User login
First SGLT1/2 inhibitor shows ‘spectacular’ phase 3 safety and efficacy in T2D
Sotagliflozin, a novel type of sodium-glucose cotransporter inhibitor, showed the diverse benefits this drug class provides along some new twists in a pair of international pivotal trials that together enrolled nearly 12,000 patients with type 2 diabetes.

Unprecedented benefits were seen for the first time with a drug, sotagliflozin (Zynquista) that produces both sodium-glucose cotransporter 2 inhibition as well as SGLT1 inhibition.
They included a big reduction in both MIs and strokes; an ability to meaningfully reduce hyperglycemia in patients with severe renal dysfunction with an estimated glomerular filtration rate (eGFR) of 25-29 mL/min per 1.73 m2; an ability to safely and effectively start in patients still hospitalized (but stable) for an acute heart failure episode; and a striking 37% relative risk reduction in cardiovascular death, heart failure hospitalizations, or an urgent outpatient visit for heart failure in 739 of the patients enrolled in both trials who had heart failure with preserved ejection fraction (HFpEF).
These studies produced for the first time evidence from controlled, prospective, randomized trials that a drug could improve the outcome of HFpEF patients.
All these novel outcomes came on top of the usual benefits clinicians have generally seen across the SGLT2 inhibitors already on the U.S. market: reductions in cardiovascular death and heart failure hospitalizations among all patients with type 2 diabetes, preservation of renal function, and hemoglobin A1c lowering among T2D patients with eGFR levels of at least 30 mL/min per 1.73 m2.
“The data look spectacular,” summed up Deepak L. Bhatt, MD, who presented the results from the two trials, SOLOIST-WHF and SCORED, in talks at the virtual scientific sessions of the American Heart Association.
“I think sotagliflozin has the potential to be the best in class” based on the several added attributes shown in the two trials, he said in an interview. “We’ve shown that it is very safe, well tolerated, and effective.”
The primary results were a significant 33% relative risk reduction with sotagliflozin treatment, compared with placebo in the rate of total cardiovascular deaths, hospitalizations for heart failure, or urgent outpatient visits for heart failure during just over 9 months of median follow-up among patients with T2D recently hospitalized for heart failure in SOLOIST-WFH. And a significant 26% relative risk reduction with sotagliflozin for the same endpoint after a median follow-up of just over 14 months in SCORED, which enrolled patients with T2D and chronic kidney disease.

“Sotagliflozin adds to the SGLT2 inhibitor story,” and the SOLOIST-WHF results “may shift our focus to vulnerable, acute heart failure patients with an opportunity to treat during the transition phase,” when these patients leave the hospital, commented Jane E. Wilcox, MD, the study’s designated discussant and a heart failure cardiologist at Northwestern Medicine in Chicago.
A dual SGLT inhibitor
What sets sotagliflozin apart from the SGLT2 inhibitors is that it not only inhibits that protein but also SGTL1, which primarily resides in the gastrointestinal tract and is the main route for gut absorption of glucose. Dr. Bhatt said that he was unaware of any other SGLT1/2 inhibitors currently in advanced clinical testing.
The activity of sotagliflozin against the SGLT1 protein likely explains its ability to cut A1c levels in patients with severe renal dysfunction, a condition that stymies glucose lowering by SGLT2 inhibitors. In SCORED, which randomized 10,584 patients with T2D at 750 study sites in 44 countries, 813 patients (8%) had an eGFR of 25-29 mL/min per 1.73 m2 at enrollment. Sotagliflozin treatment led to an average 0.6% cut in A1c in this subgroup, and by the same average amount among the patients with GFRs of 30-60 mL/min per 1.73 m2.
“This is a huge finding for endocrinologists and primary care physicians” who treat patients with T2D who have severe renal dysfunction, said Dr. Bhatt, a professor of medicine at Harvard Medical School in Boston. “It’s a good enough reason by itself to approve this drug.”
The same mechanism may also be behind another unexpected finding in SCORED. Treatment with sotagliflozin cut the rate of total episodes of cardiovascular death, nonfatal MI, or nonfatal stroke by an absolute 1.6%, compared with placebo, and by a relative 23%. This benefit was largely driven by a 32% relative risk reduction total in MIs, and a 34% relative risk reduction in total stroke, both significant differences.
“No SGLT2 inhibitor has shown a reduction in stroke, and the MI signals have been mixed. The sizable MI and stroke effects are unique to sotagliflozin,” compared with the SGLT2 inhibitors, and likely reflect one or more mechanisms that result from blocked gut SGLT1 and a cut in GI glucose uptake, said Dr. Bhatt. “Probably some novel mechanism we don’t fully understand.”
First-ever HFpEF benefit
In contrast to these two benefits that are probably unique to drugs that inhibit the SGLT1 protein, sotagliflozin showed two other notable and unprecedented benefits that are likely generalizable to the SGLT2 inhibitors.
First is the striking benefit for HFpEF. Neither SOLOIST, which enrolled 1,222 patients with T2D and just hospitalized for worsening heart failure, nor SCORED, which enrolled patients with T2D and chronic kidney disease based exclusively on an eGFR of 25-60 mL/min per 1.73 m2, excluded patients with HFpEF, defined as heart failure patients with a left ventricular ejection fraction of at least 50%. The two studies together included a total of 739 of these patients, and they split fairly evenly between treatment with sotagliflozin or placebo.
The combined analysis showed that the incidence rate for the primary endpoint in both SOLOIST and SCORED was 59% with placebo and 39% with sotagliflozin, an absolute event reduction of 11.6 events/100 patient-years, and a significant 37% relative risk reduction, with a number needed to treat to prevent 1 event per year event of 9.
Although this observation comes from a nonprespecified combined analysis, “to me this result seems real, and I think it’s a class effect that I’m willing to extrapolate to the SGLT2 inhibitors,” Dr. Bhatt said. “It will change my practice,” he added, by spurring him to more aggressively prescribe an SGLT2 inhibitor to a patient with T2D and HFpEF.
“I think there has been some hesitation to use SGLT2 inhibitors in T2D patients with HFpEF” because of the paucity of data in this population, even though labeling and society recommendations do not rule it out. “I hope this finding will move that needle, and also generally improve SGLT2 inhibitor uptake, which has been low,” he said.
Also safe soon after acute heart failure decompensation
The other finding likely generalizable to SGLT2 inhibitors stems from the design of SOLOIST-WHF, which tested the efficacy and safety of starting sotagliflozin in patients with T2D as soon as they were stable after hospitalization for acute heart failure decompensation.
“Showing safety and efficacy when started in the hospital is pretty meaningful, because its tells patients that this drug is important and they should stay on it,” which should improve adherence, predicted Dr. Bhatt, who is also executive director of Interventional Cardiovascular Programs at Brigham and Women’s Hospital in Boston. “That’s the ultimate treatment path to prevent patients from falling through the cracks” and failing to receive an SGLT2 inhibitor.
SOLOIST-WHF enrolled patients hospitalized for worsening heart failure who also required intravenous diuretic treatment but had become stable enough to transition to an oral diuretic and come off oxygen. During a median follow-up of just over 9 months (both SOLOIST-WHF and SCORED ended sooner than planned because of a change in drug company sponsorship), treatment with sotagliflozin cut the primary endpoint by a relative 33%, compared with placebo, and with an absolute reduction of 25 events per 100 patient-years for a number needed to treat of 4. Sotagliflozin produced a strikingly high level of treatment efficiency driven by the high event rate in these recently decompensated patients. The benefit also appeared quickly, with a significant cut in events discernible within 28 days.
Extrapolating this finding to the SGLT2 inhibitors is “not a huge leap of faith,” Dr. Bhatt said.
“There is a role for sotagliflozin in acute heart failure. It showed benefit in these high-risk, transition-phase patients,” said Dr. Wilcox.
Simultaneously with Dr. Bhatt’s presentation, results of SOLOIST-WHF and SCORED were published online in the New England Journal of Medicine.
The trials were sponsored initially by Sanofi, and more recently by Lexicon. Dr. Bhatt has received research funding from both companies, and also from several other companies. He also is an adviser to several companies. Dr. Wilcox has been a consultant to Boehringer Ingelheim and Medtronic.
Sotagliflozin, a novel type of sodium-glucose cotransporter inhibitor, showed the diverse benefits this drug class provides along some new twists in a pair of international pivotal trials that together enrolled nearly 12,000 patients with type 2 diabetes.
Unprecedented benefits were seen for the first time with a drug, sotagliflozin (Zynquista) that produces both sodium-glucose cotransporter 2 inhibition as well as SGLT1 inhibition.
They included a big reduction in both MIs and strokes; an ability to meaningfully reduce hyperglycemia in patients with severe renal dysfunction with an estimated glomerular filtration rate (eGFR) of 25-29 mL/min per 1.73 m2; an ability to safely and effectively start in patients still hospitalized (but stable) for an acute heart failure episode; and a striking 37% relative risk reduction in cardiovascular death, heart failure hospitalizations, or an urgent outpatient visit for heart failure in 739 of the patients enrolled in both trials who had heart failure with preserved ejection fraction (HFpEF).
These studies produced for the first time evidence from controlled, prospective, randomized trials that a drug could improve the outcome of HFpEF patients.
All these novel outcomes came on top of the usual benefits clinicians have generally seen across the SGLT2 inhibitors already on the U.S. market: reductions in cardiovascular death and heart failure hospitalizations among all patients with type 2 diabetes, preservation of renal function, and hemoglobin A1c lowering among T2D patients with eGFR levels of at least 30 mL/min per 1.73 m2.
“The data look spectacular,” summed up Deepak L. Bhatt, MD, who presented the results from the two trials, SOLOIST-WHF and SCORED, in talks at the virtual scientific sessions of the American Heart Association.
“I think sotagliflozin has the potential to be the best in class” based on the several added attributes shown in the two trials, he said in an interview. “We’ve shown that it is very safe, well tolerated, and effective.”
The primary results were a significant 33% relative risk reduction with sotagliflozin treatment, compared with placebo in the rate of total cardiovascular deaths, hospitalizations for heart failure, or urgent outpatient visits for heart failure during just over 9 months of median follow-up among patients with T2D recently hospitalized for heart failure in SOLOIST-WFH. And a significant 26% relative risk reduction with sotagliflozin for the same endpoint after a median follow-up of just over 14 months in SCORED, which enrolled patients with T2D and chronic kidney disease.
“Sotagliflozin adds to the SGLT2 inhibitor story,” and the SOLOIST-WHF results “may shift our focus to vulnerable, acute heart failure patients with an opportunity to treat during the transition phase,” when these patients leave the hospital, commented Jane E. Wilcox, MD, the study’s designated discussant and a heart failure cardiologist at Northwestern Medicine in Chicago.
A dual SGLT inhibitor
What sets sotagliflozin apart from the SGLT2 inhibitors is that it not only inhibits that protein but also SGTL1, which primarily resides in the gastrointestinal tract and is the main route for gut absorption of glucose. Dr. Bhatt said that he was unaware of any other SGLT1/2 inhibitors currently in advanced clinical testing.
The activity of sotagliflozin against the SGLT1 protein likely explains its ability to cut A1c levels in patients with severe renal dysfunction, a condition that stymies glucose lowering by SGLT2 inhibitors. In SCORED, which randomized 10,584 patients with T2D at 750 study sites in 44 countries, 813 patients (8%) had an eGFR of 25-29 mL/min per 1.73 m2 at enrollment. Sotagliflozin treatment led to an average 0.6% cut in A1c in this subgroup, and by the same average amount among the patients with GFRs of 30-60 mL/min per 1.73 m2.
“This is a huge finding for endocrinologists and primary care physicians” who treat patients with T2D who have severe renal dysfunction, said Dr. Bhatt, a professor of medicine at Harvard Medical School in Boston. “It’s a good enough reason by itself to approve this drug.”
The same mechanism may also be behind another unexpected finding in SCORED. Treatment with sotagliflozin cut the rate of total episodes of cardiovascular death, nonfatal MI, or nonfatal stroke by an absolute 1.6%, compared with placebo, and by a relative 23%. This benefit was largely driven by a 32% relative risk reduction total in MIs, and a 34% relative risk reduction in total stroke, both significant differences.
“No SGLT2 inhibitor has shown a reduction in stroke, and the MI signals have been mixed. The sizable MI and stroke effects are unique to sotagliflozin,” compared with the SGLT2 inhibitors, and likely reflect one or more mechanisms that result from blocked gut SGLT1 and a cut in GI glucose uptake, said Dr. Bhatt. “Probably some novel mechanism we don’t fully understand.”
First-ever HFpEF benefit
In contrast to these two benefits that are probably unique to drugs that inhibit the SGLT1 protein, sotagliflozin showed two other notable and unprecedented benefits that are likely generalizable to the SGLT2 inhibitors.
First is the striking benefit for HFpEF. Neither SOLOIST, which enrolled 1,222 patients with T2D and just hospitalized for worsening heart failure, nor SCORED, which enrolled patients with T2D and chronic kidney disease based exclusively on an eGFR of 25-60 mL/min per 1.73 m2, excluded patients with HFpEF, defined as heart failure patients with a left ventricular ejection fraction of at least 50%. The two studies together included a total of 739 of these patients, and they split fairly evenly between treatment with sotagliflozin or placebo.
The combined analysis showed that the incidence rate for the primary endpoint in both SOLOIST and SCORED was 59% with placebo and 39% with sotagliflozin, an absolute event reduction of 11.6 events/100 patient-years, and a significant 37% relative risk reduction, with a number needed to treat to prevent 1 event per year event of 9.
Although this observation comes from a nonprespecified combined analysis, “to me this result seems real, and I think it’s a class effect that I’m willing to extrapolate to the SGLT2 inhibitors,” Dr. Bhatt said. “It will change my practice,” he added, by spurring him to more aggressively prescribe an SGLT2 inhibitor to a patient with T2D and HFpEF.
“I think there has been some hesitation to use SGLT2 inhibitors in T2D patients with HFpEF” because of the paucity of data in this population, even though labeling and society recommendations do not rule it out. “I hope this finding will move that needle, and also generally improve SGLT2 inhibitor uptake, which has been low,” he said.
Also safe soon after acute heart failure decompensation
The other finding likely generalizable to SGLT2 inhibitors stems from the design of SOLOIST-WHF, which tested the efficacy and safety of starting sotagliflozin in patients with T2D as soon as they were stable after hospitalization for acute heart failure decompensation.
“Showing safety and efficacy when started in the hospital is pretty meaningful, because its tells patients that this drug is important and they should stay on it,” which should improve adherence, predicted Dr. Bhatt, who is also executive director of Interventional Cardiovascular Programs at Brigham and Women’s Hospital in Boston. “That’s the ultimate treatment path to prevent patients from falling through the cracks” and failing to receive an SGLT2 inhibitor.
SOLOIST-WHF enrolled patients hospitalized for worsening heart failure who also required intravenous diuretic treatment but had become stable enough to transition to an oral diuretic and come off oxygen. During a median follow-up of just over 9 months (both SOLOIST-WHF and SCORED ended sooner than planned because of a change in drug company sponsorship), treatment with sotagliflozin cut the primary endpoint by a relative 33%, compared with placebo, and with an absolute reduction of 25 events per 100 patient-years for a number needed to treat of 4. Sotagliflozin produced a strikingly high level of treatment efficiency driven by the high event rate in these recently decompensated patients. The benefit also appeared quickly, with a significant cut in events discernible within 28 days.
Extrapolating this finding to the SGLT2 inhibitors is “not a huge leap of faith,” Dr. Bhatt said.
“There is a role for sotagliflozin in acute heart failure. It showed benefit in these high-risk, transition-phase patients,” said Dr. Wilcox.
Simultaneously with Dr. Bhatt’s presentation, results of SOLOIST-WHF and SCORED were published online in the New England Journal of Medicine.
The trials were sponsored initially by Sanofi, and more recently by Lexicon. Dr. Bhatt has received research funding from both companies, and also from several other companies. He also is an adviser to several companies. Dr. Wilcox has been a consultant to Boehringer Ingelheim and Medtronic.
Sotagliflozin, a novel type of sodium-glucose cotransporter inhibitor, showed the diverse benefits this drug class provides along some new twists in a pair of international pivotal trials that together enrolled nearly 12,000 patients with type 2 diabetes.
Unprecedented benefits were seen for the first time with a drug, sotagliflozin (Zynquista) that produces both sodium-glucose cotransporter 2 inhibition as well as SGLT1 inhibition.
They included a big reduction in both MIs and strokes; an ability to meaningfully reduce hyperglycemia in patients with severe renal dysfunction with an estimated glomerular filtration rate (eGFR) of 25-29 mL/min per 1.73 m2; an ability to safely and effectively start in patients still hospitalized (but stable) for an acute heart failure episode; and a striking 37% relative risk reduction in cardiovascular death, heart failure hospitalizations, or an urgent outpatient visit for heart failure in 739 of the patients enrolled in both trials who had heart failure with preserved ejection fraction (HFpEF).
These studies produced for the first time evidence from controlled, prospective, randomized trials that a drug could improve the outcome of HFpEF patients.
All these novel outcomes came on top of the usual benefits clinicians have generally seen across the SGLT2 inhibitors already on the U.S. market: reductions in cardiovascular death and heart failure hospitalizations among all patients with type 2 diabetes, preservation of renal function, and hemoglobin A1c lowering among T2D patients with eGFR levels of at least 30 mL/min per 1.73 m2.
“The data look spectacular,” summed up Deepak L. Bhatt, MD, who presented the results from the two trials, SOLOIST-WHF and SCORED, in talks at the virtual scientific sessions of the American Heart Association.
“I think sotagliflozin has the potential to be the best in class” based on the several added attributes shown in the two trials, he said in an interview. “We’ve shown that it is very safe, well tolerated, and effective.”
The primary results were a significant 33% relative risk reduction with sotagliflozin treatment, compared with placebo in the rate of total cardiovascular deaths, hospitalizations for heart failure, or urgent outpatient visits for heart failure during just over 9 months of median follow-up among patients with T2D recently hospitalized for heart failure in SOLOIST-WFH. And a significant 26% relative risk reduction with sotagliflozin for the same endpoint after a median follow-up of just over 14 months in SCORED, which enrolled patients with T2D and chronic kidney disease.
“Sotagliflozin adds to the SGLT2 inhibitor story,” and the SOLOIST-WHF results “may shift our focus to vulnerable, acute heart failure patients with an opportunity to treat during the transition phase,” when these patients leave the hospital, commented Jane E. Wilcox, MD, the study’s designated discussant and a heart failure cardiologist at Northwestern Medicine in Chicago.
A dual SGLT inhibitor
What sets sotagliflozin apart from the SGLT2 inhibitors is that it not only inhibits that protein but also SGTL1, which primarily resides in the gastrointestinal tract and is the main route for gut absorption of glucose. Dr. Bhatt said that he was unaware of any other SGLT1/2 inhibitors currently in advanced clinical testing.
The activity of sotagliflozin against the SGLT1 protein likely explains its ability to cut A1c levels in patients with severe renal dysfunction, a condition that stymies glucose lowering by SGLT2 inhibitors. In SCORED, which randomized 10,584 patients with T2D at 750 study sites in 44 countries, 813 patients (8%) had an eGFR of 25-29 mL/min per 1.73 m2 at enrollment. Sotagliflozin treatment led to an average 0.6% cut in A1c in this subgroup, and by the same average amount among the patients with GFRs of 30-60 mL/min per 1.73 m2.
“This is a huge finding for endocrinologists and primary care physicians” who treat patients with T2D who have severe renal dysfunction, said Dr. Bhatt, a professor of medicine at Harvard Medical School in Boston. “It’s a good enough reason by itself to approve this drug.”
The same mechanism may also be behind another unexpected finding in SCORED. Treatment with sotagliflozin cut the rate of total episodes of cardiovascular death, nonfatal MI, or nonfatal stroke by an absolute 1.6%, compared with placebo, and by a relative 23%. This benefit was largely driven by a 32% relative risk reduction total in MIs, and a 34% relative risk reduction in total stroke, both significant differences.
“No SGLT2 inhibitor has shown a reduction in stroke, and the MI signals have been mixed. The sizable MI and stroke effects are unique to sotagliflozin,” compared with the SGLT2 inhibitors, and likely reflect one or more mechanisms that result from blocked gut SGLT1 and a cut in GI glucose uptake, said Dr. Bhatt. “Probably some novel mechanism we don’t fully understand.”
First-ever HFpEF benefit
In contrast to these two benefits that are probably unique to drugs that inhibit the SGLT1 protein, sotagliflozin showed two other notable and unprecedented benefits that are likely generalizable to the SGLT2 inhibitors.
First is the striking benefit for HFpEF. Neither SOLOIST, which enrolled 1,222 patients with T2D and just hospitalized for worsening heart failure, nor SCORED, which enrolled patients with T2D and chronic kidney disease based exclusively on an eGFR of 25-60 mL/min per 1.73 m2, excluded patients with HFpEF, defined as heart failure patients with a left ventricular ejection fraction of at least 50%. The two studies together included a total of 739 of these patients, and they split fairly evenly between treatment with sotagliflozin or placebo.
The combined analysis showed that the incidence rate for the primary endpoint in both SOLOIST and SCORED was 59% with placebo and 39% with sotagliflozin, an absolute event reduction of 11.6 events/100 patient-years, and a significant 37% relative risk reduction, with a number needed to treat to prevent 1 event per year event of 9.
Although this observation comes from a nonprespecified combined analysis, “to me this result seems real, and I think it’s a class effect that I’m willing to extrapolate to the SGLT2 inhibitors,” Dr. Bhatt said. “It will change my practice,” he added, by spurring him to more aggressively prescribe an SGLT2 inhibitor to a patient with T2D and HFpEF.
“I think there has been some hesitation to use SGLT2 inhibitors in T2D patients with HFpEF” because of the paucity of data in this population, even though labeling and society recommendations do not rule it out. “I hope this finding will move that needle, and also generally improve SGLT2 inhibitor uptake, which has been low,” he said.
Also safe soon after acute heart failure decompensation
The other finding likely generalizable to SGLT2 inhibitors stems from the design of SOLOIST-WHF, which tested the efficacy and safety of starting sotagliflozin in patients with T2D as soon as they were stable after hospitalization for acute heart failure decompensation.
“Showing safety and efficacy when started in the hospital is pretty meaningful, because its tells patients that this drug is important and they should stay on it,” which should improve adherence, predicted Dr. Bhatt, who is also executive director of Interventional Cardiovascular Programs at Brigham and Women’s Hospital in Boston. “That’s the ultimate treatment path to prevent patients from falling through the cracks” and failing to receive an SGLT2 inhibitor.
SOLOIST-WHF enrolled patients hospitalized for worsening heart failure who also required intravenous diuretic treatment but had become stable enough to transition to an oral diuretic and come off oxygen. During a median follow-up of just over 9 months (both SOLOIST-WHF and SCORED ended sooner than planned because of a change in drug company sponsorship), treatment with sotagliflozin cut the primary endpoint by a relative 33%, compared with placebo, and with an absolute reduction of 25 events per 100 patient-years for a number needed to treat of 4. Sotagliflozin produced a strikingly high level of treatment efficiency driven by the high event rate in these recently decompensated patients. The benefit also appeared quickly, with a significant cut in events discernible within 28 days.
Extrapolating this finding to the SGLT2 inhibitors is “not a huge leap of faith,” Dr. Bhatt said.
“There is a role for sotagliflozin in acute heart failure. It showed benefit in these high-risk, transition-phase patients,” said Dr. Wilcox.
Simultaneously with Dr. Bhatt’s presentation, results of SOLOIST-WHF and SCORED were published online in the New England Journal of Medicine.
The trials were sponsored initially by Sanofi, and more recently by Lexicon. Dr. Bhatt has received research funding from both companies, and also from several other companies. He also is an adviser to several companies. Dr. Wilcox has been a consultant to Boehringer Ingelheim and Medtronic.
FROM AHA 2020
Osteoporosis drugs don’t worsen COVID-19 risk, may help
New observational data are the first to support recommendations to continue osteoporosis medications during the COVID-19 pandemic, and even suggest that some agents may protect against the virus.
Findings from the cross-sectional study of 2,102 patients with osteoporosis, osteoarthritis, and/or fibromyalgia – so-called noninflammatory rheumatic conditions – during March 1 to May 3, 2020, were recently published in Aging by Josep Blanch-Rubió, MD, scientific clinical director of the Rheumatology Service, Hospital del Mar, Barcelona, and colleagues.
Patients taking denosumab, zoledronate, and calcium showed trends toward lower incidence of developing symptomatic presumed COVID-19 (polymerase chain reaction tests weren’t widely available at the time), as did those taking the antidepressant serotonin/norepinephrine inhibitor duloxetine.
Some analgesics, particularly pregabalin and most other antidepressants, were associated with higher incidences of COVID-19, while oral bisphosphonates, vitamin D, thiazide diuretics, antihypertensive drugs, and chronic nonsteroidal anti-inflammatory drugs had no effect on COVID-19 incidence.
These data are the first to support guidance issued in May 2020 by the American Society for Bone and Mineral Research and four other professional societies advising continuation of osteoporosis medications during the pandemic. That statement’s authors acknowledged that, lacking data, their recommendations were based primarily on expert opinion.
“There were guidelines without any scientific base. ... This is the first scientific evidence showing that indeed you should continue your osteoporosis treatment if you have COVID-19. This is the first study to provide scientific support for the guidelines,” study coauthor Rafael Maldonado, MD, PhD, of the Laboratory of Neuropharmacology, Universitat Pompeu Fabra, Barcelona, said in an interview.
And while the data don’t offer proof of benefit for any drug – all of the 95% confidence intervals crossed 1.0 – they do show trends that deserve further study, Dr. Maldonado said.
“What we observed is that there is no harm. Treatments should be continued.”
“But we obtained very interesting results with denosumab, zoledronate, calcium, and duloxetine. ... There is a clear tendency, and the message is we should promote studies to see if these four treatments provide benefit.”
Different mechanisms for each?
Asked to comment on the findings, Matthew T. Drake, MD, PhD, said in an interview, “I would agree that there’s no reason any of these medications should be stopped or discontinued since there’s no evidence that they make the risk for infection worse.”
“But how [some of them may] improve or reduce the infection risk in my mind is somewhat unclear. ... It’s hard to come up with a unifying explanation” because those mentioned as potentially beneficial “are fairly different,” he noted.
Dr. Drake, associate professor of medicine in the department of endocrinology at the Mayo Clinic, Rochester, Minn., said he agreed with the study authors that denosumab’s targeting of the RANK/RANKL system is a possible anti-COVID-19 mechanism for that drug because that system is involved in immune response.
Regarding zoledronate/zoledronic acid, both the Spanish authors and Dr. Drake pointed to a landmark study linking the intravenous drug to longer survival in patients with hip fracture. The study authors note that there could be several mechanisms for an overall survival benefit, but additionally, “zoledronate may make dendritic cells and their precursors less susceptible to SARS-CoV-2 infection, which could explain the beneficial effects here ... on COVID-19 incidence.”
And, the authors hypothesized, the reason for the lack of benefit with oral bisphosphonates might relate to the higher potency of the intravenous zoledronate. Dr. Drake added that its higher bioavailability may also play a role.
As for calcium, the authors suggest that the beneficial effect against COVID-19 could relate to its action in generating two immune cell types – T follicular helper cells and T follicular regulatory cells – which promote an appropriate immune response against infectious agents, including viruses.
Data supporting the guidelines
Of the 2,102 patients in the study by Blanch-Rubió and colleagues, 80.5% were women, and their mean age was 66.4 years. Overall, 63.7% had osteoarthritis, 43.5% had osteoporosis, and 27.2% had fibromyalgia. Treatments included vitamin D in 62%, calcium in 23.3%, denosumab in 12.6%, and intravenous zoledronate in 8.5%. Over half were taking analgesics and nearly a third antidepressants, with 9.9% taking duloxetine.
During the study period, 5.2%, or 109 individuals, were diagnosed with COVID-19 based on presenting for medical care with hallmark symptoms.
After adjustments for sex, age, diabetes, pulmonary disease, cardiovascular disease, chronic kidney disease, and active cancer or treatment, the relative risks for COVID-19 were 0.58 for denosumab, 0.62 for intravenous zoledronate, and 0.64 for calcium, all nonsignificant trends. No associations were found between COVID-19 and oral bisphosphonates, vitamin D, or thiazide diuretics. Increased but nonsignificant relative risks for COVID-19 were seen with analgesics, particularly pregabalin (1.55), gabapentin (1.39), and opioids (1.25).
Among antidepressants, there was a relative risk of 1.54 for selective serotonin reuptake inhibitors, 1.38 for amitriptyline, and 1.22 for all dual-action antidepressants together. In contrast, there was a negative association with the dual-action antidepressant duloxetine, with an adjusted relative risk of 0.68.
“The good news,” Dr. Drake said, “is that none of it appears bad.”
Dr. Blanch-Rubió has received grants or consulting fees from Amgen, Laboratorio Stada, Gedeon-Rhicter Ibérica, Lilly España, Pfizer, Gebro Pharma, and UCB Pharma. Dr. Maldonado has received research grants or consulting fees from Aelis, Almirall, Boehringer Ingelheim, BrainCo, Esteve, Ferrer, GlaxoSmithKline, Grünenthal, GW Pharmaceuticals, Janus, Lundbeck, Pharmaleads, Phytoplant, Rhodes, Sanofi, Spherium, Union de Pharmacologie Scientifique Appliquée, Upjohn, and Uriach. Dr. Drake has reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
New observational data are the first to support recommendations to continue osteoporosis medications during the COVID-19 pandemic, and even suggest that some agents may protect against the virus.
Findings from the cross-sectional study of 2,102 patients with osteoporosis, osteoarthritis, and/or fibromyalgia – so-called noninflammatory rheumatic conditions – during March 1 to May 3, 2020, were recently published in Aging by Josep Blanch-Rubió, MD, scientific clinical director of the Rheumatology Service, Hospital del Mar, Barcelona, and colleagues.
Patients taking denosumab, zoledronate, and calcium showed trends toward lower incidence of developing symptomatic presumed COVID-19 (polymerase chain reaction tests weren’t widely available at the time), as did those taking the antidepressant serotonin/norepinephrine inhibitor duloxetine.
Some analgesics, particularly pregabalin and most other antidepressants, were associated with higher incidences of COVID-19, while oral bisphosphonates, vitamin D, thiazide diuretics, antihypertensive drugs, and chronic nonsteroidal anti-inflammatory drugs had no effect on COVID-19 incidence.
These data are the first to support guidance issued in May 2020 by the American Society for Bone and Mineral Research and four other professional societies advising continuation of osteoporosis medications during the pandemic. That statement’s authors acknowledged that, lacking data, their recommendations were based primarily on expert opinion.
“There were guidelines without any scientific base. ... This is the first scientific evidence showing that indeed you should continue your osteoporosis treatment if you have COVID-19. This is the first study to provide scientific support for the guidelines,” study coauthor Rafael Maldonado, MD, PhD, of the Laboratory of Neuropharmacology, Universitat Pompeu Fabra, Barcelona, said in an interview.
And while the data don’t offer proof of benefit for any drug – all of the 95% confidence intervals crossed 1.0 – they do show trends that deserve further study, Dr. Maldonado said.
“What we observed is that there is no harm. Treatments should be continued.”
“But we obtained very interesting results with denosumab, zoledronate, calcium, and duloxetine. ... There is a clear tendency, and the message is we should promote studies to see if these four treatments provide benefit.”
Different mechanisms for each?
Asked to comment on the findings, Matthew T. Drake, MD, PhD, said in an interview, “I would agree that there’s no reason any of these medications should be stopped or discontinued since there’s no evidence that they make the risk for infection worse.”
“But how [some of them may] improve or reduce the infection risk in my mind is somewhat unclear. ... It’s hard to come up with a unifying explanation” because those mentioned as potentially beneficial “are fairly different,” he noted.
Dr. Drake, associate professor of medicine in the department of endocrinology at the Mayo Clinic, Rochester, Minn., said he agreed with the study authors that denosumab’s targeting of the RANK/RANKL system is a possible anti-COVID-19 mechanism for that drug because that system is involved in immune response.
Regarding zoledronate/zoledronic acid, both the Spanish authors and Dr. Drake pointed to a landmark study linking the intravenous drug to longer survival in patients with hip fracture. The study authors note that there could be several mechanisms for an overall survival benefit, but additionally, “zoledronate may make dendritic cells and their precursors less susceptible to SARS-CoV-2 infection, which could explain the beneficial effects here ... on COVID-19 incidence.”
And, the authors hypothesized, the reason for the lack of benefit with oral bisphosphonates might relate to the higher potency of the intravenous zoledronate. Dr. Drake added that its higher bioavailability may also play a role.
As for calcium, the authors suggest that the beneficial effect against COVID-19 could relate to its action in generating two immune cell types – T follicular helper cells and T follicular regulatory cells – which promote an appropriate immune response against infectious agents, including viruses.
Data supporting the guidelines
Of the 2,102 patients in the study by Blanch-Rubió and colleagues, 80.5% were women, and their mean age was 66.4 years. Overall, 63.7% had osteoarthritis, 43.5% had osteoporosis, and 27.2% had fibromyalgia. Treatments included vitamin D in 62%, calcium in 23.3%, denosumab in 12.6%, and intravenous zoledronate in 8.5%. Over half were taking analgesics and nearly a third antidepressants, with 9.9% taking duloxetine.
During the study period, 5.2%, or 109 individuals, were diagnosed with COVID-19 based on presenting for medical care with hallmark symptoms.
After adjustments for sex, age, diabetes, pulmonary disease, cardiovascular disease, chronic kidney disease, and active cancer or treatment, the relative risks for COVID-19 were 0.58 for denosumab, 0.62 for intravenous zoledronate, and 0.64 for calcium, all nonsignificant trends. No associations were found between COVID-19 and oral bisphosphonates, vitamin D, or thiazide diuretics. Increased but nonsignificant relative risks for COVID-19 were seen with analgesics, particularly pregabalin (1.55), gabapentin (1.39), and opioids (1.25).
Among antidepressants, there was a relative risk of 1.54 for selective serotonin reuptake inhibitors, 1.38 for amitriptyline, and 1.22 for all dual-action antidepressants together. In contrast, there was a negative association with the dual-action antidepressant duloxetine, with an adjusted relative risk of 0.68.
“The good news,” Dr. Drake said, “is that none of it appears bad.”
Dr. Blanch-Rubió has received grants or consulting fees from Amgen, Laboratorio Stada, Gedeon-Rhicter Ibérica, Lilly España, Pfizer, Gebro Pharma, and UCB Pharma. Dr. Maldonado has received research grants or consulting fees from Aelis, Almirall, Boehringer Ingelheim, BrainCo, Esteve, Ferrer, GlaxoSmithKline, Grünenthal, GW Pharmaceuticals, Janus, Lundbeck, Pharmaleads, Phytoplant, Rhodes, Sanofi, Spherium, Union de Pharmacologie Scientifique Appliquée, Upjohn, and Uriach. Dr. Drake has reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
New observational data are the first to support recommendations to continue osteoporosis medications during the COVID-19 pandemic, and even suggest that some agents may protect against the virus.
Findings from the cross-sectional study of 2,102 patients with osteoporosis, osteoarthritis, and/or fibromyalgia – so-called noninflammatory rheumatic conditions – during March 1 to May 3, 2020, were recently published in Aging by Josep Blanch-Rubió, MD, scientific clinical director of the Rheumatology Service, Hospital del Mar, Barcelona, and colleagues.
Patients taking denosumab, zoledronate, and calcium showed trends toward lower incidence of developing symptomatic presumed COVID-19 (polymerase chain reaction tests weren’t widely available at the time), as did those taking the antidepressant serotonin/norepinephrine inhibitor duloxetine.
Some analgesics, particularly pregabalin and most other antidepressants, were associated with higher incidences of COVID-19, while oral bisphosphonates, vitamin D, thiazide diuretics, antihypertensive drugs, and chronic nonsteroidal anti-inflammatory drugs had no effect on COVID-19 incidence.
These data are the first to support guidance issued in May 2020 by the American Society for Bone and Mineral Research and four other professional societies advising continuation of osteoporosis medications during the pandemic. That statement’s authors acknowledged that, lacking data, their recommendations were based primarily on expert opinion.
“There were guidelines without any scientific base. ... This is the first scientific evidence showing that indeed you should continue your osteoporosis treatment if you have COVID-19. This is the first study to provide scientific support for the guidelines,” study coauthor Rafael Maldonado, MD, PhD, of the Laboratory of Neuropharmacology, Universitat Pompeu Fabra, Barcelona, said in an interview.
And while the data don’t offer proof of benefit for any drug – all of the 95% confidence intervals crossed 1.0 – they do show trends that deserve further study, Dr. Maldonado said.
“What we observed is that there is no harm. Treatments should be continued.”
“But we obtained very interesting results with denosumab, zoledronate, calcium, and duloxetine. ... There is a clear tendency, and the message is we should promote studies to see if these four treatments provide benefit.”
Different mechanisms for each?
Asked to comment on the findings, Matthew T. Drake, MD, PhD, said in an interview, “I would agree that there’s no reason any of these medications should be stopped or discontinued since there’s no evidence that they make the risk for infection worse.”
“But how [some of them may] improve or reduce the infection risk in my mind is somewhat unclear. ... It’s hard to come up with a unifying explanation” because those mentioned as potentially beneficial “are fairly different,” he noted.
Dr. Drake, associate professor of medicine in the department of endocrinology at the Mayo Clinic, Rochester, Minn., said he agreed with the study authors that denosumab’s targeting of the RANK/RANKL system is a possible anti-COVID-19 mechanism for that drug because that system is involved in immune response.
Regarding zoledronate/zoledronic acid, both the Spanish authors and Dr. Drake pointed to a landmark study linking the intravenous drug to longer survival in patients with hip fracture. The study authors note that there could be several mechanisms for an overall survival benefit, but additionally, “zoledronate may make dendritic cells and their precursors less susceptible to SARS-CoV-2 infection, which could explain the beneficial effects here ... on COVID-19 incidence.”
And, the authors hypothesized, the reason for the lack of benefit with oral bisphosphonates might relate to the higher potency of the intravenous zoledronate. Dr. Drake added that its higher bioavailability may also play a role.
As for calcium, the authors suggest that the beneficial effect against COVID-19 could relate to its action in generating two immune cell types – T follicular helper cells and T follicular regulatory cells – which promote an appropriate immune response against infectious agents, including viruses.
Data supporting the guidelines
Of the 2,102 patients in the study by Blanch-Rubió and colleagues, 80.5% were women, and their mean age was 66.4 years. Overall, 63.7% had osteoarthritis, 43.5% had osteoporosis, and 27.2% had fibromyalgia. Treatments included vitamin D in 62%, calcium in 23.3%, denosumab in 12.6%, and intravenous zoledronate in 8.5%. Over half were taking analgesics and nearly a third antidepressants, with 9.9% taking duloxetine.
During the study period, 5.2%, or 109 individuals, were diagnosed with COVID-19 based on presenting for medical care with hallmark symptoms.
After adjustments for sex, age, diabetes, pulmonary disease, cardiovascular disease, chronic kidney disease, and active cancer or treatment, the relative risks for COVID-19 were 0.58 for denosumab, 0.62 for intravenous zoledronate, and 0.64 for calcium, all nonsignificant trends. No associations were found between COVID-19 and oral bisphosphonates, vitamin D, or thiazide diuretics. Increased but nonsignificant relative risks for COVID-19 were seen with analgesics, particularly pregabalin (1.55), gabapentin (1.39), and opioids (1.25).
Among antidepressants, there was a relative risk of 1.54 for selective serotonin reuptake inhibitors, 1.38 for amitriptyline, and 1.22 for all dual-action antidepressants together. In contrast, there was a negative association with the dual-action antidepressant duloxetine, with an adjusted relative risk of 0.68.
“The good news,” Dr. Drake said, “is that none of it appears bad.”
Dr. Blanch-Rubió has received grants or consulting fees from Amgen, Laboratorio Stada, Gedeon-Rhicter Ibérica, Lilly España, Pfizer, Gebro Pharma, and UCB Pharma. Dr. Maldonado has received research grants or consulting fees from Aelis, Almirall, Boehringer Ingelheim, BrainCo, Esteve, Ferrer, GlaxoSmithKline, Grünenthal, GW Pharmaceuticals, Janus, Lundbeck, Pharmaleads, Phytoplant, Rhodes, Sanofi, Spherium, Union de Pharmacologie Scientifique Appliquée, Upjohn, and Uriach. Dr. Drake has reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
VTEs tied to immune checkpoint inhibitor cancer treatment
Cancer patients who receive an immune checkpoint inhibitor have more than a doubled rate of venous thromboembolism during the subsequent 2 years, compared with their rate during the 2 years before treatment, according to a retrospective analysis of more than 2,800 patients treated at a single U.S. center.
The study focused on cancer patients treated with an immune checkpoint inhibitor (ICI) at Massachusetts General Hospital in Boston. It showed that during the 2 years prior to treatment with any type of ICI, the incidence of venous thromboembolic events (VTE) was 4.85/100 patient-years that then jumped to 11.75/100 patient-years during the 2 years following treatment. This translated into an incidence rate ratio of 2.43 during posttreatment follow-up, compared with pretreatment, Jingyi Gong, MD, said at the virtual American Heart Association scientific sessions.
The increased VTE rate resulted from rises in both the rate of deep vein thrombosis, which had an IRR of 3.23 during the posttreatment period, and for pulmonary embolism, which showed an IRR of 2.24, said Dr. Gong, a physician at Brigham and Women’s Hospital in Boston. She hypothesized that this effect may result from a procoagulant effect of the immune activation and inflammation triggered by ICIs.
Hypothesis-generating results
Cardiologists cautioned that these findings should only be considered hypothesis generating, but raise an important alert for clinicians to have heightened awareness of the potential for VTE following ICI treatment.
“A clear message is to be aware that there is this signal, and be vigilant for patients who might present with VTE following ICI treatment,” commented Richard J. Kovacs, MD, a cardiologist and professor at Indiana University, Indianapolis. The data that Dr. Gong reported are “moderately convincing,” he added in an interview.

“Awareness that patients who receive ICI may be at increased VTE risk is very important,” agreed Umberto Campia, MD, a cardiologist, vascular specialist, and member of the cardio-oncology group at Brigham and Women’s Hospital, who was not involved in the new study.
The potential impact of ICI treatment on VTE risk is slowly emerging, added Dr. Campia. Until recently, the literature primarily was case reports, but recently another retrospective, single-center study came out that reported a 13% incidence of VTE in cancer patients following ICI treatment. On the other hand, a recently published meta-analysis of more than 20,000 patients from 68 ICI studies failed to find a suggestion of increased VTE incidence following ICI interventions.
Attempting to assess the impact of treatment on VTE risk in cancer patients is challenging because cancer itself boosts the risk. Recommendations on the use of VTE prophylaxis in cancer patients most recently came out in 2014 from the American Society of Clinical Oncology, which said that VTE prophylaxis for ambulatory cancer patients “may be considered for highly select high-risk patients.” The impact of cancer therapy on VTE risk and the need for prophylaxis is usually assessed by applying the Khorana score, Dr. Campia said in an interview.
VTE spikes acutely after ICI treatment
Dr. Gong analyzed VTE incidence rates by time during the total 4-year period studied, and found that the rate gradually and steadily rose with time throughout the 2 years preceding treatment, spiked immediately following ICI treatment, and then gradually and steadily fell back to roughly the rate seen just before treatment, reaching that level about a year after treatment. She ran a sensitivity analysis that excluded patients who died during the first year following their ICI treatment, and in this calculation an acute spike in VTE following ICI treatment still occurred but with reduced magnitude.
She also reported the results of several subgroup analyses. The IRRs remained consistent among women and men, among patients who were aged over or under 65 years, and regardless of cancer type or treatment with corticosteroids. But the subgroup analyses identified two parameters that seemed to clearly split VTE rates.
Among patients on treatment with an anticoagulant agent at the time of their ICI treatment, roughly 10% of the patients, the IRR was 0.56, compared with a ratio of 3.86 among the other patients, suggesting possible protection. A second factor that seemed linked with VTE incidence was the number of ICI treatment cycles a patient received. Those who received more than five cycles had a risk ratio of 3.95, while those who received five or fewer cycles had a RR of 1.66.
Her analysis included 2,842 cancer patients who received treatment with an ICI at Massachusetts General Hospital. Patients averaged 64 years of age, slightly more than half were men, and 13% had a prior history of VTE. Patients received an average of 5 ICI treatment cycles, but a quarter of the patients received more than 10 cycles.
During the 2-year follow-up, 244 patients (9%) developed VTE. The patients who developed VTE were significantly younger than those who did not, with an average age of 63 years, compared with 65. And the patients who eventually developed VTE had a significantly higher prevalence of prior VTE at 18%, compared with 12% among the patients who stayed VTE free.
The cancer types patients had were non–small cell lung, 29%; melanoma, 28%; head and neck, 12%; renal genitourinary, 6%; and other, 25%. ICIs have been available for routine U.S. practice since 2011. The class includes agents such as pembrolizumab (Keytruda) and durvalumab (Imfinzi).
Researchers would need to perform a prospective, randomized study to determine whether anticoagulant prophylaxis is clearly beneficial for patients receiving ICI treatment, Dr. Gong said. But both Dr. Kovacs and Dr. Campia said that more data on this topic are first needed.
“We need to confirm that treatment with ICI is associated with VTEs. Retrospective data are not definitive,” said Dr. Campia. “We would need to prospectively assess the impact of ICI,” which will not be easy, as it’s quickly become a cornerstone for treating many cancers. “We need to become more familiar with the adverse effects of these drugs. We are still learning about their toxicities.”
The study had no commercial funding. Dr. Gong, Dr. Kovacs, and Dr. Campia had no disclosures.
Cancer patients who receive an immune checkpoint inhibitor have more than a doubled rate of venous thromboembolism during the subsequent 2 years, compared with their rate during the 2 years before treatment, according to a retrospective analysis of more than 2,800 patients treated at a single U.S. center.
The study focused on cancer patients treated with an immune checkpoint inhibitor (ICI) at Massachusetts General Hospital in Boston. It showed that during the 2 years prior to treatment with any type of ICI, the incidence of venous thromboembolic events (VTE) was 4.85/100 patient-years that then jumped to 11.75/100 patient-years during the 2 years following treatment. This translated into an incidence rate ratio of 2.43 during posttreatment follow-up, compared with pretreatment, Jingyi Gong, MD, said at the virtual American Heart Association scientific sessions.
The increased VTE rate resulted from rises in both the rate of deep vein thrombosis, which had an IRR of 3.23 during the posttreatment period, and for pulmonary embolism, which showed an IRR of 2.24, said Dr. Gong, a physician at Brigham and Women’s Hospital in Boston. She hypothesized that this effect may result from a procoagulant effect of the immune activation and inflammation triggered by ICIs.
Hypothesis-generating results
Cardiologists cautioned that these findings should only be considered hypothesis generating, but raise an important alert for clinicians to have heightened awareness of the potential for VTE following ICI treatment.
“A clear message is to be aware that there is this signal, and be vigilant for patients who might present with VTE following ICI treatment,” commented Richard J. Kovacs, MD, a cardiologist and professor at Indiana University, Indianapolis. The data that Dr. Gong reported are “moderately convincing,” he added in an interview.
“Awareness that patients who receive ICI may be at increased VTE risk is very important,” agreed Umberto Campia, MD, a cardiologist, vascular specialist, and member of the cardio-oncology group at Brigham and Women’s Hospital, who was not involved in the new study.
The potential impact of ICI treatment on VTE risk is slowly emerging, added Dr. Campia. Until recently, the literature primarily was case reports, but recently another retrospective, single-center study came out that reported a 13% incidence of VTE in cancer patients following ICI treatment. On the other hand, a recently published meta-analysis of more than 20,000 patients from 68 ICI studies failed to find a suggestion of increased VTE incidence following ICI interventions.
Attempting to assess the impact of treatment on VTE risk in cancer patients is challenging because cancer itself boosts the risk. Recommendations on the use of VTE prophylaxis in cancer patients most recently came out in 2014 from the American Society of Clinical Oncology, which said that VTE prophylaxis for ambulatory cancer patients “may be considered for highly select high-risk patients.” The impact of cancer therapy on VTE risk and the need for prophylaxis is usually assessed by applying the Khorana score, Dr. Campia said in an interview.
VTE spikes acutely after ICI treatment
Dr. Gong analyzed VTE incidence rates by time during the total 4-year period studied, and found that the rate gradually and steadily rose with time throughout the 2 years preceding treatment, spiked immediately following ICI treatment, and then gradually and steadily fell back to roughly the rate seen just before treatment, reaching that level about a year after treatment. She ran a sensitivity analysis that excluded patients who died during the first year following their ICI treatment, and in this calculation an acute spike in VTE following ICI treatment still occurred but with reduced magnitude.
She also reported the results of several subgroup analyses. The IRRs remained consistent among women and men, among patients who were aged over or under 65 years, and regardless of cancer type or treatment with corticosteroids. But the subgroup analyses identified two parameters that seemed to clearly split VTE rates.
Among patients on treatment with an anticoagulant agent at the time of their ICI treatment, roughly 10% of the patients, the IRR was 0.56, compared with a ratio of 3.86 among the other patients, suggesting possible protection. A second factor that seemed linked with VTE incidence was the number of ICI treatment cycles a patient received. Those who received more than five cycles had a risk ratio of 3.95, while those who received five or fewer cycles had a RR of 1.66.
Her analysis included 2,842 cancer patients who received treatment with an ICI at Massachusetts General Hospital. Patients averaged 64 years of age, slightly more than half were men, and 13% had a prior history of VTE. Patients received an average of 5 ICI treatment cycles, but a quarter of the patients received more than 10 cycles.
During the 2-year follow-up, 244 patients (9%) developed VTE. The patients who developed VTE were significantly younger than those who did not, with an average age of 63 years, compared with 65. And the patients who eventually developed VTE had a significantly higher prevalence of prior VTE at 18%, compared with 12% among the patients who stayed VTE free.
The cancer types patients had were non–small cell lung, 29%; melanoma, 28%; head and neck, 12%; renal genitourinary, 6%; and other, 25%. ICIs have been available for routine U.S. practice since 2011. The class includes agents such as pembrolizumab (Keytruda) and durvalumab (Imfinzi).
Researchers would need to perform a prospective, randomized study to determine whether anticoagulant prophylaxis is clearly beneficial for patients receiving ICI treatment, Dr. Gong said. But both Dr. Kovacs and Dr. Campia said that more data on this topic are first needed.
“We need to confirm that treatment with ICI is associated with VTEs. Retrospective data are not definitive,” said Dr. Campia. “We would need to prospectively assess the impact of ICI,” which will not be easy, as it’s quickly become a cornerstone for treating many cancers. “We need to become more familiar with the adverse effects of these drugs. We are still learning about their toxicities.”
The study had no commercial funding. Dr. Gong, Dr. Kovacs, and Dr. Campia had no disclosures.
Cancer patients who receive an immune checkpoint inhibitor have more than a doubled rate of venous thromboembolism during the subsequent 2 years, compared with their rate during the 2 years before treatment, according to a retrospective analysis of more than 2,800 patients treated at a single U.S. center.
The study focused on cancer patients treated with an immune checkpoint inhibitor (ICI) at Massachusetts General Hospital in Boston. It showed that during the 2 years prior to treatment with any type of ICI, the incidence of venous thromboembolic events (VTE) was 4.85/100 patient-years that then jumped to 11.75/100 patient-years during the 2 years following treatment. This translated into an incidence rate ratio of 2.43 during posttreatment follow-up, compared with pretreatment, Jingyi Gong, MD, said at the virtual American Heart Association scientific sessions.
The increased VTE rate resulted from rises in both the rate of deep vein thrombosis, which had an IRR of 3.23 during the posttreatment period, and for pulmonary embolism, which showed an IRR of 2.24, said Dr. Gong, a physician at Brigham and Women’s Hospital in Boston. She hypothesized that this effect may result from a procoagulant effect of the immune activation and inflammation triggered by ICIs.
Hypothesis-generating results
Cardiologists cautioned that these findings should only be considered hypothesis generating, but raise an important alert for clinicians to have heightened awareness of the potential for VTE following ICI treatment.
“A clear message is to be aware that there is this signal, and be vigilant for patients who might present with VTE following ICI treatment,” commented Richard J. Kovacs, MD, a cardiologist and professor at Indiana University, Indianapolis. The data that Dr. Gong reported are “moderately convincing,” he added in an interview.
“Awareness that patients who receive ICI may be at increased VTE risk is very important,” agreed Umberto Campia, MD, a cardiologist, vascular specialist, and member of the cardio-oncology group at Brigham and Women’s Hospital, who was not involved in the new study.
The potential impact of ICI treatment on VTE risk is slowly emerging, added Dr. Campia. Until recently, the literature primarily was case reports, but recently another retrospective, single-center study came out that reported a 13% incidence of VTE in cancer patients following ICI treatment. On the other hand, a recently published meta-analysis of more than 20,000 patients from 68 ICI studies failed to find a suggestion of increased VTE incidence following ICI interventions.
Attempting to assess the impact of treatment on VTE risk in cancer patients is challenging because cancer itself boosts the risk. Recommendations on the use of VTE prophylaxis in cancer patients most recently came out in 2014 from the American Society of Clinical Oncology, which said that VTE prophylaxis for ambulatory cancer patients “may be considered for highly select high-risk patients.” The impact of cancer therapy on VTE risk and the need for prophylaxis is usually assessed by applying the Khorana score, Dr. Campia said in an interview.
VTE spikes acutely after ICI treatment
Dr. Gong analyzed VTE incidence rates by time during the total 4-year period studied, and found that the rate gradually and steadily rose with time throughout the 2 years preceding treatment, spiked immediately following ICI treatment, and then gradually and steadily fell back to roughly the rate seen just before treatment, reaching that level about a year after treatment. She ran a sensitivity analysis that excluded patients who died during the first year following their ICI treatment, and in this calculation an acute spike in VTE following ICI treatment still occurred but with reduced magnitude.
She also reported the results of several subgroup analyses. The IRRs remained consistent among women and men, among patients who were aged over or under 65 years, and regardless of cancer type or treatment with corticosteroids. But the subgroup analyses identified two parameters that seemed to clearly split VTE rates.
Among patients on treatment with an anticoagulant agent at the time of their ICI treatment, roughly 10% of the patients, the IRR was 0.56, compared with a ratio of 3.86 among the other patients, suggesting possible protection. A second factor that seemed linked with VTE incidence was the number of ICI treatment cycles a patient received. Those who received more than five cycles had a risk ratio of 3.95, while those who received five or fewer cycles had a RR of 1.66.
Her analysis included 2,842 cancer patients who received treatment with an ICI at Massachusetts General Hospital. Patients averaged 64 years of age, slightly more than half were men, and 13% had a prior history of VTE. Patients received an average of 5 ICI treatment cycles, but a quarter of the patients received more than 10 cycles.
During the 2-year follow-up, 244 patients (9%) developed VTE. The patients who developed VTE were significantly younger than those who did not, with an average age of 63 years, compared with 65. And the patients who eventually developed VTE had a significantly higher prevalence of prior VTE at 18%, compared with 12% among the patients who stayed VTE free.
The cancer types patients had were non–small cell lung, 29%; melanoma, 28%; head and neck, 12%; renal genitourinary, 6%; and other, 25%. ICIs have been available for routine U.S. practice since 2011. The class includes agents such as pembrolizumab (Keytruda) and durvalumab (Imfinzi).
Researchers would need to perform a prospective, randomized study to determine whether anticoagulant prophylaxis is clearly beneficial for patients receiving ICI treatment, Dr. Gong said. But both Dr. Kovacs and Dr. Campia said that more data on this topic are first needed.
“We need to confirm that treatment with ICI is associated with VTEs. Retrospective data are not definitive,” said Dr. Campia. “We would need to prospectively assess the impact of ICI,” which will not be easy, as it’s quickly become a cornerstone for treating many cancers. “We need to become more familiar with the adverse effects of these drugs. We are still learning about their toxicities.”
The study had no commercial funding. Dr. Gong, Dr. Kovacs, and Dr. Campia had no disclosures.
FROM AHA 2020
Pembrolizumab approved for triple-negative breast cancer
The Food and Drug Administration has granted accelerated approval for pembrolizumab (Keytruda) in combination with chemotherapy to treat locally recurrent unresectable or metastatic triple-negative breast cancer (TNBC) that expresses PD-L1, as determined by a combined positive score of 10 or greater on an FDA-approved assay.
The FDA also approved a PD-L1 assay for selecting TNBC patients for pembrolizumab, the PD-L1 IHC 22C3 pharmDx.
Pembrolizumab is approved for numerous indications in the United States, but the new approval is its first breast cancer indication.
The accelerated approval for pembrolizumab in TNBC was based on progression-free survival (PFS) in the KEYNOTE-355 trial. The FDA noted that continued approval of pembrolizumab in TNBC “may be contingent upon verification and description of clinical benefit in the confirmatory trials.”
KEYNOTE-355 enrolled patients with locally recurrent unresectable or metastatic TNBC who had not received chemotherapy in the metastatic setting. Patients were randomized to chemotherapy (nab-paclitaxel, paclitaxel, or gemcitabine plus carboplatin) plus placebo (n = 281) or chemotherapy plus pembrolizumab at 200 mg on day 1 every 3 weeks (n = 562).
Among PD-L1-positive patients (n = 323), the median PFS was 5.6 months in the placebo arm and 9.7 months in the pembrolizumab arm (hazard ratio, 0.65; P = .0012).
The recommended pembrolizumab dose in TNBC is 200 mg every 3 weeks or 400 mg every 6 weeks administered prior to chemotherapy until disease progression, unacceptable toxicity, or up to 24 months.
Pembrolizumab can cause immune-mediated adverse reactions that may be severe or fatal, according to Merck, the manufacturer of pembrolizumab. These adverse reactions include pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, severe skin reactions, solid organ transplant rejection, and complications of allogeneic hematopoietic stem cell transplant.
“Based on the severity of the adverse reaction, [pembrolizumab] should be withheld or discontinued and corticosteroids administered if appropriate,” the company noted.
For more details on pembrolizumab, see the full prescribing information.
The Food and Drug Administration has granted accelerated approval for pembrolizumab (Keytruda) in combination with chemotherapy to treat locally recurrent unresectable or metastatic triple-negative breast cancer (TNBC) that expresses PD-L1, as determined by a combined positive score of 10 or greater on an FDA-approved assay.
The FDA also approved a PD-L1 assay for selecting TNBC patients for pembrolizumab, the PD-L1 IHC 22C3 pharmDx.
Pembrolizumab is approved for numerous indications in the United States, but the new approval is its first breast cancer indication.
The accelerated approval for pembrolizumab in TNBC was based on progression-free survival (PFS) in the KEYNOTE-355 trial. The FDA noted that continued approval of pembrolizumab in TNBC “may be contingent upon verification and description of clinical benefit in the confirmatory trials.”
KEYNOTE-355 enrolled patients with locally recurrent unresectable or metastatic TNBC who had not received chemotherapy in the metastatic setting. Patients were randomized to chemotherapy (nab-paclitaxel, paclitaxel, or gemcitabine plus carboplatin) plus placebo (n = 281) or chemotherapy plus pembrolizumab at 200 mg on day 1 every 3 weeks (n = 562).
Among PD-L1-positive patients (n = 323), the median PFS was 5.6 months in the placebo arm and 9.7 months in the pembrolizumab arm (hazard ratio, 0.65; P = .0012).
The recommended pembrolizumab dose in TNBC is 200 mg every 3 weeks or 400 mg every 6 weeks administered prior to chemotherapy until disease progression, unacceptable toxicity, or up to 24 months.
Pembrolizumab can cause immune-mediated adverse reactions that may be severe or fatal, according to Merck, the manufacturer of pembrolizumab. These adverse reactions include pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, severe skin reactions, solid organ transplant rejection, and complications of allogeneic hematopoietic stem cell transplant.
“Based on the severity of the adverse reaction, [pembrolizumab] should be withheld or discontinued and corticosteroids administered if appropriate,” the company noted.
For more details on pembrolizumab, see the full prescribing information.
The Food and Drug Administration has granted accelerated approval for pembrolizumab (Keytruda) in combination with chemotherapy to treat locally recurrent unresectable or metastatic triple-negative breast cancer (TNBC) that expresses PD-L1, as determined by a combined positive score of 10 or greater on an FDA-approved assay.
The FDA also approved a PD-L1 assay for selecting TNBC patients for pembrolizumab, the PD-L1 IHC 22C3 pharmDx.
Pembrolizumab is approved for numerous indications in the United States, but the new approval is its first breast cancer indication.
The accelerated approval for pembrolizumab in TNBC was based on progression-free survival (PFS) in the KEYNOTE-355 trial. The FDA noted that continued approval of pembrolizumab in TNBC “may be contingent upon verification and description of clinical benefit in the confirmatory trials.”
KEYNOTE-355 enrolled patients with locally recurrent unresectable or metastatic TNBC who had not received chemotherapy in the metastatic setting. Patients were randomized to chemotherapy (nab-paclitaxel, paclitaxel, or gemcitabine plus carboplatin) plus placebo (n = 281) or chemotherapy plus pembrolizumab at 200 mg on day 1 every 3 weeks (n = 562).
Among PD-L1-positive patients (n = 323), the median PFS was 5.6 months in the placebo arm and 9.7 months in the pembrolizumab arm (hazard ratio, 0.65; P = .0012).
The recommended pembrolizumab dose in TNBC is 200 mg every 3 weeks or 400 mg every 6 weeks administered prior to chemotherapy until disease progression, unacceptable toxicity, or up to 24 months.
Pembrolizumab can cause immune-mediated adverse reactions that may be severe or fatal, according to Merck, the manufacturer of pembrolizumab. These adverse reactions include pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, severe skin reactions, solid organ transplant rejection, and complications of allogeneic hematopoietic stem cell transplant.
“Based on the severity of the adverse reaction, [pembrolizumab] should be withheld or discontinued and corticosteroids administered if appropriate,” the company noted.
For more details on pembrolizumab, see the full prescribing information.
FROM THE FOOD AND DRUG ADMINISTRATION
Intravenous iron reduces HF readmissions: AFFIRM-AHF
Iron supplementation reduces heart failure (HF) readmissions in iron-deficient patients hospitalized for acute HF, according to results of the AFFIRM-AHF trial.
After 52 weeks, intravenous ferric carboxymaltose (Ferinject) reduced the risk of total HF hospitalizations and cardiovascular (CV) death by 21% compared with placebo (293 vs 372 events; rate ratio [RR] 0.79; 95% CI, 0.62 - 1.01).
Although the composite primary endpoint failed to achieve statistical significance, it was driven by a significant 26% reduction in the risk of total HF hospital readmissions (P = .013) without an effect on CV mortality (P =.809).
Because the management and follow-up of patients was affected by the COVID-19 pandemic, a prespecified sensitivity analysis was performed that censored patients in each country at the date when its first COVID-19 patient was reported, explained principal investigator Piotr Ponikowski, MD, PhD, Wroclaw Medical University, Wroclaw, Poland.
That analysis revealed a significant 30% reduction in total HF readmissions (P = .005) in patients receiving ferric carboxymaltose (FCM), as well as significant benefits on the primary composite and secondary endpoints.
Notably, 80% of patients required only one or two injections and HF hospitalizations were reduced irrespective of anemia status.
“Iron deficiency should be searched in patients hospitalized with acute heart failure — assessed using a simple blood test — and is now an important therapeutic target,” Ponikowski said at the virtual American Heart Association (AHA) Scientific Sessions 2020.
The results were also published simultaneously in The Lancet.
Iron deficiency is present in up to 70% of patients with acute HF and a predictor of poor outcome, independent of anemia and ejection fraction, he noted.
The FAIR-HF, CONFIRM-HF, and EFFECT-HF trials demonstrated that IV iron supplementation improves exercise capacity, symptoms, and quality of life in iron-deficient HF patients.

However, no such benefit was seen with oral IV in the IRONOUT trial. “So it seems if we are to replace iron, it needs to be done using intravenous therapy,” said John McMurray, MD, University of Glasgow, Scotland, who was invited to discuss the results.
He observed that the reduction in HF hospitalizations in AFFIRM-AHF were relatively modest and that the trial was never expected to show a benefit on CV mortality. Also, the COVID-19 sensitivity analysis providing more convincing effects is a valid approach and one recommended by regulators.
Further, the findings are supported by independent evidence in chronic kidney disease, from the PIVOTAL trial, that intravenous iron reduces HF hospitalizations, McMurray said.
“The million-dollar question, of course, is what will the results of this study mean for the guidelines: I think they probably will change the guidelines,” he said. “Certainly, I hope they will change the US guidelines, which have really given a very lukewarm recommendation for intravenous iron and I think that should probably be stronger.”
In a class IIb recommendation, the 2017 American College of Cardiology/AHA/Heart Failure Society of America heart failure guidelines say intravenous iron “might be reasonable” to improve functional status and quality of life in New York Heart Association class II and III patients with iron deficiency.
The 2016 European Society of Cardiology guidelines include a class IIa recommendation that IV iron “should be considered” in iron-deficient patients with symptomatic HF with reduced ejection fraction.
“This is the first large-scale [trial] of IV supplementation that could potentially change the way we approach patients, particularly those with hospitalized heart failure,” past AHA president Clyde Yancy, MD, MSc, Northwestern University Feinberg School of Medicine in Chicago, said during an earlier press briefing.

He pointed out that clinicians have been circumspect about the early IV iron data. “I have to congratulate you because you’ve changed the narrative,” Yancy said. “We have to start thinking about iron deficiency; we have to think about how we incorporate this in treatment protocols.”
Press briefing panelist Marc Pfeffer, MD, PhD, Brigham and Women’s Hospital and Harvard Medical School in Boston, acknowledged he was among those circumspect.
“I’m no longer a skeptic and I want to congratulate them for showing it’s a risk factor,” he said. “It’s one thing to have a risk factor; it’s another to be a modifiable risk factor and I think that’s what’s so exciting about this.”
The double-blind, phase 4 AFFIRM-AHF trial randomly assigned 1132 patients to receive a bolus injection of ferric carboxymaltose or normal saline before hospital discharge for an acute HF episode. Subsequent treatment was given, as needed, up to 24 weeks post-randomization.
At admission, all patients had left ventricular ejection fractions less than 50% and iron deficiency (serum ferritin <100 ng/mL or serum ferritin 100-299 ng/mL if transferrin saturation <20%).
The modified intention-to-treat (mITT) analysis included 558 FCM patients and 550 controls in whom study treatment was started and for whom at least one post-randomization value was available.
Press briefing discussant Nancy Sweitzer, MD, PhD, director of the University of Arizona’s Sarver Heart Center in Tucson, said AFFIRM-AHF is an “important trial likely to change guidelines” and “targeted one of the highest risk populations we have in heart failure.”
Patients with iron deficiency tend to be elderly with more comorbidities, have longer hospital lengths of stay, and higher readmission rates. “So impacting hospitalizations in this population is incredibly impactful,” she said.
“Awareness and assessment of iron deficiency are an important part of inpatient care of patients with ejection fractions less than or equal to 50% and acute decompensated heart failure, and I think all of us in the community need to pay much more attention to this issue.”
As with any new therapy, there are implementation challenges such as how to monitor patients and deliver the therapy in a cost-effective way, Sweitzer said.
The trial focused on the most vulnerable period for HF patients, but these patients should be rechecked every 3 to 4 months for iron deficiency, Ponikowski observed during the briefing.
“This is a modifiable risk factor,” he said. “We only need to remember, we only need to assess it, and we have a very, very simple tool in our hands. We just need to measure two biomarkers, transferrin saturation and ferritin — that’s all.”
Unanswered questions include the mechanism behind the reduction in hospitalization, the relationship of benefit to hemoglobin levels, and whether there is a differential benefit based on age, presence of ischemia, or sex, especially as women tend to be more severely affected by iron deficiency, Sweitzer said.
During the formal presentation, Ponikowski said the primary endpoint was consistent in subgroup analyses across baseline hemoglobin, estimated glomerular filtration rate, and N-terminal pro-brain natriuretic peptide levels, HF etiology, ejection fraction, and whether HF was diagnosed prior to the index hospitalization.
Treatment with FCM was safe, with no significant differences between the FCM and placebo groups in serious adverse events (45% vs 51%) or adverse events leading to study discontinuation (18% vs 17%), he reported. The most common adverse events were cardiac disorders (40.1% vs 44.3%) and infections (18.2% vs 22%).
AFFIRM-AHF is the first of three ongoing mortality and morbidity trials in heart failure with intravenous ferric carboxymaltose; the others are FAIR-HF2 and HEART-FID. Additional insights are also expected next year on intravenous iron isomaltoside from the Scottish-based IRONMAN trial in 1300 HF patients with iron deficiency.
The study was sponsored by Vifor International. Ponikowski has received research grants and personal fees from Vifor Pharma; and personal fees from Amgen, Bayer, Novartis, Abbott Vascular, Boehringer Ingelheim, Merck, Pfizer, Servier, AstraZeneca, Berlin Chemie, Cibiem, Renal Guard Solutions Bristol-Myers Squibb, and Impulse Dynamics.
Pfeffer reported honoraria from AstraZeneca, Corvidia, GlaxoSmithKline, Jazz, MyoKardia, Novartis, Roche, Sanofi, and Servier; other relationships with DalCor and Novo Nordisk; research grants from Novartis; and an ownership interest in DalCor. Sweitzer reported research payments from Merck and Novartis; and consulting fees from Myocardia.
McMurray reported relationships with Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Cytokinetics, Novartis, and Servier. Yancy reported a relationship with Abbott and JAMA Network.
Lancet. Published online November 13, 2020. Full text
American Heart Association Scientific Sessions 2020: Presented November 13, 2020.
A version of this article originally appeared on Medscape.com.
Iron supplementation reduces heart failure (HF) readmissions in iron-deficient patients hospitalized for acute HF, according to results of the AFFIRM-AHF trial.
After 52 weeks, intravenous ferric carboxymaltose (Ferinject) reduced the risk of total HF hospitalizations and cardiovascular (CV) death by 21% compared with placebo (293 vs 372 events; rate ratio [RR] 0.79; 95% CI, 0.62 - 1.01).
Although the composite primary endpoint failed to achieve statistical significance, it was driven by a significant 26% reduction in the risk of total HF hospital readmissions (P = .013) without an effect on CV mortality (P =.809).
Because the management and follow-up of patients was affected by the COVID-19 pandemic, a prespecified sensitivity analysis was performed that censored patients in each country at the date when its first COVID-19 patient was reported, explained principal investigator Piotr Ponikowski, MD, PhD, Wroclaw Medical University, Wroclaw, Poland.
That analysis revealed a significant 30% reduction in total HF readmissions (P = .005) in patients receiving ferric carboxymaltose (FCM), as well as significant benefits on the primary composite and secondary endpoints.
Notably, 80% of patients required only one or two injections and HF hospitalizations were reduced irrespective of anemia status.
“Iron deficiency should be searched in patients hospitalized with acute heart failure — assessed using a simple blood test — and is now an important therapeutic target,” Ponikowski said at the virtual American Heart Association (AHA) Scientific Sessions 2020.
The results were also published simultaneously in The Lancet.
Iron deficiency is present in up to 70% of patients with acute HF and a predictor of poor outcome, independent of anemia and ejection fraction, he noted.
The FAIR-HF, CONFIRM-HF, and EFFECT-HF trials demonstrated that IV iron supplementation improves exercise capacity, symptoms, and quality of life in iron-deficient HF patients.
However, no such benefit was seen with oral IV in the IRONOUT trial. “So it seems if we are to replace iron, it needs to be done using intravenous therapy,” said John McMurray, MD, University of Glasgow, Scotland, who was invited to discuss the results.
He observed that the reduction in HF hospitalizations in AFFIRM-AHF were relatively modest and that the trial was never expected to show a benefit on CV mortality. Also, the COVID-19 sensitivity analysis providing more convincing effects is a valid approach and one recommended by regulators.
Further, the findings are supported by independent evidence in chronic kidney disease, from the PIVOTAL trial, that intravenous iron reduces HF hospitalizations, McMurray said.
“The million-dollar question, of course, is what will the results of this study mean for the guidelines: I think they probably will change the guidelines,” he said. “Certainly, I hope they will change the US guidelines, which have really given a very lukewarm recommendation for intravenous iron and I think that should probably be stronger.”
In a class IIb recommendation, the 2017 American College of Cardiology/AHA/Heart Failure Society of America heart failure guidelines say intravenous iron “might be reasonable” to improve functional status and quality of life in New York Heart Association class II and III patients with iron deficiency.
The 2016 European Society of Cardiology guidelines include a class IIa recommendation that IV iron “should be considered” in iron-deficient patients with symptomatic HF with reduced ejection fraction.
“This is the first large-scale [trial] of IV supplementation that could potentially change the way we approach patients, particularly those with hospitalized heart failure,” past AHA president Clyde Yancy, MD, MSc, Northwestern University Feinberg School of Medicine in Chicago, said during an earlier press briefing.
He pointed out that clinicians have been circumspect about the early IV iron data. “I have to congratulate you because you’ve changed the narrative,” Yancy said. “We have to start thinking about iron deficiency; we have to think about how we incorporate this in treatment protocols.”
Press briefing panelist Marc Pfeffer, MD, PhD, Brigham and Women’s Hospital and Harvard Medical School in Boston, acknowledged he was among those circumspect.
“I’m no longer a skeptic and I want to congratulate them for showing it’s a risk factor,” he said. “It’s one thing to have a risk factor; it’s another to be a modifiable risk factor and I think that’s what’s so exciting about this.”
The double-blind, phase 4 AFFIRM-AHF trial randomly assigned 1132 patients to receive a bolus injection of ferric carboxymaltose or normal saline before hospital discharge for an acute HF episode. Subsequent treatment was given, as needed, up to 24 weeks post-randomization.
At admission, all patients had left ventricular ejection fractions less than 50% and iron deficiency (serum ferritin <100 ng/mL or serum ferritin 100-299 ng/mL if transferrin saturation <20%).
The modified intention-to-treat (mITT) analysis included 558 FCM patients and 550 controls in whom study treatment was started and for whom at least one post-randomization value was available.
Press briefing discussant Nancy Sweitzer, MD, PhD, director of the University of Arizona’s Sarver Heart Center in Tucson, said AFFIRM-AHF is an “important trial likely to change guidelines” and “targeted one of the highest risk populations we have in heart failure.”
Patients with iron deficiency tend to be elderly with more comorbidities, have longer hospital lengths of stay, and higher readmission rates. “So impacting hospitalizations in this population is incredibly impactful,” she said.
“Awareness and assessment of iron deficiency are an important part of inpatient care of patients with ejection fractions less than or equal to 50% and acute decompensated heart failure, and I think all of us in the community need to pay much more attention to this issue.”
As with any new therapy, there are implementation challenges such as how to monitor patients and deliver the therapy in a cost-effective way, Sweitzer said.
The trial focused on the most vulnerable period for HF patients, but these patients should be rechecked every 3 to 4 months for iron deficiency, Ponikowski observed during the briefing.
“This is a modifiable risk factor,” he said. “We only need to remember, we only need to assess it, and we have a very, very simple tool in our hands. We just need to measure two biomarkers, transferrin saturation and ferritin — that’s all.”
Unanswered questions include the mechanism behind the reduction in hospitalization, the relationship of benefit to hemoglobin levels, and whether there is a differential benefit based on age, presence of ischemia, or sex, especially as women tend to be more severely affected by iron deficiency, Sweitzer said.
During the formal presentation, Ponikowski said the primary endpoint was consistent in subgroup analyses across baseline hemoglobin, estimated glomerular filtration rate, and N-terminal pro-brain natriuretic peptide levels, HF etiology, ejection fraction, and whether HF was diagnosed prior to the index hospitalization.
Treatment with FCM was safe, with no significant differences between the FCM and placebo groups in serious adverse events (45% vs 51%) or adverse events leading to study discontinuation (18% vs 17%), he reported. The most common adverse events were cardiac disorders (40.1% vs 44.3%) and infections (18.2% vs 22%).
AFFIRM-AHF is the first of three ongoing mortality and morbidity trials in heart failure with intravenous ferric carboxymaltose; the others are FAIR-HF2 and HEART-FID. Additional insights are also expected next year on intravenous iron isomaltoside from the Scottish-based IRONMAN trial in 1300 HF patients with iron deficiency.
The study was sponsored by Vifor International. Ponikowski has received research grants and personal fees from Vifor Pharma; and personal fees from Amgen, Bayer, Novartis, Abbott Vascular, Boehringer Ingelheim, Merck, Pfizer, Servier, AstraZeneca, Berlin Chemie, Cibiem, Renal Guard Solutions Bristol-Myers Squibb, and Impulse Dynamics.
Pfeffer reported honoraria from AstraZeneca, Corvidia, GlaxoSmithKline, Jazz, MyoKardia, Novartis, Roche, Sanofi, and Servier; other relationships with DalCor and Novo Nordisk; research grants from Novartis; and an ownership interest in DalCor. Sweitzer reported research payments from Merck and Novartis; and consulting fees from Myocardia.
McMurray reported relationships with Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Cytokinetics, Novartis, and Servier. Yancy reported a relationship with Abbott and JAMA Network.
Lancet. Published online November 13, 2020. Full text
American Heart Association Scientific Sessions 2020: Presented November 13, 2020.
A version of this article originally appeared on Medscape.com.
Iron supplementation reduces heart failure (HF) readmissions in iron-deficient patients hospitalized for acute HF, according to results of the AFFIRM-AHF trial.
After 52 weeks, intravenous ferric carboxymaltose (Ferinject) reduced the risk of total HF hospitalizations and cardiovascular (CV) death by 21% compared with placebo (293 vs 372 events; rate ratio [RR] 0.79; 95% CI, 0.62 - 1.01).
Although the composite primary endpoint failed to achieve statistical significance, it was driven by a significant 26% reduction in the risk of total HF hospital readmissions (P = .013) without an effect on CV mortality (P =.809).
Because the management and follow-up of patients was affected by the COVID-19 pandemic, a prespecified sensitivity analysis was performed that censored patients in each country at the date when its first COVID-19 patient was reported, explained principal investigator Piotr Ponikowski, MD, PhD, Wroclaw Medical University, Wroclaw, Poland.
That analysis revealed a significant 30% reduction in total HF readmissions (P = .005) in patients receiving ferric carboxymaltose (FCM), as well as significant benefits on the primary composite and secondary endpoints.
Notably, 80% of patients required only one or two injections and HF hospitalizations were reduced irrespective of anemia status.
“Iron deficiency should be searched in patients hospitalized with acute heart failure — assessed using a simple blood test — and is now an important therapeutic target,” Ponikowski said at the virtual American Heart Association (AHA) Scientific Sessions 2020.
The results were also published simultaneously in The Lancet.
Iron deficiency is present in up to 70% of patients with acute HF and a predictor of poor outcome, independent of anemia and ejection fraction, he noted.
The FAIR-HF, CONFIRM-HF, and EFFECT-HF trials demonstrated that IV iron supplementation improves exercise capacity, symptoms, and quality of life in iron-deficient HF patients.
However, no such benefit was seen with oral IV in the IRONOUT trial. “So it seems if we are to replace iron, it needs to be done using intravenous therapy,” said John McMurray, MD, University of Glasgow, Scotland, who was invited to discuss the results.
He observed that the reduction in HF hospitalizations in AFFIRM-AHF were relatively modest and that the trial was never expected to show a benefit on CV mortality. Also, the COVID-19 sensitivity analysis providing more convincing effects is a valid approach and one recommended by regulators.
Further, the findings are supported by independent evidence in chronic kidney disease, from the PIVOTAL trial, that intravenous iron reduces HF hospitalizations, McMurray said.
“The million-dollar question, of course, is what will the results of this study mean for the guidelines: I think they probably will change the guidelines,” he said. “Certainly, I hope they will change the US guidelines, which have really given a very lukewarm recommendation for intravenous iron and I think that should probably be stronger.”
In a class IIb recommendation, the 2017 American College of Cardiology/AHA/Heart Failure Society of America heart failure guidelines say intravenous iron “might be reasonable” to improve functional status and quality of life in New York Heart Association class II and III patients with iron deficiency.
The 2016 European Society of Cardiology guidelines include a class IIa recommendation that IV iron “should be considered” in iron-deficient patients with symptomatic HF with reduced ejection fraction.
“This is the first large-scale [trial] of IV supplementation that could potentially change the way we approach patients, particularly those with hospitalized heart failure,” past AHA president Clyde Yancy, MD, MSc, Northwestern University Feinberg School of Medicine in Chicago, said during an earlier press briefing.
He pointed out that clinicians have been circumspect about the early IV iron data. “I have to congratulate you because you’ve changed the narrative,” Yancy said. “We have to start thinking about iron deficiency; we have to think about how we incorporate this in treatment protocols.”
Press briefing panelist Marc Pfeffer, MD, PhD, Brigham and Women’s Hospital and Harvard Medical School in Boston, acknowledged he was among those circumspect.
“I’m no longer a skeptic and I want to congratulate them for showing it’s a risk factor,” he said. “It’s one thing to have a risk factor; it’s another to be a modifiable risk factor and I think that’s what’s so exciting about this.”
The double-blind, phase 4 AFFIRM-AHF trial randomly assigned 1132 patients to receive a bolus injection of ferric carboxymaltose or normal saline before hospital discharge for an acute HF episode. Subsequent treatment was given, as needed, up to 24 weeks post-randomization.
At admission, all patients had left ventricular ejection fractions less than 50% and iron deficiency (serum ferritin <100 ng/mL or serum ferritin 100-299 ng/mL if transferrin saturation <20%).
The modified intention-to-treat (mITT) analysis included 558 FCM patients and 550 controls in whom study treatment was started and for whom at least one post-randomization value was available.
Press briefing discussant Nancy Sweitzer, MD, PhD, director of the University of Arizona’s Sarver Heart Center in Tucson, said AFFIRM-AHF is an “important trial likely to change guidelines” and “targeted one of the highest risk populations we have in heart failure.”
Patients with iron deficiency tend to be elderly with more comorbidities, have longer hospital lengths of stay, and higher readmission rates. “So impacting hospitalizations in this population is incredibly impactful,” she said.
“Awareness and assessment of iron deficiency are an important part of inpatient care of patients with ejection fractions less than or equal to 50% and acute decompensated heart failure, and I think all of us in the community need to pay much more attention to this issue.”
As with any new therapy, there are implementation challenges such as how to monitor patients and deliver the therapy in a cost-effective way, Sweitzer said.
The trial focused on the most vulnerable period for HF patients, but these patients should be rechecked every 3 to 4 months for iron deficiency, Ponikowski observed during the briefing.
“This is a modifiable risk factor,” he said. “We only need to remember, we only need to assess it, and we have a very, very simple tool in our hands. We just need to measure two biomarkers, transferrin saturation and ferritin — that’s all.”
Unanswered questions include the mechanism behind the reduction in hospitalization, the relationship of benefit to hemoglobin levels, and whether there is a differential benefit based on age, presence of ischemia, or sex, especially as women tend to be more severely affected by iron deficiency, Sweitzer said.
During the formal presentation, Ponikowski said the primary endpoint was consistent in subgroup analyses across baseline hemoglobin, estimated glomerular filtration rate, and N-terminal pro-brain natriuretic peptide levels, HF etiology, ejection fraction, and whether HF was diagnosed prior to the index hospitalization.
Treatment with FCM was safe, with no significant differences between the FCM and placebo groups in serious adverse events (45% vs 51%) or adverse events leading to study discontinuation (18% vs 17%), he reported. The most common adverse events were cardiac disorders (40.1% vs 44.3%) and infections (18.2% vs 22%).
AFFIRM-AHF is the first of three ongoing mortality and morbidity trials in heart failure with intravenous ferric carboxymaltose; the others are FAIR-HF2 and HEART-FID. Additional insights are also expected next year on intravenous iron isomaltoside from the Scottish-based IRONMAN trial in 1300 HF patients with iron deficiency.
The study was sponsored by Vifor International. Ponikowski has received research grants and personal fees from Vifor Pharma; and personal fees from Amgen, Bayer, Novartis, Abbott Vascular, Boehringer Ingelheim, Merck, Pfizer, Servier, AstraZeneca, Berlin Chemie, Cibiem, Renal Guard Solutions Bristol-Myers Squibb, and Impulse Dynamics.
Pfeffer reported honoraria from AstraZeneca, Corvidia, GlaxoSmithKline, Jazz, MyoKardia, Novartis, Roche, Sanofi, and Servier; other relationships with DalCor and Novo Nordisk; research grants from Novartis; and an ownership interest in DalCor. Sweitzer reported research payments from Merck and Novartis; and consulting fees from Myocardia.
McMurray reported relationships with Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Cytokinetics, Novartis, and Servier. Yancy reported a relationship with Abbott and JAMA Network.
Lancet. Published online November 13, 2020. Full text
American Heart Association Scientific Sessions 2020: Presented November 13, 2020.
A version of this article originally appeared on Medscape.com.
FROM AHA 2020
Escalate HIV adherence strategies amid COVID-19
"The writing is on the wall” that virtual care is not meeting the needs of people with HIV who struggled with viral suppression even before the COVID-19 pandemic, said Jason Farley, PhD, ANP-BC, AACRN, associate professor of nursing at Johns Hopkins University, Baltimore. So it’s time for HIV care teams, especially clinics in the Ryan White HIV/AIDS Program, to get creative in bringing wraparound services to patients.
That may mean reallocating the workforce so that one person serves as a community health worker. Or it could mean increasing texts and video calls; helping patients find online support groups to address problems with alcohol or drug use; and conducting an overall assessment of patients’ needs as the pandemic continues.
“The virtual patient-centered medical home may be the new normal after COVID-19, and we have to be thinking about how we use this model with patients for whom it works, but supplement this model in patients that it does not,” Farley said at the virtual Association of Nurses in AIDS Care (ANAC) 2020 Annual Meeting. That work “is essential to our being able to facilitate the best patient outcomes possible.”
Early data, tiered interventions
Farley referred to an article published in September in the Journal AIDS that confirmed unpublished data mentioned at the International AIDS Conference 2020. The article reported that viral suppression rates among people with HIV who attended San Francisco’s Ward 86 HIV clinic dropped by 31% from pre-COVID levels.
Of the 1766 people who attended the clinic, about 1 in 5 had detectable HIV viral loads at any point in 2019. But that rate was 31% higher after shelter-in-place orders were issued. And although patients participated in telemedicine visits at more or less the same rate before and after the pandemic (31% vs. 30% no-shows), viral suppression rates dropped. The impact was especially acute for homeless individuals.
“This destabilization occurred despite our population attending telemedicine visits at a higher rate than expected, given the 60% drop in ambulatory care visit volume nationwide,” the authors stated in their article. “Telehealth visits, while offering greater patient convenience, may lead to less access to clinic-based social support services essential to achieving viral suppression among vulnerable groups.”
That’s the challenge HIV clinics now face, Farley said at the ANAC meeting.
He suggested a differentiated care approach in which there are four tiers of care, starting with the standard level of outreach, which may include email, electronic health record blasts, and robo-calls to remind people of their appointments and to refill their medications. Those with sustained viral suppression may only need 90-day automatic refills of their medications. Those who are vulnerable to nonadherence may need to be contacted weekly or more often by the clinic. Such contact could be made by a social worker, a community health worker, or through some form of virtual support.
Patients at tier 4, who have labile viral suppression, need far more than that. These are the 15% of patients with HIV who struggled with viral suppression before the pandemic. They are the patients that Farley’s team focuses on at Baltimore’s John G. Bartlett Specialty Clinic for Infectious Disease.
“We’ve completely deconstructed the patient-centered medical home,” he said of the early move to virtual care. He suggested that clinicians assess their services and ask themselves some questions:
- Has someone on the team reached out to every patient and checked in to see what their biggest needs are, medical or not, during the pandemic? Have they assessed the patient’s ability to receive video calls or text messages?
- How have group-support programs that address stigma or the social determinants of health fared in the transition to virtual medicine?
- Are patients who are in recovery being supported in order that they may engage with recovery programs online?
- How well have counseling services done in engaging people in virtual care? Currently, given the overall increase in mental health challenges during the pandemic, one would expect that the use of mental health counseling is increasing. “If they’re stagnant or going down, someone needs to be reflecting on that issue internally in the clinic,” he said.
- Are patients being contacted regarding the effects that isolation is having on their lives? “The things that would normally allow us to self-mitigate and self-manage these conditions, like going to the gym, meeting with friends, religious services – all of those are being cut,” he said.
- Is there an early alert from an in-person pharmacy to trigger outreach via a community health worker for patients who haven’t picked up their medications in a week or more?
Farley pointed to a 2015 model for an enhanced e-health approach to chronic care management that called for e-support from the community and that was enhanced through virtual communities.
These are some of the approaches Farley has taken at his clinic. He leads a team that focuses specifically on patients who struggled with engagement before the pandemic. Through a grant from the US Department of Health & Human Services’ Health Resources and Services Administration – even before the pandemic – that team has been funding community health workers who have multiple contacts with patients online and virtually and are able to offer what he calls “unapologetically enabling” support for patients so that they are able to focus on their health.
He gave the following example. Before the pandemic, a community health worker on the team had been working with a patient who showed up at every scheduled visit and swore that she was taking her medications, although clearly she was not. A community health worker, who was made available through the grant, was able to recognize that the patient’s biggest challenge in her life was providing childcare for her special-needs child. The community health worker worked with the patient for months to find stable childcare for the child, paid 2 months of rent for the patient so that she would not become homeless, and helped her find transitional housing. When the pandemic hit, the community health worker was already texting and conducting video calls with the patient regularly.
For the past 9 months, that patient has had an undetectable viral load, Farley said.
“Nine months during a pandemic,” Farley reiterated, “and the community health worker keeps working with her, keeps meeting with her.”
Stigma on stigma
The need for this level of support from the clinic may be even more important for people with HIV who acquire COVID-19, said Orlando Harris, PhD, assistant professor of community health systems at the University of California, San Francisco, (UCSF) School of Nursing. HIV-related stigma is a well-known deterrent to care for people living with the virus. During the presentation, Harris asked Farley about the impact of COVID-19 stigma on people with both HIV and COVID-19.
Farley said that patients at his clinic have told him that they have “ostracized” friends who have tested positive for COVID-19. Harris remembered a person with HIV who participated in one of his trials telling the researchers that despite all his precautions – wearing a mask, staying socially distant – he still acquired COVID-19. There was nothing he could have done, Harris said, other than just not go to the grocery store.
The fear of contracting another disease that is associated with stigma, as well as the need to disclose it, can inflame memories of the trauma of being diagnosed with HIV, Harris said. And with patient-centered medical homes struggling to reconstitute their wraparound services via telehealth, he said he wonders whether clinicians should be doing more.
“I worry about people who have survived being diagnosed with HIV in the ‘80s and the ‘90s before antiretroviral therapy showed up on the scene,” he told Medscape Medical News. “I worry that the folks that survived one pandemic [may] be feeling fearful or living in that fear that this new pandemic might take them out. That’s why I’m stressing the need for us to really consider, as clinicians and also as researchers the support systems, the coping mechanisms, the counseling, or what have you to support those living with HIV and vulnerable to COVID-19.”
During telehealth visits, that can be achieved simply by asking people how they are really doing and what their coping mechanisms are.
For their part, the clinicians at San Francisco’s Ward 86 are not trying to provide that support through telehealth on the same level as they were at the beginning of the pandemic, said Matthew Spinelli, MD, assistant professor of medicine, and Monica Gandhi, MD, associate chief of the Division of HIV, Infectious Diseases and Global Medicine, who are both at UCSF and are coauthors of the study.
They still offer telemedicine appointments to patients who request them, said Spinelli. He said about one-third of his patients still prefer to receive their care virtually. The rest have gone back to face-to-face support.
“The analysis led us to promptly open up care as much as possible to our patients, with the idea that telehealth is not cutting it for vulnerable patients with HIV,” Gandhi told Medscape Medical News via email. “We don’t think it’s right for a population who relies on social support from the clinic.”
This article first appeared on Medscape.com.
"The writing is on the wall” that virtual care is not meeting the needs of people with HIV who struggled with viral suppression even before the COVID-19 pandemic, said Jason Farley, PhD, ANP-BC, AACRN, associate professor of nursing at Johns Hopkins University, Baltimore. So it’s time for HIV care teams, especially clinics in the Ryan White HIV/AIDS Program, to get creative in bringing wraparound services to patients.
That may mean reallocating the workforce so that one person serves as a community health worker. Or it could mean increasing texts and video calls; helping patients find online support groups to address problems with alcohol or drug use; and conducting an overall assessment of patients’ needs as the pandemic continues.
“The virtual patient-centered medical home may be the new normal after COVID-19, and we have to be thinking about how we use this model with patients for whom it works, but supplement this model in patients that it does not,” Farley said at the virtual Association of Nurses in AIDS Care (ANAC) 2020 Annual Meeting. That work “is essential to our being able to facilitate the best patient outcomes possible.”
Early data, tiered interventions
Farley referred to an article published in September in the Journal AIDS that confirmed unpublished data mentioned at the International AIDS Conference 2020. The article reported that viral suppression rates among people with HIV who attended San Francisco’s Ward 86 HIV clinic dropped by 31% from pre-COVID levels.
Of the 1766 people who attended the clinic, about 1 in 5 had detectable HIV viral loads at any point in 2019. But that rate was 31% higher after shelter-in-place orders were issued. And although patients participated in telemedicine visits at more or less the same rate before and after the pandemic (31% vs. 30% no-shows), viral suppression rates dropped. The impact was especially acute for homeless individuals.
“This destabilization occurred despite our population attending telemedicine visits at a higher rate than expected, given the 60% drop in ambulatory care visit volume nationwide,” the authors stated in their article. “Telehealth visits, while offering greater patient convenience, may lead to less access to clinic-based social support services essential to achieving viral suppression among vulnerable groups.”
That’s the challenge HIV clinics now face, Farley said at the ANAC meeting.
He suggested a differentiated care approach in which there are four tiers of care, starting with the standard level of outreach, which may include email, electronic health record blasts, and robo-calls to remind people of their appointments and to refill their medications. Those with sustained viral suppression may only need 90-day automatic refills of their medications. Those who are vulnerable to nonadherence may need to be contacted weekly or more often by the clinic. Such contact could be made by a social worker, a community health worker, or through some form of virtual support.
Patients at tier 4, who have labile viral suppression, need far more than that. These are the 15% of patients with HIV who struggled with viral suppression before the pandemic. They are the patients that Farley’s team focuses on at Baltimore’s John G. Bartlett Specialty Clinic for Infectious Disease.
“We’ve completely deconstructed the patient-centered medical home,” he said of the early move to virtual care. He suggested that clinicians assess their services and ask themselves some questions:
- Has someone on the team reached out to every patient and checked in to see what their biggest needs are, medical or not, during the pandemic? Have they assessed the patient’s ability to receive video calls or text messages?
- How have group-support programs that address stigma or the social determinants of health fared in the transition to virtual medicine?
- Are patients who are in recovery being supported in order that they may engage with recovery programs online?
- How well have counseling services done in engaging people in virtual care? Currently, given the overall increase in mental health challenges during the pandemic, one would expect that the use of mental health counseling is increasing. “If they’re stagnant or going down, someone needs to be reflecting on that issue internally in the clinic,” he said.
- Are patients being contacted regarding the effects that isolation is having on their lives? “The things that would normally allow us to self-mitigate and self-manage these conditions, like going to the gym, meeting with friends, religious services – all of those are being cut,” he said.
- Is there an early alert from an in-person pharmacy to trigger outreach via a community health worker for patients who haven’t picked up their medications in a week or more?
Farley pointed to a 2015 model for an enhanced e-health approach to chronic care management that called for e-support from the community and that was enhanced through virtual communities.
These are some of the approaches Farley has taken at his clinic. He leads a team that focuses specifically on patients who struggled with engagement before the pandemic. Through a grant from the US Department of Health & Human Services’ Health Resources and Services Administration – even before the pandemic – that team has been funding community health workers who have multiple contacts with patients online and virtually and are able to offer what he calls “unapologetically enabling” support for patients so that they are able to focus on their health.
He gave the following example. Before the pandemic, a community health worker on the team had been working with a patient who showed up at every scheduled visit and swore that she was taking her medications, although clearly she was not. A community health worker, who was made available through the grant, was able to recognize that the patient’s biggest challenge in her life was providing childcare for her special-needs child. The community health worker worked with the patient for months to find stable childcare for the child, paid 2 months of rent for the patient so that she would not become homeless, and helped her find transitional housing. When the pandemic hit, the community health worker was already texting and conducting video calls with the patient regularly.
For the past 9 months, that patient has had an undetectable viral load, Farley said.
“Nine months during a pandemic,” Farley reiterated, “and the community health worker keeps working with her, keeps meeting with her.”
Stigma on stigma
The need for this level of support from the clinic may be even more important for people with HIV who acquire COVID-19, said Orlando Harris, PhD, assistant professor of community health systems at the University of California, San Francisco, (UCSF) School of Nursing. HIV-related stigma is a well-known deterrent to care for people living with the virus. During the presentation, Harris asked Farley about the impact of COVID-19 stigma on people with both HIV and COVID-19.
Farley said that patients at his clinic have told him that they have “ostracized” friends who have tested positive for COVID-19. Harris remembered a person with HIV who participated in one of his trials telling the researchers that despite all his precautions – wearing a mask, staying socially distant – he still acquired COVID-19. There was nothing he could have done, Harris said, other than just not go to the grocery store.
The fear of contracting another disease that is associated with stigma, as well as the need to disclose it, can inflame memories of the trauma of being diagnosed with HIV, Harris said. And with patient-centered medical homes struggling to reconstitute their wraparound services via telehealth, he said he wonders whether clinicians should be doing more.
“I worry about people who have survived being diagnosed with HIV in the ‘80s and the ‘90s before antiretroviral therapy showed up on the scene,” he told Medscape Medical News. “I worry that the folks that survived one pandemic [may] be feeling fearful or living in that fear that this new pandemic might take them out. That’s why I’m stressing the need for us to really consider, as clinicians and also as researchers the support systems, the coping mechanisms, the counseling, or what have you to support those living with HIV and vulnerable to COVID-19.”
During telehealth visits, that can be achieved simply by asking people how they are really doing and what their coping mechanisms are.
For their part, the clinicians at San Francisco’s Ward 86 are not trying to provide that support through telehealth on the same level as they were at the beginning of the pandemic, said Matthew Spinelli, MD, assistant professor of medicine, and Monica Gandhi, MD, associate chief of the Division of HIV, Infectious Diseases and Global Medicine, who are both at UCSF and are coauthors of the study.
They still offer telemedicine appointments to patients who request them, said Spinelli. He said about one-third of his patients still prefer to receive their care virtually. The rest have gone back to face-to-face support.
“The analysis led us to promptly open up care as much as possible to our patients, with the idea that telehealth is not cutting it for vulnerable patients with HIV,” Gandhi told Medscape Medical News via email. “We don’t think it’s right for a population who relies on social support from the clinic.”
This article first appeared on Medscape.com.
"The writing is on the wall” that virtual care is not meeting the needs of people with HIV who struggled with viral suppression even before the COVID-19 pandemic, said Jason Farley, PhD, ANP-BC, AACRN, associate professor of nursing at Johns Hopkins University, Baltimore. So it’s time for HIV care teams, especially clinics in the Ryan White HIV/AIDS Program, to get creative in bringing wraparound services to patients.
That may mean reallocating the workforce so that one person serves as a community health worker. Or it could mean increasing texts and video calls; helping patients find online support groups to address problems with alcohol or drug use; and conducting an overall assessment of patients’ needs as the pandemic continues.
“The virtual patient-centered medical home may be the new normal after COVID-19, and we have to be thinking about how we use this model with patients for whom it works, but supplement this model in patients that it does not,” Farley said at the virtual Association of Nurses in AIDS Care (ANAC) 2020 Annual Meeting. That work “is essential to our being able to facilitate the best patient outcomes possible.”
Early data, tiered interventions
Farley referred to an article published in September in the Journal AIDS that confirmed unpublished data mentioned at the International AIDS Conference 2020. The article reported that viral suppression rates among people with HIV who attended San Francisco’s Ward 86 HIV clinic dropped by 31% from pre-COVID levels.
Of the 1766 people who attended the clinic, about 1 in 5 had detectable HIV viral loads at any point in 2019. But that rate was 31% higher after shelter-in-place orders were issued. And although patients participated in telemedicine visits at more or less the same rate before and after the pandemic (31% vs. 30% no-shows), viral suppression rates dropped. The impact was especially acute for homeless individuals.
“This destabilization occurred despite our population attending telemedicine visits at a higher rate than expected, given the 60% drop in ambulatory care visit volume nationwide,” the authors stated in their article. “Telehealth visits, while offering greater patient convenience, may lead to less access to clinic-based social support services essential to achieving viral suppression among vulnerable groups.”
That’s the challenge HIV clinics now face, Farley said at the ANAC meeting.
He suggested a differentiated care approach in which there are four tiers of care, starting with the standard level of outreach, which may include email, electronic health record blasts, and robo-calls to remind people of their appointments and to refill their medications. Those with sustained viral suppression may only need 90-day automatic refills of their medications. Those who are vulnerable to nonadherence may need to be contacted weekly or more often by the clinic. Such contact could be made by a social worker, a community health worker, or through some form of virtual support.
Patients at tier 4, who have labile viral suppression, need far more than that. These are the 15% of patients with HIV who struggled with viral suppression before the pandemic. They are the patients that Farley’s team focuses on at Baltimore’s John G. Bartlett Specialty Clinic for Infectious Disease.
“We’ve completely deconstructed the patient-centered medical home,” he said of the early move to virtual care. He suggested that clinicians assess their services and ask themselves some questions:
- Has someone on the team reached out to every patient and checked in to see what their biggest needs are, medical or not, during the pandemic? Have they assessed the patient’s ability to receive video calls or text messages?
- How have group-support programs that address stigma or the social determinants of health fared in the transition to virtual medicine?
- Are patients who are in recovery being supported in order that they may engage with recovery programs online?
- How well have counseling services done in engaging people in virtual care? Currently, given the overall increase in mental health challenges during the pandemic, one would expect that the use of mental health counseling is increasing. “If they’re stagnant or going down, someone needs to be reflecting on that issue internally in the clinic,” he said.
- Are patients being contacted regarding the effects that isolation is having on their lives? “The things that would normally allow us to self-mitigate and self-manage these conditions, like going to the gym, meeting with friends, religious services – all of those are being cut,” he said.
- Is there an early alert from an in-person pharmacy to trigger outreach via a community health worker for patients who haven’t picked up their medications in a week or more?
Farley pointed to a 2015 model for an enhanced e-health approach to chronic care management that called for e-support from the community and that was enhanced through virtual communities.
These are some of the approaches Farley has taken at his clinic. He leads a team that focuses specifically on patients who struggled with engagement before the pandemic. Through a grant from the US Department of Health & Human Services’ Health Resources and Services Administration – even before the pandemic – that team has been funding community health workers who have multiple contacts with patients online and virtually and are able to offer what he calls “unapologetically enabling” support for patients so that they are able to focus on their health.
He gave the following example. Before the pandemic, a community health worker on the team had been working with a patient who showed up at every scheduled visit and swore that she was taking her medications, although clearly she was not. A community health worker, who was made available through the grant, was able to recognize that the patient’s biggest challenge in her life was providing childcare for her special-needs child. The community health worker worked with the patient for months to find stable childcare for the child, paid 2 months of rent for the patient so that she would not become homeless, and helped her find transitional housing. When the pandemic hit, the community health worker was already texting and conducting video calls with the patient regularly.
For the past 9 months, that patient has had an undetectable viral load, Farley said.
“Nine months during a pandemic,” Farley reiterated, “and the community health worker keeps working with her, keeps meeting with her.”
Stigma on stigma
The need for this level of support from the clinic may be even more important for people with HIV who acquire COVID-19, said Orlando Harris, PhD, assistant professor of community health systems at the University of California, San Francisco, (UCSF) School of Nursing. HIV-related stigma is a well-known deterrent to care for people living with the virus. During the presentation, Harris asked Farley about the impact of COVID-19 stigma on people with both HIV and COVID-19.
Farley said that patients at his clinic have told him that they have “ostracized” friends who have tested positive for COVID-19. Harris remembered a person with HIV who participated in one of his trials telling the researchers that despite all his precautions – wearing a mask, staying socially distant – he still acquired COVID-19. There was nothing he could have done, Harris said, other than just not go to the grocery store.
The fear of contracting another disease that is associated with stigma, as well as the need to disclose it, can inflame memories of the trauma of being diagnosed with HIV, Harris said. And with patient-centered medical homes struggling to reconstitute their wraparound services via telehealth, he said he wonders whether clinicians should be doing more.
“I worry about people who have survived being diagnosed with HIV in the ‘80s and the ‘90s before antiretroviral therapy showed up on the scene,” he told Medscape Medical News. “I worry that the folks that survived one pandemic [may] be feeling fearful or living in that fear that this new pandemic might take them out. That’s why I’m stressing the need for us to really consider, as clinicians and also as researchers the support systems, the coping mechanisms, the counseling, or what have you to support those living with HIV and vulnerable to COVID-19.”
During telehealth visits, that can be achieved simply by asking people how they are really doing and what their coping mechanisms are.
For their part, the clinicians at San Francisco’s Ward 86 are not trying to provide that support through telehealth on the same level as they were at the beginning of the pandemic, said Matthew Spinelli, MD, assistant professor of medicine, and Monica Gandhi, MD, associate chief of the Division of HIV, Infectious Diseases and Global Medicine, who are both at UCSF and are coauthors of the study.
They still offer telemedicine appointments to patients who request them, said Spinelli. He said about one-third of his patients still prefer to receive their care virtually. The rest have gone back to face-to-face support.
“The analysis led us to promptly open up care as much as possible to our patients, with the idea that telehealth is not cutting it for vulnerable patients with HIV,” Gandhi told Medscape Medical News via email. “We don’t think it’s right for a population who relies on social support from the clinic.”
This article first appeared on Medscape.com.
Major breakthrough? Average 10% weight loss with semaglutide
In a phase 3 trial where all participants received intensive behavior therapy, investigational 2.4-mg once-weekly subcutaneous semaglutide (Novo Nordisk) resulted in a 10.3% greater average weight loss than placebo over a period of 68 weeks.
If approved, this medication could be a “potential major breakthrough” in obesity management, the investigators suggested. But other experts urged caution, as cost and uptake are important considerations.
‘Potential weight loss that patients would be happy with’
Thomas A. Wadden, PhD, presented results from the study of 611 adults with overweight or obesity but no diabetes at the virtual ObesityWeek® Interactive 2020 meeting.
“Perhaps even more impressive was the finding that 75% of patients lost 10% or more of baseline body weight,” said Dr. Wadden, of the department of psychiatry at the University of Pennsylvania, Philadelphia.
Moreover, in this trial of semaglutide, a glucagonlike peptide–1 (GLP-1) receptor agonist that is approved for treating type 2 diabetes at a weekly subcutaneous dose of 1 mg, but is being investigated at the higher dose for weight loss – 55% of patients lost ≥15% of their initial weight, and 36% lost ≥20% of their initial weight.
“These large categorical weight losses – particularly of 15% and 20% of initial weight – are potentially a major breakthrough in the management of obesity,” Dr. Wadden said in an interview.
Weight losses of this size, he added, “should confer greater improvements in cardiometabolic risk factors (such as hypertension, sleep apnea, and type 2 diabetes) as compared with losses of 5%-10% achieved with current behavioral or pharmacological approaches.” And patients are generally not satisfied with losses of less than 10% of initial weight when participating in intensive behavior programs or taking weight-loss medications.
Now, “the larger categorical weight losses will mean that a greater number of patients with obesity will be able to achieve a weight loss with which they are ... happy,” Dr. Wadden said in an interview.
According to Louis J. Aronne, MD, Weill Professor of Metabolic Research, Weill Cornell Medicine, New York, who is an investigator for another trial of semaglutide: “Even though it has the same mechanism of action [as liraglutide], the weight loss is two or more times greater [with semaglutide]. In my opinion, it’s really going to be a major advance in the treatment of obesity.”
In the discussion that followed the virtual presentation, one attendee asked about potential weight regain if a patient stopped taking the drug. Based on experience with another subcutaneous injectable GLP-1 receptor agonist, liraglutide (Saxenda), already approved for obesity, it may be that taking medicine for chronic overweight may become like taking a statin for elevated cholesterol, said Dr. Wadden.
Novo Nordisk has now completed the four trials in the STEP (Semaglutide Treatment Effect in People With Obesity) global phase 3 clinical development program, and plans to file applications with the Food and Drug Administration later this year and with the European Medicines Agency in early 2021 for review of semaglutide 2.4 mg for weight management.
“Fundamental issues need to be figured out”
Invited to comment, Scott Kahan, MD, said: “This is impressive data, confirming that semaglutide, particularly when used in concert with evidence-based counseling, is a highly effective agent for obesity management.”
However, “the real question, though, is what comes next,” stressed Dr. Kahan, director of the National Center for Weight and Wellness, Washington, DC.
“Will it be approved by the U.S. FDA? I believe so,” he said in an interview. “Yet we already have several effective obesity medications approved over the past decade – all of which are rarely used and therefore make little impact for patients in the real world.”
“Will there be insurance coverage, and therefore practical access for those who could most benefit?” he continued. “Will prescribers counsel their patients about obesity management, including the use of effective medications? Will patients utilize available options?”
“These and other fundamental issues must be figured out before we anoint any treatment option as a meaningful step forward, let alone a transformative development,” according to Dr. Kahan.
Similarly, Irl B. Hirsch, MD, stressed that, should this medication be approved for weight loss, cost would be a major factor in its uptake.
“I’m old enough to recall when we started using lovastatin in the late 1980s,” Dr. Hirsch, professor of medicine, University of Washington Medicine Diabetes Institute, Seattle, said in an interview.
“We used it without the type of evidence of statin use we have today. A pill, but in those days the statins were expensive. But over time, the evidence for statins grew and over the next 15 years it was quite clear that for both primary prevention (for those with diabetes) and secondary intervention these drugs needed to be used by millions of people. These recommendations became easier once the drugs became generic.
“Will the same thing happen for GLP-1 agonists? The problem is we need both ‘hard-outcome data’ [such as 3-point major adverse cardiovascular events] and more reasonable cost before we see this expanding to an entire population.
“In the future perhaps we could have a biosimilar GLP-1 agonist that would be more affordable than what we pay now, but even before that we need agreement from our reimbursement thought leaders that our society should reimburse these agents.
“My thinking now is the cost-benefit could be favorable, but this is all dependent on what happens to the cost of the drugs over time,” he said.
Additive effect of intensive behavior therapy plus medication
Dr. Wadden explained that intensive behavioral therapy “provides 14 or more counseling sessions in 6 months to modify diet and physical activity, through the patients’ use of behavioral strategies (such as keeping daily food and activity diaries).”
Such programs typically produce mean weight loss of 5%-8% of initial weight; less frequent (e.g., monthly) programs typically produce weight loss of only 1%-3%.
Prior studies suggest that intensive behavioral therapy and medication have additive effects. To investigate this, Dr. Wadden and colleagues randomized 611 adults (81% women) who were a mean age of 46 years and had a mean body mass index of 38 kg/m2.
All participants received 30 intensive behavior therapy sessions provided by a registered dietitian (or other qualified provider), which typically lasted 20-30 minutes and were given weekly for 12 weeks, every other week for the next 12 weeks, and then monthly.
The dietitian gave participants behavioral strategies to help them adhere to diet and physical activity goals.
During the first 8 weeks, participants were provided with a 1,000-1,200 kcal/day meal replacement diet that included liquid shakes, meal bars, and prepared entrees designed to facilitate a large initial weight loss.
They then transitioned to a diet of conventional foods (of their choosing), with a goal of 1,200-1,800 kcal/day based on body weight.
The physical activity goal was 100 minutes/week of walking or other aerobic activity in the first month, building up to 200 minutes/week by month 6.
‘More effective than current FDA-approved weight-loss medications’
At week 68, mean body weight decreased from baseline by 16.0% in the semaglutide group versus 5.7% in the placebo group (P < .0001).
In this trial, where all participants received extensive intensive behavior therapy, more participants had weight loss ≥5%, ≥10%, ≥15%, and ≥20% of their initial weight with semaglutide versus placebo (87% vs. 48%; 75% vs. 27%; 56% vs. 13%; 36% vs. 4%, respectively; all P < .0001).
From baseline to week 68, the proportion of participants with prediabetes decreased from 48% to 7% in the semaglutide group and from 53% to 26% in the placebo group.
Patients who received semaglutide had greater improvements in lipids, too.
Although the weight loss was 10.3% (10.6 kg) greater with semaglutide, Dr. Wadden noted, “additional studies have shown this net benefit to be as great as 11%-12%, which would make semaglutide 2.4 mg more effective than current [FDA-approved] weight-loss medications.”
“Naltrexone-bupropion (Contrave) with lifestyle counseling, for example,” he continued, “produces a loss that is 5 kg greater than lifestyle counseling plus placebo, liraglutide 3.0 mg (Saxenda) a loss 5.3 kg greater than placebo, and phentermine-topiramate (Qsymia) a loss that is 8.8 kg greater than placebo.”
Semaglutide was well tolerated. Gastrointestinal adverse events, the most common type, occurred in 83% of patients in the semaglutide group and 63% of patients in the placebo group.
Nausea, as well as constipation and diarrhea, are common in medications that increase GLP-1 levels, Dr. Wadden noted. Side effects can be managed by slowly increasing the medication dose over 4 months.
Dr. Wadden expects that, if approved, semaglutide 2.4 mg subcutaneous once-weekly will be recommended as an adjunct to a reduced calorie diet and increased physical activity. Additional studies suggest that monthly counseling should be sufficient to obtain similar weight losses as those seen in the current trial, which had more intensive counseling.
As well as being approved as a weekly subcutaneous injectable treatment for type 2 diabetes, semaglutide is also approved as an once-daily oral agent for the same indication (Rybelsus, Novo Nordisk) in doses of 7 mg and 14 mg to improve glycemic control along with diet and exercise. It is the first GLP-1 agonist available in tablet form.
Dr. Wadden serves on scientific advisory boards for Novo Nordisk and WW (formerly Weight Watchers), and has received grant support, on behalf of the University of Pennsylvania, from Novo Nordisk. Dr. Aronne is an investigator in a long-term trial of semaglutide and has served on scientific advisory boards for Novo Nordisk in the past. He also has other industry relationships that are not related to semaglutide.
A version of this article originally appeared on Medscape.com.
In a phase 3 trial where all participants received intensive behavior therapy, investigational 2.4-mg once-weekly subcutaneous semaglutide (Novo Nordisk) resulted in a 10.3% greater average weight loss than placebo over a period of 68 weeks.
If approved, this medication could be a “potential major breakthrough” in obesity management, the investigators suggested. But other experts urged caution, as cost and uptake are important considerations.
‘Potential weight loss that patients would be happy with’
Thomas A. Wadden, PhD, presented results from the study of 611 adults with overweight or obesity but no diabetes at the virtual ObesityWeek® Interactive 2020 meeting.
“Perhaps even more impressive was the finding that 75% of patients lost 10% or more of baseline body weight,” said Dr. Wadden, of the department of psychiatry at the University of Pennsylvania, Philadelphia.
Moreover, in this trial of semaglutide, a glucagonlike peptide–1 (GLP-1) receptor agonist that is approved for treating type 2 diabetes at a weekly subcutaneous dose of 1 mg, but is being investigated at the higher dose for weight loss – 55% of patients lost ≥15% of their initial weight, and 36% lost ≥20% of their initial weight.
“These large categorical weight losses – particularly of 15% and 20% of initial weight – are potentially a major breakthrough in the management of obesity,” Dr. Wadden said in an interview.
Weight losses of this size, he added, “should confer greater improvements in cardiometabolic risk factors (such as hypertension, sleep apnea, and type 2 diabetes) as compared with losses of 5%-10% achieved with current behavioral or pharmacological approaches.” And patients are generally not satisfied with losses of less than 10% of initial weight when participating in intensive behavior programs or taking weight-loss medications.
Now, “the larger categorical weight losses will mean that a greater number of patients with obesity will be able to achieve a weight loss with which they are ... happy,” Dr. Wadden said in an interview.
According to Louis J. Aronne, MD, Weill Professor of Metabolic Research, Weill Cornell Medicine, New York, who is an investigator for another trial of semaglutide: “Even though it has the same mechanism of action [as liraglutide], the weight loss is two or more times greater [with semaglutide]. In my opinion, it’s really going to be a major advance in the treatment of obesity.”
In the discussion that followed the virtual presentation, one attendee asked about potential weight regain if a patient stopped taking the drug. Based on experience with another subcutaneous injectable GLP-1 receptor agonist, liraglutide (Saxenda), already approved for obesity, it may be that taking medicine for chronic overweight may become like taking a statin for elevated cholesterol, said Dr. Wadden.
Novo Nordisk has now completed the four trials in the STEP (Semaglutide Treatment Effect in People With Obesity) global phase 3 clinical development program, and plans to file applications with the Food and Drug Administration later this year and with the European Medicines Agency in early 2021 for review of semaglutide 2.4 mg for weight management.
“Fundamental issues need to be figured out”
Invited to comment, Scott Kahan, MD, said: “This is impressive data, confirming that semaglutide, particularly when used in concert with evidence-based counseling, is a highly effective agent for obesity management.”
However, “the real question, though, is what comes next,” stressed Dr. Kahan, director of the National Center for Weight and Wellness, Washington, DC.
“Will it be approved by the U.S. FDA? I believe so,” he said in an interview. “Yet we already have several effective obesity medications approved over the past decade – all of which are rarely used and therefore make little impact for patients in the real world.”
“Will there be insurance coverage, and therefore practical access for those who could most benefit?” he continued. “Will prescribers counsel their patients about obesity management, including the use of effective medications? Will patients utilize available options?”
“These and other fundamental issues must be figured out before we anoint any treatment option as a meaningful step forward, let alone a transformative development,” according to Dr. Kahan.
Similarly, Irl B. Hirsch, MD, stressed that, should this medication be approved for weight loss, cost would be a major factor in its uptake.
“I’m old enough to recall when we started using lovastatin in the late 1980s,” Dr. Hirsch, professor of medicine, University of Washington Medicine Diabetes Institute, Seattle, said in an interview.
“We used it without the type of evidence of statin use we have today. A pill, but in those days the statins were expensive. But over time, the evidence for statins grew and over the next 15 years it was quite clear that for both primary prevention (for those with diabetes) and secondary intervention these drugs needed to be used by millions of people. These recommendations became easier once the drugs became generic.
“Will the same thing happen for GLP-1 agonists? The problem is we need both ‘hard-outcome data’ [such as 3-point major adverse cardiovascular events] and more reasonable cost before we see this expanding to an entire population.
“In the future perhaps we could have a biosimilar GLP-1 agonist that would be more affordable than what we pay now, but even before that we need agreement from our reimbursement thought leaders that our society should reimburse these agents.
“My thinking now is the cost-benefit could be favorable, but this is all dependent on what happens to the cost of the drugs over time,” he said.
Additive effect of intensive behavior therapy plus medication
Dr. Wadden explained that intensive behavioral therapy “provides 14 or more counseling sessions in 6 months to modify diet and physical activity, through the patients’ use of behavioral strategies (such as keeping daily food and activity diaries).”
Such programs typically produce mean weight loss of 5%-8% of initial weight; less frequent (e.g., monthly) programs typically produce weight loss of only 1%-3%.
Prior studies suggest that intensive behavioral therapy and medication have additive effects. To investigate this, Dr. Wadden and colleagues randomized 611 adults (81% women) who were a mean age of 46 years and had a mean body mass index of 38 kg/m2.
All participants received 30 intensive behavior therapy sessions provided by a registered dietitian (or other qualified provider), which typically lasted 20-30 minutes and were given weekly for 12 weeks, every other week for the next 12 weeks, and then monthly.
The dietitian gave participants behavioral strategies to help them adhere to diet and physical activity goals.
During the first 8 weeks, participants were provided with a 1,000-1,200 kcal/day meal replacement diet that included liquid shakes, meal bars, and prepared entrees designed to facilitate a large initial weight loss.
They then transitioned to a diet of conventional foods (of their choosing), with a goal of 1,200-1,800 kcal/day based on body weight.
The physical activity goal was 100 minutes/week of walking or other aerobic activity in the first month, building up to 200 minutes/week by month 6.
‘More effective than current FDA-approved weight-loss medications’
At week 68, mean body weight decreased from baseline by 16.0% in the semaglutide group versus 5.7% in the placebo group (P < .0001).
In this trial, where all participants received extensive intensive behavior therapy, more participants had weight loss ≥5%, ≥10%, ≥15%, and ≥20% of their initial weight with semaglutide versus placebo (87% vs. 48%; 75% vs. 27%; 56% vs. 13%; 36% vs. 4%, respectively; all P < .0001).
From baseline to week 68, the proportion of participants with prediabetes decreased from 48% to 7% in the semaglutide group and from 53% to 26% in the placebo group.
Patients who received semaglutide had greater improvements in lipids, too.
Although the weight loss was 10.3% (10.6 kg) greater with semaglutide, Dr. Wadden noted, “additional studies have shown this net benefit to be as great as 11%-12%, which would make semaglutide 2.4 mg more effective than current [FDA-approved] weight-loss medications.”
“Naltrexone-bupropion (Contrave) with lifestyle counseling, for example,” he continued, “produces a loss that is 5 kg greater than lifestyle counseling plus placebo, liraglutide 3.0 mg (Saxenda) a loss 5.3 kg greater than placebo, and phentermine-topiramate (Qsymia) a loss that is 8.8 kg greater than placebo.”
Semaglutide was well tolerated. Gastrointestinal adverse events, the most common type, occurred in 83% of patients in the semaglutide group and 63% of patients in the placebo group.
Nausea, as well as constipation and diarrhea, are common in medications that increase GLP-1 levels, Dr. Wadden noted. Side effects can be managed by slowly increasing the medication dose over 4 months.
Dr. Wadden expects that, if approved, semaglutide 2.4 mg subcutaneous once-weekly will be recommended as an adjunct to a reduced calorie diet and increased physical activity. Additional studies suggest that monthly counseling should be sufficient to obtain similar weight losses as those seen in the current trial, which had more intensive counseling.
As well as being approved as a weekly subcutaneous injectable treatment for type 2 diabetes, semaglutide is also approved as an once-daily oral agent for the same indication (Rybelsus, Novo Nordisk) in doses of 7 mg and 14 mg to improve glycemic control along with diet and exercise. It is the first GLP-1 agonist available in tablet form.
Dr. Wadden serves on scientific advisory boards for Novo Nordisk and WW (formerly Weight Watchers), and has received grant support, on behalf of the University of Pennsylvania, from Novo Nordisk. Dr. Aronne is an investigator in a long-term trial of semaglutide and has served on scientific advisory boards for Novo Nordisk in the past. He also has other industry relationships that are not related to semaglutide.
A version of this article originally appeared on Medscape.com.
In a phase 3 trial where all participants received intensive behavior therapy, investigational 2.4-mg once-weekly subcutaneous semaglutide (Novo Nordisk) resulted in a 10.3% greater average weight loss than placebo over a period of 68 weeks.
If approved, this medication could be a “potential major breakthrough” in obesity management, the investigators suggested. But other experts urged caution, as cost and uptake are important considerations.
‘Potential weight loss that patients would be happy with’
Thomas A. Wadden, PhD, presented results from the study of 611 adults with overweight or obesity but no diabetes at the virtual ObesityWeek® Interactive 2020 meeting.
“Perhaps even more impressive was the finding that 75% of patients lost 10% or more of baseline body weight,” said Dr. Wadden, of the department of psychiatry at the University of Pennsylvania, Philadelphia.
Moreover, in this trial of semaglutide, a glucagonlike peptide–1 (GLP-1) receptor agonist that is approved for treating type 2 diabetes at a weekly subcutaneous dose of 1 mg, but is being investigated at the higher dose for weight loss – 55% of patients lost ≥15% of their initial weight, and 36% lost ≥20% of their initial weight.
“These large categorical weight losses – particularly of 15% and 20% of initial weight – are potentially a major breakthrough in the management of obesity,” Dr. Wadden said in an interview.
Weight losses of this size, he added, “should confer greater improvements in cardiometabolic risk factors (such as hypertension, sleep apnea, and type 2 diabetes) as compared with losses of 5%-10% achieved with current behavioral or pharmacological approaches.” And patients are generally not satisfied with losses of less than 10% of initial weight when participating in intensive behavior programs or taking weight-loss medications.
Now, “the larger categorical weight losses will mean that a greater number of patients with obesity will be able to achieve a weight loss with which they are ... happy,” Dr. Wadden said in an interview.
According to Louis J. Aronne, MD, Weill Professor of Metabolic Research, Weill Cornell Medicine, New York, who is an investigator for another trial of semaglutide: “Even though it has the same mechanism of action [as liraglutide], the weight loss is two or more times greater [with semaglutide]. In my opinion, it’s really going to be a major advance in the treatment of obesity.”
In the discussion that followed the virtual presentation, one attendee asked about potential weight regain if a patient stopped taking the drug. Based on experience with another subcutaneous injectable GLP-1 receptor agonist, liraglutide (Saxenda), already approved for obesity, it may be that taking medicine for chronic overweight may become like taking a statin for elevated cholesterol, said Dr. Wadden.
Novo Nordisk has now completed the four trials in the STEP (Semaglutide Treatment Effect in People With Obesity) global phase 3 clinical development program, and plans to file applications with the Food and Drug Administration later this year and with the European Medicines Agency in early 2021 for review of semaglutide 2.4 mg for weight management.
“Fundamental issues need to be figured out”
Invited to comment, Scott Kahan, MD, said: “This is impressive data, confirming that semaglutide, particularly when used in concert with evidence-based counseling, is a highly effective agent for obesity management.”
However, “the real question, though, is what comes next,” stressed Dr. Kahan, director of the National Center for Weight and Wellness, Washington, DC.
“Will it be approved by the U.S. FDA? I believe so,” he said in an interview. “Yet we already have several effective obesity medications approved over the past decade – all of which are rarely used and therefore make little impact for patients in the real world.”
“Will there be insurance coverage, and therefore practical access for those who could most benefit?” he continued. “Will prescribers counsel their patients about obesity management, including the use of effective medications? Will patients utilize available options?”
“These and other fundamental issues must be figured out before we anoint any treatment option as a meaningful step forward, let alone a transformative development,” according to Dr. Kahan.
Similarly, Irl B. Hirsch, MD, stressed that, should this medication be approved for weight loss, cost would be a major factor in its uptake.
“I’m old enough to recall when we started using lovastatin in the late 1980s,” Dr. Hirsch, professor of medicine, University of Washington Medicine Diabetes Institute, Seattle, said in an interview.
“We used it without the type of evidence of statin use we have today. A pill, but in those days the statins were expensive. But over time, the evidence for statins grew and over the next 15 years it was quite clear that for both primary prevention (for those with diabetes) and secondary intervention these drugs needed to be used by millions of people. These recommendations became easier once the drugs became generic.
“Will the same thing happen for GLP-1 agonists? The problem is we need both ‘hard-outcome data’ [such as 3-point major adverse cardiovascular events] and more reasonable cost before we see this expanding to an entire population.
“In the future perhaps we could have a biosimilar GLP-1 agonist that would be more affordable than what we pay now, but even before that we need agreement from our reimbursement thought leaders that our society should reimburse these agents.
“My thinking now is the cost-benefit could be favorable, but this is all dependent on what happens to the cost of the drugs over time,” he said.
Additive effect of intensive behavior therapy plus medication
Dr. Wadden explained that intensive behavioral therapy “provides 14 or more counseling sessions in 6 months to modify diet and physical activity, through the patients’ use of behavioral strategies (such as keeping daily food and activity diaries).”
Such programs typically produce mean weight loss of 5%-8% of initial weight; less frequent (e.g., monthly) programs typically produce weight loss of only 1%-3%.
Prior studies suggest that intensive behavioral therapy and medication have additive effects. To investigate this, Dr. Wadden and colleagues randomized 611 adults (81% women) who were a mean age of 46 years and had a mean body mass index of 38 kg/m2.
All participants received 30 intensive behavior therapy sessions provided by a registered dietitian (or other qualified provider), which typically lasted 20-30 minutes and were given weekly for 12 weeks, every other week for the next 12 weeks, and then monthly.
The dietitian gave participants behavioral strategies to help them adhere to diet and physical activity goals.
During the first 8 weeks, participants were provided with a 1,000-1,200 kcal/day meal replacement diet that included liquid shakes, meal bars, and prepared entrees designed to facilitate a large initial weight loss.
They then transitioned to a diet of conventional foods (of their choosing), with a goal of 1,200-1,800 kcal/day based on body weight.
The physical activity goal was 100 minutes/week of walking or other aerobic activity in the first month, building up to 200 minutes/week by month 6.
‘More effective than current FDA-approved weight-loss medications’
At week 68, mean body weight decreased from baseline by 16.0% in the semaglutide group versus 5.7% in the placebo group (P < .0001).
In this trial, where all participants received extensive intensive behavior therapy, more participants had weight loss ≥5%, ≥10%, ≥15%, and ≥20% of their initial weight with semaglutide versus placebo (87% vs. 48%; 75% vs. 27%; 56% vs. 13%; 36% vs. 4%, respectively; all P < .0001).
From baseline to week 68, the proportion of participants with prediabetes decreased from 48% to 7% in the semaglutide group and from 53% to 26% in the placebo group.
Patients who received semaglutide had greater improvements in lipids, too.
Although the weight loss was 10.3% (10.6 kg) greater with semaglutide, Dr. Wadden noted, “additional studies have shown this net benefit to be as great as 11%-12%, which would make semaglutide 2.4 mg more effective than current [FDA-approved] weight-loss medications.”
“Naltrexone-bupropion (Contrave) with lifestyle counseling, for example,” he continued, “produces a loss that is 5 kg greater than lifestyle counseling plus placebo, liraglutide 3.0 mg (Saxenda) a loss 5.3 kg greater than placebo, and phentermine-topiramate (Qsymia) a loss that is 8.8 kg greater than placebo.”
Semaglutide was well tolerated. Gastrointestinal adverse events, the most common type, occurred in 83% of patients in the semaglutide group and 63% of patients in the placebo group.
Nausea, as well as constipation and diarrhea, are common in medications that increase GLP-1 levels, Dr. Wadden noted. Side effects can be managed by slowly increasing the medication dose over 4 months.
Dr. Wadden expects that, if approved, semaglutide 2.4 mg subcutaneous once-weekly will be recommended as an adjunct to a reduced calorie diet and increased physical activity. Additional studies suggest that monthly counseling should be sufficient to obtain similar weight losses as those seen in the current trial, which had more intensive counseling.
As well as being approved as a weekly subcutaneous injectable treatment for type 2 diabetes, semaglutide is also approved as an once-daily oral agent for the same indication (Rybelsus, Novo Nordisk) in doses of 7 mg and 14 mg to improve glycemic control along with diet and exercise. It is the first GLP-1 agonist available in tablet form.
Dr. Wadden serves on scientific advisory boards for Novo Nordisk and WW (formerly Weight Watchers), and has received grant support, on behalf of the University of Pennsylvania, from Novo Nordisk. Dr. Aronne is an investigator in a long-term trial of semaglutide and has served on scientific advisory boards for Novo Nordisk in the past. He also has other industry relationships that are not related to semaglutide.
A version of this article originally appeared on Medscape.com.
FDA grants emergency use authorization to Lilly’s antibody COVID-19 therapy

The monoclonal antibody therapy has emergency authorization for treating patients who have tested positive for SARS-CoV-2 infection and who are considered to be at high risk for progression to severe COVID-19 or hospitalization. To be eligible for treatment with bamlanivimab, patients must be at least 12 years of age and weigh at least 40 kg (approximately 88 lb). The agency notes that this includes patients aged 65 years and older or people with certain chronic conditions.
Bamlanivimab is not authorized for use in patients who are hospitalized or who require oxygen therapy because of COVID-19. The FDA’s action comes less than 2 weeks after Eli Lilly halted the ACTIV-3 study of the therapy for severe, hospitalized COVID-19 patients after evidence showed that adding the antibody therapy to standard care did not improve outcomes over standard care alone for patients with advanced COVID-19.
The government contract with Eli Lilly involves the purchase of 300,000 doses through December, with the option to procure another 650,000 doses through June 2021.
Because of Operation Warp Speed, “we have supplies to distribute now. Product distribution will begin this week,” US Health & Human Services (HHS) Secretary Alex Azar said at a news conference today.
“We talked about building the bridge to safe and effective vaccines” for COVID-19, Azar added. “With this therapeutic, the bridge is taking shape.”
Bamlanivimab 700 mg will be administered as a 1-hour infusion followed by a 1-hour observation period for detecting any infusion-related side effects. The authorized dose is 700 mg, which was on the lower end of the dose range evaluated in studies.
During the press conference, a reporter asked whether the lower dose was chosen in order that more doses of the antibody could be made available. “The lower dose is a rational choice in this situation because we don’t want to give more of a drug than you need,” said Janet Woodcock, MD, the therapeutics lead for Operation Warp Speed. “I think we could probably go lower.”
Bamlanivimab works by attaching to the virus and blocking its entry into the cells and possibly by helping the patients’ immune system clear the virus, said Woodcock, who is also director of the FDA’s Center for Drug Evaluation and Research.
“The goal is to treat high-risk people as soon as possible after they show symptoms and are diagnosed,” she added.
Infusions an initial challenge?
There could be some logistic challenges at first because the antibody is administered via infusion. “We expect there will initially be a challenge in administering ... these infusions and setting up infusion centers,” Woodcock said.
Outpatient intravenous infusions are normally performed at infusion centers for patients with cancer and immune disorders, she noted. “You really don’t want them mixing with people who have COVID-19 disease, so we will need to set up separate sites.”
Bamlanivimab will be provided free of cost to patients, Azar said. Patients should be aware that coinsurance may be required for the infusion.
“Fair and equitable” distribution planned
During phase 1 of distribution, the agent will first be allocated to hospitals and hospital-affiliated locations only, John Redd, MD, MPH, chief medical officer, Office of the Assistant Secretary for Preparedness and Response at HHS, said at the press conference.
During phase 2, “there will be expanded distribution to outpatient sites,” he said. In an effort to keep the process transparent, a new website features the latest updates on the distribution of bamlanivimab.
Allocation will be based on two factors: the number of new cases reported in a state or territory in the prior 7 days, and rates of COVID-19 hospitalization during the same period.
Asked why the government would determine distribution of the antibody on the basis of the number of hospitalized patients when the indication includes prevention of admission, Woodcock replied that hospitalization is a surrogate measure that can reflect risk factors in a particular state population, such as obesity, diabetes, or the proportion of older people.
Furthermore, the confirmed cases are a “leading indicator,” she said, that can help identify a steep rise in COVID-19 cases that could indicate more hospitalizations are likely soon. “We don’t want to miss that.”
Data underlying the EUA decision
A decrease in hospitalizations or emergency department visits within 28 days of treatment in preclinical studies was “the most important evidence that bamlanivimab may be effective,” the agency noted in the press release announcing the EUA. Among patients at high risk for progression, 3% required such interventions, compared with 10% of placebo-treated patients.
Potential side effects of bamlanivimab include anaphylaxis, infusion-related reactions, nausea, diarrhea, dizziness, headache, itching, and vomiting.
“As illustrated by today’s action, the FDA remains committed to expediting the development and availability of potential COVID-19 treatments and providing sick patients timely access to new therapies where appropriate,” FDA Commissioner Stephen M. Hahn, MD, said in the news release.
Healthcare providers can download a detailed FDA fact sheet on the EUA for bamlanivimab, which includes dosing instructions.
This article first appeared on Medscape.com.
The monoclonal antibody therapy has emergency authorization for treating patients who have tested positive for SARS-CoV-2 infection and who are considered to be at high risk for progression to severe COVID-19 or hospitalization. To be eligible for treatment with bamlanivimab, patients must be at least 12 years of age and weigh at least 40 kg (approximately 88 lb). The agency notes that this includes patients aged 65 years and older or people with certain chronic conditions.
Bamlanivimab is not authorized for use in patients who are hospitalized or who require oxygen therapy because of COVID-19. The FDA’s action comes less than 2 weeks after Eli Lilly halted the ACTIV-3 study of the therapy for severe, hospitalized COVID-19 patients after evidence showed that adding the antibody therapy to standard care did not improve outcomes over standard care alone for patients with advanced COVID-19.
The government contract with Eli Lilly involves the purchase of 300,000 doses through December, with the option to procure another 650,000 doses through June 2021.
Because of Operation Warp Speed, “we have supplies to distribute now. Product distribution will begin this week,” US Health & Human Services (HHS) Secretary Alex Azar said at a news conference today.
“We talked about building the bridge to safe and effective vaccines” for COVID-19, Azar added. “With this therapeutic, the bridge is taking shape.”
Bamlanivimab 700 mg will be administered as a 1-hour infusion followed by a 1-hour observation period for detecting any infusion-related side effects. The authorized dose is 700 mg, which was on the lower end of the dose range evaluated in studies.
During the press conference, a reporter asked whether the lower dose was chosen in order that more doses of the antibody could be made available. “The lower dose is a rational choice in this situation because we don’t want to give more of a drug than you need,” said Janet Woodcock, MD, the therapeutics lead for Operation Warp Speed. “I think we could probably go lower.”
Bamlanivimab works by attaching to the virus and blocking its entry into the cells and possibly by helping the patients’ immune system clear the virus, said Woodcock, who is also director of the FDA’s Center for Drug Evaluation and Research.
“The goal is to treat high-risk people as soon as possible after they show symptoms and are diagnosed,” she added.
Infusions an initial challenge?
There could be some logistic challenges at first because the antibody is administered via infusion. “We expect there will initially be a challenge in administering ... these infusions and setting up infusion centers,” Woodcock said.
Outpatient intravenous infusions are normally performed at infusion centers for patients with cancer and immune disorders, she noted. “You really don’t want them mixing with people who have COVID-19 disease, so we will need to set up separate sites.”
Bamlanivimab will be provided free of cost to patients, Azar said. Patients should be aware that coinsurance may be required for the infusion.
“Fair and equitable” distribution planned
During phase 1 of distribution, the agent will first be allocated to hospitals and hospital-affiliated locations only, John Redd, MD, MPH, chief medical officer, Office of the Assistant Secretary for Preparedness and Response at HHS, said at the press conference.
During phase 2, “there will be expanded distribution to outpatient sites,” he said. In an effort to keep the process transparent, a new website features the latest updates on the distribution of bamlanivimab.
Allocation will be based on two factors: the number of new cases reported in a state or territory in the prior 7 days, and rates of COVID-19 hospitalization during the same period.
Asked why the government would determine distribution of the antibody on the basis of the number of hospitalized patients when the indication includes prevention of admission, Woodcock replied that hospitalization is a surrogate measure that can reflect risk factors in a particular state population, such as obesity, diabetes, or the proportion of older people.
Furthermore, the confirmed cases are a “leading indicator,” she said, that can help identify a steep rise in COVID-19 cases that could indicate more hospitalizations are likely soon. “We don’t want to miss that.”
Data underlying the EUA decision
A decrease in hospitalizations or emergency department visits within 28 days of treatment in preclinical studies was “the most important evidence that bamlanivimab may be effective,” the agency noted in the press release announcing the EUA. Among patients at high risk for progression, 3% required such interventions, compared with 10% of placebo-treated patients.
Potential side effects of bamlanivimab include anaphylaxis, infusion-related reactions, nausea, diarrhea, dizziness, headache, itching, and vomiting.
“As illustrated by today’s action, the FDA remains committed to expediting the development and availability of potential COVID-19 treatments and providing sick patients timely access to new therapies where appropriate,” FDA Commissioner Stephen M. Hahn, MD, said in the news release.
Healthcare providers can download a detailed FDA fact sheet on the EUA for bamlanivimab, which includes dosing instructions.
This article first appeared on Medscape.com.
The monoclonal antibody therapy has emergency authorization for treating patients who have tested positive for SARS-CoV-2 infection and who are considered to be at high risk for progression to severe COVID-19 or hospitalization. To be eligible for treatment with bamlanivimab, patients must be at least 12 years of age and weigh at least 40 kg (approximately 88 lb). The agency notes that this includes patients aged 65 years and older or people with certain chronic conditions.
Bamlanivimab is not authorized for use in patients who are hospitalized or who require oxygen therapy because of COVID-19. The FDA’s action comes less than 2 weeks after Eli Lilly halted the ACTIV-3 study of the therapy for severe, hospitalized COVID-19 patients after evidence showed that adding the antibody therapy to standard care did not improve outcomes over standard care alone for patients with advanced COVID-19.
The government contract with Eli Lilly involves the purchase of 300,000 doses through December, with the option to procure another 650,000 doses through June 2021.
Because of Operation Warp Speed, “we have supplies to distribute now. Product distribution will begin this week,” US Health & Human Services (HHS) Secretary Alex Azar said at a news conference today.
“We talked about building the bridge to safe and effective vaccines” for COVID-19, Azar added. “With this therapeutic, the bridge is taking shape.”
Bamlanivimab 700 mg will be administered as a 1-hour infusion followed by a 1-hour observation period for detecting any infusion-related side effects. The authorized dose is 700 mg, which was on the lower end of the dose range evaluated in studies.
During the press conference, a reporter asked whether the lower dose was chosen in order that more doses of the antibody could be made available. “The lower dose is a rational choice in this situation because we don’t want to give more of a drug than you need,” said Janet Woodcock, MD, the therapeutics lead for Operation Warp Speed. “I think we could probably go lower.”
Bamlanivimab works by attaching to the virus and blocking its entry into the cells and possibly by helping the patients’ immune system clear the virus, said Woodcock, who is also director of the FDA’s Center for Drug Evaluation and Research.
“The goal is to treat high-risk people as soon as possible after they show symptoms and are diagnosed,” she added.
Infusions an initial challenge?
There could be some logistic challenges at first because the antibody is administered via infusion. “We expect there will initially be a challenge in administering ... these infusions and setting up infusion centers,” Woodcock said.
Outpatient intravenous infusions are normally performed at infusion centers for patients with cancer and immune disorders, she noted. “You really don’t want them mixing with people who have COVID-19 disease, so we will need to set up separate sites.”
Bamlanivimab will be provided free of cost to patients, Azar said. Patients should be aware that coinsurance may be required for the infusion.
“Fair and equitable” distribution planned
During phase 1 of distribution, the agent will first be allocated to hospitals and hospital-affiliated locations only, John Redd, MD, MPH, chief medical officer, Office of the Assistant Secretary for Preparedness and Response at HHS, said at the press conference.
During phase 2, “there will be expanded distribution to outpatient sites,” he said. In an effort to keep the process transparent, a new website features the latest updates on the distribution of bamlanivimab.
Allocation will be based on two factors: the number of new cases reported in a state or territory in the prior 7 days, and rates of COVID-19 hospitalization during the same period.
Asked why the government would determine distribution of the antibody on the basis of the number of hospitalized patients when the indication includes prevention of admission, Woodcock replied that hospitalization is a surrogate measure that can reflect risk factors in a particular state population, such as obesity, diabetes, or the proportion of older people.
Furthermore, the confirmed cases are a “leading indicator,” she said, that can help identify a steep rise in COVID-19 cases that could indicate more hospitalizations are likely soon. “We don’t want to miss that.”
Data underlying the EUA decision
A decrease in hospitalizations or emergency department visits within 28 days of treatment in preclinical studies was “the most important evidence that bamlanivimab may be effective,” the agency noted in the press release announcing the EUA. Among patients at high risk for progression, 3% required such interventions, compared with 10% of placebo-treated patients.
Potential side effects of bamlanivimab include anaphylaxis, infusion-related reactions, nausea, diarrhea, dizziness, headache, itching, and vomiting.
“As illustrated by today’s action, the FDA remains committed to expediting the development and availability of potential COVID-19 treatments and providing sick patients timely access to new therapies where appropriate,” FDA Commissioner Stephen M. Hahn, MD, said in the news release.
Healthcare providers can download a detailed FDA fact sheet on the EUA for bamlanivimab, which includes dosing instructions.
This article first appeared on Medscape.com.
Continued Dosing of Oritavancin for Complicated Gram-Positive Infections
Oritavancin is a lipoglycopeptide antibiotic. The US Food and Drug Administration (FDA) approved oritavancin in 2014 for adults with acute bacterial skin and skin structure infections (ABSSSI).1 The antibiotic is currently FDA approved for infections caused by Gram-positive organisms, including methicillin-resistant and methicillinsusceptible Staphylococcus aureus (MRSA, MSSA), a variety of Streptococcus species, and vancomycin-susceptible Enterococcus faecalis (VSE). Oritavancin demonstrates concentrationdependent bactericidal activity and has a half-life of 245 hours. This half-life allows for treatment of ABSSSI with a single 1,200 mg IV dose, which has been shown to be noninferior to vancomycin dosed twice daily for 7 to 10 days.1-3
Proposal for Expanded Uses
Although the approved indication for oritavancin is narrow, in vitro studies have shown that oritavancin also has activity against vancomycin-resistant enterococci (VRE), and rabbit studies have demonstrated its excellent bone penetration.4,5 These findings have raised the question of whether oritavancin can be safely and effectively used for infections such as endocarditis, osteomyelitis, and bacteremia, which are often caused by invasive Grampositive organisms. These types of invasive infections, particularly when MRSA is implicated, generally require IV antibiotic therapy for several weeks, often with vancomycin.6
To avoid long hospital stays solely for antibiotic administration, health care practitioners will often use outpatient parenteral antimicrobial therapy (OPAT). However, using OPAT presents many challenges due to the need for frequent dosing, the risk of peripheral or central-line infections, and therapeutic drug monitoring when using vancomycin; additionally, administration and line care oftentimes require caregiver support, which may not be present for all patients.7 Concerns also have been raised regarding the use of OPAT in patients with a history of IV drug use due to the potential increased risk of line infections or line abuse. Few studies have explored OPAT in this population, and the Infectious Diseases Society of America OPAT guidelines recommend that the decision to use OPAT should be made on a case-by-case basis.7 Thus, patients who are deemed inappropriate for OPAT oftentimes remain hospitalized or reside briefly in nursing facilities solely for antibiotic administration
Oritavancin’s long half-life and potent activity against Gram-positive organisms has led to increased interest in off-label use of infrequent dosing intervals, such as weekly, to treat complicated and invasive infections. Weekly rather than daily dosing would allow for less burdensome antibiotic administration regimens and shorter hospital stays especially for patients who are not candidates for OPAT.
Efficacy of Continued Dosing
This proposed weekly dosing pattern, referred to as continued dosing or a multiple-dose regimen, has gained traction in the literature. To date, no randomized controlled trials have been conducted to assess oritavancin’s efficacy in off-label indications or continued dosing, but several case reports and retrospective cohort analyses show promising outcomes.8-16 In an analysis of data from the Clinical and Historic Registry and Orbactiv Medical Evaluation (CHROME) patient registry, 32 patients received multiple doses of oritavancin for complicated Gram-positive infections with a 93.8% overall clinical success rate, including success rates of 90.9% (10/11) for general bone and joint infections and 87.5% (7/8) for patients diagnosed specifically with osteomyelitis.8
Patients received between 2 and 10 doses of 1,200 mg IV given every 6 to 14 days. Johnson and colleagues report using oritavancin 1,200 mg IV every other day for 3 doses followed by 1,200 mg IV once weekly for a patient with daptomycin- and vancomycin-resistant Enterococcus endocarditis, resulting in negative blood cultures while on therapy.9 However, source control via valve replacement and postoperative oritavancin 1,200 mg IV twice weekly for 10 weeks was required to fully clear the infection.
Schulz and colleagues published a retrospective cohort analysis of 17 patients who received multiple doses of oritavancin for complicated bacterial infections, including osteomyelitis, pneumonia, and bacteremia.10 They reported 100% of patients were either successfully cured or had demonstrable improvements in their infections by using a 1,200 mg IV loading dose followed by 800 mg IV if the second dose was given within 7 days or 1,200 mg IV if the second dose was given more than 10 days later. Patients received between 2 and 18 total doses, with 6 out of 17 (35%) receiving only 2 doses. One patient who received 18 doses was an outlier, as her treatment goal was palliative suppression due to an infected endovascular graft that could not be removed.
In a published case series, 1 of 10 patients receiving oritavancin for invasive Grampositive infections received multiple doses of oritavancin for an MSSA deep tissue infection.11 The 3 total doses (strength not reported) were separated by 19 days and 14 days and resulted in cure. Several case reports and a retrospective chart review study specifically show the effectiveness of oritavancin for osteomyelitis caused by MSSA, MRSA, and VRE.12-16 However, dosing strategies varied widely after the initial 1,200 mg IV loading dose.
Drug Interactions, Safety, and Tolerability
Oritavancin has minimal drug-drug interactions, the most notable being with anticoagulants. 1 Use of IV heparin within 120 hours of oritavancin administration can falsely elevate activated partial thromboplastin time (aPTT) levels; therefore, heparin should not be monitored with aPTT during this period. Oritavancin also can artificially prolong international normalized ratio (INR) values for up to 12 hours, and dose adjustments based on INRs during this window are not recommended. Of note, factor Xa laboratory monitoring is unaffected by oritavancin, as it does not depend on phospholipid reagents as do aPTT and INR measurements.
Oritavancin has been shown to be well tolerated when dosed according to both the package insert and continued dosing strategies. The most common adverse effects (AEs) (≥ 3%), occurring at similar rates to vancomycin, are nausea, vomiting, diarrhea, headache, and limb and subcutaneous abscesses.1 Infusion reactions also have been reported, although they are usually reversible on slowing or stopping the infusion. It is worth noting that the use of oritavancin for osteomyelitis is not recommended in the product labeling, as an increased rate of osteomyelitis was observed in the oritavancin vs IV vancomycin groups for the treatment of patients with acute bacterial skin and skin structure infection (SOLO) trials (0.6% in oritavancin group vs 0.1% in vancomycin group, statistical significance not reported).17 However, it was postulated that these osteomyelitis cases were likely present, yet not recognized, at baseline and were not the result of administering oritavancin. This conclusion is further corroborated by previously presented research demonstrating successful cure of osteomyelitis with continued dosing strategies.12-16
Many patients receiving multiple doses of oritavancin did not experience AEs or laboratory abnormalities.13,15 Four of 17 patients (24%) in one retrospective review experienced AEs, including infusion reactions, anemia, and leukopenia; all were reversible on discontinuation of oritavancin, and contributions of other antibiotics in some cases could not be ruled out.10 One patient experienced taste disturbance for several hours after each infusion, and a second had documented hearing loss after 3 doses of oritavancin in a 33-day period, though she had received 6 weeks of IV vancomycin prior to oritavancin.11,12 A patient treated for daptomycin- and vancomycinresistant Enterococcus faecium prosthetic valve endocarditis experienced nausea, anorexia, and minor liver function test (LFT) abnormalities after cumulative oritavancin exposure over 18 weeks.9 On discontinuation of the drug, nausea and anorexia improved, and LFTs normalized 11 months later. Overall, AEs reported with continued dosing of oritavancin have been minimal and largely reversible, mimicking the AEs in the product labeling for traditional dosing. This suggests that using a continued dosing strategy may not result in worse or more frequent AEs, though randomized controlled trials are needed to fully ascertain these preliminary findings.
Conclusions
The literature supporting the use of oritavancin beyond single-dose administration for ABSSSI is growing. Continued dosing regimens have been well tolerated and have resulted in clinical cure for many patients with barriers to first-line treatment and complicated or invasive infections. While randomized controlled trials are needed to concretely demonstrate the efficacy and safety of continued dosing of oritavancin, it may fill an important treatment niche in this era of growing antibiotic resistance and increasing complexity of patient cases.
1. Orbactiv [package insert]. Parsippany, NJ: The Medicines Company; 2019.
2. Corey GR, Kabler H, Mehra P, et al. Single-dose oritavancin in the treatment of acute bacterial skin infections. N Engl J Med. 2014;370(23):2180-2190. doi:10.1056/NEJMoa1310422
3. Corey GR, Good S, Jiang H, et al. Single-dose oritavancin versus 7-10 days of vancomycin in the treatment of gram-positive acute bacterial skin and skin structure infections: the SOLO II noninferiority study. Clin Infect Dis. 2015;60(2):254-262. doi:10.1093/cid/ciu778
4. Sweeney D, Stoneburner A, Shinabarger DL, et al. Comparative in vitro activity of oritavancin and other agents against vancomycin-susceptible and -resistant enterococci. J Antimicrob Chemother. 2017;72(2):622-624. doi.10.1093/jac/dkw451
5. Lehoux D, Ostiguy V, Vadieux C, et al. Oritavancin pharmacokinetics and bone penetration in rabbits. Antimicrob Agents Chemother. 2015;59(10):6501-6505. doi:10.1128/AAC.00981-15
6. Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52(3):e18-e55. doi:10.1093/cid/ciq146
7. Norris AH, Shrestha NK, Allison GM, et al. 2018 Infectious Diseases Society of America clinical practice guideline for the management of outpatient parenteral antimicrobial therapy. Clin Infect Dis. 2019;68(1):e1-e35. doi:10.1093/cid/ciy745
8. Redell M, Seirra-Hoffman M, Assi Maha, et al. The CHROME study, a real-world experience of single- and multiple-dose oritavancin for treatment of gram-positive infections. Open Forum Infect Dis. 2019;6(11):ofz479. doi:10.1093/ofid/ofz479
9. Johnson JA, Feeney ER, Kubiak DW, Corey GR. Prolonged use of oritavancin for vancomycin-resistant Enterococcus faecium prosthetic valve endocarditis. Open Forum Infect Dis. 2015;2(4):ofv156. doi:10.1093/ofid/ofv156
10. Schulz LT, Dworkin E, Dela-Pena J, Rose WE. Multipledose oritavancin evaluation in a retrospective cohort of patients with complicated infections. Pharmacotherapy. 2018;38(1):152-159. doi:10.1002/phar.2057
11. Stewart CL, Turner MS, Frens JJ, Snider CB, Smith JR. Real-world experience with oritavancin therapy in invasive gram-positive infections. Infect Dis Ther. 2017;6(2):277-289. doi:10.1007/s40121-017-0156-z
12. Delaportas DJ, Estrada SJ, Darmelio M. Successful treatment of methicillin susceptible Staphylococcus aureus osteomyelitis with oritavancin. Pharmacotherapy. 2017;37(8):e90-e92. doi:10.1002/phar.1957
13. Chastain DB, Davis A. Treatment of chronic osteomyelitis with multidose oritavancin: a case series and literature review. Int J Antimicrob Agents. 2019;53(4):429-434. doi:10.1016/j.ijantimicag.2018.11.023
14. Dahesh S, Wong B, Nizet V, Sakoulas G, Tran TT, Aitken SL. Treatment of multidrug-resistant vancomycinresistant Enterococcus faecium hardware-associated vertebral osteomyelitis with oritavancin plus ampicillin. Antimicrob Agents Chemother. 2019;63(7):e02622-18. doi:10.1128/AAC.02622-18
15. Foster RA, Philavong KP, Weissman S, Tang X, Bookstaver PB. Oritavancin for the treatment of daptomycin nonsusceptible vancomycin-resistant Enterococci osteomyelitis. Infect Dis Clin Pract. 2018;26(2):97-99. doi:10.1097/IPC.0000000000000517
16. Ruggero M, Ziegler M, Tebas P, Binkley A, Kelly B. Successful treatment of methicillin-resistant Staphylococcus aureus vertebral osteomyelitis with outpatient oritavancin therapy. Infect Dis Clin Pract. 2018;26(3):141-144. doi:10.1097/IPC.0000000000000599
17. Corey GR, Loutit J, Moeck G, et al. Single intravenous dose of oritavancin for treatment of acute skin and skin structure infections caused by gram-positive bacteria: summary of safety analysis from the phase 3 SOLO studies. Antimicrob Agents Chemother. 2018;62(4):e01919- 17. doi:10.1128/AAC.01919-17
Oritavancin is a lipoglycopeptide antibiotic. The US Food and Drug Administration (FDA) approved oritavancin in 2014 for adults with acute bacterial skin and skin structure infections (ABSSSI).1 The antibiotic is currently FDA approved for infections caused by Gram-positive organisms, including methicillin-resistant and methicillinsusceptible Staphylococcus aureus (MRSA, MSSA), a variety of Streptococcus species, and vancomycin-susceptible Enterococcus faecalis (VSE). Oritavancin demonstrates concentrationdependent bactericidal activity and has a half-life of 245 hours. This half-life allows for treatment of ABSSSI with a single 1,200 mg IV dose, which has been shown to be noninferior to vancomycin dosed twice daily for 7 to 10 days.1-3
Proposal for Expanded Uses
Although the approved indication for oritavancin is narrow, in vitro studies have shown that oritavancin also has activity against vancomycin-resistant enterococci (VRE), and rabbit studies have demonstrated its excellent bone penetration.4,5 These findings have raised the question of whether oritavancin can be safely and effectively used for infections such as endocarditis, osteomyelitis, and bacteremia, which are often caused by invasive Grampositive organisms. These types of invasive infections, particularly when MRSA is implicated, generally require IV antibiotic therapy for several weeks, often with vancomycin.6
To avoid long hospital stays solely for antibiotic administration, health care practitioners will often use outpatient parenteral antimicrobial therapy (OPAT). However, using OPAT presents many challenges due to the need for frequent dosing, the risk of peripheral or central-line infections, and therapeutic drug monitoring when using vancomycin; additionally, administration and line care oftentimes require caregiver support, which may not be present for all patients.7 Concerns also have been raised regarding the use of OPAT in patients with a history of IV drug use due to the potential increased risk of line infections or line abuse. Few studies have explored OPAT in this population, and the Infectious Diseases Society of America OPAT guidelines recommend that the decision to use OPAT should be made on a case-by-case basis.7 Thus, patients who are deemed inappropriate for OPAT oftentimes remain hospitalized or reside briefly in nursing facilities solely for antibiotic administration
Oritavancin’s long half-life and potent activity against Gram-positive organisms has led to increased interest in off-label use of infrequent dosing intervals, such as weekly, to treat complicated and invasive infections. Weekly rather than daily dosing would allow for less burdensome antibiotic administration regimens and shorter hospital stays especially for patients who are not candidates for OPAT.
Efficacy of Continued Dosing
This proposed weekly dosing pattern, referred to as continued dosing or a multiple-dose regimen, has gained traction in the literature. To date, no randomized controlled trials have been conducted to assess oritavancin’s efficacy in off-label indications or continued dosing, but several case reports and retrospective cohort analyses show promising outcomes.8-16 In an analysis of data from the Clinical and Historic Registry and Orbactiv Medical Evaluation (CHROME) patient registry, 32 patients received multiple doses of oritavancin for complicated Gram-positive infections with a 93.8% overall clinical success rate, including success rates of 90.9% (10/11) for general bone and joint infections and 87.5% (7/8) for patients diagnosed specifically with osteomyelitis.8
Patients received between 2 and 10 doses of 1,200 mg IV given every 6 to 14 days. Johnson and colleagues report using oritavancin 1,200 mg IV every other day for 3 doses followed by 1,200 mg IV once weekly for a patient with daptomycin- and vancomycin-resistant Enterococcus endocarditis, resulting in negative blood cultures while on therapy.9 However, source control via valve replacement and postoperative oritavancin 1,200 mg IV twice weekly for 10 weeks was required to fully clear the infection.
Schulz and colleagues published a retrospective cohort analysis of 17 patients who received multiple doses of oritavancin for complicated bacterial infections, including osteomyelitis, pneumonia, and bacteremia.10 They reported 100% of patients were either successfully cured or had demonstrable improvements in their infections by using a 1,200 mg IV loading dose followed by 800 mg IV if the second dose was given within 7 days or 1,200 mg IV if the second dose was given more than 10 days later. Patients received between 2 and 18 total doses, with 6 out of 17 (35%) receiving only 2 doses. One patient who received 18 doses was an outlier, as her treatment goal was palliative suppression due to an infected endovascular graft that could not be removed.
In a published case series, 1 of 10 patients receiving oritavancin for invasive Grampositive infections received multiple doses of oritavancin for an MSSA deep tissue infection.11 The 3 total doses (strength not reported) were separated by 19 days and 14 days and resulted in cure. Several case reports and a retrospective chart review study specifically show the effectiveness of oritavancin for osteomyelitis caused by MSSA, MRSA, and VRE.12-16 However, dosing strategies varied widely after the initial 1,200 mg IV loading dose.
Drug Interactions, Safety, and Tolerability
Oritavancin has minimal drug-drug interactions, the most notable being with anticoagulants. 1 Use of IV heparin within 120 hours of oritavancin administration can falsely elevate activated partial thromboplastin time (aPTT) levels; therefore, heparin should not be monitored with aPTT during this period. Oritavancin also can artificially prolong international normalized ratio (INR) values for up to 12 hours, and dose adjustments based on INRs during this window are not recommended. Of note, factor Xa laboratory monitoring is unaffected by oritavancin, as it does not depend on phospholipid reagents as do aPTT and INR measurements.
Oritavancin has been shown to be well tolerated when dosed according to both the package insert and continued dosing strategies. The most common adverse effects (AEs) (≥ 3%), occurring at similar rates to vancomycin, are nausea, vomiting, diarrhea, headache, and limb and subcutaneous abscesses.1 Infusion reactions also have been reported, although they are usually reversible on slowing or stopping the infusion. It is worth noting that the use of oritavancin for osteomyelitis is not recommended in the product labeling, as an increased rate of osteomyelitis was observed in the oritavancin vs IV vancomycin groups for the treatment of patients with acute bacterial skin and skin structure infection (SOLO) trials (0.6% in oritavancin group vs 0.1% in vancomycin group, statistical significance not reported).17 However, it was postulated that these osteomyelitis cases were likely present, yet not recognized, at baseline and were not the result of administering oritavancin. This conclusion is further corroborated by previously presented research demonstrating successful cure of osteomyelitis with continued dosing strategies.12-16
Many patients receiving multiple doses of oritavancin did not experience AEs or laboratory abnormalities.13,15 Four of 17 patients (24%) in one retrospective review experienced AEs, including infusion reactions, anemia, and leukopenia; all were reversible on discontinuation of oritavancin, and contributions of other antibiotics in some cases could not be ruled out.10 One patient experienced taste disturbance for several hours after each infusion, and a second had documented hearing loss after 3 doses of oritavancin in a 33-day period, though she had received 6 weeks of IV vancomycin prior to oritavancin.11,12 A patient treated for daptomycin- and vancomycinresistant Enterococcus faecium prosthetic valve endocarditis experienced nausea, anorexia, and minor liver function test (LFT) abnormalities after cumulative oritavancin exposure over 18 weeks.9 On discontinuation of the drug, nausea and anorexia improved, and LFTs normalized 11 months later. Overall, AEs reported with continued dosing of oritavancin have been minimal and largely reversible, mimicking the AEs in the product labeling for traditional dosing. This suggests that using a continued dosing strategy may not result in worse or more frequent AEs, though randomized controlled trials are needed to fully ascertain these preliminary findings.
Conclusions
The literature supporting the use of oritavancin beyond single-dose administration for ABSSSI is growing. Continued dosing regimens have been well tolerated and have resulted in clinical cure for many patients with barriers to first-line treatment and complicated or invasive infections. While randomized controlled trials are needed to concretely demonstrate the efficacy and safety of continued dosing of oritavancin, it may fill an important treatment niche in this era of growing antibiotic resistance and increasing complexity of patient cases.
Oritavancin is a lipoglycopeptide antibiotic. The US Food and Drug Administration (FDA) approved oritavancin in 2014 for adults with acute bacterial skin and skin structure infections (ABSSSI).1 The antibiotic is currently FDA approved for infections caused by Gram-positive organisms, including methicillin-resistant and methicillinsusceptible Staphylococcus aureus (MRSA, MSSA), a variety of Streptococcus species, and vancomycin-susceptible Enterococcus faecalis (VSE). Oritavancin demonstrates concentrationdependent bactericidal activity and has a half-life of 245 hours. This half-life allows for treatment of ABSSSI with a single 1,200 mg IV dose, which has been shown to be noninferior to vancomycin dosed twice daily for 7 to 10 days.1-3
Proposal for Expanded Uses
Although the approved indication for oritavancin is narrow, in vitro studies have shown that oritavancin also has activity against vancomycin-resistant enterococci (VRE), and rabbit studies have demonstrated its excellent bone penetration.4,5 These findings have raised the question of whether oritavancin can be safely and effectively used for infections such as endocarditis, osteomyelitis, and bacteremia, which are often caused by invasive Grampositive organisms. These types of invasive infections, particularly when MRSA is implicated, generally require IV antibiotic therapy for several weeks, often with vancomycin.6
To avoid long hospital stays solely for antibiotic administration, health care practitioners will often use outpatient parenteral antimicrobial therapy (OPAT). However, using OPAT presents many challenges due to the need for frequent dosing, the risk of peripheral or central-line infections, and therapeutic drug monitoring when using vancomycin; additionally, administration and line care oftentimes require caregiver support, which may not be present for all patients.7 Concerns also have been raised regarding the use of OPAT in patients with a history of IV drug use due to the potential increased risk of line infections or line abuse. Few studies have explored OPAT in this population, and the Infectious Diseases Society of America OPAT guidelines recommend that the decision to use OPAT should be made on a case-by-case basis.7 Thus, patients who are deemed inappropriate for OPAT oftentimes remain hospitalized or reside briefly in nursing facilities solely for antibiotic administration
Oritavancin’s long half-life and potent activity against Gram-positive organisms has led to increased interest in off-label use of infrequent dosing intervals, such as weekly, to treat complicated and invasive infections. Weekly rather than daily dosing would allow for less burdensome antibiotic administration regimens and shorter hospital stays especially for patients who are not candidates for OPAT.
Efficacy of Continued Dosing
This proposed weekly dosing pattern, referred to as continued dosing or a multiple-dose regimen, has gained traction in the literature. To date, no randomized controlled trials have been conducted to assess oritavancin’s efficacy in off-label indications or continued dosing, but several case reports and retrospective cohort analyses show promising outcomes.8-16 In an analysis of data from the Clinical and Historic Registry and Orbactiv Medical Evaluation (CHROME) patient registry, 32 patients received multiple doses of oritavancin for complicated Gram-positive infections with a 93.8% overall clinical success rate, including success rates of 90.9% (10/11) for general bone and joint infections and 87.5% (7/8) for patients diagnosed specifically with osteomyelitis.8
Patients received between 2 and 10 doses of 1,200 mg IV given every 6 to 14 days. Johnson and colleagues report using oritavancin 1,200 mg IV every other day for 3 doses followed by 1,200 mg IV once weekly for a patient with daptomycin- and vancomycin-resistant Enterococcus endocarditis, resulting in negative blood cultures while on therapy.9 However, source control via valve replacement and postoperative oritavancin 1,200 mg IV twice weekly for 10 weeks was required to fully clear the infection.
Schulz and colleagues published a retrospective cohort analysis of 17 patients who received multiple doses of oritavancin for complicated bacterial infections, including osteomyelitis, pneumonia, and bacteremia.10 They reported 100% of patients were either successfully cured or had demonstrable improvements in their infections by using a 1,200 mg IV loading dose followed by 800 mg IV if the second dose was given within 7 days or 1,200 mg IV if the second dose was given more than 10 days later. Patients received between 2 and 18 total doses, with 6 out of 17 (35%) receiving only 2 doses. One patient who received 18 doses was an outlier, as her treatment goal was palliative suppression due to an infected endovascular graft that could not be removed.
In a published case series, 1 of 10 patients receiving oritavancin for invasive Grampositive infections received multiple doses of oritavancin for an MSSA deep tissue infection.11 The 3 total doses (strength not reported) were separated by 19 days and 14 days and resulted in cure. Several case reports and a retrospective chart review study specifically show the effectiveness of oritavancin for osteomyelitis caused by MSSA, MRSA, and VRE.12-16 However, dosing strategies varied widely after the initial 1,200 mg IV loading dose.
Drug Interactions, Safety, and Tolerability
Oritavancin has minimal drug-drug interactions, the most notable being with anticoagulants. 1 Use of IV heparin within 120 hours of oritavancin administration can falsely elevate activated partial thromboplastin time (aPTT) levels; therefore, heparin should not be monitored with aPTT during this period. Oritavancin also can artificially prolong international normalized ratio (INR) values for up to 12 hours, and dose adjustments based on INRs during this window are not recommended. Of note, factor Xa laboratory monitoring is unaffected by oritavancin, as it does not depend on phospholipid reagents as do aPTT and INR measurements.
Oritavancin has been shown to be well tolerated when dosed according to both the package insert and continued dosing strategies. The most common adverse effects (AEs) (≥ 3%), occurring at similar rates to vancomycin, are nausea, vomiting, diarrhea, headache, and limb and subcutaneous abscesses.1 Infusion reactions also have been reported, although they are usually reversible on slowing or stopping the infusion. It is worth noting that the use of oritavancin for osteomyelitis is not recommended in the product labeling, as an increased rate of osteomyelitis was observed in the oritavancin vs IV vancomycin groups for the treatment of patients with acute bacterial skin and skin structure infection (SOLO) trials (0.6% in oritavancin group vs 0.1% in vancomycin group, statistical significance not reported).17 However, it was postulated that these osteomyelitis cases were likely present, yet not recognized, at baseline and were not the result of administering oritavancin. This conclusion is further corroborated by previously presented research demonstrating successful cure of osteomyelitis with continued dosing strategies.12-16
Many patients receiving multiple doses of oritavancin did not experience AEs or laboratory abnormalities.13,15 Four of 17 patients (24%) in one retrospective review experienced AEs, including infusion reactions, anemia, and leukopenia; all were reversible on discontinuation of oritavancin, and contributions of other antibiotics in some cases could not be ruled out.10 One patient experienced taste disturbance for several hours after each infusion, and a second had documented hearing loss after 3 doses of oritavancin in a 33-day period, though she had received 6 weeks of IV vancomycin prior to oritavancin.11,12 A patient treated for daptomycin- and vancomycinresistant Enterococcus faecium prosthetic valve endocarditis experienced nausea, anorexia, and minor liver function test (LFT) abnormalities after cumulative oritavancin exposure over 18 weeks.9 On discontinuation of the drug, nausea and anorexia improved, and LFTs normalized 11 months later. Overall, AEs reported with continued dosing of oritavancin have been minimal and largely reversible, mimicking the AEs in the product labeling for traditional dosing. This suggests that using a continued dosing strategy may not result in worse or more frequent AEs, though randomized controlled trials are needed to fully ascertain these preliminary findings.
Conclusions
The literature supporting the use of oritavancin beyond single-dose administration for ABSSSI is growing. Continued dosing regimens have been well tolerated and have resulted in clinical cure for many patients with barriers to first-line treatment and complicated or invasive infections. While randomized controlled trials are needed to concretely demonstrate the efficacy and safety of continued dosing of oritavancin, it may fill an important treatment niche in this era of growing antibiotic resistance and increasing complexity of patient cases.
1. Orbactiv [package insert]. Parsippany, NJ: The Medicines Company; 2019.
2. Corey GR, Kabler H, Mehra P, et al. Single-dose oritavancin in the treatment of acute bacterial skin infections. N Engl J Med. 2014;370(23):2180-2190. doi:10.1056/NEJMoa1310422
3. Corey GR, Good S, Jiang H, et al. Single-dose oritavancin versus 7-10 days of vancomycin in the treatment of gram-positive acute bacterial skin and skin structure infections: the SOLO II noninferiority study. Clin Infect Dis. 2015;60(2):254-262. doi:10.1093/cid/ciu778
4. Sweeney D, Stoneburner A, Shinabarger DL, et al. Comparative in vitro activity of oritavancin and other agents against vancomycin-susceptible and -resistant enterococci. J Antimicrob Chemother. 2017;72(2):622-624. doi.10.1093/jac/dkw451
5. Lehoux D, Ostiguy V, Vadieux C, et al. Oritavancin pharmacokinetics and bone penetration in rabbits. Antimicrob Agents Chemother. 2015;59(10):6501-6505. doi:10.1128/AAC.00981-15
6. Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52(3):e18-e55. doi:10.1093/cid/ciq146
7. Norris AH, Shrestha NK, Allison GM, et al. 2018 Infectious Diseases Society of America clinical practice guideline for the management of outpatient parenteral antimicrobial therapy. Clin Infect Dis. 2019;68(1):e1-e35. doi:10.1093/cid/ciy745
8. Redell M, Seirra-Hoffman M, Assi Maha, et al. The CHROME study, a real-world experience of single- and multiple-dose oritavancin for treatment of gram-positive infections. Open Forum Infect Dis. 2019;6(11):ofz479. doi:10.1093/ofid/ofz479
9. Johnson JA, Feeney ER, Kubiak DW, Corey GR. Prolonged use of oritavancin for vancomycin-resistant Enterococcus faecium prosthetic valve endocarditis. Open Forum Infect Dis. 2015;2(4):ofv156. doi:10.1093/ofid/ofv156
10. Schulz LT, Dworkin E, Dela-Pena J, Rose WE. Multipledose oritavancin evaluation in a retrospective cohort of patients with complicated infections. Pharmacotherapy. 2018;38(1):152-159. doi:10.1002/phar.2057
11. Stewart CL, Turner MS, Frens JJ, Snider CB, Smith JR. Real-world experience with oritavancin therapy in invasive gram-positive infections. Infect Dis Ther. 2017;6(2):277-289. doi:10.1007/s40121-017-0156-z
12. Delaportas DJ, Estrada SJ, Darmelio M. Successful treatment of methicillin susceptible Staphylococcus aureus osteomyelitis with oritavancin. Pharmacotherapy. 2017;37(8):e90-e92. doi:10.1002/phar.1957
13. Chastain DB, Davis A. Treatment of chronic osteomyelitis with multidose oritavancin: a case series and literature review. Int J Antimicrob Agents. 2019;53(4):429-434. doi:10.1016/j.ijantimicag.2018.11.023
14. Dahesh S, Wong B, Nizet V, Sakoulas G, Tran TT, Aitken SL. Treatment of multidrug-resistant vancomycinresistant Enterococcus faecium hardware-associated vertebral osteomyelitis with oritavancin plus ampicillin. Antimicrob Agents Chemother. 2019;63(7):e02622-18. doi:10.1128/AAC.02622-18
15. Foster RA, Philavong KP, Weissman S, Tang X, Bookstaver PB. Oritavancin for the treatment of daptomycin nonsusceptible vancomycin-resistant Enterococci osteomyelitis. Infect Dis Clin Pract. 2018;26(2):97-99. doi:10.1097/IPC.0000000000000517
16. Ruggero M, Ziegler M, Tebas P, Binkley A, Kelly B. Successful treatment of methicillin-resistant Staphylococcus aureus vertebral osteomyelitis with outpatient oritavancin therapy. Infect Dis Clin Pract. 2018;26(3):141-144. doi:10.1097/IPC.0000000000000599
17. Corey GR, Loutit J, Moeck G, et al. Single intravenous dose of oritavancin for treatment of acute skin and skin structure infections caused by gram-positive bacteria: summary of safety analysis from the phase 3 SOLO studies. Antimicrob Agents Chemother. 2018;62(4):e01919- 17. doi:10.1128/AAC.01919-17
1. Orbactiv [package insert]. Parsippany, NJ: The Medicines Company; 2019.
2. Corey GR, Kabler H, Mehra P, et al. Single-dose oritavancin in the treatment of acute bacterial skin infections. N Engl J Med. 2014;370(23):2180-2190. doi:10.1056/NEJMoa1310422
3. Corey GR, Good S, Jiang H, et al. Single-dose oritavancin versus 7-10 days of vancomycin in the treatment of gram-positive acute bacterial skin and skin structure infections: the SOLO II noninferiority study. Clin Infect Dis. 2015;60(2):254-262. doi:10.1093/cid/ciu778
4. Sweeney D, Stoneburner A, Shinabarger DL, et al. Comparative in vitro activity of oritavancin and other agents against vancomycin-susceptible and -resistant enterococci. J Antimicrob Chemother. 2017;72(2):622-624. doi.10.1093/jac/dkw451
5. Lehoux D, Ostiguy V, Vadieux C, et al. Oritavancin pharmacokinetics and bone penetration in rabbits. Antimicrob Agents Chemother. 2015;59(10):6501-6505. doi:10.1128/AAC.00981-15
6. Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52(3):e18-e55. doi:10.1093/cid/ciq146
7. Norris AH, Shrestha NK, Allison GM, et al. 2018 Infectious Diseases Society of America clinical practice guideline for the management of outpatient parenteral antimicrobial therapy. Clin Infect Dis. 2019;68(1):e1-e35. doi:10.1093/cid/ciy745
8. Redell M, Seirra-Hoffman M, Assi Maha, et al. The CHROME study, a real-world experience of single- and multiple-dose oritavancin for treatment of gram-positive infections. Open Forum Infect Dis. 2019;6(11):ofz479. doi:10.1093/ofid/ofz479
9. Johnson JA, Feeney ER, Kubiak DW, Corey GR. Prolonged use of oritavancin for vancomycin-resistant Enterococcus faecium prosthetic valve endocarditis. Open Forum Infect Dis. 2015;2(4):ofv156. doi:10.1093/ofid/ofv156
10. Schulz LT, Dworkin E, Dela-Pena J, Rose WE. Multipledose oritavancin evaluation in a retrospective cohort of patients with complicated infections. Pharmacotherapy. 2018;38(1):152-159. doi:10.1002/phar.2057
11. Stewart CL, Turner MS, Frens JJ, Snider CB, Smith JR. Real-world experience with oritavancin therapy in invasive gram-positive infections. Infect Dis Ther. 2017;6(2):277-289. doi:10.1007/s40121-017-0156-z
12. Delaportas DJ, Estrada SJ, Darmelio M. Successful treatment of methicillin susceptible Staphylococcus aureus osteomyelitis with oritavancin. Pharmacotherapy. 2017;37(8):e90-e92. doi:10.1002/phar.1957
13. Chastain DB, Davis A. Treatment of chronic osteomyelitis with multidose oritavancin: a case series and literature review. Int J Antimicrob Agents. 2019;53(4):429-434. doi:10.1016/j.ijantimicag.2018.11.023
14. Dahesh S, Wong B, Nizet V, Sakoulas G, Tran TT, Aitken SL. Treatment of multidrug-resistant vancomycinresistant Enterococcus faecium hardware-associated vertebral osteomyelitis with oritavancin plus ampicillin. Antimicrob Agents Chemother. 2019;63(7):e02622-18. doi:10.1128/AAC.02622-18
15. Foster RA, Philavong KP, Weissman S, Tang X, Bookstaver PB. Oritavancin for the treatment of daptomycin nonsusceptible vancomycin-resistant Enterococci osteomyelitis. Infect Dis Clin Pract. 2018;26(2):97-99. doi:10.1097/IPC.0000000000000517
16. Ruggero M, Ziegler M, Tebas P, Binkley A, Kelly B. Successful treatment of methicillin-resistant Staphylococcus aureus vertebral osteomyelitis with outpatient oritavancin therapy. Infect Dis Clin Pract. 2018;26(3):141-144. doi:10.1097/IPC.0000000000000599
17. Corey GR, Loutit J, Moeck G, et al. Single intravenous dose of oritavancin for treatment of acute skin and skin structure infections caused by gram-positive bacteria: summary of safety analysis from the phase 3 SOLO studies. Antimicrob Agents Chemother. 2018;62(4):e01919- 17. doi:10.1128/AAC.01919-17
The Effect of Radium-223 Therapy in Agent Orange-Related Prostate Carcinoma
Patients with metastatic castrate resistant prostate carcinoma (CRPC) have several treatment options, including radium-223 dichloride (Ra-223) radionuclide therapy, abiraterone, enzalutamide, and cabazitaxel. Ra-223 therapy has been reported to increase median survival in patients with bone metastatic prostate carcinoma.1,2 However, ERA 223 trial data showed an increase of bone fractures with combination of Ra-223 and abiraterone.3
Agent Orange (AO) exposure has been studied as a potential risk factor for development of prostate carcinoma. AO was a commercially manufactured defoliate that was sprayed extensively during the Vietnam War. Due to a side product of chemical manufacturing, AO was contaminated with the toxin 2,3,7,8-tetrachlorodibenzo-p-dioxin, a putative carcinogen. These dioxins can enter the food chain through soil contamination. There is enough evidence to link AO to hematologic malignancies and several solid tumors, including prostate carcinoma.4 Although no real estimates exist for what percentage of Vietnam veterans experienced AO exposure, Surveillance, Epidemiology, and End Results data showed that about 3 million veterans served in Southeast Asia where AO was used extensively in the combat theater. AO has been reported to be positively associated with a 52% increase in risk of prostate carcinoma detection at initial prostate biopsy.5
There has been no reported study of treatment efficacy in veterans with AO-related prostate carcinoma. We present a retrospective study of Ra-223 and other therapies in metastatic CRPC. The purpose of this study was to compare response to therapy and survival in veterans exposed to agent orange (AO+) vs veterans who were not exposed to (AO-) in a single US Department of Veteran Affairs (VA) medical center.
Methods
This was a retrospective study of veterans with metastatic CRPC to bones who received Ra-223 radionuclide therapy with standard dose of 50 kBq per kg of body weight and other sequential therapies at VA Pittsburgh Healthcare System (VAPHS) from January 2014 to January 2019. The purpose of this study was to measure difference in treatment outcome between AO+ veterans and AO- veterans.
Eligibility Criteria
All veterans had a history that included bone metastasis CRPC. They could have 2 to 3 small lymphadenopathies but not visceral metastasis. They received a minimum of 3 cycles and a maximum of 6 cycles of Ra-223 therapy, which was given in 4-week intervals. Pretreatment criteria was hemoglobin > 10 g/dL, platelet > 100
Statistics
Time to study was calculated from the initiation of Ra-223 therapy. Time to skeletal-related events (SRE), progression of prostate specific antigen (PSA), bone metastasis, and alkaline phosphatase (ALP) were calculated in months, using unpaired t test with 2-tailed P value. Median survival was calculated in months by Kaplan Meier R log-rank test Definition).
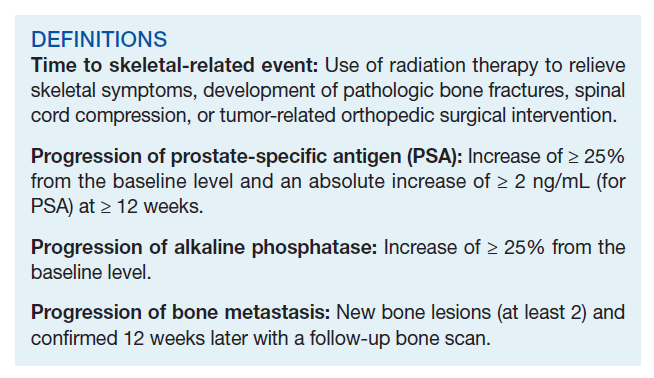
Results
Forty-eight veterans with bone metastasis CRPC received Ra-223 therapy. Of those, 34 veterans were eligible for this retrospective study: 17 AO+ veterans and 17 AO- veterans. Mean age of diagnosis was 62 years (AO+) and 69 years (AO-) (P = .005). Mean Gleason score was 8.2 (AO+) and 8.0 (AO-) (P = .705). Veterans received initial therapy at diagnosis of prostate carcinoma, including radical prostatectomy (6 AO+ and 3 AO-), localized radiation therapy (3 AO+ and 5 AO-), and ADT (8 AO+ and 9 AO-) (Table 1).
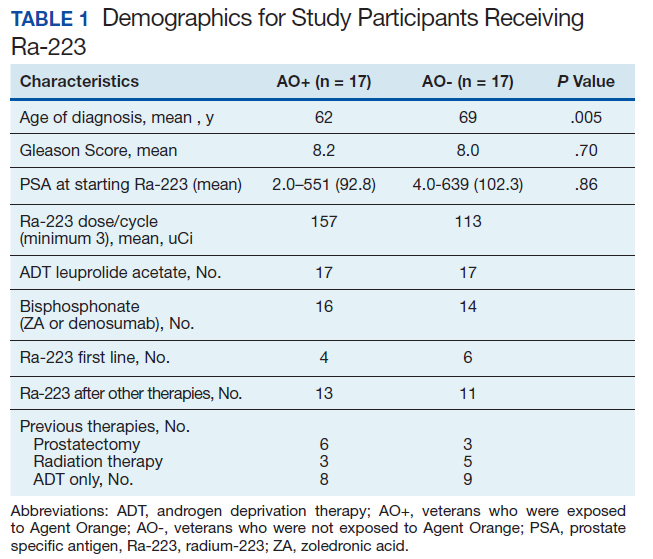
Mean PSA at the initiation of Ra-223 therapy for AO+ was 92.8 (range, 2-551) and for AO- was 102.3 (range, 4-639; P = .86). Mean Ra-223 dose per cycle for AO+ and AO- was 157 uCi and 113 uCi, respectively. All 34 veterans received ADT (leuprolide acetate), and 30 veterans (16 AO+ and 14 AO-) received bisphosphonates (zoledronic acid or denosumab). A total of 10 veterans (29%) received Ra-223 as a first-line therapy (4 AO+ and 6 AO-), and 24 veterans (71%) received Ra-223 after hormonal or chemotherapy (13 AO+ and 11 AO-).
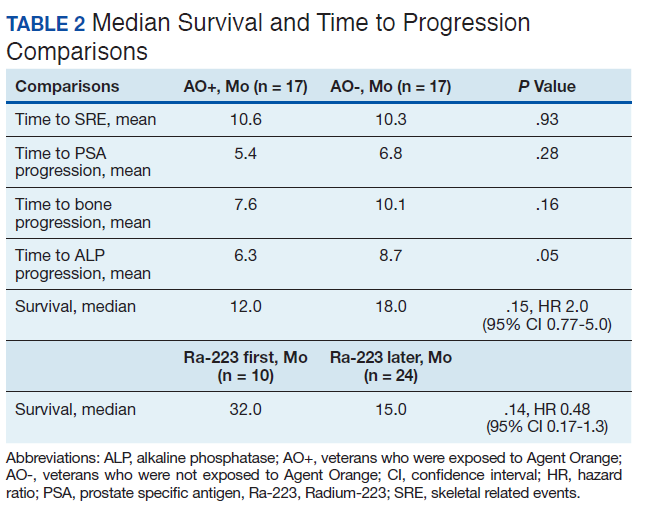
There were 12 SRE (8 AO+ and 4 AO-). Mean time to SRE for AO+ was 10.6 months and AO- was 10.3 months (P = .93). Three veterans received concurrent Ra-223 and abiraterone (participated in ERA 223 trial). Two AO+ veterans experienced SRE at 7 months and 11 months, respectively. Mean time to PSA progression for AO+ was 5.4 months and for AO- was 6.8 months (P = .28). Mean time to bone progression for AO+ and for AO- were 7.6 months and 10.1 months, respectively (P = .16). Mean time to ALP progression for AO+ and AO- were 6.3 months and 8.7 months, respectively (P = .05). (Table 2). The treatment pattern of AO+ and AO- is depicted on a swimmer plot (Figures 1 and 2).

Twenty veterans (58%) had died: 13 AO+ and 7 AO- veterans. Median survival for Ra-223 first and Ra-223 later was was 32 months and 15 months, respectively (P = .14; hazard ratio [HR], 0.48). Overall median survival for AO+ veterans and AO- veterans were 12 months and 18 months, respectively (P = .15; HR, 2.0) (Figures 3 and 4).

Discussions
There has been no reported VA study of using Ra-223 and other therapies (hormonal and chemotherapy) in veterans exposed to AO. This is the first retrospective study to compare the response and survival between AO+ and AO- veterans. Even though this study featured a small sample, it is interesting to note the difference between those 2 populations. There was 1 prior study in veterans with prostate carcinoma using radiotherapy (brachytherapy) in early-stage disease. Everly and colleagues reported that AO+ veterans were less likely to remain biochemically controlled compared with AO- and nonveteran patients with prostate carcinoma.4
Ansbaugh and colleagues reported that AO was associated with a 75% increase in the risk of Gleason ≥ 7 and a 110% increase in Gleason ≥ 8. AO+ veterans are at risk for the detection of high-grade prostate carcinoma. They also tend to have an average age of diagnosis that is 4 to 5 years younger than AO- veterans.6
Our study revealed that AO+ veterans were diagnosed at a younger age (mean 62 years) compared with that of AO- veterans (mean 69 years, P = .005). We also proved that AO veterans have a higher mean Gleason score (8.2) compared with that of AO- veterans (8.0). Veterans received therapy at the time of diagnosis of prostate carcinoma with either radical prostatectomy, radiation therapy, or ADT with leuprolide acetate. Mean PSA at the start of Ra-223 therapy for AO+ was 92.8 (range, 2-551); for AO- was 102.3 (range, 4-639), which is not statistically significant.
Ra-223, an
In a phase 3, randomized, double-blind, placebo-controlled study by Parker and colleagues (ALSYMPCA study), 921 patients who had received, were not eligible to receive, or declined docetaxel, in a 2:1 ratio, were randomized to receive 6 injections of Ra-223 or matching placebo.2 Ra-223 significantly improved overall survival (OS) (median, 14.9 months vs 11.3 months) compared with that of placebo. Ra-223 also prolonged the time to the first symptomatic SRE (median, 15.6 months vs 9.8 months), the time to an increase in the total ALP level (median 7.4 months vs 3.8 months), and the time to an increase in the PSA level (median 3.6 months vs 3.4 months).2
In our study, the mean time to SRE for AO+ was 10.6 months and AO- was 10.3 months (P = .93). Mean time to PSA progression for AO+ was 5.4 months and for AO- was 6.8 months (P = .28). Mean time to bone progression for AO+ and for AO- were 7.6 months and 10.1 months respectively (P = .16). Mean time to ALP progression for AO+ and AO- were 6.3 months and 8.7 months respectively (P = .05). There is a trend of shorter PSA progression, bone progression, and ALP progression in AO+ veterans, though these were not statistically significant due to small sample population. In our study the median survival in for AO- was 18 months and for AO+ was 12 months, which is comparable with median survival of 14.9 months from the ALSYMPCA study.
There were 12 veterans who developed SREs. All received radiation therapy due to bone progression or impending fracture. AO+ veterans developed more SREs (n = 8) when compared with AO- veterans (n = 4). There were more AO- veterans alive (n = 10) than there were AO+ veterans (n = 4). The plausible explanation of this may be due to the aggressive pattern of prostate carcinoma in AO+ veterans (younger age and higher Gleason score).
VAPHS participated in the ERA trial between 2014 and 2016. The trial enrolled 806 patients who were randomly assigned to receive first-line Ra-223 or placebo in addition to abiraterone acetate plus prednisone.3 The study was unblinded prematurely after more fractures and deaths were noted in the Ra-223 and abiraterone group than there were in the placebo and abiraterone group. Median symptomatic SRE was 22.3 months in the Ra-223 group and 26.0 months in the placebo group. Fractures (any grade) occurred in 29% in the Ra-223 group and 11% in the placebo group. It was suggested that Ra-223 could contribute to the risk of osteoporotic fractures in patients with bone metastatic prostate carcinoma. Median OS was 30.7 months in the Ra-223 group and 33.3 months in the placebo group.
We enrolled 3 veterans in the ERA clinical trial. Two AO+ veterans had SREs at 7 months and 11 months. In our study, the median OS for Ra-223 first line was 32 months, which is comparable with median survival of 30.7 months from ERA-223 study. Median survival for Ra-223 later was only 15 months. We recommend veterans with at least 2 to 3-bone metastasis receive Ra-223 in the first-line setting rather than second- or third-line setting. In this retrospective study with Ra-223 and other therapies, we proved that AO+ veterans have a worse response and OS when compared with that of AO- veterans.
Conclusions
This is the first VA study to compare the efficacy of Ra-223 and other therapies in metastatic CRPC between AO+ and AO- veterans. AO+ veterans were diagnosed at a younger age and had higher Gleason scores. There was no statistical difference between AO+ and AO- veterans in terms of time to SRE, PSA progression, and bone and ALP progression even though there was a trend of shorter duration in AO+ veterans. There was no median survival difference between veterans who received Ra-223 first vs Ra-223 later as well as between AO+ and AO- veterans, but there was a trend of worse survival in veteran who received Ra-223 later and AO+ veterans.
This study showed that AO+ veterans have a shorter duration of response to therapy and shorter median survival compared with that of AO- veterans. We recommend that veterans should get Ra-223 in the first-line setting rather than after hormonal therapy and chemotherapy because their marrows are still intact. We need to investigate further whether veterans that exposed to carcinogen 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) may have different molecular biology and as such may cause inferior efficacy in the treatment of prostate carcinoma.
1. Shore ND. Radium-223 dichloride for metastatic castration-resistant prostate cancer: the urologist’s perspective. Urology. 2015;85(4):717-724. doi:10.1016/j.urology.2014.11.031
2. Parker C, Nilsson S, Heinrich D, et al; ALSYMPCA Investigators. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369(3):213-223. doi:10.1056/NEJMoa1213755
3. Smith M, Parker C, Saad F, et al. Addition of radium-223 to abiraterone acetate and prednisone or prednisolone in patients with castration-resistant prostate cancer and bone metastases (ERA 223): a randomized, double-blind, placebo-controlled, phase 3 trial [published correction appears in Lancet Oncol. 2019 Oct;20(10):e559]. Lancet Oncol. 2019;20(3):408-419. doi:10.1016/S1470-2045(18)30860-X
4. Everly L, Merrick GS, Allen ZA, et al. Prostate cancer control and survival in Vietnam veterans exposed to Agent Orange. Brachytherapy. 2009;8(1):57-62. doi: 10.1016/j.brachy.2008.08.001
5. Altekruse S. SEER Cancer Statistics Review, 1975-2017 Bethesda, MD: National Cancer Institute. 2009. 6. Ansbaugh N, Shannon J, Mori M, Farris PE, Garzotto M. Agent Orange as a risk factor for high-grade prostate cancer. Cancer. 2013;119(13):2399-2404. doi:10.1002/cncr.27941
7. Jadvar H, Quinn DI. Targeted α-particle therapy of bone metastases in prostate cancer. Clin Nucl Med. 2013;38(12):966-971. doi:10.1097/RLU.0000000000000290
Patients with metastatic castrate resistant prostate carcinoma (CRPC) have several treatment options, including radium-223 dichloride (Ra-223) radionuclide therapy, abiraterone, enzalutamide, and cabazitaxel. Ra-223 therapy has been reported to increase median survival in patients with bone metastatic prostate carcinoma.1,2 However, ERA 223 trial data showed an increase of bone fractures with combination of Ra-223 and abiraterone.3
Agent Orange (AO) exposure has been studied as a potential risk factor for development of prostate carcinoma. AO was a commercially manufactured defoliate that was sprayed extensively during the Vietnam War. Due to a side product of chemical manufacturing, AO was contaminated with the toxin 2,3,7,8-tetrachlorodibenzo-p-dioxin, a putative carcinogen. These dioxins can enter the food chain through soil contamination. There is enough evidence to link AO to hematologic malignancies and several solid tumors, including prostate carcinoma.4 Although no real estimates exist for what percentage of Vietnam veterans experienced AO exposure, Surveillance, Epidemiology, and End Results data showed that about 3 million veterans served in Southeast Asia where AO was used extensively in the combat theater. AO has been reported to be positively associated with a 52% increase in risk of prostate carcinoma detection at initial prostate biopsy.5
There has been no reported study of treatment efficacy in veterans with AO-related prostate carcinoma. We present a retrospective study of Ra-223 and other therapies in metastatic CRPC. The purpose of this study was to compare response to therapy and survival in veterans exposed to agent orange (AO+) vs veterans who were not exposed to (AO-) in a single US Department of Veteran Affairs (VA) medical center.
Methods
This was a retrospective study of veterans with metastatic CRPC to bones who received Ra-223 radionuclide therapy with standard dose of 50 kBq per kg of body weight and other sequential therapies at VA Pittsburgh Healthcare System (VAPHS) from January 2014 to January 2019. The purpose of this study was to measure difference in treatment outcome between AO+ veterans and AO- veterans.
Eligibility Criteria
All veterans had a history that included bone metastasis CRPC. They could have 2 to 3 small lymphadenopathies but not visceral metastasis. They received a minimum of 3 cycles and a maximum of 6 cycles of Ra-223 therapy, which was given in 4-week intervals. Pretreatment criteria was hemoglobin > 10 g/dL, platelet > 100
Statistics
Time to study was calculated from the initiation of Ra-223 therapy. Time to skeletal-related events (SRE), progression of prostate specific antigen (PSA), bone metastasis, and alkaline phosphatase (ALP) were calculated in months, using unpaired t test with 2-tailed P value. Median survival was calculated in months by Kaplan Meier R log-rank test Definition).
Results
Forty-eight veterans with bone metastasis CRPC received Ra-223 therapy. Of those, 34 veterans were eligible for this retrospective study: 17 AO+ veterans and 17 AO- veterans. Mean age of diagnosis was 62 years (AO+) and 69 years (AO-) (P = .005). Mean Gleason score was 8.2 (AO+) and 8.0 (AO-) (P = .705). Veterans received initial therapy at diagnosis of prostate carcinoma, including radical prostatectomy (6 AO+ and 3 AO-), localized radiation therapy (3 AO+ and 5 AO-), and ADT (8 AO+ and 9 AO-) (Table 1).
Mean PSA at the initiation of Ra-223 therapy for AO+ was 92.8 (range, 2-551) and for AO- was 102.3 (range, 4-639; P = .86). Mean Ra-223 dose per cycle for AO+ and AO- was 157 uCi and 113 uCi, respectively. All 34 veterans received ADT (leuprolide acetate), and 30 veterans (16 AO+ and 14 AO-) received bisphosphonates (zoledronic acid or denosumab). A total of 10 veterans (29%) received Ra-223 as a first-line therapy (4 AO+ and 6 AO-), and 24 veterans (71%) received Ra-223 after hormonal or chemotherapy (13 AO+ and 11 AO-).
There were 12 SRE (8 AO+ and 4 AO-). Mean time to SRE for AO+ was 10.6 months and AO- was 10.3 months (P = .93). Three veterans received concurrent Ra-223 and abiraterone (participated in ERA 223 trial). Two AO+ veterans experienced SRE at 7 months and 11 months, respectively. Mean time to PSA progression for AO+ was 5.4 months and for AO- was 6.8 months (P = .28). Mean time to bone progression for AO+ and for AO- were 7.6 months and 10.1 months, respectively (P = .16). Mean time to ALP progression for AO+ and AO- were 6.3 months and 8.7 months, respectively (P = .05). (Table 2). The treatment pattern of AO+ and AO- is depicted on a swimmer plot (Figures 1 and 2).
Twenty veterans (58%) had died: 13 AO+ and 7 AO- veterans. Median survival for Ra-223 first and Ra-223 later was was 32 months and 15 months, respectively (P = .14; hazard ratio [HR], 0.48). Overall median survival for AO+ veterans and AO- veterans were 12 months and 18 months, respectively (P = .15; HR, 2.0) (Figures 3 and 4).
Discussions
There has been no reported VA study of using Ra-223 and other therapies (hormonal and chemotherapy) in veterans exposed to AO. This is the first retrospective study to compare the response and survival between AO+ and AO- veterans. Even though this study featured a small sample, it is interesting to note the difference between those 2 populations. There was 1 prior study in veterans with prostate carcinoma using radiotherapy (brachytherapy) in early-stage disease. Everly and colleagues reported that AO+ veterans were less likely to remain biochemically controlled compared with AO- and nonveteran patients with prostate carcinoma.4
Ansbaugh and colleagues reported that AO was associated with a 75% increase in the risk of Gleason ≥ 7 and a 110% increase in Gleason ≥ 8. AO+ veterans are at risk for the detection of high-grade prostate carcinoma. They also tend to have an average age of diagnosis that is 4 to 5 years younger than AO- veterans.6
Our study revealed that AO+ veterans were diagnosed at a younger age (mean 62 years) compared with that of AO- veterans (mean 69 years, P = .005). We also proved that AO veterans have a higher mean Gleason score (8.2) compared with that of AO- veterans (8.0). Veterans received therapy at the time of diagnosis of prostate carcinoma with either radical prostatectomy, radiation therapy, or ADT with leuprolide acetate. Mean PSA at the start of Ra-223 therapy for AO+ was 92.8 (range, 2-551); for AO- was 102.3 (range, 4-639), which is not statistically significant.
Ra-223, an
In a phase 3, randomized, double-blind, placebo-controlled study by Parker and colleagues (ALSYMPCA study), 921 patients who had received, were not eligible to receive, or declined docetaxel, in a 2:1 ratio, were randomized to receive 6 injections of Ra-223 or matching placebo.2 Ra-223 significantly improved overall survival (OS) (median, 14.9 months vs 11.3 months) compared with that of placebo. Ra-223 also prolonged the time to the first symptomatic SRE (median, 15.6 months vs 9.8 months), the time to an increase in the total ALP level (median 7.4 months vs 3.8 months), and the time to an increase in the PSA level (median 3.6 months vs 3.4 months).2
In our study, the mean time to SRE for AO+ was 10.6 months and AO- was 10.3 months (P = .93). Mean time to PSA progression for AO+ was 5.4 months and for AO- was 6.8 months (P = .28). Mean time to bone progression for AO+ and for AO- were 7.6 months and 10.1 months respectively (P = .16). Mean time to ALP progression for AO+ and AO- were 6.3 months and 8.7 months respectively (P = .05). There is a trend of shorter PSA progression, bone progression, and ALP progression in AO+ veterans, though these were not statistically significant due to small sample population. In our study the median survival in for AO- was 18 months and for AO+ was 12 months, which is comparable with median survival of 14.9 months from the ALSYMPCA study.
There were 12 veterans who developed SREs. All received radiation therapy due to bone progression or impending fracture. AO+ veterans developed more SREs (n = 8) when compared with AO- veterans (n = 4). There were more AO- veterans alive (n = 10) than there were AO+ veterans (n = 4). The plausible explanation of this may be due to the aggressive pattern of prostate carcinoma in AO+ veterans (younger age and higher Gleason score).
VAPHS participated in the ERA trial between 2014 and 2016. The trial enrolled 806 patients who were randomly assigned to receive first-line Ra-223 or placebo in addition to abiraterone acetate plus prednisone.3 The study was unblinded prematurely after more fractures and deaths were noted in the Ra-223 and abiraterone group than there were in the placebo and abiraterone group. Median symptomatic SRE was 22.3 months in the Ra-223 group and 26.0 months in the placebo group. Fractures (any grade) occurred in 29% in the Ra-223 group and 11% in the placebo group. It was suggested that Ra-223 could contribute to the risk of osteoporotic fractures in patients with bone metastatic prostate carcinoma. Median OS was 30.7 months in the Ra-223 group and 33.3 months in the placebo group.
We enrolled 3 veterans in the ERA clinical trial. Two AO+ veterans had SREs at 7 months and 11 months. In our study, the median OS for Ra-223 first line was 32 months, which is comparable with median survival of 30.7 months from ERA-223 study. Median survival for Ra-223 later was only 15 months. We recommend veterans with at least 2 to 3-bone metastasis receive Ra-223 in the first-line setting rather than second- or third-line setting. In this retrospective study with Ra-223 and other therapies, we proved that AO+ veterans have a worse response and OS when compared with that of AO- veterans.
Conclusions
This is the first VA study to compare the efficacy of Ra-223 and other therapies in metastatic CRPC between AO+ and AO- veterans. AO+ veterans were diagnosed at a younger age and had higher Gleason scores. There was no statistical difference between AO+ and AO- veterans in terms of time to SRE, PSA progression, and bone and ALP progression even though there was a trend of shorter duration in AO+ veterans. There was no median survival difference between veterans who received Ra-223 first vs Ra-223 later as well as between AO+ and AO- veterans, but there was a trend of worse survival in veteran who received Ra-223 later and AO+ veterans.
This study showed that AO+ veterans have a shorter duration of response to therapy and shorter median survival compared with that of AO- veterans. We recommend that veterans should get Ra-223 in the first-line setting rather than after hormonal therapy and chemotherapy because their marrows are still intact. We need to investigate further whether veterans that exposed to carcinogen 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) may have different molecular biology and as such may cause inferior efficacy in the treatment of prostate carcinoma.
Patients with metastatic castrate resistant prostate carcinoma (CRPC) have several treatment options, including radium-223 dichloride (Ra-223) radionuclide therapy, abiraterone, enzalutamide, and cabazitaxel. Ra-223 therapy has been reported to increase median survival in patients with bone metastatic prostate carcinoma.1,2 However, ERA 223 trial data showed an increase of bone fractures with combination of Ra-223 and abiraterone.3
Agent Orange (AO) exposure has been studied as a potential risk factor for development of prostate carcinoma. AO was a commercially manufactured defoliate that was sprayed extensively during the Vietnam War. Due to a side product of chemical manufacturing, AO was contaminated with the toxin 2,3,7,8-tetrachlorodibenzo-p-dioxin, a putative carcinogen. These dioxins can enter the food chain through soil contamination. There is enough evidence to link AO to hematologic malignancies and several solid tumors, including prostate carcinoma.4 Although no real estimates exist for what percentage of Vietnam veterans experienced AO exposure, Surveillance, Epidemiology, and End Results data showed that about 3 million veterans served in Southeast Asia where AO was used extensively in the combat theater. AO has been reported to be positively associated with a 52% increase in risk of prostate carcinoma detection at initial prostate biopsy.5
There has been no reported study of treatment efficacy in veterans with AO-related prostate carcinoma. We present a retrospective study of Ra-223 and other therapies in metastatic CRPC. The purpose of this study was to compare response to therapy and survival in veterans exposed to agent orange (AO+) vs veterans who were not exposed to (AO-) in a single US Department of Veteran Affairs (VA) medical center.
Methods
This was a retrospective study of veterans with metastatic CRPC to bones who received Ra-223 radionuclide therapy with standard dose of 50 kBq per kg of body weight and other sequential therapies at VA Pittsburgh Healthcare System (VAPHS) from January 2014 to January 2019. The purpose of this study was to measure difference in treatment outcome between AO+ veterans and AO- veterans.
Eligibility Criteria
All veterans had a history that included bone metastasis CRPC. They could have 2 to 3 small lymphadenopathies but not visceral metastasis. They received a minimum of 3 cycles and a maximum of 6 cycles of Ra-223 therapy, which was given in 4-week intervals. Pretreatment criteria was hemoglobin > 10 g/dL, platelet > 100
Statistics
Time to study was calculated from the initiation of Ra-223 therapy. Time to skeletal-related events (SRE), progression of prostate specific antigen (PSA), bone metastasis, and alkaline phosphatase (ALP) were calculated in months, using unpaired t test with 2-tailed P value. Median survival was calculated in months by Kaplan Meier R log-rank test Definition).
Results
Forty-eight veterans with bone metastasis CRPC received Ra-223 therapy. Of those, 34 veterans were eligible for this retrospective study: 17 AO+ veterans and 17 AO- veterans. Mean age of diagnosis was 62 years (AO+) and 69 years (AO-) (P = .005). Mean Gleason score was 8.2 (AO+) and 8.0 (AO-) (P = .705). Veterans received initial therapy at diagnosis of prostate carcinoma, including radical prostatectomy (6 AO+ and 3 AO-), localized radiation therapy (3 AO+ and 5 AO-), and ADT (8 AO+ and 9 AO-) (Table 1).
Mean PSA at the initiation of Ra-223 therapy for AO+ was 92.8 (range, 2-551) and for AO- was 102.3 (range, 4-639; P = .86). Mean Ra-223 dose per cycle for AO+ and AO- was 157 uCi and 113 uCi, respectively. All 34 veterans received ADT (leuprolide acetate), and 30 veterans (16 AO+ and 14 AO-) received bisphosphonates (zoledronic acid or denosumab). A total of 10 veterans (29%) received Ra-223 as a first-line therapy (4 AO+ and 6 AO-), and 24 veterans (71%) received Ra-223 after hormonal or chemotherapy (13 AO+ and 11 AO-).
There were 12 SRE (8 AO+ and 4 AO-). Mean time to SRE for AO+ was 10.6 months and AO- was 10.3 months (P = .93). Three veterans received concurrent Ra-223 and abiraterone (participated in ERA 223 trial). Two AO+ veterans experienced SRE at 7 months and 11 months, respectively. Mean time to PSA progression for AO+ was 5.4 months and for AO- was 6.8 months (P = .28). Mean time to bone progression for AO+ and for AO- were 7.6 months and 10.1 months, respectively (P = .16). Mean time to ALP progression for AO+ and AO- were 6.3 months and 8.7 months, respectively (P = .05). (Table 2). The treatment pattern of AO+ and AO- is depicted on a swimmer plot (Figures 1 and 2).
Twenty veterans (58%) had died: 13 AO+ and 7 AO- veterans. Median survival for Ra-223 first and Ra-223 later was was 32 months and 15 months, respectively (P = .14; hazard ratio [HR], 0.48). Overall median survival for AO+ veterans and AO- veterans were 12 months and 18 months, respectively (P = .15; HR, 2.0) (Figures 3 and 4).
Discussions
There has been no reported VA study of using Ra-223 and other therapies (hormonal and chemotherapy) in veterans exposed to AO. This is the first retrospective study to compare the response and survival between AO+ and AO- veterans. Even though this study featured a small sample, it is interesting to note the difference between those 2 populations. There was 1 prior study in veterans with prostate carcinoma using radiotherapy (brachytherapy) in early-stage disease. Everly and colleagues reported that AO+ veterans were less likely to remain biochemically controlled compared with AO- and nonveteran patients with prostate carcinoma.4
Ansbaugh and colleagues reported that AO was associated with a 75% increase in the risk of Gleason ≥ 7 and a 110% increase in Gleason ≥ 8. AO+ veterans are at risk for the detection of high-grade prostate carcinoma. They also tend to have an average age of diagnosis that is 4 to 5 years younger than AO- veterans.6
Our study revealed that AO+ veterans were diagnosed at a younger age (mean 62 years) compared with that of AO- veterans (mean 69 years, P = .005). We also proved that AO veterans have a higher mean Gleason score (8.2) compared with that of AO- veterans (8.0). Veterans received therapy at the time of diagnosis of prostate carcinoma with either radical prostatectomy, radiation therapy, or ADT with leuprolide acetate. Mean PSA at the start of Ra-223 therapy for AO+ was 92.8 (range, 2-551); for AO- was 102.3 (range, 4-639), which is not statistically significant.
Ra-223, an
In a phase 3, randomized, double-blind, placebo-controlled study by Parker and colleagues (ALSYMPCA study), 921 patients who had received, were not eligible to receive, or declined docetaxel, in a 2:1 ratio, were randomized to receive 6 injections of Ra-223 or matching placebo.2 Ra-223 significantly improved overall survival (OS) (median, 14.9 months vs 11.3 months) compared with that of placebo. Ra-223 also prolonged the time to the first symptomatic SRE (median, 15.6 months vs 9.8 months), the time to an increase in the total ALP level (median 7.4 months vs 3.8 months), and the time to an increase in the PSA level (median 3.6 months vs 3.4 months).2
In our study, the mean time to SRE for AO+ was 10.6 months and AO- was 10.3 months (P = .93). Mean time to PSA progression for AO+ was 5.4 months and for AO- was 6.8 months (P = .28). Mean time to bone progression for AO+ and for AO- were 7.6 months and 10.1 months respectively (P = .16). Mean time to ALP progression for AO+ and AO- were 6.3 months and 8.7 months respectively (P = .05). There is a trend of shorter PSA progression, bone progression, and ALP progression in AO+ veterans, though these were not statistically significant due to small sample population. In our study the median survival in for AO- was 18 months and for AO+ was 12 months, which is comparable with median survival of 14.9 months from the ALSYMPCA study.
There were 12 veterans who developed SREs. All received radiation therapy due to bone progression or impending fracture. AO+ veterans developed more SREs (n = 8) when compared with AO- veterans (n = 4). There were more AO- veterans alive (n = 10) than there were AO+ veterans (n = 4). The plausible explanation of this may be due to the aggressive pattern of prostate carcinoma in AO+ veterans (younger age and higher Gleason score).
VAPHS participated in the ERA trial between 2014 and 2016. The trial enrolled 806 patients who were randomly assigned to receive first-line Ra-223 or placebo in addition to abiraterone acetate plus prednisone.3 The study was unblinded prematurely after more fractures and deaths were noted in the Ra-223 and abiraterone group than there were in the placebo and abiraterone group. Median symptomatic SRE was 22.3 months in the Ra-223 group and 26.0 months in the placebo group. Fractures (any grade) occurred in 29% in the Ra-223 group and 11% in the placebo group. It was suggested that Ra-223 could contribute to the risk of osteoporotic fractures in patients with bone metastatic prostate carcinoma. Median OS was 30.7 months in the Ra-223 group and 33.3 months in the placebo group.
We enrolled 3 veterans in the ERA clinical trial. Two AO+ veterans had SREs at 7 months and 11 months. In our study, the median OS for Ra-223 first line was 32 months, which is comparable with median survival of 30.7 months from ERA-223 study. Median survival for Ra-223 later was only 15 months. We recommend veterans with at least 2 to 3-bone metastasis receive Ra-223 in the first-line setting rather than second- or third-line setting. In this retrospective study with Ra-223 and other therapies, we proved that AO+ veterans have a worse response and OS when compared with that of AO- veterans.
Conclusions
This is the first VA study to compare the efficacy of Ra-223 and other therapies in metastatic CRPC between AO+ and AO- veterans. AO+ veterans were diagnosed at a younger age and had higher Gleason scores. There was no statistical difference between AO+ and AO- veterans in terms of time to SRE, PSA progression, and bone and ALP progression even though there was a trend of shorter duration in AO+ veterans. There was no median survival difference between veterans who received Ra-223 first vs Ra-223 later as well as between AO+ and AO- veterans, but there was a trend of worse survival in veteran who received Ra-223 later and AO+ veterans.
This study showed that AO+ veterans have a shorter duration of response to therapy and shorter median survival compared with that of AO- veterans. We recommend that veterans should get Ra-223 in the first-line setting rather than after hormonal therapy and chemotherapy because their marrows are still intact. We need to investigate further whether veterans that exposed to carcinogen 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) may have different molecular biology and as such may cause inferior efficacy in the treatment of prostate carcinoma.
1. Shore ND. Radium-223 dichloride for metastatic castration-resistant prostate cancer: the urologist’s perspective. Urology. 2015;85(4):717-724. doi:10.1016/j.urology.2014.11.031
2. Parker C, Nilsson S, Heinrich D, et al; ALSYMPCA Investigators. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369(3):213-223. doi:10.1056/NEJMoa1213755
3. Smith M, Parker C, Saad F, et al. Addition of radium-223 to abiraterone acetate and prednisone or prednisolone in patients with castration-resistant prostate cancer and bone metastases (ERA 223): a randomized, double-blind, placebo-controlled, phase 3 trial [published correction appears in Lancet Oncol. 2019 Oct;20(10):e559]. Lancet Oncol. 2019;20(3):408-419. doi:10.1016/S1470-2045(18)30860-X
4. Everly L, Merrick GS, Allen ZA, et al. Prostate cancer control and survival in Vietnam veterans exposed to Agent Orange. Brachytherapy. 2009;8(1):57-62. doi: 10.1016/j.brachy.2008.08.001
5. Altekruse S. SEER Cancer Statistics Review, 1975-2017 Bethesda, MD: National Cancer Institute. 2009. 6. Ansbaugh N, Shannon J, Mori M, Farris PE, Garzotto M. Agent Orange as a risk factor for high-grade prostate cancer. Cancer. 2013;119(13):2399-2404. doi:10.1002/cncr.27941
7. Jadvar H, Quinn DI. Targeted α-particle therapy of bone metastases in prostate cancer. Clin Nucl Med. 2013;38(12):966-971. doi:10.1097/RLU.0000000000000290
1. Shore ND. Radium-223 dichloride for metastatic castration-resistant prostate cancer: the urologist’s perspective. Urology. 2015;85(4):717-724. doi:10.1016/j.urology.2014.11.031
2. Parker C, Nilsson S, Heinrich D, et al; ALSYMPCA Investigators. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369(3):213-223. doi:10.1056/NEJMoa1213755
3. Smith M, Parker C, Saad F, et al. Addition of radium-223 to abiraterone acetate and prednisone or prednisolone in patients with castration-resistant prostate cancer and bone metastases (ERA 223): a randomized, double-blind, placebo-controlled, phase 3 trial [published correction appears in Lancet Oncol. 2019 Oct;20(10):e559]. Lancet Oncol. 2019;20(3):408-419. doi:10.1016/S1470-2045(18)30860-X
4. Everly L, Merrick GS, Allen ZA, et al. Prostate cancer control and survival in Vietnam veterans exposed to Agent Orange. Brachytherapy. 2009;8(1):57-62. doi: 10.1016/j.brachy.2008.08.001
5. Altekruse S. SEER Cancer Statistics Review, 1975-2017 Bethesda, MD: National Cancer Institute. 2009. 6. Ansbaugh N, Shannon J, Mori M, Farris PE, Garzotto M. Agent Orange as a risk factor for high-grade prostate cancer. Cancer. 2013;119(13):2399-2404. doi:10.1002/cncr.27941
7. Jadvar H, Quinn DI. Targeted α-particle therapy of bone metastases in prostate cancer. Clin Nucl Med. 2013;38(12):966-971. doi:10.1097/RLU.0000000000000290