User login
MDedge conference coverage features onsite reporting of the latest study results and expert perspectives from leading researchers.
Continuous cuffless monitoring may fuel lifestyle change to lower BP
Wearing a cuffless device on the wrist to continuously monitor blood pressure was associated with a significantly lower systolic BP at 6 months among hypertensive adults, real-world results from Europe show.
“We don’t know what they did to reduce their blood pressure,” Jay Shah, MD, Division of Cardiology, Mayo Clinic Arizona, Phoenix, told this news organization.
“The idea is that because they were exposed to their data on a continual basis, that may have prompted them to do something that led to an improvement in their blood pressure, whether it be exercise more, go to their doctor, or change their medication,” said Dr. Shah, who is also chief medical officer for Aktiia.
Dr. Shah presented the study at the Hypertension Scientific Sessions, San Diego.
Empowering data
The study used the Aktiia 24/7 BP monitor; Atkiia funded the trial. The monitor passively and continually monitors BP values from photoplethysmography signals collected via optical sensors at the wrist.
After initial individualized calibration using a cuff-based reference, BP measurements are displayed on a smartphone app, allowing users to consistently monitor their own BP for long periods of time.
Aktiia received CE mark in Europe in January 2021 and is currently under review by the U.S. Food and Drug Administration.
Dr. Shah and colleagues analyzed systolic BP (SBP) trends among 838 real-world Aktiia users in Europe (age 57 ± 11 years; 14% women) who consistently used the monitor for 6 months.
Altogether, they had data on 375 (± 287) app interactions, 3,646 (± 1,417) cuffless readings per user, and 9 (± 7) cuff readings per user.
Traditional cuff SBP averages were calculated monthly and compared with the SBP average of the first month. A t-test analysis was used to detect the difference in SBP between the first and successive months.
On the basis of the mean SBP calculated over 6 months, 136 participants were hypertensive (SBP > 140 mm Hg) and the rest had SBP less than 140 mm Hg.
Hypertensive users saw a statistically significant reduction in SBP of –3.2 mm Hg (95% CI, –0.70 to –5.59; P < .02), beginning at 3 months of continual cuffless BP monitoring, which was sustained through 6 months.
Among users with SBP less than 140 mm Hg, the mean SBP remained unchanged.
“The magnitude of improvement might look modest, but even a 5 mm Hg reduction in systolic BP correlates to a 10% decrease in cardiovascular risk,” Dr. Shah told this news organization.
He noted that “one of the major hurdles is that people may not be aware they have high blood pressure because they don’t feel it. And with a regular cuff, they’ll only see that number when they actually check their blood pressure, which is extremely rare, even for people who have hypertension.”
“Having the ability to show someone their continual blood pressure picture really empowers them to do something to make changes and to be aware, [as well as] to be a more active participant in their health,” Dr. Shah said.
He said that a good analogy is diabetes management, which has transitioned from single finger-stick glucose monitoring to continuous glucose monitoring that provides a complete 24/7 picture of glucose levels.
Transforming technology
Offering perspective on the study, Harlan Krumholz, MD, SM, with Yale New Haven Hospital and Yale School of Medicine, New Haven, Conn., said that having an accurate, affordable, unobtrusive cuffless continuous BP monitor would “transform” BP management.

“This could unlock an era of precision BP management with empowerment of patients to view and act on their numbers,” Dr. Krumholz said in an interview.
“We need data to be confident in the devices – and then research to best leverage the streams of information – and strategies to optimize its use in practice,” Dr. Krumholz added.
“Like any new innovation, we need to mitigate risks and monitor for unintended adverse consequences, but I am bullish on the future of cuffless continuous BP monitors,” Dr. Krumholz said.
Dr. Krumholz said that he “applauds Aktiia for doing studies that assess the effect of the information they are producing on BP over time. We need to know that new approaches not only generate valid information but that they can improve health.”
Ready for prime time?
In June, the European Society of Hypertension issued a statement noting that cuffless BP measurement is a fast-growing and promising field with considerable potential for improving hypertension awareness, management, and control, but because the accuracy of these new devices has not yet been validated, they are not yet suitable for clinical use.
Also providing perspective, Stephen Juraschek, MD, PhD, research director, Hypertension Center of Excellence at Healthcare Associates, Beth Israel Deaconess Medical Center, Boston, said that “there is a lot of interest in cuffless BP monitors due to their ease of measurement, comfort, and ability to obtain BP measurements in multiple settings and environments, and this study showed that the monitoring improved BP over time.”
“It is believed that the increased awareness and feedback may promote healthier behaviors aimed at lowering BP. However, this result should not be conflated with the accuracy of these monitors,” Dr. Juraschek cautioned.
He also noted that there is still no formally approved validation protocol by the Association for the Advancement of Medical Instrumentation.
“While a number of cuffless devices are cleared by the FDA through its 510k mechanism (that is, demonstration of device equivalence), there is no formal stamp of approval or attestation that the measurements are accurate,” Dr. Juraschek said in an interview.
In his view, “more work is needed to understand the validity of these devices. For now, validated, cuff-based home devices are recommended for BP measurement at home, while further work is done to determine the accuracy of these cuffless technologies.”
The study was funded by Aktiia. Dr. Shah is an employee of the company. Dr. Krumholz has no relevant disclosures. Dr. Juraschek is a member of the Validate BP review committee and the AAMI sphygmomanometer committee.
A version of this article first appeared on Medscape.com.
Wearing a cuffless device on the wrist to continuously monitor blood pressure was associated with a significantly lower systolic BP at 6 months among hypertensive adults, real-world results from Europe show.
“We don’t know what they did to reduce their blood pressure,” Jay Shah, MD, Division of Cardiology, Mayo Clinic Arizona, Phoenix, told this news organization.
“The idea is that because they were exposed to their data on a continual basis, that may have prompted them to do something that led to an improvement in their blood pressure, whether it be exercise more, go to their doctor, or change their medication,” said Dr. Shah, who is also chief medical officer for Aktiia.
Dr. Shah presented the study at the Hypertension Scientific Sessions, San Diego.
Empowering data
The study used the Aktiia 24/7 BP monitor; Atkiia funded the trial. The monitor passively and continually monitors BP values from photoplethysmography signals collected via optical sensors at the wrist.
After initial individualized calibration using a cuff-based reference, BP measurements are displayed on a smartphone app, allowing users to consistently monitor their own BP for long periods of time.
Aktiia received CE mark in Europe in January 2021 and is currently under review by the U.S. Food and Drug Administration.
Dr. Shah and colleagues analyzed systolic BP (SBP) trends among 838 real-world Aktiia users in Europe (age 57 ± 11 years; 14% women) who consistently used the monitor for 6 months.
Altogether, they had data on 375 (± 287) app interactions, 3,646 (± 1,417) cuffless readings per user, and 9 (± 7) cuff readings per user.
Traditional cuff SBP averages were calculated monthly and compared with the SBP average of the first month. A t-test analysis was used to detect the difference in SBP between the first and successive months.
On the basis of the mean SBP calculated over 6 months, 136 participants were hypertensive (SBP > 140 mm Hg) and the rest had SBP less than 140 mm Hg.
Hypertensive users saw a statistically significant reduction in SBP of –3.2 mm Hg (95% CI, –0.70 to –5.59; P < .02), beginning at 3 months of continual cuffless BP monitoring, which was sustained through 6 months.
Among users with SBP less than 140 mm Hg, the mean SBP remained unchanged.
“The magnitude of improvement might look modest, but even a 5 mm Hg reduction in systolic BP correlates to a 10% decrease in cardiovascular risk,” Dr. Shah told this news organization.
He noted that “one of the major hurdles is that people may not be aware they have high blood pressure because they don’t feel it. And with a regular cuff, they’ll only see that number when they actually check their blood pressure, which is extremely rare, even for people who have hypertension.”
“Having the ability to show someone their continual blood pressure picture really empowers them to do something to make changes and to be aware, [as well as] to be a more active participant in their health,” Dr. Shah said.
He said that a good analogy is diabetes management, which has transitioned from single finger-stick glucose monitoring to continuous glucose monitoring that provides a complete 24/7 picture of glucose levels.
Transforming technology
Offering perspective on the study, Harlan Krumholz, MD, SM, with Yale New Haven Hospital and Yale School of Medicine, New Haven, Conn., said that having an accurate, affordable, unobtrusive cuffless continuous BP monitor would “transform” BP management.
“This could unlock an era of precision BP management with empowerment of patients to view and act on their numbers,” Dr. Krumholz said in an interview.
“We need data to be confident in the devices – and then research to best leverage the streams of information – and strategies to optimize its use in practice,” Dr. Krumholz added.
“Like any new innovation, we need to mitigate risks and monitor for unintended adverse consequences, but I am bullish on the future of cuffless continuous BP monitors,” Dr. Krumholz said.
Dr. Krumholz said that he “applauds Aktiia for doing studies that assess the effect of the information they are producing on BP over time. We need to know that new approaches not only generate valid information but that they can improve health.”
Ready for prime time?
In June, the European Society of Hypertension issued a statement noting that cuffless BP measurement is a fast-growing and promising field with considerable potential for improving hypertension awareness, management, and control, but because the accuracy of these new devices has not yet been validated, they are not yet suitable for clinical use.
Also providing perspective, Stephen Juraschek, MD, PhD, research director, Hypertension Center of Excellence at Healthcare Associates, Beth Israel Deaconess Medical Center, Boston, said that “there is a lot of interest in cuffless BP monitors due to their ease of measurement, comfort, and ability to obtain BP measurements in multiple settings and environments, and this study showed that the monitoring improved BP over time.”
“It is believed that the increased awareness and feedback may promote healthier behaviors aimed at lowering BP. However, this result should not be conflated with the accuracy of these monitors,” Dr. Juraschek cautioned.
He also noted that there is still no formally approved validation protocol by the Association for the Advancement of Medical Instrumentation.
“While a number of cuffless devices are cleared by the FDA through its 510k mechanism (that is, demonstration of device equivalence), there is no formal stamp of approval or attestation that the measurements are accurate,” Dr. Juraschek said in an interview.
In his view, “more work is needed to understand the validity of these devices. For now, validated, cuff-based home devices are recommended for BP measurement at home, while further work is done to determine the accuracy of these cuffless technologies.”
The study was funded by Aktiia. Dr. Shah is an employee of the company. Dr. Krumholz has no relevant disclosures. Dr. Juraschek is a member of the Validate BP review committee and the AAMI sphygmomanometer committee.
A version of this article first appeared on Medscape.com.
Wearing a cuffless device on the wrist to continuously monitor blood pressure was associated with a significantly lower systolic BP at 6 months among hypertensive adults, real-world results from Europe show.
“We don’t know what they did to reduce their blood pressure,” Jay Shah, MD, Division of Cardiology, Mayo Clinic Arizona, Phoenix, told this news organization.
“The idea is that because they were exposed to their data on a continual basis, that may have prompted them to do something that led to an improvement in their blood pressure, whether it be exercise more, go to their doctor, or change their medication,” said Dr. Shah, who is also chief medical officer for Aktiia.
Dr. Shah presented the study at the Hypertension Scientific Sessions, San Diego.
Empowering data
The study used the Aktiia 24/7 BP monitor; Atkiia funded the trial. The monitor passively and continually monitors BP values from photoplethysmography signals collected via optical sensors at the wrist.
After initial individualized calibration using a cuff-based reference, BP measurements are displayed on a smartphone app, allowing users to consistently monitor their own BP for long periods of time.
Aktiia received CE mark in Europe in January 2021 and is currently under review by the U.S. Food and Drug Administration.
Dr. Shah and colleagues analyzed systolic BP (SBP) trends among 838 real-world Aktiia users in Europe (age 57 ± 11 years; 14% women) who consistently used the monitor for 6 months.
Altogether, they had data on 375 (± 287) app interactions, 3,646 (± 1,417) cuffless readings per user, and 9 (± 7) cuff readings per user.
Traditional cuff SBP averages were calculated monthly and compared with the SBP average of the first month. A t-test analysis was used to detect the difference in SBP between the first and successive months.
On the basis of the mean SBP calculated over 6 months, 136 participants were hypertensive (SBP > 140 mm Hg) and the rest had SBP less than 140 mm Hg.
Hypertensive users saw a statistically significant reduction in SBP of –3.2 mm Hg (95% CI, –0.70 to –5.59; P < .02), beginning at 3 months of continual cuffless BP monitoring, which was sustained through 6 months.
Among users with SBP less than 140 mm Hg, the mean SBP remained unchanged.
“The magnitude of improvement might look modest, but even a 5 mm Hg reduction in systolic BP correlates to a 10% decrease in cardiovascular risk,” Dr. Shah told this news organization.
He noted that “one of the major hurdles is that people may not be aware they have high blood pressure because they don’t feel it. And with a regular cuff, they’ll only see that number when they actually check their blood pressure, which is extremely rare, even for people who have hypertension.”
“Having the ability to show someone their continual blood pressure picture really empowers them to do something to make changes and to be aware, [as well as] to be a more active participant in their health,” Dr. Shah said.
He said that a good analogy is diabetes management, which has transitioned from single finger-stick glucose monitoring to continuous glucose monitoring that provides a complete 24/7 picture of glucose levels.
Transforming technology
Offering perspective on the study, Harlan Krumholz, MD, SM, with Yale New Haven Hospital and Yale School of Medicine, New Haven, Conn., said that having an accurate, affordable, unobtrusive cuffless continuous BP monitor would “transform” BP management.
“This could unlock an era of precision BP management with empowerment of patients to view and act on their numbers,” Dr. Krumholz said in an interview.
“We need data to be confident in the devices – and then research to best leverage the streams of information – and strategies to optimize its use in practice,” Dr. Krumholz added.
“Like any new innovation, we need to mitigate risks and monitor for unintended adverse consequences, but I am bullish on the future of cuffless continuous BP monitors,” Dr. Krumholz said.
Dr. Krumholz said that he “applauds Aktiia for doing studies that assess the effect of the information they are producing on BP over time. We need to know that new approaches not only generate valid information but that they can improve health.”
Ready for prime time?
In June, the European Society of Hypertension issued a statement noting that cuffless BP measurement is a fast-growing and promising field with considerable potential for improving hypertension awareness, management, and control, but because the accuracy of these new devices has not yet been validated, they are not yet suitable for clinical use.
Also providing perspective, Stephen Juraschek, MD, PhD, research director, Hypertension Center of Excellence at Healthcare Associates, Beth Israel Deaconess Medical Center, Boston, said that “there is a lot of interest in cuffless BP monitors due to their ease of measurement, comfort, and ability to obtain BP measurements in multiple settings and environments, and this study showed that the monitoring improved BP over time.”
“It is believed that the increased awareness and feedback may promote healthier behaviors aimed at lowering BP. However, this result should not be conflated with the accuracy of these monitors,” Dr. Juraschek cautioned.
He also noted that there is still no formally approved validation protocol by the Association for the Advancement of Medical Instrumentation.
“While a number of cuffless devices are cleared by the FDA through its 510k mechanism (that is, demonstration of device equivalence), there is no formal stamp of approval or attestation that the measurements are accurate,” Dr. Juraschek said in an interview.
In his view, “more work is needed to understand the validity of these devices. For now, validated, cuff-based home devices are recommended for BP measurement at home, while further work is done to determine the accuracy of these cuffless technologies.”
The study was funded by Aktiia. Dr. Shah is an employee of the company. Dr. Krumholz has no relevant disclosures. Dr. Juraschek is a member of the Validate BP review committee and the AAMI sphygmomanometer committee.
A version of this article first appeared on Medscape.com.
FROM AHA HYPERTENSION 2022
Ultrasonic renal denervation passes 2-month test in uncontrolled HTN: RADIANCE II
Systolic blood pressure went down safely and consistently 2 months after renal denervation achieved by ultrasound ablation in patients with uncontrolled, mild to moderate hypertension (HTN) in a key sham-controlled test of the balloon-equipped catheter.
The BP reductions were significant almost regardless of how they were measured – at home, in the office, during the day, at night, or over 24 hours – and weren’t dependent on baseline BP levels.
The 224-patient RADIANCE II Pivotal Study follows two earlier successful sham-controlled trials that used the same renal denervation catheter in other types of patients with HTN. They were RADIANCE-HTN SOLO, which entered patients with mild to moderate HTN not taking medication, and RADIANCE-HTN TRIO, which included patients with HTN despite fixed-dose, single-pill, triple-antihypertensive therapy.

The consistent results of all three trials suggest that the ultrasound renal denervation (uRDN) technique “lowers blood pressure across the spectrum of hypertension,” concluded co–principal investigator Ajay J. Kirtane, MD, SM, Columbia University Irving Medical Center, New York–Presbyterian Hospital, when presenting RADIANCE II at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
RADIANCE II, the largest of the three studies, met its prespecified primary efficacy endpoint of change in daytime ambulatory systolic BP at 2 months by showing a significant 6.3–mm Hg greater reduction in the uRDN group, compared with the sham-control group. There were no major adverse events at 30 days in either group.
The trial was similarly successful for the secondary endpoints of change in systolic BP measured in various other settings, including over 24 hours. Reductions after uRDN averaged 5-7 mm Hg greater than in the control group.
Sparse top-line results of the RADIANCE II pivotal trial were announced in July by the study’s sponsor, ReCor Medical.
Dr. Kirtane stressed in an interview that uRDN and likely any form of HTN renal denervation therapy is not a substitute for standard management. “This is really for patients in whom you’ve made best efforts to do the traditional things – lifestyle modification, medications, all of that – and yet they’re still uncontrolled.” At that point, assuming denervation therapy is available in practice, “it would be something to potentially consider.”
As a panelist after Dr. Kirtane’s formal presentation of RADIANCE II at the conference, Naomi D. Fisher, MD, who was a RADIANCE-HTN TRIO investigator, described how the treatment’s perceived intended patient population evolved over time.
“We all began with the idea that we were going to treat patients with resistant hypertension, that was going to be the first target. We have learned that those patients are far fewer than we thought,” said Dr. Fisher, who directs the hypertension service at Brigham and Women’s Hospital, Boston.
Initial estimates were that such patients with the resistant form, “meaning they require more than three drugs to control their blood pressure,” would represent 15%-20% of patients with HTN.
“We learned from our TRIO data that if you give these patients one single combined pill, lo and behold, many of them become controlled,” she said. “There is so much nonadherence out there in the world, about 50% of our patients aren’t taking their pills. It’s a hard and true fact.”
Exclude patients who aren’t adherent and “our true resistance population becomes minuscule. So, I don’t think that’s going to be the main population” for renal denervation therapy.
More likely, she said, it would be “patients who are uncontrolled and unable to take their medications. So that is going to include nonadherence, intolerance. It’s a very large category of patients. And the priorities can be stacked in favor of those who have higher cardiovascular risk.”
RADIANCE II can show the persistence of uRDN’s BP-lowering effect only out to 2 months so far, but the effect’s durability based on the RADIANCE program’s combined experience appears to be at least 2 years, Dr. Kirtane said in an interview.
“The RADIANCE II pivotal trial is a powerful, well-designed study attesting to the efficacy of renal denervation in BP lowering,” Franz H. Messerli, MD, Swiss Cardiovascular Center, University Hospital Bern, said in an interview.
The trial “shows the well-known unpredictability of antihypertensive response. We cannot predict who responds to renal denervation and who does not, and who even has a paradoxical increase in BP,” Dr. Messerli, an international hypertension expert not associated with the trial, said in an interview.
“As long as we cannot predict the antihypertensive response to renal denervation therapy, potential synergism/antagonism with drug therapy remains an educated guess,” he said.
“Hypertension is a disease that lasts years and decades. As impressive as RADIANCE II’s 2-month snapshot is, I look forward to similar or better BP data 12 and 24 months after renal denervation,” Dr. Messerli added.
RADIANCE II entered patients with mild to moderate uncontrolled HTN, that is, a systolic BP at least 140/90 mm Hg and less than 180/120 mm Hg, who were receiving no more than two antihypertensive medications. They could have no history of cardiovascular or cerebrovascular events or uncontrolled diabetes, and their estimated glomerular filtration rate (eGFR) had to be at least 40 mL/min per 1.73 m2.
After a 4-week drug washout period, patients who were clinically stable with an ambulatory BP of at least 135/85 mm Hg and less than 170/105 mm Hg underwent CT and renal angiography. Then, the 224 patients still anatomically eligible for the procedure were randomly assigned 2:1 to uRDN or a sham-control procedure: 150 and 74 patients, respectively.
At 2 months, daytime ambulatory systolic BP on average fell 7.9 mm Hg in the uRDN group and 1.8 mm Hg in the sham-control group, for a drop that was steeper by 6.3 mm Hg (P < .0001) after uRDN.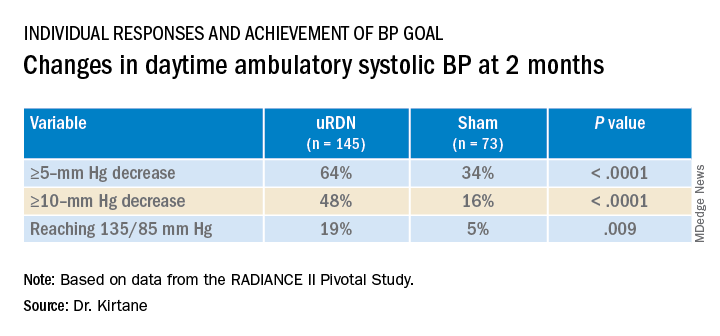
Also in the uRDN group, there was a 6.2–mm Hg larger decrease in 24-hour ambulatory systolic BP (P < .0001), a 5.8–mm Hg greater decline in nighttime ambulatory systolic BP (P < .0004), a 7.6–mm Hg steeper drop in mean home systolic BP (P < .0001), and 5.4 mm Hg more of a decrease in office-based systolic BP (P = .0035).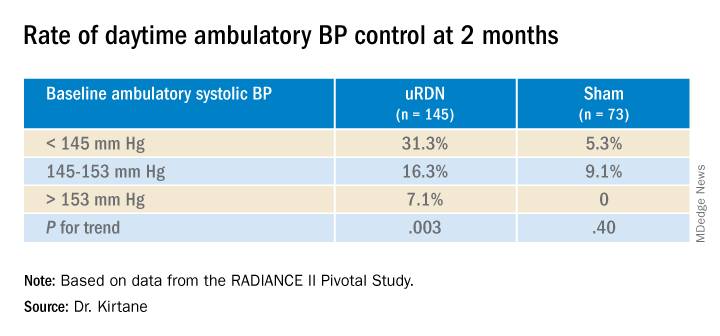
No significant differences were seen in subgroup analyses by sex, age, higher versus lower baseline systolic pressures, high versus low baseline eGFR, degree of abdominal obesity, U.S. versus European site, or whether patients entered before or during the COVID pandemic
Regulators have been accepting change in systolic BP as a surrogate for clinical endpoints in trials of antihypertensive therapy, whether pharmacologic or interventional, under consideration for approval. “That’s why safety endpoints are important to investigate” in these clinical trials, especially for invasive therapies like renal denervation, Dr. Kirtane observed.
That said, “in the longer-term follow-ups of the renal denervation therapies that are out there, including this one, there does not appear to be an appreciable decline in glomerular filtration rate, or any adverse safety signals that we see to date,” Dr. Kirtane said in an interview. “But we know that these are low-frequency events, so we have to be very vigilant, and we can’t get complacent about it.”
In RADIANCE II, there were zero adverse events within 30 days in both groups; the endpoint included death, new myocardial infarction, renal artery complications requiring invasive intervention, and hospitalization for major cardiovascular or hemodynamic-related events. Nor were there instances of new-onset renal artery stenosis greater than 70% documented by imaging at 6 months.
The ReCor uRDN catheter uses ultrasound energy to disrupt renal nerve signaling, a technology thought to deliver safer “burns,” compared with other renal denervation catheter technologies. It features an axially stabilizing balloon that transmits ultrasound energy – two to three sonications, each lasting 7 seconds, Dr. Kirtane said – outward through the arterial wall. The design is intended to ensure consistently circumferential ablation. Circulating saline within the balloon, Kirtane noted, directly cools the adjacent vessel wall to help it avoid thermal damage.
Dr. Kirtane reported receiving institutional funding from Medtronic, Boston Scientific, Abbott Vascular, Amgen, CSI, Philips, ReCor Medical, Neurotronic, Biotronik, Chiesi, Bolt Medical, Magenta Medical, Canon, SoniVie, Shockwave Medical, and Merck; consulting for IMDS; and receiving travel and meal expenses from Medtronic, Boston Scientific, Abbott Vascular, CSI, Siemens, Philips, ReCor Medical, Chiesi, OpSens, Zoll, and Regeneron. Dr. Fisher disclosed receiving honoraria or fees for consulting or serving on a speaker’s bureau for Medtronic, ReCor Medical, and Aktiia and receiving grant support or holding research contracts for Recor Medical and Aktiia. Dr. Messerli disclosed receiving honoraria from Medtronic, Menarini, Krka, and Ipca.
A version of this article first appeared on Medscape.com.
Systolic blood pressure went down safely and consistently 2 months after renal denervation achieved by ultrasound ablation in patients with uncontrolled, mild to moderate hypertension (HTN) in a key sham-controlled test of the balloon-equipped catheter.
The BP reductions were significant almost regardless of how they were measured – at home, in the office, during the day, at night, or over 24 hours – and weren’t dependent on baseline BP levels.
The 224-patient RADIANCE II Pivotal Study follows two earlier successful sham-controlled trials that used the same renal denervation catheter in other types of patients with HTN. They were RADIANCE-HTN SOLO, which entered patients with mild to moderate HTN not taking medication, and RADIANCE-HTN TRIO, which included patients with HTN despite fixed-dose, single-pill, triple-antihypertensive therapy.
The consistent results of all three trials suggest that the ultrasound renal denervation (uRDN) technique “lowers blood pressure across the spectrum of hypertension,” concluded co–principal investigator Ajay J. Kirtane, MD, SM, Columbia University Irving Medical Center, New York–Presbyterian Hospital, when presenting RADIANCE II at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
RADIANCE II, the largest of the three studies, met its prespecified primary efficacy endpoint of change in daytime ambulatory systolic BP at 2 months by showing a significant 6.3–mm Hg greater reduction in the uRDN group, compared with the sham-control group. There were no major adverse events at 30 days in either group.
The trial was similarly successful for the secondary endpoints of change in systolic BP measured in various other settings, including over 24 hours. Reductions after uRDN averaged 5-7 mm Hg greater than in the control group.
Sparse top-line results of the RADIANCE II pivotal trial were announced in July by the study’s sponsor, ReCor Medical.
Dr. Kirtane stressed in an interview that uRDN and likely any form of HTN renal denervation therapy is not a substitute for standard management. “This is really for patients in whom you’ve made best efforts to do the traditional things – lifestyle modification, medications, all of that – and yet they’re still uncontrolled.” At that point, assuming denervation therapy is available in practice, “it would be something to potentially consider.”
As a panelist after Dr. Kirtane’s formal presentation of RADIANCE II at the conference, Naomi D. Fisher, MD, who was a RADIANCE-HTN TRIO investigator, described how the treatment’s perceived intended patient population evolved over time.
“We all began with the idea that we were going to treat patients with resistant hypertension, that was going to be the first target. We have learned that those patients are far fewer than we thought,” said Dr. Fisher, who directs the hypertension service at Brigham and Women’s Hospital, Boston.
Initial estimates were that such patients with the resistant form, “meaning they require more than three drugs to control their blood pressure,” would represent 15%-20% of patients with HTN.
“We learned from our TRIO data that if you give these patients one single combined pill, lo and behold, many of them become controlled,” she said. “There is so much nonadherence out there in the world, about 50% of our patients aren’t taking their pills. It’s a hard and true fact.”
Exclude patients who aren’t adherent and “our true resistance population becomes minuscule. So, I don’t think that’s going to be the main population” for renal denervation therapy.
More likely, she said, it would be “patients who are uncontrolled and unable to take their medications. So that is going to include nonadherence, intolerance. It’s a very large category of patients. And the priorities can be stacked in favor of those who have higher cardiovascular risk.”
RADIANCE II can show the persistence of uRDN’s BP-lowering effect only out to 2 months so far, but the effect’s durability based on the RADIANCE program’s combined experience appears to be at least 2 years, Dr. Kirtane said in an interview.
“The RADIANCE II pivotal trial is a powerful, well-designed study attesting to the efficacy of renal denervation in BP lowering,” Franz H. Messerli, MD, Swiss Cardiovascular Center, University Hospital Bern, said in an interview.
The trial “shows the well-known unpredictability of antihypertensive response. We cannot predict who responds to renal denervation and who does not, and who even has a paradoxical increase in BP,” Dr. Messerli, an international hypertension expert not associated with the trial, said in an interview.
“As long as we cannot predict the antihypertensive response to renal denervation therapy, potential synergism/antagonism with drug therapy remains an educated guess,” he said.
“Hypertension is a disease that lasts years and decades. As impressive as RADIANCE II’s 2-month snapshot is, I look forward to similar or better BP data 12 and 24 months after renal denervation,” Dr. Messerli added.
RADIANCE II entered patients with mild to moderate uncontrolled HTN, that is, a systolic BP at least 140/90 mm Hg and less than 180/120 mm Hg, who were receiving no more than two antihypertensive medications. They could have no history of cardiovascular or cerebrovascular events or uncontrolled diabetes, and their estimated glomerular filtration rate (eGFR) had to be at least 40 mL/min per 1.73 m2.
After a 4-week drug washout period, patients who were clinically stable with an ambulatory BP of at least 135/85 mm Hg and less than 170/105 mm Hg underwent CT and renal angiography. Then, the 224 patients still anatomically eligible for the procedure were randomly assigned 2:1 to uRDN or a sham-control procedure: 150 and 74 patients, respectively.
At 2 months, daytime ambulatory systolic BP on average fell 7.9 mm Hg in the uRDN group and 1.8 mm Hg in the sham-control group, for a drop that was steeper by 6.3 mm Hg (P < .0001) after uRDN.
Also in the uRDN group, there was a 6.2–mm Hg larger decrease in 24-hour ambulatory systolic BP (P < .0001), a 5.8–mm Hg greater decline in nighttime ambulatory systolic BP (P < .0004), a 7.6–mm Hg steeper drop in mean home systolic BP (P < .0001), and 5.4 mm Hg more of a decrease in office-based systolic BP (P = .0035).
No significant differences were seen in subgroup analyses by sex, age, higher versus lower baseline systolic pressures, high versus low baseline eGFR, degree of abdominal obesity, U.S. versus European site, or whether patients entered before or during the COVID pandemic
Regulators have been accepting change in systolic BP as a surrogate for clinical endpoints in trials of antihypertensive therapy, whether pharmacologic or interventional, under consideration for approval. “That’s why safety endpoints are important to investigate” in these clinical trials, especially for invasive therapies like renal denervation, Dr. Kirtane observed.
That said, “in the longer-term follow-ups of the renal denervation therapies that are out there, including this one, there does not appear to be an appreciable decline in glomerular filtration rate, or any adverse safety signals that we see to date,” Dr. Kirtane said in an interview. “But we know that these are low-frequency events, so we have to be very vigilant, and we can’t get complacent about it.”
In RADIANCE II, there were zero adverse events within 30 days in both groups; the endpoint included death, new myocardial infarction, renal artery complications requiring invasive intervention, and hospitalization for major cardiovascular or hemodynamic-related events. Nor were there instances of new-onset renal artery stenosis greater than 70% documented by imaging at 6 months.
The ReCor uRDN catheter uses ultrasound energy to disrupt renal nerve signaling, a technology thought to deliver safer “burns,” compared with other renal denervation catheter technologies. It features an axially stabilizing balloon that transmits ultrasound energy – two to three sonications, each lasting 7 seconds, Dr. Kirtane said – outward through the arterial wall. The design is intended to ensure consistently circumferential ablation. Circulating saline within the balloon, Kirtane noted, directly cools the adjacent vessel wall to help it avoid thermal damage.
Dr. Kirtane reported receiving institutional funding from Medtronic, Boston Scientific, Abbott Vascular, Amgen, CSI, Philips, ReCor Medical, Neurotronic, Biotronik, Chiesi, Bolt Medical, Magenta Medical, Canon, SoniVie, Shockwave Medical, and Merck; consulting for IMDS; and receiving travel and meal expenses from Medtronic, Boston Scientific, Abbott Vascular, CSI, Siemens, Philips, ReCor Medical, Chiesi, OpSens, Zoll, and Regeneron. Dr. Fisher disclosed receiving honoraria or fees for consulting or serving on a speaker’s bureau for Medtronic, ReCor Medical, and Aktiia and receiving grant support or holding research contracts for Recor Medical and Aktiia. Dr. Messerli disclosed receiving honoraria from Medtronic, Menarini, Krka, and Ipca.
A version of this article first appeared on Medscape.com.
Systolic blood pressure went down safely and consistently 2 months after renal denervation achieved by ultrasound ablation in patients with uncontrolled, mild to moderate hypertension (HTN) in a key sham-controlled test of the balloon-equipped catheter.
The BP reductions were significant almost regardless of how they were measured – at home, in the office, during the day, at night, or over 24 hours – and weren’t dependent on baseline BP levels.
The 224-patient RADIANCE II Pivotal Study follows two earlier successful sham-controlled trials that used the same renal denervation catheter in other types of patients with HTN. They were RADIANCE-HTN SOLO, which entered patients with mild to moderate HTN not taking medication, and RADIANCE-HTN TRIO, which included patients with HTN despite fixed-dose, single-pill, triple-antihypertensive therapy.
The consistent results of all three trials suggest that the ultrasound renal denervation (uRDN) technique “lowers blood pressure across the spectrum of hypertension,” concluded co–principal investigator Ajay J. Kirtane, MD, SM, Columbia University Irving Medical Center, New York–Presbyterian Hospital, when presenting RADIANCE II at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
RADIANCE II, the largest of the three studies, met its prespecified primary efficacy endpoint of change in daytime ambulatory systolic BP at 2 months by showing a significant 6.3–mm Hg greater reduction in the uRDN group, compared with the sham-control group. There were no major adverse events at 30 days in either group.
The trial was similarly successful for the secondary endpoints of change in systolic BP measured in various other settings, including over 24 hours. Reductions after uRDN averaged 5-7 mm Hg greater than in the control group.
Sparse top-line results of the RADIANCE II pivotal trial were announced in July by the study’s sponsor, ReCor Medical.
Dr. Kirtane stressed in an interview that uRDN and likely any form of HTN renal denervation therapy is not a substitute for standard management. “This is really for patients in whom you’ve made best efforts to do the traditional things – lifestyle modification, medications, all of that – and yet they’re still uncontrolled.” At that point, assuming denervation therapy is available in practice, “it would be something to potentially consider.”
As a panelist after Dr. Kirtane’s formal presentation of RADIANCE II at the conference, Naomi D. Fisher, MD, who was a RADIANCE-HTN TRIO investigator, described how the treatment’s perceived intended patient population evolved over time.
“We all began with the idea that we were going to treat patients with resistant hypertension, that was going to be the first target. We have learned that those patients are far fewer than we thought,” said Dr. Fisher, who directs the hypertension service at Brigham and Women’s Hospital, Boston.
Initial estimates were that such patients with the resistant form, “meaning they require more than three drugs to control their blood pressure,” would represent 15%-20% of patients with HTN.
“We learned from our TRIO data that if you give these patients one single combined pill, lo and behold, many of them become controlled,” she said. “There is so much nonadherence out there in the world, about 50% of our patients aren’t taking their pills. It’s a hard and true fact.”
Exclude patients who aren’t adherent and “our true resistance population becomes minuscule. So, I don’t think that’s going to be the main population” for renal denervation therapy.
More likely, she said, it would be “patients who are uncontrolled and unable to take their medications. So that is going to include nonadherence, intolerance. It’s a very large category of patients. And the priorities can be stacked in favor of those who have higher cardiovascular risk.”
RADIANCE II can show the persistence of uRDN’s BP-lowering effect only out to 2 months so far, but the effect’s durability based on the RADIANCE program’s combined experience appears to be at least 2 years, Dr. Kirtane said in an interview.
“The RADIANCE II pivotal trial is a powerful, well-designed study attesting to the efficacy of renal denervation in BP lowering,” Franz H. Messerli, MD, Swiss Cardiovascular Center, University Hospital Bern, said in an interview.
The trial “shows the well-known unpredictability of antihypertensive response. We cannot predict who responds to renal denervation and who does not, and who even has a paradoxical increase in BP,” Dr. Messerli, an international hypertension expert not associated with the trial, said in an interview.
“As long as we cannot predict the antihypertensive response to renal denervation therapy, potential synergism/antagonism with drug therapy remains an educated guess,” he said.
“Hypertension is a disease that lasts years and decades. As impressive as RADIANCE II’s 2-month snapshot is, I look forward to similar or better BP data 12 and 24 months after renal denervation,” Dr. Messerli added.
RADIANCE II entered patients with mild to moderate uncontrolled HTN, that is, a systolic BP at least 140/90 mm Hg and less than 180/120 mm Hg, who were receiving no more than two antihypertensive medications. They could have no history of cardiovascular or cerebrovascular events or uncontrolled diabetes, and their estimated glomerular filtration rate (eGFR) had to be at least 40 mL/min per 1.73 m2.
After a 4-week drug washout period, patients who were clinically stable with an ambulatory BP of at least 135/85 mm Hg and less than 170/105 mm Hg underwent CT and renal angiography. Then, the 224 patients still anatomically eligible for the procedure were randomly assigned 2:1 to uRDN or a sham-control procedure: 150 and 74 patients, respectively.
At 2 months, daytime ambulatory systolic BP on average fell 7.9 mm Hg in the uRDN group and 1.8 mm Hg in the sham-control group, for a drop that was steeper by 6.3 mm Hg (P < .0001) after uRDN.
Also in the uRDN group, there was a 6.2–mm Hg larger decrease in 24-hour ambulatory systolic BP (P < .0001), a 5.8–mm Hg greater decline in nighttime ambulatory systolic BP (P < .0004), a 7.6–mm Hg steeper drop in mean home systolic BP (P < .0001), and 5.4 mm Hg more of a decrease in office-based systolic BP (P = .0035).
No significant differences were seen in subgroup analyses by sex, age, higher versus lower baseline systolic pressures, high versus low baseline eGFR, degree of abdominal obesity, U.S. versus European site, or whether patients entered before or during the COVID pandemic
Regulators have been accepting change in systolic BP as a surrogate for clinical endpoints in trials of antihypertensive therapy, whether pharmacologic or interventional, under consideration for approval. “That’s why safety endpoints are important to investigate” in these clinical trials, especially for invasive therapies like renal denervation, Dr. Kirtane observed.
That said, “in the longer-term follow-ups of the renal denervation therapies that are out there, including this one, there does not appear to be an appreciable decline in glomerular filtration rate, or any adverse safety signals that we see to date,” Dr. Kirtane said in an interview. “But we know that these are low-frequency events, so we have to be very vigilant, and we can’t get complacent about it.”
In RADIANCE II, there were zero adverse events within 30 days in both groups; the endpoint included death, new myocardial infarction, renal artery complications requiring invasive intervention, and hospitalization for major cardiovascular or hemodynamic-related events. Nor were there instances of new-onset renal artery stenosis greater than 70% documented by imaging at 6 months.
The ReCor uRDN catheter uses ultrasound energy to disrupt renal nerve signaling, a technology thought to deliver safer “burns,” compared with other renal denervation catheter technologies. It features an axially stabilizing balloon that transmits ultrasound energy – two to three sonications, each lasting 7 seconds, Dr. Kirtane said – outward through the arterial wall. The design is intended to ensure consistently circumferential ablation. Circulating saline within the balloon, Kirtane noted, directly cools the adjacent vessel wall to help it avoid thermal damage.
Dr. Kirtane reported receiving institutional funding from Medtronic, Boston Scientific, Abbott Vascular, Amgen, CSI, Philips, ReCor Medical, Neurotronic, Biotronik, Chiesi, Bolt Medical, Magenta Medical, Canon, SoniVie, Shockwave Medical, and Merck; consulting for IMDS; and receiving travel and meal expenses from Medtronic, Boston Scientific, Abbott Vascular, CSI, Siemens, Philips, ReCor Medical, Chiesi, OpSens, Zoll, and Regeneron. Dr. Fisher disclosed receiving honoraria or fees for consulting or serving on a speaker’s bureau for Medtronic, ReCor Medical, and Aktiia and receiving grant support or holding research contracts for Recor Medical and Aktiia. Dr. Messerli disclosed receiving honoraria from Medtronic, Menarini, Krka, and Ipca.
A version of this article first appeared on Medscape.com.
FROM TCT 2022
TAVR now used in almost 50% of younger severe aortic stenosis patients
Among patients with severe isolated aortic stenosis younger than 65, the rate of transcatheter aortic valve replacement (TAVR) now almost matches that of surgical aortic valve replacement (SAVR), despite guideline recommendations to the contrary, a study in a national U.S. population shows.
The 2020 American Heart Association/American College of Cardiology (AHA/ACC) valve guideline recommends SAVR for patients younger than 65 with severe aortic stenosis, the researchers note, but their study showed “near equal utilization between TAVR and SAVR in these younger patients by 2021,” at 48% and 52% respectively.
Toishi Sharma, MD, and colleagues presented these findings in an oral poster session at Transcatheter Cardiovascular Therapeutics 2022, and the study was simultaneously published as a Research Letter in the Journal of the American College of Cardiology (JACC).
“To our knowledge, the current findings represent the first national temporal trends study stratifying [aortic stenosis] therapies according to guideline-recommended age groups: our observations demonstrate the dramatic growth of TAVR in all age groups, including young patients,” the researchers conclude.
They analyzed changes in rates of TAVR and SAVR in a U.S. sample stratified by age: younger than 65 years, 65-80, and older than 80 years.
These findings have implications for lifetime management of younger patients who undergo TAVR, they write, “including issues related to lifetime coronary access, valve durability, and the potential for subsequent TAVR procedures over time.”
Three age groups
In a study published in JACC, this group examined changes in uptake of TAVR versus SAVR in 4,161 patients with aortic stenosis in Vermont, New Hampshire, and Maine, senior author Harold L. Dauerman, MD, said in an interview.
The greatest rate of rise of TAVR was in the group younger than 65, but that study ended in 2019, said Dr. Dauerman, from the University of Vermont Health Network, Burlington.
The 2020 guideline stratifies TAVR and SAVR recommendations such that “less than 65 should primarily be a surgical approach and greater than 80 primarily a TAVR approach, while 65 to 80 is a gray zone, and shared decision-making becomes important,” he noted.
The group hypothesized that recent trials and technology have led to a national increase in TAVR in people younger than 65.
From the Vizient clinical database, including more than 250 U.S. academic centers that perform both TAVR and SAVR, the researchers identified 142,953 patients who underwent TAVR or SAVR for isolated aortic stenosis from Oct. 1, 2015, to Dec. 31, 2021. From 2015 to 2021, the valve replacement rates in the three age groups changed as follows:
- Age less than 65: TAVR rose from 17% to 48%; SAVR fell from 83% to 52%.
- Age 65-80: TAVR rose from 46% to 87%; SAVR fell from 54% to 12%.
- Age greater than 80: TAVR rose from 83% to 99%; SAVR fell from 16% to 1%.
“All ages have grown in the last 7 years in TAVR,” Dr. Dauerman summarized. “The one that’s surprising, and in contradiction to the guideline, is the growth of TAVR in young patients less than 65.”
Among patients younger than 65, prior bypass surgery and congestive heart failure predicted the use of TAVR instead of surgery, whereas bicuspid aortic valve disease was the biggest predictor of surgery instead of TAVR.
Most studies on TAVR valve durability are limited to patients in the randomized trials who were primarily in their mid-70s to mid-80s, some of whom died before a 10-year follow-up, Dr. Dauerman noted.
European guidelines recommend surgery for patients younger than 70, and it would be interesting to see if clinicians there follow this recommendation or if TAVR is now the preferred approach, he added.
There is a need for further, longer study of TAVR in younger patients, he said, to determine whether there are long-term clinical issues of concern.
Strategy depends on more than age
The “findings are not too surprising,” John Carroll, MD, who was not involved in this research, said in an email.

“Age is only one of multiple patient characteristics that enter into consideration of TAVR versus SAVR,” said Dr. Carroll, from Anschutz Medical Campus, University of Colorado, Aurora.
“As the article reports,” he noted, “those less than 65 having TAVR are more likely to have comorbid conditions that likely made the risk of SAVR higher.”
Dr. Carroll was lead author of a review article published in 2020 based on data from the ACC–Society of Thoracic Surgeons (STS)–Transcatheter Valve Therapy (TVT) registry on 276,316 patients who had TAVR in the United States from 2011-2019.
He pointed out that Figure 2 in that review shows that “SAVR is often performed in conjunction with other surgical procedures – another major reason why SAVR remains an important treatment for valvular heart disease.”
“We are awaiting long-term data comparing TAVR to SAVR durability,” Dr. Carroll added, echoing Dr. Dauerman. “So far [there are] no major differences, but it remains a key need to fully understand TAVR and the various models in commercial use.”
“Both TAVR and SAVR used in adults are tissue valves (SAVR with mechanical valves is used in younger patients),” Dr. Carroll noted, “and all tissue valves will eventually fail if the patient lives long enough.”
Patient management strategies need to consider what treatment options exist when the first valve fails. “If the first valve is SAVR, there is now extensive experience with placing a TAVR valve inside a failing SAVR valve, so called Valve-in-Valve or TAVR-in-SAVR. This is the preferred treatment in most patients with failing SAVR valves,” he said.
“On the other hand,” he continued, “we are just beginning to see more and more patients with failing TAVR valves, and the TAVR-in-TAVR procedure is less well understood.”
“Issues such as acute coronary occlusion and long-term difficulty in accessing coronary arteries are being encountered in some patients having TAVR-in-TAVR,” Dr. Carroll noted, which he discusses in a recent editorial he coauthored about the complexities of redo TAVR, published in JACC: Cardiovascular Interventions.
The study received no funding. Dr. Dauerman has research grants and is a consultant for Medtronic and Boston Scientific. Dr. Carroll is a local principal investigator in trials sponsored by Medtronic, Abbott, and Edwards Lifesciences.
A version of this article first appeared on Medscape.com.
Among patients with severe isolated aortic stenosis younger than 65, the rate of transcatheter aortic valve replacement (TAVR) now almost matches that of surgical aortic valve replacement (SAVR), despite guideline recommendations to the contrary, a study in a national U.S. population shows.
The 2020 American Heart Association/American College of Cardiology (AHA/ACC) valve guideline recommends SAVR for patients younger than 65 with severe aortic stenosis, the researchers note, but their study showed “near equal utilization between TAVR and SAVR in these younger patients by 2021,” at 48% and 52% respectively.
Toishi Sharma, MD, and colleagues presented these findings in an oral poster session at Transcatheter Cardiovascular Therapeutics 2022, and the study was simultaneously published as a Research Letter in the Journal of the American College of Cardiology (JACC).
“To our knowledge, the current findings represent the first national temporal trends study stratifying [aortic stenosis] therapies according to guideline-recommended age groups: our observations demonstrate the dramatic growth of TAVR in all age groups, including young patients,” the researchers conclude.
They analyzed changes in rates of TAVR and SAVR in a U.S. sample stratified by age: younger than 65 years, 65-80, and older than 80 years.
These findings have implications for lifetime management of younger patients who undergo TAVR, they write, “including issues related to lifetime coronary access, valve durability, and the potential for subsequent TAVR procedures over time.”
Three age groups
In a study published in JACC, this group examined changes in uptake of TAVR versus SAVR in 4,161 patients with aortic stenosis in Vermont, New Hampshire, and Maine, senior author Harold L. Dauerman, MD, said in an interview.
The greatest rate of rise of TAVR was in the group younger than 65, but that study ended in 2019, said Dr. Dauerman, from the University of Vermont Health Network, Burlington.
The 2020 guideline stratifies TAVR and SAVR recommendations such that “less than 65 should primarily be a surgical approach and greater than 80 primarily a TAVR approach, while 65 to 80 is a gray zone, and shared decision-making becomes important,” he noted.
The group hypothesized that recent trials and technology have led to a national increase in TAVR in people younger than 65.
From the Vizient clinical database, including more than 250 U.S. academic centers that perform both TAVR and SAVR, the researchers identified 142,953 patients who underwent TAVR or SAVR for isolated aortic stenosis from Oct. 1, 2015, to Dec. 31, 2021. From 2015 to 2021, the valve replacement rates in the three age groups changed as follows:
- Age less than 65: TAVR rose from 17% to 48%; SAVR fell from 83% to 52%.
- Age 65-80: TAVR rose from 46% to 87%; SAVR fell from 54% to 12%.
- Age greater than 80: TAVR rose from 83% to 99%; SAVR fell from 16% to 1%.
“All ages have grown in the last 7 years in TAVR,” Dr. Dauerman summarized. “The one that’s surprising, and in contradiction to the guideline, is the growth of TAVR in young patients less than 65.”
Among patients younger than 65, prior bypass surgery and congestive heart failure predicted the use of TAVR instead of surgery, whereas bicuspid aortic valve disease was the biggest predictor of surgery instead of TAVR.
Most studies on TAVR valve durability are limited to patients in the randomized trials who were primarily in their mid-70s to mid-80s, some of whom died before a 10-year follow-up, Dr. Dauerman noted.
European guidelines recommend surgery for patients younger than 70, and it would be interesting to see if clinicians there follow this recommendation or if TAVR is now the preferred approach, he added.
There is a need for further, longer study of TAVR in younger patients, he said, to determine whether there are long-term clinical issues of concern.
Strategy depends on more than age
The “findings are not too surprising,” John Carroll, MD, who was not involved in this research, said in an email.
“Age is only one of multiple patient characteristics that enter into consideration of TAVR versus SAVR,” said Dr. Carroll, from Anschutz Medical Campus, University of Colorado, Aurora.
“As the article reports,” he noted, “those less than 65 having TAVR are more likely to have comorbid conditions that likely made the risk of SAVR higher.”
Dr. Carroll was lead author of a review article published in 2020 based on data from the ACC–Society of Thoracic Surgeons (STS)–Transcatheter Valve Therapy (TVT) registry on 276,316 patients who had TAVR in the United States from 2011-2019.
He pointed out that Figure 2 in that review shows that “SAVR is often performed in conjunction with other surgical procedures – another major reason why SAVR remains an important treatment for valvular heart disease.”
“We are awaiting long-term data comparing TAVR to SAVR durability,” Dr. Carroll added, echoing Dr. Dauerman. “So far [there are] no major differences, but it remains a key need to fully understand TAVR and the various models in commercial use.”
“Both TAVR and SAVR used in adults are tissue valves (SAVR with mechanical valves is used in younger patients),” Dr. Carroll noted, “and all tissue valves will eventually fail if the patient lives long enough.”
Patient management strategies need to consider what treatment options exist when the first valve fails. “If the first valve is SAVR, there is now extensive experience with placing a TAVR valve inside a failing SAVR valve, so called Valve-in-Valve or TAVR-in-SAVR. This is the preferred treatment in most patients with failing SAVR valves,” he said.
“On the other hand,” he continued, “we are just beginning to see more and more patients with failing TAVR valves, and the TAVR-in-TAVR procedure is less well understood.”
“Issues such as acute coronary occlusion and long-term difficulty in accessing coronary arteries are being encountered in some patients having TAVR-in-TAVR,” Dr. Carroll noted, which he discusses in a recent editorial he coauthored about the complexities of redo TAVR, published in JACC: Cardiovascular Interventions.
The study received no funding. Dr. Dauerman has research grants and is a consultant for Medtronic and Boston Scientific. Dr. Carroll is a local principal investigator in trials sponsored by Medtronic, Abbott, and Edwards Lifesciences.
A version of this article first appeared on Medscape.com.
Among patients with severe isolated aortic stenosis younger than 65, the rate of transcatheter aortic valve replacement (TAVR) now almost matches that of surgical aortic valve replacement (SAVR), despite guideline recommendations to the contrary, a study in a national U.S. population shows.
The 2020 American Heart Association/American College of Cardiology (AHA/ACC) valve guideline recommends SAVR for patients younger than 65 with severe aortic stenosis, the researchers note, but their study showed “near equal utilization between TAVR and SAVR in these younger patients by 2021,” at 48% and 52% respectively.
Toishi Sharma, MD, and colleagues presented these findings in an oral poster session at Transcatheter Cardiovascular Therapeutics 2022, and the study was simultaneously published as a Research Letter in the Journal of the American College of Cardiology (JACC).
“To our knowledge, the current findings represent the first national temporal trends study stratifying [aortic stenosis] therapies according to guideline-recommended age groups: our observations demonstrate the dramatic growth of TAVR in all age groups, including young patients,” the researchers conclude.
They analyzed changes in rates of TAVR and SAVR in a U.S. sample stratified by age: younger than 65 years, 65-80, and older than 80 years.
These findings have implications for lifetime management of younger patients who undergo TAVR, they write, “including issues related to lifetime coronary access, valve durability, and the potential for subsequent TAVR procedures over time.”
Three age groups
In a study published in JACC, this group examined changes in uptake of TAVR versus SAVR in 4,161 patients with aortic stenosis in Vermont, New Hampshire, and Maine, senior author Harold L. Dauerman, MD, said in an interview.
The greatest rate of rise of TAVR was in the group younger than 65, but that study ended in 2019, said Dr. Dauerman, from the University of Vermont Health Network, Burlington.
The 2020 guideline stratifies TAVR and SAVR recommendations such that “less than 65 should primarily be a surgical approach and greater than 80 primarily a TAVR approach, while 65 to 80 is a gray zone, and shared decision-making becomes important,” he noted.
The group hypothesized that recent trials and technology have led to a national increase in TAVR in people younger than 65.
From the Vizient clinical database, including more than 250 U.S. academic centers that perform both TAVR and SAVR, the researchers identified 142,953 patients who underwent TAVR or SAVR for isolated aortic stenosis from Oct. 1, 2015, to Dec. 31, 2021. From 2015 to 2021, the valve replacement rates in the three age groups changed as follows:
- Age less than 65: TAVR rose from 17% to 48%; SAVR fell from 83% to 52%.
- Age 65-80: TAVR rose from 46% to 87%; SAVR fell from 54% to 12%.
- Age greater than 80: TAVR rose from 83% to 99%; SAVR fell from 16% to 1%.
“All ages have grown in the last 7 years in TAVR,” Dr. Dauerman summarized. “The one that’s surprising, and in contradiction to the guideline, is the growth of TAVR in young patients less than 65.”
Among patients younger than 65, prior bypass surgery and congestive heart failure predicted the use of TAVR instead of surgery, whereas bicuspid aortic valve disease was the biggest predictor of surgery instead of TAVR.
Most studies on TAVR valve durability are limited to patients in the randomized trials who were primarily in their mid-70s to mid-80s, some of whom died before a 10-year follow-up, Dr. Dauerman noted.
European guidelines recommend surgery for patients younger than 70, and it would be interesting to see if clinicians there follow this recommendation or if TAVR is now the preferred approach, he added.
There is a need for further, longer study of TAVR in younger patients, he said, to determine whether there are long-term clinical issues of concern.
Strategy depends on more than age
The “findings are not too surprising,” John Carroll, MD, who was not involved in this research, said in an email.
“Age is only one of multiple patient characteristics that enter into consideration of TAVR versus SAVR,” said Dr. Carroll, from Anschutz Medical Campus, University of Colorado, Aurora.
“As the article reports,” he noted, “those less than 65 having TAVR are more likely to have comorbid conditions that likely made the risk of SAVR higher.”
Dr. Carroll was lead author of a review article published in 2020 based on data from the ACC–Society of Thoracic Surgeons (STS)–Transcatheter Valve Therapy (TVT) registry on 276,316 patients who had TAVR in the United States from 2011-2019.
He pointed out that Figure 2 in that review shows that “SAVR is often performed in conjunction with other surgical procedures – another major reason why SAVR remains an important treatment for valvular heart disease.”
“We are awaiting long-term data comparing TAVR to SAVR durability,” Dr. Carroll added, echoing Dr. Dauerman. “So far [there are] no major differences, but it remains a key need to fully understand TAVR and the various models in commercial use.”
“Both TAVR and SAVR used in adults are tissue valves (SAVR with mechanical valves is used in younger patients),” Dr. Carroll noted, “and all tissue valves will eventually fail if the patient lives long enough.”
Patient management strategies need to consider what treatment options exist when the first valve fails. “If the first valve is SAVR, there is now extensive experience with placing a TAVR valve inside a failing SAVR valve, so called Valve-in-Valve or TAVR-in-SAVR. This is the preferred treatment in most patients with failing SAVR valves,” he said.
“On the other hand,” he continued, “we are just beginning to see more and more patients with failing TAVR valves, and the TAVR-in-TAVR procedure is less well understood.”
“Issues such as acute coronary occlusion and long-term difficulty in accessing coronary arteries are being encountered in some patients having TAVR-in-TAVR,” Dr. Carroll noted, which he discusses in a recent editorial he coauthored about the complexities of redo TAVR, published in JACC: Cardiovascular Interventions.
The study received no funding. Dr. Dauerman has research grants and is a consultant for Medtronic and Boston Scientific. Dr. Carroll is a local principal investigator in trials sponsored by Medtronic, Abbott, and Edwards Lifesciences.
A version of this article first appeared on Medscape.com.
FROM TCT 2022
Obstructive sleep apnea linked to unprovoked VTE
Add unprovoked venous thromboembolic events to the list of potential consequences of severe obstructive sleep apnea.
That conclusion comes from a study showing that patients with obstructive sleep apnea (OSA) who had the longest nocturnal hypoxemia episodes had a twofold risk for venous thromboembolic events.
systems, reported Wojciech Trzepizur, MD, of Angers University Hospital, France.
Previous studies have suggested links between OSA and both cancer and cognitive decline, but this is the first study to investigate the association between OSA and the incidence of unprovoked VTE, he reported in an oral abstract session at the annual congress of the European Respiratory Society.
“We found that those who spent more than 6% of their nighttime with levels of oxygen in their blood below 90% of normal had an almost twofold risk of developing VTEs compared to patients without oxygen deprivation,” he said.
Dr. Trzepizur and colleagues conducted a retrospective study linking cohort data to an administrative health database. They identified unprovoked VTE in patients with a suspicion for OSA and no previous VTE.
They created Cox proportional hazard models to assess the association of unprovoked VTE with apnea hypopnea index (AHI) measures and nocturnal hypoxemia markers, including the time patients spent below 90% oxygen saturation (T90), oxygen desaturation index (ODI), and hypoxic burden, defined as the total area under the respiratory event-related desaturation curve.
They found that after a median follow-up of 6.3 years, 104 out of 7,355 patients had an unprovoked VTE. In an unadjusted hazard model, there were significant associations between VTE and T90, as well as with hypoxic burden, but not with either AHI or ODI.
However, in an analysis adjusted for age, gender, body mass index, alcohol intake, hypertension, depression, history of cardiovascular disease, statin use, type of sleep study, study site, and CPAP adherence, the investigators found that only T90 remained a significant independent predictor of VTE, with a hazard ratio of 1.06, P = .02.
The association between T90 and VTE strengthened as the time spent below 90% saturation increased. Patients in the highest tercile, who spent more than 6% of the time undersaturated, had an HR for VTE of 1.95 (P = .02), compared with patients with a T90 less than 1%.
There were no significant differences in VTE risk between patients who used CPAP for more than 4 hours per night and those who either used the devices for less than 4 hours or refused CPAP.
“We see that T90 seems to be a strong parameter,” said session comoderator Raphael Heinzer, MD, MPH, of Lausanne University Hospital, Switzerland.
Dr. Heinzer’s comoderator, Silke Ryan, MD, of University College Dublin, pointed out that although T90 was the main predictor of responses, Dr. Trzepizur and colleagues did not control for other pulmonary diseases.
“Obviously, there could be an influence of other hypoxic-related diseases,” she said, and recommended controlling for this in future studies.
Winfried Randerath, MD, of the Bethanien Hospital at the University of Cologne, Germany, head of the ERS specialist group on sleep disordered breathing, said that this study and others presented at the meeting “show worrying associations between obstructive sleep apnea and important diseases that affect survival and quality of life.
“While they cannot prove that OSA causes any of these health problems, people should be made aware of these links and should try to make lifestyle changes in order to reduce their risk of OSA, for instance, by maintaining a healthy weight. However, if OSA is suspected, definite diagnosis and treatment should be initiated. We look forward to further research that may help to clarify whether OSA may be causing some of the health problems seen in these studies,” said Dr. Randerath, who was not involved with the study.
The study was supported by a grant from Institut de Recherche en Santé Respiratoire des Pays de la Loire (IRSR), Beaucouzé, France. Dr. Trzepizur, Dr. Heinzer, Dr. Ryan and Dr. Randerath reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Add unprovoked venous thromboembolic events to the list of potential consequences of severe obstructive sleep apnea.
That conclusion comes from a study showing that patients with obstructive sleep apnea (OSA) who had the longest nocturnal hypoxemia episodes had a twofold risk for venous thromboembolic events.
systems, reported Wojciech Trzepizur, MD, of Angers University Hospital, France.
Previous studies have suggested links between OSA and both cancer and cognitive decline, but this is the first study to investigate the association between OSA and the incidence of unprovoked VTE, he reported in an oral abstract session at the annual congress of the European Respiratory Society.
“We found that those who spent more than 6% of their nighttime with levels of oxygen in their blood below 90% of normal had an almost twofold risk of developing VTEs compared to patients without oxygen deprivation,” he said.
Dr. Trzepizur and colleagues conducted a retrospective study linking cohort data to an administrative health database. They identified unprovoked VTE in patients with a suspicion for OSA and no previous VTE.
They created Cox proportional hazard models to assess the association of unprovoked VTE with apnea hypopnea index (AHI) measures and nocturnal hypoxemia markers, including the time patients spent below 90% oxygen saturation (T90), oxygen desaturation index (ODI), and hypoxic burden, defined as the total area under the respiratory event-related desaturation curve.
They found that after a median follow-up of 6.3 years, 104 out of 7,355 patients had an unprovoked VTE. In an unadjusted hazard model, there were significant associations between VTE and T90, as well as with hypoxic burden, but not with either AHI or ODI.
However, in an analysis adjusted for age, gender, body mass index, alcohol intake, hypertension, depression, history of cardiovascular disease, statin use, type of sleep study, study site, and CPAP adherence, the investigators found that only T90 remained a significant independent predictor of VTE, with a hazard ratio of 1.06, P = .02.
The association between T90 and VTE strengthened as the time spent below 90% saturation increased. Patients in the highest tercile, who spent more than 6% of the time undersaturated, had an HR for VTE of 1.95 (P = .02), compared with patients with a T90 less than 1%.
There were no significant differences in VTE risk between patients who used CPAP for more than 4 hours per night and those who either used the devices for less than 4 hours or refused CPAP.
“We see that T90 seems to be a strong parameter,” said session comoderator Raphael Heinzer, MD, MPH, of Lausanne University Hospital, Switzerland.
Dr. Heinzer’s comoderator, Silke Ryan, MD, of University College Dublin, pointed out that although T90 was the main predictor of responses, Dr. Trzepizur and colleagues did not control for other pulmonary diseases.
“Obviously, there could be an influence of other hypoxic-related diseases,” she said, and recommended controlling for this in future studies.
Winfried Randerath, MD, of the Bethanien Hospital at the University of Cologne, Germany, head of the ERS specialist group on sleep disordered breathing, said that this study and others presented at the meeting “show worrying associations between obstructive sleep apnea and important diseases that affect survival and quality of life.
“While they cannot prove that OSA causes any of these health problems, people should be made aware of these links and should try to make lifestyle changes in order to reduce their risk of OSA, for instance, by maintaining a healthy weight. However, if OSA is suspected, definite diagnosis and treatment should be initiated. We look forward to further research that may help to clarify whether OSA may be causing some of the health problems seen in these studies,” said Dr. Randerath, who was not involved with the study.
The study was supported by a grant from Institut de Recherche en Santé Respiratoire des Pays de la Loire (IRSR), Beaucouzé, France. Dr. Trzepizur, Dr. Heinzer, Dr. Ryan and Dr. Randerath reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Add unprovoked venous thromboembolic events to the list of potential consequences of severe obstructive sleep apnea.
That conclusion comes from a study showing that patients with obstructive sleep apnea (OSA) who had the longest nocturnal hypoxemia episodes had a twofold risk for venous thromboembolic events.
systems, reported Wojciech Trzepizur, MD, of Angers University Hospital, France.
Previous studies have suggested links between OSA and both cancer and cognitive decline, but this is the first study to investigate the association between OSA and the incidence of unprovoked VTE, he reported in an oral abstract session at the annual congress of the European Respiratory Society.
“We found that those who spent more than 6% of their nighttime with levels of oxygen in their blood below 90% of normal had an almost twofold risk of developing VTEs compared to patients without oxygen deprivation,” he said.
Dr. Trzepizur and colleagues conducted a retrospective study linking cohort data to an administrative health database. They identified unprovoked VTE in patients with a suspicion for OSA and no previous VTE.
They created Cox proportional hazard models to assess the association of unprovoked VTE with apnea hypopnea index (AHI) measures and nocturnal hypoxemia markers, including the time patients spent below 90% oxygen saturation (T90), oxygen desaturation index (ODI), and hypoxic burden, defined as the total area under the respiratory event-related desaturation curve.
They found that after a median follow-up of 6.3 years, 104 out of 7,355 patients had an unprovoked VTE. In an unadjusted hazard model, there were significant associations between VTE and T90, as well as with hypoxic burden, but not with either AHI or ODI.
However, in an analysis adjusted for age, gender, body mass index, alcohol intake, hypertension, depression, history of cardiovascular disease, statin use, type of sleep study, study site, and CPAP adherence, the investigators found that only T90 remained a significant independent predictor of VTE, with a hazard ratio of 1.06, P = .02.
The association between T90 and VTE strengthened as the time spent below 90% saturation increased. Patients in the highest tercile, who spent more than 6% of the time undersaturated, had an HR for VTE of 1.95 (P = .02), compared with patients with a T90 less than 1%.
There were no significant differences in VTE risk between patients who used CPAP for more than 4 hours per night and those who either used the devices for less than 4 hours or refused CPAP.
“We see that T90 seems to be a strong parameter,” said session comoderator Raphael Heinzer, MD, MPH, of Lausanne University Hospital, Switzerland.
Dr. Heinzer’s comoderator, Silke Ryan, MD, of University College Dublin, pointed out that although T90 was the main predictor of responses, Dr. Trzepizur and colleagues did not control for other pulmonary diseases.
“Obviously, there could be an influence of other hypoxic-related diseases,” she said, and recommended controlling for this in future studies.
Winfried Randerath, MD, of the Bethanien Hospital at the University of Cologne, Germany, head of the ERS specialist group on sleep disordered breathing, said that this study and others presented at the meeting “show worrying associations between obstructive sleep apnea and important diseases that affect survival and quality of life.
“While they cannot prove that OSA causes any of these health problems, people should be made aware of these links and should try to make lifestyle changes in order to reduce their risk of OSA, for instance, by maintaining a healthy weight. However, if OSA is suspected, definite diagnosis and treatment should be initiated. We look forward to further research that may help to clarify whether OSA may be causing some of the health problems seen in these studies,” said Dr. Randerath, who was not involved with the study.
The study was supported by a grant from Institut de Recherche en Santé Respiratoire des Pays de la Loire (IRSR), Beaucouzé, France. Dr. Trzepizur, Dr. Heinzer, Dr. Ryan and Dr. Randerath reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM ERS 2022
Atezolizumab doubles survival of NSCLC patients with poor performance status
PARIS – Patients with untreated non–small cell lung cancer (NSCLC) who could not withstand the rigors of platinum-based chemotherapy regimens had significantly better overall survival when treated with the immune checkpoint inhibitor atezolizumab (Tecentriq), compared with their counterparts treated with either vinorelbine or gemcitabine in a phase 3 randomized trial.
Among 353 patients with treatment-naive stage 3B to 4 NSCLC who were not candidates for platinum-based chemotherapy because of poor performance status (PS), advanced age, or significant comorbidities, the median overall survival (OS) was 10.3 months for patients treated with atezolizumab vs. 9.2 months for patients assigned to receive the investigator’s choice of single-agent chemotherapy.
This difference translated into a hazard ratio for death with atezolizumab of 0.78 (P = .028), Siow Ming Lee, MD, PhD, of University College London, reported at the ESMO Congress.
The 2-year OS rate with atezolizumab was 24.3%, compared with 12.4% for single-agent chemotherapy.
“When I saw the data, I was amazed. One of four patients survived for 2 years!” he said in an interview.

, those with Eastern Cooperative Oncology Group PS scores of 2 or greater, or who have substantial comorbidities that preclude their ability to receive platinum doublet or single platinum agent chemotherapy, he said.
Invited discussant Natasha Leighl, MD, MMSc, of the Princess Margaret Cancer Center, Toronto, called the study “really extraordinary. This study enrolls patients that historically are excluded or underrepresented in trials, and yet really represent the majority of patients that we diagnose and treat around the world.”
Excluded from clinical trials
“Cancer chemotherapy has changed the treatment landscape for the metastatic NSCLC population, but these treatments are mainly recommended for fit patients,” Dr. Lee said during his presentation of the data in a presidential symposium.
First-line pivotal trials for lung cancer patients comparing either single-agent immunotherapy or an immunotherapy/chemotherapy combination have all been conducted in fit patients, with ECOG PS of 0 or 1, he noted.
“In reality, we still have a large population of unfit NSCLC patients, of at least 40%, many of which we cannot treat with standard platinum chemotherapy. There are many elderly patients with poor performance status, and the elderly with many comorbidities, and they are frequently on many drug medications, which we see frequently in our clinic,” he said.
Study details
To see whether immunotherapy could improve outcomes for unfit patients, investigators designed the IPSOS trial, a phase 3 multicenter open-label study of efficacy, safety, and patient-reported outcomes with atezolizumab compared with single-agent chemotherapy.
Patients from 23 centers in North America, South America, Europe, and Asia who were ineligible for platinum-based chemotherapy because of ECOG performance status of 2 or 3, or who were aged 70 or older with performance status 0 or 1 but with multiple comorbidities or other contraindications to platinum were stratified by histology, programmed death-ligand-1 (PD-L1) expression, and brain metastases, and were then randomly assigned to receive either atezolizumab 1,200 mg intravenously every 3 weeks (302 patients), or to investigator’s choice of either vinorelbine delivered orally or intravenously, according to local practice, or intravenous gemcitabine given intravenously per local practice (151 patients).
As noted before, overall survival, the primary endpoint, was significantly better with atezolizumab, translating into a 22% reduction in risk of death compared with chemotherapy.
The 1-year OS rates were 43.7% with atezolizumab vs. 36.6% with chemotherapy, and the 2-year rates were 24.3% vs. 12.4%, respectively.
A subgroup analysis showed trends toward better benefit for immunotherapy regardless of age, sex, race, performance status, history of tobacco use, tumor histology, stage, presence of liver metastases, number of metastatic sites, or PD-L1 expression levels. The benefit of atezolizumab was also significantly better among patients without brain metastases.
The median duration of response was 14 months with ateziluzmab vs. 7.8 months with chemotherapy. Respective objective response rates were 16.9% vs. 15.5%. Median progression-free survival, a secondary endpoint, was 4.2 months with atezolizumab and 4 months with chemotherapy, a difference that was not statistically significant. Median treatment duration was 3.5 months with atezolizumab, 2.3 months with gemcitabine, and 1.8 months with vinorelbine. Treatment-related adverse events of any grade occurred in 57% of patients on immunotherapy vs. 80.3% of those on chemotherapy. Grade 3 or 4 adverse events related to therapy occurred in 16.3% vs. 33.3%, respectively. About 13% of patients in each arm had an adverse event leading to drug discontinuation. There were three treatment-related deaths among patients on atezolizumab, and four among patients on chemotherapy. Compared with chemotherapy, atezolizumab was associated with stabilizing of health-related quality-of-life domains of functioning, and significant improvement in delaying the time to deterioration of chest pain.
Age is not prognostic
“I think it’s important though to remember that in this study there are very distinct populations of patients. Poor performance status and comorbidities are prognostic, but age is not,” Dr. Leighl said in her discussion.
“In terms of current standards, performance status 3 patients are currently recommended to have best supportive care unless a targeted therapy is available for them, and while PS 2 patients have been excluded from checkpoint inhibitor trials, we treat most of these patients the same way. In this study in particular, patients had to be ineligible for platinum doublet therapy, but of course this definition was subjective,” she said.
She also commented that “if we’re now going to treat everyone with atezolizumab, I think the budget impact of this is going to be huge.”
It will be important to identify more clearly those patients aged 80 and older who might benefit from atezolizumab in this setting by better incorporating biomarkers such as PD-L1 levels to determine who can benefit from therapy and who might be spared the necessity of coming into the hospital or clinic for regular intravenous infusions, she added.
The study was supported by F. Hoffman-La Roche. Dr. Lee disclosed research funding from the company to his institution. Dr. Leighl disclosed institutional grant funding and personal fees from Roche and others.
PARIS – Patients with untreated non–small cell lung cancer (NSCLC) who could not withstand the rigors of platinum-based chemotherapy regimens had significantly better overall survival when treated with the immune checkpoint inhibitor atezolizumab (Tecentriq), compared with their counterparts treated with either vinorelbine or gemcitabine in a phase 3 randomized trial.
Among 353 patients with treatment-naive stage 3B to 4 NSCLC who were not candidates for platinum-based chemotherapy because of poor performance status (PS), advanced age, or significant comorbidities, the median overall survival (OS) was 10.3 months for patients treated with atezolizumab vs. 9.2 months for patients assigned to receive the investigator’s choice of single-agent chemotherapy.
This difference translated into a hazard ratio for death with atezolizumab of 0.78 (P = .028), Siow Ming Lee, MD, PhD, of University College London, reported at the ESMO Congress.
The 2-year OS rate with atezolizumab was 24.3%, compared with 12.4% for single-agent chemotherapy.
“When I saw the data, I was amazed. One of four patients survived for 2 years!” he said in an interview.
, those with Eastern Cooperative Oncology Group PS scores of 2 or greater, or who have substantial comorbidities that preclude their ability to receive platinum doublet or single platinum agent chemotherapy, he said.
Invited discussant Natasha Leighl, MD, MMSc, of the Princess Margaret Cancer Center, Toronto, called the study “really extraordinary. This study enrolls patients that historically are excluded or underrepresented in trials, and yet really represent the majority of patients that we diagnose and treat around the world.”
Excluded from clinical trials
“Cancer chemotherapy has changed the treatment landscape for the metastatic NSCLC population, but these treatments are mainly recommended for fit patients,” Dr. Lee said during his presentation of the data in a presidential symposium.
First-line pivotal trials for lung cancer patients comparing either single-agent immunotherapy or an immunotherapy/chemotherapy combination have all been conducted in fit patients, with ECOG PS of 0 or 1, he noted.
“In reality, we still have a large population of unfit NSCLC patients, of at least 40%, many of which we cannot treat with standard platinum chemotherapy. There are many elderly patients with poor performance status, and the elderly with many comorbidities, and they are frequently on many drug medications, which we see frequently in our clinic,” he said.
Study details
To see whether immunotherapy could improve outcomes for unfit patients, investigators designed the IPSOS trial, a phase 3 multicenter open-label study of efficacy, safety, and patient-reported outcomes with atezolizumab compared with single-agent chemotherapy.
Patients from 23 centers in North America, South America, Europe, and Asia who were ineligible for platinum-based chemotherapy because of ECOG performance status of 2 or 3, or who were aged 70 or older with performance status 0 or 1 but with multiple comorbidities or other contraindications to platinum were stratified by histology, programmed death-ligand-1 (PD-L1) expression, and brain metastases, and were then randomly assigned to receive either atezolizumab 1,200 mg intravenously every 3 weeks (302 patients), or to investigator’s choice of either vinorelbine delivered orally or intravenously, according to local practice, or intravenous gemcitabine given intravenously per local practice (151 patients).
As noted before, overall survival, the primary endpoint, was significantly better with atezolizumab, translating into a 22% reduction in risk of death compared with chemotherapy.
The 1-year OS rates were 43.7% with atezolizumab vs. 36.6% with chemotherapy, and the 2-year rates were 24.3% vs. 12.4%, respectively.
A subgroup analysis showed trends toward better benefit for immunotherapy regardless of age, sex, race, performance status, history of tobacco use, tumor histology, stage, presence of liver metastases, number of metastatic sites, or PD-L1 expression levels. The benefit of atezolizumab was also significantly better among patients without brain metastases.
The median duration of response was 14 months with ateziluzmab vs. 7.8 months with chemotherapy. Respective objective response rates were 16.9% vs. 15.5%. Median progression-free survival, a secondary endpoint, was 4.2 months with atezolizumab and 4 months with chemotherapy, a difference that was not statistically significant. Median treatment duration was 3.5 months with atezolizumab, 2.3 months with gemcitabine, and 1.8 months with vinorelbine. Treatment-related adverse events of any grade occurred in 57% of patients on immunotherapy vs. 80.3% of those on chemotherapy. Grade 3 or 4 adverse events related to therapy occurred in 16.3% vs. 33.3%, respectively. About 13% of patients in each arm had an adverse event leading to drug discontinuation. There were three treatment-related deaths among patients on atezolizumab, and four among patients on chemotherapy. Compared with chemotherapy, atezolizumab was associated with stabilizing of health-related quality-of-life domains of functioning, and significant improvement in delaying the time to deterioration of chest pain.
Age is not prognostic
“I think it’s important though to remember that in this study there are very distinct populations of patients. Poor performance status and comorbidities are prognostic, but age is not,” Dr. Leighl said in her discussion.
“In terms of current standards, performance status 3 patients are currently recommended to have best supportive care unless a targeted therapy is available for them, and while PS 2 patients have been excluded from checkpoint inhibitor trials, we treat most of these patients the same way. In this study in particular, patients had to be ineligible for platinum doublet therapy, but of course this definition was subjective,” she said.
She also commented that “if we’re now going to treat everyone with atezolizumab, I think the budget impact of this is going to be huge.”
It will be important to identify more clearly those patients aged 80 and older who might benefit from atezolizumab in this setting by better incorporating biomarkers such as PD-L1 levels to determine who can benefit from therapy and who might be spared the necessity of coming into the hospital or clinic for regular intravenous infusions, she added.
The study was supported by F. Hoffman-La Roche. Dr. Lee disclosed research funding from the company to his institution. Dr. Leighl disclosed institutional grant funding and personal fees from Roche and others.
PARIS – Patients with untreated non–small cell lung cancer (NSCLC) who could not withstand the rigors of platinum-based chemotherapy regimens had significantly better overall survival when treated with the immune checkpoint inhibitor atezolizumab (Tecentriq), compared with their counterparts treated with either vinorelbine or gemcitabine in a phase 3 randomized trial.
Among 353 patients with treatment-naive stage 3B to 4 NSCLC who were not candidates for platinum-based chemotherapy because of poor performance status (PS), advanced age, or significant comorbidities, the median overall survival (OS) was 10.3 months for patients treated with atezolizumab vs. 9.2 months for patients assigned to receive the investigator’s choice of single-agent chemotherapy.
This difference translated into a hazard ratio for death with atezolizumab of 0.78 (P = .028), Siow Ming Lee, MD, PhD, of University College London, reported at the ESMO Congress.
The 2-year OS rate with atezolizumab was 24.3%, compared with 12.4% for single-agent chemotherapy.
“When I saw the data, I was amazed. One of four patients survived for 2 years!” he said in an interview.
, those with Eastern Cooperative Oncology Group PS scores of 2 or greater, or who have substantial comorbidities that preclude their ability to receive platinum doublet or single platinum agent chemotherapy, he said.
Invited discussant Natasha Leighl, MD, MMSc, of the Princess Margaret Cancer Center, Toronto, called the study “really extraordinary. This study enrolls patients that historically are excluded or underrepresented in trials, and yet really represent the majority of patients that we diagnose and treat around the world.”
Excluded from clinical trials
“Cancer chemotherapy has changed the treatment landscape for the metastatic NSCLC population, but these treatments are mainly recommended for fit patients,” Dr. Lee said during his presentation of the data in a presidential symposium.
First-line pivotal trials for lung cancer patients comparing either single-agent immunotherapy or an immunotherapy/chemotherapy combination have all been conducted in fit patients, with ECOG PS of 0 or 1, he noted.
“In reality, we still have a large population of unfit NSCLC patients, of at least 40%, many of which we cannot treat with standard platinum chemotherapy. There are many elderly patients with poor performance status, and the elderly with many comorbidities, and they are frequently on many drug medications, which we see frequently in our clinic,” he said.
Study details
To see whether immunotherapy could improve outcomes for unfit patients, investigators designed the IPSOS trial, a phase 3 multicenter open-label study of efficacy, safety, and patient-reported outcomes with atezolizumab compared with single-agent chemotherapy.
Patients from 23 centers in North America, South America, Europe, and Asia who were ineligible for platinum-based chemotherapy because of ECOG performance status of 2 or 3, or who were aged 70 or older with performance status 0 or 1 but with multiple comorbidities or other contraindications to platinum were stratified by histology, programmed death-ligand-1 (PD-L1) expression, and brain metastases, and were then randomly assigned to receive either atezolizumab 1,200 mg intravenously every 3 weeks (302 patients), or to investigator’s choice of either vinorelbine delivered orally or intravenously, according to local practice, or intravenous gemcitabine given intravenously per local practice (151 patients).
As noted before, overall survival, the primary endpoint, was significantly better with atezolizumab, translating into a 22% reduction in risk of death compared with chemotherapy.
The 1-year OS rates were 43.7% with atezolizumab vs. 36.6% with chemotherapy, and the 2-year rates were 24.3% vs. 12.4%, respectively.
A subgroup analysis showed trends toward better benefit for immunotherapy regardless of age, sex, race, performance status, history of tobacco use, tumor histology, stage, presence of liver metastases, number of metastatic sites, or PD-L1 expression levels. The benefit of atezolizumab was also significantly better among patients without brain metastases.
The median duration of response was 14 months with ateziluzmab vs. 7.8 months with chemotherapy. Respective objective response rates were 16.9% vs. 15.5%. Median progression-free survival, a secondary endpoint, was 4.2 months with atezolizumab and 4 months with chemotherapy, a difference that was not statistically significant. Median treatment duration was 3.5 months with atezolizumab, 2.3 months with gemcitabine, and 1.8 months with vinorelbine. Treatment-related adverse events of any grade occurred in 57% of patients on immunotherapy vs. 80.3% of those on chemotherapy. Grade 3 or 4 adverse events related to therapy occurred in 16.3% vs. 33.3%, respectively. About 13% of patients in each arm had an adverse event leading to drug discontinuation. There were three treatment-related deaths among patients on atezolizumab, and four among patients on chemotherapy. Compared with chemotherapy, atezolizumab was associated with stabilizing of health-related quality-of-life domains of functioning, and significant improvement in delaying the time to deterioration of chest pain.
Age is not prognostic
“I think it’s important though to remember that in this study there are very distinct populations of patients. Poor performance status and comorbidities are prognostic, but age is not,” Dr. Leighl said in her discussion.
“In terms of current standards, performance status 3 patients are currently recommended to have best supportive care unless a targeted therapy is available for them, and while PS 2 patients have been excluded from checkpoint inhibitor trials, we treat most of these patients the same way. In this study in particular, patients had to be ineligible for platinum doublet therapy, but of course this definition was subjective,” she said.
She also commented that “if we’re now going to treat everyone with atezolizumab, I think the budget impact of this is going to be huge.”
It will be important to identify more clearly those patients aged 80 and older who might benefit from atezolizumab in this setting by better incorporating biomarkers such as PD-L1 levels to determine who can benefit from therapy and who might be spared the necessity of coming into the hospital or clinic for regular intravenous infusions, she added.
The study was supported by F. Hoffman-La Roche. Dr. Lee disclosed research funding from the company to his institution. Dr. Leighl disclosed institutional grant funding and personal fees from Roche and others.
AT ESMO CONGRESS 2022
In cardiogenic shock, edge-to-edge mitral valve repair improves outcome
In patients with severe mitral regurgitation (MR) and cardiogenic shock, successful transcatheter edge-to-edge repair (TEER) is associated with a substantial reduction in all-cause mortality and lower morbidity at 1 year, according to an analysis of registry data.
The data from this analysis also confirm that “successful reduction of MR is achievable with TEER in most patients with cardiogenic shock,” reported Mohamad A. Alkhouli, MD, an interventional cardiologist and professor of medicine at the Mayo Clinic, Rochester, Minn.

In those with device success, achieved in 85.6% of patients, all-cause mortality was about 21% lower (34.6% vs. 55.5%; P < .001) at 1 year than in those who were not successfully repaired, according to Dr. Alkhouli, who presented the findings at the Transcatheter Cardiovascular Therapeutics annual meeting in Boston. This translated into a reduction in the hazard ratio for death of nearly 50% (hazard ratio, 0.52; 95% confidence interval, 0.43-0.63).
A similar relative benefit was found for the composite endpoint of mortality and heart failure admissions at 1 year. Whether unadjusted (HR, 0.54; 95% CI, 0.45-0.66) or adjusted (HR, 0.51; 95% CI, 0.42-0.62), risk reductions with successful MR reduction, defined as greater than or equal to 1 grade improvement and a final MR grade of less than or equal to 2+, indicated that major adverse outcomes are reduced by about half.
STS/ACC TCT registry data queried
Drawn from the Society of Thoracic Surgeons/American College of Cardiology Transcatheter Valve Therapy Registry, 3,797 patients with cardiogenic shock underwent MR repair between November 2013 and December 2021. Outcomes at 1 year were evaluable in 2,773 of these patients. For inclusion, all had to meet at least one of the definitions of cardiogenic shock, such as inotrope use or mechanical circulatory support.
At baseline, 94.5% had a MR severity of at least 3+, and most of these had 4+. Thirty days after treatment, 88.8% had MR severity of 2+ or less, the majority of which had a severity of 1+.
These data address an important question not previously well studied, according to Dr. Alkhouli. In MR patients, cardiogenic shock is associated with a high risk of death, but there has been little evidence that valve repair does not exacerbate, let alone modify, this risk.
These data support the value of intervention, which was performed in almost all patients with MitraClipä (Abbott), the only device available for most of the period in which the registry was queried. However, Dr. Alkhouli cautioned that his data are best considered “hypothesis generating.”
“We need a randomized trial,” he said at the meeting sponsored by the Cardiovascular Research Foundation. He pointed out that this is a complex population for which multiple variables might have skewed results when data are analyzed retrospectively. Not least, those MR patients with cardiogenic shock in the database considered for TEER might well have been relatively healthy and not representative of an unselected population with both MR and cardiogenic shock.
The question might be better answered by the multicenter Canadian trial CAPITAL MINOS, which has just started. Described in an article in the American Heart Journal, it has a planned enrollment of about 150 MR patients with cardiogenic shock randomized to TEER or medical therapy. Results are expected in about 1 year, according to Dr. Alkhouli.
But regarding the present analysis, Dr. Alkhouli did note that sensitivity analyses conducted within his data across risk factors, such as degenerative versus nondegenerative MR, low (< 30%) versus higher left ventricular ejection fraction (LVEF), and presence or absence of an acute coronary syndrome (ACS), consistently supported a benefit from intervention.
Also, cardiogenic shock did not appear to be a factor in device failure, according to Dr. Alkhouli, addressing a potential criticism that cardiogenic shock was an underlying reason for device failure.
More than 90% in NYHA class III or IV heart failure
In this study, the mean age was 73 years. More than 90% were in class III or IV heart failure in the 2 weeks prior to TEER. More than half had established coronary artery disease. Other concomitant cardiovascular morbidities, including atrial fibrillation or flutter (65%), prior MI (39%), and prior stroke or transient ischemic attach (> 10%) were well represented.
When those with device success were compared with those with device failure, the risk profile was comparable. The predicted STS (Society of Thoracic Surgeons) mortality for mitral valve repair among these two groups was 14.8% versus 15% (P = 0.97), respectively.
However, those with device failure did have a lower baseline left ventricular ejection fraction (40.7% vs. 42.9%; P = .009) and a greater prevalence of moderate-to-severe or severe MR (96.1% vs. 84.9%; P < 0.001).
The growing experience with TEER means that benefit has now been shown in several complicated MR groups, such as those with severe ventricular dysfunction, renal insufficiency, and obstructive lung disease. This was a rationale for looking at the impact or repairing MR in patients with cardiogenic shock.
It is a pressing question, according to Dr. Alkhouli. He cited studies suggesting that up to 20% of patients hospitalized for cardiogenic shock have at least moderate-to-severe MR. Conversely, cardiogenic shock is not an uncommon finding in patients with MR.
While Dr. Alkhouli acknowledged that the many variables influencing outcome in patients with MR and cardiogenic shock will make a randomized trial “challenging,” many experts echoed this concern and even expressed some skepticism about the potential for an unbiased trial.
Data confirm MR repair is safe during shock
“These data do show that repair of MR is safe in patients safe in patients with cardiogenic shock,” said Anita W. Asgar, MD, an interventional cardiologist associated with the Montreal Heart Institute. She noted that there was a 5- to 6-day delay among the cardiogenic shock patients prior to undergoing MR repair in this analysis, potentially reflecting an elimination of those at very high risk. Similarly, she suggested that many interventionalists are likely to consider multiple variables before proceeding.
As a result, MR repair may not be amenable to randomization in a cardiogenic shock population, given that this decision is not typically undertaken out of the context of multiple variables.
“I am not sure that a clinical trial is ethical,” she said. She would expect that clinicians enrolling patients would only do so on a selective basis.
Alexandra J. Lansky, MD, Director of the Yale Heart and Vascular Research Program, Yale University, New Haven, Conn., also emphasized the difficulty of controlling for variables, such as the duration of cardiogenic shock, that influence decision-making.
Nevertheless, she called the data “very important” in that they at least lend some objective data for deciding whether to intervene a group of “challenging” patients not uncommonly faced in clinical practice.
Dr. Alkhouli reports financial relationships with Abbott Vascular, Boston Scientific, Johnson & Johnson, and Phillips. Dr. Asgar reports financial relationships with Abbott Vascular, Edwards Lifesciences, W.L. Gore & Associates, and Medtronic. Dr. Lasky reports no potential conflicts of interest.
A version of this article first appeared on Medscape.com.
In patients with severe mitral regurgitation (MR) and cardiogenic shock, successful transcatheter edge-to-edge repair (TEER) is associated with a substantial reduction in all-cause mortality and lower morbidity at 1 year, according to an analysis of registry data.
The data from this analysis also confirm that “successful reduction of MR is achievable with TEER in most patients with cardiogenic shock,” reported Mohamad A. Alkhouli, MD, an interventional cardiologist and professor of medicine at the Mayo Clinic, Rochester, Minn.
In those with device success, achieved in 85.6% of patients, all-cause mortality was about 21% lower (34.6% vs. 55.5%; P < .001) at 1 year than in those who were not successfully repaired, according to Dr. Alkhouli, who presented the findings at the Transcatheter Cardiovascular Therapeutics annual meeting in Boston. This translated into a reduction in the hazard ratio for death of nearly 50% (hazard ratio, 0.52; 95% confidence interval, 0.43-0.63).
A similar relative benefit was found for the composite endpoint of mortality and heart failure admissions at 1 year. Whether unadjusted (HR, 0.54; 95% CI, 0.45-0.66) or adjusted (HR, 0.51; 95% CI, 0.42-0.62), risk reductions with successful MR reduction, defined as greater than or equal to 1 grade improvement and a final MR grade of less than or equal to 2+, indicated that major adverse outcomes are reduced by about half.
STS/ACC TCT registry data queried
Drawn from the Society of Thoracic Surgeons/American College of Cardiology Transcatheter Valve Therapy Registry, 3,797 patients with cardiogenic shock underwent MR repair between November 2013 and December 2021. Outcomes at 1 year were evaluable in 2,773 of these patients. For inclusion, all had to meet at least one of the definitions of cardiogenic shock, such as inotrope use or mechanical circulatory support.
At baseline, 94.5% had a MR severity of at least 3+, and most of these had 4+. Thirty days after treatment, 88.8% had MR severity of 2+ or less, the majority of which had a severity of 1+.
These data address an important question not previously well studied, according to Dr. Alkhouli. In MR patients, cardiogenic shock is associated with a high risk of death, but there has been little evidence that valve repair does not exacerbate, let alone modify, this risk.
These data support the value of intervention, which was performed in almost all patients with MitraClipä (Abbott), the only device available for most of the period in which the registry was queried. However, Dr. Alkhouli cautioned that his data are best considered “hypothesis generating.”
“We need a randomized trial,” he said at the meeting sponsored by the Cardiovascular Research Foundation. He pointed out that this is a complex population for which multiple variables might have skewed results when data are analyzed retrospectively. Not least, those MR patients with cardiogenic shock in the database considered for TEER might well have been relatively healthy and not representative of an unselected population with both MR and cardiogenic shock.
The question might be better answered by the multicenter Canadian trial CAPITAL MINOS, which has just started. Described in an article in the American Heart Journal, it has a planned enrollment of about 150 MR patients with cardiogenic shock randomized to TEER or medical therapy. Results are expected in about 1 year, according to Dr. Alkhouli.
But regarding the present analysis, Dr. Alkhouli did note that sensitivity analyses conducted within his data across risk factors, such as degenerative versus nondegenerative MR, low (< 30%) versus higher left ventricular ejection fraction (LVEF), and presence or absence of an acute coronary syndrome (ACS), consistently supported a benefit from intervention.
Also, cardiogenic shock did not appear to be a factor in device failure, according to Dr. Alkhouli, addressing a potential criticism that cardiogenic shock was an underlying reason for device failure.
More than 90% in NYHA class III or IV heart failure
In this study, the mean age was 73 years. More than 90% were in class III or IV heart failure in the 2 weeks prior to TEER. More than half had established coronary artery disease. Other concomitant cardiovascular morbidities, including atrial fibrillation or flutter (65%), prior MI (39%), and prior stroke or transient ischemic attach (> 10%) were well represented.
When those with device success were compared with those with device failure, the risk profile was comparable. The predicted STS (Society of Thoracic Surgeons) mortality for mitral valve repair among these two groups was 14.8% versus 15% (P = 0.97), respectively.
However, those with device failure did have a lower baseline left ventricular ejection fraction (40.7% vs. 42.9%; P = .009) and a greater prevalence of moderate-to-severe or severe MR (96.1% vs. 84.9%; P < 0.001).
The growing experience with TEER means that benefit has now been shown in several complicated MR groups, such as those with severe ventricular dysfunction, renal insufficiency, and obstructive lung disease. This was a rationale for looking at the impact or repairing MR in patients with cardiogenic shock.
It is a pressing question, according to Dr. Alkhouli. He cited studies suggesting that up to 20% of patients hospitalized for cardiogenic shock have at least moderate-to-severe MR. Conversely, cardiogenic shock is not an uncommon finding in patients with MR.
While Dr. Alkhouli acknowledged that the many variables influencing outcome in patients with MR and cardiogenic shock will make a randomized trial “challenging,” many experts echoed this concern and even expressed some skepticism about the potential for an unbiased trial.
Data confirm MR repair is safe during shock
“These data do show that repair of MR is safe in patients safe in patients with cardiogenic shock,” said Anita W. Asgar, MD, an interventional cardiologist associated with the Montreal Heart Institute. She noted that there was a 5- to 6-day delay among the cardiogenic shock patients prior to undergoing MR repair in this analysis, potentially reflecting an elimination of those at very high risk. Similarly, she suggested that many interventionalists are likely to consider multiple variables before proceeding.
As a result, MR repair may not be amenable to randomization in a cardiogenic shock population, given that this decision is not typically undertaken out of the context of multiple variables.
“I am not sure that a clinical trial is ethical,” she said. She would expect that clinicians enrolling patients would only do so on a selective basis.
Alexandra J. Lansky, MD, Director of the Yale Heart and Vascular Research Program, Yale University, New Haven, Conn., also emphasized the difficulty of controlling for variables, such as the duration of cardiogenic shock, that influence decision-making.
Nevertheless, she called the data “very important” in that they at least lend some objective data for deciding whether to intervene a group of “challenging” patients not uncommonly faced in clinical practice.
Dr. Alkhouli reports financial relationships with Abbott Vascular, Boston Scientific, Johnson & Johnson, and Phillips. Dr. Asgar reports financial relationships with Abbott Vascular, Edwards Lifesciences, W.L. Gore & Associates, and Medtronic. Dr. Lasky reports no potential conflicts of interest.
A version of this article first appeared on Medscape.com.
In patients with severe mitral regurgitation (MR) and cardiogenic shock, successful transcatheter edge-to-edge repair (TEER) is associated with a substantial reduction in all-cause mortality and lower morbidity at 1 year, according to an analysis of registry data.
The data from this analysis also confirm that “successful reduction of MR is achievable with TEER in most patients with cardiogenic shock,” reported Mohamad A. Alkhouli, MD, an interventional cardiologist and professor of medicine at the Mayo Clinic, Rochester, Minn.
In those with device success, achieved in 85.6% of patients, all-cause mortality was about 21% lower (34.6% vs. 55.5%; P < .001) at 1 year than in those who were not successfully repaired, according to Dr. Alkhouli, who presented the findings at the Transcatheter Cardiovascular Therapeutics annual meeting in Boston. This translated into a reduction in the hazard ratio for death of nearly 50% (hazard ratio, 0.52; 95% confidence interval, 0.43-0.63).
A similar relative benefit was found for the composite endpoint of mortality and heart failure admissions at 1 year. Whether unadjusted (HR, 0.54; 95% CI, 0.45-0.66) or adjusted (HR, 0.51; 95% CI, 0.42-0.62), risk reductions with successful MR reduction, defined as greater than or equal to 1 grade improvement and a final MR grade of less than or equal to 2+, indicated that major adverse outcomes are reduced by about half.
STS/ACC TCT registry data queried
Drawn from the Society of Thoracic Surgeons/American College of Cardiology Transcatheter Valve Therapy Registry, 3,797 patients with cardiogenic shock underwent MR repair between November 2013 and December 2021. Outcomes at 1 year were evaluable in 2,773 of these patients. For inclusion, all had to meet at least one of the definitions of cardiogenic shock, such as inotrope use or mechanical circulatory support.
At baseline, 94.5% had a MR severity of at least 3+, and most of these had 4+. Thirty days after treatment, 88.8% had MR severity of 2+ or less, the majority of which had a severity of 1+.
These data address an important question not previously well studied, according to Dr. Alkhouli. In MR patients, cardiogenic shock is associated with a high risk of death, but there has been little evidence that valve repair does not exacerbate, let alone modify, this risk.
These data support the value of intervention, which was performed in almost all patients with MitraClipä (Abbott), the only device available for most of the period in which the registry was queried. However, Dr. Alkhouli cautioned that his data are best considered “hypothesis generating.”
“We need a randomized trial,” he said at the meeting sponsored by the Cardiovascular Research Foundation. He pointed out that this is a complex population for which multiple variables might have skewed results when data are analyzed retrospectively. Not least, those MR patients with cardiogenic shock in the database considered for TEER might well have been relatively healthy and not representative of an unselected population with both MR and cardiogenic shock.
The question might be better answered by the multicenter Canadian trial CAPITAL MINOS, which has just started. Described in an article in the American Heart Journal, it has a planned enrollment of about 150 MR patients with cardiogenic shock randomized to TEER or medical therapy. Results are expected in about 1 year, according to Dr. Alkhouli.
But regarding the present analysis, Dr. Alkhouli did note that sensitivity analyses conducted within his data across risk factors, such as degenerative versus nondegenerative MR, low (< 30%) versus higher left ventricular ejection fraction (LVEF), and presence or absence of an acute coronary syndrome (ACS), consistently supported a benefit from intervention.
Also, cardiogenic shock did not appear to be a factor in device failure, according to Dr. Alkhouli, addressing a potential criticism that cardiogenic shock was an underlying reason for device failure.
More than 90% in NYHA class III or IV heart failure
In this study, the mean age was 73 years. More than 90% were in class III or IV heart failure in the 2 weeks prior to TEER. More than half had established coronary artery disease. Other concomitant cardiovascular morbidities, including atrial fibrillation or flutter (65%), prior MI (39%), and prior stroke or transient ischemic attach (> 10%) were well represented.
When those with device success were compared with those with device failure, the risk profile was comparable. The predicted STS (Society of Thoracic Surgeons) mortality for mitral valve repair among these two groups was 14.8% versus 15% (P = 0.97), respectively.
However, those with device failure did have a lower baseline left ventricular ejection fraction (40.7% vs. 42.9%; P = .009) and a greater prevalence of moderate-to-severe or severe MR (96.1% vs. 84.9%; P < 0.001).
The growing experience with TEER means that benefit has now been shown in several complicated MR groups, such as those with severe ventricular dysfunction, renal insufficiency, and obstructive lung disease. This was a rationale for looking at the impact or repairing MR in patients with cardiogenic shock.
It is a pressing question, according to Dr. Alkhouli. He cited studies suggesting that up to 20% of patients hospitalized for cardiogenic shock have at least moderate-to-severe MR. Conversely, cardiogenic shock is not an uncommon finding in patients with MR.
While Dr. Alkhouli acknowledged that the many variables influencing outcome in patients with MR and cardiogenic shock will make a randomized trial “challenging,” many experts echoed this concern and even expressed some skepticism about the potential for an unbiased trial.
Data confirm MR repair is safe during shock
“These data do show that repair of MR is safe in patients safe in patients with cardiogenic shock,” said Anita W. Asgar, MD, an interventional cardiologist associated with the Montreal Heart Institute. She noted that there was a 5- to 6-day delay among the cardiogenic shock patients prior to undergoing MR repair in this analysis, potentially reflecting an elimination of those at very high risk. Similarly, she suggested that many interventionalists are likely to consider multiple variables before proceeding.
As a result, MR repair may not be amenable to randomization in a cardiogenic shock population, given that this decision is not typically undertaken out of the context of multiple variables.
“I am not sure that a clinical trial is ethical,” she said. She would expect that clinicians enrolling patients would only do so on a selective basis.
Alexandra J. Lansky, MD, Director of the Yale Heart and Vascular Research Program, Yale University, New Haven, Conn., also emphasized the difficulty of controlling for variables, such as the duration of cardiogenic shock, that influence decision-making.
Nevertheless, she called the data “very important” in that they at least lend some objective data for deciding whether to intervene a group of “challenging” patients not uncommonly faced in clinical practice.
Dr. Alkhouli reports financial relationships with Abbott Vascular, Boston Scientific, Johnson & Johnson, and Phillips. Dr. Asgar reports financial relationships with Abbott Vascular, Edwards Lifesciences, W.L. Gore & Associates, and Medtronic. Dr. Lasky reports no potential conflicts of interest.
A version of this article first appeared on Medscape.com.
FROM TCT 2022
A farewell to arms? Drug approvals based on single-arm trials can be flawed
PARIS – with results that should only be used, under certain conditions, for accelerated approvals that should then be followed by confirmatory studies.
In fact, many drugs approved over the last decade based solely on data from single-arm trials have been subsequently withdrawn when put through the rigors of a head-to-head randomized controlled trial, according to Bishal Gyawali, MD, PhD, from the department of oncology at Queen’s University, Kingston, Ont.
“Single-arm trials are not meant to provide confirmatory evidence sufficient for approval; However, that ship has sailed, and we have several drugs that are approved on the basis of single-arm trials, but we need to make sure that those approvals are accelerated or conditional approvals, not regular approval,” he said in a presentation included in a special session on drug approvals at the European Society for Medical Oncology Congress.
“We should not allow premature regular approval based on single-arm trials, because once a drug gets conditional approval, access is not an issue. Patients will have access to the drug anyway, but we should ensure that robust evidence follows, and long-term follow-up data are needed to develop confidence in the efficacy outcomes that are seen in single-arm trials,” he said.
In many cases, single-arm trials are large enough or of long enough duration that investigators could have reasonably performed a randomized controlled trial (RCT) in the first place, Dr. Gyawali added.
Why do single-arm trials?
The term “single-arm registration trial” is something of an oxymoron, he said, noting that the purpose of such trials should be whether to take the drug to a phase 3, randomized trial. But as authors of a 2019 study in JAMA Network Open showed, of a sample of phase 3 RCTs, 42% did not have a prior phase 2 trial, and 28% had a negative phase 2 trial. Single-arm trials may be acceptable for conditional drug approvals if all of the following conditions are met:
- A RCT is not possible because the disease is rare or randomization would be unethical.
- The safety of the drug is established and its potential benefits outweigh its risks.
- The drug is associated with a high and durable overall or objective response rate.
- The mechanism of action is supported by a strong scientific rationale, and if the drug may meet an unmet medical need.
Survival endpoints won’t do
Efficacy endpoints typically used in RCTs, such as progression-free survival (PFS) and overall survival (OS) can be misleading because they may be a result of the natural history of the disease and not the drug being tested, whereas ORRs are almost certainly reflective of the action of the drug itself, because spontaneous tumor regression is a rare phenomenon, Dr. Gyawali said.
He cautioned, however, that the ORR of placebo is not zero percent. For example in a 2018 study of sorafenib (Nexavar) versus placebo for advanced or refractory desmoid tumors, the ORR with the active drug was 33%, and the ORR for placebo was 20%.
It’s also open to question, he said, what constitutes an acceptably high ORR and duration of response, pointing to Food and Drug Administration accelerated approval of an indication for nivolumab (Opdivo) for treatment of patients with hepatocellular carcinoma (HCC) that had progressed on sorafenib. In the single-arm trial used as the basis for approval, the ORRs as assessed by an independent central review committee blinded to the results was 14.3%.
“So, nivolumab in hepatocellular cancer was approved on the basis of a response rate lower than that of placebo, albeit in a different tumor. But the point I’m trying to show here is we don’t have a good definition of what is a good response rate,” he said.
In July 2021, Bristol-Myers Squibb voluntarily withdrew the HCC indication for nivolumab, following negative results of the CheckMate 459 trial and a 5-4 vote against continuing the accelerated approval.
On second thought ...
Citing data compiled by Nathan I. Cherny, MD, from Shaare Zedek Medical Center, Jerusalem, Dr. Gyawali noted that 58 of 161 FDA approvals from 2017 to 2021 of drugs for adult solid tumors were based on single-arm trials. Of the 58 drugs, 39 received accelerated approvals, and 19 received regular approvals; of the 39 that received accelerated approvals, 4 were subsequently withdrawn, 8 were converted to regular approvals, and the remainder continued as accelerated approvals.
Interestingly, the median response rate among all the drugs was 40%, and did not differ between the type of approval received, suggesting that response rates are not predictive of whether a drug will receive a conditional or full-fledged go-ahead.
What’s rare and safe?
The definition of a rare disease in the United States is one that affects fewer than 40,000 per year, and in Europe it’s an incidence rate of less than 6 per 100,000 population, Dr. Gyawali noted. But he argued that even non–small cell lung cancer, the most common form of cancer in the world, could be considered rare if it is broken down into subtypes that are treated according to specific mutations that may occur in a relatively small number of patients.
He also noted that a specific drug’s safety, one of the most important criteria for granting approval to a drug based on a single-arm trial, can be difficult to judge without adequate controls for comparison.
Cherry-picking patients
Winette van der Graaf, MD, president of the European Organization for the Research and Treatment of Cancer, who attended the session where Dr. Gyawali’s presentation was played, said in an interview that clinicians should cast a critical eye on how trials are designed and conducted, including patient selection and choice of endpoints.
“One of the most obvious things to be concerned about is that we’re still having patients with good performance status enrolled, mostly PS 0 or 1, so how representative are these clinical trials for the patients we see in front of us on a daily basis?” she said.
“The other question is radiological endpoints, which we focus on with OS and PFS are most important for patients, especially if you consider that if patients may have asymptomatic disease, and we are only treating them with potentially toxic medication, what are we doing for them? Median overall survival when you look at all of these trials is only 4 months, so we really need to take into account how we affect patients in clinical trials,” she added.
Dr. van der Graaf emphasized that clinical trial investigators need to more routinely incorporate quality of life measures and other patient-reported outcomes in clinical trial results to help regulators and clinicians in practice get a better sense of the true clinical benefit of a new drug.
Dr. Gyawali did not disclose a funding source for his presentation. He reported consulting fees from Vivio Health and research grants from the American Society of Clinical Oncology. Dr. van der Graaf reported no conflicts of interest.
PARIS – with results that should only be used, under certain conditions, for accelerated approvals that should then be followed by confirmatory studies.
In fact, many drugs approved over the last decade based solely on data from single-arm trials have been subsequently withdrawn when put through the rigors of a head-to-head randomized controlled trial, according to Bishal Gyawali, MD, PhD, from the department of oncology at Queen’s University, Kingston, Ont.
“Single-arm trials are not meant to provide confirmatory evidence sufficient for approval; However, that ship has sailed, and we have several drugs that are approved on the basis of single-arm trials, but we need to make sure that those approvals are accelerated or conditional approvals, not regular approval,” he said in a presentation included in a special session on drug approvals at the European Society for Medical Oncology Congress.
“We should not allow premature regular approval based on single-arm trials, because once a drug gets conditional approval, access is not an issue. Patients will have access to the drug anyway, but we should ensure that robust evidence follows, and long-term follow-up data are needed to develop confidence in the efficacy outcomes that are seen in single-arm trials,” he said.
In many cases, single-arm trials are large enough or of long enough duration that investigators could have reasonably performed a randomized controlled trial (RCT) in the first place, Dr. Gyawali added.
Why do single-arm trials?
The term “single-arm registration trial” is something of an oxymoron, he said, noting that the purpose of such trials should be whether to take the drug to a phase 3, randomized trial. But as authors of a 2019 study in JAMA Network Open showed, of a sample of phase 3 RCTs, 42% did not have a prior phase 2 trial, and 28% had a negative phase 2 trial. Single-arm trials may be acceptable for conditional drug approvals if all of the following conditions are met:
- A RCT is not possible because the disease is rare or randomization would be unethical.
- The safety of the drug is established and its potential benefits outweigh its risks.
- The drug is associated with a high and durable overall or objective response rate.
- The mechanism of action is supported by a strong scientific rationale, and if the drug may meet an unmet medical need.
Survival endpoints won’t do
Efficacy endpoints typically used in RCTs, such as progression-free survival (PFS) and overall survival (OS) can be misleading because they may be a result of the natural history of the disease and not the drug being tested, whereas ORRs are almost certainly reflective of the action of the drug itself, because spontaneous tumor regression is a rare phenomenon, Dr. Gyawali said.
He cautioned, however, that the ORR of placebo is not zero percent. For example in a 2018 study of sorafenib (Nexavar) versus placebo for advanced or refractory desmoid tumors, the ORR with the active drug was 33%, and the ORR for placebo was 20%.
It’s also open to question, he said, what constitutes an acceptably high ORR and duration of response, pointing to Food and Drug Administration accelerated approval of an indication for nivolumab (Opdivo) for treatment of patients with hepatocellular carcinoma (HCC) that had progressed on sorafenib. In the single-arm trial used as the basis for approval, the ORRs as assessed by an independent central review committee blinded to the results was 14.3%.
“So, nivolumab in hepatocellular cancer was approved on the basis of a response rate lower than that of placebo, albeit in a different tumor. But the point I’m trying to show here is we don’t have a good definition of what is a good response rate,” he said.
In July 2021, Bristol-Myers Squibb voluntarily withdrew the HCC indication for nivolumab, following negative results of the CheckMate 459 trial and a 5-4 vote against continuing the accelerated approval.
On second thought ...
Citing data compiled by Nathan I. Cherny, MD, from Shaare Zedek Medical Center, Jerusalem, Dr. Gyawali noted that 58 of 161 FDA approvals from 2017 to 2021 of drugs for adult solid tumors were based on single-arm trials. Of the 58 drugs, 39 received accelerated approvals, and 19 received regular approvals; of the 39 that received accelerated approvals, 4 were subsequently withdrawn, 8 were converted to regular approvals, and the remainder continued as accelerated approvals.
Interestingly, the median response rate among all the drugs was 40%, and did not differ between the type of approval received, suggesting that response rates are not predictive of whether a drug will receive a conditional or full-fledged go-ahead.
What’s rare and safe?
The definition of a rare disease in the United States is one that affects fewer than 40,000 per year, and in Europe it’s an incidence rate of less than 6 per 100,000 population, Dr. Gyawali noted. But he argued that even non–small cell lung cancer, the most common form of cancer in the world, could be considered rare if it is broken down into subtypes that are treated according to specific mutations that may occur in a relatively small number of patients.
He also noted that a specific drug’s safety, one of the most important criteria for granting approval to a drug based on a single-arm trial, can be difficult to judge without adequate controls for comparison.
Cherry-picking patients
Winette van der Graaf, MD, president of the European Organization for the Research and Treatment of Cancer, who attended the session where Dr. Gyawali’s presentation was played, said in an interview that clinicians should cast a critical eye on how trials are designed and conducted, including patient selection and choice of endpoints.
“One of the most obvious things to be concerned about is that we’re still having patients with good performance status enrolled, mostly PS 0 or 1, so how representative are these clinical trials for the patients we see in front of us on a daily basis?” she said.
“The other question is radiological endpoints, which we focus on with OS and PFS are most important for patients, especially if you consider that if patients may have asymptomatic disease, and we are only treating them with potentially toxic medication, what are we doing for them? Median overall survival when you look at all of these trials is only 4 months, so we really need to take into account how we affect patients in clinical trials,” she added.
Dr. van der Graaf emphasized that clinical trial investigators need to more routinely incorporate quality of life measures and other patient-reported outcomes in clinical trial results to help regulators and clinicians in practice get a better sense of the true clinical benefit of a new drug.
Dr. Gyawali did not disclose a funding source for his presentation. He reported consulting fees from Vivio Health and research grants from the American Society of Clinical Oncology. Dr. van der Graaf reported no conflicts of interest.
PARIS – with results that should only be used, under certain conditions, for accelerated approvals that should then be followed by confirmatory studies.
In fact, many drugs approved over the last decade based solely on data from single-arm trials have been subsequently withdrawn when put through the rigors of a head-to-head randomized controlled trial, according to Bishal Gyawali, MD, PhD, from the department of oncology at Queen’s University, Kingston, Ont.
“Single-arm trials are not meant to provide confirmatory evidence sufficient for approval; However, that ship has sailed, and we have several drugs that are approved on the basis of single-arm trials, but we need to make sure that those approvals are accelerated or conditional approvals, not regular approval,” he said in a presentation included in a special session on drug approvals at the European Society for Medical Oncology Congress.
“We should not allow premature regular approval based on single-arm trials, because once a drug gets conditional approval, access is not an issue. Patients will have access to the drug anyway, but we should ensure that robust evidence follows, and long-term follow-up data are needed to develop confidence in the efficacy outcomes that are seen in single-arm trials,” he said.
In many cases, single-arm trials are large enough or of long enough duration that investigators could have reasonably performed a randomized controlled trial (RCT) in the first place, Dr. Gyawali added.
Why do single-arm trials?
The term “single-arm registration trial” is something of an oxymoron, he said, noting that the purpose of such trials should be whether to take the drug to a phase 3, randomized trial. But as authors of a 2019 study in JAMA Network Open showed, of a sample of phase 3 RCTs, 42% did not have a prior phase 2 trial, and 28% had a negative phase 2 trial. Single-arm trials may be acceptable for conditional drug approvals if all of the following conditions are met:
- A RCT is not possible because the disease is rare or randomization would be unethical.
- The safety of the drug is established and its potential benefits outweigh its risks.
- The drug is associated with a high and durable overall or objective response rate.
- The mechanism of action is supported by a strong scientific rationale, and if the drug may meet an unmet medical need.
Survival endpoints won’t do
Efficacy endpoints typically used in RCTs, such as progression-free survival (PFS) and overall survival (OS) can be misleading because they may be a result of the natural history of the disease and not the drug being tested, whereas ORRs are almost certainly reflective of the action of the drug itself, because spontaneous tumor regression is a rare phenomenon, Dr. Gyawali said.
He cautioned, however, that the ORR of placebo is not zero percent. For example in a 2018 study of sorafenib (Nexavar) versus placebo for advanced or refractory desmoid tumors, the ORR with the active drug was 33%, and the ORR for placebo was 20%.
It’s also open to question, he said, what constitutes an acceptably high ORR and duration of response, pointing to Food and Drug Administration accelerated approval of an indication for nivolumab (Opdivo) for treatment of patients with hepatocellular carcinoma (HCC) that had progressed on sorafenib. In the single-arm trial used as the basis for approval, the ORRs as assessed by an independent central review committee blinded to the results was 14.3%.
“So, nivolumab in hepatocellular cancer was approved on the basis of a response rate lower than that of placebo, albeit in a different tumor. But the point I’m trying to show here is we don’t have a good definition of what is a good response rate,” he said.
In July 2021, Bristol-Myers Squibb voluntarily withdrew the HCC indication for nivolumab, following negative results of the CheckMate 459 trial and a 5-4 vote against continuing the accelerated approval.
On second thought ...
Citing data compiled by Nathan I. Cherny, MD, from Shaare Zedek Medical Center, Jerusalem, Dr. Gyawali noted that 58 of 161 FDA approvals from 2017 to 2021 of drugs for adult solid tumors were based on single-arm trials. Of the 58 drugs, 39 received accelerated approvals, and 19 received regular approvals; of the 39 that received accelerated approvals, 4 were subsequently withdrawn, 8 were converted to regular approvals, and the remainder continued as accelerated approvals.
Interestingly, the median response rate among all the drugs was 40%, and did not differ between the type of approval received, suggesting that response rates are not predictive of whether a drug will receive a conditional or full-fledged go-ahead.
What’s rare and safe?
The definition of a rare disease in the United States is one that affects fewer than 40,000 per year, and in Europe it’s an incidence rate of less than 6 per 100,000 population, Dr. Gyawali noted. But he argued that even non–small cell lung cancer, the most common form of cancer in the world, could be considered rare if it is broken down into subtypes that are treated according to specific mutations that may occur in a relatively small number of patients.
He also noted that a specific drug’s safety, one of the most important criteria for granting approval to a drug based on a single-arm trial, can be difficult to judge without adequate controls for comparison.
Cherry-picking patients
Winette van der Graaf, MD, president of the European Organization for the Research and Treatment of Cancer, who attended the session where Dr. Gyawali’s presentation was played, said in an interview that clinicians should cast a critical eye on how trials are designed and conducted, including patient selection and choice of endpoints.
“One of the most obvious things to be concerned about is that we’re still having patients with good performance status enrolled, mostly PS 0 or 1, so how representative are these clinical trials for the patients we see in front of us on a daily basis?” she said.
“The other question is radiological endpoints, which we focus on with OS and PFS are most important for patients, especially if you consider that if patients may have asymptomatic disease, and we are only treating them with potentially toxic medication, what are we doing for them? Median overall survival when you look at all of these trials is only 4 months, so we really need to take into account how we affect patients in clinical trials,” she added.
Dr. van der Graaf emphasized that clinical trial investigators need to more routinely incorporate quality of life measures and other patient-reported outcomes in clinical trial results to help regulators and clinicians in practice get a better sense of the true clinical benefit of a new drug.
Dr. Gyawali did not disclose a funding source for his presentation. He reported consulting fees from Vivio Health and research grants from the American Society of Clinical Oncology. Dr. van der Graaf reported no conflicts of interest.
AT ESMO CONGRESS 2022
Fourth-gen transcatheter mitral valve shows clinical, procedural improvements
The design improvements introduced in the fourth-generation device for transcatheter mitral valve repair, called the MitraClip G4 (Abbott), appears to yield better outcomes than previous iterations, according to a multinational postapproval study with more than 1,000 patients.
Not least, the 1.3% all-cause mortality at 30 days in this series, called EXPAND G4, “is the lowest that has been reported to date,” reported Ralph Stephan von Bardeleben, MD, at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.

The evidence of relative advantages was based on comparisons with historical data and a similar study of the previous-generation device. That previous study, called EXPAND, evaluated the MitraClip NTR and ETR systems.
Device times shorter with new device
“There were shorter device times with MitraClip G4,” said Dr. von Bardeleben, referring to a more than 10-minute advantage over the previous generation device (35 minutes in EXPAND G4 vs. 46 min in EXPAND). Although the reduction in overall median procedure time was more modest (77 vs. 80 minutes), Dr. von Bardeleben said these are “the shortest device and procedural times reported to date.”
He also reported what appeared to be incremental advantages across multiple other endpoints, such as procedural success (96.2% vs. 95.8%) and a reduction in the mean clip rate (1.4 vs. 1.5).
Compared with historical outcomes with other devices employed in transcatheter edge-to-edge repair (TEER) of mitral valves, Dr. von Bardeleben contended that the results support the premise that the MitraClip G4 system is a meaningful advance by incorporating such features as an expanded choice of clip sizes, a greater coaptation area, and a more advanced gripper actuation for leaflet grasping.
Over 90% achieve MR 1+
Not least, it appears to increase the proportion of patients who achieve a mitral regurgitation grade of 1+ (MR1+) or lower, which is increasingly recognized as the goal of TEER, said Dr. von Bardeleben, head of the Centre of Structure Heart Disease Interventions, Heart Valve Centre, Mainz, Germany.
He said the rates of 91% achieving MR1+ or less and 98% achieved 2+ or lower compare favorably with most other series and exceeds levels achieved with surgery.
Dr. von Bardeleben also contended that, because of its design features, the MitraClip G4 “expands the spectrum of TEER-suitable patients.” He noted that 5% of the patients in this real-world series had a high risk of stenosis owing to such issues as severe annular or leaflet calcification and another 5% had factors that would predict inadequate MR reduction, such as Barlow’s disease, bi-leaflet prolapse, and severe leaflet degeneration.
The 1,164 patients in EXPAND G4 were enrolled from sites in the United States, Europe, Canada, and Japan. For the key outcome measure of procedural success, echocardiograms were assessed by an independent core laboratory. Of the 1,164 patients enrolled, 1,044 (91%) had complete follow-up data at 30 days.
The procedural success rates were reflected in improvements in New York Heart Association (NYHA) functional classes and in the Kansas City Cardiomyopathy Questionnaire (KCCQ), a quality of life instrument. Prior to treatment, 69% were in NYHA class III or greater. Following treatment, the proportion was 17% (P < .0001). The 18-point improvement in the KCCQ was characterized by Dr. von Bardeleben as “both clinically and statistically significant [P < .0001].”
There were no strokes in this series, and the 30-day incidence of myocardial infarction was 0.2%. The proportion requiring cardiovascular surgery within 30 days was less than 1%. The rate of bleeding episodes, all of which were nonserious, was 7%.
The “EXPAND G4 study confirms the safety and effectiveness of the next generation MitraClip G4 system,” according to Dr. von Bardeleben, and it did so “in a contemporary real-world setting.”
Outcome data characterized as ‘excellent’
Several invited panelists participating in a discussion following the presentation agreed.
“These results are excellent,” said Raj Makkar, MD, associate director of interventional technologies at Smidt Heart Institute, Cedars-Sinai Medical Center, Los Angeles. While he was impressed with the fact that only 2% missed the primary endpoint of MR 2+ or lower, he indicated that the 91% achieving MR 1+ or lower might be an even more apt signal that newer-generation devices are improving.
This was echoed by other panelists who appeared to form a general consensus over the premise that the target in TEER should no longer be MR 2+ for most patients.
“We should now be aiming for MR grade of 0-1,” stated panelist Stephan Windecker, MD, chairman, department of cardiology, University of Bern (Switzerland). He indicated that this goal is increasingly reasonable given the advances in device design and greater operator experience.
Dr. von Bardeleben reported financial relationships with Abbott Vascular, Edwards Lifesciences, Medtronic, and Neochord. Dr. Makkar reported financial relationships with Abbott Vascular, Cordis, Edwards Lifesciences, and Medtronic. Dr. Windecker reported financial relationships with more than 30 pharmaceutical companies, including Abbott Vascular, which manufactures MitraClip G4.
The design improvements introduced in the fourth-generation device for transcatheter mitral valve repair, called the MitraClip G4 (Abbott), appears to yield better outcomes than previous iterations, according to a multinational postapproval study with more than 1,000 patients.
Not least, the 1.3% all-cause mortality at 30 days in this series, called EXPAND G4, “is the lowest that has been reported to date,” reported Ralph Stephan von Bardeleben, MD, at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
The evidence of relative advantages was based on comparisons with historical data and a similar study of the previous-generation device. That previous study, called EXPAND, evaluated the MitraClip NTR and ETR systems.
Device times shorter with new device
“There were shorter device times with MitraClip G4,” said Dr. von Bardeleben, referring to a more than 10-minute advantage over the previous generation device (35 minutes in EXPAND G4 vs. 46 min in EXPAND). Although the reduction in overall median procedure time was more modest (77 vs. 80 minutes), Dr. von Bardeleben said these are “the shortest device and procedural times reported to date.”
He also reported what appeared to be incremental advantages across multiple other endpoints, such as procedural success (96.2% vs. 95.8%) and a reduction in the mean clip rate (1.4 vs. 1.5).
Compared with historical outcomes with other devices employed in transcatheter edge-to-edge repair (TEER) of mitral valves, Dr. von Bardeleben contended that the results support the premise that the MitraClip G4 system is a meaningful advance by incorporating such features as an expanded choice of clip sizes, a greater coaptation area, and a more advanced gripper actuation for leaflet grasping.
Over 90% achieve MR 1+
Not least, it appears to increase the proportion of patients who achieve a mitral regurgitation grade of 1+ (MR1+) or lower, which is increasingly recognized as the goal of TEER, said Dr. von Bardeleben, head of the Centre of Structure Heart Disease Interventions, Heart Valve Centre, Mainz, Germany.
He said the rates of 91% achieving MR1+ or less and 98% achieved 2+ or lower compare favorably with most other series and exceeds levels achieved with surgery.
Dr. von Bardeleben also contended that, because of its design features, the MitraClip G4 “expands the spectrum of TEER-suitable patients.” He noted that 5% of the patients in this real-world series had a high risk of stenosis owing to such issues as severe annular or leaflet calcification and another 5% had factors that would predict inadequate MR reduction, such as Barlow’s disease, bi-leaflet prolapse, and severe leaflet degeneration.
The 1,164 patients in EXPAND G4 were enrolled from sites in the United States, Europe, Canada, and Japan. For the key outcome measure of procedural success, echocardiograms were assessed by an independent core laboratory. Of the 1,164 patients enrolled, 1,044 (91%) had complete follow-up data at 30 days.
The procedural success rates were reflected in improvements in New York Heart Association (NYHA) functional classes and in the Kansas City Cardiomyopathy Questionnaire (KCCQ), a quality of life instrument. Prior to treatment, 69% were in NYHA class III or greater. Following treatment, the proportion was 17% (P < .0001). The 18-point improvement in the KCCQ was characterized by Dr. von Bardeleben as “both clinically and statistically significant [P < .0001].”
There were no strokes in this series, and the 30-day incidence of myocardial infarction was 0.2%. The proportion requiring cardiovascular surgery within 30 days was less than 1%. The rate of bleeding episodes, all of which were nonserious, was 7%.
The “EXPAND G4 study confirms the safety and effectiveness of the next generation MitraClip G4 system,” according to Dr. von Bardeleben, and it did so “in a contemporary real-world setting.”
Outcome data characterized as ‘excellent’
Several invited panelists participating in a discussion following the presentation agreed.
“These results are excellent,” said Raj Makkar, MD, associate director of interventional technologies at Smidt Heart Institute, Cedars-Sinai Medical Center, Los Angeles. While he was impressed with the fact that only 2% missed the primary endpoint of MR 2+ or lower, he indicated that the 91% achieving MR 1+ or lower might be an even more apt signal that newer-generation devices are improving.
This was echoed by other panelists who appeared to form a general consensus over the premise that the target in TEER should no longer be MR 2+ for most patients.
“We should now be aiming for MR grade of 0-1,” stated panelist Stephan Windecker, MD, chairman, department of cardiology, University of Bern (Switzerland). He indicated that this goal is increasingly reasonable given the advances in device design and greater operator experience.
Dr. von Bardeleben reported financial relationships with Abbott Vascular, Edwards Lifesciences, Medtronic, and Neochord. Dr. Makkar reported financial relationships with Abbott Vascular, Cordis, Edwards Lifesciences, and Medtronic. Dr. Windecker reported financial relationships with more than 30 pharmaceutical companies, including Abbott Vascular, which manufactures MitraClip G4.
The design improvements introduced in the fourth-generation device for transcatheter mitral valve repair, called the MitraClip G4 (Abbott), appears to yield better outcomes than previous iterations, according to a multinational postapproval study with more than 1,000 patients.
Not least, the 1.3% all-cause mortality at 30 days in this series, called EXPAND G4, “is the lowest that has been reported to date,” reported Ralph Stephan von Bardeleben, MD, at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
The evidence of relative advantages was based on comparisons with historical data and a similar study of the previous-generation device. That previous study, called EXPAND, evaluated the MitraClip NTR and ETR systems.
Device times shorter with new device
“There were shorter device times with MitraClip G4,” said Dr. von Bardeleben, referring to a more than 10-minute advantage over the previous generation device (35 minutes in EXPAND G4 vs. 46 min in EXPAND). Although the reduction in overall median procedure time was more modest (77 vs. 80 minutes), Dr. von Bardeleben said these are “the shortest device and procedural times reported to date.”
He also reported what appeared to be incremental advantages across multiple other endpoints, such as procedural success (96.2% vs. 95.8%) and a reduction in the mean clip rate (1.4 vs. 1.5).
Compared with historical outcomes with other devices employed in transcatheter edge-to-edge repair (TEER) of mitral valves, Dr. von Bardeleben contended that the results support the premise that the MitraClip G4 system is a meaningful advance by incorporating such features as an expanded choice of clip sizes, a greater coaptation area, and a more advanced gripper actuation for leaflet grasping.
Over 90% achieve MR 1+
Not least, it appears to increase the proportion of patients who achieve a mitral regurgitation grade of 1+ (MR1+) or lower, which is increasingly recognized as the goal of TEER, said Dr. von Bardeleben, head of the Centre of Structure Heart Disease Interventions, Heart Valve Centre, Mainz, Germany.
He said the rates of 91% achieving MR1+ or less and 98% achieved 2+ or lower compare favorably with most other series and exceeds levels achieved with surgery.
Dr. von Bardeleben also contended that, because of its design features, the MitraClip G4 “expands the spectrum of TEER-suitable patients.” He noted that 5% of the patients in this real-world series had a high risk of stenosis owing to such issues as severe annular or leaflet calcification and another 5% had factors that would predict inadequate MR reduction, such as Barlow’s disease, bi-leaflet prolapse, and severe leaflet degeneration.
The 1,164 patients in EXPAND G4 were enrolled from sites in the United States, Europe, Canada, and Japan. For the key outcome measure of procedural success, echocardiograms were assessed by an independent core laboratory. Of the 1,164 patients enrolled, 1,044 (91%) had complete follow-up data at 30 days.
The procedural success rates were reflected in improvements in New York Heart Association (NYHA) functional classes and in the Kansas City Cardiomyopathy Questionnaire (KCCQ), a quality of life instrument. Prior to treatment, 69% were in NYHA class III or greater. Following treatment, the proportion was 17% (P < .0001). The 18-point improvement in the KCCQ was characterized by Dr. von Bardeleben as “both clinically and statistically significant [P < .0001].”
There were no strokes in this series, and the 30-day incidence of myocardial infarction was 0.2%. The proportion requiring cardiovascular surgery within 30 days was less than 1%. The rate of bleeding episodes, all of which were nonserious, was 7%.
The “EXPAND G4 study confirms the safety and effectiveness of the next generation MitraClip G4 system,” according to Dr. von Bardeleben, and it did so “in a contemporary real-world setting.”
Outcome data characterized as ‘excellent’
Several invited panelists participating in a discussion following the presentation agreed.
“These results are excellent,” said Raj Makkar, MD, associate director of interventional technologies at Smidt Heart Institute, Cedars-Sinai Medical Center, Los Angeles. While he was impressed with the fact that only 2% missed the primary endpoint of MR 2+ or lower, he indicated that the 91% achieving MR 1+ or lower might be an even more apt signal that newer-generation devices are improving.
This was echoed by other panelists who appeared to form a general consensus over the premise that the target in TEER should no longer be MR 2+ for most patients.
“We should now be aiming for MR grade of 0-1,” stated panelist Stephan Windecker, MD, chairman, department of cardiology, University of Bern (Switzerland). He indicated that this goal is increasingly reasonable given the advances in device design and greater operator experience.
Dr. von Bardeleben reported financial relationships with Abbott Vascular, Edwards Lifesciences, Medtronic, and Neochord. Dr. Makkar reported financial relationships with Abbott Vascular, Cordis, Edwards Lifesciences, and Medtronic. Dr. Windecker reported financial relationships with more than 30 pharmaceutical companies, including Abbott Vascular, which manufactures MitraClip G4.
FROM TCT 2022
Hope shines bright for hidradenitis suppurativa treatments
in separate trials.
Around 40%-50% of patients exhibited a clinical response to these agents at 16 weeks, a leading HS expert reported at the annual congress of the European Academy of Dermatology and Venereology.
Time in the spotlight for HS
Research into HS is “an incredibly active field at this moment,” said Alexa B. Kimball, MD, MPH, professor of dermatology, Harvard Medical School, and president and chief executive officer of Harvard Medical Faculty Physicians at Beth Israel Deaconess Medical Center, Boston.
It’s “been great for advancing our understanding of the biology and the treatments that we will be able to use,” she said.

During the late-breaking sessions at the annual EADV Congress, Dr. Kimball presented data from two trials – SUNSHINE and SUNRISE – that investigated the efficacy, safety and tolerability of the interleukin (IL) 17A inhibitor secukinumab (Cosentyx) versus placebo in the treatment of moderate to severe HS.
“This is only the second phase 3 program we have ever seen in HS and the first one since 2016,” Dr. Kimball said of the trials. It’s also the largest trial program in HS conducted to date, she added, “so it really is a milestone.”
The last big development was when adalimumab, a tumor necrosis factor (TNF) blocker, gained regulatory approval for HS in 2016, observed Neil Patel, PhD, MRCP, who leads the HS service at Imperial College Healthcare NHS Trust in London.
“Adalimumab has been very helpful for many patients, but not all patients respond, and others may respond initially but then the treatment starts to fail after a year or 2,” Dr. Patel said in an interview with this news organization.
“There is definitely a huge need for alternative medication for this condition, which still has a lack of effective treatment options,” added Dr. Patel, who was not involved in either of the studies.
“One major upside for secukinumab is that its safety profile is generally very good and familiarity in the dermatologic community is already well established,” Christopher Sayed, MD, said in a separate interview.

“This will make most providers very comfortable offering it as a potential treatment option sooner rather than later given that its efficacy has now been demonstrated in phase 3 trials,” added Dr. Sayed, associate professor of dermatology at the University of North Carolina at Chapel Hill.
Two identically designed trials
Altogether, SUNSHINE and SUNRISE enrolled just over 1,000 patients at 219 sites in 33 countries. Both trials were identical in their design: A 4-week run-in phase before a randomized, double-blind treatment phase that tested two dosing regimens of secukinumab (300 mg administered subcutaneously) every 2 or 4 weeks vs. placebo for 16 weeks. The trial continued after this time, with patients in the placebo arm re–randomly assigned to treatment with one of the two secukinumab regimens out to a year.
The primary endpoint was the percentage of patients achieving a Hidradenitis Suppurativa Clinical Response (HiSCR) after 16 weeks of treatment, with key secondary endpoints, which were abscess and inflammatory nodule (AN) count, occurrence of flares, and at least a 30% reduction in Patient’s Global Assessment of Skin Pain assessed using a numeric rating scale (NPRS30).
Secukinumab superior to placebo
The HiSCR is defined as at least a 50% decrease in AN count with no increase in the number of abscesses or in the number of draining fistulas relative to baseline. This was achieved by about 42%-45% of patients who received secukinumab every 2 weeks, about 42%-46% of those who received secukinumab every 4 weeks, and about 31%-33% of those on placebo in both studies.
Of note, fewer patients treated with secukinumab (about 15%-20% among those treated every 2 weeks, and about 15% to 23% among those treated every 4 weeks) than those on placebo (27%-29%) experienced flares, defined as at least a 25% increase in AN count and at least a two-point increase relative to baseline values.
Improvement in HS pain can be a difficult parameter to meet, Dr. Kimball noted. “Pain is such an important feature of this disease as it so debilitating for the patients.” More than one-third (almost 36%-39%) of patients given secukinumab vs. just over a quarter (26.9%) given placebo achieved at least a 30% reduction in NPRS30 ratings, she reported. The difference between active and placebo treatment was significant only when secukinumab was given every 2 weeks, however.
“The placebo rates that we see in these studies are exactly parallel to what we saw in other studies, and other disease states when we had a 50% bar of improvement,” Dr. Kimball said when questioned about these results.
“HS is a highly variable disease; it’s maybe not so much the placebo rate or the scoring system used but maybe the 50% bar set for improvement is too low. It’s likely, as data start to mature and a 75% HiSCR can be calculated, that the placebo rates will drop,” she said.
There were no surprises when it came to the safety of secukinumab, being an old player in a new game, she noted. It was “well tolerated” and tolerability was “consistent with the known safety profile,” Dr. Kimball said, “so we expect it to be a new, safe, and effective add to our armamentarium in treating this disease.”
This research involves “basically borrowing drugs from other areas and trying them in HS to see what effect they may have,” Dr. Patel said, noting that drugs such as adalimumab and secukinumab already had a proven track record in other diseases, such as psoriasis. “These early data for secukinumab definitely are very exciting, but we would need to see real-life results” in patients with HS who are not enrolled in trials to see the benefits, he added.
‘Tipping point’ for HS research
“I think we will look back on this meeting and realize that it was an incredibly important tipping point for the treatment of this incredibly debilitating disease,” Dr. Kimball said.
Elsewhere at the meeting, she had presented findings from a phase 2a study that pitted three different kinase inhibitors with different modes of action against each other and compared them with placebo.
The three agents evaluated are an IL-1 receptor–associated kinase 4 inhibitor known as PF-06650833, a tyrosine kinase 2 (TYK2) JAK1 inhibitor brepocitinib, and the TYK2 inhibitor PF-06826647.
“This technique has been used in oncology,” Dr. Kimball said, noting that the ability to test multiple drugs at the same time “means we can really much more efficiently test two different things at the same time, and also put fewer patients at risk for potential problems if drugs don’t work.”
Positive signs for brepocitinib, not the other kinases tested
The results showed that though brepocitinib worked in HS, the other two novel compounds did not appear to have beneficial effects. Just over half (52%) of the 52 patients treated with brepocitinib achieved an HiSCR at 16 weeks, compared with around one-third of those given placebo, PF-06650833, or PF-06826647.
A similar benefit was seen in terms of reduction in flares for brepocitinib but not the other agents, although there was no difference between them all in terms of NPRS30 pain reduction.
“We’ve been able to test three different modalities. This tells us some things about the pathophysiology for HS, which is a very profoundly intensive inflammatory process,” which, Dr. Kimball said, “may require multiple modalities of action to get it under control.” In addition, these “general modalities seem to safe and well tolerated,” she added.
Take-homes for practice and future research
“While it is disappointing that two of the drugs tested did not clearly demonstrate efficacy, it is very possible that these mechanisms of action may be successful targets in the future as new dosing strategies and drugs targeting these pathways are developed,” Dr. Sayed said.
A case in point, he added, was that “adalimumab did not meet treatment endpoints at a dose of 40 mg every other week, but clearly has made a major impact at 40 mg weekly.”
The bottom line is that “both secukinumab and beprocitinib demonstrated efficacy over placebo and are likely to be helpful for a significant number of patients with HS,” Dr. Sayed said. “Hopefully, we’ll see head-to-head trials and more data regarding proportions of patients with deeper responses using criteria such as HiSCR75 and HiSCR90.”
Moreover, “having a larger number of drugs with a range of mechanisms of action is extraordinarily helpful given how difficult the disease can be to manage. We will hopefully continue to see creative approaches and further successes in the current wave of phase 1, 2, and 3 trials that are already underway.”
The SUNSHINE and SUNRISE studies were funded by Novartis Pharma AG, Basel, Switzerland. The phase 2A study Dr. Kimball presented was sponsored by Pfizer.
Dr. Kimball disclosed ties to both Novartis and Pfizer and acts as a consultant and investigator to AbbVie, Bristol-Myers Squibb, Janssen, Eli Lilly, Novartis, and UCB. She is an investigator for Incyte and AnaptysBio; acts as a consultant to Bayer, Boehringer Ingelheim, Ventyz, Moonlake, Lily, Concert, EvoImmune, Sonoma Bio, and Sanofi; receives fellowship funding from Janssen, and serves on the Board of Directors for Almirall.
Dr. Patel had no conflicts of interest to disclose. Dr. Sayed is the director of the HS Foundation, a nonprofit organization, and has acted as an adviser or consultant to, speaker for, and received research funding from multiple drug companies including AbbVie, ChemoCentryx, Incyte, InflaRx, Novartis, and UCB.
A version of this article first appeared on Medscape.com.
in separate trials.
Around 40%-50% of patients exhibited a clinical response to these agents at 16 weeks, a leading HS expert reported at the annual congress of the European Academy of Dermatology and Venereology.
Time in the spotlight for HS
Research into HS is “an incredibly active field at this moment,” said Alexa B. Kimball, MD, MPH, professor of dermatology, Harvard Medical School, and president and chief executive officer of Harvard Medical Faculty Physicians at Beth Israel Deaconess Medical Center, Boston.
It’s “been great for advancing our understanding of the biology and the treatments that we will be able to use,” she said.
During the late-breaking sessions at the annual EADV Congress, Dr. Kimball presented data from two trials – SUNSHINE and SUNRISE – that investigated the efficacy, safety and tolerability of the interleukin (IL) 17A inhibitor secukinumab (Cosentyx) versus placebo in the treatment of moderate to severe HS.
“This is only the second phase 3 program we have ever seen in HS and the first one since 2016,” Dr. Kimball said of the trials. It’s also the largest trial program in HS conducted to date, she added, “so it really is a milestone.”
The last big development was when adalimumab, a tumor necrosis factor (TNF) blocker, gained regulatory approval for HS in 2016, observed Neil Patel, PhD, MRCP, who leads the HS service at Imperial College Healthcare NHS Trust in London.
“Adalimumab has been very helpful for many patients, but not all patients respond, and others may respond initially but then the treatment starts to fail after a year or 2,” Dr. Patel said in an interview with this news organization.
“There is definitely a huge need for alternative medication for this condition, which still has a lack of effective treatment options,” added Dr. Patel, who was not involved in either of the studies.
“One major upside for secukinumab is that its safety profile is generally very good and familiarity in the dermatologic community is already well established,” Christopher Sayed, MD, said in a separate interview.
“This will make most providers very comfortable offering it as a potential treatment option sooner rather than later given that its efficacy has now been demonstrated in phase 3 trials,” added Dr. Sayed, associate professor of dermatology at the University of North Carolina at Chapel Hill.
Two identically designed trials
Altogether, SUNSHINE and SUNRISE enrolled just over 1,000 patients at 219 sites in 33 countries. Both trials were identical in their design: A 4-week run-in phase before a randomized, double-blind treatment phase that tested two dosing regimens of secukinumab (300 mg administered subcutaneously) every 2 or 4 weeks vs. placebo for 16 weeks. The trial continued after this time, with patients in the placebo arm re–randomly assigned to treatment with one of the two secukinumab regimens out to a year.
The primary endpoint was the percentage of patients achieving a Hidradenitis Suppurativa Clinical Response (HiSCR) after 16 weeks of treatment, with key secondary endpoints, which were abscess and inflammatory nodule (AN) count, occurrence of flares, and at least a 30% reduction in Patient’s Global Assessment of Skin Pain assessed using a numeric rating scale (NPRS30).
Secukinumab superior to placebo
The HiSCR is defined as at least a 50% decrease in AN count with no increase in the number of abscesses or in the number of draining fistulas relative to baseline. This was achieved by about 42%-45% of patients who received secukinumab every 2 weeks, about 42%-46% of those who received secukinumab every 4 weeks, and about 31%-33% of those on placebo in both studies.
Of note, fewer patients treated with secukinumab (about 15%-20% among those treated every 2 weeks, and about 15% to 23% among those treated every 4 weeks) than those on placebo (27%-29%) experienced flares, defined as at least a 25% increase in AN count and at least a two-point increase relative to baseline values.
Improvement in HS pain can be a difficult parameter to meet, Dr. Kimball noted. “Pain is such an important feature of this disease as it so debilitating for the patients.” More than one-third (almost 36%-39%) of patients given secukinumab vs. just over a quarter (26.9%) given placebo achieved at least a 30% reduction in NPRS30 ratings, she reported. The difference between active and placebo treatment was significant only when secukinumab was given every 2 weeks, however.
“The placebo rates that we see in these studies are exactly parallel to what we saw in other studies, and other disease states when we had a 50% bar of improvement,” Dr. Kimball said when questioned about these results.
“HS is a highly variable disease; it’s maybe not so much the placebo rate or the scoring system used but maybe the 50% bar set for improvement is too low. It’s likely, as data start to mature and a 75% HiSCR can be calculated, that the placebo rates will drop,” she said.
There were no surprises when it came to the safety of secukinumab, being an old player in a new game, she noted. It was “well tolerated” and tolerability was “consistent with the known safety profile,” Dr. Kimball said, “so we expect it to be a new, safe, and effective add to our armamentarium in treating this disease.”
This research involves “basically borrowing drugs from other areas and trying them in HS to see what effect they may have,” Dr. Patel said, noting that drugs such as adalimumab and secukinumab already had a proven track record in other diseases, such as psoriasis. “These early data for secukinumab definitely are very exciting, but we would need to see real-life results” in patients with HS who are not enrolled in trials to see the benefits, he added.
‘Tipping point’ for HS research
“I think we will look back on this meeting and realize that it was an incredibly important tipping point for the treatment of this incredibly debilitating disease,” Dr. Kimball said.
Elsewhere at the meeting, she had presented findings from a phase 2a study that pitted three different kinase inhibitors with different modes of action against each other and compared them with placebo.
The three agents evaluated are an IL-1 receptor–associated kinase 4 inhibitor known as PF-06650833, a tyrosine kinase 2 (TYK2) JAK1 inhibitor brepocitinib, and the TYK2 inhibitor PF-06826647.
“This technique has been used in oncology,” Dr. Kimball said, noting that the ability to test multiple drugs at the same time “means we can really much more efficiently test two different things at the same time, and also put fewer patients at risk for potential problems if drugs don’t work.”
Positive signs for brepocitinib, not the other kinases tested
The results showed that though brepocitinib worked in HS, the other two novel compounds did not appear to have beneficial effects. Just over half (52%) of the 52 patients treated with brepocitinib achieved an HiSCR at 16 weeks, compared with around one-third of those given placebo, PF-06650833, or PF-06826647.
A similar benefit was seen in terms of reduction in flares for brepocitinib but not the other agents, although there was no difference between them all in terms of NPRS30 pain reduction.
“We’ve been able to test three different modalities. This tells us some things about the pathophysiology for HS, which is a very profoundly intensive inflammatory process,” which, Dr. Kimball said, “may require multiple modalities of action to get it under control.” In addition, these “general modalities seem to safe and well tolerated,” she added.
Take-homes for practice and future research
“While it is disappointing that two of the drugs tested did not clearly demonstrate efficacy, it is very possible that these mechanisms of action may be successful targets in the future as new dosing strategies and drugs targeting these pathways are developed,” Dr. Sayed said.
A case in point, he added, was that “adalimumab did not meet treatment endpoints at a dose of 40 mg every other week, but clearly has made a major impact at 40 mg weekly.”
The bottom line is that “both secukinumab and beprocitinib demonstrated efficacy over placebo and are likely to be helpful for a significant number of patients with HS,” Dr. Sayed said. “Hopefully, we’ll see head-to-head trials and more data regarding proportions of patients with deeper responses using criteria such as HiSCR75 and HiSCR90.”
Moreover, “having a larger number of drugs with a range of mechanisms of action is extraordinarily helpful given how difficult the disease can be to manage. We will hopefully continue to see creative approaches and further successes in the current wave of phase 1, 2, and 3 trials that are already underway.”
The SUNSHINE and SUNRISE studies were funded by Novartis Pharma AG, Basel, Switzerland. The phase 2A study Dr. Kimball presented was sponsored by Pfizer.
Dr. Kimball disclosed ties to both Novartis and Pfizer and acts as a consultant and investigator to AbbVie, Bristol-Myers Squibb, Janssen, Eli Lilly, Novartis, and UCB. She is an investigator for Incyte and AnaptysBio; acts as a consultant to Bayer, Boehringer Ingelheim, Ventyz, Moonlake, Lily, Concert, EvoImmune, Sonoma Bio, and Sanofi; receives fellowship funding from Janssen, and serves on the Board of Directors for Almirall.
Dr. Patel had no conflicts of interest to disclose. Dr. Sayed is the director of the HS Foundation, a nonprofit organization, and has acted as an adviser or consultant to, speaker for, and received research funding from multiple drug companies including AbbVie, ChemoCentryx, Incyte, InflaRx, Novartis, and UCB.
A version of this article first appeared on Medscape.com.
in separate trials.
Around 40%-50% of patients exhibited a clinical response to these agents at 16 weeks, a leading HS expert reported at the annual congress of the European Academy of Dermatology and Venereology.
Time in the spotlight for HS
Research into HS is “an incredibly active field at this moment,” said Alexa B. Kimball, MD, MPH, professor of dermatology, Harvard Medical School, and president and chief executive officer of Harvard Medical Faculty Physicians at Beth Israel Deaconess Medical Center, Boston.
It’s “been great for advancing our understanding of the biology and the treatments that we will be able to use,” she said.
During the late-breaking sessions at the annual EADV Congress, Dr. Kimball presented data from two trials – SUNSHINE and SUNRISE – that investigated the efficacy, safety and tolerability of the interleukin (IL) 17A inhibitor secukinumab (Cosentyx) versus placebo in the treatment of moderate to severe HS.
“This is only the second phase 3 program we have ever seen in HS and the first one since 2016,” Dr. Kimball said of the trials. It’s also the largest trial program in HS conducted to date, she added, “so it really is a milestone.”
The last big development was when adalimumab, a tumor necrosis factor (TNF) blocker, gained regulatory approval for HS in 2016, observed Neil Patel, PhD, MRCP, who leads the HS service at Imperial College Healthcare NHS Trust in London.
“Adalimumab has been very helpful for many patients, but not all patients respond, and others may respond initially but then the treatment starts to fail after a year or 2,” Dr. Patel said in an interview with this news organization.
“There is definitely a huge need for alternative medication for this condition, which still has a lack of effective treatment options,” added Dr. Patel, who was not involved in either of the studies.
“One major upside for secukinumab is that its safety profile is generally very good and familiarity in the dermatologic community is already well established,” Christopher Sayed, MD, said in a separate interview.
“This will make most providers very comfortable offering it as a potential treatment option sooner rather than later given that its efficacy has now been demonstrated in phase 3 trials,” added Dr. Sayed, associate professor of dermatology at the University of North Carolina at Chapel Hill.
Two identically designed trials
Altogether, SUNSHINE and SUNRISE enrolled just over 1,000 patients at 219 sites in 33 countries. Both trials were identical in their design: A 4-week run-in phase before a randomized, double-blind treatment phase that tested two dosing regimens of secukinumab (300 mg administered subcutaneously) every 2 or 4 weeks vs. placebo for 16 weeks. The trial continued after this time, with patients in the placebo arm re–randomly assigned to treatment with one of the two secukinumab regimens out to a year.
The primary endpoint was the percentage of patients achieving a Hidradenitis Suppurativa Clinical Response (HiSCR) after 16 weeks of treatment, with key secondary endpoints, which were abscess and inflammatory nodule (AN) count, occurrence of flares, and at least a 30% reduction in Patient’s Global Assessment of Skin Pain assessed using a numeric rating scale (NPRS30).
Secukinumab superior to placebo
The HiSCR is defined as at least a 50% decrease in AN count with no increase in the number of abscesses or in the number of draining fistulas relative to baseline. This was achieved by about 42%-45% of patients who received secukinumab every 2 weeks, about 42%-46% of those who received secukinumab every 4 weeks, and about 31%-33% of those on placebo in both studies.
Of note, fewer patients treated with secukinumab (about 15%-20% among those treated every 2 weeks, and about 15% to 23% among those treated every 4 weeks) than those on placebo (27%-29%) experienced flares, defined as at least a 25% increase in AN count and at least a two-point increase relative to baseline values.
Improvement in HS pain can be a difficult parameter to meet, Dr. Kimball noted. “Pain is such an important feature of this disease as it so debilitating for the patients.” More than one-third (almost 36%-39%) of patients given secukinumab vs. just over a quarter (26.9%) given placebo achieved at least a 30% reduction in NPRS30 ratings, she reported. The difference between active and placebo treatment was significant only when secukinumab was given every 2 weeks, however.
“The placebo rates that we see in these studies are exactly parallel to what we saw in other studies, and other disease states when we had a 50% bar of improvement,” Dr. Kimball said when questioned about these results.
“HS is a highly variable disease; it’s maybe not so much the placebo rate or the scoring system used but maybe the 50% bar set for improvement is too low. It’s likely, as data start to mature and a 75% HiSCR can be calculated, that the placebo rates will drop,” she said.
There were no surprises when it came to the safety of secukinumab, being an old player in a new game, she noted. It was “well tolerated” and tolerability was “consistent with the known safety profile,” Dr. Kimball said, “so we expect it to be a new, safe, and effective add to our armamentarium in treating this disease.”
This research involves “basically borrowing drugs from other areas and trying them in HS to see what effect they may have,” Dr. Patel said, noting that drugs such as adalimumab and secukinumab already had a proven track record in other diseases, such as psoriasis. “These early data for secukinumab definitely are very exciting, but we would need to see real-life results” in patients with HS who are not enrolled in trials to see the benefits, he added.
‘Tipping point’ for HS research
“I think we will look back on this meeting and realize that it was an incredibly important tipping point for the treatment of this incredibly debilitating disease,” Dr. Kimball said.
Elsewhere at the meeting, she had presented findings from a phase 2a study that pitted three different kinase inhibitors with different modes of action against each other and compared them with placebo.
The three agents evaluated are an IL-1 receptor–associated kinase 4 inhibitor known as PF-06650833, a tyrosine kinase 2 (TYK2) JAK1 inhibitor brepocitinib, and the TYK2 inhibitor PF-06826647.
“This technique has been used in oncology,” Dr. Kimball said, noting that the ability to test multiple drugs at the same time “means we can really much more efficiently test two different things at the same time, and also put fewer patients at risk for potential problems if drugs don’t work.”
Positive signs for brepocitinib, not the other kinases tested
The results showed that though brepocitinib worked in HS, the other two novel compounds did not appear to have beneficial effects. Just over half (52%) of the 52 patients treated with brepocitinib achieved an HiSCR at 16 weeks, compared with around one-third of those given placebo, PF-06650833, or PF-06826647.
A similar benefit was seen in terms of reduction in flares for brepocitinib but not the other agents, although there was no difference between them all in terms of NPRS30 pain reduction.
“We’ve been able to test three different modalities. This tells us some things about the pathophysiology for HS, which is a very profoundly intensive inflammatory process,” which, Dr. Kimball said, “may require multiple modalities of action to get it under control.” In addition, these “general modalities seem to safe and well tolerated,” she added.
Take-homes for practice and future research
“While it is disappointing that two of the drugs tested did not clearly demonstrate efficacy, it is very possible that these mechanisms of action may be successful targets in the future as new dosing strategies and drugs targeting these pathways are developed,” Dr. Sayed said.
A case in point, he added, was that “adalimumab did not meet treatment endpoints at a dose of 40 mg every other week, but clearly has made a major impact at 40 mg weekly.”
The bottom line is that “both secukinumab and beprocitinib demonstrated efficacy over placebo and are likely to be helpful for a significant number of patients with HS,” Dr. Sayed said. “Hopefully, we’ll see head-to-head trials and more data regarding proportions of patients with deeper responses using criteria such as HiSCR75 and HiSCR90.”
Moreover, “having a larger number of drugs with a range of mechanisms of action is extraordinarily helpful given how difficult the disease can be to manage. We will hopefully continue to see creative approaches and further successes in the current wave of phase 1, 2, and 3 trials that are already underway.”
The SUNSHINE and SUNRISE studies were funded by Novartis Pharma AG, Basel, Switzerland. The phase 2A study Dr. Kimball presented was sponsored by Pfizer.
Dr. Kimball disclosed ties to both Novartis and Pfizer and acts as a consultant and investigator to AbbVie, Bristol-Myers Squibb, Janssen, Eli Lilly, Novartis, and UCB. She is an investigator for Incyte and AnaptysBio; acts as a consultant to Bayer, Boehringer Ingelheim, Ventyz, Moonlake, Lily, Concert, EvoImmune, Sonoma Bio, and Sanofi; receives fellowship funding from Janssen, and serves on the Board of Directors for Almirall.
Dr. Patel had no conflicts of interest to disclose. Dr. Sayed is the director of the HS Foundation, a nonprofit organization, and has acted as an adviser or consultant to, speaker for, and received research funding from multiple drug companies including AbbVie, ChemoCentryx, Incyte, InflaRx, Novartis, and UCB.
A version of this article first appeared on Medscape.com.
FROM THE EADV CONGRESS
Around 10% of back pain patients referred by chiropractors have undiagnosed SpA
GHENT, BELGIUM – Over 10% of patients referred by chiropractors to rheumatology had undiagnosed spondyloarthritis, with axial spondyloarthritis being the most common, according to new data. The U.S. study was aimed at understanding what proportion of back pain patients have undiagnosed spondyloarthritis.
The study also found that the most common cause for which patients see chiropractors is neck/cervical pain.
Atul Deodhar, MD, MRCP, rheumatologist and medical director of rheumatology clinics at Oregon Health & Science University, Portland, was senior author of the poster that was presented at the 13th International Congress of Spondyloarthritides.

“In the U.S., many people with back pain go to chiropractors, but many chiropractors are not aware of axial spondyloarthritis [axSpA] terminology, and very little – if anything – is published in chiropractic literature, “ he said in an interview.
He remarked that the study highlighted the need to develop a better strategy to identify undiagnosed patients, because the yield found in their study was poor (13%). “Patient-reported spondyloarthritis criteria are often poor, and do not match rheumatologist-inquired history,” he noted, adding that, “inflammatory back pain is in fact a poor ‘entry point.’ ”
Ulrich Weber, MD, rheumatologist from the Practice Buchsbaum in Schaffhausen, Switzerland, commented on the findings, saying he often receives delayed referrals from chiropractors, so
He added that he welcomed the study but noted, “the criteria used to identify patients in this study are broad and I’d worry that it would inundate our rheumatology practice. There remains a real need for a good method of identifying the patients.”
Referral to rheumatology
Back pain is highly prevalent in the general population, with a global mean lifetime prevalence of 38.9%. Chiropractors treat many patients with back pain of unknown cause.
“In this study, we wanted to see what percentage of patients in chiropractic practice have undiagnosed axial spondyloarthritis, and what are the common complaints. Our hypothesis was that chiropractors may be missing such patients,” Dr. Deodhar explained.
Dr. Deodhar and colleagues recruited chiropractors from four different parts of the city of Portland into the study. “We think Portland, Oregon, is a typical U.S. city, and our results could be generalized. However, this is our impression alone,” he remarked.
Adults, under the age of 45 years who attended a participating chiropractic clinics between November 2020 and November 2021 for chronic back pain and without a prior diagnosis of spondyloarthritis were eligible for inclusion.
If the patient reported at least one feature of spondyloarthritis in the screening questionnaire they were referred to a rheumatologist for a diagnostic assessment. This assessment involved taking history by telephone, both laboratory tests and imaging, and the patients were categorized as radiographic axSpA, nonradiographic axSpA, peripheral SpA, or no SpA.
The screening questionnaire included the following examples: If the patient was under 45 years and had chronic pain in back, hip or buttocks, then they were asked for more information including whether their pain was gradual (insidious) in onset; if the pain started before the age of 40; and if the pain improved with physical activities or movements. Use of drugs was investigated including whether the pain improved significantly with NSAIDs and whether the patient has current or past heel pains, particularly when waking up in the morning. They were also asked if they have experienced skin psoriasis. Other questions were asked about the presence of uveitis, iritis, family history of psoriasis, inflammatory bowel disease, or ankylosing spondylitis, and whether the patient had unexplained joint pains plus joint swelling.
Ten percent of patients referred to rheumatology
A total of 3,103 visits to chiropractor clinics were included, of which 115 patients were referred to a rheumatologist. Eventually, 63 patients were fully assessed by a rheumatologist.
Of those patients who were fully assessed, 12.7% has spondyloarthritis, with one having confirmed radiographic axSpA, five having nonradiographic SpA, and two having peripheral spondyloarthritis or psoriatic arthritis.
Based on the referral questionnaire, all patients reported at least four SpA criteria were met, said Dr. Deodhar.
Of those patients diagnosed with SpA, 14% (1) has elevated C-reactive protein (CRP) level, 14% (1) were HLA-B27 positive, and 14% (1) were identified as having both elevated CRP and HLA-B27 positivity. Sacroiliac joint inflammation was found in 14% (1) on MRI and one had sacroiliac joint inflammation according to modified New York criteria. One (14%) had both sacroiliac joint inflammation on MRI and elevated CRP, and 14% (1) had both sacroiliac joint inflammation and was HLA-B27 positive.
The top complaints reported by patients at chiropractor clinics were neck and cervical spine pain/spasm (16.8%); followed by acute low back pain (11.7%); acute upper back (7.1%); and chronic lower back pain (6.9%).
No patients with more than 10 SpA criteria
The performance of an initial diagnostic assessment based on patient reported SpA criteria, as compared with the outcome of the full diagnosis (by a rheumatologist) showed that patients with one to four SpA criteria had a sensitivity of 0.50 (95% confidence interval, 0.15-0.85), and specificity of 0.73 (95% CI, 0.61-0.84). This increased to sensitivity of 0.60 (95% CI, 0.17-1.03), and specificity of 0.61 (95% CI, 0.44-0.77) when six SpA criteria were present.
Dr. Deodhar said the results supported a need to further develop the chiropractor’s role in identifying the right patients for referral, and that the study showed that a referral strategy is required to find undiagnosed patients with spondyloarthritis from chiropractic offices. “Chiropractors need education for axSpA, when to suspect, and when to refer,” he asserted. “What referral strategy to use is for debate – the ASAS [Assessment in SpondyloArthritis international Society] strategy is too sensitive and not specific enough.”
Dr. Deodhar noted that SPARTAN (Group for Research and Assessment of Psoriasis and Psoriatic Arthritis and the Spondyloarthritis Research & Treatment Network) is working on a referral strategy that is likely to be more specific, and that more data would be forthcoming soon.
Dr. Deodhar declared affiliations with multiple companies involved in the field unrelated to the study. Dr. Weber declared no relevant disclosures.
GHENT, BELGIUM – Over 10% of patients referred by chiropractors to rheumatology had undiagnosed spondyloarthritis, with axial spondyloarthritis being the most common, according to new data. The U.S. study was aimed at understanding what proportion of back pain patients have undiagnosed spondyloarthritis.
The study also found that the most common cause for which patients see chiropractors is neck/cervical pain.
Atul Deodhar, MD, MRCP, rheumatologist and medical director of rheumatology clinics at Oregon Health & Science University, Portland, was senior author of the poster that was presented at the 13th International Congress of Spondyloarthritides.
“In the U.S., many people with back pain go to chiropractors, but many chiropractors are not aware of axial spondyloarthritis [axSpA] terminology, and very little – if anything – is published in chiropractic literature, “ he said in an interview.
He remarked that the study highlighted the need to develop a better strategy to identify undiagnosed patients, because the yield found in their study was poor (13%). “Patient-reported spondyloarthritis criteria are often poor, and do not match rheumatologist-inquired history,” he noted, adding that, “inflammatory back pain is in fact a poor ‘entry point.’ ”
Ulrich Weber, MD, rheumatologist from the Practice Buchsbaum in Schaffhausen, Switzerland, commented on the findings, saying he often receives delayed referrals from chiropractors, so
He added that he welcomed the study but noted, “the criteria used to identify patients in this study are broad and I’d worry that it would inundate our rheumatology practice. There remains a real need for a good method of identifying the patients.”
Referral to rheumatology
Back pain is highly prevalent in the general population, with a global mean lifetime prevalence of 38.9%. Chiropractors treat many patients with back pain of unknown cause.
“In this study, we wanted to see what percentage of patients in chiropractic practice have undiagnosed axial spondyloarthritis, and what are the common complaints. Our hypothesis was that chiropractors may be missing such patients,” Dr. Deodhar explained.
Dr. Deodhar and colleagues recruited chiropractors from four different parts of the city of Portland into the study. “We think Portland, Oregon, is a typical U.S. city, and our results could be generalized. However, this is our impression alone,” he remarked.
Adults, under the age of 45 years who attended a participating chiropractic clinics between November 2020 and November 2021 for chronic back pain and without a prior diagnosis of spondyloarthritis were eligible for inclusion.
If the patient reported at least one feature of spondyloarthritis in the screening questionnaire they were referred to a rheumatologist for a diagnostic assessment. This assessment involved taking history by telephone, both laboratory tests and imaging, and the patients were categorized as radiographic axSpA, nonradiographic axSpA, peripheral SpA, or no SpA.
The screening questionnaire included the following examples: If the patient was under 45 years and had chronic pain in back, hip or buttocks, then they were asked for more information including whether their pain was gradual (insidious) in onset; if the pain started before the age of 40; and if the pain improved with physical activities or movements. Use of drugs was investigated including whether the pain improved significantly with NSAIDs and whether the patient has current or past heel pains, particularly when waking up in the morning. They were also asked if they have experienced skin psoriasis. Other questions were asked about the presence of uveitis, iritis, family history of psoriasis, inflammatory bowel disease, or ankylosing spondylitis, and whether the patient had unexplained joint pains plus joint swelling.
Ten percent of patients referred to rheumatology
A total of 3,103 visits to chiropractor clinics were included, of which 115 patients were referred to a rheumatologist. Eventually, 63 patients were fully assessed by a rheumatologist.
Of those patients who were fully assessed, 12.7% has spondyloarthritis, with one having confirmed radiographic axSpA, five having nonradiographic SpA, and two having peripheral spondyloarthritis or psoriatic arthritis.
Based on the referral questionnaire, all patients reported at least four SpA criteria were met, said Dr. Deodhar.
Of those patients diagnosed with SpA, 14% (1) has elevated C-reactive protein (CRP) level, 14% (1) were HLA-B27 positive, and 14% (1) were identified as having both elevated CRP and HLA-B27 positivity. Sacroiliac joint inflammation was found in 14% (1) on MRI and one had sacroiliac joint inflammation according to modified New York criteria. One (14%) had both sacroiliac joint inflammation on MRI and elevated CRP, and 14% (1) had both sacroiliac joint inflammation and was HLA-B27 positive.
The top complaints reported by patients at chiropractor clinics were neck and cervical spine pain/spasm (16.8%); followed by acute low back pain (11.7%); acute upper back (7.1%); and chronic lower back pain (6.9%).
No patients with more than 10 SpA criteria
The performance of an initial diagnostic assessment based on patient reported SpA criteria, as compared with the outcome of the full diagnosis (by a rheumatologist) showed that patients with one to four SpA criteria had a sensitivity of 0.50 (95% confidence interval, 0.15-0.85), and specificity of 0.73 (95% CI, 0.61-0.84). This increased to sensitivity of 0.60 (95% CI, 0.17-1.03), and specificity of 0.61 (95% CI, 0.44-0.77) when six SpA criteria were present.
Dr. Deodhar said the results supported a need to further develop the chiropractor’s role in identifying the right patients for referral, and that the study showed that a referral strategy is required to find undiagnosed patients with spondyloarthritis from chiropractic offices. “Chiropractors need education for axSpA, when to suspect, and when to refer,” he asserted. “What referral strategy to use is for debate – the ASAS [Assessment in SpondyloArthritis international Society] strategy is too sensitive and not specific enough.”
Dr. Deodhar noted that SPARTAN (Group for Research and Assessment of Psoriasis and Psoriatic Arthritis and the Spondyloarthritis Research & Treatment Network) is working on a referral strategy that is likely to be more specific, and that more data would be forthcoming soon.
Dr. Deodhar declared affiliations with multiple companies involved in the field unrelated to the study. Dr. Weber declared no relevant disclosures.
GHENT, BELGIUM – Over 10% of patients referred by chiropractors to rheumatology had undiagnosed spondyloarthritis, with axial spondyloarthritis being the most common, according to new data. The U.S. study was aimed at understanding what proportion of back pain patients have undiagnosed spondyloarthritis.
The study also found that the most common cause for which patients see chiropractors is neck/cervical pain.
Atul Deodhar, MD, MRCP, rheumatologist and medical director of rheumatology clinics at Oregon Health & Science University, Portland, was senior author of the poster that was presented at the 13th International Congress of Spondyloarthritides.
“In the U.S., many people with back pain go to chiropractors, but many chiropractors are not aware of axial spondyloarthritis [axSpA] terminology, and very little – if anything – is published in chiropractic literature, “ he said in an interview.
He remarked that the study highlighted the need to develop a better strategy to identify undiagnosed patients, because the yield found in their study was poor (13%). “Patient-reported spondyloarthritis criteria are often poor, and do not match rheumatologist-inquired history,” he noted, adding that, “inflammatory back pain is in fact a poor ‘entry point.’ ”
Ulrich Weber, MD, rheumatologist from the Practice Buchsbaum in Schaffhausen, Switzerland, commented on the findings, saying he often receives delayed referrals from chiropractors, so
He added that he welcomed the study but noted, “the criteria used to identify patients in this study are broad and I’d worry that it would inundate our rheumatology practice. There remains a real need for a good method of identifying the patients.”
Referral to rheumatology
Back pain is highly prevalent in the general population, with a global mean lifetime prevalence of 38.9%. Chiropractors treat many patients with back pain of unknown cause.
“In this study, we wanted to see what percentage of patients in chiropractic practice have undiagnosed axial spondyloarthritis, and what are the common complaints. Our hypothesis was that chiropractors may be missing such patients,” Dr. Deodhar explained.
Dr. Deodhar and colleagues recruited chiropractors from four different parts of the city of Portland into the study. “We think Portland, Oregon, is a typical U.S. city, and our results could be generalized. However, this is our impression alone,” he remarked.
Adults, under the age of 45 years who attended a participating chiropractic clinics between November 2020 and November 2021 for chronic back pain and without a prior diagnosis of spondyloarthritis were eligible for inclusion.
If the patient reported at least one feature of spondyloarthritis in the screening questionnaire they were referred to a rheumatologist for a diagnostic assessment. This assessment involved taking history by telephone, both laboratory tests and imaging, and the patients were categorized as radiographic axSpA, nonradiographic axSpA, peripheral SpA, or no SpA.
The screening questionnaire included the following examples: If the patient was under 45 years and had chronic pain in back, hip or buttocks, then they were asked for more information including whether their pain was gradual (insidious) in onset; if the pain started before the age of 40; and if the pain improved with physical activities or movements. Use of drugs was investigated including whether the pain improved significantly with NSAIDs and whether the patient has current or past heel pains, particularly when waking up in the morning. They were also asked if they have experienced skin psoriasis. Other questions were asked about the presence of uveitis, iritis, family history of psoriasis, inflammatory bowel disease, or ankylosing spondylitis, and whether the patient had unexplained joint pains plus joint swelling.
Ten percent of patients referred to rheumatology
A total of 3,103 visits to chiropractor clinics were included, of which 115 patients were referred to a rheumatologist. Eventually, 63 patients were fully assessed by a rheumatologist.
Of those patients who were fully assessed, 12.7% has spondyloarthritis, with one having confirmed radiographic axSpA, five having nonradiographic SpA, and two having peripheral spondyloarthritis or psoriatic arthritis.
Based on the referral questionnaire, all patients reported at least four SpA criteria were met, said Dr. Deodhar.
Of those patients diagnosed with SpA, 14% (1) has elevated C-reactive protein (CRP) level, 14% (1) were HLA-B27 positive, and 14% (1) were identified as having both elevated CRP and HLA-B27 positivity. Sacroiliac joint inflammation was found in 14% (1) on MRI and one had sacroiliac joint inflammation according to modified New York criteria. One (14%) had both sacroiliac joint inflammation on MRI and elevated CRP, and 14% (1) had both sacroiliac joint inflammation and was HLA-B27 positive.
The top complaints reported by patients at chiropractor clinics were neck and cervical spine pain/spasm (16.8%); followed by acute low back pain (11.7%); acute upper back (7.1%); and chronic lower back pain (6.9%).
No patients with more than 10 SpA criteria
The performance of an initial diagnostic assessment based on patient reported SpA criteria, as compared with the outcome of the full diagnosis (by a rheumatologist) showed that patients with one to four SpA criteria had a sensitivity of 0.50 (95% confidence interval, 0.15-0.85), and specificity of 0.73 (95% CI, 0.61-0.84). This increased to sensitivity of 0.60 (95% CI, 0.17-1.03), and specificity of 0.61 (95% CI, 0.44-0.77) when six SpA criteria were present.
Dr. Deodhar said the results supported a need to further develop the chiropractor’s role in identifying the right patients for referral, and that the study showed that a referral strategy is required to find undiagnosed patients with spondyloarthritis from chiropractic offices. “Chiropractors need education for axSpA, when to suspect, and when to refer,” he asserted. “What referral strategy to use is for debate – the ASAS [Assessment in SpondyloArthritis international Society] strategy is too sensitive and not specific enough.”
Dr. Deodhar noted that SPARTAN (Group for Research and Assessment of Psoriasis and Psoriatic Arthritis and the Spondyloarthritis Research & Treatment Network) is working on a referral strategy that is likely to be more specific, and that more data would be forthcoming soon.
Dr. Deodhar declared affiliations with multiple companies involved in the field unrelated to the study. Dr. Weber declared no relevant disclosures.
AT THE 2022 SPA CONGRESS