User login
A less common source of dyspnea in scleroderma
A 48-year-old man reports progressive exercise intolerance, shortness of breath, fatigue, and melena over the past month. He has a long history of Raynaud phenomenon, and 5 months ago he developed severe sclerodactyly in both hands, diagnosed as limited cutaneous systemic sclerosis (scleroderma).
He has no chest pain, swelling of the lower limbs, change in weight, cough, fever, chills, or sick contacts, and he has not traveled recently.
His symptoms began as fatigue and shortness of breath, which worsened until he began having episodes of abdominal pain with melena and dizzy spills, although he never passed out.
He is currently taking long-term low-dose prednisone and mycophenolate mofetil (Cell-Cept) for the systemic sclerosis, and omeprazole (Prilosec) for gastroesophageal reflux. His father had lupus, and his grandmother had colon cancer.
An outpatient workup for sclerosis-related lung and heart involvement is negative. The workup includes computed tomography of the chest, pulmonary function tests, and Doppler echocardiography.
He is afebrile, with a blood pressure of 105/60 mm Hg and a pulse of 98. His cardiopulmonary examination results are normal. He has mild epigastric tenderness without rebound or guarding. His hemoglobin concentration at the time of hospital admission is 7.8 g/dL, down from 14.5 g/dL recorded when limited cutaneous systemic sclerosis was diagnosed. Iron studies reveal iron deficiency.
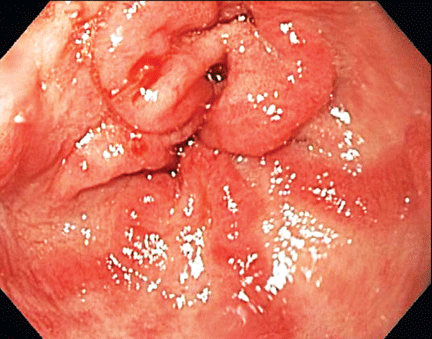
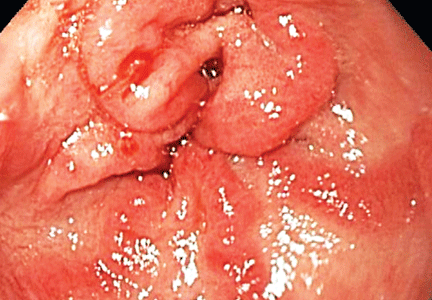
The antral ectasia is treated with argon plasma coagulation during the endoscopic examination.
Afterward, the patient's hemoglobin stabilizes, and the melena resolves. He is discharged on an oral proton pump inhibitor, with instructions to follow up for another endoscopic session in 1 month.
GASTROINTESTINAL FEATURES OF SYSTEMIC SCLEROSIS
Sclerodermal disorders have diverse manifestations that always include characteristic cutaneous signs. While there are several well-recognized symptomatic conditions commonly associated with scleroderma, attention must also be paid to the less common causes of these symptoms. Scleroderma has gastrointestinal complications that can easily be missed and may not respond to immunomodulatory or proton pump inhibitor therapy: complications can include esophageal dysmotility, hypomotility, gastric paresis, reflux esophagitis, strictures, drug-related ulcer, malabsorption, bacterial overgrowth, and pseudo-obstruction.1
This patient had an underrecognized cause of dyspnea in the setting of systemic sclerosis. Vascular symptoms of limited cutaneous systemic sclerosis are typically attributed to Raynaud phenomenon; gastrointestinal symptoms are typically attributed to esophageal dysmotility; and associated dyspnea is often considered to represent pulmonary or cardiac involvement of the sclerosis. However, gastric antral vascular ectasia should be considered in any patient with scleroderma and evidence of anemia.
The prevalence of gastric antral vascular ectasia in patients with systemic sclerosis is estimated to be about 6%.2–4 It is a relatively rare cause of upper gastrointestinal blood loss that can be clinically silent until the patient develops severe iron deficiency anemia and symptoms of dyspnea, fatigue, or congestive heart failure.
Gastric antral vascular ectasia in scleroderma usually presents as iron deficiency anemia, and only presents overtly as hematemesis or melena 10% to 14% of the time.4 Because of the often occult nature of the bleeding, the condition may be clinically silent in the early phase. Symptoms of shortness of breath and fatigue may not develop until the anemia worsens rapidly or becomes severe. Anemia is present in almost all cases of gastric antral vascular ectasia (96% to 100%) and should be a strong clinical clue for early endoscopic evaluation in patients with scleroderma, especially if there is already suspicion of upper gastrointestinal bleeding.2–5
The distinctive endoscopic streaky pattern of ectasia along the stomach antrum seen in gastric antral vascular ectasia is called “watermelon stomach”4,5 because the striped pattern recalls the stripes of a watermelon. The endoscopic appearance can vary, however, from the watermelon pattern to a coalescence of angiodysplastic lesions termed “honeycomb stomach,” which can easily be mistaken for antral gastritis.4,5 Therefore, biopsy often serves to confirm the diagnosis, with histologic features including dilated mucosal capillaries with focal fibrin thrombosis and fibromuscular hyperplasia of the lamina propria.
Gastric antral vascular ectasia often requires multiple transfusions of red blood cells, as well as repeated treatments with endoscopic argon plasma coagulation, whereby ionized argon gas is used to conduct an electric current that coagulates the surface of the mucosa to a few millimeters depth.4–6
A knowledge of the association between scleroderma and gastric antral vascular ectasia can lead to earlier recognition and treatment and can avoid unnecessary testing and complications of severe anemia.
- Forbes A, Marie I. Gastrointestinal complications: the most frequent internal complications of systemic sclerosis. Rheumatology (Oxford) 2009; 48(suppl 3):iii36–iii39.
- Ingraham KM, O’Brien MS, Shenin M, Derk CT, Steen VD. Gastric antral vascular ectasia in systemic sclerosis: demographics and disease predictors. J Rheumatol 2010; 37:603–607.
- Watson M, Hally RJ, McCue PA, Varga J, Jiménez SA. Gastric antral vascular ectasia (watermelon stomach) in patients with systemic sclerosis. Arthritis Rheum 1996; 39:341–346.
- Marie I, Ducrotte P, Antonietti M, Herve S, Levesque H. Watermelon stomach in systemic sclerosis: its incidence and management. Aliment Pharmacol Ther 2008; 28:412–421.
- Selinger CP, Ang YS. Gastric antral vascular ectasia (GAVE): an update on clinical presentation, pathophysiology and treatment. Digestion 2008; 77:131–137.
- Chaves DM, Sakai P, Oliveira CV, Cheng S, Ishioka S. Watermelon stomach: clinical aspects and treatment with argon plasma coagulation. Arq Gastroenterol 2006; 43:191–195.
A 48-year-old man reports progressive exercise intolerance, shortness of breath, fatigue, and melena over the past month. He has a long history of Raynaud phenomenon, and 5 months ago he developed severe sclerodactyly in both hands, diagnosed as limited cutaneous systemic sclerosis (scleroderma).
He has no chest pain, swelling of the lower limbs, change in weight, cough, fever, chills, or sick contacts, and he has not traveled recently.
His symptoms began as fatigue and shortness of breath, which worsened until he began having episodes of abdominal pain with melena and dizzy spills, although he never passed out.
He is currently taking long-term low-dose prednisone and mycophenolate mofetil (Cell-Cept) for the systemic sclerosis, and omeprazole (Prilosec) for gastroesophageal reflux. His father had lupus, and his grandmother had colon cancer.
An outpatient workup for sclerosis-related lung and heart involvement is negative. The workup includes computed tomography of the chest, pulmonary function tests, and Doppler echocardiography.
He is afebrile, with a blood pressure of 105/60 mm Hg and a pulse of 98. His cardiopulmonary examination results are normal. He has mild epigastric tenderness without rebound or guarding. His hemoglobin concentration at the time of hospital admission is 7.8 g/dL, down from 14.5 g/dL recorded when limited cutaneous systemic sclerosis was diagnosed. Iron studies reveal iron deficiency.
The antral ectasia is treated with argon plasma coagulation during the endoscopic examination.
Afterward, the patient's hemoglobin stabilizes, and the melena resolves. He is discharged on an oral proton pump inhibitor, with instructions to follow up for another endoscopic session in 1 month.
GASTROINTESTINAL FEATURES OF SYSTEMIC SCLEROSIS
Sclerodermal disorders have diverse manifestations that always include characteristic cutaneous signs. While there are several well-recognized symptomatic conditions commonly associated with scleroderma, attention must also be paid to the less common causes of these symptoms. Scleroderma has gastrointestinal complications that can easily be missed and may not respond to immunomodulatory or proton pump inhibitor therapy: complications can include esophageal dysmotility, hypomotility, gastric paresis, reflux esophagitis, strictures, drug-related ulcer, malabsorption, bacterial overgrowth, and pseudo-obstruction.1
This patient had an underrecognized cause of dyspnea in the setting of systemic sclerosis. Vascular symptoms of limited cutaneous systemic sclerosis are typically attributed to Raynaud phenomenon; gastrointestinal symptoms are typically attributed to esophageal dysmotility; and associated dyspnea is often considered to represent pulmonary or cardiac involvement of the sclerosis. However, gastric antral vascular ectasia should be considered in any patient with scleroderma and evidence of anemia.
The prevalence of gastric antral vascular ectasia in patients with systemic sclerosis is estimated to be about 6%.2–4 It is a relatively rare cause of upper gastrointestinal blood loss that can be clinically silent until the patient develops severe iron deficiency anemia and symptoms of dyspnea, fatigue, or congestive heart failure.
Gastric antral vascular ectasia in scleroderma usually presents as iron deficiency anemia, and only presents overtly as hematemesis or melena 10% to 14% of the time.4 Because of the often occult nature of the bleeding, the condition may be clinically silent in the early phase. Symptoms of shortness of breath and fatigue may not develop until the anemia worsens rapidly or becomes severe. Anemia is present in almost all cases of gastric antral vascular ectasia (96% to 100%) and should be a strong clinical clue for early endoscopic evaluation in patients with scleroderma, especially if there is already suspicion of upper gastrointestinal bleeding.2–5
The distinctive endoscopic streaky pattern of ectasia along the stomach antrum seen in gastric antral vascular ectasia is called “watermelon stomach”4,5 because the striped pattern recalls the stripes of a watermelon. The endoscopic appearance can vary, however, from the watermelon pattern to a coalescence of angiodysplastic lesions termed “honeycomb stomach,” which can easily be mistaken for antral gastritis.4,5 Therefore, biopsy often serves to confirm the diagnosis, with histologic features including dilated mucosal capillaries with focal fibrin thrombosis and fibromuscular hyperplasia of the lamina propria.
Gastric antral vascular ectasia often requires multiple transfusions of red blood cells, as well as repeated treatments with endoscopic argon plasma coagulation, whereby ionized argon gas is used to conduct an electric current that coagulates the surface of the mucosa to a few millimeters depth.4–6
A knowledge of the association between scleroderma and gastric antral vascular ectasia can lead to earlier recognition and treatment and can avoid unnecessary testing and complications of severe anemia.
A 48-year-old man reports progressive exercise intolerance, shortness of breath, fatigue, and melena over the past month. He has a long history of Raynaud phenomenon, and 5 months ago he developed severe sclerodactyly in both hands, diagnosed as limited cutaneous systemic sclerosis (scleroderma).
He has no chest pain, swelling of the lower limbs, change in weight, cough, fever, chills, or sick contacts, and he has not traveled recently.
His symptoms began as fatigue and shortness of breath, which worsened until he began having episodes of abdominal pain with melena and dizzy spills, although he never passed out.
He is currently taking long-term low-dose prednisone and mycophenolate mofetil (Cell-Cept) for the systemic sclerosis, and omeprazole (Prilosec) for gastroesophageal reflux. His father had lupus, and his grandmother had colon cancer.
An outpatient workup for sclerosis-related lung and heart involvement is negative. The workup includes computed tomography of the chest, pulmonary function tests, and Doppler echocardiography.
He is afebrile, with a blood pressure of 105/60 mm Hg and a pulse of 98. His cardiopulmonary examination results are normal. He has mild epigastric tenderness without rebound or guarding. His hemoglobin concentration at the time of hospital admission is 7.8 g/dL, down from 14.5 g/dL recorded when limited cutaneous systemic sclerosis was diagnosed. Iron studies reveal iron deficiency.
The antral ectasia is treated with argon plasma coagulation during the endoscopic examination.
Afterward, the patient's hemoglobin stabilizes, and the melena resolves. He is discharged on an oral proton pump inhibitor, with instructions to follow up for another endoscopic session in 1 month.
GASTROINTESTINAL FEATURES OF SYSTEMIC SCLEROSIS
Sclerodermal disorders have diverse manifestations that always include characteristic cutaneous signs. While there are several well-recognized symptomatic conditions commonly associated with scleroderma, attention must also be paid to the less common causes of these symptoms. Scleroderma has gastrointestinal complications that can easily be missed and may not respond to immunomodulatory or proton pump inhibitor therapy: complications can include esophageal dysmotility, hypomotility, gastric paresis, reflux esophagitis, strictures, drug-related ulcer, malabsorption, bacterial overgrowth, and pseudo-obstruction.1
This patient had an underrecognized cause of dyspnea in the setting of systemic sclerosis. Vascular symptoms of limited cutaneous systemic sclerosis are typically attributed to Raynaud phenomenon; gastrointestinal symptoms are typically attributed to esophageal dysmotility; and associated dyspnea is often considered to represent pulmonary or cardiac involvement of the sclerosis. However, gastric antral vascular ectasia should be considered in any patient with scleroderma and evidence of anemia.
The prevalence of gastric antral vascular ectasia in patients with systemic sclerosis is estimated to be about 6%.2–4 It is a relatively rare cause of upper gastrointestinal blood loss that can be clinically silent until the patient develops severe iron deficiency anemia and symptoms of dyspnea, fatigue, or congestive heart failure.
Gastric antral vascular ectasia in scleroderma usually presents as iron deficiency anemia, and only presents overtly as hematemesis or melena 10% to 14% of the time.4 Because of the often occult nature of the bleeding, the condition may be clinically silent in the early phase. Symptoms of shortness of breath and fatigue may not develop until the anemia worsens rapidly or becomes severe. Anemia is present in almost all cases of gastric antral vascular ectasia (96% to 100%) and should be a strong clinical clue for early endoscopic evaluation in patients with scleroderma, especially if there is already suspicion of upper gastrointestinal bleeding.2–5
The distinctive endoscopic streaky pattern of ectasia along the stomach antrum seen in gastric antral vascular ectasia is called “watermelon stomach”4,5 because the striped pattern recalls the stripes of a watermelon. The endoscopic appearance can vary, however, from the watermelon pattern to a coalescence of angiodysplastic lesions termed “honeycomb stomach,” which can easily be mistaken for antral gastritis.4,5 Therefore, biopsy often serves to confirm the diagnosis, with histologic features including dilated mucosal capillaries with focal fibrin thrombosis and fibromuscular hyperplasia of the lamina propria.
Gastric antral vascular ectasia often requires multiple transfusions of red blood cells, as well as repeated treatments with endoscopic argon plasma coagulation, whereby ionized argon gas is used to conduct an electric current that coagulates the surface of the mucosa to a few millimeters depth.4–6
A knowledge of the association between scleroderma and gastric antral vascular ectasia can lead to earlier recognition and treatment and can avoid unnecessary testing and complications of severe anemia.
- Forbes A, Marie I. Gastrointestinal complications: the most frequent internal complications of systemic sclerosis. Rheumatology (Oxford) 2009; 48(suppl 3):iii36–iii39.
- Ingraham KM, O’Brien MS, Shenin M, Derk CT, Steen VD. Gastric antral vascular ectasia in systemic sclerosis: demographics and disease predictors. J Rheumatol 2010; 37:603–607.
- Watson M, Hally RJ, McCue PA, Varga J, Jiménez SA. Gastric antral vascular ectasia (watermelon stomach) in patients with systemic sclerosis. Arthritis Rheum 1996; 39:341–346.
- Marie I, Ducrotte P, Antonietti M, Herve S, Levesque H. Watermelon stomach in systemic sclerosis: its incidence and management. Aliment Pharmacol Ther 2008; 28:412–421.
- Selinger CP, Ang YS. Gastric antral vascular ectasia (GAVE): an update on clinical presentation, pathophysiology and treatment. Digestion 2008; 77:131–137.
- Chaves DM, Sakai P, Oliveira CV, Cheng S, Ishioka S. Watermelon stomach: clinical aspects and treatment with argon plasma coagulation. Arq Gastroenterol 2006; 43:191–195.
- Forbes A, Marie I. Gastrointestinal complications: the most frequent internal complications of systemic sclerosis. Rheumatology (Oxford) 2009; 48(suppl 3):iii36–iii39.
- Ingraham KM, O’Brien MS, Shenin M, Derk CT, Steen VD. Gastric antral vascular ectasia in systemic sclerosis: demographics and disease predictors. J Rheumatol 2010; 37:603–607.
- Watson M, Hally RJ, McCue PA, Varga J, Jiménez SA. Gastric antral vascular ectasia (watermelon stomach) in patients with systemic sclerosis. Arthritis Rheum 1996; 39:341–346.
- Marie I, Ducrotte P, Antonietti M, Herve S, Levesque H. Watermelon stomach in systemic sclerosis: its incidence and management. Aliment Pharmacol Ther 2008; 28:412–421.
- Selinger CP, Ang YS. Gastric antral vascular ectasia (GAVE): an update on clinical presentation, pathophysiology and treatment. Digestion 2008; 77:131–137.
- Chaves DM, Sakai P, Oliveira CV, Cheng S, Ishioka S. Watermelon stomach: clinical aspects and treatment with argon plasma coagulation. Arq Gastroenterol 2006; 43:191–195.
Peanut Allergy Awareness
Among all persons with food allergies, those who are allergic to peanuts are at greatest risk for anaphylactic symptoms.1 About 30,000 cases of food allergy–related anaphylaxis are seen in the nation’s emergency departments (EDs) each year, and the food most commonly responsible is peanuts.2 What can primary care providers do to reduce the number of peanut allergy–associated anaphylactic reactions and fatalities, both in the ED and in the larger community?
According to a guideline from the National Institute of Allergy and Infectious Diseases (NIAID),3 prevalence of peanut allergy is about 0.6% of the US population, although in an 11-year survey involving more than 13,000 respondents, Sicherer et al4 reported allergy to peanuts, tree nuts, or both in 1.4%, possibly translating to some three million Americans; British researchers have reported peanut allergy in 1.8% of an 1,100-member children’s cohort.5 The risk of exposure to peanuts and the associated risk for severe and possibly fatal anaphylaxis present a lifelong struggle for both patient and family.
ETIOLOGY OF PEANUT ALLERGIES
Food allergy prevalence has reportedly doubled in recent decades, with a significant increase also seen in allergy severity.6 Allergies involving eggs, nuts, fish, milk, and other foods represent the leading cause of hospital-treated anaphylaxis throughout the world.1 Unlike other allergenic foods that affect only one age-group, peanuts are among the foods that trigger the “vast majority” of allergic reactions in young children, teenagers, and adults alike.3
Increases in reported episodes of peanut allergy reactions may be occurring for several reasons:
• Many people have adopted vegetarian diets, and nuts are considered a good protein source6
• Environmental exposures are increasingly common
• More people are genetically vulnerable, as the role of family history becomes clearer
• Food preparation methods (eg, shared processing equipment, contaminated raw materials, formulation errors) and inaccurate labeling lead to accidental exposures7,8
• Exposure to nuts in utero or during breastfeeding is more common.9 Nowak-Wegrzyn and Sampson6 point to the promotion of peanut butter as an economical, nutritious food source for children and for women during pregnancy and lactation; mothers’ consumption of peanuts more than once a week during pregnancy and lactation have been linked to overexposure for their children.9
Other trends that may contribute to peanut allergy prevalence are the early introduction of solid foods in the infant diet and the use of skin products that contain peanut oil.6
Environment and Genetics
The body of knowledge regarding the specific causes of peanut allergy is increasing constantly. Several known peanut proteins (Ara h1, Ara h2, Ara h3, Ara h6, Ara h7, and Ara h9; Ara h8 is a homologous allergen that may account for peanut/birch cross-reactivity) are thought to be responsible for the initial sensitization to peanuts in vulnerable persons, triggering the associated immunoglobulin E (IgE)–mediated response.10-12 Approximately 75% of known peanut-allergic patients will react to these proteins on their first ingestion after being sensitized.9
Since IgE antibodies do not cross the placenta, it is believed that sensitization to peanut proteins must occur in utero or through breast milk. This form of sensitization predisposes these patients to the initial life-threatening anaphylactic reaction.9
There is strong evidence that genetic factors may play a role in peanut allergies.2 In a study of 58 pairs of twins by Sicherer et al,13 heritability of peanut allergy was estimated at 82%, with 64% of monozygotic pairs, versus 7% of dizygotic pairs, showing concordance for peanut allergy. However, the genetic loci that may be responsible for specific food allergies have not yet been identified.2
It is believed that manifestations of food allergy are very similar to those of asthma and atopic dermatitis. According to Green and colleagues,14 82% of peanut-allergic children who visited a referral clinic also had atopic dermatitis. These conditions appear to be triggered by similar mechanisms, mediated by both environmental and genetic factors.2,14-16 Hong et al2 are optimistic about the advances being made in food allergy genetics. Increased understanding, they feel, may lead to new treatment options for potentially fatal food allergies.2
PATIENT PRESENTATION AND HISTORY
As with any IgE-mediated immune response, the patient must have been exposed to the allergen in question. Most patients present with a history of having ingested raw or boiled peanuts and/or foods produced in a facility that also processes nuts.1,18 Clinical symptoms of peanut allergy may develop within seconds of ingestion. For some patients, consumption of as little as 5 to 50 mg of peanut protein can trigger symptoms.19 (A single peanut from a jar of commercially processed peanuts contains approximately 300 mg of potentially allergenic protein.1)
Typically, the most dramatically affected patients have a medical history of asthma or other IgE-mediated immune reactions.1 In one study, young adults with IgE-mediated peanut allergy were found at especially high risk for severe anaphylaxis.6 Seventy-five percent of patients who have a reaction to peanuts do so following their first ingestion (after the initial exposure).
The mean patient age for a diagnosis of peanut allergy is about 14 months; only 20% of the patients diagnosed with a peanut allergy (most likely those with a baseline peanut-specific serum IgE level 18) will outgrow it by the time they reach school age.18,20 Those who do should be encouraged to consume peanuts on a regular basis; according to Byrne et al,21 8% of patients with allergy resolution experience recurrence, a possible result of infrequent peanut consumption.
PHYSICAL EXAMINATION
Patients with peanut allergies can present with a range of symptoms, possibly involving cutaneous, cardiovascular, gastrointestinal, and/or respiratory systems (see Table 115,22). The more notable symptoms, possibly developing within 15 minutes of exposure, are progressive upper and lower respiratory difficulties, vomiting, diarrhea, hypotension, edema of the face and hands, arrhythmia, throat tightness (in serious cases, approaching anaphylaxis), and possibly loss of consciousness. Such severe reactions often occur in the child who has ingested raw peanuts or tree nuts.22
Milder physical exam findings include erythema, pruritus, conjunctivitis, abdominal pain, nasal congestion, itchy throat, and sneezing. These reactions may have been triggered by foods produced in a facility that also processes nuts, household utensils used to prepare foods that contain nuts, or cross-contamination from another child.9,15,24
DIAGNOSTIC WORK-UP
The diagnosis of a patient with a peanut allergy is made through thorough history taking, careful physical examination, allergy testing with either a skin prick test (SPT) or serum-specific IgE, and oral food challenges. The gold standard for diagnosing food allergy is the double-blind, placebo-controlled oral food challenge,2,25-27 as this test alone can determine the amount of peanut protein needed to trigger a reaction in the given patient.9 However, this is a difficult test to administer and must be performed under strict medical supervision.21
It has been determined that a wheal size of 8.0 mm or greater on the SPT has a 95% to 100% positive predictive value for peanut allergy.1,26,27 Although conflicting results have been reported in some patients between SPT and the oral food challenge, a negative SPT result is considered useful for excluding IgE-mediated allergic responses.22
Researchers examining the peanut-specific serum IgE have demonstrated a 95% to 99% positive predictive value when serum levels exceed 15 kU/L.26,27 This cutoff value in peanut allergy patients is considered suggestive of allergic reactivity, although negative results on an oral food challenge have been reported in more than 25% of children with serum levels exceeding the cutoff.25-27 Testing may have been to whole peanut extract rather than the molecular components (eg, Ara h8).11,12
This past summer, the FDA approved a component test that detects allergen components that include Ara h1, h2, h3, h8, and h9.11,12 Another specific version of the serum IgE test has been in development, one that measures the patient’s IgE reactions to the Ara h2 and Ara h8 components in peanut protein. Johnson and colleagues10,28 have found an increasing level of serum IgE anti–Ara h2 in children who were unable to pass the oral peanut challenge, whereas serum IgE anti–Ara h8 was higher in those who did pass the challenge.28
DIAGNOSING ANAPHYLAXIS
The manifestation of anaphylaxis in patients allergic to peanuts or tree nuts can be life-threatening.29 Symptoms include intense pruritus with flushing of the skin, urticaria, and angioedema, upper-respiratory obstruction resulting from laryngeal edema, and hypotension.30 The clinical criteria for diagnosing anaphylaxis can be found in Table 2.30,31
It is important to recognize the signs and symptoms of anaphylaxis in patients with a peanut allergy; many patients who present to the ED represent first-time reactions. Among patients with life-threatening symptoms on initial reaction, 71% will have similarly severe reactions in subsequent episodes (compared with 44% of patients whose first reaction was not life-threatening).3
TREATMENT, INCLUDING PATIENT EDUCATION
Currently there is no cure for peanut allergy, and no appropriate therapies yet exist to reduce allergy severity. Modest gains have been reported in raising tolerance threshold levels through peanut oral immunotherapy—a long, painstaking process.19,21,32 For now, treatment for peanut allergy is directed at controlling symptoms, once a reaction has occurred. Therefore, the clinician’s goal is to educate peanut-allergic patients and their families on avoiding accidental peanut ingestion, recognizing signs and symptoms of an allergic reaction, and preparing an emergency plan.4
Because four in five patients can expect peanut allergy to last for a lifetime,18,20 strict avoidance of peanuts and peanut products is essential—though difficult because of accidental exposure to food allergens (for example, when dining in restaurants or purchasing bakery products22,32), cross-contamination (as can occur when a food preparation area is not properly cleaned), and allergen cross-reactivity (such as consumption of other legumes).1 Patients must be taught to read food labels carefully for possible hidden sources of peanuts (see Table 37,8); in some cases, product labels bear helpful advisory wording, such as “may contain peanuts.”34,35 US legislation mandates that listed ingredients on food packaging include the eight foods that account for 90% of allergic reactions:
• Peanuts
• Tree nuts
• Egg
• Milk
• Wheat
• Soybeans
• Fish
• Crustacean shellfish.34
Treatment for Anaphylaxis
In pediatric patients, administration of epinephrine is the definitive treatment for anaphylaxis; both the child and parents should carry an epinephrine self-injection device at all times in the event of accidental peanut ingestion. These devices are available in two strengths, based on the child’s weight, and expiration dates should be noted with care. Correct use of the epinephrine self-injection device should be reviewed at each office visit.6
Early-stage allergic reactions can be managed by oral antihistamines, such as diphenhydramine (1 mg/kg body weight up to 75 mg) and an intramuscular injection of epinephrine.1 Prompt transport to the ED should follow (see “Management of Anaphylaxis in the ED”1,9).
PREVENTION
A 2010 expert panel on diagnosis and management of food allergy sponsored by the NIAID, NIH,3 does not advise women to restrict their diet during pregnancy and lactation. Similarly, the United Kingdom’s Department of Health and the Food Standards Agency (DHFSA)36,37 does not support the belief that eating peanuts and peanut-containing foods during pregnancy correlates with a child’s potential for developing a peanut allergy.
The DHFSA does recommend breastfeeding infants for the first six months, if possible, and that mothers refrain from introducing peanut-containing foods during that time. They also recommend that foods associated with a high risk for allergy be introduced into a child’s diet one at a time, to make it easier to identify any allergenic substance.36,37
Lastly, the DHFSA advises parents with a family history of peanut allergy to introduce peanuts only after consulting with their health care provider. The same consideration is advised if a child has already been diagnosed with another allergy.34 According to the American Academy of Pediatrics,6,38 children at high risk for food allergy (eg, atopic disease in both parents or one parent and one sibling) should be breastfed or be given hypoallergenic formula until age 1 year, with no solid foods before age 6 months; peanut-containing foods should not be given before age 3 or 4 years.
CONCLUSION
Peanut allergy can present a lifelong battle for affected patients. Eating one peanut or being exposed even to minute amounts of peanut protein could mean life or death without appropriate management. Reading food labels carefully, preparing peanut-free foods, recognizing the signs and symptoms of anaphylaxis, and obtaining the necessary treatment when allergic reactions occur are essential for peanut-allergic patients and their families.
REFERENCES
1. Burks AW. Peanut allergy. Lancet. 2008;371 (9623):1538-1546.
2. Hong X, Tsai HJ, Wang X. Genetics of food allergy. Curr Opin Pediatr. 2009;21(6):770-776.
3. Boyce JA, Assa’ad A, Burks AW, et al. Guidelines for the diagnosis and management of food allergy in the United States: report of the NIAID-sponsored expert panel. J Allergy Clin Immunol. 2010;126(6 suppl):S1-S58.
4. Sicherer S, Muñoz-Furlong A, Godbold JH, Sampson HA. US prevalence of self-reported peanut, tree nut, and sesame allergy: 11-year follow-up. J Allergy Clin Immunol. 2010;125(6):1322-1326.
5. Hourihane JO, Aiken R, Briggs R, et al. The impact of government advice to pregnant mothers regarding peanut avoidance on the prevalence of peanut allergy in United Kingdom children at school entry. J Allergy Clin Immunol. 2007;312(5):1197-1202.
6. Nowak-Wegrzyn A, Sampson HA. Adverse reactions to foods. Med Clin North Am. 2006;90(1):97-127.
7. Puglisi G, Frieri M. Update on hidden food allergens and food labeling. Allergy Asthma Proc. 2007;28(6):634-639.
8. Hefle SL. Hidden food allergens. Curr Opin Allergy Clin Immunol. 2001;1(3):269-271.
9. Lee CW, Sheffer AL. Peanut allergy. Allergy Asthma Proc. 2003;24(4):259-264.
10. Boughton B. New test for peanut allergy a step forward. www.medscape.com/viewarticle/740133. Accessed November 16, 2011.
11. Asarnoj A, Movérare R, Östblom E, et al. IgE to peanut allergen components: relation to peanut symptoms and pollen sensitization in 8-year-olds. Allergy. 2010;65(9):1189-1195.
12. Codreanu F, Collignon O, Roitel O, et al. A novel immunoassay using recombinant allergens simplifies peanut allergy diagnosis. Int Arch Allergy Immunol. 2011;154(3):216-226.
13. Sicherer SH, Furlong TJ, Maes HH, et al. Genetics of peanut allergy: a twin study. J Allergy Clin Immunol. 2000;106(1 pt 1):53-56.
14. Green TD, LaBelle VS, Steele PH, et al. Clinical characteristics of peanut-allergic children: recent changes. Pediatrics. 2007;120(6):1304-1310.
15. Al-ahmed N, Alsowaidi S, Vadas P. Peanut allergy: an overview. Allergy Asthma Clin Immunol. 2008;4(4):139-143.
16. Björkstén B. Genetic and environmental risk factors for the development of food allergy. Curr Opin Allergy Clin Immunol. 2005;5(3):249-253.
17. Lack G. Epidemiologic risks for food allergy. J Allergy Clin Immunol. 2008;121(6):1331-1336.
18. Skolnick HS, Conover-Walker MK, Koerner CB, et al. The natural history of peanut allergy. J Allergy Clin Immunol. 2001;107(2):367-374.
19. Clark AT, Islam S, King Y, et al. Successful oral tolerance induction in severe peanut allergy. Allergy. 2009;64(8):1218-1220.
20. Busse PJ, Nowak-Wegrzyn AH, Noone SA, et al. Recurrent peanut allergy. N Engl J Med. 2002; 347(19):1535-1536.
21. Byrne AM, Malka-Rais J, Burks AW, Fleischer DM. How do we know when peanut and tree nut allergy have resolved, and how do we keep it resolved? Clin Exp Allergy. 2010;49(9):1303-1311.
22. Sampson HA. Update on food allergy. J Allergy Clin Immunol. 2004;113(5):805-819.
23. Furlong TJ, Desimone J, Sicherer SH. Peanut and tree nut allergic reactions in restaurants and other establishments. J Allergy Clin Immunol. 2001;108(5):866-870.
24. Nelson HS, Lahr J, Rule R, et al. Treatment of anaphylactic sensitivity to peanuts by immunotherapy with injections of aqueous peanut extract. J Allergy Clin Immunol. 1997;99(6 pt 1):744-751.
25. Du Toit G, Santos A, Roberts G, et al. The diagnosis of IgE-mediated food allergy in childhood. Pediatr Allergy Immunol. 2009;20(4):309-319.
26. Roberts G, Lack G. Diagnosing peanut allergy with skin prick and specific IgE testing. J Allergy Clin Immunol. 2005;115(6):1291-1296.
27. Wainstein BK, Yee A, Jelley D, et al. Combining skin prick, immediate skin application and specific-IgE testing in the diagnosis of peanut allergy in children. Pediatr Allergy Immunol. 2007;18(3):231-239.
28. Johnson K, Keet C, Hamilton R, Wood R. Predictive value of peanut component specific IgE in a clinical population. Presented at: 2011 Annual Meeting, American Academy of Allergy, Asthma and Immunology; March 19, 2011; San Francisco, CA. Abstract 267.
29. Sheffer AL. Allergen avoidance to reduce asthma-related morbidity. N Engl J Med. 2004;351(11):1134-1136.
30. Russell S, Monroe K, Losek JD. Anaphylaxis management in the pediatric emergency department: opportunities for improvement. Pediatr Emerg Care. 2010;26(2):71-76.
31. Sampson HA, Munoz-Furlong A, Campbell RL, et al. Second symposium on the definition and management of anaphylaxis: summary report—Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. J Allergy Clin Immunol. 2006;117(2):391-397.
32. Blumchen K, Ulbricht H, Staden U, et al. Oral peanut immunotherapy in children with peanut anaphylaxis. J Allergy Clin Immunol. 2010; 126(1):83-91.
33. Yu JW, Kagan R, Verreault N, et al. Accidental ingestions in children with peanut allergy. J Allergy Clin Immunol. 2006;118(2):466-472.
34. Taylor SL, Hefle SL. Food allergen labeling in the USA and Europe. Curr Opin Allergy Clin Immunol. 2006;6(3):186-190.
35. Sampson HA, Srivastava K, Li XM, Burks AW. New perspectives for the treatment of food allergy (peanut). Arb Paul Ehrlich Inst Bundesamt Sera Impfstoffe Frankf A M. 2003;(94):236-244.
36. McLean S, Sheikh A. Does avoidance of peanuts in early life reduce the risk of peanut allergy? BMJ. 2010 Mar 11;340:c424.
37. Department of Health. Revised government advice on consumption of peanut during pregnancy, breastfeeding, and early life and development of peanut allergy (Aug 2009). www.dh.gov.uk/en/Healthcare/Children/Maternity/Maternalandinfantnutrition/DH_104490. Accessed November 16, 2011.
38. American Academy of Pediatrics. Committee on Nutrition. Hypoallergenic infant formulas. Pediatrics. 2000;106(2):346-349.
Among all persons with food allergies, those who are allergic to peanuts are at greatest risk for anaphylactic symptoms.1 About 30,000 cases of food allergy–related anaphylaxis are seen in the nation’s emergency departments (EDs) each year, and the food most commonly responsible is peanuts.2 What can primary care providers do to reduce the number of peanut allergy–associated anaphylactic reactions and fatalities, both in the ED and in the larger community?
According to a guideline from the National Institute of Allergy and Infectious Diseases (NIAID),3 prevalence of peanut allergy is about 0.6% of the US population, although in an 11-year survey involving more than 13,000 respondents, Sicherer et al4 reported allergy to peanuts, tree nuts, or both in 1.4%, possibly translating to some three million Americans; British researchers have reported peanut allergy in 1.8% of an 1,100-member children’s cohort.5 The risk of exposure to peanuts and the associated risk for severe and possibly fatal anaphylaxis present a lifelong struggle for both patient and family.
ETIOLOGY OF PEANUT ALLERGIES
Food allergy prevalence has reportedly doubled in recent decades, with a significant increase also seen in allergy severity.6 Allergies involving eggs, nuts, fish, milk, and other foods represent the leading cause of hospital-treated anaphylaxis throughout the world.1 Unlike other allergenic foods that affect only one age-group, peanuts are among the foods that trigger the “vast majority” of allergic reactions in young children, teenagers, and adults alike.3
Increases in reported episodes of peanut allergy reactions may be occurring for several reasons:
• Many people have adopted vegetarian diets, and nuts are considered a good protein source6
• Environmental exposures are increasingly common
• More people are genetically vulnerable, as the role of family history becomes clearer
• Food preparation methods (eg, shared processing equipment, contaminated raw materials, formulation errors) and inaccurate labeling lead to accidental exposures7,8
• Exposure to nuts in utero or during breastfeeding is more common.9 Nowak-Wegrzyn and Sampson6 point to the promotion of peanut butter as an economical, nutritious food source for children and for women during pregnancy and lactation; mothers’ consumption of peanuts more than once a week during pregnancy and lactation have been linked to overexposure for their children.9
Other trends that may contribute to peanut allergy prevalence are the early introduction of solid foods in the infant diet and the use of skin products that contain peanut oil.6
Environment and Genetics
The body of knowledge regarding the specific causes of peanut allergy is increasing constantly. Several known peanut proteins (Ara h1, Ara h2, Ara h3, Ara h6, Ara h7, and Ara h9; Ara h8 is a homologous allergen that may account for peanut/birch cross-reactivity) are thought to be responsible for the initial sensitization to peanuts in vulnerable persons, triggering the associated immunoglobulin E (IgE)–mediated response.10-12 Approximately 75% of known peanut-allergic patients will react to these proteins on their first ingestion after being sensitized.9
Since IgE antibodies do not cross the placenta, it is believed that sensitization to peanut proteins must occur in utero or through breast milk. This form of sensitization predisposes these patients to the initial life-threatening anaphylactic reaction.9
There is strong evidence that genetic factors may play a role in peanut allergies.2 In a study of 58 pairs of twins by Sicherer et al,13 heritability of peanut allergy was estimated at 82%, with 64% of monozygotic pairs, versus 7% of dizygotic pairs, showing concordance for peanut allergy. However, the genetic loci that may be responsible for specific food allergies have not yet been identified.2
It is believed that manifestations of food allergy are very similar to those of asthma and atopic dermatitis. According to Green and colleagues,14 82% of peanut-allergic children who visited a referral clinic also had atopic dermatitis. These conditions appear to be triggered by similar mechanisms, mediated by both environmental and genetic factors.2,14-16 Hong et al2 are optimistic about the advances being made in food allergy genetics. Increased understanding, they feel, may lead to new treatment options for potentially fatal food allergies.2
PATIENT PRESENTATION AND HISTORY
As with any IgE-mediated immune response, the patient must have been exposed to the allergen in question. Most patients present with a history of having ingested raw or boiled peanuts and/or foods produced in a facility that also processes nuts.1,18 Clinical symptoms of peanut allergy may develop within seconds of ingestion. For some patients, consumption of as little as 5 to 50 mg of peanut protein can trigger symptoms.19 (A single peanut from a jar of commercially processed peanuts contains approximately 300 mg of potentially allergenic protein.1)
Typically, the most dramatically affected patients have a medical history of asthma or other IgE-mediated immune reactions.1 In one study, young adults with IgE-mediated peanut allergy were found at especially high risk for severe anaphylaxis.6 Seventy-five percent of patients who have a reaction to peanuts do so following their first ingestion (after the initial exposure).
The mean patient age for a diagnosis of peanut allergy is about 14 months; only 20% of the patients diagnosed with a peanut allergy (most likely those with a baseline peanut-specific serum IgE level 18) will outgrow it by the time they reach school age.18,20 Those who do should be encouraged to consume peanuts on a regular basis; according to Byrne et al,21 8% of patients with allergy resolution experience recurrence, a possible result of infrequent peanut consumption.
PHYSICAL EXAMINATION
Patients with peanut allergies can present with a range of symptoms, possibly involving cutaneous, cardiovascular, gastrointestinal, and/or respiratory systems (see Table 115,22). The more notable symptoms, possibly developing within 15 minutes of exposure, are progressive upper and lower respiratory difficulties, vomiting, diarrhea, hypotension, edema of the face and hands, arrhythmia, throat tightness (in serious cases, approaching anaphylaxis), and possibly loss of consciousness. Such severe reactions often occur in the child who has ingested raw peanuts or tree nuts.22
Milder physical exam findings include erythema, pruritus, conjunctivitis, abdominal pain, nasal congestion, itchy throat, and sneezing. These reactions may have been triggered by foods produced in a facility that also processes nuts, household utensils used to prepare foods that contain nuts, or cross-contamination from another child.9,15,24
DIAGNOSTIC WORK-UP
The diagnosis of a patient with a peanut allergy is made through thorough history taking, careful physical examination, allergy testing with either a skin prick test (SPT) or serum-specific IgE, and oral food challenges. The gold standard for diagnosing food allergy is the double-blind, placebo-controlled oral food challenge,2,25-27 as this test alone can determine the amount of peanut protein needed to trigger a reaction in the given patient.9 However, this is a difficult test to administer and must be performed under strict medical supervision.21
It has been determined that a wheal size of 8.0 mm or greater on the SPT has a 95% to 100% positive predictive value for peanut allergy.1,26,27 Although conflicting results have been reported in some patients between SPT and the oral food challenge, a negative SPT result is considered useful for excluding IgE-mediated allergic responses.22
Researchers examining the peanut-specific serum IgE have demonstrated a 95% to 99% positive predictive value when serum levels exceed 15 kU/L.26,27 This cutoff value in peanut allergy patients is considered suggestive of allergic reactivity, although negative results on an oral food challenge have been reported in more than 25% of children with serum levels exceeding the cutoff.25-27 Testing may have been to whole peanut extract rather than the molecular components (eg, Ara h8).11,12
This past summer, the FDA approved a component test that detects allergen components that include Ara h1, h2, h3, h8, and h9.11,12 Another specific version of the serum IgE test has been in development, one that measures the patient’s IgE reactions to the Ara h2 and Ara h8 components in peanut protein. Johnson and colleagues10,28 have found an increasing level of serum IgE anti–Ara h2 in children who were unable to pass the oral peanut challenge, whereas serum IgE anti–Ara h8 was higher in those who did pass the challenge.28
DIAGNOSING ANAPHYLAXIS
The manifestation of anaphylaxis in patients allergic to peanuts or tree nuts can be life-threatening.29 Symptoms include intense pruritus with flushing of the skin, urticaria, and angioedema, upper-respiratory obstruction resulting from laryngeal edema, and hypotension.30 The clinical criteria for diagnosing anaphylaxis can be found in Table 2.30,31
It is important to recognize the signs and symptoms of anaphylaxis in patients with a peanut allergy; many patients who present to the ED represent first-time reactions. Among patients with life-threatening symptoms on initial reaction, 71% will have similarly severe reactions in subsequent episodes (compared with 44% of patients whose first reaction was not life-threatening).3
TREATMENT, INCLUDING PATIENT EDUCATION
Currently there is no cure for peanut allergy, and no appropriate therapies yet exist to reduce allergy severity. Modest gains have been reported in raising tolerance threshold levels through peanut oral immunotherapy—a long, painstaking process.19,21,32 For now, treatment for peanut allergy is directed at controlling symptoms, once a reaction has occurred. Therefore, the clinician’s goal is to educate peanut-allergic patients and their families on avoiding accidental peanut ingestion, recognizing signs and symptoms of an allergic reaction, and preparing an emergency plan.4
Because four in five patients can expect peanut allergy to last for a lifetime,18,20 strict avoidance of peanuts and peanut products is essential—though difficult because of accidental exposure to food allergens (for example, when dining in restaurants or purchasing bakery products22,32), cross-contamination (as can occur when a food preparation area is not properly cleaned), and allergen cross-reactivity (such as consumption of other legumes).1 Patients must be taught to read food labels carefully for possible hidden sources of peanuts (see Table 37,8); in some cases, product labels bear helpful advisory wording, such as “may contain peanuts.”34,35 US legislation mandates that listed ingredients on food packaging include the eight foods that account for 90% of allergic reactions:
• Peanuts
• Tree nuts
• Egg
• Milk
• Wheat
• Soybeans
• Fish
• Crustacean shellfish.34
Treatment for Anaphylaxis
In pediatric patients, administration of epinephrine is the definitive treatment for anaphylaxis; both the child and parents should carry an epinephrine self-injection device at all times in the event of accidental peanut ingestion. These devices are available in two strengths, based on the child’s weight, and expiration dates should be noted with care. Correct use of the epinephrine self-injection device should be reviewed at each office visit.6
Early-stage allergic reactions can be managed by oral antihistamines, such as diphenhydramine (1 mg/kg body weight up to 75 mg) and an intramuscular injection of epinephrine.1 Prompt transport to the ED should follow (see “Management of Anaphylaxis in the ED”1,9).
PREVENTION
A 2010 expert panel on diagnosis and management of food allergy sponsored by the NIAID, NIH,3 does not advise women to restrict their diet during pregnancy and lactation. Similarly, the United Kingdom’s Department of Health and the Food Standards Agency (DHFSA)36,37 does not support the belief that eating peanuts and peanut-containing foods during pregnancy correlates with a child’s potential for developing a peanut allergy.
The DHFSA does recommend breastfeeding infants for the first six months, if possible, and that mothers refrain from introducing peanut-containing foods during that time. They also recommend that foods associated with a high risk for allergy be introduced into a child’s diet one at a time, to make it easier to identify any allergenic substance.36,37
Lastly, the DHFSA advises parents with a family history of peanut allergy to introduce peanuts only after consulting with their health care provider. The same consideration is advised if a child has already been diagnosed with another allergy.34 According to the American Academy of Pediatrics,6,38 children at high risk for food allergy (eg, atopic disease in both parents or one parent and one sibling) should be breastfed or be given hypoallergenic formula until age 1 year, with no solid foods before age 6 months; peanut-containing foods should not be given before age 3 or 4 years.
CONCLUSION
Peanut allergy can present a lifelong battle for affected patients. Eating one peanut or being exposed even to minute amounts of peanut protein could mean life or death without appropriate management. Reading food labels carefully, preparing peanut-free foods, recognizing the signs and symptoms of anaphylaxis, and obtaining the necessary treatment when allergic reactions occur are essential for peanut-allergic patients and their families.
REFERENCES
1. Burks AW. Peanut allergy. Lancet. 2008;371 (9623):1538-1546.
2. Hong X, Tsai HJ, Wang X. Genetics of food allergy. Curr Opin Pediatr. 2009;21(6):770-776.
3. Boyce JA, Assa’ad A, Burks AW, et al. Guidelines for the diagnosis and management of food allergy in the United States: report of the NIAID-sponsored expert panel. J Allergy Clin Immunol. 2010;126(6 suppl):S1-S58.
4. Sicherer S, Muñoz-Furlong A, Godbold JH, Sampson HA. US prevalence of self-reported peanut, tree nut, and sesame allergy: 11-year follow-up. J Allergy Clin Immunol. 2010;125(6):1322-1326.
5. Hourihane JO, Aiken R, Briggs R, et al. The impact of government advice to pregnant mothers regarding peanut avoidance on the prevalence of peanut allergy in United Kingdom children at school entry. J Allergy Clin Immunol. 2007;312(5):1197-1202.
6. Nowak-Wegrzyn A, Sampson HA. Adverse reactions to foods. Med Clin North Am. 2006;90(1):97-127.
7. Puglisi G, Frieri M. Update on hidden food allergens and food labeling. Allergy Asthma Proc. 2007;28(6):634-639.
8. Hefle SL. Hidden food allergens. Curr Opin Allergy Clin Immunol. 2001;1(3):269-271.
9. Lee CW, Sheffer AL. Peanut allergy. Allergy Asthma Proc. 2003;24(4):259-264.
10. Boughton B. New test for peanut allergy a step forward. www.medscape.com/viewarticle/740133. Accessed November 16, 2011.
11. Asarnoj A, Movérare R, Östblom E, et al. IgE to peanut allergen components: relation to peanut symptoms and pollen sensitization in 8-year-olds. Allergy. 2010;65(9):1189-1195.
12. Codreanu F, Collignon O, Roitel O, et al. A novel immunoassay using recombinant allergens simplifies peanut allergy diagnosis. Int Arch Allergy Immunol. 2011;154(3):216-226.
13. Sicherer SH, Furlong TJ, Maes HH, et al. Genetics of peanut allergy: a twin study. J Allergy Clin Immunol. 2000;106(1 pt 1):53-56.
14. Green TD, LaBelle VS, Steele PH, et al. Clinical characteristics of peanut-allergic children: recent changes. Pediatrics. 2007;120(6):1304-1310.
15. Al-ahmed N, Alsowaidi S, Vadas P. Peanut allergy: an overview. Allergy Asthma Clin Immunol. 2008;4(4):139-143.
16. Björkstén B. Genetic and environmental risk factors for the development of food allergy. Curr Opin Allergy Clin Immunol. 2005;5(3):249-253.
17. Lack G. Epidemiologic risks for food allergy. J Allergy Clin Immunol. 2008;121(6):1331-1336.
18. Skolnick HS, Conover-Walker MK, Koerner CB, et al. The natural history of peanut allergy. J Allergy Clin Immunol. 2001;107(2):367-374.
19. Clark AT, Islam S, King Y, et al. Successful oral tolerance induction in severe peanut allergy. Allergy. 2009;64(8):1218-1220.
20. Busse PJ, Nowak-Wegrzyn AH, Noone SA, et al. Recurrent peanut allergy. N Engl J Med. 2002; 347(19):1535-1536.
21. Byrne AM, Malka-Rais J, Burks AW, Fleischer DM. How do we know when peanut and tree nut allergy have resolved, and how do we keep it resolved? Clin Exp Allergy. 2010;49(9):1303-1311.
22. Sampson HA. Update on food allergy. J Allergy Clin Immunol. 2004;113(5):805-819.
23. Furlong TJ, Desimone J, Sicherer SH. Peanut and tree nut allergic reactions in restaurants and other establishments. J Allergy Clin Immunol. 2001;108(5):866-870.
24. Nelson HS, Lahr J, Rule R, et al. Treatment of anaphylactic sensitivity to peanuts by immunotherapy with injections of aqueous peanut extract. J Allergy Clin Immunol. 1997;99(6 pt 1):744-751.
25. Du Toit G, Santos A, Roberts G, et al. The diagnosis of IgE-mediated food allergy in childhood. Pediatr Allergy Immunol. 2009;20(4):309-319.
26. Roberts G, Lack G. Diagnosing peanut allergy with skin prick and specific IgE testing. J Allergy Clin Immunol. 2005;115(6):1291-1296.
27. Wainstein BK, Yee A, Jelley D, et al. Combining skin prick, immediate skin application and specific-IgE testing in the diagnosis of peanut allergy in children. Pediatr Allergy Immunol. 2007;18(3):231-239.
28. Johnson K, Keet C, Hamilton R, Wood R. Predictive value of peanut component specific IgE in a clinical population. Presented at: 2011 Annual Meeting, American Academy of Allergy, Asthma and Immunology; March 19, 2011; San Francisco, CA. Abstract 267.
29. Sheffer AL. Allergen avoidance to reduce asthma-related morbidity. N Engl J Med. 2004;351(11):1134-1136.
30. Russell S, Monroe K, Losek JD. Anaphylaxis management in the pediatric emergency department: opportunities for improvement. Pediatr Emerg Care. 2010;26(2):71-76.
31. Sampson HA, Munoz-Furlong A, Campbell RL, et al. Second symposium on the definition and management of anaphylaxis: summary report—Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. J Allergy Clin Immunol. 2006;117(2):391-397.
32. Blumchen K, Ulbricht H, Staden U, et al. Oral peanut immunotherapy in children with peanut anaphylaxis. J Allergy Clin Immunol. 2010; 126(1):83-91.
33. Yu JW, Kagan R, Verreault N, et al. Accidental ingestions in children with peanut allergy. J Allergy Clin Immunol. 2006;118(2):466-472.
34. Taylor SL, Hefle SL. Food allergen labeling in the USA and Europe. Curr Opin Allergy Clin Immunol. 2006;6(3):186-190.
35. Sampson HA, Srivastava K, Li XM, Burks AW. New perspectives for the treatment of food allergy (peanut). Arb Paul Ehrlich Inst Bundesamt Sera Impfstoffe Frankf A M. 2003;(94):236-244.
36. McLean S, Sheikh A. Does avoidance of peanuts in early life reduce the risk of peanut allergy? BMJ. 2010 Mar 11;340:c424.
37. Department of Health. Revised government advice on consumption of peanut during pregnancy, breastfeeding, and early life and development of peanut allergy (Aug 2009). www.dh.gov.uk/en/Healthcare/Children/Maternity/Maternalandinfantnutrition/DH_104490. Accessed November 16, 2011.
38. American Academy of Pediatrics. Committee on Nutrition. Hypoallergenic infant formulas. Pediatrics. 2000;106(2):346-349.
Among all persons with food allergies, those who are allergic to peanuts are at greatest risk for anaphylactic symptoms.1 About 30,000 cases of food allergy–related anaphylaxis are seen in the nation’s emergency departments (EDs) each year, and the food most commonly responsible is peanuts.2 What can primary care providers do to reduce the number of peanut allergy–associated anaphylactic reactions and fatalities, both in the ED and in the larger community?
According to a guideline from the National Institute of Allergy and Infectious Diseases (NIAID),3 prevalence of peanut allergy is about 0.6% of the US population, although in an 11-year survey involving more than 13,000 respondents, Sicherer et al4 reported allergy to peanuts, tree nuts, or both in 1.4%, possibly translating to some three million Americans; British researchers have reported peanut allergy in 1.8% of an 1,100-member children’s cohort.5 The risk of exposure to peanuts and the associated risk for severe and possibly fatal anaphylaxis present a lifelong struggle for both patient and family.
ETIOLOGY OF PEANUT ALLERGIES
Food allergy prevalence has reportedly doubled in recent decades, with a significant increase also seen in allergy severity.6 Allergies involving eggs, nuts, fish, milk, and other foods represent the leading cause of hospital-treated anaphylaxis throughout the world.1 Unlike other allergenic foods that affect only one age-group, peanuts are among the foods that trigger the “vast majority” of allergic reactions in young children, teenagers, and adults alike.3
Increases in reported episodes of peanut allergy reactions may be occurring for several reasons:
• Many people have adopted vegetarian diets, and nuts are considered a good protein source6
• Environmental exposures are increasingly common
• More people are genetically vulnerable, as the role of family history becomes clearer
• Food preparation methods (eg, shared processing equipment, contaminated raw materials, formulation errors) and inaccurate labeling lead to accidental exposures7,8
• Exposure to nuts in utero or during breastfeeding is more common.9 Nowak-Wegrzyn and Sampson6 point to the promotion of peanut butter as an economical, nutritious food source for children and for women during pregnancy and lactation; mothers’ consumption of peanuts more than once a week during pregnancy and lactation have been linked to overexposure for their children.9
Other trends that may contribute to peanut allergy prevalence are the early introduction of solid foods in the infant diet and the use of skin products that contain peanut oil.6
Environment and Genetics
The body of knowledge regarding the specific causes of peanut allergy is increasing constantly. Several known peanut proteins (Ara h1, Ara h2, Ara h3, Ara h6, Ara h7, and Ara h9; Ara h8 is a homologous allergen that may account for peanut/birch cross-reactivity) are thought to be responsible for the initial sensitization to peanuts in vulnerable persons, triggering the associated immunoglobulin E (IgE)–mediated response.10-12 Approximately 75% of known peanut-allergic patients will react to these proteins on their first ingestion after being sensitized.9
Since IgE antibodies do not cross the placenta, it is believed that sensitization to peanut proteins must occur in utero or through breast milk. This form of sensitization predisposes these patients to the initial life-threatening anaphylactic reaction.9
There is strong evidence that genetic factors may play a role in peanut allergies.2 In a study of 58 pairs of twins by Sicherer et al,13 heritability of peanut allergy was estimated at 82%, with 64% of monozygotic pairs, versus 7% of dizygotic pairs, showing concordance for peanut allergy. However, the genetic loci that may be responsible for specific food allergies have not yet been identified.2
It is believed that manifestations of food allergy are very similar to those of asthma and atopic dermatitis. According to Green and colleagues,14 82% of peanut-allergic children who visited a referral clinic also had atopic dermatitis. These conditions appear to be triggered by similar mechanisms, mediated by both environmental and genetic factors.2,14-16 Hong et al2 are optimistic about the advances being made in food allergy genetics. Increased understanding, they feel, may lead to new treatment options for potentially fatal food allergies.2
PATIENT PRESENTATION AND HISTORY
As with any IgE-mediated immune response, the patient must have been exposed to the allergen in question. Most patients present with a history of having ingested raw or boiled peanuts and/or foods produced in a facility that also processes nuts.1,18 Clinical symptoms of peanut allergy may develop within seconds of ingestion. For some patients, consumption of as little as 5 to 50 mg of peanut protein can trigger symptoms.19 (A single peanut from a jar of commercially processed peanuts contains approximately 300 mg of potentially allergenic protein.1)
Typically, the most dramatically affected patients have a medical history of asthma or other IgE-mediated immune reactions.1 In one study, young adults with IgE-mediated peanut allergy were found at especially high risk for severe anaphylaxis.6 Seventy-five percent of patients who have a reaction to peanuts do so following their first ingestion (after the initial exposure).
The mean patient age for a diagnosis of peanut allergy is about 14 months; only 20% of the patients diagnosed with a peanut allergy (most likely those with a baseline peanut-specific serum IgE level 18) will outgrow it by the time they reach school age.18,20 Those who do should be encouraged to consume peanuts on a regular basis; according to Byrne et al,21 8% of patients with allergy resolution experience recurrence, a possible result of infrequent peanut consumption.
PHYSICAL EXAMINATION
Patients with peanut allergies can present with a range of symptoms, possibly involving cutaneous, cardiovascular, gastrointestinal, and/or respiratory systems (see Table 115,22). The more notable symptoms, possibly developing within 15 minutes of exposure, are progressive upper and lower respiratory difficulties, vomiting, diarrhea, hypotension, edema of the face and hands, arrhythmia, throat tightness (in serious cases, approaching anaphylaxis), and possibly loss of consciousness. Such severe reactions often occur in the child who has ingested raw peanuts or tree nuts.22
Milder physical exam findings include erythema, pruritus, conjunctivitis, abdominal pain, nasal congestion, itchy throat, and sneezing. These reactions may have been triggered by foods produced in a facility that also processes nuts, household utensils used to prepare foods that contain nuts, or cross-contamination from another child.9,15,24
DIAGNOSTIC WORK-UP
The diagnosis of a patient with a peanut allergy is made through thorough history taking, careful physical examination, allergy testing with either a skin prick test (SPT) or serum-specific IgE, and oral food challenges. The gold standard for diagnosing food allergy is the double-blind, placebo-controlled oral food challenge,2,25-27 as this test alone can determine the amount of peanut protein needed to trigger a reaction in the given patient.9 However, this is a difficult test to administer and must be performed under strict medical supervision.21
It has been determined that a wheal size of 8.0 mm or greater on the SPT has a 95% to 100% positive predictive value for peanut allergy.1,26,27 Although conflicting results have been reported in some patients between SPT and the oral food challenge, a negative SPT result is considered useful for excluding IgE-mediated allergic responses.22
Researchers examining the peanut-specific serum IgE have demonstrated a 95% to 99% positive predictive value when serum levels exceed 15 kU/L.26,27 This cutoff value in peanut allergy patients is considered suggestive of allergic reactivity, although negative results on an oral food challenge have been reported in more than 25% of children with serum levels exceeding the cutoff.25-27 Testing may have been to whole peanut extract rather than the molecular components (eg, Ara h8).11,12
This past summer, the FDA approved a component test that detects allergen components that include Ara h1, h2, h3, h8, and h9.11,12 Another specific version of the serum IgE test has been in development, one that measures the patient’s IgE reactions to the Ara h2 and Ara h8 components in peanut protein. Johnson and colleagues10,28 have found an increasing level of serum IgE anti–Ara h2 in children who were unable to pass the oral peanut challenge, whereas serum IgE anti–Ara h8 was higher in those who did pass the challenge.28
DIAGNOSING ANAPHYLAXIS
The manifestation of anaphylaxis in patients allergic to peanuts or tree nuts can be life-threatening.29 Symptoms include intense pruritus with flushing of the skin, urticaria, and angioedema, upper-respiratory obstruction resulting from laryngeal edema, and hypotension.30 The clinical criteria for diagnosing anaphylaxis can be found in Table 2.30,31
It is important to recognize the signs and symptoms of anaphylaxis in patients with a peanut allergy; many patients who present to the ED represent first-time reactions. Among patients with life-threatening symptoms on initial reaction, 71% will have similarly severe reactions in subsequent episodes (compared with 44% of patients whose first reaction was not life-threatening).3
TREATMENT, INCLUDING PATIENT EDUCATION
Currently there is no cure for peanut allergy, and no appropriate therapies yet exist to reduce allergy severity. Modest gains have been reported in raising tolerance threshold levels through peanut oral immunotherapy—a long, painstaking process.19,21,32 For now, treatment for peanut allergy is directed at controlling symptoms, once a reaction has occurred. Therefore, the clinician’s goal is to educate peanut-allergic patients and their families on avoiding accidental peanut ingestion, recognizing signs and symptoms of an allergic reaction, and preparing an emergency plan.4
Because four in five patients can expect peanut allergy to last for a lifetime,18,20 strict avoidance of peanuts and peanut products is essential—though difficult because of accidental exposure to food allergens (for example, when dining in restaurants or purchasing bakery products22,32), cross-contamination (as can occur when a food preparation area is not properly cleaned), and allergen cross-reactivity (such as consumption of other legumes).1 Patients must be taught to read food labels carefully for possible hidden sources of peanuts (see Table 37,8); in some cases, product labels bear helpful advisory wording, such as “may contain peanuts.”34,35 US legislation mandates that listed ingredients on food packaging include the eight foods that account for 90% of allergic reactions:
• Peanuts
• Tree nuts
• Egg
• Milk
• Wheat
• Soybeans
• Fish
• Crustacean shellfish.34
Treatment for Anaphylaxis
In pediatric patients, administration of epinephrine is the definitive treatment for anaphylaxis; both the child and parents should carry an epinephrine self-injection device at all times in the event of accidental peanut ingestion. These devices are available in two strengths, based on the child’s weight, and expiration dates should be noted with care. Correct use of the epinephrine self-injection device should be reviewed at each office visit.6
Early-stage allergic reactions can be managed by oral antihistamines, such as diphenhydramine (1 mg/kg body weight up to 75 mg) and an intramuscular injection of epinephrine.1 Prompt transport to the ED should follow (see “Management of Anaphylaxis in the ED”1,9).
PREVENTION
A 2010 expert panel on diagnosis and management of food allergy sponsored by the NIAID, NIH,3 does not advise women to restrict their diet during pregnancy and lactation. Similarly, the United Kingdom’s Department of Health and the Food Standards Agency (DHFSA)36,37 does not support the belief that eating peanuts and peanut-containing foods during pregnancy correlates with a child’s potential for developing a peanut allergy.
The DHFSA does recommend breastfeeding infants for the first six months, if possible, and that mothers refrain from introducing peanut-containing foods during that time. They also recommend that foods associated with a high risk for allergy be introduced into a child’s diet one at a time, to make it easier to identify any allergenic substance.36,37
Lastly, the DHFSA advises parents with a family history of peanut allergy to introduce peanuts only after consulting with their health care provider. The same consideration is advised if a child has already been diagnosed with another allergy.34 According to the American Academy of Pediatrics,6,38 children at high risk for food allergy (eg, atopic disease in both parents or one parent and one sibling) should be breastfed or be given hypoallergenic formula until age 1 year, with no solid foods before age 6 months; peanut-containing foods should not be given before age 3 or 4 years.
CONCLUSION
Peanut allergy can present a lifelong battle for affected patients. Eating one peanut or being exposed even to minute amounts of peanut protein could mean life or death without appropriate management. Reading food labels carefully, preparing peanut-free foods, recognizing the signs and symptoms of anaphylaxis, and obtaining the necessary treatment when allergic reactions occur are essential for peanut-allergic patients and their families.
REFERENCES
1. Burks AW. Peanut allergy. Lancet. 2008;371 (9623):1538-1546.
2. Hong X, Tsai HJ, Wang X. Genetics of food allergy. Curr Opin Pediatr. 2009;21(6):770-776.
3. Boyce JA, Assa’ad A, Burks AW, et al. Guidelines for the diagnosis and management of food allergy in the United States: report of the NIAID-sponsored expert panel. J Allergy Clin Immunol. 2010;126(6 suppl):S1-S58.
4. Sicherer S, Muñoz-Furlong A, Godbold JH, Sampson HA. US prevalence of self-reported peanut, tree nut, and sesame allergy: 11-year follow-up. J Allergy Clin Immunol. 2010;125(6):1322-1326.
5. Hourihane JO, Aiken R, Briggs R, et al. The impact of government advice to pregnant mothers regarding peanut avoidance on the prevalence of peanut allergy in United Kingdom children at school entry. J Allergy Clin Immunol. 2007;312(5):1197-1202.
6. Nowak-Wegrzyn A, Sampson HA. Adverse reactions to foods. Med Clin North Am. 2006;90(1):97-127.
7. Puglisi G, Frieri M. Update on hidden food allergens and food labeling. Allergy Asthma Proc. 2007;28(6):634-639.
8. Hefle SL. Hidden food allergens. Curr Opin Allergy Clin Immunol. 2001;1(3):269-271.
9. Lee CW, Sheffer AL. Peanut allergy. Allergy Asthma Proc. 2003;24(4):259-264.
10. Boughton B. New test for peanut allergy a step forward. www.medscape.com/viewarticle/740133. Accessed November 16, 2011.
11. Asarnoj A, Movérare R, Östblom E, et al. IgE to peanut allergen components: relation to peanut symptoms and pollen sensitization in 8-year-olds. Allergy. 2010;65(9):1189-1195.
12. Codreanu F, Collignon O, Roitel O, et al. A novel immunoassay using recombinant allergens simplifies peanut allergy diagnosis. Int Arch Allergy Immunol. 2011;154(3):216-226.
13. Sicherer SH, Furlong TJ, Maes HH, et al. Genetics of peanut allergy: a twin study. J Allergy Clin Immunol. 2000;106(1 pt 1):53-56.
14. Green TD, LaBelle VS, Steele PH, et al. Clinical characteristics of peanut-allergic children: recent changes. Pediatrics. 2007;120(6):1304-1310.
15. Al-ahmed N, Alsowaidi S, Vadas P. Peanut allergy: an overview. Allergy Asthma Clin Immunol. 2008;4(4):139-143.
16. Björkstén B. Genetic and environmental risk factors for the development of food allergy. Curr Opin Allergy Clin Immunol. 2005;5(3):249-253.
17. Lack G. Epidemiologic risks for food allergy. J Allergy Clin Immunol. 2008;121(6):1331-1336.
18. Skolnick HS, Conover-Walker MK, Koerner CB, et al. The natural history of peanut allergy. J Allergy Clin Immunol. 2001;107(2):367-374.
19. Clark AT, Islam S, King Y, et al. Successful oral tolerance induction in severe peanut allergy. Allergy. 2009;64(8):1218-1220.
20. Busse PJ, Nowak-Wegrzyn AH, Noone SA, et al. Recurrent peanut allergy. N Engl J Med. 2002; 347(19):1535-1536.
21. Byrne AM, Malka-Rais J, Burks AW, Fleischer DM. How do we know when peanut and tree nut allergy have resolved, and how do we keep it resolved? Clin Exp Allergy. 2010;49(9):1303-1311.
22. Sampson HA. Update on food allergy. J Allergy Clin Immunol. 2004;113(5):805-819.
23. Furlong TJ, Desimone J, Sicherer SH. Peanut and tree nut allergic reactions in restaurants and other establishments. J Allergy Clin Immunol. 2001;108(5):866-870.
24. Nelson HS, Lahr J, Rule R, et al. Treatment of anaphylactic sensitivity to peanuts by immunotherapy with injections of aqueous peanut extract. J Allergy Clin Immunol. 1997;99(6 pt 1):744-751.
25. Du Toit G, Santos A, Roberts G, et al. The diagnosis of IgE-mediated food allergy in childhood. Pediatr Allergy Immunol. 2009;20(4):309-319.
26. Roberts G, Lack G. Diagnosing peanut allergy with skin prick and specific IgE testing. J Allergy Clin Immunol. 2005;115(6):1291-1296.
27. Wainstein BK, Yee A, Jelley D, et al. Combining skin prick, immediate skin application and specific-IgE testing in the diagnosis of peanut allergy in children. Pediatr Allergy Immunol. 2007;18(3):231-239.
28. Johnson K, Keet C, Hamilton R, Wood R. Predictive value of peanut component specific IgE in a clinical population. Presented at: 2011 Annual Meeting, American Academy of Allergy, Asthma and Immunology; March 19, 2011; San Francisco, CA. Abstract 267.
29. Sheffer AL. Allergen avoidance to reduce asthma-related morbidity. N Engl J Med. 2004;351(11):1134-1136.
30. Russell S, Monroe K, Losek JD. Anaphylaxis management in the pediatric emergency department: opportunities for improvement. Pediatr Emerg Care. 2010;26(2):71-76.
31. Sampson HA, Munoz-Furlong A, Campbell RL, et al. Second symposium on the definition and management of anaphylaxis: summary report—Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. J Allergy Clin Immunol. 2006;117(2):391-397.
32. Blumchen K, Ulbricht H, Staden U, et al. Oral peanut immunotherapy in children with peanut anaphylaxis. J Allergy Clin Immunol. 2010; 126(1):83-91.
33. Yu JW, Kagan R, Verreault N, et al. Accidental ingestions in children with peanut allergy. J Allergy Clin Immunol. 2006;118(2):466-472.
34. Taylor SL, Hefle SL. Food allergen labeling in the USA and Europe. Curr Opin Allergy Clin Immunol. 2006;6(3):186-190.
35. Sampson HA, Srivastava K, Li XM, Burks AW. New perspectives for the treatment of food allergy (peanut). Arb Paul Ehrlich Inst Bundesamt Sera Impfstoffe Frankf A M. 2003;(94):236-244.
36. McLean S, Sheikh A. Does avoidance of peanuts in early life reduce the risk of peanut allergy? BMJ. 2010 Mar 11;340:c424.
37. Department of Health. Revised government advice on consumption of peanut during pregnancy, breastfeeding, and early life and development of peanut allergy (Aug 2009). www.dh.gov.uk/en/Healthcare/Children/Maternity/Maternalandinfantnutrition/DH_104490. Accessed November 16, 2011.
38. American Academy of Pediatrics. Committee on Nutrition. Hypoallergenic infant formulas. Pediatrics. 2000;106(2):346-349.
An erythematous plaque on the nose
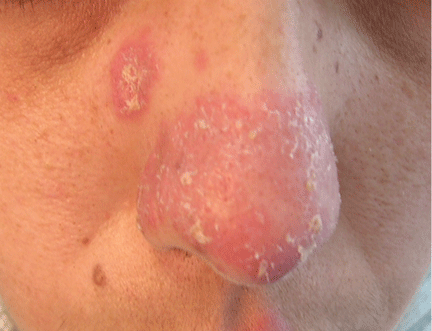
A 38-year-old woman presented with a pruriginous and erythematous lesion on her nose that appeared during periods of cold weather. She said she is completely asymptomatic during the summer months.
Q: What is the most likely diagnosis?
- Lupus pernio
- Rosacea
- Seborrheic dermatitis
- Chilblain lupus erythematosus
- Lupus vulgaris
A: The diagnosis is chilblain lupus erythematosus.
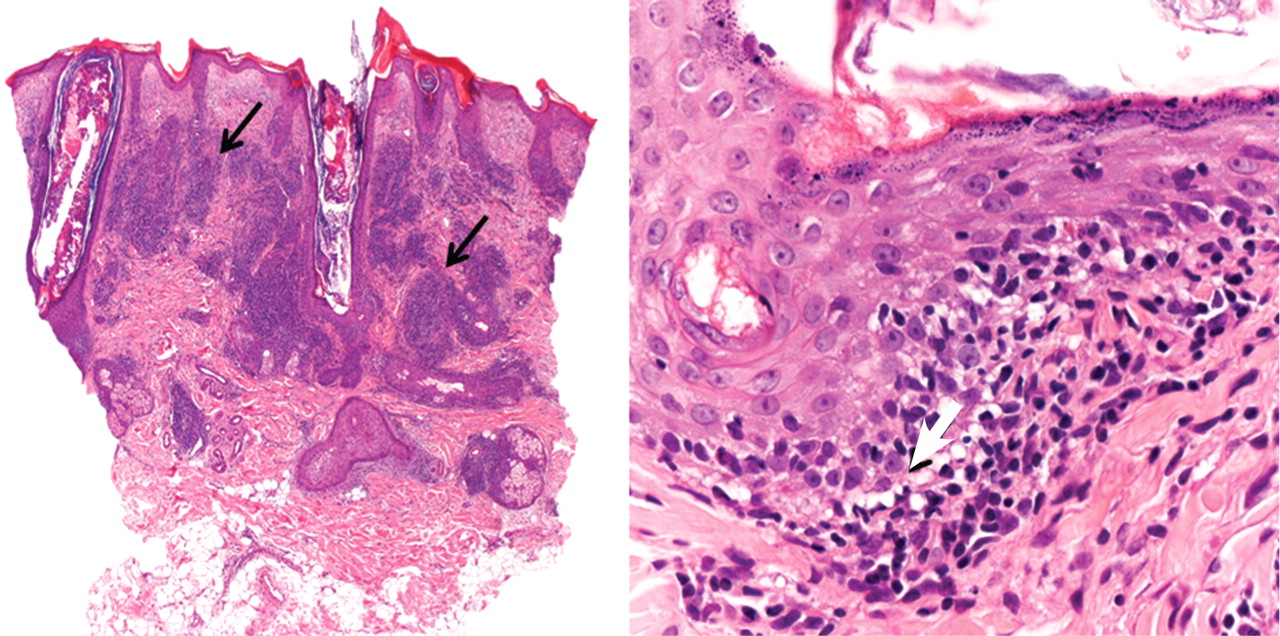
The differential diagnosis of an erythematous lesion on the nose of a middle-aged woman also includes rosacea, lupus pernio, lupus vulgaris, and seborrheic dermatitis. Some of these lesions are exacerbated by cold. Usually, the diagnosis is based on clinical findings, but in some cases histologic features on biopsy study confirm the diagnosis.
Lesions of lupus pernio (sarcoidosis) remain unaltered with changes in temperature, and biopsy study usually shows granulomas without caseous necrosis with little inflammatory infiltrate at the periphery.
Rosacea usually gets worse with heat and with alcohol consumption, although it can be exacerbated by cold. Biopsy study shows a nonspecific perivascular and perifollicular lymphohistiocytic infiltrate accompanied occasionally by multinucleated cells.
Seborrheic dermatitis is a papulosquamous disorder characterized by greasy scaling over inflamed skin on the scalp, face, and trunk. Disease activity is increased in winter and spring, with remissions commonly occurring in summer. The histologic features of seborrheic dermatitis are nonspecific; in this case, the histologic features were compatible with chilblain lupus without changes of seborrheic dermatitis.
Lupus vulgaris is a chronic form of cutaneous tuberculosis characterized by redbrown papules with central atrophy. The nose and ears are usually affected. Histologically, granulomatous tubercles with epithelioid cells and caseation necrosis are usually found.
CHILBLAIN LUPUS ERYTHEMATOSUS
Pernio, or chilblain, is a localized inflammatory lesion of the skin resulting from an abnormal response to cold.1 The cutaneous lesions of chilblain may be classified as idiopathic, autoimmune-related (as in systemic lupus erythematosus, subacute cutaneous lupus), and induced by drugs such as terbinafine (Lamisil)2 or infliximab (Remicade).,3
Chilblain lupus is a rare form of cutaneous lupus erythematosus and should not be confused with lupus pernio, which is a misleading name used for a type of cutaneous sarcoidosis.4
Chilblain lupus is characterized by reddish-purple plaques in acral areas (more often the hands and feet, but also the nose and ears) that are induced by exposure to cold—unlike other lesions of lupus erythematosus, which worsen with exposure to sunlight. The main difference from the cutaneous variety of sarcoidosis (lupus pernio) is the histopathologic appearance. In patients with chilblain lupus, epidermal atrophy, perivascular and periadnexal inflammatory infiltrates, and degeneration of the basal layer are found, whereas in lupus pernio (sarcoidosis), we observe granulomas without caseous necrosis, but with few inflammatory infiltrates on the periphery.
PROPOSED DIAGNOSTIC CRITERIA
Su et al5 have proposed diagnostic criteria for chilblain lupus. Their two major criteria are skin lesions in acral locations induced by exposure to cold or a drop in temperature, and evidence of lupus erythematosus in the skin lesions by histopathologic examination or immunofluorescence study. Both of these criteria must be met, plus one of three minor criteria: the coexistence of systemic lupus erythematosus or of skin lesions of discoid lupus erythematosus; response to lupus therapy; and negative results of testing for cryoglobulin and cold agglutinins.
CHILBLAIN LUPUS VS SYSTEMIC LUPUS
Chilblain lupus is an uncommon manifestation of systemic lupus erythematosus, and it is reported to occur in about 20% of patients with that condition.6 Often, the onset of chilblain lupus precedes the systemic disease. Patients with systemic lupus erythematosus and chilblain lupus do not usually present with renal disease, mucosal lesions, or central nervous system involvement. However, Raynaud phenomenon and photosensitivity have been reported to be more frequently associated with chilblain lupus.7
A disorder of peripheral circulation could be involved in the pathogenesis of chilblain lupus, and the association with Raynaud phenomenon, livedo reticularis, antiphospholipid syndrome, and changes in nailfold capillaries supports this hypothesis. Antinuclear antibody and anti-Ro/SS-A antibody are commonly detected in the serum of patients with chilblain lupus, and anti-Ro/SS-A antibody seems to be a major serologic marker of chilblain lupus in patients with systemic lupus erythematosus.7
TREATMENT
Protection from cold by physical measures is very important, as well as the use of topical or oral antibiotics if the lesions are infected. In severe cases unresponsive to topical corticosteroids, a calcium channel blocker is a good therapeutic option; antimalarials, commonly used in the treatment of lupus erythematosus, can also have a positive effect in patients with chilblain lupus.
CASE CONCLUDED
Our patient was advised to protect herself from the cold. Topical corticosteroids and oral hydroxychloroquine (200 mg/day) were prescribed, and they produced a good response. In severe cases, oral corticosteroids, etretinate (Tegison), mycophenolate (CellCept), or thalidomide (Thalomid) may be used.8
- Simon TD, Soep JB, Hollister JR. Pernio in pediatrics. Pediatrics 2005; 116:e472–e475.
- Bonsmann G, Schiller M, Luger TA, Ständer S. Terbinafine-induced subacute cutaneous lupus erythematosus. J Am Acad Dermatol 2001; 44:925–931.
- Richez C, Dumoulin C, Schaeverbeke T. Infliximab induced chilblain lupus in a patient with rheumatoid arthritis. J Rheumatol 2005; 32:760–761.
- Arias-Santiago SA, Girón-Prieto MS, Callejas-Rubio JL, Fernández-Pugnaire MA, Ortego-Centeno N. Lupus pernio or chilblain lupus?: two different entities. Chest 2009; 136:946–947.
- Su WP, Perniciaro C, Rogers RS, White JW. Chilblain lupus erythematosus (lupus pernio): clinical review of the Mayo Clinic experience and proposal of diagnostic criteria. Cutis 1994; 54:395–399.
- Yell JA, Mbuagbaw J, Burge SM. Cutaneous manifestations of systemic lupus erythematosus. Br J Dermatol 1996; 135:355–362.
- Franceschini F, Calzavara-Pinton P, Quinzanini M, et al. Chilblain lupus erythematosus is associated with antibodies to SSA/Ro. Lupus 1999; 8:215–219.
- Bouaziz JD, Barete S, Le Pelletier F, Amoura Z, Piette JC, Francès C. Cutaneous lesions of the digits in systemic lupus erythematosus: 50 cases. Lupus 2007; 16:163–167.
A 38-year-old woman presented with a pruriginous and erythematous lesion on her nose that appeared during periods of cold weather. She said she is completely asymptomatic during the summer months.
Q: What is the most likely diagnosis?
- Lupus pernio
- Rosacea
- Seborrheic dermatitis
- Chilblain lupus erythematosus
- Lupus vulgaris
A: The diagnosis is chilblain lupus erythematosus.
The differential diagnosis of an erythematous lesion on the nose of a middle-aged woman also includes rosacea, lupus pernio, lupus vulgaris, and seborrheic dermatitis. Some of these lesions are exacerbated by cold. Usually, the diagnosis is based on clinical findings, but in some cases histologic features on biopsy study confirm the diagnosis.
Lesions of lupus pernio (sarcoidosis) remain unaltered with changes in temperature, and biopsy study usually shows granulomas without caseous necrosis with little inflammatory infiltrate at the periphery.
Rosacea usually gets worse with heat and with alcohol consumption, although it can be exacerbated by cold. Biopsy study shows a nonspecific perivascular and perifollicular lymphohistiocytic infiltrate accompanied occasionally by multinucleated cells.
Seborrheic dermatitis is a papulosquamous disorder characterized by greasy scaling over inflamed skin on the scalp, face, and trunk. Disease activity is increased in winter and spring, with remissions commonly occurring in summer. The histologic features of seborrheic dermatitis are nonspecific; in this case, the histologic features were compatible with chilblain lupus without changes of seborrheic dermatitis.
Lupus vulgaris is a chronic form of cutaneous tuberculosis characterized by redbrown papules with central atrophy. The nose and ears are usually affected. Histologically, granulomatous tubercles with epithelioid cells and caseation necrosis are usually found.
CHILBLAIN LUPUS ERYTHEMATOSUS
Pernio, or chilblain, is a localized inflammatory lesion of the skin resulting from an abnormal response to cold.1 The cutaneous lesions of chilblain may be classified as idiopathic, autoimmune-related (as in systemic lupus erythematosus, subacute cutaneous lupus), and induced by drugs such as terbinafine (Lamisil)2 or infliximab (Remicade).,3
Chilblain lupus is a rare form of cutaneous lupus erythematosus and should not be confused with lupus pernio, which is a misleading name used for a type of cutaneous sarcoidosis.4
Chilblain lupus is characterized by reddish-purple plaques in acral areas (more often the hands and feet, but also the nose and ears) that are induced by exposure to cold—unlike other lesions of lupus erythematosus, which worsen with exposure to sunlight. The main difference from the cutaneous variety of sarcoidosis (lupus pernio) is the histopathologic appearance. In patients with chilblain lupus, epidermal atrophy, perivascular and periadnexal inflammatory infiltrates, and degeneration of the basal layer are found, whereas in lupus pernio (sarcoidosis), we observe granulomas without caseous necrosis, but with few inflammatory infiltrates on the periphery.
PROPOSED DIAGNOSTIC CRITERIA
Su et al5 have proposed diagnostic criteria for chilblain lupus. Their two major criteria are skin lesions in acral locations induced by exposure to cold or a drop in temperature, and evidence of lupus erythematosus in the skin lesions by histopathologic examination or immunofluorescence study. Both of these criteria must be met, plus one of three minor criteria: the coexistence of systemic lupus erythematosus or of skin lesions of discoid lupus erythematosus; response to lupus therapy; and negative results of testing for cryoglobulin and cold agglutinins.
CHILBLAIN LUPUS VS SYSTEMIC LUPUS
Chilblain lupus is an uncommon manifestation of systemic lupus erythematosus, and it is reported to occur in about 20% of patients with that condition.6 Often, the onset of chilblain lupus precedes the systemic disease. Patients with systemic lupus erythematosus and chilblain lupus do not usually present with renal disease, mucosal lesions, or central nervous system involvement. However, Raynaud phenomenon and photosensitivity have been reported to be more frequently associated with chilblain lupus.7
A disorder of peripheral circulation could be involved in the pathogenesis of chilblain lupus, and the association with Raynaud phenomenon, livedo reticularis, antiphospholipid syndrome, and changes in nailfold capillaries supports this hypothesis. Antinuclear antibody and anti-Ro/SS-A antibody are commonly detected in the serum of patients with chilblain lupus, and anti-Ro/SS-A antibody seems to be a major serologic marker of chilblain lupus in patients with systemic lupus erythematosus.7
TREATMENT
Protection from cold by physical measures is very important, as well as the use of topical or oral antibiotics if the lesions are infected. In severe cases unresponsive to topical corticosteroids, a calcium channel blocker is a good therapeutic option; antimalarials, commonly used in the treatment of lupus erythematosus, can also have a positive effect in patients with chilblain lupus.
CASE CONCLUDED
Our patient was advised to protect herself from the cold. Topical corticosteroids and oral hydroxychloroquine (200 mg/day) were prescribed, and they produced a good response. In severe cases, oral corticosteroids, etretinate (Tegison), mycophenolate (CellCept), or thalidomide (Thalomid) may be used.8
A 38-year-old woman presented with a pruriginous and erythematous lesion on her nose that appeared during periods of cold weather. She said she is completely asymptomatic during the summer months.
Q: What is the most likely diagnosis?
- Lupus pernio
- Rosacea
- Seborrheic dermatitis
- Chilblain lupus erythematosus
- Lupus vulgaris
A: The diagnosis is chilblain lupus erythematosus.
The differential diagnosis of an erythematous lesion on the nose of a middle-aged woman also includes rosacea, lupus pernio, lupus vulgaris, and seborrheic dermatitis. Some of these lesions are exacerbated by cold. Usually, the diagnosis is based on clinical findings, but in some cases histologic features on biopsy study confirm the diagnosis.
Lesions of lupus pernio (sarcoidosis) remain unaltered with changes in temperature, and biopsy study usually shows granulomas without caseous necrosis with little inflammatory infiltrate at the periphery.
Rosacea usually gets worse with heat and with alcohol consumption, although it can be exacerbated by cold. Biopsy study shows a nonspecific perivascular and perifollicular lymphohistiocytic infiltrate accompanied occasionally by multinucleated cells.
Seborrheic dermatitis is a papulosquamous disorder characterized by greasy scaling over inflamed skin on the scalp, face, and trunk. Disease activity is increased in winter and spring, with remissions commonly occurring in summer. The histologic features of seborrheic dermatitis are nonspecific; in this case, the histologic features were compatible with chilblain lupus without changes of seborrheic dermatitis.
Lupus vulgaris is a chronic form of cutaneous tuberculosis characterized by redbrown papules with central atrophy. The nose and ears are usually affected. Histologically, granulomatous tubercles with epithelioid cells and caseation necrosis are usually found.
CHILBLAIN LUPUS ERYTHEMATOSUS
Pernio, or chilblain, is a localized inflammatory lesion of the skin resulting from an abnormal response to cold.1 The cutaneous lesions of chilblain may be classified as idiopathic, autoimmune-related (as in systemic lupus erythematosus, subacute cutaneous lupus), and induced by drugs such as terbinafine (Lamisil)2 or infliximab (Remicade).,3
Chilblain lupus is a rare form of cutaneous lupus erythematosus and should not be confused with lupus pernio, which is a misleading name used for a type of cutaneous sarcoidosis.4
Chilblain lupus is characterized by reddish-purple plaques in acral areas (more often the hands and feet, but also the nose and ears) that are induced by exposure to cold—unlike other lesions of lupus erythematosus, which worsen with exposure to sunlight. The main difference from the cutaneous variety of sarcoidosis (lupus pernio) is the histopathologic appearance. In patients with chilblain lupus, epidermal atrophy, perivascular and periadnexal inflammatory infiltrates, and degeneration of the basal layer are found, whereas in lupus pernio (sarcoidosis), we observe granulomas without caseous necrosis, but with few inflammatory infiltrates on the periphery.
PROPOSED DIAGNOSTIC CRITERIA
Su et al5 have proposed diagnostic criteria for chilblain lupus. Their two major criteria are skin lesions in acral locations induced by exposure to cold or a drop in temperature, and evidence of lupus erythematosus in the skin lesions by histopathologic examination or immunofluorescence study. Both of these criteria must be met, plus one of three minor criteria: the coexistence of systemic lupus erythematosus or of skin lesions of discoid lupus erythematosus; response to lupus therapy; and negative results of testing for cryoglobulin and cold agglutinins.
CHILBLAIN LUPUS VS SYSTEMIC LUPUS
Chilblain lupus is an uncommon manifestation of systemic lupus erythematosus, and it is reported to occur in about 20% of patients with that condition.6 Often, the onset of chilblain lupus precedes the systemic disease. Patients with systemic lupus erythematosus and chilblain lupus do not usually present with renal disease, mucosal lesions, or central nervous system involvement. However, Raynaud phenomenon and photosensitivity have been reported to be more frequently associated with chilblain lupus.7
A disorder of peripheral circulation could be involved in the pathogenesis of chilblain lupus, and the association with Raynaud phenomenon, livedo reticularis, antiphospholipid syndrome, and changes in nailfold capillaries supports this hypothesis. Antinuclear antibody and anti-Ro/SS-A antibody are commonly detected in the serum of patients with chilblain lupus, and anti-Ro/SS-A antibody seems to be a major serologic marker of chilblain lupus in patients with systemic lupus erythematosus.7
TREATMENT
Protection from cold by physical measures is very important, as well as the use of topical or oral antibiotics if the lesions are infected. In severe cases unresponsive to topical corticosteroids, a calcium channel blocker is a good therapeutic option; antimalarials, commonly used in the treatment of lupus erythematosus, can also have a positive effect in patients with chilblain lupus.
CASE CONCLUDED
Our patient was advised to protect herself from the cold. Topical corticosteroids and oral hydroxychloroquine (200 mg/day) were prescribed, and they produced a good response. In severe cases, oral corticosteroids, etretinate (Tegison), mycophenolate (CellCept), or thalidomide (Thalomid) may be used.8
- Simon TD, Soep JB, Hollister JR. Pernio in pediatrics. Pediatrics 2005; 116:e472–e475.
- Bonsmann G, Schiller M, Luger TA, Ständer S. Terbinafine-induced subacute cutaneous lupus erythematosus. J Am Acad Dermatol 2001; 44:925–931.
- Richez C, Dumoulin C, Schaeverbeke T. Infliximab induced chilblain lupus in a patient with rheumatoid arthritis. J Rheumatol 2005; 32:760–761.
- Arias-Santiago SA, Girón-Prieto MS, Callejas-Rubio JL, Fernández-Pugnaire MA, Ortego-Centeno N. Lupus pernio or chilblain lupus?: two different entities. Chest 2009; 136:946–947.
- Su WP, Perniciaro C, Rogers RS, White JW. Chilblain lupus erythematosus (lupus pernio): clinical review of the Mayo Clinic experience and proposal of diagnostic criteria. Cutis 1994; 54:395–399.
- Yell JA, Mbuagbaw J, Burge SM. Cutaneous manifestations of systemic lupus erythematosus. Br J Dermatol 1996; 135:355–362.
- Franceschini F, Calzavara-Pinton P, Quinzanini M, et al. Chilblain lupus erythematosus is associated with antibodies to SSA/Ro. Lupus 1999; 8:215–219.
- Bouaziz JD, Barete S, Le Pelletier F, Amoura Z, Piette JC, Francès C. Cutaneous lesions of the digits in systemic lupus erythematosus: 50 cases. Lupus 2007; 16:163–167.
- Simon TD, Soep JB, Hollister JR. Pernio in pediatrics. Pediatrics 2005; 116:e472–e475.
- Bonsmann G, Schiller M, Luger TA, Ständer S. Terbinafine-induced subacute cutaneous lupus erythematosus. J Am Acad Dermatol 2001; 44:925–931.
- Richez C, Dumoulin C, Schaeverbeke T. Infliximab induced chilblain lupus in a patient with rheumatoid arthritis. J Rheumatol 2005; 32:760–761.
- Arias-Santiago SA, Girón-Prieto MS, Callejas-Rubio JL, Fernández-Pugnaire MA, Ortego-Centeno N. Lupus pernio or chilblain lupus?: two different entities. Chest 2009; 136:946–947.
- Su WP, Perniciaro C, Rogers RS, White JW. Chilblain lupus erythematosus (lupus pernio): clinical review of the Mayo Clinic experience and proposal of diagnostic criteria. Cutis 1994; 54:395–399.
- Yell JA, Mbuagbaw J, Burge SM. Cutaneous manifestations of systemic lupus erythematosus. Br J Dermatol 1996; 135:355–362.
- Franceschini F, Calzavara-Pinton P, Quinzanini M, et al. Chilblain lupus erythematosus is associated with antibodies to SSA/Ro. Lupus 1999; 8:215–219.
- Bouaziz JD, Barete S, Le Pelletier F, Amoura Z, Piette JC, Francès C. Cutaneous lesions of the digits in systemic lupus erythematosus: 50 cases. Lupus 2007; 16:163–167.
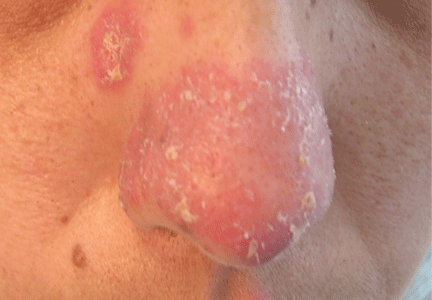
The bittersweet of steroid therapy

For many of those long-term effects, such as osteoporosis, cushingoid features, skin fragility, and cataracts, all we can do is hope that they don’t occur, since there is little we can do to screen for or prevent them. We have previously discussed steroid-associated osteoporosis in the Journal,1 and strategies for preventing it have been proposed by specialty societies.2 For other complications such as hypertension, weight gain, and glucose intolerance, we can offer common-sense protective suggestions, monitor for them, and intervene if they occur.
In this issue, Dr. M. Cecilia Lansang and Ms. Leighanne Kramer Hustak3 discuss the management of steroid-induced adrenal suppression and diabetes. They offer practical management suggestions but also point out that the evidence base for our treatment decisions is surprisingly limited.
Nearly all patients chronically receiving high-dose glucocorticoid therapy develop glucose intolerance, but knowing when that is happening is not always easy. In patients destined to develop type 2 diabetes, the laboratory or clinical signs of hyperglycemia appear only when the pancreas can no longer maintain the insulin production necessary to overcome peripheral insulin resistance. Steroid-induced diabetes is characterized by increased gluconeogenesis, insulin resistance, and excessive postprandial surges, so fasting glucose levels are not sensitive for this clinical syndrome.
The degree and duration of the chronic hyperinsulinemia and hyperglycemia dictates the risk of microvascular complications and thus will be linked to duration of steroid therapy (unless the steroid is unmasking preexisting mild diabetes). Although issues surrounding tight control of blood glucose levels in the acute setting remain unresolved, I believe that even short-term significant steroid-induced hyperglycemia should be prevented when reasonably possible, at the least keeping in mind the additive ill effects of hyperglycemia and steroid therapy on the risk of nuisance infections such as oral and vaginal candidiasis and urinary tract infections that, in the setting of high-dose steroid therapy, can rapidly turn nasty.
- Dore RK. How to prevent glucocorticoid-induced osteoporosis. Cleve Clin J Med 2010; 77:529–536.
- American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. Arthritis Rheum 2001; 44:1496–1503.
- Lansang MC, Hustak LK. Glucocorticoid-induced diabetes and adrenal suppression: how to detect and manage them. Cleve Clin J Med 2011; 78:748–756.
For many of those long-term effects, such as osteoporosis, cushingoid features, skin fragility, and cataracts, all we can do is hope that they don’t occur, since there is little we can do to screen for or prevent them. We have previously discussed steroid-associated osteoporosis in the Journal,1 and strategies for preventing it have been proposed by specialty societies.2 For other complications such as hypertension, weight gain, and glucose intolerance, we can offer common-sense protective suggestions, monitor for them, and intervene if they occur.
In this issue, Dr. M. Cecilia Lansang and Ms. Leighanne Kramer Hustak3 discuss the management of steroid-induced adrenal suppression and diabetes. They offer practical management suggestions but also point out that the evidence base for our treatment decisions is surprisingly limited.
Nearly all patients chronically receiving high-dose glucocorticoid therapy develop glucose intolerance, but knowing when that is happening is not always easy. In patients destined to develop type 2 diabetes, the laboratory or clinical signs of hyperglycemia appear only when the pancreas can no longer maintain the insulin production necessary to overcome peripheral insulin resistance. Steroid-induced diabetes is characterized by increased gluconeogenesis, insulin resistance, and excessive postprandial surges, so fasting glucose levels are not sensitive for this clinical syndrome.
The degree and duration of the chronic hyperinsulinemia and hyperglycemia dictates the risk of microvascular complications and thus will be linked to duration of steroid therapy (unless the steroid is unmasking preexisting mild diabetes). Although issues surrounding tight control of blood glucose levels in the acute setting remain unresolved, I believe that even short-term significant steroid-induced hyperglycemia should be prevented when reasonably possible, at the least keeping in mind the additive ill effects of hyperglycemia and steroid therapy on the risk of nuisance infections such as oral and vaginal candidiasis and urinary tract infections that, in the setting of high-dose steroid therapy, can rapidly turn nasty.
For many of those long-term effects, such as osteoporosis, cushingoid features, skin fragility, and cataracts, all we can do is hope that they don’t occur, since there is little we can do to screen for or prevent them. We have previously discussed steroid-associated osteoporosis in the Journal,1 and strategies for preventing it have been proposed by specialty societies.2 For other complications such as hypertension, weight gain, and glucose intolerance, we can offer common-sense protective suggestions, monitor for them, and intervene if they occur.
In this issue, Dr. M. Cecilia Lansang and Ms. Leighanne Kramer Hustak3 discuss the management of steroid-induced adrenal suppression and diabetes. They offer practical management suggestions but also point out that the evidence base for our treatment decisions is surprisingly limited.
Nearly all patients chronically receiving high-dose glucocorticoid therapy develop glucose intolerance, but knowing when that is happening is not always easy. In patients destined to develop type 2 diabetes, the laboratory or clinical signs of hyperglycemia appear only when the pancreas can no longer maintain the insulin production necessary to overcome peripheral insulin resistance. Steroid-induced diabetes is characterized by increased gluconeogenesis, insulin resistance, and excessive postprandial surges, so fasting glucose levels are not sensitive for this clinical syndrome.
The degree and duration of the chronic hyperinsulinemia and hyperglycemia dictates the risk of microvascular complications and thus will be linked to duration of steroid therapy (unless the steroid is unmasking preexisting mild diabetes). Although issues surrounding tight control of blood glucose levels in the acute setting remain unresolved, I believe that even short-term significant steroid-induced hyperglycemia should be prevented when reasonably possible, at the least keeping in mind the additive ill effects of hyperglycemia and steroid therapy on the risk of nuisance infections such as oral and vaginal candidiasis and urinary tract infections that, in the setting of high-dose steroid therapy, can rapidly turn nasty.
- Dore RK. How to prevent glucocorticoid-induced osteoporosis. Cleve Clin J Med 2010; 77:529–536.
- American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. Arthritis Rheum 2001; 44:1496–1503.
- Lansang MC, Hustak LK. Glucocorticoid-induced diabetes and adrenal suppression: how to detect and manage them. Cleve Clin J Med 2011; 78:748–756.
- Dore RK. How to prevent glucocorticoid-induced osteoporosis. Cleve Clin J Med 2010; 77:529–536.
- American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. Arthritis Rheum 2001; 44:1496–1503.
- Lansang MC, Hustak LK. Glucocorticoid-induced diabetes and adrenal suppression: how to detect and manage them. Cleve Clin J Med 2011; 78:748–756.
Glucocorticoid-induced diabetes and adrenal suppression: How to detect and manage them
Glucocorticoids are commonly prescribed by primary care physicians and specialists alike for multiple medical problems, acute as well as chronic.
However, these useful drugs have adverse effects on multiple endocrine systems, effects that include diabetes (or worsening of hyperglycemia in those with known diabetes), Cushing syndrome, adrenal suppression, osteoporosis (reviewed in the Cleveland Clinic Journal of Medicine in August 2010),1 and dyslipidemia. In addition, suppression of gonadotropins, growth hormone, and, acutely, thyrotropin can ensue.
The focus of this review is on the diabetogenic and adrenal suppressive effects of glucocorticoids and their management. We describe the rationale for choosing specific drugs to counter hyperglycemia, tests for determining adrenal suppression and systemic glucocorticoid absorption, and how and why to taper these drugs.
WIDELY USED DRUGS
Although glucocorticoids (often simply called steroids or corticosteroids, although not all steroids are corticosteroids, and not all corticosteroids are glucocorticoids) are the core treatment for adrenal insufficiency, in most cases they are prescribed for their anti-inflammatory effects. They act through multiple pathways at the cellular and molecular levels, suppressing the cascades that would otherwise result in inflammation and promoting pathways that produce anti-inflammatory proteins.2
In addition to formulations that are intended to have systemic effects, other, “local” formulations are made for specific conditions, such as intra-articular injections for arthritis, epidural injections for lumbar disk pain, eye drops for uveitis, nasal sprays for allergic rhinitis, inhalers for asthma, and topical ointments and creams for eczema. However, as we will discuss, even these preparations can have systemic effects.
GLUCOCORTICOID-INDUCED DIABETES IS COMMON
Glucocorticoids are the most common cause of drug-induced diabetes. Though the exact prevalence is not known, a few observations suggest that glucocorticoid-induced diabetes or hyperglycemia is common:
- In patients with rheumatoid arthritis, mean age 62 years, nearly 9% developed diabetes in the 2 years after starting glucocorticoid treatment, which was a higher rate than expected.3
- In nondiabetic patients with primary renal disease treated with prednisolone 0.75 mg/kg/day, 42% were found to have 2-hour post-lunch plasma glucose concentrations higher than 200 mg/dL but normal fasting glucose levels.4
- In a case-control study, the odds ratio of starting an oral hypoglycemic agent or insulin was 1.77 for patients receiving a hydrocortisone-equivalent dose of 1 to 39 mg/day, 3.02 for 40 to 79 mg/day, 5.82 for 80 to 119 mg/day, and 10.34 for 120 mg/day or more.5 (For a full discussion of glucocorticoid equivalents, see the section below on Cushing syndrome and adrenal suppression.)
- In patients with type 1 diabetes, prednisone 60 mg/day raised the blood glucose levels starting 6 hours after the prednisone dose.6
- Diabetic ketoacidosis and hyperosmolar nonketotic syndrome have been reported as a result of glucocorticoid treatment.7–9
GLUCOCORTICOIDS CAUSE DIABETES MAINLY VIA INSULIN RESISTANCE
The mechanism by which glucocorticoids cause diabetes predominantly involves insulin resistance rather than decreased insulin production. In fact, in a study in healthy volunteers, 10 hydrocortisone infusion resulted in higher insulin production than saline infusion did. (In high doses, however, glucocorticoids have been shown to decrease insulin secretion.11)
Normally, in response to insulin, the liver decreases its output of glucose. Glucocorticoids decrease the liver’s sensitivity to insulin, thereby increasing hepatic glucose output.12 They also inhibit glucose uptake in muscle and fat, reducing insulin sensitivity as much as 60% in healthy volunteers. This seems primarily due to a postreceptor effect, ie, inhibition of glucose transport.13–15
THE PEAK EFFECT OCCURS 4 TO 6 HOURS AFTER DOSING
To understand the optimal time for checking plasma glucose and to apply appropriate treatment, we should consider the pharmacokinetic profile of glucocorticoids.
Studied using the whole-blood lymphocyte proliferation technique, prednisone shows a peak effect at about 4 to 6 hours and a duration of action of 13 to 16 hours.16 This closely resembles what we see in terms of glucose excursion with this drug.17 Two studies of intravenous dexamethasone 10 mg showed that glucose levels rose within 4 hours of injection, but did not pursue this beyond that time frame.18,19
PATIENTS WITHOUT A PREVIOUS DIAGNOSIS OF DIABETES
Be alert for new-onset diabetes
For most diseases treated with glucocorticoids, clinicians can estimate in advance how long the patient will need to take the drug. We can arbitrarily classify the projected exposure as either short-term (3 to 4 weeks or less, such as a 6-day course of methylprednisolone for allergic conditions) or long-term (such as in transplant recipients to prevent rejection or to treat graft-vs-host disease). Hyperglycemia is a potential concern with both short-term and long-term treatment. However, guidelines on checking blood sugar levels, as opposed to relying on symptoms alone, are available only for long-term glucocorticoid treatment.
Patients beginning treatment should be warned of typical diabetes symptoms such as thirst and increased urination and, should these occur, to seek medical attention to have their blood glucose level checked. It is also reasonable to have them return in a week for a random postprandial plasma glucose test in the mid-afternoon.
Why this timing? In most once-daily regimens, glucocorticoids are given in the morning to prevent adrenal suppression (discussed below). In our experience, glucose levels start to rise mid-morning and continue to increase until bedtime. Measuring glucose levels 1 to 2 hours after lunch allows for both the glucocorticoid action and the carbohydrate absorption from lunch to reach their peaks. If hyperglycemia is going to happen, it should be detectable by then. A glucose level of 200 mg/dL or higher should prompt the practitioner to pursue this further.
If glucocorticoid treatment is to continue beyond 3 to 4 weeks, the only population for which there are published guidelines on managing glucocorticoid-related diabetes is transplant recipients. International consensus guidelines, published in 2003, suggest checking the fasting plasma glucose level once a week for the first 4 weeks after transplantation, then at 3 months, at 6 months, and then once a year.20
Though practical, this suggestion does not reflect the fact that glucocorticoids often do not affect fasting plasma glucose, especially if given once daily in the morning at doses of 30 mg or less of prednisone or its equivalent. These guidelines thus may not be applicable to other populations with glucocorticoid-induced diabetes.
The transplant guidelines do mention that an oral glucose tolerance test may be more sensitive, but this is often cumbersome to perform. We believe that checking random postprandial plasma glucose levels is helpful in this regard.

If the patient was at risk of developing diabetes even before receiving a glucocorticoid (for example, if he or she is overweight, has a family history of diabetes, or had a previous hemoglobin A1c of 5.7% or higher), then a fasting plasma glucose level of 126 mg/dL or higher or a hemoglobin A1c of 6.5% or higher might suffice to diagnose diabetes. Results should be confirmed on a separate day in the absence of unequivocal hyperglycemia. Fasting hyperglycemia can also be seen in patients receiving higher once-daily glucocorticoid doses—in our experience, an equivalent of prednisone 40 mg once a day in the morning— or twice-daily dosing.
A hemoglobin A1c checked less than 2 to 3 months after starting glucocorticoid treatment will not be sensitive in picking up glucocorticoid-induced diabetes if the patient did not have underlying diabetes.
Diet and exercise may not be practical
Though diet and exercise are important in managing diabetes, the condition for which the patient is receiving a glucocorticoid may prevent him or her from exercising, at least in the acute phase of the illness.
In addition, though the exact mechanism is not known, glucocorticoids increase hunger, and so decreasing food intake is not easy either. Nonetheless, patients should be familiarized with what carbohydrates are and should be advised to reduce their intake of them.
For suspected type 1 diabetes, start insulin
If type 1 diabetes is suspected, for example, in patients who are lean, younger than 30 years, or who had presented with diabetic ketoacidosis, then insulin should be started. In equivocal cases, insulin therapy can commence while testing is done for C-peptide, glutamic acid decarboxylase antibodies, islet cell antibodies, and insulinoma-associated protein antibodies.

Starting oral antidiabetic drugs
Some patients may have contraindications to specific drugs. For example, metformin (Glucophage) is contraindicated if the serum creatinine level is elevated, an abnormality that renal transplant patients may continue to have.
If the patient has no such contraindications, we have found the following medications suitable in view of their efficacy, low risk of hypoglycemia, or lack of distressing side effects. They will often lower glucose levels enough to achieve capillary blood glucose or fingerstick goals (discussed below). None of them has been specifically approved by the US Food and Drug Administration for glucocorticoid-induced diabetes, but they are approved for type 2 diabetes.
Guidelines from the American Association of Clinical Endocrinologists for type 2 diabetes call for starting monotherapy if the hemoglobin A1c is 6.5% to 7.5%, dual therapy if it is 7.6% to 9%, triple therapy if it is higher than 9% and the patient has no symptoms, and insulin if it is higher than 9% and the patient does have symptoms.22
In terms of estimated average glucose levels, these categories correspond to 140 to 169 mg/dL for monotherapy, 171 to 212 mg/dL for dual therapy, and higher than 212 mg/dL for triple therapy or insulin. Since estimated average levels also include fasting glucose levels (which are lower in glucocorticoid-induced diabetes compared with nonfasting levels), and because we use the American Diabetes Association general hemoglobin A1c goal of less than 7%, we believe that our suggestions below are reasonable.
We divide our recommendations according to initial random (ideally, 1- to 2-hour postprandial) plasma glucose levels.
If the random or 1- to 2-hour post-meal plasma glucose is lower than 220 mg/dL
In this situation the choices are:
- Metformin
- Dipeptidyl peptidase-4 (DPP-4) inhibitors (“gliptins”)
- Meglitinides (“glinides”). The guidelines on new-onset diabetes after transplantation point out that meglitinides may be the safest agents apart from insulin in the renal transplant population, but does acknowledge that efficacies of different oral agents have not been compared in this group.20
- Glucagon-like protein-1 (GLP-1) agonists
- Sulfonylureas. However, the longer-acting forms such as glimepiride (Amaryl) are not suitable if the fasting plasma glucose is not affected.
We have not used thiazolidinediones (“glitazones”) routinely because they can cause weight gain and edema—problems that are already seen with the use of steroids—and have a slower onset of action.
If the random or 1- to 2-hour post-meal plasma glucose is 220 to 300 mg/dL
Often, a combination of drugs or insulin (see below) is needed. However, you can start with one agent and add a second agent within 2 or 3 months (as is recommended for type 2 diabetes).22,23 The following combinations of the agents listed above are supported by published guidelines for type 2 diabetes:
- Metformin plus a sulfonylurea22,23
- Metformin plus a glinide22
- Metformin plus a GLP-1 agonist23
- Metformin plus a DPP-4 inhibitor.22
If the random or 1- to 2-hour post-meal plasma glucose is higher than 300 mg/dL
In our experience, if their plasma glucose levels are this high, patients are experiencing frank symptoms of hyperglycemia.
Insulin addresses those symptoms and avoids the prolonged wait that often results from unsuccessfully starting one agent and then adding another. Of all the available drugs, insulin is the only one that can be used despite multiple underlying illnesses; it does not cause a lot of drug interactions, and the dose can be adjusted upward and downward in increments to fit the patient’s needs, especially when a larger glucocorticoid load is given up front and then is tapered either slowly or rapidly. However, it can cause hypoglycemia and weight gain.
The initial total daily dose of insulin can be based on the patient’s weight. A starting total daily dose of 0.15 to 0.3 U/kg is reasonable— on the lower end if only the postprandial glucose levels are elevated, and on the higher end if both fasting and postprandial glucose levels are affected.
If fasting glucose levels are not elevated, then Neutral Protamine Hagedorn insulin (which is intermediate-acting) or a premixed combination of an intermediate-acting plus a fast- or short-acting insulin can be given once a day before breakfast, or even before lunch if the glucose levels start to rise only after lunch.
If both the fasting and the postprandial glucose levels are elevated, regimens similar to those for type 1 or insulin-requiring type 2 diabetes can be used, except that the ratios of the doses are tilted more toward covering postprandial than fasting hyperglycemia:
- Long-acting insulin plus prandial insulin, in a ratio of 30:70 to 50:50. As glucocorticoids are tapered, the long-acting insulin may have to be discontinued while the prandial doses are continued, since the fasting glucose level decreases first.
- Premixed insulins, with one-half to two-thirds of the dose given before breakfast and the rest before the evening meal, with the possibility of a third injection before lunch. As glucocorticoids are tapered, the evening dose is tapered first.
- Intermediate-acting insulin plus short- or fast-acting insulin in the morning (these two will make up one-half to two-thirds of the total daily dose), short- or fast-acting insulin before the evening meal, and intermediate-acting insulin at bedtime. As glucocorticoids are tapered, the bedtime insulin is tapered first.
Capillary blood glucose (fingerstick) checks
The timing and frequency of fingerstick checks depend on the treatment.
Though postprandial testing is ideal, it is often not practical or convenient. Before lunch, before dinner, and at bedtime are good alternatives since they reflect the pattern of glucose rise throughout the day. For patients on diet and exercise with or without agents other than insulin, testing once or twice a day is reasonable, rotating times before meals (including fasting if this time is affected) and at bedtime.
For patients on insulin, checking two to four times a day initially would help match insulin doses with glucose excursions. For continued care, the American Diabetes Association recommends fingerstick checks three times daily in patients on multiple insulin injections, but it has no specific recommendations for those on once-a-day insulin.21 We have been recommending that our patients on once-daily insulin check at least twice a day.
Goal fingerstick glucose levels that we use are in accordance with the American Diabetes Association guidelines for diabetes in general21:
- Before meals 70 to 130 mg/dL or
- 1 to 2 hours after meals < 180 mg/dL.
During steroid taper, if the glucocorticoid dose is in the lower range (eg, a prednisone-equivalent dose of approximately 7.5 mg per day or less), the fingerstick glucose levels are at the lower end of the target range, and the patient is on a single antidiabetic agent that does not often cause hypoglycemia (eg, metformin), then it is reasonable to ask the patient to not take the antidiabetic medication for 3 to 7 days while continuing to check fingersticks to see if it needs to be resumed. Patients on agents that can cause hypoglycemia need to check more often during the 1 to 3 days after the glucocorticoid dose reduction, as it may take this much time for the glycemic effect to diminish and to adjust the diabetes medication to the appropriate dose.
STARTING GLUCOCORTICOIDS IN PATIENTS WITH KNOWN DIABETES
Fingerstick checks more often
Most patients will already have a glucose meter. They should be instructed to check as discussed above if they do not have a previous diagnosis of diabetes, or to continue as they are doing if they are already checking more often. Patients who have been checking only fasting levels should be instructed to check later in the day, either before or 1 to 2 hours after meals, as discussed above. Patients on oral medications may need additional oral agents or insulin.
Adjust medications if glucose is not at goal
Patients with type 2 diabetes treated with diet and exercise alone can be started on the medications discussed above if their fingerstick readings are not at goal.
If they are already on insulin, we advise them to increase the short- or fast-acting insulins and the morning intermediate-acting insulin by at least 10% to 20% as soon as an elevation in glucose is detected. Long-acting insulin or nighttime intermediate-acting insulin should be increased if fasting glucose levels are affected.
Insulin requirements can double depending on the glucocorticoid dose. In patients with type 1 diabetes who were given prednisone 60 mg orally for 3 days, mean blood glucose levels increased from a baseline of 110 mg/dL at baseline to 149 mg/dL on the days on prednisone.6 The average blood glucose level remained elevated at 141 mg/dL on the day after the last dose of prednisone. The insulin dose increased by 31% to 102% (mean 69%).
CUSHING SYNDROME AND ADRENAL SUPPRESSION
Unlike glucocorticoid-induced diabetes, in which the dilemma is often when to initiate antidiabetic treatment, the question for patients in whom Cushing syndrome or adrenal suppression has developed is when to discontinue glucocorticoids.
Adrenal suppression for the most part goes hand in hand with exogenous Cushing syndrome. If cushingoid features develop, we can infer that the dose of exogenous glucocorticoid exceeds the physiologic needs. This supraphysiologic dosing also leads to suppression of endogenous cortisol production. The suppression occurs at the level of the hypothalamus and pituitary gland, with subsequent atrophy of the part of the adrenal cortex that produces endogenous glucocorticoids.
To understand further the concept of supraphysiologic dosing, the following interconversion of systemic glucocorticoid effects is helpful24,25:

However, there is not much information on interconversion for the local preparations (intra-articular, epidural, inhaled, topical).
Moreover, the definition of supraphysiologic dosing seems to be evolving. Though a total hydrocortisone-equivalent dose of 30 mg/day is still often touted as physiologic replacement, many patients require less. Several studies in the early 1990s, mostly in children and adolescents, showed the mean daily cortisol production rate to be 4.8 to 6.8 mg/m2/day, or closer to 10 to 15 mg/day.26–28 For purposes of this discussion, a physiologic dose will be defined as up to 30 mg hydrocortisone per day or its equivalent.
Adrenal suppression vs insufficiency
Adrenal suppression is often confused with adrenal insufficiency.
Adrenal suppression occurs when cortisol production is decreased because of the presence of exogenous glucocorticoids or other drugs, such as megestrol acetate (Megace), that act on the glucocorticoid receptor. Another situation beyond the scope of this review is excess endogenous cortisol production by an adrenal adenoma or adrenal carcinoma that causes suppression of the contralateral adrenal gland.29
In contrast, adrenal insufficiency is caused by failure of the adrenal gland to produce cortisol as a result of an innate disorder of the adrenal gland (eg, Addison disease) or pituitary gland (eg, pituitary surgery).
Hence, endogenous cortisol production in a patient taking supraphysiologic doses of exogenous glucocorticoids may be suppressed. Recovery of endogenous cortisol production is expected after stopping the exogenous glucocorticoid, though the time to recovery can vary and the patient can be adrenally insufficient if the glucocorticoid is stopped abruptly.
In addition, during times of intercurrent illness, a patient with adrenal suppression may be relatively adrenally insufficient and may need larger doses (“stress doses”) of glucocorticoids, since the adrenal glands may be unable to mount a stress response.29
Local steroids can suppress the adrenal glands
Glucocorticoids are the most common cause of Cushing syndrome. Oral formulations such as dexamethasone, prednisone, and hydrocortisone taken in supraphysiologic doses and for prolonged durations are easily recognized as obvious causes of Cushing syndrome. However, intra-articular, epidural, inhaled, nasal, ocular, and topical steroids—so-called local preparations—have also been linked to Cushing syndrome, and physicians are less likely to recognize them as causes.30–38
In a study in 16 pediatric patients with asthma and 48 controls, inhaled beclomethasone dipropionate (Qvar) 300 to 500 μg daily resulted in adrenal suppression in 100% of patients after 6 to 42 months, as determined by an insulin tolerance test.30
The topical steroid betamethasone (Diprosone) carries a warning that systemic absorption of topical steroids can cause adrenal suppression.39 Intra-articular, intranasal, epidural, and ocular routes are also reported to cause adrenal suppression.32–38
When is adrenal suppression more likely?
Adrenal suppression is more likely in the following situations:
- Longer duration of treatment. Studies have shown that exposure to supraphysiologic steroid doses for 2 weeks or less might already suppress the adrenal glands, but the clinical significance of this is unclear since some recovery already occurs a few days after the glucocorticoids are discontinued.31,40
- Supraphysiologic doses, stronger formulations, longer-acting formulations.41
When is adrenal suppression less likely?
Adrenal suppression is less likely in the following situations:
- Regimens that mimic the diurnal rhythm of cortisol (higher dose in the morning, lower dose in the afternoon)42
- Alternate-day dosing of steroids.43
Steroid withdrawal vs adrenal insufficiency
Another phenomenon that can be confused with adrenal insufficiency or glucocorticoid insufficiency is steroid withdrawal, in which patients experience lethargy, muscle aches, nausea, vomiting, and postural hypotension as glucocorticoids are tapered and their effects wane.42 Increasing the glucocorticoid dose for presumed adrenal insufficiency may delay recovery of the adrenal function and would have to be weighed against the patient’s symptoms.
The following may help distinguish the two: if the patient is on supraphysiologic glucocorticoid doses, then he or she is not glucocorticoid-deficient and is likely suffering from steroid withdrawal. At this point, patients may just need reassurance, symptomatic treatment, or if necessary, a brief (1-week) increase of the previous lowest dose, followed by reevaluation.
With local glucocorticoid preparations that may be systemically absorbed, however, there is no good way of estimating dose equivalence. In these situations, the decision to simply reassure the patient or give symptomatic treatment—as opposed to giving low-dose oral glucocorticoids such as hydrocortisone 5 to 10 mg daily for a week followed by reevaluation— depends on the severity of symptoms and whether the patient has quick access to medical attention should he or she develop an intercurrent illness.
Identifying patients at risk of adrenal suppression
Patients presenting with weight gain or symptoms suggesting Cushing syndrome should be asked about steroid intake and should be prompted to recall possible nonoral routes. In addition, patients presenting with muscle aches and fatigue—symptoms of steroid withdrawal— may have received unrecognized local glucocorticoids that were systemically absorbed, now with diminishing effects.
The ACTH stimulation test for adrenal recovery
Testing can be done to see if the adrenal glands have recovered and glucocorticoid therapy can be discontinued (see Tapering from glucocorticoids, below).
The test most often used is the corticotropin (ACTH) stimulation test. Since the suppression is at the level of the hypothalamus and the pituitary gland, the ACTH stimulation test is an indirect method of assessing hypothalamic and pituitary function in the context of glucocorticoid-induced adrenal suppression. It has good correlation with the insulin tolerance test, the gold-standard test for an intact hypothalamic-pituitary-adrenal axis.
The synthetic ACTH cosyntropin (Cortrosyn) 250 μg is injected intravenously or intramuscularly, and a cortisol level is drawn at baseline and 30 and 60 minutes later. Other doses such as 1 μg or 10 μg have been reported but are not yet widely accepted. A cortisol level of greater than 18 to 20 μg/dL at any time point shows that the adrenals have regained function and the steroids may be discontinued.42 If adrenal suppression persists, weaning from steroids should continue.
In reality, it may not be possible or practical to do an ACTH stimulation test, as not all physicians’ offices have a supply of cosyntropin or the manpower to perform the test correctly. In these cases, weaning can progress with monitoring of symptoms.
Testing for synthetic glucocorticoids in the urine and serum can demonstrate systemic absorption and may be helpful in patients who do not recall receiving steroids.33
Tapering from glucocorticoids
Several tapering schedules have been suggested (although not necessarily validated). Whether and how to taper depend on how long the glucocorticoid has been taken.
If taken for less than 1 week, glucocorticoids can be stopped without tapering, regardless of the dose.
If taken for 1 to 3 weeks, the decision to taper depends on the clinician’s assessment of the patient’s general health or constitution and the illness for which the glucocorticoid was prescribed. For example, if the underlying disease is less likely to flare with a gradual dose reduction, then tapering would be suitable.44
If taken for more than 3 weeks, the practice has been a more rapid taper at the beginning until a physiologic dose is reached. How quickly to reduce the dose depends on whether the underlying illness is expected to flare up, or if the patient might experience steroid withdrawal symptoms.
One schedule is to lower the glucocorticoid dose by an amount equivalent to prednisolone 2.5 mg every 3 to 4 days when above the physiologic dose, then to taper more slowly by 1 mg every 2 to 4 weeks.44 Once the physiologic dose is reached, one can switch to the equivalent dose of hydrocortisone and decrease the dose by 2.5 mg a week until a daily dose of 10 mg a day is reached and maintained for 2 to 3 months, and then perform a test of adrenal function (see above).44 Passing the test implies that the adrenal glands have recovered and the glucocorticoid can be stopped.
Another option is to switch to alternate-day therapy once a physiologic dose is reached and to test 8:00 am cortisol levels, continuing the glucocorticoid and retesting in 4 to 6 weeks if the value is less than 3 μg/dL; stopping the glucocorticoid if the value is higher than 20 μg/dL; and performing an ACTH stimulation test for values in between.45
A review of other tapering regimens for chronic diseases, mostly pulmonary, did not find enough evidence to recommend one particular schedule over another.46 The tapering schedule may have to be adjusted to prevent disease flare and symptoms of steroid withdrawal.
Locally administered steroids. Since the equivalence of systemically absorbed local glucocorticoids is not known, these patients are likely to present when they have symptoms of steroid withdrawal. In this situation, testing adrenal function will help.
- Dore RK. How to prevent glucocorticoid-induced osteoporosis. Cleve Clin J Med 2010; 77:529–536.
- Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med 2005; 353:1711–1723.
- Panthakalam S, Bhatnagar D, Klimiuk P. The prevalence and management of hyperglycaemia in patients with rheumatoid arthritis on corticosteroid therapy. Scott Med J 2004; 49:139–141.
- Uzu T, Harada T, Sakaguchi M, et al. Glucocorticoid-induced diabetes mellitus: prevalence and risk factors in primary renal diseases. Nephron Clin Pract 2007; 105:c54–c57.
- Gurwitz JH, Bohn RL, Glynn RJ, Monane M, Mogun H, Avorn J. Glucocorticoids and the risk for initiation of hypoglycemic therapy. Arch Intern Med 1994; 154:97–101.
- Bevier WC, Zisser HC, Jovanovic L, et al. Use of continuous glucose monitoring to estimate insulin requirements in patients with type 1 diabetes mellitus during a short course of prednisone. J Diabetes Sci Technol 2008; 2:578–583.
- Cagdas DN, Paç FA, Cakal E. Glucocorticoid-induced diabetic ketoacidosis in acute rheumatic fever. J Cardiovasc Pharmacol Ther 2008; 13:298–300.
- Bedalov A, Balasubramanyam A. Glucocorticoid-induced ketoacidosis in gestational diabetes: sequela of the acute treatment of preterm labor. A case report. Diabetes Care 1997; 20:922–924.
- Yang JY, Cui XL, He XJ. Non-ketotic hyperosmolar coma complicating steroid treatment in childhood nephrosis. Pediatr Nephrol 1995; 9:621–622.
- Nielsen MF, Caumo A, Chandramouli V, et al. Impaired basal glucose effectiveness but unaltered fasting glucose release and gluconeogenesis during short-term hypercortisolemia in healthy subjects. Am J Physiol Endocrinol Metab 2004; 286:E102–E110.
- Matsumoto K, Yamasaki H, Akazawa S, et al. High-dose but not low-dose dexamethasone impairs glucose tolerance by inducing compensatory failure of pancreatic beta-cells in normal men. J Clin Endocrinol Metab 1996; 81:2621–2626.
- Rizza RA, Mandarino LJ, Gerich JE. Cortisol-induced insulin resistance in man: impaired suppression of glucose production and stimulation of glucose utilization due to a postreceptor detect of insulin action. J Clin Endocrinol Metab 1982; 54:131–138.
- Meyuhas O, Reshef L, Gunn JM, Hanson RW, Ballard FJ. Regulation of phosphoenolpyruvate carboxykinase (GTP) in adipose tissue in vivo by glucocorticoids and insulin. Biochem J 1976; 158:1–7.
- Tappy L, Randin D, Vollenweider P, et al. Mechanisms of dexamethasone-induced insulin resistance in healthy humans. J Clin Endocrinol Metab 1994; 79:1063–1069.
- Pagano G, Cavallo-Perin P, Cassader M, et al. An in vivo and in vitro study of the mechanism of prednisone-induced insulin resistance in healthy subjects. J Clin Invest 1983; 72:1814–1820.
- Magee MH, Blum RA, Lates CD, Jusko WJ. Pharmacokinetic/pharmaco-dynamic model for prednisolone inhibition of whole blood lymphocyte proliferation. Br J Clin Pharmacol 2002; 53:474–484.
- Burt MG, Roberts GW, Aguilar-Loza NR, Frith P, Stranks SN. Continuous monitoring of circadian glycemic patterns in patients receiving prednisolone for COPD. J Clin Endocrinol Metab 2011; 96:1789–1796.
- Hans P, Vanthuyne A, Dewandre PY, Brichant JF, Bonhomme V. Blood glucose concentration profile after 10 mg dexamethasone in non-diabetic and type 2 diabetic patients undergoing abdominal surgery. Br J Anaesth 2006; 97:164–170.
- Pasternak JJ, McGregor DG, Lanier WL. Effect of single-dose dexamethasone on blood glucose concentration in patients undergoing craniotomy. J Neurosurg Anesthesiol 2004; 16:122–125.
- Davidson J, Wilkinson A, Dantal J, et al; International Expert Panel. New-onset diabetes after transplantation: 2003 international consensus guidelines. Proceedings of an international expert panel meeting. Barcelona, Spain, 19 February 2003. Transplantation 2003; 75(suppl 10):SS3–SS24.
- American Diabetes Association. Standards of medical care in diabetes— 2011. Diabetes Care 2011; 34(suppl 1):S11–S61.
- Rodbard HW, Jellinger PS, Davidson JA, et al. Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on type 2 diabetes mellitus: an algorithm for glycemic control. Endocr Pract 2009; 15:540–559.
- Nathan DM, Buse JB, Davidson MB, et al; American Diabetes Association; European Association for Study of Diabetes. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009; 32:193–203.
- Axelrod L. Corticosteroid therapy. In:Becker KL, editor. Principles and Practice of Endocrinology and Metabolism. 3rd ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2000:752–763.
- Ferri FF, editor. Practical Guide to the Care of the Medical Patient. 8th ed. Philadelphia, PA: Mosby/Elsevier; 2011.
- Kerrigan JR, Veldhuis JD, Leyo SA, Iranmanesh A, Rogol AD. Estimation of daily cortisol production and clearance rates in normal pubertal males by deconvolution analysis. J Clin Endocrinol Metab 1993; 76:1505–1510.
- Linder BL, Esteban NV, Yergey AL, Winterer JC, Loriaux DL, Cassorla F. Cortisol production rate in childhood and adolescence. J Pediatr 1990; 117:892–896.
- Esteban NV, Loughlin T, Yergey AL, et al. Daily cortisol production rate in man determined by stable isotope dilution/mass spectrometry. J Clin Endocrinol Metab 1991; 72:39–45.
- Lansang MC, Quinn SL. Adrenal suppression. BMJ BestPractice 2010. http://bestpractice.bmj.com/best-practice/monograph/863/diagnosis/stepby-step.html. Accessed August 19, 2011.
- Zöllner EW. Hypothalamic-pituitary-adrenal axis suppression in asthmatic children on inhaled corticosteroids (part 2)—the risk as determined by gold standard adrenal function tests: a systematic review. Pediatr Allergy Immunol 2007; 18:469–474.
- Schuetz P, Christ-Crain M, Schild U, et al. Effect of a 14-day course of systemic corticosteroids on the hypothalamic-pituitary-adrenal-axis in patients with acute exacerbation of chronic obstructive pulmonary disease. BMC Pulm Med 2008; 8:1.
- Kay J, Findling JW, Raff H. Epidural triamcinolone suppresses the pituitary-adrenal axis in human subjects. Anesth Analg 1994; 79:501–505.
- Lansang MC, Farmer T, Kennedy L. Diagnosing the unrecognized systemic absorption of intra-articular and epidural steroid injections. Endocr Pract 2009; 15:225–228.
- Duclos M, Guinot M, Colsy M, et al. High risk of adrenal insufficiency after a single articular steroid injection in athletes. Med Sci Sports Exerc 2007; 39:1036–1043.
- Bong JL, Connell JM, Lever R. Intranasal betamethasone induced acne and adrenal suppression. Br J Dermatol 2000; 142:579–580.
- Atabek ME, Pirgon O, Unal E. Pituitary-adrenal axis suppression due to topical steroid administration in an infant. Pediatr Int 2007; 49:242–244.
- Ozerdem U, Levi L, Cheng L, Song MK, Scher C, Freeman WR. Systemic toxicity of topical and periocular corticosteroid therapy in an 11-year-old male with posterior uveitis. Am J Ophthalmol 2000; 130:240–241.
- Chiang MY, Sarkar M, Koppens JM, Milles J, Shah P. Exogenous Cushing’s syndrome and topical ocular steroids. Eye (Lond) 2006; 20:725–727.
- Diprolene prescribing information. Schering Corp 2005. www.theodora.com/drugs/diprolene_gel_005_schering.html. Accessed September 27, 2011.
- Villabona CV, Koh C, Panergo J, Reddy A, Fogelfeld L. Adrenocorticotropic hormone stimulation test during high-dose glucocorticoid therapy. Endocr Pract 2009; 15:122–127.
- Ortega E, Rodriguez C, Strand LJ, Segre E. Effects of cloprednol and other corticosteroids on hypothalamic-pituitary-adrenal axis function. J Int Med Res 1976; 4:326–337.
- Axelrod L. Glucocorticoid therapy. Medicine (Baltimore) 1976; 55:39–65.
- Schürmeyer TH, Tsokos GC, Avgerinos PC, et al. Pituitary-adrenal responsiveness to corticotropin-releasing hormone in patients receiving chronic, alternate day glucocorticoid therapy. J Clin Endocrinol Metab 1985; 61:22–27.
- Stewart PM. The adrenal cortex. In:Kronenberg HM, editor. Williams Textbook of Endocrinology. 11th ed. Philadelphia, PA: Saunders/Elsevier; 2008.
- Hopkins RL, Leinung MC. Exogenous Cushing’s syndrome and glucocorticoid withdrawal. Endocrinol Metab Clin North Am 2005; 34:371–384.
- Richter B, Neises G, Clar C. Glucocorticoid withdrawal schemes in chronic medical disorders. A systematic review. Endocrinol Metab Clin North Am 2002; 31:751–778.
Glucocorticoids are commonly prescribed by primary care physicians and specialists alike for multiple medical problems, acute as well as chronic.
However, these useful drugs have adverse effects on multiple endocrine systems, effects that include diabetes (or worsening of hyperglycemia in those with known diabetes), Cushing syndrome, adrenal suppression, osteoporosis (reviewed in the Cleveland Clinic Journal of Medicine in August 2010),1 and dyslipidemia. In addition, suppression of gonadotropins, growth hormone, and, acutely, thyrotropin can ensue.
The focus of this review is on the diabetogenic and adrenal suppressive effects of glucocorticoids and their management. We describe the rationale for choosing specific drugs to counter hyperglycemia, tests for determining adrenal suppression and systemic glucocorticoid absorption, and how and why to taper these drugs.
WIDELY USED DRUGS
Although glucocorticoids (often simply called steroids or corticosteroids, although not all steroids are corticosteroids, and not all corticosteroids are glucocorticoids) are the core treatment for adrenal insufficiency, in most cases they are prescribed for their anti-inflammatory effects. They act through multiple pathways at the cellular and molecular levels, suppressing the cascades that would otherwise result in inflammation and promoting pathways that produce anti-inflammatory proteins.2
In addition to formulations that are intended to have systemic effects, other, “local” formulations are made for specific conditions, such as intra-articular injections for arthritis, epidural injections for lumbar disk pain, eye drops for uveitis, nasal sprays for allergic rhinitis, inhalers for asthma, and topical ointments and creams for eczema. However, as we will discuss, even these preparations can have systemic effects.
GLUCOCORTICOID-INDUCED DIABETES IS COMMON
Glucocorticoids are the most common cause of drug-induced diabetes. Though the exact prevalence is not known, a few observations suggest that glucocorticoid-induced diabetes or hyperglycemia is common:
- In patients with rheumatoid arthritis, mean age 62 years, nearly 9% developed diabetes in the 2 years after starting glucocorticoid treatment, which was a higher rate than expected.3
- In nondiabetic patients with primary renal disease treated with prednisolone 0.75 mg/kg/day, 42% were found to have 2-hour post-lunch plasma glucose concentrations higher than 200 mg/dL but normal fasting glucose levels.4
- In a case-control study, the odds ratio of starting an oral hypoglycemic agent or insulin was 1.77 for patients receiving a hydrocortisone-equivalent dose of 1 to 39 mg/day, 3.02 for 40 to 79 mg/day, 5.82 for 80 to 119 mg/day, and 10.34 for 120 mg/day or more.5 (For a full discussion of glucocorticoid equivalents, see the section below on Cushing syndrome and adrenal suppression.)
- In patients with type 1 diabetes, prednisone 60 mg/day raised the blood glucose levels starting 6 hours after the prednisone dose.6
- Diabetic ketoacidosis and hyperosmolar nonketotic syndrome have been reported as a result of glucocorticoid treatment.7–9
GLUCOCORTICOIDS CAUSE DIABETES MAINLY VIA INSULIN RESISTANCE
The mechanism by which glucocorticoids cause diabetes predominantly involves insulin resistance rather than decreased insulin production. In fact, in a study in healthy volunteers, 10 hydrocortisone infusion resulted in higher insulin production than saline infusion did. (In high doses, however, glucocorticoids have been shown to decrease insulin secretion.11)
Normally, in response to insulin, the liver decreases its output of glucose. Glucocorticoids decrease the liver’s sensitivity to insulin, thereby increasing hepatic glucose output.12 They also inhibit glucose uptake in muscle and fat, reducing insulin sensitivity as much as 60% in healthy volunteers. This seems primarily due to a postreceptor effect, ie, inhibition of glucose transport.13–15
THE PEAK EFFECT OCCURS 4 TO 6 HOURS AFTER DOSING
To understand the optimal time for checking plasma glucose and to apply appropriate treatment, we should consider the pharmacokinetic profile of glucocorticoids.
Studied using the whole-blood lymphocyte proliferation technique, prednisone shows a peak effect at about 4 to 6 hours and a duration of action of 13 to 16 hours.16 This closely resembles what we see in terms of glucose excursion with this drug.17 Two studies of intravenous dexamethasone 10 mg showed that glucose levels rose within 4 hours of injection, but did not pursue this beyond that time frame.18,19
PATIENTS WITHOUT A PREVIOUS DIAGNOSIS OF DIABETES
Be alert for new-onset diabetes
For most diseases treated with glucocorticoids, clinicians can estimate in advance how long the patient will need to take the drug. We can arbitrarily classify the projected exposure as either short-term (3 to 4 weeks or less, such as a 6-day course of methylprednisolone for allergic conditions) or long-term (such as in transplant recipients to prevent rejection or to treat graft-vs-host disease). Hyperglycemia is a potential concern with both short-term and long-term treatment. However, guidelines on checking blood sugar levels, as opposed to relying on symptoms alone, are available only for long-term glucocorticoid treatment.
Patients beginning treatment should be warned of typical diabetes symptoms such as thirst and increased urination and, should these occur, to seek medical attention to have their blood glucose level checked. It is also reasonable to have them return in a week for a random postprandial plasma glucose test in the mid-afternoon.
Why this timing? In most once-daily regimens, glucocorticoids are given in the morning to prevent adrenal suppression (discussed below). In our experience, glucose levels start to rise mid-morning and continue to increase until bedtime. Measuring glucose levels 1 to 2 hours after lunch allows for both the glucocorticoid action and the carbohydrate absorption from lunch to reach their peaks. If hyperglycemia is going to happen, it should be detectable by then. A glucose level of 200 mg/dL or higher should prompt the practitioner to pursue this further.
If glucocorticoid treatment is to continue beyond 3 to 4 weeks, the only population for which there are published guidelines on managing glucocorticoid-related diabetes is transplant recipients. International consensus guidelines, published in 2003, suggest checking the fasting plasma glucose level once a week for the first 4 weeks after transplantation, then at 3 months, at 6 months, and then once a year.20
Though practical, this suggestion does not reflect the fact that glucocorticoids often do not affect fasting plasma glucose, especially if given once daily in the morning at doses of 30 mg or less of prednisone or its equivalent. These guidelines thus may not be applicable to other populations with glucocorticoid-induced diabetes.
The transplant guidelines do mention that an oral glucose tolerance test may be more sensitive, but this is often cumbersome to perform. We believe that checking random postprandial plasma glucose levels is helpful in this regard.
If the patient was at risk of developing diabetes even before receiving a glucocorticoid (for example, if he or she is overweight, has a family history of diabetes, or had a previous hemoglobin A1c of 5.7% or higher), then a fasting plasma glucose level of 126 mg/dL or higher or a hemoglobin A1c of 6.5% or higher might suffice to diagnose diabetes. Results should be confirmed on a separate day in the absence of unequivocal hyperglycemia. Fasting hyperglycemia can also be seen in patients receiving higher once-daily glucocorticoid doses—in our experience, an equivalent of prednisone 40 mg once a day in the morning— or twice-daily dosing.
A hemoglobin A1c checked less than 2 to 3 months after starting glucocorticoid treatment will not be sensitive in picking up glucocorticoid-induced diabetes if the patient did not have underlying diabetes.
Diet and exercise may not be practical
Though diet and exercise are important in managing diabetes, the condition for which the patient is receiving a glucocorticoid may prevent him or her from exercising, at least in the acute phase of the illness.
In addition, though the exact mechanism is not known, glucocorticoids increase hunger, and so decreasing food intake is not easy either. Nonetheless, patients should be familiarized with what carbohydrates are and should be advised to reduce their intake of them.
For suspected type 1 diabetes, start insulin
If type 1 diabetes is suspected, for example, in patients who are lean, younger than 30 years, or who had presented with diabetic ketoacidosis, then insulin should be started. In equivocal cases, insulin therapy can commence while testing is done for C-peptide, glutamic acid decarboxylase antibodies, islet cell antibodies, and insulinoma-associated protein antibodies.
Starting oral antidiabetic drugs
Some patients may have contraindications to specific drugs. For example, metformin (Glucophage) is contraindicated if the serum creatinine level is elevated, an abnormality that renal transplant patients may continue to have.
If the patient has no such contraindications, we have found the following medications suitable in view of their efficacy, low risk of hypoglycemia, or lack of distressing side effects. They will often lower glucose levels enough to achieve capillary blood glucose or fingerstick goals (discussed below). None of them has been specifically approved by the US Food and Drug Administration for glucocorticoid-induced diabetes, but they are approved for type 2 diabetes.
Guidelines from the American Association of Clinical Endocrinologists for type 2 diabetes call for starting monotherapy if the hemoglobin A1c is 6.5% to 7.5%, dual therapy if it is 7.6% to 9%, triple therapy if it is higher than 9% and the patient has no symptoms, and insulin if it is higher than 9% and the patient does have symptoms.22
In terms of estimated average glucose levels, these categories correspond to 140 to 169 mg/dL for monotherapy, 171 to 212 mg/dL for dual therapy, and higher than 212 mg/dL for triple therapy or insulin. Since estimated average levels also include fasting glucose levels (which are lower in glucocorticoid-induced diabetes compared with nonfasting levels), and because we use the American Diabetes Association general hemoglobin A1c goal of less than 7%, we believe that our suggestions below are reasonable.
We divide our recommendations according to initial random (ideally, 1- to 2-hour postprandial) plasma glucose levels.
If the random or 1- to 2-hour post-meal plasma glucose is lower than 220 mg/dL
In this situation the choices are:
- Metformin
- Dipeptidyl peptidase-4 (DPP-4) inhibitors (“gliptins”)
- Meglitinides (“glinides”). The guidelines on new-onset diabetes after transplantation point out that meglitinides may be the safest agents apart from insulin in the renal transplant population, but does acknowledge that efficacies of different oral agents have not been compared in this group.20
- Glucagon-like protein-1 (GLP-1) agonists
- Sulfonylureas. However, the longer-acting forms such as glimepiride (Amaryl) are not suitable if the fasting plasma glucose is not affected.
We have not used thiazolidinediones (“glitazones”) routinely because they can cause weight gain and edema—problems that are already seen with the use of steroids—and have a slower onset of action.
If the random or 1- to 2-hour post-meal plasma glucose is 220 to 300 mg/dL
Often, a combination of drugs or insulin (see below) is needed. However, you can start with one agent and add a second agent within 2 or 3 months (as is recommended for type 2 diabetes).22,23 The following combinations of the agents listed above are supported by published guidelines for type 2 diabetes:
- Metformin plus a sulfonylurea22,23
- Metformin plus a glinide22
- Metformin plus a GLP-1 agonist23
- Metformin plus a DPP-4 inhibitor.22
If the random or 1- to 2-hour post-meal plasma glucose is higher than 300 mg/dL
In our experience, if their plasma glucose levels are this high, patients are experiencing frank symptoms of hyperglycemia.
Insulin addresses those symptoms and avoids the prolonged wait that often results from unsuccessfully starting one agent and then adding another. Of all the available drugs, insulin is the only one that can be used despite multiple underlying illnesses; it does not cause a lot of drug interactions, and the dose can be adjusted upward and downward in increments to fit the patient’s needs, especially when a larger glucocorticoid load is given up front and then is tapered either slowly or rapidly. However, it can cause hypoglycemia and weight gain.
The initial total daily dose of insulin can be based on the patient’s weight. A starting total daily dose of 0.15 to 0.3 U/kg is reasonable— on the lower end if only the postprandial glucose levels are elevated, and on the higher end if both fasting and postprandial glucose levels are affected.
If fasting glucose levels are not elevated, then Neutral Protamine Hagedorn insulin (which is intermediate-acting) or a premixed combination of an intermediate-acting plus a fast- or short-acting insulin can be given once a day before breakfast, or even before lunch if the glucose levels start to rise only after lunch.
If both the fasting and the postprandial glucose levels are elevated, regimens similar to those for type 1 or insulin-requiring type 2 diabetes can be used, except that the ratios of the doses are tilted more toward covering postprandial than fasting hyperglycemia:
- Long-acting insulin plus prandial insulin, in a ratio of 30:70 to 50:50. As glucocorticoids are tapered, the long-acting insulin may have to be discontinued while the prandial doses are continued, since the fasting glucose level decreases first.
- Premixed insulins, with one-half to two-thirds of the dose given before breakfast and the rest before the evening meal, with the possibility of a third injection before lunch. As glucocorticoids are tapered, the evening dose is tapered first.
- Intermediate-acting insulin plus short- or fast-acting insulin in the morning (these two will make up one-half to two-thirds of the total daily dose), short- or fast-acting insulin before the evening meal, and intermediate-acting insulin at bedtime. As glucocorticoids are tapered, the bedtime insulin is tapered first.
Capillary blood glucose (fingerstick) checks
The timing and frequency of fingerstick checks depend on the treatment.
Though postprandial testing is ideal, it is often not practical or convenient. Before lunch, before dinner, and at bedtime are good alternatives since they reflect the pattern of glucose rise throughout the day. For patients on diet and exercise with or without agents other than insulin, testing once or twice a day is reasonable, rotating times before meals (including fasting if this time is affected) and at bedtime.
For patients on insulin, checking two to four times a day initially would help match insulin doses with glucose excursions. For continued care, the American Diabetes Association recommends fingerstick checks three times daily in patients on multiple insulin injections, but it has no specific recommendations for those on once-a-day insulin.21 We have been recommending that our patients on once-daily insulin check at least twice a day.
Goal fingerstick glucose levels that we use are in accordance with the American Diabetes Association guidelines for diabetes in general21:
- Before meals 70 to 130 mg/dL or
- 1 to 2 hours after meals < 180 mg/dL.
During steroid taper, if the glucocorticoid dose is in the lower range (eg, a prednisone-equivalent dose of approximately 7.5 mg per day or less), the fingerstick glucose levels are at the lower end of the target range, and the patient is on a single antidiabetic agent that does not often cause hypoglycemia (eg, metformin), then it is reasonable to ask the patient to not take the antidiabetic medication for 3 to 7 days while continuing to check fingersticks to see if it needs to be resumed. Patients on agents that can cause hypoglycemia need to check more often during the 1 to 3 days after the glucocorticoid dose reduction, as it may take this much time for the glycemic effect to diminish and to adjust the diabetes medication to the appropriate dose.
STARTING GLUCOCORTICOIDS IN PATIENTS WITH KNOWN DIABETES
Fingerstick checks more often
Most patients will already have a glucose meter. They should be instructed to check as discussed above if they do not have a previous diagnosis of diabetes, or to continue as they are doing if they are already checking more often. Patients who have been checking only fasting levels should be instructed to check later in the day, either before or 1 to 2 hours after meals, as discussed above. Patients on oral medications may need additional oral agents or insulin.
Adjust medications if glucose is not at goal
Patients with type 2 diabetes treated with diet and exercise alone can be started on the medications discussed above if their fingerstick readings are not at goal.
If they are already on insulin, we advise them to increase the short- or fast-acting insulins and the morning intermediate-acting insulin by at least 10% to 20% as soon as an elevation in glucose is detected. Long-acting insulin or nighttime intermediate-acting insulin should be increased if fasting glucose levels are affected.
Insulin requirements can double depending on the glucocorticoid dose. In patients with type 1 diabetes who were given prednisone 60 mg orally for 3 days, mean blood glucose levels increased from a baseline of 110 mg/dL at baseline to 149 mg/dL on the days on prednisone.6 The average blood glucose level remained elevated at 141 mg/dL on the day after the last dose of prednisone. The insulin dose increased by 31% to 102% (mean 69%).
CUSHING SYNDROME AND ADRENAL SUPPRESSION
Unlike glucocorticoid-induced diabetes, in which the dilemma is often when to initiate antidiabetic treatment, the question for patients in whom Cushing syndrome or adrenal suppression has developed is when to discontinue glucocorticoids.
Adrenal suppression for the most part goes hand in hand with exogenous Cushing syndrome. If cushingoid features develop, we can infer that the dose of exogenous glucocorticoid exceeds the physiologic needs. This supraphysiologic dosing also leads to suppression of endogenous cortisol production. The suppression occurs at the level of the hypothalamus and pituitary gland, with subsequent atrophy of the part of the adrenal cortex that produces endogenous glucocorticoids.
To understand further the concept of supraphysiologic dosing, the following interconversion of systemic glucocorticoid effects is helpful24,25:
However, there is not much information on interconversion for the local preparations (intra-articular, epidural, inhaled, topical).
Moreover, the definition of supraphysiologic dosing seems to be evolving. Though a total hydrocortisone-equivalent dose of 30 mg/day is still often touted as physiologic replacement, many patients require less. Several studies in the early 1990s, mostly in children and adolescents, showed the mean daily cortisol production rate to be 4.8 to 6.8 mg/m2/day, or closer to 10 to 15 mg/day.26–28 For purposes of this discussion, a physiologic dose will be defined as up to 30 mg hydrocortisone per day or its equivalent.
Adrenal suppression vs insufficiency
Adrenal suppression is often confused with adrenal insufficiency.
Adrenal suppression occurs when cortisol production is decreased because of the presence of exogenous glucocorticoids or other drugs, such as megestrol acetate (Megace), that act on the glucocorticoid receptor. Another situation beyond the scope of this review is excess endogenous cortisol production by an adrenal adenoma or adrenal carcinoma that causes suppression of the contralateral adrenal gland.29
In contrast, adrenal insufficiency is caused by failure of the adrenal gland to produce cortisol as a result of an innate disorder of the adrenal gland (eg, Addison disease) or pituitary gland (eg, pituitary surgery).
Hence, endogenous cortisol production in a patient taking supraphysiologic doses of exogenous glucocorticoids may be suppressed. Recovery of endogenous cortisol production is expected after stopping the exogenous glucocorticoid, though the time to recovery can vary and the patient can be adrenally insufficient if the glucocorticoid is stopped abruptly.
In addition, during times of intercurrent illness, a patient with adrenal suppression may be relatively adrenally insufficient and may need larger doses (“stress doses”) of glucocorticoids, since the adrenal glands may be unable to mount a stress response.29
Local steroids can suppress the adrenal glands
Glucocorticoids are the most common cause of Cushing syndrome. Oral formulations such as dexamethasone, prednisone, and hydrocortisone taken in supraphysiologic doses and for prolonged durations are easily recognized as obvious causes of Cushing syndrome. However, intra-articular, epidural, inhaled, nasal, ocular, and topical steroids—so-called local preparations—have also been linked to Cushing syndrome, and physicians are less likely to recognize them as causes.30–38
In a study in 16 pediatric patients with asthma and 48 controls, inhaled beclomethasone dipropionate (Qvar) 300 to 500 μg daily resulted in adrenal suppression in 100% of patients after 6 to 42 months, as determined by an insulin tolerance test.30
The topical steroid betamethasone (Diprosone) carries a warning that systemic absorption of topical steroids can cause adrenal suppression.39 Intra-articular, intranasal, epidural, and ocular routes are also reported to cause adrenal suppression.32–38
When is adrenal suppression more likely?
Adrenal suppression is more likely in the following situations:
- Longer duration of treatment. Studies have shown that exposure to supraphysiologic steroid doses for 2 weeks or less might already suppress the adrenal glands, but the clinical significance of this is unclear since some recovery already occurs a few days after the glucocorticoids are discontinued.31,40
- Supraphysiologic doses, stronger formulations, longer-acting formulations.41
When is adrenal suppression less likely?
Adrenal suppression is less likely in the following situations:
- Regimens that mimic the diurnal rhythm of cortisol (higher dose in the morning, lower dose in the afternoon)42
- Alternate-day dosing of steroids.43
Steroid withdrawal vs adrenal insufficiency
Another phenomenon that can be confused with adrenal insufficiency or glucocorticoid insufficiency is steroid withdrawal, in which patients experience lethargy, muscle aches, nausea, vomiting, and postural hypotension as glucocorticoids are tapered and their effects wane.42 Increasing the glucocorticoid dose for presumed adrenal insufficiency may delay recovery of the adrenal function and would have to be weighed against the patient’s symptoms.
The following may help distinguish the two: if the patient is on supraphysiologic glucocorticoid doses, then he or she is not glucocorticoid-deficient and is likely suffering from steroid withdrawal. At this point, patients may just need reassurance, symptomatic treatment, or if necessary, a brief (1-week) increase of the previous lowest dose, followed by reevaluation.
With local glucocorticoid preparations that may be systemically absorbed, however, there is no good way of estimating dose equivalence. In these situations, the decision to simply reassure the patient or give symptomatic treatment—as opposed to giving low-dose oral glucocorticoids such as hydrocortisone 5 to 10 mg daily for a week followed by reevaluation— depends on the severity of symptoms and whether the patient has quick access to medical attention should he or she develop an intercurrent illness.
Identifying patients at risk of adrenal suppression
Patients presenting with weight gain or symptoms suggesting Cushing syndrome should be asked about steroid intake and should be prompted to recall possible nonoral routes. In addition, patients presenting with muscle aches and fatigue—symptoms of steroid withdrawal— may have received unrecognized local glucocorticoids that were systemically absorbed, now with diminishing effects.
The ACTH stimulation test for adrenal recovery
Testing can be done to see if the adrenal glands have recovered and glucocorticoid therapy can be discontinued (see Tapering from glucocorticoids, below).
The test most often used is the corticotropin (ACTH) stimulation test. Since the suppression is at the level of the hypothalamus and the pituitary gland, the ACTH stimulation test is an indirect method of assessing hypothalamic and pituitary function in the context of glucocorticoid-induced adrenal suppression. It has good correlation with the insulin tolerance test, the gold-standard test for an intact hypothalamic-pituitary-adrenal axis.
The synthetic ACTH cosyntropin (Cortrosyn) 250 μg is injected intravenously or intramuscularly, and a cortisol level is drawn at baseline and 30 and 60 minutes later. Other doses such as 1 μg or 10 μg have been reported but are not yet widely accepted. A cortisol level of greater than 18 to 20 μg/dL at any time point shows that the adrenals have regained function and the steroids may be discontinued.42 If adrenal suppression persists, weaning from steroids should continue.
In reality, it may not be possible or practical to do an ACTH stimulation test, as not all physicians’ offices have a supply of cosyntropin or the manpower to perform the test correctly. In these cases, weaning can progress with monitoring of symptoms.
Testing for synthetic glucocorticoids in the urine and serum can demonstrate systemic absorption and may be helpful in patients who do not recall receiving steroids.33
Tapering from glucocorticoids
Several tapering schedules have been suggested (although not necessarily validated). Whether and how to taper depend on how long the glucocorticoid has been taken.
If taken for less than 1 week, glucocorticoids can be stopped without tapering, regardless of the dose.
If taken for 1 to 3 weeks, the decision to taper depends on the clinician’s assessment of the patient’s general health or constitution and the illness for which the glucocorticoid was prescribed. For example, if the underlying disease is less likely to flare with a gradual dose reduction, then tapering would be suitable.44
If taken for more than 3 weeks, the practice has been a more rapid taper at the beginning until a physiologic dose is reached. How quickly to reduce the dose depends on whether the underlying illness is expected to flare up, or if the patient might experience steroid withdrawal symptoms.
One schedule is to lower the glucocorticoid dose by an amount equivalent to prednisolone 2.5 mg every 3 to 4 days when above the physiologic dose, then to taper more slowly by 1 mg every 2 to 4 weeks.44 Once the physiologic dose is reached, one can switch to the equivalent dose of hydrocortisone and decrease the dose by 2.5 mg a week until a daily dose of 10 mg a day is reached and maintained for 2 to 3 months, and then perform a test of adrenal function (see above).44 Passing the test implies that the adrenal glands have recovered and the glucocorticoid can be stopped.
Another option is to switch to alternate-day therapy once a physiologic dose is reached and to test 8:00 am cortisol levels, continuing the glucocorticoid and retesting in 4 to 6 weeks if the value is less than 3 μg/dL; stopping the glucocorticoid if the value is higher than 20 μg/dL; and performing an ACTH stimulation test for values in between.45
A review of other tapering regimens for chronic diseases, mostly pulmonary, did not find enough evidence to recommend one particular schedule over another.46 The tapering schedule may have to be adjusted to prevent disease flare and symptoms of steroid withdrawal.
Locally administered steroids. Since the equivalence of systemically absorbed local glucocorticoids is not known, these patients are likely to present when they have symptoms of steroid withdrawal. In this situation, testing adrenal function will help.
Glucocorticoids are commonly prescribed by primary care physicians and specialists alike for multiple medical problems, acute as well as chronic.
However, these useful drugs have adverse effects on multiple endocrine systems, effects that include diabetes (or worsening of hyperglycemia in those with known diabetes), Cushing syndrome, adrenal suppression, osteoporosis (reviewed in the Cleveland Clinic Journal of Medicine in August 2010),1 and dyslipidemia. In addition, suppression of gonadotropins, growth hormone, and, acutely, thyrotropin can ensue.
The focus of this review is on the diabetogenic and adrenal suppressive effects of glucocorticoids and their management. We describe the rationale for choosing specific drugs to counter hyperglycemia, tests for determining adrenal suppression and systemic glucocorticoid absorption, and how and why to taper these drugs.
WIDELY USED DRUGS
Although glucocorticoids (often simply called steroids or corticosteroids, although not all steroids are corticosteroids, and not all corticosteroids are glucocorticoids) are the core treatment for adrenal insufficiency, in most cases they are prescribed for their anti-inflammatory effects. They act through multiple pathways at the cellular and molecular levels, suppressing the cascades that would otherwise result in inflammation and promoting pathways that produce anti-inflammatory proteins.2
In addition to formulations that are intended to have systemic effects, other, “local” formulations are made for specific conditions, such as intra-articular injections for arthritis, epidural injections for lumbar disk pain, eye drops for uveitis, nasal sprays for allergic rhinitis, inhalers for asthma, and topical ointments and creams for eczema. However, as we will discuss, even these preparations can have systemic effects.
GLUCOCORTICOID-INDUCED DIABETES IS COMMON
Glucocorticoids are the most common cause of drug-induced diabetes. Though the exact prevalence is not known, a few observations suggest that glucocorticoid-induced diabetes or hyperglycemia is common:
- In patients with rheumatoid arthritis, mean age 62 years, nearly 9% developed diabetes in the 2 years after starting glucocorticoid treatment, which was a higher rate than expected.3
- In nondiabetic patients with primary renal disease treated with prednisolone 0.75 mg/kg/day, 42% were found to have 2-hour post-lunch plasma glucose concentrations higher than 200 mg/dL but normal fasting glucose levels.4
- In a case-control study, the odds ratio of starting an oral hypoglycemic agent or insulin was 1.77 for patients receiving a hydrocortisone-equivalent dose of 1 to 39 mg/day, 3.02 for 40 to 79 mg/day, 5.82 for 80 to 119 mg/day, and 10.34 for 120 mg/day or more.5 (For a full discussion of glucocorticoid equivalents, see the section below on Cushing syndrome and adrenal suppression.)
- In patients with type 1 diabetes, prednisone 60 mg/day raised the blood glucose levels starting 6 hours after the prednisone dose.6
- Diabetic ketoacidosis and hyperosmolar nonketotic syndrome have been reported as a result of glucocorticoid treatment.7–9
GLUCOCORTICOIDS CAUSE DIABETES MAINLY VIA INSULIN RESISTANCE
The mechanism by which glucocorticoids cause diabetes predominantly involves insulin resistance rather than decreased insulin production. In fact, in a study in healthy volunteers, 10 hydrocortisone infusion resulted in higher insulin production than saline infusion did. (In high doses, however, glucocorticoids have been shown to decrease insulin secretion.11)
Normally, in response to insulin, the liver decreases its output of glucose. Glucocorticoids decrease the liver’s sensitivity to insulin, thereby increasing hepatic glucose output.12 They also inhibit glucose uptake in muscle and fat, reducing insulin sensitivity as much as 60% in healthy volunteers. This seems primarily due to a postreceptor effect, ie, inhibition of glucose transport.13–15
THE PEAK EFFECT OCCURS 4 TO 6 HOURS AFTER DOSING
To understand the optimal time for checking plasma glucose and to apply appropriate treatment, we should consider the pharmacokinetic profile of glucocorticoids.
Studied using the whole-blood lymphocyte proliferation technique, prednisone shows a peak effect at about 4 to 6 hours and a duration of action of 13 to 16 hours.16 This closely resembles what we see in terms of glucose excursion with this drug.17 Two studies of intravenous dexamethasone 10 mg showed that glucose levels rose within 4 hours of injection, but did not pursue this beyond that time frame.18,19
PATIENTS WITHOUT A PREVIOUS DIAGNOSIS OF DIABETES
Be alert for new-onset diabetes
For most diseases treated with glucocorticoids, clinicians can estimate in advance how long the patient will need to take the drug. We can arbitrarily classify the projected exposure as either short-term (3 to 4 weeks or less, such as a 6-day course of methylprednisolone for allergic conditions) or long-term (such as in transplant recipients to prevent rejection or to treat graft-vs-host disease). Hyperglycemia is a potential concern with both short-term and long-term treatment. However, guidelines on checking blood sugar levels, as opposed to relying on symptoms alone, are available only for long-term glucocorticoid treatment.
Patients beginning treatment should be warned of typical diabetes symptoms such as thirst and increased urination and, should these occur, to seek medical attention to have their blood glucose level checked. It is also reasonable to have them return in a week for a random postprandial plasma glucose test in the mid-afternoon.
Why this timing? In most once-daily regimens, glucocorticoids are given in the morning to prevent adrenal suppression (discussed below). In our experience, glucose levels start to rise mid-morning and continue to increase until bedtime. Measuring glucose levels 1 to 2 hours after lunch allows for both the glucocorticoid action and the carbohydrate absorption from lunch to reach their peaks. If hyperglycemia is going to happen, it should be detectable by then. A glucose level of 200 mg/dL or higher should prompt the practitioner to pursue this further.
If glucocorticoid treatment is to continue beyond 3 to 4 weeks, the only population for which there are published guidelines on managing glucocorticoid-related diabetes is transplant recipients. International consensus guidelines, published in 2003, suggest checking the fasting plasma glucose level once a week for the first 4 weeks after transplantation, then at 3 months, at 6 months, and then once a year.20
Though practical, this suggestion does not reflect the fact that glucocorticoids often do not affect fasting plasma glucose, especially if given once daily in the morning at doses of 30 mg or less of prednisone or its equivalent. These guidelines thus may not be applicable to other populations with glucocorticoid-induced diabetes.
The transplant guidelines do mention that an oral glucose tolerance test may be more sensitive, but this is often cumbersome to perform. We believe that checking random postprandial plasma glucose levels is helpful in this regard.
If the patient was at risk of developing diabetes even before receiving a glucocorticoid (for example, if he or she is overweight, has a family history of diabetes, or had a previous hemoglobin A1c of 5.7% or higher), then a fasting plasma glucose level of 126 mg/dL or higher or a hemoglobin A1c of 6.5% or higher might suffice to diagnose diabetes. Results should be confirmed on a separate day in the absence of unequivocal hyperglycemia. Fasting hyperglycemia can also be seen in patients receiving higher once-daily glucocorticoid doses—in our experience, an equivalent of prednisone 40 mg once a day in the morning— or twice-daily dosing.
A hemoglobin A1c checked less than 2 to 3 months after starting glucocorticoid treatment will not be sensitive in picking up glucocorticoid-induced diabetes if the patient did not have underlying diabetes.
Diet and exercise may not be practical
Though diet and exercise are important in managing diabetes, the condition for which the patient is receiving a glucocorticoid may prevent him or her from exercising, at least in the acute phase of the illness.
In addition, though the exact mechanism is not known, glucocorticoids increase hunger, and so decreasing food intake is not easy either. Nonetheless, patients should be familiarized with what carbohydrates are and should be advised to reduce their intake of them.
For suspected type 1 diabetes, start insulin
If type 1 diabetes is suspected, for example, in patients who are lean, younger than 30 years, or who had presented with diabetic ketoacidosis, then insulin should be started. In equivocal cases, insulin therapy can commence while testing is done for C-peptide, glutamic acid decarboxylase antibodies, islet cell antibodies, and insulinoma-associated protein antibodies.
Starting oral antidiabetic drugs
Some patients may have contraindications to specific drugs. For example, metformin (Glucophage) is contraindicated if the serum creatinine level is elevated, an abnormality that renal transplant patients may continue to have.
If the patient has no such contraindications, we have found the following medications suitable in view of their efficacy, low risk of hypoglycemia, or lack of distressing side effects. They will often lower glucose levels enough to achieve capillary blood glucose or fingerstick goals (discussed below). None of them has been specifically approved by the US Food and Drug Administration for glucocorticoid-induced diabetes, but they are approved for type 2 diabetes.
Guidelines from the American Association of Clinical Endocrinologists for type 2 diabetes call for starting monotherapy if the hemoglobin A1c is 6.5% to 7.5%, dual therapy if it is 7.6% to 9%, triple therapy if it is higher than 9% and the patient has no symptoms, and insulin if it is higher than 9% and the patient does have symptoms.22
In terms of estimated average glucose levels, these categories correspond to 140 to 169 mg/dL for monotherapy, 171 to 212 mg/dL for dual therapy, and higher than 212 mg/dL for triple therapy or insulin. Since estimated average levels also include fasting glucose levels (which are lower in glucocorticoid-induced diabetes compared with nonfasting levels), and because we use the American Diabetes Association general hemoglobin A1c goal of less than 7%, we believe that our suggestions below are reasonable.
We divide our recommendations according to initial random (ideally, 1- to 2-hour postprandial) plasma glucose levels.
If the random or 1- to 2-hour post-meal plasma glucose is lower than 220 mg/dL
In this situation the choices are:
- Metformin
- Dipeptidyl peptidase-4 (DPP-4) inhibitors (“gliptins”)
- Meglitinides (“glinides”). The guidelines on new-onset diabetes after transplantation point out that meglitinides may be the safest agents apart from insulin in the renal transplant population, but does acknowledge that efficacies of different oral agents have not been compared in this group.20
- Glucagon-like protein-1 (GLP-1) agonists
- Sulfonylureas. However, the longer-acting forms such as glimepiride (Amaryl) are not suitable if the fasting plasma glucose is not affected.
We have not used thiazolidinediones (“glitazones”) routinely because they can cause weight gain and edema—problems that are already seen with the use of steroids—and have a slower onset of action.
If the random or 1- to 2-hour post-meal plasma glucose is 220 to 300 mg/dL
Often, a combination of drugs or insulin (see below) is needed. However, you can start with one agent and add a second agent within 2 or 3 months (as is recommended for type 2 diabetes).22,23 The following combinations of the agents listed above are supported by published guidelines for type 2 diabetes:
- Metformin plus a sulfonylurea22,23
- Metformin plus a glinide22
- Metformin plus a GLP-1 agonist23
- Metformin plus a DPP-4 inhibitor.22
If the random or 1- to 2-hour post-meal plasma glucose is higher than 300 mg/dL
In our experience, if their plasma glucose levels are this high, patients are experiencing frank symptoms of hyperglycemia.
Insulin addresses those symptoms and avoids the prolonged wait that often results from unsuccessfully starting one agent and then adding another. Of all the available drugs, insulin is the only one that can be used despite multiple underlying illnesses; it does not cause a lot of drug interactions, and the dose can be adjusted upward and downward in increments to fit the patient’s needs, especially when a larger glucocorticoid load is given up front and then is tapered either slowly or rapidly. However, it can cause hypoglycemia and weight gain.
The initial total daily dose of insulin can be based on the patient’s weight. A starting total daily dose of 0.15 to 0.3 U/kg is reasonable— on the lower end if only the postprandial glucose levels are elevated, and on the higher end if both fasting and postprandial glucose levels are affected.
If fasting glucose levels are not elevated, then Neutral Protamine Hagedorn insulin (which is intermediate-acting) or a premixed combination of an intermediate-acting plus a fast- or short-acting insulin can be given once a day before breakfast, or even before lunch if the glucose levels start to rise only after lunch.
If both the fasting and the postprandial glucose levels are elevated, regimens similar to those for type 1 or insulin-requiring type 2 diabetes can be used, except that the ratios of the doses are tilted more toward covering postprandial than fasting hyperglycemia:
- Long-acting insulin plus prandial insulin, in a ratio of 30:70 to 50:50. As glucocorticoids are tapered, the long-acting insulin may have to be discontinued while the prandial doses are continued, since the fasting glucose level decreases first.
- Premixed insulins, with one-half to two-thirds of the dose given before breakfast and the rest before the evening meal, with the possibility of a third injection before lunch. As glucocorticoids are tapered, the evening dose is tapered first.
- Intermediate-acting insulin plus short- or fast-acting insulin in the morning (these two will make up one-half to two-thirds of the total daily dose), short- or fast-acting insulin before the evening meal, and intermediate-acting insulin at bedtime. As glucocorticoids are tapered, the bedtime insulin is tapered first.
Capillary blood glucose (fingerstick) checks
The timing and frequency of fingerstick checks depend on the treatment.
Though postprandial testing is ideal, it is often not practical or convenient. Before lunch, before dinner, and at bedtime are good alternatives since they reflect the pattern of glucose rise throughout the day. For patients on diet and exercise with or without agents other than insulin, testing once or twice a day is reasonable, rotating times before meals (including fasting if this time is affected) and at bedtime.
For patients on insulin, checking two to four times a day initially would help match insulin doses with glucose excursions. For continued care, the American Diabetes Association recommends fingerstick checks three times daily in patients on multiple insulin injections, but it has no specific recommendations for those on once-a-day insulin.21 We have been recommending that our patients on once-daily insulin check at least twice a day.
Goal fingerstick glucose levels that we use are in accordance with the American Diabetes Association guidelines for diabetes in general21:
- Before meals 70 to 130 mg/dL or
- 1 to 2 hours after meals < 180 mg/dL.
During steroid taper, if the glucocorticoid dose is in the lower range (eg, a prednisone-equivalent dose of approximately 7.5 mg per day or less), the fingerstick glucose levels are at the lower end of the target range, and the patient is on a single antidiabetic agent that does not often cause hypoglycemia (eg, metformin), then it is reasonable to ask the patient to not take the antidiabetic medication for 3 to 7 days while continuing to check fingersticks to see if it needs to be resumed. Patients on agents that can cause hypoglycemia need to check more often during the 1 to 3 days after the glucocorticoid dose reduction, as it may take this much time for the glycemic effect to diminish and to adjust the diabetes medication to the appropriate dose.
STARTING GLUCOCORTICOIDS IN PATIENTS WITH KNOWN DIABETES
Fingerstick checks more often
Most patients will already have a glucose meter. They should be instructed to check as discussed above if they do not have a previous diagnosis of diabetes, or to continue as they are doing if they are already checking more often. Patients who have been checking only fasting levels should be instructed to check later in the day, either before or 1 to 2 hours after meals, as discussed above. Patients on oral medications may need additional oral agents or insulin.
Adjust medications if glucose is not at goal
Patients with type 2 diabetes treated with diet and exercise alone can be started on the medications discussed above if their fingerstick readings are not at goal.
If they are already on insulin, we advise them to increase the short- or fast-acting insulins and the morning intermediate-acting insulin by at least 10% to 20% as soon as an elevation in glucose is detected. Long-acting insulin or nighttime intermediate-acting insulin should be increased if fasting glucose levels are affected.
Insulin requirements can double depending on the glucocorticoid dose. In patients with type 1 diabetes who were given prednisone 60 mg orally for 3 days, mean blood glucose levels increased from a baseline of 110 mg/dL at baseline to 149 mg/dL on the days on prednisone.6 The average blood glucose level remained elevated at 141 mg/dL on the day after the last dose of prednisone. The insulin dose increased by 31% to 102% (mean 69%).
CUSHING SYNDROME AND ADRENAL SUPPRESSION
Unlike glucocorticoid-induced diabetes, in which the dilemma is often when to initiate antidiabetic treatment, the question for patients in whom Cushing syndrome or adrenal suppression has developed is when to discontinue glucocorticoids.
Adrenal suppression for the most part goes hand in hand with exogenous Cushing syndrome. If cushingoid features develop, we can infer that the dose of exogenous glucocorticoid exceeds the physiologic needs. This supraphysiologic dosing also leads to suppression of endogenous cortisol production. The suppression occurs at the level of the hypothalamus and pituitary gland, with subsequent atrophy of the part of the adrenal cortex that produces endogenous glucocorticoids.
To understand further the concept of supraphysiologic dosing, the following interconversion of systemic glucocorticoid effects is helpful24,25:
However, there is not much information on interconversion for the local preparations (intra-articular, epidural, inhaled, topical).
Moreover, the definition of supraphysiologic dosing seems to be evolving. Though a total hydrocortisone-equivalent dose of 30 mg/day is still often touted as physiologic replacement, many patients require less. Several studies in the early 1990s, mostly in children and adolescents, showed the mean daily cortisol production rate to be 4.8 to 6.8 mg/m2/day, or closer to 10 to 15 mg/day.26–28 For purposes of this discussion, a physiologic dose will be defined as up to 30 mg hydrocortisone per day or its equivalent.
Adrenal suppression vs insufficiency
Adrenal suppression is often confused with adrenal insufficiency.
Adrenal suppression occurs when cortisol production is decreased because of the presence of exogenous glucocorticoids or other drugs, such as megestrol acetate (Megace), that act on the glucocorticoid receptor. Another situation beyond the scope of this review is excess endogenous cortisol production by an adrenal adenoma or adrenal carcinoma that causes suppression of the contralateral adrenal gland.29
In contrast, adrenal insufficiency is caused by failure of the adrenal gland to produce cortisol as a result of an innate disorder of the adrenal gland (eg, Addison disease) or pituitary gland (eg, pituitary surgery).
Hence, endogenous cortisol production in a patient taking supraphysiologic doses of exogenous glucocorticoids may be suppressed. Recovery of endogenous cortisol production is expected after stopping the exogenous glucocorticoid, though the time to recovery can vary and the patient can be adrenally insufficient if the glucocorticoid is stopped abruptly.
In addition, during times of intercurrent illness, a patient with adrenal suppression may be relatively adrenally insufficient and may need larger doses (“stress doses”) of glucocorticoids, since the adrenal glands may be unable to mount a stress response.29
Local steroids can suppress the adrenal glands
Glucocorticoids are the most common cause of Cushing syndrome. Oral formulations such as dexamethasone, prednisone, and hydrocortisone taken in supraphysiologic doses and for prolonged durations are easily recognized as obvious causes of Cushing syndrome. However, intra-articular, epidural, inhaled, nasal, ocular, and topical steroids—so-called local preparations—have also been linked to Cushing syndrome, and physicians are less likely to recognize them as causes.30–38
In a study in 16 pediatric patients with asthma and 48 controls, inhaled beclomethasone dipropionate (Qvar) 300 to 500 μg daily resulted in adrenal suppression in 100% of patients after 6 to 42 months, as determined by an insulin tolerance test.30
The topical steroid betamethasone (Diprosone) carries a warning that systemic absorption of topical steroids can cause adrenal suppression.39 Intra-articular, intranasal, epidural, and ocular routes are also reported to cause adrenal suppression.32–38
When is adrenal suppression more likely?
Adrenal suppression is more likely in the following situations:
- Longer duration of treatment. Studies have shown that exposure to supraphysiologic steroid doses for 2 weeks or less might already suppress the adrenal glands, but the clinical significance of this is unclear since some recovery already occurs a few days after the glucocorticoids are discontinued.31,40
- Supraphysiologic doses, stronger formulations, longer-acting formulations.41
When is adrenal suppression less likely?
Adrenal suppression is less likely in the following situations:
- Regimens that mimic the diurnal rhythm of cortisol (higher dose in the morning, lower dose in the afternoon)42
- Alternate-day dosing of steroids.43
Steroid withdrawal vs adrenal insufficiency
Another phenomenon that can be confused with adrenal insufficiency or glucocorticoid insufficiency is steroid withdrawal, in which patients experience lethargy, muscle aches, nausea, vomiting, and postural hypotension as glucocorticoids are tapered and their effects wane.42 Increasing the glucocorticoid dose for presumed adrenal insufficiency may delay recovery of the adrenal function and would have to be weighed against the patient’s symptoms.
The following may help distinguish the two: if the patient is on supraphysiologic glucocorticoid doses, then he or she is not glucocorticoid-deficient and is likely suffering from steroid withdrawal. At this point, patients may just need reassurance, symptomatic treatment, or if necessary, a brief (1-week) increase of the previous lowest dose, followed by reevaluation.
With local glucocorticoid preparations that may be systemically absorbed, however, there is no good way of estimating dose equivalence. In these situations, the decision to simply reassure the patient or give symptomatic treatment—as opposed to giving low-dose oral glucocorticoids such as hydrocortisone 5 to 10 mg daily for a week followed by reevaluation— depends on the severity of symptoms and whether the patient has quick access to medical attention should he or she develop an intercurrent illness.
Identifying patients at risk of adrenal suppression
Patients presenting with weight gain or symptoms suggesting Cushing syndrome should be asked about steroid intake and should be prompted to recall possible nonoral routes. In addition, patients presenting with muscle aches and fatigue—symptoms of steroid withdrawal— may have received unrecognized local glucocorticoids that were systemically absorbed, now with diminishing effects.
The ACTH stimulation test for adrenal recovery
Testing can be done to see if the adrenal glands have recovered and glucocorticoid therapy can be discontinued (see Tapering from glucocorticoids, below).
The test most often used is the corticotropin (ACTH) stimulation test. Since the suppression is at the level of the hypothalamus and the pituitary gland, the ACTH stimulation test is an indirect method of assessing hypothalamic and pituitary function in the context of glucocorticoid-induced adrenal suppression. It has good correlation with the insulin tolerance test, the gold-standard test for an intact hypothalamic-pituitary-adrenal axis.
The synthetic ACTH cosyntropin (Cortrosyn) 250 μg is injected intravenously or intramuscularly, and a cortisol level is drawn at baseline and 30 and 60 minutes later. Other doses such as 1 μg or 10 μg have been reported but are not yet widely accepted. A cortisol level of greater than 18 to 20 μg/dL at any time point shows that the adrenals have regained function and the steroids may be discontinued.42 If adrenal suppression persists, weaning from steroids should continue.
In reality, it may not be possible or practical to do an ACTH stimulation test, as not all physicians’ offices have a supply of cosyntropin or the manpower to perform the test correctly. In these cases, weaning can progress with monitoring of symptoms.
Testing for synthetic glucocorticoids in the urine and serum can demonstrate systemic absorption and may be helpful in patients who do not recall receiving steroids.33
Tapering from glucocorticoids
Several tapering schedules have been suggested (although not necessarily validated). Whether and how to taper depend on how long the glucocorticoid has been taken.
If taken for less than 1 week, glucocorticoids can be stopped without tapering, regardless of the dose.
If taken for 1 to 3 weeks, the decision to taper depends on the clinician’s assessment of the patient’s general health or constitution and the illness for which the glucocorticoid was prescribed. For example, if the underlying disease is less likely to flare with a gradual dose reduction, then tapering would be suitable.44
If taken for more than 3 weeks, the practice has been a more rapid taper at the beginning until a physiologic dose is reached. How quickly to reduce the dose depends on whether the underlying illness is expected to flare up, or if the patient might experience steroid withdrawal symptoms.
One schedule is to lower the glucocorticoid dose by an amount equivalent to prednisolone 2.5 mg every 3 to 4 days when above the physiologic dose, then to taper more slowly by 1 mg every 2 to 4 weeks.44 Once the physiologic dose is reached, one can switch to the equivalent dose of hydrocortisone and decrease the dose by 2.5 mg a week until a daily dose of 10 mg a day is reached and maintained for 2 to 3 months, and then perform a test of adrenal function (see above).44 Passing the test implies that the adrenal glands have recovered and the glucocorticoid can be stopped.
Another option is to switch to alternate-day therapy once a physiologic dose is reached and to test 8:00 am cortisol levels, continuing the glucocorticoid and retesting in 4 to 6 weeks if the value is less than 3 μg/dL; stopping the glucocorticoid if the value is higher than 20 μg/dL; and performing an ACTH stimulation test for values in between.45
A review of other tapering regimens for chronic diseases, mostly pulmonary, did not find enough evidence to recommend one particular schedule over another.46 The tapering schedule may have to be adjusted to prevent disease flare and symptoms of steroid withdrawal.
Locally administered steroids. Since the equivalence of systemically absorbed local glucocorticoids is not known, these patients are likely to present when they have symptoms of steroid withdrawal. In this situation, testing adrenal function will help.
- Dore RK. How to prevent glucocorticoid-induced osteoporosis. Cleve Clin J Med 2010; 77:529–536.
- Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med 2005; 353:1711–1723.
- Panthakalam S, Bhatnagar D, Klimiuk P. The prevalence and management of hyperglycaemia in patients with rheumatoid arthritis on corticosteroid therapy. Scott Med J 2004; 49:139–141.
- Uzu T, Harada T, Sakaguchi M, et al. Glucocorticoid-induced diabetes mellitus: prevalence and risk factors in primary renal diseases. Nephron Clin Pract 2007; 105:c54–c57.
- Gurwitz JH, Bohn RL, Glynn RJ, Monane M, Mogun H, Avorn J. Glucocorticoids and the risk for initiation of hypoglycemic therapy. Arch Intern Med 1994; 154:97–101.
- Bevier WC, Zisser HC, Jovanovic L, et al. Use of continuous glucose monitoring to estimate insulin requirements in patients with type 1 diabetes mellitus during a short course of prednisone. J Diabetes Sci Technol 2008; 2:578–583.
- Cagdas DN, Paç FA, Cakal E. Glucocorticoid-induced diabetic ketoacidosis in acute rheumatic fever. J Cardiovasc Pharmacol Ther 2008; 13:298–300.
- Bedalov A, Balasubramanyam A. Glucocorticoid-induced ketoacidosis in gestational diabetes: sequela of the acute treatment of preterm labor. A case report. Diabetes Care 1997; 20:922–924.
- Yang JY, Cui XL, He XJ. Non-ketotic hyperosmolar coma complicating steroid treatment in childhood nephrosis. Pediatr Nephrol 1995; 9:621–622.
- Nielsen MF, Caumo A, Chandramouli V, et al. Impaired basal glucose effectiveness but unaltered fasting glucose release and gluconeogenesis during short-term hypercortisolemia in healthy subjects. Am J Physiol Endocrinol Metab 2004; 286:E102–E110.
- Matsumoto K, Yamasaki H, Akazawa S, et al. High-dose but not low-dose dexamethasone impairs glucose tolerance by inducing compensatory failure of pancreatic beta-cells in normal men. J Clin Endocrinol Metab 1996; 81:2621–2626.
- Rizza RA, Mandarino LJ, Gerich JE. Cortisol-induced insulin resistance in man: impaired suppression of glucose production and stimulation of glucose utilization due to a postreceptor detect of insulin action. J Clin Endocrinol Metab 1982; 54:131–138.
- Meyuhas O, Reshef L, Gunn JM, Hanson RW, Ballard FJ. Regulation of phosphoenolpyruvate carboxykinase (GTP) in adipose tissue in vivo by glucocorticoids and insulin. Biochem J 1976; 158:1–7.
- Tappy L, Randin D, Vollenweider P, et al. Mechanisms of dexamethasone-induced insulin resistance in healthy humans. J Clin Endocrinol Metab 1994; 79:1063–1069.
- Pagano G, Cavallo-Perin P, Cassader M, et al. An in vivo and in vitro study of the mechanism of prednisone-induced insulin resistance in healthy subjects. J Clin Invest 1983; 72:1814–1820.
- Magee MH, Blum RA, Lates CD, Jusko WJ. Pharmacokinetic/pharmaco-dynamic model for prednisolone inhibition of whole blood lymphocyte proliferation. Br J Clin Pharmacol 2002; 53:474–484.
- Burt MG, Roberts GW, Aguilar-Loza NR, Frith P, Stranks SN. Continuous monitoring of circadian glycemic patterns in patients receiving prednisolone for COPD. J Clin Endocrinol Metab 2011; 96:1789–1796.
- Hans P, Vanthuyne A, Dewandre PY, Brichant JF, Bonhomme V. Blood glucose concentration profile after 10 mg dexamethasone in non-diabetic and type 2 diabetic patients undergoing abdominal surgery. Br J Anaesth 2006; 97:164–170.
- Pasternak JJ, McGregor DG, Lanier WL. Effect of single-dose dexamethasone on blood glucose concentration in patients undergoing craniotomy. J Neurosurg Anesthesiol 2004; 16:122–125.
- Davidson J, Wilkinson A, Dantal J, et al; International Expert Panel. New-onset diabetes after transplantation: 2003 international consensus guidelines. Proceedings of an international expert panel meeting. Barcelona, Spain, 19 February 2003. Transplantation 2003; 75(suppl 10):SS3–SS24.
- American Diabetes Association. Standards of medical care in diabetes— 2011. Diabetes Care 2011; 34(suppl 1):S11–S61.
- Rodbard HW, Jellinger PS, Davidson JA, et al. Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on type 2 diabetes mellitus: an algorithm for glycemic control. Endocr Pract 2009; 15:540–559.
- Nathan DM, Buse JB, Davidson MB, et al; American Diabetes Association; European Association for Study of Diabetes. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009; 32:193–203.
- Axelrod L. Corticosteroid therapy. In:Becker KL, editor. Principles and Practice of Endocrinology and Metabolism. 3rd ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2000:752–763.
- Ferri FF, editor. Practical Guide to the Care of the Medical Patient. 8th ed. Philadelphia, PA: Mosby/Elsevier; 2011.
- Kerrigan JR, Veldhuis JD, Leyo SA, Iranmanesh A, Rogol AD. Estimation of daily cortisol production and clearance rates in normal pubertal males by deconvolution analysis. J Clin Endocrinol Metab 1993; 76:1505–1510.
- Linder BL, Esteban NV, Yergey AL, Winterer JC, Loriaux DL, Cassorla F. Cortisol production rate in childhood and adolescence. J Pediatr 1990; 117:892–896.
- Esteban NV, Loughlin T, Yergey AL, et al. Daily cortisol production rate in man determined by stable isotope dilution/mass spectrometry. J Clin Endocrinol Metab 1991; 72:39–45.
- Lansang MC, Quinn SL. Adrenal suppression. BMJ BestPractice 2010. http://bestpractice.bmj.com/best-practice/monograph/863/diagnosis/stepby-step.html. Accessed August 19, 2011.
- Zöllner EW. Hypothalamic-pituitary-adrenal axis suppression in asthmatic children on inhaled corticosteroids (part 2)—the risk as determined by gold standard adrenal function tests: a systematic review. Pediatr Allergy Immunol 2007; 18:469–474.
- Schuetz P, Christ-Crain M, Schild U, et al. Effect of a 14-day course of systemic corticosteroids on the hypothalamic-pituitary-adrenal-axis in patients with acute exacerbation of chronic obstructive pulmonary disease. BMC Pulm Med 2008; 8:1.
- Kay J, Findling JW, Raff H. Epidural triamcinolone suppresses the pituitary-adrenal axis in human subjects. Anesth Analg 1994; 79:501–505.
- Lansang MC, Farmer T, Kennedy L. Diagnosing the unrecognized systemic absorption of intra-articular and epidural steroid injections. Endocr Pract 2009; 15:225–228.
- Duclos M, Guinot M, Colsy M, et al. High risk of adrenal insufficiency after a single articular steroid injection in athletes. Med Sci Sports Exerc 2007; 39:1036–1043.
- Bong JL, Connell JM, Lever R. Intranasal betamethasone induced acne and adrenal suppression. Br J Dermatol 2000; 142:579–580.
- Atabek ME, Pirgon O, Unal E. Pituitary-adrenal axis suppression due to topical steroid administration in an infant. Pediatr Int 2007; 49:242–244.
- Ozerdem U, Levi L, Cheng L, Song MK, Scher C, Freeman WR. Systemic toxicity of topical and periocular corticosteroid therapy in an 11-year-old male with posterior uveitis. Am J Ophthalmol 2000; 130:240–241.
- Chiang MY, Sarkar M, Koppens JM, Milles J, Shah P. Exogenous Cushing’s syndrome and topical ocular steroids. Eye (Lond) 2006; 20:725–727.
- Diprolene prescribing information. Schering Corp 2005. www.theodora.com/drugs/diprolene_gel_005_schering.html. Accessed September 27, 2011.
- Villabona CV, Koh C, Panergo J, Reddy A, Fogelfeld L. Adrenocorticotropic hormone stimulation test during high-dose glucocorticoid therapy. Endocr Pract 2009; 15:122–127.
- Ortega E, Rodriguez C, Strand LJ, Segre E. Effects of cloprednol and other corticosteroids on hypothalamic-pituitary-adrenal axis function. J Int Med Res 1976; 4:326–337.
- Axelrod L. Glucocorticoid therapy. Medicine (Baltimore) 1976; 55:39–65.
- Schürmeyer TH, Tsokos GC, Avgerinos PC, et al. Pituitary-adrenal responsiveness to corticotropin-releasing hormone in patients receiving chronic, alternate day glucocorticoid therapy. J Clin Endocrinol Metab 1985; 61:22–27.
- Stewart PM. The adrenal cortex. In:Kronenberg HM, editor. Williams Textbook of Endocrinology. 11th ed. Philadelphia, PA: Saunders/Elsevier; 2008.
- Hopkins RL, Leinung MC. Exogenous Cushing’s syndrome and glucocorticoid withdrawal. Endocrinol Metab Clin North Am 2005; 34:371–384.
- Richter B, Neises G, Clar C. Glucocorticoid withdrawal schemes in chronic medical disorders. A systematic review. Endocrinol Metab Clin North Am 2002; 31:751–778.
- Dore RK. How to prevent glucocorticoid-induced osteoporosis. Cleve Clin J Med 2010; 77:529–536.
- Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med 2005; 353:1711–1723.
- Panthakalam S, Bhatnagar D, Klimiuk P. The prevalence and management of hyperglycaemia in patients with rheumatoid arthritis on corticosteroid therapy. Scott Med J 2004; 49:139–141.
- Uzu T, Harada T, Sakaguchi M, et al. Glucocorticoid-induced diabetes mellitus: prevalence and risk factors in primary renal diseases. Nephron Clin Pract 2007; 105:c54–c57.
- Gurwitz JH, Bohn RL, Glynn RJ, Monane M, Mogun H, Avorn J. Glucocorticoids and the risk for initiation of hypoglycemic therapy. Arch Intern Med 1994; 154:97–101.
- Bevier WC, Zisser HC, Jovanovic L, et al. Use of continuous glucose monitoring to estimate insulin requirements in patients with type 1 diabetes mellitus during a short course of prednisone. J Diabetes Sci Technol 2008; 2:578–583.
- Cagdas DN, Paç FA, Cakal E. Glucocorticoid-induced diabetic ketoacidosis in acute rheumatic fever. J Cardiovasc Pharmacol Ther 2008; 13:298–300.
- Bedalov A, Balasubramanyam A. Glucocorticoid-induced ketoacidosis in gestational diabetes: sequela of the acute treatment of preterm labor. A case report. Diabetes Care 1997; 20:922–924.
- Yang JY, Cui XL, He XJ. Non-ketotic hyperosmolar coma complicating steroid treatment in childhood nephrosis. Pediatr Nephrol 1995; 9:621–622.
- Nielsen MF, Caumo A, Chandramouli V, et al. Impaired basal glucose effectiveness but unaltered fasting glucose release and gluconeogenesis during short-term hypercortisolemia in healthy subjects. Am J Physiol Endocrinol Metab 2004; 286:E102–E110.
- Matsumoto K, Yamasaki H, Akazawa S, et al. High-dose but not low-dose dexamethasone impairs glucose tolerance by inducing compensatory failure of pancreatic beta-cells in normal men. J Clin Endocrinol Metab 1996; 81:2621–2626.
- Rizza RA, Mandarino LJ, Gerich JE. Cortisol-induced insulin resistance in man: impaired suppression of glucose production and stimulation of glucose utilization due to a postreceptor detect of insulin action. J Clin Endocrinol Metab 1982; 54:131–138.
- Meyuhas O, Reshef L, Gunn JM, Hanson RW, Ballard FJ. Regulation of phosphoenolpyruvate carboxykinase (GTP) in adipose tissue in vivo by glucocorticoids and insulin. Biochem J 1976; 158:1–7.
- Tappy L, Randin D, Vollenweider P, et al. Mechanisms of dexamethasone-induced insulin resistance in healthy humans. J Clin Endocrinol Metab 1994; 79:1063–1069.
- Pagano G, Cavallo-Perin P, Cassader M, et al. An in vivo and in vitro study of the mechanism of prednisone-induced insulin resistance in healthy subjects. J Clin Invest 1983; 72:1814–1820.
- Magee MH, Blum RA, Lates CD, Jusko WJ. Pharmacokinetic/pharmaco-dynamic model for prednisolone inhibition of whole blood lymphocyte proliferation. Br J Clin Pharmacol 2002; 53:474–484.
- Burt MG, Roberts GW, Aguilar-Loza NR, Frith P, Stranks SN. Continuous monitoring of circadian glycemic patterns in patients receiving prednisolone for COPD. J Clin Endocrinol Metab 2011; 96:1789–1796.
- Hans P, Vanthuyne A, Dewandre PY, Brichant JF, Bonhomme V. Blood glucose concentration profile after 10 mg dexamethasone in non-diabetic and type 2 diabetic patients undergoing abdominal surgery. Br J Anaesth 2006; 97:164–170.
- Pasternak JJ, McGregor DG, Lanier WL. Effect of single-dose dexamethasone on blood glucose concentration in patients undergoing craniotomy. J Neurosurg Anesthesiol 2004; 16:122–125.
- Davidson J, Wilkinson A, Dantal J, et al; International Expert Panel. New-onset diabetes after transplantation: 2003 international consensus guidelines. Proceedings of an international expert panel meeting. Barcelona, Spain, 19 February 2003. Transplantation 2003; 75(suppl 10):SS3–SS24.
- American Diabetes Association. Standards of medical care in diabetes— 2011. Diabetes Care 2011; 34(suppl 1):S11–S61.
- Rodbard HW, Jellinger PS, Davidson JA, et al. Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on type 2 diabetes mellitus: an algorithm for glycemic control. Endocr Pract 2009; 15:540–559.
- Nathan DM, Buse JB, Davidson MB, et al; American Diabetes Association; European Association for Study of Diabetes. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009; 32:193–203.
- Axelrod L. Corticosteroid therapy. In:Becker KL, editor. Principles and Practice of Endocrinology and Metabolism. 3rd ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2000:752–763.
- Ferri FF, editor. Practical Guide to the Care of the Medical Patient. 8th ed. Philadelphia, PA: Mosby/Elsevier; 2011.
- Kerrigan JR, Veldhuis JD, Leyo SA, Iranmanesh A, Rogol AD. Estimation of daily cortisol production and clearance rates in normal pubertal males by deconvolution analysis. J Clin Endocrinol Metab 1993; 76:1505–1510.
- Linder BL, Esteban NV, Yergey AL, Winterer JC, Loriaux DL, Cassorla F. Cortisol production rate in childhood and adolescence. J Pediatr 1990; 117:892–896.
- Esteban NV, Loughlin T, Yergey AL, et al. Daily cortisol production rate in man determined by stable isotope dilution/mass spectrometry. J Clin Endocrinol Metab 1991; 72:39–45.
- Lansang MC, Quinn SL. Adrenal suppression. BMJ BestPractice 2010. http://bestpractice.bmj.com/best-practice/monograph/863/diagnosis/stepby-step.html. Accessed August 19, 2011.
- Zöllner EW. Hypothalamic-pituitary-adrenal axis suppression in asthmatic children on inhaled corticosteroids (part 2)—the risk as determined by gold standard adrenal function tests: a systematic review. Pediatr Allergy Immunol 2007; 18:469–474.
- Schuetz P, Christ-Crain M, Schild U, et al. Effect of a 14-day course of systemic corticosteroids on the hypothalamic-pituitary-adrenal-axis in patients with acute exacerbation of chronic obstructive pulmonary disease. BMC Pulm Med 2008; 8:1.
- Kay J, Findling JW, Raff H. Epidural triamcinolone suppresses the pituitary-adrenal axis in human subjects. Anesth Analg 1994; 79:501–505.
- Lansang MC, Farmer T, Kennedy L. Diagnosing the unrecognized systemic absorption of intra-articular and epidural steroid injections. Endocr Pract 2009; 15:225–228.
- Duclos M, Guinot M, Colsy M, et al. High risk of adrenal insufficiency after a single articular steroid injection in athletes. Med Sci Sports Exerc 2007; 39:1036–1043.
- Bong JL, Connell JM, Lever R. Intranasal betamethasone induced acne and adrenal suppression. Br J Dermatol 2000; 142:579–580.
- Atabek ME, Pirgon O, Unal E. Pituitary-adrenal axis suppression due to topical steroid administration in an infant. Pediatr Int 2007; 49:242–244.
- Ozerdem U, Levi L, Cheng L, Song MK, Scher C, Freeman WR. Systemic toxicity of topical and periocular corticosteroid therapy in an 11-year-old male with posterior uveitis. Am J Ophthalmol 2000; 130:240–241.
- Chiang MY, Sarkar M, Koppens JM, Milles J, Shah P. Exogenous Cushing’s syndrome and topical ocular steroids. Eye (Lond) 2006; 20:725–727.
- Diprolene prescribing information. Schering Corp 2005. www.theodora.com/drugs/diprolene_gel_005_schering.html. Accessed September 27, 2011.
- Villabona CV, Koh C, Panergo J, Reddy A, Fogelfeld L. Adrenocorticotropic hormone stimulation test during high-dose glucocorticoid therapy. Endocr Pract 2009; 15:122–127.
- Ortega E, Rodriguez C, Strand LJ, Segre E. Effects of cloprednol and other corticosteroids on hypothalamic-pituitary-adrenal axis function. J Int Med Res 1976; 4:326–337.
- Axelrod L. Glucocorticoid therapy. Medicine (Baltimore) 1976; 55:39–65.
- Schürmeyer TH, Tsokos GC, Avgerinos PC, et al. Pituitary-adrenal responsiveness to corticotropin-releasing hormone in patients receiving chronic, alternate day glucocorticoid therapy. J Clin Endocrinol Metab 1985; 61:22–27.
- Stewart PM. The adrenal cortex. In:Kronenberg HM, editor. Williams Textbook of Endocrinology. 11th ed. Philadelphia, PA: Saunders/Elsevier; 2008.
- Hopkins RL, Leinung MC. Exogenous Cushing’s syndrome and glucocorticoid withdrawal. Endocrinol Metab Clin North Am 2005; 34:371–384.
- Richter B, Neises G, Clar C. Glucocorticoid withdrawal schemes in chronic medical disorders. A systematic review. Endocrinol Metab Clin North Am 2002; 31:751–778.
KEY POINTS
- Nonfasting plasma glucose levels are more sensitive than fasting levels for detecting glucocorticoid-induced diabetes, and antidiabetic agents that have greater effects on random postprandial plasma glucose levels are more suitable than those that mostly affect fasting levels.
- Even those glucocorticoid formulations that are not intended to have systemic effects (eg, eye drops, inhaled corticosteroids, creams, intra-articular injections) can cause adrenal suppression and, therefore, if they are discontinued, steroid withdrawal and adrenal insufficiency.
- Needed are studies comparing antidiabetic regimens for glucocorticoid-induced hyperglycemia and studies comparing glucocorticoid tapering schedules for adrenal suppression to determine the best way to manage these adverse effects.
Allergy blood testing: A practical guide for clinicians
Health care providers often need to evaluate allergic disorders such as allergic rhinoconjunctivitis, asthma, and allergies to foods, drugs, latex, and venom, both in the hospital and in the clinic.
Unfortunately, some symptoms, such as chronic nasal symptoms, can occur in both allergic and nonallergic disorders, and this overlap can confound the diagnosis and therapy. Studies suggest that when clinicians use the history and physical examination alone in evaluating possible allergic disease, the accuracy of their diagnoses rarely exceeds 50%.1
Blood tests are now available that measure immunoglobulin E (IgE) directed against specific antigens. These in vitro tests can be important tools in assessing a patient whose history suggests an allergic disease.2 However, neither allergy skin testing nor these blood tests are intended to be used for screening: they may be most useful as confirmatory diagnostic tests in cases in which the pretest clinical impression of allergic disease is high.
ALLERGY IS MEDIATED BY IgE
In susceptible people, IgE is produced by B cells in response to specific antigens such as foods, pollens, latex, and drugs. This antigen-specific (or allergen-specific) IgE circulates in the serum and binds to high-affinity IgE receptors on immune effector cells such as mast cells located throughout the body.
Upon subsequent exposure to the same allergen, IgE receptors cross-link and initiate downstream signaling events that trigger mast cell degranulation and an immediate allergic response—hence the term immediate (or Gell-Coombs type I) hypersensitivity.3
Common manifestations of type I hypersensitivity reactions include signs and symptoms that can be:
- Cutaneous (eg, acute urticaria, angioedema)
- Respiratory (eg, acute bronchospasm, rhinoconjunctivitis)
- Cardiovascular (eg, tachycardia, hypotension)
- Gastrointestinal (eg, vomiting, diarrhea)
- Generalized (eg, anaphylactic shock). By definition, anaphylaxis is a life-threatening reaction that occurs on exposure to an allergen and involves acute respiratory distress, cardiovascular failure, or involvement of two or more organ systems.4
MOST IgE BLOOD TESTS ARE IMMUNOASSAYS
The blood tests for allergic disease are immunoassays that measure the level of IgE specific to a particular allergen. The tests can be used to evaluate sensitivity to various allergens, for example, to common inhalants such as dust mites and pollens and to foods, drugs, venom, and latex.
Types of immunoassays include enzyme-linked immunosorbent assays (ELISAs), fluorescent enzyme immunoassays (FEIAs), and radioallergosorbent assays (RASTs). At present, most commercial laboratories use one of three autoanalyzer systems to measure specific IgE:
- ImmunoCAP (Phadia AB, Uppsala, Sweden)
- Immulite (Siemens AG, Berlin, Germany)
- HYTEC-288 (Hycor/Agilent, Garden Grove, CA).
These systems use a solid-phase polymer (cellulose or avidin) in which the antigen is embedded. The polymer also facilitates binding of IgE and, therefore, increases the sensitivity of the test.5 Specific IgE from the patient’s serum binds to the allergen embedded in the polymer, and then unbound antibodies are washed off.
Despite the term “RAST,” these systems do not use radiation. A fluorescent antibody is added that binds to the patient’s IgE, and the amount of IgE present is calculated from the amount of fluorescence.6 Results are reported in kilounits of antibody per liter (kU/L) or nanograms per milliliter (ng/mL).5–7
INTERPRETATION IS INDIVIDUALIZED
In general, the sensitivity of these tests ranges from 60% to 95% and their specificity from 30% to 95%, with a concordance among different immunoassays of 75% to 90%.8
Levels of IgE for a particular allergen are also divided into semiquantitative classes, from class I to class V or VI. In general, class I and class II correlate with a low level of allergen sensitization and, often, with a low likelihood of a clinical reaction. On the other hand, classes V and VI reflect higher degrees of sensitization and generally correlate with IgE-mediated clinical reactions upon allergen exposure.
The interpretation of a positive (ie, “nonzero”) test result must be individualized on the basis of clinical presentation and risk factors. A specialist can make an important contribution by helping to interpret any positive test result or a negative test result that does not correlate with the patient’s history.
ADVANTAGES OF ALLERGY BLOOD TESTING
Allergy blood testing is convenient, since it involves only a standard blood draw.
In theory, allergy blood testing may be safer, since it does not expose the patient to any allergens. On the other hand, many patients experience bruising from venipuncture performed for any reason: 16% in one survey.9 In another survey,10 adverse reactions of any type occurred in 0.49% of patients undergoing venipuncture but only in 0.04% of those undergoing allergy skin testing. Therefore, allergy blood testing may be most appropriate in situations in which a patient’s history suggests that he or she may be at risk of a systemic reaction from a traditional skin test or in cases in which skin testing is not possible (eg, extensive eczema).
Another advantage of allergy blood testing is that it is not affected by drugs such as antihistamines or tricyclic antidepressants that suppress the histamine response, which is a problem with skin testing.
Allergy blood testing may also be useful in patients on long-term glucocorticoid therapy, although the data conflict. Prolonged oral glucocorticoid use is associated with a decrease in mast cell density and histamine content in the skin,11,12 although in one study a corticosteroid was found not to affect the results of skin-prick testing for allergy.13 Thus, allergy blood testing can be performed in patients who have severe eczema or dermatographism or who cannot safely suspend taking antihistamines or tricyclic antidepressants.
LIMITATIONS OF THESE TESTS
A limitation of allergy blood tests is that there is no gold-standard test for many allergic conditions. (Double-blind, placebo-controlled oral food challenge testing has been proposed as the gold-standard test for food allergy, and nasal allergen provocation challenge has been proposed for allergic rhinitis.)
Also, allergy blood tests can give false-positive results because of nonspecific binding of antibody in the assay.
Of note: evidence of sensitization to a particular allergen (ie, a positive blood test result) is not synonymous with clinically relevant disease (ie, clinical sensitivity).
Conversely, these tests can give false-negative results in patients who have true IgE-mediated disease as confirmed by skin testing or allergen challenge. The sensitivity of blood allergy testing is approximately 25% to 30% lower than that of skin testing, based on comparative studies.2 The blood tests are usually considered positive if the allergen-specific IgE level is greater than 0.35 kU/L; however, sensitization to certain inhalant allergens can occur at levels as low as 0.12 kU/L.14
Specific IgE levels measured by different commercial assays are not always interchangeable or equivalent, so a clinician should consistently select the same immunoassay if possible when assessing any given patient over time.15
Levels of specific IgE have been shown to depend on age, allergen specificity, total serum IgE, and, with inhalant allergens, the season of the year.15,16
Other limitations of blood testing are its cost and a delay of several days to a week in obtaining the results.17
WHEN TO ORDER ALLERGY BLOOD TESTING
The allergy evaluation should begin with a thorough history to look for possible triggers for the patient’s symptoms.
For example, respiratory conditions such as asthma and rhinitis may be exacerbated during particular times of the year when certain pollens are commonly present. For patients with this pattern, blood testing for allergy to common inhalants, including pollens, may be appropriate. Similarly, peanut allergy evaluation is indicated for a child who has suffered an anaphylactic reaction after consuming peanut butter. Blood testing is also indicated in patients with a history of venom anaphylaxis, especially if venom skin testing was negative.
In cases in which the patient does not have a clear history of sensitization, blood testing for allergy to multiple foods may find evidence of sensitization that does not necessarily correlate with clinical disease.18
Likewise, blood tests are not likely to be clinically relevant in conditions not mediated by IgE, such as food intolerances (eg, lactose intolerance), celiac disease, the DRESS syndrome (drug rash, eosinophilia, and systemic symptoms), Stevens-Johnson syndrome, toxic epidermal necrolysis, or other types of drug hypersensitivity reactions, such as serum sickness.3
INTERPRETING COMMONLY ORDERED BLOOD TESTS FOR ALLERGY
Tests for allergy to hundreds of substances are available.
Foods
Milk, eggs, soy, wheat, peanuts, tree nuts, fish, and shellfish account for most cases of food allergy in the United States.18
IgE-mediated hypersensitivity to milk, eggs, and peanuts tends to be more common in children, whereas peanuts, tree nuts, fish, and shellfish are more commonly associated with reactions in adults.18 Children are more likely to outgrow allergy to milk, soy, wheat, and eggs than allergy to peanuts, tree nuts, fish, and shellfish—only about 20% of children outgrow peanut allergy.18
Patients with an IgE-mediated reaction to foods should be closely followed by a specialist, who can best help determine the appropriateness of additional testing (such as an oral challenge under observation), avoidance recommendations, and the introduction of foods back into the diet.19
Specific IgE tests for allergy to a variety of foods are available and can be very useful for diagnosis when used in the appropriate setting.
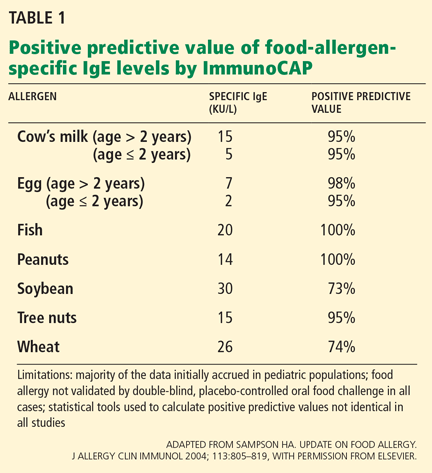
One caveat about these studies is that many were initially performed in children with a history of food allergy, many of whom had atopic dermatitis, and the findings have not been systematically reexamined in larger studies in more heterogeneous populations.
For example, at least eight studies tried to identify a diagnostic IgE level for cow’s milk allergy. The 95% confidence intervals varied widely, depending on the study design, the age of the study population, the prevalence of food allergy in the population, and the statistical method used for analysis.5 For most other foods for which blood tests are available, few studies have been performed to establish predictive values similar to those in Table 1.
Thus, slight elevations in antigen-specific IgE (> 0.35 kU/L) may correlate only with in vitro sensitization in a patient who has no clinical reactivity upon oral exposure to a particular antigen.
Broad food panels have been shown to have false-positive rates higher than 50%—ie, in more than half of cases, positive results have no clinical relevance. Therefore, these large food panels should not be used for screening.19 Instead, it is recommended that tests be limited to relevant foods based on the patient’s history when evaluating symptoms consistent with an IgE-mediated reaction to a particular food.
Food-specific IgE evaluation is also not helpful in evaluating non-IgE adverse reactions to foods (eg, intolerances).
Therefore, the patient’s history remains the most important tool for evaluation of food allergy. In cases in which the patient’s history suggests a food-associated IgE-mediated reaction and the blood test is negative, the patient should be referred to a specialist for skin testing with commercial extracts or even fresh food extracts, given the higher sensitivity of in vivo testing.20
Inhalants
Common aeroallergens associated with allergic rhinitis, allergic conjunctivitis, and allergic asthma include dust mites, animal dander, cockroach debris, molds, trees, grasses, weeds, and ragweed. Dust mites, animal dander, and mold spores are perennial allergens and may trigger symptoms year-round. Pollen, including pollen from trees, grasses, and weeds, is generally present in a seasonal pattern in many parts of the United States.
A positive blood test for an inhalant allergen can reinforce the physician’s clinical impression in making a diagnosis of allergic rhinoconjunctivitis. Interestingly, studies have suggested a high rate of false-positives based on history alone when in vivo and in vitro allergy testing were negative for IgE-mediated respiratory disease.21
Various studies have aimed to establish threshold values of aeroallergen-specific IgE that predict the likelihood of clinically relevant disease. Unfortunately, other factors also contribute to clinical symptoms of rhinoconjunctivitis; these include concurrent inflammation, infection, physical stress, psychological stress, exposure to irritants, and hormonal changes. These factors introduce variability and make specific IgE cutoffs for inhalant allergens unreliable.22
Prospective studies have suggested that skin testing correlates better with nasal allergen challenge (the gold standard) than blood testing for the diagnosis of inhalant allergy, though more recent studies using modern technologies demonstrate reasonable concordance (67%) between skin testing and blood testing (specifically, ImmunoCAP).23,24 According to current guidelines, skin tests are the preferred method for diagnosing IgE-mediated sensitivity to inhalants.25
Compared with skin prick tests as the gold standard, the sensitivity of specific IgE immunoassays is approximately 70% to 75%.25 Nevertheless, specific IgE values greater than 0.35 kU/L are generally considered positive for aeroallergen sensitization, although lower levels of dog-specific IgE have recently been shown to correlate with clinical disease.14
Drugs, including penicillins
A variety of clinical reactions can occur in response to oral, intravenous, or topical medications.
At present, blood tests are available for the evaluation of IgE-mediated adverse reactions to only a limited number of drugs. Reactions involving other mechanisms, such as those related to the drug’s metabolism, intolerances (eg, nausea), idiosyncratic reactions (eg, Stevens-Johnson syndrome, the DRESS syndrome), or other types of reactions can be diagnosed only by history and physical examination.
The development of specific IgE tests for sensitivity to medications has been limited by incomplete characterization of metabolic products and the possibility that a single medication can have different epitopes or IgE binding sites in different individuals.26
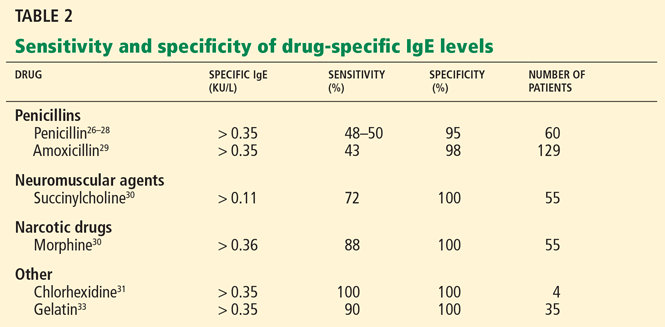
In vitro allergy testing has been most studied for beta-lactam antibiotics (eg, penicillin) and not so much for other drugs.
Table 2 summarizes the sensitivity and specificity of blood allergy tests that are commercially available for drugs.
Penicillin, a beta-lactam antibiotic, is degraded into various metabolites known as the major determinant (penicilloyl) and the minor determinants (eg, benzylpenicilloate and benzylpenilloate), which act as haptens. Specific IgE testing is not available for all these determinants.
The sensitivity of blood tests for allergy to penicilloyl (penicillin) and amino-penicillins such as amoxicilloyl (amoxicillin) is reported as between 32% and 50%, and the specificity as 96% to 98%.29
By definition, any nonzero level of IgE specific for penicillin or its derivatives is considered a positive result and may be associated with a higher risk of IgE-mediated reaction to penicillins. However, in a situation analogous to that in people with food allergy who have a food-specific IgE titer lower than the empirically established threshold value (Table 1), low-titer values to penicillin may not predict anaphylactic sensitivity in a penicillin oral challenge.28 Further studies are needed to determine if there is a threshold level of penicillin-specific IgE above which a patient has a higher likelihood of an IgE-mediated systemic reaction.
Other drugs. Specific IgE blood tests are also available for certain neuromuscular agents, insulin, cefaclor (Ceclor), chlorhexidine (contained in various antiseptic products), and gelatin (Table 2). These substances have not been as well studied as penicillins, and the sensitivity and specificity data reported in Table 2 are limited by few studies and small study sizes.
Neuromuscular blocking agents. Tests for IgE against neuromuscular blocking agents are reported to have low sensitivity (30%–60%) using a cutoff value of 0.35 kU/L.30 In small studies, the sensitivity was higher (68% to 92%) when threshold values for rocuronium-specific IgE were lowered from 0.35 to 0.13 kU/L.29
Chlorhexidine, an antiseptic commonly used in surgery, has been linked to IgE-mediated reactions.31 Chlorhexidine-specific IgE levels greater than 0.35 kU/L are considered positive, based on very limited data.
Insulin. Blood tests for allergy to insulin are also commercially available. However, studies have shown a significant overlap in the range of insulin-specific IgE in patients with a clinical history consistent with insulin allergy and in controls. Therefore, this test has a very limited ability to distinguish people who do not have a history of a reaction to insulin.32 More research is needed to determine the clinical utility of insulin-specific IgE testing.
Gelatin. IgE-mediated reactions have occurred after exposure to gelatin (from either cows or pigs) contained in foods and vaccines, including measles-mumps-rubella and yellow fever. One study identified gelatin-specific IgE in 10 of 11 children with a history of systemic reaction to measles or mumps vaccine.33 In the same study, gelatin-specific IgE levels were negative in 24 children who had developed non-IgE-mediated reactions to the vaccine.33
Tests for IgE against bovine gelatin are commercially available; results are considered positive for values higher than 0.35 kU/L. A negative test result does not exclude the possibility of an allergic reaction to porcine gelatin, which can also be found in foods and vaccines, but tests for anti-porcine gelatin IgE are not commercially available.
Latex
Latex, obtained from the rubber tree Hevea brasiliensis, has 13 known polypeptides (allergens Hev b 1–13) that cause IgE-mediated reactions, particularly in health care workers and patients with spina bifida.34 Overall, the incidence of latex allergy has decreased in the United States as most medical institutions have implemented a latex-free environment.
In vitro testing is the only mode of evaluation for allergy to latex approved by the US Food and Drug Administration (FDA).35 Its sensitivity is 80% and its specificity is 95%.36
In a 2007 study, 145 people at risk for latex allergy, including 104 health care workers, 31 patients with spina bifida, and 10 patients requiring multiple surgeries, underwent latex-specific IgE analysis for sensitivity to various recombinant and native latex allergens.34 The three groups differed in their latex allergy profiles, highlighting the diversity of clinical response to latex in high-risk groups and our current inability to establish specific cutoff points for quantitative latex-specific IgE. Thus, at present, any nonzero latex-specific IgE value is considered positive.
A formal evaluation for allergy is recommended for patients who have a strong history of an IgE-mediated reaction to latex and a latex-specific IgE value of zero. Blood tests for allergy to some native or recombinant latex allergens are available; these allergens may be underrepresented in the native total latex extract.33 Skin testing for allergy to latex, although not FDA-approved or standardized, can also be useful in this setting.37
Insect venom
Type I hypersensitivity reactions can occur from the stings of Vespidae (vespids), Apidae (bees), and Formicidae (fire ants). Large localized reactions after an insect sting are not infrequent and typically do not predict anaphylactic sensitivity with future stings, even though they are considered mild IgE-mediated reactions. However, systemic reactions are considered life-threatening and warrant allergy testing.38
The level of venom-specific IgE usually increases weeks to months after a sting.39 Therefore, blood tests can be falsely negative if performed within a short time of the sting.
Patients who have suffered a systemic reaction to venom and have evidence of sensitization by either in vitro or in vivo allergy testing are candidates for venom immunotherapy.40
At present, any nonzero venom-specific IgE test is considered positive, as there is no specific value for venom-specific IgE that predicts clinical risk.
A negative blood test does not exclude the possibility of an IgE-mediated reaction.41 In cases in which a patient has a clinical history compatible with venom allergy but the blood test is negative, the patient should be referred to an allergist for further evaluation, including venom skin testing and possibly repeat blood testing at a later time.
Conversely, specific IgE testing to venom is recommended when a patient has a history consistent with venom allergy and negative skin test results.38
As mentioned previously, in vitro test performance can vary with the laboratory and testing method used, and sending samples directly to a reference laboratory could be considered.41
TESTING FOR IgG AGAINST FOODS IS UNVALIDATED AND INAPPROPRIATE
In recent years, some practitioners of alternative medicine have started testing for allergen-specific IgG or IgG4 as part of evaluations for hypersensitivity, especially in cases in which patients describe atypical gastrointestinal, neurologic, or other symptoms after eating specific foods.19
However, this testing often finds IgG or IgG4 against foods that are well tolerated. At present, allergen-specific IgG testing lacks scientific evidence to support its clinical use in the evaluation of allergic disease.5,19
- Williams PB, Ahlstedt S, Barnes JH, Söderström L, Portnoy J. Are our impressions of allergy test performances correct? Ann Allergy Asthma Immunol 2003; 91:26–33.
- Bernstein IL, Li JT, Bernstein DI, et al; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology. Allergy diagnostic testing: an updated practice parameter. Ann Allergy Asthma Immunol 2008; 100(suppl 3):S1–S148.
- Pichler WJ. Immune mechanism of drug hypersensitivity. Immunol Allergy Clin North Am 2004; 24:373–397.
- Lieberman P, Nicklas RA, Oppenheimer J, et al. The diagnosis and management of anaphylaxis practice parameter: 2010 update. J Allergy Clin Immunol 2010; 126:477–480.
- Hamilton RG. Clinical laboratory assessment of immediate-type hypersensitivity. J Allergy Clin Immunol 2010; 125(suppl 2):S284–S296.
- Cox L, Williams B, Sicherer S, et al; American College of Allergy, Asthma and Immunology Test Task Force; American Academy of Allergy, Asthma and Immunology Specific IgE Test Task Force. Pearls and pitfalls of allergy diagnostic testing: report from the American College of Allergy, Asthma and Immunology/American Academy of Allergy, Asthma and Immunology Specific IgE Test Task Force. Ann Allergy Asthma Immunol 2008; 101:580–592.
- Hamilton RG, Franklin Adkinson N. In vitro assays for the diagnosis of IgE-mediated disorders. J Allergy Clin Immunol 2004; 114:213–225.
- Williams PB, Dolen WK, Koepke JW, Selner JC. Comparison of skin testing and three in vitro assays for specific IgE in the clinical evaluation of immediate hypersensitivity. Ann Allergy 1992; 68:35–45.
- Howanitz PJ, Cembrowski GS, Bachner P. Laboratory phlebotomy. College of American Pathologists Q-Probe study of patient satisfaction and complications in 23,783 patients. Arch Pathol Lab Med 1991; 115:867–872.
- Turkeltaub PC, Gergen PJ. The risk of adverse reactions from percutaneous prick-puncture allergen skin testing, venipuncture, and body measurements: data from the second National Health and Nutrition Examination Survey 1976–80 (NHANES II). J Allergy Clin Immunol 1989; 84:886–890.
- Pipkorn U, Hammarlund A, Enerbäck L. Prolonged treatment with topical glucocorticoids results in an inhibition of the allergen-induced weal-and-flare response and a reduction in skin mast cell numbers and histamine content. Clin Exp Allergy 1989; 19:19–25.
- Cole ZA, Clough GF, Church MK. Inhibition by glucocorticoids of the mast cell-dependent weal and flare response in human skin in vivo. Br J Pharmacol 2001; 132:286–292.
- Des Roches A, Paradis L, Bougeard YH, Godard P, Bousquet J, Chanez P. Long-term oral corticosteroid therapy does not alter the results of immediate-type allergy skin prick tests. J Allergy Clin Immunol 1996; 98:522–527.
- Linden CC, Misiak RT, Wegienka G, et al. Analysis of allergen specific IgE cut points to cat and dog in the Childhood Allergy Study. Ann Allergy Asthma Immunol 2011; 106:153–158.
- Hamilton RG, Williams PB; Specific IgE Testing Task Force of the American Academy of Allergy, Asthma & Immunology; American College of Allergy, Asthma and Immunology. Human IgE antibody serology: a primer for the practicing North American allergist/immunologist. J Allergy Clin Immunol 2010; 126:33–38.
- Somville MA, Machiels J, Gilles JG, Saint-Remy JM. Seasonal variation in specific IgE antibodies of grass-pollen hypersensitive patients depends on the steady state IgE concentration and is not related to clinical symptoms. J Allergy Clin Immunol 1989; 83( 2 Pt 1):486–494.
- Poon AW, Goodman CS, Rubin RJ. In vitro and skin testing for allergy: comparable clinical utility and costs. Am J Manag Care 1998; 4:969–985.
- Sampson HA. Update on food allergy. J Allergy Clin Immunol 2004; 113:805–819.
- Boyce JA, Assa’ad A, Burks AW, et al; NIAID-Sponsored Expert Panel. Guidelines for the diagnosis and management of food allergy in the United States: summary of the NIAID-sponsored expert panel report. J Allergy Clin Immunol 2010; 126:1105–1118.
- Rosen JP, Selcow JE, Mendelson LM, Grodofsky MP, Factor JM, Sampson HA. Skin testing with natural foods in patients suspected of having food allergies: is it a necessity? J Allergy Clin Immunol 1994; 93:1068–1070.
- Williams PB, Siegel C, Portnoy J. Efficacy of a single diagnostic test for sensitization to common inhalant allergens. Ann Allergy Asthma Immunol 2001; 86:196–202.
- Söderström L, Kober A, Ahlstedt S, et al. A further evaluation of the clinical use of specific IgE antibody testing in allergic diseases. Allergy 2003; 58:921–928.
- Bousquet J, Lebel B, Dhivert H, Bataille Y, Martinot B, Michel FB. Nasal challenge with pollen grains, skin-prick tests and specific IgE in patients with grass pollen allergy. Clin Allergy 1987; 17:529–536.
- Nepper-Christensen S, Backer V, DuBuske LM, Nolte H. In vitro diagnostic evaluation of patients with inhalant allergies: summary of probability outcomes comparing results of CLA- and CAP-specific immunoglobulin E test systems. Allergy Asthma Proc 2003; 24:253–258.
- Wallace DV, Dykewicz MS, Bernstein DI, et al; Joint Task Force on Practice; American Academy of Allergy; Asthma & Immunology; Joint Council of Allergy, Asthma and Immunology. The diagnosis and management of rhinitis: an updated practice parameter. J Allergy Clin Immunol 2008; 122(suppl 2):S1–S84.
- Mayorga C, Sanz ML, Gamboa PM, et al; Immunology Committee of the Spanish Society of Allergology and Clinical Immunology of the SEAIC. In vitro diagnosis of immediate allergic reactions to drugs: an update. J Investig Allergol Clin Immunol 2010; 20:103–109.
- Garcia JJ, Blanca M, Moreno F, et al. Determination of IgE antibodies to the benzylpenicilloyl determinant: a comparison of the sensitivity and specificity of three radio allergo sorbent test methods. J Clin Lab Anal 1997; 11:251–257.
- Macy E, Goldberg B, Poon KY. Use of commercial anti-penicillin IgE fluorometric enzyme immunoassays to diagnose penicillin allergy. Ann Allergy Asthma Immunol 2010; 105:136–141.
- Blanca M, Mayorga C, Torres MJ, et al. Clinical evaluation of Pharmacia CAP System RAST FEIA amoxicilloyl and benzylpenicilloyl in patients with penicillin allergy. Allergy 2001; 56:862–870.
- Ebo DG, Venemalm L, Bridts CH, et al. Immunoglobulin E antibodies to rocuronium: a new diagnostic tool. Anesthesiology 2007; 107:253–259.
- Ebo DG, Bridts CH, Stevens WJ. IgE-mediated anaphylaxis from chlorhexidine: diagnostic possibilities. Contact Dermatitis 2006; 55:301–302.
- deShazo RD, Mather P, Grant W, et al. Evaluation of patients with local reactions to insulin with skin tests and in vitro techniques. Diabetes Care 1987; 10:330–336.
- Sakaguchi M, Ogura H, Inouye S. IgE antibody to gelatin in children with immediate-type reactions to measles and mumps vaccines. J Allergy Clin Immunol 1995; 96:563–565.
- Raulf-Heimsoth M, Rihs HP, Rozynek P, et al. Quantitative analysis of immunoglobulin E reactivity profiles in patients allergic or sensitized to natural rubber latex (Hevea brasiliensis). Clin Exp Allergy 2007; 37:1657–1667.
- Biagini RE, MacKenzie BA, Sammons DL, et al. Latex specific IgE: performance characteristics of the IMMULITE 2000 3gAllergy assay compared with skin testing. Ann Allergy Asthma Immunol 2006; 97:196–202.
- Hamilton RG, Peterson EL, Ownby DR. Clinical and laboratory-based methods in the diagnosis of natural rubber latex allergy. J Allergy Clin Immunol 2002; 110(suppl 2):S47–S56.
- Safadi GS, Corey EC, Taylor JS, Wagner WO, Pien LC, Melton AL. Latex hypersensitivity in emergency medical service providers. Ann Allergy Asthma Immunol 1996; 77:39–42.
- Moffitt JE, Golden DB, Reisman RE, et al. Stinging insect hypersensitivity: a practice parameter update. J Allergy Clin Immunol 2004; 114:869–886.
- Biló BM, Rueff F, Mosbech H, Bonifazi F, Oude-Elberink JN; EAACI Interest Group on Insect Venom Hypersensitivity. Diagnosis of Hymenoptera venom allergy. Allergy 2005; 60:1339–1349.
- Cox L, Nelson H, Lockey R, et al. Allergen immunotherapy: a practice parameter third update. J Allergy Clin Immunol 2011; 127(suppl 1):S1–S55.
- Golden DB, Kagey-Sobotka A, Norman PS, Hamilton RG, Lichtenstein LM. Insect sting allergy with negative venom skin test responses. J Allergy Clin Immunol 2001; 107:897–901.
Health care providers often need to evaluate allergic disorders such as allergic rhinoconjunctivitis, asthma, and allergies to foods, drugs, latex, and venom, both in the hospital and in the clinic.
Unfortunately, some symptoms, such as chronic nasal symptoms, can occur in both allergic and nonallergic disorders, and this overlap can confound the diagnosis and therapy. Studies suggest that when clinicians use the history and physical examination alone in evaluating possible allergic disease, the accuracy of their diagnoses rarely exceeds 50%.1
Blood tests are now available that measure immunoglobulin E (IgE) directed against specific antigens. These in vitro tests can be important tools in assessing a patient whose history suggests an allergic disease.2 However, neither allergy skin testing nor these blood tests are intended to be used for screening: they may be most useful as confirmatory diagnostic tests in cases in which the pretest clinical impression of allergic disease is high.
ALLERGY IS MEDIATED BY IgE
In susceptible people, IgE is produced by B cells in response to specific antigens such as foods, pollens, latex, and drugs. This antigen-specific (or allergen-specific) IgE circulates in the serum and binds to high-affinity IgE receptors on immune effector cells such as mast cells located throughout the body.
Upon subsequent exposure to the same allergen, IgE receptors cross-link and initiate downstream signaling events that trigger mast cell degranulation and an immediate allergic response—hence the term immediate (or Gell-Coombs type I) hypersensitivity.3
Common manifestations of type I hypersensitivity reactions include signs and symptoms that can be:
- Cutaneous (eg, acute urticaria, angioedema)
- Respiratory (eg, acute bronchospasm, rhinoconjunctivitis)
- Cardiovascular (eg, tachycardia, hypotension)
- Gastrointestinal (eg, vomiting, diarrhea)
- Generalized (eg, anaphylactic shock). By definition, anaphylaxis is a life-threatening reaction that occurs on exposure to an allergen and involves acute respiratory distress, cardiovascular failure, or involvement of two or more organ systems.4
MOST IgE BLOOD TESTS ARE IMMUNOASSAYS
The blood tests for allergic disease are immunoassays that measure the level of IgE specific to a particular allergen. The tests can be used to evaluate sensitivity to various allergens, for example, to common inhalants such as dust mites and pollens and to foods, drugs, venom, and latex.
Types of immunoassays include enzyme-linked immunosorbent assays (ELISAs), fluorescent enzyme immunoassays (FEIAs), and radioallergosorbent assays (RASTs). At present, most commercial laboratories use one of three autoanalyzer systems to measure specific IgE:
- ImmunoCAP (Phadia AB, Uppsala, Sweden)
- Immulite (Siemens AG, Berlin, Germany)
- HYTEC-288 (Hycor/Agilent, Garden Grove, CA).
These systems use a solid-phase polymer (cellulose or avidin) in which the antigen is embedded. The polymer also facilitates binding of IgE and, therefore, increases the sensitivity of the test.5 Specific IgE from the patient’s serum binds to the allergen embedded in the polymer, and then unbound antibodies are washed off.
Despite the term “RAST,” these systems do not use radiation. A fluorescent antibody is added that binds to the patient’s IgE, and the amount of IgE present is calculated from the amount of fluorescence.6 Results are reported in kilounits of antibody per liter (kU/L) or nanograms per milliliter (ng/mL).5–7
INTERPRETATION IS INDIVIDUALIZED
In general, the sensitivity of these tests ranges from 60% to 95% and their specificity from 30% to 95%, with a concordance among different immunoassays of 75% to 90%.8
Levels of IgE for a particular allergen are also divided into semiquantitative classes, from class I to class V or VI. In general, class I and class II correlate with a low level of allergen sensitization and, often, with a low likelihood of a clinical reaction. On the other hand, classes V and VI reflect higher degrees of sensitization and generally correlate with IgE-mediated clinical reactions upon allergen exposure.
The interpretation of a positive (ie, “nonzero”) test result must be individualized on the basis of clinical presentation and risk factors. A specialist can make an important contribution by helping to interpret any positive test result or a negative test result that does not correlate with the patient’s history.
ADVANTAGES OF ALLERGY BLOOD TESTING
Allergy blood testing is convenient, since it involves only a standard blood draw.
In theory, allergy blood testing may be safer, since it does not expose the patient to any allergens. On the other hand, many patients experience bruising from venipuncture performed for any reason: 16% in one survey.9 In another survey,10 adverse reactions of any type occurred in 0.49% of patients undergoing venipuncture but only in 0.04% of those undergoing allergy skin testing. Therefore, allergy blood testing may be most appropriate in situations in which a patient’s history suggests that he or she may be at risk of a systemic reaction from a traditional skin test or in cases in which skin testing is not possible (eg, extensive eczema).
Another advantage of allergy blood testing is that it is not affected by drugs such as antihistamines or tricyclic antidepressants that suppress the histamine response, which is a problem with skin testing.
Allergy blood testing may also be useful in patients on long-term glucocorticoid therapy, although the data conflict. Prolonged oral glucocorticoid use is associated with a decrease in mast cell density and histamine content in the skin,11,12 although in one study a corticosteroid was found not to affect the results of skin-prick testing for allergy.13 Thus, allergy blood testing can be performed in patients who have severe eczema or dermatographism or who cannot safely suspend taking antihistamines or tricyclic antidepressants.
LIMITATIONS OF THESE TESTS
A limitation of allergy blood tests is that there is no gold-standard test for many allergic conditions. (Double-blind, placebo-controlled oral food challenge testing has been proposed as the gold-standard test for food allergy, and nasal allergen provocation challenge has been proposed for allergic rhinitis.)
Also, allergy blood tests can give false-positive results because of nonspecific binding of antibody in the assay.
Of note: evidence of sensitization to a particular allergen (ie, a positive blood test result) is not synonymous with clinically relevant disease (ie, clinical sensitivity).
Conversely, these tests can give false-negative results in patients who have true IgE-mediated disease as confirmed by skin testing or allergen challenge. The sensitivity of blood allergy testing is approximately 25% to 30% lower than that of skin testing, based on comparative studies.2 The blood tests are usually considered positive if the allergen-specific IgE level is greater than 0.35 kU/L; however, sensitization to certain inhalant allergens can occur at levels as low as 0.12 kU/L.14
Specific IgE levels measured by different commercial assays are not always interchangeable or equivalent, so a clinician should consistently select the same immunoassay if possible when assessing any given patient over time.15
Levels of specific IgE have been shown to depend on age, allergen specificity, total serum IgE, and, with inhalant allergens, the season of the year.15,16
Other limitations of blood testing are its cost and a delay of several days to a week in obtaining the results.17
WHEN TO ORDER ALLERGY BLOOD TESTING
The allergy evaluation should begin with a thorough history to look for possible triggers for the patient’s symptoms.
For example, respiratory conditions such as asthma and rhinitis may be exacerbated during particular times of the year when certain pollens are commonly present. For patients with this pattern, blood testing for allergy to common inhalants, including pollens, may be appropriate. Similarly, peanut allergy evaluation is indicated for a child who has suffered an anaphylactic reaction after consuming peanut butter. Blood testing is also indicated in patients with a history of venom anaphylaxis, especially if venom skin testing was negative.
In cases in which the patient does not have a clear history of sensitization, blood testing for allergy to multiple foods may find evidence of sensitization that does not necessarily correlate with clinical disease.18
Likewise, blood tests are not likely to be clinically relevant in conditions not mediated by IgE, such as food intolerances (eg, lactose intolerance), celiac disease, the DRESS syndrome (drug rash, eosinophilia, and systemic symptoms), Stevens-Johnson syndrome, toxic epidermal necrolysis, or other types of drug hypersensitivity reactions, such as serum sickness.3
INTERPRETING COMMONLY ORDERED BLOOD TESTS FOR ALLERGY
Tests for allergy to hundreds of substances are available.
Foods
Milk, eggs, soy, wheat, peanuts, tree nuts, fish, and shellfish account for most cases of food allergy in the United States.18
IgE-mediated hypersensitivity to milk, eggs, and peanuts tends to be more common in children, whereas peanuts, tree nuts, fish, and shellfish are more commonly associated with reactions in adults.18 Children are more likely to outgrow allergy to milk, soy, wheat, and eggs than allergy to peanuts, tree nuts, fish, and shellfish—only about 20% of children outgrow peanut allergy.18
Patients with an IgE-mediated reaction to foods should be closely followed by a specialist, who can best help determine the appropriateness of additional testing (such as an oral challenge under observation), avoidance recommendations, and the introduction of foods back into the diet.19
Specific IgE tests for allergy to a variety of foods are available and can be very useful for diagnosis when used in the appropriate setting.
One caveat about these studies is that many were initially performed in children with a history of food allergy, many of whom had atopic dermatitis, and the findings have not been systematically reexamined in larger studies in more heterogeneous populations.
For example, at least eight studies tried to identify a diagnostic IgE level for cow’s milk allergy. The 95% confidence intervals varied widely, depending on the study design, the age of the study population, the prevalence of food allergy in the population, and the statistical method used for analysis.5 For most other foods for which blood tests are available, few studies have been performed to establish predictive values similar to those in Table 1.
Thus, slight elevations in antigen-specific IgE (> 0.35 kU/L) may correlate only with in vitro sensitization in a patient who has no clinical reactivity upon oral exposure to a particular antigen.
Broad food panels have been shown to have false-positive rates higher than 50%—ie, in more than half of cases, positive results have no clinical relevance. Therefore, these large food panels should not be used for screening.19 Instead, it is recommended that tests be limited to relevant foods based on the patient’s history when evaluating symptoms consistent with an IgE-mediated reaction to a particular food.
Food-specific IgE evaluation is also not helpful in evaluating non-IgE adverse reactions to foods (eg, intolerances).
Therefore, the patient’s history remains the most important tool for evaluation of food allergy. In cases in which the patient’s history suggests a food-associated IgE-mediated reaction and the blood test is negative, the patient should be referred to a specialist for skin testing with commercial extracts or even fresh food extracts, given the higher sensitivity of in vivo testing.20
Inhalants
Common aeroallergens associated with allergic rhinitis, allergic conjunctivitis, and allergic asthma include dust mites, animal dander, cockroach debris, molds, trees, grasses, weeds, and ragweed. Dust mites, animal dander, and mold spores are perennial allergens and may trigger symptoms year-round. Pollen, including pollen from trees, grasses, and weeds, is generally present in a seasonal pattern in many parts of the United States.
A positive blood test for an inhalant allergen can reinforce the physician’s clinical impression in making a diagnosis of allergic rhinoconjunctivitis. Interestingly, studies have suggested a high rate of false-positives based on history alone when in vivo and in vitro allergy testing were negative for IgE-mediated respiratory disease.21
Various studies have aimed to establish threshold values of aeroallergen-specific IgE that predict the likelihood of clinically relevant disease. Unfortunately, other factors also contribute to clinical symptoms of rhinoconjunctivitis; these include concurrent inflammation, infection, physical stress, psychological stress, exposure to irritants, and hormonal changes. These factors introduce variability and make specific IgE cutoffs for inhalant allergens unreliable.22
Prospective studies have suggested that skin testing correlates better with nasal allergen challenge (the gold standard) than blood testing for the diagnosis of inhalant allergy, though more recent studies using modern technologies demonstrate reasonable concordance (67%) between skin testing and blood testing (specifically, ImmunoCAP).23,24 According to current guidelines, skin tests are the preferred method for diagnosing IgE-mediated sensitivity to inhalants.25
Compared with skin prick tests as the gold standard, the sensitivity of specific IgE immunoassays is approximately 70% to 75%.25 Nevertheless, specific IgE values greater than 0.35 kU/L are generally considered positive for aeroallergen sensitization, although lower levels of dog-specific IgE have recently been shown to correlate with clinical disease.14
Drugs, including penicillins
A variety of clinical reactions can occur in response to oral, intravenous, or topical medications.
At present, blood tests are available for the evaluation of IgE-mediated adverse reactions to only a limited number of drugs. Reactions involving other mechanisms, such as those related to the drug’s metabolism, intolerances (eg, nausea), idiosyncratic reactions (eg, Stevens-Johnson syndrome, the DRESS syndrome), or other types of reactions can be diagnosed only by history and physical examination.
The development of specific IgE tests for sensitivity to medications has been limited by incomplete characterization of metabolic products and the possibility that a single medication can have different epitopes or IgE binding sites in different individuals.26
In vitro allergy testing has been most studied for beta-lactam antibiotics (eg, penicillin) and not so much for other drugs.
Table 2 summarizes the sensitivity and specificity of blood allergy tests that are commercially available for drugs.
Penicillin, a beta-lactam antibiotic, is degraded into various metabolites known as the major determinant (penicilloyl) and the minor determinants (eg, benzylpenicilloate and benzylpenilloate), which act as haptens. Specific IgE testing is not available for all these determinants.
The sensitivity of blood tests for allergy to penicilloyl (penicillin) and amino-penicillins such as amoxicilloyl (amoxicillin) is reported as between 32% and 50%, and the specificity as 96% to 98%.29
By definition, any nonzero level of IgE specific for penicillin or its derivatives is considered a positive result and may be associated with a higher risk of IgE-mediated reaction to penicillins. However, in a situation analogous to that in people with food allergy who have a food-specific IgE titer lower than the empirically established threshold value (Table 1), low-titer values to penicillin may not predict anaphylactic sensitivity in a penicillin oral challenge.28 Further studies are needed to determine if there is a threshold level of penicillin-specific IgE above which a patient has a higher likelihood of an IgE-mediated systemic reaction.
Other drugs. Specific IgE blood tests are also available for certain neuromuscular agents, insulin, cefaclor (Ceclor), chlorhexidine (contained in various antiseptic products), and gelatin (Table 2). These substances have not been as well studied as penicillins, and the sensitivity and specificity data reported in Table 2 are limited by few studies and small study sizes.
Neuromuscular blocking agents. Tests for IgE against neuromuscular blocking agents are reported to have low sensitivity (30%–60%) using a cutoff value of 0.35 kU/L.30 In small studies, the sensitivity was higher (68% to 92%) when threshold values for rocuronium-specific IgE were lowered from 0.35 to 0.13 kU/L.29
Chlorhexidine, an antiseptic commonly used in surgery, has been linked to IgE-mediated reactions.31 Chlorhexidine-specific IgE levels greater than 0.35 kU/L are considered positive, based on very limited data.
Insulin. Blood tests for allergy to insulin are also commercially available. However, studies have shown a significant overlap in the range of insulin-specific IgE in patients with a clinical history consistent with insulin allergy and in controls. Therefore, this test has a very limited ability to distinguish people who do not have a history of a reaction to insulin.32 More research is needed to determine the clinical utility of insulin-specific IgE testing.
Gelatin. IgE-mediated reactions have occurred after exposure to gelatin (from either cows or pigs) contained in foods and vaccines, including measles-mumps-rubella and yellow fever. One study identified gelatin-specific IgE in 10 of 11 children with a history of systemic reaction to measles or mumps vaccine.33 In the same study, gelatin-specific IgE levels were negative in 24 children who had developed non-IgE-mediated reactions to the vaccine.33
Tests for IgE against bovine gelatin are commercially available; results are considered positive for values higher than 0.35 kU/L. A negative test result does not exclude the possibility of an allergic reaction to porcine gelatin, which can also be found in foods and vaccines, but tests for anti-porcine gelatin IgE are not commercially available.
Latex
Latex, obtained from the rubber tree Hevea brasiliensis, has 13 known polypeptides (allergens Hev b 1–13) that cause IgE-mediated reactions, particularly in health care workers and patients with spina bifida.34 Overall, the incidence of latex allergy has decreased in the United States as most medical institutions have implemented a latex-free environment.
In vitro testing is the only mode of evaluation for allergy to latex approved by the US Food and Drug Administration (FDA).35 Its sensitivity is 80% and its specificity is 95%.36
In a 2007 study, 145 people at risk for latex allergy, including 104 health care workers, 31 patients with spina bifida, and 10 patients requiring multiple surgeries, underwent latex-specific IgE analysis for sensitivity to various recombinant and native latex allergens.34 The three groups differed in their latex allergy profiles, highlighting the diversity of clinical response to latex in high-risk groups and our current inability to establish specific cutoff points for quantitative latex-specific IgE. Thus, at present, any nonzero latex-specific IgE value is considered positive.
A formal evaluation for allergy is recommended for patients who have a strong history of an IgE-mediated reaction to latex and a latex-specific IgE value of zero. Blood tests for allergy to some native or recombinant latex allergens are available; these allergens may be underrepresented in the native total latex extract.33 Skin testing for allergy to latex, although not FDA-approved or standardized, can also be useful in this setting.37
Insect venom
Type I hypersensitivity reactions can occur from the stings of Vespidae (vespids), Apidae (bees), and Formicidae (fire ants). Large localized reactions after an insect sting are not infrequent and typically do not predict anaphylactic sensitivity with future stings, even though they are considered mild IgE-mediated reactions. However, systemic reactions are considered life-threatening and warrant allergy testing.38
The level of venom-specific IgE usually increases weeks to months after a sting.39 Therefore, blood tests can be falsely negative if performed within a short time of the sting.
Patients who have suffered a systemic reaction to venom and have evidence of sensitization by either in vitro or in vivo allergy testing are candidates for venom immunotherapy.40
At present, any nonzero venom-specific IgE test is considered positive, as there is no specific value for venom-specific IgE that predicts clinical risk.
A negative blood test does not exclude the possibility of an IgE-mediated reaction.41 In cases in which a patient has a clinical history compatible with venom allergy but the blood test is negative, the patient should be referred to an allergist for further evaluation, including venom skin testing and possibly repeat blood testing at a later time.
Conversely, specific IgE testing to venom is recommended when a patient has a history consistent with venom allergy and negative skin test results.38
As mentioned previously, in vitro test performance can vary with the laboratory and testing method used, and sending samples directly to a reference laboratory could be considered.41
TESTING FOR IgG AGAINST FOODS IS UNVALIDATED AND INAPPROPRIATE
In recent years, some practitioners of alternative medicine have started testing for allergen-specific IgG or IgG4 as part of evaluations for hypersensitivity, especially in cases in which patients describe atypical gastrointestinal, neurologic, or other symptoms after eating specific foods.19
However, this testing often finds IgG or IgG4 against foods that are well tolerated. At present, allergen-specific IgG testing lacks scientific evidence to support its clinical use in the evaluation of allergic disease.5,19
Health care providers often need to evaluate allergic disorders such as allergic rhinoconjunctivitis, asthma, and allergies to foods, drugs, latex, and venom, both in the hospital and in the clinic.
Unfortunately, some symptoms, such as chronic nasal symptoms, can occur in both allergic and nonallergic disorders, and this overlap can confound the diagnosis and therapy. Studies suggest that when clinicians use the history and physical examination alone in evaluating possible allergic disease, the accuracy of their diagnoses rarely exceeds 50%.1
Blood tests are now available that measure immunoglobulin E (IgE) directed against specific antigens. These in vitro tests can be important tools in assessing a patient whose history suggests an allergic disease.2 However, neither allergy skin testing nor these blood tests are intended to be used for screening: they may be most useful as confirmatory diagnostic tests in cases in which the pretest clinical impression of allergic disease is high.
ALLERGY IS MEDIATED BY IgE
In susceptible people, IgE is produced by B cells in response to specific antigens such as foods, pollens, latex, and drugs. This antigen-specific (or allergen-specific) IgE circulates in the serum and binds to high-affinity IgE receptors on immune effector cells such as mast cells located throughout the body.
Upon subsequent exposure to the same allergen, IgE receptors cross-link and initiate downstream signaling events that trigger mast cell degranulation and an immediate allergic response—hence the term immediate (or Gell-Coombs type I) hypersensitivity.3
Common manifestations of type I hypersensitivity reactions include signs and symptoms that can be:
- Cutaneous (eg, acute urticaria, angioedema)
- Respiratory (eg, acute bronchospasm, rhinoconjunctivitis)
- Cardiovascular (eg, tachycardia, hypotension)
- Gastrointestinal (eg, vomiting, diarrhea)
- Generalized (eg, anaphylactic shock). By definition, anaphylaxis is a life-threatening reaction that occurs on exposure to an allergen and involves acute respiratory distress, cardiovascular failure, or involvement of two or more organ systems.4
MOST IgE BLOOD TESTS ARE IMMUNOASSAYS
The blood tests for allergic disease are immunoassays that measure the level of IgE specific to a particular allergen. The tests can be used to evaluate sensitivity to various allergens, for example, to common inhalants such as dust mites and pollens and to foods, drugs, venom, and latex.
Types of immunoassays include enzyme-linked immunosorbent assays (ELISAs), fluorescent enzyme immunoassays (FEIAs), and radioallergosorbent assays (RASTs). At present, most commercial laboratories use one of three autoanalyzer systems to measure specific IgE:
- ImmunoCAP (Phadia AB, Uppsala, Sweden)
- Immulite (Siemens AG, Berlin, Germany)
- HYTEC-288 (Hycor/Agilent, Garden Grove, CA).
These systems use a solid-phase polymer (cellulose or avidin) in which the antigen is embedded. The polymer also facilitates binding of IgE and, therefore, increases the sensitivity of the test.5 Specific IgE from the patient’s serum binds to the allergen embedded in the polymer, and then unbound antibodies are washed off.
Despite the term “RAST,” these systems do not use radiation. A fluorescent antibody is added that binds to the patient’s IgE, and the amount of IgE present is calculated from the amount of fluorescence.6 Results are reported in kilounits of antibody per liter (kU/L) or nanograms per milliliter (ng/mL).5–7
INTERPRETATION IS INDIVIDUALIZED
In general, the sensitivity of these tests ranges from 60% to 95% and their specificity from 30% to 95%, with a concordance among different immunoassays of 75% to 90%.8
Levels of IgE for a particular allergen are also divided into semiquantitative classes, from class I to class V or VI. In general, class I and class II correlate with a low level of allergen sensitization and, often, with a low likelihood of a clinical reaction. On the other hand, classes V and VI reflect higher degrees of sensitization and generally correlate with IgE-mediated clinical reactions upon allergen exposure.
The interpretation of a positive (ie, “nonzero”) test result must be individualized on the basis of clinical presentation and risk factors. A specialist can make an important contribution by helping to interpret any positive test result or a negative test result that does not correlate with the patient’s history.
ADVANTAGES OF ALLERGY BLOOD TESTING
Allergy blood testing is convenient, since it involves only a standard blood draw.
In theory, allergy blood testing may be safer, since it does not expose the patient to any allergens. On the other hand, many patients experience bruising from venipuncture performed for any reason: 16% in one survey.9 In another survey,10 adverse reactions of any type occurred in 0.49% of patients undergoing venipuncture but only in 0.04% of those undergoing allergy skin testing. Therefore, allergy blood testing may be most appropriate in situations in which a patient’s history suggests that he or she may be at risk of a systemic reaction from a traditional skin test or in cases in which skin testing is not possible (eg, extensive eczema).
Another advantage of allergy blood testing is that it is not affected by drugs such as antihistamines or tricyclic antidepressants that suppress the histamine response, which is a problem with skin testing.
Allergy blood testing may also be useful in patients on long-term glucocorticoid therapy, although the data conflict. Prolonged oral glucocorticoid use is associated with a decrease in mast cell density and histamine content in the skin,11,12 although in one study a corticosteroid was found not to affect the results of skin-prick testing for allergy.13 Thus, allergy blood testing can be performed in patients who have severe eczema or dermatographism or who cannot safely suspend taking antihistamines or tricyclic antidepressants.
LIMITATIONS OF THESE TESTS
A limitation of allergy blood tests is that there is no gold-standard test for many allergic conditions. (Double-blind, placebo-controlled oral food challenge testing has been proposed as the gold-standard test for food allergy, and nasal allergen provocation challenge has been proposed for allergic rhinitis.)
Also, allergy blood tests can give false-positive results because of nonspecific binding of antibody in the assay.
Of note: evidence of sensitization to a particular allergen (ie, a positive blood test result) is not synonymous with clinically relevant disease (ie, clinical sensitivity).
Conversely, these tests can give false-negative results in patients who have true IgE-mediated disease as confirmed by skin testing or allergen challenge. The sensitivity of blood allergy testing is approximately 25% to 30% lower than that of skin testing, based on comparative studies.2 The blood tests are usually considered positive if the allergen-specific IgE level is greater than 0.35 kU/L; however, sensitization to certain inhalant allergens can occur at levels as low as 0.12 kU/L.14
Specific IgE levels measured by different commercial assays are not always interchangeable or equivalent, so a clinician should consistently select the same immunoassay if possible when assessing any given patient over time.15
Levels of specific IgE have been shown to depend on age, allergen specificity, total serum IgE, and, with inhalant allergens, the season of the year.15,16
Other limitations of blood testing are its cost and a delay of several days to a week in obtaining the results.17
WHEN TO ORDER ALLERGY BLOOD TESTING
The allergy evaluation should begin with a thorough history to look for possible triggers for the patient’s symptoms.
For example, respiratory conditions such as asthma and rhinitis may be exacerbated during particular times of the year when certain pollens are commonly present. For patients with this pattern, blood testing for allergy to common inhalants, including pollens, may be appropriate. Similarly, peanut allergy evaluation is indicated for a child who has suffered an anaphylactic reaction after consuming peanut butter. Blood testing is also indicated in patients with a history of venom anaphylaxis, especially if venom skin testing was negative.
In cases in which the patient does not have a clear history of sensitization, blood testing for allergy to multiple foods may find evidence of sensitization that does not necessarily correlate with clinical disease.18
Likewise, blood tests are not likely to be clinically relevant in conditions not mediated by IgE, such as food intolerances (eg, lactose intolerance), celiac disease, the DRESS syndrome (drug rash, eosinophilia, and systemic symptoms), Stevens-Johnson syndrome, toxic epidermal necrolysis, or other types of drug hypersensitivity reactions, such as serum sickness.3
INTERPRETING COMMONLY ORDERED BLOOD TESTS FOR ALLERGY
Tests for allergy to hundreds of substances are available.
Foods
Milk, eggs, soy, wheat, peanuts, tree nuts, fish, and shellfish account for most cases of food allergy in the United States.18
IgE-mediated hypersensitivity to milk, eggs, and peanuts tends to be more common in children, whereas peanuts, tree nuts, fish, and shellfish are more commonly associated with reactions in adults.18 Children are more likely to outgrow allergy to milk, soy, wheat, and eggs than allergy to peanuts, tree nuts, fish, and shellfish—only about 20% of children outgrow peanut allergy.18
Patients with an IgE-mediated reaction to foods should be closely followed by a specialist, who can best help determine the appropriateness of additional testing (such as an oral challenge under observation), avoidance recommendations, and the introduction of foods back into the diet.19
Specific IgE tests for allergy to a variety of foods are available and can be very useful for diagnosis when used in the appropriate setting.
One caveat about these studies is that many were initially performed in children with a history of food allergy, many of whom had atopic dermatitis, and the findings have not been systematically reexamined in larger studies in more heterogeneous populations.
For example, at least eight studies tried to identify a diagnostic IgE level for cow’s milk allergy. The 95% confidence intervals varied widely, depending on the study design, the age of the study population, the prevalence of food allergy in the population, and the statistical method used for analysis.5 For most other foods for which blood tests are available, few studies have been performed to establish predictive values similar to those in Table 1.
Thus, slight elevations in antigen-specific IgE (> 0.35 kU/L) may correlate only with in vitro sensitization in a patient who has no clinical reactivity upon oral exposure to a particular antigen.
Broad food panels have been shown to have false-positive rates higher than 50%—ie, in more than half of cases, positive results have no clinical relevance. Therefore, these large food panels should not be used for screening.19 Instead, it is recommended that tests be limited to relevant foods based on the patient’s history when evaluating symptoms consistent with an IgE-mediated reaction to a particular food.
Food-specific IgE evaluation is also not helpful in evaluating non-IgE adverse reactions to foods (eg, intolerances).
Therefore, the patient’s history remains the most important tool for evaluation of food allergy. In cases in which the patient’s history suggests a food-associated IgE-mediated reaction and the blood test is negative, the patient should be referred to a specialist for skin testing with commercial extracts or even fresh food extracts, given the higher sensitivity of in vivo testing.20
Inhalants
Common aeroallergens associated with allergic rhinitis, allergic conjunctivitis, and allergic asthma include dust mites, animal dander, cockroach debris, molds, trees, grasses, weeds, and ragweed. Dust mites, animal dander, and mold spores are perennial allergens and may trigger symptoms year-round. Pollen, including pollen from trees, grasses, and weeds, is generally present in a seasonal pattern in many parts of the United States.
A positive blood test for an inhalant allergen can reinforce the physician’s clinical impression in making a diagnosis of allergic rhinoconjunctivitis. Interestingly, studies have suggested a high rate of false-positives based on history alone when in vivo and in vitro allergy testing were negative for IgE-mediated respiratory disease.21
Various studies have aimed to establish threshold values of aeroallergen-specific IgE that predict the likelihood of clinically relevant disease. Unfortunately, other factors also contribute to clinical symptoms of rhinoconjunctivitis; these include concurrent inflammation, infection, physical stress, psychological stress, exposure to irritants, and hormonal changes. These factors introduce variability and make specific IgE cutoffs for inhalant allergens unreliable.22
Prospective studies have suggested that skin testing correlates better with nasal allergen challenge (the gold standard) than blood testing for the diagnosis of inhalant allergy, though more recent studies using modern technologies demonstrate reasonable concordance (67%) between skin testing and blood testing (specifically, ImmunoCAP).23,24 According to current guidelines, skin tests are the preferred method for diagnosing IgE-mediated sensitivity to inhalants.25
Compared with skin prick tests as the gold standard, the sensitivity of specific IgE immunoassays is approximately 70% to 75%.25 Nevertheless, specific IgE values greater than 0.35 kU/L are generally considered positive for aeroallergen sensitization, although lower levels of dog-specific IgE have recently been shown to correlate with clinical disease.14
Drugs, including penicillins
A variety of clinical reactions can occur in response to oral, intravenous, or topical medications.
At present, blood tests are available for the evaluation of IgE-mediated adverse reactions to only a limited number of drugs. Reactions involving other mechanisms, such as those related to the drug’s metabolism, intolerances (eg, nausea), idiosyncratic reactions (eg, Stevens-Johnson syndrome, the DRESS syndrome), or other types of reactions can be diagnosed only by history and physical examination.
The development of specific IgE tests for sensitivity to medications has been limited by incomplete characterization of metabolic products and the possibility that a single medication can have different epitopes or IgE binding sites in different individuals.26
In vitro allergy testing has been most studied for beta-lactam antibiotics (eg, penicillin) and not so much for other drugs.
Table 2 summarizes the sensitivity and specificity of blood allergy tests that are commercially available for drugs.
Penicillin, a beta-lactam antibiotic, is degraded into various metabolites known as the major determinant (penicilloyl) and the minor determinants (eg, benzylpenicilloate and benzylpenilloate), which act as haptens. Specific IgE testing is not available for all these determinants.
The sensitivity of blood tests for allergy to penicilloyl (penicillin) and amino-penicillins such as amoxicilloyl (amoxicillin) is reported as between 32% and 50%, and the specificity as 96% to 98%.29
By definition, any nonzero level of IgE specific for penicillin or its derivatives is considered a positive result and may be associated with a higher risk of IgE-mediated reaction to penicillins. However, in a situation analogous to that in people with food allergy who have a food-specific IgE titer lower than the empirically established threshold value (Table 1), low-titer values to penicillin may not predict anaphylactic sensitivity in a penicillin oral challenge.28 Further studies are needed to determine if there is a threshold level of penicillin-specific IgE above which a patient has a higher likelihood of an IgE-mediated systemic reaction.
Other drugs. Specific IgE blood tests are also available for certain neuromuscular agents, insulin, cefaclor (Ceclor), chlorhexidine (contained in various antiseptic products), and gelatin (Table 2). These substances have not been as well studied as penicillins, and the sensitivity and specificity data reported in Table 2 are limited by few studies and small study sizes.
Neuromuscular blocking agents. Tests for IgE against neuromuscular blocking agents are reported to have low sensitivity (30%–60%) using a cutoff value of 0.35 kU/L.30 In small studies, the sensitivity was higher (68% to 92%) when threshold values for rocuronium-specific IgE were lowered from 0.35 to 0.13 kU/L.29
Chlorhexidine, an antiseptic commonly used in surgery, has been linked to IgE-mediated reactions.31 Chlorhexidine-specific IgE levels greater than 0.35 kU/L are considered positive, based on very limited data.
Insulin. Blood tests for allergy to insulin are also commercially available. However, studies have shown a significant overlap in the range of insulin-specific IgE in patients with a clinical history consistent with insulin allergy and in controls. Therefore, this test has a very limited ability to distinguish people who do not have a history of a reaction to insulin.32 More research is needed to determine the clinical utility of insulin-specific IgE testing.
Gelatin. IgE-mediated reactions have occurred after exposure to gelatin (from either cows or pigs) contained in foods and vaccines, including measles-mumps-rubella and yellow fever. One study identified gelatin-specific IgE in 10 of 11 children with a history of systemic reaction to measles or mumps vaccine.33 In the same study, gelatin-specific IgE levels were negative in 24 children who had developed non-IgE-mediated reactions to the vaccine.33
Tests for IgE against bovine gelatin are commercially available; results are considered positive for values higher than 0.35 kU/L. A negative test result does not exclude the possibility of an allergic reaction to porcine gelatin, which can also be found in foods and vaccines, but tests for anti-porcine gelatin IgE are not commercially available.
Latex
Latex, obtained from the rubber tree Hevea brasiliensis, has 13 known polypeptides (allergens Hev b 1–13) that cause IgE-mediated reactions, particularly in health care workers and patients with spina bifida.34 Overall, the incidence of latex allergy has decreased in the United States as most medical institutions have implemented a latex-free environment.
In vitro testing is the only mode of evaluation for allergy to latex approved by the US Food and Drug Administration (FDA).35 Its sensitivity is 80% and its specificity is 95%.36
In a 2007 study, 145 people at risk for latex allergy, including 104 health care workers, 31 patients with spina bifida, and 10 patients requiring multiple surgeries, underwent latex-specific IgE analysis for sensitivity to various recombinant and native latex allergens.34 The three groups differed in their latex allergy profiles, highlighting the diversity of clinical response to latex in high-risk groups and our current inability to establish specific cutoff points for quantitative latex-specific IgE. Thus, at present, any nonzero latex-specific IgE value is considered positive.
A formal evaluation for allergy is recommended for patients who have a strong history of an IgE-mediated reaction to latex and a latex-specific IgE value of zero. Blood tests for allergy to some native or recombinant latex allergens are available; these allergens may be underrepresented in the native total latex extract.33 Skin testing for allergy to latex, although not FDA-approved or standardized, can also be useful in this setting.37
Insect venom
Type I hypersensitivity reactions can occur from the stings of Vespidae (vespids), Apidae (bees), and Formicidae (fire ants). Large localized reactions after an insect sting are not infrequent and typically do not predict anaphylactic sensitivity with future stings, even though they are considered mild IgE-mediated reactions. However, systemic reactions are considered life-threatening and warrant allergy testing.38
The level of venom-specific IgE usually increases weeks to months after a sting.39 Therefore, blood tests can be falsely negative if performed within a short time of the sting.
Patients who have suffered a systemic reaction to venom and have evidence of sensitization by either in vitro or in vivo allergy testing are candidates for venom immunotherapy.40
At present, any nonzero venom-specific IgE test is considered positive, as there is no specific value for venom-specific IgE that predicts clinical risk.
A negative blood test does not exclude the possibility of an IgE-mediated reaction.41 In cases in which a patient has a clinical history compatible with venom allergy but the blood test is negative, the patient should be referred to an allergist for further evaluation, including venom skin testing and possibly repeat blood testing at a later time.
Conversely, specific IgE testing to venom is recommended when a patient has a history consistent with venom allergy and negative skin test results.38
As mentioned previously, in vitro test performance can vary with the laboratory and testing method used, and sending samples directly to a reference laboratory could be considered.41
TESTING FOR IgG AGAINST FOODS IS UNVALIDATED AND INAPPROPRIATE
In recent years, some practitioners of alternative medicine have started testing for allergen-specific IgG or IgG4 as part of evaluations for hypersensitivity, especially in cases in which patients describe atypical gastrointestinal, neurologic, or other symptoms after eating specific foods.19
However, this testing often finds IgG or IgG4 against foods that are well tolerated. At present, allergen-specific IgG testing lacks scientific evidence to support its clinical use in the evaluation of allergic disease.5,19
- Williams PB, Ahlstedt S, Barnes JH, Söderström L, Portnoy J. Are our impressions of allergy test performances correct? Ann Allergy Asthma Immunol 2003; 91:26–33.
- Bernstein IL, Li JT, Bernstein DI, et al; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology. Allergy diagnostic testing: an updated practice parameter. Ann Allergy Asthma Immunol 2008; 100(suppl 3):S1–S148.
- Pichler WJ. Immune mechanism of drug hypersensitivity. Immunol Allergy Clin North Am 2004; 24:373–397.
- Lieberman P, Nicklas RA, Oppenheimer J, et al. The diagnosis and management of anaphylaxis practice parameter: 2010 update. J Allergy Clin Immunol 2010; 126:477–480.
- Hamilton RG. Clinical laboratory assessment of immediate-type hypersensitivity. J Allergy Clin Immunol 2010; 125(suppl 2):S284–S296.
- Cox L, Williams B, Sicherer S, et al; American College of Allergy, Asthma and Immunology Test Task Force; American Academy of Allergy, Asthma and Immunology Specific IgE Test Task Force. Pearls and pitfalls of allergy diagnostic testing: report from the American College of Allergy, Asthma and Immunology/American Academy of Allergy, Asthma and Immunology Specific IgE Test Task Force. Ann Allergy Asthma Immunol 2008; 101:580–592.
- Hamilton RG, Franklin Adkinson N. In vitro assays for the diagnosis of IgE-mediated disorders. J Allergy Clin Immunol 2004; 114:213–225.
- Williams PB, Dolen WK, Koepke JW, Selner JC. Comparison of skin testing and three in vitro assays for specific IgE in the clinical evaluation of immediate hypersensitivity. Ann Allergy 1992; 68:35–45.
- Howanitz PJ, Cembrowski GS, Bachner P. Laboratory phlebotomy. College of American Pathologists Q-Probe study of patient satisfaction and complications in 23,783 patients. Arch Pathol Lab Med 1991; 115:867–872.
- Turkeltaub PC, Gergen PJ. The risk of adverse reactions from percutaneous prick-puncture allergen skin testing, venipuncture, and body measurements: data from the second National Health and Nutrition Examination Survey 1976–80 (NHANES II). J Allergy Clin Immunol 1989; 84:886–890.
- Pipkorn U, Hammarlund A, Enerbäck L. Prolonged treatment with topical glucocorticoids results in an inhibition of the allergen-induced weal-and-flare response and a reduction in skin mast cell numbers and histamine content. Clin Exp Allergy 1989; 19:19–25.
- Cole ZA, Clough GF, Church MK. Inhibition by glucocorticoids of the mast cell-dependent weal and flare response in human skin in vivo. Br J Pharmacol 2001; 132:286–292.
- Des Roches A, Paradis L, Bougeard YH, Godard P, Bousquet J, Chanez P. Long-term oral corticosteroid therapy does not alter the results of immediate-type allergy skin prick tests. J Allergy Clin Immunol 1996; 98:522–527.
- Linden CC, Misiak RT, Wegienka G, et al. Analysis of allergen specific IgE cut points to cat and dog in the Childhood Allergy Study. Ann Allergy Asthma Immunol 2011; 106:153–158.
- Hamilton RG, Williams PB; Specific IgE Testing Task Force of the American Academy of Allergy, Asthma & Immunology; American College of Allergy, Asthma and Immunology. Human IgE antibody serology: a primer for the practicing North American allergist/immunologist. J Allergy Clin Immunol 2010; 126:33–38.
- Somville MA, Machiels J, Gilles JG, Saint-Remy JM. Seasonal variation in specific IgE antibodies of grass-pollen hypersensitive patients depends on the steady state IgE concentration and is not related to clinical symptoms. J Allergy Clin Immunol 1989; 83( 2 Pt 1):486–494.
- Poon AW, Goodman CS, Rubin RJ. In vitro and skin testing for allergy: comparable clinical utility and costs. Am J Manag Care 1998; 4:969–985.
- Sampson HA. Update on food allergy. J Allergy Clin Immunol 2004; 113:805–819.
- Boyce JA, Assa’ad A, Burks AW, et al; NIAID-Sponsored Expert Panel. Guidelines for the diagnosis and management of food allergy in the United States: summary of the NIAID-sponsored expert panel report. J Allergy Clin Immunol 2010; 126:1105–1118.
- Rosen JP, Selcow JE, Mendelson LM, Grodofsky MP, Factor JM, Sampson HA. Skin testing with natural foods in patients suspected of having food allergies: is it a necessity? J Allergy Clin Immunol 1994; 93:1068–1070.
- Williams PB, Siegel C, Portnoy J. Efficacy of a single diagnostic test for sensitization to common inhalant allergens. Ann Allergy Asthma Immunol 2001; 86:196–202.
- Söderström L, Kober A, Ahlstedt S, et al. A further evaluation of the clinical use of specific IgE antibody testing in allergic diseases. Allergy 2003; 58:921–928.
- Bousquet J, Lebel B, Dhivert H, Bataille Y, Martinot B, Michel FB. Nasal challenge with pollen grains, skin-prick tests and specific IgE in patients with grass pollen allergy. Clin Allergy 1987; 17:529–536.
- Nepper-Christensen S, Backer V, DuBuske LM, Nolte H. In vitro diagnostic evaluation of patients with inhalant allergies: summary of probability outcomes comparing results of CLA- and CAP-specific immunoglobulin E test systems. Allergy Asthma Proc 2003; 24:253–258.
- Wallace DV, Dykewicz MS, Bernstein DI, et al; Joint Task Force on Practice; American Academy of Allergy; Asthma & Immunology; Joint Council of Allergy, Asthma and Immunology. The diagnosis and management of rhinitis: an updated practice parameter. J Allergy Clin Immunol 2008; 122(suppl 2):S1–S84.
- Mayorga C, Sanz ML, Gamboa PM, et al; Immunology Committee of the Spanish Society of Allergology and Clinical Immunology of the SEAIC. In vitro diagnosis of immediate allergic reactions to drugs: an update. J Investig Allergol Clin Immunol 2010; 20:103–109.
- Garcia JJ, Blanca M, Moreno F, et al. Determination of IgE antibodies to the benzylpenicilloyl determinant: a comparison of the sensitivity and specificity of three radio allergo sorbent test methods. J Clin Lab Anal 1997; 11:251–257.
- Macy E, Goldberg B, Poon KY. Use of commercial anti-penicillin IgE fluorometric enzyme immunoassays to diagnose penicillin allergy. Ann Allergy Asthma Immunol 2010; 105:136–141.
- Blanca M, Mayorga C, Torres MJ, et al. Clinical evaluation of Pharmacia CAP System RAST FEIA amoxicilloyl and benzylpenicilloyl in patients with penicillin allergy. Allergy 2001; 56:862–870.
- Ebo DG, Venemalm L, Bridts CH, et al. Immunoglobulin E antibodies to rocuronium: a new diagnostic tool. Anesthesiology 2007; 107:253–259.
- Ebo DG, Bridts CH, Stevens WJ. IgE-mediated anaphylaxis from chlorhexidine: diagnostic possibilities. Contact Dermatitis 2006; 55:301–302.
- deShazo RD, Mather P, Grant W, et al. Evaluation of patients with local reactions to insulin with skin tests and in vitro techniques. Diabetes Care 1987; 10:330–336.
- Sakaguchi M, Ogura H, Inouye S. IgE antibody to gelatin in children with immediate-type reactions to measles and mumps vaccines. J Allergy Clin Immunol 1995; 96:563–565.
- Raulf-Heimsoth M, Rihs HP, Rozynek P, et al. Quantitative analysis of immunoglobulin E reactivity profiles in patients allergic or sensitized to natural rubber latex (Hevea brasiliensis). Clin Exp Allergy 2007; 37:1657–1667.
- Biagini RE, MacKenzie BA, Sammons DL, et al. Latex specific IgE: performance characteristics of the IMMULITE 2000 3gAllergy assay compared with skin testing. Ann Allergy Asthma Immunol 2006; 97:196–202.
- Hamilton RG, Peterson EL, Ownby DR. Clinical and laboratory-based methods in the diagnosis of natural rubber latex allergy. J Allergy Clin Immunol 2002; 110(suppl 2):S47–S56.
- Safadi GS, Corey EC, Taylor JS, Wagner WO, Pien LC, Melton AL. Latex hypersensitivity in emergency medical service providers. Ann Allergy Asthma Immunol 1996; 77:39–42.
- Moffitt JE, Golden DB, Reisman RE, et al. Stinging insect hypersensitivity: a practice parameter update. J Allergy Clin Immunol 2004; 114:869–886.
- Biló BM, Rueff F, Mosbech H, Bonifazi F, Oude-Elberink JN; EAACI Interest Group on Insect Venom Hypersensitivity. Diagnosis of Hymenoptera venom allergy. Allergy 2005; 60:1339–1349.
- Cox L, Nelson H, Lockey R, et al. Allergen immunotherapy: a practice parameter third update. J Allergy Clin Immunol 2011; 127(suppl 1):S1–S55.
- Golden DB, Kagey-Sobotka A, Norman PS, Hamilton RG, Lichtenstein LM. Insect sting allergy with negative venom skin test responses. J Allergy Clin Immunol 2001; 107:897–901.
- Williams PB, Ahlstedt S, Barnes JH, Söderström L, Portnoy J. Are our impressions of allergy test performances correct? Ann Allergy Asthma Immunol 2003; 91:26–33.
- Bernstein IL, Li JT, Bernstein DI, et al; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology. Allergy diagnostic testing: an updated practice parameter. Ann Allergy Asthma Immunol 2008; 100(suppl 3):S1–S148.
- Pichler WJ. Immune mechanism of drug hypersensitivity. Immunol Allergy Clin North Am 2004; 24:373–397.
- Lieberman P, Nicklas RA, Oppenheimer J, et al. The diagnosis and management of anaphylaxis practice parameter: 2010 update. J Allergy Clin Immunol 2010; 126:477–480.
- Hamilton RG. Clinical laboratory assessment of immediate-type hypersensitivity. J Allergy Clin Immunol 2010; 125(suppl 2):S284–S296.
- Cox L, Williams B, Sicherer S, et al; American College of Allergy, Asthma and Immunology Test Task Force; American Academy of Allergy, Asthma and Immunology Specific IgE Test Task Force. Pearls and pitfalls of allergy diagnostic testing: report from the American College of Allergy, Asthma and Immunology/American Academy of Allergy, Asthma and Immunology Specific IgE Test Task Force. Ann Allergy Asthma Immunol 2008; 101:580–592.
- Hamilton RG, Franklin Adkinson N. In vitro assays for the diagnosis of IgE-mediated disorders. J Allergy Clin Immunol 2004; 114:213–225.
- Williams PB, Dolen WK, Koepke JW, Selner JC. Comparison of skin testing and three in vitro assays for specific IgE in the clinical evaluation of immediate hypersensitivity. Ann Allergy 1992; 68:35–45.
- Howanitz PJ, Cembrowski GS, Bachner P. Laboratory phlebotomy. College of American Pathologists Q-Probe study of patient satisfaction and complications in 23,783 patients. Arch Pathol Lab Med 1991; 115:867–872.
- Turkeltaub PC, Gergen PJ. The risk of adverse reactions from percutaneous prick-puncture allergen skin testing, venipuncture, and body measurements: data from the second National Health and Nutrition Examination Survey 1976–80 (NHANES II). J Allergy Clin Immunol 1989; 84:886–890.
- Pipkorn U, Hammarlund A, Enerbäck L. Prolonged treatment with topical glucocorticoids results in an inhibition of the allergen-induced weal-and-flare response and a reduction in skin mast cell numbers and histamine content. Clin Exp Allergy 1989; 19:19–25.
- Cole ZA, Clough GF, Church MK. Inhibition by glucocorticoids of the mast cell-dependent weal and flare response in human skin in vivo. Br J Pharmacol 2001; 132:286–292.
- Des Roches A, Paradis L, Bougeard YH, Godard P, Bousquet J, Chanez P. Long-term oral corticosteroid therapy does not alter the results of immediate-type allergy skin prick tests. J Allergy Clin Immunol 1996; 98:522–527.
- Linden CC, Misiak RT, Wegienka G, et al. Analysis of allergen specific IgE cut points to cat and dog in the Childhood Allergy Study. Ann Allergy Asthma Immunol 2011; 106:153–158.
- Hamilton RG, Williams PB; Specific IgE Testing Task Force of the American Academy of Allergy, Asthma & Immunology; American College of Allergy, Asthma and Immunology. Human IgE antibody serology: a primer for the practicing North American allergist/immunologist. J Allergy Clin Immunol 2010; 126:33–38.
- Somville MA, Machiels J, Gilles JG, Saint-Remy JM. Seasonal variation in specific IgE antibodies of grass-pollen hypersensitive patients depends on the steady state IgE concentration and is not related to clinical symptoms. J Allergy Clin Immunol 1989; 83( 2 Pt 1):486–494.
- Poon AW, Goodman CS, Rubin RJ. In vitro and skin testing for allergy: comparable clinical utility and costs. Am J Manag Care 1998; 4:969–985.
- Sampson HA. Update on food allergy. J Allergy Clin Immunol 2004; 113:805–819.
- Boyce JA, Assa’ad A, Burks AW, et al; NIAID-Sponsored Expert Panel. Guidelines for the diagnosis and management of food allergy in the United States: summary of the NIAID-sponsored expert panel report. J Allergy Clin Immunol 2010; 126:1105–1118.
- Rosen JP, Selcow JE, Mendelson LM, Grodofsky MP, Factor JM, Sampson HA. Skin testing with natural foods in patients suspected of having food allergies: is it a necessity? J Allergy Clin Immunol 1994; 93:1068–1070.
- Williams PB, Siegel C, Portnoy J. Efficacy of a single diagnostic test for sensitization to common inhalant allergens. Ann Allergy Asthma Immunol 2001; 86:196–202.
- Söderström L, Kober A, Ahlstedt S, et al. A further evaluation of the clinical use of specific IgE antibody testing in allergic diseases. Allergy 2003; 58:921–928.
- Bousquet J, Lebel B, Dhivert H, Bataille Y, Martinot B, Michel FB. Nasal challenge with pollen grains, skin-prick tests and specific IgE in patients with grass pollen allergy. Clin Allergy 1987; 17:529–536.
- Nepper-Christensen S, Backer V, DuBuske LM, Nolte H. In vitro diagnostic evaluation of patients with inhalant allergies: summary of probability outcomes comparing results of CLA- and CAP-specific immunoglobulin E test systems. Allergy Asthma Proc 2003; 24:253–258.
- Wallace DV, Dykewicz MS, Bernstein DI, et al; Joint Task Force on Practice; American Academy of Allergy; Asthma & Immunology; Joint Council of Allergy, Asthma and Immunology. The diagnosis and management of rhinitis: an updated practice parameter. J Allergy Clin Immunol 2008; 122(suppl 2):S1–S84.
- Mayorga C, Sanz ML, Gamboa PM, et al; Immunology Committee of the Spanish Society of Allergology and Clinical Immunology of the SEAIC. In vitro diagnosis of immediate allergic reactions to drugs: an update. J Investig Allergol Clin Immunol 2010; 20:103–109.
- Garcia JJ, Blanca M, Moreno F, et al. Determination of IgE antibodies to the benzylpenicilloyl determinant: a comparison of the sensitivity and specificity of three radio allergo sorbent test methods. J Clin Lab Anal 1997; 11:251–257.
- Macy E, Goldberg B, Poon KY. Use of commercial anti-penicillin IgE fluorometric enzyme immunoassays to diagnose penicillin allergy. Ann Allergy Asthma Immunol 2010; 105:136–141.
- Blanca M, Mayorga C, Torres MJ, et al. Clinical evaluation of Pharmacia CAP System RAST FEIA amoxicilloyl and benzylpenicilloyl in patients with penicillin allergy. Allergy 2001; 56:862–870.
- Ebo DG, Venemalm L, Bridts CH, et al. Immunoglobulin E antibodies to rocuronium: a new diagnostic tool. Anesthesiology 2007; 107:253–259.
- Ebo DG, Bridts CH, Stevens WJ. IgE-mediated anaphylaxis from chlorhexidine: diagnostic possibilities. Contact Dermatitis 2006; 55:301–302.
- deShazo RD, Mather P, Grant W, et al. Evaluation of patients with local reactions to insulin with skin tests and in vitro techniques. Diabetes Care 1987; 10:330–336.
- Sakaguchi M, Ogura H, Inouye S. IgE antibody to gelatin in children with immediate-type reactions to measles and mumps vaccines. J Allergy Clin Immunol 1995; 96:563–565.
- Raulf-Heimsoth M, Rihs HP, Rozynek P, et al. Quantitative analysis of immunoglobulin E reactivity profiles in patients allergic or sensitized to natural rubber latex (Hevea brasiliensis). Clin Exp Allergy 2007; 37:1657–1667.
- Biagini RE, MacKenzie BA, Sammons DL, et al. Latex specific IgE: performance characteristics of the IMMULITE 2000 3gAllergy assay compared with skin testing. Ann Allergy Asthma Immunol 2006; 97:196–202.
- Hamilton RG, Peterson EL, Ownby DR. Clinical and laboratory-based methods in the diagnosis of natural rubber latex allergy. J Allergy Clin Immunol 2002; 110(suppl 2):S47–S56.
- Safadi GS, Corey EC, Taylor JS, Wagner WO, Pien LC, Melton AL. Latex hypersensitivity in emergency medical service providers. Ann Allergy Asthma Immunol 1996; 77:39–42.
- Moffitt JE, Golden DB, Reisman RE, et al. Stinging insect hypersensitivity: a practice parameter update. J Allergy Clin Immunol 2004; 114:869–886.
- Biló BM, Rueff F, Mosbech H, Bonifazi F, Oude-Elberink JN; EAACI Interest Group on Insect Venom Hypersensitivity. Diagnosis of Hymenoptera venom allergy. Allergy 2005; 60:1339–1349.
- Cox L, Nelson H, Lockey R, et al. Allergen immunotherapy: a practice parameter third update. J Allergy Clin Immunol 2011; 127(suppl 1):S1–S55.
- Golden DB, Kagey-Sobotka A, Norman PS, Hamilton RG, Lichtenstein LM. Insect sting allergy with negative venom skin test responses. J Allergy Clin Immunol 2001; 107:897–901.
KEY POINTS
- Specific IgE levels higher than 0.35 kU/L suggest sensitization, but that is not synonymous with clinical disease.
- Prospective studies have identified IgE levels that can predict clinical reactivity with greater than 95% certainty for certain foods, but similar studies have not been performed for most other foods, drugs, latex, or venom.
- The likelihood of an IgE-mediated clinical reaction often increases with the level of specific IgE, but these levels do not predict severity or guarantee a reaction will occur.
- The sensitivity of allergy blood tests ranges from 60% to 95%, and the specificity ranges from 30% to 95%.
- In the appropriate setting, these tests can help in identifying specific allergens and assessing allergic disease.
- Neither allergy blood testing nor skin testing should be used for screening: they may be most useful as confirmatory tests when the patient’s history is compatible with an IgE-mediated reaction.
In reply: Giant cell arteritis
In Reply: We know from autopsy studies that most patients with giant cell arteritis, if not all, develop aortitis at some point during the course of their disease, but we don’t know (and no study yet has completely addressed) the following questions:
- What is the most clinically appropriate and cost-effective method of screening?
- How often should we be screening these patients?
Given the high cost of the most accurate and detailed available test, ie, magnetic resonance angiography of the aorta, annual chest radiography has been recommended by some experts in the field.
Although the high frequency of thoracic aneurysm justifies high clinical vigilance, we don’t know the most adequate and cost-effective test for screening for aortic aneurysm. Until we have an answer to these questions it is difficult to formulate specific guidelines, and different experts will continue to have different practices that are based on their own experience.
At this time, I carefully listen for bruits and murmurs on physical examination and check the blood pressure in all four extremities during patient follow-up visits. If I detect any abnormalities suggesting pathology of the aorta or major branches, I order magnetic resonance angiography of the entire aorta and its main branches.
In Reply: We know from autopsy studies that most patients with giant cell arteritis, if not all, develop aortitis at some point during the course of their disease, but we don’t know (and no study yet has completely addressed) the following questions:
- What is the most clinically appropriate and cost-effective method of screening?
- How often should we be screening these patients?
Given the high cost of the most accurate and detailed available test, ie, magnetic resonance angiography of the aorta, annual chest radiography has been recommended by some experts in the field.
Although the high frequency of thoracic aneurysm justifies high clinical vigilance, we don’t know the most adequate and cost-effective test for screening for aortic aneurysm. Until we have an answer to these questions it is difficult to formulate specific guidelines, and different experts will continue to have different practices that are based on their own experience.
At this time, I carefully listen for bruits and murmurs on physical examination and check the blood pressure in all four extremities during patient follow-up visits. If I detect any abnormalities suggesting pathology of the aorta or major branches, I order magnetic resonance angiography of the entire aorta and its main branches.
In Reply: We know from autopsy studies that most patients with giant cell arteritis, if not all, develop aortitis at some point during the course of their disease, but we don’t know (and no study yet has completely addressed) the following questions:
- What is the most clinically appropriate and cost-effective method of screening?
- How often should we be screening these patients?
Given the high cost of the most accurate and detailed available test, ie, magnetic resonance angiography of the aorta, annual chest radiography has been recommended by some experts in the field.
Although the high frequency of thoracic aneurysm justifies high clinical vigilance, we don’t know the most adequate and cost-effective test for screening for aortic aneurysm. Until we have an answer to these questions it is difficult to formulate specific guidelines, and different experts will continue to have different practices that are based on their own experience.
At this time, I carefully listen for bruits and murmurs on physical examination and check the blood pressure in all four extremities during patient follow-up visits. If I detect any abnormalities suggesting pathology of the aorta or major branches, I order magnetic resonance angiography of the entire aorta and its main branches.
Giant cell arteritis
To the Editor: As a practicing internist, I found Dr. Alexandra Villa-Forte’s review of giant-cell arteritis (Cleve Clin J Med 2011; 78:265–270) both interesting and useful, as usual for the Cleveland Clinic Journal of Medicine. However, she did not mention the recommendation by some experts that patients who have had temporal arteritis should receive annual chest x-rays, for a decade or longer, to screen for the development of thoracic aortic aneurysm. Does she agree with this precaution? Is it advisable, in addition, to screen for abdominal aortic aneurysm by means of abdominal ultrasonography? If so, at what time intervals should this be done?
To the Editor: As a practicing internist, I found Dr. Alexandra Villa-Forte’s review of giant-cell arteritis (Cleve Clin J Med 2011; 78:265–270) both interesting and useful, as usual for the Cleveland Clinic Journal of Medicine. However, she did not mention the recommendation by some experts that patients who have had temporal arteritis should receive annual chest x-rays, for a decade or longer, to screen for the development of thoracic aortic aneurysm. Does she agree with this precaution? Is it advisable, in addition, to screen for abdominal aortic aneurysm by means of abdominal ultrasonography? If so, at what time intervals should this be done?
To the Editor: As a practicing internist, I found Dr. Alexandra Villa-Forte’s review of giant-cell arteritis (Cleve Clin J Med 2011; 78:265–270) both interesting and useful, as usual for the Cleveland Clinic Journal of Medicine. However, she did not mention the recommendation by some experts that patients who have had temporal arteritis should receive annual chest x-rays, for a decade or longer, to screen for the development of thoracic aortic aneurysm. Does she agree with this precaution? Is it advisable, in addition, to screen for abdominal aortic aneurysm by means of abdominal ultrasonography? If so, at what time intervals should this be done?
Immunizing the Adult Patient
Reported incidence rates of certain vaccine-preventable diseases—measles, rubella, diphtheria, polio, and tetanus—are low in the United States.1 However, as was demonstrated during the 2009-2010 flu season and the outbreak of H1N1 influenza,2 we cannot afford to be complacent in our attitudes toward vaccines and vaccination. New virus strains exist and can become endemic quickly and ravenously. Furthermore, certain vaccine-preventable illnesses are frequently reported among adult patients, including hepatitis B, herpes zoster, human papillomavirus infection, influenza, pertussis, and pneumococcal infection.3
Vigilance regarding vaccination of children and adults was addressed by the CDC’s Office of Disease Prevention and Health Promotion in Healthy People 2010.4 These target objectives are proposed to be retained in Healthy People 2020,5 as the objectives from 2010 have not been met. The Healthy People 2010 target for administration of the pneumococcal vaccine in adults ages 65 and older, for example, is 90%. As of 2008, research has shown, only 60% of that population was immunized.6
There are subgroups of immunocompromised people who will probably never achieve adequate antibody levels to ensure immunity to vaccine-preventable diseases (for example, measles and influenza can be deadly to the immunocompromised person, as they can be to the very young and the very old). This is an important reason why vaccination of the healthy population is essential: the concept of herd immunity.7 Herd immunity (or community immunity) suggests that if most people around you are immune to an infection and do not become ill, then there is no one who can infect you—even if you are not immune to the infection.
WHY SOME ADULTS MIGHT NEED VACCINES
Adults who were vaccinated as children may incorrectly assume that they are protected from disease for life. In the case of some diseases, this may be true. However:
• Some adults were never vaccinated as children
• Newer vaccines have been developed since many adults were children
• Immunity can begin to fade over time
• As we age, we become more susceptible to serious disease caused by common infections (eg, influenza, pneumococcus).8
Barriers to Vaccination
Barriers to vaccination are varied, but none is insurmountable. Some of these barriers include8-15:
Missed opportunities. Providers should address vaccination needs for both adults and children at each visit or encounter. According to the CDC, studies have shown that eliminating missed opportunities could increase vaccination coverage by as much as 20%.8,11
Provider misconceptions regarding vaccine contraindications, schedules, and simultaneous vaccine administration. These misconceptions may prompt providers to forego an opportunity to vaccinate. Up-to-date information about vaccinations and ongoing provider education are imperative to improve immunity among both adults and children against vaccine-preventable disease.8 Numerous Web sites and publications are instrumental and essential in furnishing the health care provider with the most current information about vaccinations (see Table 1, above, and Table 2,10,12-14).

A belief on patients’ part that they are fully vaccinated when they are not. It is important to provide a vaccination record and a return date at every vaccination encounter, even if just one vaccination has been administered on a given day. Participating in Immunization Information System,15 if one is available, is an efficient way to access computerized vaccine records easily at the point of contact.
Just as parents should be encouraged to bring their child’s vaccine record with them to every health care visit, adults are also called upon to maintain a record of all their vaccinations. Each entry in the immunization record should include:
• The type of vaccine and dose
• The site and route of administration
• The date that the vaccine was administered
• The date that the next dose is due
• The manufacturer and lot number
• The name, address, and title of the person who administered the vaccine.15
PRINCIPLES OF VACCINATION
There are two ways to acquire immunity: active and passive.
Active immunity is produced by the individual’s own immune system and usually represents a permanent immunity toward the antigen.10,16
Passive immunity is produced when the individual receives products of immunity made by another animal or a human and transferred to the host. Passive immunity can be accomplished by injection of these products or through the placenta in infants. This type of immunity is not permanent and wanes over time—usually within weeks or months.10,16
This article will concentrate on active immunity, acquired through the administration of vaccines.
CLASSIFICATION OF VACCINES
Vaccines are classified as either live, attenuated vaccine (viral or bacterial) or inactivated vaccine.
Live, attenuated vaccines are derived from “wild” or disease-causing viruses and bacteria. Through procedures conducted in the laboratory, these wild organisms are weakened or attenuated. The live, attenuated vaccine must grow and replicate in the vaccinated person in order to stimulate an immune response. However, because the organism has been weakened, it usually does not cause disease or illness.10,16
The immune response to a live, attenuated vaccine is virtually identical to a response produced by natural infection. In rare instances, however, live, attenuated vaccines may cause severe or fatal reactions as a result of uncontrolled replication of the organism. This occurs only in individuals who are significantly immunocompromised.10,16
Inactivated vaccines are produced by growing the viral or bacterial organism in a culture medium, then using heat and/or chemicals to inactivate the organism. Because inactivated vaccines are not alive, they cannot replicate, and therefore cannot cause disease, even in an immunodeficient patient. The immune response to an inactivated vaccine is mostly humoral (in contrast to the natural infection response of a live vaccine), and little or no cellular immunity is produced.10,16
Inactivated vaccines always require multiple doses, gradually building up a protective immune response. The antibody titers diminish over time, so some inactivated vaccines may require periodic doses to “boost” or increase the titers.16
Some vaccines, such as hepatitis B vaccine, lead to the development of immune memory, which stays intact for at least 20 years following immunization. Immune memory occurs during replication of B cells and T cells; some cells will become long-lived memory cells that will “remember” the pathogen and produce an immune response if the pathogen is detected again. In this case, boosters are not recommended.10
Toxoids are a type of vaccine made from the inactivated toxin of a bacterium—not the bacterium itself. Tetanus and diphtheria vaccines are examples of toxoid vaccines.10,16
Subunit and conjugate vaccines are segments of the pathogen. A subunit vaccine can be created via genetic engineering. The end result is a recombinant vaccine that can stimulate cell memory (eg, hepatitis B vaccine).10,16 Conjugate vaccines, which are similar to recombinant vaccines, are made by combining two different components to prompt a more powerful, combined immune response.10
Spacing of Live-Virus and Inactivated Vaccines
There are almost no spacing requirements between two or more inactivated vaccines8 (see Table 38,14 for spacing recommendations from the Advisory Committee on Immunization Practices [ACIP]). The only vaccines that must be spaced at least four weeks apart are live-virus vaccines—that is, if they are not given on the same day. Studies have shown that the immune response to a live-virus vaccine may be impaired if it is administered within 30 days of another live-virus vaccine. Inactivated vaccines, on the other hand, may usually be administered at any time after or before a live-virus vaccine.8
One exception to this statement is the administration of Zostavax (zoster live-virus vaccine) with Pneumovax 23 (inactivated pneumococcal vaccine, polyvalent, MSD).17-19 The manufacturer of the two vaccines recommends a spacing of at least four weeks between them, based on research showing that concomitant use may result in reduced immunogenicity for Zostavax.20 However, as of this writing, ACIP has not revised its statement that both vaccines can be given at the same time or at any time before or after each other.21
Live-virus vaccines currently licensed in the US provide protection against diseases including measles/mumps/rubella, varicella, zoster (ie, shingles), influenza, and yellow fever.
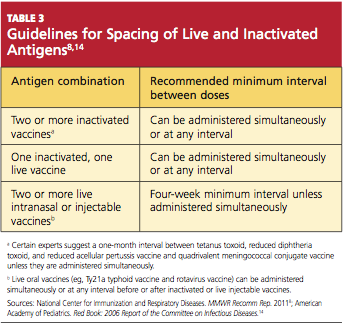
ADULT VACCINATION HIGHLIGHTS
A summary of 2011 recommendations for adult immunization from ACIP is shown in Table 412,21. The following information is specific to each of the vaccine-preventable illnesses of concern in adults.12,13,22
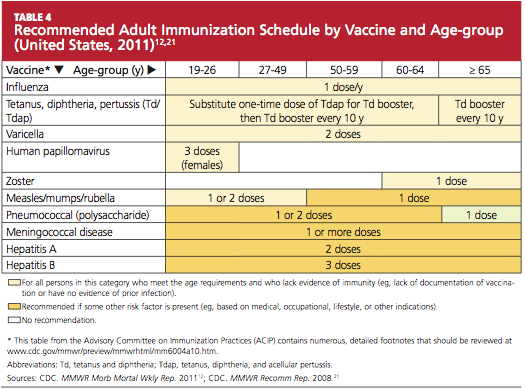
Seasonal Influenza
The options to protect the adult patient against seasonal influenza are a trivalent, inactivated influenza vaccine (TIV; Fluzone, high-dose Fluzone for adults ages 65 and older, Fluvirin, Fluarix, FluLaval, Afluria, Agriflu) or a live, attenuated influenza vaccine (LAIV; FluMist).2,23 Dosage of TIV for adults is 0.5 mL IM in the deltoid once annually. For adults ages 49 and younger, LAIV is administered at 0.2 mL intranasally, once per year.
ACIP now recommends universal influenza vaccination for all persons ages 6 months and older with no contraindications.2,8 Strong consideration should be given to concurrent administration of influenza vaccine and pneumococcal vaccine to high-risk persons not previously vaccinated against pneumococcal disease.12
Note: When influenza and pneumococcal vaccines are given at the same time, they should be administered in opposite arms to reduce the risk of adverse reactions or a decreased antibody response to either vaccine.18,19
Pneumococcal Polysaccharide (PPSV)
Pneumovax 2319 is administered as a 0.5-mL dose IM in the deltoid or subcutaneously in the upper arm. The vaccine is recommended for8,19,24:
• Adults ages 65 and older who have not been previously vaccinated
• Adults now 65 and older who received PPSV vaccine at least five years ago and were younger than 65 at that time
• Adults ages 19 through 64 years who have asthma or who smoke24
• Any adult with the following underlying medical conditions: chronic heart or lung disease, diabetes mellitus, cerebrospinal fluid leaks, cochlear implants, chronic liver disease, cirrhosis, chronic alcoholism, functional or anatomic asplenia, and immunocompromising conditions (HIV infection, diseases that require immunosuppressive therapy, chemotherapy, or radiation therapy; congenital immunodeficiency).24
A one-time revaccination is recommended after five years for persons ages 19 to 64 who have chronic renal failure, nephrotic syndrome, or functional or anatomic asplenia, and for those who are immunocompromised.24
Note: According to the manufacturer of Zostavax18 and Pneumovax,23,19 these vaccines should not be given at the same time, as research has shown that Zostavax immunogenicity is reduced as a result.18-20
Zoster
For adults ages 60 and older, Zostavax18 is administered in a single 0.65-mL dose, subcutaneously in the upper arm. Providers are not required to ask about varicella vaccination history or history of varicella disease before administering the vaccine. Adults ages 60 and older who have previously had shingles can still be vaccinated during a routine health care visit.10,21,22
Immunization is contraindicated in adults with a previous anaphylactic reaction to neomycin or gelatin, although a nonanaphylactic reaction to neomycin (most commonly, contact dermatitis) is not considered a contraindication.8
Any adult patient who has close household or occupational contact with persons at risk for severe varicella (eg, infants) need not take precautions after receiving the zoster vaccine, except in the rare case in which a varicella-like rash develops.10,21,22
Note: Review the note appearing in “Pneumococcal Polysaccharide (PPSV),” above, regarding coadministration of Zostavax and Pneumovax 23.18-20
Varicella
Adults who were born in the US before 1980 are considered immune to varicella and don’t need to be vaccinated, with the exception of health care workers, pregnant women, and immunocompromised persons. Nonimmune healthy adults who have not previously undergone vaccination should receive two 0.5-mL doses of Varivax, administered subcutaneously, four to eight weeks apart.25
Immunization is contraindicated in adults with a previous anaphylactic reaction to neomycin or gelatin, although a nonanaphylactic reaction to neomycin (eg, contact dermatitis) is not considered a contraindication.8
Measles, Mumps, Rubella (MMR)
The MMR vaccine is administered at 0.5 mL, given subcutaneously in the posterolateral fat of the upper arm.8
MMR-susceptible adults who were born during or since 1957 and are not at increased risk (see below) need only one dose of the MMR vaccine; those considered at increased risk need two doses, and a second dose can also be considered during an outbreak. Adults who require two doses should wait at least four weeks between the first and second doses.12
The following factors place adults at increased risk for MMR:
• Anticipated international travel
• Being a student in a post–secondary educational setting
• Working in a health care facility
• Recent exposure to measles, or an outbreak of measles or mumps
• Previous vaccination with killed measles vaccine
• Previous vaccination with an unknown measles vaccine between 1963 and 1967.
Also at risk are health care workers born before 1957 who have no evidence of immunity, and women who plan to become pregnant and have no evidence of immunity.8,12
Tetanus, Diphtheria, Pertussis
Options for adults include a vaccine against tetanus and diphtheria (Td; Decavac); or a vaccine that protects against tetanus, diphtheria, and acellular pertussis (Tdap; Adacel, Boostrix). Adults who have not been previously vaccinated should receive one dose of Tdap and two doses of Td (the first, one month after Tdap; the second at six to 12 months after the Tdap). Each is administered as a 0.5-mL dose IM in the deltoid. A booster dose is recommended every 10 years but can be given earlier in patients who sustain wounds or who anticipate international travel.8,12
Adults ages 19 through 64 should receive a single dose of Tdap in place of a booster dose if the last Td dose was administered at least 10 years earlier and the patient has not previously received Tdap. Additionally, a dose of Tdap (if not previously given) is recommended for postpartum women, close contacts of infants younger than 12 months, and all health care workers with direct patient contact. An interval as short as two years from the last Td is suggested; shorter intervals may be appropriate.8,12
According to the new 2011 recommendations, persons ages 65 and older who have close contact with an infant younger than 12 months should be vaccinated with Tdap, and any person age 65 or older may be vaccinated with Tdap. Also added is a recommendation to administer Tdap, regardless of the interval since the patient received his or her most recent Td-containing vaccine.8,12
Human Papillomavirus (HPV)
Gardasil26 protects both female and male patients against HPV infection; Cervarix27 is indicated only for female patients. Either quadrivalent vaccine or bivalent vaccine is recommended for female patients.12
In women ages 26 and younger, Gardasil (0.5 mL IM, administered in the deltoid at 0 month, 2 months, and 6 months) provides protection against diseases caused by HPV types 6, 11, 16, and 18 (including cervical, vaginal, and vulvar cancer caused by HPV types 16 and 18). In men ages 26 and younger, Gardasil provides protection against genital warts caused by HPV types 6 and 11.26
Cervarix,27 administered at 0 month, 1 month, and 6 months (0.5 mL IM in the deltoid), provides protection for women ages 25 and younger against cervical cancer and precancerous lesions caused by HPV types 16 and 18.
Caution: Patients should be advised to sit or lie down when the HPV vaccine is administered, and they should be observed for the subsequent 15 minutes. Syncope can occur after vaccination—most commonly among adolescents and young adults.28 Convulsive syncope has been reported.
Meningococcal Disease
Two vaccines are available to protect against meningococcal disease: Menactra29 (meningococcal groups A, C, Y, and W-135 polysaccharide diphtheria toxoids conjugate vaccine); and Menveo30 (meningococcal groups A, C, Y, and W-135 oligosaccharide diphtheria CRM197 conjugate vaccine). Both are administered in the deltoid, 0.5 mL IM.
The following patients should be considered for vaccination:
• College freshmen living in dormitories, as well as college students with immune deficiencies, as they are at higher risk for meningococcal disease
• Patients who travel to or reside in countries in which Neisseria meningitidis is epidemic (particularly those with the potential for prolonged contact with the local population)
• Travelers to Saudi Arabia for pilgrimage to Mecca (the Hajj)
• Patients with anatomical or functional asplenia (two-dose series).
A two-dose series of meningococcal conjugate vaccine is also recommended for adults with persistent complement component deficiencies, and for those with HIV infection who are vaccinated.12
Hepatitis A
Two hepatitis A vaccines (both inactivated) can be used interchangeably: Havrix31 and Vaqta.32 Dosing for both vaccines in 18-year-old patients is 0.5 mL IM in the deltoid at 0 months, then at 6 to 12 months. In patients ages 19 and older, administration is the same, with the exception of increased dosing (1.0 mL IM).
Vaccination against hepatitis A is recommended for men who have sex with men, and for all adult patients who12:
• Travel to or work in areas where risk for hepatitis A transmission is high (especially those who take frequent trips or experience prolonged stays)
• Use injection drugs
• Have chronic liver disease
• Receive clotting factor concentrates for treatment of a blood-clotting disorder
• May have been exposed to hepatitis A in the previous two weeks
• Wish to be vaccinated against hepatitis A to avoid future infection.
Hepatitis B
Recombivax HB33 and Engerix-B34 are the two vaccines available to protect patients against hepatitis B (HBV), and they can be used interchangeably.12 In patients from birth through age 19, Recombivax HB33 or Engerix-B34 is given as 0.5 mL IM in the deltoid at 0, 1, and 6 months; patients ages 20 and older receive an increased dose (1.0 mL IM), with administration otherwise the same. According to the manufacturer of Recombivax HB,33 patients age 11 through 15 may be given either three doses of 0.5 mL or two doses of 1.0 mL.
The following adults are advised to undergo vaccination for HBV:
• At-risk, unvaccinated adults
• Those requesting protection against HBV infection
• Those planning to travel to areas where HBV is common
• Household contacts of a patient with chronic HBV infection, and sexual partners of a patient with HBV infection
• Adults with chronic liver disease
• Men who have sex with men
• Sexually active adults who are not in a long-term, mutually monogamous relationship
• Adults who are being evaluated or treated for a sexually transmitted disease, including HIV infection
• Health care or public safety workers who may be exposed to blood or blood-contaminated body fluids
• Workers and residents in facilities for developmentally disabled persons
• Patients undergoing or anticipating dialysis
• Adults who inject illegal drugs or who have done so recently.12
CONTRAINDICATIONS AND PRECAUTIONS FOR VACCINES COMMONLY USED IN ADULTS
See Table 5,8,22 for a summary of contraindications and precautions from ACIP and the Immunization Action Coalition that are associated with vaccinations mentioned in this article. A more complete summary can be found at www.im munize.org/catg.d/p3072a.pdf.
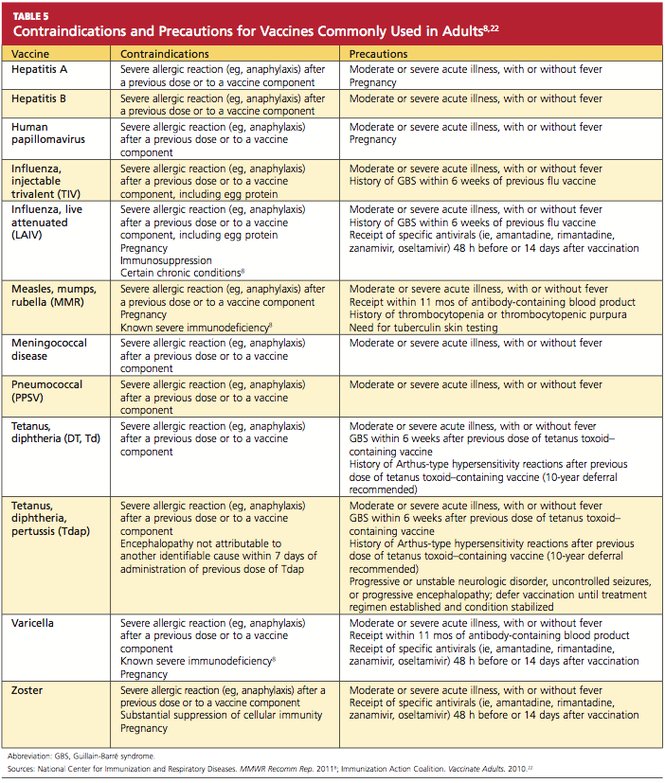
Conclusion
The adult patient’s vaccination status should be addressed at each health care encounter, and current recommendations should be followed. The duration of efficacy for vaccines is not an exact science. Many vaccines licensed in the US are relatively new, and recommendations for boosters for some of these vaccines will be forthcoming as more data are gathered. For example, the recommendation that a booster dose of Tdap be given to adults resulted from the recent increase in reported pertussis cases.35
Providers armed with the most current information and resources represent the forefront in ensuring that the US adult population is adequately immunized.
REFERENCES
1. World Health Organization. Immunization surveillance, assessment, and monitoring (2010). www.who.int/immunization_monitor ing/en. Accessed May 12, 2011.
2. Fiore AE, Uyeki TM, Broder K, et al; Centers for Disease Control and Prevention. Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Morb Mortal Wkly Rep. 2010;59(RR-08);1-62.
3. Schaffner W. Update on vaccine-preventable diseases: are adults in your community adequately protected? J Fam Pract. 2008;57(4 suppl):S1-S11.
4. CDC. Healthy People 2010: Objectives for Improving Health. www.healthypeople.gov. Accessed May 6, 2011.
5. US Department of Health and Human Services. Developing Healthy People 2020: immunization and infectious diseases. www.healthy people.gov/2020. Accessed May 12, 2011.
6. Lu PJ, Nuorti JP. Pneumococcal polysaccharide vaccination among adults aged 65 years and older, United States, 1989-2008. Am J Prev Med. 2010;39(4):287-295.
7. National Institute of Allergy and Infectious Diseases, NIH. Community immunity (“herd” immunity) (2010). www.niaid.nih.gov/topics/pages/communityimmunity.aspx. Accessed May 12, 2011.
8. National Center for Immunization and Respiratory Diseases. General recommendations on immunization: recommendations of the Advisory Committee on Immunization Practices (ADIP). MMWR Recomm Rep. 2011;60(2):1-64.
9. High KP. Overcoming barriers to adult immunization. J Am Osteopath Assoc. 2009;109(6): 525-528.
10. CDC; Atkinson W, Wolfe S, Hamborsky J, eds. Epidemiology and Prevention of Vaccine-Preventable Diseases (Pink Book). 12th ed. Washington, DC: Public Health Foundation, 2011.
11. CDC. Vaccine-preventable diseases: improving vaccination coverage in children, adolescents, and adults: a report on recommendations from the Task Force on Community Preventive Services. MMWR Recomm Rep. 1999;48(RR-8):1-15.
12. CDC. Recommended adult immunization schedule: United States, 2011. MMWR Morb Mortal Wkly Rep. 2011;60(4):1-4.
13. Thompson RF. Travel & Routine Immunizations: A Practical Guide for the Medical Office. 19th ed. Milwaukee, WI: Shoreland, Inc: 2001.
14. American Academy of Pediatrics. Pertussis. In: Pickering LK, Backer, CJ, Long SS, McMillan J, eds. Red Book: 2006 Report of the Committee on Infectious Diseases. 27th ed. Elk Grove Village, IL: American Academy of Pediatrics.
15. CDC. Immunization information systems (IIS). www.cdc.gov/vaccines/programs/iis/default.htm. Accessed May 12, 2011.
16. College of Physicians of Philadelphia. The history of vaccines: a project of the College of Physicians of Philadelphia (2011). www.history ofvaccines.org/content/articles/different-types-vaccines. Accessed May 12, 2011.
17. US Food and Drug Administration. Vaccines, blood, and biologics: December 18, 2009 Approval Letter—Zostavax. www.fda.gov/
BiologicsBloodVaccines/Vaccines/Approved Products/ucm195993.htm. Accessed May 12, 2011.
18. Merck & Co, Inc. Zostavax® (zoster vaccine live; product insert). www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/Approved Products/UCM132831.pdf. Accessed May 12, 2011.
19. Merck & Co, Inc. Pneumovax® 23 (pneumococcal vaccine, polyvalent, MSD; product information). www.merck.com/product/usa/pi_circulars/p/pneumovax_23/pneumovax_pi.pdf. Accessed May 16, 2011.
20. Macintyre CR, Egerton T, McCaughey M, et al. Concomitant administration of zoster and pneumococcal vaccines in adults ≥60 years old. Hum Vaccin. 2010;6(11):18-26.
21. CDC. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2008;57(RR-5):1–30.
22. Immunization Action Coalition. Vaccinate Adults. 2010 Aug;14(5). www.immunize.org/va. Accessed May 12, 2011.
23. CDC. Update: recommendations of the Advisory Committee on Immunization Practices (ACIP) regarding use of CSL seasonal influenza vaccine (Afluria) in the United States during 2010–2011. MMWR Morb Mortal Wkly Rep. 2010;59(31);989-992.
24. CDC. Updated recommendations for prevention of invasive pneumococcal disease among adults using the 23-valent pneumococcal polysaccharide vaccine (PPSV23). MMWR Morb Mortal Wkly Rep. 2010;59(RR-34):1102-1106.
25. Merck & Co, Inc. Varivax® varicella virus vaccine live (product information). www.merck
.com/product/usa/pi_circulars/v/varivax/varivax_pi.pdf. Accessed May 12, 2011.
26. Merck & Co, Inc. Gardasil® (human papillomavirus quadrivalent [types 6, 11, 16, and 18] vaccine, recombinant; product information). www.merck.com/product/usa/pi_circulars/g/gardasil/gardasil_ppi.pdf. Accessed May 12, 2011.
27. GlaxoSmithKline Biologicals. Cervarix (human papillomavirus bivalent [types 16 and 18] vaccine, recombinant; product information). http://us.gsk.com/products/assets/us_cervarix.pdf. Accessed May 12, 2011.
28. CDC. Syncope after vaccination—United States, January 2005-July 2007. MMWR Morb Mortal Wkly Rep. 2008;57(17):457-460.
29. Sanofi Pasteur. Meningococcal (groups A, C, Y, and W-135) polysaccharide diphtheria
toxoids conjugate vaccine Menactra® for intramuscular injection. www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/Approved
Products/UCM131170.pdf. Accessed May 12, 2011.
30. Novartis Vaccines and Diagnostics, Inc. Menveo® (meningococcal [groups A, C, Y and W-135] oligosaccharide diphtheria CRM197 conjugate vaccine solution for intramuscular injection; prescribing information highlights). www .fda.gov/downloads/biologicsbloodvaccines/vaccines/approvedproducts/ucm201349.pdf. Accessed May 12, 2011.
31. GlaxoSmithKline Biologicals. Havrix (hepatitis A vaccine, suspension for intramuscular injection; prescribing information highlights). http://us.gsk.com/products/assets/us_havrix .pdf. Accessed May 12, 2011.
32. Merck & Co, Inc. Vaqta (hepatitis A vaccine, inactivated; suspension for intramuscular injection; highlighted prescribing information). www.merck.com/product/usa/pi_circulars/v/vaqta/vaqta_pi.pdf. Accessed May 12, 2011.
33. Merck & Co, Inc. Recombivax HB® hepatitis B vaccine (recombinant; product information). www.merck.com/product/usa/pi_circulars/r/recombivax_hb/recombivax_pi.pdf. Accessed May 12, 2011.
34. GlaxoSmithKline Biologicals. Engerix-B® (hepatitis B vaccine, recombinant; prescribing information). http://us.gsk.com/products/assets/us_engerixb.pdf. Accessed May 12, 2011.
35. CDC. Tetanus and pertussis vaccination coverage among adults aged ≥ 18 years—United States, 1999 and 2008. MMWR Morb Mortal Wkly Rep. 2010;59(40):1302-1306.
Reported incidence rates of certain vaccine-preventable diseases—measles, rubella, diphtheria, polio, and tetanus—are low in the United States.1 However, as was demonstrated during the 2009-2010 flu season and the outbreak of H1N1 influenza,2 we cannot afford to be complacent in our attitudes toward vaccines and vaccination. New virus strains exist and can become endemic quickly and ravenously. Furthermore, certain vaccine-preventable illnesses are frequently reported among adult patients, including hepatitis B, herpes zoster, human papillomavirus infection, influenza, pertussis, and pneumococcal infection.3
Vigilance regarding vaccination of children and adults was addressed by the CDC’s Office of Disease Prevention and Health Promotion in Healthy People 2010.4 These target objectives are proposed to be retained in Healthy People 2020,5 as the objectives from 2010 have not been met. The Healthy People 2010 target for administration of the pneumococcal vaccine in adults ages 65 and older, for example, is 90%. As of 2008, research has shown, only 60% of that population was immunized.6
There are subgroups of immunocompromised people who will probably never achieve adequate antibody levels to ensure immunity to vaccine-preventable diseases (for example, measles and influenza can be deadly to the immunocompromised person, as they can be to the very young and the very old). This is an important reason why vaccination of the healthy population is essential: the concept of herd immunity.7 Herd immunity (or community immunity) suggests that if most people around you are immune to an infection and do not become ill, then there is no one who can infect you—even if you are not immune to the infection.
WHY SOME ADULTS MIGHT NEED VACCINES
Adults who were vaccinated as children may incorrectly assume that they are protected from disease for life. In the case of some diseases, this may be true. However:
• Some adults were never vaccinated as children
• Newer vaccines have been developed since many adults were children
• Immunity can begin to fade over time
• As we age, we become more susceptible to serious disease caused by common infections (eg, influenza, pneumococcus).8
Barriers to Vaccination
Barriers to vaccination are varied, but none is insurmountable. Some of these barriers include8-15:
Missed opportunities. Providers should address vaccination needs for both adults and children at each visit or encounter. According to the CDC, studies have shown that eliminating missed opportunities could increase vaccination coverage by as much as 20%.8,11
Provider misconceptions regarding vaccine contraindications, schedules, and simultaneous vaccine administration. These misconceptions may prompt providers to forego an opportunity to vaccinate. Up-to-date information about vaccinations and ongoing provider education are imperative to improve immunity among both adults and children against vaccine-preventable disease.8 Numerous Web sites and publications are instrumental and essential in furnishing the health care provider with the most current information about vaccinations (see Table 1, above, and Table 2,10,12-14).
A belief on patients’ part that they are fully vaccinated when they are not. It is important to provide a vaccination record and a return date at every vaccination encounter, even if just one vaccination has been administered on a given day. Participating in Immunization Information System,15 if one is available, is an efficient way to access computerized vaccine records easily at the point of contact.
Just as parents should be encouraged to bring their child’s vaccine record with them to every health care visit, adults are also called upon to maintain a record of all their vaccinations. Each entry in the immunization record should include:
• The type of vaccine and dose
• The site and route of administration
• The date that the vaccine was administered
• The date that the next dose is due
• The manufacturer and lot number
• The name, address, and title of the person who administered the vaccine.15
PRINCIPLES OF VACCINATION
There are two ways to acquire immunity: active and passive.
Active immunity is produced by the individual’s own immune system and usually represents a permanent immunity toward the antigen.10,16
Passive immunity is produced when the individual receives products of immunity made by another animal or a human and transferred to the host. Passive immunity can be accomplished by injection of these products or through the placenta in infants. This type of immunity is not permanent and wanes over time—usually within weeks or months.10,16
This article will concentrate on active immunity, acquired through the administration of vaccines.
CLASSIFICATION OF VACCINES
Vaccines are classified as either live, attenuated vaccine (viral or bacterial) or inactivated vaccine.
Live, attenuated vaccines are derived from “wild” or disease-causing viruses and bacteria. Through procedures conducted in the laboratory, these wild organisms are weakened or attenuated. The live, attenuated vaccine must grow and replicate in the vaccinated person in order to stimulate an immune response. However, because the organism has been weakened, it usually does not cause disease or illness.10,16
The immune response to a live, attenuated vaccine is virtually identical to a response produced by natural infection. In rare instances, however, live, attenuated vaccines may cause severe or fatal reactions as a result of uncontrolled replication of the organism. This occurs only in individuals who are significantly immunocompromised.10,16
Inactivated vaccines are produced by growing the viral or bacterial organism in a culture medium, then using heat and/or chemicals to inactivate the organism. Because inactivated vaccines are not alive, they cannot replicate, and therefore cannot cause disease, even in an immunodeficient patient. The immune response to an inactivated vaccine is mostly humoral (in contrast to the natural infection response of a live vaccine), and little or no cellular immunity is produced.10,16
Inactivated vaccines always require multiple doses, gradually building up a protective immune response. The antibody titers diminish over time, so some inactivated vaccines may require periodic doses to “boost” or increase the titers.16
Some vaccines, such as hepatitis B vaccine, lead to the development of immune memory, which stays intact for at least 20 years following immunization. Immune memory occurs during replication of B cells and T cells; some cells will become long-lived memory cells that will “remember” the pathogen and produce an immune response if the pathogen is detected again. In this case, boosters are not recommended.10
Toxoids are a type of vaccine made from the inactivated toxin of a bacterium—not the bacterium itself. Tetanus and diphtheria vaccines are examples of toxoid vaccines.10,16
Subunit and conjugate vaccines are segments of the pathogen. A subunit vaccine can be created via genetic engineering. The end result is a recombinant vaccine that can stimulate cell memory (eg, hepatitis B vaccine).10,16 Conjugate vaccines, which are similar to recombinant vaccines, are made by combining two different components to prompt a more powerful, combined immune response.10
Spacing of Live-Virus and Inactivated Vaccines
There are almost no spacing requirements between two or more inactivated vaccines8 (see Table 38,14 for spacing recommendations from the Advisory Committee on Immunization Practices [ACIP]). The only vaccines that must be spaced at least four weeks apart are live-virus vaccines—that is, if they are not given on the same day. Studies have shown that the immune response to a live-virus vaccine may be impaired if it is administered within 30 days of another live-virus vaccine. Inactivated vaccines, on the other hand, may usually be administered at any time after or before a live-virus vaccine.8
One exception to this statement is the administration of Zostavax (zoster live-virus vaccine) with Pneumovax 23 (inactivated pneumococcal vaccine, polyvalent, MSD).17-19 The manufacturer of the two vaccines recommends a spacing of at least four weeks between them, based on research showing that concomitant use may result in reduced immunogenicity for Zostavax.20 However, as of this writing, ACIP has not revised its statement that both vaccines can be given at the same time or at any time before or after each other.21
Live-virus vaccines currently licensed in the US provide protection against diseases including measles/mumps/rubella, varicella, zoster (ie, shingles), influenza, and yellow fever.
ADULT VACCINATION HIGHLIGHTS
A summary of 2011 recommendations for adult immunization from ACIP is shown in Table 412,21. The following information is specific to each of the vaccine-preventable illnesses of concern in adults.12,13,22
Seasonal Influenza
The options to protect the adult patient against seasonal influenza are a trivalent, inactivated influenza vaccine (TIV; Fluzone, high-dose Fluzone for adults ages 65 and older, Fluvirin, Fluarix, FluLaval, Afluria, Agriflu) or a live, attenuated influenza vaccine (LAIV; FluMist).2,23 Dosage of TIV for adults is 0.5 mL IM in the deltoid once annually. For adults ages 49 and younger, LAIV is administered at 0.2 mL intranasally, once per year.
ACIP now recommends universal influenza vaccination for all persons ages 6 months and older with no contraindications.2,8 Strong consideration should be given to concurrent administration of influenza vaccine and pneumococcal vaccine to high-risk persons not previously vaccinated against pneumococcal disease.12
Note: When influenza and pneumococcal vaccines are given at the same time, they should be administered in opposite arms to reduce the risk of adverse reactions or a decreased antibody response to either vaccine.18,19
Pneumococcal Polysaccharide (PPSV)
Pneumovax 2319 is administered as a 0.5-mL dose IM in the deltoid or subcutaneously in the upper arm. The vaccine is recommended for8,19,24:
• Adults ages 65 and older who have not been previously vaccinated
• Adults now 65 and older who received PPSV vaccine at least five years ago and were younger than 65 at that time
• Adults ages 19 through 64 years who have asthma or who smoke24
• Any adult with the following underlying medical conditions: chronic heart or lung disease, diabetes mellitus, cerebrospinal fluid leaks, cochlear implants, chronic liver disease, cirrhosis, chronic alcoholism, functional or anatomic asplenia, and immunocompromising conditions (HIV infection, diseases that require immunosuppressive therapy, chemotherapy, or radiation therapy; congenital immunodeficiency).24
A one-time revaccination is recommended after five years for persons ages 19 to 64 who have chronic renal failure, nephrotic syndrome, or functional or anatomic asplenia, and for those who are immunocompromised.24
Note: According to the manufacturer of Zostavax18 and Pneumovax,23,19 these vaccines should not be given at the same time, as research has shown that Zostavax immunogenicity is reduced as a result.18-20
Zoster
For adults ages 60 and older, Zostavax18 is administered in a single 0.65-mL dose, subcutaneously in the upper arm. Providers are not required to ask about varicella vaccination history or history of varicella disease before administering the vaccine. Adults ages 60 and older who have previously had shingles can still be vaccinated during a routine health care visit.10,21,22
Immunization is contraindicated in adults with a previous anaphylactic reaction to neomycin or gelatin, although a nonanaphylactic reaction to neomycin (most commonly, contact dermatitis) is not considered a contraindication.8
Any adult patient who has close household or occupational contact with persons at risk for severe varicella (eg, infants) need not take precautions after receiving the zoster vaccine, except in the rare case in which a varicella-like rash develops.10,21,22
Note: Review the note appearing in “Pneumococcal Polysaccharide (PPSV),” above, regarding coadministration of Zostavax and Pneumovax 23.18-20
Varicella
Adults who were born in the US before 1980 are considered immune to varicella and don’t need to be vaccinated, with the exception of health care workers, pregnant women, and immunocompromised persons. Nonimmune healthy adults who have not previously undergone vaccination should receive two 0.5-mL doses of Varivax, administered subcutaneously, four to eight weeks apart.25
Immunization is contraindicated in adults with a previous anaphylactic reaction to neomycin or gelatin, although a nonanaphylactic reaction to neomycin (eg, contact dermatitis) is not considered a contraindication.8
Measles, Mumps, Rubella (MMR)
The MMR vaccine is administered at 0.5 mL, given subcutaneously in the posterolateral fat of the upper arm.8
MMR-susceptible adults who were born during or since 1957 and are not at increased risk (see below) need only one dose of the MMR vaccine; those considered at increased risk need two doses, and a second dose can also be considered during an outbreak. Adults who require two doses should wait at least four weeks between the first and second doses.12
The following factors place adults at increased risk for MMR:
• Anticipated international travel
• Being a student in a post–secondary educational setting
• Working in a health care facility
• Recent exposure to measles, or an outbreak of measles or mumps
• Previous vaccination with killed measles vaccine
• Previous vaccination with an unknown measles vaccine between 1963 and 1967.
Also at risk are health care workers born before 1957 who have no evidence of immunity, and women who plan to become pregnant and have no evidence of immunity.8,12
Tetanus, Diphtheria, Pertussis
Options for adults include a vaccine against tetanus and diphtheria (Td; Decavac); or a vaccine that protects against tetanus, diphtheria, and acellular pertussis (Tdap; Adacel, Boostrix). Adults who have not been previously vaccinated should receive one dose of Tdap and two doses of Td (the first, one month after Tdap; the second at six to 12 months after the Tdap). Each is administered as a 0.5-mL dose IM in the deltoid. A booster dose is recommended every 10 years but can be given earlier in patients who sustain wounds or who anticipate international travel.8,12
Adults ages 19 through 64 should receive a single dose of Tdap in place of a booster dose if the last Td dose was administered at least 10 years earlier and the patient has not previously received Tdap. Additionally, a dose of Tdap (if not previously given) is recommended for postpartum women, close contacts of infants younger than 12 months, and all health care workers with direct patient contact. An interval as short as two years from the last Td is suggested; shorter intervals may be appropriate.8,12
According to the new 2011 recommendations, persons ages 65 and older who have close contact with an infant younger than 12 months should be vaccinated with Tdap, and any person age 65 or older may be vaccinated with Tdap. Also added is a recommendation to administer Tdap, regardless of the interval since the patient received his or her most recent Td-containing vaccine.8,12
Human Papillomavirus (HPV)
Gardasil26 protects both female and male patients against HPV infection; Cervarix27 is indicated only for female patients. Either quadrivalent vaccine or bivalent vaccine is recommended for female patients.12
In women ages 26 and younger, Gardasil (0.5 mL IM, administered in the deltoid at 0 month, 2 months, and 6 months) provides protection against diseases caused by HPV types 6, 11, 16, and 18 (including cervical, vaginal, and vulvar cancer caused by HPV types 16 and 18). In men ages 26 and younger, Gardasil provides protection against genital warts caused by HPV types 6 and 11.26
Cervarix,27 administered at 0 month, 1 month, and 6 months (0.5 mL IM in the deltoid), provides protection for women ages 25 and younger against cervical cancer and precancerous lesions caused by HPV types 16 and 18.
Caution: Patients should be advised to sit or lie down when the HPV vaccine is administered, and they should be observed for the subsequent 15 minutes. Syncope can occur after vaccination—most commonly among adolescents and young adults.28 Convulsive syncope has been reported.
Meningococcal Disease
Two vaccines are available to protect against meningococcal disease: Menactra29 (meningococcal groups A, C, Y, and W-135 polysaccharide diphtheria toxoids conjugate vaccine); and Menveo30 (meningococcal groups A, C, Y, and W-135 oligosaccharide diphtheria CRM197 conjugate vaccine). Both are administered in the deltoid, 0.5 mL IM.
The following patients should be considered for vaccination:
• College freshmen living in dormitories, as well as college students with immune deficiencies, as they are at higher risk for meningococcal disease
• Patients who travel to or reside in countries in which Neisseria meningitidis is epidemic (particularly those with the potential for prolonged contact with the local population)
• Travelers to Saudi Arabia for pilgrimage to Mecca (the Hajj)
• Patients with anatomical or functional asplenia (two-dose series).
A two-dose series of meningococcal conjugate vaccine is also recommended for adults with persistent complement component deficiencies, and for those with HIV infection who are vaccinated.12
Hepatitis A
Two hepatitis A vaccines (both inactivated) can be used interchangeably: Havrix31 and Vaqta.32 Dosing for both vaccines in 18-year-old patients is 0.5 mL IM in the deltoid at 0 months, then at 6 to 12 months. In patients ages 19 and older, administration is the same, with the exception of increased dosing (1.0 mL IM).
Vaccination against hepatitis A is recommended for men who have sex with men, and for all adult patients who12:
• Travel to or work in areas where risk for hepatitis A transmission is high (especially those who take frequent trips or experience prolonged stays)
• Use injection drugs
• Have chronic liver disease
• Receive clotting factor concentrates for treatment of a blood-clotting disorder
• May have been exposed to hepatitis A in the previous two weeks
• Wish to be vaccinated against hepatitis A to avoid future infection.
Hepatitis B
Recombivax HB33 and Engerix-B34 are the two vaccines available to protect patients against hepatitis B (HBV), and they can be used interchangeably.12 In patients from birth through age 19, Recombivax HB33 or Engerix-B34 is given as 0.5 mL IM in the deltoid at 0, 1, and 6 months; patients ages 20 and older receive an increased dose (1.0 mL IM), with administration otherwise the same. According to the manufacturer of Recombivax HB,33 patients age 11 through 15 may be given either three doses of 0.5 mL or two doses of 1.0 mL.
The following adults are advised to undergo vaccination for HBV:
• At-risk, unvaccinated adults
• Those requesting protection against HBV infection
• Those planning to travel to areas where HBV is common
• Household contacts of a patient with chronic HBV infection, and sexual partners of a patient with HBV infection
• Adults with chronic liver disease
• Men who have sex with men
• Sexually active adults who are not in a long-term, mutually monogamous relationship
• Adults who are being evaluated or treated for a sexually transmitted disease, including HIV infection
• Health care or public safety workers who may be exposed to blood or blood-contaminated body fluids
• Workers and residents in facilities for developmentally disabled persons
• Patients undergoing or anticipating dialysis
• Adults who inject illegal drugs or who have done so recently.12
CONTRAINDICATIONS AND PRECAUTIONS FOR VACCINES COMMONLY USED IN ADULTS
See Table 5,8,22 for a summary of contraindications and precautions from ACIP and the Immunization Action Coalition that are associated with vaccinations mentioned in this article. A more complete summary can be found at www.im munize.org/catg.d/p3072a.pdf.
Conclusion
The adult patient’s vaccination status should be addressed at each health care encounter, and current recommendations should be followed. The duration of efficacy for vaccines is not an exact science. Many vaccines licensed in the US are relatively new, and recommendations for boosters for some of these vaccines will be forthcoming as more data are gathered. For example, the recommendation that a booster dose of Tdap be given to adults resulted from the recent increase in reported pertussis cases.35
Providers armed with the most current information and resources represent the forefront in ensuring that the US adult population is adequately immunized.
REFERENCES
1. World Health Organization. Immunization surveillance, assessment, and monitoring (2010). www.who.int/immunization_monitor ing/en. Accessed May 12, 2011.
2. Fiore AE, Uyeki TM, Broder K, et al; Centers for Disease Control and Prevention. Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Morb Mortal Wkly Rep. 2010;59(RR-08);1-62.
3. Schaffner W. Update on vaccine-preventable diseases: are adults in your community adequately protected? J Fam Pract. 2008;57(4 suppl):S1-S11.
4. CDC. Healthy People 2010: Objectives for Improving Health. www.healthypeople.gov. Accessed May 6, 2011.
5. US Department of Health and Human Services. Developing Healthy People 2020: immunization and infectious diseases. www.healthy people.gov/2020. Accessed May 12, 2011.
6. Lu PJ, Nuorti JP. Pneumococcal polysaccharide vaccination among adults aged 65 years and older, United States, 1989-2008. Am J Prev Med. 2010;39(4):287-295.
7. National Institute of Allergy and Infectious Diseases, NIH. Community immunity (“herd” immunity) (2010). www.niaid.nih.gov/topics/pages/communityimmunity.aspx. Accessed May 12, 2011.
8. National Center for Immunization and Respiratory Diseases. General recommendations on immunization: recommendations of the Advisory Committee on Immunization Practices (ADIP). MMWR Recomm Rep. 2011;60(2):1-64.
9. High KP. Overcoming barriers to adult immunization. J Am Osteopath Assoc. 2009;109(6): 525-528.
10. CDC; Atkinson W, Wolfe S, Hamborsky J, eds. Epidemiology and Prevention of Vaccine-Preventable Diseases (Pink Book). 12th ed. Washington, DC: Public Health Foundation, 2011.
11. CDC. Vaccine-preventable diseases: improving vaccination coverage in children, adolescents, and adults: a report on recommendations from the Task Force on Community Preventive Services. MMWR Recomm Rep. 1999;48(RR-8):1-15.
12. CDC. Recommended adult immunization schedule: United States, 2011. MMWR Morb Mortal Wkly Rep. 2011;60(4):1-4.
13. Thompson RF. Travel & Routine Immunizations: A Practical Guide for the Medical Office. 19th ed. Milwaukee, WI: Shoreland, Inc: 2001.
14. American Academy of Pediatrics. Pertussis. In: Pickering LK, Backer, CJ, Long SS, McMillan J, eds. Red Book: 2006 Report of the Committee on Infectious Diseases. 27th ed. Elk Grove Village, IL: American Academy of Pediatrics.
15. CDC. Immunization information systems (IIS). www.cdc.gov/vaccines/programs/iis/default.htm. Accessed May 12, 2011.
16. College of Physicians of Philadelphia. The history of vaccines: a project of the College of Physicians of Philadelphia (2011). www.history ofvaccines.org/content/articles/different-types-vaccines. Accessed May 12, 2011.
17. US Food and Drug Administration. Vaccines, blood, and biologics: December 18, 2009 Approval Letter—Zostavax. www.fda.gov/
BiologicsBloodVaccines/Vaccines/Approved Products/ucm195993.htm. Accessed May 12, 2011.
18. Merck & Co, Inc. Zostavax® (zoster vaccine live; product insert). www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/Approved Products/UCM132831.pdf. Accessed May 12, 2011.
19. Merck & Co, Inc. Pneumovax® 23 (pneumococcal vaccine, polyvalent, MSD; product information). www.merck.com/product/usa/pi_circulars/p/pneumovax_23/pneumovax_pi.pdf. Accessed May 16, 2011.
20. Macintyre CR, Egerton T, McCaughey M, et al. Concomitant administration of zoster and pneumococcal vaccines in adults ≥60 years old. Hum Vaccin. 2010;6(11):18-26.
21. CDC. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2008;57(RR-5):1–30.
22. Immunization Action Coalition. Vaccinate Adults. 2010 Aug;14(5). www.immunize.org/va. Accessed May 12, 2011.
23. CDC. Update: recommendations of the Advisory Committee on Immunization Practices (ACIP) regarding use of CSL seasonal influenza vaccine (Afluria) in the United States during 2010–2011. MMWR Morb Mortal Wkly Rep. 2010;59(31);989-992.
24. CDC. Updated recommendations for prevention of invasive pneumococcal disease among adults using the 23-valent pneumococcal polysaccharide vaccine (PPSV23). MMWR Morb Mortal Wkly Rep. 2010;59(RR-34):1102-1106.
25. Merck & Co, Inc. Varivax® varicella virus vaccine live (product information). www.merck
.com/product/usa/pi_circulars/v/varivax/varivax_pi.pdf. Accessed May 12, 2011.
26. Merck & Co, Inc. Gardasil® (human papillomavirus quadrivalent [types 6, 11, 16, and 18] vaccine, recombinant; product information). www.merck.com/product/usa/pi_circulars/g/gardasil/gardasil_ppi.pdf. Accessed May 12, 2011.
27. GlaxoSmithKline Biologicals. Cervarix (human papillomavirus bivalent [types 16 and 18] vaccine, recombinant; product information). http://us.gsk.com/products/assets/us_cervarix.pdf. Accessed May 12, 2011.
28. CDC. Syncope after vaccination—United States, January 2005-July 2007. MMWR Morb Mortal Wkly Rep. 2008;57(17):457-460.
29. Sanofi Pasteur. Meningococcal (groups A, C, Y, and W-135) polysaccharide diphtheria
toxoids conjugate vaccine Menactra® for intramuscular injection. www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/Approved
Products/UCM131170.pdf. Accessed May 12, 2011.
30. Novartis Vaccines and Diagnostics, Inc. Menveo® (meningococcal [groups A, C, Y and W-135] oligosaccharide diphtheria CRM197 conjugate vaccine solution for intramuscular injection; prescribing information highlights). www .fda.gov/downloads/biologicsbloodvaccines/vaccines/approvedproducts/ucm201349.pdf. Accessed May 12, 2011.
31. GlaxoSmithKline Biologicals. Havrix (hepatitis A vaccine, suspension for intramuscular injection; prescribing information highlights). http://us.gsk.com/products/assets/us_havrix .pdf. Accessed May 12, 2011.
32. Merck & Co, Inc. Vaqta (hepatitis A vaccine, inactivated; suspension for intramuscular injection; highlighted prescribing information). www.merck.com/product/usa/pi_circulars/v/vaqta/vaqta_pi.pdf. Accessed May 12, 2011.
33. Merck & Co, Inc. Recombivax HB® hepatitis B vaccine (recombinant; product information). www.merck.com/product/usa/pi_circulars/r/recombivax_hb/recombivax_pi.pdf. Accessed May 12, 2011.
34. GlaxoSmithKline Biologicals. Engerix-B® (hepatitis B vaccine, recombinant; prescribing information). http://us.gsk.com/products/assets/us_engerixb.pdf. Accessed May 12, 2011.
35. CDC. Tetanus and pertussis vaccination coverage among adults aged ≥ 18 years—United States, 1999 and 2008. MMWR Morb Mortal Wkly Rep. 2010;59(40):1302-1306.
Reported incidence rates of certain vaccine-preventable diseases—measles, rubella, diphtheria, polio, and tetanus—are low in the United States.1 However, as was demonstrated during the 2009-2010 flu season and the outbreak of H1N1 influenza,2 we cannot afford to be complacent in our attitudes toward vaccines and vaccination. New virus strains exist and can become endemic quickly and ravenously. Furthermore, certain vaccine-preventable illnesses are frequently reported among adult patients, including hepatitis B, herpes zoster, human papillomavirus infection, influenza, pertussis, and pneumococcal infection.3
Vigilance regarding vaccination of children and adults was addressed by the CDC’s Office of Disease Prevention and Health Promotion in Healthy People 2010.4 These target objectives are proposed to be retained in Healthy People 2020,5 as the objectives from 2010 have not been met. The Healthy People 2010 target for administration of the pneumococcal vaccine in adults ages 65 and older, for example, is 90%. As of 2008, research has shown, only 60% of that population was immunized.6
There are subgroups of immunocompromised people who will probably never achieve adequate antibody levels to ensure immunity to vaccine-preventable diseases (for example, measles and influenza can be deadly to the immunocompromised person, as they can be to the very young and the very old). This is an important reason why vaccination of the healthy population is essential: the concept of herd immunity.7 Herd immunity (or community immunity) suggests that if most people around you are immune to an infection and do not become ill, then there is no one who can infect you—even if you are not immune to the infection.
WHY SOME ADULTS MIGHT NEED VACCINES
Adults who were vaccinated as children may incorrectly assume that they are protected from disease for life. In the case of some diseases, this may be true. However:
• Some adults were never vaccinated as children
• Newer vaccines have been developed since many adults were children
• Immunity can begin to fade over time
• As we age, we become more susceptible to serious disease caused by common infections (eg, influenza, pneumococcus).8
Barriers to Vaccination
Barriers to vaccination are varied, but none is insurmountable. Some of these barriers include8-15:
Missed opportunities. Providers should address vaccination needs for both adults and children at each visit or encounter. According to the CDC, studies have shown that eliminating missed opportunities could increase vaccination coverage by as much as 20%.8,11
Provider misconceptions regarding vaccine contraindications, schedules, and simultaneous vaccine administration. These misconceptions may prompt providers to forego an opportunity to vaccinate. Up-to-date information about vaccinations and ongoing provider education are imperative to improve immunity among both adults and children against vaccine-preventable disease.8 Numerous Web sites and publications are instrumental and essential in furnishing the health care provider with the most current information about vaccinations (see Table 1, above, and Table 2,10,12-14).
A belief on patients’ part that they are fully vaccinated when they are not. It is important to provide a vaccination record and a return date at every vaccination encounter, even if just one vaccination has been administered on a given day. Participating in Immunization Information System,15 if one is available, is an efficient way to access computerized vaccine records easily at the point of contact.
Just as parents should be encouraged to bring their child’s vaccine record with them to every health care visit, adults are also called upon to maintain a record of all their vaccinations. Each entry in the immunization record should include:
• The type of vaccine and dose
• The site and route of administration
• The date that the vaccine was administered
• The date that the next dose is due
• The manufacturer and lot number
• The name, address, and title of the person who administered the vaccine.15
PRINCIPLES OF VACCINATION
There are two ways to acquire immunity: active and passive.
Active immunity is produced by the individual’s own immune system and usually represents a permanent immunity toward the antigen.10,16
Passive immunity is produced when the individual receives products of immunity made by another animal or a human and transferred to the host. Passive immunity can be accomplished by injection of these products or through the placenta in infants. This type of immunity is not permanent and wanes over time—usually within weeks or months.10,16
This article will concentrate on active immunity, acquired through the administration of vaccines.
CLASSIFICATION OF VACCINES
Vaccines are classified as either live, attenuated vaccine (viral or bacterial) or inactivated vaccine.
Live, attenuated vaccines are derived from “wild” or disease-causing viruses and bacteria. Through procedures conducted in the laboratory, these wild organisms are weakened or attenuated. The live, attenuated vaccine must grow and replicate in the vaccinated person in order to stimulate an immune response. However, because the organism has been weakened, it usually does not cause disease or illness.10,16
The immune response to a live, attenuated vaccine is virtually identical to a response produced by natural infection. In rare instances, however, live, attenuated vaccines may cause severe or fatal reactions as a result of uncontrolled replication of the organism. This occurs only in individuals who are significantly immunocompromised.10,16
Inactivated vaccines are produced by growing the viral or bacterial organism in a culture medium, then using heat and/or chemicals to inactivate the organism. Because inactivated vaccines are not alive, they cannot replicate, and therefore cannot cause disease, even in an immunodeficient patient. The immune response to an inactivated vaccine is mostly humoral (in contrast to the natural infection response of a live vaccine), and little or no cellular immunity is produced.10,16
Inactivated vaccines always require multiple doses, gradually building up a protective immune response. The antibody titers diminish over time, so some inactivated vaccines may require periodic doses to “boost” or increase the titers.16
Some vaccines, such as hepatitis B vaccine, lead to the development of immune memory, which stays intact for at least 20 years following immunization. Immune memory occurs during replication of B cells and T cells; some cells will become long-lived memory cells that will “remember” the pathogen and produce an immune response if the pathogen is detected again. In this case, boosters are not recommended.10
Toxoids are a type of vaccine made from the inactivated toxin of a bacterium—not the bacterium itself. Tetanus and diphtheria vaccines are examples of toxoid vaccines.10,16
Subunit and conjugate vaccines are segments of the pathogen. A subunit vaccine can be created via genetic engineering. The end result is a recombinant vaccine that can stimulate cell memory (eg, hepatitis B vaccine).10,16 Conjugate vaccines, which are similar to recombinant vaccines, are made by combining two different components to prompt a more powerful, combined immune response.10
Spacing of Live-Virus and Inactivated Vaccines
There are almost no spacing requirements between two or more inactivated vaccines8 (see Table 38,14 for spacing recommendations from the Advisory Committee on Immunization Practices [ACIP]). The only vaccines that must be spaced at least four weeks apart are live-virus vaccines—that is, if they are not given on the same day. Studies have shown that the immune response to a live-virus vaccine may be impaired if it is administered within 30 days of another live-virus vaccine. Inactivated vaccines, on the other hand, may usually be administered at any time after or before a live-virus vaccine.8
One exception to this statement is the administration of Zostavax (zoster live-virus vaccine) with Pneumovax 23 (inactivated pneumococcal vaccine, polyvalent, MSD).17-19 The manufacturer of the two vaccines recommends a spacing of at least four weeks between them, based on research showing that concomitant use may result in reduced immunogenicity for Zostavax.20 However, as of this writing, ACIP has not revised its statement that both vaccines can be given at the same time or at any time before or after each other.21
Live-virus vaccines currently licensed in the US provide protection against diseases including measles/mumps/rubella, varicella, zoster (ie, shingles), influenza, and yellow fever.
ADULT VACCINATION HIGHLIGHTS
A summary of 2011 recommendations for adult immunization from ACIP is shown in Table 412,21. The following information is specific to each of the vaccine-preventable illnesses of concern in adults.12,13,22
Seasonal Influenza
The options to protect the adult patient against seasonal influenza are a trivalent, inactivated influenza vaccine (TIV; Fluzone, high-dose Fluzone for adults ages 65 and older, Fluvirin, Fluarix, FluLaval, Afluria, Agriflu) or a live, attenuated influenza vaccine (LAIV; FluMist).2,23 Dosage of TIV for adults is 0.5 mL IM in the deltoid once annually. For adults ages 49 and younger, LAIV is administered at 0.2 mL intranasally, once per year.
ACIP now recommends universal influenza vaccination for all persons ages 6 months and older with no contraindications.2,8 Strong consideration should be given to concurrent administration of influenza vaccine and pneumococcal vaccine to high-risk persons not previously vaccinated against pneumococcal disease.12
Note: When influenza and pneumococcal vaccines are given at the same time, they should be administered in opposite arms to reduce the risk of adverse reactions or a decreased antibody response to either vaccine.18,19
Pneumococcal Polysaccharide (PPSV)
Pneumovax 2319 is administered as a 0.5-mL dose IM in the deltoid or subcutaneously in the upper arm. The vaccine is recommended for8,19,24:
• Adults ages 65 and older who have not been previously vaccinated
• Adults now 65 and older who received PPSV vaccine at least five years ago and were younger than 65 at that time
• Adults ages 19 through 64 years who have asthma or who smoke24
• Any adult with the following underlying medical conditions: chronic heart or lung disease, diabetes mellitus, cerebrospinal fluid leaks, cochlear implants, chronic liver disease, cirrhosis, chronic alcoholism, functional or anatomic asplenia, and immunocompromising conditions (HIV infection, diseases that require immunosuppressive therapy, chemotherapy, or radiation therapy; congenital immunodeficiency).24
A one-time revaccination is recommended after five years for persons ages 19 to 64 who have chronic renal failure, nephrotic syndrome, or functional or anatomic asplenia, and for those who are immunocompromised.24
Note: According to the manufacturer of Zostavax18 and Pneumovax,23,19 these vaccines should not be given at the same time, as research has shown that Zostavax immunogenicity is reduced as a result.18-20
Zoster
For adults ages 60 and older, Zostavax18 is administered in a single 0.65-mL dose, subcutaneously in the upper arm. Providers are not required to ask about varicella vaccination history or history of varicella disease before administering the vaccine. Adults ages 60 and older who have previously had shingles can still be vaccinated during a routine health care visit.10,21,22
Immunization is contraindicated in adults with a previous anaphylactic reaction to neomycin or gelatin, although a nonanaphylactic reaction to neomycin (most commonly, contact dermatitis) is not considered a contraindication.8
Any adult patient who has close household or occupational contact with persons at risk for severe varicella (eg, infants) need not take precautions after receiving the zoster vaccine, except in the rare case in which a varicella-like rash develops.10,21,22
Note: Review the note appearing in “Pneumococcal Polysaccharide (PPSV),” above, regarding coadministration of Zostavax and Pneumovax 23.18-20
Varicella
Adults who were born in the US before 1980 are considered immune to varicella and don’t need to be vaccinated, with the exception of health care workers, pregnant women, and immunocompromised persons. Nonimmune healthy adults who have not previously undergone vaccination should receive two 0.5-mL doses of Varivax, administered subcutaneously, four to eight weeks apart.25
Immunization is contraindicated in adults with a previous anaphylactic reaction to neomycin or gelatin, although a nonanaphylactic reaction to neomycin (eg, contact dermatitis) is not considered a contraindication.8
Measles, Mumps, Rubella (MMR)
The MMR vaccine is administered at 0.5 mL, given subcutaneously in the posterolateral fat of the upper arm.8
MMR-susceptible adults who were born during or since 1957 and are not at increased risk (see below) need only one dose of the MMR vaccine; those considered at increased risk need two doses, and a second dose can also be considered during an outbreak. Adults who require two doses should wait at least four weeks between the first and second doses.12
The following factors place adults at increased risk for MMR:
• Anticipated international travel
• Being a student in a post–secondary educational setting
• Working in a health care facility
• Recent exposure to measles, or an outbreak of measles or mumps
• Previous vaccination with killed measles vaccine
• Previous vaccination with an unknown measles vaccine between 1963 and 1967.
Also at risk are health care workers born before 1957 who have no evidence of immunity, and women who plan to become pregnant and have no evidence of immunity.8,12
Tetanus, Diphtheria, Pertussis
Options for adults include a vaccine against tetanus and diphtheria (Td; Decavac); or a vaccine that protects against tetanus, diphtheria, and acellular pertussis (Tdap; Adacel, Boostrix). Adults who have not been previously vaccinated should receive one dose of Tdap and two doses of Td (the first, one month after Tdap; the second at six to 12 months after the Tdap). Each is administered as a 0.5-mL dose IM in the deltoid. A booster dose is recommended every 10 years but can be given earlier in patients who sustain wounds or who anticipate international travel.8,12
Adults ages 19 through 64 should receive a single dose of Tdap in place of a booster dose if the last Td dose was administered at least 10 years earlier and the patient has not previously received Tdap. Additionally, a dose of Tdap (if not previously given) is recommended for postpartum women, close contacts of infants younger than 12 months, and all health care workers with direct patient contact. An interval as short as two years from the last Td is suggested; shorter intervals may be appropriate.8,12
According to the new 2011 recommendations, persons ages 65 and older who have close contact with an infant younger than 12 months should be vaccinated with Tdap, and any person age 65 or older may be vaccinated with Tdap. Also added is a recommendation to administer Tdap, regardless of the interval since the patient received his or her most recent Td-containing vaccine.8,12
Human Papillomavirus (HPV)
Gardasil26 protects both female and male patients against HPV infection; Cervarix27 is indicated only for female patients. Either quadrivalent vaccine or bivalent vaccine is recommended for female patients.12
In women ages 26 and younger, Gardasil (0.5 mL IM, administered in the deltoid at 0 month, 2 months, and 6 months) provides protection against diseases caused by HPV types 6, 11, 16, and 18 (including cervical, vaginal, and vulvar cancer caused by HPV types 16 and 18). In men ages 26 and younger, Gardasil provides protection against genital warts caused by HPV types 6 and 11.26
Cervarix,27 administered at 0 month, 1 month, and 6 months (0.5 mL IM in the deltoid), provides protection for women ages 25 and younger against cervical cancer and precancerous lesions caused by HPV types 16 and 18.
Caution: Patients should be advised to sit or lie down when the HPV vaccine is administered, and they should be observed for the subsequent 15 minutes. Syncope can occur after vaccination—most commonly among adolescents and young adults.28 Convulsive syncope has been reported.
Meningococcal Disease
Two vaccines are available to protect against meningococcal disease: Menactra29 (meningococcal groups A, C, Y, and W-135 polysaccharide diphtheria toxoids conjugate vaccine); and Menveo30 (meningococcal groups A, C, Y, and W-135 oligosaccharide diphtheria CRM197 conjugate vaccine). Both are administered in the deltoid, 0.5 mL IM.
The following patients should be considered for vaccination:
• College freshmen living in dormitories, as well as college students with immune deficiencies, as they are at higher risk for meningococcal disease
• Patients who travel to or reside in countries in which Neisseria meningitidis is epidemic (particularly those with the potential for prolonged contact with the local population)
• Travelers to Saudi Arabia for pilgrimage to Mecca (the Hajj)
• Patients with anatomical or functional asplenia (two-dose series).
A two-dose series of meningococcal conjugate vaccine is also recommended for adults with persistent complement component deficiencies, and for those with HIV infection who are vaccinated.12
Hepatitis A
Two hepatitis A vaccines (both inactivated) can be used interchangeably: Havrix31 and Vaqta.32 Dosing for both vaccines in 18-year-old patients is 0.5 mL IM in the deltoid at 0 months, then at 6 to 12 months. In patients ages 19 and older, administration is the same, with the exception of increased dosing (1.0 mL IM).
Vaccination against hepatitis A is recommended for men who have sex with men, and for all adult patients who12:
• Travel to or work in areas where risk for hepatitis A transmission is high (especially those who take frequent trips or experience prolonged stays)
• Use injection drugs
• Have chronic liver disease
• Receive clotting factor concentrates for treatment of a blood-clotting disorder
• May have been exposed to hepatitis A in the previous two weeks
• Wish to be vaccinated against hepatitis A to avoid future infection.
Hepatitis B
Recombivax HB33 and Engerix-B34 are the two vaccines available to protect patients against hepatitis B (HBV), and they can be used interchangeably.12 In patients from birth through age 19, Recombivax HB33 or Engerix-B34 is given as 0.5 mL IM in the deltoid at 0, 1, and 6 months; patients ages 20 and older receive an increased dose (1.0 mL IM), with administration otherwise the same. According to the manufacturer of Recombivax HB,33 patients age 11 through 15 may be given either three doses of 0.5 mL or two doses of 1.0 mL.
The following adults are advised to undergo vaccination for HBV:
• At-risk, unvaccinated adults
• Those requesting protection against HBV infection
• Those planning to travel to areas where HBV is common
• Household contacts of a patient with chronic HBV infection, and sexual partners of a patient with HBV infection
• Adults with chronic liver disease
• Men who have sex with men
• Sexually active adults who are not in a long-term, mutually monogamous relationship
• Adults who are being evaluated or treated for a sexually transmitted disease, including HIV infection
• Health care or public safety workers who may be exposed to blood or blood-contaminated body fluids
• Workers and residents in facilities for developmentally disabled persons
• Patients undergoing or anticipating dialysis
• Adults who inject illegal drugs or who have done so recently.12
CONTRAINDICATIONS AND PRECAUTIONS FOR VACCINES COMMONLY USED IN ADULTS
See Table 5,8,22 for a summary of contraindications and precautions from ACIP and the Immunization Action Coalition that are associated with vaccinations mentioned in this article. A more complete summary can be found at www.im munize.org/catg.d/p3072a.pdf.
Conclusion
The adult patient’s vaccination status should be addressed at each health care encounter, and current recommendations should be followed. The duration of efficacy for vaccines is not an exact science. Many vaccines licensed in the US are relatively new, and recommendations for boosters for some of these vaccines will be forthcoming as more data are gathered. For example, the recommendation that a booster dose of Tdap be given to adults resulted from the recent increase in reported pertussis cases.35
Providers armed with the most current information and resources represent the forefront in ensuring that the US adult population is adequately immunized.
REFERENCES
1. World Health Organization. Immunization surveillance, assessment, and monitoring (2010). www.who.int/immunization_monitor ing/en. Accessed May 12, 2011.
2. Fiore AE, Uyeki TM, Broder K, et al; Centers for Disease Control and Prevention. Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Morb Mortal Wkly Rep. 2010;59(RR-08);1-62.
3. Schaffner W. Update on vaccine-preventable diseases: are adults in your community adequately protected? J Fam Pract. 2008;57(4 suppl):S1-S11.
4. CDC. Healthy People 2010: Objectives for Improving Health. www.healthypeople.gov. Accessed May 6, 2011.
5. US Department of Health and Human Services. Developing Healthy People 2020: immunization and infectious diseases. www.healthy people.gov/2020. Accessed May 12, 2011.
6. Lu PJ, Nuorti JP. Pneumococcal polysaccharide vaccination among adults aged 65 years and older, United States, 1989-2008. Am J Prev Med. 2010;39(4):287-295.
7. National Institute of Allergy and Infectious Diseases, NIH. Community immunity (“herd” immunity) (2010). www.niaid.nih.gov/topics/pages/communityimmunity.aspx. Accessed May 12, 2011.
8. National Center for Immunization and Respiratory Diseases. General recommendations on immunization: recommendations of the Advisory Committee on Immunization Practices (ADIP). MMWR Recomm Rep. 2011;60(2):1-64.
9. High KP. Overcoming barriers to adult immunization. J Am Osteopath Assoc. 2009;109(6): 525-528.
10. CDC; Atkinson W, Wolfe S, Hamborsky J, eds. Epidemiology and Prevention of Vaccine-Preventable Diseases (Pink Book). 12th ed. Washington, DC: Public Health Foundation, 2011.
11. CDC. Vaccine-preventable diseases: improving vaccination coverage in children, adolescents, and adults: a report on recommendations from the Task Force on Community Preventive Services. MMWR Recomm Rep. 1999;48(RR-8):1-15.
12. CDC. Recommended adult immunization schedule: United States, 2011. MMWR Morb Mortal Wkly Rep. 2011;60(4):1-4.
13. Thompson RF. Travel & Routine Immunizations: A Practical Guide for the Medical Office. 19th ed. Milwaukee, WI: Shoreland, Inc: 2001.
14. American Academy of Pediatrics. Pertussis. In: Pickering LK, Backer, CJ, Long SS, McMillan J, eds. Red Book: 2006 Report of the Committee on Infectious Diseases. 27th ed. Elk Grove Village, IL: American Academy of Pediatrics.
15. CDC. Immunization information systems (IIS). www.cdc.gov/vaccines/programs/iis/default.htm. Accessed May 12, 2011.
16. College of Physicians of Philadelphia. The history of vaccines: a project of the College of Physicians of Philadelphia (2011). www.history ofvaccines.org/content/articles/different-types-vaccines. Accessed May 12, 2011.
17. US Food and Drug Administration. Vaccines, blood, and biologics: December 18, 2009 Approval Letter—Zostavax. www.fda.gov/
BiologicsBloodVaccines/Vaccines/Approved Products/ucm195993.htm. Accessed May 12, 2011.
18. Merck & Co, Inc. Zostavax® (zoster vaccine live; product insert). www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/Approved Products/UCM132831.pdf. Accessed May 12, 2011.
19. Merck & Co, Inc. Pneumovax® 23 (pneumococcal vaccine, polyvalent, MSD; product information). www.merck.com/product/usa/pi_circulars/p/pneumovax_23/pneumovax_pi.pdf. Accessed May 16, 2011.
20. Macintyre CR, Egerton T, McCaughey M, et al. Concomitant administration of zoster and pneumococcal vaccines in adults ≥60 years old. Hum Vaccin. 2010;6(11):18-26.
21. CDC. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2008;57(RR-5):1–30.
22. Immunization Action Coalition. Vaccinate Adults. 2010 Aug;14(5). www.immunize.org/va. Accessed May 12, 2011.
23. CDC. Update: recommendations of the Advisory Committee on Immunization Practices (ACIP) regarding use of CSL seasonal influenza vaccine (Afluria) in the United States during 2010–2011. MMWR Morb Mortal Wkly Rep. 2010;59(31);989-992.
24. CDC. Updated recommendations for prevention of invasive pneumococcal disease among adults using the 23-valent pneumococcal polysaccharide vaccine (PPSV23). MMWR Morb Mortal Wkly Rep. 2010;59(RR-34):1102-1106.
25. Merck & Co, Inc. Varivax® varicella virus vaccine live (product information). www.merck
.com/product/usa/pi_circulars/v/varivax/varivax_pi.pdf. Accessed May 12, 2011.
26. Merck & Co, Inc. Gardasil® (human papillomavirus quadrivalent [types 6, 11, 16, and 18] vaccine, recombinant; product information). www.merck.com/product/usa/pi_circulars/g/gardasil/gardasil_ppi.pdf. Accessed May 12, 2011.
27. GlaxoSmithKline Biologicals. Cervarix (human papillomavirus bivalent [types 16 and 18] vaccine, recombinant; product information). http://us.gsk.com/products/assets/us_cervarix.pdf. Accessed May 12, 2011.
28. CDC. Syncope after vaccination—United States, January 2005-July 2007. MMWR Morb Mortal Wkly Rep. 2008;57(17):457-460.
29. Sanofi Pasteur. Meningococcal (groups A, C, Y, and W-135) polysaccharide diphtheria
toxoids conjugate vaccine Menactra® for intramuscular injection. www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/Approved
Products/UCM131170.pdf. Accessed May 12, 2011.
30. Novartis Vaccines and Diagnostics, Inc. Menveo® (meningococcal [groups A, C, Y and W-135] oligosaccharide diphtheria CRM197 conjugate vaccine solution for intramuscular injection; prescribing information highlights). www .fda.gov/downloads/biologicsbloodvaccines/vaccines/approvedproducts/ucm201349.pdf. Accessed May 12, 2011.
31. GlaxoSmithKline Biologicals. Havrix (hepatitis A vaccine, suspension for intramuscular injection; prescribing information highlights). http://us.gsk.com/products/assets/us_havrix .pdf. Accessed May 12, 2011.
32. Merck & Co, Inc. Vaqta (hepatitis A vaccine, inactivated; suspension for intramuscular injection; highlighted prescribing information). www.merck.com/product/usa/pi_circulars/v/vaqta/vaqta_pi.pdf. Accessed May 12, 2011.
33. Merck & Co, Inc. Recombivax HB® hepatitis B vaccine (recombinant; product information). www.merck.com/product/usa/pi_circulars/r/recombivax_hb/recombivax_pi.pdf. Accessed May 12, 2011.
34. GlaxoSmithKline Biologicals. Engerix-B® (hepatitis B vaccine, recombinant; prescribing information). http://us.gsk.com/products/assets/us_engerixb.pdf. Accessed May 12, 2011.
35. CDC. Tetanus and pertussis vaccination coverage among adults aged ≥ 18 years—United States, 1999 and 2008. MMWR Morb Mortal Wkly Rep. 2010;59(40):1302-1306.
Immune thrombocytopenia: No longer ‘idiopathic’
Once regarded as idiopathic, immune thrombocytopenia (ITP) is now understood to have a complex pathogenesis, involving the evolution of antibodies against multiple platelet antigens leading to reduced platelet survival as well as impaired platelet production. For this reason, multiple therapies with different mechanisms of action are available to treat ITP, though not all of them are effective for individual patients.
In this article, I discuss the pathogenesis, demographics, manifestations, diagnosis, and management of ITP.
THE NAME AND THE CUTOFF HAVE CHANGED
The term ITP formerly was used to refer to “idiopathic” or “immune” thrombocytopenic purpura. However, although not all aspects of the pathogenesis of ITP are understood, the disease can no longer be considered idiopathic. In addition, many patients do not have purpura at the time of diagnosis. Though the abbreviation “ITP” remains the same, it now refers to immune thrombocytopenia, which can be either primary or secondary.1
ITP is defined as a platelet count of less than 100 × 109/L (100,000/μL) with no evidence of leukopenia or anemia. This cutoff point is new: in the past, ITP was defined as a platelet count of less than 150 × 109/L, which is the threshold for a normal platelet count in most laboratories.
The platelet threshold of 100 × 109/L was based on a study by Stasi et al,2 who followed 217 otherwise healthy people who had an incidental finding of mild thrombocytopenia (platelet count 100–150 × 109/L). Within 6 months, the platelet count rose to more than 150 × 109/L in 23, while three had either worsening thrombocytopenia or were diagnosed with other conditions. During long-term follow-up (median 64 months), 109 of the remaining 191 individuals remained stable, 13 developed counts greater than 150 × 109/L, 12 developed ITP, 13 developed an autoimmune disorder, 18 developed other disorders, and 26 were lost to follow-up. The 10-year probability of developing ITP, defined as a platelet count persistently below 100 × 109/L, was only 6.9%, indicating that the chances are small that a person with an isolated finding of mild, stable thrombocytopenia will develop ITP.
Categories of ITP
An international working group designated to standardize terminology has divided ITP into two major diagnostic categories.1 The proportion of patients within each is not well established and varies by region and demographic characteristics.
Primary ITP accounts for the majority of cases in most studies; other conditions associated with thrombocytopenia are absent.
Secondary ITP can be due to infection with a number of agents, including hepatitis C virus (HCV), human immunodeficiency virus (HIV), and Helicobacter pylori. Other causes include underlying autoimmune and lymphoproliferative disorders such as systemic lupus erythematosus, Wiskott-Aldrich syndrome, chronic lymphocytic leukemia, antiphospholipid syndrome, and common variable immunodeficiency, as well as drugs such as quinine and trimethoprim-sulfamethoxazole.
Categories of ITP have also been established to facilitate management decisions, as follows:
Newly diagnosed ITP refers to ITP diagnosed within the preceding 3 months.
Persistent ITP refers to ITP diagnosed 3 to 12 months previously, and includes ITP in patients not reaching spontaneous remission and in those not maintaining a complete response off therapy. (When ITP spontaneously remits in adults, it usually does so within the first 12 months after the condition is diagnosed.)
Chronic ITP: Lasting for more than 12 months.
Severe ITP is defined by bleeding at presentation sufficient to mandate treatment, or new bleeding symptoms requiring additional therapeutic intervention with a different platelet-enhancing agent or an increased dosage of a current agent.
ITP IS COMMON IN OLDER ADULTS
We previously believed that ITP was a disorder that primarily affected women in their third and fourth decades. However, this was not borne out in recent epidemiologic studies, which have demonstrated that the highest age-specific incidence of ITP occurs in the elderly. This may potentially reflect the development of immune dysregulation as a consequence of aging. There is a female preponderance in the incidence of ITP throughout adulthood until around age 60, after which the overall incidence increases in both sexes, and the ratio of affected women to men is about equal.3,4 Thus, even though thrombocytopenia in the elderly may reflect myelodysplasia in some individuals, ITP is much more common than previously appreciated.
Previous guidelines from the American Society of Hematology suggested that a bone marrow examination be strongly considered in patients over age 60 with suspected ITP. With the realization that ITP occurs more commonly in the elderly, it is apparent that bone marrow examination is not necessary in this group if there are no other cytopenias present and the physical examination and blood smear are consistent with ITP.
In children, ITP has a peak incidence between ages 5 and 6, and behaves differently from the adult syndrome. ITP in children usually follows an apparent viral infection and tends to be self-limited, with approximately 80% of cases resolving spontaneously within 6 months. In contrast, adult ITP usually develops into a chronic disease.
BLEEDING MAY NOT BE PRESENT AT DIAGNOSIS
ITP is now recognized as a diverse syndrome with a constellation of signs and symptoms.
Petechiae are pinpoint microvascular hemorrhages that do not blanch with pressure. This distinguishes them from small hemangiomas, which look similar but blanch transiently with pressure. Petechiae tend to occur on dependent areas, particularly the hands and feet, when the platelet count drops below approximately 15 × 109/L.
Ecchymoses (dry purpura) appear as large bruises.
Mucosal bleeding (wet purpura) involves the oral mucosa. Particularly in children, wet purpura tends to be associated with systemic bleeding complications, involving the gastrointestinal tract for example. The incidence of intracranial hemorrhage, though very low, may also be increased in patients with wet purpura.
Other bleeding manifestations may include heavy menstrual bleeding, oral bleeding, and epistaxis.
Bleeding is generally but not entirely proportional to the platelet count. In a study of adults with newly diagnosed ITP and a platelet count of less than 50 × 109/L,4 the presenting symptom was hemorrhage in 12% and purpura in 58%.4 Remarkably, 28% of cases were asymptomatic, with some patients remaining free of symptoms for years despite very low platelet counts. More than half of patients with a platelet count of 30 to 50 × 109/L have no symptoms.3,4
A PARADOXICAL RISK OF THROMBOSIS
Although ITP is primarily a bleeding disorder, it is paradoxically also associated with thrombosis. Sarpatwari et al,5 in a study in the United Kingdom, found that the 4-year incidence of thromboembolic events was about 1.3 times higher in patients with ITP than in matched controls.
The reason for the increased risk of thrombosis is not clear. It is possible that in some patients, antiphospholipid antibodies may contribute to the development of thrombosis, although this has not been confirmed in all studies.
A DIAGNOSIS OF EXCLUSION
The evaluation of any patient suspected of having ITP should include the following:
- Personal history, with special attention to drugs and to medical conditions that could cause thrombocytopenia.
- Family history. ITP may occasionally be mistaken for an inherited cause of thrombocytopenia. The presence of the latter can often be confirmed by review of the peripheral blood film of the patient as well as other family members with thrombocytopenia. ITP is generally not considered to be an inherited disorder, although some HLA alleles may be more prevalent in ITP patients.
- Physical examination, with special attention to lymphadenopathy or splenomegaly, which may suggest an underlying malignancy such as a lymphoproliferative disorder. In general, patients with ITP have a normal physical examination, except for signs of bleeding or bruising in some.
- Laboratory tests, including a complete blood cell count, blood smear, reticulocyte count, Rh typing, and direct antiglobulin (Coombs) test.
In ITP, the peripheral blood smear should appear normal except for the presence of thrombocytopenia, although platelets may be mildly enlarged in some individuals. Red cell and leukocyte morphology is normal. It is important to exclude the presence of schistocytes (red cell fragments) and nucleated red blood cells, which often indicate a microangiopathic hemolytic anemia caused by disorders such as thrombotic thrombocytopenic purpura.
International guidelines suggest that testing for reduced immunoglobulin levels (as seen in common variable hypogammaglobulinemia) and HIV, HCV, and H pylori infections should also be considered. Coincident HCV infection is particularly high in some regions. Other cytopenias or abnormalities in the history or physical examination may prompt bone marrow examination. Testing for antiphospholipid antibodies, antinuclear antibodies, parvovirus, and cytomegalovirus may also be indicated in specific individuals. Testing for antiplatelet antibodies is not commonly performed in the current era because of its relatively low sensitivity and specificity.
Ultimately, the diagnosis of ITP is clinical, however, and cannot be established by any specific laboratory assay. Perhaps the best diagnostic study is assessment of the patient’s response to ITP therapy.
ITP INVOLVES ACCELERATED PLATELET DESTRUCTION
In 1951, William Harrington, a fellow at Washington University, infused blood from a patient with ITP into himself and, subsequently, into normal volunteers.6 The majority of recipients demonstrated significant reductions in the platelet count, sometimes severe. This fascinating and bold experiment provided the first demonstration that ITP was caused by a factor that circulates in blood. What is often not emphasized, however, is that some recipients did not develop thrombocytopenia, suggesting an alternative mechanism.
Later, Luiken et al7 and Hirschman and Shulman8 demonstrated that the transmissible agent in the blood was immunoglobulin, primarily immunoglobulin G (IgG). We now understand that much of the pathogenesis of ITP is caused by antibodies against platelet glycoproteins, most commonly platelet glycoprotein IIb/IIIa, the platelet fibrinogen receptor. Most patients, especially those with chronic ITP, also have antibodies against other platelet glycoproteins, including glycoprotein Ib/IX (the receptor for von Willebrand factor), and glycoprotein Ia/IIa, a collagen receptor. It is commonly believed that ITP may begin with antibodies against a single glycoprotein, which leads to accelerated clearance of antibody-coated platelets in the spleen. Degradation of cleared platelets by splenic macrophages leads to the release and subsequent presentation of antigenic peptides from proteolyzed platelet components, including glycoproteins, on the macrophage or dendritic cell. This may lead to recruitment and activation of specific T cells that in turn interact with and stimulate B cells to produce new antibodies against the platelet-derived peptides. This phenomenon, known as epitope spreading, may be responsible for the fact that most patients with long-standing, chronic ITP develop autoantibodies against multiple platelet glycoprotein targets.9
Several agents used in the treatment of ITP may work by impairing clearance of antibody-coated platelets by the reticuloendothelial system. One of many potential mechanisms underlying the therapeutic efficacy of intravenous immunoglobulin (IVIG) may be its ability to interact with a specific type of Fc gamma receptor, Fc gamma RIIb. IVIG therapy stimulates increased expression of this receptor, which in turn may impair the function of other “activating” Fc gamma receptors responsible for platelet clearance.10,11
ITP associated with infection may arise due to molecular mimicry. HCV, HIV, and H pylori contain amino acid sequences that may have structural similarity to regions within platelet glycoproteins. Thus, antibodies directed against the pathogen may cross-react with the glycoprotein, leading to thrombocytopenia.12–15
HCV has been found in up to one-third of cases of ITP in some centers.16–20H pylori-associated ITP is very common in some regions, particularly in Japan, and may often resolve after eradication of the infection. However, in the United States, eradication of H pylori generally does not improve the course of ITP. This may reflect antigen mimicry, in particular the fact that different cagA proteins are expressed by different strains of H pylori in certain regions of the world.
Our understanding of the immunologic basis of ITP has greatly expanded over the last decade. Although it has long been known that B cells produce autoantibodies, T cells have more recently been shown to play a critical role in regulating B-cell-mediated autoantibody production in ITP. In some situations, T cells may directly lyse platelets, or suppress megakaryopoiesis. This may explain why some patients who do not respond to standard B-cell-targeted therapy may respond to cyclosporine or other T-cell-directed agents.
ANOTHER MECHANISM OF ITP: REDUCED PLATELET PRODUCTION
In addition to accelerated platelet destruction, ITP is also associated with decreased platelet production by megakaryocytes in the bone marrow.21–25
Increased platelet destruction and reduced platelet production are likely two ends of a spectrum of ITP, and most patients likely have some degree of both processes. This concept helps explain why different drug strategies are more effective in some patients than in others.
ARE THE RISKS OF THERAPY JUSTIFIED?
It is important to understand the natural history of ITP to determine whether the risks of therapy are justified.
A 2001 study from the Netherlands26 followed 134 patients with primary ITP for 10 years: 90% had taken prednisone or had had a splenectomy. Within 2 years, 85% of these patients had platelet counts above 30 × 109/L off therapy. Although this group likely experienced more bleeding and bruising than the general population, the mortality rate was not increased. Another 6% also achieved a platelet count above 30 × 109/L, but required chronic maintenance therapy (usually steroids) to do so. This group led a nearly normal life but had more hospitalizations. The remaining 9% of patients had refractory ITP, with platelet counts remaining below 30 × 109/L despite therapy. This group had a death rate 4.2 times that of age-matched controls. About half died of bleeding and the others died of opportunistic infections to which they were susceptible because of long-term steroid therapy.
This study was influential in the general opinion that 30 × 109/L is a reasonable cutoff for treating ITP. An international consensus report states that treatment is rarely indicated in patients with platelet counts above 50 × 109/L in the absence of bleeding due to platelet dysfunction or other hemostatic defect, trauma, or surgery.27 Although this number is not supported by evidence-based data, it is a reasonable threshold endorsed by an international working group.27 Individual factors must be weighed heavily: for example, an athlete involved in contact sports requires a higher platelet count in order to play safely.

FIRST-LINE THERAPIES
First-line therapies for ITP include corticosteroids, IVIG, and anti-Rho(D) immune globulin (WinRho).27
Corticosteroids are standard therapy
Corticosteroids can be given in one of two ways:
Standard prednisone therapy, ie, 1 to 2 mg/kg per day, is given until a response is seen, and then tapered. Some maintain therapy for an additional week before tapering. There are no guidelines on how to taper: some decrease the dosage by 50% per week, although many recommend going more slowly, particularly at the lower range of dosing.
Up to 85% of patients achieve a clinical response, usually within 7 to 10 days, with platelet counts peaking in 2 to 4 weeks. Unfortunately, only about 15% of patients maintain the response over the subsequent 6 to 12 months. Restarting prednisone often initiates a vicious circle and makes patients vulnerable to steroid toxicities.
“Pulse” dexamethasone therapy consists of 40 mg per day for 4 days for one to three cycles. (Dexamethasone 1 mg is equivalent to about 10 mg of prednisone.)
Pulse dexamethasone therapy as an initial approach to ITP has been developed during the past decade and has been used primarily in research studies. This regimen evolved from studies of patients with multiple myelomas and has the potential to induce more durable remissions in some patients with newly diagnosed ITP.29 However, high-dose corticosteroids may be associated with increased toxicity, at least in the short term, and should be used cautiously. A study to address the role of high-dose vs standard-dose steroid therapy has recently been opened under the guidance of the Transfusion Medicine–Hemostasis Clinical Trials Network of the National Heart, Lung, and Blood Institute.
Immunoglobulin is useful for very low platelet counts and bleeding
Another primary therapy for ITP is IVIG 0.5 to 2.0 g/kg over 2 to 5 days. Its efficacy is similar to that of prednisone: about 65% of patients achieve a platelet count above 100 × 109/L, and 85% achieve 50 × 109/L. However, most responses are transient, and a significant minority of cases become refractory to IVIG after repeated infusions.
IVIG is associated with numerous adverse effects, including thrombosis, renal insufficiency, headache, and anaphylaxis in IgA-deficient patients. It also converts the direct antiglobulin test to positive. IVIG is expensive, is inconvenient to administer, and may require lengthy infusions depending on the formulation.
Although IVIG is not a good long-term therapy, it can help raise the platelet count relatively quickly in patients who present with severe thrombocytopenia accompanied by bleeding. Such patients should be treated with high-dose steroids, IVIG, and platelet transfusions. IVIG may also be useful to increase platelet counts prior to interventional procedures.
Intravenous anti-Rho(D)
Anti-Rho(D) is an alternative to IVIG in patients who are Rho(D)-positive and have an intact spleen. Anti-Rho(D) binds to Rh-positive red blood cells, causing them to be cleared in the reticuloendothelial system and blocking the clearance of antibody-coated platelets. In effect, red cells are sacrificed to save platelets, but because there are many more red cells than platelets, the benefits usually outweigh the risks.
The initial dose is 50 μg/kg given intravenously over 2 to 5 minutes. Anti-Rho(D) should not be given to patients whose hemoglobin level is less than 10 g/dL or who have compromised bone marrow function. It is ineffective in Rh-negative patients or those who have undergone splenectomy.
Accelerated hemolysis is a rare but severe possible adverse event associated with this therapy, occurring in slightly more than 1 in 1,000 infusions. About 1 out of every 20,000 patients develops disseminated intravascular coagulation.30 Its cause is poorly understood, and it is probably an accelerated extravascular rather than an intravascular event. The US Food and Drug Administration has recently issued a black-box warning cautioning that patients who receive anti-Rho(D) should remain in a health care setting for 8 hours after treatment, although most cases of accelerated hemolysis occur within 4 hours. Moreover, it is possible that many of these cases can be avoided by appropriate patient selection.
SECOND-LINE THERAPIES
Second-line therapies, as designated by the international working group, include azathioprine (Imuran), cyclosporine A, cyclophosphamide (Cytoxan), danazol (Danocrine), dapsone, mycophenolate mofetil (CellCept), rituximab (Rituxan), splenectomy, thrombopoietin receptor agonists, and vinca alkaloids.27 Only the most commonly used therapies will be briefly discussed below.
The evidence for efficacy of the cytotoxic agents, ie, cyclophosphamide, the vinca alkaloids, and azathioprine, comes from small, nonrandomized studies.31 Although these agents are useful in some patients, they may be associated with significant toxicities, and they are used less commonly than in the past.
Splenectomy has a high success rate
Splenectomy probably offers the best response of any treatment for ITP. About 80% of patients with ITP respond rapidly—often within 1 week. Of those, 15% relapse within the first year, and after 10 years, two-thirds remain in remission.32,33
Because there is no well-accepted predictor of a short- or long-term response to splenectomy, and because more medical options are currently available, the use of splenectomy has declined over the past 10 years. Nevertheless, splenectomy remains a useful option for therapy of ITP.
Whether and which second-line drugs should be tried before splenectomy is still controversial and should be determined on a case-by-case basis. Some patients are poor candidates for splenectomy because of comorbidities. If possible, splenectomy should be delayed until at least a year after diagnosis to allow an opportunity for spontaneous remission.
Splenectomy increases the risk of subsequent infection by encapsulated organisms, and patients should be immunized with pneumococcal, Haemophilus influenzae type B, and meningococcal vaccines, preferably at least 3 weeks before the spleen is removed.
Splenectomy is associated with pulmonary hypertension and thrombosis, primarily in patients who have had their spleens removed because of accelerated red cell destruction. Whether these risks are applicable to patients with ITP is unknown, but if so they are probably much lower than in patients with red cell disorders.
Rituximab
Rituximab, a humanized monoclonal antibody against the CD20 antigen on B lymphocytes, was developed for treating lymphoma. However, it has been found to have significant activity in a number of immunohematologic disorders. Although many studies of rituximab for ITP have been published,34–38 it has never been tested in a randomized controlled study. The response rate is generally around 50%, and it is effective in patients with or without a spleen.
In one study,39 44 (32%) of 137 patients with chronic ITP who were given rituximab achieved a complete remission that was sustained 1 year. After more than 5 years, 63% of this group (ie, approximately 20% of the original group) were still in remission.
Potential drawbacks of rituximab include its expense as well as the risk of first-infusion reactions, which may be severe or, rarely, fatal. Rituxan has also been associated with rare cases of progressive multifocal leukoencephalopathy, usually in patients heavily treated with other immunosuppressive agents; however, very rare cases of progressive multifocal leukoencephalopathy have been reported in patients with ITP who received rituximab.
Thrombopoietin receptor agonists increase platelet production
Thrombopoietin receptor agonists are approved for patients with chronic ITP who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy. Rather than inhibit platelet destruction, as do all the other ITP therapies, they enhance platelet production.
Diseases involving bone marrow failure that also involve a low platelet count tend to be associated with very high levels of serum thrombopoietin, which is produced constitutively by the liver. In ITP, thrombopoeitin levels tend to be close to normal and not significantly elevated, most likely because of accelerated thrombopoietin clearance when bound to antibody-coated platelets.40 This provides a rationale for the use of thrombopoietic agents in the treatment of ITP.
Earlier-generation thrombopoietic drugs had significant amino acid homology with natural thrombopoietin, and some patients who were treated with these drugs developed antibodies against them that cross-reacted with endogenous thrombopoietin. In some cases, this led to severe, refractory thrombocytopenia. Because the newer thrombopoietic agents have no sequence homology to natural thrombopoietin, antibody production has not been a significant problem.
Two drugs in this class are currently available for treating ITP:
Romiplostim (Nplate) is a peptibody (comprising an IgG Fc region and four peptidometic regions that interact with the thrombopoietin receptor, c-mpl) that is given subcutaneously once a week.
Romiplostim performed well in several phase I clinical trials.41 In a 24-week phase III trial that compared romiplostim against placebo in patients with ITP that had been refractory to other primary treatments, 79% of splenectomized patients and 88% of nonsplenectomized patients had an overall response (defined as a platelet count > 50 × 109/L for 4 weeks during the study period), and 38% of splenectomized patients and 61% of nonsplenectomized patients had a durable response (platelet count > 50 × 109/L for 6 of the last 8 weeks of the study).42
In an ongoing long-term extension study of romiplostim that allows dose adjustments to maintain a platelet count between 50 and 200 × 109/L, romiplostim dosage and efficacy have remained stable over 5 years.42,43
Eltrombopag (Promacta) is a nonpeptide small-molecule c-mpl agonist that is taken orally once daily. A recent randomized, placebo-controlled study in patients with ITP refractory to other primary treatments found that eltrombopag was highly effective in raising platelet counts over the 6 months of the study.44 Like romiplostim, it was effective in both splenectomized and nonsplenectomized patients.
Although eltrombopag has not been studied for as long as romiplostim, data over 3 years indicate that increased platelet counts are maintained without the emergence of drug resistance or cumulative toxicity.45
Several other drugs in this class are currently in development.
Adverse effects of thrombopoietic agents
Thrombopoietic agents have several associated toxicities:
Rebound thrombocytopenia occurs in up to 10% of patients following treatment with either romiplostim or eltrombopag. Rebound thrombocytopenia is defined as a fall in the platelet count that occurs following discontinuation of a thrombopoietic agent that may result in more severe thrombocytopenia, transiently, than before the drug was initiated. Thus, the platelet count must be closely monitored after treatment with these drugs is discontinued.
Bone marrow fibrosis, which consists primarily of increased marrow reticulin content, occurs in less than 10% of treated patients, and all patients on therapy must be monitored for this potential complication by close examination of the peripheral blood film on a frequent basis. Appearance of abnormalities such as teardrop cells or nucleated red blood cells in the peripheral blood smear should prompt at least temporary discontinuation of the drug and consideration of bone marrow examination. There have been no cases of actual irreversible myelofibrosis in which thrombopoietic agents have been clearly implicated in causation. Interestingly, some reports suggest that increased reticulin is a common finding in marrow from ITP patients who have not been treated with thrombopoietic agents.46
Thrombosis must be considered a risk of treatment with thrombopoietic agents, which increase the platelet count in a disease that may already be thrombogenic. However, in the placebo-controlled studies, a significantly increased incidence of thrombosis was not observed in the treatment arms vs placebo. Moreover, even in treated patients who developed thrombosis, there was no clear association with the degree of elevation in the platelet count. Nevertheless, thrombopoietic agents should be used according to the manufacturer’s recommendations, to increase the platelet count to a range of 50 to 200 × 109/L, but not to exceed that.
Progression of hematologic malignancies. Thrombopoietin receptor agonists act not only on megakaryocytes but also on stem cells and other hematopoieic precursors. Although trials for treating patients with hematologic malignancies and bone marrow failure with thrombopoietic agents are ongoing, there is concern that they could worsen certain hematologic malignancies, though there are no controlled data to either support or refute this concern at present. At this time, these drugs are approved only for ITP and should not be used for other conditions.
Hepatotoxicity has been seen with eltrombopag, but it is usually reversible and may resolve with continued therapy. Nevertheless, close monitoring for this potential complication is indicated.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393.
- Stasi R, Amadori S, Osborn J, Newland AC, Provan D. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006; 3:e24.
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytoenic purpura among adults: a population-based study and literature review. Eur Haematol 2009; 83:83–89.
- Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR; Northern Region Haematology Group. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol 2003; 122:966–974.
- Sarpatwari A, Bennett D, Logie JW, et al. Thromboembolic events among adult patients with primary immune thrombocytopenia in the United Kingdom General Practice Research Database. Haematologica 2010; 95:1167–1175.
- Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med 1951; 38:1–10.
- Luiken GA, McMillan R, Lightsey AL, et al. Platelet-associated IgG in immune thrombocytopenic purpura. Blood 1977; 50:317–325.
- Hirschman RJ, Schulman NR. Utilization of the platelet release reaction to measure ITP factor and platelet antibodies. Trans Assoc Am Physicians 1972; 85:325–334.
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995–1008.
- Karpatkin S. Autoimmune (idiopathic) thrombocytopenic purpura. Lancet 1997; 349:1531–1536.
- Psaila B, Bussel JB. Fc receptors in immune thrombocytopenias: a target for immunomodulation? J Clin Invest 2008; 118:2677–2681.
- Aster RH. Molecular mimicry and immune thrombocytopenia (comment). Blood 2009; 113:3887–3888.
- Takahashi T, Yujiri T, Shinohara K, et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori-associated chronic idiopathic thrombocytopenic purpura. Br J Haematol 2004; 124:91–96.
- Nardi MA, Liu LX, Karpatkin S. GPIIIa-(49-66) is a major pathophysiologically relevant antigenic determinant for anti-platelet GPIIIa of HIV-1-related immunologic thrombocytopenia. Proc Natl Acad Sci U S A 1997; 94:7589–7594.
- Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009; 113:4086–4093.
- Pivetti S, Novarino A, Merico F, et al. High prevalence of autoimmune phenomena in hepatitis C virus antibody positive patients with lymphoproliferative and connective tissue disorders. Br J Haematol 1996; 95:204–211.
- Pawlotsky JM, Bouvier M, Fromont P, et al. Hepatitis C virus infection and autoimmune thrombocytopenic purpura. J Hepatol 1995; 23:635–639.
- Sakuraya M, Murakami H, Uchiumi H, et al. Steroid-refractory chronic idiopathic thrombocytopenic purpura associated with hepatitis C virus infection. Eur J Haematol 2002; 68:49–53.
- García-Suárez J, Burgaleta C, Hernanz N, Albarran F, Tobaruela P, Alvarez-Mon M. HCV-associated thrombocytopenia: clinical characteristics and platelet response after recombinant alpha2b-interferon therapy. Br J Haematol 2000; 110:98–103.
- Rajan SK, Espina BM, Liebman HA. Hepatitis C virus-related thrombocytopenia: clinical and laboratory characteristics compared with chronic immune thrombocytopenic purpura. Br J Haematol 2005; 129:818–824.
- Harker LA. Thrombokinetics in idiopathic thrombocytopenic purpura. Br J Haematol 1970; 19:95–104.
- Branehög I, Kutti J, Weinfeld A. Platelet survival and platelet production in idiopathic thrombocytopenic purpura (ITP). Br J Haematol 1974; 27:127–143.
- Stoll D, Cines DB, Aster RH, Murphy S. Platelet kinetics in patients with idiopathic thrombocytopenic purpura and moderate thrombocytopenia. Blood 1985; 65:584–588.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 1987; 80:33–40.
- McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004; 103:1364–1369.
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 97:2549–2554.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186.
- British Committee for Standards in Haematology General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol 2003; 120:574–596.
- Mazzucconi MG, Fazi P, Bernasconi S, et al; Gruppo Italiano Malattie Ematoligiche dell’Adulto (GIMEMA) Thrombocytopenia Working Party. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 2007; 109:1401–1407.
- Gaines AR. Disseminated intravascular coagulation associated with acute hemoglobinemia or hemoglobinuria following RH9O)(D) immune globulin intravenous administration for immune thrombocytopenic purpura. Blood 2005; 106:1532–1537.
- George JN, Kojouri K, Perdue JJ, Vesely SK. Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Semin Hematol 2000; 37:290–298.
- Schwartz J, Leber MD, Gillis S, Giunta A, Eldor A, Bussel JB. Long term follow-up after splenectomy performed for immune thrombocytopenic purpura (ITP). Am J Hematol 2003; 72:94–98.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004; 104:2623–2634.
- Stasi R, Provan D. Management of immune thrombocytopenic purpura in adults. Mayo Clin Proc 2004; 79:504–522.
- Giagounidis AA, Anhuf J, Schneider P, et al. Treatment of relapsed idiopathic thrombocytopenic purpura with the anti-CD20 monoclonal antibody rituximab: a pilot study. Eur J Haematol 2002; 69:95–100.
- Stasi R, Stipa E, Forte V, Meo P, Amadori S. Variable patterns of response to rituximab treatment in adults with chronic idiopathic thrombocytopenic purpura (letter). Blood 2002; 99:3872–3873.
- Cooper N, Stasi R, Cunningham-Rundles S, et al. The efficacy and safetyof B-cell depletion with anti-CD20 monocloncal antibody in adults with chronic immune thrombocytopenic purpura. Br J Haematol 2004; 125:232–239.
- Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytoopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 2003; 78:1340–1346.
- Patel V, Mihatov N, Cooper N, Stasi R, Cunningham-Rundles S, Bussel JB. Long term follow-up of patients with immune thrombocytopenic purpura (ITP) whose initial response to rituximab lasted a minimum of 1 year (abstract). Blood (ASH Annual Meeting Abstracts): 2006;108:Abstract 479.
- Mukai HY, Kojima H, Todokoro K, et al. Serum thrombopoietin (TPO) levels in patients with amegakaryocytic thrombocytopenia are much higher than those with immune thrombocytopenic purpura. Thromb Haemost 1996; 76:675–678.
- Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006; 355:1672–1681. (Published correction in N Engl J Med 2006; 355:2054.)
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008; 371:395–403.
- Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost 2010; 8:1372–1382.
- Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2010 Aug 23(Epub ahead of print).
- Saleh MN, Bussel JB, Cheng G, et al. Long-term treatment of chronic immune thrombocytopenic purpura with oral eltrombopag. Abstract #682 presented at the 51st American Society of Hematology Annual Meeting and Exposition, New Orleans, LA, December 5–8, 2009; http://ash.confex.com/ash/2009/webprogram/Paper24081.html. Accessed April 26, 2011.
- Kuter DJ, Mufti GJ, Bain BJ, Hasserjian RP, Davis W, Rutstein M. Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood 2009; 114:3748–3756.
Once regarded as idiopathic, immune thrombocytopenia (ITP) is now understood to have a complex pathogenesis, involving the evolution of antibodies against multiple platelet antigens leading to reduced platelet survival as well as impaired platelet production. For this reason, multiple therapies with different mechanisms of action are available to treat ITP, though not all of them are effective for individual patients.
In this article, I discuss the pathogenesis, demographics, manifestations, diagnosis, and management of ITP.
THE NAME AND THE CUTOFF HAVE CHANGED
The term ITP formerly was used to refer to “idiopathic” or “immune” thrombocytopenic purpura. However, although not all aspects of the pathogenesis of ITP are understood, the disease can no longer be considered idiopathic. In addition, many patients do not have purpura at the time of diagnosis. Though the abbreviation “ITP” remains the same, it now refers to immune thrombocytopenia, which can be either primary or secondary.1
ITP is defined as a platelet count of less than 100 × 109/L (100,000/μL) with no evidence of leukopenia or anemia. This cutoff point is new: in the past, ITP was defined as a platelet count of less than 150 × 109/L, which is the threshold for a normal platelet count in most laboratories.
The platelet threshold of 100 × 109/L was based on a study by Stasi et al,2 who followed 217 otherwise healthy people who had an incidental finding of mild thrombocytopenia (platelet count 100–150 × 109/L). Within 6 months, the platelet count rose to more than 150 × 109/L in 23, while three had either worsening thrombocytopenia or were diagnosed with other conditions. During long-term follow-up (median 64 months), 109 of the remaining 191 individuals remained stable, 13 developed counts greater than 150 × 109/L, 12 developed ITP, 13 developed an autoimmune disorder, 18 developed other disorders, and 26 were lost to follow-up. The 10-year probability of developing ITP, defined as a platelet count persistently below 100 × 109/L, was only 6.9%, indicating that the chances are small that a person with an isolated finding of mild, stable thrombocytopenia will develop ITP.
Categories of ITP
An international working group designated to standardize terminology has divided ITP into two major diagnostic categories.1 The proportion of patients within each is not well established and varies by region and demographic characteristics.
Primary ITP accounts for the majority of cases in most studies; other conditions associated with thrombocytopenia are absent.
Secondary ITP can be due to infection with a number of agents, including hepatitis C virus (HCV), human immunodeficiency virus (HIV), and Helicobacter pylori. Other causes include underlying autoimmune and lymphoproliferative disorders such as systemic lupus erythematosus, Wiskott-Aldrich syndrome, chronic lymphocytic leukemia, antiphospholipid syndrome, and common variable immunodeficiency, as well as drugs such as quinine and trimethoprim-sulfamethoxazole.
Categories of ITP have also been established to facilitate management decisions, as follows:
Newly diagnosed ITP refers to ITP diagnosed within the preceding 3 months.
Persistent ITP refers to ITP diagnosed 3 to 12 months previously, and includes ITP in patients not reaching spontaneous remission and in those not maintaining a complete response off therapy. (When ITP spontaneously remits in adults, it usually does so within the first 12 months after the condition is diagnosed.)
Chronic ITP: Lasting for more than 12 months.
Severe ITP is defined by bleeding at presentation sufficient to mandate treatment, or new bleeding symptoms requiring additional therapeutic intervention with a different platelet-enhancing agent or an increased dosage of a current agent.
ITP IS COMMON IN OLDER ADULTS
We previously believed that ITP was a disorder that primarily affected women in their third and fourth decades. However, this was not borne out in recent epidemiologic studies, which have demonstrated that the highest age-specific incidence of ITP occurs in the elderly. This may potentially reflect the development of immune dysregulation as a consequence of aging. There is a female preponderance in the incidence of ITP throughout adulthood until around age 60, after which the overall incidence increases in both sexes, and the ratio of affected women to men is about equal.3,4 Thus, even though thrombocytopenia in the elderly may reflect myelodysplasia in some individuals, ITP is much more common than previously appreciated.
Previous guidelines from the American Society of Hematology suggested that a bone marrow examination be strongly considered in patients over age 60 with suspected ITP. With the realization that ITP occurs more commonly in the elderly, it is apparent that bone marrow examination is not necessary in this group if there are no other cytopenias present and the physical examination and blood smear are consistent with ITP.
In children, ITP has a peak incidence between ages 5 and 6, and behaves differently from the adult syndrome. ITP in children usually follows an apparent viral infection and tends to be self-limited, with approximately 80% of cases resolving spontaneously within 6 months. In contrast, adult ITP usually develops into a chronic disease.
BLEEDING MAY NOT BE PRESENT AT DIAGNOSIS
ITP is now recognized as a diverse syndrome with a constellation of signs and symptoms.
Petechiae are pinpoint microvascular hemorrhages that do not blanch with pressure. This distinguishes them from small hemangiomas, which look similar but blanch transiently with pressure. Petechiae tend to occur on dependent areas, particularly the hands and feet, when the platelet count drops below approximately 15 × 109/L.
Ecchymoses (dry purpura) appear as large bruises.
Mucosal bleeding (wet purpura) involves the oral mucosa. Particularly in children, wet purpura tends to be associated with systemic bleeding complications, involving the gastrointestinal tract for example. The incidence of intracranial hemorrhage, though very low, may also be increased in patients with wet purpura.
Other bleeding manifestations may include heavy menstrual bleeding, oral bleeding, and epistaxis.
Bleeding is generally but not entirely proportional to the platelet count. In a study of adults with newly diagnosed ITP and a platelet count of less than 50 × 109/L,4 the presenting symptom was hemorrhage in 12% and purpura in 58%.4 Remarkably, 28% of cases were asymptomatic, with some patients remaining free of symptoms for years despite very low platelet counts. More than half of patients with a platelet count of 30 to 50 × 109/L have no symptoms.3,4
A PARADOXICAL RISK OF THROMBOSIS
Although ITP is primarily a bleeding disorder, it is paradoxically also associated with thrombosis. Sarpatwari et al,5 in a study in the United Kingdom, found that the 4-year incidence of thromboembolic events was about 1.3 times higher in patients with ITP than in matched controls.
The reason for the increased risk of thrombosis is not clear. It is possible that in some patients, antiphospholipid antibodies may contribute to the development of thrombosis, although this has not been confirmed in all studies.
A DIAGNOSIS OF EXCLUSION
The evaluation of any patient suspected of having ITP should include the following:
- Personal history, with special attention to drugs and to medical conditions that could cause thrombocytopenia.
- Family history. ITP may occasionally be mistaken for an inherited cause of thrombocytopenia. The presence of the latter can often be confirmed by review of the peripheral blood film of the patient as well as other family members with thrombocytopenia. ITP is generally not considered to be an inherited disorder, although some HLA alleles may be more prevalent in ITP patients.
- Physical examination, with special attention to lymphadenopathy or splenomegaly, which may suggest an underlying malignancy such as a lymphoproliferative disorder. In general, patients with ITP have a normal physical examination, except for signs of bleeding or bruising in some.
- Laboratory tests, including a complete blood cell count, blood smear, reticulocyte count, Rh typing, and direct antiglobulin (Coombs) test.
In ITP, the peripheral blood smear should appear normal except for the presence of thrombocytopenia, although platelets may be mildly enlarged in some individuals. Red cell and leukocyte morphology is normal. It is important to exclude the presence of schistocytes (red cell fragments) and nucleated red blood cells, which often indicate a microangiopathic hemolytic anemia caused by disorders such as thrombotic thrombocytopenic purpura.
International guidelines suggest that testing for reduced immunoglobulin levels (as seen in common variable hypogammaglobulinemia) and HIV, HCV, and H pylori infections should also be considered. Coincident HCV infection is particularly high in some regions. Other cytopenias or abnormalities in the history or physical examination may prompt bone marrow examination. Testing for antiphospholipid antibodies, antinuclear antibodies, parvovirus, and cytomegalovirus may also be indicated in specific individuals. Testing for antiplatelet antibodies is not commonly performed in the current era because of its relatively low sensitivity and specificity.
Ultimately, the diagnosis of ITP is clinical, however, and cannot be established by any specific laboratory assay. Perhaps the best diagnostic study is assessment of the patient’s response to ITP therapy.
ITP INVOLVES ACCELERATED PLATELET DESTRUCTION
In 1951, William Harrington, a fellow at Washington University, infused blood from a patient with ITP into himself and, subsequently, into normal volunteers.6 The majority of recipients demonstrated significant reductions in the platelet count, sometimes severe. This fascinating and bold experiment provided the first demonstration that ITP was caused by a factor that circulates in blood. What is often not emphasized, however, is that some recipients did not develop thrombocytopenia, suggesting an alternative mechanism.
Later, Luiken et al7 and Hirschman and Shulman8 demonstrated that the transmissible agent in the blood was immunoglobulin, primarily immunoglobulin G (IgG). We now understand that much of the pathogenesis of ITP is caused by antibodies against platelet glycoproteins, most commonly platelet glycoprotein IIb/IIIa, the platelet fibrinogen receptor. Most patients, especially those with chronic ITP, also have antibodies against other platelet glycoproteins, including glycoprotein Ib/IX (the receptor for von Willebrand factor), and glycoprotein Ia/IIa, a collagen receptor. It is commonly believed that ITP may begin with antibodies against a single glycoprotein, which leads to accelerated clearance of antibody-coated platelets in the spleen. Degradation of cleared platelets by splenic macrophages leads to the release and subsequent presentation of antigenic peptides from proteolyzed platelet components, including glycoproteins, on the macrophage or dendritic cell. This may lead to recruitment and activation of specific T cells that in turn interact with and stimulate B cells to produce new antibodies against the platelet-derived peptides. This phenomenon, known as epitope spreading, may be responsible for the fact that most patients with long-standing, chronic ITP develop autoantibodies against multiple platelet glycoprotein targets.9
Several agents used in the treatment of ITP may work by impairing clearance of antibody-coated platelets by the reticuloendothelial system. One of many potential mechanisms underlying the therapeutic efficacy of intravenous immunoglobulin (IVIG) may be its ability to interact with a specific type of Fc gamma receptor, Fc gamma RIIb. IVIG therapy stimulates increased expression of this receptor, which in turn may impair the function of other “activating” Fc gamma receptors responsible for platelet clearance.10,11
ITP associated with infection may arise due to molecular mimicry. HCV, HIV, and H pylori contain amino acid sequences that may have structural similarity to regions within platelet glycoproteins. Thus, antibodies directed against the pathogen may cross-react with the glycoprotein, leading to thrombocytopenia.12–15
HCV has been found in up to one-third of cases of ITP in some centers.16–20H pylori-associated ITP is very common in some regions, particularly in Japan, and may often resolve after eradication of the infection. However, in the United States, eradication of H pylori generally does not improve the course of ITP. This may reflect antigen mimicry, in particular the fact that different cagA proteins are expressed by different strains of H pylori in certain regions of the world.
Our understanding of the immunologic basis of ITP has greatly expanded over the last decade. Although it has long been known that B cells produce autoantibodies, T cells have more recently been shown to play a critical role in regulating B-cell-mediated autoantibody production in ITP. In some situations, T cells may directly lyse platelets, or suppress megakaryopoiesis. This may explain why some patients who do not respond to standard B-cell-targeted therapy may respond to cyclosporine or other T-cell-directed agents.
ANOTHER MECHANISM OF ITP: REDUCED PLATELET PRODUCTION
In addition to accelerated platelet destruction, ITP is also associated with decreased platelet production by megakaryocytes in the bone marrow.21–25
Increased platelet destruction and reduced platelet production are likely two ends of a spectrum of ITP, and most patients likely have some degree of both processes. This concept helps explain why different drug strategies are more effective in some patients than in others.
ARE THE RISKS OF THERAPY JUSTIFIED?
It is important to understand the natural history of ITP to determine whether the risks of therapy are justified.
A 2001 study from the Netherlands26 followed 134 patients with primary ITP for 10 years: 90% had taken prednisone or had had a splenectomy. Within 2 years, 85% of these patients had platelet counts above 30 × 109/L off therapy. Although this group likely experienced more bleeding and bruising than the general population, the mortality rate was not increased. Another 6% also achieved a platelet count above 30 × 109/L, but required chronic maintenance therapy (usually steroids) to do so. This group led a nearly normal life but had more hospitalizations. The remaining 9% of patients had refractory ITP, with platelet counts remaining below 30 × 109/L despite therapy. This group had a death rate 4.2 times that of age-matched controls. About half died of bleeding and the others died of opportunistic infections to which they were susceptible because of long-term steroid therapy.
This study was influential in the general opinion that 30 × 109/L is a reasonable cutoff for treating ITP. An international consensus report states that treatment is rarely indicated in patients with platelet counts above 50 × 109/L in the absence of bleeding due to platelet dysfunction or other hemostatic defect, trauma, or surgery.27 Although this number is not supported by evidence-based data, it is a reasonable threshold endorsed by an international working group.27 Individual factors must be weighed heavily: for example, an athlete involved in contact sports requires a higher platelet count in order to play safely.
FIRST-LINE THERAPIES
First-line therapies for ITP include corticosteroids, IVIG, and anti-Rho(D) immune globulin (WinRho).27
Corticosteroids are standard therapy
Corticosteroids can be given in one of two ways:
Standard prednisone therapy, ie, 1 to 2 mg/kg per day, is given until a response is seen, and then tapered. Some maintain therapy for an additional week before tapering. There are no guidelines on how to taper: some decrease the dosage by 50% per week, although many recommend going more slowly, particularly at the lower range of dosing.
Up to 85% of patients achieve a clinical response, usually within 7 to 10 days, with platelet counts peaking in 2 to 4 weeks. Unfortunately, only about 15% of patients maintain the response over the subsequent 6 to 12 months. Restarting prednisone often initiates a vicious circle and makes patients vulnerable to steroid toxicities.
“Pulse” dexamethasone therapy consists of 40 mg per day for 4 days for one to three cycles. (Dexamethasone 1 mg is equivalent to about 10 mg of prednisone.)
Pulse dexamethasone therapy as an initial approach to ITP has been developed during the past decade and has been used primarily in research studies. This regimen evolved from studies of patients with multiple myelomas and has the potential to induce more durable remissions in some patients with newly diagnosed ITP.29 However, high-dose corticosteroids may be associated with increased toxicity, at least in the short term, and should be used cautiously. A study to address the role of high-dose vs standard-dose steroid therapy has recently been opened under the guidance of the Transfusion Medicine–Hemostasis Clinical Trials Network of the National Heart, Lung, and Blood Institute.
Immunoglobulin is useful for very low platelet counts and bleeding
Another primary therapy for ITP is IVIG 0.5 to 2.0 g/kg over 2 to 5 days. Its efficacy is similar to that of prednisone: about 65% of patients achieve a platelet count above 100 × 109/L, and 85% achieve 50 × 109/L. However, most responses are transient, and a significant minority of cases become refractory to IVIG after repeated infusions.
IVIG is associated with numerous adverse effects, including thrombosis, renal insufficiency, headache, and anaphylaxis in IgA-deficient patients. It also converts the direct antiglobulin test to positive. IVIG is expensive, is inconvenient to administer, and may require lengthy infusions depending on the formulation.
Although IVIG is not a good long-term therapy, it can help raise the platelet count relatively quickly in patients who present with severe thrombocytopenia accompanied by bleeding. Such patients should be treated with high-dose steroids, IVIG, and platelet transfusions. IVIG may also be useful to increase platelet counts prior to interventional procedures.
Intravenous anti-Rho(D)
Anti-Rho(D) is an alternative to IVIG in patients who are Rho(D)-positive and have an intact spleen. Anti-Rho(D) binds to Rh-positive red blood cells, causing them to be cleared in the reticuloendothelial system and blocking the clearance of antibody-coated platelets. In effect, red cells are sacrificed to save platelets, but because there are many more red cells than platelets, the benefits usually outweigh the risks.
The initial dose is 50 μg/kg given intravenously over 2 to 5 minutes. Anti-Rho(D) should not be given to patients whose hemoglobin level is less than 10 g/dL or who have compromised bone marrow function. It is ineffective in Rh-negative patients or those who have undergone splenectomy.
Accelerated hemolysis is a rare but severe possible adverse event associated with this therapy, occurring in slightly more than 1 in 1,000 infusions. About 1 out of every 20,000 patients develops disseminated intravascular coagulation.30 Its cause is poorly understood, and it is probably an accelerated extravascular rather than an intravascular event. The US Food and Drug Administration has recently issued a black-box warning cautioning that patients who receive anti-Rho(D) should remain in a health care setting for 8 hours after treatment, although most cases of accelerated hemolysis occur within 4 hours. Moreover, it is possible that many of these cases can be avoided by appropriate patient selection.
SECOND-LINE THERAPIES
Second-line therapies, as designated by the international working group, include azathioprine (Imuran), cyclosporine A, cyclophosphamide (Cytoxan), danazol (Danocrine), dapsone, mycophenolate mofetil (CellCept), rituximab (Rituxan), splenectomy, thrombopoietin receptor agonists, and vinca alkaloids.27 Only the most commonly used therapies will be briefly discussed below.
The evidence for efficacy of the cytotoxic agents, ie, cyclophosphamide, the vinca alkaloids, and azathioprine, comes from small, nonrandomized studies.31 Although these agents are useful in some patients, they may be associated with significant toxicities, and they are used less commonly than in the past.
Splenectomy has a high success rate
Splenectomy probably offers the best response of any treatment for ITP. About 80% of patients with ITP respond rapidly—often within 1 week. Of those, 15% relapse within the first year, and after 10 years, two-thirds remain in remission.32,33
Because there is no well-accepted predictor of a short- or long-term response to splenectomy, and because more medical options are currently available, the use of splenectomy has declined over the past 10 years. Nevertheless, splenectomy remains a useful option for therapy of ITP.
Whether and which second-line drugs should be tried before splenectomy is still controversial and should be determined on a case-by-case basis. Some patients are poor candidates for splenectomy because of comorbidities. If possible, splenectomy should be delayed until at least a year after diagnosis to allow an opportunity for spontaneous remission.
Splenectomy increases the risk of subsequent infection by encapsulated organisms, and patients should be immunized with pneumococcal, Haemophilus influenzae type B, and meningococcal vaccines, preferably at least 3 weeks before the spleen is removed.
Splenectomy is associated with pulmonary hypertension and thrombosis, primarily in patients who have had their spleens removed because of accelerated red cell destruction. Whether these risks are applicable to patients with ITP is unknown, but if so they are probably much lower than in patients with red cell disorders.
Rituximab
Rituximab, a humanized monoclonal antibody against the CD20 antigen on B lymphocytes, was developed for treating lymphoma. However, it has been found to have significant activity in a number of immunohematologic disorders. Although many studies of rituximab for ITP have been published,34–38 it has never been tested in a randomized controlled study. The response rate is generally around 50%, and it is effective in patients with or without a spleen.
In one study,39 44 (32%) of 137 patients with chronic ITP who were given rituximab achieved a complete remission that was sustained 1 year. After more than 5 years, 63% of this group (ie, approximately 20% of the original group) were still in remission.
Potential drawbacks of rituximab include its expense as well as the risk of first-infusion reactions, which may be severe or, rarely, fatal. Rituxan has also been associated with rare cases of progressive multifocal leukoencephalopathy, usually in patients heavily treated with other immunosuppressive agents; however, very rare cases of progressive multifocal leukoencephalopathy have been reported in patients with ITP who received rituximab.
Thrombopoietin receptor agonists increase platelet production
Thrombopoietin receptor agonists are approved for patients with chronic ITP who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy. Rather than inhibit platelet destruction, as do all the other ITP therapies, they enhance platelet production.
Diseases involving bone marrow failure that also involve a low platelet count tend to be associated with very high levels of serum thrombopoietin, which is produced constitutively by the liver. In ITP, thrombopoeitin levels tend to be close to normal and not significantly elevated, most likely because of accelerated thrombopoietin clearance when bound to antibody-coated platelets.40 This provides a rationale for the use of thrombopoietic agents in the treatment of ITP.
Earlier-generation thrombopoietic drugs had significant amino acid homology with natural thrombopoietin, and some patients who were treated with these drugs developed antibodies against them that cross-reacted with endogenous thrombopoietin. In some cases, this led to severe, refractory thrombocytopenia. Because the newer thrombopoietic agents have no sequence homology to natural thrombopoietin, antibody production has not been a significant problem.
Two drugs in this class are currently available for treating ITP:
Romiplostim (Nplate) is a peptibody (comprising an IgG Fc region and four peptidometic regions that interact with the thrombopoietin receptor, c-mpl) that is given subcutaneously once a week.
Romiplostim performed well in several phase I clinical trials.41 In a 24-week phase III trial that compared romiplostim against placebo in patients with ITP that had been refractory to other primary treatments, 79% of splenectomized patients and 88% of nonsplenectomized patients had an overall response (defined as a platelet count > 50 × 109/L for 4 weeks during the study period), and 38% of splenectomized patients and 61% of nonsplenectomized patients had a durable response (platelet count > 50 × 109/L for 6 of the last 8 weeks of the study).42
In an ongoing long-term extension study of romiplostim that allows dose adjustments to maintain a platelet count between 50 and 200 × 109/L, romiplostim dosage and efficacy have remained stable over 5 years.42,43
Eltrombopag (Promacta) is a nonpeptide small-molecule c-mpl agonist that is taken orally once daily. A recent randomized, placebo-controlled study in patients with ITP refractory to other primary treatments found that eltrombopag was highly effective in raising platelet counts over the 6 months of the study.44 Like romiplostim, it was effective in both splenectomized and nonsplenectomized patients.
Although eltrombopag has not been studied for as long as romiplostim, data over 3 years indicate that increased platelet counts are maintained without the emergence of drug resistance or cumulative toxicity.45
Several other drugs in this class are currently in development.
Adverse effects of thrombopoietic agents
Thrombopoietic agents have several associated toxicities:
Rebound thrombocytopenia occurs in up to 10% of patients following treatment with either romiplostim or eltrombopag. Rebound thrombocytopenia is defined as a fall in the platelet count that occurs following discontinuation of a thrombopoietic agent that may result in more severe thrombocytopenia, transiently, than before the drug was initiated. Thus, the platelet count must be closely monitored after treatment with these drugs is discontinued.
Bone marrow fibrosis, which consists primarily of increased marrow reticulin content, occurs in less than 10% of treated patients, and all patients on therapy must be monitored for this potential complication by close examination of the peripheral blood film on a frequent basis. Appearance of abnormalities such as teardrop cells or nucleated red blood cells in the peripheral blood smear should prompt at least temporary discontinuation of the drug and consideration of bone marrow examination. There have been no cases of actual irreversible myelofibrosis in which thrombopoietic agents have been clearly implicated in causation. Interestingly, some reports suggest that increased reticulin is a common finding in marrow from ITP patients who have not been treated with thrombopoietic agents.46
Thrombosis must be considered a risk of treatment with thrombopoietic agents, which increase the platelet count in a disease that may already be thrombogenic. However, in the placebo-controlled studies, a significantly increased incidence of thrombosis was not observed in the treatment arms vs placebo. Moreover, even in treated patients who developed thrombosis, there was no clear association with the degree of elevation in the platelet count. Nevertheless, thrombopoietic agents should be used according to the manufacturer’s recommendations, to increase the platelet count to a range of 50 to 200 × 109/L, but not to exceed that.
Progression of hematologic malignancies. Thrombopoietin receptor agonists act not only on megakaryocytes but also on stem cells and other hematopoieic precursors. Although trials for treating patients with hematologic malignancies and bone marrow failure with thrombopoietic agents are ongoing, there is concern that they could worsen certain hematologic malignancies, though there are no controlled data to either support or refute this concern at present. At this time, these drugs are approved only for ITP and should not be used for other conditions.
Hepatotoxicity has been seen with eltrombopag, but it is usually reversible and may resolve with continued therapy. Nevertheless, close monitoring for this potential complication is indicated.
Once regarded as idiopathic, immune thrombocytopenia (ITP) is now understood to have a complex pathogenesis, involving the evolution of antibodies against multiple platelet antigens leading to reduced platelet survival as well as impaired platelet production. For this reason, multiple therapies with different mechanisms of action are available to treat ITP, though not all of them are effective for individual patients.
In this article, I discuss the pathogenesis, demographics, manifestations, diagnosis, and management of ITP.
THE NAME AND THE CUTOFF HAVE CHANGED
The term ITP formerly was used to refer to “idiopathic” or “immune” thrombocytopenic purpura. However, although not all aspects of the pathogenesis of ITP are understood, the disease can no longer be considered idiopathic. In addition, many patients do not have purpura at the time of diagnosis. Though the abbreviation “ITP” remains the same, it now refers to immune thrombocytopenia, which can be either primary or secondary.1
ITP is defined as a platelet count of less than 100 × 109/L (100,000/μL) with no evidence of leukopenia or anemia. This cutoff point is new: in the past, ITP was defined as a platelet count of less than 150 × 109/L, which is the threshold for a normal platelet count in most laboratories.
The platelet threshold of 100 × 109/L was based on a study by Stasi et al,2 who followed 217 otherwise healthy people who had an incidental finding of mild thrombocytopenia (platelet count 100–150 × 109/L). Within 6 months, the platelet count rose to more than 150 × 109/L in 23, while three had either worsening thrombocytopenia or were diagnosed with other conditions. During long-term follow-up (median 64 months), 109 of the remaining 191 individuals remained stable, 13 developed counts greater than 150 × 109/L, 12 developed ITP, 13 developed an autoimmune disorder, 18 developed other disorders, and 26 were lost to follow-up. The 10-year probability of developing ITP, defined as a platelet count persistently below 100 × 109/L, was only 6.9%, indicating that the chances are small that a person with an isolated finding of mild, stable thrombocytopenia will develop ITP.
Categories of ITP
An international working group designated to standardize terminology has divided ITP into two major diagnostic categories.1 The proportion of patients within each is not well established and varies by region and demographic characteristics.
Primary ITP accounts for the majority of cases in most studies; other conditions associated with thrombocytopenia are absent.
Secondary ITP can be due to infection with a number of agents, including hepatitis C virus (HCV), human immunodeficiency virus (HIV), and Helicobacter pylori. Other causes include underlying autoimmune and lymphoproliferative disorders such as systemic lupus erythematosus, Wiskott-Aldrich syndrome, chronic lymphocytic leukemia, antiphospholipid syndrome, and common variable immunodeficiency, as well as drugs such as quinine and trimethoprim-sulfamethoxazole.
Categories of ITP have also been established to facilitate management decisions, as follows:
Newly diagnosed ITP refers to ITP diagnosed within the preceding 3 months.
Persistent ITP refers to ITP diagnosed 3 to 12 months previously, and includes ITP in patients not reaching spontaneous remission and in those not maintaining a complete response off therapy. (When ITP spontaneously remits in adults, it usually does so within the first 12 months after the condition is diagnosed.)
Chronic ITP: Lasting for more than 12 months.
Severe ITP is defined by bleeding at presentation sufficient to mandate treatment, or new bleeding symptoms requiring additional therapeutic intervention with a different platelet-enhancing agent or an increased dosage of a current agent.
ITP IS COMMON IN OLDER ADULTS
We previously believed that ITP was a disorder that primarily affected women in their third and fourth decades. However, this was not borne out in recent epidemiologic studies, which have demonstrated that the highest age-specific incidence of ITP occurs in the elderly. This may potentially reflect the development of immune dysregulation as a consequence of aging. There is a female preponderance in the incidence of ITP throughout adulthood until around age 60, after which the overall incidence increases in both sexes, and the ratio of affected women to men is about equal.3,4 Thus, even though thrombocytopenia in the elderly may reflect myelodysplasia in some individuals, ITP is much more common than previously appreciated.
Previous guidelines from the American Society of Hematology suggested that a bone marrow examination be strongly considered in patients over age 60 with suspected ITP. With the realization that ITP occurs more commonly in the elderly, it is apparent that bone marrow examination is not necessary in this group if there are no other cytopenias present and the physical examination and blood smear are consistent with ITP.
In children, ITP has a peak incidence between ages 5 and 6, and behaves differently from the adult syndrome. ITP in children usually follows an apparent viral infection and tends to be self-limited, with approximately 80% of cases resolving spontaneously within 6 months. In contrast, adult ITP usually develops into a chronic disease.
BLEEDING MAY NOT BE PRESENT AT DIAGNOSIS
ITP is now recognized as a diverse syndrome with a constellation of signs and symptoms.
Petechiae are pinpoint microvascular hemorrhages that do not blanch with pressure. This distinguishes them from small hemangiomas, which look similar but blanch transiently with pressure. Petechiae tend to occur on dependent areas, particularly the hands and feet, when the platelet count drops below approximately 15 × 109/L.
Ecchymoses (dry purpura) appear as large bruises.
Mucosal bleeding (wet purpura) involves the oral mucosa. Particularly in children, wet purpura tends to be associated with systemic bleeding complications, involving the gastrointestinal tract for example. The incidence of intracranial hemorrhage, though very low, may also be increased in patients with wet purpura.
Other bleeding manifestations may include heavy menstrual bleeding, oral bleeding, and epistaxis.
Bleeding is generally but not entirely proportional to the platelet count. In a study of adults with newly diagnosed ITP and a platelet count of less than 50 × 109/L,4 the presenting symptom was hemorrhage in 12% and purpura in 58%.4 Remarkably, 28% of cases were asymptomatic, with some patients remaining free of symptoms for years despite very low platelet counts. More than half of patients with a platelet count of 30 to 50 × 109/L have no symptoms.3,4
A PARADOXICAL RISK OF THROMBOSIS
Although ITP is primarily a bleeding disorder, it is paradoxically also associated with thrombosis. Sarpatwari et al,5 in a study in the United Kingdom, found that the 4-year incidence of thromboembolic events was about 1.3 times higher in patients with ITP than in matched controls.
The reason for the increased risk of thrombosis is not clear. It is possible that in some patients, antiphospholipid antibodies may contribute to the development of thrombosis, although this has not been confirmed in all studies.
A DIAGNOSIS OF EXCLUSION
The evaluation of any patient suspected of having ITP should include the following:
- Personal history, with special attention to drugs and to medical conditions that could cause thrombocytopenia.
- Family history. ITP may occasionally be mistaken for an inherited cause of thrombocytopenia. The presence of the latter can often be confirmed by review of the peripheral blood film of the patient as well as other family members with thrombocytopenia. ITP is generally not considered to be an inherited disorder, although some HLA alleles may be more prevalent in ITP patients.
- Physical examination, with special attention to lymphadenopathy or splenomegaly, which may suggest an underlying malignancy such as a lymphoproliferative disorder. In general, patients with ITP have a normal physical examination, except for signs of bleeding or bruising in some.
- Laboratory tests, including a complete blood cell count, blood smear, reticulocyte count, Rh typing, and direct antiglobulin (Coombs) test.
In ITP, the peripheral blood smear should appear normal except for the presence of thrombocytopenia, although platelets may be mildly enlarged in some individuals. Red cell and leukocyte morphology is normal. It is important to exclude the presence of schistocytes (red cell fragments) and nucleated red blood cells, which often indicate a microangiopathic hemolytic anemia caused by disorders such as thrombotic thrombocytopenic purpura.
International guidelines suggest that testing for reduced immunoglobulin levels (as seen in common variable hypogammaglobulinemia) and HIV, HCV, and H pylori infections should also be considered. Coincident HCV infection is particularly high in some regions. Other cytopenias or abnormalities in the history or physical examination may prompt bone marrow examination. Testing for antiphospholipid antibodies, antinuclear antibodies, parvovirus, and cytomegalovirus may also be indicated in specific individuals. Testing for antiplatelet antibodies is not commonly performed in the current era because of its relatively low sensitivity and specificity.
Ultimately, the diagnosis of ITP is clinical, however, and cannot be established by any specific laboratory assay. Perhaps the best diagnostic study is assessment of the patient’s response to ITP therapy.
ITP INVOLVES ACCELERATED PLATELET DESTRUCTION
In 1951, William Harrington, a fellow at Washington University, infused blood from a patient with ITP into himself and, subsequently, into normal volunteers.6 The majority of recipients demonstrated significant reductions in the platelet count, sometimes severe. This fascinating and bold experiment provided the first demonstration that ITP was caused by a factor that circulates in blood. What is often not emphasized, however, is that some recipients did not develop thrombocytopenia, suggesting an alternative mechanism.
Later, Luiken et al7 and Hirschman and Shulman8 demonstrated that the transmissible agent in the blood was immunoglobulin, primarily immunoglobulin G (IgG). We now understand that much of the pathogenesis of ITP is caused by antibodies against platelet glycoproteins, most commonly platelet glycoprotein IIb/IIIa, the platelet fibrinogen receptor. Most patients, especially those with chronic ITP, also have antibodies against other platelet glycoproteins, including glycoprotein Ib/IX (the receptor for von Willebrand factor), and glycoprotein Ia/IIa, a collagen receptor. It is commonly believed that ITP may begin with antibodies against a single glycoprotein, which leads to accelerated clearance of antibody-coated platelets in the spleen. Degradation of cleared platelets by splenic macrophages leads to the release and subsequent presentation of antigenic peptides from proteolyzed platelet components, including glycoproteins, on the macrophage or dendritic cell. This may lead to recruitment and activation of specific T cells that in turn interact with and stimulate B cells to produce new antibodies against the platelet-derived peptides. This phenomenon, known as epitope spreading, may be responsible for the fact that most patients with long-standing, chronic ITP develop autoantibodies against multiple platelet glycoprotein targets.9
Several agents used in the treatment of ITP may work by impairing clearance of antibody-coated platelets by the reticuloendothelial system. One of many potential mechanisms underlying the therapeutic efficacy of intravenous immunoglobulin (IVIG) may be its ability to interact with a specific type of Fc gamma receptor, Fc gamma RIIb. IVIG therapy stimulates increased expression of this receptor, which in turn may impair the function of other “activating” Fc gamma receptors responsible for platelet clearance.10,11
ITP associated with infection may arise due to molecular mimicry. HCV, HIV, and H pylori contain amino acid sequences that may have structural similarity to regions within platelet glycoproteins. Thus, antibodies directed against the pathogen may cross-react with the glycoprotein, leading to thrombocytopenia.12–15
HCV has been found in up to one-third of cases of ITP in some centers.16–20H pylori-associated ITP is very common in some regions, particularly in Japan, and may often resolve after eradication of the infection. However, in the United States, eradication of H pylori generally does not improve the course of ITP. This may reflect antigen mimicry, in particular the fact that different cagA proteins are expressed by different strains of H pylori in certain regions of the world.
Our understanding of the immunologic basis of ITP has greatly expanded over the last decade. Although it has long been known that B cells produce autoantibodies, T cells have more recently been shown to play a critical role in regulating B-cell-mediated autoantibody production in ITP. In some situations, T cells may directly lyse platelets, or suppress megakaryopoiesis. This may explain why some patients who do not respond to standard B-cell-targeted therapy may respond to cyclosporine or other T-cell-directed agents.
ANOTHER MECHANISM OF ITP: REDUCED PLATELET PRODUCTION
In addition to accelerated platelet destruction, ITP is also associated with decreased platelet production by megakaryocytes in the bone marrow.21–25
Increased platelet destruction and reduced platelet production are likely two ends of a spectrum of ITP, and most patients likely have some degree of both processes. This concept helps explain why different drug strategies are more effective in some patients than in others.
ARE THE RISKS OF THERAPY JUSTIFIED?
It is important to understand the natural history of ITP to determine whether the risks of therapy are justified.
A 2001 study from the Netherlands26 followed 134 patients with primary ITP for 10 years: 90% had taken prednisone or had had a splenectomy. Within 2 years, 85% of these patients had platelet counts above 30 × 109/L off therapy. Although this group likely experienced more bleeding and bruising than the general population, the mortality rate was not increased. Another 6% also achieved a platelet count above 30 × 109/L, but required chronic maintenance therapy (usually steroids) to do so. This group led a nearly normal life but had more hospitalizations. The remaining 9% of patients had refractory ITP, with platelet counts remaining below 30 × 109/L despite therapy. This group had a death rate 4.2 times that of age-matched controls. About half died of bleeding and the others died of opportunistic infections to which they were susceptible because of long-term steroid therapy.
This study was influential in the general opinion that 30 × 109/L is a reasonable cutoff for treating ITP. An international consensus report states that treatment is rarely indicated in patients with platelet counts above 50 × 109/L in the absence of bleeding due to platelet dysfunction or other hemostatic defect, trauma, or surgery.27 Although this number is not supported by evidence-based data, it is a reasonable threshold endorsed by an international working group.27 Individual factors must be weighed heavily: for example, an athlete involved in contact sports requires a higher platelet count in order to play safely.
FIRST-LINE THERAPIES
First-line therapies for ITP include corticosteroids, IVIG, and anti-Rho(D) immune globulin (WinRho).27
Corticosteroids are standard therapy
Corticosteroids can be given in one of two ways:
Standard prednisone therapy, ie, 1 to 2 mg/kg per day, is given until a response is seen, and then tapered. Some maintain therapy for an additional week before tapering. There are no guidelines on how to taper: some decrease the dosage by 50% per week, although many recommend going more slowly, particularly at the lower range of dosing.
Up to 85% of patients achieve a clinical response, usually within 7 to 10 days, with platelet counts peaking in 2 to 4 weeks. Unfortunately, only about 15% of patients maintain the response over the subsequent 6 to 12 months. Restarting prednisone often initiates a vicious circle and makes patients vulnerable to steroid toxicities.
“Pulse” dexamethasone therapy consists of 40 mg per day for 4 days for one to three cycles. (Dexamethasone 1 mg is equivalent to about 10 mg of prednisone.)
Pulse dexamethasone therapy as an initial approach to ITP has been developed during the past decade and has been used primarily in research studies. This regimen evolved from studies of patients with multiple myelomas and has the potential to induce more durable remissions in some patients with newly diagnosed ITP.29 However, high-dose corticosteroids may be associated with increased toxicity, at least in the short term, and should be used cautiously. A study to address the role of high-dose vs standard-dose steroid therapy has recently been opened under the guidance of the Transfusion Medicine–Hemostasis Clinical Trials Network of the National Heart, Lung, and Blood Institute.
Immunoglobulin is useful for very low platelet counts and bleeding
Another primary therapy for ITP is IVIG 0.5 to 2.0 g/kg over 2 to 5 days. Its efficacy is similar to that of prednisone: about 65% of patients achieve a platelet count above 100 × 109/L, and 85% achieve 50 × 109/L. However, most responses are transient, and a significant minority of cases become refractory to IVIG after repeated infusions.
IVIG is associated with numerous adverse effects, including thrombosis, renal insufficiency, headache, and anaphylaxis in IgA-deficient patients. It also converts the direct antiglobulin test to positive. IVIG is expensive, is inconvenient to administer, and may require lengthy infusions depending on the formulation.
Although IVIG is not a good long-term therapy, it can help raise the platelet count relatively quickly in patients who present with severe thrombocytopenia accompanied by bleeding. Such patients should be treated with high-dose steroids, IVIG, and platelet transfusions. IVIG may also be useful to increase platelet counts prior to interventional procedures.
Intravenous anti-Rho(D)
Anti-Rho(D) is an alternative to IVIG in patients who are Rho(D)-positive and have an intact spleen. Anti-Rho(D) binds to Rh-positive red blood cells, causing them to be cleared in the reticuloendothelial system and blocking the clearance of antibody-coated platelets. In effect, red cells are sacrificed to save platelets, but because there are many more red cells than platelets, the benefits usually outweigh the risks.
The initial dose is 50 μg/kg given intravenously over 2 to 5 minutes. Anti-Rho(D) should not be given to patients whose hemoglobin level is less than 10 g/dL or who have compromised bone marrow function. It is ineffective in Rh-negative patients or those who have undergone splenectomy.
Accelerated hemolysis is a rare but severe possible adverse event associated with this therapy, occurring in slightly more than 1 in 1,000 infusions. About 1 out of every 20,000 patients develops disseminated intravascular coagulation.30 Its cause is poorly understood, and it is probably an accelerated extravascular rather than an intravascular event. The US Food and Drug Administration has recently issued a black-box warning cautioning that patients who receive anti-Rho(D) should remain in a health care setting for 8 hours after treatment, although most cases of accelerated hemolysis occur within 4 hours. Moreover, it is possible that many of these cases can be avoided by appropriate patient selection.
SECOND-LINE THERAPIES
Second-line therapies, as designated by the international working group, include azathioprine (Imuran), cyclosporine A, cyclophosphamide (Cytoxan), danazol (Danocrine), dapsone, mycophenolate mofetil (CellCept), rituximab (Rituxan), splenectomy, thrombopoietin receptor agonists, and vinca alkaloids.27 Only the most commonly used therapies will be briefly discussed below.
The evidence for efficacy of the cytotoxic agents, ie, cyclophosphamide, the vinca alkaloids, and azathioprine, comes from small, nonrandomized studies.31 Although these agents are useful in some patients, they may be associated with significant toxicities, and they are used less commonly than in the past.
Splenectomy has a high success rate
Splenectomy probably offers the best response of any treatment for ITP. About 80% of patients with ITP respond rapidly—often within 1 week. Of those, 15% relapse within the first year, and after 10 years, two-thirds remain in remission.32,33
Because there is no well-accepted predictor of a short- or long-term response to splenectomy, and because more medical options are currently available, the use of splenectomy has declined over the past 10 years. Nevertheless, splenectomy remains a useful option for therapy of ITP.
Whether and which second-line drugs should be tried before splenectomy is still controversial and should be determined on a case-by-case basis. Some patients are poor candidates for splenectomy because of comorbidities. If possible, splenectomy should be delayed until at least a year after diagnosis to allow an opportunity for spontaneous remission.
Splenectomy increases the risk of subsequent infection by encapsulated organisms, and patients should be immunized with pneumococcal, Haemophilus influenzae type B, and meningococcal vaccines, preferably at least 3 weeks before the spleen is removed.
Splenectomy is associated with pulmonary hypertension and thrombosis, primarily in patients who have had their spleens removed because of accelerated red cell destruction. Whether these risks are applicable to patients with ITP is unknown, but if so they are probably much lower than in patients with red cell disorders.
Rituximab
Rituximab, a humanized monoclonal antibody against the CD20 antigen on B lymphocytes, was developed for treating lymphoma. However, it has been found to have significant activity in a number of immunohematologic disorders. Although many studies of rituximab for ITP have been published,34–38 it has never been tested in a randomized controlled study. The response rate is generally around 50%, and it is effective in patients with or without a spleen.
In one study,39 44 (32%) of 137 patients with chronic ITP who were given rituximab achieved a complete remission that was sustained 1 year. After more than 5 years, 63% of this group (ie, approximately 20% of the original group) were still in remission.
Potential drawbacks of rituximab include its expense as well as the risk of first-infusion reactions, which may be severe or, rarely, fatal. Rituxan has also been associated with rare cases of progressive multifocal leukoencephalopathy, usually in patients heavily treated with other immunosuppressive agents; however, very rare cases of progressive multifocal leukoencephalopathy have been reported in patients with ITP who received rituximab.
Thrombopoietin receptor agonists increase platelet production
Thrombopoietin receptor agonists are approved for patients with chronic ITP who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy. Rather than inhibit platelet destruction, as do all the other ITP therapies, they enhance platelet production.
Diseases involving bone marrow failure that also involve a low platelet count tend to be associated with very high levels of serum thrombopoietin, which is produced constitutively by the liver. In ITP, thrombopoeitin levels tend to be close to normal and not significantly elevated, most likely because of accelerated thrombopoietin clearance when bound to antibody-coated platelets.40 This provides a rationale for the use of thrombopoietic agents in the treatment of ITP.
Earlier-generation thrombopoietic drugs had significant amino acid homology with natural thrombopoietin, and some patients who were treated with these drugs developed antibodies against them that cross-reacted with endogenous thrombopoietin. In some cases, this led to severe, refractory thrombocytopenia. Because the newer thrombopoietic agents have no sequence homology to natural thrombopoietin, antibody production has not been a significant problem.
Two drugs in this class are currently available for treating ITP:
Romiplostim (Nplate) is a peptibody (comprising an IgG Fc region and four peptidometic regions that interact with the thrombopoietin receptor, c-mpl) that is given subcutaneously once a week.
Romiplostim performed well in several phase I clinical trials.41 In a 24-week phase III trial that compared romiplostim against placebo in patients with ITP that had been refractory to other primary treatments, 79% of splenectomized patients and 88% of nonsplenectomized patients had an overall response (defined as a platelet count > 50 × 109/L for 4 weeks during the study period), and 38% of splenectomized patients and 61% of nonsplenectomized patients had a durable response (platelet count > 50 × 109/L for 6 of the last 8 weeks of the study).42
In an ongoing long-term extension study of romiplostim that allows dose adjustments to maintain a platelet count between 50 and 200 × 109/L, romiplostim dosage and efficacy have remained stable over 5 years.42,43
Eltrombopag (Promacta) is a nonpeptide small-molecule c-mpl agonist that is taken orally once daily. A recent randomized, placebo-controlled study in patients with ITP refractory to other primary treatments found that eltrombopag was highly effective in raising platelet counts over the 6 months of the study.44 Like romiplostim, it was effective in both splenectomized and nonsplenectomized patients.
Although eltrombopag has not been studied for as long as romiplostim, data over 3 years indicate that increased platelet counts are maintained without the emergence of drug resistance or cumulative toxicity.45
Several other drugs in this class are currently in development.
Adverse effects of thrombopoietic agents
Thrombopoietic agents have several associated toxicities:
Rebound thrombocytopenia occurs in up to 10% of patients following treatment with either romiplostim or eltrombopag. Rebound thrombocytopenia is defined as a fall in the platelet count that occurs following discontinuation of a thrombopoietic agent that may result in more severe thrombocytopenia, transiently, than before the drug was initiated. Thus, the platelet count must be closely monitored after treatment with these drugs is discontinued.
Bone marrow fibrosis, which consists primarily of increased marrow reticulin content, occurs in less than 10% of treated patients, and all patients on therapy must be monitored for this potential complication by close examination of the peripheral blood film on a frequent basis. Appearance of abnormalities such as teardrop cells or nucleated red blood cells in the peripheral blood smear should prompt at least temporary discontinuation of the drug and consideration of bone marrow examination. There have been no cases of actual irreversible myelofibrosis in which thrombopoietic agents have been clearly implicated in causation. Interestingly, some reports suggest that increased reticulin is a common finding in marrow from ITP patients who have not been treated with thrombopoietic agents.46
Thrombosis must be considered a risk of treatment with thrombopoietic agents, which increase the platelet count in a disease that may already be thrombogenic. However, in the placebo-controlled studies, a significantly increased incidence of thrombosis was not observed in the treatment arms vs placebo. Moreover, even in treated patients who developed thrombosis, there was no clear association with the degree of elevation in the platelet count. Nevertheless, thrombopoietic agents should be used according to the manufacturer’s recommendations, to increase the platelet count to a range of 50 to 200 × 109/L, but not to exceed that.
Progression of hematologic malignancies. Thrombopoietin receptor agonists act not only on megakaryocytes but also on stem cells and other hematopoieic precursors. Although trials for treating patients with hematologic malignancies and bone marrow failure with thrombopoietic agents are ongoing, there is concern that they could worsen certain hematologic malignancies, though there are no controlled data to either support or refute this concern at present. At this time, these drugs are approved only for ITP and should not be used for other conditions.
Hepatotoxicity has been seen with eltrombopag, but it is usually reversible and may resolve with continued therapy. Nevertheless, close monitoring for this potential complication is indicated.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393.
- Stasi R, Amadori S, Osborn J, Newland AC, Provan D. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006; 3:e24.
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytoenic purpura among adults: a population-based study and literature review. Eur Haematol 2009; 83:83–89.
- Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR; Northern Region Haematology Group. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol 2003; 122:966–974.
- Sarpatwari A, Bennett D, Logie JW, et al. Thromboembolic events among adult patients with primary immune thrombocytopenia in the United Kingdom General Practice Research Database. Haematologica 2010; 95:1167–1175.
- Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med 1951; 38:1–10.
- Luiken GA, McMillan R, Lightsey AL, et al. Platelet-associated IgG in immune thrombocytopenic purpura. Blood 1977; 50:317–325.
- Hirschman RJ, Schulman NR. Utilization of the platelet release reaction to measure ITP factor and platelet antibodies. Trans Assoc Am Physicians 1972; 85:325–334.
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995–1008.
- Karpatkin S. Autoimmune (idiopathic) thrombocytopenic purpura. Lancet 1997; 349:1531–1536.
- Psaila B, Bussel JB. Fc receptors in immune thrombocytopenias: a target for immunomodulation? J Clin Invest 2008; 118:2677–2681.
- Aster RH. Molecular mimicry and immune thrombocytopenia (comment). Blood 2009; 113:3887–3888.
- Takahashi T, Yujiri T, Shinohara K, et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori-associated chronic idiopathic thrombocytopenic purpura. Br J Haematol 2004; 124:91–96.
- Nardi MA, Liu LX, Karpatkin S. GPIIIa-(49-66) is a major pathophysiologically relevant antigenic determinant for anti-platelet GPIIIa of HIV-1-related immunologic thrombocytopenia. Proc Natl Acad Sci U S A 1997; 94:7589–7594.
- Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009; 113:4086–4093.
- Pivetti S, Novarino A, Merico F, et al. High prevalence of autoimmune phenomena in hepatitis C virus antibody positive patients with lymphoproliferative and connective tissue disorders. Br J Haematol 1996; 95:204–211.
- Pawlotsky JM, Bouvier M, Fromont P, et al. Hepatitis C virus infection and autoimmune thrombocytopenic purpura. J Hepatol 1995; 23:635–639.
- Sakuraya M, Murakami H, Uchiumi H, et al. Steroid-refractory chronic idiopathic thrombocytopenic purpura associated with hepatitis C virus infection. Eur J Haematol 2002; 68:49–53.
- García-Suárez J, Burgaleta C, Hernanz N, Albarran F, Tobaruela P, Alvarez-Mon M. HCV-associated thrombocytopenia: clinical characteristics and platelet response after recombinant alpha2b-interferon therapy. Br J Haematol 2000; 110:98–103.
- Rajan SK, Espina BM, Liebman HA. Hepatitis C virus-related thrombocytopenia: clinical and laboratory characteristics compared with chronic immune thrombocytopenic purpura. Br J Haematol 2005; 129:818–824.
- Harker LA. Thrombokinetics in idiopathic thrombocytopenic purpura. Br J Haematol 1970; 19:95–104.
- Branehög I, Kutti J, Weinfeld A. Platelet survival and platelet production in idiopathic thrombocytopenic purpura (ITP). Br J Haematol 1974; 27:127–143.
- Stoll D, Cines DB, Aster RH, Murphy S. Platelet kinetics in patients with idiopathic thrombocytopenic purpura and moderate thrombocytopenia. Blood 1985; 65:584–588.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 1987; 80:33–40.
- McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004; 103:1364–1369.
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 97:2549–2554.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186.
- British Committee for Standards in Haematology General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol 2003; 120:574–596.
- Mazzucconi MG, Fazi P, Bernasconi S, et al; Gruppo Italiano Malattie Ematoligiche dell’Adulto (GIMEMA) Thrombocytopenia Working Party. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 2007; 109:1401–1407.
- Gaines AR. Disseminated intravascular coagulation associated with acute hemoglobinemia or hemoglobinuria following RH9O)(D) immune globulin intravenous administration for immune thrombocytopenic purpura. Blood 2005; 106:1532–1537.
- George JN, Kojouri K, Perdue JJ, Vesely SK. Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Semin Hematol 2000; 37:290–298.
- Schwartz J, Leber MD, Gillis S, Giunta A, Eldor A, Bussel JB. Long term follow-up after splenectomy performed for immune thrombocytopenic purpura (ITP). Am J Hematol 2003; 72:94–98.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004; 104:2623–2634.
- Stasi R, Provan D. Management of immune thrombocytopenic purpura in adults. Mayo Clin Proc 2004; 79:504–522.
- Giagounidis AA, Anhuf J, Schneider P, et al. Treatment of relapsed idiopathic thrombocytopenic purpura with the anti-CD20 monoclonal antibody rituximab: a pilot study. Eur J Haematol 2002; 69:95–100.
- Stasi R, Stipa E, Forte V, Meo P, Amadori S. Variable patterns of response to rituximab treatment in adults with chronic idiopathic thrombocytopenic purpura (letter). Blood 2002; 99:3872–3873.
- Cooper N, Stasi R, Cunningham-Rundles S, et al. The efficacy and safetyof B-cell depletion with anti-CD20 monocloncal antibody in adults with chronic immune thrombocytopenic purpura. Br J Haematol 2004; 125:232–239.
- Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytoopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 2003; 78:1340–1346.
- Patel V, Mihatov N, Cooper N, Stasi R, Cunningham-Rundles S, Bussel JB. Long term follow-up of patients with immune thrombocytopenic purpura (ITP) whose initial response to rituximab lasted a minimum of 1 year (abstract). Blood (ASH Annual Meeting Abstracts): 2006;108:Abstract 479.
- Mukai HY, Kojima H, Todokoro K, et al. Serum thrombopoietin (TPO) levels in patients with amegakaryocytic thrombocytopenia are much higher than those with immune thrombocytopenic purpura. Thromb Haemost 1996; 76:675–678.
- Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006; 355:1672–1681. (Published correction in N Engl J Med 2006; 355:2054.)
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008; 371:395–403.
- Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost 2010; 8:1372–1382.
- Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2010 Aug 23(Epub ahead of print).
- Saleh MN, Bussel JB, Cheng G, et al. Long-term treatment of chronic immune thrombocytopenic purpura with oral eltrombopag. Abstract #682 presented at the 51st American Society of Hematology Annual Meeting and Exposition, New Orleans, LA, December 5–8, 2009; http://ash.confex.com/ash/2009/webprogram/Paper24081.html. Accessed April 26, 2011.
- Kuter DJ, Mufti GJ, Bain BJ, Hasserjian RP, Davis W, Rutstein M. Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood 2009; 114:3748–3756.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393.
- Stasi R, Amadori S, Osborn J, Newland AC, Provan D. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006; 3:e24.
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytoenic purpura among adults: a population-based study and literature review. Eur Haematol 2009; 83:83–89.
- Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR; Northern Region Haematology Group. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol 2003; 122:966–974.
- Sarpatwari A, Bennett D, Logie JW, et al. Thromboembolic events among adult patients with primary immune thrombocytopenia in the United Kingdom General Practice Research Database. Haematologica 2010; 95:1167–1175.
- Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med 1951; 38:1–10.
- Luiken GA, McMillan R, Lightsey AL, et al. Platelet-associated IgG in immune thrombocytopenic purpura. Blood 1977; 50:317–325.
- Hirschman RJ, Schulman NR. Utilization of the platelet release reaction to measure ITP factor and platelet antibodies. Trans Assoc Am Physicians 1972; 85:325–334.
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995–1008.
- Karpatkin S. Autoimmune (idiopathic) thrombocytopenic purpura. Lancet 1997; 349:1531–1536.
- Psaila B, Bussel JB. Fc receptors in immune thrombocytopenias: a target for immunomodulation? J Clin Invest 2008; 118:2677–2681.
- Aster RH. Molecular mimicry and immune thrombocytopenia (comment). Blood 2009; 113:3887–3888.
- Takahashi T, Yujiri T, Shinohara K, et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori-associated chronic idiopathic thrombocytopenic purpura. Br J Haematol 2004; 124:91–96.
- Nardi MA, Liu LX, Karpatkin S. GPIIIa-(49-66) is a major pathophysiologically relevant antigenic determinant for anti-platelet GPIIIa of HIV-1-related immunologic thrombocytopenia. Proc Natl Acad Sci U S A 1997; 94:7589–7594.
- Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009; 113:4086–4093.
- Pivetti S, Novarino A, Merico F, et al. High prevalence of autoimmune phenomena in hepatitis C virus antibody positive patients with lymphoproliferative and connective tissue disorders. Br J Haematol 1996; 95:204–211.
- Pawlotsky JM, Bouvier M, Fromont P, et al. Hepatitis C virus infection and autoimmune thrombocytopenic purpura. J Hepatol 1995; 23:635–639.
- Sakuraya M, Murakami H, Uchiumi H, et al. Steroid-refractory chronic idiopathic thrombocytopenic purpura associated with hepatitis C virus infection. Eur J Haematol 2002; 68:49–53.
- García-Suárez J, Burgaleta C, Hernanz N, Albarran F, Tobaruela P, Alvarez-Mon M. HCV-associated thrombocytopenia: clinical characteristics and platelet response after recombinant alpha2b-interferon therapy. Br J Haematol 2000; 110:98–103.
- Rajan SK, Espina BM, Liebman HA. Hepatitis C virus-related thrombocytopenia: clinical and laboratory characteristics compared with chronic immune thrombocytopenic purpura. Br J Haematol 2005; 129:818–824.
- Harker LA. Thrombokinetics in idiopathic thrombocytopenic purpura. Br J Haematol 1970; 19:95–104.
- Branehög I, Kutti J, Weinfeld A. Platelet survival and platelet production in idiopathic thrombocytopenic purpura (ITP). Br J Haematol 1974; 27:127–143.
- Stoll D, Cines DB, Aster RH, Murphy S. Platelet kinetics in patients with idiopathic thrombocytopenic purpura and moderate thrombocytopenia. Blood 1985; 65:584–588.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 1987; 80:33–40.
- McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004; 103:1364–1369.
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 97:2549–2554.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186.
- British Committee for Standards in Haematology General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol 2003; 120:574–596.
- Mazzucconi MG, Fazi P, Bernasconi S, et al; Gruppo Italiano Malattie Ematoligiche dell’Adulto (GIMEMA) Thrombocytopenia Working Party. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 2007; 109:1401–1407.
- Gaines AR. Disseminated intravascular coagulation associated with acute hemoglobinemia or hemoglobinuria following RH9O)(D) immune globulin intravenous administration for immune thrombocytopenic purpura. Blood 2005; 106:1532–1537.
- George JN, Kojouri K, Perdue JJ, Vesely SK. Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Semin Hematol 2000; 37:290–298.
- Schwartz J, Leber MD, Gillis S, Giunta A, Eldor A, Bussel JB. Long term follow-up after splenectomy performed for immune thrombocytopenic purpura (ITP). Am J Hematol 2003; 72:94–98.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004; 104:2623–2634.
- Stasi R, Provan D. Management of immune thrombocytopenic purpura in adults. Mayo Clin Proc 2004; 79:504–522.
- Giagounidis AA, Anhuf J, Schneider P, et al. Treatment of relapsed idiopathic thrombocytopenic purpura with the anti-CD20 monoclonal antibody rituximab: a pilot study. Eur J Haematol 2002; 69:95–100.
- Stasi R, Stipa E, Forte V, Meo P, Amadori S. Variable patterns of response to rituximab treatment in adults with chronic idiopathic thrombocytopenic purpura (letter). Blood 2002; 99:3872–3873.
- Cooper N, Stasi R, Cunningham-Rundles S, et al. The efficacy and safetyof B-cell depletion with anti-CD20 monocloncal antibody in adults with chronic immune thrombocytopenic purpura. Br J Haematol 2004; 125:232–239.
- Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytoopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 2003; 78:1340–1346.
- Patel V, Mihatov N, Cooper N, Stasi R, Cunningham-Rundles S, Bussel JB. Long term follow-up of patients with immune thrombocytopenic purpura (ITP) whose initial response to rituximab lasted a minimum of 1 year (abstract). Blood (ASH Annual Meeting Abstracts): 2006;108:Abstract 479.
- Mukai HY, Kojima H, Todokoro K, et al. Serum thrombopoietin (TPO) levels in patients with amegakaryocytic thrombocytopenia are much higher than those with immune thrombocytopenic purpura. Thromb Haemost 1996; 76:675–678.
- Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006; 355:1672–1681. (Published correction in N Engl J Med 2006; 355:2054.)
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008; 371:395–403.
- Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost 2010; 8:1372–1382.
- Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2010 Aug 23(Epub ahead of print).
- Saleh MN, Bussel JB, Cheng G, et al. Long-term treatment of chronic immune thrombocytopenic purpura with oral eltrombopag. Abstract #682 presented at the 51st American Society of Hematology Annual Meeting and Exposition, New Orleans, LA, December 5–8, 2009; http://ash.confex.com/ash/2009/webprogram/Paper24081.html. Accessed April 26, 2011.
- Kuter DJ, Mufti GJ, Bain BJ, Hasserjian RP, Davis W, Rutstein M. Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood 2009; 114:3748–3756.
KEY POINTS
- ITP is defined as an isolated platelet count of less than 100 × 109/L (100,000/μL) and usually presents without symptoms.
- Patients without symptoms who have a platelet count above 30 × 109/L should generally not be treated unless they have an increased risk of bleeding.
- Recent studies suggest that viruses and other pathogens play an important role in secondary ITP.
- Initially, corticosteroids are usually given as prednisone (1–2 mg/kg/day, then tapered), though recent studies suggest that dexamethasone pulses (40 mg/day for 4 days) may provide more durable responses when used in this setting.
- Thrombopoietic agents are important new treatments, although their place in the overall therapy of ITP has not been established.