J Clin Gastroenterol 2009(Epub ahead of print)
- Spergel JM, Andrews T, Brown-Whitehorn TF, Beausoleil JL, Liacouras CA. Treatment of eosinophilic esophagitis with specific food elimination diet directed by a combination of skin prick and patch tests. Ann Allergy Asthma Immunol 2005; 95:336–343.
- Spergel JM, Brown-Whitehorn T, Beausoleil JL, Shuker M, Liacouras CA. Predictive values for skin prick test and atopy patch test for eosinophilic esophagitis. J Allergy Clin Immunol 2007; 119:509–511.
KEY POINTS
- Food allergies can be classified as mediated by immunoglobulin E (IgE-mediated), non-IgE-mediated, or mixed. Their clinical presentation can vary from life-threatening anaphylaxis in IgE-mediated reactions to chronic, delayed symptoms as seen in eosinophilic esophagitis (a mixed reaction).
- The diagnosis of an IgE-mediated food allergy is made by taking a complete history and performing directed testing—skin-prick testing or measurement of foodspecific IgE levels in the serum, or both.
- Despite promising developments, food allergies continue to be treated primarily by telling patients to avoid allergens and to initiate therapy if ingestion occurs.
- Because most patients with eosinophilic esophagitis have a strong history of atopic disease and respond to allergen-free diets, a complete evaluation by a specialist in allergy and immunology is recommended.
Lupus update: Perspective and clinical pearls
Many questions about systemic lupus erythematosus (SLE, lupus) remain unanswered. Why is this disease so difficult to diagnose even for rheumatologists? Why does lupus tend to develop in previously healthy young women? Why does the disease manifest in so many ways? Why are our current treatments suboptimal?
This article addresses these questions in a brief overview and update of SLE, with an emphasis on clinical pearls regarding prevention and treatment that are relevant to any physician who sees patients with this disease.
WOMEN AND MINORITIES ARE OVERREPRESENTED
Women have a much higher prevalence of almost all autoimmune diseases. SLE has a 12:1 female-to-male ratio during the ages of 15 to 45 years, but when disease develops in either children or the elderly, the female-to-male ratio is only 2:1.
African Americans, Asian Americans, and Hispanics have about a three to four times higher frequency of lupus than white non-Hispanics and often have more severe disease.
WHY IS SLE SO DIFFICULT TO DIAGNOSE?
SLE is frequently overlooked; patients spend an average of 4 years and see three physicians before the disease is correctly diagnosed. Part of the problem is that presentations of the disease vary so widely between patients and that signs and symptoms evolve over time. Often, physicians do not consider SLE in the differential diagnosis.
On the other hand, SLE is also often over-diagnosed. Narain et al1 evaluated 263 patients who had a presumptive diagnosis of SLE. Only about half of the patients had a confirmed diagnosis; about 5% had a different autoimmune disease, such as scleroderma, systemic sclerosis, Sjögren syndrome, or polymyositis; 5% had fibromyalgia; 29% tested positive for ANA but did not have an autoimmune disease; and 10% had a nonrheumatic disease, such as a hematologic malignancy with rheumatic disease manifestations. For patients referred by a community rheumatologist, the diagnostic accuracy was better, about 80%.
The traditional classification criteria for SLE2,3 are problematic. Some criteria are very specific for SLE but are not very sensitive—eg, anti-double-stranded DNA is present in about half of patients with SLE. Others tests, like ANA, are sensitive but not specific—although ANA is present in 95% of patients with SLE, the positive predictive value of the test for SLE for any given patient is only 11%.
Other criteria are highly subjective, including oral ulcers and photosensitivity. These signs may be present in normal individuals who get an occasional aphthous ulcer or who are fair-skinned and burn easily with prolonged sun exposure. It takes a trained clinician to distinguish these from the photosensitivity and oral ulcers associated with lupus.
Many diseases can mimic SLE
Fibromyalgia frequently presents in women and may include joint and muscle aches, fatigue, and occasionally a positive ANA. ANA may be seen in about 15% of healthy women.
Sjögren syndrome can also present with arthritis, fatigue, and a positive ANA; it is commonly overlooked because physicians do not often think to ask about the classic symptoms of dry eyes and dry mouth.
Dermatomyositis causes rashes that have many features in common with SLE. Even the skin biopsy is often indistinguishable from SLE.
Hematologic problems, such as idiopathic or thrombotic thrombocytopenic purpura, primary antiphospholipid syndrome, and hematologic neoplasms, can cause serologic changes, a positive ANA, and other manifestations seen in SLE.
Drug-induced lupus should always be considered in older patients presenting with SLE-like disease. Now with the use of minocycline (Minocin) and other related agents for the treatment of acne, we are seeing younger women with drug-induced lupus.
PATIENTS ASK ‘WHY ME?’
Lupus typically develops in a young woman who was previously healthy. Such patients inevitably wonder, why me?
Lupus is like a puzzle, with genetics, gender, and the environment being important pieces of the puzzle. If all the pieces come together, people develop defective immune regulation and a break in self-tolerance. Everyone generates antibodies to self, but these low-affinity, nonpathologic antibodies are inconsequential. In SLE, autoantibodies lead to the formation of immune complexes, complement activation, and tissue damage.
Genetics plays an important role
Genetics plays an important role but is clearly not the only determining factor. Clustering in families has been shown, although a patient with lupus is more likely to have a relative with another autoimmune disease, especially autoimmune thyroid disease, than with SLE. The likelihood of an identical twin of a patient with SLE having the disease is only 25% to 30%, and is only about 5% for a fraternal twin.
In the first few months of 2008, four major studies were published that shed light on the genetics of SLE.4–7 Together, the studies evaluated more than 5,000 patients with SLE using genome-wide association scans and identified areas of the genome that are frequently different in patients with lupus than in healthy controls. Three of the four studies identified the same genetic area as important and supported the concept that B cells and complement activation play important roles in the disease pathogenesis.
Although over 95% of cases of SLE cannot be attributed to a single gene, there are rare cases of lupus that may provide important clues to mechanisms of disease. For example a homozygous deficiency of C1q (an early component of complement) is extremely rare in lupus but is associated with the highest risk (nearly 90%) of developing the disease. Deficiencies in other components of the complement cascade also carry a high risk of disease development.
Investigators discovered that C1q plays an important role in clearing away apoptotic cellular debris. If a person is deficient in C1q, clearance of this debris is impaired. In a person genetically predisposed to getting lupus, the immune system now has an opportunity to react to self-antigens exposed during apoptosis that are not being cleared away.
Even though lupus cases cannot be explained by an absence of C1q, a defect in the clearance of apoptotic cells is a common, unifying feature of the disease.
Immune response is enhanced by environmental factors
Environmental factors, especially sun exposure, are also important. Following sunburn, skin cells undergo massive cell death, and patients with lupus have a huge release of self-antigens that can be recognized by the immune system. Sunburn is like having a booster vaccine of self-antigen to stimulate autoantibody production. Not only does the skin flare, but internal organs can also flare after intense sun exposure.
LUPUS SURVIVAL HAS IMPROVED; DISEASES OF AGING NOW A FOCUS
In 1950, only 50% of patients with SLE survived 5 years after diagnosis; now, thanks to better treatment and earlier diagnosis, 80% to 90% survive at least 10 years.
Early on, patients tend to die of active disease (manifestations of vasculitis, pulmonary hemorrhage, kidney problems) or infection. Over time, cardiovascular disease and osteoporosis become more of a problem. Patients also have a higher risk of cancer throughout life.
Lupus has an unpredictable course, with flares and remissions. But underlying the reversible inflammatory changes is irreversible organ damage caused by the disease itself and, possibly, by treatment. Preventing bone disease, heart disease, and cancer now play more prominent roles in managing SLE.
Increased bone disease
Fracture rates are higher than expected in women with lupus; Ramsey-Goldman et al8 calculated the rate as five times higher than in the general population. The increased risk of osteoporosis is partly due to treatment with corticosteroids, but it is also likely caused by inflammation from lupus. Even controlling for steroid use, increased bone loss is still evident in patients with SLE.
African American women with lupus are not exempt. Lee et al9 found that, after adjusting for body mass, steroid use, thyroid disease, and menopausal status, African American women with SLE had more than five times the risk of low bone mineral density in the spine than white women with the disease.
Increased cancer risk
Patients with SLE have an increased risk of hematologic cancer and possibly lung and hepatobiliary cancers.
Bernatsky et al10 evaluated cancer risk in an international cohort of patients with SLE from 23 sites. Among patients with SLE, for all cancers combined, the standardized incidence ratio was 1.2; for hematologic cancers the ratio was 2.8; and for non-Hodgkin lymphoma it was 2.4. Surprisingly, although SLE is primarily a disease of women, reproductive cancer rates in patients with SLE did not differ from background rates. Bernatsky et al did not compare rates of cervical cancer, as many cancer registries do not record it. However, studies from the National Institutes of Health indicate that cervical dysplasia is common in patients with lupus.
Other interesting findings included an increased risk of hepatobiliary cancer, especially among men with SLE. Lung cancers were also increased, which has been observed in patients with other autoimmune diseases such as scleroderma and polymyositis. Smoking is a strong predictor for developing autoimmune conditions and may play a role in the observed increased cancer risk.
Early and advanced cardiovascular disease
Patients with SLE are at high risk of atherosclerotic cardiovascular disease. At the University of Pittsburgh Medical Center from 1980 to 1993, we compared the incidence of myocardial infarction in nearly 500 women with SLE and more than 2,000 women of similar age in the Framingham Offspring Study. At ages 15 to 24, women with lupus had a rate of 6.33 per 1,000 person-years; at age 25 to 34, the rate was 3.66 per 1,000 person-years. None of the Framingham women in those age groups had events.
Women ages 35 to 44 with lupus had a risk of heart attack 50 times higher than women in the Framingham cohort, and women in older age groups had a risk 2.5 to 4 times higher.11
Subclinical cardiovascular disease is also increased in women with SLE. Asanuma et al12 used electron-beam computed tomography to screen for coronary artery calcification in 65 patients with SLE and 69 control subjects with no history of coronary artery disease matched for age, sex, and race. Calcification was present in 31% of patients with lupus vs 9% of controls (P = .002). Roman et al13 performed carotid ultrasonography on 197 patients with lupus and 197 matched controls and found more plaque in patients with lupus (37%) than in controls (15%, P < .001).
Other data also suggest that women with lupus have advanced cardiovascular disease and develop it early, with most studies finding the greatest relative risk of cardiovascular disease between ages 18 and 45 years.
Traditional risk factors for cardiovascular disease cannot fully explain the increased risk. Many patients with lupus have metabolic syndrome, hypertension, and renal disease, but even after adjusting for these risk factors, patients with lupus still have about a 7 to 10 times higher risk of nonfatal coronary heart disease and a 17 times higher risk of fatal coronary heart disease.14
Many investigators are now exploring the role of immune dysfunction and inflammation in cardiovascular disease. A number of biomarkers have been proposed for predicting risk of cardiovascular disease in the general population. The list includes many inflammatory factors that rheumatologists have been studying for decades, including myeloperoxidase, autoantibodies, inflammatory cytokines, tumor necrosis factor alpha, and adhesion molecules, many of which also play a role in autoimmunity.
In our patients with SLE, we found that about 90% had three or more modifiable cardiovascular risk factors that were not being addressed appropriately (unpublished data). Lipid management was the least often addressed by rheumatologists and primary caregivers.
There is reason to believe that lupus patients are a high-risk group that merit aggressive risk-factor management, but no formal recommendations can be made without clear evidence that this approach improves outcomes.
SLE INCREASES THE RISK OF ADVERSE PREGNANCY OUTCOMES
Women with SLE more commonly miscarry and deliver low-birth-weight babies than do other women. A history of renal disease is the factor most predictive of poor pregnancy outcome, and the presence of certain autoantibodies increases the risk of neonatal lupus.
We recommend that women with lupus have inactive disease for at least 6 months before becoming pregnant.
ORAL CONTRACEPTIVES AND HORMONE REPLACEMENT
Hormone replacement therapy and oral contraceptives do not increase the risk of significant disease activity flares in lupus. However, women with lupus have an increased risk of cardiovascular disease and thrombosis.
Buyon et al15 randomly assigned 351 menopausal women with inactive or stable active SLE to receive either hormone replacement therapy or placebo for 12 months. No significant increase in severe flares of the disease was observed, although the treatment group had a mild increase in minor flares.
Petri et al16 randomly assigned 183 women with inactive or stable active SLE to receive either combined oral contraceptives or placebo for 12 months and found similar rates of disease activity between the two groups.
A weakness of these trials is that women with antiphospholipid antibodies in high titers or who had previous thrombotic events were excluded.
TREATMENTS ON THE HORIZON?
In the past 50 years, only three drug treatments have been approved for lupus: corticosteroids, hydroxychloroquine, and aspirin. Fortunately, research in autoimmune diseases has rapidly expanded, and new drugs are on the horizon.
Mycophenolate mofetil (CellCept) may be a reasonable alternative to cyclophosphamide (Cytoxan) for lupus nephritis and may be appropriate as maintenance therapy after induction with cyclophosphamide.
Ginzler et al,17 in a randomized, open-label trial in 140 patients with active lupus nephritis, gave either oral mycophenolate mofetil (initial dosage 1,000 mg/day, increased to 3,000 mg/day) or monthly intravenous cyclophosphamide (0.5 g/m2, increased to 1.0 g/m2). Mycophenolate mofetil was more effective in inducing remission than cyclophosphamide and had a better safety profile.
The Aspreva Lupus Management Study was designed to assess the efficacy of oral mycophenolate mofetil compared with intravenous cyclophosphamide in the initial treatment of patients with active class III–V lupus nephritis and to assess the long-term efficacy of mycophenolate mofetil compared with azathioprine in maintaining remission and renal function. It was the largest study of mycophenolate mofetil in lupus nephritis to date. There were 370 patients with SLE enrolled. In the 24-week induction phase, patients were randomized to receive open-label mycophenolate mofetil with a target dose of 3 g/day or intravenous cyclophosphamide 0.5 to 1.0 g/m2 in monthly pulses. Both groups received prednisone. Response to treatment was defined as a decrease in proteinuria (as measured by the urinary protein-creatinine ratio) and improvement or stabilization in serum creatinine.
The results (presented at the American College of Rheumatology Meeting, November 6–11, 2007, in Boston, MA) showed that 104 (56%) of the 185 patients treated with mycophenolate mofetil responded, compared with 98 (53%) of the 185 patients treated with intravenous cyclophosphamide (P = .575). The study therefore did not meet its primary objective of showing a superior response rate with mycophenolate mofetil compared with cyclophosphamide. There was no difference in adverse events. It is this author’s opinion that having an agent that is at least as good as cyclphosphamide in treating lupus nephritis is a major step forward.
Mycophenolate mofetil can cause fetal harm and should not be used during pregnancy. It is recommended that the drug be stopped for 3 to 6 months before a woman tries to conceive.
New drugs target B cells
Many new drugs for lupus target B cells.
Rituximab (Rituxan) is a monoclonal antibody that depletes B cells by targeting the B-cell-specific antigen CD20. It has been studied for treating lupus in several open-label studies that altogether have included more than 400 patients.18–21 Regimens included either those used in oncology for treatment of lymphoma or those used in rheumatoid arthritis, coupled with high-dose corticosteroids and cyclophosphamide. In early studies, nearly 80% of treated patients entered at least partial remission, and 25% to 50% are still in remission more than 12 months later.
The first randomized controlled trial of rituximab vs placebo was recently completed and presented at the American College of Rheumatology meeting, October 24–28, 2008, in Boston, MA. The EXPLORER trial (sponsored by Genentech) included 257 patients with moderate to severe disease activity. The results showed that there was no difference in major or partial clinical response (based on a change in the British Isles Lupus Assessment Group index) in those on rituximab (28.4%) vs placebo (29.6%) at 12 months (P = .97). Overall, adverse events were balanced between the groups. It is this author’s opinion that the bar for “response” was set very high in this study, considering that all patients who entered were fairly sick and received significant doses of corticosteroids that were tapered over the course of the trial.
B-cell toleragens, which render B cells incapable of presenting specific antigens, are also of interest.
Other experimental drugs target the soluble cytokine BLyS, which normally binds to a B-cell receptor and prolongs B-cell survival. It may also be possible to inhibit costimulatory pathways (which are normally important for inducing activation, proliferation, and class-switching of B cells) with the use of cytotoxic T-lymphocyte-associated antigen 4 immunoglobulin inhibitor (CTLA4Ig) and anti-CD40 ligand.
The results of a 12-month exploratory, phase II trial of abatacept (Bristol-Myers Squibb) in patients with SLE and active polyarthritis, serositis, or discoid lesions were presented at the American College of Rheumatology meeting in 2008. The primary and secondary end points (based on an adjudicated British Isles Lupus Assessment Group index) were not met. There were no differences in adverse events. Post hoc analyses of other clinical end points and biomarkers suggested that abatacept may have benefit in lupus. Further studies are under way.
Downstream blockade may also be useful, with drugs that inhibit inflammatory cytokines, particularly interferon alfa. This is now being tested in clinical trials.
- Narain S, Richards HB, Satoh M, et al. Diagnostic accuracy for lupus and other systemic autoimmune diseases in the community setting. Arch Intern Med 2004; 164:2435–2441.
- Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982; 25:1271–1277.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter]. Arthritis Rheum 1997; 40:1725.
- Hom G, Graham RR, Modrek B, et al. Association of systemic lupus erythematosus with C8orf13 BLK and ITGAM ITGAX. N Engl J Med 2008; 358:900–909.
- Kozyrev SV, Abelson AK, Wojcik J, et al. Functional variants in the B cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet 2008; 40:211–216.
- International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN), Harley JB, Alarcón-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 2008; 40:204–210.
- Nath SK, Han S, Kim Howard X, et al. A nonsynonymous functional variant in integrin alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet 2008; 40:152–154.
- Ramsey-Goldman R, Dunn JE, Huang CF, et al. Frequency of fractures in women with systemic lupus erythematosus: comparison with United States population data. Arthritis Rheum 1999; 42:882–890.
- Lee C, Almagor O, Dunlop DD, et al. Association between African-American race/ethnicity and low bone mineral density in women with systemic lupus erythematosus. Arthritis Rheum 2007; 57:585–592.
- Bernatsky S, Boivin JF, Joseph L, et al. An international cohort study of cancer in systemic lupus erythematosus. Arthritis Rheum 2005; 52:1481–1490.
- Manzi S, Meilahn EN, Rairie JE, et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am J Epidemiol 1997; 145:408–415.
- Asanuma Y, Oeser A, Shintani AK, et al. Premature coronary artery atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003; 349:2407–2415.
- Roman MJ, Shanker BA, Davis A, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003; 349:2399–2406. Erratum in: N Engl J Med 2006; 355:1746.
- Esdaile JM, Abrahamowicz M, Grodzicky T, et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum 2001; 44:2331–2337.
- Buyon JP, Petri MA, Kim MY, et al. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: a randomized trial. Ann Intern Med 2005; 142:953–962.
- Petri M, Kim MY, Kalunian KC, et al; OC SELENA Trial. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med 2005; 353:2550–2558.
- Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005; 353:2219–2228.
- Anolik JH, Barnard J, Cappione A, et al. Rituximab improves peripheral B cell abnormalities in human systemic lupus erythematosus. Arthritis Rheum 2004; 50:3580–3590.
- Looney RJ, Anolik JH, Campbell D, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose escalation trial of rituximab. Arthritis Rheum 2004; 50:2580–2589.
- Leandro MJ, Edwards JC, Cambridge G, Ehrenstein MR, Isenberg DA. An open study of B lymphocyte depletion in systemic lupus erythematosus. Arthritis Rheum 2002; 46:2673–2677.
- Cambridge G, Stohl W, Leandro MJ, Migone TS, Hilbert DM, Edwards JC. Circulating levels of B lymphocyte stimulator in patients with rheumatoid arthritis following rituximab treatment: relationships with B cell depletion, circulating antibodies, and clinical relapse. Arthritis Rheum 2006; 54:723–732.
Many questions about systemic lupus erythematosus (SLE, lupus) remain unanswered. Why is this disease so difficult to diagnose even for rheumatologists? Why does lupus tend to develop in previously healthy young women? Why does the disease manifest in so many ways? Why are our current treatments suboptimal?
This article addresses these questions in a brief overview and update of SLE, with an emphasis on clinical pearls regarding prevention and treatment that are relevant to any physician who sees patients with this disease.
WOMEN AND MINORITIES ARE OVERREPRESENTED
Women have a much higher prevalence of almost all autoimmune diseases. SLE has a 12:1 female-to-male ratio during the ages of 15 to 45 years, but when disease develops in either children or the elderly, the female-to-male ratio is only 2:1.
African Americans, Asian Americans, and Hispanics have about a three to four times higher frequency of lupus than white non-Hispanics and often have more severe disease.
WHY IS SLE SO DIFFICULT TO DIAGNOSE?
SLE is frequently overlooked; patients spend an average of 4 years and see three physicians before the disease is correctly diagnosed. Part of the problem is that presentations of the disease vary so widely between patients and that signs and symptoms evolve over time. Often, physicians do not consider SLE in the differential diagnosis.
On the other hand, SLE is also often over-diagnosed. Narain et al1 evaluated 263 patients who had a presumptive diagnosis of SLE. Only about half of the patients had a confirmed diagnosis; about 5% had a different autoimmune disease, such as scleroderma, systemic sclerosis, Sjögren syndrome, or polymyositis; 5% had fibromyalgia; 29% tested positive for ANA but did not have an autoimmune disease; and 10% had a nonrheumatic disease, such as a hematologic malignancy with rheumatic disease manifestations. For patients referred by a community rheumatologist, the diagnostic accuracy was better, about 80%.
The traditional classification criteria for SLE2,3 are problematic. Some criteria are very specific for SLE but are not very sensitive—eg, anti-double-stranded DNA is present in about half of patients with SLE. Others tests, like ANA, are sensitive but not specific—although ANA is present in 95% of patients with SLE, the positive predictive value of the test for SLE for any given patient is only 11%.
Other criteria are highly subjective, including oral ulcers and photosensitivity. These signs may be present in normal individuals who get an occasional aphthous ulcer or who are fair-skinned and burn easily with prolonged sun exposure. It takes a trained clinician to distinguish these from the photosensitivity and oral ulcers associated with lupus.
Many diseases can mimic SLE
Fibromyalgia frequently presents in women and may include joint and muscle aches, fatigue, and occasionally a positive ANA. ANA may be seen in about 15% of healthy women.
Sjögren syndrome can also present with arthritis, fatigue, and a positive ANA; it is commonly overlooked because physicians do not often think to ask about the classic symptoms of dry eyes and dry mouth.
Dermatomyositis causes rashes that have many features in common with SLE. Even the skin biopsy is often indistinguishable from SLE.
Hematologic problems, such as idiopathic or thrombotic thrombocytopenic purpura, primary antiphospholipid syndrome, and hematologic neoplasms, can cause serologic changes, a positive ANA, and other manifestations seen in SLE.
Drug-induced lupus should always be considered in older patients presenting with SLE-like disease. Now with the use of minocycline (Minocin) and other related agents for the treatment of acne, we are seeing younger women with drug-induced lupus.
PATIENTS ASK ‘WHY ME?’
Lupus typically develops in a young woman who was previously healthy. Such patients inevitably wonder, why me?
Lupus is like a puzzle, with genetics, gender, and the environment being important pieces of the puzzle. If all the pieces come together, people develop defective immune regulation and a break in self-tolerance. Everyone generates antibodies to self, but these low-affinity, nonpathologic antibodies are inconsequential. In SLE, autoantibodies lead to the formation of immune complexes, complement activation, and tissue damage.
Genetics plays an important role
Genetics plays an important role but is clearly not the only determining factor. Clustering in families has been shown, although a patient with lupus is more likely to have a relative with another autoimmune disease, especially autoimmune thyroid disease, than with SLE. The likelihood of an identical twin of a patient with SLE having the disease is only 25% to 30%, and is only about 5% for a fraternal twin.
In the first few months of 2008, four major studies were published that shed light on the genetics of SLE.4–7 Together, the studies evaluated more than 5,000 patients with SLE using genome-wide association scans and identified areas of the genome that are frequently different in patients with lupus than in healthy controls. Three of the four studies identified the same genetic area as important and supported the concept that B cells and complement activation play important roles in the disease pathogenesis.
Although over 95% of cases of SLE cannot be attributed to a single gene, there are rare cases of lupus that may provide important clues to mechanisms of disease. For example a homozygous deficiency of C1q (an early component of complement) is extremely rare in lupus but is associated with the highest risk (nearly 90%) of developing the disease. Deficiencies in other components of the complement cascade also carry a high risk of disease development.
Investigators discovered that C1q plays an important role in clearing away apoptotic cellular debris. If a person is deficient in C1q, clearance of this debris is impaired. In a person genetically predisposed to getting lupus, the immune system now has an opportunity to react to self-antigens exposed during apoptosis that are not being cleared away.
Even though lupus cases cannot be explained by an absence of C1q, a defect in the clearance of apoptotic cells is a common, unifying feature of the disease.
Immune response is enhanced by environmental factors
Environmental factors, especially sun exposure, are also important. Following sunburn, skin cells undergo massive cell death, and patients with lupus have a huge release of self-antigens that can be recognized by the immune system. Sunburn is like having a booster vaccine of self-antigen to stimulate autoantibody production. Not only does the skin flare, but internal organs can also flare after intense sun exposure.
LUPUS SURVIVAL HAS IMPROVED; DISEASES OF AGING NOW A FOCUS
In 1950, only 50% of patients with SLE survived 5 years after diagnosis; now, thanks to better treatment and earlier diagnosis, 80% to 90% survive at least 10 years.
Early on, patients tend to die of active disease (manifestations of vasculitis, pulmonary hemorrhage, kidney problems) or infection. Over time, cardiovascular disease and osteoporosis become more of a problem. Patients also have a higher risk of cancer throughout life.
Lupus has an unpredictable course, with flares and remissions. But underlying the reversible inflammatory changes is irreversible organ damage caused by the disease itself and, possibly, by treatment. Preventing bone disease, heart disease, and cancer now play more prominent roles in managing SLE.
Increased bone disease
Fracture rates are higher than expected in women with lupus; Ramsey-Goldman et al8 calculated the rate as five times higher than in the general population. The increased risk of osteoporosis is partly due to treatment with corticosteroids, but it is also likely caused by inflammation from lupus. Even controlling for steroid use, increased bone loss is still evident in patients with SLE.
African American women with lupus are not exempt. Lee et al9 found that, after adjusting for body mass, steroid use, thyroid disease, and menopausal status, African American women with SLE had more than five times the risk of low bone mineral density in the spine than white women with the disease.
Increased cancer risk
Patients with SLE have an increased risk of hematologic cancer and possibly lung and hepatobiliary cancers.
Bernatsky et al10 evaluated cancer risk in an international cohort of patients with SLE from 23 sites. Among patients with SLE, for all cancers combined, the standardized incidence ratio was 1.2; for hematologic cancers the ratio was 2.8; and for non-Hodgkin lymphoma it was 2.4. Surprisingly, although SLE is primarily a disease of women, reproductive cancer rates in patients with SLE did not differ from background rates. Bernatsky et al did not compare rates of cervical cancer, as many cancer registries do not record it. However, studies from the National Institutes of Health indicate that cervical dysplasia is common in patients with lupus.
Other interesting findings included an increased risk of hepatobiliary cancer, especially among men with SLE. Lung cancers were also increased, which has been observed in patients with other autoimmune diseases such as scleroderma and polymyositis. Smoking is a strong predictor for developing autoimmune conditions and may play a role in the observed increased cancer risk.
Early and advanced cardiovascular disease
Patients with SLE are at high risk of atherosclerotic cardiovascular disease. At the University of Pittsburgh Medical Center from 1980 to 1993, we compared the incidence of myocardial infarction in nearly 500 women with SLE and more than 2,000 women of similar age in the Framingham Offspring Study. At ages 15 to 24, women with lupus had a rate of 6.33 per 1,000 person-years; at age 25 to 34, the rate was 3.66 per 1,000 person-years. None of the Framingham women in those age groups had events.
Women ages 35 to 44 with lupus had a risk of heart attack 50 times higher than women in the Framingham cohort, and women in older age groups had a risk 2.5 to 4 times higher.11
Subclinical cardiovascular disease is also increased in women with SLE. Asanuma et al12 used electron-beam computed tomography to screen for coronary artery calcification in 65 patients with SLE and 69 control subjects with no history of coronary artery disease matched for age, sex, and race. Calcification was present in 31% of patients with lupus vs 9% of controls (P = .002). Roman et al13 performed carotid ultrasonography on 197 patients with lupus and 197 matched controls and found more plaque in patients with lupus (37%) than in controls (15%, P < .001).
Other data also suggest that women with lupus have advanced cardiovascular disease and develop it early, with most studies finding the greatest relative risk of cardiovascular disease between ages 18 and 45 years.
Traditional risk factors for cardiovascular disease cannot fully explain the increased risk. Many patients with lupus have metabolic syndrome, hypertension, and renal disease, but even after adjusting for these risk factors, patients with lupus still have about a 7 to 10 times higher risk of nonfatal coronary heart disease and a 17 times higher risk of fatal coronary heart disease.14
Many investigators are now exploring the role of immune dysfunction and inflammation in cardiovascular disease. A number of biomarkers have been proposed for predicting risk of cardiovascular disease in the general population. The list includes many inflammatory factors that rheumatologists have been studying for decades, including myeloperoxidase, autoantibodies, inflammatory cytokines, tumor necrosis factor alpha, and adhesion molecules, many of which also play a role in autoimmunity.
In our patients with SLE, we found that about 90% had three or more modifiable cardiovascular risk factors that were not being addressed appropriately (unpublished data). Lipid management was the least often addressed by rheumatologists and primary caregivers.
There is reason to believe that lupus patients are a high-risk group that merit aggressive risk-factor management, but no formal recommendations can be made without clear evidence that this approach improves outcomes.
SLE INCREASES THE RISK OF ADVERSE PREGNANCY OUTCOMES
Women with SLE more commonly miscarry and deliver low-birth-weight babies than do other women. A history of renal disease is the factor most predictive of poor pregnancy outcome, and the presence of certain autoantibodies increases the risk of neonatal lupus.
We recommend that women with lupus have inactive disease for at least 6 months before becoming pregnant.
ORAL CONTRACEPTIVES AND HORMONE REPLACEMENT
Hormone replacement therapy and oral contraceptives do not increase the risk of significant disease activity flares in lupus. However, women with lupus have an increased risk of cardiovascular disease and thrombosis.
Buyon et al15 randomly assigned 351 menopausal women with inactive or stable active SLE to receive either hormone replacement therapy or placebo for 12 months. No significant increase in severe flares of the disease was observed, although the treatment group had a mild increase in minor flares.
Petri et al16 randomly assigned 183 women with inactive or stable active SLE to receive either combined oral contraceptives or placebo for 12 months and found similar rates of disease activity between the two groups.
A weakness of these trials is that women with antiphospholipid antibodies in high titers or who had previous thrombotic events were excluded.
TREATMENTS ON THE HORIZON?
In the past 50 years, only three drug treatments have been approved for lupus: corticosteroids, hydroxychloroquine, and aspirin. Fortunately, research in autoimmune diseases has rapidly expanded, and new drugs are on the horizon.
Mycophenolate mofetil (CellCept) may be a reasonable alternative to cyclophosphamide (Cytoxan) for lupus nephritis and may be appropriate as maintenance therapy after induction with cyclophosphamide.
Ginzler et al,17 in a randomized, open-label trial in 140 patients with active lupus nephritis, gave either oral mycophenolate mofetil (initial dosage 1,000 mg/day, increased to 3,000 mg/day) or monthly intravenous cyclophosphamide (0.5 g/m2, increased to 1.0 g/m2). Mycophenolate mofetil was more effective in inducing remission than cyclophosphamide and had a better safety profile.
The Aspreva Lupus Management Study was designed to assess the efficacy of oral mycophenolate mofetil compared with intravenous cyclophosphamide in the initial treatment of patients with active class III–V lupus nephritis and to assess the long-term efficacy of mycophenolate mofetil compared with azathioprine in maintaining remission and renal function. It was the largest study of mycophenolate mofetil in lupus nephritis to date. There were 370 patients with SLE enrolled. In the 24-week induction phase, patients were randomized to receive open-label mycophenolate mofetil with a target dose of 3 g/day or intravenous cyclophosphamide 0.5 to 1.0 g/m2 in monthly pulses. Both groups received prednisone. Response to treatment was defined as a decrease in proteinuria (as measured by the urinary protein-creatinine ratio) and improvement or stabilization in serum creatinine.
The results (presented at the American College of Rheumatology Meeting, November 6–11, 2007, in Boston, MA) showed that 104 (56%) of the 185 patients treated with mycophenolate mofetil responded, compared with 98 (53%) of the 185 patients treated with intravenous cyclophosphamide (P = .575). The study therefore did not meet its primary objective of showing a superior response rate with mycophenolate mofetil compared with cyclophosphamide. There was no difference in adverse events. It is this author’s opinion that having an agent that is at least as good as cyclphosphamide in treating lupus nephritis is a major step forward.
Mycophenolate mofetil can cause fetal harm and should not be used during pregnancy. It is recommended that the drug be stopped for 3 to 6 months before a woman tries to conceive.
New drugs target B cells
Many new drugs for lupus target B cells.
Rituximab (Rituxan) is a monoclonal antibody that depletes B cells by targeting the B-cell-specific antigen CD20. It has been studied for treating lupus in several open-label studies that altogether have included more than 400 patients.18–21 Regimens included either those used in oncology for treatment of lymphoma or those used in rheumatoid arthritis, coupled with high-dose corticosteroids and cyclophosphamide. In early studies, nearly 80% of treated patients entered at least partial remission, and 25% to 50% are still in remission more than 12 months later.
The first randomized controlled trial of rituximab vs placebo was recently completed and presented at the American College of Rheumatology meeting, October 24–28, 2008, in Boston, MA. The EXPLORER trial (sponsored by Genentech) included 257 patients with moderate to severe disease activity. The results showed that there was no difference in major or partial clinical response (based on a change in the British Isles Lupus Assessment Group index) in those on rituximab (28.4%) vs placebo (29.6%) at 12 months (P = .97). Overall, adverse events were balanced between the groups. It is this author’s opinion that the bar for “response” was set very high in this study, considering that all patients who entered were fairly sick and received significant doses of corticosteroids that were tapered over the course of the trial.
B-cell toleragens, which render B cells incapable of presenting specific antigens, are also of interest.
Other experimental drugs target the soluble cytokine BLyS, which normally binds to a B-cell receptor and prolongs B-cell survival. It may also be possible to inhibit costimulatory pathways (which are normally important for inducing activation, proliferation, and class-switching of B cells) with the use of cytotoxic T-lymphocyte-associated antigen 4 immunoglobulin inhibitor (CTLA4Ig) and anti-CD40 ligand.
The results of a 12-month exploratory, phase II trial of abatacept (Bristol-Myers Squibb) in patients with SLE and active polyarthritis, serositis, or discoid lesions were presented at the American College of Rheumatology meeting in 2008. The primary and secondary end points (based on an adjudicated British Isles Lupus Assessment Group index) were not met. There were no differences in adverse events. Post hoc analyses of other clinical end points and biomarkers suggested that abatacept may have benefit in lupus. Further studies are under way.
Downstream blockade may also be useful, with drugs that inhibit inflammatory cytokines, particularly interferon alfa. This is now being tested in clinical trials.
Many questions about systemic lupus erythematosus (SLE, lupus) remain unanswered. Why is this disease so difficult to diagnose even for rheumatologists? Why does lupus tend to develop in previously healthy young women? Why does the disease manifest in so many ways? Why are our current treatments suboptimal?
This article addresses these questions in a brief overview and update of SLE, with an emphasis on clinical pearls regarding prevention and treatment that are relevant to any physician who sees patients with this disease.
WOMEN AND MINORITIES ARE OVERREPRESENTED
Women have a much higher prevalence of almost all autoimmune diseases. SLE has a 12:1 female-to-male ratio during the ages of 15 to 45 years, but when disease develops in either children or the elderly, the female-to-male ratio is only 2:1.
African Americans, Asian Americans, and Hispanics have about a three to four times higher frequency of lupus than white non-Hispanics and often have more severe disease.
WHY IS SLE SO DIFFICULT TO DIAGNOSE?
SLE is frequently overlooked; patients spend an average of 4 years and see three physicians before the disease is correctly diagnosed. Part of the problem is that presentations of the disease vary so widely between patients and that signs and symptoms evolve over time. Often, physicians do not consider SLE in the differential diagnosis.
On the other hand, SLE is also often over-diagnosed. Narain et al1 evaluated 263 patients who had a presumptive diagnosis of SLE. Only about half of the patients had a confirmed diagnosis; about 5% had a different autoimmune disease, such as scleroderma, systemic sclerosis, Sjögren syndrome, or polymyositis; 5% had fibromyalgia; 29% tested positive for ANA but did not have an autoimmune disease; and 10% had a nonrheumatic disease, such as a hematologic malignancy with rheumatic disease manifestations. For patients referred by a community rheumatologist, the diagnostic accuracy was better, about 80%.
The traditional classification criteria for SLE2,3 are problematic. Some criteria are very specific for SLE but are not very sensitive—eg, anti-double-stranded DNA is present in about half of patients with SLE. Others tests, like ANA, are sensitive but not specific—although ANA is present in 95% of patients with SLE, the positive predictive value of the test for SLE for any given patient is only 11%.
Other criteria are highly subjective, including oral ulcers and photosensitivity. These signs may be present in normal individuals who get an occasional aphthous ulcer or who are fair-skinned and burn easily with prolonged sun exposure. It takes a trained clinician to distinguish these from the photosensitivity and oral ulcers associated with lupus.
Many diseases can mimic SLE
Fibromyalgia frequently presents in women and may include joint and muscle aches, fatigue, and occasionally a positive ANA. ANA may be seen in about 15% of healthy women.
Sjögren syndrome can also present with arthritis, fatigue, and a positive ANA; it is commonly overlooked because physicians do not often think to ask about the classic symptoms of dry eyes and dry mouth.
Dermatomyositis causes rashes that have many features in common with SLE. Even the skin biopsy is often indistinguishable from SLE.
Hematologic problems, such as idiopathic or thrombotic thrombocytopenic purpura, primary antiphospholipid syndrome, and hematologic neoplasms, can cause serologic changes, a positive ANA, and other manifestations seen in SLE.
Drug-induced lupus should always be considered in older patients presenting with SLE-like disease. Now with the use of minocycline (Minocin) and other related agents for the treatment of acne, we are seeing younger women with drug-induced lupus.
PATIENTS ASK ‘WHY ME?’
Lupus typically develops in a young woman who was previously healthy. Such patients inevitably wonder, why me?
Lupus is like a puzzle, with genetics, gender, and the environment being important pieces of the puzzle. If all the pieces come together, people develop defective immune regulation and a break in self-tolerance. Everyone generates antibodies to self, but these low-affinity, nonpathologic antibodies are inconsequential. In SLE, autoantibodies lead to the formation of immune complexes, complement activation, and tissue damage.
Genetics plays an important role
Genetics plays an important role but is clearly not the only determining factor. Clustering in families has been shown, although a patient with lupus is more likely to have a relative with another autoimmune disease, especially autoimmune thyroid disease, than with SLE. The likelihood of an identical twin of a patient with SLE having the disease is only 25% to 30%, and is only about 5% for a fraternal twin.
In the first few months of 2008, four major studies were published that shed light on the genetics of SLE.4–7 Together, the studies evaluated more than 5,000 patients with SLE using genome-wide association scans and identified areas of the genome that are frequently different in patients with lupus than in healthy controls. Three of the four studies identified the same genetic area as important and supported the concept that B cells and complement activation play important roles in the disease pathogenesis.
Although over 95% of cases of SLE cannot be attributed to a single gene, there are rare cases of lupus that may provide important clues to mechanisms of disease. For example a homozygous deficiency of C1q (an early component of complement) is extremely rare in lupus but is associated with the highest risk (nearly 90%) of developing the disease. Deficiencies in other components of the complement cascade also carry a high risk of disease development.
Investigators discovered that C1q plays an important role in clearing away apoptotic cellular debris. If a person is deficient in C1q, clearance of this debris is impaired. In a person genetically predisposed to getting lupus, the immune system now has an opportunity to react to self-antigens exposed during apoptosis that are not being cleared away.
Even though lupus cases cannot be explained by an absence of C1q, a defect in the clearance of apoptotic cells is a common, unifying feature of the disease.
Immune response is enhanced by environmental factors
Environmental factors, especially sun exposure, are also important. Following sunburn, skin cells undergo massive cell death, and patients with lupus have a huge release of self-antigens that can be recognized by the immune system. Sunburn is like having a booster vaccine of self-antigen to stimulate autoantibody production. Not only does the skin flare, but internal organs can also flare after intense sun exposure.
LUPUS SURVIVAL HAS IMPROVED; DISEASES OF AGING NOW A FOCUS
In 1950, only 50% of patients with SLE survived 5 years after diagnosis; now, thanks to better treatment and earlier diagnosis, 80% to 90% survive at least 10 years.
Early on, patients tend to die of active disease (manifestations of vasculitis, pulmonary hemorrhage, kidney problems) or infection. Over time, cardiovascular disease and osteoporosis become more of a problem. Patients also have a higher risk of cancer throughout life.
Lupus has an unpredictable course, with flares and remissions. But underlying the reversible inflammatory changes is irreversible organ damage caused by the disease itself and, possibly, by treatment. Preventing bone disease, heart disease, and cancer now play more prominent roles in managing SLE.
Increased bone disease
Fracture rates are higher than expected in women with lupus; Ramsey-Goldman et al8 calculated the rate as five times higher than in the general population. The increased risk of osteoporosis is partly due to treatment with corticosteroids, but it is also likely caused by inflammation from lupus. Even controlling for steroid use, increased bone loss is still evident in patients with SLE.
African American women with lupus are not exempt. Lee et al9 found that, after adjusting for body mass, steroid use, thyroid disease, and menopausal status, African American women with SLE had more than five times the risk of low bone mineral density in the spine than white women with the disease.
Increased cancer risk
Patients with SLE have an increased risk of hematologic cancer and possibly lung and hepatobiliary cancers.
Bernatsky et al10 evaluated cancer risk in an international cohort of patients with SLE from 23 sites. Among patients with SLE, for all cancers combined, the standardized incidence ratio was 1.2; for hematologic cancers the ratio was 2.8; and for non-Hodgkin lymphoma it was 2.4. Surprisingly, although SLE is primarily a disease of women, reproductive cancer rates in patients with SLE did not differ from background rates. Bernatsky et al did not compare rates of cervical cancer, as many cancer registries do not record it. However, studies from the National Institutes of Health indicate that cervical dysplasia is common in patients with lupus.
Other interesting findings included an increased risk of hepatobiliary cancer, especially among men with SLE. Lung cancers were also increased, which has been observed in patients with other autoimmune diseases such as scleroderma and polymyositis. Smoking is a strong predictor for developing autoimmune conditions and may play a role in the observed increased cancer risk.
Early and advanced cardiovascular disease
Patients with SLE are at high risk of atherosclerotic cardiovascular disease. At the University of Pittsburgh Medical Center from 1980 to 1993, we compared the incidence of myocardial infarction in nearly 500 women with SLE and more than 2,000 women of similar age in the Framingham Offspring Study. At ages 15 to 24, women with lupus had a rate of 6.33 per 1,000 person-years; at age 25 to 34, the rate was 3.66 per 1,000 person-years. None of the Framingham women in those age groups had events.
Women ages 35 to 44 with lupus had a risk of heart attack 50 times higher than women in the Framingham cohort, and women in older age groups had a risk 2.5 to 4 times higher.11
Subclinical cardiovascular disease is also increased in women with SLE. Asanuma et al12 used electron-beam computed tomography to screen for coronary artery calcification in 65 patients with SLE and 69 control subjects with no history of coronary artery disease matched for age, sex, and race. Calcification was present in 31% of patients with lupus vs 9% of controls (P = .002). Roman et al13 performed carotid ultrasonography on 197 patients with lupus and 197 matched controls and found more plaque in patients with lupus (37%) than in controls (15%, P < .001).
Other data also suggest that women with lupus have advanced cardiovascular disease and develop it early, with most studies finding the greatest relative risk of cardiovascular disease between ages 18 and 45 years.
Traditional risk factors for cardiovascular disease cannot fully explain the increased risk. Many patients with lupus have metabolic syndrome, hypertension, and renal disease, but even after adjusting for these risk factors, patients with lupus still have about a 7 to 10 times higher risk of nonfatal coronary heart disease and a 17 times higher risk of fatal coronary heart disease.14
Many investigators are now exploring the role of immune dysfunction and inflammation in cardiovascular disease. A number of biomarkers have been proposed for predicting risk of cardiovascular disease in the general population. The list includes many inflammatory factors that rheumatologists have been studying for decades, including myeloperoxidase, autoantibodies, inflammatory cytokines, tumor necrosis factor alpha, and adhesion molecules, many of which also play a role in autoimmunity.
In our patients with SLE, we found that about 90% had three or more modifiable cardiovascular risk factors that were not being addressed appropriately (unpublished data). Lipid management was the least often addressed by rheumatologists and primary caregivers.
There is reason to believe that lupus patients are a high-risk group that merit aggressive risk-factor management, but no formal recommendations can be made without clear evidence that this approach improves outcomes.
SLE INCREASES THE RISK OF ADVERSE PREGNANCY OUTCOMES
Women with SLE more commonly miscarry and deliver low-birth-weight babies than do other women. A history of renal disease is the factor most predictive of poor pregnancy outcome, and the presence of certain autoantibodies increases the risk of neonatal lupus.
We recommend that women with lupus have inactive disease for at least 6 months before becoming pregnant.
ORAL CONTRACEPTIVES AND HORMONE REPLACEMENT
Hormone replacement therapy and oral contraceptives do not increase the risk of significant disease activity flares in lupus. However, women with lupus have an increased risk of cardiovascular disease and thrombosis.
Buyon et al15 randomly assigned 351 menopausal women with inactive or stable active SLE to receive either hormone replacement therapy or placebo for 12 months. No significant increase in severe flares of the disease was observed, although the treatment group had a mild increase in minor flares.
Petri et al16 randomly assigned 183 women with inactive or stable active SLE to receive either combined oral contraceptives or placebo for 12 months and found similar rates of disease activity between the two groups.
A weakness of these trials is that women with antiphospholipid antibodies in high titers or who had previous thrombotic events were excluded.
TREATMENTS ON THE HORIZON?
In the past 50 years, only three drug treatments have been approved for lupus: corticosteroids, hydroxychloroquine, and aspirin. Fortunately, research in autoimmune diseases has rapidly expanded, and new drugs are on the horizon.
Mycophenolate mofetil (CellCept) may be a reasonable alternative to cyclophosphamide (Cytoxan) for lupus nephritis and may be appropriate as maintenance therapy after induction with cyclophosphamide.
Ginzler et al,17 in a randomized, open-label trial in 140 patients with active lupus nephritis, gave either oral mycophenolate mofetil (initial dosage 1,000 mg/day, increased to 3,000 mg/day) or monthly intravenous cyclophosphamide (0.5 g/m2, increased to 1.0 g/m2). Mycophenolate mofetil was more effective in inducing remission than cyclophosphamide and had a better safety profile.
The Aspreva Lupus Management Study was designed to assess the efficacy of oral mycophenolate mofetil compared with intravenous cyclophosphamide in the initial treatment of patients with active class III–V lupus nephritis and to assess the long-term efficacy of mycophenolate mofetil compared with azathioprine in maintaining remission and renal function. It was the largest study of mycophenolate mofetil in lupus nephritis to date. There were 370 patients with SLE enrolled. In the 24-week induction phase, patients were randomized to receive open-label mycophenolate mofetil with a target dose of 3 g/day or intravenous cyclophosphamide 0.5 to 1.0 g/m2 in monthly pulses. Both groups received prednisone. Response to treatment was defined as a decrease in proteinuria (as measured by the urinary protein-creatinine ratio) and improvement or stabilization in serum creatinine.
The results (presented at the American College of Rheumatology Meeting, November 6–11, 2007, in Boston, MA) showed that 104 (56%) of the 185 patients treated with mycophenolate mofetil responded, compared with 98 (53%) of the 185 patients treated with intravenous cyclophosphamide (P = .575). The study therefore did not meet its primary objective of showing a superior response rate with mycophenolate mofetil compared with cyclophosphamide. There was no difference in adverse events. It is this author’s opinion that having an agent that is at least as good as cyclphosphamide in treating lupus nephritis is a major step forward.
Mycophenolate mofetil can cause fetal harm and should not be used during pregnancy. It is recommended that the drug be stopped for 3 to 6 months before a woman tries to conceive.
New drugs target B cells
Many new drugs for lupus target B cells.
Rituximab (Rituxan) is a monoclonal antibody that depletes B cells by targeting the B-cell-specific antigen CD20. It has been studied for treating lupus in several open-label studies that altogether have included more than 400 patients.18–21 Regimens included either those used in oncology for treatment of lymphoma or those used in rheumatoid arthritis, coupled with high-dose corticosteroids and cyclophosphamide. In early studies, nearly 80% of treated patients entered at least partial remission, and 25% to 50% are still in remission more than 12 months later.
The first randomized controlled trial of rituximab vs placebo was recently completed and presented at the American College of Rheumatology meeting, October 24–28, 2008, in Boston, MA. The EXPLORER trial (sponsored by Genentech) included 257 patients with moderate to severe disease activity. The results showed that there was no difference in major or partial clinical response (based on a change in the British Isles Lupus Assessment Group index) in those on rituximab (28.4%) vs placebo (29.6%) at 12 months (P = .97). Overall, adverse events were balanced between the groups. It is this author’s opinion that the bar for “response” was set very high in this study, considering that all patients who entered were fairly sick and received significant doses of corticosteroids that were tapered over the course of the trial.
B-cell toleragens, which render B cells incapable of presenting specific antigens, are also of interest.
Other experimental drugs target the soluble cytokine BLyS, which normally binds to a B-cell receptor and prolongs B-cell survival. It may also be possible to inhibit costimulatory pathways (which are normally important for inducing activation, proliferation, and class-switching of B cells) with the use of cytotoxic T-lymphocyte-associated antigen 4 immunoglobulin inhibitor (CTLA4Ig) and anti-CD40 ligand.
The results of a 12-month exploratory, phase II trial of abatacept (Bristol-Myers Squibb) in patients with SLE and active polyarthritis, serositis, or discoid lesions were presented at the American College of Rheumatology meeting in 2008. The primary and secondary end points (based on an adjudicated British Isles Lupus Assessment Group index) were not met. There were no differences in adverse events. Post hoc analyses of other clinical end points and biomarkers suggested that abatacept may have benefit in lupus. Further studies are under way.
Downstream blockade may also be useful, with drugs that inhibit inflammatory cytokines, particularly interferon alfa. This is now being tested in clinical trials.
- Narain S, Richards HB, Satoh M, et al. Diagnostic accuracy for lupus and other systemic autoimmune diseases in the community setting. Arch Intern Med 2004; 164:2435–2441.
- Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982; 25:1271–1277.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter]. Arthritis Rheum 1997; 40:1725.
- Hom G, Graham RR, Modrek B, et al. Association of systemic lupus erythematosus with C8orf13 BLK and ITGAM ITGAX. N Engl J Med 2008; 358:900–909.
- Kozyrev SV, Abelson AK, Wojcik J, et al. Functional variants in the B cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet 2008; 40:211–216.
- International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN), Harley JB, Alarcón-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 2008; 40:204–210.
- Nath SK, Han S, Kim Howard X, et al. A nonsynonymous functional variant in integrin alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet 2008; 40:152–154.
- Ramsey-Goldman R, Dunn JE, Huang CF, et al. Frequency of fractures in women with systemic lupus erythematosus: comparison with United States population data. Arthritis Rheum 1999; 42:882–890.
- Lee C, Almagor O, Dunlop DD, et al. Association between African-American race/ethnicity and low bone mineral density in women with systemic lupus erythematosus. Arthritis Rheum 2007; 57:585–592.
- Bernatsky S, Boivin JF, Joseph L, et al. An international cohort study of cancer in systemic lupus erythematosus. Arthritis Rheum 2005; 52:1481–1490.
- Manzi S, Meilahn EN, Rairie JE, et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am J Epidemiol 1997; 145:408–415.
- Asanuma Y, Oeser A, Shintani AK, et al. Premature coronary artery atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003; 349:2407–2415.
- Roman MJ, Shanker BA, Davis A, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003; 349:2399–2406. Erratum in: N Engl J Med 2006; 355:1746.
- Esdaile JM, Abrahamowicz M, Grodzicky T, et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum 2001; 44:2331–2337.
- Buyon JP, Petri MA, Kim MY, et al. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: a randomized trial. Ann Intern Med 2005; 142:953–962.
- Petri M, Kim MY, Kalunian KC, et al; OC SELENA Trial. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med 2005; 353:2550–2558.
- Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005; 353:2219–2228.
- Anolik JH, Barnard J, Cappione A, et al. Rituximab improves peripheral B cell abnormalities in human systemic lupus erythematosus. Arthritis Rheum 2004; 50:3580–3590.
- Looney RJ, Anolik JH, Campbell D, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose escalation trial of rituximab. Arthritis Rheum 2004; 50:2580–2589.
- Leandro MJ, Edwards JC, Cambridge G, Ehrenstein MR, Isenberg DA. An open study of B lymphocyte depletion in systemic lupus erythematosus. Arthritis Rheum 2002; 46:2673–2677.
- Cambridge G, Stohl W, Leandro MJ, Migone TS, Hilbert DM, Edwards JC. Circulating levels of B lymphocyte stimulator in patients with rheumatoid arthritis following rituximab treatment: relationships with B cell depletion, circulating antibodies, and clinical relapse. Arthritis Rheum 2006; 54:723–732.
- Narain S, Richards HB, Satoh M, et al. Diagnostic accuracy for lupus and other systemic autoimmune diseases in the community setting. Arch Intern Med 2004; 164:2435–2441.
- Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982; 25:1271–1277.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter]. Arthritis Rheum 1997; 40:1725.
- Hom G, Graham RR, Modrek B, et al. Association of systemic lupus erythematosus with C8orf13 BLK and ITGAM ITGAX. N Engl J Med 2008; 358:900–909.
- Kozyrev SV, Abelson AK, Wojcik J, et al. Functional variants in the B cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet 2008; 40:211–216.
- International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN), Harley JB, Alarcón-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 2008; 40:204–210.
- Nath SK, Han S, Kim Howard X, et al. A nonsynonymous functional variant in integrin alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet 2008; 40:152–154.
- Ramsey-Goldman R, Dunn JE, Huang CF, et al. Frequency of fractures in women with systemic lupus erythematosus: comparison with United States population data. Arthritis Rheum 1999; 42:882–890.
- Lee C, Almagor O, Dunlop DD, et al. Association between African-American race/ethnicity and low bone mineral density in women with systemic lupus erythematosus. Arthritis Rheum 2007; 57:585–592.
- Bernatsky S, Boivin JF, Joseph L, et al. An international cohort study of cancer in systemic lupus erythematosus. Arthritis Rheum 2005; 52:1481–1490.
- Manzi S, Meilahn EN, Rairie JE, et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am J Epidemiol 1997; 145:408–415.
- Asanuma Y, Oeser A, Shintani AK, et al. Premature coronary artery atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003; 349:2407–2415.
- Roman MJ, Shanker BA, Davis A, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med 2003; 349:2399–2406. Erratum in: N Engl J Med 2006; 355:1746.
- Esdaile JM, Abrahamowicz M, Grodzicky T, et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum 2001; 44:2331–2337.
- Buyon JP, Petri MA, Kim MY, et al. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: a randomized trial. Ann Intern Med 2005; 142:953–962.
- Petri M, Kim MY, Kalunian KC, et al; OC SELENA Trial. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med 2005; 353:2550–2558.
- Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005; 353:2219–2228.
- Anolik JH, Barnard J, Cappione A, et al. Rituximab improves peripheral B cell abnormalities in human systemic lupus erythematosus. Arthritis Rheum 2004; 50:3580–3590.
- Looney RJ, Anolik JH, Campbell D, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose escalation trial of rituximab. Arthritis Rheum 2004; 50:2580–2589.
- Leandro MJ, Edwards JC, Cambridge G, Ehrenstein MR, Isenberg DA. An open study of B lymphocyte depletion in systemic lupus erythematosus. Arthritis Rheum 2002; 46:2673–2677.
- Cambridge G, Stohl W, Leandro MJ, Migone TS, Hilbert DM, Edwards JC. Circulating levels of B lymphocyte stimulator in patients with rheumatoid arthritis following rituximab treatment: relationships with B cell depletion, circulating antibodies, and clinical relapse. Arthritis Rheum 2006; 54:723–732.
KEY POINTS
- Lupus is often misdiagnosed. A person may be given a diagnosis based on a positive antinuclear antibody (ANA) test, a finding that alone is not sufficient to establish the diagnosis. In contrast, some patients with lupus may go several years and see numerous physicians before the proper diagnosis is made.
- One of the major mechanisms for lupus involves defective clearance of apoptotic cells, which act as a source of self-antigens. Because sun exposure can result in massive cell death of keratinocytes (skin cells), protection from the damaging effects of ultraviolet light plays an important role in the management of lupus.
- Patients at any age with SLE should have their modifiable cardiovascular risk factors managed.
- Drugs on the horizon for treating SLE inactivate B cells or their actions.
Saline irrigation spells relief for sinusitis sufferers
ILLUSTRATIVE CASE
A 45-year-old woman presents to your office with an 8-month history of nasal congestion and thick nasal discharge. Her symptoms have waxed and waned, the patient reports. She’s tried decongestants, antibiotics, and nasal steroids, with limited success. The patient has not had a recent respiratory infection, has never had sinus surgery, and does not want to be on long-term medication. You wonder if there’s an alternative treatment you can offer.
Rhinosinusitis is one of the most common conditions seen by primary care physicians in the United States, and its incidence and prevalence are increasing.2,3 While acute rhinosinusitis is usually self-limiting and resolves within a month, some patients develop chronic—and hard to treat—sinonasal symptoms.
No single cause, no definitive treatment
We’ve moved away from the notion that chronic rhinosinusitis is always a manifestation of persistent bacterial infection, and now recognize that there’s an inflammatory, nonbacterial component.4 In any given patient, several mechanisms—acting either simultaneously or independently—may contribute to sinonasal symptoms.3
Chronic sinusitis is treated in a variety of ways, including medications, immunotherapy, and surgery. Despite their limited efficacy, antibiotics and nasal steroids have been the mainstays of treatment.5 Treating underlying allergies, when they exist, may be helpful. But regardless of which treatment patients receive for chronic rhinosinusitis, many remain symptomatic.6
Benefits of saline irrigation extend beyond postop care
Otolaryngologists recommend saline irrigation after sinus surgery to clear secretions, debris, and crusts; reduce the risk of postoperative mucosal adhesions; and expedite mucosal healing.7,8 Saline irrigation is also gaining popularity as an alternative approach to chronic sinusitis symptom relief, and several randomized controlled trials (RCTs) have demonstrated both objective and subjective efficacy of this treatment for sinonasal disease.8-11
In 2007, the Cochrane Collaboration reviewed evidence for the effectiveness of nasal saline irrigation for symptoms of chronic rhinosinusitis. The reviewers concluded that it is well tolerated and beneficial, whether used alone or as an adjunctive treatment.12
Nasal saline sprays are often recommended because they’re thought to be better tolerated than other delivery modes.13 There have, however, been no comparisons of the relative efficacy of different means of saline delivery, until now.
STUDY SUMMARY: Nasal irrigation and spray go head-to-head
This study was a high-quality, prospective RCT comparing nasal spray and nasal irrigation.1 Subjects were recruited from the general population. To be eligible, participants had to be 18 years of age or older and have reported at least one of the following chronic rhinosinusitis symptoms on 4 or more days each week in the preceding 2 weeks:
- nasal stuffiness
- nasal dryness or crusting
- nasal congestion
- thick or discolored nasal discharge.
In addition, the symptoms must have been present on at least 15 of the preceding 30 days. Exclusion criteria included recent sinus surgery, a respiratory infection within the past 2 weeks, and the use of nasal saline within the past month.
Researchers enrolled 127 patients in the study; 63 were randomized to the nasal spray group and 64 to the large-volume, low-pressure irrigation group. Demographic and baseline characteristics of the groups were similar. The average ages of those in the irrigation and spray groups were 45 and 48 years, respectively. Most patients were nonsmokers and had been symptomatic for 7 to 12 months.
Twice-daily treatment. Researchers asked the patients to perform the assigned treatment twice daily for 8 weeks, but the patients were also permitted to continue using their usual medications. Symptom severity and disease-specific quality of life were assessed with the Sino-Nasal Outcome Test (SNOT-20), a 20-item survey that measures physical problems, emotional consequences, and functional limitations of sinusitis.14
The SNOT-20 is a validated, self-administered survey that asks patients to score items such as runny nose, postnasal discharge, need to blow the nose, reduced productivity, and embarrassment, on a 0- to 5-point scale (0=never, 5=always). A SNOT-20 score of 100 indicates the worst possible symptoms.
As a measure of chronicity of symptoms, patients were also asked to estimate how many months they’d had these symptoms during the last year. In addition, they were instructed to keep a diary to document treatment compliance and the use of other medications for sinonasal symptoms.
To measure outcomes, the researchers provided patients with mail-in packets so they could send in their completed SNOT-20 questionnaire and the medication diary completed at 2, 4, and 8 weeks after randomization.
Biggest improvements seen in irrigation group
Severity of symptoms. In each outcome measurement period, the saline irrigation group had lower SNOT-20 scores than the nasal spray group. At 2 weeks, the irrigation group scores were 4.4 points lower than the spray group (P=.02); at 4 weeks, the scores were 8.2 points lower (P<.001), and at 8 weeks the scores were 6.4 points lower (P=.002). Those in the irrigation group also had a significantly greater change from baseline than the patients in the spray group at 4 weeks (16.2 vs 7.4, P=.002) and at 8 weeks (15.0 vs 8.5, P=.04). The difference was marginally significant at 2 weeks (12.2 vs 6.7, P=.05).
Frequency of symptoms. At 8 weeks, 40% of the irrigation group and 61% of the nasal spray group reported nasal or sinus symptoms “often or always.” The absolute risk reduction in symptom frequency with saline irrigations, therefore, was 0.21; 95% confidence interval, 0.02-0.38 (P=.01). The odds of frequent nasal symptoms were 50% lower in the irrigation group compared to the spray group.
WHAT’S NEW: One delivery method is better than another
Prior studies had proven the effectiveness of nasal saline for reduction of rhinosinusitis symptoms. This RCT demonstrated that large-volume, low-pressure nasal irrigation brings greater symptom relief than nasal spray.
The researchers found little difference between the 2 groups in the rate of adverse effects, and reported that nasal irrigation appears to be well accepted once patients become accustomed to it. The fact that the participants were recruited from the general population further suggests that the results will be generalizable to primary care patients.
CAVEATS: High dropout rate in irrigation group
The absence of a control group prevents us from knowing the effect of saline nasal spray or irrigation compared with no treatment. In prior studies, however, nasal saline spray was found to be more effective than placebo in reducing rhinosinusitis symptoms.8,15
It is notable that a significant portion (21%) of the irrigation group abandoned this treatment by 8 weeks; in comparison, just 7% of the nasal spray group discontinued treatment.
This lower rate of adherence makes the beneficial effects of the irrigation group even more impressive. But it also suggests that a significant portion of patients are unlikely to stay with this recommended regimen. For those who try saline irrigation and choose not to continue it or are unwilling even to try it, saline spray is a reasonable alternative.
It should be noted that financial support for this study was provided by NeilMed Pharmaceuticals, a manufacturer of nasal saline solution and irrigation devices. However, the sponsor was not involved in the design or conduct of the study, in data collection or analysis, or in the preparation of the manuscript.
CHALLENGES TO IMPLEMENTATION: Tx may “scare away” some patients
Despite its effectiveness in reducing rhinosinusitis symptoms, performing large-volume, low-pressure nasal saline irrigation is not intuitive—and may sound downright scary to some patients. The need to learn how to perform nasal irrigation effectively, overcome the fear of water in the nasal cavity, and find the time to perform irrigation regularly can be barriers to this treatment.
A little bit of coaching can go a long way
A study by Rabago et al16 found that coached practice and patient education are effective tools in mastery of the technique ( PATIENT HANDOUT ).10,17 The researchers also found that several home strategies—incorporating nasal irrigation into the daily hygiene routine, placing the materials in a convenient location, and using warm water—facilitate regular use.
There is evidence, too, that patients who successfully use large-volume, low-pressure saline irrigation gain more than symptom relief. Rabago et al also found that effective use of this technique was associated with a sense of empowerment, and led to improved self-management skills, as well as a rapid, and long-term, improvement in quality of life.16
Saline nasal irrigation Your step-by-step guide
STEP 1: GATHER THE SUPPLIES
- - Salt (kosher, canning, or pickling salt)
- - Baking soda
- - Nasal irrigation pot (available at most pharmacies)
- - Measuring spoons
- - Container with lid
OR
- - An irrigation kit that includes the device and premixed saline packets
STEP 2: PREPARE THE SOLUTION
- - Put 1 tsp salt and ½ tsp baking soda into the container.
- - Add 1 pint of lukewarm tap water.
- - Mix contents.
- - Fill the nasal pot.
STEP 3: POSITION YOUR HEAD
- - Lean over the sink; rotate your head to one side.
- - Insert the spout of the irrigation device into the uppermost nostril.
- - Breathe through your mouth.
- - Raise the handle of the nasal pot so the solution flows into the upper nostril; in a few moments, the solution will begin to drain from the lower nostril.
- - Continue until the pot is empty, then exhale gently through both nostrils and gently blow your nose.
- - Refill the nasal pot, turn your head to the opposite side, and repeat with the other nostril.
- - Do this twice a day or as directed.
STEP 4: CLEAN AND PUT AWAY THE EQUIPMENT
- - Wash the nasal pot daily with warm water and dish detergent; rinse thoroughly.
- - Store unused saline solution in the sealed container; it can be kept at room temperature and reused for 2 days.
Adapted from: University of Wisconsin Department of Family Medicine.17
Acknowledgement
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
PURLs methodology
This study was selected and evaluated using FPIN’s Priority Updates from the Research Literature (PURL) Surveillance System methodology. The criteria and findings leading to the selection of this study as a PURL can be accessed at www.jfponline.com/purls.
Click here to view PURL METHODOLOGY
1. Pynnonen MA, Mukerji SS, Kim HM, et al. Nasal saline for chronic sinonasal symptoms: a randomized controlled trial. Arch Otolaryngol Head Neck Surg. 2007;133:11115-1120.
2. Gliklich RE, Metson R. Economic implications of chronic sinusitis. Otolaryngol Head Neck Surg. 1998;118(3 Pt 1):344-349.
3. International Rhinosinusitis Advisory Board. Infectious rhinosinusitis in adults classification, etiology and management. Ear Nose Throat J. 1997;76(12 suppl):s5-s22.
4. Lanza DC, Kennedy DW. Adult rhinosinusitis defined. Otolaryngol Head Neck Surg. 1997;117(3 Pt 2):s1-s7.
5. Sharp HJ, Denman D, Puumala S, et al. Treatment of acute and chronic rhinosinusitis in the United States, 1999-2002. Arch Otolaryngol Head Neck Surg. 2007;133:260-265.
6. Subramanian HN, Schechtman KB, Hamilos DL. A retrospective analysis of treatment outcomes and time to relapse after intensive medical treatment for chronic sinusitis. Am J Rhinol. 2002;16:303-312.
7. Druce HM. Adjuncts to medical management of sinusitis. Otolaryngol Head Neck Surg. 1990;103(5 Pt 2):880-883.
8. Tomooka LT, Murphy C, Davidson TM. Clinical study and literature review of nasal irrigation. Laryngoscope. 2000;110:1189-1193.
9. Heatley DG, McConnell KE, Kille TL, et al. Nasal irrigation for the alleviation of sinonasal symptoms. Otolaryngol Head Neck Surg. 2001;125:44-48.
10. Rabago D, Zgierska A, Mundt M, et al. Efficacy of daily hypertonic saline nasal irrigation among patients with sinusitis: a randomized controlled trial. J Fam Pract. 2002;51:1049-1055.
11. Taccariello M, Parikh A, Darby Y, et al. Nasal douching as a valuable adjunct in the management of chronic rhinosinusitis. Rhinology. 1999;37:29-32.
12. Harvey R, Hannan SA, Badia L, Scadding G. Nasal saline irrigations for the symptoms of chronic rhinosinusitis. Cochrane Database Syst Rev. 2007;(3):CD006394.-
13. Keojampa BK, Nguyen MH, Ryan MW. Effects of buffered saline solution on nasal mucociliary clearance and nasal airway patency. Otolaryngol Head Neck Surg. 2004;131:679-682.
14. Piccirillo JF, Merritt MG, Jr, Richards ML. Psychometric and clinimetric validity of the 20-Item Sino-Nasal Outcome Test (SNOT-20). Otolaryngol Head Neck Surg. 2002;126:41-47.
15. Hauptman G, Ryan MW. The effect of saline solutions on nasal patency and mucociliary clearance in rhinosinusitis patients. Otolaryngol Head Neck Surg. 2007;137:815-821.
16. Rabago D, Barrett B, Marchand L, et al. Qualitative aspects of nasal irrigation use by patients with chronic sinus disease in a multimethod study. Ann Fam Med. 2006;4:295-301.
17. University of Wisconsin Department of Family Medicine. Nasal Irrigation Instructions. Available at: http://www.fammed.wisc.edu/files/webfm-uploads/documents/research/nasalirrigationinstructions.pdf. Accessed December 1, 2008.
ILLUSTRATIVE CASE
A 45-year-old woman presents to your office with an 8-month history of nasal congestion and thick nasal discharge. Her symptoms have waxed and waned, the patient reports. She’s tried decongestants, antibiotics, and nasal steroids, with limited success. The patient has not had a recent respiratory infection, has never had sinus surgery, and does not want to be on long-term medication. You wonder if there’s an alternative treatment you can offer.
Rhinosinusitis is one of the most common conditions seen by primary care physicians in the United States, and its incidence and prevalence are increasing.2,3 While acute rhinosinusitis is usually self-limiting and resolves within a month, some patients develop chronic—and hard to treat—sinonasal symptoms.
No single cause, no definitive treatment
We’ve moved away from the notion that chronic rhinosinusitis is always a manifestation of persistent bacterial infection, and now recognize that there’s an inflammatory, nonbacterial component.4 In any given patient, several mechanisms—acting either simultaneously or independently—may contribute to sinonasal symptoms.3
Chronic sinusitis is treated in a variety of ways, including medications, immunotherapy, and surgery. Despite their limited efficacy, antibiotics and nasal steroids have been the mainstays of treatment.5 Treating underlying allergies, when they exist, may be helpful. But regardless of which treatment patients receive for chronic rhinosinusitis, many remain symptomatic.6
Benefits of saline irrigation extend beyond postop care
Otolaryngologists recommend saline irrigation after sinus surgery to clear secretions, debris, and crusts; reduce the risk of postoperative mucosal adhesions; and expedite mucosal healing.7,8 Saline irrigation is also gaining popularity as an alternative approach to chronic sinusitis symptom relief, and several randomized controlled trials (RCTs) have demonstrated both objective and subjective efficacy of this treatment for sinonasal disease.8-11
In 2007, the Cochrane Collaboration reviewed evidence for the effectiveness of nasal saline irrigation for symptoms of chronic rhinosinusitis. The reviewers concluded that it is well tolerated and beneficial, whether used alone or as an adjunctive treatment.12
Nasal saline sprays are often recommended because they’re thought to be better tolerated than other delivery modes.13 There have, however, been no comparisons of the relative efficacy of different means of saline delivery, until now.
STUDY SUMMARY: Nasal irrigation and spray go head-to-head
This study was a high-quality, prospective RCT comparing nasal spray and nasal irrigation.1 Subjects were recruited from the general population. To be eligible, participants had to be 18 years of age or older and have reported at least one of the following chronic rhinosinusitis symptoms on 4 or more days each week in the preceding 2 weeks:
- nasal stuffiness
- nasal dryness or crusting
- nasal congestion
- thick or discolored nasal discharge.
In addition, the symptoms must have been present on at least 15 of the preceding 30 days. Exclusion criteria included recent sinus surgery, a respiratory infection within the past 2 weeks, and the use of nasal saline within the past month.
Researchers enrolled 127 patients in the study; 63 were randomized to the nasal spray group and 64 to the large-volume, low-pressure irrigation group. Demographic and baseline characteristics of the groups were similar. The average ages of those in the irrigation and spray groups were 45 and 48 years, respectively. Most patients were nonsmokers and had been symptomatic for 7 to 12 months.
Twice-daily treatment. Researchers asked the patients to perform the assigned treatment twice daily for 8 weeks, but the patients were also permitted to continue using their usual medications. Symptom severity and disease-specific quality of life were assessed with the Sino-Nasal Outcome Test (SNOT-20), a 20-item survey that measures physical problems, emotional consequences, and functional limitations of sinusitis.14
The SNOT-20 is a validated, self-administered survey that asks patients to score items such as runny nose, postnasal discharge, need to blow the nose, reduced productivity, and embarrassment, on a 0- to 5-point scale (0=never, 5=always). A SNOT-20 score of 100 indicates the worst possible symptoms.
As a measure of chronicity of symptoms, patients were also asked to estimate how many months they’d had these symptoms during the last year. In addition, they were instructed to keep a diary to document treatment compliance and the use of other medications for sinonasal symptoms.
To measure outcomes, the researchers provided patients with mail-in packets so they could send in their completed SNOT-20 questionnaire and the medication diary completed at 2, 4, and 8 weeks after randomization.
Biggest improvements seen in irrigation group
Severity of symptoms. In each outcome measurement period, the saline irrigation group had lower SNOT-20 scores than the nasal spray group. At 2 weeks, the irrigation group scores were 4.4 points lower than the spray group (P=.02); at 4 weeks, the scores were 8.2 points lower (P<.001), and at 8 weeks the scores were 6.4 points lower (P=.002). Those in the irrigation group also had a significantly greater change from baseline than the patients in the spray group at 4 weeks (16.2 vs 7.4, P=.002) and at 8 weeks (15.0 vs 8.5, P=.04). The difference was marginally significant at 2 weeks (12.2 vs 6.7, P=.05).
Frequency of symptoms. At 8 weeks, 40% of the irrigation group and 61% of the nasal spray group reported nasal or sinus symptoms “often or always.” The absolute risk reduction in symptom frequency with saline irrigations, therefore, was 0.21; 95% confidence interval, 0.02-0.38 (P=.01). The odds of frequent nasal symptoms were 50% lower in the irrigation group compared to the spray group.
WHAT’S NEW: One delivery method is better than another
Prior studies had proven the effectiveness of nasal saline for reduction of rhinosinusitis symptoms. This RCT demonstrated that large-volume, low-pressure nasal irrigation brings greater symptom relief than nasal spray.
The researchers found little difference between the 2 groups in the rate of adverse effects, and reported that nasal irrigation appears to be well accepted once patients become accustomed to it. The fact that the participants were recruited from the general population further suggests that the results will be generalizable to primary care patients.
CAVEATS: High dropout rate in irrigation group
The absence of a control group prevents us from knowing the effect of saline nasal spray or irrigation compared with no treatment. In prior studies, however, nasal saline spray was found to be more effective than placebo in reducing rhinosinusitis symptoms.8,15
It is notable that a significant portion (21%) of the irrigation group abandoned this treatment by 8 weeks; in comparison, just 7% of the nasal spray group discontinued treatment.
This lower rate of adherence makes the beneficial effects of the irrigation group even more impressive. But it also suggests that a significant portion of patients are unlikely to stay with this recommended regimen. For those who try saline irrigation and choose not to continue it or are unwilling even to try it, saline spray is a reasonable alternative.
It should be noted that financial support for this study was provided by NeilMed Pharmaceuticals, a manufacturer of nasal saline solution and irrigation devices. However, the sponsor was not involved in the design or conduct of the study, in data collection or analysis, or in the preparation of the manuscript.
CHALLENGES TO IMPLEMENTATION: Tx may “scare away” some patients
Despite its effectiveness in reducing rhinosinusitis symptoms, performing large-volume, low-pressure nasal saline irrigation is not intuitive—and may sound downright scary to some patients. The need to learn how to perform nasal irrigation effectively, overcome the fear of water in the nasal cavity, and find the time to perform irrigation regularly can be barriers to this treatment.
A little bit of coaching can go a long way
A study by Rabago et al16 found that coached practice and patient education are effective tools in mastery of the technique ( PATIENT HANDOUT ).10,17 The researchers also found that several home strategies—incorporating nasal irrigation into the daily hygiene routine, placing the materials in a convenient location, and using warm water—facilitate regular use.
There is evidence, too, that patients who successfully use large-volume, low-pressure saline irrigation gain more than symptom relief. Rabago et al also found that effective use of this technique was associated with a sense of empowerment, and led to improved self-management skills, as well as a rapid, and long-term, improvement in quality of life.16
Saline nasal irrigation Your step-by-step guide
STEP 1: GATHER THE SUPPLIES
- - Salt (kosher, canning, or pickling salt)
- - Baking soda
- - Nasal irrigation pot (available at most pharmacies)
- - Measuring spoons
- - Container with lid
OR
- - An irrigation kit that includes the device and premixed saline packets
STEP 2: PREPARE THE SOLUTION
- - Put 1 tsp salt and ½ tsp baking soda into the container.
- - Add 1 pint of lukewarm tap water.
- - Mix contents.
- - Fill the nasal pot.
STEP 3: POSITION YOUR HEAD
- - Lean over the sink; rotate your head to one side.
- - Insert the spout of the irrigation device into the uppermost nostril.
- - Breathe through your mouth.
- - Raise the handle of the nasal pot so the solution flows into the upper nostril; in a few moments, the solution will begin to drain from the lower nostril.
- - Continue until the pot is empty, then exhale gently through both nostrils and gently blow your nose.
- - Refill the nasal pot, turn your head to the opposite side, and repeat with the other nostril.
- - Do this twice a day or as directed.
STEP 4: CLEAN AND PUT AWAY THE EQUIPMENT
- - Wash the nasal pot daily with warm water and dish detergent; rinse thoroughly.
- - Store unused saline solution in the sealed container; it can be kept at room temperature and reused for 2 days.
Adapted from: University of Wisconsin Department of Family Medicine.17
Acknowledgement
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
PURLs methodology
This study was selected and evaluated using FPIN’s Priority Updates from the Research Literature (PURL) Surveillance System methodology. The criteria and findings leading to the selection of this study as a PURL can be accessed at www.jfponline.com/purls.
Click here to view PURL METHODOLOGY
ILLUSTRATIVE CASE
A 45-year-old woman presents to your office with an 8-month history of nasal congestion and thick nasal discharge. Her symptoms have waxed and waned, the patient reports. She’s tried decongestants, antibiotics, and nasal steroids, with limited success. The patient has not had a recent respiratory infection, has never had sinus surgery, and does not want to be on long-term medication. You wonder if there’s an alternative treatment you can offer.
Rhinosinusitis is one of the most common conditions seen by primary care physicians in the United States, and its incidence and prevalence are increasing.2,3 While acute rhinosinusitis is usually self-limiting and resolves within a month, some patients develop chronic—and hard to treat—sinonasal symptoms.
No single cause, no definitive treatment
We’ve moved away from the notion that chronic rhinosinusitis is always a manifestation of persistent bacterial infection, and now recognize that there’s an inflammatory, nonbacterial component.4 In any given patient, several mechanisms—acting either simultaneously or independently—may contribute to sinonasal symptoms.3
Chronic sinusitis is treated in a variety of ways, including medications, immunotherapy, and surgery. Despite their limited efficacy, antibiotics and nasal steroids have been the mainstays of treatment.5 Treating underlying allergies, when they exist, may be helpful. But regardless of which treatment patients receive for chronic rhinosinusitis, many remain symptomatic.6
Benefits of saline irrigation extend beyond postop care
Otolaryngologists recommend saline irrigation after sinus surgery to clear secretions, debris, and crusts; reduce the risk of postoperative mucosal adhesions; and expedite mucosal healing.7,8 Saline irrigation is also gaining popularity as an alternative approach to chronic sinusitis symptom relief, and several randomized controlled trials (RCTs) have demonstrated both objective and subjective efficacy of this treatment for sinonasal disease.8-11
In 2007, the Cochrane Collaboration reviewed evidence for the effectiveness of nasal saline irrigation for symptoms of chronic rhinosinusitis. The reviewers concluded that it is well tolerated and beneficial, whether used alone or as an adjunctive treatment.12
Nasal saline sprays are often recommended because they’re thought to be better tolerated than other delivery modes.13 There have, however, been no comparisons of the relative efficacy of different means of saline delivery, until now.
STUDY SUMMARY: Nasal irrigation and spray go head-to-head
This study was a high-quality, prospective RCT comparing nasal spray and nasal irrigation.1 Subjects were recruited from the general population. To be eligible, participants had to be 18 years of age or older and have reported at least one of the following chronic rhinosinusitis symptoms on 4 or more days each week in the preceding 2 weeks:
- nasal stuffiness
- nasal dryness or crusting
- nasal congestion
- thick or discolored nasal discharge.
In addition, the symptoms must have been present on at least 15 of the preceding 30 days. Exclusion criteria included recent sinus surgery, a respiratory infection within the past 2 weeks, and the use of nasal saline within the past month.
Researchers enrolled 127 patients in the study; 63 were randomized to the nasal spray group and 64 to the large-volume, low-pressure irrigation group. Demographic and baseline characteristics of the groups were similar. The average ages of those in the irrigation and spray groups were 45 and 48 years, respectively. Most patients were nonsmokers and had been symptomatic for 7 to 12 months.
Twice-daily treatment. Researchers asked the patients to perform the assigned treatment twice daily for 8 weeks, but the patients were also permitted to continue using their usual medications. Symptom severity and disease-specific quality of life were assessed with the Sino-Nasal Outcome Test (SNOT-20), a 20-item survey that measures physical problems, emotional consequences, and functional limitations of sinusitis.14
The SNOT-20 is a validated, self-administered survey that asks patients to score items such as runny nose, postnasal discharge, need to blow the nose, reduced productivity, and embarrassment, on a 0- to 5-point scale (0=never, 5=always). A SNOT-20 score of 100 indicates the worst possible symptoms.
As a measure of chronicity of symptoms, patients were also asked to estimate how many months they’d had these symptoms during the last year. In addition, they were instructed to keep a diary to document treatment compliance and the use of other medications for sinonasal symptoms.
To measure outcomes, the researchers provided patients with mail-in packets so they could send in their completed SNOT-20 questionnaire and the medication diary completed at 2, 4, and 8 weeks after randomization.
Biggest improvements seen in irrigation group
Severity of symptoms. In each outcome measurement period, the saline irrigation group had lower SNOT-20 scores than the nasal spray group. At 2 weeks, the irrigation group scores were 4.4 points lower than the spray group (P=.02); at 4 weeks, the scores were 8.2 points lower (P<.001), and at 8 weeks the scores were 6.4 points lower (P=.002). Those in the irrigation group also had a significantly greater change from baseline than the patients in the spray group at 4 weeks (16.2 vs 7.4, P=.002) and at 8 weeks (15.0 vs 8.5, P=.04). The difference was marginally significant at 2 weeks (12.2 vs 6.7, P=.05).
Frequency of symptoms. At 8 weeks, 40% of the irrigation group and 61% of the nasal spray group reported nasal or sinus symptoms “often or always.” The absolute risk reduction in symptom frequency with saline irrigations, therefore, was 0.21; 95% confidence interval, 0.02-0.38 (P=.01). The odds of frequent nasal symptoms were 50% lower in the irrigation group compared to the spray group.
WHAT’S NEW: One delivery method is better than another
Prior studies had proven the effectiveness of nasal saline for reduction of rhinosinusitis symptoms. This RCT demonstrated that large-volume, low-pressure nasal irrigation brings greater symptom relief than nasal spray.
The researchers found little difference between the 2 groups in the rate of adverse effects, and reported that nasal irrigation appears to be well accepted once patients become accustomed to it. The fact that the participants were recruited from the general population further suggests that the results will be generalizable to primary care patients.
CAVEATS: High dropout rate in irrigation group
The absence of a control group prevents us from knowing the effect of saline nasal spray or irrigation compared with no treatment. In prior studies, however, nasal saline spray was found to be more effective than placebo in reducing rhinosinusitis symptoms.8,15
It is notable that a significant portion (21%) of the irrigation group abandoned this treatment by 8 weeks; in comparison, just 7% of the nasal spray group discontinued treatment.
This lower rate of adherence makes the beneficial effects of the irrigation group even more impressive. But it also suggests that a significant portion of patients are unlikely to stay with this recommended regimen. For those who try saline irrigation and choose not to continue it or are unwilling even to try it, saline spray is a reasonable alternative.
It should be noted that financial support for this study was provided by NeilMed Pharmaceuticals, a manufacturer of nasal saline solution and irrigation devices. However, the sponsor was not involved in the design or conduct of the study, in data collection or analysis, or in the preparation of the manuscript.
CHALLENGES TO IMPLEMENTATION: Tx may “scare away” some patients
Despite its effectiveness in reducing rhinosinusitis symptoms, performing large-volume, low-pressure nasal saline irrigation is not intuitive—and may sound downright scary to some patients. The need to learn how to perform nasal irrigation effectively, overcome the fear of water in the nasal cavity, and find the time to perform irrigation regularly can be barriers to this treatment.
A little bit of coaching can go a long way
A study by Rabago et al16 found that coached practice and patient education are effective tools in mastery of the technique ( PATIENT HANDOUT ).10,17 The researchers also found that several home strategies—incorporating nasal irrigation into the daily hygiene routine, placing the materials in a convenient location, and using warm water—facilitate regular use.
There is evidence, too, that patients who successfully use large-volume, low-pressure saline irrigation gain more than symptom relief. Rabago et al also found that effective use of this technique was associated with a sense of empowerment, and led to improved self-management skills, as well as a rapid, and long-term, improvement in quality of life.16
Saline nasal irrigation Your step-by-step guide
STEP 1: GATHER THE SUPPLIES
- - Salt (kosher, canning, or pickling salt)
- - Baking soda
- - Nasal irrigation pot (available at most pharmacies)
- - Measuring spoons
- - Container with lid
OR
- - An irrigation kit that includes the device and premixed saline packets
STEP 2: PREPARE THE SOLUTION
- - Put 1 tsp salt and ½ tsp baking soda into the container.
- - Add 1 pint of lukewarm tap water.
- - Mix contents.
- - Fill the nasal pot.
STEP 3: POSITION YOUR HEAD
- - Lean over the sink; rotate your head to one side.
- - Insert the spout of the irrigation device into the uppermost nostril.
- - Breathe through your mouth.
- - Raise the handle of the nasal pot so the solution flows into the upper nostril; in a few moments, the solution will begin to drain from the lower nostril.
- - Continue until the pot is empty, then exhale gently through both nostrils and gently blow your nose.
- - Refill the nasal pot, turn your head to the opposite side, and repeat with the other nostril.
- - Do this twice a day or as directed.
STEP 4: CLEAN AND PUT AWAY THE EQUIPMENT
- - Wash the nasal pot daily with warm water and dish detergent; rinse thoroughly.
- - Store unused saline solution in the sealed container; it can be kept at room temperature and reused for 2 days.
Adapted from: University of Wisconsin Department of Family Medicine.17
Acknowledgement
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
PURLs methodology
This study was selected and evaluated using FPIN’s Priority Updates from the Research Literature (PURL) Surveillance System methodology. The criteria and findings leading to the selection of this study as a PURL can be accessed at www.jfponline.com/purls.
Click here to view PURL METHODOLOGY
1. Pynnonen MA, Mukerji SS, Kim HM, et al. Nasal saline for chronic sinonasal symptoms: a randomized controlled trial. Arch Otolaryngol Head Neck Surg. 2007;133:11115-1120.
2. Gliklich RE, Metson R. Economic implications of chronic sinusitis. Otolaryngol Head Neck Surg. 1998;118(3 Pt 1):344-349.
3. International Rhinosinusitis Advisory Board. Infectious rhinosinusitis in adults classification, etiology and management. Ear Nose Throat J. 1997;76(12 suppl):s5-s22.
4. Lanza DC, Kennedy DW. Adult rhinosinusitis defined. Otolaryngol Head Neck Surg. 1997;117(3 Pt 2):s1-s7.
5. Sharp HJ, Denman D, Puumala S, et al. Treatment of acute and chronic rhinosinusitis in the United States, 1999-2002. Arch Otolaryngol Head Neck Surg. 2007;133:260-265.
6. Subramanian HN, Schechtman KB, Hamilos DL. A retrospective analysis of treatment outcomes and time to relapse after intensive medical treatment for chronic sinusitis. Am J Rhinol. 2002;16:303-312.
7. Druce HM. Adjuncts to medical management of sinusitis. Otolaryngol Head Neck Surg. 1990;103(5 Pt 2):880-883.
8. Tomooka LT, Murphy C, Davidson TM. Clinical study and literature review of nasal irrigation. Laryngoscope. 2000;110:1189-1193.
9. Heatley DG, McConnell KE, Kille TL, et al. Nasal irrigation for the alleviation of sinonasal symptoms. Otolaryngol Head Neck Surg. 2001;125:44-48.
10. Rabago D, Zgierska A, Mundt M, et al. Efficacy of daily hypertonic saline nasal irrigation among patients with sinusitis: a randomized controlled trial. J Fam Pract. 2002;51:1049-1055.
11. Taccariello M, Parikh A, Darby Y, et al. Nasal douching as a valuable adjunct in the management of chronic rhinosinusitis. Rhinology. 1999;37:29-32.
12. Harvey R, Hannan SA, Badia L, Scadding G. Nasal saline irrigations for the symptoms of chronic rhinosinusitis. Cochrane Database Syst Rev. 2007;(3):CD006394.-
13. Keojampa BK, Nguyen MH, Ryan MW. Effects of buffered saline solution on nasal mucociliary clearance and nasal airway patency. Otolaryngol Head Neck Surg. 2004;131:679-682.
14. Piccirillo JF, Merritt MG, Jr, Richards ML. Psychometric and clinimetric validity of the 20-Item Sino-Nasal Outcome Test (SNOT-20). Otolaryngol Head Neck Surg. 2002;126:41-47.
15. Hauptman G, Ryan MW. The effect of saline solutions on nasal patency and mucociliary clearance in rhinosinusitis patients. Otolaryngol Head Neck Surg. 2007;137:815-821.
16. Rabago D, Barrett B, Marchand L, et al. Qualitative aspects of nasal irrigation use by patients with chronic sinus disease in a multimethod study. Ann Fam Med. 2006;4:295-301.
17. University of Wisconsin Department of Family Medicine. Nasal Irrigation Instructions. Available at: http://www.fammed.wisc.edu/files/webfm-uploads/documents/research/nasalirrigationinstructions.pdf. Accessed December 1, 2008.
1. Pynnonen MA, Mukerji SS, Kim HM, et al. Nasal saline for chronic sinonasal symptoms: a randomized controlled trial. Arch Otolaryngol Head Neck Surg. 2007;133:11115-1120.
2. Gliklich RE, Metson R. Economic implications of chronic sinusitis. Otolaryngol Head Neck Surg. 1998;118(3 Pt 1):344-349.
3. International Rhinosinusitis Advisory Board. Infectious rhinosinusitis in adults classification, etiology and management. Ear Nose Throat J. 1997;76(12 suppl):s5-s22.
4. Lanza DC, Kennedy DW. Adult rhinosinusitis defined. Otolaryngol Head Neck Surg. 1997;117(3 Pt 2):s1-s7.
5. Sharp HJ, Denman D, Puumala S, et al. Treatment of acute and chronic rhinosinusitis in the United States, 1999-2002. Arch Otolaryngol Head Neck Surg. 2007;133:260-265.
6. Subramanian HN, Schechtman KB, Hamilos DL. A retrospective analysis of treatment outcomes and time to relapse after intensive medical treatment for chronic sinusitis. Am J Rhinol. 2002;16:303-312.
7. Druce HM. Adjuncts to medical management of sinusitis. Otolaryngol Head Neck Surg. 1990;103(5 Pt 2):880-883.
8. Tomooka LT, Murphy C, Davidson TM. Clinical study and literature review of nasal irrigation. Laryngoscope. 2000;110:1189-1193.
9. Heatley DG, McConnell KE, Kille TL, et al. Nasal irrigation for the alleviation of sinonasal symptoms. Otolaryngol Head Neck Surg. 2001;125:44-48.
10. Rabago D, Zgierska A, Mundt M, et al. Efficacy of daily hypertonic saline nasal irrigation among patients with sinusitis: a randomized controlled trial. J Fam Pract. 2002;51:1049-1055.
11. Taccariello M, Parikh A, Darby Y, et al. Nasal douching as a valuable adjunct in the management of chronic rhinosinusitis. Rhinology. 1999;37:29-32.
12. Harvey R, Hannan SA, Badia L, Scadding G. Nasal saline irrigations for the symptoms of chronic rhinosinusitis. Cochrane Database Syst Rev. 2007;(3):CD006394.-
13. Keojampa BK, Nguyen MH, Ryan MW. Effects of buffered saline solution on nasal mucociliary clearance and nasal airway patency. Otolaryngol Head Neck Surg. 2004;131:679-682.
14. Piccirillo JF, Merritt MG, Jr, Richards ML. Psychometric and clinimetric validity of the 20-Item Sino-Nasal Outcome Test (SNOT-20). Otolaryngol Head Neck Surg. 2002;126:41-47.
15. Hauptman G, Ryan MW. The effect of saline solutions on nasal patency and mucociliary clearance in rhinosinusitis patients. Otolaryngol Head Neck Surg. 2007;137:815-821.
16. Rabago D, Barrett B, Marchand L, et al. Qualitative aspects of nasal irrigation use by patients with chronic sinus disease in a multimethod study. Ann Fam Med. 2006;4:295-301.
17. University of Wisconsin Department of Family Medicine. Nasal Irrigation Instructions. Available at: http://www.fammed.wisc.edu/files/webfm-uploads/documents/research/nasalirrigationinstructions.pdf. Accessed December 1, 2008.
Copyright © 2009 The Family Physicians Inquiries Network.
All rights reserved.
New asthma guidelines emphasize control, regular monitoring
This review focuses on several elements in the National Asthma Education and Prevention Program’s new guidelines, the third Expert Panel Report (EPR3),1 that differ substantially from those in EPR2,2 issued in 1997 and updated in 2002.3 These differences in approach to the management of asthma described in EPR3 offer a clear potential for reducing the gap between optimal asthma care outcomes as described in guidelines and normative asthma care outcomes in the “real world.”
GREATER EMPHASIS ON CONTROL
The EPR2 guidelines2 recommended that asthma management be carried out in an algorithmic manner. Patients were classified into four severity categories: mild intermittent, mild persistent, moderate persistent, and severe persistent asthma, based on assessment of the level of symptoms (day/night), reliance on “reliever” medication, and lung function at the time of presentation. Pharmacologic management was then assigned according to each respective categorization in an evidence-based fashion.
In an ideal world, this would result in patients with asthma receiving appropriate pharmacotherapeutic agents associated with favorable asthma care outcomes, which were also advantageous from both cost- and risk-benefit standpoints. In the real world, however, this paradigm was flawed, as it relied on accurate categorization of patients in order for pharmacotherapy to be prescribed appropriately. Both providers and patients are prone to underestimate asthma severity,4,5 and for this reason many patients managed on the basis of this paradigm were undertreated.
A new paradigm, based on the assessment of asthma control, has been encouraged in the EPR3 guidelines.1
Severity and control are not synonymous
More than a decade ago, Cockroft and Swystun6 pointed out that asthma control (or lack thereof) is often used inappropriately to define asthma severity: ie, well-controlled asthma is seen as synonymous with mild asthma, and poorly controlled asthma with severe asthma.
Asthma severity can be defined as the intrinsic intensity of the disease process, while asthma control is the degree to which the manifestations of asthma are minimized. Asthma severity is clearly a determinant of asthma control, but its impact is affected by a variety of factors, including but not limited to:
- Whether appropriate medication is prescribed
- Patterns of therapeutic adherence
- The degree to which recommended measures for avoiding for clinically relevant aeroallergens are pursued.
Health care utilization, including hospitalizations and emergency department visits, correlates more closely with asthma control than with asthma severity.7–9 Indeed, a patient with severe persistent asthma who is treated appropriately with multiple “controller” medications and who takes his or her medications and avoids allergens as directed can achieve well-controlled or totally controlled asthma, and is not likely to require hospitalization or emergency department management, to miss school or work, or to experience nocturnal awakening or limitation in routine activities due to asthma. This patient has severe persistent asthma that is well controlled.
In contrast, a patient with mild or moderate persistent asthma who does not receive appropriate instructions for avoiding allergens or taking controller medication regularly or who is poorly adherent will likely have poor asthma control. This patient is more likely to require hospitalization or emergency department management, to miss school or work, and to experience nocturnal awakening or limitation in routine activities due to asthma. This patient has mild persistent asthma that is poorly controlled.
Assess asthma severity in the first visit, and control in subsequent visits
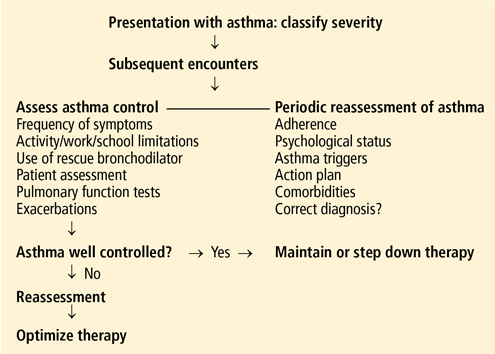
How to assess severity
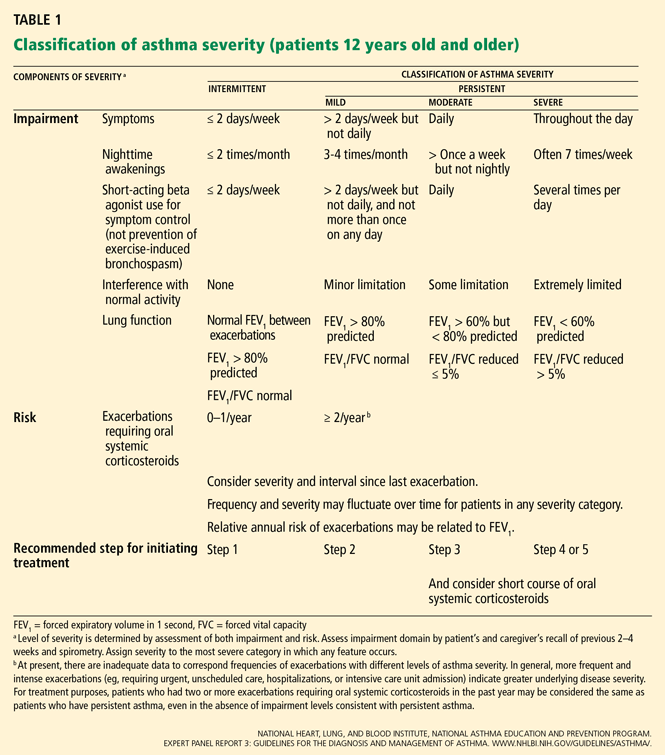
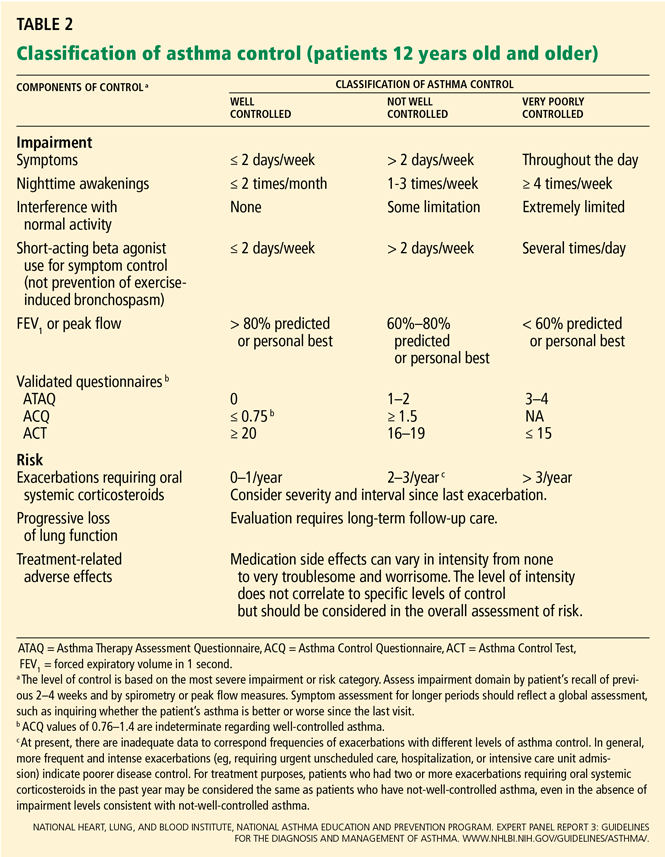
How to measure control
For all patients with asthma, regardless of severity, the goal is the same: to achieve control by reducing both impairment and risk. Asthma is classified as well controlled, not well controlled, or poorly controlled (Table 2).1
Validated tests are available to assess control
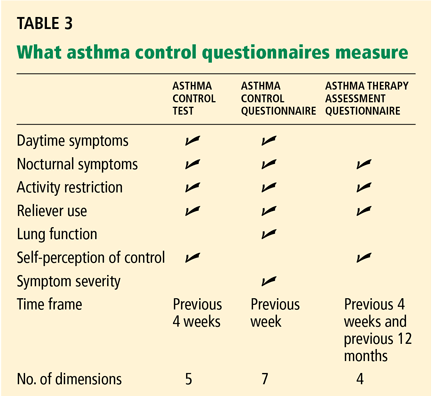
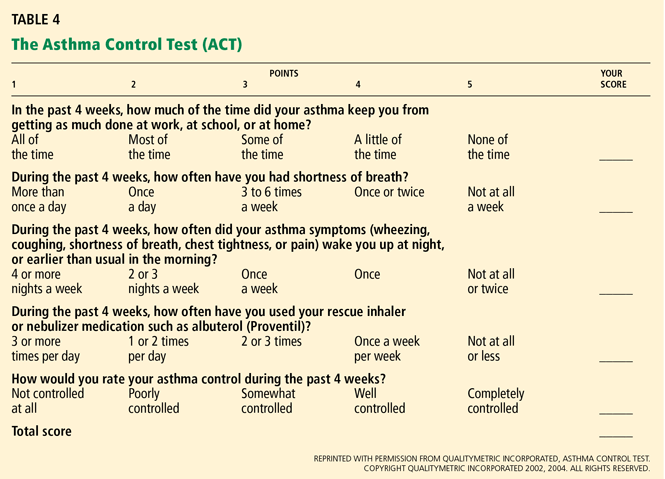
Serial testing as a quality indicator
Serial ACT scores give an objective measure of the degree to which the goals of management1 are being achieved, and in so doing can encourage optimal outcomes.14
Another use of these tests is to document whether asthma control improves over time when patients receive care from a particular physician or group. This use may become increasingly important in view of efforts underway to implement a pay-for-performance model for asthma care, in which providers will be financially rewarded for improved patient care outcomes and adherence to standards of practice based on Health Plan Employer Data and Information Set measures.15
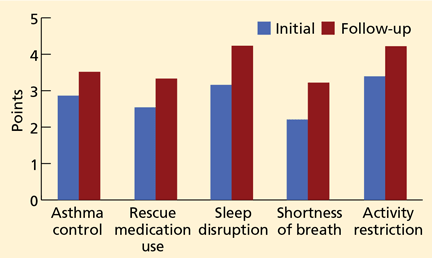
We have used the ACT in the Section of Allergy/Immunology at Cleveland Clinic for 3 years on a routine basis. All patients with asthma being seen either for the first time or as follow-up complete the ACT, which has been entered in a flow sheet in our electronic medical record, at the same time they undergo spirometry. We have shown that care in the Section of Allergy/Immunology is associated with improvement in asthma control over time, in patients who have completed serial ACT measurements at initial visits and at follow-up visits (Figure 2).
Objective measurement of lung function is also important
Serial monitoring of lung function at every patient visit with spirometry is also important, as some patients may be “poor perceivers,”16 ie, they may have little or no subjective awareness of moderate or even severe ventilatory impairment. A number of studies17,18 support the contention that symptoms and lung function are separate and independent dimensions of asthma control, and that both of them need to be assessed.
Responding to changes in control

THE STEP 3 CONTROVERSY
Salmeterol Multicenter Asthma Research Trial
In the Salmeterol Multicenter Asthma Research Trial (SMART), patients randomized to the long-acting beta agonist (LABA) salmeterol (Serevent)—particularly African Americans—had a statistically significant increase in the risk of untoward asthma care outcomes.20
SMART was launched in 1996. Patients were randomized in a double-blind fashion to receive either salmeterol 42 μg twice a day or placebo in addition to their usual asthma therapy for 28 weeks. The rate of the primary outcome (respiratory-related deaths or life-threatening experiences) was not significantly different with salmeterol than with placebo (relative risk [RR] = 1.40, 95% confidence interval [CI] 0.91–2.14). However, in 2003, the study was halted prematurely because of difficulty enrolling the targeted number of 60,000 patients, and an interim analysis that revealed significantly higher rates of secondary outcomes in subjects randomized to salmeterol. Compared with the placebo group, the salmeterol group had significantly higher rates of respiratory-related deaths (RR 2.16, 95% CI 1.06–4.41), asthma-related deaths (RR = 4.37, 95% CI = 1.25–15.34), and combined asthma-related deaths or life-threatening experiences (RR = 1.71, 95% CI 1.01–2.89). There were 13 asthma-related deaths and 37 combined asthma-related deaths or life-threatening experiences in the salmeterol group, compared with 3 and 22, respectively, in the placebo group. Of the 16 asthma deaths in the study, 13 (81%) occurred in the initial phase of SMART, when patients were recruited via print, radio, and television advertising; afterward, patients were recruited directly by investigators.
Statistically significant differences in outcomes occurred primarily in African Americans. African Americans who received salmeterol had higher rates of respiratory death or life-threatening experiences (RR = 4.10, 95% CI 1.54–10.90), the primary end point for the study, as well as higher rates of combined asthma-related deaths or life-threatening experiences (RR = 10.46, 95% CI 1.34–81.58), a secondary end point. No statistically significant differences were observed in white patients randomized to salmeterol with respect to the primary end point (RR = 1.05, 95% = 0.62–1.76); the secondary end point of combined asthma-related deaths or life-threatening experiences (RR = 1.08, 95% CI 0.55–2.14); or other end points.
Medication exposures were not tracked during the study, and allocation to inhaled corticosteroids combined with salmeterol was not randomized, so the effect of concomitant inhaled corticosteroid use cannot be determined from these data.
As a result of SMART, medications that contain either of the two LABAs, salmeterol or formoterol (Foradil), carry a black-box warning.
LABAs: Risks and benefits
Two studies21,22 have suggested that asthmatic patients who are homozygous for Arg/Arg at codon 16 of the beta-2 adrenergic receptor are predisposed to untoward asthma outcomes with regular exposure to LABAs. However, other data23–25 do not support the contention that B16 Arg/Arg patients experience adverse asthma outcomes with LABA exposure. In two recently published studies, no difference in rates of exacerbations, severe exacerbations, lung function, frequency of reliance on SABA, or nocturnal awakenings was observed in patients receiving formoterol combined with budesonide24 or salmeterol combined with fluticasone25 according to genotype. A prospective study26 also found no statistically significant difference in exacerbation rates according to beta adrenergic receptor genotype in individuals randomized to LABA monotherapy, or LABA combined with inhaled corticosteroids.
The updated EPR2 asthma guidelines,3 published in November 2002, stipulated that LABAs were the preferred controller agent to “add on” to low-dose inhaled corticosteroids for patients with moderate persistent asthma, and that the combination of low-dose inhaled corticosteroids and LABA was associated with superior outcomes: reduction of symptoms, including nocturnal awakening, increase in lung function, improvement in health-related quality of life, decreased use of “rescue” medication, and reduced rate of exacerbations and severe exacerbations, compared with higher-dose inhaled corticosteroid monotherapy. This management recommendation was categorized as level A, on the basis of data from multiple randomized, controlled, double-blinded trials.27–29 Additional evidence14,30 and data from two meta-analyses31,32 have provided further support for this recommendation, while no evidence linking LABA exposure to risk for fatal or near-fatal asthma has been found in cohort or case-control studies.33–38
Based on safety concerns, the EPR3 guidelines1 recommend that medium-dose inhaled corticosteroids be regarded as equivalent to adding LABAs to low-dose inhaled corticosteroids, and state: “the established, beneficial effects of LABA for the great majority of patients whose asthma is not well controlled with [inhaled corticosteroids] alone should be weighed against the increased risk for severe exacerbations, although uncommon, associated with daily use of LABA.”1
There is currently an honest difference of opinion39,40 among asthma specialists as to how this management recommendation for moderate persistent asthma—now depicted at “step 3” in the EPR3 guidelines (Table 4)—should be implemented. The LABA controversy was reviewed previously in the Cleveland Clinic Journal of Medicine.41
THE ROLE OF OMALIZUMAB: WEIGHING COST VS BENEFIT
The 2002 update to the EPR2 guidelines3 was issued before omalizumab (Xolair) was approved in June 2003.
Patients with severe persistent asthma are categorized in steps 5 or 6 in the EPR3 guidelines (Table 5).1 Preferred management for these patients includes inhaled corticosteroids in high doses combined with long-acting beta agonists and, for step 6 patients, oral corticosteroids.
Omalizumab was approved for management of patients with moderate or severe persistent asthma who are not achieving the goals of asthma management on inhaled corticosteroids, who exhibit a wheal-flare reaction to a perennial allergen, and whose immunoglobulin E (IgE) level is in the range of 30 to 700 IU/mL.42 Omalizumab dosing is based on the serum IgE level and on body weight.
Omalizumab, an anti-IgE monoclonal antibody
Omalizumab is a recombinant, humanized, monoclonal anti-IgE antibody that binds to IgE at the same Fc site as the high-affinity IgE receptor. Its primary mechanism of action is the binding of free IgE in the circulation, forming biologically inert, small complexes that do not activate complement and are cleared by the reticuloendothelial system.42 Its secondary mechanism of action entails a reduction in the number of high-affinity receptors on basophils, from approximately 220,000 to 8,300 receptors per cell. The latter effect was associated with a 90% reduction in histamine release from basophils in response to ex vivo challenge with dust mite allergen.43
Benefit in randomized trials
Omalizumab has been associated with statistically and clinically significant benefit in randomized, double-blind, placebo-controlled trials.44,45
Humbert et al46 randomized 419 patients whose asthma was not adequately controlled on high-dose inhaled corticosteroids and long-acting beta agonists, who were 12 to 75 years old, with reduced lung function and a history of recent asthma exacerbation, to treatment with omalizumab or placebo. Omalizumab was associated with a statistically significant reduction in the rate of asthma exacerbations and severe asthma exacerbations, as well as statistically significant improvements in asthma-related quality of life, morning peak expiratory flow rate, and asthma symptom scores.
These data support the recommendation in EPR3 to consider a trial of omalizumab in properly selected patients with severe, persistent allergic asthma.
Omalizumab is cost-beneficial in properly selected patients
The current wholesale acquisition cost of omalizumab is $532 for one 150-mg vial (David Zito, personal communication). The cost of treatment varies based on body weight and IgE level but may range from a wholesale cost of $6,388 to $38,326 per year.
However, as asthma severity increases, both direct and indirect medical expenditures increase substantially.47,48 Annual costs are approximately four times higher for severe asthma compared with mild asthma49; not only are treatment and exacerbation costs higher, but indirect costs are also disproportionately greater. Annual costs for severe asthma are significantly greater if the disease is inadequately controlled.50 For these reasons, an intervention that leads to improved outcomes for severe, poorly controlled asthma carries the potential for the greatest cost-utility for society, as it can lower direct costs by reducing the frequency and severity of exacerbations, in addition to reducing indirect medical expenditures on the basis of increased productivity and fewer days of missed work or school. The cost of omalizumab in quality-adjusted life years compares favorably with that of biologicals used in managing rheumatoid arthritis, Crohn disease, and multiple sclerosis.50
Adverse effects of omalizumab
In pivotal trials,43,44 omalizumab was associated with a substantial rate of local reactions. The rate of anaphylaxis was slightly less than 1 in 1,000, and this has been confirmed by surveillance data recorded since approval of the drug in 2003. Based on the observed risk of anaphylaxis, in July 2007, the US Food and Drug Administration added a black-box warning to the omalizumab label and stipulated that a medication guide should be provided for patients.51 The warning indicates that health care providers administering omalizumab should be prepared to manage anaphylaxis and that patients should be closely observed for an appropriate period after omalizumab administration.
The package insert also describes a numerical, but not statistically significant, increase in the rate of malignancy in patients receiving omalizumab.42 Malignancy developed in 0.5% of patients receiving omalizumab, compared with 0.2% of patients who received placebo. Because these malignancies were diagnosed over a shorter period than the time required for oncogenesis (ie, 6 months in 60% of cases), and because a heterogeneous variety of tumors was observed, there is reason to doubt these tumors were causally associated with omalizumab.
Postmarketing surveillance studies are in progress that will provide more definitive data on the potential relationship between malignancy and omalizumab exposure.
Omalizumab: Guideline recommendations
The EPR3 guidelines1 state that omalizumab is the only adjunctive therapy to demonstrate efficacy when added to high-dose inhaled corticosteroids plus long-acting beta agonists in patients with severe, persistent, allergic asthma and that evidence does not support use of the following agents, which in some cases are approved for managing other conditions and have been advocated for management of severe, refractory asthma: methotrexate, soluble interleukin (IL)-4 receptor, anti-IL-5, anti-IL-12, cyclosporine A, intravenous immune globulin, gold, troleandomycin, and colchicine. The data supporting use of macrolides were characterized as “encouraging but insufficient to support a recommendation.”
The strength of evidence for the use of omalizumab for patients in steps 5 and 6 who fulfill the criteria for its use (see above) was classified in the EPR3 guidelines1 as category B. The guidelines also say that omalizumab may be considered for adjunctive therapy in properly selected patients in step 4, as a means to avoid higher doses of inhaled corticosteroids, but that additional studies are needed to establish its utility for such patients. This recommendation was classified as category D because of the lack of published comparator trials.
ALLERGEN IMMUNOTHERAPY FOR PATIENTS WITH ASTHMA
Many patients with asthma have clinically relevant, IgE-mediated (allergic) potential to inhaled allergens.1 For patients with persistent asthma (steps 2–6 in Table 5), allergic reactions can contribute to airway inflammation, provoke symptoms, and lead to more use of medications. For this reason, identification and management of clinically relevant allergy merits consideration.52
The EPR3 guidelines1 recommend considering allergen immunotherapy for patients with mild or moderate persistent asthma (steps 2–4) who have a clinically relevant component of allergy to inhaled substances.
Changing the immune response
Allergen immunotherapy entails the incremental administration of inhalant allergens by subcutaneous injection for the purpose of inducing immune system changes in the host response. The goal of immunotherapy is to protect against allergic reactions that can be expected to occur with ongoing exposure to clinically relevant allergens.53
The immunologic changes that develop with allergen immunotherapy are complex.53,54 Successful immunotherapy results in generation of a population of CD4+/CD25+ T lymphocytes producing IL-10, transforming growth factor beta, or both. Allergen immunotherapy has been shown to block the immediate- and late-phase allergic response; to decrease recruitment of mast cells, basophils, and eosinophils on provocation or natural exposure to allergens in the skin, nose, eye, and bronchial mucosa; to blunt the seasonal rise in specific IgE; and to suppress late-phase inflammatory responses in the skin and respiratory tract. However, the efficacy of immunotherapy in relation to these immunologic changes is not completely understood.54
Many patients need skin testing
Allergen immunotherapy may be considered for patients with asthma for whom a clear relationship exists between symptoms and exposure to an allergen to which the patient is sensitive.53 Because it is often not possible to determine whether a patient is sensitive to a perennial indoor allergen (eg, dust mite) on the basis of the medical history alone,55 many patients with asthma benefit from immediate hypersensitivity skin testing to objectively assess or rule out allergy to common inhalants. In certain situations, in vitro testing may be performed, but skin testing has a higher negative predictive value and is recommended as a better screening test.56
Benefits of allergen immunotherapy
Numerous randomized, double-blind, placebo-controlled trials have shown that allergen immunotherapy is associated with benefit for reducing symptoms and medication reliance.57–63
A meta-analysis of 75 randomized, placebo-controlled studies confirmed the effectiveness of immunotherapy in asthma, with a significant reduction in asthma symptoms and medication use and with improvement in bronchial hyperreactivity.64 This meta-analysis included 36 trials of dust mite allergen, 20 of pollen, and 10 of animal dander. Immunotherapy is efficacious for pollen, mold, dust mite, cockroach, and animal allergens; however, its effectiveness is more established for dust mite, animal dander, and pollen allergens, as fewer studies have been published demonstrating efficacy using mold and cockroach allergens.53
In addition, several studies have found that children with allergic rhinitis who receive allergen immunotherapy are significantly less likely to develop asthma.65–67 Immunotherapy has also been associated with a statistically significant reduction in future sensitization to other aeroallergens.68,69
Risk of systemic reaction from allergen immunotherapy
The decision to begin allergen immunotherapy should be individualized on the basis of symptom severity, relative benefit compared with drug therapy, and whether comorbid conditions such as cardiovascular disease or beta-blocker exposure are present. These comorbid conditions are associated with heightened risk of (more serious) anaphylaxis—the major hazard of allergen immunotherapy.70 Systemic reactions during allergen immunotherapy occur at a rate of approximately 3 to 5 per 1,000 injections; for this reason, allergen immunotherapy should only be administered in a medical facility where personnel, supplies, and equipment are available to treat anaphylaxis.5
- National Heart, Lung, and Blood institute, National Asthma education and Prevention Program. Expert Panel Report 3: guidelines for the diagnosis and management of asthma. www.nhlbi.nih.gov/guidelines/asthma. Accessed 8/7/08.
- Expert Panel Report 2: Guidelines for the diagnosis and management of asthma. U.S. Department of Health and Human Services. Publication No. 97-4051; 1997.
- Expert Panel Report: Guidelines for the diagnosis and management of asthma. Update on Selected Topics—2002. J Allergy Clin Immunol 2002; 110:S141–S207.
- FitzGerald JM, Boulet LP, McIvor RA, Zimmerman S, Chapman KR. Asthma control in Canada remains suboptimal: the Reality of Asthma Control (TRAC) study. Can Respir J 2006; 13:253–259.
- Braganza S, Sharif I, Ozuah P. Documenting asthma severity: do we get it right? J Asthma 2003; 40:661–665.
- Cockcroft DW, Swystun VA. Asthma control versus asthma severity. J Allergy Clin Immunol 1996; 98:1016–1018.
- Peters SP, Jones CA, Haselkorn T, Mink DR, Valacer DJ, Weiss ST. Real-world Evaluation of Asthma Control and Treatment (REACT): findings from a national Web-based survey. J Allergy Clin Immunol. 2007; 119:1454–1461.
- Osborne ML, Vollmer WM, Pedula KL, Wilkins J, Buist AS, O’Hollaren M. Lack of correlation of symptoms with specialist-assessed long-term asthma severity. Chest 1999; 115:85–91.
- Li JT, Oppenheimer J, Bernstein IL, et al. Attaining optimal asthma control: a practice parameter. J Allergy Clin Immunol 2005; 116:S3–S11.
- Nathan RA, Sorkness C, Kosinski M, et al. Development of the Asthma Control Test: a survey for assessing asthma control. J Allergy Clin Immunol 2004; 113:59–65.
- Schatz M, Zeiger RS, Drane A, et al. Reliability and predictive validity of the Asthma Control Test administered by telephone calls using speech recognition technology. J Allergy Clin Immunol 2007; 119:336–343.
- Peters D, Chen C, Markson LE, Allen-Ramey FC, Vollmer WM. Using an asthma control questionnaire and administrative data to predict healthcare utilization. Chest 2006; 129:918–924.
- Schatz M, Sorkness C, Li JT, et al. Asthma Control Test: reliability, validity, and responsiveness in patients not previously followed by asthma specialists. J Allergy Clin Immunol 2006; 117:549–556.
- Bateman E, Boushey H, Bousquet J, et al. Can guideline-defined asthma control be achieved? Am J Respir Crit Care Med 2004; 170:836–844.
- Davies TJ, Bunn WB, Fromer L, Gelfand EW, Colice GL. A focus on the asthma HEDIS measure and its implications for clinical practice. Manag Care Interface 2006; 19:29–36.
- Rubinfeld AR, Pain MC. Perception of asthma. Lancet 1976; 1:882–884.
- Teeter J, Bleecker E. Relationship between airway obstruction and respiratory symptoms in adult asthmatics. Chest 1998; 113:272–277.
- Shingo S, Zhang J, Reiss T. Correlation of airway obstruction and patient reported endpoints in clinical studies. Eur Resp J 2001; 17:220–224.
- Juniper EF, Bousquet J, Abetz L, Bateman ED; GOAL Committee. Identifying ‘well-controlled’ and ‘not well-controlled’ asthma using the Asthma Control Questionnaire. Respir Med 2006; 100:616–621.
- Nelson H, Weiss S, Bleecker E, Yancey S, Dorinsky P. The Salmeterol Multicenter Asthma Research Trial. Chest 2006; 129:15–26.
- Wechsler M, Lehman E, Lazarus S, et al. ß-Adrenergic receptor polymorphisms and response to salmeterol. Am J Respir Crit Care Med 2006; 173:519–526.
- Palmer CNA, Lipworth BJ, Lee S, Ismail T, MacGregor DF, Mukhopadhyay S. Arginine-16 beta-2 adrenoceptor genotype predisposes to exacerbations in young asthmatics taking regular salmeterol. Thorax 2006; 61:940–944.
- Taylor DR, Drazen JM, Herbison GP, Yandava CN, Hancox RJ, Town GI. Asthma exacerbations during long term beta agonist use: influence of beta 2 adrenoceptor polymorphism. Thorax 2000; 55:762–727.
- Bleecker E, Postma D, Lawrance R, Meyers D, Ambrose H, Goldman M. Effect of ADRB2 polymorphisms on response to long-acting beta2-agonist therapy: a pharmacogenetic analysis of two randomized studies. Lancet 2007; 370:2118–2125.
- Bleecker E, Yancey S, Baitinger L, et al. Salmeterol response is not affected by beta-2 adrenergic receptor genotype in subjects with persistent asthma. J Allergy Clin Immunol 2006; 118:809–816.
- Nelson H, Bleecker E, Corren J, et al. Characterization of asthma exacerbations by Arg16Gly genotype in subjects with asthma receiving salmeterol alone or with fluticasone propionate. J Allergy Clin Immunol 2008; 121:S131.
- O’Byrne P, Barnes P, Rodriguez-Roisin R, et al. Low dose Inhaled budesonide and formoterol in mild persistent asthma. The OPTIMA Randomized Trial. Am J Respir Crit Care Med 2001; 164:1392–1397.
- Greening AP, Ind PW, Northfield M, Shaw G. Added salmeterol versus higher dose corticosteroid in asthma patients with symptoms on existing inhaled corticosteroid. Lancet 1994; 344:219–224.
- Woolcock A, Lundback B, Ringdal N, Jacques LA. Comparison of addition of salmeterol to inhaled steroids with doubling of the dose of inhaled steroids. Am J Respir Crit Care Med 1996; 153:1481–1488.
- Walters EH, Walters JAE, Gibson MDP. Long-acting beta2-agonists for stable chronic asthma. Cochrane Database Syst Rev 2003; (3):CD001385. doi:10.1002/14651858.CD001385.
- Masoli M, Weatherall M, Holt S, Beasley R. Moderate dose inhaled corticosteroids plus salmeterol versus higher doses of inhaled corticosteroid in symptomatic asthma. Thorax 2005; 60:730–734.
- Sin DD, Man J, Sharpe H, Gan WQ, Man SFP. Pharmacological management to reduce exacerbations in adults with asthma. A systematic review and meta-analysis. JAMA 2004; 292:367–376.
- Mann RD, Kubota K, Pearce G, Wilton L. Salmeterol: a study by prescription event monitoring in a UK cohort of 15,407 patients. J Clin Epidemiol 1996; 49:247–250.
- Lanes S, Lanza L, Wentworth C. Risk of emergency care, hospitalization, and ICU stays for acute asthma among recipients of salmeterol. Am J Respir Crit Care Med 1998; 158:857–861.
- Meier CR, Jick H. Drug use and pulmonary death rates in increasingly symptomatic asthma patients in the UK. Thorax 1997; 52:612–617.
- Williams C, Crossland L, Finnerty J, et al. A case control study of salmeterol and near-fatal attacks of asthma. Thorax 1998; 53:7–13.
- Lanes S, Garcia Rodriguez LA, Herta C. Respiratory medications and risk of asthma death. Thorax 2002; 57:683–686.
- Anderson HR, Ayres JG, Sturdy PM, et al. Bronchodilator treatment and deaths from asthma: case control study. Br Med J 2005; 330:117–124.
- Martinez FD. Safety of long-acting beta agonists—an urgent need to clear the air. N Engl J Med 2005; 353:2637–2639.
- Nelson HS. Long-acting beta-agonists in adult asthma: evidence that these drugs are safe. Prim Care Respir J 2006; 15:271–277.
- Lang DM. The long-acting beta agonist controversy: a critical examination of the evidence. Cleve Clin J Med 2006; 73:973–992.
- Rambasek T, Lang DM, Kavuru M. Omalizumab: where does it fit in current asthma management? Cleve Clin J Med 2004; 71:251–261.
- McGlashan D, Bochner B, Adelman D, et al. Down regulation of Fc(epsilon)RI expression on human basophils during in vivo treatment of atopic patients with anti-IgE antibody. J Immunol 1997; 158:1438–1445.
- Busse W, Corren J, Lanier B, et al. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol 2001; 108:184–190.
- Soler M, Matz J, Townley R, et al. The anti-IgE antibody omalizumab reduces asthma exacerbations and steroid requirement in allergic asthmatics. Eur Respir J 2001; 18:254–261.
- Humbert M, Beasley R, Ayres J, et al. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy 2005; 60:309–316.
- Van Ganse E, Antonicelli L, Zhang Q, et al. Asthma-related resource use and cost by GINA classification of severity in three European countries. Respir Med 2006; 100:140–147.
- Godard P, Chanez P, Siraudin L, Nicoloyannis N, Duru G. Costs of asthma are correlated with severity: a 1-yr prospective study. Eur Respir J 2002; 19:61–67.
- Cisternas MG, Blanc PH, Yen IH, et al. A comprehensive study of the direct and indirect costs of adult asthma. J Allergy Clin Immunol 2003; 111:1212–1218.
- Sullivan S, Turk F. An evaluation of the cost effectiveness of omalizumab for the treatment of severe persistent asthma. Allergy 2008; 63:670–684.
- US Food and Drug Administration. Omalizumab (marketed as Xolair) information. www.fda.gov/cder/drug/infopage/omalizumab/default.htm. Accessed August 31, 2007.
- Williams SG, Schmidt DK, Redd SC, Storms W. Key clinical activities for quality asthma care. Recommendations of the National Asthma Education and Prevention Program. MMWR Recomm Rep 2003; 52 RR-6:1–8.
- Cox L, Li J, Nelson H, Lockey R, et al. Allergy Immunotherapy: a practice parameter second update. J Allergy Clin Immunol 2007; 120:S25–S85.
- Akdis M, Akdis CA. Mechanisms of allergen-specific immunotherapy. J Allergy Clin Immunol 2007; 119:780–789.
- Murray AB, Milner RA. The accuracy of features in the clinical history for predicting atopic sensitization to airborne allergens in children. J Allergy Clin Immunol 1995; 96:588–596.
- Bernstein IL, Li JT, Bernstein DI, et al. Allergy diagnostic testing: an updated practice parameter. Ann Allergy Asthma Immunol 2008; 100 suppl 3:1S–148S.
- Walker S, Pajno GB, Lima MT, Wilson DR, Durham SR. Grass pollen immunotherapy for seasonal rhinitis and asthma: a randomized, controlled trial. J Allergy Clin Immunol 2001; 107:87–93.
- Varney VA, Edwards J, Tabbah K, Brewster H, Mavroleon G, Frew AJ. Clinical efficacy of specific immunotherapy to cat dander: a double-blind placebo-controlled trial. Clin Exp Allergy 1997; 27:860–867.
- Cantani A, Arcese G, Lucenti P, Gagliesi D, Bartolucci M. A three-year prospective study of specific immunotherapy to inhalant allergens: evidence of safety and efficacy in 300 children with allergic asthma. J Investig Allergol Clin Immunol 1997; 7:90–97.
- Hedlin G, Wille S, Browaldh L, et al. Immunotherapy in children with allergic asthma: effect on bronchial hyperreactivity and pharmacotherapy. J Allergy Clin Immunol 1999; 103:609–614.
- Arvidsson MB, Löwhagen O, Rak S. Allergen specific immunotherapy attenuates early and late phase reactions in lower airways of birch pollen asthmatic patients: a double blind placebo-controlled study. Allergy 2004; 59:74–80.
- Pichler CE, Helbling A, Pichler WJ. Three years of specific immunotherapy with house-dust-mite extracts in patients with rhinitis and asthma: significant improvement of allergen-specific parameters and of nonspecific bronchial hyperreactivity. Allergy 2001; 56:301–306.
- Mirone C, Albert F, Tosi A, et al. Efficacy and safety of subcutaneous immunotherapy with a biologically standardized extract of Ambrosia artemisiifolia pollen: a double-blind, placebo-controlled study. Clin Exp Allergy 2004; 34:1408–1414.
- Abramson MJ, Puy RM, Weiner JM. Allergen immunotherapy for asthma. Cochrane Database Syst Rev 2003; (4):CD001186.
- Jacobsen L. Preventive aspects of immunotherapy: prevention for children at risk of developing asthma. Ann Allergy Asthma Immunol 2001; 87:43–46.
- Moller C, Dreborg S, Ferdousi HA, et al. Pollen immunotherapy reduces the development of asthma in children with seasonal rhinoconjunctivitis (the PAT study). J Allergy Clin Immunol 2002; 109:251–256.
- Niggemann B, Jacobsen L, Dreborg S, et al; PAT Investigator Group. Five year follow-up on the PAT study: specific immunotherapy and long-term prevention of asthma in children. Allergy 2006: 61:855–859.
- Des Roches A, Paradis L, Menardo JL, et al. Immunotherapy with a standardized Dermatophagoides pteronyssinus extract VI: specific immunotherapy prevents the onset of new sensitizations in children. J Allergy Clin Immunol 1997; 99:450–453.
- Pajno GB, Barberio G, DeLuca F, et al. Prevention of new sensitizations in asthmatic children monosensitized to the house dust mite by specific immunotherapy: a six year follow up study. Clin Exp Allergy 2001; 31:1392–1397.
- Lang DM. Do beta blockers really enhance the risk of anaphylaxis during immunotherapy? Curr Allergy Asthma Rep 2008; 8:37–44.
This review focuses on several elements in the National Asthma Education and Prevention Program’s new guidelines, the third Expert Panel Report (EPR3),1 that differ substantially from those in EPR2,2 issued in 1997 and updated in 2002.3 These differences in approach to the management of asthma described in EPR3 offer a clear potential for reducing the gap between optimal asthma care outcomes as described in guidelines and normative asthma care outcomes in the “real world.”
GREATER EMPHASIS ON CONTROL
The EPR2 guidelines2 recommended that asthma management be carried out in an algorithmic manner. Patients were classified into four severity categories: mild intermittent, mild persistent, moderate persistent, and severe persistent asthma, based on assessment of the level of symptoms (day/night), reliance on “reliever” medication, and lung function at the time of presentation. Pharmacologic management was then assigned according to each respective categorization in an evidence-based fashion.
In an ideal world, this would result in patients with asthma receiving appropriate pharmacotherapeutic agents associated with favorable asthma care outcomes, which were also advantageous from both cost- and risk-benefit standpoints. In the real world, however, this paradigm was flawed, as it relied on accurate categorization of patients in order for pharmacotherapy to be prescribed appropriately. Both providers and patients are prone to underestimate asthma severity,4,5 and for this reason many patients managed on the basis of this paradigm were undertreated.
A new paradigm, based on the assessment of asthma control, has been encouraged in the EPR3 guidelines.1
Severity and control are not synonymous
More than a decade ago, Cockroft and Swystun6 pointed out that asthma control (or lack thereof) is often used inappropriately to define asthma severity: ie, well-controlled asthma is seen as synonymous with mild asthma, and poorly controlled asthma with severe asthma.
Asthma severity can be defined as the intrinsic intensity of the disease process, while asthma control is the degree to which the manifestations of asthma are minimized. Asthma severity is clearly a determinant of asthma control, but its impact is affected by a variety of factors, including but not limited to:
- Whether appropriate medication is prescribed
- Patterns of therapeutic adherence
- The degree to which recommended measures for avoiding for clinically relevant aeroallergens are pursued.
Health care utilization, including hospitalizations and emergency department visits, correlates more closely with asthma control than with asthma severity.7–9 Indeed, a patient with severe persistent asthma who is treated appropriately with multiple “controller” medications and who takes his or her medications and avoids allergens as directed can achieve well-controlled or totally controlled asthma, and is not likely to require hospitalization or emergency department management, to miss school or work, or to experience nocturnal awakening or limitation in routine activities due to asthma. This patient has severe persistent asthma that is well controlled.
In contrast, a patient with mild or moderate persistent asthma who does not receive appropriate instructions for avoiding allergens or taking controller medication regularly or who is poorly adherent will likely have poor asthma control. This patient is more likely to require hospitalization or emergency department management, to miss school or work, and to experience nocturnal awakening or limitation in routine activities due to asthma. This patient has mild persistent asthma that is poorly controlled.
Assess asthma severity in the first visit, and control in subsequent visits
How to assess severity
How to measure control
For all patients with asthma, regardless of severity, the goal is the same: to achieve control by reducing both impairment and risk. Asthma is classified as well controlled, not well controlled, or poorly controlled (Table 2).1
Validated tests are available to assess control
Serial testing as a quality indicator
Serial ACT scores give an objective measure of the degree to which the goals of management1 are being achieved, and in so doing can encourage optimal outcomes.14
Another use of these tests is to document whether asthma control improves over time when patients receive care from a particular physician or group. This use may become increasingly important in view of efforts underway to implement a pay-for-performance model for asthma care, in which providers will be financially rewarded for improved patient care outcomes and adherence to standards of practice based on Health Plan Employer Data and Information Set measures.15
We have used the ACT in the Section of Allergy/Immunology at Cleveland Clinic for 3 years on a routine basis. All patients with asthma being seen either for the first time or as follow-up complete the ACT, which has been entered in a flow sheet in our electronic medical record, at the same time they undergo spirometry. We have shown that care in the Section of Allergy/Immunology is associated with improvement in asthma control over time, in patients who have completed serial ACT measurements at initial visits and at follow-up visits (Figure 2).
Objective measurement of lung function is also important
Serial monitoring of lung function at every patient visit with spirometry is also important, as some patients may be “poor perceivers,”16 ie, they may have little or no subjective awareness of moderate or even severe ventilatory impairment. A number of studies17,18 support the contention that symptoms and lung function are separate and independent dimensions of asthma control, and that both of them need to be assessed.
Responding to changes in control
THE STEP 3 CONTROVERSY
Salmeterol Multicenter Asthma Research Trial
In the Salmeterol Multicenter Asthma Research Trial (SMART), patients randomized to the long-acting beta agonist (LABA) salmeterol (Serevent)—particularly African Americans—had a statistically significant increase in the risk of untoward asthma care outcomes.20
SMART was launched in 1996. Patients were randomized in a double-blind fashion to receive either salmeterol 42 μg twice a day or placebo in addition to their usual asthma therapy for 28 weeks. The rate of the primary outcome (respiratory-related deaths or life-threatening experiences) was not significantly different with salmeterol than with placebo (relative risk [RR] = 1.40, 95% confidence interval [CI] 0.91–2.14). However, in 2003, the study was halted prematurely because of difficulty enrolling the targeted number of 60,000 patients, and an interim analysis that revealed significantly higher rates of secondary outcomes in subjects randomized to salmeterol. Compared with the placebo group, the salmeterol group had significantly higher rates of respiratory-related deaths (RR 2.16, 95% CI 1.06–4.41), asthma-related deaths (RR = 4.37, 95% CI = 1.25–15.34), and combined asthma-related deaths or life-threatening experiences (RR = 1.71, 95% CI 1.01–2.89). There were 13 asthma-related deaths and 37 combined asthma-related deaths or life-threatening experiences in the salmeterol group, compared with 3 and 22, respectively, in the placebo group. Of the 16 asthma deaths in the study, 13 (81%) occurred in the initial phase of SMART, when patients were recruited via print, radio, and television advertising; afterward, patients were recruited directly by investigators.
Statistically significant differences in outcomes occurred primarily in African Americans. African Americans who received salmeterol had higher rates of respiratory death or life-threatening experiences (RR = 4.10, 95% CI 1.54–10.90), the primary end point for the study, as well as higher rates of combined asthma-related deaths or life-threatening experiences (RR = 10.46, 95% CI 1.34–81.58), a secondary end point. No statistically significant differences were observed in white patients randomized to salmeterol with respect to the primary end point (RR = 1.05, 95% = 0.62–1.76); the secondary end point of combined asthma-related deaths or life-threatening experiences (RR = 1.08, 95% CI 0.55–2.14); or other end points.
Medication exposures were not tracked during the study, and allocation to inhaled corticosteroids combined with salmeterol was not randomized, so the effect of concomitant inhaled corticosteroid use cannot be determined from these data.
As a result of SMART, medications that contain either of the two LABAs, salmeterol or formoterol (Foradil), carry a black-box warning.
LABAs: Risks and benefits
Two studies21,22 have suggested that asthmatic patients who are homozygous for Arg/Arg at codon 16 of the beta-2 adrenergic receptor are predisposed to untoward asthma outcomes with regular exposure to LABAs. However, other data23–25 do not support the contention that B16 Arg/Arg patients experience adverse asthma outcomes with LABA exposure. In two recently published studies, no difference in rates of exacerbations, severe exacerbations, lung function, frequency of reliance on SABA, or nocturnal awakenings was observed in patients receiving formoterol combined with budesonide24 or salmeterol combined with fluticasone25 according to genotype. A prospective study26 also found no statistically significant difference in exacerbation rates according to beta adrenergic receptor genotype in individuals randomized to LABA monotherapy, or LABA combined with inhaled corticosteroids.
The updated EPR2 asthma guidelines,3 published in November 2002, stipulated that LABAs were the preferred controller agent to “add on” to low-dose inhaled corticosteroids for patients with moderate persistent asthma, and that the combination of low-dose inhaled corticosteroids and LABA was associated with superior outcomes: reduction of symptoms, including nocturnal awakening, increase in lung function, improvement in health-related quality of life, decreased use of “rescue” medication, and reduced rate of exacerbations and severe exacerbations, compared with higher-dose inhaled corticosteroid monotherapy. This management recommendation was categorized as level A, on the basis of data from multiple randomized, controlled, double-blinded trials.27–29 Additional evidence14,30 and data from two meta-analyses31,32 have provided further support for this recommendation, while no evidence linking LABA exposure to risk for fatal or near-fatal asthma has been found in cohort or case-control studies.33–38
Based on safety concerns, the EPR3 guidelines1 recommend that medium-dose inhaled corticosteroids be regarded as equivalent to adding LABAs to low-dose inhaled corticosteroids, and state: “the established, beneficial effects of LABA for the great majority of patients whose asthma is not well controlled with [inhaled corticosteroids] alone should be weighed against the increased risk for severe exacerbations, although uncommon, associated with daily use of LABA.”1
There is currently an honest difference of opinion39,40 among asthma specialists as to how this management recommendation for moderate persistent asthma—now depicted at “step 3” in the EPR3 guidelines (Table 4)—should be implemented. The LABA controversy was reviewed previously in the Cleveland Clinic Journal of Medicine.41
THE ROLE OF OMALIZUMAB: WEIGHING COST VS BENEFIT
The 2002 update to the EPR2 guidelines3 was issued before omalizumab (Xolair) was approved in June 2003.
Patients with severe persistent asthma are categorized in steps 5 or 6 in the EPR3 guidelines (Table 5).1 Preferred management for these patients includes inhaled corticosteroids in high doses combined with long-acting beta agonists and, for step 6 patients, oral corticosteroids.
Omalizumab was approved for management of patients with moderate or severe persistent asthma who are not achieving the goals of asthma management on inhaled corticosteroids, who exhibit a wheal-flare reaction to a perennial allergen, and whose immunoglobulin E (IgE) level is in the range of 30 to 700 IU/mL.42 Omalizumab dosing is based on the serum IgE level and on body weight.
Omalizumab, an anti-IgE monoclonal antibody
Omalizumab is a recombinant, humanized, monoclonal anti-IgE antibody that binds to IgE at the same Fc site as the high-affinity IgE receptor. Its primary mechanism of action is the binding of free IgE in the circulation, forming biologically inert, small complexes that do not activate complement and are cleared by the reticuloendothelial system.42 Its secondary mechanism of action entails a reduction in the number of high-affinity receptors on basophils, from approximately 220,000 to 8,300 receptors per cell. The latter effect was associated with a 90% reduction in histamine release from basophils in response to ex vivo challenge with dust mite allergen.43
Benefit in randomized trials
Omalizumab has been associated with statistically and clinically significant benefit in randomized, double-blind, placebo-controlled trials.44,45
Humbert et al46 randomized 419 patients whose asthma was not adequately controlled on high-dose inhaled corticosteroids and long-acting beta agonists, who were 12 to 75 years old, with reduced lung function and a history of recent asthma exacerbation, to treatment with omalizumab or placebo. Omalizumab was associated with a statistically significant reduction in the rate of asthma exacerbations and severe asthma exacerbations, as well as statistically significant improvements in asthma-related quality of life, morning peak expiratory flow rate, and asthma symptom scores.
These data support the recommendation in EPR3 to consider a trial of omalizumab in properly selected patients with severe, persistent allergic asthma.
Omalizumab is cost-beneficial in properly selected patients
The current wholesale acquisition cost of omalizumab is $532 for one 150-mg vial (David Zito, personal communication). The cost of treatment varies based on body weight and IgE level but may range from a wholesale cost of $6,388 to $38,326 per year.
However, as asthma severity increases, both direct and indirect medical expenditures increase substantially.47,48 Annual costs are approximately four times higher for severe asthma compared with mild asthma49; not only are treatment and exacerbation costs higher, but indirect costs are also disproportionately greater. Annual costs for severe asthma are significantly greater if the disease is inadequately controlled.50 For these reasons, an intervention that leads to improved outcomes for severe, poorly controlled asthma carries the potential for the greatest cost-utility for society, as it can lower direct costs by reducing the frequency and severity of exacerbations, in addition to reducing indirect medical expenditures on the basis of increased productivity and fewer days of missed work or school. The cost of omalizumab in quality-adjusted life years compares favorably with that of biologicals used in managing rheumatoid arthritis, Crohn disease, and multiple sclerosis.50
Adverse effects of omalizumab
In pivotal trials,43,44 omalizumab was associated with a substantial rate of local reactions. The rate of anaphylaxis was slightly less than 1 in 1,000, and this has been confirmed by surveillance data recorded since approval of the drug in 2003. Based on the observed risk of anaphylaxis, in July 2007, the US Food and Drug Administration added a black-box warning to the omalizumab label and stipulated that a medication guide should be provided for patients.51 The warning indicates that health care providers administering omalizumab should be prepared to manage anaphylaxis and that patients should be closely observed for an appropriate period after omalizumab administration.
The package insert also describes a numerical, but not statistically significant, increase in the rate of malignancy in patients receiving omalizumab.42 Malignancy developed in 0.5% of patients receiving omalizumab, compared with 0.2% of patients who received placebo. Because these malignancies were diagnosed over a shorter period than the time required for oncogenesis (ie, 6 months in 60% of cases), and because a heterogeneous variety of tumors was observed, there is reason to doubt these tumors were causally associated with omalizumab.
Postmarketing surveillance studies are in progress that will provide more definitive data on the potential relationship between malignancy and omalizumab exposure.
Omalizumab: Guideline recommendations
The EPR3 guidelines1 state that omalizumab is the only adjunctive therapy to demonstrate efficacy when added to high-dose inhaled corticosteroids plus long-acting beta agonists in patients with severe, persistent, allergic asthma and that evidence does not support use of the following agents, which in some cases are approved for managing other conditions and have been advocated for management of severe, refractory asthma: methotrexate, soluble interleukin (IL)-4 receptor, anti-IL-5, anti-IL-12, cyclosporine A, intravenous immune globulin, gold, troleandomycin, and colchicine. The data supporting use of macrolides were characterized as “encouraging but insufficient to support a recommendation.”
The strength of evidence for the use of omalizumab for patients in steps 5 and 6 who fulfill the criteria for its use (see above) was classified in the EPR3 guidelines1 as category B. The guidelines also say that omalizumab may be considered for adjunctive therapy in properly selected patients in step 4, as a means to avoid higher doses of inhaled corticosteroids, but that additional studies are needed to establish its utility for such patients. This recommendation was classified as category D because of the lack of published comparator trials.
ALLERGEN IMMUNOTHERAPY FOR PATIENTS WITH ASTHMA
Many patients with asthma have clinically relevant, IgE-mediated (allergic) potential to inhaled allergens.1 For patients with persistent asthma (steps 2–6 in Table 5), allergic reactions can contribute to airway inflammation, provoke symptoms, and lead to more use of medications. For this reason, identification and management of clinically relevant allergy merits consideration.52
The EPR3 guidelines1 recommend considering allergen immunotherapy for patients with mild or moderate persistent asthma (steps 2–4) who have a clinically relevant component of allergy to inhaled substances.
Changing the immune response
Allergen immunotherapy entails the incremental administration of inhalant allergens by subcutaneous injection for the purpose of inducing immune system changes in the host response. The goal of immunotherapy is to protect against allergic reactions that can be expected to occur with ongoing exposure to clinically relevant allergens.53
The immunologic changes that develop with allergen immunotherapy are complex.53,54 Successful immunotherapy results in generation of a population of CD4+/CD25+ T lymphocytes producing IL-10, transforming growth factor beta, or both. Allergen immunotherapy has been shown to block the immediate- and late-phase allergic response; to decrease recruitment of mast cells, basophils, and eosinophils on provocation or natural exposure to allergens in the skin, nose, eye, and bronchial mucosa; to blunt the seasonal rise in specific IgE; and to suppress late-phase inflammatory responses in the skin and respiratory tract. However, the efficacy of immunotherapy in relation to these immunologic changes is not completely understood.54
Many patients need skin testing
Allergen immunotherapy may be considered for patients with asthma for whom a clear relationship exists between symptoms and exposure to an allergen to which the patient is sensitive.53 Because it is often not possible to determine whether a patient is sensitive to a perennial indoor allergen (eg, dust mite) on the basis of the medical history alone,55 many patients with asthma benefit from immediate hypersensitivity skin testing to objectively assess or rule out allergy to common inhalants. In certain situations, in vitro testing may be performed, but skin testing has a higher negative predictive value and is recommended as a better screening test.56
Benefits of allergen immunotherapy
Numerous randomized, double-blind, placebo-controlled trials have shown that allergen immunotherapy is associated with benefit for reducing symptoms and medication reliance.57–63
A meta-analysis of 75 randomized, placebo-controlled studies confirmed the effectiveness of immunotherapy in asthma, with a significant reduction in asthma symptoms and medication use and with improvement in bronchial hyperreactivity.64 This meta-analysis included 36 trials of dust mite allergen, 20 of pollen, and 10 of animal dander. Immunotherapy is efficacious for pollen, mold, dust mite, cockroach, and animal allergens; however, its effectiveness is more established for dust mite, animal dander, and pollen allergens, as fewer studies have been published demonstrating efficacy using mold and cockroach allergens.53
In addition, several studies have found that children with allergic rhinitis who receive allergen immunotherapy are significantly less likely to develop asthma.65–67 Immunotherapy has also been associated with a statistically significant reduction in future sensitization to other aeroallergens.68,69
Risk of systemic reaction from allergen immunotherapy
The decision to begin allergen immunotherapy should be individualized on the basis of symptom severity, relative benefit compared with drug therapy, and whether comorbid conditions such as cardiovascular disease or beta-blocker exposure are present. These comorbid conditions are associated with heightened risk of (more serious) anaphylaxis—the major hazard of allergen immunotherapy.70 Systemic reactions during allergen immunotherapy occur at a rate of approximately 3 to 5 per 1,000 injections; for this reason, allergen immunotherapy should only be administered in a medical facility where personnel, supplies, and equipment are available to treat anaphylaxis.5
This review focuses on several elements in the National Asthma Education and Prevention Program’s new guidelines, the third Expert Panel Report (EPR3),1 that differ substantially from those in EPR2,2 issued in 1997 and updated in 2002.3 These differences in approach to the management of asthma described in EPR3 offer a clear potential for reducing the gap between optimal asthma care outcomes as described in guidelines and normative asthma care outcomes in the “real world.”
GREATER EMPHASIS ON CONTROL
The EPR2 guidelines2 recommended that asthma management be carried out in an algorithmic manner. Patients were classified into four severity categories: mild intermittent, mild persistent, moderate persistent, and severe persistent asthma, based on assessment of the level of symptoms (day/night), reliance on “reliever” medication, and lung function at the time of presentation. Pharmacologic management was then assigned according to each respective categorization in an evidence-based fashion.
In an ideal world, this would result in patients with asthma receiving appropriate pharmacotherapeutic agents associated with favorable asthma care outcomes, which were also advantageous from both cost- and risk-benefit standpoints. In the real world, however, this paradigm was flawed, as it relied on accurate categorization of patients in order for pharmacotherapy to be prescribed appropriately. Both providers and patients are prone to underestimate asthma severity,4,5 and for this reason many patients managed on the basis of this paradigm were undertreated.
A new paradigm, based on the assessment of asthma control, has been encouraged in the EPR3 guidelines.1
Severity and control are not synonymous
More than a decade ago, Cockroft and Swystun6 pointed out that asthma control (or lack thereof) is often used inappropriately to define asthma severity: ie, well-controlled asthma is seen as synonymous with mild asthma, and poorly controlled asthma with severe asthma.
Asthma severity can be defined as the intrinsic intensity of the disease process, while asthma control is the degree to which the manifestations of asthma are minimized. Asthma severity is clearly a determinant of asthma control, but its impact is affected by a variety of factors, including but not limited to:
- Whether appropriate medication is prescribed
- Patterns of therapeutic adherence
- The degree to which recommended measures for avoiding for clinically relevant aeroallergens are pursued.
Health care utilization, including hospitalizations and emergency department visits, correlates more closely with asthma control than with asthma severity.7–9 Indeed, a patient with severe persistent asthma who is treated appropriately with multiple “controller” medications and who takes his or her medications and avoids allergens as directed can achieve well-controlled or totally controlled asthma, and is not likely to require hospitalization or emergency department management, to miss school or work, or to experience nocturnal awakening or limitation in routine activities due to asthma. This patient has severe persistent asthma that is well controlled.
In contrast, a patient with mild or moderate persistent asthma who does not receive appropriate instructions for avoiding allergens or taking controller medication regularly or who is poorly adherent will likely have poor asthma control. This patient is more likely to require hospitalization or emergency department management, to miss school or work, and to experience nocturnal awakening or limitation in routine activities due to asthma. This patient has mild persistent asthma that is poorly controlled.
Assess asthma severity in the first visit, and control in subsequent visits
How to assess severity
How to measure control
For all patients with asthma, regardless of severity, the goal is the same: to achieve control by reducing both impairment and risk. Asthma is classified as well controlled, not well controlled, or poorly controlled (Table 2).1
Validated tests are available to assess control
Serial testing as a quality indicator
Serial ACT scores give an objective measure of the degree to which the goals of management1 are being achieved, and in so doing can encourage optimal outcomes.14
Another use of these tests is to document whether asthma control improves over time when patients receive care from a particular physician or group. This use may become increasingly important in view of efforts underway to implement a pay-for-performance model for asthma care, in which providers will be financially rewarded for improved patient care outcomes and adherence to standards of practice based on Health Plan Employer Data and Information Set measures.15
We have used the ACT in the Section of Allergy/Immunology at Cleveland Clinic for 3 years on a routine basis. All patients with asthma being seen either for the first time or as follow-up complete the ACT, which has been entered in a flow sheet in our electronic medical record, at the same time they undergo spirometry. We have shown that care in the Section of Allergy/Immunology is associated with improvement in asthma control over time, in patients who have completed serial ACT measurements at initial visits and at follow-up visits (Figure 2).
Objective measurement of lung function is also important
Serial monitoring of lung function at every patient visit with spirometry is also important, as some patients may be “poor perceivers,”16 ie, they may have little or no subjective awareness of moderate or even severe ventilatory impairment. A number of studies17,18 support the contention that symptoms and lung function are separate and independent dimensions of asthma control, and that both of them need to be assessed.
Responding to changes in control
THE STEP 3 CONTROVERSY
Salmeterol Multicenter Asthma Research Trial
In the Salmeterol Multicenter Asthma Research Trial (SMART), patients randomized to the long-acting beta agonist (LABA) salmeterol (Serevent)—particularly African Americans—had a statistically significant increase in the risk of untoward asthma care outcomes.20
SMART was launched in 1996. Patients were randomized in a double-blind fashion to receive either salmeterol 42 μg twice a day or placebo in addition to their usual asthma therapy for 28 weeks. The rate of the primary outcome (respiratory-related deaths or life-threatening experiences) was not significantly different with salmeterol than with placebo (relative risk [RR] = 1.40, 95% confidence interval [CI] 0.91–2.14). However, in 2003, the study was halted prematurely because of difficulty enrolling the targeted number of 60,000 patients, and an interim analysis that revealed significantly higher rates of secondary outcomes in subjects randomized to salmeterol. Compared with the placebo group, the salmeterol group had significantly higher rates of respiratory-related deaths (RR 2.16, 95% CI 1.06–4.41), asthma-related deaths (RR = 4.37, 95% CI = 1.25–15.34), and combined asthma-related deaths or life-threatening experiences (RR = 1.71, 95% CI 1.01–2.89). There were 13 asthma-related deaths and 37 combined asthma-related deaths or life-threatening experiences in the salmeterol group, compared with 3 and 22, respectively, in the placebo group. Of the 16 asthma deaths in the study, 13 (81%) occurred in the initial phase of SMART, when patients were recruited via print, radio, and television advertising; afterward, patients were recruited directly by investigators.
Statistically significant differences in outcomes occurred primarily in African Americans. African Americans who received salmeterol had higher rates of respiratory death or life-threatening experiences (RR = 4.10, 95% CI 1.54–10.90), the primary end point for the study, as well as higher rates of combined asthma-related deaths or life-threatening experiences (RR = 10.46, 95% CI 1.34–81.58), a secondary end point. No statistically significant differences were observed in white patients randomized to salmeterol with respect to the primary end point (RR = 1.05, 95% = 0.62–1.76); the secondary end point of combined asthma-related deaths or life-threatening experiences (RR = 1.08, 95% CI 0.55–2.14); or other end points.
Medication exposures were not tracked during the study, and allocation to inhaled corticosteroids combined with salmeterol was not randomized, so the effect of concomitant inhaled corticosteroid use cannot be determined from these data.
As a result of SMART, medications that contain either of the two LABAs, salmeterol or formoterol (Foradil), carry a black-box warning.
LABAs: Risks and benefits
Two studies21,22 have suggested that asthmatic patients who are homozygous for Arg/Arg at codon 16 of the beta-2 adrenergic receptor are predisposed to untoward asthma outcomes with regular exposure to LABAs. However, other data23–25 do not support the contention that B16 Arg/Arg patients experience adverse asthma outcomes with LABA exposure. In two recently published studies, no difference in rates of exacerbations, severe exacerbations, lung function, frequency of reliance on SABA, or nocturnal awakenings was observed in patients receiving formoterol combined with budesonide24 or salmeterol combined with fluticasone25 according to genotype. A prospective study26 also found no statistically significant difference in exacerbation rates according to beta adrenergic receptor genotype in individuals randomized to LABA monotherapy, or LABA combined with inhaled corticosteroids.
The updated EPR2 asthma guidelines,3 published in November 2002, stipulated that LABAs were the preferred controller agent to “add on” to low-dose inhaled corticosteroids for patients with moderate persistent asthma, and that the combination of low-dose inhaled corticosteroids and LABA was associated with superior outcomes: reduction of symptoms, including nocturnal awakening, increase in lung function, improvement in health-related quality of life, decreased use of “rescue” medication, and reduced rate of exacerbations and severe exacerbations, compared with higher-dose inhaled corticosteroid monotherapy. This management recommendation was categorized as level A, on the basis of data from multiple randomized, controlled, double-blinded trials.27–29 Additional evidence14,30 and data from two meta-analyses31,32 have provided further support for this recommendation, while no evidence linking LABA exposure to risk for fatal or near-fatal asthma has been found in cohort or case-control studies.33–38
Based on safety concerns, the EPR3 guidelines1 recommend that medium-dose inhaled corticosteroids be regarded as equivalent to adding LABAs to low-dose inhaled corticosteroids, and state: “the established, beneficial effects of LABA for the great majority of patients whose asthma is not well controlled with [inhaled corticosteroids] alone should be weighed against the increased risk for severe exacerbations, although uncommon, associated with daily use of LABA.”1
There is currently an honest difference of opinion39,40 among asthma specialists as to how this management recommendation for moderate persistent asthma—now depicted at “step 3” in the EPR3 guidelines (Table 4)—should be implemented. The LABA controversy was reviewed previously in the Cleveland Clinic Journal of Medicine.41
THE ROLE OF OMALIZUMAB: WEIGHING COST VS BENEFIT
The 2002 update to the EPR2 guidelines3 was issued before omalizumab (Xolair) was approved in June 2003.
Patients with severe persistent asthma are categorized in steps 5 or 6 in the EPR3 guidelines (Table 5).1 Preferred management for these patients includes inhaled corticosteroids in high doses combined with long-acting beta agonists and, for step 6 patients, oral corticosteroids.
Omalizumab was approved for management of patients with moderate or severe persistent asthma who are not achieving the goals of asthma management on inhaled corticosteroids, who exhibit a wheal-flare reaction to a perennial allergen, and whose immunoglobulin E (IgE) level is in the range of 30 to 700 IU/mL.42 Omalizumab dosing is based on the serum IgE level and on body weight.
Omalizumab, an anti-IgE monoclonal antibody
Omalizumab is a recombinant, humanized, monoclonal anti-IgE antibody that binds to IgE at the same Fc site as the high-affinity IgE receptor. Its primary mechanism of action is the binding of free IgE in the circulation, forming biologically inert, small complexes that do not activate complement and are cleared by the reticuloendothelial system.42 Its secondary mechanism of action entails a reduction in the number of high-affinity receptors on basophils, from approximately 220,000 to 8,300 receptors per cell. The latter effect was associated with a 90% reduction in histamine release from basophils in response to ex vivo challenge with dust mite allergen.43
Benefit in randomized trials
Omalizumab has been associated with statistically and clinically significant benefit in randomized, double-blind, placebo-controlled trials.44,45
Humbert et al46 randomized 419 patients whose asthma was not adequately controlled on high-dose inhaled corticosteroids and long-acting beta agonists, who were 12 to 75 years old, with reduced lung function and a history of recent asthma exacerbation, to treatment with omalizumab or placebo. Omalizumab was associated with a statistically significant reduction in the rate of asthma exacerbations and severe asthma exacerbations, as well as statistically significant improvements in asthma-related quality of life, morning peak expiratory flow rate, and asthma symptom scores.
These data support the recommendation in EPR3 to consider a trial of omalizumab in properly selected patients with severe, persistent allergic asthma.
Omalizumab is cost-beneficial in properly selected patients
The current wholesale acquisition cost of omalizumab is $532 for one 150-mg vial (David Zito, personal communication). The cost of treatment varies based on body weight and IgE level but may range from a wholesale cost of $6,388 to $38,326 per year.
However, as asthma severity increases, both direct and indirect medical expenditures increase substantially.47,48 Annual costs are approximately four times higher for severe asthma compared with mild asthma49; not only are treatment and exacerbation costs higher, but indirect costs are also disproportionately greater. Annual costs for severe asthma are significantly greater if the disease is inadequately controlled.50 For these reasons, an intervention that leads to improved outcomes for severe, poorly controlled asthma carries the potential for the greatest cost-utility for society, as it can lower direct costs by reducing the frequency and severity of exacerbations, in addition to reducing indirect medical expenditures on the basis of increased productivity and fewer days of missed work or school. The cost of omalizumab in quality-adjusted life years compares favorably with that of biologicals used in managing rheumatoid arthritis, Crohn disease, and multiple sclerosis.50
Adverse effects of omalizumab
In pivotal trials,43,44 omalizumab was associated with a substantial rate of local reactions. The rate of anaphylaxis was slightly less than 1 in 1,000, and this has been confirmed by surveillance data recorded since approval of the drug in 2003. Based on the observed risk of anaphylaxis, in July 2007, the US Food and Drug Administration added a black-box warning to the omalizumab label and stipulated that a medication guide should be provided for patients.51 The warning indicates that health care providers administering omalizumab should be prepared to manage anaphylaxis and that patients should be closely observed for an appropriate period after omalizumab administration.
The package insert also describes a numerical, but not statistically significant, increase in the rate of malignancy in patients receiving omalizumab.42 Malignancy developed in 0.5% of patients receiving omalizumab, compared with 0.2% of patients who received placebo. Because these malignancies were diagnosed over a shorter period than the time required for oncogenesis (ie, 6 months in 60% of cases), and because a heterogeneous variety of tumors was observed, there is reason to doubt these tumors were causally associated with omalizumab.
Postmarketing surveillance studies are in progress that will provide more definitive data on the potential relationship between malignancy and omalizumab exposure.
Omalizumab: Guideline recommendations
The EPR3 guidelines1 state that omalizumab is the only adjunctive therapy to demonstrate efficacy when added to high-dose inhaled corticosteroids plus long-acting beta agonists in patients with severe, persistent, allergic asthma and that evidence does not support use of the following agents, which in some cases are approved for managing other conditions and have been advocated for management of severe, refractory asthma: methotrexate, soluble interleukin (IL)-4 receptor, anti-IL-5, anti-IL-12, cyclosporine A, intravenous immune globulin, gold, troleandomycin, and colchicine. The data supporting use of macrolides were characterized as “encouraging but insufficient to support a recommendation.”
The strength of evidence for the use of omalizumab for patients in steps 5 and 6 who fulfill the criteria for its use (see above) was classified in the EPR3 guidelines1 as category B. The guidelines also say that omalizumab may be considered for adjunctive therapy in properly selected patients in step 4, as a means to avoid higher doses of inhaled corticosteroids, but that additional studies are needed to establish its utility for such patients. This recommendation was classified as category D because of the lack of published comparator trials.
ALLERGEN IMMUNOTHERAPY FOR PATIENTS WITH ASTHMA
Many patients with asthma have clinically relevant, IgE-mediated (allergic) potential to inhaled allergens.1 For patients with persistent asthma (steps 2–6 in Table 5), allergic reactions can contribute to airway inflammation, provoke symptoms, and lead to more use of medications. For this reason, identification and management of clinically relevant allergy merits consideration.52
The EPR3 guidelines1 recommend considering allergen immunotherapy for patients with mild or moderate persistent asthma (steps 2–4) who have a clinically relevant component of allergy to inhaled substances.
Changing the immune response
Allergen immunotherapy entails the incremental administration of inhalant allergens by subcutaneous injection for the purpose of inducing immune system changes in the host response. The goal of immunotherapy is to protect against allergic reactions that can be expected to occur with ongoing exposure to clinically relevant allergens.53
The immunologic changes that develop with allergen immunotherapy are complex.53,54 Successful immunotherapy results in generation of a population of CD4+/CD25+ T lymphocytes producing IL-10, transforming growth factor beta, or both. Allergen immunotherapy has been shown to block the immediate- and late-phase allergic response; to decrease recruitment of mast cells, basophils, and eosinophils on provocation or natural exposure to allergens in the skin, nose, eye, and bronchial mucosa; to blunt the seasonal rise in specific IgE; and to suppress late-phase inflammatory responses in the skin and respiratory tract. However, the efficacy of immunotherapy in relation to these immunologic changes is not completely understood.54
Many patients need skin testing
Allergen immunotherapy may be considered for patients with asthma for whom a clear relationship exists between symptoms and exposure to an allergen to which the patient is sensitive.53 Because it is often not possible to determine whether a patient is sensitive to a perennial indoor allergen (eg, dust mite) on the basis of the medical history alone,55 many patients with asthma benefit from immediate hypersensitivity skin testing to objectively assess or rule out allergy to common inhalants. In certain situations, in vitro testing may be performed, but skin testing has a higher negative predictive value and is recommended as a better screening test.56
Benefits of allergen immunotherapy
Numerous randomized, double-blind, placebo-controlled trials have shown that allergen immunotherapy is associated with benefit for reducing symptoms and medication reliance.57–63
A meta-analysis of 75 randomized, placebo-controlled studies confirmed the effectiveness of immunotherapy in asthma, with a significant reduction in asthma symptoms and medication use and with improvement in bronchial hyperreactivity.64 This meta-analysis included 36 trials of dust mite allergen, 20 of pollen, and 10 of animal dander. Immunotherapy is efficacious for pollen, mold, dust mite, cockroach, and animal allergens; however, its effectiveness is more established for dust mite, animal dander, and pollen allergens, as fewer studies have been published demonstrating efficacy using mold and cockroach allergens.53
In addition, several studies have found that children with allergic rhinitis who receive allergen immunotherapy are significantly less likely to develop asthma.65–67 Immunotherapy has also been associated with a statistically significant reduction in future sensitization to other aeroallergens.68,69
Risk of systemic reaction from allergen immunotherapy
The decision to begin allergen immunotherapy should be individualized on the basis of symptom severity, relative benefit compared with drug therapy, and whether comorbid conditions such as cardiovascular disease or beta-blocker exposure are present. These comorbid conditions are associated with heightened risk of (more serious) anaphylaxis—the major hazard of allergen immunotherapy.70 Systemic reactions during allergen immunotherapy occur at a rate of approximately 3 to 5 per 1,000 injections; for this reason, allergen immunotherapy should only be administered in a medical facility where personnel, supplies, and equipment are available to treat anaphylaxis.5
- National Heart, Lung, and Blood institute, National Asthma education and Prevention Program. Expert Panel Report 3: guidelines for the diagnosis and management of asthma. www.nhlbi.nih.gov/guidelines/asthma. Accessed 8/7/08.
- Expert Panel Report 2: Guidelines for the diagnosis and management of asthma. U.S. Department of Health and Human Services. Publication No. 97-4051; 1997.
- Expert Panel Report: Guidelines for the diagnosis and management of asthma. Update on Selected Topics—2002. J Allergy Clin Immunol 2002; 110:S141–S207.
- FitzGerald JM, Boulet LP, McIvor RA, Zimmerman S, Chapman KR. Asthma control in Canada remains suboptimal: the Reality of Asthma Control (TRAC) study. Can Respir J 2006; 13:253–259.
- Braganza S, Sharif I, Ozuah P. Documenting asthma severity: do we get it right? J Asthma 2003; 40:661–665.
- Cockcroft DW, Swystun VA. Asthma control versus asthma severity. J Allergy Clin Immunol 1996; 98:1016–1018.
- Peters SP, Jones CA, Haselkorn T, Mink DR, Valacer DJ, Weiss ST. Real-world Evaluation of Asthma Control and Treatment (REACT): findings from a national Web-based survey. J Allergy Clin Immunol. 2007; 119:1454–1461.
- Osborne ML, Vollmer WM, Pedula KL, Wilkins J, Buist AS, O’Hollaren M. Lack of correlation of symptoms with specialist-assessed long-term asthma severity. Chest 1999; 115:85–91.
- Li JT, Oppenheimer J, Bernstein IL, et al. Attaining optimal asthma control: a practice parameter. J Allergy Clin Immunol 2005; 116:S3–S11.
- Nathan RA, Sorkness C, Kosinski M, et al. Development of the Asthma Control Test: a survey for assessing asthma control. J Allergy Clin Immunol 2004; 113:59–65.
- Schatz M, Zeiger RS, Drane A, et al. Reliability and predictive validity of the Asthma Control Test administered by telephone calls using speech recognition technology. J Allergy Clin Immunol 2007; 119:336–343.
- Peters D, Chen C, Markson LE, Allen-Ramey FC, Vollmer WM. Using an asthma control questionnaire and administrative data to predict healthcare utilization. Chest 2006; 129:918–924.
- Schatz M, Sorkness C, Li JT, et al. Asthma Control Test: reliability, validity, and responsiveness in patients not previously followed by asthma specialists. J Allergy Clin Immunol 2006; 117:549–556.
- Bateman E, Boushey H, Bousquet J, et al. Can guideline-defined asthma control be achieved? Am J Respir Crit Care Med 2004; 170:836–844.
- Davies TJ, Bunn WB, Fromer L, Gelfand EW, Colice GL. A focus on the asthma HEDIS measure and its implications for clinical practice. Manag Care Interface 2006; 19:29–36.
- Rubinfeld AR, Pain MC. Perception of asthma. Lancet 1976; 1:882–884.
- Teeter J, Bleecker E. Relationship between airway obstruction and respiratory symptoms in adult asthmatics. Chest 1998; 113:272–277.
- Shingo S, Zhang J, Reiss T. Correlation of airway obstruction and patient reported endpoints in clinical studies. Eur Resp J 2001; 17:220–224.
- Juniper EF, Bousquet J, Abetz L, Bateman ED; GOAL Committee. Identifying ‘well-controlled’ and ‘not well-controlled’ asthma using the Asthma Control Questionnaire. Respir Med 2006; 100:616–621.
- Nelson H, Weiss S, Bleecker E, Yancey S, Dorinsky P. The Salmeterol Multicenter Asthma Research Trial. Chest 2006; 129:15–26.
- Wechsler M, Lehman E, Lazarus S, et al. ß-Adrenergic receptor polymorphisms and response to salmeterol. Am J Respir Crit Care Med 2006; 173:519–526.
- Palmer CNA, Lipworth BJ, Lee S, Ismail T, MacGregor DF, Mukhopadhyay S. Arginine-16 beta-2 adrenoceptor genotype predisposes to exacerbations in young asthmatics taking regular salmeterol. Thorax 2006; 61:940–944.
- Taylor DR, Drazen JM, Herbison GP, Yandava CN, Hancox RJ, Town GI. Asthma exacerbations during long term beta agonist use: influence of beta 2 adrenoceptor polymorphism. Thorax 2000; 55:762–727.
- Bleecker E, Postma D, Lawrance R, Meyers D, Ambrose H, Goldman M. Effect of ADRB2 polymorphisms on response to long-acting beta2-agonist therapy: a pharmacogenetic analysis of two randomized studies. Lancet 2007; 370:2118–2125.
- Bleecker E, Yancey S, Baitinger L, et al. Salmeterol response is not affected by beta-2 adrenergic receptor genotype in subjects with persistent asthma. J Allergy Clin Immunol 2006; 118:809–816.
- Nelson H, Bleecker E, Corren J, et al. Characterization of asthma exacerbations by Arg16Gly genotype in subjects with asthma receiving salmeterol alone or with fluticasone propionate. J Allergy Clin Immunol 2008; 121:S131.
- O’Byrne P, Barnes P, Rodriguez-Roisin R, et al. Low dose Inhaled budesonide and formoterol in mild persistent asthma. The OPTIMA Randomized Trial. Am J Respir Crit Care Med 2001; 164:1392–1397.
- Greening AP, Ind PW, Northfield M, Shaw G. Added salmeterol versus higher dose corticosteroid in asthma patients with symptoms on existing inhaled corticosteroid. Lancet 1994; 344:219–224.
- Woolcock A, Lundback B, Ringdal N, Jacques LA. Comparison of addition of salmeterol to inhaled steroids with doubling of the dose of inhaled steroids. Am J Respir Crit Care Med 1996; 153:1481–1488.
- Walters EH, Walters JAE, Gibson MDP. Long-acting beta2-agonists for stable chronic asthma. Cochrane Database Syst Rev 2003; (3):CD001385. doi:10.1002/14651858.CD001385.
- Masoli M, Weatherall M, Holt S, Beasley R. Moderate dose inhaled corticosteroids plus salmeterol versus higher doses of inhaled corticosteroid in symptomatic asthma. Thorax 2005; 60:730–734.
- Sin DD, Man J, Sharpe H, Gan WQ, Man SFP. Pharmacological management to reduce exacerbations in adults with asthma. A systematic review and meta-analysis. JAMA 2004; 292:367–376.
- Mann RD, Kubota K, Pearce G, Wilton L. Salmeterol: a study by prescription event monitoring in a UK cohort of 15,407 patients. J Clin Epidemiol 1996; 49:247–250.
- Lanes S, Lanza L, Wentworth C. Risk of emergency care, hospitalization, and ICU stays for acute asthma among recipients of salmeterol. Am J Respir Crit Care Med 1998; 158:857–861.
- Meier CR, Jick H. Drug use and pulmonary death rates in increasingly symptomatic asthma patients in the UK. Thorax 1997; 52:612–617.
- Williams C, Crossland L, Finnerty J, et al. A case control study of salmeterol and near-fatal attacks of asthma. Thorax 1998; 53:7–13.
- Lanes S, Garcia Rodriguez LA, Herta C. Respiratory medications and risk of asthma death. Thorax 2002; 57:683–686.
- Anderson HR, Ayres JG, Sturdy PM, et al. Bronchodilator treatment and deaths from asthma: case control study. Br Med J 2005; 330:117–124.
- Martinez FD. Safety of long-acting beta agonists—an urgent need to clear the air. N Engl J Med 2005; 353:2637–2639.
- Nelson HS. Long-acting beta-agonists in adult asthma: evidence that these drugs are safe. Prim Care Respir J 2006; 15:271–277.
- Lang DM. The long-acting beta agonist controversy: a critical examination of the evidence. Cleve Clin J Med 2006; 73:973–992.
- Rambasek T, Lang DM, Kavuru M. Omalizumab: where does it fit in current asthma management? Cleve Clin J Med 2004; 71:251–261.
- McGlashan D, Bochner B, Adelman D, et al. Down regulation of Fc(epsilon)RI expression on human basophils during in vivo treatment of atopic patients with anti-IgE antibody. J Immunol 1997; 158:1438–1445.
- Busse W, Corren J, Lanier B, et al. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol 2001; 108:184–190.
- Soler M, Matz J, Townley R, et al. The anti-IgE antibody omalizumab reduces asthma exacerbations and steroid requirement in allergic asthmatics. Eur Respir J 2001; 18:254–261.
- Humbert M, Beasley R, Ayres J, et al. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy 2005; 60:309–316.
- Van Ganse E, Antonicelli L, Zhang Q, et al. Asthma-related resource use and cost by GINA classification of severity in three European countries. Respir Med 2006; 100:140–147.
- Godard P, Chanez P, Siraudin L, Nicoloyannis N, Duru G. Costs of asthma are correlated with severity: a 1-yr prospective study. Eur Respir J 2002; 19:61–67.
- Cisternas MG, Blanc PH, Yen IH, et al. A comprehensive study of the direct and indirect costs of adult asthma. J Allergy Clin Immunol 2003; 111:1212–1218.
- Sullivan S, Turk F. An evaluation of the cost effectiveness of omalizumab for the treatment of severe persistent asthma. Allergy 2008; 63:670–684.
- US Food and Drug Administration. Omalizumab (marketed as Xolair) information. www.fda.gov/cder/drug/infopage/omalizumab/default.htm. Accessed August 31, 2007.
- Williams SG, Schmidt DK, Redd SC, Storms W. Key clinical activities for quality asthma care. Recommendations of the National Asthma Education and Prevention Program. MMWR Recomm Rep 2003; 52 RR-6:1–8.
- Cox L, Li J, Nelson H, Lockey R, et al. Allergy Immunotherapy: a practice parameter second update. J Allergy Clin Immunol 2007; 120:S25–S85.
- Akdis M, Akdis CA. Mechanisms of allergen-specific immunotherapy. J Allergy Clin Immunol 2007; 119:780–789.
- Murray AB, Milner RA. The accuracy of features in the clinical history for predicting atopic sensitization to airborne allergens in children. J Allergy Clin Immunol 1995; 96:588–596.
- Bernstein IL, Li JT, Bernstein DI, et al. Allergy diagnostic testing: an updated practice parameter. Ann Allergy Asthma Immunol 2008; 100 suppl 3:1S–148S.
- Walker S, Pajno GB, Lima MT, Wilson DR, Durham SR. Grass pollen immunotherapy for seasonal rhinitis and asthma: a randomized, controlled trial. J Allergy Clin Immunol 2001; 107:87–93.
- Varney VA, Edwards J, Tabbah K, Brewster H, Mavroleon G, Frew AJ. Clinical efficacy of specific immunotherapy to cat dander: a double-blind placebo-controlled trial. Clin Exp Allergy 1997; 27:860–867.
- Cantani A, Arcese G, Lucenti P, Gagliesi D, Bartolucci M. A three-year prospective study of specific immunotherapy to inhalant allergens: evidence of safety and efficacy in 300 children with allergic asthma. J Investig Allergol Clin Immunol 1997; 7:90–97.
- Hedlin G, Wille S, Browaldh L, et al. Immunotherapy in children with allergic asthma: effect on bronchial hyperreactivity and pharmacotherapy. J Allergy Clin Immunol 1999; 103:609–614.
- Arvidsson MB, Löwhagen O, Rak S. Allergen specific immunotherapy attenuates early and late phase reactions in lower airways of birch pollen asthmatic patients: a double blind placebo-controlled study. Allergy 2004; 59:74–80.
- Pichler CE, Helbling A, Pichler WJ. Three years of specific immunotherapy with house-dust-mite extracts in patients with rhinitis and asthma: significant improvement of allergen-specific parameters and of nonspecific bronchial hyperreactivity. Allergy 2001; 56:301–306.
- Mirone C, Albert F, Tosi A, et al. Efficacy and safety of subcutaneous immunotherapy with a biologically standardized extract of Ambrosia artemisiifolia pollen: a double-blind, placebo-controlled study. Clin Exp Allergy 2004; 34:1408–1414.
- Abramson MJ, Puy RM, Weiner JM. Allergen immunotherapy for asthma. Cochrane Database Syst Rev 2003; (4):CD001186.
- Jacobsen L. Preventive aspects of immunotherapy: prevention for children at risk of developing asthma. Ann Allergy Asthma Immunol 2001; 87:43–46.
- Moller C, Dreborg S, Ferdousi HA, et al. Pollen immunotherapy reduces the development of asthma in children with seasonal rhinoconjunctivitis (the PAT study). J Allergy Clin Immunol 2002; 109:251–256.
- Niggemann B, Jacobsen L, Dreborg S, et al; PAT Investigator Group. Five year follow-up on the PAT study: specific immunotherapy and long-term prevention of asthma in children. Allergy 2006: 61:855–859.
- Des Roches A, Paradis L, Menardo JL, et al. Immunotherapy with a standardized Dermatophagoides pteronyssinus extract VI: specific immunotherapy prevents the onset of new sensitizations in children. J Allergy Clin Immunol 1997; 99:450–453.
- Pajno GB, Barberio G, DeLuca F, et al. Prevention of new sensitizations in asthmatic children monosensitized to the house dust mite by specific immunotherapy: a six year follow up study. Clin Exp Allergy 2001; 31:1392–1397.
- Lang DM. Do beta blockers really enhance the risk of anaphylaxis during immunotherapy? Curr Allergy Asthma Rep 2008; 8:37–44.
- National Heart, Lung, and Blood institute, National Asthma education and Prevention Program. Expert Panel Report 3: guidelines for the diagnosis and management of asthma. www.nhlbi.nih.gov/guidelines/asthma. Accessed 8/7/08.
- Expert Panel Report 2: Guidelines for the diagnosis and management of asthma. U.S. Department of Health and Human Services. Publication No. 97-4051; 1997.
- Expert Panel Report: Guidelines for the diagnosis and management of asthma. Update on Selected Topics—2002. J Allergy Clin Immunol 2002; 110:S141–S207.
- FitzGerald JM, Boulet LP, McIvor RA, Zimmerman S, Chapman KR. Asthma control in Canada remains suboptimal: the Reality of Asthma Control (TRAC) study. Can Respir J 2006; 13:253–259.
- Braganza S, Sharif I, Ozuah P. Documenting asthma severity: do we get it right? J Asthma 2003; 40:661–665.
- Cockcroft DW, Swystun VA. Asthma control versus asthma severity. J Allergy Clin Immunol 1996; 98:1016–1018.
- Peters SP, Jones CA, Haselkorn T, Mink DR, Valacer DJ, Weiss ST. Real-world Evaluation of Asthma Control and Treatment (REACT): findings from a national Web-based survey. J Allergy Clin Immunol. 2007; 119:1454–1461.
- Osborne ML, Vollmer WM, Pedula KL, Wilkins J, Buist AS, O’Hollaren M. Lack of correlation of symptoms with specialist-assessed long-term asthma severity. Chest 1999; 115:85–91.
- Li JT, Oppenheimer J, Bernstein IL, et al. Attaining optimal asthma control: a practice parameter. J Allergy Clin Immunol 2005; 116:S3–S11.
- Nathan RA, Sorkness C, Kosinski M, et al. Development of the Asthma Control Test: a survey for assessing asthma control. J Allergy Clin Immunol 2004; 113:59–65.
- Schatz M, Zeiger RS, Drane A, et al. Reliability and predictive validity of the Asthma Control Test administered by telephone calls using speech recognition technology. J Allergy Clin Immunol 2007; 119:336–343.
- Peters D, Chen C, Markson LE, Allen-Ramey FC, Vollmer WM. Using an asthma control questionnaire and administrative data to predict healthcare utilization. Chest 2006; 129:918–924.
- Schatz M, Sorkness C, Li JT, et al. Asthma Control Test: reliability, validity, and responsiveness in patients not previously followed by asthma specialists. J Allergy Clin Immunol 2006; 117:549–556.
- Bateman E, Boushey H, Bousquet J, et al. Can guideline-defined asthma control be achieved? Am J Respir Crit Care Med 2004; 170:836–844.
- Davies TJ, Bunn WB, Fromer L, Gelfand EW, Colice GL. A focus on the asthma HEDIS measure and its implications for clinical practice. Manag Care Interface 2006; 19:29–36.
- Rubinfeld AR, Pain MC. Perception of asthma. Lancet 1976; 1:882–884.
- Teeter J, Bleecker E. Relationship between airway obstruction and respiratory symptoms in adult asthmatics. Chest 1998; 113:272–277.
- Shingo S, Zhang J, Reiss T. Correlation of airway obstruction and patient reported endpoints in clinical studies. Eur Resp J 2001; 17:220–224.
- Juniper EF, Bousquet J, Abetz L, Bateman ED; GOAL Committee. Identifying ‘well-controlled’ and ‘not well-controlled’ asthma using the Asthma Control Questionnaire. Respir Med 2006; 100:616–621.
- Nelson H, Weiss S, Bleecker E, Yancey S, Dorinsky P. The Salmeterol Multicenter Asthma Research Trial. Chest 2006; 129:15–26.
- Wechsler M, Lehman E, Lazarus S, et al. ß-Adrenergic receptor polymorphisms and response to salmeterol. Am J Respir Crit Care Med 2006; 173:519–526.
- Palmer CNA, Lipworth BJ, Lee S, Ismail T, MacGregor DF, Mukhopadhyay S. Arginine-16 beta-2 adrenoceptor genotype predisposes to exacerbations in young asthmatics taking regular salmeterol. Thorax 2006; 61:940–944.
- Taylor DR, Drazen JM, Herbison GP, Yandava CN, Hancox RJ, Town GI. Asthma exacerbations during long term beta agonist use: influence of beta 2 adrenoceptor polymorphism. Thorax 2000; 55:762–727.
- Bleecker E, Postma D, Lawrance R, Meyers D, Ambrose H, Goldman M. Effect of ADRB2 polymorphisms on response to long-acting beta2-agonist therapy: a pharmacogenetic analysis of two randomized studies. Lancet 2007; 370:2118–2125.
- Bleecker E, Yancey S, Baitinger L, et al. Salmeterol response is not affected by beta-2 adrenergic receptor genotype in subjects with persistent asthma. J Allergy Clin Immunol 2006; 118:809–816.
- Nelson H, Bleecker E, Corren J, et al. Characterization of asthma exacerbations by Arg16Gly genotype in subjects with asthma receiving salmeterol alone or with fluticasone propionate. J Allergy Clin Immunol 2008; 121:S131.
- O’Byrne P, Barnes P, Rodriguez-Roisin R, et al. Low dose Inhaled budesonide and formoterol in mild persistent asthma. The OPTIMA Randomized Trial. Am J Respir Crit Care Med 2001; 164:1392–1397.
- Greening AP, Ind PW, Northfield M, Shaw G. Added salmeterol versus higher dose corticosteroid in asthma patients with symptoms on existing inhaled corticosteroid. Lancet 1994; 344:219–224.
- Woolcock A, Lundback B, Ringdal N, Jacques LA. Comparison of addition of salmeterol to inhaled steroids with doubling of the dose of inhaled steroids. Am J Respir Crit Care Med 1996; 153:1481–1488.
- Walters EH, Walters JAE, Gibson MDP. Long-acting beta2-agonists for stable chronic asthma. Cochrane Database Syst Rev 2003; (3):CD001385. doi:10.1002/14651858.CD001385.
- Masoli M, Weatherall M, Holt S, Beasley R. Moderate dose inhaled corticosteroids plus salmeterol versus higher doses of inhaled corticosteroid in symptomatic asthma. Thorax 2005; 60:730–734.
- Sin DD, Man J, Sharpe H, Gan WQ, Man SFP. Pharmacological management to reduce exacerbations in adults with asthma. A systematic review and meta-analysis. JAMA 2004; 292:367–376.
- Mann RD, Kubota K, Pearce G, Wilton L. Salmeterol: a study by prescription event monitoring in a UK cohort of 15,407 patients. J Clin Epidemiol 1996; 49:247–250.
- Lanes S, Lanza L, Wentworth C. Risk of emergency care, hospitalization, and ICU stays for acute asthma among recipients of salmeterol. Am J Respir Crit Care Med 1998; 158:857–861.
- Meier CR, Jick H. Drug use and pulmonary death rates in increasingly symptomatic asthma patients in the UK. Thorax 1997; 52:612–617.
- Williams C, Crossland L, Finnerty J, et al. A case control study of salmeterol and near-fatal attacks of asthma. Thorax 1998; 53:7–13.
- Lanes S, Garcia Rodriguez LA, Herta C. Respiratory medications and risk of asthma death. Thorax 2002; 57:683–686.
- Anderson HR, Ayres JG, Sturdy PM, et al. Bronchodilator treatment and deaths from asthma: case control study. Br Med J 2005; 330:117–124.
- Martinez FD. Safety of long-acting beta agonists—an urgent need to clear the air. N Engl J Med 2005; 353:2637–2639.
- Nelson HS. Long-acting beta-agonists in adult asthma: evidence that these drugs are safe. Prim Care Respir J 2006; 15:271–277.
- Lang DM. The long-acting beta agonist controversy: a critical examination of the evidence. Cleve Clin J Med 2006; 73:973–992.
- Rambasek T, Lang DM, Kavuru M. Omalizumab: where does it fit in current asthma management? Cleve Clin J Med 2004; 71:251–261.
- McGlashan D, Bochner B, Adelman D, et al. Down regulation of Fc(epsilon)RI expression on human basophils during in vivo treatment of atopic patients with anti-IgE antibody. J Immunol 1997; 158:1438–1445.
- Busse W, Corren J, Lanier B, et al. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol 2001; 108:184–190.
- Soler M, Matz J, Townley R, et al. The anti-IgE antibody omalizumab reduces asthma exacerbations and steroid requirement in allergic asthmatics. Eur Respir J 2001; 18:254–261.
- Humbert M, Beasley R, Ayres J, et al. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy 2005; 60:309–316.
- Van Ganse E, Antonicelli L, Zhang Q, et al. Asthma-related resource use and cost by GINA classification of severity in three European countries. Respir Med 2006; 100:140–147.
- Godard P, Chanez P, Siraudin L, Nicoloyannis N, Duru G. Costs of asthma are correlated with severity: a 1-yr prospective study. Eur Respir J 2002; 19:61–67.
- Cisternas MG, Blanc PH, Yen IH, et al. A comprehensive study of the direct and indirect costs of adult asthma. J Allergy Clin Immunol 2003; 111:1212–1218.
- Sullivan S, Turk F. An evaluation of the cost effectiveness of omalizumab for the treatment of severe persistent asthma. Allergy 2008; 63:670–684.
- US Food and Drug Administration. Omalizumab (marketed as Xolair) information. www.fda.gov/cder/drug/infopage/omalizumab/default.htm. Accessed August 31, 2007.
- Williams SG, Schmidt DK, Redd SC, Storms W. Key clinical activities for quality asthma care. Recommendations of the National Asthma Education and Prevention Program. MMWR Recomm Rep 2003; 52 RR-6:1–8.
- Cox L, Li J, Nelson H, Lockey R, et al. Allergy Immunotherapy: a practice parameter second update. J Allergy Clin Immunol 2007; 120:S25–S85.
- Akdis M, Akdis CA. Mechanisms of allergen-specific immunotherapy. J Allergy Clin Immunol 2007; 119:780–789.
- Murray AB, Milner RA. The accuracy of features in the clinical history for predicting atopic sensitization to airborne allergens in children. J Allergy Clin Immunol 1995; 96:588–596.
- Bernstein IL, Li JT, Bernstein DI, et al. Allergy diagnostic testing: an updated practice parameter. Ann Allergy Asthma Immunol 2008; 100 suppl 3:1S–148S.
- Walker S, Pajno GB, Lima MT, Wilson DR, Durham SR. Grass pollen immunotherapy for seasonal rhinitis and asthma: a randomized, controlled trial. J Allergy Clin Immunol 2001; 107:87–93.
- Varney VA, Edwards J, Tabbah K, Brewster H, Mavroleon G, Frew AJ. Clinical efficacy of specific immunotherapy to cat dander: a double-blind placebo-controlled trial. Clin Exp Allergy 1997; 27:860–867.
- Cantani A, Arcese G, Lucenti P, Gagliesi D, Bartolucci M. A three-year prospective study of specific immunotherapy to inhalant allergens: evidence of safety and efficacy in 300 children with allergic asthma. J Investig Allergol Clin Immunol 1997; 7:90–97.
- Hedlin G, Wille S, Browaldh L, et al. Immunotherapy in children with allergic asthma: effect on bronchial hyperreactivity and pharmacotherapy. J Allergy Clin Immunol 1999; 103:609–614.
- Arvidsson MB, Löwhagen O, Rak S. Allergen specific immunotherapy attenuates early and late phase reactions in lower airways of birch pollen asthmatic patients: a double blind placebo-controlled study. Allergy 2004; 59:74–80.
- Pichler CE, Helbling A, Pichler WJ. Three years of specific immunotherapy with house-dust-mite extracts in patients with rhinitis and asthma: significant improvement of allergen-specific parameters and of nonspecific bronchial hyperreactivity. Allergy 2001; 56:301–306.
- Mirone C, Albert F, Tosi A, et al. Efficacy and safety of subcutaneous immunotherapy with a biologically standardized extract of Ambrosia artemisiifolia pollen: a double-blind, placebo-controlled study. Clin Exp Allergy 2004; 34:1408–1414.
- Abramson MJ, Puy RM, Weiner JM. Allergen immunotherapy for asthma. Cochrane Database Syst Rev 2003; (4):CD001186.
- Jacobsen L. Preventive aspects of immunotherapy: prevention for children at risk of developing asthma. Ann Allergy Asthma Immunol 2001; 87:43–46.
- Moller C, Dreborg S, Ferdousi HA, et al. Pollen immunotherapy reduces the development of asthma in children with seasonal rhinoconjunctivitis (the PAT study). J Allergy Clin Immunol 2002; 109:251–256.
- Niggemann B, Jacobsen L, Dreborg S, et al; PAT Investigator Group. Five year follow-up on the PAT study: specific immunotherapy and long-term prevention of asthma in children. Allergy 2006: 61:855–859.
- Des Roches A, Paradis L, Menardo JL, et al. Immunotherapy with a standardized Dermatophagoides pteronyssinus extract VI: specific immunotherapy prevents the onset of new sensitizations in children. J Allergy Clin Immunol 1997; 99:450–453.
- Pajno GB, Barberio G, DeLuca F, et al. Prevention of new sensitizations in asthmatic children monosensitized to the house dust mite by specific immunotherapy: a six year follow up study. Clin Exp Allergy 2001; 31:1392–1397.
- Lang DM. Do beta blockers really enhance the risk of anaphylaxis during immunotherapy? Curr Allergy Asthma Rep 2008; 8:37–44.
KEY POINTS
- The EPR3 recommends that management decisions be based initially on asthma severity, and subsequently on asthma control as assessed serially by validated tests.
- Omalizumab, a monoclonal antibody against immunoglobulin E, is the only adjunctive therapy to demonstrate efficacy when added to high-dose inhaled corticosteroids plus long-acting beta agonists in patients with severe, persistent, allergic asthma.
- The EPR3 guidelines recommend consideration of allergen immunotherapy for patients with mild or moderate persistent allergic asthma.
A 51-year-old man with nodular lesions
A 51-year-old diabetic man presents with a 1-year history of episodic pain, swelling, and stiffness in some of the metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints of his fingers. During these episodes, he has significant morning stiffness. He says he has no other joint problems or back pain. A review of systems is otherwise unremarkable.
WHAT IS THE MOST LIKELY DIAGNOSIS?
- Gouty tophi
- Rheumatoid nodulosis
- Calcinosis cutis
- Tuberous xanthomas
GOUTY TOPHI: OUR INITIAL IMPRESSION
RHEUMATOID NODULOSIS: THE TRUE DIAGNOSIS
The patient’s nodules kept growing, and new ones kept developing, causing significant impairment of hand function. Hence, some of the larger nodules were surgically removed. The resected specimens revealed a yellow nodular cut surface on sectioning. Histopathologic analysis revealed multiple necrobiotic nodules, consistent with rheumatoid nodulosis. Urate crystals were not seen on histology, although crystals can be dissolved in some tissue preparations, and gouty tophi provoke pathologically a granulomatous inflammatory reaction. However, unlike what is expected with gouty tophi, material aspirated from the nodules did not reveal monosodium urate crystals on polarized light microscopy. A repeat rheumatoid factor test 2 years after his initial presentation became positive at 57 IU/ mL (normal < 20 IU/mL).
Comment. Rheumatoid nodules are one of the most common extra-articular manifestations of rheumatoid arthritis, seen in 20% to 25% of cases, and they are usually associated with seropositivity for rheumatoid factor and with more aggressive disease.1 Rheumatoid nodulosis, on the other hand, usually runs a more benign clinical course.2 It was first described in 1949,3 and the diagnostic criteria were developed in 1988 by Couret et al.4 Patients develop nontender subcutaneous rheumatoid nodules, usually around areas of repeated microtrauma.2 Often there is a history of attacks of palindromic rheumatism, characterized by recurrent, self-limited episodes of monoarthritis or polyarthritis without an alternative explanation, as in this patient. However, systemic manifestations of rheumatoid arthritis and radiologic evidence of joint damage are often not seen. Rheumatoid factor positivity is also not a requirement. Over time, some patients progress to full-blown rheumatoid arthritis. Methotrexate use has been associated with accelerated rheumatoid nodulosis in some rheumatoid arthritis patients.2
Rheumatoid nodulosis can be progressive and difficult to treat. Hydroxychloroquine has induced complete resolution in some cases.5 Surgical removal of the nodules may be considered if they limit joint motion.6 A placebo-controlled, double-blind trial of intralesional corticosteroid injection has demonstrated efficacy in reducing nodule size.7
In our patient, treatment with hydroxychloroquine, sulfasalazine, and methotrexate did not relieve the joint pain, nor did these drugs stop the nodules from growing. He was started on the tumor necrosis factor antagonist etanercept (Enbrel), which significantly helped the joint pain, but the nodules continued to progress relentlessly. Some of the larger nodules were later injected with triamcinolone (Kenalog), which led to significant shrinkage in nodule size.
THE OTHER DIAGNOSTIC CHOICES
The other two choices are unlikely.
Calcinosis cutis results from the cutaneous deposition of insoluble compounds of calcium (hydroxyapatite or amorphous calcium phosphate), due to local or systemic factors, or both. This can be the result either of ectopic calcification in normal tissue in the setting of hypercalcemia or hyperphosphatemia, or of dystrophic calcification in damaged tissue. They appear as multiple, firm, whitish dermal papules, plaques, nodules, or subcutaneous nodules, which can sometimes ulcerate. They are radio-opaque. On biopsy, dermal deposits of calcium are seen, with or without a surrounding foreign-body giant-cell reaction. Calcium deposition may be confirmed on Von Kossa and alizarin red stains.
Tuberous xanthomas are firm, painless, red-yellow nodules that usually develop in pressure areas such as the extensor surfaces of the knees, the elbows, and the buttocks. They can be associated with familial dysbetalipoproteinemia, familial hypercholesterolemia, and even some of the secondary dyslipidemias. Histologic study shows accumulations of vacuolated lipid-laden macrophages (foamy histiocytes) and sometimes multinucleated histiocytes (Touton giant cells). The lipid droplets are dissolved during routine histologic processing, but lipid stains on frozen sections can be useful.
- Ziff M. The rheumatoid nodule. Arthritis Rheum 1990; 33:761–767.
- Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg 2007; 26:100–107.
- Bywaters EGL. A variant of rheumatoid arthritis characterized by recurrent digital pad nodules and palmar fasciitis, closely resembling palindromic rheumatism. Ann Rheum Dis 1949; 8:2–30.
- Couret M, Combe B, Chuong VT, et al. Rheumatoid nodulosis: report of two new cases and discussion of diagnostic criteria. J Rheumatol 1988; 15:1427–1430.
- McCarty DJ. Complete reversal of rheumatoid nodulosis. J Rheumatol 1991; 18:736–737.
- Kai Y, Anzai S, Shibuya H, et al. A case of rheumatoid nodulosis successfully treated with surgery. J Dermatol 2004; 31:910–915.
- Ching DW, Petrie JP, Klemp P, Jones JG. Injection therapy of superficial rheumatoid nodules. Br J Rheumatol 1992; 31:775–777.
A 51-year-old diabetic man presents with a 1-year history of episodic pain, swelling, and stiffness in some of the metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints of his fingers. During these episodes, he has significant morning stiffness. He says he has no other joint problems or back pain. A review of systems is otherwise unremarkable.
WHAT IS THE MOST LIKELY DIAGNOSIS?
- Gouty tophi
- Rheumatoid nodulosis
- Calcinosis cutis
- Tuberous xanthomas
GOUTY TOPHI: OUR INITIAL IMPRESSION
RHEUMATOID NODULOSIS: THE TRUE DIAGNOSIS
The patient’s nodules kept growing, and new ones kept developing, causing significant impairment of hand function. Hence, some of the larger nodules were surgically removed. The resected specimens revealed a yellow nodular cut surface on sectioning. Histopathologic analysis revealed multiple necrobiotic nodules, consistent with rheumatoid nodulosis. Urate crystals were not seen on histology, although crystals can be dissolved in some tissue preparations, and gouty tophi provoke pathologically a granulomatous inflammatory reaction. However, unlike what is expected with gouty tophi, material aspirated from the nodules did not reveal monosodium urate crystals on polarized light microscopy. A repeat rheumatoid factor test 2 years after his initial presentation became positive at 57 IU/ mL (normal < 20 IU/mL).
Comment. Rheumatoid nodules are one of the most common extra-articular manifestations of rheumatoid arthritis, seen in 20% to 25% of cases, and they are usually associated with seropositivity for rheumatoid factor and with more aggressive disease.1 Rheumatoid nodulosis, on the other hand, usually runs a more benign clinical course.2 It was first described in 1949,3 and the diagnostic criteria were developed in 1988 by Couret et al.4 Patients develop nontender subcutaneous rheumatoid nodules, usually around areas of repeated microtrauma.2 Often there is a history of attacks of palindromic rheumatism, characterized by recurrent, self-limited episodes of monoarthritis or polyarthritis without an alternative explanation, as in this patient. However, systemic manifestations of rheumatoid arthritis and radiologic evidence of joint damage are often not seen. Rheumatoid factor positivity is also not a requirement. Over time, some patients progress to full-blown rheumatoid arthritis. Methotrexate use has been associated with accelerated rheumatoid nodulosis in some rheumatoid arthritis patients.2
Rheumatoid nodulosis can be progressive and difficult to treat. Hydroxychloroquine has induced complete resolution in some cases.5 Surgical removal of the nodules may be considered if they limit joint motion.6 A placebo-controlled, double-blind trial of intralesional corticosteroid injection has demonstrated efficacy in reducing nodule size.7
In our patient, treatment with hydroxychloroquine, sulfasalazine, and methotrexate did not relieve the joint pain, nor did these drugs stop the nodules from growing. He was started on the tumor necrosis factor antagonist etanercept (Enbrel), which significantly helped the joint pain, but the nodules continued to progress relentlessly. Some of the larger nodules were later injected with triamcinolone (Kenalog), which led to significant shrinkage in nodule size.
THE OTHER DIAGNOSTIC CHOICES
The other two choices are unlikely.
Calcinosis cutis results from the cutaneous deposition of insoluble compounds of calcium (hydroxyapatite or amorphous calcium phosphate), due to local or systemic factors, or both. This can be the result either of ectopic calcification in normal tissue in the setting of hypercalcemia or hyperphosphatemia, or of dystrophic calcification in damaged tissue. They appear as multiple, firm, whitish dermal papules, plaques, nodules, or subcutaneous nodules, which can sometimes ulcerate. They are radio-opaque. On biopsy, dermal deposits of calcium are seen, with or without a surrounding foreign-body giant-cell reaction. Calcium deposition may be confirmed on Von Kossa and alizarin red stains.
Tuberous xanthomas are firm, painless, red-yellow nodules that usually develop in pressure areas such as the extensor surfaces of the knees, the elbows, and the buttocks. They can be associated with familial dysbetalipoproteinemia, familial hypercholesterolemia, and even some of the secondary dyslipidemias. Histologic study shows accumulations of vacuolated lipid-laden macrophages (foamy histiocytes) and sometimes multinucleated histiocytes (Touton giant cells). The lipid droplets are dissolved during routine histologic processing, but lipid stains on frozen sections can be useful.
A 51-year-old diabetic man presents with a 1-year history of episodic pain, swelling, and stiffness in some of the metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints of his fingers. During these episodes, he has significant morning stiffness. He says he has no other joint problems or back pain. A review of systems is otherwise unremarkable.
WHAT IS THE MOST LIKELY DIAGNOSIS?
- Gouty tophi
- Rheumatoid nodulosis
- Calcinosis cutis
- Tuberous xanthomas
GOUTY TOPHI: OUR INITIAL IMPRESSION
RHEUMATOID NODULOSIS: THE TRUE DIAGNOSIS
The patient’s nodules kept growing, and new ones kept developing, causing significant impairment of hand function. Hence, some of the larger nodules were surgically removed. The resected specimens revealed a yellow nodular cut surface on sectioning. Histopathologic analysis revealed multiple necrobiotic nodules, consistent with rheumatoid nodulosis. Urate crystals were not seen on histology, although crystals can be dissolved in some tissue preparations, and gouty tophi provoke pathologically a granulomatous inflammatory reaction. However, unlike what is expected with gouty tophi, material aspirated from the nodules did not reveal monosodium urate crystals on polarized light microscopy. A repeat rheumatoid factor test 2 years after his initial presentation became positive at 57 IU/ mL (normal < 20 IU/mL).
Comment. Rheumatoid nodules are one of the most common extra-articular manifestations of rheumatoid arthritis, seen in 20% to 25% of cases, and they are usually associated with seropositivity for rheumatoid factor and with more aggressive disease.1 Rheumatoid nodulosis, on the other hand, usually runs a more benign clinical course.2 It was first described in 1949,3 and the diagnostic criteria were developed in 1988 by Couret et al.4 Patients develop nontender subcutaneous rheumatoid nodules, usually around areas of repeated microtrauma.2 Often there is a history of attacks of palindromic rheumatism, characterized by recurrent, self-limited episodes of monoarthritis or polyarthritis without an alternative explanation, as in this patient. However, systemic manifestations of rheumatoid arthritis and radiologic evidence of joint damage are often not seen. Rheumatoid factor positivity is also not a requirement. Over time, some patients progress to full-blown rheumatoid arthritis. Methotrexate use has been associated with accelerated rheumatoid nodulosis in some rheumatoid arthritis patients.2
Rheumatoid nodulosis can be progressive and difficult to treat. Hydroxychloroquine has induced complete resolution in some cases.5 Surgical removal of the nodules may be considered if they limit joint motion.6 A placebo-controlled, double-blind trial of intralesional corticosteroid injection has demonstrated efficacy in reducing nodule size.7
In our patient, treatment with hydroxychloroquine, sulfasalazine, and methotrexate did not relieve the joint pain, nor did these drugs stop the nodules from growing. He was started on the tumor necrosis factor antagonist etanercept (Enbrel), which significantly helped the joint pain, but the nodules continued to progress relentlessly. Some of the larger nodules were later injected with triamcinolone (Kenalog), which led to significant shrinkage in nodule size.
THE OTHER DIAGNOSTIC CHOICES
The other two choices are unlikely.
Calcinosis cutis results from the cutaneous deposition of insoluble compounds of calcium (hydroxyapatite or amorphous calcium phosphate), due to local or systemic factors, or both. This can be the result either of ectopic calcification in normal tissue in the setting of hypercalcemia or hyperphosphatemia, or of dystrophic calcification in damaged tissue. They appear as multiple, firm, whitish dermal papules, plaques, nodules, or subcutaneous nodules, which can sometimes ulcerate. They are radio-opaque. On biopsy, dermal deposits of calcium are seen, with or without a surrounding foreign-body giant-cell reaction. Calcium deposition may be confirmed on Von Kossa and alizarin red stains.
Tuberous xanthomas are firm, painless, red-yellow nodules that usually develop in pressure areas such as the extensor surfaces of the knees, the elbows, and the buttocks. They can be associated with familial dysbetalipoproteinemia, familial hypercholesterolemia, and even some of the secondary dyslipidemias. Histologic study shows accumulations of vacuolated lipid-laden macrophages (foamy histiocytes) and sometimes multinucleated histiocytes (Touton giant cells). The lipid droplets are dissolved during routine histologic processing, but lipid stains on frozen sections can be useful.
- Ziff M. The rheumatoid nodule. Arthritis Rheum 1990; 33:761–767.
- Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg 2007; 26:100–107.
- Bywaters EGL. A variant of rheumatoid arthritis characterized by recurrent digital pad nodules and palmar fasciitis, closely resembling palindromic rheumatism. Ann Rheum Dis 1949; 8:2–30.
- Couret M, Combe B, Chuong VT, et al. Rheumatoid nodulosis: report of two new cases and discussion of diagnostic criteria. J Rheumatol 1988; 15:1427–1430.
- McCarty DJ. Complete reversal of rheumatoid nodulosis. J Rheumatol 1991; 18:736–737.
- Kai Y, Anzai S, Shibuya H, et al. A case of rheumatoid nodulosis successfully treated with surgery. J Dermatol 2004; 31:910–915.
- Ching DW, Petrie JP, Klemp P, Jones JG. Injection therapy of superficial rheumatoid nodules. Br J Rheumatol 1992; 31:775–777.
- Ziff M. The rheumatoid nodule. Arthritis Rheum 1990; 33:761–767.
- Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg 2007; 26:100–107.
- Bywaters EGL. A variant of rheumatoid arthritis characterized by recurrent digital pad nodules and palmar fasciitis, closely resembling palindromic rheumatism. Ann Rheum Dis 1949; 8:2–30.
- Couret M, Combe B, Chuong VT, et al. Rheumatoid nodulosis: report of two new cases and discussion of diagnostic criteria. J Rheumatol 1988; 15:1427–1430.
- McCarty DJ. Complete reversal of rheumatoid nodulosis. J Rheumatol 1991; 18:736–737.
- Kai Y, Anzai S, Shibuya H, et al. A case of rheumatoid nodulosis successfully treated with surgery. J Dermatol 2004; 31:910–915.
- Ching DW, Petrie JP, Klemp P, Jones JG. Injection therapy of superficial rheumatoid nodules. Br J Rheumatol 1992; 31:775–777.
Eosinophilic esophagitis: An increasingly recognized cause of dysphagia, food impaction, and refractory heartburn
Abundant eosinophils in the esophagus were first described in 1977 in a 51-year-old man with dysphagia, chest pain, and a personal history of severe asthma and marked peripheral eosinophilia.1 In 1983, a similar case was reported in an adolescent with dysphagia.2 In both patients, large numbers of eosinophils were also noted in the duodenum, suggesting that these findings were part of a systemic hypereosinophilic syndrome.
Increased numbers of eosinophils in the gastrointestinal tract have been described in a number of diseases, including Crohn disease, connective tissue disorders, malignancy, various infections, and drug hypersensitivity reactions. However, not until 1993 was eosinophilic esophagitis described as a distinct clinical entity, consisting of isolated esophageal eosinophilia (typically more than 15 eosinophils per high-power field) in patients with dysphagia.3
Now, epidemiologic studies suggest that eosinophilic esophagitis may be as common as inflammatory bowel disease. In a study of children in Cincinnati, OH,4 the incidence was estimated at 10 per 100,000 children per year and the prevalence was estimated at 43 per 100,000. Of interest, 97% of cases were diagnosed after the year 2000.
RISING INCIDENCE, OR INCREASED RECOGNITION?
Over the last several years, the number of reported cases has increased substantially as interest in this disease has grown. The increase has been attributed in part to heightened awareness of this condition among clinicians and, hence, more esophageal biopsies being performed. Similarly, pathologists may have previously attributed esophageal eosinophilia to gastroesophageal reflux disease (GERD). However, the prevalence of eosinophilic esophagitis increased 10-fold between 1989 and 2003 in a fixed and stable adult population in Olten, Switzerland, suggesting that more than just increased awareness is responsible for this dramatic rise.5
PATHOGENESIS: SIMILAR TO OTHER ALLERGIC DISEASES?
The growing incidence of eosinophilic esophagitis parallels that of asthma, eczema, allergic rhinitis, and other atopic diseases, raising the possibility that these disorders share common environmental exposures and similar inflammatory pathways.6 The pathologic mechanisms of eosinophilic esophagitis are unknown, but emerging evidence suggests that, like other allergic diseases, it is an immune response mediated by type 2 T helper cells.
Several animal studies support this hypothesis. Mice sensitized and then exposed to aeroallergens developed both allergic airway inflammation and eosinophilic esophagitis. Interleukin 5, a cytokine involved in asthma, also helps recruit eosinophils into the esophagus, as transgenic mice deficient in interleukin 5 do not develop esophageal eosinophilia upon allergen exposure.7
Recently, eotaxin-3, a potent attractant for eosinophils, was shown to be markedly overexpressed in children with eosinophilic esophagitis compared with controls.8
Acid reflux does not appear to be a causative factor in most patients. However, reflux may play a secondary role, as some patients have experienced symptomatic, endoscopic, and histologic resolution of eosinophilic esophagitis after treatment with a proton pump inhibitor.9
GERD AND EOSINOPHILIC ESOPHAGITIS: WHAT IS THE RELATIONSHIP?
Given the high prevalence of GERD in the general population, much time and effort have been spent on comparing eosinophilic esophagitis with GERD. In fact, some endoscopic features typically seen in eosinophilic esophagitis were previously attributed to acid reflux.10
Both diseases share varying degrees of esophageal eosinophilia, and some have speculated on the relationship of eosinophilic esophagitis and GERD. Spechler et al11 recently suggested that the mucosal injury caused by acid reflux may allow swallowed allergens to penetrate an esophageal layer that is otherwise impermeable to most proteins, thereby causing mild eosinophilia. Conversely, the intense degranulation of activated eosinophils seen in eosinophilic esophagitis can trigger changes in the lower esophageal sphincter that could predispose to acid reflux.
Although their clinical and pathologic features may overlap, GERD and eosinophilic esophagitis appear to have different genetic profiles. In a recent pediatric study, Blanchard et al8 found that genes up-regulated in eosinophilic esophagitis were markedly different than those in chronic esophagitis. This suggests that while the two diseases share a constellation of symptoms, they have a different pathogenesis. Nevertheless, because of this possible overlap, the diagnosis of eosinophilic esophagitis should be made after acid reflux has been either treated or excluded with pH testing (see below).
THE ROLE OF ENVIRONMENTAL ALLERGENS AND GENETICS
Studies in children suggest that food allergies are a major contributor to eosinophilic esophagitis. In children, a strict amino-acid elemental diet has led to complete resolution of symptoms and a marked decrease in esophageal eosinophils. However, symptoms tend to recur once patients resume a regular diet.12
It is unclear if dietary modification is effective in adults. In six adults with eosinophilic esophagitis and a history of wheat and rye allergies, symptoms did not improve when these foods were eliminated and did not worsen when they were reintroduced.13
Of interest, there may be a seasonal variation of eosinophilic esophagitis, as suggested by a case report of a 21-year-old woman who had eosinophilic esophagitis that worsened symptomatically and histologically during the pollen season but resolved during winter. This is another example of the role aeroallergens may play in this disease.14
Evidence of a genetic predisposition to this disease is also growing, with a number of case reports describing multiple affected family members spanning generations.15
NEW CONSENSUS ON DIAGNOSTIC CRITERIA
The diagnosis of eosinophilic esophagitis is made histologically, with “marked” eosinophilia on esophageal biopsies, ie, usually 15 or more eosinophils per high-power field. In contrast, a normal esophagus contains almost no eosinophils,16 and esophageal biopsies of patients with GERD usually have fewer than 10 eosinophils per high-power field, with eosinophils limited to the distal esophagus.17
However, a recent systematic review of the literature found 10 different histologic definitions of eosinophilic esophagitis, ranging from more than 5 to more than 30 eosinophils, and more than one-third of the articles included in the review did not contain any specific diagnostic criteria. Similarly, a lack of consensus on the size of a high-power field (ranging from 0.12 to 0.44 mm2) resulted in a 23-fold variability in the description of eosinophil density. Moreover, the biopsy protocols were reported in only 39% of the articles.18
In view of the growing interest in this disease, its increasing recognition, the diagnostic ambiguity described above, and concern about the role of acid reflux, consensus recommendations for its diagnosis and treatment in adults and children have recently been published.19 The current consensus definition for eosinophilic esophagitis is:
- Clinical symptoms of esophageal dysfunction (eg, dysphagia, food impaction);
- At least 15 eosinophils per high-power field; and
- Either no response to a high-dose proton pump inhibitor or normal results on pH monitoring of the distal esophagus.
CLINICAL PRESENTATION
Eosinophilic esophagitis predominantly affects men between the ages of 20 and 40, but cases in women and in younger and older patients have also been reported. Recent systematic reviews found a male-to-female ratio of approximately 3:1.
More than 90% of adults with eosinophilic esophagitis present with intermittent difficulty swallowing solids, while food impaction occurs in more than 60%. Heartburn is the only manifestation in 24% of patients. Noncardiac chest pain, vomiting, and abdominal pain have also been seen, but less frequently.
Up to 80% of patients with eosinophilic esophagitis have a history of atopic disease such as asthma, allergic rhinitis, or allergies to food or medicine. One-third to one-half of patients have peripheral eosinophilia, and up to 55% have increased serum levels of immunoglobulin E (IgE).21
In children, presenting symptoms vary with age and include feeding disorders, vomiting, abdominal pain, and dysphagia. Moreover, children with eosinophilic esophagitis have a higher frequency of atopic symptoms and peripheral eosinophilia than do adults.5,22
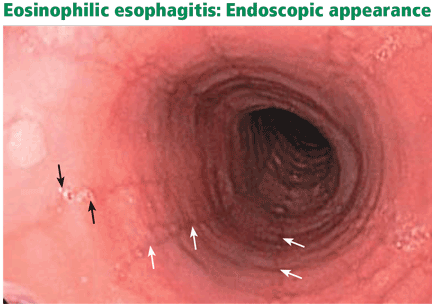
Although motor abnormalities are common in patients with eosinophilic esophagitis (up to 40% of patients have esophageal manometric abnormalities, including uncoordinated contractions and ineffective peristalsis),21 esophageal manometry is of limited diagnostic value and so is not recommended as a routine test.19
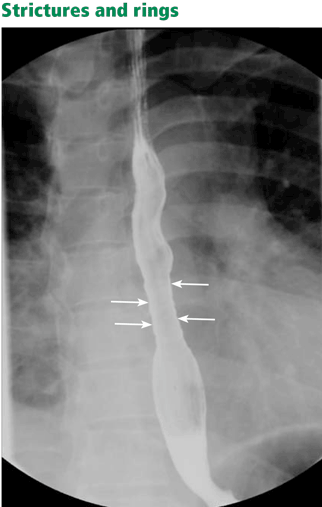
These findings support the theory that inflammation can lead to submucosal fibrosis, remodeling, narrowing, and eventually symptoms. Furthermore, two recent studies found that children with eosinophilic esophagitis had increased subepithelial collagen deposition in their biopsy specimens,24 suggesting increased potential for fibrosis. Also increased are transforming growth factor beta (a profibrotic cytokine) and vascular cell adhesion molecule 1, which is implicated in angiogenesis.25
Although many patients with eosinophilic esophagitis have abnormal findings on barium radiography, the test is most useful before esophagogastroduodenoscopy to determine whether a stricture is present and potentially to guide endoscopic dilation.19
NATURAL HISTORY: CHRONIC, RELAPSING, AND MOST LIKELY BENIGN
Our understanding of the natural history of eosinophilic esophagitis is limited, but the available evidence suggests that its prognosis is favorable.
Thirty adults followed for up to 11.5 years remained in good health, maintained their weight, and had no evidence of nutritional deficiencies.26 However, all but 1 patient continued to have dysphagia, with the overall intensity of dysphagia increasing in 7 (23%), remaining stable and persistent in 11 (37%), and decreasing in the remainder. In over half of these patients, the disease impaired quality of life. The only treatment offered was endoscopic dilation, which 11 patients required. Patients with peripheral blood eosinophilia and those with more pronounced findings on endoscopy were more likely to have symptoms at follow-up.
Although dysphagia persisted, the number of eosinophils in esophageal biopsy specimens decreased significantly over time, suggesting that the intense eosinophilic infiltration seen earlier in the disease may evolve into fibrosis and remodeling, similar to that seen in asthma and other chronic atopic diseases. Unlike in Barrett esophagus, a premalignant complication of longstanding GERD, there appeared to be no increased risk of esophageal cancer in these patients with eosinophilic esophagitis during the follow-up period.26
TREATMENT
Dietary therapy
Strict elemental amino-acid diets have resulted in complete symptomatic and histologic resolution of eosinophilic esophagitis in children. However, these elemental diets often have to be given by nasogastric tube because they are unpalatable, and the disease tends to return once the diet is discontinued.27
Elimination diets, based either on avoiding the six foods most commonly associated with allergy (egg, wheat, soy, cow’s milk protein, seafood, peanuts) or on allergy testing such as skin prick testing or atopy patch testing, have shown promise in children.12,28 However, similar large-scale studies of elimination diets in adults have not been conducted.
Allergy evaluation
The recent consensus recommendations devoted considerable attention to the role of allergy evaluation.19 Between 50% and 80% of patients with eosinophilic esophagitis have a coexisting atopic disease such as atopic dermatitis, eczema, allergic rhinitis, or asthma, with a higher prevalence in children than in adults. In these patients, evidence suggests that allergy testing may predict response to therapy. Therefore, the current recommendation is for all patients with eosinophilic esophagitis to undergo a complete evaluation by an experienced allergist.
Checking the peripheral blood eosinophil count before and after treatment is reasonable, as many patients have elevated eosinophil counts that decrease after treatment.
Similarly, many patients with eosinophilic esophagitis have elevated serum total IgE levels, which suggests a concomitant atopic disease. Therefore, total IgE levels should also be checked before and after treatment. Checking for IgE against specific aeroallergens is recommended, but checking for IgE against specific food antigens has not proven beneficial at this time. Similarly, skin prick testing for aeroallergens may be useful, but not for food allergens.
Data on atopy patch testing in eosinophilic esophagitis are currently limited but promising.19
Medical therapy
Swallowed fluticasone (Flonase, using an inhaler) is the mainstay of therapy for both children and adults.
In one case series, 21 adult patients with eosinophilic esophagitis received a 6-week course of swallowed fluticasone 220 μg/puff, two to four puffs twice daily. Symptoms completely resolved in all patients for at least 4 months, and no patient needed endoscopic dilation.29
In another study, 19 patients treated with fluticasone for 4 weeks showed dramatic improvement both symptomatically and histologically. However, after 3 months, 14 (74%) of the 19 patients had a recurrence of symptoms, pointing to the chronic relapsing nature of this disease.30
The only randomized placebo-controlled trial of fluticasone to date has been in children. Konikoff et al31 found that a 3-month course of fluticasone induced remission, defined as less than one eosinophil per high-power field, in 50% of patients, compared with 9% in the placebo group.
Swallowed fluticasone is generally well tolerated, although cases of esophageal candidiasis have been reported.30
Acid suppression still has an unclear role in the treatment of eosinophilic esophagitis. As mentioned above, the disease is defined as the presence or persistence of esophageal eosinophilia after acid reflux has been maximally treated or ruled out. Most patients referred for further evaluation of eosinophilic esophagitis have tried twice-daily proton pump inhibitor therapy without success. The impact of concomitant therapy with a proton pump inhibitor has not yet been determined, but the recent guidelines suggest that these drugs are reasonable as co-therapy in patients who also have GERD symptoms.19
In patients whose symptoms do not improve with fluticasone, several other medications have been used:
Systemic corticosteroids have been used with success in both adults and children with hypereosinophilic syndromes, as well as in patients with refractory eosinophilic esophagitis, but adverse effects limit their routine and long-term use.
Cromolyn sodium (NasalCrom, Intal), a mast cell stabilizer, and montelukast (Singulair), a leukotriene inhibitor, have been used with limited success.32
Mepolizumab (Bosatria), a humanized monoclonal antibody to human interleukin 5, decreased the number of eosinophils in the esophagus and peripheral blood and improved clinical symptoms in patients with refractory eosinophilic esophagitis in a recent open-label trial.33 Further studies with mepolizumab and other biologic agents are expected.
Endoscopic dilation
Endoscopic dilation with either a guidewire or a balloon technique is often used to treat strictures and a diffusely narrowed esophagus in patients with eosinophilic esophagitis.
As mentioned above, a common endoscopic feature is mucosal fragility, which has been described as resembling crepe paper. Shearing and longitudinal splitting of this fragile mucosa may occur after dilation therapy.
Although esophageal dilation may be done safely in patients with eosinophilic esophagitis, the risk of perforation appears to be greater than in those with other indications for dilation.
Nevertheless, immediate symptomatic improvement has been reported in 83% of patients after dilation, with symptoms recurring in 20% within 3 to 8 months.34 Current recommendations suggest that dilation should be done cautiously in patients who have documented esophageal narrowing for which drug therapy has failed.
RECOMMENDED APPROACH
The approach to diagnosing and treating eosinophilic esophagitis begins with being aware of its prevalence. One should suspect it more in younger patients presenting with intermittent dysphagia, food impaction, or heartburn that does not respond to maximal doses of a proton pump inhibitor. Special attention should be paid to a personal or family history of allergic diseases or similar symptoms.
According to the consensus recommendations, barium esophagography is useful if the presentation suggests long-standing disease and associated esophageal stricture.
Upper endoscopy is performed, with biopsies obtained in the proximal, middle, and distal esophagus regardless of the appearance of the esophageal mucosa. Biopsies of the stomach and duodenum are also recommended to rule out eosinophilic gastroenteritis.19
After biopsy confirms the diagnosis, a trial of a proton pump inhibitor in maximum doses (usually twice daily) for 8 weeks is recommended if not already tried. If there is evidence of eosinophilic esophagitis on repeat endoscopy and biopsy studies after proton pump inhibitor therapy, the next step is swallowed fluticasone (220 μg, up to four puffs twice daily) for 6 to 8 weeks, with follow-up visits to confirm resolution of symptoms. Without a spacer, the fluticasone is swallowed after maximal expiration. Patients are instructed to avoid food and liquids for at least 30 minutes after use.
Optimal strategies for monitoring in adults have yet to be established, and following symptoms alone may or may not be sufficient.19 Our approach is to follow for symptomatic improvement after treatment is completed, and to consider repeat endoscopy with biopsy if the patient’s symptoms do not improve or if the patient has a recurrence after treatment.
In patients with evidence of long-standing esophageal narrowing or poor response to drug therapy, esophageal dilation can be performed after careful consideration.
Although data are limited as to the role of specific allergens in adult eosinophilic esophagitis, patients with eosinophilic esophagitis are referred to an allergist for allergy testing. Offending food or aeroallergens are removed for a period of time and patients are followed for changes in symptoms.
For patients who do not respond to swallowed fluticasone, proton pump inhibitors, or both, other medications such as systemic steroids, montelukast, or cromolyn can be considered. In the near future, anti-interleukin 5 therapy may be another option.
Patients are asked to return periodically for evaluation after treatment. Due to the chronic and relapsing nature of eosinophilic esophagitis, various therapies (especially fluticasone) are often restarted or continued because of symptom recurrence.
- Dobbins JW, Sheahan DG, Behar J. Eosinophilic gastroenteritis with esophageal involvement. Gastroenterology 1977; 72:1312–1316.
- Matzinger MA, Daneman A. Esophageal involvement in eosinophilic gastroenteritis. Pediatr Radiol 1983; 13:35–38.
- Attwood SE, Smyrk TC, Demeester TR, Jones JB. Esophageal eosinophilia with dysphagia. Dig Dis Sci 1993; 38:109–116.
- Noel RJ, Putnam PE, Rothenberg ME. Eosinophilic esophagitis. N Engl J Med 2004; 351:940–941.
- Straumann A, Simon HU. Eosinophilic esophagitis: escalating epidemiology? J Allergy Clin Immunol 2005; 115:418–419.
- Rothenberg ME. Eosinophilic gastrointestinal disorders (EGID). J Allergy Clin Immunol 2004; 113:11–28.
- Mishra A, Rothenberg ME. Intratracheal IL-13 induces eosinophilic esophagitis by an IL-5, eotaxin-1, and STAT6-dependent mechanism. Gastroenterology 2003; 125:1419–1427.
- Blanchard C, Wang N, Stringer KF, et al. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest 2006; 116:536–547.
- Ngo P, Furuta G, Antonioli D, Fox V. Eosinophils in the esophagus—peptic or allergic eosinophilic esophagitis? Case series of three patients with esophageal eosinophilia. Am J Gastroenterol 2006; 101:1666–1670.
- Morrow JB, Vargo JJ, Goldblum JR, Richter JE. The ringed esophagus—histologic features of GERD. Am J Gastroenterol 2001; 96:984–989.
- Spechler SJ, Genta RM, Souza RF. Thoughts on the complex relationship between gastroesophageal reflux disease and eosinophilic esophagitis. Am J Gastroenterol 2007; 102:1301–1306.
- Markowitz JE, Spergel JM, Ruchelli E, Liacouras CA. Elemental diet is an effective treatment for eosinophilic esophagitis in children and adolescents. Am J Gastroenterol 2003; 98:777–782.
- Simon D, Straumann A, Wenk A, et al. Eosinophilic esophagitis in adults: no clinical relevance of wheat and rye sensitizations. Allergy 2006; 61:1480–1483.
- Fogg MI, Ruchelli E, Spergel JM. Pollen and eosinophilic esophagitis. J Allergy Clin Immunol 2003; 112:796–797.
- Zink DA, Amin M, Gebara S, Desai TK. Familial dysphagia and eosinophilia. Gastrointest Endoscop 2007; 65:330–334.
- Dellon ES, Aderoju A, Woosely JT, et al. Variability in diagnostic criteria for eosinophilic esophagitis: a systematic review. Am J Gastroenterol 2007; 102:2300–2313.
- Furuta GT, Liacouras CA, Collins MH, et al. Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology 2007; 133:1342–1363.
- Parfitt JR, Gregor JC, Suskin NG, Jawa HA. Eosinophilic esophagitis in adults: distinguishing features from gastroesophageal reflux disease: a study of 41 patients. Mod Pathol 2006; 19:90–96.
- Kato M, Kephart GM, Talley NJ, et al. Eosinophil infiltration and degranulation in normal human tissue. Anat Rec 1998; 242:418–425.
- Steiner SJ, Gupta SK, Croffie JM, Fitzgerald JF. Correlation between number of eosinophils and reflux index on same day esophageal biopsy and 24 hour esophageal pH monitoring. Am J Gastroenterol 2004; 99:801–805.
- Sgouros SN, Bergele C, Mantides A. Eosinophilic esophagitis in adults: a systematic review. Eur J Gastroenterol Hepatol 2006; 18:211–217.
- Liacouras CA, Spergel JM, Ruchelli E, Verma R. Eosinophilic esophagitis: a 10-year experience in 381 children. Clin Gastroenterol Hepatol 2005; 3:1198–1206.
- Zimmerman SL, Levine MS, Rubesin SE, et al. Idiopathic eosino-philic esophagitis in adults: the ringed esophagus. Radiology 2005; 236:159–165.
- Chehade M, Sampson HA, Morotti RA, Magrid MS. Esophageal sub-epithelial fibrosis in children with eosinophilic esophagitis. J Pediatr Gastroenterol Nutr 2007; 45:319–328.
- Aceves SS, Newbury RO, Dohil R, et al. Esophageal remodeling in pediatric eosinophilic esophagitis. J Allergy Clin Immunol 2007; 119:206–212.
- Straumann A, Spichtin HP, Grize L, et al. Natural history of primary eosinophilic esophagitis: a follow-up of 30 adult patients for up to 11.5 years. Gastroenterology 2003; 125:1660–1669.
- Kelly KJ, Lazenby AJ, Rowe PC, et al. Eosinophilic esophagitis attributed to gastroesophageal reflux: improvement with an amino acid-based formula. Gastroenterology 1995; 109:1503–1512.
- Kagalwalla AF, Sentongo TA, Ritz S, et al. Effect of six-food elimination diet on clinical and histologic outcomes in eosinophilic esophagitis. Clin Gastroenterol Hepatol 2006; 4:1097–1102.
- Arora AS, Perrault J, Smyrk TC. Topical corticosteroid treatment of dysphagia due to eosinophilic esophagitis in adults. Mayo Clin Proc 2003; 78:830–835.
- Remedios M, Campbell C, Jones DM, Kerlin P. Eosinophilic esophagitis in adults: clinical, endoscopic, histologic findings, and response to treatment with fluticasone propionate. Gastrointest Endoscop 2006; 63:3–12.
- Konikoff MR, Noel RJ, Blanchard C, et al. A randomized, double-blind, placebo-controlled trial of fluticasone propionate for pediatric eosinophilic esophagitis. Gastroenterology 2006; 131:1381–1391.
- Attwood SE, Lewis CJ, Bronder CS, et al. Eosinophilic oesophagitis: a novel treatment using montelukast. Gut 2003; 52:181–185.
- Stein ML, Collins MH, Villanueva JM, et al. Anti-IL-5 (mepolizumab) therapy for eosinophilic esophagitis. J Allergy Clin Immunol 2006; 118:1312–1319.
- Sgouros SN, Bergele C, Mantides A. Eosinophilic esophagitis in adults: what is the clinical significance? Endoscopy 2006; 38:512–520.
Abundant eosinophils in the esophagus were first described in 1977 in a 51-year-old man with dysphagia, chest pain, and a personal history of severe asthma and marked peripheral eosinophilia.1 In 1983, a similar case was reported in an adolescent with dysphagia.2 In both patients, large numbers of eosinophils were also noted in the duodenum, suggesting that these findings were part of a systemic hypereosinophilic syndrome.
Increased numbers of eosinophils in the gastrointestinal tract have been described in a number of diseases, including Crohn disease, connective tissue disorders, malignancy, various infections, and drug hypersensitivity reactions. However, not until 1993 was eosinophilic esophagitis described as a distinct clinical entity, consisting of isolated esophageal eosinophilia (typically more than 15 eosinophils per high-power field) in patients with dysphagia.3
Now, epidemiologic studies suggest that eosinophilic esophagitis may be as common as inflammatory bowel disease. In a study of children in Cincinnati, OH,4 the incidence was estimated at 10 per 100,000 children per year and the prevalence was estimated at 43 per 100,000. Of interest, 97% of cases were diagnosed after the year 2000.
RISING INCIDENCE, OR INCREASED RECOGNITION?
Over the last several years, the number of reported cases has increased substantially as interest in this disease has grown. The increase has been attributed in part to heightened awareness of this condition among clinicians and, hence, more esophageal biopsies being performed. Similarly, pathologists may have previously attributed esophageal eosinophilia to gastroesophageal reflux disease (GERD). However, the prevalence of eosinophilic esophagitis increased 10-fold between 1989 and 2003 in a fixed and stable adult population in Olten, Switzerland, suggesting that more than just increased awareness is responsible for this dramatic rise.5
PATHOGENESIS: SIMILAR TO OTHER ALLERGIC DISEASES?
The growing incidence of eosinophilic esophagitis parallels that of asthma, eczema, allergic rhinitis, and other atopic diseases, raising the possibility that these disorders share common environmental exposures and similar inflammatory pathways.6 The pathologic mechanisms of eosinophilic esophagitis are unknown, but emerging evidence suggests that, like other allergic diseases, it is an immune response mediated by type 2 T helper cells.
Several animal studies support this hypothesis. Mice sensitized and then exposed to aeroallergens developed both allergic airway inflammation and eosinophilic esophagitis. Interleukin 5, a cytokine involved in asthma, also helps recruit eosinophils into the esophagus, as transgenic mice deficient in interleukin 5 do not develop esophageal eosinophilia upon allergen exposure.7
Recently, eotaxin-3, a potent attractant for eosinophils, was shown to be markedly overexpressed in children with eosinophilic esophagitis compared with controls.8
Acid reflux does not appear to be a causative factor in most patients. However, reflux may play a secondary role, as some patients have experienced symptomatic, endoscopic, and histologic resolution of eosinophilic esophagitis after treatment with a proton pump inhibitor.9
GERD AND EOSINOPHILIC ESOPHAGITIS: WHAT IS THE RELATIONSHIP?
Given the high prevalence of GERD in the general population, much time and effort have been spent on comparing eosinophilic esophagitis with GERD. In fact, some endoscopic features typically seen in eosinophilic esophagitis were previously attributed to acid reflux.10
Both diseases share varying degrees of esophageal eosinophilia, and some have speculated on the relationship of eosinophilic esophagitis and GERD. Spechler et al11 recently suggested that the mucosal injury caused by acid reflux may allow swallowed allergens to penetrate an esophageal layer that is otherwise impermeable to most proteins, thereby causing mild eosinophilia. Conversely, the intense degranulation of activated eosinophils seen in eosinophilic esophagitis can trigger changes in the lower esophageal sphincter that could predispose to acid reflux.
Although their clinical and pathologic features may overlap, GERD and eosinophilic esophagitis appear to have different genetic profiles. In a recent pediatric study, Blanchard et al8 found that genes up-regulated in eosinophilic esophagitis were markedly different than those in chronic esophagitis. This suggests that while the two diseases share a constellation of symptoms, they have a different pathogenesis. Nevertheless, because of this possible overlap, the diagnosis of eosinophilic esophagitis should be made after acid reflux has been either treated or excluded with pH testing (see below).
THE ROLE OF ENVIRONMENTAL ALLERGENS AND GENETICS
Studies in children suggest that food allergies are a major contributor to eosinophilic esophagitis. In children, a strict amino-acid elemental diet has led to complete resolution of symptoms and a marked decrease in esophageal eosinophils. However, symptoms tend to recur once patients resume a regular diet.12
It is unclear if dietary modification is effective in adults. In six adults with eosinophilic esophagitis and a history of wheat and rye allergies, symptoms did not improve when these foods were eliminated and did not worsen when they were reintroduced.13
Of interest, there may be a seasonal variation of eosinophilic esophagitis, as suggested by a case report of a 21-year-old woman who had eosinophilic esophagitis that worsened symptomatically and histologically during the pollen season but resolved during winter. This is another example of the role aeroallergens may play in this disease.14
Evidence of a genetic predisposition to this disease is also growing, with a number of case reports describing multiple affected family members spanning generations.15
NEW CONSENSUS ON DIAGNOSTIC CRITERIA
The diagnosis of eosinophilic esophagitis is made histologically, with “marked” eosinophilia on esophageal biopsies, ie, usually 15 or more eosinophils per high-power field. In contrast, a normal esophagus contains almost no eosinophils,16 and esophageal biopsies of patients with GERD usually have fewer than 10 eosinophils per high-power field, with eosinophils limited to the distal esophagus.17
However, a recent systematic review of the literature found 10 different histologic definitions of eosinophilic esophagitis, ranging from more than 5 to more than 30 eosinophils, and more than one-third of the articles included in the review did not contain any specific diagnostic criteria. Similarly, a lack of consensus on the size of a high-power field (ranging from 0.12 to 0.44 mm2) resulted in a 23-fold variability in the description of eosinophil density. Moreover, the biopsy protocols were reported in only 39% of the articles.18
In view of the growing interest in this disease, its increasing recognition, the diagnostic ambiguity described above, and concern about the role of acid reflux, consensus recommendations for its diagnosis and treatment in adults and children have recently been published.19 The current consensus definition for eosinophilic esophagitis is:
- Clinical symptoms of esophageal dysfunction (eg, dysphagia, food impaction);
- At least 15 eosinophils per high-power field; and
- Either no response to a high-dose proton pump inhibitor or normal results on pH monitoring of the distal esophagus.
CLINICAL PRESENTATION
Eosinophilic esophagitis predominantly affects men between the ages of 20 and 40, but cases in women and in younger and older patients have also been reported. Recent systematic reviews found a male-to-female ratio of approximately 3:1.
More than 90% of adults with eosinophilic esophagitis present with intermittent difficulty swallowing solids, while food impaction occurs in more than 60%. Heartburn is the only manifestation in 24% of patients. Noncardiac chest pain, vomiting, and abdominal pain have also been seen, but less frequently.
Up to 80% of patients with eosinophilic esophagitis have a history of atopic disease such as asthma, allergic rhinitis, or allergies to food or medicine. One-third to one-half of patients have peripheral eosinophilia, and up to 55% have increased serum levels of immunoglobulin E (IgE).21
In children, presenting symptoms vary with age and include feeding disorders, vomiting, abdominal pain, and dysphagia. Moreover, children with eosinophilic esophagitis have a higher frequency of atopic symptoms and peripheral eosinophilia than do adults.5,22
Although motor abnormalities are common in patients with eosinophilic esophagitis (up to 40% of patients have esophageal manometric abnormalities, including uncoordinated contractions and ineffective peristalsis),21 esophageal manometry is of limited diagnostic value and so is not recommended as a routine test.19
These findings support the theory that inflammation can lead to submucosal fibrosis, remodeling, narrowing, and eventually symptoms. Furthermore, two recent studies found that children with eosinophilic esophagitis had increased subepithelial collagen deposition in their biopsy specimens,24 suggesting increased potential for fibrosis. Also increased are transforming growth factor beta (a profibrotic cytokine) and vascular cell adhesion molecule 1, which is implicated in angiogenesis.25
Although many patients with eosinophilic esophagitis have abnormal findings on barium radiography, the test is most useful before esophagogastroduodenoscopy to determine whether a stricture is present and potentially to guide endoscopic dilation.19
NATURAL HISTORY: CHRONIC, RELAPSING, AND MOST LIKELY BENIGN
Our understanding of the natural history of eosinophilic esophagitis is limited, but the available evidence suggests that its prognosis is favorable.
Thirty adults followed for up to 11.5 years remained in good health, maintained their weight, and had no evidence of nutritional deficiencies.26 However, all but 1 patient continued to have dysphagia, with the overall intensity of dysphagia increasing in 7 (23%), remaining stable and persistent in 11 (37%), and decreasing in the remainder. In over half of these patients, the disease impaired quality of life. The only treatment offered was endoscopic dilation, which 11 patients required. Patients with peripheral blood eosinophilia and those with more pronounced findings on endoscopy were more likely to have symptoms at follow-up.
Although dysphagia persisted, the number of eosinophils in esophageal biopsy specimens decreased significantly over time, suggesting that the intense eosinophilic infiltration seen earlier in the disease may evolve into fibrosis and remodeling, similar to that seen in asthma and other chronic atopic diseases. Unlike in Barrett esophagus, a premalignant complication of longstanding GERD, there appeared to be no increased risk of esophageal cancer in these patients with eosinophilic esophagitis during the follow-up period.26
TREATMENT
Dietary therapy
Strict elemental amino-acid diets have resulted in complete symptomatic and histologic resolution of eosinophilic esophagitis in children. However, these elemental diets often have to be given by nasogastric tube because they are unpalatable, and the disease tends to return once the diet is discontinued.27
Elimination diets, based either on avoiding the six foods most commonly associated with allergy (egg, wheat, soy, cow’s milk protein, seafood, peanuts) or on allergy testing such as skin prick testing or atopy patch testing, have shown promise in children.12,28 However, similar large-scale studies of elimination diets in adults have not been conducted.
Allergy evaluation
The recent consensus recommendations devoted considerable attention to the role of allergy evaluation.19 Between 50% and 80% of patients with eosinophilic esophagitis have a coexisting atopic disease such as atopic dermatitis, eczema, allergic rhinitis, or asthma, with a higher prevalence in children than in adults. In these patients, evidence suggests that allergy testing may predict response to therapy. Therefore, the current recommendation is for all patients with eosinophilic esophagitis to undergo a complete evaluation by an experienced allergist.
Checking the peripheral blood eosinophil count before and after treatment is reasonable, as many patients have elevated eosinophil counts that decrease after treatment.
Similarly, many patients with eosinophilic esophagitis have elevated serum total IgE levels, which suggests a concomitant atopic disease. Therefore, total IgE levels should also be checked before and after treatment. Checking for IgE against specific aeroallergens is recommended, but checking for IgE against specific food antigens has not proven beneficial at this time. Similarly, skin prick testing for aeroallergens may be useful, but not for food allergens.
Data on atopy patch testing in eosinophilic esophagitis are currently limited but promising.19
Medical therapy
Swallowed fluticasone (Flonase, using an inhaler) is the mainstay of therapy for both children and adults.
In one case series, 21 adult patients with eosinophilic esophagitis received a 6-week course of swallowed fluticasone 220 μg/puff, two to four puffs twice daily. Symptoms completely resolved in all patients for at least 4 months, and no patient needed endoscopic dilation.29
In another study, 19 patients treated with fluticasone for 4 weeks showed dramatic improvement both symptomatically and histologically. However, after 3 months, 14 (74%) of the 19 patients had a recurrence of symptoms, pointing to the chronic relapsing nature of this disease.30
The only randomized placebo-controlled trial of fluticasone to date has been in children. Konikoff et al31 found that a 3-month course of fluticasone induced remission, defined as less than one eosinophil per high-power field, in 50% of patients, compared with 9% in the placebo group.
Swallowed fluticasone is generally well tolerated, although cases of esophageal candidiasis have been reported.30
Acid suppression still has an unclear role in the treatment of eosinophilic esophagitis. As mentioned above, the disease is defined as the presence or persistence of esophageal eosinophilia after acid reflux has been maximally treated or ruled out. Most patients referred for further evaluation of eosinophilic esophagitis have tried twice-daily proton pump inhibitor therapy without success. The impact of concomitant therapy with a proton pump inhibitor has not yet been determined, but the recent guidelines suggest that these drugs are reasonable as co-therapy in patients who also have GERD symptoms.19
In patients whose symptoms do not improve with fluticasone, several other medications have been used:
Systemic corticosteroids have been used with success in both adults and children with hypereosinophilic syndromes, as well as in patients with refractory eosinophilic esophagitis, but adverse effects limit their routine and long-term use.
Cromolyn sodium (NasalCrom, Intal), a mast cell stabilizer, and montelukast (Singulair), a leukotriene inhibitor, have been used with limited success.32
Mepolizumab (Bosatria), a humanized monoclonal antibody to human interleukin 5, decreased the number of eosinophils in the esophagus and peripheral blood and improved clinical symptoms in patients with refractory eosinophilic esophagitis in a recent open-label trial.33 Further studies with mepolizumab and other biologic agents are expected.
Endoscopic dilation
Endoscopic dilation with either a guidewire or a balloon technique is often used to treat strictures and a diffusely narrowed esophagus in patients with eosinophilic esophagitis.
As mentioned above, a common endoscopic feature is mucosal fragility, which has been described as resembling crepe paper. Shearing and longitudinal splitting of this fragile mucosa may occur after dilation therapy.
Although esophageal dilation may be done safely in patients with eosinophilic esophagitis, the risk of perforation appears to be greater than in those with other indications for dilation.
Nevertheless, immediate symptomatic improvement has been reported in 83% of patients after dilation, with symptoms recurring in 20% within 3 to 8 months.34 Current recommendations suggest that dilation should be done cautiously in patients who have documented esophageal narrowing for which drug therapy has failed.
RECOMMENDED APPROACH
The approach to diagnosing and treating eosinophilic esophagitis begins with being aware of its prevalence. One should suspect it more in younger patients presenting with intermittent dysphagia, food impaction, or heartburn that does not respond to maximal doses of a proton pump inhibitor. Special attention should be paid to a personal or family history of allergic diseases or similar symptoms.
According to the consensus recommendations, barium esophagography is useful if the presentation suggests long-standing disease and associated esophageal stricture.
Upper endoscopy is performed, with biopsies obtained in the proximal, middle, and distal esophagus regardless of the appearance of the esophageal mucosa. Biopsies of the stomach and duodenum are also recommended to rule out eosinophilic gastroenteritis.19
After biopsy confirms the diagnosis, a trial of a proton pump inhibitor in maximum doses (usually twice daily) for 8 weeks is recommended if not already tried. If there is evidence of eosinophilic esophagitis on repeat endoscopy and biopsy studies after proton pump inhibitor therapy, the next step is swallowed fluticasone (220 μg, up to four puffs twice daily) for 6 to 8 weeks, with follow-up visits to confirm resolution of symptoms. Without a spacer, the fluticasone is swallowed after maximal expiration. Patients are instructed to avoid food and liquids for at least 30 minutes after use.
Optimal strategies for monitoring in adults have yet to be established, and following symptoms alone may or may not be sufficient.19 Our approach is to follow for symptomatic improvement after treatment is completed, and to consider repeat endoscopy with biopsy if the patient’s symptoms do not improve or if the patient has a recurrence after treatment.
In patients with evidence of long-standing esophageal narrowing or poor response to drug therapy, esophageal dilation can be performed after careful consideration.
Although data are limited as to the role of specific allergens in adult eosinophilic esophagitis, patients with eosinophilic esophagitis are referred to an allergist for allergy testing. Offending food or aeroallergens are removed for a period of time and patients are followed for changes in symptoms.
For patients who do not respond to swallowed fluticasone, proton pump inhibitors, or both, other medications such as systemic steroids, montelukast, or cromolyn can be considered. In the near future, anti-interleukin 5 therapy may be another option.
Patients are asked to return periodically for evaluation after treatment. Due to the chronic and relapsing nature of eosinophilic esophagitis, various therapies (especially fluticasone) are often restarted or continued because of symptom recurrence.
Abundant eosinophils in the esophagus were first described in 1977 in a 51-year-old man with dysphagia, chest pain, and a personal history of severe asthma and marked peripheral eosinophilia.1 In 1983, a similar case was reported in an adolescent with dysphagia.2 In both patients, large numbers of eosinophils were also noted in the duodenum, suggesting that these findings were part of a systemic hypereosinophilic syndrome.
Increased numbers of eosinophils in the gastrointestinal tract have been described in a number of diseases, including Crohn disease, connective tissue disorders, malignancy, various infections, and drug hypersensitivity reactions. However, not until 1993 was eosinophilic esophagitis described as a distinct clinical entity, consisting of isolated esophageal eosinophilia (typically more than 15 eosinophils per high-power field) in patients with dysphagia.3
Now, epidemiologic studies suggest that eosinophilic esophagitis may be as common as inflammatory bowel disease. In a study of children in Cincinnati, OH,4 the incidence was estimated at 10 per 100,000 children per year and the prevalence was estimated at 43 per 100,000. Of interest, 97% of cases were diagnosed after the year 2000.
RISING INCIDENCE, OR INCREASED RECOGNITION?
Over the last several years, the number of reported cases has increased substantially as interest in this disease has grown. The increase has been attributed in part to heightened awareness of this condition among clinicians and, hence, more esophageal biopsies being performed. Similarly, pathologists may have previously attributed esophageal eosinophilia to gastroesophageal reflux disease (GERD). However, the prevalence of eosinophilic esophagitis increased 10-fold between 1989 and 2003 in a fixed and stable adult population in Olten, Switzerland, suggesting that more than just increased awareness is responsible for this dramatic rise.5
PATHOGENESIS: SIMILAR TO OTHER ALLERGIC DISEASES?
The growing incidence of eosinophilic esophagitis parallels that of asthma, eczema, allergic rhinitis, and other atopic diseases, raising the possibility that these disorders share common environmental exposures and similar inflammatory pathways.6 The pathologic mechanisms of eosinophilic esophagitis are unknown, but emerging evidence suggests that, like other allergic diseases, it is an immune response mediated by type 2 T helper cells.
Several animal studies support this hypothesis. Mice sensitized and then exposed to aeroallergens developed both allergic airway inflammation and eosinophilic esophagitis. Interleukin 5, a cytokine involved in asthma, also helps recruit eosinophils into the esophagus, as transgenic mice deficient in interleukin 5 do not develop esophageal eosinophilia upon allergen exposure.7
Recently, eotaxin-3, a potent attractant for eosinophils, was shown to be markedly overexpressed in children with eosinophilic esophagitis compared with controls.8
Acid reflux does not appear to be a causative factor in most patients. However, reflux may play a secondary role, as some patients have experienced symptomatic, endoscopic, and histologic resolution of eosinophilic esophagitis after treatment with a proton pump inhibitor.9
GERD AND EOSINOPHILIC ESOPHAGITIS: WHAT IS THE RELATIONSHIP?
Given the high prevalence of GERD in the general population, much time and effort have been spent on comparing eosinophilic esophagitis with GERD. In fact, some endoscopic features typically seen in eosinophilic esophagitis were previously attributed to acid reflux.10
Both diseases share varying degrees of esophageal eosinophilia, and some have speculated on the relationship of eosinophilic esophagitis and GERD. Spechler et al11 recently suggested that the mucosal injury caused by acid reflux may allow swallowed allergens to penetrate an esophageal layer that is otherwise impermeable to most proteins, thereby causing mild eosinophilia. Conversely, the intense degranulation of activated eosinophils seen in eosinophilic esophagitis can trigger changes in the lower esophageal sphincter that could predispose to acid reflux.
Although their clinical and pathologic features may overlap, GERD and eosinophilic esophagitis appear to have different genetic profiles. In a recent pediatric study, Blanchard et al8 found that genes up-regulated in eosinophilic esophagitis were markedly different than those in chronic esophagitis. This suggests that while the two diseases share a constellation of symptoms, they have a different pathogenesis. Nevertheless, because of this possible overlap, the diagnosis of eosinophilic esophagitis should be made after acid reflux has been either treated or excluded with pH testing (see below).
THE ROLE OF ENVIRONMENTAL ALLERGENS AND GENETICS
Studies in children suggest that food allergies are a major contributor to eosinophilic esophagitis. In children, a strict amino-acid elemental diet has led to complete resolution of symptoms and a marked decrease in esophageal eosinophils. However, symptoms tend to recur once patients resume a regular diet.12
It is unclear if dietary modification is effective in adults. In six adults with eosinophilic esophagitis and a history of wheat and rye allergies, symptoms did not improve when these foods were eliminated and did not worsen when they were reintroduced.13
Of interest, there may be a seasonal variation of eosinophilic esophagitis, as suggested by a case report of a 21-year-old woman who had eosinophilic esophagitis that worsened symptomatically and histologically during the pollen season but resolved during winter. This is another example of the role aeroallergens may play in this disease.14
Evidence of a genetic predisposition to this disease is also growing, with a number of case reports describing multiple affected family members spanning generations.15
NEW CONSENSUS ON DIAGNOSTIC CRITERIA
The diagnosis of eosinophilic esophagitis is made histologically, with “marked” eosinophilia on esophageal biopsies, ie, usually 15 or more eosinophils per high-power field. In contrast, a normal esophagus contains almost no eosinophils,16 and esophageal biopsies of patients with GERD usually have fewer than 10 eosinophils per high-power field, with eosinophils limited to the distal esophagus.17
However, a recent systematic review of the literature found 10 different histologic definitions of eosinophilic esophagitis, ranging from more than 5 to more than 30 eosinophils, and more than one-third of the articles included in the review did not contain any specific diagnostic criteria. Similarly, a lack of consensus on the size of a high-power field (ranging from 0.12 to 0.44 mm2) resulted in a 23-fold variability in the description of eosinophil density. Moreover, the biopsy protocols were reported in only 39% of the articles.18
In view of the growing interest in this disease, its increasing recognition, the diagnostic ambiguity described above, and concern about the role of acid reflux, consensus recommendations for its diagnosis and treatment in adults and children have recently been published.19 The current consensus definition for eosinophilic esophagitis is:
- Clinical symptoms of esophageal dysfunction (eg, dysphagia, food impaction);
- At least 15 eosinophils per high-power field; and
- Either no response to a high-dose proton pump inhibitor or normal results on pH monitoring of the distal esophagus.
CLINICAL PRESENTATION
Eosinophilic esophagitis predominantly affects men between the ages of 20 and 40, but cases in women and in younger and older patients have also been reported. Recent systematic reviews found a male-to-female ratio of approximately 3:1.
More than 90% of adults with eosinophilic esophagitis present with intermittent difficulty swallowing solids, while food impaction occurs in more than 60%. Heartburn is the only manifestation in 24% of patients. Noncardiac chest pain, vomiting, and abdominal pain have also been seen, but less frequently.
Up to 80% of patients with eosinophilic esophagitis have a history of atopic disease such as asthma, allergic rhinitis, or allergies to food or medicine. One-third to one-half of patients have peripheral eosinophilia, and up to 55% have increased serum levels of immunoglobulin E (IgE).21
In children, presenting symptoms vary with age and include feeding disorders, vomiting, abdominal pain, and dysphagia. Moreover, children with eosinophilic esophagitis have a higher frequency of atopic symptoms and peripheral eosinophilia than do adults.5,22
Although motor abnormalities are common in patients with eosinophilic esophagitis (up to 40% of patients have esophageal manometric abnormalities, including uncoordinated contractions and ineffective peristalsis),21 esophageal manometry is of limited diagnostic value and so is not recommended as a routine test.19
These findings support the theory that inflammation can lead to submucosal fibrosis, remodeling, narrowing, and eventually symptoms. Furthermore, two recent studies found that children with eosinophilic esophagitis had increased subepithelial collagen deposition in their biopsy specimens,24 suggesting increased potential for fibrosis. Also increased are transforming growth factor beta (a profibrotic cytokine) and vascular cell adhesion molecule 1, which is implicated in angiogenesis.25
Although many patients with eosinophilic esophagitis have abnormal findings on barium radiography, the test is most useful before esophagogastroduodenoscopy to determine whether a stricture is present and potentially to guide endoscopic dilation.19
NATURAL HISTORY: CHRONIC, RELAPSING, AND MOST LIKELY BENIGN
Our understanding of the natural history of eosinophilic esophagitis is limited, but the available evidence suggests that its prognosis is favorable.
Thirty adults followed for up to 11.5 years remained in good health, maintained their weight, and had no evidence of nutritional deficiencies.26 However, all but 1 patient continued to have dysphagia, with the overall intensity of dysphagia increasing in 7 (23%), remaining stable and persistent in 11 (37%), and decreasing in the remainder. In over half of these patients, the disease impaired quality of life. The only treatment offered was endoscopic dilation, which 11 patients required. Patients with peripheral blood eosinophilia and those with more pronounced findings on endoscopy were more likely to have symptoms at follow-up.
Although dysphagia persisted, the number of eosinophils in esophageal biopsy specimens decreased significantly over time, suggesting that the intense eosinophilic infiltration seen earlier in the disease may evolve into fibrosis and remodeling, similar to that seen in asthma and other chronic atopic diseases. Unlike in Barrett esophagus, a premalignant complication of longstanding GERD, there appeared to be no increased risk of esophageal cancer in these patients with eosinophilic esophagitis during the follow-up period.26
TREATMENT
Dietary therapy
Strict elemental amino-acid diets have resulted in complete symptomatic and histologic resolution of eosinophilic esophagitis in children. However, these elemental diets often have to be given by nasogastric tube because they are unpalatable, and the disease tends to return once the diet is discontinued.27
Elimination diets, based either on avoiding the six foods most commonly associated with allergy (egg, wheat, soy, cow’s milk protein, seafood, peanuts) or on allergy testing such as skin prick testing or atopy patch testing, have shown promise in children.12,28 However, similar large-scale studies of elimination diets in adults have not been conducted.
Allergy evaluation
The recent consensus recommendations devoted considerable attention to the role of allergy evaluation.19 Between 50% and 80% of patients with eosinophilic esophagitis have a coexisting atopic disease such as atopic dermatitis, eczema, allergic rhinitis, or asthma, with a higher prevalence in children than in adults. In these patients, evidence suggests that allergy testing may predict response to therapy. Therefore, the current recommendation is for all patients with eosinophilic esophagitis to undergo a complete evaluation by an experienced allergist.
Checking the peripheral blood eosinophil count before and after treatment is reasonable, as many patients have elevated eosinophil counts that decrease after treatment.
Similarly, many patients with eosinophilic esophagitis have elevated serum total IgE levels, which suggests a concomitant atopic disease. Therefore, total IgE levels should also be checked before and after treatment. Checking for IgE against specific aeroallergens is recommended, but checking for IgE against specific food antigens has not proven beneficial at this time. Similarly, skin prick testing for aeroallergens may be useful, but not for food allergens.
Data on atopy patch testing in eosinophilic esophagitis are currently limited but promising.19
Medical therapy
Swallowed fluticasone (Flonase, using an inhaler) is the mainstay of therapy for both children and adults.
In one case series, 21 adult patients with eosinophilic esophagitis received a 6-week course of swallowed fluticasone 220 μg/puff, two to four puffs twice daily. Symptoms completely resolved in all patients for at least 4 months, and no patient needed endoscopic dilation.29
In another study, 19 patients treated with fluticasone for 4 weeks showed dramatic improvement both symptomatically and histologically. However, after 3 months, 14 (74%) of the 19 patients had a recurrence of symptoms, pointing to the chronic relapsing nature of this disease.30
The only randomized placebo-controlled trial of fluticasone to date has been in children. Konikoff et al31 found that a 3-month course of fluticasone induced remission, defined as less than one eosinophil per high-power field, in 50% of patients, compared with 9% in the placebo group.
Swallowed fluticasone is generally well tolerated, although cases of esophageal candidiasis have been reported.30
Acid suppression still has an unclear role in the treatment of eosinophilic esophagitis. As mentioned above, the disease is defined as the presence or persistence of esophageal eosinophilia after acid reflux has been maximally treated or ruled out. Most patients referred for further evaluation of eosinophilic esophagitis have tried twice-daily proton pump inhibitor therapy without success. The impact of concomitant therapy with a proton pump inhibitor has not yet been determined, but the recent guidelines suggest that these drugs are reasonable as co-therapy in patients who also have GERD symptoms.19
In patients whose symptoms do not improve with fluticasone, several other medications have been used:
Systemic corticosteroids have been used with success in both adults and children with hypereosinophilic syndromes, as well as in patients with refractory eosinophilic esophagitis, but adverse effects limit their routine and long-term use.
Cromolyn sodium (NasalCrom, Intal), a mast cell stabilizer, and montelukast (Singulair), a leukotriene inhibitor, have been used with limited success.32
Mepolizumab (Bosatria), a humanized monoclonal antibody to human interleukin 5, decreased the number of eosinophils in the esophagus and peripheral blood and improved clinical symptoms in patients with refractory eosinophilic esophagitis in a recent open-label trial.33 Further studies with mepolizumab and other biologic agents are expected.
Endoscopic dilation
Endoscopic dilation with either a guidewire or a balloon technique is often used to treat strictures and a diffusely narrowed esophagus in patients with eosinophilic esophagitis.
As mentioned above, a common endoscopic feature is mucosal fragility, which has been described as resembling crepe paper. Shearing and longitudinal splitting of this fragile mucosa may occur after dilation therapy.
Although esophageal dilation may be done safely in patients with eosinophilic esophagitis, the risk of perforation appears to be greater than in those with other indications for dilation.
Nevertheless, immediate symptomatic improvement has been reported in 83% of patients after dilation, with symptoms recurring in 20% within 3 to 8 months.34 Current recommendations suggest that dilation should be done cautiously in patients who have documented esophageal narrowing for which drug therapy has failed.
RECOMMENDED APPROACH
The approach to diagnosing and treating eosinophilic esophagitis begins with being aware of its prevalence. One should suspect it more in younger patients presenting with intermittent dysphagia, food impaction, or heartburn that does not respond to maximal doses of a proton pump inhibitor. Special attention should be paid to a personal or family history of allergic diseases or similar symptoms.
According to the consensus recommendations, barium esophagography is useful if the presentation suggests long-standing disease and associated esophageal stricture.
Upper endoscopy is performed, with biopsies obtained in the proximal, middle, and distal esophagus regardless of the appearance of the esophageal mucosa. Biopsies of the stomach and duodenum are also recommended to rule out eosinophilic gastroenteritis.19
After biopsy confirms the diagnosis, a trial of a proton pump inhibitor in maximum doses (usually twice daily) for 8 weeks is recommended if not already tried. If there is evidence of eosinophilic esophagitis on repeat endoscopy and biopsy studies after proton pump inhibitor therapy, the next step is swallowed fluticasone (220 μg, up to four puffs twice daily) for 6 to 8 weeks, with follow-up visits to confirm resolution of symptoms. Without a spacer, the fluticasone is swallowed after maximal expiration. Patients are instructed to avoid food and liquids for at least 30 minutes after use.
Optimal strategies for monitoring in adults have yet to be established, and following symptoms alone may or may not be sufficient.19 Our approach is to follow for symptomatic improvement after treatment is completed, and to consider repeat endoscopy with biopsy if the patient’s symptoms do not improve or if the patient has a recurrence after treatment.
In patients with evidence of long-standing esophageal narrowing or poor response to drug therapy, esophageal dilation can be performed after careful consideration.
Although data are limited as to the role of specific allergens in adult eosinophilic esophagitis, patients with eosinophilic esophagitis are referred to an allergist for allergy testing. Offending food or aeroallergens are removed for a period of time and patients are followed for changes in symptoms.
For patients who do not respond to swallowed fluticasone, proton pump inhibitors, or both, other medications such as systemic steroids, montelukast, or cromolyn can be considered. In the near future, anti-interleukin 5 therapy may be another option.
Patients are asked to return periodically for evaluation after treatment. Due to the chronic and relapsing nature of eosinophilic esophagitis, various therapies (especially fluticasone) are often restarted or continued because of symptom recurrence.
- Dobbins JW, Sheahan DG, Behar J. Eosinophilic gastroenteritis with esophageal involvement. Gastroenterology 1977; 72:1312–1316.
- Matzinger MA, Daneman A. Esophageal involvement in eosinophilic gastroenteritis. Pediatr Radiol 1983; 13:35–38.
- Attwood SE, Smyrk TC, Demeester TR, Jones JB. Esophageal eosinophilia with dysphagia. Dig Dis Sci 1993; 38:109–116.
- Noel RJ, Putnam PE, Rothenberg ME. Eosinophilic esophagitis. N Engl J Med 2004; 351:940–941.
- Straumann A, Simon HU. Eosinophilic esophagitis: escalating epidemiology? J Allergy Clin Immunol 2005; 115:418–419.
- Rothenberg ME. Eosinophilic gastrointestinal disorders (EGID). J Allergy Clin Immunol 2004; 113:11–28.
- Mishra A, Rothenberg ME. Intratracheal IL-13 induces eosinophilic esophagitis by an IL-5, eotaxin-1, and STAT6-dependent mechanism. Gastroenterology 2003; 125:1419–1427.
- Blanchard C, Wang N, Stringer KF, et al. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest 2006; 116:536–547.
- Ngo P, Furuta G, Antonioli D, Fox V. Eosinophils in the esophagus—peptic or allergic eosinophilic esophagitis? Case series of three patients with esophageal eosinophilia. Am J Gastroenterol 2006; 101:1666–1670.
- Morrow JB, Vargo JJ, Goldblum JR, Richter JE. The ringed esophagus—histologic features of GERD. Am J Gastroenterol 2001; 96:984–989.
- Spechler SJ, Genta RM, Souza RF. Thoughts on the complex relationship between gastroesophageal reflux disease and eosinophilic esophagitis. Am J Gastroenterol 2007; 102:1301–1306.
- Markowitz JE, Spergel JM, Ruchelli E, Liacouras CA. Elemental diet is an effective treatment for eosinophilic esophagitis in children and adolescents. Am J Gastroenterol 2003; 98:777–782.
- Simon D, Straumann A, Wenk A, et al. Eosinophilic esophagitis in adults: no clinical relevance of wheat and rye sensitizations. Allergy 2006; 61:1480–1483.
- Fogg MI, Ruchelli E, Spergel JM. Pollen and eosinophilic esophagitis. J Allergy Clin Immunol 2003; 112:796–797.
- Zink DA, Amin M, Gebara S, Desai TK. Familial dysphagia and eosinophilia. Gastrointest Endoscop 2007; 65:330–334.
- Dellon ES, Aderoju A, Woosely JT, et al. Variability in diagnostic criteria for eosinophilic esophagitis: a systematic review. Am J Gastroenterol 2007; 102:2300–2313.
- Furuta GT, Liacouras CA, Collins MH, et al. Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology 2007; 133:1342–1363.
- Parfitt JR, Gregor JC, Suskin NG, Jawa HA. Eosinophilic esophagitis in adults: distinguishing features from gastroesophageal reflux disease: a study of 41 patients. Mod Pathol 2006; 19:90–96.
- Kato M, Kephart GM, Talley NJ, et al. Eosinophil infiltration and degranulation in normal human tissue. Anat Rec 1998; 242:418–425.
- Steiner SJ, Gupta SK, Croffie JM, Fitzgerald JF. Correlation between number of eosinophils and reflux index on same day esophageal biopsy and 24 hour esophageal pH monitoring. Am J Gastroenterol 2004; 99:801–805.
- Sgouros SN, Bergele C, Mantides A. Eosinophilic esophagitis in adults: a systematic review. Eur J Gastroenterol Hepatol 2006; 18:211–217.
- Liacouras CA, Spergel JM, Ruchelli E, Verma R. Eosinophilic esophagitis: a 10-year experience in 381 children. Clin Gastroenterol Hepatol 2005; 3:1198–1206.
- Zimmerman SL, Levine MS, Rubesin SE, et al. Idiopathic eosino-philic esophagitis in adults: the ringed esophagus. Radiology 2005; 236:159–165.
- Chehade M, Sampson HA, Morotti RA, Magrid MS. Esophageal sub-epithelial fibrosis in children with eosinophilic esophagitis. J Pediatr Gastroenterol Nutr 2007; 45:319–328.
- Aceves SS, Newbury RO, Dohil R, et al. Esophageal remodeling in pediatric eosinophilic esophagitis. J Allergy Clin Immunol 2007; 119:206–212.
- Straumann A, Spichtin HP, Grize L, et al. Natural history of primary eosinophilic esophagitis: a follow-up of 30 adult patients for up to 11.5 years. Gastroenterology 2003; 125:1660–1669.
- Kelly KJ, Lazenby AJ, Rowe PC, et al. Eosinophilic esophagitis attributed to gastroesophageal reflux: improvement with an amino acid-based formula. Gastroenterology 1995; 109:1503–1512.
- Kagalwalla AF, Sentongo TA, Ritz S, et al. Effect of six-food elimination diet on clinical and histologic outcomes in eosinophilic esophagitis. Clin Gastroenterol Hepatol 2006; 4:1097–1102.
- Arora AS, Perrault J, Smyrk TC. Topical corticosteroid treatment of dysphagia due to eosinophilic esophagitis in adults. Mayo Clin Proc 2003; 78:830–835.
- Remedios M, Campbell C, Jones DM, Kerlin P. Eosinophilic esophagitis in adults: clinical, endoscopic, histologic findings, and response to treatment with fluticasone propionate. Gastrointest Endoscop 2006; 63:3–12.
- Konikoff MR, Noel RJ, Blanchard C, et al. A randomized, double-blind, placebo-controlled trial of fluticasone propionate for pediatric eosinophilic esophagitis. Gastroenterology 2006; 131:1381–1391.
- Attwood SE, Lewis CJ, Bronder CS, et al. Eosinophilic oesophagitis: a novel treatment using montelukast. Gut 2003; 52:181–185.
- Stein ML, Collins MH, Villanueva JM, et al. Anti-IL-5 (mepolizumab) therapy for eosinophilic esophagitis. J Allergy Clin Immunol 2006; 118:1312–1319.
- Sgouros SN, Bergele C, Mantides A. Eosinophilic esophagitis in adults: what is the clinical significance? Endoscopy 2006; 38:512–520.
- Dobbins JW, Sheahan DG, Behar J. Eosinophilic gastroenteritis with esophageal involvement. Gastroenterology 1977; 72:1312–1316.
- Matzinger MA, Daneman A. Esophageal involvement in eosinophilic gastroenteritis. Pediatr Radiol 1983; 13:35–38.
- Attwood SE, Smyrk TC, Demeester TR, Jones JB. Esophageal eosinophilia with dysphagia. Dig Dis Sci 1993; 38:109–116.
- Noel RJ, Putnam PE, Rothenberg ME. Eosinophilic esophagitis. N Engl J Med 2004; 351:940–941.
- Straumann A, Simon HU. Eosinophilic esophagitis: escalating epidemiology? J Allergy Clin Immunol 2005; 115:418–419.
- Rothenberg ME. Eosinophilic gastrointestinal disorders (EGID). J Allergy Clin Immunol 2004; 113:11–28.
- Mishra A, Rothenberg ME. Intratracheal IL-13 induces eosinophilic esophagitis by an IL-5, eotaxin-1, and STAT6-dependent mechanism. Gastroenterology 2003; 125:1419–1427.
- Blanchard C, Wang N, Stringer KF, et al. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest 2006; 116:536–547.
- Ngo P, Furuta G, Antonioli D, Fox V. Eosinophils in the esophagus—peptic or allergic eosinophilic esophagitis? Case series of three patients with esophageal eosinophilia. Am J Gastroenterol 2006; 101:1666–1670.
- Morrow JB, Vargo JJ, Goldblum JR, Richter JE. The ringed esophagus—histologic features of GERD. Am J Gastroenterol 2001; 96:984–989.
- Spechler SJ, Genta RM, Souza RF. Thoughts on the complex relationship between gastroesophageal reflux disease and eosinophilic esophagitis. Am J Gastroenterol 2007; 102:1301–1306.
- Markowitz JE, Spergel JM, Ruchelli E, Liacouras CA. Elemental diet is an effective treatment for eosinophilic esophagitis in children and adolescents. Am J Gastroenterol 2003; 98:777–782.
- Simon D, Straumann A, Wenk A, et al. Eosinophilic esophagitis in adults: no clinical relevance of wheat and rye sensitizations. Allergy 2006; 61:1480–1483.
- Fogg MI, Ruchelli E, Spergel JM. Pollen and eosinophilic esophagitis. J Allergy Clin Immunol 2003; 112:796–797.
- Zink DA, Amin M, Gebara S, Desai TK. Familial dysphagia and eosinophilia. Gastrointest Endoscop 2007; 65:330–334.
- Dellon ES, Aderoju A, Woosely JT, et al. Variability in diagnostic criteria for eosinophilic esophagitis: a systematic review. Am J Gastroenterol 2007; 102:2300–2313.
- Furuta GT, Liacouras CA, Collins MH, et al. Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology 2007; 133:1342–1363.
- Parfitt JR, Gregor JC, Suskin NG, Jawa HA. Eosinophilic esophagitis in adults: distinguishing features from gastroesophageal reflux disease: a study of 41 patients. Mod Pathol 2006; 19:90–96.
- Kato M, Kephart GM, Talley NJ, et al. Eosinophil infiltration and degranulation in normal human tissue. Anat Rec 1998; 242:418–425.
- Steiner SJ, Gupta SK, Croffie JM, Fitzgerald JF. Correlation between number of eosinophils and reflux index on same day esophageal biopsy and 24 hour esophageal pH monitoring. Am J Gastroenterol 2004; 99:801–805.
- Sgouros SN, Bergele C, Mantides A. Eosinophilic esophagitis in adults: a systematic review. Eur J Gastroenterol Hepatol 2006; 18:211–217.
- Liacouras CA, Spergel JM, Ruchelli E, Verma R. Eosinophilic esophagitis: a 10-year experience in 381 children. Clin Gastroenterol Hepatol 2005; 3:1198–1206.
- Zimmerman SL, Levine MS, Rubesin SE, et al. Idiopathic eosino-philic esophagitis in adults: the ringed esophagus. Radiology 2005; 236:159–165.
- Chehade M, Sampson HA, Morotti RA, Magrid MS. Esophageal sub-epithelial fibrosis in children with eosinophilic esophagitis. J Pediatr Gastroenterol Nutr 2007; 45:319–328.
- Aceves SS, Newbury RO, Dohil R, et al. Esophageal remodeling in pediatric eosinophilic esophagitis. J Allergy Clin Immunol 2007; 119:206–212.
- Straumann A, Spichtin HP, Grize L, et al. Natural history of primary eosinophilic esophagitis: a follow-up of 30 adult patients for up to 11.5 years. Gastroenterology 2003; 125:1660–1669.
- Kelly KJ, Lazenby AJ, Rowe PC, et al. Eosinophilic esophagitis attributed to gastroesophageal reflux: improvement with an amino acid-based formula. Gastroenterology 1995; 109:1503–1512.
- Kagalwalla AF, Sentongo TA, Ritz S, et al. Effect of six-food elimination diet on clinical and histologic outcomes in eosinophilic esophagitis. Clin Gastroenterol Hepatol 2006; 4:1097–1102.
- Arora AS, Perrault J, Smyrk TC. Topical corticosteroid treatment of dysphagia due to eosinophilic esophagitis in adults. Mayo Clin Proc 2003; 78:830–835.
- Remedios M, Campbell C, Jones DM, Kerlin P. Eosinophilic esophagitis in adults: clinical, endoscopic, histologic findings, and response to treatment with fluticasone propionate. Gastrointest Endoscop 2006; 63:3–12.
- Konikoff MR, Noel RJ, Blanchard C, et al. A randomized, double-blind, placebo-controlled trial of fluticasone propionate for pediatric eosinophilic esophagitis. Gastroenterology 2006; 131:1381–1391.
- Attwood SE, Lewis CJ, Bronder CS, et al. Eosinophilic oesophagitis: a novel treatment using montelukast. Gut 2003; 52:181–185.
- Stein ML, Collins MH, Villanueva JM, et al. Anti-IL-5 (mepolizumab) therapy for eosinophilic esophagitis. J Allergy Clin Immunol 2006; 118:1312–1319.
- Sgouros SN, Bergele C, Mantides A. Eosinophilic esophagitis in adults: what is the clinical significance? Endoscopy 2006; 38:512–520.
KEY POINTS
- The diagnosis is made with upper endoscopy and esophageal biopsies that show diffuse infiltration of eosinophils.
- Current treatment in adults is limited and consists of either swallowed fluticasone (Flonase) or a proton pump inhibitor.
- Because many patients with eosinophilic esophagitis have atopic disease, a complete evaluation for dietary allergens and aeroallergens is recommended, as avoidance of these allergens may be helpful in some adults.
- Cautious endoscopic dilation is a treatment option in patients with evidence of esophageal stenosis. Systemic corticosteroids and novel biologic therapy have been used in refractory cases.
IgA nephropathy: Challenges and opportunities
Much progress has been made in the 40 years since immunoglobulin A (IgA) nephropathy was first described. We now have a reasonably complete understanding of the pathogenesis and mediation of this disease, but its etiology remains obscure and mysterious. New data on its epidemiology continue to emerge that will undoubtedly have clinical significance. We are beginning to perceive—but only dimly—the genetic predisposition to the disease.
Prognostication remains an imperfect science, but we are clearly making progress. The role of pathology in estimating prognosis in individual patients needs to be thoroughly reexamined, based on a uniformly agreed-upon classification scheme. Such work is currently in progress.
Therapy has certainly advanced, and we now have the rudiments of an evidence-based approach to management. However, much more needs to be done to refine these strategies so that they can be better matched to the characteristics of the patients, and there is a great need for novel therapeutic approaches and more information on multidrug regimens in selected patients. Many opportunities exist for improvement in the control of this common cause of chronic kidney disease, but we should not underestimate the challenges that present themselves in the field of IgA nephropathy in 2008 and beyond.
THE SCOPE OF THE PROBLEM
IgA nephropathy, also called Berger disease, is the most common form of primary glomerular disease in the developed world.1,2 Morphologically, it is characterized by diffuse deposition of IgA in the glomerular mesangium and by various degrees of damage of the glomerular capillary network seen on light microscopy.3,4 By some estimates, as many as 5% to 15% (averaging about 10%) of the general population may have IgA deposits in the glomerular mesangium, but only about 1 in 50 people with IgA deposits will actually have some abnormal clinical manifestation (principally recurring bouts of hematuria, with or without accompanying proteinuria) that brings them to the attention of a physician.5
Although not all patients with IgA nephropathy have progressive renal disease, IgA nephropathy is a significant contributor to the incidence of end-stage renal disease (ESRD) in many countries.1–4
DIAGNOSTIC AND PROGNOSTIC CHALLENGES
Since 1968, when IgA nephropathy was first described,6 great strides have been made in clarifying its epidemiology, its pathogenesis, the prognostic factors involved in its progression to ESRD, and its treatment. However, many gaps in our knowledge remain, particularly regarding its etiology, the genetic factors predisposing to it, its therapy, and the problem of recurrent disease in renal transplant recipients.
Can IgA nephropathy be diagnosed without a renal biopsy?
While renal biopsy and immunochemical analysis of renal tissue remain the gold standard for diagnosing IgA nephropathy, new sensitive and reasonably specific noninvasive tests are emerging and may provide another diagnostic approach. One of the most promising new tests is for abnormal circulating levels of abnormally glycosylated IgA subclass 1 (IgA1), which appears to be involved in the pathogenesis of the disease (see below).7 If noninvasive diagnostic techniques can be simplified and their accuracy validated across diverse populations, they offer great promise for use in epidemiologic and genetic studies, in which routine renal biopsy for diagnosis is impractical.
Signs and symptoms of IgA nephropathy are nonspecific
The most common clinical presentation of IgA nephropathy is recurring bouts of macroscopic hematuria, often but not invariably accompanied by proteinuria.2 Persistent asymptomatic hematuria without any detectable proteinuria (so-called isolated hematuria) affects a minority of patients. The red cells in the urine are typically dysmorphic (altered in size and shape compared with normal red cells), as they are in many other glomerulonephritic diseases.
Because low-grade fever and pain in the loins may accompany these bouts of hematuria, the disorder is often initially mistaken for urinary tract infection or urolithiasis. Careful microscopic examination of the urinary sediment for the characteristic dysmorphic erythrocytes that indicate a glomerular disease often provides the crucial clue that a glomerular disorder is the cause of the hematuria.8
However, a somewhat similar presentation may also be seen in thin basement membrane nephropathy, Alport syndrome (hereditary nephritis), and membranoproliferative glomerulonephritis,2 although these disorders can be readily distinguished from IgA nephropathy on examination of renal biopsy material under light, immunofluorescence, and electron microscopy. In addition, serum complement levels are typically reduced in membranoproliferative glomerulonephritis, and a family history of nephritis (without father-to-son transmission), often with deafness, can be obtained in the X-linked form of Alport syndrome. IgA nephropathy can be reliably distinguished from thin basement membrane nephropathy only by renal biopsy and electron microscopy.
Can we better predict which patients with IgA nephropathy will develop renal failure?
Although the rate of progression is very slow, and in only about 50% (or less) of patients does IgA nephropathy progress to ESRD within 25 years of diagnosis, the risk varies considerably among populations.9 Spontaneous clinical remissions are relatively uncommon in adults but much more common among children.
Several factors, if present at the time of discovery or developing within a relatively short time thereafter (usually within 6 months to 1 year), appear to predict a progressive course and, eventually, ESRD.9,10 We need to characterize and validate these risk factors in detail to be able to design and carry out appropriately powered, randomized, controlled clinical trials of treatment.
Unfortunately, cumulatively, the risk factors identified so far explain less than 50% of the variation in observed outcome of IgA nephropathy. Many of the risk factors identified so far are primarily indicators of the extent of disease at a particular time, and it is therefore not surprising that they would have some ability to predict the later behavior of the disease.
Clinical and pathologic risk factors in IgA nephropathy
Although imperfect, the major risk factors auguring a poor prognosis are:
- Proteinuria (> 500 mg/day) that persists for more than 6 months
- Elevated serum creatinine at diagnosis
- Microscopic hematuria that persists for more than 6 months
- Poorly controlled hypertension
- Extensive glomerulosclerosis or interstitial fibrosis or both on renal biopsy.7,10
Extensive crescentic disease also confers a worse short-term prognosis, often accompanied by a rapidly progressive loss of renal function.
Are clinical risk factors more useful than pathologic risk factors in IgA nephropathy?
Of importance, clinical factors, such as persistent proteinuria or declining renal function on follow-up appear to have greater predictive power than pathologic factors for long-term outcome.9–12 Clinical factors, such as decreasing estimated glomerular filtration rate (GFR) after short-term follow-up, persistent moderate to marked proteinuria (500–1,000 mg/day, or more), hyperuricemia, hyperlipidemia, concomitant obesity, poorly controlled hypertension, absence of treatment with angiotensin II inhibitors, and, possibly, persistent micro-hematuria are the most consistent factors independently associated with a poor prognosis in multivariate analysis. Pathologic changes noted in the original diagnostic renal biopsy do not consistently add greatly to the precision of prognosis beyond the analysis of these clinical and laboratory factors.11
A detailed and uniform immunologic and morphologic approach to classifying the pathology of IgA nephropathy may yet uncover some new and very useful prognostic factors, independent of those generated by simple clinical assessment. Efforts are under way, and such a development would greatly improve the accuracy and precision of outcome prediction and reduce the amount of unexplained variation in prognosis observed in groups of patients with IgA nephropathy.
At present, the heterogeneity of participants in clinical trials of therapy, the tendency for the disease to progress slowly, and the variation in prognosis due to unexplained factors pose major challenges in designing and carrying out randomized controlled trials of therapy in IgA nephropathy. If we can find new risk factors that can predict progressive disease earlier, the knowledge will help us in designing future clinical trials, which will be vital if progress is to be made towards controlling IgA nephropathy.
Prognosis in individual patients vs populations with IgA nephropathy
At present, we need a way to determine the prognosis more precisely in individual patients rather than in groups of patients. After all, physicians are called upon to determine the likely outcome in single patients, not in a population. Several prediction formulas have been devised, most of them based on relatively simple clinical factors present at discovery or short-term follow-up.12,13
Conventional pathologic observations have limited utility in such individualized prognostic formulations.12 This is not to say that renal biopsy only offers diagnostic utility and has little if any value as a prognostic tool. However, the challenge is to enhance the prognostic usefulness of renal biopsy by refining the examination of the tissue specimens using modern approaches and to conduct the appropriate correlative studies to confirm the value of new pathologic criteria in prognostication, independent of clinical features alone.
For example, the risk of ESRD is greater if the patient has very extensive (> 50%) crescentic glomerular involvement with a rapidly progressive glomerulonephritic evolution. The risk is less if there are minimal glomerular changes with nephrotic-range proteinuria. Extensive interstitial fibrosis and glomerulosclerosis in the original “diagnostic” renal biopsy merely highlight the existence of prior progressive disease that is likely to continue. The significance of persistent focal necrotizing glomerular lesions (capillaritis) in IgA nephropathy, often associated with persistent microhematuria, is not entirely clear and needs to be specifically explored, especially as it pertains to the need for immunosuppressive therapy added to treatment for hypertension, proteinuria, or both with inhibitors of the renin-angiotensin system (see below).
At present, the most powerful prognostic factor in IgA nephropathy is moderate to severe proteinuria that persists for 6 months or longer.9,10,12 The relationship between the level of proteinuria and the outcome is continuous, ie, the greater the proteinuria, the worse the prognosis. Compared with some other primary glomerular diseases (such as membranous nephropathy or focal and segmental glomerulosclerosis), progressive disease in IgA nephropathy is associated with lower levels of persistent proteinuria (usually 500 mg to 3 g/day).
The estimated GFR at the time IgA nephropathy is discovered is a rather weak independent predictor of outcome (up to a point; see below). Many patients have stable (but reduced) renal function in the long term, especially if they receive angiotensin II inhibitor therapy and can keep their systolic blood pressure between 110 and 120 mm Hg.
How can IgA nephropathy be diagnosed and treated before the ‘point of no return’?
For patients at risk of developing ESRD, the two most critical goals of treatment are to:
- Control blood pressure rigorously, preferably with an angiotensin-converting enzyme (ACE) inhibitor, an angiotensin II receptor antagonist (ARB), or both, and
- Reduce proteinuria to less than 500 mg/day.
If these two goals can be met without undue side effects and if the patient remains compliant in the long term, many patients can avoid ESRD. Patients who cannot achieve these goals despite vigorous attempts become candidates for adjunctive therapy, such as pulse intravenous methylprednisolone (Solu-Medrol) combined with oral prednisone, or in some cases a cytotoxic drug combined with prednisone. Small randomized controlled trials suggest these adjunctive treatments are effective and safe.
Unfortunately, IgA nephropathy can progress silently, and many patients do not receive the diagnosis until late in its course. In such patients, the disease may relentlessly progress even with optimal therapy. The “point of no return” appears to be an estimated GFR of about 30 mL/min/1.73 m2 (stage 4 chronic kidney disease).14
These observations underscore the need for early diagnosis and treatment based on factors that accurately predict an unfavorable outcome. Finding these factors will not be easy, because it will require detailed observation of homogeneous groups of patients over prolonged periods of time. New findings show great promise for identifying patients earlier in the course of disease who are more or less likely to progress to ESRD. The challenge is to translate these findings into rational, safe, and effective therapies applicable across a broad spectrum of disease.
OPPORTUNITIES: GENETICS, PROTEOMICS, NEW TESTS AND TREATMENTS
Genetic studies may lead to novel treatments for IgA nephropathy
Susceptibility to IgA nephropathy has a genetic component to varying degrees, depending on geography and the existence of “founder effects.” Familial forms of IgA nephropathy are more common in northern Italy and in eastern Kentucky. The familial cases may derive from a mutation of a specific gene occurring in a founder many hundreds of years ago. Several genetic loci are strongly associated with IgA nephropathy (usually as an autosomal-dominant trait with highly variable penetrance).15 Familial IgA nephropathy is most likely genetically heterogeneous, and many cases of IgA nephropathy that are believed to be sporadic may actually have a less apparent genetic basis, with skipped generations, lanthanic (covert) disease, and incomplete penetrance.
At present, genetic testing based on genomic or transcriptosomic analysis does not appear to have much diagnostic value except in clearly familial cases, because many loci are involved. Many asymptomatic people have mesangial IgA deposits that could be detected by renal biopsy but not by genetic analysis, and this inability is a major obstacle for genetic susceptibility studies. Indeed, most current genetic studies actually examine susceptibility to the clinical expression of disease rather than susceptibility to the mesangial IgA deposition that underlies the disease.5
The opportunity that lies ahead in genetic testing of IgA nephropathy (including haplotype analysis) appears to be primarily in the elucidation of potential pathogenetic pathways and in the refinement of prognosis and the definition of treatment responsiveness (pharmacogenomics).
If a gene (or group of genes) can be identified that is strongly and consistently associated with IgA nephropathy across diverse populations, its protein product isolated and characterized, and its role in pathogenesis elucidated, then a new era in targeted therapy of IgA nephropathy will be unleashed, much in the same way as the identification of tyrosine phosphatases played a role in the design of targeted therapy in chronic myelogenous leukemia. Early progress is being made in this area, but many obstacles lie in the way.
Proteomics may prove useful in diagnosis and prognosis of IgA nephropathy
Proteomics—the characterization and analysis of the patient’s entire complement of serum and urinary proteins—is a new, exciting, and largely unexplored area in IgA nephropathy. Preliminary studies have shown that this technique may provide a novel noninvasive means of diagnosing IgA nephropathy, and it may have additional value as a prognostic tool.16
Much work needs to be done to standardize how specimens are collected, stored, and shipped and to verify the precision and accuracy of proteomics in diverse populations of patients with IgA nephropathy, patients with other glomerular diseases, and normal subjects to ascertain this technique’s false-negative and false-positive rates.
IgA1 testing may help detect IgA nephropathy early in its course
Abnormally undergalactosylated and oversialyted epitopes at the hinge region of the IgA1 molecule play a critical role in the pathogenesis of sporadic IgA nephropathy.17 This discovery provides a great opportunity for profiling patients suspected of having IgA nephropathy on the basis of sensitive determination of the serum level of these abnormal IgA1 molecules.7
It may be that pathogenic IgA1 molecules (and autoantibodies to them) arise many months or even years before the onset of clinical manifestations of overt IgA nephropathy, similar to the situation known to occur in systemic lupus erythematosus. It is also possible that an abnormality of the disposal of immune complexes created by the interaction of autoantibodies with the abnormally glycosylated IgA1 creates the opportunity for preferential glomerular mesangial deposition of polymeric IgA.
Clearly, the greatest opportunity lies with understanding the fundamental abnormality leading to defective O-linked galactosylation of the serine/threonine residues at the hinge region of IgA1 in IgA nephropathy. In addition, it would be very useful to know if this is a generalized and acquired abnormality or whether it is focal in distribution (eg, in the tonsils, bone marrow, or lymphoid tissue in the gut).
Knowledge of secondary mediators may also lead to new treatments for IgA nephropathy
Detailed knowledge of the participation of specific cell types and the “cytokine milieu” (eg, interleukin 4, interferon) in directing the abnormality toward defective glycosylation would also be very important in designing new approaches to diagnosis and therapy.
A better understanding is slowly emerging of the pathways by which pathogenic immune complexes containing IgA are deposited and cleared, and of the secondary mediator systems evoked by their formation and tissue localization. Interference with these secondary mediator processes, such as alternative or mannose-dependent complement activation, platelet-derived growth factor or transforming growth factor stimulation, also offers a new approach to therapy.
We lack a suitable animal model of IgA nephropathy that mimics all aspects of the human condition, which has impeded progress in this area. A fully humanized mouse model of disease would be a welcome addition to the investigative toolkit.
Prognostic biopsy analysis may be improved in IgA nephropathy
As discussed above, the science of prognostication and stratification of patients with IgA nephropathy into those at high or low risk of ESRD has clearly advanced but is still quite incomplete, especially with respect to individual patients.
Great opportunities lie in refining the value of renal biopsy in prognostication. Although the “snapshot” nature and potential sampling errors intrinsic to diagnostic renal biopsy cannot easily be overcome, at least not without performing multiple and repeated renal biopsies (a very impractical approach to prognostication), refinements in the laboratory seem to offer numerous opportunities for advancement. Much better clinicopathological correlations, especially with respect to outcomes, among well-characterized patients with IgA nephropathy are greatly needed. New nonconventional markers of progression, such as “tubulitis,” deposition of fibroblast-specific proteins, and the proteome of the deposited immunoglobulins and complement show much promise.18
Immunosuppressive therapy could be added to ACE inhibitors or ARBs in IgA nephropathy
The management of IgA nephropathy has clearly advanced over the last several decades, largely as the result of randomized clinical trials.3,19 However, these trials had serious limitations: the numbers of patients were relatively small, follow-up was relatively short, and the findings may not apply to the IgA nephropathy population at large or to specific patients having features that diverge from those in the patients enrolled in the studies.
The value of initial therapy with an ACE inhibitor, an ARB, or both in combination appears well established. However, details of dosage, duration of therapy, and the relative values of monotherapy and combined therapy remain uncertain.
Many opportunities for combining angiotensin II inhibition and immunosuppressive therapy are being explored. By and large, all current therapies are empiric and their long-term effects relatively uncertain, owing to small study size and short duration.
Oral and parenteral glucocorticoids,20 combined regimens of cyclophosphamide (Cytoxan) and azathioprine (Imuran),21 omega-3 fatty acids,22 and anticoagulants and anti-thrombotics3 each have their advocates and their specified target populations.
Tonsillectomy as a treatment has been particularly controversial. While no controlled studies have been performed yet, observational studies (most of them conducted in some prefectures in Japan) have suggested a higher rate of clinical remission with tonsillectomy than with steroid treatment alone.5 However, long-term observations have not shown any consistent effect of tonsillectomy on progression to ESRD.
We hope that a better understanding of the fundamental mechanisms of disease and its mediation will provide an impetus for development of more rational targeted therapy. Evaluating potentially promising targeted therapies will be very difficult. Evaluation of safety and efficacy with long-term use will be a key requirement for a successful novel therapeutic agent.
FOR NOW, AN EMPIRIC APPROACH TO IGA NEPHROPATHY
Start with an angiotensin II inhibitor
The current body of evidence for choosing a particular therapeutic approach for a given patient with IgA nephropathy cannot be regarded as definitive, owing to limitations in the quality and strength of the trials serving as the basis of the evidence. Nonetheless, patients with IgA nephropathy and abnormal protein excretion (> 500 mg/day) should probably always be given angiotensin II inhibitor therapy (an ACE inhibitor, an ARB, or both) if they have no contraindications to it such as a hypersensitivity reaction or pregnancy, as a base for future monitoring and adjuvant therapy.
A response, tentatively defined as a 30% to 50% decline in proteinuria from baseline levels or a decrease to less than 500 mg/day, would be a reason to continue this conservative approach. Lack of a response after several months of observation at maximal tolerated dosage (plus salt restriction or a diuretic) would be a reason for considering adjuvant therapy.
If the patient does not respond to an ACE inhibitor or ARB and his or her estimated GFR is over 70 mL/min/1.73 m2, a trial of oral and parenteral glucocorticoids might be undertaken, as suggested by Pozzi and coworkers.20
On the other hand, if the estimated GFR is in the range of 30 to 70 mL/min/1.73 m2 and declining at a rate that predicts that ESRD will develop in less than 5 to 7 years, this would be a possible indication for low-dose oral cyclo-phosphamide and then azathioprine, as suggested by Ballardie and Roberts.21 Omega-3 fatty acids (Omacor) could also be considered as add-on therapy, particularly for patients with very heavy proteinuria (> 3.0 g/d) and reduced estimated GFR.22
Patients with an estimated GFR of less than 30 mL/min/1.73 m2 and chronic (irreversible) changes on renal biopsy—the point of no return—probably will not respond to any therapy other than an ACE inhibitor, an ARB, or both.
The role of more aggressive immunosuppression
At present, the evidence for using mycophenolate mofetil (CellCept) or calcineurin inhibitors (such as cyclosporin or tacrolimus) is fragmentary or contradictory.3,19,23 Similarly, the benefits of long-term azathioprine therapy are based on observational data only and so it cannot be recommended as evidence-based.24 Opportunities exist for combined therapy (eg, an ACE inhibitor or an ARB or both, combined with omega-3 fatty acids and azathioprine or mycophenolate mofetil), but at present, controlled trials are lacking. Crescentic disease and rapidly progressive glomerulonephritis should probably be treated with combined cyclophosphamide and parental and oral corticosteroids, based on observational data. Patients with IgA nephropathy and minimal change disease with nephrotic syndrome should be treated with oral steroids, but the only data available are observational. Low-protein diets could be tried in the presence of slowly progressive renal disease with estimated GFR less than 30 mL/min/1.73 m2, but there are no controlled trials demonstrating efficacy for this approach in IgA neph-ropathy.
Renal transplantation is very successful
Renal transplantation is a very suitable alternative for patients with IgA nephropathy that progresses to ESRD. Overall success rates are as good or better than those in other primary glomerular diseases. Unfortunately, the disease recurs in the majority of renal grafts and may in some cases lead to loss of the graft.25,26 We need much more information on the factors that predict such recurrences and their undesirable effects on transplantation outcomes.
MUCH WORK TO BE DONE
Much work needs to be done in the field of therapeutics in IgA nephropathy. Much of this effort will hinge on the interests of the pharmaceutical industry in IgA nephropathy as a potential therapeutic market. At present, the prospects for the development of a safe and effective novel therapy for IgA nephropathy (eg, approvable by the US Food and Drug Administration) do not appear great, but this may be overly pessimistic. The nature of the disease mandates long-term observation, agents that are very safe (with low rates of ESRD, death, and transplantation), and dependency on surrogate markers of efficacy. Therefore, designing and executing studies will not be easy.
- Tomino Y. IgA nephropathy today. Contrib Nephrol 2007; 157:1–255.
- D’Amico G. The commonest glomerulonephritis in the world: IgA nephropathy. Quart J Med 1987; 245:709–727.
- Lee G, Glassock RJ. Immunoglobulin A nephropathy. In:Ponticelli C, Glassock R, editors. Treatment of Primary Glomerulonephritis. Oxford: Oxford Medical Publication, 1997:187–217.
- Donadio JV, Grande JP. IgA nephropathy. N Engl J Med 2002; 347:738–748.
- Glassock RJ. Concluding remarks. IgA nephropathy today. Contrib Nephrol 2002; 157:169–173.
- Berger J, Hinglais N. Les dépots intercapillaries d’IgA-IgG. J Urol Nephrol (Paris) 1968; 74:694–700.
- Moldoveanu Z, Wyatt RJ, Lee JY, et al. Patients with IgA nephropa- levels. Kidney Int thy have increased serum galactose deficient IgA1. 2002; 71:1148–1154.
- Kincaid-Smith P, Fairley K. The investigation of hematuria. Semin Nephrol 2005; 25:127–135.
- Coppo R, D’Amico G. Factors predicting progression of IgA nephropathies. J Nephrol 2005; 18:503–512.
- Donadio JV, Bergstralh EJ, Grande JP, Rademcher DM. Proteinuria patterns and their association with subsequent end-stage renal disease in IgA nephropathy. Nephrol Dial Transplant 2002; 17:1197–1203.
- Cook T. Interpretation of renal biopsies in IgA nephropathy. Contrib Nephrol 2007; 157:44–49.
- Bartosik LP, Lajole G, Sugar L, Cattran D. Predicting progression in IgA nephropathy. Am J Kidney Dis 2001; 58:551–553.
- Rauta V, Finne P, Fagerudd J, et al. Factors associated with progression of IgA nephropathy are related to renal function—a model for estimating risk of progression in mild disease. Clin Nephrol 2002; 58:85–94.
- Komatsu H, Fujimoto S, Sato Y, et al. “Point of no return (PNR)” in progressive IgA nephropathy: significance of blood pressure and proteinuria management up to PNR”. J Nephrol 2005; 18:690–695.
- Schena FP, Cerullo G, Torres DD, et al European IgA Nephropathy Consortium. Searching for IgA nephropathy candidate genes: genetic studies combined with high throughput innovative investigations. Contrib Nephrol 2007; 157:80–89.
- Haubitz M, Wittke S, Weissinger EM, et al. Urine protein patterns can serve as a diagnostic tools in patients with IgA nephropathy. Kidney Int 2005; 67:2313–2320.
- Barratt J, Feehally J, Smith AC. The pathogenesis of IgA nephropathy. Semin Nephrol 2004; 24:197–217.
- Nishitani Y, Iwano M, Yamaguchi Y, et al. Fibroblast-specific protein 1 is a specific prognostic marker for renal survival in patients with IgAN. Kidney Int 2005; 68:1078–1085.
- Barratt J, Feehally J. Treatment of IgA nephropathy. Kidney Int 2006; 69:1934–1938.
- Pozzi C, Andrulli S, Del Vecchio L, et al. Corticosteroid effectiveness in IgA nephropathy: long-term follow-up of a randomized, controlled trial. J Am Soc Nephrol 2004; 15:157–163.
- Ballardie FW, Roberts IS. Controlled prospective trial of prednisolone and cytotoxics in progressive IgA nephropathy. J Am Soc Nephrol 2002; 13:142–148.
- Donadio JV, Grande JP. The role of fish oil/omega-3 fatty acid in the treatment of IgA nephropathy. Semin Nephrol 2004; 24:225–243.
- Maes BD, Oyen R, Claes K, et al. Mycophenolate mofetil in IgA nephropathy: results of a 3-year prospective placebo-controlled randomized study. Kidney Int 2004; 65:1842–1849.
- Goumenous DS, Davlouros P, El Nahas AM, et al. Prednis-olone and azathioprine in IgA nephropathy—a ten year follow-up study. Nephron Clin Pract 2003; 93:c58–c68.
- Soler MG, Mir M, Rodriguez E, et al. Recurrence of IgA nephropathy and Henoch-Schönlein purpura after kidney transplantation: risk factors and graft survival. Transplant Proc 2005; 37:3705–3709.
- Floege J. Recurrent IgA nephropathy after renal transplantation. Semin Nephrol 2004; 24:287–291.
Much progress has been made in the 40 years since immunoglobulin A (IgA) nephropathy was first described. We now have a reasonably complete understanding of the pathogenesis and mediation of this disease, but its etiology remains obscure and mysterious. New data on its epidemiology continue to emerge that will undoubtedly have clinical significance. We are beginning to perceive—but only dimly—the genetic predisposition to the disease.
Prognostication remains an imperfect science, but we are clearly making progress. The role of pathology in estimating prognosis in individual patients needs to be thoroughly reexamined, based on a uniformly agreed-upon classification scheme. Such work is currently in progress.
Therapy has certainly advanced, and we now have the rudiments of an evidence-based approach to management. However, much more needs to be done to refine these strategies so that they can be better matched to the characteristics of the patients, and there is a great need for novel therapeutic approaches and more information on multidrug regimens in selected patients. Many opportunities exist for improvement in the control of this common cause of chronic kidney disease, but we should not underestimate the challenges that present themselves in the field of IgA nephropathy in 2008 and beyond.
THE SCOPE OF THE PROBLEM
IgA nephropathy, also called Berger disease, is the most common form of primary glomerular disease in the developed world.1,2 Morphologically, it is characterized by diffuse deposition of IgA in the glomerular mesangium and by various degrees of damage of the glomerular capillary network seen on light microscopy.3,4 By some estimates, as many as 5% to 15% (averaging about 10%) of the general population may have IgA deposits in the glomerular mesangium, but only about 1 in 50 people with IgA deposits will actually have some abnormal clinical manifestation (principally recurring bouts of hematuria, with or without accompanying proteinuria) that brings them to the attention of a physician.5
Although not all patients with IgA nephropathy have progressive renal disease, IgA nephropathy is a significant contributor to the incidence of end-stage renal disease (ESRD) in many countries.1–4
DIAGNOSTIC AND PROGNOSTIC CHALLENGES
Since 1968, when IgA nephropathy was first described,6 great strides have been made in clarifying its epidemiology, its pathogenesis, the prognostic factors involved in its progression to ESRD, and its treatment. However, many gaps in our knowledge remain, particularly regarding its etiology, the genetic factors predisposing to it, its therapy, and the problem of recurrent disease in renal transplant recipients.
Can IgA nephropathy be diagnosed without a renal biopsy?
While renal biopsy and immunochemical analysis of renal tissue remain the gold standard for diagnosing IgA nephropathy, new sensitive and reasonably specific noninvasive tests are emerging and may provide another diagnostic approach. One of the most promising new tests is for abnormal circulating levels of abnormally glycosylated IgA subclass 1 (IgA1), which appears to be involved in the pathogenesis of the disease (see below).7 If noninvasive diagnostic techniques can be simplified and their accuracy validated across diverse populations, they offer great promise for use in epidemiologic and genetic studies, in which routine renal biopsy for diagnosis is impractical.
Signs and symptoms of IgA nephropathy are nonspecific
The most common clinical presentation of IgA nephropathy is recurring bouts of macroscopic hematuria, often but not invariably accompanied by proteinuria.2 Persistent asymptomatic hematuria without any detectable proteinuria (so-called isolated hematuria) affects a minority of patients. The red cells in the urine are typically dysmorphic (altered in size and shape compared with normal red cells), as they are in many other glomerulonephritic diseases.
Because low-grade fever and pain in the loins may accompany these bouts of hematuria, the disorder is often initially mistaken for urinary tract infection or urolithiasis. Careful microscopic examination of the urinary sediment for the characteristic dysmorphic erythrocytes that indicate a glomerular disease often provides the crucial clue that a glomerular disorder is the cause of the hematuria.8
However, a somewhat similar presentation may also be seen in thin basement membrane nephropathy, Alport syndrome (hereditary nephritis), and membranoproliferative glomerulonephritis,2 although these disorders can be readily distinguished from IgA nephropathy on examination of renal biopsy material under light, immunofluorescence, and electron microscopy. In addition, serum complement levels are typically reduced in membranoproliferative glomerulonephritis, and a family history of nephritis (without father-to-son transmission), often with deafness, can be obtained in the X-linked form of Alport syndrome. IgA nephropathy can be reliably distinguished from thin basement membrane nephropathy only by renal biopsy and electron microscopy.
Can we better predict which patients with IgA nephropathy will develop renal failure?
Although the rate of progression is very slow, and in only about 50% (or less) of patients does IgA nephropathy progress to ESRD within 25 years of diagnosis, the risk varies considerably among populations.9 Spontaneous clinical remissions are relatively uncommon in adults but much more common among children.
Several factors, if present at the time of discovery or developing within a relatively short time thereafter (usually within 6 months to 1 year), appear to predict a progressive course and, eventually, ESRD.9,10 We need to characterize and validate these risk factors in detail to be able to design and carry out appropriately powered, randomized, controlled clinical trials of treatment.
Unfortunately, cumulatively, the risk factors identified so far explain less than 50% of the variation in observed outcome of IgA nephropathy. Many of the risk factors identified so far are primarily indicators of the extent of disease at a particular time, and it is therefore not surprising that they would have some ability to predict the later behavior of the disease.
Clinical and pathologic risk factors in IgA nephropathy
Although imperfect, the major risk factors auguring a poor prognosis are:
- Proteinuria (> 500 mg/day) that persists for more than 6 months
- Elevated serum creatinine at diagnosis
- Microscopic hematuria that persists for more than 6 months
- Poorly controlled hypertension
- Extensive glomerulosclerosis or interstitial fibrosis or both on renal biopsy.7,10
Extensive crescentic disease also confers a worse short-term prognosis, often accompanied by a rapidly progressive loss of renal function.
Are clinical risk factors more useful than pathologic risk factors in IgA nephropathy?
Of importance, clinical factors, such as persistent proteinuria or declining renal function on follow-up appear to have greater predictive power than pathologic factors for long-term outcome.9–12 Clinical factors, such as decreasing estimated glomerular filtration rate (GFR) after short-term follow-up, persistent moderate to marked proteinuria (500–1,000 mg/day, or more), hyperuricemia, hyperlipidemia, concomitant obesity, poorly controlled hypertension, absence of treatment with angiotensin II inhibitors, and, possibly, persistent micro-hematuria are the most consistent factors independently associated with a poor prognosis in multivariate analysis. Pathologic changes noted in the original diagnostic renal biopsy do not consistently add greatly to the precision of prognosis beyond the analysis of these clinical and laboratory factors.11
A detailed and uniform immunologic and morphologic approach to classifying the pathology of IgA nephropathy may yet uncover some new and very useful prognostic factors, independent of those generated by simple clinical assessment. Efforts are under way, and such a development would greatly improve the accuracy and precision of outcome prediction and reduce the amount of unexplained variation in prognosis observed in groups of patients with IgA nephropathy.
At present, the heterogeneity of participants in clinical trials of therapy, the tendency for the disease to progress slowly, and the variation in prognosis due to unexplained factors pose major challenges in designing and carrying out randomized controlled trials of therapy in IgA nephropathy. If we can find new risk factors that can predict progressive disease earlier, the knowledge will help us in designing future clinical trials, which will be vital if progress is to be made towards controlling IgA nephropathy.
Prognosis in individual patients vs populations with IgA nephropathy
At present, we need a way to determine the prognosis more precisely in individual patients rather than in groups of patients. After all, physicians are called upon to determine the likely outcome in single patients, not in a population. Several prediction formulas have been devised, most of them based on relatively simple clinical factors present at discovery or short-term follow-up.12,13
Conventional pathologic observations have limited utility in such individualized prognostic formulations.12 This is not to say that renal biopsy only offers diagnostic utility and has little if any value as a prognostic tool. However, the challenge is to enhance the prognostic usefulness of renal biopsy by refining the examination of the tissue specimens using modern approaches and to conduct the appropriate correlative studies to confirm the value of new pathologic criteria in prognostication, independent of clinical features alone.
For example, the risk of ESRD is greater if the patient has very extensive (> 50%) crescentic glomerular involvement with a rapidly progressive glomerulonephritic evolution. The risk is less if there are minimal glomerular changes with nephrotic-range proteinuria. Extensive interstitial fibrosis and glomerulosclerosis in the original “diagnostic” renal biopsy merely highlight the existence of prior progressive disease that is likely to continue. The significance of persistent focal necrotizing glomerular lesions (capillaritis) in IgA nephropathy, often associated with persistent microhematuria, is not entirely clear and needs to be specifically explored, especially as it pertains to the need for immunosuppressive therapy added to treatment for hypertension, proteinuria, or both with inhibitors of the renin-angiotensin system (see below).
At present, the most powerful prognostic factor in IgA nephropathy is moderate to severe proteinuria that persists for 6 months or longer.9,10,12 The relationship between the level of proteinuria and the outcome is continuous, ie, the greater the proteinuria, the worse the prognosis. Compared with some other primary glomerular diseases (such as membranous nephropathy or focal and segmental glomerulosclerosis), progressive disease in IgA nephropathy is associated with lower levels of persistent proteinuria (usually 500 mg to 3 g/day).
The estimated GFR at the time IgA nephropathy is discovered is a rather weak independent predictor of outcome (up to a point; see below). Many patients have stable (but reduced) renal function in the long term, especially if they receive angiotensin II inhibitor therapy and can keep their systolic blood pressure between 110 and 120 mm Hg.
How can IgA nephropathy be diagnosed and treated before the ‘point of no return’?
For patients at risk of developing ESRD, the two most critical goals of treatment are to:
- Control blood pressure rigorously, preferably with an angiotensin-converting enzyme (ACE) inhibitor, an angiotensin II receptor antagonist (ARB), or both, and
- Reduce proteinuria to less than 500 mg/day.
If these two goals can be met without undue side effects and if the patient remains compliant in the long term, many patients can avoid ESRD. Patients who cannot achieve these goals despite vigorous attempts become candidates for adjunctive therapy, such as pulse intravenous methylprednisolone (Solu-Medrol) combined with oral prednisone, or in some cases a cytotoxic drug combined with prednisone. Small randomized controlled trials suggest these adjunctive treatments are effective and safe.
Unfortunately, IgA nephropathy can progress silently, and many patients do not receive the diagnosis until late in its course. In such patients, the disease may relentlessly progress even with optimal therapy. The “point of no return” appears to be an estimated GFR of about 30 mL/min/1.73 m2 (stage 4 chronic kidney disease).14
These observations underscore the need for early diagnosis and treatment based on factors that accurately predict an unfavorable outcome. Finding these factors will not be easy, because it will require detailed observation of homogeneous groups of patients over prolonged periods of time. New findings show great promise for identifying patients earlier in the course of disease who are more or less likely to progress to ESRD. The challenge is to translate these findings into rational, safe, and effective therapies applicable across a broad spectrum of disease.
OPPORTUNITIES: GENETICS, PROTEOMICS, NEW TESTS AND TREATMENTS
Genetic studies may lead to novel treatments for IgA nephropathy
Susceptibility to IgA nephropathy has a genetic component to varying degrees, depending on geography and the existence of “founder effects.” Familial forms of IgA nephropathy are more common in northern Italy and in eastern Kentucky. The familial cases may derive from a mutation of a specific gene occurring in a founder many hundreds of years ago. Several genetic loci are strongly associated with IgA nephropathy (usually as an autosomal-dominant trait with highly variable penetrance).15 Familial IgA nephropathy is most likely genetically heterogeneous, and many cases of IgA nephropathy that are believed to be sporadic may actually have a less apparent genetic basis, with skipped generations, lanthanic (covert) disease, and incomplete penetrance.
At present, genetic testing based on genomic or transcriptosomic analysis does not appear to have much diagnostic value except in clearly familial cases, because many loci are involved. Many asymptomatic people have mesangial IgA deposits that could be detected by renal biopsy but not by genetic analysis, and this inability is a major obstacle for genetic susceptibility studies. Indeed, most current genetic studies actually examine susceptibility to the clinical expression of disease rather than susceptibility to the mesangial IgA deposition that underlies the disease.5
The opportunity that lies ahead in genetic testing of IgA nephropathy (including haplotype analysis) appears to be primarily in the elucidation of potential pathogenetic pathways and in the refinement of prognosis and the definition of treatment responsiveness (pharmacogenomics).
If a gene (or group of genes) can be identified that is strongly and consistently associated with IgA nephropathy across diverse populations, its protein product isolated and characterized, and its role in pathogenesis elucidated, then a new era in targeted therapy of IgA nephropathy will be unleashed, much in the same way as the identification of tyrosine phosphatases played a role in the design of targeted therapy in chronic myelogenous leukemia. Early progress is being made in this area, but many obstacles lie in the way.
Proteomics may prove useful in diagnosis and prognosis of IgA nephropathy
Proteomics—the characterization and analysis of the patient’s entire complement of serum and urinary proteins—is a new, exciting, and largely unexplored area in IgA nephropathy. Preliminary studies have shown that this technique may provide a novel noninvasive means of diagnosing IgA nephropathy, and it may have additional value as a prognostic tool.16
Much work needs to be done to standardize how specimens are collected, stored, and shipped and to verify the precision and accuracy of proteomics in diverse populations of patients with IgA nephropathy, patients with other glomerular diseases, and normal subjects to ascertain this technique’s false-negative and false-positive rates.
IgA1 testing may help detect IgA nephropathy early in its course
Abnormally undergalactosylated and oversialyted epitopes at the hinge region of the IgA1 molecule play a critical role in the pathogenesis of sporadic IgA nephropathy.17 This discovery provides a great opportunity for profiling patients suspected of having IgA nephropathy on the basis of sensitive determination of the serum level of these abnormal IgA1 molecules.7
It may be that pathogenic IgA1 molecules (and autoantibodies to them) arise many months or even years before the onset of clinical manifestations of overt IgA nephropathy, similar to the situation known to occur in systemic lupus erythematosus. It is also possible that an abnormality of the disposal of immune complexes created by the interaction of autoantibodies with the abnormally glycosylated IgA1 creates the opportunity for preferential glomerular mesangial deposition of polymeric IgA.
Clearly, the greatest opportunity lies with understanding the fundamental abnormality leading to defective O-linked galactosylation of the serine/threonine residues at the hinge region of IgA1 in IgA nephropathy. In addition, it would be very useful to know if this is a generalized and acquired abnormality or whether it is focal in distribution (eg, in the tonsils, bone marrow, or lymphoid tissue in the gut).
Knowledge of secondary mediators may also lead to new treatments for IgA nephropathy
Detailed knowledge of the participation of specific cell types and the “cytokine milieu” (eg, interleukin 4, interferon) in directing the abnormality toward defective glycosylation would also be very important in designing new approaches to diagnosis and therapy.
A better understanding is slowly emerging of the pathways by which pathogenic immune complexes containing IgA are deposited and cleared, and of the secondary mediator systems evoked by their formation and tissue localization. Interference with these secondary mediator processes, such as alternative or mannose-dependent complement activation, platelet-derived growth factor or transforming growth factor stimulation, also offers a new approach to therapy.
We lack a suitable animal model of IgA nephropathy that mimics all aspects of the human condition, which has impeded progress in this area. A fully humanized mouse model of disease would be a welcome addition to the investigative toolkit.
Prognostic biopsy analysis may be improved in IgA nephropathy
As discussed above, the science of prognostication and stratification of patients with IgA nephropathy into those at high or low risk of ESRD has clearly advanced but is still quite incomplete, especially with respect to individual patients.
Great opportunities lie in refining the value of renal biopsy in prognostication. Although the “snapshot” nature and potential sampling errors intrinsic to diagnostic renal biopsy cannot easily be overcome, at least not without performing multiple and repeated renal biopsies (a very impractical approach to prognostication), refinements in the laboratory seem to offer numerous opportunities for advancement. Much better clinicopathological correlations, especially with respect to outcomes, among well-characterized patients with IgA nephropathy are greatly needed. New nonconventional markers of progression, such as “tubulitis,” deposition of fibroblast-specific proteins, and the proteome of the deposited immunoglobulins and complement show much promise.18
Immunosuppressive therapy could be added to ACE inhibitors or ARBs in IgA nephropathy
The management of IgA nephropathy has clearly advanced over the last several decades, largely as the result of randomized clinical trials.3,19 However, these trials had serious limitations: the numbers of patients were relatively small, follow-up was relatively short, and the findings may not apply to the IgA nephropathy population at large or to specific patients having features that diverge from those in the patients enrolled in the studies.
The value of initial therapy with an ACE inhibitor, an ARB, or both in combination appears well established. However, details of dosage, duration of therapy, and the relative values of monotherapy and combined therapy remain uncertain.
Many opportunities for combining angiotensin II inhibition and immunosuppressive therapy are being explored. By and large, all current therapies are empiric and their long-term effects relatively uncertain, owing to small study size and short duration.
Oral and parenteral glucocorticoids,20 combined regimens of cyclophosphamide (Cytoxan) and azathioprine (Imuran),21 omega-3 fatty acids,22 and anticoagulants and anti-thrombotics3 each have their advocates and their specified target populations.
Tonsillectomy as a treatment has been particularly controversial. While no controlled studies have been performed yet, observational studies (most of them conducted in some prefectures in Japan) have suggested a higher rate of clinical remission with tonsillectomy than with steroid treatment alone.5 However, long-term observations have not shown any consistent effect of tonsillectomy on progression to ESRD.
We hope that a better understanding of the fundamental mechanisms of disease and its mediation will provide an impetus for development of more rational targeted therapy. Evaluating potentially promising targeted therapies will be very difficult. Evaluation of safety and efficacy with long-term use will be a key requirement for a successful novel therapeutic agent.
FOR NOW, AN EMPIRIC APPROACH TO IGA NEPHROPATHY
Start with an angiotensin II inhibitor
The current body of evidence for choosing a particular therapeutic approach for a given patient with IgA nephropathy cannot be regarded as definitive, owing to limitations in the quality and strength of the trials serving as the basis of the evidence. Nonetheless, patients with IgA nephropathy and abnormal protein excretion (> 500 mg/day) should probably always be given angiotensin II inhibitor therapy (an ACE inhibitor, an ARB, or both) if they have no contraindications to it such as a hypersensitivity reaction or pregnancy, as a base for future monitoring and adjuvant therapy.
A response, tentatively defined as a 30% to 50% decline in proteinuria from baseline levels or a decrease to less than 500 mg/day, would be a reason to continue this conservative approach. Lack of a response after several months of observation at maximal tolerated dosage (plus salt restriction or a diuretic) would be a reason for considering adjuvant therapy.
If the patient does not respond to an ACE inhibitor or ARB and his or her estimated GFR is over 70 mL/min/1.73 m2, a trial of oral and parenteral glucocorticoids might be undertaken, as suggested by Pozzi and coworkers.20
On the other hand, if the estimated GFR is in the range of 30 to 70 mL/min/1.73 m2 and declining at a rate that predicts that ESRD will develop in less than 5 to 7 years, this would be a possible indication for low-dose oral cyclo-phosphamide and then azathioprine, as suggested by Ballardie and Roberts.21 Omega-3 fatty acids (Omacor) could also be considered as add-on therapy, particularly for patients with very heavy proteinuria (> 3.0 g/d) and reduced estimated GFR.22
Patients with an estimated GFR of less than 30 mL/min/1.73 m2 and chronic (irreversible) changes on renal biopsy—the point of no return—probably will not respond to any therapy other than an ACE inhibitor, an ARB, or both.
The role of more aggressive immunosuppression
At present, the evidence for using mycophenolate mofetil (CellCept) or calcineurin inhibitors (such as cyclosporin or tacrolimus) is fragmentary or contradictory.3,19,23 Similarly, the benefits of long-term azathioprine therapy are based on observational data only and so it cannot be recommended as evidence-based.24 Opportunities exist for combined therapy (eg, an ACE inhibitor or an ARB or both, combined with omega-3 fatty acids and azathioprine or mycophenolate mofetil), but at present, controlled trials are lacking. Crescentic disease and rapidly progressive glomerulonephritis should probably be treated with combined cyclophosphamide and parental and oral corticosteroids, based on observational data. Patients with IgA nephropathy and minimal change disease with nephrotic syndrome should be treated with oral steroids, but the only data available are observational. Low-protein diets could be tried in the presence of slowly progressive renal disease with estimated GFR less than 30 mL/min/1.73 m2, but there are no controlled trials demonstrating efficacy for this approach in IgA neph-ropathy.
Renal transplantation is very successful
Renal transplantation is a very suitable alternative for patients with IgA nephropathy that progresses to ESRD. Overall success rates are as good or better than those in other primary glomerular diseases. Unfortunately, the disease recurs in the majority of renal grafts and may in some cases lead to loss of the graft.25,26 We need much more information on the factors that predict such recurrences and their undesirable effects on transplantation outcomes.
MUCH WORK TO BE DONE
Much work needs to be done in the field of therapeutics in IgA nephropathy. Much of this effort will hinge on the interests of the pharmaceutical industry in IgA nephropathy as a potential therapeutic market. At present, the prospects for the development of a safe and effective novel therapy for IgA nephropathy (eg, approvable by the US Food and Drug Administration) do not appear great, but this may be overly pessimistic. The nature of the disease mandates long-term observation, agents that are very safe (with low rates of ESRD, death, and transplantation), and dependency on surrogate markers of efficacy. Therefore, designing and executing studies will not be easy.
Much progress has been made in the 40 years since immunoglobulin A (IgA) nephropathy was first described. We now have a reasonably complete understanding of the pathogenesis and mediation of this disease, but its etiology remains obscure and mysterious. New data on its epidemiology continue to emerge that will undoubtedly have clinical significance. We are beginning to perceive—but only dimly—the genetic predisposition to the disease.
Prognostication remains an imperfect science, but we are clearly making progress. The role of pathology in estimating prognosis in individual patients needs to be thoroughly reexamined, based on a uniformly agreed-upon classification scheme. Such work is currently in progress.
Therapy has certainly advanced, and we now have the rudiments of an evidence-based approach to management. However, much more needs to be done to refine these strategies so that they can be better matched to the characteristics of the patients, and there is a great need for novel therapeutic approaches and more information on multidrug regimens in selected patients. Many opportunities exist for improvement in the control of this common cause of chronic kidney disease, but we should not underestimate the challenges that present themselves in the field of IgA nephropathy in 2008 and beyond.
THE SCOPE OF THE PROBLEM
IgA nephropathy, also called Berger disease, is the most common form of primary glomerular disease in the developed world.1,2 Morphologically, it is characterized by diffuse deposition of IgA in the glomerular mesangium and by various degrees of damage of the glomerular capillary network seen on light microscopy.3,4 By some estimates, as many as 5% to 15% (averaging about 10%) of the general population may have IgA deposits in the glomerular mesangium, but only about 1 in 50 people with IgA deposits will actually have some abnormal clinical manifestation (principally recurring bouts of hematuria, with or without accompanying proteinuria) that brings them to the attention of a physician.5
Although not all patients with IgA nephropathy have progressive renal disease, IgA nephropathy is a significant contributor to the incidence of end-stage renal disease (ESRD) in many countries.1–4
DIAGNOSTIC AND PROGNOSTIC CHALLENGES
Since 1968, when IgA nephropathy was first described,6 great strides have been made in clarifying its epidemiology, its pathogenesis, the prognostic factors involved in its progression to ESRD, and its treatment. However, many gaps in our knowledge remain, particularly regarding its etiology, the genetic factors predisposing to it, its therapy, and the problem of recurrent disease in renal transplant recipients.
Can IgA nephropathy be diagnosed without a renal biopsy?
While renal biopsy and immunochemical analysis of renal tissue remain the gold standard for diagnosing IgA nephropathy, new sensitive and reasonably specific noninvasive tests are emerging and may provide another diagnostic approach. One of the most promising new tests is for abnormal circulating levels of abnormally glycosylated IgA subclass 1 (IgA1), which appears to be involved in the pathogenesis of the disease (see below).7 If noninvasive diagnostic techniques can be simplified and their accuracy validated across diverse populations, they offer great promise for use in epidemiologic and genetic studies, in which routine renal biopsy for diagnosis is impractical.
Signs and symptoms of IgA nephropathy are nonspecific
The most common clinical presentation of IgA nephropathy is recurring bouts of macroscopic hematuria, often but not invariably accompanied by proteinuria.2 Persistent asymptomatic hematuria without any detectable proteinuria (so-called isolated hematuria) affects a minority of patients. The red cells in the urine are typically dysmorphic (altered in size and shape compared with normal red cells), as they are in many other glomerulonephritic diseases.
Because low-grade fever and pain in the loins may accompany these bouts of hematuria, the disorder is often initially mistaken for urinary tract infection or urolithiasis. Careful microscopic examination of the urinary sediment for the characteristic dysmorphic erythrocytes that indicate a glomerular disease often provides the crucial clue that a glomerular disorder is the cause of the hematuria.8
However, a somewhat similar presentation may also be seen in thin basement membrane nephropathy, Alport syndrome (hereditary nephritis), and membranoproliferative glomerulonephritis,2 although these disorders can be readily distinguished from IgA nephropathy on examination of renal biopsy material under light, immunofluorescence, and electron microscopy. In addition, serum complement levels are typically reduced in membranoproliferative glomerulonephritis, and a family history of nephritis (without father-to-son transmission), often with deafness, can be obtained in the X-linked form of Alport syndrome. IgA nephropathy can be reliably distinguished from thin basement membrane nephropathy only by renal biopsy and electron microscopy.
Can we better predict which patients with IgA nephropathy will develop renal failure?
Although the rate of progression is very slow, and in only about 50% (or less) of patients does IgA nephropathy progress to ESRD within 25 years of diagnosis, the risk varies considerably among populations.9 Spontaneous clinical remissions are relatively uncommon in adults but much more common among children.
Several factors, if present at the time of discovery or developing within a relatively short time thereafter (usually within 6 months to 1 year), appear to predict a progressive course and, eventually, ESRD.9,10 We need to characterize and validate these risk factors in detail to be able to design and carry out appropriately powered, randomized, controlled clinical trials of treatment.
Unfortunately, cumulatively, the risk factors identified so far explain less than 50% of the variation in observed outcome of IgA nephropathy. Many of the risk factors identified so far are primarily indicators of the extent of disease at a particular time, and it is therefore not surprising that they would have some ability to predict the later behavior of the disease.
Clinical and pathologic risk factors in IgA nephropathy
Although imperfect, the major risk factors auguring a poor prognosis are:
- Proteinuria (> 500 mg/day) that persists for more than 6 months
- Elevated serum creatinine at diagnosis
- Microscopic hematuria that persists for more than 6 months
- Poorly controlled hypertension
- Extensive glomerulosclerosis or interstitial fibrosis or both on renal biopsy.7,10
Extensive crescentic disease also confers a worse short-term prognosis, often accompanied by a rapidly progressive loss of renal function.
Are clinical risk factors more useful than pathologic risk factors in IgA nephropathy?
Of importance, clinical factors, such as persistent proteinuria or declining renal function on follow-up appear to have greater predictive power than pathologic factors for long-term outcome.9–12 Clinical factors, such as decreasing estimated glomerular filtration rate (GFR) after short-term follow-up, persistent moderate to marked proteinuria (500–1,000 mg/day, or more), hyperuricemia, hyperlipidemia, concomitant obesity, poorly controlled hypertension, absence of treatment with angiotensin II inhibitors, and, possibly, persistent micro-hematuria are the most consistent factors independently associated with a poor prognosis in multivariate analysis. Pathologic changes noted in the original diagnostic renal biopsy do not consistently add greatly to the precision of prognosis beyond the analysis of these clinical and laboratory factors.11
A detailed and uniform immunologic and morphologic approach to classifying the pathology of IgA nephropathy may yet uncover some new and very useful prognostic factors, independent of those generated by simple clinical assessment. Efforts are under way, and such a development would greatly improve the accuracy and precision of outcome prediction and reduce the amount of unexplained variation in prognosis observed in groups of patients with IgA nephropathy.
At present, the heterogeneity of participants in clinical trials of therapy, the tendency for the disease to progress slowly, and the variation in prognosis due to unexplained factors pose major challenges in designing and carrying out randomized controlled trials of therapy in IgA nephropathy. If we can find new risk factors that can predict progressive disease earlier, the knowledge will help us in designing future clinical trials, which will be vital if progress is to be made towards controlling IgA nephropathy.
Prognosis in individual patients vs populations with IgA nephropathy
At present, we need a way to determine the prognosis more precisely in individual patients rather than in groups of patients. After all, physicians are called upon to determine the likely outcome in single patients, not in a population. Several prediction formulas have been devised, most of them based on relatively simple clinical factors present at discovery or short-term follow-up.12,13
Conventional pathologic observations have limited utility in such individualized prognostic formulations.12 This is not to say that renal biopsy only offers diagnostic utility and has little if any value as a prognostic tool. However, the challenge is to enhance the prognostic usefulness of renal biopsy by refining the examination of the tissue specimens using modern approaches and to conduct the appropriate correlative studies to confirm the value of new pathologic criteria in prognostication, independent of clinical features alone.
For example, the risk of ESRD is greater if the patient has very extensive (> 50%) crescentic glomerular involvement with a rapidly progressive glomerulonephritic evolution. The risk is less if there are minimal glomerular changes with nephrotic-range proteinuria. Extensive interstitial fibrosis and glomerulosclerosis in the original “diagnostic” renal biopsy merely highlight the existence of prior progressive disease that is likely to continue. The significance of persistent focal necrotizing glomerular lesions (capillaritis) in IgA nephropathy, often associated with persistent microhematuria, is not entirely clear and needs to be specifically explored, especially as it pertains to the need for immunosuppressive therapy added to treatment for hypertension, proteinuria, or both with inhibitors of the renin-angiotensin system (see below).
At present, the most powerful prognostic factor in IgA nephropathy is moderate to severe proteinuria that persists for 6 months or longer.9,10,12 The relationship between the level of proteinuria and the outcome is continuous, ie, the greater the proteinuria, the worse the prognosis. Compared with some other primary glomerular diseases (such as membranous nephropathy or focal and segmental glomerulosclerosis), progressive disease in IgA nephropathy is associated with lower levels of persistent proteinuria (usually 500 mg to 3 g/day).
The estimated GFR at the time IgA nephropathy is discovered is a rather weak independent predictor of outcome (up to a point; see below). Many patients have stable (but reduced) renal function in the long term, especially if they receive angiotensin II inhibitor therapy and can keep their systolic blood pressure between 110 and 120 mm Hg.
How can IgA nephropathy be diagnosed and treated before the ‘point of no return’?
For patients at risk of developing ESRD, the two most critical goals of treatment are to:
- Control blood pressure rigorously, preferably with an angiotensin-converting enzyme (ACE) inhibitor, an angiotensin II receptor antagonist (ARB), or both, and
- Reduce proteinuria to less than 500 mg/day.
If these two goals can be met without undue side effects and if the patient remains compliant in the long term, many patients can avoid ESRD. Patients who cannot achieve these goals despite vigorous attempts become candidates for adjunctive therapy, such as pulse intravenous methylprednisolone (Solu-Medrol) combined with oral prednisone, or in some cases a cytotoxic drug combined with prednisone. Small randomized controlled trials suggest these adjunctive treatments are effective and safe.
Unfortunately, IgA nephropathy can progress silently, and many patients do not receive the diagnosis until late in its course. In such patients, the disease may relentlessly progress even with optimal therapy. The “point of no return” appears to be an estimated GFR of about 30 mL/min/1.73 m2 (stage 4 chronic kidney disease).14
These observations underscore the need for early diagnosis and treatment based on factors that accurately predict an unfavorable outcome. Finding these factors will not be easy, because it will require detailed observation of homogeneous groups of patients over prolonged periods of time. New findings show great promise for identifying patients earlier in the course of disease who are more or less likely to progress to ESRD. The challenge is to translate these findings into rational, safe, and effective therapies applicable across a broad spectrum of disease.
OPPORTUNITIES: GENETICS, PROTEOMICS, NEW TESTS AND TREATMENTS
Genetic studies may lead to novel treatments for IgA nephropathy
Susceptibility to IgA nephropathy has a genetic component to varying degrees, depending on geography and the existence of “founder effects.” Familial forms of IgA nephropathy are more common in northern Italy and in eastern Kentucky. The familial cases may derive from a mutation of a specific gene occurring in a founder many hundreds of years ago. Several genetic loci are strongly associated with IgA nephropathy (usually as an autosomal-dominant trait with highly variable penetrance).15 Familial IgA nephropathy is most likely genetically heterogeneous, and many cases of IgA nephropathy that are believed to be sporadic may actually have a less apparent genetic basis, with skipped generations, lanthanic (covert) disease, and incomplete penetrance.
At present, genetic testing based on genomic or transcriptosomic analysis does not appear to have much diagnostic value except in clearly familial cases, because many loci are involved. Many asymptomatic people have mesangial IgA deposits that could be detected by renal biopsy but not by genetic analysis, and this inability is a major obstacle for genetic susceptibility studies. Indeed, most current genetic studies actually examine susceptibility to the clinical expression of disease rather than susceptibility to the mesangial IgA deposition that underlies the disease.5
The opportunity that lies ahead in genetic testing of IgA nephropathy (including haplotype analysis) appears to be primarily in the elucidation of potential pathogenetic pathways and in the refinement of prognosis and the definition of treatment responsiveness (pharmacogenomics).
If a gene (or group of genes) can be identified that is strongly and consistently associated with IgA nephropathy across diverse populations, its protein product isolated and characterized, and its role in pathogenesis elucidated, then a new era in targeted therapy of IgA nephropathy will be unleashed, much in the same way as the identification of tyrosine phosphatases played a role in the design of targeted therapy in chronic myelogenous leukemia. Early progress is being made in this area, but many obstacles lie in the way.
Proteomics may prove useful in diagnosis and prognosis of IgA nephropathy
Proteomics—the characterization and analysis of the patient’s entire complement of serum and urinary proteins—is a new, exciting, and largely unexplored area in IgA nephropathy. Preliminary studies have shown that this technique may provide a novel noninvasive means of diagnosing IgA nephropathy, and it may have additional value as a prognostic tool.16
Much work needs to be done to standardize how specimens are collected, stored, and shipped and to verify the precision and accuracy of proteomics in diverse populations of patients with IgA nephropathy, patients with other glomerular diseases, and normal subjects to ascertain this technique’s false-negative and false-positive rates.
IgA1 testing may help detect IgA nephropathy early in its course
Abnormally undergalactosylated and oversialyted epitopes at the hinge region of the IgA1 molecule play a critical role in the pathogenesis of sporadic IgA nephropathy.17 This discovery provides a great opportunity for profiling patients suspected of having IgA nephropathy on the basis of sensitive determination of the serum level of these abnormal IgA1 molecules.7
It may be that pathogenic IgA1 molecules (and autoantibodies to them) arise many months or even years before the onset of clinical manifestations of overt IgA nephropathy, similar to the situation known to occur in systemic lupus erythematosus. It is also possible that an abnormality of the disposal of immune complexes created by the interaction of autoantibodies with the abnormally glycosylated IgA1 creates the opportunity for preferential glomerular mesangial deposition of polymeric IgA.
Clearly, the greatest opportunity lies with understanding the fundamental abnormality leading to defective O-linked galactosylation of the serine/threonine residues at the hinge region of IgA1 in IgA nephropathy. In addition, it would be very useful to know if this is a generalized and acquired abnormality or whether it is focal in distribution (eg, in the tonsils, bone marrow, or lymphoid tissue in the gut).
Knowledge of secondary mediators may also lead to new treatments for IgA nephropathy
Detailed knowledge of the participation of specific cell types and the “cytokine milieu” (eg, interleukin 4, interferon) in directing the abnormality toward defective glycosylation would also be very important in designing new approaches to diagnosis and therapy.
A better understanding is slowly emerging of the pathways by which pathogenic immune complexes containing IgA are deposited and cleared, and of the secondary mediator systems evoked by their formation and tissue localization. Interference with these secondary mediator processes, such as alternative or mannose-dependent complement activation, platelet-derived growth factor or transforming growth factor stimulation, also offers a new approach to therapy.
We lack a suitable animal model of IgA nephropathy that mimics all aspects of the human condition, which has impeded progress in this area. A fully humanized mouse model of disease would be a welcome addition to the investigative toolkit.
Prognostic biopsy analysis may be improved in IgA nephropathy
As discussed above, the science of prognostication and stratification of patients with IgA nephropathy into those at high or low risk of ESRD has clearly advanced but is still quite incomplete, especially with respect to individual patients.
Great opportunities lie in refining the value of renal biopsy in prognostication. Although the “snapshot” nature and potential sampling errors intrinsic to diagnostic renal biopsy cannot easily be overcome, at least not without performing multiple and repeated renal biopsies (a very impractical approach to prognostication), refinements in the laboratory seem to offer numerous opportunities for advancement. Much better clinicopathological correlations, especially with respect to outcomes, among well-characterized patients with IgA nephropathy are greatly needed. New nonconventional markers of progression, such as “tubulitis,” deposition of fibroblast-specific proteins, and the proteome of the deposited immunoglobulins and complement show much promise.18
Immunosuppressive therapy could be added to ACE inhibitors or ARBs in IgA nephropathy
The management of IgA nephropathy has clearly advanced over the last several decades, largely as the result of randomized clinical trials.3,19 However, these trials had serious limitations: the numbers of patients were relatively small, follow-up was relatively short, and the findings may not apply to the IgA nephropathy population at large or to specific patients having features that diverge from those in the patients enrolled in the studies.
The value of initial therapy with an ACE inhibitor, an ARB, or both in combination appears well established. However, details of dosage, duration of therapy, and the relative values of monotherapy and combined therapy remain uncertain.
Many opportunities for combining angiotensin II inhibition and immunosuppressive therapy are being explored. By and large, all current therapies are empiric and their long-term effects relatively uncertain, owing to small study size and short duration.
Oral and parenteral glucocorticoids,20 combined regimens of cyclophosphamide (Cytoxan) and azathioprine (Imuran),21 omega-3 fatty acids,22 and anticoagulants and anti-thrombotics3 each have their advocates and their specified target populations.
Tonsillectomy as a treatment has been particularly controversial. While no controlled studies have been performed yet, observational studies (most of them conducted in some prefectures in Japan) have suggested a higher rate of clinical remission with tonsillectomy than with steroid treatment alone.5 However, long-term observations have not shown any consistent effect of tonsillectomy on progression to ESRD.
We hope that a better understanding of the fundamental mechanisms of disease and its mediation will provide an impetus for development of more rational targeted therapy. Evaluating potentially promising targeted therapies will be very difficult. Evaluation of safety and efficacy with long-term use will be a key requirement for a successful novel therapeutic agent.
FOR NOW, AN EMPIRIC APPROACH TO IGA NEPHROPATHY
Start with an angiotensin II inhibitor
The current body of evidence for choosing a particular therapeutic approach for a given patient with IgA nephropathy cannot be regarded as definitive, owing to limitations in the quality and strength of the trials serving as the basis of the evidence. Nonetheless, patients with IgA nephropathy and abnormal protein excretion (> 500 mg/day) should probably always be given angiotensin II inhibitor therapy (an ACE inhibitor, an ARB, or both) if they have no contraindications to it such as a hypersensitivity reaction or pregnancy, as a base for future monitoring and adjuvant therapy.
A response, tentatively defined as a 30% to 50% decline in proteinuria from baseline levels or a decrease to less than 500 mg/day, would be a reason to continue this conservative approach. Lack of a response after several months of observation at maximal tolerated dosage (plus salt restriction or a diuretic) would be a reason for considering adjuvant therapy.
If the patient does not respond to an ACE inhibitor or ARB and his or her estimated GFR is over 70 mL/min/1.73 m2, a trial of oral and parenteral glucocorticoids might be undertaken, as suggested by Pozzi and coworkers.20
On the other hand, if the estimated GFR is in the range of 30 to 70 mL/min/1.73 m2 and declining at a rate that predicts that ESRD will develop in less than 5 to 7 years, this would be a possible indication for low-dose oral cyclo-phosphamide and then azathioprine, as suggested by Ballardie and Roberts.21 Omega-3 fatty acids (Omacor) could also be considered as add-on therapy, particularly for patients with very heavy proteinuria (> 3.0 g/d) and reduced estimated GFR.22
Patients with an estimated GFR of less than 30 mL/min/1.73 m2 and chronic (irreversible) changes on renal biopsy—the point of no return—probably will not respond to any therapy other than an ACE inhibitor, an ARB, or both.
The role of more aggressive immunosuppression
At present, the evidence for using mycophenolate mofetil (CellCept) or calcineurin inhibitors (such as cyclosporin or tacrolimus) is fragmentary or contradictory.3,19,23 Similarly, the benefits of long-term azathioprine therapy are based on observational data only and so it cannot be recommended as evidence-based.24 Opportunities exist for combined therapy (eg, an ACE inhibitor or an ARB or both, combined with omega-3 fatty acids and azathioprine or mycophenolate mofetil), but at present, controlled trials are lacking. Crescentic disease and rapidly progressive glomerulonephritis should probably be treated with combined cyclophosphamide and parental and oral corticosteroids, based on observational data. Patients with IgA nephropathy and minimal change disease with nephrotic syndrome should be treated with oral steroids, but the only data available are observational. Low-protein diets could be tried in the presence of slowly progressive renal disease with estimated GFR less than 30 mL/min/1.73 m2, but there are no controlled trials demonstrating efficacy for this approach in IgA neph-ropathy.
Renal transplantation is very successful
Renal transplantation is a very suitable alternative for patients with IgA nephropathy that progresses to ESRD. Overall success rates are as good or better than those in other primary glomerular diseases. Unfortunately, the disease recurs in the majority of renal grafts and may in some cases lead to loss of the graft.25,26 We need much more information on the factors that predict such recurrences and their undesirable effects on transplantation outcomes.
MUCH WORK TO BE DONE
Much work needs to be done in the field of therapeutics in IgA nephropathy. Much of this effort will hinge on the interests of the pharmaceutical industry in IgA nephropathy as a potential therapeutic market. At present, the prospects for the development of a safe and effective novel therapy for IgA nephropathy (eg, approvable by the US Food and Drug Administration) do not appear great, but this may be overly pessimistic. The nature of the disease mandates long-term observation, agents that are very safe (with low rates of ESRD, death, and transplantation), and dependency on surrogate markers of efficacy. Therefore, designing and executing studies will not be easy.
- Tomino Y. IgA nephropathy today. Contrib Nephrol 2007; 157:1–255.
- D’Amico G. The commonest glomerulonephritis in the world: IgA nephropathy. Quart J Med 1987; 245:709–727.
- Lee G, Glassock RJ. Immunoglobulin A nephropathy. In:Ponticelli C, Glassock R, editors. Treatment of Primary Glomerulonephritis. Oxford: Oxford Medical Publication, 1997:187–217.
- Donadio JV, Grande JP. IgA nephropathy. N Engl J Med 2002; 347:738–748.
- Glassock RJ. Concluding remarks. IgA nephropathy today. Contrib Nephrol 2002; 157:169–173.
- Berger J, Hinglais N. Les dépots intercapillaries d’IgA-IgG. J Urol Nephrol (Paris) 1968; 74:694–700.
- Moldoveanu Z, Wyatt RJ, Lee JY, et al. Patients with IgA nephropa- levels. Kidney Int thy have increased serum galactose deficient IgA1. 2002; 71:1148–1154.
- Kincaid-Smith P, Fairley K. The investigation of hematuria. Semin Nephrol 2005; 25:127–135.
- Coppo R, D’Amico G. Factors predicting progression of IgA nephropathies. J Nephrol 2005; 18:503–512.
- Donadio JV, Bergstralh EJ, Grande JP, Rademcher DM. Proteinuria patterns and their association with subsequent end-stage renal disease in IgA nephropathy. Nephrol Dial Transplant 2002; 17:1197–1203.
- Cook T. Interpretation of renal biopsies in IgA nephropathy. Contrib Nephrol 2007; 157:44–49.
- Bartosik LP, Lajole G, Sugar L, Cattran D. Predicting progression in IgA nephropathy. Am J Kidney Dis 2001; 58:551–553.
- Rauta V, Finne P, Fagerudd J, et al. Factors associated with progression of IgA nephropathy are related to renal function—a model for estimating risk of progression in mild disease. Clin Nephrol 2002; 58:85–94.
- Komatsu H, Fujimoto S, Sato Y, et al. “Point of no return (PNR)” in progressive IgA nephropathy: significance of blood pressure and proteinuria management up to PNR”. J Nephrol 2005; 18:690–695.
- Schena FP, Cerullo G, Torres DD, et al European IgA Nephropathy Consortium. Searching for IgA nephropathy candidate genes: genetic studies combined with high throughput innovative investigations. Contrib Nephrol 2007; 157:80–89.
- Haubitz M, Wittke S, Weissinger EM, et al. Urine protein patterns can serve as a diagnostic tools in patients with IgA nephropathy. Kidney Int 2005; 67:2313–2320.
- Barratt J, Feehally J, Smith AC. The pathogenesis of IgA nephropathy. Semin Nephrol 2004; 24:197–217.
- Nishitani Y, Iwano M, Yamaguchi Y, et al. Fibroblast-specific protein 1 is a specific prognostic marker for renal survival in patients with IgAN. Kidney Int 2005; 68:1078–1085.
- Barratt J, Feehally J. Treatment of IgA nephropathy. Kidney Int 2006; 69:1934–1938.
- Pozzi C, Andrulli S, Del Vecchio L, et al. Corticosteroid effectiveness in IgA nephropathy: long-term follow-up of a randomized, controlled trial. J Am Soc Nephrol 2004; 15:157–163.
- Ballardie FW, Roberts IS. Controlled prospective trial of prednisolone and cytotoxics in progressive IgA nephropathy. J Am Soc Nephrol 2002; 13:142–148.
- Donadio JV, Grande JP. The role of fish oil/omega-3 fatty acid in the treatment of IgA nephropathy. Semin Nephrol 2004; 24:225–243.
- Maes BD, Oyen R, Claes K, et al. Mycophenolate mofetil in IgA nephropathy: results of a 3-year prospective placebo-controlled randomized study. Kidney Int 2004; 65:1842–1849.
- Goumenous DS, Davlouros P, El Nahas AM, et al. Prednis-olone and azathioprine in IgA nephropathy—a ten year follow-up study. Nephron Clin Pract 2003; 93:c58–c68.
- Soler MG, Mir M, Rodriguez E, et al. Recurrence of IgA nephropathy and Henoch-Schönlein purpura after kidney transplantation: risk factors and graft survival. Transplant Proc 2005; 37:3705–3709.
- Floege J. Recurrent IgA nephropathy after renal transplantation. Semin Nephrol 2004; 24:287–291.
- Tomino Y. IgA nephropathy today. Contrib Nephrol 2007; 157:1–255.
- D’Amico G. The commonest glomerulonephritis in the world: IgA nephropathy. Quart J Med 1987; 245:709–727.
- Lee G, Glassock RJ. Immunoglobulin A nephropathy. In:Ponticelli C, Glassock R, editors. Treatment of Primary Glomerulonephritis. Oxford: Oxford Medical Publication, 1997:187–217.
- Donadio JV, Grande JP. IgA nephropathy. N Engl J Med 2002; 347:738–748.
- Glassock RJ. Concluding remarks. IgA nephropathy today. Contrib Nephrol 2002; 157:169–173.
- Berger J, Hinglais N. Les dépots intercapillaries d’IgA-IgG. J Urol Nephrol (Paris) 1968; 74:694–700.
- Moldoveanu Z, Wyatt RJ, Lee JY, et al. Patients with IgA nephropa- levels. Kidney Int thy have increased serum galactose deficient IgA1. 2002; 71:1148–1154.
- Kincaid-Smith P, Fairley K. The investigation of hematuria. Semin Nephrol 2005; 25:127–135.
- Coppo R, D’Amico G. Factors predicting progression of IgA nephropathies. J Nephrol 2005; 18:503–512.
- Donadio JV, Bergstralh EJ, Grande JP, Rademcher DM. Proteinuria patterns and their association with subsequent end-stage renal disease in IgA nephropathy. Nephrol Dial Transplant 2002; 17:1197–1203.
- Cook T. Interpretation of renal biopsies in IgA nephropathy. Contrib Nephrol 2007; 157:44–49.
- Bartosik LP, Lajole G, Sugar L, Cattran D. Predicting progression in IgA nephropathy. Am J Kidney Dis 2001; 58:551–553.
- Rauta V, Finne P, Fagerudd J, et al. Factors associated with progression of IgA nephropathy are related to renal function—a model for estimating risk of progression in mild disease. Clin Nephrol 2002; 58:85–94.
- Komatsu H, Fujimoto S, Sato Y, et al. “Point of no return (PNR)” in progressive IgA nephropathy: significance of blood pressure and proteinuria management up to PNR”. J Nephrol 2005; 18:690–695.
- Schena FP, Cerullo G, Torres DD, et al European IgA Nephropathy Consortium. Searching for IgA nephropathy candidate genes: genetic studies combined with high throughput innovative investigations. Contrib Nephrol 2007; 157:80–89.
- Haubitz M, Wittke S, Weissinger EM, et al. Urine protein patterns can serve as a diagnostic tools in patients with IgA nephropathy. Kidney Int 2005; 67:2313–2320.
- Barratt J, Feehally J, Smith AC. The pathogenesis of IgA nephropathy. Semin Nephrol 2004; 24:197–217.
- Nishitani Y, Iwano M, Yamaguchi Y, et al. Fibroblast-specific protein 1 is a specific prognostic marker for renal survival in patients with IgAN. Kidney Int 2005; 68:1078–1085.
- Barratt J, Feehally J. Treatment of IgA nephropathy. Kidney Int 2006; 69:1934–1938.
- Pozzi C, Andrulli S, Del Vecchio L, et al. Corticosteroid effectiveness in IgA nephropathy: long-term follow-up of a randomized, controlled trial. J Am Soc Nephrol 2004; 15:157–163.
- Ballardie FW, Roberts IS. Controlled prospective trial of prednisolone and cytotoxics in progressive IgA nephropathy. J Am Soc Nephrol 2002; 13:142–148.
- Donadio JV, Grande JP. The role of fish oil/omega-3 fatty acid in the treatment of IgA nephropathy. Semin Nephrol 2004; 24:225–243.
- Maes BD, Oyen R, Claes K, et al. Mycophenolate mofetil in IgA nephropathy: results of a 3-year prospective placebo-controlled randomized study. Kidney Int 2004; 65:1842–1849.
- Goumenous DS, Davlouros P, El Nahas AM, et al. Prednis-olone and azathioprine in IgA nephropathy—a ten year follow-up study. Nephron Clin Pract 2003; 93:c58–c68.
- Soler MG, Mir M, Rodriguez E, et al. Recurrence of IgA nephropathy and Henoch-Schönlein purpura after kidney transplantation: risk factors and graft survival. Transplant Proc 2005; 37:3705–3709.
- Floege J. Recurrent IgA nephropathy after renal transplantation. Semin Nephrol 2004; 24:287–291.
KEY POINTS
- IgA nephropathy tends to progress slowly, and in only about half of patients does it progress to end-stage renal disease within 25 years.
- At present, the factors that predict an accelerated course and progression to end-stage renal disease are persistent proteinuria, elevated serum creatinine at diagnosis, persistent microscopic hematuria, poorly controlled hypertension, and extensive glomerulosclerosis or interstitial fibrosis, or both, on renal biopsy.
- Needed are better diagnostic and prognostic tests and therapies that address the mechanism of the disease.
- The value of treatment with an angiotensin-converting enzyme inhibitor, an angiotensin receptor blocker, or both is well established. If protein excretion does not decrease with this therapy, one can consider adding immunosuppressive therapy in selected patients, but this strategy is still empiric and unproven.