User login
Rifabutin-based triple therapy for H. pylori gets high marks
SAN ANTONIO – David Y. Graham, MD, asserted at the annual meeting of the American College of Gastroenterology.

The drug, recently approved as Talicia, is a rifabutin-based triple therapy. Each capsule contains 50 mg of rifabutin, 1,000 mg of amoxicillin, and 40 mg of omeprazole. As in the pivotal phase 3 trial led by Dr. Graham, the approved treatment regimen calls for adults to take four capsules every 8 hours for 14 days.
The impetus for developing the new therapy centers on the growing problem of resistance to long-standard agents for H. pylori eradication, including metronidazole and clarithromycin. The World Health Organization has declared H. pylori eradication to be a high priority for therapeutic development. Rifabutin resistance is rare: In one study, 413 of 414 strains of H. pylori were sensitive to the antibiotic, noted Dr. Graham, professor of medicine at Baylor College of Medicine, Houston.
He presented the results of the pivotal phase 3, double-blind, multicenter, active comparator trial, known as ERADICATE Hp2, in which 455 participants with confirmed H. pylori infection were randomized to a course of the all-in-one-capsule triple drug combo or to dual therapy with four capsules, each containing 1,000 mg of amoxicillin and 40 mg of omeprazole, every 8 hours for 14 days.
The primary endpoint was H. pylori eradication as documented by a negative urea breath test obtained 4-6 weeks after completing 14 days of treatment. The rate was 84% with the rifabutin-based combo, compared with 58% seen with the high-dose dual therapy. Moreover, in a prespecified secondary analysis restricted to the 391 participants who were confirmed to be actually taking their medication as evidenced by a positive blood level measured on day 13, the eradication rates rose to 90% and 65%, respectively.
The antimicrobial resistance rates documented in this study were eye opening: 17% of patients’ strains were resistant to clarithromycin, 44% to metronidazole, and 10.5% to both. Of concern, 6.4% of participants’ strains were amoxicillin resistant.
“For the first time we saw a low level – but a definite level – of amoxicillin resistance. That’s something we had not seen previously,” Dr. Graham said.
No rifabutin resistance was detected before or after treatment.
The side effect profiles of the two treatment regimens were similar. Diarrhea was reported by 9% of participants, headache by 7%, and nausea by 5%. No serious adverse events occurred in the 14-day study.
The efficacy of the rifabutin-based therapy wasn’t affected by metronidazole or clarithromycin resistance.
The ERADICATE Hp2 trial was sponsored by RedHill Biopharma of Tel Aviv. Dr. Graham reported having no financial conflicts.
SAN ANTONIO – David Y. Graham, MD, asserted at the annual meeting of the American College of Gastroenterology.
The drug, recently approved as Talicia, is a rifabutin-based triple therapy. Each capsule contains 50 mg of rifabutin, 1,000 mg of amoxicillin, and 40 mg of omeprazole. As in the pivotal phase 3 trial led by Dr. Graham, the approved treatment regimen calls for adults to take four capsules every 8 hours for 14 days.
The impetus for developing the new therapy centers on the growing problem of resistance to long-standard agents for H. pylori eradication, including metronidazole and clarithromycin. The World Health Organization has declared H. pylori eradication to be a high priority for therapeutic development. Rifabutin resistance is rare: In one study, 413 of 414 strains of H. pylori were sensitive to the antibiotic, noted Dr. Graham, professor of medicine at Baylor College of Medicine, Houston.
He presented the results of the pivotal phase 3, double-blind, multicenter, active comparator trial, known as ERADICATE Hp2, in which 455 participants with confirmed H. pylori infection were randomized to a course of the all-in-one-capsule triple drug combo or to dual therapy with four capsules, each containing 1,000 mg of amoxicillin and 40 mg of omeprazole, every 8 hours for 14 days.
The primary endpoint was H. pylori eradication as documented by a negative urea breath test obtained 4-6 weeks after completing 14 days of treatment. The rate was 84% with the rifabutin-based combo, compared with 58% seen with the high-dose dual therapy. Moreover, in a prespecified secondary analysis restricted to the 391 participants who were confirmed to be actually taking their medication as evidenced by a positive blood level measured on day 13, the eradication rates rose to 90% and 65%, respectively.
The antimicrobial resistance rates documented in this study were eye opening: 17% of patients’ strains were resistant to clarithromycin, 44% to metronidazole, and 10.5% to both. Of concern, 6.4% of participants’ strains were amoxicillin resistant.
“For the first time we saw a low level – but a definite level – of amoxicillin resistance. That’s something we had not seen previously,” Dr. Graham said.
No rifabutin resistance was detected before or after treatment.
The side effect profiles of the two treatment regimens were similar. Diarrhea was reported by 9% of participants, headache by 7%, and nausea by 5%. No serious adverse events occurred in the 14-day study.
The efficacy of the rifabutin-based therapy wasn’t affected by metronidazole or clarithromycin resistance.
The ERADICATE Hp2 trial was sponsored by RedHill Biopharma of Tel Aviv. Dr. Graham reported having no financial conflicts.
SAN ANTONIO – David Y. Graham, MD, asserted at the annual meeting of the American College of Gastroenterology.
The drug, recently approved as Talicia, is a rifabutin-based triple therapy. Each capsule contains 50 mg of rifabutin, 1,000 mg of amoxicillin, and 40 mg of omeprazole. As in the pivotal phase 3 trial led by Dr. Graham, the approved treatment regimen calls for adults to take four capsules every 8 hours for 14 days.
The impetus for developing the new therapy centers on the growing problem of resistance to long-standard agents for H. pylori eradication, including metronidazole and clarithromycin. The World Health Organization has declared H. pylori eradication to be a high priority for therapeutic development. Rifabutin resistance is rare: In one study, 413 of 414 strains of H. pylori were sensitive to the antibiotic, noted Dr. Graham, professor of medicine at Baylor College of Medicine, Houston.
He presented the results of the pivotal phase 3, double-blind, multicenter, active comparator trial, known as ERADICATE Hp2, in which 455 participants with confirmed H. pylori infection were randomized to a course of the all-in-one-capsule triple drug combo or to dual therapy with four capsules, each containing 1,000 mg of amoxicillin and 40 mg of omeprazole, every 8 hours for 14 days.
The primary endpoint was H. pylori eradication as documented by a negative urea breath test obtained 4-6 weeks after completing 14 days of treatment. The rate was 84% with the rifabutin-based combo, compared with 58% seen with the high-dose dual therapy. Moreover, in a prespecified secondary analysis restricted to the 391 participants who were confirmed to be actually taking their medication as evidenced by a positive blood level measured on day 13, the eradication rates rose to 90% and 65%, respectively.
The antimicrobial resistance rates documented in this study were eye opening: 17% of patients’ strains were resistant to clarithromycin, 44% to metronidazole, and 10.5% to both. Of concern, 6.4% of participants’ strains were amoxicillin resistant.
“For the first time we saw a low level – but a definite level – of amoxicillin resistance. That’s something we had not seen previously,” Dr. Graham said.
No rifabutin resistance was detected before or after treatment.
The side effect profiles of the two treatment regimens were similar. Diarrhea was reported by 9% of participants, headache by 7%, and nausea by 5%. No serious adverse events occurred in the 14-day study.
The efficacy of the rifabutin-based therapy wasn’t affected by metronidazole or clarithromycin resistance.
The ERADICATE Hp2 trial was sponsored by RedHill Biopharma of Tel Aviv. Dr. Graham reported having no financial conflicts.
REPORTING FROM ACG 2019
Lorlatinib induces deep responses in ROS1-positive NSCLC
The tyrosine kinase inhibitor (TKI) lorlatinib showed deep responses and intracranial activity in both TKI-pretreated and TKI-naive patients with advanced ROS1-positive non–small cell lung cancer (NSCLC), according to results from a phase 1-2 trial.
“We investigated the antitumour activity and safety of lorlatinib in advanced, ROS1-positive NSCLC,” wrote Alice T. Shaw, MD, PhD, of Massachusetts General Hospital Cancer Center, Boston, and colleagues. Their report is in The Lancet Oncology.
The single-arm, open-label study included 69 patients with advanced ROS1-positive disease with or without CNS involvement. The effects of lorlatinib were evaluated across 28 institutions in 12 different countries around the globe.
At baseline, the median age of study participants was 54 years (range, 44-61 years), and 57% were positive for brain metastases.
Study participants received 100 mg of oral lorlatinib once daily in repeated 21-day cycles. Drug therapy was continued until death, disease progression, unacceptable toxicity, or withdrawal of consent.
The primary outcome measured was intracranial and overall response. Activity outcomes were evaluated in subjects given a minimum of one dose of lorlatinib.
A total of 58% of patients were previously treated with crizotinib, while 30% of patients were TKI-naive. Among 40 crizotinib-pretreated patients, 14 patients (35%) had an objective response, with a median duration of response and PFS of 13.8 and 8.5 months, respectively.
Among 21 TKI-naive patients, 13 patients (62%) had an objective response, with a median duration of response and PFS of 25.3 and 21 months, respectively.
“Intracranial responses were achieved in seven (64%) of 11 TKI-naive patients and 12 (50%) of 24 previous crizotinib-only patients,” they reported.
With respect to safety, serious lorlatinib-related adverse events were observed in 7% of patients, with no therapy-related deaths reported. The most frequently seen grade 3-4 TEAEs were hypertriglyceridemia (19%) and hypercholesterolemia (14%).
The researchers noted a key limitation of the study was the small sample size; however, due to the rare nature of ROS1 rearrangements in patients with NSCLC, increasing enrollment for future studies could be challenging.
“Because crizotinib-refractory patients have few treatment options, lorlatinib could represent an important next-line targeted agent,” they concluded.
Pfizer funded the study. The authors reported financial affiliations with Ariad, Blueprint Medicines, Chugai Pharmaceutical, Daiichi Sankyo, EMD Serono, Pfizer, KSQ Therapeutics, Servier, TP Therapeutics, and other companies.
SOURCE: Shaw AT et al. Lancet Oncol. 2019 Oct 25. doi: 10.1016/S1470-2045(19)30655-2.
The tyrosine kinase inhibitor (TKI) crizotinib was recently established as an optimal first-line treatment option for patients with ROS1-positive non–small cell lung cancer (NSCLC). Despite strong efficacy seen in clinical trials, disease progression can still occur in patients on crizotinib, often through the development of resistance, which is largely the result of on-target mutations, such as Gly2032Arg.
Early results suggest the novel oral TKI candidate, lorlatinib, a potent inhibitor of the Gly2032Arg mutation, may be a treatment of choice in patients with crizotinib-resistance. Recent phase 1 data showed lorlatinib had antitumor activity in ROS1-positive patients.
Correspondingly, the deep and durable responses reported by Dr. Shaw and colleagues represents a significant milestone for lorlatinib, particularly in the setting of crizotinib resistance, where a paucity of later-line treatment options exist. In comparison to platinum-pemetrexed chemotherapy, lorlatinib is better tolerated and has demonstrated potent intracranial activity, which may prevent or delay CNS progression in the disease.
One question that remains from the current study is whether other ROS1 TKI drug candidates, such as repotrectinib and entrectinib, will show similar results to lorlatinib. Several trials are presently ongoing in an attempt to help answer this, and other remaining questions.
Michaël Duruisseaux, MD, PhD, is affiliated with the Hospices Civils de Lyon (France), Universit é Claude Bernard Lyon. Dr. Duruisseaux reported financial affiliations with Boehringer Ingelheim, Bristol-Myers Squibb, Roche, and Takeda. These comments are adapted from his editorial (Lancet Oncol. 2019 Oct 25. doi: 10.1016/S1470-2045[19]30716-8 ).
The tyrosine kinase inhibitor (TKI) crizotinib was recently established as an optimal first-line treatment option for patients with ROS1-positive non–small cell lung cancer (NSCLC). Despite strong efficacy seen in clinical trials, disease progression can still occur in patients on crizotinib, often through the development of resistance, which is largely the result of on-target mutations, such as Gly2032Arg.
Early results suggest the novel oral TKI candidate, lorlatinib, a potent inhibitor of the Gly2032Arg mutation, may be a treatment of choice in patients with crizotinib-resistance. Recent phase 1 data showed lorlatinib had antitumor activity in ROS1-positive patients.
Correspondingly, the deep and durable responses reported by Dr. Shaw and colleagues represents a significant milestone for lorlatinib, particularly in the setting of crizotinib resistance, where a paucity of later-line treatment options exist. In comparison to platinum-pemetrexed chemotherapy, lorlatinib is better tolerated and has demonstrated potent intracranial activity, which may prevent or delay CNS progression in the disease.
One question that remains from the current study is whether other ROS1 TKI drug candidates, such as repotrectinib and entrectinib, will show similar results to lorlatinib. Several trials are presently ongoing in an attempt to help answer this, and other remaining questions.
Michaël Duruisseaux, MD, PhD, is affiliated with the Hospices Civils de Lyon (France), Universit é Claude Bernard Lyon. Dr. Duruisseaux reported financial affiliations with Boehringer Ingelheim, Bristol-Myers Squibb, Roche, and Takeda. These comments are adapted from his editorial (Lancet Oncol. 2019 Oct 25. doi: 10.1016/S1470-2045[19]30716-8 ).
The tyrosine kinase inhibitor (TKI) crizotinib was recently established as an optimal first-line treatment option for patients with ROS1-positive non–small cell lung cancer (NSCLC). Despite strong efficacy seen in clinical trials, disease progression can still occur in patients on crizotinib, often through the development of resistance, which is largely the result of on-target mutations, such as Gly2032Arg.
Early results suggest the novel oral TKI candidate, lorlatinib, a potent inhibitor of the Gly2032Arg mutation, may be a treatment of choice in patients with crizotinib-resistance. Recent phase 1 data showed lorlatinib had antitumor activity in ROS1-positive patients.
Correspondingly, the deep and durable responses reported by Dr. Shaw and colleagues represents a significant milestone for lorlatinib, particularly in the setting of crizotinib resistance, where a paucity of later-line treatment options exist. In comparison to platinum-pemetrexed chemotherapy, lorlatinib is better tolerated and has demonstrated potent intracranial activity, which may prevent or delay CNS progression in the disease.
One question that remains from the current study is whether other ROS1 TKI drug candidates, such as repotrectinib and entrectinib, will show similar results to lorlatinib. Several trials are presently ongoing in an attempt to help answer this, and other remaining questions.
Michaël Duruisseaux, MD, PhD, is affiliated with the Hospices Civils de Lyon (France), Universit é Claude Bernard Lyon. Dr. Duruisseaux reported financial affiliations with Boehringer Ingelheim, Bristol-Myers Squibb, Roche, and Takeda. These comments are adapted from his editorial (Lancet Oncol. 2019 Oct 25. doi: 10.1016/S1470-2045[19]30716-8 ).
The tyrosine kinase inhibitor (TKI) lorlatinib showed deep responses and intracranial activity in both TKI-pretreated and TKI-naive patients with advanced ROS1-positive non–small cell lung cancer (NSCLC), according to results from a phase 1-2 trial.
“We investigated the antitumour activity and safety of lorlatinib in advanced, ROS1-positive NSCLC,” wrote Alice T. Shaw, MD, PhD, of Massachusetts General Hospital Cancer Center, Boston, and colleagues. Their report is in The Lancet Oncology.
The single-arm, open-label study included 69 patients with advanced ROS1-positive disease with or without CNS involvement. The effects of lorlatinib were evaluated across 28 institutions in 12 different countries around the globe.
At baseline, the median age of study participants was 54 years (range, 44-61 years), and 57% were positive for brain metastases.
Study participants received 100 mg of oral lorlatinib once daily in repeated 21-day cycles. Drug therapy was continued until death, disease progression, unacceptable toxicity, or withdrawal of consent.
The primary outcome measured was intracranial and overall response. Activity outcomes were evaluated in subjects given a minimum of one dose of lorlatinib.
A total of 58% of patients were previously treated with crizotinib, while 30% of patients were TKI-naive. Among 40 crizotinib-pretreated patients, 14 patients (35%) had an objective response, with a median duration of response and PFS of 13.8 and 8.5 months, respectively.
Among 21 TKI-naive patients, 13 patients (62%) had an objective response, with a median duration of response and PFS of 25.3 and 21 months, respectively.
“Intracranial responses were achieved in seven (64%) of 11 TKI-naive patients and 12 (50%) of 24 previous crizotinib-only patients,” they reported.
With respect to safety, serious lorlatinib-related adverse events were observed in 7% of patients, with no therapy-related deaths reported. The most frequently seen grade 3-4 TEAEs were hypertriglyceridemia (19%) and hypercholesterolemia (14%).
The researchers noted a key limitation of the study was the small sample size; however, due to the rare nature of ROS1 rearrangements in patients with NSCLC, increasing enrollment for future studies could be challenging.
“Because crizotinib-refractory patients have few treatment options, lorlatinib could represent an important next-line targeted agent,” they concluded.
Pfizer funded the study. The authors reported financial affiliations with Ariad, Blueprint Medicines, Chugai Pharmaceutical, Daiichi Sankyo, EMD Serono, Pfizer, KSQ Therapeutics, Servier, TP Therapeutics, and other companies.
SOURCE: Shaw AT et al. Lancet Oncol. 2019 Oct 25. doi: 10.1016/S1470-2045(19)30655-2.
The tyrosine kinase inhibitor (TKI) lorlatinib showed deep responses and intracranial activity in both TKI-pretreated and TKI-naive patients with advanced ROS1-positive non–small cell lung cancer (NSCLC), according to results from a phase 1-2 trial.
“We investigated the antitumour activity and safety of lorlatinib in advanced, ROS1-positive NSCLC,” wrote Alice T. Shaw, MD, PhD, of Massachusetts General Hospital Cancer Center, Boston, and colleagues. Their report is in The Lancet Oncology.
The single-arm, open-label study included 69 patients with advanced ROS1-positive disease with or without CNS involvement. The effects of lorlatinib were evaluated across 28 institutions in 12 different countries around the globe.
At baseline, the median age of study participants was 54 years (range, 44-61 years), and 57% were positive for brain metastases.
Study participants received 100 mg of oral lorlatinib once daily in repeated 21-day cycles. Drug therapy was continued until death, disease progression, unacceptable toxicity, or withdrawal of consent.
The primary outcome measured was intracranial and overall response. Activity outcomes were evaluated in subjects given a minimum of one dose of lorlatinib.
A total of 58% of patients were previously treated with crizotinib, while 30% of patients were TKI-naive. Among 40 crizotinib-pretreated patients, 14 patients (35%) had an objective response, with a median duration of response and PFS of 13.8 and 8.5 months, respectively.
Among 21 TKI-naive patients, 13 patients (62%) had an objective response, with a median duration of response and PFS of 25.3 and 21 months, respectively.
“Intracranial responses were achieved in seven (64%) of 11 TKI-naive patients and 12 (50%) of 24 previous crizotinib-only patients,” they reported.
With respect to safety, serious lorlatinib-related adverse events were observed in 7% of patients, with no therapy-related deaths reported. The most frequently seen grade 3-4 TEAEs were hypertriglyceridemia (19%) and hypercholesterolemia (14%).
The researchers noted a key limitation of the study was the small sample size; however, due to the rare nature of ROS1 rearrangements in patients with NSCLC, increasing enrollment for future studies could be challenging.
“Because crizotinib-refractory patients have few treatment options, lorlatinib could represent an important next-line targeted agent,” they concluded.
Pfizer funded the study. The authors reported financial affiliations with Ariad, Blueprint Medicines, Chugai Pharmaceutical, Daiichi Sankyo, EMD Serono, Pfizer, KSQ Therapeutics, Servier, TP Therapeutics, and other companies.
SOURCE: Shaw AT et al. Lancet Oncol. 2019 Oct 25. doi: 10.1016/S1470-2045(19)30655-2.
FROM THE LANCET ONCOLOGY
Cilofexor passes phase 2 for primary biliary cholangitis
BOSTON – Cilofexor, a nonsteroidal farnesoid X receptor (FXR) agonist, can improve disease biomarkers in patients with primary biliary cholangitis (PBC), based on results of a phase 2 trial.

Compared with placebo, patients treated with cilofexor had significant reductions in serum alkaline phosphatase (ALP), gamma-glutamyltransferase (GGT), C-reactive protein (CRP), and primary bile acids, reported lead author Kris V. Kowdley, MD, of Swedish Medical Center in Seattle, and colleagues.
Dr. Kowdley, who presented findings at the annual meeting of the American Association for the Study of Liver Diseases, began by offering some context for the trial.
“There’s a strong rationale for FXR agonist therapy in PBC,” he said. “FXR is the key regulator of bile acid homeostasis, and FXR agonists have shown favorable effects on fibrosis, inflammatory activity, bile acid export and synthesis, as well as possibly effects on the microbiome and downstream in the gut.” He went on to explain that cilofexor may benefit patients with PBC, primary sclerosing cholangitis, or nonalcoholic steatohepatitis (NASH), noting preclinical data that have demonstrated reductions in bile acids, inflammation, fibrosis, and portal pressure.
The present trial involved 71 patients with PBC who lacked cirrhosis and had a serum ALP level that was at least 1.67 times greater than the upper limit of normal, and an elevated serum total bilirubin that was less than 2 times the upper limit of normal. Patients were randomized to receive either cilofexor 30 mg, cilofexor 100 mg, or placebo, once daily for 12 weeks. Stratification was based on use of ursodeoxycholic acid, which was stable for at least the preceding year. Safety and efficacy were evaluated, with the latter based on liver biochemistry, serum C4, bile acids, and serum fibrosis markers.
Across the entire population, baseline median serum bilirubin was 0.6 mg/dL and median serum ALP was 286 U/L. After 12 weeks, compared with placebo, patients treated with cilofexor, particularly those who received the 100-mg dose, showed significant improvements across multiple measures of liver health. Specifically, patients in the 100-mg group achieved median reductions in ALP (–13.8%; P = .005), GGT (–47.7%; P less than .001), CRP (–33.6%; P = .03), and primary bile acids (–30.5%; P = .008). These patients also exhibited trends toward reduced aspartate aminotransferase and aminoterminal propeptide of type III procollagen; Dr. Kowdley attributed the lack of statistical significance to insufficient population size.
Highlighting magnitude of ALP improvement, Dr. Kowdley noted that reductions in ALP greater than 25% were observed in 17% and 18% of patients in the 100-mg and 30-mg cilofexor groups, respectively, versus 0% of patients in the placebo group.
Although the 100-mg dose of cilofexor appeared more effective, the higher dose did come with some trade-offs in tolerability; grade 2 or 3 pruritus was more common in patients treated with the higher dose than in those who received the 30-mg dose (39% vs. 10%). As such, 7% of patients in the 100-mg group discontinued therapy because of the pruritus, compared with no patients in the 30-mg or placebo group.
Responding to a question from a conference attendee, Dr. Kowdley said that ALP reductions to below the 1.67-fold threshold were achieved by 9% and 14% of patients who received the 30-mg dose and 100-mg dose of cilofexor, respectively.
“We believe these data support further evaluation of cilofexor for the treatment of cholestatic liver disorders,” Dr. Kowdley concluded.
The study was funded by Gilead. The investigators disclosed additional relationships with Allergan, Novartis, GlaxoSmithKline, and others.
SOURCE: Kowdley KV et al. The Liver Meeting 2019. Abstract 45.
BOSTON – Cilofexor, a nonsteroidal farnesoid X receptor (FXR) agonist, can improve disease biomarkers in patients with primary biliary cholangitis (PBC), based on results of a phase 2 trial.
Compared with placebo, patients treated with cilofexor had significant reductions in serum alkaline phosphatase (ALP), gamma-glutamyltransferase (GGT), C-reactive protein (CRP), and primary bile acids, reported lead author Kris V. Kowdley, MD, of Swedish Medical Center in Seattle, and colleagues.
Dr. Kowdley, who presented findings at the annual meeting of the American Association for the Study of Liver Diseases, began by offering some context for the trial.
“There’s a strong rationale for FXR agonist therapy in PBC,” he said. “FXR is the key regulator of bile acid homeostasis, and FXR agonists have shown favorable effects on fibrosis, inflammatory activity, bile acid export and synthesis, as well as possibly effects on the microbiome and downstream in the gut.” He went on to explain that cilofexor may benefit patients with PBC, primary sclerosing cholangitis, or nonalcoholic steatohepatitis (NASH), noting preclinical data that have demonstrated reductions in bile acids, inflammation, fibrosis, and portal pressure.
The present trial involved 71 patients with PBC who lacked cirrhosis and had a serum ALP level that was at least 1.67 times greater than the upper limit of normal, and an elevated serum total bilirubin that was less than 2 times the upper limit of normal. Patients were randomized to receive either cilofexor 30 mg, cilofexor 100 mg, or placebo, once daily for 12 weeks. Stratification was based on use of ursodeoxycholic acid, which was stable for at least the preceding year. Safety and efficacy were evaluated, with the latter based on liver biochemistry, serum C4, bile acids, and serum fibrosis markers.
Across the entire population, baseline median serum bilirubin was 0.6 mg/dL and median serum ALP was 286 U/L. After 12 weeks, compared with placebo, patients treated with cilofexor, particularly those who received the 100-mg dose, showed significant improvements across multiple measures of liver health. Specifically, patients in the 100-mg group achieved median reductions in ALP (–13.8%; P = .005), GGT (–47.7%; P less than .001), CRP (–33.6%; P = .03), and primary bile acids (–30.5%; P = .008). These patients also exhibited trends toward reduced aspartate aminotransferase and aminoterminal propeptide of type III procollagen; Dr. Kowdley attributed the lack of statistical significance to insufficient population size.
Highlighting magnitude of ALP improvement, Dr. Kowdley noted that reductions in ALP greater than 25% were observed in 17% and 18% of patients in the 100-mg and 30-mg cilofexor groups, respectively, versus 0% of patients in the placebo group.
Although the 100-mg dose of cilofexor appeared more effective, the higher dose did come with some trade-offs in tolerability; grade 2 or 3 pruritus was more common in patients treated with the higher dose than in those who received the 30-mg dose (39% vs. 10%). As such, 7% of patients in the 100-mg group discontinued therapy because of the pruritus, compared with no patients in the 30-mg or placebo group.
Responding to a question from a conference attendee, Dr. Kowdley said that ALP reductions to below the 1.67-fold threshold were achieved by 9% and 14% of patients who received the 30-mg dose and 100-mg dose of cilofexor, respectively.
“We believe these data support further evaluation of cilofexor for the treatment of cholestatic liver disorders,” Dr. Kowdley concluded.
The study was funded by Gilead. The investigators disclosed additional relationships with Allergan, Novartis, GlaxoSmithKline, and others.
SOURCE: Kowdley KV et al. The Liver Meeting 2019. Abstract 45.
BOSTON – Cilofexor, a nonsteroidal farnesoid X receptor (FXR) agonist, can improve disease biomarkers in patients with primary biliary cholangitis (PBC), based on results of a phase 2 trial.
Compared with placebo, patients treated with cilofexor had significant reductions in serum alkaline phosphatase (ALP), gamma-glutamyltransferase (GGT), C-reactive protein (CRP), and primary bile acids, reported lead author Kris V. Kowdley, MD, of Swedish Medical Center in Seattle, and colleagues.
Dr. Kowdley, who presented findings at the annual meeting of the American Association for the Study of Liver Diseases, began by offering some context for the trial.
“There’s a strong rationale for FXR agonist therapy in PBC,” he said. “FXR is the key regulator of bile acid homeostasis, and FXR agonists have shown favorable effects on fibrosis, inflammatory activity, bile acid export and synthesis, as well as possibly effects on the microbiome and downstream in the gut.” He went on to explain that cilofexor may benefit patients with PBC, primary sclerosing cholangitis, or nonalcoholic steatohepatitis (NASH), noting preclinical data that have demonstrated reductions in bile acids, inflammation, fibrosis, and portal pressure.
The present trial involved 71 patients with PBC who lacked cirrhosis and had a serum ALP level that was at least 1.67 times greater than the upper limit of normal, and an elevated serum total bilirubin that was less than 2 times the upper limit of normal. Patients were randomized to receive either cilofexor 30 mg, cilofexor 100 mg, or placebo, once daily for 12 weeks. Stratification was based on use of ursodeoxycholic acid, which was stable for at least the preceding year. Safety and efficacy were evaluated, with the latter based on liver biochemistry, serum C4, bile acids, and serum fibrosis markers.
Across the entire population, baseline median serum bilirubin was 0.6 mg/dL and median serum ALP was 286 U/L. After 12 weeks, compared with placebo, patients treated with cilofexor, particularly those who received the 100-mg dose, showed significant improvements across multiple measures of liver health. Specifically, patients in the 100-mg group achieved median reductions in ALP (–13.8%; P = .005), GGT (–47.7%; P less than .001), CRP (–33.6%; P = .03), and primary bile acids (–30.5%; P = .008). These patients also exhibited trends toward reduced aspartate aminotransferase and aminoterminal propeptide of type III procollagen; Dr. Kowdley attributed the lack of statistical significance to insufficient population size.
Highlighting magnitude of ALP improvement, Dr. Kowdley noted that reductions in ALP greater than 25% were observed in 17% and 18% of patients in the 100-mg and 30-mg cilofexor groups, respectively, versus 0% of patients in the placebo group.
Although the 100-mg dose of cilofexor appeared more effective, the higher dose did come with some trade-offs in tolerability; grade 2 or 3 pruritus was more common in patients treated with the higher dose than in those who received the 30-mg dose (39% vs. 10%). As such, 7% of patients in the 100-mg group discontinued therapy because of the pruritus, compared with no patients in the 30-mg or placebo group.
Responding to a question from a conference attendee, Dr. Kowdley said that ALP reductions to below the 1.67-fold threshold were achieved by 9% and 14% of patients who received the 30-mg dose and 100-mg dose of cilofexor, respectively.
“We believe these data support further evaluation of cilofexor for the treatment of cholestatic liver disorders,” Dr. Kowdley concluded.
The study was funded by Gilead. The investigators disclosed additional relationships with Allergan, Novartis, GlaxoSmithKline, and others.
SOURCE: Kowdley KV et al. The Liver Meeting 2019. Abstract 45.
REPORTING FROM THE LIVER MEETING 2019
Direct-acting antiviral treatment linked to lower mortality in patients with HCC history
BOSTON – For patients with a complete response following treatment for hepatitis C virus (HCV)–related hepatocellular carcinoma (HCC), treatment with direct-acting antiviral therapy was linked to significantly reduced mortality compared with no such treatment, according to results of a large cohort study.

The mortality benefit associated with direct-acting antiviral (DAA) therapy was consistent across most subgroups, suggesting that the association was driven by achieving sustained virological response (SVR), according to Amit G. Singal, MD, associate professor of medicine at UT Southwestern Medical Center, Dallas.
Those results suggest that DAA therapy in patients with a history of HCC is not only safe, but is also beneficial, Dr. Singal said at the annual meeting of the American Association for the Study of Liver Diseases.
“To be slightly controversial, I think that this changes the paradigm in this subgroup of patients from ‘can be treated’ for their hepatitis C to ‘should be treated,’ ” he concluded in an oral presentation of the results.
While DAA treatment is proven to reduce risk of incident HCC in patients with cirrhosis, the risk-benefit ratio is “less clear” in patients with a history of HCC following complete response, according to Dr. Singal.
Moreover, concerns were raised about the safety of DAA therapy in patients with an HCC history, after early data suggested a potentially higher recurrence risk, he added.
In the current multicenter, retrospective North American cohort study, Dr. Singal and colleagues reviewed data for 797 patients with HCV–related HCC who achieved complete response following ablation, resection, radiotherapy, transarterial chemoembolization, or transarterial radioembolization.
Treatment with DAA therapy was associated with improved overall survival, according to results of multivariable analysis, with a hazard ratio of 0.54 (95% confidence interval, 0.33-0.90). Median time from HCC complete response to death was 25.7 months for the DAA treatment group, versus 11.5 months for the untreated group.
The association between DAA treatment and death was apparently driven by SVR, as reduced mortality was seen in the DAA-treated patients who did achieve SVR, but not in those without SVR, Dr. Singal reported.
While these findings together suggest that DAA treatment is linked to reduced mortality after HCC complete response, prospective studies are needed to confirm this association, Dr. Singal said.
Dr. Singal reported disclosures related to AbbVie, Bayer, BMS, Eisai, Exact Sciences, Exelixis, Genentech, Gilead FOCUS, Glycotest, GRAIL, Merck, Roche, TARGET Pharmasolutions, and Wako Diagnostics.
SOURCE: Singal AG et al. The Liver Meeting 2019, Abstract 199.
BOSTON – For patients with a complete response following treatment for hepatitis C virus (HCV)–related hepatocellular carcinoma (HCC), treatment with direct-acting antiviral therapy was linked to significantly reduced mortality compared with no such treatment, according to results of a large cohort study.
The mortality benefit associated with direct-acting antiviral (DAA) therapy was consistent across most subgroups, suggesting that the association was driven by achieving sustained virological response (SVR), according to Amit G. Singal, MD, associate professor of medicine at UT Southwestern Medical Center, Dallas.
Those results suggest that DAA therapy in patients with a history of HCC is not only safe, but is also beneficial, Dr. Singal said at the annual meeting of the American Association for the Study of Liver Diseases.
“To be slightly controversial, I think that this changes the paradigm in this subgroup of patients from ‘can be treated’ for their hepatitis C to ‘should be treated,’ ” he concluded in an oral presentation of the results.
While DAA treatment is proven to reduce risk of incident HCC in patients with cirrhosis, the risk-benefit ratio is “less clear” in patients with a history of HCC following complete response, according to Dr. Singal.
Moreover, concerns were raised about the safety of DAA therapy in patients with an HCC history, after early data suggested a potentially higher recurrence risk, he added.
In the current multicenter, retrospective North American cohort study, Dr. Singal and colleagues reviewed data for 797 patients with HCV–related HCC who achieved complete response following ablation, resection, radiotherapy, transarterial chemoembolization, or transarterial radioembolization.
Treatment with DAA therapy was associated with improved overall survival, according to results of multivariable analysis, with a hazard ratio of 0.54 (95% confidence interval, 0.33-0.90). Median time from HCC complete response to death was 25.7 months for the DAA treatment group, versus 11.5 months for the untreated group.
The association between DAA treatment and death was apparently driven by SVR, as reduced mortality was seen in the DAA-treated patients who did achieve SVR, but not in those without SVR, Dr. Singal reported.
While these findings together suggest that DAA treatment is linked to reduced mortality after HCC complete response, prospective studies are needed to confirm this association, Dr. Singal said.
Dr. Singal reported disclosures related to AbbVie, Bayer, BMS, Eisai, Exact Sciences, Exelixis, Genentech, Gilead FOCUS, Glycotest, GRAIL, Merck, Roche, TARGET Pharmasolutions, and Wako Diagnostics.
SOURCE: Singal AG et al. The Liver Meeting 2019, Abstract 199.
BOSTON – For patients with a complete response following treatment for hepatitis C virus (HCV)–related hepatocellular carcinoma (HCC), treatment with direct-acting antiviral therapy was linked to significantly reduced mortality compared with no such treatment, according to results of a large cohort study.
The mortality benefit associated with direct-acting antiviral (DAA) therapy was consistent across most subgroups, suggesting that the association was driven by achieving sustained virological response (SVR), according to Amit G. Singal, MD, associate professor of medicine at UT Southwestern Medical Center, Dallas.
Those results suggest that DAA therapy in patients with a history of HCC is not only safe, but is also beneficial, Dr. Singal said at the annual meeting of the American Association for the Study of Liver Diseases.
“To be slightly controversial, I think that this changes the paradigm in this subgroup of patients from ‘can be treated’ for their hepatitis C to ‘should be treated,’ ” he concluded in an oral presentation of the results.
While DAA treatment is proven to reduce risk of incident HCC in patients with cirrhosis, the risk-benefit ratio is “less clear” in patients with a history of HCC following complete response, according to Dr. Singal.
Moreover, concerns were raised about the safety of DAA therapy in patients with an HCC history, after early data suggested a potentially higher recurrence risk, he added.
In the current multicenter, retrospective North American cohort study, Dr. Singal and colleagues reviewed data for 797 patients with HCV–related HCC who achieved complete response following ablation, resection, radiotherapy, transarterial chemoembolization, or transarterial radioembolization.
Treatment with DAA therapy was associated with improved overall survival, according to results of multivariable analysis, with a hazard ratio of 0.54 (95% confidence interval, 0.33-0.90). Median time from HCC complete response to death was 25.7 months for the DAA treatment group, versus 11.5 months for the untreated group.
The association between DAA treatment and death was apparently driven by SVR, as reduced mortality was seen in the DAA-treated patients who did achieve SVR, but not in those without SVR, Dr. Singal reported.
While these findings together suggest that DAA treatment is linked to reduced mortality after HCC complete response, prospective studies are needed to confirm this association, Dr. Singal said.
Dr. Singal reported disclosures related to AbbVie, Bayer, BMS, Eisai, Exact Sciences, Exelixis, Genentech, Gilead FOCUS, Glycotest, GRAIL, Merck, Roche, TARGET Pharmasolutions, and Wako Diagnostics.
SOURCE: Singal AG et al. The Liver Meeting 2019, Abstract 199.
REPORTING FROM THE LIVER MEETING 2019
Kratom: Botanical with opiate-like effects increasingly blamed for liver injury
BOSTON – Kratom, a botanical product with opioid-like activity, is increasingly responsible for cases of liver injury in the United States, according to investigators.

Kratom-associated liver damage involves a mixed pattern of hepatocellular and cholestatic injury that typically occurs after about 2-6 weeks of use, reported lead author Victor J. Navarro, MD, division head of gastroenterology at Einstein Healthcare Network in Philadelphia, and colleagues.
“I think it’s important for clinicians to have heightened awareness of the abuse potential [of kratom], because it is an opioid agonist and [because of] its capacity to cause liver injury,” Dr. Navarro said.
Kratom acts as a stimulant at low doses, while higher doses have sedating and narcotic properties. These effects are attributed to several alkaloids found in kratom’s source plant, Mitragyna speciose, of which mitragynine, a suspected opioid agonist, is most common.
Presenting at the annual meeting of the American Association for the Study of Liver Diseases, Dr. Navarro cited figures from the National Poison Data System that suggest an upward trend in kratom usage in the United States, from very little use in 2011 to 1 exposure per million people in 2014 and more recently to slightly more than 2.5 exposures per million people in 2017, predominantly among individuals aged 20 years and older. According to the Centers for Disease Control and Prevention, more than 90 kratom-associated deaths occurred between July 2016 and December 2017. Because of growing concerns, the Food and Drug Administration has issued multiple public warnings about kratom, ranging from products contaminated with Salmonella and heavy metals, to adverse effects such as seizures and liver toxicity.
The present study aimed to characterize kratom-associated liver injury through a case series analysis. First, the investigators reviewed 404 cases of herbal and dietary supplement-associated liver injury from the Drug-Induced Liver Injury Network prospective study. They found 11 suspected cases of kratom-related liver injury, with an upward trend in recent years. At this time, seven of the cases have been adjudicated by an expert panel and confirmed to be highly likely or probably associated with kratom.
Of these seven cases, all patients were hospitalized, although all recovered without need for liver transplant. Patients presented after a median of 15 days of kratom use, with a 28-day symptom latency period. However, Dr. Navarro noted that some cases presented after just 5 days of use. The most common presenting symptom was itching (86%), followed by jaundice (71%), abdominal pain (71%), nausea (57%), and fever (43%). Blood work revealed a mixed hepatocellular and cholestatic pattern. Median peak ALT was 362 U/L, peak alkaline phosphatase was 294 U/L, and peak total bilirubin was 20.1 mg/dL. Despite these changes, patients did not have significant liver dysfunction, such as coagulopathy.
Following this clinical characterization, Dr. Navarro reviewed existing toxicity data. Rat studies suggest that kratom is safe at doses between 1-10 mg/kg, while toxicity occurs after prolonged exposure to more than 100 mg/kg. A cross-sectional human study reported that kratom was safe at doses up to 75 mg/day. However, in the present case series, some patients presented after ingesting as little as 0.66 mg/day, and Dr. Navarro pointed out wide variations in product concentrations of mitragynine.
“Certainly, we need more human toxicity studies to determine what a safe dose really is, because this product is not going away,” Dr. Navarro said.
The investigators disclosed relationships with Gilead, Bristol-Myers Squibb, Sanofi, and others.
SOURCE: Navarro VJ et al. The Liver Meeting 2019, Abstract 212.
BOSTON – Kratom, a botanical product with opioid-like activity, is increasingly responsible for cases of liver injury in the United States, according to investigators.
Kratom-associated liver damage involves a mixed pattern of hepatocellular and cholestatic injury that typically occurs after about 2-6 weeks of use, reported lead author Victor J. Navarro, MD, division head of gastroenterology at Einstein Healthcare Network in Philadelphia, and colleagues.
“I think it’s important for clinicians to have heightened awareness of the abuse potential [of kratom], because it is an opioid agonist and [because of] its capacity to cause liver injury,” Dr. Navarro said.
Kratom acts as a stimulant at low doses, while higher doses have sedating and narcotic properties. These effects are attributed to several alkaloids found in kratom’s source plant, Mitragyna speciose, of which mitragynine, a suspected opioid agonist, is most common.
Presenting at the annual meeting of the American Association for the Study of Liver Diseases, Dr. Navarro cited figures from the National Poison Data System that suggest an upward trend in kratom usage in the United States, from very little use in 2011 to 1 exposure per million people in 2014 and more recently to slightly more than 2.5 exposures per million people in 2017, predominantly among individuals aged 20 years and older. According to the Centers for Disease Control and Prevention, more than 90 kratom-associated deaths occurred between July 2016 and December 2017. Because of growing concerns, the Food and Drug Administration has issued multiple public warnings about kratom, ranging from products contaminated with Salmonella and heavy metals, to adverse effects such as seizures and liver toxicity.
The present study aimed to characterize kratom-associated liver injury through a case series analysis. First, the investigators reviewed 404 cases of herbal and dietary supplement-associated liver injury from the Drug-Induced Liver Injury Network prospective study. They found 11 suspected cases of kratom-related liver injury, with an upward trend in recent years. At this time, seven of the cases have been adjudicated by an expert panel and confirmed to be highly likely or probably associated with kratom.
Of these seven cases, all patients were hospitalized, although all recovered without need for liver transplant. Patients presented after a median of 15 days of kratom use, with a 28-day symptom latency period. However, Dr. Navarro noted that some cases presented after just 5 days of use. The most common presenting symptom was itching (86%), followed by jaundice (71%), abdominal pain (71%), nausea (57%), and fever (43%). Blood work revealed a mixed hepatocellular and cholestatic pattern. Median peak ALT was 362 U/L, peak alkaline phosphatase was 294 U/L, and peak total bilirubin was 20.1 mg/dL. Despite these changes, patients did not have significant liver dysfunction, such as coagulopathy.
Following this clinical characterization, Dr. Navarro reviewed existing toxicity data. Rat studies suggest that kratom is safe at doses between 1-10 mg/kg, while toxicity occurs after prolonged exposure to more than 100 mg/kg. A cross-sectional human study reported that kratom was safe at doses up to 75 mg/day. However, in the present case series, some patients presented after ingesting as little as 0.66 mg/day, and Dr. Navarro pointed out wide variations in product concentrations of mitragynine.
“Certainly, we need more human toxicity studies to determine what a safe dose really is, because this product is not going away,” Dr. Navarro said.
The investigators disclosed relationships with Gilead, Bristol-Myers Squibb, Sanofi, and others.
SOURCE: Navarro VJ et al. The Liver Meeting 2019, Abstract 212.
BOSTON – Kratom, a botanical product with opioid-like activity, is increasingly responsible for cases of liver injury in the United States, according to investigators.
Kratom-associated liver damage involves a mixed pattern of hepatocellular and cholestatic injury that typically occurs after about 2-6 weeks of use, reported lead author Victor J. Navarro, MD, division head of gastroenterology at Einstein Healthcare Network in Philadelphia, and colleagues.
“I think it’s important for clinicians to have heightened awareness of the abuse potential [of kratom], because it is an opioid agonist and [because of] its capacity to cause liver injury,” Dr. Navarro said.
Kratom acts as a stimulant at low doses, while higher doses have sedating and narcotic properties. These effects are attributed to several alkaloids found in kratom’s source plant, Mitragyna speciose, of which mitragynine, a suspected opioid agonist, is most common.
Presenting at the annual meeting of the American Association for the Study of Liver Diseases, Dr. Navarro cited figures from the National Poison Data System that suggest an upward trend in kratom usage in the United States, from very little use in 2011 to 1 exposure per million people in 2014 and more recently to slightly more than 2.5 exposures per million people in 2017, predominantly among individuals aged 20 years and older. According to the Centers for Disease Control and Prevention, more than 90 kratom-associated deaths occurred between July 2016 and December 2017. Because of growing concerns, the Food and Drug Administration has issued multiple public warnings about kratom, ranging from products contaminated with Salmonella and heavy metals, to adverse effects such as seizures and liver toxicity.
The present study aimed to characterize kratom-associated liver injury through a case series analysis. First, the investigators reviewed 404 cases of herbal and dietary supplement-associated liver injury from the Drug-Induced Liver Injury Network prospective study. They found 11 suspected cases of kratom-related liver injury, with an upward trend in recent years. At this time, seven of the cases have been adjudicated by an expert panel and confirmed to be highly likely or probably associated with kratom.
Of these seven cases, all patients were hospitalized, although all recovered without need for liver transplant. Patients presented after a median of 15 days of kratom use, with a 28-day symptom latency period. However, Dr. Navarro noted that some cases presented after just 5 days of use. The most common presenting symptom was itching (86%), followed by jaundice (71%), abdominal pain (71%), nausea (57%), and fever (43%). Blood work revealed a mixed hepatocellular and cholestatic pattern. Median peak ALT was 362 U/L, peak alkaline phosphatase was 294 U/L, and peak total bilirubin was 20.1 mg/dL. Despite these changes, patients did not have significant liver dysfunction, such as coagulopathy.
Following this clinical characterization, Dr. Navarro reviewed existing toxicity data. Rat studies suggest that kratom is safe at doses between 1-10 mg/kg, while toxicity occurs after prolonged exposure to more than 100 mg/kg. A cross-sectional human study reported that kratom was safe at doses up to 75 mg/day. However, in the present case series, some patients presented after ingesting as little as 0.66 mg/day, and Dr. Navarro pointed out wide variations in product concentrations of mitragynine.
“Certainly, we need more human toxicity studies to determine what a safe dose really is, because this product is not going away,” Dr. Navarro said.
The investigators disclosed relationships with Gilead, Bristol-Myers Squibb, Sanofi, and others.
SOURCE: Navarro VJ et al. The Liver Meeting 2019, Abstract 212.
REPORTING FROM THE LIVER MEETING 2019
Fentanyl-related deaths show strong regional pattern
Fentanyl was involved in more overdose deaths than any other drug in 2017, and the death rate in New England was 15 times higher than in regions of the Midwest and West, according to the National Center for Health Statistics.
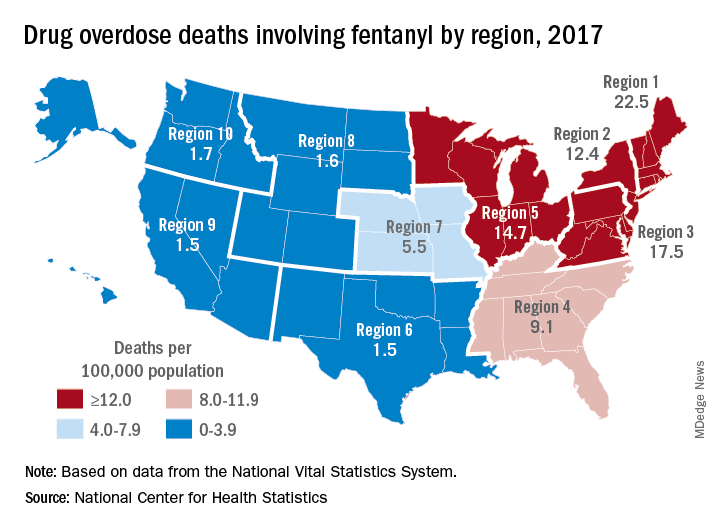
Nationally, fentanyl was involved in 39% of all drug overdose deaths and had an age-adjusted death rate of 8.7/100,000 standard population in 2017. In 2016, when fentanyl also was the most involved drug in the United States, the corresponding figures were 29% and 5.9/100,000, the agency said in a recent report.
Fentanyl was the most involved drug in overdose deaths for 6 of the country’s 10 public health regions in 2017, with a clear pattern of decreasing use from east to west. The highest death rate (22.5/100,000) occurred in Region 1 (New England) and the lowest rates (1.5/100,000) came in Region 6 (Arkansas, Louisiana, New Mexico, Oklahoma, and Texas) and Region 9 (Arizona, California, Hawaii, and Nevada), the researchers said.
A somewhat similar pattern was seen for heroin, which was second nationally on the list of drugs most frequently involved in overdose deaths (23%), except that New England was somewhat below three other regions in the East and upper Midwest. The highest heroin death rate (8.6/100,000) was seen in Region 2 (New Jersey and New York) and the lowest (2.2) occurred in Region 9, they said, based on data from the National Vital Statistics System’s mortality files.
The fentanyl pattern was even more closely repeated with cocaine, third in involvement nationally at 21% of overdose deaths in 2017. The high in overdose deaths (9.5/100,000) came in Region 1 again, and the low in Region 9 (1.3), along with Region 7 (Iowa, Kansas, Missouri, and Nebraska) and Region 10 (Alaska, Idaho, Oregon, and Washington), the report showed.
The regional pattern of overdose deaths for methamphetamine, which was fourth nationally in involvement (13.3%), basically reversed the other three drugs: highest in the West and lowest in the Northeast. Region 9 had the highest death rate (5.2/100,000) and Region 2 the lowest (0.4), with Region 1 just ahead at 0.6.
Fentanyl was involved in more overdose deaths than any other drug in 2017, and the death rate in New England was 15 times higher than in regions of the Midwest and West, according to the National Center for Health Statistics.
Nationally, fentanyl was involved in 39% of all drug overdose deaths and had an age-adjusted death rate of 8.7/100,000 standard population in 2017. In 2016, when fentanyl also was the most involved drug in the United States, the corresponding figures were 29% and 5.9/100,000, the agency said in a recent report.
Fentanyl was the most involved drug in overdose deaths for 6 of the country’s 10 public health regions in 2017, with a clear pattern of decreasing use from east to west. The highest death rate (22.5/100,000) occurred in Region 1 (New England) and the lowest rates (1.5/100,000) came in Region 6 (Arkansas, Louisiana, New Mexico, Oklahoma, and Texas) and Region 9 (Arizona, California, Hawaii, and Nevada), the researchers said.
A somewhat similar pattern was seen for heroin, which was second nationally on the list of drugs most frequently involved in overdose deaths (23%), except that New England was somewhat below three other regions in the East and upper Midwest. The highest heroin death rate (8.6/100,000) was seen in Region 2 (New Jersey and New York) and the lowest (2.2) occurred in Region 9, they said, based on data from the National Vital Statistics System’s mortality files.
The fentanyl pattern was even more closely repeated with cocaine, third in involvement nationally at 21% of overdose deaths in 2017. The high in overdose deaths (9.5/100,000) came in Region 1 again, and the low in Region 9 (1.3), along with Region 7 (Iowa, Kansas, Missouri, and Nebraska) and Region 10 (Alaska, Idaho, Oregon, and Washington), the report showed.
The regional pattern of overdose deaths for methamphetamine, which was fourth nationally in involvement (13.3%), basically reversed the other three drugs: highest in the West and lowest in the Northeast. Region 9 had the highest death rate (5.2/100,000) and Region 2 the lowest (0.4), with Region 1 just ahead at 0.6.
Fentanyl was involved in more overdose deaths than any other drug in 2017, and the death rate in New England was 15 times higher than in regions of the Midwest and West, according to the National Center for Health Statistics.
Nationally, fentanyl was involved in 39% of all drug overdose deaths and had an age-adjusted death rate of 8.7/100,000 standard population in 2017. In 2016, when fentanyl also was the most involved drug in the United States, the corresponding figures were 29% and 5.9/100,000, the agency said in a recent report.
Fentanyl was the most involved drug in overdose deaths for 6 of the country’s 10 public health regions in 2017, with a clear pattern of decreasing use from east to west. The highest death rate (22.5/100,000) occurred in Region 1 (New England) and the lowest rates (1.5/100,000) came in Region 6 (Arkansas, Louisiana, New Mexico, Oklahoma, and Texas) and Region 9 (Arizona, California, Hawaii, and Nevada), the researchers said.
A somewhat similar pattern was seen for heroin, which was second nationally on the list of drugs most frequently involved in overdose deaths (23%), except that New England was somewhat below three other regions in the East and upper Midwest. The highest heroin death rate (8.6/100,000) was seen in Region 2 (New Jersey and New York) and the lowest (2.2) occurred in Region 9, they said, based on data from the National Vital Statistics System’s mortality files.
The fentanyl pattern was even more closely repeated with cocaine, third in involvement nationally at 21% of overdose deaths in 2017. The high in overdose deaths (9.5/100,000) came in Region 1 again, and the low in Region 9 (1.3), along with Region 7 (Iowa, Kansas, Missouri, and Nebraska) and Region 10 (Alaska, Idaho, Oregon, and Washington), the report showed.
The regional pattern of overdose deaths for methamphetamine, which was fourth nationally in involvement (13.3%), basically reversed the other three drugs: highest in the West and lowest in the Northeast. Region 9 had the highest death rate (5.2/100,000) and Region 2 the lowest (0.4), with Region 1 just ahead at 0.6.
Combo shows promise for checkpoint inhibitor-refractory urothelial carcinoma
NATIONAL HARBOR, MD. – Sitravatinib may “restore or enhance” the activity of anti-PD-1 therapy in patients with checkpoint inhibitor–refractory urothelial carcinoma, an investigator reported at the annual meeting of the Society for Immunotherapy of Cancer.

Clinical activity was observed with combination sitravatinib and nivolumab in patients with urothelial carcinoma who had disease progression on or after an immune checkpoint inhibitor and were previously treated with platinum-based chemotherapy.
“Up until a few years ago, the only therapies we had [for urothelial carcinoma] were cytotoxic, platinum-based chemotherapies,” said Pavlos Msaouel, MD, PhD, of the University of Texas MD Anderson Cancer Center, Houston.
“Thankfully, since 2016, immune checkpoint therapy has become part of our toolbox. But even with single-agent, approved immune checkpoint therapies, anti-PD-1/anti-PD-L1, the response rates are still low, around 20%, and durable responses are only seen in a subset of patients. So we have to do better, if possible, potentially by combining immune checkpoint therapies with other immunotherapies such as sitravatinib.”
Dr. Msaouel explained that sitravatinib inhibits a spectrum of related receptor tyrosine kinases, including TAM family receptors (TYRO3, Axl, and Mer), split family receptors (VEGFR2/PDGFR and c-KIT), and c-Met. Researchers are investigating sitravatinib in combination with nivolumab in a phase 2 trial of patients with urothelial carcinoma (NCT03606174). Dr. Msaouel presented results from one cohort on this trial – 33 patients who had previously received platinum-based chemotherapy and a PD-1/PD-L1 inhibitor.
At baseline, the patients’ median age was 68 years (range, 47-83 years), and 70% were male. Patients had metastatic (n = 30) or locally advanced (n = 3) disease. They had received a median of two (range, one to four) prior systemic therapies.
For this study, patients received oral sitravatinib at 120 mg daily and intravenous nivolumab at 240 mg every 2 weeks or 480 mg every 4 weeks on continuous 28-day cycles. Tumor assessments were performed every 8 weeks.
Results
Of the 22 patients evaluable for efficacy, 1 patient achieved a complete response, 5 had a partial response, 15 had stable disease, and 1 progressed. Eight patients had tumor regression greater than 30%.
Treatment duration exceeded 26 weeks in six patients. Nine patients, including four responders, were still on study at the data cutoff in mid-October.
“This ongoing trial continues to show promising clinical activity, including tumor regression and prolonged duration on treatment,” Dr. Msaouel said.
He added that combination sitravatinib and nivolumab has “an acceptable side effect profile, with manageable adverse events.”
Common treatment-related adverse events, in all 33 patients, were fatigue (58%), diarrhea (48%), decreased appetite (33%), dysphonia (33%), nausea (33%), and alanine aminotransferase increase (21%).
Grade 3 treatment-related adverse events included fatigue (12%), hypertension (12%), diarrhea (9%), lipase increase (9%), decreased appetite (3%), and palmar-plantar erythrodysesthesia syndrome (3%). There were no grade 4 or 5 treatment-related events.
Mirati Therapeutics sponsored the trial. Dr. Msaouel disclosed relationships with Mirati, Bristol-Myers Squibb, Exelixis, Pfizer, and Takeda.
SOURCE: Msaouel P et al. SITC 2019. Abstract O23.
NATIONAL HARBOR, MD. – Sitravatinib may “restore or enhance” the activity of anti-PD-1 therapy in patients with checkpoint inhibitor–refractory urothelial carcinoma, an investigator reported at the annual meeting of the Society for Immunotherapy of Cancer.
Clinical activity was observed with combination sitravatinib and nivolumab in patients with urothelial carcinoma who had disease progression on or after an immune checkpoint inhibitor and were previously treated with platinum-based chemotherapy.
“Up until a few years ago, the only therapies we had [for urothelial carcinoma] were cytotoxic, platinum-based chemotherapies,” said Pavlos Msaouel, MD, PhD, of the University of Texas MD Anderson Cancer Center, Houston.
“Thankfully, since 2016, immune checkpoint therapy has become part of our toolbox. But even with single-agent, approved immune checkpoint therapies, anti-PD-1/anti-PD-L1, the response rates are still low, around 20%, and durable responses are only seen in a subset of patients. So we have to do better, if possible, potentially by combining immune checkpoint therapies with other immunotherapies such as sitravatinib.”
Dr. Msaouel explained that sitravatinib inhibits a spectrum of related receptor tyrosine kinases, including TAM family receptors (TYRO3, Axl, and Mer), split family receptors (VEGFR2/PDGFR and c-KIT), and c-Met. Researchers are investigating sitravatinib in combination with nivolumab in a phase 2 trial of patients with urothelial carcinoma (NCT03606174). Dr. Msaouel presented results from one cohort on this trial – 33 patients who had previously received platinum-based chemotherapy and a PD-1/PD-L1 inhibitor.
At baseline, the patients’ median age was 68 years (range, 47-83 years), and 70% were male. Patients had metastatic (n = 30) or locally advanced (n = 3) disease. They had received a median of two (range, one to four) prior systemic therapies.
For this study, patients received oral sitravatinib at 120 mg daily and intravenous nivolumab at 240 mg every 2 weeks or 480 mg every 4 weeks on continuous 28-day cycles. Tumor assessments were performed every 8 weeks.
Results
Of the 22 patients evaluable for efficacy, 1 patient achieved a complete response, 5 had a partial response, 15 had stable disease, and 1 progressed. Eight patients had tumor regression greater than 30%.
Treatment duration exceeded 26 weeks in six patients. Nine patients, including four responders, were still on study at the data cutoff in mid-October.
“This ongoing trial continues to show promising clinical activity, including tumor regression and prolonged duration on treatment,” Dr. Msaouel said.
He added that combination sitravatinib and nivolumab has “an acceptable side effect profile, with manageable adverse events.”
Common treatment-related adverse events, in all 33 patients, were fatigue (58%), diarrhea (48%), decreased appetite (33%), dysphonia (33%), nausea (33%), and alanine aminotransferase increase (21%).
Grade 3 treatment-related adverse events included fatigue (12%), hypertension (12%), diarrhea (9%), lipase increase (9%), decreased appetite (3%), and palmar-plantar erythrodysesthesia syndrome (3%). There were no grade 4 or 5 treatment-related events.
Mirati Therapeutics sponsored the trial. Dr. Msaouel disclosed relationships with Mirati, Bristol-Myers Squibb, Exelixis, Pfizer, and Takeda.
SOURCE: Msaouel P et al. SITC 2019. Abstract O23.
NATIONAL HARBOR, MD. – Sitravatinib may “restore or enhance” the activity of anti-PD-1 therapy in patients with checkpoint inhibitor–refractory urothelial carcinoma, an investigator reported at the annual meeting of the Society for Immunotherapy of Cancer.
Clinical activity was observed with combination sitravatinib and nivolumab in patients with urothelial carcinoma who had disease progression on or after an immune checkpoint inhibitor and were previously treated with platinum-based chemotherapy.
“Up until a few years ago, the only therapies we had [for urothelial carcinoma] were cytotoxic, platinum-based chemotherapies,” said Pavlos Msaouel, MD, PhD, of the University of Texas MD Anderson Cancer Center, Houston.
“Thankfully, since 2016, immune checkpoint therapy has become part of our toolbox. But even with single-agent, approved immune checkpoint therapies, anti-PD-1/anti-PD-L1, the response rates are still low, around 20%, and durable responses are only seen in a subset of patients. So we have to do better, if possible, potentially by combining immune checkpoint therapies with other immunotherapies such as sitravatinib.”
Dr. Msaouel explained that sitravatinib inhibits a spectrum of related receptor tyrosine kinases, including TAM family receptors (TYRO3, Axl, and Mer), split family receptors (VEGFR2/PDGFR and c-KIT), and c-Met. Researchers are investigating sitravatinib in combination with nivolumab in a phase 2 trial of patients with urothelial carcinoma (NCT03606174). Dr. Msaouel presented results from one cohort on this trial – 33 patients who had previously received platinum-based chemotherapy and a PD-1/PD-L1 inhibitor.
At baseline, the patients’ median age was 68 years (range, 47-83 years), and 70% were male. Patients had metastatic (n = 30) or locally advanced (n = 3) disease. They had received a median of two (range, one to four) prior systemic therapies.
For this study, patients received oral sitravatinib at 120 mg daily and intravenous nivolumab at 240 mg every 2 weeks or 480 mg every 4 weeks on continuous 28-day cycles. Tumor assessments were performed every 8 weeks.
Results
Of the 22 patients evaluable for efficacy, 1 patient achieved a complete response, 5 had a partial response, 15 had stable disease, and 1 progressed. Eight patients had tumor regression greater than 30%.
Treatment duration exceeded 26 weeks in six patients. Nine patients, including four responders, were still on study at the data cutoff in mid-October.
“This ongoing trial continues to show promising clinical activity, including tumor regression and prolonged duration on treatment,” Dr. Msaouel said.
He added that combination sitravatinib and nivolumab has “an acceptable side effect profile, with manageable adverse events.”
Common treatment-related adverse events, in all 33 patients, were fatigue (58%), diarrhea (48%), decreased appetite (33%), dysphonia (33%), nausea (33%), and alanine aminotransferase increase (21%).
Grade 3 treatment-related adverse events included fatigue (12%), hypertension (12%), diarrhea (9%), lipase increase (9%), decreased appetite (3%), and palmar-plantar erythrodysesthesia syndrome (3%). There were no grade 4 or 5 treatment-related events.
Mirati Therapeutics sponsored the trial. Dr. Msaouel disclosed relationships with Mirati, Bristol-Myers Squibb, Exelixis, Pfizer, and Takeda.
SOURCE: Msaouel P et al. SITC 2019. Abstract O23.
REPORTING FROM SITC 2019
Short-course DAA therapy may prevent hepatitis transmission in transplant patients
BOSTON – A short course of results of a recent study show.
The regimen, given right before transplantation and for 7 days afterward, reduced the cost of direct-acting antiviral (DAA) therapy and allowed patients to complete hepatitis C virus (HCV) therapy before hospital discharge, according to authors of the study, which was presented at the annual meeting of the American Association for the Study of Liver Diseases.
If confirmed in subsequent studies, this regimen could become the standard of care for donor-positive, recipient-negative transplantation, said lead study author Jordan J. Feld, MD, R. Phelan Chair in translational liver disease research at the University of Toronto and research director at the Toronto Centre for Liver Disease.
“Transplant recipients are understandably nervous about accepting organs from people with HCV infection,” said Dr. Feld in a press release. “This very short therapy allows them to leave hospital free of HCV, which is a huge benefit. Not only is it cheaper and likely safer, but the patients really prefer not having to worry about HCV with all of the other challenges after a transplant.”
Results of this study come at a time when the proportion of overdose death organ donors is on the rise, from just 1% in 2000 to 15% in 2016, according to Dr. Feld. Overdose deaths account for the largest percentage of HCV-infected donors, most of whom are young and often otherwise healthy, he added.
Recipients of HCV-infected organs can be cured after transplant as a number of studies have previously shown. However, preventing transmission would be better than cure, Dr. Feld said, in part because of issues with drug-drug interactions, potential for relapse, and issues with procuring the drugs after transplant.
Accordingly, Dr. Feld and colleagues sought to evaluate “preemptive” treatment with DAA therapy combined with ezetimibe, which they said has been shown to inhibit HCV entry blockers. The recipients, who were listed for heart, lung, kidney, or kidney-pancreas transplant, were given glecaprevir/pibrentasvir plus ezetimibe starting 6-12 hours prior to transplantation, and then daily for 7 days.
The median age was 36 years for the 16 donors reported, and 61 years for the 25 recipients. Most recipients (12 patients) had a lung transplant, while 8 had a heart transplant, 4 had a kidney transplant, and 1 had a kidney-pancreas transplant.
There were no virologic failures, according to the investigators, with sustained virologic response (SVR) after 6 weeks in 7 patients, and SVR after 12 weeks in the remaining 18. Three recipients did have detectable HCV RNA, though all cleared and had SVR at 6 weeks in one case, and SVR at 12 weeks in the other two, according to the investigators’ report.
Of 22 serious adverse events noted in the study, 1 was considered treatment related, according to the report, and there were 2 deaths among lung transplant patients, caused by sepsis in 1 case to sepsis and subarachnoid hemorrhage in another.
It’s not clear whether ezetimibe is needed in this short-duration regimen, but in any case, it is well tolerated and inexpensive, and so there is “minimal downside” to include it, Dr. Feld and coinvestigators wrote in their report.
Dr. Feld reported disclosures related to Abbvie, Abbott, Enanta Pharmaceuticals, Gilead, Janssen, Merck, and Roche.
SOURCE: Feld JJ et al. The Liver Meeting 2019, Abstract 38.
BOSTON – A short course of results of a recent study show.
The regimen, given right before transplantation and for 7 days afterward, reduced the cost of direct-acting antiviral (DAA) therapy and allowed patients to complete hepatitis C virus (HCV) therapy before hospital discharge, according to authors of the study, which was presented at the annual meeting of the American Association for the Study of Liver Diseases.
If confirmed in subsequent studies, this regimen could become the standard of care for donor-positive, recipient-negative transplantation, said lead study author Jordan J. Feld, MD, R. Phelan Chair in translational liver disease research at the University of Toronto and research director at the Toronto Centre for Liver Disease.
“Transplant recipients are understandably nervous about accepting organs from people with HCV infection,” said Dr. Feld in a press release. “This very short therapy allows them to leave hospital free of HCV, which is a huge benefit. Not only is it cheaper and likely safer, but the patients really prefer not having to worry about HCV with all of the other challenges after a transplant.”
Results of this study come at a time when the proportion of overdose death organ donors is on the rise, from just 1% in 2000 to 15% in 2016, according to Dr. Feld. Overdose deaths account for the largest percentage of HCV-infected donors, most of whom are young and often otherwise healthy, he added.
Recipients of HCV-infected organs can be cured after transplant as a number of studies have previously shown. However, preventing transmission would be better than cure, Dr. Feld said, in part because of issues with drug-drug interactions, potential for relapse, and issues with procuring the drugs after transplant.
Accordingly, Dr. Feld and colleagues sought to evaluate “preemptive” treatment with DAA therapy combined with ezetimibe, which they said has been shown to inhibit HCV entry blockers. The recipients, who were listed for heart, lung, kidney, or kidney-pancreas transplant, were given glecaprevir/pibrentasvir plus ezetimibe starting 6-12 hours prior to transplantation, and then daily for 7 days.
The median age was 36 years for the 16 donors reported, and 61 years for the 25 recipients. Most recipients (12 patients) had a lung transplant, while 8 had a heart transplant, 4 had a kidney transplant, and 1 had a kidney-pancreas transplant.
There were no virologic failures, according to the investigators, with sustained virologic response (SVR) after 6 weeks in 7 patients, and SVR after 12 weeks in the remaining 18. Three recipients did have detectable HCV RNA, though all cleared and had SVR at 6 weeks in one case, and SVR at 12 weeks in the other two, according to the investigators’ report.
Of 22 serious adverse events noted in the study, 1 was considered treatment related, according to the report, and there were 2 deaths among lung transplant patients, caused by sepsis in 1 case to sepsis and subarachnoid hemorrhage in another.
It’s not clear whether ezetimibe is needed in this short-duration regimen, but in any case, it is well tolerated and inexpensive, and so there is “minimal downside” to include it, Dr. Feld and coinvestigators wrote in their report.
Dr. Feld reported disclosures related to Abbvie, Abbott, Enanta Pharmaceuticals, Gilead, Janssen, Merck, and Roche.
SOURCE: Feld JJ et al. The Liver Meeting 2019, Abstract 38.
BOSTON – A short course of results of a recent study show.
The regimen, given right before transplantation and for 7 days afterward, reduced the cost of direct-acting antiviral (DAA) therapy and allowed patients to complete hepatitis C virus (HCV) therapy before hospital discharge, according to authors of the study, which was presented at the annual meeting of the American Association for the Study of Liver Diseases.
If confirmed in subsequent studies, this regimen could become the standard of care for donor-positive, recipient-negative transplantation, said lead study author Jordan J. Feld, MD, R. Phelan Chair in translational liver disease research at the University of Toronto and research director at the Toronto Centre for Liver Disease.
“Transplant recipients are understandably nervous about accepting organs from people with HCV infection,” said Dr. Feld in a press release. “This very short therapy allows them to leave hospital free of HCV, which is a huge benefit. Not only is it cheaper and likely safer, but the patients really prefer not having to worry about HCV with all of the other challenges after a transplant.”
Results of this study come at a time when the proportion of overdose death organ donors is on the rise, from just 1% in 2000 to 15% in 2016, according to Dr. Feld. Overdose deaths account for the largest percentage of HCV-infected donors, most of whom are young and often otherwise healthy, he added.
Recipients of HCV-infected organs can be cured after transplant as a number of studies have previously shown. However, preventing transmission would be better than cure, Dr. Feld said, in part because of issues with drug-drug interactions, potential for relapse, and issues with procuring the drugs after transplant.
Accordingly, Dr. Feld and colleagues sought to evaluate “preemptive” treatment with DAA therapy combined with ezetimibe, which they said has been shown to inhibit HCV entry blockers. The recipients, who were listed for heart, lung, kidney, or kidney-pancreas transplant, were given glecaprevir/pibrentasvir plus ezetimibe starting 6-12 hours prior to transplantation, and then daily for 7 days.
The median age was 36 years for the 16 donors reported, and 61 years for the 25 recipients. Most recipients (12 patients) had a lung transplant, while 8 had a heart transplant, 4 had a kidney transplant, and 1 had a kidney-pancreas transplant.
There were no virologic failures, according to the investigators, with sustained virologic response (SVR) after 6 weeks in 7 patients, and SVR after 12 weeks in the remaining 18. Three recipients did have detectable HCV RNA, though all cleared and had SVR at 6 weeks in one case, and SVR at 12 weeks in the other two, according to the investigators’ report.
Of 22 serious adverse events noted in the study, 1 was considered treatment related, according to the report, and there were 2 deaths among lung transplant patients, caused by sepsis in 1 case to sepsis and subarachnoid hemorrhage in another.
It’s not clear whether ezetimibe is needed in this short-duration regimen, but in any case, it is well tolerated and inexpensive, and so there is “minimal downside” to include it, Dr. Feld and coinvestigators wrote in their report.
Dr. Feld reported disclosures related to Abbvie, Abbott, Enanta Pharmaceuticals, Gilead, Janssen, Merck, and Roche.
SOURCE: Feld JJ et al. The Liver Meeting 2019, Abstract 38.
REPORTING FROM THE LIVER MEETING 2019
Expert shares tips for TNF-alpha inhibitor use in special populations
LAS VEGAS – Francisco A. Kerdel, BSc, MBBS, said at the Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.

Dr. Kerdel, professor and vice chair of the department of dermatology at Florida International University, Miami, noted that, while tumor necrosis factor (TNF)–alpha inhibitors are category B drugs, inadequate data exist regarding lactation and exposure throughout pregnancy. “Rates of malformations and spontaneous abortions with therapy are similar to those in the general population, higher concentrations of infliximab and adalimumab have been found in infant and cord blood, compared with certolizumab pegol,” an anti-TNF biologic, he said.
In a prospective, postmarketing, multicenter pharmacokinetic study, researchers found a lack of placental transfer of certolizumab pegol during pregnancy (Ann Rheum Dis. 2018;77:228-33). Specifically, certolizumab levels were below the lower limit of quantification (less than 0.032 mcg/mL) in 13 of 14 infant samples at birth and in all infant samples at weeks 4 and 8. Only one infant had a minimal certolizumab level at birth (infant/mother ratio of 0.0009). No antibodies were detected at any time point during the study. Safety data in mothers were in line with the known safety profile of certolizumab and pregnancy profile of these underlying diseases. Adverse events experienced by the infants did not show any patterns or clusters of events suggesting a specific safety signal in children.
In a separate postmarketing pharmacokinetic study, investigators evaluated the transfer of certolizumab into breast milk (Ann Rheum Dis. 2017;76:1890-6). They found that the average daily infant dose of certolizumab was minimal. Specifically, the highest concentration of certolizumab in breast milk (0.0758 mcg/mL) was less than 1% of the expected mean plasma trough concentration of a therapeutic dose.
How do TNF-alpha inhibitors fare in the pediatric population? In a retrospective study of 390 children with psoriasis treated at 20 centers in the United States, Canada, and Europe, researchers evaluated the safety of systemic agents (JAMA Dermatol. 2017;153[11]:1147-57). Most (69%) were prescribed methotrexate, followed by biologics, acitretin, cyclosporine, and fumaric acid. Drug discontinuation (because of adverse events), which is sometimes used as an efficacy parameter, occurred in 12% of those who were on methotrexate, compared with 3% of those on biologics, 67% of those on acitretin, and 68% of those on fumaric acid.
At the other end of the age spectrum, biologic therapy is generally effective and well tolerated in elderly patients. “Sometimes, they may be more effective than other traditional drugs,” Dr. Kerdel said. “We’re a little bit concerned about immunosenescence, which can increase the risk for severe infections and malignancies. And, 90% of elderly patients with psoriasis may have comorbidities that need to be taken into account when treating psoriasis.”
Other factors come into play when choosing the right anti-TNF agent, including weight. While clinical trials show efficacy across weight groups, infliximab has weight-based dosing, “which may make it a better choice,” Dr. Kerdel said. “Patients taking etanercept may need a biweekly dose.”
Treatment flexibility also comes into play. For example, stopping therapy because of an infection or surgery may be problematic in drugs with a long half-life. Then there’s the issue of patient preference. “Some people don’t want to be injected frequently,” he said. “Some people don’t want to be injected at all and may require a simpler dosing regimen.”
Optimizing anti-TNF-alpha treatment starts with recognizing that there is a loss of response over time, Dr. Kerdel said, “or there may not be a response at all.” Contributing factors may include immunogenicity, suboptimal dosing, and poor patient adherence. In order to optimize treatment, clinicians can try switching agents or combination therapy, and explore continuous versus intermittent dosing.
“We really don’t have good data on the best protocol for switching treatment after failure of an anti-TNF-alpha agent,” he added. In cases of primary and secondary treatment failure, there is no consensus or guidelines on which second-line agent to use, nor good data on which measures to use.
No evidence-based guidelines are available for screening and monitoring patients receiving biologic therapy for psoriasis, either. “Evidence is strongest [grade B] for tuberculosis screening in patients treated with biologic agents,” Dr. Kerdel said. “Among known hepatitis B virus carriers, consider monitoring liver function tests and viral load [grade C]. High-grade evidence is lacking to support other routine testing. Physicians should use clinical judgment when screening and monitoring patients.”
He concluded his presentation by noting that there are a number of biosimilar agents available or in the pipeline for infliximab, adalimumab, and etanercept. This raises a number of questions for current and future consideration. For one, “will biosimilars show the same long-term efficacy and safety as the innovator products?” he asked. “Real-world, postmarketing, and registry data are needed. Will biosimilar agents offer significant cost benefits? Will biosimilar labeling be adequately transparent? Will we find biomarkers to help us target biologic agents to specific patients and subtypes of psoriasis?”
Dr. Kerdel reported that he is a member of the speaker’s bureau for AbbVie, Amgen, Celgene, Janssen, Novartis, Lilly, Leo, Ortho, and Novartis. He has also received grant/research support from AbbVie, Amgen, AstraZeneca, Celgene, Janssen, Leo, Lilly, Menlo Therapeutics, Novartis, Pfizer, and XBiotech.
SDEF and this news organization are owned by the same parent company.
LAS VEGAS – Francisco A. Kerdel, BSc, MBBS, said at the Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
Dr. Kerdel, professor and vice chair of the department of dermatology at Florida International University, Miami, noted that, while tumor necrosis factor (TNF)–alpha inhibitors are category B drugs, inadequate data exist regarding lactation and exposure throughout pregnancy. “Rates of malformations and spontaneous abortions with therapy are similar to those in the general population, higher concentrations of infliximab and adalimumab have been found in infant and cord blood, compared with certolizumab pegol,” an anti-TNF biologic, he said.
In a prospective, postmarketing, multicenter pharmacokinetic study, researchers found a lack of placental transfer of certolizumab pegol during pregnancy (Ann Rheum Dis. 2018;77:228-33). Specifically, certolizumab levels were below the lower limit of quantification (less than 0.032 mcg/mL) in 13 of 14 infant samples at birth and in all infant samples at weeks 4 and 8. Only one infant had a minimal certolizumab level at birth (infant/mother ratio of 0.0009). No antibodies were detected at any time point during the study. Safety data in mothers were in line with the known safety profile of certolizumab and pregnancy profile of these underlying diseases. Adverse events experienced by the infants did not show any patterns or clusters of events suggesting a specific safety signal in children.
In a separate postmarketing pharmacokinetic study, investigators evaluated the transfer of certolizumab into breast milk (Ann Rheum Dis. 2017;76:1890-6). They found that the average daily infant dose of certolizumab was minimal. Specifically, the highest concentration of certolizumab in breast milk (0.0758 mcg/mL) was less than 1% of the expected mean plasma trough concentration of a therapeutic dose.
How do TNF-alpha inhibitors fare in the pediatric population? In a retrospective study of 390 children with psoriasis treated at 20 centers in the United States, Canada, and Europe, researchers evaluated the safety of systemic agents (JAMA Dermatol. 2017;153[11]:1147-57). Most (69%) were prescribed methotrexate, followed by biologics, acitretin, cyclosporine, and fumaric acid. Drug discontinuation (because of adverse events), which is sometimes used as an efficacy parameter, occurred in 12% of those who were on methotrexate, compared with 3% of those on biologics, 67% of those on acitretin, and 68% of those on fumaric acid.
At the other end of the age spectrum, biologic therapy is generally effective and well tolerated in elderly patients. “Sometimes, they may be more effective than other traditional drugs,” Dr. Kerdel said. “We’re a little bit concerned about immunosenescence, which can increase the risk for severe infections and malignancies. And, 90% of elderly patients with psoriasis may have comorbidities that need to be taken into account when treating psoriasis.”
Other factors come into play when choosing the right anti-TNF agent, including weight. While clinical trials show efficacy across weight groups, infliximab has weight-based dosing, “which may make it a better choice,” Dr. Kerdel said. “Patients taking etanercept may need a biweekly dose.”
Treatment flexibility also comes into play. For example, stopping therapy because of an infection or surgery may be problematic in drugs with a long half-life. Then there’s the issue of patient preference. “Some people don’t want to be injected frequently,” he said. “Some people don’t want to be injected at all and may require a simpler dosing regimen.”
Optimizing anti-TNF-alpha treatment starts with recognizing that there is a loss of response over time, Dr. Kerdel said, “or there may not be a response at all.” Contributing factors may include immunogenicity, suboptimal dosing, and poor patient adherence. In order to optimize treatment, clinicians can try switching agents or combination therapy, and explore continuous versus intermittent dosing.
“We really don’t have good data on the best protocol for switching treatment after failure of an anti-TNF-alpha agent,” he added. In cases of primary and secondary treatment failure, there is no consensus or guidelines on which second-line agent to use, nor good data on which measures to use.
No evidence-based guidelines are available for screening and monitoring patients receiving biologic therapy for psoriasis, either. “Evidence is strongest [grade B] for tuberculosis screening in patients treated with biologic agents,” Dr. Kerdel said. “Among known hepatitis B virus carriers, consider monitoring liver function tests and viral load [grade C]. High-grade evidence is lacking to support other routine testing. Physicians should use clinical judgment when screening and monitoring patients.”
He concluded his presentation by noting that there are a number of biosimilar agents available or in the pipeline for infliximab, adalimumab, and etanercept. This raises a number of questions for current and future consideration. For one, “will biosimilars show the same long-term efficacy and safety as the innovator products?” he asked. “Real-world, postmarketing, and registry data are needed. Will biosimilar agents offer significant cost benefits? Will biosimilar labeling be adequately transparent? Will we find biomarkers to help us target biologic agents to specific patients and subtypes of psoriasis?”
Dr. Kerdel reported that he is a member of the speaker’s bureau for AbbVie, Amgen, Celgene, Janssen, Novartis, Lilly, Leo, Ortho, and Novartis. He has also received grant/research support from AbbVie, Amgen, AstraZeneca, Celgene, Janssen, Leo, Lilly, Menlo Therapeutics, Novartis, Pfizer, and XBiotech.
SDEF and this news organization are owned by the same parent company.
LAS VEGAS – Francisco A. Kerdel, BSc, MBBS, said at the Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
Dr. Kerdel, professor and vice chair of the department of dermatology at Florida International University, Miami, noted that, while tumor necrosis factor (TNF)–alpha inhibitors are category B drugs, inadequate data exist regarding lactation and exposure throughout pregnancy. “Rates of malformations and spontaneous abortions with therapy are similar to those in the general population, higher concentrations of infliximab and adalimumab have been found in infant and cord blood, compared with certolizumab pegol,” an anti-TNF biologic, he said.
In a prospective, postmarketing, multicenter pharmacokinetic study, researchers found a lack of placental transfer of certolizumab pegol during pregnancy (Ann Rheum Dis. 2018;77:228-33). Specifically, certolizumab levels were below the lower limit of quantification (less than 0.032 mcg/mL) in 13 of 14 infant samples at birth and in all infant samples at weeks 4 and 8. Only one infant had a minimal certolizumab level at birth (infant/mother ratio of 0.0009). No antibodies were detected at any time point during the study. Safety data in mothers were in line with the known safety profile of certolizumab and pregnancy profile of these underlying diseases. Adverse events experienced by the infants did not show any patterns or clusters of events suggesting a specific safety signal in children.
In a separate postmarketing pharmacokinetic study, investigators evaluated the transfer of certolizumab into breast milk (Ann Rheum Dis. 2017;76:1890-6). They found that the average daily infant dose of certolizumab was minimal. Specifically, the highest concentration of certolizumab in breast milk (0.0758 mcg/mL) was less than 1% of the expected mean plasma trough concentration of a therapeutic dose.
How do TNF-alpha inhibitors fare in the pediatric population? In a retrospective study of 390 children with psoriasis treated at 20 centers in the United States, Canada, and Europe, researchers evaluated the safety of systemic agents (JAMA Dermatol. 2017;153[11]:1147-57). Most (69%) were prescribed methotrexate, followed by biologics, acitretin, cyclosporine, and fumaric acid. Drug discontinuation (because of adverse events), which is sometimes used as an efficacy parameter, occurred in 12% of those who were on methotrexate, compared with 3% of those on biologics, 67% of those on acitretin, and 68% of those on fumaric acid.
At the other end of the age spectrum, biologic therapy is generally effective and well tolerated in elderly patients. “Sometimes, they may be more effective than other traditional drugs,” Dr. Kerdel said. “We’re a little bit concerned about immunosenescence, which can increase the risk for severe infections and malignancies. And, 90% of elderly patients with psoriasis may have comorbidities that need to be taken into account when treating psoriasis.”
Other factors come into play when choosing the right anti-TNF agent, including weight. While clinical trials show efficacy across weight groups, infliximab has weight-based dosing, “which may make it a better choice,” Dr. Kerdel said. “Patients taking etanercept may need a biweekly dose.”
Treatment flexibility also comes into play. For example, stopping therapy because of an infection or surgery may be problematic in drugs with a long half-life. Then there’s the issue of patient preference. “Some people don’t want to be injected frequently,” he said. “Some people don’t want to be injected at all and may require a simpler dosing regimen.”
Optimizing anti-TNF-alpha treatment starts with recognizing that there is a loss of response over time, Dr. Kerdel said, “or there may not be a response at all.” Contributing factors may include immunogenicity, suboptimal dosing, and poor patient adherence. In order to optimize treatment, clinicians can try switching agents or combination therapy, and explore continuous versus intermittent dosing.
“We really don’t have good data on the best protocol for switching treatment after failure of an anti-TNF-alpha agent,” he added. In cases of primary and secondary treatment failure, there is no consensus or guidelines on which second-line agent to use, nor good data on which measures to use.
No evidence-based guidelines are available for screening and monitoring patients receiving biologic therapy for psoriasis, either. “Evidence is strongest [grade B] for tuberculosis screening in patients treated with biologic agents,” Dr. Kerdel said. “Among known hepatitis B virus carriers, consider monitoring liver function tests and viral load [grade C]. High-grade evidence is lacking to support other routine testing. Physicians should use clinical judgment when screening and monitoring patients.”
He concluded his presentation by noting that there are a number of biosimilar agents available or in the pipeline for infliximab, adalimumab, and etanercept. This raises a number of questions for current and future consideration. For one, “will biosimilars show the same long-term efficacy and safety as the innovator products?” he asked. “Real-world, postmarketing, and registry data are needed. Will biosimilar agents offer significant cost benefits? Will biosimilar labeling be adequately transparent? Will we find biomarkers to help us target biologic agents to specific patients and subtypes of psoriasis?”
Dr. Kerdel reported that he is a member of the speaker’s bureau for AbbVie, Amgen, Celgene, Janssen, Novartis, Lilly, Leo, Ortho, and Novartis. He has also received grant/research support from AbbVie, Amgen, AstraZeneca, Celgene, Janssen, Leo, Lilly, Menlo Therapeutics, Novartis, Pfizer, and XBiotech.
SDEF and this news organization are owned by the same parent company.
EXPERT ANALYSIS FROM THE SDEF LAS VEGAS DERMATOLOGY SEMINAR
Cancer pain management inadequate in opioid-saturated areas
Patients with cancer who live in regions with high levels of opioid misuse may be undertreated for pain, according to investigators who studied opioid prescription patterns and cancer incidence in rural southwest Virginia.
Among 4,324 patients with cancer, only 22.16% were prescribed a Controlled Schedule II (C-II) prescription opioid medication at least 3 times in 1 year, from prescribers likely to be treating cancer pain. More than 60% of patients never received a C-II opioid prescription, reported Virginia T. LeBaron, PhD, of the University of Virginia School of Nursing in Charlottesville, and colleagues.
“A clearer view of geographic patterns and predictors of both POM [prescription opioid medication] prescribing and potential harms can inform targeted interventions and policy initiatives that achieve a balanced approach to POMs – ensuring access for patients in need while reducing risk to both patients and communities. Our research makes an important contribution by exploring how the current ‘opioid epidemic’ relates to rural patients with cancer,” they wrote. Their report is in Journal of Oncology Practice.
The investigators studied the confluence of disproportionately high cancer mortality rates and opioid fatality rates in rural southwest Virginia, in the heart of Appalachia.
They conducted a longitudinal, exploratory secondary analysis of data from the Commonwealth of Virginia All Payer Claims database to look at opioid prescribing patterns and explore whether concerns about opioid misuse could result in undertreatment of pain in cancer patients.
They looked at prescribing patterns at the patient, provider, and insurance claim levels, predictors of opioid prescription frequency, opioid-related harms and patterns related to opioid prescribing, cancer incidence, and fatalities.
They identified 4,324 patients with cancer, 958 of whom (22.16%) received a C-II opioid at least three times in any study year. The majority of patients were in the 45-64 age range, and approximately 88% were diagnosed with solid malignancies, with breast cancer and lung cancer being the most frequent diagnoses.
As noted, more than 60% of patients never received a C-II prescription.
“The large percentages of cancer patients never prescribed a C-II are concerning for a number of reasons, especially when we consider the results per year,” the investigators wrote. “First, the ‘no C-II’ patients remain over 80% of the total sample, each year, even after accounting for the upscheduling (from C-III to C-II) of commonly-prescribed hydrocodone products in 2014. Second, anecdotal data and emerging empirical evidence demonstrate that patients with legitimate pain needs, including patients with cancer, experience significant difficulty accessing POMs.”
They noted that regulations regarding opioid prescriptions have become increasingly strict since the end date of their analysis in 2015, suggesting that the number of patients with cancer who are not receiving C-II opioids today may be even higher.
They also pointed to evidence of prescription practices suggesting suboptimal pain management or potential patient harm, such as frequent prescription of opioid-acetaminophen combinations that are dose-limited due to acetaminophen toxicity; coprescription of opioids and benzodiazepines, which is not recommended under current prescribing guidelines; and infrequent use of deterrent formulations of C-II opioids such as crush-resistant tablets.
The study was supported by the University of Virginia Cancer Center, Cancer Control & Population Health Division and the Virginia Tobacco Region Revitalization Commission. The authors reported having no disclaimers or conflicts of interest.
SOURCE: LeBaron VT et al. J Oncol Pract. 2019 Nov. 4. doi: 10.1200/JOP.19.00149.
Patients with cancer who live in regions with high levels of opioid misuse may be undertreated for pain, according to investigators who studied opioid prescription patterns and cancer incidence in rural southwest Virginia.
Among 4,324 patients with cancer, only 22.16% were prescribed a Controlled Schedule II (C-II) prescription opioid medication at least 3 times in 1 year, from prescribers likely to be treating cancer pain. More than 60% of patients never received a C-II opioid prescription, reported Virginia T. LeBaron, PhD, of the University of Virginia School of Nursing in Charlottesville, and colleagues.
“A clearer view of geographic patterns and predictors of both POM [prescription opioid medication] prescribing and potential harms can inform targeted interventions and policy initiatives that achieve a balanced approach to POMs – ensuring access for patients in need while reducing risk to both patients and communities. Our research makes an important contribution by exploring how the current ‘opioid epidemic’ relates to rural patients with cancer,” they wrote. Their report is in Journal of Oncology Practice.
The investigators studied the confluence of disproportionately high cancer mortality rates and opioid fatality rates in rural southwest Virginia, in the heart of Appalachia.
They conducted a longitudinal, exploratory secondary analysis of data from the Commonwealth of Virginia All Payer Claims database to look at opioid prescribing patterns and explore whether concerns about opioid misuse could result in undertreatment of pain in cancer patients.
They looked at prescribing patterns at the patient, provider, and insurance claim levels, predictors of opioid prescription frequency, opioid-related harms and patterns related to opioid prescribing, cancer incidence, and fatalities.
They identified 4,324 patients with cancer, 958 of whom (22.16%) received a C-II opioid at least three times in any study year. The majority of patients were in the 45-64 age range, and approximately 88% were diagnosed with solid malignancies, with breast cancer and lung cancer being the most frequent diagnoses.
As noted, more than 60% of patients never received a C-II prescription.
“The large percentages of cancer patients never prescribed a C-II are concerning for a number of reasons, especially when we consider the results per year,” the investigators wrote. “First, the ‘no C-II’ patients remain over 80% of the total sample, each year, even after accounting for the upscheduling (from C-III to C-II) of commonly-prescribed hydrocodone products in 2014. Second, anecdotal data and emerging empirical evidence demonstrate that patients with legitimate pain needs, including patients with cancer, experience significant difficulty accessing POMs.”
They noted that regulations regarding opioid prescriptions have become increasingly strict since the end date of their analysis in 2015, suggesting that the number of patients with cancer who are not receiving C-II opioids today may be even higher.
They also pointed to evidence of prescription practices suggesting suboptimal pain management or potential patient harm, such as frequent prescription of opioid-acetaminophen combinations that are dose-limited due to acetaminophen toxicity; coprescription of opioids and benzodiazepines, which is not recommended under current prescribing guidelines; and infrequent use of deterrent formulations of C-II opioids such as crush-resistant tablets.
The study was supported by the University of Virginia Cancer Center, Cancer Control & Population Health Division and the Virginia Tobacco Region Revitalization Commission. The authors reported having no disclaimers or conflicts of interest.
SOURCE: LeBaron VT et al. J Oncol Pract. 2019 Nov. 4. doi: 10.1200/JOP.19.00149.
Patients with cancer who live in regions with high levels of opioid misuse may be undertreated for pain, according to investigators who studied opioid prescription patterns and cancer incidence in rural southwest Virginia.
Among 4,324 patients with cancer, only 22.16% were prescribed a Controlled Schedule II (C-II) prescription opioid medication at least 3 times in 1 year, from prescribers likely to be treating cancer pain. More than 60% of patients never received a C-II opioid prescription, reported Virginia T. LeBaron, PhD, of the University of Virginia School of Nursing in Charlottesville, and colleagues.
“A clearer view of geographic patterns and predictors of both POM [prescription opioid medication] prescribing and potential harms can inform targeted interventions and policy initiatives that achieve a balanced approach to POMs – ensuring access for patients in need while reducing risk to both patients and communities. Our research makes an important contribution by exploring how the current ‘opioid epidemic’ relates to rural patients with cancer,” they wrote. Their report is in Journal of Oncology Practice.
The investigators studied the confluence of disproportionately high cancer mortality rates and opioid fatality rates in rural southwest Virginia, in the heart of Appalachia.
They conducted a longitudinal, exploratory secondary analysis of data from the Commonwealth of Virginia All Payer Claims database to look at opioid prescribing patterns and explore whether concerns about opioid misuse could result in undertreatment of pain in cancer patients.
They looked at prescribing patterns at the patient, provider, and insurance claim levels, predictors of opioid prescription frequency, opioid-related harms and patterns related to opioid prescribing, cancer incidence, and fatalities.
They identified 4,324 patients with cancer, 958 of whom (22.16%) received a C-II opioid at least three times in any study year. The majority of patients were in the 45-64 age range, and approximately 88% were diagnosed with solid malignancies, with breast cancer and lung cancer being the most frequent diagnoses.
As noted, more than 60% of patients never received a C-II prescription.
“The large percentages of cancer patients never prescribed a C-II are concerning for a number of reasons, especially when we consider the results per year,” the investigators wrote. “First, the ‘no C-II’ patients remain over 80% of the total sample, each year, even after accounting for the upscheduling (from C-III to C-II) of commonly-prescribed hydrocodone products in 2014. Second, anecdotal data and emerging empirical evidence demonstrate that patients with legitimate pain needs, including patients with cancer, experience significant difficulty accessing POMs.”
They noted that regulations regarding opioid prescriptions have become increasingly strict since the end date of their analysis in 2015, suggesting that the number of patients with cancer who are not receiving C-II opioids today may be even higher.
They also pointed to evidence of prescription practices suggesting suboptimal pain management or potential patient harm, such as frequent prescription of opioid-acetaminophen combinations that are dose-limited due to acetaminophen toxicity; coprescription of opioids and benzodiazepines, which is not recommended under current prescribing guidelines; and infrequent use of deterrent formulations of C-II opioids such as crush-resistant tablets.
The study was supported by the University of Virginia Cancer Center, Cancer Control & Population Health Division and the Virginia Tobacco Region Revitalization Commission. The authors reported having no disclaimers or conflicts of interest.
SOURCE: LeBaron VT et al. J Oncol Pract. 2019 Nov. 4. doi: 10.1200/JOP.19.00149.
FROM JOURNAL OF ONCOLOGY PRACTICE