User login
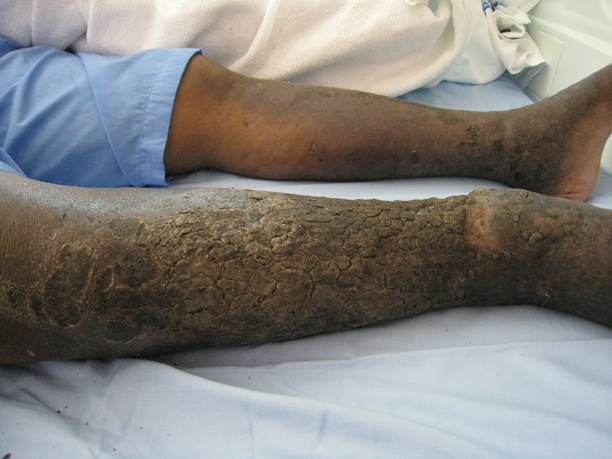
Rapidly Enlarging Noduloulcerative Lesions
The Diagnosis: Lues Maligna
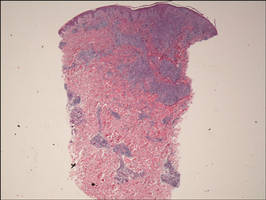
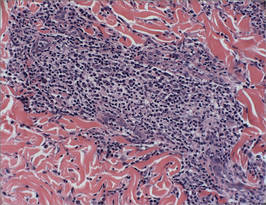
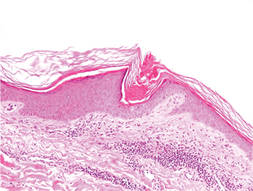
Biopsy revealed dense nodular aggregates of lymphocytes, histiocytes, and abundant plasma cells in both the superficial and deep dermis (Figure 1). There were perivascular and periadnexal aggregates of lymphocytes, histiocytes, and numerous plasma cells (Figure 2). Special stains for organisms, including Warthin-Starry silver, Giemsa, acid-fast bacilli, Gomori methenamine-silver, and Brown-Brenn stains were negative. Immunoperoxidase stain for Treponema pallidum also was negative. The patient’s rapid plasma reagin titer at the time of the fourth biopsy was 1:256, and appropriate treatment with penicillin resulted in complete clearance of the lesions in 3 to 4 weeks.
|
| Figure 1. Superficial and deep perivascular and periadnexal lymphohistiocytic infiltrate (H&E, original magnification ×2). |
|
| Figure 2. Perivascular and periadnexal aggregates of lymphocytes, histiocytes, and numerous plasma cells (H&E, original magnification ×20). |
Syphilis is caused by T pallidum. Three stages typically are identified in immunocompetent hosts: primary, secondary, and tertiary syphilis. Immunocompromised patients with human immunodeficiency virus (HIV) infection may have unusual presentations.
Lues maligna is used to describe a rare noduloulcerative form of secondary syphilis.1 It was first described in 18592 and has been associated with other disorders such as diabetes mellitus3 and chronic alcoholism.4 Patients usually are gravely ill and develop polymorphic ulcerating lesions. Facial and scalp involvement are common, but patients typically do not have palmoplantar involvement in conventional presentations of secondary syphilis.
A scanning view of a punch biopsy from our patient revealed irregular acanthosis of the epidermis with long and thin rete pegs, a bandlike infiltrate at the dermoepidermal junction, and a dense superficial and deep perivascular and periadnexal infiltrate. The histologic differential diagnosis includes pyoderma gangrenosum, vasculitis, lymphoma, leishmaniasis, leprosy, yaws, and mycobacterial or fungal infections.
The Centers for Disease Control and Prevention recommends screening of all HIV-positive indivi-duals for syphilis, and all sexually active individuals with syphilis should be screened for HIV.5 If clinical examination and findings suggest syphilis in the presence of negative serologic testing, then direct fluorescence assay for T pallidum staining of lesions, exudates or biopsy, or dark-field microscopic examination should be performed. In our case, dark-field microscopy was not performed and serologic tests were negative at presentation. Silver stains can detect T pallidum in tissue specimens, though detection may not be possible late in the course of disease.6
The morphology and rapid response to treatment confirmed the diagnosis in our patient. The incidence of syphilis in HIV-positive patients has risen substantially in the last 2 decades. This case illustrates an uncommon presentation that is increasing in prevalence.
1. Fisher DA, Chang LW, Tuffanelli DL. Lues maligna: presentation of a case and review of the literature. Arch Dermatol. 1969;99:70-73.
2. Passoni LF, de Menezes JA, Ribeiro SR, et al. Lues maligna in an HIV-infected patient [published online ahead of print March 30, 2005]. Rev Soc Bras Med Trop. 2005;38:181-184.
3. Hofmann UB, Hund M, Bröcker EB, et al. Lues maligna in a female patient with diabetes [in German]. J Dtsch Dermatol Ges. 2005;3:780-782.
4. Bayramgürler D, Bilen N, Yildiz K, et al. Lues maligna in a chronic alcoholic patient. J Dermatol. 2005;32:217-219.
5. Centers for Disease Control. Recommendations for diagnosing and treating syphilis in HIV-infected patients. MMWR Morb Mortal Wkly Rep. 1988;37:600-602.
6. Mannara GM. Bilateral secondary syphilis of the tonsil. J Laryngol Otol. 1999;113:1125-1127.
The Diagnosis: Lues Maligna
Biopsy revealed dense nodular aggregates of lymphocytes, histiocytes, and abundant plasma cells in both the superficial and deep dermis (Figure 1). There were perivascular and periadnexal aggregates of lymphocytes, histiocytes, and numerous plasma cells (Figure 2). Special stains for organisms, including Warthin-Starry silver, Giemsa, acid-fast bacilli, Gomori methenamine-silver, and Brown-Brenn stains were negative. Immunoperoxidase stain for Treponema pallidum also was negative. The patient’s rapid plasma reagin titer at the time of the fourth biopsy was 1:256, and appropriate treatment with penicillin resulted in complete clearance of the lesions in 3 to 4 weeks.
|
|
| Figure 1. Superficial and deep perivascular and periadnexal lymphohistiocytic infiltrate (H&E, original magnification ×2). |
|
|
| Figure 2. Perivascular and periadnexal aggregates of lymphocytes, histiocytes, and numerous plasma cells (H&E, original magnification ×20). |
Syphilis is caused by T pallidum. Three stages typically are identified in immunocompetent hosts: primary, secondary, and tertiary syphilis. Immunocompromised patients with human immunodeficiency virus (HIV) infection may have unusual presentations.
Lues maligna is used to describe a rare noduloulcerative form of secondary syphilis.1 It was first described in 18592 and has been associated with other disorders such as diabetes mellitus3 and chronic alcoholism.4 Patients usually are gravely ill and develop polymorphic ulcerating lesions. Facial and scalp involvement are common, but patients typically do not have palmoplantar involvement in conventional presentations of secondary syphilis.
A scanning view of a punch biopsy from our patient revealed irregular acanthosis of the epidermis with long and thin rete pegs, a bandlike infiltrate at the dermoepidermal junction, and a dense superficial and deep perivascular and periadnexal infiltrate. The histologic differential diagnosis includes pyoderma gangrenosum, vasculitis, lymphoma, leishmaniasis, leprosy, yaws, and mycobacterial or fungal infections.
The Centers for Disease Control and Prevention recommends screening of all HIV-positive indivi-duals for syphilis, and all sexually active individuals with syphilis should be screened for HIV.5 If clinical examination and findings suggest syphilis in the presence of negative serologic testing, then direct fluorescence assay for T pallidum staining of lesions, exudates or biopsy, or dark-field microscopic examination should be performed. In our case, dark-field microscopy was not performed and serologic tests were negative at presentation. Silver stains can detect T pallidum in tissue specimens, though detection may not be possible late in the course of disease.6
The morphology and rapid response to treatment confirmed the diagnosis in our patient. The incidence of syphilis in HIV-positive patients has risen substantially in the last 2 decades. This case illustrates an uncommon presentation that is increasing in prevalence.
The Diagnosis: Lues Maligna
Biopsy revealed dense nodular aggregates of lymphocytes, histiocytes, and abundant plasma cells in both the superficial and deep dermis (Figure 1). There were perivascular and periadnexal aggregates of lymphocytes, histiocytes, and numerous plasma cells (Figure 2). Special stains for organisms, including Warthin-Starry silver, Giemsa, acid-fast bacilli, Gomori methenamine-silver, and Brown-Brenn stains were negative. Immunoperoxidase stain for Treponema pallidum also was negative. The patient’s rapid plasma reagin titer at the time of the fourth biopsy was 1:256, and appropriate treatment with penicillin resulted in complete clearance of the lesions in 3 to 4 weeks.
|
|
| Figure 1. Superficial and deep perivascular and periadnexal lymphohistiocytic infiltrate (H&E, original magnification ×2). |
|
|
| Figure 2. Perivascular and periadnexal aggregates of lymphocytes, histiocytes, and numerous plasma cells (H&E, original magnification ×20). |
Syphilis is caused by T pallidum. Three stages typically are identified in immunocompetent hosts: primary, secondary, and tertiary syphilis. Immunocompromised patients with human immunodeficiency virus (HIV) infection may have unusual presentations.
Lues maligna is used to describe a rare noduloulcerative form of secondary syphilis.1 It was first described in 18592 and has been associated with other disorders such as diabetes mellitus3 and chronic alcoholism.4 Patients usually are gravely ill and develop polymorphic ulcerating lesions. Facial and scalp involvement are common, but patients typically do not have palmoplantar involvement in conventional presentations of secondary syphilis.
A scanning view of a punch biopsy from our patient revealed irregular acanthosis of the epidermis with long and thin rete pegs, a bandlike infiltrate at the dermoepidermal junction, and a dense superficial and deep perivascular and periadnexal infiltrate. The histologic differential diagnosis includes pyoderma gangrenosum, vasculitis, lymphoma, leishmaniasis, leprosy, yaws, and mycobacterial or fungal infections.
The Centers for Disease Control and Prevention recommends screening of all HIV-positive indivi-duals for syphilis, and all sexually active individuals with syphilis should be screened for HIV.5 If clinical examination and findings suggest syphilis in the presence of negative serologic testing, then direct fluorescence assay for T pallidum staining of lesions, exudates or biopsy, or dark-field microscopic examination should be performed. In our case, dark-field microscopy was not performed and serologic tests were negative at presentation. Silver stains can detect T pallidum in tissue specimens, though detection may not be possible late in the course of disease.6
The morphology and rapid response to treatment confirmed the diagnosis in our patient. The incidence of syphilis in HIV-positive patients has risen substantially in the last 2 decades. This case illustrates an uncommon presentation that is increasing in prevalence.
1. Fisher DA, Chang LW, Tuffanelli DL. Lues maligna: presentation of a case and review of the literature. Arch Dermatol. 1969;99:70-73.
2. Passoni LF, de Menezes JA, Ribeiro SR, et al. Lues maligna in an HIV-infected patient [published online ahead of print March 30, 2005]. Rev Soc Bras Med Trop. 2005;38:181-184.
3. Hofmann UB, Hund M, Bröcker EB, et al. Lues maligna in a female patient with diabetes [in German]. J Dtsch Dermatol Ges. 2005;3:780-782.
4. Bayramgürler D, Bilen N, Yildiz K, et al. Lues maligna in a chronic alcoholic patient. J Dermatol. 2005;32:217-219.
5. Centers for Disease Control. Recommendations for diagnosing and treating syphilis in HIV-infected patients. MMWR Morb Mortal Wkly Rep. 1988;37:600-602.
6. Mannara GM. Bilateral secondary syphilis of the tonsil. J Laryngol Otol. 1999;113:1125-1127.
1. Fisher DA, Chang LW, Tuffanelli DL. Lues maligna: presentation of a case and review of the literature. Arch Dermatol. 1969;99:70-73.
2. Passoni LF, de Menezes JA, Ribeiro SR, et al. Lues maligna in an HIV-infected patient [published online ahead of print March 30, 2005]. Rev Soc Bras Med Trop. 2005;38:181-184.
3. Hofmann UB, Hund M, Bröcker EB, et al. Lues maligna in a female patient with diabetes [in German]. J Dtsch Dermatol Ges. 2005;3:780-782.
4. Bayramgürler D, Bilen N, Yildiz K, et al. Lues maligna in a chronic alcoholic patient. J Dermatol. 2005;32:217-219.
5. Centers for Disease Control. Recommendations for diagnosing and treating syphilis in HIV-infected patients. MMWR Morb Mortal Wkly Rep. 1988;37:600-602.
6. Mannara GM. Bilateral secondary syphilis of the tonsil. J Laryngol Otol. 1999;113:1125-1127.
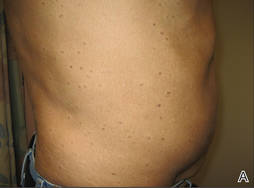
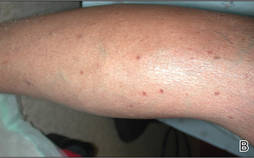
A 43-year-old man presented with a rapidly enlarging ulcerated nodule on the right ankle with a necrotic and crusted center. He also had multiple red-brown papules on the trunk and extremities. Some of these lesions had central erosions, while others had surface scale. He was known to be human immunodeficiency virus positive but had no lymphadenopathy. The CD4+ lymphocyte count was 153 cells/mm3 (reference range, 400–1600 cells/mm3) and he was on highly active antiretroviral therapy.
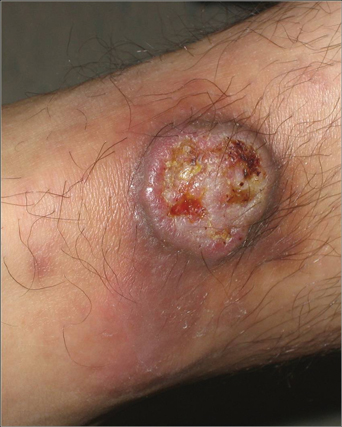
What Is Your Diagnosis? Verruciform Xanthoma

An 82-year-old man presented for evaluation of a solitary asymptomatic, pink, velvety, exophytic nodule on the scrotum that had been present for approximately 1 month. His medical history was notable for Parkinson disease, hypertension, and hyperlipidemia. His current medications included carbidopa-levodopa, lisinopril-hydrochlorothiazide, amlodipine, and simvastatin.
The Diagnosis: Verruciform Xanthoma
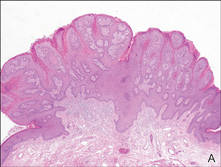
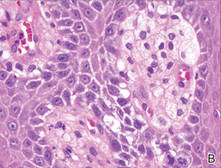
Our patient presented with a solitary asymptomatic, pink, velvety, exophytic nodule on the scrotum that had been present for approximately 1 month (Figure 1). Biopsy of the specimen showed a lesion with a verrucalike configuration. There was hyperkeratosis, focal parakeratosis, and verrucous acanthosis without atypia. The papillary dermis was filled with large histiocytes with foamy cytoplasm (xanthoma cells). A few lymphocytes, neutrophils, eosinophils, and plasma cells also were present (Figure 2).

|
| Figure 1. A solitary pink, velvety, exophytic nodule on the scrotum. |
|
|
| Figure 2. Biopsy showed a lesion with a verrucalike configuration (A)(H&E, original magnification ×20). The papillary dermis was filled with xanthoma cells (B)(H&E, original magnification ×400). |
Clinically, the differential diagnosis of this lesion includes verruciform xanthoma (VX), verruca vulgaris, condyloma, verrucous carcinoma, and squamous cell carcinoma; however, the histologic finding of xanthoma cells in the papillary dermis helped to confirm the diagnosis of VX. Specimens must be evaluated closely to rule out associated or concurrent disease processes.
Verruciform xanthoma is a rare lesion first described by Shafer1 in 1971. It usually appears as a solitary lesion most commonly presenting on the oral mucosa. In 2003, a worldwide survey of the literature profiled a total of 282 oral and 46 extraoral VX lesions.2 Extraoral cutaneous lesions are less common and usually present in the anogenital region, including the penis, scrotum, vulva, and anus. Isolated cases have been reported on the extremities and other sites.3,4 The lesions often are described as pale yellow to pink verrucous papules or plaques. Typical histologic features include hyperkeratosis, focal parakeratosis, acanthosis without atypia, and xanthoma cells in the papillary dermis that usually do not extend beyond the rete ridges.3,5 A mild inflammatory infiltrate is common with some lymphocytes, plasma cells, neutrophils, and eosinophils.3
Several reports have noted that VX does not occur in the presence of any lipid abnormalities or detectable human papillomavirus.3,6-9 Cases may occur sporadically without any apparent associated disease but also have been seen in the setting of atypia, malignancy, and inflammatory processes. There are at least 2 reports of VX lesions occurring with squamous cell carcinoma.5,10 Verruciform xanthoma also has presented in the setting of epidermal nevi, actinic keratosis, seborrheic keratosis, lichen planus, lichen sclerosus, pemphigus, discoid lupus erythematosus, lymphedema, CHILD (congenital hemidysplasia with ichthyosiform erythroderma and limb defects) syndrome, and recessive dystrophic epidermolysis bullosa.6,7 The association of VX with these other diseases has led many authors to believe that it is a reactive phenomenon to chronic inflammation or cutaneous trauma in which degenerating keratinocytes are phagocytosed by dermal macrophages that then become lipid-laden foam cells. Rapid epidermal growth may be a result of cytokines released during this process.4
A Japanese report of several cases of VX with lesions on the scrotum suggested that this rare finding may be caused by chronic pressure associated with the Japanese tradition of sitting on the floor.11 One case occurred with lesions on the penis and perineum 4 years after an incidence of necrotizing fasciitis and skin grafting in the same area. The authors suggested that the severe cutaneous trauma predisposed the patient to forming VX lesions.4 There also have been at least 6 cases of VX reported in immunocompromised patients, specifically in the setting of bone marrow transplantation, human immunodeficiency virus infection, common variable immunodeficiency, and renal transplantation.8 The proportion of immunocompromised cases is greater than expected considering the relatively low prevalence of cutaneous
VX lesions. A possible explanation that is consistent with the proposed reactive mechanism for VX formation is that immunocompromised patients may have a lower number of epidermal Langerhans cells, which results in decreased removal of degenerated keratinocytes and increased dermal macrophage phagocytosis.8
Management of VX lesions usually consists of simple excision. Intraoral excisions have been well documented as curative with rare recurrence2,9; however, there are limited reports in the literature documenting successful excision of cutaneous lesions. Despite the availability of several treatment modalities (eg, wire loop electrosection, pulsed dye laser, radiation therapy, chloroxylenol scrub, wide surgical resection), lesions may reoccur.9 Because VX lesions have occurred in the setting of other cutaneous conditions, patients should be assessed for other concurrent diseases. Biopsy specimens should be carefully analyzed, as a few VX lesions have been associated with epidermal atypia and invasive squamous cell carcinoma.5,10
1. Shafer WG. Verruciform xanthoma. Oral Surg Oral Med Oral Pathol. 1971;31:784-789.
2. Philipsen HP, Reichart PA, Takata T, et al. Verruciform xanthoma—biological profile of 282 oral lesions based on a literature survey with nine new cases from Japan. Oral Oncol. 2003;39:325-336.
3. Sopena J, Gamo R, Iglesias L, et al. Disseminated verruciform xanthoma. Br J Dermatol. 2004;151:717-719.
4. Cumberland L, Dana A, Resh B, et al. Verruciform xanthoma in the setting of cutaneous trauma and chronic inflammation: report of a patient and a brief review of the literature. J Cutan Pathol. 2009;37:895-900.
5. Mannes KD, Dekle CL, Requena L, et al. Verruciform xanthoma associated with squamous cell carcinoma. Am J Dermatopathol. 1999;21:66-69.
6. Orpin SD, Scott IC, Rajaratnam R, et al. A rare case of recessive dystrophic epidermolysis bullosa and verruciform xanthoma. Clin Exp Dermatol. 2009;34:49-51.
7. Wu Y, Hsiao P, Lin Y. Verruciform xanthoma-like phenomenon in seborrheic keratosis. J Cutan Pathol. 2006;33:373-377.
8. Kanitakis J, Euvrard S, Butnaru AC, et al. Verruciform xanthoma of the scrotum in a renal transplant patient. Br J Dermatol. 2004;150:161-163.
9. Connolly SB, Lewis EJ, Lindholm JS, et al. Management of cutaneous verruciform xanthoma. J Am Acad Dermatol. 2000;42:343-347.
10. Takiwaki H, Yokota M, Ahsan K, et al. Squamous cell carcinoma associated with verruciform xanthoma of the penis. Am J Dermatopathol. 1996;18:551-554.
11. Nakamura S, Kanamori S, Nakayama K, et al. Verruciform xanthoma on the scrotum. J Dermatol. 1989;16:397-401.
An 82-year-old man presented for evaluation of a solitary asymptomatic, pink, velvety, exophytic nodule on the scrotum that had been present for approximately 1 month. His medical history was notable for Parkinson disease, hypertension, and hyperlipidemia. His current medications included carbidopa-levodopa, lisinopril-hydrochlorothiazide, amlodipine, and simvastatin.
The Diagnosis: Verruciform Xanthoma
Our patient presented with a solitary asymptomatic, pink, velvety, exophytic nodule on the scrotum that had been present for approximately 1 month (Figure 1). Biopsy of the specimen showed a lesion with a verrucalike configuration. There was hyperkeratosis, focal parakeratosis, and verrucous acanthosis without atypia. The papillary dermis was filled with large histiocytes with foamy cytoplasm (xanthoma cells). A few lymphocytes, neutrophils, eosinophils, and plasma cells also were present (Figure 2).
|
|
| Figure 1. A solitary pink, velvety, exophytic nodule on the scrotum. |
|
|
|
|
| Figure 2. Biopsy showed a lesion with a verrucalike configuration (A)(H&E, original magnification ×20). The papillary dermis was filled with xanthoma cells (B)(H&E, original magnification ×400). |
Clinically, the differential diagnosis of this lesion includes verruciform xanthoma (VX), verruca vulgaris, condyloma, verrucous carcinoma, and squamous cell carcinoma; however, the histologic finding of xanthoma cells in the papillary dermis helped to confirm the diagnosis of VX. Specimens must be evaluated closely to rule out associated or concurrent disease processes.
Verruciform xanthoma is a rare lesion first described by Shafer1 in 1971. It usually appears as a solitary lesion most commonly presenting on the oral mucosa. In 2003, a worldwide survey of the literature profiled a total of 282 oral and 46 extraoral VX lesions.2 Extraoral cutaneous lesions are less common and usually present in the anogenital region, including the penis, scrotum, vulva, and anus. Isolated cases have been reported on the extremities and other sites.3,4 The lesions often are described as pale yellow to pink verrucous papules or plaques. Typical histologic features include hyperkeratosis, focal parakeratosis, acanthosis without atypia, and xanthoma cells in the papillary dermis that usually do not extend beyond the rete ridges.3,5 A mild inflammatory infiltrate is common with some lymphocytes, plasma cells, neutrophils, and eosinophils.3
Several reports have noted that VX does not occur in the presence of any lipid abnormalities or detectable human papillomavirus.3,6-9 Cases may occur sporadically without any apparent associated disease but also have been seen in the setting of atypia, malignancy, and inflammatory processes. There are at least 2 reports of VX lesions occurring with squamous cell carcinoma.5,10 Verruciform xanthoma also has presented in the setting of epidermal nevi, actinic keratosis, seborrheic keratosis, lichen planus, lichen sclerosus, pemphigus, discoid lupus erythematosus, lymphedema, CHILD (congenital hemidysplasia with ichthyosiform erythroderma and limb defects) syndrome, and recessive dystrophic epidermolysis bullosa.6,7 The association of VX with these other diseases has led many authors to believe that it is a reactive phenomenon to chronic inflammation or cutaneous trauma in which degenerating keratinocytes are phagocytosed by dermal macrophages that then become lipid-laden foam cells. Rapid epidermal growth may be a result of cytokines released during this process.4
A Japanese report of several cases of VX with lesions on the scrotum suggested that this rare finding may be caused by chronic pressure associated with the Japanese tradition of sitting on the floor.11 One case occurred with lesions on the penis and perineum 4 years after an incidence of necrotizing fasciitis and skin grafting in the same area. The authors suggested that the severe cutaneous trauma predisposed the patient to forming VX lesions.4 There also have been at least 6 cases of VX reported in immunocompromised patients, specifically in the setting of bone marrow transplantation, human immunodeficiency virus infection, common variable immunodeficiency, and renal transplantation.8 The proportion of immunocompromised cases is greater than expected considering the relatively low prevalence of cutaneous
VX lesions. A possible explanation that is consistent with the proposed reactive mechanism for VX formation is that immunocompromised patients may have a lower number of epidermal Langerhans cells, which results in decreased removal of degenerated keratinocytes and increased dermal macrophage phagocytosis.8
Management of VX lesions usually consists of simple excision. Intraoral excisions have been well documented as curative with rare recurrence2,9; however, there are limited reports in the literature documenting successful excision of cutaneous lesions. Despite the availability of several treatment modalities (eg, wire loop electrosection, pulsed dye laser, radiation therapy, chloroxylenol scrub, wide surgical resection), lesions may reoccur.9 Because VX lesions have occurred in the setting of other cutaneous conditions, patients should be assessed for other concurrent diseases. Biopsy specimens should be carefully analyzed, as a few VX lesions have been associated with epidermal atypia and invasive squamous cell carcinoma.5,10
An 82-year-old man presented for evaluation of a solitary asymptomatic, pink, velvety, exophytic nodule on the scrotum that had been present for approximately 1 month. His medical history was notable for Parkinson disease, hypertension, and hyperlipidemia. His current medications included carbidopa-levodopa, lisinopril-hydrochlorothiazide, amlodipine, and simvastatin.
The Diagnosis: Verruciform Xanthoma
Our patient presented with a solitary asymptomatic, pink, velvety, exophytic nodule on the scrotum that had been present for approximately 1 month (Figure 1). Biopsy of the specimen showed a lesion with a verrucalike configuration. There was hyperkeratosis, focal parakeratosis, and verrucous acanthosis without atypia. The papillary dermis was filled with large histiocytes with foamy cytoplasm (xanthoma cells). A few lymphocytes, neutrophils, eosinophils, and plasma cells also were present (Figure 2).
|
|
| Figure 1. A solitary pink, velvety, exophytic nodule on the scrotum. |
|
|
|
|
| Figure 2. Biopsy showed a lesion with a verrucalike configuration (A)(H&E, original magnification ×20). The papillary dermis was filled with xanthoma cells (B)(H&E, original magnification ×400). |
Clinically, the differential diagnosis of this lesion includes verruciform xanthoma (VX), verruca vulgaris, condyloma, verrucous carcinoma, and squamous cell carcinoma; however, the histologic finding of xanthoma cells in the papillary dermis helped to confirm the diagnosis of VX. Specimens must be evaluated closely to rule out associated or concurrent disease processes.
Verruciform xanthoma is a rare lesion first described by Shafer1 in 1971. It usually appears as a solitary lesion most commonly presenting on the oral mucosa. In 2003, a worldwide survey of the literature profiled a total of 282 oral and 46 extraoral VX lesions.2 Extraoral cutaneous lesions are less common and usually present in the anogenital region, including the penis, scrotum, vulva, and anus. Isolated cases have been reported on the extremities and other sites.3,4 The lesions often are described as pale yellow to pink verrucous papules or plaques. Typical histologic features include hyperkeratosis, focal parakeratosis, acanthosis without atypia, and xanthoma cells in the papillary dermis that usually do not extend beyond the rete ridges.3,5 A mild inflammatory infiltrate is common with some lymphocytes, plasma cells, neutrophils, and eosinophils.3
Several reports have noted that VX does not occur in the presence of any lipid abnormalities or detectable human papillomavirus.3,6-9 Cases may occur sporadically without any apparent associated disease but also have been seen in the setting of atypia, malignancy, and inflammatory processes. There are at least 2 reports of VX lesions occurring with squamous cell carcinoma.5,10 Verruciform xanthoma also has presented in the setting of epidermal nevi, actinic keratosis, seborrheic keratosis, lichen planus, lichen sclerosus, pemphigus, discoid lupus erythematosus, lymphedema, CHILD (congenital hemidysplasia with ichthyosiform erythroderma and limb defects) syndrome, and recessive dystrophic epidermolysis bullosa.6,7 The association of VX with these other diseases has led many authors to believe that it is a reactive phenomenon to chronic inflammation or cutaneous trauma in which degenerating keratinocytes are phagocytosed by dermal macrophages that then become lipid-laden foam cells. Rapid epidermal growth may be a result of cytokines released during this process.4
A Japanese report of several cases of VX with lesions on the scrotum suggested that this rare finding may be caused by chronic pressure associated with the Japanese tradition of sitting on the floor.11 One case occurred with lesions on the penis and perineum 4 years after an incidence of necrotizing fasciitis and skin grafting in the same area. The authors suggested that the severe cutaneous trauma predisposed the patient to forming VX lesions.4 There also have been at least 6 cases of VX reported in immunocompromised patients, specifically in the setting of bone marrow transplantation, human immunodeficiency virus infection, common variable immunodeficiency, and renal transplantation.8 The proportion of immunocompromised cases is greater than expected considering the relatively low prevalence of cutaneous
VX lesions. A possible explanation that is consistent with the proposed reactive mechanism for VX formation is that immunocompromised patients may have a lower number of epidermal Langerhans cells, which results in decreased removal of degenerated keratinocytes and increased dermal macrophage phagocytosis.8
Management of VX lesions usually consists of simple excision. Intraoral excisions have been well documented as curative with rare recurrence2,9; however, there are limited reports in the literature documenting successful excision of cutaneous lesions. Despite the availability of several treatment modalities (eg, wire loop electrosection, pulsed dye laser, radiation therapy, chloroxylenol scrub, wide surgical resection), lesions may reoccur.9 Because VX lesions have occurred in the setting of other cutaneous conditions, patients should be assessed for other concurrent diseases. Biopsy specimens should be carefully analyzed, as a few VX lesions have been associated with epidermal atypia and invasive squamous cell carcinoma.5,10
1. Shafer WG. Verruciform xanthoma. Oral Surg Oral Med Oral Pathol. 1971;31:784-789.
2. Philipsen HP, Reichart PA, Takata T, et al. Verruciform xanthoma—biological profile of 282 oral lesions based on a literature survey with nine new cases from Japan. Oral Oncol. 2003;39:325-336.
3. Sopena J, Gamo R, Iglesias L, et al. Disseminated verruciform xanthoma. Br J Dermatol. 2004;151:717-719.
4. Cumberland L, Dana A, Resh B, et al. Verruciform xanthoma in the setting of cutaneous trauma and chronic inflammation: report of a patient and a brief review of the literature. J Cutan Pathol. 2009;37:895-900.
5. Mannes KD, Dekle CL, Requena L, et al. Verruciform xanthoma associated with squamous cell carcinoma. Am J Dermatopathol. 1999;21:66-69.
6. Orpin SD, Scott IC, Rajaratnam R, et al. A rare case of recessive dystrophic epidermolysis bullosa and verruciform xanthoma. Clin Exp Dermatol. 2009;34:49-51.
7. Wu Y, Hsiao P, Lin Y. Verruciform xanthoma-like phenomenon in seborrheic keratosis. J Cutan Pathol. 2006;33:373-377.
8. Kanitakis J, Euvrard S, Butnaru AC, et al. Verruciform xanthoma of the scrotum in a renal transplant patient. Br J Dermatol. 2004;150:161-163.
9. Connolly SB, Lewis EJ, Lindholm JS, et al. Management of cutaneous verruciform xanthoma. J Am Acad Dermatol. 2000;42:343-347.
10. Takiwaki H, Yokota M, Ahsan K, et al. Squamous cell carcinoma associated with verruciform xanthoma of the penis. Am J Dermatopathol. 1996;18:551-554.
11. Nakamura S, Kanamori S, Nakayama K, et al. Verruciform xanthoma on the scrotum. J Dermatol. 1989;16:397-401.
1. Shafer WG. Verruciform xanthoma. Oral Surg Oral Med Oral Pathol. 1971;31:784-789.
2. Philipsen HP, Reichart PA, Takata T, et al. Verruciform xanthoma—biological profile of 282 oral lesions based on a literature survey with nine new cases from Japan. Oral Oncol. 2003;39:325-336.
3. Sopena J, Gamo R, Iglesias L, et al. Disseminated verruciform xanthoma. Br J Dermatol. 2004;151:717-719.
4. Cumberland L, Dana A, Resh B, et al. Verruciform xanthoma in the setting of cutaneous trauma and chronic inflammation: report of a patient and a brief review of the literature. J Cutan Pathol. 2009;37:895-900.
5. Mannes KD, Dekle CL, Requena L, et al. Verruciform xanthoma associated with squamous cell carcinoma. Am J Dermatopathol. 1999;21:66-69.
6. Orpin SD, Scott IC, Rajaratnam R, et al. A rare case of recessive dystrophic epidermolysis bullosa and verruciform xanthoma. Clin Exp Dermatol. 2009;34:49-51.
7. Wu Y, Hsiao P, Lin Y. Verruciform xanthoma-like phenomenon in seborrheic keratosis. J Cutan Pathol. 2006;33:373-377.
8. Kanitakis J, Euvrard S, Butnaru AC, et al. Verruciform xanthoma of the scrotum in a renal transplant patient. Br J Dermatol. 2004;150:161-163.
9. Connolly SB, Lewis EJ, Lindholm JS, et al. Management of cutaneous verruciform xanthoma. J Am Acad Dermatol. 2000;42:343-347.
10. Takiwaki H, Yokota M, Ahsan K, et al. Squamous cell carcinoma associated with verruciform xanthoma of the penis. Am J Dermatopathol. 1996;18:551-554.
11. Nakamura S, Kanamori S, Nakayama K, et al. Verruciform xanthoma on the scrotum. J Dermatol. 1989;16:397-401.

Yellowish Papulonodular Periorbital Eruption
The Diagnosis: Adult-Onset Xanthogranuloma
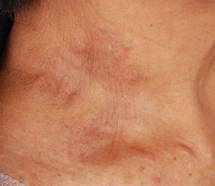
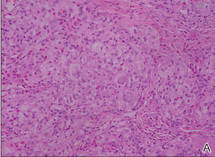
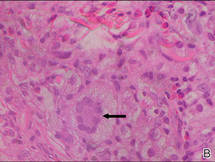
Biopsies of the lesions on the neck (Figure 1) and back were performed. Histologic analyses revealed a diffuse dermatitis consisting of foamy histiocytes admixed with a few Touton-type giant cells in the dermis (Figure 2), which was associated with an inflammatory infiltrate of eosinophils and lymphocytes. Laboratory investigations revealed mild thrombocytopenia with a platelet count of 134×109/L (reference range, 140–440×109/L). Other investigations including biochemistry, lipid, serum electrophoresis, and chest radiogram were normal. A bone marrow trephine biopsy and flow cytometry were performed and were normal. Magnetic resonance imaging revealed periorbital soft-tissue masses that did not extend into the orbits.
|
| Figure 1. Firm plaques with a yellowish tinge over the side of the neck |
|
|
| Figure 2. A dense infiltrate of foamy histiocytes in the dermis (A) associated with a Touton-type giant cell (arrow) and an inflammatory infiltrate consisting of eosinophils and lymphocytes (B)(both H&E, original magnifications ×200 and ×400). |
Adult-onset xanthogranuloma (AXG) is a rare disease entity, usually presenting in the third to fourth decades of life. The condition typically presents as a red to yellow-brown nodular cutaneous lesion located on the scalp, face, neck, trunk, or limbs. The presentation typically consists of a solitary lesion, occurring in 70% to 89% of cases,1 but more rarely, as in this case, lesions can be multiple or even disseminated.
Histologically, AXG presents as a dense, well-circumscribed, histiocytic infiltrate consisting of lipophages possessing foamy cytoplasm and giant cells. The presence of histiocytic giant cells differentiates AXG from xanthelasma, a clinical differential diagnosis in this case, and xanthoma. In AXG, there are 4 main types of histiocytes: xanthomatized, spindle shaped, vacuolated, and oncocytic.2 They can be seen in variable proportions, together with different types of giant cells (eg, Touton, foreign body, ground glass, nonspecific). A mixed infiltrate of eosinophils, lymphocytes, plasma cells, and neutrophils also may be seen scattered throughout the lesion.2
Correlating with the clinical and histological features of xanthogranuloma, the firm plaques and nodules represent the dense dermal infiltration of histiocytes that may extend into the subcutis. The lesions demonstrate a time-dependent progression both clinically and histologically. Early lesions are comprised of a dense monomorphous nonlipid histiocytic inflammatory infiltrate, and they clinically appear more erythematous. In mature lesions, as in our patient, the infiltrate is predominantly composed of lipid-laden histiocytes with associated Touton giant cells. They appear more yellowish on clinical presentation.
Adult-onset xanthogranuloma is part of a rare heterogenous group of disorders termed adult orbital xanthogranulomatous disease, which includes 3 other syndromes: necrobiotic xanthogranuloma, adult-onset asthma and periocular xanthogranuloma, and Erdheim-Chester disease.
Necrobiotic xanthogranuloma is clinically characterized by the presence of subcutaneous lesions that ulcerate in approximately 40% to 50% of cases and is histologically characterized by necrobiosis with palisading epithelioid histiocytes. It also is systemically associated with paraproteinemia and multiple myeloma.3
Adult-onset asthma and periocular xanthogranuloma is characterized by yellowish papules and nodules predominantly over the lower eyelids that are histologically comprised of lymphoid follicles with reactive germinal centers. It is associated with asthma, which normally is severe and often occurs almost simultaneously with the periorbital lesions.4
There are no definite diagnostic criteria for Erdheim-Chester disease and the diagnosis is usually based on radiologic findings of osteosclerosis and histopathologic evidence of foamy histiocytic infiltration. Systemic manifestations are common with lymphohistiocytic infiltration of the heart, lungs, pericardium, bones, and intestines. Prognosis is uniformly dismal.
Based on the clinical presentation of a nonulcerative papulonodular eruption and the absence of systemic involvement including asthma, we made the diagnosis of AXG. In view of the heterogeneity among these clinical entities as well as the time-based evolution of the lesions involved, we continued to monitor the patient for 2 years and there was no development of other systemic manifestations and hematologic abnormalities.
In contrast to the more common form of juvenile-onset xanthogranuloma, the adult type is not associated with widespread visceral lesions. Hence extensive screening investigations for systemic disease generally are not necessary. Another difference is that AXG has been associated with hematologic abnormalities, including essential thrombocytosis, chronic lymphocytic leukemia, large B-cell lymphoma, and monoclonal gammopathy.5,6 In our patient, the presence of thrombocytopenia and older age caused us to be concerned about an associated hematologic malignancy; therefore, a bone marrow biopsy and flow cytometry were performed.
Adult-onset xanthogranuloma typically is an asymptomatic and self-healing disease and therefore treatment usually is not required. Spontaneous regression of xanthogranuloma was observed to occur in 54% of cases with a median duration of 22 months,7 though lesions were noted to last as long as 15 years.8 When treatment is necessary, a combination of local and systemic corticosteroids, cytotoxic agents, and radiotherapy have been routinely used. In particular, intralesional corticosteroid therapy has been found to be efficacious in controlling symptoms and signs of AXG affecting the eyelids and orbits while avoiding the systemic side effects of other agents.9
Because our patient’s lesions were longstanding and disfiguring, we opted for active intervention with intralesional triamcinolone, which resulted in only a slight reduction in size of the lesions. The lesions remain largely unchanged in 2 years of follow-up.
1. Chang SE, Cho S, Choi JC, et al. Clinicohistopathologic comparison of adult type and juvenile type xanthogranulomas in Korea. J Dermatol. 2001;28:413-418.
2. Gelmetti C. Non-Langerhans cell histiocytosis. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
3. Mehregan DA, Winkelmann RK. Necrobiotic xanthogranuloma. Arch Dermatol. 1992;128:94-100.
4. Jakobiec FA, Mills MD, Hidayat AA, et al. Periocular xanthogranulomas associated with severe adult-onset asthma. Trans Am Ophthalmol Soc. 1993;91:99-125.
5. Shoo BA, Shinkai K, McCalmont TH, et al. Xanthogranulomas associated with hematologic malignancy in adulthood. J Am Acad Dermatol. 2008;59:488-493.
6. Chiou CC, Wang PN, Yang LC, et al. Disseminated xanthogranulomas associated with adult T-cell leukaemia/lymphoma: a case report and review the association of haematologic malignancies. J Eur Acad Dermatol Venereol. 2007;21:532-535.
7. Robinson HM, Harmon CE, Firminger HI. Multiple lipoidal histiocytomas with regression. Arch Dermatol. 1963;88:660-667.
8. Winkelmann RK. Cutaneous syndromes of non-X histiocytosis: a review of the macrophage-histiocyte diseases of the skin. Arch Dermatol. 1981;117:667-672.
9. Elner VM, Mintz R, Demirci H, et al. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Ophthal Plast Reconstr Surg. 2006;22:36-40.
The Diagnosis: Adult-Onset Xanthogranuloma
Biopsies of the lesions on the neck (Figure 1) and back were performed. Histologic analyses revealed a diffuse dermatitis consisting of foamy histiocytes admixed with a few Touton-type giant cells in the dermis (Figure 2), which was associated with an inflammatory infiltrate of eosinophils and lymphocytes. Laboratory investigations revealed mild thrombocytopenia with a platelet count of 134×109/L (reference range, 140–440×109/L). Other investigations including biochemistry, lipid, serum electrophoresis, and chest radiogram were normal. A bone marrow trephine biopsy and flow cytometry were performed and were normal. Magnetic resonance imaging revealed periorbital soft-tissue masses that did not extend into the orbits.
|
|
| Figure 1. Firm plaques with a yellowish tinge over the side of the neck |
|
|
|
|
| Figure 2. A dense infiltrate of foamy histiocytes in the dermis (A) associated with a Touton-type giant cell (arrow) and an inflammatory infiltrate consisting of eosinophils and lymphocytes (B)(both H&E, original magnifications ×200 and ×400). |
Adult-onset xanthogranuloma (AXG) is a rare disease entity, usually presenting in the third to fourth decades of life. The condition typically presents as a red to yellow-brown nodular cutaneous lesion located on the scalp, face, neck, trunk, or limbs. The presentation typically consists of a solitary lesion, occurring in 70% to 89% of cases,1 but more rarely, as in this case, lesions can be multiple or even disseminated.
Histologically, AXG presents as a dense, well-circumscribed, histiocytic infiltrate consisting of lipophages possessing foamy cytoplasm and giant cells. The presence of histiocytic giant cells differentiates AXG from xanthelasma, a clinical differential diagnosis in this case, and xanthoma. In AXG, there are 4 main types of histiocytes: xanthomatized, spindle shaped, vacuolated, and oncocytic.2 They can be seen in variable proportions, together with different types of giant cells (eg, Touton, foreign body, ground glass, nonspecific). A mixed infiltrate of eosinophils, lymphocytes, plasma cells, and neutrophils also may be seen scattered throughout the lesion.2
Correlating with the clinical and histological features of xanthogranuloma, the firm plaques and nodules represent the dense dermal infiltration of histiocytes that may extend into the subcutis. The lesions demonstrate a time-dependent progression both clinically and histologically. Early lesions are comprised of a dense monomorphous nonlipid histiocytic inflammatory infiltrate, and they clinically appear more erythematous. In mature lesions, as in our patient, the infiltrate is predominantly composed of lipid-laden histiocytes with associated Touton giant cells. They appear more yellowish on clinical presentation.
Adult-onset xanthogranuloma is part of a rare heterogenous group of disorders termed adult orbital xanthogranulomatous disease, which includes 3 other syndromes: necrobiotic xanthogranuloma, adult-onset asthma and periocular xanthogranuloma, and Erdheim-Chester disease.
Necrobiotic xanthogranuloma is clinically characterized by the presence of subcutaneous lesions that ulcerate in approximately 40% to 50% of cases and is histologically characterized by necrobiosis with palisading epithelioid histiocytes. It also is systemically associated with paraproteinemia and multiple myeloma.3
Adult-onset asthma and periocular xanthogranuloma is characterized by yellowish papules and nodules predominantly over the lower eyelids that are histologically comprised of lymphoid follicles with reactive germinal centers. It is associated with asthma, which normally is severe and often occurs almost simultaneously with the periorbital lesions.4
There are no definite diagnostic criteria for Erdheim-Chester disease and the diagnosis is usually based on radiologic findings of osteosclerosis and histopathologic evidence of foamy histiocytic infiltration. Systemic manifestations are common with lymphohistiocytic infiltration of the heart, lungs, pericardium, bones, and intestines. Prognosis is uniformly dismal.
Based on the clinical presentation of a nonulcerative papulonodular eruption and the absence of systemic involvement including asthma, we made the diagnosis of AXG. In view of the heterogeneity among these clinical entities as well as the time-based evolution of the lesions involved, we continued to monitor the patient for 2 years and there was no development of other systemic manifestations and hematologic abnormalities.
In contrast to the more common form of juvenile-onset xanthogranuloma, the adult type is not associated with widespread visceral lesions. Hence extensive screening investigations for systemic disease generally are not necessary. Another difference is that AXG has been associated with hematologic abnormalities, including essential thrombocytosis, chronic lymphocytic leukemia, large B-cell lymphoma, and monoclonal gammopathy.5,6 In our patient, the presence of thrombocytopenia and older age caused us to be concerned about an associated hematologic malignancy; therefore, a bone marrow biopsy and flow cytometry were performed.
Adult-onset xanthogranuloma typically is an asymptomatic and self-healing disease and therefore treatment usually is not required. Spontaneous regression of xanthogranuloma was observed to occur in 54% of cases with a median duration of 22 months,7 though lesions were noted to last as long as 15 years.8 When treatment is necessary, a combination of local and systemic corticosteroids, cytotoxic agents, and radiotherapy have been routinely used. In particular, intralesional corticosteroid therapy has been found to be efficacious in controlling symptoms and signs of AXG affecting the eyelids and orbits while avoiding the systemic side effects of other agents.9
Because our patient’s lesions were longstanding and disfiguring, we opted for active intervention with intralesional triamcinolone, which resulted in only a slight reduction in size of the lesions. The lesions remain largely unchanged in 2 years of follow-up.
The Diagnosis: Adult-Onset Xanthogranuloma
Biopsies of the lesions on the neck (Figure 1) and back were performed. Histologic analyses revealed a diffuse dermatitis consisting of foamy histiocytes admixed with a few Touton-type giant cells in the dermis (Figure 2), which was associated with an inflammatory infiltrate of eosinophils and lymphocytes. Laboratory investigations revealed mild thrombocytopenia with a platelet count of 134×109/L (reference range, 140–440×109/L). Other investigations including biochemistry, lipid, serum electrophoresis, and chest radiogram were normal. A bone marrow trephine biopsy and flow cytometry were performed and were normal. Magnetic resonance imaging revealed periorbital soft-tissue masses that did not extend into the orbits.
|
|
| Figure 1. Firm plaques with a yellowish tinge over the side of the neck |
|
|
|
|
| Figure 2. A dense infiltrate of foamy histiocytes in the dermis (A) associated with a Touton-type giant cell (arrow) and an inflammatory infiltrate consisting of eosinophils and lymphocytes (B)(both H&E, original magnifications ×200 and ×400). |
Adult-onset xanthogranuloma (AXG) is a rare disease entity, usually presenting in the third to fourth decades of life. The condition typically presents as a red to yellow-brown nodular cutaneous lesion located on the scalp, face, neck, trunk, or limbs. The presentation typically consists of a solitary lesion, occurring in 70% to 89% of cases,1 but more rarely, as in this case, lesions can be multiple or even disseminated.
Histologically, AXG presents as a dense, well-circumscribed, histiocytic infiltrate consisting of lipophages possessing foamy cytoplasm and giant cells. The presence of histiocytic giant cells differentiates AXG from xanthelasma, a clinical differential diagnosis in this case, and xanthoma. In AXG, there are 4 main types of histiocytes: xanthomatized, spindle shaped, vacuolated, and oncocytic.2 They can be seen in variable proportions, together with different types of giant cells (eg, Touton, foreign body, ground glass, nonspecific). A mixed infiltrate of eosinophils, lymphocytes, plasma cells, and neutrophils also may be seen scattered throughout the lesion.2
Correlating with the clinical and histological features of xanthogranuloma, the firm plaques and nodules represent the dense dermal infiltration of histiocytes that may extend into the subcutis. The lesions demonstrate a time-dependent progression both clinically and histologically. Early lesions are comprised of a dense monomorphous nonlipid histiocytic inflammatory infiltrate, and they clinically appear more erythematous. In mature lesions, as in our patient, the infiltrate is predominantly composed of lipid-laden histiocytes with associated Touton giant cells. They appear more yellowish on clinical presentation.
Adult-onset xanthogranuloma is part of a rare heterogenous group of disorders termed adult orbital xanthogranulomatous disease, which includes 3 other syndromes: necrobiotic xanthogranuloma, adult-onset asthma and periocular xanthogranuloma, and Erdheim-Chester disease.
Necrobiotic xanthogranuloma is clinically characterized by the presence of subcutaneous lesions that ulcerate in approximately 40% to 50% of cases and is histologically characterized by necrobiosis with palisading epithelioid histiocytes. It also is systemically associated with paraproteinemia and multiple myeloma.3
Adult-onset asthma and periocular xanthogranuloma is characterized by yellowish papules and nodules predominantly over the lower eyelids that are histologically comprised of lymphoid follicles with reactive germinal centers. It is associated with asthma, which normally is severe and often occurs almost simultaneously with the periorbital lesions.4
There are no definite diagnostic criteria for Erdheim-Chester disease and the diagnosis is usually based on radiologic findings of osteosclerosis and histopathologic evidence of foamy histiocytic infiltration. Systemic manifestations are common with lymphohistiocytic infiltration of the heart, lungs, pericardium, bones, and intestines. Prognosis is uniformly dismal.
Based on the clinical presentation of a nonulcerative papulonodular eruption and the absence of systemic involvement including asthma, we made the diagnosis of AXG. In view of the heterogeneity among these clinical entities as well as the time-based evolution of the lesions involved, we continued to monitor the patient for 2 years and there was no development of other systemic manifestations and hematologic abnormalities.
In contrast to the more common form of juvenile-onset xanthogranuloma, the adult type is not associated with widespread visceral lesions. Hence extensive screening investigations for systemic disease generally are not necessary. Another difference is that AXG has been associated with hematologic abnormalities, including essential thrombocytosis, chronic lymphocytic leukemia, large B-cell lymphoma, and monoclonal gammopathy.5,6 In our patient, the presence of thrombocytopenia and older age caused us to be concerned about an associated hematologic malignancy; therefore, a bone marrow biopsy and flow cytometry were performed.
Adult-onset xanthogranuloma typically is an asymptomatic and self-healing disease and therefore treatment usually is not required. Spontaneous regression of xanthogranuloma was observed to occur in 54% of cases with a median duration of 22 months,7 though lesions were noted to last as long as 15 years.8 When treatment is necessary, a combination of local and systemic corticosteroids, cytotoxic agents, and radiotherapy have been routinely used. In particular, intralesional corticosteroid therapy has been found to be efficacious in controlling symptoms and signs of AXG affecting the eyelids and orbits while avoiding the systemic side effects of other agents.9
Because our patient’s lesions were longstanding and disfiguring, we opted for active intervention with intralesional triamcinolone, which resulted in only a slight reduction in size of the lesions. The lesions remain largely unchanged in 2 years of follow-up.
1. Chang SE, Cho S, Choi JC, et al. Clinicohistopathologic comparison of adult type and juvenile type xanthogranulomas in Korea. J Dermatol. 2001;28:413-418.
2. Gelmetti C. Non-Langerhans cell histiocytosis. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
3. Mehregan DA, Winkelmann RK. Necrobiotic xanthogranuloma. Arch Dermatol. 1992;128:94-100.
4. Jakobiec FA, Mills MD, Hidayat AA, et al. Periocular xanthogranulomas associated with severe adult-onset asthma. Trans Am Ophthalmol Soc. 1993;91:99-125.
5. Shoo BA, Shinkai K, McCalmont TH, et al. Xanthogranulomas associated with hematologic malignancy in adulthood. J Am Acad Dermatol. 2008;59:488-493.
6. Chiou CC, Wang PN, Yang LC, et al. Disseminated xanthogranulomas associated with adult T-cell leukaemia/lymphoma: a case report and review the association of haematologic malignancies. J Eur Acad Dermatol Venereol. 2007;21:532-535.
7. Robinson HM, Harmon CE, Firminger HI. Multiple lipoidal histiocytomas with regression. Arch Dermatol. 1963;88:660-667.
8. Winkelmann RK. Cutaneous syndromes of non-X histiocytosis: a review of the macrophage-histiocyte diseases of the skin. Arch Dermatol. 1981;117:667-672.
9. Elner VM, Mintz R, Demirci H, et al. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Ophthal Plast Reconstr Surg. 2006;22:36-40.
1. Chang SE, Cho S, Choi JC, et al. Clinicohistopathologic comparison of adult type and juvenile type xanthogranulomas in Korea. J Dermatol. 2001;28:413-418.
2. Gelmetti C. Non-Langerhans cell histiocytosis. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
3. Mehregan DA, Winkelmann RK. Necrobiotic xanthogranuloma. Arch Dermatol. 1992;128:94-100.
4. Jakobiec FA, Mills MD, Hidayat AA, et al. Periocular xanthogranulomas associated with severe adult-onset asthma. Trans Am Ophthalmol Soc. 1993;91:99-125.
5. Shoo BA, Shinkai K, McCalmont TH, et al. Xanthogranulomas associated with hematologic malignancy in adulthood. J Am Acad Dermatol. 2008;59:488-493.
6. Chiou CC, Wang PN, Yang LC, et al. Disseminated xanthogranulomas associated with adult T-cell leukaemia/lymphoma: a case report and review the association of haematologic malignancies. J Eur Acad Dermatol Venereol. 2007;21:532-535.
7. Robinson HM, Harmon CE, Firminger HI. Multiple lipoidal histiocytomas with regression. Arch Dermatol. 1963;88:660-667.
8. Winkelmann RK. Cutaneous syndromes of non-X histiocytosis: a review of the macrophage-histiocyte diseases of the skin. Arch Dermatol. 1981;117:667-672.
9. Elner VM, Mintz R, Demirci H, et al. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Ophthal Plast Reconstr Surg. 2006;22:36-40.
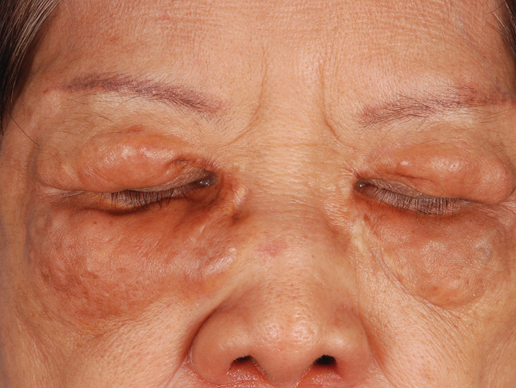
A 66-year-old woman with a history of type 2 diabetes mellitus and mild dyslipidemia presented with persistent lesions over the eyelids and cheeks of 10 years’ duration. Systemic review was unremarkable. There was no family or personal history of atopy, asthma, or other dermatologic disorders. Physical examination revealed confluent yellowish plaques and nodules over the periorbital regions as well as yellowish plaques over the neck and back. The lesions were firm to palpation and the epidermis appeared unaffected. The ophthalmic examination was normal and other mucosal surfaces were unaffected.
Papular Eruption Following Excessive Tanning Bed Use
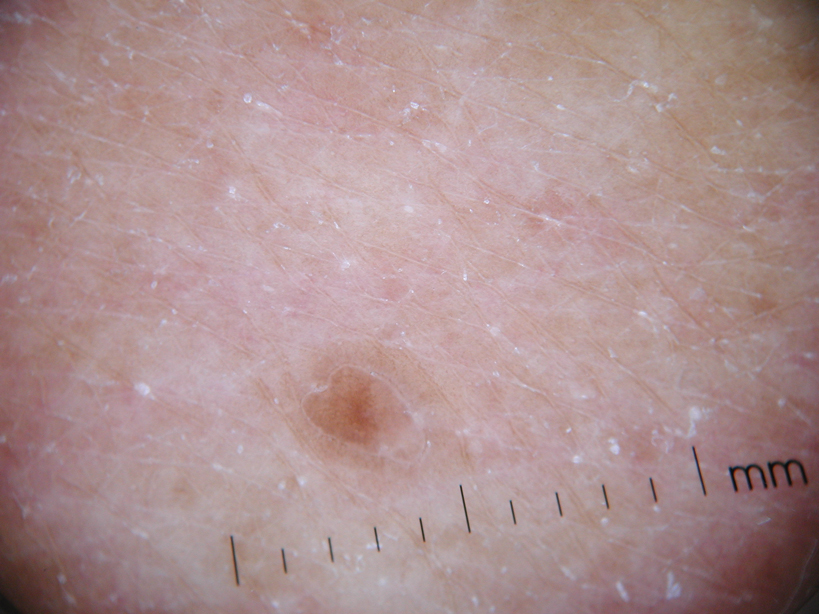
The Diagnosis: Disseminated Superficial Actinic Porokeratosis
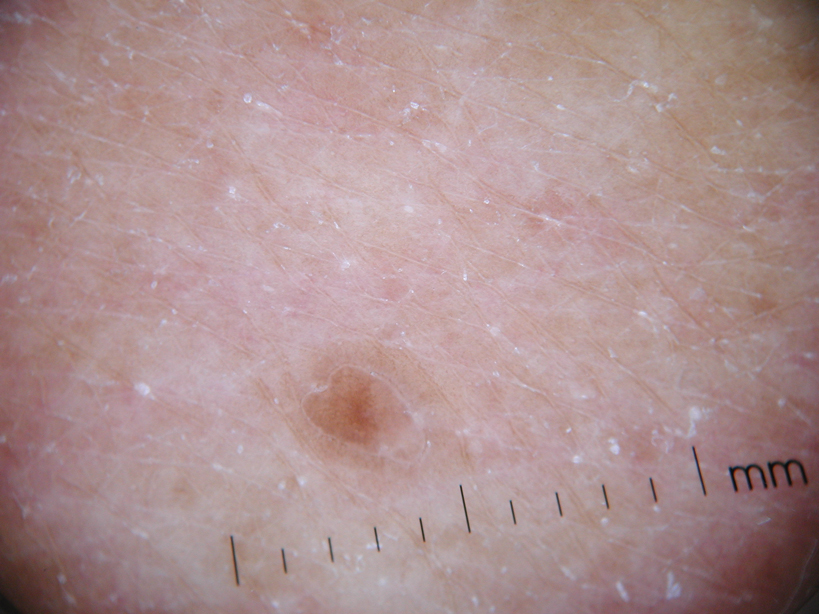
Physical examination after 7 years of tanning salon use showed a tanned white man with multiple 4- to 5-mm, discrete, round to oval, reddish brown papules on the chest, back, abdomen, arms, and legs that were rough to palpate (Figure 1), with a peripheral rim of scale seen more prominently on dermoscopy. There were no lesions on the palms or soles. A subsequent 4-mm punch biopsy was done on the right abdomen and right thigh, which showed focal thinning of the epidermis, loss of the granular layer, a discrete column of parakeratosis and a characteristic feature of a coronoid lamella in the epidermis (Figure 2). The patient received 14 narrowband UVB (NB-UVB) treatments; however, he could not continue due to transportation issues. He visited the clinic sporadically for 6 months thereafter and reportedly went to tanning salons daily. The patient was subsequently lost to follow-up.
|
|
| Figure 1. Multiple 4- to 5-mm, discrete, round to oval, reddish brown papules on the abdomen (A) and leg (B) that were rough to palpate. |
Disseminated porokeratosis, or disseminated superficial actinic porokeratosis (DSAP), was first described in 1967 by Chernosky and Freeman.1 It is the most common variant of porokeratosis. Other variants include Mibelli type, porokeratosis palmaris et plantaris disseminata, punctuate porokeratosis, and linear porokeratosis.2-4 Porokeratosis is inherited in an autosomal-dominant fashion, presenting in the third or fourth decades of life; however, most cases are sporadic.5 Pruritus is a common symptom and can be debilitating.5
Disseminated superficial actinic porokeratosis can be precipitated by excessive sun exposure, with a reported increase in lesions during summer months and resolution during the winter months.6 The lesions of DSAP can be experimentally induced by exposure to daily use of artificial UV sunlamps.7 Patients with psoriasis undergoing psoralen plus UVA and NB-UVB treatments also have been reported to trigger DSAP.8 A study by Neumann et al6 suggested that a combination of both UVB and UVA wavelengths may be most effective in inducing DSAP. Exposure to UVA and UVB light may explain an increased number of DSAP lesions in patients who excessively visit tanning salons, as the bulbs emit a combination of wavelengths with UVA in much greater amounts than UVB.
Our patient developed DSAP secondary to artificial UV light exposure from excessive tanning salon use. Medications (allopurinol and lisinopril) were initially thought to be etiologic agents for the eruption and also corroborated with histologic findings of a drug eruption on the initial biopsy. However, new lesions continued to develop even after cessation of medications and NB-UVB treatments. A subsequent biopsy and further history of daily tanning salon use confirmed the diagnosis of DSAP.
Therapies for this condition are limited with variable degrees of success. Cryotherapy, 5-fluorouracil cream, imiquimod cream 5%, Q-switched ruby laser, diclofenac gel 3%, and acitretin for more widespread or refractory lesions have been used with partial to complete resolution of DSAP.9
We present this case to highlight the occurrence of DSAP secondary to UV light exposure from excessive tanning salon use.
1. Chernosky ME, Freeman RG. Disseminated superficial actinic porokeratosis (DSAP). Arch Dermatol. 1967;96:611-624.
2. Guss SB, Osbourn RA, Lutzner MA. Porokeratosis plantaris, palmaris et disseminata: a third type of porokeratosis. Arch Dermatol. 1971;104:366-373.
3. Brown FC. Punctate keratoderma. Arch Dermatol. 1971;104:682-683.
4. Eyre WG, Carson WE. Linear porokeratosis of Mibelli. Arch Dermatol. 1972;105:426-429.
5. Anderson DE, Chernosky ME. Disseminated superficial actinic porokeratosis. Arch Dermatol. 1969;99:408-412.
6. Neumann RA, Knobler RM, Jurecka W, et al. Disseminated superficial actinic porokeratosis: experimental induction and exacerbation of skin lesions. J Am Acad Dermatol. 1989;21:1182-1188.
7. Chernosky ME, Anderson DE. Disseminated superficial actinic porokeratosis: clinical studies and experimental production of lesions. Arch Dermatol. 1969;99:401-407.
8. Allen LA, Glaser DA. Disseminated superficial actinic porokeratosis associated with topical PUVA. J Am Acad Dermatol. 2000;43:720-722.
9. Arun B, Pearson J, Chalmers R. Disseminated superficial actinic porokeratosis treated effectively with topical imiquimod 5% cream. Clin Exp Dermatol. 2011;36:509-511.
The Diagnosis: Disseminated Superficial Actinic Porokeratosis
Physical examination after 7 years of tanning salon use showed a tanned white man with multiple 4- to 5-mm, discrete, round to oval, reddish brown papules on the chest, back, abdomen, arms, and legs that were rough to palpate (Figure 1), with a peripheral rim of scale seen more prominently on dermoscopy. There were no lesions on the palms or soles. A subsequent 4-mm punch biopsy was done on the right abdomen and right thigh, which showed focal thinning of the epidermis, loss of the granular layer, a discrete column of parakeratosis and a characteristic feature of a coronoid lamella in the epidermis (Figure 2). The patient received 14 narrowband UVB (NB-UVB) treatments; however, he could not continue due to transportation issues. He visited the clinic sporadically for 6 months thereafter and reportedly went to tanning salons daily. The patient was subsequently lost to follow-up.
|
|
|
|
| Figure 1. Multiple 4- to 5-mm, discrete, round to oval, reddish brown papules on the abdomen (A) and leg (B) that were rough to palpate. |
Disseminated porokeratosis, or disseminated superficial actinic porokeratosis (DSAP), was first described in 1967 by Chernosky and Freeman.1 It is the most common variant of porokeratosis. Other variants include Mibelli type, porokeratosis palmaris et plantaris disseminata, punctuate porokeratosis, and linear porokeratosis.2-4 Porokeratosis is inherited in an autosomal-dominant fashion, presenting in the third or fourth decades of life; however, most cases are sporadic.5 Pruritus is a common symptom and can be debilitating.5
Disseminated superficial actinic porokeratosis can be precipitated by excessive sun exposure, with a reported increase in lesions during summer months and resolution during the winter months.6 The lesions of DSAP can be experimentally induced by exposure to daily use of artificial UV sunlamps.7 Patients with psoriasis undergoing psoralen plus UVA and NB-UVB treatments also have been reported to trigger DSAP.8 A study by Neumann et al6 suggested that a combination of both UVB and UVA wavelengths may be most effective in inducing DSAP. Exposure to UVA and UVB light may explain an increased number of DSAP lesions in patients who excessively visit tanning salons, as the bulbs emit a combination of wavelengths with UVA in much greater amounts than UVB.
Our patient developed DSAP secondary to artificial UV light exposure from excessive tanning salon use. Medications (allopurinol and lisinopril) were initially thought to be etiologic agents for the eruption and also corroborated with histologic findings of a drug eruption on the initial biopsy. However, new lesions continued to develop even after cessation of medications and NB-UVB treatments. A subsequent biopsy and further history of daily tanning salon use confirmed the diagnosis of DSAP.
Therapies for this condition are limited with variable degrees of success. Cryotherapy, 5-fluorouracil cream, imiquimod cream 5%, Q-switched ruby laser, diclofenac gel 3%, and acitretin for more widespread or refractory lesions have been used with partial to complete resolution of DSAP.9
We present this case to highlight the occurrence of DSAP secondary to UV light exposure from excessive tanning salon use.
The Diagnosis: Disseminated Superficial Actinic Porokeratosis
Physical examination after 7 years of tanning salon use showed a tanned white man with multiple 4- to 5-mm, discrete, round to oval, reddish brown papules on the chest, back, abdomen, arms, and legs that were rough to palpate (Figure 1), with a peripheral rim of scale seen more prominently on dermoscopy. There were no lesions on the palms or soles. A subsequent 4-mm punch biopsy was done on the right abdomen and right thigh, which showed focal thinning of the epidermis, loss of the granular layer, a discrete column of parakeratosis and a characteristic feature of a coronoid lamella in the epidermis (Figure 2). The patient received 14 narrowband UVB (NB-UVB) treatments; however, he could not continue due to transportation issues. He visited the clinic sporadically for 6 months thereafter and reportedly went to tanning salons daily. The patient was subsequently lost to follow-up.
|
|
|
|
| Figure 1. Multiple 4- to 5-mm, discrete, round to oval, reddish brown papules on the abdomen (A) and leg (B) that were rough to palpate. |
Disseminated porokeratosis, or disseminated superficial actinic porokeratosis (DSAP), was first described in 1967 by Chernosky and Freeman.1 It is the most common variant of porokeratosis. Other variants include Mibelli type, porokeratosis palmaris et plantaris disseminata, punctuate porokeratosis, and linear porokeratosis.2-4 Porokeratosis is inherited in an autosomal-dominant fashion, presenting in the third or fourth decades of life; however, most cases are sporadic.5 Pruritus is a common symptom and can be debilitating.5
Disseminated superficial actinic porokeratosis can be precipitated by excessive sun exposure, with a reported increase in lesions during summer months and resolution during the winter months.6 The lesions of DSAP can be experimentally induced by exposure to daily use of artificial UV sunlamps.7 Patients with psoriasis undergoing psoralen plus UVA and NB-UVB treatments also have been reported to trigger DSAP.8 A study by Neumann et al6 suggested that a combination of both UVB and UVA wavelengths may be most effective in inducing DSAP. Exposure to UVA and UVB light may explain an increased number of DSAP lesions in patients who excessively visit tanning salons, as the bulbs emit a combination of wavelengths with UVA in much greater amounts than UVB.
Our patient developed DSAP secondary to artificial UV light exposure from excessive tanning salon use. Medications (allopurinol and lisinopril) were initially thought to be etiologic agents for the eruption and also corroborated with histologic findings of a drug eruption on the initial biopsy. However, new lesions continued to develop even after cessation of medications and NB-UVB treatments. A subsequent biopsy and further history of daily tanning salon use confirmed the diagnosis of DSAP.
Therapies for this condition are limited with variable degrees of success. Cryotherapy, 5-fluorouracil cream, imiquimod cream 5%, Q-switched ruby laser, diclofenac gel 3%, and acitretin for more widespread or refractory lesions have been used with partial to complete resolution of DSAP.9
We present this case to highlight the occurrence of DSAP secondary to UV light exposure from excessive tanning salon use.
1. Chernosky ME, Freeman RG. Disseminated superficial actinic porokeratosis (DSAP). Arch Dermatol. 1967;96:611-624.
2. Guss SB, Osbourn RA, Lutzner MA. Porokeratosis plantaris, palmaris et disseminata: a third type of porokeratosis. Arch Dermatol. 1971;104:366-373.
3. Brown FC. Punctate keratoderma. Arch Dermatol. 1971;104:682-683.
4. Eyre WG, Carson WE. Linear porokeratosis of Mibelli. Arch Dermatol. 1972;105:426-429.
5. Anderson DE, Chernosky ME. Disseminated superficial actinic porokeratosis. Arch Dermatol. 1969;99:408-412.
6. Neumann RA, Knobler RM, Jurecka W, et al. Disseminated superficial actinic porokeratosis: experimental induction and exacerbation of skin lesions. J Am Acad Dermatol. 1989;21:1182-1188.
7. Chernosky ME, Anderson DE. Disseminated superficial actinic porokeratosis: clinical studies and experimental production of lesions. Arch Dermatol. 1969;99:401-407.
8. Allen LA, Glaser DA. Disseminated superficial actinic porokeratosis associated with topical PUVA. J Am Acad Dermatol. 2000;43:720-722.
9. Arun B, Pearson J, Chalmers R. Disseminated superficial actinic porokeratosis treated effectively with topical imiquimod 5% cream. Clin Exp Dermatol. 2011;36:509-511.
1. Chernosky ME, Freeman RG. Disseminated superficial actinic porokeratosis (DSAP). Arch Dermatol. 1967;96:611-624.
2. Guss SB, Osbourn RA, Lutzner MA. Porokeratosis plantaris, palmaris et disseminata: a third type of porokeratosis. Arch Dermatol. 1971;104:366-373.
3. Brown FC. Punctate keratoderma. Arch Dermatol. 1971;104:682-683.
4. Eyre WG, Carson WE. Linear porokeratosis of Mibelli. Arch Dermatol. 1972;105:426-429.
5. Anderson DE, Chernosky ME. Disseminated superficial actinic porokeratosis. Arch Dermatol. 1969;99:408-412.
6. Neumann RA, Knobler RM, Jurecka W, et al. Disseminated superficial actinic porokeratosis: experimental induction and exacerbation of skin lesions. J Am Acad Dermatol. 1989;21:1182-1188.
7. Chernosky ME, Anderson DE. Disseminated superficial actinic porokeratosis: clinical studies and experimental production of lesions. Arch Dermatol. 1969;99:401-407.
8. Allen LA, Glaser DA. Disseminated superficial actinic porokeratosis associated with topical PUVA. J Am Acad Dermatol. 2000;43:720-722.
9. Arun B, Pearson J, Chalmers R. Disseminated superficial actinic porokeratosis treated effectively with topical imiquimod 5% cream. Clin Exp Dermatol. 2011;36:509-511.
A 78-year-old man with Fitzpatrick skin type III presented to the dermatology department for evaluation of a pruritic, erythematous, papular eruption on the chest, back, abdomen, arms, and legs of 5 years’ duration. His medications include clonazepam, lisinopril, allopurinol, omeprazole, tramadol, and mirtazapine. The lesions did not respond to topical corticosteroids; however, the pruritus improved with narrowband UVB (NB-UVB) treatments. Review of systems did not reveal any abnormalities. The patient’s medical history included gout, hypertension, anxiety, esophageal stricture, and emphysema. He reported a history of tanning salon use at least 3 times weekly for 7 years. After initial consultation, the patient was treated with clobetasol propionate cream 0.05% twice daily and hydroxyzine 10 mg 3 times daily. Following 1 month of treatment, the eruption did not improve. A 4-mm punch biopsy of the left upper arm revealed a dense infiltrate in the upper dermis with prominent parakeratosis, lymphocytes, and numerous eosinophils, suggestive of a drug eruption. As a result, allopurinol was discontinued as a causative agent; however, the eruption presented prior to taking allopurinol. Because the patient experienced intense pruritus, he was started on NB-UVB treatments. After 14 treatments of NB-UVB 3 times weekly, the patient noticed some improvement with respect to pruritus, but the lesions did not resolve. A complete blood cell count indicated 7.6% eosinophils (reference range, 0%–5%). Liver function tests, complete metabolic profile, and renal function were within reference range. Lisinopril was then discontinued as a likely culprit for persistent drug eruption; however, new lesions continued to develop.
What Is Your Diagnosis? Granulocytic Sarcoma
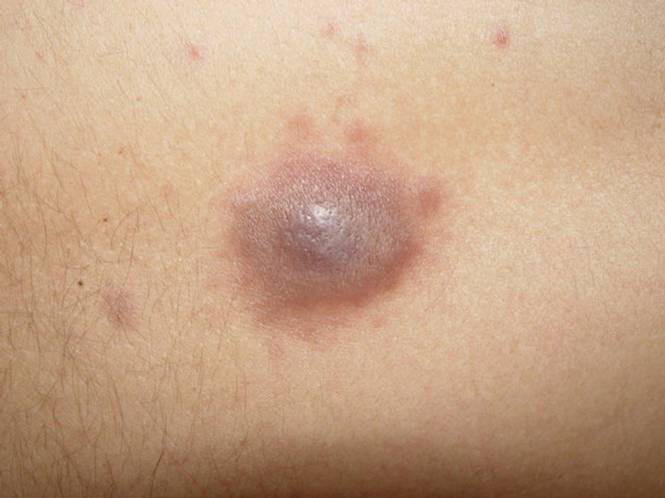
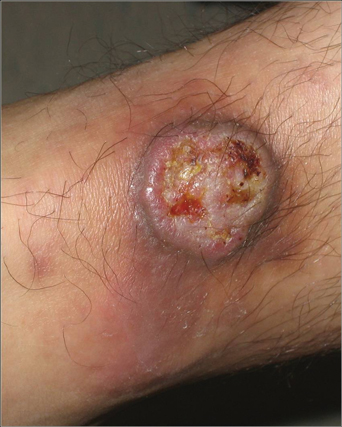
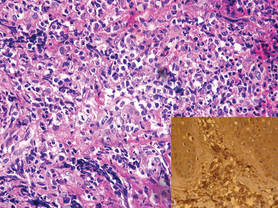
A 24-year-old man presented with a 5-cm asymptomatic, indurated, well-defined nodule with a violaceous center surrounded by a greenish halo on the lower back of 3 months’ duration, along with lymphadenopathy. There was no history of trauma, weight loss, bleeding, or systemic illness. A biopsy revealed sheets of pleomorphic cells with lobulated nuclei and detectable granular cytoplasm. Immunohistochemistry showed strong myeloperoxidase activity in neoplastic cells that were CD13- and CD56-.
The Diagnosis: Granulocytic Sarcoma
Granulocytic sarcoma (GS), sometimes known as leukemia cutis, myeloid sarcoma, or an extramedullary myeloid tumor, is a rare localized collection of immature cells of granulocytic series or the cells of each maturation step in extramedullary sites. Originally described in 1811, the condition was noted after identifying an extramedullary collection of immature granulocytes involving the orbit. Then in 1853 the term chloroma was coined because of the greenish appearance of the tumor when exposed to light due to myeloperoxidase activity in the tumor cells. Finally, in 1967 the term granulocytic sarcoma was introduced because the greenish appearance was not observed in all cases, providing a more correct term.1
Granulocytic sarcoma often manifests in patients with acute myeloid leukemia, chronic myelogenous leukemia, chronic idiopathic myelofibrosis, eosinophilia, refractory anemia, or polycythemia vera, and as an isolated form in patients without apparent hematologic disorder.2
Cutaneous manifestations of leukemia can be specific (eg, GS) or nonspecific (eg, panniculitis, generalized pruritus).2,3 The tumor often appears as a solitary mass (Figure 1); however, in rare instances it can be multifocal. It usually presents as a rapidly growing nodular mass with solid consistence, and it is seen in patients of any age but most often those aged 45 to 55 years as well as in patients younger than 15 years.4
Granulocytic sarcoma is most commonly associated with acute myeloid leukemia. However, it is interesting that the chloroma appears before the onset of leukemia. Occasionally GS is associated with acute myeloid leukemia as a presenting symptom or as a manifestation of recurrence of the disease that was usually related to poor prognosis.5 In patients with GS that is associated with myeloproliferative neoplasms, the occurrence of GS frequently is associated with acute transformation of the disease and therefore a poor prognosis.6
The most common sites of involvement are bone, soft tissue, lymph nodes, and skin. The prognosis does not vary according to location.
Cutaneous B-cell lymphoma should be considered in the differential diagnosis, as it may be clinically observed as single or multiple violaceous nodules involving the head, neck, and/or trunk. Histologically, there generally is a bottom-heavy infiltrate of lymphocytes sparing the epidermis (Figure 2). Immunohistochemistry, flow cytometry, or cytochemical stains often are needed to exclude lymphoma. Pseudochloroma is a rare disease characterized by a collection of normal hematopoietic tissue in locations other than GS. It can be distinguished from GS by its constituent of mature cells.7
Granulocytic sarcoma implies a therapeutic challenge for the clinician. It should always be considered as part of a systemic disease rather than an isolated skin disorder. In patients with evidence of leukemia and GS, systemic chemotherapy is the first line of treatment. Other effective treatment regimens include radiotherapy (electron beam radiation), either alone or in combination with systemic chemotherapy (eg, hydroxyurea, methotrexate).8 If GS persists after chemotherapy, local treatment (eg, radiotherapy, surgery) should be considered with variable results.2
1. Meis JM, Butler JJ, Osborne BM, et al. Granulocytic sarcoma in nonleukemic patients. Cancer. 1986;58:2697-2709.
2. Sun NCJ, Ellis R. Granulocytic sarcoma of the skin. Arch Dermatol. 1980;116:800-802.
3. Campidelli C, Agostinelli C, Stitson R, et al. Myeloid sarcoma. Extramedullary manifestation of myeloid disorders. Am J Clin Pathol. 2009;132:426-437.
4. Pulsoni A, Falcucci P, Anghel G, et al. Isolated granulocytic sarcoma of the skin in an elderly patient: good response to treatment with local radiotherapy and low dose methotrexate. J Eur Acad Dermatol Venereol. 2000;14:216-218.
5. Neiman RS, Barcos M, Berard C, et al. Granulocytic sarcoma: a clinicopathologic study of 61 biopsied cases. Cancer. 1981;48:1426-1437.
6. Byrd JC, Edenfield WJ, Dow NS, et al. Extramedullary myeloid cell tumors in myelodysplastic syndromes: not a true indication of impending acute myeloid leukemia. Leukemia. 1996;21:153-159.
7. Beswick SJ, Jones EL, Mahendra P, et al. Chloroma (aleukaemic leukaemia cutis) initially diagnosed as a cutaneous lymphoma. Clin Exp Dermatol. 2002;27:272-274.
8. Chen WY, Wang CW, Chang CH, et al. Clinicopathologic features and responses to radiotherapy of myeloid sarcoma. Radiat Oncol. 2013;8:245.
A 24-year-old man presented with a 5-cm asymptomatic, indurated, well-defined nodule with a violaceous center surrounded by a greenish halo on the lower back of 3 months’ duration, along with lymphadenopathy. There was no history of trauma, weight loss, bleeding, or systemic illness. A biopsy revealed sheets of pleomorphic cells with lobulated nuclei and detectable granular cytoplasm. Immunohistochemistry showed strong myeloperoxidase activity in neoplastic cells that were CD13- and CD56-.
The Diagnosis: Granulocytic Sarcoma
Granulocytic sarcoma (GS), sometimes known as leukemia cutis, myeloid sarcoma, or an extramedullary myeloid tumor, is a rare localized collection of immature cells of granulocytic series or the cells of each maturation step in extramedullary sites. Originally described in 1811, the condition was noted after identifying an extramedullary collection of immature granulocytes involving the orbit. Then in 1853 the term chloroma was coined because of the greenish appearance of the tumor when exposed to light due to myeloperoxidase activity in the tumor cells. Finally, in 1967 the term granulocytic sarcoma was introduced because the greenish appearance was not observed in all cases, providing a more correct term.1
Granulocytic sarcoma often manifests in patients with acute myeloid leukemia, chronic myelogenous leukemia, chronic idiopathic myelofibrosis, eosinophilia, refractory anemia, or polycythemia vera, and as an isolated form in patients without apparent hematologic disorder.2
Cutaneous manifestations of leukemia can be specific (eg, GS) or nonspecific (eg, panniculitis, generalized pruritus).2,3 The tumor often appears as a solitary mass (Figure 1); however, in rare instances it can be multifocal. It usually presents as a rapidly growing nodular mass with solid consistence, and it is seen in patients of any age but most often those aged 45 to 55 years as well as in patients younger than 15 years.4
Granulocytic sarcoma is most commonly associated with acute myeloid leukemia. However, it is interesting that the chloroma appears before the onset of leukemia. Occasionally GS is associated with acute myeloid leukemia as a presenting symptom or as a manifestation of recurrence of the disease that was usually related to poor prognosis.5 In patients with GS that is associated with myeloproliferative neoplasms, the occurrence of GS frequently is associated with acute transformation of the disease and therefore a poor prognosis.6
The most common sites of involvement are bone, soft tissue, lymph nodes, and skin. The prognosis does not vary according to location.
Cutaneous B-cell lymphoma should be considered in the differential diagnosis, as it may be clinically observed as single or multiple violaceous nodules involving the head, neck, and/or trunk. Histologically, there generally is a bottom-heavy infiltrate of lymphocytes sparing the epidermis (Figure 2). Immunohistochemistry, flow cytometry, or cytochemical stains often are needed to exclude lymphoma. Pseudochloroma is a rare disease characterized by a collection of normal hematopoietic tissue in locations other than GS. It can be distinguished from GS by its constituent of mature cells.7
Granulocytic sarcoma implies a therapeutic challenge for the clinician. It should always be considered as part of a systemic disease rather than an isolated skin disorder. In patients with evidence of leukemia and GS, systemic chemotherapy is the first line of treatment. Other effective treatment regimens include radiotherapy (electron beam radiation), either alone or in combination with systemic chemotherapy (eg, hydroxyurea, methotrexate).8 If GS persists after chemotherapy, local treatment (eg, radiotherapy, surgery) should be considered with variable results.2
A 24-year-old man presented with a 5-cm asymptomatic, indurated, well-defined nodule with a violaceous center surrounded by a greenish halo on the lower back of 3 months’ duration, along with lymphadenopathy. There was no history of trauma, weight loss, bleeding, or systemic illness. A biopsy revealed sheets of pleomorphic cells with lobulated nuclei and detectable granular cytoplasm. Immunohistochemistry showed strong myeloperoxidase activity in neoplastic cells that were CD13- and CD56-.
The Diagnosis: Granulocytic Sarcoma
Granulocytic sarcoma (GS), sometimes known as leukemia cutis, myeloid sarcoma, or an extramedullary myeloid tumor, is a rare localized collection of immature cells of granulocytic series or the cells of each maturation step in extramedullary sites. Originally described in 1811, the condition was noted after identifying an extramedullary collection of immature granulocytes involving the orbit. Then in 1853 the term chloroma was coined because of the greenish appearance of the tumor when exposed to light due to myeloperoxidase activity in the tumor cells. Finally, in 1967 the term granulocytic sarcoma was introduced because the greenish appearance was not observed in all cases, providing a more correct term.1
Granulocytic sarcoma often manifests in patients with acute myeloid leukemia, chronic myelogenous leukemia, chronic idiopathic myelofibrosis, eosinophilia, refractory anemia, or polycythemia vera, and as an isolated form in patients without apparent hematologic disorder.2
Cutaneous manifestations of leukemia can be specific (eg, GS) or nonspecific (eg, panniculitis, generalized pruritus).2,3 The tumor often appears as a solitary mass (Figure 1); however, in rare instances it can be multifocal. It usually presents as a rapidly growing nodular mass with solid consistence, and it is seen in patients of any age but most often those aged 45 to 55 years as well as in patients younger than 15 years.4
Granulocytic sarcoma is most commonly associated with acute myeloid leukemia. However, it is interesting that the chloroma appears before the onset of leukemia. Occasionally GS is associated with acute myeloid leukemia as a presenting symptom or as a manifestation of recurrence of the disease that was usually related to poor prognosis.5 In patients with GS that is associated with myeloproliferative neoplasms, the occurrence of GS frequently is associated with acute transformation of the disease and therefore a poor prognosis.6
The most common sites of involvement are bone, soft tissue, lymph nodes, and skin. The prognosis does not vary according to location.
Cutaneous B-cell lymphoma should be considered in the differential diagnosis, as it may be clinically observed as single or multiple violaceous nodules involving the head, neck, and/or trunk. Histologically, there generally is a bottom-heavy infiltrate of lymphocytes sparing the epidermis (Figure 2). Immunohistochemistry, flow cytometry, or cytochemical stains often are needed to exclude lymphoma. Pseudochloroma is a rare disease characterized by a collection of normal hematopoietic tissue in locations other than GS. It can be distinguished from GS by its constituent of mature cells.7
Granulocytic sarcoma implies a therapeutic challenge for the clinician. It should always be considered as part of a systemic disease rather than an isolated skin disorder. In patients with evidence of leukemia and GS, systemic chemotherapy is the first line of treatment. Other effective treatment regimens include radiotherapy (electron beam radiation), either alone or in combination with systemic chemotherapy (eg, hydroxyurea, methotrexate).8 If GS persists after chemotherapy, local treatment (eg, radiotherapy, surgery) should be considered with variable results.2
1. Meis JM, Butler JJ, Osborne BM, et al. Granulocytic sarcoma in nonleukemic patients. Cancer. 1986;58:2697-2709.
2. Sun NCJ, Ellis R. Granulocytic sarcoma of the skin. Arch Dermatol. 1980;116:800-802.
3. Campidelli C, Agostinelli C, Stitson R, et al. Myeloid sarcoma. Extramedullary manifestation of myeloid disorders. Am J Clin Pathol. 2009;132:426-437.
4. Pulsoni A, Falcucci P, Anghel G, et al. Isolated granulocytic sarcoma of the skin in an elderly patient: good response to treatment with local radiotherapy and low dose methotrexate. J Eur Acad Dermatol Venereol. 2000;14:216-218.
5. Neiman RS, Barcos M, Berard C, et al. Granulocytic sarcoma: a clinicopathologic study of 61 biopsied cases. Cancer. 1981;48:1426-1437.
6. Byrd JC, Edenfield WJ, Dow NS, et al. Extramedullary myeloid cell tumors in myelodysplastic syndromes: not a true indication of impending acute myeloid leukemia. Leukemia. 1996;21:153-159.
7. Beswick SJ, Jones EL, Mahendra P, et al. Chloroma (aleukaemic leukaemia cutis) initially diagnosed as a cutaneous lymphoma. Clin Exp Dermatol. 2002;27:272-274.
8. Chen WY, Wang CW, Chang CH, et al. Clinicopathologic features and responses to radiotherapy of myeloid sarcoma. Radiat Oncol. 2013;8:245.
1. Meis JM, Butler JJ, Osborne BM, et al. Granulocytic sarcoma in nonleukemic patients. Cancer. 1986;58:2697-2709.
2. Sun NCJ, Ellis R. Granulocytic sarcoma of the skin. Arch Dermatol. 1980;116:800-802.
3. Campidelli C, Agostinelli C, Stitson R, et al. Myeloid sarcoma. Extramedullary manifestation of myeloid disorders. Am J Clin Pathol. 2009;132:426-437.
4. Pulsoni A, Falcucci P, Anghel G, et al. Isolated granulocytic sarcoma of the skin in an elderly patient: good response to treatment with local radiotherapy and low dose methotrexate. J Eur Acad Dermatol Venereol. 2000;14:216-218.
5. Neiman RS, Barcos M, Berard C, et al. Granulocytic sarcoma: a clinicopathologic study of 61 biopsied cases. Cancer. 1981;48:1426-1437.
6. Byrd JC, Edenfield WJ, Dow NS, et al. Extramedullary myeloid cell tumors in myelodysplastic syndromes: not a true indication of impending acute myeloid leukemia. Leukemia. 1996;21:153-159.
7. Beswick SJ, Jones EL, Mahendra P, et al. Chloroma (aleukaemic leukaemia cutis) initially diagnosed as a cutaneous lymphoma. Clin Exp Dermatol. 2002;27:272-274.
8. Chen WY, Wang CW, Chang CH, et al. Clinicopathologic features and responses to radiotherapy of myeloid sarcoma. Radiat Oncol. 2013;8:245.
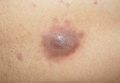
Nontender Nodules on the Lower Lip
The Diagnosis: Primary Systemic Amyloidosis
Our patient presented with multiple firm waxy nodules on the mucosal surface of the lower lip. Excision biopsy showed thickening of blood vessel walls with abundant amorphous material that was consistent with amyloid. Further staining with Congo red demonstrated brick red amorphous material within the vessel walls on routine light microscopy (Figure 1), and crystal violet stain showed metachromasia (Figure 2). Fine needle aspiration of the abdominal fat-pad showed amyloid. The final diagnosis was primary systemic amyloidosis (PSA).
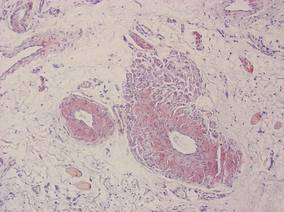
|
| Figure 1. Congo red staining showed a brick red appearance of amyloid within the vessel walls on routine light microscopy (original magnification ×200). |
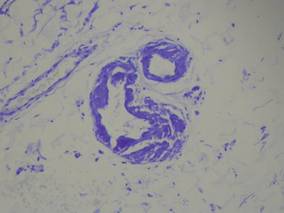
|
| Figure 2. Crystal violet staining around the blood vessels showed metachromasia (original magnification ×100). |
Amyloid is an ubiquitous fibrillar protein arranged in a cross-beta-pleated sheet that is confirmed with x-ray crystallography.1,2 More than 25 variants of amyloid have been identified.3 Pathologic deposition of amyloid-derived material results in a variable spectrum of clinical findings, collectively known as amyloidosis, with presentations ranging from nonspecific fever or fatigue to frank organ failure, depending on the organ involved. In PSA, immunoglobulin light chains are deposited throughout the body. Associated conditions include malignant or benign monoclonal gammopathy, multiple myeloma, Waldenström macroglobulinemia, malignant lymphoma, heavy chain disease, and chronic lymphocytic leukemia.2,4 The most commonly involved organ systems are the heart, lungs, liver, and kidneys. When patients present with unexplained heart failure, orthostatic hypotension, hepatomegaly, peripheral neuropathy, carpal tunnel syndrome, or renal insufficiency, amyloidosis should always be considered in the differential diagnosis.3
Cutaneous lesions of PSA tend to be vascular due to amyloid infiltration of blood vessel walls, manifesting as petechiae, purpura, ecchymoses, or nonhealing ulcers. Pinch purpura frequently are seen in the periorbital region after minor trauma and are recognized as a clinical indicator of PSA.2,4 Xerostomia from amyloid infiltrates in salivary glands is extremely common, and cases of amyloid in the eyes, bones, and thyroid gland have been reported.2 Macroglossia is seen in 12% to 40% of cases; coupled with xerostomia, it can lead to oropharyngeal dysphagia.2,5
Systemic amyloidosis can be further divided into primary (idiopathic or multiple myeloma associated) or secondary to chronic inflammatory conditions or infections; the key difference is the protein from which the abnormal amyloid is derived.1,4 The presence of cutaneous amyloidosis renders the need to rule out systemic disease because amyloidosis may be a purely localized or systemic process.4 Nodular amyloidosis is a localized form of amyloid that also has immunoglobulin light chain deposits and clinically appears exactly the same as PSA; however, the deposits are restricted to the skin.1,2,4,5
Characteristic biopsy findings in cutaneous amyloidosis include amorphous orange-red amyloid deposits on hematoxylin and eosin–stained sections. The gold standard for amyloid detection is apple green birefringence under polarized light with Congo red stain or electron microscopy.6,7 Other stains used to identify amyloid include crystal violet, methyl violet, periodic acid–Schiff, Sirius red, pagoda red, Dylon stain, and thioflavine T.1,2 Confirmation of systemic disease can be accomplished by fine needle aspiration of abdominal fat-pads or rectal mucosal biopsies.1,3,4 Biopsy of accessory salivary glands also has been reported to be very sensitive and specific.2
Treatment options remain limited; localized cutaneous disease may respond to topical corticosteroids, calcineurin inhibitors, or phototherapy. Primary systemic amyloidosis can be treated with a combination of steroids, melphalan, or colchicine often followed by autologous stem cell transplantation8; however, these regimens are not always curative and patients often have a poor prognosis.1
1. Black MM, Upjohn E, Albert S. Amyloidosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 1. 2nd ed. Spain: Mosby Elsevier; 2008:623-631.
2. Steciuk A, Dompmartin A, Troussard X, et al. Cutaneous amyloidosis and possible association with systemic amyloidosis. Int J Dermatol. 2002;41:127-132.
3. Picken MM. Amyloidosis-where are we now and where are we heading? Arch Pathol Lab Med. 2010;134:545-551.
4. Schreml S, Szeimies RM, Vogt T, et al. Cutaneous amyloidoses and systemic amyloidoses with cutaneous involvement. Eur J Dermatol. 2010;20:152-160.
5. Breathnach SM. Amyloid and amyloidosis. J Am Acad Dermatol. 1988;18(1, pt 1):1-16.
6. Li WM. Histopathology of primary cutaneous amyloidoses and systemic amyloidosis. Clin Dermatol. 1990;8:30-35.
7. Lin CS, Wong CK. Electron microscopy of primary and secondary cutaneous amyloidoses and systemic amyloidosis. Clin Dermatol. 1990;8:36-45.
8. Dember LM. Modern treatment of amyloidosis: unresolved questions. J Am Soc Nephrol. 2009;20:469-472.
The Diagnosis: Primary Systemic Amyloidosis
Our patient presented with multiple firm waxy nodules on the mucosal surface of the lower lip. Excision biopsy showed thickening of blood vessel walls with abundant amorphous material that was consistent with amyloid. Further staining with Congo red demonstrated brick red amorphous material within the vessel walls on routine light microscopy (Figure 1), and crystal violet stain showed metachromasia (Figure 2). Fine needle aspiration of the abdominal fat-pad showed amyloid. The final diagnosis was primary systemic amyloidosis (PSA).
|
|
| Figure 1. Congo red staining showed a brick red appearance of amyloid within the vessel walls on routine light microscopy (original magnification ×200). |
|
|
| Figure 2. Crystal violet staining around the blood vessels showed metachromasia (original magnification ×100). |
Amyloid is an ubiquitous fibrillar protein arranged in a cross-beta-pleated sheet that is confirmed with x-ray crystallography.1,2 More than 25 variants of amyloid have been identified.3 Pathologic deposition of amyloid-derived material results in a variable spectrum of clinical findings, collectively known as amyloidosis, with presentations ranging from nonspecific fever or fatigue to frank organ failure, depending on the organ involved. In PSA, immunoglobulin light chains are deposited throughout the body. Associated conditions include malignant or benign monoclonal gammopathy, multiple myeloma, Waldenström macroglobulinemia, malignant lymphoma, heavy chain disease, and chronic lymphocytic leukemia.2,4 The most commonly involved organ systems are the heart, lungs, liver, and kidneys. When patients present with unexplained heart failure, orthostatic hypotension, hepatomegaly, peripheral neuropathy, carpal tunnel syndrome, or renal insufficiency, amyloidosis should always be considered in the differential diagnosis.3
Cutaneous lesions of PSA tend to be vascular due to amyloid infiltration of blood vessel walls, manifesting as petechiae, purpura, ecchymoses, or nonhealing ulcers. Pinch purpura frequently are seen in the periorbital region after minor trauma and are recognized as a clinical indicator of PSA.2,4 Xerostomia from amyloid infiltrates in salivary glands is extremely common, and cases of amyloid in the eyes, bones, and thyroid gland have been reported.2 Macroglossia is seen in 12% to 40% of cases; coupled with xerostomia, it can lead to oropharyngeal dysphagia.2,5
Systemic amyloidosis can be further divided into primary (idiopathic or multiple myeloma associated) or secondary to chronic inflammatory conditions or infections; the key difference is the protein from which the abnormal amyloid is derived.1,4 The presence of cutaneous amyloidosis renders the need to rule out systemic disease because amyloidosis may be a purely localized or systemic process.4 Nodular amyloidosis is a localized form of amyloid that also has immunoglobulin light chain deposits and clinically appears exactly the same as PSA; however, the deposits are restricted to the skin.1,2,4,5
Characteristic biopsy findings in cutaneous amyloidosis include amorphous orange-red amyloid deposits on hematoxylin and eosin–stained sections. The gold standard for amyloid detection is apple green birefringence under polarized light with Congo red stain or electron microscopy.6,7 Other stains used to identify amyloid include crystal violet, methyl violet, periodic acid–Schiff, Sirius red, pagoda red, Dylon stain, and thioflavine T.1,2 Confirmation of systemic disease can be accomplished by fine needle aspiration of abdominal fat-pads or rectal mucosal biopsies.1,3,4 Biopsy of accessory salivary glands also has been reported to be very sensitive and specific.2
Treatment options remain limited; localized cutaneous disease may respond to topical corticosteroids, calcineurin inhibitors, or phototherapy. Primary systemic amyloidosis can be treated with a combination of steroids, melphalan, or colchicine often followed by autologous stem cell transplantation8; however, these regimens are not always curative and patients often have a poor prognosis.1
The Diagnosis: Primary Systemic Amyloidosis
Our patient presented with multiple firm waxy nodules on the mucosal surface of the lower lip. Excision biopsy showed thickening of blood vessel walls with abundant amorphous material that was consistent with amyloid. Further staining with Congo red demonstrated brick red amorphous material within the vessel walls on routine light microscopy (Figure 1), and crystal violet stain showed metachromasia (Figure 2). Fine needle aspiration of the abdominal fat-pad showed amyloid. The final diagnosis was primary systemic amyloidosis (PSA).
|
|
| Figure 1. Congo red staining showed a brick red appearance of amyloid within the vessel walls on routine light microscopy (original magnification ×200). |
|
|
| Figure 2. Crystal violet staining around the blood vessels showed metachromasia (original magnification ×100). |
Amyloid is an ubiquitous fibrillar protein arranged in a cross-beta-pleated sheet that is confirmed with x-ray crystallography.1,2 More than 25 variants of amyloid have been identified.3 Pathologic deposition of amyloid-derived material results in a variable spectrum of clinical findings, collectively known as amyloidosis, with presentations ranging from nonspecific fever or fatigue to frank organ failure, depending on the organ involved. In PSA, immunoglobulin light chains are deposited throughout the body. Associated conditions include malignant or benign monoclonal gammopathy, multiple myeloma, Waldenström macroglobulinemia, malignant lymphoma, heavy chain disease, and chronic lymphocytic leukemia.2,4 The most commonly involved organ systems are the heart, lungs, liver, and kidneys. When patients present with unexplained heart failure, orthostatic hypotension, hepatomegaly, peripheral neuropathy, carpal tunnel syndrome, or renal insufficiency, amyloidosis should always be considered in the differential diagnosis.3
Cutaneous lesions of PSA tend to be vascular due to amyloid infiltration of blood vessel walls, manifesting as petechiae, purpura, ecchymoses, or nonhealing ulcers. Pinch purpura frequently are seen in the periorbital region after minor trauma and are recognized as a clinical indicator of PSA.2,4 Xerostomia from amyloid infiltrates in salivary glands is extremely common, and cases of amyloid in the eyes, bones, and thyroid gland have been reported.2 Macroglossia is seen in 12% to 40% of cases; coupled with xerostomia, it can lead to oropharyngeal dysphagia.2,5
Systemic amyloidosis can be further divided into primary (idiopathic or multiple myeloma associated) or secondary to chronic inflammatory conditions or infections; the key difference is the protein from which the abnormal amyloid is derived.1,4 The presence of cutaneous amyloidosis renders the need to rule out systemic disease because amyloidosis may be a purely localized or systemic process.4 Nodular amyloidosis is a localized form of amyloid that also has immunoglobulin light chain deposits and clinically appears exactly the same as PSA; however, the deposits are restricted to the skin.1,2,4,5
Characteristic biopsy findings in cutaneous amyloidosis include amorphous orange-red amyloid deposits on hematoxylin and eosin–stained sections. The gold standard for amyloid detection is apple green birefringence under polarized light with Congo red stain or electron microscopy.6,7 Other stains used to identify amyloid include crystal violet, methyl violet, periodic acid–Schiff, Sirius red, pagoda red, Dylon stain, and thioflavine T.1,2 Confirmation of systemic disease can be accomplished by fine needle aspiration of abdominal fat-pads or rectal mucosal biopsies.1,3,4 Biopsy of accessory salivary glands also has been reported to be very sensitive and specific.2
Treatment options remain limited; localized cutaneous disease may respond to topical corticosteroids, calcineurin inhibitors, or phototherapy. Primary systemic amyloidosis can be treated with a combination of steroids, melphalan, or colchicine often followed by autologous stem cell transplantation8; however, these regimens are not always curative and patients often have a poor prognosis.1
1. Black MM, Upjohn E, Albert S. Amyloidosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 1. 2nd ed. Spain: Mosby Elsevier; 2008:623-631.
2. Steciuk A, Dompmartin A, Troussard X, et al. Cutaneous amyloidosis and possible association with systemic amyloidosis. Int J Dermatol. 2002;41:127-132.
3. Picken MM. Amyloidosis-where are we now and where are we heading? Arch Pathol Lab Med. 2010;134:545-551.
4. Schreml S, Szeimies RM, Vogt T, et al. Cutaneous amyloidoses and systemic amyloidoses with cutaneous involvement. Eur J Dermatol. 2010;20:152-160.
5. Breathnach SM. Amyloid and amyloidosis. J Am Acad Dermatol. 1988;18(1, pt 1):1-16.
6. Li WM. Histopathology of primary cutaneous amyloidoses and systemic amyloidosis. Clin Dermatol. 1990;8:30-35.
7. Lin CS, Wong CK. Electron microscopy of primary and secondary cutaneous amyloidoses and systemic amyloidosis. Clin Dermatol. 1990;8:36-45.
8. Dember LM. Modern treatment of amyloidosis: unresolved questions. J Am Soc Nephrol. 2009;20:469-472.
1. Black MM, Upjohn E, Albert S. Amyloidosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 1. 2nd ed. Spain: Mosby Elsevier; 2008:623-631.
2. Steciuk A, Dompmartin A, Troussard X, et al. Cutaneous amyloidosis and possible association with systemic amyloidosis. Int J Dermatol. 2002;41:127-132.
3. Picken MM. Amyloidosis-where are we now and where are we heading? Arch Pathol Lab Med. 2010;134:545-551.
4. Schreml S, Szeimies RM, Vogt T, et al. Cutaneous amyloidoses and systemic amyloidoses with cutaneous involvement. Eur J Dermatol. 2010;20:152-160.
5. Breathnach SM. Amyloid and amyloidosis. J Am Acad Dermatol. 1988;18(1, pt 1):1-16.
6. Li WM. Histopathology of primary cutaneous amyloidoses and systemic amyloidosis. Clin Dermatol. 1990;8:30-35.
7. Lin CS, Wong CK. Electron microscopy of primary and secondary cutaneous amyloidoses and systemic amyloidosis. Clin Dermatol. 1990;8:36-45.
8. Dember LM. Modern treatment of amyloidosis: unresolved questions. J Am Soc Nephrol. 2009;20:469-472.
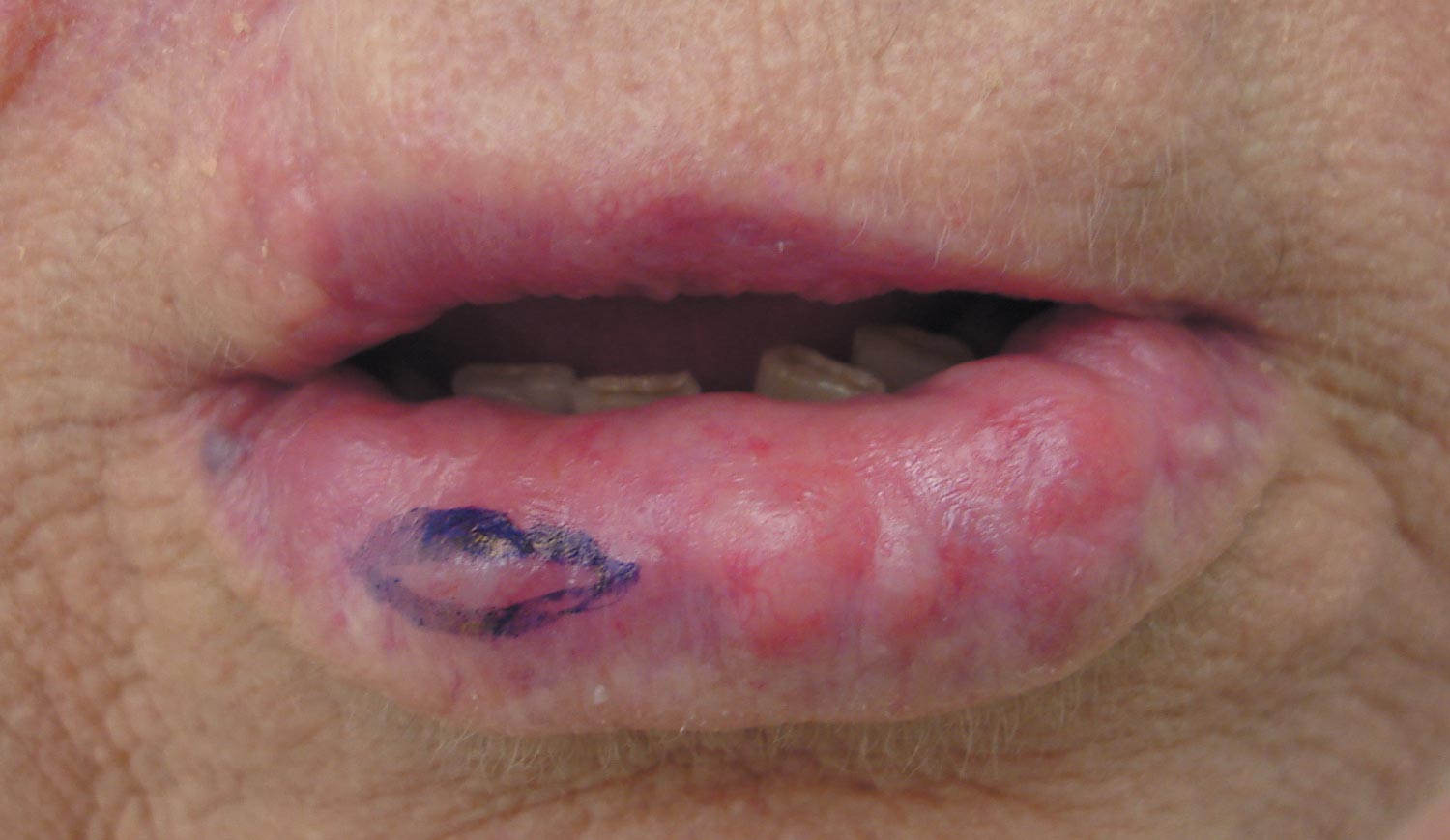
A 71-year-old woman presented with multiple 3×3-mm, firm, nontender nodules of 3 years’ duration on the mucosal surface of the lower lip that gradually enlarged. There was no macroglossia on presentation. The lip nodules were asymptomatic; however, they did interfere with eating and drinking, necessitating the use of a straw. Her medical history was remarkable for emphysema that required supplemental oxygen, rheumatoid arthritis, orthostatic hypotension, renal insufficiency, autonomic nervous system dysfunction, and Sjögren syndrome. She also had a recent thyroidectomy due to multiple thyroid nodules. An excision biopsy was performed of the lip nodules.
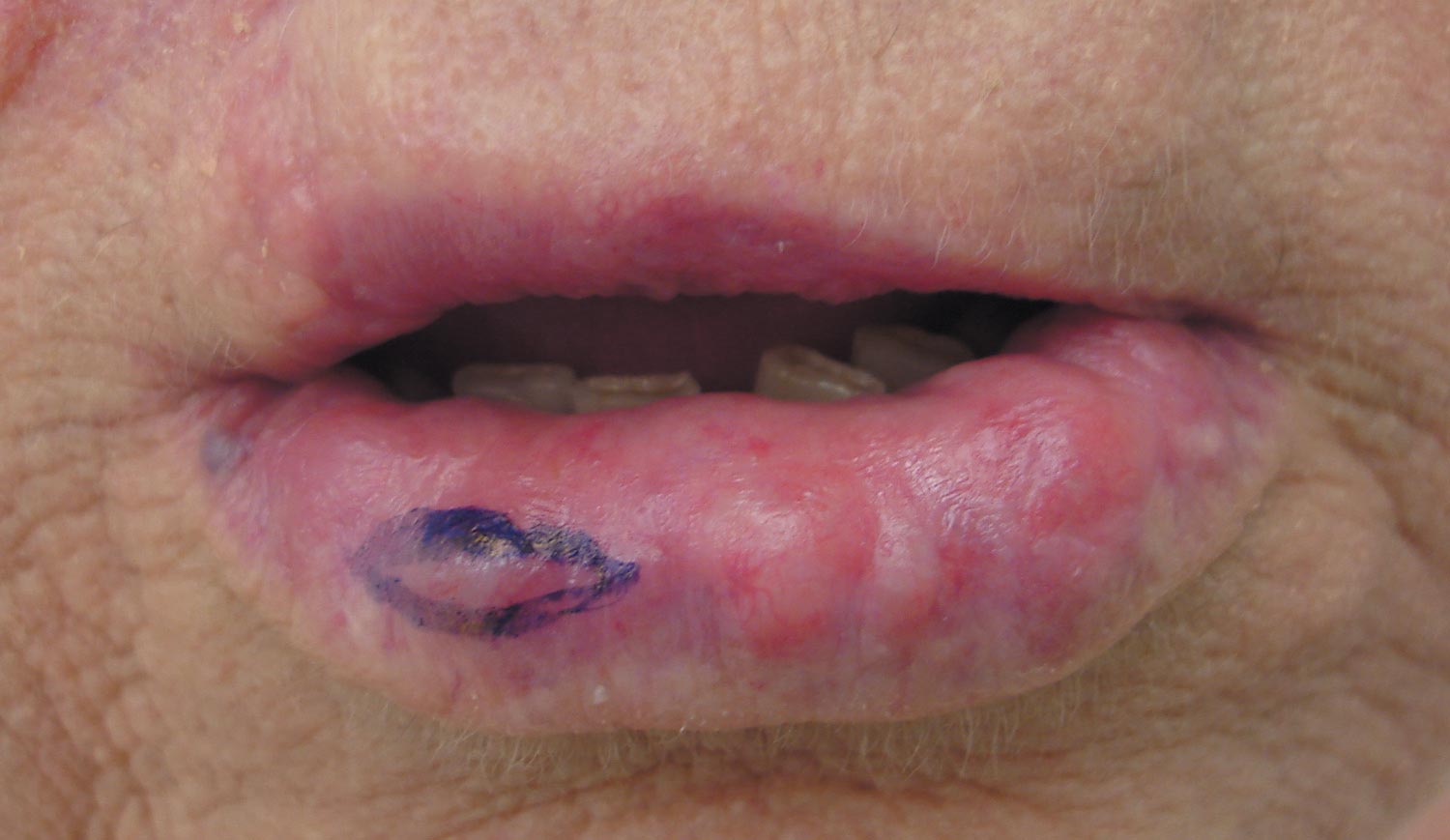
Tender Subcutaneous Nodules on the Back and Shoulders
The Diagnosis: Prostate Cancer Metastases
Cutaneous involvement of visceral tumors can occur indirectly, with the tumor causing changes in the skin without the presence of tumor cells (ie, paraneoplastic processes), or directly, with the presence of tumor cells in the skin (ie, metastasis).1 Cutaneous metastases from solid primary tumors are uncommon. The incidence of metastasis to the skin from visceral malignancies ranges from 0.3% to 9.0%.2 The most common primary internal tumors to metastasize to the skin are those arising from the breasts, lungs, colon, ovaries, and head and neck.3 Although prostate cancer is the most common cancer in males (making up 28% of new cancer diagnoses), excluding basal and squamous cell skin cancers, it rarely metastasizes to the skin, representing less than 1% of all cutaneous metastasis.4,5
There are 4 proposed mechanisms for metastatic dissemination to the skin: (1) direct invasion from underlying neoplasm; (2) implantation from a surgical scar; (3) spread through the lymphatics; and/or (4) hematogenous spread. The most common sites of prostate cancer cutaneous metastasis are the inguinal area, penis, and lower abdomen,2 which is likely due to spread via the lymphatic drainage. Cutaneous metastasis to distant sites such as the scalp or chest, as in our patient, may be secondary to hematogenous spread.
Generally, cutaneous metastases from visceral malignancies manifest as urticaria or a nonspecific macular rash.2 However, prostate cancer cutaneous metastases commonly present as papules or subcutaneous nodules but also can have inflammatory, cicatricial, sclerodermoid, telangiectatic, or zosteriform morphologies.2,5,6 The lesions of prostate cancer cutaneous metastases are typically well-circumscribed, flesh-colored or pink to red, oval plaques or nodules ranging in size from several millimeters to a few centimeters.2 The cutaneous lesions generally are multiple and can be localized or diffuse in distribution. The clinical differential diagnosis is broad in patients undergoing systemic treatment of prostate cancer and often includes drug reaction, cutaneous lymphoma, atypical mycobacterial or deep fungal infection, or paraneoplastic dermatosis.
Laboratory studies often reveal an elevated serum prostate-specific antigen (PSA) level, a glycoprotein that functions in the liquefaction of seminal fluid made primarily by the prostate, often in benign hypertrophic and malignant prostatic processes.7 Definitive diagnosis of prostate cancer cutaneous metastasis can be established by histologic examination. Excisional or punch biopsy is preferred over superficial shave biopsy. Metastasis from prostate adenocarcinoma often reveals infiltrative growth of disorganized atypical epithelial cells along collagen bundles in the dermis and subcutis (Figure 1).2,8 The presence of lymphovascular invasion further increases the suspicion of metastasis. Metastatic lesions may resemble the primary lesion; however, they are often poorly differentiated, making histologic comparison difficult.
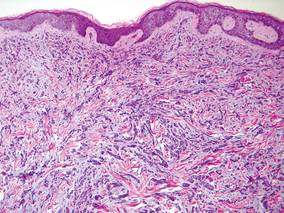
|
| Figure 1. Biopsy revealed cords of atypical epithelioid cells dissecting through dermal collagen bundles, accompanied by abundant mucin. Atypical cells displayed nuclear pleomorphism, and multiple mitotic figures were present (H&E, original magnification ×100). |
|
| Figure 2. Immunohistochemistry was strongly positive for prostate-specific antigen and focally positive for prostate-specific acid phosphatase (original magnification ×100). |
The diagnosis is confirmed immunohistochemically with PSA and/or prostate-specific acid phosphatase immunostaining (Figure 2), which together have nearly 100% specificity for prostate adenocarcinomas.9 Although rare, PSA and or prostate-specific acid phosphatase may be weakly positive in nephrogenic adenoma.10 Another biomarker, urinary prostate cancer antigen 3, is being evaluated to identify men with indolent prostate cancer.11 It is unknown how useful this marker will be in the immunohistochemical identification of prostate cancer cutaneous metastases.
A large review of reported cases of cutaneous metastases from internal malignancies revealed that the majority of patients had known systemic disease but enjoyed a good performance status at the time of diagnosis.2 As such, skin metastases may serve as the initial indicator of visceral recurrence. Prostate cancer is second only to lung cancer as the deadliest cancer in males,4 and cutaneous metastasis garners a particularly poor prognosis. The mean survival time after diagnosis of cutaneous metastasis is 7 months.8 Therefore, treatment of cutaneous metastases is largely palliative, including local excision and intralesional chemotherapy.
1. Thiers BH, Sahn RE, Callen JP. Cutaneous manifestations of internal malignancy. CA Cancer J Clin 2009;59:73-98.
2. Mueller TJ, Wu H, Greenberg RE, et al. Cutaneous metastases from genitourinary malignancies. Urology. 2004;63:1021-1026.
3. Brownstein MH, Helwig EB. Metastatic tumors of the skin. Cancer. 1972;29:1298-1307.
4. Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277-300.
5. Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33:161-182.
6. Reddy S, Bang RH, Contreras ME. Telangiectatic cutaneous metastasis from carcinoma of the prostate. Br J Dermatol. 2007;156:598-600.
7. Tosoian J, Loeb S. PSA and beyond: the past, present, and future of investigative biomarkers for prostate cancer. ScientificWorldJournal. 2010;10:1919-1931.
8. Wang SQ, Mecca PS, Myskowski PL, et al. Scrotal and penile papules and plaques as the initial manifestation of a cutaneous metastasis of adenocarcinoma of the prostate: case report and review of the literature. J Cutan Pathol. 2008;35:681-684.
9. Nadji M, Tabei SZ, Castro A, et al. Prostate-specific antigen: an immunohistochemical marker for prostatic neoplasms. Cancer. 1981;48:1229-1232.
10. Paner GP, Luthringer DJ, Amin MB. Best practice in diagnostic immunohistochemistry: prostate carcinoma and its mimics in needle core biopsies. Arch Pathol Lab Med. 2008;132:1388-1396.
11. Cooperberg MR, Carroll PR, Klotz L. Active surveillance for prostate cancer: progress and promise. J Clin Oncol. 2011;29:3669-3676.
The Diagnosis: Prostate Cancer Metastases
Cutaneous involvement of visceral tumors can occur indirectly, with the tumor causing changes in the skin without the presence of tumor cells (ie, paraneoplastic processes), or directly, with the presence of tumor cells in the skin (ie, metastasis).1 Cutaneous metastases from solid primary tumors are uncommon. The incidence of metastasis to the skin from visceral malignancies ranges from 0.3% to 9.0%.2 The most common primary internal tumors to metastasize to the skin are those arising from the breasts, lungs, colon, ovaries, and head and neck.3 Although prostate cancer is the most common cancer in males (making up 28% of new cancer diagnoses), excluding basal and squamous cell skin cancers, it rarely metastasizes to the skin, representing less than 1% of all cutaneous metastasis.4,5
There are 4 proposed mechanisms for metastatic dissemination to the skin: (1) direct invasion from underlying neoplasm; (2) implantation from a surgical scar; (3) spread through the lymphatics; and/or (4) hematogenous spread. The most common sites of prostate cancer cutaneous metastasis are the inguinal area, penis, and lower abdomen,2 which is likely due to spread via the lymphatic drainage. Cutaneous metastasis to distant sites such as the scalp or chest, as in our patient, may be secondary to hematogenous spread.
Generally, cutaneous metastases from visceral malignancies manifest as urticaria or a nonspecific macular rash.2 However, prostate cancer cutaneous metastases commonly present as papules or subcutaneous nodules but also can have inflammatory, cicatricial, sclerodermoid, telangiectatic, or zosteriform morphologies.2,5,6 The lesions of prostate cancer cutaneous metastases are typically well-circumscribed, flesh-colored or pink to red, oval plaques or nodules ranging in size from several millimeters to a few centimeters.2 The cutaneous lesions generally are multiple and can be localized or diffuse in distribution. The clinical differential diagnosis is broad in patients undergoing systemic treatment of prostate cancer and often includes drug reaction, cutaneous lymphoma, atypical mycobacterial or deep fungal infection, or paraneoplastic dermatosis.
Laboratory studies often reveal an elevated serum prostate-specific antigen (PSA) level, a glycoprotein that functions in the liquefaction of seminal fluid made primarily by the prostate, often in benign hypertrophic and malignant prostatic processes.7 Definitive diagnosis of prostate cancer cutaneous metastasis can be established by histologic examination. Excisional or punch biopsy is preferred over superficial shave biopsy. Metastasis from prostate adenocarcinoma often reveals infiltrative growth of disorganized atypical epithelial cells along collagen bundles in the dermis and subcutis (Figure 1).2,8 The presence of lymphovascular invasion further increases the suspicion of metastasis. Metastatic lesions may resemble the primary lesion; however, they are often poorly differentiated, making histologic comparison difficult.
|
|
| Figure 1. Biopsy revealed cords of atypical epithelioid cells dissecting through dermal collagen bundles, accompanied by abundant mucin. Atypical cells displayed nuclear pleomorphism, and multiple mitotic figures were present (H&E, original magnification ×100). |
|
|
| Figure 2. Immunohistochemistry was strongly positive for prostate-specific antigen and focally positive for prostate-specific acid phosphatase (original magnification ×100). |
The diagnosis is confirmed immunohistochemically with PSA and/or prostate-specific acid phosphatase immunostaining (Figure 2), which together have nearly 100% specificity for prostate adenocarcinomas.9 Although rare, PSA and or prostate-specific acid phosphatase may be weakly positive in nephrogenic adenoma.10 Another biomarker, urinary prostate cancer antigen 3, is being evaluated to identify men with indolent prostate cancer.11 It is unknown how useful this marker will be in the immunohistochemical identification of prostate cancer cutaneous metastases.
A large review of reported cases of cutaneous metastases from internal malignancies revealed that the majority of patients had known systemic disease but enjoyed a good performance status at the time of diagnosis.2 As such, skin metastases may serve as the initial indicator of visceral recurrence. Prostate cancer is second only to lung cancer as the deadliest cancer in males,4 and cutaneous metastasis garners a particularly poor prognosis. The mean survival time after diagnosis of cutaneous metastasis is 7 months.8 Therefore, treatment of cutaneous metastases is largely palliative, including local excision and intralesional chemotherapy.
The Diagnosis: Prostate Cancer Metastases
Cutaneous involvement of visceral tumors can occur indirectly, with the tumor causing changes in the skin without the presence of tumor cells (ie, paraneoplastic processes), or directly, with the presence of tumor cells in the skin (ie, metastasis).1 Cutaneous metastases from solid primary tumors are uncommon. The incidence of metastasis to the skin from visceral malignancies ranges from 0.3% to 9.0%.2 The most common primary internal tumors to metastasize to the skin are those arising from the breasts, lungs, colon, ovaries, and head and neck.3 Although prostate cancer is the most common cancer in males (making up 28% of new cancer diagnoses), excluding basal and squamous cell skin cancers, it rarely metastasizes to the skin, representing less than 1% of all cutaneous metastasis.4,5
There are 4 proposed mechanisms for metastatic dissemination to the skin: (1) direct invasion from underlying neoplasm; (2) implantation from a surgical scar; (3) spread through the lymphatics; and/or (4) hematogenous spread. The most common sites of prostate cancer cutaneous metastasis are the inguinal area, penis, and lower abdomen,2 which is likely due to spread via the lymphatic drainage. Cutaneous metastasis to distant sites such as the scalp or chest, as in our patient, may be secondary to hematogenous spread.
Generally, cutaneous metastases from visceral malignancies manifest as urticaria or a nonspecific macular rash.2 However, prostate cancer cutaneous metastases commonly present as papules or subcutaneous nodules but also can have inflammatory, cicatricial, sclerodermoid, telangiectatic, or zosteriform morphologies.2,5,6 The lesions of prostate cancer cutaneous metastases are typically well-circumscribed, flesh-colored or pink to red, oval plaques or nodules ranging in size from several millimeters to a few centimeters.2 The cutaneous lesions generally are multiple and can be localized or diffuse in distribution. The clinical differential diagnosis is broad in patients undergoing systemic treatment of prostate cancer and often includes drug reaction, cutaneous lymphoma, atypical mycobacterial or deep fungal infection, or paraneoplastic dermatosis.
Laboratory studies often reveal an elevated serum prostate-specific antigen (PSA) level, a glycoprotein that functions in the liquefaction of seminal fluid made primarily by the prostate, often in benign hypertrophic and malignant prostatic processes.7 Definitive diagnosis of prostate cancer cutaneous metastasis can be established by histologic examination. Excisional or punch biopsy is preferred over superficial shave biopsy. Metastasis from prostate adenocarcinoma often reveals infiltrative growth of disorganized atypical epithelial cells along collagen bundles in the dermis and subcutis (Figure 1).2,8 The presence of lymphovascular invasion further increases the suspicion of metastasis. Metastatic lesions may resemble the primary lesion; however, they are often poorly differentiated, making histologic comparison difficult.
|
|
| Figure 1. Biopsy revealed cords of atypical epithelioid cells dissecting through dermal collagen bundles, accompanied by abundant mucin. Atypical cells displayed nuclear pleomorphism, and multiple mitotic figures were present (H&E, original magnification ×100). |
|
|
| Figure 2. Immunohistochemistry was strongly positive for prostate-specific antigen and focally positive for prostate-specific acid phosphatase (original magnification ×100). |
The diagnosis is confirmed immunohistochemically with PSA and/or prostate-specific acid phosphatase immunostaining (Figure 2), which together have nearly 100% specificity for prostate adenocarcinomas.9 Although rare, PSA and or prostate-specific acid phosphatase may be weakly positive in nephrogenic adenoma.10 Another biomarker, urinary prostate cancer antigen 3, is being evaluated to identify men with indolent prostate cancer.11 It is unknown how useful this marker will be in the immunohistochemical identification of prostate cancer cutaneous metastases.
A large review of reported cases of cutaneous metastases from internal malignancies revealed that the majority of patients had known systemic disease but enjoyed a good performance status at the time of diagnosis.2 As such, skin metastases may serve as the initial indicator of visceral recurrence. Prostate cancer is second only to lung cancer as the deadliest cancer in males,4 and cutaneous metastasis garners a particularly poor prognosis. The mean survival time after diagnosis of cutaneous metastasis is 7 months.8 Therefore, treatment of cutaneous metastases is largely palliative, including local excision and intralesional chemotherapy.
1. Thiers BH, Sahn RE, Callen JP. Cutaneous manifestations of internal malignancy. CA Cancer J Clin 2009;59:73-98.
2. Mueller TJ, Wu H, Greenberg RE, et al. Cutaneous metastases from genitourinary malignancies. Urology. 2004;63:1021-1026.
3. Brownstein MH, Helwig EB. Metastatic tumors of the skin. Cancer. 1972;29:1298-1307.
4. Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277-300.
5. Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33:161-182.
6. Reddy S, Bang RH, Contreras ME. Telangiectatic cutaneous metastasis from carcinoma of the prostate. Br J Dermatol. 2007;156:598-600.
7. Tosoian J, Loeb S. PSA and beyond: the past, present, and future of investigative biomarkers for prostate cancer. ScientificWorldJournal. 2010;10:1919-1931.
8. Wang SQ, Mecca PS, Myskowski PL, et al. Scrotal and penile papules and plaques as the initial manifestation of a cutaneous metastasis of adenocarcinoma of the prostate: case report and review of the literature. J Cutan Pathol. 2008;35:681-684.
9. Nadji M, Tabei SZ, Castro A, et al. Prostate-specific antigen: an immunohistochemical marker for prostatic neoplasms. Cancer. 1981;48:1229-1232.
10. Paner GP, Luthringer DJ, Amin MB. Best practice in diagnostic immunohistochemistry: prostate carcinoma and its mimics in needle core biopsies. Arch Pathol Lab Med. 2008;132:1388-1396.
11. Cooperberg MR, Carroll PR, Klotz L. Active surveillance for prostate cancer: progress and promise. J Clin Oncol. 2011;29:3669-3676.
1. Thiers BH, Sahn RE, Callen JP. Cutaneous manifestations of internal malignancy. CA Cancer J Clin 2009;59:73-98.
2. Mueller TJ, Wu H, Greenberg RE, et al. Cutaneous metastases from genitourinary malignancies. Urology. 2004;63:1021-1026.
3. Brownstein MH, Helwig EB. Metastatic tumors of the skin. Cancer. 1972;29:1298-1307.
4. Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277-300.
5. Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33:161-182.
6. Reddy S, Bang RH, Contreras ME. Telangiectatic cutaneous metastasis from carcinoma of the prostate. Br J Dermatol. 2007;156:598-600.
7. Tosoian J, Loeb S. PSA and beyond: the past, present, and future of investigative biomarkers for prostate cancer. ScientificWorldJournal. 2010;10:1919-1931.
8. Wang SQ, Mecca PS, Myskowski PL, et al. Scrotal and penile papules and plaques as the initial manifestation of a cutaneous metastasis of adenocarcinoma of the prostate: case report and review of the literature. J Cutan Pathol. 2008;35:681-684.
9. Nadji M, Tabei SZ, Castro A, et al. Prostate-specific antigen: an immunohistochemical marker for prostatic neoplasms. Cancer. 1981;48:1229-1232.
10. Paner GP, Luthringer DJ, Amin MB. Best practice in diagnostic immunohistochemistry: prostate carcinoma and its mimics in needle core biopsies. Arch Pathol Lab Med. 2008;132:1388-1396.
11. Cooperberg MR, Carroll PR, Klotz L. Active surveillance for prostate cancer: progress and promise. J Clin Oncol. 2011;29:3669-3676.
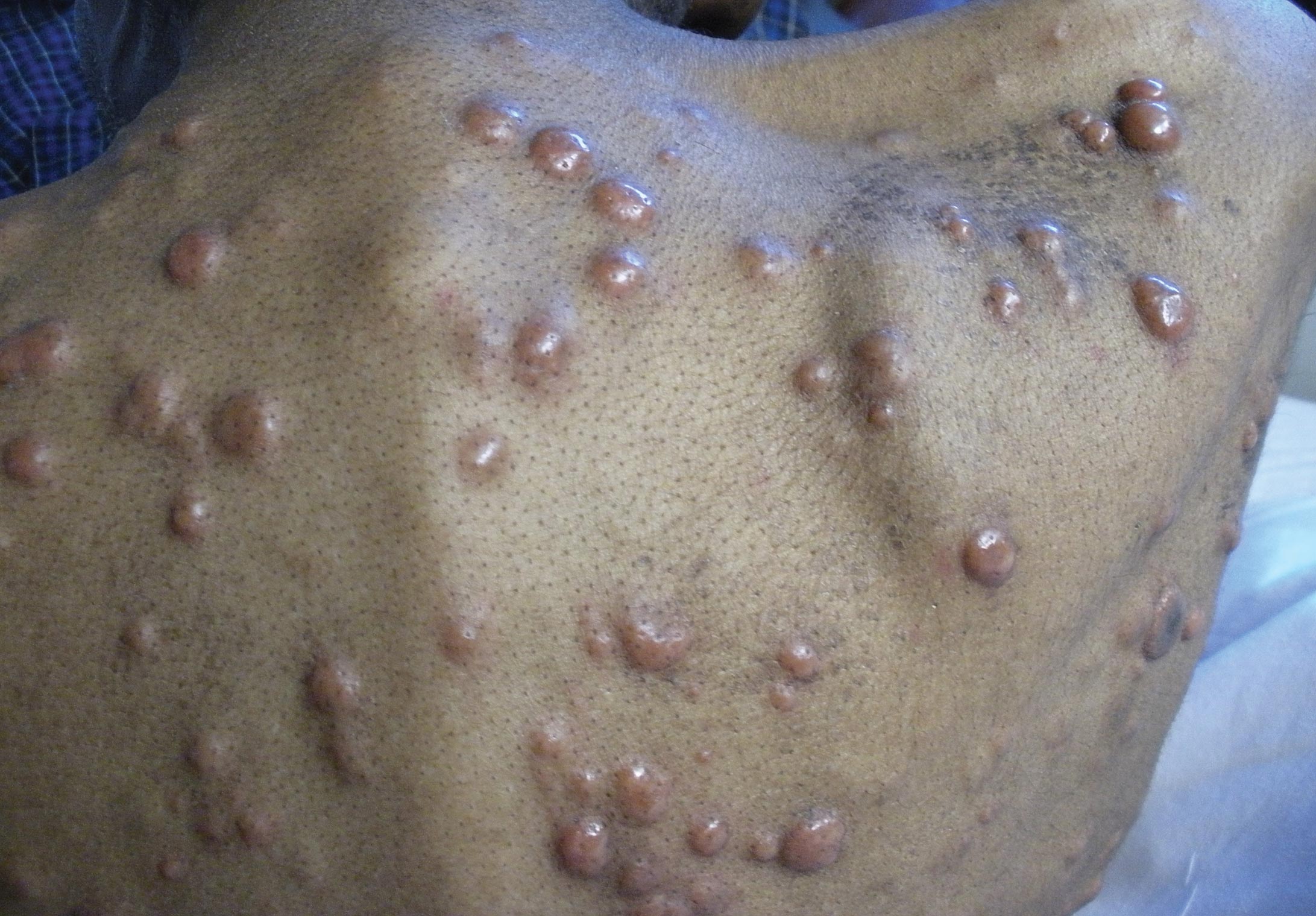
A 57-year-old cachectic man with a history of metastatic, hormone-refractory adenocarcinoma of the prostate presented with multiple tender subcutaneous nodules on the back and shoulders that developed over the course of 12 months. During that time, he was treated with cyclophosphamide, leuprolide acetate, bicalutamide, zoledronic acid, and filgrastim. A punch biopsy specimen obtained from the left shoulder revealed cords of atypical epithelioid cells dissecting through dermal collagen bundles, accompanied by abundant mucin. The atypical cells displayed nuclear pleomorphism, and multiple mitotic figures were observed.
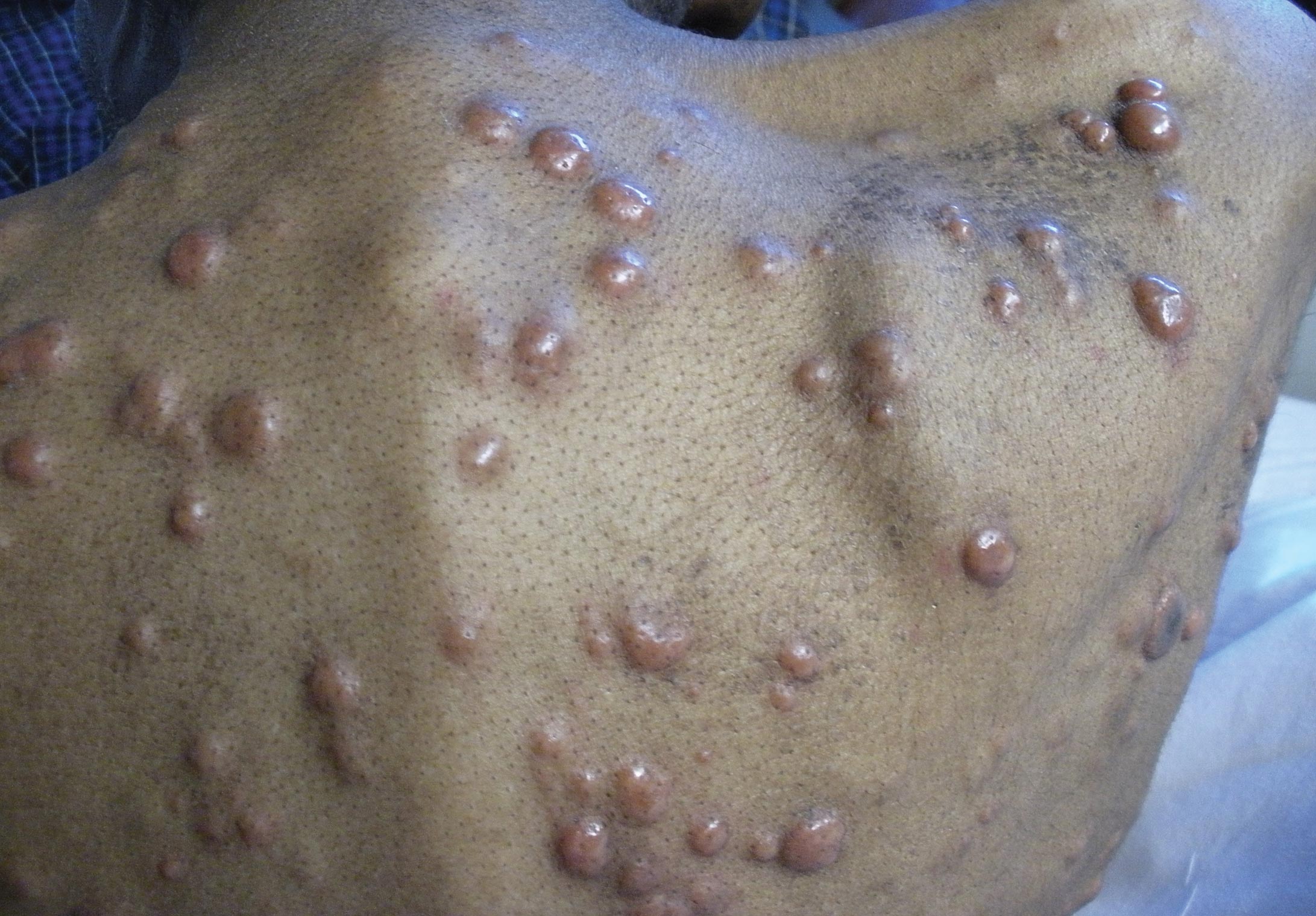
Shedding of the Fingernails
The Diagnosis: Onychomadesis
The nail changes were characteristic of onychomadesis. Systemic illness in this patient most likely resulted in temporary arrest of nail matrix activity, leading to separation of the proximal nail plate from the proximal nail fold, which gave rise to a deep transverse sulcus.1 Conversely, Beau lines are characterized by transverse grooves that move distally as the nail grows. Onychomadesis also is seen in pemphigus vulgaris, which could be due to an autoimmune disease inhibiting normal nail plate growth and development of blisters beneath the nail causing detachment of the nail plate.2 Drug-induced Beau lines or onychomadesis are most frequently caused by chemotherapeutic agents (taxanes) and retinoids, which reflect an arrest in epithelial proliferation.3 Familial cases also have been described.4 Management of the nail abnormality should focus on the underlying medical problem or triggering factor. In our patient, hypertension and kidney disease were managed by a low-salt diet, oral antihypertensives, and iron replacement.
1. Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2008.
2. Engineer L, Norton LA, Ahmed AR. Nail involvement in pemphigus vulgaris. J Am Acad Dermatol. 2000;43:529-535.
3. Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;14:333-337.
4. Mehra A, Murphy RJ, Wilson BB. Idiopathic familial onychomadesis. J Am Acad Dermatol. 2000;43(2, pt 2):349-350.
The Diagnosis: Onychomadesis
The nail changes were characteristic of onychomadesis. Systemic illness in this patient most likely resulted in temporary arrest of nail matrix activity, leading to separation of the proximal nail plate from the proximal nail fold, which gave rise to a deep transverse sulcus.1 Conversely, Beau lines are characterized by transverse grooves that move distally as the nail grows. Onychomadesis also is seen in pemphigus vulgaris, which could be due to an autoimmune disease inhibiting normal nail plate growth and development of blisters beneath the nail causing detachment of the nail plate.2 Drug-induced Beau lines or onychomadesis are most frequently caused by chemotherapeutic agents (taxanes) and retinoids, which reflect an arrest in epithelial proliferation.3 Familial cases also have been described.4 Management of the nail abnormality should focus on the underlying medical problem or triggering factor. In our patient, hypertension and kidney disease were managed by a low-salt diet, oral antihypertensives, and iron replacement.
The Diagnosis: Onychomadesis
The nail changes were characteristic of onychomadesis. Systemic illness in this patient most likely resulted in temporary arrest of nail matrix activity, leading to separation of the proximal nail plate from the proximal nail fold, which gave rise to a deep transverse sulcus.1 Conversely, Beau lines are characterized by transverse grooves that move distally as the nail grows. Onychomadesis also is seen in pemphigus vulgaris, which could be due to an autoimmune disease inhibiting normal nail plate growth and development of blisters beneath the nail causing detachment of the nail plate.2 Drug-induced Beau lines or onychomadesis are most frequently caused by chemotherapeutic agents (taxanes) and retinoids, which reflect an arrest in epithelial proliferation.3 Familial cases also have been described.4 Management of the nail abnormality should focus on the underlying medical problem or triggering factor. In our patient, hypertension and kidney disease were managed by a low-salt diet, oral antihypertensives, and iron replacement.
1. Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2008.
2. Engineer L, Norton LA, Ahmed AR. Nail involvement in pemphigus vulgaris. J Am Acad Dermatol. 2000;43:529-535.
3. Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;14:333-337.
4. Mehra A, Murphy RJ, Wilson BB. Idiopathic familial onychomadesis. J Am Acad Dermatol. 2000;43(2, pt 2):349-350.
1. Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2008.
2. Engineer L, Norton LA, Ahmed AR. Nail involvement in pemphigus vulgaris. J Am Acad Dermatol. 2000;43:529-535.
3. Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;14:333-337.
4. Mehra A, Murphy RJ, Wilson BB. Idiopathic familial onychomadesis. J Am Acad Dermatol. 2000;43(2, pt 2):349-350.
A 70-year-old woman was referred to the dermatology department with abnormal-appearing fingernails of 6 months’ duration. Clinical examination showed complete shedding of the proximal nail plate and separation from the nail bed involving all the fingernails. There also was thickening of the distal nail plate. The patient also had diffuse thinning of the hair on the scalp. She had chronic kidney disease, likely from hypertensive nephrosclerosis, that was complicated by iron-deficient anemia. No new systemic medication had been given.
Nonpruritic Papular Facial Eruption
The Diagnosis: Granulomatous Periorificial Dermatitis
Review of the prior biopsy from the lower cutaneous lip revealed granulomatous perifolliculitis. The patient’s age combined with the morphology, distribution, and histopathologic features (Figure) of his eruption were characteristic of granulomatous periorificial dermatitis (GPD) of childhood. The patient was treated with oral erythromycin and topical metronidazole with immediate improvement, particularly with resolution of the erythema.
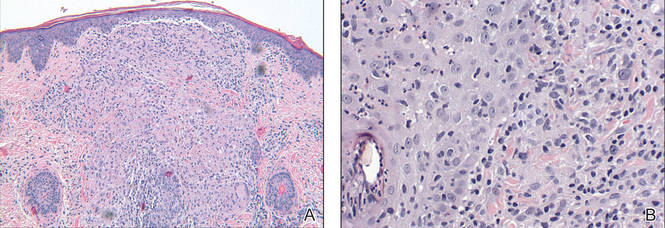
Granulomatous periorificial dermatitis is a benign, self-limited eruption in healthy prepubertal children that is characterized by coalescent, asymptomatic, dome-shaped, yellow-brown to erythematous papules.1,2 The monomorphous lesions are firm, measure 1 to 3 mm in diameter, and are most commonly located around the mouth.2,3 Other areas of involvement include the nostrils and alar creases as well as the periocular skin.4 Less commonly, GPD has been described on the scalp, ears, neck, trunk, extremities, and genital region.5 Slight peripheral scaling or erythema and small pitted scarring are variable.4,5 There may be a history of failed topical corticosteroid treatment that either caused no change or a flare in the rash.1-5 Patients have no systemic findings.3,5
Biopsy of GPD shows characteristic noncaseating granulomas in the dermis, typified by perifollicular localization.5 The granulomatous infiltrate consists of epithelioid histiocytes and multinucleated giant cells surrounded by lymphocytes, with focal collections of neutrophils and occasionally overlying parakeratosis.2,3
The nomenclature of GPD has varied since the 1970s and has included Gianotti-type perioral dermatitis,6 GPD of childhood,7 rosacealike eruption in children,8 FACE (facial Afro-Caribbean childhood eruption),9 and sarcoidlike granulomatous dermatitis.10
The etiology of GPD is unknown, though it has been associated with use of topical, inhaled, or systemic steroids; a personal history of skin problems; a family history of atopy; vaccination; and local reactions to allergens such as cosmetic preparations, antiseptic solutions, tartar control toothpaste, and bubble gum.4,5,11-13 Granulomatous periorificial dermatitis may represent a pediatric variant of granulomatous rosacea or a granulomatous variant of perioral dermatitis, but importantly, it is not related to sarcoidosis.1,4,5 Granulomatous periorificial dermatitis typically affects children of dark-skinned, African Caribbean ancestry, though it has been described in both white and Asian populations.1,2,5 Genders are equally affected.
Although generally a benign condition that spontaneously remits within a few months to 3 years,5 GPD can be quite disruptive to a patient’s self-image, necessitating therapy to hasten resolution. Prior to initiation of treatment, any topical corticosteroids being applied to the affected region should be discontinued. For children older than 9 years, a suggested regimen is oral tetracycline 250 mg twice daily; for those younger than 9 years, erythromycin 30 to 40 mg/kg daily in 2 divided doses is advised.14 Metronidazole 0.75% cream and gel also have shown efficacy in GPD and represent a topical adjunct or alternative to oral therapy.1,14,15
- Knautz MA, Lesher JL. Childhood granulomatous periorificial dermatitis. Pediatr Dermatol. 1996;13:131-134.
- Choi YL, Lee KJ, Cho HJ, et al. Case of childhood granulomatous periorificial dermatitis in a Korean boy treated by oral erythromycin. J Dermatol. 2006;33:806-808.
- Tarm K, Creel NB, Krivda SJ, et al. Granulomatous periorificial dermatitis. Cutis. 2004;73:399-402.
- Nguyen V, Eichenfield LF. Periorificial dermatitis in children and adolescents. J Am Acad Dermatol. 2006;55:781-785.
- Urbatsch AJ, Frieden I, Williams ML, et al. Extrafacial and generalized granulomatous periorificial dermatitis. Arch Dermatol. 2002;138:1354-1358.
- Gianotti F, Ermacora E, Bennelli MG, et al. Particuliere dermatite peri-orale infantile: observations sur cinq cas. Bull Soc Fr Dermatol Syph. 1970;77:341.
- Frieden IJ, Prose NS, Fletcher V, et al. Granulomatous perioral dermatitis in children. Arch Dermatol. 1989;125:369-373.
- Savin JA, Alexander S, Marks R. A rosacea-like eruption of children. Br J Dermatol. 1972;87:425-429.
- Marten RH, Presbury DG, Adamson JE, et al. An unusual popular and acneiform facial eruption in the Negro child. Br J Dermatol. 1974;91:435-438.
- Falk ES. Sarcoid-like granulomatous periocular dermatitis treated with tetracycline. Acta Derm Venereol. 1985;65:270-272.
- Hafeez ZH. Perioral dermatitis: an update. Int J Dermatol. 2003;42:514-517.
- Ferlito TA. Tartar-control toothpaste and perioral dermatitis. J Clin Oncol. 1992;27:43-44.
- Georgouras K, Kocsard E. Micropapular sarcoidal facial eruption in a child. Acta Derm Venereol. 1978;48:433-436.
- Laude TA, Salvemini JN. Perioral dermatitis in children. Semin Cutan Med Surg. 1999;18:206-209.
- Miller SR, Shalita AR. Topical metronidazole gel (0.75%) for the treatment of perioral dermatitis in children. J Am Acad Dermatol. 1994;31:847-848.
The Diagnosis: Granulomatous Periorificial Dermatitis
Review of the prior biopsy from the lower cutaneous lip revealed granulomatous perifolliculitis. The patient’s age combined with the morphology, distribution, and histopathologic features (Figure) of his eruption were characteristic of granulomatous periorificial dermatitis (GPD) of childhood. The patient was treated with oral erythromycin and topical metronidazole with immediate improvement, particularly with resolution of the erythema.
Granulomatous periorificial dermatitis is a benign, self-limited eruption in healthy prepubertal children that is characterized by coalescent, asymptomatic, dome-shaped, yellow-brown to erythematous papules.1,2 The monomorphous lesions are firm, measure 1 to 3 mm in diameter, and are most commonly located around the mouth.2,3 Other areas of involvement include the nostrils and alar creases as well as the periocular skin.4 Less commonly, GPD has been described on the scalp, ears, neck, trunk, extremities, and genital region.5 Slight peripheral scaling or erythema and small pitted scarring are variable.4,5 There may be a history of failed topical corticosteroid treatment that either caused no change or a flare in the rash.1-5 Patients have no systemic findings.3,5
Biopsy of GPD shows characteristic noncaseating granulomas in the dermis, typified by perifollicular localization.5 The granulomatous infiltrate consists of epithelioid histiocytes and multinucleated giant cells surrounded by lymphocytes, with focal collections of neutrophils and occasionally overlying parakeratosis.2,3
The nomenclature of GPD has varied since the 1970s and has included Gianotti-type perioral dermatitis,6 GPD of childhood,7 rosacealike eruption in children,8 FACE (facial Afro-Caribbean childhood eruption),9 and sarcoidlike granulomatous dermatitis.10
The etiology of GPD is unknown, though it has been associated with use of topical, inhaled, or systemic steroids; a personal history of skin problems; a family history of atopy; vaccination; and local reactions to allergens such as cosmetic preparations, antiseptic solutions, tartar control toothpaste, and bubble gum.4,5,11-13 Granulomatous periorificial dermatitis may represent a pediatric variant of granulomatous rosacea or a granulomatous variant of perioral dermatitis, but importantly, it is not related to sarcoidosis.1,4,5 Granulomatous periorificial dermatitis typically affects children of dark-skinned, African Caribbean ancestry, though it has been described in both white and Asian populations.1,2,5 Genders are equally affected.
Although generally a benign condition that spontaneously remits within a few months to 3 years,5 GPD can be quite disruptive to a patient’s self-image, necessitating therapy to hasten resolution. Prior to initiation of treatment, any topical corticosteroids being applied to the affected region should be discontinued. For children older than 9 years, a suggested regimen is oral tetracycline 250 mg twice daily; for those younger than 9 years, erythromycin 30 to 40 mg/kg daily in 2 divided doses is advised.14 Metronidazole 0.75% cream and gel also have shown efficacy in GPD and represent a topical adjunct or alternative to oral therapy.1,14,15
The Diagnosis: Granulomatous Periorificial Dermatitis
Review of the prior biopsy from the lower cutaneous lip revealed granulomatous perifolliculitis. The patient’s age combined with the morphology, distribution, and histopathologic features (Figure) of his eruption were characteristic of granulomatous periorificial dermatitis (GPD) of childhood. The patient was treated with oral erythromycin and topical metronidazole with immediate improvement, particularly with resolution of the erythema.
Granulomatous periorificial dermatitis is a benign, self-limited eruption in healthy prepubertal children that is characterized by coalescent, asymptomatic, dome-shaped, yellow-brown to erythematous papules.1,2 The monomorphous lesions are firm, measure 1 to 3 mm in diameter, and are most commonly located around the mouth.2,3 Other areas of involvement include the nostrils and alar creases as well as the periocular skin.4 Less commonly, GPD has been described on the scalp, ears, neck, trunk, extremities, and genital region.5 Slight peripheral scaling or erythema and small pitted scarring are variable.4,5 There may be a history of failed topical corticosteroid treatment that either caused no change or a flare in the rash.1-5 Patients have no systemic findings.3,5
Biopsy of GPD shows characteristic noncaseating granulomas in the dermis, typified by perifollicular localization.5 The granulomatous infiltrate consists of epithelioid histiocytes and multinucleated giant cells surrounded by lymphocytes, with focal collections of neutrophils and occasionally overlying parakeratosis.2,3
The nomenclature of GPD has varied since the 1970s and has included Gianotti-type perioral dermatitis,6 GPD of childhood,7 rosacealike eruption in children,8 FACE (facial Afro-Caribbean childhood eruption),9 and sarcoidlike granulomatous dermatitis.10
The etiology of GPD is unknown, though it has been associated with use of topical, inhaled, or systemic steroids; a personal history of skin problems; a family history of atopy; vaccination; and local reactions to allergens such as cosmetic preparations, antiseptic solutions, tartar control toothpaste, and bubble gum.4,5,11-13 Granulomatous periorificial dermatitis may represent a pediatric variant of granulomatous rosacea or a granulomatous variant of perioral dermatitis, but importantly, it is not related to sarcoidosis.1,4,5 Granulomatous periorificial dermatitis typically affects children of dark-skinned, African Caribbean ancestry, though it has been described in both white and Asian populations.1,2,5 Genders are equally affected.
Although generally a benign condition that spontaneously remits within a few months to 3 years,5 GPD can be quite disruptive to a patient’s self-image, necessitating therapy to hasten resolution. Prior to initiation of treatment, any topical corticosteroids being applied to the affected region should be discontinued. For children older than 9 years, a suggested regimen is oral tetracycline 250 mg twice daily; for those younger than 9 years, erythromycin 30 to 40 mg/kg daily in 2 divided doses is advised.14 Metronidazole 0.75% cream and gel also have shown efficacy in GPD and represent a topical adjunct or alternative to oral therapy.1,14,15
- Knautz MA, Lesher JL. Childhood granulomatous periorificial dermatitis. Pediatr Dermatol. 1996;13:131-134.
- Choi YL, Lee KJ, Cho HJ, et al. Case of childhood granulomatous periorificial dermatitis in a Korean boy treated by oral erythromycin. J Dermatol. 2006;33:806-808.
- Tarm K, Creel NB, Krivda SJ, et al. Granulomatous periorificial dermatitis. Cutis. 2004;73:399-402.
- Nguyen V, Eichenfield LF. Periorificial dermatitis in children and adolescents. J Am Acad Dermatol. 2006;55:781-785.
- Urbatsch AJ, Frieden I, Williams ML, et al. Extrafacial and generalized granulomatous periorificial dermatitis. Arch Dermatol. 2002;138:1354-1358.
- Gianotti F, Ermacora E, Bennelli MG, et al. Particuliere dermatite peri-orale infantile: observations sur cinq cas. Bull Soc Fr Dermatol Syph. 1970;77:341.
- Frieden IJ, Prose NS, Fletcher V, et al. Granulomatous perioral dermatitis in children. Arch Dermatol. 1989;125:369-373.
- Savin JA, Alexander S, Marks R. A rosacea-like eruption of children. Br J Dermatol. 1972;87:425-429.
- Marten RH, Presbury DG, Adamson JE, et al. An unusual popular and acneiform facial eruption in the Negro child. Br J Dermatol. 1974;91:435-438.
- Falk ES. Sarcoid-like granulomatous periocular dermatitis treated with tetracycline. Acta Derm Venereol. 1985;65:270-272.
- Hafeez ZH. Perioral dermatitis: an update. Int J Dermatol. 2003;42:514-517.
- Ferlito TA. Tartar-control toothpaste and perioral dermatitis. J Clin Oncol. 1992;27:43-44.
- Georgouras K, Kocsard E. Micropapular sarcoidal facial eruption in a child. Acta Derm Venereol. 1978;48:433-436.
- Laude TA, Salvemini JN. Perioral dermatitis in children. Semin Cutan Med Surg. 1999;18:206-209.
- Miller SR, Shalita AR. Topical metronidazole gel (0.75%) for the treatment of perioral dermatitis in children. J Am Acad Dermatol. 1994;31:847-848.
- Knautz MA, Lesher JL. Childhood granulomatous periorificial dermatitis. Pediatr Dermatol. 1996;13:131-134.
- Choi YL, Lee KJ, Cho HJ, et al. Case of childhood granulomatous periorificial dermatitis in a Korean boy treated by oral erythromycin. J Dermatol. 2006;33:806-808.
- Tarm K, Creel NB, Krivda SJ, et al. Granulomatous periorificial dermatitis. Cutis. 2004;73:399-402.
- Nguyen V, Eichenfield LF. Periorificial dermatitis in children and adolescents. J Am Acad Dermatol. 2006;55:781-785.
- Urbatsch AJ, Frieden I, Williams ML, et al. Extrafacial and generalized granulomatous periorificial dermatitis. Arch Dermatol. 2002;138:1354-1358.
- Gianotti F, Ermacora E, Bennelli MG, et al. Particuliere dermatite peri-orale infantile: observations sur cinq cas. Bull Soc Fr Dermatol Syph. 1970;77:341.
- Frieden IJ, Prose NS, Fletcher V, et al. Granulomatous perioral dermatitis in children. Arch Dermatol. 1989;125:369-373.
- Savin JA, Alexander S, Marks R. A rosacea-like eruption of children. Br J Dermatol. 1972;87:425-429.
- Marten RH, Presbury DG, Adamson JE, et al. An unusual popular and acneiform facial eruption in the Negro child. Br J Dermatol. 1974;91:435-438.
- Falk ES. Sarcoid-like granulomatous periocular dermatitis treated with tetracycline. Acta Derm Venereol. 1985;65:270-272.
- Hafeez ZH. Perioral dermatitis: an update. Int J Dermatol. 2003;42:514-517.
- Ferlito TA. Tartar-control toothpaste and perioral dermatitis. J Clin Oncol. 1992;27:43-44.
- Georgouras K, Kocsard E. Micropapular sarcoidal facial eruption in a child. Acta Derm Venereol. 1978;48:433-436.
- Laude TA, Salvemini JN. Perioral dermatitis in children. Semin Cutan Med Surg. 1999;18:206-209.
- Miller SR, Shalita AR. Topical metronidazole gel (0.75%) for the treatment of perioral dermatitis in children. J Am Acad Dermatol. 1994;31:847-848.
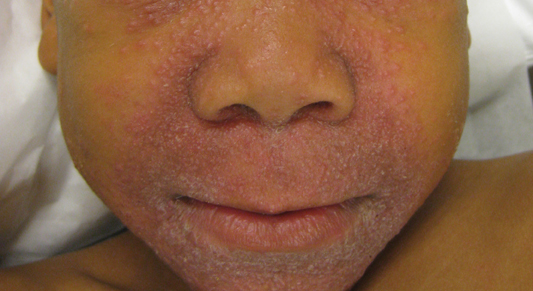
A 7-year-old boy was referred to the dermatology department with a red, bumpy, nonpruritic facial rash of 6 months’ duration. There was no identifiable trigger. The lesions were grouped around the nose and mouth with some extension onto the neck. He was treated with pimecrolimus cream 1% for presumed atopic dermatitis with good response, but the rash recurred soon thereafter. A biopsy performed at an outside institution shortly after rash onset showed dermal granulomas, leading to a diagnosis of cutaneous sarcoidosis (lupus pernio). Prior to presenting to our clinic, treatment with topical and oral corticosteroids failed. He had a normal chest radiograph, ophthalmologic examination, and angiotensin-converting enzyme level to exclude extracutaneous sarcoidosis. On physical examination the patient had innumerable, monomorphic, flesh-colored to erythematous papules confluent over the medial eyelids and canthi, perinasal skin, cutaneous lips, and preauricular skin extending onto the lateral aspect of the neck. There was superimposed scale around the mouth.
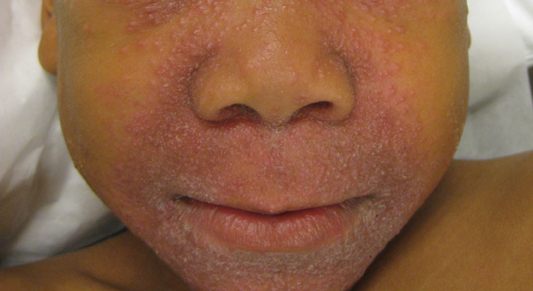
Verrucous Cobblestonelike Papules and Nodules of the Right Lower Limb
The Diagnosis: Elephantiasis Nostras Verrucosa
Elephantiasis nostras verrucosa (ENV) is a chronic deforming disorder characterized by hyperkeratosis and papillomatosis of the epidermis with underlying woody fibrosis of the dermis and the subcutaneous tissue in the setting of chronic nonfilarial lymphedema. Mossy papules, plaques, and cobblestonelike nodules are classic clinical features of ENV (Figure). The cobblestone lesion often is foul smelling and may be colonized with multiple bacteria and fungi.1
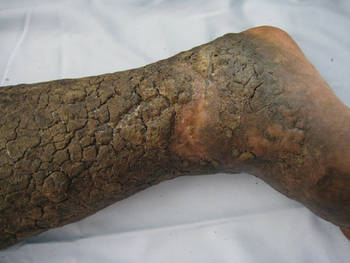
The differential diagnosis includes filariasis, chromoblastomycosis, and venous stasis dermatitis. Filariasis is infection with the filarial worms Wuchereria bancrofti, Brugia malayi, or Brugia timori. These parasites are transmitted to humans through the bite of an infected mosquito and develop into adult worms in the lymphatic vessels, causing lymphedema. Elephantiasis, the classic sign of late-stage disease, also causes swelling of the legs and genital organs that can be disfiguring. Thus travel history to Southeast Asia and Africa regions is particularly important.
Chromoblastomycosis is a chronic fungal infection of the skin and subcutaneous tissue caused by traumatic inoculation of a specific group of dematiaceous fungi, usually Fonsecaea pedrosoi, Phialophora verrucosa, Cladosporium carrionii, or Fonsecaea compacta. Chromoblastomycosis-induced lymphedema may mimic elephantiasis. Thus it is important to perform fungal culture and fungal scraping, which will show typical thick-walled, cigar-colored, sclerotic cells (known as Medlar bodies).
The skin changes of venous stasis dermatitis include edema, varicosities, hyperpigmentation, atrophie blanche, diffuse red-brown discoloration representing deep dermal deposits of hemosiderin, and lipodermatosclerosis. Venous stasis dermatitis is caused by chronic venous insufficiency. It is one of the predispositions to ENV, as it can cause repeated infections and cellulitis leading to blockage of lymphatic drainage and secondary lymphedema. A study of 21 cases highlighted that chronic venous insufficiency may be an underappreciated risk factor in the pathogenesis of ENV.2
The development of verrucous cobblestonelike plaques in ENV occurs with chronic lymphedema. Chronic lymphedema can be caused by a variety of etiologies including infection, chronic venous stasis, congestive heart failure, obesity, trauma, tumor obstruction, and radiation. Our patient was obese with chronic venous stasis, congestive heart failure, and recurrent cellulitis that can lead to secondary lymphatic obstruction and edema, all contributing to the picture of ENV. Because of the unilateral presentation in our patient, we believe that recurrent infections and inflammation are the important risk factors that lead to fibrosis of the dermis and lymph channels, ultimately resulting in ENV.
The diagnosis of ENV is based on the patient’s history and characteristic skin changes. However, a biopsy should be performed if the diagnosis is not clinically apparent, if the lesion looks suspicious for malignancy, or if areas of chronic ulceration exist. Histologic findings include hyperkeratosis as well as a collarette of acanthotic epidermis extending around dilated lymph vessels in the upper dermis. Sometimes a few dilated lymph vessels present in the deeper or subcutaneous tissue. Bacterial and fungal culture should be done to exclude concomitant infection. Ultrasonography can be used to evaluate the lymphatic and venous systems.
Treatment of ENV aims at improving lymphedema and preventing infection. Physical therapy to improve lymphedema includes manual lymphatic drainage, massage, compression stockings, elastic bandages, and leg elevation. In cases associated with obesity, weight loss is recommended. In cases of recurrent cellulitis or lymphangitis, long-term antibiotic therapy with agents such as penicillins or cephalosporins is sometimes used.3 Success has been reported with the use of oral and topical retinoids, such as acitretin and tazarotene, which are thought to decrease epidermal proliferation, fibrogenesis, and inflammation.4,5 Our patient was started on tretinoin cream. Surgery is reserved for cases of lymphedema resistant to medical therapies, including debridement,6 lymphatic and lymphovenous anastomosis,7 lymphatic transplantation, and amputation for severe cases.
Malignancies can develop in areas of chronic lymphedema such as lymphangiosarcoma, squamous cell carcinoma, Kaposi sarcoma, B-cell lymphoma, and malignant fibrous histiocytoma. The morbidity and mortality usually are from the underlying medical problems rather than ENV itself. Our patient died 1 month after the diagnosis of ENV from his poorly controlled heart failure.
- Schissel DJ, Hivnor C, Elston DM. Elephantiasis nostras verrucosa. Cutis. 1998;62:77-80.
- Dean SM, Zirwas MJ, Horst AV. Elephantiasis nostras verrucosa: an institutional analysis of 21 cases. J Am Acad Dermatol. 2011;64:1104-1110.
- Olszewski WL, Jamal S, Manokaran G, et al. The effectiveness of long-acting penicillin (penidur) in preventing recurrences of dermatolymphangioadenitis (DLA) and controlling skin, deep tissues, and lymph bacterial flora in patients with “filarial” lymphedema. Lymphology. 2005;38:66-80.
- Feind-Koopmans A, van de Kerkhof PC. Successful treatment of papillomatosis cutis lymphostatica with acitretin. Acta Derm Venereol. 1995;75:411.
- Boyd J, Sloan S, Meffert J. Elephantiasis nostrum verrucosa of the abdomen: clinical results with tazarotene. J Drugs Dermatol. 2004;3:446-448.
- Iwao F, Sato-Matsumura KC, Sawamura D, et al. Elephantiasis nostras verrucosa successfully treated by surgical debridement. Dermatol Surg. 2004;30:939-41.
- Motegi S, Tamura A, Okada E, et al. Successful treatment with lymphaticovenular anastomosis for secondary skin lesions of chronic lymphedema. Dermatology. 2007;215:147-151.
The Diagnosis: Elephantiasis Nostras Verrucosa
Elephantiasis nostras verrucosa (ENV) is a chronic deforming disorder characterized by hyperkeratosis and papillomatosis of the epidermis with underlying woody fibrosis of the dermis and the subcutaneous tissue in the setting of chronic nonfilarial lymphedema. Mossy papules, plaques, and cobblestonelike nodules are classic clinical features of ENV (Figure). The cobblestone lesion often is foul smelling and may be colonized with multiple bacteria and fungi.1
The differential diagnosis includes filariasis, chromoblastomycosis, and venous stasis dermatitis. Filariasis is infection with the filarial worms Wuchereria bancrofti, Brugia malayi, or Brugia timori. These parasites are transmitted to humans through the bite of an infected mosquito and develop into adult worms in the lymphatic vessels, causing lymphedema. Elephantiasis, the classic sign of late-stage disease, also causes swelling of the legs and genital organs that can be disfiguring. Thus travel history to Southeast Asia and Africa regions is particularly important.
Chromoblastomycosis is a chronic fungal infection of the skin and subcutaneous tissue caused by traumatic inoculation of a specific group of dematiaceous fungi, usually Fonsecaea pedrosoi, Phialophora verrucosa, Cladosporium carrionii, or Fonsecaea compacta. Chromoblastomycosis-induced lymphedema may mimic elephantiasis. Thus it is important to perform fungal culture and fungal scraping, which will show typical thick-walled, cigar-colored, sclerotic cells (known as Medlar bodies).
The skin changes of venous stasis dermatitis include edema, varicosities, hyperpigmentation, atrophie blanche, diffuse red-brown discoloration representing deep dermal deposits of hemosiderin, and lipodermatosclerosis. Venous stasis dermatitis is caused by chronic venous insufficiency. It is one of the predispositions to ENV, as it can cause repeated infections and cellulitis leading to blockage of lymphatic drainage and secondary lymphedema. A study of 21 cases highlighted that chronic venous insufficiency may be an underappreciated risk factor in the pathogenesis of ENV.2
The development of verrucous cobblestonelike plaques in ENV occurs with chronic lymphedema. Chronic lymphedema can be caused by a variety of etiologies including infection, chronic venous stasis, congestive heart failure, obesity, trauma, tumor obstruction, and radiation. Our patient was obese with chronic venous stasis, congestive heart failure, and recurrent cellulitis that can lead to secondary lymphatic obstruction and edema, all contributing to the picture of ENV. Because of the unilateral presentation in our patient, we believe that recurrent infections and inflammation are the important risk factors that lead to fibrosis of the dermis and lymph channels, ultimately resulting in ENV.
The diagnosis of ENV is based on the patient’s history and characteristic skin changes. However, a biopsy should be performed if the diagnosis is not clinically apparent, if the lesion looks suspicious for malignancy, or if areas of chronic ulceration exist. Histologic findings include hyperkeratosis as well as a collarette of acanthotic epidermis extending around dilated lymph vessels in the upper dermis. Sometimes a few dilated lymph vessels present in the deeper or subcutaneous tissue. Bacterial and fungal culture should be done to exclude concomitant infection. Ultrasonography can be used to evaluate the lymphatic and venous systems.
Treatment of ENV aims at improving lymphedema and preventing infection. Physical therapy to improve lymphedema includes manual lymphatic drainage, massage, compression stockings, elastic bandages, and leg elevation. In cases associated with obesity, weight loss is recommended. In cases of recurrent cellulitis or lymphangitis, long-term antibiotic therapy with agents such as penicillins or cephalosporins is sometimes used.3 Success has been reported with the use of oral and topical retinoids, such as acitretin and tazarotene, which are thought to decrease epidermal proliferation, fibrogenesis, and inflammation.4,5 Our patient was started on tretinoin cream. Surgery is reserved for cases of lymphedema resistant to medical therapies, including debridement,6 lymphatic and lymphovenous anastomosis,7 lymphatic transplantation, and amputation for severe cases.
Malignancies can develop in areas of chronic lymphedema such as lymphangiosarcoma, squamous cell carcinoma, Kaposi sarcoma, B-cell lymphoma, and malignant fibrous histiocytoma. The morbidity and mortality usually are from the underlying medical problems rather than ENV itself. Our patient died 1 month after the diagnosis of ENV from his poorly controlled heart failure.
The Diagnosis: Elephantiasis Nostras Verrucosa
Elephantiasis nostras verrucosa (ENV) is a chronic deforming disorder characterized by hyperkeratosis and papillomatosis of the epidermis with underlying woody fibrosis of the dermis and the subcutaneous tissue in the setting of chronic nonfilarial lymphedema. Mossy papules, plaques, and cobblestonelike nodules are classic clinical features of ENV (Figure). The cobblestone lesion often is foul smelling and may be colonized with multiple bacteria and fungi.1
The differential diagnosis includes filariasis, chromoblastomycosis, and venous stasis dermatitis. Filariasis is infection with the filarial worms Wuchereria bancrofti, Brugia malayi, or Brugia timori. These parasites are transmitted to humans through the bite of an infected mosquito and develop into adult worms in the lymphatic vessels, causing lymphedema. Elephantiasis, the classic sign of late-stage disease, also causes swelling of the legs and genital organs that can be disfiguring. Thus travel history to Southeast Asia and Africa regions is particularly important.
Chromoblastomycosis is a chronic fungal infection of the skin and subcutaneous tissue caused by traumatic inoculation of a specific group of dematiaceous fungi, usually Fonsecaea pedrosoi, Phialophora verrucosa, Cladosporium carrionii, or Fonsecaea compacta. Chromoblastomycosis-induced lymphedema may mimic elephantiasis. Thus it is important to perform fungal culture and fungal scraping, which will show typical thick-walled, cigar-colored, sclerotic cells (known as Medlar bodies).
The skin changes of venous stasis dermatitis include edema, varicosities, hyperpigmentation, atrophie blanche, diffuse red-brown discoloration representing deep dermal deposits of hemosiderin, and lipodermatosclerosis. Venous stasis dermatitis is caused by chronic venous insufficiency. It is one of the predispositions to ENV, as it can cause repeated infections and cellulitis leading to blockage of lymphatic drainage and secondary lymphedema. A study of 21 cases highlighted that chronic venous insufficiency may be an underappreciated risk factor in the pathogenesis of ENV.2
The development of verrucous cobblestonelike plaques in ENV occurs with chronic lymphedema. Chronic lymphedema can be caused by a variety of etiologies including infection, chronic venous stasis, congestive heart failure, obesity, trauma, tumor obstruction, and radiation. Our patient was obese with chronic venous stasis, congestive heart failure, and recurrent cellulitis that can lead to secondary lymphatic obstruction and edema, all contributing to the picture of ENV. Because of the unilateral presentation in our patient, we believe that recurrent infections and inflammation are the important risk factors that lead to fibrosis of the dermis and lymph channels, ultimately resulting in ENV.
The diagnosis of ENV is based on the patient’s history and characteristic skin changes. However, a biopsy should be performed if the diagnosis is not clinically apparent, if the lesion looks suspicious for malignancy, or if areas of chronic ulceration exist. Histologic findings include hyperkeratosis as well as a collarette of acanthotic epidermis extending around dilated lymph vessels in the upper dermis. Sometimes a few dilated lymph vessels present in the deeper or subcutaneous tissue. Bacterial and fungal culture should be done to exclude concomitant infection. Ultrasonography can be used to evaluate the lymphatic and venous systems.
Treatment of ENV aims at improving lymphedema and preventing infection. Physical therapy to improve lymphedema includes manual lymphatic drainage, massage, compression stockings, elastic bandages, and leg elevation. In cases associated with obesity, weight loss is recommended. In cases of recurrent cellulitis or lymphangitis, long-term antibiotic therapy with agents such as penicillins or cephalosporins is sometimes used.3 Success has been reported with the use of oral and topical retinoids, such as acitretin and tazarotene, which are thought to decrease epidermal proliferation, fibrogenesis, and inflammation.4,5 Our patient was started on tretinoin cream. Surgery is reserved for cases of lymphedema resistant to medical therapies, including debridement,6 lymphatic and lymphovenous anastomosis,7 lymphatic transplantation, and amputation for severe cases.
Malignancies can develop in areas of chronic lymphedema such as lymphangiosarcoma, squamous cell carcinoma, Kaposi sarcoma, B-cell lymphoma, and malignant fibrous histiocytoma. The morbidity and mortality usually are from the underlying medical problems rather than ENV itself. Our patient died 1 month after the diagnosis of ENV from his poorly controlled heart failure.
- Schissel DJ, Hivnor C, Elston DM. Elephantiasis nostras verrucosa. Cutis. 1998;62:77-80.
- Dean SM, Zirwas MJ, Horst AV. Elephantiasis nostras verrucosa: an institutional analysis of 21 cases. J Am Acad Dermatol. 2011;64:1104-1110.
- Olszewski WL, Jamal S, Manokaran G, et al. The effectiveness of long-acting penicillin (penidur) in preventing recurrences of dermatolymphangioadenitis (DLA) and controlling skin, deep tissues, and lymph bacterial flora in patients with “filarial” lymphedema. Lymphology. 2005;38:66-80.
- Feind-Koopmans A, van de Kerkhof PC. Successful treatment of papillomatosis cutis lymphostatica with acitretin. Acta Derm Venereol. 1995;75:411.
- Boyd J, Sloan S, Meffert J. Elephantiasis nostrum verrucosa of the abdomen: clinical results with tazarotene. J Drugs Dermatol. 2004;3:446-448.
- Iwao F, Sato-Matsumura KC, Sawamura D, et al. Elephantiasis nostras verrucosa successfully treated by surgical debridement. Dermatol Surg. 2004;30:939-41.
- Motegi S, Tamura A, Okada E, et al. Successful treatment with lymphaticovenular anastomosis for secondary skin lesions of chronic lymphedema. Dermatology. 2007;215:147-151.
- Schissel DJ, Hivnor C, Elston DM. Elephantiasis nostras verrucosa. Cutis. 1998;62:77-80.
- Dean SM, Zirwas MJ, Horst AV. Elephantiasis nostras verrucosa: an institutional analysis of 21 cases. J Am Acad Dermatol. 2011;64:1104-1110.
- Olszewski WL, Jamal S, Manokaran G, et al. The effectiveness of long-acting penicillin (penidur) in preventing recurrences of dermatolymphangioadenitis (DLA) and controlling skin, deep tissues, and lymph bacterial flora in patients with “filarial” lymphedema. Lymphology. 2005;38:66-80.
- Feind-Koopmans A, van de Kerkhof PC. Successful treatment of papillomatosis cutis lymphostatica with acitretin. Acta Derm Venereol. 1995;75:411.
- Boyd J, Sloan S, Meffert J. Elephantiasis nostrum verrucosa of the abdomen: clinical results with tazarotene. J Drugs Dermatol. 2004;3:446-448.
- Iwao F, Sato-Matsumura KC, Sawamura D, et al. Elephantiasis nostras verrucosa successfully treated by surgical debridement. Dermatol Surg. 2004;30:939-41.
- Motegi S, Tamura A, Okada E, et al. Successful treatment with lymphaticovenular anastomosis for secondary skin lesions of chronic lymphedema. Dermatology. 2007;215:147-151.
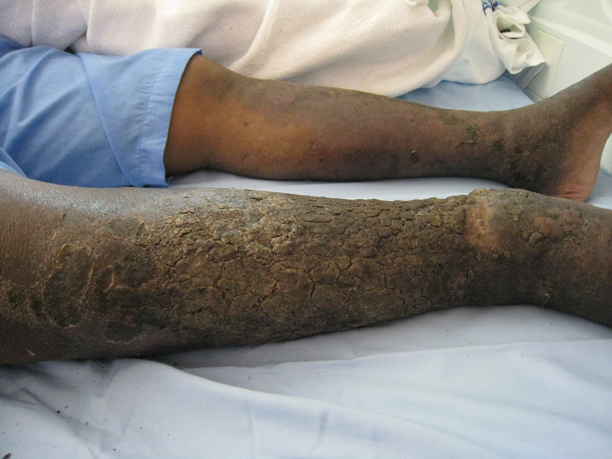
An obese 58-year-old man was admitted to the cardiology department for poorly controlled congestive heart failure. He was referred to the dermatology department with progressive painless swelling of the right lower limb of a year’s duration. He had chronic right lower limb insufficiency of 3 years’ duration with a history of a recurrent right medial malleolus ulcer and cellulitis. There was no notable travel or family history. Physical examination revealed woody edema of the right lower limb with verrucous cobblestonelike papules and nodules, foul-smelling odor, and thick crusts that were easily removed.