User login
MDedge conference coverage features onsite reporting of the latest study results and expert perspectives from leading researchers.
Twelve-month overall survival benefit with ribociclib for metastatic breast cancer
“Based on these results, ribociclib and letrozole should be considered the preferred treatment option,” said lead investigator Gabriel N. Hortobagyi, MD, a breast cancer medical oncologist at the University of Texas MD Anderson Cancer Center, Houston.
He presented the definitive overall survival results from MONALESSA-2 which randomized 668 patients equally and in the first line to either ribociclib or placebo on a background of standard dose letrozole.
At a median follow up of 6.6 years, median overall survival with ribociclib was 63.9 months versus 51.4 months in the placebo arm, a 24% reduction in the relative risk of death (P = .004).
It was the first report of a median overall survival (OS) exceeding 5 years in a phase 3 trial for advanced breast cancer. The estimated 6-year OS rate was 44.2% for ribociclib versus 32.0% with placebo.
“These are really impressive results” and support the use of CDK 4/6 inhibitors in the front-line setting,” said study discussant Gonzalo Gomez Abuin, MD, a medical oncologist at Hospital Alemán in Bueno Aires.
Ribociclib and other CDK 4/6 inhibitors have shown consistent progression-free survival benefit for metastatic disease, but ribociclib is the first of the major phase 3 trials with definitive overall survival results. They have “been long awaited,” Dr. Abuin said.
The overall survival benefit in MONALESSA-2 began to emerge at around 20 months and continued to increase over time.
Women had no prior CDK4/6 inhibitor treatment, chemotherapy, or endocrine therapy for metastatic disease. “They represented a pure first-line population,” Dr. Hortobagyi said.
Among other benefits, the time to first chemotherapy was a median of 50.6 months with ribociclib versus 38.9 months with placebo, so patients “had an extra year of delay before chemotherapy was utilized,” he said.
In general, Dr. Abuin said, we “see a consistent benefit with CDK 4/6 inhibitors in metastatic breast cancer across different settings.”
However, “it’s a little intriguing” that in a subgroup analysis of non–de novo disease, the overall survival benefit with ribociclib had a hazard ratio of 0.91, whereas the progression-free survival benefit was robust and statistically significant in an earlier report.
“This has been an important question, but I would caution all of us not to make too much out of the forest plot,” Dr. Hortobagyi said.
“There are a number of hypotheses one could come up with that could explain why the de novo and non–de novo populations faired differently in overall survival as opposed to progression-free survival, but there is also the simple possibility that this is a statistical fluke,” he said.
“We are in the process of analyzing this particular observation. In the meantime, I think we should just take the overall survival results of the entire population as the lead answer, and not follow the subgroup analysis until further information is available,” Dr. Hortobagyi said.
No new ribociclib safety signals were observed in the trial. The most common adverse events were neutropenia and liver function abnormalities, but they were “largely asymptomatic laboratory findings and completely reversible,” he said.
Twice as many patients treated with ribociclib developed prolonged QT intervals, but again, “no clinical consequences of this EKG finding were detected,” Dr. Hortobagyi said.
Less than 1% of patients in the ribociclib arm developed interstitial lung disease. The majority of safety events occurred in the first 12 months of treatment.
The work was funded by Novartis, maker of both ribociclib and letrozole. Dr. Hortobagyi reported receiving an institutional grant from the company and personal fees related to the trial. Other investigators disclosed ties to Novartis. Dr. Abuin reported relationships with many companies, including Novartis.
This article was updated 9/24/21.
“Based on these results, ribociclib and letrozole should be considered the preferred treatment option,” said lead investigator Gabriel N. Hortobagyi, MD, a breast cancer medical oncologist at the University of Texas MD Anderson Cancer Center, Houston.
He presented the definitive overall survival results from MONALESSA-2 which randomized 668 patients equally and in the first line to either ribociclib or placebo on a background of standard dose letrozole.
At a median follow up of 6.6 years, median overall survival with ribociclib was 63.9 months versus 51.4 months in the placebo arm, a 24% reduction in the relative risk of death (P = .004).
It was the first report of a median overall survival (OS) exceeding 5 years in a phase 3 trial for advanced breast cancer. The estimated 6-year OS rate was 44.2% for ribociclib versus 32.0% with placebo.
“These are really impressive results” and support the use of CDK 4/6 inhibitors in the front-line setting,” said study discussant Gonzalo Gomez Abuin, MD, a medical oncologist at Hospital Alemán in Bueno Aires.
Ribociclib and other CDK 4/6 inhibitors have shown consistent progression-free survival benefit for metastatic disease, but ribociclib is the first of the major phase 3 trials with definitive overall survival results. They have “been long awaited,” Dr. Abuin said.
The overall survival benefit in MONALESSA-2 began to emerge at around 20 months and continued to increase over time.
Women had no prior CDK4/6 inhibitor treatment, chemotherapy, or endocrine therapy for metastatic disease. “They represented a pure first-line population,” Dr. Hortobagyi said.
Among other benefits, the time to first chemotherapy was a median of 50.6 months with ribociclib versus 38.9 months with placebo, so patients “had an extra year of delay before chemotherapy was utilized,” he said.
In general, Dr. Abuin said, we “see a consistent benefit with CDK 4/6 inhibitors in metastatic breast cancer across different settings.”
However, “it’s a little intriguing” that in a subgroup analysis of non–de novo disease, the overall survival benefit with ribociclib had a hazard ratio of 0.91, whereas the progression-free survival benefit was robust and statistically significant in an earlier report.
“This has been an important question, but I would caution all of us not to make too much out of the forest plot,” Dr. Hortobagyi said.
“There are a number of hypotheses one could come up with that could explain why the de novo and non–de novo populations faired differently in overall survival as opposed to progression-free survival, but there is also the simple possibility that this is a statistical fluke,” he said.
“We are in the process of analyzing this particular observation. In the meantime, I think we should just take the overall survival results of the entire population as the lead answer, and not follow the subgroup analysis until further information is available,” Dr. Hortobagyi said.
No new ribociclib safety signals were observed in the trial. The most common adverse events were neutropenia and liver function abnormalities, but they were “largely asymptomatic laboratory findings and completely reversible,” he said.
Twice as many patients treated with ribociclib developed prolonged QT intervals, but again, “no clinical consequences of this EKG finding were detected,” Dr. Hortobagyi said.
Less than 1% of patients in the ribociclib arm developed interstitial lung disease. The majority of safety events occurred in the first 12 months of treatment.
The work was funded by Novartis, maker of both ribociclib and letrozole. Dr. Hortobagyi reported receiving an institutional grant from the company and personal fees related to the trial. Other investigators disclosed ties to Novartis. Dr. Abuin reported relationships with many companies, including Novartis.
This article was updated 9/24/21.
“Based on these results, ribociclib and letrozole should be considered the preferred treatment option,” said lead investigator Gabriel N. Hortobagyi, MD, a breast cancer medical oncologist at the University of Texas MD Anderson Cancer Center, Houston.
He presented the definitive overall survival results from MONALESSA-2 which randomized 668 patients equally and in the first line to either ribociclib or placebo on a background of standard dose letrozole.
At a median follow up of 6.6 years, median overall survival with ribociclib was 63.9 months versus 51.4 months in the placebo arm, a 24% reduction in the relative risk of death (P = .004).
It was the first report of a median overall survival (OS) exceeding 5 years in a phase 3 trial for advanced breast cancer. The estimated 6-year OS rate was 44.2% for ribociclib versus 32.0% with placebo.
“These are really impressive results” and support the use of CDK 4/6 inhibitors in the front-line setting,” said study discussant Gonzalo Gomez Abuin, MD, a medical oncologist at Hospital Alemán in Bueno Aires.
Ribociclib and other CDK 4/6 inhibitors have shown consistent progression-free survival benefit for metastatic disease, but ribociclib is the first of the major phase 3 trials with definitive overall survival results. They have “been long awaited,” Dr. Abuin said.
The overall survival benefit in MONALESSA-2 began to emerge at around 20 months and continued to increase over time.
Women had no prior CDK4/6 inhibitor treatment, chemotherapy, or endocrine therapy for metastatic disease. “They represented a pure first-line population,” Dr. Hortobagyi said.
Among other benefits, the time to first chemotherapy was a median of 50.6 months with ribociclib versus 38.9 months with placebo, so patients “had an extra year of delay before chemotherapy was utilized,” he said.
In general, Dr. Abuin said, we “see a consistent benefit with CDK 4/6 inhibitors in metastatic breast cancer across different settings.”
However, “it’s a little intriguing” that in a subgroup analysis of non–de novo disease, the overall survival benefit with ribociclib had a hazard ratio of 0.91, whereas the progression-free survival benefit was robust and statistically significant in an earlier report.
“This has been an important question, but I would caution all of us not to make too much out of the forest plot,” Dr. Hortobagyi said.
“There are a number of hypotheses one could come up with that could explain why the de novo and non–de novo populations faired differently in overall survival as opposed to progression-free survival, but there is also the simple possibility that this is a statistical fluke,” he said.
“We are in the process of analyzing this particular observation. In the meantime, I think we should just take the overall survival results of the entire population as the lead answer, and not follow the subgroup analysis until further information is available,” Dr. Hortobagyi said.
No new ribociclib safety signals were observed in the trial. The most common adverse events were neutropenia and liver function abnormalities, but they were “largely asymptomatic laboratory findings and completely reversible,” he said.
Twice as many patients treated with ribociclib developed prolonged QT intervals, but again, “no clinical consequences of this EKG finding were detected,” Dr. Hortobagyi said.
Less than 1% of patients in the ribociclib arm developed interstitial lung disease. The majority of safety events occurred in the first 12 months of treatment.
The work was funded by Novartis, maker of both ribociclib and letrozole. Dr. Hortobagyi reported receiving an institutional grant from the company and personal fees related to the trial. Other investigators disclosed ties to Novartis. Dr. Abuin reported relationships with many companies, including Novartis.
This article was updated 9/24/21.
FROM ESMO 2021
Sublingual film well tolerated for Parkinson ‘off’ episodes
new research shows.
“The bottom line was that the majority of patients did not have dose-limiting nausea or vomiting,” said coinvestigator William Ondo, MD, from Houston Methodist Neurological Institute. “And although it really did not compare in a prospective, placebo-controlled manner use of [trimethobenzamide antiemetic] ... versus not using [it], anecdotally and based on historic data, nausea really seemed to be about the same even without the antinausea medication.”
The findings were presented at the International Congress of Parkinson’s Disease and Movement Disorders.
This study was the dose-titration phase to determine the effective and tolerable dose of the drug as part of a longer study looking at safety and efficacy.
Only 13% of patients experienced nausea and/or vomiting, and of those, 74% cases were of mild severity and 26% were of moderate severity. These rates of nausea/vomiting were lower than those seen when trimethobenzamide (Tigan, Pfizer) was needed to be administered during the titration period, at the discretion of the investigator.
This multicenter, ongoing, open-label, phase 3 study enrolled 176 patients (mean age, 64.4 years) who had idiopathic Parkinson’s disease for a mean of 8.0 years and had no prior exposure to SL-apo, with modified Hoehn and Yahr stage 1-3 disease (83% stage 2 or 2.5 during “on” time).
Study participants had Mini-Mental State Examination scores greater than 25, were receiving stable doses of levodopa/carbidopa, and had 1 or more (mean, 4.2) “off” episodes per day with a total daily “off” time of 2 hours or more. Patients with mouth cankers or sores within 30 days of screening were excluded.
Open-label dose titration occurred during sequential office visits while patients were “off,” with escalating doses of 10-35 mg in 5-mg increments to determine a tolerable dose leading to a full “on” period within 45 minutes. Patients self-administered this achieved dose of SL-apo for up to five “off” episodes per day with a minimum of 2 hours between doses for the full 48-week study period.
The study protocol prohibited antiemetic use except when clinically warranted at the investigator’s discretion. Of the 176 patients, 31 (18%) received the antiemetic trimethobenzamide and 145 (82%) did not.
Of the 176 patients, 76% received their effective and tolerated dose within the first three doses. Just over half (55%) received 10 mg or 15 mg. Only 24% received the highest doses of 25 mg or 30 mg.
About 52%of patients who received trimethobenzamide experienced treatment-related nausea and 13% experienced vomiting; in comparison, 13% not receiving trimethobenzamide had nausea and 1% had vomiting. About 10%of patients in the former group and none in the latter discontinued the study because of nausea and/or vomiting.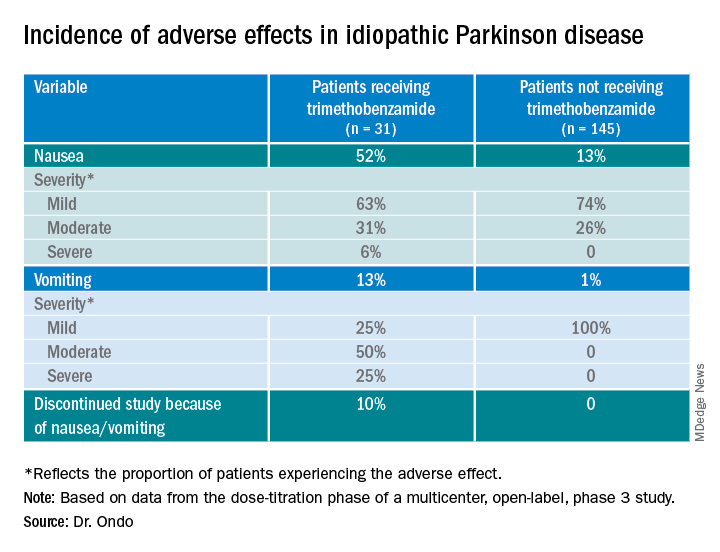
The apomorphine sublingual film has “the advantage of ease of use compared to the injectable form,” Dr. Ondo said. “I think the injectable form, purely based on anecdotal experience, might start to work a minute or 2 faster than the sublingual form, but overall I would say efficacy as far as potency of turning ‘on’ and consistency of turning ‘on’ is comparable.”
In addition to the known adverse effects of nausea, vomiting, and hypotension with the use of any apomorphine, he said that long-term use of the sublingual form can lead to gingival irritation. Two recommendations are to place the film in a different site and to use a more basic toothpaste, such as one containing baking powder, because irritation may result from the acidity of the apomorphine.
Good news
Commenting on the study, Ludy Shih, MD, MMSc, from Boston University, noted that the drug label reports that “13%-15% had oropharyngeal soft tissue swelling or pain ... and 7% had oral ulcers and stomatitis.”
In addition, oral trimethobenzamide has been discontinued, although an injectable form is still available. This situation may present a problem, she said. “Most antinausea drugs block dopamine, so ... I would say they’re contraindicated for treating people with Parkinson’s disease. But trimethobenzamide in particular is one that we often reach for. ... But that appears to be constrained and may, in fact, be expensive for patients.”
Turning to the study findings, she said they suggest that “not everyone needs prophylactic use of trimethobenzamide before they take the apomorphine sublingual film, which is good news that helps doctors try to decide whether or not it’s reasonable to recommend people trying it without the trimethobenzamide.”
Although some patients did experience mild nausea, she said the fact that no needle is involved may attract some patients. Moreover, taking this medication may be easier than administering an injection during an “off” episode.
Dr. Ondo is a consultant for Sunovion Pharmaceuticals, which sponsored the study. Dr. Shih had no relevant disclosures.
A version of this article first appeared on Medscape.com.
new research shows.
“The bottom line was that the majority of patients did not have dose-limiting nausea or vomiting,” said coinvestigator William Ondo, MD, from Houston Methodist Neurological Institute. “And although it really did not compare in a prospective, placebo-controlled manner use of [trimethobenzamide antiemetic] ... versus not using [it], anecdotally and based on historic data, nausea really seemed to be about the same even without the antinausea medication.”
The findings were presented at the International Congress of Parkinson’s Disease and Movement Disorders.
This study was the dose-titration phase to determine the effective and tolerable dose of the drug as part of a longer study looking at safety and efficacy.
Only 13% of patients experienced nausea and/or vomiting, and of those, 74% cases were of mild severity and 26% were of moderate severity. These rates of nausea/vomiting were lower than those seen when trimethobenzamide (Tigan, Pfizer) was needed to be administered during the titration period, at the discretion of the investigator.
This multicenter, ongoing, open-label, phase 3 study enrolled 176 patients (mean age, 64.4 years) who had idiopathic Parkinson’s disease for a mean of 8.0 years and had no prior exposure to SL-apo, with modified Hoehn and Yahr stage 1-3 disease (83% stage 2 or 2.5 during “on” time).
Study participants had Mini-Mental State Examination scores greater than 25, were receiving stable doses of levodopa/carbidopa, and had 1 or more (mean, 4.2) “off” episodes per day with a total daily “off” time of 2 hours or more. Patients with mouth cankers or sores within 30 days of screening were excluded.
Open-label dose titration occurred during sequential office visits while patients were “off,” with escalating doses of 10-35 mg in 5-mg increments to determine a tolerable dose leading to a full “on” period within 45 minutes. Patients self-administered this achieved dose of SL-apo for up to five “off” episodes per day with a minimum of 2 hours between doses for the full 48-week study period.
The study protocol prohibited antiemetic use except when clinically warranted at the investigator’s discretion. Of the 176 patients, 31 (18%) received the antiemetic trimethobenzamide and 145 (82%) did not.
Of the 176 patients, 76% received their effective and tolerated dose within the first three doses. Just over half (55%) received 10 mg or 15 mg. Only 24% received the highest doses of 25 mg or 30 mg.
About 52%of patients who received trimethobenzamide experienced treatment-related nausea and 13% experienced vomiting; in comparison, 13% not receiving trimethobenzamide had nausea and 1% had vomiting. About 10%of patients in the former group and none in the latter discontinued the study because of nausea and/or vomiting.
The apomorphine sublingual film has “the advantage of ease of use compared to the injectable form,” Dr. Ondo said. “I think the injectable form, purely based on anecdotal experience, might start to work a minute or 2 faster than the sublingual form, but overall I would say efficacy as far as potency of turning ‘on’ and consistency of turning ‘on’ is comparable.”
In addition to the known adverse effects of nausea, vomiting, and hypotension with the use of any apomorphine, he said that long-term use of the sublingual form can lead to gingival irritation. Two recommendations are to place the film in a different site and to use a more basic toothpaste, such as one containing baking powder, because irritation may result from the acidity of the apomorphine.
Good news
Commenting on the study, Ludy Shih, MD, MMSc, from Boston University, noted that the drug label reports that “13%-15% had oropharyngeal soft tissue swelling or pain ... and 7% had oral ulcers and stomatitis.”
In addition, oral trimethobenzamide has been discontinued, although an injectable form is still available. This situation may present a problem, she said. “Most antinausea drugs block dopamine, so ... I would say they’re contraindicated for treating people with Parkinson’s disease. But trimethobenzamide in particular is one that we often reach for. ... But that appears to be constrained and may, in fact, be expensive for patients.”
Turning to the study findings, she said they suggest that “not everyone needs prophylactic use of trimethobenzamide before they take the apomorphine sublingual film, which is good news that helps doctors try to decide whether or not it’s reasonable to recommend people trying it without the trimethobenzamide.”
Although some patients did experience mild nausea, she said the fact that no needle is involved may attract some patients. Moreover, taking this medication may be easier than administering an injection during an “off” episode.
Dr. Ondo is a consultant for Sunovion Pharmaceuticals, which sponsored the study. Dr. Shih had no relevant disclosures.
A version of this article first appeared on Medscape.com.
new research shows.
“The bottom line was that the majority of patients did not have dose-limiting nausea or vomiting,” said coinvestigator William Ondo, MD, from Houston Methodist Neurological Institute. “And although it really did not compare in a prospective, placebo-controlled manner use of [trimethobenzamide antiemetic] ... versus not using [it], anecdotally and based on historic data, nausea really seemed to be about the same even without the antinausea medication.”
The findings were presented at the International Congress of Parkinson’s Disease and Movement Disorders.
This study was the dose-titration phase to determine the effective and tolerable dose of the drug as part of a longer study looking at safety and efficacy.
Only 13% of patients experienced nausea and/or vomiting, and of those, 74% cases were of mild severity and 26% were of moderate severity. These rates of nausea/vomiting were lower than those seen when trimethobenzamide (Tigan, Pfizer) was needed to be administered during the titration period, at the discretion of the investigator.
This multicenter, ongoing, open-label, phase 3 study enrolled 176 patients (mean age, 64.4 years) who had idiopathic Parkinson’s disease for a mean of 8.0 years and had no prior exposure to SL-apo, with modified Hoehn and Yahr stage 1-3 disease (83% stage 2 or 2.5 during “on” time).
Study participants had Mini-Mental State Examination scores greater than 25, were receiving stable doses of levodopa/carbidopa, and had 1 or more (mean, 4.2) “off” episodes per day with a total daily “off” time of 2 hours or more. Patients with mouth cankers or sores within 30 days of screening were excluded.
Open-label dose titration occurred during sequential office visits while patients were “off,” with escalating doses of 10-35 mg in 5-mg increments to determine a tolerable dose leading to a full “on” period within 45 minutes. Patients self-administered this achieved dose of SL-apo for up to five “off” episodes per day with a minimum of 2 hours between doses for the full 48-week study period.
The study protocol prohibited antiemetic use except when clinically warranted at the investigator’s discretion. Of the 176 patients, 31 (18%) received the antiemetic trimethobenzamide and 145 (82%) did not.
Of the 176 patients, 76% received their effective and tolerated dose within the first three doses. Just over half (55%) received 10 mg or 15 mg. Only 24% received the highest doses of 25 mg or 30 mg.
About 52%of patients who received trimethobenzamide experienced treatment-related nausea and 13% experienced vomiting; in comparison, 13% not receiving trimethobenzamide had nausea and 1% had vomiting. About 10%of patients in the former group and none in the latter discontinued the study because of nausea and/or vomiting.
The apomorphine sublingual film has “the advantage of ease of use compared to the injectable form,” Dr. Ondo said. “I think the injectable form, purely based on anecdotal experience, might start to work a minute or 2 faster than the sublingual form, but overall I would say efficacy as far as potency of turning ‘on’ and consistency of turning ‘on’ is comparable.”
In addition to the known adverse effects of nausea, vomiting, and hypotension with the use of any apomorphine, he said that long-term use of the sublingual form can lead to gingival irritation. Two recommendations are to place the film in a different site and to use a more basic toothpaste, such as one containing baking powder, because irritation may result from the acidity of the apomorphine.
Good news
Commenting on the study, Ludy Shih, MD, MMSc, from Boston University, noted that the drug label reports that “13%-15% had oropharyngeal soft tissue swelling or pain ... and 7% had oral ulcers and stomatitis.”
In addition, oral trimethobenzamide has been discontinued, although an injectable form is still available. This situation may present a problem, she said. “Most antinausea drugs block dopamine, so ... I would say they’re contraindicated for treating people with Parkinson’s disease. But trimethobenzamide in particular is one that we often reach for. ... But that appears to be constrained and may, in fact, be expensive for patients.”
Turning to the study findings, she said they suggest that “not everyone needs prophylactic use of trimethobenzamide before they take the apomorphine sublingual film, which is good news that helps doctors try to decide whether or not it’s reasonable to recommend people trying it without the trimethobenzamide.”
Although some patients did experience mild nausea, she said the fact that no needle is involved may attract some patients. Moreover, taking this medication may be easier than administering an injection during an “off” episode.
Dr. Ondo is a consultant for Sunovion Pharmaceuticals, which sponsored the study. Dr. Shih had no relevant disclosures.
A version of this article first appeared on Medscape.com.
FROM MDS VIRTUAL CONGRESS 2021
Friedreich’s ataxia treatment shows extended benefit
according to results of a clinical trial presented as a late-breaking abstract at the International Congress of Parkinson’s Disease and Movement Disorders.

The study, labeled the Delayed-Start Study, is an extension study of the two-part MOXIE phase 2 trial of omaveloxolone.
“This study shows two things,” said David Lynch, MD, PhD, of Children’s Hospital of Philadelphia. “It doesn’t matter when you started omaveloxolone for you to see a benefit; and that the benefit that the active group saw in the first part of the study was maintained as they went into the delayed-start part. So in fact omaveloxolone does modify the long-term behavior of the disease.”
Friedreich’s ataxia only affects about 22,000 people worldwide, and children typically present between the ages of 5 and 15, Dr. Lynch said.
The extension study included 73 patients who completed either of the first two parts of the MOXIe trial. The MOXIe trial randomized patients on a 3:1 basis to either omaveloxolone 2.5-300 mg or placebo for 12 weeks in the first part. The second part was a double-blind trial of 103 patients randomized on a 1:1 basis to 150 mg omaveloxolone or placebo for 48 weeks. Participants had a baseline modified Friedreich’s ataxia scale (mFARS) of 20-80 and were aged 16-40 years.
Patients in the extension study did not have severe pes cavus. The extension study was a 72-week evaluation of patients who were in either the treatment or placebo groups in the first two parts. There was a 4-week off-treatment period between the end of MOXIe part 2 and the beginning of the extension study, in which all patients received omaveloxolone.
At the end of the placebo-controlled study, patients taking omaveloxolone showed a –2.18-point (±0.96) difference in improvement in mFARS score (P = .027), compared with the placebo group, which was preserved at the end of the delayed-start period, with a –2.92-point (±2.13) improvement (P = .179), Dr. Lynch said.
In the extension study, former placebo patients who went on omaveloxolone had annualized mFARS slopes similar to the previously treated patients – 0.29 (±0.68) and 0.17 (±0.61), respectively (P = .85) – from weeks 48 to 144, Dr. Lynch said.
“This study showed that, when analyzed in a delayed-start fashion, it does not matter when you start omaveloxolone to see a benefit: Each cohort benefited almost equally once they started the drug,” Dr. Lynch said in an interview. “Also, in both groups, once they started omaveloxolone, they changed slower than people in natural history studies.”
A clinically meaningful difference?
Reached for comment, Massimo Pandolfo, MD, a neurologist at McGill University, Montreal, noted that the Delayed-Start Study included only patients without pes cavus, an indication that the patients had less severe disease. “It would be important to see how overall patients with Friedreich’s ataxia would have responded to the medication without this kind of selection,” Dr. Pandolfo said in an interview.
He also noted that the seemingly modest improvement in mFARS score could be an issue. “It’s a very difficult question: What is a clinically meaningful difference in this kind of rating scale? I would argue that probably 2 points is not a huge difference by itself, but it may be meaningful and one indicator of that is that if it was accompanied by also a significant difference in activities of daily living scale.”
In any event, Dr. Pandolfo said this is the first medication for Friedreich’s ataxia that has “survived” a randomized clinical trial.
Dr. Lynch said the study sponsor, Reata, may prepare a new drug application for omaveloxolone in patients ages 16 and older. “That would leave a need for investigation in younger FA patients.”
Dr. Lynch disclosed that his institution receives a grant from trial sponsor Reata to conduct the MOXIe trial. Dr. Pandolfo reports financial relationships with Design Therapeutics, Exicure and Voyager Therapeutics.
according to results of a clinical trial presented as a late-breaking abstract at the International Congress of Parkinson’s Disease and Movement Disorders.
The study, labeled the Delayed-Start Study, is an extension study of the two-part MOXIE phase 2 trial of omaveloxolone.
“This study shows two things,” said David Lynch, MD, PhD, of Children’s Hospital of Philadelphia. “It doesn’t matter when you started omaveloxolone for you to see a benefit; and that the benefit that the active group saw in the first part of the study was maintained as they went into the delayed-start part. So in fact omaveloxolone does modify the long-term behavior of the disease.”
Friedreich’s ataxia only affects about 22,000 people worldwide, and children typically present between the ages of 5 and 15, Dr. Lynch said.
The extension study included 73 patients who completed either of the first two parts of the MOXIe trial. The MOXIe trial randomized patients on a 3:1 basis to either omaveloxolone 2.5-300 mg or placebo for 12 weeks in the first part. The second part was a double-blind trial of 103 patients randomized on a 1:1 basis to 150 mg omaveloxolone or placebo for 48 weeks. Participants had a baseline modified Friedreich’s ataxia scale (mFARS) of 20-80 and were aged 16-40 years.
Patients in the extension study did not have severe pes cavus. The extension study was a 72-week evaluation of patients who were in either the treatment or placebo groups in the first two parts. There was a 4-week off-treatment period between the end of MOXIe part 2 and the beginning of the extension study, in which all patients received omaveloxolone.
At the end of the placebo-controlled study, patients taking omaveloxolone showed a –2.18-point (±0.96) difference in improvement in mFARS score (P = .027), compared with the placebo group, which was preserved at the end of the delayed-start period, with a –2.92-point (±2.13) improvement (P = .179), Dr. Lynch said.
In the extension study, former placebo patients who went on omaveloxolone had annualized mFARS slopes similar to the previously treated patients – 0.29 (±0.68) and 0.17 (±0.61), respectively (P = .85) – from weeks 48 to 144, Dr. Lynch said.
“This study showed that, when analyzed in a delayed-start fashion, it does not matter when you start omaveloxolone to see a benefit: Each cohort benefited almost equally once they started the drug,” Dr. Lynch said in an interview. “Also, in both groups, once they started omaveloxolone, they changed slower than people in natural history studies.”
A clinically meaningful difference?
Reached for comment, Massimo Pandolfo, MD, a neurologist at McGill University, Montreal, noted that the Delayed-Start Study included only patients without pes cavus, an indication that the patients had less severe disease. “It would be important to see how overall patients with Friedreich’s ataxia would have responded to the medication without this kind of selection,” Dr. Pandolfo said in an interview.
He also noted that the seemingly modest improvement in mFARS score could be an issue. “It’s a very difficult question: What is a clinically meaningful difference in this kind of rating scale? I would argue that probably 2 points is not a huge difference by itself, but it may be meaningful and one indicator of that is that if it was accompanied by also a significant difference in activities of daily living scale.”
In any event, Dr. Pandolfo said this is the first medication for Friedreich’s ataxia that has “survived” a randomized clinical trial.
Dr. Lynch said the study sponsor, Reata, may prepare a new drug application for omaveloxolone in patients ages 16 and older. “That would leave a need for investigation in younger FA patients.”
Dr. Lynch disclosed that his institution receives a grant from trial sponsor Reata to conduct the MOXIe trial. Dr. Pandolfo reports financial relationships with Design Therapeutics, Exicure and Voyager Therapeutics.
according to results of a clinical trial presented as a late-breaking abstract at the International Congress of Parkinson’s Disease and Movement Disorders.
The study, labeled the Delayed-Start Study, is an extension study of the two-part MOXIE phase 2 trial of omaveloxolone.
“This study shows two things,” said David Lynch, MD, PhD, of Children’s Hospital of Philadelphia. “It doesn’t matter when you started omaveloxolone for you to see a benefit; and that the benefit that the active group saw in the first part of the study was maintained as they went into the delayed-start part. So in fact omaveloxolone does modify the long-term behavior of the disease.”
Friedreich’s ataxia only affects about 22,000 people worldwide, and children typically present between the ages of 5 and 15, Dr. Lynch said.
The extension study included 73 patients who completed either of the first two parts of the MOXIe trial. The MOXIe trial randomized patients on a 3:1 basis to either omaveloxolone 2.5-300 mg or placebo for 12 weeks in the first part. The second part was a double-blind trial of 103 patients randomized on a 1:1 basis to 150 mg omaveloxolone or placebo for 48 weeks. Participants had a baseline modified Friedreich’s ataxia scale (mFARS) of 20-80 and were aged 16-40 years.
Patients in the extension study did not have severe pes cavus. The extension study was a 72-week evaluation of patients who were in either the treatment or placebo groups in the first two parts. There was a 4-week off-treatment period between the end of MOXIe part 2 and the beginning of the extension study, in which all patients received omaveloxolone.
At the end of the placebo-controlled study, patients taking omaveloxolone showed a –2.18-point (±0.96) difference in improvement in mFARS score (P = .027), compared with the placebo group, which was preserved at the end of the delayed-start period, with a –2.92-point (±2.13) improvement (P = .179), Dr. Lynch said.
In the extension study, former placebo patients who went on omaveloxolone had annualized mFARS slopes similar to the previously treated patients – 0.29 (±0.68) and 0.17 (±0.61), respectively (P = .85) – from weeks 48 to 144, Dr. Lynch said.
“This study showed that, when analyzed in a delayed-start fashion, it does not matter when you start omaveloxolone to see a benefit: Each cohort benefited almost equally once they started the drug,” Dr. Lynch said in an interview. “Also, in both groups, once they started omaveloxolone, they changed slower than people in natural history studies.”
A clinically meaningful difference?
Reached for comment, Massimo Pandolfo, MD, a neurologist at McGill University, Montreal, noted that the Delayed-Start Study included only patients without pes cavus, an indication that the patients had less severe disease. “It would be important to see how overall patients with Friedreich’s ataxia would have responded to the medication without this kind of selection,” Dr. Pandolfo said in an interview.
He also noted that the seemingly modest improvement in mFARS score could be an issue. “It’s a very difficult question: What is a clinically meaningful difference in this kind of rating scale? I would argue that probably 2 points is not a huge difference by itself, but it may be meaningful and one indicator of that is that if it was accompanied by also a significant difference in activities of daily living scale.”
In any event, Dr. Pandolfo said this is the first medication for Friedreich’s ataxia that has “survived” a randomized clinical trial.
Dr. Lynch said the study sponsor, Reata, may prepare a new drug application for omaveloxolone in patients ages 16 and older. “That would leave a need for investigation in younger FA patients.”
Dr. Lynch disclosed that his institution receives a grant from trial sponsor Reata to conduct the MOXIe trial. Dr. Pandolfo reports financial relationships with Design Therapeutics, Exicure and Voyager Therapeutics.
FROM MDS VIRTUAL CONGRESS 2021
Survey identifies clinicians’ unease with genetic testing
Before getting to work on developing guidelines for genetic testing in Parkinson’s disease, a task force of the Movement Disorders Society surveyed members worldwide to identify concerns they have about using genetic testing in practice. In results presented as a late-breaking abstract at the International Congress of Parkinson’s Disease and Movement Disorders,
“Some of the major outstanding issues are the clinical actionability of genetic testing – and this was highlighted by some survey participants,” senior study author Rachel Saunders-Pullman, MD, MPH, professor of neurology at the Icahn School of Medicine at Mount Sinai, New York, said in an interview. The issue is “dynamic,” and will change even more radically when genetic therapies for Parkinson’s disease become available. “It is planned that, in the development of the MDS Task Force guidelines, scenarios which outline the changes in consideration of testing will depend on the availability of clinically actionable data,” she said.
Barriers to genetic testing
The MDS Task Force for Genetic Testing in Parkinson Disease conducted the survey, completed online by 568 MDS members. Respondents were from the four regions from which the MDS draws members: Africa, Europe, Asia/Oceania, and Pan-America. Half of the respondents considered themselves movement disorder specialists and 31% as general neurologists, said Maggie Markgraf, research coordinator at Mount Sinai Beth Israel in New York, who presented the survey findings.
Barriers to genetic testing that the clinicians cited included cost (57%), lack of availability of genetic counseling (37%), time for testing (20%) or time for counseling (17%). About 14%also cited a lack of knowledge, and only 8.5 % said they saw no barriers for genetic testing. Other concerns included a lack of therapeutic options if tests are positive and low overall positivity rates.
“Perceived barriers for general neurologists differed slightly, with limited knowledge being the most widely reported barrier, followed closely by cost and access to testing and genetic counseling,” Ms. Markgraf said.
Respondents were also asked to identify what they thought their patients perceived as barriers to genetic testing. The major one was cost (65%), followed by limited knowledge about genetics (43%), lack of access to genetic counseling (34%), and lack of access to testing separate from cost (30%). “Across all MDS regions, the perceived level of a patient’s knowledge about genetic testing is considered to be exceedingly low,” Ms. Markgraf said.
Europe had the highest availability to genetic tests, with 41.8% saying they’re accessible to general neurologists, followed by Asia/Oceania (31%) and Pan-America (30%).
“The area of most unmet need when it comes to PD genetic testing was cost for each MDS region, although the intertwined issue of access was also high, and over 50% reported that knowledge was an unmet need in their region,” Dr. Saunders-Pullman said.
Insurance coverage was another issue the survey respondents identified. In Europe, 53.6% said insurance or government programs cover genetic testing for PD, while only 14% in Pan-America and 10.3% in Asia/Oceania (and 0% in Africa) said such coverage was available.
“While there are limitations to this study, greater awareness of availability and barriers to genetic testing and counseling across different regions, as well as disparities among regions, will help inform development of the MDS Task Force guidelines,” Dr. Saunders-Pullman said.
Unmet needs
Connie Marras, MD, PhD, a professor of neurology at the University of Toronto, noted the survey suggested neurologists exhibit a “lack of comfort or lack of time” with genetic testing and counseling for Parkinson’s disease. “Even if we make genetic testing more widely available, we need health care providers that are comfortable and available to counsel patients before and after the testing, and clearly these are unmet needs,” Dr. Marras said in an interview.
“To date, pharmacologic treatment of Parkinson’s disease did not depend on genetics,” Dr. Marras said. “This may well change in the near future with treatments specifically targeting mechanisms related to two of the most common genetic risk factors for PD: LRRK2 and GBA gene variants being in clinical trials.” These developments may soon raise the urgency to reduce barriers to genetic testing.
Dr. Saunders-Pullman and Dr. Marras have no relevant relationships to disclose.
Before getting to work on developing guidelines for genetic testing in Parkinson’s disease, a task force of the Movement Disorders Society surveyed members worldwide to identify concerns they have about using genetic testing in practice. In results presented as a late-breaking abstract at the International Congress of Parkinson’s Disease and Movement Disorders,
“Some of the major outstanding issues are the clinical actionability of genetic testing – and this was highlighted by some survey participants,” senior study author Rachel Saunders-Pullman, MD, MPH, professor of neurology at the Icahn School of Medicine at Mount Sinai, New York, said in an interview. The issue is “dynamic,” and will change even more radically when genetic therapies for Parkinson’s disease become available. “It is planned that, in the development of the MDS Task Force guidelines, scenarios which outline the changes in consideration of testing will depend on the availability of clinically actionable data,” she said.
Barriers to genetic testing
The MDS Task Force for Genetic Testing in Parkinson Disease conducted the survey, completed online by 568 MDS members. Respondents were from the four regions from which the MDS draws members: Africa, Europe, Asia/Oceania, and Pan-America. Half of the respondents considered themselves movement disorder specialists and 31% as general neurologists, said Maggie Markgraf, research coordinator at Mount Sinai Beth Israel in New York, who presented the survey findings.
Barriers to genetic testing that the clinicians cited included cost (57%), lack of availability of genetic counseling (37%), time for testing (20%) or time for counseling (17%). About 14%also cited a lack of knowledge, and only 8.5 % said they saw no barriers for genetic testing. Other concerns included a lack of therapeutic options if tests are positive and low overall positivity rates.
“Perceived barriers for general neurologists differed slightly, with limited knowledge being the most widely reported barrier, followed closely by cost and access to testing and genetic counseling,” Ms. Markgraf said.
Respondents were also asked to identify what they thought their patients perceived as barriers to genetic testing. The major one was cost (65%), followed by limited knowledge about genetics (43%), lack of access to genetic counseling (34%), and lack of access to testing separate from cost (30%). “Across all MDS regions, the perceived level of a patient’s knowledge about genetic testing is considered to be exceedingly low,” Ms. Markgraf said.
Europe had the highest availability to genetic tests, with 41.8% saying they’re accessible to general neurologists, followed by Asia/Oceania (31%) and Pan-America (30%).
“The area of most unmet need when it comes to PD genetic testing was cost for each MDS region, although the intertwined issue of access was also high, and over 50% reported that knowledge was an unmet need in their region,” Dr. Saunders-Pullman said.
Insurance coverage was another issue the survey respondents identified. In Europe, 53.6% said insurance or government programs cover genetic testing for PD, while only 14% in Pan-America and 10.3% in Asia/Oceania (and 0% in Africa) said such coverage was available.
“While there are limitations to this study, greater awareness of availability and barriers to genetic testing and counseling across different regions, as well as disparities among regions, will help inform development of the MDS Task Force guidelines,” Dr. Saunders-Pullman said.
Unmet needs
Connie Marras, MD, PhD, a professor of neurology at the University of Toronto, noted the survey suggested neurologists exhibit a “lack of comfort or lack of time” with genetic testing and counseling for Parkinson’s disease. “Even if we make genetic testing more widely available, we need health care providers that are comfortable and available to counsel patients before and after the testing, and clearly these are unmet needs,” Dr. Marras said in an interview.
“To date, pharmacologic treatment of Parkinson’s disease did not depend on genetics,” Dr. Marras said. “This may well change in the near future with treatments specifically targeting mechanisms related to two of the most common genetic risk factors for PD: LRRK2 and GBA gene variants being in clinical trials.” These developments may soon raise the urgency to reduce barriers to genetic testing.
Dr. Saunders-Pullman and Dr. Marras have no relevant relationships to disclose.
Before getting to work on developing guidelines for genetic testing in Parkinson’s disease, a task force of the Movement Disorders Society surveyed members worldwide to identify concerns they have about using genetic testing in practice. In results presented as a late-breaking abstract at the International Congress of Parkinson’s Disease and Movement Disorders,
“Some of the major outstanding issues are the clinical actionability of genetic testing – and this was highlighted by some survey participants,” senior study author Rachel Saunders-Pullman, MD, MPH, professor of neurology at the Icahn School of Medicine at Mount Sinai, New York, said in an interview. The issue is “dynamic,” and will change even more radically when genetic therapies for Parkinson’s disease become available. “It is planned that, in the development of the MDS Task Force guidelines, scenarios which outline the changes in consideration of testing will depend on the availability of clinically actionable data,” she said.
Barriers to genetic testing
The MDS Task Force for Genetic Testing in Parkinson Disease conducted the survey, completed online by 568 MDS members. Respondents were from the four regions from which the MDS draws members: Africa, Europe, Asia/Oceania, and Pan-America. Half of the respondents considered themselves movement disorder specialists and 31% as general neurologists, said Maggie Markgraf, research coordinator at Mount Sinai Beth Israel in New York, who presented the survey findings.
Barriers to genetic testing that the clinicians cited included cost (57%), lack of availability of genetic counseling (37%), time for testing (20%) or time for counseling (17%). About 14%also cited a lack of knowledge, and only 8.5 % said they saw no barriers for genetic testing. Other concerns included a lack of therapeutic options if tests are positive and low overall positivity rates.
“Perceived barriers for general neurologists differed slightly, with limited knowledge being the most widely reported barrier, followed closely by cost and access to testing and genetic counseling,” Ms. Markgraf said.
Respondents were also asked to identify what they thought their patients perceived as barriers to genetic testing. The major one was cost (65%), followed by limited knowledge about genetics (43%), lack of access to genetic counseling (34%), and lack of access to testing separate from cost (30%). “Across all MDS regions, the perceived level of a patient’s knowledge about genetic testing is considered to be exceedingly low,” Ms. Markgraf said.
Europe had the highest availability to genetic tests, with 41.8% saying they’re accessible to general neurologists, followed by Asia/Oceania (31%) and Pan-America (30%).
“The area of most unmet need when it comes to PD genetic testing was cost for each MDS region, although the intertwined issue of access was also high, and over 50% reported that knowledge was an unmet need in their region,” Dr. Saunders-Pullman said.
Insurance coverage was another issue the survey respondents identified. In Europe, 53.6% said insurance or government programs cover genetic testing for PD, while only 14% in Pan-America and 10.3% in Asia/Oceania (and 0% in Africa) said such coverage was available.
“While there are limitations to this study, greater awareness of availability and barriers to genetic testing and counseling across different regions, as well as disparities among regions, will help inform development of the MDS Task Force guidelines,” Dr. Saunders-Pullman said.
Unmet needs
Connie Marras, MD, PhD, a professor of neurology at the University of Toronto, noted the survey suggested neurologists exhibit a “lack of comfort or lack of time” with genetic testing and counseling for Parkinson’s disease. “Even if we make genetic testing more widely available, we need health care providers that are comfortable and available to counsel patients before and after the testing, and clearly these are unmet needs,” Dr. Marras said in an interview.
“To date, pharmacologic treatment of Parkinson’s disease did not depend on genetics,” Dr. Marras said. “This may well change in the near future with treatments specifically targeting mechanisms related to two of the most common genetic risk factors for PD: LRRK2 and GBA gene variants being in clinical trials.” These developments may soon raise the urgency to reduce barriers to genetic testing.
Dr. Saunders-Pullman and Dr. Marras have no relevant relationships to disclose.
FROM MDS VIRTUAL CONGRESS 2021
EMPEROR-Preserved: Empagliflozin’s HFpEF efficacy catalyzes a heart failure redefinition
Groundbreaking results from the EMPEROR-Preserved trial did more than establish for the first time that a drug, empagliflozin, has clearly proven efficacy for treating patients with heart failure with preserved ejection fraction (HFpEF). The results also helped catalyze a paradigm shift in how heart failure thought leaders think about the role of ejection fraction for making important distinctions among patients with heart failure.

EMPEROR-Preserved may also be the final nail in the coffin for defining patients with heart failure as having HFpEF or heart failure with reduced ejection fraction (HFrEF).
This new consensus essentially throws out left ventricular ejection fraction (EF) as the key metric for matching patients to heart failure treatments. Experts have instead begun suggesting a more unified treatment approach for all heart failure patients regardless of their EF.
‘Forget about ejection fraction’
“We encourage you to forget about ejection fraction,” declared Milton Packer, MD, during discussion at a session of the annual scientific meeting of the Heart Failure Society of America. “We certainly encourage you to forget about an ejection fraction of less than 40%” as having special significance,” added Dr. Packer, a lead investigator for both the EMPEROR-Reduced and EMPEROR-Preserved trials (which researchers combined in a unified analysis with a total of 9,718 patients with heart failure called EMPEROR-Pooled), and a heart failure researcher at Baylor University Medical Center in Dallas.
“The 40% ejection fraction divide is artificial. It was created in 2003 as part of a trial design, but it has no physiological significance,” Dr. Packer explained. A much better way to distinguish systolic and diastolic heart failure is by strain assessment rather than by ejection fraction. “Strain is a measure of myocardial shortening, a measure of what the heart does. Ejection fraction is a measure of volume,” said Dr. Packer. “Sign me up to get rid of ejection fraction,” he added.
“Ejection fraction is not as valuable as we thought for distinguishing the therapeutic benefit” of heart failure drugs, agreed Marvin A. Konstam, MD, professor of medicine at Tufts University and chief physician executive of the CardioVascular Center of Tufts Medical Center, both in Boston, who spoke during a different session at the meeting.

“It would easier if we didn’t spend time parsing this number,” ejection fraction, commented Clyde W. Yancy, MD, professor of medicine and chief of cardiology at Northwestern Medicine in Chicago. “Wouldn’t it be easier if we said that every patient with heart failure needs to receive one agent from each of the four [pillar] drug classes, and put them in a polypill” at reduced dosages, he proposed, envisioning one potential consequence of jettisoning ejection fraction.
The four pillar drug classes, recently identified as essential for patients with HFrEF but until now not endorsed for patients with HFpEF, are the sodium-glucose cotransporter 2 (SGLT2) inhibitors, such as empagliflozin (Jardiance); an angiotensin receptor blocker neprilysin inhibitor compound such as sacubitril/valsartan (Entresto); beta-blockers; and mineralocorticoid receptor antagonists such as spironolactone and eplerenone.
An opportunity for ‘simpler and easier’ treatments
“This is an opportunity to disrupt the way we’ve been doing things and think about something that is simpler and easier,” said Dr. Yancy, who chaired some of the panels serially formed by the American Heart Association and American College of Cardiology to write guidelines for treating heart failure. “An approach that would be easier to implement without worrying about staggering the start of each drug class and an incessant focus on titrating individual elements and taking 6 months to get to a certain place.”
Results from EMPEROR-Preserved and the combined EMPEROR-Pooled analysis triggered these paradigm-shifting sentiments by showing clear evidence that treatment with empagliflozin exerts consistent benefit – and is consistently safe – for patients with heart failure across a spectrum of EFs, from less than 25% to 64%, though its performance in patients with HFpEF and EFs of 65% or greater in the EMPEROR-Preserved trial remains unclear.
The consequence is that clinicians should feel comfortable prescribing empagliflozin to most patients with heart failure without regard to EF, even patients with EF values in the mid-60% range.
The EMPEROR-Preserved results showed a clear signal of attenuated benefit among patients with an EF of 65% or greater “on a population basis,” stressed Dr. Packer. “But on an individual basis, ejection fraction is not that reproducible, so measuring ejection fraction will not help you determine whom to treat or not treat. “

“There is significant variability” measuring EF using the most common modality, echocardiography, noted Javed Butler, MD, an EMPEROR coinvestigator who also spoke at the meeting session. A person with a measured EF of 65% could actually have a value that may be as low as 58% or as high as about 72%, noted Dr. Butler, who is professor and chair of medicine at the University of Mississippi, Jackson. The upshot is that any patient diagnosed with heart failure should receive an SGLT2 inhibitor “irrespective of their ejection fraction,” Dr. Butler advised.
“Ejection fraction is very crude, and probably not sufficient to identify a phenotype,” for treatment, said Dr. Yancy. “The real takeaway may be that we need to revisit what we call HFrEF, and then let that be the new standard for treatment.”
“Is [an EF of] 60% the new 40%?” asked Dr. Packer, implying that the answer was yes.
Results from several trials suggest redefining HFrEF
The idea that patients without traditionally defined HFrEF – an EF of 40% or less – could also benefit from other classes of heart failure drugs has been gestating for a while, and then rose to a new level with the August 2021 report of results from EMPEROR-Preserved. Two years ago, in September 2019, Dr. Butler, Dr. Packer, and a third colleague advanced the notion of redefining HFrEF by raising the ejection fraction ceiling in a published commentary.
They cited the experience with the angiotensin receptor blocker candesartan in a post hoc analysis of data collected in the CHARM-Preserved trial, which showed a strong signal of benefit in the subgroup of patients with EFs of 41%-49%, but not in those with an EF of 50% or higher. This finding prompted Dr. Konstam to express doubts about relying on EF to define heart failure subgroups in trials and guide management in a commentary published more than 3 years ago.
Another crack in the traditional EF framework came from analysis of results from the TOPCAT trial that tested spironolactone as a treatment for patients with HFpEF, according to the 2019 opinion published by Dr. Butler and Dr. Packer. Once again a post hoc analysis, this time using data from TOPCAT, suggested a benefit from the mineralocorticoid receptor antagonist spironolactone in patients with heart failure and an EF of 45%-49% (45% was the minimum EF for enrollment into the study).
Recently, data from a third trial that tested sacubitril/valsartan in patients with HFpEF, PARAGON-HF, showed benefit among patients with EFs below the study median of 57%. This finding led the Food and Drug Administration in February 2021 to amend its initial approval for sacubitril/valsartan by removing a specific EF ceiling from the drug’s indication and instead saying that patient’s receiving the drug should have a “below normal” EF.
Writing in a recent commentary, Dr. Yancy called the FDA’s action on sacubitril/valsartan “reasonable,” and that the subgroup assessment of data from the PARAGON-HF trial creates a “new, reasonably evidence-based therapy for HFpEF.” He also predicted that guideline-writing panels will “likely align with a permissive statement of indication” for sacubitril/valsartan in patients with HFpEF, especially those with EFs of less than 57%.
The idea of using an SGLT2 inhibitor like empagliflozin on all heart failure patients, and also adding agents like sacubitril/valsartan and spironolactone in patients with HFpEF and EFs in the mid-50% range or lower may take some time to catch on, but it already has one influential advocate.
“If a patient has HFpEF with an EF of less than 55%, use quadruple-class therapy,” summed up Dr. Butler during the HFSA session, while also suggesting prescribing an SGLT2 inhibitor to essentially all patients with heart failure regardless of their EF.
The EMPEROR-Preserved and EMPEROR-Reduced trials and the EMPEROR-Pooled analysis were sponsored by Boehringer Ingelheim and Lilly, the companies that jointly market empagliflozin (Jardiance). Dr. Packer has had financial relationships with BI and Lilly and numerous other companies. Dr. Konstam has served on data monitoring committees for trials funded by Boehringer Ingelheim and by Amgen, Luitpold, and Pfizer, and has been a consultant to Arena, LivaNova, Merck, SC Pharma, and Takeda. Dr. Yancy had no disclosures. Dr. Butler has had financial relationships with Boehringer Ingelheim and numerous other companies.
Groundbreaking results from the EMPEROR-Preserved trial did more than establish for the first time that a drug, empagliflozin, has clearly proven efficacy for treating patients with heart failure with preserved ejection fraction (HFpEF). The results also helped catalyze a paradigm shift in how heart failure thought leaders think about the role of ejection fraction for making important distinctions among patients with heart failure.
EMPEROR-Preserved may also be the final nail in the coffin for defining patients with heart failure as having HFpEF or heart failure with reduced ejection fraction (HFrEF).
This new consensus essentially throws out left ventricular ejection fraction (EF) as the key metric for matching patients to heart failure treatments. Experts have instead begun suggesting a more unified treatment approach for all heart failure patients regardless of their EF.
‘Forget about ejection fraction’
“We encourage you to forget about ejection fraction,” declared Milton Packer, MD, during discussion at a session of the annual scientific meeting of the Heart Failure Society of America. “We certainly encourage you to forget about an ejection fraction of less than 40%” as having special significance,” added Dr. Packer, a lead investigator for both the EMPEROR-Reduced and EMPEROR-Preserved trials (which researchers combined in a unified analysis with a total of 9,718 patients with heart failure called EMPEROR-Pooled), and a heart failure researcher at Baylor University Medical Center in Dallas.
“The 40% ejection fraction divide is artificial. It was created in 2003 as part of a trial design, but it has no physiological significance,” Dr. Packer explained. A much better way to distinguish systolic and diastolic heart failure is by strain assessment rather than by ejection fraction. “Strain is a measure of myocardial shortening, a measure of what the heart does. Ejection fraction is a measure of volume,” said Dr. Packer. “Sign me up to get rid of ejection fraction,” he added.
“Ejection fraction is not as valuable as we thought for distinguishing the therapeutic benefit” of heart failure drugs, agreed Marvin A. Konstam, MD, professor of medicine at Tufts University and chief physician executive of the CardioVascular Center of Tufts Medical Center, both in Boston, who spoke during a different session at the meeting.
“It would easier if we didn’t spend time parsing this number,” ejection fraction, commented Clyde W. Yancy, MD, professor of medicine and chief of cardiology at Northwestern Medicine in Chicago. “Wouldn’t it be easier if we said that every patient with heart failure needs to receive one agent from each of the four [pillar] drug classes, and put them in a polypill” at reduced dosages, he proposed, envisioning one potential consequence of jettisoning ejection fraction.
The four pillar drug classes, recently identified as essential for patients with HFrEF but until now not endorsed for patients with HFpEF, are the sodium-glucose cotransporter 2 (SGLT2) inhibitors, such as empagliflozin (Jardiance); an angiotensin receptor blocker neprilysin inhibitor compound such as sacubitril/valsartan (Entresto); beta-blockers; and mineralocorticoid receptor antagonists such as spironolactone and eplerenone.
An opportunity for ‘simpler and easier’ treatments
“This is an opportunity to disrupt the way we’ve been doing things and think about something that is simpler and easier,” said Dr. Yancy, who chaired some of the panels serially formed by the American Heart Association and American College of Cardiology to write guidelines for treating heart failure. “An approach that would be easier to implement without worrying about staggering the start of each drug class and an incessant focus on titrating individual elements and taking 6 months to get to a certain place.”
Results from EMPEROR-Preserved and the combined EMPEROR-Pooled analysis triggered these paradigm-shifting sentiments by showing clear evidence that treatment with empagliflozin exerts consistent benefit – and is consistently safe – for patients with heart failure across a spectrum of EFs, from less than 25% to 64%, though its performance in patients with HFpEF and EFs of 65% or greater in the EMPEROR-Preserved trial remains unclear.
The consequence is that clinicians should feel comfortable prescribing empagliflozin to most patients with heart failure without regard to EF, even patients with EF values in the mid-60% range.
The EMPEROR-Preserved results showed a clear signal of attenuated benefit among patients with an EF of 65% or greater “on a population basis,” stressed Dr. Packer. “But on an individual basis, ejection fraction is not that reproducible, so measuring ejection fraction will not help you determine whom to treat or not treat. “
“There is significant variability” measuring EF using the most common modality, echocardiography, noted Javed Butler, MD, an EMPEROR coinvestigator who also spoke at the meeting session. A person with a measured EF of 65% could actually have a value that may be as low as 58% or as high as about 72%, noted Dr. Butler, who is professor and chair of medicine at the University of Mississippi, Jackson. The upshot is that any patient diagnosed with heart failure should receive an SGLT2 inhibitor “irrespective of their ejection fraction,” Dr. Butler advised.
“Ejection fraction is very crude, and probably not sufficient to identify a phenotype,” for treatment, said Dr. Yancy. “The real takeaway may be that we need to revisit what we call HFrEF, and then let that be the new standard for treatment.”
“Is [an EF of] 60% the new 40%?” asked Dr. Packer, implying that the answer was yes.
Results from several trials suggest redefining HFrEF
The idea that patients without traditionally defined HFrEF – an EF of 40% or less – could also benefit from other classes of heart failure drugs has been gestating for a while, and then rose to a new level with the August 2021 report of results from EMPEROR-Preserved. Two years ago, in September 2019, Dr. Butler, Dr. Packer, and a third colleague advanced the notion of redefining HFrEF by raising the ejection fraction ceiling in a published commentary.
They cited the experience with the angiotensin receptor blocker candesartan in a post hoc analysis of data collected in the CHARM-Preserved trial, which showed a strong signal of benefit in the subgroup of patients with EFs of 41%-49%, but not in those with an EF of 50% or higher. This finding prompted Dr. Konstam to express doubts about relying on EF to define heart failure subgroups in trials and guide management in a commentary published more than 3 years ago.
Another crack in the traditional EF framework came from analysis of results from the TOPCAT trial that tested spironolactone as a treatment for patients with HFpEF, according to the 2019 opinion published by Dr. Butler and Dr. Packer. Once again a post hoc analysis, this time using data from TOPCAT, suggested a benefit from the mineralocorticoid receptor antagonist spironolactone in patients with heart failure and an EF of 45%-49% (45% was the minimum EF for enrollment into the study).
Recently, data from a third trial that tested sacubitril/valsartan in patients with HFpEF, PARAGON-HF, showed benefit among patients with EFs below the study median of 57%. This finding led the Food and Drug Administration in February 2021 to amend its initial approval for sacubitril/valsartan by removing a specific EF ceiling from the drug’s indication and instead saying that patient’s receiving the drug should have a “below normal” EF.
Writing in a recent commentary, Dr. Yancy called the FDA’s action on sacubitril/valsartan “reasonable,” and that the subgroup assessment of data from the PARAGON-HF trial creates a “new, reasonably evidence-based therapy for HFpEF.” He also predicted that guideline-writing panels will “likely align with a permissive statement of indication” for sacubitril/valsartan in patients with HFpEF, especially those with EFs of less than 57%.
The idea of using an SGLT2 inhibitor like empagliflozin on all heart failure patients, and also adding agents like sacubitril/valsartan and spironolactone in patients with HFpEF and EFs in the mid-50% range or lower may take some time to catch on, but it already has one influential advocate.
“If a patient has HFpEF with an EF of less than 55%, use quadruple-class therapy,” summed up Dr. Butler during the HFSA session, while also suggesting prescribing an SGLT2 inhibitor to essentially all patients with heart failure regardless of their EF.
The EMPEROR-Preserved and EMPEROR-Reduced trials and the EMPEROR-Pooled analysis were sponsored by Boehringer Ingelheim and Lilly, the companies that jointly market empagliflozin (Jardiance). Dr. Packer has had financial relationships with BI and Lilly and numerous other companies. Dr. Konstam has served on data monitoring committees for trials funded by Boehringer Ingelheim and by Amgen, Luitpold, and Pfizer, and has been a consultant to Arena, LivaNova, Merck, SC Pharma, and Takeda. Dr. Yancy had no disclosures. Dr. Butler has had financial relationships with Boehringer Ingelheim and numerous other companies.
Groundbreaking results from the EMPEROR-Preserved trial did more than establish for the first time that a drug, empagliflozin, has clearly proven efficacy for treating patients with heart failure with preserved ejection fraction (HFpEF). The results also helped catalyze a paradigm shift in how heart failure thought leaders think about the role of ejection fraction for making important distinctions among patients with heart failure.
EMPEROR-Preserved may also be the final nail in the coffin for defining patients with heart failure as having HFpEF or heart failure with reduced ejection fraction (HFrEF).
This new consensus essentially throws out left ventricular ejection fraction (EF) as the key metric for matching patients to heart failure treatments. Experts have instead begun suggesting a more unified treatment approach for all heart failure patients regardless of their EF.
‘Forget about ejection fraction’
“We encourage you to forget about ejection fraction,” declared Milton Packer, MD, during discussion at a session of the annual scientific meeting of the Heart Failure Society of America. “We certainly encourage you to forget about an ejection fraction of less than 40%” as having special significance,” added Dr. Packer, a lead investigator for both the EMPEROR-Reduced and EMPEROR-Preserved trials (which researchers combined in a unified analysis with a total of 9,718 patients with heart failure called EMPEROR-Pooled), and a heart failure researcher at Baylor University Medical Center in Dallas.
“The 40% ejection fraction divide is artificial. It was created in 2003 as part of a trial design, but it has no physiological significance,” Dr. Packer explained. A much better way to distinguish systolic and diastolic heart failure is by strain assessment rather than by ejection fraction. “Strain is a measure of myocardial shortening, a measure of what the heart does. Ejection fraction is a measure of volume,” said Dr. Packer. “Sign me up to get rid of ejection fraction,” he added.
“Ejection fraction is not as valuable as we thought for distinguishing the therapeutic benefit” of heart failure drugs, agreed Marvin A. Konstam, MD, professor of medicine at Tufts University and chief physician executive of the CardioVascular Center of Tufts Medical Center, both in Boston, who spoke during a different session at the meeting.
“It would easier if we didn’t spend time parsing this number,” ejection fraction, commented Clyde W. Yancy, MD, professor of medicine and chief of cardiology at Northwestern Medicine in Chicago. “Wouldn’t it be easier if we said that every patient with heart failure needs to receive one agent from each of the four [pillar] drug classes, and put them in a polypill” at reduced dosages, he proposed, envisioning one potential consequence of jettisoning ejection fraction.
The four pillar drug classes, recently identified as essential for patients with HFrEF but until now not endorsed for patients with HFpEF, are the sodium-glucose cotransporter 2 (SGLT2) inhibitors, such as empagliflozin (Jardiance); an angiotensin receptor blocker neprilysin inhibitor compound such as sacubitril/valsartan (Entresto); beta-blockers; and mineralocorticoid receptor antagonists such as spironolactone and eplerenone.
An opportunity for ‘simpler and easier’ treatments
“This is an opportunity to disrupt the way we’ve been doing things and think about something that is simpler and easier,” said Dr. Yancy, who chaired some of the panels serially formed by the American Heart Association and American College of Cardiology to write guidelines for treating heart failure. “An approach that would be easier to implement without worrying about staggering the start of each drug class and an incessant focus on titrating individual elements and taking 6 months to get to a certain place.”
Results from EMPEROR-Preserved and the combined EMPEROR-Pooled analysis triggered these paradigm-shifting sentiments by showing clear evidence that treatment with empagliflozin exerts consistent benefit – and is consistently safe – for patients with heart failure across a spectrum of EFs, from less than 25% to 64%, though its performance in patients with HFpEF and EFs of 65% or greater in the EMPEROR-Preserved trial remains unclear.
The consequence is that clinicians should feel comfortable prescribing empagliflozin to most patients with heart failure without regard to EF, even patients with EF values in the mid-60% range.
The EMPEROR-Preserved results showed a clear signal of attenuated benefit among patients with an EF of 65% or greater “on a population basis,” stressed Dr. Packer. “But on an individual basis, ejection fraction is not that reproducible, so measuring ejection fraction will not help you determine whom to treat or not treat. “
“There is significant variability” measuring EF using the most common modality, echocardiography, noted Javed Butler, MD, an EMPEROR coinvestigator who also spoke at the meeting session. A person with a measured EF of 65% could actually have a value that may be as low as 58% or as high as about 72%, noted Dr. Butler, who is professor and chair of medicine at the University of Mississippi, Jackson. The upshot is that any patient diagnosed with heart failure should receive an SGLT2 inhibitor “irrespective of their ejection fraction,” Dr. Butler advised.
“Ejection fraction is very crude, and probably not sufficient to identify a phenotype,” for treatment, said Dr. Yancy. “The real takeaway may be that we need to revisit what we call HFrEF, and then let that be the new standard for treatment.”
“Is [an EF of] 60% the new 40%?” asked Dr. Packer, implying that the answer was yes.
Results from several trials suggest redefining HFrEF
The idea that patients without traditionally defined HFrEF – an EF of 40% or less – could also benefit from other classes of heart failure drugs has been gestating for a while, and then rose to a new level with the August 2021 report of results from EMPEROR-Preserved. Two years ago, in September 2019, Dr. Butler, Dr. Packer, and a third colleague advanced the notion of redefining HFrEF by raising the ejection fraction ceiling in a published commentary.
They cited the experience with the angiotensin receptor blocker candesartan in a post hoc analysis of data collected in the CHARM-Preserved trial, which showed a strong signal of benefit in the subgroup of patients with EFs of 41%-49%, but not in those with an EF of 50% or higher. This finding prompted Dr. Konstam to express doubts about relying on EF to define heart failure subgroups in trials and guide management in a commentary published more than 3 years ago.
Another crack in the traditional EF framework came from analysis of results from the TOPCAT trial that tested spironolactone as a treatment for patients with HFpEF, according to the 2019 opinion published by Dr. Butler and Dr. Packer. Once again a post hoc analysis, this time using data from TOPCAT, suggested a benefit from the mineralocorticoid receptor antagonist spironolactone in patients with heart failure and an EF of 45%-49% (45% was the minimum EF for enrollment into the study).
Recently, data from a third trial that tested sacubitril/valsartan in patients with HFpEF, PARAGON-HF, showed benefit among patients with EFs below the study median of 57%. This finding led the Food and Drug Administration in February 2021 to amend its initial approval for sacubitril/valsartan by removing a specific EF ceiling from the drug’s indication and instead saying that patient’s receiving the drug should have a “below normal” EF.
Writing in a recent commentary, Dr. Yancy called the FDA’s action on sacubitril/valsartan “reasonable,” and that the subgroup assessment of data from the PARAGON-HF trial creates a “new, reasonably evidence-based therapy for HFpEF.” He also predicted that guideline-writing panels will “likely align with a permissive statement of indication” for sacubitril/valsartan in patients with HFpEF, especially those with EFs of less than 57%.
The idea of using an SGLT2 inhibitor like empagliflozin on all heart failure patients, and also adding agents like sacubitril/valsartan and spironolactone in patients with HFpEF and EFs in the mid-50% range or lower may take some time to catch on, but it already has one influential advocate.
“If a patient has HFpEF with an EF of less than 55%, use quadruple-class therapy,” summed up Dr. Butler during the HFSA session, while also suggesting prescribing an SGLT2 inhibitor to essentially all patients with heart failure regardless of their EF.
The EMPEROR-Preserved and EMPEROR-Reduced trials and the EMPEROR-Pooled analysis were sponsored by Boehringer Ingelheim and Lilly, the companies that jointly market empagliflozin (Jardiance). Dr. Packer has had financial relationships with BI and Lilly and numerous other companies. Dr. Konstam has served on data monitoring committees for trials funded by Boehringer Ingelheim and by Amgen, Luitpold, and Pfizer, and has been a consultant to Arena, LivaNova, Merck, SC Pharma, and Takeda. Dr. Yancy had no disclosures. Dr. Butler has had financial relationships with Boehringer Ingelheim and numerous other companies.
FROM HFSA 2021
Targeting IL-23 could still be important for axial spondyloarthritis treatment
Interleukin (IL)–23 inhibition may still have a role to play in the treatment of patients with axial spondyloarthritis (SpA), suggests research presented at the 12th International Congress on Spondyloarthritides.
There is a strong rationale for using IL-23 inhibitors in patients with axial SpA, and the IL-23/IL-17 axis has been proposed as a critical player in the pathophysiology of the disease. But around 2018 it became clear from randomized, controlled trials that IL-23 inhibition was ineffective at improving key clinical outcomes, at least in patients with axial disease.
Although the overall results of a systematic review and meta-analysis that was presented at the meeting corroborated the negative results seen with IL-23–inhibiting agents in clinical trials, there were some data showing benefits of the IL-23 inhibitor risankizumab on secondary outcomes in one trial.
To look at the available evidence, Louise Vanhoutte, a 2nd-year internal medicine student at University Hospitals Leuven (Belgium) worked under the guidance of Rik Lories, MD, PhD, head of the division of rheumatology at University Hospitals Leuven. Together they searched known databases for randomized, controlled trials investigating the use of IL-23 and IL-17 inhibitors for the treatment of adults with axial SpA or psoriatic arthritis. Studies could be either phase 2 or phase 3, but had to have included a placebo and used the ASAS40 (40% Improvement in Assessment of SpondyloArthritis International Society Response criteria), ASAS20, Bath Ankylosing Spondylitis Disease Activity Index, or SPARCC (Spondyloarthritis Research Consortium of Canada) index score to assess outcomes.
The systematic review whittled the number of clinical trials in the meta-analysis to 12, which concerned the use of ustekinumab, an IL-12/23 inhibitor, and risankizumab, along with two IL-17 inhibitors, ixekizumab and secukinumab. Data for the IL-23 inhibitors guselkumab and tildrakizumab were not available.
“To no surprise, Forest plots showed that there was a lack of efficacy for IL-23 agents in the treatment of axial spondyloarthritis and a superior efficacy for IL-17 inhibitors in the treatment of axial spondyloarthritis,” Ms. Vanhoutte reported.
The respective odds ratios for IL-23 and IL-17 inhibitors in getting patients to meet ASAS40 response criteria in comparison to baseline were 1.51 (95% confidence interval, 0.98-2.31) and 2.54 (95% CI, 2.02-3.19).
“Does this mean it is a dead-end street for all IL-23 inhibition?” she asked. Not necessarily. In the meta-analysis, not only did risankizumab lower the Ankylosing Spondylitis Disease Activity Score based on C-reactive protein (ASDAS-CRP) by a mean difference (MD) of –0.30 (95% CI, –0.41 to –0.19) from baseline values, but it also led to statistically significant reductions in SPARCC index score for the spine (MD, –3.10; 95% CI, –4.50 to –1.70) and high-sensitivity CRP (MD, –2.10; 95% CI, –2.56 to –1.64). The risankizumab findings might suggest there are potential disease-modifying properties for specifically targeting IL-23p19. There could also be a window of opportunity to use IL-23 inhibitors earlier.
“These are only results from one randomized, controlled trial in a small sample size where outcomes were reported as medians and interquartile ranges, so they had to be converted to means and standard deviations to have an odds ratio in the end,” she explained.
“Also, these were results from a radiographic axial spondyloarthritis population and not a nonradiographic axial spondyloarthritis population,” she added.
While that might limit the interpretation of the findings, “what we see here is both reduction in inflammation and reduction in structural disease progression as [measured] by SPARCC,” Ms. Vanhoutte said.
“Since IL-23 is an upstream molecule from IL-17 it’s probable that IL-23 is present in the prephase of the disease, in a prephase inflammation state,” she hypothesized. “This is especially interesting because there are very few randomized, controlled trials that examine therapeutic agents in nonradiographic axial spondyloarthritis,” she observed. Looking at IL-23 in radiographic, or established, disease therefore may not be as useful.
“I’m thinking you’re making actually a very important point for us,” commented Robert Landewé, MD, PhD, of Amsterdam University Medical Center.
“We are discussing whether or not IL-23 is important in inhibiting the disease activity of patients with axial spondyloarthritis, and we are surprised that it is not shown in RCTs.
“Why is it completely ineffective in axial spondyloarthritis? You show us that that is probably not the case,” Dr. Landewé suggested.
“What you make very clear here is that indeed there is some efficacy, and from a pathophysiological way of thinking it might be slightly different as compared with what most clinicians nowadays think.”
The study had no specific funding, and no disclosures were reported.
Interleukin (IL)–23 inhibition may still have a role to play in the treatment of patients with axial spondyloarthritis (SpA), suggests research presented at the 12th International Congress on Spondyloarthritides.
There is a strong rationale for using IL-23 inhibitors in patients with axial SpA, and the IL-23/IL-17 axis has been proposed as a critical player in the pathophysiology of the disease. But around 2018 it became clear from randomized, controlled trials that IL-23 inhibition was ineffective at improving key clinical outcomes, at least in patients with axial disease.
Although the overall results of a systematic review and meta-analysis that was presented at the meeting corroborated the negative results seen with IL-23–inhibiting agents in clinical trials, there were some data showing benefits of the IL-23 inhibitor risankizumab on secondary outcomes in one trial.
To look at the available evidence, Louise Vanhoutte, a 2nd-year internal medicine student at University Hospitals Leuven (Belgium) worked under the guidance of Rik Lories, MD, PhD, head of the division of rheumatology at University Hospitals Leuven. Together they searched known databases for randomized, controlled trials investigating the use of IL-23 and IL-17 inhibitors for the treatment of adults with axial SpA or psoriatic arthritis. Studies could be either phase 2 or phase 3, but had to have included a placebo and used the ASAS40 (40% Improvement in Assessment of SpondyloArthritis International Society Response criteria), ASAS20, Bath Ankylosing Spondylitis Disease Activity Index, or SPARCC (Spondyloarthritis Research Consortium of Canada) index score to assess outcomes.
The systematic review whittled the number of clinical trials in the meta-analysis to 12, which concerned the use of ustekinumab, an IL-12/23 inhibitor, and risankizumab, along with two IL-17 inhibitors, ixekizumab and secukinumab. Data for the IL-23 inhibitors guselkumab and tildrakizumab were not available.
“To no surprise, Forest plots showed that there was a lack of efficacy for IL-23 agents in the treatment of axial spondyloarthritis and a superior efficacy for IL-17 inhibitors in the treatment of axial spondyloarthritis,” Ms. Vanhoutte reported.
The respective odds ratios for IL-23 and IL-17 inhibitors in getting patients to meet ASAS40 response criteria in comparison to baseline were 1.51 (95% confidence interval, 0.98-2.31) and 2.54 (95% CI, 2.02-3.19).
“Does this mean it is a dead-end street for all IL-23 inhibition?” she asked. Not necessarily. In the meta-analysis, not only did risankizumab lower the Ankylosing Spondylitis Disease Activity Score based on C-reactive protein (ASDAS-CRP) by a mean difference (MD) of –0.30 (95% CI, –0.41 to –0.19) from baseline values, but it also led to statistically significant reductions in SPARCC index score for the spine (MD, –3.10; 95% CI, –4.50 to –1.70) and high-sensitivity CRP (MD, –2.10; 95% CI, –2.56 to –1.64). The risankizumab findings might suggest there are potential disease-modifying properties for specifically targeting IL-23p19. There could also be a window of opportunity to use IL-23 inhibitors earlier.
“These are only results from one randomized, controlled trial in a small sample size where outcomes were reported as medians and interquartile ranges, so they had to be converted to means and standard deviations to have an odds ratio in the end,” she explained.
“Also, these were results from a radiographic axial spondyloarthritis population and not a nonradiographic axial spondyloarthritis population,” she added.
While that might limit the interpretation of the findings, “what we see here is both reduction in inflammation and reduction in structural disease progression as [measured] by SPARCC,” Ms. Vanhoutte said.
“Since IL-23 is an upstream molecule from IL-17 it’s probable that IL-23 is present in the prephase of the disease, in a prephase inflammation state,” she hypothesized. “This is especially interesting because there are very few randomized, controlled trials that examine therapeutic agents in nonradiographic axial spondyloarthritis,” she observed. Looking at IL-23 in radiographic, or established, disease therefore may not be as useful.
“I’m thinking you’re making actually a very important point for us,” commented Robert Landewé, MD, PhD, of Amsterdam University Medical Center.
“We are discussing whether or not IL-23 is important in inhibiting the disease activity of patients with axial spondyloarthritis, and we are surprised that it is not shown in RCTs.
“Why is it completely ineffective in axial spondyloarthritis? You show us that that is probably not the case,” Dr. Landewé suggested.
“What you make very clear here is that indeed there is some efficacy, and from a pathophysiological way of thinking it might be slightly different as compared with what most clinicians nowadays think.”
The study had no specific funding, and no disclosures were reported.
Interleukin (IL)–23 inhibition may still have a role to play in the treatment of patients with axial spondyloarthritis (SpA), suggests research presented at the 12th International Congress on Spondyloarthritides.
There is a strong rationale for using IL-23 inhibitors in patients with axial SpA, and the IL-23/IL-17 axis has been proposed as a critical player in the pathophysiology of the disease. But around 2018 it became clear from randomized, controlled trials that IL-23 inhibition was ineffective at improving key clinical outcomes, at least in patients with axial disease.
Although the overall results of a systematic review and meta-analysis that was presented at the meeting corroborated the negative results seen with IL-23–inhibiting agents in clinical trials, there were some data showing benefits of the IL-23 inhibitor risankizumab on secondary outcomes in one trial.
To look at the available evidence, Louise Vanhoutte, a 2nd-year internal medicine student at University Hospitals Leuven (Belgium) worked under the guidance of Rik Lories, MD, PhD, head of the division of rheumatology at University Hospitals Leuven. Together they searched known databases for randomized, controlled trials investigating the use of IL-23 and IL-17 inhibitors for the treatment of adults with axial SpA or psoriatic arthritis. Studies could be either phase 2 or phase 3, but had to have included a placebo and used the ASAS40 (40% Improvement in Assessment of SpondyloArthritis International Society Response criteria), ASAS20, Bath Ankylosing Spondylitis Disease Activity Index, or SPARCC (Spondyloarthritis Research Consortium of Canada) index score to assess outcomes.
The systematic review whittled the number of clinical trials in the meta-analysis to 12, which concerned the use of ustekinumab, an IL-12/23 inhibitor, and risankizumab, along with two IL-17 inhibitors, ixekizumab and secukinumab. Data for the IL-23 inhibitors guselkumab and tildrakizumab were not available.
“To no surprise, Forest plots showed that there was a lack of efficacy for IL-23 agents in the treatment of axial spondyloarthritis and a superior efficacy for IL-17 inhibitors in the treatment of axial spondyloarthritis,” Ms. Vanhoutte reported.
The respective odds ratios for IL-23 and IL-17 inhibitors in getting patients to meet ASAS40 response criteria in comparison to baseline were 1.51 (95% confidence interval, 0.98-2.31) and 2.54 (95% CI, 2.02-3.19).
“Does this mean it is a dead-end street for all IL-23 inhibition?” she asked. Not necessarily. In the meta-analysis, not only did risankizumab lower the Ankylosing Spondylitis Disease Activity Score based on C-reactive protein (ASDAS-CRP) by a mean difference (MD) of –0.30 (95% CI, –0.41 to –0.19) from baseline values, but it also led to statistically significant reductions in SPARCC index score for the spine (MD, –3.10; 95% CI, –4.50 to –1.70) and high-sensitivity CRP (MD, –2.10; 95% CI, –2.56 to –1.64). The risankizumab findings might suggest there are potential disease-modifying properties for specifically targeting IL-23p19. There could also be a window of opportunity to use IL-23 inhibitors earlier.
“These are only results from one randomized, controlled trial in a small sample size where outcomes were reported as medians and interquartile ranges, so they had to be converted to means and standard deviations to have an odds ratio in the end,” she explained.
“Also, these were results from a radiographic axial spondyloarthritis population and not a nonradiographic axial spondyloarthritis population,” she added.
While that might limit the interpretation of the findings, “what we see here is both reduction in inflammation and reduction in structural disease progression as [measured] by SPARCC,” Ms. Vanhoutte said.
“Since IL-23 is an upstream molecule from IL-17 it’s probable that IL-23 is present in the prephase of the disease, in a prephase inflammation state,” she hypothesized. “This is especially interesting because there are very few randomized, controlled trials that examine therapeutic agents in nonradiographic axial spondyloarthritis,” she observed. Looking at IL-23 in radiographic, or established, disease therefore may not be as useful.
“I’m thinking you’re making actually a very important point for us,” commented Robert Landewé, MD, PhD, of Amsterdam University Medical Center.
“We are discussing whether or not IL-23 is important in inhibiting the disease activity of patients with axial spondyloarthritis, and we are surprised that it is not shown in RCTs.
“Why is it completely ineffective in axial spondyloarthritis? You show us that that is probably not the case,” Dr. Landewé suggested.
“What you make very clear here is that indeed there is some efficacy, and from a pathophysiological way of thinking it might be slightly different as compared with what most clinicians nowadays think.”
The study had no specific funding, and no disclosures were reported.
FROM THE 2021 SPA CONGRESS
Durvalumab combos beat monotherapy for unresectable stage 3 NSCLC
Both combinations – durvalumab plus either the anti-CD73 monoclonal antibody oleclumab or the anti-NKG2A mAb monalizumab – also numerically improved objective response rate. “Safety profiles were consistent across arms and no new safety signals were identified,” said Alexandre Martinez-Marti, MD, the lead investigator on the phase 2 trial, dubbed COAST, which he presented (abstract LBA42) at the 2021 European Society for Medical Oncology Congress on Sept. 17.
“These data support further evaluations of these combinations,” said Dr. Martinez-Marti, also a thoracic medical oncologist at the Vall d’Hebron Institute of Oncology in Barcelona.
Durvalumab is already established as a standard of care option for patients with unresectable stage 3 NSCLC who don’t progress after concurrent chemoradiation. There’s been preliminary data suggesting the benefits might be greater with oleclumab or monalizumab, so the study team looked into the issue.
They randomized 66 patients with unresectable stage 3 NSCLC and no progression after chemoradiotherapy to durvalumab 1,500 mg IV every 4 weeks; 59 others to durvalumab at the same dosage plus oleclumab 3,000 mg IV every 2 weeks for the first two cycles then every 4 weeks, and 61 were randomized to durvalumab plus monalizumab 750 mg IV every 2 weeks.
Patients were treated for up to 12 months, and they started treatment no later than 42 days after completing chemoradiation.
Over a median follow-up of 11.5 months, median progression-free survival (PFS) was 6.3 months in the durvalumab arm, but 15.1 months with the monalizumab combination (PFS hazard ratio versus durvalumab alone, 0.65; 95% CI, 0.49-0.85), and not reached in the oleclumab arm (PFS HR, 0.44; 95% CI, 0.26-0.75).
There were only a few complete responders across the study groups. Partial responses rates were 22.4% of the durvalumab alone arm, 36.7% in the oleclumab group, and 32.3% in the monalizumab arm.
The investigator assessed objective response rate was 25.4% with durvalumab alone, 38.3% in the oleclumab group, and 37.1% with the monalizumab combination. Curves started to separate from durvalumab monotherapy at around 2-4 months.
“Overall, the safety profiles of the two combinations were generally similar to the safety profile of durvalumab alone,” Dr. Martinez-Marti said. The rate of grade 3 or higher treatment-emergent events incidence was 39.4% with durvalumab, 40.7% the oleclumab combination, and 27.9% with monalizumab.
The most common grade 3/4 events were pneumonia (5.9%) and decreased lymphocyte count (3.2%); both were less common with the monalizumab combination.
Combined rates of pneumonitis and radiation pneumonitis of any grade were 21.2% with durvalumab, 28.8% in the oleclumab group, and 21.3% with monalizumab.
The groups were generally well balanced at baseline. The majority of subjects were men, White, and former smokers. Most subjects had stage 3A or 3B disease.
The work was funded by AstraZeneca. The investigators disclosed numerous ties to the company, including Dr. Martinez-Marti, who reported personal fees, travel expenses, and other connections.
This article was updated 9/24/21.
Both combinations – durvalumab plus either the anti-CD73 monoclonal antibody oleclumab or the anti-NKG2A mAb monalizumab – also numerically improved objective response rate. “Safety profiles were consistent across arms and no new safety signals were identified,” said Alexandre Martinez-Marti, MD, the lead investigator on the phase 2 trial, dubbed COAST, which he presented (abstract LBA42) at the 2021 European Society for Medical Oncology Congress on Sept. 17.
“These data support further evaluations of these combinations,” said Dr. Martinez-Marti, also a thoracic medical oncologist at the Vall d’Hebron Institute of Oncology in Barcelona.
Durvalumab is already established as a standard of care option for patients with unresectable stage 3 NSCLC who don’t progress after concurrent chemoradiation. There’s been preliminary data suggesting the benefits might be greater with oleclumab or monalizumab, so the study team looked into the issue.
They randomized 66 patients with unresectable stage 3 NSCLC and no progression after chemoradiotherapy to durvalumab 1,500 mg IV every 4 weeks; 59 others to durvalumab at the same dosage plus oleclumab 3,000 mg IV every 2 weeks for the first two cycles then every 4 weeks, and 61 were randomized to durvalumab plus monalizumab 750 mg IV every 2 weeks.
Patients were treated for up to 12 months, and they started treatment no later than 42 days after completing chemoradiation.
Over a median follow-up of 11.5 months, median progression-free survival (PFS) was 6.3 months in the durvalumab arm, but 15.1 months with the monalizumab combination (PFS hazard ratio versus durvalumab alone, 0.65; 95% CI, 0.49-0.85), and not reached in the oleclumab arm (PFS HR, 0.44; 95% CI, 0.26-0.75).
There were only a few complete responders across the study groups. Partial responses rates were 22.4% of the durvalumab alone arm, 36.7% in the oleclumab group, and 32.3% in the monalizumab arm.
The investigator assessed objective response rate was 25.4% with durvalumab alone, 38.3% in the oleclumab group, and 37.1% with the monalizumab combination. Curves started to separate from durvalumab monotherapy at around 2-4 months.
“Overall, the safety profiles of the two combinations were generally similar to the safety profile of durvalumab alone,” Dr. Martinez-Marti said. The rate of grade 3 or higher treatment-emergent events incidence was 39.4% with durvalumab, 40.7% the oleclumab combination, and 27.9% with monalizumab.
The most common grade 3/4 events were pneumonia (5.9%) and decreased lymphocyte count (3.2%); both were less common with the monalizumab combination.
Combined rates of pneumonitis and radiation pneumonitis of any grade were 21.2% with durvalumab, 28.8% in the oleclumab group, and 21.3% with monalizumab.
The groups were generally well balanced at baseline. The majority of subjects were men, White, and former smokers. Most subjects had stage 3A or 3B disease.
The work was funded by AstraZeneca. The investigators disclosed numerous ties to the company, including Dr. Martinez-Marti, who reported personal fees, travel expenses, and other connections.
This article was updated 9/24/21.
Both combinations – durvalumab plus either the anti-CD73 monoclonal antibody oleclumab or the anti-NKG2A mAb monalizumab – also numerically improved objective response rate. “Safety profiles were consistent across arms and no new safety signals were identified,” said Alexandre Martinez-Marti, MD, the lead investigator on the phase 2 trial, dubbed COAST, which he presented (abstract LBA42) at the 2021 European Society for Medical Oncology Congress on Sept. 17.
“These data support further evaluations of these combinations,” said Dr. Martinez-Marti, also a thoracic medical oncologist at the Vall d’Hebron Institute of Oncology in Barcelona.
Durvalumab is already established as a standard of care option for patients with unresectable stage 3 NSCLC who don’t progress after concurrent chemoradiation. There’s been preliminary data suggesting the benefits might be greater with oleclumab or monalizumab, so the study team looked into the issue.
They randomized 66 patients with unresectable stage 3 NSCLC and no progression after chemoradiotherapy to durvalumab 1,500 mg IV every 4 weeks; 59 others to durvalumab at the same dosage plus oleclumab 3,000 mg IV every 2 weeks for the first two cycles then every 4 weeks, and 61 were randomized to durvalumab plus monalizumab 750 mg IV every 2 weeks.
Patients were treated for up to 12 months, and they started treatment no later than 42 days after completing chemoradiation.
Over a median follow-up of 11.5 months, median progression-free survival (PFS) was 6.3 months in the durvalumab arm, but 15.1 months with the monalizumab combination (PFS hazard ratio versus durvalumab alone, 0.65; 95% CI, 0.49-0.85), and not reached in the oleclumab arm (PFS HR, 0.44; 95% CI, 0.26-0.75).
There were only a few complete responders across the study groups. Partial responses rates were 22.4% of the durvalumab alone arm, 36.7% in the oleclumab group, and 32.3% in the monalizumab arm.
The investigator assessed objective response rate was 25.4% with durvalumab alone, 38.3% in the oleclumab group, and 37.1% with the monalizumab combination. Curves started to separate from durvalumab monotherapy at around 2-4 months.
“Overall, the safety profiles of the two combinations were generally similar to the safety profile of durvalumab alone,” Dr. Martinez-Marti said. The rate of grade 3 or higher treatment-emergent events incidence was 39.4% with durvalumab, 40.7% the oleclumab combination, and 27.9% with monalizumab.
The most common grade 3/4 events were pneumonia (5.9%) and decreased lymphocyte count (3.2%); both were less common with the monalizumab combination.
Combined rates of pneumonitis and radiation pneumonitis of any grade were 21.2% with durvalumab, 28.8% in the oleclumab group, and 21.3% with monalizumab.
The groups were generally well balanced at baseline. The majority of subjects were men, White, and former smokers. Most subjects had stage 3A or 3B disease.
The work was funded by AstraZeneca. The investigators disclosed numerous ties to the company, including Dr. Martinez-Marti, who reported personal fees, travel expenses, and other connections.
This article was updated 9/24/21.
FROM ESMO CONGRESS 2021
Mediastinal relapse risk lower with PORT, but no survival benefit
The report was an update on the LungART international clinical trial, which, at the 2020 ESMO meeting, was shown not to improve disease-free survival over the course of 3 years. The update showed that, in addition to the lack of disease-free survival benefit, there was also no difference in metastases, and patients randomized to PORT had higher rates of death and grade 3/4 cardiopulmonary toxicity. The investigators returned this year to expand on another finding from the trial, a 51% reduction in the risk of mediastinal relapse with postoperative radiotherapy. The new analysis suggests there might still be a role for PORT in select patients, perhaps those with heavy nodal involvement, said lead investigator and presenter Cecile Le Pechoux, MD, radiation oncologist at the Gustave Roussy cancer treatment center in Villejuif, France.
For now, “personalized prescription of PORT should be based on prognostic factors of relapse and joint assessment of toxicity and efficacy,” she said.
Study discussant Pilar Garrido, MD, PhD, head of thoracic tumors Ramon y Cajal University Hospital, Madrid, agreed that there might still be a benefit for people with multiple N2 nodal station involvement, but at present, she said, “for me PORT cannot be the standard of care ... given the toxicity and mortality among PORT patients in LungART.”
The trial randomized 501 patients with non–small cell lung cancer with mediastinal involvement to either PORT at 54 Gy over 5.5 weeks or no further treatment following complete resection. Neoadjuvant or adjuvant chemotherapy were allowed.
The 3-year mediastinal relapse-free survival was 72.26% in the control arm but 86.06% with PORT (hazard ratio, 0.45; 95% confidence interval, 0.3-0.69).
“There is a significant difference” when it comes to mediastinal relapse, and “patients who have PORT do better. If we look at the location of mediastinal relapse, most [patients] relapse within the initially involved node. This is important information,” Dr. Le Pechoux said.
For left-sided tumors, the most frequent sites of mediastinal relapse were thoracic lymph node stations 7, 4L, and 4R. For right sided tumors, the most frequent stations were 4R, 2R and 7.
Prognostic factors for disease-free survival included quality of resection, extent of mediastinal involvement, and lymph node ratio (involved/explored). Nodal involvement was a significant prognostic factor for overall survival, but PORT was not (HR, 0.98; 95% CI, 0.7-1.4).
Mediastinal involvement with more than two node stations and less than an RO, or microscopically margin-negative resection, increased the risk of relapse.
The work was funded by the French National Cancer Institute, French Health Ministry, Institute Gustave Roussy, and Cancer Research UK. Dr. Pechoux and Dr. Garido disclosed ties to AstraZeneca, Roche, Amgen, and other companies.
This article was updated 9/24/21.
The report was an update on the LungART international clinical trial, which, at the 2020 ESMO meeting, was shown not to improve disease-free survival over the course of 3 years. The update showed that, in addition to the lack of disease-free survival benefit, there was also no difference in metastases, and patients randomized to PORT had higher rates of death and grade 3/4 cardiopulmonary toxicity. The investigators returned this year to expand on another finding from the trial, a 51% reduction in the risk of mediastinal relapse with postoperative radiotherapy. The new analysis suggests there might still be a role for PORT in select patients, perhaps those with heavy nodal involvement, said lead investigator and presenter Cecile Le Pechoux, MD, radiation oncologist at the Gustave Roussy cancer treatment center in Villejuif, France.
For now, “personalized prescription of PORT should be based on prognostic factors of relapse and joint assessment of toxicity and efficacy,” she said.
Study discussant Pilar Garrido, MD, PhD, head of thoracic tumors Ramon y Cajal University Hospital, Madrid, agreed that there might still be a benefit for people with multiple N2 nodal station involvement, but at present, she said, “for me PORT cannot be the standard of care ... given the toxicity and mortality among PORT patients in LungART.”
The trial randomized 501 patients with non–small cell lung cancer with mediastinal involvement to either PORT at 54 Gy over 5.5 weeks or no further treatment following complete resection. Neoadjuvant or adjuvant chemotherapy were allowed.
The 3-year mediastinal relapse-free survival was 72.26% in the control arm but 86.06% with PORT (hazard ratio, 0.45; 95% confidence interval, 0.3-0.69).
“There is a significant difference” when it comes to mediastinal relapse, and “patients who have PORT do better. If we look at the location of mediastinal relapse, most [patients] relapse within the initially involved node. This is important information,” Dr. Le Pechoux said.
For left-sided tumors, the most frequent sites of mediastinal relapse were thoracic lymph node stations 7, 4L, and 4R. For right sided tumors, the most frequent stations were 4R, 2R and 7.
Prognostic factors for disease-free survival included quality of resection, extent of mediastinal involvement, and lymph node ratio (involved/explored). Nodal involvement was a significant prognostic factor for overall survival, but PORT was not (HR, 0.98; 95% CI, 0.7-1.4).
Mediastinal involvement with more than two node stations and less than an RO, or microscopically margin-negative resection, increased the risk of relapse.
The work was funded by the French National Cancer Institute, French Health Ministry, Institute Gustave Roussy, and Cancer Research UK. Dr. Pechoux and Dr. Garido disclosed ties to AstraZeneca, Roche, Amgen, and other companies.
This article was updated 9/24/21.
The report was an update on the LungART international clinical trial, which, at the 2020 ESMO meeting, was shown not to improve disease-free survival over the course of 3 years. The update showed that, in addition to the lack of disease-free survival benefit, there was also no difference in metastases, and patients randomized to PORT had higher rates of death and grade 3/4 cardiopulmonary toxicity. The investigators returned this year to expand on another finding from the trial, a 51% reduction in the risk of mediastinal relapse with postoperative radiotherapy. The new analysis suggests there might still be a role for PORT in select patients, perhaps those with heavy nodal involvement, said lead investigator and presenter Cecile Le Pechoux, MD, radiation oncologist at the Gustave Roussy cancer treatment center in Villejuif, France.
For now, “personalized prescription of PORT should be based on prognostic factors of relapse and joint assessment of toxicity and efficacy,” she said.
Study discussant Pilar Garrido, MD, PhD, head of thoracic tumors Ramon y Cajal University Hospital, Madrid, agreed that there might still be a benefit for people with multiple N2 nodal station involvement, but at present, she said, “for me PORT cannot be the standard of care ... given the toxicity and mortality among PORT patients in LungART.”
The trial randomized 501 patients with non–small cell lung cancer with mediastinal involvement to either PORT at 54 Gy over 5.5 weeks or no further treatment following complete resection. Neoadjuvant or adjuvant chemotherapy were allowed.
The 3-year mediastinal relapse-free survival was 72.26% in the control arm but 86.06% with PORT (hazard ratio, 0.45; 95% confidence interval, 0.3-0.69).
“There is a significant difference” when it comes to mediastinal relapse, and “patients who have PORT do better. If we look at the location of mediastinal relapse, most [patients] relapse within the initially involved node. This is important information,” Dr. Le Pechoux said.
For left-sided tumors, the most frequent sites of mediastinal relapse were thoracic lymph node stations 7, 4L, and 4R. For right sided tumors, the most frequent stations were 4R, 2R and 7.
Prognostic factors for disease-free survival included quality of resection, extent of mediastinal involvement, and lymph node ratio (involved/explored). Nodal involvement was a significant prognostic factor for overall survival, but PORT was not (HR, 0.98; 95% CI, 0.7-1.4).
Mediastinal involvement with more than two node stations and less than an RO, or microscopically margin-negative resection, increased the risk of relapse.
The work was funded by the French National Cancer Institute, French Health Ministry, Institute Gustave Roussy, and Cancer Research UK. Dr. Pechoux and Dr. Garido disclosed ties to AstraZeneca, Roche, Amgen, and other companies.
This article was updated 9/24/21.
FROM ESMO CONGRESS 2021
‘New first-line standard of care’ in cervical cancer
That declaration was made by Raza Mirza, MD, chief oncologist at Copenhagen University Hospital in Denmark, who was invited to discuss the pros and cons of the KEYNOTE-826 trial at the European Society for Medical Oncology (ESMO) Congress 2021.
The trial showed that adding the checkpoint inhibitor pembrolizumab (Keytruda) to standard chemotherapy — with or without bevacizumab — resulted in about a one third reduction in the risk for both disease progression and death compared with chemotherapy alone.
The benefit of adding pembrolizumab was seen both in the overall study population and in patients with higher levels of programmed death ligand-1 (PD-L1), but not in those with biomarker-negative tumors, reported investigator Nicoletta Colombo, MD, PhD, from the University of Milan-Bicocca, Italy.
“Overall, data from KEYNOTE-826 suggest that pembrolizumab plus platinum-based chemotherapy with or without bevacizumab may be a new first-line standard of care,” she said in a late-breaking oral abstract presentation. The study was also simultaneously published online in The New England Journal of Medicine.
Since 2014, the standard of care for treating patients with recurrent, persistent, or metastatic cervical cancer has been chemotherapy with a platinum compound, paclitaxel, plus bevacizumab, based on the results of the GOG 240 study.
Immunotherapy with PD-1 inhibitors have shown efficacy as monotherapy in second- or later-line therapy for women with cervical cancer, but until now no data about the addition of these agents to chemotherapy were available, Dr. Colombo noted.
Dr. Mirza noted that there is sound rationale for using checkpoint inhibitors targeted against PD-1 in patients with cervical cancer, because PD-L1 has been shown to be a consistent biomarker for infection of the cervix with human papillomavirus (HPV), which is responsible for more than 90% of cervical cancers.
“PD-L1 is significantly upregulated in cervical cancer and detectable by immunohistochemistry,” he said. “PD-L1 expression reduces the immune response since it is able to bind to PD-1 on T-cell lymphocytes, thereby inhibiting their function. These findings suggest that targeting the PD-1/PD-L1 pathway may be therapeutically effective and should be considered in the treatment of cervical cancer.”
KEYNOTE-826 details
This was a double-blind trial conducted in 617 patients stratified by metastatic disease status at diagnosis; PD-L1 combined positive score (CPS) either < 1, 1 to < 10, or ≥ 10. They were randomized in a 1:1 ratio to receive pembrolizumab 200 mg or placebo every 3 weeks for up to 35 cycles plus platinum-based chemotherapy, with bevacizumab added at the investigator’s discretion.
The dual primary endpoints of progression-free survival (PFS) and overall survival (OS) were each tested sequentially in patients with a PD-L1 CPS ≥ 1 in both the intention-to-treat (ITT) or “all-comers” population, and in patients with a PD-L1 CPS ≥ 10.
Patient characteristics were generally well balanced between the treatment groups, except for a slightly higher proportion of patients with squamous cell histology in the pembrolizumab versus the placebo group (76.3% vs 68.3%).
PFS and OS results
The addition of pembrolizumab was associated with improved PFS across most protocol-specified subgroups, Dr. Colombo and colleagues noted.
After a median follow-up of 22 months, the 12-month PFS rate in the biomarker-selected population (all patients with a PD-L1 CPS ≥ 1) was 45.5% for patients in the pembrolizumab group versus 34.1% in the placebo group. This translated into a hazard ratio (HR) for progression on pembrolizumab of 0.62 (P < .001).
The respective PFS rates in the ITT population were 44.7% and 33.5%, with an HR for progression of 0.65 (P < .001) with the checkpoint inhibitor.
In patients with PD-L1 CPS ≥ 10, the respective rates of PFS and the HR were 44.6%, 33.5%, and 0.58 (P < .001).
OS rates were also significantly improved, he noted.
The 12-month and 24-month OS rates in all patients with PD-L1 CPS ≥ 1 were 75.3% and 53%, respectively, for patients assigned to pembrolizumab versus 63.1% and 41.7% in patients assigned to placebo, translating to an HR for death with pembrolizumab in this group of 0.64 (P < .001).
In the all-comers (ITT) population, respective 12- and 24-month OS rates were 74.8% and 50.4% with pembrolizumab versus 63.6% and 40.4% with placebo. This difference translated into an HR for death with anti-PD-1 of 0.67 (P < .001).
Among patients with the higher PD-L1 levels (≥ CPS 10), the respective OS rates were 75.7% and 54.4% with pembrolizumab versus 61.5% and 44.6% with placebo (HR 0.61, P < .001).
Dr. Mirza emphasized that “we did not see any efficacy of pembrolizumab in the biomarker-negative population,” with an HR for PFS of 0.94 and HR for OS of 1.0 in this subgroup.
The most common grade ≥ 3 adverse events were anemia, which occurred in 30.3% of patients assigned to pembrolizumab compared with 26.9% in the placebo group, and neutropenias, which occurred in 12.4% and 9.7% of patients, respectively. One patient in the pembrolizumab group died from an immune-related event, encephalitis.
Despite his enthusiasm for the regimen, Dr. Mirza tempered it by pointing out that there was an imbalance in the sample sizes regarding histology, and a potential bias introduced by the failure to stratify by tumor histology.
He noted that in other studies checkpoint inhibitors have had only modest activity against adenocarcinomas, which were more frequent in the placebo group in KEYNOTE-826, resulting in a potential positive bias in favor of pembrolizumab.
KEYNOTE-826 is funded by MSD. Dr. Colombo has disclosed consultant, research, and promotional speaking activities for multiple companies. Dr. Mirza has disclosed personal financial interests with Merck and other companies.
A version of this article was first published on Medscape.com.
That declaration was made by Raza Mirza, MD, chief oncologist at Copenhagen University Hospital in Denmark, who was invited to discuss the pros and cons of the KEYNOTE-826 trial at the European Society for Medical Oncology (ESMO) Congress 2021.
The trial showed that adding the checkpoint inhibitor pembrolizumab (Keytruda) to standard chemotherapy — with or without bevacizumab — resulted in about a one third reduction in the risk for both disease progression and death compared with chemotherapy alone.
The benefit of adding pembrolizumab was seen both in the overall study population and in patients with higher levels of programmed death ligand-1 (PD-L1), but not in those with biomarker-negative tumors, reported investigator Nicoletta Colombo, MD, PhD, from the University of Milan-Bicocca, Italy.
“Overall, data from KEYNOTE-826 suggest that pembrolizumab plus platinum-based chemotherapy with or without bevacizumab may be a new first-line standard of care,” she said in a late-breaking oral abstract presentation. The study was also simultaneously published online in The New England Journal of Medicine.
Since 2014, the standard of care for treating patients with recurrent, persistent, or metastatic cervical cancer has been chemotherapy with a platinum compound, paclitaxel, plus bevacizumab, based on the results of the GOG 240 study.
Immunotherapy with PD-1 inhibitors have shown efficacy as monotherapy in second- or later-line therapy for women with cervical cancer, but until now no data about the addition of these agents to chemotherapy were available, Dr. Colombo noted.
Dr. Mirza noted that there is sound rationale for using checkpoint inhibitors targeted against PD-1 in patients with cervical cancer, because PD-L1 has been shown to be a consistent biomarker for infection of the cervix with human papillomavirus (HPV), which is responsible for more than 90% of cervical cancers.
“PD-L1 is significantly upregulated in cervical cancer and detectable by immunohistochemistry,” he said. “PD-L1 expression reduces the immune response since it is able to bind to PD-1 on T-cell lymphocytes, thereby inhibiting their function. These findings suggest that targeting the PD-1/PD-L1 pathway may be therapeutically effective and should be considered in the treatment of cervical cancer.”
KEYNOTE-826 details
This was a double-blind trial conducted in 617 patients stratified by metastatic disease status at diagnosis; PD-L1 combined positive score (CPS) either < 1, 1 to < 10, or ≥ 10. They were randomized in a 1:1 ratio to receive pembrolizumab 200 mg or placebo every 3 weeks for up to 35 cycles plus platinum-based chemotherapy, with bevacizumab added at the investigator’s discretion.
The dual primary endpoints of progression-free survival (PFS) and overall survival (OS) were each tested sequentially in patients with a PD-L1 CPS ≥ 1 in both the intention-to-treat (ITT) or “all-comers” population, and in patients with a PD-L1 CPS ≥ 10.
Patient characteristics were generally well balanced between the treatment groups, except for a slightly higher proportion of patients with squamous cell histology in the pembrolizumab versus the placebo group (76.3% vs 68.3%).
PFS and OS results
The addition of pembrolizumab was associated with improved PFS across most protocol-specified subgroups, Dr. Colombo and colleagues noted.
After a median follow-up of 22 months, the 12-month PFS rate in the biomarker-selected population (all patients with a PD-L1 CPS ≥ 1) was 45.5% for patients in the pembrolizumab group versus 34.1% in the placebo group. This translated into a hazard ratio (HR) for progression on pembrolizumab of 0.62 (P < .001).
The respective PFS rates in the ITT population were 44.7% and 33.5%, with an HR for progression of 0.65 (P < .001) with the checkpoint inhibitor.
In patients with PD-L1 CPS ≥ 10, the respective rates of PFS and the HR were 44.6%, 33.5%, and 0.58 (P < .001).
OS rates were also significantly improved, he noted.
The 12-month and 24-month OS rates in all patients with PD-L1 CPS ≥ 1 were 75.3% and 53%, respectively, for patients assigned to pembrolizumab versus 63.1% and 41.7% in patients assigned to placebo, translating to an HR for death with pembrolizumab in this group of 0.64 (P < .001).
In the all-comers (ITT) population, respective 12- and 24-month OS rates were 74.8% and 50.4% with pembrolizumab versus 63.6% and 40.4% with placebo. This difference translated into an HR for death with anti-PD-1 of 0.67 (P < .001).
Among patients with the higher PD-L1 levels (≥ CPS 10), the respective OS rates were 75.7% and 54.4% with pembrolizumab versus 61.5% and 44.6% with placebo (HR 0.61, P < .001).
Dr. Mirza emphasized that “we did not see any efficacy of pembrolizumab in the biomarker-negative population,” with an HR for PFS of 0.94 and HR for OS of 1.0 in this subgroup.
The most common grade ≥ 3 adverse events were anemia, which occurred in 30.3% of patients assigned to pembrolizumab compared with 26.9% in the placebo group, and neutropenias, which occurred in 12.4% and 9.7% of patients, respectively. One patient in the pembrolizumab group died from an immune-related event, encephalitis.
Despite his enthusiasm for the regimen, Dr. Mirza tempered it by pointing out that there was an imbalance in the sample sizes regarding histology, and a potential bias introduced by the failure to stratify by tumor histology.
He noted that in other studies checkpoint inhibitors have had only modest activity against adenocarcinomas, which were more frequent in the placebo group in KEYNOTE-826, resulting in a potential positive bias in favor of pembrolizumab.
KEYNOTE-826 is funded by MSD. Dr. Colombo has disclosed consultant, research, and promotional speaking activities for multiple companies. Dr. Mirza has disclosed personal financial interests with Merck and other companies.
A version of this article was first published on Medscape.com.
That declaration was made by Raza Mirza, MD, chief oncologist at Copenhagen University Hospital in Denmark, who was invited to discuss the pros and cons of the KEYNOTE-826 trial at the European Society for Medical Oncology (ESMO) Congress 2021.
The trial showed that adding the checkpoint inhibitor pembrolizumab (Keytruda) to standard chemotherapy — with or without bevacizumab — resulted in about a one third reduction in the risk for both disease progression and death compared with chemotherapy alone.
The benefit of adding pembrolizumab was seen both in the overall study population and in patients with higher levels of programmed death ligand-1 (PD-L1), but not in those with biomarker-negative tumors, reported investigator Nicoletta Colombo, MD, PhD, from the University of Milan-Bicocca, Italy.
“Overall, data from KEYNOTE-826 suggest that pembrolizumab plus platinum-based chemotherapy with or without bevacizumab may be a new first-line standard of care,” she said in a late-breaking oral abstract presentation. The study was also simultaneously published online in The New England Journal of Medicine.
Since 2014, the standard of care for treating patients with recurrent, persistent, or metastatic cervical cancer has been chemotherapy with a platinum compound, paclitaxel, plus bevacizumab, based on the results of the GOG 240 study.
Immunotherapy with PD-1 inhibitors have shown efficacy as monotherapy in second- or later-line therapy for women with cervical cancer, but until now no data about the addition of these agents to chemotherapy were available, Dr. Colombo noted.
Dr. Mirza noted that there is sound rationale for using checkpoint inhibitors targeted against PD-1 in patients with cervical cancer, because PD-L1 has been shown to be a consistent biomarker for infection of the cervix with human papillomavirus (HPV), which is responsible for more than 90% of cervical cancers.
“PD-L1 is significantly upregulated in cervical cancer and detectable by immunohistochemistry,” he said. “PD-L1 expression reduces the immune response since it is able to bind to PD-1 on T-cell lymphocytes, thereby inhibiting their function. These findings suggest that targeting the PD-1/PD-L1 pathway may be therapeutically effective and should be considered in the treatment of cervical cancer.”
KEYNOTE-826 details
This was a double-blind trial conducted in 617 patients stratified by metastatic disease status at diagnosis; PD-L1 combined positive score (CPS) either < 1, 1 to < 10, or ≥ 10. They were randomized in a 1:1 ratio to receive pembrolizumab 200 mg or placebo every 3 weeks for up to 35 cycles plus platinum-based chemotherapy, with bevacizumab added at the investigator’s discretion.
The dual primary endpoints of progression-free survival (PFS) and overall survival (OS) were each tested sequentially in patients with a PD-L1 CPS ≥ 1 in both the intention-to-treat (ITT) or “all-comers” population, and in patients with a PD-L1 CPS ≥ 10.
Patient characteristics were generally well balanced between the treatment groups, except for a slightly higher proportion of patients with squamous cell histology in the pembrolizumab versus the placebo group (76.3% vs 68.3%).
PFS and OS results
The addition of pembrolizumab was associated with improved PFS across most protocol-specified subgroups, Dr. Colombo and colleagues noted.
After a median follow-up of 22 months, the 12-month PFS rate in the biomarker-selected population (all patients with a PD-L1 CPS ≥ 1) was 45.5% for patients in the pembrolizumab group versus 34.1% in the placebo group. This translated into a hazard ratio (HR) for progression on pembrolizumab of 0.62 (P < .001).
The respective PFS rates in the ITT population were 44.7% and 33.5%, with an HR for progression of 0.65 (P < .001) with the checkpoint inhibitor.
In patients with PD-L1 CPS ≥ 10, the respective rates of PFS and the HR were 44.6%, 33.5%, and 0.58 (P < .001).
OS rates were also significantly improved, he noted.
The 12-month and 24-month OS rates in all patients with PD-L1 CPS ≥ 1 were 75.3% and 53%, respectively, for patients assigned to pembrolizumab versus 63.1% and 41.7% in patients assigned to placebo, translating to an HR for death with pembrolizumab in this group of 0.64 (P < .001).
In the all-comers (ITT) population, respective 12- and 24-month OS rates were 74.8% and 50.4% with pembrolizumab versus 63.6% and 40.4% with placebo. This difference translated into an HR for death with anti-PD-1 of 0.67 (P < .001).
Among patients with the higher PD-L1 levels (≥ CPS 10), the respective OS rates were 75.7% and 54.4% with pembrolizumab versus 61.5% and 44.6% with placebo (HR 0.61, P < .001).
Dr. Mirza emphasized that “we did not see any efficacy of pembrolizumab in the biomarker-negative population,” with an HR for PFS of 0.94 and HR for OS of 1.0 in this subgroup.
The most common grade ≥ 3 adverse events were anemia, which occurred in 30.3% of patients assigned to pembrolizumab compared with 26.9% in the placebo group, and neutropenias, which occurred in 12.4% and 9.7% of patients, respectively. One patient in the pembrolizumab group died from an immune-related event, encephalitis.
Despite his enthusiasm for the regimen, Dr. Mirza tempered it by pointing out that there was an imbalance in the sample sizes regarding histology, and a potential bias introduced by the failure to stratify by tumor histology.
He noted that in other studies checkpoint inhibitors have had only modest activity against adenocarcinomas, which were more frequent in the placebo group in KEYNOTE-826, resulting in a potential positive bias in favor of pembrolizumab.
KEYNOTE-826 is funded by MSD. Dr. Colombo has disclosed consultant, research, and promotional speaking activities for multiple companies. Dr. Mirza has disclosed personal financial interests with Merck and other companies.
A version of this article was first published on Medscape.com.
Adjuvant pembro success in early melanoma raises questions
However, the results raise many questions, says an expert invited to discuss the new data.
Adjuvant pembrolizumab is already approved in the United States for use in patients with melanoma with lymph node involvement following complete resection, having been shown to prolong both recurrence-free and distant metastasis-free survival (DMFS) in stage 3 melanoma.
This latest trial involved patients with slightly earlier disease, those with resected stage 2B and 2C melanoma. These patients are at “high risk” of disease recurrence and have similar outcomes to stage 3A and 3B melanoma patients, explained study presenter Jason J. Luke, MD, director of the Cancer Immunotherapeutics Center at UPMC Hillman Cancer Center, Pittsburgh.
Results from the KEYNOTE-716 trial showed that adjuvant pembrolizumab is also beneficial in this earlier stage disease: it improved recurrence-free survival (RFS) by 35% and improved distant metastasis-free survival by 40% compared with placebo.
Adjuvant pembrolizumab is an “effective treatment option with a favorable benefit-risk profile for patients with high-risk stage 2 melanoma,” Dr. Luke concluded.
The manufacturer, Merck, has said that these new results have already been accepted for priority review by the U.S. Food and Drug Administration, making it likely that the indication will be extended to include patients with earlier disease.
Dr. Luke presented the results at the European Society of Medical Oncology 2021 annual meeting.
Invited discussant Omid Hamid, MD, chief of research/immuno-oncology, the Angeles Clinic and Research Institute, a Cedars-Sinai Affiliate, Los Angeles, said that Dr. Luke’s presentation was “amazing.”
However, these new results have “sabotaged how we think about how we treat our patients and how we’re going to think about what we do in the future.”
Dr. Hamid noted that the incidence of stage 2B and 2C melanoma is “equal” to that of stage 3 disease, “so with a proposed approval” of pembrolizumab in this earlier setting, “we will have a lot more patients” to treat earlier in their disease course.
Of course, this raises the inevitable question of how to treat these patients when they relapse, and how to treat these patients in the metastatic setting “having already exhausted single-agent PD-1 therapy,” he commented.
Dr. Hamid said that the current results also reveal the “current problem” with adjuvant therapy, which is that “we don’t know who benefits,” and there is a subset patients who “never recur” even if they are untreated.
So the questions are: “How come all get treated? What about the risks of toxicity? The costs? And where do we fit these patients into clinic?”
As with so many presentations of immunotherapy trial data, the need for biomarkers was raised, with Dr. Hamid emphasizing the need for predictive biomarkers that could exclude patients, and save them from toxicity.
He noted that there were data with another checkpoint inhibitor, nivolumab (Opdivo), in the adjuvant setting (from the CheckMate 238 trial) that suggested higher tumor mutation burden and tumor interferon-gamma levels could play a role, and he hopes that similar data may be available from this latest trial.
Also, there are ongoing and upcoming trials in patients with stage 2B and 2C melanoma that may answer some of the outstanding questions, including a study of neoadjuvant PD-1 blockade before resection, and the DETECTION trial, which is exploring circulating tumor DNA-guided therapy postsurgery.
Then there is the NivoMela trial that will look at nivolumab in stage 2A as well as 2B and 2C disease, while the REFINE trial will assess whether giving immunotherapy less often to patients with advanced cancer, including those with melanoma, results in fewer side effects while continuing to be effective.
The current results also raise the question of whether to go “earlier and earlier” with adjuvant immunotherapy into “poor risk” stage 1 melanoma, which is already being tried in the United States, although there is “no clear understanding of what to do for those patients.”
Overall, Dr. Hamid said that the results of KEYNOTE-716 have “created more questions than answers,” including its impact on the inclusion criteria for phase 3/4 clinical trials, “which now exclude patients who have received adjuvant therapy within 6 months.”
“That will have to change,” he suggested.
Some of the questions raised by Dr. Hamid were discussed on social media, sparking a lively Twitter debate on how best to take the results forward and into the clinic.
Florentia Dimitriou, MD, a dermatology consultant in the Skin Cancer Clinic, University Hospital Zurich, Switzerland, said the data were “great” but she was “still unclear” over who needs adjuvant immunotherapy in this setting.
She also emphasized that, for her, the greater RFS benefit seen in T3b than in T4b disease “doesn’t make sense,” and she also highlighted the finding of long-term toxicity in approximately 18% of patients.
Dr. Luke replied that he agrees that the T3b/T4b results are puzzling but he said the event rate was “low” and the data are “immature,” and that he hopes to have “more info soon.”
He acknowledged that around 18% of patients taking pembrolizumab went on to receive hormone therapy for adverse events, including 13.9% due to hypothyroidism, and others including hypophysitis, adrenal sufficiency, and type 1 diabetes. However, he also pointed out that about 5% of patients in this study had background thyroid issues. The risks and benefits of treatment need to be discussed with patients, he added.
Over a series of tweets, Rebecca J. Lee, PhD, NIHR clinical lecturer in medical oncology at the University of Manchester, United Kingdom, said, “we need to know more” about the distant metastasis-free survival results, and that results for overall survival are “really” needed.
She also emphasized the need for biomarkers to identify those patients who are likely to benefit, and whether benefit can be upfront or early on in treatment. Dr. Lee added that, as endocrine thyroid toxicity occurs after a median of 3.3 months, “pretreatment biomarkers will be more important than on-treatment biomarkers in this setting.”
Details of the results in earlier stage disease
The KEYNOTE-716 trial enrolled patients with newly diagnosed, resected, high-risk stage 2 melanoma aged ≥ 12 years and a good performance status. The majority (~64%) had stage 2B melanoma, and the rest had stage 2C. T3b disease was present in 41% of patients, 23% had T4a disease, and 35% had T4b disease.
Patients were randomized to receive pembrolizumab or placebo.
In a subsequent part of the study, patients with recurrence will be unblended, with either crossover from the placebo to active treatment group or rechallenge with pembrolizumab for up to 2 years.
Presenting the first part, Dr. Luke said that, of 487 patients assigned to pembrolizumab, 483 started treatment, of whom 206 have completed treatment, 133 are still on therapy, and 144 have discontinued.
In the placebo group, 489 patients were assigned and 486 began treatment. Of those, 229 completed treatment, 152 are still ongoing, and 105 discontinued.
The two groups were well balanced in terms of baseline characteristics. The median age was approximately 60 years, with only one patient enrolled who was aged 12-17 years.
At 12 months, the study met its primary endpoint.
Relapse-fee survival was 90.5% in patients treated with pembrolizumab versus 83.1% in the placebo group, at a hazard ratio for recurrence of 0.65 (P = .00658).
“Despite this trial hitting this primary endpoint very early, there are a number of patients who are censored later in the curves,” Dr. Luke said, adding that “we will continue to see these data mature.”
“In fact, it’s our full expectation that curves will continue to separate over time.”
When looking at key subgroups, Dr. Luke showed that the results favored pembrolizumab when stratifying patients by age, gender, race, and performance status.
Interestingly, patients with T3b disease did a lot better on pembrolizumab compared with those who had T4b disease, at a hazard ratio for recurrence of 0.44 versus 0.94.
Data on recurrence patterns revealed that 11.1% of patients taking pembrolizumab had an event, with 6.4% experiencing skin and/or lymph node regional recurrence and 4.7% distant recurrence.
In the placebo group, 16.8% of patients had a recurrence event, with 8.4% having a loco-regional recurrence and 7.8% a distant recurrence.
Dr. Luke explained that this equates to an approximate 40% reduction in distant recurrence with pembrolizumab over placebo.
Finally, the researchers examined change in global health status on the EORTC QLQ-C30 quality of life score. Examining mean change over time, they found that there were no clinically meaningful changes, and the scores in the pembrolizumab and placebo groups tracked each other during the course of follow-up.
Quality of life was, therefore, “only minimally changed,” Dr. Luke said.
The study was funded by MSD. Dr. Luke and Dr. Hamid have declared relationships with multiple companies.
A version of this article first appeared on Medscape.com.
However, the results raise many questions, says an expert invited to discuss the new data.
Adjuvant pembrolizumab is already approved in the United States for use in patients with melanoma with lymph node involvement following complete resection, having been shown to prolong both recurrence-free and distant metastasis-free survival (DMFS) in stage 3 melanoma.
This latest trial involved patients with slightly earlier disease, those with resected stage 2B and 2C melanoma. These patients are at “high risk” of disease recurrence and have similar outcomes to stage 3A and 3B melanoma patients, explained study presenter Jason J. Luke, MD, director of the Cancer Immunotherapeutics Center at UPMC Hillman Cancer Center, Pittsburgh.
Results from the KEYNOTE-716 trial showed that adjuvant pembrolizumab is also beneficial in this earlier stage disease: it improved recurrence-free survival (RFS) by 35% and improved distant metastasis-free survival by 40% compared with placebo.
Adjuvant pembrolizumab is an “effective treatment option with a favorable benefit-risk profile for patients with high-risk stage 2 melanoma,” Dr. Luke concluded.
The manufacturer, Merck, has said that these new results have already been accepted for priority review by the U.S. Food and Drug Administration, making it likely that the indication will be extended to include patients with earlier disease.
Dr. Luke presented the results at the European Society of Medical Oncology 2021 annual meeting.
Invited discussant Omid Hamid, MD, chief of research/immuno-oncology, the Angeles Clinic and Research Institute, a Cedars-Sinai Affiliate, Los Angeles, said that Dr. Luke’s presentation was “amazing.”
However, these new results have “sabotaged how we think about how we treat our patients and how we’re going to think about what we do in the future.”
Dr. Hamid noted that the incidence of stage 2B and 2C melanoma is “equal” to that of stage 3 disease, “so with a proposed approval” of pembrolizumab in this earlier setting, “we will have a lot more patients” to treat earlier in their disease course.
Of course, this raises the inevitable question of how to treat these patients when they relapse, and how to treat these patients in the metastatic setting “having already exhausted single-agent PD-1 therapy,” he commented.
Dr. Hamid said that the current results also reveal the “current problem” with adjuvant therapy, which is that “we don’t know who benefits,” and there is a subset patients who “never recur” even if they are untreated.
So the questions are: “How come all get treated? What about the risks of toxicity? The costs? And where do we fit these patients into clinic?”
As with so many presentations of immunotherapy trial data, the need for biomarkers was raised, with Dr. Hamid emphasizing the need for predictive biomarkers that could exclude patients, and save them from toxicity.
He noted that there were data with another checkpoint inhibitor, nivolumab (Opdivo), in the adjuvant setting (from the CheckMate 238 trial) that suggested higher tumor mutation burden and tumor interferon-gamma levels could play a role, and he hopes that similar data may be available from this latest trial.
Also, there are ongoing and upcoming trials in patients with stage 2B and 2C melanoma that may answer some of the outstanding questions, including a study of neoadjuvant PD-1 blockade before resection, and the DETECTION trial, which is exploring circulating tumor DNA-guided therapy postsurgery.
Then there is the NivoMela trial that will look at nivolumab in stage 2A as well as 2B and 2C disease, while the REFINE trial will assess whether giving immunotherapy less often to patients with advanced cancer, including those with melanoma, results in fewer side effects while continuing to be effective.
The current results also raise the question of whether to go “earlier and earlier” with adjuvant immunotherapy into “poor risk” stage 1 melanoma, which is already being tried in the United States, although there is “no clear understanding of what to do for those patients.”
Overall, Dr. Hamid said that the results of KEYNOTE-716 have “created more questions than answers,” including its impact on the inclusion criteria for phase 3/4 clinical trials, “which now exclude patients who have received adjuvant therapy within 6 months.”
“That will have to change,” he suggested.
Some of the questions raised by Dr. Hamid were discussed on social media, sparking a lively Twitter debate on how best to take the results forward and into the clinic.
Florentia Dimitriou, MD, a dermatology consultant in the Skin Cancer Clinic, University Hospital Zurich, Switzerland, said the data were “great” but she was “still unclear” over who needs adjuvant immunotherapy in this setting.
She also emphasized that, for her, the greater RFS benefit seen in T3b than in T4b disease “doesn’t make sense,” and she also highlighted the finding of long-term toxicity in approximately 18% of patients.
Dr. Luke replied that he agrees that the T3b/T4b results are puzzling but he said the event rate was “low” and the data are “immature,” and that he hopes to have “more info soon.”
He acknowledged that around 18% of patients taking pembrolizumab went on to receive hormone therapy for adverse events, including 13.9% due to hypothyroidism, and others including hypophysitis, adrenal sufficiency, and type 1 diabetes. However, he also pointed out that about 5% of patients in this study had background thyroid issues. The risks and benefits of treatment need to be discussed with patients, he added.
Over a series of tweets, Rebecca J. Lee, PhD, NIHR clinical lecturer in medical oncology at the University of Manchester, United Kingdom, said, “we need to know more” about the distant metastasis-free survival results, and that results for overall survival are “really” needed.
She also emphasized the need for biomarkers to identify those patients who are likely to benefit, and whether benefit can be upfront or early on in treatment. Dr. Lee added that, as endocrine thyroid toxicity occurs after a median of 3.3 months, “pretreatment biomarkers will be more important than on-treatment biomarkers in this setting.”
Details of the results in earlier stage disease
The KEYNOTE-716 trial enrolled patients with newly diagnosed, resected, high-risk stage 2 melanoma aged ≥ 12 years and a good performance status. The majority (~64%) had stage 2B melanoma, and the rest had stage 2C. T3b disease was present in 41% of patients, 23% had T4a disease, and 35% had T4b disease.
Patients were randomized to receive pembrolizumab or placebo.
In a subsequent part of the study, patients with recurrence will be unblended, with either crossover from the placebo to active treatment group or rechallenge with pembrolizumab for up to 2 years.
Presenting the first part, Dr. Luke said that, of 487 patients assigned to pembrolizumab, 483 started treatment, of whom 206 have completed treatment, 133 are still on therapy, and 144 have discontinued.
In the placebo group, 489 patients were assigned and 486 began treatment. Of those, 229 completed treatment, 152 are still ongoing, and 105 discontinued.
The two groups were well balanced in terms of baseline characteristics. The median age was approximately 60 years, with only one patient enrolled who was aged 12-17 years.
At 12 months, the study met its primary endpoint.
Relapse-fee survival was 90.5% in patients treated with pembrolizumab versus 83.1% in the placebo group, at a hazard ratio for recurrence of 0.65 (P = .00658).
“Despite this trial hitting this primary endpoint very early, there are a number of patients who are censored later in the curves,” Dr. Luke said, adding that “we will continue to see these data mature.”
“In fact, it’s our full expectation that curves will continue to separate over time.”
When looking at key subgroups, Dr. Luke showed that the results favored pembrolizumab when stratifying patients by age, gender, race, and performance status.
Interestingly, patients with T3b disease did a lot better on pembrolizumab compared with those who had T4b disease, at a hazard ratio for recurrence of 0.44 versus 0.94.
Data on recurrence patterns revealed that 11.1% of patients taking pembrolizumab had an event, with 6.4% experiencing skin and/or lymph node regional recurrence and 4.7% distant recurrence.
In the placebo group, 16.8% of patients had a recurrence event, with 8.4% having a loco-regional recurrence and 7.8% a distant recurrence.
Dr. Luke explained that this equates to an approximate 40% reduction in distant recurrence with pembrolizumab over placebo.
Finally, the researchers examined change in global health status on the EORTC QLQ-C30 quality of life score. Examining mean change over time, they found that there were no clinically meaningful changes, and the scores in the pembrolizumab and placebo groups tracked each other during the course of follow-up.
Quality of life was, therefore, “only minimally changed,” Dr. Luke said.
The study was funded by MSD. Dr. Luke and Dr. Hamid have declared relationships with multiple companies.
A version of this article first appeared on Medscape.com.
However, the results raise many questions, says an expert invited to discuss the new data.
Adjuvant pembrolizumab is already approved in the United States for use in patients with melanoma with lymph node involvement following complete resection, having been shown to prolong both recurrence-free and distant metastasis-free survival (DMFS) in stage 3 melanoma.
This latest trial involved patients with slightly earlier disease, those with resected stage 2B and 2C melanoma. These patients are at “high risk” of disease recurrence and have similar outcomes to stage 3A and 3B melanoma patients, explained study presenter Jason J. Luke, MD, director of the Cancer Immunotherapeutics Center at UPMC Hillman Cancer Center, Pittsburgh.
Results from the KEYNOTE-716 trial showed that adjuvant pembrolizumab is also beneficial in this earlier stage disease: it improved recurrence-free survival (RFS) by 35% and improved distant metastasis-free survival by 40% compared with placebo.
Adjuvant pembrolizumab is an “effective treatment option with a favorable benefit-risk profile for patients with high-risk stage 2 melanoma,” Dr. Luke concluded.
The manufacturer, Merck, has said that these new results have already been accepted for priority review by the U.S. Food and Drug Administration, making it likely that the indication will be extended to include patients with earlier disease.
Dr. Luke presented the results at the European Society of Medical Oncology 2021 annual meeting.
Invited discussant Omid Hamid, MD, chief of research/immuno-oncology, the Angeles Clinic and Research Institute, a Cedars-Sinai Affiliate, Los Angeles, said that Dr. Luke’s presentation was “amazing.”
However, these new results have “sabotaged how we think about how we treat our patients and how we’re going to think about what we do in the future.”
Dr. Hamid noted that the incidence of stage 2B and 2C melanoma is “equal” to that of stage 3 disease, “so with a proposed approval” of pembrolizumab in this earlier setting, “we will have a lot more patients” to treat earlier in their disease course.
Of course, this raises the inevitable question of how to treat these patients when they relapse, and how to treat these patients in the metastatic setting “having already exhausted single-agent PD-1 therapy,” he commented.
Dr. Hamid said that the current results also reveal the “current problem” with adjuvant therapy, which is that “we don’t know who benefits,” and there is a subset patients who “never recur” even if they are untreated.
So the questions are: “How come all get treated? What about the risks of toxicity? The costs? And where do we fit these patients into clinic?”
As with so many presentations of immunotherapy trial data, the need for biomarkers was raised, with Dr. Hamid emphasizing the need for predictive biomarkers that could exclude patients, and save them from toxicity.
He noted that there were data with another checkpoint inhibitor, nivolumab (Opdivo), in the adjuvant setting (from the CheckMate 238 trial) that suggested higher tumor mutation burden and tumor interferon-gamma levels could play a role, and he hopes that similar data may be available from this latest trial.
Also, there are ongoing and upcoming trials in patients with stage 2B and 2C melanoma that may answer some of the outstanding questions, including a study of neoadjuvant PD-1 blockade before resection, and the DETECTION trial, which is exploring circulating tumor DNA-guided therapy postsurgery.
Then there is the NivoMela trial that will look at nivolumab in stage 2A as well as 2B and 2C disease, while the REFINE trial will assess whether giving immunotherapy less often to patients with advanced cancer, including those with melanoma, results in fewer side effects while continuing to be effective.
The current results also raise the question of whether to go “earlier and earlier” with adjuvant immunotherapy into “poor risk” stage 1 melanoma, which is already being tried in the United States, although there is “no clear understanding of what to do for those patients.”
Overall, Dr. Hamid said that the results of KEYNOTE-716 have “created more questions than answers,” including its impact on the inclusion criteria for phase 3/4 clinical trials, “which now exclude patients who have received adjuvant therapy within 6 months.”
“That will have to change,” he suggested.
Some of the questions raised by Dr. Hamid were discussed on social media, sparking a lively Twitter debate on how best to take the results forward and into the clinic.
Florentia Dimitriou, MD, a dermatology consultant in the Skin Cancer Clinic, University Hospital Zurich, Switzerland, said the data were “great” but she was “still unclear” over who needs adjuvant immunotherapy in this setting.
She also emphasized that, for her, the greater RFS benefit seen in T3b than in T4b disease “doesn’t make sense,” and she also highlighted the finding of long-term toxicity in approximately 18% of patients.
Dr. Luke replied that he agrees that the T3b/T4b results are puzzling but he said the event rate was “low” and the data are “immature,” and that he hopes to have “more info soon.”
He acknowledged that around 18% of patients taking pembrolizumab went on to receive hormone therapy for adverse events, including 13.9% due to hypothyroidism, and others including hypophysitis, adrenal sufficiency, and type 1 diabetes. However, he also pointed out that about 5% of patients in this study had background thyroid issues. The risks and benefits of treatment need to be discussed with patients, he added.
Over a series of tweets, Rebecca J. Lee, PhD, NIHR clinical lecturer in medical oncology at the University of Manchester, United Kingdom, said, “we need to know more” about the distant metastasis-free survival results, and that results for overall survival are “really” needed.
She also emphasized the need for biomarkers to identify those patients who are likely to benefit, and whether benefit can be upfront or early on in treatment. Dr. Lee added that, as endocrine thyroid toxicity occurs after a median of 3.3 months, “pretreatment biomarkers will be more important than on-treatment biomarkers in this setting.”
Details of the results in earlier stage disease
The KEYNOTE-716 trial enrolled patients with newly diagnosed, resected, high-risk stage 2 melanoma aged ≥ 12 years and a good performance status. The majority (~64%) had stage 2B melanoma, and the rest had stage 2C. T3b disease was present in 41% of patients, 23% had T4a disease, and 35% had T4b disease.
Patients were randomized to receive pembrolizumab or placebo.
In a subsequent part of the study, patients with recurrence will be unblended, with either crossover from the placebo to active treatment group or rechallenge with pembrolizumab for up to 2 years.
Presenting the first part, Dr. Luke said that, of 487 patients assigned to pembrolizumab, 483 started treatment, of whom 206 have completed treatment, 133 are still on therapy, and 144 have discontinued.
In the placebo group, 489 patients were assigned and 486 began treatment. Of those, 229 completed treatment, 152 are still ongoing, and 105 discontinued.
The two groups were well balanced in terms of baseline characteristics. The median age was approximately 60 years, with only one patient enrolled who was aged 12-17 years.
At 12 months, the study met its primary endpoint.
Relapse-fee survival was 90.5% in patients treated with pembrolizumab versus 83.1% in the placebo group, at a hazard ratio for recurrence of 0.65 (P = .00658).
“Despite this trial hitting this primary endpoint very early, there are a number of patients who are censored later in the curves,” Dr. Luke said, adding that “we will continue to see these data mature.”
“In fact, it’s our full expectation that curves will continue to separate over time.”
When looking at key subgroups, Dr. Luke showed that the results favored pembrolizumab when stratifying patients by age, gender, race, and performance status.
Interestingly, patients with T3b disease did a lot better on pembrolizumab compared with those who had T4b disease, at a hazard ratio for recurrence of 0.44 versus 0.94.
Data on recurrence patterns revealed that 11.1% of patients taking pembrolizumab had an event, with 6.4% experiencing skin and/or lymph node regional recurrence and 4.7% distant recurrence.
In the placebo group, 16.8% of patients had a recurrence event, with 8.4% having a loco-regional recurrence and 7.8% a distant recurrence.
Dr. Luke explained that this equates to an approximate 40% reduction in distant recurrence with pembrolizumab over placebo.
Finally, the researchers examined change in global health status on the EORTC QLQ-C30 quality of life score. Examining mean change over time, they found that there were no clinically meaningful changes, and the scores in the pembrolizumab and placebo groups tracked each other during the course of follow-up.
Quality of life was, therefore, “only minimally changed,” Dr. Luke said.
The study was funded by MSD. Dr. Luke and Dr. Hamid have declared relationships with multiple companies.
A version of this article first appeared on Medscape.com.