User login
Acute Generalized Exanthematous Pustulosis Caused by Pantoprazole
To the Editor:
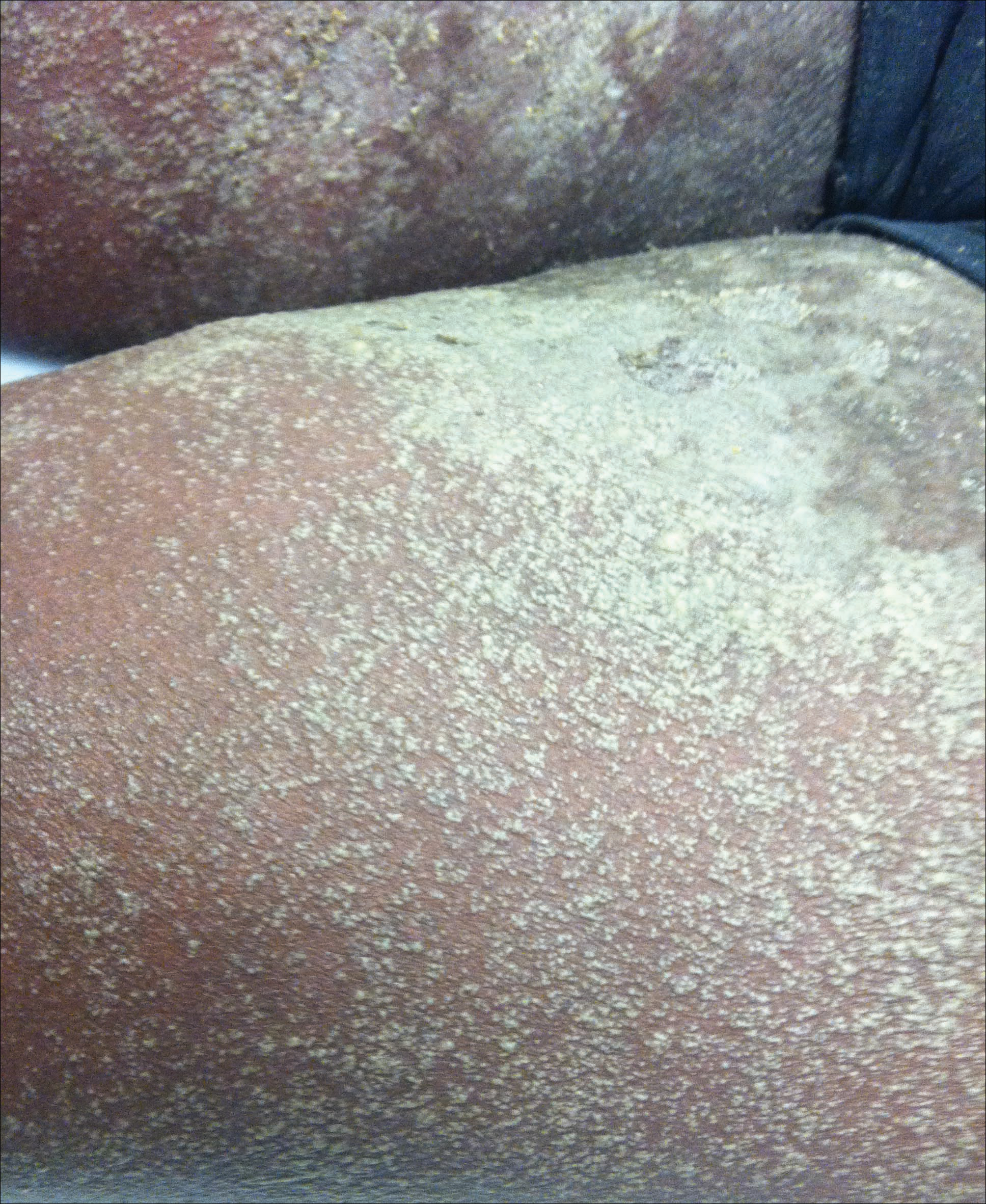
A 34-year-old woman presented with a generalized pustular eruption with subjective fevers, chills, night sweats, and light-headedness. Ten days prior to admission she developed a generalized erythematous and pruritic rash; she had started pantoprazole for reflux 4 days prior to the rash. On admission, skin examination revealed facial edema and diffuse erythema covering 80% of the total body surface area with multiple 1- to 4-mm pustules coalescing into lakes of pus on the trunk as well as bilateral upper and lower arms and legs sparing the palms and soles. Desquamation and serous drainage with crust were observed on the skin of the head, upper trunk, and thighs (Figure 1). Vital signs were notable for hypotension. Laboratory tests on admission were remarkable for leukocytosis (white blood cell count: 22.5×103/μL [reference range, 4.5–11×103/μL]) with absolute eosinophilia but no neutrophilia. C-reactive protein (CRP) was elevated (237.9 mg/L [reference range, 5.0–9.9 mg/L]). Renal and hepatic functions were normal. Blood cultures grew methicillin-sensitive Staphylococcus aureus (MSSA). Further infectious disease workup for viral and fungal pathogens was negative.
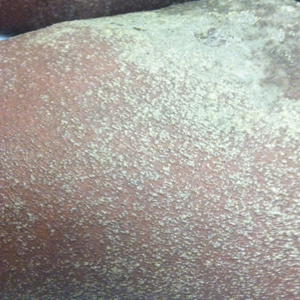
Skin biopsy from the left thigh revealed subcorneal, pustular, acute spongiotic dermatitis with marked intraepidermal spongiosis and papillary edema; exocytosis of eosinophils; and single cell necrosis of keratinocytes (Figure 2). These findings were consistent with acute generalized exanthematous pustulosis (AGEP). Pantoprazole was discontinued, and cardiovascular support and antibiotic therapy for MSSA bacteremia were initiated. Respiratory, kidney, and liver functions remained normal throughout the 11-day hospitalization, and the pustular dermatitis, MSSA bacteremia, and cardiovascular symptoms resolved within 10 days.

Acute generalized exanthematous pustulosis is an uncommon, self-limited, generalized sterile pustular eruption notable for the usual absence of systemic symptoms and extracutaneous organ involvement. Hotz et al1 found that mean peripheral neutrophil counts (mean, 21.5×103/μL) and CRP levels (mean, 241.6 mg/L) were notably elevated in patients with systemic (ie, hepatic, pulmonary, renal, bone marrow) involvement. In our patient, only the CRP approached the elevated value reported by Hotz et al.1 However, the patient exhibited only cardiovascular instability in the context of secondary bacteremia and no other systemic symptoms. The combination of highly elevated neutrophilia and CRP may be a better marker for AGEP-precipitated extracutaneous organ involvement.
Although infectious pathogens such as Epstein-Barr virus and cytomegalovirus have been implicated, the majority of AGEP cases are adverse reactions (ARs) to medications, such as β-lactam antibiotics. In our patient, the widely prescribed proton pump inhibitor (PPI) pantoprazole was the most likely cause. Acute generalized exanthematous pustulosis was reported in a patient taking another PPI, omeprazole.2 However, PPIs are recognized to cause many cutaneous and other organ ARs, though prevalence of ARs is still low. In Thailand, Chularojanamontri et al3 reported 13.8 per 100,000 individuals developed a cutaneous AR to PPIs, and the ARs most frequently were attributed to omeprazole. They found that drug exanthems were the most common cutaneous ARs.3 However, more severe hypersensitivity reactions have been reported, including Stevens-Johnson syndrome, toxic epidermal necrolysis, and autoimmune eruptions such as cutaneous lupus erythematosus.3,4 Other systemic reactions to PPIs include increased risks for urticaria, pneumonia, Clostridium difficile infections, and acute interstitial nephritis.4,5
- Hotz C, Valeyrie-Allanore L, Haddad C, et al. Systemic involvement of acute generalized exanthematous pustulosis: a retrospective study on 58 patients. Br J Dermatol. 2013;169:1223-1232.
- Nantes Castillejo O, Zozaya Urmeneta JM, Valcayo Peñalba A, et al. Acute generalized exanthematous pustulosis induced by omeprazole [in Spanish]. Gastroenterol Hepatol. 2008;31:295-298.
- Chularojanamontri L, Jiamton S, Manapajon A, et al. Cutaneous reactions to proton pump inhibitors: a case-control study. J Drugs Dermatol. 2012;11:E43-E47.
- Chang YS. Hypersensitivity reactions to proton pump inhibitors. Curr Opin Allergy Clin Immunol. 2012;12:348-353.
- Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6:443-551.
To the Editor:
A 34-year-old woman presented with a generalized pustular eruption with subjective fevers, chills, night sweats, and light-headedness. Ten days prior to admission she developed a generalized erythematous and pruritic rash; she had started pantoprazole for reflux 4 days prior to the rash. On admission, skin examination revealed facial edema and diffuse erythema covering 80% of the total body surface area with multiple 1- to 4-mm pustules coalescing into lakes of pus on the trunk as well as bilateral upper and lower arms and legs sparing the palms and soles. Desquamation and serous drainage with crust were observed on the skin of the head, upper trunk, and thighs (Figure 1). Vital signs were notable for hypotension. Laboratory tests on admission were remarkable for leukocytosis (white blood cell count: 22.5×103/μL [reference range, 4.5–11×103/μL]) with absolute eosinophilia but no neutrophilia. C-reactive protein (CRP) was elevated (237.9 mg/L [reference range, 5.0–9.9 mg/L]). Renal and hepatic functions were normal. Blood cultures grew methicillin-sensitive Staphylococcus aureus (MSSA). Further infectious disease workup for viral and fungal pathogens was negative.
Skin biopsy from the left thigh revealed subcorneal, pustular, acute spongiotic dermatitis with marked intraepidermal spongiosis and papillary edema; exocytosis of eosinophils; and single cell necrosis of keratinocytes (Figure 2). These findings were consistent with acute generalized exanthematous pustulosis (AGEP). Pantoprazole was discontinued, and cardiovascular support and antibiotic therapy for MSSA bacteremia were initiated. Respiratory, kidney, and liver functions remained normal throughout the 11-day hospitalization, and the pustular dermatitis, MSSA bacteremia, and cardiovascular symptoms resolved within 10 days.
Acute generalized exanthematous pustulosis is an uncommon, self-limited, generalized sterile pustular eruption notable for the usual absence of systemic symptoms and extracutaneous organ involvement. Hotz et al1 found that mean peripheral neutrophil counts (mean, 21.5×103/μL) and CRP levels (mean, 241.6 mg/L) were notably elevated in patients with systemic (ie, hepatic, pulmonary, renal, bone marrow) involvement. In our patient, only the CRP approached the elevated value reported by Hotz et al.1 However, the patient exhibited only cardiovascular instability in the context of secondary bacteremia and no other systemic symptoms. The combination of highly elevated neutrophilia and CRP may be a better marker for AGEP-precipitated extracutaneous organ involvement.
Although infectious pathogens such as Epstein-Barr virus and cytomegalovirus have been implicated, the majority of AGEP cases are adverse reactions (ARs) to medications, such as β-lactam antibiotics. In our patient, the widely prescribed proton pump inhibitor (PPI) pantoprazole was the most likely cause. Acute generalized exanthematous pustulosis was reported in a patient taking another PPI, omeprazole.2 However, PPIs are recognized to cause many cutaneous and other organ ARs, though prevalence of ARs is still low. In Thailand, Chularojanamontri et al3 reported 13.8 per 100,000 individuals developed a cutaneous AR to PPIs, and the ARs most frequently were attributed to omeprazole. They found that drug exanthems were the most common cutaneous ARs.3 However, more severe hypersensitivity reactions have been reported, including Stevens-Johnson syndrome, toxic epidermal necrolysis, and autoimmune eruptions such as cutaneous lupus erythematosus.3,4 Other systemic reactions to PPIs include increased risks for urticaria, pneumonia, Clostridium difficile infections, and acute interstitial nephritis.4,5
To the Editor:
A 34-year-old woman presented with a generalized pustular eruption with subjective fevers, chills, night sweats, and light-headedness. Ten days prior to admission she developed a generalized erythematous and pruritic rash; she had started pantoprazole for reflux 4 days prior to the rash. On admission, skin examination revealed facial edema and diffuse erythema covering 80% of the total body surface area with multiple 1- to 4-mm pustules coalescing into lakes of pus on the trunk as well as bilateral upper and lower arms and legs sparing the palms and soles. Desquamation and serous drainage with crust were observed on the skin of the head, upper trunk, and thighs (Figure 1). Vital signs were notable for hypotension. Laboratory tests on admission were remarkable for leukocytosis (white blood cell count: 22.5×103/μL [reference range, 4.5–11×103/μL]) with absolute eosinophilia but no neutrophilia. C-reactive protein (CRP) was elevated (237.9 mg/L [reference range, 5.0–9.9 mg/L]). Renal and hepatic functions were normal. Blood cultures grew methicillin-sensitive Staphylococcus aureus (MSSA). Further infectious disease workup for viral and fungal pathogens was negative.
Skin biopsy from the left thigh revealed subcorneal, pustular, acute spongiotic dermatitis with marked intraepidermal spongiosis and papillary edema; exocytosis of eosinophils; and single cell necrosis of keratinocytes (Figure 2). These findings were consistent with acute generalized exanthematous pustulosis (AGEP). Pantoprazole was discontinued, and cardiovascular support and antibiotic therapy for MSSA bacteremia were initiated. Respiratory, kidney, and liver functions remained normal throughout the 11-day hospitalization, and the pustular dermatitis, MSSA bacteremia, and cardiovascular symptoms resolved within 10 days.
Acute generalized exanthematous pustulosis is an uncommon, self-limited, generalized sterile pustular eruption notable for the usual absence of systemic symptoms and extracutaneous organ involvement. Hotz et al1 found that mean peripheral neutrophil counts (mean, 21.5×103/μL) and CRP levels (mean, 241.6 mg/L) were notably elevated in patients with systemic (ie, hepatic, pulmonary, renal, bone marrow) involvement. In our patient, only the CRP approached the elevated value reported by Hotz et al.1 However, the patient exhibited only cardiovascular instability in the context of secondary bacteremia and no other systemic symptoms. The combination of highly elevated neutrophilia and CRP may be a better marker for AGEP-precipitated extracutaneous organ involvement.
Although infectious pathogens such as Epstein-Barr virus and cytomegalovirus have been implicated, the majority of AGEP cases are adverse reactions (ARs) to medications, such as β-lactam antibiotics. In our patient, the widely prescribed proton pump inhibitor (PPI) pantoprazole was the most likely cause. Acute generalized exanthematous pustulosis was reported in a patient taking another PPI, omeprazole.2 However, PPIs are recognized to cause many cutaneous and other organ ARs, though prevalence of ARs is still low. In Thailand, Chularojanamontri et al3 reported 13.8 per 100,000 individuals developed a cutaneous AR to PPIs, and the ARs most frequently were attributed to omeprazole. They found that drug exanthems were the most common cutaneous ARs.3 However, more severe hypersensitivity reactions have been reported, including Stevens-Johnson syndrome, toxic epidermal necrolysis, and autoimmune eruptions such as cutaneous lupus erythematosus.3,4 Other systemic reactions to PPIs include increased risks for urticaria, pneumonia, Clostridium difficile infections, and acute interstitial nephritis.4,5
- Hotz C, Valeyrie-Allanore L, Haddad C, et al. Systemic involvement of acute generalized exanthematous pustulosis: a retrospective study on 58 patients. Br J Dermatol. 2013;169:1223-1232.
- Nantes Castillejo O, Zozaya Urmeneta JM, Valcayo Peñalba A, et al. Acute generalized exanthematous pustulosis induced by omeprazole [in Spanish]. Gastroenterol Hepatol. 2008;31:295-298.
- Chularojanamontri L, Jiamton S, Manapajon A, et al. Cutaneous reactions to proton pump inhibitors: a case-control study. J Drugs Dermatol. 2012;11:E43-E47.
- Chang YS. Hypersensitivity reactions to proton pump inhibitors. Curr Opin Allergy Clin Immunol. 2012;12:348-353.
- Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6:443-551.
- Hotz C, Valeyrie-Allanore L, Haddad C, et al. Systemic involvement of acute generalized exanthematous pustulosis: a retrospective study on 58 patients. Br J Dermatol. 2013;169:1223-1232.
- Nantes Castillejo O, Zozaya Urmeneta JM, Valcayo Peñalba A, et al. Acute generalized exanthematous pustulosis induced by omeprazole [in Spanish]. Gastroenterol Hepatol. 2008;31:295-298.
- Chularojanamontri L, Jiamton S, Manapajon A, et al. Cutaneous reactions to proton pump inhibitors: a case-control study. J Drugs Dermatol. 2012;11:E43-E47.
- Chang YS. Hypersensitivity reactions to proton pump inhibitors. Curr Opin Allergy Clin Immunol. 2012;12:348-353.
- Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6:443-551.
Interstitial Granulomatous Dermatitis and Palisaded Neutrophilic Granulomatous Dermatitis
To the Editor:
Palisaded neutrophilic granulomatous dermatitis (PNGD) is a rare disorder that often is associated with systemic disease. It has been shown to manifest in the presence of systemic lupus erythematosus; rheumatoid arthritis; Wegener granulomatosis; and other diseases, mainly autoimmune conditions. Interstitial granulomatous dermatitis (IGD) associated with arthritis was first described by Ackerman et al1 in 1993. In 1994, IGD was placed among the spectrum of PNGD by Chu et al.2 The disease entities included in the spectrum of PNGD of the immune complex disease are Churg-Strauss granuloma, cutaneous extravascular necrotizing granuloma, rheumatoid papules, superficial ulcerating rheumatoid necrobiosis, and IGD with arthritis.2 It has been suggested that IGD has a distinct clinical presentation with associated histopathology, while others suggest it still is part of the PNGD spectrum.2,3 We present 2 cases of granulomatous dermatitis and their findings related to IGD and PNGD.
A 58-year-old woman presented with recurrent painful lesions on the trunk, arms, and legs of 2 years’ duration. The lesions spontaneously resolved without scarring or hyperpigmentation but would recur in different areas on the trunk. She was diagnosed with rheumatoid arthritis following a recent autoimmune workup. At presentation, physical examination revealed tender erythematous edematous plaques on the bilateral upper back (Figure 1) and erythematous nodules on the bilateral upper arms. The patient previously had an antinuclear antibody titer of 1:320 with a speckled pattern. A repeat antinuclear antibody titer taken 1 year later was negative. Her rheumatoid factor initially was positive and remained positive upon repeat testing. Punch biopsies were performed for histologic evaluation of the lesions and immunofluorescence. Biopsies examined with hematoxylin and eosin stain revealed perivascular and interstitial mixed (lymphocytic, neutrophilic, eosinophilic) bottom-heavy inflammation with nuclear dust and basophilic degeneration of collagen (Figure 2). Immunofluorescence studies were negative. The patient deferred treatment.

.")
A 74-year-old man presented with a rash on the flank and back with associated pruritus and occasional pain of 2 months’ duration. His primary care physician prescribed a course of cephalexin, but the rash did not improve. Review of systems was positive for intermittent swelling of the hands, feet, and lips, and negative for arthritis. His medical history included 2 episodes of rheumatic fever, one complicated by pneumonia. His medications included finasteride, simvastatin, bisoprolol-hydrochlorothiazide, aspirin, tiotropium, vitamin D, and fish oil. At presentation, physical examination revealed tender violaceous plaques with induration and central clearing distributed on the left side of the back, left side of the flank, and left axilla. The lesion on the axilla measured 30.0×3.5 cm and the lesions on the left side of the back measured 30.0×9.0 cm. The rims of the lesions were elevated and consistent with the rope sign (Figure 3). A punch biopsy of the lesion on the left axilla showed perivascular and interstitial infiltrate of lymphocytes, neutrophils, histiocytes, and eosinophils. There was no evidence of fibrin deposition in the blood vessels. Small areas of necrobiotic collagen surrounded by multinucleated giant cells and lymphocytes were noted (Figure 4). The rash improved spontaneously at the time of suture removal. No treatment was initiated.

.")
Granulomatous dermatitis in the presence of an autoimmune disorder can present as IGD or PNGD. Both forms of granulomatous dermatitis are rare conditions and considered to be part of the same clinicopathological spectrum. These conditions can be difficult to distinguish clinically but are histologically unique.
Interstitial granulomatous dermatitis and PNGD can have a variable clinical expression. Palisaded neutrophilic granulomatous dermatitis generally presents as flesh-colored to erythematous papules or plaques, most commonly located on the upper arms. The lesions may have a central umbilication with perforation and ulceration.4 Interstitial granulomatous dermatitis most commonly presents as erythematous plaques and papules. The lesions are symmetric and asymptomatic. They most commonly appear on the trunk, axillae, buttocks, thighs, and groin. Subcutaneous linear cords (the rope sign) is a characteristic associated with IGD.3,5 However, the rope sign also has been reported in a patient with PNGD with systemic lupus,6 which further demonstrates the overlapping spectrum of clinical expression seen in these 2 forms of granulomatous dermatitis. Therefore, a diagnosis cannot be made by clinical expression alone; histologic findings are needed for confirmation.
When differentiating IGD and PNGD histologically, it is important to keep in mind that these features exist on a spectrum and depend on the age of the lesion. Deposition of the immune complex around the dermal blood vessel initiates the pathogenesis. Early lesions of PNGD show a neutrophilic infiltrate, focal leukocytoclastic vasculitis, and dense nuclear dust. Developed lesions show zones of basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris.2 The histologic pattern of IGD features smaller areas of palisading histiocytes surrounding foci of degenerated collagen. Neutrophils and eosinophils are seen among the degenerated collagen. There is no evidence of vasculitis and dermal mucin usually is absent.7
Palisaded neutrophilic granulomatous dermatitis has been reported to improve with systemic steroids and dapsone.8 Th
Some authors have disputed the spectrum that Chu et al2 had determined in their study and proposed IGD is a separate entity from the PNGD spectrum. Verneuil et al9 stated that the clinical presentations in Chu et al’s2 study (symmetric papules of the extremities) had not been reported in a patient with IGD. However, in a study of IGD by Peroni et al,3 7 of 12 patients presented with symmetrical papules of the extremities. We believe that the spectrum proposed by Chu et al2 still holds true.
These 2 reports demonstrate the diverse presentation of IGD and PNGD. It is important for dermatologists to keep in mind the PNGD spectrum when a patient presents with granulomatous dermatitis in the presence of an autoimmune disorder.
- Ackerman AB, Guo Y, Vitale P. Clues to diagnosis in dermatopathology. Am Society Clin Pathol. 1993;3:309-312.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Hantash BM, Chiang D, Kohler S, et al. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661-664.
- Garcia-Rabasco A, Esteve-Martinez A, Zaragoza-Ninet V, et al. Interstitial granulomatous dermatitis in a patient with lupus erythematosus. Am J Dermatopathol. 2011;33:871-872.
- Gulati A, Paige D, Yaqoob M, et al. Palisaded neutrophilic granulomatous dermatitis associated with systemic lupus erythematosus presenting with the burning rope sign. J Am Acad Dermatol. 2009;61:711-714.
- Tomasini C, Pippione M. Interstitial granulomatous dermatitis with plaques. J Am Acad Dermatol. 2002;46:892-899.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad Dermatol. 2011;65:E92-E93.
- Verneuil L, Dompmartin A, Comoz F, et al. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: a disorder associated with autoantibodies. J Am Acad Dermatol. 2001;45:286-291.
To the Editor:
Palisaded neutrophilic granulomatous dermatitis (PNGD) is a rare disorder that often is associated with systemic disease. It has been shown to manifest in the presence of systemic lupus erythematosus; rheumatoid arthritis; Wegener granulomatosis; and other diseases, mainly autoimmune conditions. Interstitial granulomatous dermatitis (IGD) associated with arthritis was first described by Ackerman et al1 in 1993. In 1994, IGD was placed among the spectrum of PNGD by Chu et al.2 The disease entities included in the spectrum of PNGD of the immune complex disease are Churg-Strauss granuloma, cutaneous extravascular necrotizing granuloma, rheumatoid papules, superficial ulcerating rheumatoid necrobiosis, and IGD with arthritis.2 It has been suggested that IGD has a distinct clinical presentation with associated histopathology, while others suggest it still is part of the PNGD spectrum.2,3 We present 2 cases of granulomatous dermatitis and their findings related to IGD and PNGD.
A 58-year-old woman presented with recurrent painful lesions on the trunk, arms, and legs of 2 years’ duration. The lesions spontaneously resolved without scarring or hyperpigmentation but would recur in different areas on the trunk. She was diagnosed with rheumatoid arthritis following a recent autoimmune workup. At presentation, physical examination revealed tender erythematous edematous plaques on the bilateral upper back (Figure 1) and erythematous nodules on the bilateral upper arms. The patient previously had an antinuclear antibody titer of 1:320 with a speckled pattern. A repeat antinuclear antibody titer taken 1 year later was negative. Her rheumatoid factor initially was positive and remained positive upon repeat testing. Punch biopsies were performed for histologic evaluation of the lesions and immunofluorescence. Biopsies examined with hematoxylin and eosin stain revealed perivascular and interstitial mixed (lymphocytic, neutrophilic, eosinophilic) bottom-heavy inflammation with nuclear dust and basophilic degeneration of collagen (Figure 2). Immunofluorescence studies were negative. The patient deferred treatment.
A 74-year-old man presented with a rash on the flank and back with associated pruritus and occasional pain of 2 months’ duration. His primary care physician prescribed a course of cephalexin, but the rash did not improve. Review of systems was positive for intermittent swelling of the hands, feet, and lips, and negative for arthritis. His medical history included 2 episodes of rheumatic fever, one complicated by pneumonia. His medications included finasteride, simvastatin, bisoprolol-hydrochlorothiazide, aspirin, tiotropium, vitamin D, and fish oil. At presentation, physical examination revealed tender violaceous plaques with induration and central clearing distributed on the left side of the back, left side of the flank, and left axilla. The lesion on the axilla measured 30.0×3.5 cm and the lesions on the left side of the back measured 30.0×9.0 cm. The rims of the lesions were elevated and consistent with the rope sign (Figure 3). A punch biopsy of the lesion on the left axilla showed perivascular and interstitial infiltrate of lymphocytes, neutrophils, histiocytes, and eosinophils. There was no evidence of fibrin deposition in the blood vessels. Small areas of necrobiotic collagen surrounded by multinucleated giant cells and lymphocytes were noted (Figure 4). The rash improved spontaneously at the time of suture removal. No treatment was initiated.
Granulomatous dermatitis in the presence of an autoimmune disorder can present as IGD or PNGD. Both forms of granulomatous dermatitis are rare conditions and considered to be part of the same clinicopathological spectrum. These conditions can be difficult to distinguish clinically but are histologically unique.
Interstitial granulomatous dermatitis and PNGD can have a variable clinical expression. Palisaded neutrophilic granulomatous dermatitis generally presents as flesh-colored to erythematous papules or plaques, most commonly located on the upper arms. The lesions may have a central umbilication with perforation and ulceration.4 Interstitial granulomatous dermatitis most commonly presents as erythematous plaques and papules. The lesions are symmetric and asymptomatic. They most commonly appear on the trunk, axillae, buttocks, thighs, and groin. Subcutaneous linear cords (the rope sign) is a characteristic associated with IGD.3,5 However, the rope sign also has been reported in a patient with PNGD with systemic lupus,6 which further demonstrates the overlapping spectrum of clinical expression seen in these 2 forms of granulomatous dermatitis. Therefore, a diagnosis cannot be made by clinical expression alone; histologic findings are needed for confirmation.
When differentiating IGD and PNGD histologically, it is important to keep in mind that these features exist on a spectrum and depend on the age of the lesion. Deposition of the immune complex around the dermal blood vessel initiates the pathogenesis. Early lesions of PNGD show a neutrophilic infiltrate, focal leukocytoclastic vasculitis, and dense nuclear dust. Developed lesions show zones of basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris.2 The histologic pattern of IGD features smaller areas of palisading histiocytes surrounding foci of degenerated collagen. Neutrophils and eosinophils are seen among the degenerated collagen. There is no evidence of vasculitis and dermal mucin usually is absent.7
Palisaded neutrophilic granulomatous dermatitis has been reported to improve with systemic steroids and dapsone.8 Th
Some authors have disputed the spectrum that Chu et al2 had determined in their study and proposed IGD is a separate entity from the PNGD spectrum. Verneuil et al9 stated that the clinical presentations in Chu et al’s2 study (symmetric papules of the extremities) had not been reported in a patient with IGD. However, in a study of IGD by Peroni et al,3 7 of 12 patients presented with symmetrical papules of the extremities. We believe that the spectrum proposed by Chu et al2 still holds true.
These 2 reports demonstrate the diverse presentation of IGD and PNGD. It is important for dermatologists to keep in mind the PNGD spectrum when a patient presents with granulomatous dermatitis in the presence of an autoimmune disorder.
To the Editor:
Palisaded neutrophilic granulomatous dermatitis (PNGD) is a rare disorder that often is associated with systemic disease. It has been shown to manifest in the presence of systemic lupus erythematosus; rheumatoid arthritis; Wegener granulomatosis; and other diseases, mainly autoimmune conditions. Interstitial granulomatous dermatitis (IGD) associated with arthritis was first described by Ackerman et al1 in 1993. In 1994, IGD was placed among the spectrum of PNGD by Chu et al.2 The disease entities included in the spectrum of PNGD of the immune complex disease are Churg-Strauss granuloma, cutaneous extravascular necrotizing granuloma, rheumatoid papules, superficial ulcerating rheumatoid necrobiosis, and IGD with arthritis.2 It has been suggested that IGD has a distinct clinical presentation with associated histopathology, while others suggest it still is part of the PNGD spectrum.2,3 We present 2 cases of granulomatous dermatitis and their findings related to IGD and PNGD.
A 58-year-old woman presented with recurrent painful lesions on the trunk, arms, and legs of 2 years’ duration. The lesions spontaneously resolved without scarring or hyperpigmentation but would recur in different areas on the trunk. She was diagnosed with rheumatoid arthritis following a recent autoimmune workup. At presentation, physical examination revealed tender erythematous edematous plaques on the bilateral upper back (Figure 1) and erythematous nodules on the bilateral upper arms. The patient previously had an antinuclear antibody titer of 1:320 with a speckled pattern. A repeat antinuclear antibody titer taken 1 year later was negative. Her rheumatoid factor initially was positive and remained positive upon repeat testing. Punch biopsies were performed for histologic evaluation of the lesions and immunofluorescence. Biopsies examined with hematoxylin and eosin stain revealed perivascular and interstitial mixed (lymphocytic, neutrophilic, eosinophilic) bottom-heavy inflammation with nuclear dust and basophilic degeneration of collagen (Figure 2). Immunofluorescence studies were negative. The patient deferred treatment.
A 74-year-old man presented with a rash on the flank and back with associated pruritus and occasional pain of 2 months’ duration. His primary care physician prescribed a course of cephalexin, but the rash did not improve. Review of systems was positive for intermittent swelling of the hands, feet, and lips, and negative for arthritis. His medical history included 2 episodes of rheumatic fever, one complicated by pneumonia. His medications included finasteride, simvastatin, bisoprolol-hydrochlorothiazide, aspirin, tiotropium, vitamin D, and fish oil. At presentation, physical examination revealed tender violaceous plaques with induration and central clearing distributed on the left side of the back, left side of the flank, and left axilla. The lesion on the axilla measured 30.0×3.5 cm and the lesions on the left side of the back measured 30.0×9.0 cm. The rims of the lesions were elevated and consistent with the rope sign (Figure 3). A punch biopsy of the lesion on the left axilla showed perivascular and interstitial infiltrate of lymphocytes, neutrophils, histiocytes, and eosinophils. There was no evidence of fibrin deposition in the blood vessels. Small areas of necrobiotic collagen surrounded by multinucleated giant cells and lymphocytes were noted (Figure 4). The rash improved spontaneously at the time of suture removal. No treatment was initiated.
Granulomatous dermatitis in the presence of an autoimmune disorder can present as IGD or PNGD. Both forms of granulomatous dermatitis are rare conditions and considered to be part of the same clinicopathological spectrum. These conditions can be difficult to distinguish clinically but are histologically unique.
Interstitial granulomatous dermatitis and PNGD can have a variable clinical expression. Palisaded neutrophilic granulomatous dermatitis generally presents as flesh-colored to erythematous papules or plaques, most commonly located on the upper arms. The lesions may have a central umbilication with perforation and ulceration.4 Interstitial granulomatous dermatitis most commonly presents as erythematous plaques and papules. The lesions are symmetric and asymptomatic. They most commonly appear on the trunk, axillae, buttocks, thighs, and groin. Subcutaneous linear cords (the rope sign) is a characteristic associated with IGD.3,5 However, the rope sign also has been reported in a patient with PNGD with systemic lupus,6 which further demonstrates the overlapping spectrum of clinical expression seen in these 2 forms of granulomatous dermatitis. Therefore, a diagnosis cannot be made by clinical expression alone; histologic findings are needed for confirmation.
When differentiating IGD and PNGD histologically, it is important to keep in mind that these features exist on a spectrum and depend on the age of the lesion. Deposition of the immune complex around the dermal blood vessel initiates the pathogenesis. Early lesions of PNGD show a neutrophilic infiltrate, focal leukocytoclastic vasculitis, and dense nuclear dust. Developed lesions show zones of basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris.2 The histologic pattern of IGD features smaller areas of palisading histiocytes surrounding foci of degenerated collagen. Neutrophils and eosinophils are seen among the degenerated collagen. There is no evidence of vasculitis and dermal mucin usually is absent.7
Palisaded neutrophilic granulomatous dermatitis has been reported to improve with systemic steroids and dapsone.8 Th
Some authors have disputed the spectrum that Chu et al2 had determined in their study and proposed IGD is a separate entity from the PNGD spectrum. Verneuil et al9 stated that the clinical presentations in Chu et al’s2 study (symmetric papules of the extremities) had not been reported in a patient with IGD. However, in a study of IGD by Peroni et al,3 7 of 12 patients presented with symmetrical papules of the extremities. We believe that the spectrum proposed by Chu et al2 still holds true.
These 2 reports demonstrate the diverse presentation of IGD and PNGD. It is important for dermatologists to keep in mind the PNGD spectrum when a patient presents with granulomatous dermatitis in the presence of an autoimmune disorder.
- Ackerman AB, Guo Y, Vitale P. Clues to diagnosis in dermatopathology. Am Society Clin Pathol. 1993;3:309-312.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Hantash BM, Chiang D, Kohler S, et al. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661-664.
- Garcia-Rabasco A, Esteve-Martinez A, Zaragoza-Ninet V, et al. Interstitial granulomatous dermatitis in a patient with lupus erythematosus. Am J Dermatopathol. 2011;33:871-872.
- Gulati A, Paige D, Yaqoob M, et al. Palisaded neutrophilic granulomatous dermatitis associated with systemic lupus erythematosus presenting with the burning rope sign. J Am Acad Dermatol. 2009;61:711-714.
- Tomasini C, Pippione M. Interstitial granulomatous dermatitis with plaques. J Am Acad Dermatol. 2002;46:892-899.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad Dermatol. 2011;65:E92-E93.
- Verneuil L, Dompmartin A, Comoz F, et al. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: a disorder associated with autoantibodies. J Am Acad Dermatol. 2001;45:286-291.
- Ackerman AB, Guo Y, Vitale P. Clues to diagnosis in dermatopathology. Am Society Clin Pathol. 1993;3:309-312.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Hantash BM, Chiang D, Kohler S, et al. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661-664.
- Garcia-Rabasco A, Esteve-Martinez A, Zaragoza-Ninet V, et al. Interstitial granulomatous dermatitis in a patient with lupus erythematosus. Am J Dermatopathol. 2011;33:871-872.
- Gulati A, Paige D, Yaqoob M, et al. Palisaded neutrophilic granulomatous dermatitis associated with systemic lupus erythematosus presenting with the burning rope sign. J Am Acad Dermatol. 2009;61:711-714.
- Tomasini C, Pippione M. Interstitial granulomatous dermatitis with plaques. J Am Acad Dermatol. 2002;46:892-899.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad Dermatol. 2011;65:E92-E93.
- Verneuil L, Dompmartin A, Comoz F, et al. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: a disorder associated with autoantibodies. J Am Acad Dermatol. 2001;45:286-291.
Practice Points
- The clinical features of interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis exist on a spectrum, and these is considerable overlap between the features of these 2 clinicopathologic entities.
- Interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis may respond to systemic steroids or treatment of the underlying systemic disease. Some cases spontaneously resolve.
Solitary Angiokeratoma of the Vulva Mimicking Malignant Melanoma
To the Editor:
Angiokeratoma is a benign vascular tumor characterized by several dilated vessels in the superficial dermis accompanied by epidermal hyperplasia and hyperkeratosis.1 Angiokeratoma of the vulva is a rare clinical finding, usually involving multiple lesions as part of the Fordyce type.2 Solitary angiokeratoma occurs predominantly on the lower legs,3 and although other locations have been described, the presence of a solitary angiokeratoma on the vulva is rare.4 We report 2 cases of solitary angiokeratoma on the vulva that was misdiagnosed as malignant melanoma. Both patients were referred to our center for evaluation and excision.
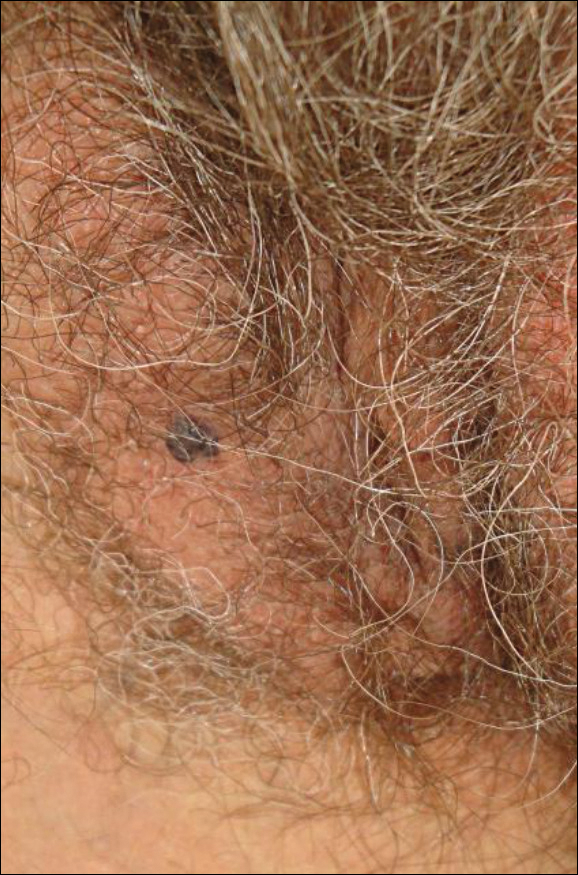
A 65-year-old woman (patient 1) and a 67-year-old woman (patient 2) presented with a bluish black, growing, asymptomatic lesion on the right (Figure 1) and left labia majora, respectively. Both patients were referred by outside physicians for excision because of suspected malignant melanoma. Physical examinations revealed bluish black globular nodules that measured 0.5 and 0.3 cm in diameter, respectively. Dermoscopy (patient 1) revealed dark lacunae. Histopathologic examination of the vulvar lesion (patient 2) showed dilated, blood-filled, vascular spaces in the papillary dermis, accompanied by overlying acanthosis, hyperkeratosis, and papillomatosis that was consistent with angiokeratoma (Figure 2).
Angiokeratoma, particularly the solitary type, often is misdiagnosed. Clinical differential diagnoses may include a wide range of pathologic conditions, including condyloma acuminata, basal cell carcinoma, pyogenic granuloma, lymphangioma, nevi, condyloma lata, nodular prurigo, seborrheic keratosis, granuloma inguinale, and deep fungal infection.2,5 However, due to its quickly growing nature and its dark complexion, malignant melanoma often is initially diagnosed. Because patients affected by angiokeratoma of the vulva usually are aged 20 to 40 years,5 and vulvar melanoma is typical for middle-aged women (median age, 68 years),6 this misdiagnosis is more likely in older patients. It should be noted that a high index of suspicion for melanoma often is present when examining the vulva, considering that this area is difficult to monitor, and there is an especially poor prognosis of vulvar melanoma due to its late detection.6,7
In the past, biopsy was considered mandatory for confirming the diagnosis of vulvar angiokeratoma.5,8,9 However, dermoscopy has emerged as a valuable tool for diagnosis of angiokeratoma10 and also was helpful as a diagnostic aid in one of our patients (patient 1). Therefore, we believe that dermoscopy should be performed prior to a biopsy of angiokeratomas of the vulva.
- Requena L, Sangueza OP. Cutaneous vascular anomalies. part I. hamartomas, malformations, and dilation of preexisting vessels. J Am Acad Dermatol. 1997;37:523-549.
- Schiller PI, Itin PH. Angiokeratomas: an update. Dermatology. 1996;193:275-282.
- Gomi H, Eriyama Y, Horikawa E, et al. Solitary angiokeratoma. J Dermatol. 1988;15:349-350.
- Yamazaki M, Hiruma M, Irie H, et al. Angiokeratoma of the clitoris: a subtype of angiokeratoma vulvae. J Dermatol. 1992;19:553-555.
- Cohen PR, Young AW Jr, Tovell HM. Angiokeratoma of the vulva: diagnosis and review of the literature. Obstet Gynecol Surv. 1989;44:339-346.
- Sugiyama VE, Chan JK, Shin JY, et al. Vulvar melanoma: a multivariable analysis of 644 patients. Obstet Gynecol. 2007;110:296-301.
- De Simone P, Silipo V, Buccini P, et al. Vulvar melanoma: a report of 10 cases and review of the literature. Melanoma Res. 2008;18:127-133.
- Novick NL. Angiokeratoma vulvae. J Am Acad Dermatol. 1985;12:561-563.
- Yigiter M, Arda IS, Tosun E, et al. Angiokeratoma of clitoris: a rare lesion in an adolescent girl. Urology. 2008;71:604-606.
- Zaballos P, Daufi C, Puig S, et al. Dermoscopy of solitary angiokeratomas: a morphological study. Arch Dermatol. 2007;143:318-325.
To the Editor:
Angiokeratoma is a benign vascular tumor characterized by several dilated vessels in the superficial dermis accompanied by epidermal hyperplasia and hyperkeratosis.1 Angiokeratoma of the vulva is a rare clinical finding, usually involving multiple lesions as part of the Fordyce type.2 Solitary angiokeratoma occurs predominantly on the lower legs,3 and although other locations have been described, the presence of a solitary angiokeratoma on the vulva is rare.4 We report 2 cases of solitary angiokeratoma on the vulva that was misdiagnosed as malignant melanoma. Both patients were referred to our center for evaluation and excision.
A 65-year-old woman (patient 1) and a 67-year-old woman (patient 2) presented with a bluish black, growing, asymptomatic lesion on the right (Figure 1) and left labia majora, respectively. Both patients were referred by outside physicians for excision because of suspected malignant melanoma. Physical examinations revealed bluish black globular nodules that measured 0.5 and 0.3 cm in diameter, respectively. Dermoscopy (patient 1) revealed dark lacunae. Histopathologic examination of the vulvar lesion (patient 2) showed dilated, blood-filled, vascular spaces in the papillary dermis, accompanied by overlying acanthosis, hyperkeratosis, and papillomatosis that was consistent with angiokeratoma (Figure 2).
Angiokeratoma, particularly the solitary type, often is misdiagnosed. Clinical differential diagnoses may include a wide range of pathologic conditions, including condyloma acuminata, basal cell carcinoma, pyogenic granuloma, lymphangioma, nevi, condyloma lata, nodular prurigo, seborrheic keratosis, granuloma inguinale, and deep fungal infection.2,5 However, due to its quickly growing nature and its dark complexion, malignant melanoma often is initially diagnosed. Because patients affected by angiokeratoma of the vulva usually are aged 20 to 40 years,5 and vulvar melanoma is typical for middle-aged women (median age, 68 years),6 this misdiagnosis is more likely in older patients. It should be noted that a high index of suspicion for melanoma often is present when examining the vulva, considering that this area is difficult to monitor, and there is an especially poor prognosis of vulvar melanoma due to its late detection.6,7
In the past, biopsy was considered mandatory for confirming the diagnosis of vulvar angiokeratoma.5,8,9 However, dermoscopy has emerged as a valuable tool for diagnosis of angiokeratoma10 and also was helpful as a diagnostic aid in one of our patients (patient 1). Therefore, we believe that dermoscopy should be performed prior to a biopsy of angiokeratomas of the vulva.
To the Editor:
Angiokeratoma is a benign vascular tumor characterized by several dilated vessels in the superficial dermis accompanied by epidermal hyperplasia and hyperkeratosis.1 Angiokeratoma of the vulva is a rare clinical finding, usually involving multiple lesions as part of the Fordyce type.2 Solitary angiokeratoma occurs predominantly on the lower legs,3 and although other locations have been described, the presence of a solitary angiokeratoma on the vulva is rare.4 We report 2 cases of solitary angiokeratoma on the vulva that was misdiagnosed as malignant melanoma. Both patients were referred to our center for evaluation and excision.
A 65-year-old woman (patient 1) and a 67-year-old woman (patient 2) presented with a bluish black, growing, asymptomatic lesion on the right (Figure 1) and left labia majora, respectively. Both patients were referred by outside physicians for excision because of suspected malignant melanoma. Physical examinations revealed bluish black globular nodules that measured 0.5 and 0.3 cm in diameter, respectively. Dermoscopy (patient 1) revealed dark lacunae. Histopathologic examination of the vulvar lesion (patient 2) showed dilated, blood-filled, vascular spaces in the papillary dermis, accompanied by overlying acanthosis, hyperkeratosis, and papillomatosis that was consistent with angiokeratoma (Figure 2).
Angiokeratoma, particularly the solitary type, often is misdiagnosed. Clinical differential diagnoses may include a wide range of pathologic conditions, including condyloma acuminata, basal cell carcinoma, pyogenic granuloma, lymphangioma, nevi, condyloma lata, nodular prurigo, seborrheic keratosis, granuloma inguinale, and deep fungal infection.2,5 However, due to its quickly growing nature and its dark complexion, malignant melanoma often is initially diagnosed. Because patients affected by angiokeratoma of the vulva usually are aged 20 to 40 years,5 and vulvar melanoma is typical for middle-aged women (median age, 68 years),6 this misdiagnosis is more likely in older patients. It should be noted that a high index of suspicion for melanoma often is present when examining the vulva, considering that this area is difficult to monitor, and there is an especially poor prognosis of vulvar melanoma due to its late detection.6,7
In the past, biopsy was considered mandatory for confirming the diagnosis of vulvar angiokeratoma.5,8,9 However, dermoscopy has emerged as a valuable tool for diagnosis of angiokeratoma10 and also was helpful as a diagnostic aid in one of our patients (patient 1). Therefore, we believe that dermoscopy should be performed prior to a biopsy of angiokeratomas of the vulva.
- Requena L, Sangueza OP. Cutaneous vascular anomalies. part I. hamartomas, malformations, and dilation of preexisting vessels. J Am Acad Dermatol. 1997;37:523-549.
- Schiller PI, Itin PH. Angiokeratomas: an update. Dermatology. 1996;193:275-282.
- Gomi H, Eriyama Y, Horikawa E, et al. Solitary angiokeratoma. J Dermatol. 1988;15:349-350.
- Yamazaki M, Hiruma M, Irie H, et al. Angiokeratoma of the clitoris: a subtype of angiokeratoma vulvae. J Dermatol. 1992;19:553-555.
- Cohen PR, Young AW Jr, Tovell HM. Angiokeratoma of the vulva: diagnosis and review of the literature. Obstet Gynecol Surv. 1989;44:339-346.
- Sugiyama VE, Chan JK, Shin JY, et al. Vulvar melanoma: a multivariable analysis of 644 patients. Obstet Gynecol. 2007;110:296-301.
- De Simone P, Silipo V, Buccini P, et al. Vulvar melanoma: a report of 10 cases and review of the literature. Melanoma Res. 2008;18:127-133.
- Novick NL. Angiokeratoma vulvae. J Am Acad Dermatol. 1985;12:561-563.
- Yigiter M, Arda IS, Tosun E, et al. Angiokeratoma of clitoris: a rare lesion in an adolescent girl. Urology. 2008;71:604-606.
- Zaballos P, Daufi C, Puig S, et al. Dermoscopy of solitary angiokeratomas: a morphological study. Arch Dermatol. 2007;143:318-325.
- Requena L, Sangueza OP. Cutaneous vascular anomalies. part I. hamartomas, malformations, and dilation of preexisting vessels. J Am Acad Dermatol. 1997;37:523-549.
- Schiller PI, Itin PH. Angiokeratomas: an update. Dermatology. 1996;193:275-282.
- Gomi H, Eriyama Y, Horikawa E, et al. Solitary angiokeratoma. J Dermatol. 1988;15:349-350.
- Yamazaki M, Hiruma M, Irie H, et al. Angiokeratoma of the clitoris: a subtype of angiokeratoma vulvae. J Dermatol. 1992;19:553-555.
- Cohen PR, Young AW Jr, Tovell HM. Angiokeratoma of the vulva: diagnosis and review of the literature. Obstet Gynecol Surv. 1989;44:339-346.
- Sugiyama VE, Chan JK, Shin JY, et al. Vulvar melanoma: a multivariable analysis of 644 patients. Obstet Gynecol. 2007;110:296-301.
- De Simone P, Silipo V, Buccini P, et al. Vulvar melanoma: a report of 10 cases and review of the literature. Melanoma Res. 2008;18:127-133.
- Novick NL. Angiokeratoma vulvae. J Am Acad Dermatol. 1985;12:561-563.
- Yigiter M, Arda IS, Tosun E, et al. Angiokeratoma of clitoris: a rare lesion in an adolescent girl. Urology. 2008;71:604-606.
- Zaballos P, Daufi C, Puig S, et al. Dermoscopy of solitary angiokeratomas: a morphological study. Arch Dermatol. 2007;143:318-325.
Practice Points
- Solitary angiokeratoma of the vulva often is misdiagnosed as malignant melanoma due to its rapid growth and dark color.
- Dermoscopy is a valuable tool for diagnosing vulvar angiokeratoma to avoid unnecessary excisions.

VIDEO: Skin exam crucial in rheumatic diseases, expert says
SANDESTIN, FLA. – Even when you know a patient’s serology and hear their symptoms and think you have a bead on their rheumatic disease, you might not. It’s vital to check the skin in patients with rheumatic disease to be sure the right disease is being treated and that they don’t actually have a more severe condition that might progress suddenly if left unchecked, said Alisa Femia, MD, assistant professor of dermatology at the annual Congress of Clinical Rheumatology.
In a session filled with pearls for rheumatologists on what to look for on their patients’ skin to help guide diagnosis and treatment, she told the story of a woman whom a rheumatologist colleague had correctly diagnosed with dermatomyositis. She was started on prednisone and mycophenolate mofetil, but her skin disease did not clear.
After examining her skin, Dr. Femia became immediately concerned.
“Despite prednisone, despite mycophenolate, here not only does she have Gottron’s papules, but she has erosions within her Gottron’s papules,” Dr. Femia said. The woman also had erosions within papules on her palms.
These were telltale signs of MDA5-associated dermatomyositis, which studies have found to be linked with interstitial lung disease (J Am Acad Dermatol. 2011 Jul;65[1]:25-34). Under her care, these patients ideally undergo lung monitoring every 3 months, Dr. Femia said.
“That is a form of dermatomyositis that you cannot miss,” she said.
The effects of discoid lupus are another reason to take special care in skin examination. Once the disease, which involves a scaling of the skin, is obvious, there can be permanent aesthetic effects that could have been avoided with earlier detection and treatment, Dr. Femia said.
Clinicians should also be on the lookout for volume loss, or contour change, in discoid lupus patients, because that’s a sign of lupus panniculitis, which involves deeper lesions mainly to fatty areas such as the cheeks or thighs. The disease can progress fast, with sudden, massive loss of body volume, so therapy should be escalated quickly, she said.
“We want to treat these patients aggressively in order to avoid this.”
SOURCE: Femia A. CCR 2018.
SANDESTIN, FLA. – Even when you know a patient’s serology and hear their symptoms and think you have a bead on their rheumatic disease, you might not. It’s vital to check the skin in patients with rheumatic disease to be sure the right disease is being treated and that they don’t actually have a more severe condition that might progress suddenly if left unchecked, said Alisa Femia, MD, assistant professor of dermatology at the annual Congress of Clinical Rheumatology.
In a session filled with pearls for rheumatologists on what to look for on their patients’ skin to help guide diagnosis and treatment, she told the story of a woman whom a rheumatologist colleague had correctly diagnosed with dermatomyositis. She was started on prednisone and mycophenolate mofetil, but her skin disease did not clear.
After examining her skin, Dr. Femia became immediately concerned.
“Despite prednisone, despite mycophenolate, here not only does she have Gottron’s papules, but she has erosions within her Gottron’s papules,” Dr. Femia said. The woman also had erosions within papules on her palms.
These were telltale signs of MDA5-associated dermatomyositis, which studies have found to be linked with interstitial lung disease (J Am Acad Dermatol. 2011 Jul;65[1]:25-34). Under her care, these patients ideally undergo lung monitoring every 3 months, Dr. Femia said.
“That is a form of dermatomyositis that you cannot miss,” she said.
The effects of discoid lupus are another reason to take special care in skin examination. Once the disease, which involves a scaling of the skin, is obvious, there can be permanent aesthetic effects that could have been avoided with earlier detection and treatment, Dr. Femia said.
Clinicians should also be on the lookout for volume loss, or contour change, in discoid lupus patients, because that’s a sign of lupus panniculitis, which involves deeper lesions mainly to fatty areas such as the cheeks or thighs. The disease can progress fast, with sudden, massive loss of body volume, so therapy should be escalated quickly, she said.
“We want to treat these patients aggressively in order to avoid this.”
SOURCE: Femia A. CCR 2018.
SANDESTIN, FLA. – Even when you know a patient’s serology and hear their symptoms and think you have a bead on their rheumatic disease, you might not. It’s vital to check the skin in patients with rheumatic disease to be sure the right disease is being treated and that they don’t actually have a more severe condition that might progress suddenly if left unchecked, said Alisa Femia, MD, assistant professor of dermatology at the annual Congress of Clinical Rheumatology.
In a session filled with pearls for rheumatologists on what to look for on their patients’ skin to help guide diagnosis and treatment, she told the story of a woman whom a rheumatologist colleague had correctly diagnosed with dermatomyositis. She was started on prednisone and mycophenolate mofetil, but her skin disease did not clear.
After examining her skin, Dr. Femia became immediately concerned.
“Despite prednisone, despite mycophenolate, here not only does she have Gottron’s papules, but she has erosions within her Gottron’s papules,” Dr. Femia said. The woman also had erosions within papules on her palms.
These were telltale signs of MDA5-associated dermatomyositis, which studies have found to be linked with interstitial lung disease (J Am Acad Dermatol. 2011 Jul;65[1]:25-34). Under her care, these patients ideally undergo lung monitoring every 3 months, Dr. Femia said.
“That is a form of dermatomyositis that you cannot miss,” she said.
The effects of discoid lupus are another reason to take special care in skin examination. Once the disease, which involves a scaling of the skin, is obvious, there can be permanent aesthetic effects that could have been avoided with earlier detection and treatment, Dr. Femia said.
Clinicians should also be on the lookout for volume loss, or contour change, in discoid lupus patients, because that’s a sign of lupus panniculitis, which involves deeper lesions mainly to fatty areas such as the cheeks or thighs. The disease can progress fast, with sudden, massive loss of body volume, so therapy should be escalated quickly, she said.
“We want to treat these patients aggressively in order to avoid this.”
SOURCE: Femia A. CCR 2018.
EXPERT ANALYSIS AT CCR 18
Progressive Widespread Telangiectasias
The Diagnosis: Cutaneous Collagenous Vasculopathy
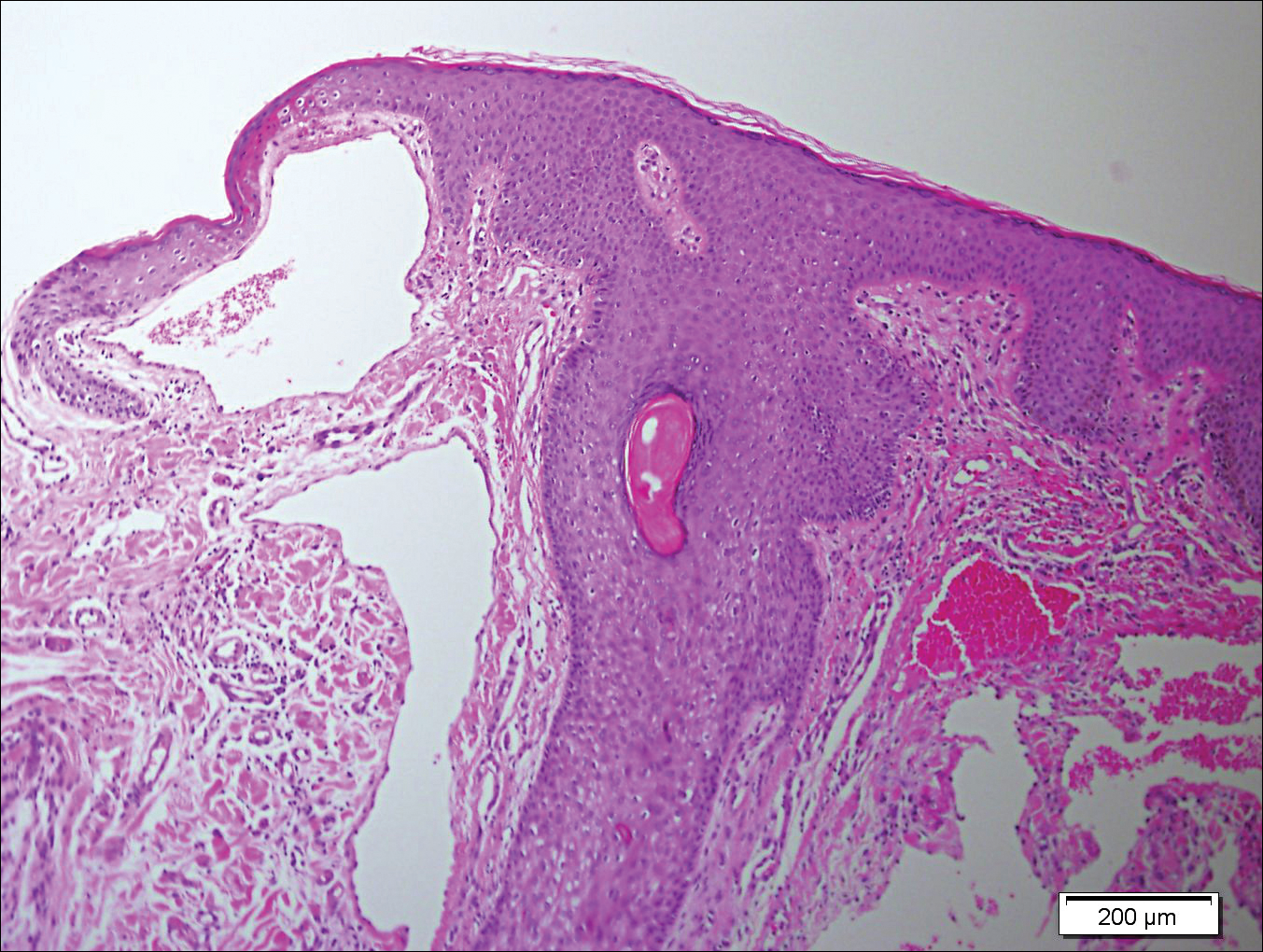
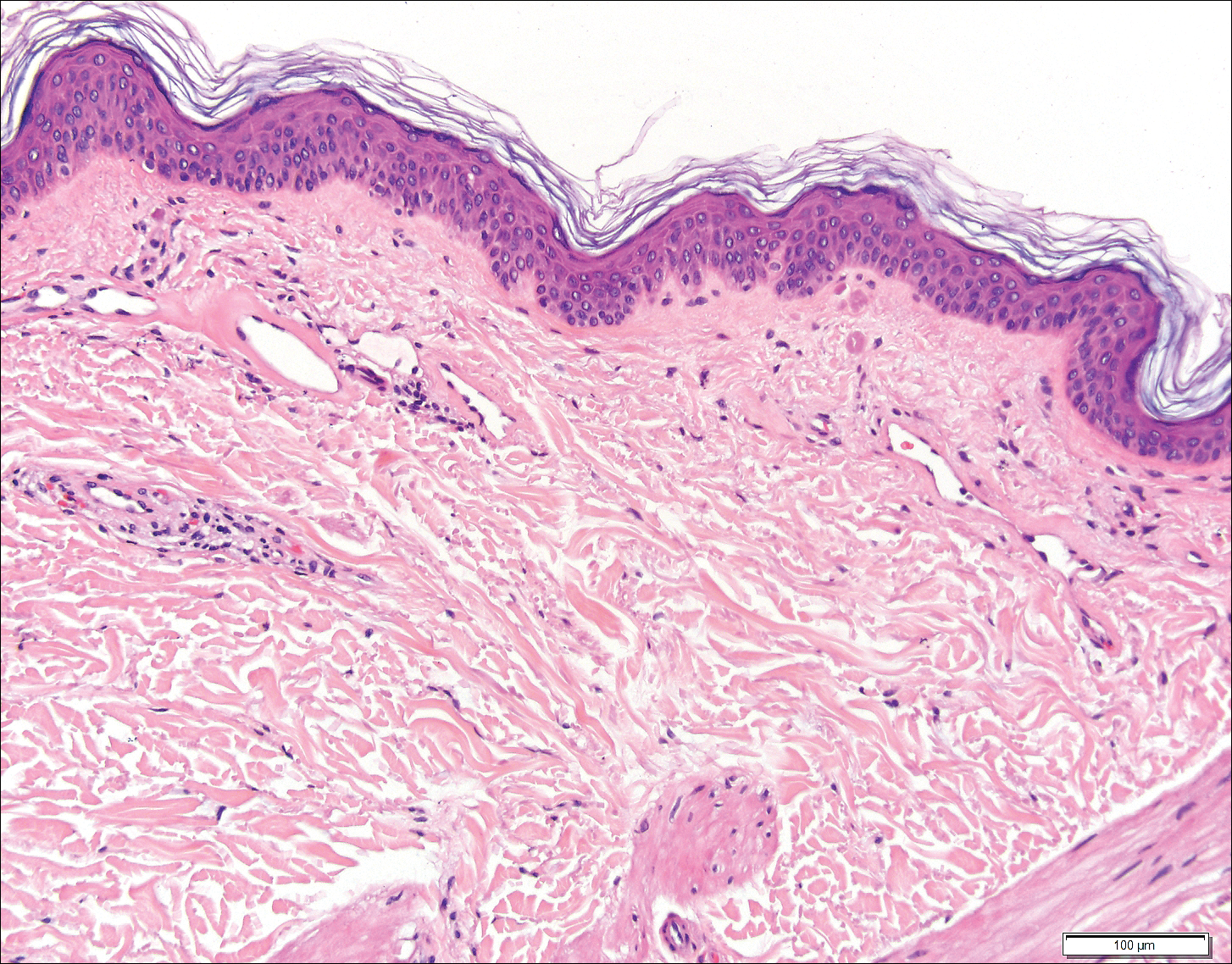
Histopathologic examination revealed ectatic blood vessels lined with unremarkable endothelial cells and thickened, hyalinized vessel walls scattered within the papillary dermis (Figure 1). The epidermis was unremarkable. There was minimal associated inflammation and no extravasation of erythrocytes. The hyalinized material was weakly positive on periodic acid-Schiff staining (Figure 2) and negative on Congo red staining, which supported of a diagnosis of cutaneous collagenous vasculopathy (CCV).
The patient previously had been given a suspected diagnosis of generalized essential telangiectasia by an outside dermatologist several years prior to the current presentation, as CCV had yet to be recognized as its own entity and therefore few cases had been described in the literature. She had a known history of obesity, hypertension, hyperlipidemia, and type 2 diabetes mellitus, which are associated with the condition. Multiple specialists concluded that the disease was too extensive for laser treatment. A review of PubMed articles indexed for MEDLINE yielded no established treatment options.
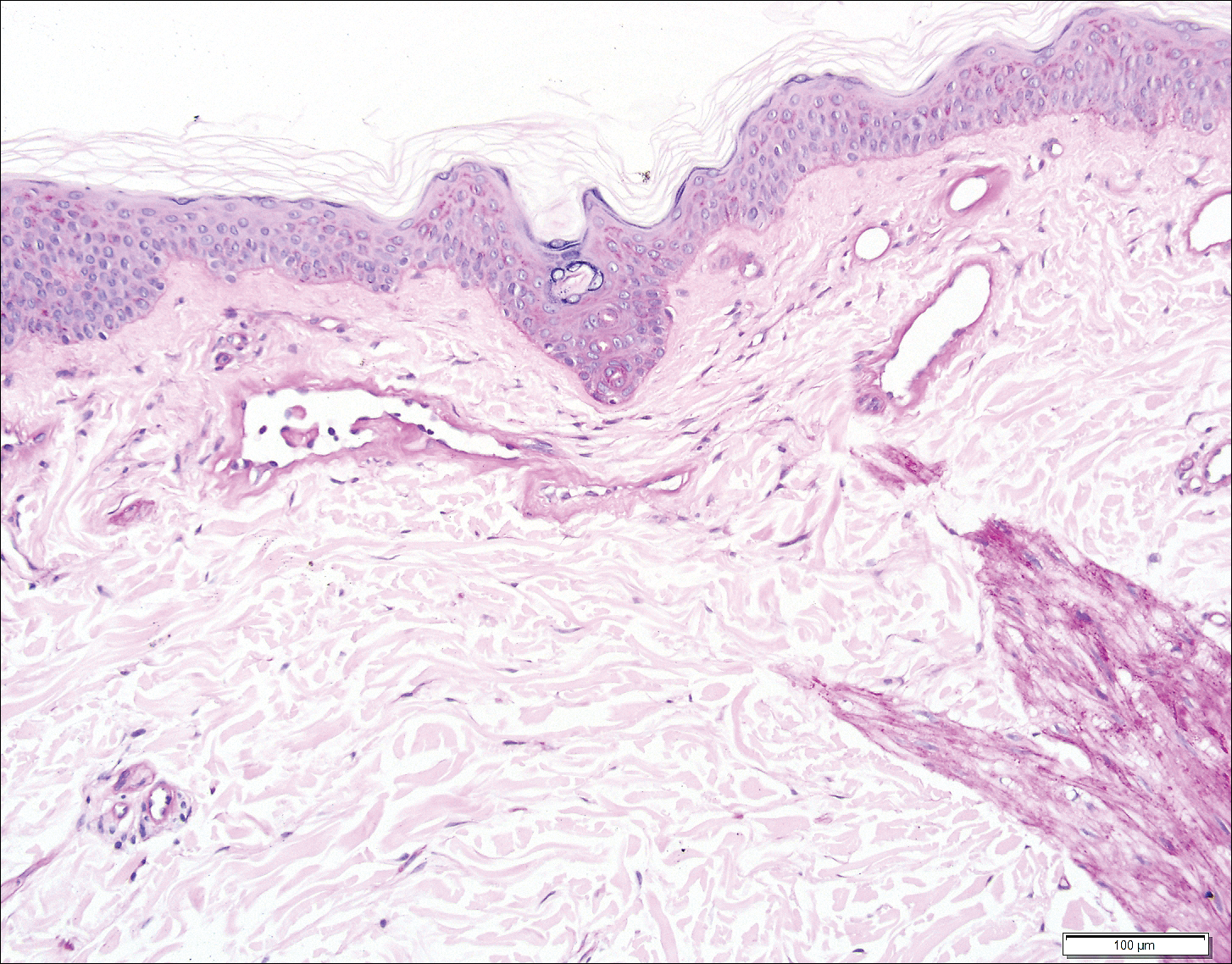
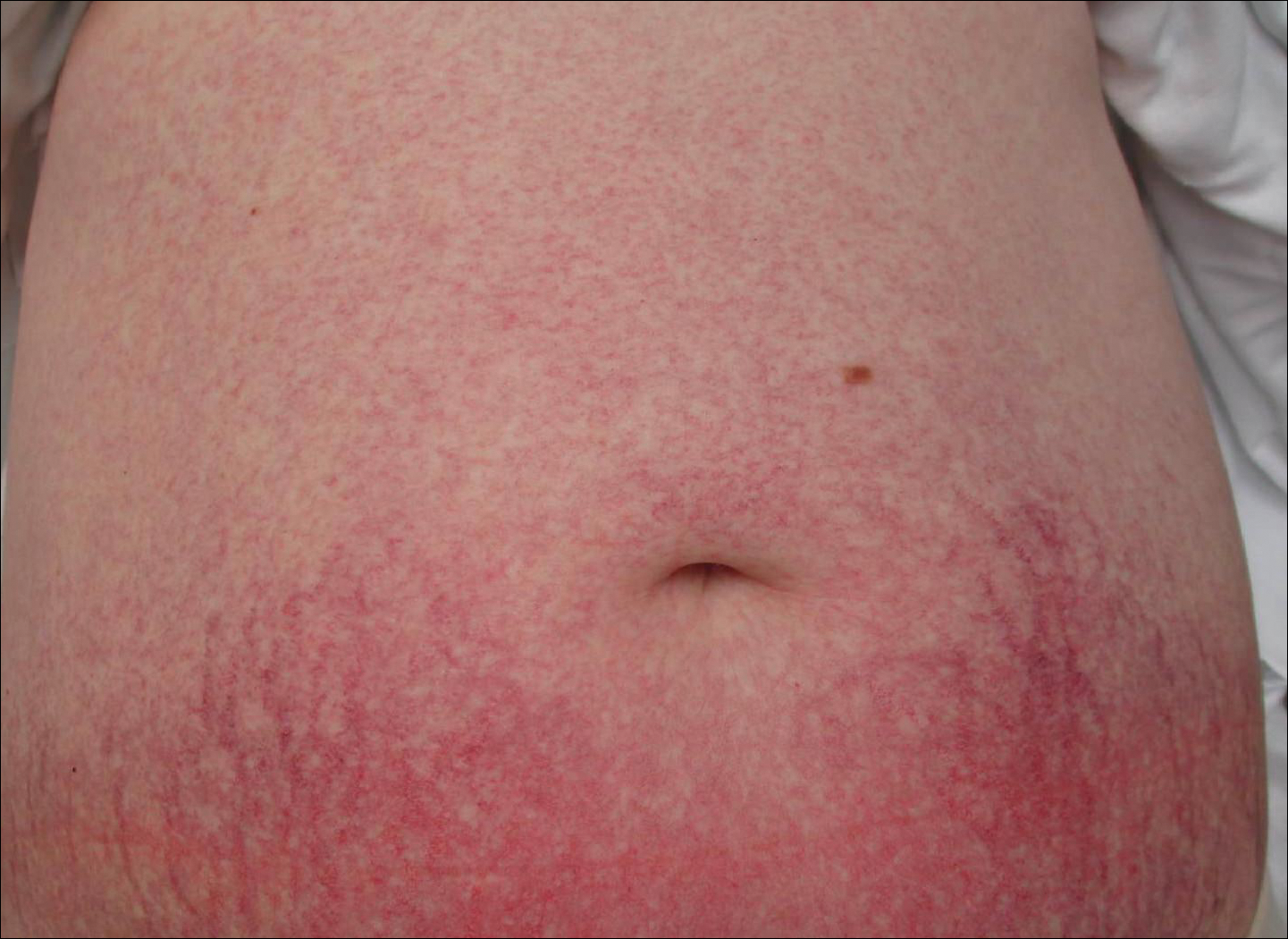
Cutaneous collagenous vasculopathy is a rare acquired microangiopathy involving the small vessels of the skin. Its clinical presentation is indistinguishable from that of generalized essential telangiectasia (GET). Patients generally present with asymptomatic, widespread, blanching, symmetric telangiectasias that classically begin on the legs and steadily progress upward with classic sparing of the face (Figure 3). Whereas GET has been reported to involve the oral and conjunctival mucosa, mucosal involvement is not typically observed in CCV and is considered to be a distinguishing factor between the 2 conditions.1,2 However, our patient reported oral symptoms, and oral erosions were seen on multiple physical examinations; therefore, ours is a rare case of mucosal involvement in conjunction with CCV. Given this finding, it is possible that more cases of CCV with mucosal involvement may exist but have been clinically misdiagnosed as GET.
First described by Salama and Rosenthal3 in 2000, CCV remains a rarely reported entity, with approximately 33 reported cases in the worldwide literature.2,4-7 The condition typically arises in adults with an equal predilection for males and females.2 The true incidence of CCV is unknown and likely is underreported given its close similarities to GET, which often is diagnosed clinically. The unique histopathologic finding of superficial ectatic vascular spaces with eosinophilic hyalinized vessel walls in CCV is key to distinguishing these similar entities, and even this finding can be subtle and is easily overlooked. Inflammation is sparse to absent. Deposited material is positive on periodic acid-Schiff and cytokeratin IV staining (representing reduplicated basement membrane-type collagen) and is diastase resistant. Smooth muscle actin staining is diminished or absent. Ultrastructural examination reveals reduplicated, laminated basement membrane; Luse bodies (abnormally long, widely spaced collagen fibers); and a decrease in or loss of pericytes. Of note, Luse bodies are nonspecific and their absence does not exclude a diagnosis of CCV.1
The etiology of CCV is unclear, and multiple pathogenetic mechanisms have been proposed. Ultimately, this entity is thought to arise from repeated endothelial cell damage, although the trigger for the endothelial cell injury is not completely understood. Diabetes mellitus sometimes is associated with microangiopathy and may be a confounding but not causative factor in some cases.1 Some investigators believe CCV is caused by a genetic defect that alters collagen production in the small vessels of the skin.5 Others have hypothesized that it is a secondary manifestation of an underlying disease or is associated with a medication; however, no disease or drug has been convincingly implicated in CCV.8
Cutaneous collagenous vasculopathy is limited to the skin, with no known reports of systemic involvement in the literature.7 There are no recommended laboratory studies to aid in diagnosis.1 It is critical to exclude hereditary hemorrhagic telangiectasia (HHT), as these patients can have life-threatening systemic involvement. Patients with CCV generally have no history of a bleeding diathesis, patients with HHT classically report recurrent epistaxis and gastrointestinal bleeding.7 A family history of HHT also is helpful for diagnosis, as the condition is autosomal dominant.1 Neither HHT or telangiectasia macularis eruptiva perstans, which also can be included in the differential diagnosis, demonstrate vessel wall hyalinization.
Treatment options for CCV are limited. Basso et al6 reported notable improvement in a patient with CCV treated with a combined 595-nm pulsed dye laser and 1064-nm Nd:YAG laser and optimized pulsed light. In one patient, treatment with a 585-nm pulsed dye laser produced a blanching response, suggesting that this may be a potential treatment option.7 Treatment with sclerotherapy has been ineffective.2
It is critical for both dermatologists and dermatopathologists to recognize and report this newly described entity, as the unique finding of vessel wall hyalinization in CCV may be indicative of a certain pathogenetic mechanism and effective treatment avenue that has yet to be established due to the relatively few number of reports that currently exist in the literature.
- Burdick LM, Lohser S, Somach SC, et al. Cutaneous collagenous vasculopathy: a rare cutaneous microangiopathy. J Cutan Pathol. 2012;39:741-746.
- Brady BG, Ortleb M, Boyd AS, et al. Cutaneous collagenous vasculopathy. J Clin Aesthet Dermatol. 2015;8:49-52.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and structural study. J Cutan Pathol. 2000;27:40-48.
- Toda-Brito H, Resende C, Catorze G, et al. Cutaneous collagenous vasculopathy: a rare cause of generalised cutaneous telangiectasia. BMJ Case Rep. 2015. doi: 10.1136/bcr-2015-210635.
- Ma DL, Vano-Galvan S. Images in clinical medicine: cutaneous collagenous vasculopathy. N Engl J Med. 2015;373:E14.
- Basso D, Ribero S, Blazek C, et al. Cutaneous collagenous vasculopathy: a rare form of microangiopathy successfully treated with a combination of multiplex laser and optimized pulsed light with a review of the literature. Dermatology. 2016;232:107-111.
- Moteqi SI, Yasuda M, Yamanaka M, et al. Cutaneous collagenous vasculopathy: report of first Japanese case and review of the literature. Australas J Dermatol. 2017;58:145-149.
- González Fernández D, Gómez Bernal S, Vivanco Allende B, et al. Cutaneous collagenous vasculopathy: description of two new cases in elderly women and review of the literature. Dermatology. 2012;225:1-8.
The Diagnosis: Cutaneous Collagenous Vasculopathy
Histopathologic examination revealed ectatic blood vessels lined with unremarkable endothelial cells and thickened, hyalinized vessel walls scattered within the papillary dermis (Figure 1). The epidermis was unremarkable. There was minimal associated inflammation and no extravasation of erythrocytes. The hyalinized material was weakly positive on periodic acid-Schiff staining (Figure 2) and negative on Congo red staining, which supported of a diagnosis of cutaneous collagenous vasculopathy (CCV).
The patient previously had been given a suspected diagnosis of generalized essential telangiectasia by an outside dermatologist several years prior to the current presentation, as CCV had yet to be recognized as its own entity and therefore few cases had been described in the literature. She had a known history of obesity, hypertension, hyperlipidemia, and type 2 diabetes mellitus, which are associated with the condition. Multiple specialists concluded that the disease was too extensive for laser treatment. A review of PubMed articles indexed for MEDLINE yielded no established treatment options.
Cutaneous collagenous vasculopathy is a rare acquired microangiopathy involving the small vessels of the skin. Its clinical presentation is indistinguishable from that of generalized essential telangiectasia (GET). Patients generally present with asymptomatic, widespread, blanching, symmetric telangiectasias that classically begin on the legs and steadily progress upward with classic sparing of the face (Figure 3). Whereas GET has been reported to involve the oral and conjunctival mucosa, mucosal involvement is not typically observed in CCV and is considered to be a distinguishing factor between the 2 conditions.1,2 However, our patient reported oral symptoms, and oral erosions were seen on multiple physical examinations; therefore, ours is a rare case of mucosal involvement in conjunction with CCV. Given this finding, it is possible that more cases of CCV with mucosal involvement may exist but have been clinically misdiagnosed as GET.
First described by Salama and Rosenthal3 in 2000, CCV remains a rarely reported entity, with approximately 33 reported cases in the worldwide literature.2,4-7 The condition typically arises in adults with an equal predilection for males and females.2 The true incidence of CCV is unknown and likely is underreported given its close similarities to GET, which often is diagnosed clinically. The unique histopathologic finding of superficial ectatic vascular spaces with eosinophilic hyalinized vessel walls in CCV is key to distinguishing these similar entities, and even this finding can be subtle and is easily overlooked. Inflammation is sparse to absent. Deposited material is positive on periodic acid-Schiff and cytokeratin IV staining (representing reduplicated basement membrane-type collagen) and is diastase resistant. Smooth muscle actin staining is diminished or absent. Ultrastructural examination reveals reduplicated, laminated basement membrane; Luse bodies (abnormally long, widely spaced collagen fibers); and a decrease in or loss of pericytes. Of note, Luse bodies are nonspecific and their absence does not exclude a diagnosis of CCV.1
The etiology of CCV is unclear, and multiple pathogenetic mechanisms have been proposed. Ultimately, this entity is thought to arise from repeated endothelial cell damage, although the trigger for the endothelial cell injury is not completely understood. Diabetes mellitus sometimes is associated with microangiopathy and may be a confounding but not causative factor in some cases.1 Some investigators believe CCV is caused by a genetic defect that alters collagen production in the small vessels of the skin.5 Others have hypothesized that it is a secondary manifestation of an underlying disease or is associated with a medication; however, no disease or drug has been convincingly implicated in CCV.8
Cutaneous collagenous vasculopathy is limited to the skin, with no known reports of systemic involvement in the literature.7 There are no recommended laboratory studies to aid in diagnosis.1 It is critical to exclude hereditary hemorrhagic telangiectasia (HHT), as these patients can have life-threatening systemic involvement. Patients with CCV generally have no history of a bleeding diathesis, patients with HHT classically report recurrent epistaxis and gastrointestinal bleeding.7 A family history of HHT also is helpful for diagnosis, as the condition is autosomal dominant.1 Neither HHT or telangiectasia macularis eruptiva perstans, which also can be included in the differential diagnosis, demonstrate vessel wall hyalinization.
Treatment options for CCV are limited. Basso et al6 reported notable improvement in a patient with CCV treated with a combined 595-nm pulsed dye laser and 1064-nm Nd:YAG laser and optimized pulsed light. In one patient, treatment with a 585-nm pulsed dye laser produced a blanching response, suggesting that this may be a potential treatment option.7 Treatment with sclerotherapy has been ineffective.2
It is critical for both dermatologists and dermatopathologists to recognize and report this newly described entity, as the unique finding of vessel wall hyalinization in CCV may be indicative of a certain pathogenetic mechanism and effective treatment avenue that has yet to be established due to the relatively few number of reports that currently exist in the literature.
The Diagnosis: Cutaneous Collagenous Vasculopathy
Histopathologic examination revealed ectatic blood vessels lined with unremarkable endothelial cells and thickened, hyalinized vessel walls scattered within the papillary dermis (Figure 1). The epidermis was unremarkable. There was minimal associated inflammation and no extravasation of erythrocytes. The hyalinized material was weakly positive on periodic acid-Schiff staining (Figure 2) and negative on Congo red staining, which supported of a diagnosis of cutaneous collagenous vasculopathy (CCV).
The patient previously had been given a suspected diagnosis of generalized essential telangiectasia by an outside dermatologist several years prior to the current presentation, as CCV had yet to be recognized as its own entity and therefore few cases had been described in the literature. She had a known history of obesity, hypertension, hyperlipidemia, and type 2 diabetes mellitus, which are associated with the condition. Multiple specialists concluded that the disease was too extensive for laser treatment. A review of PubMed articles indexed for MEDLINE yielded no established treatment options.
Cutaneous collagenous vasculopathy is a rare acquired microangiopathy involving the small vessels of the skin. Its clinical presentation is indistinguishable from that of generalized essential telangiectasia (GET). Patients generally present with asymptomatic, widespread, blanching, symmetric telangiectasias that classically begin on the legs and steadily progress upward with classic sparing of the face (Figure 3). Whereas GET has been reported to involve the oral and conjunctival mucosa, mucosal involvement is not typically observed in CCV and is considered to be a distinguishing factor between the 2 conditions.1,2 However, our patient reported oral symptoms, and oral erosions were seen on multiple physical examinations; therefore, ours is a rare case of mucosal involvement in conjunction with CCV. Given this finding, it is possible that more cases of CCV with mucosal involvement may exist but have been clinically misdiagnosed as GET.
First described by Salama and Rosenthal3 in 2000, CCV remains a rarely reported entity, with approximately 33 reported cases in the worldwide literature.2,4-7 The condition typically arises in adults with an equal predilection for males and females.2 The true incidence of CCV is unknown and likely is underreported given its close similarities to GET, which often is diagnosed clinically. The unique histopathologic finding of superficial ectatic vascular spaces with eosinophilic hyalinized vessel walls in CCV is key to distinguishing these similar entities, and even this finding can be subtle and is easily overlooked. Inflammation is sparse to absent. Deposited material is positive on periodic acid-Schiff and cytokeratin IV staining (representing reduplicated basement membrane-type collagen) and is diastase resistant. Smooth muscle actin staining is diminished or absent. Ultrastructural examination reveals reduplicated, laminated basement membrane; Luse bodies (abnormally long, widely spaced collagen fibers); and a decrease in or loss of pericytes. Of note, Luse bodies are nonspecific and their absence does not exclude a diagnosis of CCV.1
The etiology of CCV is unclear, and multiple pathogenetic mechanisms have been proposed. Ultimately, this entity is thought to arise from repeated endothelial cell damage, although the trigger for the endothelial cell injury is not completely understood. Diabetes mellitus sometimes is associated with microangiopathy and may be a confounding but not causative factor in some cases.1 Some investigators believe CCV is caused by a genetic defect that alters collagen production in the small vessels of the skin.5 Others have hypothesized that it is a secondary manifestation of an underlying disease or is associated with a medication; however, no disease or drug has been convincingly implicated in CCV.8
Cutaneous collagenous vasculopathy is limited to the skin, with no known reports of systemic involvement in the literature.7 There are no recommended laboratory studies to aid in diagnosis.1 It is critical to exclude hereditary hemorrhagic telangiectasia (HHT), as these patients can have life-threatening systemic involvement. Patients with CCV generally have no history of a bleeding diathesis, patients with HHT classically report recurrent epistaxis and gastrointestinal bleeding.7 A family history of HHT also is helpful for diagnosis, as the condition is autosomal dominant.1 Neither HHT or telangiectasia macularis eruptiva perstans, which also can be included in the differential diagnosis, demonstrate vessel wall hyalinization.
Treatment options for CCV are limited. Basso et al6 reported notable improvement in a patient with CCV treated with a combined 595-nm pulsed dye laser and 1064-nm Nd:YAG laser and optimized pulsed light. In one patient, treatment with a 585-nm pulsed dye laser produced a blanching response, suggesting that this may be a potential treatment option.7 Treatment with sclerotherapy has been ineffective.2
It is critical for both dermatologists and dermatopathologists to recognize and report this newly described entity, as the unique finding of vessel wall hyalinization in CCV may be indicative of a certain pathogenetic mechanism and effective treatment avenue that has yet to be established due to the relatively few number of reports that currently exist in the literature.
- Burdick LM, Lohser S, Somach SC, et al. Cutaneous collagenous vasculopathy: a rare cutaneous microangiopathy. J Cutan Pathol. 2012;39:741-746.
- Brady BG, Ortleb M, Boyd AS, et al. Cutaneous collagenous vasculopathy. J Clin Aesthet Dermatol. 2015;8:49-52.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and structural study. J Cutan Pathol. 2000;27:40-48.
- Toda-Brito H, Resende C, Catorze G, et al. Cutaneous collagenous vasculopathy: a rare cause of generalised cutaneous telangiectasia. BMJ Case Rep. 2015. doi: 10.1136/bcr-2015-210635.
- Ma DL, Vano-Galvan S. Images in clinical medicine: cutaneous collagenous vasculopathy. N Engl J Med. 2015;373:E14.
- Basso D, Ribero S, Blazek C, et al. Cutaneous collagenous vasculopathy: a rare form of microangiopathy successfully treated with a combination of multiplex laser and optimized pulsed light with a review of the literature. Dermatology. 2016;232:107-111.
- Moteqi SI, Yasuda M, Yamanaka M, et al. Cutaneous collagenous vasculopathy: report of first Japanese case and review of the literature. Australas J Dermatol. 2017;58:145-149.
- González Fernández D, Gómez Bernal S, Vivanco Allende B, et al. Cutaneous collagenous vasculopathy: description of two new cases in elderly women and review of the literature. Dermatology. 2012;225:1-8.
- Burdick LM, Lohser S, Somach SC, et al. Cutaneous collagenous vasculopathy: a rare cutaneous microangiopathy. J Cutan Pathol. 2012;39:741-746.
- Brady BG, Ortleb M, Boyd AS, et al. Cutaneous collagenous vasculopathy. J Clin Aesthet Dermatol. 2015;8:49-52.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and structural study. J Cutan Pathol. 2000;27:40-48.
- Toda-Brito H, Resende C, Catorze G, et al. Cutaneous collagenous vasculopathy: a rare cause of generalised cutaneous telangiectasia. BMJ Case Rep. 2015. doi: 10.1136/bcr-2015-210635.
- Ma DL, Vano-Galvan S. Images in clinical medicine: cutaneous collagenous vasculopathy. N Engl J Med. 2015;373:E14.
- Basso D, Ribero S, Blazek C, et al. Cutaneous collagenous vasculopathy: a rare form of microangiopathy successfully treated with a combination of multiplex laser and optimized pulsed light with a review of the literature. Dermatology. 2016;232:107-111.
- Moteqi SI, Yasuda M, Yamanaka M, et al. Cutaneous collagenous vasculopathy: report of first Japanese case and review of the literature. Australas J Dermatol. 2017;58:145-149.
- González Fernández D, Gómez Bernal S, Vivanco Allende B, et al. Cutaneous collagenous vasculopathy: description of two new cases in elderly women and review of the literature. Dermatology. 2012;225:1-8.
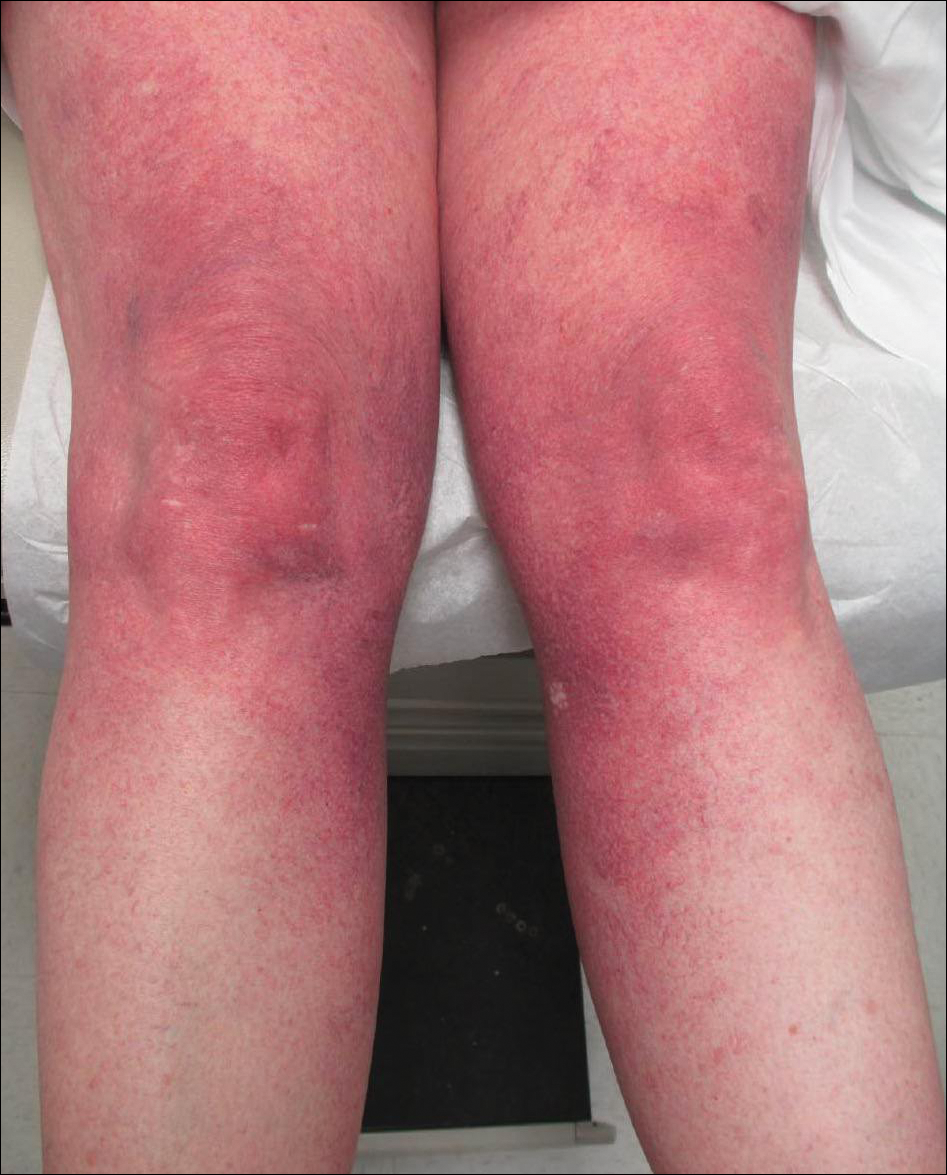
A 55-year-old woman presented for evaluation of widespread asymptomatic telangiectasias of several years' duration that first appeared on the legs and steadily progressed to involve the trunk and arms. A review of systems was remarkable for episodic glossitis and oral erosions that developed at the same time as the eruption. The patient had no history of bleeding diasthesis, and her family history was unremarkable. A laboratory workup (including autoimmune screening) and a malignancy workup were negative. Physical examination revealed confluent sheets of erythematous and purple blanching telangiectasias scattered symmetrically on the trunk, bilateral arms and legs, buttocks, and dorsal aspects of the feet with sparing of the palms, soles, and head and neck regions. A small, shallow erosion was present on the lateral aspect of the tongue. A 4-mm punch biopsy of a thigh lesion revealed ectatic blood vessels with hyalinized walls.
Perianal Extramammary Paget Disease Treated With Topical Imiquimod and Oral Cimetidine
Case Report
A 56-year-old woman with well-controlled hypertension, hyperlipidemia, and gastroesophageal reflux disease initially presented with itching and a rash in the perianal region of 1 year’s duration. She had been treated intermittently by her primary care physician over the past year for presumed hemorrhoids and a perianal fungal infection without improvement. Physical examination at the time of intitial presentation revealed a single, well-demarcated, scaly, pink plaque on the perianal area on the right buttock extending toward the anal canal (Figure 1).
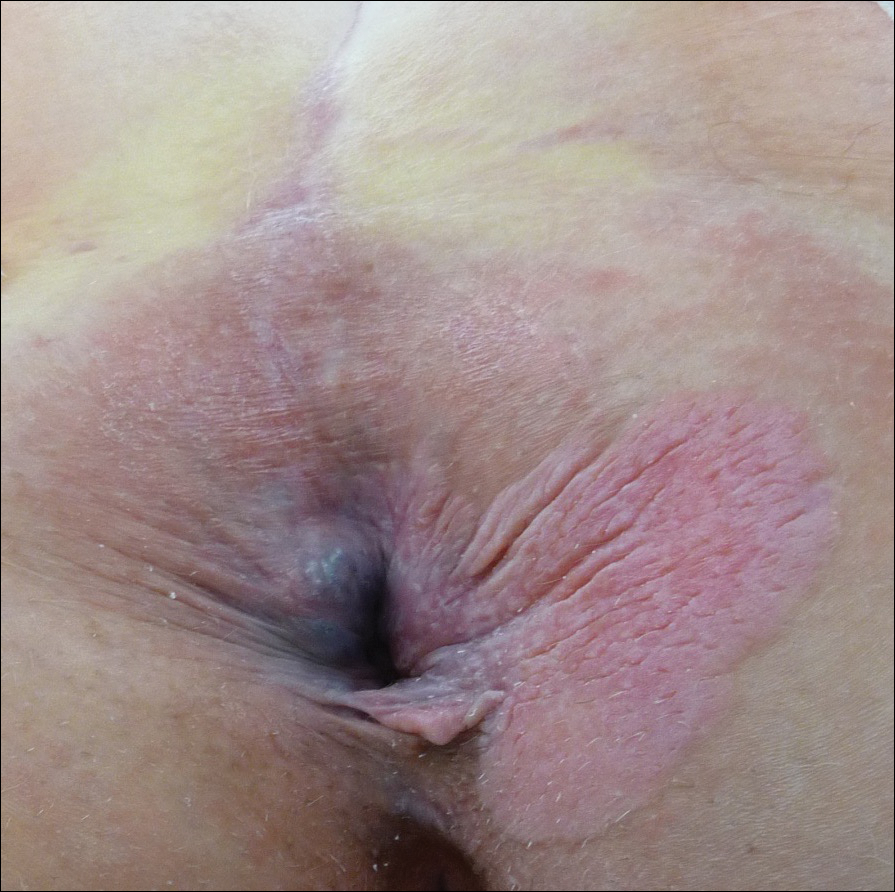
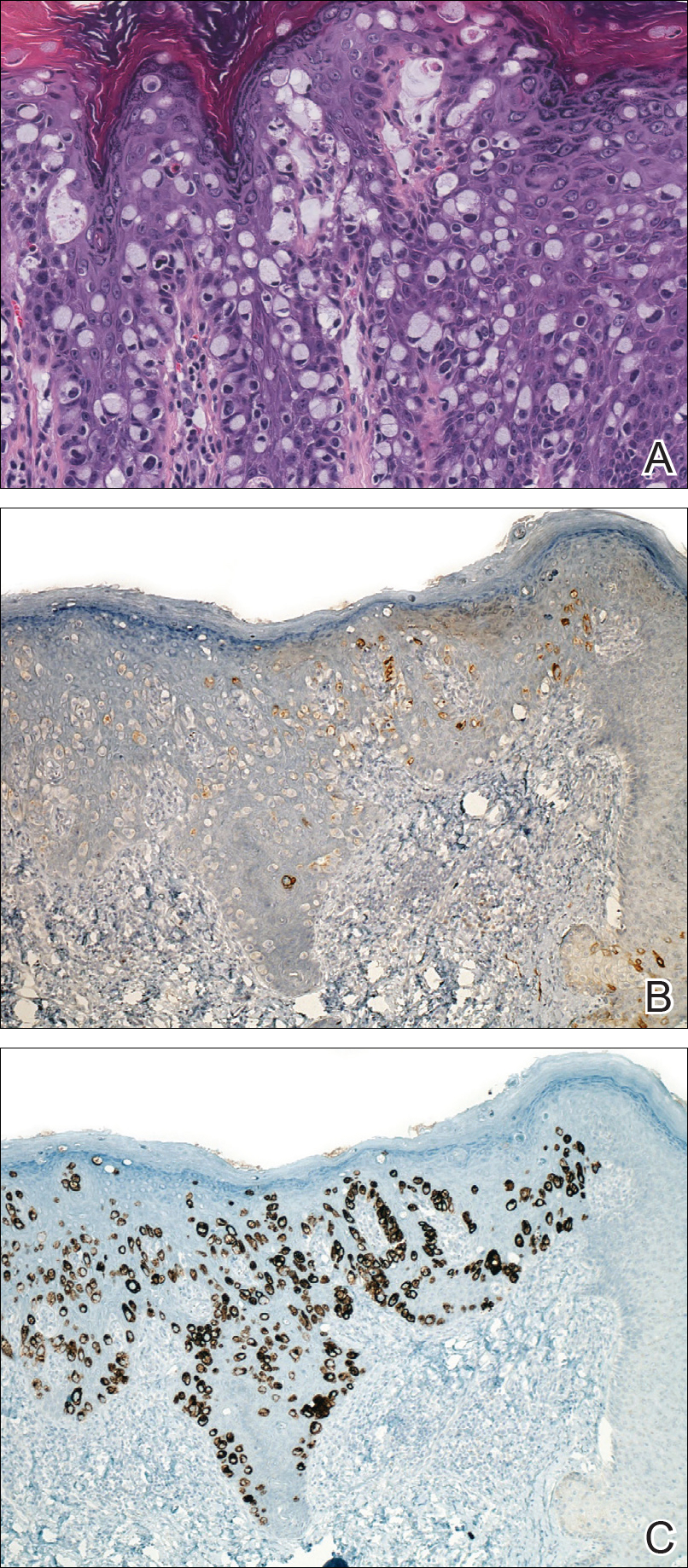
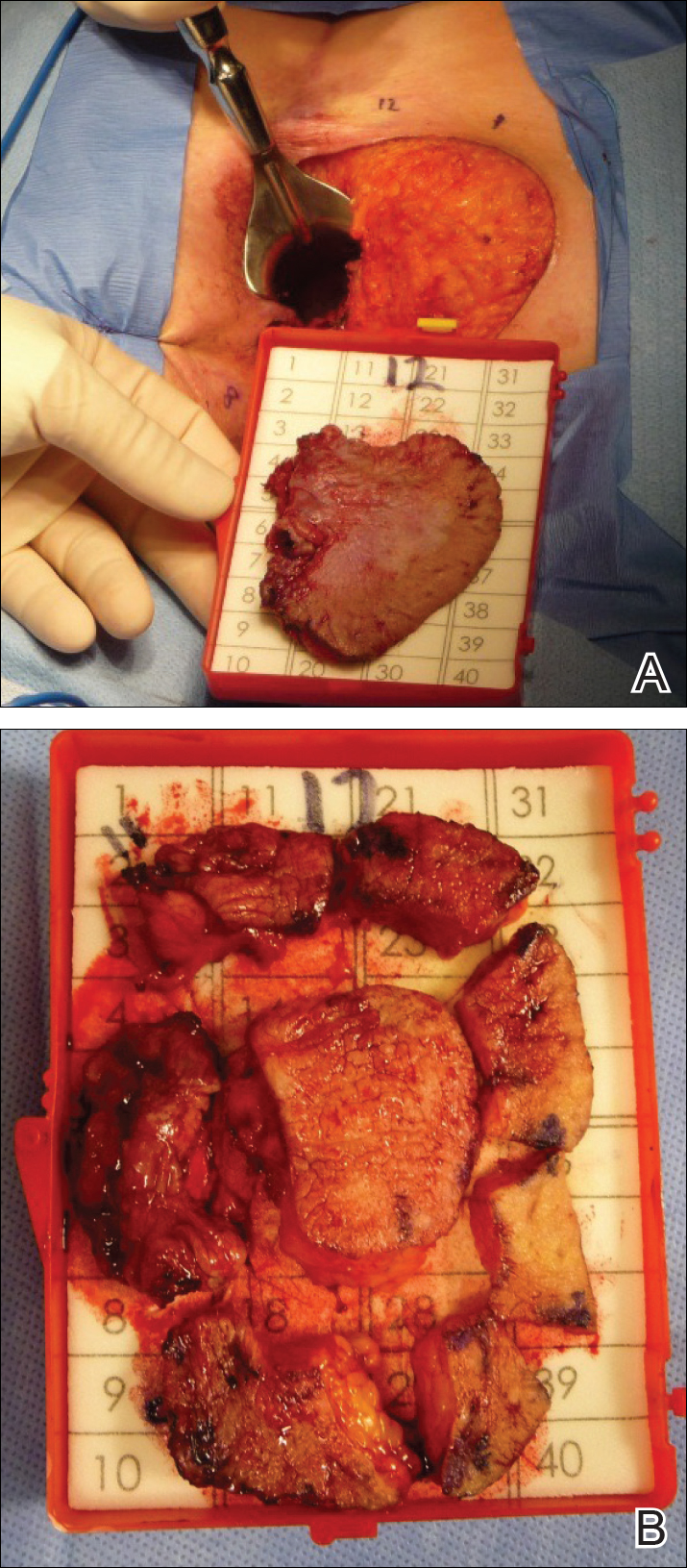
Four years later, the patient returned with new symptoms of bleeding when wiping the perianal region, pruritus, and fecal urgency of 3 to 4 months’ duration. Physical examination revealed scaly patches on the anus that were suspicious for recurrence of EMPD. Biopsies from the anal margin and anal canal confirmed recurrent EMPD involving the anal canal. Repeat evaluation for internal malignancy was negative.
Given the involvement of the anal canal, repeat wide local excision would have required anal resection and would therefore have been functionally impairing. The patient refused further surgical intervention as well as radiotherapy. Rather, a novel 16-week immunomodulatory regimen involving imiquimod cream 5% cream and low-dose oral cimetidine was started. To address the anal involvement, the patient was instructed to lubricate glycerin suppositories with the imiquimod cream and insert intra-anally once weekly. Dosing was adjusted based on the patient’s inflammatory response and tolerability, as she did initially report some flulike symptoms with the first few weeks of treatment. For most of the 16-week course, she applied 250 mg of imiquimod cream 5% to the perianal area 3 times weekly and 250 mg into the anal canal once weekly. Oral cimetidine initially was dosed at 800 mg twice daily as tolerated, but due to stomach irritation, the patient self-reduced her intake to 800 mg 3 times weekly.
To determine treatment response, scouting biopsies of the anal margin and anal canal were obtained 4 weeks after treatment cessation and demonstrated no evidence of residual disease. The patient resumed topical imiquimod applied once weekly into the anal canal and around the anus for a planned prolonged course of at least 1 year. To reduce the risk of recurrence, the patient continued taking oral cimetidine 800 mg 3 times weekly. Recommended follow-up included annual anoscopy or colonoscopy, serum carcinoembryonic antigen evaluation, and regular clinical monitoring by the dermatology and colorectal surgery teams.
Six months after completing the combination therapy, she was seen by the dermatology department and remained clinically free of disease (Figure 4). Anoscopy examination by the colorectal surgery department 4 months later showed no clinical evidence of malignancy.
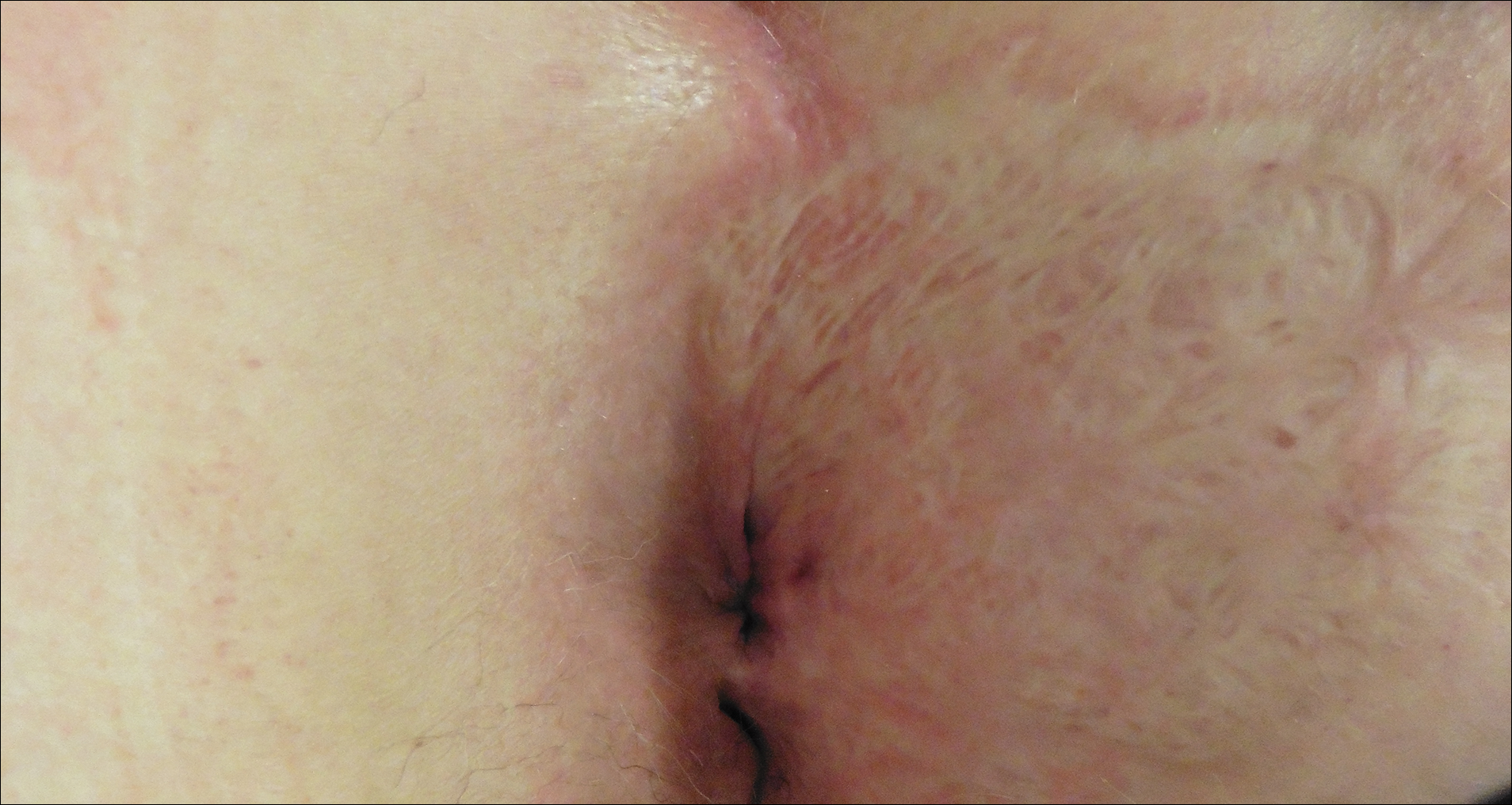
Comment
Extramammary Paget disease is a rare intraepithelial adenocarcinoma with a predilection for white females and an average age of onset of 50 to 80 years.1-3 The vulva, perianal region, scrotum, penis, and perineum are the most commonly affected sites.1-3 Clinically, EMPD presents as a chronic, well-demarcated, scaly, and often expanding plaque. The incidence of EMPD is unknown, as there are only a few hundred cases reported in the literature.2
Extramammary Paget disease can occur primarily, arising in the epidermis at the sweat-gland level or from primitive epidermal basal cells, or secondarily due to pagetoid spread of malignant cells from an adjacent or contiguous underlying adnexal adenocarcinoma or visceral malignancy.2 While primary EMPD is not associated with an underlying adenocarcinoma, it may become invasive, infiltrate the dermis, or metastasize via the lymphatics.2 Secondary EMPD is associated with underlying malignancy most often originating in the gastrointestinal or genitourinary tracts.1,2
Currently, treatment of primary EMPD typically is surgical with wide local excision or Mohs micrographic surgery.1,2 However, margins often are positive, and the local recurrence rate is high (ie, 33%–66%).2,3 There are a variety of other therapies that have been reported in the literature, including radiation, topical chemotherapeutics (eg, imiquimod, 5-fluorouracil, bleomycin), photodynamic therapy, and CO2 laser ablation.1,3 To our knowledge, there are no randomized controlled trials that compare surgery with other treatment options for EMPD.
Despite recurrence of EMPD with involvement of the anal canal, our patient refused further surgical intervention, as it would have required anal resection and radiotherapy due to the potentially negative impact on sphincter function. While investigating minimally invasive treatment options, we found several citations in the literature highlighting positive response with imiquimod cream 5% in patients with vulvar and periscrotal EMPD.4,5 A large, systematic review that analyzed 63 cases of vulvar EMPD—nearly half of which were recurrences of a prior malignancy—reported a response rate of 52% to 80% following treatment with imiquimod.5 Almost 70% of patients achieved complete clearance while applying imiquimod 3 to 4 times weekly for a median of 4 months; however, little has been written about the effectiveness of topical imiquimod in EMPD. Knight et al6 reported the case of a 40-year-old woman with perianal EMPD who was treated with imiquimod 3 times weekly for 16 weeks. At the end of treatment, the patient was completely clear of disease both clinically and histologically on random biopsies of the perianal skin; however, the EMPD later recurred with lymph node metastasis 18 months after stopping treatment.6
Given the growing evidence demonstrating disease control of EMPD with topical imiquimod, we elected to utilize this agent in combination with oral cimetidine in our patient. Cimetidine, an H2 receptor antagonist, has been shown to have antineoplastic properties in a broad range of preclinical and clinical studies for a number of different malignancies.7 Four distinct mechanisms of action have been shown. Cimetidine, which blocks the histamine pathway, has been shown to have a direct antiproliferative action on cancer cells.7 Histamine has been associated with increased regulatory T-cell activity, decreased antigen-presenting activity of dendritic cells, reduced natural killer cell activity, and increased myeloid-derived suppressor cell activity, which create an immunosuppressive tumor microenvironment in the setting of cancer. By blocking histamine and thus reversing this immunosuppressive environment, cimetidine demonstrates immunomodulatory effects.7 Cimetidine also has demonstrated an inhibitory effect on cancer cell adhesion to endothelial cells, which is noted to be independent of histamine-blocking activity.7 Finally, an antiangiogenic action is attributed to blocking of the upregulation of vascular endothelial growth factor that is normally induced by histamine.7
Cimetidine’s antineoplastic properties, specifically in the setting of colorectal cancer,8 were particularly compelling given our patient’s EMPD involvement of the anal canal. The most impressive clinical trial data showed a dramatically increased survival rate for colorectal cancer patients treated with oral cimetidine (800 mg once daily) and oral 5-fluorouracil (200 mg once daily) for 1 year following curative resection. The cimetidine-treated group had a 10-year survival rate of 84.6% versus 49.8% for the 5-fluorouracil–only group.8
Conclusion
We present this case of recurrent perianal and anal EMPD treated successfully with imiquimod cream 5% and oral cimetidine to highlight a potential alternative treatment regimen for poor surgical candidates with EMPD.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. London, England: Elsevier Saunders; 2012.
- Lam C, Funaro D. Extramammary Paget’s disease: summary of current knowledge. Dermatol Clin. 2010;28:807-826.
- Vergati M, Filingeri V, Palmieri G, et al. Perianal Paget’s disease: a case report and literature review. Anticancer Res. 2012;32:4461-4465.
- Liau MM, Yang SS, Tan KB, et al. Topical imiquimod in the treatment of extramammary Paget’s disease: a 10 year retrospective analysis in an Asian tertiary centre. Dermatol Ther. 2016;29:459-462.
- Machida H, Moeini A, Roman LD, et al. Effects of imiquimod on vulvar Paget’s disease: a systematic review of literature. Gynecol Oncol. 2015;139:165-171.
- Knight SR, Proby C, Ziyaie D, et al. Extramammary Paget disease of the perianal region: the potential role of imiquimod in achieving disease control. J Surg Case Rep. 2016;8:1-3.
- Pantziarka P, Bouche G, Meheus L, et al. Repurposing drugs in oncology (ReDO)—cimetidine as an anti-cancer agent. Ecancermedicalscience. 2014;8:485.
- Matsumoto S, Imaeda Y, Umemoto S, et al. Cimetidine increases survival of colorectal cancer patients with high levels of sialyl Lewis-X and sialyl Lewis-A epitope expression on tumour cells. Br J Cancer. 2002;86:161-167.
Case Report
A 56-year-old woman with well-controlled hypertension, hyperlipidemia, and gastroesophageal reflux disease initially presented with itching and a rash in the perianal region of 1 year’s duration. She had been treated intermittently by her primary care physician over the past year for presumed hemorrhoids and a perianal fungal infection without improvement. Physical examination at the time of intitial presentation revealed a single, well-demarcated, scaly, pink plaque on the perianal area on the right buttock extending toward the anal canal (Figure 1).
Four years later, the patient returned with new symptoms of bleeding when wiping the perianal region, pruritus, and fecal urgency of 3 to 4 months’ duration. Physical examination revealed scaly patches on the anus that were suspicious for recurrence of EMPD. Biopsies from the anal margin and anal canal confirmed recurrent EMPD involving the anal canal. Repeat evaluation for internal malignancy was negative.
Given the involvement of the anal canal, repeat wide local excision would have required anal resection and would therefore have been functionally impairing. The patient refused further surgical intervention as well as radiotherapy. Rather, a novel 16-week immunomodulatory regimen involving imiquimod cream 5% cream and low-dose oral cimetidine was started. To address the anal involvement, the patient was instructed to lubricate glycerin suppositories with the imiquimod cream and insert intra-anally once weekly. Dosing was adjusted based on the patient’s inflammatory response and tolerability, as she did initially report some flulike symptoms with the first few weeks of treatment. For most of the 16-week course, she applied 250 mg of imiquimod cream 5% to the perianal area 3 times weekly and 250 mg into the anal canal once weekly. Oral cimetidine initially was dosed at 800 mg twice daily as tolerated, but due to stomach irritation, the patient self-reduced her intake to 800 mg 3 times weekly.
To determine treatment response, scouting biopsies of the anal margin and anal canal were obtained 4 weeks after treatment cessation and demonstrated no evidence of residual disease. The patient resumed topical imiquimod applied once weekly into the anal canal and around the anus for a planned prolonged course of at least 1 year. To reduce the risk of recurrence, the patient continued taking oral cimetidine 800 mg 3 times weekly. Recommended follow-up included annual anoscopy or colonoscopy, serum carcinoembryonic antigen evaluation, and regular clinical monitoring by the dermatology and colorectal surgery teams.
Six months after completing the combination therapy, she was seen by the dermatology department and remained clinically free of disease (Figure 4). Anoscopy examination by the colorectal surgery department 4 months later showed no clinical evidence of malignancy.
Comment
Extramammary Paget disease is a rare intraepithelial adenocarcinoma with a predilection for white females and an average age of onset of 50 to 80 years.1-3 The vulva, perianal region, scrotum, penis, and perineum are the most commonly affected sites.1-3 Clinically, EMPD presents as a chronic, well-demarcated, scaly, and often expanding plaque. The incidence of EMPD is unknown, as there are only a few hundred cases reported in the literature.2
Extramammary Paget disease can occur primarily, arising in the epidermis at the sweat-gland level or from primitive epidermal basal cells, or secondarily due to pagetoid spread of malignant cells from an adjacent or contiguous underlying adnexal adenocarcinoma or visceral malignancy.2 While primary EMPD is not associated with an underlying adenocarcinoma, it may become invasive, infiltrate the dermis, or metastasize via the lymphatics.2 Secondary EMPD is associated with underlying malignancy most often originating in the gastrointestinal or genitourinary tracts.1,2
Currently, treatment of primary EMPD typically is surgical with wide local excision or Mohs micrographic surgery.1,2 However, margins often are positive, and the local recurrence rate is high (ie, 33%–66%).2,3 There are a variety of other therapies that have been reported in the literature, including radiation, topical chemotherapeutics (eg, imiquimod, 5-fluorouracil, bleomycin), photodynamic therapy, and CO2 laser ablation.1,3 To our knowledge, there are no randomized controlled trials that compare surgery with other treatment options for EMPD.
Despite recurrence of EMPD with involvement of the anal canal, our patient refused further surgical intervention, as it would have required anal resection and radiotherapy due to the potentially negative impact on sphincter function. While investigating minimally invasive treatment options, we found several citations in the literature highlighting positive response with imiquimod cream 5% in patients with vulvar and periscrotal EMPD.4,5 A large, systematic review that analyzed 63 cases of vulvar EMPD—nearly half of which were recurrences of a prior malignancy—reported a response rate of 52% to 80% following treatment with imiquimod.5 Almost 70% of patients achieved complete clearance while applying imiquimod 3 to 4 times weekly for a median of 4 months; however, little has been written about the effectiveness of topical imiquimod in EMPD. Knight et al6 reported the case of a 40-year-old woman with perianal EMPD who was treated with imiquimod 3 times weekly for 16 weeks. At the end of treatment, the patient was completely clear of disease both clinically and histologically on random biopsies of the perianal skin; however, the EMPD later recurred with lymph node metastasis 18 months after stopping treatment.6
Given the growing evidence demonstrating disease control of EMPD with topical imiquimod, we elected to utilize this agent in combination with oral cimetidine in our patient. Cimetidine, an H2 receptor antagonist, has been shown to have antineoplastic properties in a broad range of preclinical and clinical studies for a number of different malignancies.7 Four distinct mechanisms of action have been shown. Cimetidine, which blocks the histamine pathway, has been shown to have a direct antiproliferative action on cancer cells.7 Histamine has been associated with increased regulatory T-cell activity, decreased antigen-presenting activity of dendritic cells, reduced natural killer cell activity, and increased myeloid-derived suppressor cell activity, which create an immunosuppressive tumor microenvironment in the setting of cancer. By blocking histamine and thus reversing this immunosuppressive environment, cimetidine demonstrates immunomodulatory effects.7 Cimetidine also has demonstrated an inhibitory effect on cancer cell adhesion to endothelial cells, which is noted to be independent of histamine-blocking activity.7 Finally, an antiangiogenic action is attributed to blocking of the upregulation of vascular endothelial growth factor that is normally induced by histamine.7
Cimetidine’s antineoplastic properties, specifically in the setting of colorectal cancer,8 were particularly compelling given our patient’s EMPD involvement of the anal canal. The most impressive clinical trial data showed a dramatically increased survival rate for colorectal cancer patients treated with oral cimetidine (800 mg once daily) and oral 5-fluorouracil (200 mg once daily) for 1 year following curative resection. The cimetidine-treated group had a 10-year survival rate of 84.6% versus 49.8% for the 5-fluorouracil–only group.8
Conclusion
We present this case of recurrent perianal and anal EMPD treated successfully with imiquimod cream 5% and oral cimetidine to highlight a potential alternative treatment regimen for poor surgical candidates with EMPD.
Case Report
A 56-year-old woman with well-controlled hypertension, hyperlipidemia, and gastroesophageal reflux disease initially presented with itching and a rash in the perianal region of 1 year’s duration. She had been treated intermittently by her primary care physician over the past year for presumed hemorrhoids and a perianal fungal infection without improvement. Physical examination at the time of intitial presentation revealed a single, well-demarcated, scaly, pink plaque on the perianal area on the right buttock extending toward the anal canal (Figure 1).
Four years later, the patient returned with new symptoms of bleeding when wiping the perianal region, pruritus, and fecal urgency of 3 to 4 months’ duration. Physical examination revealed scaly patches on the anus that were suspicious for recurrence of EMPD. Biopsies from the anal margin and anal canal confirmed recurrent EMPD involving the anal canal. Repeat evaluation for internal malignancy was negative.
Given the involvement of the anal canal, repeat wide local excision would have required anal resection and would therefore have been functionally impairing. The patient refused further surgical intervention as well as radiotherapy. Rather, a novel 16-week immunomodulatory regimen involving imiquimod cream 5% cream and low-dose oral cimetidine was started. To address the anal involvement, the patient was instructed to lubricate glycerin suppositories with the imiquimod cream and insert intra-anally once weekly. Dosing was adjusted based on the patient’s inflammatory response and tolerability, as she did initially report some flulike symptoms with the first few weeks of treatment. For most of the 16-week course, she applied 250 mg of imiquimod cream 5% to the perianal area 3 times weekly and 250 mg into the anal canal once weekly. Oral cimetidine initially was dosed at 800 mg twice daily as tolerated, but due to stomach irritation, the patient self-reduced her intake to 800 mg 3 times weekly.
To determine treatment response, scouting biopsies of the anal margin and anal canal were obtained 4 weeks after treatment cessation and demonstrated no evidence of residual disease. The patient resumed topical imiquimod applied once weekly into the anal canal and around the anus for a planned prolonged course of at least 1 year. To reduce the risk of recurrence, the patient continued taking oral cimetidine 800 mg 3 times weekly. Recommended follow-up included annual anoscopy or colonoscopy, serum carcinoembryonic antigen evaluation, and regular clinical monitoring by the dermatology and colorectal surgery teams.
Six months after completing the combination therapy, she was seen by the dermatology department and remained clinically free of disease (Figure 4). Anoscopy examination by the colorectal surgery department 4 months later showed no clinical evidence of malignancy.
Comment
Extramammary Paget disease is a rare intraepithelial adenocarcinoma with a predilection for white females and an average age of onset of 50 to 80 years.1-3 The vulva, perianal region, scrotum, penis, and perineum are the most commonly affected sites.1-3 Clinically, EMPD presents as a chronic, well-demarcated, scaly, and often expanding plaque. The incidence of EMPD is unknown, as there are only a few hundred cases reported in the literature.2
Extramammary Paget disease can occur primarily, arising in the epidermis at the sweat-gland level or from primitive epidermal basal cells, or secondarily due to pagetoid spread of malignant cells from an adjacent or contiguous underlying adnexal adenocarcinoma or visceral malignancy.2 While primary EMPD is not associated with an underlying adenocarcinoma, it may become invasive, infiltrate the dermis, or metastasize via the lymphatics.2 Secondary EMPD is associated with underlying malignancy most often originating in the gastrointestinal or genitourinary tracts.1,2
Currently, treatment of primary EMPD typically is surgical with wide local excision or Mohs micrographic surgery.1,2 However, margins often are positive, and the local recurrence rate is high (ie, 33%–66%).2,3 There are a variety of other therapies that have been reported in the literature, including radiation, topical chemotherapeutics (eg, imiquimod, 5-fluorouracil, bleomycin), photodynamic therapy, and CO2 laser ablation.1,3 To our knowledge, there are no randomized controlled trials that compare surgery with other treatment options for EMPD.
Despite recurrence of EMPD with involvement of the anal canal, our patient refused further surgical intervention, as it would have required anal resection and radiotherapy due to the potentially negative impact on sphincter function. While investigating minimally invasive treatment options, we found several citations in the literature highlighting positive response with imiquimod cream 5% in patients with vulvar and periscrotal EMPD.4,5 A large, systematic review that analyzed 63 cases of vulvar EMPD—nearly half of which were recurrences of a prior malignancy—reported a response rate of 52% to 80% following treatment with imiquimod.5 Almost 70% of patients achieved complete clearance while applying imiquimod 3 to 4 times weekly for a median of 4 months; however, little has been written about the effectiveness of topical imiquimod in EMPD. Knight et al6 reported the case of a 40-year-old woman with perianal EMPD who was treated with imiquimod 3 times weekly for 16 weeks. At the end of treatment, the patient was completely clear of disease both clinically and histologically on random biopsies of the perianal skin; however, the EMPD later recurred with lymph node metastasis 18 months after stopping treatment.6
Given the growing evidence demonstrating disease control of EMPD with topical imiquimod, we elected to utilize this agent in combination with oral cimetidine in our patient. Cimetidine, an H2 receptor antagonist, has been shown to have antineoplastic properties in a broad range of preclinical and clinical studies for a number of different malignancies.7 Four distinct mechanisms of action have been shown. Cimetidine, which blocks the histamine pathway, has been shown to have a direct antiproliferative action on cancer cells.7 Histamine has been associated with increased regulatory T-cell activity, decreased antigen-presenting activity of dendritic cells, reduced natural killer cell activity, and increased myeloid-derived suppressor cell activity, which create an immunosuppressive tumor microenvironment in the setting of cancer. By blocking histamine and thus reversing this immunosuppressive environment, cimetidine demonstrates immunomodulatory effects.7 Cimetidine also has demonstrated an inhibitory effect on cancer cell adhesion to endothelial cells, which is noted to be independent of histamine-blocking activity.7 Finally, an antiangiogenic action is attributed to blocking of the upregulation of vascular endothelial growth factor that is normally induced by histamine.7
Cimetidine’s antineoplastic properties, specifically in the setting of colorectal cancer,8 were particularly compelling given our patient’s EMPD involvement of the anal canal. The most impressive clinical trial data showed a dramatically increased survival rate for colorectal cancer patients treated with oral cimetidine (800 mg once daily) and oral 5-fluorouracil (200 mg once daily) for 1 year following curative resection. The cimetidine-treated group had a 10-year survival rate of 84.6% versus 49.8% for the 5-fluorouracil–only group.8
Conclusion
We present this case of recurrent perianal and anal EMPD treated successfully with imiquimod cream 5% and oral cimetidine to highlight a potential alternative treatment regimen for poor surgical candidates with EMPD.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. London, England: Elsevier Saunders; 2012.
- Lam C, Funaro D. Extramammary Paget’s disease: summary of current knowledge. Dermatol Clin. 2010;28:807-826.
- Vergati M, Filingeri V, Palmieri G, et al. Perianal Paget’s disease: a case report and literature review. Anticancer Res. 2012;32:4461-4465.
- Liau MM, Yang SS, Tan KB, et al. Topical imiquimod in the treatment of extramammary Paget’s disease: a 10 year retrospective analysis in an Asian tertiary centre. Dermatol Ther. 2016;29:459-462.
- Machida H, Moeini A, Roman LD, et al. Effects of imiquimod on vulvar Paget’s disease: a systematic review of literature. Gynecol Oncol. 2015;139:165-171.
- Knight SR, Proby C, Ziyaie D, et al. Extramammary Paget disease of the perianal region: the potential role of imiquimod in achieving disease control. J Surg Case Rep. 2016;8:1-3.
- Pantziarka P, Bouche G, Meheus L, et al. Repurposing drugs in oncology (ReDO)—cimetidine as an anti-cancer agent. Ecancermedicalscience. 2014;8:485.
- Matsumoto S, Imaeda Y, Umemoto S, et al. Cimetidine increases survival of colorectal cancer patients with high levels of sialyl Lewis-X and sialyl Lewis-A epitope expression on tumour cells. Br J Cancer. 2002;86:161-167.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. London, England: Elsevier Saunders; 2012.
- Lam C, Funaro D. Extramammary Paget’s disease: summary of current knowledge. Dermatol Clin. 2010;28:807-826.
- Vergati M, Filingeri V, Palmieri G, et al. Perianal Paget’s disease: a case report and literature review. Anticancer Res. 2012;32:4461-4465.
- Liau MM, Yang SS, Tan KB, et al. Topical imiquimod in the treatment of extramammary Paget’s disease: a 10 year retrospective analysis in an Asian tertiary centre. Dermatol Ther. 2016;29:459-462.
- Machida H, Moeini A, Roman LD, et al. Effects of imiquimod on vulvar Paget’s disease: a systematic review of literature. Gynecol Oncol. 2015;139:165-171.
- Knight SR, Proby C, Ziyaie D, et al. Extramammary Paget disease of the perianal region: the potential role of imiquimod in achieving disease control. J Surg Case Rep. 2016;8:1-3.
- Pantziarka P, Bouche G, Meheus L, et al. Repurposing drugs in oncology (ReDO)—cimetidine as an anti-cancer agent. Ecancermedicalscience. 2014;8:485.
- Matsumoto S, Imaeda Y, Umemoto S, et al. Cimetidine increases survival of colorectal cancer patients with high levels of sialyl Lewis-X and sialyl Lewis-A epitope expression on tumour cells. Br J Cancer. 2002;86:161-167.
Resident Pearls
- Topical imiquimod cream 5% and oral cimetidine can be a potential alternative treatment regimen for poor surgical candidates with perianal extramammary Paget disease (EMPD).
- Its antineoplastic and immunomodulatory properties may suggest a role for oral cimetidine as an adjuvant therapy in the treatment of perianal EMPD.

Painful Mouth Ulcers
The Diagnosis: Paraneoplastic Pemphigus
A workup for infectious organisms and vasculitis was negative. The patient reported unintentional weight loss despite taking oral steroids prescribed by her pulmonologist for severe obstructive lung disease that appeared to develop around the same time as the mouth ulcers.
Computed tomography of the abdomen revealed an 8.1-cm pelvic mass that a subsequent biopsy revealed to be a follicular dendritic cell sarcoma. Biopsies of the mouth ulcers showed a mildly hyperplastic mucosa with acantholysis and interface change with dyskeratosis. Direct immunofluorescence of the perilesional mucosa showed IgG and complement C3 in an intercellular distribution (Figure 1). The pathologic findings were consistent with a diagnosis of paraneoplastic pemphigus (PNP). Serologic testing via enzyme-linked immunosorbent assay, immunoblotting, and indirect immunofluorescence were not performed. The patient died within a few months after the initial presentation from bronchiolitis obliterans, a potentially fatal complication of PNP.
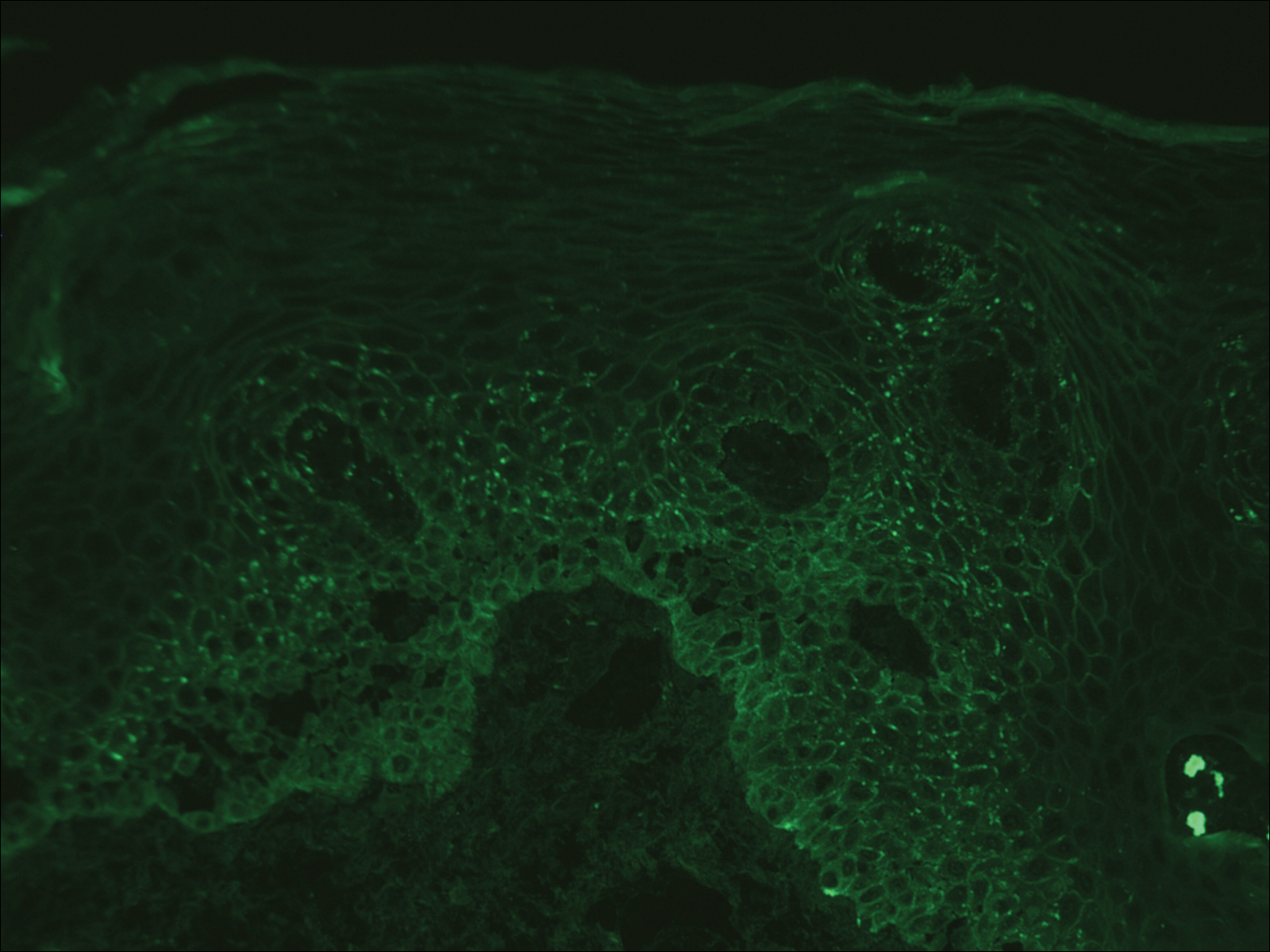
Paraneoplastic pemphigus is an autoimmune blistering disease associated with neoplasia, particularly lymphoproliferative disorders and thymoma.1 Oral mucosal erosions and crusting along the lips commonly is seen along with cutaneous involvement. The main histologic features are interface changes with dyskeratosis and a lichenoid infiltrate and variable acantholysis.2
Direct immunofluorescence of perilesional skin classically shows IgG and complement C3 in an intercellular distribution, usually in a granular or linear distribution along the basement membrane. This same pattern of direct immunofluorescence is seen in pemphigus erythematosus; however, pemphigus erythematosus is clinically distinct from PNP, lacking mucosal involvement and affecting the face and/or seborrheic areas with an appearance more similar to seborrheic dermatitis or lupus erythematosus, depending on the patient.3 Indirect immunofluorescence with rat bladder epithelium typically is positive in PNP and can be a helpful feature in distinguishing PNP from other autoimmune blistering diseases (eg, pemphigus erythematosus, pemphigus vulgaris, pemphigus foliaceus).2
Immunoblotting assays via serology often detect numerous antigens in patients with PNP, including but not limited to plectin, desmoplakin, bullous pemphigoid antigens, envoplakin, desmoplakin II, and desmogleins 1 and 3.4 Some of these autoantibodies have been identified in tumors associated with paraneoplastic pemphigus, particularly Castleman disease and follicular dendritic cell sarcoma.
Acute graft-versus-host disease (GVHD) can have a similar histologic appearance to PNP with prominent dyskeratosis and characteristically shows satellite cell necrosis consisting of dyskeratosis with surrounding lymphocytes (Figure 2). Unlike PNP, acantholysis is not a feature of GVHD. Direct immunofluorescence typically is negative; however, nonspecific IgM and complement C3 deposition at the dermoepidermal junction and around the superficial vasculature has been reported in 39% of cases.5 Early chronic GVHD often shows retained lichenoid interface change, but late chronic GVHD has a sclerodermoid morphology that is easily distinguished histologically from PNP. Patients also have a history of either a bone marrow or solid organ transplant.6

Lichen planus also shows interface change with dyskeratosis and a lichenoid infiltrate; however, acantholysis typically is not seen and, there often is prominent hypergranulosis (Figure 3). Mucosal lesions often show more subtle features with decreased hyperkeratosis, more subtle hypergranulosis, and decreased interface change with plasma cells in the inflammatory infiltrate.6 Additionally, direct immunofluorescence is either negative or shows IgM-positive colloid bodies and/or an irregular band of fibrinogen at the dermoepidermal junction. The characteristic intercellular and granular/linear IgG positivity at the dermoepidermal junction of PNP is not seen.
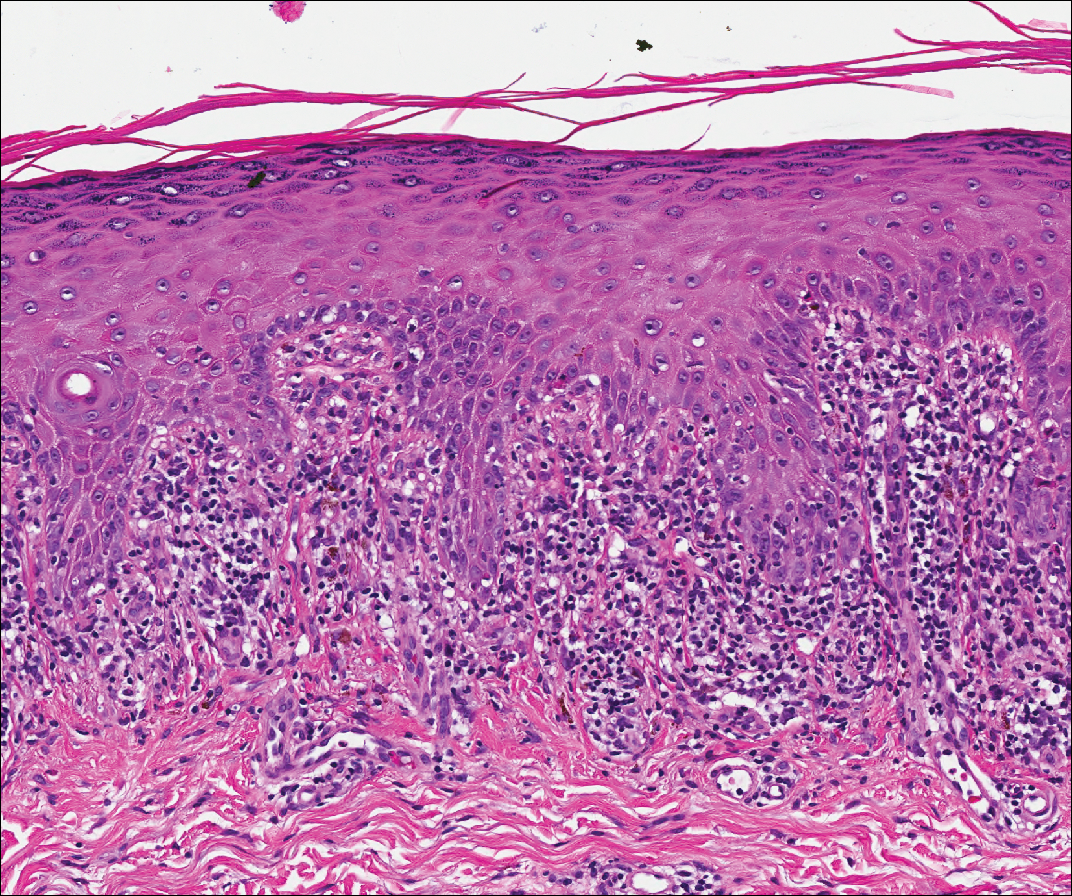
Lupus erythematosus is an interface dermatitis with histologic features that can overlap with PNP, in addition to positive direct immunofluorescence, which has been seen in 50% to 94% of cases and can vary depending on previous steroid treatment and timing of the biopsy in the disease process.7 Unlike PNP, lupus erythematosus has a full-house pattern on direct immunofluorescence with IgG, IgM, IgA, and complement C3 deposition in a granular pattern at the dermoepidermal junction. While PNP also typically shows granular deposition of IgG and complement C3 at the dermoepidermal junction, there also is intercellular positivity without a full-house pattern. While both conditions show interface change, histologic features that distinguish lupus erythematosus from PNP are a superficial and deep perivascular lymphocytic infiltrate, basement membrane thickening, follicular plugging, and increased dermal mucin (Figure 4). Subacute lupus erythematosus and discoid lupus erythematosus can have similar histologic features, and definitive distinction on biopsy is not always possible; however, subacute lupus erythematosus shows milder follicular plugging and milder to absent basement membrane thickening, and the inflammatory infiltrate typically is sparser than in discoid lupus erythematosus.7 Subacute lupus erythematosus also can show anti-Ro/Sjögren syndrome antigen A antibodies, which typically are not seen in discoid lupus eythematosus.8
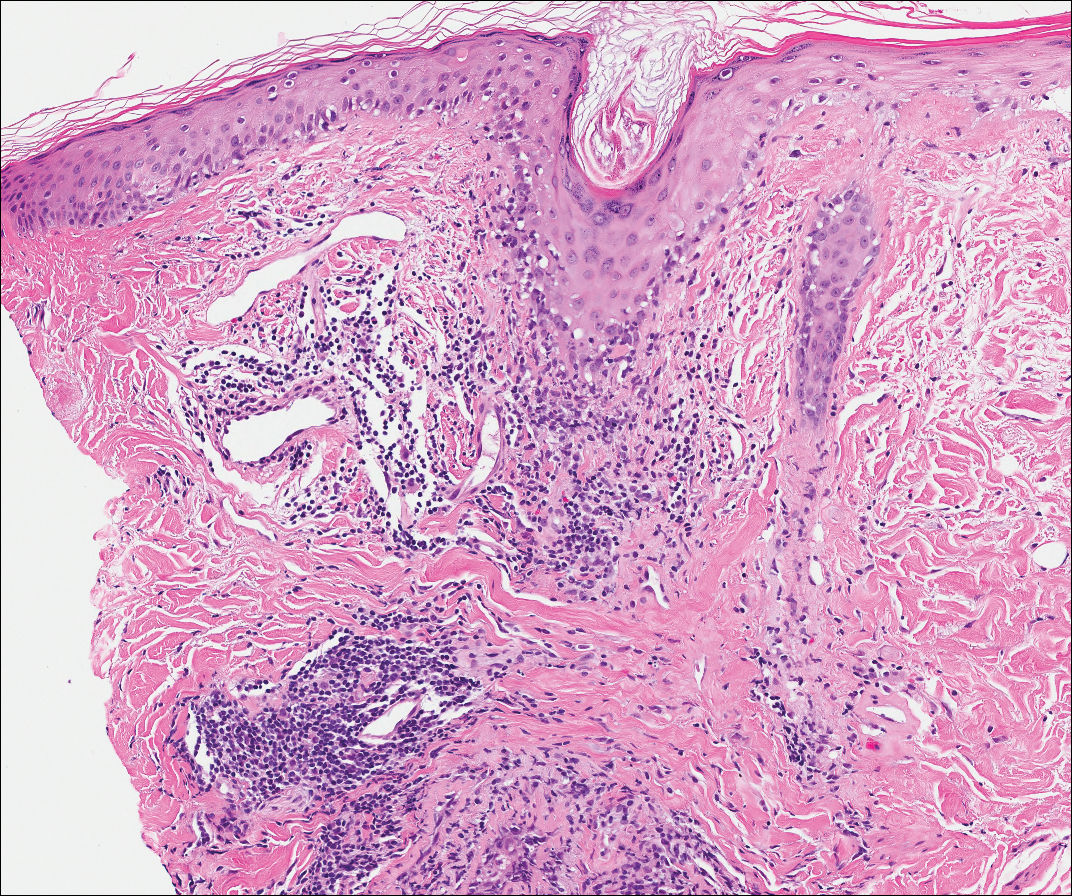
Stevens-Johnson syndrome (SJS) is on a spectrum with toxic epidermal necrolysis, with SJS involving less than 10% and toxic epidermal necrolysis involving 30% or more of the body surface area.5 Erythema multiforme also is on the histologic spectrum of SJS and toxic epidermal necrolysis; however, erythema multiforme typically is more inflammatory than SJS and toxic epidermal necrolysis. Stevens-Johnson syndrome typically affects older adults and shows both cutaneous and mucosal involvement; however, isolated mucosal involvement can be seen in children.5 Drugs, particularly sulfonamide antibiotics, usually are implicated as causative agents, but infections from Mycoplasma and other pathogens also may be the cause. There is notable clinical (with a combination of mucosal and cutaneous lesions) as well as histologic overlap between SJS and PNP. The density of the lichenoid infiltrate is variable, with dyskeratosis, basal cell hydropic degeneration, and occasional formation of subepidermal clefts (Figure 5). Unlike PNP, acantholysis is not a characteristic feature of SJS, and direct immunofluorescence generally is negative.
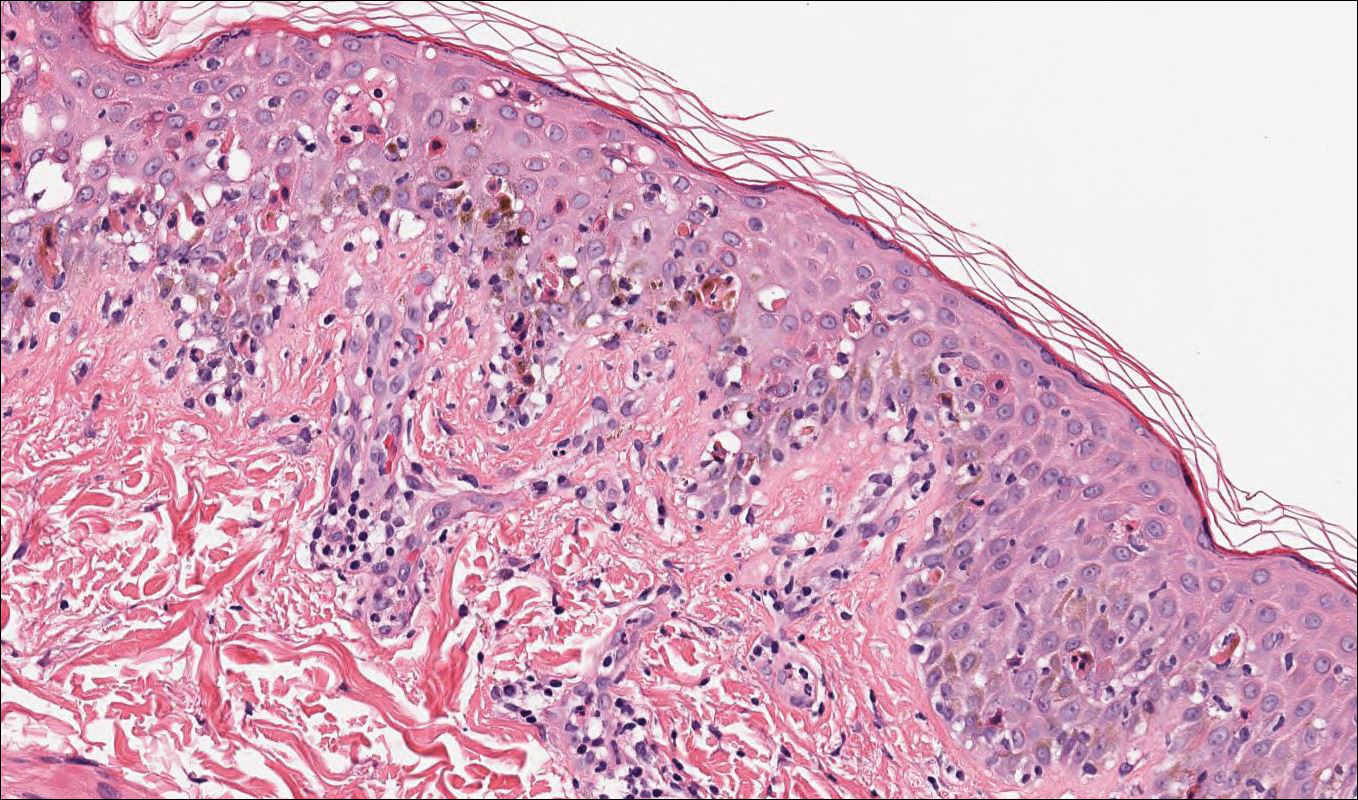
- Camisa C, Helm TN. Paraneoplastic pemphigus is a distinct neoplasia-induced autoimmune disease. Arch Dermatol. 1993;129:883-886.
- Joly P, Richard C, Gilbert D, et al. Sensitivity and specificity of clinical, histologic, and immunologic features in the diagnosis of paraneoplastic pemphigus. J Am Acad Dermatol. 2000;43:619-626.
- Calonje E, Brenn T, Lazar A. Acantholytic disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:151-179.
- Billet ES, Grando AS, Pittelkow MR. Paraneoplastic autoimmune multiorgan syndrome: review of the literature and support for a cytotoxic role in pathogenesis. Autoimmunity. 2006;36:617-630.
- Calonje E, Brenn T, Lazar A. Lichenoid and interface dermatitis. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:219-255.
- Billings SD, Cotton J. Inflammatory Dermatopathology: A Pathologist's Survival Guide. 2nd ed. Switzerland: Springer International Publishing; 2016.
- Calonje E, Brenn T, Lazar A. Idiopathic connective tissue disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:711-757.
- Lee LA, Roberts CM, Frank MB, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262-1268.
The Diagnosis: Paraneoplastic Pemphigus
A workup for infectious organisms and vasculitis was negative. The patient reported unintentional weight loss despite taking oral steroids prescribed by her pulmonologist for severe obstructive lung disease that appeared to develop around the same time as the mouth ulcers.
Computed tomography of the abdomen revealed an 8.1-cm pelvic mass that a subsequent biopsy revealed to be a follicular dendritic cell sarcoma. Biopsies of the mouth ulcers showed a mildly hyperplastic mucosa with acantholysis and interface change with dyskeratosis. Direct immunofluorescence of the perilesional mucosa showed IgG and complement C3 in an intercellular distribution (Figure 1). The pathologic findings were consistent with a diagnosis of paraneoplastic pemphigus (PNP). Serologic testing via enzyme-linked immunosorbent assay, immunoblotting, and indirect immunofluorescence were not performed. The patient died within a few months after the initial presentation from bronchiolitis obliterans, a potentially fatal complication of PNP.
Paraneoplastic pemphigus is an autoimmune blistering disease associated with neoplasia, particularly lymphoproliferative disorders and thymoma.1 Oral mucosal erosions and crusting along the lips commonly is seen along with cutaneous involvement. The main histologic features are interface changes with dyskeratosis and a lichenoid infiltrate and variable acantholysis.2
Direct immunofluorescence of perilesional skin classically shows IgG and complement C3 in an intercellular distribution, usually in a granular or linear distribution along the basement membrane. This same pattern of direct immunofluorescence is seen in pemphigus erythematosus; however, pemphigus erythematosus is clinically distinct from PNP, lacking mucosal involvement and affecting the face and/or seborrheic areas with an appearance more similar to seborrheic dermatitis or lupus erythematosus, depending on the patient.3 Indirect immunofluorescence with rat bladder epithelium typically is positive in PNP and can be a helpful feature in distinguishing PNP from other autoimmune blistering diseases (eg, pemphigus erythematosus, pemphigus vulgaris, pemphigus foliaceus).2
Immunoblotting assays via serology often detect numerous antigens in patients with PNP, including but not limited to plectin, desmoplakin, bullous pemphigoid antigens, envoplakin, desmoplakin II, and desmogleins 1 and 3.4 Some of these autoantibodies have been identified in tumors associated with paraneoplastic pemphigus, particularly Castleman disease and follicular dendritic cell sarcoma.
Acute graft-versus-host disease (GVHD) can have a similar histologic appearance to PNP with prominent dyskeratosis and characteristically shows satellite cell necrosis consisting of dyskeratosis with surrounding lymphocytes (Figure 2). Unlike PNP, acantholysis is not a feature of GVHD. Direct immunofluorescence typically is negative; however, nonspecific IgM and complement C3 deposition at the dermoepidermal junction and around the superficial vasculature has been reported in 39% of cases.5 Early chronic GVHD often shows retained lichenoid interface change, but late chronic GVHD has a sclerodermoid morphology that is easily distinguished histologically from PNP. Patients also have a history of either a bone marrow or solid organ transplant.6
Lichen planus also shows interface change with dyskeratosis and a lichenoid infiltrate; however, acantholysis typically is not seen and, there often is prominent hypergranulosis (Figure 3). Mucosal lesions often show more subtle features with decreased hyperkeratosis, more subtle hypergranulosis, and decreased interface change with plasma cells in the inflammatory infiltrate.6 Additionally, direct immunofluorescence is either negative or shows IgM-positive colloid bodies and/or an irregular band of fibrinogen at the dermoepidermal junction. The characteristic intercellular and granular/linear IgG positivity at the dermoepidermal junction of PNP is not seen.
Lupus erythematosus is an interface dermatitis with histologic features that can overlap with PNP, in addition to positive direct immunofluorescence, which has been seen in 50% to 94% of cases and can vary depending on previous steroid treatment and timing of the biopsy in the disease process.7 Unlike PNP, lupus erythematosus has a full-house pattern on direct immunofluorescence with IgG, IgM, IgA, and complement C3 deposition in a granular pattern at the dermoepidermal junction. While PNP also typically shows granular deposition of IgG and complement C3 at the dermoepidermal junction, there also is intercellular positivity without a full-house pattern. While both conditions show interface change, histologic features that distinguish lupus erythematosus from PNP are a superficial and deep perivascular lymphocytic infiltrate, basement membrane thickening, follicular plugging, and increased dermal mucin (Figure 4). Subacute lupus erythematosus and discoid lupus erythematosus can have similar histologic features, and definitive distinction on biopsy is not always possible; however, subacute lupus erythematosus shows milder follicular plugging and milder to absent basement membrane thickening, and the inflammatory infiltrate typically is sparser than in discoid lupus erythematosus.7 Subacute lupus erythematosus also can show anti-Ro/Sjögren syndrome antigen A antibodies, which typically are not seen in discoid lupus eythematosus.8
Stevens-Johnson syndrome (SJS) is on a spectrum with toxic epidermal necrolysis, with SJS involving less than 10% and toxic epidermal necrolysis involving 30% or more of the body surface area.5 Erythema multiforme also is on the histologic spectrum of SJS and toxic epidermal necrolysis; however, erythema multiforme typically is more inflammatory than SJS and toxic epidermal necrolysis. Stevens-Johnson syndrome typically affects older adults and shows both cutaneous and mucosal involvement; however, isolated mucosal involvement can be seen in children.5 Drugs, particularly sulfonamide antibiotics, usually are implicated as causative agents, but infections from Mycoplasma and other pathogens also may be the cause. There is notable clinical (with a combination of mucosal and cutaneous lesions) as well as histologic overlap between SJS and PNP. The density of the lichenoid infiltrate is variable, with dyskeratosis, basal cell hydropic degeneration, and occasional formation of subepidermal clefts (Figure 5). Unlike PNP, acantholysis is not a characteristic feature of SJS, and direct immunofluorescence generally is negative.
The Diagnosis: Paraneoplastic Pemphigus
A workup for infectious organisms and vasculitis was negative. The patient reported unintentional weight loss despite taking oral steroids prescribed by her pulmonologist for severe obstructive lung disease that appeared to develop around the same time as the mouth ulcers.
Computed tomography of the abdomen revealed an 8.1-cm pelvic mass that a subsequent biopsy revealed to be a follicular dendritic cell sarcoma. Biopsies of the mouth ulcers showed a mildly hyperplastic mucosa with acantholysis and interface change with dyskeratosis. Direct immunofluorescence of the perilesional mucosa showed IgG and complement C3 in an intercellular distribution (Figure 1). The pathologic findings were consistent with a diagnosis of paraneoplastic pemphigus (PNP). Serologic testing via enzyme-linked immunosorbent assay, immunoblotting, and indirect immunofluorescence were not performed. The patient died within a few months after the initial presentation from bronchiolitis obliterans, a potentially fatal complication of PNP.
Paraneoplastic pemphigus is an autoimmune blistering disease associated with neoplasia, particularly lymphoproliferative disorders and thymoma.1 Oral mucosal erosions and crusting along the lips commonly is seen along with cutaneous involvement. The main histologic features are interface changes with dyskeratosis and a lichenoid infiltrate and variable acantholysis.2
Direct immunofluorescence of perilesional skin classically shows IgG and complement C3 in an intercellular distribution, usually in a granular or linear distribution along the basement membrane. This same pattern of direct immunofluorescence is seen in pemphigus erythematosus; however, pemphigus erythematosus is clinically distinct from PNP, lacking mucosal involvement and affecting the face and/or seborrheic areas with an appearance more similar to seborrheic dermatitis or lupus erythematosus, depending on the patient.3 Indirect immunofluorescence with rat bladder epithelium typically is positive in PNP and can be a helpful feature in distinguishing PNP from other autoimmune blistering diseases (eg, pemphigus erythematosus, pemphigus vulgaris, pemphigus foliaceus).2
Immunoblotting assays via serology often detect numerous antigens in patients with PNP, including but not limited to plectin, desmoplakin, bullous pemphigoid antigens, envoplakin, desmoplakin II, and desmogleins 1 and 3.4 Some of these autoantibodies have been identified in tumors associated with paraneoplastic pemphigus, particularly Castleman disease and follicular dendritic cell sarcoma.
Acute graft-versus-host disease (GVHD) can have a similar histologic appearance to PNP with prominent dyskeratosis and characteristically shows satellite cell necrosis consisting of dyskeratosis with surrounding lymphocytes (Figure 2). Unlike PNP, acantholysis is not a feature of GVHD. Direct immunofluorescence typically is negative; however, nonspecific IgM and complement C3 deposition at the dermoepidermal junction and around the superficial vasculature has been reported in 39% of cases.5 Early chronic GVHD often shows retained lichenoid interface change, but late chronic GVHD has a sclerodermoid morphology that is easily distinguished histologically from PNP. Patients also have a history of either a bone marrow or solid organ transplant.6
Lichen planus also shows interface change with dyskeratosis and a lichenoid infiltrate; however, acantholysis typically is not seen and, there often is prominent hypergranulosis (Figure 3). Mucosal lesions often show more subtle features with decreased hyperkeratosis, more subtle hypergranulosis, and decreased interface change with plasma cells in the inflammatory infiltrate.6 Additionally, direct immunofluorescence is either negative or shows IgM-positive colloid bodies and/or an irregular band of fibrinogen at the dermoepidermal junction. The characteristic intercellular and granular/linear IgG positivity at the dermoepidermal junction of PNP is not seen.
Lupus erythematosus is an interface dermatitis with histologic features that can overlap with PNP, in addition to positive direct immunofluorescence, which has been seen in 50% to 94% of cases and can vary depending on previous steroid treatment and timing of the biopsy in the disease process.7 Unlike PNP, lupus erythematosus has a full-house pattern on direct immunofluorescence with IgG, IgM, IgA, and complement C3 deposition in a granular pattern at the dermoepidermal junction. While PNP also typically shows granular deposition of IgG and complement C3 at the dermoepidermal junction, there also is intercellular positivity without a full-house pattern. While both conditions show interface change, histologic features that distinguish lupus erythematosus from PNP are a superficial and deep perivascular lymphocytic infiltrate, basement membrane thickening, follicular plugging, and increased dermal mucin (Figure 4). Subacute lupus erythematosus and discoid lupus erythematosus can have similar histologic features, and definitive distinction on biopsy is not always possible; however, subacute lupus erythematosus shows milder follicular plugging and milder to absent basement membrane thickening, and the inflammatory infiltrate typically is sparser than in discoid lupus erythematosus.7 Subacute lupus erythematosus also can show anti-Ro/Sjögren syndrome antigen A antibodies, which typically are not seen in discoid lupus eythematosus.8
Stevens-Johnson syndrome (SJS) is on a spectrum with toxic epidermal necrolysis, with SJS involving less than 10% and toxic epidermal necrolysis involving 30% or more of the body surface area.5 Erythema multiforme also is on the histologic spectrum of SJS and toxic epidermal necrolysis; however, erythema multiforme typically is more inflammatory than SJS and toxic epidermal necrolysis. Stevens-Johnson syndrome typically affects older adults and shows both cutaneous and mucosal involvement; however, isolated mucosal involvement can be seen in children.5 Drugs, particularly sulfonamide antibiotics, usually are implicated as causative agents, but infections from Mycoplasma and other pathogens also may be the cause. There is notable clinical (with a combination of mucosal and cutaneous lesions) as well as histologic overlap between SJS and PNP. The density of the lichenoid infiltrate is variable, with dyskeratosis, basal cell hydropic degeneration, and occasional formation of subepidermal clefts (Figure 5). Unlike PNP, acantholysis is not a characteristic feature of SJS, and direct immunofluorescence generally is negative.
- Camisa C, Helm TN. Paraneoplastic pemphigus is a distinct neoplasia-induced autoimmune disease. Arch Dermatol. 1993;129:883-886.
- Joly P, Richard C, Gilbert D, et al. Sensitivity and specificity of clinical, histologic, and immunologic features in the diagnosis of paraneoplastic pemphigus. J Am Acad Dermatol. 2000;43:619-626.
- Calonje E, Brenn T, Lazar A. Acantholytic disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:151-179.
- Billet ES, Grando AS, Pittelkow MR. Paraneoplastic autoimmune multiorgan syndrome: review of the literature and support for a cytotoxic role in pathogenesis. Autoimmunity. 2006;36:617-630.
- Calonje E, Brenn T, Lazar A. Lichenoid and interface dermatitis. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:219-255.
- Billings SD, Cotton J. Inflammatory Dermatopathology: A Pathologist's Survival Guide. 2nd ed. Switzerland: Springer International Publishing; 2016.
- Calonje E, Brenn T, Lazar A. Idiopathic connective tissue disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:711-757.
- Lee LA, Roberts CM, Frank MB, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262-1268.
- Camisa C, Helm TN. Paraneoplastic pemphigus is a distinct neoplasia-induced autoimmune disease. Arch Dermatol. 1993;129:883-886.
- Joly P, Richard C, Gilbert D, et al. Sensitivity and specificity of clinical, histologic, and immunologic features in the diagnosis of paraneoplastic pemphigus. J Am Acad Dermatol. 2000;43:619-626.
- Calonje E, Brenn T, Lazar A. Acantholytic disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:151-179.
- Billet ES, Grando AS, Pittelkow MR. Paraneoplastic autoimmune multiorgan syndrome: review of the literature and support for a cytotoxic role in pathogenesis. Autoimmunity. 2006;36:617-630.
- Calonje E, Brenn T, Lazar A. Lichenoid and interface dermatitis. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:219-255.
- Billings SD, Cotton J. Inflammatory Dermatopathology: A Pathologist's Survival Guide. 2nd ed. Switzerland: Springer International Publishing; 2016.
- Calonje E, Brenn T, Lazar A. Idiopathic connective tissue disorders. McKee's Pathology of the Skin With Clinical Correlations. 4th ed. Philadelphia, PA: Elsevier; 2011:711-757.
- Lee LA, Roberts CM, Frank MB, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262-1268.
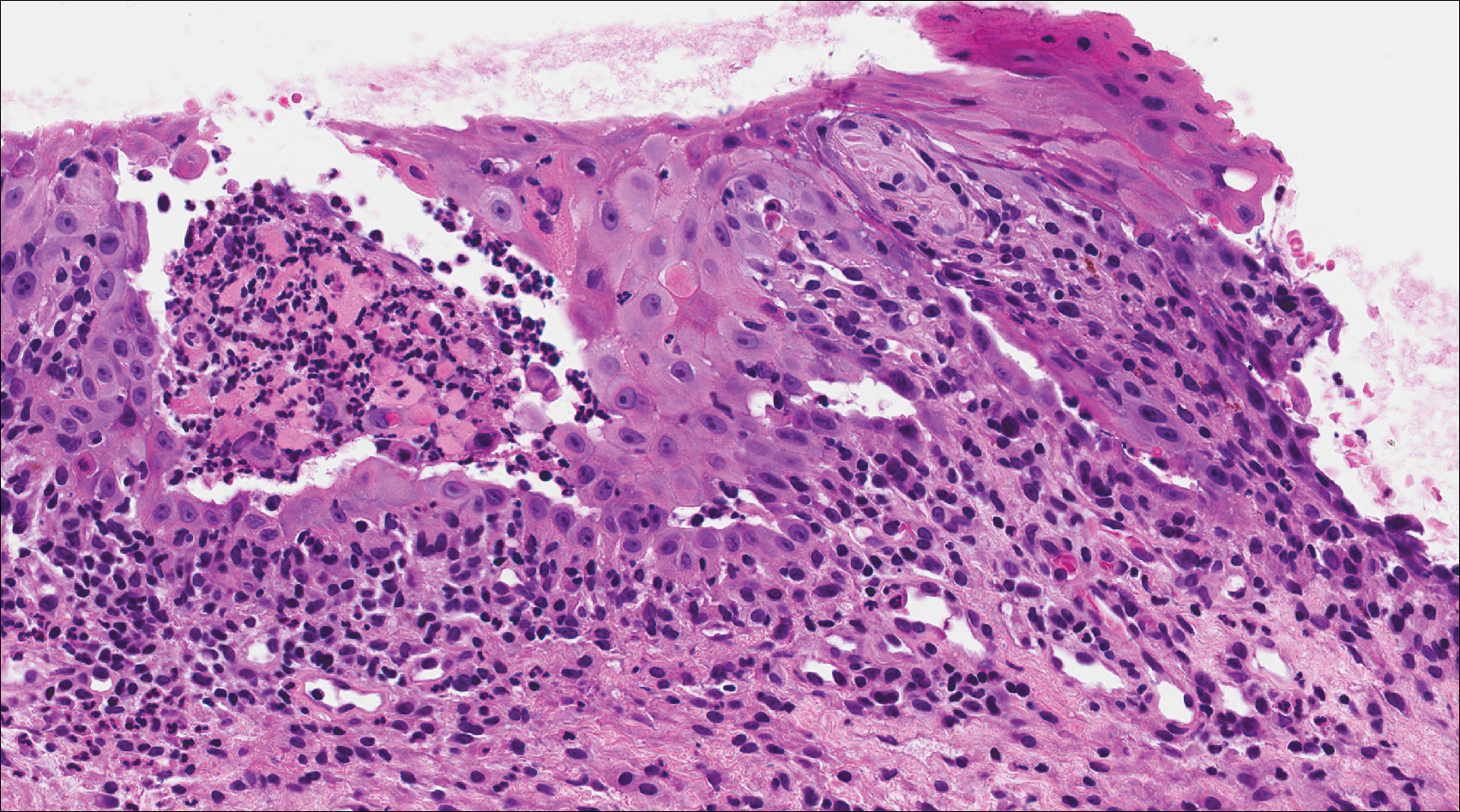
A 41-year-old woman presented with painful ulcers on the oral mucosa of 2 months' duration that were unresponsive to treatment with acyclovir. She had been diagnosed with a pelvic tumor a few weeks prior to the development of the mouth ulcers. Direct immunofluorescence of the perilesional mucosa showed positive IgG and complement C3 with an intercellular distribution. A biopsy of an oral lesion was performed.
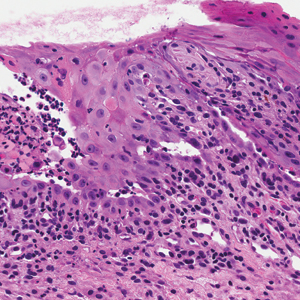
Intralymphatic Histiocytosis Treated With Intralesional Triamcinolone Acetonide and Pressure Bandage
Intralymphatic histiocytosis was first described in 1994.1 To date, at least 70 cases have been reported in the English-language literature, the majority being associated with systemic or local inflammatory conditions such as rheumatoid arthritis (RA), malignancy, and metal prostheses. The remaining cases arose independent of any detectable disease process.2 The clinical lesion localizes to areas around surgical scars or inflamed joints and generally presents with erythematous livedoid papules and plaques. Because of its rarity, pathologists and clinicians may be unfamiliar with this entity, leading to delayed or missed diagnoses.
Although the pathogenesis of intralymphatic histiocytosis remains unclear, it may be related to dysregulated immune signaling. The condition follows a chronic, relapsing-remitting course that has shown variable response to topical and systemic treatments. We present a rare case of intralymphatic histiocytosis associated with joint replacement/metal prosthesis3-14 that was responsive to a novel treatment with intralesional steroid injection and pressure bandage.
Case Report
An 89-year-old woman presented with a relapsing and remitting rash on the right calf and popliteal fossa of 11 months’ duration. It was becoming more painful over time and recently began to hurt when walking. Her medical history was remarkable for deep vein thromboses of the bilateral legs, Factor V Leiden deficiency, osteoarthritis, and a popliteal (Baker) cyst on the right leg that ruptured 22 months prior to presentation. Her surgical history included bilateral knee replacements (10 years and 2 years prior to the current presentation for the right and left knees, respectively). Her international normalized ratio (2.0) was therapeutic on warfarin.
Initially, swelling, pain, and redness developed in the right calf, and recurrent right-leg deep venous thrombosis was ruled out by Doppler ultrasound. The findings were considered to be secondary to inflammation from a popliteal cyst. Symptoms persisted despite application of warm compresses, leg elevation, and compression stockings. Treatment with doxycycline prescribed by the patient’s primary care physician 9 months prior for presumed cellulitis produced little improvement. Physical examination revealed a well-healed vertical scar on the right calf from an incisional biopsy within an 8-cm, tender, erythematous, indurated, sclerotic plaque with erythematous streaks radiating from the center of the plaque (Figure 1). There also was red-brown, indurated discoloration on the right shin.
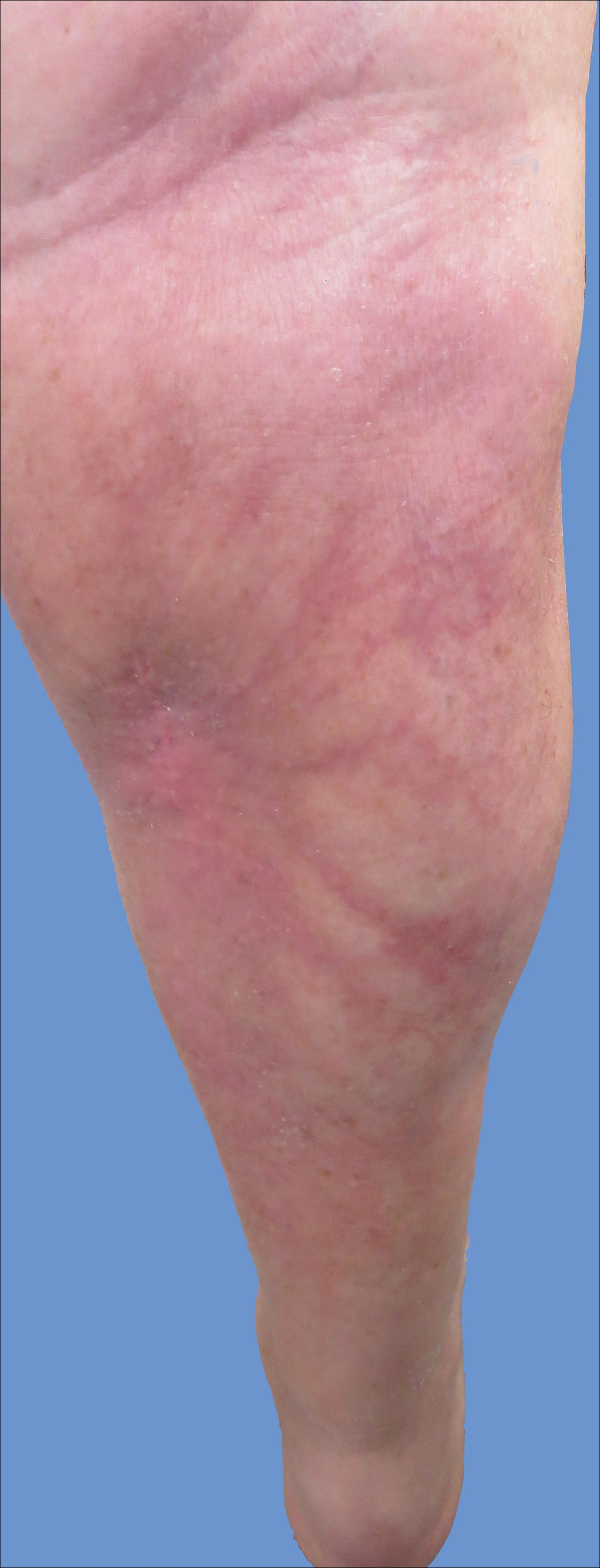
Fine-needle aspiration of the lesion revealed red blood cells and histiocytes. Laboratory studies showed an elevated erythrocyte sedimentation rate of 74 mm/h (reference range, 0–30 mm/h) and a C-reactive protein level of 39 mg/L (reference range, 0–10 mg/L). An incisional biopsy including the muscular fascia showed dense dermal fibrosis with chronic inflammation and scarring. A dermatopathologist (G. A. S.) reviewed the case and confirmed variable fibrosis and chronic inflammation associated with edema in the dermis and epidermal acanthosis. Inspection of vessels in the mid to upper dermis in one area revealed stellate, thin-walled, vascular structures that contained bland epithelioid cells lining the lumen as well as packed within the vessels. The epithelioid cells did not show atypia or mitotic figures, and they did not show intracytoplasmic vacuoles (Figure 2). Immunocytochemical staining for D2-40 was strongly positive in cells lining the vessels, consistent with lymphatics (Figure 3). CD68 immunohistochemistry for histiocytes stained the cells within the lymphatics (Figure 4). A diagnosis of intralymphatic histiocytosis was made.
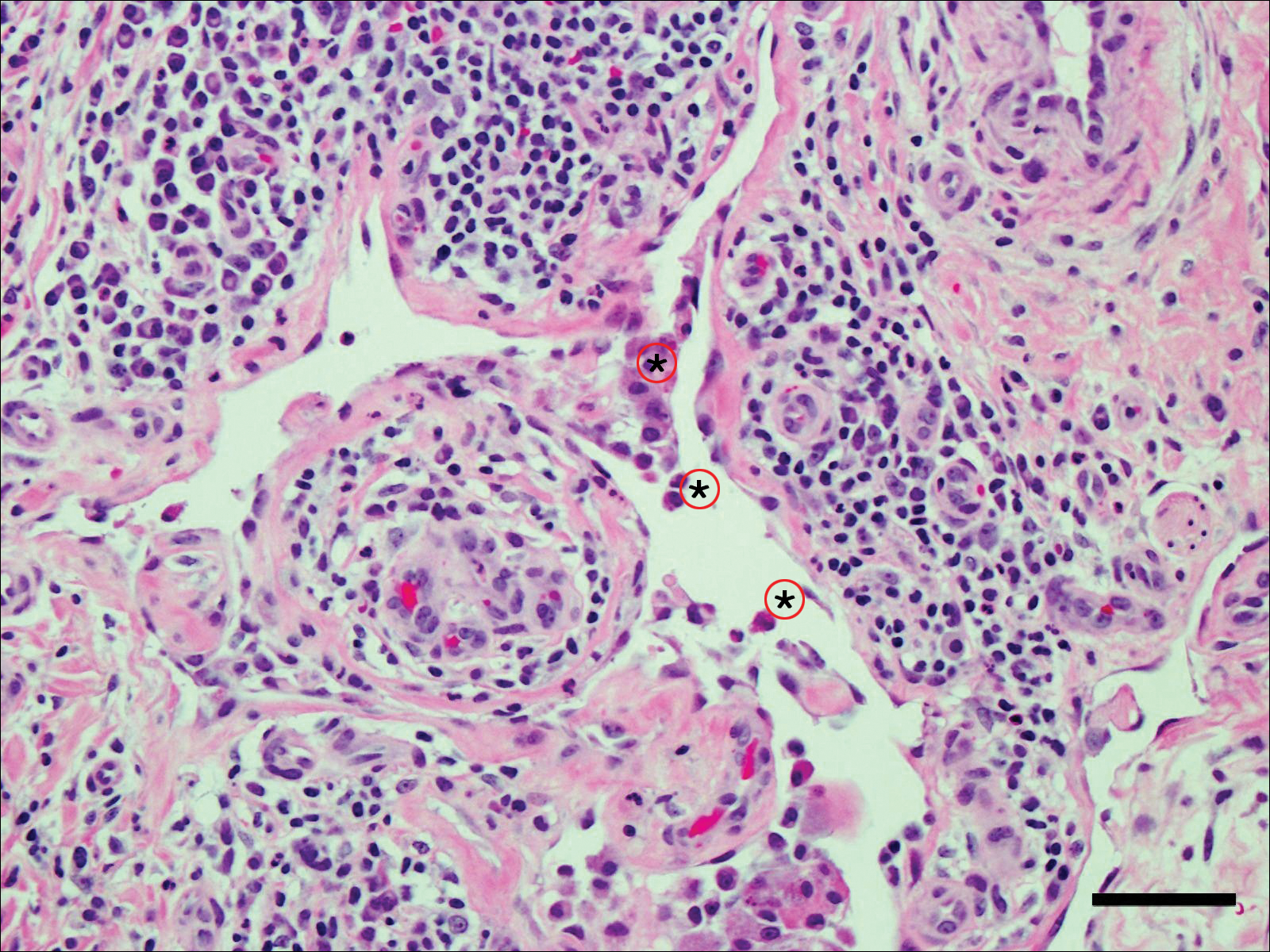
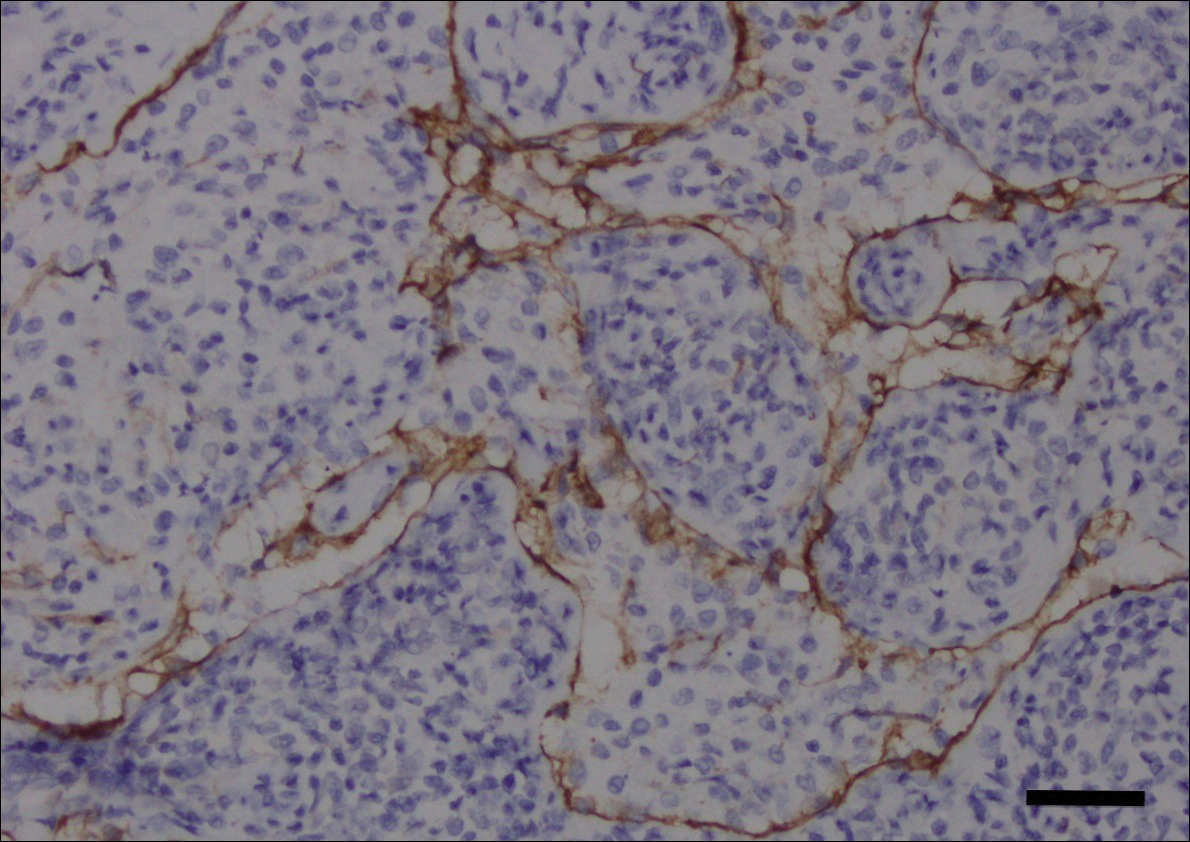
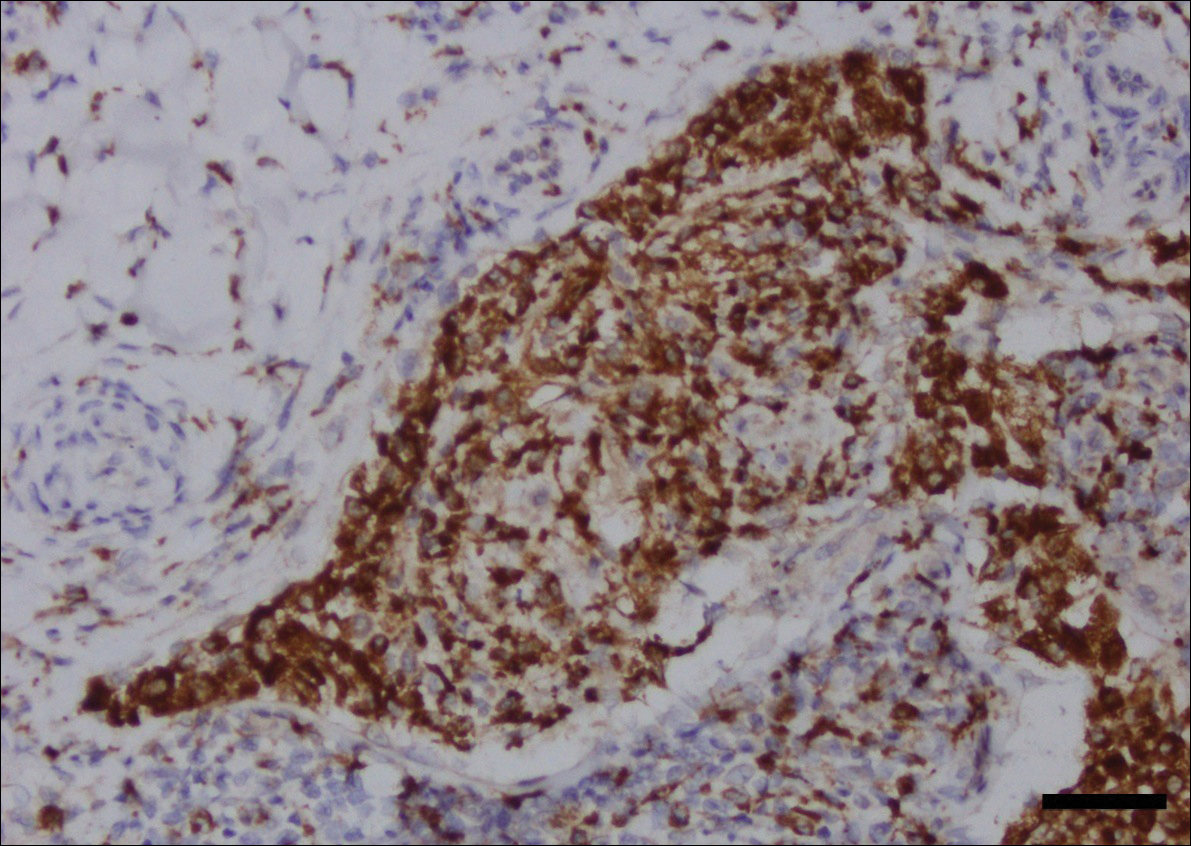
Intralesional triamcinolone acetonide 10 mg/cc×1.6 cc was injected into the plaque once monthly for 2 consecutive months, and daily compression with a pressure bandage of the right lower leg was initiated. Four months after the first treatment with this regimen, the plaque was smaller and no longer sclerotic or painful, and the erythema was markedly reduced (Figure 5). Clinical and symptomatic improvement continued at 1-year follow-up.
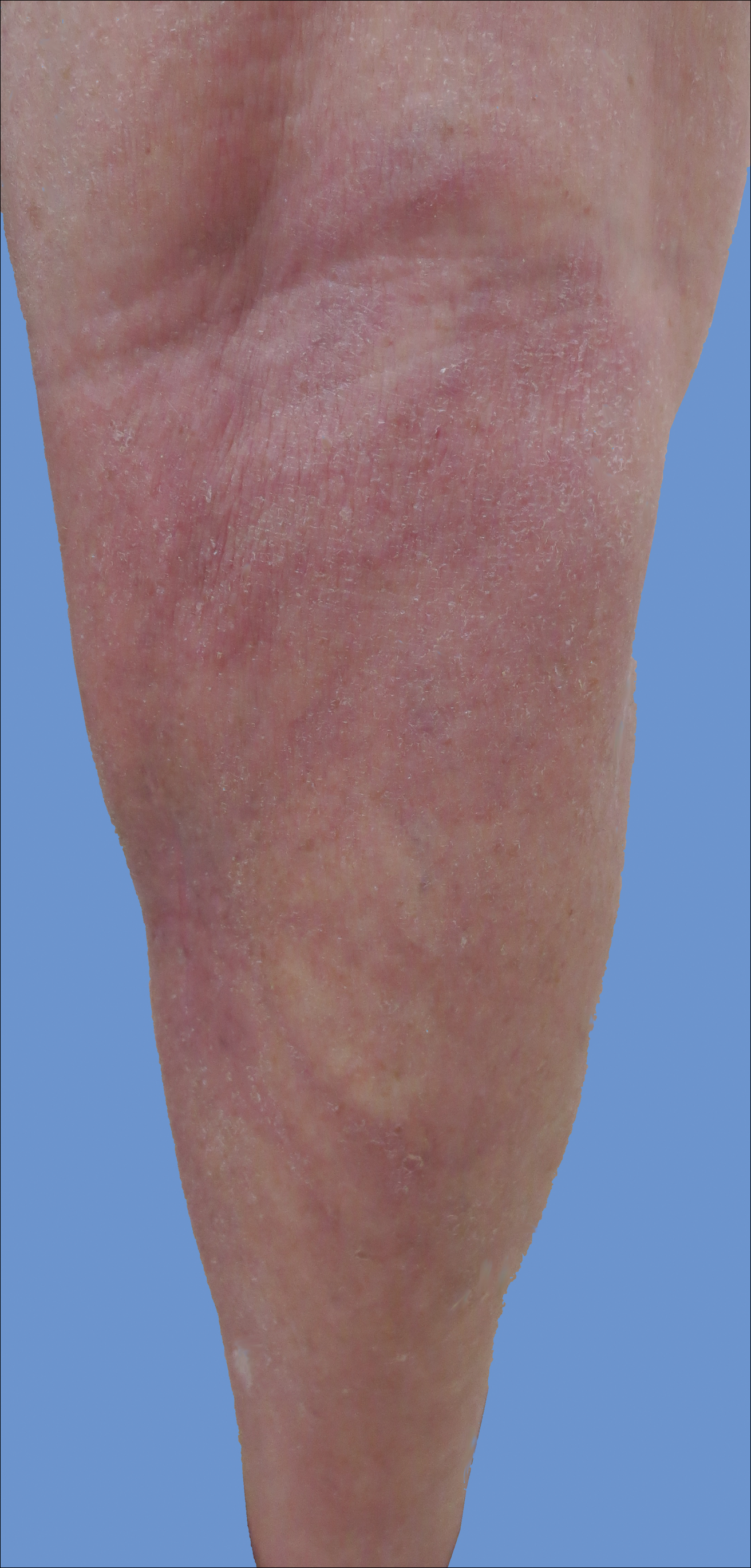
Comment
Intralymphatic histiocytosis is a rare cutaneous disorder defined histologically by histiocytes within the lumina of lymphatics. In addition to the current case, our review of PubMed articles indexed for MEDLINE using the search term intralymphatic histiocytosis yielded more than 70 total cases. The condition has a slight female predominance and typically is seen in individuals over the age of 60 years (age range, 16–89 years).12 Many cases are associated with RA/elevated rheumatoid factor.2,4,8,15-30 At least 9 cases of intralymphatic histiocytosis were associated with premalignant or malignant conditions (ie, adenocarcinoma of the breasts, lungs, and colon; Merkel cell carcinoma; melanoma; melanoma in situ; Mullerian carcinoma, gammopathy).4,15,31-34 Primary disease, defined as occurring in patients who are otherwise healthy, was noted in at least 10 cases.1,2,4,12,35,36 Finally, intralymphatic histiocytosis was identified in areas adjacent to metal implants and joint replacements or exploration in approximately 15 cases (including the current case).3-14,29,37
The condition presents with papules, plaques, and nodules in the setting of characteristic livedoid discoloration; however, some patients present with nonspecific nodules or plaques. Lesions may be symptomatic (eg, pruritic, tender) or asymptomatic. The histologic features of intralymphatic histiocytosis are distinctive but may be focal, as in our case, and the diagnosis is easily missed. The histologic differential diagnosis includes diseases in which intravascular accumulations of cells may be seen, including intravascular B-cell lymphoma, which can be excluded with stains that detect B cells (CD20/CD79a), and reactive angioendotheliomatosis, a benign proliferation of endothelial cells, which may be excluded with stains against endothelial markers (CD31/CD34). The course typically is chronic, and treatment with topical steroids,3,9,15,22,26 cyclophosphamide,15 local radiation,1 thalidomide,35 pentoxifylline,7 and RA medications (eg, prednisolone, methotrexate, nonsteroidal anti-inflammatory drugs, hydroxychloroquine) generally are ineffective.2,16,20,25 Symptoms may improve with joint replacement,4 excision of the involved lesion, treatment of an associated malignancy/infection,33,36,38,39 nonsteroidal anti-inflammatory drugs, intra-articular steroid injection,18 amoxicillin and aspirin,19 infliximab,25 pressure bandage application,26 steroid-containing adhesive application,18 arthrocentesis,3,27 oral pentoxifylline,21 tacrolimus,29 CO2 laser,40 prednisolone,41 and tocilizumab.28 Treatment of associated RA is beneficial in rare cases.2,15,20,25,26
The pathogenesis of intralymphatic histiocytosis has not been elucidated with certainty but may represent an abnormal proliferative response of histiocytes and vessels in response to chronic systemic or local inflammation. Lymphangiectasis caused by lymphatic obstruction secondary to trauma, surgical manipulation, or chronic inflammation can promote lymphostasis and slowed clearance of antigens producing an accumulation of histiocytes and subsequent local immunologic reactions, thus an “immunocompromised district” is formed.42 It also is thought that rheumatic or prosthetic joints produce inflammatory mediator–rich (namely tumor necrosis factor α) synovial fluid that drains and collects within the dilated lymphatics, creating a nidus for histiocytes.1,5 In one case, treatment with an anti–tumor necrosis factor antibody (infliximab) improved the skin presentation and rheumatoid joint pain.25 Bakr et al2 noted an association with increased intralymphatic macrophage HLA-DR expression. This T-cell surface receptor typically is upregulated in cases of chronic antigen stimulation and autoimmune conditions.
Conclusion
Our patient had a history of a joint prosthesis and a popliteal cyst, which could have altered lymphatic drainage promoting abnormal immune cell trafficking contributing to the development of intralymphatic histiocytosis. The response to intralesional steroids supports this pathogenic hypothesis. Specifically, direct injection of the area suppressed the immune dysregulation, while compression lessened the degree of lymphostasis. In light of previously reported cases of intralymphatic histiocytosis in association with metal implants,3-9 we suggest that the condition should be considered in patients with chronic painful livedoid nodules or plaques around an affected joint, even in the absence of RA. The dermatopathologist should be warned to search carefully for the subtle but distinctive histologic features of the disease that confirm the diagnosis. Treatment with intralesional triamcinolone acetonide with an overlying pressure wrap has minimal side effects and can work quickly with sustained benefits.
- O’Grady JT, Shahidullah H, Doherty VR, et al. Intravascular histiocytosis. Histopathology. 1994;24:265-268.
- Bakr F, Webber N, Fassihi H, et al. Primary and secondary intralymphatic histiocytosis [published online January 17, 2014]. J Am Acad Dermatol. 2014;70:927-933.
- Watanabe T, Yamada N, Yoshida Y, et al. Intralymphatic histiocytosis with granuloma formation associated with orthopaedic metal implants [published online November 10, 2007]. Br J Dermatol. 2008;158:402-404.
- Requena L, El-Shabrawi-Caelen L, Walsh SN, et al. Intralymphatic histiocytosis. a clinicopathologic study of 16 cases. Am J Dermatopathol. 2009;31:140-151.
- Grekin S, Mesfin M, Kang S, et al. Intralymphatic histiocytosis following placement of a metal implant. J Cutan Pathol. 2011;38:351-353.
- Rossari S, Scatena C, Gori A, et al. Intralymphatic histiocytosis: cutaneous nodules and metal implants [published online March 6, 2011]. J Cutan Pathol. 2011;38:534-535.
- de Unamuno Bustos B, García Rabasco A, Ballester Sánchez R, et al. Erythematous indurated plaque on the right upper limb. intralymphatic histiocytosis (IH) associated with orthopedic metal implant. Int J Dermatol. 2013;52:547-549.
- Chiu YE, Maloney JE, Bengana C. Erythematous patch overlying a swollen knee—quiz case. intralymphatic histiocytosis. Arch Dermatol. 2010;146:1037-1042.
- Saggar S, Lee B, Krivo J, et al. Intralymphatic histiocytosis associated with orthopedic implants. J Drugs Dermatol. 2011;10:1208-1209.
- Bidier M, Hamsch C, Kutzner H, et al. Two cases of intralymphatic histiocytosis following hip replacement [published online June 9, 2015]. J Dtsch Dermatol Ges. 2015;13:700-702.
- Darling MD, Akin R, Tarbox MB, et al. Intralymphatic histiocytosis overlying hip implantation treated with pentoxifilline. J Biol Regul Homeost Agents. 2015;29(1 suppl):117-121.
- Demirkesen C, Kran T, Leblebici C, et al. Intravascular/intralymphatic histiocytosis: a report of 3 cases. Am J Dermatopathol. 2015;37:783-789.
- Gómez-Sánchez ME, Azaña-Defez JM, Martínez-Martínez ML, et al. Intralymphatic histiocytosis: a report of 2 cases. Actas Dermosifiliogr. 2018;109:E1-E5.
- Haitz KA, Chapman MS, Seidel GD. Intralymphatic histiocytosis associated with an orthopedic metal implant. Cutis. 2016;97:E12-E14.
- Rieger E, Soyer HP, Leboit PE, et al. Reactive angioendotheliomatosis or intravascular histiocytosis? an immunohistochemical and ultrastructural study in two cases of intravascular histiocytic cell proliferation. Br J Dermatol. 1999;140:497-504.
- Pruim B, Strutton G, Congdon S, et al. Cutaneous histiocytic lymphangitis: an unusual manifestation of rheumatoid arthritis. Australas J Dermatol. 2000;41:101-105.
- Magro CM, Crowson AN. The spectrum of cutaneous lesions in rheumatoid arthritis: a clinical and pathological study of 43 patients. J Cutan Pathol. 2003;30:1-10.
- Takiwaki H, Adachi A, Kohno H, et al. Intravascular or intralymphatic histiocytosis associated with rheumatoid arthritis: a report of 4 cases.J Am Acad Dermatol. 2004;50:585-590.
- Mensing CH, Krengel S, Tronnier M, et al. Reactive angioendotheliomatosis: is it “intravascular histiocytosis”? J Eur Acad Dermatol Venereol. 2005;19:216-219.
- Okazaki A, Asada H, Niizeki H, et al. Intravascular histiocytosis associated with rheumatoid arthritis: report of a case with lymphatic endothelial proliferation. Br J Dermatol. 2005;152:1385-1387.
- Catalina-Fernández I, Alvárez AC, Martin FC, et al. Cutaneous intralymphatic histiocytosis associated with rheumatoid arthritis: report of a case and review of the literature. Am J Dermatopathol. 2007;29:165-168.
- Nishie W, Sawamura D, Iitoyo M, et al. Intravascular histiocytosis associated with rheumatoid arthritis. Dermatology. 2008;217:144-145.
- Okamoto N, Tanioka M, Yamamoto T, et al. Intralymphatic histiocytosis associated with rheumatoid arthritis. Clin Exp Dermatol. 2008;33:516-518.
- Huang H-Y, Liang C-W, Hu S-L, et al. Cutaneous intravascular histiocytosis associated with rheumatoid arthritis: a case report and review of the literature. Clin Exp Dermatol. 2009;34:E302-E303.
- Sakaguchi M, Nagai H, Tsuji G, et al. Effectiveness of infliximab for intralymphatic histiocytosis with rheumatoid arthritis. Arch Dermatol. 2011;147:131-133.
- Washio K, Nakata K, Nakamura A, et al. Pressure bandage as an effective treatment for intralymphatic histiocytosis associated with rheumatoid arthritis. Dermatology. 2011;223:20-24.
- Kaneko T, Takeuchi S, Nakano H, et al. Intralymphatic histiocytosis with rheumatoid arthritis: possible association with the joint involvement. Case Reports Clin Med. 2014;3:149-152.
- Nakajima T, Kawabata D, Nakabo S, et al. Successful treatment with tocilizumab in a case of intralymphatic histiocytosis associated with rheumatoid arthritis. Intern Med. 2014;53:2255-2258.
- Tsujiwaki M, Hata H, Miyauchi T, et al. Warty intralymphatic histiocytosis successfully treated with topical tacrolimus. J Eur Acad Dermatol Venereol. 2015;29:2267-2269.
- Tanaka M, Funasaka Y, Tsuruta K, et al. Intralymphatic histiocytosis with massive interstitial granulomatous foci in a patient with rheumatoid arthritis. Ann Dermatol. 2017;29:237-238.
- Cornejo KM, Cosar EF, O’Donnell P. Cutaneous intralymphatic histiocytosis associated with lung adenocarcinoma. Am J Dermatopathol. 2016;38:568-570.
- Tran TAN, Tran Q, Carlson JA. Intralymphatic histiocytosis of the appendix and fallopian tube associated with primary peritoneal high-grade, poorly differentiated adenocarcinoma of Müllerian origin. Int J Surg Pathol. 2017;25:357-364.
- Echeverría-García B, Botella-Estrada R, Requena C, et al. Intralymphatic histiocytosis and cancer of the colon [in Spanish]. Actas Dermosifiliogr. 2010;101:257-262.
- Ergen EN, Zwerner JP. Cover image: intralymphatic histiocytosis with giant blanching violaceous plaques. Br J Dermatol. 2017;177:325-326.
- Wang Y, Yang H, Tu P. Upper facial swelling: an uncommon manifestation of intralymphatic histiocytosis. Eur J Dermatol. 2012;22:814-815.
- Rhee D-Y, Lee D-W, Chang S-E, et al. Intravascular histiocytosis without rheumatoid arthritis. J Dermatol. 2008;35:691-693.
- Gilchrest BA, Eller MS, Geller AC, et al. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 1999;340:1341-1348.
- Asagoe K, Torigoe R, Ofuji R, et al. Reactive intravascular histiocytosis associated with tonsillitis. Br J Dermatol. 2006;154:560-563.
- Pouryazdanparast P, Yu L, Dalton VK, et al. Intravascular histiocytosis presenting with extensive vulvar necrosis. J Cutan Pathol. 2009;(36 suppl 1):1-7.
- Reznitsky M, Daugaard S, Charabi BW. Two rare cases of laryngeal intralymphatic histiocytosis. Eur Arch Otorhinolaryngol. 2016;273:783-788.
- Fujimoto N, Nakanishi G, Manabe T, et al. Intralymphatic histiocytosis comprises M2 macrophages in superficial dermal lymphatics with or without smooth muscles. J Cutan Pathol. 2016;43:898-902.
- Piccolo V, Ruocco E, Russo T, et al. A possible relationship between metal implant-induced intralymphatic histiocytosis and the concept of the immunocompromised district. Int J Dermatol. 2014;53:E365.
Intralymphatic histiocytosis was first described in 1994.1 To date, at least 70 cases have been reported in the English-language literature, the majority being associated with systemic or local inflammatory conditions such as rheumatoid arthritis (RA), malignancy, and metal prostheses. The remaining cases arose independent of any detectable disease process.2 The clinical lesion localizes to areas around surgical scars or inflamed joints and generally presents with erythematous livedoid papules and plaques. Because of its rarity, pathologists and clinicians may be unfamiliar with this entity, leading to delayed or missed diagnoses.
Although the pathogenesis of intralymphatic histiocytosis remains unclear, it may be related to dysregulated immune signaling. The condition follows a chronic, relapsing-remitting course that has shown variable response to topical and systemic treatments. We present a rare case of intralymphatic histiocytosis associated with joint replacement/metal prosthesis3-14 that was responsive to a novel treatment with intralesional steroid injection and pressure bandage.
Case Report
An 89-year-old woman presented with a relapsing and remitting rash on the right calf and popliteal fossa of 11 months’ duration. It was becoming more painful over time and recently began to hurt when walking. Her medical history was remarkable for deep vein thromboses of the bilateral legs, Factor V Leiden deficiency, osteoarthritis, and a popliteal (Baker) cyst on the right leg that ruptured 22 months prior to presentation. Her surgical history included bilateral knee replacements (10 years and 2 years prior to the current presentation for the right and left knees, respectively). Her international normalized ratio (2.0) was therapeutic on warfarin.
Initially, swelling, pain, and redness developed in the right calf, and recurrent right-leg deep venous thrombosis was ruled out by Doppler ultrasound. The findings were considered to be secondary to inflammation from a popliteal cyst. Symptoms persisted despite application of warm compresses, leg elevation, and compression stockings. Treatment with doxycycline prescribed by the patient’s primary care physician 9 months prior for presumed cellulitis produced little improvement. Physical examination revealed a well-healed vertical scar on the right calf from an incisional biopsy within an 8-cm, tender, erythematous, indurated, sclerotic plaque with erythematous streaks radiating from the center of the plaque (Figure 1). There also was red-brown, indurated discoloration on the right shin.
Fine-needle aspiration of the lesion revealed red blood cells and histiocytes. Laboratory studies showed an elevated erythrocyte sedimentation rate of 74 mm/h (reference range, 0–30 mm/h) and a C-reactive protein level of 39 mg/L (reference range, 0–10 mg/L). An incisional biopsy including the muscular fascia showed dense dermal fibrosis with chronic inflammation and scarring. A dermatopathologist (G. A. S.) reviewed the case and confirmed variable fibrosis and chronic inflammation associated with edema in the dermis and epidermal acanthosis. Inspection of vessels in the mid to upper dermis in one area revealed stellate, thin-walled, vascular structures that contained bland epithelioid cells lining the lumen as well as packed within the vessels. The epithelioid cells did not show atypia or mitotic figures, and they did not show intracytoplasmic vacuoles (Figure 2). Immunocytochemical staining for D2-40 was strongly positive in cells lining the vessels, consistent with lymphatics (Figure 3). CD68 immunohistochemistry for histiocytes stained the cells within the lymphatics (Figure 4). A diagnosis of intralymphatic histiocytosis was made.
Intralesional triamcinolone acetonide 10 mg/cc×1.6 cc was injected into the plaque once monthly for 2 consecutive months, and daily compression with a pressure bandage of the right lower leg was initiated. Four months after the first treatment with this regimen, the plaque was smaller and no longer sclerotic or painful, and the erythema was markedly reduced (Figure 5). Clinical and symptomatic improvement continued at 1-year follow-up.
Comment
Intralymphatic histiocytosis is a rare cutaneous disorder defined histologically by histiocytes within the lumina of lymphatics. In addition to the current case, our review of PubMed articles indexed for MEDLINE using the search term intralymphatic histiocytosis yielded more than 70 total cases. The condition has a slight female predominance and typically is seen in individuals over the age of 60 years (age range, 16–89 years).12 Many cases are associated with RA/elevated rheumatoid factor.2,4,8,15-30 At least 9 cases of intralymphatic histiocytosis were associated with premalignant or malignant conditions (ie, adenocarcinoma of the breasts, lungs, and colon; Merkel cell carcinoma; melanoma; melanoma in situ; Mullerian carcinoma, gammopathy).4,15,31-34 Primary disease, defined as occurring in patients who are otherwise healthy, was noted in at least 10 cases.1,2,4,12,35,36 Finally, intralymphatic histiocytosis was identified in areas adjacent to metal implants and joint replacements or exploration in approximately 15 cases (including the current case).3-14,29,37
The condition presents with papules, plaques, and nodules in the setting of characteristic livedoid discoloration; however, some patients present with nonspecific nodules or plaques. Lesions may be symptomatic (eg, pruritic, tender) or asymptomatic. The histologic features of intralymphatic histiocytosis are distinctive but may be focal, as in our case, and the diagnosis is easily missed. The histologic differential diagnosis includes diseases in which intravascular accumulations of cells may be seen, including intravascular B-cell lymphoma, which can be excluded with stains that detect B cells (CD20/CD79a), and reactive angioendotheliomatosis, a benign proliferation of endothelial cells, which may be excluded with stains against endothelial markers (CD31/CD34). The course typically is chronic, and treatment with topical steroids,3,9,15,22,26 cyclophosphamide,15 local radiation,1 thalidomide,35 pentoxifylline,7 and RA medications (eg, prednisolone, methotrexate, nonsteroidal anti-inflammatory drugs, hydroxychloroquine) generally are ineffective.2,16,20,25 Symptoms may improve with joint replacement,4 excision of the involved lesion, treatment of an associated malignancy/infection,33,36,38,39 nonsteroidal anti-inflammatory drugs, intra-articular steroid injection,18 amoxicillin and aspirin,19 infliximab,25 pressure bandage application,26 steroid-containing adhesive application,18 arthrocentesis,3,27 oral pentoxifylline,21 tacrolimus,29 CO2 laser,40 prednisolone,41 and tocilizumab.28 Treatment of associated RA is beneficial in rare cases.2,15,20,25,26
The pathogenesis of intralymphatic histiocytosis has not been elucidated with certainty but may represent an abnormal proliferative response of histiocytes and vessels in response to chronic systemic or local inflammation. Lymphangiectasis caused by lymphatic obstruction secondary to trauma, surgical manipulation, or chronic inflammation can promote lymphostasis and slowed clearance of antigens producing an accumulation of histiocytes and subsequent local immunologic reactions, thus an “immunocompromised district” is formed.42 It also is thought that rheumatic or prosthetic joints produce inflammatory mediator–rich (namely tumor necrosis factor α) synovial fluid that drains and collects within the dilated lymphatics, creating a nidus for histiocytes.1,5 In one case, treatment with an anti–tumor necrosis factor antibody (infliximab) improved the skin presentation and rheumatoid joint pain.25 Bakr et al2 noted an association with increased intralymphatic macrophage HLA-DR expression. This T-cell surface receptor typically is upregulated in cases of chronic antigen stimulation and autoimmune conditions.
Conclusion
Our patient had a history of a joint prosthesis and a popliteal cyst, which could have altered lymphatic drainage promoting abnormal immune cell trafficking contributing to the development of intralymphatic histiocytosis. The response to intralesional steroids supports this pathogenic hypothesis. Specifically, direct injection of the area suppressed the immune dysregulation, while compression lessened the degree of lymphostasis. In light of previously reported cases of intralymphatic histiocytosis in association with metal implants,3-9 we suggest that the condition should be considered in patients with chronic painful livedoid nodules or plaques around an affected joint, even in the absence of RA. The dermatopathologist should be warned to search carefully for the subtle but distinctive histologic features of the disease that confirm the diagnosis. Treatment with intralesional triamcinolone acetonide with an overlying pressure wrap has minimal side effects and can work quickly with sustained benefits.
Intralymphatic histiocytosis was first described in 1994.1 To date, at least 70 cases have been reported in the English-language literature, the majority being associated with systemic or local inflammatory conditions such as rheumatoid arthritis (RA), malignancy, and metal prostheses. The remaining cases arose independent of any detectable disease process.2 The clinical lesion localizes to areas around surgical scars or inflamed joints and generally presents with erythematous livedoid papules and plaques. Because of its rarity, pathologists and clinicians may be unfamiliar with this entity, leading to delayed or missed diagnoses.
Although the pathogenesis of intralymphatic histiocytosis remains unclear, it may be related to dysregulated immune signaling. The condition follows a chronic, relapsing-remitting course that has shown variable response to topical and systemic treatments. We present a rare case of intralymphatic histiocytosis associated with joint replacement/metal prosthesis3-14 that was responsive to a novel treatment with intralesional steroid injection and pressure bandage.
Case Report
An 89-year-old woman presented with a relapsing and remitting rash on the right calf and popliteal fossa of 11 months’ duration. It was becoming more painful over time and recently began to hurt when walking. Her medical history was remarkable for deep vein thromboses of the bilateral legs, Factor V Leiden deficiency, osteoarthritis, and a popliteal (Baker) cyst on the right leg that ruptured 22 months prior to presentation. Her surgical history included bilateral knee replacements (10 years and 2 years prior to the current presentation for the right and left knees, respectively). Her international normalized ratio (2.0) was therapeutic on warfarin.
Initially, swelling, pain, and redness developed in the right calf, and recurrent right-leg deep venous thrombosis was ruled out by Doppler ultrasound. The findings were considered to be secondary to inflammation from a popliteal cyst. Symptoms persisted despite application of warm compresses, leg elevation, and compression stockings. Treatment with doxycycline prescribed by the patient’s primary care physician 9 months prior for presumed cellulitis produced little improvement. Physical examination revealed a well-healed vertical scar on the right calf from an incisional biopsy within an 8-cm, tender, erythematous, indurated, sclerotic plaque with erythematous streaks radiating from the center of the plaque (Figure 1). There also was red-brown, indurated discoloration on the right shin.
Fine-needle aspiration of the lesion revealed red blood cells and histiocytes. Laboratory studies showed an elevated erythrocyte sedimentation rate of 74 mm/h (reference range, 0–30 mm/h) and a C-reactive protein level of 39 mg/L (reference range, 0–10 mg/L). An incisional biopsy including the muscular fascia showed dense dermal fibrosis with chronic inflammation and scarring. A dermatopathologist (G. A. S.) reviewed the case and confirmed variable fibrosis and chronic inflammation associated with edema in the dermis and epidermal acanthosis. Inspection of vessels in the mid to upper dermis in one area revealed stellate, thin-walled, vascular structures that contained bland epithelioid cells lining the lumen as well as packed within the vessels. The epithelioid cells did not show atypia or mitotic figures, and they did not show intracytoplasmic vacuoles (Figure 2). Immunocytochemical staining for D2-40 was strongly positive in cells lining the vessels, consistent with lymphatics (Figure 3). CD68 immunohistochemistry for histiocytes stained the cells within the lymphatics (Figure 4). A diagnosis of intralymphatic histiocytosis was made.
Intralesional triamcinolone acetonide 10 mg/cc×1.6 cc was injected into the plaque once monthly for 2 consecutive months, and daily compression with a pressure bandage of the right lower leg was initiated. Four months after the first treatment with this regimen, the plaque was smaller and no longer sclerotic or painful, and the erythema was markedly reduced (Figure 5). Clinical and symptomatic improvement continued at 1-year follow-up.
Comment
Intralymphatic histiocytosis is a rare cutaneous disorder defined histologically by histiocytes within the lumina of lymphatics. In addition to the current case, our review of PubMed articles indexed for MEDLINE using the search term intralymphatic histiocytosis yielded more than 70 total cases. The condition has a slight female predominance and typically is seen in individuals over the age of 60 years (age range, 16–89 years).12 Many cases are associated with RA/elevated rheumatoid factor.2,4,8,15-30 At least 9 cases of intralymphatic histiocytosis were associated with premalignant or malignant conditions (ie, adenocarcinoma of the breasts, lungs, and colon; Merkel cell carcinoma; melanoma; melanoma in situ; Mullerian carcinoma, gammopathy).4,15,31-34 Primary disease, defined as occurring in patients who are otherwise healthy, was noted in at least 10 cases.1,2,4,12,35,36 Finally, intralymphatic histiocytosis was identified in areas adjacent to metal implants and joint replacements or exploration in approximately 15 cases (including the current case).3-14,29,37
The condition presents with papules, plaques, and nodules in the setting of characteristic livedoid discoloration; however, some patients present with nonspecific nodules or plaques. Lesions may be symptomatic (eg, pruritic, tender) or asymptomatic. The histologic features of intralymphatic histiocytosis are distinctive but may be focal, as in our case, and the diagnosis is easily missed. The histologic differential diagnosis includes diseases in which intravascular accumulations of cells may be seen, including intravascular B-cell lymphoma, which can be excluded with stains that detect B cells (CD20/CD79a), and reactive angioendotheliomatosis, a benign proliferation of endothelial cells, which may be excluded with stains against endothelial markers (CD31/CD34). The course typically is chronic, and treatment with topical steroids,3,9,15,22,26 cyclophosphamide,15 local radiation,1 thalidomide,35 pentoxifylline,7 and RA medications (eg, prednisolone, methotrexate, nonsteroidal anti-inflammatory drugs, hydroxychloroquine) generally are ineffective.2,16,20,25 Symptoms may improve with joint replacement,4 excision of the involved lesion, treatment of an associated malignancy/infection,33,36,38,39 nonsteroidal anti-inflammatory drugs, intra-articular steroid injection,18 amoxicillin and aspirin,19 infliximab,25 pressure bandage application,26 steroid-containing adhesive application,18 arthrocentesis,3,27 oral pentoxifylline,21 tacrolimus,29 CO2 laser,40 prednisolone,41 and tocilizumab.28 Treatment of associated RA is beneficial in rare cases.2,15,20,25,26
The pathogenesis of intralymphatic histiocytosis has not been elucidated with certainty but may represent an abnormal proliferative response of histiocytes and vessels in response to chronic systemic or local inflammation. Lymphangiectasis caused by lymphatic obstruction secondary to trauma, surgical manipulation, or chronic inflammation can promote lymphostasis and slowed clearance of antigens producing an accumulation of histiocytes and subsequent local immunologic reactions, thus an “immunocompromised district” is formed.42 It also is thought that rheumatic or prosthetic joints produce inflammatory mediator–rich (namely tumor necrosis factor α) synovial fluid that drains and collects within the dilated lymphatics, creating a nidus for histiocytes.1,5 In one case, treatment with an anti–tumor necrosis factor antibody (infliximab) improved the skin presentation and rheumatoid joint pain.25 Bakr et al2 noted an association with increased intralymphatic macrophage HLA-DR expression. This T-cell surface receptor typically is upregulated in cases of chronic antigen stimulation and autoimmune conditions.
Conclusion
Our patient had a history of a joint prosthesis and a popliteal cyst, which could have altered lymphatic drainage promoting abnormal immune cell trafficking contributing to the development of intralymphatic histiocytosis. The response to intralesional steroids supports this pathogenic hypothesis. Specifically, direct injection of the area suppressed the immune dysregulation, while compression lessened the degree of lymphostasis. In light of previously reported cases of intralymphatic histiocytosis in association with metal implants,3-9 we suggest that the condition should be considered in patients with chronic painful livedoid nodules or plaques around an affected joint, even in the absence of RA. The dermatopathologist should be warned to search carefully for the subtle but distinctive histologic features of the disease that confirm the diagnosis. Treatment with intralesional triamcinolone acetonide with an overlying pressure wrap has minimal side effects and can work quickly with sustained benefits.
- O’Grady JT, Shahidullah H, Doherty VR, et al. Intravascular histiocytosis. Histopathology. 1994;24:265-268.
- Bakr F, Webber N, Fassihi H, et al. Primary and secondary intralymphatic histiocytosis [published online January 17, 2014]. J Am Acad Dermatol. 2014;70:927-933.
- Watanabe T, Yamada N, Yoshida Y, et al. Intralymphatic histiocytosis with granuloma formation associated with orthopaedic metal implants [published online November 10, 2007]. Br J Dermatol. 2008;158:402-404.
- Requena L, El-Shabrawi-Caelen L, Walsh SN, et al. Intralymphatic histiocytosis. a clinicopathologic study of 16 cases. Am J Dermatopathol. 2009;31:140-151.
- Grekin S, Mesfin M, Kang S, et al. Intralymphatic histiocytosis following placement of a metal implant. J Cutan Pathol. 2011;38:351-353.
- Rossari S, Scatena C, Gori A, et al. Intralymphatic histiocytosis: cutaneous nodules and metal implants [published online March 6, 2011]. J Cutan Pathol. 2011;38:534-535.
- de Unamuno Bustos B, García Rabasco A, Ballester Sánchez R, et al. Erythematous indurated plaque on the right upper limb. intralymphatic histiocytosis (IH) associated with orthopedic metal implant. Int J Dermatol. 2013;52:547-549.
- Chiu YE, Maloney JE, Bengana C. Erythematous patch overlying a swollen knee—quiz case. intralymphatic histiocytosis. Arch Dermatol. 2010;146:1037-1042.
- Saggar S, Lee B, Krivo J, et al. Intralymphatic histiocytosis associated with orthopedic implants. J Drugs Dermatol. 2011;10:1208-1209.
- Bidier M, Hamsch C, Kutzner H, et al. Two cases of intralymphatic histiocytosis following hip replacement [published online June 9, 2015]. J Dtsch Dermatol Ges. 2015;13:700-702.
- Darling MD, Akin R, Tarbox MB, et al. Intralymphatic histiocytosis overlying hip implantation treated with pentoxifilline. J Biol Regul Homeost Agents. 2015;29(1 suppl):117-121.
- Demirkesen C, Kran T, Leblebici C, et al. Intravascular/intralymphatic histiocytosis: a report of 3 cases. Am J Dermatopathol. 2015;37:783-789.
- Gómez-Sánchez ME, Azaña-Defez JM, Martínez-Martínez ML, et al. Intralymphatic histiocytosis: a report of 2 cases. Actas Dermosifiliogr. 2018;109:E1-E5.
- Haitz KA, Chapman MS, Seidel GD. Intralymphatic histiocytosis associated with an orthopedic metal implant. Cutis. 2016;97:E12-E14.
- Rieger E, Soyer HP, Leboit PE, et al. Reactive angioendotheliomatosis or intravascular histiocytosis? an immunohistochemical and ultrastructural study in two cases of intravascular histiocytic cell proliferation. Br J Dermatol. 1999;140:497-504.
- Pruim B, Strutton G, Congdon S, et al. Cutaneous histiocytic lymphangitis: an unusual manifestation of rheumatoid arthritis. Australas J Dermatol. 2000;41:101-105.
- Magro CM, Crowson AN. The spectrum of cutaneous lesions in rheumatoid arthritis: a clinical and pathological study of 43 patients. J Cutan Pathol. 2003;30:1-10.
- Takiwaki H, Adachi A, Kohno H, et al. Intravascular or intralymphatic histiocytosis associated with rheumatoid arthritis: a report of 4 cases.J Am Acad Dermatol. 2004;50:585-590.
- Mensing CH, Krengel S, Tronnier M, et al. Reactive angioendotheliomatosis: is it “intravascular histiocytosis”? J Eur Acad Dermatol Venereol. 2005;19:216-219.
- Okazaki A, Asada H, Niizeki H, et al. Intravascular histiocytosis associated with rheumatoid arthritis: report of a case with lymphatic endothelial proliferation. Br J Dermatol. 2005;152:1385-1387.
- Catalina-Fernández I, Alvárez AC, Martin FC, et al. Cutaneous intralymphatic histiocytosis associated with rheumatoid arthritis: report of a case and review of the literature. Am J Dermatopathol. 2007;29:165-168.
- Nishie W, Sawamura D, Iitoyo M, et al. Intravascular histiocytosis associated with rheumatoid arthritis. Dermatology. 2008;217:144-145.
- Okamoto N, Tanioka M, Yamamoto T, et al. Intralymphatic histiocytosis associated with rheumatoid arthritis. Clin Exp Dermatol. 2008;33:516-518.
- Huang H-Y, Liang C-W, Hu S-L, et al. Cutaneous intravascular histiocytosis associated with rheumatoid arthritis: a case report and review of the literature. Clin Exp Dermatol. 2009;34:E302-E303.
- Sakaguchi M, Nagai H, Tsuji G, et al. Effectiveness of infliximab for intralymphatic histiocytosis with rheumatoid arthritis. Arch Dermatol. 2011;147:131-133.
- Washio K, Nakata K, Nakamura A, et al. Pressure bandage as an effective treatment for intralymphatic histiocytosis associated with rheumatoid arthritis. Dermatology. 2011;223:20-24.
- Kaneko T, Takeuchi S, Nakano H, et al. Intralymphatic histiocytosis with rheumatoid arthritis: possible association with the joint involvement. Case Reports Clin Med. 2014;3:149-152.
- Nakajima T, Kawabata D, Nakabo S, et al. Successful treatment with tocilizumab in a case of intralymphatic histiocytosis associated with rheumatoid arthritis. Intern Med. 2014;53:2255-2258.
- Tsujiwaki M, Hata H, Miyauchi T, et al. Warty intralymphatic histiocytosis successfully treated with topical tacrolimus. J Eur Acad Dermatol Venereol. 2015;29:2267-2269.
- Tanaka M, Funasaka Y, Tsuruta K, et al. Intralymphatic histiocytosis with massive interstitial granulomatous foci in a patient with rheumatoid arthritis. Ann Dermatol. 2017;29:237-238.
- Cornejo KM, Cosar EF, O’Donnell P. Cutaneous intralymphatic histiocytosis associated with lung adenocarcinoma. Am J Dermatopathol. 2016;38:568-570.
- Tran TAN, Tran Q, Carlson JA. Intralymphatic histiocytosis of the appendix and fallopian tube associated with primary peritoneal high-grade, poorly differentiated adenocarcinoma of Müllerian origin. Int J Surg Pathol. 2017;25:357-364.
- Echeverría-García B, Botella-Estrada R, Requena C, et al. Intralymphatic histiocytosis and cancer of the colon [in Spanish]. Actas Dermosifiliogr. 2010;101:257-262.
- Ergen EN, Zwerner JP. Cover image: intralymphatic histiocytosis with giant blanching violaceous plaques. Br J Dermatol. 2017;177:325-326.
- Wang Y, Yang H, Tu P. Upper facial swelling: an uncommon manifestation of intralymphatic histiocytosis. Eur J Dermatol. 2012;22:814-815.
- Rhee D-Y, Lee D-W, Chang S-E, et al. Intravascular histiocytosis without rheumatoid arthritis. J Dermatol. 2008;35:691-693.
- Gilchrest BA, Eller MS, Geller AC, et al. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 1999;340:1341-1348.
- Asagoe K, Torigoe R, Ofuji R, et al. Reactive intravascular histiocytosis associated with tonsillitis. Br J Dermatol. 2006;154:560-563.
- Pouryazdanparast P, Yu L, Dalton VK, et al. Intravascular histiocytosis presenting with extensive vulvar necrosis. J Cutan Pathol. 2009;(36 suppl 1):1-7.
- Reznitsky M, Daugaard S, Charabi BW. Two rare cases of laryngeal intralymphatic histiocytosis. Eur Arch Otorhinolaryngol. 2016;273:783-788.
- Fujimoto N, Nakanishi G, Manabe T, et al. Intralymphatic histiocytosis comprises M2 macrophages in superficial dermal lymphatics with or without smooth muscles. J Cutan Pathol. 2016;43:898-902.
- Piccolo V, Ruocco E, Russo T, et al. A possible relationship between metal implant-induced intralymphatic histiocytosis and the concept of the immunocompromised district. Int J Dermatol. 2014;53:E365.
- O’Grady JT, Shahidullah H, Doherty VR, et al. Intravascular histiocytosis. Histopathology. 1994;24:265-268.
- Bakr F, Webber N, Fassihi H, et al. Primary and secondary intralymphatic histiocytosis [published online January 17, 2014]. J Am Acad Dermatol. 2014;70:927-933.
- Watanabe T, Yamada N, Yoshida Y, et al. Intralymphatic histiocytosis with granuloma formation associated with orthopaedic metal implants [published online November 10, 2007]. Br J Dermatol. 2008;158:402-404.
- Requena L, El-Shabrawi-Caelen L, Walsh SN, et al. Intralymphatic histiocytosis. a clinicopathologic study of 16 cases. Am J Dermatopathol. 2009;31:140-151.
- Grekin S, Mesfin M, Kang S, et al. Intralymphatic histiocytosis following placement of a metal implant. J Cutan Pathol. 2011;38:351-353.
- Rossari S, Scatena C, Gori A, et al. Intralymphatic histiocytosis: cutaneous nodules and metal implants [published online March 6, 2011]. J Cutan Pathol. 2011;38:534-535.
- de Unamuno Bustos B, García Rabasco A, Ballester Sánchez R, et al. Erythematous indurated plaque on the right upper limb. intralymphatic histiocytosis (IH) associated with orthopedic metal implant. Int J Dermatol. 2013;52:547-549.
- Chiu YE, Maloney JE, Bengana C. Erythematous patch overlying a swollen knee—quiz case. intralymphatic histiocytosis. Arch Dermatol. 2010;146:1037-1042.
- Saggar S, Lee B, Krivo J, et al. Intralymphatic histiocytosis associated with orthopedic implants. J Drugs Dermatol. 2011;10:1208-1209.
- Bidier M, Hamsch C, Kutzner H, et al. Two cases of intralymphatic histiocytosis following hip replacement [published online June 9, 2015]. J Dtsch Dermatol Ges. 2015;13:700-702.
- Darling MD, Akin R, Tarbox MB, et al. Intralymphatic histiocytosis overlying hip implantation treated with pentoxifilline. J Biol Regul Homeost Agents. 2015;29(1 suppl):117-121.
- Demirkesen C, Kran T, Leblebici C, et al. Intravascular/intralymphatic histiocytosis: a report of 3 cases. Am J Dermatopathol. 2015;37:783-789.
- Gómez-Sánchez ME, Azaña-Defez JM, Martínez-Martínez ML, et al. Intralymphatic histiocytosis: a report of 2 cases. Actas Dermosifiliogr. 2018;109:E1-E5.
- Haitz KA, Chapman MS, Seidel GD. Intralymphatic histiocytosis associated with an orthopedic metal implant. Cutis. 2016;97:E12-E14.
- Rieger E, Soyer HP, Leboit PE, et al. Reactive angioendotheliomatosis or intravascular histiocytosis? an immunohistochemical and ultrastructural study in two cases of intravascular histiocytic cell proliferation. Br J Dermatol. 1999;140:497-504.
- Pruim B, Strutton G, Congdon S, et al. Cutaneous histiocytic lymphangitis: an unusual manifestation of rheumatoid arthritis. Australas J Dermatol. 2000;41:101-105.
- Magro CM, Crowson AN. The spectrum of cutaneous lesions in rheumatoid arthritis: a clinical and pathological study of 43 patients. J Cutan Pathol. 2003;30:1-10.
- Takiwaki H, Adachi A, Kohno H, et al. Intravascular or intralymphatic histiocytosis associated with rheumatoid arthritis: a report of 4 cases.J Am Acad Dermatol. 2004;50:585-590.
- Mensing CH, Krengel S, Tronnier M, et al. Reactive angioendotheliomatosis: is it “intravascular histiocytosis”? J Eur Acad Dermatol Venereol. 2005;19:216-219.
- Okazaki A, Asada H, Niizeki H, et al. Intravascular histiocytosis associated with rheumatoid arthritis: report of a case with lymphatic endothelial proliferation. Br J Dermatol. 2005;152:1385-1387.
- Catalina-Fernández I, Alvárez AC, Martin FC, et al. Cutaneous intralymphatic histiocytosis associated with rheumatoid arthritis: report of a case and review of the literature. Am J Dermatopathol. 2007;29:165-168.
- Nishie W, Sawamura D, Iitoyo M, et al. Intravascular histiocytosis associated with rheumatoid arthritis. Dermatology. 2008;217:144-145.
- Okamoto N, Tanioka M, Yamamoto T, et al. Intralymphatic histiocytosis associated with rheumatoid arthritis. Clin Exp Dermatol. 2008;33:516-518.
- Huang H-Y, Liang C-W, Hu S-L, et al. Cutaneous intravascular histiocytosis associated with rheumatoid arthritis: a case report and review of the literature. Clin Exp Dermatol. 2009;34:E302-E303.
- Sakaguchi M, Nagai H, Tsuji G, et al. Effectiveness of infliximab for intralymphatic histiocytosis with rheumatoid arthritis. Arch Dermatol. 2011;147:131-133.
- Washio K, Nakata K, Nakamura A, et al. Pressure bandage as an effective treatment for intralymphatic histiocytosis associated with rheumatoid arthritis. Dermatology. 2011;223:20-24.
- Kaneko T, Takeuchi S, Nakano H, et al. Intralymphatic histiocytosis with rheumatoid arthritis: possible association with the joint involvement. Case Reports Clin Med. 2014;3:149-152.
- Nakajima T, Kawabata D, Nakabo S, et al. Successful treatment with tocilizumab in a case of intralymphatic histiocytosis associated with rheumatoid arthritis. Intern Med. 2014;53:2255-2258.
- Tsujiwaki M, Hata H, Miyauchi T, et al. Warty intralymphatic histiocytosis successfully treated with topical tacrolimus. J Eur Acad Dermatol Venereol. 2015;29:2267-2269.
- Tanaka M, Funasaka Y, Tsuruta K, et al. Intralymphatic histiocytosis with massive interstitial granulomatous foci in a patient with rheumatoid arthritis. Ann Dermatol. 2017;29:237-238.
- Cornejo KM, Cosar EF, O’Donnell P. Cutaneous intralymphatic histiocytosis associated with lung adenocarcinoma. Am J Dermatopathol. 2016;38:568-570.
- Tran TAN, Tran Q, Carlson JA. Intralymphatic histiocytosis of the appendix and fallopian tube associated with primary peritoneal high-grade, poorly differentiated adenocarcinoma of Müllerian origin. Int J Surg Pathol. 2017;25:357-364.
- Echeverría-García B, Botella-Estrada R, Requena C, et al. Intralymphatic histiocytosis and cancer of the colon [in Spanish]. Actas Dermosifiliogr. 2010;101:257-262.
- Ergen EN, Zwerner JP. Cover image: intralymphatic histiocytosis with giant blanching violaceous plaques. Br J Dermatol. 2017;177:325-326.
- Wang Y, Yang H, Tu P. Upper facial swelling: an uncommon manifestation of intralymphatic histiocytosis. Eur J Dermatol. 2012;22:814-815.
- Rhee D-Y, Lee D-W, Chang S-E, et al. Intravascular histiocytosis without rheumatoid arthritis. J Dermatol. 2008;35:691-693.
- Gilchrest BA, Eller MS, Geller AC, et al. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 1999;340:1341-1348.
- Asagoe K, Torigoe R, Ofuji R, et al. Reactive intravascular histiocytosis associated with tonsillitis. Br J Dermatol. 2006;154:560-563.
- Pouryazdanparast P, Yu L, Dalton VK, et al. Intravascular histiocytosis presenting with extensive vulvar necrosis. J Cutan Pathol. 2009;(36 suppl 1):1-7.
- Reznitsky M, Daugaard S, Charabi BW. Two rare cases of laryngeal intralymphatic histiocytosis. Eur Arch Otorhinolaryngol. 2016;273:783-788.
- Fujimoto N, Nakanishi G, Manabe T, et al. Intralymphatic histiocytosis comprises M2 macrophages in superficial dermal lymphatics with or without smooth muscles. J Cutan Pathol. 2016;43:898-902.
- Piccolo V, Ruocco E, Russo T, et al. A possible relationship between metal implant-induced intralymphatic histiocytosis and the concept of the immunocompromised district. Int J Dermatol. 2014;53:E365.
Practice Points
- Intralymphatic histiocytosis is a rare disorder often associated with rheumatic arthritis and joint prostheses.
- The diagnosis is made by histopathology as well as D2-40 and CD68 immunostaining.
- While there is no gold standard of treatment for intralymphatic histiocytosis, intralesional triamcinolone proved efficacious in this case with prolonged results.
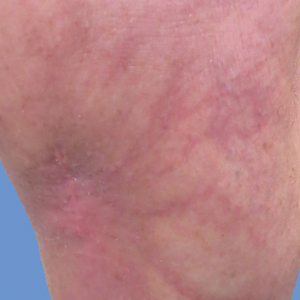
Pigmented Peduncule on the Leg
The Diagnosis: Polypoid Dermatofibroma
Histologic examination revealed a poorly demarcated lesion localized in the dermis that was composed of an admixture of fibroblastlike cells, histiocytes, siderophages, multinucleated giant cells, and hemorrhage (Figure). Based on these findings, a diagnosis of polypoid dermatofibroma (DF) was made. No further treatment was necessary because the lesion was completely excised.
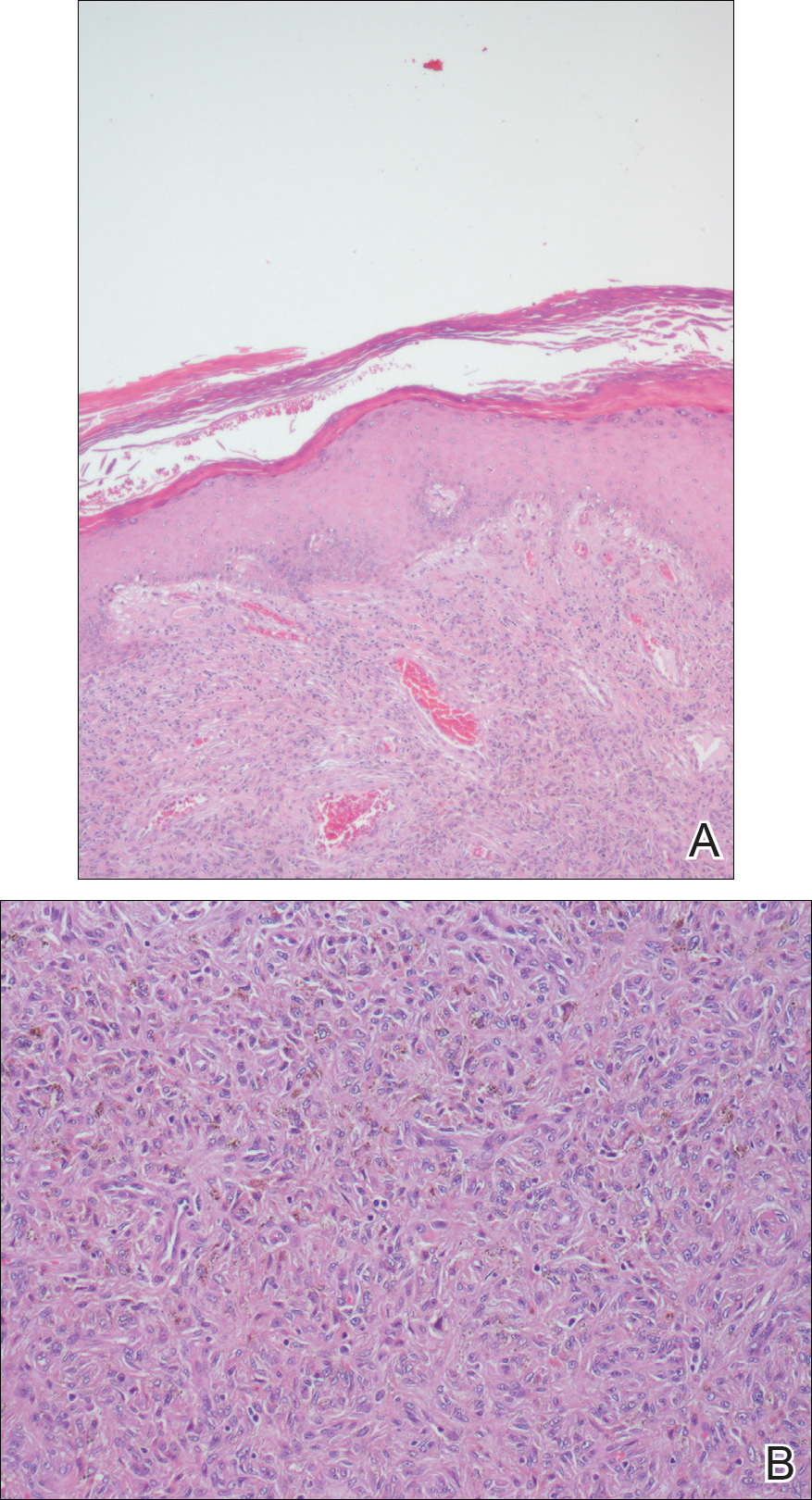
Dermatofibromas, also known as benign fibrous histiocytomas, are common, benign, mesenchymal, fibrosing tumors of the skin that appear predominantly on the legs in in women, but any part of the body in either sex can be affected. Clinically, DFs show the dimple sign when laterally squeezed and can be painful spontaneously or when rubbed. Histologically, DFs are poorly demarcated lesions composed of a variable admixture of fibroblastlike cells, histiocytes (some of which may be xanthomatous or multinucleated), and blood vessels. The etiology still remains unclear. Most investigators consider DF to be a reactive process, but some think that it is a benign mesenchymal tumor.1,2
Many subtypes of DF have been described based on their unique architectural, cellular, stromal, and clinical features.2,3 Polypoid DF is a rare variant that comprises only 3% of reported cases4 and tends to be larger than other DF subtypes. Requena et al5 reported 12 cases of giant DF, another clinical subtype, that were larger than 5 cm in diameter, most of which had a polypoid appearance.
Moreover, 3 distinct variants of DF with unique histologic features tend to show polypoid morphology.3,4 In epithelioid fibrous histiocytoma, also known as epithelioid cell histiocytoma, at least 50% of the lesion is composed of rounded or polygonal epithelioid cells with abundant eosinophilic cytoplasm and round to oval nuclei containing small eosinophilic nucleoli.4 A grenz zone generally is lacking and numerous small blood vessels are a constant feature of epithelioid fibrous histiocytoma. The other variant of DF that also tends to have a polypoid appearance is lipidized fibrous histiocytoma, or ankle-type fibrous histiocytoma, which usually arises below the knee, especially around the ankle, and often becomes larger than common DF.4 Lastly, atypical polypoid DF is a benign, polyp-shaped lesion that shows hypercellularity with striking nuclear atypism and scattered mitotic figures in addition to the ordinary histologic features of DF.3
Acquired fibrokeratomas manifest as solitary dome-shaped, flesh-colored protrusions usually located on the feet and hands. Sclerotic fibroma is a rare cutaneous neoplasm that presents as a solitary, translucent or waxy nodule or as multiple nodules when it is part of Cowden disease. Fibromas, also known as skin tags, are common cutaneous tumors that appear in intertriginous areas and frequently adopt a pedunculated morphology. Although clinically some of these lesions may resemble polypoid DF, the differential diagnosis is made by histologic examination.
Dermatofibroma is a common cutaneous tumor that follows a benign course. It can adopt multiple morphologies. Awareness of this rare variant may aid in its appropriate diagnosis and management. Dermatofibromas are benign neoplasms, and therefore they usually do not require treatment. If they become symptomatic or are bothersome to the patient, the treatment of choice is surgical removal.
- Luzar B, Calonje E. Cutaneous fibrohistiocytic tumours--an update. Histopathology. 2010;56:148-165.
- Santos-Briz A, Llamas-Velasco M, Arango L, et al. Cutaneous adenodermatofibroma: report of 2 cases. Am J Dermatopathol. 2013;35:E103-E105.
- Sogabe Y, Takahashi A, Tamura A, et al. A case of polypoid dermatofibroma. J Dermatol. 2002;29:786-789.
- Kai H, Fujita H, Yamamoto M, et al. Polypoid dermatofibroma with a slim pedicle: a case report. Dermatol Online J. 2012;18:16.
- Requena L, Farina K, Fuente C, et al. Giant dermatofibroma. a little known clinical variant of dermatofibroma. J Am Acad Dermatol. 1994;30:714-718.
The Diagnosis: Polypoid Dermatofibroma
Histologic examination revealed a poorly demarcated lesion localized in the dermis that was composed of an admixture of fibroblastlike cells, histiocytes, siderophages, multinucleated giant cells, and hemorrhage (Figure). Based on these findings, a diagnosis of polypoid dermatofibroma (DF) was made. No further treatment was necessary because the lesion was completely excised.
Dermatofibromas, also known as benign fibrous histiocytomas, are common, benign, mesenchymal, fibrosing tumors of the skin that appear predominantly on the legs in in women, but any part of the body in either sex can be affected. Clinically, DFs show the dimple sign when laterally squeezed and can be painful spontaneously or when rubbed. Histologically, DFs are poorly demarcated lesions composed of a variable admixture of fibroblastlike cells, histiocytes (some of which may be xanthomatous or multinucleated), and blood vessels. The etiology still remains unclear. Most investigators consider DF to be a reactive process, but some think that it is a benign mesenchymal tumor.1,2
Many subtypes of DF have been described based on their unique architectural, cellular, stromal, and clinical features.2,3 Polypoid DF is a rare variant that comprises only 3% of reported cases4 and tends to be larger than other DF subtypes. Requena et al5 reported 12 cases of giant DF, another clinical subtype, that were larger than 5 cm in diameter, most of which had a polypoid appearance.
Moreover, 3 distinct variants of DF with unique histologic features tend to show polypoid morphology.3,4 In epithelioid fibrous histiocytoma, also known as epithelioid cell histiocytoma, at least 50% of the lesion is composed of rounded or polygonal epithelioid cells with abundant eosinophilic cytoplasm and round to oval nuclei containing small eosinophilic nucleoli.4 A grenz zone generally is lacking and numerous small blood vessels are a constant feature of epithelioid fibrous histiocytoma. The other variant of DF that also tends to have a polypoid appearance is lipidized fibrous histiocytoma, or ankle-type fibrous histiocytoma, which usually arises below the knee, especially around the ankle, and often becomes larger than common DF.4 Lastly, atypical polypoid DF is a benign, polyp-shaped lesion that shows hypercellularity with striking nuclear atypism and scattered mitotic figures in addition to the ordinary histologic features of DF.3
Acquired fibrokeratomas manifest as solitary dome-shaped, flesh-colored protrusions usually located on the feet and hands. Sclerotic fibroma is a rare cutaneous neoplasm that presents as a solitary, translucent or waxy nodule or as multiple nodules when it is part of Cowden disease. Fibromas, also known as skin tags, are common cutaneous tumors that appear in intertriginous areas and frequently adopt a pedunculated morphology. Although clinically some of these lesions may resemble polypoid DF, the differential diagnosis is made by histologic examination.
Dermatofibroma is a common cutaneous tumor that follows a benign course. It can adopt multiple morphologies. Awareness of this rare variant may aid in its appropriate diagnosis and management. Dermatofibromas are benign neoplasms, and therefore they usually do not require treatment. If they become symptomatic or are bothersome to the patient, the treatment of choice is surgical removal.
The Diagnosis: Polypoid Dermatofibroma
Histologic examination revealed a poorly demarcated lesion localized in the dermis that was composed of an admixture of fibroblastlike cells, histiocytes, siderophages, multinucleated giant cells, and hemorrhage (Figure). Based on these findings, a diagnosis of polypoid dermatofibroma (DF) was made. No further treatment was necessary because the lesion was completely excised.
Dermatofibromas, also known as benign fibrous histiocytomas, are common, benign, mesenchymal, fibrosing tumors of the skin that appear predominantly on the legs in in women, but any part of the body in either sex can be affected. Clinically, DFs show the dimple sign when laterally squeezed and can be painful spontaneously or when rubbed. Histologically, DFs are poorly demarcated lesions composed of a variable admixture of fibroblastlike cells, histiocytes (some of which may be xanthomatous or multinucleated), and blood vessels. The etiology still remains unclear. Most investigators consider DF to be a reactive process, but some think that it is a benign mesenchymal tumor.1,2
Many subtypes of DF have been described based on their unique architectural, cellular, stromal, and clinical features.2,3 Polypoid DF is a rare variant that comprises only 3% of reported cases4 and tends to be larger than other DF subtypes. Requena et al5 reported 12 cases of giant DF, another clinical subtype, that were larger than 5 cm in diameter, most of which had a polypoid appearance.
Moreover, 3 distinct variants of DF with unique histologic features tend to show polypoid morphology.3,4 In epithelioid fibrous histiocytoma, also known as epithelioid cell histiocytoma, at least 50% of the lesion is composed of rounded or polygonal epithelioid cells with abundant eosinophilic cytoplasm and round to oval nuclei containing small eosinophilic nucleoli.4 A grenz zone generally is lacking and numerous small blood vessels are a constant feature of epithelioid fibrous histiocytoma. The other variant of DF that also tends to have a polypoid appearance is lipidized fibrous histiocytoma, or ankle-type fibrous histiocytoma, which usually arises below the knee, especially around the ankle, and often becomes larger than common DF.4 Lastly, atypical polypoid DF is a benign, polyp-shaped lesion that shows hypercellularity with striking nuclear atypism and scattered mitotic figures in addition to the ordinary histologic features of DF.3
Acquired fibrokeratomas manifest as solitary dome-shaped, flesh-colored protrusions usually located on the feet and hands. Sclerotic fibroma is a rare cutaneous neoplasm that presents as a solitary, translucent or waxy nodule or as multiple nodules when it is part of Cowden disease. Fibromas, also known as skin tags, are common cutaneous tumors that appear in intertriginous areas and frequently adopt a pedunculated morphology. Although clinically some of these lesions may resemble polypoid DF, the differential diagnosis is made by histologic examination.
Dermatofibroma is a common cutaneous tumor that follows a benign course. It can adopt multiple morphologies. Awareness of this rare variant may aid in its appropriate diagnosis and management. Dermatofibromas are benign neoplasms, and therefore they usually do not require treatment. If they become symptomatic or are bothersome to the patient, the treatment of choice is surgical removal.
- Luzar B, Calonje E. Cutaneous fibrohistiocytic tumours--an update. Histopathology. 2010;56:148-165.
- Santos-Briz A, Llamas-Velasco M, Arango L, et al. Cutaneous adenodermatofibroma: report of 2 cases. Am J Dermatopathol. 2013;35:E103-E105.
- Sogabe Y, Takahashi A, Tamura A, et al. A case of polypoid dermatofibroma. J Dermatol. 2002;29:786-789.
- Kai H, Fujita H, Yamamoto M, et al. Polypoid dermatofibroma with a slim pedicle: a case report. Dermatol Online J. 2012;18:16.
- Requena L, Farina K, Fuente C, et al. Giant dermatofibroma. a little known clinical variant of dermatofibroma. J Am Acad Dermatol. 1994;30:714-718.
- Luzar B, Calonje E. Cutaneous fibrohistiocytic tumours--an update. Histopathology. 2010;56:148-165.
- Santos-Briz A, Llamas-Velasco M, Arango L, et al. Cutaneous adenodermatofibroma: report of 2 cases. Am J Dermatopathol. 2013;35:E103-E105.
- Sogabe Y, Takahashi A, Tamura A, et al. A case of polypoid dermatofibroma. J Dermatol. 2002;29:786-789.
- Kai H, Fujita H, Yamamoto M, et al. Polypoid dermatofibroma with a slim pedicle: a case report. Dermatol Online J. 2012;18:16.
- Requena L, Farina K, Fuente C, et al. Giant dermatofibroma. a little known clinical variant of dermatofibroma. J Am Acad Dermatol. 1994;30:714-718.
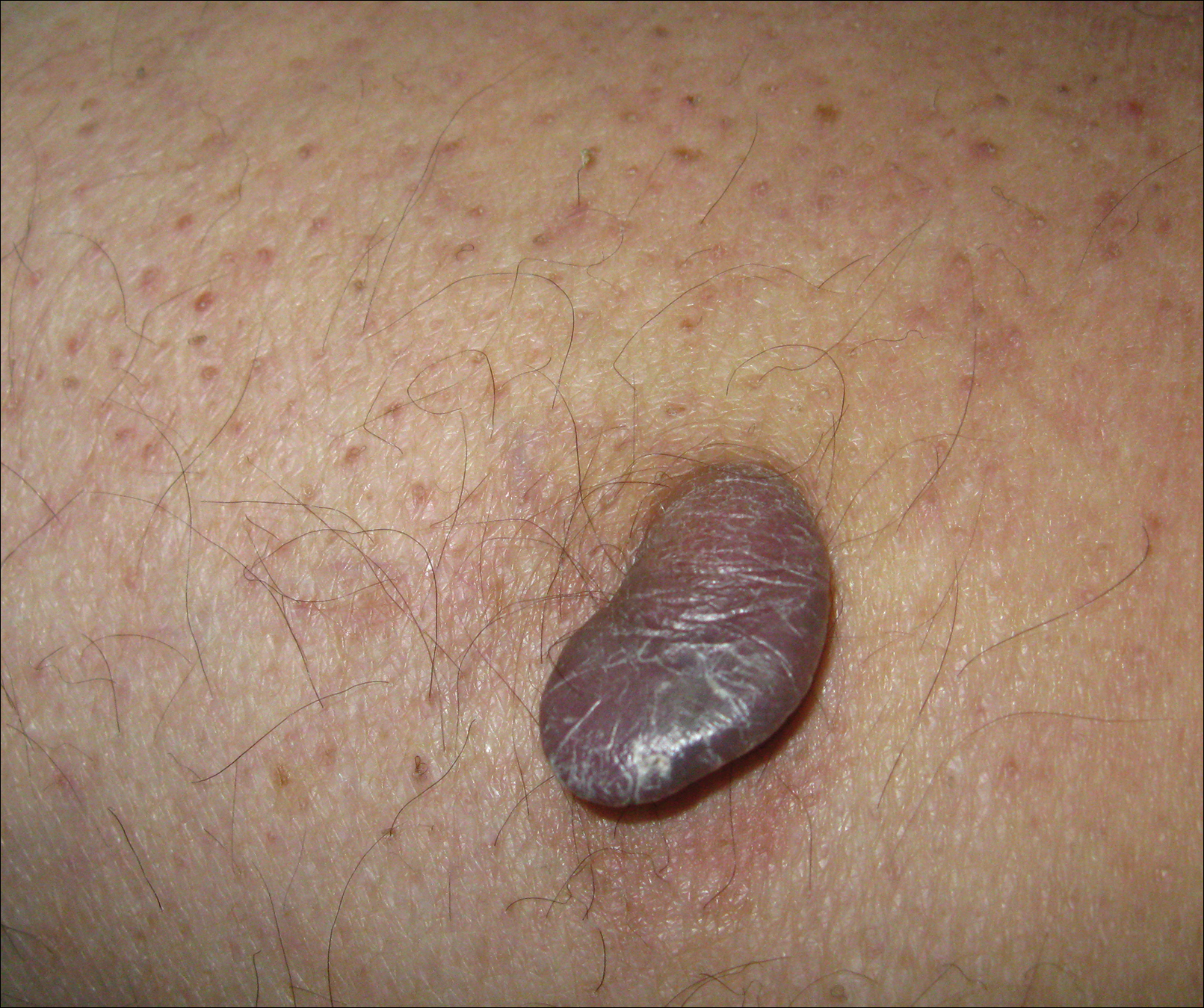
A 68-year-old man with a history of type 2 diabetes mellitus and hypercholesterolemia presented to the dermatology department with a cutaneous lesion on the posterior aspect of the right thigh of 2 years' duration. The lesion had become larger during the 4 months prior to presentation and was mostly asymptomatic but became tender when subjected to trauma. Physical examination revealed a firm, 2-cm, slightly pigmented peduncule on the posterior right thigh. No lymphadenopathies were noted. The lesion was completely excised for histologic examination.
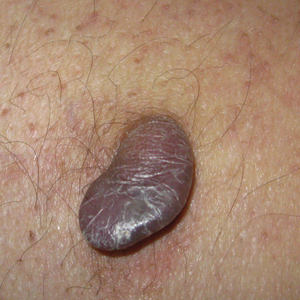
Recently approved SK treatment spares melanocytes in preclinical study
A study using an ex vivo model to evaluate a seborrheic keratosis (SK) treatment shows that a topical application of 40% hydrogen peroxide is gentler on skin than a 5- or 10-second treatment with liquid nitrogen, particularly with respect to melanocytes, suggesting that the former may be less likely to produce disfiguring damage.
The 40% hydrogen peroxide solution (Eskata), also known as A-101, received Food and Drug Administration approval for the treatment of “seborrheic keratoses that are raised” in December, 2017. The study was published online in the Journal of the American Academy of Dermatology.
Melanocyte damage can lead to significant dyschromia, a poor cosmetic outcome that can have a tremendous impact on quality of life for dark-skinned patients, in whom it produces white spots. “A lot of these destructive approaches, especially liquid nitrogen, can leave more disfigurement upon treatment than the lesion itself,” the study’s lead author Adam Friedman, MD, said in an interview.
Melanocytes are particularly vulnerable to the effects of cold, so the destructive potential of liquid nitrogen is no surprise. But Dr. Friedman of the department of dermatology, George Washington University, Washington, wanted to get a better understanding of the impact of the new treatment on different skin cell types and the toxicity profile, so he approached the manufacturer, Aclaris Therapeutics, to do a study.
His team tested 40% hydrogen peroxide treatment and liquid nitrogen cryosurgery on a validated ex vivo human reconstituted full-thickness model derived from Fitzpatrick V skin, with 5 or 10 seconds of cryosurgery or 1 or 2 mcL of 40% hydrogen peroxide.
Using standard a hematoxylin and eosin stain as well as immunohistochemical staining to examine the architecture and cells types of the skin model following both treatments, the researchers found that 5- and 10-second cryosurgery resulted in significant thinning of the epidermis and increased cell death. There was also separation at the dermal-epidermal junction, which was more prominent in the 10-second cryosurgery group, although present even with a 5-second freeze cycle.
The hydrogen peroxide–treated groups showed acanthosis of the epidermis and mild pallor, but this was less noticeable than in the cryosurgery specimens. There was no epidermal clefting in the hydrogen peroxide–treated samples.
Tunel staining revealed 16.4 (plus or minus 0.6424) apoptotic cells per high-powered field in the 5-second cryosurgery experiment and 20.6 (plus or minus 0.6424) in the 10-second procedure. For the hydrogen peroxide treatment, those numbers were 8.65 (plus or minus 0.4122) in the 1 mcL experiment and 12.4 (plus or minus 0.3728) in the 2 mcL experiment.
As expected, melanocytes fared better with the hydrogen peroxide treatment. In the untreated samples, there were 2.5 melanocytes (plus or minus 0.1987) in the untreated sample and 2.0 (plus or minus 0.5000) melanocytes in the vehicle-treated sample. In the 5-second cryosurgery sample, there were 0.45 melanocytes (plus or minus 0.1535), and in the 10 second cryosurgery sample there were 0.2 (plus or minus 0.0918) melanocytes. In contrast, with the 1-mcL hydrogen peroxide-treated sample, there were 1.95 melanocytes in both the 1-mcL and 2-mcL samples (plus or minus 0.1535 for both groups).
“,” Dr. Friedman said. These results, he added, “offer us a lot of insight in terms of how damaging liquid nitrogen is, and it’s good to be reminded of that so that we don’t cause too much harm.”
The authors noted that a clinical trial evaluating the risk of hypopigmentation and hyperpigmentation with 40% hydrogen peroxide in people with darker skin types is underway. In the study, hydrogen peroxide is used to treat dermatosis papulosa nigra.
The study was funded by Aclaris Therapeutics. Senior author Adam Friedman, MD, is a consultant for Aclaris. Dr. Friedman is on the editorial board of Dermatology News.
SOURCE: Kao S et al. J Am Acad Dermatol. 2018 Mar 27. doi: 10.1016/j.jaad.2018.03.034.
A study using an ex vivo model to evaluate a seborrheic keratosis (SK) treatment shows that a topical application of 40% hydrogen peroxide is gentler on skin than a 5- or 10-second treatment with liquid nitrogen, particularly with respect to melanocytes, suggesting that the former may be less likely to produce disfiguring damage.
The 40% hydrogen peroxide solution (Eskata), also known as A-101, received Food and Drug Administration approval for the treatment of “seborrheic keratoses that are raised” in December, 2017. The study was published online in the Journal of the American Academy of Dermatology.
Melanocyte damage can lead to significant dyschromia, a poor cosmetic outcome that can have a tremendous impact on quality of life for dark-skinned patients, in whom it produces white spots. “A lot of these destructive approaches, especially liquid nitrogen, can leave more disfigurement upon treatment than the lesion itself,” the study’s lead author Adam Friedman, MD, said in an interview.
Melanocytes are particularly vulnerable to the effects of cold, so the destructive potential of liquid nitrogen is no surprise. But Dr. Friedman of the department of dermatology, George Washington University, Washington, wanted to get a better understanding of the impact of the new treatment on different skin cell types and the toxicity profile, so he approached the manufacturer, Aclaris Therapeutics, to do a study.
His team tested 40% hydrogen peroxide treatment and liquid nitrogen cryosurgery on a validated ex vivo human reconstituted full-thickness model derived from Fitzpatrick V skin, with 5 or 10 seconds of cryosurgery or 1 or 2 mcL of 40% hydrogen peroxide.
Using standard a hematoxylin and eosin stain as well as immunohistochemical staining to examine the architecture and cells types of the skin model following both treatments, the researchers found that 5- and 10-second cryosurgery resulted in significant thinning of the epidermis and increased cell death. There was also separation at the dermal-epidermal junction, which was more prominent in the 10-second cryosurgery group, although present even with a 5-second freeze cycle.
The hydrogen peroxide–treated groups showed acanthosis of the epidermis and mild pallor, but this was less noticeable than in the cryosurgery specimens. There was no epidermal clefting in the hydrogen peroxide–treated samples.
Tunel staining revealed 16.4 (plus or minus 0.6424) apoptotic cells per high-powered field in the 5-second cryosurgery experiment and 20.6 (plus or minus 0.6424) in the 10-second procedure. For the hydrogen peroxide treatment, those numbers were 8.65 (plus or minus 0.4122) in the 1 mcL experiment and 12.4 (plus or minus 0.3728) in the 2 mcL experiment.
As expected, melanocytes fared better with the hydrogen peroxide treatment. In the untreated samples, there were 2.5 melanocytes (plus or minus 0.1987) in the untreated sample and 2.0 (plus or minus 0.5000) melanocytes in the vehicle-treated sample. In the 5-second cryosurgery sample, there were 0.45 melanocytes (plus or minus 0.1535), and in the 10 second cryosurgery sample there were 0.2 (plus or minus 0.0918) melanocytes. In contrast, with the 1-mcL hydrogen peroxide-treated sample, there were 1.95 melanocytes in both the 1-mcL and 2-mcL samples (plus or minus 0.1535 for both groups).
“,” Dr. Friedman said. These results, he added, “offer us a lot of insight in terms of how damaging liquid nitrogen is, and it’s good to be reminded of that so that we don’t cause too much harm.”
The authors noted that a clinical trial evaluating the risk of hypopigmentation and hyperpigmentation with 40% hydrogen peroxide in people with darker skin types is underway. In the study, hydrogen peroxide is used to treat dermatosis papulosa nigra.
The study was funded by Aclaris Therapeutics. Senior author Adam Friedman, MD, is a consultant for Aclaris. Dr. Friedman is on the editorial board of Dermatology News.
SOURCE: Kao S et al. J Am Acad Dermatol. 2018 Mar 27. doi: 10.1016/j.jaad.2018.03.034.
A study using an ex vivo model to evaluate a seborrheic keratosis (SK) treatment shows that a topical application of 40% hydrogen peroxide is gentler on skin than a 5- or 10-second treatment with liquid nitrogen, particularly with respect to melanocytes, suggesting that the former may be less likely to produce disfiguring damage.
The 40% hydrogen peroxide solution (Eskata), also known as A-101, received Food and Drug Administration approval for the treatment of “seborrheic keratoses that are raised” in December, 2017. The study was published online in the Journal of the American Academy of Dermatology.
Melanocyte damage can lead to significant dyschromia, a poor cosmetic outcome that can have a tremendous impact on quality of life for dark-skinned patients, in whom it produces white spots. “A lot of these destructive approaches, especially liquid nitrogen, can leave more disfigurement upon treatment than the lesion itself,” the study’s lead author Adam Friedman, MD, said in an interview.
Melanocytes are particularly vulnerable to the effects of cold, so the destructive potential of liquid nitrogen is no surprise. But Dr. Friedman of the department of dermatology, George Washington University, Washington, wanted to get a better understanding of the impact of the new treatment on different skin cell types and the toxicity profile, so he approached the manufacturer, Aclaris Therapeutics, to do a study.
His team tested 40% hydrogen peroxide treatment and liquid nitrogen cryosurgery on a validated ex vivo human reconstituted full-thickness model derived from Fitzpatrick V skin, with 5 or 10 seconds of cryosurgery or 1 or 2 mcL of 40% hydrogen peroxide.
Using standard a hematoxylin and eosin stain as well as immunohistochemical staining to examine the architecture and cells types of the skin model following both treatments, the researchers found that 5- and 10-second cryosurgery resulted in significant thinning of the epidermis and increased cell death. There was also separation at the dermal-epidermal junction, which was more prominent in the 10-second cryosurgery group, although present even with a 5-second freeze cycle.
The hydrogen peroxide–treated groups showed acanthosis of the epidermis and mild pallor, but this was less noticeable than in the cryosurgery specimens. There was no epidermal clefting in the hydrogen peroxide–treated samples.
Tunel staining revealed 16.4 (plus or minus 0.6424) apoptotic cells per high-powered field in the 5-second cryosurgery experiment and 20.6 (plus or minus 0.6424) in the 10-second procedure. For the hydrogen peroxide treatment, those numbers were 8.65 (plus or minus 0.4122) in the 1 mcL experiment and 12.4 (plus or minus 0.3728) in the 2 mcL experiment.
As expected, melanocytes fared better with the hydrogen peroxide treatment. In the untreated samples, there were 2.5 melanocytes (plus or minus 0.1987) in the untreated sample and 2.0 (plus or minus 0.5000) melanocytes in the vehicle-treated sample. In the 5-second cryosurgery sample, there were 0.45 melanocytes (plus or minus 0.1535), and in the 10 second cryosurgery sample there were 0.2 (plus or minus 0.0918) melanocytes. In contrast, with the 1-mcL hydrogen peroxide-treated sample, there were 1.95 melanocytes in both the 1-mcL and 2-mcL samples (plus or minus 0.1535 for both groups).
“,” Dr. Friedman said. These results, he added, “offer us a lot of insight in terms of how damaging liquid nitrogen is, and it’s good to be reminded of that so that we don’t cause too much harm.”
The authors noted that a clinical trial evaluating the risk of hypopigmentation and hyperpigmentation with 40% hydrogen peroxide in people with darker skin types is underway. In the study, hydrogen peroxide is used to treat dermatosis papulosa nigra.
The study was funded by Aclaris Therapeutics. Senior author Adam Friedman, MD, is a consultant for Aclaris. Dr. Friedman is on the editorial board of Dermatology News.
SOURCE: Kao S et al. J Am Acad Dermatol. 2018 Mar 27. doi: 10.1016/j.jaad.2018.03.034.
FROM JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Key clinical point: The results of a preclinical study using an ex vivo model of darker skin found that a 40% hydrogen peroxide solution for treating SKs was less damaging to the skin.
Major finding: The topical hydrogen peroxide treatment left 1.95 melanocytes in place, compared with 0.2-0.4 in the liquid nitrogen samples. As expected, melanocytes fared better with the hydrogen peroxide treatment. In the untreated samples, there were 2.5 melanocytes (plus or minus 0.1987) in the untreated sample and 2.0 (plus or minus 0.5000) melanocytes in the vehicle-treated sample. In the 5-second cryosurgery sample, there were 0.45 melanocytes (plus or minus 0.1535), and in the 10-second cryosurgery sample there were 0.2 (plus or minus 0.0918) melanocytes.
Study details: The study compared the cytotoxic effects and impact on melanocytes of liquid nitrogen and 40% hydrogen peroxide solution using ex vivo human reconstituted full-thickness model.
Disclosures: The study was funded by Aclaris Therapeutics. Senior author Adam Friedman, MD, is a consultant for Aclaris.
Source: Kao S. et al. J Am Acad Dermatol. 2018 Mar 27. doi: 10.1016/j.jaad.2018.03.034.