User login
Recalcitrant Ulcer on the Lower Leg
The Diagnosis: Nonuremic Calciphylaxis
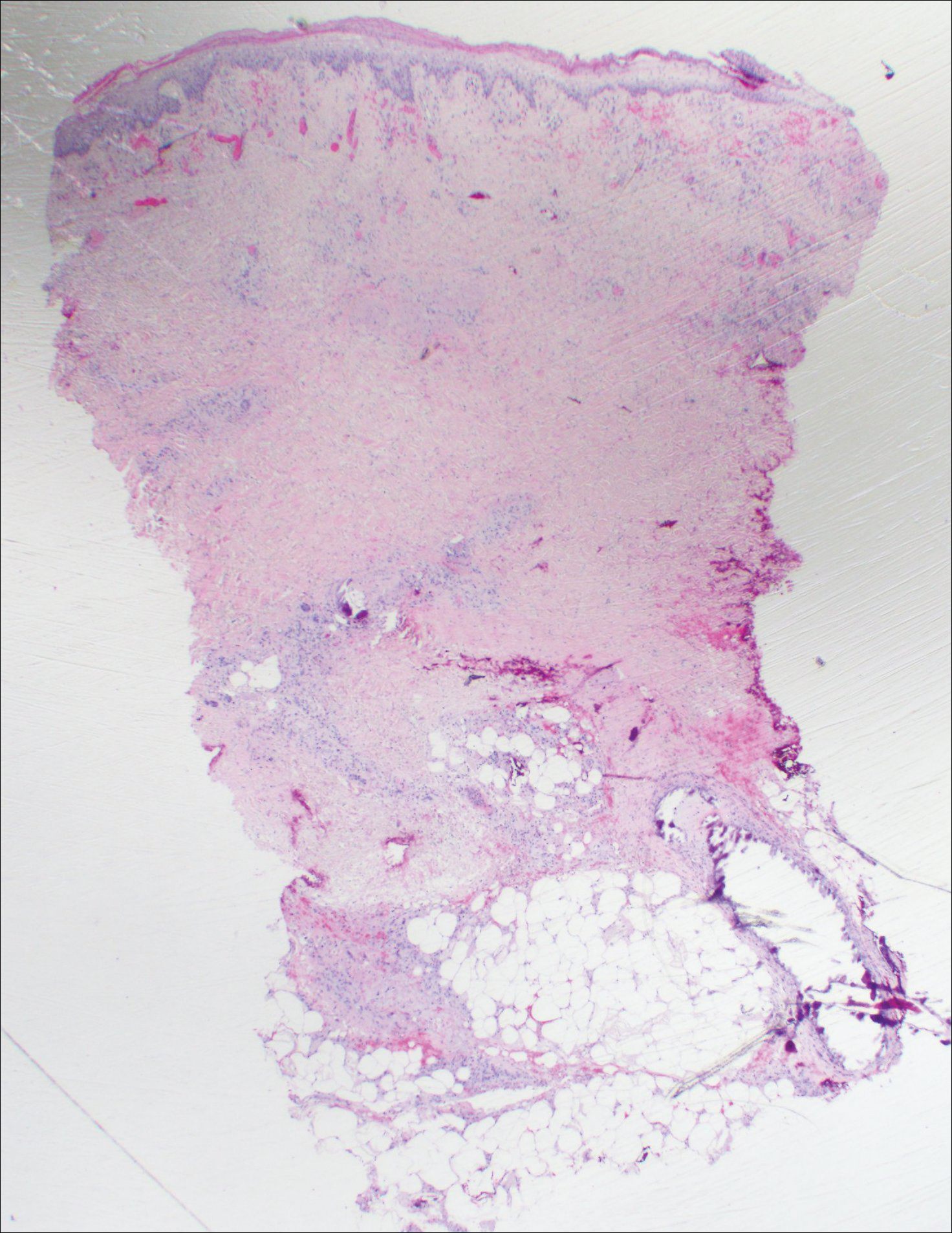
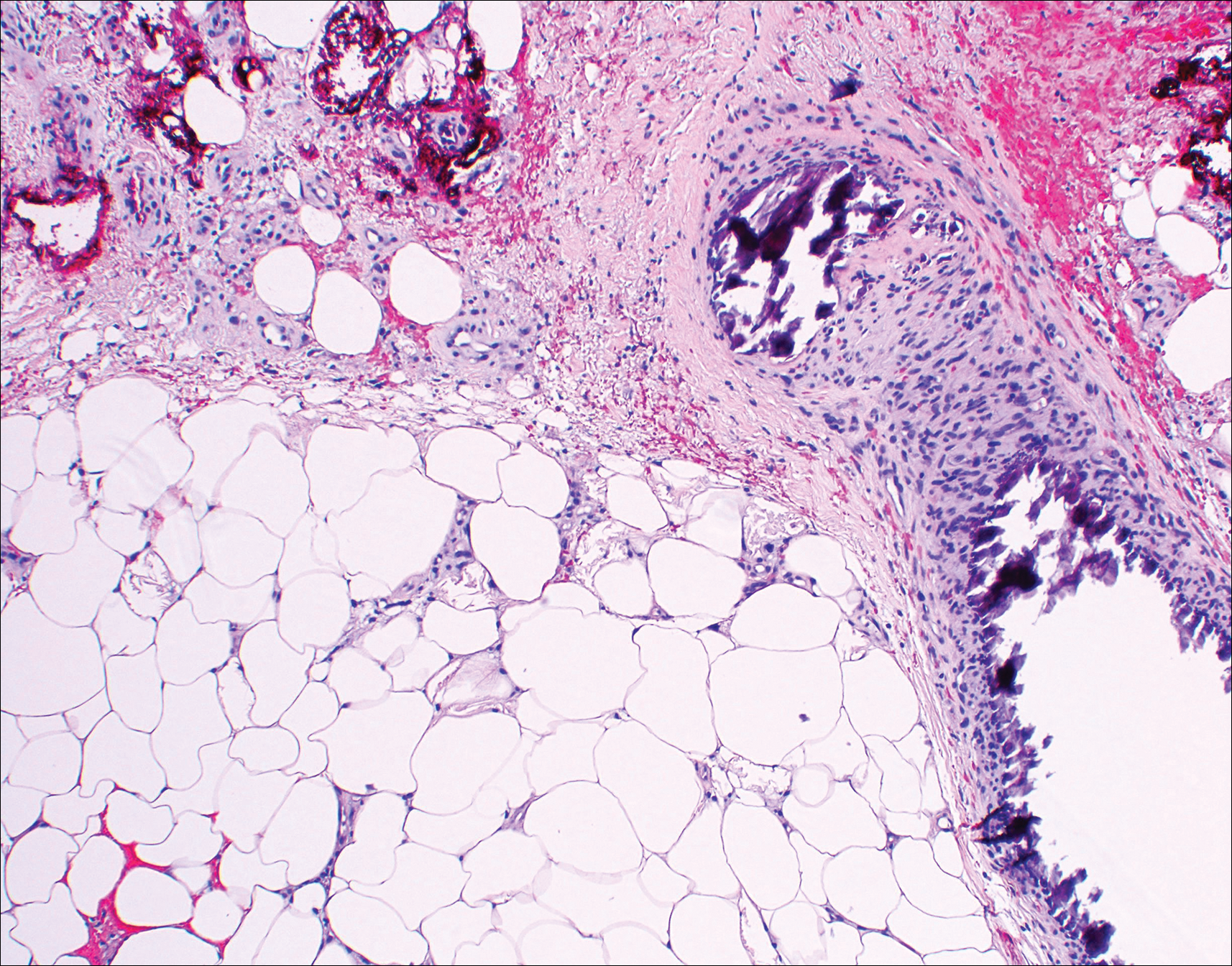
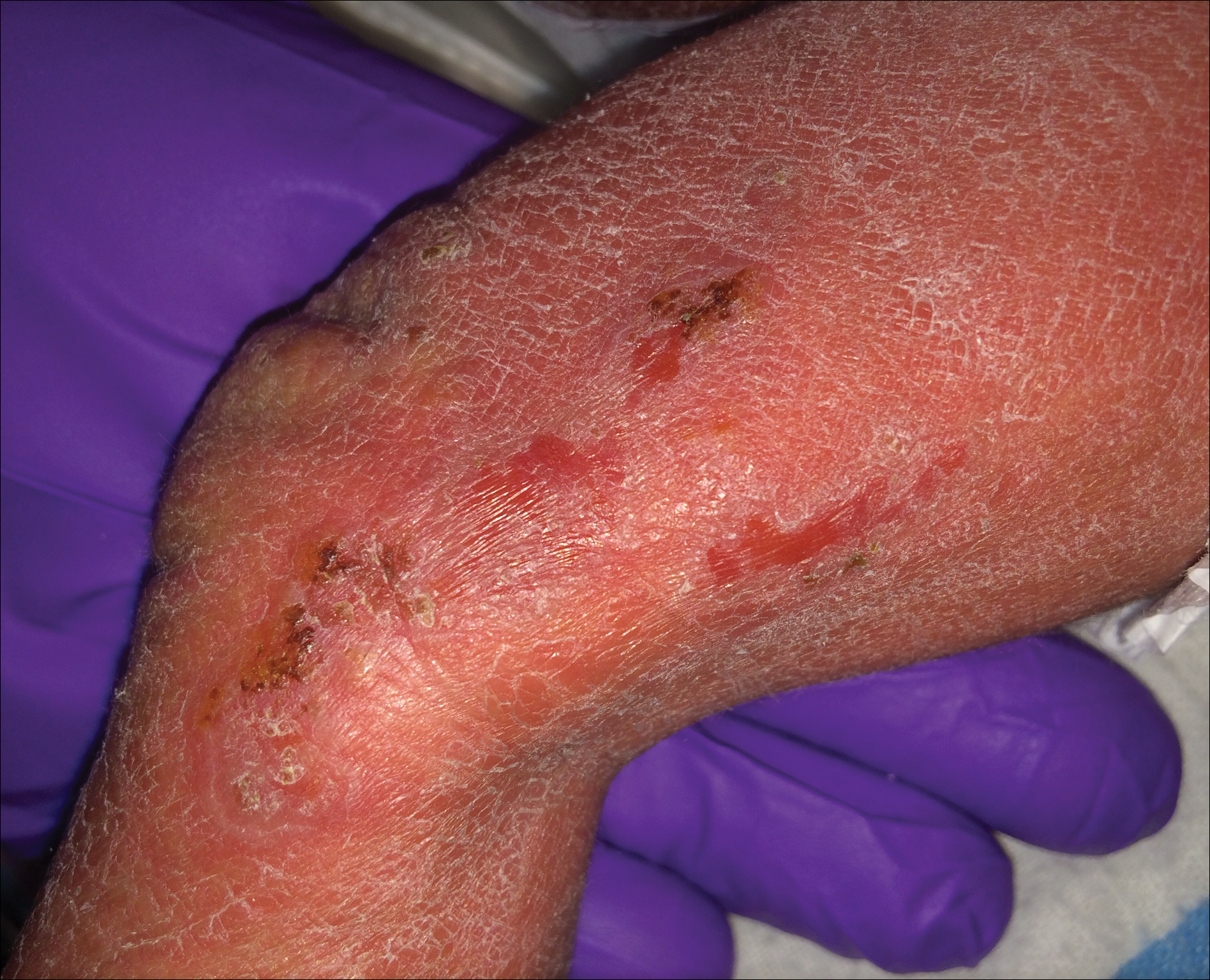
Histopathologic findings revealed ischemic necrosis and a subepidermal blister (Figure 1) with arteriosclerotic changes and fat necrosis. Foci of calcification were noted within the fat lobules. Arterioles within the deeper dermis and subcutis showed thickened hyalinized walls, narrowed lumina, and medial calcification (Figure 2). Multiple sections did not reveal any granulomatous inflammation. Periodic acid-Schiff and Gram stains were negative for fungal and bacterial elements, respectively. No dense neutrophilic infiltrate was seen. Multifocal calcific deposits within fat lobules and vessel walls (endothelium highlighted by the CD31 stain) suggested calciphylaxis.
Laboratory test results revealed a normal white blood cell count, international normalized ratio level of 4 (on warfarin), and an elevated sedimentation rate at 72 mm/h (reference range, 0-20 mm/h). Serum creatinine was 1.1 mg/dL (reference range, 0.6-1.2 mg/dL) and the calcium-phosphorous product was 40.8 mg2/dL (reference range, <55 mg2/dL). Hemoglobin A1C (glycated hemoglobin) was 8.2% (reference range, 4%-7%). Wound cultures grew Proteus mirabilis sensitive to cefazolin. Acid-fast bacilli and fungal cultures were negative. Computed tomography of the left lower leg without contrast showed no evidence of osteomyelitis. Of note, the popliteal arteries and distal vessels showed moderate vascular calcification.
Histopathology findings as well as a clinical picture of painful ulceration on the distal extremities and uncontrolled diabetes with normal renal function favored a diagnosis of nonuremic calciphylaxis (NUC). The patient was treated with intravenous infusions of sodium thiosulfate 25 mg 3 times weekly and oral cefazolin for superadded bacterial infection. Local wound care included collagenase dressings with light compression. Warfarin was discontinued, as it can worsen calciphylaxis. Complete reepithelialization of the ulcer along with substantial reduction in pain was noted within 4 weeks.
Ulceration of the lower legs is a relatively common condition in the Western world, the prevalence of which increases up to 5% in patients older than 65 years.1 Of the myriad of causes that lead to ulceration of the distal aspect of the leg, NUC is a rare but known phenomenon. The pathogenesis of NUC is complicated based on theories of derangement of receptor activator of nuclear factor κβ, receptor activator of nuclear factor κβ ligand, and osteoprotegerin, leading to calcium deposits in the media of the arteries.2 This deposition precipitates vascular occlusion coupled with ischemic necrosis of the subcutaneous tissue and skin.3 Some of the more common causes of NUC are primary hyperparathyroidism, malignancy, and rheumatoid arthritis. Type 2 diabetes mellitus is a less common cause but often is found in association with NUC, as noted by Nigwekar et al.2 According to their study, the laboratory parameters commonly found in NUC included a calcium-phosphorous product greater than 50 mg2/dL and serum creatinine of 1.2 mg/dL or less.2
Our patient displayed these laboratory findings. However, distinguishing NUC from other atypical lower extremity ulcers such as Martorell hypertensive ischemic ulcer, pyoderma gangrenosum, and warfarin necrosis can pose a challenge to the dermatologist. Martorell hypertensive ischemic ulcer is excruciatingly painful and occurs more frequently near the Achilles tendon, responding well to surgical debridement. Histopathologically, medial calcinosis and arteriosclerosis are seen.4
Pyoderma gangrenosum is a neutrophilic dermatosis wherein the classical ulcerative variant is painful. It occurs mostly on the pretibial area and worsens after debridement.5 Clinically and histopathologically, it is a diagnosis of exclusion in which a dense neutrophilic to mixed lymphocytic infiltrate is seen with necrosis of dermal vessels.6
Warfarin necrosis is extremely rare, affecting 0.01% to 0.1% of patients on warfarin-derived anticoagulant therapy.7 Necrosis occurs mostly on fat-bearing areas such as the breasts, abdomen, and thighs 3 to 5 days after initiating treatment. Histologically, fibrin deposits occlude dermal vessels without perivascular inflammation.8
Necrobiosis lipoidica is a rare cutaneous entity seen in 0.3% of diabetic patients.9 The exact pathogenesis is unknown; however, microangiopathy in collaboration with cross-linking of abnormal collagen fibers play a role. These lesions appear as erythematous plaques with a slightly depressed to atrophic center, ultimately taking on a waxy porcelain appearance. Although most of these lesions either resolve or become chronically persistent, approximately 15% undergo ulceration, which can be painful. Histologically, with hematoxylin and eosin staining, areas of necrobiosis are seen surrounded by an inflammatory infiltrate comprised mainly of histiocytes along with lymphocytes and plasma cells.9
Nonuremic calciphylaxis can mimic the aforementioned conditions to a greater extent in female patients with obesity, diabetes mellitus, and hypertension. However, microscopic calcium deposition in the media of dermal arterioles, extravascular calcification within fat lobules, and cutaneous necrosis, along with remarkable response to intravenous sodium thiosulfate, confirmed a diagnosis of NUC in our patient. Sodium thiosulfate scavenges reactive oxygen species and promotes nitric oxygen generation, thereby reducing endothelial damage.10 Although there are no randomized controlled trials to support its use, sodium thiosulfate has been successfully used to treat established cases of NUC.11
- Spentzouris G, Labropoulos N. The evaluation of lower-extremity ulcers. Semin Intervent Radiol. 2009;26:286-295.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Bardin T. Musculoskeletal manifestations of chronic renal failure. Curr Opin Rheumatol. 2003;15:48-54.
- Hafner J, Nobbe S, Partsch H, et al. Martorell hypertensive ischemic leg ulcer: a model of ischemic subcutaneous arteriolosclerosis. Arch Dermatol. 2010;146:961-968.
- Sedda S, Caruso R, Marafini I, et al. Pyoderma gangrenosum in refractory celiac disease: a case report. BMC Gastroenterol. 2013;13:162.
- Su WP, Davis MD, Weenig RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Breakey W, Hall C, Vann Jones S, et al. Warfarin-induced skin necrosis progressing to calciphylaxis. J Plast Reconstr Aesthet Surg. 2014;67:244-246.
- Kakagia DD, Papanas N, Karadimas E, et al. Warfarin-induced skin necrosis. Ann Dermatol. 2014;26:96-98.
- Kota SK, Jammula S, Kota SK, et al. Necrobiosis lipoidica diabeticorum: a case-based review of literature. Indian J Endocrinol Metab. 2012;16:614-620.
- Hayden MR, Goldsmith DJ. Sodium thiosulfate: new hope for the treatment of calciphylaxis. Semin Dial. 2010;23:258-262.
- Ning MS, Dahir KM, Castellanos EH, et al. Sodium thiosulfate in the treatment of non-uremic calciphylaxis. J Dermatol. 2013;40:649-652.
The Diagnosis: Nonuremic Calciphylaxis
Histopathologic findings revealed ischemic necrosis and a subepidermal blister (Figure 1) with arteriosclerotic changes and fat necrosis. Foci of calcification were noted within the fat lobules. Arterioles within the deeper dermis and subcutis showed thickened hyalinized walls, narrowed lumina, and medial calcification (Figure 2). Multiple sections did not reveal any granulomatous inflammation. Periodic acid-Schiff and Gram stains were negative for fungal and bacterial elements, respectively. No dense neutrophilic infiltrate was seen. Multifocal calcific deposits within fat lobules and vessel walls (endothelium highlighted by the CD31 stain) suggested calciphylaxis.
Laboratory test results revealed a normal white blood cell count, international normalized ratio level of 4 (on warfarin), and an elevated sedimentation rate at 72 mm/h (reference range, 0-20 mm/h). Serum creatinine was 1.1 mg/dL (reference range, 0.6-1.2 mg/dL) and the calcium-phosphorous product was 40.8 mg2/dL (reference range, <55 mg2/dL). Hemoglobin A1C (glycated hemoglobin) was 8.2% (reference range, 4%-7%). Wound cultures grew Proteus mirabilis sensitive to cefazolin. Acid-fast bacilli and fungal cultures were negative. Computed tomography of the left lower leg without contrast showed no evidence of osteomyelitis. Of note, the popliteal arteries and distal vessels showed moderate vascular calcification.
Histopathology findings as well as a clinical picture of painful ulceration on the distal extremities and uncontrolled diabetes with normal renal function favored a diagnosis of nonuremic calciphylaxis (NUC). The patient was treated with intravenous infusions of sodium thiosulfate 25 mg 3 times weekly and oral cefazolin for superadded bacterial infection. Local wound care included collagenase dressings with light compression. Warfarin was discontinued, as it can worsen calciphylaxis. Complete reepithelialization of the ulcer along with substantial reduction in pain was noted within 4 weeks.
Ulceration of the lower legs is a relatively common condition in the Western world, the prevalence of which increases up to 5% in patients older than 65 years.1 Of the myriad of causes that lead to ulceration of the distal aspect of the leg, NUC is a rare but known phenomenon. The pathogenesis of NUC is complicated based on theories of derangement of receptor activator of nuclear factor κβ, receptor activator of nuclear factor κβ ligand, and osteoprotegerin, leading to calcium deposits in the media of the arteries.2 This deposition precipitates vascular occlusion coupled with ischemic necrosis of the subcutaneous tissue and skin.3 Some of the more common causes of NUC are primary hyperparathyroidism, malignancy, and rheumatoid arthritis. Type 2 diabetes mellitus is a less common cause but often is found in association with NUC, as noted by Nigwekar et al.2 According to their study, the laboratory parameters commonly found in NUC included a calcium-phosphorous product greater than 50 mg2/dL and serum creatinine of 1.2 mg/dL or less.2
Our patient displayed these laboratory findings. However, distinguishing NUC from other atypical lower extremity ulcers such as Martorell hypertensive ischemic ulcer, pyoderma gangrenosum, and warfarin necrosis can pose a challenge to the dermatologist. Martorell hypertensive ischemic ulcer is excruciatingly painful and occurs more frequently near the Achilles tendon, responding well to surgical debridement. Histopathologically, medial calcinosis and arteriosclerosis are seen.4
Pyoderma gangrenosum is a neutrophilic dermatosis wherein the classical ulcerative variant is painful. It occurs mostly on the pretibial area and worsens after debridement.5 Clinically and histopathologically, it is a diagnosis of exclusion in which a dense neutrophilic to mixed lymphocytic infiltrate is seen with necrosis of dermal vessels.6
Warfarin necrosis is extremely rare, affecting 0.01% to 0.1% of patients on warfarin-derived anticoagulant therapy.7 Necrosis occurs mostly on fat-bearing areas such as the breasts, abdomen, and thighs 3 to 5 days after initiating treatment. Histologically, fibrin deposits occlude dermal vessels without perivascular inflammation.8
Necrobiosis lipoidica is a rare cutaneous entity seen in 0.3% of diabetic patients.9 The exact pathogenesis is unknown; however, microangiopathy in collaboration with cross-linking of abnormal collagen fibers play a role. These lesions appear as erythematous plaques with a slightly depressed to atrophic center, ultimately taking on a waxy porcelain appearance. Although most of these lesions either resolve or become chronically persistent, approximately 15% undergo ulceration, which can be painful. Histologically, with hematoxylin and eosin staining, areas of necrobiosis are seen surrounded by an inflammatory infiltrate comprised mainly of histiocytes along with lymphocytes and plasma cells.9
Nonuremic calciphylaxis can mimic the aforementioned conditions to a greater extent in female patients with obesity, diabetes mellitus, and hypertension. However, microscopic calcium deposition in the media of dermal arterioles, extravascular calcification within fat lobules, and cutaneous necrosis, along with remarkable response to intravenous sodium thiosulfate, confirmed a diagnosis of NUC in our patient. Sodium thiosulfate scavenges reactive oxygen species and promotes nitric oxygen generation, thereby reducing endothelial damage.10 Although there are no randomized controlled trials to support its use, sodium thiosulfate has been successfully used to treat established cases of NUC.11
The Diagnosis: Nonuremic Calciphylaxis
Histopathologic findings revealed ischemic necrosis and a subepidermal blister (Figure 1) with arteriosclerotic changes and fat necrosis. Foci of calcification were noted within the fat lobules. Arterioles within the deeper dermis and subcutis showed thickened hyalinized walls, narrowed lumina, and medial calcification (Figure 2). Multiple sections did not reveal any granulomatous inflammation. Periodic acid-Schiff and Gram stains were negative for fungal and bacterial elements, respectively. No dense neutrophilic infiltrate was seen. Multifocal calcific deposits within fat lobules and vessel walls (endothelium highlighted by the CD31 stain) suggested calciphylaxis.
Laboratory test results revealed a normal white blood cell count, international normalized ratio level of 4 (on warfarin), and an elevated sedimentation rate at 72 mm/h (reference range, 0-20 mm/h). Serum creatinine was 1.1 mg/dL (reference range, 0.6-1.2 mg/dL) and the calcium-phosphorous product was 40.8 mg2/dL (reference range, <55 mg2/dL). Hemoglobin A1C (glycated hemoglobin) was 8.2% (reference range, 4%-7%). Wound cultures grew Proteus mirabilis sensitive to cefazolin. Acid-fast bacilli and fungal cultures were negative. Computed tomography of the left lower leg without contrast showed no evidence of osteomyelitis. Of note, the popliteal arteries and distal vessels showed moderate vascular calcification.
Histopathology findings as well as a clinical picture of painful ulceration on the distal extremities and uncontrolled diabetes with normal renal function favored a diagnosis of nonuremic calciphylaxis (NUC). The patient was treated with intravenous infusions of sodium thiosulfate 25 mg 3 times weekly and oral cefazolin for superadded bacterial infection. Local wound care included collagenase dressings with light compression. Warfarin was discontinued, as it can worsen calciphylaxis. Complete reepithelialization of the ulcer along with substantial reduction in pain was noted within 4 weeks.
Ulceration of the lower legs is a relatively common condition in the Western world, the prevalence of which increases up to 5% in patients older than 65 years.1 Of the myriad of causes that lead to ulceration of the distal aspect of the leg, NUC is a rare but known phenomenon. The pathogenesis of NUC is complicated based on theories of derangement of receptor activator of nuclear factor κβ, receptor activator of nuclear factor κβ ligand, and osteoprotegerin, leading to calcium deposits in the media of the arteries.2 This deposition precipitates vascular occlusion coupled with ischemic necrosis of the subcutaneous tissue and skin.3 Some of the more common causes of NUC are primary hyperparathyroidism, malignancy, and rheumatoid arthritis. Type 2 diabetes mellitus is a less common cause but often is found in association with NUC, as noted by Nigwekar et al.2 According to their study, the laboratory parameters commonly found in NUC included a calcium-phosphorous product greater than 50 mg2/dL and serum creatinine of 1.2 mg/dL or less.2
Our patient displayed these laboratory findings. However, distinguishing NUC from other atypical lower extremity ulcers such as Martorell hypertensive ischemic ulcer, pyoderma gangrenosum, and warfarin necrosis can pose a challenge to the dermatologist. Martorell hypertensive ischemic ulcer is excruciatingly painful and occurs more frequently near the Achilles tendon, responding well to surgical debridement. Histopathologically, medial calcinosis and arteriosclerosis are seen.4
Pyoderma gangrenosum is a neutrophilic dermatosis wherein the classical ulcerative variant is painful. It occurs mostly on the pretibial area and worsens after debridement.5 Clinically and histopathologically, it is a diagnosis of exclusion in which a dense neutrophilic to mixed lymphocytic infiltrate is seen with necrosis of dermal vessels.6
Warfarin necrosis is extremely rare, affecting 0.01% to 0.1% of patients on warfarin-derived anticoagulant therapy.7 Necrosis occurs mostly on fat-bearing areas such as the breasts, abdomen, and thighs 3 to 5 days after initiating treatment. Histologically, fibrin deposits occlude dermal vessels without perivascular inflammation.8
Necrobiosis lipoidica is a rare cutaneous entity seen in 0.3% of diabetic patients.9 The exact pathogenesis is unknown; however, microangiopathy in collaboration with cross-linking of abnormal collagen fibers play a role. These lesions appear as erythematous plaques with a slightly depressed to atrophic center, ultimately taking on a waxy porcelain appearance. Although most of these lesions either resolve or become chronically persistent, approximately 15% undergo ulceration, which can be painful. Histologically, with hematoxylin and eosin staining, areas of necrobiosis are seen surrounded by an inflammatory infiltrate comprised mainly of histiocytes along with lymphocytes and plasma cells.9
Nonuremic calciphylaxis can mimic the aforementioned conditions to a greater extent in female patients with obesity, diabetes mellitus, and hypertension. However, microscopic calcium deposition in the media of dermal arterioles, extravascular calcification within fat lobules, and cutaneous necrosis, along with remarkable response to intravenous sodium thiosulfate, confirmed a diagnosis of NUC in our patient. Sodium thiosulfate scavenges reactive oxygen species and promotes nitric oxygen generation, thereby reducing endothelial damage.10 Although there are no randomized controlled trials to support its use, sodium thiosulfate has been successfully used to treat established cases of NUC.11
- Spentzouris G, Labropoulos N. The evaluation of lower-extremity ulcers. Semin Intervent Radiol. 2009;26:286-295.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Bardin T. Musculoskeletal manifestations of chronic renal failure. Curr Opin Rheumatol. 2003;15:48-54.
- Hafner J, Nobbe S, Partsch H, et al. Martorell hypertensive ischemic leg ulcer: a model of ischemic subcutaneous arteriolosclerosis. Arch Dermatol. 2010;146:961-968.
- Sedda S, Caruso R, Marafini I, et al. Pyoderma gangrenosum in refractory celiac disease: a case report. BMC Gastroenterol. 2013;13:162.
- Su WP, Davis MD, Weenig RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Breakey W, Hall C, Vann Jones S, et al. Warfarin-induced skin necrosis progressing to calciphylaxis. J Plast Reconstr Aesthet Surg. 2014;67:244-246.
- Kakagia DD, Papanas N, Karadimas E, et al. Warfarin-induced skin necrosis. Ann Dermatol. 2014;26:96-98.
- Kota SK, Jammula S, Kota SK, et al. Necrobiosis lipoidica diabeticorum: a case-based review of literature. Indian J Endocrinol Metab. 2012;16:614-620.
- Hayden MR, Goldsmith DJ. Sodium thiosulfate: new hope for the treatment of calciphylaxis. Semin Dial. 2010;23:258-262.
- Ning MS, Dahir KM, Castellanos EH, et al. Sodium thiosulfate in the treatment of non-uremic calciphylaxis. J Dermatol. 2013;40:649-652.
- Spentzouris G, Labropoulos N. The evaluation of lower-extremity ulcers. Semin Intervent Radiol. 2009;26:286-295.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Bardin T. Musculoskeletal manifestations of chronic renal failure. Curr Opin Rheumatol. 2003;15:48-54.
- Hafner J, Nobbe S, Partsch H, et al. Martorell hypertensive ischemic leg ulcer: a model of ischemic subcutaneous arteriolosclerosis. Arch Dermatol. 2010;146:961-968.
- Sedda S, Caruso R, Marafini I, et al. Pyoderma gangrenosum in refractory celiac disease: a case report. BMC Gastroenterol. 2013;13:162.
- Su WP, Davis MD, Weenig RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Breakey W, Hall C, Vann Jones S, et al. Warfarin-induced skin necrosis progressing to calciphylaxis. J Plast Reconstr Aesthet Surg. 2014;67:244-246.
- Kakagia DD, Papanas N, Karadimas E, et al. Warfarin-induced skin necrosis. Ann Dermatol. 2014;26:96-98.
- Kota SK, Jammula S, Kota SK, et al. Necrobiosis lipoidica diabeticorum: a case-based review of literature. Indian J Endocrinol Metab. 2012;16:614-620.
- Hayden MR, Goldsmith DJ. Sodium thiosulfate: new hope for the treatment of calciphylaxis. Semin Dial. 2010;23:258-262.
- Ning MS, Dahir KM, Castellanos EH, et al. Sodium thiosulfate in the treatment of non-uremic calciphylaxis. J Dermatol. 2013;40:649-652.
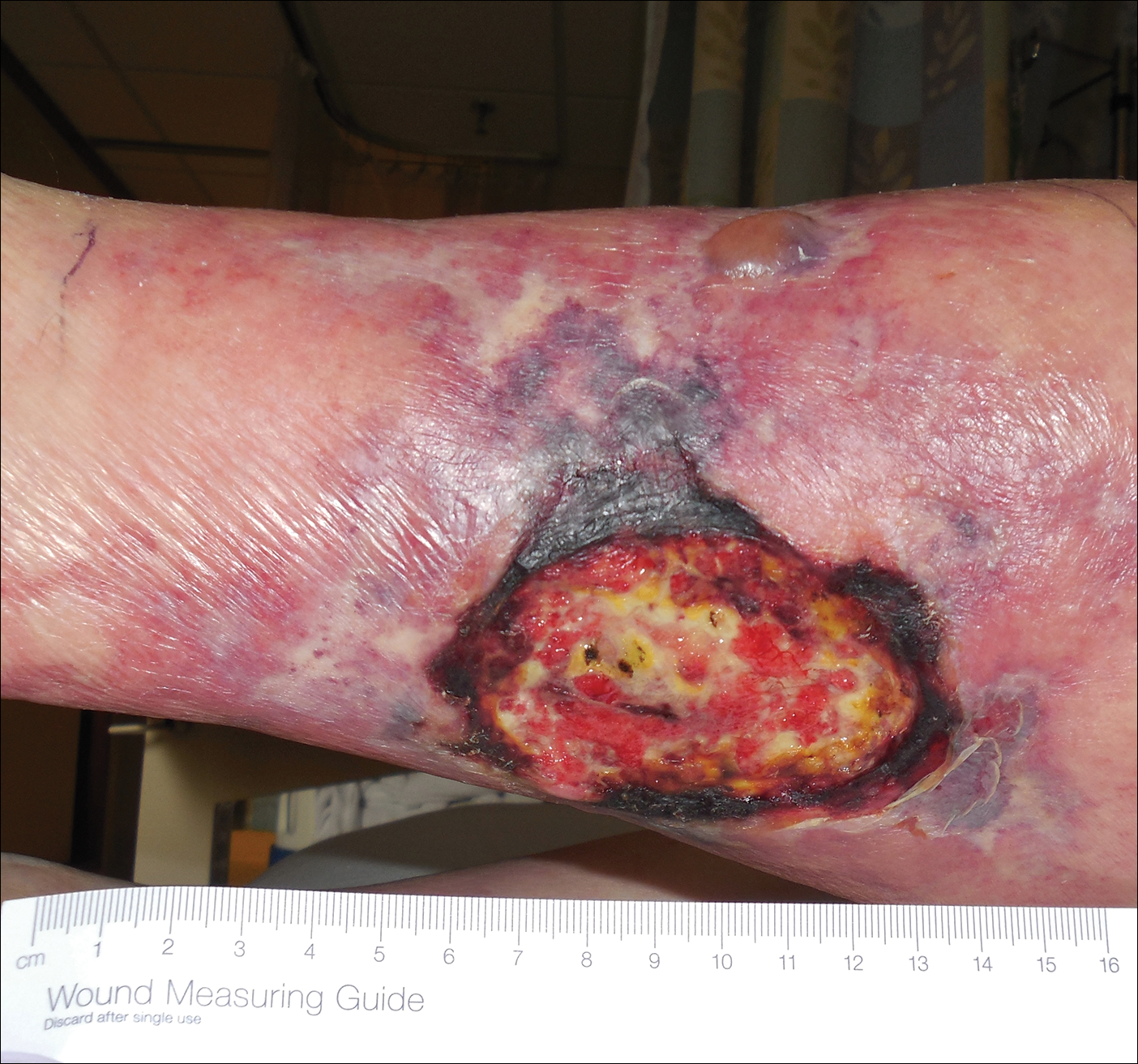
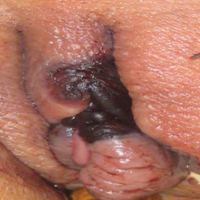
An 80-year-old woman with a medical history notable for obesity (body mass index, 31.2), type 2 diabetes mellitus, hypertension, and chronic atrial fibrillation treated with warfarin presented with a chronic painful wound on the left lower calf of 1 month's duration. A 7×7-cm ulcer on the posterior aspect of the left calf with necrotic debris was seen surrounded by skin of mottled purple discoloration. The edge of the ulcer was not undermined. There were tense nonhemorrhagic bullae on the medial aspect of the left leg and on bilateral anterior tibial areas. Two punch biopsy specimens were obtained from the anterior tibial bulla and the edge of the ulcer.
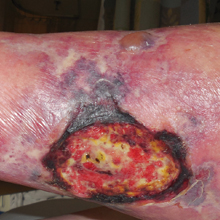
Tender Edematous Nodules on the Hand
The Diagnosis: Ecthyma Contagiosum (Orf)
Orf, or ecthyma contagiosum, is a zoonotic cutaneous infection caused by the orf DNA virus of the genus Parapoxvirus of the family Poxviridae. It is transmitted to humans through direct contact with infected animals, namely sheep and goats, and as such is most commonly seen in patients with occupational exposure to these animals such as butchers, farmers, veterinarians, and shepherds.1,2 Human-to-human transmission is exceedingly rare in immunocompetent patients.2,3 In affected animals, lesions usually are found around the mouth, muzzle, and eyes. In humans, hands are the most commonly affected site, and lesions occur 3 to 10 days after contact. Clinically, the lesions are nonspecific, and our patient presented with tender, erythematous, edematous nodules on the left hand. The differential diagnosis is broad and includes a milker's nodule, pyogenic granuloma, tularemia, anthrax, atypical mycobacterial infection, and sporotrichosis.1,4,5
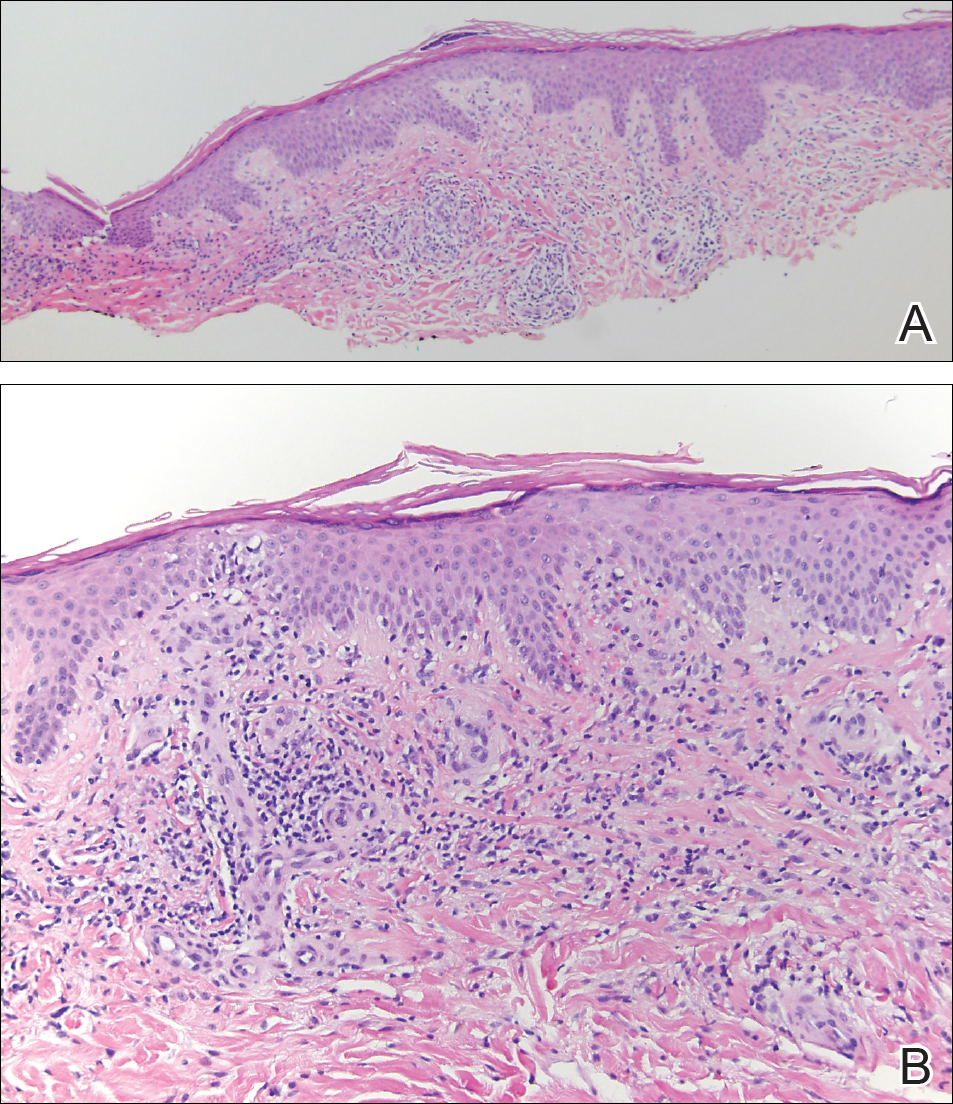
The diagnosis usually is made with a thorough history and examination, but in cases of uncertainty, routine pathology with hematoxylin and eosin staining, electron microscopy, or real-time polymerase chain reaction may be used.2-4 Histopathologically, lesions demonstrate intraepidermal vesicles, vacuolization of keratinocytes of the upper epidermis with characteristic cytoplasmic inclusion bodies, rete ridge elongation, and dilated vessels in the intervening dermal papillae. Central necrosis may occur in well-developed lesions.2,6 Interestingly, our patient's biopsy exhibited all of these findings (Figure). Immunostains for cytomegalovirus and herpes simplex virus were negative, and Grocott-Gomori methenamine-silver and acid-fast bacillus stains also were negative.

Our patient also developed lymphangitic streaking suggestive of a bacterial superinfection and was treated with a course of intravenous antibiotics. She eventually was discharged with reassurance, wound care instructions, and outpatient antibiotics. She returned to an outside institution's emergency department for further evaluation, and she was admitted for workup. A lesional swab was sent for real-time polymerase chain reaction, which confirmed the diagnosis as orf. When the patient was contacted for follow-up 1 week after biopsy, the hand lesions had notably improved.
Orf is self-limited and typically resolves within 4 to 8 weeks after undergoing evolution through 5 described stages. The maculopapular stage is denoted by enlarging erythematous macule. The targetoid stage is described by a red center within a white halo surrounded by a broader red halo. The nodular stage is self-descriptive. The regenerative and regression stages describe the progressively improving, drier, and crusted nodules.3
Because orf is self-limited, no treatment is required, and patients should be counseled that their lesions should resolve within weeks. Complications include lymphangitis, secondary bacterial infection, and erythema multiforme.1,2,4,5 Immunocompromised patients may develop recalcitrant, giant, or multiple lesions that may be treated with topical imiquimod, topical cidofovir, intralesional interferon alfa, or surgical excision.1,2,4,7
We present a case of orf to remind practitioners of this rare entity. Although the disease is endemic worldwide, it likely is underreported due to its self-limited nature.2,4 A careful history may reveal the diagnosis, and overtreatment with antibiotics, many of which have their own significant side-effect profile, can then be avoided.
Acknowledgment
We thank Eric Behling, MD (Camden, New Jersey), for his contributions in obtaining the histologic images.
- Veraldi S, Nazzaro G, Vaira F, et al. Presentation of orf (ecthyma contagiosum) after sheep slaughtering for religious feasts. Infection. 2014;42:767-769.
- Al-Salam S, Nowotny N, Sohail MR, et al. Ecthyma contagiosum (orf)--report of a human case from the United Arab Emirates and review of the literature. J Cutan Pathol. 2008;35:603-607.
- Thurman RJ, Fitch RW. Images in clinical medicine. contagious ecthyma. N Engl J Med. 2015;372:E12.
- Meier R, Sommacal A, Stahel A, et al. Orf--an orphan disease? JRSM Open. 2015;6:2054270415593718.
- Joseph RH, Haddad FA, Matthews AL, et al. Erythema multiforme after orf virus infection: a report of two cases and literature review. Epidemiol Infect. 2015;143:385-390.
- Xu X, Yun SJ, Erikson L, et al. Diseases caused by viruses. In: Elder DE, Elenitsas R, Rosenbach M, eds. Lever's Histopathology of the Skin. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015:781-815.
- Koufakis T, Katsaitis P, Gabranis I. Orf disease: a report of a case. Braz J Infect Dis. 2014;18:568-569.
The Diagnosis: Ecthyma Contagiosum (Orf)
Orf, or ecthyma contagiosum, is a zoonotic cutaneous infection caused by the orf DNA virus of the genus Parapoxvirus of the family Poxviridae. It is transmitted to humans through direct contact with infected animals, namely sheep and goats, and as such is most commonly seen in patients with occupational exposure to these animals such as butchers, farmers, veterinarians, and shepherds.1,2 Human-to-human transmission is exceedingly rare in immunocompetent patients.2,3 In affected animals, lesions usually are found around the mouth, muzzle, and eyes. In humans, hands are the most commonly affected site, and lesions occur 3 to 10 days after contact. Clinically, the lesions are nonspecific, and our patient presented with tender, erythematous, edematous nodules on the left hand. The differential diagnosis is broad and includes a milker's nodule, pyogenic granuloma, tularemia, anthrax, atypical mycobacterial infection, and sporotrichosis.1,4,5
The diagnosis usually is made with a thorough history and examination, but in cases of uncertainty, routine pathology with hematoxylin and eosin staining, electron microscopy, or real-time polymerase chain reaction may be used.2-4 Histopathologically, lesions demonstrate intraepidermal vesicles, vacuolization of keratinocytes of the upper epidermis with characteristic cytoplasmic inclusion bodies, rete ridge elongation, and dilated vessels in the intervening dermal papillae. Central necrosis may occur in well-developed lesions.2,6 Interestingly, our patient's biopsy exhibited all of these findings (Figure). Immunostains for cytomegalovirus and herpes simplex virus were negative, and Grocott-Gomori methenamine-silver and acid-fast bacillus stains also were negative.
Our patient also developed lymphangitic streaking suggestive of a bacterial superinfection and was treated with a course of intravenous antibiotics. She eventually was discharged with reassurance, wound care instructions, and outpatient antibiotics. She returned to an outside institution's emergency department for further evaluation, and she was admitted for workup. A lesional swab was sent for real-time polymerase chain reaction, which confirmed the diagnosis as orf. When the patient was contacted for follow-up 1 week after biopsy, the hand lesions had notably improved.
Orf is self-limited and typically resolves within 4 to 8 weeks after undergoing evolution through 5 described stages. The maculopapular stage is denoted by enlarging erythematous macule. The targetoid stage is described by a red center within a white halo surrounded by a broader red halo. The nodular stage is self-descriptive. The regenerative and regression stages describe the progressively improving, drier, and crusted nodules.3
Because orf is self-limited, no treatment is required, and patients should be counseled that their lesions should resolve within weeks. Complications include lymphangitis, secondary bacterial infection, and erythema multiforme.1,2,4,5 Immunocompromised patients may develop recalcitrant, giant, or multiple lesions that may be treated with topical imiquimod, topical cidofovir, intralesional interferon alfa, or surgical excision.1,2,4,7
We present a case of orf to remind practitioners of this rare entity. Although the disease is endemic worldwide, it likely is underreported due to its self-limited nature.2,4 A careful history may reveal the diagnosis, and overtreatment with antibiotics, many of which have their own significant side-effect profile, can then be avoided.
Acknowledgment
We thank Eric Behling, MD (Camden, New Jersey), for his contributions in obtaining the histologic images.
The Diagnosis: Ecthyma Contagiosum (Orf)
Orf, or ecthyma contagiosum, is a zoonotic cutaneous infection caused by the orf DNA virus of the genus Parapoxvirus of the family Poxviridae. It is transmitted to humans through direct contact with infected animals, namely sheep and goats, and as such is most commonly seen in patients with occupational exposure to these animals such as butchers, farmers, veterinarians, and shepherds.1,2 Human-to-human transmission is exceedingly rare in immunocompetent patients.2,3 In affected animals, lesions usually are found around the mouth, muzzle, and eyes. In humans, hands are the most commonly affected site, and lesions occur 3 to 10 days after contact. Clinically, the lesions are nonspecific, and our patient presented with tender, erythematous, edematous nodules on the left hand. The differential diagnosis is broad and includes a milker's nodule, pyogenic granuloma, tularemia, anthrax, atypical mycobacterial infection, and sporotrichosis.1,4,5
The diagnosis usually is made with a thorough history and examination, but in cases of uncertainty, routine pathology with hematoxylin and eosin staining, electron microscopy, or real-time polymerase chain reaction may be used.2-4 Histopathologically, lesions demonstrate intraepidermal vesicles, vacuolization of keratinocytes of the upper epidermis with characteristic cytoplasmic inclusion bodies, rete ridge elongation, and dilated vessels in the intervening dermal papillae. Central necrosis may occur in well-developed lesions.2,6 Interestingly, our patient's biopsy exhibited all of these findings (Figure). Immunostains for cytomegalovirus and herpes simplex virus were negative, and Grocott-Gomori methenamine-silver and acid-fast bacillus stains also were negative.
Our patient also developed lymphangitic streaking suggestive of a bacterial superinfection and was treated with a course of intravenous antibiotics. She eventually was discharged with reassurance, wound care instructions, and outpatient antibiotics. She returned to an outside institution's emergency department for further evaluation, and she was admitted for workup. A lesional swab was sent for real-time polymerase chain reaction, which confirmed the diagnosis as orf. When the patient was contacted for follow-up 1 week after biopsy, the hand lesions had notably improved.
Orf is self-limited and typically resolves within 4 to 8 weeks after undergoing evolution through 5 described stages. The maculopapular stage is denoted by enlarging erythematous macule. The targetoid stage is described by a red center within a white halo surrounded by a broader red halo. The nodular stage is self-descriptive. The regenerative and regression stages describe the progressively improving, drier, and crusted nodules.3
Because orf is self-limited, no treatment is required, and patients should be counseled that their lesions should resolve within weeks. Complications include lymphangitis, secondary bacterial infection, and erythema multiforme.1,2,4,5 Immunocompromised patients may develop recalcitrant, giant, or multiple lesions that may be treated with topical imiquimod, topical cidofovir, intralesional interferon alfa, or surgical excision.1,2,4,7
We present a case of orf to remind practitioners of this rare entity. Although the disease is endemic worldwide, it likely is underreported due to its self-limited nature.2,4 A careful history may reveal the diagnosis, and overtreatment with antibiotics, many of which have their own significant side-effect profile, can then be avoided.
Acknowledgment
We thank Eric Behling, MD (Camden, New Jersey), for his contributions in obtaining the histologic images.
- Veraldi S, Nazzaro G, Vaira F, et al. Presentation of orf (ecthyma contagiosum) after sheep slaughtering for religious feasts. Infection. 2014;42:767-769.
- Al-Salam S, Nowotny N, Sohail MR, et al. Ecthyma contagiosum (orf)--report of a human case from the United Arab Emirates and review of the literature. J Cutan Pathol. 2008;35:603-607.
- Thurman RJ, Fitch RW. Images in clinical medicine. contagious ecthyma. N Engl J Med. 2015;372:E12.
- Meier R, Sommacal A, Stahel A, et al. Orf--an orphan disease? JRSM Open. 2015;6:2054270415593718.
- Joseph RH, Haddad FA, Matthews AL, et al. Erythema multiforme after orf virus infection: a report of two cases and literature review. Epidemiol Infect. 2015;143:385-390.
- Xu X, Yun SJ, Erikson L, et al. Diseases caused by viruses. In: Elder DE, Elenitsas R, Rosenbach M, eds. Lever's Histopathology of the Skin. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015:781-815.
- Koufakis T, Katsaitis P, Gabranis I. Orf disease: a report of a case. Braz J Infect Dis. 2014;18:568-569.
- Veraldi S, Nazzaro G, Vaira F, et al. Presentation of orf (ecthyma contagiosum) after sheep slaughtering for religious feasts. Infection. 2014;42:767-769.
- Al-Salam S, Nowotny N, Sohail MR, et al. Ecthyma contagiosum (orf)--report of a human case from the United Arab Emirates and review of the literature. J Cutan Pathol. 2008;35:603-607.
- Thurman RJ, Fitch RW. Images in clinical medicine. contagious ecthyma. N Engl J Med. 2015;372:E12.
- Meier R, Sommacal A, Stahel A, et al. Orf--an orphan disease? JRSM Open. 2015;6:2054270415593718.
- Joseph RH, Haddad FA, Matthews AL, et al. Erythema multiforme after orf virus infection: a report of two cases and literature review. Epidemiol Infect. 2015;143:385-390.
- Xu X, Yun SJ, Erikson L, et al. Diseases caused by viruses. In: Elder DE, Elenitsas R, Rosenbach M, eds. Lever's Histopathology of the Skin. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015:781-815.
- Koufakis T, Katsaitis P, Gabranis I. Orf disease: a report of a case. Braz J Infect Dis. 2014;18:568-569.
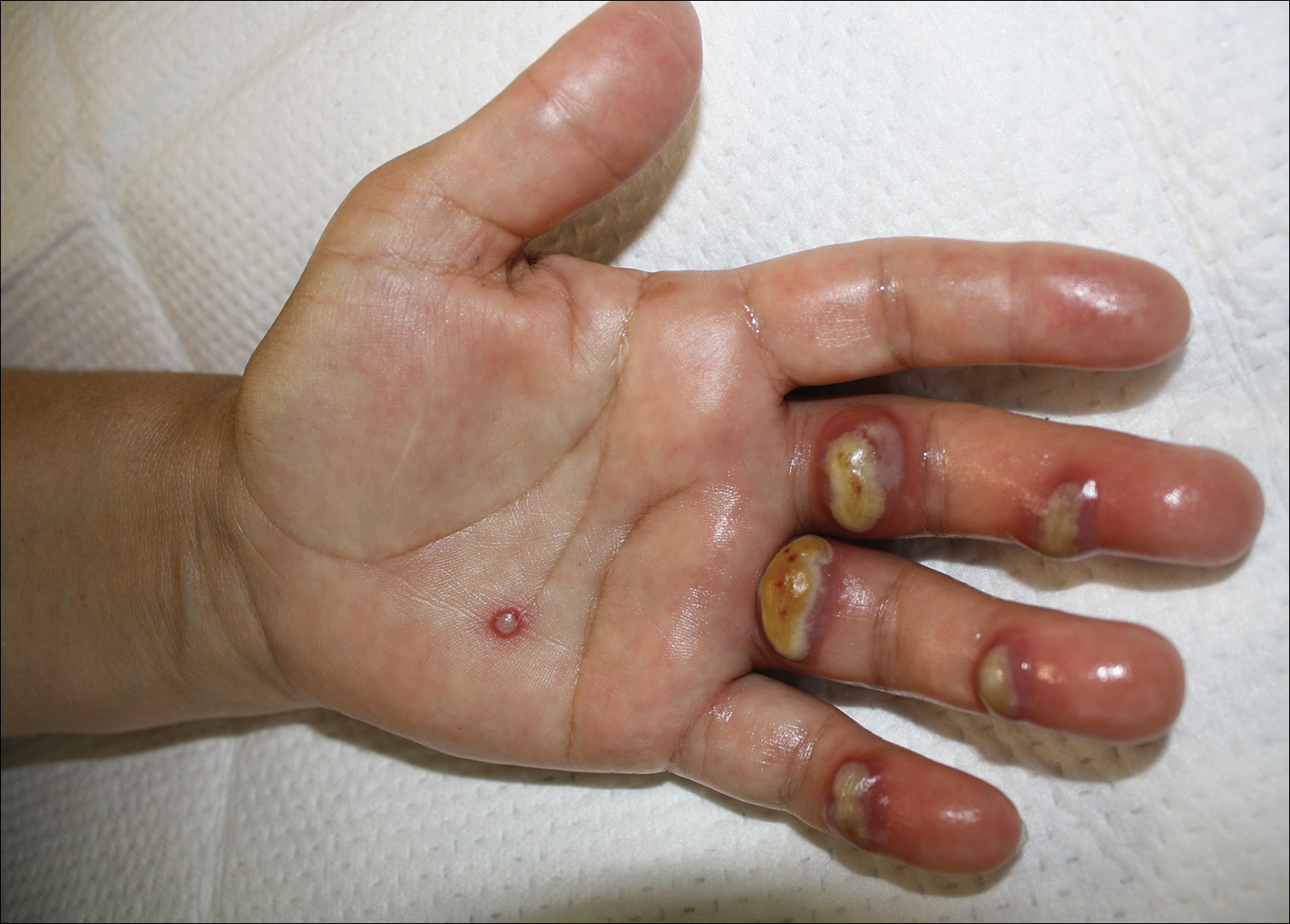
A 57-year-old woman presented to the emergency department (ED) for evaluation of a rash on the left hand of 2 weeks' duration. She described pinpoint red lesions on the left palm, as well as the third, fourth, and fifth fingers, which gradually enlarged and became painful. She denied any specific trauma but recalled cutting her hand on a piece of metal in the ground prior to the onset of the rash. She worked on a farm and bottle-fed sheep and chickens. Physical examination revealed tender edematous nodules with central gray pustules, and the left axillary lymph node was enlarged and tender. Ulceration was not appreciated. Various antibiotics including cephalexin, trimethoprim-sulfamethoxazole, and clindamycin were prescribed during prior ED visits, but she reported no improvement with these medications. She remained afebrile throughout the course of the hand rash, and laboratory workup was consistently unremarkable. Two sets of herpes simplex virus cultures from the ED visits showed no growth, and a hand radiograph also was normal. Medical history included coronary artery disease, myocardial infarction, mitral regurgitation, and hyperlipidemia.
Hyperpigmented Patch on the Leg
The Diagnosis: Lichen Aureus
The clinicopathological findings were diagnostic of lichen aureus (LA). Microscopic examination revealed a relatively sparse, superficial, perivascular and interstitial lymphohistiocytic infiltrate with scattered siderophages in the upper dermis. Extravasation of red blood cells also was noted (Figure 1). An immunohistochemical stain for Melan-A highlighted a normal number and distribution of single melanocytes at the dermoepidermal junction with no evidence of pagetoid scatter. A Perls Prussian blue stain for iron demonstrated abundant hemosiderin in the dermis (Figure 2).
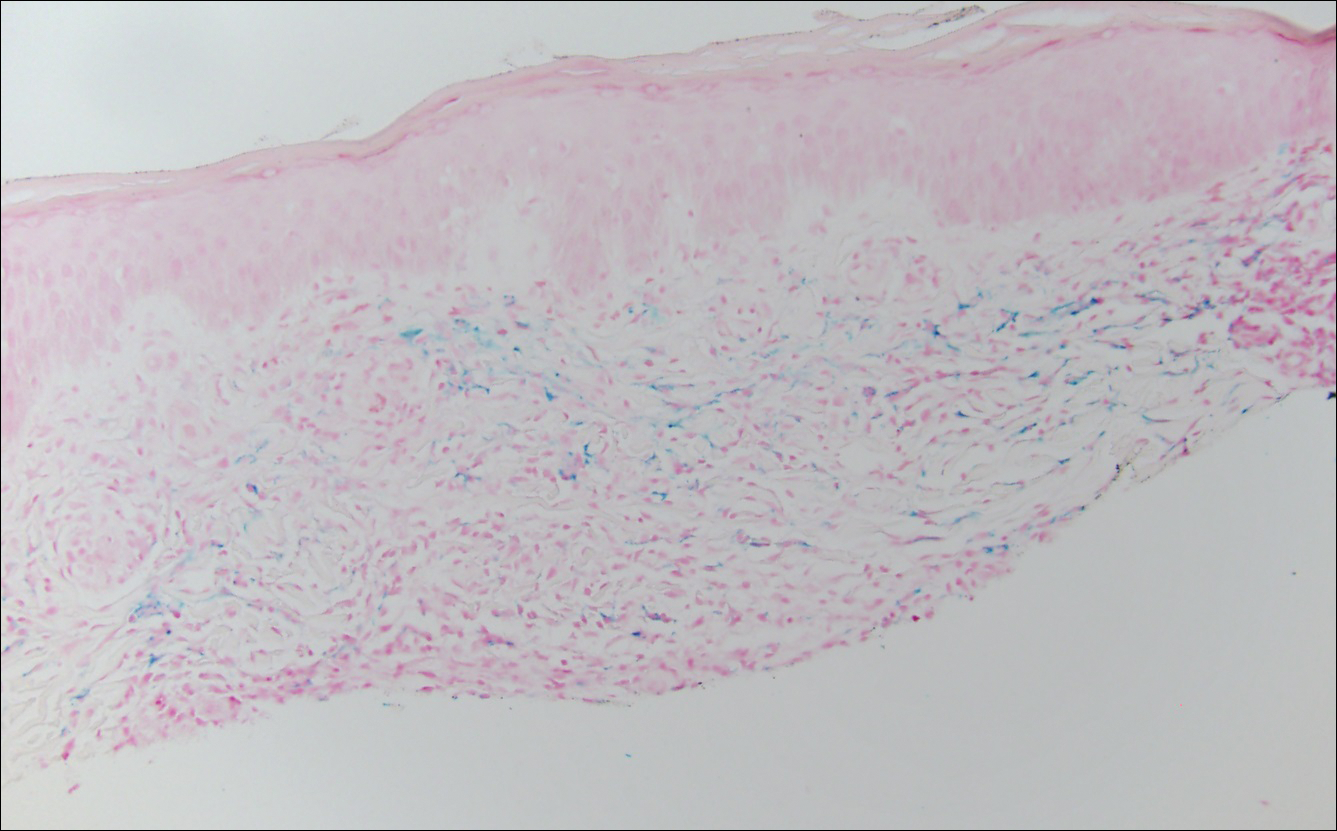
Pigmented purpuric dermatosis (PPD) describes a group of cutaneous lesions that are characterized by petechiae and pigmentary changes. These lesions most commonly present on the lower limbs; however, other sites have been reported.1 This group includes several major clinical forms such as Schamberg disease, LA, purpura annularis telangiectodes of Majocchi, eczematidlike purpura of Doucas and Kapetanakis, and lichenoid PPD of Gougerot and Blum. Lesions typically demonstrate a striking golden brown color clinically and by definition occur in the absence of platelet defects or vasculitis.1
Factors implicated in the pathogenesis of pigmented purpura include gravitational dependency, venous stasis, infection, and drugs.2 It is suggested that cellular immunity may play a role in the development of the disease based on the presence of CD4+ T lymphocytes in the infiltrate and the expression of HLA-DR by these lymphocytes and the keratinocytes.3 Lichen aureus differs in that it relates to increased intravascular pressure from an incompetent valve in an underlying perforating vein.4
Lichen aureus, also referred to as lichen purpuricus, is one major variant of PPD. The name reflects both the characteristic golden brown color and the histopathologic pattern of inflammation.1 Lichen aureus usually presents as a unilateral, asymptomatic, confined single lesion located mainly on the leg,1 though it can develop at other sites or as a localized group of lesions. Extensive lesions have been reported5 and cases with a segmental distribution have been described.6 In contrast, Schamberg disease demonstrates pinhead-sized reddish lesions giving the characteristic cayenne pepper pigmentation. These lesions coalesce to form thumbprint patches that progress proximally.1 Majocchi purpura is annular and telangiectatic, while lichenoid purpura of Gougerot and Blum presents with flat-topped, polygonal, violaceous papules that turn brown over time.
Some authors have championed a role for dermoscopy in diagnosis of LA.7 By dermoscopy, LA demonstrates a diffuse copper background reflecting the lymphohistiocytic dermal infiltrate, red dots and globules representing the extravasated red blood cells and the dilated swollen vessels, and grey dots that reflect the hemosiderin present in the dermis.8
Histologically, LA demonstrates a superficial perivascular infiltrate composed mainly of CD4+ lymphocytes surrounding the superficial capillaries. Over time, red cell extravasation leads to the formation of hemosiderin-laden macrophages, which can be highlighted with Perls Prussian blue stain. A bandlike infiltrate with thin strands of collagen separating it from the epidermis also may be noted.9
An important consideration in the differential diagnosis of PPD is mycosis fungoides (MF). Mycosis fungoides is a cutaneous T-cell lymphoma that clinically presents as a single or multiple hypopigmented or hyperpigmented patches or as erythematous scaly lesions in the patch or plaque stage. These lesions eventually may evolve into tumor stage.10 Mycosis fungoides may mimic PPD clinically and/or histopathologically, and rarely PPD also may precede MF.11 Involvement of the trunk, especially the lower abdomen and buttock region, favors a diagnosis of MF. Typically, histopathologic examination of MF demonstrates an epidermotropic lymphocytic infiltrate composed of atypical cerebriform lymphocytes overlying papillary dermal fibrosis. Although classic MF would be difficult to confuse with PPD, the atrophic lichenoid pattern of MF may show remarkable overlap with PPD.12 Such cases require clinicopathologic correlation, immunophenotyping of the epidermotropic lymphocytes, and occasionally T-cell clonality studies.
Lichen aureus is a chronic persistent disease unless the underlying incompetent perforator vessel is ligated. Various treatments have been used for other forms of pigmented purpura including topical corticosteroids, topical tacrolimus, systemic vasodilators such as prostacyclin and pentoxifylline, and phototherapy.1 Clinical follow-up is recommended for lesions that show some clinical or histopathological overlap with MF. Additional biopsies also may prove useful in establishing a definitive diagnosis in ambiguous cases.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165-169.
- Aiba S, Tagami H. Immunohistologic studies in Schamberg's disease. evidence for cellular immune reaction in lesional skin. Arch Dermatol. 1988;124:1058-1062.
- English J. Lichen aureus. J Am Acad Dermatol. 1985;12(2, pt 1):377-379.
- Duhra P, Tan CY. Lichen aureus. Br J Dermatol. 1986;114:395.
- Moche J, Glassman S, Modi D, et al. Segmental lichen aureus: a report of two cases treated with methylprednisolone aceponate. Australas J Dermatol. 2011;52:E15-E18.
- Zaballos P, Puig S, Malvehy J. Dermoscopy of pigmented purpuric dermatoses (lichen aureus): a useful tool for clinical diagnosis. Arch Dermatol. 2004;140:1290-1291.
- Portela PS, Melo DF, Ormiga P, et al. Dermoscopy of lichen aureus. An Bras Dermatol. 2013;88:253-255.
- Smoller BR, Kamel OW. Pigmented purpuric eruptions: immunopathologic studies supportive of a common immunophenotype. J Cutan Pathol. 1991;18:423-427.
- Jaffe ES, Harris NL, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. a progress report. Am J Clin Pathol. 1999;111(1 suppl 1):S8-S12.
- Hanna S, Walsh N, D'Intino Y, et al. Mycosis fungoides presenting as pigmented purpuric dermatitis. Pediatr Dermatol. 2006;23:350-354.
- Toro JR, Sander CA, LeBoit PE. Persistent pigmented purpuric dermatitis and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
The Diagnosis: Lichen Aureus
The clinicopathological findings were diagnostic of lichen aureus (LA). Microscopic examination revealed a relatively sparse, superficial, perivascular and interstitial lymphohistiocytic infiltrate with scattered siderophages in the upper dermis. Extravasation of red blood cells also was noted (Figure 1). An immunohistochemical stain for Melan-A highlighted a normal number and distribution of single melanocytes at the dermoepidermal junction with no evidence of pagetoid scatter. A Perls Prussian blue stain for iron demonstrated abundant hemosiderin in the dermis (Figure 2).
Pigmented purpuric dermatosis (PPD) describes a group of cutaneous lesions that are characterized by petechiae and pigmentary changes. These lesions most commonly present on the lower limbs; however, other sites have been reported.1 This group includes several major clinical forms such as Schamberg disease, LA, purpura annularis telangiectodes of Majocchi, eczematidlike purpura of Doucas and Kapetanakis, and lichenoid PPD of Gougerot and Blum. Lesions typically demonstrate a striking golden brown color clinically and by definition occur in the absence of platelet defects or vasculitis.1
Factors implicated in the pathogenesis of pigmented purpura include gravitational dependency, venous stasis, infection, and drugs.2 It is suggested that cellular immunity may play a role in the development of the disease based on the presence of CD4+ T lymphocytes in the infiltrate and the expression of HLA-DR by these lymphocytes and the keratinocytes.3 Lichen aureus differs in that it relates to increased intravascular pressure from an incompetent valve in an underlying perforating vein.4
Lichen aureus, also referred to as lichen purpuricus, is one major variant of PPD. The name reflects both the characteristic golden brown color and the histopathologic pattern of inflammation.1 Lichen aureus usually presents as a unilateral, asymptomatic, confined single lesion located mainly on the leg,1 though it can develop at other sites or as a localized group of lesions. Extensive lesions have been reported5 and cases with a segmental distribution have been described.6 In contrast, Schamberg disease demonstrates pinhead-sized reddish lesions giving the characteristic cayenne pepper pigmentation. These lesions coalesce to form thumbprint patches that progress proximally.1 Majocchi purpura is annular and telangiectatic, while lichenoid purpura of Gougerot and Blum presents with flat-topped, polygonal, violaceous papules that turn brown over time.
Some authors have championed a role for dermoscopy in diagnosis of LA.7 By dermoscopy, LA demonstrates a diffuse copper background reflecting the lymphohistiocytic dermal infiltrate, red dots and globules representing the extravasated red blood cells and the dilated swollen vessels, and grey dots that reflect the hemosiderin present in the dermis.8
Histologically, LA demonstrates a superficial perivascular infiltrate composed mainly of CD4+ lymphocytes surrounding the superficial capillaries. Over time, red cell extravasation leads to the formation of hemosiderin-laden macrophages, which can be highlighted with Perls Prussian blue stain. A bandlike infiltrate with thin strands of collagen separating it from the epidermis also may be noted.9
An important consideration in the differential diagnosis of PPD is mycosis fungoides (MF). Mycosis fungoides is a cutaneous T-cell lymphoma that clinically presents as a single or multiple hypopigmented or hyperpigmented patches or as erythematous scaly lesions in the patch or plaque stage. These lesions eventually may evolve into tumor stage.10 Mycosis fungoides may mimic PPD clinically and/or histopathologically, and rarely PPD also may precede MF.11 Involvement of the trunk, especially the lower abdomen and buttock region, favors a diagnosis of MF. Typically, histopathologic examination of MF demonstrates an epidermotropic lymphocytic infiltrate composed of atypical cerebriform lymphocytes overlying papillary dermal fibrosis. Although classic MF would be difficult to confuse with PPD, the atrophic lichenoid pattern of MF may show remarkable overlap with PPD.12 Such cases require clinicopathologic correlation, immunophenotyping of the epidermotropic lymphocytes, and occasionally T-cell clonality studies.
Lichen aureus is a chronic persistent disease unless the underlying incompetent perforator vessel is ligated. Various treatments have been used for other forms of pigmented purpura including topical corticosteroids, topical tacrolimus, systemic vasodilators such as prostacyclin and pentoxifylline, and phototherapy.1 Clinical follow-up is recommended for lesions that show some clinical or histopathological overlap with MF. Additional biopsies also may prove useful in establishing a definitive diagnosis in ambiguous cases.
The Diagnosis: Lichen Aureus
The clinicopathological findings were diagnostic of lichen aureus (LA). Microscopic examination revealed a relatively sparse, superficial, perivascular and interstitial lymphohistiocytic infiltrate with scattered siderophages in the upper dermis. Extravasation of red blood cells also was noted (Figure 1). An immunohistochemical stain for Melan-A highlighted a normal number and distribution of single melanocytes at the dermoepidermal junction with no evidence of pagetoid scatter. A Perls Prussian blue stain for iron demonstrated abundant hemosiderin in the dermis (Figure 2).
Pigmented purpuric dermatosis (PPD) describes a group of cutaneous lesions that are characterized by petechiae and pigmentary changes. These lesions most commonly present on the lower limbs; however, other sites have been reported.1 This group includes several major clinical forms such as Schamberg disease, LA, purpura annularis telangiectodes of Majocchi, eczematidlike purpura of Doucas and Kapetanakis, and lichenoid PPD of Gougerot and Blum. Lesions typically demonstrate a striking golden brown color clinically and by definition occur in the absence of platelet defects or vasculitis.1
Factors implicated in the pathogenesis of pigmented purpura include gravitational dependency, venous stasis, infection, and drugs.2 It is suggested that cellular immunity may play a role in the development of the disease based on the presence of CD4+ T lymphocytes in the infiltrate and the expression of HLA-DR by these lymphocytes and the keratinocytes.3 Lichen aureus differs in that it relates to increased intravascular pressure from an incompetent valve in an underlying perforating vein.4
Lichen aureus, also referred to as lichen purpuricus, is one major variant of PPD. The name reflects both the characteristic golden brown color and the histopathologic pattern of inflammation.1 Lichen aureus usually presents as a unilateral, asymptomatic, confined single lesion located mainly on the leg,1 though it can develop at other sites or as a localized group of lesions. Extensive lesions have been reported5 and cases with a segmental distribution have been described.6 In contrast, Schamberg disease demonstrates pinhead-sized reddish lesions giving the characteristic cayenne pepper pigmentation. These lesions coalesce to form thumbprint patches that progress proximally.1 Majocchi purpura is annular and telangiectatic, while lichenoid purpura of Gougerot and Blum presents with flat-topped, polygonal, violaceous papules that turn brown over time.
Some authors have championed a role for dermoscopy in diagnosis of LA.7 By dermoscopy, LA demonstrates a diffuse copper background reflecting the lymphohistiocytic dermal infiltrate, red dots and globules representing the extravasated red blood cells and the dilated swollen vessels, and grey dots that reflect the hemosiderin present in the dermis.8
Histologically, LA demonstrates a superficial perivascular infiltrate composed mainly of CD4+ lymphocytes surrounding the superficial capillaries. Over time, red cell extravasation leads to the formation of hemosiderin-laden macrophages, which can be highlighted with Perls Prussian blue stain. A bandlike infiltrate with thin strands of collagen separating it from the epidermis also may be noted.9
An important consideration in the differential diagnosis of PPD is mycosis fungoides (MF). Mycosis fungoides is a cutaneous T-cell lymphoma that clinically presents as a single or multiple hypopigmented or hyperpigmented patches or as erythematous scaly lesions in the patch or plaque stage. These lesions eventually may evolve into tumor stage.10 Mycosis fungoides may mimic PPD clinically and/or histopathologically, and rarely PPD also may precede MF.11 Involvement of the trunk, especially the lower abdomen and buttock region, favors a diagnosis of MF. Typically, histopathologic examination of MF demonstrates an epidermotropic lymphocytic infiltrate composed of atypical cerebriform lymphocytes overlying papillary dermal fibrosis. Although classic MF would be difficult to confuse with PPD, the atrophic lichenoid pattern of MF may show remarkable overlap with PPD.12 Such cases require clinicopathologic correlation, immunophenotyping of the epidermotropic lymphocytes, and occasionally T-cell clonality studies.
Lichen aureus is a chronic persistent disease unless the underlying incompetent perforator vessel is ligated. Various treatments have been used for other forms of pigmented purpura including topical corticosteroids, topical tacrolimus, systemic vasodilators such as prostacyclin and pentoxifylline, and phototherapy.1 Clinical follow-up is recommended for lesions that show some clinical or histopathological overlap with MF. Additional biopsies also may prove useful in establishing a definitive diagnosis in ambiguous cases.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165-169.
- Aiba S, Tagami H. Immunohistologic studies in Schamberg's disease. evidence for cellular immune reaction in lesional skin. Arch Dermatol. 1988;124:1058-1062.
- English J. Lichen aureus. J Am Acad Dermatol. 1985;12(2, pt 1):377-379.
- Duhra P, Tan CY. Lichen aureus. Br J Dermatol. 1986;114:395.
- Moche J, Glassman S, Modi D, et al. Segmental lichen aureus: a report of two cases treated with methylprednisolone aceponate. Australas J Dermatol. 2011;52:E15-E18.
- Zaballos P, Puig S, Malvehy J. Dermoscopy of pigmented purpuric dermatoses (lichen aureus): a useful tool for clinical diagnosis. Arch Dermatol. 2004;140:1290-1291.
- Portela PS, Melo DF, Ormiga P, et al. Dermoscopy of lichen aureus. An Bras Dermatol. 2013;88:253-255.
- Smoller BR, Kamel OW. Pigmented purpuric eruptions: immunopathologic studies supportive of a common immunophenotype. J Cutan Pathol. 1991;18:423-427.
- Jaffe ES, Harris NL, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. a progress report. Am J Clin Pathol. 1999;111(1 suppl 1):S8-S12.
- Hanna S, Walsh N, D'Intino Y, et al. Mycosis fungoides presenting as pigmented purpuric dermatitis. Pediatr Dermatol. 2006;23:350-354.
- Toro JR, Sander CA, LeBoit PE. Persistent pigmented purpuric dermatitis and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165-169.
- Aiba S, Tagami H. Immunohistologic studies in Schamberg's disease. evidence for cellular immune reaction in lesional skin. Arch Dermatol. 1988;124:1058-1062.
- English J. Lichen aureus. J Am Acad Dermatol. 1985;12(2, pt 1):377-379.
- Duhra P, Tan CY. Lichen aureus. Br J Dermatol. 1986;114:395.
- Moche J, Glassman S, Modi D, et al. Segmental lichen aureus: a report of two cases treated with methylprednisolone aceponate. Australas J Dermatol. 2011;52:E15-E18.
- Zaballos P, Puig S, Malvehy J. Dermoscopy of pigmented purpuric dermatoses (lichen aureus): a useful tool for clinical diagnosis. Arch Dermatol. 2004;140:1290-1291.
- Portela PS, Melo DF, Ormiga P, et al. Dermoscopy of lichen aureus. An Bras Dermatol. 2013;88:253-255.
- Smoller BR, Kamel OW. Pigmented purpuric eruptions: immunopathologic studies supportive of a common immunophenotype. J Cutan Pathol. 1991;18:423-427.
- Jaffe ES, Harris NL, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. a progress report. Am J Clin Pathol. 1999;111(1 suppl 1):S8-S12.
- Hanna S, Walsh N, D'Intino Y, et al. Mycosis fungoides presenting as pigmented purpuric dermatitis. Pediatr Dermatol. 2006;23:350-354.
- Toro JR, Sander CA, LeBoit PE. Persistent pigmented purpuric dermatitis and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
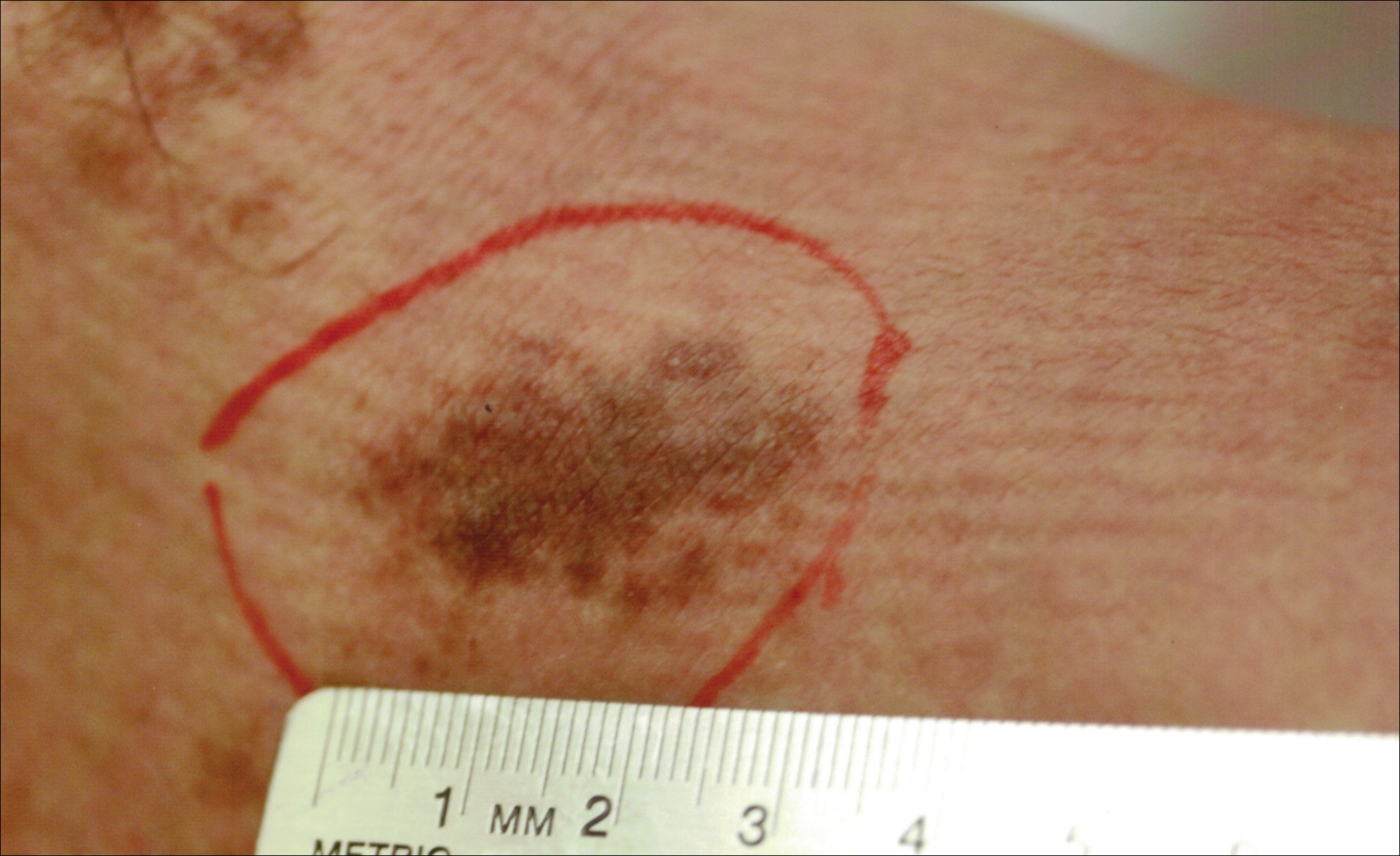
A 32-year-old man presented with an asymptomatic pigmented lesion on the left foot that developed over the course of 4 months. Physical examination revealed a 4-cm asymmetrical, deeply pigmented macule on the left foot. A shave biopsy of the lesion was performed.
Bullous Lesions in a Neonate
The Diagnosis: Incontinentia Pigmenti
The infant's mother was noted to have diffuse hypopigmented patches over the trunk, arms, and legs (present since adolescence) with whorled cicatricial alopecia of the vertex scalp and peg-shaped teeth (Figure). Together, these findings suggested incontinentia pigmenti (IP), which the mother revealed she had been diagnosed with in childhood. The infant's characteristic lesions in the setting of her mother's diagnosed genodermatosis confirmed the diagnosis of IP.
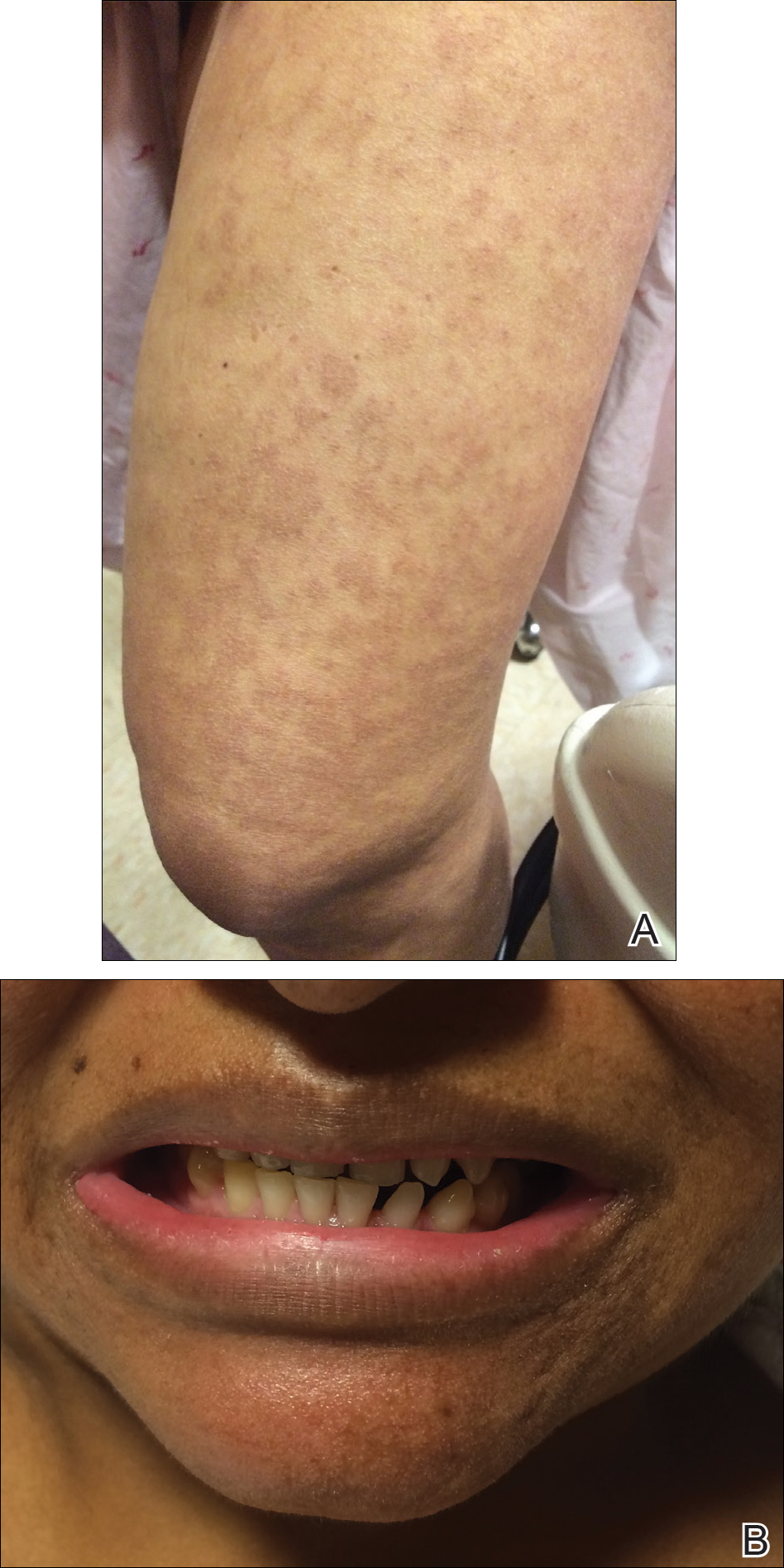
Incontinentia pigmenti is an X-linked dominant disorder that presents with many classic dermatologic, dental, neurologic, and ophthalmologic findings. The causative mutation occurs in IKBKG/NEMO (inhibitor of κ polypeptide gene enhancer in B-cells, kinase γ/nuclear factor-κB essential modulator) gene on Xq28, disabling the resultant protein that normally protects cells from tumor necrosis factor family-induced apoptosis.1 Incontinentia pigmenti usually is lethal in males and causes an unbalanced X-inactivation in surviving female IP patients. Occurring at a rate of 1.2 per 100,000 births,2 IP typically presents in female infants with skin lesions patterned along Blaschko lines that evolve in 4 stages over a lifetime.3 Stage I, presenting in the neonatal period, manifests as vesiculobullous eruptions on the limbs and scalp. Stages II to IV vary in duration from months to years and are comprised of a verrucous stage, a hyperpigmented stage, and a hypopigmented stage, respectively.3 All stages of IP can overlap and coexist.
The vesiculobullous findings in infants with IP may be mistakenly attributed to other diseases with prominent vesicular or bullous components including herpes simplex virus, epidermolysis bullosa, and infantile acropustulosis. With neonatal herpes simplex virus infection, vesicular skin or mucocutaneous lesions occur 9 to 11 days after birth and can be confirmed by specimen culture or qualitative polymerase chain reaction, while stage I of IP appears within the first 6 to 8 weeks of life and can be present at birth.4 The hallmark of epidermolysis bullosa, caused by mutations in keratins 5 and 14, is blistering erosions of the skin in response to frictional stress,1 thus these lesions do not follow Blaschko lines. Infantile acropustulosis, a nonheritable vesiculopustular eruption of the hands and feet, rarely occurs in the immediate newborn period; it most often appears in the 3- to 6-month age range with recurrent eruptions at 3- to 4-week intervals.5 Focal dermal hypoplasia is another X-linked dominant disorder with blaschkolinear findings at birth that presents with pink or red, angular, atrophic macules, in contrast to the bullous lesions of IP.6
Incontinentia pigmenti may encompass a wide range of systemic symptoms in addition to the classic dermatologic findings. Notably, central nervous system defects are concurrent in up to 40% of IP cases, with seizures, mental retardation, and spastic paresis being the most common sequelae.7 Teeth defects, seen in 35% of patients, include delayed primary dentition and peg-shaped teeth. Many patients will experience ophthalmologic defects including vision problems (16%) and retinopathy (15%).7
The cutaneous eruptions of IP may be treated with topical corticosteroids or topical tacrolimus, and vesicles should be left intact and monitored for signs of infection.8,9 Seizures, if present, should be treated with anticonvulsants, and regular neuropsychiatric monitoring and physical rehabilitation may be warranted. Patients should be regularly monitored for retinopathy beginning at the time of diagnosis. Retinal fibrovascular proliferation is treated with xenon laser photocoagulation to reduce the high risk for retinal detachment in this population.10,11 Older and younger at-risk relatives must be evaluated by genetic testing or thorough physical examination to clarify their disease status and determine the need for additional genetic counseling.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012.
- Prevalence and incidence of rare diseases: bibliographic data. Orphanet Report Series, Rare Diseases collection. http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf. Published June 2017. Accessed July 13, 2017.
- Scheuerle AE, Ursini MV. Incontinentia pigmenti. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. Seattle, WA: University of Washington; 2015. http://www.ncbi.nlm.nih.gov/books/NBK1472/. Accessed July 25, 2017.
- James SH, Kimberlin DW. Neonatal herpes simplex virus infection. Infect Dis Clin North Am. 2015;29:391-400.
- Eichenfield LF, Frieden IJ, Mathes E, et al, eds. Neonatal and Infant Dermatology. Philadelphia, PA: Saunders; 2015.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Fusco F, Paciolla M, Conte MI, et al. Incontinentia pigmenti: report on data from 2000 to 2013. Orphanet J Rare Dis. 2014;9:93.
- Jessup CJ, Morgan SC, Cohen LM, et al. Incontinentia pigmenti: treatment of IP with topical tacrolimus. J Drugs Dermatol. 2009;8:944-946.
- Kaya TI, Tursen U, Ikizoglu G. Therapeutic use of topical corticosteroids in the vesiculobullous lesions of incontinentia pigmenti [published online June 1, 2009]. Clin Exp Dermatol. 2009;34:E611-E613.
- Nguyen JK, Brady-Mccreery KM. Laser photocoagulation in preproliferative retinopathy of incontinentia pigmenti. J AAPOS. 2001;5:258-259.
- Chen CJ, Han IC, Tian J, et al. Extended follow-up of treated and untreated retinopathy in incontinentia pigmenti: analysis of peripheral vascular changes and incidence of retinal detachment. JAMA Ophthalmol. 2015;133:542-548.
The Diagnosis: Incontinentia Pigmenti
The infant's mother was noted to have diffuse hypopigmented patches over the trunk, arms, and legs (present since adolescence) with whorled cicatricial alopecia of the vertex scalp and peg-shaped teeth (Figure). Together, these findings suggested incontinentia pigmenti (IP), which the mother revealed she had been diagnosed with in childhood. The infant's characteristic lesions in the setting of her mother's diagnosed genodermatosis confirmed the diagnosis of IP.
Incontinentia pigmenti is an X-linked dominant disorder that presents with many classic dermatologic, dental, neurologic, and ophthalmologic findings. The causative mutation occurs in IKBKG/NEMO (inhibitor of κ polypeptide gene enhancer in B-cells, kinase γ/nuclear factor-κB essential modulator) gene on Xq28, disabling the resultant protein that normally protects cells from tumor necrosis factor family-induced apoptosis.1 Incontinentia pigmenti usually is lethal in males and causes an unbalanced X-inactivation in surviving female IP patients. Occurring at a rate of 1.2 per 100,000 births,2 IP typically presents in female infants with skin lesions patterned along Blaschko lines that evolve in 4 stages over a lifetime.3 Stage I, presenting in the neonatal period, manifests as vesiculobullous eruptions on the limbs and scalp. Stages II to IV vary in duration from months to years and are comprised of a verrucous stage, a hyperpigmented stage, and a hypopigmented stage, respectively.3 All stages of IP can overlap and coexist.
The vesiculobullous findings in infants with IP may be mistakenly attributed to other diseases with prominent vesicular or bullous components including herpes simplex virus, epidermolysis bullosa, and infantile acropustulosis. With neonatal herpes simplex virus infection, vesicular skin or mucocutaneous lesions occur 9 to 11 days after birth and can be confirmed by specimen culture or qualitative polymerase chain reaction, while stage I of IP appears within the first 6 to 8 weeks of life and can be present at birth.4 The hallmark of epidermolysis bullosa, caused by mutations in keratins 5 and 14, is blistering erosions of the skin in response to frictional stress,1 thus these lesions do not follow Blaschko lines. Infantile acropustulosis, a nonheritable vesiculopustular eruption of the hands and feet, rarely occurs in the immediate newborn period; it most often appears in the 3- to 6-month age range with recurrent eruptions at 3- to 4-week intervals.5 Focal dermal hypoplasia is another X-linked dominant disorder with blaschkolinear findings at birth that presents with pink or red, angular, atrophic macules, in contrast to the bullous lesions of IP.6
Incontinentia pigmenti may encompass a wide range of systemic symptoms in addition to the classic dermatologic findings. Notably, central nervous system defects are concurrent in up to 40% of IP cases, with seizures, mental retardation, and spastic paresis being the most common sequelae.7 Teeth defects, seen in 35% of patients, include delayed primary dentition and peg-shaped teeth. Many patients will experience ophthalmologic defects including vision problems (16%) and retinopathy (15%).7
The cutaneous eruptions of IP may be treated with topical corticosteroids or topical tacrolimus, and vesicles should be left intact and monitored for signs of infection.8,9 Seizures, if present, should be treated with anticonvulsants, and regular neuropsychiatric monitoring and physical rehabilitation may be warranted. Patients should be regularly monitored for retinopathy beginning at the time of diagnosis. Retinal fibrovascular proliferation is treated with xenon laser photocoagulation to reduce the high risk for retinal detachment in this population.10,11 Older and younger at-risk relatives must be evaluated by genetic testing or thorough physical examination to clarify their disease status and determine the need for additional genetic counseling.
The Diagnosis: Incontinentia Pigmenti
The infant's mother was noted to have diffuse hypopigmented patches over the trunk, arms, and legs (present since adolescence) with whorled cicatricial alopecia of the vertex scalp and peg-shaped teeth (Figure). Together, these findings suggested incontinentia pigmenti (IP), which the mother revealed she had been diagnosed with in childhood. The infant's characteristic lesions in the setting of her mother's diagnosed genodermatosis confirmed the diagnosis of IP.
Incontinentia pigmenti is an X-linked dominant disorder that presents with many classic dermatologic, dental, neurologic, and ophthalmologic findings. The causative mutation occurs in IKBKG/NEMO (inhibitor of κ polypeptide gene enhancer in B-cells, kinase γ/nuclear factor-κB essential modulator) gene on Xq28, disabling the resultant protein that normally protects cells from tumor necrosis factor family-induced apoptosis.1 Incontinentia pigmenti usually is lethal in males and causes an unbalanced X-inactivation in surviving female IP patients. Occurring at a rate of 1.2 per 100,000 births,2 IP typically presents in female infants with skin lesions patterned along Blaschko lines that evolve in 4 stages over a lifetime.3 Stage I, presenting in the neonatal period, manifests as vesiculobullous eruptions on the limbs and scalp. Stages II to IV vary in duration from months to years and are comprised of a verrucous stage, a hyperpigmented stage, and a hypopigmented stage, respectively.3 All stages of IP can overlap and coexist.
The vesiculobullous findings in infants with IP may be mistakenly attributed to other diseases with prominent vesicular or bullous components including herpes simplex virus, epidermolysis bullosa, and infantile acropustulosis. With neonatal herpes simplex virus infection, vesicular skin or mucocutaneous lesions occur 9 to 11 days after birth and can be confirmed by specimen culture or qualitative polymerase chain reaction, while stage I of IP appears within the first 6 to 8 weeks of life and can be present at birth.4 The hallmark of epidermolysis bullosa, caused by mutations in keratins 5 and 14, is blistering erosions of the skin in response to frictional stress,1 thus these lesions do not follow Blaschko lines. Infantile acropustulosis, a nonheritable vesiculopustular eruption of the hands and feet, rarely occurs in the immediate newborn period; it most often appears in the 3- to 6-month age range with recurrent eruptions at 3- to 4-week intervals.5 Focal dermal hypoplasia is another X-linked dominant disorder with blaschkolinear findings at birth that presents with pink or red, angular, atrophic macules, in contrast to the bullous lesions of IP.6
Incontinentia pigmenti may encompass a wide range of systemic symptoms in addition to the classic dermatologic findings. Notably, central nervous system defects are concurrent in up to 40% of IP cases, with seizures, mental retardation, and spastic paresis being the most common sequelae.7 Teeth defects, seen in 35% of patients, include delayed primary dentition and peg-shaped teeth. Many patients will experience ophthalmologic defects including vision problems (16%) and retinopathy (15%).7
The cutaneous eruptions of IP may be treated with topical corticosteroids or topical tacrolimus, and vesicles should be left intact and monitored for signs of infection.8,9 Seizures, if present, should be treated with anticonvulsants, and regular neuropsychiatric monitoring and physical rehabilitation may be warranted. Patients should be regularly monitored for retinopathy beginning at the time of diagnosis. Retinal fibrovascular proliferation is treated with xenon laser photocoagulation to reduce the high risk for retinal detachment in this population.10,11 Older and younger at-risk relatives must be evaluated by genetic testing or thorough physical examination to clarify their disease status and determine the need for additional genetic counseling.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012.
- Prevalence and incidence of rare diseases: bibliographic data. Orphanet Report Series, Rare Diseases collection. http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf. Published June 2017. Accessed July 13, 2017.
- Scheuerle AE, Ursini MV. Incontinentia pigmenti. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. Seattle, WA: University of Washington; 2015. http://www.ncbi.nlm.nih.gov/books/NBK1472/. Accessed July 25, 2017.
- James SH, Kimberlin DW. Neonatal herpes simplex virus infection. Infect Dis Clin North Am. 2015;29:391-400.
- Eichenfield LF, Frieden IJ, Mathes E, et al, eds. Neonatal and Infant Dermatology. Philadelphia, PA: Saunders; 2015.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Fusco F, Paciolla M, Conte MI, et al. Incontinentia pigmenti: report on data from 2000 to 2013. Orphanet J Rare Dis. 2014;9:93.
- Jessup CJ, Morgan SC, Cohen LM, et al. Incontinentia pigmenti: treatment of IP with topical tacrolimus. J Drugs Dermatol. 2009;8:944-946.
- Kaya TI, Tursen U, Ikizoglu G. Therapeutic use of topical corticosteroids in the vesiculobullous lesions of incontinentia pigmenti [published online June 1, 2009]. Clin Exp Dermatol. 2009;34:E611-E613.
- Nguyen JK, Brady-Mccreery KM. Laser photocoagulation in preproliferative retinopathy of incontinentia pigmenti. J AAPOS. 2001;5:258-259.
- Chen CJ, Han IC, Tian J, et al. Extended follow-up of treated and untreated retinopathy in incontinentia pigmenti: analysis of peripheral vascular changes and incidence of retinal detachment. JAMA Ophthalmol. 2015;133:542-548.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012.
- Prevalence and incidence of rare diseases: bibliographic data. Orphanet Report Series, Rare Diseases collection. http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf. Published June 2017. Accessed July 13, 2017.
- Scheuerle AE, Ursini MV. Incontinentia pigmenti. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. Seattle, WA: University of Washington; 2015. http://www.ncbi.nlm.nih.gov/books/NBK1472/. Accessed July 25, 2017.
- James SH, Kimberlin DW. Neonatal herpes simplex virus infection. Infect Dis Clin North Am. 2015;29:391-400.
- Eichenfield LF, Frieden IJ, Mathes E, et al, eds. Neonatal and Infant Dermatology. Philadelphia, PA: Saunders; 2015.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Fusco F, Paciolla M, Conte MI, et al. Incontinentia pigmenti: report on data from 2000 to 2013. Orphanet J Rare Dis. 2014;9:93.
- Jessup CJ, Morgan SC, Cohen LM, et al. Incontinentia pigmenti: treatment of IP with topical tacrolimus. J Drugs Dermatol. 2009;8:944-946.
- Kaya TI, Tursen U, Ikizoglu G. Therapeutic use of topical corticosteroids in the vesiculobullous lesions of incontinentia pigmenti [published online June 1, 2009]. Clin Exp Dermatol. 2009;34:E611-E613.
- Nguyen JK, Brady-Mccreery KM. Laser photocoagulation in preproliferative retinopathy of incontinentia pigmenti. J AAPOS. 2001;5:258-259.
- Chen CJ, Han IC, Tian J, et al. Extended follow-up of treated and untreated retinopathy in incontinentia pigmenti: analysis of peripheral vascular changes and incidence of retinal detachment. JAMA Ophthalmol. 2015;133:542-548.
A 1-day-old Hispanic female infant was born via uncomplicated vaginal delivery at 41 weeks' gestation after a normal pregnancy. Linear plaques containing multiple ruptured vesicles and bullae following Blaschko lines were noted on the right medial thigh and anterior arm. The infant was afebrile and generally well-appearing.
Solitary Nodule With White Hairs
The Diagnosis: Trichofolliculoma
Microscopic examination revealed a dilated cystic follicle that communicated with the skin surface (Figure). The follicle was lined with squamous epithelium and surrounded by numerous secondary follicles, many of which contained a hair shaft. A diagnosis of trichofolliculoma was made.
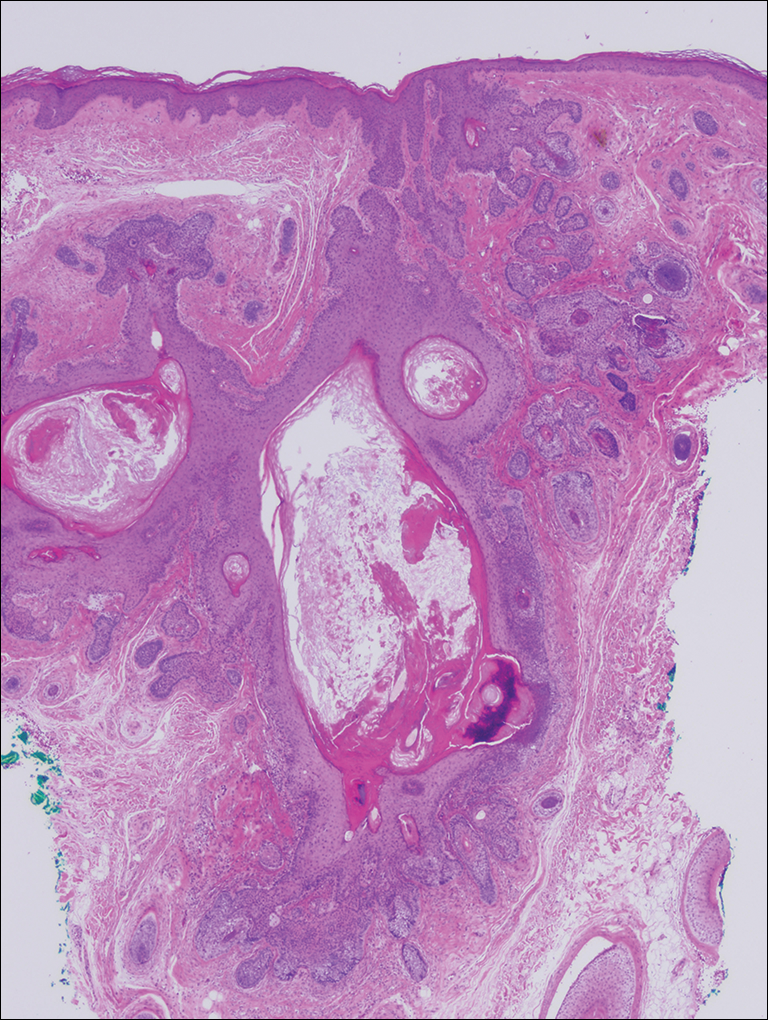
Clinically, the differential diagnosis of a flesh-colored papule on the scalp with prominent follicle includes dilated pore of Winer, epidermoid cyst, pilar sheath acanthoma, and trichoepithelioma.1,2 Multiple hair shafts present in a single follicle may be seen in pili multigemini, tufted folliculitis, trichostasis spinulosa, and trichofolliculoma. On histopathologic examination, a dilated central follicle surrounded with smaller secondary follicles was identified, consistent with trichofolliculoma.
Trichofolliculoma is a rare follicular hamartoma typically occurring on the face, scalp, or trunk as a solitary papule or nodule due to the proliferation of abnormal hair follicle stem cells.3,4 It may present as a flesh-colored nodule with a central pore that may drain sebum or contain white vellus hairs. Trichofolliculoma is considered a benign entity, despite one case report of malignant transformation.5 Biopsy is diagnostic and no further treatment is needed. Recurrence rarely occurs at the primary site after surgical excision, which may be performed for cosmetic purposes or to alleviate functional impairment.
- Ghosh SK, Bandyopadhyay D, Barma KD. Perifollicular nodule on the face of a young man. Indian J Dermatol Venereol Leprol. 2011;77:531-533.
- Gokalp H, Gurer MA, Alan S. Trichofolliculoma: a rare variant of hair follicle hamartoma. Dermatol Online J. 2013;19:19264.
- Choi CM, Lew BL, Sim WY. Multiple trichofolliculomas on unusual sites: a case report and review of the literature. Int J Dermatol. 2013;52:87-89.
- Misago N, Kimura T, Toda S, et al. A revaluation of trichofolliculoma: the histopathological and immunohistochemical features. Am J Dermatopathol. 2010;32:35-43.
- Stem JB, Stout DA. Trichofolliculoma showing perineural invasion. trichofolliculocarcinoma? Arch Dermatol. 1979;115:1003-1004.
The Diagnosis: Trichofolliculoma
Microscopic examination revealed a dilated cystic follicle that communicated with the skin surface (Figure). The follicle was lined with squamous epithelium and surrounded by numerous secondary follicles, many of which contained a hair shaft. A diagnosis of trichofolliculoma was made.
Clinically, the differential diagnosis of a flesh-colored papule on the scalp with prominent follicle includes dilated pore of Winer, epidermoid cyst, pilar sheath acanthoma, and trichoepithelioma.1,2 Multiple hair shafts present in a single follicle may be seen in pili multigemini, tufted folliculitis, trichostasis spinulosa, and trichofolliculoma. On histopathologic examination, a dilated central follicle surrounded with smaller secondary follicles was identified, consistent with trichofolliculoma.
Trichofolliculoma is a rare follicular hamartoma typically occurring on the face, scalp, or trunk as a solitary papule or nodule due to the proliferation of abnormal hair follicle stem cells.3,4 It may present as a flesh-colored nodule with a central pore that may drain sebum or contain white vellus hairs. Trichofolliculoma is considered a benign entity, despite one case report of malignant transformation.5 Biopsy is diagnostic and no further treatment is needed. Recurrence rarely occurs at the primary site after surgical excision, which may be performed for cosmetic purposes or to alleviate functional impairment.
The Diagnosis: Trichofolliculoma
Microscopic examination revealed a dilated cystic follicle that communicated with the skin surface (Figure). The follicle was lined with squamous epithelium and surrounded by numerous secondary follicles, many of which contained a hair shaft. A diagnosis of trichofolliculoma was made.
Clinically, the differential diagnosis of a flesh-colored papule on the scalp with prominent follicle includes dilated pore of Winer, epidermoid cyst, pilar sheath acanthoma, and trichoepithelioma.1,2 Multiple hair shafts present in a single follicle may be seen in pili multigemini, tufted folliculitis, trichostasis spinulosa, and trichofolliculoma. On histopathologic examination, a dilated central follicle surrounded with smaller secondary follicles was identified, consistent with trichofolliculoma.
Trichofolliculoma is a rare follicular hamartoma typically occurring on the face, scalp, or trunk as a solitary papule or nodule due to the proliferation of abnormal hair follicle stem cells.3,4 It may present as a flesh-colored nodule with a central pore that may drain sebum or contain white vellus hairs. Trichofolliculoma is considered a benign entity, despite one case report of malignant transformation.5 Biopsy is diagnostic and no further treatment is needed. Recurrence rarely occurs at the primary site after surgical excision, which may be performed for cosmetic purposes or to alleviate functional impairment.
- Ghosh SK, Bandyopadhyay D, Barma KD. Perifollicular nodule on the face of a young man. Indian J Dermatol Venereol Leprol. 2011;77:531-533.
- Gokalp H, Gurer MA, Alan S. Trichofolliculoma: a rare variant of hair follicle hamartoma. Dermatol Online J. 2013;19:19264.
- Choi CM, Lew BL, Sim WY. Multiple trichofolliculomas on unusual sites: a case report and review of the literature. Int J Dermatol. 2013;52:87-89.
- Misago N, Kimura T, Toda S, et al. A revaluation of trichofolliculoma: the histopathological and immunohistochemical features. Am J Dermatopathol. 2010;32:35-43.
- Stem JB, Stout DA. Trichofolliculoma showing perineural invasion. trichofolliculocarcinoma? Arch Dermatol. 1979;115:1003-1004.
- Ghosh SK, Bandyopadhyay D, Barma KD. Perifollicular nodule on the face of a young man. Indian J Dermatol Venereol Leprol. 2011;77:531-533.
- Gokalp H, Gurer MA, Alan S. Trichofolliculoma: a rare variant of hair follicle hamartoma. Dermatol Online J. 2013;19:19264.
- Choi CM, Lew BL, Sim WY. Multiple trichofolliculomas on unusual sites: a case report and review of the literature. Int J Dermatol. 2013;52:87-89.
- Misago N, Kimura T, Toda S, et al. A revaluation of trichofolliculoma: the histopathological and immunohistochemical features. Am J Dermatopathol. 2010;32:35-43.
- Stem JB, Stout DA. Trichofolliculoma showing perineural invasion. trichofolliculocarcinoma? Arch Dermatol. 1979;115:1003-1004.
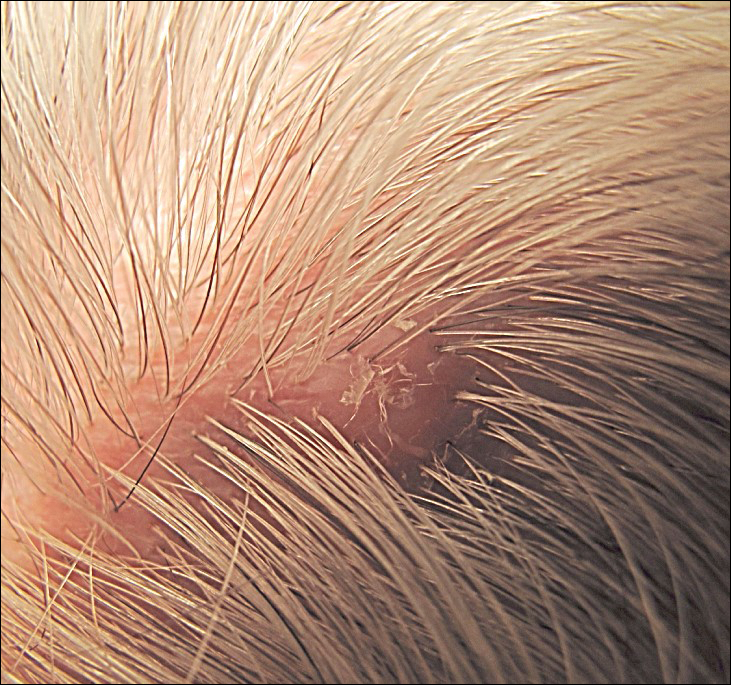
A 72-year-old man presented with a new asymptomatic 0.7-cm flesh-colored papule with a central tuft of white hairs on the posterior scalp. The remainder of the physical examination was unremarkable. Biopsy for histopathologic examination was performed to confirm diagnosis.
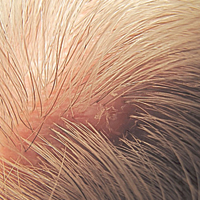
Necrotic Ulcer on the Thigh
The Diagnosis: Disseminated Cryptococcosis
Histopathologic examination of a 3-mm punch biopsy showed a diffuse dermal neutrophilic infiltrate with necrosis and subcutaneous tissue with round yeast surrounded by a prominent halo staining bright red with mucicarmine, representing a thick mucinous capsule (Figure). Grocott-Gomori methenamine-silver and periodic acid-Schiff stains also demonstrated fungal spores morphologically. Cerebrospinal fluid culture grew Cryptococcus neoformans, and cryptococcal antigen titers were positive in both serum and cerebrospinal fluid samples (>1:4096). The patient had autolytic debridement of the ulcer after completing a 4-week induction course of intravenous liposomal amphotericin B with oral flucytosine. He was transitioned to oral fluconazole for the consolidation phase of treatment.
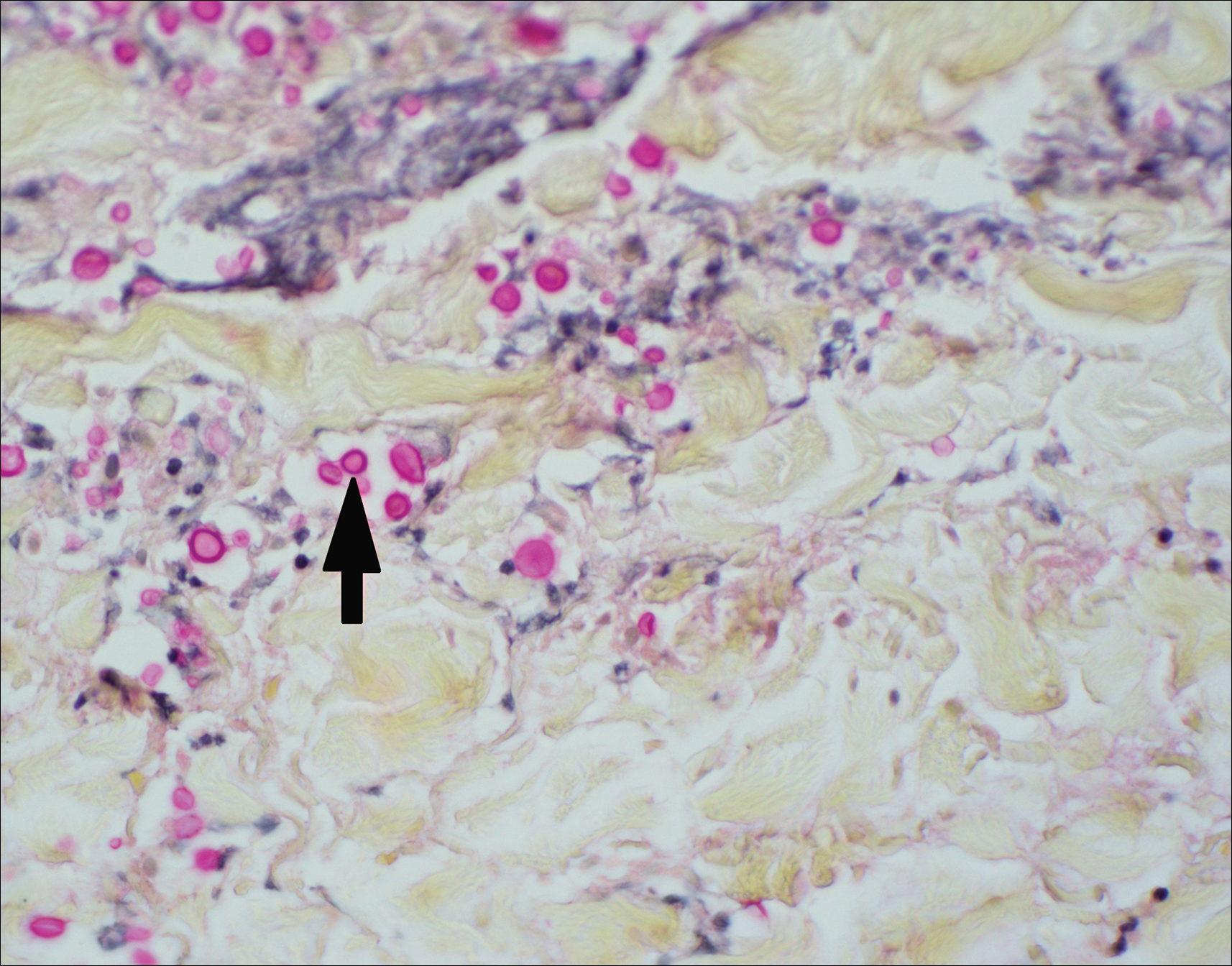
Cryptococcus is an opportunistic basidiomycetous yeast with worldwide distribution and 2 primary pathogenic species in humans: C neoformans and Cryptococcus gattii. It is associated with bird feces, composted food, and decayed wood.1,2 A predilection toward an immunosuppressed host is recognized in 70% to 90% of the infections caused by C neoformans; however, C gattii commonly affects individuals with apparently intact immune systems.1,3 Risk factors for infection include advanced human immunodeficiency virus infection, solid organ transplantation, chronic liver disease, autoimmune disease, hematological malignancy, and underlying genetic susceptibility.1,2
Initial exposure is through the respiratory tract with formation of latent reservoirs in the pulmonary lymph nodes with subsequent reactivation that can result in hematogenous dissemination.1,2 Cutaneous involvement was described in 108 patients (5%) in a large review of 1974 cases in France.4 Among those with cutaneous involvement, disseminated disease was diagnosed in 80 cases (74%), and 28 cases (26%) were considered primary cutaneous cryptococcosis. Primary cutaneous cryptococcosis typically presents as a single lesion, predominantly on the hand, with whitlow and more rarely with extensive cellulitis or necrotizing fasciitis.4 In disseminated cutaneous disease, there is no pathognomonic single lesion; however, it is commonly associated with multiple cutaneous lesions predominantly involving the head and neck. Plaques, abscesses, nodules, and pustular or umbilicated papules have been reported.1,5 There are few case reports that describe a single isolated necrotic ulcer with disseminated disease similar to our presented case, and more typically the necrotic ulcer is seen in transplanted patients.6 The differential diagnosis of a necrotic thigh ulcer includes pseudomonal ecthyma gangrenosum, cutaneous anthrax and aspergillosis, fusariosis, and a bite from the brown recluse spider.7 Our patient had an increased susceptibility to infection from his ongoing chemotherapy, a risk previously described in oncology patients with cell-mediated immunosuppression.8
Management for disseminated cryptococcosis is a 3-phase therapy including induction with intravenous amphotericin B and oral flucytosine for a minimum of 2 weeks, with consolidation and maintenance phases both with oral fluconazole for a length depending on underlying immunosuppression.9
- Chen SC, Meyer W, Sorrell TC. Cryptococcus gattii infections. Clin Microbiol Rev. 2014;27:980-1024.
- Williamson PR, Jarvis JN, Panackal AA, et al. Cryptococcal meningitis: epidemiology, immunology, diagnosis, and therapy [published online November 25, 2016]. Nat Rev Neurol. 2017;13:13-24.
- Speed B, Dunt D. Clinical and host differences between infections with the two varieties of Cryptococcus neoformans. Clin Infect Dis. 1995;21:28-34.
- Neuville S, Dromer F, Morin O, et al; French Cryptococcosis Study Group. Primary cutaneous cryptococcosis: a distinct clinical entity [published online January 17, 2003]. Clin Infect Dis. 2003;36:337-347.
- Murakawa GJ, Kerschmann R, Berger T. Cutaneous cryptococcus infection and AIDS: report of 12 cases and review of the literature. JAMA Dermatol. 1996;132:545-548.
- Sun HY, Alexander BD, Lortholary O, et al. Cutaneous cryptococcosis in solid organ transplant recipients. Med Mycol. 2010;48:785-791.
- Grossman ME, Fox LP, Kovarik C, et al. Cutaneous Manifestations of Infection in the Immunocompromised Host. Baltimore, MD: Williams & Wilkins; 2012.
- Korfel A, Menssen HD, Schwartz S, et al. Cryptococcosis in Hodgkin's disease: description of two cases and review of the literature. Ann Hematol. 1998;76:283-286.
- Perfect JR, Dismukes WE, Dromer F. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis. 2010;50:291-322.
The Diagnosis: Disseminated Cryptococcosis
Histopathologic examination of a 3-mm punch biopsy showed a diffuse dermal neutrophilic infiltrate with necrosis and subcutaneous tissue with round yeast surrounded by a prominent halo staining bright red with mucicarmine, representing a thick mucinous capsule (Figure). Grocott-Gomori methenamine-silver and periodic acid-Schiff stains also demonstrated fungal spores morphologically. Cerebrospinal fluid culture grew Cryptococcus neoformans, and cryptococcal antigen titers were positive in both serum and cerebrospinal fluid samples (>1:4096). The patient had autolytic debridement of the ulcer after completing a 4-week induction course of intravenous liposomal amphotericin B with oral flucytosine. He was transitioned to oral fluconazole for the consolidation phase of treatment.
Cryptococcus is an opportunistic basidiomycetous yeast with worldwide distribution and 2 primary pathogenic species in humans: C neoformans and Cryptococcus gattii. It is associated with bird feces, composted food, and decayed wood.1,2 A predilection toward an immunosuppressed host is recognized in 70% to 90% of the infections caused by C neoformans; however, C gattii commonly affects individuals with apparently intact immune systems.1,3 Risk factors for infection include advanced human immunodeficiency virus infection, solid organ transplantation, chronic liver disease, autoimmune disease, hematological malignancy, and underlying genetic susceptibility.1,2
Initial exposure is through the respiratory tract with formation of latent reservoirs in the pulmonary lymph nodes with subsequent reactivation that can result in hematogenous dissemination.1,2 Cutaneous involvement was described in 108 patients (5%) in a large review of 1974 cases in France.4 Among those with cutaneous involvement, disseminated disease was diagnosed in 80 cases (74%), and 28 cases (26%) were considered primary cutaneous cryptococcosis. Primary cutaneous cryptococcosis typically presents as a single lesion, predominantly on the hand, with whitlow and more rarely with extensive cellulitis or necrotizing fasciitis.4 In disseminated cutaneous disease, there is no pathognomonic single lesion; however, it is commonly associated with multiple cutaneous lesions predominantly involving the head and neck. Plaques, abscesses, nodules, and pustular or umbilicated papules have been reported.1,5 There are few case reports that describe a single isolated necrotic ulcer with disseminated disease similar to our presented case, and more typically the necrotic ulcer is seen in transplanted patients.6 The differential diagnosis of a necrotic thigh ulcer includes pseudomonal ecthyma gangrenosum, cutaneous anthrax and aspergillosis, fusariosis, and a bite from the brown recluse spider.7 Our patient had an increased susceptibility to infection from his ongoing chemotherapy, a risk previously described in oncology patients with cell-mediated immunosuppression.8
Management for disseminated cryptococcosis is a 3-phase therapy including induction with intravenous amphotericin B and oral flucytosine for a minimum of 2 weeks, with consolidation and maintenance phases both with oral fluconazole for a length depending on underlying immunosuppression.9
The Diagnosis: Disseminated Cryptococcosis
Histopathologic examination of a 3-mm punch biopsy showed a diffuse dermal neutrophilic infiltrate with necrosis and subcutaneous tissue with round yeast surrounded by a prominent halo staining bright red with mucicarmine, representing a thick mucinous capsule (Figure). Grocott-Gomori methenamine-silver and periodic acid-Schiff stains also demonstrated fungal spores morphologically. Cerebrospinal fluid culture grew Cryptococcus neoformans, and cryptococcal antigen titers were positive in both serum and cerebrospinal fluid samples (>1:4096). The patient had autolytic debridement of the ulcer after completing a 4-week induction course of intravenous liposomal amphotericin B with oral flucytosine. He was transitioned to oral fluconazole for the consolidation phase of treatment.
Cryptococcus is an opportunistic basidiomycetous yeast with worldwide distribution and 2 primary pathogenic species in humans: C neoformans and Cryptococcus gattii. It is associated with bird feces, composted food, and decayed wood.1,2 A predilection toward an immunosuppressed host is recognized in 70% to 90% of the infections caused by C neoformans; however, C gattii commonly affects individuals with apparently intact immune systems.1,3 Risk factors for infection include advanced human immunodeficiency virus infection, solid organ transplantation, chronic liver disease, autoimmune disease, hematological malignancy, and underlying genetic susceptibility.1,2
Initial exposure is through the respiratory tract with formation of latent reservoirs in the pulmonary lymph nodes with subsequent reactivation that can result in hematogenous dissemination.1,2 Cutaneous involvement was described in 108 patients (5%) in a large review of 1974 cases in France.4 Among those with cutaneous involvement, disseminated disease was diagnosed in 80 cases (74%), and 28 cases (26%) were considered primary cutaneous cryptococcosis. Primary cutaneous cryptococcosis typically presents as a single lesion, predominantly on the hand, with whitlow and more rarely with extensive cellulitis or necrotizing fasciitis.4 In disseminated cutaneous disease, there is no pathognomonic single lesion; however, it is commonly associated with multiple cutaneous lesions predominantly involving the head and neck. Plaques, abscesses, nodules, and pustular or umbilicated papules have been reported.1,5 There are few case reports that describe a single isolated necrotic ulcer with disseminated disease similar to our presented case, and more typically the necrotic ulcer is seen in transplanted patients.6 The differential diagnosis of a necrotic thigh ulcer includes pseudomonal ecthyma gangrenosum, cutaneous anthrax and aspergillosis, fusariosis, and a bite from the brown recluse spider.7 Our patient had an increased susceptibility to infection from his ongoing chemotherapy, a risk previously described in oncology patients with cell-mediated immunosuppression.8
Management for disseminated cryptococcosis is a 3-phase therapy including induction with intravenous amphotericin B and oral flucytosine for a minimum of 2 weeks, with consolidation and maintenance phases both with oral fluconazole for a length depending on underlying immunosuppression.9
- Chen SC, Meyer W, Sorrell TC. Cryptococcus gattii infections. Clin Microbiol Rev. 2014;27:980-1024.
- Williamson PR, Jarvis JN, Panackal AA, et al. Cryptococcal meningitis: epidemiology, immunology, diagnosis, and therapy [published online November 25, 2016]. Nat Rev Neurol. 2017;13:13-24.
- Speed B, Dunt D. Clinical and host differences between infections with the two varieties of Cryptococcus neoformans. Clin Infect Dis. 1995;21:28-34.
- Neuville S, Dromer F, Morin O, et al; French Cryptococcosis Study Group. Primary cutaneous cryptococcosis: a distinct clinical entity [published online January 17, 2003]. Clin Infect Dis. 2003;36:337-347.
- Murakawa GJ, Kerschmann R, Berger T. Cutaneous cryptococcus infection and AIDS: report of 12 cases and review of the literature. JAMA Dermatol. 1996;132:545-548.
- Sun HY, Alexander BD, Lortholary O, et al. Cutaneous cryptococcosis in solid organ transplant recipients. Med Mycol. 2010;48:785-791.
- Grossman ME, Fox LP, Kovarik C, et al. Cutaneous Manifestations of Infection in the Immunocompromised Host. Baltimore, MD: Williams & Wilkins; 2012.
- Korfel A, Menssen HD, Schwartz S, et al. Cryptococcosis in Hodgkin's disease: description of two cases and review of the literature. Ann Hematol. 1998;76:283-286.
- Perfect JR, Dismukes WE, Dromer F. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis. 2010;50:291-322.
- Chen SC, Meyer W, Sorrell TC. Cryptococcus gattii infections. Clin Microbiol Rev. 2014;27:980-1024.
- Williamson PR, Jarvis JN, Panackal AA, et al. Cryptococcal meningitis: epidemiology, immunology, diagnosis, and therapy [published online November 25, 2016]. Nat Rev Neurol. 2017;13:13-24.
- Speed B, Dunt D. Clinical and host differences between infections with the two varieties of Cryptococcus neoformans. Clin Infect Dis. 1995;21:28-34.
- Neuville S, Dromer F, Morin O, et al; French Cryptococcosis Study Group. Primary cutaneous cryptococcosis: a distinct clinical entity [published online January 17, 2003]. Clin Infect Dis. 2003;36:337-347.
- Murakawa GJ, Kerschmann R, Berger T. Cutaneous cryptococcus infection and AIDS: report of 12 cases and review of the literature. JAMA Dermatol. 1996;132:545-548.
- Sun HY, Alexander BD, Lortholary O, et al. Cutaneous cryptococcosis in solid organ transplant recipients. Med Mycol. 2010;48:785-791.
- Grossman ME, Fox LP, Kovarik C, et al. Cutaneous Manifestations of Infection in the Immunocompromised Host. Baltimore, MD: Williams & Wilkins; 2012.
- Korfel A, Menssen HD, Schwartz S, et al. Cryptococcosis in Hodgkin's disease: description of two cases and review of the literature. Ann Hematol. 1998;76:283-286.
- Perfect JR, Dismukes WE, Dromer F. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis. 2010;50:291-322.
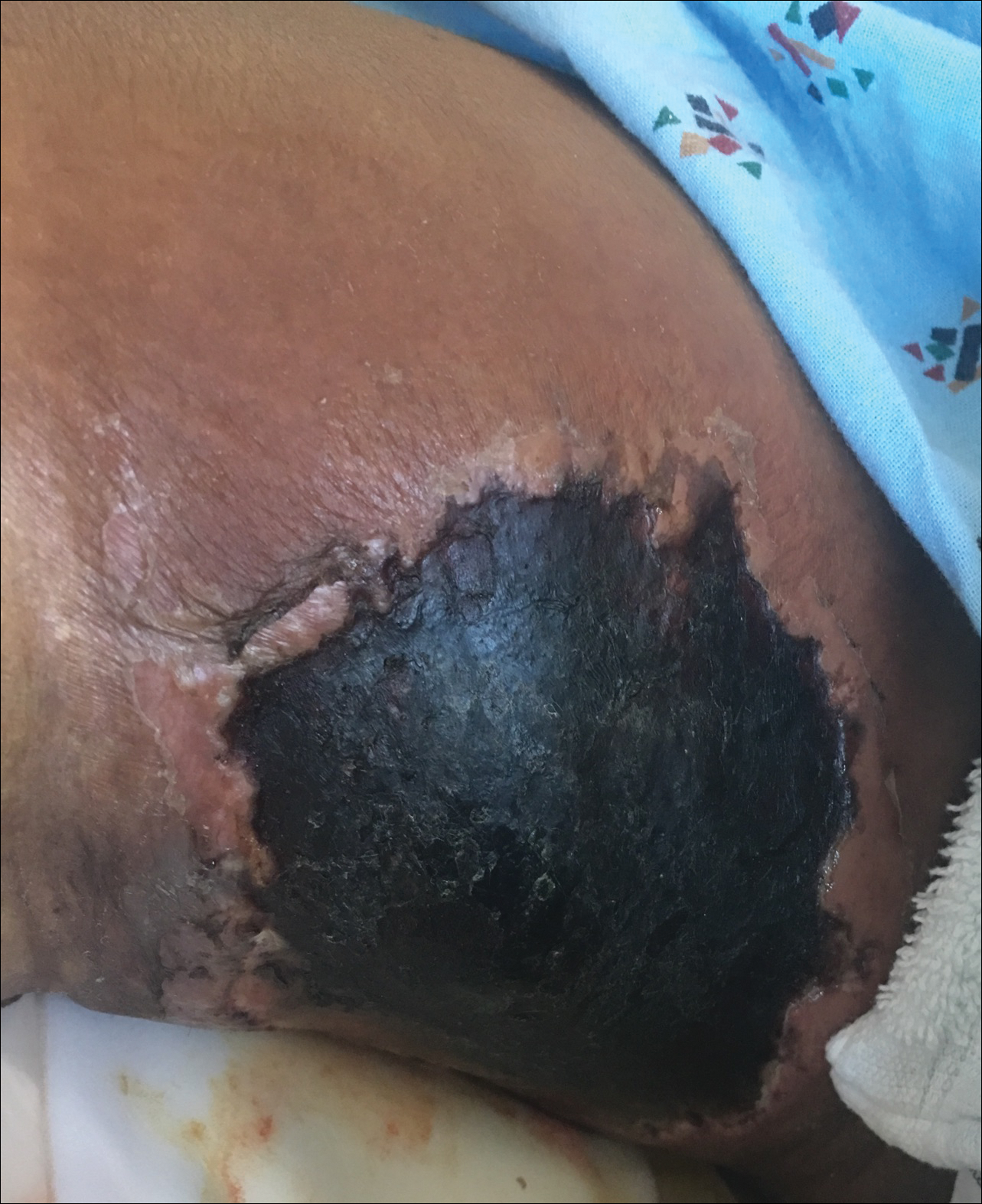
A 29-year-old man with a history of acute lymphoblastic leukemia was admitted for acute encephalopathy and a necrotic ulcer on the right thigh of 2 weeks' duration. He had received chemotherapy with pegaspargase and vincristine 6 weeks prior to admission. He reported headache with nausea and vomiting of 2 weeks' duration and had sustained a fall in the bathtub a week prior that initially resulted in a right thigh abrasion. He denied recent travel, unusual food consumption, animal exposure, exposure to sick persons, and alcohol or other drug use. On examination the patient was alert but was not oriented to person, place, or time. A 10.2 ×10-cm necrotic ulcer with surrounding mild erythema and tenderness was noted on the right inner thigh.
Chronic Diffuse Erythematous Papulonodules
The Diagnosis: Lymphomatoid Papulosis
A shave biopsy of an established lesion on the volar aspect of the left wrist was performed (Figure 1). The biopsy showed an ulcerated nodular lesion characterized by a dense mixed inflammatory cell infiltrate in the dermis composed of lymphocytes, histiocytes, scattered neutrophils, and numerous eosinophils (Figure 2). Notably there was a minor population of large atypical cells with immunoblastic and anaplastic morphology present individually and in small clusters most prominently within the upper dermis (Figures 3 and 4). Immunohistochemistry of the anaplastic cells revealed a CD30+, CD3−, CD4+, CD5−, CD8−, CD2−, CD7−, CD56−, ALK1− (anaplastic lymphoma kinase-1), PAX5− (paired box protein-5), CD20−, and CD15− phenotype. These morphologic and immunohistochemical features suggested a CD30+ cutaneous lymphoproliferative disorder. The clinical history of recurrent self-healing papulonodules in an otherwise-healthy patient established the diagnosis of lymphomatoid papulosis (LyP).

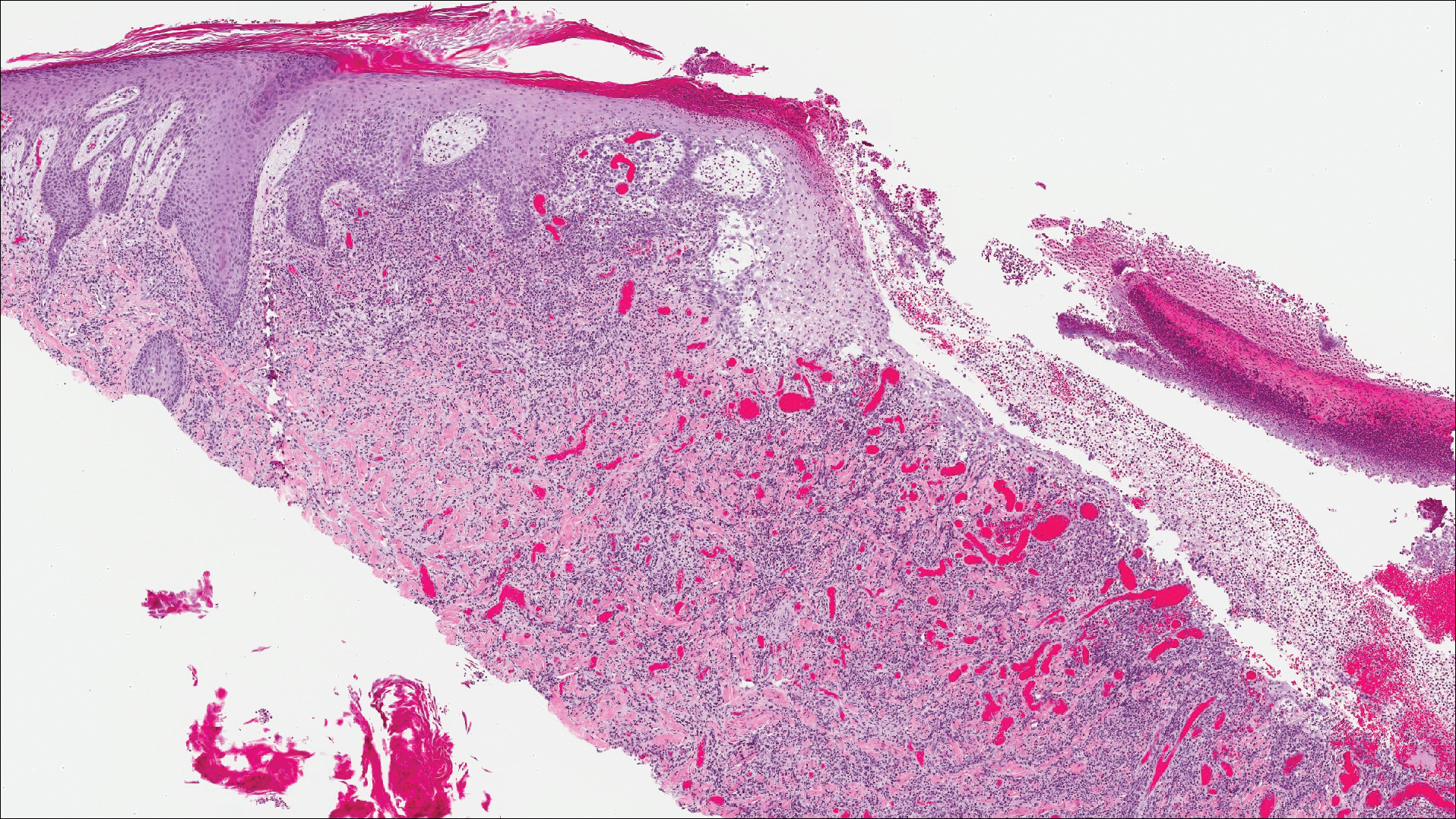
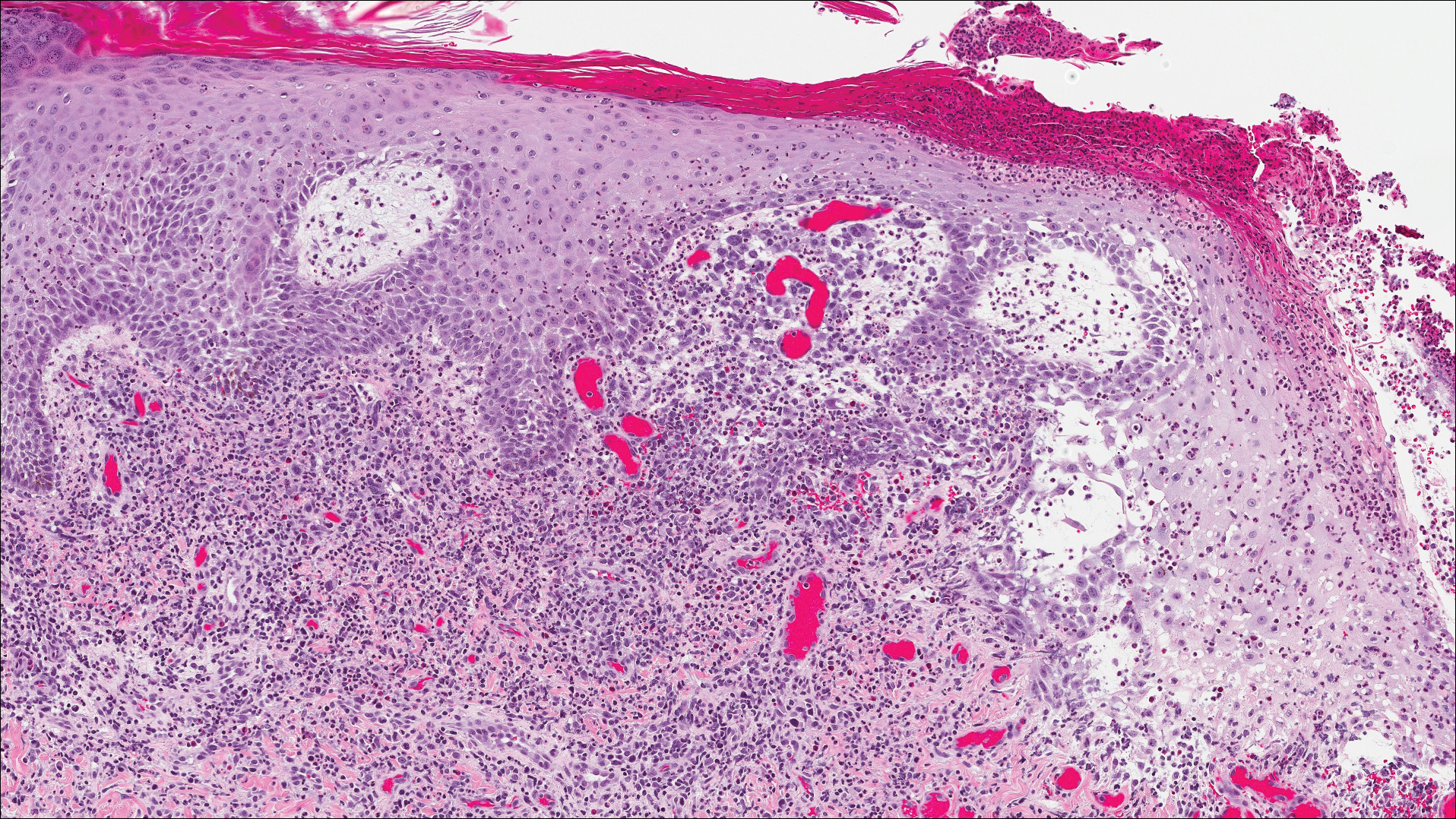
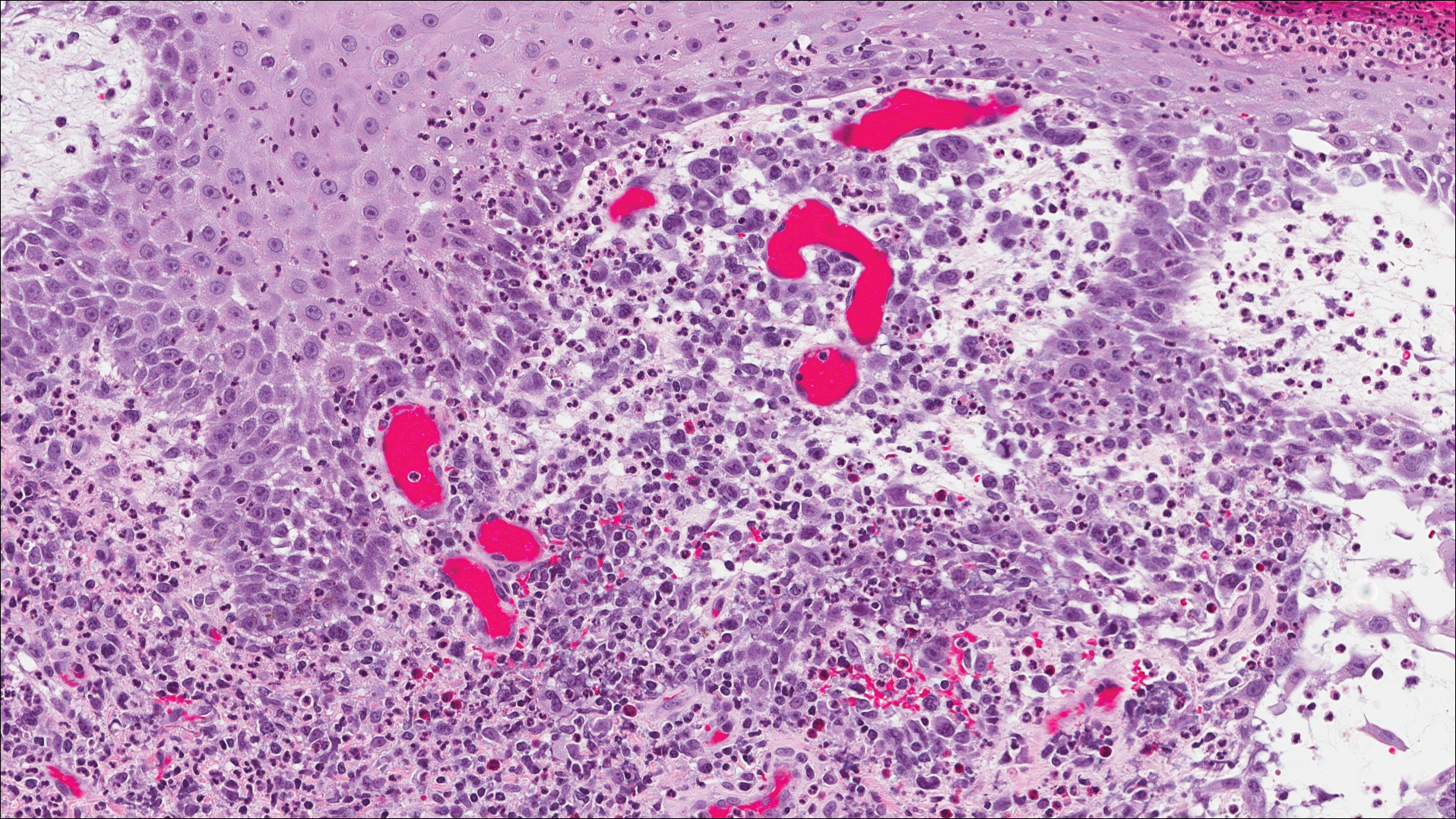
Lymphomatoid papulosis is a lymphoproliferative disorder characterized by recurrent crops of self-resolving eruptive papulonodular skin lesions that may show a variety of histologic features including a CD30+ malignant T-cell lymphoma.1 Lymphomatoid papulosis was first described in 19681 but debate continues whether the condition should be considered malignant or benign.2 Although the prognosis is excellent, LyP is characterized by a protracted course, often lasting many years. Additionally, these patients have a lifelong increased risk for development of a second cutaneous or systemic lymphoma such as mycosis fungoides (MF), cutaneous or nodal anaplastic large cell lymphoma (ALCL), or Hodgkin lymphoma, among others.
Lymphomatoid papulosis is a rare disease occurring in all ethnic groups and at any age, though most commonly presenting in the fifth decade of life. Finding large atypical T cells expressing CD30 in recurring skin lesions is highly suggestive of LyP; however, large CD30+ cells also can be seen in numerous benign reactive processes such as arthropod assault, drug eruption, viral skin infections, and other dermatoses, thus clinical correlation is always paramount. The cause of LyP is largely unknown; however, spontaneous regression may be explained by CD30-CD30 ligand interaction3 as well as an increased proapoptotic milieu.4 Specific translocations such as interferon regulatory factor-4 have been hypothesized as a risk factor for malignant progression.5-7 Additionally, an inactivating gene mutation resulting in loss of transforming growth factor β1 receptor expression and subsequent unresponsiveness to the growth inhibitory effect of transforming growth factor β may play a role in progression of LyP to ALCL.8
Clinically, LyP consists of red-brown papules and nodules generally smaller than 2 cm, often with central hemorrhage, necrosis, and crusting. Lesions are at different stages of eruption and resolution. They are often grouped but may be disseminated. Spontaneous regression typically occurs within 3 to 8 weeks. Pruritus or mild tenderness may occur as well as residual hyperpigmentation or scarring. Systemic symptoms are notably absent.
The histologic features of LyP vary according to the age of the lesion and subtype.2 Early lesions may only show a few inflammatory cells, but as lesions evolve, larger immunoblastlike CD30+ atypical cells accumulate that may resemble the Reed-Sternberg cells of Hodgkin lymphoma. Of the 5 subtypes, the most common is type A. It is characterized by a wedge-shaped infiltrate with a mixed population of scattered or clustered, large, atypical CD30+ cells, lymphocytes, neutrophils, eosinophils, and histiocytes.9 Frequent mitoses often are seen. Type B appears similar to MF due to a predominantly epidermotropic infiltrate of CD3+ and often CD30− atypical cells. Spontaneously regressing papules favor LyP, whereas persistent patches or plaques favor MF. Type C appears identical to ALCL with diffuse sheets of large atypical CD30+ cells and relatively few inflammatory cells, but spontaneously regressing lesions again favor LyP, whereas persistent tumors favor ALCL. Type D appears similar to primary cutaneous aggressive epidermotropic CD8+ cytotoxic T cell lymphoma due to a markedly epidermotropic infiltrate of small atypical CD8+ and CD30+ lymphocytes, often TIA-1+ (T-cell intracytoplasmic antigen-1) or granzyme B+, but CD30 positivity and self-resolving lesions favor LyP. Type E mimics extranodal natural killer/T cell lymphoma (nasal type) due to angioinvasive CD30+ and beta F1+ T lymphocytes, often CD8+ and/or TIA-1+, but self-resolving lesions again favor LyP, as well as absence of Epstein-Barr virus and CD56−.9
The most common therapeutic approaches to LyP include topical steroids, phototherapy, and low-dose methotrexate.10 However, treatment does not change overall disease course or reduce the future risk for developing an associated lymphoma. Accordingly, abstaining from active therapeutic intervention is reasonable, especially in patients with only a few asymptomatic lesions.
- Macaulay WL. Lymphomatoid papulosis: a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Slater DN. The new World Health Organization-European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas: a practical marriage of two giants. Br J Dermatol. 2005;153:874-880.
- Mori M, Manuelli C, Pimpinelli N, et al. CD30-CD30 ligand interaction in primary cutaneous CD30(+) T-cell lymphomas: a clue to the pathophysiology of clinical regression. Blood. 1999;94:3077-3083.
- Greisser J, Doebbeling U, Roos M, et al. Apoptosis in CD30-positive lymphoproliferative disorders of the skin. Exp Dermatol. 2005;14:380-385.
- Kiran T, Demirkesen C, Eker C, et al. The significance of MUM1/IRF4 protein expression and IRF4 translocation of CD30(+) cutaneous T-cell lymphoproliferative disorders: a study of 53 cases. Leuk Res. 2013;37:396-400.
- Wada DA, Law ME, Hsi ED, et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Mod Pathol. 2011;24:596-605.
- Pham-Ledard A, Prochazkova-Carlotti M, Laharanne E, et al. IRF4 gene rearrangements define a subgroup of CD30-positive cutaneous T-cell lymphoma: a study of 54 cases. J Invest Dermatol. 2010;130:816-825.
- Schiemann WP, Pfeifer WM, Levi E, et al. A deletion in the gene for transforming growth factor β type I receptor abolishes growth regulation by transforming growth factor β in a cutaneous T-cell lymphoma. Blood. 1999;94:2854-2861.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
The Diagnosis: Lymphomatoid Papulosis
A shave biopsy of an established lesion on the volar aspect of the left wrist was performed (Figure 1). The biopsy showed an ulcerated nodular lesion characterized by a dense mixed inflammatory cell infiltrate in the dermis composed of lymphocytes, histiocytes, scattered neutrophils, and numerous eosinophils (Figure 2). Notably there was a minor population of large atypical cells with immunoblastic and anaplastic morphology present individually and in small clusters most prominently within the upper dermis (Figures 3 and 4). Immunohistochemistry of the anaplastic cells revealed a CD30+, CD3−, CD4+, CD5−, CD8−, CD2−, CD7−, CD56−, ALK1− (anaplastic lymphoma kinase-1), PAX5− (paired box protein-5), CD20−, and CD15− phenotype. These morphologic and immunohistochemical features suggested a CD30+ cutaneous lymphoproliferative disorder. The clinical history of recurrent self-healing papulonodules in an otherwise-healthy patient established the diagnosis of lymphomatoid papulosis (LyP).
Lymphomatoid papulosis is a lymphoproliferative disorder characterized by recurrent crops of self-resolving eruptive papulonodular skin lesions that may show a variety of histologic features including a CD30+ malignant T-cell lymphoma.1 Lymphomatoid papulosis was first described in 19681 but debate continues whether the condition should be considered malignant or benign.2 Although the prognosis is excellent, LyP is characterized by a protracted course, often lasting many years. Additionally, these patients have a lifelong increased risk for development of a second cutaneous or systemic lymphoma such as mycosis fungoides (MF), cutaneous or nodal anaplastic large cell lymphoma (ALCL), or Hodgkin lymphoma, among others.
Lymphomatoid papulosis is a rare disease occurring in all ethnic groups and at any age, though most commonly presenting in the fifth decade of life. Finding large atypical T cells expressing CD30 in recurring skin lesions is highly suggestive of LyP; however, large CD30+ cells also can be seen in numerous benign reactive processes such as arthropod assault, drug eruption, viral skin infections, and other dermatoses, thus clinical correlation is always paramount. The cause of LyP is largely unknown; however, spontaneous regression may be explained by CD30-CD30 ligand interaction3 as well as an increased proapoptotic milieu.4 Specific translocations such as interferon regulatory factor-4 have been hypothesized as a risk factor for malignant progression.5-7 Additionally, an inactivating gene mutation resulting in loss of transforming growth factor β1 receptor expression and subsequent unresponsiveness to the growth inhibitory effect of transforming growth factor β may play a role in progression of LyP to ALCL.8
Clinically, LyP consists of red-brown papules and nodules generally smaller than 2 cm, often with central hemorrhage, necrosis, and crusting. Lesions are at different stages of eruption and resolution. They are often grouped but may be disseminated. Spontaneous regression typically occurs within 3 to 8 weeks. Pruritus or mild tenderness may occur as well as residual hyperpigmentation or scarring. Systemic symptoms are notably absent.
The histologic features of LyP vary according to the age of the lesion and subtype.2 Early lesions may only show a few inflammatory cells, but as lesions evolve, larger immunoblastlike CD30+ atypical cells accumulate that may resemble the Reed-Sternberg cells of Hodgkin lymphoma. Of the 5 subtypes, the most common is type A. It is characterized by a wedge-shaped infiltrate with a mixed population of scattered or clustered, large, atypical CD30+ cells, lymphocytes, neutrophils, eosinophils, and histiocytes.9 Frequent mitoses often are seen. Type B appears similar to MF due to a predominantly epidermotropic infiltrate of CD3+ and often CD30− atypical cells. Spontaneously regressing papules favor LyP, whereas persistent patches or plaques favor MF. Type C appears identical to ALCL with diffuse sheets of large atypical CD30+ cells and relatively few inflammatory cells, but spontaneously regressing lesions again favor LyP, whereas persistent tumors favor ALCL. Type D appears similar to primary cutaneous aggressive epidermotropic CD8+ cytotoxic T cell lymphoma due to a markedly epidermotropic infiltrate of small atypical CD8+ and CD30+ lymphocytes, often TIA-1+ (T-cell intracytoplasmic antigen-1) or granzyme B+, but CD30 positivity and self-resolving lesions favor LyP. Type E mimics extranodal natural killer/T cell lymphoma (nasal type) due to angioinvasive CD30+ and beta F1+ T lymphocytes, often CD8+ and/or TIA-1+, but self-resolving lesions again favor LyP, as well as absence of Epstein-Barr virus and CD56−.9
The most common therapeutic approaches to LyP include topical steroids, phototherapy, and low-dose methotrexate.10 However, treatment does not change overall disease course or reduce the future risk for developing an associated lymphoma. Accordingly, abstaining from active therapeutic intervention is reasonable, especially in patients with only a few asymptomatic lesions.
The Diagnosis: Lymphomatoid Papulosis
A shave biopsy of an established lesion on the volar aspect of the left wrist was performed (Figure 1). The biopsy showed an ulcerated nodular lesion characterized by a dense mixed inflammatory cell infiltrate in the dermis composed of lymphocytes, histiocytes, scattered neutrophils, and numerous eosinophils (Figure 2). Notably there was a minor population of large atypical cells with immunoblastic and anaplastic morphology present individually and in small clusters most prominently within the upper dermis (Figures 3 and 4). Immunohistochemistry of the anaplastic cells revealed a CD30+, CD3−, CD4+, CD5−, CD8−, CD2−, CD7−, CD56−, ALK1− (anaplastic lymphoma kinase-1), PAX5− (paired box protein-5), CD20−, and CD15− phenotype. These morphologic and immunohistochemical features suggested a CD30+ cutaneous lymphoproliferative disorder. The clinical history of recurrent self-healing papulonodules in an otherwise-healthy patient established the diagnosis of lymphomatoid papulosis (LyP).
Lymphomatoid papulosis is a lymphoproliferative disorder characterized by recurrent crops of self-resolving eruptive papulonodular skin lesions that may show a variety of histologic features including a CD30+ malignant T-cell lymphoma.1 Lymphomatoid papulosis was first described in 19681 but debate continues whether the condition should be considered malignant or benign.2 Although the prognosis is excellent, LyP is characterized by a protracted course, often lasting many years. Additionally, these patients have a lifelong increased risk for development of a second cutaneous or systemic lymphoma such as mycosis fungoides (MF), cutaneous or nodal anaplastic large cell lymphoma (ALCL), or Hodgkin lymphoma, among others.
Lymphomatoid papulosis is a rare disease occurring in all ethnic groups and at any age, though most commonly presenting in the fifth decade of life. Finding large atypical T cells expressing CD30 in recurring skin lesions is highly suggestive of LyP; however, large CD30+ cells also can be seen in numerous benign reactive processes such as arthropod assault, drug eruption, viral skin infections, and other dermatoses, thus clinical correlation is always paramount. The cause of LyP is largely unknown; however, spontaneous regression may be explained by CD30-CD30 ligand interaction3 as well as an increased proapoptotic milieu.4 Specific translocations such as interferon regulatory factor-4 have been hypothesized as a risk factor for malignant progression.5-7 Additionally, an inactivating gene mutation resulting in loss of transforming growth factor β1 receptor expression and subsequent unresponsiveness to the growth inhibitory effect of transforming growth factor β may play a role in progression of LyP to ALCL.8
Clinically, LyP consists of red-brown papules and nodules generally smaller than 2 cm, often with central hemorrhage, necrosis, and crusting. Lesions are at different stages of eruption and resolution. They are often grouped but may be disseminated. Spontaneous regression typically occurs within 3 to 8 weeks. Pruritus or mild tenderness may occur as well as residual hyperpigmentation or scarring. Systemic symptoms are notably absent.
The histologic features of LyP vary according to the age of the lesion and subtype.2 Early lesions may only show a few inflammatory cells, but as lesions evolve, larger immunoblastlike CD30+ atypical cells accumulate that may resemble the Reed-Sternberg cells of Hodgkin lymphoma. Of the 5 subtypes, the most common is type A. It is characterized by a wedge-shaped infiltrate with a mixed population of scattered or clustered, large, atypical CD30+ cells, lymphocytes, neutrophils, eosinophils, and histiocytes.9 Frequent mitoses often are seen. Type B appears similar to MF due to a predominantly epidermotropic infiltrate of CD3+ and often CD30− atypical cells. Spontaneously regressing papules favor LyP, whereas persistent patches or plaques favor MF. Type C appears identical to ALCL with diffuse sheets of large atypical CD30+ cells and relatively few inflammatory cells, but spontaneously regressing lesions again favor LyP, whereas persistent tumors favor ALCL. Type D appears similar to primary cutaneous aggressive epidermotropic CD8+ cytotoxic T cell lymphoma due to a markedly epidermotropic infiltrate of small atypical CD8+ and CD30+ lymphocytes, often TIA-1+ (T-cell intracytoplasmic antigen-1) or granzyme B+, but CD30 positivity and self-resolving lesions favor LyP. Type E mimics extranodal natural killer/T cell lymphoma (nasal type) due to angioinvasive CD30+ and beta F1+ T lymphocytes, often CD8+ and/or TIA-1+, but self-resolving lesions again favor LyP, as well as absence of Epstein-Barr virus and CD56−.9
The most common therapeutic approaches to LyP include topical steroids, phototherapy, and low-dose methotrexate.10 However, treatment does not change overall disease course or reduce the future risk for developing an associated lymphoma. Accordingly, abstaining from active therapeutic intervention is reasonable, especially in patients with only a few asymptomatic lesions.
- Macaulay WL. Lymphomatoid papulosis: a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Slater DN. The new World Health Organization-European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas: a practical marriage of two giants. Br J Dermatol. 2005;153:874-880.
- Mori M, Manuelli C, Pimpinelli N, et al. CD30-CD30 ligand interaction in primary cutaneous CD30(+) T-cell lymphomas: a clue to the pathophysiology of clinical regression. Blood. 1999;94:3077-3083.
- Greisser J, Doebbeling U, Roos M, et al. Apoptosis in CD30-positive lymphoproliferative disorders of the skin. Exp Dermatol. 2005;14:380-385.
- Kiran T, Demirkesen C, Eker C, et al. The significance of MUM1/IRF4 protein expression and IRF4 translocation of CD30(+) cutaneous T-cell lymphoproliferative disorders: a study of 53 cases. Leuk Res. 2013;37:396-400.
- Wada DA, Law ME, Hsi ED, et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Mod Pathol. 2011;24:596-605.
- Pham-Ledard A, Prochazkova-Carlotti M, Laharanne E, et al. IRF4 gene rearrangements define a subgroup of CD30-positive cutaneous T-cell lymphoma: a study of 54 cases. J Invest Dermatol. 2010;130:816-825.
- Schiemann WP, Pfeifer WM, Levi E, et al. A deletion in the gene for transforming growth factor β type I receptor abolishes growth regulation by transforming growth factor β in a cutaneous T-cell lymphoma. Blood. 1999;94:2854-2861.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
- Macaulay WL. Lymphomatoid papulosis: a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Slater DN. The new World Health Organization-European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas: a practical marriage of two giants. Br J Dermatol. 2005;153:874-880.
- Mori M, Manuelli C, Pimpinelli N, et al. CD30-CD30 ligand interaction in primary cutaneous CD30(+) T-cell lymphomas: a clue to the pathophysiology of clinical regression. Blood. 1999;94:3077-3083.
- Greisser J, Doebbeling U, Roos M, et al. Apoptosis in CD30-positive lymphoproliferative disorders of the skin. Exp Dermatol. 2005;14:380-385.
- Kiran T, Demirkesen C, Eker C, et al. The significance of MUM1/IRF4 protein expression and IRF4 translocation of CD30(+) cutaneous T-cell lymphoproliferative disorders: a study of 53 cases. Leuk Res. 2013;37:396-400.
- Wada DA, Law ME, Hsi ED, et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Mod Pathol. 2011;24:596-605.
- Pham-Ledard A, Prochazkova-Carlotti M, Laharanne E, et al. IRF4 gene rearrangements define a subgroup of CD30-positive cutaneous T-cell lymphoma: a study of 54 cases. J Invest Dermatol. 2010;130:816-825.
- Schiemann WP, Pfeifer WM, Levi E, et al. A deletion in the gene for transforming growth factor β type I receptor abolishes growth regulation by transforming growth factor β in a cutaneous T-cell lymphoma. Blood. 1999;94:2854-2861.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
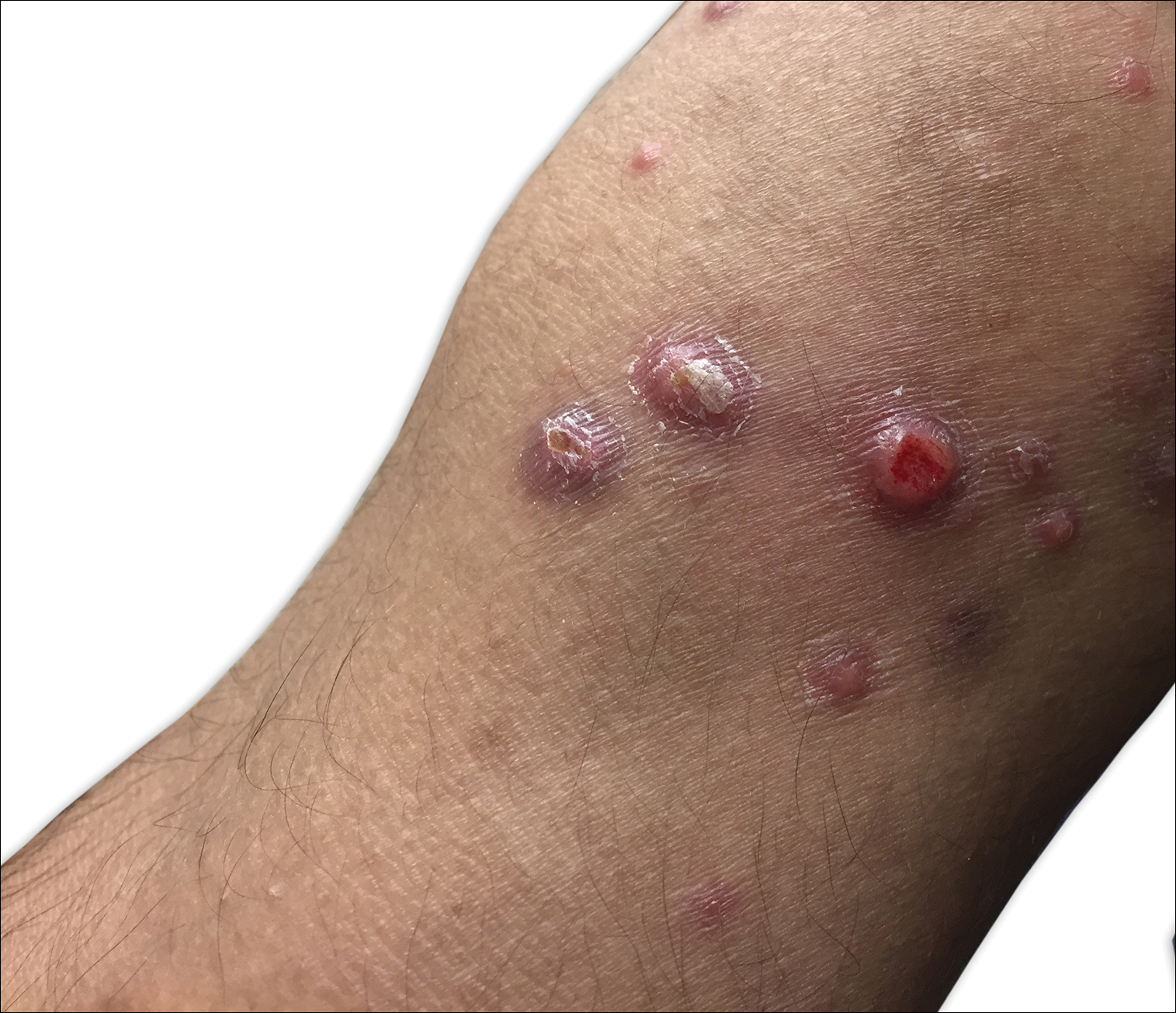
A 29-year-old man from Saudi Arabia presented with slightly tender skin lesions occurring in crops every few months over the last 7 years. The lesions typically would occur on the inguinal area, lower abdomen, buttocks, thighs, or arms, resolving within a few weeks despite no treatment. The patient denied having systemic symptoms such as fevers, chills, sweats, chest pain, shortness of breath, or unexpected weight loss. Physical examination revealed multiple erythematous papulonodules, some ulcerated with a superficial crust, grouped predominantly on the medial aspect of the right upper arm and left lower inguinal region. Isolated lesions also were present on the forearms, dorsal aspects of the hands, abdomen, and thighs. The grouped papulonodules were intermixed with faint hyperpigmented macules indicative of prior lesions. No oral lesions were noted, and there was no marked axillary or inguinal lymphadenopathy.
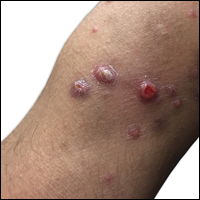
Eroded Plaque on the Lower Lip
The Diagnosis: Squamous Cell Carcinoma
The initial clinical presentation suggested a diagnosis of herpes simplex labialis. The patient reported no response to topical acyclovir, and because the plaque persisted, a biopsy was performed. Pathology demonstrated squamous cell carcinoma (SCC) that was moderately well differentiated and invasive (Figure).
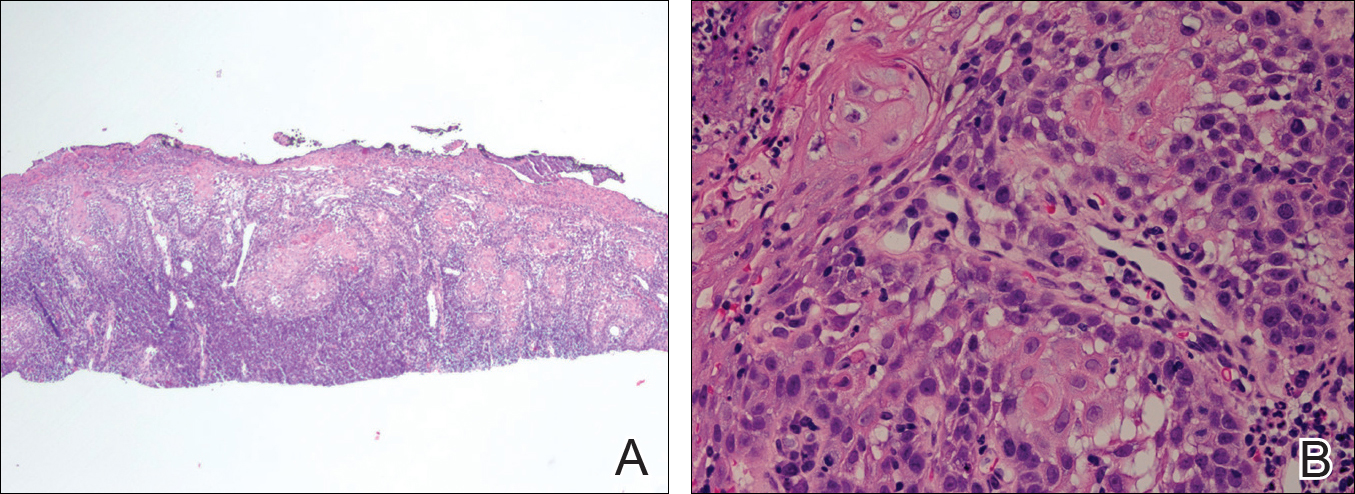
Approximately 38% of all oral SCCs in the United States occur on the lower lip and typically are solar-related cancers developing within the epidermis.1 Oral lesions initially may be asymptomatic and may not be of concern to the patient; however, it is important to recognize SCC early, as invasive lesions have the potential to metastasize. Some factors that increase the chance for the development of metastases include tumor size larger than 2 cm; location on the ear, lip, or other sites on the head and neck; and history of prior unsuccessful treatment.2 Any solitary ulcer, lump, wound, or lesion that will not heal and persists for more than 3 weeks should be regarded as cancer until proven otherwise. Although few oral SCCs are detected by clinicians at an early stage, diagnostic aids such as vital staining and molecular markers in tissues and saliva may be implemented.3 Toluidine blue is a simple, fast, and inexpensive technique that stains the nuclear material of malignant lesions, but not normal mucosa, and may be a worthwhile diagnostic adjunct to clinical inspection.4
Our patient presented with a lesion that clinically looked herpetic, though he reported no prodromal signs of tingling, burning, or pain before the occurrence of the lesion. Due to the persistence of the lesion and lack of response to treatment, a biopsy was indicated. The differential diagnoses include aphthous ulcers, which may occasionally extend on to the vermilion border of the lip and exhibit nondiagnostic histology.5 Bullous oral lichen planus is the least common variant of oral lichen planus, is unlikely to present as a solitary lesion, and is rarely seen on the lips. Histologically, the lesion demonstrated lichenoid inflammation.6 Solitary keratoacanthoma, though histologically similar to SCC, typically presents as a rapidly growing crateriform nodule without erosion or ulceration.7 The differential diagnoses are summarized in the Table.

The patient underwent wide excision with repair by mucosal advancement flap. He continues to be regularly seen in the clinic for monitoring of other skin cancers and is doing well. Clinicians encountering any wound or ulcer that does not show signs of healing should be wary of underlying malignancy and be prompted to perform a biopsy.
- Fehrenbach MJ. Extraoral and intraoral clinical assessment. In: Darby ML, Walsh MM, eds. Dental Hygiene: Theory and Practice. 4th ed. St Louis, MO: Elsevier; 2014:214-233.
- Hawrot A, Alam M, Ratner D. Squamous cell carcinoma. Curr Probl Dermatol. 2003;15:91-133.
- Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral Oncol. 2009;45:301-308.
- Chhabra N, Chhabra S, Sapra N. Diagnostic modalities for squamous cell carcinoma: an extensive review of literature considering toluidine blue as a useful adjunct. J Oral Maxillofac Surg. 2015;14:188-200.
- Porter SR, Scully C, Pedersen A. Recurrent aphthous stomatitis. Crit Rev Oral Biol Med. 2003;9:1499-1505.
- Bricker SL. Oral lichen planus: a review. Semin Dermatol. 1994;13:87-90.
- Cabrijan L, Lipozencic´ J, Batinac T, et al. Differences between keratoacanthoma and squamous cell carcinoma using TGF-alpha. Coll Antropol. 2013;37:147-150.
- Douglas GD, Couch RB. A prospective study of chronic herpes simplex virus infection and recurrent herpes labialis in humans. J Immunol. 1970;104:289-295.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med. 2001;344:976-983.
- van Tuyll van Serooskerken AM, van Marion AM, de Zwart-Storm E, et al. Lichen planus with bullous manifestation on the lip. Int J Dermatol. 2007;46(suppl 3):25-26.
- Messadi DV, Younai F. Apthous ulcers. Dermatol Ther. 2010;23:281-290.
- Ko CJ. Keratoacanthoma: facts and controversies. Clin Dermatol. 2010;28:254-261.
The Diagnosis: Squamous Cell Carcinoma
The initial clinical presentation suggested a diagnosis of herpes simplex labialis. The patient reported no response to topical acyclovir, and because the plaque persisted, a biopsy was performed. Pathology demonstrated squamous cell carcinoma (SCC) that was moderately well differentiated and invasive (Figure).
Approximately 38% of all oral SCCs in the United States occur on the lower lip and typically are solar-related cancers developing within the epidermis.1 Oral lesions initially may be asymptomatic and may not be of concern to the patient; however, it is important to recognize SCC early, as invasive lesions have the potential to metastasize. Some factors that increase the chance for the development of metastases include tumor size larger than 2 cm; location on the ear, lip, or other sites on the head and neck; and history of prior unsuccessful treatment.2 Any solitary ulcer, lump, wound, or lesion that will not heal and persists for more than 3 weeks should be regarded as cancer until proven otherwise. Although few oral SCCs are detected by clinicians at an early stage, diagnostic aids such as vital staining and molecular markers in tissues and saliva may be implemented.3 Toluidine blue is a simple, fast, and inexpensive technique that stains the nuclear material of malignant lesions, but not normal mucosa, and may be a worthwhile diagnostic adjunct to clinical inspection.4
Our patient presented with a lesion that clinically looked herpetic, though he reported no prodromal signs of tingling, burning, or pain before the occurrence of the lesion. Due to the persistence of the lesion and lack of response to treatment, a biopsy was indicated. The differential diagnoses include aphthous ulcers, which may occasionally extend on to the vermilion border of the lip and exhibit nondiagnostic histology.5 Bullous oral lichen planus is the least common variant of oral lichen planus, is unlikely to present as a solitary lesion, and is rarely seen on the lips. Histologically, the lesion demonstrated lichenoid inflammation.6 Solitary keratoacanthoma, though histologically similar to SCC, typically presents as a rapidly growing crateriform nodule without erosion or ulceration.7 The differential diagnoses are summarized in the Table.
The patient underwent wide excision with repair by mucosal advancement flap. He continues to be regularly seen in the clinic for monitoring of other skin cancers and is doing well. Clinicians encountering any wound or ulcer that does not show signs of healing should be wary of underlying malignancy and be prompted to perform a biopsy.
The Diagnosis: Squamous Cell Carcinoma
The initial clinical presentation suggested a diagnosis of herpes simplex labialis. The patient reported no response to topical acyclovir, and because the plaque persisted, a biopsy was performed. Pathology demonstrated squamous cell carcinoma (SCC) that was moderately well differentiated and invasive (Figure).
Approximately 38% of all oral SCCs in the United States occur on the lower lip and typically are solar-related cancers developing within the epidermis.1 Oral lesions initially may be asymptomatic and may not be of concern to the patient; however, it is important to recognize SCC early, as invasive lesions have the potential to metastasize. Some factors that increase the chance for the development of metastases include tumor size larger than 2 cm; location on the ear, lip, or other sites on the head and neck; and history of prior unsuccessful treatment.2 Any solitary ulcer, lump, wound, or lesion that will not heal and persists for more than 3 weeks should be regarded as cancer until proven otherwise. Although few oral SCCs are detected by clinicians at an early stage, diagnostic aids such as vital staining and molecular markers in tissues and saliva may be implemented.3 Toluidine blue is a simple, fast, and inexpensive technique that stains the nuclear material of malignant lesions, but not normal mucosa, and may be a worthwhile diagnostic adjunct to clinical inspection.4
Our patient presented with a lesion that clinically looked herpetic, though he reported no prodromal signs of tingling, burning, or pain before the occurrence of the lesion. Due to the persistence of the lesion and lack of response to treatment, a biopsy was indicated. The differential diagnoses include aphthous ulcers, which may occasionally extend on to the vermilion border of the lip and exhibit nondiagnostic histology.5 Bullous oral lichen planus is the least common variant of oral lichen planus, is unlikely to present as a solitary lesion, and is rarely seen on the lips. Histologically, the lesion demonstrated lichenoid inflammation.6 Solitary keratoacanthoma, though histologically similar to SCC, typically presents as a rapidly growing crateriform nodule without erosion or ulceration.7 The differential diagnoses are summarized in the Table.
The patient underwent wide excision with repair by mucosal advancement flap. He continues to be regularly seen in the clinic for monitoring of other skin cancers and is doing well. Clinicians encountering any wound or ulcer that does not show signs of healing should be wary of underlying malignancy and be prompted to perform a biopsy.
- Fehrenbach MJ. Extraoral and intraoral clinical assessment. In: Darby ML, Walsh MM, eds. Dental Hygiene: Theory and Practice. 4th ed. St Louis, MO: Elsevier; 2014:214-233.
- Hawrot A, Alam M, Ratner D. Squamous cell carcinoma. Curr Probl Dermatol. 2003;15:91-133.
- Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral Oncol. 2009;45:301-308.
- Chhabra N, Chhabra S, Sapra N. Diagnostic modalities for squamous cell carcinoma: an extensive review of literature considering toluidine blue as a useful adjunct. J Oral Maxillofac Surg. 2015;14:188-200.
- Porter SR, Scully C, Pedersen A. Recurrent aphthous stomatitis. Crit Rev Oral Biol Med. 2003;9:1499-1505.
- Bricker SL. Oral lichen planus: a review. Semin Dermatol. 1994;13:87-90.
- Cabrijan L, Lipozencic´ J, Batinac T, et al. Differences between keratoacanthoma and squamous cell carcinoma using TGF-alpha. Coll Antropol. 2013;37:147-150.
- Douglas GD, Couch RB. A prospective study of chronic herpes simplex virus infection and recurrent herpes labialis in humans. J Immunol. 1970;104:289-295.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med. 2001;344:976-983.
- van Tuyll van Serooskerken AM, van Marion AM, de Zwart-Storm E, et al. Lichen planus with bullous manifestation on the lip. Int J Dermatol. 2007;46(suppl 3):25-26.
- Messadi DV, Younai F. Apthous ulcers. Dermatol Ther. 2010;23:281-290.
- Ko CJ. Keratoacanthoma: facts and controversies. Clin Dermatol. 2010;28:254-261.
- Fehrenbach MJ. Extraoral and intraoral clinical assessment. In: Darby ML, Walsh MM, eds. Dental Hygiene: Theory and Practice. 4th ed. St Louis, MO: Elsevier; 2014:214-233.
- Hawrot A, Alam M, Ratner D. Squamous cell carcinoma. Curr Probl Dermatol. 2003;15:91-133.
- Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral Oncol. 2009;45:301-308.
- Chhabra N, Chhabra S, Sapra N. Diagnostic modalities for squamous cell carcinoma: an extensive review of literature considering toluidine blue as a useful adjunct. J Oral Maxillofac Surg. 2015;14:188-200.
- Porter SR, Scully C, Pedersen A. Recurrent aphthous stomatitis. Crit Rev Oral Biol Med. 2003;9:1499-1505.
- Bricker SL. Oral lichen planus: a review. Semin Dermatol. 1994;13:87-90.
- Cabrijan L, Lipozencic´ J, Batinac T, et al. Differences between keratoacanthoma and squamous cell carcinoma using TGF-alpha. Coll Antropol. 2013;37:147-150.
- Douglas GD, Couch RB. A prospective study of chronic herpes simplex virus infection and recurrent herpes labialis in humans. J Immunol. 1970;104:289-295.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med. 2001;344:976-983.
- van Tuyll van Serooskerken AM, van Marion AM, de Zwart-Storm E, et al. Lichen planus with bullous manifestation on the lip. Int J Dermatol. 2007;46(suppl 3):25-26.
- Messadi DV, Younai F. Apthous ulcers. Dermatol Ther. 2010;23:281-290.
- Ko CJ. Keratoacanthoma: facts and controversies. Clin Dermatol. 2010;28:254-261.
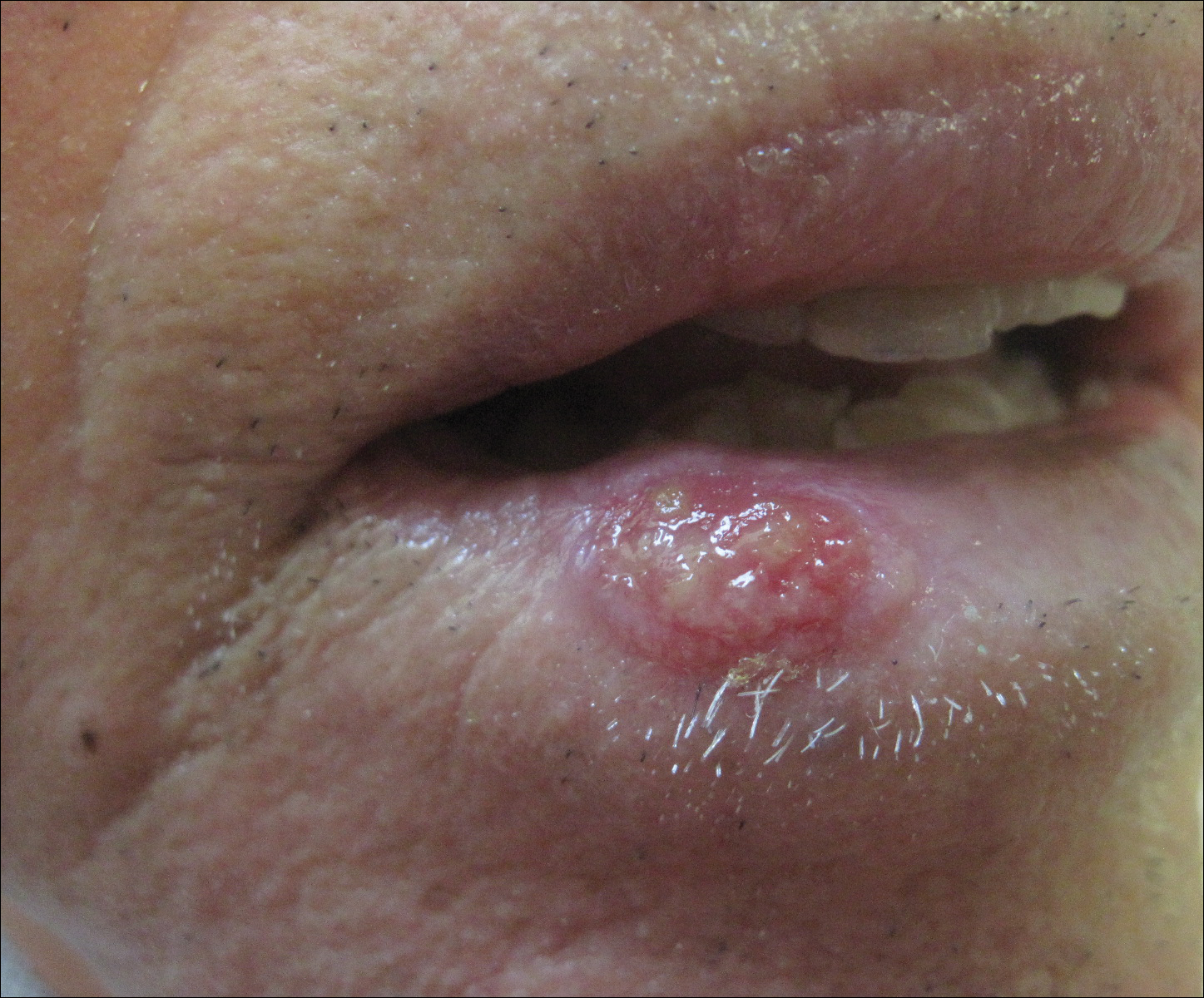
An 83-year-old man presented with a new-onset 1.2-cm eroded plaque on the vermilion border of the right lower lip that reportedly developed 2 weeks prior and was increasing in size. The plaque was moist and was composed of confluent glistening papules. Medical history was notable for the presence of both basal cell and squamous cell carcinomas.
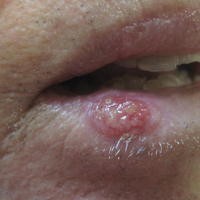
Black Adherence Nodules on the Scalp Hair Shaft
The Diagnosis: Piedra
Microscopic examination of the hair shafts revealed brown to black, firmly adherent concretions (Figure 1). Scanning electron microscopy of the nodules was performed, which allowed for greater definition of the constituent hyphae and arthrospores (Figure 2).


Fungal cultures grew Trichosporon inkin along with other dematiaceous molds. The patient initially was treated with a combination of ketoconazole shampoo and weekly application of topical terbinafine. She trimmed 15.2 cm of the hair of her own volition. At 2-month follow-up the nodules were still present, though smaller and less numerous. Repeat cultures were obtained, which again grew T inkin. She then began taking oral terbinafine 250 mg daily for 6 weeks.
This case of piedra is unique in that our patient presented with black nodules clinically, but cultures grew only the causative agent of white piedra, T inkin. A search of PubMed articles indexed for MEDLINE using the terms black piedra, white piedra, or piedra, and mixed infection or coinfection yielded one other similar case.1 Kanitakis et al1 speculated that perhaps there was coinfection of black and white piedra and that Piedraia hortae, the causative agent of black piedra, was unable to flourish in culture facing competition from other fungi. This scenario also could apply to our patient. However, the original culture taken from our patient also grew other dematiaceous molds including Cladosporium and Exophiala species. It also is possible that these other fungi could have contributed pigment to the nodules, giving it the appearance of black piedra when only T inkin was present as the true pathogen.
White piedra is a rare fungal infection of the hair shaft caused by organisms of the genus Trichosporon, with Trichosporon ovoides most likely to infect the scalp.2 Black piedra is a similar fungal infection caused by P hortae. Piedra means stone in Spanish, reflecting the appearance of these organisms on the hair shaft. It is common in tropical regions of the world such as Southeast Asia and South America, flourishing in the high temperatures and humidity.2 Both infectious agents are found in the soil or in standing water.3 White piedra most commonly is found in facial, axilla, or pubic hair, while black piedra most often is found in the hair of the scalp.2,4 Local cultural practices may contribute to transfer of Trichosporon or P hortae to the scalp, including the use of Brazilian plant oils in the hair or tying a veil or hijab to wet hair. Interestingly, some groups intentionally introduce the fungus to their hair for cosmetic reasons in endemic areas.2,3,5
Patients with white or black piedra generally are asymptomatic.4 Some may notice a rough texture to the hair or hear a characteristic metallic rattling sound as the nodules make contact with brush bristles.2,3 On inspection of the scalp, white piedra will appear to be white to light brown nodules, while black piedra presents as brown to black in color. The nodules are often firm on palpation.2,3 The nodules of white piedra generally are easy to remove in contrast to black piedra, which involves nodules that securely attach to the hair shaft but can be removed with pressure.3,5 Piedra has natural keratolytic activities and with prolonged infection can penetrate the hair cuticle, causing weakness and eventual breakage of the hair. This invasion into the hair cortex also can complicate treatment regimens, contributing to the chronic course of these infections.6
Diagnosis is based on clinical and microscopic findings. Nodules on hair shafts can be prepared with potassium hydroxide and placed on glass slides for examination.4 Dyes such as toluidine blue or chlorazol black E stain can be used to assist in identifying fungal structures.2 Sabouraud agar with cycloheximide may be the best choice for culture medium.2 Black piedra slowly grows into small dome-shaped colonies. White piedra will grow more quickly into cream-colored colonies with wrinkles and sometimes mucinous characteristics.3
The best treatment of black or white piedra is to cut the hair, thereby eliminating the fungi,7 which is not an easy option for many patients, such as ours, because of the aesthetic implications. Alternative treatments include azole shampoos such as ketoconazole.2,4 Treatment with oral terbinafine 250 mg daily for 6 weeks has been successfully used for black piedra.7 Patients must be careful to thoroughly clean or discard hairbrushes, as they can serve as reservoirs of fungi to reinfect patients or spread to others.5,7
- Kanitakis J, Persat F, Piens MA, et al. Black piedra: report of a French case associated with Trichosporon asahii. Int J Dermatol. 2006;45:1258-1260.
- Schwartz RA. Superficial fungal infections. Lancet. 2004;364:1173-1182.
- Khatu SS, Poojary SA, Nagpur NG. Nodules on the hair: a rare case of mixed piedra. Int J Trichology. 2013;5:220-223.
- Elewski BE, Hughey LC, Sobera JO, et al. Fungal diseases. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Health Sciences; 2012:1251-1284.
- Desai DH, Nadkarni NJ. Piedra: an ethnicity-related trichosis? Int J Dermatol. 2013;53:1008-1011.
- Figueras M, Guarro J, Zaror L. New findings in black piedra infection. Br J Dermatol. 1996;135:157-158.
- Gip L. Black piedra: the first case treated with terbinafine (Lamisil). Br J Dermatol. 1994;130(suppl 43):26-28.
The Diagnosis: Piedra
Microscopic examination of the hair shafts revealed brown to black, firmly adherent concretions (Figure 1). Scanning electron microscopy of the nodules was performed, which allowed for greater definition of the constituent hyphae and arthrospores (Figure 2).
Fungal cultures grew Trichosporon inkin along with other dematiaceous molds. The patient initially was treated with a combination of ketoconazole shampoo and weekly application of topical terbinafine. She trimmed 15.2 cm of the hair of her own volition. At 2-month follow-up the nodules were still present, though smaller and less numerous. Repeat cultures were obtained, which again grew T inkin. She then began taking oral terbinafine 250 mg daily for 6 weeks.
This case of piedra is unique in that our patient presented with black nodules clinically, but cultures grew only the causative agent of white piedra, T inkin. A search of PubMed articles indexed for MEDLINE using the terms black piedra, white piedra, or piedra, and mixed infection or coinfection yielded one other similar case.1 Kanitakis et al1 speculated that perhaps there was coinfection of black and white piedra and that Piedraia hortae, the causative agent of black piedra, was unable to flourish in culture facing competition from other fungi. This scenario also could apply to our patient. However, the original culture taken from our patient also grew other dematiaceous molds including Cladosporium and Exophiala species. It also is possible that these other fungi could have contributed pigment to the nodules, giving it the appearance of black piedra when only T inkin was present as the true pathogen.
White piedra is a rare fungal infection of the hair shaft caused by organisms of the genus Trichosporon, with Trichosporon ovoides most likely to infect the scalp.2 Black piedra is a similar fungal infection caused by P hortae. Piedra means stone in Spanish, reflecting the appearance of these organisms on the hair shaft. It is common in tropical regions of the world such as Southeast Asia and South America, flourishing in the high temperatures and humidity.2 Both infectious agents are found in the soil or in standing water.3 White piedra most commonly is found in facial, axilla, or pubic hair, while black piedra most often is found in the hair of the scalp.2,4 Local cultural practices may contribute to transfer of Trichosporon or P hortae to the scalp, including the use of Brazilian plant oils in the hair or tying a veil or hijab to wet hair. Interestingly, some groups intentionally introduce the fungus to their hair for cosmetic reasons in endemic areas.2,3,5
Patients with white or black piedra generally are asymptomatic.4 Some may notice a rough texture to the hair or hear a characteristic metallic rattling sound as the nodules make contact with brush bristles.2,3 On inspection of the scalp, white piedra will appear to be white to light brown nodules, while black piedra presents as brown to black in color. The nodules are often firm on palpation.2,3 The nodules of white piedra generally are easy to remove in contrast to black piedra, which involves nodules that securely attach to the hair shaft but can be removed with pressure.3,5 Piedra has natural keratolytic activities and with prolonged infection can penetrate the hair cuticle, causing weakness and eventual breakage of the hair. This invasion into the hair cortex also can complicate treatment regimens, contributing to the chronic course of these infections.6
Diagnosis is based on clinical and microscopic findings. Nodules on hair shafts can be prepared with potassium hydroxide and placed on glass slides for examination.4 Dyes such as toluidine blue or chlorazol black E stain can be used to assist in identifying fungal structures.2 Sabouraud agar with cycloheximide may be the best choice for culture medium.2 Black piedra slowly grows into small dome-shaped colonies. White piedra will grow more quickly into cream-colored colonies with wrinkles and sometimes mucinous characteristics.3
The best treatment of black or white piedra is to cut the hair, thereby eliminating the fungi,7 which is not an easy option for many patients, such as ours, because of the aesthetic implications. Alternative treatments include azole shampoos such as ketoconazole.2,4 Treatment with oral terbinafine 250 mg daily for 6 weeks has been successfully used for black piedra.7 Patients must be careful to thoroughly clean or discard hairbrushes, as they can serve as reservoirs of fungi to reinfect patients or spread to others.5,7
The Diagnosis: Piedra
Microscopic examination of the hair shafts revealed brown to black, firmly adherent concretions (Figure 1). Scanning electron microscopy of the nodules was performed, which allowed for greater definition of the constituent hyphae and arthrospores (Figure 2).
Fungal cultures grew Trichosporon inkin along with other dematiaceous molds. The patient initially was treated with a combination of ketoconazole shampoo and weekly application of topical terbinafine. She trimmed 15.2 cm of the hair of her own volition. At 2-month follow-up the nodules were still present, though smaller and less numerous. Repeat cultures were obtained, which again grew T inkin. She then began taking oral terbinafine 250 mg daily for 6 weeks.
This case of piedra is unique in that our patient presented with black nodules clinically, but cultures grew only the causative agent of white piedra, T inkin. A search of PubMed articles indexed for MEDLINE using the terms black piedra, white piedra, or piedra, and mixed infection or coinfection yielded one other similar case.1 Kanitakis et al1 speculated that perhaps there was coinfection of black and white piedra and that Piedraia hortae, the causative agent of black piedra, was unable to flourish in culture facing competition from other fungi. This scenario also could apply to our patient. However, the original culture taken from our patient also grew other dematiaceous molds including Cladosporium and Exophiala species. It also is possible that these other fungi could have contributed pigment to the nodules, giving it the appearance of black piedra when only T inkin was present as the true pathogen.
White piedra is a rare fungal infection of the hair shaft caused by organisms of the genus Trichosporon, with Trichosporon ovoides most likely to infect the scalp.2 Black piedra is a similar fungal infection caused by P hortae. Piedra means stone in Spanish, reflecting the appearance of these organisms on the hair shaft. It is common in tropical regions of the world such as Southeast Asia and South America, flourishing in the high temperatures and humidity.2 Both infectious agents are found in the soil or in standing water.3 White piedra most commonly is found in facial, axilla, or pubic hair, while black piedra most often is found in the hair of the scalp.2,4 Local cultural practices may contribute to transfer of Trichosporon or P hortae to the scalp, including the use of Brazilian plant oils in the hair or tying a veil or hijab to wet hair. Interestingly, some groups intentionally introduce the fungus to their hair for cosmetic reasons in endemic areas.2,3,5
Patients with white or black piedra generally are asymptomatic.4 Some may notice a rough texture to the hair or hear a characteristic metallic rattling sound as the nodules make contact with brush bristles.2,3 On inspection of the scalp, white piedra will appear to be white to light brown nodules, while black piedra presents as brown to black in color. The nodules are often firm on palpation.2,3 The nodules of white piedra generally are easy to remove in contrast to black piedra, which involves nodules that securely attach to the hair shaft but can be removed with pressure.3,5 Piedra has natural keratolytic activities and with prolonged infection can penetrate the hair cuticle, causing weakness and eventual breakage of the hair. This invasion into the hair cortex also can complicate treatment regimens, contributing to the chronic course of these infections.6
Diagnosis is based on clinical and microscopic findings. Nodules on hair shafts can be prepared with potassium hydroxide and placed on glass slides for examination.4 Dyes such as toluidine blue or chlorazol black E stain can be used to assist in identifying fungal structures.2 Sabouraud agar with cycloheximide may be the best choice for culture medium.2 Black piedra slowly grows into small dome-shaped colonies. White piedra will grow more quickly into cream-colored colonies with wrinkles and sometimes mucinous characteristics.3
The best treatment of black or white piedra is to cut the hair, thereby eliminating the fungi,7 which is not an easy option for many patients, such as ours, because of the aesthetic implications. Alternative treatments include azole shampoos such as ketoconazole.2,4 Treatment with oral terbinafine 250 mg daily for 6 weeks has been successfully used for black piedra.7 Patients must be careful to thoroughly clean or discard hairbrushes, as they can serve as reservoirs of fungi to reinfect patients or spread to others.5,7
- Kanitakis J, Persat F, Piens MA, et al. Black piedra: report of a French case associated with Trichosporon asahii. Int J Dermatol. 2006;45:1258-1260.
- Schwartz RA. Superficial fungal infections. Lancet. 2004;364:1173-1182.
- Khatu SS, Poojary SA, Nagpur NG. Nodules on the hair: a rare case of mixed piedra. Int J Trichology. 2013;5:220-223.
- Elewski BE, Hughey LC, Sobera JO, et al. Fungal diseases. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Health Sciences; 2012:1251-1284.
- Desai DH, Nadkarni NJ. Piedra: an ethnicity-related trichosis? Int J Dermatol. 2013;53:1008-1011.
- Figueras M, Guarro J, Zaror L. New findings in black piedra infection. Br J Dermatol. 1996;135:157-158.
- Gip L. Black piedra: the first case treated with terbinafine (Lamisil). Br J Dermatol. 1994;130(suppl 43):26-28.
- Kanitakis J, Persat F, Piens MA, et al. Black piedra: report of a French case associated with Trichosporon asahii. Int J Dermatol. 2006;45:1258-1260.
- Schwartz RA. Superficial fungal infections. Lancet. 2004;364:1173-1182.
- Khatu SS, Poojary SA, Nagpur NG. Nodules on the hair: a rare case of mixed piedra. Int J Trichology. 2013;5:220-223.
- Elewski BE, Hughey LC, Sobera JO, et al. Fungal diseases. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Health Sciences; 2012:1251-1284.
- Desai DH, Nadkarni NJ. Piedra: an ethnicity-related trichosis? Int J Dermatol. 2013;53:1008-1011.
- Figueras M, Guarro J, Zaror L. New findings in black piedra infection. Br J Dermatol. 1996;135:157-158.
- Gip L. Black piedra: the first case treated with terbinafine (Lamisil). Br J Dermatol. 1994;130(suppl 43):26-28.

A 21-year-old woman presented to the dermatology clinic with what she described as small black dots in her hair that she first noted 3 months prior to presentation. The black nodules were asymptomatic, but the patient noted that they seemed to be moving up the hair shaft. They were firmly attached and great effort was required to remove them. The patient's sister recently developed similar nodules. The patient and her sister work as missionaries and had spent time in India, Southeast Asia, and Central America within the last few years. Physical examination revealed firmly adherent black nodules involving the mid to distal portions of the hair shafts on the scalp. There were no nail or skin findings. Cultures were obtained, and microscopic examination was performed.

Painful Necrotic Ulcer on the Vulva
The Diagnosis: Mucormycosis
Skin biopsy and histology revealed broad, wide-angle, branched, nonseptate hyphae suggestive of mucormycosis infection (Figure 1). Computed tomography of the abdomen and pelvis revealed marked stranding in the vulvar region and urothelial thickening and enhancement suggestive of infection (Figure 2). Computed tomography of the chest demonstrated multiple irregular nodules in the bilateral upper lobes consistent with disseminated mucormycosis (Figure 3). The patient was started on intravenous amphotericin B and posaconazole. Surgery was not pursued given the poor prognosis of her refractory acute lymphoblastic leukemia, pancytopenia, and disseminated fungal infection. The patient was discharged home with hospice care.
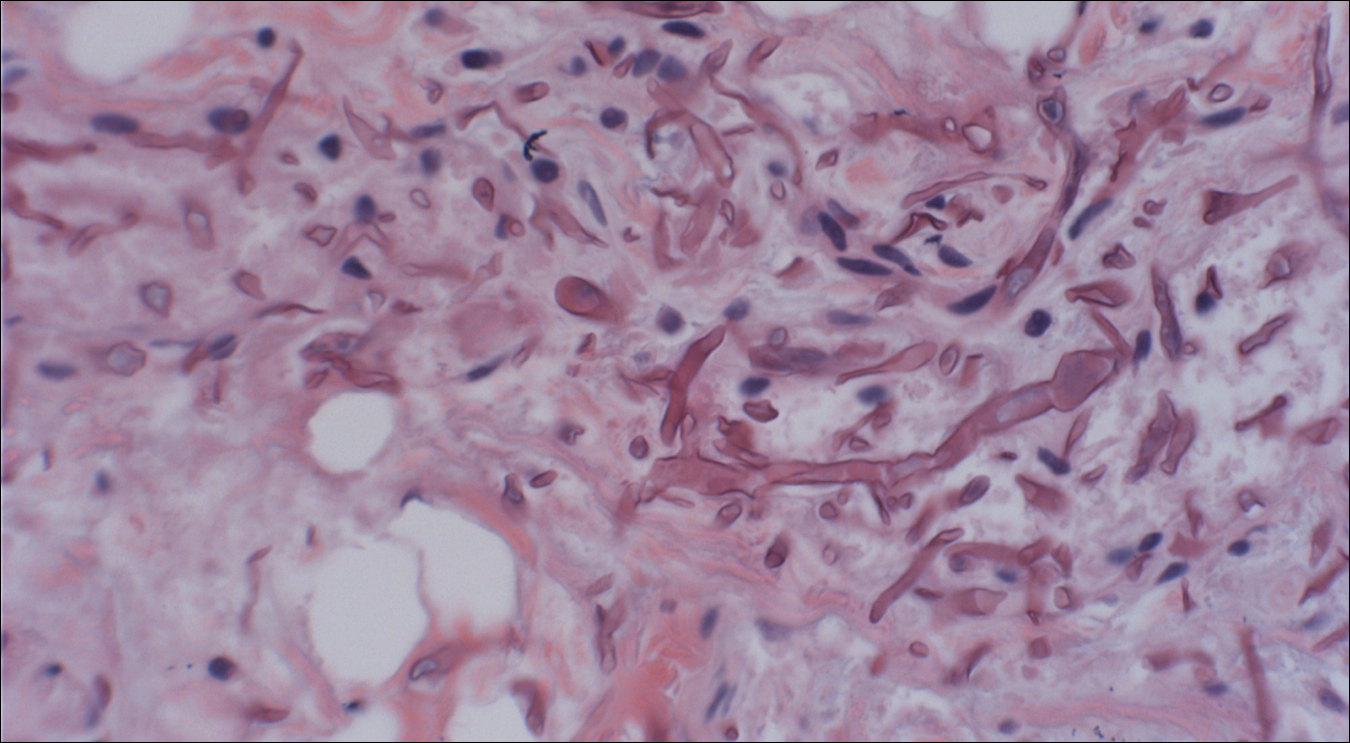
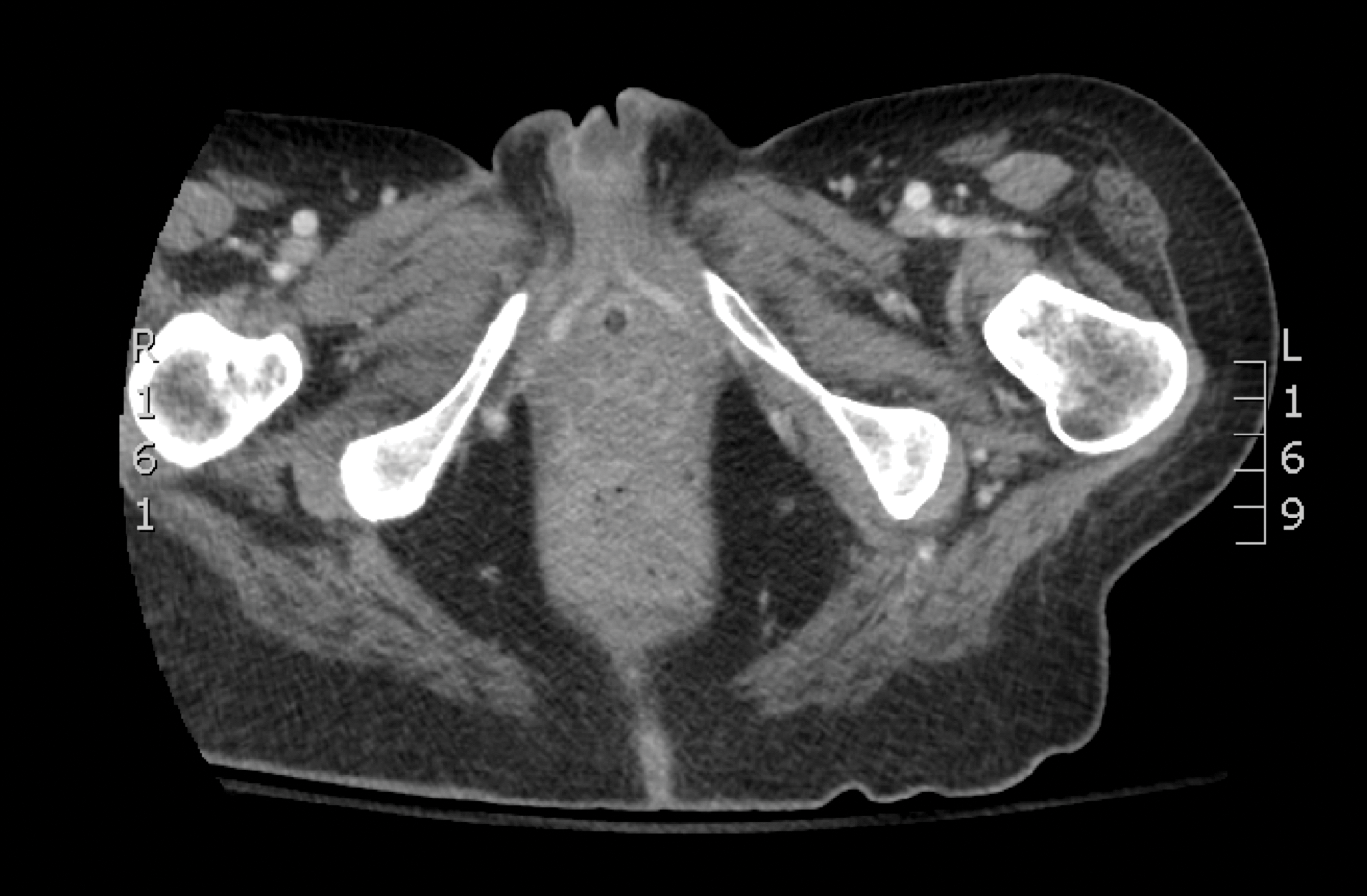
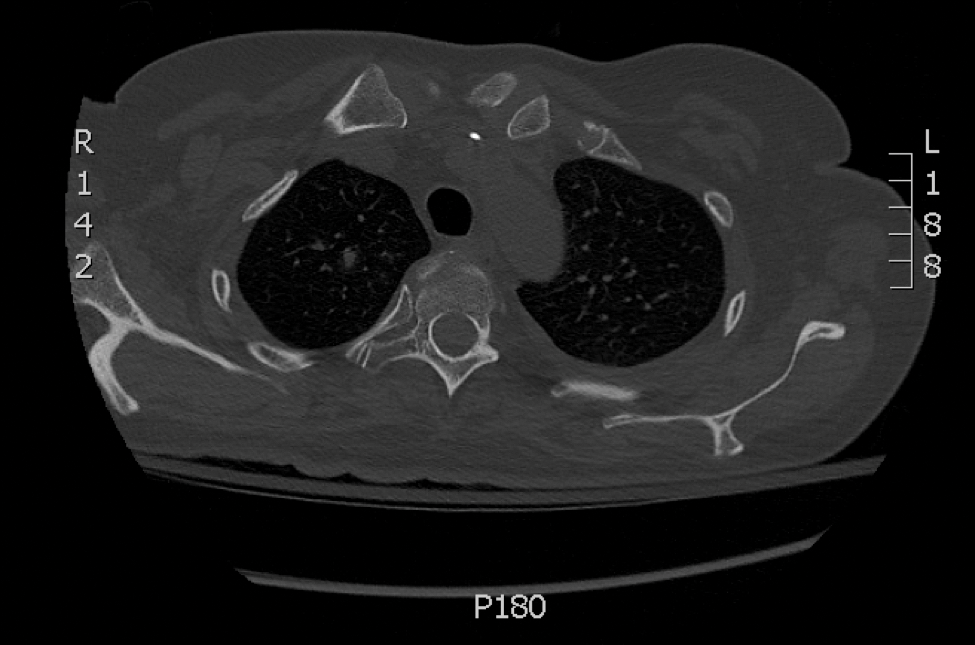
Mucormycosis is an infection caused by fungi that belong to the order Mucorales. The most common genera responsible for human disease are Rhizopus, Mucor, and Rhizomucor, which are organisms ubiquitous in nature and found in soil.1 Mucorales hyphae are widely branched and primarily nonseptate, which distinguishes them from hyphae of ascomycetous molds such as Aspergillus, which are narrowly branched and septate.
Mucormycosis primarily affects immunocompromised individuals. The overall incidence of mucormycosis is difficult to estimate, and the risk for infection varies based on the patient population. For example, the incidence of mucormycosis in hematologic malignancy ranges from 1% to 8% and from 0.4% to 16.0% in solid organ transplant recipients.2 One large series of 929 cases noted that the most common risk factors were associated with impaired immune function including diabetes mellitus and diabetic ketoacidosis (36% of cases), hematologic malignancy (17%), and solid organ (7%) or bone marrow transplantation (5%). Other risk factors include neutropenia, steroid therapy, and other immunocompromising conditions.3 Healthy individuals have a strong natural immunity to mucormycosis and rarely are affected by the disease.2
The host response to Mucorales is primarily driven by phagocyte-mediated killing via oxidative metabolites and cationic peptides called defensins.1 Thus, severely neutropenic patients are at high risk for developing mucormycosis.1 In contrast, it appears as though AIDS patients are not at increased risk for mucormycosis, supporting the theory that T lymphocytes are not involved in the host response.1 The conditions of diabetic ketoacidosis leave patients susceptible to mucormycosis for several reasons. First, hyperglycemia and low pH induce phagocyte dysfunction and thus inhibit the host response to Mucorales.4 Second, these organisms have an active ketone reductase system that may allow them to grow more readily in high glucose, acidic conditions.1 Third, diabetic ketoacidosis conditions increase serum free iron, and Mucorales utilizes host iron for cell growth and development.1 Individuals such as hemodialysis patients receiving the iron chelator deferoxamine also are at risk for mucormycosis, as Rhizopus can bind to this molecule and transport the bound iron intracellularly for growth utilization.1
Mucormycosis infection is characterized by infarction and rapid necrosis of host tissues resulting from vascular infiltration by fungal hyphae. The most common site of infection is rhino-orbital-cerebral (39%), followed by lungs (24%) and skin (19%).3 Dissemination occurs in 23% of cases.3 Inoculation most commonly occurs via inhalation of airborne fungal spores by an immunocompromised host with resultant fungal proliferation in the paranasal sinuses, bronchioles, or alveoli. Gastrointestinal tract infection is presumed to occur via ingestion of spores.5
Cutaneous infection, as in our patient, occurs via the inoculation of spores into the dermis through breaks in the skin such as from intravenous lines, urinary catheters, injection sites, surgical sites, and traumatic wounds. Cutaneous infections typically present as a single erythematous, painful, indurated papule that rapidly progresses to a necrotic ulcer with overlying black eschar. In some cases, the progression may be more indolent over the course of several weeks.2 There are few reported cases of primary vulvar mucormycosis, as in our patient.6,7 The previously reported cases involved severely immunocompromised patients who developed large necrotic lesions over the vulva that demonstrated widely branching, nonseptate hyphae on histologic examination. Each patient required extensive surgical debridement with systemic antifungal treatment.6,7
A timely diagnosis of mucormycosis often hinges on a high index of suspicion on behalf of the clinician. A fungal etiology always should be considered for an infection in an immunocompromised patient. Furthermore, nonresponse to antibiotic treatment should be an important diagnostic clue that the infection could be fungal in origin. The definitive diagnosis of mucormycosis is confirmed by tissue biopsy and the presence of broad, widely branching, nonseptate hyphae seen on histopathologic examination.
Treatment involves aggressive surgical debridement of all necrotic tissues and elimination of predisposing factors for infection such as hyperglycemia, metabolic acidosis, deferoxamine administration, and immunosuppressive medications. Early initiation of antifungal therapy with the lipid formulation of amphotericin B is recommended. Oral posaconazole or isavuconazole typically are used as step-down therapy after a favorable clinical response with initial amphotericin B treatment. Deferasirox, in contrast to deferoxamine, is an iron chelator that may reduce the pathogenicity of Mucorales and may help as an adjunctive therapy.8 In addition, hyperbaric oxygen therapy may have limited benefit in some cases.9 In spite of these treatments, the overall mortality of mucormycosis is 50% or higher and approaches nearly 100% in cases of disseminated disease, such as in our patient.1,3
- Ibrahim AS, Spellberg B, Walsh TJ, et al. Pathogenesis of mucormycosis. Clin Infect Dis. 2012;54(suppl 1):S16-S22.
- Petrikkos G, Skiada A, Lortholary O, et al. Epidemiology and clinical manifestations of mucormycosis. Clin Infect Dis. 2012;54(suppl 1):S23-S34.
- Roden MM, Zaoutis TE, Buchanan WL, et al. Epidemiology and outcome of zygomycosis: a review of 929 reported cases. Clin Infect Dis. 2005;41:634-653.
- Chinn RY, Diamond RD. Generation of chemotactic factors by Rhizopus oryzae in the presence and absence of serum: relationship to hyphal damage mediated by human neutrophils and effects of hyperglycemia and ketoacidosis. Infect Immun. 1982;38:1123-1129.
- Cheng VC, Chan JF, Ngan AH, et al. Outbreak of intestinal infection due to Rhizopus microsporus [published online July 29, 2009]. J Clin Microbiol. 2009;47:2834-2843.
- Colon M, Romaguera J, Mendez K, et al. Mucormycosis of the vulva in an immunocompromised pediatric patient. Bol Asoc Med P R. 2013;105:65-67.
- Nomura J, Ruskin J, Sahebi F, et al. Mucormycosis of the vulva following bone marrow transplantation. Bone Marrow Transplant. 1997;19:859-860.
- Spellberg B, Andes D, Perez M, et al. Safety and outcomes of open-label deferasirox iron chelation therapy for mucormycosis. Antimicrob Agents Chemother. 2009;53:3122-3125.
- Ferguson BJ, Mitchell TG, Moon R, et al. Adjunctive hyperbaric oxygen for treatment of rhinocerebral mucormycosis. Rev Infect Dis. 1988;10:551-559.
The Diagnosis: Mucormycosis
Skin biopsy and histology revealed broad, wide-angle, branched, nonseptate hyphae suggestive of mucormycosis infection (Figure 1). Computed tomography of the abdomen and pelvis revealed marked stranding in the vulvar region and urothelial thickening and enhancement suggestive of infection (Figure 2). Computed tomography of the chest demonstrated multiple irregular nodules in the bilateral upper lobes consistent with disseminated mucormycosis (Figure 3). The patient was started on intravenous amphotericin B and posaconazole. Surgery was not pursued given the poor prognosis of her refractory acute lymphoblastic leukemia, pancytopenia, and disseminated fungal infection. The patient was discharged home with hospice care.
Mucormycosis is an infection caused by fungi that belong to the order Mucorales. The most common genera responsible for human disease are Rhizopus, Mucor, and Rhizomucor, which are organisms ubiquitous in nature and found in soil.1 Mucorales hyphae are widely branched and primarily nonseptate, which distinguishes them from hyphae of ascomycetous molds such as Aspergillus, which are narrowly branched and septate.
Mucormycosis primarily affects immunocompromised individuals. The overall incidence of mucormycosis is difficult to estimate, and the risk for infection varies based on the patient population. For example, the incidence of mucormycosis in hematologic malignancy ranges from 1% to 8% and from 0.4% to 16.0% in solid organ transplant recipients.2 One large series of 929 cases noted that the most common risk factors were associated with impaired immune function including diabetes mellitus and diabetic ketoacidosis (36% of cases), hematologic malignancy (17%), and solid organ (7%) or bone marrow transplantation (5%). Other risk factors include neutropenia, steroid therapy, and other immunocompromising conditions.3 Healthy individuals have a strong natural immunity to mucormycosis and rarely are affected by the disease.2
The host response to Mucorales is primarily driven by phagocyte-mediated killing via oxidative metabolites and cationic peptides called defensins.1 Thus, severely neutropenic patients are at high risk for developing mucormycosis.1 In contrast, it appears as though AIDS patients are not at increased risk for mucormycosis, supporting the theory that T lymphocytes are not involved in the host response.1 The conditions of diabetic ketoacidosis leave patients susceptible to mucormycosis for several reasons. First, hyperglycemia and low pH induce phagocyte dysfunction and thus inhibit the host response to Mucorales.4 Second, these organisms have an active ketone reductase system that may allow them to grow more readily in high glucose, acidic conditions.1 Third, diabetic ketoacidosis conditions increase serum free iron, and Mucorales utilizes host iron for cell growth and development.1 Individuals such as hemodialysis patients receiving the iron chelator deferoxamine also are at risk for mucormycosis, as Rhizopus can bind to this molecule and transport the bound iron intracellularly for growth utilization.1
Mucormycosis infection is characterized by infarction and rapid necrosis of host tissues resulting from vascular infiltration by fungal hyphae. The most common site of infection is rhino-orbital-cerebral (39%), followed by lungs (24%) and skin (19%).3 Dissemination occurs in 23% of cases.3 Inoculation most commonly occurs via inhalation of airborne fungal spores by an immunocompromised host with resultant fungal proliferation in the paranasal sinuses, bronchioles, or alveoli. Gastrointestinal tract infection is presumed to occur via ingestion of spores.5
Cutaneous infection, as in our patient, occurs via the inoculation of spores into the dermis through breaks in the skin such as from intravenous lines, urinary catheters, injection sites, surgical sites, and traumatic wounds. Cutaneous infections typically present as a single erythematous, painful, indurated papule that rapidly progresses to a necrotic ulcer with overlying black eschar. In some cases, the progression may be more indolent over the course of several weeks.2 There are few reported cases of primary vulvar mucormycosis, as in our patient.6,7 The previously reported cases involved severely immunocompromised patients who developed large necrotic lesions over the vulva that demonstrated widely branching, nonseptate hyphae on histologic examination. Each patient required extensive surgical debridement with systemic antifungal treatment.6,7
A timely diagnosis of mucormycosis often hinges on a high index of suspicion on behalf of the clinician. A fungal etiology always should be considered for an infection in an immunocompromised patient. Furthermore, nonresponse to antibiotic treatment should be an important diagnostic clue that the infection could be fungal in origin. The definitive diagnosis of mucormycosis is confirmed by tissue biopsy and the presence of broad, widely branching, nonseptate hyphae seen on histopathologic examination.
Treatment involves aggressive surgical debridement of all necrotic tissues and elimination of predisposing factors for infection such as hyperglycemia, metabolic acidosis, deferoxamine administration, and immunosuppressive medications. Early initiation of antifungal therapy with the lipid formulation of amphotericin B is recommended. Oral posaconazole or isavuconazole typically are used as step-down therapy after a favorable clinical response with initial amphotericin B treatment. Deferasirox, in contrast to deferoxamine, is an iron chelator that may reduce the pathogenicity of Mucorales and may help as an adjunctive therapy.8 In addition, hyperbaric oxygen therapy may have limited benefit in some cases.9 In spite of these treatments, the overall mortality of mucormycosis is 50% or higher and approaches nearly 100% in cases of disseminated disease, such as in our patient.1,3
The Diagnosis: Mucormycosis
Skin biopsy and histology revealed broad, wide-angle, branched, nonseptate hyphae suggestive of mucormycosis infection (Figure 1). Computed tomography of the abdomen and pelvis revealed marked stranding in the vulvar region and urothelial thickening and enhancement suggestive of infection (Figure 2). Computed tomography of the chest demonstrated multiple irregular nodules in the bilateral upper lobes consistent with disseminated mucormycosis (Figure 3). The patient was started on intravenous amphotericin B and posaconazole. Surgery was not pursued given the poor prognosis of her refractory acute lymphoblastic leukemia, pancytopenia, and disseminated fungal infection. The patient was discharged home with hospice care.
Mucormycosis is an infection caused by fungi that belong to the order Mucorales. The most common genera responsible for human disease are Rhizopus, Mucor, and Rhizomucor, which are organisms ubiquitous in nature and found in soil.1 Mucorales hyphae are widely branched and primarily nonseptate, which distinguishes them from hyphae of ascomycetous molds such as Aspergillus, which are narrowly branched and septate.
Mucormycosis primarily affects immunocompromised individuals. The overall incidence of mucormycosis is difficult to estimate, and the risk for infection varies based on the patient population. For example, the incidence of mucormycosis in hematologic malignancy ranges from 1% to 8% and from 0.4% to 16.0% in solid organ transplant recipients.2 One large series of 929 cases noted that the most common risk factors were associated with impaired immune function including diabetes mellitus and diabetic ketoacidosis (36% of cases), hematologic malignancy (17%), and solid organ (7%) or bone marrow transplantation (5%). Other risk factors include neutropenia, steroid therapy, and other immunocompromising conditions.3 Healthy individuals have a strong natural immunity to mucormycosis and rarely are affected by the disease.2
The host response to Mucorales is primarily driven by phagocyte-mediated killing via oxidative metabolites and cationic peptides called defensins.1 Thus, severely neutropenic patients are at high risk for developing mucormycosis.1 In contrast, it appears as though AIDS patients are not at increased risk for mucormycosis, supporting the theory that T lymphocytes are not involved in the host response.1 The conditions of diabetic ketoacidosis leave patients susceptible to mucormycosis for several reasons. First, hyperglycemia and low pH induce phagocyte dysfunction and thus inhibit the host response to Mucorales.4 Second, these organisms have an active ketone reductase system that may allow them to grow more readily in high glucose, acidic conditions.1 Third, diabetic ketoacidosis conditions increase serum free iron, and Mucorales utilizes host iron for cell growth and development.1 Individuals such as hemodialysis patients receiving the iron chelator deferoxamine also are at risk for mucormycosis, as Rhizopus can bind to this molecule and transport the bound iron intracellularly for growth utilization.1
Mucormycosis infection is characterized by infarction and rapid necrosis of host tissues resulting from vascular infiltration by fungal hyphae. The most common site of infection is rhino-orbital-cerebral (39%), followed by lungs (24%) and skin (19%).3 Dissemination occurs in 23% of cases.3 Inoculation most commonly occurs via inhalation of airborne fungal spores by an immunocompromised host with resultant fungal proliferation in the paranasal sinuses, bronchioles, or alveoli. Gastrointestinal tract infection is presumed to occur via ingestion of spores.5
Cutaneous infection, as in our patient, occurs via the inoculation of spores into the dermis through breaks in the skin such as from intravenous lines, urinary catheters, injection sites, surgical sites, and traumatic wounds. Cutaneous infections typically present as a single erythematous, painful, indurated papule that rapidly progresses to a necrotic ulcer with overlying black eschar. In some cases, the progression may be more indolent over the course of several weeks.2 There are few reported cases of primary vulvar mucormycosis, as in our patient.6,7 The previously reported cases involved severely immunocompromised patients who developed large necrotic lesions over the vulva that demonstrated widely branching, nonseptate hyphae on histologic examination. Each patient required extensive surgical debridement with systemic antifungal treatment.6,7
A timely diagnosis of mucormycosis often hinges on a high index of suspicion on behalf of the clinician. A fungal etiology always should be considered for an infection in an immunocompromised patient. Furthermore, nonresponse to antibiotic treatment should be an important diagnostic clue that the infection could be fungal in origin. The definitive diagnosis of mucormycosis is confirmed by tissue biopsy and the presence of broad, widely branching, nonseptate hyphae seen on histopathologic examination.
Treatment involves aggressive surgical debridement of all necrotic tissues and elimination of predisposing factors for infection such as hyperglycemia, metabolic acidosis, deferoxamine administration, and immunosuppressive medications. Early initiation of antifungal therapy with the lipid formulation of amphotericin B is recommended. Oral posaconazole or isavuconazole typically are used as step-down therapy after a favorable clinical response with initial amphotericin B treatment. Deferasirox, in contrast to deferoxamine, is an iron chelator that may reduce the pathogenicity of Mucorales and may help as an adjunctive therapy.8 In addition, hyperbaric oxygen therapy may have limited benefit in some cases.9 In spite of these treatments, the overall mortality of mucormycosis is 50% or higher and approaches nearly 100% in cases of disseminated disease, such as in our patient.1,3
- Ibrahim AS, Spellberg B, Walsh TJ, et al. Pathogenesis of mucormycosis. Clin Infect Dis. 2012;54(suppl 1):S16-S22.
- Petrikkos G, Skiada A, Lortholary O, et al. Epidemiology and clinical manifestations of mucormycosis. Clin Infect Dis. 2012;54(suppl 1):S23-S34.
- Roden MM, Zaoutis TE, Buchanan WL, et al. Epidemiology and outcome of zygomycosis: a review of 929 reported cases. Clin Infect Dis. 2005;41:634-653.
- Chinn RY, Diamond RD. Generation of chemotactic factors by Rhizopus oryzae in the presence and absence of serum: relationship to hyphal damage mediated by human neutrophils and effects of hyperglycemia and ketoacidosis. Infect Immun. 1982;38:1123-1129.
- Cheng VC, Chan JF, Ngan AH, et al. Outbreak of intestinal infection due to Rhizopus microsporus [published online July 29, 2009]. J Clin Microbiol. 2009;47:2834-2843.
- Colon M, Romaguera J, Mendez K, et al. Mucormycosis of the vulva in an immunocompromised pediatric patient. Bol Asoc Med P R. 2013;105:65-67.
- Nomura J, Ruskin J, Sahebi F, et al. Mucormycosis of the vulva following bone marrow transplantation. Bone Marrow Transplant. 1997;19:859-860.
- Spellberg B, Andes D, Perez M, et al. Safety and outcomes of open-label deferasirox iron chelation therapy for mucormycosis. Antimicrob Agents Chemother. 2009;53:3122-3125.
- Ferguson BJ, Mitchell TG, Moon R, et al. Adjunctive hyperbaric oxygen for treatment of rhinocerebral mucormycosis. Rev Infect Dis. 1988;10:551-559.
- Ibrahim AS, Spellberg B, Walsh TJ, et al. Pathogenesis of mucormycosis. Clin Infect Dis. 2012;54(suppl 1):S16-S22.
- Petrikkos G, Skiada A, Lortholary O, et al. Epidemiology and clinical manifestations of mucormycosis. Clin Infect Dis. 2012;54(suppl 1):S23-S34.
- Roden MM, Zaoutis TE, Buchanan WL, et al. Epidemiology and outcome of zygomycosis: a review of 929 reported cases. Clin Infect Dis. 2005;41:634-653.
- Chinn RY, Diamond RD. Generation of chemotactic factors by Rhizopus oryzae in the presence and absence of serum: relationship to hyphal damage mediated by human neutrophils and effects of hyperglycemia and ketoacidosis. Infect Immun. 1982;38:1123-1129.
- Cheng VC, Chan JF, Ngan AH, et al. Outbreak of intestinal infection due to Rhizopus microsporus [published online July 29, 2009]. J Clin Microbiol. 2009;47:2834-2843.
- Colon M, Romaguera J, Mendez K, et al. Mucormycosis of the vulva in an immunocompromised pediatric patient. Bol Asoc Med P R. 2013;105:65-67.
- Nomura J, Ruskin J, Sahebi F, et al. Mucormycosis of the vulva following bone marrow transplantation. Bone Marrow Transplant. 1997;19:859-860.
- Spellberg B, Andes D, Perez M, et al. Safety and outcomes of open-label deferasirox iron chelation therapy for mucormycosis. Antimicrob Agents Chemother. 2009;53:3122-3125.
- Ferguson BJ, Mitchell TG, Moon R, et al. Adjunctive hyperbaric oxygen for treatment of rhinocerebral mucormycosis. Rev Infect Dis. 1988;10:551-559.
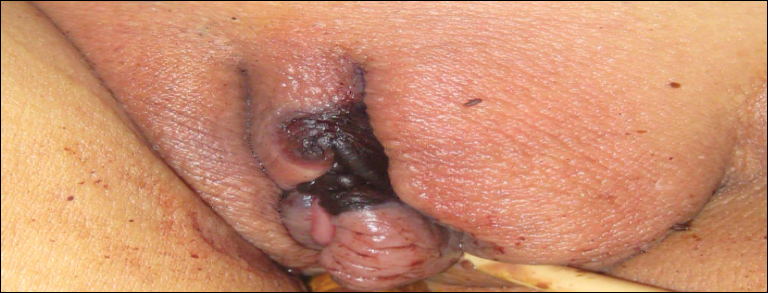
A 48-year-old woman with relapsed T-cell acute lymphoblastic leukemia was admitted to the oncology service for salvage chemotherapy and allogeneic stem cell transplant. Her admission was complicated by extended-spectrum β-lactamase-producing Escherichia coli sepsis and persistent pancytopenia, which required transfer to the intensive care unit. After 2 weeks and while still in the intensive care unit, she developed a painful necrotic vulvar ulcer over the right labia and clitoris that progressed and formed an overlying black eschar.