User login
Lomitapide manufacturer will plead guilty to two misdemeanor charges of misbranding
Juxtapid was misbranded because Aegerion failed to comply with the requirements of the Juxtapid Risk Evaluation and Mitigation Strategy (REMS) program and because the drug’s labeling lacked adequate directions for all of Juxtapid’s intended uses, according to the charges. Aegerion also agreed to a comprehensive compliance program and legal tools for the FDA to ensure that Aegerion complies with the law, subject to judicial oversight.
Rather than following the REMS requirement to distribute Juxtapid only for the narrow indication of homozygous familial hypercholesterolemia, Aegerion portrayed the definition of the rare disorder as vague and indefinite in order to extend its use to lower-risk patients. Further, Aegerion filed a misleading REMS assessment report to the FDA in which the company failed to disclose that it was distributing Juxtapid using this definition, which was inconsistent with Aegerion’s preapproval filings and peer-reviewed clinical standards of diagnosis, according to the FDA release.
Once entered by the court, the plea and consent decree will be part of a global resolution of multiple government investigations into Aegerion’s conduct with respect to the marketing and distribution of Juxtapid. This resolution was the result of a coordinated effort by the U.S. Department of Justice and several government agencies, including the FDA, the press release stated.
Juxtapid was approved in December 2012 as an adjunct therapy to treat homozygous familial hypercholesterolemia. The Juxtapid REMS requires Aegerion to educate prescribers about the risks of hepatotoxicity and the need to monitor patients treated with Juxtapid and to ensure that Juxtapid is prescribed and dispensed only to those patients with a clinical or laboratory diagnosis consistent with homozygous familial hypercholesterolemia.
Juxtapid was misbranded because Aegerion failed to comply with the requirements of the Juxtapid Risk Evaluation and Mitigation Strategy (REMS) program and because the drug’s labeling lacked adequate directions for all of Juxtapid’s intended uses, according to the charges. Aegerion also agreed to a comprehensive compliance program and legal tools for the FDA to ensure that Aegerion complies with the law, subject to judicial oversight.
Rather than following the REMS requirement to distribute Juxtapid only for the narrow indication of homozygous familial hypercholesterolemia, Aegerion portrayed the definition of the rare disorder as vague and indefinite in order to extend its use to lower-risk patients. Further, Aegerion filed a misleading REMS assessment report to the FDA in which the company failed to disclose that it was distributing Juxtapid using this definition, which was inconsistent with Aegerion’s preapproval filings and peer-reviewed clinical standards of diagnosis, according to the FDA release.
Once entered by the court, the plea and consent decree will be part of a global resolution of multiple government investigations into Aegerion’s conduct with respect to the marketing and distribution of Juxtapid. This resolution was the result of a coordinated effort by the U.S. Department of Justice and several government agencies, including the FDA, the press release stated.
Juxtapid was approved in December 2012 as an adjunct therapy to treat homozygous familial hypercholesterolemia. The Juxtapid REMS requires Aegerion to educate prescribers about the risks of hepatotoxicity and the need to monitor patients treated with Juxtapid and to ensure that Juxtapid is prescribed and dispensed only to those patients with a clinical or laboratory diagnosis consistent with homozygous familial hypercholesterolemia.
Juxtapid was misbranded because Aegerion failed to comply with the requirements of the Juxtapid Risk Evaluation and Mitigation Strategy (REMS) program and because the drug’s labeling lacked adequate directions for all of Juxtapid’s intended uses, according to the charges. Aegerion also agreed to a comprehensive compliance program and legal tools for the FDA to ensure that Aegerion complies with the law, subject to judicial oversight.
Rather than following the REMS requirement to distribute Juxtapid only for the narrow indication of homozygous familial hypercholesterolemia, Aegerion portrayed the definition of the rare disorder as vague and indefinite in order to extend its use to lower-risk patients. Further, Aegerion filed a misleading REMS assessment report to the FDA in which the company failed to disclose that it was distributing Juxtapid using this definition, which was inconsistent with Aegerion’s preapproval filings and peer-reviewed clinical standards of diagnosis, according to the FDA release.
Once entered by the court, the plea and consent decree will be part of a global resolution of multiple government investigations into Aegerion’s conduct with respect to the marketing and distribution of Juxtapid. This resolution was the result of a coordinated effort by the U.S. Department of Justice and several government agencies, including the FDA, the press release stated.
Juxtapid was approved in December 2012 as an adjunct therapy to treat homozygous familial hypercholesterolemia. The Juxtapid REMS requires Aegerion to educate prescribers about the risks of hepatotoxicity and the need to monitor patients treated with Juxtapid and to ensure that Juxtapid is prescribed and dispensed only to those patients with a clinical or laboratory diagnosis consistent with homozygous familial hypercholesterolemia.
Do not withhold opioid addiction drugs from patients taking benzodiazepines
The opioid addiction medications buprenorphine and methadone should not be withheld from patients who are taking benzodiazepines or other drugs that depress the central nervous system, the Food and Drug Administration advised in a safety alert posted Sept. 20.
The combined use of these medication-assisted treatment (MAT) drugs and central nervous system (CNS) depressants can lead to serious side effects. But the harm associated with opioid addiction that remains untreated usually outweighs those risks, the agency said. After reviewing this issue, the FDA said, it is requiring that this information be added to the drug labels of buprenorphine and methadone in addition to “recommendations for minimizing the use of [MAT] drugs and benzodiazepines together.”
Discontinuing MAT drugs is a goal, but patients might need medication-assisted treatment indefinitely. “Use should continue for as long as patients are benefiting and their use contributes to the intended treatment goals,” the alert said.
The opioid addiction medications buprenorphine and methadone should not be withheld from patients who are taking benzodiazepines or other drugs that depress the central nervous system, the Food and Drug Administration advised in a safety alert posted Sept. 20.
The combined use of these medication-assisted treatment (MAT) drugs and central nervous system (CNS) depressants can lead to serious side effects. But the harm associated with opioid addiction that remains untreated usually outweighs those risks, the agency said. After reviewing this issue, the FDA said, it is requiring that this information be added to the drug labels of buprenorphine and methadone in addition to “recommendations for minimizing the use of [MAT] drugs and benzodiazepines together.”
Discontinuing MAT drugs is a goal, but patients might need medication-assisted treatment indefinitely. “Use should continue for as long as patients are benefiting and their use contributes to the intended treatment goals,” the alert said.
The opioid addiction medications buprenorphine and methadone should not be withheld from patients who are taking benzodiazepines or other drugs that depress the central nervous system, the Food and Drug Administration advised in a safety alert posted Sept. 20.
The combined use of these medication-assisted treatment (MAT) drugs and central nervous system (CNS) depressants can lead to serious side effects. But the harm associated with opioid addiction that remains untreated usually outweighs those risks, the agency said. After reviewing this issue, the FDA said, it is requiring that this information be added to the drug labels of buprenorphine and methadone in addition to “recommendations for minimizing the use of [MAT] drugs and benzodiazepines together.”
Discontinuing MAT drugs is a goal, but patients might need medication-assisted treatment indefinitely. “Use should continue for as long as patients are benefiting and their use contributes to the intended treatment goals,” the alert said.
FDA approves triple-therapy inhaler for COPD
The Food and Drug Administration has approved Trelegy Ellipta (fluticasone furoate/umeclidinium/vilanterol), a triple-therapy inhaler for the treatment of chronic obstructive pulmonary disease (COPD) in adult patients, according to a press release from GlaxoSmithKline and Innoviva.
Trelegy Ellipta combines an inhaled corticosteroid, a long-acting muscarinic antagonist, and a long-acting beta2-adrenergic agonist into an inhaler meant for once-daily use in people with COPD. Chronic bronchitis and/or emphysema patients are also indicated for treatment. The FDA-approved dosage is 100 mcg of fluticasone furoate, 62.5 mcg of umeclidinium, and 25 mcg of vilanterol.
“This approval represents a significant therapeutic convenience for those appropriate patients already on Breo Ellipta, that require additional bronchodilation or for those patients already on a combination of Breo Ellipta and Incruse Ellipta,” Mike Aguiar, CEO of Innoviva said in the press release.
In results supporting the FDA approval, the IMPACT study, a 52-week phase 3 clinical trial including 10,355 COPD patients sponsored by GSK, found that patients receiving Trelegy Ellipta experienced a 25% reduction in moderate to severe exacerbations compared to patients receiving Anoro Ellipta, and a 15% reduction in moderate to severe exacerbations, compared with patients receiving Relvar/Breo Ellipta. Change from baseline FEV1, change from baseline scores on the St George’s Respiratory Questionnaire, and time to first moderate/severe COPD exacerbation also were improved in the Trelegy Ellipta study group compared to the others.
“This is the first study to report a comparison of a single inhaler triple therapy with two dual therapies, providing much needed clinical evidence about the ability of a single inhaler triple therapy to reduce exacerbations,” Patrick Vallance, President of R&D at GSK, noted in a press release announcing the results of the IMPACT study.

The Food and Drug Administration has approved Trelegy Ellipta (fluticasone furoate/umeclidinium/vilanterol), a triple-therapy inhaler for the treatment of chronic obstructive pulmonary disease (COPD) in adult patients, according to a press release from GlaxoSmithKline and Innoviva.
Trelegy Ellipta combines an inhaled corticosteroid, a long-acting muscarinic antagonist, and a long-acting beta2-adrenergic agonist into an inhaler meant for once-daily use in people with COPD. Chronic bronchitis and/or emphysema patients are also indicated for treatment. The FDA-approved dosage is 100 mcg of fluticasone furoate, 62.5 mcg of umeclidinium, and 25 mcg of vilanterol.
“This approval represents a significant therapeutic convenience for those appropriate patients already on Breo Ellipta, that require additional bronchodilation or for those patients already on a combination of Breo Ellipta and Incruse Ellipta,” Mike Aguiar, CEO of Innoviva said in the press release.
In results supporting the FDA approval, the IMPACT study, a 52-week phase 3 clinical trial including 10,355 COPD patients sponsored by GSK, found that patients receiving Trelegy Ellipta experienced a 25% reduction in moderate to severe exacerbations compared to patients receiving Anoro Ellipta, and a 15% reduction in moderate to severe exacerbations, compared with patients receiving Relvar/Breo Ellipta. Change from baseline FEV1, change from baseline scores on the St George’s Respiratory Questionnaire, and time to first moderate/severe COPD exacerbation also were improved in the Trelegy Ellipta study group compared to the others.
“This is the first study to report a comparison of a single inhaler triple therapy with two dual therapies, providing much needed clinical evidence about the ability of a single inhaler triple therapy to reduce exacerbations,” Patrick Vallance, President of R&D at GSK, noted in a press release announcing the results of the IMPACT study.
The Food and Drug Administration has approved Trelegy Ellipta (fluticasone furoate/umeclidinium/vilanterol), a triple-therapy inhaler for the treatment of chronic obstructive pulmonary disease (COPD) in adult patients, according to a press release from GlaxoSmithKline and Innoviva.
Trelegy Ellipta combines an inhaled corticosteroid, a long-acting muscarinic antagonist, and a long-acting beta2-adrenergic agonist into an inhaler meant for once-daily use in people with COPD. Chronic bronchitis and/or emphysema patients are also indicated for treatment. The FDA-approved dosage is 100 mcg of fluticasone furoate, 62.5 mcg of umeclidinium, and 25 mcg of vilanterol.
“This approval represents a significant therapeutic convenience for those appropriate patients already on Breo Ellipta, that require additional bronchodilation or for those patients already on a combination of Breo Ellipta and Incruse Ellipta,” Mike Aguiar, CEO of Innoviva said in the press release.
In results supporting the FDA approval, the IMPACT study, a 52-week phase 3 clinical trial including 10,355 COPD patients sponsored by GSK, found that patients receiving Trelegy Ellipta experienced a 25% reduction in moderate to severe exacerbations compared to patients receiving Anoro Ellipta, and a 15% reduction in moderate to severe exacerbations, compared with patients receiving Relvar/Breo Ellipta. Change from baseline FEV1, change from baseline scores on the St George’s Respiratory Questionnaire, and time to first moderate/severe COPD exacerbation also were improved in the Trelegy Ellipta study group compared to the others.
“This is the first study to report a comparison of a single inhaler triple therapy with two dual therapies, providing much needed clinical evidence about the ability of a single inhaler triple therapy to reduce exacerbations,” Patrick Vallance, President of R&D at GSK, noted in a press release announcing the results of the IMPACT study.
E-cigarettes most popular among youngest adults
Over 15% of adults have used electronic cigarettes at some time, and about 3% reported current use when they were surveyed in 2016, according to the Centers for Disease Control and Prevention.
When those numbers are broken down by age group, the youngest adults are the most likely e-cigarette users: 23.5% of those aged 18-24 years had ever vaped and 4.5% were currently vaping either every day or on some days, the CDC reported (MMWR. 2017;66[33]:892).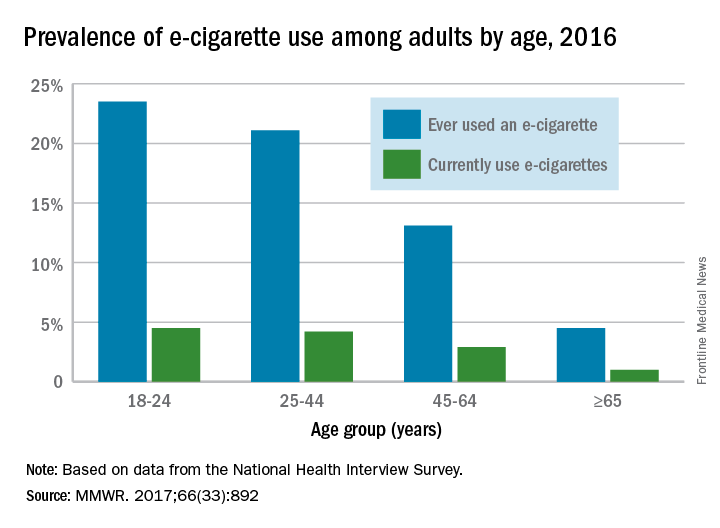
Over 15% of adults have used electronic cigarettes at some time, and about 3% reported current use when they were surveyed in 2016, according to the Centers for Disease Control and Prevention.
When those numbers are broken down by age group, the youngest adults are the most likely e-cigarette users: 23.5% of those aged 18-24 years had ever vaped and 4.5% were currently vaping either every day or on some days, the CDC reported (MMWR. 2017;66[33]:892).
Over 15% of adults have used electronic cigarettes at some time, and about 3% reported current use when they were surveyed in 2016, according to the Centers for Disease Control and Prevention.
When those numbers are broken down by age group, the youngest adults are the most likely e-cigarette users: 23.5% of those aged 18-24 years had ever vaped and 4.5% were currently vaping either every day or on some days, the CDC reported (MMWR. 2017;66[33]:892).
FROM MMWR
FDA gives nod to first mobile app for substance use disorders
The Food and Drug Administration has permitted marketing of the first mobile medical application aimed at helping to treat substance use disorders (SUDs) in adults, the agency announced Sept. 14.
“This is an example of how innovative digital technologies can help provide patients access to additional tools during their treatment,” said Carlos Peña, PhD, director of the division of neurological and physical medicine devices in the FDA’s Center for Devices and Radiological Health, in a statement.
“More therapy tools means a greater potential to help improve outcomes ... for patients with substance use disorder,” Dr. Peña added.
The agency’s permission is based on data reviewed from a multisite, unblinded 12-week clinical trial sponsored by the National Institute on Drug Abuse of 399 patients who received either standard treatment or standard treatment with the addition of a desktop-based version of reSET. According to the data, adherence to abstinence for patients with alcohol, cocaine, marijuana, and stimulant SUD who used reSET increased 40.3%, compared with an increase of 17.6% in adherence to abstinence for patients who did not use the system.
No side effects are associated with the reSET application. Adverse events reported in clinical trials were in line with those found in patients with SUD, including cardiovascular disease, gastrointestinal events, depression, mania, suicidal behavior and ideation, and suicide attempts.
Edward V. Nunes, MD, said in a statement issued by the company that developed reSET, Pear Therapeutics, that the clinical outcomes found in the pivotal study were remarkable. “Clinically validated digital therapeutics may become a cornerstone of future treatment,” said Dr. Nunes, lead investigator in the study submitted to the FDA and a professor of psychiatry at Columbia University in New York.
For more information about the reSET medical application system, click here.
acruz@frontlinemedcom.com
On Twitter @abbbbeeeyyy
The Food and Drug Administration has permitted marketing of the first mobile medical application aimed at helping to treat substance use disorders (SUDs) in adults, the agency announced Sept. 14.
“This is an example of how innovative digital technologies can help provide patients access to additional tools during their treatment,” said Carlos Peña, PhD, director of the division of neurological and physical medicine devices in the FDA’s Center for Devices and Radiological Health, in a statement.
“More therapy tools means a greater potential to help improve outcomes ... for patients with substance use disorder,” Dr. Peña added.
The agency’s permission is based on data reviewed from a multisite, unblinded 12-week clinical trial sponsored by the National Institute on Drug Abuse of 399 patients who received either standard treatment or standard treatment with the addition of a desktop-based version of reSET. According to the data, adherence to abstinence for patients with alcohol, cocaine, marijuana, and stimulant SUD who used reSET increased 40.3%, compared with an increase of 17.6% in adherence to abstinence for patients who did not use the system.
No side effects are associated with the reSET application. Adverse events reported in clinical trials were in line with those found in patients with SUD, including cardiovascular disease, gastrointestinal events, depression, mania, suicidal behavior and ideation, and suicide attempts.
Edward V. Nunes, MD, said in a statement issued by the company that developed reSET, Pear Therapeutics, that the clinical outcomes found in the pivotal study were remarkable. “Clinically validated digital therapeutics may become a cornerstone of future treatment,” said Dr. Nunes, lead investigator in the study submitted to the FDA and a professor of psychiatry at Columbia University in New York.
For more information about the reSET medical application system, click here.
acruz@frontlinemedcom.com
On Twitter @abbbbeeeyyy
The Food and Drug Administration has permitted marketing of the first mobile medical application aimed at helping to treat substance use disorders (SUDs) in adults, the agency announced Sept. 14.
“This is an example of how innovative digital technologies can help provide patients access to additional tools during their treatment,” said Carlos Peña, PhD, director of the division of neurological and physical medicine devices in the FDA’s Center for Devices and Radiological Health, in a statement.
“More therapy tools means a greater potential to help improve outcomes ... for patients with substance use disorder,” Dr. Peña added.
The agency’s permission is based on data reviewed from a multisite, unblinded 12-week clinical trial sponsored by the National Institute on Drug Abuse of 399 patients who received either standard treatment or standard treatment with the addition of a desktop-based version of reSET. According to the data, adherence to abstinence for patients with alcohol, cocaine, marijuana, and stimulant SUD who used reSET increased 40.3%, compared with an increase of 17.6% in adherence to abstinence for patients who did not use the system.
No side effects are associated with the reSET application. Adverse events reported in clinical trials were in line with those found in patients with SUD, including cardiovascular disease, gastrointestinal events, depression, mania, suicidal behavior and ideation, and suicide attempts.
Edward V. Nunes, MD, said in a statement issued by the company that developed reSET, Pear Therapeutics, that the clinical outcomes found in the pivotal study were remarkable. “Clinically validated digital therapeutics may become a cornerstone of future treatment,” said Dr. Nunes, lead investigator in the study submitted to the FDA and a professor of psychiatry at Columbia University in New York.
For more information about the reSET medical application system, click here.
acruz@frontlinemedcom.com
On Twitter @abbbbeeeyyy
FDA approves biosimilar to bevacizumab
The Food and Drug Administration has approved a biosimilar to bevacizumab (Avastin) for the treatment of certain colorectal, lung, brain, kidney, and cervical cancers.
Bevacizumab-awwb is the first biosimilar approved in the United States for the treatment of cancer, the FDA said in a press release.
Approval is based on structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamics data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrate bevacizumab-awwb is biosimilar to bevacizumab, the FDA said.
• Metastatic colorectal cancer, in combination with intravenous 5-fluorouracil-based chemotherapy for first- or second-line treatment.
• Metastatic colorectal cancer, in combination with fluoropyrimidine-irinotecan–based or fluoropyrimidine-oxaliplatin–based chemotherapy for the second-line treatment of patients who have progressed on a first-line bevacizumab product–containing regimen.
• Non-squamous non–small cell lung cancer, in combination with carboplatin and paclitaxel for first line treatment of unresectable, locally advanced, recurrent, or metastatic disease.
• Glioblastoma with progressive disease following prior therapy, based on improvement in objective response rate.
• Metastatic renal cell carcinoma, in combination with interferon alfa.
• Cervical cancer that is persistent, recurrent, or metastatic, in combination with paclitaxel and cisplatin or paclitaxel and topotecan.
Common expected side effects of the biosimilar include epistaxis, headache, hypertension, rhinitis, proteinuria, taste alteration, dry skin, hemorrhage, lacrimation disorder, back pain, and exfoliative dermatitis.
Serious expected side effects include perforation or fistula, arterial and venous thromboembolic events, hypertension, posterior reversible encephalopathy syndrome, proteinuria, infusion-related reactions, and ovarian failure. Women who are pregnant should not take bevacizumab-awwb.
The biosimilar to bevacizumab carries a similar boxed warning regarding the increased risk of gastrointestinal perforations; surgery and wound healing complications; and severe or fatal pulmonary, gastrointestinal, central nervous system, and vaginal hemorrhage.
The biosimilar approval was granted to Amgen, which will market the drug under the trade name Mvasi.
The Food and Drug Administration has approved a biosimilar to bevacizumab (Avastin) for the treatment of certain colorectal, lung, brain, kidney, and cervical cancers.
Bevacizumab-awwb is the first biosimilar approved in the United States for the treatment of cancer, the FDA said in a press release.
Approval is based on structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamics data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrate bevacizumab-awwb is biosimilar to bevacizumab, the FDA said.
• Metastatic colorectal cancer, in combination with intravenous 5-fluorouracil-based chemotherapy for first- or second-line treatment.
• Metastatic colorectal cancer, in combination with fluoropyrimidine-irinotecan–based or fluoropyrimidine-oxaliplatin–based chemotherapy for the second-line treatment of patients who have progressed on a first-line bevacizumab product–containing regimen.
• Non-squamous non–small cell lung cancer, in combination with carboplatin and paclitaxel for first line treatment of unresectable, locally advanced, recurrent, or metastatic disease.
• Glioblastoma with progressive disease following prior therapy, based on improvement in objective response rate.
• Metastatic renal cell carcinoma, in combination with interferon alfa.
• Cervical cancer that is persistent, recurrent, or metastatic, in combination with paclitaxel and cisplatin or paclitaxel and topotecan.
Common expected side effects of the biosimilar include epistaxis, headache, hypertension, rhinitis, proteinuria, taste alteration, dry skin, hemorrhage, lacrimation disorder, back pain, and exfoliative dermatitis.
Serious expected side effects include perforation or fistula, arterial and venous thromboembolic events, hypertension, posterior reversible encephalopathy syndrome, proteinuria, infusion-related reactions, and ovarian failure. Women who are pregnant should not take bevacizumab-awwb.
The biosimilar to bevacizumab carries a similar boxed warning regarding the increased risk of gastrointestinal perforations; surgery and wound healing complications; and severe or fatal pulmonary, gastrointestinal, central nervous system, and vaginal hemorrhage.
The biosimilar approval was granted to Amgen, which will market the drug under the trade name Mvasi.
The Food and Drug Administration has approved a biosimilar to bevacizumab (Avastin) for the treatment of certain colorectal, lung, brain, kidney, and cervical cancers.
Bevacizumab-awwb is the first biosimilar approved in the United States for the treatment of cancer, the FDA said in a press release.
Approval is based on structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamics data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrate bevacizumab-awwb is biosimilar to bevacizumab, the FDA said.
• Metastatic colorectal cancer, in combination with intravenous 5-fluorouracil-based chemotherapy for first- or second-line treatment.
• Metastatic colorectal cancer, in combination with fluoropyrimidine-irinotecan–based or fluoropyrimidine-oxaliplatin–based chemotherapy for the second-line treatment of patients who have progressed on a first-line bevacizumab product–containing regimen.
• Non-squamous non–small cell lung cancer, in combination with carboplatin and paclitaxel for first line treatment of unresectable, locally advanced, recurrent, or metastatic disease.
• Glioblastoma with progressive disease following prior therapy, based on improvement in objective response rate.
• Metastatic renal cell carcinoma, in combination with interferon alfa.
• Cervical cancer that is persistent, recurrent, or metastatic, in combination with paclitaxel and cisplatin or paclitaxel and topotecan.
Common expected side effects of the biosimilar include epistaxis, headache, hypertension, rhinitis, proteinuria, taste alteration, dry skin, hemorrhage, lacrimation disorder, back pain, and exfoliative dermatitis.
Serious expected side effects include perforation or fistula, arterial and venous thromboembolic events, hypertension, posterior reversible encephalopathy syndrome, proteinuria, infusion-related reactions, and ovarian failure. Women who are pregnant should not take bevacizumab-awwb.
The biosimilar to bevacizumab carries a similar boxed warning regarding the increased risk of gastrointestinal perforations; surgery and wound healing complications; and severe or fatal pulmonary, gastrointestinal, central nervous system, and vaginal hemorrhage.
The biosimilar approval was granted to Amgen, which will market the drug under the trade name Mvasi.
Adjuvant-boosted shingles vaccine earns FDA panel’s unanimous nod
A new vaccine for herpes zoster is both safe and effective in preventing herpes zoster, and in reducing the incidence of postherpetic neuralgia in older adults, according to a Food and Drug Administration advisory committee, which voted unanimously to recommend the vaccine.
The FDA generally follows the recommendations of its advisory committees.
The recombinant vaccine, dubbed HZ/su during the trial phase, showed efficacy of 97.2% against herpes zoster infection in adults aged 50 years and older, and 91.3% in adults aged 70 years and older. The effect persisted for up to the 4 years of study follow-up.
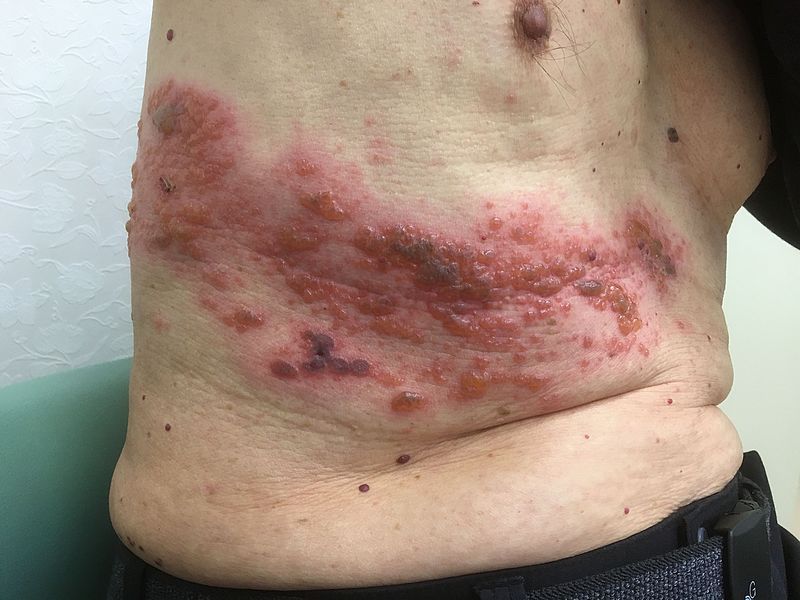
HZ/su had a generally favorable safety profile, though early constitutional symptoms and local site reactions were common, according to data presented by GlaxoSmithKline. HZ/su uses an adjuvant not found in any other U.S.-approved vaccine.
The incidence of postherpetic neuralgia, a common, persistent, and costly complication of herpes zoster, was 0.1 per 1,000 person-years in those receiving vaccine, compared with 0.9-1.2 per 1,000 person-years for those receiving placebo in the pivotal clinical trials for a median follow-up of 4 years.
In the vaccine’s pivotal clinical trials, efficacy was significantly higher than the levels seen for the only currently approved zoster live vaccine, Zostavax, especially for older populations. Zostavax’s efficacy for those aged 50-59 years is 69.8%, dropping to 18% for those aged 80 years or older.
The results of the two pivotal clinical trials were presented and analyzed by the sponsor and by FDA staff during a meeting of the Vaccines and Related Biological Products Advisory Committee of the FDA’s Center for Biologics Evaluation and Research (CBER).
During pre-vote discussions, committee members were unanimous in noting with favor the high and sustained efficacy seen for HZ/su in the trial data, especially for older populations. However, some participants wondered about the generalizability of both safety and efficacy data to all populations, given the very low trial enrollment numbers for Africans, African Americans, and individuals of Hispanic origin.
The two studies, Zoster-006 and Zoster-022, were similar in design and were conducted in parallel across 18 countries; data were able to be pooled for key efficacy and safety outcomes. Study Zoster-006 enrolled patients aged 50 years and older, while study Zoster-022 began enrollment at age 70. Patients were randomized to receive vaccine or placebo, and were followed for a median of 3.1 years for efficacy in Zoster-006 and a median of 3.9 years for Zoster-022. Safety data were obtained for a median 4.4 years for both studies.
The primary outcome measure for both studies in pooled analysis was the vaccine’s effectiveness against herpes zoster and postherpetic neuropathy in adults aged 70 and over. Safety was also assessed using pooled data.
The United States was represented by 3,934 of more than 29,000 patients enrolled globally. The remainder were primarily in Western Europe, with some sites in Australia and eastern Asia, Canada, and Latin America.
The vaccine consists of a recombinant, lyophilized truncated form of the varicella zoster virus (VZV) glycoprotein E (gE) antigen protein that, at the time of administration, is reconstituted with a novel adjuvant suspension. The antigen selection was based on the fact that gE is expressed on the surface of infected cells and is the target of both humoral and cellular immune responses in the host, said GSK’s Arnaud Didierlaurent, PhD, director and head of the adjuvant platform for GSK Vaccine’s Belgium research and development division.
The adjuvant, termed ASO1B, is not currently in use for any U.S.-approved vaccine, though it was developed more than 20 years ago, said Dr. Didierlaurent. Its combination with recombinant VZV gE was found to significantly boost the antigen’s immunogenicity during GSK’s vaccine development program. The adjuvant enhances a transient innate response in the first 3 days after administration that later helps maintain durably high levels of gE-specific antibodies and strengthens gE-specific cell-mediated immunity.
Mechanistically, the robust initial innate response is responsible for the constitutional symptoms and local site reactions seen in pooled data from the two pivotal clinical trials: 70%-85% of participants receiving HZ/su reported injection site pain, 38% of participants receiving HZ/su reported redness, and about a quarter reported swelling. By comparison, 9%-13% of those receiving placebo reported injection site pain, and about 1% reported redness and swelling.
Fatigue, headache, mild fever, myalgia, and shivering were all more common in those receiving HZ/su; both local and generalized symptoms were more common in younger recipients.
“I think this is a very good case for the first licensure of this adjuvant in the United States, because the efficacy seems pretty compelling, the disease is morbid, and there are a lot of people whose lives would be changed,” said committee member Sarah Long, MD, professor of pediatrics at Drexel University, Philadelphia.
Both the GSK and FDA presentations were in agreement that serious adverse events were in the range to be expected for an older population, and balanced across study arms. However, particular attention will be given to certain potential complications during the proposed pharmacovigilance plan.
“An imbalance toward vaccine versus placebo was observed” for gout, optic ischemic neuropathy, amyotrophic lateral sclerosis, osteonecrosis, convulsion-type reactions, and supraventricular tachycardias. “All are an adverse event of interest and will be included in planned targeted safety study,” said Dr. Didierlaurent.
Several committee members remarked on the difficulty of evaluating vaccine safety in an older population, where analysis takes place against the backdrop of more comorbidities and acute illnesses than in the younger population.
“There has been a thoughtful job both by the sponsor and by CBER in looking at complicated data,” said Melinda Wharton, MD, the director of the immunization services division of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention, Atlanta.
The committee’s chair, Kathryn Edwards, MD, agreed. “I applaud the comprehensive analysis of all these safety signals. Both the sponsor and the FDA have done a wonderful job of drilling down and answering these questions,” she said. Dr. Edwards is the Sarah H. Sell and Cornelius Vanderbilt chair in pediatrics at Vanderbilt University, Nashville, Tenn.
Herpes zoster, a reactivation of the varicella virus that lies dormant in dorsal root or cranial nerve ganglia from earlier infection, is seen in about 1 million cases per year in the United States, with about 100,000 to 200,000 cases of postherpetic neuralgia occurring, said Jeffrey Cohen, MD, chief of the laboratory of infectious diseases at the National Institute of Allergy and Infectious Diseases, Bethesda, Md. The rates of herpes zoster are increasing in the United States for unknown reasons, and direct medical costs may currently exceed $1 billion annually, he said.
Each 0.5 mL dose of the HZ/su vaccine contains 50 mcg each of the recombinant VZV gE antigen and each of the two component parts of the ASO1B adjuvant. Two doses of the vaccine are administered intramuscularly 2-6 months apart. Dose-ranging studies were conducted before the pivotal clinical trials to ascertain the optimal dose of all of the vaccine components, the need for two doses, and the optimal spacing between doses.
All committee participants submitted conflict of interest statements to the FDA, and any potential conflicts were resolved before the hearing.
koakes@frontlinemedcom.com
On Twitter @karioakes
A new vaccine for herpes zoster is both safe and effective in preventing herpes zoster, and in reducing the incidence of postherpetic neuralgia in older adults, according to a Food and Drug Administration advisory committee, which voted unanimously to recommend the vaccine.
The FDA generally follows the recommendations of its advisory committees.
The recombinant vaccine, dubbed HZ/su during the trial phase, showed efficacy of 97.2% against herpes zoster infection in adults aged 50 years and older, and 91.3% in adults aged 70 years and older. The effect persisted for up to the 4 years of study follow-up.
HZ/su had a generally favorable safety profile, though early constitutional symptoms and local site reactions were common, according to data presented by GlaxoSmithKline. HZ/su uses an adjuvant not found in any other U.S.-approved vaccine.
The incidence of postherpetic neuralgia, a common, persistent, and costly complication of herpes zoster, was 0.1 per 1,000 person-years in those receiving vaccine, compared with 0.9-1.2 per 1,000 person-years for those receiving placebo in the pivotal clinical trials for a median follow-up of 4 years.
In the vaccine’s pivotal clinical trials, efficacy was significantly higher than the levels seen for the only currently approved zoster live vaccine, Zostavax, especially for older populations. Zostavax’s efficacy for those aged 50-59 years is 69.8%, dropping to 18% for those aged 80 years or older.
The results of the two pivotal clinical trials were presented and analyzed by the sponsor and by FDA staff during a meeting of the Vaccines and Related Biological Products Advisory Committee of the FDA’s Center for Biologics Evaluation and Research (CBER).
During pre-vote discussions, committee members were unanimous in noting with favor the high and sustained efficacy seen for HZ/su in the trial data, especially for older populations. However, some participants wondered about the generalizability of both safety and efficacy data to all populations, given the very low trial enrollment numbers for Africans, African Americans, and individuals of Hispanic origin.
The two studies, Zoster-006 and Zoster-022, were similar in design and were conducted in parallel across 18 countries; data were able to be pooled for key efficacy and safety outcomes. Study Zoster-006 enrolled patients aged 50 years and older, while study Zoster-022 began enrollment at age 70. Patients were randomized to receive vaccine or placebo, and were followed for a median of 3.1 years for efficacy in Zoster-006 and a median of 3.9 years for Zoster-022. Safety data were obtained for a median 4.4 years for both studies.
The primary outcome measure for both studies in pooled analysis was the vaccine’s effectiveness against herpes zoster and postherpetic neuropathy in adults aged 70 and over. Safety was also assessed using pooled data.
The United States was represented by 3,934 of more than 29,000 patients enrolled globally. The remainder were primarily in Western Europe, with some sites in Australia and eastern Asia, Canada, and Latin America.
The vaccine consists of a recombinant, lyophilized truncated form of the varicella zoster virus (VZV) glycoprotein E (gE) antigen protein that, at the time of administration, is reconstituted with a novel adjuvant suspension. The antigen selection was based on the fact that gE is expressed on the surface of infected cells and is the target of both humoral and cellular immune responses in the host, said GSK’s Arnaud Didierlaurent, PhD, director and head of the adjuvant platform for GSK Vaccine’s Belgium research and development division.
The adjuvant, termed ASO1B, is not currently in use for any U.S.-approved vaccine, though it was developed more than 20 years ago, said Dr. Didierlaurent. Its combination with recombinant VZV gE was found to significantly boost the antigen’s immunogenicity during GSK’s vaccine development program. The adjuvant enhances a transient innate response in the first 3 days after administration that later helps maintain durably high levels of gE-specific antibodies and strengthens gE-specific cell-mediated immunity.
Mechanistically, the robust initial innate response is responsible for the constitutional symptoms and local site reactions seen in pooled data from the two pivotal clinical trials: 70%-85% of participants receiving HZ/su reported injection site pain, 38% of participants receiving HZ/su reported redness, and about a quarter reported swelling. By comparison, 9%-13% of those receiving placebo reported injection site pain, and about 1% reported redness and swelling.
Fatigue, headache, mild fever, myalgia, and shivering were all more common in those receiving HZ/su; both local and generalized symptoms were more common in younger recipients.
“I think this is a very good case for the first licensure of this adjuvant in the United States, because the efficacy seems pretty compelling, the disease is morbid, and there are a lot of people whose lives would be changed,” said committee member Sarah Long, MD, professor of pediatrics at Drexel University, Philadelphia.
Both the GSK and FDA presentations were in agreement that serious adverse events were in the range to be expected for an older population, and balanced across study arms. However, particular attention will be given to certain potential complications during the proposed pharmacovigilance plan.
“An imbalance toward vaccine versus placebo was observed” for gout, optic ischemic neuropathy, amyotrophic lateral sclerosis, osteonecrosis, convulsion-type reactions, and supraventricular tachycardias. “All are an adverse event of interest and will be included in planned targeted safety study,” said Dr. Didierlaurent.
Several committee members remarked on the difficulty of evaluating vaccine safety in an older population, where analysis takes place against the backdrop of more comorbidities and acute illnesses than in the younger population.
“There has been a thoughtful job both by the sponsor and by CBER in looking at complicated data,” said Melinda Wharton, MD, the director of the immunization services division of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention, Atlanta.
The committee’s chair, Kathryn Edwards, MD, agreed. “I applaud the comprehensive analysis of all these safety signals. Both the sponsor and the FDA have done a wonderful job of drilling down and answering these questions,” she said. Dr. Edwards is the Sarah H. Sell and Cornelius Vanderbilt chair in pediatrics at Vanderbilt University, Nashville, Tenn.
Herpes zoster, a reactivation of the varicella virus that lies dormant in dorsal root or cranial nerve ganglia from earlier infection, is seen in about 1 million cases per year in the United States, with about 100,000 to 200,000 cases of postherpetic neuralgia occurring, said Jeffrey Cohen, MD, chief of the laboratory of infectious diseases at the National Institute of Allergy and Infectious Diseases, Bethesda, Md. The rates of herpes zoster are increasing in the United States for unknown reasons, and direct medical costs may currently exceed $1 billion annually, he said.
Each 0.5 mL dose of the HZ/su vaccine contains 50 mcg each of the recombinant VZV gE antigen and each of the two component parts of the ASO1B adjuvant. Two doses of the vaccine are administered intramuscularly 2-6 months apart. Dose-ranging studies were conducted before the pivotal clinical trials to ascertain the optimal dose of all of the vaccine components, the need for two doses, and the optimal spacing between doses.
All committee participants submitted conflict of interest statements to the FDA, and any potential conflicts were resolved before the hearing.
koakes@frontlinemedcom.com
On Twitter @karioakes
A new vaccine for herpes zoster is both safe and effective in preventing herpes zoster, and in reducing the incidence of postherpetic neuralgia in older adults, according to a Food and Drug Administration advisory committee, which voted unanimously to recommend the vaccine.
The FDA generally follows the recommendations of its advisory committees.
The recombinant vaccine, dubbed HZ/su during the trial phase, showed efficacy of 97.2% against herpes zoster infection in adults aged 50 years and older, and 91.3% in adults aged 70 years and older. The effect persisted for up to the 4 years of study follow-up.
HZ/su had a generally favorable safety profile, though early constitutional symptoms and local site reactions were common, according to data presented by GlaxoSmithKline. HZ/su uses an adjuvant not found in any other U.S.-approved vaccine.
The incidence of postherpetic neuralgia, a common, persistent, and costly complication of herpes zoster, was 0.1 per 1,000 person-years in those receiving vaccine, compared with 0.9-1.2 per 1,000 person-years for those receiving placebo in the pivotal clinical trials for a median follow-up of 4 years.
In the vaccine’s pivotal clinical trials, efficacy was significantly higher than the levels seen for the only currently approved zoster live vaccine, Zostavax, especially for older populations. Zostavax’s efficacy for those aged 50-59 years is 69.8%, dropping to 18% for those aged 80 years or older.
The results of the two pivotal clinical trials were presented and analyzed by the sponsor and by FDA staff during a meeting of the Vaccines and Related Biological Products Advisory Committee of the FDA’s Center for Biologics Evaluation and Research (CBER).
During pre-vote discussions, committee members were unanimous in noting with favor the high and sustained efficacy seen for HZ/su in the trial data, especially for older populations. However, some participants wondered about the generalizability of both safety and efficacy data to all populations, given the very low trial enrollment numbers for Africans, African Americans, and individuals of Hispanic origin.
The two studies, Zoster-006 and Zoster-022, were similar in design and were conducted in parallel across 18 countries; data were able to be pooled for key efficacy and safety outcomes. Study Zoster-006 enrolled patients aged 50 years and older, while study Zoster-022 began enrollment at age 70. Patients were randomized to receive vaccine or placebo, and were followed for a median of 3.1 years for efficacy in Zoster-006 and a median of 3.9 years for Zoster-022. Safety data were obtained for a median 4.4 years for both studies.
The primary outcome measure for both studies in pooled analysis was the vaccine’s effectiveness against herpes zoster and postherpetic neuropathy in adults aged 70 and over. Safety was also assessed using pooled data.
The United States was represented by 3,934 of more than 29,000 patients enrolled globally. The remainder were primarily in Western Europe, with some sites in Australia and eastern Asia, Canada, and Latin America.
The vaccine consists of a recombinant, lyophilized truncated form of the varicella zoster virus (VZV) glycoprotein E (gE) antigen protein that, at the time of administration, is reconstituted with a novel adjuvant suspension. The antigen selection was based on the fact that gE is expressed on the surface of infected cells and is the target of both humoral and cellular immune responses in the host, said GSK’s Arnaud Didierlaurent, PhD, director and head of the adjuvant platform for GSK Vaccine’s Belgium research and development division.
The adjuvant, termed ASO1B, is not currently in use for any U.S.-approved vaccine, though it was developed more than 20 years ago, said Dr. Didierlaurent. Its combination with recombinant VZV gE was found to significantly boost the antigen’s immunogenicity during GSK’s vaccine development program. The adjuvant enhances a transient innate response in the first 3 days after administration that later helps maintain durably high levels of gE-specific antibodies and strengthens gE-specific cell-mediated immunity.
Mechanistically, the robust initial innate response is responsible for the constitutional symptoms and local site reactions seen in pooled data from the two pivotal clinical trials: 70%-85% of participants receiving HZ/su reported injection site pain, 38% of participants receiving HZ/su reported redness, and about a quarter reported swelling. By comparison, 9%-13% of those receiving placebo reported injection site pain, and about 1% reported redness and swelling.
Fatigue, headache, mild fever, myalgia, and shivering were all more common in those receiving HZ/su; both local and generalized symptoms were more common in younger recipients.
“I think this is a very good case for the first licensure of this adjuvant in the United States, because the efficacy seems pretty compelling, the disease is morbid, and there are a lot of people whose lives would be changed,” said committee member Sarah Long, MD, professor of pediatrics at Drexel University, Philadelphia.
Both the GSK and FDA presentations were in agreement that serious adverse events were in the range to be expected for an older population, and balanced across study arms. However, particular attention will be given to certain potential complications during the proposed pharmacovigilance plan.
“An imbalance toward vaccine versus placebo was observed” for gout, optic ischemic neuropathy, amyotrophic lateral sclerosis, osteonecrosis, convulsion-type reactions, and supraventricular tachycardias. “All are an adverse event of interest and will be included in planned targeted safety study,” said Dr. Didierlaurent.
Several committee members remarked on the difficulty of evaluating vaccine safety in an older population, where analysis takes place against the backdrop of more comorbidities and acute illnesses than in the younger population.
“There has been a thoughtful job both by the sponsor and by CBER in looking at complicated data,” said Melinda Wharton, MD, the director of the immunization services division of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention, Atlanta.
The committee’s chair, Kathryn Edwards, MD, agreed. “I applaud the comprehensive analysis of all these safety signals. Both the sponsor and the FDA have done a wonderful job of drilling down and answering these questions,” she said. Dr. Edwards is the Sarah H. Sell and Cornelius Vanderbilt chair in pediatrics at Vanderbilt University, Nashville, Tenn.
Herpes zoster, a reactivation of the varicella virus that lies dormant in dorsal root or cranial nerve ganglia from earlier infection, is seen in about 1 million cases per year in the United States, with about 100,000 to 200,000 cases of postherpetic neuralgia occurring, said Jeffrey Cohen, MD, chief of the laboratory of infectious diseases at the National Institute of Allergy and Infectious Diseases, Bethesda, Md. The rates of herpes zoster are increasing in the United States for unknown reasons, and direct medical costs may currently exceed $1 billion annually, he said.
Each 0.5 mL dose of the HZ/su vaccine contains 50 mcg each of the recombinant VZV gE antigen and each of the two component parts of the ASO1B adjuvant. Two doses of the vaccine are administered intramuscularly 2-6 months apart. Dose-ranging studies were conducted before the pivotal clinical trials to ascertain the optimal dose of all of the vaccine components, the need for two doses, and the optimal spacing between doses.
All committee participants submitted conflict of interest statements to the FDA, and any potential conflicts were resolved before the hearing.
koakes@frontlinemedcom.com
On Twitter @karioakes
FDA advisory committee rejects opioids in children’s cough syrup
ROCKVILLE, MD – The majority of
The voting was broken into multiple votes based on age range of patients and the specific opioid present in the cough syrup. Unlike other advisory committee meetings, this meeting did not focus on a the treatment of a disease state, but rather on the treatment of a symptom.
On Sept. 11, 2017, the FDA’s Pediatric Advisory Committee voted 21 no, 2 yes, with one abstention, that the benefit versus the risk of opioid cough suppressants for pediatric patients was not favorable.
This vote was preceded by two previous votes specifically questioning the use of codeine and hydrocodone in medications for pediatric patients. For codeine, the committee voted unanimously with 24 against that the benefit versus risk was not favorable in pediatric patients aged 12 years to less than 18 years. 
For hydrocodone, the committee asked two questions: 1) Was the benefit versus risk favorable for pediatric patients aged 6 years to less than 12 years? and 2) Was the benefit versus risk favorable for pediatric patients aged 12 years to less than 18 years? On the vote for patients aged 6 years to less than 12 years, the committee voted 23 no, 1 yes with no abstention. For the patients aged 12 years to less than 18, the committee voted 23 no, 1 yes, with no abstention.
According to Sharon Levy, MD, MPH, adolescents are the most at-risk population for opioid misuse. This susceptibility is due to the developmental neurobiology of adolescent brains. A region of the brain associated with the reward pathway, nucleus accumbens, is developing in adolescents and plays a role in salience. Salience, or the differentiation between important vs. unimportant rewards, varies widely by age group. Young children show little salience with rewards, and treat rewards equivocally. Adults have a proportional response to rewards with accurate salience. Adolescents, on the other hand, are unhappy with small rewards, but receive a massive return with large rewards. This type of neurobiological feedback makes adolescents “vulnerable to develop substance use disorders.”
Dr. Levy also noted a correlation between prescribed opioid use and alcohol, marijuana, and tobacco use as contributing factors to opioid misuse. When opioids are prescribed for pain management, there is an adjusted odds ratio (AOR) of 1.33, indicating a high likelihood of misuse. Similar AORs are seen in adolescents who have used marijuana, cigarettes, and alcohol: 2.44, 1.25, and 1.23, respectively.
Sovereign pharmaceuticals representative Leonard Lawrence presented the findings of a pharmacokinetic study for hydrocodone and guaifenesin in 25-35 pediatric patients evenly divided into groups aged 6 years to less than 12 years, and 12 years to less than 18 years. According to Mr. Lawrence, codeine appears “to be a greater risk in children younger than 12 years, and should not be used” because of difficulty breathing. Mr Lawrence went on to say that these effects were exacerbated in obese children with lung disease or obstructive sleep apnea.
Victor S. Sloan, MD, of UCB in Brussels, presented an internal review of Tussionex, a combination cough medicine (hydrocodone/chlorpheniramine). This review took into account modern pharmacovigilance methods, changes in clinical practice, and a literature review. “Upon annual review, UCB determined that benefit risk balance for use of Tussionex for cough in children was no longer favorable,” said Dr. Sloan. Based on the results of the review, UCB has filed a label supplement to limit use of Tussionex to patients aged 18 years or older.
“Codeine, in particular, is an antiquated drug,” said Kathleen Neville, MD, pediatrics and clinical pharmacology section chief of Arkansas Children’s Hospital, Little Rock. Many of the committee members echoed Dr. Neville’s opinion.
The committee members had no relevant financial disclosures.
ilacy@frontlinemedcom.com
On Twitter @ilacy_19
ROCKVILLE, MD – The majority of
The voting was broken into multiple votes based on age range of patients and the specific opioid present in the cough syrup. Unlike other advisory committee meetings, this meeting did not focus on a the treatment of a disease state, but rather on the treatment of a symptom.
On Sept. 11, 2017, the FDA’s Pediatric Advisory Committee voted 21 no, 2 yes, with one abstention, that the benefit versus the risk of opioid cough suppressants for pediatric patients was not favorable.
This vote was preceded by two previous votes specifically questioning the use of codeine and hydrocodone in medications for pediatric patients. For codeine, the committee voted unanimously with 24 against that the benefit versus risk was not favorable in pediatric patients aged 12 years to less than 18 years.
For hydrocodone, the committee asked two questions: 1) Was the benefit versus risk favorable for pediatric patients aged 6 years to less than 12 years? and 2) Was the benefit versus risk favorable for pediatric patients aged 12 years to less than 18 years? On the vote for patients aged 6 years to less than 12 years, the committee voted 23 no, 1 yes with no abstention. For the patients aged 12 years to less than 18, the committee voted 23 no, 1 yes, with no abstention.
According to Sharon Levy, MD, MPH, adolescents are the most at-risk population for opioid misuse. This susceptibility is due to the developmental neurobiology of adolescent brains. A region of the brain associated with the reward pathway, nucleus accumbens, is developing in adolescents and plays a role in salience. Salience, or the differentiation between important vs. unimportant rewards, varies widely by age group. Young children show little salience with rewards, and treat rewards equivocally. Adults have a proportional response to rewards with accurate salience. Adolescents, on the other hand, are unhappy with small rewards, but receive a massive return with large rewards. This type of neurobiological feedback makes adolescents “vulnerable to develop substance use disorders.”
Dr. Levy also noted a correlation between prescribed opioid use and alcohol, marijuana, and tobacco use as contributing factors to opioid misuse. When opioids are prescribed for pain management, there is an adjusted odds ratio (AOR) of 1.33, indicating a high likelihood of misuse. Similar AORs are seen in adolescents who have used marijuana, cigarettes, and alcohol: 2.44, 1.25, and 1.23, respectively.
Sovereign pharmaceuticals representative Leonard Lawrence presented the findings of a pharmacokinetic study for hydrocodone and guaifenesin in 25-35 pediatric patients evenly divided into groups aged 6 years to less than 12 years, and 12 years to less than 18 years. According to Mr. Lawrence, codeine appears “to be a greater risk in children younger than 12 years, and should not be used” because of difficulty breathing. Mr Lawrence went on to say that these effects were exacerbated in obese children with lung disease or obstructive sleep apnea.
Victor S. Sloan, MD, of UCB in Brussels, presented an internal review of Tussionex, a combination cough medicine (hydrocodone/chlorpheniramine). This review took into account modern pharmacovigilance methods, changes in clinical practice, and a literature review. “Upon annual review, UCB determined that benefit risk balance for use of Tussionex for cough in children was no longer favorable,” said Dr. Sloan. Based on the results of the review, UCB has filed a label supplement to limit use of Tussionex to patients aged 18 years or older.
“Codeine, in particular, is an antiquated drug,” said Kathleen Neville, MD, pediatrics and clinical pharmacology section chief of Arkansas Children’s Hospital, Little Rock. Many of the committee members echoed Dr. Neville’s opinion.
The committee members had no relevant financial disclosures.
ilacy@frontlinemedcom.com
On Twitter @ilacy_19
ROCKVILLE, MD – The majority of
The voting was broken into multiple votes based on age range of patients and the specific opioid present in the cough syrup. Unlike other advisory committee meetings, this meeting did not focus on a the treatment of a disease state, but rather on the treatment of a symptom.
On Sept. 11, 2017, the FDA’s Pediatric Advisory Committee voted 21 no, 2 yes, with one abstention, that the benefit versus the risk of opioid cough suppressants for pediatric patients was not favorable.
This vote was preceded by two previous votes specifically questioning the use of codeine and hydrocodone in medications for pediatric patients. For codeine, the committee voted unanimously with 24 against that the benefit versus risk was not favorable in pediatric patients aged 12 years to less than 18 years.
For hydrocodone, the committee asked two questions: 1) Was the benefit versus risk favorable for pediatric patients aged 6 years to less than 12 years? and 2) Was the benefit versus risk favorable for pediatric patients aged 12 years to less than 18 years? On the vote for patients aged 6 years to less than 12 years, the committee voted 23 no, 1 yes with no abstention. For the patients aged 12 years to less than 18, the committee voted 23 no, 1 yes, with no abstention.
According to Sharon Levy, MD, MPH, adolescents are the most at-risk population for opioid misuse. This susceptibility is due to the developmental neurobiology of adolescent brains. A region of the brain associated with the reward pathway, nucleus accumbens, is developing in adolescents and plays a role in salience. Salience, or the differentiation between important vs. unimportant rewards, varies widely by age group. Young children show little salience with rewards, and treat rewards equivocally. Adults have a proportional response to rewards with accurate salience. Adolescents, on the other hand, are unhappy with small rewards, but receive a massive return with large rewards. This type of neurobiological feedback makes adolescents “vulnerable to develop substance use disorders.”
Dr. Levy also noted a correlation between prescribed opioid use and alcohol, marijuana, and tobacco use as contributing factors to opioid misuse. When opioids are prescribed for pain management, there is an adjusted odds ratio (AOR) of 1.33, indicating a high likelihood of misuse. Similar AORs are seen in adolescents who have used marijuana, cigarettes, and alcohol: 2.44, 1.25, and 1.23, respectively.
Sovereign pharmaceuticals representative Leonard Lawrence presented the findings of a pharmacokinetic study for hydrocodone and guaifenesin in 25-35 pediatric patients evenly divided into groups aged 6 years to less than 12 years, and 12 years to less than 18 years. According to Mr. Lawrence, codeine appears “to be a greater risk in children younger than 12 years, and should not be used” because of difficulty breathing. Mr Lawrence went on to say that these effects were exacerbated in obese children with lung disease or obstructive sleep apnea.
Victor S. Sloan, MD, of UCB in Brussels, presented an internal review of Tussionex, a combination cough medicine (hydrocodone/chlorpheniramine). This review took into account modern pharmacovigilance methods, changes in clinical practice, and a literature review. “Upon annual review, UCB determined that benefit risk balance for use of Tussionex for cough in children was no longer favorable,” said Dr. Sloan. Based on the results of the review, UCB has filed a label supplement to limit use of Tussionex to patients aged 18 years or older.
“Codeine, in particular, is an antiquated drug,” said Kathleen Neville, MD, pediatrics and clinical pharmacology section chief of Arkansas Children’s Hospital, Little Rock. Many of the committee members echoed Dr. Neville’s opinion.
The committee members had no relevant financial disclosures.
ilacy@frontlinemedcom.com
On Twitter @ilacy_19
AT AN FDA PEDIATRIC ADVISORY COMMITTEE MEETING
Sleep issues vary by menopausal status
Perimenopausal women aged 40-59 years were less likely than were others in the same age group to average at least 7 hours’ sleep each night in 2015, according to the National Center for Health Statistics.
Among the perimenopausal women in that age group, 56% said that they slept less than 7 hours, on average, in a 24-hour period, compared with 40.5% of postmenopausal women and 32.5% of those who were premenopausal. Overall, 35.1% of women aged 40-59 did not average at least 7 hours of sleep per night, the NCHS reported in a data brief released Sept. 7.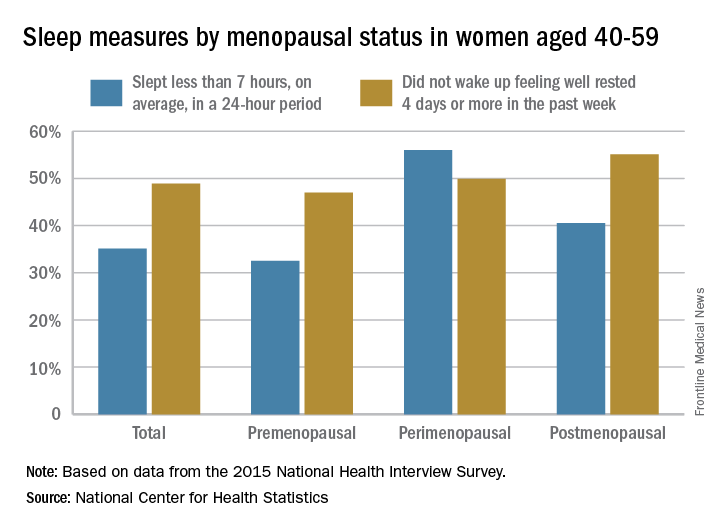
For this analysis, about 74% of the women included were premenopausal (still had a menstrual cycle), 4% were perimenopausal (last menstrual cycle was 1 year before or less), and 22% were postmenopausal (no menstrual cycle for more than 1 year or surgical menopause after removal of their ovaries).
Perimenopausal women aged 40-59 years were less likely than were others in the same age group to average at least 7 hours’ sleep each night in 2015, according to the National Center for Health Statistics.
Among the perimenopausal women in that age group, 56% said that they slept less than 7 hours, on average, in a 24-hour period, compared with 40.5% of postmenopausal women and 32.5% of those who were premenopausal. Overall, 35.1% of women aged 40-59 did not average at least 7 hours of sleep per night, the NCHS reported in a data brief released Sept. 7.
For this analysis, about 74% of the women included were premenopausal (still had a menstrual cycle), 4% were perimenopausal (last menstrual cycle was 1 year before or less), and 22% were postmenopausal (no menstrual cycle for more than 1 year or surgical menopause after removal of their ovaries).
Perimenopausal women aged 40-59 years were less likely than were others in the same age group to average at least 7 hours’ sleep each night in 2015, according to the National Center for Health Statistics.
Among the perimenopausal women in that age group, 56% said that they slept less than 7 hours, on average, in a 24-hour period, compared with 40.5% of postmenopausal women and 32.5% of those who were premenopausal. Overall, 35.1% of women aged 40-59 did not average at least 7 hours of sleep per night, the NCHS reported in a data brief released Sept. 7.
For this analysis, about 74% of the women included were premenopausal (still had a menstrual cycle), 4% were perimenopausal (last menstrual cycle was 1 year before or less), and 22% were postmenopausal (no menstrual cycle for more than 1 year or surgical menopause after removal of their ovaries).
Recent upturn seen in stroke death rate
A recent increase in the death rate from stroke resulted in more than 32,000 more deaths than would have occurred had the previous long-term decline continued, according to the Centers for Disease Control and Prevention.
The overall age-standardized stroke death rate among adults aged 35 years and older declined from 118.4 per 100,000 in 2000 to 73.3 per 100,000 in 2015, for an average annual percent change of –3.1%. That long-term rate, however, includes a more recent, but statistically nonsignificant, increase of 2.5% a year in 2013-2015, which produced an estimated 32,593 excess deaths based on the previous rate of decline, the CDC investigators said (MMWR 2017 Sep 6;66[early release]:1-7).
“Reasons for the slowing, stalling, and reversing in declines in stroke death rates are not clear. … Recent studies have reported that younger adults have experienced a significant increase in both stroke hospitalizations and in associated stroke risk factors (e.g., hypertension, obesity, diabetes, lipid disorder, and tobacco use). … These changes in modifiable stroke risk factors might present new challenges for stroke prevention and for maintaining a sustained decline in stroke mortality in the United States,” the investigators wrote.
A recent increase in the death rate from stroke resulted in more than 32,000 more deaths than would have occurred had the previous long-term decline continued, according to the Centers for Disease Control and Prevention.
The overall age-standardized stroke death rate among adults aged 35 years and older declined from 118.4 per 100,000 in 2000 to 73.3 per 100,000 in 2015, for an average annual percent change of –3.1%. That long-term rate, however, includes a more recent, but statistically nonsignificant, increase of 2.5% a year in 2013-2015, which produced an estimated 32,593 excess deaths based on the previous rate of decline, the CDC investigators said (MMWR 2017 Sep 6;66[early release]:1-7).
“Reasons for the slowing, stalling, and reversing in declines in stroke death rates are not clear. … Recent studies have reported that younger adults have experienced a significant increase in both stroke hospitalizations and in associated stroke risk factors (e.g., hypertension, obesity, diabetes, lipid disorder, and tobacco use). … These changes in modifiable stroke risk factors might present new challenges for stroke prevention and for maintaining a sustained decline in stroke mortality in the United States,” the investigators wrote.
A recent increase in the death rate from stroke resulted in more than 32,000 more deaths than would have occurred had the previous long-term decline continued, according to the Centers for Disease Control and Prevention.
The overall age-standardized stroke death rate among adults aged 35 years and older declined from 118.4 per 100,000 in 2000 to 73.3 per 100,000 in 2015, for an average annual percent change of –3.1%. That long-term rate, however, includes a more recent, but statistically nonsignificant, increase of 2.5% a year in 2013-2015, which produced an estimated 32,593 excess deaths based on the previous rate of decline, the CDC investigators said (MMWR 2017 Sep 6;66[early release]:1-7).
“Reasons for the slowing, stalling, and reversing in declines in stroke death rates are not clear. … Recent studies have reported that younger adults have experienced a significant increase in both stroke hospitalizations and in associated stroke risk factors (e.g., hypertension, obesity, diabetes, lipid disorder, and tobacco use). … These changes in modifiable stroke risk factors might present new challenges for stroke prevention and for maintaining a sustained decline in stroke mortality in the United States,” the investigators wrote.
FROM MMWR