User login
What’s new in the latest melanoma guidelines
KAUAI, HAWAII – Melanoma , resulting in an evidence-based improved prognosis for many of them, Laura Korb Ferris, MD, PhD, said at the Hawaii Dermatology Seminar provided by Skin Disease Education Foundation/Global Academy for Medical Education.
Dr. Ferris, of the department of dermatology, University of Pittsburgh, highlighted some of the key changes in the eighth edition of the AJCC staging manual, which is now in effect. She also described the clinical implications of important updates introduced in the 2018 National Comprehensive Cancer Network (NCCN) guidelines for the diagnosis and management of melanoma.
The AJCC eighth edition
The eighth edition is built upon an AJCC database of more than 46,000 patients with stage I-III melanoma diagnosed since 1998 at 10 academic medical centers. The AJCC panel made no changes in stage IV melanoma guidance because the newer targeted therapies have rapidly changed treatment outcomes in that setting and longer follow-up is needed to assess the full impact. 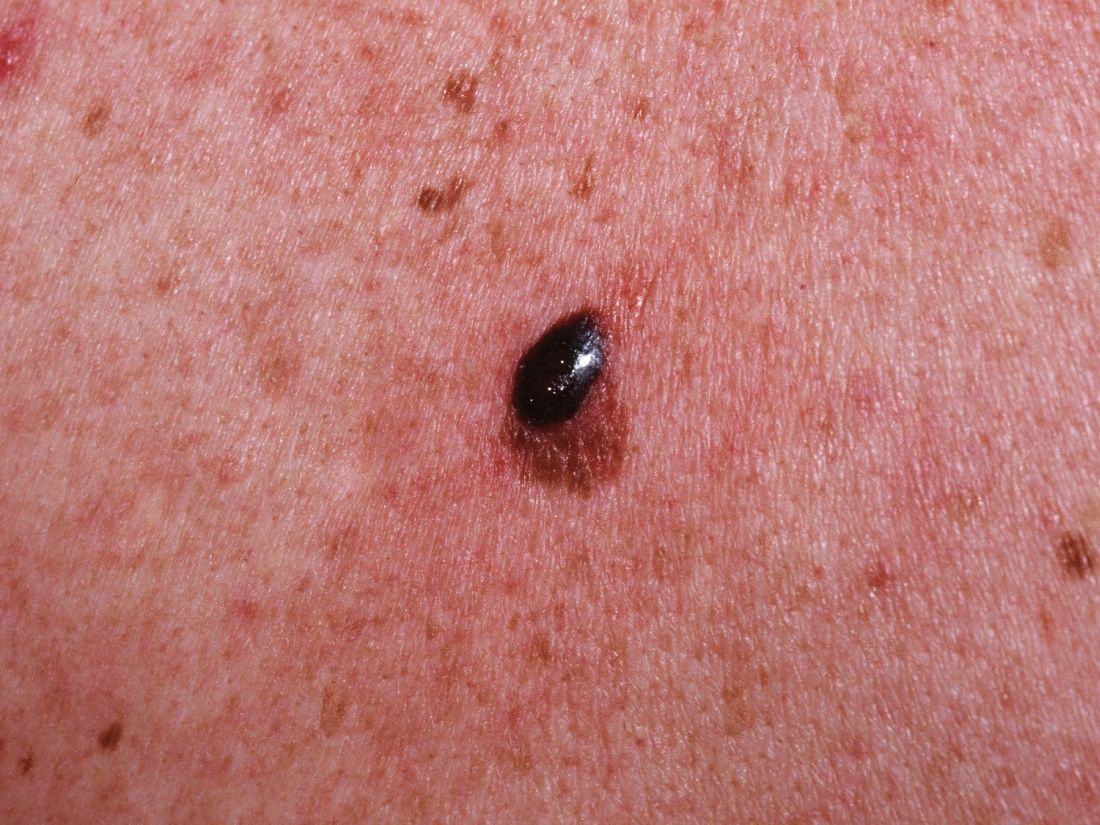
The current edition of the AJCC melanoma staging manual creates a new subcategory within pathologic stage III. In the melanoma staging world, that’s exciting news, especially because this change has important implications for prognosis.
This fourth subcategory, stage IIID, is for melanomas, which in the Tumor, Nodes, Metastasis (TNM) classification scheme, are primary tumor stage T4b, meaning greater than 4.0 mm in thickness and with ulceration; regional lymph node N3a, b, or c, based upon the number of metastatic nodes involved and whether they were clinically occult nodal metastases detected by sentinel lymph node biopsy (SLNB) or clinically detected; and M0, meaning no distant metastatic disease. In the 8th edition, the AJCC staging system can be applied in patients with T2 through T4 primary melanoma only if they have undergone SLNB.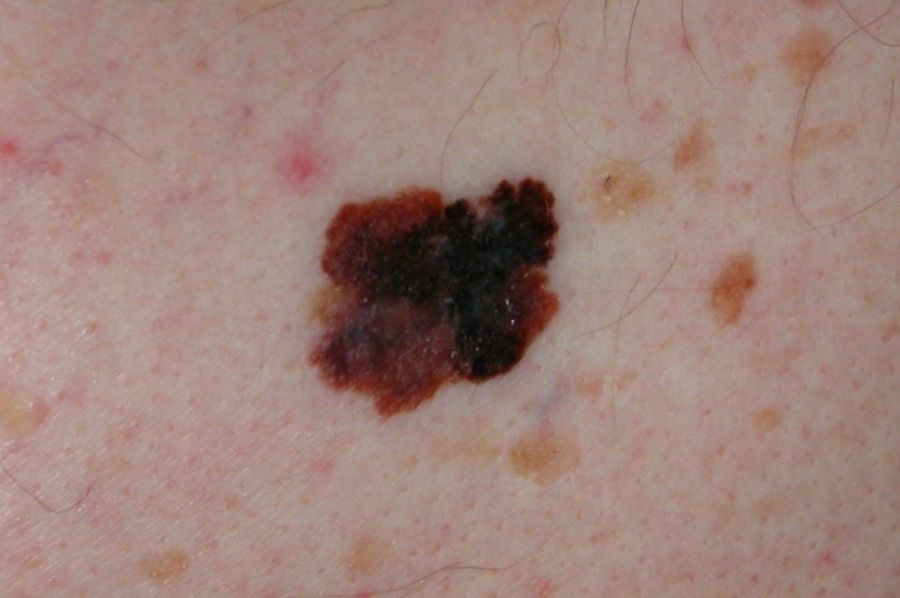
This new approach to stage III disease makes for more homogeneous patient subgroups, which in turn provides much better stratification of prognosis than was possible in the seventh edition of the AJCC staging manual, which dates back to 2010. Most strikingly, the 5-year melanoma-specific survival rate for patients with stage IIIA disease was 78% in the seventh edition of AJCC, but it climbs to 93% in the eighth edition. For patients with stage IIIB melanoma, 5-year melanoma-specific survival improved from 59% in the seventh edition to 83% in the current iteration, while in stage IIIC, the jump is from 40% to 69%. All this is made possible because the eighth edition separates out patients with the new stage IIID, whose 5-year melanoma-specific survival is only 32%, Dr. Ferris explained.
Among the other key points to remember about the eighth edition of AJCC:
- Tumor thickness is now measured to the nearest 0.1 mm rather than to the nearest 0.01 mm, as previously. Thus, a 0.75-mm-thick melanoma is now rounded up to 0.8 mm, while a 0.74-mm melanoma becomes a 0.7-mm tumor.
- Based upon recent evidence, tumors that are 0.8-1.0 mm thick, with or without ulceration, are now classified at T1b. So are ulcerated lesions that are less than 0.8 mm.
- Dermal mitotic rate is no longer used in staging T1 tumors, although it’s still supposed to be included in pathology reports.
- The T category definitions of primary tumors have been clarified in the eighth edition. A tumor is now classified as T0 only if there is no evidence of a primary tumor. Tx is employed when the primary tumor thickness can’t be determined, as for example when the biopsy specimen was obtained by curettage. Tis is utilized for melanoma in situ.
- The N subcategory definitions of regional nodal status have been revised. Microsatellites, clinical satellites, and in-transit metastases are now categorized as N1c, N2c, or N3c based upon the number of tumor-involved regional lymph nodes. These features are no longer defined by their size or distance from the primary tumor.
2018 NCCN melanoma guidelines
The guidelines have been revised to recommend against SLNB if a patient’s pretest probability of finding a positive SLN is less than 5%. This includes patients who have a clinical stage IA/T1a melanoma with a Breslow thickness of less than 0.8 mm without ulceration.
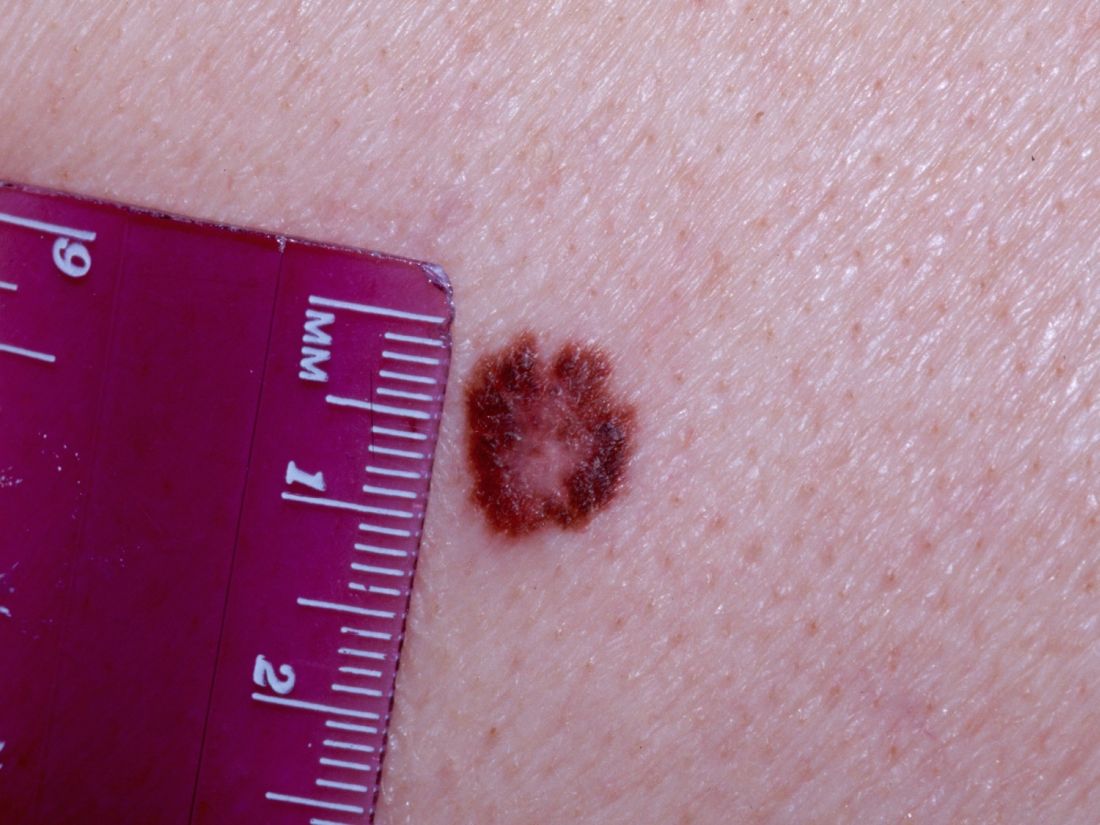
There is to be no SLNB in patients with microsatellites, clinical satellites, or in-transit metastases because SLN status has no prognostic significance in this situation.

Routine ordering of prognostic genetic tests for BRAF or the multigene test panels that are now commercially available is not recommended except to guide systemic therapy or to determine if a patient is a candidate for a specific clinical trial. “Basically, there is not a place to use this information in the NCCN guidelines,” according to the dermatologist.
What about completion lymphadenectomy in the SLN-positive melanoma patient?
Completion lymph node dissection looks increasingly like a procedure in search of an indication. Results of the National Cancer Institute–sponsored Multicenter Selective Lymphadenectomy Trial–II (MSLT-II) demonstrated not even a hint of a difference in 3-year melanoma-specific survival in 1,934 melanoma patients with sentinel lymph node metastases regardless of whether they were randomized to immediate completion lymph node dissection or ultrasound-based nodal monitoring. Moreover, completion lymphadenectomy was associated with significant morbidity: a 24.1% incidence of lymphedema, compared with a 6.3% rate in the observation group (N Engl J Med. 2017 Jun 8;376[23]:2211-22).
On the other hand, Dr. Ferris noted that many newer drugs are being approved for the treatment of stage III melanoma, and in all the pivotal clinical trials, patients had to have undergone completion lymph node dissection as a condition of participation. So the surgery becomes a consideration if physicians want to use the newer agents the way they were used successfully in the trials.
The full eighth edition of the AJCC cancer staging manual is available for purchase. For physicians with a specific interest in melanoma, Dr. Ferris recommended as an extremely useful alternative the AJCC expert writing panel’s free downloadable summary of the evidence-based changes made in melanoma staging (CA Cancer J Clin. 2017 Nov;67[6]:472-92). The 2018 NCCN guidelines (Melanoma. Version 1.2018 Oct. 11, 2017) are available for free (www.NCCN.org).
Dr. Ferris reported serving as a consultant to DermTech.
SDEF/Global Academy for Medical Education and this news organization are owned by the same parent company.
KAUAI, HAWAII – Melanoma , resulting in an evidence-based improved prognosis for many of them, Laura Korb Ferris, MD, PhD, said at the Hawaii Dermatology Seminar provided by Skin Disease Education Foundation/Global Academy for Medical Education.
Dr. Ferris, of the department of dermatology, University of Pittsburgh, highlighted some of the key changes in the eighth edition of the AJCC staging manual, which is now in effect. She also described the clinical implications of important updates introduced in the 2018 National Comprehensive Cancer Network (NCCN) guidelines for the diagnosis and management of melanoma.
The AJCC eighth edition
The eighth edition is built upon an AJCC database of more than 46,000 patients with stage I-III melanoma diagnosed since 1998 at 10 academic medical centers. The AJCC panel made no changes in stage IV melanoma guidance because the newer targeted therapies have rapidly changed treatment outcomes in that setting and longer follow-up is needed to assess the full impact.
The current edition of the AJCC melanoma staging manual creates a new subcategory within pathologic stage III. In the melanoma staging world, that’s exciting news, especially because this change has important implications for prognosis.
This fourth subcategory, stage IIID, is for melanomas, which in the Tumor, Nodes, Metastasis (TNM) classification scheme, are primary tumor stage T4b, meaning greater than 4.0 mm in thickness and with ulceration; regional lymph node N3a, b, or c, based upon the number of metastatic nodes involved and whether they were clinically occult nodal metastases detected by sentinel lymph node biopsy (SLNB) or clinically detected; and M0, meaning no distant metastatic disease. In the 8th edition, the AJCC staging system can be applied in patients with T2 through T4 primary melanoma only if they have undergone SLNB.
This new approach to stage III disease makes for more homogeneous patient subgroups, which in turn provides much better stratification of prognosis than was possible in the seventh edition of the AJCC staging manual, which dates back to 2010. Most strikingly, the 5-year melanoma-specific survival rate for patients with stage IIIA disease was 78% in the seventh edition of AJCC, but it climbs to 93% in the eighth edition. For patients with stage IIIB melanoma, 5-year melanoma-specific survival improved from 59% in the seventh edition to 83% in the current iteration, while in stage IIIC, the jump is from 40% to 69%. All this is made possible because the eighth edition separates out patients with the new stage IIID, whose 5-year melanoma-specific survival is only 32%, Dr. Ferris explained.
Among the other key points to remember about the eighth edition of AJCC:
- Tumor thickness is now measured to the nearest 0.1 mm rather than to the nearest 0.01 mm, as previously. Thus, a 0.75-mm-thick melanoma is now rounded up to 0.8 mm, while a 0.74-mm melanoma becomes a 0.7-mm tumor.
- Based upon recent evidence, tumors that are 0.8-1.0 mm thick, with or without ulceration, are now classified at T1b. So are ulcerated lesions that are less than 0.8 mm.
- Dermal mitotic rate is no longer used in staging T1 tumors, although it’s still supposed to be included in pathology reports.
- The T category definitions of primary tumors have been clarified in the eighth edition. A tumor is now classified as T0 only if there is no evidence of a primary tumor. Tx is employed when the primary tumor thickness can’t be determined, as for example when the biopsy specimen was obtained by curettage. Tis is utilized for melanoma in situ.
- The N subcategory definitions of regional nodal status have been revised. Microsatellites, clinical satellites, and in-transit metastases are now categorized as N1c, N2c, or N3c based upon the number of tumor-involved regional lymph nodes. These features are no longer defined by their size or distance from the primary tumor.
2018 NCCN melanoma guidelines
The guidelines have been revised to recommend against SLNB if a patient’s pretest probability of finding a positive SLN is less than 5%. This includes patients who have a clinical stage IA/T1a melanoma with a Breslow thickness of less than 0.8 mm without ulceration.
There is to be no SLNB in patients with microsatellites, clinical satellites, or in-transit metastases because SLN status has no prognostic significance in this situation.
Routine ordering of prognostic genetic tests for BRAF or the multigene test panels that are now commercially available is not recommended except to guide systemic therapy or to determine if a patient is a candidate for a specific clinical trial. “Basically, there is not a place to use this information in the NCCN guidelines,” according to the dermatologist.
What about completion lymphadenectomy in the SLN-positive melanoma patient?
Completion lymph node dissection looks increasingly like a procedure in search of an indication. Results of the National Cancer Institute–sponsored Multicenter Selective Lymphadenectomy Trial–II (MSLT-II) demonstrated not even a hint of a difference in 3-year melanoma-specific survival in 1,934 melanoma patients with sentinel lymph node metastases regardless of whether they were randomized to immediate completion lymph node dissection or ultrasound-based nodal monitoring. Moreover, completion lymphadenectomy was associated with significant morbidity: a 24.1% incidence of lymphedema, compared with a 6.3% rate in the observation group (N Engl J Med. 2017 Jun 8;376[23]:2211-22).
On the other hand, Dr. Ferris noted that many newer drugs are being approved for the treatment of stage III melanoma, and in all the pivotal clinical trials, patients had to have undergone completion lymph node dissection as a condition of participation. So the surgery becomes a consideration if physicians want to use the newer agents the way they were used successfully in the trials.
The full eighth edition of the AJCC cancer staging manual is available for purchase. For physicians with a specific interest in melanoma, Dr. Ferris recommended as an extremely useful alternative the AJCC expert writing panel’s free downloadable summary of the evidence-based changes made in melanoma staging (CA Cancer J Clin. 2017 Nov;67[6]:472-92). The 2018 NCCN guidelines (Melanoma. Version 1.2018 Oct. 11, 2017) are available for free (www.NCCN.org).
Dr. Ferris reported serving as a consultant to DermTech.
SDEF/Global Academy for Medical Education and this news organization are owned by the same parent company.
KAUAI, HAWAII – Melanoma , resulting in an evidence-based improved prognosis for many of them, Laura Korb Ferris, MD, PhD, said at the Hawaii Dermatology Seminar provided by Skin Disease Education Foundation/Global Academy for Medical Education.
Dr. Ferris, of the department of dermatology, University of Pittsburgh, highlighted some of the key changes in the eighth edition of the AJCC staging manual, which is now in effect. She also described the clinical implications of important updates introduced in the 2018 National Comprehensive Cancer Network (NCCN) guidelines for the diagnosis and management of melanoma.
The AJCC eighth edition
The eighth edition is built upon an AJCC database of more than 46,000 patients with stage I-III melanoma diagnosed since 1998 at 10 academic medical centers. The AJCC panel made no changes in stage IV melanoma guidance because the newer targeted therapies have rapidly changed treatment outcomes in that setting and longer follow-up is needed to assess the full impact.
The current edition of the AJCC melanoma staging manual creates a new subcategory within pathologic stage III. In the melanoma staging world, that’s exciting news, especially because this change has important implications for prognosis.
This fourth subcategory, stage IIID, is for melanomas, which in the Tumor, Nodes, Metastasis (TNM) classification scheme, are primary tumor stage T4b, meaning greater than 4.0 mm in thickness and with ulceration; regional lymph node N3a, b, or c, based upon the number of metastatic nodes involved and whether they were clinically occult nodal metastases detected by sentinel lymph node biopsy (SLNB) or clinically detected; and M0, meaning no distant metastatic disease. In the 8th edition, the AJCC staging system can be applied in patients with T2 through T4 primary melanoma only if they have undergone SLNB.
This new approach to stage III disease makes for more homogeneous patient subgroups, which in turn provides much better stratification of prognosis than was possible in the seventh edition of the AJCC staging manual, which dates back to 2010. Most strikingly, the 5-year melanoma-specific survival rate for patients with stage IIIA disease was 78% in the seventh edition of AJCC, but it climbs to 93% in the eighth edition. For patients with stage IIIB melanoma, 5-year melanoma-specific survival improved from 59% in the seventh edition to 83% in the current iteration, while in stage IIIC, the jump is from 40% to 69%. All this is made possible because the eighth edition separates out patients with the new stage IIID, whose 5-year melanoma-specific survival is only 32%, Dr. Ferris explained.
Among the other key points to remember about the eighth edition of AJCC:
- Tumor thickness is now measured to the nearest 0.1 mm rather than to the nearest 0.01 mm, as previously. Thus, a 0.75-mm-thick melanoma is now rounded up to 0.8 mm, while a 0.74-mm melanoma becomes a 0.7-mm tumor.
- Based upon recent evidence, tumors that are 0.8-1.0 mm thick, with or without ulceration, are now classified at T1b. So are ulcerated lesions that are less than 0.8 mm.
- Dermal mitotic rate is no longer used in staging T1 tumors, although it’s still supposed to be included in pathology reports.
- The T category definitions of primary tumors have been clarified in the eighth edition. A tumor is now classified as T0 only if there is no evidence of a primary tumor. Tx is employed when the primary tumor thickness can’t be determined, as for example when the biopsy specimen was obtained by curettage. Tis is utilized for melanoma in situ.
- The N subcategory definitions of regional nodal status have been revised. Microsatellites, clinical satellites, and in-transit metastases are now categorized as N1c, N2c, or N3c based upon the number of tumor-involved regional lymph nodes. These features are no longer defined by their size or distance from the primary tumor.
2018 NCCN melanoma guidelines
The guidelines have been revised to recommend against SLNB if a patient’s pretest probability of finding a positive SLN is less than 5%. This includes patients who have a clinical stage IA/T1a melanoma with a Breslow thickness of less than 0.8 mm without ulceration.
There is to be no SLNB in patients with microsatellites, clinical satellites, or in-transit metastases because SLN status has no prognostic significance in this situation.
Routine ordering of prognostic genetic tests for BRAF or the multigene test panels that are now commercially available is not recommended except to guide systemic therapy or to determine if a patient is a candidate for a specific clinical trial. “Basically, there is not a place to use this information in the NCCN guidelines,” according to the dermatologist.
What about completion lymphadenectomy in the SLN-positive melanoma patient?
Completion lymph node dissection looks increasingly like a procedure in search of an indication. Results of the National Cancer Institute–sponsored Multicenter Selective Lymphadenectomy Trial–II (MSLT-II) demonstrated not even a hint of a difference in 3-year melanoma-specific survival in 1,934 melanoma patients with sentinel lymph node metastases regardless of whether they were randomized to immediate completion lymph node dissection or ultrasound-based nodal monitoring. Moreover, completion lymphadenectomy was associated with significant morbidity: a 24.1% incidence of lymphedema, compared with a 6.3% rate in the observation group (N Engl J Med. 2017 Jun 8;376[23]:2211-22).
On the other hand, Dr. Ferris noted that many newer drugs are being approved for the treatment of stage III melanoma, and in all the pivotal clinical trials, patients had to have undergone completion lymph node dissection as a condition of participation. So the surgery becomes a consideration if physicians want to use the newer agents the way they were used successfully in the trials.
The full eighth edition of the AJCC cancer staging manual is available for purchase. For physicians with a specific interest in melanoma, Dr. Ferris recommended as an extremely useful alternative the AJCC expert writing panel’s free downloadable summary of the evidence-based changes made in melanoma staging (CA Cancer J Clin. 2017 Nov;67[6]:472-92). The 2018 NCCN guidelines (Melanoma. Version 1.2018 Oct. 11, 2017) are available for free (www.NCCN.org).
Dr. Ferris reported serving as a consultant to DermTech.
SDEF/Global Academy for Medical Education and this news organization are owned by the same parent company.
EXPERT ANALYSIS FROM SDEF HAWAII DERMATOLOGY SEMINAR
Gone Fishing: A Unique Histologic Pattern in Cutaneous Angiosarcoma
Cutaneous angiosarcoma is a rare malignant tumor of vascular endothelial cells that has the propensity to arise in various clinical settings. This tumor predominantly occurs in the head and neck region in elderly patients, but it also has been reported to develop postradiotherapy or in the setting of chronic lymphedema in the extremities.1-3 In all settings, the diagnosis carries a very poor prognosis with a high likelihood of local recurrence and rapid dissemination. The mortality rate typically is 80% or higher.2,4-6
Making the correct clinical diagnosis of cutaneous angiosarcoma may be difficult given the variety of patient symptoms and clinical appearances that can be demonstrated on presentation. Lesions can appear as bluish or violaceous plaques, macules, or nodules, and ulceration may be present in some advanced cases.5,7 Clinical misdiagnosis is common, as cutaneous angiosarcomas may be mistaken for infectious processes, benign vascular malformations, and other cutaneous malignancies.1 Biopsy often is delayed given the initial benign appearance of the lesions, and this frequently results in aggressive and extensive disease at the time of diagnosis, which is unfortunate given that small tumor size has been shown to be one of the only favorable prognostic indicators in cutaneous angiosarcoma.1,2,6,8
Microscopically, diagnosis of cutaneous angiosarcoma can present a challenge, as the histology varies between a well-differentiated vascular neoplasm and a considerably anaplastic and poorly differentiated malignancy. On low power, some areas may appear as benign hemangiomas with other areas showing frank sarcomatous features.9 As a result, these tumors can be mistaken for a variety of other diseases including melanomas, carcinomas, or other vascular tumors.6,8,9 Previously, electron microscopy has been utilized on undifferentiated tumors to help distinguish cutaneous angiosarcomas from other potential diagnoses. The atypical tumor cells of cutaneous angiosarcoma display common features of endothelial cells (eg, pinocytotic vesicles, tubulated bodies).7 Historically, it has been noted that the histologic findings and tumor grade provide little evidence regarding the aggressiveness of the tumor, and all cutaneous angiosarcoma diagnoses receive a poor prognosis.6,8
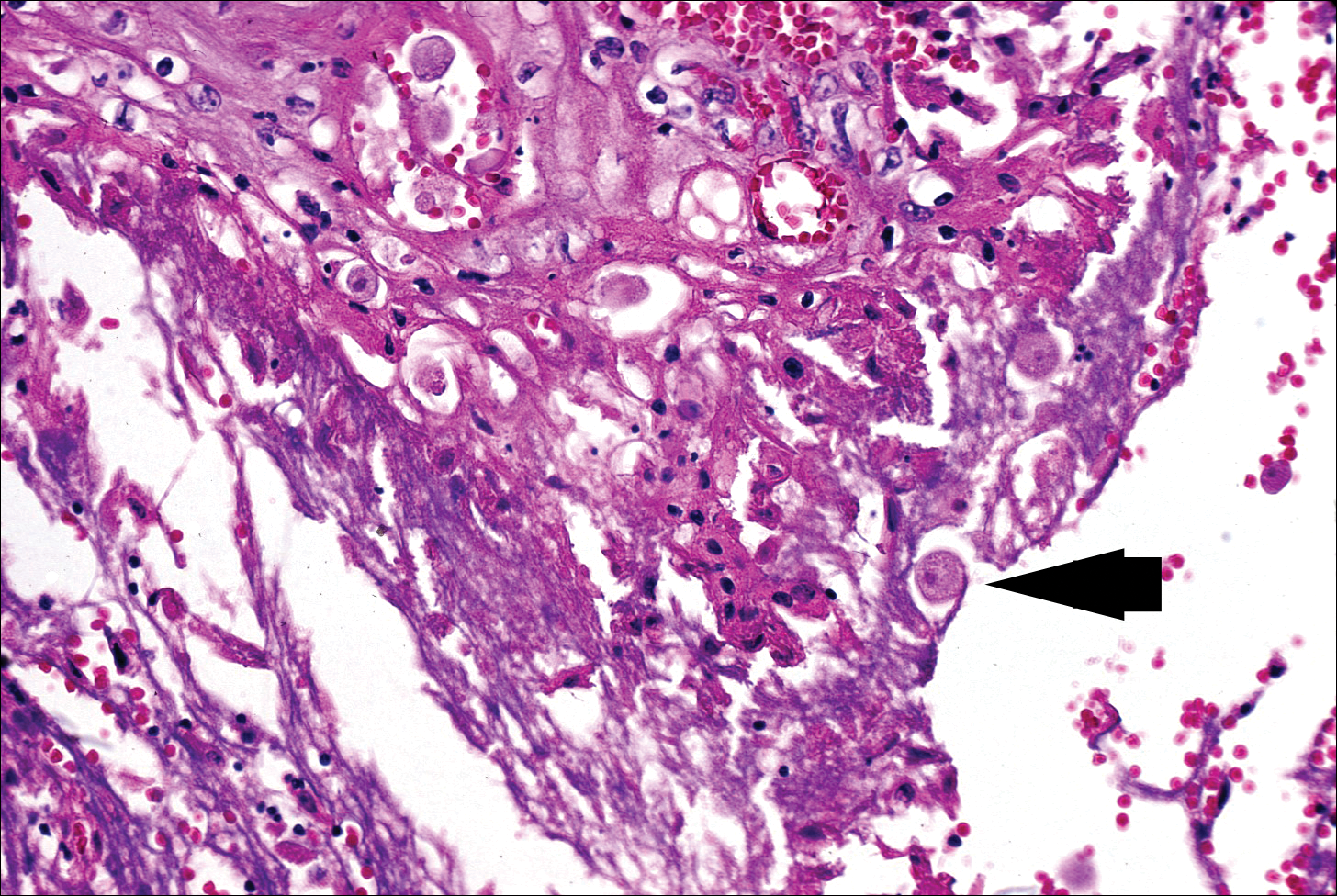
Classically, the histologic findings of cutaneous angiosarcoma include a highly infiltrative neoplasm forming irregular vascular channels that penetrate through the cutaneous soft tissues and frequently extend into the subcutaneous fat. The vascular spaces are lined by hyperchromatic endothelial cells with varying degrees of atypia.1,2,4,6,7,10 Occasionally, prominent endothelial cells lining a papillary structure within the lumen of the neoformed vessel may also be observed. Currently, immunohistochemical staining for MYC, Ki-67, D2-40, and various other markers complement the histologic findings to aid in the diagnosis of cutaneous angiosarcoma.11,12 An additional diagnostic clue that has been described in cases of postirradiation cutaneous angiosarcoma shows free-floating or tufted pleomorphic spindle cells within the vascular lumen (Figure). This finding has been described as “fish in the creek.”11 In this study, we aimed to determine the frequency and subsequent diagnostic utility of the fish-in-the-creek finding in cases of cutaneous angiosarcoma.

Methods
A natural language search of our institutional archives over a 20-year period (1997–2017) using the term angiosarcoma was performed. Fifteen cases of cutaneous angiosarcoma were identified. Fifteen additional benign and malignant vascular tumors with cutaneous angiosarco
Results
The histologic pattern of fish in the creek was identified in all 15 cases of cutaneous angiosarcoma and was absent in the other 15 malignancies examined in this study. This finding shows the potential for the fish-in-the-creek pattern to be used as an additional diagnostic tool for dermatopathologists.
Comment
Cutaneous angiosarcoma is a rare but aggressive malignancy that proves difficult to diagnose both clinically and histologically as well as to treat effectively.1,5-8 Our results indicate that fish in the creek may be a useful and salient histologic feature in cutaneous angiosarcoma. It is important to recognize, however, that this finding should not be the sole feature upon which a diagnosis of cutaneous angiosarcoma is made, as it requires corroboration with positivity of MYC and D2-40 as well as a high Ki-67 proliferation index (>20%).11,12 Finding a fish-in-the-creek pattern should prompt dermatopathologists to consider a diagnosis of cutaneous angiosarcoma in the appropriate clinical and histologic settings.
The chief limitation of this study was the small sample size, with only 15 cases of cutaneous angiosarcoma available in the last 20 years at our institution. The limited sample size did not allow us to make claims on sensitivity and specificity regarding this histologic feature; however, with a larger sample size, the true diagnostic potential could be elucidated. Although the pathologists were blinded to the original diagnoses as they examined it for fish in the creek, it is possible they were able to make the correct diagnosis based on other histopathologic clues and therefore were biased.
Although the fish-in-the-creek pattern is present in cutaneous angiosarcoma, there may be other mimickers to consider. Intraluminal papillary projections lined by endothelial cells may be sectioned in a manner imitating this finding.3 In such a case, these endothelial cells must be differentiated from the free-floating or tufted spindle cells in order to have a positive finding for fish in the creek. There can be confusion if the biopsy cuts through a section of spindled cells, resulting in difficulty differentiating cutaneous angiosarcoma from other spindle tumors such as spindle cell melanoma or spindle cell squamous cell carcinoma.6 In such cases, immunohistochemistry may be helpful, as spindle cell melanoma would stain positive for S100 and SOX10 and spindle cell squamous cell carcinoma would stain positive for p63 and cytokeratin.
Various treatment strategies for cutaneous angiosarcoma have been employed, with the majority still resulting in poor outcomes.2,4-6 The recommended treatment is radical surgical excision of the primary tumor with lymph node clearance if possible. Following excision, the patient should undergo high-dose, wide-field radiotherapy to the region.5,8 Cutaneous angiosarcomas also have the ability to spread extensively through the dermis and can result in subclinical or clinically obvious widespread disease with multifocal or satellite lesions present. Distant metastases occur most frequently in the cervical lymph nodes and lungs.7 In cases where the disease is too extensive for surgery, palliative radiation monotherapy can be used.5,6
As atypical vascular lesions are considered to be a precursor to cutaneous angiosarcoma, it is important to note that the fish-in-the-creek feature was absent in all 6 of the atypical vascular lesions observed in the study. The differentiation generally is made based on MYC, which is present in cutaneous angiosarcomas and absent in atypical vascular lesions.10 The feature of fish in the creek may now be an additional clue for dermatopathologists to differentiate between angiosarcomas and other similar-appearing tumors.
Conclusion
Our study aimed to highlight an important histologic feature of cutaneous angiosarcomas that can aid in the diagnosis of this deceptive malignancy. Our findings warrant further study of the fish-in-the-creek histologic pattern in a larger sample size to determine its success as a diagnostic tool for cutaneous angiosarcomas. As noted previously, tumor grade does not impact survival outcome, but small tumor size has been one of the only features found to result in a more favorable prognosis.1,6,8 Future studies to identify a correlation between the histologic finding of fish in the creek and disease outcome in cutaneous angiosarcoma may be helpful to determine if these histologic findings provide prognostic significance in cases of cutaneous angiosarcoma.
- Aust MR, Olsen KD, Lewis JE, et al. Angiosarcomas of the head and neck: clinical and pathologic characteristics. Ann Otol Rhinol Laryngol. 1997;106:943-951.
- Holden CA, Spittle MF, Jones EW. Angiosarcoma of the face and scalp, prognosis and treatment. Cancer. 1987;59:1046-1057.
- Woodward AH, Ivins JC, Soule EH. Lymphangiosarcoma arising in chronic lymphedematous extremities. Cancer. 1972;30:562-572.
- Calonje E, Brenn T, McKee PH, et al. McKee’s Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier Saunders; 2012.
- Morrison WH, Byers RM, Garden AS, et al. Cutaneous angiosarcoma of the head and neck. a therapeutic dilemma. Cancer. 1995;76:319-327.
- Hodgkinson DJ, Soule EH, Woods JE. Cutaneous angiosarcoma of the head and neck. Cancer. 1979;44:1106-1113.
- Rosai J, Sumner HW, Kostianovsky M, et al. Angiosarcoma of the skin: a clinicopathologic and fine structural study. Hum Pathol. 1976;7:83-109.
- Pawlik TM, Paulino AF, Mcginn CJ, et al. Cutaneous angiosarcoma of the scalp: a multidisciplinary approach. Cancer. 2003;98:1716-1726.
- Haustein UF. Angiosarcoma of the face and scalp. Int J Dermatol. 1991;30:851-856.
- Elston DM, Ferringer T, Ko C, et al. Dermatopathology. 2nd ed. Edinburgh, Scotland: Saunders Elsevier; 2014.
- Requena L, Kutzner H. Cutaneous Soft Tissue Tumors. Philadelphia, PA: Wolters Kluwer; 2015.
- Cuda J, Mirzamani N, Kantipudi R, et al. Diagnostic utility of Fli-1 and D2-40 in distinguishing atypical fibroxanthoma from angiosarcoma. Am J Dermatopathol. 2013;35:316-318.
Cutaneous angiosarcoma is a rare malignant tumor of vascular endothelial cells that has the propensity to arise in various clinical settings. This tumor predominantly occurs in the head and neck region in elderly patients, but it also has been reported to develop postradiotherapy or in the setting of chronic lymphedema in the extremities.1-3 In all settings, the diagnosis carries a very poor prognosis with a high likelihood of local recurrence and rapid dissemination. The mortality rate typically is 80% or higher.2,4-6
Making the correct clinical diagnosis of cutaneous angiosarcoma may be difficult given the variety of patient symptoms and clinical appearances that can be demonstrated on presentation. Lesions can appear as bluish or violaceous plaques, macules, or nodules, and ulceration may be present in some advanced cases.5,7 Clinical misdiagnosis is common, as cutaneous angiosarcomas may be mistaken for infectious processes, benign vascular malformations, and other cutaneous malignancies.1 Biopsy often is delayed given the initial benign appearance of the lesions, and this frequently results in aggressive and extensive disease at the time of diagnosis, which is unfortunate given that small tumor size has been shown to be one of the only favorable prognostic indicators in cutaneous angiosarcoma.1,2,6,8
Microscopically, diagnosis of cutaneous angiosarcoma can present a challenge, as the histology varies between a well-differentiated vascular neoplasm and a considerably anaplastic and poorly differentiated malignancy. On low power, some areas may appear as benign hemangiomas with other areas showing frank sarcomatous features.9 As a result, these tumors can be mistaken for a variety of other diseases including melanomas, carcinomas, or other vascular tumors.6,8,9 Previously, electron microscopy has been utilized on undifferentiated tumors to help distinguish cutaneous angiosarcomas from other potential diagnoses. The atypical tumor cells of cutaneous angiosarcoma display common features of endothelial cells (eg, pinocytotic vesicles, tubulated bodies).7 Historically, it has been noted that the histologic findings and tumor grade provide little evidence regarding the aggressiveness of the tumor, and all cutaneous angiosarcoma diagnoses receive a poor prognosis.6,8
Classically, the histologic findings of cutaneous angiosarcoma include a highly infiltrative neoplasm forming irregular vascular channels that penetrate through the cutaneous soft tissues and frequently extend into the subcutaneous fat. The vascular spaces are lined by hyperchromatic endothelial cells with varying degrees of atypia.1,2,4,6,7,10 Occasionally, prominent endothelial cells lining a papillary structure within the lumen of the neoformed vessel may also be observed. Currently, immunohistochemical staining for MYC, Ki-67, D2-40, and various other markers complement the histologic findings to aid in the diagnosis of cutaneous angiosarcoma.11,12 An additional diagnostic clue that has been described in cases of postirradiation cutaneous angiosarcoma shows free-floating or tufted pleomorphic spindle cells within the vascular lumen (Figure). This finding has been described as “fish in the creek.”11 In this study, we aimed to determine the frequency and subsequent diagnostic utility of the fish-in-the-creek finding in cases of cutaneous angiosarcoma.
Methods
A natural language search of our institutional archives over a 20-year period (1997–2017) using the term angiosarcoma was performed. Fifteen cases of cutaneous angiosarcoma were identified. Fifteen additional benign and malignant vascular tumors with cutaneous angiosarco
Results
The histologic pattern of fish in the creek was identified in all 15 cases of cutaneous angiosarcoma and was absent in the other 15 malignancies examined in this study. This finding shows the potential for the fish-in-the-creek pattern to be used as an additional diagnostic tool for dermatopathologists.
Comment
Cutaneous angiosarcoma is a rare but aggressive malignancy that proves difficult to diagnose both clinically and histologically as well as to treat effectively.1,5-8 Our results indicate that fish in the creek may be a useful and salient histologic feature in cutaneous angiosarcoma. It is important to recognize, however, that this finding should not be the sole feature upon which a diagnosis of cutaneous angiosarcoma is made, as it requires corroboration with positivity of MYC and D2-40 as well as a high Ki-67 proliferation index (>20%).11,12 Finding a fish-in-the-creek pattern should prompt dermatopathologists to consider a diagnosis of cutaneous angiosarcoma in the appropriate clinical and histologic settings.
The chief limitation of this study was the small sample size, with only 15 cases of cutaneous angiosarcoma available in the last 20 years at our institution. The limited sample size did not allow us to make claims on sensitivity and specificity regarding this histologic feature; however, with a larger sample size, the true diagnostic potential could be elucidated. Although the pathologists were blinded to the original diagnoses as they examined it for fish in the creek, it is possible they were able to make the correct diagnosis based on other histopathologic clues and therefore were biased.
Although the fish-in-the-creek pattern is present in cutaneous angiosarcoma, there may be other mimickers to consider. Intraluminal papillary projections lined by endothelial cells may be sectioned in a manner imitating this finding.3 In such a case, these endothelial cells must be differentiated from the free-floating or tufted spindle cells in order to have a positive finding for fish in the creek. There can be confusion if the biopsy cuts through a section of spindled cells, resulting in difficulty differentiating cutaneous angiosarcoma from other spindle tumors such as spindle cell melanoma or spindle cell squamous cell carcinoma.6 In such cases, immunohistochemistry may be helpful, as spindle cell melanoma would stain positive for S100 and SOX10 and spindle cell squamous cell carcinoma would stain positive for p63 and cytokeratin.
Various treatment strategies for cutaneous angiosarcoma have been employed, with the majority still resulting in poor outcomes.2,4-6 The recommended treatment is radical surgical excision of the primary tumor with lymph node clearance if possible. Following excision, the patient should undergo high-dose, wide-field radiotherapy to the region.5,8 Cutaneous angiosarcomas also have the ability to spread extensively through the dermis and can result in subclinical or clinically obvious widespread disease with multifocal or satellite lesions present. Distant metastases occur most frequently in the cervical lymph nodes and lungs.7 In cases where the disease is too extensive for surgery, palliative radiation monotherapy can be used.5,6
As atypical vascular lesions are considered to be a precursor to cutaneous angiosarcoma, it is important to note that the fish-in-the-creek feature was absent in all 6 of the atypical vascular lesions observed in the study. The differentiation generally is made based on MYC, which is present in cutaneous angiosarcomas and absent in atypical vascular lesions.10 The feature of fish in the creek may now be an additional clue for dermatopathologists to differentiate between angiosarcomas and other similar-appearing tumors.
Conclusion
Our study aimed to highlight an important histologic feature of cutaneous angiosarcomas that can aid in the diagnosis of this deceptive malignancy. Our findings warrant further study of the fish-in-the-creek histologic pattern in a larger sample size to determine its success as a diagnostic tool for cutaneous angiosarcomas. As noted previously, tumor grade does not impact survival outcome, but small tumor size has been one of the only features found to result in a more favorable prognosis.1,6,8 Future studies to identify a correlation between the histologic finding of fish in the creek and disease outcome in cutaneous angiosarcoma may be helpful to determine if these histologic findings provide prognostic significance in cases of cutaneous angiosarcoma.
Cutaneous angiosarcoma is a rare malignant tumor of vascular endothelial cells that has the propensity to arise in various clinical settings. This tumor predominantly occurs in the head and neck region in elderly patients, but it also has been reported to develop postradiotherapy or in the setting of chronic lymphedema in the extremities.1-3 In all settings, the diagnosis carries a very poor prognosis with a high likelihood of local recurrence and rapid dissemination. The mortality rate typically is 80% or higher.2,4-6
Making the correct clinical diagnosis of cutaneous angiosarcoma may be difficult given the variety of patient symptoms and clinical appearances that can be demonstrated on presentation. Lesions can appear as bluish or violaceous plaques, macules, or nodules, and ulceration may be present in some advanced cases.5,7 Clinical misdiagnosis is common, as cutaneous angiosarcomas may be mistaken for infectious processes, benign vascular malformations, and other cutaneous malignancies.1 Biopsy often is delayed given the initial benign appearance of the lesions, and this frequently results in aggressive and extensive disease at the time of diagnosis, which is unfortunate given that small tumor size has been shown to be one of the only favorable prognostic indicators in cutaneous angiosarcoma.1,2,6,8
Microscopically, diagnosis of cutaneous angiosarcoma can present a challenge, as the histology varies between a well-differentiated vascular neoplasm and a considerably anaplastic and poorly differentiated malignancy. On low power, some areas may appear as benign hemangiomas with other areas showing frank sarcomatous features.9 As a result, these tumors can be mistaken for a variety of other diseases including melanomas, carcinomas, or other vascular tumors.6,8,9 Previously, electron microscopy has been utilized on undifferentiated tumors to help distinguish cutaneous angiosarcomas from other potential diagnoses. The atypical tumor cells of cutaneous angiosarcoma display common features of endothelial cells (eg, pinocytotic vesicles, tubulated bodies).7 Historically, it has been noted that the histologic findings and tumor grade provide little evidence regarding the aggressiveness of the tumor, and all cutaneous angiosarcoma diagnoses receive a poor prognosis.6,8
Classically, the histologic findings of cutaneous angiosarcoma include a highly infiltrative neoplasm forming irregular vascular channels that penetrate through the cutaneous soft tissues and frequently extend into the subcutaneous fat. The vascular spaces are lined by hyperchromatic endothelial cells with varying degrees of atypia.1,2,4,6,7,10 Occasionally, prominent endothelial cells lining a papillary structure within the lumen of the neoformed vessel may also be observed. Currently, immunohistochemical staining for MYC, Ki-67, D2-40, and various other markers complement the histologic findings to aid in the diagnosis of cutaneous angiosarcoma.11,12 An additional diagnostic clue that has been described in cases of postirradiation cutaneous angiosarcoma shows free-floating or tufted pleomorphic spindle cells within the vascular lumen (Figure). This finding has been described as “fish in the creek.”11 In this study, we aimed to determine the frequency and subsequent diagnostic utility of the fish-in-the-creek finding in cases of cutaneous angiosarcoma.
Methods
A natural language search of our institutional archives over a 20-year period (1997–2017) using the term angiosarcoma was performed. Fifteen cases of cutaneous angiosarcoma were identified. Fifteen additional benign and malignant vascular tumors with cutaneous angiosarco
Results
The histologic pattern of fish in the creek was identified in all 15 cases of cutaneous angiosarcoma and was absent in the other 15 malignancies examined in this study. This finding shows the potential for the fish-in-the-creek pattern to be used as an additional diagnostic tool for dermatopathologists.
Comment
Cutaneous angiosarcoma is a rare but aggressive malignancy that proves difficult to diagnose both clinically and histologically as well as to treat effectively.1,5-8 Our results indicate that fish in the creek may be a useful and salient histologic feature in cutaneous angiosarcoma. It is important to recognize, however, that this finding should not be the sole feature upon which a diagnosis of cutaneous angiosarcoma is made, as it requires corroboration with positivity of MYC and D2-40 as well as a high Ki-67 proliferation index (>20%).11,12 Finding a fish-in-the-creek pattern should prompt dermatopathologists to consider a diagnosis of cutaneous angiosarcoma in the appropriate clinical and histologic settings.
The chief limitation of this study was the small sample size, with only 15 cases of cutaneous angiosarcoma available in the last 20 years at our institution. The limited sample size did not allow us to make claims on sensitivity and specificity regarding this histologic feature; however, with a larger sample size, the true diagnostic potential could be elucidated. Although the pathologists were blinded to the original diagnoses as they examined it for fish in the creek, it is possible they were able to make the correct diagnosis based on other histopathologic clues and therefore were biased.
Although the fish-in-the-creek pattern is present in cutaneous angiosarcoma, there may be other mimickers to consider. Intraluminal papillary projections lined by endothelial cells may be sectioned in a manner imitating this finding.3 In such a case, these endothelial cells must be differentiated from the free-floating or tufted spindle cells in order to have a positive finding for fish in the creek. There can be confusion if the biopsy cuts through a section of spindled cells, resulting in difficulty differentiating cutaneous angiosarcoma from other spindle tumors such as spindle cell melanoma or spindle cell squamous cell carcinoma.6 In such cases, immunohistochemistry may be helpful, as spindle cell melanoma would stain positive for S100 and SOX10 and spindle cell squamous cell carcinoma would stain positive for p63 and cytokeratin.
Various treatment strategies for cutaneous angiosarcoma have been employed, with the majority still resulting in poor outcomes.2,4-6 The recommended treatment is radical surgical excision of the primary tumor with lymph node clearance if possible. Following excision, the patient should undergo high-dose, wide-field radiotherapy to the region.5,8 Cutaneous angiosarcomas also have the ability to spread extensively through the dermis and can result in subclinical or clinically obvious widespread disease with multifocal or satellite lesions present. Distant metastases occur most frequently in the cervical lymph nodes and lungs.7 In cases where the disease is too extensive for surgery, palliative radiation monotherapy can be used.5,6
As atypical vascular lesions are considered to be a precursor to cutaneous angiosarcoma, it is important to note that the fish-in-the-creek feature was absent in all 6 of the atypical vascular lesions observed in the study. The differentiation generally is made based on MYC, which is present in cutaneous angiosarcomas and absent in atypical vascular lesions.10 The feature of fish in the creek may now be an additional clue for dermatopathologists to differentiate between angiosarcomas and other similar-appearing tumors.
Conclusion
Our study aimed to highlight an important histologic feature of cutaneous angiosarcomas that can aid in the diagnosis of this deceptive malignancy. Our findings warrant further study of the fish-in-the-creek histologic pattern in a larger sample size to determine its success as a diagnostic tool for cutaneous angiosarcomas. As noted previously, tumor grade does not impact survival outcome, but small tumor size has been one of the only features found to result in a more favorable prognosis.1,6,8 Future studies to identify a correlation between the histologic finding of fish in the creek and disease outcome in cutaneous angiosarcoma may be helpful to determine if these histologic findings provide prognostic significance in cases of cutaneous angiosarcoma.
- Aust MR, Olsen KD, Lewis JE, et al. Angiosarcomas of the head and neck: clinical and pathologic characteristics. Ann Otol Rhinol Laryngol. 1997;106:943-951.
- Holden CA, Spittle MF, Jones EW. Angiosarcoma of the face and scalp, prognosis and treatment. Cancer. 1987;59:1046-1057.
- Woodward AH, Ivins JC, Soule EH. Lymphangiosarcoma arising in chronic lymphedematous extremities. Cancer. 1972;30:562-572.
- Calonje E, Brenn T, McKee PH, et al. McKee’s Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier Saunders; 2012.
- Morrison WH, Byers RM, Garden AS, et al. Cutaneous angiosarcoma of the head and neck. a therapeutic dilemma. Cancer. 1995;76:319-327.
- Hodgkinson DJ, Soule EH, Woods JE. Cutaneous angiosarcoma of the head and neck. Cancer. 1979;44:1106-1113.
- Rosai J, Sumner HW, Kostianovsky M, et al. Angiosarcoma of the skin: a clinicopathologic and fine structural study. Hum Pathol. 1976;7:83-109.
- Pawlik TM, Paulino AF, Mcginn CJ, et al. Cutaneous angiosarcoma of the scalp: a multidisciplinary approach. Cancer. 2003;98:1716-1726.
- Haustein UF. Angiosarcoma of the face and scalp. Int J Dermatol. 1991;30:851-856.
- Elston DM, Ferringer T, Ko C, et al. Dermatopathology. 2nd ed. Edinburgh, Scotland: Saunders Elsevier; 2014.
- Requena L, Kutzner H. Cutaneous Soft Tissue Tumors. Philadelphia, PA: Wolters Kluwer; 2015.
- Cuda J, Mirzamani N, Kantipudi R, et al. Diagnostic utility of Fli-1 and D2-40 in distinguishing atypical fibroxanthoma from angiosarcoma. Am J Dermatopathol. 2013;35:316-318.
- Aust MR, Olsen KD, Lewis JE, et al. Angiosarcomas of the head and neck: clinical and pathologic characteristics. Ann Otol Rhinol Laryngol. 1997;106:943-951.
- Holden CA, Spittle MF, Jones EW. Angiosarcoma of the face and scalp, prognosis and treatment. Cancer. 1987;59:1046-1057.
- Woodward AH, Ivins JC, Soule EH. Lymphangiosarcoma arising in chronic lymphedematous extremities. Cancer. 1972;30:562-572.
- Calonje E, Brenn T, McKee PH, et al. McKee’s Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier Saunders; 2012.
- Morrison WH, Byers RM, Garden AS, et al. Cutaneous angiosarcoma of the head and neck. a therapeutic dilemma. Cancer. 1995;76:319-327.
- Hodgkinson DJ, Soule EH, Woods JE. Cutaneous angiosarcoma of the head and neck. Cancer. 1979;44:1106-1113.
- Rosai J, Sumner HW, Kostianovsky M, et al. Angiosarcoma of the skin: a clinicopathologic and fine structural study. Hum Pathol. 1976;7:83-109.
- Pawlik TM, Paulino AF, Mcginn CJ, et al. Cutaneous angiosarcoma of the scalp: a multidisciplinary approach. Cancer. 2003;98:1716-1726.
- Haustein UF. Angiosarcoma of the face and scalp. Int J Dermatol. 1991;30:851-856.
- Elston DM, Ferringer T, Ko C, et al. Dermatopathology. 2nd ed. Edinburgh, Scotland: Saunders Elsevier; 2014.
- Requena L, Kutzner H. Cutaneous Soft Tissue Tumors. Philadelphia, PA: Wolters Kluwer; 2015.
- Cuda J, Mirzamani N, Kantipudi R, et al. Diagnostic utility of Fli-1 and D2-40 in distinguishing atypical fibroxanthoma from angiosarcoma. Am J Dermatopathol. 2013;35:316-318.
Practice Points
- The histologic finding of “fish in the creek” is characterized by free-floating or tufted pleomorphic spindle cells within the vascular lumen.
- Fish in the creek has only been demonstrated in cutaneous angiosarcoma when compared to histologic findings of other similar vascular malignancies.
- The fish-in-the-creek finding may be an additional diagnostic tool in cases of cutaneous angiosarcoma.
Perianal Condyloma Acuminatum-like Plaque
The Diagnosis: Metastatic Crohn Disease
Crohn disease (CD), a chronic inflammatory granulomatous disease of the gastrointestinal tract, has a wide spectrum of presentations.1 The condition may affect the vulva, perineum, or perianal skin by direct extension from the gastrointestinal tract or may appear as a separate and distinct cutaneous focus of disease referred to as metastatic Crohn disease (MCD).2
Cutaneous lesions of MCD include ulcers, fissures, sinus tracts, abscesses, and vegetative plaques, which typically extend in continuity with sites of intra-abdominal disease to the perineum, buttocks, or abdominal wall, as well as ostomy sites or incisional scars. Erythema nodosum and pyoderma gangrenosum are the most common nonspecific cutaneous manifestations. Other cutaneous lesions described in CD include polyarteritis nodosa, psoriasis, erythema multiforme, erythema elevatum diutinum, epidermolysis bullosa acquisita, acne fulminans, pyoderma faciale, neutrophilic lobular panniculitis, granulomatous vasculitis, and porokeratosis.3
Perianal skin is the most common site of cutaneous involvement in individuals with CD. It is a marker of more severe disease and is associated with multiple surgical interventions and frequent relapses and has been reported in 22% of patients with CD.4 Most already had an existing diagnosis of gastrointestinal CD, which was active in one-third of individuals; however, 20% presented with disease at nongastrointestinal sites 2 months to 4 years prior to developing the gastrointestinal CD manifestations.5 Our patient presented with lesions on the perianal skin of 2 years' duration and a 6-month history of diarrhea. A colonoscopy demonstrated shallow ulcers involving the ileocecal portion of the gut, colon, and rectum. A biopsy from intestinal mucosal tissue showed acute and chronic inflammation with necrosis mixed with granulomatous inflammation, suggestive of CD.
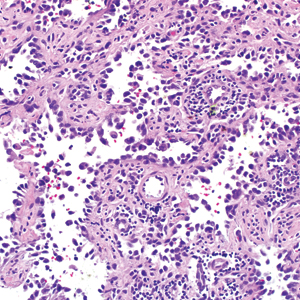
Microscopically, the dominant histologic features of MCD are similar to those of bowel lesions, including an inflammatory infiltrate commonly consisting of sterile noncaseating sarcoidal granulomas, foreign body and Langhans giant cells, epithelioid histiocytes, and plasma cells surrounded by numerous mononuclear cells within the dermis with occasional extension into the subcutis (quiz image). Less common features include collagen degeneration, an infiltrate rich in eosinophils, dermal edema, and mixed lichenoid and granulomatous dermatitis.6
Metastatic CD often is misdiagnosed. A detailed history and physical examination may help narrow the differential; however, biopsy is necessary to establish a diagnosis of MCD. The histologic differential diagnosis of sarcoidal granulomatous inflammation of genital skin includes sarcoidosis, rheumatoid arthritis, leprosy or other mycobacterial and parasitic infection, granulomatosis with polyangiitis (GPA), and granulomatous infiltrate associated with certain exogenous material (eg, silica, zirconium, beryllium, tattoo pigment).
Sarcoidosis is a multiorgan disease that most frequently affects the lungs, skin, and lymph nodes. Its etiopathogenesis has not been clearly elucidated.7 Cutaneous lesions are present in 20% to 35% of patients.8 Given the wide variability of clinical manifestations, cutaneous sarcoidosis is another one of the great imitators. Cutaneous lesions are classified as specific and nonspecific depending on the presence of noncaseating granulomas on histologic studies and include maculopapules, plaques, nodules, lupus pernio, scar infiltration, alopecia, ulcerative lesions, and hypopigmentation. The most common nonspecific lesion of cutaneous sarcoidosis is erythema nodosum. Other manifestations include calcifications, prurigo, erythema multiforme, nail clubbing, and Sweet syndrome.9
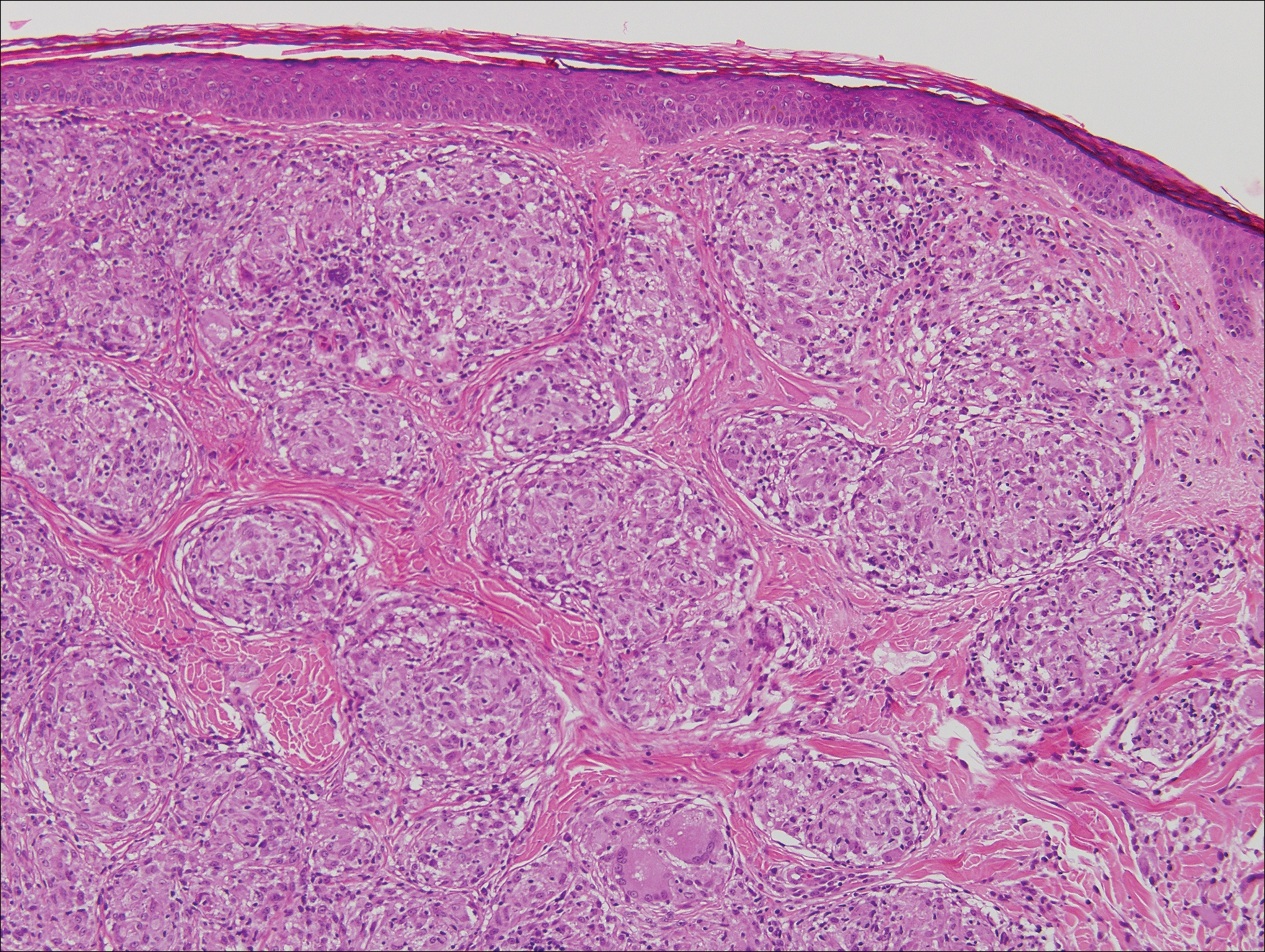
Histologic findings in sarcoidosis generally are independent of the respective organ and clinical disease presentation. The epidermis usually remains unchanged, whereas the dermis shows a superficial and deep nodular granulomatous infiltrate. Granulomas consist of epithelioid cells with only few giant cells and no surrounding lymphocytes or a very sparse lymphocytic infiltrate ("naked" granuloma)(Figure 1). Foreign bodies, including silica, are known to be able to induce sarcoid granulomas, especially in patients with sarcoidosis. A sarcoidal reaction in long-standing scar tissue points to a diagnosis of sarcoidosis.10
Cutaneous tuberculosis primarily is caused by Mycobacterium tuberculosis and less frequently Mycobacterium bovis.11,12 The manifestations of cutaneous tuberculosis depends on various factors such as the type of infection, mode of dissemination, host immunity, and whether it is a first-time infection or a recurrence. In Europe, the head and neck regions are most frequently affected.13 Lesions present as red-brown papules coalescing into a plaque. The tissue, especially in central parts of the lesion, is fragile (probe phenomenon). Diascopy shows the typical apple jelly-like color.
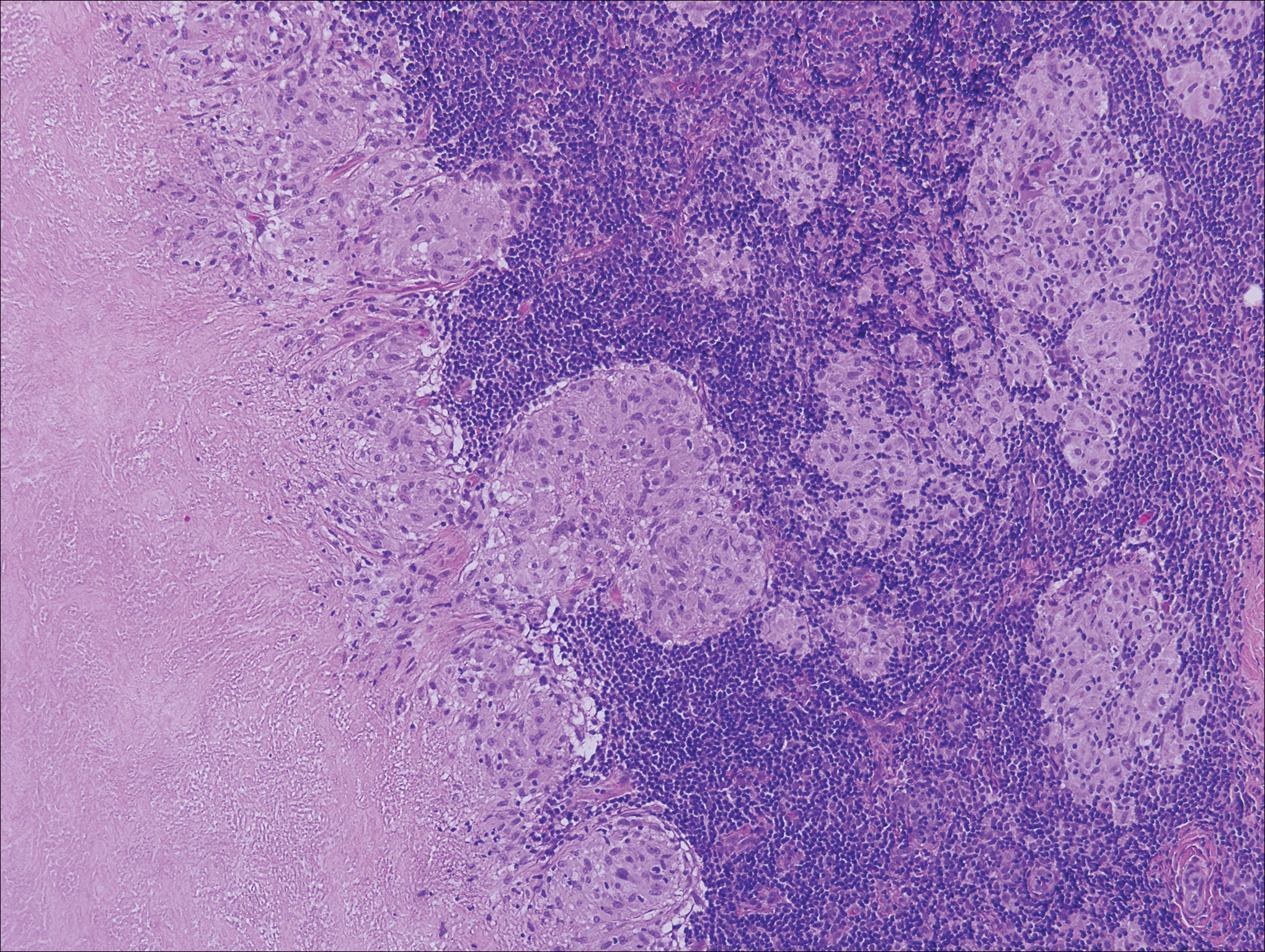
Histologically, cutaneous tuberculosis is characterized by typical tuberculoid granulomas with epithelioid cells and Langhans giant cells at the center surrounded by lymphocytes (Figure 2). Caseous necrosis as well as fibrosis may occur,14,15 and the granulomas tend to coalesce.
Granulomatosis with polyangiitis, formerly known as Wegener granulomatosis, is a complex, multisystemic disease with varying manifestations. The condition has been defined as a necrotizing granulomatous inflammation usually involving the upper and lower respiratory tracts and necrotizing vasculitis affecting predominantly small- to medium-sized vessels.16 The etiology of GPA is thought to be linked to environmental and infectious triggers inciting onset of disease in genetically predisposed individuals. Antineutrophil cytoplasmic antibodies play an important role in the pathogenesis of this disease. Cutaneous vasculitis secondary to GPA can present as papules, nodules, palpable purpura, ulcers resembling pyoderma gangrenosum, or necrotizing lesions leading to gangrene.17
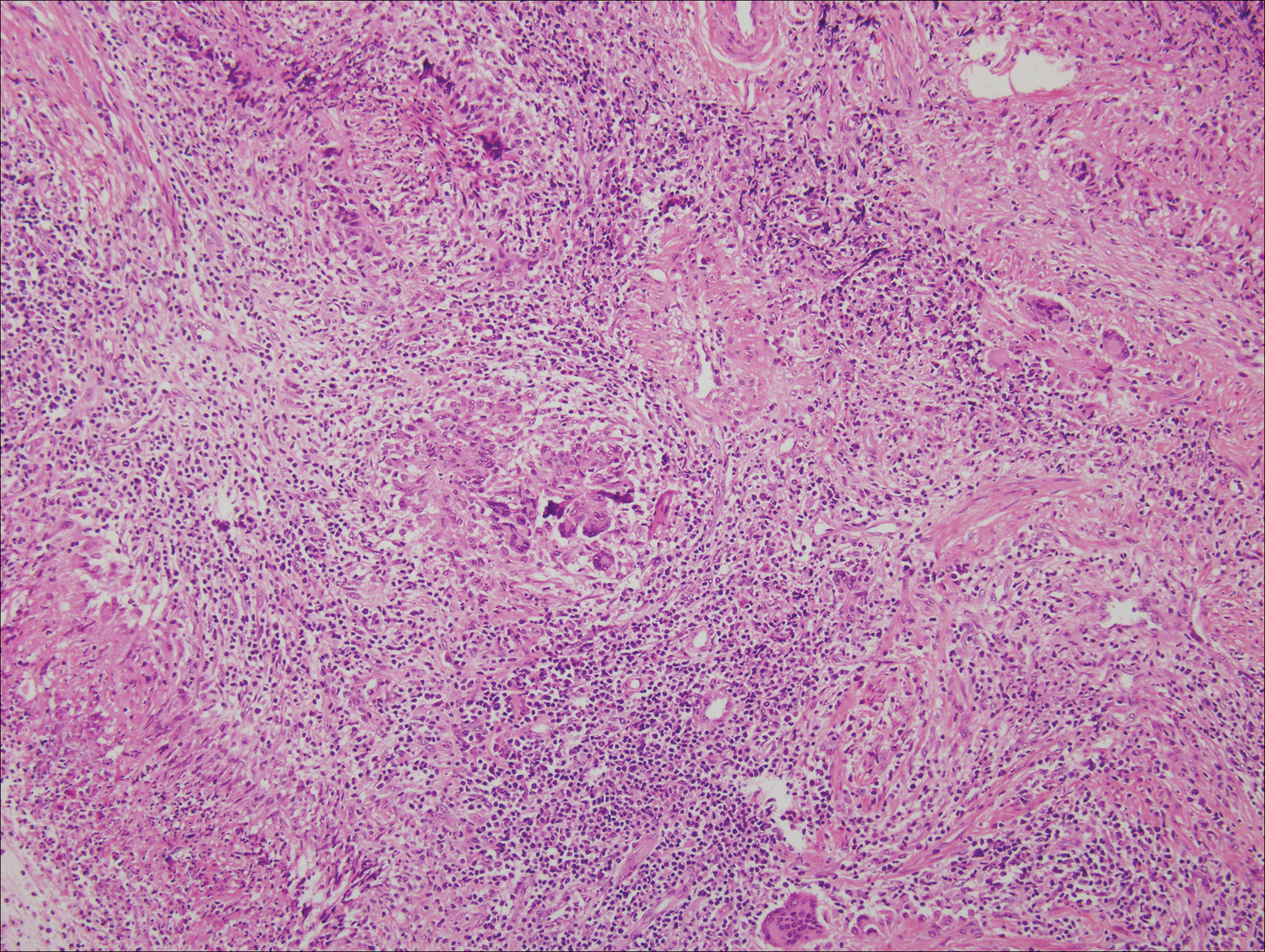
The predominant histopathologic pattern in cutaneous lesions of GPA is leukocytoclastic vasculitis, which is present in up to 50% of biopsies.18 Characteristic findings that aid in establishing the diagnosis include histologic evidence of focal necrosis, fibrinoid degeneration, palisading granuloma surrounding neutrophils (Figure 3), and granulomatous vasculitis involving muscular vessel walls.19 Nonpalisading foci of necrosis or fibrinoid degeneration may precede the development of the typical palisading granuloma.20
The typical histopathologic pattern of cutaneous amebiasis is ulceration with vascular necrosis (Figure 4).21 The organisms have prominent round nuclei and nucleoli and the cytoplasm may have a scalloped border.
- Crohn BB, Ginzburg L, Oppenheimer GD. Landmark article Oct 25, 1932. regional ileitis. a pathologic and clinical entity. by Burril B. Crohn, Leon Gonzburg and Gordon D. Oppenheimer. JAMA. 1984;251:73-79.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Weedon D. Miscellaneous conditions. Skin Pathology. 2nd ed. London, England: Churchill Livingstone; 2002:554.
- Samitz MH, Dana Jr AS, Rosenberg P. Cutaneous vasculitis in association with Crohn's disease. Cutis. 1970;6:51-56.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Aberumand B, Howard J, Howard J. Metastatic Crohn's disease: an approach to an uncommon but important cutaneous disorder: a review [published online January 3, 2017]. BioMed Res Int. 2017;2017:8192150.
- Mahony J, Helms SE, Brodell RT. The sarcoidal granuloma: a unifying hypothesis for an enigmatic response. Clin Dermatol. 2014;32:654-659.
- Freedberg IM, Eisen AZ, Wolf K, et al. Fitzpatrick's Dermatology in General Medicine. 6th ed. New York, NY: McGraw Hill; 2003.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
- Walsh NM, Hanly JG, Tremaine R, et al. Cutaneous sarcoidosis and foreign bodies. Am J Dermatopathol. 1993;15:203-207.
- Semaan R, Traboulsi R, Kanj S. Primary Mycobacterium tuberculosis complex cutaneous infection: report of two cases and literature review. Int J Infect Dis. 2008;12:472-477.
- Lai-Cheong JE, Perez A, Tang V, et al. Cutaneous manifestations of tuberculosis. Clin Exp Dermatol. 2007;32:461-466.
- Marcoval J, Servitje O, Moreno A, et al. Lupus vulgaris. clinical, histopathologic, and bacteriologic study of 10 cases. J Am Acad Dermatol. 1992;26:404-407.
- Tronnier M, Wolff H. Dermatosen mit granulomatöser Entzündung. Histopathologie der Haut. In: Kerl H, Garbe C, Cerroni L, et al, eds. New York, NY: Springer; 2003.
- Min KW, Ko JY, Park CK. Histopathological spectrum of cutaneous tuberculosis and non-tuberculous mycobacterial infections. J Cutan Pathol. 2012;39:582-595.
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1-11.
- Comfere NI, Macaron NC, Gibson LE. Cutaneous manifestations of Wegener's granulomatosis: a clinicopathologic study of 17 patients and correlation to antineutrophil cytoplasmic antibody status. J Cutan Pathol. 2007;34:739-747.
- Marzano AV, Vezzoli P, Berti E. Skin involvement in cutaneous and systemic vasculitis. Autoimmun Rev. 2012;12:467-476.
- Bramsiepe I, Danz B, Heine R, et al. Primary cutaneous manifestation of Wegener's granulomatosis [in German]. Dtsch Med Wochenschr. 2008;27:1429-1432.
- Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener's granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
- Guidry JA, Downing C, Tyring SK. Deep fungal infections, blastomycosis-like pyoderma, and granulomatous sexually transmitted infections. Dermatol Clin. 2015;33:595-607.
The Diagnosis: Metastatic Crohn Disease
Crohn disease (CD), a chronic inflammatory granulomatous disease of the gastrointestinal tract, has a wide spectrum of presentations.1 The condition may affect the vulva, perineum, or perianal skin by direct extension from the gastrointestinal tract or may appear as a separate and distinct cutaneous focus of disease referred to as metastatic Crohn disease (MCD).2
Cutaneous lesions of MCD include ulcers, fissures, sinus tracts, abscesses, and vegetative plaques, which typically extend in continuity with sites of intra-abdominal disease to the perineum, buttocks, or abdominal wall, as well as ostomy sites or incisional scars. Erythema nodosum and pyoderma gangrenosum are the most common nonspecific cutaneous manifestations. Other cutaneous lesions described in CD include polyarteritis nodosa, psoriasis, erythema multiforme, erythema elevatum diutinum, epidermolysis bullosa acquisita, acne fulminans, pyoderma faciale, neutrophilic lobular panniculitis, granulomatous vasculitis, and porokeratosis.3
Perianal skin is the most common site of cutaneous involvement in individuals with CD. It is a marker of more severe disease and is associated with multiple surgical interventions and frequent relapses and has been reported in 22% of patients with CD.4 Most already had an existing diagnosis of gastrointestinal CD, which was active in one-third of individuals; however, 20% presented with disease at nongastrointestinal sites 2 months to 4 years prior to developing the gastrointestinal CD manifestations.5 Our patient presented with lesions on the perianal skin of 2 years' duration and a 6-month history of diarrhea. A colonoscopy demonstrated shallow ulcers involving the ileocecal portion of the gut, colon, and rectum. A biopsy from intestinal mucosal tissue showed acute and chronic inflammation with necrosis mixed with granulomatous inflammation, suggestive of CD.
Microscopically, the dominant histologic features of MCD are similar to those of bowel lesions, including an inflammatory infiltrate commonly consisting of sterile noncaseating sarcoidal granulomas, foreign body and Langhans giant cells, epithelioid histiocytes, and plasma cells surrounded by numerous mononuclear cells within the dermis with occasional extension into the subcutis (quiz image). Less common features include collagen degeneration, an infiltrate rich in eosinophils, dermal edema, and mixed lichenoid and granulomatous dermatitis.6
Metastatic CD often is misdiagnosed. A detailed history and physical examination may help narrow the differential; however, biopsy is necessary to establish a diagnosis of MCD. The histologic differential diagnosis of sarcoidal granulomatous inflammation of genital skin includes sarcoidosis, rheumatoid arthritis, leprosy or other mycobacterial and parasitic infection, granulomatosis with polyangiitis (GPA), and granulomatous infiltrate associated with certain exogenous material (eg, silica, zirconium, beryllium, tattoo pigment).
Sarcoidosis is a multiorgan disease that most frequently affects the lungs, skin, and lymph nodes. Its etiopathogenesis has not been clearly elucidated.7 Cutaneous lesions are present in 20% to 35% of patients.8 Given the wide variability of clinical manifestations, cutaneous sarcoidosis is another one of the great imitators. Cutaneous lesions are classified as specific and nonspecific depending on the presence of noncaseating granulomas on histologic studies and include maculopapules, plaques, nodules, lupus pernio, scar infiltration, alopecia, ulcerative lesions, and hypopigmentation. The most common nonspecific lesion of cutaneous sarcoidosis is erythema nodosum. Other manifestations include calcifications, prurigo, erythema multiforme, nail clubbing, and Sweet syndrome.9
Histologic findings in sarcoidosis generally are independent of the respective organ and clinical disease presentation. The epidermis usually remains unchanged, whereas the dermis shows a superficial and deep nodular granulomatous infiltrate. Granulomas consist of epithelioid cells with only few giant cells and no surrounding lymphocytes or a very sparse lymphocytic infiltrate ("naked" granuloma)(Figure 1). Foreign bodies, including silica, are known to be able to induce sarcoid granulomas, especially in patients with sarcoidosis. A sarcoidal reaction in long-standing scar tissue points to a diagnosis of sarcoidosis.10
Cutaneous tuberculosis primarily is caused by Mycobacterium tuberculosis and less frequently Mycobacterium bovis.11,12 The manifestations of cutaneous tuberculosis depends on various factors such as the type of infection, mode of dissemination, host immunity, and whether it is a first-time infection or a recurrence. In Europe, the head and neck regions are most frequently affected.13 Lesions present as red-brown papules coalescing into a plaque. The tissue, especially in central parts of the lesion, is fragile (probe phenomenon). Diascopy shows the typical apple jelly-like color.
Histologically, cutaneous tuberculosis is characterized by typical tuberculoid granulomas with epithelioid cells and Langhans giant cells at the center surrounded by lymphocytes (Figure 2). Caseous necrosis as well as fibrosis may occur,14,15 and the granulomas tend to coalesce.
Granulomatosis with polyangiitis, formerly known as Wegener granulomatosis, is a complex, multisystemic disease with varying manifestations. The condition has been defined as a necrotizing granulomatous inflammation usually involving the upper and lower respiratory tracts and necrotizing vasculitis affecting predominantly small- to medium-sized vessels.16 The etiology of GPA is thought to be linked to environmental and infectious triggers inciting onset of disease in genetically predisposed individuals. Antineutrophil cytoplasmic antibodies play an important role in the pathogenesis of this disease. Cutaneous vasculitis secondary to GPA can present as papules, nodules, palpable purpura, ulcers resembling pyoderma gangrenosum, or necrotizing lesions leading to gangrene.17
The predominant histopathologic pattern in cutaneous lesions of GPA is leukocytoclastic vasculitis, which is present in up to 50% of biopsies.18 Characteristic findings that aid in establishing the diagnosis include histologic evidence of focal necrosis, fibrinoid degeneration, palisading granuloma surrounding neutrophils (Figure 3), and granulomatous vasculitis involving muscular vessel walls.19 Nonpalisading foci of necrosis or fibrinoid degeneration may precede the development of the typical palisading granuloma.20
The typical histopathologic pattern of cutaneous amebiasis is ulceration with vascular necrosis (Figure 4).21 The organisms have prominent round nuclei and nucleoli and the cytoplasm may have a scalloped border.
The Diagnosis: Metastatic Crohn Disease
Crohn disease (CD), a chronic inflammatory granulomatous disease of the gastrointestinal tract, has a wide spectrum of presentations.1 The condition may affect the vulva, perineum, or perianal skin by direct extension from the gastrointestinal tract or may appear as a separate and distinct cutaneous focus of disease referred to as metastatic Crohn disease (MCD).2
Cutaneous lesions of MCD include ulcers, fissures, sinus tracts, abscesses, and vegetative plaques, which typically extend in continuity with sites of intra-abdominal disease to the perineum, buttocks, or abdominal wall, as well as ostomy sites or incisional scars. Erythema nodosum and pyoderma gangrenosum are the most common nonspecific cutaneous manifestations. Other cutaneous lesions described in CD include polyarteritis nodosa, psoriasis, erythema multiforme, erythema elevatum diutinum, epidermolysis bullosa acquisita, acne fulminans, pyoderma faciale, neutrophilic lobular panniculitis, granulomatous vasculitis, and porokeratosis.3
Perianal skin is the most common site of cutaneous involvement in individuals with CD. It is a marker of more severe disease and is associated with multiple surgical interventions and frequent relapses and has been reported in 22% of patients with CD.4 Most already had an existing diagnosis of gastrointestinal CD, which was active in one-third of individuals; however, 20% presented with disease at nongastrointestinal sites 2 months to 4 years prior to developing the gastrointestinal CD manifestations.5 Our patient presented with lesions on the perianal skin of 2 years' duration and a 6-month history of diarrhea. A colonoscopy demonstrated shallow ulcers involving the ileocecal portion of the gut, colon, and rectum. A biopsy from intestinal mucosal tissue showed acute and chronic inflammation with necrosis mixed with granulomatous inflammation, suggestive of CD.
Microscopically, the dominant histologic features of MCD are similar to those of bowel lesions, including an inflammatory infiltrate commonly consisting of sterile noncaseating sarcoidal granulomas, foreign body and Langhans giant cells, epithelioid histiocytes, and plasma cells surrounded by numerous mononuclear cells within the dermis with occasional extension into the subcutis (quiz image). Less common features include collagen degeneration, an infiltrate rich in eosinophils, dermal edema, and mixed lichenoid and granulomatous dermatitis.6
Metastatic CD often is misdiagnosed. A detailed history and physical examination may help narrow the differential; however, biopsy is necessary to establish a diagnosis of MCD. The histologic differential diagnosis of sarcoidal granulomatous inflammation of genital skin includes sarcoidosis, rheumatoid arthritis, leprosy or other mycobacterial and parasitic infection, granulomatosis with polyangiitis (GPA), and granulomatous infiltrate associated with certain exogenous material (eg, silica, zirconium, beryllium, tattoo pigment).
Sarcoidosis is a multiorgan disease that most frequently affects the lungs, skin, and lymph nodes. Its etiopathogenesis has not been clearly elucidated.7 Cutaneous lesions are present in 20% to 35% of patients.8 Given the wide variability of clinical manifestations, cutaneous sarcoidosis is another one of the great imitators. Cutaneous lesions are classified as specific and nonspecific depending on the presence of noncaseating granulomas on histologic studies and include maculopapules, plaques, nodules, lupus pernio, scar infiltration, alopecia, ulcerative lesions, and hypopigmentation. The most common nonspecific lesion of cutaneous sarcoidosis is erythema nodosum. Other manifestations include calcifications, prurigo, erythema multiforme, nail clubbing, and Sweet syndrome.9
Histologic findings in sarcoidosis generally are independent of the respective organ and clinical disease presentation. The epidermis usually remains unchanged, whereas the dermis shows a superficial and deep nodular granulomatous infiltrate. Granulomas consist of epithelioid cells with only few giant cells and no surrounding lymphocytes or a very sparse lymphocytic infiltrate ("naked" granuloma)(Figure 1). Foreign bodies, including silica, are known to be able to induce sarcoid granulomas, especially in patients with sarcoidosis. A sarcoidal reaction in long-standing scar tissue points to a diagnosis of sarcoidosis.10
Cutaneous tuberculosis primarily is caused by Mycobacterium tuberculosis and less frequently Mycobacterium bovis.11,12 The manifestations of cutaneous tuberculosis depends on various factors such as the type of infection, mode of dissemination, host immunity, and whether it is a first-time infection or a recurrence. In Europe, the head and neck regions are most frequently affected.13 Lesions present as red-brown papules coalescing into a plaque. The tissue, especially in central parts of the lesion, is fragile (probe phenomenon). Diascopy shows the typical apple jelly-like color.
Histologically, cutaneous tuberculosis is characterized by typical tuberculoid granulomas with epithelioid cells and Langhans giant cells at the center surrounded by lymphocytes (Figure 2). Caseous necrosis as well as fibrosis may occur,14,15 and the granulomas tend to coalesce.
Granulomatosis with polyangiitis, formerly known as Wegener granulomatosis, is a complex, multisystemic disease with varying manifestations. The condition has been defined as a necrotizing granulomatous inflammation usually involving the upper and lower respiratory tracts and necrotizing vasculitis affecting predominantly small- to medium-sized vessels.16 The etiology of GPA is thought to be linked to environmental and infectious triggers inciting onset of disease in genetically predisposed individuals. Antineutrophil cytoplasmic antibodies play an important role in the pathogenesis of this disease. Cutaneous vasculitis secondary to GPA can present as papules, nodules, palpable purpura, ulcers resembling pyoderma gangrenosum, or necrotizing lesions leading to gangrene.17
The predominant histopathologic pattern in cutaneous lesions of GPA is leukocytoclastic vasculitis, which is present in up to 50% of biopsies.18 Characteristic findings that aid in establishing the diagnosis include histologic evidence of focal necrosis, fibrinoid degeneration, palisading granuloma surrounding neutrophils (Figure 3), and granulomatous vasculitis involving muscular vessel walls.19 Nonpalisading foci of necrosis or fibrinoid degeneration may precede the development of the typical palisading granuloma.20
The typical histopathologic pattern of cutaneous amebiasis is ulceration with vascular necrosis (Figure 4).21 The organisms have prominent round nuclei and nucleoli and the cytoplasm may have a scalloped border.
- Crohn BB, Ginzburg L, Oppenheimer GD. Landmark article Oct 25, 1932. regional ileitis. a pathologic and clinical entity. by Burril B. Crohn, Leon Gonzburg and Gordon D. Oppenheimer. JAMA. 1984;251:73-79.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Weedon D. Miscellaneous conditions. Skin Pathology. 2nd ed. London, England: Churchill Livingstone; 2002:554.
- Samitz MH, Dana Jr AS, Rosenberg P. Cutaneous vasculitis in association with Crohn's disease. Cutis. 1970;6:51-56.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Aberumand B, Howard J, Howard J. Metastatic Crohn's disease: an approach to an uncommon but important cutaneous disorder: a review [published online January 3, 2017]. BioMed Res Int. 2017;2017:8192150.
- Mahony J, Helms SE, Brodell RT. The sarcoidal granuloma: a unifying hypothesis for an enigmatic response. Clin Dermatol. 2014;32:654-659.
- Freedberg IM, Eisen AZ, Wolf K, et al. Fitzpatrick's Dermatology in General Medicine. 6th ed. New York, NY: McGraw Hill; 2003.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
- Walsh NM, Hanly JG, Tremaine R, et al. Cutaneous sarcoidosis and foreign bodies. Am J Dermatopathol. 1993;15:203-207.
- Semaan R, Traboulsi R, Kanj S. Primary Mycobacterium tuberculosis complex cutaneous infection: report of two cases and literature review. Int J Infect Dis. 2008;12:472-477.
- Lai-Cheong JE, Perez A, Tang V, et al. Cutaneous manifestations of tuberculosis. Clin Exp Dermatol. 2007;32:461-466.
- Marcoval J, Servitje O, Moreno A, et al. Lupus vulgaris. clinical, histopathologic, and bacteriologic study of 10 cases. J Am Acad Dermatol. 1992;26:404-407.
- Tronnier M, Wolff H. Dermatosen mit granulomatöser Entzündung. Histopathologie der Haut. In: Kerl H, Garbe C, Cerroni L, et al, eds. New York, NY: Springer; 2003.
- Min KW, Ko JY, Park CK. Histopathological spectrum of cutaneous tuberculosis and non-tuberculous mycobacterial infections. J Cutan Pathol. 2012;39:582-595.
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1-11.
- Comfere NI, Macaron NC, Gibson LE. Cutaneous manifestations of Wegener's granulomatosis: a clinicopathologic study of 17 patients and correlation to antineutrophil cytoplasmic antibody status. J Cutan Pathol. 2007;34:739-747.
- Marzano AV, Vezzoli P, Berti E. Skin involvement in cutaneous and systemic vasculitis. Autoimmun Rev. 2012;12:467-476.
- Bramsiepe I, Danz B, Heine R, et al. Primary cutaneous manifestation of Wegener's granulomatosis [in German]. Dtsch Med Wochenschr. 2008;27:1429-1432.
- Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener's granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
- Guidry JA, Downing C, Tyring SK. Deep fungal infections, blastomycosis-like pyoderma, and granulomatous sexually transmitted infections. Dermatol Clin. 2015;33:595-607.
- Crohn BB, Ginzburg L, Oppenheimer GD. Landmark article Oct 25, 1932. regional ileitis. a pathologic and clinical entity. by Burril B. Crohn, Leon Gonzburg and Gordon D. Oppenheimer. JAMA. 1984;251:73-79.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Weedon D. Miscellaneous conditions. Skin Pathology. 2nd ed. London, England: Churchill Livingstone; 2002:554.
- Samitz MH, Dana Jr AS, Rosenberg P. Cutaneous vasculitis in association with Crohn's disease. Cutis. 1970;6:51-56.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Aberumand B, Howard J, Howard J. Metastatic Crohn's disease: an approach to an uncommon but important cutaneous disorder: a review [published online January 3, 2017]. BioMed Res Int. 2017;2017:8192150.
- Mahony J, Helms SE, Brodell RT. The sarcoidal granuloma: a unifying hypothesis for an enigmatic response. Clin Dermatol. 2014;32:654-659.
- Freedberg IM, Eisen AZ, Wolf K, et al. Fitzpatrick's Dermatology in General Medicine. 6th ed. New York, NY: McGraw Hill; 2003.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
- Walsh NM, Hanly JG, Tremaine R, et al. Cutaneous sarcoidosis and foreign bodies. Am J Dermatopathol. 1993;15:203-207.
- Semaan R, Traboulsi R, Kanj S. Primary Mycobacterium tuberculosis complex cutaneous infection: report of two cases and literature review. Int J Infect Dis. 2008;12:472-477.
- Lai-Cheong JE, Perez A, Tang V, et al. Cutaneous manifestations of tuberculosis. Clin Exp Dermatol. 2007;32:461-466.
- Marcoval J, Servitje O, Moreno A, et al. Lupus vulgaris. clinical, histopathologic, and bacteriologic study of 10 cases. J Am Acad Dermatol. 1992;26:404-407.
- Tronnier M, Wolff H. Dermatosen mit granulomatöser Entzündung. Histopathologie der Haut. In: Kerl H, Garbe C, Cerroni L, et al, eds. New York, NY: Springer; 2003.
- Min KW, Ko JY, Park CK. Histopathological spectrum of cutaneous tuberculosis and non-tuberculous mycobacterial infections. J Cutan Pathol. 2012;39:582-595.
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1-11.
- Comfere NI, Macaron NC, Gibson LE. Cutaneous manifestations of Wegener's granulomatosis: a clinicopathologic study of 17 patients and correlation to antineutrophil cytoplasmic antibody status. J Cutan Pathol. 2007;34:739-747.
- Marzano AV, Vezzoli P, Berti E. Skin involvement in cutaneous and systemic vasculitis. Autoimmun Rev. 2012;12:467-476.
- Bramsiepe I, Danz B, Heine R, et al. Primary cutaneous manifestation of Wegener's granulomatosis [in German]. Dtsch Med Wochenschr. 2008;27:1429-1432.
- Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener's granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
- Guidry JA, Downing C, Tyring SK. Deep fungal infections, blastomycosis-like pyoderma, and granulomatous sexually transmitted infections. Dermatol Clin. 2015;33:595-607.
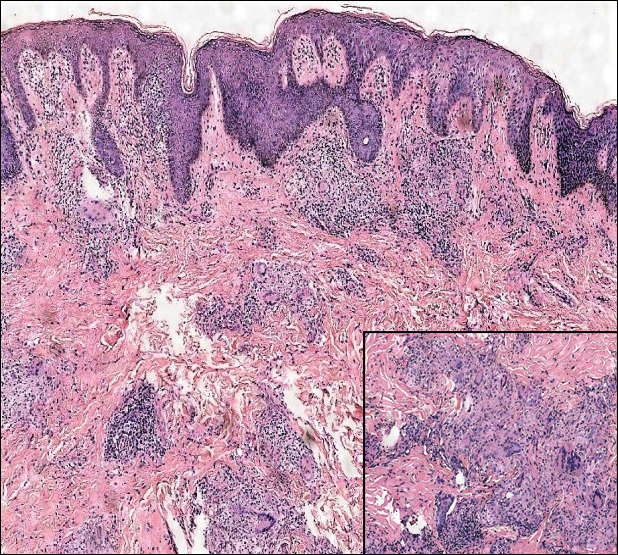
A 19-year-old man presented with a perianal condyloma acuminatum-like plaque of 2 years' duration and a 6-month history of diarrhea.
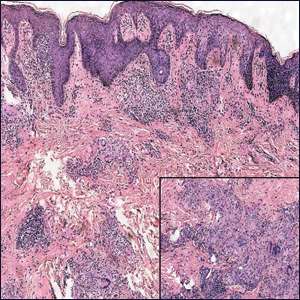
Disseminated Vesicles and Necrotic Papules
The Diagnosis: Lues Maligna
Laboratory evaluation demonstrated a total CD4 count of 26 cells/μL (reference range, 443-1471 cells/μL) with a viral load of 1,770,111 copies/mL (reference range, 0 copies/mL), as well as a positive rapid plasma reagin (RPR) test with a titer of 1:8 (reference range, nonreactive). A reactive treponemal antibody test confirmed a true positive RPR test result. Viral culture as well as direct fluorescence antibodies for varicella-zoster virus and an active vesicle of herpes simplex virus (HSV) were negative. Serum immunoglobulin titers for varicella-zoster virus demonstrated low IgM with a positive IgG demonstrating immunity without recent infection. Blood and lesional skin tissue cultures were negative for additional infectious etiologies including bacterial and fungal elements. A lumbar puncture was not performed.
Biopsy of a papulonodule on the left arm demonstrated a lichenoid lymphohistiocytic infiltrate with superficial and deep inflammation (Figure 1). Neutrophils also were noted within a follicle with ballooning and acantholysis within the follicular epithelium. Additional staining for Mycobacterium, HSV-1, HSV-2, and Treponema were negative. In the clinical setting, this histologic pattern was most consistent with secondary syphilis. Pityriasis lichenoides et varioliformis acuta also was included in the histopathologic differential diagnosis by a dermatopathologist (M.C.).
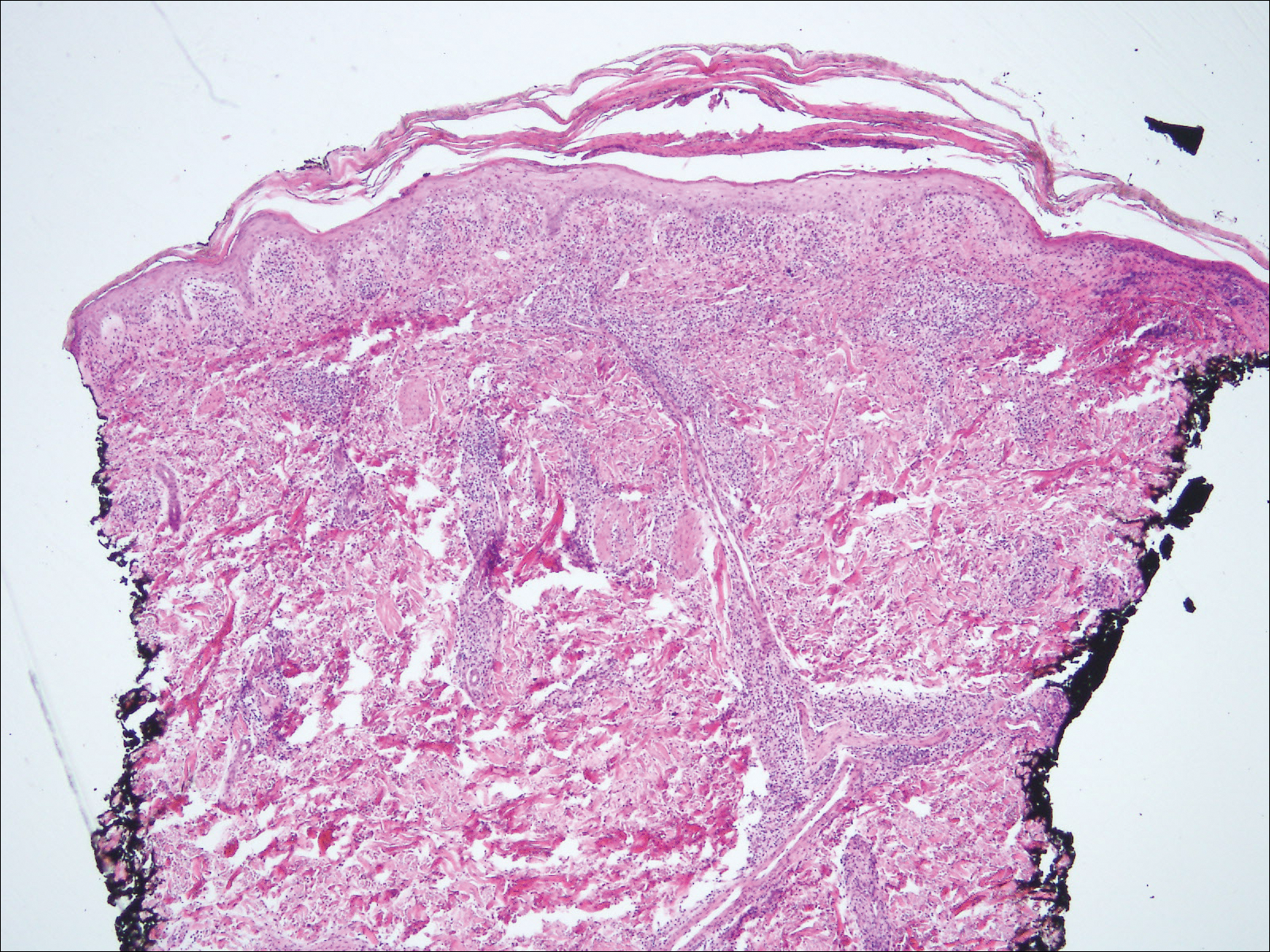
Based on the clinical, microbiologic, and histopathologic findings, a diagnosis of lues maligna (cutaneous secondary syphilis) with a vesiculonecrotic presentation was made. The patient's low RPR titer was attributed to profound immunosuppression, while a confirmation of syphilis infection was made with treponemal antibody testing. Histopathologic examination was consistent with lues maligna and did not demonstrate evidence of any other infectious etiologies.
Following 7 days of intravenous penicillin, the patient demonstrated dramatic improvement of all skin lesions and was discharged receiving once-weekly intramuscular penicillin for 4 weeks. In accordance with the diagnosis, the patient demonstrated rapid improvement of the lesions following appropriate antibiotic therapy.
After the diagnosis of lues maligna was made, the patient disclosed a sexual encounter with a male partner 6 weeks prior to the current presentation, after which he developed a self-resolving genital ulcer suspicious for a primary chancre.
Increasing rates of syphilis transmission have been attributed to males aged 15 to 44 years who have sexual encounters with other males.1 Although syphilis commonly is known as the great mimicker, syphilology texts state that lesions are not associated with syphilis if vesicles are part of the cutaneous eruption in an adult.2 However, rare reports of secondary syphilis presenting as vesicles, pustules, bullae, and pityriasis lichenoides et varioliformis acuta-like eruptions also have been documented.2-4
Initial screening for suspected syphilis involves sensitive, but not specific, nontreponemal RPR testing reported in the form of a titer. Nontreponemal titers in human immunodeficiency virus-positive individuals can be unusually high or low, fluctuate rapidly, and/or be unresponsive to antibiotic therapy.1
Lues maligna is a rare form of malignant secondary syphilis that most commonly presents in human immunodeficiency virus-positive hosts.5 Although lues maligna often presents with ulceronodular lesions, 2 cases presenting with vesiculonecrotic lesions also have been reported.6 Patients often experience systemic symptoms including fever, fatigue, and joint pain. Rapid plasma reagin titers can range from 1:8 to 1:128 in affected individuals.6 Diagnosis is dependent on serologic and histologic confirmation while ruling out viral, fungal, and bacterial etiologies. Characteristic red-brown lesions of secondary syphilis involving the palms and soles (Figure 2) alsoaid in diagnosis.1 Additionally, identification of the Jarisch-Herxheimer reaction following treatment and rapid response to antibiotic therapy are helpful diagnostic findings.6,7 While histopathologic examination of lues maligna typically does not reveal evidence of spirochetes, it also is important to rule out other infectious etiologies.7

Our case emphasizes the importance of early recognition and treatment of the variable clinical, laboratory, and histologic presentations of lues maligna.
- Syphilis fact sheet. Centers for Disease Control and Prevention website. https://www.cdc.gov/std/syphilis/stdfact-syphilis.htm. Updated June 13, 2017. Accessed March 22, 2018.
- Lawrence P, Saxe N. Bullous secondary syphilis. Clin Exp Dermatol. 1992;17:44-46.
- Pastuszczak M, Woz´niak W, Jaworek AK, et al. Pityriasis lichenoides-like secondary syphilis and neurosyphilis in a HIV-infected patient. Postepy Dermatol Alergol. 2013;30:127-130.
- Schnirring-Judge M, Gustaferro C, Terol C. Vesiculobullous syphilis: a case involving an unusual cutaneous manifestation of secondary syphilis [published online November 24, 2010]. J Foot Ankle Surg. 2011;50:96-101.
- Pföhler C, Koerner R, von Müller L, et al. Lues maligna in a patient with unknown HIV infection. BMJ Case Rep. 2011. pii: bcr0520114221. doi: 10.1136/bcr.05.2011.4221.
- Don PC, Rubinstein R, Christie S. Malignant syphilis (lues maligna) and concurrent infection with HIV. Int J Dermatol. 1995;34:403-407.
- Tucker JD, Shah S, Jarell AD, et al. Lues maligna in early HIV infection case report and review of the literature. Sex Transm Dis. 2009;36:512-514.
The Diagnosis: Lues Maligna
Laboratory evaluation demonstrated a total CD4 count of 26 cells/μL (reference range, 443-1471 cells/μL) with a viral load of 1,770,111 copies/mL (reference range, 0 copies/mL), as well as a positive rapid plasma reagin (RPR) test with a titer of 1:8 (reference range, nonreactive). A reactive treponemal antibody test confirmed a true positive RPR test result. Viral culture as well as direct fluorescence antibodies for varicella-zoster virus and an active vesicle of herpes simplex virus (HSV) were negative. Serum immunoglobulin titers for varicella-zoster virus demonstrated low IgM with a positive IgG demonstrating immunity without recent infection. Blood and lesional skin tissue cultures were negative for additional infectious etiologies including bacterial and fungal elements. A lumbar puncture was not performed.
Biopsy of a papulonodule on the left arm demonstrated a lichenoid lymphohistiocytic infiltrate with superficial and deep inflammation (Figure 1). Neutrophils also were noted within a follicle with ballooning and acantholysis within the follicular epithelium. Additional staining for Mycobacterium, HSV-1, HSV-2, and Treponema were negative. In the clinical setting, this histologic pattern was most consistent with secondary syphilis. Pityriasis lichenoides et varioliformis acuta also was included in the histopathologic differential diagnosis by a dermatopathologist (M.C.).
Based on the clinical, microbiologic, and histopathologic findings, a diagnosis of lues maligna (cutaneous secondary syphilis) with a vesiculonecrotic presentation was made. The patient's low RPR titer was attributed to profound immunosuppression, while a confirmation of syphilis infection was made with treponemal antibody testing. Histopathologic examination was consistent with lues maligna and did not demonstrate evidence of any other infectious etiologies.
Following 7 days of intravenous penicillin, the patient demonstrated dramatic improvement of all skin lesions and was discharged receiving once-weekly intramuscular penicillin for 4 weeks. In accordance with the diagnosis, the patient demonstrated rapid improvement of the lesions following appropriate antibiotic therapy.
After the diagnosis of lues maligna was made, the patient disclosed a sexual encounter with a male partner 6 weeks prior to the current presentation, after which he developed a self-resolving genital ulcer suspicious for a primary chancre.
Increasing rates of syphilis transmission have been attributed to males aged 15 to 44 years who have sexual encounters with other males.1 Although syphilis commonly is known as the great mimicker, syphilology texts state that lesions are not associated with syphilis if vesicles are part of the cutaneous eruption in an adult.2 However, rare reports of secondary syphilis presenting as vesicles, pustules, bullae, and pityriasis lichenoides et varioliformis acuta-like eruptions also have been documented.2-4
Initial screening for suspected syphilis involves sensitive, but not specific, nontreponemal RPR testing reported in the form of a titer. Nontreponemal titers in human immunodeficiency virus-positive individuals can be unusually high or low, fluctuate rapidly, and/or be unresponsive to antibiotic therapy.1
Lues maligna is a rare form of malignant secondary syphilis that most commonly presents in human immunodeficiency virus-positive hosts.5 Although lues maligna often presents with ulceronodular lesions, 2 cases presenting with vesiculonecrotic lesions also have been reported.6 Patients often experience systemic symptoms including fever, fatigue, and joint pain. Rapid plasma reagin titers can range from 1:8 to 1:128 in affected individuals.6 Diagnosis is dependent on serologic and histologic confirmation while ruling out viral, fungal, and bacterial etiologies. Characteristic red-brown lesions of secondary syphilis involving the palms and soles (Figure 2) alsoaid in diagnosis.1 Additionally, identification of the Jarisch-Herxheimer reaction following treatment and rapid response to antibiotic therapy are helpful diagnostic findings.6,7 While histopathologic examination of lues maligna typically does not reveal evidence of spirochetes, it also is important to rule out other infectious etiologies.7
Our case emphasizes the importance of early recognition and treatment of the variable clinical, laboratory, and histologic presentations of lues maligna.
The Diagnosis: Lues Maligna
Laboratory evaluation demonstrated a total CD4 count of 26 cells/μL (reference range, 443-1471 cells/μL) with a viral load of 1,770,111 copies/mL (reference range, 0 copies/mL), as well as a positive rapid plasma reagin (RPR) test with a titer of 1:8 (reference range, nonreactive). A reactive treponemal antibody test confirmed a true positive RPR test result. Viral culture as well as direct fluorescence antibodies for varicella-zoster virus and an active vesicle of herpes simplex virus (HSV) were negative. Serum immunoglobulin titers for varicella-zoster virus demonstrated low IgM with a positive IgG demonstrating immunity without recent infection. Blood and lesional skin tissue cultures were negative for additional infectious etiologies including bacterial and fungal elements. A lumbar puncture was not performed.
Biopsy of a papulonodule on the left arm demonstrated a lichenoid lymphohistiocytic infiltrate with superficial and deep inflammation (Figure 1). Neutrophils also were noted within a follicle with ballooning and acantholysis within the follicular epithelium. Additional staining for Mycobacterium, HSV-1, HSV-2, and Treponema were negative. In the clinical setting, this histologic pattern was most consistent with secondary syphilis. Pityriasis lichenoides et varioliformis acuta also was included in the histopathologic differential diagnosis by a dermatopathologist (M.C.).
Based on the clinical, microbiologic, and histopathologic findings, a diagnosis of lues maligna (cutaneous secondary syphilis) with a vesiculonecrotic presentation was made. The patient's low RPR titer was attributed to profound immunosuppression, while a confirmation of syphilis infection was made with treponemal antibody testing. Histopathologic examination was consistent with lues maligna and did not demonstrate evidence of any other infectious etiologies.
Following 7 days of intravenous penicillin, the patient demonstrated dramatic improvement of all skin lesions and was discharged receiving once-weekly intramuscular penicillin for 4 weeks. In accordance with the diagnosis, the patient demonstrated rapid improvement of the lesions following appropriate antibiotic therapy.
After the diagnosis of lues maligna was made, the patient disclosed a sexual encounter with a male partner 6 weeks prior to the current presentation, after which he developed a self-resolving genital ulcer suspicious for a primary chancre.
Increasing rates of syphilis transmission have been attributed to males aged 15 to 44 years who have sexual encounters with other males.1 Although syphilis commonly is known as the great mimicker, syphilology texts state that lesions are not associated with syphilis if vesicles are part of the cutaneous eruption in an adult.2 However, rare reports of secondary syphilis presenting as vesicles, pustules, bullae, and pityriasis lichenoides et varioliformis acuta-like eruptions also have been documented.2-4
Initial screening for suspected syphilis involves sensitive, but not specific, nontreponemal RPR testing reported in the form of a titer. Nontreponemal titers in human immunodeficiency virus-positive individuals can be unusually high or low, fluctuate rapidly, and/or be unresponsive to antibiotic therapy.1
Lues maligna is a rare form of malignant secondary syphilis that most commonly presents in human immunodeficiency virus-positive hosts.5 Although lues maligna often presents with ulceronodular lesions, 2 cases presenting with vesiculonecrotic lesions also have been reported.6 Patients often experience systemic symptoms including fever, fatigue, and joint pain. Rapid plasma reagin titers can range from 1:8 to 1:128 in affected individuals.6 Diagnosis is dependent on serologic and histologic confirmation while ruling out viral, fungal, and bacterial etiologies. Characteristic red-brown lesions of secondary syphilis involving the palms and soles (Figure 2) alsoaid in diagnosis.1 Additionally, identification of the Jarisch-Herxheimer reaction following treatment and rapid response to antibiotic therapy are helpful diagnostic findings.6,7 While histopathologic examination of lues maligna typically does not reveal evidence of spirochetes, it also is important to rule out other infectious etiologies.7
Our case emphasizes the importance of early recognition and treatment of the variable clinical, laboratory, and histologic presentations of lues maligna.
- Syphilis fact sheet. Centers for Disease Control and Prevention website. https://www.cdc.gov/std/syphilis/stdfact-syphilis.htm. Updated June 13, 2017. Accessed March 22, 2018.
- Lawrence P, Saxe N. Bullous secondary syphilis. Clin Exp Dermatol. 1992;17:44-46.
- Pastuszczak M, Woz´niak W, Jaworek AK, et al. Pityriasis lichenoides-like secondary syphilis and neurosyphilis in a HIV-infected patient. Postepy Dermatol Alergol. 2013;30:127-130.
- Schnirring-Judge M, Gustaferro C, Terol C. Vesiculobullous syphilis: a case involving an unusual cutaneous manifestation of secondary syphilis [published online November 24, 2010]. J Foot Ankle Surg. 2011;50:96-101.
- Pföhler C, Koerner R, von Müller L, et al. Lues maligna in a patient with unknown HIV infection. BMJ Case Rep. 2011. pii: bcr0520114221. doi: 10.1136/bcr.05.2011.4221.
- Don PC, Rubinstein R, Christie S. Malignant syphilis (lues maligna) and concurrent infection with HIV. Int J Dermatol. 1995;34:403-407.
- Tucker JD, Shah S, Jarell AD, et al. Lues maligna in early HIV infection case report and review of the literature. Sex Transm Dis. 2009;36:512-514.
- Syphilis fact sheet. Centers for Disease Control and Prevention website. https://www.cdc.gov/std/syphilis/stdfact-syphilis.htm. Updated June 13, 2017. Accessed March 22, 2018.
- Lawrence P, Saxe N. Bullous secondary syphilis. Clin Exp Dermatol. 1992;17:44-46.
- Pastuszczak M, Woz´niak W, Jaworek AK, et al. Pityriasis lichenoides-like secondary syphilis and neurosyphilis in a HIV-infected patient. Postepy Dermatol Alergol. 2013;30:127-130.
- Schnirring-Judge M, Gustaferro C, Terol C. Vesiculobullous syphilis: a case involving an unusual cutaneous manifestation of secondary syphilis [published online November 24, 2010]. J Foot Ankle Surg. 2011;50:96-101.
- Pföhler C, Koerner R, von Müller L, et al. Lues maligna in a patient with unknown HIV infection. BMJ Case Rep. 2011. pii: bcr0520114221. doi: 10.1136/bcr.05.2011.4221.
- Don PC, Rubinstein R, Christie S. Malignant syphilis (lues maligna) and concurrent infection with HIV. Int J Dermatol. 1995;34:403-407.
- Tucker JD, Shah S, Jarell AD, et al. Lues maligna in early HIV infection case report and review of the literature. Sex Transm Dis. 2009;36:512-514.

A 30-year-old man who had contracted human immunodeficiency virus from a male sexual partner 4 years prior presented to the emergency department with fevers, chills, night sweats, and rhinorrhea of 2 weeks' duration. He reported that he had been off highly active antiretroviral therapy for 2 years. Physical examination revealed numerous erythematous, papulonecrotic, crusted lesions on the face, neck, chest, back, arms, and legs that had developed over the past 4 days. Fluid-filled vesicles also were noted on the arms and legs, while erythematous, indurated nodules with overlying scaling were noted on the bilateral palms and soles. The patient reported that he had been vaccinated for varicella-zoster virus as a child without primary infection.
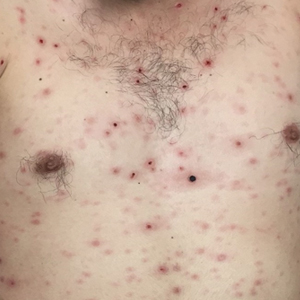
A Recalcitrant Case of Toxic Epidermal Necrolysis
One of the most severe complications of systemic medications is the development of a life-threatening rash, especially toxic epidermal necrolysis (TEN). Most patients can expect a full recovery if the complicating medication is discontinued early on in its course.1 When suspected TEN does not improve despite discontinuation of the detrimental medication, other diseases must be considered, particularly immunobullous and infectious etiologies. Treatment of these diseases differs substantially; therefore, a quick diagnosis is crucial. We present a case of a patient with an acute blistering eruption that was initially diagnosed and managed as TEN but physical examination and histopathologic confirmed another diagnosis. We review key examination findings that can help differentiate the causes of an acute blistering eruption with mucosal involvement, allowing for earlier diagnosis and treatment of these patients.
Case Report
An 85-year-old immunocompetent man was admitted to an outside hospital with a pruritic blistering eruption associated with myalgia, weakness, and fatigue of 3 weeks’ duration. The eruption initiated on the scalp and face and then spread down to the trunk and proximal arms and legs, with oral erosions also reported. An outside dermatologist was consulted on admission and performed a skin biopsy; the initial pathology was read as TEN. The patient was admitted to our institution on the same day, and all potentially complicating medications were stopped. He was treated with intravenous (IV)
At that time, physical examination revealed numerous confluent erosions with honey-colored crust involving the entire face (Figure 1A) and sharp demarcation at the cutaneous lip (Figure 1B). There was a large erosion on the dorsal aspect of the tongue, but the rest of the oral mucosa was spared. The trunk and proximal extremities showed numerous grouped, punched-out erosions with scalloped borders (Figure 1C). A repeat skin biopsy showed an ulcer with viral cytopathic changes. Immunoperoxidase studies demonstrated positive staining for herpes simplex virus (HSV) type 1 (Figure 2). The original slides were a frozen section from an outside facility and could not be obtained. A tissue culture and direct fluorescent antibody also confirmed HSV-1, and the patient was diagnosed with disseminated herpes. He was rapidly tapered off of the steroids and started on IV acyclovir 10 mg/kg every 8 hours for 21 days. All prior erosions reepithelialized within 7 days of treatment (Figure 3). The patient had an otherwise uncomplicated hospital course and was discharged on hospital day 21.
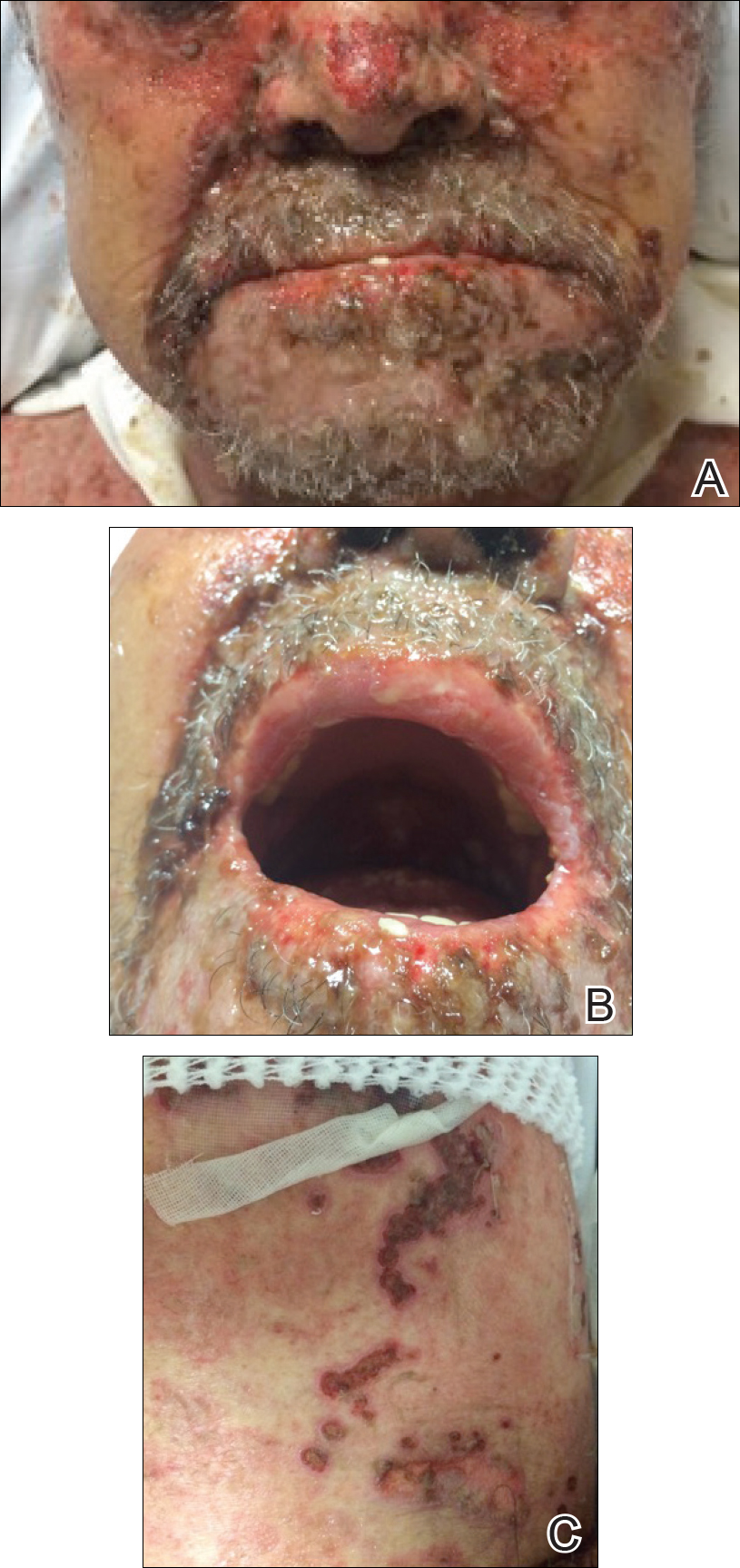
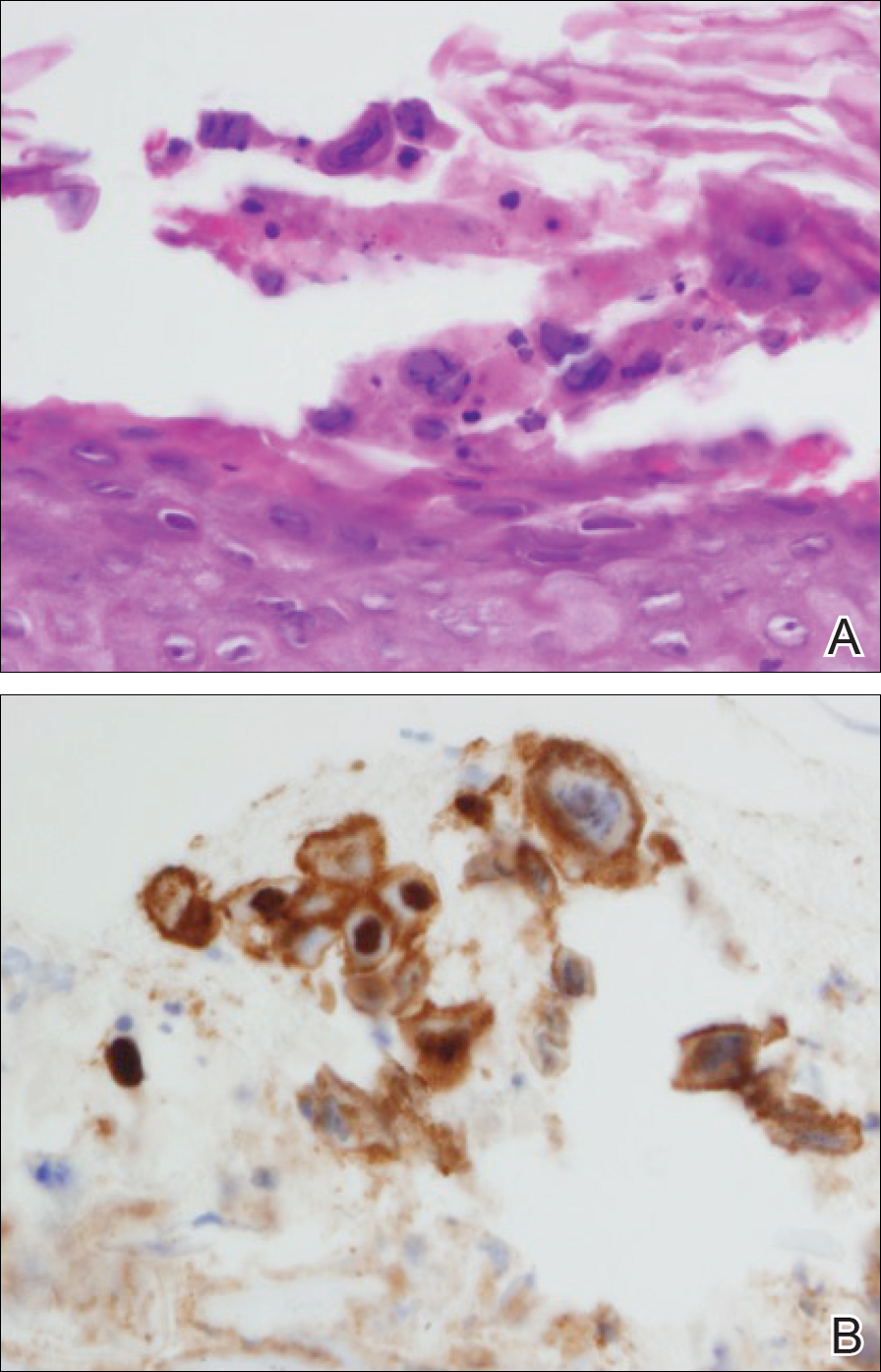
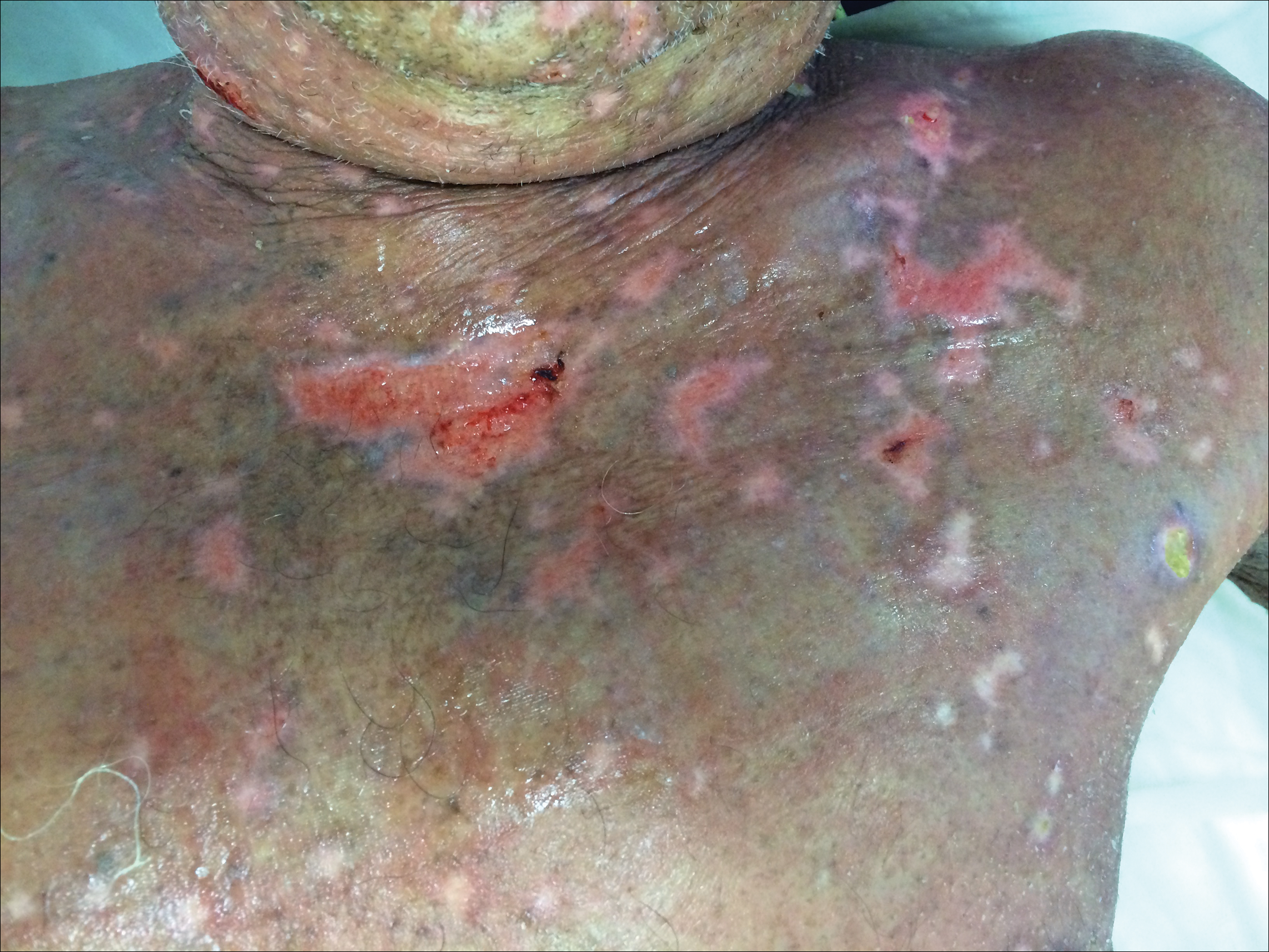
Comment
A patient with an acute generalized blistering eruption requires urgent workup and treatment given the potentially devastating sequelae. Toxic epidermal necrolysis and immunobullous diseases often are the first diagnoses to be ruled out. Certainly infections such as HSV can cause a vesicular and erosive eruption, especially in the setting of a poorly controlled dermatitis, but they typically are not in the same differential as the other diagnoses.
Clinical Presentation
This case highlights 2 key physical examination findings that can alert the clinician to a possible underlying herpetic infection. First, the distribution of this patient’s oral lesions was telling. In most cases of TEN or pemphigus vulgaris, there is notable involvement of the oral mucosa, particularly the buccal and labial mucosa. Although herpes can involve any mucocutaneous surface, it does have a predilection for keratinized tissue, with the tongue and cutaneous lip commonly involved.2,3 Our patient had a solitary linear erosion on the dorsal aspect of the tongue, but the rest of the oral cavity was strikingly spared. In addition, the erosions around the mouth stopped right at the cutaneous lip, sparing the labial mucosa (Figure 1B).
Second, the configuration of the erosions on the trunk, arms, and legs was diagnostic. Herpes classically presents as a cluster of vesicles overlying an erythematous base. When these vesicles rupture, punched-out erosions are left behind. Because these vesicles often are grouped, they can develop a scalloped border, which is a helpful indicator of HSV (Figure 1C). When these erosions become more confluent and irregular, the distinction from other conditions may not be as clear. A careful skin examination often can show areas that have preserved this herpetiform configuration.
Immune Compromise
Additionally, this case is illustrative of how immunosuppression and immunocompromise can affect the clinical presentation of HSV infection. Herpetic infections in the immunocompromised host tend to have a more protracted course, with chronic enlarging ulcers involving multiple sites.
Conclusion
This case is a good reminder that not everything that blisters and involves the mucosa is due to a hypersensitivity state such as TEN and Stevens-Johnson syndrome or an immunobullous disorder such as pemphigus vulgaris and pemphigus vegetans. The fact that this patient was worsening despite drug cessation, high-dose steroids, and IV immunoglobulin should have indicated a misdiagnosis. This case also shows that the early histopathologic findings of disseminated HSV and TEN can be nonspecific, and viral cytopathic changes may not always be obvious early in the disease.
Disseminated HSV should be considered in the differential diagnosis of a patient with an acute blistering eruption with mucosal involvement, and careful history and physical examination should be taken to rule out a viral etiology.
- Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis: part I. introduction, history, classification, clinical features, systemic manifestations, etiology, and immunopathogenesis. J Am Acad Dermatol. 2013;69:173.e1-173.e13.
- Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. New York, NY: Mosby; 2008.
- Woo SB, Lee SF. Oral recrudescent herpes simplex virus infection. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;83:239-243.
One of the most severe complications of systemic medications is the development of a life-threatening rash, especially toxic epidermal necrolysis (TEN). Most patients can expect a full recovery if the complicating medication is discontinued early on in its course.1 When suspected TEN does not improve despite discontinuation of the detrimental medication, other diseases must be considered, particularly immunobullous and infectious etiologies. Treatment of these diseases differs substantially; therefore, a quick diagnosis is crucial. We present a case of a patient with an acute blistering eruption that was initially diagnosed and managed as TEN but physical examination and histopathologic confirmed another diagnosis. We review key examination findings that can help differentiate the causes of an acute blistering eruption with mucosal involvement, allowing for earlier diagnosis and treatment of these patients.
Case Report
An 85-year-old immunocompetent man was admitted to an outside hospital with a pruritic blistering eruption associated with myalgia, weakness, and fatigue of 3 weeks’ duration. The eruption initiated on the scalp and face and then spread down to the trunk and proximal arms and legs, with oral erosions also reported. An outside dermatologist was consulted on admission and performed a skin biopsy; the initial pathology was read as TEN. The patient was admitted to our institution on the same day, and all potentially complicating medications were stopped. He was treated with intravenous (IV)
At that time, physical examination revealed numerous confluent erosions with honey-colored crust involving the entire face (Figure 1A) and sharp demarcation at the cutaneous lip (Figure 1B). There was a large erosion on the dorsal aspect of the tongue, but the rest of the oral mucosa was spared. The trunk and proximal extremities showed numerous grouped, punched-out erosions with scalloped borders (Figure 1C). A repeat skin biopsy showed an ulcer with viral cytopathic changes. Immunoperoxidase studies demonstrated positive staining for herpes simplex virus (HSV) type 1 (Figure 2). The original slides were a frozen section from an outside facility and could not be obtained. A tissue culture and direct fluorescent antibody also confirmed HSV-1, and the patient was diagnosed with disseminated herpes. He was rapidly tapered off of the steroids and started on IV acyclovir 10 mg/kg every 8 hours for 21 days. All prior erosions reepithelialized within 7 days of treatment (Figure 3). The patient had an otherwise uncomplicated hospital course and was discharged on hospital day 21.
Comment
A patient with an acute generalized blistering eruption requires urgent workup and treatment given the potentially devastating sequelae. Toxic epidermal necrolysis and immunobullous diseases often are the first diagnoses to be ruled out. Certainly infections such as HSV can cause a vesicular and erosive eruption, especially in the setting of a poorly controlled dermatitis, but they typically are not in the same differential as the other diagnoses.
Clinical Presentation
This case highlights 2 key physical examination findings that can alert the clinician to a possible underlying herpetic infection. First, the distribution of this patient’s oral lesions was telling. In most cases of TEN or pemphigus vulgaris, there is notable involvement of the oral mucosa, particularly the buccal and labial mucosa. Although herpes can involve any mucocutaneous surface, it does have a predilection for keratinized tissue, with the tongue and cutaneous lip commonly involved.2,3 Our patient had a solitary linear erosion on the dorsal aspect of the tongue, but the rest of the oral cavity was strikingly spared. In addition, the erosions around the mouth stopped right at the cutaneous lip, sparing the labial mucosa (Figure 1B).
Second, the configuration of the erosions on the trunk, arms, and legs was diagnostic. Herpes classically presents as a cluster of vesicles overlying an erythematous base. When these vesicles rupture, punched-out erosions are left behind. Because these vesicles often are grouped, they can develop a scalloped border, which is a helpful indicator of HSV (Figure 1C). When these erosions become more confluent and irregular, the distinction from other conditions may not be as clear. A careful skin examination often can show areas that have preserved this herpetiform configuration.
Immune Compromise
Additionally, this case is illustrative of how immunosuppression and immunocompromise can affect the clinical presentation of HSV infection. Herpetic infections in the immunocompromised host tend to have a more protracted course, with chronic enlarging ulcers involving multiple sites.
Conclusion
This case is a good reminder that not everything that blisters and involves the mucosa is due to a hypersensitivity state such as TEN and Stevens-Johnson syndrome or an immunobullous disorder such as pemphigus vulgaris and pemphigus vegetans. The fact that this patient was worsening despite drug cessation, high-dose steroids, and IV immunoglobulin should have indicated a misdiagnosis. This case also shows that the early histopathologic findings of disseminated HSV and TEN can be nonspecific, and viral cytopathic changes may not always be obvious early in the disease.
Disseminated HSV should be considered in the differential diagnosis of a patient with an acute blistering eruption with mucosal involvement, and careful history and physical examination should be taken to rule out a viral etiology.
One of the most severe complications of systemic medications is the development of a life-threatening rash, especially toxic epidermal necrolysis (TEN). Most patients can expect a full recovery if the complicating medication is discontinued early on in its course.1 When suspected TEN does not improve despite discontinuation of the detrimental medication, other diseases must be considered, particularly immunobullous and infectious etiologies. Treatment of these diseases differs substantially; therefore, a quick diagnosis is crucial. We present a case of a patient with an acute blistering eruption that was initially diagnosed and managed as TEN but physical examination and histopathologic confirmed another diagnosis. We review key examination findings that can help differentiate the causes of an acute blistering eruption with mucosal involvement, allowing for earlier diagnosis and treatment of these patients.
Case Report
An 85-year-old immunocompetent man was admitted to an outside hospital with a pruritic blistering eruption associated with myalgia, weakness, and fatigue of 3 weeks’ duration. The eruption initiated on the scalp and face and then spread down to the trunk and proximal arms and legs, with oral erosions also reported. An outside dermatologist was consulted on admission and performed a skin biopsy; the initial pathology was read as TEN. The patient was admitted to our institution on the same day, and all potentially complicating medications were stopped. He was treated with intravenous (IV)
At that time, physical examination revealed numerous confluent erosions with honey-colored crust involving the entire face (Figure 1A) and sharp demarcation at the cutaneous lip (Figure 1B). There was a large erosion on the dorsal aspect of the tongue, but the rest of the oral mucosa was spared. The trunk and proximal extremities showed numerous grouped, punched-out erosions with scalloped borders (Figure 1C). A repeat skin biopsy showed an ulcer with viral cytopathic changes. Immunoperoxidase studies demonstrated positive staining for herpes simplex virus (HSV) type 1 (Figure 2). The original slides were a frozen section from an outside facility and could not be obtained. A tissue culture and direct fluorescent antibody also confirmed HSV-1, and the patient was diagnosed with disseminated herpes. He was rapidly tapered off of the steroids and started on IV acyclovir 10 mg/kg every 8 hours for 21 days. All prior erosions reepithelialized within 7 days of treatment (Figure 3). The patient had an otherwise uncomplicated hospital course and was discharged on hospital day 21.
Comment
A patient with an acute generalized blistering eruption requires urgent workup and treatment given the potentially devastating sequelae. Toxic epidermal necrolysis and immunobullous diseases often are the first diagnoses to be ruled out. Certainly infections such as HSV can cause a vesicular and erosive eruption, especially in the setting of a poorly controlled dermatitis, but they typically are not in the same differential as the other diagnoses.
Clinical Presentation
This case highlights 2 key physical examination findings that can alert the clinician to a possible underlying herpetic infection. First, the distribution of this patient’s oral lesions was telling. In most cases of TEN or pemphigus vulgaris, there is notable involvement of the oral mucosa, particularly the buccal and labial mucosa. Although herpes can involve any mucocutaneous surface, it does have a predilection for keratinized tissue, with the tongue and cutaneous lip commonly involved.2,3 Our patient had a solitary linear erosion on the dorsal aspect of the tongue, but the rest of the oral cavity was strikingly spared. In addition, the erosions around the mouth stopped right at the cutaneous lip, sparing the labial mucosa (Figure 1B).
Second, the configuration of the erosions on the trunk, arms, and legs was diagnostic. Herpes classically presents as a cluster of vesicles overlying an erythematous base. When these vesicles rupture, punched-out erosions are left behind. Because these vesicles often are grouped, they can develop a scalloped border, which is a helpful indicator of HSV (Figure 1C). When these erosions become more confluent and irregular, the distinction from other conditions may not be as clear. A careful skin examination often can show areas that have preserved this herpetiform configuration.
Immune Compromise
Additionally, this case is illustrative of how immunosuppression and immunocompromise can affect the clinical presentation of HSV infection. Herpetic infections in the immunocompromised host tend to have a more protracted course, with chronic enlarging ulcers involving multiple sites.
Conclusion
This case is a good reminder that not everything that blisters and involves the mucosa is due to a hypersensitivity state such as TEN and Stevens-Johnson syndrome or an immunobullous disorder such as pemphigus vulgaris and pemphigus vegetans. The fact that this patient was worsening despite drug cessation, high-dose steroids, and IV immunoglobulin should have indicated a misdiagnosis. This case also shows that the early histopathologic findings of disseminated HSV and TEN can be nonspecific, and viral cytopathic changes may not always be obvious early in the disease.
Disseminated HSV should be considered in the differential diagnosis of a patient with an acute blistering eruption with mucosal involvement, and careful history and physical examination should be taken to rule out a viral etiology.
- Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis: part I. introduction, history, classification, clinical features, systemic manifestations, etiology, and immunopathogenesis. J Am Acad Dermatol. 2013;69:173.e1-173.e13.
- Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. New York, NY: Mosby; 2008.
- Woo SB, Lee SF. Oral recrudescent herpes simplex virus infection. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;83:239-243.
- Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis: part I. introduction, history, classification, clinical features, systemic manifestations, etiology, and immunopathogenesis. J Am Acad Dermatol. 2013;69:173.e1-173.e13.
- Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. New York, NY: Mosby; 2008.
- Woo SB, Lee SF. Oral recrudescent herpes simplex virus infection. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;83:239-243.
Practice Points
- Toxic epidermal necrolysis can be difficult to diagnose and treat.
- Patients who are refractory to treatment should prompt further management considerations.
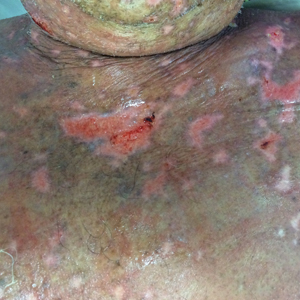
Brown-Black Papulonodules on the Arm
The Diagnosis: Glochid Dermatitis
Biopsy of a nodule on the upper right arm showed chronic granulomatous inflammation and polarizable foreign material consistent with plant cellulose (Figure). A diagnosis of glochid dermatitis was made. The treatment plan included follow-up skin evaluation and punch excision of persistent papules 1 month after the initial presentation. The patient reported the rash began after he fell on a cactus plant while chasing his grandson. He was seen by various clinicians and was given hydrocortisone and clobetasol, which helped with pruritis but did not resolve the rash. His grandson developed a similar rash at the site of contact with the cactus plant. The patient and his grandson did not detect the presence of any cactus spines.
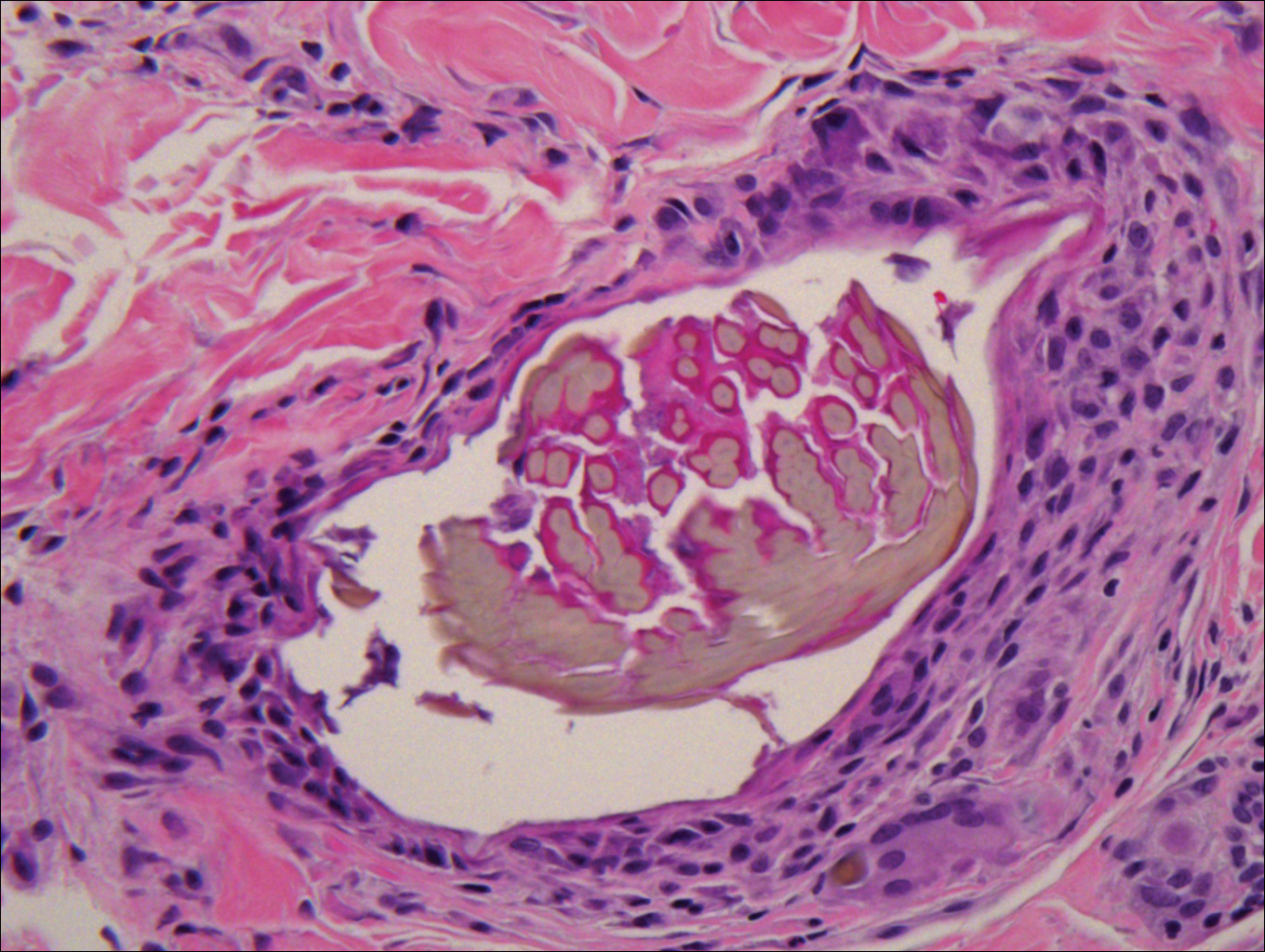
Injuries from cactus glochids most often occur due to accidental falls on cactus plants, but glochids also may be transferred from clothing to other individuals. The thin, hairlike glochids easily detach from the stem of the cactus and can become deeply embedded with virtually no pressure.1
Glochid implantation from the prickly pear cactus commonly presents as a pruritic papular eruption known as glochid dermatitis. These penetrating injuries can lead to inoculation of Clostridium tetani and Staphylococcus aureus. Additionally, unrecognized and unremoved cactus spines may be highly inflammatory and may cause chronic granulomatous inflammation.2
Initially, acute glochid dermatitis occurs due to mechanical damage caused by the detatched cactus spine and may not resolve for up to 4 months. Granuloma formation has been reported several weeks after exposure and may persist for more than 8 months.3 Although an immune mechanism has been suggested, the literature has indicated that delayed hypersensitivity reactions are a more probable cause of the granulomatous inflammation after glochid exposure.3 Madkan et al4 reported that relatively few patients developed granulomas after implantation of glochids in the skin, thus suggesting that granuloma formation is an allergic response.
With regard to the pathogenesis of glochid dermatitis, the initial response to foreign plant matter in the dermis involves a neutrophilic infiltrate, which later is replaced by histiocytes; however, the foreign material remains undegraded in the macrophage cytoplasm.5 Activated macrophages secrete cytokines that intensify the inflammatory response, resulting in formation of a granuloma around the foreign body. The granuloma acts as a wall to isolate the foreign matter from the rest of the body.5
Regarding treatment of chronic granulomas, Madkan et al4 reported a case that showed some improvement with clobetasol ointment; however, clinical lesions resolved only after punch biopsies were performed to confirm the diagnosis of cactus spine granuloma. In a controlled study in rabbits, glochids were successfully removed by first detaching the larger clumps with tweezers then applying glue and gauze to the affected area.6 After the glue dried, the gauze was peeled off, resulting in the removal of 95% of the implanted glochids. Overall, removal of embedded spines is difficult because the glochids typically radiate in several directions.7 Treatment of foreign body granulomas caused by cactus spines can be achieved by expulsion of plant matter remnants and symptomatic treatment using midpotency topical steroids twice daily.4 Uncovering and performing punch biopsies of papules also can result in rapid healing of the lesions. Without manual removal of the glochid, lesions can persist for 2 to 8 months until gradual resolution with possible postinflammatory hyperpigmentation.4
- Suzuki H, Baba S. Cactus granuloma of the skin. J Dermatol. 1993;20:424-427.
- Suárez A, Freeman S, Puls L, et al. Unusual presentation of cactus spines in the flank of an elderly man: a case report. J Med Case Rep. 2010;4:152.
- Spoerke DG, Spoerke SE. Granuloma formation induced by spines of the cactus, Opuntia acanthocarpa. Vet Hum Toxicol. 1991;33:342-344.
- Madkan VK, Abraham T, Lesher JL Jr. Cactus spine granuloma. Cutis. 2007;79:208-210.
- Molina-Ruiz AM, Requena L. Foreign body granulomas. Dermatol Clin. 2015;33:497-523.
- McGovern TW, Barkley TM. Botanical dermatology. Int J Dermatol. 1998;37:321-334.
- Lindsey D, Lindsey WE. Cactus spine injuries. Am J Emerg Med. 1988;6:362-369.
The Diagnosis: Glochid Dermatitis
Biopsy of a nodule on the upper right arm showed chronic granulomatous inflammation and polarizable foreign material consistent with plant cellulose (Figure). A diagnosis of glochid dermatitis was made. The treatment plan included follow-up skin evaluation and punch excision of persistent papules 1 month after the initial presentation. The patient reported the rash began after he fell on a cactus plant while chasing his grandson. He was seen by various clinicians and was given hydrocortisone and clobetasol, which helped with pruritis but did not resolve the rash. His grandson developed a similar rash at the site of contact with the cactus plant. The patient and his grandson did not detect the presence of any cactus spines.
Injuries from cactus glochids most often occur due to accidental falls on cactus plants, but glochids also may be transferred from clothing to other individuals. The thin, hairlike glochids easily detach from the stem of the cactus and can become deeply embedded with virtually no pressure.1
Glochid implantation from the prickly pear cactus commonly presents as a pruritic papular eruption known as glochid dermatitis. These penetrating injuries can lead to inoculation of Clostridium tetani and Staphylococcus aureus. Additionally, unrecognized and unremoved cactus spines may be highly inflammatory and may cause chronic granulomatous inflammation.2
Initially, acute glochid dermatitis occurs due to mechanical damage caused by the detatched cactus spine and may not resolve for up to 4 months. Granuloma formation has been reported several weeks after exposure and may persist for more than 8 months.3 Although an immune mechanism has been suggested, the literature has indicated that delayed hypersensitivity reactions are a more probable cause of the granulomatous inflammation after glochid exposure.3 Madkan et al4 reported that relatively few patients developed granulomas after implantation of glochids in the skin, thus suggesting that granuloma formation is an allergic response.
With regard to the pathogenesis of glochid dermatitis, the initial response to foreign plant matter in the dermis involves a neutrophilic infiltrate, which later is replaced by histiocytes; however, the foreign material remains undegraded in the macrophage cytoplasm.5 Activated macrophages secrete cytokines that intensify the inflammatory response, resulting in formation of a granuloma around the foreign body. The granuloma acts as a wall to isolate the foreign matter from the rest of the body.5
Regarding treatment of chronic granulomas, Madkan et al4 reported a case that showed some improvement with clobetasol ointment; however, clinical lesions resolved only after punch biopsies were performed to confirm the diagnosis of cactus spine granuloma. In a controlled study in rabbits, glochids were successfully removed by first detaching the larger clumps with tweezers then applying glue and gauze to the affected area.6 After the glue dried, the gauze was peeled off, resulting in the removal of 95% of the implanted glochids. Overall, removal of embedded spines is difficult because the glochids typically radiate in several directions.7 Treatment of foreign body granulomas caused by cactus spines can be achieved by expulsion of plant matter remnants and symptomatic treatment using midpotency topical steroids twice daily.4 Uncovering and performing punch biopsies of papules also can result in rapid healing of the lesions. Without manual removal of the glochid, lesions can persist for 2 to 8 months until gradual resolution with possible postinflammatory hyperpigmentation.4
The Diagnosis: Glochid Dermatitis
Biopsy of a nodule on the upper right arm showed chronic granulomatous inflammation and polarizable foreign material consistent with plant cellulose (Figure). A diagnosis of glochid dermatitis was made. The treatment plan included follow-up skin evaluation and punch excision of persistent papules 1 month after the initial presentation. The patient reported the rash began after he fell on a cactus plant while chasing his grandson. He was seen by various clinicians and was given hydrocortisone and clobetasol, which helped with pruritis but did not resolve the rash. His grandson developed a similar rash at the site of contact with the cactus plant. The patient and his grandson did not detect the presence of any cactus spines.
Injuries from cactus glochids most often occur due to accidental falls on cactus plants, but glochids also may be transferred from clothing to other individuals. The thin, hairlike glochids easily detach from the stem of the cactus and can become deeply embedded with virtually no pressure.1
Glochid implantation from the prickly pear cactus commonly presents as a pruritic papular eruption known as glochid dermatitis. These penetrating injuries can lead to inoculation of Clostridium tetani and Staphylococcus aureus. Additionally, unrecognized and unremoved cactus spines may be highly inflammatory and may cause chronic granulomatous inflammation.2
Initially, acute glochid dermatitis occurs due to mechanical damage caused by the detatched cactus spine and may not resolve for up to 4 months. Granuloma formation has been reported several weeks after exposure and may persist for more than 8 months.3 Although an immune mechanism has been suggested, the literature has indicated that delayed hypersensitivity reactions are a more probable cause of the granulomatous inflammation after glochid exposure.3 Madkan et al4 reported that relatively few patients developed granulomas after implantation of glochids in the skin, thus suggesting that granuloma formation is an allergic response.
With regard to the pathogenesis of glochid dermatitis, the initial response to foreign plant matter in the dermis involves a neutrophilic infiltrate, which later is replaced by histiocytes; however, the foreign material remains undegraded in the macrophage cytoplasm.5 Activated macrophages secrete cytokines that intensify the inflammatory response, resulting in formation of a granuloma around the foreign body. The granuloma acts as a wall to isolate the foreign matter from the rest of the body.5
Regarding treatment of chronic granulomas, Madkan et al4 reported a case that showed some improvement with clobetasol ointment; however, clinical lesions resolved only after punch biopsies were performed to confirm the diagnosis of cactus spine granuloma. In a controlled study in rabbits, glochids were successfully removed by first detaching the larger clumps with tweezers then applying glue and gauze to the affected area.6 After the glue dried, the gauze was peeled off, resulting in the removal of 95% of the implanted glochids. Overall, removal of embedded spines is difficult because the glochids typically radiate in several directions.7 Treatment of foreign body granulomas caused by cactus spines can be achieved by expulsion of plant matter remnants and symptomatic treatment using midpotency topical steroids twice daily.4 Uncovering and performing punch biopsies of papules also can result in rapid healing of the lesions. Without manual removal of the glochid, lesions can persist for 2 to 8 months until gradual resolution with possible postinflammatory hyperpigmentation.4
- Suzuki H, Baba S. Cactus granuloma of the skin. J Dermatol. 1993;20:424-427.
- Suárez A, Freeman S, Puls L, et al. Unusual presentation of cactus spines in the flank of an elderly man: a case report. J Med Case Rep. 2010;4:152.
- Spoerke DG, Spoerke SE. Granuloma formation induced by spines of the cactus, Opuntia acanthocarpa. Vet Hum Toxicol. 1991;33:342-344.
- Madkan VK, Abraham T, Lesher JL Jr. Cactus spine granuloma. Cutis. 2007;79:208-210.
- Molina-Ruiz AM, Requena L. Foreign body granulomas. Dermatol Clin. 2015;33:497-523.
- McGovern TW, Barkley TM. Botanical dermatology. Int J Dermatol. 1998;37:321-334.
- Lindsey D, Lindsey WE. Cactus spine injuries. Am J Emerg Med. 1988;6:362-369.
- Suzuki H, Baba S. Cactus granuloma of the skin. J Dermatol. 1993;20:424-427.
- Suárez A, Freeman S, Puls L, et al. Unusual presentation of cactus spines in the flank of an elderly man: a case report. J Med Case Rep. 2010;4:152.
- Spoerke DG, Spoerke SE. Granuloma formation induced by spines of the cactus, Opuntia acanthocarpa. Vet Hum Toxicol. 1991;33:342-344.
- Madkan VK, Abraham T, Lesher JL Jr. Cactus spine granuloma. Cutis. 2007;79:208-210.
- Molina-Ruiz AM, Requena L. Foreign body granulomas. Dermatol Clin. 2015;33:497-523.
- McGovern TW, Barkley TM. Botanical dermatology. Int J Dermatol. 1998;37:321-334.
- Lindsey D, Lindsey WE. Cactus spine injuries. Am J Emerg Med. 1988;6:362-369.
A 63-year-old man presented with a pruritic rash on the right arm of approximately 3 months' duration. On physical examination, several discrete, 4- to 5-mm, brown-black papulonodules with a central punctum were identified along the extensor aspects of the upper and lower right arm. No foreign bodies were appreciated. Biopsies of nodules on the right upper arm were performed (sites marked with letters).
Asymptomatic Subcutaneous Nodule on the Cheek
The Diagnosis: Lymphoepitheliomalike Carcinoma of the Skin
The term lymphoepitheliomalike carcinoma of the skin (LELCS) initially was proposed by Swanson et al1 in 1988 when they described 5 patients with cutaneous neoplasms histologically resembling nasopharyngeal carcinoma, also known as lymphoepithelioma. A PubMed search of articles indexed for MEDLINE using the term lymphoepitheliomalike carcinoma of the skin revealed over 60 cases of LELCS since 1988. However, unlike nasopharyngeal carcinoma, LELCS has not been associated with Epstein-Barr virus, with the exception of 1 known reported case.2 The clinical appearance of LELCS is nonspecific but usually presents as a flesh-colored to erythematous nodule, as was seen in the current case. Lesions commonly are found on the head and neck in middle-aged to elderly patients with a slight male predominance.2
On histology, LELCS is characterized by aggregations of large, atypical epithelioid cells surrounded by a dense lymphoplasmocytic infiltrate (right quiz image). The neoplasm tends to reside within the deep dermis and/or subcutis1 without appreciable epidermal involvement (left quiz image). The atypical epithelioid cells demonstrate positive immunoreactivity for cytokeratins (right quiz image inset), p40/p63, and epithelial membrane antigen,3 and the surrounding lymphocytic infiltrate stains positively for leukocyte common antigen. The tumor histogenesis still is unknown, although an epidermal origin has been suggested given its staining pattern.2 Other investigators have postulated on an adnexal origin, citing the tumor's dermal location along with case reports describing possible glandular, sebaceous, or follicular differentiation.2,4
Treatment for LELCS can include either standard surgical excision or Mohs micrographic surgery, with radiation reserved for lymph node involvement, tumor recurrence, or poor surgical candidates.2,3,5 With appropriate therapy, prognosis may be considered favorable. Data from 49 LELCS patients presenting from 1988 and 2008 showed that 36 (73.5%) had no evidence of recurrence after treatment with standard surgical excision, 4 (8.2%) had local recurrence, and 6 (12.2%) developed lymph node metastasis, which led to death in 1 (2.0%) patient.2
Given the histologic similarity of LELCS to nasopharyngeal carcinoma, it is important to rule out the possibility of cutaneous metastasis, which can be done by testing for Epstein-Barr virus and performing either computed tomography imaging or comprehensive laryngoscopic examination of the head and neck region. In the current case, the patient was referred for laryngoscopy, at which time no suspicious lesions were identified. He subsequently underwent treatment with Mohs micrographic surgery, and the tumor was cleared after 2 surgical stages. At 5-month follow-up, the patient continued to do well with no signs of clinical recurrence.
Cutaneous lymphadenoma may be included in the differential diagnosis for LELCS on histopathology. This neoplasm is characterized by a well-circumscribed dermal proliferation of basaloid tumor islands within a fibrotic stroma (Figure 1). The basaloid cells may display peripheral palisading, and lymphocytes often are seen infiltrating the tumor lobules and the surrounding stroma (Figure 1 inset). Clinically, cutaneous lymphadenomas are slowly growing nodules that typically occur in young to middle-aged patients,4,6 unlike LELCS, which is more commonly observed in middle-aged to elderly patients.2
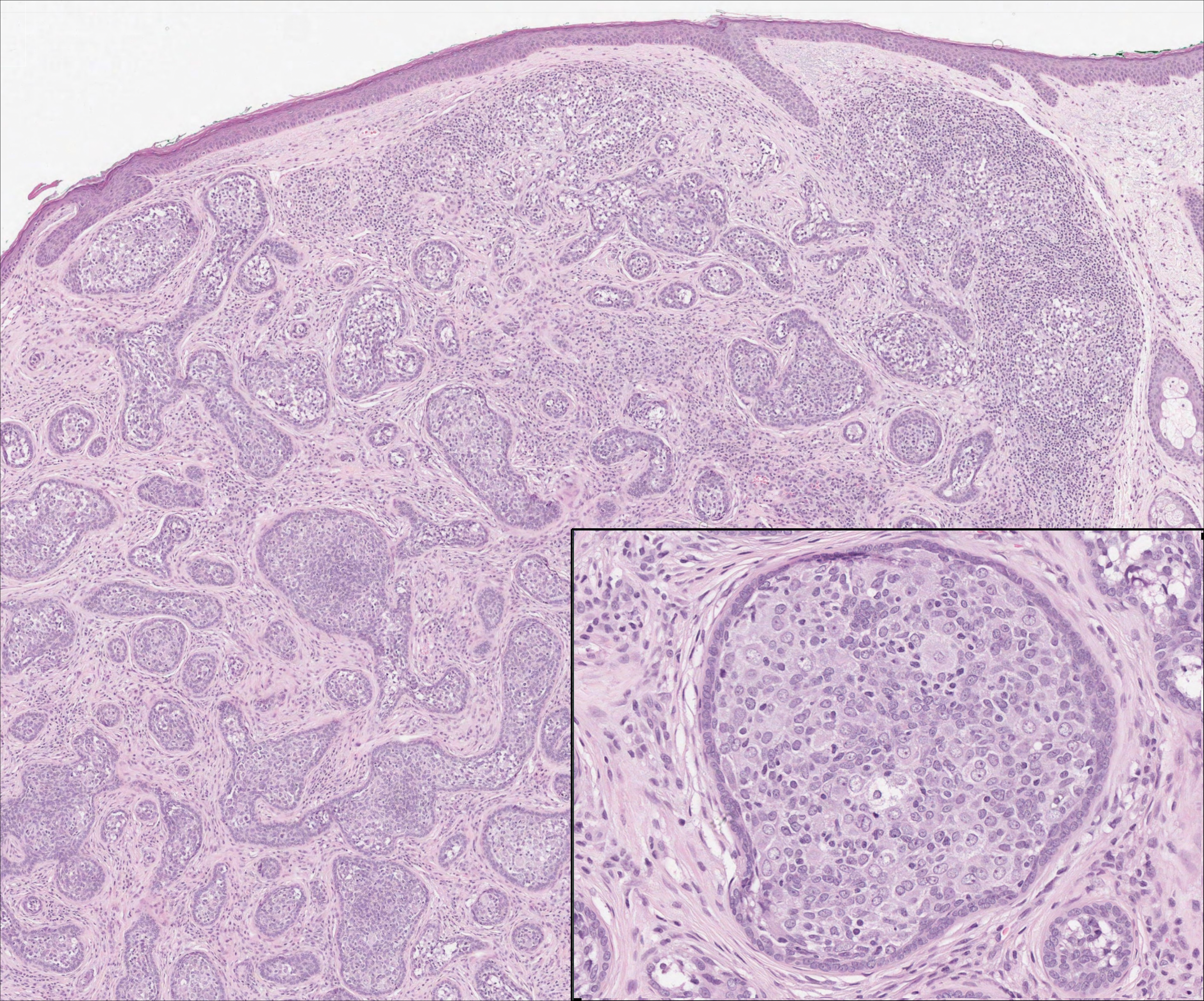
The dense lymphocytic infiltrate seen in LELCS may obscure the neoplastic epithelioid cells and in doing so may mimic a lymphoproliferative disorder, such as lymphomatoid papulosis (LyP). Lymphomatoid papulosis is a chronic CD30+ lymphoproliferative disorder consisting of recurrent crops of self-resolving papulonodules occurring on the trunk, arms, and legs. The average age of onset is in the third to fourth decades of life. Histology is dependent on the subtype; type A, the most common subtype, displays a wedge-shaped dermal infiltrate consisting of small lymphocytes (Figure 2) admixed with larger CD30+ atypical lymphocytes with prominent nucleoli (Figure 2 inset).7 Bizarre, binucleated forms resembling Reed-Sternberg cells also may be observed along with hallmark cells, which contain a horseshoe-shaped nucleus. The presence of admixed neutrophils and eosinophils also are common in type A LyP, a feature that is not characteristic of LELCS. Moreover, the atypical cells in LyP would not stain positively for epithelial markers as they would in LELCS.
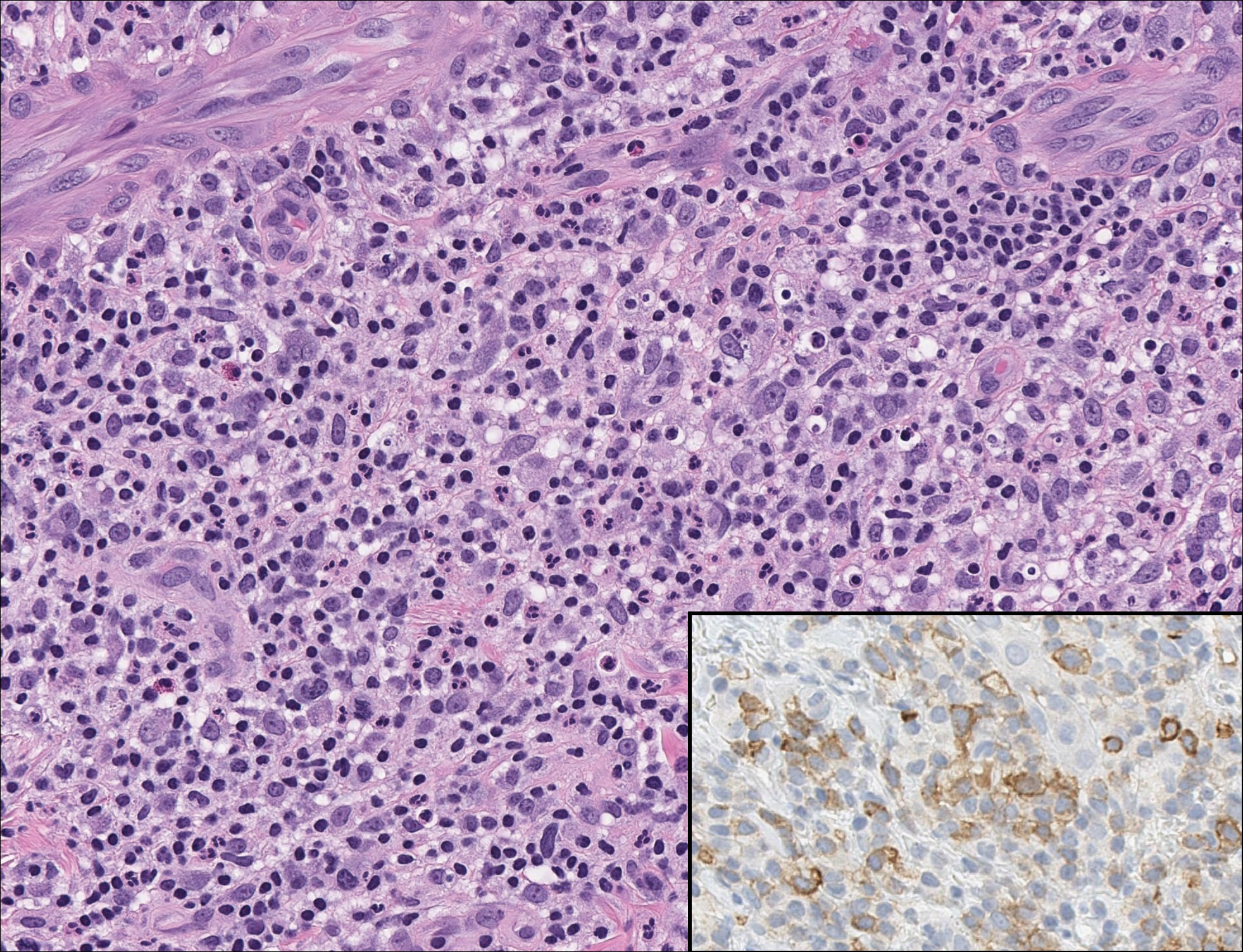
Rosai-Dorfman disease is a rare condition that usually presents with painless cervical lymphadenopathy, typically in the first and second decades of life. Skin involvement can be seen in a small subset of extranodal cases, but cutaneous involvement alone is uncommon. On histopathology, cutaneous lesions are characterized by a dense dermal infiltrate of atypical histiocytes with vesicular nuclei and pale cytoplasm admixed with inflammatory cells, including lymphocytes, neutrophils, and plasma cells (Figure 3). Intracytoplasmic inflammatory cells or emperipolesis often is appreciated (Figure 3 inset).8,9 The atypical histiocytes stain positively for S100 and negatively for CD1a.
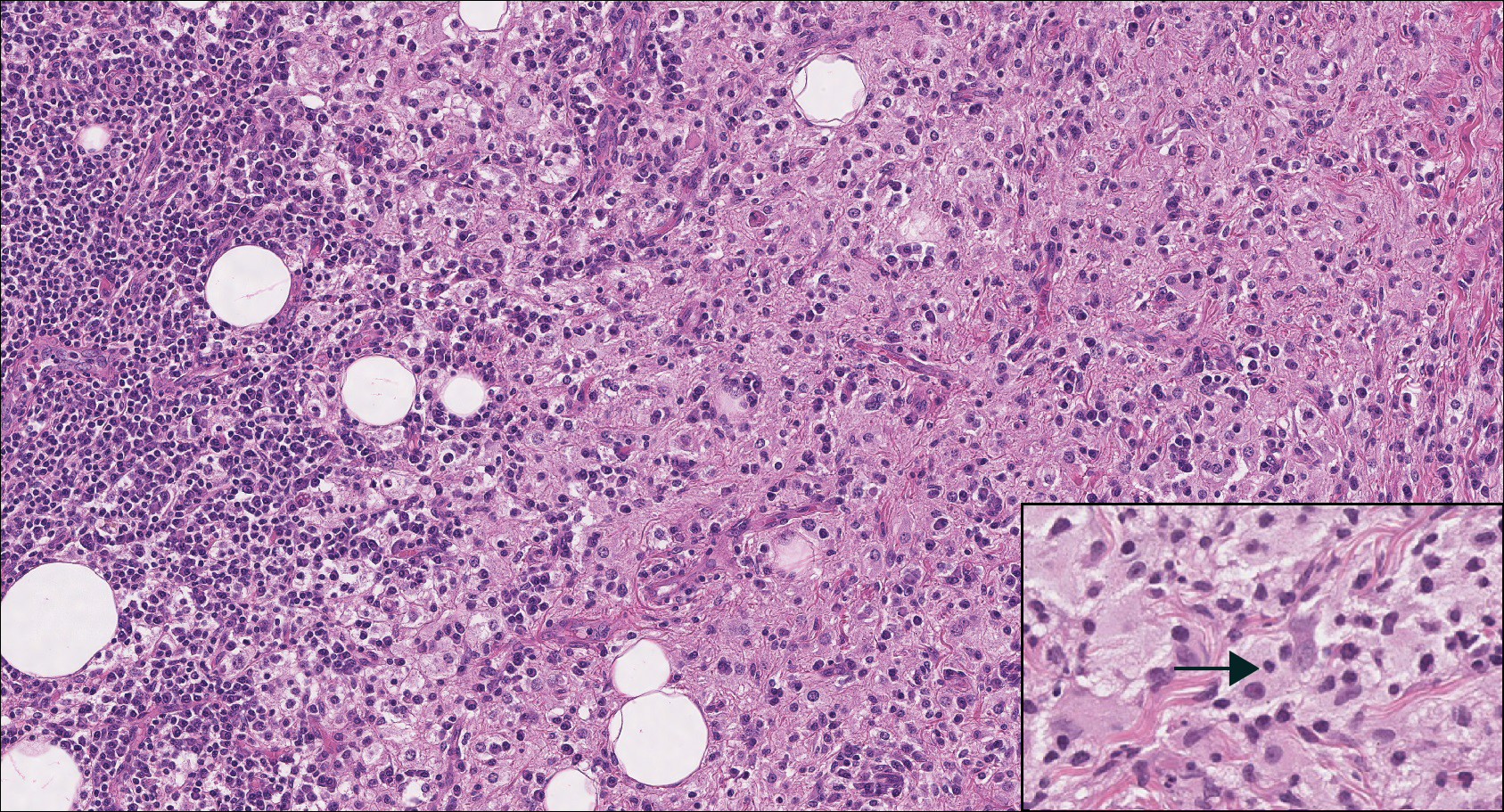
Lymphoepitheliomalike carcinoma of the skin sometimes is considered to be a poorly differentiated, inflamed variant of squamous cell carcinoma (SCC).10 A number of features may allow distinction of a primary cutaneous SCC from LELCS; for instance, SCC is more likely to have an epidermal connection and at least focal signs of squamous differentiation,11 which can include the presence of poorly differentiated epithelial cells with mitoses (Figure 4), keratin pearls, dyskeratotic cells, or intercellular bridges.12 Moreover, SCCs have a more variable surrounding inflammatory infiltrate compared to LELCS.
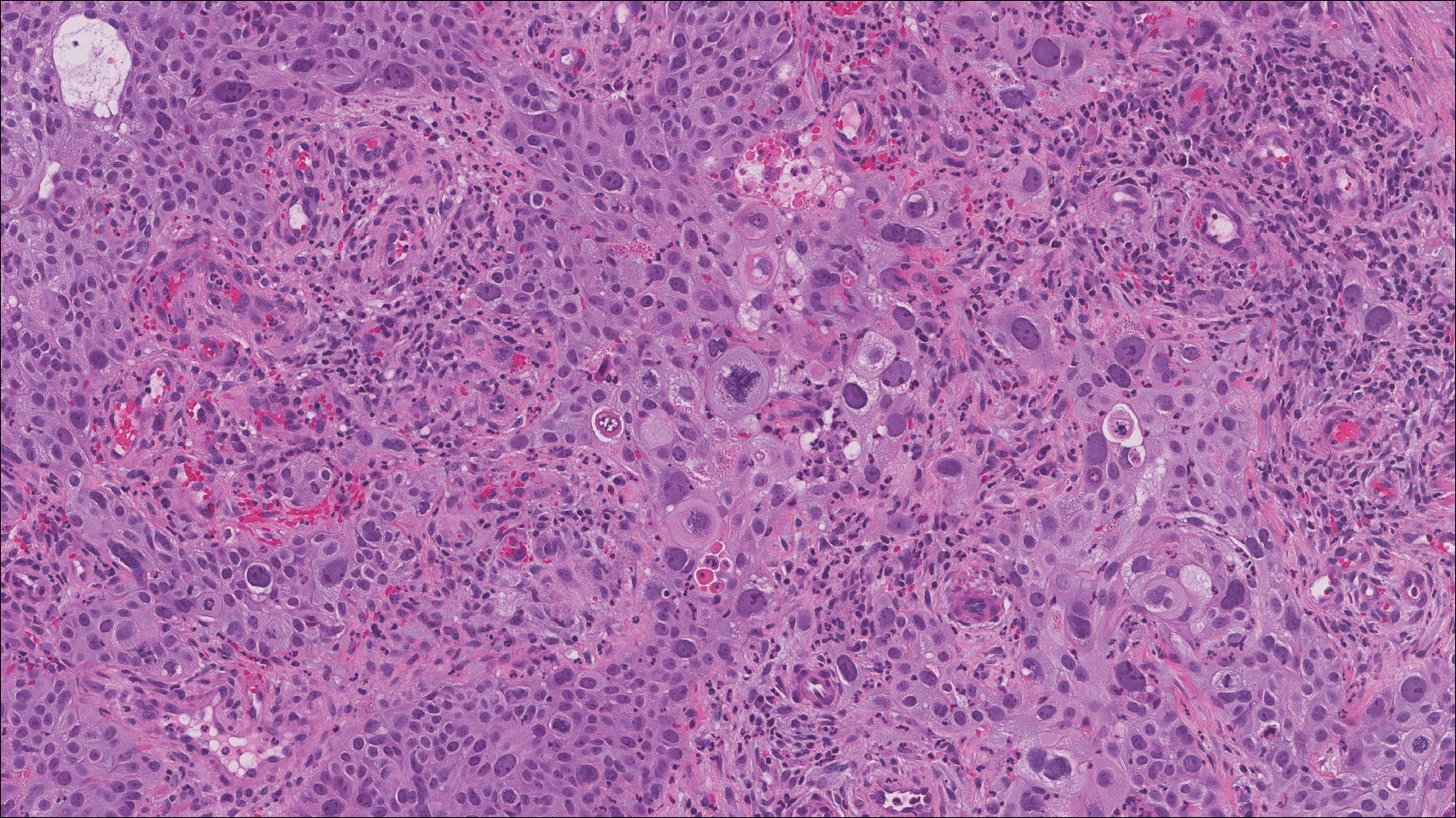
- Swanson SA, Cooper PH, Mills SE, et al. Lymphoepithelioma-like carcinoma of the skin. Mod Pathol. 1988;1:359-365.
- Aoki R, Mitsui H, Harada K, et al. A case of lymphoepithelioma-like carcinoma of the skin associated with Epstein-Barr virus infection. J Am Acad Dermatol. 2010;62:681-684.
- Morteza Abedi S, Salama S, Alowami S. Lymphoepithelioma-like carcinoma of the skin: case report and approach to surgical pathology sign out. Rare Tumors. 2013;5:E47.
- Requena L, Sánchez Yus E, Jiménez E, et al. Lymphoepithelioma-like carcinoma of the skin: a light-microscopic and immunohistochemical study. J Cutan Pathol. 1994;21:541-548.
- Welch PQ, Williams SB, Foss RD, et al. Lymphoepithelioma-like carcinoma of head and neck skin: a systematic analysis of 11 cases and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:78-86.
- Santa Cruz DJ, Barr RJ, Headington JT. Cutaneous lymphadenoma. Am J Surg Pathol. 1991;15:101-110.
- Patterson JW. Cutaneous infiltrates--lymphomatous and leukemic. In: Patterson JW, Hosler GA, eds. Weedon's Skin Pathology. 4th ed. London, United Kingdom: Churchill Livingstone; 2016:1186-1189.
- Patterson JW. Cutaneous infiltrates--nonlymphoid. In: Patterson JW, Hosler GA, eds. Weedon's Skin Pathology. 4th ed. London, United Kingdom: Churchill Livingstone; 2016:1158.
- Skiljo M, Garcia-Lora E, Tercedor J, et al. Purely cutaneous Rosai-Dorfman disease. Dermatology. 1995;191:49-51.
- Wang G, Bordeaux JS, Rowe DJ, et al. Lymphoepithelioma-like carcinoma vs inflamed squamous cell carcinoma of the skin. JAMA Dermatol. 2014;150:1367-1368.
- Hall G, Duncan A, Azurdia R, et al. Lymphoepithelioma-like carcinoma of the skin: a case with lymph node metastases at presentation. Am J Dermatopathol. 2006;28:211-215.
- Lind AC, Breer WA, Wick MR. Lymphoepithelioma-like carcinoma of the skin with apparent origin in the epidermis--a pattern or an entity? a case report. Cancer. 1999;85:884-890.
The Diagnosis: Lymphoepitheliomalike Carcinoma of the Skin
The term lymphoepitheliomalike carcinoma of the skin (LELCS) initially was proposed by Swanson et al1 in 1988 when they described 5 patients with cutaneous neoplasms histologically resembling nasopharyngeal carcinoma, also known as lymphoepithelioma. A PubMed search of articles indexed for MEDLINE using the term lymphoepitheliomalike carcinoma of the skin revealed over 60 cases of LELCS since 1988. However, unlike nasopharyngeal carcinoma, LELCS has not been associated with Epstein-Barr virus, with the exception of 1 known reported case.2 The clinical appearance of LELCS is nonspecific but usually presents as a flesh-colored to erythematous nodule, as was seen in the current case. Lesions commonly are found on the head and neck in middle-aged to elderly patients with a slight male predominance.2
On histology, LELCS is characterized by aggregations of large, atypical epithelioid cells surrounded by a dense lymphoplasmocytic infiltrate (right quiz image). The neoplasm tends to reside within the deep dermis and/or subcutis1 without appreciable epidermal involvement (left quiz image). The atypical epithelioid cells demonstrate positive immunoreactivity for cytokeratins (right quiz image inset), p40/p63, and epithelial membrane antigen,3 and the surrounding lymphocytic infiltrate stains positively for leukocyte common antigen. The tumor histogenesis still is unknown, although an epidermal origin has been suggested given its staining pattern.2 Other investigators have postulated on an adnexal origin, citing the tumor's dermal location along with case reports describing possible glandular, sebaceous, or follicular differentiation.2,4
Treatment for LELCS can include either standard surgical excision or Mohs micrographic surgery, with radiation reserved for lymph node involvement, tumor recurrence, or poor surgical candidates.2,3,5 With appropriate therapy, prognosis may be considered favorable. Data from 49 LELCS patients presenting from 1988 and 2008 showed that 36 (73.5%) had no evidence of recurrence after treatment with standard surgical excision, 4 (8.2%) had local recurrence, and 6 (12.2%) developed lymph node metastasis, which led to death in 1 (2.0%) patient.2
Given the histologic similarity of LELCS to nasopharyngeal carcinoma, it is important to rule out the possibility of cutaneous metastasis, which can be done by testing for Epstein-Barr virus and performing either computed tomography imaging or comprehensive laryngoscopic examination of the head and neck region. In the current case, the patient was referred for laryngoscopy, at which time no suspicious lesions were identified. He subsequently underwent treatment with Mohs micrographic surgery, and the tumor was cleared after 2 surgical stages. At 5-month follow-up, the patient continued to do well with no signs of clinical recurrence.
Cutaneous lymphadenoma may be included in the differential diagnosis for LELCS on histopathology. This neoplasm is characterized by a well-circumscribed dermal proliferation of basaloid tumor islands within a fibrotic stroma (Figure 1). The basaloid cells may display peripheral palisading, and lymphocytes often are seen infiltrating the tumor lobules and the surrounding stroma (Figure 1 inset). Clinically, cutaneous lymphadenomas are slowly growing nodules that typically occur in young to middle-aged patients,4,6 unlike LELCS, which is more commonly observed in middle-aged to elderly patients.2
The dense lymphocytic infiltrate seen in LELCS may obscure the neoplastic epithelioid cells and in doing so may mimic a lymphoproliferative disorder, such as lymphomatoid papulosis (LyP). Lymphomatoid papulosis is a chronic CD30+ lymphoproliferative disorder consisting of recurrent crops of self-resolving papulonodules occurring on the trunk, arms, and legs. The average age of onset is in the third to fourth decades of life. Histology is dependent on the subtype; type A, the most common subtype, displays a wedge-shaped dermal infiltrate consisting of small lymphocytes (Figure 2) admixed with larger CD30+ atypical lymphocytes with prominent nucleoli (Figure 2 inset).7 Bizarre, binucleated forms resembling Reed-Sternberg cells also may be observed along with hallmark cells, which contain a horseshoe-shaped nucleus. The presence of admixed neutrophils and eosinophils also are common in type A LyP, a feature that is not characteristic of LELCS. Moreover, the atypical cells in LyP would not stain positively for epithelial markers as they would in LELCS.
Rosai-Dorfman disease is a rare condition that usually presents with painless cervical lymphadenopathy, typically in the first and second decades of life. Skin involvement can be seen in a small subset of extranodal cases, but cutaneous involvement alone is uncommon. On histopathology, cutaneous lesions are characterized by a dense dermal infiltrate of atypical histiocytes with vesicular nuclei and pale cytoplasm admixed with inflammatory cells, including lymphocytes, neutrophils, and plasma cells (Figure 3). Intracytoplasmic inflammatory cells or emperipolesis often is appreciated (Figure 3 inset).8,9 The atypical histiocytes stain positively for S100 and negatively for CD1a.
Lymphoepitheliomalike carcinoma of the skin sometimes is considered to be a poorly differentiated, inflamed variant of squamous cell carcinoma (SCC).10 A number of features may allow distinction of a primary cutaneous SCC from LELCS; for instance, SCC is more likely to have an epidermal connection and at least focal signs of squamous differentiation,11 which can include the presence of poorly differentiated epithelial cells with mitoses (Figure 4), keratin pearls, dyskeratotic cells, or intercellular bridges.12 Moreover, SCCs have a more variable surrounding inflammatory infiltrate compared to LELCS.
The Diagnosis: Lymphoepitheliomalike Carcinoma of the Skin
The term lymphoepitheliomalike carcinoma of the skin (LELCS) initially was proposed by Swanson et al1 in 1988 when they described 5 patients with cutaneous neoplasms histologically resembling nasopharyngeal carcinoma, also known as lymphoepithelioma. A PubMed search of articles indexed for MEDLINE using the term lymphoepitheliomalike carcinoma of the skin revealed over 60 cases of LELCS since 1988. However, unlike nasopharyngeal carcinoma, LELCS has not been associated with Epstein-Barr virus, with the exception of 1 known reported case.2 The clinical appearance of LELCS is nonspecific but usually presents as a flesh-colored to erythematous nodule, as was seen in the current case. Lesions commonly are found on the head and neck in middle-aged to elderly patients with a slight male predominance.2
On histology, LELCS is characterized by aggregations of large, atypical epithelioid cells surrounded by a dense lymphoplasmocytic infiltrate (right quiz image). The neoplasm tends to reside within the deep dermis and/or subcutis1 without appreciable epidermal involvement (left quiz image). The atypical epithelioid cells demonstrate positive immunoreactivity for cytokeratins (right quiz image inset), p40/p63, and epithelial membrane antigen,3 and the surrounding lymphocytic infiltrate stains positively for leukocyte common antigen. The tumor histogenesis still is unknown, although an epidermal origin has been suggested given its staining pattern.2 Other investigators have postulated on an adnexal origin, citing the tumor's dermal location along with case reports describing possible glandular, sebaceous, or follicular differentiation.2,4
Treatment for LELCS can include either standard surgical excision or Mohs micrographic surgery, with radiation reserved for lymph node involvement, tumor recurrence, or poor surgical candidates.2,3,5 With appropriate therapy, prognosis may be considered favorable. Data from 49 LELCS patients presenting from 1988 and 2008 showed that 36 (73.5%) had no evidence of recurrence after treatment with standard surgical excision, 4 (8.2%) had local recurrence, and 6 (12.2%) developed lymph node metastasis, which led to death in 1 (2.0%) patient.2
Given the histologic similarity of LELCS to nasopharyngeal carcinoma, it is important to rule out the possibility of cutaneous metastasis, which can be done by testing for Epstein-Barr virus and performing either computed tomography imaging or comprehensive laryngoscopic examination of the head and neck region. In the current case, the patient was referred for laryngoscopy, at which time no suspicious lesions were identified. He subsequently underwent treatment with Mohs micrographic surgery, and the tumor was cleared after 2 surgical stages. At 5-month follow-up, the patient continued to do well with no signs of clinical recurrence.
Cutaneous lymphadenoma may be included in the differential diagnosis for LELCS on histopathology. This neoplasm is characterized by a well-circumscribed dermal proliferation of basaloid tumor islands within a fibrotic stroma (Figure 1). The basaloid cells may display peripheral palisading, and lymphocytes often are seen infiltrating the tumor lobules and the surrounding stroma (Figure 1 inset). Clinically, cutaneous lymphadenomas are slowly growing nodules that typically occur in young to middle-aged patients,4,6 unlike LELCS, which is more commonly observed in middle-aged to elderly patients.2
The dense lymphocytic infiltrate seen in LELCS may obscure the neoplastic epithelioid cells and in doing so may mimic a lymphoproliferative disorder, such as lymphomatoid papulosis (LyP). Lymphomatoid papulosis is a chronic CD30+ lymphoproliferative disorder consisting of recurrent crops of self-resolving papulonodules occurring on the trunk, arms, and legs. The average age of onset is in the third to fourth decades of life. Histology is dependent on the subtype; type A, the most common subtype, displays a wedge-shaped dermal infiltrate consisting of small lymphocytes (Figure 2) admixed with larger CD30+ atypical lymphocytes with prominent nucleoli (Figure 2 inset).7 Bizarre, binucleated forms resembling Reed-Sternberg cells also may be observed along with hallmark cells, which contain a horseshoe-shaped nucleus. The presence of admixed neutrophils and eosinophils also are common in type A LyP, a feature that is not characteristic of LELCS. Moreover, the atypical cells in LyP would not stain positively for epithelial markers as they would in LELCS.
Rosai-Dorfman disease is a rare condition that usually presents with painless cervical lymphadenopathy, typically in the first and second decades of life. Skin involvement can be seen in a small subset of extranodal cases, but cutaneous involvement alone is uncommon. On histopathology, cutaneous lesions are characterized by a dense dermal infiltrate of atypical histiocytes with vesicular nuclei and pale cytoplasm admixed with inflammatory cells, including lymphocytes, neutrophils, and plasma cells (Figure 3). Intracytoplasmic inflammatory cells or emperipolesis often is appreciated (Figure 3 inset).8,9 The atypical histiocytes stain positively for S100 and negatively for CD1a.
Lymphoepitheliomalike carcinoma of the skin sometimes is considered to be a poorly differentiated, inflamed variant of squamous cell carcinoma (SCC).10 A number of features may allow distinction of a primary cutaneous SCC from LELCS; for instance, SCC is more likely to have an epidermal connection and at least focal signs of squamous differentiation,11 which can include the presence of poorly differentiated epithelial cells with mitoses (Figure 4), keratin pearls, dyskeratotic cells, or intercellular bridges.12 Moreover, SCCs have a more variable surrounding inflammatory infiltrate compared to LELCS.
- Swanson SA, Cooper PH, Mills SE, et al. Lymphoepithelioma-like carcinoma of the skin. Mod Pathol. 1988;1:359-365.
- Aoki R, Mitsui H, Harada K, et al. A case of lymphoepithelioma-like carcinoma of the skin associated with Epstein-Barr virus infection. J Am Acad Dermatol. 2010;62:681-684.
- Morteza Abedi S, Salama S, Alowami S. Lymphoepithelioma-like carcinoma of the skin: case report and approach to surgical pathology sign out. Rare Tumors. 2013;5:E47.
- Requena L, Sánchez Yus E, Jiménez E, et al. Lymphoepithelioma-like carcinoma of the skin: a light-microscopic and immunohistochemical study. J Cutan Pathol. 1994;21:541-548.
- Welch PQ, Williams SB, Foss RD, et al. Lymphoepithelioma-like carcinoma of head and neck skin: a systematic analysis of 11 cases and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:78-86.
- Santa Cruz DJ, Barr RJ, Headington JT. Cutaneous lymphadenoma. Am J Surg Pathol. 1991;15:101-110.
- Patterson JW. Cutaneous infiltrates--lymphomatous and leukemic. In: Patterson JW, Hosler GA, eds. Weedon's Skin Pathology. 4th ed. London, United Kingdom: Churchill Livingstone; 2016:1186-1189.
- Patterson JW. Cutaneous infiltrates--nonlymphoid. In: Patterson JW, Hosler GA, eds. Weedon's Skin Pathology. 4th ed. London, United Kingdom: Churchill Livingstone; 2016:1158.
- Skiljo M, Garcia-Lora E, Tercedor J, et al. Purely cutaneous Rosai-Dorfman disease. Dermatology. 1995;191:49-51.
- Wang G, Bordeaux JS, Rowe DJ, et al. Lymphoepithelioma-like carcinoma vs inflamed squamous cell carcinoma of the skin. JAMA Dermatol. 2014;150:1367-1368.
- Hall G, Duncan A, Azurdia R, et al. Lymphoepithelioma-like carcinoma of the skin: a case with lymph node metastases at presentation. Am J Dermatopathol. 2006;28:211-215.
- Lind AC, Breer WA, Wick MR. Lymphoepithelioma-like carcinoma of the skin with apparent origin in the epidermis--a pattern or an entity? a case report. Cancer. 1999;85:884-890.
- Swanson SA, Cooper PH, Mills SE, et al. Lymphoepithelioma-like carcinoma of the skin. Mod Pathol. 1988;1:359-365.
- Aoki R, Mitsui H, Harada K, et al. A case of lymphoepithelioma-like carcinoma of the skin associated with Epstein-Barr virus infection. J Am Acad Dermatol. 2010;62:681-684.
- Morteza Abedi S, Salama S, Alowami S. Lymphoepithelioma-like carcinoma of the skin: case report and approach to surgical pathology sign out. Rare Tumors. 2013;5:E47.
- Requena L, Sánchez Yus E, Jiménez E, et al. Lymphoepithelioma-like carcinoma of the skin: a light-microscopic and immunohistochemical study. J Cutan Pathol. 1994;21:541-548.
- Welch PQ, Williams SB, Foss RD, et al. Lymphoepithelioma-like carcinoma of head and neck skin: a systematic analysis of 11 cases and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:78-86.
- Santa Cruz DJ, Barr RJ, Headington JT. Cutaneous lymphadenoma. Am J Surg Pathol. 1991;15:101-110.
- Patterson JW. Cutaneous infiltrates--lymphomatous and leukemic. In: Patterson JW, Hosler GA, eds. Weedon's Skin Pathology. 4th ed. London, United Kingdom: Churchill Livingstone; 2016:1186-1189.
- Patterson JW. Cutaneous infiltrates--nonlymphoid. In: Patterson JW, Hosler GA, eds. Weedon's Skin Pathology. 4th ed. London, United Kingdom: Churchill Livingstone; 2016:1158.
- Skiljo M, Garcia-Lora E, Tercedor J, et al. Purely cutaneous Rosai-Dorfman disease. Dermatology. 1995;191:49-51.
- Wang G, Bordeaux JS, Rowe DJ, et al. Lymphoepithelioma-like carcinoma vs inflamed squamous cell carcinoma of the skin. JAMA Dermatol. 2014;150:1367-1368.
- Hall G, Duncan A, Azurdia R, et al. Lymphoepithelioma-like carcinoma of the skin: a case with lymph node metastases at presentation. Am J Dermatopathol. 2006;28:211-215.
- Lind AC, Breer WA, Wick MR. Lymphoepithelioma-like carcinoma of the skin with apparent origin in the epidermis--a pattern or an entity? a case report. Cancer. 1999;85:884-890.
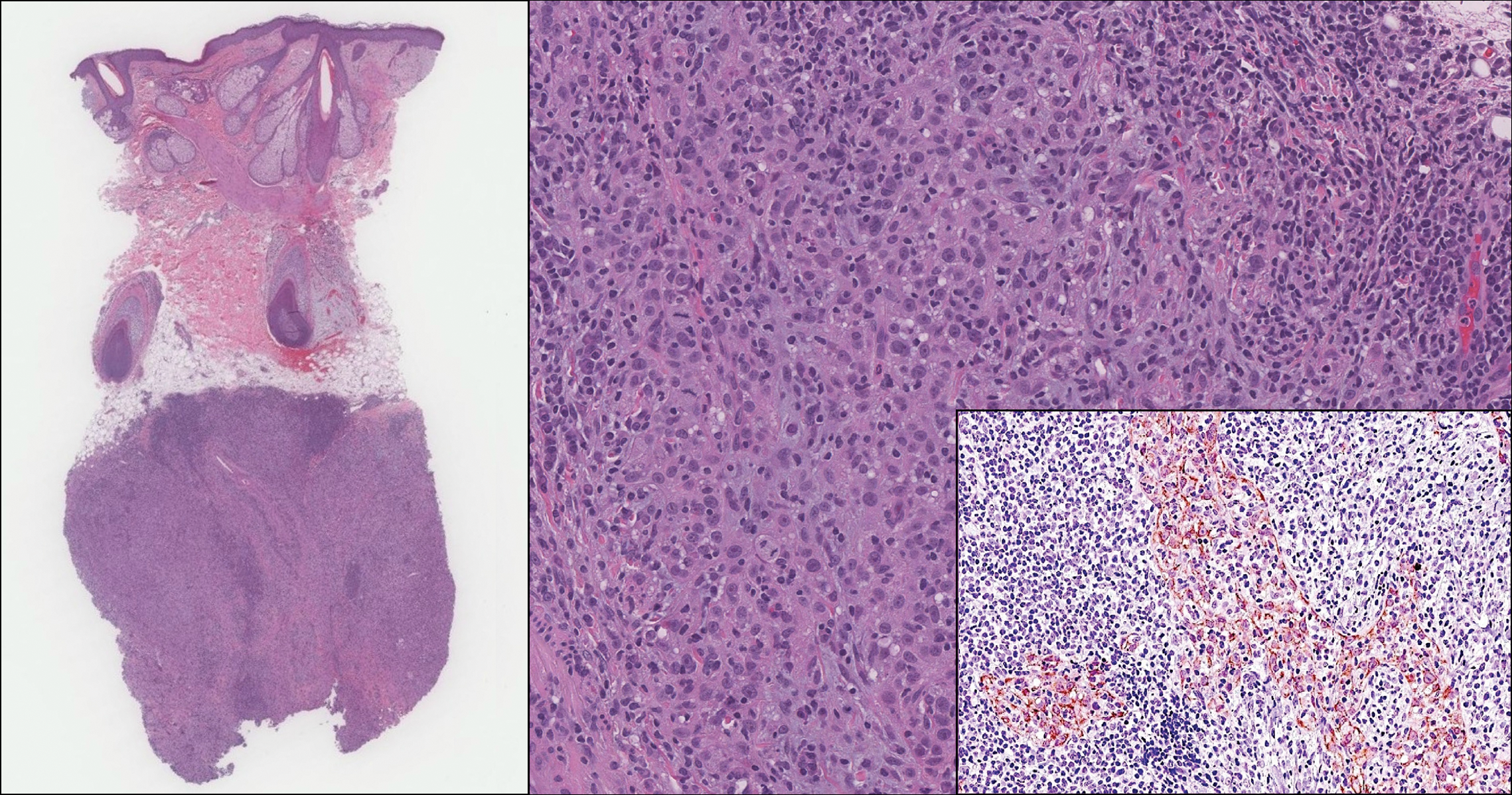
An 81-year-old man with history of melanoma and nonmelanoma skin cancer presented with a subcutaneous nodule on the left cheek of 3 months' duration. The lesion was reportedly asymptomatic and measured 2.6×2.9 cm. A punch biopsy of the lesion was obtained for histopathologic evaluation.
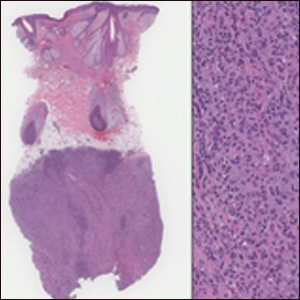
Asymptomatic Erythematous Plaques on the Scalp and Face
The Diagnosis: Granuloma Faciale
A biopsy from a scalp lesion showed an intense mixed inflammatory infiltrate mainly consisting of eosinophils, but lymphocytes, histiocytes, neutrophils, and plasma cells also were present. A grenz zone was observed between the dermal infiltrate and epidermis. Perivascular infiltrates were penetrating vessel walls, and hyalinization of the vessel walls also was seen (Figure 1). Direct immunofluorescence demonstrated IgG positivity on vessel walls (Figure 2). A diagnosis of granuloma faciale with extrafacial lesions was made. Twice daily application of tacrolimus ointment 0.1% was started, but after a 10-month course of treatment, there was no notable difference in the lesions.
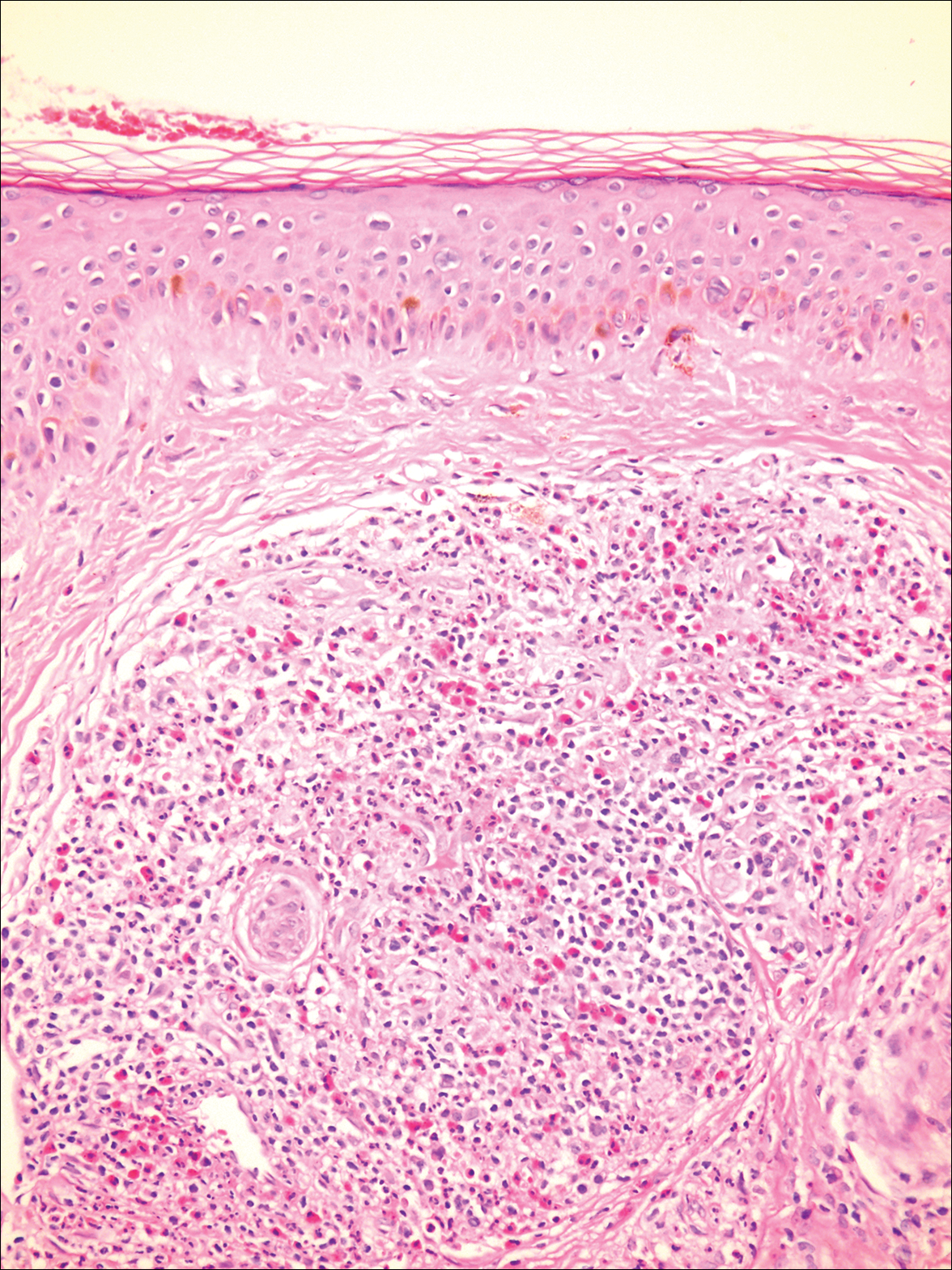
Granuloma faciale (GF) is an uncommon benign dermatosis of unknown pathogenesis characterized by erythematous, brown, or violaceous papules, plaques, or nodules. Granuloma faciale lesions can be solitary or multiple as well as disseminated and most often occur on the face. Predilection sites include the nose, periauricular area, cheeks, forehead, eyelids, and ears; however, lesions also have been reported to occur in extrafacial areas such as the trunk, arms, and legs.1-4 In our patient, multiple plaques were seen on the scalp. Facial lesions usually precede extrafacial lesions, which may present months to several years after the appearance of facial disease; however, according to our patient's history his scalp lesions appeared before the facial lesions.
The differential diagnoses for GF mainly include erythema elevatum diutinum, cutaneous sarcoidosis, cutaneous lymphoma, lupus, basal cell carcinoma, and cutaneous pseudolymphoma.5 Diagnosis may be established based on a combination of clinical features and skin biopsy results. On histopathologic examination, small-vessel vasculitis usually is present with an infiltrate predominantly consisting of neutrophils and eosinophils.6
It has been suggested that actinic damage plays a role in the etiology of GF.7 The pathogenesis is uncertain, but it is thought that immunophenotypic and molecular analysis of the dermal infiltrate in GF reveals that most lymphocytes are clonally expanded and the process is mediated by interferon gamma.7 Tacrolimus acts by binding and inactivating calcineurin and thus blocking T-cell activation and proliferation, so it is not surprising that topical tacrolimus has been shown to be useful in the management of this condition.8
- Leite I, Moreira A, Guedes R, et al. Granuloma faciale of the scalp. Dermatol Online J. 2011;17:6.
- De D, Kanwar AJ, Radotra BD, et al. Extrafacial granuloma faciale: report of a case. J Eur Acad Dermatol Venereol. 2007;21:1284-1286.
- Castellano-Howard L, Fairbee SI, Hogan DJ, et al. Extrafacial granuloma faciale: report of a case and response to treatment. Cutis. 2001;67:413-415.
- Inanir I, Alvur Y. Granuloma faciale with extrafacial lesions. Br J Dermatol. 2001;145:360-362.
- Ortonne N, Wechsler J, Bagot M, et al. Granuloma faciale: a clinicopathologic study of 66 patients. J Am Acad Dermatol. 2005;53:1002-1009.
- LeBoit PE. Granuloma faciale: a diagnosis deserving of dignity. Am J Dermatopathol. 2002;24:440-443.
- Koplon BS, Wood MG. Granuloma faciale. first reported case in a Negro. Arch Dermatol. 1967;96:188-192.
- Ludwig E, Allam JP, Bieber T, et al. New treatment modalities for granuloma faciale. Br J Dermatol. 2003;149:634-637.
The Diagnosis: Granuloma Faciale
A biopsy from a scalp lesion showed an intense mixed inflammatory infiltrate mainly consisting of eosinophils, but lymphocytes, histiocytes, neutrophils, and plasma cells also were present. A grenz zone was observed between the dermal infiltrate and epidermis. Perivascular infiltrates were penetrating vessel walls, and hyalinization of the vessel walls also was seen (Figure 1). Direct immunofluorescence demonstrated IgG positivity on vessel walls (Figure 2). A diagnosis of granuloma faciale with extrafacial lesions was made. Twice daily application of tacrolimus ointment 0.1% was started, but after a 10-month course of treatment, there was no notable difference in the lesions.
Granuloma faciale (GF) is an uncommon benign dermatosis of unknown pathogenesis characterized by erythematous, brown, or violaceous papules, plaques, or nodules. Granuloma faciale lesions can be solitary or multiple as well as disseminated and most often occur on the face. Predilection sites include the nose, periauricular area, cheeks, forehead, eyelids, and ears; however, lesions also have been reported to occur in extrafacial areas such as the trunk, arms, and legs.1-4 In our patient, multiple plaques were seen on the scalp. Facial lesions usually precede extrafacial lesions, which may present months to several years after the appearance of facial disease; however, according to our patient's history his scalp lesions appeared before the facial lesions.
The differential diagnoses for GF mainly include erythema elevatum diutinum, cutaneous sarcoidosis, cutaneous lymphoma, lupus, basal cell carcinoma, and cutaneous pseudolymphoma.5 Diagnosis may be established based on a combination of clinical features and skin biopsy results. On histopathologic examination, small-vessel vasculitis usually is present with an infiltrate predominantly consisting of neutrophils and eosinophils.6
It has been suggested that actinic damage plays a role in the etiology of GF.7 The pathogenesis is uncertain, but it is thought that immunophenotypic and molecular analysis of the dermal infiltrate in GF reveals that most lymphocytes are clonally expanded and the process is mediated by interferon gamma.7 Tacrolimus acts by binding and inactivating calcineurin and thus blocking T-cell activation and proliferation, so it is not surprising that topical tacrolimus has been shown to be useful in the management of this condition.8
The Diagnosis: Granuloma Faciale
A biopsy from a scalp lesion showed an intense mixed inflammatory infiltrate mainly consisting of eosinophils, but lymphocytes, histiocytes, neutrophils, and plasma cells also were present. A grenz zone was observed between the dermal infiltrate and epidermis. Perivascular infiltrates were penetrating vessel walls, and hyalinization of the vessel walls also was seen (Figure 1). Direct immunofluorescence demonstrated IgG positivity on vessel walls (Figure 2). A diagnosis of granuloma faciale with extrafacial lesions was made. Twice daily application of tacrolimus ointment 0.1% was started, but after a 10-month course of treatment, there was no notable difference in the lesions.
Granuloma faciale (GF) is an uncommon benign dermatosis of unknown pathogenesis characterized by erythematous, brown, or violaceous papules, plaques, or nodules. Granuloma faciale lesions can be solitary or multiple as well as disseminated and most often occur on the face. Predilection sites include the nose, periauricular area, cheeks, forehead, eyelids, and ears; however, lesions also have been reported to occur in extrafacial areas such as the trunk, arms, and legs.1-4 In our patient, multiple plaques were seen on the scalp. Facial lesions usually precede extrafacial lesions, which may present months to several years after the appearance of facial disease; however, according to our patient's history his scalp lesions appeared before the facial lesions.
The differential diagnoses for GF mainly include erythema elevatum diutinum, cutaneous sarcoidosis, cutaneous lymphoma, lupus, basal cell carcinoma, and cutaneous pseudolymphoma.5 Diagnosis may be established based on a combination of clinical features and skin biopsy results. On histopathologic examination, small-vessel vasculitis usually is present with an infiltrate predominantly consisting of neutrophils and eosinophils.6
It has been suggested that actinic damage plays a role in the etiology of GF.7 The pathogenesis is uncertain, but it is thought that immunophenotypic and molecular analysis of the dermal infiltrate in GF reveals that most lymphocytes are clonally expanded and the process is mediated by interferon gamma.7 Tacrolimus acts by binding and inactivating calcineurin and thus blocking T-cell activation and proliferation, so it is not surprising that topical tacrolimus has been shown to be useful in the management of this condition.8
- Leite I, Moreira A, Guedes R, et al. Granuloma faciale of the scalp. Dermatol Online J. 2011;17:6.
- De D, Kanwar AJ, Radotra BD, et al. Extrafacial granuloma faciale: report of a case. J Eur Acad Dermatol Venereol. 2007;21:1284-1286.
- Castellano-Howard L, Fairbee SI, Hogan DJ, et al. Extrafacial granuloma faciale: report of a case and response to treatment. Cutis. 2001;67:413-415.
- Inanir I, Alvur Y. Granuloma faciale with extrafacial lesions. Br J Dermatol. 2001;145:360-362.
- Ortonne N, Wechsler J, Bagot M, et al. Granuloma faciale: a clinicopathologic study of 66 patients. J Am Acad Dermatol. 2005;53:1002-1009.
- LeBoit PE. Granuloma faciale: a diagnosis deserving of dignity. Am J Dermatopathol. 2002;24:440-443.
- Koplon BS, Wood MG. Granuloma faciale. first reported case in a Negro. Arch Dermatol. 1967;96:188-192.
- Ludwig E, Allam JP, Bieber T, et al. New treatment modalities for granuloma faciale. Br J Dermatol. 2003;149:634-637.
- Leite I, Moreira A, Guedes R, et al. Granuloma faciale of the scalp. Dermatol Online J. 2011;17:6.
- De D, Kanwar AJ, Radotra BD, et al. Extrafacial granuloma faciale: report of a case. J Eur Acad Dermatol Venereol. 2007;21:1284-1286.
- Castellano-Howard L, Fairbee SI, Hogan DJ, et al. Extrafacial granuloma faciale: report of a case and response to treatment. Cutis. 2001;67:413-415.
- Inanir I, Alvur Y. Granuloma faciale with extrafacial lesions. Br J Dermatol. 2001;145:360-362.
- Ortonne N, Wechsler J, Bagot M, et al. Granuloma faciale: a clinicopathologic study of 66 patients. J Am Acad Dermatol. 2005;53:1002-1009.
- LeBoit PE. Granuloma faciale: a diagnosis deserving of dignity. Am J Dermatopathol. 2002;24:440-443.
- Koplon BS, Wood MG. Granuloma faciale. first reported case in a Negro. Arch Dermatol. 1967;96:188-192.
- Ludwig E, Allam JP, Bieber T, et al. New treatment modalities for granuloma faciale. Br J Dermatol. 2003;149:634-637.
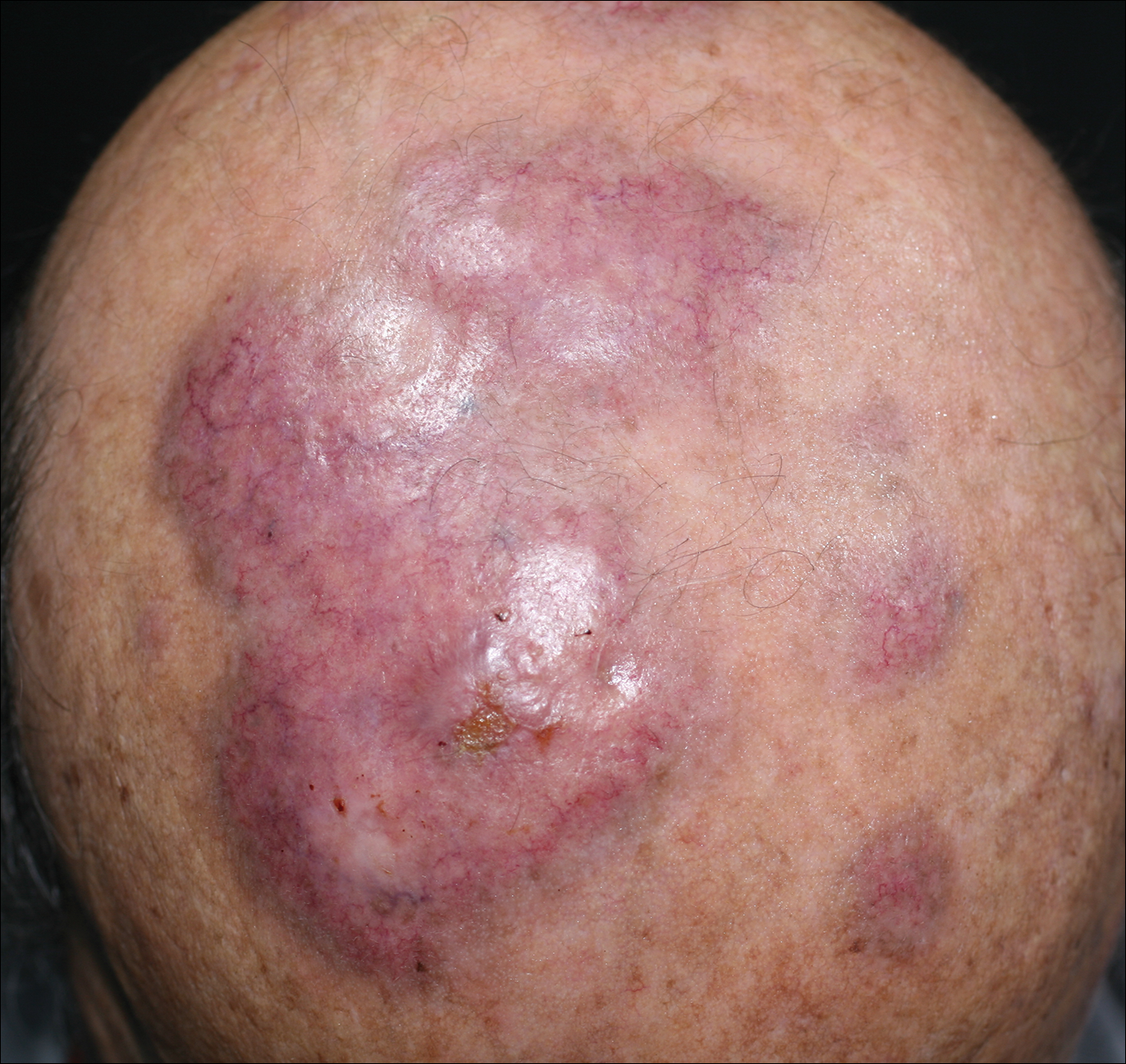
An 84-year-old man presented with gradually enlarging, asymptomatic, erythematous to violaceous plaques on the face and scalp of 11 years' duration ranging in size from 0.5×0.5 cm to 10×8 cm. The plaques were unresponsive to treatment with topical steroids. The lesions were nontender with no associated bleeding, burning, or pruritus. The patient denied any trauma to the sites or systemic symptoms. He had a history of essential hypertension and benign prostatic hyperplasia and had been taking ramipril, tamsulosin, and dutasteride for 5 years. His medical history was otherwise unremarkable, and routine laboratory findings were within normal range.
Enlarging Red Papulonodule on the Chest
The Diagnosis: Metastatic Renal Cell Carcinoma
Histopathologic examination of the punch biopsy demonstrated epithelioid cells with abundant clear cytoplasm and numerous chicken wire-like vascular channels consistent with a diagnosis of cutaneous metastasis of renal cell carcinoma (RCC)(Figure). Collateral history revealed that 8 years prior, the patient had been diagnosed with clear cell RCC, stage III (T3aN0M0). At that time, he was treated with radical nephrectomy, which was considered curative. He remained disease free until several months prior to the development of the cutaneous lesion when he was found to have pulmonary and cerebral metastases with biopsies showing metastatic RCC. He was treated with lobectomy and Gamma Knife radiation for the lung and cerebral metastases, respectively. His oncologist planned to initiate therapy with the multikinase inhibitor sunitinib, which inhibits vascular endothelial growth factor (VEGF) signaling. Unfortunately, the patient died prior to treatment due to overwhelming tumor burden.
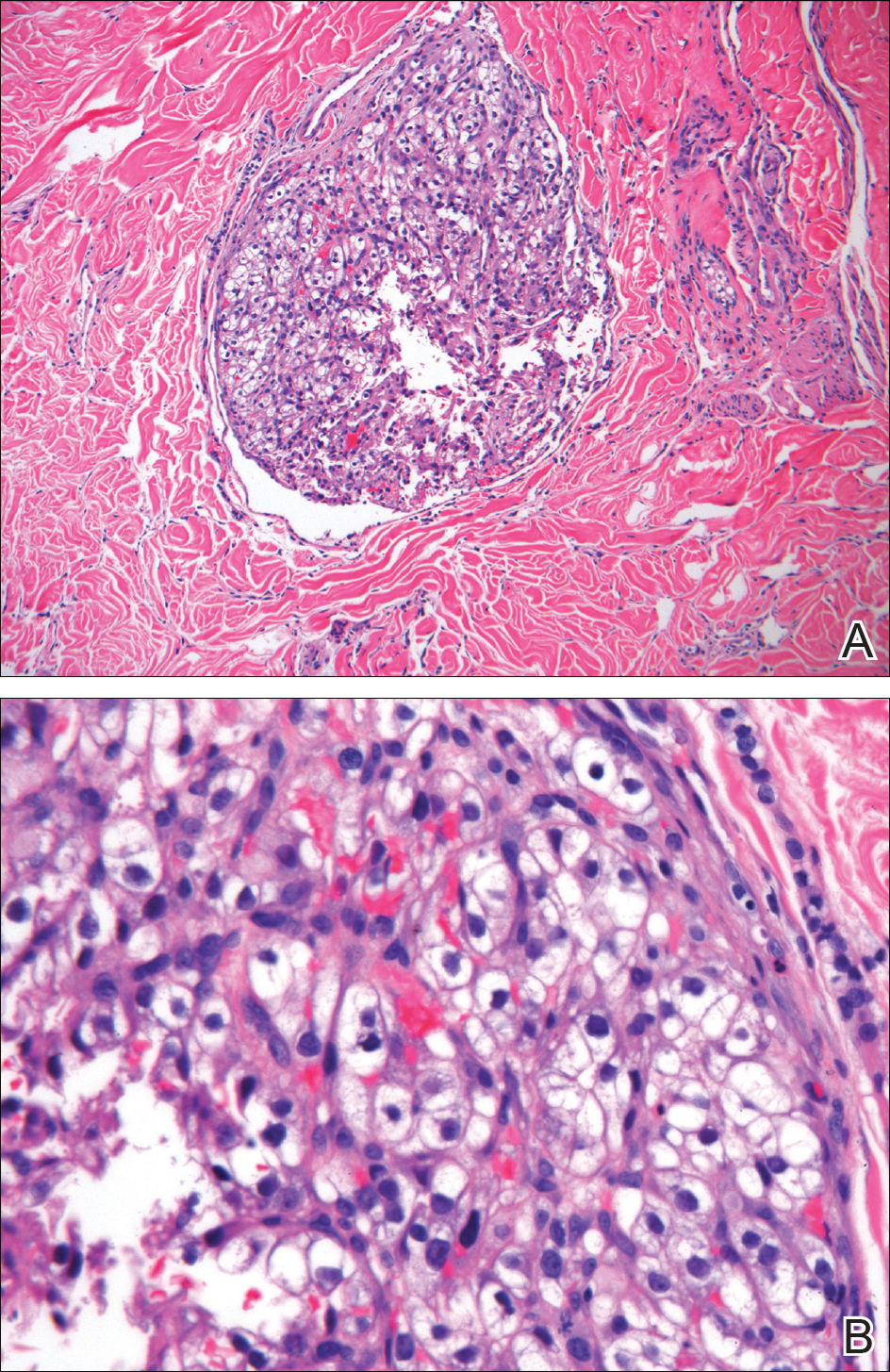
Clear cell RCC, the most common renal malignancy, presents with metastatic disease at the time of diagnosis in 21% of patients.1 An additional 20% of patients with localized disease develop metastases within several years of receiving a nephrectomy without adjuvant therapy, which is standard treatment for stage I to stage III disease.1,2 Metastatic RCC most frequently targets the lungs, bone, liver, and brain, though virtually any organ can be involved. Cutaneous involvement is estimated to occur in 3.3% of RCC cases,3 accounting for only 1.4% of cutaneous metastases overall.4 The risk for developing cutaneous metastases is greatest within 3 years following nephrectomy.3 However, our patient demonstrates that metastasis of RCC to skin can be long delayed (>5 years) despite an initial diagnosis of localized disease.
Cutaneous RCC classically presents as a painless firm papulonodule with a deep red or purple color due to its high vascularity.4 Several retrospective studies have identified the scalp as the most frequent site of cutaneous involvement, followed by the chest, abdomen, and nephrectomy scar.3,4 The differential diagnosis includes other vascular lesions such as pyogenic granuloma, hemangioma, angiosarcoma, bacillary angiomatosis, and Kaposi sarcoma. Diagnosis usually is easily confirmed histologically. Proliferative nests of epithelioid cells with clear cell morphology are surrounded by delicately branching vessels referred to as chicken wire-like vasculature. Immunohistochemical studies demonstrate positivity for pan-cytokeratin, vimentin, and CD-10, and negativity for p63 and cytokeratins 5 and 6, helping to confirm the diagnosis in more challenging cases, especially when there is no known history of primary RCC.5
If cutaneous metastasis of RCC is diagnosed, a chest and abdominal computed tomography scan as well as serum alkaline phosphatase test are warranted, as up to 90% of patients with RCC in the skin have additional lesions in at least 1 other site such as the lungs, bones, or liver.3 Management of metastatic RCC includes surgical excision if a single metastasis is found and either immunotherapy with high-dose IL-2 or an anti-programmed cell death inhibitor. Patients with progressive disease also may receive targeted anti-VEGF inhibitors (eg, axitinib, pazopanib, sunitinib), which have been shown to increase progression-free survival in metastatic RCC.6-8 Interestingly, some evidence suggests severely delayed recurrence of RCC (>5 years following nephrectomy) may predict better response to systemic therapy.9
This case of severely delayed metastasis of RCC 8 years after nephrectomy raises the question of whether routine surveillance for RCC recurrence should continue beyond 5 years. It also underscores the need for further studies to determine the utility of postsurgical adjuvant therapy for localized disease (stages I-III). A randomized clinical trial showed no significant difference in disease-free survival when the multikinase inhibitors sunitinib and sorafenib were used as adjuvant therapy.10 The randomized, placebo-controlled PROTECT trial showed no significant difference in disease-free survival between the VEGF inhibitor pazopanib and placebo when used as adjuvant therapy.11 However, trials are ongoing to investigate a potential survival advantage of adjuvant therapy with the VEGF receptor inhibitor axitinib and the mammalian target of rapamycin inhibitor everolimus.
- Dabestani S, Thorstenson A, Lindblad P, et al. Renal cell carcinoma recurrences and metastases in primary non-metastatic patients: a population-based study. World J Urol. 2016;34:1081-1086.
- Ljungberg B, Campbell SC, Choi HY, et al. The epidemiology of renal cell carcinoma. Eur Urol. 2011;60:615-621.
- Dorairajan LN, Hemal AK, Aron M, et al. Cutaneous metastases in renal cell carcinoma. Urol Int. 1999;63:164-167.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2, pt 1):228-236.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061-1068.
- Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:3584-3590.
- Rini BI, Grunwald V, Fishman MN, et al. Axitinib for first-line metastatic renal cell carcinoma (mRCC): overall efficacy and pharmacokinetic (PK) analyses from a randomized phase II study. J Clin Oncol. 2012;30(suppl). doi:10.1200/jco.2012.30.15_suppl.4503.
- Ficarra V, Novara G. Characterizing late recurrence of renal cell carcinoma. Nat Rev Urol. 2013;10:687-689.
- Haas NB, Manola J, Uzzo RG, et al. Adjuvant sunitinib or sorafenib for high-risk, non-metastatic renal-cell carcinoma (ECOG-ACRIN E2805): a double-blind, placebo-controlled, randomised, phase 3 trial [published online March 9, 2016]. Lancet. 2016;387:2008-2016.
- Motzer RJ, Haas NB, Donskov F, et al; PROTECT investigators. Randomized phase III trial of adjuvant pazopanib versus placebo after nephrectomy in patients with localized or locally advanced renal cell carcinoma [published online September 13, 2017]. J Clin Oncol. 2017;35:3916-3923.
The Diagnosis: Metastatic Renal Cell Carcinoma
Histopathologic examination of the punch biopsy demonstrated epithelioid cells with abundant clear cytoplasm and numerous chicken wire-like vascular channels consistent with a diagnosis of cutaneous metastasis of renal cell carcinoma (RCC)(Figure). Collateral history revealed that 8 years prior, the patient had been diagnosed with clear cell RCC, stage III (T3aN0M0). At that time, he was treated with radical nephrectomy, which was considered curative. He remained disease free until several months prior to the development of the cutaneous lesion when he was found to have pulmonary and cerebral metastases with biopsies showing metastatic RCC. He was treated with lobectomy and Gamma Knife radiation for the lung and cerebral metastases, respectively. His oncologist planned to initiate therapy with the multikinase inhibitor sunitinib, which inhibits vascular endothelial growth factor (VEGF) signaling. Unfortunately, the patient died prior to treatment due to overwhelming tumor burden.
Clear cell RCC, the most common renal malignancy, presents with metastatic disease at the time of diagnosis in 21% of patients.1 An additional 20% of patients with localized disease develop metastases within several years of receiving a nephrectomy without adjuvant therapy, which is standard treatment for stage I to stage III disease.1,2 Metastatic RCC most frequently targets the lungs, bone, liver, and brain, though virtually any organ can be involved. Cutaneous involvement is estimated to occur in 3.3% of RCC cases,3 accounting for only 1.4% of cutaneous metastases overall.4 The risk for developing cutaneous metastases is greatest within 3 years following nephrectomy.3 However, our patient demonstrates that metastasis of RCC to skin can be long delayed (>5 years) despite an initial diagnosis of localized disease.
Cutaneous RCC classically presents as a painless firm papulonodule with a deep red or purple color due to its high vascularity.4 Several retrospective studies have identified the scalp as the most frequent site of cutaneous involvement, followed by the chest, abdomen, and nephrectomy scar.3,4 The differential diagnosis includes other vascular lesions such as pyogenic granuloma, hemangioma, angiosarcoma, bacillary angiomatosis, and Kaposi sarcoma. Diagnosis usually is easily confirmed histologically. Proliferative nests of epithelioid cells with clear cell morphology are surrounded by delicately branching vessels referred to as chicken wire-like vasculature. Immunohistochemical studies demonstrate positivity for pan-cytokeratin, vimentin, and CD-10, and negativity for p63 and cytokeratins 5 and 6, helping to confirm the diagnosis in more challenging cases, especially when there is no known history of primary RCC.5
If cutaneous metastasis of RCC is diagnosed, a chest and abdominal computed tomography scan as well as serum alkaline phosphatase test are warranted, as up to 90% of patients with RCC in the skin have additional lesions in at least 1 other site such as the lungs, bones, or liver.3 Management of metastatic RCC includes surgical excision if a single metastasis is found and either immunotherapy with high-dose IL-2 or an anti-programmed cell death inhibitor. Patients with progressive disease also may receive targeted anti-VEGF inhibitors (eg, axitinib, pazopanib, sunitinib), which have been shown to increase progression-free survival in metastatic RCC.6-8 Interestingly, some evidence suggests severely delayed recurrence of RCC (>5 years following nephrectomy) may predict better response to systemic therapy.9
This case of severely delayed metastasis of RCC 8 years after nephrectomy raises the question of whether routine surveillance for RCC recurrence should continue beyond 5 years. It also underscores the need for further studies to determine the utility of postsurgical adjuvant therapy for localized disease (stages I-III). A randomized clinical trial showed no significant difference in disease-free survival when the multikinase inhibitors sunitinib and sorafenib were used as adjuvant therapy.10 The randomized, placebo-controlled PROTECT trial showed no significant difference in disease-free survival between the VEGF inhibitor pazopanib and placebo when used as adjuvant therapy.11 However, trials are ongoing to investigate a potential survival advantage of adjuvant therapy with the VEGF receptor inhibitor axitinib and the mammalian target of rapamycin inhibitor everolimus.
The Diagnosis: Metastatic Renal Cell Carcinoma
Histopathologic examination of the punch biopsy demonstrated epithelioid cells with abundant clear cytoplasm and numerous chicken wire-like vascular channels consistent with a diagnosis of cutaneous metastasis of renal cell carcinoma (RCC)(Figure). Collateral history revealed that 8 years prior, the patient had been diagnosed with clear cell RCC, stage III (T3aN0M0). At that time, he was treated with radical nephrectomy, which was considered curative. He remained disease free until several months prior to the development of the cutaneous lesion when he was found to have pulmonary and cerebral metastases with biopsies showing metastatic RCC. He was treated with lobectomy and Gamma Knife radiation for the lung and cerebral metastases, respectively. His oncologist planned to initiate therapy with the multikinase inhibitor sunitinib, which inhibits vascular endothelial growth factor (VEGF) signaling. Unfortunately, the patient died prior to treatment due to overwhelming tumor burden.
Clear cell RCC, the most common renal malignancy, presents with metastatic disease at the time of diagnosis in 21% of patients.1 An additional 20% of patients with localized disease develop metastases within several years of receiving a nephrectomy without adjuvant therapy, which is standard treatment for stage I to stage III disease.1,2 Metastatic RCC most frequently targets the lungs, bone, liver, and brain, though virtually any organ can be involved. Cutaneous involvement is estimated to occur in 3.3% of RCC cases,3 accounting for only 1.4% of cutaneous metastases overall.4 The risk for developing cutaneous metastases is greatest within 3 years following nephrectomy.3 However, our patient demonstrates that metastasis of RCC to skin can be long delayed (>5 years) despite an initial diagnosis of localized disease.
Cutaneous RCC classically presents as a painless firm papulonodule with a deep red or purple color due to its high vascularity.4 Several retrospective studies have identified the scalp as the most frequent site of cutaneous involvement, followed by the chest, abdomen, and nephrectomy scar.3,4 The differential diagnosis includes other vascular lesions such as pyogenic granuloma, hemangioma, angiosarcoma, bacillary angiomatosis, and Kaposi sarcoma. Diagnosis usually is easily confirmed histologically. Proliferative nests of epithelioid cells with clear cell morphology are surrounded by delicately branching vessels referred to as chicken wire-like vasculature. Immunohistochemical studies demonstrate positivity for pan-cytokeratin, vimentin, and CD-10, and negativity for p63 and cytokeratins 5 and 6, helping to confirm the diagnosis in more challenging cases, especially when there is no known history of primary RCC.5
If cutaneous metastasis of RCC is diagnosed, a chest and abdominal computed tomography scan as well as serum alkaline phosphatase test are warranted, as up to 90% of patients with RCC in the skin have additional lesions in at least 1 other site such as the lungs, bones, or liver.3 Management of metastatic RCC includes surgical excision if a single metastasis is found and either immunotherapy with high-dose IL-2 or an anti-programmed cell death inhibitor. Patients with progressive disease also may receive targeted anti-VEGF inhibitors (eg, axitinib, pazopanib, sunitinib), which have been shown to increase progression-free survival in metastatic RCC.6-8 Interestingly, some evidence suggests severely delayed recurrence of RCC (>5 years following nephrectomy) may predict better response to systemic therapy.9
This case of severely delayed metastasis of RCC 8 years after nephrectomy raises the question of whether routine surveillance for RCC recurrence should continue beyond 5 years. It also underscores the need for further studies to determine the utility of postsurgical adjuvant therapy for localized disease (stages I-III). A randomized clinical trial showed no significant difference in disease-free survival when the multikinase inhibitors sunitinib and sorafenib were used as adjuvant therapy.10 The randomized, placebo-controlled PROTECT trial showed no significant difference in disease-free survival between the VEGF inhibitor pazopanib and placebo when used as adjuvant therapy.11 However, trials are ongoing to investigate a potential survival advantage of adjuvant therapy with the VEGF receptor inhibitor axitinib and the mammalian target of rapamycin inhibitor everolimus.
- Dabestani S, Thorstenson A, Lindblad P, et al. Renal cell carcinoma recurrences and metastases in primary non-metastatic patients: a population-based study. World J Urol. 2016;34:1081-1086.
- Ljungberg B, Campbell SC, Choi HY, et al. The epidemiology of renal cell carcinoma. Eur Urol. 2011;60:615-621.
- Dorairajan LN, Hemal AK, Aron M, et al. Cutaneous metastases in renal cell carcinoma. Urol Int. 1999;63:164-167.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2, pt 1):228-236.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061-1068.
- Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:3584-3590.
- Rini BI, Grunwald V, Fishman MN, et al. Axitinib for first-line metastatic renal cell carcinoma (mRCC): overall efficacy and pharmacokinetic (PK) analyses from a randomized phase II study. J Clin Oncol. 2012;30(suppl). doi:10.1200/jco.2012.30.15_suppl.4503.
- Ficarra V, Novara G. Characterizing late recurrence of renal cell carcinoma. Nat Rev Urol. 2013;10:687-689.
- Haas NB, Manola J, Uzzo RG, et al. Adjuvant sunitinib or sorafenib for high-risk, non-metastatic renal-cell carcinoma (ECOG-ACRIN E2805): a double-blind, placebo-controlled, randomised, phase 3 trial [published online March 9, 2016]. Lancet. 2016;387:2008-2016.
- Motzer RJ, Haas NB, Donskov F, et al; PROTECT investigators. Randomized phase III trial of adjuvant pazopanib versus placebo after nephrectomy in patients with localized or locally advanced renal cell carcinoma [published online September 13, 2017]. J Clin Oncol. 2017;35:3916-3923.
- Dabestani S, Thorstenson A, Lindblad P, et al. Renal cell carcinoma recurrences and metastases in primary non-metastatic patients: a population-based study. World J Urol. 2016;34:1081-1086.
- Ljungberg B, Campbell SC, Choi HY, et al. The epidemiology of renal cell carcinoma. Eur Urol. 2011;60:615-621.
- Dorairajan LN, Hemal AK, Aron M, et al. Cutaneous metastases in renal cell carcinoma. Urol Int. 1999;63:164-167.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2, pt 1):228-236.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061-1068.
- Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:3584-3590.
- Rini BI, Grunwald V, Fishman MN, et al. Axitinib for first-line metastatic renal cell carcinoma (mRCC): overall efficacy and pharmacokinetic (PK) analyses from a randomized phase II study. J Clin Oncol. 2012;30(suppl). doi:10.1200/jco.2012.30.15_suppl.4503.
- Ficarra V, Novara G. Characterizing late recurrence of renal cell carcinoma. Nat Rev Urol. 2013;10:687-689.
- Haas NB, Manola J, Uzzo RG, et al. Adjuvant sunitinib or sorafenib for high-risk, non-metastatic renal-cell carcinoma (ECOG-ACRIN E2805): a double-blind, placebo-controlled, randomised, phase 3 trial [published online March 9, 2016]. Lancet. 2016;387:2008-2016.
- Motzer RJ, Haas NB, Donskov F, et al; PROTECT investigators. Randomized phase III trial of adjuvant pazopanib versus placebo after nephrectomy in patients with localized or locally advanced renal cell carcinoma [published online September 13, 2017]. J Clin Oncol. 2017;35:3916-3923.
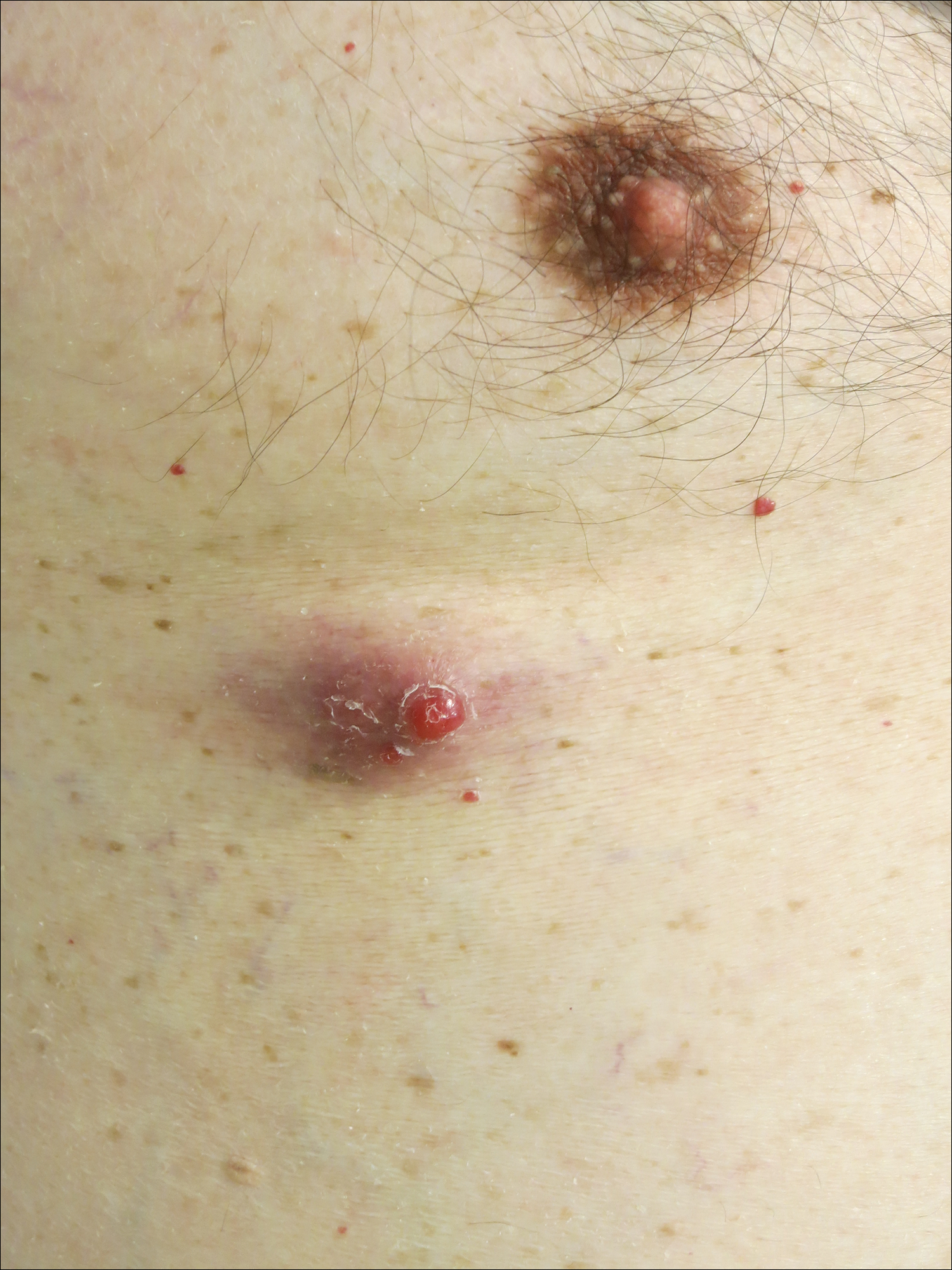
A man in his 60s presented with a subcutaneous nodule on the right side of the chest. Due to impaired mental status, he was unable to describe the precise age of the lesion, but his wife reported it had been present at least several weeks. She recently noted a new, bright red growth on top of the nodule. The lesion was asymptomatic but seemed to be growing in size. Physical examination revealed a 3-cm firm fixed nodule on the right side of the chest with an overlying, exophytic bright red papule. No similar lesions were found elsewhere on physical examination. A punch biopsy of the lesion was performed.
Growing Nodule on the Arm
The Diagnosis: Primary Cutaneous Anaplastic Large Cell Lymphoma
Primary cutaneous CD30+ lymphoproliferative disorders encompass lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma (PCALCL) as well as borderline cases. Primary cutaneous anaplastic large cell lymphoma is a rare disease that is more common in white patients with slight male predominance and median age at diagnosis of 61 years.1 Prognosis is excellent, with a 90% survival rate at 10 years. Although lesions spontaneously regress in 6% to 22% of cases, complete resolution is rare.2 Clinically, the classic presentation is a solitary, rapidly growing, flesh-colored, erythematous nodule or plaque on the arms and legs or trunk, often with ulceration. Proper diagnosis requires clinical, histopathologic, and immunophenotypic correlation.
Histopathologic examination of PCALCL typically reveals large, atypical, Reed-Sternberg-like cells most commonly with anaplastic cytomorphology, but pleomorphic or immunoblastic morphology is not uncommon. Cells are in sheets or nodules, diffusely occupying the dermis and often the subcutaneous fat, with more than 75% of large cells expressing CD30.3 In addition to CD30 positivity, immunophenotype is classically CD4+, cutaneous lymphocyte-associated antigen positive, epithelial membrane antigen negative, and anaplastic lymphoma kinase negative; CD2, CD5, and CD3 expression is variable. Interestingly, in our case, there was a minor population of CD8+ cells. CD8 expression is seen in less than 5% of PCALCL cases; this phenotype is associated with an indolent disease with favorable prognosis.3 Of note, anaplastic lymphoma kinase positivity corresponding to a t(2;5) translocation is more suggestive of systemic anaplastic large cell lymphoma with secondary skin involvement and more commonly is seen in children. For reasons possibly related to mediators such as epidermal growth factor or transforming growth factor α from CD30+ cells, epidermal hyperplasia can be seen in PCALCL.4 The subsequent hyperkeratosis, crusting, and ulceration can be difficult to distinguish from lesions such as pyoderma gangrenosum, squamous cell carcinoma, arthropod bite, leukemia cutis, Merkel cell carcinoma (MCC), and metastatic breast cancer.
Skin involvement with leukemia is rare but most commonly is seen in acute myelogenous leukemia, specifically more mature forms such as acute myelomonocytic leukemia and acute monocytic leukemia. Approximately 10% to 20% of acute myelomonocytic leukemia cases have cutaneous involvement.5 Although there is a variety of potential skin lesions, the most common is a red-purple papule or nodule, sometimes with hemorrhage or ulceration, on the head, neck, and trunk. Leukemic infiltrates may arise from sites of prior trauma. Histopathology depends on the type of leukemia; however, general features include a normal epidermis without epidermotropism and perivascular, nodular, or diffuse infiltrate of neoplastic cells in the dermis, often with a Grenz zone (Figure 1). Compared to PCALCL, leukemia cutis shows sparing of the papillary dermis (Grenz zone), and the cells have more cytoplasm and show a different immunophenotype. The cells often are fragile and show crush artifact. Acute myelogenous leukemia often will show cytoplasmic granules; however, immature precursor cells may not have granules. The myeloid cells will stain with myeloperoxidase and chloroacetate. Positivity is seen for CD13, CD33, and CD68. Clinical correlation is important because other diseases with nodular or diffuse infiltrates of small cell infiltrates, such as extramedullary hematopoiesis and lymphoma, appear similar. Acute myelogenous leukemia is associated with neutrophilic dermatoses such as Sweet syndrome and pyoderma gangrenosum. Cutaneous eruption resolves with successful treatment of the leukemia.
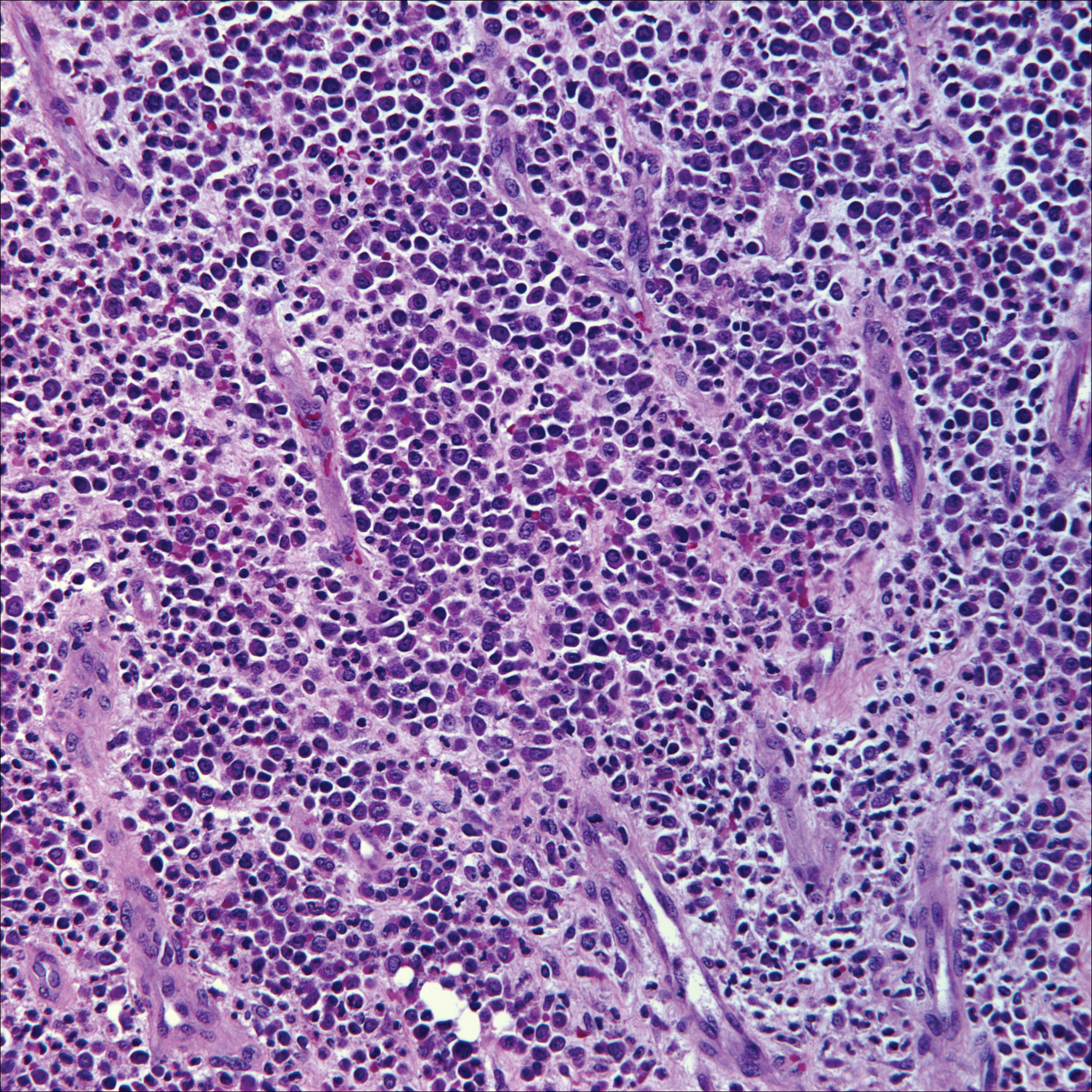
Breast cancer is the most common cancer to metastasize to the skin in women, accounting for 73% of cutaneous metastases, followed by melanoma, which is responsible for 11%.5 The classic presentation is an erythematous patch with spreading borders or a nodule on the trunk. Many cases of metastatic breast cancer with skin involvement may represent direct extension of the cancer into the skin. General histologic clues to cutaneous metastasis include well-circumscribed dermal or subcutaneous nodules of atypical cells with an increase in mitotic activity without connection to the epidermis. Tumor cells may show diffuse, nodular, or single file pattern and may exhibit areas of necrosis. Ductal carcinoma additionally may show ductal or glandular differentiation with surrounding desmoplasia (Figure 2). Immunohistochemistry typically is positive for cytokeratin (CK) 7, estrogen receptor/progesterone receptor, mammaglobin, and gross cystic disease fluid protein-15, and negative for CK20, CK5/6, and thyroid transcription factor-1.
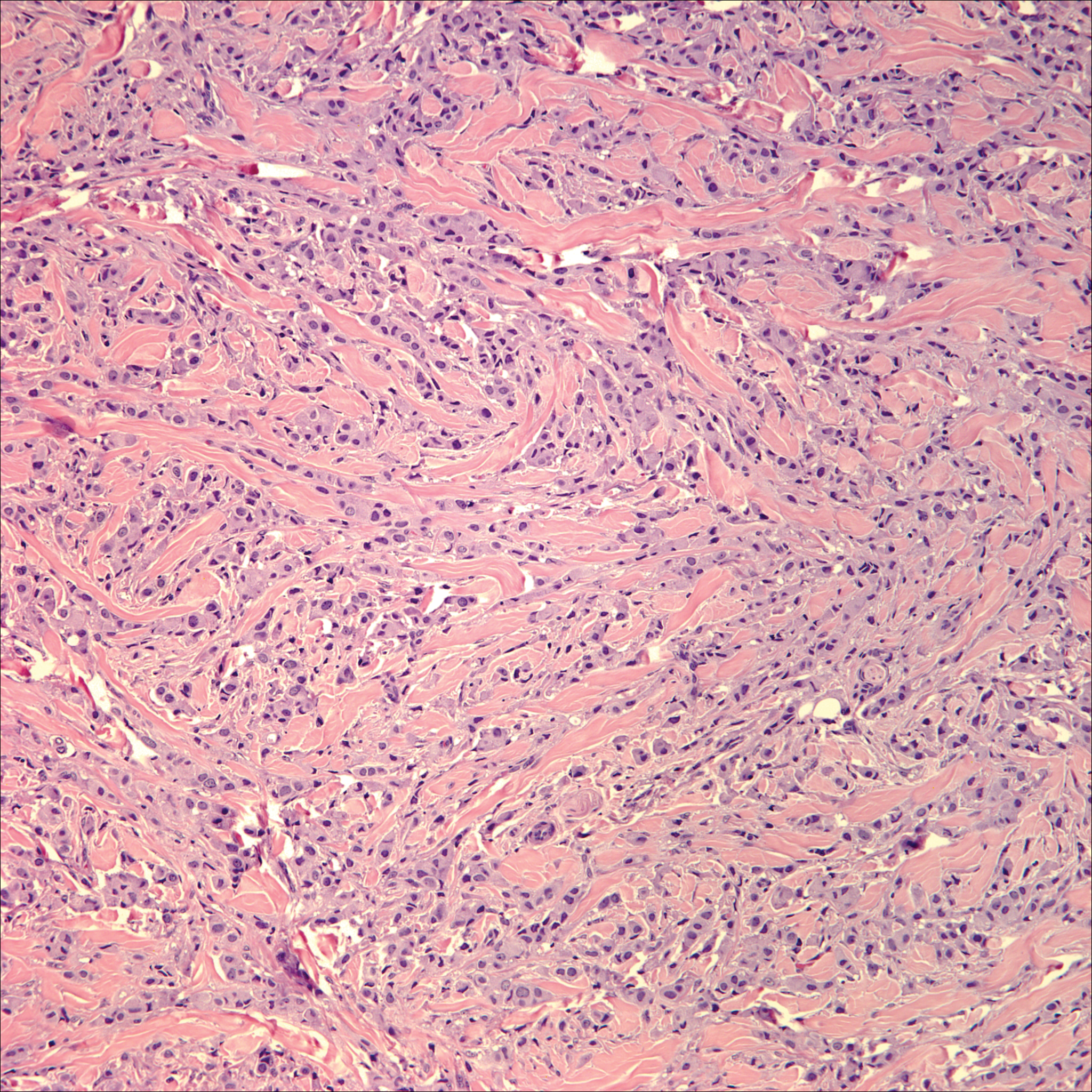
Papulovesicular and nodular lesions appearing as an arthropod bite have been noted in hematologic malignancies, underscoring the importance of histopathology and clinical correlation. Arthropod bites commonly present as red papules, nodules, vesicles, or pustules at the site of the bite. Pseudolymphomatous nodules occasionally develop. Excoriations and further progression to persistent prurigo also may occur. Histopathology shows variable epidermal features including spongiosis, acanthosis, parakeratosis, dermal edema, and superficial and deep perivascular neutrophils (Figure 3). Additionally, lymphocytes sometimes with CD30 positivity may be seen. The presence of eosinophils in interstitial areas, especially in the deep dermis, is a useful clue.
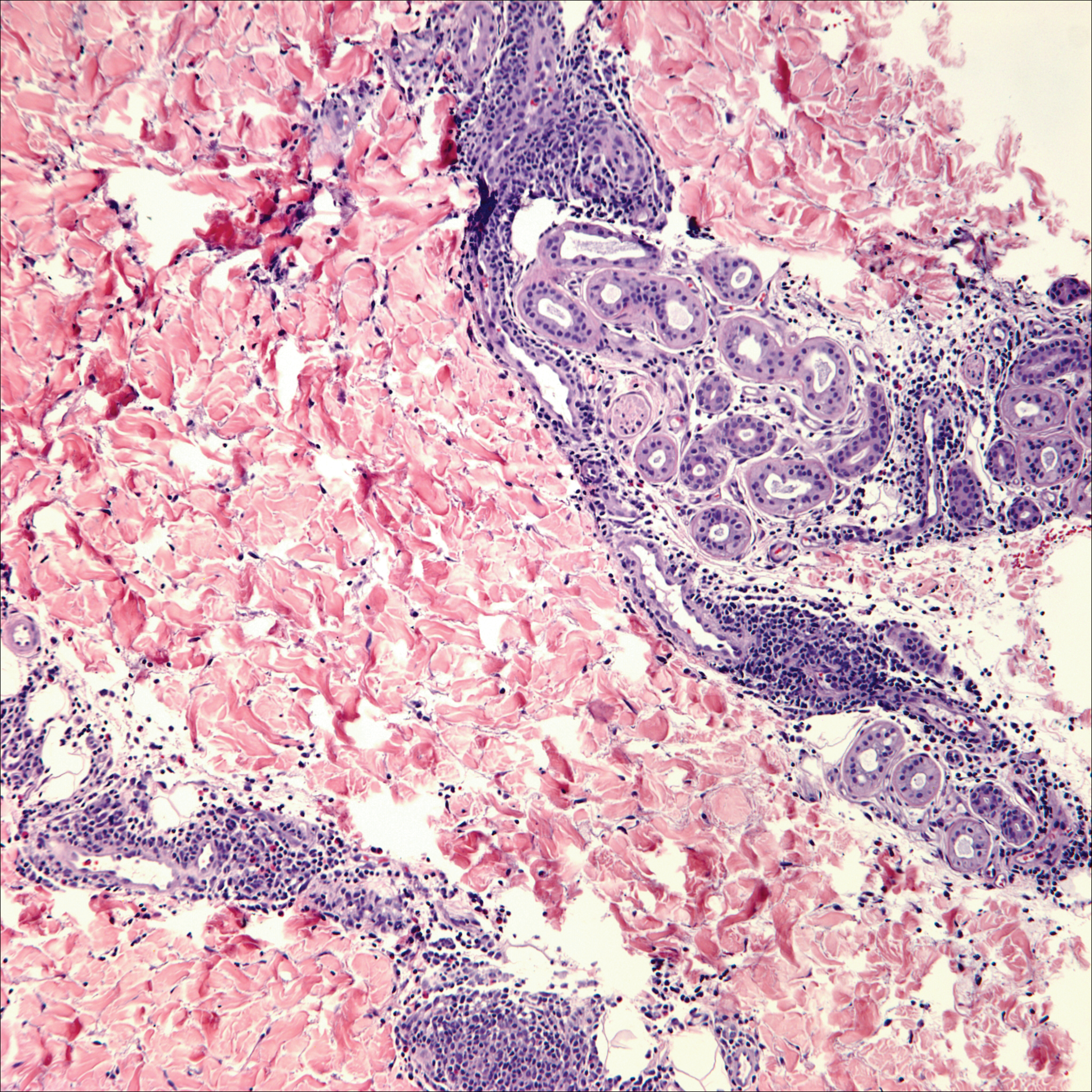
Lack of staining for epithelial and neuroendocrine markers differentiates PCALCL from MCC; specifically CK20, an epithelial marker positive in more than 90% of MCC cases, excludes lymphoma.6 Merkel cell carcinoma presents as a solitary, quickly growing, red and often ulcerated nodule or plaque on the head, neck, or legs of elderly patients. The lesions often are in areas of sun damage. Histopathology classically shows a diffuse dermal infiltrate of monotonous round blue cells with a scant cytoplasmic rim and multiple inconspicuous nucleoli in nests, rosettes, or strands in the dermis. There are frequent mitotic figures. The cells are uniform and 2 to 3 times larger than mature lymphocytes. Single-cell necrosis and crush artifact is common. Epidermotropism or coexisting Bowenoid change also may be observed (Figure 4). The term primary neuroendocrine carcinoma of the skin is preferred over Merkel cell carcinoma because the tumor cells share similar morphology to the specialized touch receptor of the basal layer (Merkel cell), but no direct histogenetic relationship has been established.7,8
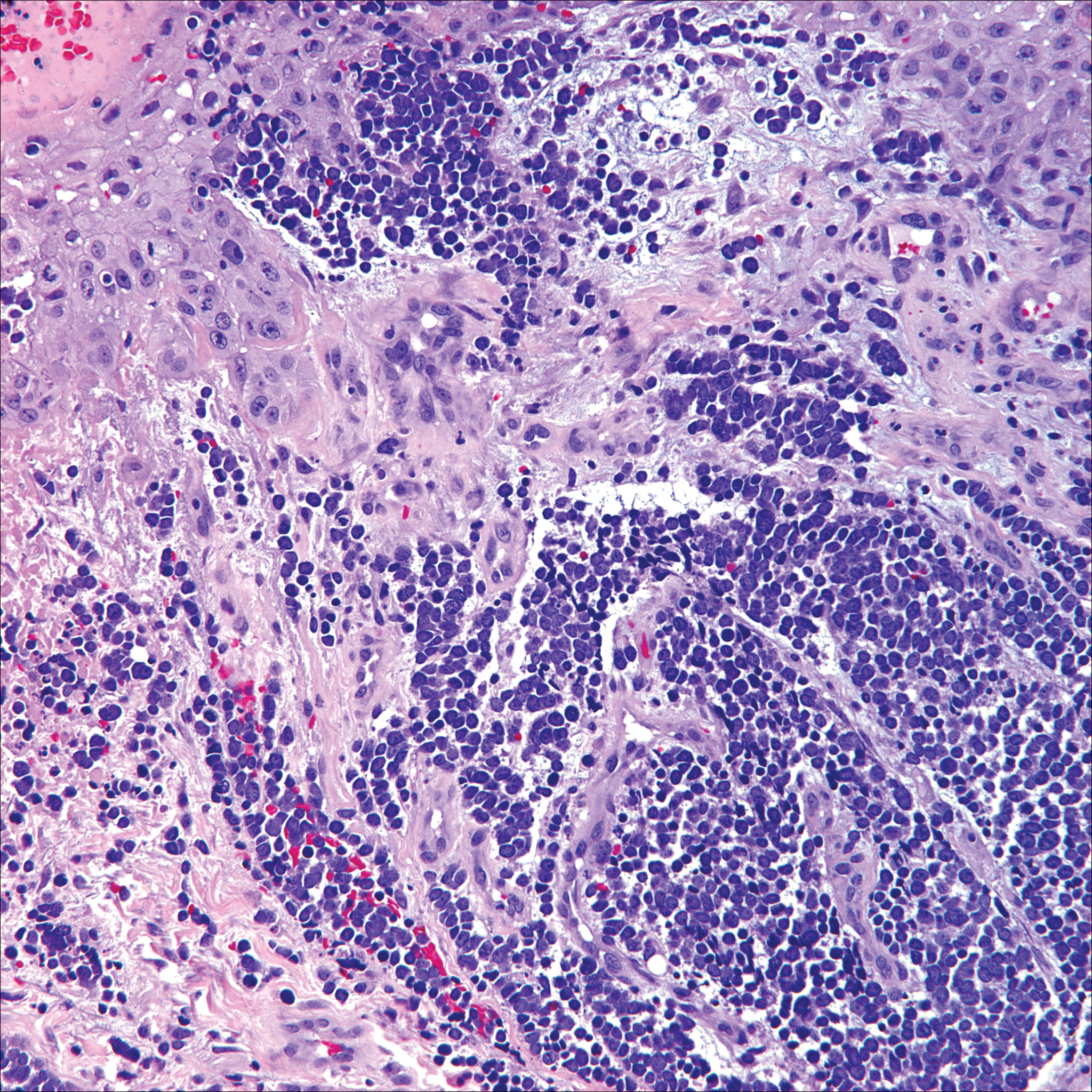
Immunohistochemistry is key to diagnosis because MCC stains for both epithelial and neuroendocrine markers. Positivity is seen for neuron-specific enolase, epithelial membrane antigen, neurofilament, synaptophysin, and chromogranin. Because the histology of MCC may resemble small cell carcinoma of the lung, staining for low-molecular-weight keratin such as CK20 and CK7 help to distinguish MCC. Merkel cell carcinoma typically is CK20+ and CK7-, while small cell carcinoma of the lung is the opposite.9 The tumor grows aggressively and metastasis is common, thus surgery is the primary approach, but adjuvant chemotherapy and radiation often are given in addition.
- Yu J, Blitzblau R, Decker R, et al. Analysis of primary CD30+ cutaneous lymphoproliferative disease and survival from the Surveillance, Epidemiology, and End Results database. J Clin Oncol. 2008;26:1483-1488.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Nasit JG, Patel SC. Primary cutaneous CD8(+) CD30(+) anaplastic large cell lymphoma: an unusual case with a high Ki-67 index--a short review. Indian J Dermatol. 2015;60:373-377.
- Park J, Lee J, Lim Y, et al. Synchronous occurrence of primary cutaneous anaplastic large cell lymphoma and squamous cell carcinoma. Ann Dermatol. 2016;28:491-494.
- Marks JG Jr, Miller JJ. Lookingbill and Marks' Principles of Dermatology. 5th ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Kudchadkar R, Gonzalez R, Lewis K, et al. A case of Merkel cell carcinoma. Oncology. 2008;22:322-328.
- Ratner D, Nelson BR, Brown MD, et al. Merkel cell carcinoma. J Am Acad Dermatol. 1993;29:143-156.
- Zur Hausen A, Rennspiess D, Winnepenninckx V, et al. Early B-cell differentiation in Merkel cell carcinomas: clues to cellular ancestry [published online April 10, 2013]. Cancer Res. 2013;73:4982-4987.
- Sidiropoulos M, Hanna W, Raphael SJ, et al. Expression of TdT in Merkel cell carcinoma and small cell lung carcinoma. Am J Clin Pathol. 2011;135:831-838.
The Diagnosis: Primary Cutaneous Anaplastic Large Cell Lymphoma
Primary cutaneous CD30+ lymphoproliferative disorders encompass lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma (PCALCL) as well as borderline cases. Primary cutaneous anaplastic large cell lymphoma is a rare disease that is more common in white patients with slight male predominance and median age at diagnosis of 61 years.1 Prognosis is excellent, with a 90% survival rate at 10 years. Although lesions spontaneously regress in 6% to 22% of cases, complete resolution is rare.2 Clinically, the classic presentation is a solitary, rapidly growing, flesh-colored, erythematous nodule or plaque on the arms and legs or trunk, often with ulceration. Proper diagnosis requires clinical, histopathologic, and immunophenotypic correlation.
Histopathologic examination of PCALCL typically reveals large, atypical, Reed-Sternberg-like cells most commonly with anaplastic cytomorphology, but pleomorphic or immunoblastic morphology is not uncommon. Cells are in sheets or nodules, diffusely occupying the dermis and often the subcutaneous fat, with more than 75% of large cells expressing CD30.3 In addition to CD30 positivity, immunophenotype is classically CD4+, cutaneous lymphocyte-associated antigen positive, epithelial membrane antigen negative, and anaplastic lymphoma kinase negative; CD2, CD5, and CD3 expression is variable. Interestingly, in our case, there was a minor population of CD8+ cells. CD8 expression is seen in less than 5% of PCALCL cases; this phenotype is associated with an indolent disease with favorable prognosis.3 Of note, anaplastic lymphoma kinase positivity corresponding to a t(2;5) translocation is more suggestive of systemic anaplastic large cell lymphoma with secondary skin involvement and more commonly is seen in children. For reasons possibly related to mediators such as epidermal growth factor or transforming growth factor α from CD30+ cells, epidermal hyperplasia can be seen in PCALCL.4 The subsequent hyperkeratosis, crusting, and ulceration can be difficult to distinguish from lesions such as pyoderma gangrenosum, squamous cell carcinoma, arthropod bite, leukemia cutis, Merkel cell carcinoma (MCC), and metastatic breast cancer.
Skin involvement with leukemia is rare but most commonly is seen in acute myelogenous leukemia, specifically more mature forms such as acute myelomonocytic leukemia and acute monocytic leukemia. Approximately 10% to 20% of acute myelomonocytic leukemia cases have cutaneous involvement.5 Although there is a variety of potential skin lesions, the most common is a red-purple papule or nodule, sometimes with hemorrhage or ulceration, on the head, neck, and trunk. Leukemic infiltrates may arise from sites of prior trauma. Histopathology depends on the type of leukemia; however, general features include a normal epidermis without epidermotropism and perivascular, nodular, or diffuse infiltrate of neoplastic cells in the dermis, often with a Grenz zone (Figure 1). Compared to PCALCL, leukemia cutis shows sparing of the papillary dermis (Grenz zone), and the cells have more cytoplasm and show a different immunophenotype. The cells often are fragile and show crush artifact. Acute myelogenous leukemia often will show cytoplasmic granules; however, immature precursor cells may not have granules. The myeloid cells will stain with myeloperoxidase and chloroacetate. Positivity is seen for CD13, CD33, and CD68. Clinical correlation is important because other diseases with nodular or diffuse infiltrates of small cell infiltrates, such as extramedullary hematopoiesis and lymphoma, appear similar. Acute myelogenous leukemia is associated with neutrophilic dermatoses such as Sweet syndrome and pyoderma gangrenosum. Cutaneous eruption resolves with successful treatment of the leukemia.
Breast cancer is the most common cancer to metastasize to the skin in women, accounting for 73% of cutaneous metastases, followed by melanoma, which is responsible for 11%.5 The classic presentation is an erythematous patch with spreading borders or a nodule on the trunk. Many cases of metastatic breast cancer with skin involvement may represent direct extension of the cancer into the skin. General histologic clues to cutaneous metastasis include well-circumscribed dermal or subcutaneous nodules of atypical cells with an increase in mitotic activity without connection to the epidermis. Tumor cells may show diffuse, nodular, or single file pattern and may exhibit areas of necrosis. Ductal carcinoma additionally may show ductal or glandular differentiation with surrounding desmoplasia (Figure 2). Immunohistochemistry typically is positive for cytokeratin (CK) 7, estrogen receptor/progesterone receptor, mammaglobin, and gross cystic disease fluid protein-15, and negative for CK20, CK5/6, and thyroid transcription factor-1.
Papulovesicular and nodular lesions appearing as an arthropod bite have been noted in hematologic malignancies, underscoring the importance of histopathology and clinical correlation. Arthropod bites commonly present as red papules, nodules, vesicles, or pustules at the site of the bite. Pseudolymphomatous nodules occasionally develop. Excoriations and further progression to persistent prurigo also may occur. Histopathology shows variable epidermal features including spongiosis, acanthosis, parakeratosis, dermal edema, and superficial and deep perivascular neutrophils (Figure 3). Additionally, lymphocytes sometimes with CD30 positivity may be seen. The presence of eosinophils in interstitial areas, especially in the deep dermis, is a useful clue.
Lack of staining for epithelial and neuroendocrine markers differentiates PCALCL from MCC; specifically CK20, an epithelial marker positive in more than 90% of MCC cases, excludes lymphoma.6 Merkel cell carcinoma presents as a solitary, quickly growing, red and often ulcerated nodule or plaque on the head, neck, or legs of elderly patients. The lesions often are in areas of sun damage. Histopathology classically shows a diffuse dermal infiltrate of monotonous round blue cells with a scant cytoplasmic rim and multiple inconspicuous nucleoli in nests, rosettes, or strands in the dermis. There are frequent mitotic figures. The cells are uniform and 2 to 3 times larger than mature lymphocytes. Single-cell necrosis and crush artifact is common. Epidermotropism or coexisting Bowenoid change also may be observed (Figure 4). The term primary neuroendocrine carcinoma of the skin is preferred over Merkel cell carcinoma because the tumor cells share similar morphology to the specialized touch receptor of the basal layer (Merkel cell), but no direct histogenetic relationship has been established.7,8
Immunohistochemistry is key to diagnosis because MCC stains for both epithelial and neuroendocrine markers. Positivity is seen for neuron-specific enolase, epithelial membrane antigen, neurofilament, synaptophysin, and chromogranin. Because the histology of MCC may resemble small cell carcinoma of the lung, staining for low-molecular-weight keratin such as CK20 and CK7 help to distinguish MCC. Merkel cell carcinoma typically is CK20+ and CK7-, while small cell carcinoma of the lung is the opposite.9 The tumor grows aggressively and metastasis is common, thus surgery is the primary approach, but adjuvant chemotherapy and radiation often are given in addition.
The Diagnosis: Primary Cutaneous Anaplastic Large Cell Lymphoma
Primary cutaneous CD30+ lymphoproliferative disorders encompass lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma (PCALCL) as well as borderline cases. Primary cutaneous anaplastic large cell lymphoma is a rare disease that is more common in white patients with slight male predominance and median age at diagnosis of 61 years.1 Prognosis is excellent, with a 90% survival rate at 10 years. Although lesions spontaneously regress in 6% to 22% of cases, complete resolution is rare.2 Clinically, the classic presentation is a solitary, rapidly growing, flesh-colored, erythematous nodule or plaque on the arms and legs or trunk, often with ulceration. Proper diagnosis requires clinical, histopathologic, and immunophenotypic correlation.
Histopathologic examination of PCALCL typically reveals large, atypical, Reed-Sternberg-like cells most commonly with anaplastic cytomorphology, but pleomorphic or immunoblastic morphology is not uncommon. Cells are in sheets or nodules, diffusely occupying the dermis and often the subcutaneous fat, with more than 75% of large cells expressing CD30.3 In addition to CD30 positivity, immunophenotype is classically CD4+, cutaneous lymphocyte-associated antigen positive, epithelial membrane antigen negative, and anaplastic lymphoma kinase negative; CD2, CD5, and CD3 expression is variable. Interestingly, in our case, there was a minor population of CD8+ cells. CD8 expression is seen in less than 5% of PCALCL cases; this phenotype is associated with an indolent disease with favorable prognosis.3 Of note, anaplastic lymphoma kinase positivity corresponding to a t(2;5) translocation is more suggestive of systemic anaplastic large cell lymphoma with secondary skin involvement and more commonly is seen in children. For reasons possibly related to mediators such as epidermal growth factor or transforming growth factor α from CD30+ cells, epidermal hyperplasia can be seen in PCALCL.4 The subsequent hyperkeratosis, crusting, and ulceration can be difficult to distinguish from lesions such as pyoderma gangrenosum, squamous cell carcinoma, arthropod bite, leukemia cutis, Merkel cell carcinoma (MCC), and metastatic breast cancer.
Skin involvement with leukemia is rare but most commonly is seen in acute myelogenous leukemia, specifically more mature forms such as acute myelomonocytic leukemia and acute monocytic leukemia. Approximately 10% to 20% of acute myelomonocytic leukemia cases have cutaneous involvement.5 Although there is a variety of potential skin lesions, the most common is a red-purple papule or nodule, sometimes with hemorrhage or ulceration, on the head, neck, and trunk. Leukemic infiltrates may arise from sites of prior trauma. Histopathology depends on the type of leukemia; however, general features include a normal epidermis without epidermotropism and perivascular, nodular, or diffuse infiltrate of neoplastic cells in the dermis, often with a Grenz zone (Figure 1). Compared to PCALCL, leukemia cutis shows sparing of the papillary dermis (Grenz zone), and the cells have more cytoplasm and show a different immunophenotype. The cells often are fragile and show crush artifact. Acute myelogenous leukemia often will show cytoplasmic granules; however, immature precursor cells may not have granules. The myeloid cells will stain with myeloperoxidase and chloroacetate. Positivity is seen for CD13, CD33, and CD68. Clinical correlation is important because other diseases with nodular or diffuse infiltrates of small cell infiltrates, such as extramedullary hematopoiesis and lymphoma, appear similar. Acute myelogenous leukemia is associated with neutrophilic dermatoses such as Sweet syndrome and pyoderma gangrenosum. Cutaneous eruption resolves with successful treatment of the leukemia.
Breast cancer is the most common cancer to metastasize to the skin in women, accounting for 73% of cutaneous metastases, followed by melanoma, which is responsible for 11%.5 The classic presentation is an erythematous patch with spreading borders or a nodule on the trunk. Many cases of metastatic breast cancer with skin involvement may represent direct extension of the cancer into the skin. General histologic clues to cutaneous metastasis include well-circumscribed dermal or subcutaneous nodules of atypical cells with an increase in mitotic activity without connection to the epidermis. Tumor cells may show diffuse, nodular, or single file pattern and may exhibit areas of necrosis. Ductal carcinoma additionally may show ductal or glandular differentiation with surrounding desmoplasia (Figure 2). Immunohistochemistry typically is positive for cytokeratin (CK) 7, estrogen receptor/progesterone receptor, mammaglobin, and gross cystic disease fluid protein-15, and negative for CK20, CK5/6, and thyroid transcription factor-1.
Papulovesicular and nodular lesions appearing as an arthropod bite have been noted in hematologic malignancies, underscoring the importance of histopathology and clinical correlation. Arthropod bites commonly present as red papules, nodules, vesicles, or pustules at the site of the bite. Pseudolymphomatous nodules occasionally develop. Excoriations and further progression to persistent prurigo also may occur. Histopathology shows variable epidermal features including spongiosis, acanthosis, parakeratosis, dermal edema, and superficial and deep perivascular neutrophils (Figure 3). Additionally, lymphocytes sometimes with CD30 positivity may be seen. The presence of eosinophils in interstitial areas, especially in the deep dermis, is a useful clue.
Lack of staining for epithelial and neuroendocrine markers differentiates PCALCL from MCC; specifically CK20, an epithelial marker positive in more than 90% of MCC cases, excludes lymphoma.6 Merkel cell carcinoma presents as a solitary, quickly growing, red and often ulcerated nodule or plaque on the head, neck, or legs of elderly patients. The lesions often are in areas of sun damage. Histopathology classically shows a diffuse dermal infiltrate of monotonous round blue cells with a scant cytoplasmic rim and multiple inconspicuous nucleoli in nests, rosettes, or strands in the dermis. There are frequent mitotic figures. The cells are uniform and 2 to 3 times larger than mature lymphocytes. Single-cell necrosis and crush artifact is common. Epidermotropism or coexisting Bowenoid change also may be observed (Figure 4). The term primary neuroendocrine carcinoma of the skin is preferred over Merkel cell carcinoma because the tumor cells share similar morphology to the specialized touch receptor of the basal layer (Merkel cell), but no direct histogenetic relationship has been established.7,8
Immunohistochemistry is key to diagnosis because MCC stains for both epithelial and neuroendocrine markers. Positivity is seen for neuron-specific enolase, epithelial membrane antigen, neurofilament, synaptophysin, and chromogranin. Because the histology of MCC may resemble small cell carcinoma of the lung, staining for low-molecular-weight keratin such as CK20 and CK7 help to distinguish MCC. Merkel cell carcinoma typically is CK20+ and CK7-, while small cell carcinoma of the lung is the opposite.9 The tumor grows aggressively and metastasis is common, thus surgery is the primary approach, but adjuvant chemotherapy and radiation often are given in addition.
- Yu J, Blitzblau R, Decker R, et al. Analysis of primary CD30+ cutaneous lymphoproliferative disease and survival from the Surveillance, Epidemiology, and End Results database. J Clin Oncol. 2008;26:1483-1488.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Nasit JG, Patel SC. Primary cutaneous CD8(+) CD30(+) anaplastic large cell lymphoma: an unusual case with a high Ki-67 index--a short review. Indian J Dermatol. 2015;60:373-377.
- Park J, Lee J, Lim Y, et al. Synchronous occurrence of primary cutaneous anaplastic large cell lymphoma and squamous cell carcinoma. Ann Dermatol. 2016;28:491-494.
- Marks JG Jr, Miller JJ. Lookingbill and Marks' Principles of Dermatology. 5th ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Kudchadkar R, Gonzalez R, Lewis K, et al. A case of Merkel cell carcinoma. Oncology. 2008;22:322-328.
- Ratner D, Nelson BR, Brown MD, et al. Merkel cell carcinoma. J Am Acad Dermatol. 1993;29:143-156.
- Zur Hausen A, Rennspiess D, Winnepenninckx V, et al. Early B-cell differentiation in Merkel cell carcinomas: clues to cellular ancestry [published online April 10, 2013]. Cancer Res. 2013;73:4982-4987.
- Sidiropoulos M, Hanna W, Raphael SJ, et al. Expression of TdT in Merkel cell carcinoma and small cell lung carcinoma. Am J Clin Pathol. 2011;135:831-838.
- Yu J, Blitzblau R, Decker R, et al. Analysis of primary CD30+ cutaneous lymphoproliferative disease and survival from the Surveillance, Epidemiology, and End Results database. J Clin Oncol. 2008;26:1483-1488.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Nasit JG, Patel SC. Primary cutaneous CD8(+) CD30(+) anaplastic large cell lymphoma: an unusual case with a high Ki-67 index--a short review. Indian J Dermatol. 2015;60:373-377.
- Park J, Lee J, Lim Y, et al. Synchronous occurrence of primary cutaneous anaplastic large cell lymphoma and squamous cell carcinoma. Ann Dermatol. 2016;28:491-494.
- Marks JG Jr, Miller JJ. Lookingbill and Marks' Principles of Dermatology. 5th ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Kudchadkar R, Gonzalez R, Lewis K, et al. A case of Merkel cell carcinoma. Oncology. 2008;22:322-328.
- Ratner D, Nelson BR, Brown MD, et al. Merkel cell carcinoma. J Am Acad Dermatol. 1993;29:143-156.
- Zur Hausen A, Rennspiess D, Winnepenninckx V, et al. Early B-cell differentiation in Merkel cell carcinomas: clues to cellular ancestry [published online April 10, 2013]. Cancer Res. 2013;73:4982-4987.
- Sidiropoulos M, Hanna W, Raphael SJ, et al. Expression of TdT in Merkel cell carcinoma and small cell lung carcinoma. Am J Clin Pathol. 2011;135:831-838.
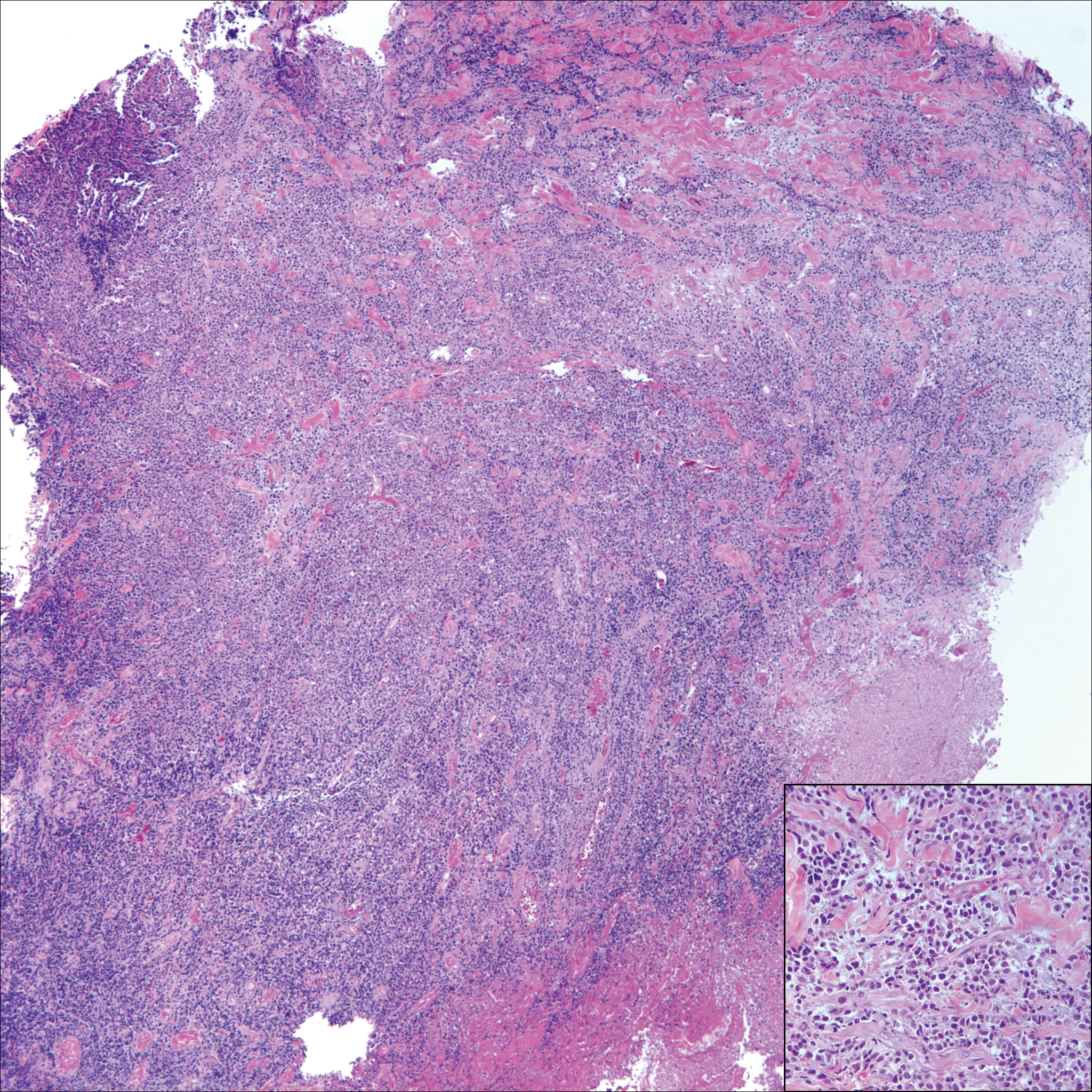
A 65-year-old white woman presented with an asymptomatic bump on the left upper arm of 4 months' duration that arose following a cat scratch. Physical examination was notable for a 35×30-mm, firm, ulcerated, exophytic nodule. Histologic examination demonstrated an ulcerated epidermis and a dense basophilic infiltrate occupying the entire dermis and extending to the subcutaneous tissue. Higher magnification (inset) demonstrated a pleomorphic population of medium- to large-sized discohesive round cells containing variable amounts of slightly eosinophilic cytoplasm, irregular nuclear contours, and prominent nucleoli. Scattered atypical mitotic figures were identified. CD30, CD4, leukocyte common antigen, and Ki-67 immunostains were strongly and diffusely positive. Notable negative stains included anaplastic lymphoma kinase, synaptophysin, epithelial membrane antigen, neuron-specific enolase, CD20, and S-100.