User login
Chromoblastomycosis
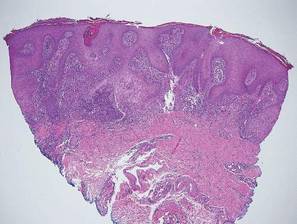
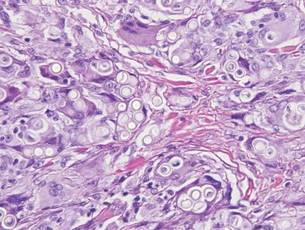
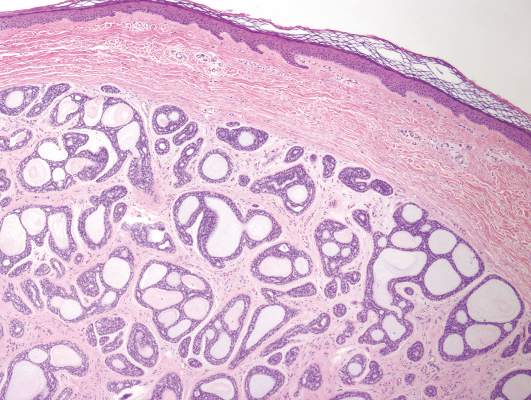
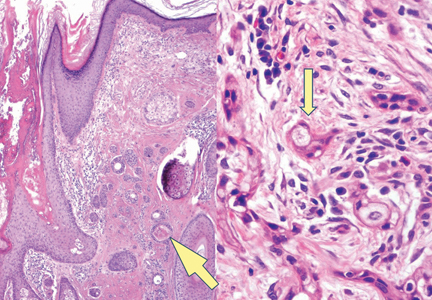
Chromoblastomycosis is a chronic fungal infection of the skin and subcutaneous tissues that demonstrates characteristic Medlar or sclerotic bodies that resemble copper pennies on histopathology.1 Cutaneous infection often results from direct inoculation, such as from a wood splinter. Clinically, the lesion typically is a pink papule that progresses to a verrucous plaque on the legs of farmers or rural workers in the tropics or subtropics. There usually are no associated constitutional symptoms. Several dematiaceous (darkly pigmented) fungi cause chromoblastomycosis, including Fonsecaea compacta, Cladophialophora carrionii, Rhinocladiella aquaspersa, Phialophora verrucosa, and Fonsecaea pedrosoi. Cellular division occurs by internal septation rather than budding. Skin biopsy can confirm the diagnosis.1 Chromoblastomycosis is histopathologically characterized by pseudoepitheli- omatous hyperplasia (Figure 1) with histiocytes and neutrophils surrounding distinct copper-colored Medlar bodies (6–12 μm)(Figure 2), which are fungal spores.1-3 Several conditions demonstrate pseudoepitheliomatous hyperplasia with intraepidermal pustules and can be remembered by the mnemonic “here come big green leafy vegetables”: halogenoderma, chromoblastomycosis, blastomycosis, granuloma inguinale, leishmaniasis, and pemphigus vegetans.2 Treatment of chromoblastomycosis can be challenging, as no standard treatment has been established and therapy can be complicated by low cure rates and high relapse rates, especially in chronic and extensive disease. Treatment can include cryotherapy or surgical excision for small lesions in combination with systemic antifungals.4 Itraconazole (200–400 mg daily) for at least 6 months has been reported to have up to a 90% cure rate with mild to moderate disease and 44% with severe disease.5 Combination oral antifungal treatment with itraconazole and terbinafine has been recommended.6 There are reports of progression of chromoblastomycosis to squamous cell carcinoma, which is rare and occurred after long-standing, inadequately treated lesions.7

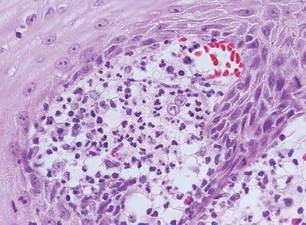
Blastomycosis also presents with pseudoepitheliomatous hyperplasia, as seen in chromoblastomycosis, but organisms typically are few in number and demonstrate a thick, asymmetrical, refractile wall and a dark nucleus. Although chromoblastomycosis and blastomycosis are similar in size (8–15 μm), the broad-based budding of blastomycosis (Figure 3) is a key feature and the yeast are not pigmented.1-3 Blastomycosis is caused by Blastomyces dermatitidis and is endemic to the Mississippi and Ohio River valleys, Great Lakes region, and Southeastern United States. Cutaneous infection typically occurs from inhalation of the dimorphic fungi into the lungs and occasional dissemination involving the skin, causing papulopustules and thick, crusted, warty plaques with central ulceration. Rarely, primary cutaneous blastomycosis can occur from direct inoculation, typically in a laboratory. Treatment of disseminated blastomycosis includes systemic antifungals.1
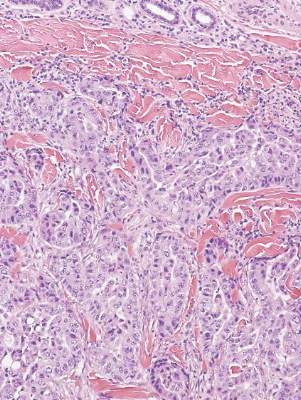
Coccidioidomycosis is characterized by large spherules (10–80 μm) with refractile walls and granular gray cytoplasm.2,3 Coccidioidomycosis spherules occasionally contain endospores2 and often are noticeably larger than surrounding histiocyte nuclei (Figure 4), whereas chromoblastomycosis, blastomycosis, cryptococcosis, and lobomycosis are more similar in size to histiocyte nuclei. Coccidioidomycosis is caused by Coccidioides immitis, a highly virulent dimorphic fungus found in the Southwestern United States, northern Mexico, and Central and South America. Pulmonary infection occurs by inhalation of arthroconidia, often from soil, and is asymptomatic in most patients; however, immunocompromised patients are predisposed to disseminated cutaneous infection. Facial lesions are most common and can present as papules, pustules, plaques, abscesses, sinus tracts, and/or ulcerations. Treatment of disseminated infection requires systemic antifungals; amphotericin B has proven most effective.1
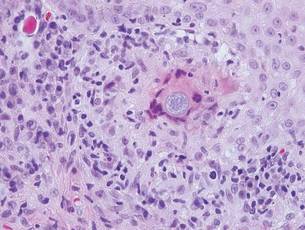
Cryptococcosis is characterized by vacuoles with small (2–20 μm), central, pleomorphic yeast (Figure 5). The vacuole is due to a gelati- nous capsule that stains red with mucicarmine and blue with Alcian blue.2,3 Cryptococcosis is caused by Cryptococcus neoformans and is associated with pigeon droppings. Disseminated infection in patients with human immunodefi- ciency virus often presents as umbilicated molluscumlike lesions and portends a poor prognosis with a mortality rate of up to 80%.8 Disseminated infection necessitates aggressive treatment with systemic antifungals.1
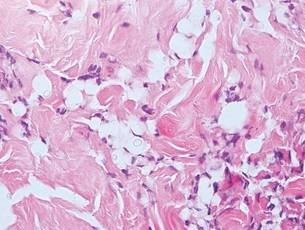
Lobomycosis demonstrates thick-walled, refractile spherules with surrounding histiocytes and multinucleated giant cells. The yeast of lobomycosis (6–12 μm) is of similar size to chromoblastomycosis and blastomycosis, but linear chains resembling a child’s pop beads are characteristic of this condition (Figure 6).2,3 Lobomycosis is caused by Lacazia loboi and is acquired most frequently through contact with dolphins in Central and South America. Clinically, lesions present as slow-growing, keloidlike nodules, often on the face, ears, and distal extremities. Surgical treatment may be required given that oral antifungals typically are ineffective.1
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Fernandez-Flores A, Saeb-Lima M, Arenas-Guzman R. Morphological findings of deep cutaneous fungal infections. Am J Dermatopathol. 2014;36:531-556.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Queiroz-Telles F, McGinnis MR, Salkin I, et al. Subcutaneous mycoses. Infect Dis Clin North Am. 2003;17:59-85.
- Bonifaz A, Paredes-Solís, Saúl A. Treating chromoblastomycosis with systemic antifungals. Expert Opin Pharmacother. 2004;5:247-254.
- Rojas OC, González GM, Moreno-Treviño M, et al. Chromoblastomycosis by Cladophialophora carrionii associated with squamous cell carcinoma and review of published reports. Mycopathologia. 2015;179:153-157.
- Durden FM, Elewski B. Cutaneous involvement with Cryptococcus neoformans in AIDS. J Am Acad Dermatol. 1994;30:844-848.
Chromoblastomycosis is a chronic fungal infection of the skin and subcutaneous tissues that demonstrates characteristic Medlar or sclerotic bodies that resemble copper pennies on histopathology.1 Cutaneous infection often results from direct inoculation, such as from a wood splinter. Clinically, the lesion typically is a pink papule that progresses to a verrucous plaque on the legs of farmers or rural workers in the tropics or subtropics. There usually are no associated constitutional symptoms. Several dematiaceous (darkly pigmented) fungi cause chromoblastomycosis, including Fonsecaea compacta, Cladophialophora carrionii, Rhinocladiella aquaspersa, Phialophora verrucosa, and Fonsecaea pedrosoi. Cellular division occurs by internal septation rather than budding. Skin biopsy can confirm the diagnosis.1 Chromoblastomycosis is histopathologically characterized by pseudoepitheli- omatous hyperplasia (Figure 1) with histiocytes and neutrophils surrounding distinct copper-colored Medlar bodies (6–12 μm)(Figure 2), which are fungal spores.1-3 Several conditions demonstrate pseudoepitheliomatous hyperplasia with intraepidermal pustules and can be remembered by the mnemonic “here come big green leafy vegetables”: halogenoderma, chromoblastomycosis, blastomycosis, granuloma inguinale, leishmaniasis, and pemphigus vegetans.2 Treatment of chromoblastomycosis can be challenging, as no standard treatment has been established and therapy can be complicated by low cure rates and high relapse rates, especially in chronic and extensive disease. Treatment can include cryotherapy or surgical excision for small lesions in combination with systemic antifungals.4 Itraconazole (200–400 mg daily) for at least 6 months has been reported to have up to a 90% cure rate with mild to moderate disease and 44% with severe disease.5 Combination oral antifungal treatment with itraconazole and terbinafine has been recommended.6 There are reports of progression of chromoblastomycosis to squamous cell carcinoma, which is rare and occurred after long-standing, inadequately treated lesions.7
Blastomycosis also presents with pseudoepitheliomatous hyperplasia, as seen in chromoblastomycosis, but organisms typically are few in number and demonstrate a thick, asymmetrical, refractile wall and a dark nucleus. Although chromoblastomycosis and blastomycosis are similar in size (8–15 μm), the broad-based budding of blastomycosis (Figure 3) is a key feature and the yeast are not pigmented.1-3 Blastomycosis is caused by Blastomyces dermatitidis and is endemic to the Mississippi and Ohio River valleys, Great Lakes region, and Southeastern United States. Cutaneous infection typically occurs from inhalation of the dimorphic fungi into the lungs and occasional dissemination involving the skin, causing papulopustules and thick, crusted, warty plaques with central ulceration. Rarely, primary cutaneous blastomycosis can occur from direct inoculation, typically in a laboratory. Treatment of disseminated blastomycosis includes systemic antifungals.1
Coccidioidomycosis is characterized by large spherules (10–80 μm) with refractile walls and granular gray cytoplasm.2,3 Coccidioidomycosis spherules occasionally contain endospores2 and often are noticeably larger than surrounding histiocyte nuclei (Figure 4), whereas chromoblastomycosis, blastomycosis, cryptococcosis, and lobomycosis are more similar in size to histiocyte nuclei. Coccidioidomycosis is caused by Coccidioides immitis, a highly virulent dimorphic fungus found in the Southwestern United States, northern Mexico, and Central and South America. Pulmonary infection occurs by inhalation of arthroconidia, often from soil, and is asymptomatic in most patients; however, immunocompromised patients are predisposed to disseminated cutaneous infection. Facial lesions are most common and can present as papules, pustules, plaques, abscesses, sinus tracts, and/or ulcerations. Treatment of disseminated infection requires systemic antifungals; amphotericin B has proven most effective.1
Cryptococcosis is characterized by vacuoles with small (2–20 μm), central, pleomorphic yeast (Figure 5). The vacuole is due to a gelati- nous capsule that stains red with mucicarmine and blue with Alcian blue.2,3 Cryptococcosis is caused by Cryptococcus neoformans and is associated with pigeon droppings. Disseminated infection in patients with human immunodefi- ciency virus often presents as umbilicated molluscumlike lesions and portends a poor prognosis with a mortality rate of up to 80%.8 Disseminated infection necessitates aggressive treatment with systemic antifungals.1
Lobomycosis demonstrates thick-walled, refractile spherules with surrounding histiocytes and multinucleated giant cells. The yeast of lobomycosis (6–12 μm) is of similar size to chromoblastomycosis and blastomycosis, but linear chains resembling a child’s pop beads are characteristic of this condition (Figure 6).2,3 Lobomycosis is caused by Lacazia loboi and is acquired most frequently through contact with dolphins in Central and South America. Clinically, lesions present as slow-growing, keloidlike nodules, often on the face, ears, and distal extremities. Surgical treatment may be required given that oral antifungals typically are ineffective.1
Chromoblastomycosis is a chronic fungal infection of the skin and subcutaneous tissues that demonstrates characteristic Medlar or sclerotic bodies that resemble copper pennies on histopathology.1 Cutaneous infection often results from direct inoculation, such as from a wood splinter. Clinically, the lesion typically is a pink papule that progresses to a verrucous plaque on the legs of farmers or rural workers in the tropics or subtropics. There usually are no associated constitutional symptoms. Several dematiaceous (darkly pigmented) fungi cause chromoblastomycosis, including Fonsecaea compacta, Cladophialophora carrionii, Rhinocladiella aquaspersa, Phialophora verrucosa, and Fonsecaea pedrosoi. Cellular division occurs by internal septation rather than budding. Skin biopsy can confirm the diagnosis.1 Chromoblastomycosis is histopathologically characterized by pseudoepitheli- omatous hyperplasia (Figure 1) with histiocytes and neutrophils surrounding distinct copper-colored Medlar bodies (6–12 μm)(Figure 2), which are fungal spores.1-3 Several conditions demonstrate pseudoepitheliomatous hyperplasia with intraepidermal pustules and can be remembered by the mnemonic “here come big green leafy vegetables”: halogenoderma, chromoblastomycosis, blastomycosis, granuloma inguinale, leishmaniasis, and pemphigus vegetans.2 Treatment of chromoblastomycosis can be challenging, as no standard treatment has been established and therapy can be complicated by low cure rates and high relapse rates, especially in chronic and extensive disease. Treatment can include cryotherapy or surgical excision for small lesions in combination with systemic antifungals.4 Itraconazole (200–400 mg daily) for at least 6 months has been reported to have up to a 90% cure rate with mild to moderate disease and 44% with severe disease.5 Combination oral antifungal treatment with itraconazole and terbinafine has been recommended.6 There are reports of progression of chromoblastomycosis to squamous cell carcinoma, which is rare and occurred after long-standing, inadequately treated lesions.7
Blastomycosis also presents with pseudoepitheliomatous hyperplasia, as seen in chromoblastomycosis, but organisms typically are few in number and demonstrate a thick, asymmetrical, refractile wall and a dark nucleus. Although chromoblastomycosis and blastomycosis are similar in size (8–15 μm), the broad-based budding of blastomycosis (Figure 3) is a key feature and the yeast are not pigmented.1-3 Blastomycosis is caused by Blastomyces dermatitidis and is endemic to the Mississippi and Ohio River valleys, Great Lakes region, and Southeastern United States. Cutaneous infection typically occurs from inhalation of the dimorphic fungi into the lungs and occasional dissemination involving the skin, causing papulopustules and thick, crusted, warty plaques with central ulceration. Rarely, primary cutaneous blastomycosis can occur from direct inoculation, typically in a laboratory. Treatment of disseminated blastomycosis includes systemic antifungals.1
Coccidioidomycosis is characterized by large spherules (10–80 μm) with refractile walls and granular gray cytoplasm.2,3 Coccidioidomycosis spherules occasionally contain endospores2 and often are noticeably larger than surrounding histiocyte nuclei (Figure 4), whereas chromoblastomycosis, blastomycosis, cryptococcosis, and lobomycosis are more similar in size to histiocyte nuclei. Coccidioidomycosis is caused by Coccidioides immitis, a highly virulent dimorphic fungus found in the Southwestern United States, northern Mexico, and Central and South America. Pulmonary infection occurs by inhalation of arthroconidia, often from soil, and is asymptomatic in most patients; however, immunocompromised patients are predisposed to disseminated cutaneous infection. Facial lesions are most common and can present as papules, pustules, plaques, abscesses, sinus tracts, and/or ulcerations. Treatment of disseminated infection requires systemic antifungals; amphotericin B has proven most effective.1
Cryptococcosis is characterized by vacuoles with small (2–20 μm), central, pleomorphic yeast (Figure 5). The vacuole is due to a gelati- nous capsule that stains red with mucicarmine and blue with Alcian blue.2,3 Cryptococcosis is caused by Cryptococcus neoformans and is associated with pigeon droppings. Disseminated infection in patients with human immunodefi- ciency virus often presents as umbilicated molluscumlike lesions and portends a poor prognosis with a mortality rate of up to 80%.8 Disseminated infection necessitates aggressive treatment with systemic antifungals.1
Lobomycosis demonstrates thick-walled, refractile spherules with surrounding histiocytes and multinucleated giant cells. The yeast of lobomycosis (6–12 μm) is of similar size to chromoblastomycosis and blastomycosis, but linear chains resembling a child’s pop beads are characteristic of this condition (Figure 6).2,3 Lobomycosis is caused by Lacazia loboi and is acquired most frequently through contact with dolphins in Central and South America. Clinically, lesions present as slow-growing, keloidlike nodules, often on the face, ears, and distal extremities. Surgical treatment may be required given that oral antifungals typically are ineffective.1
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Fernandez-Flores A, Saeb-Lima M, Arenas-Guzman R. Morphological findings of deep cutaneous fungal infections. Am J Dermatopathol. 2014;36:531-556.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Queiroz-Telles F, McGinnis MR, Salkin I, et al. Subcutaneous mycoses. Infect Dis Clin North Am. 2003;17:59-85.
- Bonifaz A, Paredes-Solís, Saúl A. Treating chromoblastomycosis with systemic antifungals. Expert Opin Pharmacother. 2004;5:247-254.
- Rojas OC, González GM, Moreno-Treviño M, et al. Chromoblastomycosis by Cladophialophora carrionii associated with squamous cell carcinoma and review of published reports. Mycopathologia. 2015;179:153-157.
- Durden FM, Elewski B. Cutaneous involvement with Cryptococcus neoformans in AIDS. J Am Acad Dermatol. 1994;30:844-848.
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Fernandez-Flores A, Saeb-Lima M, Arenas-Guzman R. Morphological findings of deep cutaneous fungal infections. Am J Dermatopathol. 2014;36:531-556.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Queiroz-Telles F, McGinnis MR, Salkin I, et al. Subcutaneous mycoses. Infect Dis Clin North Am. 2003;17:59-85.
- Bonifaz A, Paredes-Solís, Saúl A. Treating chromoblastomycosis with systemic antifungals. Expert Opin Pharmacother. 2004;5:247-254.
- Rojas OC, González GM, Moreno-Treviño M, et al. Chromoblastomycosis by Cladophialophora carrionii associated with squamous cell carcinoma and review of published reports. Mycopathologia. 2015;179:153-157.
- Durden FM, Elewski B. Cutaneous involvement with Cryptococcus neoformans in AIDS. J Am Acad Dermatol. 1994;30:844-848.
Syringoid Eccrine Carcinoma
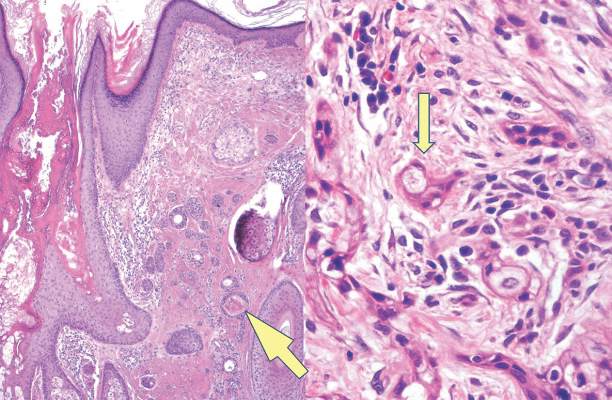
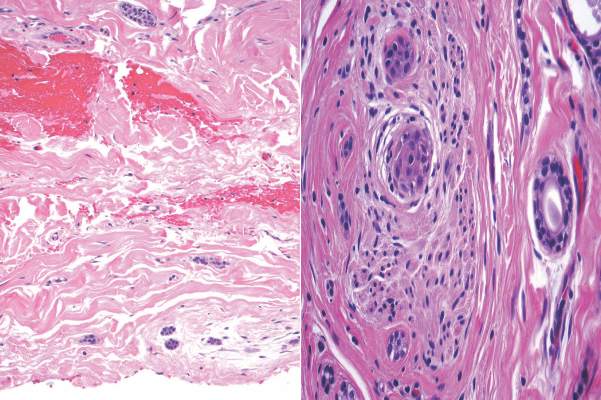
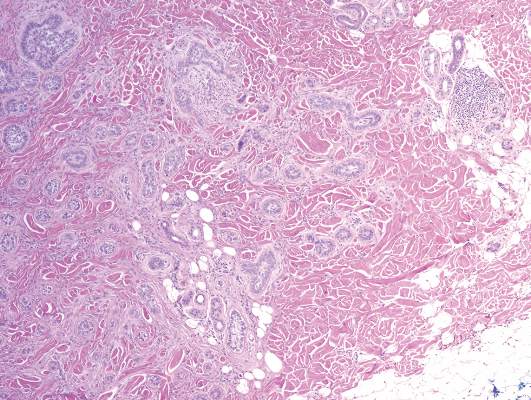
Syringoid eccrine carcinoma is a rare malignant adnexal tumor with eccrine differentiation that histologically resembles a syringoma.1 Originally described as eccrine epithelioma by Freeman and Winklemann2 in 1969, syringoid eccrine carcinoma has been reported in the literature as eccrine carcinoma, eccrine syringomatous carcinoma, and sclerosing sweat duct carcinoma.3 Clinically, syringoid eccrine carcinoma most commonly presents as a tender plaque or nodule on the scalp, and histologic examination generally reveals a dermal-based lesion that rarely shows epidermal connection. It demonstrates syringomalike tadpole morphology (epithelial strands with lumen formation) composed of basaloid epithelium with uniform hyperchromatic nuclei (Figure 1). There usually is an infiltrative growth pattern to the subcutis (Figure 2 [left]) or skeletal muscle as well as remarkable perineural invasion (Figure 2 [right]). Mitotic activity is minimal to absent. The tumor cells of syringoid eccrine carcinoma typically show positive immuno-staining for high- and low-molecular-weight cytokeratin, while the lumina are highlighted by epithelial membrane antigen and carcinoembryonic antigen.4 However, immunohistochemistry often is not contributory in diagnosing primary eccrine carcinomas.
The differential diagnosis of syringoid eccrine carcinoma includes cutaneous adenoid cystic carcinoma, metastatic adenocarcinoma, sclerosing basal cell carcinoma, and syringoma. Cutaneous adenoid cystic carcinoma is a rare, slow-growing, flesh-colored tumor that consists of lobules, islands, and cords of basaloid cells with prominent cystic cribriforming (Figure 3). The tumor cells typically are small, cuboidal, and monomorphic. Metastatic adenoid cystic carcinoma, such as from a primary tumor of the salivary glands or breasts, must be excluded before rendering a diagnosis of primary cutaneous disease.
Metastatic adenocarcinoma of the skin usually presents in patients with a clinical history of preexisting disease. The breasts, colon, stomach, and ovaries are common origins of metastases. The histopathologic and immunohistochemical findings depend on the particular site of origin of the metastasis. Compared with primary eccrine carcinomas, metastatic adenocarcinomas of the skin generally are high-grade lesions with prominent atypia, mitosis, and necrosis (Figure 4).
Sclerosing basal cell carcinoma shows basaloid tumor cells with deep infiltration. Unlike syringoid eccrine carcinoma, basal cell carcinoma is an epidermal tumor that does not have true lumen formation. Furthermore, other variants of basal cell carcinoma, including nodular, micronodular, or superficial multicentric tumors, often coexist with the sclerosing variant in the same lesion and constitute a useful diagnostic clue (Figure 5). Staining for epithelial membrane antigen may be useful in identifying the absence of lumen formation, and Ber-EP4 highlights the epidermal origin of the lesion.5
Syringomas most commonly present as multiple small flesh-colored papules on the eyelids. On histology, syringomas present as small superficial dermal lesions composed of small ducts that may form tadpolelike structures in a fibrotic stroma (Figure 6). The ducts are lined by benign cuboidal cells. In contrast to syringoid eccrine carcinomas, syringomas usually present as multiple lesions that are microscopically superficial without perineural involvement.
1. Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
2. Freeman RG, Winklemann RK. Basal cell tumor with eccrine differentiations (eccrine epithelioma). Arch Dermatol. 1969;100:234-242.
3. Nishizawa A, Nakanishi Y, Sasajima Y, et al. Syringoid carcinoma with apparently aggressive transformation: case report and review of the literature. Int J Dermatol. 2006;45:1218-1221.
4. Urso C, Bondi R, Paglierani M, et al. Carcinomas of sweat glands: report of 60 cases. Arch Pathol Lab Med. 2001;125:498-505.
5. Cassarino D. Diagnostic Pathology: Neoplastic Dermatopathology. Salt Lake City, UT: Amirsys Publishing Inc; 2012.
Syringoid eccrine carcinoma is a rare malignant adnexal tumor with eccrine differentiation that histologically resembles a syringoma.1 Originally described as eccrine epithelioma by Freeman and Winklemann2 in 1969, syringoid eccrine carcinoma has been reported in the literature as eccrine carcinoma, eccrine syringomatous carcinoma, and sclerosing sweat duct carcinoma.3 Clinically, syringoid eccrine carcinoma most commonly presents as a tender plaque or nodule on the scalp, and histologic examination generally reveals a dermal-based lesion that rarely shows epidermal connection. It demonstrates syringomalike tadpole morphology (epithelial strands with lumen formation) composed of basaloid epithelium with uniform hyperchromatic nuclei (Figure 1). There usually is an infiltrative growth pattern to the subcutis (Figure 2 [left]) or skeletal muscle as well as remarkable perineural invasion (Figure 2 [right]). Mitotic activity is minimal to absent. The tumor cells of syringoid eccrine carcinoma typically show positive immuno-staining for high- and low-molecular-weight cytokeratin, while the lumina are highlighted by epithelial membrane antigen and carcinoembryonic antigen.4 However, immunohistochemistry often is not contributory in diagnosing primary eccrine carcinomas.
The differential diagnosis of syringoid eccrine carcinoma includes cutaneous adenoid cystic carcinoma, metastatic adenocarcinoma, sclerosing basal cell carcinoma, and syringoma. Cutaneous adenoid cystic carcinoma is a rare, slow-growing, flesh-colored tumor that consists of lobules, islands, and cords of basaloid cells with prominent cystic cribriforming (Figure 3). The tumor cells typically are small, cuboidal, and monomorphic. Metastatic adenoid cystic carcinoma, such as from a primary tumor of the salivary glands or breasts, must be excluded before rendering a diagnosis of primary cutaneous disease.
Metastatic adenocarcinoma of the skin usually presents in patients with a clinical history of preexisting disease. The breasts, colon, stomach, and ovaries are common origins of metastases. The histopathologic and immunohistochemical findings depend on the particular site of origin of the metastasis. Compared with primary eccrine carcinomas, metastatic adenocarcinomas of the skin generally are high-grade lesions with prominent atypia, mitosis, and necrosis (Figure 4).
Sclerosing basal cell carcinoma shows basaloid tumor cells with deep infiltration. Unlike syringoid eccrine carcinoma, basal cell carcinoma is an epidermal tumor that does not have true lumen formation. Furthermore, other variants of basal cell carcinoma, including nodular, micronodular, or superficial multicentric tumors, often coexist with the sclerosing variant in the same lesion and constitute a useful diagnostic clue (Figure 5). Staining for epithelial membrane antigen may be useful in identifying the absence of lumen formation, and Ber-EP4 highlights the epidermal origin of the lesion.5
Syringomas most commonly present as multiple small flesh-colored papules on the eyelids. On histology, syringomas present as small superficial dermal lesions composed of small ducts that may form tadpolelike structures in a fibrotic stroma (Figure 6). The ducts are lined by benign cuboidal cells. In contrast to syringoid eccrine carcinomas, syringomas usually present as multiple lesions that are microscopically superficial without perineural involvement.
Syringoid eccrine carcinoma is a rare malignant adnexal tumor with eccrine differentiation that histologically resembles a syringoma.1 Originally described as eccrine epithelioma by Freeman and Winklemann2 in 1969, syringoid eccrine carcinoma has been reported in the literature as eccrine carcinoma, eccrine syringomatous carcinoma, and sclerosing sweat duct carcinoma.3 Clinically, syringoid eccrine carcinoma most commonly presents as a tender plaque or nodule on the scalp, and histologic examination generally reveals a dermal-based lesion that rarely shows epidermal connection. It demonstrates syringomalike tadpole morphology (epithelial strands with lumen formation) composed of basaloid epithelium with uniform hyperchromatic nuclei (Figure 1). There usually is an infiltrative growth pattern to the subcutis (Figure 2 [left]) or skeletal muscle as well as remarkable perineural invasion (Figure 2 [right]). Mitotic activity is minimal to absent. The tumor cells of syringoid eccrine carcinoma typically show positive immuno-staining for high- and low-molecular-weight cytokeratin, while the lumina are highlighted by epithelial membrane antigen and carcinoembryonic antigen.4 However, immunohistochemistry often is not contributory in diagnosing primary eccrine carcinomas.
The differential diagnosis of syringoid eccrine carcinoma includes cutaneous adenoid cystic carcinoma, metastatic adenocarcinoma, sclerosing basal cell carcinoma, and syringoma. Cutaneous adenoid cystic carcinoma is a rare, slow-growing, flesh-colored tumor that consists of lobules, islands, and cords of basaloid cells with prominent cystic cribriforming (Figure 3). The tumor cells typically are small, cuboidal, and monomorphic. Metastatic adenoid cystic carcinoma, such as from a primary tumor of the salivary glands or breasts, must be excluded before rendering a diagnosis of primary cutaneous disease.
Metastatic adenocarcinoma of the skin usually presents in patients with a clinical history of preexisting disease. The breasts, colon, stomach, and ovaries are common origins of metastases. The histopathologic and immunohistochemical findings depend on the particular site of origin of the metastasis. Compared with primary eccrine carcinomas, metastatic adenocarcinomas of the skin generally are high-grade lesions with prominent atypia, mitosis, and necrosis (Figure 4).
Sclerosing basal cell carcinoma shows basaloid tumor cells with deep infiltration. Unlike syringoid eccrine carcinoma, basal cell carcinoma is an epidermal tumor that does not have true lumen formation. Furthermore, other variants of basal cell carcinoma, including nodular, micronodular, or superficial multicentric tumors, often coexist with the sclerosing variant in the same lesion and constitute a useful diagnostic clue (Figure 5). Staining for epithelial membrane antigen may be useful in identifying the absence of lumen formation, and Ber-EP4 highlights the epidermal origin of the lesion.5
Syringomas most commonly present as multiple small flesh-colored papules on the eyelids. On histology, syringomas present as small superficial dermal lesions composed of small ducts that may form tadpolelike structures in a fibrotic stroma (Figure 6). The ducts are lined by benign cuboidal cells. In contrast to syringoid eccrine carcinomas, syringomas usually present as multiple lesions that are microscopically superficial without perineural involvement.
1. Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
2. Freeman RG, Winklemann RK. Basal cell tumor with eccrine differentiations (eccrine epithelioma). Arch Dermatol. 1969;100:234-242.
3. Nishizawa A, Nakanishi Y, Sasajima Y, et al. Syringoid carcinoma with apparently aggressive transformation: case report and review of the literature. Int J Dermatol. 2006;45:1218-1221.
4. Urso C, Bondi R, Paglierani M, et al. Carcinomas of sweat glands: report of 60 cases. Arch Pathol Lab Med. 2001;125:498-505.
5. Cassarino D. Diagnostic Pathology: Neoplastic Dermatopathology. Salt Lake City, UT: Amirsys Publishing Inc; 2012.
1. Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
2. Freeman RG, Winklemann RK. Basal cell tumor with eccrine differentiations (eccrine epithelioma). Arch Dermatol. 1969;100:234-242.
3. Nishizawa A, Nakanishi Y, Sasajima Y, et al. Syringoid carcinoma with apparently aggressive transformation: case report and review of the literature. Int J Dermatol. 2006;45:1218-1221.
4. Urso C, Bondi R, Paglierani M, et al. Carcinomas of sweat glands: report of 60 cases. Arch Pathol Lab Med. 2001;125:498-505.
5. Cassarino D. Diagnostic Pathology: Neoplastic Dermatopathology. Salt Lake City, UT: Amirsys Publishing Inc; 2012.
Granulomatous Cheilitis: A Stiff Upper Lip
To the Editor:
A 51-year-old woman presented to her dermatologist with recurrent and progressive upper lip swelling of 2 years’ duration. Her condition was previously evaluated by several other physicians without a diagnosis or resolution of the symptoms. The swelling began on the right side of the upper lip and right cheek; however, over the course of 2 years, the swelling had progressed to involve the entire upper lip with complete sparing of the lower lip. She denied pain but reported numbness of the upper lip. The patient visited her dentist who ruled out periodontal infection as the cause of the swelling. Diphenhydramine provided no relief; however, the cheek swelling resolved after a course of antibiotics prescribed by an ear, nose, and throat physician.
She consulted her primary care physician and was subsequently referred to a neurologist and allergist who were unable to provide a definitive diagnosis or complete relief of the symptoms. She denied any history of hypersensitivity reactions, odontogenic infections, gastrointestinal concerns, or any other signs or symptoms of systemic granulomatous disease.
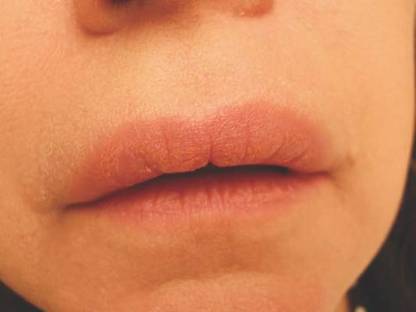
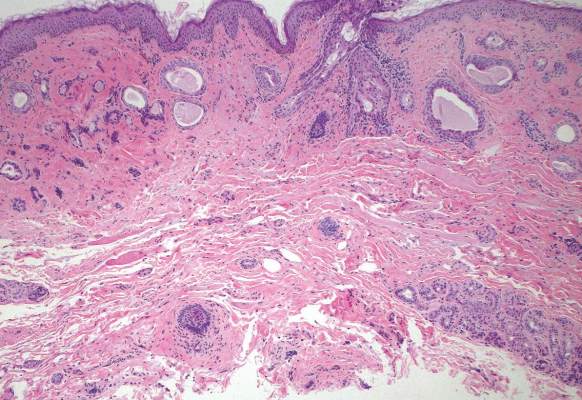
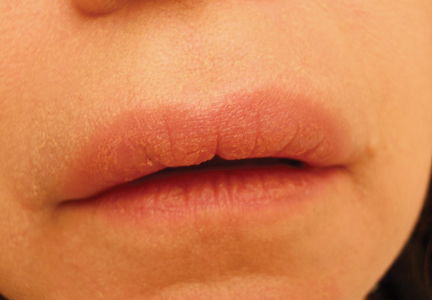
On physical examination, the upper lip was swollen symmetrically without evidence of ulceration, fissuring, or scaling (Figure 1). Palpation of the upper lip was notable for firm, nontender, nonpitting edema without nodularity. The oral mucosa did not appear swollen or erythematous. Examination did not reveal ulceration or a cobblestone appearance.
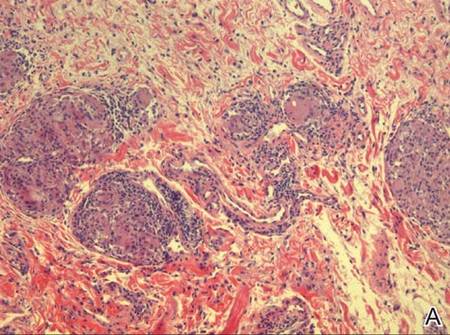
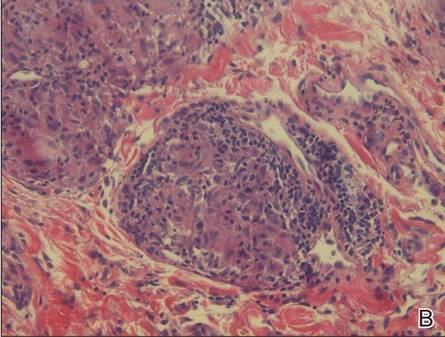
A full-thickness skin biopsy of the upper lip was performed. Histopathology revealed perivascular nonnecrotizing granulomas adjacent to ectatic vascular channels with associated lymphoplasmacytic infiltrate (Figure 2). Periodic acid–Schiff stain was negative for fungal hyphae, tissue Gram stain was negative for bacteria, Fite and acid-fast bacillus stains were both negative for acid-fast organisms, and polariscopy was negative for polarizable foreign material. In this clinical context, the morphologic findings were consistent with the diagnosis of granulomatous cheilitis (GC).
|
Figure 2. Upper lip biopsy showed dermal edema, vascular ectasia, perivascular nonnecrotizing granulomas, and perivascular lymphocyte predominant inflammatory infiltrate (A)(H&E, original magnification ×100). Higher magnification of granulomas with perivascular lymphoplasmacytic infiltrate (B)(H&E, original magnification ×200). |
Granulomatous cheilitis is a rare disorder of the lips and orofacial mucosa that was first described by Meischer1 in 1945 as persistent or recurrent orofacial swelling secondary to lymphatic obstruction by granulomatous proliferation. It often has been described as a monosymptomatic form of Melkersson-Rosenthal syndrome (MRS). In its entirety, MRS constitutes a triad of GC, facial nerve palsy, and lingua plicata (also known as fissured tongue).2,3 Although many authors agree that GC is associated with MRS, some believe that GC is a distinct entity because the majority of patients who present with GC subsequently do not develop MRS.4 Despite its relationship to MRS, the true incidence of GC largely is unknown. The onset of disease usually occurs in early adulthood but can present in middle-aged or older individuals.
The typical course of GC is relapsing and remitting, nontender and nonpitting swelling of the lips that eventually becomes permanent, leading to possible facial distortion and disability. Involvement of the upper lip is the most common, followed by (in order of decreasing frequency) the lower lip and cheeks.5 The swelling may be unilateral or bilateral and generally is not associated with ulceration, fissuring, or scaling; however, these complications have been reported in the terminal stages of the disease in which the macrocheilia has become permanent.
Despite the controversy over the etiology, pathophysiology, and classification of GC, it largely is accepted that when a patient presents clinically with a history of recurrent or persistent lip swelling, a full-thickness skin biopsy of the involved oral mucosa should be taken. Conditions that are considered in the differential diagnosis of orofacial granulomatosis are systemic granulomatous diseases that are known to have oral manifestations including Crohn disease, sarcoidosis, and mycobacterial infections. Given the many causes of orofacial and labial swelling, GC is a diagnosis of exclusion based on a thorough history and physical examination as well as appropriate diagnostic studies, with the cornerstone of the diagnosis resting on the histologic appearance of the lesion. Histologically, the diagnosis lies in the demonstration of granuloma formation, consisting of collections of epithelioid histiocytes and Langerhans giant cells. Once granuloma formation is documented, special stains are used to rule out other granulomatous diseases.
Intralesional steroids have been reported to provide the greatest improvement; however, in the majority of patients, multiple treatments are required.6,7 Allen et al8 suggested that the efficacy of intralesional therapy increases when preceded by local anesthesia of the lip, thus allowing larger doses of triamcinolone to be tolerated by the patient. Systemic corticosteroids also have been used with moderate success, but the side effects of long-term systemic corticosteroid therapy make this treatment option less appealing.9 Other agents with known anti-inflammatory properties also have been used that may offer better side-effect profiles when used for long-term suppressive therapy, including clofazimine, dapsone, sulfapyridine, danazol, hydroxychloroquine, and antibiotics such as doxycycline and metronidazole.10
In severe or recalcitrant cases, surgical intervention by way of a reduction cheiloplasty is considered by some to be an appropriate next step in therapy but is rarely needed. Postoperative intralesional steroid injections are necessary due to reported cases of worsening disease when injections are discontinued after cheiloplasty.11,12
Our patient was treated with 5 mg of intralesional triamcinolone acetonide with 10 separate injections of 0.5 cc each along the affected portions of the upper lip. She also was given doxycycline 100 mg once daily for 30 days. The patient reported complete resolution of the upper lip swelling 7 days after the initiation of therapy. At 1-month follow-up, she reported that the swelling had completely resolved. However, 1 day prior to the scheduled visit, shortly after finishing the course of doxycycline, she noted recurrent swelling. Due to the concomitant initial administration of both the steroid injections and doxycycline, it was unclear which treatment had provided relief. To avoid, or at least delay, the need for chronic intralesional steroid injections, another course of 40 mg doxycycline daily was prescribed. After 2 weeks, the patient reported that the swelling had markedly improved. The patient has maintained remission of the symptoms for approximately 6 months on daily suppressive therapy with 40 mg of doxycycline.
The recurrence of lip swelling after therapy, as in our patient, is typical of GC, and most cases require multiple follow-up visits and frequent alterations in therapy, which is often frustrating for both the patient and physician. However, awareness of this disease entity, its natural course, and the therapeutic options will allow physicians to more appropriately counsel and educate patients of this uncommon disease process.
1. Meischer G. Über essentielle granulomatöse makrocheilie (cheilitis granulomatosa). Dermatologica. 1945;91:57-85.
2. Melkersson E. Ett Fall av recidiverande facialispares i samband med angioneurotiskt ödem. Hygiea (Stockh). 1928;90:737-741.
3. Rosenthal C. Klinish-erbbiologischer beitrag zur konstitutionspathologie: gemeinsames auftreten von (rezidiverender familiärer) facialislähmung, angioneurotischem gesichtsödem und lingua plicata in arthritismus-familien. Z Ges Neurol Psychiat. 1931;131:475-501.
4. van der Waal RI, Schulten EA, van der Meij EH, et al. Cheilitis granulomatosa: overview of 13 patients with long-term follow up–results of management. Int J Dermatol. 2002;41:225-229.
5. Worsaae N, Christensen KC, Schiødt M, et al. Melkersson-Rosenthal syndrome and cheilitis granulomatosa. a clinical pathological study of thirty-three patients with special reference to their oral lesions. Oral Surg Oral Med Oral Pathol. 1982;54:404-413.
6. El-Hakim M, Chauvin P. Orofacial granulomatosis presenting as persistent lip swelling: review of 6 new cases. J Oral Maxillofac Surg. 2004;62:1114-1117.
7. Williams PM, Greenberg MS. Management of cheilitis granulomatosa. Oral Surg Oral Med Oral Pathol. 1991;72:436-439.
8. Allen CM, Camisa C, Hamzeh S, et al. Cheilitis granulomatosa: report of six cases and review of the literature. J Am Acad Dermatol. 1990;23(3, pt 1):444-450.
9. Banks T, Gada S. A comprehensive review of current treatments for granulomatous cheilitis. Br J Dermatol. 2012;166:934-937.
10. Sciubba JJ, Said-Al-Naief N. Orofacial granulomatosis: presentation, pathology and management of 13 cases. J Oral Pathol Med. 2003;32:576-585.
11. Glickman LT, Gruss JS, Birt BD, et al. The surgical management of Melkersson-Rosenthal syndrome. Plast Reconstr Surg. 1992;89:815-821.
12. Krutchkoff D, James R. Cheilitis granulomatosa. successful treatment with combined local triamcinolone injections and surgery. Arch Dermatol. 1978;114:1203-1206.
To the Editor:
A 51-year-old woman presented to her dermatologist with recurrent and progressive upper lip swelling of 2 years’ duration. Her condition was previously evaluated by several other physicians without a diagnosis or resolution of the symptoms. The swelling began on the right side of the upper lip and right cheek; however, over the course of 2 years, the swelling had progressed to involve the entire upper lip with complete sparing of the lower lip. She denied pain but reported numbness of the upper lip. The patient visited her dentist who ruled out periodontal infection as the cause of the swelling. Diphenhydramine provided no relief; however, the cheek swelling resolved after a course of antibiotics prescribed by an ear, nose, and throat physician.
She consulted her primary care physician and was subsequently referred to a neurologist and allergist who were unable to provide a definitive diagnosis or complete relief of the symptoms. She denied any history of hypersensitivity reactions, odontogenic infections, gastrointestinal concerns, or any other signs or symptoms of systemic granulomatous disease.
On physical examination, the upper lip was swollen symmetrically without evidence of ulceration, fissuring, or scaling (Figure 1). Palpation of the upper lip was notable for firm, nontender, nonpitting edema without nodularity. The oral mucosa did not appear swollen or erythematous. Examination did not reveal ulceration or a cobblestone appearance.
A full-thickness skin biopsy of the upper lip was performed. Histopathology revealed perivascular nonnecrotizing granulomas adjacent to ectatic vascular channels with associated lymphoplasmacytic infiltrate (Figure 2). Periodic acid–Schiff stain was negative for fungal hyphae, tissue Gram stain was negative for bacteria, Fite and acid-fast bacillus stains were both negative for acid-fast organisms, and polariscopy was negative for polarizable foreign material. In this clinical context, the morphologic findings were consistent with the diagnosis of granulomatous cheilitis (GC).
|
|
Figure 2. Upper lip biopsy showed dermal edema, vascular ectasia, perivascular nonnecrotizing granulomas, and perivascular lymphocyte predominant inflammatory infiltrate (A)(H&E, original magnification ×100). Higher magnification of granulomas with perivascular lymphoplasmacytic infiltrate (B)(H&E, original magnification ×200). |
Granulomatous cheilitis is a rare disorder of the lips and orofacial mucosa that was first described by Meischer1 in 1945 as persistent or recurrent orofacial swelling secondary to lymphatic obstruction by granulomatous proliferation. It often has been described as a monosymptomatic form of Melkersson-Rosenthal syndrome (MRS). In its entirety, MRS constitutes a triad of GC, facial nerve palsy, and lingua plicata (also known as fissured tongue).2,3 Although many authors agree that GC is associated with MRS, some believe that GC is a distinct entity because the majority of patients who present with GC subsequently do not develop MRS.4 Despite its relationship to MRS, the true incidence of GC largely is unknown. The onset of disease usually occurs in early adulthood but can present in middle-aged or older individuals.
The typical course of GC is relapsing and remitting, nontender and nonpitting swelling of the lips that eventually becomes permanent, leading to possible facial distortion and disability. Involvement of the upper lip is the most common, followed by (in order of decreasing frequency) the lower lip and cheeks.5 The swelling may be unilateral or bilateral and generally is not associated with ulceration, fissuring, or scaling; however, these complications have been reported in the terminal stages of the disease in which the macrocheilia has become permanent.
Despite the controversy over the etiology, pathophysiology, and classification of GC, it largely is accepted that when a patient presents clinically with a history of recurrent or persistent lip swelling, a full-thickness skin biopsy of the involved oral mucosa should be taken. Conditions that are considered in the differential diagnosis of orofacial granulomatosis are systemic granulomatous diseases that are known to have oral manifestations including Crohn disease, sarcoidosis, and mycobacterial infections. Given the many causes of orofacial and labial swelling, GC is a diagnosis of exclusion based on a thorough history and physical examination as well as appropriate diagnostic studies, with the cornerstone of the diagnosis resting on the histologic appearance of the lesion. Histologically, the diagnosis lies in the demonstration of granuloma formation, consisting of collections of epithelioid histiocytes and Langerhans giant cells. Once granuloma formation is documented, special stains are used to rule out other granulomatous diseases.
Intralesional steroids have been reported to provide the greatest improvement; however, in the majority of patients, multiple treatments are required.6,7 Allen et al8 suggested that the efficacy of intralesional therapy increases when preceded by local anesthesia of the lip, thus allowing larger doses of triamcinolone to be tolerated by the patient. Systemic corticosteroids also have been used with moderate success, but the side effects of long-term systemic corticosteroid therapy make this treatment option less appealing.9 Other agents with known anti-inflammatory properties also have been used that may offer better side-effect profiles when used for long-term suppressive therapy, including clofazimine, dapsone, sulfapyridine, danazol, hydroxychloroquine, and antibiotics such as doxycycline and metronidazole.10
In severe or recalcitrant cases, surgical intervention by way of a reduction cheiloplasty is considered by some to be an appropriate next step in therapy but is rarely needed. Postoperative intralesional steroid injections are necessary due to reported cases of worsening disease when injections are discontinued after cheiloplasty.11,12
Our patient was treated with 5 mg of intralesional triamcinolone acetonide with 10 separate injections of 0.5 cc each along the affected portions of the upper lip. She also was given doxycycline 100 mg once daily for 30 days. The patient reported complete resolution of the upper lip swelling 7 days after the initiation of therapy. At 1-month follow-up, she reported that the swelling had completely resolved. However, 1 day prior to the scheduled visit, shortly after finishing the course of doxycycline, she noted recurrent swelling. Due to the concomitant initial administration of both the steroid injections and doxycycline, it was unclear which treatment had provided relief. To avoid, or at least delay, the need for chronic intralesional steroid injections, another course of 40 mg doxycycline daily was prescribed. After 2 weeks, the patient reported that the swelling had markedly improved. The patient has maintained remission of the symptoms for approximately 6 months on daily suppressive therapy with 40 mg of doxycycline.
The recurrence of lip swelling after therapy, as in our patient, is typical of GC, and most cases require multiple follow-up visits and frequent alterations in therapy, which is often frustrating for both the patient and physician. However, awareness of this disease entity, its natural course, and the therapeutic options will allow physicians to more appropriately counsel and educate patients of this uncommon disease process.
To the Editor:
A 51-year-old woman presented to her dermatologist with recurrent and progressive upper lip swelling of 2 years’ duration. Her condition was previously evaluated by several other physicians without a diagnosis or resolution of the symptoms. The swelling began on the right side of the upper lip and right cheek; however, over the course of 2 years, the swelling had progressed to involve the entire upper lip with complete sparing of the lower lip. She denied pain but reported numbness of the upper lip. The patient visited her dentist who ruled out periodontal infection as the cause of the swelling. Diphenhydramine provided no relief; however, the cheek swelling resolved after a course of antibiotics prescribed by an ear, nose, and throat physician.
She consulted her primary care physician and was subsequently referred to a neurologist and allergist who were unable to provide a definitive diagnosis or complete relief of the symptoms. She denied any history of hypersensitivity reactions, odontogenic infections, gastrointestinal concerns, or any other signs or symptoms of systemic granulomatous disease.
On physical examination, the upper lip was swollen symmetrically without evidence of ulceration, fissuring, or scaling (Figure 1). Palpation of the upper lip was notable for firm, nontender, nonpitting edema without nodularity. The oral mucosa did not appear swollen or erythematous. Examination did not reveal ulceration or a cobblestone appearance.
A full-thickness skin biopsy of the upper lip was performed. Histopathology revealed perivascular nonnecrotizing granulomas adjacent to ectatic vascular channels with associated lymphoplasmacytic infiltrate (Figure 2). Periodic acid–Schiff stain was negative for fungal hyphae, tissue Gram stain was negative for bacteria, Fite and acid-fast bacillus stains were both negative for acid-fast organisms, and polariscopy was negative for polarizable foreign material. In this clinical context, the morphologic findings were consistent with the diagnosis of granulomatous cheilitis (GC).
|
|
Figure 2. Upper lip biopsy showed dermal edema, vascular ectasia, perivascular nonnecrotizing granulomas, and perivascular lymphocyte predominant inflammatory infiltrate (A)(H&E, original magnification ×100). Higher magnification of granulomas with perivascular lymphoplasmacytic infiltrate (B)(H&E, original magnification ×200). |
Granulomatous cheilitis is a rare disorder of the lips and orofacial mucosa that was first described by Meischer1 in 1945 as persistent or recurrent orofacial swelling secondary to lymphatic obstruction by granulomatous proliferation. It often has been described as a monosymptomatic form of Melkersson-Rosenthal syndrome (MRS). In its entirety, MRS constitutes a triad of GC, facial nerve palsy, and lingua plicata (also known as fissured tongue).2,3 Although many authors agree that GC is associated with MRS, some believe that GC is a distinct entity because the majority of patients who present with GC subsequently do not develop MRS.4 Despite its relationship to MRS, the true incidence of GC largely is unknown. The onset of disease usually occurs in early adulthood but can present in middle-aged or older individuals.
The typical course of GC is relapsing and remitting, nontender and nonpitting swelling of the lips that eventually becomes permanent, leading to possible facial distortion and disability. Involvement of the upper lip is the most common, followed by (in order of decreasing frequency) the lower lip and cheeks.5 The swelling may be unilateral or bilateral and generally is not associated with ulceration, fissuring, or scaling; however, these complications have been reported in the terminal stages of the disease in which the macrocheilia has become permanent.
Despite the controversy over the etiology, pathophysiology, and classification of GC, it largely is accepted that when a patient presents clinically with a history of recurrent or persistent lip swelling, a full-thickness skin biopsy of the involved oral mucosa should be taken. Conditions that are considered in the differential diagnosis of orofacial granulomatosis are systemic granulomatous diseases that are known to have oral manifestations including Crohn disease, sarcoidosis, and mycobacterial infections. Given the many causes of orofacial and labial swelling, GC is a diagnosis of exclusion based on a thorough history and physical examination as well as appropriate diagnostic studies, with the cornerstone of the diagnosis resting on the histologic appearance of the lesion. Histologically, the diagnosis lies in the demonstration of granuloma formation, consisting of collections of epithelioid histiocytes and Langerhans giant cells. Once granuloma formation is documented, special stains are used to rule out other granulomatous diseases.
Intralesional steroids have been reported to provide the greatest improvement; however, in the majority of patients, multiple treatments are required.6,7 Allen et al8 suggested that the efficacy of intralesional therapy increases when preceded by local anesthesia of the lip, thus allowing larger doses of triamcinolone to be tolerated by the patient. Systemic corticosteroids also have been used with moderate success, but the side effects of long-term systemic corticosteroid therapy make this treatment option less appealing.9 Other agents with known anti-inflammatory properties also have been used that may offer better side-effect profiles when used for long-term suppressive therapy, including clofazimine, dapsone, sulfapyridine, danazol, hydroxychloroquine, and antibiotics such as doxycycline and metronidazole.10
In severe or recalcitrant cases, surgical intervention by way of a reduction cheiloplasty is considered by some to be an appropriate next step in therapy but is rarely needed. Postoperative intralesional steroid injections are necessary due to reported cases of worsening disease when injections are discontinued after cheiloplasty.11,12
Our patient was treated with 5 mg of intralesional triamcinolone acetonide with 10 separate injections of 0.5 cc each along the affected portions of the upper lip. She also was given doxycycline 100 mg once daily for 30 days. The patient reported complete resolution of the upper lip swelling 7 days after the initiation of therapy. At 1-month follow-up, she reported that the swelling had completely resolved. However, 1 day prior to the scheduled visit, shortly after finishing the course of doxycycline, she noted recurrent swelling. Due to the concomitant initial administration of both the steroid injections and doxycycline, it was unclear which treatment had provided relief. To avoid, or at least delay, the need for chronic intralesional steroid injections, another course of 40 mg doxycycline daily was prescribed. After 2 weeks, the patient reported that the swelling had markedly improved. The patient has maintained remission of the symptoms for approximately 6 months on daily suppressive therapy with 40 mg of doxycycline.
The recurrence of lip swelling after therapy, as in our patient, is typical of GC, and most cases require multiple follow-up visits and frequent alterations in therapy, which is often frustrating for both the patient and physician. However, awareness of this disease entity, its natural course, and the therapeutic options will allow physicians to more appropriately counsel and educate patients of this uncommon disease process.
1. Meischer G. Über essentielle granulomatöse makrocheilie (cheilitis granulomatosa). Dermatologica. 1945;91:57-85.
2. Melkersson E. Ett Fall av recidiverande facialispares i samband med angioneurotiskt ödem. Hygiea (Stockh). 1928;90:737-741.
3. Rosenthal C. Klinish-erbbiologischer beitrag zur konstitutionspathologie: gemeinsames auftreten von (rezidiverender familiärer) facialislähmung, angioneurotischem gesichtsödem und lingua plicata in arthritismus-familien. Z Ges Neurol Psychiat. 1931;131:475-501.
4. van der Waal RI, Schulten EA, van der Meij EH, et al. Cheilitis granulomatosa: overview of 13 patients with long-term follow up–results of management. Int J Dermatol. 2002;41:225-229.
5. Worsaae N, Christensen KC, Schiødt M, et al. Melkersson-Rosenthal syndrome and cheilitis granulomatosa. a clinical pathological study of thirty-three patients with special reference to their oral lesions. Oral Surg Oral Med Oral Pathol. 1982;54:404-413.
6. El-Hakim M, Chauvin P. Orofacial granulomatosis presenting as persistent lip swelling: review of 6 new cases. J Oral Maxillofac Surg. 2004;62:1114-1117.
7. Williams PM, Greenberg MS. Management of cheilitis granulomatosa. Oral Surg Oral Med Oral Pathol. 1991;72:436-439.
8. Allen CM, Camisa C, Hamzeh S, et al. Cheilitis granulomatosa: report of six cases and review of the literature. J Am Acad Dermatol. 1990;23(3, pt 1):444-450.
9. Banks T, Gada S. A comprehensive review of current treatments for granulomatous cheilitis. Br J Dermatol. 2012;166:934-937.
10. Sciubba JJ, Said-Al-Naief N. Orofacial granulomatosis: presentation, pathology and management of 13 cases. J Oral Pathol Med. 2003;32:576-585.
11. Glickman LT, Gruss JS, Birt BD, et al. The surgical management of Melkersson-Rosenthal syndrome. Plast Reconstr Surg. 1992;89:815-821.
12. Krutchkoff D, James R. Cheilitis granulomatosa. successful treatment with combined local triamcinolone injections and surgery. Arch Dermatol. 1978;114:1203-1206.
1. Meischer G. Über essentielle granulomatöse makrocheilie (cheilitis granulomatosa). Dermatologica. 1945;91:57-85.
2. Melkersson E. Ett Fall av recidiverande facialispares i samband med angioneurotiskt ödem. Hygiea (Stockh). 1928;90:737-741.
3. Rosenthal C. Klinish-erbbiologischer beitrag zur konstitutionspathologie: gemeinsames auftreten von (rezidiverender familiärer) facialislähmung, angioneurotischem gesichtsödem und lingua plicata in arthritismus-familien. Z Ges Neurol Psychiat. 1931;131:475-501.
4. van der Waal RI, Schulten EA, van der Meij EH, et al. Cheilitis granulomatosa: overview of 13 patients with long-term follow up–results of management. Int J Dermatol. 2002;41:225-229.
5. Worsaae N, Christensen KC, Schiødt M, et al. Melkersson-Rosenthal syndrome and cheilitis granulomatosa. a clinical pathological study of thirty-three patients with special reference to their oral lesions. Oral Surg Oral Med Oral Pathol. 1982;54:404-413.
6. El-Hakim M, Chauvin P. Orofacial granulomatosis presenting as persistent lip swelling: review of 6 new cases. J Oral Maxillofac Surg. 2004;62:1114-1117.
7. Williams PM, Greenberg MS. Management of cheilitis granulomatosa. Oral Surg Oral Med Oral Pathol. 1991;72:436-439.
8. Allen CM, Camisa C, Hamzeh S, et al. Cheilitis granulomatosa: report of six cases and review of the literature. J Am Acad Dermatol. 1990;23(3, pt 1):444-450.
9. Banks T, Gada S. A comprehensive review of current treatments for granulomatous cheilitis. Br J Dermatol. 2012;166:934-937.
10. Sciubba JJ, Said-Al-Naief N. Orofacial granulomatosis: presentation, pathology and management of 13 cases. J Oral Pathol Med. 2003;32:576-585.
11. Glickman LT, Gruss JS, Birt BD, et al. The surgical management of Melkersson-Rosenthal syndrome. Plast Reconstr Surg. 1992;89:815-821.
12. Krutchkoff D, James R. Cheilitis granulomatosa. successful treatment with combined local triamcinolone injections and surgery. Arch Dermatol. 1978;114:1203-1206.
What Is Your Diagnosis? Verrucous Carcinoma
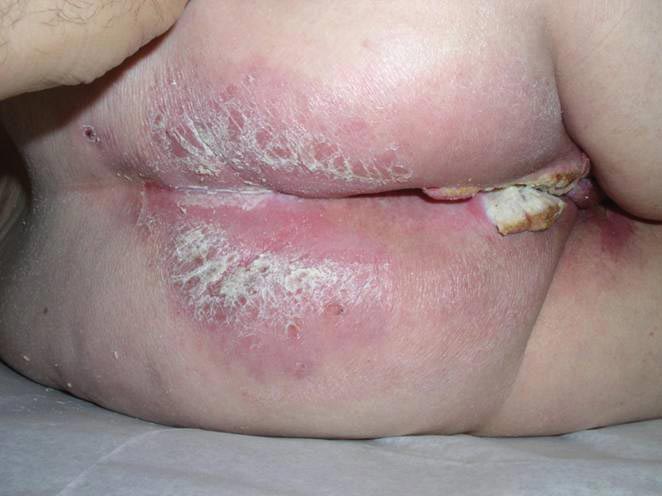
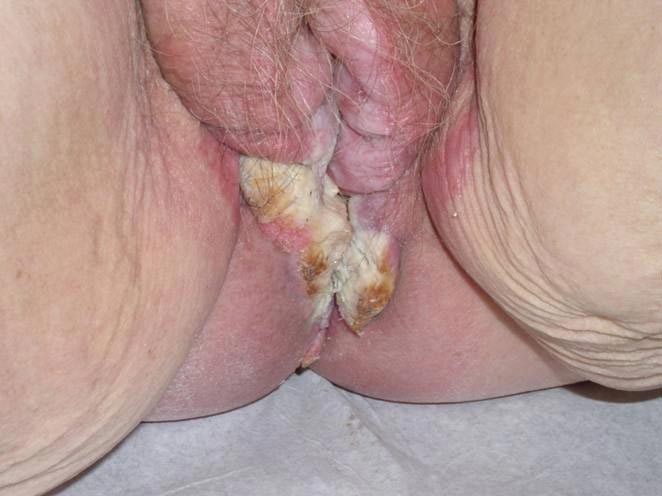
An 81-year-old woman presented for evaluation of a nodule on the right labia majora that had been present for 1 year. She had a history of intertriginous psoriasis, and several biopsies were performed at an outside facility over the last 5 years that revealed psoriasis but were otherwise noncontributory. Physical examination revealed erythema and scaling on the buttocks with maceration in the intertriginous area (top) and the perineum associated with a verrucous nodule (bottom).
The Diagnosis: Verrucous Carcinoma
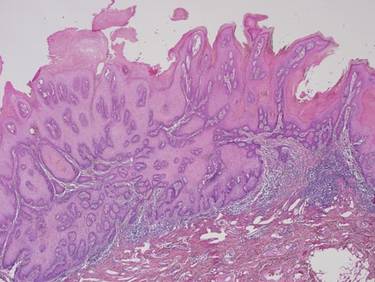
Biopsies of early lesions often may be difficult to interpret without clinicopathological correlation. Our patient’s tumor was associated with intertriginous psoriasis, which was the only abnormality previously noted on superficial biopsies performed at an outside facility. The patient was scheduled for an excisional biopsy due to the large tumor size and clinical suspicion that the prior biopsies were inadequate and failed to demonstrate the primary underlying pathology. Excisional biopsy of the verrucous tumor revealed epithelium composed of keratinocytes with glassy cytoplasm. Papillomatosis was noted along with an endophytic component of well-differentiated epithelial cells extending into the dermis in a bulbous pattern consistent with the verrucous carcinoma variant of squamous cell carcinoma (SCC)(Figure). Verrucous carcinoma often requires correlation with both the clinical and histopathologic findings for definitive diagnosis, as keratinocytes often appear to be well differentiated.1
Verrucous carcinoma may begin as an innocuous papule that slowly grows into a large fungating tumor. Verrucous carcinomas typically are slow growing, exophytic, and low grade. The etiology of verrucous carcinoma is not clear, and the role of human papillomavirus (HPV) infection is controversial.2 Best classified as a well-differentiated SCC, verrucous carcinoma rarely metastasizes but may invade adjacent tissues.
Differential diagnoses include a giant inflamed seborrheic keratosis, condyloma acuminatum, rupioid psoriasis, and inflammatory linear verrucous epidermal nevus (ILVEN). Although large and inflamed seborrheic keratoses may have squamous eddies that mimic SCC, seborrheic keratoses do not invade the dermis and typically have a well-circumscribed stuck-on appearance. Abnormal mitotic figures are not identified. Condylomas are genital warts caused by HPV infection that often are clustered, well circumscribed, and exophytic. Large lesions can be difficult to distinguish from verrucous carcinomas, and biopsy generally reveals koilocytes identified by perinuclear clearing and raisinlike nuclei. Immunohistochemical staining and in situ hybridization studies can be of value in diagnosis and in identifying those lesions that are at high risk for malignant transformation. High-risk condylomas are associated with HPV-16, HPV-18, HPV-31, HPV-33, HPV-35, and HPV-39, as well as other types, whereas low-risk condylomas are associated with HPV-6, HPV-11, HPV-42, and others.2 Differentiating squamous cell hyperplasia from squamous cell carcinoma in situ also can be aided by immunohistochemistry. Squamous cell hyperplasia is usually negative for INK4 p16Ink4A and p53 and exhibits variable Ki-67 staining. Differentiated squamous cell carcinoma in situ exhibits a profile that is p16Ink4A negative, Ki-67 positive, and exhibits variable p53 staining.3 Basaloid and warty intraepithelial neoplasia is consistently p16Ink4A positive, Ki-67 positive, and variably positive for p53.3 Therefore, p16 staining of high-grade areas is a useful biomarker that can help establish diagnosis of associated squamous cell carcinoma.4 The role of papillomaviruses in the development of nonmelanoma skin cancer is an area of active study, and research suggests that papillomaviruses may have a much greater role than previously suspected.5
At times, psoriasis may be markedly hyperkeratotic, clinically mimicking a verrucous neoplasm. This hyperkeratotic type of psoriasis is known as rupioid psoriasis. However, these psoriatic lesions are exophytic, are associated with spongiform pustules, and lack the atypia and endophytic pattern typically seen with verrucous carcinoma. An ILVEN also lacks atypia and an endophytic pattern and usually presents in childhood as a persistent linear plaque, rather than the verrucous plaque noted in our patient. Squamous cell carcinoma has been reported to arise in the setting of verrucoid ILVEN but is exceptionally uncommon.6
Successful treatment of verrucous carcinoma is best achieved by complete excision. Oral retinoids and immunomodulators such as imiquimod also may be of value.7 Our patient’s tumor qualifies as T2N0M0 because it was greater than 2 cm in size.8 A Breslow thickness of 2 mm or greater and Clark level IV are high-risk features associated with a worse prognosis, but clinical evaluation of our patient’s lymph nodes was unremarkable and no distant metastases were identified. Our patient continues to do well with no evidence of recurrence.
1. Bambao C, Nofech-Mozes S, Shier M. Giant condyloma versus verrucous carcinoma: a case report. J Low Genit Tract Dis. 2010;14:230-233.
2. Asiaf A, Ahmad ST, Mohannad SO, et al. Review of the current knowledge on the epidemiology, pathogenesis, and prevention of human papillomavirus infection. Eur J Cancer Prev. 2014;23:206-224.
3. Chaux A, Pfannl R, Rodríguez IM, et al. Distinctive immunohistochemical profile of penile intraepithelial lesions: a study of 74 cases. Am J Surg Pathol. 2011;35:553-562.
4. Darragh TM, Colgan TJ, Cox JT, et al. The lower anogenital squamous terminology standardization project for HPV-associated lesions: background and consensus recommendations from the College of American Pathologists and the American Society for Colposcopy and Cervical Pathology. Arch Pathol Lab Med. 2012;136:1266-1297.
5. Aldabagh B, Angeles J, Cardones AR, et al. Cutaneous squamous cell carcinoma and human papillomavirus: is there an association? Dermatol Surg. 2013;39:1-23.
6. Turk BG, Ertam I, Urkmez A, et al. Development of squamous cell carcinoma on an inflammatory linear verrucous epidermal nevus in the genital area. Cutis. 2012;89:273-275.
7. Erkek E, Basar H, Bozdogan O, et al. Giant condyloma acuminata of Buschke-Löwenstein: successful treatment with a combination of surgical excision, oral acitretin and topical imiquimod. Clin Exp Dermatol. 2009;34:366-368.
8. Cutaneous squamous cell carcinoma and other cutaneous carcinomas. In: Edge SB, Byrd DR, Compton CC, et al, eds. AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer; 2010:301-314.
An 81-year-old woman presented for evaluation of a nodule on the right labia majora that had been present for 1 year. She had a history of intertriginous psoriasis, and several biopsies were performed at an outside facility over the last 5 years that revealed psoriasis but were otherwise noncontributory. Physical examination revealed erythema and scaling on the buttocks with maceration in the intertriginous area (top) and the perineum associated with a verrucous nodule (bottom).
The Diagnosis: Verrucous Carcinoma
Biopsies of early lesions often may be difficult to interpret without clinicopathological correlation. Our patient’s tumor was associated with intertriginous psoriasis, which was the only abnormality previously noted on superficial biopsies performed at an outside facility. The patient was scheduled for an excisional biopsy due to the large tumor size and clinical suspicion that the prior biopsies were inadequate and failed to demonstrate the primary underlying pathology. Excisional biopsy of the verrucous tumor revealed epithelium composed of keratinocytes with glassy cytoplasm. Papillomatosis was noted along with an endophytic component of well-differentiated epithelial cells extending into the dermis in a bulbous pattern consistent with the verrucous carcinoma variant of squamous cell carcinoma (SCC)(Figure). Verrucous carcinoma often requires correlation with both the clinical and histopathologic findings for definitive diagnosis, as keratinocytes often appear to be well differentiated.1
Verrucous carcinoma may begin as an innocuous papule that slowly grows into a large fungating tumor. Verrucous carcinomas typically are slow growing, exophytic, and low grade. The etiology of verrucous carcinoma is not clear, and the role of human papillomavirus (HPV) infection is controversial.2 Best classified as a well-differentiated SCC, verrucous carcinoma rarely metastasizes but may invade adjacent tissues.
Differential diagnoses include a giant inflamed seborrheic keratosis, condyloma acuminatum, rupioid psoriasis, and inflammatory linear verrucous epidermal nevus (ILVEN). Although large and inflamed seborrheic keratoses may have squamous eddies that mimic SCC, seborrheic keratoses do not invade the dermis and typically have a well-circumscribed stuck-on appearance. Abnormal mitotic figures are not identified. Condylomas are genital warts caused by HPV infection that often are clustered, well circumscribed, and exophytic. Large lesions can be difficult to distinguish from verrucous carcinomas, and biopsy generally reveals koilocytes identified by perinuclear clearing and raisinlike nuclei. Immunohistochemical staining and in situ hybridization studies can be of value in diagnosis and in identifying those lesions that are at high risk for malignant transformation. High-risk condylomas are associated with HPV-16, HPV-18, HPV-31, HPV-33, HPV-35, and HPV-39, as well as other types, whereas low-risk condylomas are associated with HPV-6, HPV-11, HPV-42, and others.2 Differentiating squamous cell hyperplasia from squamous cell carcinoma in situ also can be aided by immunohistochemistry. Squamous cell hyperplasia is usually negative for INK4 p16Ink4A and p53 and exhibits variable Ki-67 staining. Differentiated squamous cell carcinoma in situ exhibits a profile that is p16Ink4A negative, Ki-67 positive, and exhibits variable p53 staining.3 Basaloid and warty intraepithelial neoplasia is consistently p16Ink4A positive, Ki-67 positive, and variably positive for p53.3 Therefore, p16 staining of high-grade areas is a useful biomarker that can help establish diagnosis of associated squamous cell carcinoma.4 The role of papillomaviruses in the development of nonmelanoma skin cancer is an area of active study, and research suggests that papillomaviruses may have a much greater role than previously suspected.5
At times, psoriasis may be markedly hyperkeratotic, clinically mimicking a verrucous neoplasm. This hyperkeratotic type of psoriasis is known as rupioid psoriasis. However, these psoriatic lesions are exophytic, are associated with spongiform pustules, and lack the atypia and endophytic pattern typically seen with verrucous carcinoma. An ILVEN also lacks atypia and an endophytic pattern and usually presents in childhood as a persistent linear plaque, rather than the verrucous plaque noted in our patient. Squamous cell carcinoma has been reported to arise in the setting of verrucoid ILVEN but is exceptionally uncommon.6
Successful treatment of verrucous carcinoma is best achieved by complete excision. Oral retinoids and immunomodulators such as imiquimod also may be of value.7 Our patient’s tumor qualifies as T2N0M0 because it was greater than 2 cm in size.8 A Breslow thickness of 2 mm or greater and Clark level IV are high-risk features associated with a worse prognosis, but clinical evaluation of our patient’s lymph nodes was unremarkable and no distant metastases were identified. Our patient continues to do well with no evidence of recurrence.
An 81-year-old woman presented for evaluation of a nodule on the right labia majora that had been present for 1 year. She had a history of intertriginous psoriasis, and several biopsies were performed at an outside facility over the last 5 years that revealed psoriasis but were otherwise noncontributory. Physical examination revealed erythema and scaling on the buttocks with maceration in the intertriginous area (top) and the perineum associated with a verrucous nodule (bottom).
The Diagnosis: Verrucous Carcinoma
Biopsies of early lesions often may be difficult to interpret without clinicopathological correlation. Our patient’s tumor was associated with intertriginous psoriasis, which was the only abnormality previously noted on superficial biopsies performed at an outside facility. The patient was scheduled for an excisional biopsy due to the large tumor size and clinical suspicion that the prior biopsies were inadequate and failed to demonstrate the primary underlying pathology. Excisional biopsy of the verrucous tumor revealed epithelium composed of keratinocytes with glassy cytoplasm. Papillomatosis was noted along with an endophytic component of well-differentiated epithelial cells extending into the dermis in a bulbous pattern consistent with the verrucous carcinoma variant of squamous cell carcinoma (SCC)(Figure). Verrucous carcinoma often requires correlation with both the clinical and histopathologic findings for definitive diagnosis, as keratinocytes often appear to be well differentiated.1
Verrucous carcinoma may begin as an innocuous papule that slowly grows into a large fungating tumor. Verrucous carcinomas typically are slow growing, exophytic, and low grade. The etiology of verrucous carcinoma is not clear, and the role of human papillomavirus (HPV) infection is controversial.2 Best classified as a well-differentiated SCC, verrucous carcinoma rarely metastasizes but may invade adjacent tissues.
Differential diagnoses include a giant inflamed seborrheic keratosis, condyloma acuminatum, rupioid psoriasis, and inflammatory linear verrucous epidermal nevus (ILVEN). Although large and inflamed seborrheic keratoses may have squamous eddies that mimic SCC, seborrheic keratoses do not invade the dermis and typically have a well-circumscribed stuck-on appearance. Abnormal mitotic figures are not identified. Condylomas are genital warts caused by HPV infection that often are clustered, well circumscribed, and exophytic. Large lesions can be difficult to distinguish from verrucous carcinomas, and biopsy generally reveals koilocytes identified by perinuclear clearing and raisinlike nuclei. Immunohistochemical staining and in situ hybridization studies can be of value in diagnosis and in identifying those lesions that are at high risk for malignant transformation. High-risk condylomas are associated with HPV-16, HPV-18, HPV-31, HPV-33, HPV-35, and HPV-39, as well as other types, whereas low-risk condylomas are associated with HPV-6, HPV-11, HPV-42, and others.2 Differentiating squamous cell hyperplasia from squamous cell carcinoma in situ also can be aided by immunohistochemistry. Squamous cell hyperplasia is usually negative for INK4 p16Ink4A and p53 and exhibits variable Ki-67 staining. Differentiated squamous cell carcinoma in situ exhibits a profile that is p16Ink4A negative, Ki-67 positive, and exhibits variable p53 staining.3 Basaloid and warty intraepithelial neoplasia is consistently p16Ink4A positive, Ki-67 positive, and variably positive for p53.3 Therefore, p16 staining of high-grade areas is a useful biomarker that can help establish diagnosis of associated squamous cell carcinoma.4 The role of papillomaviruses in the development of nonmelanoma skin cancer is an area of active study, and research suggests that papillomaviruses may have a much greater role than previously suspected.5
At times, psoriasis may be markedly hyperkeratotic, clinically mimicking a verrucous neoplasm. This hyperkeratotic type of psoriasis is known as rupioid psoriasis. However, these psoriatic lesions are exophytic, are associated with spongiform pustules, and lack the atypia and endophytic pattern typically seen with verrucous carcinoma. An ILVEN also lacks atypia and an endophytic pattern and usually presents in childhood as a persistent linear plaque, rather than the verrucous plaque noted in our patient. Squamous cell carcinoma has been reported to arise in the setting of verrucoid ILVEN but is exceptionally uncommon.6
Successful treatment of verrucous carcinoma is best achieved by complete excision. Oral retinoids and immunomodulators such as imiquimod also may be of value.7 Our patient’s tumor qualifies as T2N0M0 because it was greater than 2 cm in size.8 A Breslow thickness of 2 mm or greater and Clark level IV are high-risk features associated with a worse prognosis, but clinical evaluation of our patient’s lymph nodes was unremarkable and no distant metastases were identified. Our patient continues to do well with no evidence of recurrence.
1. Bambao C, Nofech-Mozes S, Shier M. Giant condyloma versus verrucous carcinoma: a case report. J Low Genit Tract Dis. 2010;14:230-233.
2. Asiaf A, Ahmad ST, Mohannad SO, et al. Review of the current knowledge on the epidemiology, pathogenesis, and prevention of human papillomavirus infection. Eur J Cancer Prev. 2014;23:206-224.
3. Chaux A, Pfannl R, Rodríguez IM, et al. Distinctive immunohistochemical profile of penile intraepithelial lesions: a study of 74 cases. Am J Surg Pathol. 2011;35:553-562.
4. Darragh TM, Colgan TJ, Cox JT, et al. The lower anogenital squamous terminology standardization project for HPV-associated lesions: background and consensus recommendations from the College of American Pathologists and the American Society for Colposcopy and Cervical Pathology. Arch Pathol Lab Med. 2012;136:1266-1297.
5. Aldabagh B, Angeles J, Cardones AR, et al. Cutaneous squamous cell carcinoma and human papillomavirus: is there an association? Dermatol Surg. 2013;39:1-23.
6. Turk BG, Ertam I, Urkmez A, et al. Development of squamous cell carcinoma on an inflammatory linear verrucous epidermal nevus in the genital area. Cutis. 2012;89:273-275.
7. Erkek E, Basar H, Bozdogan O, et al. Giant condyloma acuminata of Buschke-Löwenstein: successful treatment with a combination of surgical excision, oral acitretin and topical imiquimod. Clin Exp Dermatol. 2009;34:366-368.
8. Cutaneous squamous cell carcinoma and other cutaneous carcinomas. In: Edge SB, Byrd DR, Compton CC, et al, eds. AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer; 2010:301-314.
1. Bambao C, Nofech-Mozes S, Shier M. Giant condyloma versus verrucous carcinoma: a case report. J Low Genit Tract Dis. 2010;14:230-233.
2. Asiaf A, Ahmad ST, Mohannad SO, et al. Review of the current knowledge on the epidemiology, pathogenesis, and prevention of human papillomavirus infection. Eur J Cancer Prev. 2014;23:206-224.
3. Chaux A, Pfannl R, Rodríguez IM, et al. Distinctive immunohistochemical profile of penile intraepithelial lesions: a study of 74 cases. Am J Surg Pathol. 2011;35:553-562.
4. Darragh TM, Colgan TJ, Cox JT, et al. The lower anogenital squamous terminology standardization project for HPV-associated lesions: background and consensus recommendations from the College of American Pathologists and the American Society for Colposcopy and Cervical Pathology. Arch Pathol Lab Med. 2012;136:1266-1297.
5. Aldabagh B, Angeles J, Cardones AR, et al. Cutaneous squamous cell carcinoma and human papillomavirus: is there an association? Dermatol Surg. 2013;39:1-23.
6. Turk BG, Ertam I, Urkmez A, et al. Development of squamous cell carcinoma on an inflammatory linear verrucous epidermal nevus in the genital area. Cutis. 2012;89:273-275.
7. Erkek E, Basar H, Bozdogan O, et al. Giant condyloma acuminata of Buschke-Löwenstein: successful treatment with a combination of surgical excision, oral acitretin and topical imiquimod. Clin Exp Dermatol. 2009;34:366-368.
8. Cutaneous squamous cell carcinoma and other cutaneous carcinomas. In: Edge SB, Byrd DR, Compton CC, et al, eds. AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer; 2010:301-314.
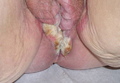
Trichilemmoma
Trichilemmomas are benign follicular neoplasms that exhibit differentiation toward the outer root sheath of the pilosebaceous follicular epithelium.1 Trichilemmomas clinically present as individual or multiple, slowly growing, verrucous papules appearing most commonly on the face or neck. The lesions may coalesce to form small plaques. Although trichilemmomas typically are isolated, patients with multiple trichilemmomas require a cancer screening workup due to their association with Cowden disease, which results from a mutation in the phosphatase and tensin homolog tumor suppressor gene, PTEN.2 An easy way to remember the association between trichilemmomas and Cowden disease is to alter the spelling to “trichile-moo-moo,” using the “moo moo” sound of an animal cow as a clue linking the tumor to Cowden disease.
Histologically, trichilemmomas exhibit a lobular epidermal downgrowth into the dermis (Figure 1). The surface of the lesion may be hyperkeratotic and somewhat papillomatous. Cells toward the center of the lobule are pale staining, periodic acid–Schiff positive, and diastase labile due to high levels of intracellular glycogen (Figure 2). Cells toward the periphery of the lobule usually appear basophilic with a palisading arrangement of the peripheral cells. The entire lobule is enclosed within an eosinophilic basement membrane that stains positively with periodic acid–Schiff (Figure 2).1 Consistent with the tumor’s differentiation toward the outer root sheath of the hair follicle, trichilemmomas have been reported to express CD34 focally or diffusely.3
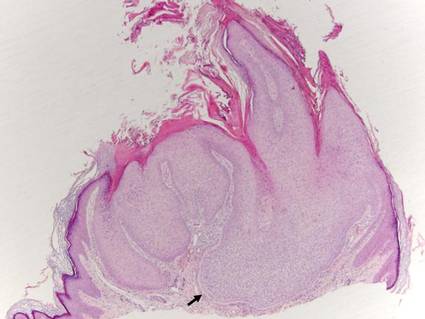
| 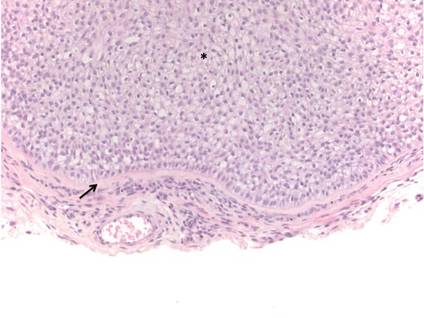
|
Similar to trichilemmoma, inverted follicular keratosis (IFK) commonly presents as a solitary asymptomatic papule on the face. Inverted follicular keratosis is a somewhat controversial entity, with some authorities arguing IFK is a variant of verruca vulgaris or seborrheic keratosis. Histologically, IFKs can be differentiated by the presence of squamous eddies (concentric layers of squamous cells in a whorled pattern), which are diagnostic, and central longitudinal crypts that contain keratin and are lined by squamous epithelium.4 Basaloid cells can be seen at the periphery of the tumors; however, IFKs lack an eosinophilic basement membrane surrounding the tumor (Figure 3).

Squamous cell carcinoma in situ classically appears as an erythematous hyperkeratotic papule or plaque on sun-exposed sites that can become crusted or ulcerated. Microscopically, squamous cell carcinoma in situ displays full-thickness disorderly maturation of keratinocytes. The keratinocytes exhibit nuclear pleomorphism. Atypical mitotic figures and dyskeratotic keratinocytes also can be seen throughout the full thickness of the epidermis (Figure 4).5
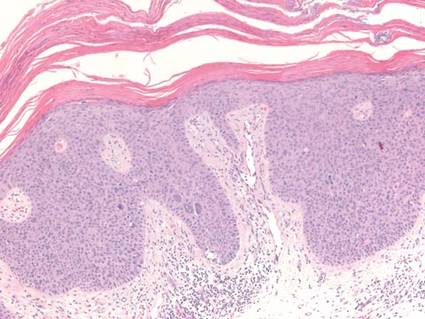
Verruca vulgaris (Figure 5) histologically demonstrates hyperkeratosis with tiers of parakeratosis, digitated epidermal hyperplasia, and dilated tortuous capillaries within the dermal papillae. At the edges of the lesion there often is inward turning of elongated rete ridges,6,7 which can be thought of as the rete reaching out for a hug of sorts to spread the human papillomavirus infection. Although the surface of a trichilemmoma can bear resemblance to a verruca vulgaris, the remainder of the histologic features can be used to help differentiate these tumors. Additionally, there has been no evidence suggestive of a viral etiology for trichilemmomas.8

Warty dyskeratoma features an umbilicated papule, usually on the face, head, or neck, that is associated with a follicular unit. The papule shows a cup-shaped, keratin-filled invagination; suprabasilar clefting; and acantholytic dyskeratotic cells, which are features that are not seen in trichilemmomas (Figure 6).9

Acknowledgment—The authors would like to thank Brandon Litzner, MD, St Louis, Missouri, for proofreading the manuscript.
1. Brownstein MH, Shapiro L. Trichilemmoma: analysis of 40 new cases. Arch Dermatol. 1973;107:866-869.
2. Al-Zaid T, Ditelberg J, Prieto V, et al. Trichilemmomas show loss of PTEN in Cowden syndrome but only rarely in sporadic tumors. J Cutan Pathol. 2012;39:493-499.
3. Tardío JC. CD34-reactive tumors of the skin. an updated review of an ever-growing list of lesions. J Cutan Pathol. 2009;36:89-102.
4. Mehregan A. Inverted follicular keratosis is a distinct follicular tumor. Am J Dermatopathol. 1983;5:467-470.
5. Cockerell CJ. Histopathology of incipient intraepidermal squamous cell carcinoma (“actinic keratosis”). J Am Acad Dermatol. 2000;42(1, pt 2):11-17.
6. Jabłonska S, Majewski S, Obalek S, et al. Cutaneous warts. Clin Dermatol. 1997;15:309-319.
7. Hardin J, Gardner J, Colome M, et al. Verrucous cyst with melanocytic and sebaceous differentiation. Arch Path Lab Med. 2013;137:576-579.
8. Johnson BL, Kramer EM, Lavker RM. The keratotic tumors of Cowden’s disease: an electron microscopy study. J Cutan Pathol. 1987;14:291-298.
9. Kaddu S, Dong H, Mayer G, et al. Warty dyskeratoma—“follicular dyskeratoma”: analysis of clinicopathologic features of a distinctive follicular adnexal neoplasm. J Am Acad Dermatol. 2002;47:423-428.
Trichilemmomas are benign follicular neoplasms that exhibit differentiation toward the outer root sheath of the pilosebaceous follicular epithelium.1 Trichilemmomas clinically present as individual or multiple, slowly growing, verrucous papules appearing most commonly on the face or neck. The lesions may coalesce to form small plaques. Although trichilemmomas typically are isolated, patients with multiple trichilemmomas require a cancer screening workup due to their association with Cowden disease, which results from a mutation in the phosphatase and tensin homolog tumor suppressor gene, PTEN.2 An easy way to remember the association between trichilemmomas and Cowden disease is to alter the spelling to “trichile-moo-moo,” using the “moo moo” sound of an animal cow as a clue linking the tumor to Cowden disease.
Histologically, trichilemmomas exhibit a lobular epidermal downgrowth into the dermis (Figure 1). The surface of the lesion may be hyperkeratotic and somewhat papillomatous. Cells toward the center of the lobule are pale staining, periodic acid–Schiff positive, and diastase labile due to high levels of intracellular glycogen (Figure 2). Cells toward the periphery of the lobule usually appear basophilic with a palisading arrangement of the peripheral cells. The entire lobule is enclosed within an eosinophilic basement membrane that stains positively with periodic acid–Schiff (Figure 2).1 Consistent with the tumor’s differentiation toward the outer root sheath of the hair follicle, trichilemmomas have been reported to express CD34 focally or diffusely.3
|
|
|
Similar to trichilemmoma, inverted follicular keratosis (IFK) commonly presents as a solitary asymptomatic papule on the face. Inverted follicular keratosis is a somewhat controversial entity, with some authorities arguing IFK is a variant of verruca vulgaris or seborrheic keratosis. Histologically, IFKs can be differentiated by the presence of squamous eddies (concentric layers of squamous cells in a whorled pattern), which are diagnostic, and central longitudinal crypts that contain keratin and are lined by squamous epithelium.4 Basaloid cells can be seen at the periphery of the tumors; however, IFKs lack an eosinophilic basement membrane surrounding the tumor (Figure 3).
Squamous cell carcinoma in situ classically appears as an erythematous hyperkeratotic papule or plaque on sun-exposed sites that can become crusted or ulcerated. Microscopically, squamous cell carcinoma in situ displays full-thickness disorderly maturation of keratinocytes. The keratinocytes exhibit nuclear pleomorphism. Atypical mitotic figures and dyskeratotic keratinocytes also can be seen throughout the full thickness of the epidermis (Figure 4).5
Verruca vulgaris (Figure 5) histologically demonstrates hyperkeratosis with tiers of parakeratosis, digitated epidermal hyperplasia, and dilated tortuous capillaries within the dermal papillae. At the edges of the lesion there often is inward turning of elongated rete ridges,6,7 which can be thought of as the rete reaching out for a hug of sorts to spread the human papillomavirus infection. Although the surface of a trichilemmoma can bear resemblance to a verruca vulgaris, the remainder of the histologic features can be used to help differentiate these tumors. Additionally, there has been no evidence suggestive of a viral etiology for trichilemmomas.8
Warty dyskeratoma features an umbilicated papule, usually on the face, head, or neck, that is associated with a follicular unit. The papule shows a cup-shaped, keratin-filled invagination; suprabasilar clefting; and acantholytic dyskeratotic cells, which are features that are not seen in trichilemmomas (Figure 6).9
Acknowledgment—The authors would like to thank Brandon Litzner, MD, St Louis, Missouri, for proofreading the manuscript.
Trichilemmomas are benign follicular neoplasms that exhibit differentiation toward the outer root sheath of the pilosebaceous follicular epithelium.1 Trichilemmomas clinically present as individual or multiple, slowly growing, verrucous papules appearing most commonly on the face or neck. The lesions may coalesce to form small plaques. Although trichilemmomas typically are isolated, patients with multiple trichilemmomas require a cancer screening workup due to their association with Cowden disease, which results from a mutation in the phosphatase and tensin homolog tumor suppressor gene, PTEN.2 An easy way to remember the association between trichilemmomas and Cowden disease is to alter the spelling to “trichile-moo-moo,” using the “moo moo” sound of an animal cow as a clue linking the tumor to Cowden disease.
Histologically, trichilemmomas exhibit a lobular epidermal downgrowth into the dermis (Figure 1). The surface of the lesion may be hyperkeratotic and somewhat papillomatous. Cells toward the center of the lobule are pale staining, periodic acid–Schiff positive, and diastase labile due to high levels of intracellular glycogen (Figure 2). Cells toward the periphery of the lobule usually appear basophilic with a palisading arrangement of the peripheral cells. The entire lobule is enclosed within an eosinophilic basement membrane that stains positively with periodic acid–Schiff (Figure 2).1 Consistent with the tumor’s differentiation toward the outer root sheath of the hair follicle, trichilemmomas have been reported to express CD34 focally or diffusely.3
|
|
|
Similar to trichilemmoma, inverted follicular keratosis (IFK) commonly presents as a solitary asymptomatic papule on the face. Inverted follicular keratosis is a somewhat controversial entity, with some authorities arguing IFK is a variant of verruca vulgaris or seborrheic keratosis. Histologically, IFKs can be differentiated by the presence of squamous eddies (concentric layers of squamous cells in a whorled pattern), which are diagnostic, and central longitudinal crypts that contain keratin and are lined by squamous epithelium.4 Basaloid cells can be seen at the periphery of the tumors; however, IFKs lack an eosinophilic basement membrane surrounding the tumor (Figure 3).
Squamous cell carcinoma in situ classically appears as an erythematous hyperkeratotic papule or plaque on sun-exposed sites that can become crusted or ulcerated. Microscopically, squamous cell carcinoma in situ displays full-thickness disorderly maturation of keratinocytes. The keratinocytes exhibit nuclear pleomorphism. Atypical mitotic figures and dyskeratotic keratinocytes also can be seen throughout the full thickness of the epidermis (Figure 4).5
Verruca vulgaris (Figure 5) histologically demonstrates hyperkeratosis with tiers of parakeratosis, digitated epidermal hyperplasia, and dilated tortuous capillaries within the dermal papillae. At the edges of the lesion there often is inward turning of elongated rete ridges,6,7 which can be thought of as the rete reaching out for a hug of sorts to spread the human papillomavirus infection. Although the surface of a trichilemmoma can bear resemblance to a verruca vulgaris, the remainder of the histologic features can be used to help differentiate these tumors. Additionally, there has been no evidence suggestive of a viral etiology for trichilemmomas.8
Warty dyskeratoma features an umbilicated papule, usually on the face, head, or neck, that is associated with a follicular unit. The papule shows a cup-shaped, keratin-filled invagination; suprabasilar clefting; and acantholytic dyskeratotic cells, which are features that are not seen in trichilemmomas (Figure 6).9
Acknowledgment—The authors would like to thank Brandon Litzner, MD, St Louis, Missouri, for proofreading the manuscript.
1. Brownstein MH, Shapiro L. Trichilemmoma: analysis of 40 new cases. Arch Dermatol. 1973;107:866-869.
2. Al-Zaid T, Ditelberg J, Prieto V, et al. Trichilemmomas show loss of PTEN in Cowden syndrome but only rarely in sporadic tumors. J Cutan Pathol. 2012;39:493-499.
3. Tardío JC. CD34-reactive tumors of the skin. an updated review of an ever-growing list of lesions. J Cutan Pathol. 2009;36:89-102.
4. Mehregan A. Inverted follicular keratosis is a distinct follicular tumor. Am J Dermatopathol. 1983;5:467-470.
5. Cockerell CJ. Histopathology of incipient intraepidermal squamous cell carcinoma (“actinic keratosis”). J Am Acad Dermatol. 2000;42(1, pt 2):11-17.
6. Jabłonska S, Majewski S, Obalek S, et al. Cutaneous warts. Clin Dermatol. 1997;15:309-319.
7. Hardin J, Gardner J, Colome M, et al. Verrucous cyst with melanocytic and sebaceous differentiation. Arch Path Lab Med. 2013;137:576-579.
8. Johnson BL, Kramer EM, Lavker RM. The keratotic tumors of Cowden’s disease: an electron microscopy study. J Cutan Pathol. 1987;14:291-298.
9. Kaddu S, Dong H, Mayer G, et al. Warty dyskeratoma—“follicular dyskeratoma”: analysis of clinicopathologic features of a distinctive follicular adnexal neoplasm. J Am Acad Dermatol. 2002;47:423-428.
1. Brownstein MH, Shapiro L. Trichilemmoma: analysis of 40 new cases. Arch Dermatol. 1973;107:866-869.
2. Al-Zaid T, Ditelberg J, Prieto V, et al. Trichilemmomas show loss of PTEN in Cowden syndrome but only rarely in sporadic tumors. J Cutan Pathol. 2012;39:493-499.
3. Tardío JC. CD34-reactive tumors of the skin. an updated review of an ever-growing list of lesions. J Cutan Pathol. 2009;36:89-102.
4. Mehregan A. Inverted follicular keratosis is a distinct follicular tumor. Am J Dermatopathol. 1983;5:467-470.
5. Cockerell CJ. Histopathology of incipient intraepidermal squamous cell carcinoma (“actinic keratosis”). J Am Acad Dermatol. 2000;42(1, pt 2):11-17.
6. Jabłonska S, Majewski S, Obalek S, et al. Cutaneous warts. Clin Dermatol. 1997;15:309-319.
7. Hardin J, Gardner J, Colome M, et al. Verrucous cyst with melanocytic and sebaceous differentiation. Arch Path Lab Med. 2013;137:576-579.
8. Johnson BL, Kramer EM, Lavker RM. The keratotic tumors of Cowden’s disease: an electron microscopy study. J Cutan Pathol. 1987;14:291-298.
9. Kaddu S, Dong H, Mayer G, et al. Warty dyskeratoma—“follicular dyskeratoma”: analysis of clinicopathologic features of a distinctive follicular adnexal neoplasm. J Am Acad Dermatol. 2002;47:423-428.
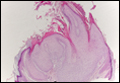
Perianal North American Blastomycosis
Cutaneous North American blastomycosis is a deep fungal infection caused by Blastomyces dermatitidis, a thermally dimorphic fungus that is endemic to the Great Lakes region as well as the Mississippi and Ohio River valleys where it thrives in moist acidic soil enriched with organic material.1,2 In humans, the annual incidence rate is estimated to be 0.6 cases per million,3 though it may be as high as 42 cases per 100,000 in endemic areas.4 Infection typically results from the inhalation of conidia and manifests as either acute or chronic pneumonia.5 Most patients with acute disease present with nonspecific flulike symptoms and a nonproductive cough.
Dissemination occurs in approximately 25% of cases,6 most commonly affecting the skin. Other potential sites of dissemination include bone, the genitourinary tract, and the central nervous system. Cutaneous lesions, which may be either verrucous or ulcerative plaques, often occur on or around orifices contiguous to the respiratory tract.7 Verrucous lesions tend to have an irregular shape with well-defined borders and surface crusting. Ulcerative lesions have heaped-up borders and often have an exudative base.8 The differential diagnosis of cutaneous North American blastomycosis lesions includes squamous cell carcinoma, giant keratoacanthoma, verrucae, basal cell carcinoma, scrofuloderma, lupus vulgaris, nocardiosis, syphilis, bromoderma, iododerma, granuloma inguinale, tuberculosis verrucosa cutis, mycetoma, and actinomycosis.7,8
Although periorificial cutaneous manifestations of disseminated blastomycosis are common, perianal lesions are rare. The differential diagnosis of perianal verrucous plaques includes condyloma acuminatum, squamous cell carcinoma, adenocarcinoma, Buschke-Löwenstein tumor, actinomycosis, and localized fungal infections such as blastomycosis.9
Case Report
A 57-year-old man presented with a palpable perianal mass that produced small amounts of blood in his underwear and on toilet paper. The patient reported no history of hemorrhoids, anoreceptive intercourse, or sexually transmitted disease. Four months prior to presentation, he had a prolonged upper respiratory tract illness with a subjective fever and productive cough of 2 months’ duration. The patient described himself as an avid outdoorsman who worked at a summer resort and spent a great deal of time in the forests of central Wisconsin last autumn. Physical examination revealed a well-demarcated, firm, moist plaque with a verrucous surface that measured 3.5×2.7 cm and extended from the anal verge to the perianal skin (Figure 1).
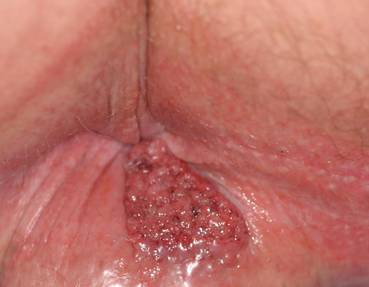
Potassium hydroxide preparation of a biopsy specimen (Figure 2), a punch biopsy of the lesion (Figure 3), and Gomori methenamine-silver staining (Figure 4) revealed scattered yeast spores, some demonstrating broad-based budding, with pseudoepitheliomatous hyperplasia, dermal neutrophils, and intraepithelial microabscesses. The patient’s urine was positive for Blastomyces antigen (1.04 ng/mL). Chest radiography demonstrated a localized infiltrate in the right hilum with possible mass effect. Computed tomography showed a consolidative opacity measuring 4.0×3.4 cm in the upper lobe of the right lung (Figure 5).
| 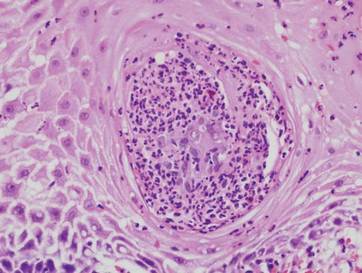
|
The patient was diagnosed with cutaneous North American blastomycosis and prescribed a 6-month course of oral itraconazole 200 mg twice daily. At his 3-month follow-up visit, the perianal plaque hadalmost completely resolved (Figure 6). However, because the patient had increasing lower extremity edema, subjective hearing loss, and abnormal liver function tests, itraconazole treatment was discontinued and replaced with oral fluconazole 400 mg daily for the next 3 months. The right hilar mass had visibly improved on follow-up chest radiography 2 months after the patient started antifungal therapy with itraconazole and had resolved within another 3 months of treatment.
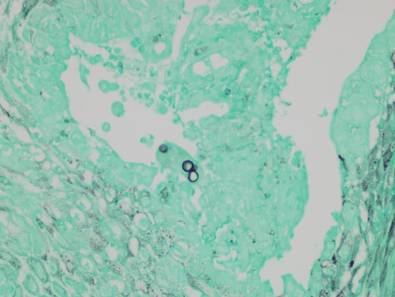
| 
|
Comment
Cutaneous blastomycosis results most often from the hematogenous spread of B dermatitidis from the lungs and rarely from direct inoculation.5,10 Skin lesions tend to occur on exposed areas, such as the face, scalp, hands, wrists, feet, and ankles.7,11-13 Dissemination to the perianal skin is rare, though it has been reported in 2 other patients; both patients, similar to our patient, had evidence of pulmonary involvement at some point in their clinical course.9,14
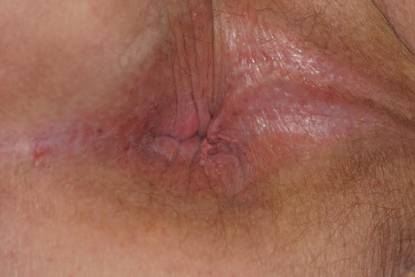
Diagnosis is based on identification of B dermatitidis by microscopy or culture. Potassium hydroxide preparation of biopsy specimens typically shows broad-based budding yeast.13 Characteristic findings of histopathologic studies include pseudo-epitheliomatous hyperplasia, intraepidermal abscesses, and a dermal infiltrate of polymorphonuclear leukocytes.15 On fungal culture, B dermatitidis is slow growing and may require a 2- to 4-week incubation period. Serologic tests are available, but sensitivity is low, at 9%, 28%, and 77% for complement fixation, immunodiffusion, and enzyme immunoassay, respectively.16
Conclusion
North American blastomycosis should be considered in patients who have verrucous or ulcerative perianal lesions and have lived in or traveled to endemic regions, especially if they have recent or ongoing pulmonary symptoms. Potassium hydroxide preparation and fungal staining of biopsy specimens can aid in diagnosis.
Acknowledgment
The authors thank the Marshfield Clinic Research Foundation’s Office of Scientific Writing and Publication (Marshfield, Wisconsin) for editorial assistance in the preparation of this manuscript.
1. Klein BS, Vergeront JM, Davis JP. Epidemiologic aspects of blastomycosis, the enigmatic systemic mycosis. Semin Respir Infect. 1986;1:29-39.
2. Klein BS, Vergeront JM, Weeks RJ, et al. Isolation of Blastomyces dermatitidis in soil associated with a large outbreak of blastomycosis in Wisconsin. N Engl J Med. 1986;314:529-534.
3. Reingold AL, Lu XD, Plikaytis BD, et al. Systemic mycoses in the United States, 1980-1982. J Med Vet Mycol. 1986;24:433-436.
4. Centers for Disease Control and Prevention (CDC). Blastomycosis—Wisconsin, 1986-1995. MMWR Morb Mortal Wkly Rep. 1996;45:601-603.
5. Smith JA, Kauffman CA. Blastomycosis. Proc Am Thorac Soc. 2010;7:173-180.
6. Goldman M, Johnson PC, Sarosi GA. Fungal pneumonias. the endemic mycoses. Clin Chest Med. 1999;20:507-519.
7. Mercurio MG, Elewski BE. Cutaneous blastomycosis. Cutis. 1992;50:422-424.
8. Saccente M, Woods GL. Clinical and laboratory update on blastomycosis. Clin Microbiol Rev. 2010;23:367-381.
9. Ricciardi R, Alavi K, Filice GA, et al. Blastomyces dermatitidis of the perianal skin: report of a case. Dis Colon Rectum. 2007;50:118-121.
10. Gray NA, Baddour LM. Cutaneous inoculation blastomycosis [published online ahead of print April 17, 2002]. Clin Infect Dis. 2002;34:e44-e49.
11. Kisso B, Mahmoud F, Thakkar JR. Blastomycosis presenting as recurrent tender cutaneous nodules. S D Med. 2006;59:255-259.
12. Mandell GL, Bennett JE, Dolin R. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2010.
13. Mason AR, Cortes GY, Cook J, et al. Cutaneous blastomycosis: a diagnostic challenge. Int J Dermatol. 2008;47:824-830.
14. Linn JE. Pseudo-epitheliomatous lesions of the perirectal tissue: report of a case of squamous epithelioma due to blastomycosis. South Med J. 1958;51:1101-1104.
15. Woofter MJ, Cripps DJ, Warner TF. Verrucous plaques on the face. North American blastomycosis. Arch Dermatol. 2000;136:547, 550.
16. Klein BS, Vergeront JM, Kaufman L, et al. Serological tests for blastomycosis: assessments during a large point-source outbreak in Wisconsin. J Infect Dis. 1987;155:262-268.
Cutaneous North American blastomycosis is a deep fungal infection caused by Blastomyces dermatitidis, a thermally dimorphic fungus that is endemic to the Great Lakes region as well as the Mississippi and Ohio River valleys where it thrives in moist acidic soil enriched with organic material.1,2 In humans, the annual incidence rate is estimated to be 0.6 cases per million,3 though it may be as high as 42 cases per 100,000 in endemic areas.4 Infection typically results from the inhalation of conidia and manifests as either acute or chronic pneumonia.5 Most patients with acute disease present with nonspecific flulike symptoms and a nonproductive cough.
Dissemination occurs in approximately 25% of cases,6 most commonly affecting the skin. Other potential sites of dissemination include bone, the genitourinary tract, and the central nervous system. Cutaneous lesions, which may be either verrucous or ulcerative plaques, often occur on or around orifices contiguous to the respiratory tract.7 Verrucous lesions tend to have an irregular shape with well-defined borders and surface crusting. Ulcerative lesions have heaped-up borders and often have an exudative base.8 The differential diagnosis of cutaneous North American blastomycosis lesions includes squamous cell carcinoma, giant keratoacanthoma, verrucae, basal cell carcinoma, scrofuloderma, lupus vulgaris, nocardiosis, syphilis, bromoderma, iododerma, granuloma inguinale, tuberculosis verrucosa cutis, mycetoma, and actinomycosis.7,8
Although periorificial cutaneous manifestations of disseminated blastomycosis are common, perianal lesions are rare. The differential diagnosis of perianal verrucous plaques includes condyloma acuminatum, squamous cell carcinoma, adenocarcinoma, Buschke-Löwenstein tumor, actinomycosis, and localized fungal infections such as blastomycosis.9
Case Report
A 57-year-old man presented with a palpable perianal mass that produced small amounts of blood in his underwear and on toilet paper. The patient reported no history of hemorrhoids, anoreceptive intercourse, or sexually transmitted disease. Four months prior to presentation, he had a prolonged upper respiratory tract illness with a subjective fever and productive cough of 2 months’ duration. The patient described himself as an avid outdoorsman who worked at a summer resort and spent a great deal of time in the forests of central Wisconsin last autumn. Physical examination revealed a well-demarcated, firm, moist plaque with a verrucous surface that measured 3.5×2.7 cm and extended from the anal verge to the perianal skin (Figure 1).
Potassium hydroxide preparation of a biopsy specimen (Figure 2), a punch biopsy of the lesion (Figure 3), and Gomori methenamine-silver staining (Figure 4) revealed scattered yeast spores, some demonstrating broad-based budding, with pseudoepitheliomatous hyperplasia, dermal neutrophils, and intraepithelial microabscesses. The patient’s urine was positive for Blastomyces antigen (1.04 ng/mL). Chest radiography demonstrated a localized infiltrate in the right hilum with possible mass effect. Computed tomography showed a consolidative opacity measuring 4.0×3.4 cm in the upper lobe of the right lung (Figure 5).
|
|
|
The patient was diagnosed with cutaneous North American blastomycosis and prescribed a 6-month course of oral itraconazole 200 mg twice daily. At his 3-month follow-up visit, the perianal plaque hadalmost completely resolved (Figure 6). However, because the patient had increasing lower extremity edema, subjective hearing loss, and abnormal liver function tests, itraconazole treatment was discontinued and replaced with oral fluconazole 400 mg daily for the next 3 months. The right hilar mass had visibly improved on follow-up chest radiography 2 months after the patient started antifungal therapy with itraconazole and had resolved within another 3 months of treatment.
|
|
|
Comment
Cutaneous blastomycosis results most often from the hematogenous spread of B dermatitidis from the lungs and rarely from direct inoculation.5,10 Skin lesions tend to occur on exposed areas, such as the face, scalp, hands, wrists, feet, and ankles.7,11-13 Dissemination to the perianal skin is rare, though it has been reported in 2 other patients; both patients, similar to our patient, had evidence of pulmonary involvement at some point in their clinical course.9,14
Diagnosis is based on identification of B dermatitidis by microscopy or culture. Potassium hydroxide preparation of biopsy specimens typically shows broad-based budding yeast.13 Characteristic findings of histopathologic studies include pseudo-epitheliomatous hyperplasia, intraepidermal abscesses, and a dermal infiltrate of polymorphonuclear leukocytes.15 On fungal culture, B dermatitidis is slow growing and may require a 2- to 4-week incubation period. Serologic tests are available, but sensitivity is low, at 9%, 28%, and 77% for complement fixation, immunodiffusion, and enzyme immunoassay, respectively.16
Conclusion
North American blastomycosis should be considered in patients who have verrucous or ulcerative perianal lesions and have lived in or traveled to endemic regions, especially if they have recent or ongoing pulmonary symptoms. Potassium hydroxide preparation and fungal staining of biopsy specimens can aid in diagnosis.
Acknowledgment
The authors thank the Marshfield Clinic Research Foundation’s Office of Scientific Writing and Publication (Marshfield, Wisconsin) for editorial assistance in the preparation of this manuscript.
Cutaneous North American blastomycosis is a deep fungal infection caused by Blastomyces dermatitidis, a thermally dimorphic fungus that is endemic to the Great Lakes region as well as the Mississippi and Ohio River valleys where it thrives in moist acidic soil enriched with organic material.1,2 In humans, the annual incidence rate is estimated to be 0.6 cases per million,3 though it may be as high as 42 cases per 100,000 in endemic areas.4 Infection typically results from the inhalation of conidia and manifests as either acute or chronic pneumonia.5 Most patients with acute disease present with nonspecific flulike symptoms and a nonproductive cough.
Dissemination occurs in approximately 25% of cases,6 most commonly affecting the skin. Other potential sites of dissemination include bone, the genitourinary tract, and the central nervous system. Cutaneous lesions, which may be either verrucous or ulcerative plaques, often occur on or around orifices contiguous to the respiratory tract.7 Verrucous lesions tend to have an irregular shape with well-defined borders and surface crusting. Ulcerative lesions have heaped-up borders and often have an exudative base.8 The differential diagnosis of cutaneous North American blastomycosis lesions includes squamous cell carcinoma, giant keratoacanthoma, verrucae, basal cell carcinoma, scrofuloderma, lupus vulgaris, nocardiosis, syphilis, bromoderma, iododerma, granuloma inguinale, tuberculosis verrucosa cutis, mycetoma, and actinomycosis.7,8
Although periorificial cutaneous manifestations of disseminated blastomycosis are common, perianal lesions are rare. The differential diagnosis of perianal verrucous plaques includes condyloma acuminatum, squamous cell carcinoma, adenocarcinoma, Buschke-Löwenstein tumor, actinomycosis, and localized fungal infections such as blastomycosis.9
Case Report
A 57-year-old man presented with a palpable perianal mass that produced small amounts of blood in his underwear and on toilet paper. The patient reported no history of hemorrhoids, anoreceptive intercourse, or sexually transmitted disease. Four months prior to presentation, he had a prolonged upper respiratory tract illness with a subjective fever and productive cough of 2 months’ duration. The patient described himself as an avid outdoorsman who worked at a summer resort and spent a great deal of time in the forests of central Wisconsin last autumn. Physical examination revealed a well-demarcated, firm, moist plaque with a verrucous surface that measured 3.5×2.7 cm and extended from the anal verge to the perianal skin (Figure 1).
Potassium hydroxide preparation of a biopsy specimen (Figure 2), a punch biopsy of the lesion (Figure 3), and Gomori methenamine-silver staining (Figure 4) revealed scattered yeast spores, some demonstrating broad-based budding, with pseudoepitheliomatous hyperplasia, dermal neutrophils, and intraepithelial microabscesses. The patient’s urine was positive for Blastomyces antigen (1.04 ng/mL). Chest radiography demonstrated a localized infiltrate in the right hilum with possible mass effect. Computed tomography showed a consolidative opacity measuring 4.0×3.4 cm in the upper lobe of the right lung (Figure 5).
|
|
|
The patient was diagnosed with cutaneous North American blastomycosis and prescribed a 6-month course of oral itraconazole 200 mg twice daily. At his 3-month follow-up visit, the perianal plaque hadalmost completely resolved (Figure 6). However, because the patient had increasing lower extremity edema, subjective hearing loss, and abnormal liver function tests, itraconazole treatment was discontinued and replaced with oral fluconazole 400 mg daily for the next 3 months. The right hilar mass had visibly improved on follow-up chest radiography 2 months after the patient started antifungal therapy with itraconazole and had resolved within another 3 months of treatment.
|
|
|
Comment
Cutaneous blastomycosis results most often from the hematogenous spread of B dermatitidis from the lungs and rarely from direct inoculation.5,10 Skin lesions tend to occur on exposed areas, such as the face, scalp, hands, wrists, feet, and ankles.7,11-13 Dissemination to the perianal skin is rare, though it has been reported in 2 other patients; both patients, similar to our patient, had evidence of pulmonary involvement at some point in their clinical course.9,14
Diagnosis is based on identification of B dermatitidis by microscopy or culture. Potassium hydroxide preparation of biopsy specimens typically shows broad-based budding yeast.13 Characteristic findings of histopathologic studies include pseudo-epitheliomatous hyperplasia, intraepidermal abscesses, and a dermal infiltrate of polymorphonuclear leukocytes.15 On fungal culture, B dermatitidis is slow growing and may require a 2- to 4-week incubation period. Serologic tests are available, but sensitivity is low, at 9%, 28%, and 77% for complement fixation, immunodiffusion, and enzyme immunoassay, respectively.16
Conclusion
North American blastomycosis should be considered in patients who have verrucous or ulcerative perianal lesions and have lived in or traveled to endemic regions, especially if they have recent or ongoing pulmonary symptoms. Potassium hydroxide preparation and fungal staining of biopsy specimens can aid in diagnosis.
Acknowledgment
The authors thank the Marshfield Clinic Research Foundation’s Office of Scientific Writing and Publication (Marshfield, Wisconsin) for editorial assistance in the preparation of this manuscript.
1. Klein BS, Vergeront JM, Davis JP. Epidemiologic aspects of blastomycosis, the enigmatic systemic mycosis. Semin Respir Infect. 1986;1:29-39.
2. Klein BS, Vergeront JM, Weeks RJ, et al. Isolation of Blastomyces dermatitidis in soil associated with a large outbreak of blastomycosis in Wisconsin. N Engl J Med. 1986;314:529-534.
3. Reingold AL, Lu XD, Plikaytis BD, et al. Systemic mycoses in the United States, 1980-1982. J Med Vet Mycol. 1986;24:433-436.
4. Centers for Disease Control and Prevention (CDC). Blastomycosis—Wisconsin, 1986-1995. MMWR Morb Mortal Wkly Rep. 1996;45:601-603.
5. Smith JA, Kauffman CA. Blastomycosis. Proc Am Thorac Soc. 2010;7:173-180.
6. Goldman M, Johnson PC, Sarosi GA. Fungal pneumonias. the endemic mycoses. Clin Chest Med. 1999;20:507-519.
7. Mercurio MG, Elewski BE. Cutaneous blastomycosis. Cutis. 1992;50:422-424.
8. Saccente M, Woods GL. Clinical and laboratory update on blastomycosis. Clin Microbiol Rev. 2010;23:367-381.
9. Ricciardi R, Alavi K, Filice GA, et al. Blastomyces dermatitidis of the perianal skin: report of a case. Dis Colon Rectum. 2007;50:118-121.
10. Gray NA, Baddour LM. Cutaneous inoculation blastomycosis [published online ahead of print April 17, 2002]. Clin Infect Dis. 2002;34:e44-e49.
11. Kisso B, Mahmoud F, Thakkar JR. Blastomycosis presenting as recurrent tender cutaneous nodules. S D Med. 2006;59:255-259.
12. Mandell GL, Bennett JE, Dolin R. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2010.
13. Mason AR, Cortes GY, Cook J, et al. Cutaneous blastomycosis: a diagnostic challenge. Int J Dermatol. 2008;47:824-830.
14. Linn JE. Pseudo-epitheliomatous lesions of the perirectal tissue: report of a case of squamous epithelioma due to blastomycosis. South Med J. 1958;51:1101-1104.
15. Woofter MJ, Cripps DJ, Warner TF. Verrucous plaques on the face. North American blastomycosis. Arch Dermatol. 2000;136:547, 550.
16. Klein BS, Vergeront JM, Kaufman L, et al. Serological tests for blastomycosis: assessments during a large point-source outbreak in Wisconsin. J Infect Dis. 1987;155:262-268.
1. Klein BS, Vergeront JM, Davis JP. Epidemiologic aspects of blastomycosis, the enigmatic systemic mycosis. Semin Respir Infect. 1986;1:29-39.
2. Klein BS, Vergeront JM, Weeks RJ, et al. Isolation of Blastomyces dermatitidis in soil associated with a large outbreak of blastomycosis in Wisconsin. N Engl J Med. 1986;314:529-534.
3. Reingold AL, Lu XD, Plikaytis BD, et al. Systemic mycoses in the United States, 1980-1982. J Med Vet Mycol. 1986;24:433-436.
4. Centers for Disease Control and Prevention (CDC). Blastomycosis—Wisconsin, 1986-1995. MMWR Morb Mortal Wkly Rep. 1996;45:601-603.
5. Smith JA, Kauffman CA. Blastomycosis. Proc Am Thorac Soc. 2010;7:173-180.
6. Goldman M, Johnson PC, Sarosi GA. Fungal pneumonias. the endemic mycoses. Clin Chest Med. 1999;20:507-519.
7. Mercurio MG, Elewski BE. Cutaneous blastomycosis. Cutis. 1992;50:422-424.
8. Saccente M, Woods GL. Clinical and laboratory update on blastomycosis. Clin Microbiol Rev. 2010;23:367-381.
9. Ricciardi R, Alavi K, Filice GA, et al. Blastomyces dermatitidis of the perianal skin: report of a case. Dis Colon Rectum. 2007;50:118-121.
10. Gray NA, Baddour LM. Cutaneous inoculation blastomycosis [published online ahead of print April 17, 2002]. Clin Infect Dis. 2002;34:e44-e49.
11. Kisso B, Mahmoud F, Thakkar JR. Blastomycosis presenting as recurrent tender cutaneous nodules. S D Med. 2006;59:255-259.
12. Mandell GL, Bennett JE, Dolin R. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2010.
13. Mason AR, Cortes GY, Cook J, et al. Cutaneous blastomycosis: a diagnostic challenge. Int J Dermatol. 2008;47:824-830.
14. Linn JE. Pseudo-epitheliomatous lesions of the perirectal tissue: report of a case of squamous epithelioma due to blastomycosis. South Med J. 1958;51:1101-1104.
15. Woofter MJ, Cripps DJ, Warner TF. Verrucous plaques on the face. North American blastomycosis. Arch Dermatol. 2000;136:547, 550.
16. Klein BS, Vergeront JM, Kaufman L, et al. Serological tests for blastomycosis: assessments during a large point-source outbreak in Wisconsin. J Infect Dis. 1987;155:262-268.
Practice Points
- Cutaneous North American blastomycosis usually occurs in a periorificial distribution.
- The perianal region should be included in the periorificial regions considered in North American blastomycosis infections.
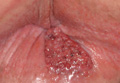
Nodular Scleroderma in a Patient With Chronic Hepatitis C Virus Infection: A Coexistent or Causal Infection?
Case Report
A 63-year-old woman was referred to our clinic for evaluation of multiple papules and nodules on the neck and trunk that had been present for 2 years. Three years prior to presentation she had been diagnosed with systemic sclerosis (SSc) after developing progressive diffuse cutaneous sclerosis, Raynaud phenomenon with digital pitted scarring, esophageal dysmotility, myositis, pericardial effusion, and interstitial lung disease. Serologic test results were positive for anti-Scl-70 antibodies. Antinuclear antibody test results were negative for anti–double-stranded DNA, anti-nRNP, anti-Ro/La, anti-Sm, and anti-Jo-1 antibodies. The patient was treated with prednisolone 7.5 mg daily, nifedipine 15 mg daily, valsartan 80 mg daily, manidipine 20 mg daily, omeprazole 20 mg daily, and beraprost 80 mg daily. One year later, numerous asymptomatic flesh-colored papules and nodules developed on the neck, chest, abdomen, and back. There was no history of trauma or surgery at any of the affected sites.
On further investigation, anti–hepatitis C virus (HCV) antibodies were identified and confirmed by HCV ribonucleic acid polymerase chain reaction at the same time that the diagnosis of SSc was established. Hepatitis C virus genotype 3a was noted, and the patient’s viral load was 378,000 IU/mL. Therefore, a diagnosis of chronic HCV infection was established. The patient was initially unable to receive medical treatment due to lack of finances. A year and a half following the diagnosis of HCV infection, with worsening liver function tests and increasing viral load (1,369,113 IU/mL), the patient began therapy with peginterferon alfa-2b 80 mg weekly and ribavirin 800 mg daily. However, the medications were discontinued after 2 months when she developed severe hemolytic anemia related to ribavirin.
On physical examination, the patient was noted to have a masklike facies with a pinched nose and constricted opening of the mouth. Her skin was tightened and stiff extending from the fingers to the proximal extremities. Numerous well-circumscribed, flesh-colored, firm papules and nodules ranging from 2 to 20 mm in diameter were present on the neck (Figure 1), chest, abdomen (Figure 2), and back.
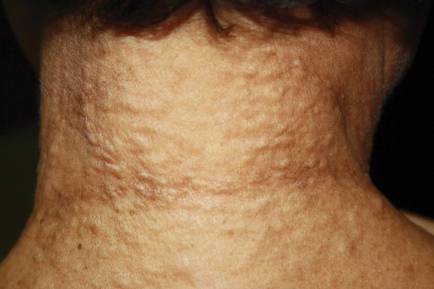
| 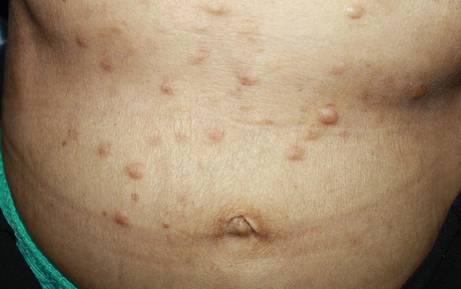
|
Two 4-mm punch biopsy samples obtained from a papule on the neck and a nodule on the abdomen revealed homogenized collagen bundles with scattered plump fibroblasts in the lower reticular dermis. Clinicopathologic correlation of the biopsy findings with the cutaneous examination resulted in a diagnosis of nodular scleroderma (Figures 3 and 4).
|
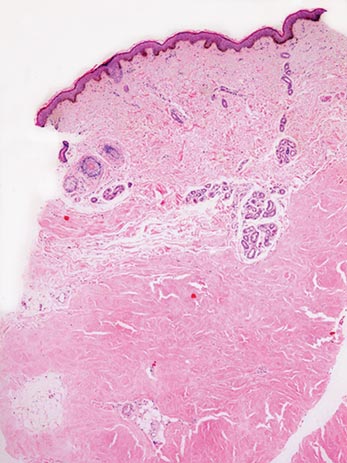
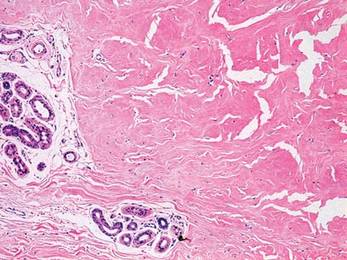
The patient began treatment with intralesional injections of triamcinolone 5 to 10 mg/mL for nodules as well as an ultrapotent corticosteroid cream, clobetasol propionate 0.05%, for small papules. Injections were performed at 4- to 8-week intervals and resulted in modest clinical improvement.
Comment
Scleroderma may be present only in the skin (morphea) or as a systemic disease (systemic scleroderma). Rarely, cutaneous involvement can exhibit a nodular or hypertrophic morphology, which has been described in the literature as nodular or keloidal scleroderma in a patient with known SSc1-10 and as nodular or keloidal morphea in localized cutaneous scleroderma.3,11-13
Histopathology
The distinction between the terms nodular scleroderma and keloidal scleroderma is not clear, and they are not necessarily interchangeable. To provide clarity, we find it useful to delineate specific histologic findings associated with the diagnoses of keloid, scleroderma, and the uncommon keloid/scleroderma overlap. The histopathologic findings of keloids include a fibrotic dermis and broad dispersed bundles of eosinophilic hyalinized collagen. The histopathologic findings of scleroderma include broad sclerotic bands of collagen throughout the dermis with loss of perieccrine fat. In the overlapping keloid/scleroderma condition, which is a variant of scleroderma, hyalinized collagen fibers and keloidal collagen appear in the same specimen.3,4
To distinguish these conditions, Barzilai et al5 proposed that only cases showing both clinical and histologic characteristics of a keloid should be referred to as keloidal morphea/scleroderma. They further stated that the terms nodular morphea or nodular scleroderma ought to be used only for cases that are indistinguishable histologically from scleroderma. The term morphea is appropriate when only a limited amount of skin disease is present, while scleroderma implies association with systemic disease.5 Likely, there is a histologic continuum in this variant of scleroderma, in which nodular morphea/scleroderma exists at one end and keloidal morphea/scleroderma exists at the other end.5,13
In the case of our patient, papulonodular lesions developed 1 year after the diagnosis of SSc was made, and the histopathologic examination revealed classic findings of scleroderma. As a result, our patient is most appropriately classified as having nodular scleroderma.
Clinical Features
Nodular scleroderma mostly affects young and middle-aged women and is clinically characterized by solitary or multiple firm, long-lasting papules or nodules on the upper trunk and chest, neck, and proximal extremities.1-4,6
Etiology and Pathogenesis
The triggers and cellular mechanisms of nodular scleroderma are unclear. Some authors have implicated matricellular protein and growth factors such as tenascin, connective tissue growth factor, and epidermal growth factor in nodule formation.7,8,11 Yamamoto et al9 cited chemical exposure to a silica-containing abrasive as the cause of nodular scleroderma in a worker.
Possible HCV Association
Some reports have indicated an association between nodular scleroderma and pathogens such as acid-fast bacteria10 and HCV.6 Of note, many extrahepatic conditions have been associated with HCV infection, such as membranoproliferative glomerulonephritis, cutaneous vasculitis, lichen planus, and porphyria cutanea tarda.14
The association of HCV infection with systemic autoimmune disease (SAD) has been described in a number of instances; cryoglobulinemia has most commonly been linked to HCV.15 Although the association between HCV and other SADs is less clear, there is growing interest in a possible relationship between them. To that end, physicians of the HISPAMEC (Hispanoamerican Study Group of Autoimmune Manifestations Associated With Hepatitis C Virus) study group described the clinical and immunologic characteristics of 1020 patients with SAD and associated chronic HCV infection. The 3 most frequent SADs (>90% of cases) were Sjögren syndrome, rheumatoid arthritis, and systemic lupus erythematosus.16 However, the strength of association differs for each SAD based on existing descriptions.16,17 Less commonly, there may be a causal relationship between HCV infection and SSc. It should be noted that most of these data are based on small series and case reports.6,16-19
The role of HCV in the pathogenesis of systemic scleroderma and other autoimmune diseases is unknown. It is also possible that the replication of HCV outside the liver, particularly in mononuclear cells, may suppress immune tolerance in genetically predisposed individuals.20
Conclusion
Nodular scleroderma associated with HCV infection is a rare entity. At present, it cannot be determined whether there is an etiopathologic association between HCV infection and SSc or whether the simultaneous diagnosis may be coincidental. Routine determination of HCV serology in scleroderma patients may help to clarify this issue.
1. Krell JM, Solomon AR, Glavey CM, et al. Nodular scleroderma. J Am Acad Dermatol. 1995;32:343-345.
2. Cannick L 3rd, Douglas G, Crater S, et al. Nodular scleroderma: case report and literature review. J Rheumatol. 2003;30:2500-2502.
3. Rencic A, Brinster NK, Nousari CH. Keloid morphea and nodular scleroderma: two distinct clinical variants of scleroderma? J Cutan Med Surg. 2003;7:20-24.
4. Wriston CC, Rubin AI, Elenitsas R, et al. Nodular scleroderma: a report of 2 cases. Am J Dermatopathol. 2008;30:385-388.
5. Barzilai A, Lyakhovitsky A, Horowitz A, et al. Keloid-like scleroderma. Am J Dermatopathol. 2003;25:327-330.
6. Melani L, Caproni M, Cardinali C, et al. A case of nodular scleroderma. J Dermatol. 2005;32:1028-1031.
7. Mizutani H, Taniguchi H, Sakakura T, et al. Nodular scleroderma: focally increased tenascin expression differing from that in the surrounding scleroderma skin. J Dermatol. 1995;22:267-271.
8. Yamamoto T, Sawada Y, Katayama I, et al. Nodular scleroderma: increased expression of connective tissue growth factor. Dermatology. 2005;211:218-223.
9. Yamamoto T, Furuse Y, Katayama I, et al. Nodular scleroderma in a worker using a silica-containing abrasive. J Dermatol. 1994;21:751-754.
10. Cantwell AR Jr, Rowe L, Kelso DW. Nodular scleroderma and pleomorphic acid-fast bacteria. Arch Dermatol. 1980;116:1283-1290.
11. Yamamoto T, Sakashita S, Sawada Y, et al. Possible role of epidermal growth factor in the lesional skin of nodular morphea. Acta Derm Venereol. 1998;78:312-313.
12. Jain K, Dayal S, Jain VK, et al. Blaschko linear nodular morphea with dermal mucinosis. Arch Dermatol. 2007;143:953-955.
13. Kauer F, Simon JC, Sticherling M. Nodular morphea. Dermatology. 2009;218:63-66.
14. Gumber SC, Chopra S. Hepatitis C: a multifaceted disease. review of extrahepatic manifestations. Ann Intern Med. 1995;123:615-620.
15. Ferri C, Greco F, Longombardo G, et al. Antibodies to hepatitis C virus in patients with mixed cryoglobulinemia. Arthritis Rheum. 1991;34:1606-1610.
16. Ramos-Casals M, Munoz S, Medina F, et al. Systemic autoimmune diseases in patients with hepatitis C virus infection: characterization of 1020 cases (The HISPAMEC Registry). J Rheumatol. 2009;36:1442-1448.
17. Ramos-Casals M, Jara LJ, Medina F, et al. Systemic autoimmune diseases co-existing with chronic hepatitis C virus infection (the HISPAMEC Registry): patterns of clinical and immunological expression in 180 cases. J Intern Med. 2005;257:549-557.
18. Abu-Shakra M, Sukenik S, Buskila D. Systemic sclerosis: another rheumatic disease associated with hepatitis C virus infection. Clin Rheumatol. 2000;19:378-380.
19. Yamamoto M, Yamamoto T, Tsuboi R. Discoid lupus erythematosus in a patient with scleroderma and hepatitis C virus infection. Rheumatol Int. 2010;30:969-971.
20. Abu-Shakra M, Shoenfeld Y. Chronic infections and autoimmunity. Immunol Ser. 1992;55:285-313.
Case Report
A 63-year-old woman was referred to our clinic for evaluation of multiple papules and nodules on the neck and trunk that had been present for 2 years. Three years prior to presentation she had been diagnosed with systemic sclerosis (SSc) after developing progressive diffuse cutaneous sclerosis, Raynaud phenomenon with digital pitted scarring, esophageal dysmotility, myositis, pericardial effusion, and interstitial lung disease. Serologic test results were positive for anti-Scl-70 antibodies. Antinuclear antibody test results were negative for anti–double-stranded DNA, anti-nRNP, anti-Ro/La, anti-Sm, and anti-Jo-1 antibodies. The patient was treated with prednisolone 7.5 mg daily, nifedipine 15 mg daily, valsartan 80 mg daily, manidipine 20 mg daily, omeprazole 20 mg daily, and beraprost 80 mg daily. One year later, numerous asymptomatic flesh-colored papules and nodules developed on the neck, chest, abdomen, and back. There was no history of trauma or surgery at any of the affected sites.
On further investigation, anti–hepatitis C virus (HCV) antibodies were identified and confirmed by HCV ribonucleic acid polymerase chain reaction at the same time that the diagnosis of SSc was established. Hepatitis C virus genotype 3a was noted, and the patient’s viral load was 378,000 IU/mL. Therefore, a diagnosis of chronic HCV infection was established. The patient was initially unable to receive medical treatment due to lack of finances. A year and a half following the diagnosis of HCV infection, with worsening liver function tests and increasing viral load (1,369,113 IU/mL), the patient began therapy with peginterferon alfa-2b 80 mg weekly and ribavirin 800 mg daily. However, the medications were discontinued after 2 months when she developed severe hemolytic anemia related to ribavirin.
On physical examination, the patient was noted to have a masklike facies with a pinched nose and constricted opening of the mouth. Her skin was tightened and stiff extending from the fingers to the proximal extremities. Numerous well-circumscribed, flesh-colored, firm papules and nodules ranging from 2 to 20 mm in diameter were present on the neck (Figure 1), chest, abdomen (Figure 2), and back.
|
|
|
Two 4-mm punch biopsy samples obtained from a papule on the neck and a nodule on the abdomen revealed homogenized collagen bundles with scattered plump fibroblasts in the lower reticular dermis. Clinicopathologic correlation of the biopsy findings with the cutaneous examination resulted in a diagnosis of nodular scleroderma (Figures 3 and 4).
|
The patient began treatment with intralesional injections of triamcinolone 5 to 10 mg/mL for nodules as well as an ultrapotent corticosteroid cream, clobetasol propionate 0.05%, for small papules. Injections were performed at 4- to 8-week intervals and resulted in modest clinical improvement.
Comment
Scleroderma may be present only in the skin (morphea) or as a systemic disease (systemic scleroderma). Rarely, cutaneous involvement can exhibit a nodular or hypertrophic morphology, which has been described in the literature as nodular or keloidal scleroderma in a patient with known SSc1-10 and as nodular or keloidal morphea in localized cutaneous scleroderma.3,11-13
Histopathology
The distinction between the terms nodular scleroderma and keloidal scleroderma is not clear, and they are not necessarily interchangeable. To provide clarity, we find it useful to delineate specific histologic findings associated with the diagnoses of keloid, scleroderma, and the uncommon keloid/scleroderma overlap. The histopathologic findings of keloids include a fibrotic dermis and broad dispersed bundles of eosinophilic hyalinized collagen. The histopathologic findings of scleroderma include broad sclerotic bands of collagen throughout the dermis with loss of perieccrine fat. In the overlapping keloid/scleroderma condition, which is a variant of scleroderma, hyalinized collagen fibers and keloidal collagen appear in the same specimen.3,4
To distinguish these conditions, Barzilai et al5 proposed that only cases showing both clinical and histologic characteristics of a keloid should be referred to as keloidal morphea/scleroderma. They further stated that the terms nodular morphea or nodular scleroderma ought to be used only for cases that are indistinguishable histologically from scleroderma. The term morphea is appropriate when only a limited amount of skin disease is present, while scleroderma implies association with systemic disease.5 Likely, there is a histologic continuum in this variant of scleroderma, in which nodular morphea/scleroderma exists at one end and keloidal morphea/scleroderma exists at the other end.5,13
In the case of our patient, papulonodular lesions developed 1 year after the diagnosis of SSc was made, and the histopathologic examination revealed classic findings of scleroderma. As a result, our patient is most appropriately classified as having nodular scleroderma.
Clinical Features
Nodular scleroderma mostly affects young and middle-aged women and is clinically characterized by solitary or multiple firm, long-lasting papules or nodules on the upper trunk and chest, neck, and proximal extremities.1-4,6
Etiology and Pathogenesis
The triggers and cellular mechanisms of nodular scleroderma are unclear. Some authors have implicated matricellular protein and growth factors such as tenascin, connective tissue growth factor, and epidermal growth factor in nodule formation.7,8,11 Yamamoto et al9 cited chemical exposure to a silica-containing abrasive as the cause of nodular scleroderma in a worker.
Possible HCV Association
Some reports have indicated an association between nodular scleroderma and pathogens such as acid-fast bacteria10 and HCV.6 Of note, many extrahepatic conditions have been associated with HCV infection, such as membranoproliferative glomerulonephritis, cutaneous vasculitis, lichen planus, and porphyria cutanea tarda.14
The association of HCV infection with systemic autoimmune disease (SAD) has been described in a number of instances; cryoglobulinemia has most commonly been linked to HCV.15 Although the association between HCV and other SADs is less clear, there is growing interest in a possible relationship between them. To that end, physicians of the HISPAMEC (Hispanoamerican Study Group of Autoimmune Manifestations Associated With Hepatitis C Virus) study group described the clinical and immunologic characteristics of 1020 patients with SAD and associated chronic HCV infection. The 3 most frequent SADs (>90% of cases) were Sjögren syndrome, rheumatoid arthritis, and systemic lupus erythematosus.16 However, the strength of association differs for each SAD based on existing descriptions.16,17 Less commonly, there may be a causal relationship between HCV infection and SSc. It should be noted that most of these data are based on small series and case reports.6,16-19
The role of HCV in the pathogenesis of systemic scleroderma and other autoimmune diseases is unknown. It is also possible that the replication of HCV outside the liver, particularly in mononuclear cells, may suppress immune tolerance in genetically predisposed individuals.20
Conclusion
Nodular scleroderma associated with HCV infection is a rare entity. At present, it cannot be determined whether there is an etiopathologic association between HCV infection and SSc or whether the simultaneous diagnosis may be coincidental. Routine determination of HCV serology in scleroderma patients may help to clarify this issue.
Case Report
A 63-year-old woman was referred to our clinic for evaluation of multiple papules and nodules on the neck and trunk that had been present for 2 years. Three years prior to presentation she had been diagnosed with systemic sclerosis (SSc) after developing progressive diffuse cutaneous sclerosis, Raynaud phenomenon with digital pitted scarring, esophageal dysmotility, myositis, pericardial effusion, and interstitial lung disease. Serologic test results were positive for anti-Scl-70 antibodies. Antinuclear antibody test results were negative for anti–double-stranded DNA, anti-nRNP, anti-Ro/La, anti-Sm, and anti-Jo-1 antibodies. The patient was treated with prednisolone 7.5 mg daily, nifedipine 15 mg daily, valsartan 80 mg daily, manidipine 20 mg daily, omeprazole 20 mg daily, and beraprost 80 mg daily. One year later, numerous asymptomatic flesh-colored papules and nodules developed on the neck, chest, abdomen, and back. There was no history of trauma or surgery at any of the affected sites.
On further investigation, anti–hepatitis C virus (HCV) antibodies were identified and confirmed by HCV ribonucleic acid polymerase chain reaction at the same time that the diagnosis of SSc was established. Hepatitis C virus genotype 3a was noted, and the patient’s viral load was 378,000 IU/mL. Therefore, a diagnosis of chronic HCV infection was established. The patient was initially unable to receive medical treatment due to lack of finances. A year and a half following the diagnosis of HCV infection, with worsening liver function tests and increasing viral load (1,369,113 IU/mL), the patient began therapy with peginterferon alfa-2b 80 mg weekly and ribavirin 800 mg daily. However, the medications were discontinued after 2 months when she developed severe hemolytic anemia related to ribavirin.
On physical examination, the patient was noted to have a masklike facies with a pinched nose and constricted opening of the mouth. Her skin was tightened and stiff extending from the fingers to the proximal extremities. Numerous well-circumscribed, flesh-colored, firm papules and nodules ranging from 2 to 20 mm in diameter were present on the neck (Figure 1), chest, abdomen (Figure 2), and back.
|
|
|
Two 4-mm punch biopsy samples obtained from a papule on the neck and a nodule on the abdomen revealed homogenized collagen bundles with scattered plump fibroblasts in the lower reticular dermis. Clinicopathologic correlation of the biopsy findings with the cutaneous examination resulted in a diagnosis of nodular scleroderma (Figures 3 and 4).
|
The patient began treatment with intralesional injections of triamcinolone 5 to 10 mg/mL for nodules as well as an ultrapotent corticosteroid cream, clobetasol propionate 0.05%, for small papules. Injections were performed at 4- to 8-week intervals and resulted in modest clinical improvement.
Comment
Scleroderma may be present only in the skin (morphea) or as a systemic disease (systemic scleroderma). Rarely, cutaneous involvement can exhibit a nodular or hypertrophic morphology, which has been described in the literature as nodular or keloidal scleroderma in a patient with known SSc1-10 and as nodular or keloidal morphea in localized cutaneous scleroderma.3,11-13
Histopathology
The distinction between the terms nodular scleroderma and keloidal scleroderma is not clear, and they are not necessarily interchangeable. To provide clarity, we find it useful to delineate specific histologic findings associated with the diagnoses of keloid, scleroderma, and the uncommon keloid/scleroderma overlap. The histopathologic findings of keloids include a fibrotic dermis and broad dispersed bundles of eosinophilic hyalinized collagen. The histopathologic findings of scleroderma include broad sclerotic bands of collagen throughout the dermis with loss of perieccrine fat. In the overlapping keloid/scleroderma condition, which is a variant of scleroderma, hyalinized collagen fibers and keloidal collagen appear in the same specimen.3,4
To distinguish these conditions, Barzilai et al5 proposed that only cases showing both clinical and histologic characteristics of a keloid should be referred to as keloidal morphea/scleroderma. They further stated that the terms nodular morphea or nodular scleroderma ought to be used only for cases that are indistinguishable histologically from scleroderma. The term morphea is appropriate when only a limited amount of skin disease is present, while scleroderma implies association with systemic disease.5 Likely, there is a histologic continuum in this variant of scleroderma, in which nodular morphea/scleroderma exists at one end and keloidal morphea/scleroderma exists at the other end.5,13
In the case of our patient, papulonodular lesions developed 1 year after the diagnosis of SSc was made, and the histopathologic examination revealed classic findings of scleroderma. As a result, our patient is most appropriately classified as having nodular scleroderma.
Clinical Features
Nodular scleroderma mostly affects young and middle-aged women and is clinically characterized by solitary or multiple firm, long-lasting papules or nodules on the upper trunk and chest, neck, and proximal extremities.1-4,6
Etiology and Pathogenesis
The triggers and cellular mechanisms of nodular scleroderma are unclear. Some authors have implicated matricellular protein and growth factors such as tenascin, connective tissue growth factor, and epidermal growth factor in nodule formation.7,8,11 Yamamoto et al9 cited chemical exposure to a silica-containing abrasive as the cause of nodular scleroderma in a worker.
Possible HCV Association
Some reports have indicated an association between nodular scleroderma and pathogens such as acid-fast bacteria10 and HCV.6 Of note, many extrahepatic conditions have been associated with HCV infection, such as membranoproliferative glomerulonephritis, cutaneous vasculitis, lichen planus, and porphyria cutanea tarda.14
The association of HCV infection with systemic autoimmune disease (SAD) has been described in a number of instances; cryoglobulinemia has most commonly been linked to HCV.15 Although the association between HCV and other SADs is less clear, there is growing interest in a possible relationship between them. To that end, physicians of the HISPAMEC (Hispanoamerican Study Group of Autoimmune Manifestations Associated With Hepatitis C Virus) study group described the clinical and immunologic characteristics of 1020 patients with SAD and associated chronic HCV infection. The 3 most frequent SADs (>90% of cases) were Sjögren syndrome, rheumatoid arthritis, and systemic lupus erythematosus.16 However, the strength of association differs for each SAD based on existing descriptions.16,17 Less commonly, there may be a causal relationship between HCV infection and SSc. It should be noted that most of these data are based on small series and case reports.6,16-19
The role of HCV in the pathogenesis of systemic scleroderma and other autoimmune diseases is unknown. It is also possible that the replication of HCV outside the liver, particularly in mononuclear cells, may suppress immune tolerance in genetically predisposed individuals.20
Conclusion
Nodular scleroderma associated with HCV infection is a rare entity. At present, it cannot be determined whether there is an etiopathologic association between HCV infection and SSc or whether the simultaneous diagnosis may be coincidental. Routine determination of HCV serology in scleroderma patients may help to clarify this issue.
1. Krell JM, Solomon AR, Glavey CM, et al. Nodular scleroderma. J Am Acad Dermatol. 1995;32:343-345.
2. Cannick L 3rd, Douglas G, Crater S, et al. Nodular scleroderma: case report and literature review. J Rheumatol. 2003;30:2500-2502.
3. Rencic A, Brinster NK, Nousari CH. Keloid morphea and nodular scleroderma: two distinct clinical variants of scleroderma? J Cutan Med Surg. 2003;7:20-24.
4. Wriston CC, Rubin AI, Elenitsas R, et al. Nodular scleroderma: a report of 2 cases. Am J Dermatopathol. 2008;30:385-388.
5. Barzilai A, Lyakhovitsky A, Horowitz A, et al. Keloid-like scleroderma. Am J Dermatopathol. 2003;25:327-330.
6. Melani L, Caproni M, Cardinali C, et al. A case of nodular scleroderma. J Dermatol. 2005;32:1028-1031.
7. Mizutani H, Taniguchi H, Sakakura T, et al. Nodular scleroderma: focally increased tenascin expression differing from that in the surrounding scleroderma skin. J Dermatol. 1995;22:267-271.
8. Yamamoto T, Sawada Y, Katayama I, et al. Nodular scleroderma: increased expression of connective tissue growth factor. Dermatology. 2005;211:218-223.
9. Yamamoto T, Furuse Y, Katayama I, et al. Nodular scleroderma in a worker using a silica-containing abrasive. J Dermatol. 1994;21:751-754.
10. Cantwell AR Jr, Rowe L, Kelso DW. Nodular scleroderma and pleomorphic acid-fast bacteria. Arch Dermatol. 1980;116:1283-1290.
11. Yamamoto T, Sakashita S, Sawada Y, et al. Possible role of epidermal growth factor in the lesional skin of nodular morphea. Acta Derm Venereol. 1998;78:312-313.
12. Jain K, Dayal S, Jain VK, et al. Blaschko linear nodular morphea with dermal mucinosis. Arch Dermatol. 2007;143:953-955.
13. Kauer F, Simon JC, Sticherling M. Nodular morphea. Dermatology. 2009;218:63-66.
14. Gumber SC, Chopra S. Hepatitis C: a multifaceted disease. review of extrahepatic manifestations. Ann Intern Med. 1995;123:615-620.
15. Ferri C, Greco F, Longombardo G, et al. Antibodies to hepatitis C virus in patients with mixed cryoglobulinemia. Arthritis Rheum. 1991;34:1606-1610.
16. Ramos-Casals M, Munoz S, Medina F, et al. Systemic autoimmune diseases in patients with hepatitis C virus infection: characterization of 1020 cases (The HISPAMEC Registry). J Rheumatol. 2009;36:1442-1448.
17. Ramos-Casals M, Jara LJ, Medina F, et al. Systemic autoimmune diseases co-existing with chronic hepatitis C virus infection (the HISPAMEC Registry): patterns of clinical and immunological expression in 180 cases. J Intern Med. 2005;257:549-557.
18. Abu-Shakra M, Sukenik S, Buskila D. Systemic sclerosis: another rheumatic disease associated with hepatitis C virus infection. Clin Rheumatol. 2000;19:378-380.
19. Yamamoto M, Yamamoto T, Tsuboi R. Discoid lupus erythematosus in a patient with scleroderma and hepatitis C virus infection. Rheumatol Int. 2010;30:969-971.
20. Abu-Shakra M, Shoenfeld Y. Chronic infections and autoimmunity. Immunol Ser. 1992;55:285-313.
1. Krell JM, Solomon AR, Glavey CM, et al. Nodular scleroderma. J Am Acad Dermatol. 1995;32:343-345.
2. Cannick L 3rd, Douglas G, Crater S, et al. Nodular scleroderma: case report and literature review. J Rheumatol. 2003;30:2500-2502.
3. Rencic A, Brinster NK, Nousari CH. Keloid morphea and nodular scleroderma: two distinct clinical variants of scleroderma? J Cutan Med Surg. 2003;7:20-24.
4. Wriston CC, Rubin AI, Elenitsas R, et al. Nodular scleroderma: a report of 2 cases. Am J Dermatopathol. 2008;30:385-388.
5. Barzilai A, Lyakhovitsky A, Horowitz A, et al. Keloid-like scleroderma. Am J Dermatopathol. 2003;25:327-330.
6. Melani L, Caproni M, Cardinali C, et al. A case of nodular scleroderma. J Dermatol. 2005;32:1028-1031.
7. Mizutani H, Taniguchi H, Sakakura T, et al. Nodular scleroderma: focally increased tenascin expression differing from that in the surrounding scleroderma skin. J Dermatol. 1995;22:267-271.
8. Yamamoto T, Sawada Y, Katayama I, et al. Nodular scleroderma: increased expression of connective tissue growth factor. Dermatology. 2005;211:218-223.
9. Yamamoto T, Furuse Y, Katayama I, et al. Nodular scleroderma in a worker using a silica-containing abrasive. J Dermatol. 1994;21:751-754.
10. Cantwell AR Jr, Rowe L, Kelso DW. Nodular scleroderma and pleomorphic acid-fast bacteria. Arch Dermatol. 1980;116:1283-1290.
11. Yamamoto T, Sakashita S, Sawada Y, et al. Possible role of epidermal growth factor in the lesional skin of nodular morphea. Acta Derm Venereol. 1998;78:312-313.
12. Jain K, Dayal S, Jain VK, et al. Blaschko linear nodular morphea with dermal mucinosis. Arch Dermatol. 2007;143:953-955.
13. Kauer F, Simon JC, Sticherling M. Nodular morphea. Dermatology. 2009;218:63-66.
14. Gumber SC, Chopra S. Hepatitis C: a multifaceted disease. review of extrahepatic manifestations. Ann Intern Med. 1995;123:615-620.
15. Ferri C, Greco F, Longombardo G, et al. Antibodies to hepatitis C virus in patients with mixed cryoglobulinemia. Arthritis Rheum. 1991;34:1606-1610.
16. Ramos-Casals M, Munoz S, Medina F, et al. Systemic autoimmune diseases in patients with hepatitis C virus infection: characterization of 1020 cases (The HISPAMEC Registry). J Rheumatol. 2009;36:1442-1448.
17. Ramos-Casals M, Jara LJ, Medina F, et al. Systemic autoimmune diseases co-existing with chronic hepatitis C virus infection (the HISPAMEC Registry): patterns of clinical and immunological expression in 180 cases. J Intern Med. 2005;257:549-557.
18. Abu-Shakra M, Sukenik S, Buskila D. Systemic sclerosis: another rheumatic disease associated with hepatitis C virus infection. Clin Rheumatol. 2000;19:378-380.
19. Yamamoto M, Yamamoto T, Tsuboi R. Discoid lupus erythematosus in a patient with scleroderma and hepatitis C virus infection. Rheumatol Int. 2010;30:969-971.
20. Abu-Shakra M, Shoenfeld Y. Chronic infections and autoimmunity. Immunol Ser. 1992;55:285-313.
Practice Points
- Nodular scleroderma is a rare form of cutaneous scleroderma that can occur in association with systemic scleroderma or localized morphea.
- The clinical features are characterized by solitary or multiple, firm, long-lasting papules or nodules on the neck, upper trunk, and proximal extremities.
- The pathogenesis is still unclear. Some reports have suggested that matricellular protein and growth factor, acid-fast bacteria, organic solvents, or the hepatitis C virus may be involved.
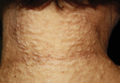
Sweet Syndrome Presenting With an Unusual Morphology
To the Editor:
Sweet syndrome is a neutrophilic dermatosis that typically presents as an acute onset of multiple, painful, sharply demarcated, small (measuring a few centimeters), raised, red plaques that occasionally present with superimposed pustules, vesicles, or bullae on the face, neck, upper chest, back, and extremities. Patients are often febrile and may have mucosal and systemic involvement.1 Although 71% of cases are idiopathic, others are associated with malignancy; autoimmune disorders; infections; pregnancy; and rarely medications, especially all-trans-retinoic acid, granulocyte colony-stimulating factor, vaccines, and antibiotics.1,2 We present a case of Sweet syndrome induced by trimethoprim-sulfamethoxazole (TMP-SMX) with an unusual clinical presentation.
A 71-year-old man with a medical history of nonmelanoma skin cancer initiated a course of TMP-SMX for a wound infection of the lower leg following Mohs micrographic surgery. Eight days later, he developed a painful eruption preceded by 1 day of fever, malaise, blurry vision, and myalgia. Trimethoprim-sulfamethoxazole was discontinued. Physical examination revealed ill-defined, discrete and coalescing, 1- to 6-mm edematous erythematous papules studded with pustules involving the scalp, face, neck, back (Figure 1), and extremities. The patient also had conjunctival erythema and an elevated temperature (38.3°C). Laboratory workup revealed an elevated white blood cell count (11,300/mL [reference range, 4500–11,000/µL]), blood urea nitrogen level (33 mg/µL [reference range, 7–20 mg/dL]), and creatinine level (2.00 mg/dL [reference range, 0.6–1.2 mg/dL]). Liver function tests were normal. A biopsy demonstrated marked papillary dermal edema with a dense, bandlike, superficial dermal neutrophilic infiltrate (Figure 2). A few neutrophils were present in the epidermis with formation of minute intraepidermal pustules. The patient was diagnosed with Sweet syndrome and treated with intravenous methylprednisolone 60 mg 3 times daily (1.5 mg/kg body weight) tapered over 17 days and triamcinolone acetonide ointment 0.1% twice daily. His fever and leukocytosis resolved within 1 day and the eruption improved within 2 days with residual desquamation that cleared by 3 weeks.
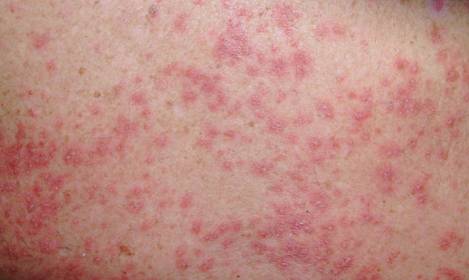
| 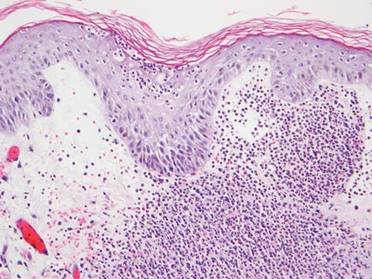
|
Morphologically, our case resembled acute generalized exanthematous pustulosis (AGEP), which presents with edematous erythema studded with pustules.3 Although fever and leukocytosis are often present in both AGEP and Sweet syndrome, our patient’s pain, malaise, and myalgia favored Sweet syndrome, as did his conjunctivitis, which is unusual in AGEP.1,3 Histologically, our case was characteristic for Sweet syndrome, which presents with marked papillary dermal edema and a dense neutrophilic dermal infiltrate with neutrophil exocytosis and spongiform pustules in 21% of cases.1 Acute generalized exanthematous pustulosis, characterized by spongiform pustules and a perivascular neutrophilic infiltrate, does not exhibit the dense dermal neutrophilic infiltrate of Sweet syndrome.3 Mecca et al4 also reported a case displaying overlapping features of Sweet syndrome and AGEP. The patient presented with photodistributed papules and pinpoint pustules on an erythematous base favoring a diagnosis of AGEP with histologic findings compatible with Sweet syndrome. The authors suggested a clinicopathologic continuum may exist among drug-related neutrophilic dermatoses.4
In conclusion, we present a case of TMP-SMX–induced Sweet syndrome that morphologically resembled AGEP. It is important to recognize that Sweet syndrome may present in this unusual manner, as it may have notable internal involvement, and responds rapidly to systemic steroids, whereas AGEP has minimal systemic involvement and clears spontaneously.
1. von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556.
2. Kluger N, Marque M, Stoebner PE, et al. Possible drug-induced Sweet’s syndrome due to trimethoprim-sulfamethoxazole. Acta Derm Venereol. 2008;88:637-638.
3. Sidoroff A, Halevy S, Bavinck JN, et al. Acute generalized exanthematous pustulosis (AGEP)—a clinical reaction pattern. J Cutan Pathol. 2001;28:113-119.
4. Mecca P, Tobin E, Andrew Carlson J. Photo-distributed neutrophilic drug eruption and adult respiratory distress syndrome associated with antidepressant therapy. J Cutan Pathol. 2004;31:189-194.
To the Editor:
Sweet syndrome is a neutrophilic dermatosis that typically presents as an acute onset of multiple, painful, sharply demarcated, small (measuring a few centimeters), raised, red plaques that occasionally present with superimposed pustules, vesicles, or bullae on the face, neck, upper chest, back, and extremities. Patients are often febrile and may have mucosal and systemic involvement.1 Although 71% of cases are idiopathic, others are associated with malignancy; autoimmune disorders; infections; pregnancy; and rarely medications, especially all-trans-retinoic acid, granulocyte colony-stimulating factor, vaccines, and antibiotics.1,2 We present a case of Sweet syndrome induced by trimethoprim-sulfamethoxazole (TMP-SMX) with an unusual clinical presentation.
A 71-year-old man with a medical history of nonmelanoma skin cancer initiated a course of TMP-SMX for a wound infection of the lower leg following Mohs micrographic surgery. Eight days later, he developed a painful eruption preceded by 1 day of fever, malaise, blurry vision, and myalgia. Trimethoprim-sulfamethoxazole was discontinued. Physical examination revealed ill-defined, discrete and coalescing, 1- to 6-mm edematous erythematous papules studded with pustules involving the scalp, face, neck, back (Figure 1), and extremities. The patient also had conjunctival erythema and an elevated temperature (38.3°C). Laboratory workup revealed an elevated white blood cell count (11,300/mL [reference range, 4500–11,000/µL]), blood urea nitrogen level (33 mg/µL [reference range, 7–20 mg/dL]), and creatinine level (2.00 mg/dL [reference range, 0.6–1.2 mg/dL]). Liver function tests were normal. A biopsy demonstrated marked papillary dermal edema with a dense, bandlike, superficial dermal neutrophilic infiltrate (Figure 2). A few neutrophils were present in the epidermis with formation of minute intraepidermal pustules. The patient was diagnosed with Sweet syndrome and treated with intravenous methylprednisolone 60 mg 3 times daily (1.5 mg/kg body weight) tapered over 17 days and triamcinolone acetonide ointment 0.1% twice daily. His fever and leukocytosis resolved within 1 day and the eruption improved within 2 days with residual desquamation that cleared by 3 weeks.
|
|
|
Morphologically, our case resembled acute generalized exanthematous pustulosis (AGEP), which presents with edematous erythema studded with pustules.3 Although fever and leukocytosis are often present in both AGEP and Sweet syndrome, our patient’s pain, malaise, and myalgia favored Sweet syndrome, as did his conjunctivitis, which is unusual in AGEP.1,3 Histologically, our case was characteristic for Sweet syndrome, which presents with marked papillary dermal edema and a dense neutrophilic dermal infiltrate with neutrophil exocytosis and spongiform pustules in 21% of cases.1 Acute generalized exanthematous pustulosis, characterized by spongiform pustules and a perivascular neutrophilic infiltrate, does not exhibit the dense dermal neutrophilic infiltrate of Sweet syndrome.3 Mecca et al4 also reported a case displaying overlapping features of Sweet syndrome and AGEP. The patient presented with photodistributed papules and pinpoint pustules on an erythematous base favoring a diagnosis of AGEP with histologic findings compatible with Sweet syndrome. The authors suggested a clinicopathologic continuum may exist among drug-related neutrophilic dermatoses.4
In conclusion, we present a case of TMP-SMX–induced Sweet syndrome that morphologically resembled AGEP. It is important to recognize that Sweet syndrome may present in this unusual manner, as it may have notable internal involvement, and responds rapidly to systemic steroids, whereas AGEP has minimal systemic involvement and clears spontaneously.
To the Editor:
Sweet syndrome is a neutrophilic dermatosis that typically presents as an acute onset of multiple, painful, sharply demarcated, small (measuring a few centimeters), raised, red plaques that occasionally present with superimposed pustules, vesicles, or bullae on the face, neck, upper chest, back, and extremities. Patients are often febrile and may have mucosal and systemic involvement.1 Although 71% of cases are idiopathic, others are associated with malignancy; autoimmune disorders; infections; pregnancy; and rarely medications, especially all-trans-retinoic acid, granulocyte colony-stimulating factor, vaccines, and antibiotics.1,2 We present a case of Sweet syndrome induced by trimethoprim-sulfamethoxazole (TMP-SMX) with an unusual clinical presentation.
A 71-year-old man with a medical history of nonmelanoma skin cancer initiated a course of TMP-SMX for a wound infection of the lower leg following Mohs micrographic surgery. Eight days later, he developed a painful eruption preceded by 1 day of fever, malaise, blurry vision, and myalgia. Trimethoprim-sulfamethoxazole was discontinued. Physical examination revealed ill-defined, discrete and coalescing, 1- to 6-mm edematous erythematous papules studded with pustules involving the scalp, face, neck, back (Figure 1), and extremities. The patient also had conjunctival erythema and an elevated temperature (38.3°C). Laboratory workup revealed an elevated white blood cell count (11,300/mL [reference range, 4500–11,000/µL]), blood urea nitrogen level (33 mg/µL [reference range, 7–20 mg/dL]), and creatinine level (2.00 mg/dL [reference range, 0.6–1.2 mg/dL]). Liver function tests were normal. A biopsy demonstrated marked papillary dermal edema with a dense, bandlike, superficial dermal neutrophilic infiltrate (Figure 2). A few neutrophils were present in the epidermis with formation of minute intraepidermal pustules. The patient was diagnosed with Sweet syndrome and treated with intravenous methylprednisolone 60 mg 3 times daily (1.5 mg/kg body weight) tapered over 17 days and triamcinolone acetonide ointment 0.1% twice daily. His fever and leukocytosis resolved within 1 day and the eruption improved within 2 days with residual desquamation that cleared by 3 weeks.
|
|
|
Morphologically, our case resembled acute generalized exanthematous pustulosis (AGEP), which presents with edematous erythema studded with pustules.3 Although fever and leukocytosis are often present in both AGEP and Sweet syndrome, our patient’s pain, malaise, and myalgia favored Sweet syndrome, as did his conjunctivitis, which is unusual in AGEP.1,3 Histologically, our case was characteristic for Sweet syndrome, which presents with marked papillary dermal edema and a dense neutrophilic dermal infiltrate with neutrophil exocytosis and spongiform pustules in 21% of cases.1 Acute generalized exanthematous pustulosis, characterized by spongiform pustules and a perivascular neutrophilic infiltrate, does not exhibit the dense dermal neutrophilic infiltrate of Sweet syndrome.3 Mecca et al4 also reported a case displaying overlapping features of Sweet syndrome and AGEP. The patient presented with photodistributed papules and pinpoint pustules on an erythematous base favoring a diagnosis of AGEP with histologic findings compatible with Sweet syndrome. The authors suggested a clinicopathologic continuum may exist among drug-related neutrophilic dermatoses.4
In conclusion, we present a case of TMP-SMX–induced Sweet syndrome that morphologically resembled AGEP. It is important to recognize that Sweet syndrome may present in this unusual manner, as it may have notable internal involvement, and responds rapidly to systemic steroids, whereas AGEP has minimal systemic involvement and clears spontaneously.
1. von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556.
2. Kluger N, Marque M, Stoebner PE, et al. Possible drug-induced Sweet’s syndrome due to trimethoprim-sulfamethoxazole. Acta Derm Venereol. 2008;88:637-638.
3. Sidoroff A, Halevy S, Bavinck JN, et al. Acute generalized exanthematous pustulosis (AGEP)—a clinical reaction pattern. J Cutan Pathol. 2001;28:113-119.
4. Mecca P, Tobin E, Andrew Carlson J. Photo-distributed neutrophilic drug eruption and adult respiratory distress syndrome associated with antidepressant therapy. J Cutan Pathol. 2004;31:189-194.
1. von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556.
2. Kluger N, Marque M, Stoebner PE, et al. Possible drug-induced Sweet’s syndrome due to trimethoprim-sulfamethoxazole. Acta Derm Venereol. 2008;88:637-638.
3. Sidoroff A, Halevy S, Bavinck JN, et al. Acute generalized exanthematous pustulosis (AGEP)—a clinical reaction pattern. J Cutan Pathol. 2001;28:113-119.
4. Mecca P, Tobin E, Andrew Carlson J. Photo-distributed neutrophilic drug eruption and adult respiratory distress syndrome associated with antidepressant therapy. J Cutan Pathol. 2004;31:189-194.
Bilateral Auricular Swelling: Marginal Zone Lymphoma With Cutaneous Involvement
To the Editor:
A 66-year-old man with hypertension presented with asymptomatic, edematous, swelling plaques without local heat on the bilateral auricles of 2 months’ duration (Figure 1). Topical corticosteroids and multiple oral antihistamines were prescribed without any improvement. He reported no history of trauma or use of any topical agents except topical corticosteroids. There was no sensory defect or numbness.
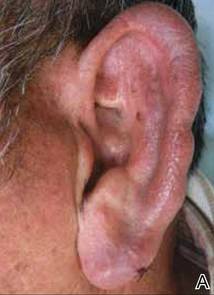
|
Laboratory results revealed leukocytosis with a white blood cell count of 13,900/mL (reference range, 3500–9900/μL) and 55.8% lymphocytes (reference range, 20%–40%). Biochemistry and tumor markers data were normal. No palpable neck lymphadenopathy was found. A skin biopsy was performed on the left earlobe showing a grenz zone between the tumor infiltrate and epidermis and a dense neoplastic lymphoid proliferation with a bottom-heavy configuration in the reticular dermis (Figure 2A). These lymphoid cells were small to medium sized with indented and irregular nuclei and abundant pale cytoplasm (Figure 2B). Immunohistochemical staining showed positivity for CD20 and BCL2; stains for CD5, CD10, CD23, and BCL6 were negative. Positron emission tomography scan showed bilateral auricular infiltration and bilateral neck lymph node involvement. A bone marrow biopsy was performed during hospitalization and was positive for lymphoma involvement. On the basis of histologic and immunohistochemical findings, a diagnosis of malignant nodal marginal zone lymphoma (MZL) with cutaneous involvement was made. The patient underwent chemotherapy with R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone).
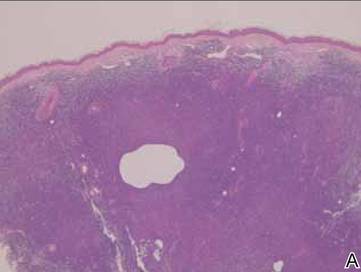
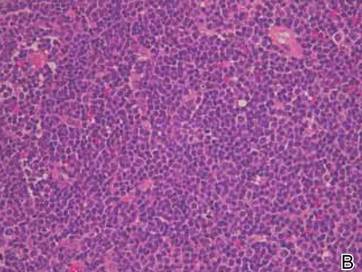
Figure 2. A skin biopsy showed basket weave hyperkeratosis, a grenz zone between the tumor infiltrate and epidermis, and dense lymphoid proliferation with a bottom-heavy configuration in the reticular dermis (A)(H&E, original magnification ×40). Small- to medium-sized lymphoid cells with indented and irregular nuclei and abundant pale cytoplasm were seen (B)(H&E, original magnification ×400). |
Cutaneous MZL may be a primary cutaneous condition or the result of secondary involvement from noncutaneous MZL. The histologic and immunophenotypic changes in skin lesions from secondary cutaneous MZL may be indistinguishable from those in primary cutaneous MZL. Primary cutaneous MZL may be seen in younger patients and favors the trunk and extremities, whereas MZL secondarily involves the skin, favors the head and neck regions, and is limited to older patients.1 Histologic aspects include a dense, nodular, deep-seated infiltrate containing various proportions of small cells displaying a centrocytelike, plasmacytoid, or monocytoid appearance.2 Chronic antigen stimulation is a key player in the pathogenesis and involves deregulation of the nuclear factor κb pathway. While Helicobacter pylori and Epstein-Barr virus do not seem to be implicated in primary cutaneous MZL, the role of Borrelia burgdorferi is still a matter of debate with discordant results.3,4
Treatment may include excision, curative or adjunctive radiotherapy, topical or intralesional corticosteroids, interferon or intralesional rituximab, or systemic therapies such as chemotherapy and/or intravenous rituximab depending on disease stage and tumor burden.5
Cutaneous presentation of MZL as bilateral auricular swelling is unique. Because there may be considerable overlap in the clinical presentations for patients with primary and secondary cutaneous MZL, it is imperative to perform a systemic evaluation. Clinicians should be aware of possible hematologic malignancy in patients with unexplained and refractory bilateral auricular swelling.
1. Gerami P, Wickless SC, Querfeld C, et al. Cutaneous involvement with marginal zone lymphoma [published online ahead of print May 11, 2010]. J Am Acad Dermatol. 2010;63:142-145.
2. de la Fouchardière A, Balme B, Chouvet B, et al. Primary cutaneous marginal zone B-cell lymphoma: a report of 9 cases. J Am Acad Dermatol. 1999;41(2, pt 1):181-188.
3. Dalle S, Thomas L, Balme B, et al. Primary cutaneous marginal zone lymphoma [published online ahead of print October 12, 2009]. Crit Rev Oncol Hematol. 2010;74:156-162.
4. Li C, Inagaki H, Kuo TT, et al. Primary cutaneous marginal zone B-cell lymphoma: a molecular and clinicopathologic study of 24 Asian cases. Am J Surg Pathol. 2003;27:1061-1069.
5. Grange F, D’Incan M, Ortonne N, et al. Management of cutaneous B-cell lymphoma: recommendations of the French cutaneous lymphoma study group [published online ahead of print June 18, 2010]. Ann Dermatol Venereol. 2010;137:523-531.
To the Editor:
A 66-year-old man with hypertension presented with asymptomatic, edematous, swelling plaques without local heat on the bilateral auricles of 2 months’ duration (Figure 1). Topical corticosteroids and multiple oral antihistamines were prescribed without any improvement. He reported no history of trauma or use of any topical agents except topical corticosteroids. There was no sensory defect or numbness.
|
|
Laboratory results revealed leukocytosis with a white blood cell count of 13,900/mL (reference range, 3500–9900/μL) and 55.8% lymphocytes (reference range, 20%–40%). Biochemistry and tumor markers data were normal. No palpable neck lymphadenopathy was found. A skin biopsy was performed on the left earlobe showing a grenz zone between the tumor infiltrate and epidermis and a dense neoplastic lymphoid proliferation with a bottom-heavy configuration in the reticular dermis (Figure 2A). These lymphoid cells were small to medium sized with indented and irregular nuclei and abundant pale cytoplasm (Figure 2B). Immunohistochemical staining showed positivity for CD20 and BCL2; stains for CD5, CD10, CD23, and BCL6 were negative. Positron emission tomography scan showed bilateral auricular infiltration and bilateral neck lymph node involvement. A bone marrow biopsy was performed during hospitalization and was positive for lymphoma involvement. On the basis of histologic and immunohistochemical findings, a diagnosis of malignant nodal marginal zone lymphoma (MZL) with cutaneous involvement was made. The patient underwent chemotherapy with R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone).
|
Figure 2. A skin biopsy showed basket weave hyperkeratosis, a grenz zone between the tumor infiltrate and epidermis, and dense lymphoid proliferation with a bottom-heavy configuration in the reticular dermis (A)(H&E, original magnification ×40). Small- to medium-sized lymphoid cells with indented and irregular nuclei and abundant pale cytoplasm were seen (B)(H&E, original magnification ×400). |
Cutaneous MZL may be a primary cutaneous condition or the result of secondary involvement from noncutaneous MZL. The histologic and immunophenotypic changes in skin lesions from secondary cutaneous MZL may be indistinguishable from those in primary cutaneous MZL. Primary cutaneous MZL may be seen in younger patients and favors the trunk and extremities, whereas MZL secondarily involves the skin, favors the head and neck regions, and is limited to older patients.1 Histologic aspects include a dense, nodular, deep-seated infiltrate containing various proportions of small cells displaying a centrocytelike, plasmacytoid, or monocytoid appearance.2 Chronic antigen stimulation is a key player in the pathogenesis and involves deregulation of the nuclear factor κb pathway. While Helicobacter pylori and Epstein-Barr virus do not seem to be implicated in primary cutaneous MZL, the role of Borrelia burgdorferi is still a matter of debate with discordant results.3,4
Treatment may include excision, curative or adjunctive radiotherapy, topical or intralesional corticosteroids, interferon or intralesional rituximab, or systemic therapies such as chemotherapy and/or intravenous rituximab depending on disease stage and tumor burden.5
Cutaneous presentation of MZL as bilateral auricular swelling is unique. Because there may be considerable overlap in the clinical presentations for patients with primary and secondary cutaneous MZL, it is imperative to perform a systemic evaluation. Clinicians should be aware of possible hematologic malignancy in patients with unexplained and refractory bilateral auricular swelling.
To the Editor:
A 66-year-old man with hypertension presented with asymptomatic, edematous, swelling plaques without local heat on the bilateral auricles of 2 months’ duration (Figure 1). Topical corticosteroids and multiple oral antihistamines were prescribed without any improvement. He reported no history of trauma or use of any topical agents except topical corticosteroids. There was no sensory defect or numbness.
|
|
Laboratory results revealed leukocytosis with a white blood cell count of 13,900/mL (reference range, 3500–9900/μL) and 55.8% lymphocytes (reference range, 20%–40%). Biochemistry and tumor markers data were normal. No palpable neck lymphadenopathy was found. A skin biopsy was performed on the left earlobe showing a grenz zone between the tumor infiltrate and epidermis and a dense neoplastic lymphoid proliferation with a bottom-heavy configuration in the reticular dermis (Figure 2A). These lymphoid cells were small to medium sized with indented and irregular nuclei and abundant pale cytoplasm (Figure 2B). Immunohistochemical staining showed positivity for CD20 and BCL2; stains for CD5, CD10, CD23, and BCL6 were negative. Positron emission tomography scan showed bilateral auricular infiltration and bilateral neck lymph node involvement. A bone marrow biopsy was performed during hospitalization and was positive for lymphoma involvement. On the basis of histologic and immunohistochemical findings, a diagnosis of malignant nodal marginal zone lymphoma (MZL) with cutaneous involvement was made. The patient underwent chemotherapy with R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone).
|
Figure 2. A skin biopsy showed basket weave hyperkeratosis, a grenz zone between the tumor infiltrate and epidermis, and dense lymphoid proliferation with a bottom-heavy configuration in the reticular dermis (A)(H&E, original magnification ×40). Small- to medium-sized lymphoid cells with indented and irregular nuclei and abundant pale cytoplasm were seen (B)(H&E, original magnification ×400). |
Cutaneous MZL may be a primary cutaneous condition or the result of secondary involvement from noncutaneous MZL. The histologic and immunophenotypic changes in skin lesions from secondary cutaneous MZL may be indistinguishable from those in primary cutaneous MZL. Primary cutaneous MZL may be seen in younger patients and favors the trunk and extremities, whereas MZL secondarily involves the skin, favors the head and neck regions, and is limited to older patients.1 Histologic aspects include a dense, nodular, deep-seated infiltrate containing various proportions of small cells displaying a centrocytelike, plasmacytoid, or monocytoid appearance.2 Chronic antigen stimulation is a key player in the pathogenesis and involves deregulation of the nuclear factor κb pathway. While Helicobacter pylori and Epstein-Barr virus do not seem to be implicated in primary cutaneous MZL, the role of Borrelia burgdorferi is still a matter of debate with discordant results.3,4
Treatment may include excision, curative or adjunctive radiotherapy, topical or intralesional corticosteroids, interferon or intralesional rituximab, or systemic therapies such as chemotherapy and/or intravenous rituximab depending on disease stage and tumor burden.5
Cutaneous presentation of MZL as bilateral auricular swelling is unique. Because there may be considerable overlap in the clinical presentations for patients with primary and secondary cutaneous MZL, it is imperative to perform a systemic evaluation. Clinicians should be aware of possible hematologic malignancy in patients with unexplained and refractory bilateral auricular swelling.
1. Gerami P, Wickless SC, Querfeld C, et al. Cutaneous involvement with marginal zone lymphoma [published online ahead of print May 11, 2010]. J Am Acad Dermatol. 2010;63:142-145.
2. de la Fouchardière A, Balme B, Chouvet B, et al. Primary cutaneous marginal zone B-cell lymphoma: a report of 9 cases. J Am Acad Dermatol. 1999;41(2, pt 1):181-188.
3. Dalle S, Thomas L, Balme B, et al. Primary cutaneous marginal zone lymphoma [published online ahead of print October 12, 2009]. Crit Rev Oncol Hematol. 2010;74:156-162.
4. Li C, Inagaki H, Kuo TT, et al. Primary cutaneous marginal zone B-cell lymphoma: a molecular and clinicopathologic study of 24 Asian cases. Am J Surg Pathol. 2003;27:1061-1069.
5. Grange F, D’Incan M, Ortonne N, et al. Management of cutaneous B-cell lymphoma: recommendations of the French cutaneous lymphoma study group [published online ahead of print June 18, 2010]. Ann Dermatol Venereol. 2010;137:523-531.
1. Gerami P, Wickless SC, Querfeld C, et al. Cutaneous involvement with marginal zone lymphoma [published online ahead of print May 11, 2010]. J Am Acad Dermatol. 2010;63:142-145.
2. de la Fouchardière A, Balme B, Chouvet B, et al. Primary cutaneous marginal zone B-cell lymphoma: a report of 9 cases. J Am Acad Dermatol. 1999;41(2, pt 1):181-188.
3. Dalle S, Thomas L, Balme B, et al. Primary cutaneous marginal zone lymphoma [published online ahead of print October 12, 2009]. Crit Rev Oncol Hematol. 2010;74:156-162.
4. Li C, Inagaki H, Kuo TT, et al. Primary cutaneous marginal zone B-cell lymphoma: a molecular and clinicopathologic study of 24 Asian cases. Am J Surg Pathol. 2003;27:1061-1069.
5. Grange F, D’Incan M, Ortonne N, et al. Management of cutaneous B-cell lymphoma: recommendations of the French cutaneous lymphoma study group [published online ahead of print June 18, 2010]. Ann Dermatol Venereol. 2010;137:523-531.

High-Yield Biopsy Technique for Subepidermal Blisters
The traditional approach for confirming the diagnosis of subepidermal blistering diseases such as bullous pemphigoid (BP), epidermolysis bullosa acquisita (EBA), dermatitis herpetiformis (DH), linear IgA bullous dermatosis (LABD) requires 2 punch biopsies: one from perilesional skin for direct immunofluorescence (DIF) and one from lesional skin for light microscopy using hematoxylin and eosin (H&E) stain.1-4 These conditions are distinguished by a combination of features appreciated on H&E-stained sections, DIF, and indirect immunofluorescence for a subset of disorders. Additional information may be provided by DIF or indirect immunofluorescence utilizing the salt-split skin technique to differentiate BP (in which linear IgG deposition is identified by immunofluorescence on the roof of salt-split skin) from EBA and antiepiligrin cicatricial BP (in which linear IgG deposition is identified by immunofluorescence along the floor of the salt-split skin), which is more rare.4 One bisected punch biopsy of a subepidermal blister yields salt-split skin–like information through standard DIF and supersedes the need for the more cumbersome salt-split skin technique.
Serologic tests for the presence of circulating antibodies to BP180 and BP230 represent an emerging technology that can confirm the diagnosis of BP, but it has been difficult to identify clinically useful autoantibodies to confirm diagnoses of EBA and LABD.5-7 Serologic tests for tissue transglutaminase IgA antibodies may be useful in the diagnosis of DH.8 We present a cost-effective approach to biopsy in the diagnosis of subepidermal blistering diseases that provides the necessary diagnostic information to distinguish relevant disease processes.
Subepidermal Blistering Diseases
Bullous pemphigoid commonly presents with widespread tense bullae of varying sizes on an erythematous base or on otherwise normal skin.9 Some cases of BP present not with bullae but with pruritic, urticarial, plaquelike, or papular lesions. Bullous pemphigoid commonly involves flexural surfaces and the trunk but can appear anywhere on the skin. The induction of blisters by shearing with mechanical pressure on perilesional skin (Nikolsky sign) is not characteristically present in BP as it is in pemphigus vulgaris.10 Epidermolysis bullosa acquisita can mimic BP in the development of widespread tense bullae, but blisters typically appear on areas of the skin that are prone to trauma (eg, toes, knees, elbows, hands). Crusted erosions, scarring, and milia also are clinical manifestations of EBA.11 Dermatitis herpetiformis presents with grouped vesicles, papulovesicles, plaques, and excoriations that are symmetrically distributed on extensor surfaces of the skin but also can occur on the buttocks, scalp, and other areas of the skin.12,13 Although it may mimic both BP and DH, LABD frequently is less pruritic than these other conditions.14,15 Linear IgA bullous dermatosis also demonstrates the characteristic finding of multiple bullae that form concentrically around a crusted area of skin. This physical finding is known as a string of pearls. Linear IgA bullous dermatosis typically occurs in childhood and may resolve without treatment in months to years.16
Traditional Biopsy Approach
A review of several articles from the literature and multiple dermatology and dermatopathology textbooks revealed uniform recommendations for biopsy of subepidermal blistering conditions that manifest as tense blisters.1-4,9-23 A biopsy of early lesional skin or of a blister for light microscopy with H&E stain and biopsy of perilesional skin for DIF is recommended.1-4,9-23 Three review articles specifically suggested biopsy of “perilesional skin” for DIF.1-3 The majority of textbooks we reviewed also suggested that perilesional skin, or skin adjacent to a zone of erythema in the case of DH, should be sampled for DIF to assist in the diagnosis of BP, EBA, DH, and LABD.4,9-21 Biopsy of adjacent or nonlesional skin or skin around the lesion for DIF also was recommended by other textbooks for diagnosis of subepidermal blistering diseases.22,23 Perilesional skin is chosen because it is critical that the epidermis be included for adequate immunofluorescence studies.5,20 Biopsy of healed and crusted lesions should be avoided.24
Recommended Alternative Approach
A single punch biopsy produces the best possible specimen for H&E and DIF if it is obtained via one of 2 methods.
The first method involves choosing a small, 1- to 2-mm tense blister.25 Use an 8-mm punch centered on the blister that includes at least 3 mm of circumferential perilesional skin (Figures 1 and 2).20 Holding the specimen with forceps, use a no. 15 scalpel blade to bisect the blister with a sawing motion. Place half of the specimen in formalin for H&E staining and the other half in Zeus (or Michel) medium for DIF (Figure 3).
The second method is to choose any intact tense blister20,24 and, utilizing a surgical marker, draw a line from the roof of the blister onto the adjacent perilesional skin (Figure 1). After blotting with an alcohol pad so as not to remove the mark, anesthetize the site with lidocaine 1% with epinephrine,24 then take an 8-mm punch biopsy encompassing 75% perilesional skin and 25% of the blister centered on the line (Figure 2). After separating the punch specimen from the subcutaneous tissue with surgical scissors, hold the tissue with forceps and bisect the specimen with a no. 15 scalpel blade. Use a sawing motion along the line drawn in the prior steps (Figure 3). Submit half of the biopsy for H&E staining in formalin and the other half for DIF in Zeus medium.
Advantages
This approach offers several advantages. First, the technique requires only 1 invasive procedure, not 2 separate biopsies, so that the procedure can be done quickly and efficiently with the least morbidity and scarring. Secondly, because the patient is billed for 1 biopsy instead of 2, the single punch biopsy technique is more cost effective.
The bisected specimen resulting from complete excision of a small blister or from biopsy of a larger blister that includes 75% perilesional skin and 25% from the blister cavity also provides the best tissue specimen for interpretation of the subepidermal blistering processes via H&E staining.4,20,24 When traditional unmarked punch specimens of a blister margin are sent to the laboratory in formalin for H&E staining, the technician that grosses the specimen may or may not bisect the specimen showing the “take-off” point of the blister.
Finally, when the DIF specimen is prepared using either of these 2 approaches, the immunoprecipitants can be seen at the dermoepidermal junction or in the papillary dermis in the perilesional portion of the specimen.2,4 Additionally, the immunoprecipitant may be identified on the roof or floor of the blister. Although this approach has not been studied in a systematic fashion, we believe this technique provides “bonus” information to help differentiate BP and EBA correlating with salt-split skin blisters produced for indirect immunofluorescence.5,6
Limitations
It is critical for the pathologist or technician grossing these specimens to understand the techniques that are being employed and to ensure that the submitted half punch specimens are embedded so that the flat surface is cut so that the edge of the blister is properly sectioned for both H&E and DIF specimens. Additionally, with either recommended technique, if the portion of perilesional skin is not sufficient and the epidermis completely separates from the dermis, interpretation of both the H&E staining and DIF sections is substantially compromised.20 Therefore, an 8-mm disposable punch is recommended to avoid mangling the specimens when they are bisected and to insure that the epithelium is not lost. This technique is less suitable for blistering processes with a positive Nikolsky sign, such as pemphigus and toxic epidermal necrolysis, because the small area of perilesional skin adjacent to the blister may detach completely, requiring the epidermis and dermis to be evaluated separately or, in the worst-case scenario, the epidermis may be lost in processing.
Conclusion
Bisecting a single punch biopsy on subepidermal blisters provides the best specimen for H&E staining and DIF. The single punch biopsy technique also differentiates BP and EBA without utilizing salt-split skin immunofluorescence studies. This technique is more efficient and cost effective than the traditional approach of multiple biopsies on subepidermal blisters.
The traditional approach for confirming the diagnosis of subepidermal blistering diseases such as bullous pemphigoid (BP), epidermolysis bullosa acquisita (EBA), dermatitis herpetiformis (DH), linear IgA bullous dermatosis (LABD) requires 2 punch biopsies: one from perilesional skin for direct immunofluorescence (DIF) and one from lesional skin for light microscopy using hematoxylin and eosin (H&E) stain.1-4 These conditions are distinguished by a combination of features appreciated on H&E-stained sections, DIF, and indirect immunofluorescence for a subset of disorders. Additional information may be provided by DIF or indirect immunofluorescence utilizing the salt-split skin technique to differentiate BP (in which linear IgG deposition is identified by immunofluorescence on the roof of salt-split skin) from EBA and antiepiligrin cicatricial BP (in which linear IgG deposition is identified by immunofluorescence along the floor of the salt-split skin), which is more rare.4 One bisected punch biopsy of a subepidermal blister yields salt-split skin–like information through standard DIF and supersedes the need for the more cumbersome salt-split skin technique.
Serologic tests for the presence of circulating antibodies to BP180 and BP230 represent an emerging technology that can confirm the diagnosis of BP, but it has been difficult to identify clinically useful autoantibodies to confirm diagnoses of EBA and LABD.5-7 Serologic tests for tissue transglutaminase IgA antibodies may be useful in the diagnosis of DH.8 We present a cost-effective approach to biopsy in the diagnosis of subepidermal blistering diseases that provides the necessary diagnostic information to distinguish relevant disease processes.
Subepidermal Blistering Diseases
Bullous pemphigoid commonly presents with widespread tense bullae of varying sizes on an erythematous base or on otherwise normal skin.9 Some cases of BP present not with bullae but with pruritic, urticarial, plaquelike, or papular lesions. Bullous pemphigoid commonly involves flexural surfaces and the trunk but can appear anywhere on the skin. The induction of blisters by shearing with mechanical pressure on perilesional skin (Nikolsky sign) is not characteristically present in BP as it is in pemphigus vulgaris.10 Epidermolysis bullosa acquisita can mimic BP in the development of widespread tense bullae, but blisters typically appear on areas of the skin that are prone to trauma (eg, toes, knees, elbows, hands). Crusted erosions, scarring, and milia also are clinical manifestations of EBA.11 Dermatitis herpetiformis presents with grouped vesicles, papulovesicles, plaques, and excoriations that are symmetrically distributed on extensor surfaces of the skin but also can occur on the buttocks, scalp, and other areas of the skin.12,13 Although it may mimic both BP and DH, LABD frequently is less pruritic than these other conditions.14,15 Linear IgA bullous dermatosis also demonstrates the characteristic finding of multiple bullae that form concentrically around a crusted area of skin. This physical finding is known as a string of pearls. Linear IgA bullous dermatosis typically occurs in childhood and may resolve without treatment in months to years.16
Traditional Biopsy Approach
A review of several articles from the literature and multiple dermatology and dermatopathology textbooks revealed uniform recommendations for biopsy of subepidermal blistering conditions that manifest as tense blisters.1-4,9-23 A biopsy of early lesional skin or of a blister for light microscopy with H&E stain and biopsy of perilesional skin for DIF is recommended.1-4,9-23 Three review articles specifically suggested biopsy of “perilesional skin” for DIF.1-3 The majority of textbooks we reviewed also suggested that perilesional skin, or skin adjacent to a zone of erythema in the case of DH, should be sampled for DIF to assist in the diagnosis of BP, EBA, DH, and LABD.4,9-21 Biopsy of adjacent or nonlesional skin or skin around the lesion for DIF also was recommended by other textbooks for diagnosis of subepidermal blistering diseases.22,23 Perilesional skin is chosen because it is critical that the epidermis be included for adequate immunofluorescence studies.5,20 Biopsy of healed and crusted lesions should be avoided.24
Recommended Alternative Approach
A single punch biopsy produces the best possible specimen for H&E and DIF if it is obtained via one of 2 methods.
The first method involves choosing a small, 1- to 2-mm tense blister.25 Use an 8-mm punch centered on the blister that includes at least 3 mm of circumferential perilesional skin (Figures 1 and 2).20 Holding the specimen with forceps, use a no. 15 scalpel blade to bisect the blister with a sawing motion. Place half of the specimen in formalin for H&E staining and the other half in Zeus (or Michel) medium for DIF (Figure 3).
The second method is to choose any intact tense blister20,24 and, utilizing a surgical marker, draw a line from the roof of the blister onto the adjacent perilesional skin (Figure 1). After blotting with an alcohol pad so as not to remove the mark, anesthetize the site with lidocaine 1% with epinephrine,24 then take an 8-mm punch biopsy encompassing 75% perilesional skin and 25% of the blister centered on the line (Figure 2). After separating the punch specimen from the subcutaneous tissue with surgical scissors, hold the tissue with forceps and bisect the specimen with a no. 15 scalpel blade. Use a sawing motion along the line drawn in the prior steps (Figure 3). Submit half of the biopsy for H&E staining in formalin and the other half for DIF in Zeus medium.
Advantages
This approach offers several advantages. First, the technique requires only 1 invasive procedure, not 2 separate biopsies, so that the procedure can be done quickly and efficiently with the least morbidity and scarring. Secondly, because the patient is billed for 1 biopsy instead of 2, the single punch biopsy technique is more cost effective.
The bisected specimen resulting from complete excision of a small blister or from biopsy of a larger blister that includes 75% perilesional skin and 25% from the blister cavity also provides the best tissue specimen for interpretation of the subepidermal blistering processes via H&E staining.4,20,24 When traditional unmarked punch specimens of a blister margin are sent to the laboratory in formalin for H&E staining, the technician that grosses the specimen may or may not bisect the specimen showing the “take-off” point of the blister.
Finally, when the DIF specimen is prepared using either of these 2 approaches, the immunoprecipitants can be seen at the dermoepidermal junction or in the papillary dermis in the perilesional portion of the specimen.2,4 Additionally, the immunoprecipitant may be identified on the roof or floor of the blister. Although this approach has not been studied in a systematic fashion, we believe this technique provides “bonus” information to help differentiate BP and EBA correlating with salt-split skin blisters produced for indirect immunofluorescence.5,6
Limitations
It is critical for the pathologist or technician grossing these specimens to understand the techniques that are being employed and to ensure that the submitted half punch specimens are embedded so that the flat surface is cut so that the edge of the blister is properly sectioned for both H&E and DIF specimens. Additionally, with either recommended technique, if the portion of perilesional skin is not sufficient and the epidermis completely separates from the dermis, interpretation of both the H&E staining and DIF sections is substantially compromised.20 Therefore, an 8-mm disposable punch is recommended to avoid mangling the specimens when they are bisected and to insure that the epithelium is not lost. This technique is less suitable for blistering processes with a positive Nikolsky sign, such as pemphigus and toxic epidermal necrolysis, because the small area of perilesional skin adjacent to the blister may detach completely, requiring the epidermis and dermis to be evaluated separately or, in the worst-case scenario, the epidermis may be lost in processing.
Conclusion
Bisecting a single punch biopsy on subepidermal blisters provides the best specimen for H&E staining and DIF. The single punch biopsy technique also differentiates BP and EBA without utilizing salt-split skin immunofluorescence studies. This technique is more efficient and cost effective than the traditional approach of multiple biopsies on subepidermal blisters.
The traditional approach for confirming the diagnosis of subepidermal blistering diseases such as bullous pemphigoid (BP), epidermolysis bullosa acquisita (EBA), dermatitis herpetiformis (DH), linear IgA bullous dermatosis (LABD) requires 2 punch biopsies: one from perilesional skin for direct immunofluorescence (DIF) and one from lesional skin for light microscopy using hematoxylin and eosin (H&E) stain.1-4 These conditions are distinguished by a combination of features appreciated on H&E-stained sections, DIF, and indirect immunofluorescence for a subset of disorders. Additional information may be provided by DIF or indirect immunofluorescence utilizing the salt-split skin technique to differentiate BP (in which linear IgG deposition is identified by immunofluorescence on the roof of salt-split skin) from EBA and antiepiligrin cicatricial BP (in which linear IgG deposition is identified by immunofluorescence along the floor of the salt-split skin), which is more rare.4 One bisected punch biopsy of a subepidermal blister yields salt-split skin–like information through standard DIF and supersedes the need for the more cumbersome salt-split skin technique.
Serologic tests for the presence of circulating antibodies to BP180 and BP230 represent an emerging technology that can confirm the diagnosis of BP, but it has been difficult to identify clinically useful autoantibodies to confirm diagnoses of EBA and LABD.5-7 Serologic tests for tissue transglutaminase IgA antibodies may be useful in the diagnosis of DH.8 We present a cost-effective approach to biopsy in the diagnosis of subepidermal blistering diseases that provides the necessary diagnostic information to distinguish relevant disease processes.
Subepidermal Blistering Diseases
Bullous pemphigoid commonly presents with widespread tense bullae of varying sizes on an erythematous base or on otherwise normal skin.9 Some cases of BP present not with bullae but with pruritic, urticarial, plaquelike, or papular lesions. Bullous pemphigoid commonly involves flexural surfaces and the trunk but can appear anywhere on the skin. The induction of blisters by shearing with mechanical pressure on perilesional skin (Nikolsky sign) is not characteristically present in BP as it is in pemphigus vulgaris.10 Epidermolysis bullosa acquisita can mimic BP in the development of widespread tense bullae, but blisters typically appear on areas of the skin that are prone to trauma (eg, toes, knees, elbows, hands). Crusted erosions, scarring, and milia also are clinical manifestations of EBA.11 Dermatitis herpetiformis presents with grouped vesicles, papulovesicles, plaques, and excoriations that are symmetrically distributed on extensor surfaces of the skin but also can occur on the buttocks, scalp, and other areas of the skin.12,13 Although it may mimic both BP and DH, LABD frequently is less pruritic than these other conditions.14,15 Linear IgA bullous dermatosis also demonstrates the characteristic finding of multiple bullae that form concentrically around a crusted area of skin. This physical finding is known as a string of pearls. Linear IgA bullous dermatosis typically occurs in childhood and may resolve without treatment in months to years.16
Traditional Biopsy Approach
A review of several articles from the literature and multiple dermatology and dermatopathology textbooks revealed uniform recommendations for biopsy of subepidermal blistering conditions that manifest as tense blisters.1-4,9-23 A biopsy of early lesional skin or of a blister for light microscopy with H&E stain and biopsy of perilesional skin for DIF is recommended.1-4,9-23 Three review articles specifically suggested biopsy of “perilesional skin” for DIF.1-3 The majority of textbooks we reviewed also suggested that perilesional skin, or skin adjacent to a zone of erythema in the case of DH, should be sampled for DIF to assist in the diagnosis of BP, EBA, DH, and LABD.4,9-21 Biopsy of adjacent or nonlesional skin or skin around the lesion for DIF also was recommended by other textbooks for diagnosis of subepidermal blistering diseases.22,23 Perilesional skin is chosen because it is critical that the epidermis be included for adequate immunofluorescence studies.5,20 Biopsy of healed and crusted lesions should be avoided.24
Recommended Alternative Approach
A single punch biopsy produces the best possible specimen for H&E and DIF if it is obtained via one of 2 methods.
The first method involves choosing a small, 1- to 2-mm tense blister.25 Use an 8-mm punch centered on the blister that includes at least 3 mm of circumferential perilesional skin (Figures 1 and 2).20 Holding the specimen with forceps, use a no. 15 scalpel blade to bisect the blister with a sawing motion. Place half of the specimen in formalin for H&E staining and the other half in Zeus (or Michel) medium for DIF (Figure 3).
The second method is to choose any intact tense blister20,24 and, utilizing a surgical marker, draw a line from the roof of the blister onto the adjacent perilesional skin (Figure 1). After blotting with an alcohol pad so as not to remove the mark, anesthetize the site with lidocaine 1% with epinephrine,24 then take an 8-mm punch biopsy encompassing 75% perilesional skin and 25% of the blister centered on the line (Figure 2). After separating the punch specimen from the subcutaneous tissue with surgical scissors, hold the tissue with forceps and bisect the specimen with a no. 15 scalpel blade. Use a sawing motion along the line drawn in the prior steps (Figure 3). Submit half of the biopsy for H&E staining in formalin and the other half for DIF in Zeus medium.
Advantages
This approach offers several advantages. First, the technique requires only 1 invasive procedure, not 2 separate biopsies, so that the procedure can be done quickly and efficiently with the least morbidity and scarring. Secondly, because the patient is billed for 1 biopsy instead of 2, the single punch biopsy technique is more cost effective.
The bisected specimen resulting from complete excision of a small blister or from biopsy of a larger blister that includes 75% perilesional skin and 25% from the blister cavity also provides the best tissue specimen for interpretation of the subepidermal blistering processes via H&E staining.4,20,24 When traditional unmarked punch specimens of a blister margin are sent to the laboratory in formalin for H&E staining, the technician that grosses the specimen may or may not bisect the specimen showing the “take-off” point of the blister.
Finally, when the DIF specimen is prepared using either of these 2 approaches, the immunoprecipitants can be seen at the dermoepidermal junction or in the papillary dermis in the perilesional portion of the specimen.2,4 Additionally, the immunoprecipitant may be identified on the roof or floor of the blister. Although this approach has not been studied in a systematic fashion, we believe this technique provides “bonus” information to help differentiate BP and EBA correlating with salt-split skin blisters produced for indirect immunofluorescence.5,6
Limitations
It is critical for the pathologist or technician grossing these specimens to understand the techniques that are being employed and to ensure that the submitted half punch specimens are embedded so that the flat surface is cut so that the edge of the blister is properly sectioned for both H&E and DIF specimens. Additionally, with either recommended technique, if the portion of perilesional skin is not sufficient and the epidermis completely separates from the dermis, interpretation of both the H&E staining and DIF sections is substantially compromised.20 Therefore, an 8-mm disposable punch is recommended to avoid mangling the specimens when they are bisected and to insure that the epithelium is not lost. This technique is less suitable for blistering processes with a positive Nikolsky sign, such as pemphigus and toxic epidermal necrolysis, because the small area of perilesional skin adjacent to the blister may detach completely, requiring the epidermis and dermis to be evaluated separately or, in the worst-case scenario, the epidermis may be lost in processing.
Conclusion
Bisecting a single punch biopsy on subepidermal blisters provides the best specimen for H&E staining and DIF. The single punch biopsy technique also differentiates BP and EBA without utilizing salt-split skin immunofluorescence studies. This technique is more efficient and cost effective than the traditional approach of multiple biopsies on subepidermal blisters.
This is purlsCopyright text added for testing purposes.
