User login
Hyperkeratotic Nummular Plaques on the Upper Trunk
The Diagnosis: Extragenital Lichen Sclerosus Et Atrophicus
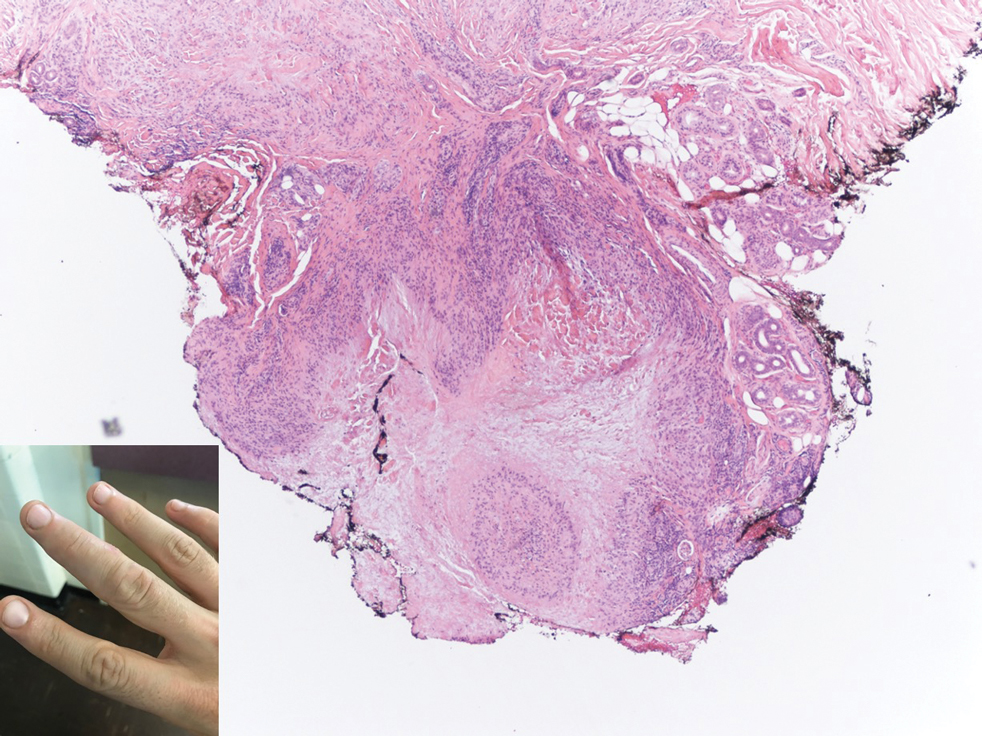
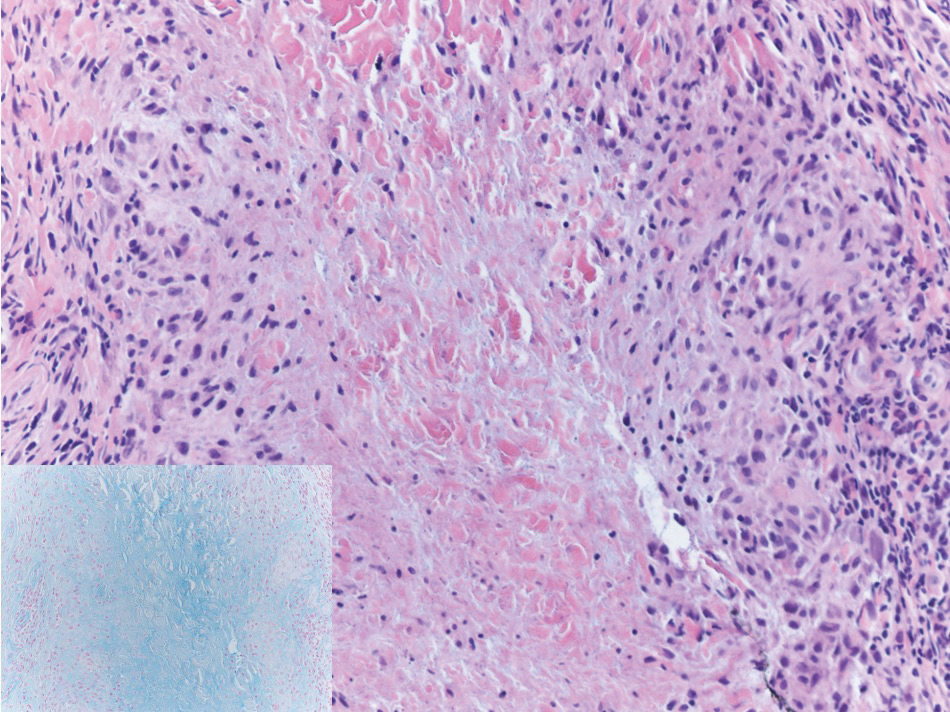
Histopathologic evaluation revealed hyperkeratosis, follicular plugging, epidermal atrophy, and homogenization of papillary dermal collagen with an underlying lymphocytic infiltrate (Figure 1). Direct immunofluorescence of a plaque with a superimposed bulla was negative for deposition of C3, IgG, IgA, IgM, or fibrinogen. Accordingly, clinicopathologic correlation supported a diagnosis of extragenital lichen sclerosus et atrophicus (LSA). Of note, the patient's history of genital irritation was due to genital LSA that preceded the extragenital manifestations.
Lichen sclerosus et atrophicus is an inflammatory dermatosis that typically presents as atrophic white papules of the anogenital area that coalesce into pruritic plaques; the exact etiology remains to be elucidated, yet various circulating autoantibodies have been identified, suggesting a role for autoimmunity.1,2 Lichen sclerosus et atrophicus is more common in women than in men, with a bimodal peak in the age of onset affecting postmenopausal and prepubertal populations.1 In women, affected areas include the labia minora and majora, clitoris, perineum, and perianal skin; LSA spares the mucosal surfaces of the vagina and cervix.2 In men, uncircumscribed genital skin more commonly is affected. Involvement is localized to the foreskin and glans with occasional urethral involvement.2
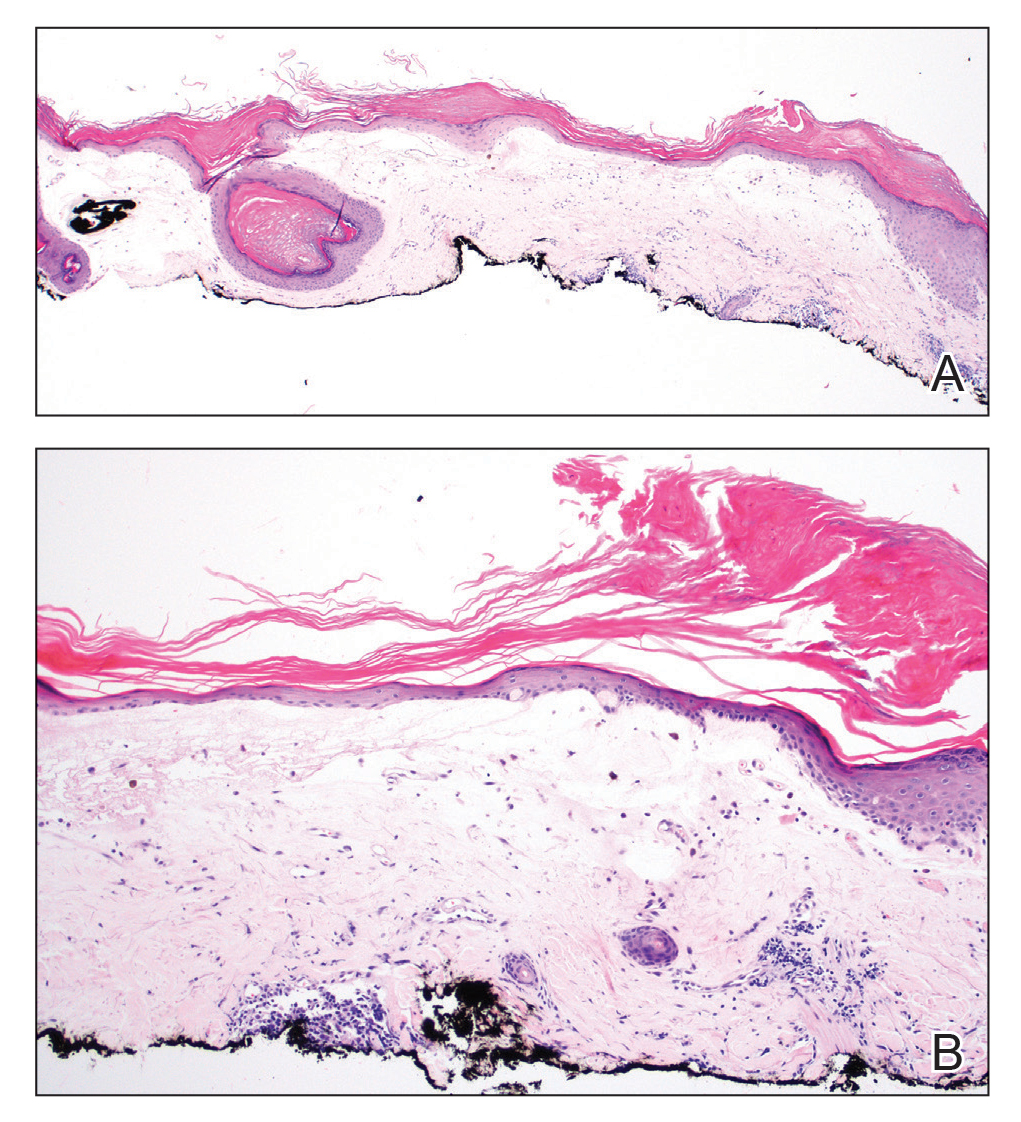
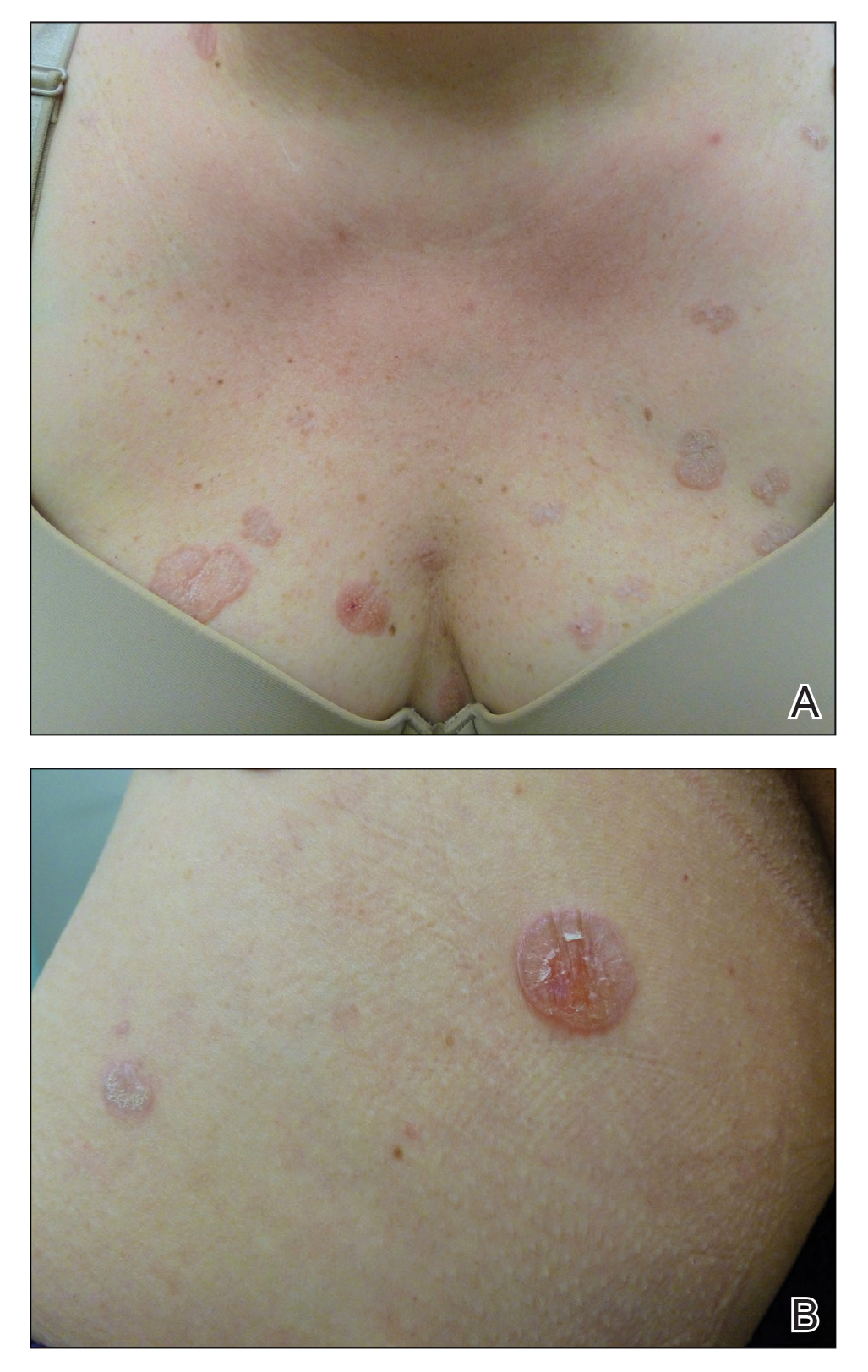
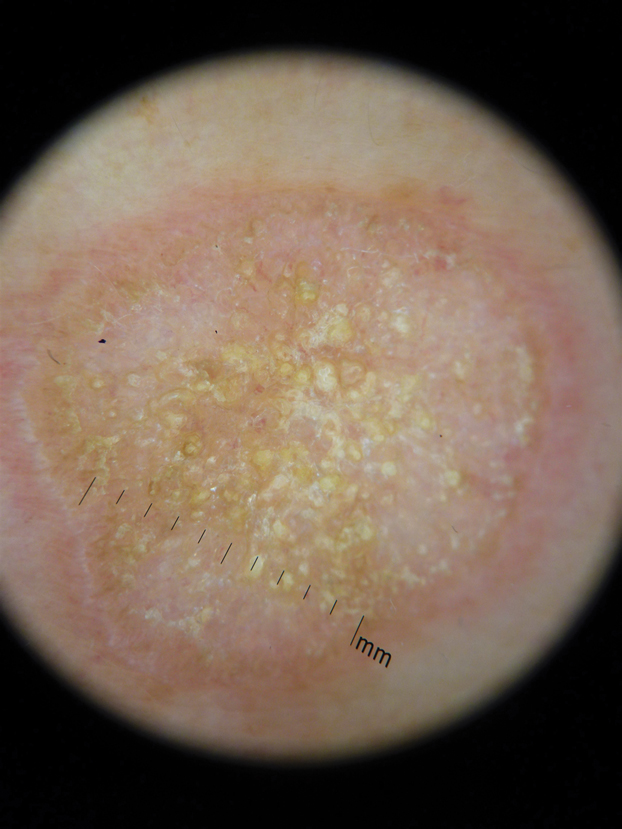
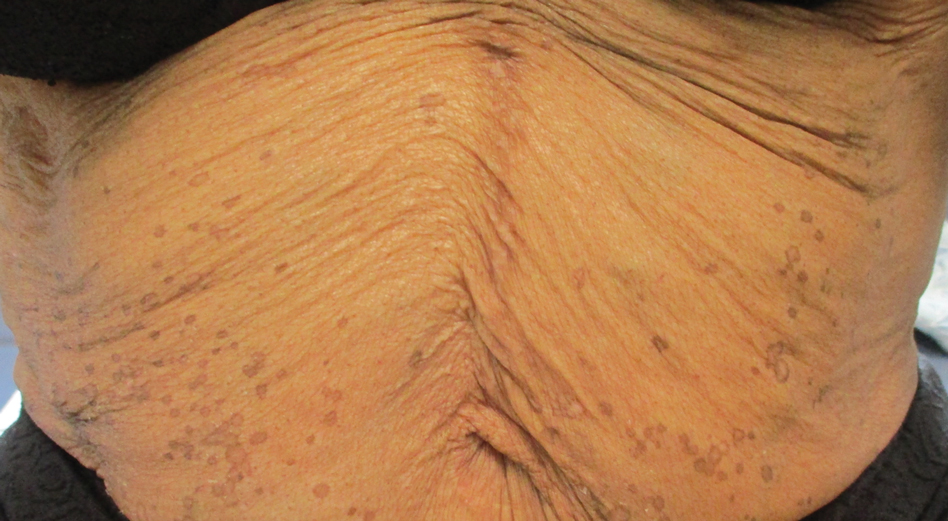
In contrast, extragenital LSA tends to present as asymptomatic papules and plaques that develop atrophy with time, involving the back, shoulders, neck, chest, thighs, axillae, and flexural wrists2,3; an erythematous rim often is present,4 and hyperkeratosis with follicular plugging may be prominent.5 Our patient's case emphasizes the predilection of plaques for the chest and intermammary skin (Figure 2A). Approximately 15% of LSA cases have extragenital involvement, and extragenital-limited disease accounts for roughly 5% of cases.6,7 Unlike genital LSA, extragenital disease has not been associated with an increased risk for squamous cell carcinoma.1 Bullae formation within plaques of genital or extragenital LSA has been reported3,8 and is exemplified in our patient (Figure 2B). Intralesional bullae formation likely is due to a combination of internal and external factors, mainly the inability to withstand shear forces due to an atrophic epidermis with basal vacuolar injury overlying an edematous papillary dermis with altered collagen.8 Dermatoscopic findings may aid in recognizing extragenital LSA9,10; our patient's plaques demonstrated the characteristic findings of comedolike openings, structureless white areas, and pink borders (Figure 3).
The clinical differential diagnosis for well-demarcated, pink, scaly plaques is broad. Nummular eczema usually presents as coin-shaped eczematous plaques on the dorsal aspects of the hands or lower extremities, and histology shows epidermal spongiosis.11 Nummular eczema may be considered due to the striking round morphology of various plaques, yet our patient's presentation was better served by a consideration of several papulosquamous disorders.
Lichen planus (LP) presents as intensely pruritic, violaceous, polygonal, flat-topped papules with overlying reticular white lines, or Wickham striae, that favor the flexural wrists, lower back, and lower extremities. Lichen planus also may have oral and genital mucosal involvement. Similar to LSA, LP is more common in women and preferentially affects the postmenopausal population.12 Additionally, hypertrophic LP may obscure Wickham striae and mimic extragenital LSA; distinguishing features of hypertrophic LP are intense pruritus and a predilection for the shins. Histology is defined by orthohyperkeratosis, hypergranulosis, sawtooth acanthosis, and vacuolar degeneration of the basal layer with Civatte bodies or dyskeratotic basal keratinocytes overlying a characteristic bandlike infiltrate of lymphocytes.12
Lichen simplex chronicus (LSC) is characterized by intense pruritus and presents as hyperkeratotic plaques with a predilection for accessible regions such as the posterior neck and extremities.13 The striking annular demarcation of this case makes LSC unlikely. Comparable to LSA and LP, LSC also may present with both genital and extragenital findings. Histology of LSC is characterized by irregular acanthosis or thickening of the epidermis with vertical streaking of collagen and vascular bundles of the papillary dermis.13
Subacute cutaneous lupus erythematosus (SCLE) is important to consider for a new papulosquamous eruption with a predilection for the sun-exposed skin of a middle-aged woman. The presence of papules on the volar wrist and history of genital irritation, however, make this entity less likely. Similar to LSA, histologic examination of SCLE reveals epidermal atrophy, basal layer degeneration, and papillary dermal edema with lymphocytic inflammation. However, SCLE lacks the band of inflammation underlying pale homogenized papillary dermal collagen, the most distinguishing feature of LSA; instead, SCLE shows superficial and deep perivascular and periadnexal lymphocytes and mucin in the dermis.14
Lichen sclerosus et atrophicus may be chronic and progressive in nature or cycle through remissions and relapses.2 Treatment is not curative, and management is directed to alleviating symptoms and preventing the progression of disease. First-line management of extragenital LSA is potent topical steroids.1 Adjuvant topical calcineurin inhibitors may be used as steroid-sparing agents.2 Phototherapy is a second-line therapy and even narrowband UVB phototherapy has demonstrated efficacy in managing extragenital LSA.15,16 Our patient was started on mometasone ointment and calcipotriene cream with slight improvement after a 6-month trial. Ongoing management is focused on optimizing application of topical therapies.
- Powell JJ, Wojnarowska F. Lichen sclerosus. Lancet. 1999;353:1777-1783.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus. Am J Clin Dermatol. 2013;14:27-47.
- Meffert JJ, Davis BM, Grimwood RE. Lichen sclerosus. J Am Acad Dermatol. 1995;32:393-416.
- Surkan M, Hull P. A case of lichen sclerosus et atrophicus with distinct erythematous borders. J Cutan Med Surg. 2015;19:600-603.
- Kimura A, Kambe N, Satoh T, et al. Follicular keratosis and bullous formation are typical signs of extragenital lichen sclerosus. J Dermatol. 2011;38:834-836.
- Meyrick Thomas RH, Ridley CM, McGibbon DH, et al. Lichen sclerosus et atrophicus and autoimmunity: a study of 350 women. Br J Dermatol. 1988;118:41-46.
- Wallace HJ. Lichen sclerosus et atrophicus. Trans St Johns Hosp Dermatol Soc. 1971;57:9-30.
- Hallel-Halevy D, Grunwald MH, Yerushalmi J, et al. Bullous lichen sclerosus et atrophicus. J Am Acad Dermatol. 1998;39:500-501.
- Garrido-Ríos AA, Álvarez-Garrido H, Sanz-Muñoz C, et al. Dermoscopy of extragenital lichen sclerosus. Arch Dermatol. 2009;145:1468.
- Larre Borges A, Tiodorovic-Zivkovic D, Lallas A, et al. Clinical, dermoscopic and histopathologic features of genital and extragenital lichen sclerosus. J Eur Acad Dermatol Venereol. 2013;27:1433-1439.
- Rudikoff D. Differential diagnosis of round or discoid lesions. Clin Dermatol. 2011;29:489-497.
- Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol. 1991;25:593-619.
- Shaffer B, Beerman H. Lichen simplex chronicus and its variants: a discussion of certain psychodynamic mechanisms and clinical and histopathologic correlations. AMA Arch Derm Syphilol. 1951;64:340-351.
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus. Am J Clin Dermatol. 2009;10:365-381.
- Sauder MB, Linzon-Smith J, Beecker J. Extragenital bullous lichen sclerosus. J Am Acad Dermatol. 2014;71:981-984.
- Colbert RL, Chiang MP, Carlin CS, et al. Progressive extragenital lichen sclerosus successfully treated with narrowband UV-B phototherapy. Arch Dermatol. 2007;143:19-20.
The Diagnosis: Extragenital Lichen Sclerosus Et Atrophicus
Histopathologic evaluation revealed hyperkeratosis, follicular plugging, epidermal atrophy, and homogenization of papillary dermal collagen with an underlying lymphocytic infiltrate (Figure 1). Direct immunofluorescence of a plaque with a superimposed bulla was negative for deposition of C3, IgG, IgA, IgM, or fibrinogen. Accordingly, clinicopathologic correlation supported a diagnosis of extragenital lichen sclerosus et atrophicus (LSA). Of note, the patient's history of genital irritation was due to genital LSA that preceded the extragenital manifestations.
Lichen sclerosus et atrophicus is an inflammatory dermatosis that typically presents as atrophic white papules of the anogenital area that coalesce into pruritic plaques; the exact etiology remains to be elucidated, yet various circulating autoantibodies have been identified, suggesting a role for autoimmunity.1,2 Lichen sclerosus et atrophicus is more common in women than in men, with a bimodal peak in the age of onset affecting postmenopausal and prepubertal populations.1 In women, affected areas include the labia minora and majora, clitoris, perineum, and perianal skin; LSA spares the mucosal surfaces of the vagina and cervix.2 In men, uncircumscribed genital skin more commonly is affected. Involvement is localized to the foreskin and glans with occasional urethral involvement.2
In contrast, extragenital LSA tends to present as asymptomatic papules and plaques that develop atrophy with time, involving the back, shoulders, neck, chest, thighs, axillae, and flexural wrists2,3; an erythematous rim often is present,4 and hyperkeratosis with follicular plugging may be prominent.5 Our patient's case emphasizes the predilection of plaques for the chest and intermammary skin (Figure 2A). Approximately 15% of LSA cases have extragenital involvement, and extragenital-limited disease accounts for roughly 5% of cases.6,7 Unlike genital LSA, extragenital disease has not been associated with an increased risk for squamous cell carcinoma.1 Bullae formation within plaques of genital or extragenital LSA has been reported3,8 and is exemplified in our patient (Figure 2B). Intralesional bullae formation likely is due to a combination of internal and external factors, mainly the inability to withstand shear forces due to an atrophic epidermis with basal vacuolar injury overlying an edematous papillary dermis with altered collagen.8 Dermatoscopic findings may aid in recognizing extragenital LSA9,10; our patient's plaques demonstrated the characteristic findings of comedolike openings, structureless white areas, and pink borders (Figure 3).
The clinical differential diagnosis for well-demarcated, pink, scaly plaques is broad. Nummular eczema usually presents as coin-shaped eczematous plaques on the dorsal aspects of the hands or lower extremities, and histology shows epidermal spongiosis.11 Nummular eczema may be considered due to the striking round morphology of various plaques, yet our patient's presentation was better served by a consideration of several papulosquamous disorders.
Lichen planus (LP) presents as intensely pruritic, violaceous, polygonal, flat-topped papules with overlying reticular white lines, or Wickham striae, that favor the flexural wrists, lower back, and lower extremities. Lichen planus also may have oral and genital mucosal involvement. Similar to LSA, LP is more common in women and preferentially affects the postmenopausal population.12 Additionally, hypertrophic LP may obscure Wickham striae and mimic extragenital LSA; distinguishing features of hypertrophic LP are intense pruritus and a predilection for the shins. Histology is defined by orthohyperkeratosis, hypergranulosis, sawtooth acanthosis, and vacuolar degeneration of the basal layer with Civatte bodies or dyskeratotic basal keratinocytes overlying a characteristic bandlike infiltrate of lymphocytes.12
Lichen simplex chronicus (LSC) is characterized by intense pruritus and presents as hyperkeratotic plaques with a predilection for accessible regions such as the posterior neck and extremities.13 The striking annular demarcation of this case makes LSC unlikely. Comparable to LSA and LP, LSC also may present with both genital and extragenital findings. Histology of LSC is characterized by irregular acanthosis or thickening of the epidermis with vertical streaking of collagen and vascular bundles of the papillary dermis.13
Subacute cutaneous lupus erythematosus (SCLE) is important to consider for a new papulosquamous eruption with a predilection for the sun-exposed skin of a middle-aged woman. The presence of papules on the volar wrist and history of genital irritation, however, make this entity less likely. Similar to LSA, histologic examination of SCLE reveals epidermal atrophy, basal layer degeneration, and papillary dermal edema with lymphocytic inflammation. However, SCLE lacks the band of inflammation underlying pale homogenized papillary dermal collagen, the most distinguishing feature of LSA; instead, SCLE shows superficial and deep perivascular and periadnexal lymphocytes and mucin in the dermis.14
Lichen sclerosus et atrophicus may be chronic and progressive in nature or cycle through remissions and relapses.2 Treatment is not curative, and management is directed to alleviating symptoms and preventing the progression of disease. First-line management of extragenital LSA is potent topical steroids.1 Adjuvant topical calcineurin inhibitors may be used as steroid-sparing agents.2 Phototherapy is a second-line therapy and even narrowband UVB phototherapy has demonstrated efficacy in managing extragenital LSA.15,16 Our patient was started on mometasone ointment and calcipotriene cream with slight improvement after a 6-month trial. Ongoing management is focused on optimizing application of topical therapies.
The Diagnosis: Extragenital Lichen Sclerosus Et Atrophicus
Histopathologic evaluation revealed hyperkeratosis, follicular plugging, epidermal atrophy, and homogenization of papillary dermal collagen with an underlying lymphocytic infiltrate (Figure 1). Direct immunofluorescence of a plaque with a superimposed bulla was negative for deposition of C3, IgG, IgA, IgM, or fibrinogen. Accordingly, clinicopathologic correlation supported a diagnosis of extragenital lichen sclerosus et atrophicus (LSA). Of note, the patient's history of genital irritation was due to genital LSA that preceded the extragenital manifestations.
Lichen sclerosus et atrophicus is an inflammatory dermatosis that typically presents as atrophic white papules of the anogenital area that coalesce into pruritic plaques; the exact etiology remains to be elucidated, yet various circulating autoantibodies have been identified, suggesting a role for autoimmunity.1,2 Lichen sclerosus et atrophicus is more common in women than in men, with a bimodal peak in the age of onset affecting postmenopausal and prepubertal populations.1 In women, affected areas include the labia minora and majora, clitoris, perineum, and perianal skin; LSA spares the mucosal surfaces of the vagina and cervix.2 In men, uncircumscribed genital skin more commonly is affected. Involvement is localized to the foreskin and glans with occasional urethral involvement.2
In contrast, extragenital LSA tends to present as asymptomatic papules and plaques that develop atrophy with time, involving the back, shoulders, neck, chest, thighs, axillae, and flexural wrists2,3; an erythematous rim often is present,4 and hyperkeratosis with follicular plugging may be prominent.5 Our patient's case emphasizes the predilection of plaques for the chest and intermammary skin (Figure 2A). Approximately 15% of LSA cases have extragenital involvement, and extragenital-limited disease accounts for roughly 5% of cases.6,7 Unlike genital LSA, extragenital disease has not been associated with an increased risk for squamous cell carcinoma.1 Bullae formation within plaques of genital or extragenital LSA has been reported3,8 and is exemplified in our patient (Figure 2B). Intralesional bullae formation likely is due to a combination of internal and external factors, mainly the inability to withstand shear forces due to an atrophic epidermis with basal vacuolar injury overlying an edematous papillary dermis with altered collagen.8 Dermatoscopic findings may aid in recognizing extragenital LSA9,10; our patient's plaques demonstrated the characteristic findings of comedolike openings, structureless white areas, and pink borders (Figure 3).
The clinical differential diagnosis for well-demarcated, pink, scaly plaques is broad. Nummular eczema usually presents as coin-shaped eczematous plaques on the dorsal aspects of the hands or lower extremities, and histology shows epidermal spongiosis.11 Nummular eczema may be considered due to the striking round morphology of various plaques, yet our patient's presentation was better served by a consideration of several papulosquamous disorders.
Lichen planus (LP) presents as intensely pruritic, violaceous, polygonal, flat-topped papules with overlying reticular white lines, or Wickham striae, that favor the flexural wrists, lower back, and lower extremities. Lichen planus also may have oral and genital mucosal involvement. Similar to LSA, LP is more common in women and preferentially affects the postmenopausal population.12 Additionally, hypertrophic LP may obscure Wickham striae and mimic extragenital LSA; distinguishing features of hypertrophic LP are intense pruritus and a predilection for the shins. Histology is defined by orthohyperkeratosis, hypergranulosis, sawtooth acanthosis, and vacuolar degeneration of the basal layer with Civatte bodies or dyskeratotic basal keratinocytes overlying a characteristic bandlike infiltrate of lymphocytes.12
Lichen simplex chronicus (LSC) is characterized by intense pruritus and presents as hyperkeratotic plaques with a predilection for accessible regions such as the posterior neck and extremities.13 The striking annular demarcation of this case makes LSC unlikely. Comparable to LSA and LP, LSC also may present with both genital and extragenital findings. Histology of LSC is characterized by irregular acanthosis or thickening of the epidermis with vertical streaking of collagen and vascular bundles of the papillary dermis.13
Subacute cutaneous lupus erythematosus (SCLE) is important to consider for a new papulosquamous eruption with a predilection for the sun-exposed skin of a middle-aged woman. The presence of papules on the volar wrist and history of genital irritation, however, make this entity less likely. Similar to LSA, histologic examination of SCLE reveals epidermal atrophy, basal layer degeneration, and papillary dermal edema with lymphocytic inflammation. However, SCLE lacks the band of inflammation underlying pale homogenized papillary dermal collagen, the most distinguishing feature of LSA; instead, SCLE shows superficial and deep perivascular and periadnexal lymphocytes and mucin in the dermis.14
Lichen sclerosus et atrophicus may be chronic and progressive in nature or cycle through remissions and relapses.2 Treatment is not curative, and management is directed to alleviating symptoms and preventing the progression of disease. First-line management of extragenital LSA is potent topical steroids.1 Adjuvant topical calcineurin inhibitors may be used as steroid-sparing agents.2 Phototherapy is a second-line therapy and even narrowband UVB phototherapy has demonstrated efficacy in managing extragenital LSA.15,16 Our patient was started on mometasone ointment and calcipotriene cream with slight improvement after a 6-month trial. Ongoing management is focused on optimizing application of topical therapies.
- Powell JJ, Wojnarowska F. Lichen sclerosus. Lancet. 1999;353:1777-1783.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus. Am J Clin Dermatol. 2013;14:27-47.
- Meffert JJ, Davis BM, Grimwood RE. Lichen sclerosus. J Am Acad Dermatol. 1995;32:393-416.
- Surkan M, Hull P. A case of lichen sclerosus et atrophicus with distinct erythematous borders. J Cutan Med Surg. 2015;19:600-603.
- Kimura A, Kambe N, Satoh T, et al. Follicular keratosis and bullous formation are typical signs of extragenital lichen sclerosus. J Dermatol. 2011;38:834-836.
- Meyrick Thomas RH, Ridley CM, McGibbon DH, et al. Lichen sclerosus et atrophicus and autoimmunity: a study of 350 women. Br J Dermatol. 1988;118:41-46.
- Wallace HJ. Lichen sclerosus et atrophicus. Trans St Johns Hosp Dermatol Soc. 1971;57:9-30.
- Hallel-Halevy D, Grunwald MH, Yerushalmi J, et al. Bullous lichen sclerosus et atrophicus. J Am Acad Dermatol. 1998;39:500-501.
- Garrido-Ríos AA, Álvarez-Garrido H, Sanz-Muñoz C, et al. Dermoscopy of extragenital lichen sclerosus. Arch Dermatol. 2009;145:1468.
- Larre Borges A, Tiodorovic-Zivkovic D, Lallas A, et al. Clinical, dermoscopic and histopathologic features of genital and extragenital lichen sclerosus. J Eur Acad Dermatol Venereol. 2013;27:1433-1439.
- Rudikoff D. Differential diagnosis of round or discoid lesions. Clin Dermatol. 2011;29:489-497.
- Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol. 1991;25:593-619.
- Shaffer B, Beerman H. Lichen simplex chronicus and its variants: a discussion of certain psychodynamic mechanisms and clinical and histopathologic correlations. AMA Arch Derm Syphilol. 1951;64:340-351.
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus. Am J Clin Dermatol. 2009;10:365-381.
- Sauder MB, Linzon-Smith J, Beecker J. Extragenital bullous lichen sclerosus. J Am Acad Dermatol. 2014;71:981-984.
- Colbert RL, Chiang MP, Carlin CS, et al. Progressive extragenital lichen sclerosus successfully treated with narrowband UV-B phototherapy. Arch Dermatol. 2007;143:19-20.
- Powell JJ, Wojnarowska F. Lichen sclerosus. Lancet. 1999;353:1777-1783.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus. Am J Clin Dermatol. 2013;14:27-47.
- Meffert JJ, Davis BM, Grimwood RE. Lichen sclerosus. J Am Acad Dermatol. 1995;32:393-416.
- Surkan M, Hull P. A case of lichen sclerosus et atrophicus with distinct erythematous borders. J Cutan Med Surg. 2015;19:600-603.
- Kimura A, Kambe N, Satoh T, et al. Follicular keratosis and bullous formation are typical signs of extragenital lichen sclerosus. J Dermatol. 2011;38:834-836.
- Meyrick Thomas RH, Ridley CM, McGibbon DH, et al. Lichen sclerosus et atrophicus and autoimmunity: a study of 350 women. Br J Dermatol. 1988;118:41-46.
- Wallace HJ. Lichen sclerosus et atrophicus. Trans St Johns Hosp Dermatol Soc. 1971;57:9-30.
- Hallel-Halevy D, Grunwald MH, Yerushalmi J, et al. Bullous lichen sclerosus et atrophicus. J Am Acad Dermatol. 1998;39:500-501.
- Garrido-Ríos AA, Álvarez-Garrido H, Sanz-Muñoz C, et al. Dermoscopy of extragenital lichen sclerosus. Arch Dermatol. 2009;145:1468.
- Larre Borges A, Tiodorovic-Zivkovic D, Lallas A, et al. Clinical, dermoscopic and histopathologic features of genital and extragenital lichen sclerosus. J Eur Acad Dermatol Venereol. 2013;27:1433-1439.
- Rudikoff D. Differential diagnosis of round or discoid lesions. Clin Dermatol. 2011;29:489-497.
- Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol. 1991;25:593-619.
- Shaffer B, Beerman H. Lichen simplex chronicus and its variants: a discussion of certain psychodynamic mechanisms and clinical and histopathologic correlations. AMA Arch Derm Syphilol. 1951;64:340-351.
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus. Am J Clin Dermatol. 2009;10:365-381.
- Sauder MB, Linzon-Smith J, Beecker J. Extragenital bullous lichen sclerosus. J Am Acad Dermatol. 2014;71:981-984.
- Colbert RL, Chiang MP, Carlin CS, et al. Progressive extragenital lichen sclerosus successfully treated with narrowband UV-B phototherapy. Arch Dermatol. 2007;143:19-20.
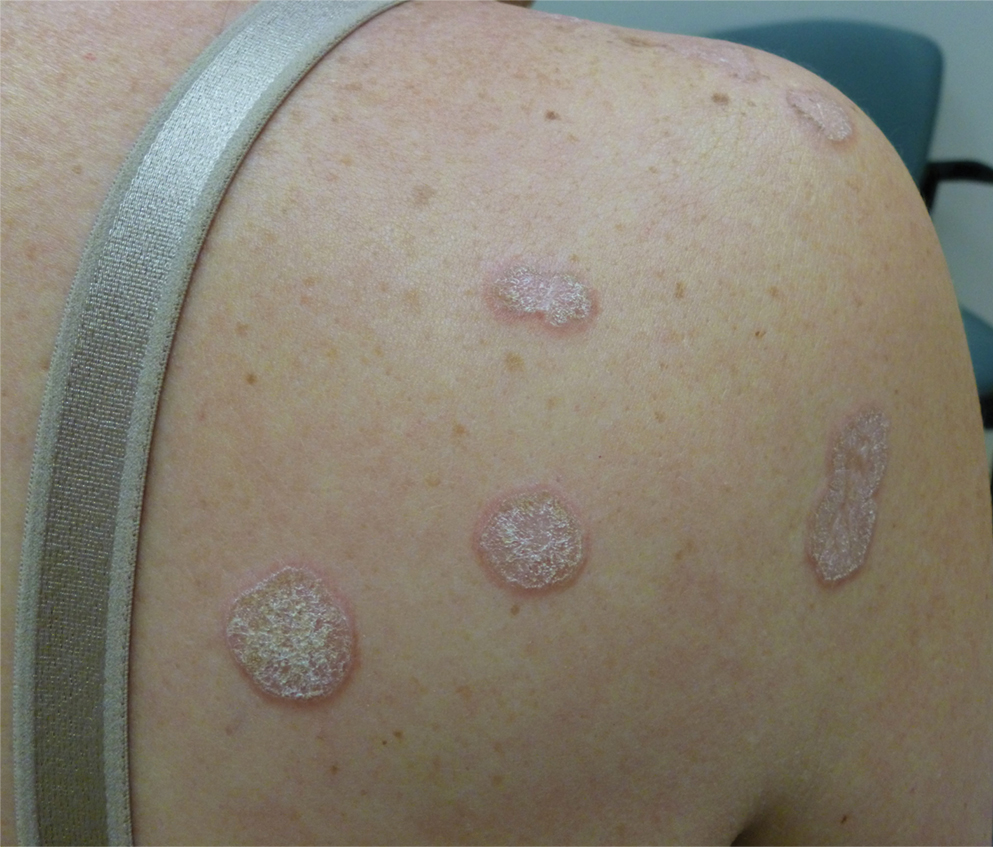
A 48-year-old woman with a history of type 2 diabetes mellitus and nonalcoholic steatohepatitis presented with papules and plaques on the upper trunk, proximal extremities, and volar wrists. Clear fluid–filled bullae occasionally developed within the plaques and subsequently ruptured and healed. Aside from intermittent lesion tenderness and irritation with the formation and rupture of the bullae, the patient’s plaques were asymptomatic, and she specifically denied pruritus. A review of systems revealed a history of genital irritation evaluated by a gynecologist; nystatin–triamcinolone cream 0.1% applied as needed provided relief. The patient denied any recent flares or any new or changing oral mucosa findings or symptoms, preceding medications, or family history of similar lesions. Physical examination revealed well-demarcated, round, pink plaques with keratotic scale scattered across the upper trunk and central chest. The bilateral volar wrists were surfaced by well-circumscribed, thin, pink to violaceous, hyperkeratotic papules.
Painless Mobile Nodule on the Shoulder
The Diagnosis: Cutaneous Metaplastic Synovial Cyst
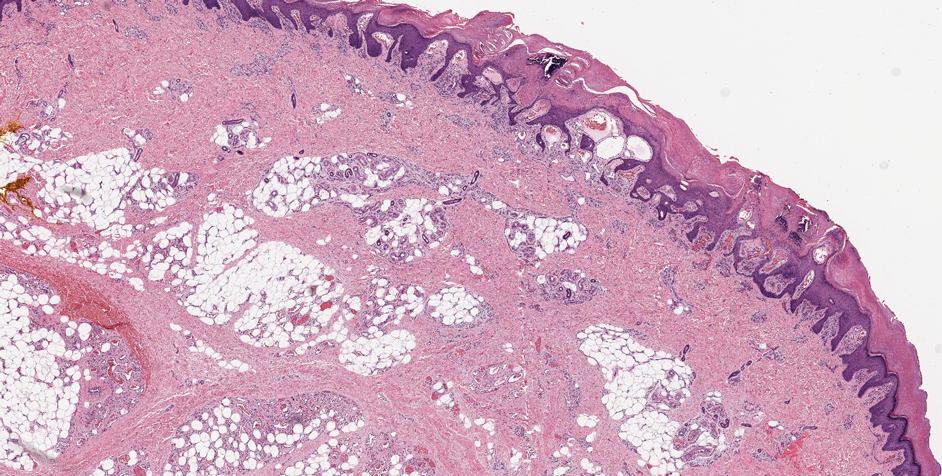
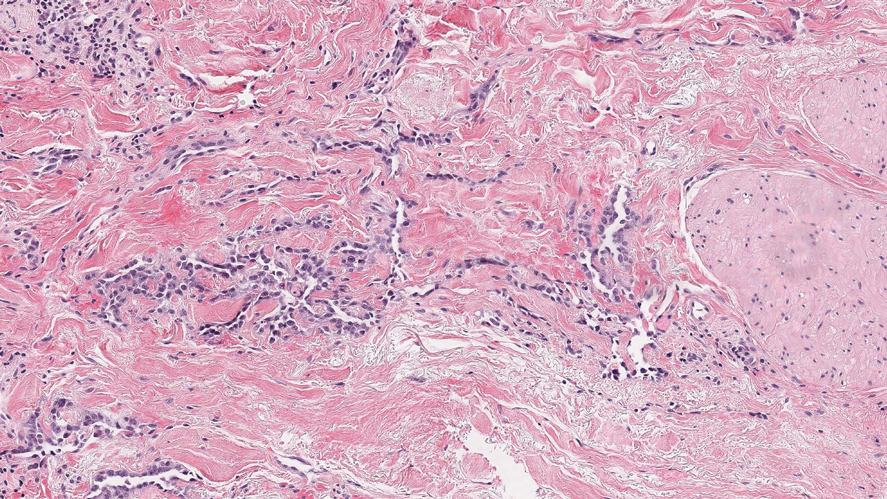
Gross examination of the excised nodule revealed a 2.5×1.2×1.0-cm, intact, gray-white, thin-walled, smooth-lined nodule filled with clear mucinouslike material. Hematoxylin and eosin-stained sections demonstrated a dermal-based cystlike structure composed of a lining of connective tissue with hyalinized material and fibrin as well as spindle and epithelioid cells with a mild mixed inflammatory infiltrate (Figure). These histopathologic findings led to the diagnosis of cutaneous metaplastic synovial cyst (CMSC).
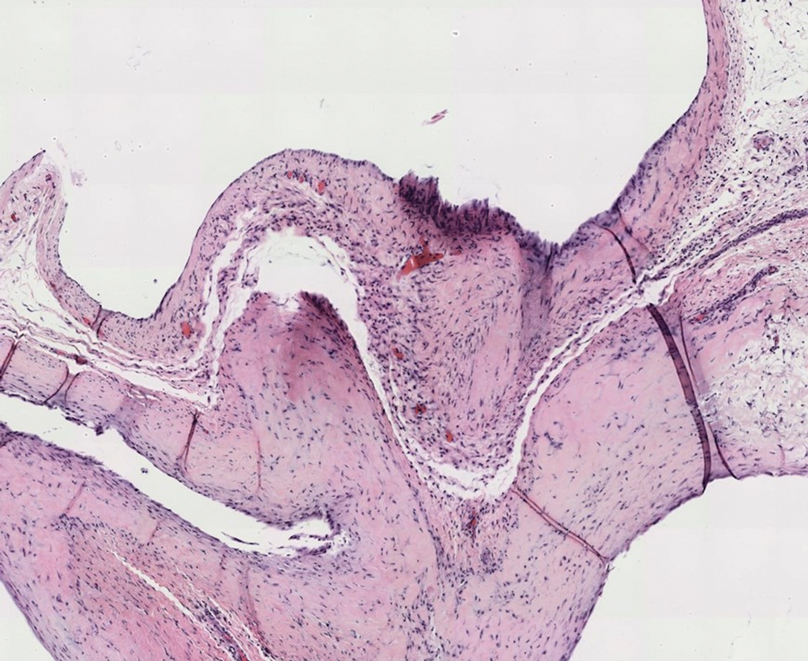
Cutaneous metaplastic synovial cyst, also known as synovial metaplasia of the skin, is an uncommon benign cystic lesion that was first reported by Gonzalez et al1 in 1987. Histologically, CMSC lacks an epithelial lining and therefore is not a true cyst but rather a pseudocyst.2 Clinically, the lesion typically presents as a solitary subcutaneous nodule that may be tender or painless. In a literature review of CMSC cases performed by Fukuyama et al,3 distribution of reported cases according to body site varied; however, limbs were found to be the most commonly involved area. A PubMed search of articles indexed for MEDLINE as well as a Google Scholar search using the term cutaneous metaplastic synovial cyst revealed at least 37 cases reported in the English-language literature,3-9 including our present case. The pathogenesis remains uncertain; however, a majority of previously reported cases of CMSC characteristically have been associated with a pre-existing lesion, with most presentations developing at surgical scar sites secondary to operation or trauma.5 Relative tissue fragility secondary to rheumatoid arthritis10 and Ehlers-Danlos syndrome9,11,12 has been linked to CMSC in some documented reports, while a minority of cases report no antecedent events triggering formation of the lesion.13-15
As evidenced by our patient, CMSC clinically mimics several other benign entities; histopathologic examination is necessary to confirm the diagnosis. Although nodular hidradenoma also may clinically present as a solitary firm intradermal nodule, microscopy reveals a dermal-based lobulated tumor containing cystic spaces and solid areas composed of basophilic polyhedral cells and round glycogen-filled clear cells.16 Epidermoid cysts are differentiated from CMSC by the presence of a cyst wall lining composed of stratified squamous epithelium and associated laminated keratin within the lumen,17 which corresponds to its pearly white appearance on gross examination. Cutaneous ciliated cysts predominantly occur on the lower extremities of young women and are lined by simple cuboidal or columnar ciliated cells that resemble müllerian epithelium.18 Similar to CMSC, ganglion cysts are pseudocysts that lack a true epithelial lining but differ in appearance due to their mucin-filled synovial-lined sac.19 Additionally, ganglion cysts most often occur on the dorsal and volar aspects of the wrist.
Excisional biopsy is indicated as the preferred treatment of CMSC, given the lesion's benign behavior and low recurrence rate.6 Our case highlights this rare entity and reinforces its inclusion in the differential diagnosis of subcutaneous mobile nodules, especially in the setting of prior tissue injury secondary to trauma, surgical procedures, or conditions such as rheumatoid arthritis or Ehlers-Danlos syndrome. Unlike most previously reported cases, our patient reported no preceding tissue injury associated with formation of the lesion, and she was largely asymptomatic on presentation. Considering the limited number of CMSC cases demonstrated in the literature, it is important to continue reporting new cases to better understand characteristics and presentations of this uncommon lesion.
- Gonzalez JG, Ghiselli RW, Santa Cruz DJ. Synovial metaplasia of the skin. Am J Surg Pathol. 1987;11:343-350.
- Calonje E, Brenn T, Lazar A, et al. Cutaneous cysts. In: Calonje E, Brenn T, Lazar A, et al. McKee's Pathology of the Skin. 5th ed. Elsevier Limited; 2020:1680-1697.
- Fukuyama M, Sato Y, Hayakawa J, et al. Cutaneous metaplastic synovial cyst: case report and literature review from the dermatological point of view. Keio J Med. 2016;66:9-13.
- Karaytug K, Kapicioglu M, Can N, et al. Unprecedented recurrence of carpal tunnel syndrome by metaplastic synovial cyst in the carpal tunnel. Acta Orthop Traumatol Turc. 2019;53:230-232.
- Martelli SJ, Silveira FM, Carvalho PH, et al. Asymptomatic subcutaneous swelling of lower face. Oral Surg Oral Med Oral Pathol Oral Radiol. 2019;128:101-105.
- Majdi M, Saffar H, Ghanadan A. Cutaneous metaplastic synovial cyst: a case report. Iran J Pathol. 2016;11:423-426.
- Ramachandra S, Rao L, Al-Kindi M. Cutaneous metaplastic synovial cyst. Sultan Qaboos Univ Med J. 2016;16:E117-E118.
- Heidarian A, Xie Q, Banihashemi A. Cutaneous metaplastic synovial cyst presenting as an axillary mass after modified mastectomy and adjuvant radiotherapy. Am J Clin Pathol. 2016;146:S2.
- Fernandez-Flores A, Barja-Lopez JM. Cutaneous metaplastic synovial cyst in Ehlers-Danlos syndrome. J Cutan Pathol. 2020;47:729-733.
- Choonhakarn C, Tang S. Cutaneous metaplastic synovial cyst. J Dermatol. 2003;30:480-484.
- Guala A, Viglio S, Ottinetti A, et al. Cutaneous metaplastic synovial cyst in Ehlers-Danlos syndrome: report of a second case. Am J Dermatopathol. 2008;30:59-61.
- Nieto S, Buezo GF, Jones-Caballero M, et al. Cutaneous metaplastic synovial cyst in an Ehlers-Danlos patient. Am J Dermatopathol. 1997;19:407-410.
- Goiriz R, Rios-Buceta L, Alonso-Perez A, et al. Cutaneous metaplastic synovial cyst. J Am Acad Dermatol. 2005;53:180-181.
- Kim BC, Choi WJ, Park EJ, et al. Cutaneous metaplastic synovial cyst of the first metatarsal head area. Ann Dermatol. 2011;23(suppl 2):S165-S168.
- Yang HC, Tsai YJ, Hu SL, et al. Cutaneous metaplastic synovial cyst--a case report and review of literature. Dermatol Sinica. 2003;21:275-279.
- Kataria SP, Singh G, Batra A, et al. Nodular hidradenoma: a series of five cases in male subjects and review of literature. Adv Cytol Pathol. 2018;3:46-47.
- Mohamed Haflah N, Mohd Kassim A, Hassan Shukur M. Giant epidermoid cyst of the thigh. Malays Orthop J. 2011;5:17-19.
- Torisu-Itakura H, Itakura E, Horiuchi R, et al. Cutaneous ciliated cyst on the leg of a woman of menopausal age. Acta Derm Venereol. 2009;89:323-324.
- Fullen DR. Cysts and sinuses. In: Busam K, ed. Dermatopathology. Saunders; 2010:300-330.
The Diagnosis: Cutaneous Metaplastic Synovial Cyst
Gross examination of the excised nodule revealed a 2.5×1.2×1.0-cm, intact, gray-white, thin-walled, smooth-lined nodule filled with clear mucinouslike material. Hematoxylin and eosin-stained sections demonstrated a dermal-based cystlike structure composed of a lining of connective tissue with hyalinized material and fibrin as well as spindle and epithelioid cells with a mild mixed inflammatory infiltrate (Figure). These histopathologic findings led to the diagnosis of cutaneous metaplastic synovial cyst (CMSC).
Cutaneous metaplastic synovial cyst, also known as synovial metaplasia of the skin, is an uncommon benign cystic lesion that was first reported by Gonzalez et al1 in 1987. Histologically, CMSC lacks an epithelial lining and therefore is not a true cyst but rather a pseudocyst.2 Clinically, the lesion typically presents as a solitary subcutaneous nodule that may be tender or painless. In a literature review of CMSC cases performed by Fukuyama et al,3 distribution of reported cases according to body site varied; however, limbs were found to be the most commonly involved area. A PubMed search of articles indexed for MEDLINE as well as a Google Scholar search using the term cutaneous metaplastic synovial cyst revealed at least 37 cases reported in the English-language literature,3-9 including our present case. The pathogenesis remains uncertain; however, a majority of previously reported cases of CMSC characteristically have been associated with a pre-existing lesion, with most presentations developing at surgical scar sites secondary to operation or trauma.5 Relative tissue fragility secondary to rheumatoid arthritis10 and Ehlers-Danlos syndrome9,11,12 has been linked to CMSC in some documented reports, while a minority of cases report no antecedent events triggering formation of the lesion.13-15
As evidenced by our patient, CMSC clinically mimics several other benign entities; histopathologic examination is necessary to confirm the diagnosis. Although nodular hidradenoma also may clinically present as a solitary firm intradermal nodule, microscopy reveals a dermal-based lobulated tumor containing cystic spaces and solid areas composed of basophilic polyhedral cells and round glycogen-filled clear cells.16 Epidermoid cysts are differentiated from CMSC by the presence of a cyst wall lining composed of stratified squamous epithelium and associated laminated keratin within the lumen,17 which corresponds to its pearly white appearance on gross examination. Cutaneous ciliated cysts predominantly occur on the lower extremities of young women and are lined by simple cuboidal or columnar ciliated cells that resemble müllerian epithelium.18 Similar to CMSC, ganglion cysts are pseudocysts that lack a true epithelial lining but differ in appearance due to their mucin-filled synovial-lined sac.19 Additionally, ganglion cysts most often occur on the dorsal and volar aspects of the wrist.
Excisional biopsy is indicated as the preferred treatment of CMSC, given the lesion's benign behavior and low recurrence rate.6 Our case highlights this rare entity and reinforces its inclusion in the differential diagnosis of subcutaneous mobile nodules, especially in the setting of prior tissue injury secondary to trauma, surgical procedures, or conditions such as rheumatoid arthritis or Ehlers-Danlos syndrome. Unlike most previously reported cases, our patient reported no preceding tissue injury associated with formation of the lesion, and she was largely asymptomatic on presentation. Considering the limited number of CMSC cases demonstrated in the literature, it is important to continue reporting new cases to better understand characteristics and presentations of this uncommon lesion.
The Diagnosis: Cutaneous Metaplastic Synovial Cyst
Gross examination of the excised nodule revealed a 2.5×1.2×1.0-cm, intact, gray-white, thin-walled, smooth-lined nodule filled with clear mucinouslike material. Hematoxylin and eosin-stained sections demonstrated a dermal-based cystlike structure composed of a lining of connective tissue with hyalinized material and fibrin as well as spindle and epithelioid cells with a mild mixed inflammatory infiltrate (Figure). These histopathologic findings led to the diagnosis of cutaneous metaplastic synovial cyst (CMSC).
Cutaneous metaplastic synovial cyst, also known as synovial metaplasia of the skin, is an uncommon benign cystic lesion that was first reported by Gonzalez et al1 in 1987. Histologically, CMSC lacks an epithelial lining and therefore is not a true cyst but rather a pseudocyst.2 Clinically, the lesion typically presents as a solitary subcutaneous nodule that may be tender or painless. In a literature review of CMSC cases performed by Fukuyama et al,3 distribution of reported cases according to body site varied; however, limbs were found to be the most commonly involved area. A PubMed search of articles indexed for MEDLINE as well as a Google Scholar search using the term cutaneous metaplastic synovial cyst revealed at least 37 cases reported in the English-language literature,3-9 including our present case. The pathogenesis remains uncertain; however, a majority of previously reported cases of CMSC characteristically have been associated with a pre-existing lesion, with most presentations developing at surgical scar sites secondary to operation or trauma.5 Relative tissue fragility secondary to rheumatoid arthritis10 and Ehlers-Danlos syndrome9,11,12 has been linked to CMSC in some documented reports, while a minority of cases report no antecedent events triggering formation of the lesion.13-15
As evidenced by our patient, CMSC clinically mimics several other benign entities; histopathologic examination is necessary to confirm the diagnosis. Although nodular hidradenoma also may clinically present as a solitary firm intradermal nodule, microscopy reveals a dermal-based lobulated tumor containing cystic spaces and solid areas composed of basophilic polyhedral cells and round glycogen-filled clear cells.16 Epidermoid cysts are differentiated from CMSC by the presence of a cyst wall lining composed of stratified squamous epithelium and associated laminated keratin within the lumen,17 which corresponds to its pearly white appearance on gross examination. Cutaneous ciliated cysts predominantly occur on the lower extremities of young women and are lined by simple cuboidal or columnar ciliated cells that resemble müllerian epithelium.18 Similar to CMSC, ganglion cysts are pseudocysts that lack a true epithelial lining but differ in appearance due to their mucin-filled synovial-lined sac.19 Additionally, ganglion cysts most often occur on the dorsal and volar aspects of the wrist.
Excisional biopsy is indicated as the preferred treatment of CMSC, given the lesion's benign behavior and low recurrence rate.6 Our case highlights this rare entity and reinforces its inclusion in the differential diagnosis of subcutaneous mobile nodules, especially in the setting of prior tissue injury secondary to trauma, surgical procedures, or conditions such as rheumatoid arthritis or Ehlers-Danlos syndrome. Unlike most previously reported cases, our patient reported no preceding tissue injury associated with formation of the lesion, and she was largely asymptomatic on presentation. Considering the limited number of CMSC cases demonstrated in the literature, it is important to continue reporting new cases to better understand characteristics and presentations of this uncommon lesion.
- Gonzalez JG, Ghiselli RW, Santa Cruz DJ. Synovial metaplasia of the skin. Am J Surg Pathol. 1987;11:343-350.
- Calonje E, Brenn T, Lazar A, et al. Cutaneous cysts. In: Calonje E, Brenn T, Lazar A, et al. McKee's Pathology of the Skin. 5th ed. Elsevier Limited; 2020:1680-1697.
- Fukuyama M, Sato Y, Hayakawa J, et al. Cutaneous metaplastic synovial cyst: case report and literature review from the dermatological point of view. Keio J Med. 2016;66:9-13.
- Karaytug K, Kapicioglu M, Can N, et al. Unprecedented recurrence of carpal tunnel syndrome by metaplastic synovial cyst in the carpal tunnel. Acta Orthop Traumatol Turc. 2019;53:230-232.
- Martelli SJ, Silveira FM, Carvalho PH, et al. Asymptomatic subcutaneous swelling of lower face. Oral Surg Oral Med Oral Pathol Oral Radiol. 2019;128:101-105.
- Majdi M, Saffar H, Ghanadan A. Cutaneous metaplastic synovial cyst: a case report. Iran J Pathol. 2016;11:423-426.
- Ramachandra S, Rao L, Al-Kindi M. Cutaneous metaplastic synovial cyst. Sultan Qaboos Univ Med J. 2016;16:E117-E118.
- Heidarian A, Xie Q, Banihashemi A. Cutaneous metaplastic synovial cyst presenting as an axillary mass after modified mastectomy and adjuvant radiotherapy. Am J Clin Pathol. 2016;146:S2.
- Fernandez-Flores A, Barja-Lopez JM. Cutaneous metaplastic synovial cyst in Ehlers-Danlos syndrome. J Cutan Pathol. 2020;47:729-733.
- Choonhakarn C, Tang S. Cutaneous metaplastic synovial cyst. J Dermatol. 2003;30:480-484.
- Guala A, Viglio S, Ottinetti A, et al. Cutaneous metaplastic synovial cyst in Ehlers-Danlos syndrome: report of a second case. Am J Dermatopathol. 2008;30:59-61.
- Nieto S, Buezo GF, Jones-Caballero M, et al. Cutaneous metaplastic synovial cyst in an Ehlers-Danlos patient. Am J Dermatopathol. 1997;19:407-410.
- Goiriz R, Rios-Buceta L, Alonso-Perez A, et al. Cutaneous metaplastic synovial cyst. J Am Acad Dermatol. 2005;53:180-181.
- Kim BC, Choi WJ, Park EJ, et al. Cutaneous metaplastic synovial cyst of the first metatarsal head area. Ann Dermatol. 2011;23(suppl 2):S165-S168.
- Yang HC, Tsai YJ, Hu SL, et al. Cutaneous metaplastic synovial cyst--a case report and review of literature. Dermatol Sinica. 2003;21:275-279.
- Kataria SP, Singh G, Batra A, et al. Nodular hidradenoma: a series of five cases in male subjects and review of literature. Adv Cytol Pathol. 2018;3:46-47.
- Mohamed Haflah N, Mohd Kassim A, Hassan Shukur M. Giant epidermoid cyst of the thigh. Malays Orthop J. 2011;5:17-19.
- Torisu-Itakura H, Itakura E, Horiuchi R, et al. Cutaneous ciliated cyst on the leg of a woman of menopausal age. Acta Derm Venereol. 2009;89:323-324.
- Fullen DR. Cysts and sinuses. In: Busam K, ed. Dermatopathology. Saunders; 2010:300-330.
- Gonzalez JG, Ghiselli RW, Santa Cruz DJ. Synovial metaplasia of the skin. Am J Surg Pathol. 1987;11:343-350.
- Calonje E, Brenn T, Lazar A, et al. Cutaneous cysts. In: Calonje E, Brenn T, Lazar A, et al. McKee's Pathology of the Skin. 5th ed. Elsevier Limited; 2020:1680-1697.
- Fukuyama M, Sato Y, Hayakawa J, et al. Cutaneous metaplastic synovial cyst: case report and literature review from the dermatological point of view. Keio J Med. 2016;66:9-13.
- Karaytug K, Kapicioglu M, Can N, et al. Unprecedented recurrence of carpal tunnel syndrome by metaplastic synovial cyst in the carpal tunnel. Acta Orthop Traumatol Turc. 2019;53:230-232.
- Martelli SJ, Silveira FM, Carvalho PH, et al. Asymptomatic subcutaneous swelling of lower face. Oral Surg Oral Med Oral Pathol Oral Radiol. 2019;128:101-105.
- Majdi M, Saffar H, Ghanadan A. Cutaneous metaplastic synovial cyst: a case report. Iran J Pathol. 2016;11:423-426.
- Ramachandra S, Rao L, Al-Kindi M. Cutaneous metaplastic synovial cyst. Sultan Qaboos Univ Med J. 2016;16:E117-E118.
- Heidarian A, Xie Q, Banihashemi A. Cutaneous metaplastic synovial cyst presenting as an axillary mass after modified mastectomy and adjuvant radiotherapy. Am J Clin Pathol. 2016;146:S2.
- Fernandez-Flores A, Barja-Lopez JM. Cutaneous metaplastic synovial cyst in Ehlers-Danlos syndrome. J Cutan Pathol. 2020;47:729-733.
- Choonhakarn C, Tang S. Cutaneous metaplastic synovial cyst. J Dermatol. 2003;30:480-484.
- Guala A, Viglio S, Ottinetti A, et al. Cutaneous metaplastic synovial cyst in Ehlers-Danlos syndrome: report of a second case. Am J Dermatopathol. 2008;30:59-61.
- Nieto S, Buezo GF, Jones-Caballero M, et al. Cutaneous metaplastic synovial cyst in an Ehlers-Danlos patient. Am J Dermatopathol. 1997;19:407-410.
- Goiriz R, Rios-Buceta L, Alonso-Perez A, et al. Cutaneous metaplastic synovial cyst. J Am Acad Dermatol. 2005;53:180-181.
- Kim BC, Choi WJ, Park EJ, et al. Cutaneous metaplastic synovial cyst of the first metatarsal head area. Ann Dermatol. 2011;23(suppl 2):S165-S168.
- Yang HC, Tsai YJ, Hu SL, et al. Cutaneous metaplastic synovial cyst--a case report and review of literature. Dermatol Sinica. 2003;21:275-279.
- Kataria SP, Singh G, Batra A, et al. Nodular hidradenoma: a series of five cases in male subjects and review of literature. Adv Cytol Pathol. 2018;3:46-47.
- Mohamed Haflah N, Mohd Kassim A, Hassan Shukur M. Giant epidermoid cyst of the thigh. Malays Orthop J. 2011;5:17-19.
- Torisu-Itakura H, Itakura E, Horiuchi R, et al. Cutaneous ciliated cyst on the leg of a woman of menopausal age. Acta Derm Venereol. 2009;89:323-324.
- Fullen DR. Cysts and sinuses. In: Busam K, ed. Dermatopathology. Saunders; 2010:300-330.
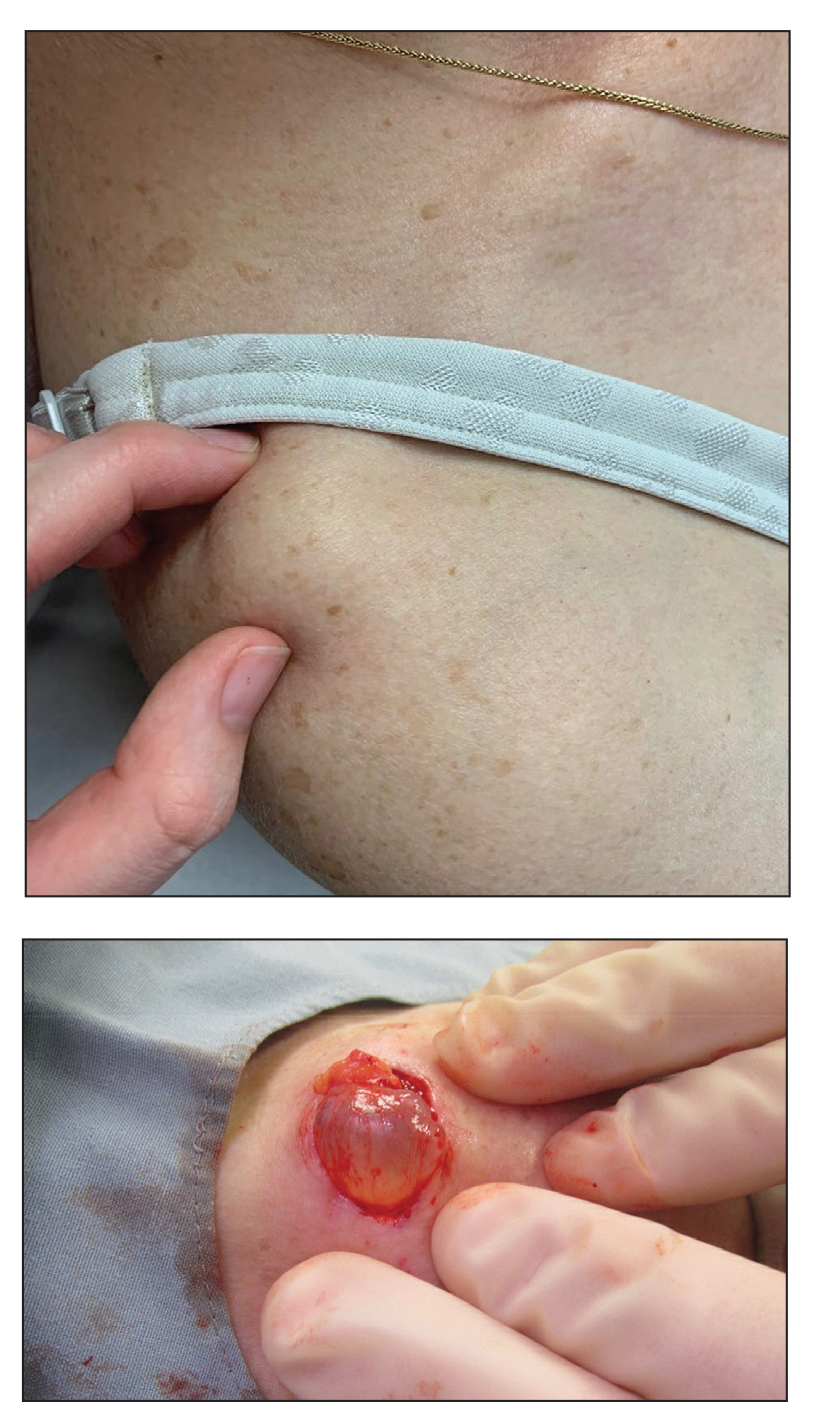
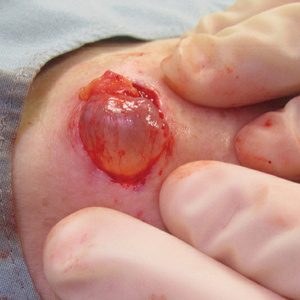
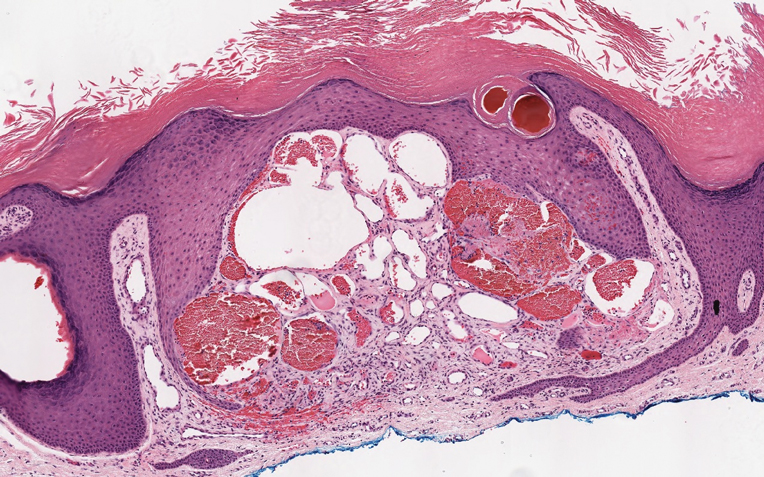
A 70-year-old woman presented to the outpatient dermatology clinic with an acute-onset lesion on the right shoulder. She first noticed a “cyst” developing in the area approximately 3 weeks prior but noted that it may have been present longer. The lesion was bothersome when her undergarments rubbed against it, but she otherwise denied pain, increase in size, or drainage from the site. Her medical history was remarkable for a proliferating trichilemmal tumor on the right parietal scalp treated with Mohs surgery approximately 13 years prior to presentation. She had no personal or family history of skin cancer. Physical examination revealed a 2.5-cm, mobile, nontender, flesh-colored subcutaneous nodule on the right shoulder (top); no ulceration, bleeding, or drainage was present. The surrounding skin demonstrated no clinical changes. The patient was scheduled for outpatient surgical excision of the nodule, which initially was suspected to be a lipoma. During the excision, a translucent cystlike nodule (bottom) was gently dissected and sent for histopathologic examination.
Eruptive Annular Papules on the Trunk of an Organ Transplant Recipient
The Diagnosis: Epidermodysplasia Verruciformis
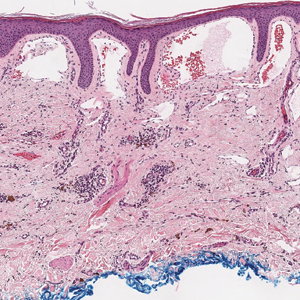
Histopathologic examination of our patient's biopsy specimen revealed mild acanthosis with prominent hypergranulosis and enlarged keratinocytes with blue-gray cytoplasm (Figure). A diagnosis of acquired epidermodysplasia verruciformis (EV) was rendered. The patient was treated with photodynamic therapy utilizing 5-aminolevulinic acid.
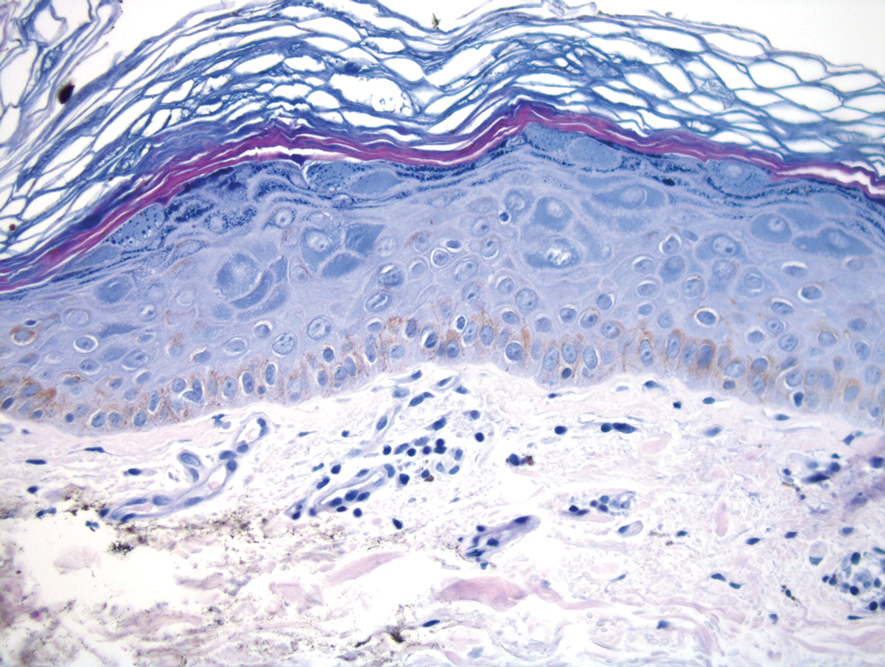
Epidermodysplasia verruciformis is characterized by susceptibility to human papillomavirus (HPV) infections via a defect in cellular immunity. Epidermodysplasia verruciformis was first described as an autosomal-recessive genodermatosis, but it can be acquired in immunosuppressed states with an atypical clinical appearance.1 There are few case reports in skin of color. Acquired EV appears in patients with acquired immunodeficiencies that are susceptible to EV-causing HPVs via a similar mechanism found in inherited EV.2 The most common HPV serotypes involved in EV are HPV-5 and HPV-8. The duration of immunosuppression has been found to be positively correlated with the risk for EV development, with the majority of patients developing lesions after 5 years of immunosuppression.3 There is an approximately 60% risk of malignant transformation of EV lesions into nonmelanoma skin cancer.2 This risk is believed to be lower in patients with darker skin.4
Preventative measures including sun protection and annual surveillance are crucial in EV patients given the high rate of malignant transformation in sun-exposed lesions.5 Treatment options for EV are anecdotal and have variable results, ranging from topicals including 5-fluorouracil and imiquimod to systemic medications including acitretin and interferon.3 Photodynamic therapy can be used for extensive EV. Surgical modalities and other destructive methods also have been tried.6
Epidermodysplasia verruciformis often can be confused with similar dermatoses. Porokeratosis appears as annular pink papules with waferlike peripheral scales. Tinea versicolor is a dermatophyte infection caused by Malassezia furfur and presents as multiple dyspigmented, finely scaling, thin papules and plaques. Subacute cutaneous lupus erythematosus presents as pink, scaly, annular or psoriasiform papules and plaques most commonly on the trunk. Discoid lupus erythematosus presents as pink, hypopigmented or depigmented, atrophic plaques with a peripheral rim of erythema that indicates activity. Secondary syphilis, commonly denoted as the "great mimicker," presents as psoriasiform papules and plaques among other variable morphologies.
- Sa NB, Guerini MB, Barbato MT, et al. Epidermodysplasia verruciformis: clinical presentation with varied forms of lesions. An Bras Dermatol. 2011;86(4 suppl 1):S57-S60.
- Rogers HD, Macgregor JL, Nord KM, et al. Acquired epidermodysplasia verruciformis. J Am Acad Dermatol. 2009;60:315-320.
- Henley JK, Hossler EW. Acquired epidermodysplasia verruciformis occurring in a renal transplant recipient. Cutis. 2017;99:E9-E12.
- Jacyk WK, De Villiers EM. Epidermodysplasia verruciformis in Africans. Int J Dermatol. 1993;32:806-810.
- Fox SH, Elston DM. Epidermodysplasia verruciformis and the risk for malignancy. Cutis. 2016;98:E10-E12.
- Shruti S, Siraj F, Singh A, et al. Epidermodysplasia verruciformis: three case reports and a brief review. Acta Dermatovenerol Alp Pannonica Adriat. 2017;26:59-61.
The Diagnosis: Epidermodysplasia Verruciformis
Histopathologic examination of our patient's biopsy specimen revealed mild acanthosis with prominent hypergranulosis and enlarged keratinocytes with blue-gray cytoplasm (Figure). A diagnosis of acquired epidermodysplasia verruciformis (EV) was rendered. The patient was treated with photodynamic therapy utilizing 5-aminolevulinic acid.
Epidermodysplasia verruciformis is characterized by susceptibility to human papillomavirus (HPV) infections via a defect in cellular immunity. Epidermodysplasia verruciformis was first described as an autosomal-recessive genodermatosis, but it can be acquired in immunosuppressed states with an atypical clinical appearance.1 There are few case reports in skin of color. Acquired EV appears in patients with acquired immunodeficiencies that are susceptible to EV-causing HPVs via a similar mechanism found in inherited EV.2 The most common HPV serotypes involved in EV are HPV-5 and HPV-8. The duration of immunosuppression has been found to be positively correlated with the risk for EV development, with the majority of patients developing lesions after 5 years of immunosuppression.3 There is an approximately 60% risk of malignant transformation of EV lesions into nonmelanoma skin cancer.2 This risk is believed to be lower in patients with darker skin.4
Preventative measures including sun protection and annual surveillance are crucial in EV patients given the high rate of malignant transformation in sun-exposed lesions.5 Treatment options for EV are anecdotal and have variable results, ranging from topicals including 5-fluorouracil and imiquimod to systemic medications including acitretin and interferon.3 Photodynamic therapy can be used for extensive EV. Surgical modalities and other destructive methods also have been tried.6
Epidermodysplasia verruciformis often can be confused with similar dermatoses. Porokeratosis appears as annular pink papules with waferlike peripheral scales. Tinea versicolor is a dermatophyte infection caused by Malassezia furfur and presents as multiple dyspigmented, finely scaling, thin papules and plaques. Subacute cutaneous lupus erythematosus presents as pink, scaly, annular or psoriasiform papules and plaques most commonly on the trunk. Discoid lupus erythematosus presents as pink, hypopigmented or depigmented, atrophic plaques with a peripheral rim of erythema that indicates activity. Secondary syphilis, commonly denoted as the "great mimicker," presents as psoriasiform papules and plaques among other variable morphologies.
The Diagnosis: Epidermodysplasia Verruciformis
Histopathologic examination of our patient's biopsy specimen revealed mild acanthosis with prominent hypergranulosis and enlarged keratinocytes with blue-gray cytoplasm (Figure). A diagnosis of acquired epidermodysplasia verruciformis (EV) was rendered. The patient was treated with photodynamic therapy utilizing 5-aminolevulinic acid.
Epidermodysplasia verruciformis is characterized by susceptibility to human papillomavirus (HPV) infections via a defect in cellular immunity. Epidermodysplasia verruciformis was first described as an autosomal-recessive genodermatosis, but it can be acquired in immunosuppressed states with an atypical clinical appearance.1 There are few case reports in skin of color. Acquired EV appears in patients with acquired immunodeficiencies that are susceptible to EV-causing HPVs via a similar mechanism found in inherited EV.2 The most common HPV serotypes involved in EV are HPV-5 and HPV-8. The duration of immunosuppression has been found to be positively correlated with the risk for EV development, with the majority of patients developing lesions after 5 years of immunosuppression.3 There is an approximately 60% risk of malignant transformation of EV lesions into nonmelanoma skin cancer.2 This risk is believed to be lower in patients with darker skin.4
Preventative measures including sun protection and annual surveillance are crucial in EV patients given the high rate of malignant transformation in sun-exposed lesions.5 Treatment options for EV are anecdotal and have variable results, ranging from topicals including 5-fluorouracil and imiquimod to systemic medications including acitretin and interferon.3 Photodynamic therapy can be used for extensive EV. Surgical modalities and other destructive methods also have been tried.6
Epidermodysplasia verruciformis often can be confused with similar dermatoses. Porokeratosis appears as annular pink papules with waferlike peripheral scales. Tinea versicolor is a dermatophyte infection caused by Malassezia furfur and presents as multiple dyspigmented, finely scaling, thin papules and plaques. Subacute cutaneous lupus erythematosus presents as pink, scaly, annular or psoriasiform papules and plaques most commonly on the trunk. Discoid lupus erythematosus presents as pink, hypopigmented or depigmented, atrophic plaques with a peripheral rim of erythema that indicates activity. Secondary syphilis, commonly denoted as the "great mimicker," presents as psoriasiform papules and plaques among other variable morphologies.
- Sa NB, Guerini MB, Barbato MT, et al. Epidermodysplasia verruciformis: clinical presentation with varied forms of lesions. An Bras Dermatol. 2011;86(4 suppl 1):S57-S60.
- Rogers HD, Macgregor JL, Nord KM, et al. Acquired epidermodysplasia verruciformis. J Am Acad Dermatol. 2009;60:315-320.
- Henley JK, Hossler EW. Acquired epidermodysplasia verruciformis occurring in a renal transplant recipient. Cutis. 2017;99:E9-E12.
- Jacyk WK, De Villiers EM. Epidermodysplasia verruciformis in Africans. Int J Dermatol. 1993;32:806-810.
- Fox SH, Elston DM. Epidermodysplasia verruciformis and the risk for malignancy. Cutis. 2016;98:E10-E12.
- Shruti S, Siraj F, Singh A, et al. Epidermodysplasia verruciformis: three case reports and a brief review. Acta Dermatovenerol Alp Pannonica Adriat. 2017;26:59-61.
- Sa NB, Guerini MB, Barbato MT, et al. Epidermodysplasia verruciformis: clinical presentation with varied forms of lesions. An Bras Dermatol. 2011;86(4 suppl 1):S57-S60.
- Rogers HD, Macgregor JL, Nord KM, et al. Acquired epidermodysplasia verruciformis. J Am Acad Dermatol. 2009;60:315-320.
- Henley JK, Hossler EW. Acquired epidermodysplasia verruciformis occurring in a renal transplant recipient. Cutis. 2017;99:E9-E12.
- Jacyk WK, De Villiers EM. Epidermodysplasia verruciformis in Africans. Int J Dermatol. 1993;32:806-810.
- Fox SH, Elston DM. Epidermodysplasia verruciformis and the risk for malignancy. Cutis. 2016;98:E10-E12.
- Shruti S, Siraj F, Singh A, et al. Epidermodysplasia verruciformis: three case reports and a brief review. Acta Dermatovenerol Alp Pannonica Adriat. 2017;26:59-61.
A 50-year-old Black woman with systemic lupus erythematosus and a renal transplant 15 years prior due to lupus nephritis presented with a nonpruritic rash on the abdomen of 1 year’s duration. Her immunosuppressive regimen consisted of tacrolimus, azathioprine, and prednisone. Physical examination revealed numerous monomorphic, annular, hyperpigmented, and thin papules with central clearing present on the abdomen extending to the flanks and groin. The patient denied any family history of similar lesions. A 4-mm punch biopsy of an abdominal lesion was performed.
Violaceous Papule With an Erythematous Rim
The Diagnosis: Targetoid Hemosiderotic Hemangioma
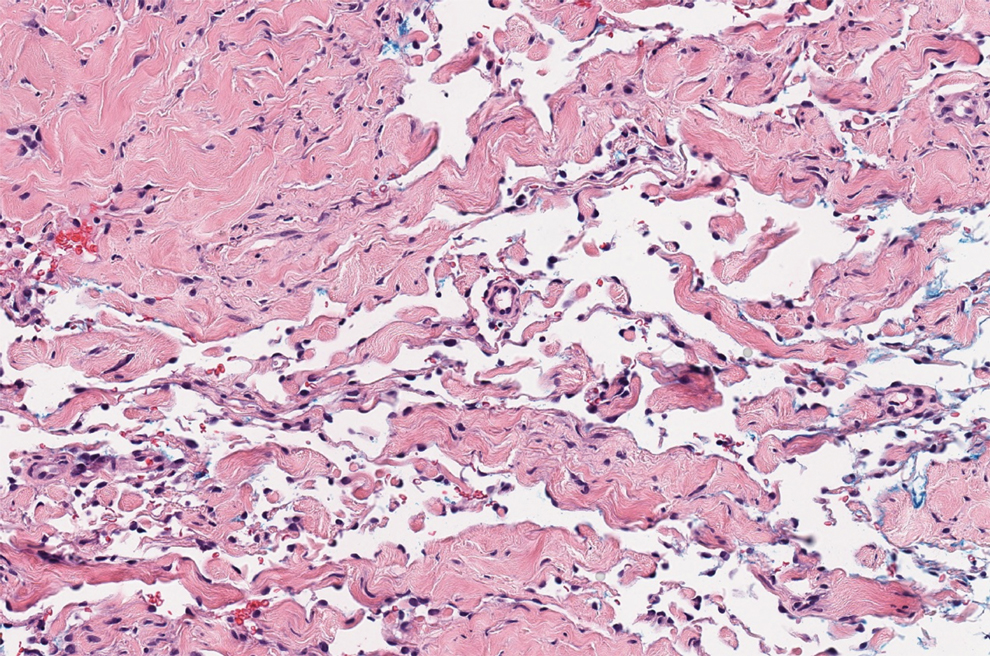
Targetoid hemosiderotic hemangioma (THH), also known as hobnail hemangioma, is a benign vascular tumor that usually occurs in young or middle-aged adults. It most commonly presents on the extremities or trunk as an isolated red-brown plaque or papule.1,2 Histologically, THH is characterized by superficial dilated ectatic vessels with underlying proliferating vascular channels lined by plump hobnail endothelial cells.1 Targetoid hemosiderotic hemangioma typically involves the dermis and spares the subcutis. The vascular channels may contain erythrocytes as well as pale eosinophilic lymph, as seen in our patient (quiz image). The deeper dermis contains vascular spaces that are more angulated and smaller and appear to be dissecting through the collagen bundles or collapsed.1,3 A variable amount of hemosiderin deposition and extravasated erythrocytes are seen.2,3 Histologic features evolve with the age of the lesion. Increasing amounts of hemosiderin deposition and erythrocyte extravasation may correspond histologically to the recent clinical color change reported by the patient.
Verrucous hemangioma is a rare congenital vascular abnormality that is characterized by dilated vessels in the papillary dermis along with acanthosis, hyperkeratosis, and irregular papillomatosis, as seen in angiokeratoma.4 However, the vascular proliferation composed of variably sized, thin-walled capillaries extends into the deep dermis as well as the subcutis (Figure 1). Verrucous hemangioma most commonly is reported on the legs and generally starts as a violaceous patch that progresses into a hyperkeratotic verrucous plaque or nodule.5,6
Angiokeratoma is characterized by superficial vascular ectasia of the papillary dermis in association with overlying acanthosis, hyperkeratosis, and rete elongation.7 The dilated vascular spaces appear encircled by the epidermis (Figure 2). Intravascular thrombosis can be seen within the ectatic vessels.7 In contrast to verrucous hemangioma, angiokeratoma is limited to the papillary dermis. Therefore, obtaining a biopsy of sufficient depth is necessary for differentiation.8 There are 5 clinical presentations of angiokeratoma: sporadic, angiokeratoma of Mibelli, angiokeratoma of Fordyce, angiokeratoma circumscriptum, and angiokeratoma corporis diffusum (Fabry disease). Angiokeratomas may present on the lower extremities, tongue, trunk, and scrotum as hyperkeratotic, dark red to purple or black papules.7
There are 3 clinical stages of Kaposi sarcoma: patch, plaque, and nodular stages. The patch stage is characterized histologically by vascular channels that dissect through the dermis and extend around native vessels (the promontory sign)(Figure 3).9,10 These features can show histologic overlap with THH. The plaque stage shows a more diffuse dermal vascular proliferation, increased cellularity of spindle cells, and possible extension into the subcutis.9,10 Focal plasma cells, hemosiderin, and extravasated red blood cells can be seen. The nodular stage is characterized by a proliferation of spindle cells with red blood cells squeezed between slitlike vascular spaces, hyaline globules, and scattered mitotic figures, but not atypical forms.10 In this stage, plasma cells and hemosiderin are more readily identifiable. A biopsy from the nodular stage is unlikely to enter the histologic differential diagnosis with THH. Clinically, there are 4 variants of Kaposi sarcoma: the classic or sporadic form, an endemic form, iatrogenic, and AIDS associated. Overall, it is more common in males and can occur at any age.10 Human herpesvirus 8 is seen in all forms, and infected cells can be highlighted by the immunohistochemical stain for latent nuclear antigen 1.9,10
Angiosarcoma is a malignant endothelial tumor of soft tissue, skin, bone, and visceral organs.11,12 Clinically, cutaneous angiosarcoma can present in a variety of ways, including single or multiple bluish red lesions that can ulcerate or bleed; violaceous nodules or plaques; and hematomalike lesions that can mimic epithelial neoplasms including squamous cell carcinoma, basal cell carcinoma, and malignant melanoma.11,13,14 The cutaneous lesions most commonly occur on sun-exposed skin, particularly on the face and scalp.12 Other clinical variants that are important to recognize are postradiation angiosarcoma, characterized by MYC gene amplification, and lymphedema-associated angiosarcoma (Stewart-Treves syndrome). Angiosarcoma can have a variety of morphologic features, ranging from well to poorly differentiated. Classically, angiosarcoma is characterized by infiltrating vascular spaces lined by atypical endothelial cells (Figure 4). Poorly differentiated angiosarcoma can demonstrate spindle, epithelioid, or polygonal cells with increased mitotic activity, pleomorphism, and irregular vascular spaces.11 Endothelial markers such as ERG (erythroblast transformation specific-related gene)(nuclear) and CD31 (membranous) can be used to aid in the diagnosis of a poorly differentiated lesion. Epithelioid angiosarcoma also occasionally stains with cytokeratins.13,14
- Joyce JC, Keith PJ, Szabo S, et al. Superficial hemosiderotic lymphovascular malformation (hobnail hemangioma): a report of six cases. Pediatr Dermatol. 2014;31:281-285.
- Sahin MT, Demir MA, Gunduz K, et al. Targetoid haemosiderotic haemangioma: dermoscopic monitoring of three cases and review of the literature. Clin Exp Dermatol. 2005;30:672-676.
- Kakizaki P, Valente NY, Paiva DL, et al. Targetoid hemosiderotic hemangioma--case report. An Bras Dermatol. 2014;89:956-959.
- Oppermann K, Boff AL, Bonamigo RR. Verrucous hemangioma and histopathological differential diagnosis with angiokeratoma circumscriptum neviforme. An Bras Dermatol. 2018;93:712-715.
- Boccara, O, Ariche-Maman, S, Hadj-Rabia, S, et al. Verrucous hemangioma (also known as verrucous venous malformation): a vascular anomaly frequently misdiagnosed as a lymphatic malformation. Pediatr Dermatol. 2018;35:E378-E381.
- Mestre T, Amaro C, Freitas I. Verrucous haemangioma: a diagnosis to consider [published online June 4, 2014]. BMJ Case Rep. doi:10.1136/bcr-2014-204612
- Ivy H, Julian CA. Angiokeratoma circumscriptum. StatPearls. StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK549769/
- Shetty S, Geetha V, Rao R, et al. Verrucous hemangioma: importance of a deeper biopsy. Indian J Dermatopathol Diagn Dermatol. 2014;1:99-100.
- Bishop BN, Lynch DT. Cancer, Kaposi sarcoma. StatPearls. StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK534839/
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Cao J, Wang J, He C, et al. Angiosarcoma: a review of diagnosis and current treatment. Am J Cancer Res. 2019;9:2303-2313.
- Papke DJ Jr, Hornick JL. What is new in endothelial neoplasia? Virchows Arch. 2020;476:17-28.
- Ambujam S, Audhya M, Reddy A, et al. Cutaneous angiosarcoma of the head, neck, and face of the elderly in type 5 skin. J Cutan Aesthet Surg. 2013;6:45-47.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.
The Diagnosis: Targetoid Hemosiderotic Hemangioma
Targetoid hemosiderotic hemangioma (THH), also known as hobnail hemangioma, is a benign vascular tumor that usually occurs in young or middle-aged adults. It most commonly presents on the extremities or trunk as an isolated red-brown plaque or papule.1,2 Histologically, THH is characterized by superficial dilated ectatic vessels with underlying proliferating vascular channels lined by plump hobnail endothelial cells.1 Targetoid hemosiderotic hemangioma typically involves the dermis and spares the subcutis. The vascular channels may contain erythrocytes as well as pale eosinophilic lymph, as seen in our patient (quiz image). The deeper dermis contains vascular spaces that are more angulated and smaller and appear to be dissecting through the collagen bundles or collapsed.1,3 A variable amount of hemosiderin deposition and extravasated erythrocytes are seen.2,3 Histologic features evolve with the age of the lesion. Increasing amounts of hemosiderin deposition and erythrocyte extravasation may correspond histologically to the recent clinical color change reported by the patient.
Verrucous hemangioma is a rare congenital vascular abnormality that is characterized by dilated vessels in the papillary dermis along with acanthosis, hyperkeratosis, and irregular papillomatosis, as seen in angiokeratoma.4 However, the vascular proliferation composed of variably sized, thin-walled capillaries extends into the deep dermis as well as the subcutis (Figure 1). Verrucous hemangioma most commonly is reported on the legs and generally starts as a violaceous patch that progresses into a hyperkeratotic verrucous plaque or nodule.5,6
Angiokeratoma is characterized by superficial vascular ectasia of the papillary dermis in association with overlying acanthosis, hyperkeratosis, and rete elongation.7 The dilated vascular spaces appear encircled by the epidermis (Figure 2). Intravascular thrombosis can be seen within the ectatic vessels.7 In contrast to verrucous hemangioma, angiokeratoma is limited to the papillary dermis. Therefore, obtaining a biopsy of sufficient depth is necessary for differentiation.8 There are 5 clinical presentations of angiokeratoma: sporadic, angiokeratoma of Mibelli, angiokeratoma of Fordyce, angiokeratoma circumscriptum, and angiokeratoma corporis diffusum (Fabry disease). Angiokeratomas may present on the lower extremities, tongue, trunk, and scrotum as hyperkeratotic, dark red to purple or black papules.7
There are 3 clinical stages of Kaposi sarcoma: patch, plaque, and nodular stages. The patch stage is characterized histologically by vascular channels that dissect through the dermis and extend around native vessels (the promontory sign)(Figure 3).9,10 These features can show histologic overlap with THH. The plaque stage shows a more diffuse dermal vascular proliferation, increased cellularity of spindle cells, and possible extension into the subcutis.9,10 Focal plasma cells, hemosiderin, and extravasated red blood cells can be seen. The nodular stage is characterized by a proliferation of spindle cells with red blood cells squeezed between slitlike vascular spaces, hyaline globules, and scattered mitotic figures, but not atypical forms.10 In this stage, plasma cells and hemosiderin are more readily identifiable. A biopsy from the nodular stage is unlikely to enter the histologic differential diagnosis with THH. Clinically, there are 4 variants of Kaposi sarcoma: the classic or sporadic form, an endemic form, iatrogenic, and AIDS associated. Overall, it is more common in males and can occur at any age.10 Human herpesvirus 8 is seen in all forms, and infected cells can be highlighted by the immunohistochemical stain for latent nuclear antigen 1.9,10
Angiosarcoma is a malignant endothelial tumor of soft tissue, skin, bone, and visceral organs.11,12 Clinically, cutaneous angiosarcoma can present in a variety of ways, including single or multiple bluish red lesions that can ulcerate or bleed; violaceous nodules or plaques; and hematomalike lesions that can mimic epithelial neoplasms including squamous cell carcinoma, basal cell carcinoma, and malignant melanoma.11,13,14 The cutaneous lesions most commonly occur on sun-exposed skin, particularly on the face and scalp.12 Other clinical variants that are important to recognize are postradiation angiosarcoma, characterized by MYC gene amplification, and lymphedema-associated angiosarcoma (Stewart-Treves syndrome). Angiosarcoma can have a variety of morphologic features, ranging from well to poorly differentiated. Classically, angiosarcoma is characterized by infiltrating vascular spaces lined by atypical endothelial cells (Figure 4). Poorly differentiated angiosarcoma can demonstrate spindle, epithelioid, or polygonal cells with increased mitotic activity, pleomorphism, and irregular vascular spaces.11 Endothelial markers such as ERG (erythroblast transformation specific-related gene)(nuclear) and CD31 (membranous) can be used to aid in the diagnosis of a poorly differentiated lesion. Epithelioid angiosarcoma also occasionally stains with cytokeratins.13,14
The Diagnosis: Targetoid Hemosiderotic Hemangioma
Targetoid hemosiderotic hemangioma (THH), also known as hobnail hemangioma, is a benign vascular tumor that usually occurs in young or middle-aged adults. It most commonly presents on the extremities or trunk as an isolated red-brown plaque or papule.1,2 Histologically, THH is characterized by superficial dilated ectatic vessels with underlying proliferating vascular channels lined by plump hobnail endothelial cells.1 Targetoid hemosiderotic hemangioma typically involves the dermis and spares the subcutis. The vascular channels may contain erythrocytes as well as pale eosinophilic lymph, as seen in our patient (quiz image). The deeper dermis contains vascular spaces that are more angulated and smaller and appear to be dissecting through the collagen bundles or collapsed.1,3 A variable amount of hemosiderin deposition and extravasated erythrocytes are seen.2,3 Histologic features evolve with the age of the lesion. Increasing amounts of hemosiderin deposition and erythrocyte extravasation may correspond histologically to the recent clinical color change reported by the patient.
Verrucous hemangioma is a rare congenital vascular abnormality that is characterized by dilated vessels in the papillary dermis along with acanthosis, hyperkeratosis, and irregular papillomatosis, as seen in angiokeratoma.4 However, the vascular proliferation composed of variably sized, thin-walled capillaries extends into the deep dermis as well as the subcutis (Figure 1). Verrucous hemangioma most commonly is reported on the legs and generally starts as a violaceous patch that progresses into a hyperkeratotic verrucous plaque or nodule.5,6
Angiokeratoma is characterized by superficial vascular ectasia of the papillary dermis in association with overlying acanthosis, hyperkeratosis, and rete elongation.7 The dilated vascular spaces appear encircled by the epidermis (Figure 2). Intravascular thrombosis can be seen within the ectatic vessels.7 In contrast to verrucous hemangioma, angiokeratoma is limited to the papillary dermis. Therefore, obtaining a biopsy of sufficient depth is necessary for differentiation.8 There are 5 clinical presentations of angiokeratoma: sporadic, angiokeratoma of Mibelli, angiokeratoma of Fordyce, angiokeratoma circumscriptum, and angiokeratoma corporis diffusum (Fabry disease). Angiokeratomas may present on the lower extremities, tongue, trunk, and scrotum as hyperkeratotic, dark red to purple or black papules.7
There are 3 clinical stages of Kaposi sarcoma: patch, plaque, and nodular stages. The patch stage is characterized histologically by vascular channels that dissect through the dermis and extend around native vessels (the promontory sign)(Figure 3).9,10 These features can show histologic overlap with THH. The plaque stage shows a more diffuse dermal vascular proliferation, increased cellularity of spindle cells, and possible extension into the subcutis.9,10 Focal plasma cells, hemosiderin, and extravasated red blood cells can be seen. The nodular stage is characterized by a proliferation of spindle cells with red blood cells squeezed between slitlike vascular spaces, hyaline globules, and scattered mitotic figures, but not atypical forms.10 In this stage, plasma cells and hemosiderin are more readily identifiable. A biopsy from the nodular stage is unlikely to enter the histologic differential diagnosis with THH. Clinically, there are 4 variants of Kaposi sarcoma: the classic or sporadic form, an endemic form, iatrogenic, and AIDS associated. Overall, it is more common in males and can occur at any age.10 Human herpesvirus 8 is seen in all forms, and infected cells can be highlighted by the immunohistochemical stain for latent nuclear antigen 1.9,10
Angiosarcoma is a malignant endothelial tumor of soft tissue, skin, bone, and visceral organs.11,12 Clinically, cutaneous angiosarcoma can present in a variety of ways, including single or multiple bluish red lesions that can ulcerate or bleed; violaceous nodules or plaques; and hematomalike lesions that can mimic epithelial neoplasms including squamous cell carcinoma, basal cell carcinoma, and malignant melanoma.11,13,14 The cutaneous lesions most commonly occur on sun-exposed skin, particularly on the face and scalp.12 Other clinical variants that are important to recognize are postradiation angiosarcoma, characterized by MYC gene amplification, and lymphedema-associated angiosarcoma (Stewart-Treves syndrome). Angiosarcoma can have a variety of morphologic features, ranging from well to poorly differentiated. Classically, angiosarcoma is characterized by infiltrating vascular spaces lined by atypical endothelial cells (Figure 4). Poorly differentiated angiosarcoma can demonstrate spindle, epithelioid, or polygonal cells with increased mitotic activity, pleomorphism, and irregular vascular spaces.11 Endothelial markers such as ERG (erythroblast transformation specific-related gene)(nuclear) and CD31 (membranous) can be used to aid in the diagnosis of a poorly differentiated lesion. Epithelioid angiosarcoma also occasionally stains with cytokeratins.13,14
- Joyce JC, Keith PJ, Szabo S, et al. Superficial hemosiderotic lymphovascular malformation (hobnail hemangioma): a report of six cases. Pediatr Dermatol. 2014;31:281-285.
- Sahin MT, Demir MA, Gunduz K, et al. Targetoid haemosiderotic haemangioma: dermoscopic monitoring of three cases and review of the literature. Clin Exp Dermatol. 2005;30:672-676.
- Kakizaki P, Valente NY, Paiva DL, et al. Targetoid hemosiderotic hemangioma--case report. An Bras Dermatol. 2014;89:956-959.
- Oppermann K, Boff AL, Bonamigo RR. Verrucous hemangioma and histopathological differential diagnosis with angiokeratoma circumscriptum neviforme. An Bras Dermatol. 2018;93:712-715.
- Boccara, O, Ariche-Maman, S, Hadj-Rabia, S, et al. Verrucous hemangioma (also known as verrucous venous malformation): a vascular anomaly frequently misdiagnosed as a lymphatic malformation. Pediatr Dermatol. 2018;35:E378-E381.
- Mestre T, Amaro C, Freitas I. Verrucous haemangioma: a diagnosis to consider [published online June 4, 2014]. BMJ Case Rep. doi:10.1136/bcr-2014-204612
- Ivy H, Julian CA. Angiokeratoma circumscriptum. StatPearls. StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK549769/
- Shetty S, Geetha V, Rao R, et al. Verrucous hemangioma: importance of a deeper biopsy. Indian J Dermatopathol Diagn Dermatol. 2014;1:99-100.
- Bishop BN, Lynch DT. Cancer, Kaposi sarcoma. StatPearls. StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK534839/
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Cao J, Wang J, He C, et al. Angiosarcoma: a review of diagnosis and current treatment. Am J Cancer Res. 2019;9:2303-2313.
- Papke DJ Jr, Hornick JL. What is new in endothelial neoplasia? Virchows Arch. 2020;476:17-28.
- Ambujam S, Audhya M, Reddy A, et al. Cutaneous angiosarcoma of the head, neck, and face of the elderly in type 5 skin. J Cutan Aesthet Surg. 2013;6:45-47.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.
- Joyce JC, Keith PJ, Szabo S, et al. Superficial hemosiderotic lymphovascular malformation (hobnail hemangioma): a report of six cases. Pediatr Dermatol. 2014;31:281-285.
- Sahin MT, Demir MA, Gunduz K, et al. Targetoid haemosiderotic haemangioma: dermoscopic monitoring of three cases and review of the literature. Clin Exp Dermatol. 2005;30:672-676.
- Kakizaki P, Valente NY, Paiva DL, et al. Targetoid hemosiderotic hemangioma--case report. An Bras Dermatol. 2014;89:956-959.
- Oppermann K, Boff AL, Bonamigo RR. Verrucous hemangioma and histopathological differential diagnosis with angiokeratoma circumscriptum neviforme. An Bras Dermatol. 2018;93:712-715.
- Boccara, O, Ariche-Maman, S, Hadj-Rabia, S, et al. Verrucous hemangioma (also known as verrucous venous malformation): a vascular anomaly frequently misdiagnosed as a lymphatic malformation. Pediatr Dermatol. 2018;35:E378-E381.
- Mestre T, Amaro C, Freitas I. Verrucous haemangioma: a diagnosis to consider [published online June 4, 2014]. BMJ Case Rep. doi:10.1136/bcr-2014-204612
- Ivy H, Julian CA. Angiokeratoma circumscriptum. StatPearls. StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK549769/
- Shetty S, Geetha V, Rao R, et al. Verrucous hemangioma: importance of a deeper biopsy. Indian J Dermatopathol Diagn Dermatol. 2014;1:99-100.
- Bishop BN, Lynch DT. Cancer, Kaposi sarcoma. StatPearls. StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK534839/
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Cao J, Wang J, He C, et al. Angiosarcoma: a review of diagnosis and current treatment. Am J Cancer Res. 2019;9:2303-2313.
- Papke DJ Jr, Hornick JL. What is new in endothelial neoplasia? Virchows Arch. 2020;476:17-28.
- Ambujam S, Audhya M, Reddy A, et al. Cutaneous angiosarcoma of the head, neck, and face of the elderly in type 5 skin. J Cutan Aesthet Surg. 2013;6:45-47.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.

A 35-year-old man presented with a reddish brown papule on the left upper chest of 1 year’s duration that had changed color to reddish purple. Physical examination revealed a 6-mm violaceous papule with an erythematous rim.
Hair Follicle Bulb Region: A Potential Nidus for the Formation of Osteoma Cutis
The term osteoma cutis (OC) is defined as the ossification or bone formation either in the dermis or hypodermis. 1 It is heterotopic in nature, referring to extraneous bone formation in soft tissue. Osteoma cutis was first described in 1858 2,3 ; in 1868, the multiple miliary form on the face was described. 4 Cutaneous ossification can take many forms, ranging from occurrence in a nevus (nevus of Nanta) to its association with rare genetic disorders, such as fibrodysplasia ossificans progressiva and Albright hereditary osteodystrophy.
Some of these ossifications are classified as primary; others are secondary, depending on the presence of a preexisting lesion (eg, pilomatricoma, basal cell carcinoma). However, certain conditions, such as multiple miliary osteoma of the face, can be difficult to classify due to the presence or absence of a history of acne or dermabrasion, or both. The secondary forms more commonly are encountered due to their incidental association with an excised lesion, such as pilomatricoma.
A precursor of OC has been neglected in the literature despite its common occurrence. It may have been peripherally alluded to in the literature in reference to the miliary form of OC.5,6 The cases reported here demonstrate small round nodules of calcification or ossification, or both, in punch biopsies and excision specimens from hair-bearing areas of skin, especially from the head and neck. These lesions are mainly observed in the peripilar location or more specifically in the approximate location of the hair bulb.
This article reviews a possible mechanism of formation of these osteocalcific micronodules. These often-encountered micronodules are small osteocalcific lesions without typical bone or well-formed OC, such as trabeculae formation or fatty marrow, and may represent earliest stages in the formation of OC.
Clinical Observations
During routine dermatopathologic practice, I observed incidental small osteocalcific micronodules in close proximity to the lower part of the hair follicle in multiple cases. These nodules were not related to the main lesion in the specimen and were not the reason for the biopsy or excision. Most of the time, these micronodules were noted in excision or re-excision specimens or in a punch biopsy.
In my review of multiple unrelated cases over time, incidental osteocalcific micronodules were observed occasionally in punch biopsies and excision specimens during routine practice. These micronodules were mainly located in the vicinity of a hair bulb (Figure 1). If the hair bulb was not present in the sections, these micronodules were noted near or within the fibrous tract (Figure 2) or beneath a sebaceous lobule (Figure 3). In an exceptional case, a small round deposit of osteoid was seen forming just above the dermal papilla of the hair bulb (Figure 4).
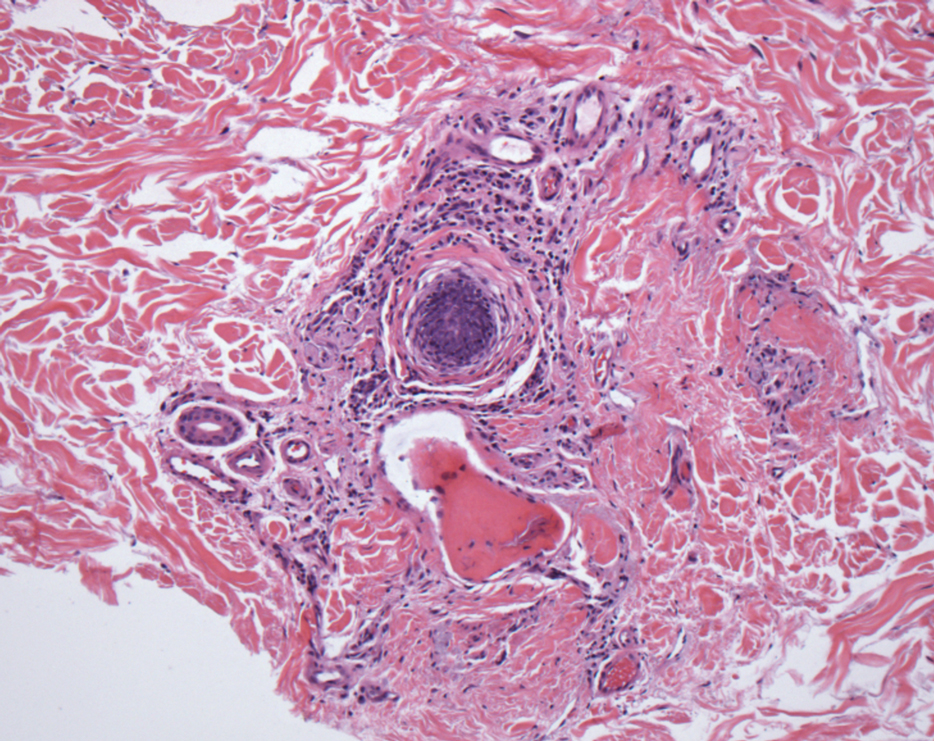
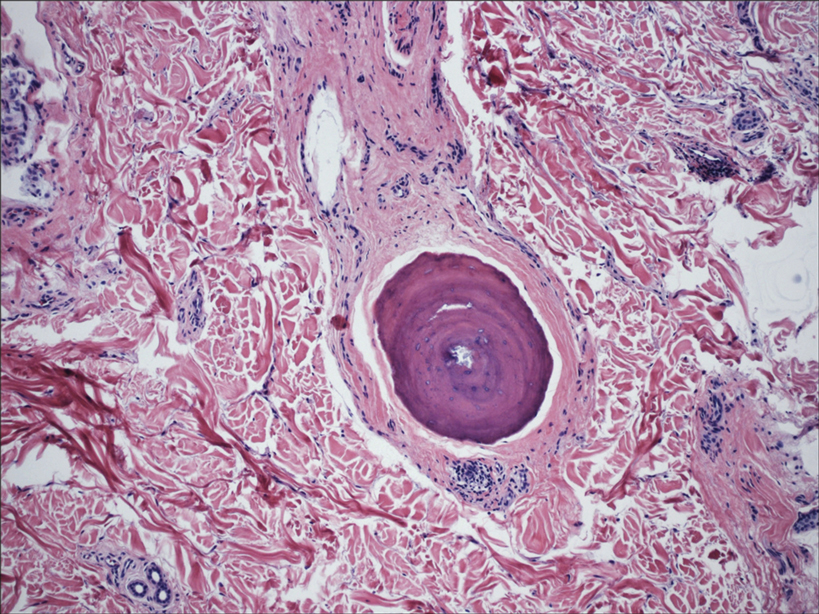
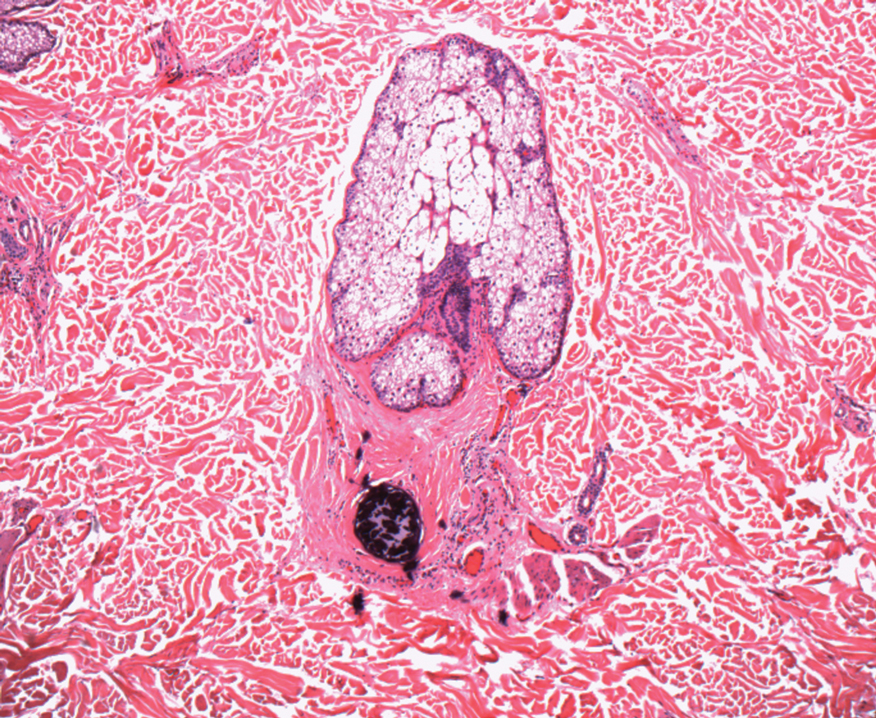
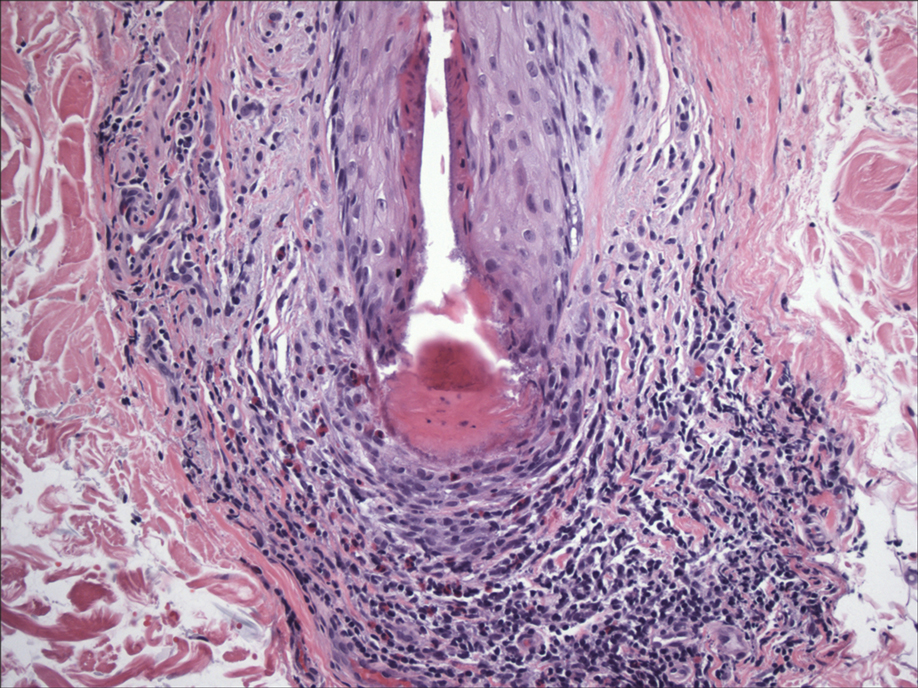
Multiple osteocalcific micronodules were identified in a case of cicatricial alopecia. These micronodules were observed in sections taken at the levels of hair bulbs, and more or less corresponded to the size of the bulb (Figure 5A). Fortuitously, the patient was dark-skinned; the remnants of melanin within the micronodules provided evidence that the micronodules were formed within hair bulbs. Melanin staining confirmed the presence of melanin within some of the micronodules (Figure 5B).
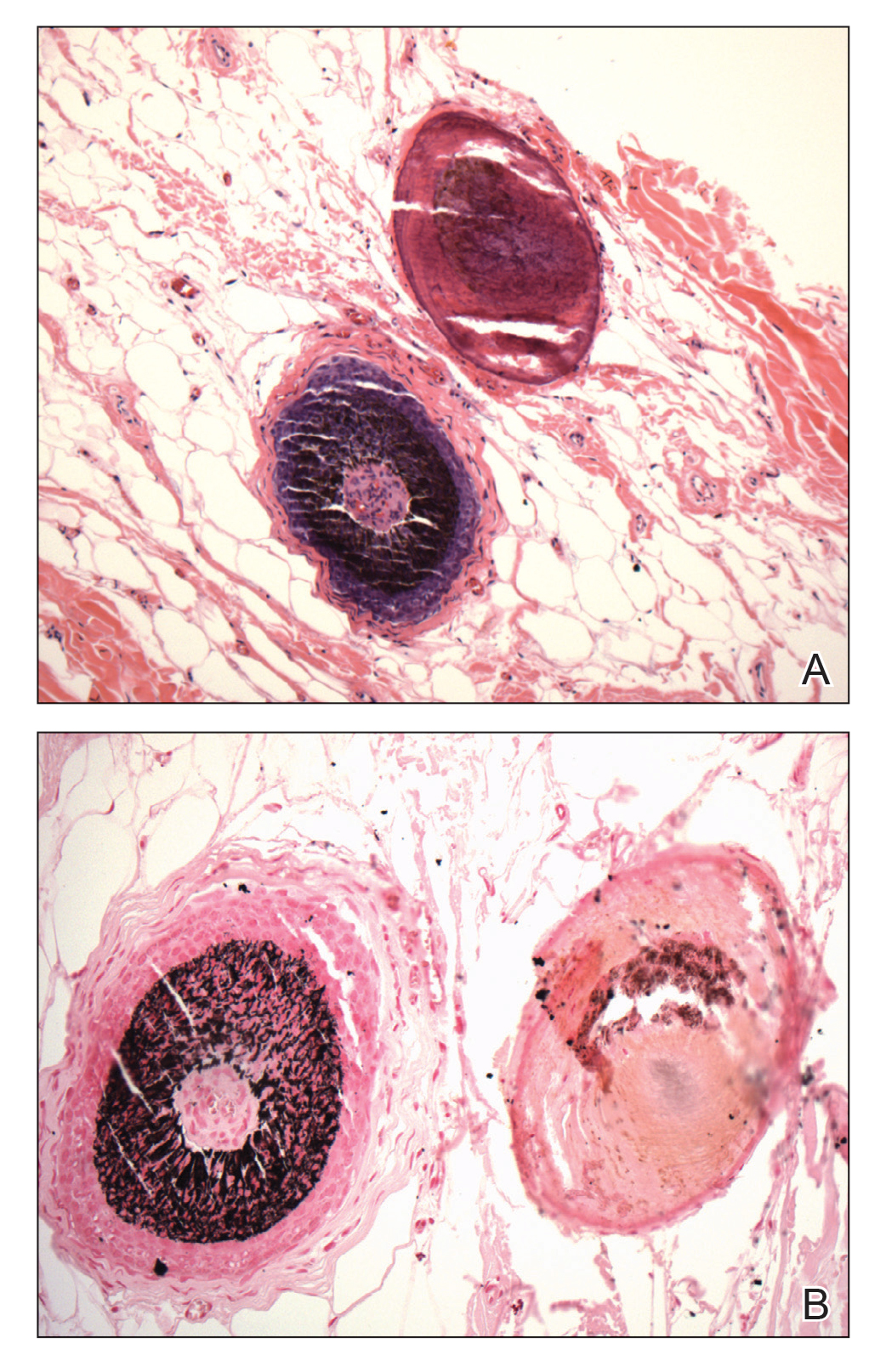
Comment
Skeletogenesis in humans takes place by 2 methods: endochondral ossification and intramembranous ossification. In contrast to endochondral ossification, intramembranous ossification does not require a preexisting cartilaginous template. Instead, there is condensation of mesenchymal cells, which differentiate into osteoblasts and lay down osteoid, thus forming an ossification center. Little is known about the mechanism of formation of OC or the nidus of formation of the primary form.
Incidental micronodules of calcification and ossification are routinely encountered during histopathologic review of specimens from hair-bearing areas of the skin in dermatopathology practice. A review of the literature, however, does not reveal any specific dermatopathologic term ascribed to this phenomenon. These lesions might be similar to those described by Hopkins5 in 1928 in the setting of miliary OC of the face secondary to acne. Rossman and Freeman6 also described the same lesions when referring to facial OC as a “stage of pre-osseous calcification.”
When these osteocalcific micronodules are encountered, it usually is in close proximity to a hair follicle bulb. When a hair bulb is not seen in the sections, the micronodules are noted near fibrous tracts, arrector pili muscles, or sebaceous lobules, suggesting a close peripilar or peribulbar location. The micronodules are approximately 0.5 mm in diameter—roughly the size of a hair bulb. Due to the close anatomic association of micronodules and the hair bulb, these lesions can be called pilar osteocalcific nodules (PONs).
The role of bone morphogenetic protein (BMP) signaling in the maintenance of the hair cycle is well established. Bone morphogenetic proteins are extracellular cytokines that belong to the transforming growth factor β family. The hair bulb microenvironment is rich in BMPs
As the name implies, BMPs were discovered in relation to their important role in osteogenesis and tissue homeostasis. More than 20 BMPs have been identified, many of which promote bone formation and repair of bone fracture. Osteoinductive BMPs include BMP-2 and BMP-4 through BMP-10; BMP-2 and BMP-4 are expressed in the hair matrix and BMP-4 and BMP-6 are expressed in the FDP.8,9 All bone-inducing BMPs can cause mesenchymal stem cells to differentiate into osteoblasts in vitro.10
Overactive BMP signaling has been shown to cause heterotopic ossification in patients with fibrodysplasia ossificans progressiva.8 Immunohistochemical expression of BMP-2 has been demonstrated in shadow cells of pilomatricoma.11 Calcification and ossification are seen in as many as 20% of pilomatricomas. Both BMP-2 and BMP-4 have been shown to induce osteogenic differentiation of mouse skin−derived fibroblasts and FDP cells.12
Myllylä et al13 described 4 cases of multiple miliary osteoma cutis (MMOC). They also found 47 reported cases of MMOC, in which there was a history of acne in 55% (26/47). Only 15% (7/47) of these cases were extrafacial on the neck, chest, back, and arms. Osteomas in these cases were not associated with folliculosebaceous units or other adnexal structures, which may have been due to replacement by acne scarring, as all 4 patients had a history of acne vulgaris. The authors postulated a role for the GNAS gene mutation in the morphogenesis of MMOC; however, no supporting evidence was found for this claim. They also postulated a role for BMPs in the formation of MMOC.13
Some disturbance or imbalance in hair bulb homeostasis leads to overactivity of BMP signaling, causing osteoinduction in the hair bulb region and formation of PONs. The cause of the disturbance could be a traumatic or inflammatory injury to the hair follicle, as in the case of the secondary form of MMOC in association with chronic acne. In the primary form of osteoma cutis, the trigger could be more subtle or subclinical.
Trauma and inflammation are the main initiating factors involved in ossification in patients with fibrodysplasia ossificans progressiva due to ectopic activity of BMPs.9 The primary form of ossification appears to be similar to the mechanism by which intramembranous ossification is laid down (ie, by differentiation of mesenchymal cells into osteoblasts). In the proposed scenario, the cells of FDP, under the influence of BMPs, differentiate into osteoblasts and lay down osteoid, forming a limited-capacity “ossification center” or pilar osteocalcific nodule.
It is difficult to know the exact relationship of PONs or OC to the hair bulb due to the 2-dimensional nature of histologic sections. However, considering the finding of a rare case of osteoid forming within the bulb and in another the presence of melanin within the osteocalcific nodule, it is likely that these lesions are formed within the hair bulb or in situations in which the conditions replicate the biochemical characteristics of the hair bulb (eg, pilomatricoma).
The formation of PONs might act as a terminal phase in the hair cycle that is rarely induced to provide an exit for damaged hair follicles from cyclical perpetuity. An unspecified event or injury might render a hair follicle unable to continue its cyclical growth and cause BMPs to induce premature calcification in or around the hair bulb, which would probably be the only known quasiphysiological mechanism for a damaged hair follicle to exit the hair cycle.
Another interesting aspect of osteoma formation in human skin is the similarity to osteoderms or the integumentary skeleton of vertebrates.14 Early in evolution, the dermal skeleton was the predominant skeletal system in some lineages. Phylogenetically, osteoderms are not uniformly distributed, and show a latent ability to manifest in some groups or lay dormant or disappear in others. The occurrence of primary osteomas in the human integument might be a vestigial manifestation of deep homology,15 a latent ability to form structures that have been lost. The embryologic formation of osteoderms in the dermis of vertebrates is thought to depend on the interaction or cross-talk between ectomesenchymal cells of neural crest origin and cells of the stratum basalis of epidermis, which is somewhat similar to the formation of the hair follicles.
Conclusion
Under certain conditions, the bulb region of a hair follicle might provide a nidus for the formation of OC. The hair bulb region contains both the precursor cellular element (mesenchymal cells of FDP) and the trigger cytokine (BMP) for the induction of osteogenic metaplasia.
- Burgdorf W, Nasemann T. Cutaneous osteomas: a clinical and histopathologic review. Arch Dermatol Res. 1977;260:121-135.
- Essing M. Osteoma cutis of the forehead. HNO. 1985;33:548-550.
- Bouraoui S, Mlika M, Kort R, et al. Miliary osteoma cutis of the face. J Dermatol Case Rep. 2011;5:77-81.
- Virchow R. Die krankhaften Geschwülste. Vol 2. Hirschwald; 1864.
- Hopkins JG. Multiple miliary osteomas of the skin: report of a case. Arch Derm Syphilol. 1928;18:706-715.
- Rossman RE, Freeman RG. Osteoma cutis, a stage of preosseous calcification. Arch Dermatol. 1964;89:68-73.
- Guha U, Mecklenburg L, Cowin P, et al. Bone morphogenetic protein signaling regulates postnatal hair follicle differentiation and cycling. Am J Pathol. 2004;165:729-740.
- Rendl M, Polak L, Fuchs E. BMP signaling in dermal papilla cells is required for their hair follicle-inductive properties. Genes Dev. 2008;22:543-557.
- Shi S, de Gorter DJJ, Hoogaars WMH, et al. Overactive bone morphogenetic protein signaling in heterotopic ossification and Duchenne muscular dystrophy. Cell Mol Life Sci. 2013;70:407-423.
- Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem. 2010;147:35-51.
- Kurokawa I, Kusumoto K, Bessho K. Immunohistochemical expression of bone morphogenetic protein-2 in pilomatricoma. Br J Dermatol. 2000;143:754-758.
- Myllylä RM, Haapasaari K-M, Lehenkari P, et al. Bone morphogenetic proteins 4 and 2/7 induce osteogenic differentiation of mouse skin derived fibroblast and dermal papilla cells. Cell Tissue Res. 2014;355:463-470.
- Myllylä RM, Haapasaari KM, Palatsi R, et al. Multiple miliary osteoma cutis is a distinct disease entity: four case reports and review of the literature. Br J Dermatol. 2011;164:544-552.
- Vickaryous MK, Sire J-Y. The integumentary skeleton of tetrapods: origin, evolution, and development. J Anat. 2009;214:441-464.
- Vickaryous MK, Hall BK. Development of the dermal skeleton in Alligator mississippiensis (Archosauria, Crocodylia) with comments on the homology of osteoderms. J Morphol. 2008;269:398-422.
The term osteoma cutis (OC) is defined as the ossification or bone formation either in the dermis or hypodermis. 1 It is heterotopic in nature, referring to extraneous bone formation in soft tissue. Osteoma cutis was first described in 1858 2,3 ; in 1868, the multiple miliary form on the face was described. 4 Cutaneous ossification can take many forms, ranging from occurrence in a nevus (nevus of Nanta) to its association with rare genetic disorders, such as fibrodysplasia ossificans progressiva and Albright hereditary osteodystrophy.
Some of these ossifications are classified as primary; others are secondary, depending on the presence of a preexisting lesion (eg, pilomatricoma, basal cell carcinoma). However, certain conditions, such as multiple miliary osteoma of the face, can be difficult to classify due to the presence or absence of a history of acne or dermabrasion, or both. The secondary forms more commonly are encountered due to their incidental association with an excised lesion, such as pilomatricoma.
A precursor of OC has been neglected in the literature despite its common occurrence. It may have been peripherally alluded to in the literature in reference to the miliary form of OC.5,6 The cases reported here demonstrate small round nodules of calcification or ossification, or both, in punch biopsies and excision specimens from hair-bearing areas of skin, especially from the head and neck. These lesions are mainly observed in the peripilar location or more specifically in the approximate location of the hair bulb.
This article reviews a possible mechanism of formation of these osteocalcific micronodules. These often-encountered micronodules are small osteocalcific lesions without typical bone or well-formed OC, such as trabeculae formation or fatty marrow, and may represent earliest stages in the formation of OC.
Clinical Observations
During routine dermatopathologic practice, I observed incidental small osteocalcific micronodules in close proximity to the lower part of the hair follicle in multiple cases. These nodules were not related to the main lesion in the specimen and were not the reason for the biopsy or excision. Most of the time, these micronodules were noted in excision or re-excision specimens or in a punch biopsy.
In my review of multiple unrelated cases over time, incidental osteocalcific micronodules were observed occasionally in punch biopsies and excision specimens during routine practice. These micronodules were mainly located in the vicinity of a hair bulb (Figure 1). If the hair bulb was not present in the sections, these micronodules were noted near or within the fibrous tract (Figure 2) or beneath a sebaceous lobule (Figure 3). In an exceptional case, a small round deposit of osteoid was seen forming just above the dermal papilla of the hair bulb (Figure 4).
Multiple osteocalcific micronodules were identified in a case of cicatricial alopecia. These micronodules were observed in sections taken at the levels of hair bulbs, and more or less corresponded to the size of the bulb (Figure 5A). Fortuitously, the patient was dark-skinned; the remnants of melanin within the micronodules provided evidence that the micronodules were formed within hair bulbs. Melanin staining confirmed the presence of melanin within some of the micronodules (Figure 5B).
Comment
Skeletogenesis in humans takes place by 2 methods: endochondral ossification and intramembranous ossification. In contrast to endochondral ossification, intramembranous ossification does not require a preexisting cartilaginous template. Instead, there is condensation of mesenchymal cells, which differentiate into osteoblasts and lay down osteoid, thus forming an ossification center. Little is known about the mechanism of formation of OC or the nidus of formation of the primary form.
Incidental micronodules of calcification and ossification are routinely encountered during histopathologic review of specimens from hair-bearing areas of the skin in dermatopathology practice. A review of the literature, however, does not reveal any specific dermatopathologic term ascribed to this phenomenon. These lesions might be similar to those described by Hopkins5 in 1928 in the setting of miliary OC of the face secondary to acne. Rossman and Freeman6 also described the same lesions when referring to facial OC as a “stage of pre-osseous calcification.”
When these osteocalcific micronodules are encountered, it usually is in close proximity to a hair follicle bulb. When a hair bulb is not seen in the sections, the micronodules are noted near fibrous tracts, arrector pili muscles, or sebaceous lobules, suggesting a close peripilar or peribulbar location. The micronodules are approximately 0.5 mm in diameter—roughly the size of a hair bulb. Due to the close anatomic association of micronodules and the hair bulb, these lesions can be called pilar osteocalcific nodules (PONs).
The role of bone morphogenetic protein (BMP) signaling in the maintenance of the hair cycle is well established. Bone morphogenetic proteins are extracellular cytokines that belong to the transforming growth factor β family. The hair bulb microenvironment is rich in BMPs
As the name implies, BMPs were discovered in relation to their important role in osteogenesis and tissue homeostasis. More than 20 BMPs have been identified, many of which promote bone formation and repair of bone fracture. Osteoinductive BMPs include BMP-2 and BMP-4 through BMP-10; BMP-2 and BMP-4 are expressed in the hair matrix and BMP-4 and BMP-6 are expressed in the FDP.8,9 All bone-inducing BMPs can cause mesenchymal stem cells to differentiate into osteoblasts in vitro.10
Overactive BMP signaling has been shown to cause heterotopic ossification in patients with fibrodysplasia ossificans progressiva.8 Immunohistochemical expression of BMP-2 has been demonstrated in shadow cells of pilomatricoma.11 Calcification and ossification are seen in as many as 20% of pilomatricomas. Both BMP-2 and BMP-4 have been shown to induce osteogenic differentiation of mouse skin−derived fibroblasts and FDP cells.12
Myllylä et al13 described 4 cases of multiple miliary osteoma cutis (MMOC). They also found 47 reported cases of MMOC, in which there was a history of acne in 55% (26/47). Only 15% (7/47) of these cases were extrafacial on the neck, chest, back, and arms. Osteomas in these cases were not associated with folliculosebaceous units or other adnexal structures, which may have been due to replacement by acne scarring, as all 4 patients had a history of acne vulgaris. The authors postulated a role for the GNAS gene mutation in the morphogenesis of MMOC; however, no supporting evidence was found for this claim. They also postulated a role for BMPs in the formation of MMOC.13
Some disturbance or imbalance in hair bulb homeostasis leads to overactivity of BMP signaling, causing osteoinduction in the hair bulb region and formation of PONs. The cause of the disturbance could be a traumatic or inflammatory injury to the hair follicle, as in the case of the secondary form of MMOC in association with chronic acne. In the primary form of osteoma cutis, the trigger could be more subtle or subclinical.
Trauma and inflammation are the main initiating factors involved in ossification in patients with fibrodysplasia ossificans progressiva due to ectopic activity of BMPs.9 The primary form of ossification appears to be similar to the mechanism by which intramembranous ossification is laid down (ie, by differentiation of mesenchymal cells into osteoblasts). In the proposed scenario, the cells of FDP, under the influence of BMPs, differentiate into osteoblasts and lay down osteoid, forming a limited-capacity “ossification center” or pilar osteocalcific nodule.
It is difficult to know the exact relationship of PONs or OC to the hair bulb due to the 2-dimensional nature of histologic sections. However, considering the finding of a rare case of osteoid forming within the bulb and in another the presence of melanin within the osteocalcific nodule, it is likely that these lesions are formed within the hair bulb or in situations in which the conditions replicate the biochemical characteristics of the hair bulb (eg, pilomatricoma).
The formation of PONs might act as a terminal phase in the hair cycle that is rarely induced to provide an exit for damaged hair follicles from cyclical perpetuity. An unspecified event or injury might render a hair follicle unable to continue its cyclical growth and cause BMPs to induce premature calcification in or around the hair bulb, which would probably be the only known quasiphysiological mechanism for a damaged hair follicle to exit the hair cycle.
Another interesting aspect of osteoma formation in human skin is the similarity to osteoderms or the integumentary skeleton of vertebrates.14 Early in evolution, the dermal skeleton was the predominant skeletal system in some lineages. Phylogenetically, osteoderms are not uniformly distributed, and show a latent ability to manifest in some groups or lay dormant or disappear in others. The occurrence of primary osteomas in the human integument might be a vestigial manifestation of deep homology,15 a latent ability to form structures that have been lost. The embryologic formation of osteoderms in the dermis of vertebrates is thought to depend on the interaction or cross-talk between ectomesenchymal cells of neural crest origin and cells of the stratum basalis of epidermis, which is somewhat similar to the formation of the hair follicles.
Conclusion
Under certain conditions, the bulb region of a hair follicle might provide a nidus for the formation of OC. The hair bulb region contains both the precursor cellular element (mesenchymal cells of FDP) and the trigger cytokine (BMP) for the induction of osteogenic metaplasia.
The term osteoma cutis (OC) is defined as the ossification or bone formation either in the dermis or hypodermis. 1 It is heterotopic in nature, referring to extraneous bone formation in soft tissue. Osteoma cutis was first described in 1858 2,3 ; in 1868, the multiple miliary form on the face was described. 4 Cutaneous ossification can take many forms, ranging from occurrence in a nevus (nevus of Nanta) to its association with rare genetic disorders, such as fibrodysplasia ossificans progressiva and Albright hereditary osteodystrophy.
Some of these ossifications are classified as primary; others are secondary, depending on the presence of a preexisting lesion (eg, pilomatricoma, basal cell carcinoma). However, certain conditions, such as multiple miliary osteoma of the face, can be difficult to classify due to the presence or absence of a history of acne or dermabrasion, or both. The secondary forms more commonly are encountered due to their incidental association with an excised lesion, such as pilomatricoma.
A precursor of OC has been neglected in the literature despite its common occurrence. It may have been peripherally alluded to in the literature in reference to the miliary form of OC.5,6 The cases reported here demonstrate small round nodules of calcification or ossification, or both, in punch biopsies and excision specimens from hair-bearing areas of skin, especially from the head and neck. These lesions are mainly observed in the peripilar location or more specifically in the approximate location of the hair bulb.
This article reviews a possible mechanism of formation of these osteocalcific micronodules. These often-encountered micronodules are small osteocalcific lesions without typical bone or well-formed OC, such as trabeculae formation or fatty marrow, and may represent earliest stages in the formation of OC.
Clinical Observations
During routine dermatopathologic practice, I observed incidental small osteocalcific micronodules in close proximity to the lower part of the hair follicle in multiple cases. These nodules were not related to the main lesion in the specimen and were not the reason for the biopsy or excision. Most of the time, these micronodules were noted in excision or re-excision specimens or in a punch biopsy.
In my review of multiple unrelated cases over time, incidental osteocalcific micronodules were observed occasionally in punch biopsies and excision specimens during routine practice. These micronodules were mainly located in the vicinity of a hair bulb (Figure 1). If the hair bulb was not present in the sections, these micronodules were noted near or within the fibrous tract (Figure 2) or beneath a sebaceous lobule (Figure 3). In an exceptional case, a small round deposit of osteoid was seen forming just above the dermal papilla of the hair bulb (Figure 4).
Multiple osteocalcific micronodules were identified in a case of cicatricial alopecia. These micronodules were observed in sections taken at the levels of hair bulbs, and more or less corresponded to the size of the bulb (Figure 5A). Fortuitously, the patient was dark-skinned; the remnants of melanin within the micronodules provided evidence that the micronodules were formed within hair bulbs. Melanin staining confirmed the presence of melanin within some of the micronodules (Figure 5B).
Comment
Skeletogenesis in humans takes place by 2 methods: endochondral ossification and intramembranous ossification. In contrast to endochondral ossification, intramembranous ossification does not require a preexisting cartilaginous template. Instead, there is condensation of mesenchymal cells, which differentiate into osteoblasts and lay down osteoid, thus forming an ossification center. Little is known about the mechanism of formation of OC or the nidus of formation of the primary form.
Incidental micronodules of calcification and ossification are routinely encountered during histopathologic review of specimens from hair-bearing areas of the skin in dermatopathology practice. A review of the literature, however, does not reveal any specific dermatopathologic term ascribed to this phenomenon. These lesions might be similar to those described by Hopkins5 in 1928 in the setting of miliary OC of the face secondary to acne. Rossman and Freeman6 also described the same lesions when referring to facial OC as a “stage of pre-osseous calcification.”
When these osteocalcific micronodules are encountered, it usually is in close proximity to a hair follicle bulb. When a hair bulb is not seen in the sections, the micronodules are noted near fibrous tracts, arrector pili muscles, or sebaceous lobules, suggesting a close peripilar or peribulbar location. The micronodules are approximately 0.5 mm in diameter—roughly the size of a hair bulb. Due to the close anatomic association of micronodules and the hair bulb, these lesions can be called pilar osteocalcific nodules (PONs).
The role of bone morphogenetic protein (BMP) signaling in the maintenance of the hair cycle is well established. Bone morphogenetic proteins are extracellular cytokines that belong to the transforming growth factor β family. The hair bulb microenvironment is rich in BMPs
As the name implies, BMPs were discovered in relation to their important role in osteogenesis and tissue homeostasis. More than 20 BMPs have been identified, many of which promote bone formation and repair of bone fracture. Osteoinductive BMPs include BMP-2 and BMP-4 through BMP-10; BMP-2 and BMP-4 are expressed in the hair matrix and BMP-4 and BMP-6 are expressed in the FDP.8,9 All bone-inducing BMPs can cause mesenchymal stem cells to differentiate into osteoblasts in vitro.10
Overactive BMP signaling has been shown to cause heterotopic ossification in patients with fibrodysplasia ossificans progressiva.8 Immunohistochemical expression of BMP-2 has been demonstrated in shadow cells of pilomatricoma.11 Calcification and ossification are seen in as many as 20% of pilomatricomas. Both BMP-2 and BMP-4 have been shown to induce osteogenic differentiation of mouse skin−derived fibroblasts and FDP cells.12
Myllylä et al13 described 4 cases of multiple miliary osteoma cutis (MMOC). They also found 47 reported cases of MMOC, in which there was a history of acne in 55% (26/47). Only 15% (7/47) of these cases were extrafacial on the neck, chest, back, and arms. Osteomas in these cases were not associated with folliculosebaceous units or other adnexal structures, which may have been due to replacement by acne scarring, as all 4 patients had a history of acne vulgaris. The authors postulated a role for the GNAS gene mutation in the morphogenesis of MMOC; however, no supporting evidence was found for this claim. They also postulated a role for BMPs in the formation of MMOC.13
Some disturbance or imbalance in hair bulb homeostasis leads to overactivity of BMP signaling, causing osteoinduction in the hair bulb region and formation of PONs. The cause of the disturbance could be a traumatic or inflammatory injury to the hair follicle, as in the case of the secondary form of MMOC in association with chronic acne. In the primary form of osteoma cutis, the trigger could be more subtle or subclinical.
Trauma and inflammation are the main initiating factors involved in ossification in patients with fibrodysplasia ossificans progressiva due to ectopic activity of BMPs.9 The primary form of ossification appears to be similar to the mechanism by which intramembranous ossification is laid down (ie, by differentiation of mesenchymal cells into osteoblasts). In the proposed scenario, the cells of FDP, under the influence of BMPs, differentiate into osteoblasts and lay down osteoid, forming a limited-capacity “ossification center” or pilar osteocalcific nodule.
It is difficult to know the exact relationship of PONs or OC to the hair bulb due to the 2-dimensional nature of histologic sections. However, considering the finding of a rare case of osteoid forming within the bulb and in another the presence of melanin within the osteocalcific nodule, it is likely that these lesions are formed within the hair bulb or in situations in which the conditions replicate the biochemical characteristics of the hair bulb (eg, pilomatricoma).
The formation of PONs might act as a terminal phase in the hair cycle that is rarely induced to provide an exit for damaged hair follicles from cyclical perpetuity. An unspecified event or injury might render a hair follicle unable to continue its cyclical growth and cause BMPs to induce premature calcification in or around the hair bulb, which would probably be the only known quasiphysiological mechanism for a damaged hair follicle to exit the hair cycle.
Another interesting aspect of osteoma formation in human skin is the similarity to osteoderms or the integumentary skeleton of vertebrates.14 Early in evolution, the dermal skeleton was the predominant skeletal system in some lineages. Phylogenetically, osteoderms are not uniformly distributed, and show a latent ability to manifest in some groups or lay dormant or disappear in others. The occurrence of primary osteomas in the human integument might be a vestigial manifestation of deep homology,15 a latent ability to form structures that have been lost. The embryologic formation of osteoderms in the dermis of vertebrates is thought to depend on the interaction or cross-talk between ectomesenchymal cells of neural crest origin and cells of the stratum basalis of epidermis, which is somewhat similar to the formation of the hair follicles.
Conclusion
Under certain conditions, the bulb region of a hair follicle might provide a nidus for the formation of OC. The hair bulb region contains both the precursor cellular element (mesenchymal cells of FDP) and the trigger cytokine (BMP) for the induction of osteogenic metaplasia.
- Burgdorf W, Nasemann T. Cutaneous osteomas: a clinical and histopathologic review. Arch Dermatol Res. 1977;260:121-135.
- Essing M. Osteoma cutis of the forehead. HNO. 1985;33:548-550.
- Bouraoui S, Mlika M, Kort R, et al. Miliary osteoma cutis of the face. J Dermatol Case Rep. 2011;5:77-81.
- Virchow R. Die krankhaften Geschwülste. Vol 2. Hirschwald; 1864.
- Hopkins JG. Multiple miliary osteomas of the skin: report of a case. Arch Derm Syphilol. 1928;18:706-715.
- Rossman RE, Freeman RG. Osteoma cutis, a stage of preosseous calcification. Arch Dermatol. 1964;89:68-73.
- Guha U, Mecklenburg L, Cowin P, et al. Bone morphogenetic protein signaling regulates postnatal hair follicle differentiation and cycling. Am J Pathol. 2004;165:729-740.
- Rendl M, Polak L, Fuchs E. BMP signaling in dermal papilla cells is required for their hair follicle-inductive properties. Genes Dev. 2008;22:543-557.
- Shi S, de Gorter DJJ, Hoogaars WMH, et al. Overactive bone morphogenetic protein signaling in heterotopic ossification and Duchenne muscular dystrophy. Cell Mol Life Sci. 2013;70:407-423.
- Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem. 2010;147:35-51.
- Kurokawa I, Kusumoto K, Bessho K. Immunohistochemical expression of bone morphogenetic protein-2 in pilomatricoma. Br J Dermatol. 2000;143:754-758.
- Myllylä RM, Haapasaari K-M, Lehenkari P, et al. Bone morphogenetic proteins 4 and 2/7 induce osteogenic differentiation of mouse skin derived fibroblast and dermal papilla cells. Cell Tissue Res. 2014;355:463-470.
- Myllylä RM, Haapasaari KM, Palatsi R, et al. Multiple miliary osteoma cutis is a distinct disease entity: four case reports and review of the literature. Br J Dermatol. 2011;164:544-552.
- Vickaryous MK, Sire J-Y. The integumentary skeleton of tetrapods: origin, evolution, and development. J Anat. 2009;214:441-464.
- Vickaryous MK, Hall BK. Development of the dermal skeleton in Alligator mississippiensis (Archosauria, Crocodylia) with comments on the homology of osteoderms. J Morphol. 2008;269:398-422.
- Burgdorf W, Nasemann T. Cutaneous osteomas: a clinical and histopathologic review. Arch Dermatol Res. 1977;260:121-135.
- Essing M. Osteoma cutis of the forehead. HNO. 1985;33:548-550.
- Bouraoui S, Mlika M, Kort R, et al. Miliary osteoma cutis of the face. J Dermatol Case Rep. 2011;5:77-81.
- Virchow R. Die krankhaften Geschwülste. Vol 2. Hirschwald; 1864.
- Hopkins JG. Multiple miliary osteomas of the skin: report of a case. Arch Derm Syphilol. 1928;18:706-715.
- Rossman RE, Freeman RG. Osteoma cutis, a stage of preosseous calcification. Arch Dermatol. 1964;89:68-73.
- Guha U, Mecklenburg L, Cowin P, et al. Bone morphogenetic protein signaling regulates postnatal hair follicle differentiation and cycling. Am J Pathol. 2004;165:729-740.
- Rendl M, Polak L, Fuchs E. BMP signaling in dermal papilla cells is required for their hair follicle-inductive properties. Genes Dev. 2008;22:543-557.
- Shi S, de Gorter DJJ, Hoogaars WMH, et al. Overactive bone morphogenetic protein signaling in heterotopic ossification and Duchenne muscular dystrophy. Cell Mol Life Sci. 2013;70:407-423.
- Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem. 2010;147:35-51.
- Kurokawa I, Kusumoto K, Bessho K. Immunohistochemical expression of bone morphogenetic protein-2 in pilomatricoma. Br J Dermatol. 2000;143:754-758.
- Myllylä RM, Haapasaari K-M, Lehenkari P, et al. Bone morphogenetic proteins 4 and 2/7 induce osteogenic differentiation of mouse skin derived fibroblast and dermal papilla cells. Cell Tissue Res. 2014;355:463-470.
- Myllylä RM, Haapasaari KM, Palatsi R, et al. Multiple miliary osteoma cutis is a distinct disease entity: four case reports and review of the literature. Br J Dermatol. 2011;164:544-552.
- Vickaryous MK, Sire J-Y. The integumentary skeleton of tetrapods: origin, evolution, and development. J Anat. 2009;214:441-464.
- Vickaryous MK, Hall BK. Development of the dermal skeleton in Alligator mississippiensis (Archosauria, Crocodylia) with comments on the homology of osteoderms. J Morphol. 2008;269:398-422.
Practice Points
- Understanding the pathogenesis of osteoma cutis (OC) can help physicians devise management of these disfiguring lesions.
- Small osteocalcific nodules in close proximity to the lower aspect of the hair bulb may be an important precursor to OC.
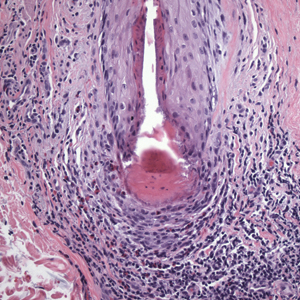
Erythema, Blisters, and Scars on the Elbows, Knees, and Legs
The Diagnosis: Epidermolysis Bullosa Acquisita
The diagnosis of epidermolysis bullosa acquisita (EBA) was made based on the clinical and pathologic findings. A blistering disorder that resolves with milia is characteristic of EBA. Hematoxylin and eosin staining demonstrated a pauci-inflammatory separation between the epidermis and dermis (Figure 1). Direct immunofluorescence studies showed linear IgG deposition along the basement membrane zone while C3 was negative (Figure 2). Salt-split skin was essential, as it revealed IgG deposition to the floor of the split (Figure 3), a pattern seen in EBA and not bullous pemphigoid (BP).1
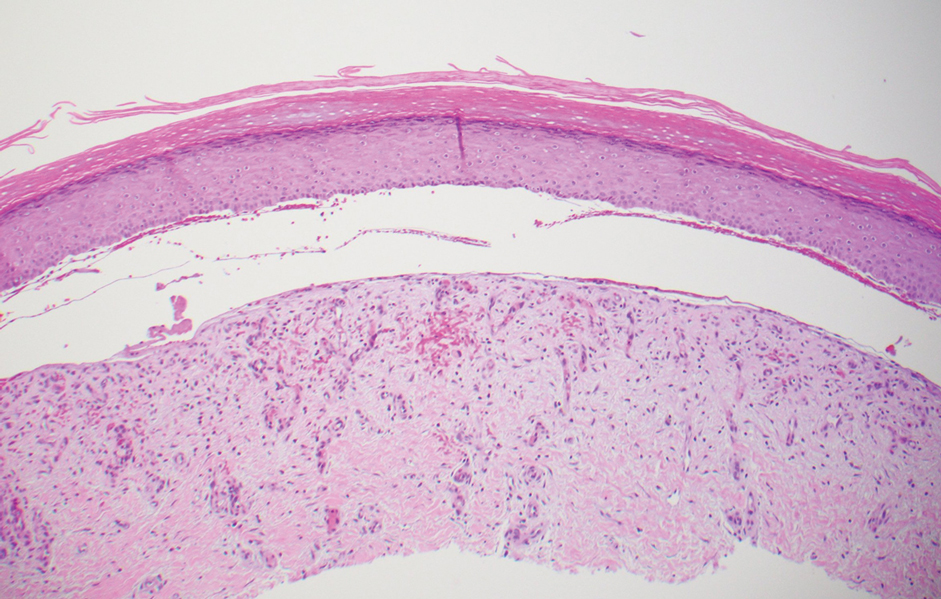
Epidermolysis bullosa acquisita is an acquired autoimmune bullous disorder that results from antibodies to type VII collagen, an anchoring fibril that attaches the lamina densa to the dermis. The epidemiology and etiology of the trigger that leads to antibody production are not well known, but an association between EBA and inflammatory bowel disease has been described.2 Although this disease may present in childhood, EBA most commonly is a disorder seen in adults and the elderly. A classic noninflammatory mechanobullous form as well as an inflammatory BP-like form are the most commonly encountered presentations. Light microscopy demonstrates subepidermal cleavage without acantholysis. In the inflammatory BP-like subtype, an inflammatory infiltrate may be present. Direct immunofluorescence is remarkable for a linear band of IgG deposits along the basement membrane zone, with or without C3 deposition in a similar pattern.1
Bullous pemphigoid is within the differential of EBA. It can be difficult to differentiate clinically, especially when a patient has the BP-like variant of EBA because, as the name implies, it mimics BP. Patients with BP often will report a pruritic patch that will then develop into an urticarial plaque. Scarring and milia rarely are seen in BP but can be observed in the multiple presentations of EBA. Hematoxylin and eosin staining and direct immunofluorescence may be almost identical, and differentiating between the 2 disorders can be a challenge. Immunodeposition in EBA occurs in a U-shaped, serrated pattern, while the pattern in BP is N-shaped and serrated.3 Although the U-shaped, serrated pattern is relatively specific, it is not always easy to interpret and requires a high-quality biopsy specimen, which can be difficult to discern with certainty in suboptimal preparations. Another way to differentiate between the 2 entities is to utilize the salt-split skin technique, as performed in our patient. With salt-split skin, the biopsy is placed into a solution of 1 mol/L sodium chloride and incubated at 4 °C (39 °F) for 18 to 24 hours. A blister is then produced at the level of the lamina lucida, which allows for the staining of immunoreactants to occur either above or below that split (commonly referred to as staining on the roof or floor of the blister cavity). With EBA, there is immunoreactant deposition on the floor of the blister, while the opposite occurs in BP.4
Epidermolysis bullosa simplex is the most common type of epidermolysis bullosa, with keratin genes KRT5 and KRT14 as frequent mutations. Patients develop blisters, vesicles, bullae, and milia on traumatized areas of the body such as the hands, elbows, knees, and feet. This disease presents early in childhood. Histology exhibits a cell-poor subepidermal blister.5 With porphyria cutanea tarda, reduced activity of uroporphyrinogen decarboxylase, a major enzyme in the heme synthesis pathway, leads to blisters with erosions and milia on sun-exposed areas of the body. Histologic evaluation reveals a subepidermal pauci-inflammatory vesicle with festooning of the dermal papillae and amphophilic basement membrane within the epidermis. Direct immunofluorescence of porphyria cutanea tarda demonstrates IgM and C3 in the vessels.6 Sweet syndrome is a neutrophilic dermatosis that presents as erythematous, edematous, hot, and tender plaques along with fever and leukocytosis. It is associated with myeloproliferative disorders. Biopsy demonstrates papillary dermal edema along with diffuse neutrophilic infiltrate.7
Numerous medications have been recommended for the treatment of EBA, ranging from steroids to steroid-sparing drugs such as colchicine and dapsone.8,9 Our patient was educated on physical precautions and was started on dapsone alone due to comorbid diabetes mellitus and renal disease. Within a few weeks of initiating dapsone, he observed a reduction in erythema, and within months he experienced a decrease in blister eruption frequency.
- Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. 2017;13:157-169.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-230.
- Vodegel RM, Jonkman MF, Pas HH, et al. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol. 2004;151:112-118.
- Gardner KM, Crawford RI. Distinguishing epidermolysis bullosa acquisita from bullous pemphigoid without direct immunofluorescence. J Cutan Med Surg. 2018;22:22-24.
- Sprecher E. Epidermolysis bullosa simplex. Dermatol Clin. 2010;28:23-32.
- Maynard B, Peters MS. Histologic and immunofluorescence study of cutaneous porphyrias. J Cutan Pathol. 1992;19:40-47.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018:79:987-1006.
- Kirtschig G, Murrell D, Wojnarowska F, et al. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database Syst Rev. 2003;1:CD004056
- Gürcan HM, Ahmed AR. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin Pharmacother. 2011;12:1259-1268.
The Diagnosis: Epidermolysis Bullosa Acquisita
The diagnosis of epidermolysis bullosa acquisita (EBA) was made based on the clinical and pathologic findings. A blistering disorder that resolves with milia is characteristic of EBA. Hematoxylin and eosin staining demonstrated a pauci-inflammatory separation between the epidermis and dermis (Figure 1). Direct immunofluorescence studies showed linear IgG deposition along the basement membrane zone while C3 was negative (Figure 2). Salt-split skin was essential, as it revealed IgG deposition to the floor of the split (Figure 3), a pattern seen in EBA and not bullous pemphigoid (BP).1
Epidermolysis bullosa acquisita is an acquired autoimmune bullous disorder that results from antibodies to type VII collagen, an anchoring fibril that attaches the lamina densa to the dermis. The epidemiology and etiology of the trigger that leads to antibody production are not well known, but an association between EBA and inflammatory bowel disease has been described.2 Although this disease may present in childhood, EBA most commonly is a disorder seen in adults and the elderly. A classic noninflammatory mechanobullous form as well as an inflammatory BP-like form are the most commonly encountered presentations. Light microscopy demonstrates subepidermal cleavage without acantholysis. In the inflammatory BP-like subtype, an inflammatory infiltrate may be present. Direct immunofluorescence is remarkable for a linear band of IgG deposits along the basement membrane zone, with or without C3 deposition in a similar pattern.1
Bullous pemphigoid is within the differential of EBA. It can be difficult to differentiate clinically, especially when a patient has the BP-like variant of EBA because, as the name implies, it mimics BP. Patients with BP often will report a pruritic patch that will then develop into an urticarial plaque. Scarring and milia rarely are seen in BP but can be observed in the multiple presentations of EBA. Hematoxylin and eosin staining and direct immunofluorescence may be almost identical, and differentiating between the 2 disorders can be a challenge. Immunodeposition in EBA occurs in a U-shaped, serrated pattern, while the pattern in BP is N-shaped and serrated.3 Although the U-shaped, serrated pattern is relatively specific, it is not always easy to interpret and requires a high-quality biopsy specimen, which can be difficult to discern with certainty in suboptimal preparations. Another way to differentiate between the 2 entities is to utilize the salt-split skin technique, as performed in our patient. With salt-split skin, the biopsy is placed into a solution of 1 mol/L sodium chloride and incubated at 4 °C (39 °F) for 18 to 24 hours. A blister is then produced at the level of the lamina lucida, which allows for the staining of immunoreactants to occur either above or below that split (commonly referred to as staining on the roof or floor of the blister cavity). With EBA, there is immunoreactant deposition on the floor of the blister, while the opposite occurs in BP.4
Epidermolysis bullosa simplex is the most common type of epidermolysis bullosa, with keratin genes KRT5 and KRT14 as frequent mutations. Patients develop blisters, vesicles, bullae, and milia on traumatized areas of the body such as the hands, elbows, knees, and feet. This disease presents early in childhood. Histology exhibits a cell-poor subepidermal blister.5 With porphyria cutanea tarda, reduced activity of uroporphyrinogen decarboxylase, a major enzyme in the heme synthesis pathway, leads to blisters with erosions and milia on sun-exposed areas of the body. Histologic evaluation reveals a subepidermal pauci-inflammatory vesicle with festooning of the dermal papillae and amphophilic basement membrane within the epidermis. Direct immunofluorescence of porphyria cutanea tarda demonstrates IgM and C3 in the vessels.6 Sweet syndrome is a neutrophilic dermatosis that presents as erythematous, edematous, hot, and tender plaques along with fever and leukocytosis. It is associated with myeloproliferative disorders. Biopsy demonstrates papillary dermal edema along with diffuse neutrophilic infiltrate.7
Numerous medications have been recommended for the treatment of EBA, ranging from steroids to steroid-sparing drugs such as colchicine and dapsone.8,9 Our patient was educated on physical precautions and was started on dapsone alone due to comorbid diabetes mellitus and renal disease. Within a few weeks of initiating dapsone, he observed a reduction in erythema, and within months he experienced a decrease in blister eruption frequency.
The Diagnosis: Epidermolysis Bullosa Acquisita
The diagnosis of epidermolysis bullosa acquisita (EBA) was made based on the clinical and pathologic findings. A blistering disorder that resolves with milia is characteristic of EBA. Hematoxylin and eosin staining demonstrated a pauci-inflammatory separation between the epidermis and dermis (Figure 1). Direct immunofluorescence studies showed linear IgG deposition along the basement membrane zone while C3 was negative (Figure 2). Salt-split skin was essential, as it revealed IgG deposition to the floor of the split (Figure 3), a pattern seen in EBA and not bullous pemphigoid (BP).1
Epidermolysis bullosa acquisita is an acquired autoimmune bullous disorder that results from antibodies to type VII collagen, an anchoring fibril that attaches the lamina densa to the dermis. The epidemiology and etiology of the trigger that leads to antibody production are not well known, but an association between EBA and inflammatory bowel disease has been described.2 Although this disease may present in childhood, EBA most commonly is a disorder seen in adults and the elderly. A classic noninflammatory mechanobullous form as well as an inflammatory BP-like form are the most commonly encountered presentations. Light microscopy demonstrates subepidermal cleavage without acantholysis. In the inflammatory BP-like subtype, an inflammatory infiltrate may be present. Direct immunofluorescence is remarkable for a linear band of IgG deposits along the basement membrane zone, with or without C3 deposition in a similar pattern.1
Bullous pemphigoid is within the differential of EBA. It can be difficult to differentiate clinically, especially when a patient has the BP-like variant of EBA because, as the name implies, it mimics BP. Patients with BP often will report a pruritic patch that will then develop into an urticarial plaque. Scarring and milia rarely are seen in BP but can be observed in the multiple presentations of EBA. Hematoxylin and eosin staining and direct immunofluorescence may be almost identical, and differentiating between the 2 disorders can be a challenge. Immunodeposition in EBA occurs in a U-shaped, serrated pattern, while the pattern in BP is N-shaped and serrated.3 Although the U-shaped, serrated pattern is relatively specific, it is not always easy to interpret and requires a high-quality biopsy specimen, which can be difficult to discern with certainty in suboptimal preparations. Another way to differentiate between the 2 entities is to utilize the salt-split skin technique, as performed in our patient. With salt-split skin, the biopsy is placed into a solution of 1 mol/L sodium chloride and incubated at 4 °C (39 °F) for 18 to 24 hours. A blister is then produced at the level of the lamina lucida, which allows for the staining of immunoreactants to occur either above or below that split (commonly referred to as staining on the roof or floor of the blister cavity). With EBA, there is immunoreactant deposition on the floor of the blister, while the opposite occurs in BP.4
Epidermolysis bullosa simplex is the most common type of epidermolysis bullosa, with keratin genes KRT5 and KRT14 as frequent mutations. Patients develop blisters, vesicles, bullae, and milia on traumatized areas of the body such as the hands, elbows, knees, and feet. This disease presents early in childhood. Histology exhibits a cell-poor subepidermal blister.5 With porphyria cutanea tarda, reduced activity of uroporphyrinogen decarboxylase, a major enzyme in the heme synthesis pathway, leads to blisters with erosions and milia on sun-exposed areas of the body. Histologic evaluation reveals a subepidermal pauci-inflammatory vesicle with festooning of the dermal papillae and amphophilic basement membrane within the epidermis. Direct immunofluorescence of porphyria cutanea tarda demonstrates IgM and C3 in the vessels.6 Sweet syndrome is a neutrophilic dermatosis that presents as erythematous, edematous, hot, and tender plaques along with fever and leukocytosis. It is associated with myeloproliferative disorders. Biopsy demonstrates papillary dermal edema along with diffuse neutrophilic infiltrate.7
Numerous medications have been recommended for the treatment of EBA, ranging from steroids to steroid-sparing drugs such as colchicine and dapsone.8,9 Our patient was educated on physical precautions and was started on dapsone alone due to comorbid diabetes mellitus and renal disease. Within a few weeks of initiating dapsone, he observed a reduction in erythema, and within months he experienced a decrease in blister eruption frequency.
- Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. 2017;13:157-169.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-230.
- Vodegel RM, Jonkman MF, Pas HH, et al. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol. 2004;151:112-118.
- Gardner KM, Crawford RI. Distinguishing epidermolysis bullosa acquisita from bullous pemphigoid without direct immunofluorescence. J Cutan Med Surg. 2018;22:22-24.
- Sprecher E. Epidermolysis bullosa simplex. Dermatol Clin. 2010;28:23-32.
- Maynard B, Peters MS. Histologic and immunofluorescence study of cutaneous porphyrias. J Cutan Pathol. 1992;19:40-47.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018:79:987-1006.
- Kirtschig G, Murrell D, Wojnarowska F, et al. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database Syst Rev. 2003;1:CD004056
- Gürcan HM, Ahmed AR. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin Pharmacother. 2011;12:1259-1268.
- Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. 2017;13:157-169.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-230.
- Vodegel RM, Jonkman MF, Pas HH, et al. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol. 2004;151:112-118.
- Gardner KM, Crawford RI. Distinguishing epidermolysis bullosa acquisita from bullous pemphigoid without direct immunofluorescence. J Cutan Med Surg. 2018;22:22-24.
- Sprecher E. Epidermolysis bullosa simplex. Dermatol Clin. 2010;28:23-32.
- Maynard B, Peters MS. Histologic and immunofluorescence study of cutaneous porphyrias. J Cutan Pathol. 1992;19:40-47.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018:79:987-1006.
- Kirtschig G, Murrell D, Wojnarowska F, et al. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database Syst Rev. 2003;1:CD004056
- Gürcan HM, Ahmed AR. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin Pharmacother. 2011;12:1259-1268.
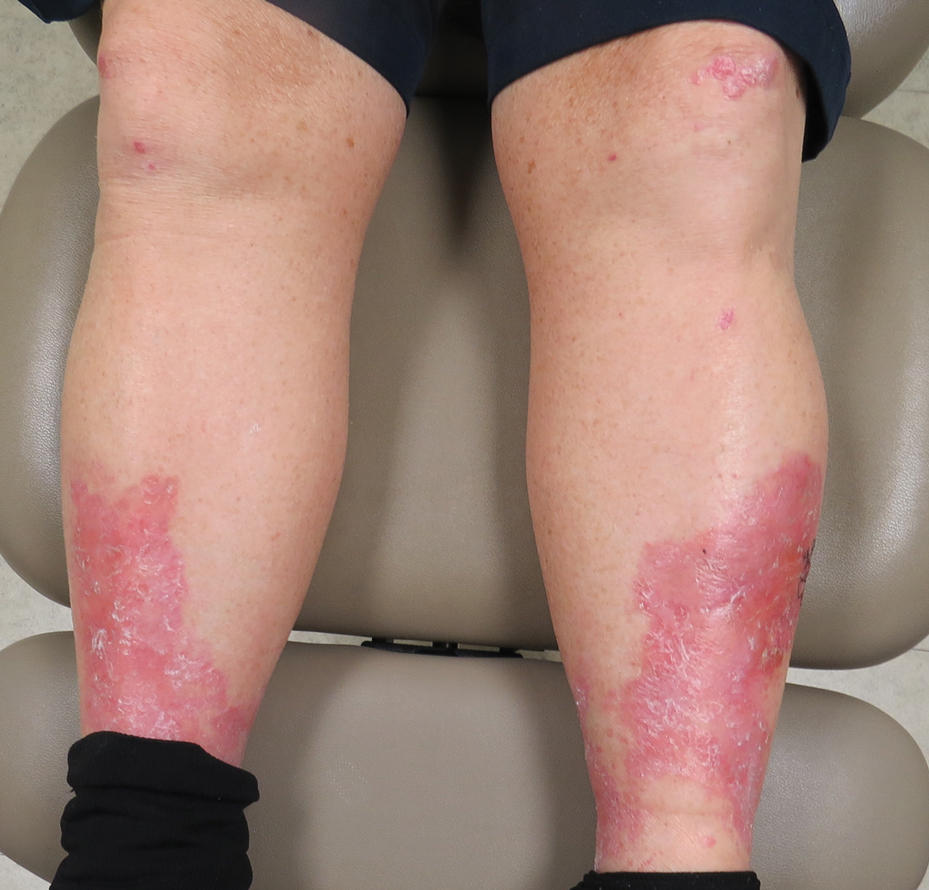
A 69-year-old man presented with an asymptomatic rash on the extensor surfaces of 2 years' duration. He reported recurrent blisters that would then scar over. The lesions did not occur in relation to any known trauma. The patient's medical history revealed dialysis-dependent end-stage renal disease secondary to type 2 diabetes mellitus. His medications were noncontributory, and there was no family history of blistering disorders. He had tried triamcinolone cream without any improvement. Physical examination was remarkable for erythematous blisters and bullae with scales and milia on the elbows, knees, and lower legs. The oral mucosa was unremarkable. Shave biopsies of the skin for direct immunofluorescence and salt-split skin studies were obtained.
Cutaneous Insulin-Derived Amyloidosis Presenting as Hyperkeratotic Nodules
Amyloidosis consists of approximately 30 protein-folding disorders sharing the common feature of abnormal extracellular amyloid deposition. In each condition, a specific soluble precursor protein aggregates to form the insoluble fibrils of amyloid, characterized by the beta-pleated sheet structure.1 Amyloidosis occurs as either a systemic or localized process. Insulin-derived (AIns) amyloidosis, a localized process occurring at insulin injection sites, was first reported in 1983.2 There were fewer than 20 reported cases until 2014, when 57 additional cases were reported by just 2 institutions,3,4 indicating that AIns amyloidosis may be more common than previously thought.3,5
Despite the increasing prevalence of diabetes mellitus and insulin use, there is a paucity of published cases of AIns amyloidosis. The lack of awareness of this condition among both dermatologists and general practitioners may be in part due to its variable clinical manifestations. We describe 2 patients with unique presentations of localized amyloidosis at repeated insulin injection sites.
Case Reports
Patient 1
A 39-year-old man with a history of type 1 diabetes mellitus presented with 4 asymptomatic nodules on the lateral thighs in areas of previous insulin injection. He first noticed the lesions 9 months prior to presentation and subsequently switched the injection site to the abdomen without development of new nodules. Despite being compliant with his insulin regimen, he had a long history of irregular glucose control, including frequent hypoglycemic episodes. The patient was using regular and neutral protamine hagedorn insulin.
On physical examination, 2 soft, nontender, exophytic nodules were noted on each upper thigh with surrounding hyperpigmented and hyperkeratotic collarettes (Figure 1). The nodules ranged in size from 2 to 3.5 cm in diameter.
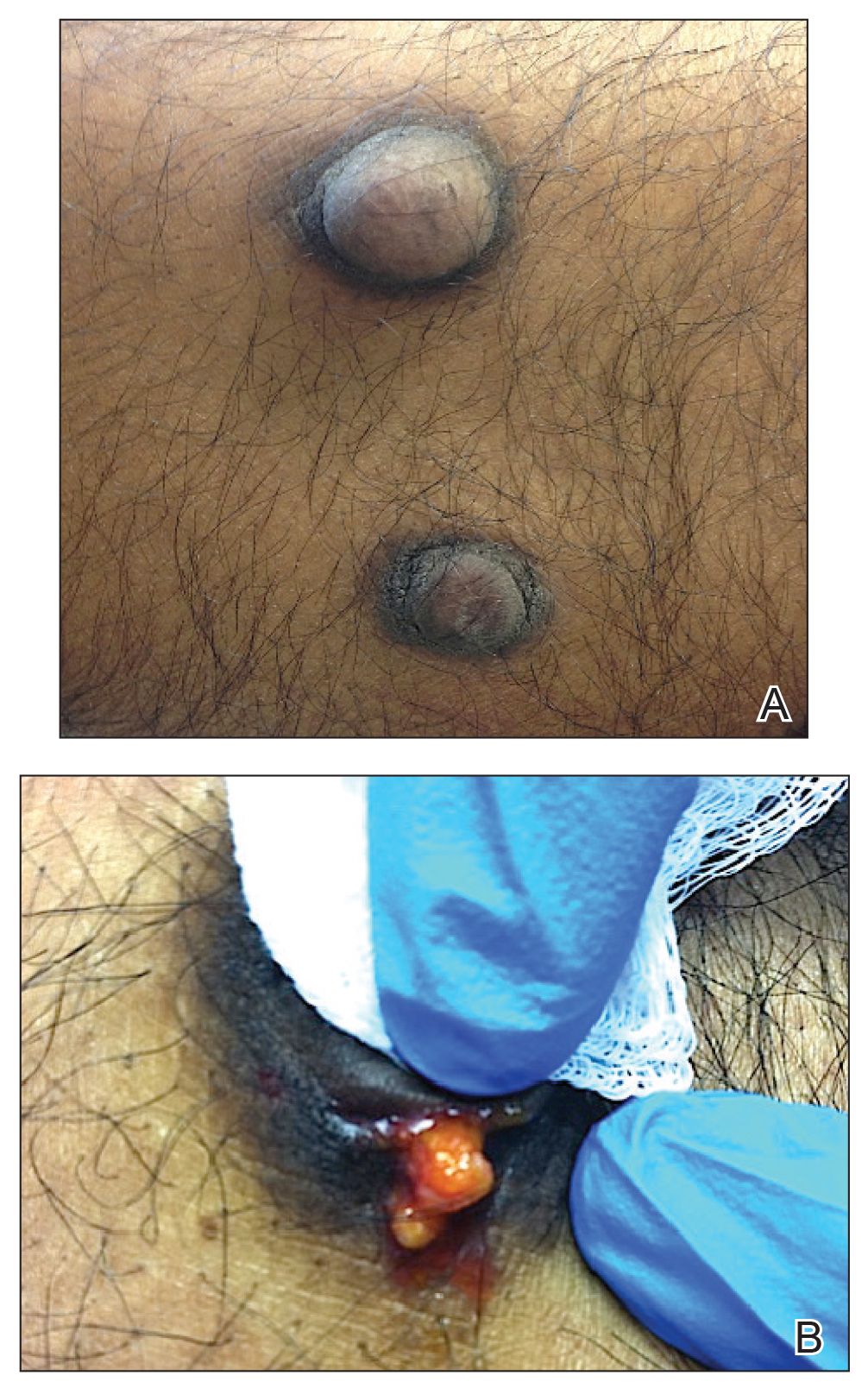
Remarkable laboratory data included a fasting glucose level of 207 mg/dL (reference range, 70–110 mg/dL) and a glycohemoglobin of 8.8% (reference range, <5.7%). Serum protein electrophoresis and immunofixation were normal. Histopathology of the lesions demonstrated diffuse deposition of pink amorphous material associated with prominent papillomatosis, hyperkeratosis, and acanthosis (Figure 2). Congo red staining was positive with green birefringence under polarized light, indicative of amyloid deposits (Figure 3). Liquid chromatography–tandem mass spectrometry of the specimens was consistent with deposition of AIns amyloidosis.
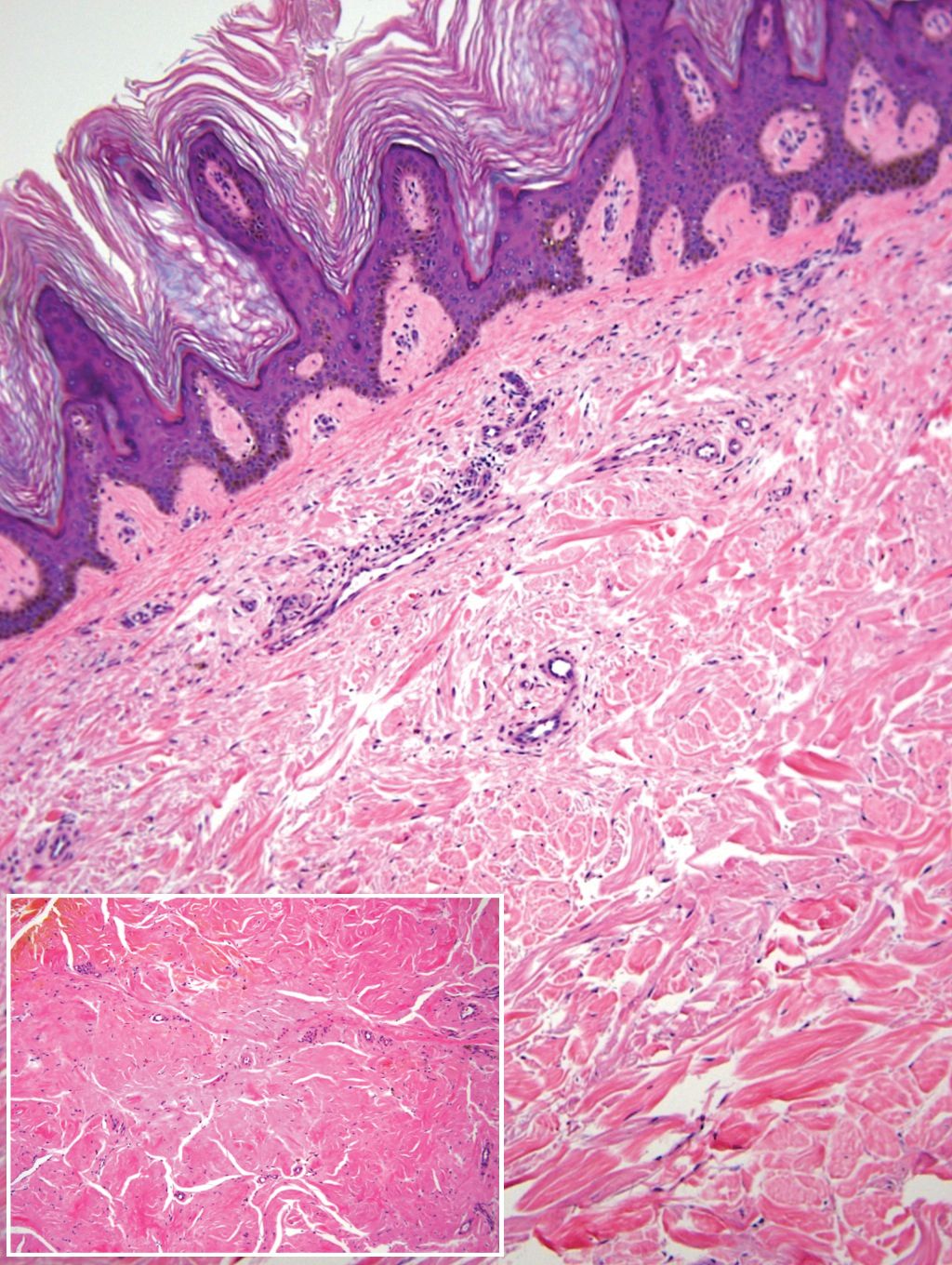
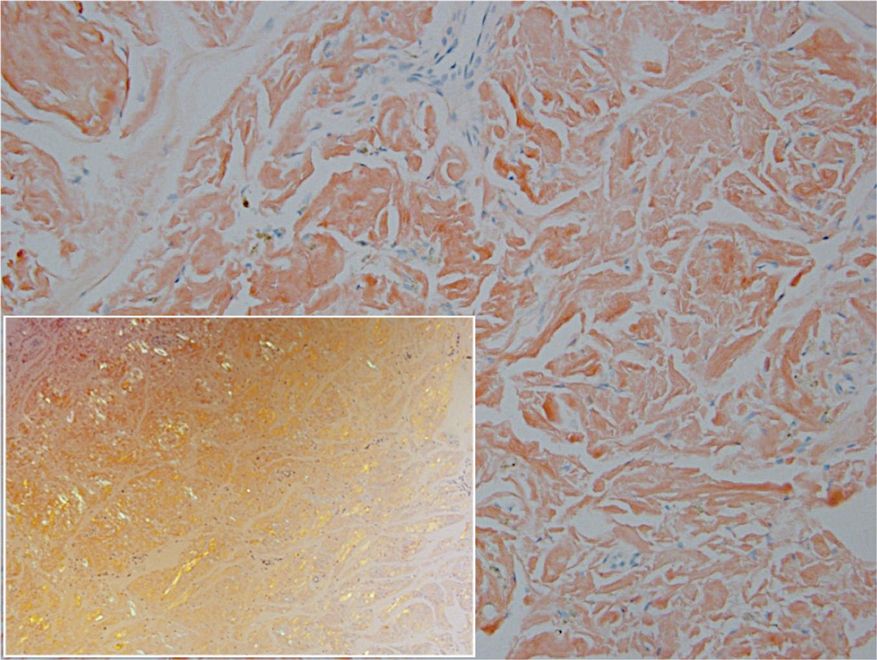
Due to the size and persistent nature of the lesions, the nodules were removed by tangential excision. In addition, the patient was advised to continue rotating injection sites frequently. His blood glucose levels are now well controlled, and he has not developed any new nodules.
Patient 2
A 53-year-old woman with a history of type 2 diabetes mellitus presented with painful subcutaneous nodules on the lower abdomen at sites of previous insulin injections. The nodules developed approximately 1 month after she started treatment with neutral protamine hagedorn insulin and had been slowly enlarging over the past year. She tried switching injection sites after noticing the lesions, but the nodules persisted. The patient had a long history of poor glucose control with chronically elevated glycohemoglobin and blood glucose levels.
On physical examination, 2 hyperpigmented, exophytic, smooth nodules were noted on the right and left lower abdomen, ranging in size from 2.5 to 5.5 cm in diameter (Figure 4).
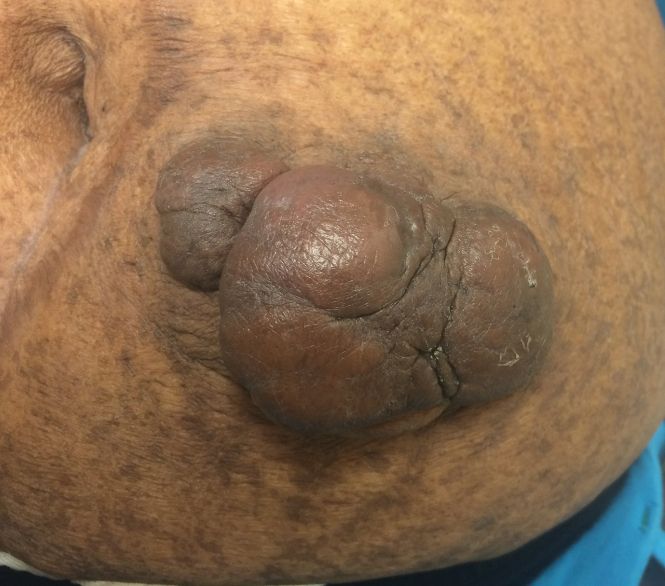
Relevant laboratory data included a fasting glucose level of 197 mg/dL and a glycohemoglobin of 9.3%. A biopsy of the lesion on the left lower abdomen revealed eosinophilic amorphous deposits with fissuring in the dermis (Figure 5). Congo red stain was positive with green birefringence under polarized light. Liquid chromatography–tandem mass spectrometry of the specimen showed deposition of AIns amyloid. The patient began injecting away from the amyloid nodules without development of any new lesions. The original nodules have persisted, and surgical excision is planned.
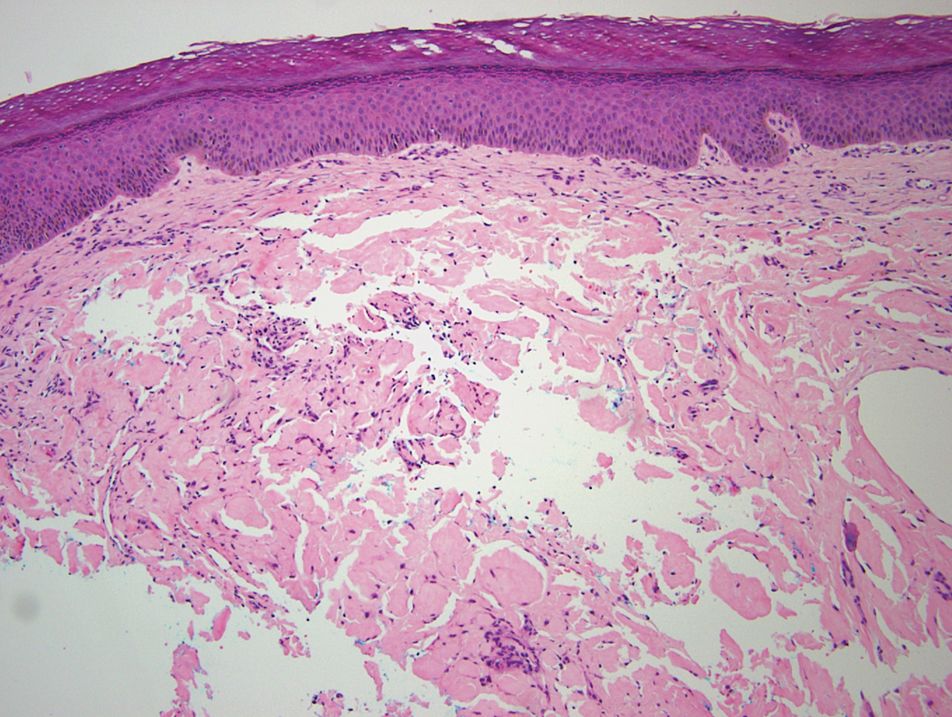
Comment
Insulin is the suspected precursor protein in AIns amyloidosis, but the exact pathogenesis is unknown. The protein that is derived from insulin in these tumors is now identified as AIns amyloidosis.5,6 It is hypothesized that insulin accumulates locally and is converted to amyloid by an unknown mechanism.7 Other potential contributory factors include chronic inflammation and foreign body reactions developing around amyloid deposits, as well as repeated trauma from injections into a single site.4,5 It appears that lesions may derive from a wide range of insulin types and occur after variable time periods.
A majority of cases of iatrogenic amyloid have been described as single, firm, subcutaneous masses at an injection site that commonly are misdiagnosed as lipomas or lipohypertrophy.7-11 To our knowledge, none of the reported cases resembled the multiple, discrete, exophytic nodules seen in our patients.3,4 The surrounding hyperkeratosis noted in patient 1 is another uncommon feature of AIns amyloidosis (Figures 1 and 2). Only 3 AIns amyloidosis cases described lesions with acanthosis nigricans–like changes, only 1 of which provided a clinical image.6,7,12The mechanism for the acanthosis nigricans–like changes may have been due to the high levels of insulin at the injection site. It has been suggested that the activation of insulinlike growth factor receptor by insulin leads to the proliferation of keratinocytes and fibroblasts.6 Histologic examination of AIns amyloidosis lesions generally demonstrates deposition of homogenous eosinophilic material consistent with amyloid, as well as positive Congo red staining with green birefringence by polarization. Immunohistologic staining with insulin antibody with or without proteomic analysis of the amyloid deposits can confirm the diagnosis. In both of our patients’ specimens, liquid chromatography–tandem mass spectrometry was performed for proteomic analysis, and results were consistent with AIns amyloidosis.
Reports in the literature have suggested that the deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.4,11,13 Hence, injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis. In their study of 4 patients, Nagase et al4 compared serum insulin levels after insulin injection into amyloid nodules vs insulin levels after injection into normal skin. Insulin absorption at the amyloid sites was 34% of that at normal sites. Given these results, patients should be instructed to inject away from the amyloid deposit once it is identified.6 Glucose levels should be monitored closely when patients first inject away from the amyloid mass, as injection of the same dosage to an area of normal skin can lead to increased insulin absorption and hypoglycemia.4,6 It is possible that the frequent hypoglycemic episodes noted in patient 1 were due to increased insulin sensitivity after switching to injection sites away from amyloid lesions.
Conclusion
Our patients demonstrate unique presentations of localized cutaneous amyloidosis at repeated insulin injection sites. We report these cases to complement the current data of iatrogenic amyloidosis and provide insight into this likely underreported phenomenon.
- Hazenberg BPC. Amyloidosis: a clinical overview. Rheum Dis Clin North Am. 2013;39:323-345.
- Storkel S, Schneider HM, Muntefering H, et al. Iatrogenic, insulin-dependent, local amyloidosis. Lab Invest. 1983;48:108-111.
- D’souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Nagase T, Iwaya K, Iwaki Y, et al. Insulin-derived amyloidosis and poor glycemic control: a case series. Am J Med. 2014;127:450-454.
- Gupta Y, Singla G, Singla R. Insulin-derived amyloidosis. Indian J Endocrinol Metab. 2015;19:174-177.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Yumlu S, Barany R, Eriksson M, et al. Localized insulin-derived amyloidosis in patients with diabetes mellitus: a case report. Hum Pathol. 2009;40:1655-1660.
- Okamura S, Hayashino Y, Kore-Eda S, et al. Localized amyloidosis at the site of repeated insulin injection in a patient with type 2 diabetes. Diabetes Care. 2013;36:E200.
- Dische FE, Wernstedt C, Westermark GT, et al. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia. 1988;31:158-161.
- Swift B, Hawkins PN, Richards C, et al. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabetic Med. 2002;19:881-882.
- Albert SG, Obadiah J, Parseghian SA, et al. Severe insulin resistance associated with subcutaneous amyloid deposition. Diabetes Res Clin Pract. 2007;75:374-376.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Endo JO, Rocken C, Lamb S, et al. Nodular amyloidosis in a diabetic patient with frequent hypoglycemia: sequelae of repeatedly injecting insulin without site rotation. J Am Acad Dermatol. 2010;63:E113-E114.
Amyloidosis consists of approximately 30 protein-folding disorders sharing the common feature of abnormal extracellular amyloid deposition. In each condition, a specific soluble precursor protein aggregates to form the insoluble fibrils of amyloid, characterized by the beta-pleated sheet structure.1 Amyloidosis occurs as either a systemic or localized process. Insulin-derived (AIns) amyloidosis, a localized process occurring at insulin injection sites, was first reported in 1983.2 There were fewer than 20 reported cases until 2014, when 57 additional cases were reported by just 2 institutions,3,4 indicating that AIns amyloidosis may be more common than previously thought.3,5
Despite the increasing prevalence of diabetes mellitus and insulin use, there is a paucity of published cases of AIns amyloidosis. The lack of awareness of this condition among both dermatologists and general practitioners may be in part due to its variable clinical manifestations. We describe 2 patients with unique presentations of localized amyloidosis at repeated insulin injection sites.
Case Reports
Patient 1
A 39-year-old man with a history of type 1 diabetes mellitus presented with 4 asymptomatic nodules on the lateral thighs in areas of previous insulin injection. He first noticed the lesions 9 months prior to presentation and subsequently switched the injection site to the abdomen without development of new nodules. Despite being compliant with his insulin regimen, he had a long history of irregular glucose control, including frequent hypoglycemic episodes. The patient was using regular and neutral protamine hagedorn insulin.
On physical examination, 2 soft, nontender, exophytic nodules were noted on each upper thigh with surrounding hyperpigmented and hyperkeratotic collarettes (Figure 1). The nodules ranged in size from 2 to 3.5 cm in diameter.
Remarkable laboratory data included a fasting glucose level of 207 mg/dL (reference range, 70–110 mg/dL) and a glycohemoglobin of 8.8% (reference range, <5.7%). Serum protein electrophoresis and immunofixation were normal. Histopathology of the lesions demonstrated diffuse deposition of pink amorphous material associated with prominent papillomatosis, hyperkeratosis, and acanthosis (Figure 2). Congo red staining was positive with green birefringence under polarized light, indicative of amyloid deposits (Figure 3). Liquid chromatography–tandem mass spectrometry of the specimens was consistent with deposition of AIns amyloidosis.
Due to the size and persistent nature of the lesions, the nodules were removed by tangential excision. In addition, the patient was advised to continue rotating injection sites frequently. His blood glucose levels are now well controlled, and he has not developed any new nodules.
Patient 2
A 53-year-old woman with a history of type 2 diabetes mellitus presented with painful subcutaneous nodules on the lower abdomen at sites of previous insulin injections. The nodules developed approximately 1 month after she started treatment with neutral protamine hagedorn insulin and had been slowly enlarging over the past year. She tried switching injection sites after noticing the lesions, but the nodules persisted. The patient had a long history of poor glucose control with chronically elevated glycohemoglobin and blood glucose levels.
On physical examination, 2 hyperpigmented, exophytic, smooth nodules were noted on the right and left lower abdomen, ranging in size from 2.5 to 5.5 cm in diameter (Figure 4).
Relevant laboratory data included a fasting glucose level of 197 mg/dL and a glycohemoglobin of 9.3%. A biopsy of the lesion on the left lower abdomen revealed eosinophilic amorphous deposits with fissuring in the dermis (Figure 5). Congo red stain was positive with green birefringence under polarized light. Liquid chromatography–tandem mass spectrometry of the specimen showed deposition of AIns amyloid. The patient began injecting away from the amyloid nodules without development of any new lesions. The original nodules have persisted, and surgical excision is planned.
Comment
Insulin is the suspected precursor protein in AIns amyloidosis, but the exact pathogenesis is unknown. The protein that is derived from insulin in these tumors is now identified as AIns amyloidosis.5,6 It is hypothesized that insulin accumulates locally and is converted to amyloid by an unknown mechanism.7 Other potential contributory factors include chronic inflammation and foreign body reactions developing around amyloid deposits, as well as repeated trauma from injections into a single site.4,5 It appears that lesions may derive from a wide range of insulin types and occur after variable time periods.
A majority of cases of iatrogenic amyloid have been described as single, firm, subcutaneous masses at an injection site that commonly are misdiagnosed as lipomas or lipohypertrophy.7-11 To our knowledge, none of the reported cases resembled the multiple, discrete, exophytic nodules seen in our patients.3,4 The surrounding hyperkeratosis noted in patient 1 is another uncommon feature of AIns amyloidosis (Figures 1 and 2). Only 3 AIns amyloidosis cases described lesions with acanthosis nigricans–like changes, only 1 of which provided a clinical image.6,7,12The mechanism for the acanthosis nigricans–like changes may have been due to the high levels of insulin at the injection site. It has been suggested that the activation of insulinlike growth factor receptor by insulin leads to the proliferation of keratinocytes and fibroblasts.6 Histologic examination of AIns amyloidosis lesions generally demonstrates deposition of homogenous eosinophilic material consistent with amyloid, as well as positive Congo red staining with green birefringence by polarization. Immunohistologic staining with insulin antibody with or without proteomic analysis of the amyloid deposits can confirm the diagnosis. In both of our patients’ specimens, liquid chromatography–tandem mass spectrometry was performed for proteomic analysis, and results were consistent with AIns amyloidosis.
Reports in the literature have suggested that the deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.4,11,13 Hence, injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis. In their study of 4 patients, Nagase et al4 compared serum insulin levels after insulin injection into amyloid nodules vs insulin levels after injection into normal skin. Insulin absorption at the amyloid sites was 34% of that at normal sites. Given these results, patients should be instructed to inject away from the amyloid deposit once it is identified.6 Glucose levels should be monitored closely when patients first inject away from the amyloid mass, as injection of the same dosage to an area of normal skin can lead to increased insulin absorption and hypoglycemia.4,6 It is possible that the frequent hypoglycemic episodes noted in patient 1 were due to increased insulin sensitivity after switching to injection sites away from amyloid lesions.
Conclusion
Our patients demonstrate unique presentations of localized cutaneous amyloidosis at repeated insulin injection sites. We report these cases to complement the current data of iatrogenic amyloidosis and provide insight into this likely underreported phenomenon.
Amyloidosis consists of approximately 30 protein-folding disorders sharing the common feature of abnormal extracellular amyloid deposition. In each condition, a specific soluble precursor protein aggregates to form the insoluble fibrils of amyloid, characterized by the beta-pleated sheet structure.1 Amyloidosis occurs as either a systemic or localized process. Insulin-derived (AIns) amyloidosis, a localized process occurring at insulin injection sites, was first reported in 1983.2 There were fewer than 20 reported cases until 2014, when 57 additional cases were reported by just 2 institutions,3,4 indicating that AIns amyloidosis may be more common than previously thought.3,5
Despite the increasing prevalence of diabetes mellitus and insulin use, there is a paucity of published cases of AIns amyloidosis. The lack of awareness of this condition among both dermatologists and general practitioners may be in part due to its variable clinical manifestations. We describe 2 patients with unique presentations of localized amyloidosis at repeated insulin injection sites.
Case Reports
Patient 1
A 39-year-old man with a history of type 1 diabetes mellitus presented with 4 asymptomatic nodules on the lateral thighs in areas of previous insulin injection. He first noticed the lesions 9 months prior to presentation and subsequently switched the injection site to the abdomen without development of new nodules. Despite being compliant with his insulin regimen, he had a long history of irregular glucose control, including frequent hypoglycemic episodes. The patient was using regular and neutral protamine hagedorn insulin.
On physical examination, 2 soft, nontender, exophytic nodules were noted on each upper thigh with surrounding hyperpigmented and hyperkeratotic collarettes (Figure 1). The nodules ranged in size from 2 to 3.5 cm in diameter.
Remarkable laboratory data included a fasting glucose level of 207 mg/dL (reference range, 70–110 mg/dL) and a glycohemoglobin of 8.8% (reference range, <5.7%). Serum protein electrophoresis and immunofixation were normal. Histopathology of the lesions demonstrated diffuse deposition of pink amorphous material associated with prominent papillomatosis, hyperkeratosis, and acanthosis (Figure 2). Congo red staining was positive with green birefringence under polarized light, indicative of amyloid deposits (Figure 3). Liquid chromatography–tandem mass spectrometry of the specimens was consistent with deposition of AIns amyloidosis.
Due to the size and persistent nature of the lesions, the nodules were removed by tangential excision. In addition, the patient was advised to continue rotating injection sites frequently. His blood glucose levels are now well controlled, and he has not developed any new nodules.
Patient 2
A 53-year-old woman with a history of type 2 diabetes mellitus presented with painful subcutaneous nodules on the lower abdomen at sites of previous insulin injections. The nodules developed approximately 1 month after she started treatment with neutral protamine hagedorn insulin and had been slowly enlarging over the past year. She tried switching injection sites after noticing the lesions, but the nodules persisted. The patient had a long history of poor glucose control with chronically elevated glycohemoglobin and blood glucose levels.
On physical examination, 2 hyperpigmented, exophytic, smooth nodules were noted on the right and left lower abdomen, ranging in size from 2.5 to 5.5 cm in diameter (Figure 4).
Relevant laboratory data included a fasting glucose level of 197 mg/dL and a glycohemoglobin of 9.3%. A biopsy of the lesion on the left lower abdomen revealed eosinophilic amorphous deposits with fissuring in the dermis (Figure 5). Congo red stain was positive with green birefringence under polarized light. Liquid chromatography–tandem mass spectrometry of the specimen showed deposition of AIns amyloid. The patient began injecting away from the amyloid nodules without development of any new lesions. The original nodules have persisted, and surgical excision is planned.
Comment
Insulin is the suspected precursor protein in AIns amyloidosis, but the exact pathogenesis is unknown. The protein that is derived from insulin in these tumors is now identified as AIns amyloidosis.5,6 It is hypothesized that insulin accumulates locally and is converted to amyloid by an unknown mechanism.7 Other potential contributory factors include chronic inflammation and foreign body reactions developing around amyloid deposits, as well as repeated trauma from injections into a single site.4,5 It appears that lesions may derive from a wide range of insulin types and occur after variable time periods.
A majority of cases of iatrogenic amyloid have been described as single, firm, subcutaneous masses at an injection site that commonly are misdiagnosed as lipomas or lipohypertrophy.7-11 To our knowledge, none of the reported cases resembled the multiple, discrete, exophytic nodules seen in our patients.3,4 The surrounding hyperkeratosis noted in patient 1 is another uncommon feature of AIns amyloidosis (Figures 1 and 2). Only 3 AIns amyloidosis cases described lesions with acanthosis nigricans–like changes, only 1 of which provided a clinical image.6,7,12The mechanism for the acanthosis nigricans–like changes may have been due to the high levels of insulin at the injection site. It has been suggested that the activation of insulinlike growth factor receptor by insulin leads to the proliferation of keratinocytes and fibroblasts.6 Histologic examination of AIns amyloidosis lesions generally demonstrates deposition of homogenous eosinophilic material consistent with amyloid, as well as positive Congo red staining with green birefringence by polarization. Immunohistologic staining with insulin antibody with or without proteomic analysis of the amyloid deposits can confirm the diagnosis. In both of our patients’ specimens, liquid chromatography–tandem mass spectrometry was performed for proteomic analysis, and results were consistent with AIns amyloidosis.
Reports in the literature have suggested that the deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.4,11,13 Hence, injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis. In their study of 4 patients, Nagase et al4 compared serum insulin levels after insulin injection into amyloid nodules vs insulin levels after injection into normal skin. Insulin absorption at the amyloid sites was 34% of that at normal sites. Given these results, patients should be instructed to inject away from the amyloid deposit once it is identified.6 Glucose levels should be monitored closely when patients first inject away from the amyloid mass, as injection of the same dosage to an area of normal skin can lead to increased insulin absorption and hypoglycemia.4,6 It is possible that the frequent hypoglycemic episodes noted in patient 1 were due to increased insulin sensitivity after switching to injection sites away from amyloid lesions.
Conclusion
Our patients demonstrate unique presentations of localized cutaneous amyloidosis at repeated insulin injection sites. We report these cases to complement the current data of iatrogenic amyloidosis and provide insight into this likely underreported phenomenon.
- Hazenberg BPC. Amyloidosis: a clinical overview. Rheum Dis Clin North Am. 2013;39:323-345.
- Storkel S, Schneider HM, Muntefering H, et al. Iatrogenic, insulin-dependent, local amyloidosis. Lab Invest. 1983;48:108-111.
- D’souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Nagase T, Iwaya K, Iwaki Y, et al. Insulin-derived amyloidosis and poor glycemic control: a case series. Am J Med. 2014;127:450-454.
- Gupta Y, Singla G, Singla R. Insulin-derived amyloidosis. Indian J Endocrinol Metab. 2015;19:174-177.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Yumlu S, Barany R, Eriksson M, et al. Localized insulin-derived amyloidosis in patients with diabetes mellitus: a case report. Hum Pathol. 2009;40:1655-1660.
- Okamura S, Hayashino Y, Kore-Eda S, et al. Localized amyloidosis at the site of repeated insulin injection in a patient with type 2 diabetes. Diabetes Care. 2013;36:E200.
- Dische FE, Wernstedt C, Westermark GT, et al. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia. 1988;31:158-161.
- Swift B, Hawkins PN, Richards C, et al. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabetic Med. 2002;19:881-882.
- Albert SG, Obadiah J, Parseghian SA, et al. Severe insulin resistance associated with subcutaneous amyloid deposition. Diabetes Res Clin Pract. 2007;75:374-376.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Endo JO, Rocken C, Lamb S, et al. Nodular amyloidosis in a diabetic patient with frequent hypoglycemia: sequelae of repeatedly injecting insulin without site rotation. J Am Acad Dermatol. 2010;63:E113-E114.
- Hazenberg BPC. Amyloidosis: a clinical overview. Rheum Dis Clin North Am. 2013;39:323-345.
- Storkel S, Schneider HM, Muntefering H, et al. Iatrogenic, insulin-dependent, local amyloidosis. Lab Invest. 1983;48:108-111.
- D’souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Nagase T, Iwaya K, Iwaki Y, et al. Insulin-derived amyloidosis and poor glycemic control: a case series. Am J Med. 2014;127:450-454.
- Gupta Y, Singla G, Singla R. Insulin-derived amyloidosis. Indian J Endocrinol Metab. 2015;19:174-177.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Yumlu S, Barany R, Eriksson M, et al. Localized insulin-derived amyloidosis in patients with diabetes mellitus: a case report. Hum Pathol. 2009;40:1655-1660.
- Okamura S, Hayashino Y, Kore-Eda S, et al. Localized amyloidosis at the site of repeated insulin injection in a patient with type 2 diabetes. Diabetes Care. 2013;36:E200.
- Dische FE, Wernstedt C, Westermark GT, et al. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia. 1988;31:158-161.
- Swift B, Hawkins PN, Richards C, et al. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabetic Med. 2002;19:881-882.
- Albert SG, Obadiah J, Parseghian SA, et al. Severe insulin resistance associated with subcutaneous amyloid deposition. Diabetes Res Clin Pract. 2007;75:374-376.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Endo JO, Rocken C, Lamb S, et al. Nodular amyloidosis in a diabetic patient with frequent hypoglycemia: sequelae of repeatedly injecting insulin without site rotation. J Am Acad Dermatol. 2010;63:E113-E114.
Practice Points
- Deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.
- Patients with insulin-derived (AIns) amyloidosis may initially present after noticing nodular deposits.
- Insulin injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis.
Tender Soft Tissue Mass on the Base of the Neck
The Diagnosis: Subcutaneous Panniculitislike T-cell Lymphoma
Subcutaneous panniculitislike T-cell lymphoma (SPTCL) is a rare form of cutaneous lymphoma of mature cytotoxic T cells simulating panniculitis and preferentially infiltrating the subcutaneous tissue.1 Subcutaneous panniculitislike T-cell lymphoma can affect all ages but predominantly affects younger individuals, with 20% being younger than 20 years.2 It is a rare lymphoma that accounts for less than 1% of all non-Hodgkin lymphomas.3 It presents clinically as multiple subcutaneous masses, nodules, or plaques generally on the trunk or extremities.1,2 The skin surrounding the nodules may be erythematous, and the nodules may become necrotic; however, ulceration typically is not seen. Systemic symptoms such as fever, night sweats, and chills are present in half of cases.1 According to the World Health Organization, cytopenia and elevated liver function tests are common, and a hemophagocytic syndrome may be present in 15% to 20% of cases.3 The presence of a hemophagocytic syndrome yields a poor prognosis.1,3 Current guidelines denote that SPTCL T-cell receptor (TCR) αβ; is a distinct entity from the TCRγδ; phenotype, known as cutaneous γδ-positive T-cell lymphoma.3,4 Cutaneous γδ-positive T-cell lymphoma is associated with rapid decline and a worse prognosis.4
Histology of SPTCL is characteristic for a lobular panniculitislike infiltrate.1 The heavy subcutaneous lymphoid infiltrate is composed of atypical small- to medium-sized lymphocytes with mature chromatin and inconspicuous nucleoli lining adipocytes. The dense inflammatory infiltrate composed predominantly of neoplastic T cells and macrophages may diffusely invade into the subcutaneous tissue.1 Admixed histocytes and karyorrhectic debris as well as rimming of the lymphocytes around the fat cells is typical and was seen in our patient (quiz image). The T cells of SPTCL have the following immunophenotype: TCR-beta F1+, CD3+, CD4-, CD8+, CD56-. They can express numerous cytotoxic proteins, such as T1a-1, granzyme B, and perforin.2,3 Although the CD8+ T cells may be sparse, they generally surround the adipocytes in a rimming manner and may distort the adipocyte membrane.1
Lupus erythematosus profundus (LEP) is a form of chronic cutaneous lupus that affects the deep dermis and fat.5 It also can present clinically as tender plaques or nodules. It most frequently involves the upper arms, shoulders, face, or buttocks--areas that are less commonly involved in other panniculitides.6 Histologically, LEP is similar to chronic discoid lupus with features such as epidermal atrophy, interface changes, and a thickened basement membrane (Figure 1). Lupus erythematosus profundus can present as a lobular panniculitis with mucin as well as a superficial and deep lymphocytic infiltrate that can involve the septa.5 Some cases of LEP have a predominantly lobular lymphocytic panniculitis in the absence of the typical epidermal or dermal changes of lupus erythematosus. Lymphoid follicles with germinal center formation are present in half of cases and reportedly are characteristic of LEP.6,7 The lymphoid follicles often have plasma cells, can extend into the septa as well as in between collagen bundles, and may have nuclear fragmentation.5 Another characteristic feature of LEP is hyaline sclerosis of lobules with focal extension into the interlobular septa. Immunofluorescence studies usually show linear deposition of IgM and C3 at the dermoepidermal junction. Antinuclear antibodies can be present in patients who have LEP but are not entirely specific.6
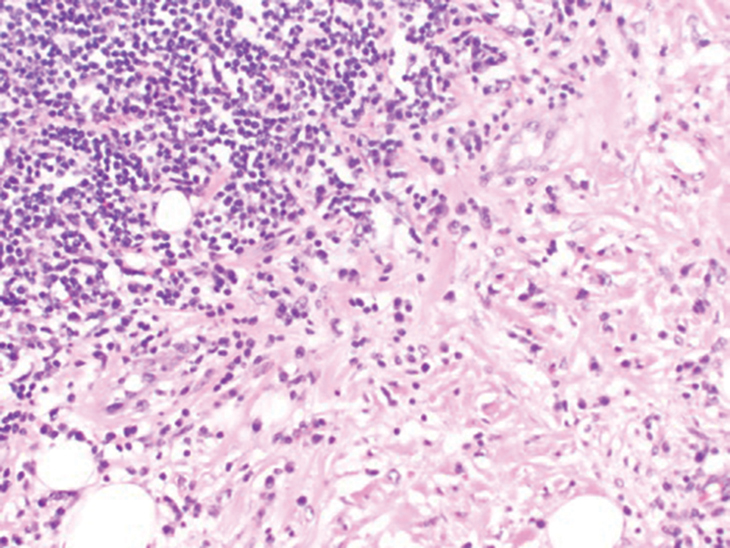
Lupus erythematosus profundus and SPTCL are part of a spectrum and may have overlapping clinical and histopathologic characteristics; therefore, distinguishing them may be difficult.6-8 It is important to monitor these patients closely, as their disease may progress to lymphoma.6 Patients with SPTCL are more likely to present with advanced symptoms such as fever and hepatosplenomegaly and to succumb to hemophagocytic syndrome than patients with LEP.9
Although SPTCL usually is clonal, several cases of LEP with clonality also have been described. Clonal LEP cases generally are identified in patients who present with fever and cytopenia.8 Lymphoid atypia and morphologic abnormalities may be seen in cases of LEP, further complicating the distinction between LEP and SPTCL. An elevated Ki67 level may be seen in cases of SPTCL with periadipocytic rimming.9 LeBlanc et al10 used Ki67 "hot spots" along with CD8 immunohistochemistry to identify atypical lymphocytes associated with SPTCL. Lymphocyte rimming was defined by the presence of CD8+ lymphocytes with an elevated Ki67 index. Clinical, histopathologic, and molecular findings all should be used when dealing with challenging cases.
Fat necrosis can occur in any part of the body where trauma has occurred and can be associated with many disease processes. Patients typically present with a palpable mass, but a clinical history of trauma is not always present. Histopathologic findings include necrotic fat alongside lipid-laden foamy macrophages and scattered inflammatory cells (Figure 2).11 Fragments of normal as well as degenerating adipose tissue and multinucleated giant cells can be present.
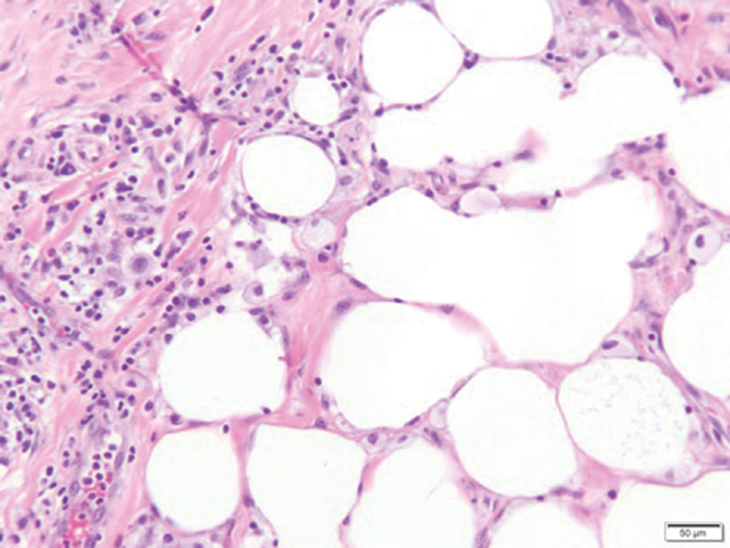
Erythema nodosum (EN) is the most frequently encountered panniculitis and usually is seen in women in early adulthood.12 Patients present with several tender subcutaneous nodules and plaques that most commonly are present on the anterior surface of the legs.12,13 Patients may have a constellation of symptoms including fever and leukocytosis, but the disorder generally is self-limited.12 Erythema nodosum may be associated with a variety of diseases or infections including sarcoidosis, inflammatory bowel disease, and malignancy.14 The etiology of EN is diverse; therefore, a proper clinical workup may be necessary. Histopathology is that of a septal panniculitis with lymphocytes, histiocytes, and occasional eosinophils (Figure 3).13
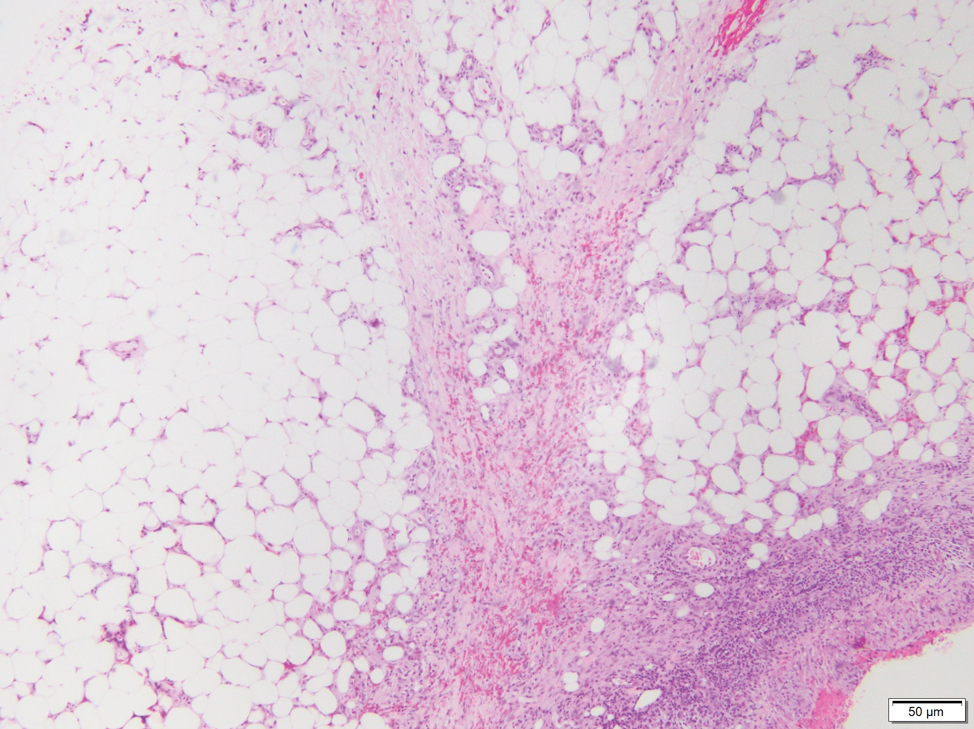
Lipodermatosclerosis also occurs on the legs, most commonly in patients with venous insufficiency.12,15 Patients present clinically with pain, induration, redness, or swelling of the legs. Histopathology predominantly is characterized by membranous fat necrosis, fibrosis, and fatty microcysts that may be lined by a thickened hyaline membrane (Figure 4). Lipodermatosclerosis lesions generally do not resolve spontaneously and may need to be treated.16
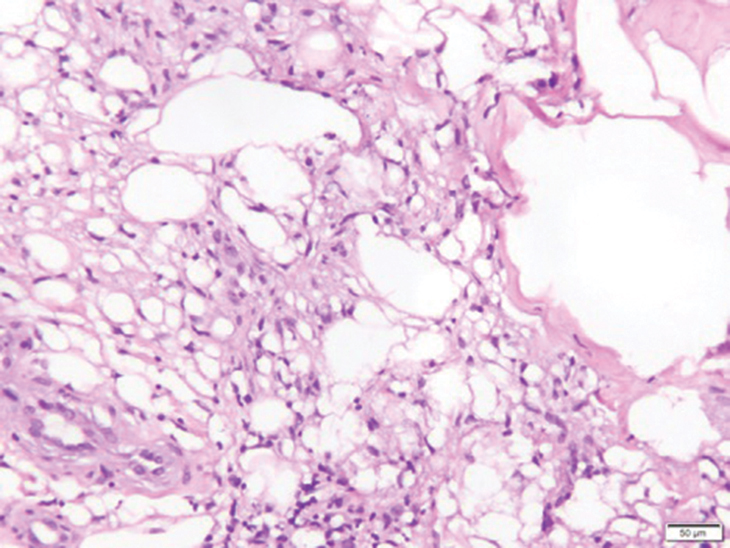
- Musick SR, Lynch DT. Subcutaneous Panniculitis Like T-cell Lymphoma. StatPearls Publishing; 2020.
- Guenova E, Schanz S, Hoetzenecker W, et al. Systemic corticosteroids for subcutaneous panniculitis-like T-cell lymphoma. Br J Dermatol. 2014;171:891-894.
- Swerdlow SH. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. International Agency for Research on Cancer; 2017.
- Bagheri F, Cervellione KL, Delgado B, et al. An illustrative case of subcutaneous panniculitis-like T-cell lymphoma [published online March 3, 2011]. J Skin Cancer. doi:10.1155/2011/824528
- Kogame T, Yamashita R, Hirata M, et al. Analysis of possible structures of inducible skin‐associated lymphoid tissue in lupus erythematosus profundus. J Dermatol. 2018;45:1117-1121.
- Arps DP, Patel RM. Lupus profundus (panniculitis): a potential mimic of subcutaneous panniculitis-like T-cell lymphoma. Arch Pathol Lab Med. 2013;137:1211-1215.
- Alberti-Violetti S, Berti E. Lymphocytic lobular panniculitis: a diagnostic challenge. Dermatopathology. 2018;5:30-33.
- Magro CM, Crowson AN, Kovatich AJ, et al. Lupus profundus, indeterminate lymphocytic lobular panniculitis and subcutaneous T-cell lymphoma: a spectrum of subcuticular T-cell lymphoid dyscrasia. J Cutan Pathol. 2001;28:235-247.
- Sitthinamsuwan P, Pattanaprichakul P, Treetipsatit J, et al. Subcutaneous panniculitis-like T-cell lymphoma versus lupus erythematosus panniculitis: distinction by means of the periadipocytic cell proliferation index. Am J Dermatopathol. 2018;40:567-574.
- LeBlanc RE, Tavallaee M, Kim YH, et al. Useful parameters for distinguishing subcutaneous panniculitis-like T-cell lymphoma from lupus erythematosus panniculitis. Am J Surg Pathol. 2016;40:745-754.
- Burkholz KJ, Roberts CC, Lidner TK. Posttraumatic pseudolipoma (fat necrosis) mimicking atypical lipoma or liposarcoma on MRI. Radiol Case Rep. 2015;2:56-60.
- Wick MR. Panniculitis: a summary. Semin Diagn Pathol. 2017;34:261-272.
- Thurber S, Kohler S. Histopathologic spectrum of erythema nodosum. J Cutan Pathol. 2006;33:18-26.
- Requena L, Requena C. Erythema nodosum. Dermatol Online J. 2002;8:4.
- Choonhakarn C, Chaowattanapanit S, Julanon N. Lipodermatosclerosis: a clinicopathologic correlation. Int J Dermatol. 2016;55:303-308.
- Huang TM, Lee JY. Lipodermatosclerosis: a clinicopathologic study of 17 cases and differential diagnosis from erythema nodosum. J Cutan Pathol. 2009;36:453-460.
The Diagnosis: Subcutaneous Panniculitislike T-cell Lymphoma
Subcutaneous panniculitislike T-cell lymphoma (SPTCL) is a rare form of cutaneous lymphoma of mature cytotoxic T cells simulating panniculitis and preferentially infiltrating the subcutaneous tissue.1 Subcutaneous panniculitislike T-cell lymphoma can affect all ages but predominantly affects younger individuals, with 20% being younger than 20 years.2 It is a rare lymphoma that accounts for less than 1% of all non-Hodgkin lymphomas.3 It presents clinically as multiple subcutaneous masses, nodules, or plaques generally on the trunk or extremities.1,2 The skin surrounding the nodules may be erythematous, and the nodules may become necrotic; however, ulceration typically is not seen. Systemic symptoms such as fever, night sweats, and chills are present in half of cases.1 According to the World Health Organization, cytopenia and elevated liver function tests are common, and a hemophagocytic syndrome may be present in 15% to 20% of cases.3 The presence of a hemophagocytic syndrome yields a poor prognosis.1,3 Current guidelines denote that SPTCL T-cell receptor (TCR) αβ; is a distinct entity from the TCRγδ; phenotype, known as cutaneous γδ-positive T-cell lymphoma.3,4 Cutaneous γδ-positive T-cell lymphoma is associated with rapid decline and a worse prognosis.4
Histology of SPTCL is characteristic for a lobular panniculitislike infiltrate.1 The heavy subcutaneous lymphoid infiltrate is composed of atypical small- to medium-sized lymphocytes with mature chromatin and inconspicuous nucleoli lining adipocytes. The dense inflammatory infiltrate composed predominantly of neoplastic T cells and macrophages may diffusely invade into the subcutaneous tissue.1 Admixed histocytes and karyorrhectic debris as well as rimming of the lymphocytes around the fat cells is typical and was seen in our patient (quiz image). The T cells of SPTCL have the following immunophenotype: TCR-beta F1+, CD3+, CD4-, CD8+, CD56-. They can express numerous cytotoxic proteins, such as T1a-1, granzyme B, and perforin.2,3 Although the CD8+ T cells may be sparse, they generally surround the adipocytes in a rimming manner and may distort the adipocyte membrane.1
Lupus erythematosus profundus (LEP) is a form of chronic cutaneous lupus that affects the deep dermis and fat.5 It also can present clinically as tender plaques or nodules. It most frequently involves the upper arms, shoulders, face, or buttocks--areas that are less commonly involved in other panniculitides.6 Histologically, LEP is similar to chronic discoid lupus with features such as epidermal atrophy, interface changes, and a thickened basement membrane (Figure 1). Lupus erythematosus profundus can present as a lobular panniculitis with mucin as well as a superficial and deep lymphocytic infiltrate that can involve the septa.5 Some cases of LEP have a predominantly lobular lymphocytic panniculitis in the absence of the typical epidermal or dermal changes of lupus erythematosus. Lymphoid follicles with germinal center formation are present in half of cases and reportedly are characteristic of LEP.6,7 The lymphoid follicles often have plasma cells, can extend into the septa as well as in between collagen bundles, and may have nuclear fragmentation.5 Another characteristic feature of LEP is hyaline sclerosis of lobules with focal extension into the interlobular septa. Immunofluorescence studies usually show linear deposition of IgM and C3 at the dermoepidermal junction. Antinuclear antibodies can be present in patients who have LEP but are not entirely specific.6
Lupus erythematosus profundus and SPTCL are part of a spectrum and may have overlapping clinical and histopathologic characteristics; therefore, distinguishing them may be difficult.6-8 It is important to monitor these patients closely, as their disease may progress to lymphoma.6 Patients with SPTCL are more likely to present with advanced symptoms such as fever and hepatosplenomegaly and to succumb to hemophagocytic syndrome than patients with LEP.9
Although SPTCL usually is clonal, several cases of LEP with clonality also have been described. Clonal LEP cases generally are identified in patients who present with fever and cytopenia.8 Lymphoid atypia and morphologic abnormalities may be seen in cases of LEP, further complicating the distinction between LEP and SPTCL. An elevated Ki67 level may be seen in cases of SPTCL with periadipocytic rimming.9 LeBlanc et al10 used Ki67 "hot spots" along with CD8 immunohistochemistry to identify atypical lymphocytes associated with SPTCL. Lymphocyte rimming was defined by the presence of CD8+ lymphocytes with an elevated Ki67 index. Clinical, histopathologic, and molecular findings all should be used when dealing with challenging cases.
Fat necrosis can occur in any part of the body where trauma has occurred and can be associated with many disease processes. Patients typically present with a palpable mass, but a clinical history of trauma is not always present. Histopathologic findings include necrotic fat alongside lipid-laden foamy macrophages and scattered inflammatory cells (Figure 2).11 Fragments of normal as well as degenerating adipose tissue and multinucleated giant cells can be present.
Erythema nodosum (EN) is the most frequently encountered panniculitis and usually is seen in women in early adulthood.12 Patients present with several tender subcutaneous nodules and plaques that most commonly are present on the anterior surface of the legs.12,13 Patients may have a constellation of symptoms including fever and leukocytosis, but the disorder generally is self-limited.12 Erythema nodosum may be associated with a variety of diseases or infections including sarcoidosis, inflammatory bowel disease, and malignancy.14 The etiology of EN is diverse; therefore, a proper clinical workup may be necessary. Histopathology is that of a septal panniculitis with lymphocytes, histiocytes, and occasional eosinophils (Figure 3).13
Lipodermatosclerosis also occurs on the legs, most commonly in patients with venous insufficiency.12,15 Patients present clinically with pain, induration, redness, or swelling of the legs. Histopathology predominantly is characterized by membranous fat necrosis, fibrosis, and fatty microcysts that may be lined by a thickened hyaline membrane (Figure 4). Lipodermatosclerosis lesions generally do not resolve spontaneously and may need to be treated.16
The Diagnosis: Subcutaneous Panniculitislike T-cell Lymphoma
Subcutaneous panniculitislike T-cell lymphoma (SPTCL) is a rare form of cutaneous lymphoma of mature cytotoxic T cells simulating panniculitis and preferentially infiltrating the subcutaneous tissue.1 Subcutaneous panniculitislike T-cell lymphoma can affect all ages but predominantly affects younger individuals, with 20% being younger than 20 years.2 It is a rare lymphoma that accounts for less than 1% of all non-Hodgkin lymphomas.3 It presents clinically as multiple subcutaneous masses, nodules, or plaques generally on the trunk or extremities.1,2 The skin surrounding the nodules may be erythematous, and the nodules may become necrotic; however, ulceration typically is not seen. Systemic symptoms such as fever, night sweats, and chills are present in half of cases.1 According to the World Health Organization, cytopenia and elevated liver function tests are common, and a hemophagocytic syndrome may be present in 15% to 20% of cases.3 The presence of a hemophagocytic syndrome yields a poor prognosis.1,3 Current guidelines denote that SPTCL T-cell receptor (TCR) αβ; is a distinct entity from the TCRγδ; phenotype, known as cutaneous γδ-positive T-cell lymphoma.3,4 Cutaneous γδ-positive T-cell lymphoma is associated with rapid decline and a worse prognosis.4
Histology of SPTCL is characteristic for a lobular panniculitislike infiltrate.1 The heavy subcutaneous lymphoid infiltrate is composed of atypical small- to medium-sized lymphocytes with mature chromatin and inconspicuous nucleoli lining adipocytes. The dense inflammatory infiltrate composed predominantly of neoplastic T cells and macrophages may diffusely invade into the subcutaneous tissue.1 Admixed histocytes and karyorrhectic debris as well as rimming of the lymphocytes around the fat cells is typical and was seen in our patient (quiz image). The T cells of SPTCL have the following immunophenotype: TCR-beta F1+, CD3+, CD4-, CD8+, CD56-. They can express numerous cytotoxic proteins, such as T1a-1, granzyme B, and perforin.2,3 Although the CD8+ T cells may be sparse, they generally surround the adipocytes in a rimming manner and may distort the adipocyte membrane.1
Lupus erythematosus profundus (LEP) is a form of chronic cutaneous lupus that affects the deep dermis and fat.5 It also can present clinically as tender plaques or nodules. It most frequently involves the upper arms, shoulders, face, or buttocks--areas that are less commonly involved in other panniculitides.6 Histologically, LEP is similar to chronic discoid lupus with features such as epidermal atrophy, interface changes, and a thickened basement membrane (Figure 1). Lupus erythematosus profundus can present as a lobular panniculitis with mucin as well as a superficial and deep lymphocytic infiltrate that can involve the septa.5 Some cases of LEP have a predominantly lobular lymphocytic panniculitis in the absence of the typical epidermal or dermal changes of lupus erythematosus. Lymphoid follicles with germinal center formation are present in half of cases and reportedly are characteristic of LEP.6,7 The lymphoid follicles often have plasma cells, can extend into the septa as well as in between collagen bundles, and may have nuclear fragmentation.5 Another characteristic feature of LEP is hyaline sclerosis of lobules with focal extension into the interlobular septa. Immunofluorescence studies usually show linear deposition of IgM and C3 at the dermoepidermal junction. Antinuclear antibodies can be present in patients who have LEP but are not entirely specific.6
Lupus erythematosus profundus and SPTCL are part of a spectrum and may have overlapping clinical and histopathologic characteristics; therefore, distinguishing them may be difficult.6-8 It is important to monitor these patients closely, as their disease may progress to lymphoma.6 Patients with SPTCL are more likely to present with advanced symptoms such as fever and hepatosplenomegaly and to succumb to hemophagocytic syndrome than patients with LEP.9
Although SPTCL usually is clonal, several cases of LEP with clonality also have been described. Clonal LEP cases generally are identified in patients who present with fever and cytopenia.8 Lymphoid atypia and morphologic abnormalities may be seen in cases of LEP, further complicating the distinction between LEP and SPTCL. An elevated Ki67 level may be seen in cases of SPTCL with periadipocytic rimming.9 LeBlanc et al10 used Ki67 "hot spots" along with CD8 immunohistochemistry to identify atypical lymphocytes associated with SPTCL. Lymphocyte rimming was defined by the presence of CD8+ lymphocytes with an elevated Ki67 index. Clinical, histopathologic, and molecular findings all should be used when dealing with challenging cases.
Fat necrosis can occur in any part of the body where trauma has occurred and can be associated with many disease processes. Patients typically present with a palpable mass, but a clinical history of trauma is not always present. Histopathologic findings include necrotic fat alongside lipid-laden foamy macrophages and scattered inflammatory cells (Figure 2).11 Fragments of normal as well as degenerating adipose tissue and multinucleated giant cells can be present.
Erythema nodosum (EN) is the most frequently encountered panniculitis and usually is seen in women in early adulthood.12 Patients present with several tender subcutaneous nodules and plaques that most commonly are present on the anterior surface of the legs.12,13 Patients may have a constellation of symptoms including fever and leukocytosis, but the disorder generally is self-limited.12 Erythema nodosum may be associated with a variety of diseases or infections including sarcoidosis, inflammatory bowel disease, and malignancy.14 The etiology of EN is diverse; therefore, a proper clinical workup may be necessary. Histopathology is that of a septal panniculitis with lymphocytes, histiocytes, and occasional eosinophils (Figure 3).13
Lipodermatosclerosis also occurs on the legs, most commonly in patients with venous insufficiency.12,15 Patients present clinically with pain, induration, redness, or swelling of the legs. Histopathology predominantly is characterized by membranous fat necrosis, fibrosis, and fatty microcysts that may be lined by a thickened hyaline membrane (Figure 4). Lipodermatosclerosis lesions generally do not resolve spontaneously and may need to be treated.16
- Musick SR, Lynch DT. Subcutaneous Panniculitis Like T-cell Lymphoma. StatPearls Publishing; 2020.
- Guenova E, Schanz S, Hoetzenecker W, et al. Systemic corticosteroids for subcutaneous panniculitis-like T-cell lymphoma. Br J Dermatol. 2014;171:891-894.
- Swerdlow SH. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. International Agency for Research on Cancer; 2017.
- Bagheri F, Cervellione KL, Delgado B, et al. An illustrative case of subcutaneous panniculitis-like T-cell lymphoma [published online March 3, 2011]. J Skin Cancer. doi:10.1155/2011/824528
- Kogame T, Yamashita R, Hirata M, et al. Analysis of possible structures of inducible skin‐associated lymphoid tissue in lupus erythematosus profundus. J Dermatol. 2018;45:1117-1121.
- Arps DP, Patel RM. Lupus profundus (panniculitis): a potential mimic of subcutaneous panniculitis-like T-cell lymphoma. Arch Pathol Lab Med. 2013;137:1211-1215.
- Alberti-Violetti S, Berti E. Lymphocytic lobular panniculitis: a diagnostic challenge. Dermatopathology. 2018;5:30-33.
- Magro CM, Crowson AN, Kovatich AJ, et al. Lupus profundus, indeterminate lymphocytic lobular panniculitis and subcutaneous T-cell lymphoma: a spectrum of subcuticular T-cell lymphoid dyscrasia. J Cutan Pathol. 2001;28:235-247.
- Sitthinamsuwan P, Pattanaprichakul P, Treetipsatit J, et al. Subcutaneous panniculitis-like T-cell lymphoma versus lupus erythematosus panniculitis: distinction by means of the periadipocytic cell proliferation index. Am J Dermatopathol. 2018;40:567-574.
- LeBlanc RE, Tavallaee M, Kim YH, et al. Useful parameters for distinguishing subcutaneous panniculitis-like T-cell lymphoma from lupus erythematosus panniculitis. Am J Surg Pathol. 2016;40:745-754.
- Burkholz KJ, Roberts CC, Lidner TK. Posttraumatic pseudolipoma (fat necrosis) mimicking atypical lipoma or liposarcoma on MRI. Radiol Case Rep. 2015;2:56-60.
- Wick MR. Panniculitis: a summary. Semin Diagn Pathol. 2017;34:261-272.
- Thurber S, Kohler S. Histopathologic spectrum of erythema nodosum. J Cutan Pathol. 2006;33:18-26.
- Requena L, Requena C. Erythema nodosum. Dermatol Online J. 2002;8:4.
- Choonhakarn C, Chaowattanapanit S, Julanon N. Lipodermatosclerosis: a clinicopathologic correlation. Int J Dermatol. 2016;55:303-308.
- Huang TM, Lee JY. Lipodermatosclerosis: a clinicopathologic study of 17 cases and differential diagnosis from erythema nodosum. J Cutan Pathol. 2009;36:453-460.
- Musick SR, Lynch DT. Subcutaneous Panniculitis Like T-cell Lymphoma. StatPearls Publishing; 2020.
- Guenova E, Schanz S, Hoetzenecker W, et al. Systemic corticosteroids for subcutaneous panniculitis-like T-cell lymphoma. Br J Dermatol. 2014;171:891-894.
- Swerdlow SH. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. International Agency for Research on Cancer; 2017.
- Bagheri F, Cervellione KL, Delgado B, et al. An illustrative case of subcutaneous panniculitis-like T-cell lymphoma [published online March 3, 2011]. J Skin Cancer. doi:10.1155/2011/824528
- Kogame T, Yamashita R, Hirata M, et al. Analysis of possible structures of inducible skin‐associated lymphoid tissue in lupus erythematosus profundus. J Dermatol. 2018;45:1117-1121.
- Arps DP, Patel RM. Lupus profundus (panniculitis): a potential mimic of subcutaneous panniculitis-like T-cell lymphoma. Arch Pathol Lab Med. 2013;137:1211-1215.
- Alberti-Violetti S, Berti E. Lymphocytic lobular panniculitis: a diagnostic challenge. Dermatopathology. 2018;5:30-33.
- Magro CM, Crowson AN, Kovatich AJ, et al. Lupus profundus, indeterminate lymphocytic lobular panniculitis and subcutaneous T-cell lymphoma: a spectrum of subcuticular T-cell lymphoid dyscrasia. J Cutan Pathol. 2001;28:235-247.
- Sitthinamsuwan P, Pattanaprichakul P, Treetipsatit J, et al. Subcutaneous panniculitis-like T-cell lymphoma versus lupus erythematosus panniculitis: distinction by means of the periadipocytic cell proliferation index. Am J Dermatopathol. 2018;40:567-574.
- LeBlanc RE, Tavallaee M, Kim YH, et al. Useful parameters for distinguishing subcutaneous panniculitis-like T-cell lymphoma from lupus erythematosus panniculitis. Am J Surg Pathol. 2016;40:745-754.
- Burkholz KJ, Roberts CC, Lidner TK. Posttraumatic pseudolipoma (fat necrosis) mimicking atypical lipoma or liposarcoma on MRI. Radiol Case Rep. 2015;2:56-60.
- Wick MR. Panniculitis: a summary. Semin Diagn Pathol. 2017;34:261-272.
- Thurber S, Kohler S. Histopathologic spectrum of erythema nodosum. J Cutan Pathol. 2006;33:18-26.
- Requena L, Requena C. Erythema nodosum. Dermatol Online J. 2002;8:4.
- Choonhakarn C, Chaowattanapanit S, Julanon N. Lipodermatosclerosis: a clinicopathologic correlation. Int J Dermatol. 2016;55:303-308.
- Huang TM, Lee JY. Lipodermatosclerosis: a clinicopathologic study of 17 cases and differential diagnosis from erythema nodosum. J Cutan Pathol. 2009;36:453-460.
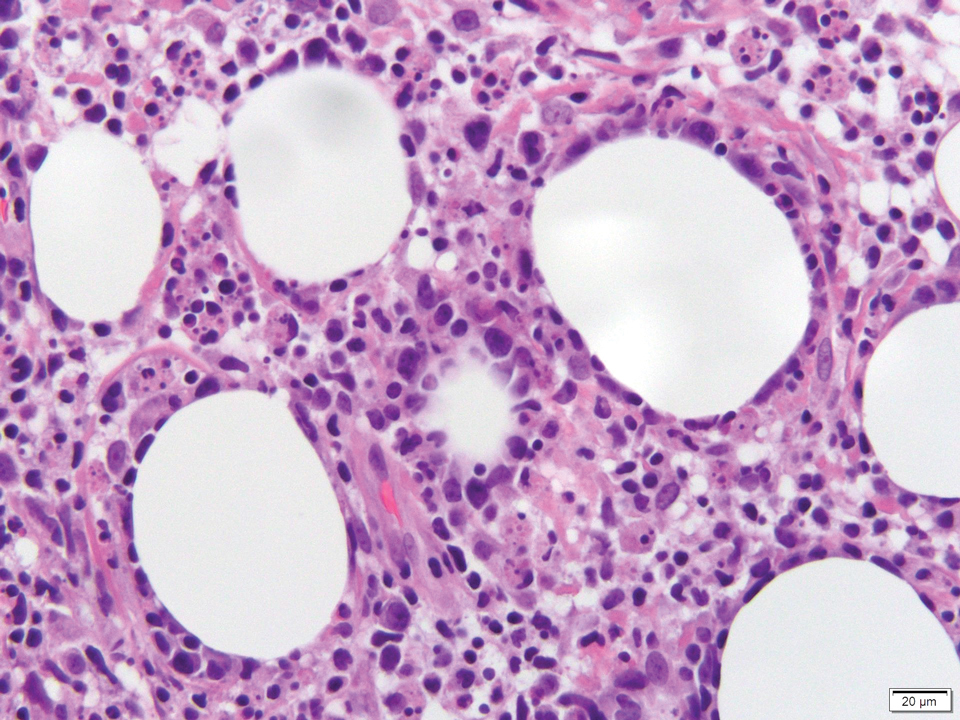
A 47-year-old man presented with a tender soft tissue mass on the upper back with increasing discomfort over the last 4 weeks. He noted that he felt feverish a few times. Physical examination revealed a 3×4-cm area of induration involving the upper mid back with faint erythema of the overlying skin; no drainage was noted. A prominent left posterior cervical lymph node also was appreciated, and a punch biopsy of the mass was performed.
Telangiectatic Patch on the Forehead
The Diagnosis: Cutaneous B-cell Lymphoma
Histopathology was suggestive of cutaneous B-cell lymphoma (Figure). Further immunohistochemical studies including Bcl-6 positivity and Bcl-2 negativity in the large atypical cells supported a diagnosis of primary cutaneous follicle center lymphoma (PCFCL). The designation of primary cutaneous B-cell lymphoma includes several different types of lymphoma, including marginal zone lymphoma, diffuse large B-cell lymphoma, and intravascular lymphoma. To be considered a primary cutaneous lymphoma, there must be evidence of the lymphoma in the skin without concomitant evidence of systemic involvement, as determined through a full staging workup. Primary cutaneous follicle center lymphoma is an indolent lymphoma that most commonly presents as solitary or grouped, pink to plum-colored papules, plaques, nodules, and tumors on the scalp, forehead, or back.1 The lesions often are biopsied as suspected basal cell carcinomas or Merkel cell carcinomas (MCCs). Lesions on the face or scalp may easily evade diagnosis, as they initially may mimic rosacea or insect bites. Less common presentations include infiltrative lesions that cause rhinophymatous changes or scarring alopecia. Multifocal or disseminated lesions rarely can be observed. This case presentation is unique in its patchy appearance that clinically resembled angiosarcoma.2 When identified and treated, the disease-specific 5-year survival rate for PCFCL is greater than 95%.3
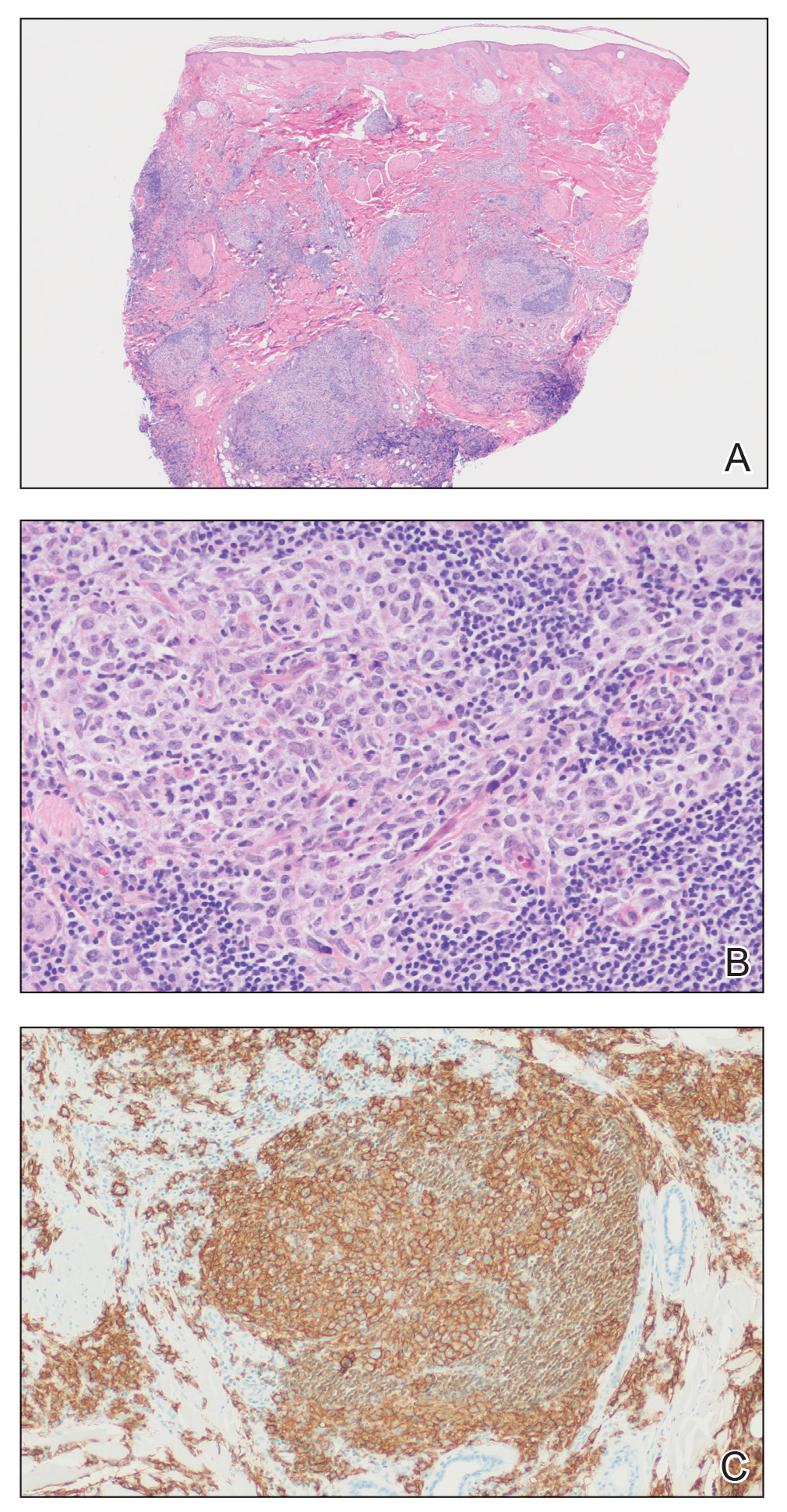
Merkel cell carcinoma was first described in 1972 and has been diagnosed with increasing frequency each year.4 It generally presents as an erythematous or violaceous, tender, indurated nodule on sun-exposed skin of the head or neck in elderly White men. However, other presentations have been reported, including papules, plaques, cystlike structures, pruritic tumors, pedunculated lesions, subcutaneous masses, and telangiectatic papules.5 Histopathologically, MCC is characterized by dermal nests and sheets of basaloid cells with finely granular salt and pepper-like chromatin. The histologic features can resemble other small blue cell tumors; therefore, the differential diagnosis can be broad.5 Immunohistochemistry that can confirm the diagnosis of MCC generally will be positive for cytokeratin 20 and neuroendocrine markers but negative for cytokeratin 7 and thyroid transcription factor 1. Merkel cell carcinoma is an aggressive tumor with a high risk for local recurrence and distant metastasis that carries a generally poor prognosis, especially when there is evidence of metastatic disease at presentation.5,6
Rosacea can appear as telangiectatic patches, though generally not as one discrete patch limited to the forehead, as in our patient. Histologic features vary based on the age of the lesion and clinical variant. In early lesions there is a mild perivascular lymphoplasmacytic infiltrate within the dermis, while older lesions can have a mixed infiltrate crowded around vessels and adnexal structures. Granulomas often are seen near hair follicles and interspersed throughout the dermis with ectatic vessels and dermal edema.7
Angiosarcoma is divided into 3 clinicopathological subtypes: idiopathic angiosarcoma of the head and neck, angiosarcoma in the setting of lymphedema, and postirradiation angiosarcoma.7 Idiopathic angiosarcoma most closely mimics PCFCL, as it can present as single or multifocal nodules, plaques, or patches. Histologically, the 3 groups appear similar with poorly circumscribed, infiltrative, dermal tumors. The neoplastic endothelial cells have large hyperchromatic nuclei that protrude into vascular lumens. The prognosis for idiopathic angiosarcoma of the head and neck is poor, with a 5-year survival rate of 15% to 34%, which often is due to delayed diagnosis.7
Pigmented purpuric dermatoses (PPDs) are chronic skin disorders characterized by purpura due to extravasation of blood from capillaries; the resulting hemosiderin deposition leads to pigmentation.7 There are various forms of PPD, which are classified into groups based on clinical appearance including Schamberg disease, purpura annularis telangiectodes of Majocchi, pigmented purpuric lichenoid dermatosis of Gougerot and Blum, lichen aureus, and others including eczematid and itching variants, which some consider to be distinct entities. Purpura annularis telangiectodes of Majocchi is the specific PPD that should be included in the clinical differential for PCFCL because it presents as annular patches with telangiectasias. Histologically, PPDs are characterized by a CD4+ lymphocytic infiltrate in the upper dermis with extravasated red blood cells and the presence of hemosiderin mostly within macrophages and a lack of true vasculitis. Clonality of the T cells has been shown, and there is some evidence that PPD may overlap with mycosis fungoides. However, this overlap mainly has been seen in patients with widespread lesions and would not apply to this case. In general, patients with PPD can be reassured of the benign process. In cases of widespread PPD, patients should be followed clinically to assess for progression to mycosis fungoides, though the likelihood is low.7
Our patient underwent a full staging workup, which confirmed the diagnosis of PCFCL. He was treated with radiation to the forehead that resulted in clearance of the lesion. Approximately 2 years after the initial diagnosis, the patient was alive and well with no evidence of recurrence of PCFCL.
In conclusion, it is imperative to identify unusual, macular, vascular-appearing patches, especially on the head and neck in older individuals. Because the clinical presentations of PCFCL, angiosarcoma, rosacea, MCC, and PPD can overlap with one another as well as with other entities, it is necessary to have a high level of suspicion and low threshold to biopsy these types of lesions, as outcomes can be drastically different.
- Goyal A, LeBlanc RE, Carter JB. Cutaneous B-cell lymphoma. Hematol Oncol Clin North Am. 2019;33:149-161.
- Massone C, Fink-Puches R, Cerroni L. Atypical clinical presentation of primary and secondary cutaneous follicle center lymphoma (FCL) on the head characterized by macular lesions. J Am Acad Dermatol. 2016;75:1000-1006.
- Wilcox RA. Cutaneous B-cell lymphomas: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:1052-1055.
- Conic RRZ, Ko J, Saridakis S, et al. Sentinel lymph node biopsy in Merkel cell carcinoma: predictors of sentinel lymph node positivity and association with overall survival. J Am Acad Dermatol. 2019;81:364-372
- Coggshall K, Tello TL, North JP, et al. Merkel cell carcinoma: an update and review: pathogenesis, diagnosis, and staging. J Am Acad Dermatol. 2018;78:433-442.
- Tello TL, Coggshall K, Yom SS, et al. Merkel cell carcinoma: an update and review: current and future therapy. J Am Acad Dermatol. 2018;78:445-454.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
The Diagnosis: Cutaneous B-cell Lymphoma
Histopathology was suggestive of cutaneous B-cell lymphoma (Figure). Further immunohistochemical studies including Bcl-6 positivity and Bcl-2 negativity in the large atypical cells supported a diagnosis of primary cutaneous follicle center lymphoma (PCFCL). The designation of primary cutaneous B-cell lymphoma includes several different types of lymphoma, including marginal zone lymphoma, diffuse large B-cell lymphoma, and intravascular lymphoma. To be considered a primary cutaneous lymphoma, there must be evidence of the lymphoma in the skin without concomitant evidence of systemic involvement, as determined through a full staging workup. Primary cutaneous follicle center lymphoma is an indolent lymphoma that most commonly presents as solitary or grouped, pink to plum-colored papules, plaques, nodules, and tumors on the scalp, forehead, or back.1 The lesions often are biopsied as suspected basal cell carcinomas or Merkel cell carcinomas (MCCs). Lesions on the face or scalp may easily evade diagnosis, as they initially may mimic rosacea or insect bites. Less common presentations include infiltrative lesions that cause rhinophymatous changes or scarring alopecia. Multifocal or disseminated lesions rarely can be observed. This case presentation is unique in its patchy appearance that clinically resembled angiosarcoma.2 When identified and treated, the disease-specific 5-year survival rate for PCFCL is greater than 95%.3
Merkel cell carcinoma was first described in 1972 and has been diagnosed with increasing frequency each year.4 It generally presents as an erythematous or violaceous, tender, indurated nodule on sun-exposed skin of the head or neck in elderly White men. However, other presentations have been reported, including papules, plaques, cystlike structures, pruritic tumors, pedunculated lesions, subcutaneous masses, and telangiectatic papules.5 Histopathologically, MCC is characterized by dermal nests and sheets of basaloid cells with finely granular salt and pepper-like chromatin. The histologic features can resemble other small blue cell tumors; therefore, the differential diagnosis can be broad.5 Immunohistochemistry that can confirm the diagnosis of MCC generally will be positive for cytokeratin 20 and neuroendocrine markers but negative for cytokeratin 7 and thyroid transcription factor 1. Merkel cell carcinoma is an aggressive tumor with a high risk for local recurrence and distant metastasis that carries a generally poor prognosis, especially when there is evidence of metastatic disease at presentation.5,6
Rosacea can appear as telangiectatic patches, though generally not as one discrete patch limited to the forehead, as in our patient. Histologic features vary based on the age of the lesion and clinical variant. In early lesions there is a mild perivascular lymphoplasmacytic infiltrate within the dermis, while older lesions can have a mixed infiltrate crowded around vessels and adnexal structures. Granulomas often are seen near hair follicles and interspersed throughout the dermis with ectatic vessels and dermal edema.7
Angiosarcoma is divided into 3 clinicopathological subtypes: idiopathic angiosarcoma of the head and neck, angiosarcoma in the setting of lymphedema, and postirradiation angiosarcoma.7 Idiopathic angiosarcoma most closely mimics PCFCL, as it can present as single or multifocal nodules, plaques, or patches. Histologically, the 3 groups appear similar with poorly circumscribed, infiltrative, dermal tumors. The neoplastic endothelial cells have large hyperchromatic nuclei that protrude into vascular lumens. The prognosis for idiopathic angiosarcoma of the head and neck is poor, with a 5-year survival rate of 15% to 34%, which often is due to delayed diagnosis.7
Pigmented purpuric dermatoses (PPDs) are chronic skin disorders characterized by purpura due to extravasation of blood from capillaries; the resulting hemosiderin deposition leads to pigmentation.7 There are various forms of PPD, which are classified into groups based on clinical appearance including Schamberg disease, purpura annularis telangiectodes of Majocchi, pigmented purpuric lichenoid dermatosis of Gougerot and Blum, lichen aureus, and others including eczematid and itching variants, which some consider to be distinct entities. Purpura annularis telangiectodes of Majocchi is the specific PPD that should be included in the clinical differential for PCFCL because it presents as annular patches with telangiectasias. Histologically, PPDs are characterized by a CD4+ lymphocytic infiltrate in the upper dermis with extravasated red blood cells and the presence of hemosiderin mostly within macrophages and a lack of true vasculitis. Clonality of the T cells has been shown, and there is some evidence that PPD may overlap with mycosis fungoides. However, this overlap mainly has been seen in patients with widespread lesions and would not apply to this case. In general, patients with PPD can be reassured of the benign process. In cases of widespread PPD, patients should be followed clinically to assess for progression to mycosis fungoides, though the likelihood is low.7
Our patient underwent a full staging workup, which confirmed the diagnosis of PCFCL. He was treated with radiation to the forehead that resulted in clearance of the lesion. Approximately 2 years after the initial diagnosis, the patient was alive and well with no evidence of recurrence of PCFCL.
In conclusion, it is imperative to identify unusual, macular, vascular-appearing patches, especially on the head and neck in older individuals. Because the clinical presentations of PCFCL, angiosarcoma, rosacea, MCC, and PPD can overlap with one another as well as with other entities, it is necessary to have a high level of suspicion and low threshold to biopsy these types of lesions, as outcomes can be drastically different.
The Diagnosis: Cutaneous B-cell Lymphoma
Histopathology was suggestive of cutaneous B-cell lymphoma (Figure). Further immunohistochemical studies including Bcl-6 positivity and Bcl-2 negativity in the large atypical cells supported a diagnosis of primary cutaneous follicle center lymphoma (PCFCL). The designation of primary cutaneous B-cell lymphoma includes several different types of lymphoma, including marginal zone lymphoma, diffuse large B-cell lymphoma, and intravascular lymphoma. To be considered a primary cutaneous lymphoma, there must be evidence of the lymphoma in the skin without concomitant evidence of systemic involvement, as determined through a full staging workup. Primary cutaneous follicle center lymphoma is an indolent lymphoma that most commonly presents as solitary or grouped, pink to plum-colored papules, plaques, nodules, and tumors on the scalp, forehead, or back.1 The lesions often are biopsied as suspected basal cell carcinomas or Merkel cell carcinomas (MCCs). Lesions on the face or scalp may easily evade diagnosis, as they initially may mimic rosacea or insect bites. Less common presentations include infiltrative lesions that cause rhinophymatous changes or scarring alopecia. Multifocal or disseminated lesions rarely can be observed. This case presentation is unique in its patchy appearance that clinically resembled angiosarcoma.2 When identified and treated, the disease-specific 5-year survival rate for PCFCL is greater than 95%.3
Merkel cell carcinoma was first described in 1972 and has been diagnosed with increasing frequency each year.4 It generally presents as an erythematous or violaceous, tender, indurated nodule on sun-exposed skin of the head or neck in elderly White men. However, other presentations have been reported, including papules, plaques, cystlike structures, pruritic tumors, pedunculated lesions, subcutaneous masses, and telangiectatic papules.5 Histopathologically, MCC is characterized by dermal nests and sheets of basaloid cells with finely granular salt and pepper-like chromatin. The histologic features can resemble other small blue cell tumors; therefore, the differential diagnosis can be broad.5 Immunohistochemistry that can confirm the diagnosis of MCC generally will be positive for cytokeratin 20 and neuroendocrine markers but negative for cytokeratin 7 and thyroid transcription factor 1. Merkel cell carcinoma is an aggressive tumor with a high risk for local recurrence and distant metastasis that carries a generally poor prognosis, especially when there is evidence of metastatic disease at presentation.5,6
Rosacea can appear as telangiectatic patches, though generally not as one discrete patch limited to the forehead, as in our patient. Histologic features vary based on the age of the lesion and clinical variant. In early lesions there is a mild perivascular lymphoplasmacytic infiltrate within the dermis, while older lesions can have a mixed infiltrate crowded around vessels and adnexal structures. Granulomas often are seen near hair follicles and interspersed throughout the dermis with ectatic vessels and dermal edema.7
Angiosarcoma is divided into 3 clinicopathological subtypes: idiopathic angiosarcoma of the head and neck, angiosarcoma in the setting of lymphedema, and postirradiation angiosarcoma.7 Idiopathic angiosarcoma most closely mimics PCFCL, as it can present as single or multifocal nodules, plaques, or patches. Histologically, the 3 groups appear similar with poorly circumscribed, infiltrative, dermal tumors. The neoplastic endothelial cells have large hyperchromatic nuclei that protrude into vascular lumens. The prognosis for idiopathic angiosarcoma of the head and neck is poor, with a 5-year survival rate of 15% to 34%, which often is due to delayed diagnosis.7
Pigmented purpuric dermatoses (PPDs) are chronic skin disorders characterized by purpura due to extravasation of blood from capillaries; the resulting hemosiderin deposition leads to pigmentation.7 There are various forms of PPD, which are classified into groups based on clinical appearance including Schamberg disease, purpura annularis telangiectodes of Majocchi, pigmented purpuric lichenoid dermatosis of Gougerot and Blum, lichen aureus, and others including eczematid and itching variants, which some consider to be distinct entities. Purpura annularis telangiectodes of Majocchi is the specific PPD that should be included in the clinical differential for PCFCL because it presents as annular patches with telangiectasias. Histologically, PPDs are characterized by a CD4+ lymphocytic infiltrate in the upper dermis with extravasated red blood cells and the presence of hemosiderin mostly within macrophages and a lack of true vasculitis. Clonality of the T cells has been shown, and there is some evidence that PPD may overlap with mycosis fungoides. However, this overlap mainly has been seen in patients with widespread lesions and would not apply to this case. In general, patients with PPD can be reassured of the benign process. In cases of widespread PPD, patients should be followed clinically to assess for progression to mycosis fungoides, though the likelihood is low.7
Our patient underwent a full staging workup, which confirmed the diagnosis of PCFCL. He was treated with radiation to the forehead that resulted in clearance of the lesion. Approximately 2 years after the initial diagnosis, the patient was alive and well with no evidence of recurrence of PCFCL.
In conclusion, it is imperative to identify unusual, macular, vascular-appearing patches, especially on the head and neck in older individuals. Because the clinical presentations of PCFCL, angiosarcoma, rosacea, MCC, and PPD can overlap with one another as well as with other entities, it is necessary to have a high level of suspicion and low threshold to biopsy these types of lesions, as outcomes can be drastically different.
- Goyal A, LeBlanc RE, Carter JB. Cutaneous B-cell lymphoma. Hematol Oncol Clin North Am. 2019;33:149-161.
- Massone C, Fink-Puches R, Cerroni L. Atypical clinical presentation of primary and secondary cutaneous follicle center lymphoma (FCL) on the head characterized by macular lesions. J Am Acad Dermatol. 2016;75:1000-1006.
- Wilcox RA. Cutaneous B-cell lymphomas: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:1052-1055.
- Conic RRZ, Ko J, Saridakis S, et al. Sentinel lymph node biopsy in Merkel cell carcinoma: predictors of sentinel lymph node positivity and association with overall survival. J Am Acad Dermatol. 2019;81:364-372
- Coggshall K, Tello TL, North JP, et al. Merkel cell carcinoma: an update and review: pathogenesis, diagnosis, and staging. J Am Acad Dermatol. 2018;78:433-442.
- Tello TL, Coggshall K, Yom SS, et al. Merkel cell carcinoma: an update and review: current and future therapy. J Am Acad Dermatol. 2018;78:445-454.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
- Goyal A, LeBlanc RE, Carter JB. Cutaneous B-cell lymphoma. Hematol Oncol Clin North Am. 2019;33:149-161.
- Massone C, Fink-Puches R, Cerroni L. Atypical clinical presentation of primary and secondary cutaneous follicle center lymphoma (FCL) on the head characterized by macular lesions. J Am Acad Dermatol. 2016;75:1000-1006.
- Wilcox RA. Cutaneous B-cell lymphomas: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:1052-1055.
- Conic RRZ, Ko J, Saridakis S, et al. Sentinel lymph node biopsy in Merkel cell carcinoma: predictors of sentinel lymph node positivity and association with overall survival. J Am Acad Dermatol. 2019;81:364-372
- Coggshall K, Tello TL, North JP, et al. Merkel cell carcinoma: an update and review: pathogenesis, diagnosis, and staging. J Am Acad Dermatol. 2018;78:433-442.
- Tello TL, Coggshall K, Yom SS, et al. Merkel cell carcinoma: an update and review: current and future therapy. J Am Acad Dermatol. 2018;78:445-454.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
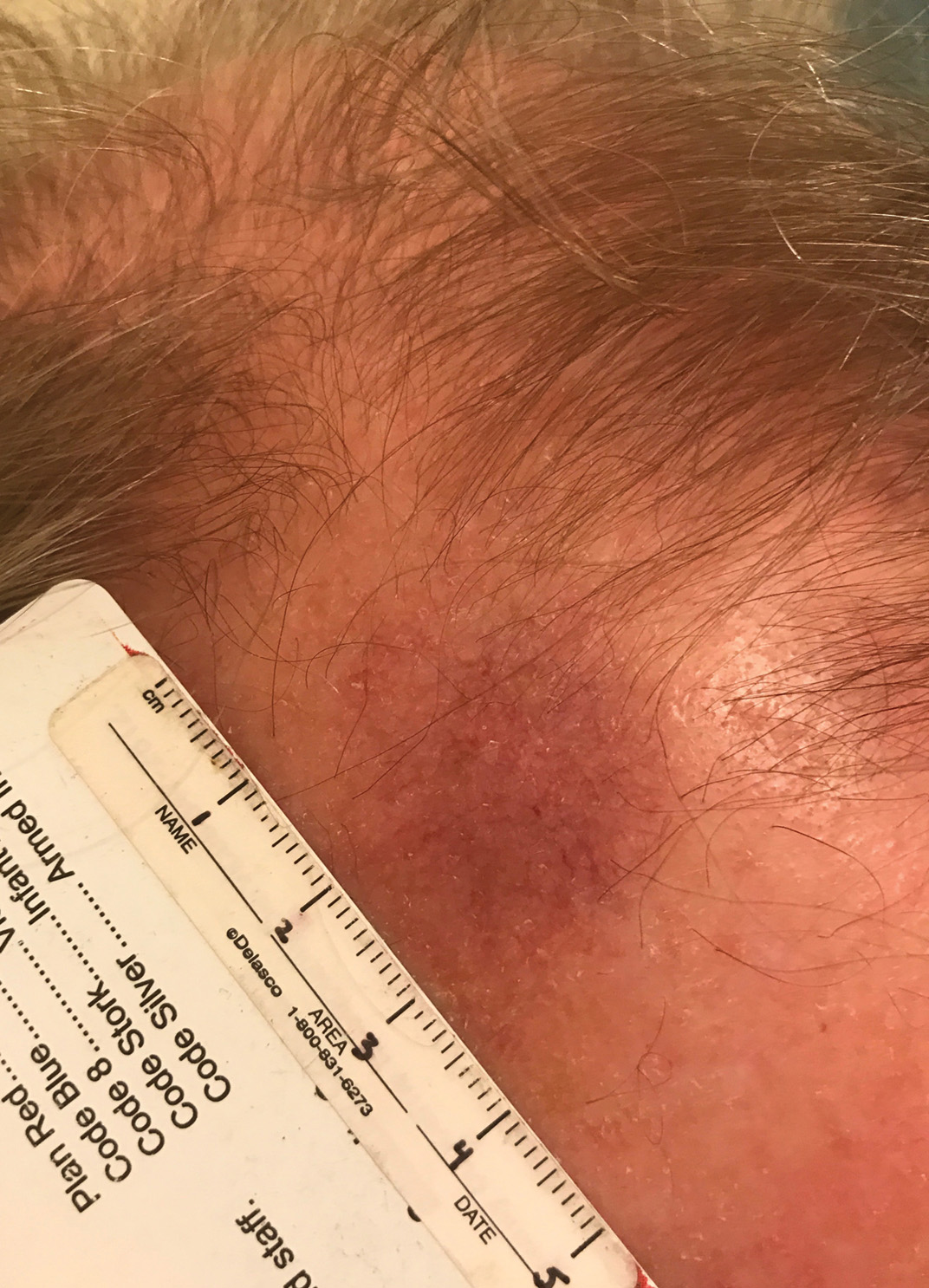
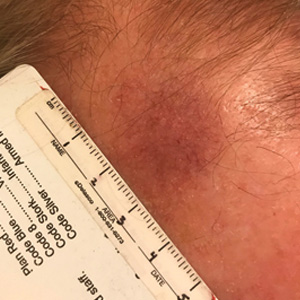
Multiple Nontender Subcutaneous Nodules on the Finger
The Diagnosis: Subcutaneous Granuloma Annulare
Subcutaneous granuloma annulare (SGA), also known as deep GA, is a rare variant of GA that usually occurs in children and young adults. It presents as single or multiple, nontender, deep dermal and/or subcutaneous nodules with normal-appearing skin usually on the anterior lower legs, dorsal aspects of the hands and fingers, scalp, or buttocks.1-3 The pathogenesis of SGA as well as GA is not fully understood, and proposed inciting factors include trauma, insect bite reactions, tuberculin skin testing, vaccines, UV exposure, medications, and viral infections.3-6 A cell-mediated, delayed-type hypersensitivity reaction to an unknown antigen also has been postulated as a possible mechanism.7 Treatment usually is not necessary, as the nature of the condition is benign and the course often is self-limited. Spontaneous resolution occurs within 2 years in 50% of patients with localized GA.4,8 Surgery usually is not recommended due to the high recurrence rate (40%-75%).4,9
Absence of epidermal change in this entity obfuscates clinical recognition, and accurate diagnosis often depends on punch or excisional biopsies revealing characteristic histopathology. The histology of SGA consists of palisaded granulomas with central areas of necrobiosis composed of degenerated collagen, mucin deposition, and nuclear dust from neutrophils that extend into the deep dermis and subcutis.2 The periphery of the granulomas is lined by palisading epithelioid histiocytes with occasional multinucleated giant cells.10,11 Eosinophils often are present.12 Colloidal iron and Alcian blue stains can be used to highlight the abundant connective tissue mucin of the granulomas.4
The histologic differential diagnosis of SGA includes rheumatoid nodule, necrobiosis lipoidica, epithelioid sarcoma, and tophaceous gout.2 Rheumatoid nodules are the most common dermatologic presentation of rheumatoid arthritis and are found in up to 30% to 40% of patients with the disease.13-15 They present as firm, painless, subcutaneous papulonodules on the extensor surfaces and at sites of trauma or pressure. Histologically, rheumatoid nodules exhibit a homogenous and eosinophilic central area of necrobiosis with fibrin deposition and absent mucin deep within the dermis and subcutaneous tissue (Figure 1). In contrast, granulomas in SGA usually are pale and basophilic with abundant mucin.2
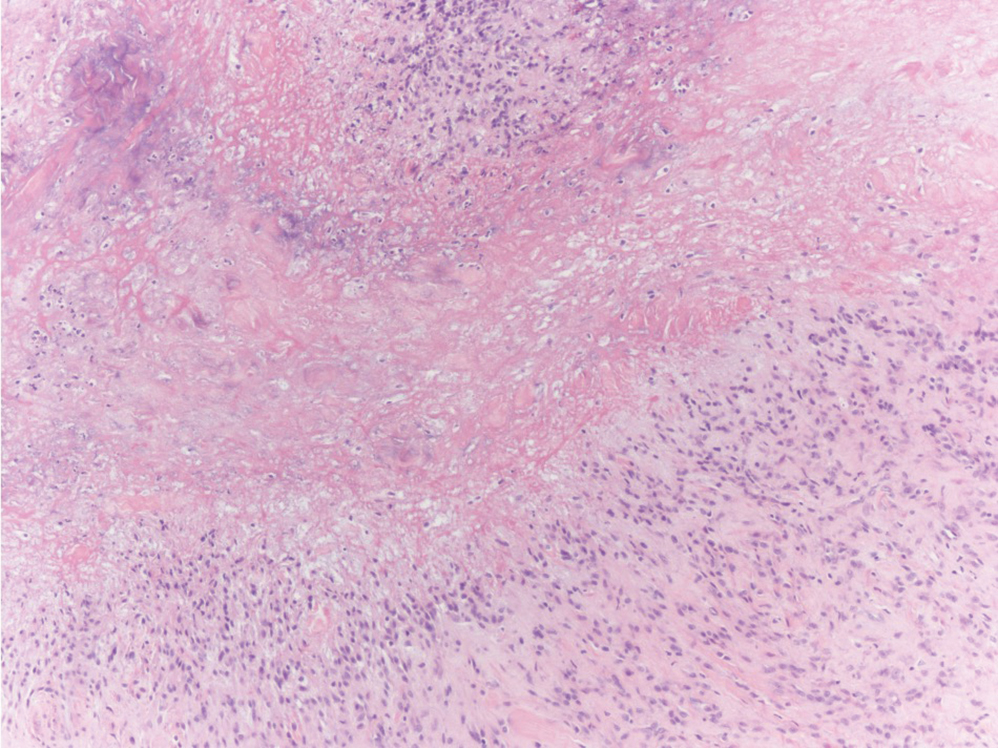
Necrobiosis lipoidica is a rare chronic granulomatous disease of the skin that most commonly occurs in young to middle-aged adults and is strongly associated with diabetes mellitus.16 It clinically presents as yellow to red-brown papules and plaques with a peripheral erythematous to violaceous rim usually on the pretibial area. Over time, lesions become yellowish atrophic patches and plaques that sometimes can ulcerate. Histopathology reveals areas of horizontally arranged, palisaded, and interstitial granulomatous dermatitis intermixed with areas of degenerated collagen and widespread fibrosis extending from the superficial dermis into the subcutis (Figure 2).2 These areas lack mucin and have an increased number of plasma cells. Eosinophils and/or lymphoid nodules occasionally can be seen.17,18
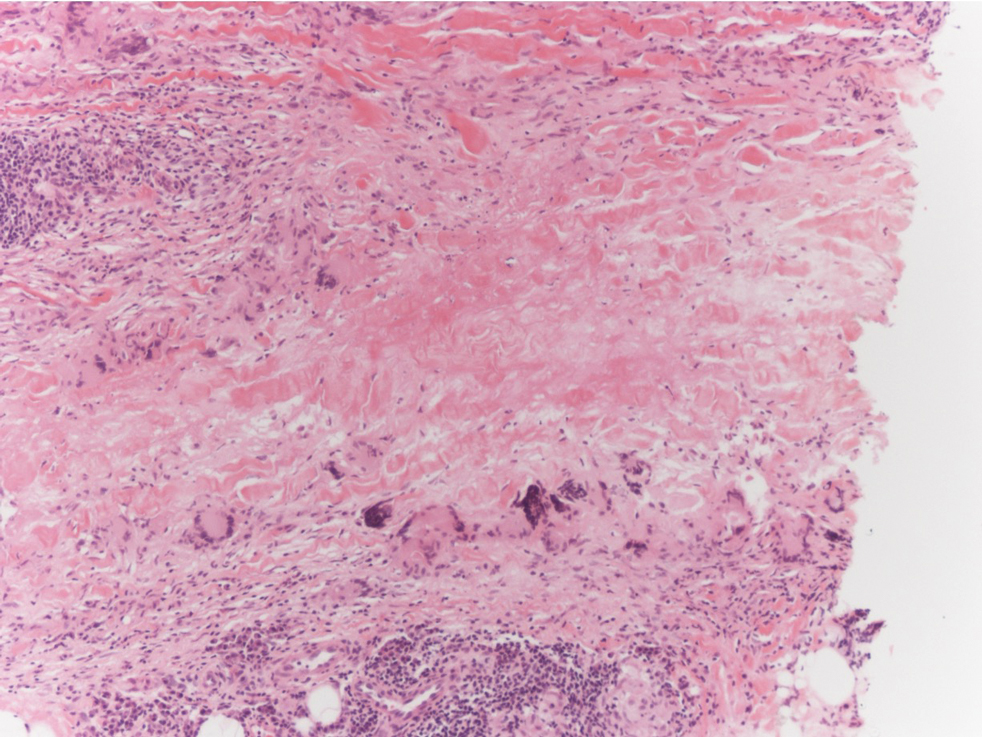
Epithelioid sarcoma is a rare malignant soft tissue sarcoma that tends to occur on the distal extremities in younger patients, typically aged 20 to 40 years, often with preceding trauma to the area. It usually presents as a solitary, poorly defined, hard, subcutaneous nodule. Histologic analysis shows central areas of necrosis and degenerated collagen surrounded by epithelioid and spindle cells with hyperchromatic and pleomorphic nuclei and mitoses (Figure 3).2 These tumor cells express positivity for keratins, vimentin, epithelial membrane antigen, and CD34, while they usually are negative for desmin, S-100, and FLI-1 nuclear transcription factor.2,4,19
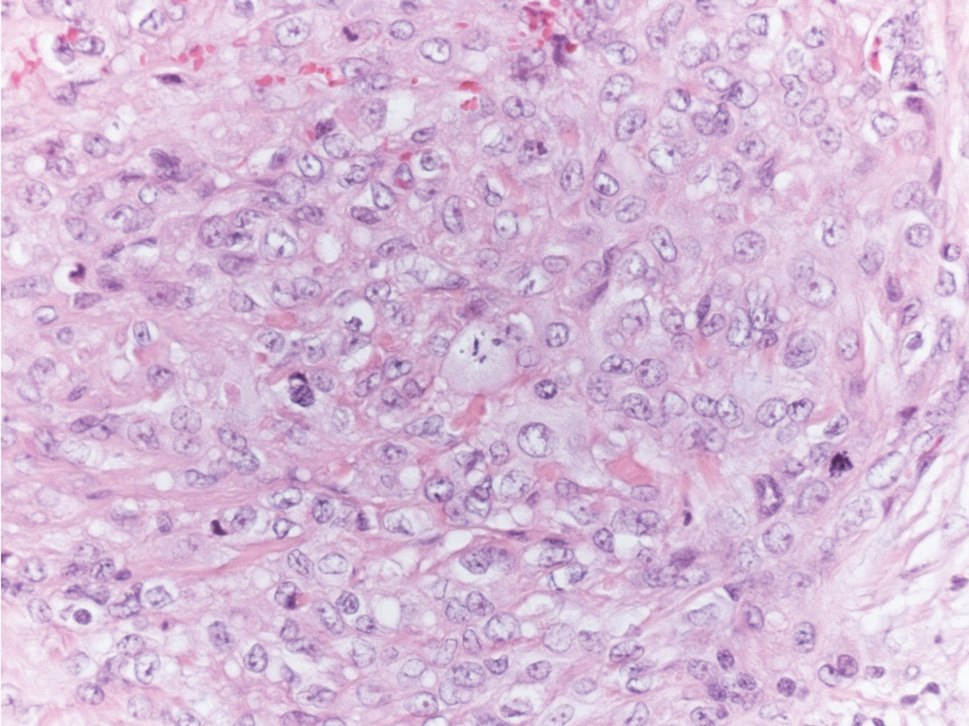
Tophaceous gout results from the accumulation of monosodium urate crystals in the skin. It clinically presents as firm, white-yellow, dermal and subcutaneous papulonodules on the helix of the ear and the skin overlying joints. Histopathology reveals palisaded granulomas surrounding an amorphous feathery material that corresponds to the urate crystals that were destroyed with formalin fixation (Figure 4). When the tissue is fixed with ethanol or is incompletely fixed in formalin, birefringent urate crystals are evident with polarization.20
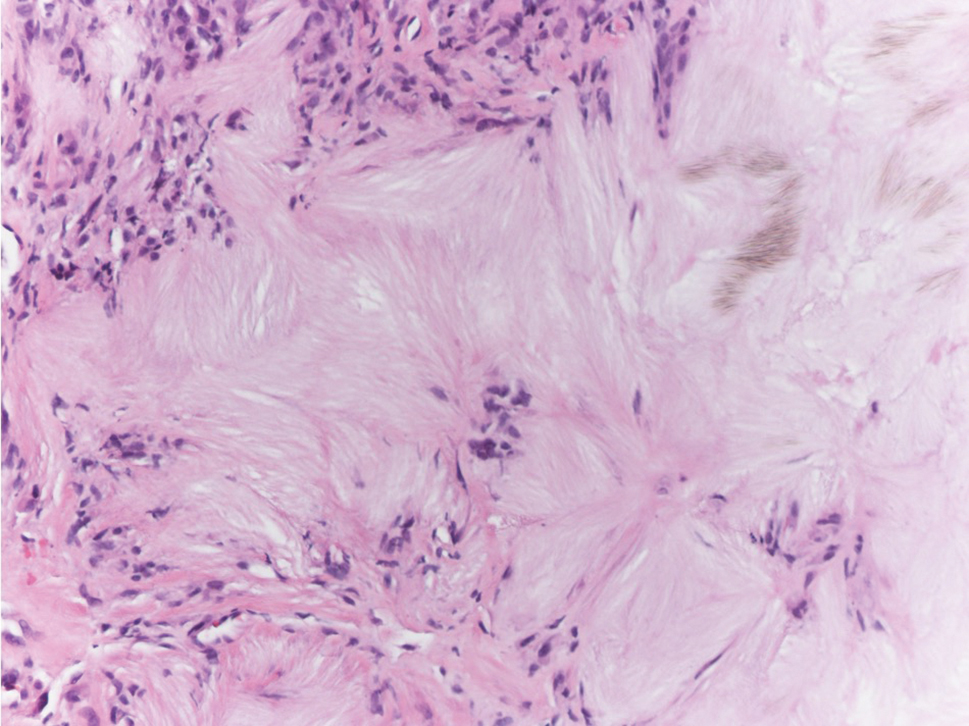
- Felner EI, Steinberg JB, Weinberg AG. Subcutaneous granuloma annulare: a review of 47 cases. Pediatrics. 1997;100:965-967.
- Requena L, Fernández-Figueras MT. Subcutaneous granuloma annulare. Semin Cutan Med Surg. 2007;26:96-99.
- Taranu T, Grigorovici M, Constantin M, et al. Subcutaneous granuloma annulare. Acta Dermatovenerol Croat. 2017;25:292-294.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1644-1663.
- Mills A, Chetty R. Auricular granuloma annulare: a consequence of trauma? Am J Dermatopathol. 1992;14:431-433.
- Muhlbauer JE. Granuloma annulare. J Am Acad Dermatol. 1980;3:217-230.
- Buechner SA, Winkelmann RK, Banks PM. Identification of T-cell subpopulations in granuloma annulare. Arch Dermatol. 1983;119:125-128.
- Wells RS, Smith MA. The natural history of granuloma annulare. Br J Dermatol. 1963;75:199.
- Davids JR, Kolman BH, Billman GF, et al. Subcutaneous granuloma annulare: recognition and treatment. J Pediatr Orthop. 1993;13:582-586.
- Evans MJ, Blessing K, Gray ES. Pseudorheumatoid nodule (deep granuloma annulare) of childhood: clinicopathologic features of twenty patients. Pediatr Dermatol. 1994;11:6-9.
- Patterson JW. Rheumatoid nodule and subcutaneous granuloma annulare: a comparative histologic study. Am J Dermatopathol. 1988;10:1-8.
- Weedon D. Granuloma annulare. Skin Pathology. Edinburgh, Scotland: Churchill-Livingstone; 1997:167-170.
- Sayah A, English JC 3rd. Rheumatoid arthritis: a review of the cutaneous manifestations. J Am Acad Dermatol. 2005;53:191-209.
- Highton J, Hessian PA, Stamp L. The rheumatoid nodule: peripheral or central to rheumatoid arthritis? Rheumatology (Oxford). 2007;46:1385-1387.
- Turesson C, Jacobsson LT. Epidemiology of extra-articular manifestations in rheumatoid arthritis. Scand J Rheumatol. 2004;33:65-72.
- Erfurt-Berge C, Dissemond J, Schwede K, et al. Updated results of 100 patients on clinical features and therapeutic options in necrobiosis lipoidica in a retrospective multicenter study. Eur J Dermatol. 2015;25:595-601.
- Kota SK, Jammula S, Kota SK, et al. Necrobiosis lipoidica diabeticorum: a case-based review of literature. Indian J Endocrinol Metab. 2012;16:614-620.
- Alegre VA, Winkelmann RK. A new histopathologic feature of necrobiosis lipoidica diabeticorum: lymphoid nodules. J Cutan Pathol. 1988;15:75-77.
- Armah HB, Parwani AV. Epithelioid sarcoma. Arch Pathol Lab Med. 2009;133:814-819.
- Shidham V, Chivukula M, Basir Z, et al. Evaluation of crystals in formalin-fixed, paraffin-embedded tissue sections for the differential diagnosis pseudogout, gout, and tumoral calcinosis. Mod Pathol. 2001;14:806-810.
The Diagnosis: Subcutaneous Granuloma Annulare
Subcutaneous granuloma annulare (SGA), also known as deep GA, is a rare variant of GA that usually occurs in children and young adults. It presents as single or multiple, nontender, deep dermal and/or subcutaneous nodules with normal-appearing skin usually on the anterior lower legs, dorsal aspects of the hands and fingers, scalp, or buttocks.1-3 The pathogenesis of SGA as well as GA is not fully understood, and proposed inciting factors include trauma, insect bite reactions, tuberculin skin testing, vaccines, UV exposure, medications, and viral infections.3-6 A cell-mediated, delayed-type hypersensitivity reaction to an unknown antigen also has been postulated as a possible mechanism.7 Treatment usually is not necessary, as the nature of the condition is benign and the course often is self-limited. Spontaneous resolution occurs within 2 years in 50% of patients with localized GA.4,8 Surgery usually is not recommended due to the high recurrence rate (40%-75%).4,9
Absence of epidermal change in this entity obfuscates clinical recognition, and accurate diagnosis often depends on punch or excisional biopsies revealing characteristic histopathology. The histology of SGA consists of palisaded granulomas with central areas of necrobiosis composed of degenerated collagen, mucin deposition, and nuclear dust from neutrophils that extend into the deep dermis and subcutis.2 The periphery of the granulomas is lined by palisading epithelioid histiocytes with occasional multinucleated giant cells.10,11 Eosinophils often are present.12 Colloidal iron and Alcian blue stains can be used to highlight the abundant connective tissue mucin of the granulomas.4
The histologic differential diagnosis of SGA includes rheumatoid nodule, necrobiosis lipoidica, epithelioid sarcoma, and tophaceous gout.2 Rheumatoid nodules are the most common dermatologic presentation of rheumatoid arthritis and are found in up to 30% to 40% of patients with the disease.13-15 They present as firm, painless, subcutaneous papulonodules on the extensor surfaces and at sites of trauma or pressure. Histologically, rheumatoid nodules exhibit a homogenous and eosinophilic central area of necrobiosis with fibrin deposition and absent mucin deep within the dermis and subcutaneous tissue (Figure 1). In contrast, granulomas in SGA usually are pale and basophilic with abundant mucin.2
Necrobiosis lipoidica is a rare chronic granulomatous disease of the skin that most commonly occurs in young to middle-aged adults and is strongly associated with diabetes mellitus.16 It clinically presents as yellow to red-brown papules and plaques with a peripheral erythematous to violaceous rim usually on the pretibial area. Over time, lesions become yellowish atrophic patches and plaques that sometimes can ulcerate. Histopathology reveals areas of horizontally arranged, palisaded, and interstitial granulomatous dermatitis intermixed with areas of degenerated collagen and widespread fibrosis extending from the superficial dermis into the subcutis (Figure 2).2 These areas lack mucin and have an increased number of plasma cells. Eosinophils and/or lymphoid nodules occasionally can be seen.17,18
Epithelioid sarcoma is a rare malignant soft tissue sarcoma that tends to occur on the distal extremities in younger patients, typically aged 20 to 40 years, often with preceding trauma to the area. It usually presents as a solitary, poorly defined, hard, subcutaneous nodule. Histologic analysis shows central areas of necrosis and degenerated collagen surrounded by epithelioid and spindle cells with hyperchromatic and pleomorphic nuclei and mitoses (Figure 3).2 These tumor cells express positivity for keratins, vimentin, epithelial membrane antigen, and CD34, while they usually are negative for desmin, S-100, and FLI-1 nuclear transcription factor.2,4,19
Tophaceous gout results from the accumulation of monosodium urate crystals in the skin. It clinically presents as firm, white-yellow, dermal and subcutaneous papulonodules on the helix of the ear and the skin overlying joints. Histopathology reveals palisaded granulomas surrounding an amorphous feathery material that corresponds to the urate crystals that were destroyed with formalin fixation (Figure 4). When the tissue is fixed with ethanol or is incompletely fixed in formalin, birefringent urate crystals are evident with polarization.20
The Diagnosis: Subcutaneous Granuloma Annulare
Subcutaneous granuloma annulare (SGA), also known as deep GA, is a rare variant of GA that usually occurs in children and young adults. It presents as single or multiple, nontender, deep dermal and/or subcutaneous nodules with normal-appearing skin usually on the anterior lower legs, dorsal aspects of the hands and fingers, scalp, or buttocks.1-3 The pathogenesis of SGA as well as GA is not fully understood, and proposed inciting factors include trauma, insect bite reactions, tuberculin skin testing, vaccines, UV exposure, medications, and viral infections.3-6 A cell-mediated, delayed-type hypersensitivity reaction to an unknown antigen also has been postulated as a possible mechanism.7 Treatment usually is not necessary, as the nature of the condition is benign and the course often is self-limited. Spontaneous resolution occurs within 2 years in 50% of patients with localized GA.4,8 Surgery usually is not recommended due to the high recurrence rate (40%-75%).4,9
Absence of epidermal change in this entity obfuscates clinical recognition, and accurate diagnosis often depends on punch or excisional biopsies revealing characteristic histopathology. The histology of SGA consists of palisaded granulomas with central areas of necrobiosis composed of degenerated collagen, mucin deposition, and nuclear dust from neutrophils that extend into the deep dermis and subcutis.2 The periphery of the granulomas is lined by palisading epithelioid histiocytes with occasional multinucleated giant cells.10,11 Eosinophils often are present.12 Colloidal iron and Alcian blue stains can be used to highlight the abundant connective tissue mucin of the granulomas.4
The histologic differential diagnosis of SGA includes rheumatoid nodule, necrobiosis lipoidica, epithelioid sarcoma, and tophaceous gout.2 Rheumatoid nodules are the most common dermatologic presentation of rheumatoid arthritis and are found in up to 30% to 40% of patients with the disease.13-15 They present as firm, painless, subcutaneous papulonodules on the extensor surfaces and at sites of trauma or pressure. Histologically, rheumatoid nodules exhibit a homogenous and eosinophilic central area of necrobiosis with fibrin deposition and absent mucin deep within the dermis and subcutaneous tissue (Figure 1). In contrast, granulomas in SGA usually are pale and basophilic with abundant mucin.2
Necrobiosis lipoidica is a rare chronic granulomatous disease of the skin that most commonly occurs in young to middle-aged adults and is strongly associated with diabetes mellitus.16 It clinically presents as yellow to red-brown papules and plaques with a peripheral erythematous to violaceous rim usually on the pretibial area. Over time, lesions become yellowish atrophic patches and plaques that sometimes can ulcerate. Histopathology reveals areas of horizontally arranged, palisaded, and interstitial granulomatous dermatitis intermixed with areas of degenerated collagen and widespread fibrosis extending from the superficial dermis into the subcutis (Figure 2).2 These areas lack mucin and have an increased number of plasma cells. Eosinophils and/or lymphoid nodules occasionally can be seen.17,18
Epithelioid sarcoma is a rare malignant soft tissue sarcoma that tends to occur on the distal extremities in younger patients, typically aged 20 to 40 years, often with preceding trauma to the area. It usually presents as a solitary, poorly defined, hard, subcutaneous nodule. Histologic analysis shows central areas of necrosis and degenerated collagen surrounded by epithelioid and spindle cells with hyperchromatic and pleomorphic nuclei and mitoses (Figure 3).2 These tumor cells express positivity for keratins, vimentin, epithelial membrane antigen, and CD34, while they usually are negative for desmin, S-100, and FLI-1 nuclear transcription factor.2,4,19
Tophaceous gout results from the accumulation of monosodium urate crystals in the skin. It clinically presents as firm, white-yellow, dermal and subcutaneous papulonodules on the helix of the ear and the skin overlying joints. Histopathology reveals palisaded granulomas surrounding an amorphous feathery material that corresponds to the urate crystals that were destroyed with formalin fixation (Figure 4). When the tissue is fixed with ethanol or is incompletely fixed in formalin, birefringent urate crystals are evident with polarization.20
- Felner EI, Steinberg JB, Weinberg AG. Subcutaneous granuloma annulare: a review of 47 cases. Pediatrics. 1997;100:965-967.
- Requena L, Fernández-Figueras MT. Subcutaneous granuloma annulare. Semin Cutan Med Surg. 2007;26:96-99.
- Taranu T, Grigorovici M, Constantin M, et al. Subcutaneous granuloma annulare. Acta Dermatovenerol Croat. 2017;25:292-294.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1644-1663.
- Mills A, Chetty R. Auricular granuloma annulare: a consequence of trauma? Am J Dermatopathol. 1992;14:431-433.
- Muhlbauer JE. Granuloma annulare. J Am Acad Dermatol. 1980;3:217-230.
- Buechner SA, Winkelmann RK, Banks PM. Identification of T-cell subpopulations in granuloma annulare. Arch Dermatol. 1983;119:125-128.
- Wells RS, Smith MA. The natural history of granuloma annulare. Br J Dermatol. 1963;75:199.
- Davids JR, Kolman BH, Billman GF, et al. Subcutaneous granuloma annulare: recognition and treatment. J Pediatr Orthop. 1993;13:582-586.
- Evans MJ, Blessing K, Gray ES. Pseudorheumatoid nodule (deep granuloma annulare) of childhood: clinicopathologic features of twenty patients. Pediatr Dermatol. 1994;11:6-9.
- Patterson JW. Rheumatoid nodule and subcutaneous granuloma annulare: a comparative histologic study. Am J Dermatopathol. 1988;10:1-8.
- Weedon D. Granuloma annulare. Skin Pathology. Edinburgh, Scotland: Churchill-Livingstone; 1997:167-170.
- Sayah A, English JC 3rd. Rheumatoid arthritis: a review of the cutaneous manifestations. J Am Acad Dermatol. 2005;53:191-209.
- Highton J, Hessian PA, Stamp L. The rheumatoid nodule: peripheral or central to rheumatoid arthritis? Rheumatology (Oxford). 2007;46:1385-1387.
- Turesson C, Jacobsson LT. Epidemiology of extra-articular manifestations in rheumatoid arthritis. Scand J Rheumatol. 2004;33:65-72.
- Erfurt-Berge C, Dissemond J, Schwede K, et al. Updated results of 100 patients on clinical features and therapeutic options in necrobiosis lipoidica in a retrospective multicenter study. Eur J Dermatol. 2015;25:595-601.
- Kota SK, Jammula S, Kota SK, et al. Necrobiosis lipoidica diabeticorum: a case-based review of literature. Indian J Endocrinol Metab. 2012;16:614-620.
- Alegre VA, Winkelmann RK. A new histopathologic feature of necrobiosis lipoidica diabeticorum: lymphoid nodules. J Cutan Pathol. 1988;15:75-77.
- Armah HB, Parwani AV. Epithelioid sarcoma. Arch Pathol Lab Med. 2009;133:814-819.
- Shidham V, Chivukula M, Basir Z, et al. Evaluation of crystals in formalin-fixed, paraffin-embedded tissue sections for the differential diagnosis pseudogout, gout, and tumoral calcinosis. Mod Pathol. 2001;14:806-810.
- Felner EI, Steinberg JB, Weinberg AG. Subcutaneous granuloma annulare: a review of 47 cases. Pediatrics. 1997;100:965-967.
- Requena L, Fernández-Figueras MT. Subcutaneous granuloma annulare. Semin Cutan Med Surg. 2007;26:96-99.
- Taranu T, Grigorovici M, Constantin M, et al. Subcutaneous granuloma annulare. Acta Dermatovenerol Croat. 2017;25:292-294.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1644-1663.
- Mills A, Chetty R. Auricular granuloma annulare: a consequence of trauma? Am J Dermatopathol. 1992;14:431-433.
- Muhlbauer JE. Granuloma annulare. J Am Acad Dermatol. 1980;3:217-230.
- Buechner SA, Winkelmann RK, Banks PM. Identification of T-cell subpopulations in granuloma annulare. Arch Dermatol. 1983;119:125-128.
- Wells RS, Smith MA. The natural history of granuloma annulare. Br J Dermatol. 1963;75:199.
- Davids JR, Kolman BH, Billman GF, et al. Subcutaneous granuloma annulare: recognition and treatment. J Pediatr Orthop. 1993;13:582-586.
- Evans MJ, Blessing K, Gray ES. Pseudorheumatoid nodule (deep granuloma annulare) of childhood: clinicopathologic features of twenty patients. Pediatr Dermatol. 1994;11:6-9.
- Patterson JW. Rheumatoid nodule and subcutaneous granuloma annulare: a comparative histologic study. Am J Dermatopathol. 1988;10:1-8.
- Weedon D. Granuloma annulare. Skin Pathology. Edinburgh, Scotland: Churchill-Livingstone; 1997:167-170.
- Sayah A, English JC 3rd. Rheumatoid arthritis: a review of the cutaneous manifestations. J Am Acad Dermatol. 2005;53:191-209.
- Highton J, Hessian PA, Stamp L. The rheumatoid nodule: peripheral or central to rheumatoid arthritis? Rheumatology (Oxford). 2007;46:1385-1387.
- Turesson C, Jacobsson LT. Epidemiology of extra-articular manifestations in rheumatoid arthritis. Scand J Rheumatol. 2004;33:65-72.
- Erfurt-Berge C, Dissemond J, Schwede K, et al. Updated results of 100 patients on clinical features and therapeutic options in necrobiosis lipoidica in a retrospective multicenter study. Eur J Dermatol. 2015;25:595-601.
- Kota SK, Jammula S, Kota SK, et al. Necrobiosis lipoidica diabeticorum: a case-based review of literature. Indian J Endocrinol Metab. 2012;16:614-620.
- Alegre VA, Winkelmann RK. A new histopathologic feature of necrobiosis lipoidica diabeticorum: lymphoid nodules. J Cutan Pathol. 1988;15:75-77.
- Armah HB, Parwani AV. Epithelioid sarcoma. Arch Pathol Lab Med. 2009;133:814-819.
- Shidham V, Chivukula M, Basir Z, et al. Evaluation of crystals in formalin-fixed, paraffin-embedded tissue sections for the differential diagnosis pseudogout, gout, and tumoral calcinosis. Mod Pathol. 2001;14:806-810.