User login
Acute Onset of Vancomycin Anaphylaxis With Disseminated Intravascular Coagulation in an Orthopedic Patient Despite Prior Repeated Exposure
Vancomycin is a glycopeptide antibiotic that exhibits bactericidal activity against gram-positive cocci. It is commonly recommended for surgical prophylaxis in cases of suspected bacterial resistance or penicillin allergy.1 Two main types of hypersensitivity reactions associated with vancomycin can have similar presentations. Red man syndrome is an anaphylactoid reaction caused by direct release of histamine from mast cells via a nonimmunologic mechanism, and is the more common of the 2 reactions. The second type is an anaphylactic reaction, which is an immunoglobulin E (IgE)–mediated systemic event and requires exposure to become sensitized.2,3
We present a patient who had received vancomycin on at least 12 occasions without incident. On this occasion, however, she developed a true anaphylactic reaction causing acute hemodynamic collapse that she survived after extensive resuscitation. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 55-year-old woman had a history of metastatic giant cell tumor of the right proximal tibia. She was originally treated 27 years ago for proximal tibial resection and reconstruction with a custom proximal tibial prosthesis. Four months later, she underwent resection of multiple pulmonary metastases via bilateral thoracotomies in a single surgical setting. After this, the patient had no evidence of recurrent metastatic disease. In subsequent years, the patient underwent multiple revision surgeries for problems such as hardware failure, patellar maltracking, and infection. The patient underwent 19 operations, including several nonorthopedic procedures. Because the patient had a rash after receiving penicillin as a child, she was thought to be allergic to penicillin. Consequently, she received vancomycin as antibiotic prophylaxis for the majority of these procedures. She also received extended courses of vancomycin of at least 6 weeks on 2 separate occasions. During her most recent revision procedure, 6 weeks prior to the procedure under discussion, the patient took vancomycin without incident. She was then found to have a prosthetic infection with Staphylococcus epidermidis, the same organism isolated in her previous infections, and she was advised to undergo a staged revision.
After a preoperative medical evaluation by her primary care physician, the patient was taken to the operating room for prosthesis removal and antibiotic spacer placement. She was anemic with a hemoglobin level of 8.8 g/dL; her erythrocyte sedimentation rate (ESR) was 102 mm/h (normal, <22 mm/h) and her C-reactive protein (CRP) was 38 mg/L (normal, <3 mg/L), but, otherwise, her laboratory values were normal, including a white blood cell count (WBC) of 8100/µL. Her electrocardiogram showed a normal sinus rhythm with nonspecific ST- and T-wave changes. Antibiotics were held until after cultures were taken. General endotracheal tube anesthesia was induced with 2 mg midazolam, 100 µg fentanyl, 180 mg propofol, and 140 mg succinylcholine, followed by 10 mg vecuronium, and maintained with desflurane. A tourniquet was not used per the surgeon’s routine. Dissection was carried down to the prosthesis and showed a small amount of purulent fluid. Transfusion of 1 unit of packed red blood cells (pRBC) was started during the approach owing to relatively low preoperative hemoglobin and significant blood loss. Approximately 500 mL of blood was lost during the approach secondary to the extensive dissection and the local inflammatory response from infection and recent surgery. After cultures were taken, and approximately 10 minutes after blood transfusion began, infusion of 1 g vancomycin in 250 mL normal saline was started via an infusion pump to run over 1 hour.
After infusion of 5 mL vancomycin, the patient’s blood pressure dropped from 117/63 mm Hg to 63/30 mm Hg; her pulse concurrently dropped from 90 to 50 beats/min. Vancomycin infusion was immediately stopped, anesthesia gasses were turned off, and patient received a bolus of normal saline with a second unit of pRBC. Patient received boluses of 0.5 mg to 1.0 mg epinephrine and 100 µg phenylephrine without sustained increase in blood pressure, which had dropped to 54/24 mm Hg, although the patient became tachycardic to ~120 beats/min after epinephrine. A sudden drop in end-tidal CO2 from 40s mm Hg to 20s mm Hg was also noted, indicating continuous but significantly decreased perfusion of the lungs.
We elected to abort the procedure, and a vacuum-assisted closure (VAC) dressing was applied to the open wound. After 15 minutes, the patient’s pulses, which had been faint, became impalpable, and cardiopulmonary resuscitation was initiated for about 7 minutes. The patient received 40 units vasopressin with repeated boluses of 0.5 mg epinephrine; a norepinephrine continuous infusion was started with the return of pulses. The patient also received 50 mg diphenhydramine, 125 mg methylprednisolone, and 20 mg famotidine for suspected anaphylaxis. A central venous line and arterial line were placed, and blood was drawn for laboratory analysis. The patient was noted to have clear breath sounds with no obvious rash, and her urine remained clear. Blood gas showed a profound metabolic acidosis, with pH of 7.09, base deficit of 5.9, and lactate of 8.9. The patient was treated with bicarbonate infusion. The patient was noted to ooze significantly during central venous line and arterial line placement, despite apparently normal coagulation during the surgical approach. Coagulation values were consistent with disseminated intravascular coagulation (DIC): prothrombin time, 57 s (international normalized ratio, 6.7); partial thromboplastin time, >200 s; thrombin time, 110 s; D-dimer, >10,000 ng/mL (normal, 0-200 ng/mL); and fibrinogen, <60 mg/dL (normal, 222-475 mg/dL). The patient’s thromboelastogram showed a flat line indicating an absence of clotting. Interestingly, the platelet count remained near the preoperative level at 338×103/µL. The patient’s blood pressure remained labile and was responsive primarily to epinephrine boluses, of which she received a total of 5 mg. After 1 hour of resuscitation, during which time the patient received a total of 5 L crystalloid and 3 units pRBC, the patient was transferred to the intensive care unit (ICU), intubated, and started on a titrated epinephrine infusion.
Upon arrival in the ICU, the patient quickly stabilized hemodynamically. She was weaned from all inotropic support within 2 hours of arrival. The patient lost 800 mL of blood through wound VAC over the first 12 hours postoperatively and required a total of 11 units of pRBC, 6 units fresh frozen plasma, and 3 units of pooled cryoprecipitate, all of which were compatible. Laboratory values, including arterial pH, lactic acid, and coagulation studies, normalized on the evening of surgery, and, by the next morning, the patient was alert and was extubated without difficulty. Steroids were tapered without hemodynamic compromise while the patient was in the ICU. Cardiology examination revealed no abnormalities. Because of the temporal association of blood transfusion with cardiovascular collapse, pRBC units were retested for antibodies and cultured. Both of these investigations were negative. Wound cultures again were positive for Staphylococcus epidermidis, and blood cultures were negative. The patient was started on daptomycin based on susceptibility profiles. Serum histamine levels taken during initial resuscitation in the operating room were normal. The serum tryptase level obtained at the same time was markedly elevated at >700 ng/mL (normal, <11.5 ng/mL), although this information was not available until several days later.
The patient underwent 2 additional surgeries during the same admission, including the prosthesis removal and tobramycin cement spacer placement, without incident. She was discharged home, again without incident. The patient was later evaluated by an outside allergist and underwent skin puncture and intradermal allergy testing. The results were consistent with a strong IgE-mediated hypersensitivity. Interestingly, she was found not to have a penicillin allergy.
Discussion
Vancomycin hypersensitivity reactions include the anaphylactoid reaction red man syndrome and a true IgE-mediated anaphylactic reaction. Red man syndrome is much more common, with reported rates in infected patients from 3.7% to 47%,4,5 when vancomycin is given at the suggested rate of 1 g over 1 hour. The reaction occurs because of histamine release from mast cells and basophils, and does not require previous sensitization.3 The rate of infusion is directly related to the development of symptoms, with 100% of patients developing symptoms in 1 study with rapid infusion (1 g over 10 min).6 Red man syndrome can typically be prevented by slowing the rate of infusion or by giving an H1 blocker.3 Anaphylaxis is more rare but can occur.7 Anaphylaxis is mediated by vancomycin-specific IgE, which requires previous exposure, as was the case with our patient. Interestingly, the patient had received vancomycin many times without any signs of a hypersensitivity reaction. Antihistamines are not effective in treating anaphylaxis, and epinephrine is the first-line agent.3 This was clearly demonstrated in this case, as there was a significant hemodynamic response to epinephrine and a negligible response to other vasopressors, specifically norepinephrine and vasopressin.
Most hypersensitivity reactions during the course of a surgical procedure occur with induction of anesthesia, with neuromuscular blocking agents and antibiotics being the most common causes.8 In our case, antibiotics were held until after deep cultures were taken. Given the time from induction to the anaphylactic reaction, it is unlikely the reaction resulted from the induction agents or the neuromuscular blocking agent. The possibility of a transfusion reaction was also investigated, since a unit of pRBC was still being transfused when symptoms began. An acute hemolytic transfusion reaction has the classic triad of fever, flank pain, and hemoglobinuria, and can also present as DIC.9 Under anesthesia, DIC can often be the presenting sign. In this case, a hemolytic transfusion reaction appeared very unlikely. All of the blood components the patient received were rechecked and found to be compatible, posttransfusion analysis showed no evidence of hemolysis in any sample, and the direct antiglobulin test was negative in all components.
To our knowledge, there are no reported cases of vancomycin-induced anaphylaxis with concomitant DIC. Symptoms of anaphylaxis after exposure to a possible antigen include rapid onset of hypotension or rapid onset of signs in at least 2 organ systems, including cutaneous, gastrointestinal, respiratory, and cardiovascular.10 Anaphylaxis with DIC is rare after exposure to any substance but has been reported.11 In fact, induction of systemic anaphylaxis in mice is known to cause DIC, with platelet-activating factor suggested as an important common mediator. A similar mechanism is suspected in humans.12
Confirmation of, and, certainly, prediction of, a vancomycin hypersensitivity reaction is difficult. Histamine levels can be used as a measure of mast-cell degranulation, but serum levels peak within 5 minutes and quickly return to baseline, limiting its diagnostic usefulness.3 Tryptase is an enzyme found in the secretory granules of mast cells. It has become an accepted marker of acute anaphylaxis, and, in vancomycin hypersensitivity reactions, can also distinguish between anaphylactic and anaphylactoid reactions.13 Tryptase levels peak 1 to 2 hours after the reaction, making this easier to measure than histamine, but results may not be available for several days, making it useful only in retrospect, as in our case. Skin testing is probably the best way to confirm a hypersensitivity reaction, although even this has been questioned with vancomycin because some find a high false-positive rate3, while others think the false-negative rate is likely too high.7 In this case, we were able to confirm our initial clinical suspicion with both an elevated tryptase level and a positive skin test.
Conclusion
We present a rare case of vancomycin anaphylaxis with DIC after repeated and prolonged previous exposure, which was treated acutely with hemodynamic resuscitation, replacement of blood components, steroids, and, most importantly, repeated boluses of epinephrine. Although several papers have described successful vancomycin desensitization7, this was fortunately not necessary in this case because the causative organism was sensitive to other acceptable antibiotics. The patient has been treated with systemic daptomycin and a tobramycin cement spacer without further incident.
1. Recommendation for the use of intravenous antibiotic prophylaxis in primary total joint arthroplasty. AAOS Information Statement 1027. American Academy of Orthopaedic Surgeons website. http://www.aaos.org/about/papers/advistmt/1027.asp. Published June 2004. Accessed October 28, 2015.
2. Duffy BL. Vancomycin reaction during spinal anesthesia. Anaesth Intensive Case. 2002;30(3):364-366.
3. Wazny LD, Daghigh B. Desensitization protocols for vancomycin hypersensitivity. Ann Pharmacother. 2001;35(11):1458-1464.
4. O’Sullivan TL, Ruffing MJ, Lamp KC, Warbasse LH, Rybak MJ. Prospective evaluation of red man syndrome in patients receiving vancomycin. J Infect Dis. 1993;168(3):773-776.
5. Wallace MR, Mascola JR, Oldfield EC 3rd. Red man syndrome: incidence, etiology, and prophylaxis. J Infect Dis. 1991;164(6):1180-1185.
6. Renz CL, Thurn JD, Finn HA, Lynch JP, Moss J. Antihistamine prophylaxis permits rapid vancomycin infusion. Crit Care Med. 1999;27(9):1732-1737.
7. Kupstaite R, Baranauskaite A, Pileckyte M, Sveikata A, Kadusevicius E, Muckiene G. Severe vancomycin-induced anaphylactic reaction. Medicina (Kaunas). 2010;46(1):30-33.
8. Lobera T, Audicana MT, Pozo MD, et al. Study of hypersensitivity reactions and anaphylaxis during anesthesia in Spain. J Investig Allergol Clin Immunol. 2008;18(5):350-356.
9. Berséus O, Boman K, Nessen SC, Westerberg LA. Risks of hemolysis due to anti-A and anti-B caused by the transfusion of blood or blood components containing ABO-incompatible plasma. Transfusion. 2013;53(suppl 1):114S-123S.
10. Schwartz LB. Systemic anaphylaxis, food allergy, and insect sting allergy. In: Goldman L, Schafer AI, eds. Goldman’s Cecil Medicine. 24th ed. Philadelphia, PA: Elsevier; 2011:1633-1638.
11. Jung JW, Jeon EJ, Kim JW, et al. A fatal case of intravascular coagulation after bee sting acupuncture. Allergy Asthma Immunol Res. 2012;4(2):107-109.
12. Choi IH, Ha TY, Lee DG, et al. Occurrence of disseminated intravascular coagulation (DIC) in active systemic anaphylaxis: role of platelet-activating factor. Clin Exp Immunol. 1995;100(3):390-394.
13. Renz CL, Laroche D, Thurn JD, et al. Tryptase levels are not increased during vancomycin-induced anaphylactoid reactions. Anesthesiology. 1998;89(3):620-625.
Vancomycin is a glycopeptide antibiotic that exhibits bactericidal activity against gram-positive cocci. It is commonly recommended for surgical prophylaxis in cases of suspected bacterial resistance or penicillin allergy.1 Two main types of hypersensitivity reactions associated with vancomycin can have similar presentations. Red man syndrome is an anaphylactoid reaction caused by direct release of histamine from mast cells via a nonimmunologic mechanism, and is the more common of the 2 reactions. The second type is an anaphylactic reaction, which is an immunoglobulin E (IgE)–mediated systemic event and requires exposure to become sensitized.2,3
We present a patient who had received vancomycin on at least 12 occasions without incident. On this occasion, however, she developed a true anaphylactic reaction causing acute hemodynamic collapse that she survived after extensive resuscitation. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 55-year-old woman had a history of metastatic giant cell tumor of the right proximal tibia. She was originally treated 27 years ago for proximal tibial resection and reconstruction with a custom proximal tibial prosthesis. Four months later, she underwent resection of multiple pulmonary metastases via bilateral thoracotomies in a single surgical setting. After this, the patient had no evidence of recurrent metastatic disease. In subsequent years, the patient underwent multiple revision surgeries for problems such as hardware failure, patellar maltracking, and infection. The patient underwent 19 operations, including several nonorthopedic procedures. Because the patient had a rash after receiving penicillin as a child, she was thought to be allergic to penicillin. Consequently, she received vancomycin as antibiotic prophylaxis for the majority of these procedures. She also received extended courses of vancomycin of at least 6 weeks on 2 separate occasions. During her most recent revision procedure, 6 weeks prior to the procedure under discussion, the patient took vancomycin without incident. She was then found to have a prosthetic infection with Staphylococcus epidermidis, the same organism isolated in her previous infections, and she was advised to undergo a staged revision.
After a preoperative medical evaluation by her primary care physician, the patient was taken to the operating room for prosthesis removal and antibiotic spacer placement. She was anemic with a hemoglobin level of 8.8 g/dL; her erythrocyte sedimentation rate (ESR) was 102 mm/h (normal, <22 mm/h) and her C-reactive protein (CRP) was 38 mg/L (normal, <3 mg/L), but, otherwise, her laboratory values were normal, including a white blood cell count (WBC) of 8100/µL. Her electrocardiogram showed a normal sinus rhythm with nonspecific ST- and T-wave changes. Antibiotics were held until after cultures were taken. General endotracheal tube anesthesia was induced with 2 mg midazolam, 100 µg fentanyl, 180 mg propofol, and 140 mg succinylcholine, followed by 10 mg vecuronium, and maintained with desflurane. A tourniquet was not used per the surgeon’s routine. Dissection was carried down to the prosthesis and showed a small amount of purulent fluid. Transfusion of 1 unit of packed red blood cells (pRBC) was started during the approach owing to relatively low preoperative hemoglobin and significant blood loss. Approximately 500 mL of blood was lost during the approach secondary to the extensive dissection and the local inflammatory response from infection and recent surgery. After cultures were taken, and approximately 10 minutes after blood transfusion began, infusion of 1 g vancomycin in 250 mL normal saline was started via an infusion pump to run over 1 hour.
After infusion of 5 mL vancomycin, the patient’s blood pressure dropped from 117/63 mm Hg to 63/30 mm Hg; her pulse concurrently dropped from 90 to 50 beats/min. Vancomycin infusion was immediately stopped, anesthesia gasses were turned off, and patient received a bolus of normal saline with a second unit of pRBC. Patient received boluses of 0.5 mg to 1.0 mg epinephrine and 100 µg phenylephrine without sustained increase in blood pressure, which had dropped to 54/24 mm Hg, although the patient became tachycardic to ~120 beats/min after epinephrine. A sudden drop in end-tidal CO2 from 40s mm Hg to 20s mm Hg was also noted, indicating continuous but significantly decreased perfusion of the lungs.
We elected to abort the procedure, and a vacuum-assisted closure (VAC) dressing was applied to the open wound. After 15 minutes, the patient’s pulses, which had been faint, became impalpable, and cardiopulmonary resuscitation was initiated for about 7 minutes. The patient received 40 units vasopressin with repeated boluses of 0.5 mg epinephrine; a norepinephrine continuous infusion was started with the return of pulses. The patient also received 50 mg diphenhydramine, 125 mg methylprednisolone, and 20 mg famotidine for suspected anaphylaxis. A central venous line and arterial line were placed, and blood was drawn for laboratory analysis. The patient was noted to have clear breath sounds with no obvious rash, and her urine remained clear. Blood gas showed a profound metabolic acidosis, with pH of 7.09, base deficit of 5.9, and lactate of 8.9. The patient was treated with bicarbonate infusion. The patient was noted to ooze significantly during central venous line and arterial line placement, despite apparently normal coagulation during the surgical approach. Coagulation values were consistent with disseminated intravascular coagulation (DIC): prothrombin time, 57 s (international normalized ratio, 6.7); partial thromboplastin time, >200 s; thrombin time, 110 s; D-dimer, >10,000 ng/mL (normal, 0-200 ng/mL); and fibrinogen, <60 mg/dL (normal, 222-475 mg/dL). The patient’s thromboelastogram showed a flat line indicating an absence of clotting. Interestingly, the platelet count remained near the preoperative level at 338×103/µL. The patient’s blood pressure remained labile and was responsive primarily to epinephrine boluses, of which she received a total of 5 mg. After 1 hour of resuscitation, during which time the patient received a total of 5 L crystalloid and 3 units pRBC, the patient was transferred to the intensive care unit (ICU), intubated, and started on a titrated epinephrine infusion.
Upon arrival in the ICU, the patient quickly stabilized hemodynamically. She was weaned from all inotropic support within 2 hours of arrival. The patient lost 800 mL of blood through wound VAC over the first 12 hours postoperatively and required a total of 11 units of pRBC, 6 units fresh frozen plasma, and 3 units of pooled cryoprecipitate, all of which were compatible. Laboratory values, including arterial pH, lactic acid, and coagulation studies, normalized on the evening of surgery, and, by the next morning, the patient was alert and was extubated without difficulty. Steroids were tapered without hemodynamic compromise while the patient was in the ICU. Cardiology examination revealed no abnormalities. Because of the temporal association of blood transfusion with cardiovascular collapse, pRBC units were retested for antibodies and cultured. Both of these investigations were negative. Wound cultures again were positive for Staphylococcus epidermidis, and blood cultures were negative. The patient was started on daptomycin based on susceptibility profiles. Serum histamine levels taken during initial resuscitation in the operating room were normal. The serum tryptase level obtained at the same time was markedly elevated at >700 ng/mL (normal, <11.5 ng/mL), although this information was not available until several days later.
The patient underwent 2 additional surgeries during the same admission, including the prosthesis removal and tobramycin cement spacer placement, without incident. She was discharged home, again without incident. The patient was later evaluated by an outside allergist and underwent skin puncture and intradermal allergy testing. The results were consistent with a strong IgE-mediated hypersensitivity. Interestingly, she was found not to have a penicillin allergy.
Discussion
Vancomycin hypersensitivity reactions include the anaphylactoid reaction red man syndrome and a true IgE-mediated anaphylactic reaction. Red man syndrome is much more common, with reported rates in infected patients from 3.7% to 47%,4,5 when vancomycin is given at the suggested rate of 1 g over 1 hour. The reaction occurs because of histamine release from mast cells and basophils, and does not require previous sensitization.3 The rate of infusion is directly related to the development of symptoms, with 100% of patients developing symptoms in 1 study with rapid infusion (1 g over 10 min).6 Red man syndrome can typically be prevented by slowing the rate of infusion or by giving an H1 blocker.3 Anaphylaxis is more rare but can occur.7 Anaphylaxis is mediated by vancomycin-specific IgE, which requires previous exposure, as was the case with our patient. Interestingly, the patient had received vancomycin many times without any signs of a hypersensitivity reaction. Antihistamines are not effective in treating anaphylaxis, and epinephrine is the first-line agent.3 This was clearly demonstrated in this case, as there was a significant hemodynamic response to epinephrine and a negligible response to other vasopressors, specifically norepinephrine and vasopressin.
Most hypersensitivity reactions during the course of a surgical procedure occur with induction of anesthesia, with neuromuscular blocking agents and antibiotics being the most common causes.8 In our case, antibiotics were held until after deep cultures were taken. Given the time from induction to the anaphylactic reaction, it is unlikely the reaction resulted from the induction agents or the neuromuscular blocking agent. The possibility of a transfusion reaction was also investigated, since a unit of pRBC was still being transfused when symptoms began. An acute hemolytic transfusion reaction has the classic triad of fever, flank pain, and hemoglobinuria, and can also present as DIC.9 Under anesthesia, DIC can often be the presenting sign. In this case, a hemolytic transfusion reaction appeared very unlikely. All of the blood components the patient received were rechecked and found to be compatible, posttransfusion analysis showed no evidence of hemolysis in any sample, and the direct antiglobulin test was negative in all components.
To our knowledge, there are no reported cases of vancomycin-induced anaphylaxis with concomitant DIC. Symptoms of anaphylaxis after exposure to a possible antigen include rapid onset of hypotension or rapid onset of signs in at least 2 organ systems, including cutaneous, gastrointestinal, respiratory, and cardiovascular.10 Anaphylaxis with DIC is rare after exposure to any substance but has been reported.11 In fact, induction of systemic anaphylaxis in mice is known to cause DIC, with platelet-activating factor suggested as an important common mediator. A similar mechanism is suspected in humans.12
Confirmation of, and, certainly, prediction of, a vancomycin hypersensitivity reaction is difficult. Histamine levels can be used as a measure of mast-cell degranulation, but serum levels peak within 5 minutes and quickly return to baseline, limiting its diagnostic usefulness.3 Tryptase is an enzyme found in the secretory granules of mast cells. It has become an accepted marker of acute anaphylaxis, and, in vancomycin hypersensitivity reactions, can also distinguish between anaphylactic and anaphylactoid reactions.13 Tryptase levels peak 1 to 2 hours after the reaction, making this easier to measure than histamine, but results may not be available for several days, making it useful only in retrospect, as in our case. Skin testing is probably the best way to confirm a hypersensitivity reaction, although even this has been questioned with vancomycin because some find a high false-positive rate3, while others think the false-negative rate is likely too high.7 In this case, we were able to confirm our initial clinical suspicion with both an elevated tryptase level and a positive skin test.
Conclusion
We present a rare case of vancomycin anaphylaxis with DIC after repeated and prolonged previous exposure, which was treated acutely with hemodynamic resuscitation, replacement of blood components, steroids, and, most importantly, repeated boluses of epinephrine. Although several papers have described successful vancomycin desensitization7, this was fortunately not necessary in this case because the causative organism was sensitive to other acceptable antibiotics. The patient has been treated with systemic daptomycin and a tobramycin cement spacer without further incident.
Vancomycin is a glycopeptide antibiotic that exhibits bactericidal activity against gram-positive cocci. It is commonly recommended for surgical prophylaxis in cases of suspected bacterial resistance or penicillin allergy.1 Two main types of hypersensitivity reactions associated with vancomycin can have similar presentations. Red man syndrome is an anaphylactoid reaction caused by direct release of histamine from mast cells via a nonimmunologic mechanism, and is the more common of the 2 reactions. The second type is an anaphylactic reaction, which is an immunoglobulin E (IgE)–mediated systemic event and requires exposure to become sensitized.2,3
We present a patient who had received vancomycin on at least 12 occasions without incident. On this occasion, however, she developed a true anaphylactic reaction causing acute hemodynamic collapse that she survived after extensive resuscitation. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 55-year-old woman had a history of metastatic giant cell tumor of the right proximal tibia. She was originally treated 27 years ago for proximal tibial resection and reconstruction with a custom proximal tibial prosthesis. Four months later, she underwent resection of multiple pulmonary metastases via bilateral thoracotomies in a single surgical setting. After this, the patient had no evidence of recurrent metastatic disease. In subsequent years, the patient underwent multiple revision surgeries for problems such as hardware failure, patellar maltracking, and infection. The patient underwent 19 operations, including several nonorthopedic procedures. Because the patient had a rash after receiving penicillin as a child, she was thought to be allergic to penicillin. Consequently, she received vancomycin as antibiotic prophylaxis for the majority of these procedures. She also received extended courses of vancomycin of at least 6 weeks on 2 separate occasions. During her most recent revision procedure, 6 weeks prior to the procedure under discussion, the patient took vancomycin without incident. She was then found to have a prosthetic infection with Staphylococcus epidermidis, the same organism isolated in her previous infections, and she was advised to undergo a staged revision.
After a preoperative medical evaluation by her primary care physician, the patient was taken to the operating room for prosthesis removal and antibiotic spacer placement. She was anemic with a hemoglobin level of 8.8 g/dL; her erythrocyte sedimentation rate (ESR) was 102 mm/h (normal, <22 mm/h) and her C-reactive protein (CRP) was 38 mg/L (normal, <3 mg/L), but, otherwise, her laboratory values were normal, including a white blood cell count (WBC) of 8100/µL. Her electrocardiogram showed a normal sinus rhythm with nonspecific ST- and T-wave changes. Antibiotics were held until after cultures were taken. General endotracheal tube anesthesia was induced with 2 mg midazolam, 100 µg fentanyl, 180 mg propofol, and 140 mg succinylcholine, followed by 10 mg vecuronium, and maintained with desflurane. A tourniquet was not used per the surgeon’s routine. Dissection was carried down to the prosthesis and showed a small amount of purulent fluid. Transfusion of 1 unit of packed red blood cells (pRBC) was started during the approach owing to relatively low preoperative hemoglobin and significant blood loss. Approximately 500 mL of blood was lost during the approach secondary to the extensive dissection and the local inflammatory response from infection and recent surgery. After cultures were taken, and approximately 10 minutes after blood transfusion began, infusion of 1 g vancomycin in 250 mL normal saline was started via an infusion pump to run over 1 hour.
After infusion of 5 mL vancomycin, the patient’s blood pressure dropped from 117/63 mm Hg to 63/30 mm Hg; her pulse concurrently dropped from 90 to 50 beats/min. Vancomycin infusion was immediately stopped, anesthesia gasses were turned off, and patient received a bolus of normal saline with a second unit of pRBC. Patient received boluses of 0.5 mg to 1.0 mg epinephrine and 100 µg phenylephrine without sustained increase in blood pressure, which had dropped to 54/24 mm Hg, although the patient became tachycardic to ~120 beats/min after epinephrine. A sudden drop in end-tidal CO2 from 40s mm Hg to 20s mm Hg was also noted, indicating continuous but significantly decreased perfusion of the lungs.
We elected to abort the procedure, and a vacuum-assisted closure (VAC) dressing was applied to the open wound. After 15 minutes, the patient’s pulses, which had been faint, became impalpable, and cardiopulmonary resuscitation was initiated for about 7 minutes. The patient received 40 units vasopressin with repeated boluses of 0.5 mg epinephrine; a norepinephrine continuous infusion was started with the return of pulses. The patient also received 50 mg diphenhydramine, 125 mg methylprednisolone, and 20 mg famotidine for suspected anaphylaxis. A central venous line and arterial line were placed, and blood was drawn for laboratory analysis. The patient was noted to have clear breath sounds with no obvious rash, and her urine remained clear. Blood gas showed a profound metabolic acidosis, with pH of 7.09, base deficit of 5.9, and lactate of 8.9. The patient was treated with bicarbonate infusion. The patient was noted to ooze significantly during central venous line and arterial line placement, despite apparently normal coagulation during the surgical approach. Coagulation values were consistent with disseminated intravascular coagulation (DIC): prothrombin time, 57 s (international normalized ratio, 6.7); partial thromboplastin time, >200 s; thrombin time, 110 s; D-dimer, >10,000 ng/mL (normal, 0-200 ng/mL); and fibrinogen, <60 mg/dL (normal, 222-475 mg/dL). The patient’s thromboelastogram showed a flat line indicating an absence of clotting. Interestingly, the platelet count remained near the preoperative level at 338×103/µL. The patient’s blood pressure remained labile and was responsive primarily to epinephrine boluses, of which she received a total of 5 mg. After 1 hour of resuscitation, during which time the patient received a total of 5 L crystalloid and 3 units pRBC, the patient was transferred to the intensive care unit (ICU), intubated, and started on a titrated epinephrine infusion.
Upon arrival in the ICU, the patient quickly stabilized hemodynamically. She was weaned from all inotropic support within 2 hours of arrival. The patient lost 800 mL of blood through wound VAC over the first 12 hours postoperatively and required a total of 11 units of pRBC, 6 units fresh frozen plasma, and 3 units of pooled cryoprecipitate, all of which were compatible. Laboratory values, including arterial pH, lactic acid, and coagulation studies, normalized on the evening of surgery, and, by the next morning, the patient was alert and was extubated without difficulty. Steroids were tapered without hemodynamic compromise while the patient was in the ICU. Cardiology examination revealed no abnormalities. Because of the temporal association of blood transfusion with cardiovascular collapse, pRBC units were retested for antibodies and cultured. Both of these investigations were negative. Wound cultures again were positive for Staphylococcus epidermidis, and blood cultures were negative. The patient was started on daptomycin based on susceptibility profiles. Serum histamine levels taken during initial resuscitation in the operating room were normal. The serum tryptase level obtained at the same time was markedly elevated at >700 ng/mL (normal, <11.5 ng/mL), although this information was not available until several days later.
The patient underwent 2 additional surgeries during the same admission, including the prosthesis removal and tobramycin cement spacer placement, without incident. She was discharged home, again without incident. The patient was later evaluated by an outside allergist and underwent skin puncture and intradermal allergy testing. The results were consistent with a strong IgE-mediated hypersensitivity. Interestingly, she was found not to have a penicillin allergy.
Discussion
Vancomycin hypersensitivity reactions include the anaphylactoid reaction red man syndrome and a true IgE-mediated anaphylactic reaction. Red man syndrome is much more common, with reported rates in infected patients from 3.7% to 47%,4,5 when vancomycin is given at the suggested rate of 1 g over 1 hour. The reaction occurs because of histamine release from mast cells and basophils, and does not require previous sensitization.3 The rate of infusion is directly related to the development of symptoms, with 100% of patients developing symptoms in 1 study with rapid infusion (1 g over 10 min).6 Red man syndrome can typically be prevented by slowing the rate of infusion or by giving an H1 blocker.3 Anaphylaxis is more rare but can occur.7 Anaphylaxis is mediated by vancomycin-specific IgE, which requires previous exposure, as was the case with our patient. Interestingly, the patient had received vancomycin many times without any signs of a hypersensitivity reaction. Antihistamines are not effective in treating anaphylaxis, and epinephrine is the first-line agent.3 This was clearly demonstrated in this case, as there was a significant hemodynamic response to epinephrine and a negligible response to other vasopressors, specifically norepinephrine and vasopressin.
Most hypersensitivity reactions during the course of a surgical procedure occur with induction of anesthesia, with neuromuscular blocking agents and antibiotics being the most common causes.8 In our case, antibiotics were held until after deep cultures were taken. Given the time from induction to the anaphylactic reaction, it is unlikely the reaction resulted from the induction agents or the neuromuscular blocking agent. The possibility of a transfusion reaction was also investigated, since a unit of pRBC was still being transfused when symptoms began. An acute hemolytic transfusion reaction has the classic triad of fever, flank pain, and hemoglobinuria, and can also present as DIC.9 Under anesthesia, DIC can often be the presenting sign. In this case, a hemolytic transfusion reaction appeared very unlikely. All of the blood components the patient received were rechecked and found to be compatible, posttransfusion analysis showed no evidence of hemolysis in any sample, and the direct antiglobulin test was negative in all components.
To our knowledge, there are no reported cases of vancomycin-induced anaphylaxis with concomitant DIC. Symptoms of anaphylaxis after exposure to a possible antigen include rapid onset of hypotension or rapid onset of signs in at least 2 organ systems, including cutaneous, gastrointestinal, respiratory, and cardiovascular.10 Anaphylaxis with DIC is rare after exposure to any substance but has been reported.11 In fact, induction of systemic anaphylaxis in mice is known to cause DIC, with platelet-activating factor suggested as an important common mediator. A similar mechanism is suspected in humans.12
Confirmation of, and, certainly, prediction of, a vancomycin hypersensitivity reaction is difficult. Histamine levels can be used as a measure of mast-cell degranulation, but serum levels peak within 5 minutes and quickly return to baseline, limiting its diagnostic usefulness.3 Tryptase is an enzyme found in the secretory granules of mast cells. It has become an accepted marker of acute anaphylaxis, and, in vancomycin hypersensitivity reactions, can also distinguish between anaphylactic and anaphylactoid reactions.13 Tryptase levels peak 1 to 2 hours after the reaction, making this easier to measure than histamine, but results may not be available for several days, making it useful only in retrospect, as in our case. Skin testing is probably the best way to confirm a hypersensitivity reaction, although even this has been questioned with vancomycin because some find a high false-positive rate3, while others think the false-negative rate is likely too high.7 In this case, we were able to confirm our initial clinical suspicion with both an elevated tryptase level and a positive skin test.
Conclusion
We present a rare case of vancomycin anaphylaxis with DIC after repeated and prolonged previous exposure, which was treated acutely with hemodynamic resuscitation, replacement of blood components, steroids, and, most importantly, repeated boluses of epinephrine. Although several papers have described successful vancomycin desensitization7, this was fortunately not necessary in this case because the causative organism was sensitive to other acceptable antibiotics. The patient has been treated with systemic daptomycin and a tobramycin cement spacer without further incident.
1. Recommendation for the use of intravenous antibiotic prophylaxis in primary total joint arthroplasty. AAOS Information Statement 1027. American Academy of Orthopaedic Surgeons website. http://www.aaos.org/about/papers/advistmt/1027.asp. Published June 2004. Accessed October 28, 2015.
2. Duffy BL. Vancomycin reaction during spinal anesthesia. Anaesth Intensive Case. 2002;30(3):364-366.
3. Wazny LD, Daghigh B. Desensitization protocols for vancomycin hypersensitivity. Ann Pharmacother. 2001;35(11):1458-1464.
4. O’Sullivan TL, Ruffing MJ, Lamp KC, Warbasse LH, Rybak MJ. Prospective evaluation of red man syndrome in patients receiving vancomycin. J Infect Dis. 1993;168(3):773-776.
5. Wallace MR, Mascola JR, Oldfield EC 3rd. Red man syndrome: incidence, etiology, and prophylaxis. J Infect Dis. 1991;164(6):1180-1185.
6. Renz CL, Thurn JD, Finn HA, Lynch JP, Moss J. Antihistamine prophylaxis permits rapid vancomycin infusion. Crit Care Med. 1999;27(9):1732-1737.
7. Kupstaite R, Baranauskaite A, Pileckyte M, Sveikata A, Kadusevicius E, Muckiene G. Severe vancomycin-induced anaphylactic reaction. Medicina (Kaunas). 2010;46(1):30-33.
8. Lobera T, Audicana MT, Pozo MD, et al. Study of hypersensitivity reactions and anaphylaxis during anesthesia in Spain. J Investig Allergol Clin Immunol. 2008;18(5):350-356.
9. Berséus O, Boman K, Nessen SC, Westerberg LA. Risks of hemolysis due to anti-A and anti-B caused by the transfusion of blood or blood components containing ABO-incompatible plasma. Transfusion. 2013;53(suppl 1):114S-123S.
10. Schwartz LB. Systemic anaphylaxis, food allergy, and insect sting allergy. In: Goldman L, Schafer AI, eds. Goldman’s Cecil Medicine. 24th ed. Philadelphia, PA: Elsevier; 2011:1633-1638.
11. Jung JW, Jeon EJ, Kim JW, et al. A fatal case of intravascular coagulation after bee sting acupuncture. Allergy Asthma Immunol Res. 2012;4(2):107-109.
12. Choi IH, Ha TY, Lee DG, et al. Occurrence of disseminated intravascular coagulation (DIC) in active systemic anaphylaxis: role of platelet-activating factor. Clin Exp Immunol. 1995;100(3):390-394.
13. Renz CL, Laroche D, Thurn JD, et al. Tryptase levels are not increased during vancomycin-induced anaphylactoid reactions. Anesthesiology. 1998;89(3):620-625.
1. Recommendation for the use of intravenous antibiotic prophylaxis in primary total joint arthroplasty. AAOS Information Statement 1027. American Academy of Orthopaedic Surgeons website. http://www.aaos.org/about/papers/advistmt/1027.asp. Published June 2004. Accessed October 28, 2015.
2. Duffy BL. Vancomycin reaction during spinal anesthesia. Anaesth Intensive Case. 2002;30(3):364-366.
3. Wazny LD, Daghigh B. Desensitization protocols for vancomycin hypersensitivity. Ann Pharmacother. 2001;35(11):1458-1464.
4. O’Sullivan TL, Ruffing MJ, Lamp KC, Warbasse LH, Rybak MJ. Prospective evaluation of red man syndrome in patients receiving vancomycin. J Infect Dis. 1993;168(3):773-776.
5. Wallace MR, Mascola JR, Oldfield EC 3rd. Red man syndrome: incidence, etiology, and prophylaxis. J Infect Dis. 1991;164(6):1180-1185.
6. Renz CL, Thurn JD, Finn HA, Lynch JP, Moss J. Antihistamine prophylaxis permits rapid vancomycin infusion. Crit Care Med. 1999;27(9):1732-1737.
7. Kupstaite R, Baranauskaite A, Pileckyte M, Sveikata A, Kadusevicius E, Muckiene G. Severe vancomycin-induced anaphylactic reaction. Medicina (Kaunas). 2010;46(1):30-33.
8. Lobera T, Audicana MT, Pozo MD, et al. Study of hypersensitivity reactions and anaphylaxis during anesthesia in Spain. J Investig Allergol Clin Immunol. 2008;18(5):350-356.
9. Berséus O, Boman K, Nessen SC, Westerberg LA. Risks of hemolysis due to anti-A and anti-B caused by the transfusion of blood or blood components containing ABO-incompatible plasma. Transfusion. 2013;53(suppl 1):114S-123S.
10. Schwartz LB. Systemic anaphylaxis, food allergy, and insect sting allergy. In: Goldman L, Schafer AI, eds. Goldman’s Cecil Medicine. 24th ed. Philadelphia, PA: Elsevier; 2011:1633-1638.
11. Jung JW, Jeon EJ, Kim JW, et al. A fatal case of intravascular coagulation after bee sting acupuncture. Allergy Asthma Immunol Res. 2012;4(2):107-109.
12. Choi IH, Ha TY, Lee DG, et al. Occurrence of disseminated intravascular coagulation (DIC) in active systemic anaphylaxis: role of platelet-activating factor. Clin Exp Immunol. 1995;100(3):390-394.
13. Renz CL, Laroche D, Thurn JD, et al. Tryptase levels are not increased during vancomycin-induced anaphylactoid reactions. Anesthesiology. 1998;89(3):620-625.
Necrotizing Fasciitis Caused by Cryptococcus gattii
Necrotizing fasciitis (NF) is a severe, rapidly spreading soft-tissue infection with high morbidity and mortality. Bacteriology in NF may be varied, and the etiology is often polymicrobial. It is important to consider the potential for fungal involvement despite its rarity. Cryptococcal NF has been reported in immunocompromised patients, with Cryptococcus neoformans being the most common offending organism.1-4
C neoformans is a basidiomycotic yeast that was previously considered a homogenous species.5,6 From the antigenic properties of its polysaccharide capsule, 3 main variants were described: C neoformans var. grubii, C neoformans var. neoformans, and C neoformans var. gattii. Subsequently, C neoformans var. gattii was found to be genetically and biochemically different from C neoformans. This discovery led to the distinction of C neoformans var. gattii as a separate species and it being renamed C gattii.6
C gattii was first recognized on Vancouver Island in 2001.7 Although C gattii is predominantly restricted to tropical and subtropical climates, its true epidemiology has been limited by diagnostic methods. C gattii can be diagnosed with laboratory culture media such as birdseed agars and L-canavanine-glycine-bromothymol (CGB) agar.6 However, most reports of Cryptococcus NF do not specify the culture media used to isolate Cryptococcus. In addition to culture media, molecular genotyping studies also allow for confirmation of the diagnosis of C gattii and have the added benefit of enabling identification of the molecular genotype. Nonetheless, in many clinical microbiology laboratories, Cryptococcus is not identified to the species level, much less to the molecular genotype.7 Given these diagnostic limitations and the fact that C gattii was only recently identified as a separate species, it is possible that any pre-2006 cases of NF attributed to C neoformans could in fact have been caused by C gattii.
In this article, we review the literature and report a case of NF of the hand that was caused by C gattii in a patient with diabetes. To our knowledge, this is the first reported case of NF caused by C gattii. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 73-year-old man was admitted with a 1-week history of swelling and pain in the dorsum of the left hand. He had been sitting in an outdoor eatery in Singapore when an insect bit the hand over the dorsum. Two days later, he consulted his family physician, who began treatment with oral amoxicillin/clavulanic acid. After 4 days of treatment, there was clinical progression of increased swelling and pain in the hand. Six days after initial injury, the patient presented to the department of orthopedic surgery.
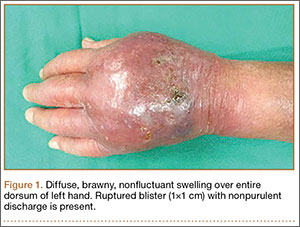
Physical examination revealed diffuse, brawny, nonfluctuant swelling over the entire dorsum of the left hand (Figure 1). There was a 1×1-cm ruptured blister with some nonpurulent discharge just distal to the wrist joint. Neurovascular status and the extensor mechanism of the fingers were intact. The wrist joint had full range of motion. There was no fever.
Laboratory testing revealed an elevated white blood cell count (16.6×109/L), a C-reactive protein (CRP) level of 237 nmol/L, a random blood glucose level of 12.6 mmol/L, and a LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score of 7.8
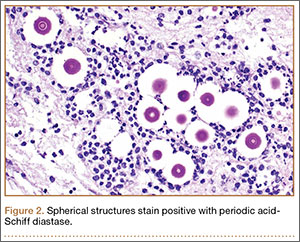
Given the severe swelling, intravenous amoxicillin/clavulanic acid was started. The patient received a total of 3 doses before operative débridement of the left hand. Operative findings were NF of the hand, grayish necrotic fascia, and foul-smelling “dishwater” fluid. A single specimen of fascia from the surgical site was sent for examination. Histopathologic examination of formalin-fixed, paraffin-embedded tissue revealed necrotizing suppurative inflammation with fungal organisms present (Figures 2, 3).

Tissue cultures were obtained during surgery. The organism grew as scanty, small, wet-looking colonies on sheep blood agar after 48 hours of incubation. Microscopy revealed an oval yeast. The organism was identified and reported as C gattii by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS; Biotyper 2.0.1 software; Bruker Daltonics), with a score of 1.914.9 All other intraoperative cultures for aerobic and anaerobic bacteria were negative. Molecular genotyping was performed with polymerase chain reaction assay to identify the molecular subtype.10C gattii genotype VGII was isolated. A cryptococcal serum antigen assay was positive at 1:256.
A series of tests was performed to screen for disseminated disease. Blood cultures were negative for fungus. Chest radiography and computed tomography of the brain did not show any pulmonary or cerebral involvement. Cerebrospinal fluid was not available for examination, as the patient declined lumbar puncture. Blood tests included a negative result for human immunodeficiency virus (HIV). The patient was found to have previously undiagnosed diabetes mellitus (hemoglobin A1c, 7.9%). T-cell counts and ratios were normal.
The patient was started on intravenous amphotericin B 60 mg/d and flucytosine 500 mg every 6 hours for 3 weeks. Oral fluconazole 400 mg every morning was also given (intended duration, 6 mo). Given that diabetes was newly diagnosed, the patient was treated with metformin; his capillary blood glucose level remained stable during his inpatient stay.
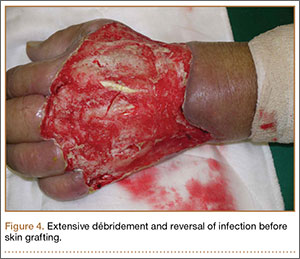
Four débridements of the dorsal hand wound were performed—the first on day of admission and the other 3 on hospitalization days 3, 7, and 18 (Figure 4). Subsequent wound resurfacing with a split skin graft harvested from the forearm was performed on hospitalization day 22. After surgery, the hand was dressed with a bulky cotton dressing. Five days after the patient was discharged, during review in the outpatient clinic, the skin graft was noted to be taking well. The patient did not attend postoperative physical therapy. He was maintained on metformin and given a follow-up clinic appointment for his diabetes. Four months after surgery, the wound was completely healed, and normal functional use of the hand recovered.
Discussion
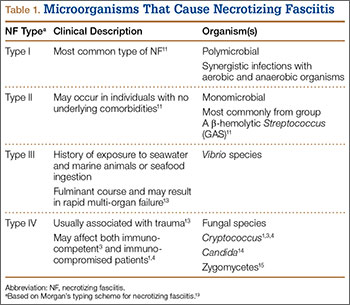
NF is a severe soft-tissue infection with potential for rapid progression. Surgical débridement should be performed urgently to reduce the chance of morbidity and mortality.11 The initial classification by Giuliano and colleagues12 was based on bacteriology and included type I (anaerobic species in combination with a facultative species) and type II (monomicrobial usually involving group A β-hemolytic Streptococcus). This classification was modified by Morgan13 to include gram-negative organisms as well as fungal organisms (Table 1).
Fungal NF is rare, with Candida, Apophysomyces, and Cryptococcus described in the literature.1,14,15 Fungal infections tend to occur in immunocompromised patients; risk factors are steroid immunosuppression, poorly controlled diabetes, and peripheral vascular disease.16 Some zygomycetes may also affect immunocompetent patients.15
C gattii is an encapsulated yeast organism that is genetically and biochemically distinct from C neoformans. It is endemic to tropical parts of Africa and Australia. Its main environmental sources are eucalyptus trees (Eucalyptus camaldulensis, Eucalyptus tereticornis) and decaying hollows in living trees.17 In addition, there have been reports of isolation of C gattii from insect frass,18 which would make infection by an insect bite a possible transmission route. Worldwide distribution of this pathogen has increased recently, with outbreaks noted on Vancouver Island and in areas in Canada and the northwest United States.7
The true incidence of NF secondary to C gattii is difficult to determine. C gattii was only recently identified as a separate species, and pre-2006 cases of NF attributed to C neoformans may instead have been caused by C gattii. Misidentification has been compounded by the fact that the tests required for accurate diagnosis of C gattii infection may not be readily available in many clinical microbiology laboratories. Cryptococcus can be identified with various methods, including direct microscopy, culturing of tissue or fluid samples, and measurement of cryptococcal serum antigen. However, tests such as specific culture media, mass spectrometry, and molecular typing studies are required to determine cryptococcal species. L-canavanine-glycine-bromothymol blue (CGB) agar is a medium that is often used to differentiate C gattii from C neoformans because of the ability of C gattii to produce a blue appearance.6 Modern techniques, such as MALDI-TOF MS, have also been used to successfully distinguish between C gattii and C neoformans.9 MALDI-TOF MS identifies species on the basis of characteristic protein spectra extracted from whole cells. Using commercial and supplemental reference libraries, the system compares signal matches in the reference spectrum with Cryptococcus entries in the library—allowing rapid and accurate identification of cryptococcal species. However, this diagnostic method is limited by availability of adequate Cryptococcus entries in the reference library and by the high cost of acquiring the machine.
Serotyping is based on the antigenic property of the capsule and was once used to differentiate C neoformans into its 3 main varieties: var. neoformans, var. grubii, var. gattii. However, when it was realized that the antigenic property of the strain can be unstable and that there are hybrids containing more than 1 serotype, serotyping was abandoned as a species-differentiation test.6 The current gold standard for species differentiation is molecular genotyping. Molecular genotyping studies can confirm the diagnosis of C gattii infection and allow differentiation of C gattii into its 4 main molecular types: VGI, VGII, VGIII, VGIV. Using methods such as polymerase chain reaction (PCR) and restriction fragment length polymorphism (RFLP) analysis, molecular typing allows for specific epidemiology charting of C gattii genotypes.7
Although the transmission route for cryptococcal infection is mainly respiratory, direct inoculation has been reported as well.19 Cutaneous lesions, which occur in 5% to 20% of cryptococcal infections, often present in the head and neck.2,20,21 Primary cutaneous infections from cryptococcosis are rare, and cutaneous manifestations are often a sign of disseminated disease. Disseminated disease is defined as the involvement of 2 or more noncontiguous sites or evidence of high fungal burden based on cryptococcal antigen titer of more than 1:512.12 It is important to exclude disseminated disease in all cases of cryptococcosis, as it may be fatal.20 The neural and pulmonary systems should be screened.22 Cellulitis from cryptococcosis is almost always limited to immunocompromised patients, though there are reports of crytococcal cutaneous disease in immunocompetent patients.3,15 Interestingly, though C neoformans often affects immunocompromised patients, the emerging pathogen of C gattii affects immunocompetent patients.7,17,23 Our patient’s undiagnosed diabetes may have been a risk factor for cryptococcal infection. His cryptococcal antigen titer was 1:256, with no evidence of other sites of involvement. We therefore believe this to be a rare case of direct inoculation secondary to an insect bite.
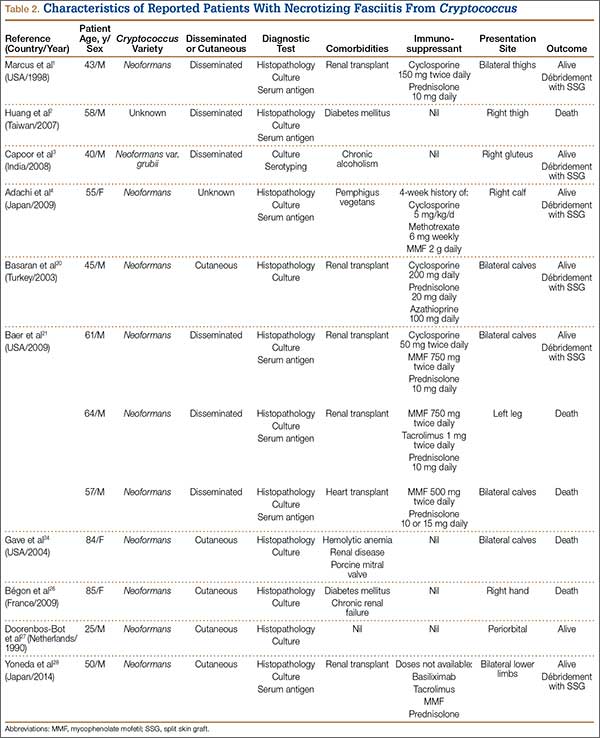
The literature includes 12 reported cases of NF secondary to Cryptococcus (Table 2), all C neoformans. Of these cases, 9 involved immunosuppression, and most of these patients were on long-term steroid treatment after organ transplantation. The most common infection site was the lower extremity. These cases of cryptococcal NF show that immunosuppression, and long-term steroid use in particular, is an important risk factor. The mortality rate for these reviewed cases was 41.6% (5/12). According to the literature, the mortality rates for patients with cryptococcal soft-tissue infections24 and posttransplant patients with cryptococcal NF21 were 37.5% and 60%, respectively. We believe the mortality rate in our reviewed cases likely was confounded by the fact that most of the patients were posttransplant patients on long-term immunosuppression.
Of the 12 patients, 5 had primary cutaneous disease. There seems to be no relationship between outcome and dissemination of disease. In addition, there is a paucity of literature on the effect of disseminated disease and cryptococcal soft-tissue infections. Therefore, no firm conclusions can be drawn regarding the effects of disseminated disease on severity of cryptococcal soft-tissue infection.
Treatment of cryptococcal NF involves a combination of surgical débridement and long-term antifungal therapy. Surgical débridement of NF includes delineating the extent of infection with complete surgical excision of the affected tissue.25 The aims of surgery should be to remove all unhealthy tissue, identify the offending organism, and plan for resurfacing or reconstruction of the afflicted extremity. Intraoperative-tissue histology should be performed to confirm the diagnosis of NF. Histology can be used to demonstrate cryptococcal infection. The diagnosis of cryptococcal infection can be aided with fungal cultures, and therefore we recommend that tissue cultures be sent not only for routine aerobic/anaerobic bacteria but also for mycobacteria and fungal organisms. Laboratory tests that aid in diagnosis include serum cryptococcal antigen titer.
The current treatment recommendation for cryptococcal disease in patients who are not HIV-positive or transplant hosts is amphotericin B deoxycholate 0.7 to 1.0 mg/kg/d plus flucytosine 100 mg/kg/d for at least 4 weeks.22 The regimen period may be shortened to 14 days for patients at low risk of treatment failure. Fluconazole should be given as maintenance therapy (200 mg/d) for 6 to 12 months. There is no compelling evidence for immunoglobulin therapy for cryptococcal disease.22
Conclusion
NF caused by Cryptococcus is rare. A high level of suspicion, and intraoperative specimens for histology and fungal microscopy and culture, can help in establishing the diagnosis. Molecular genotyping remains the diagnostic method of choice for NF secondary to Cryptococcus. Effective treatment consists of aggressive surgical débridement and antifungal therapy.
1. Marcus JR, Hussong JW, Gonzalez C, Dumanian GA. Risk factors in necrotizing fasciitis: a case involving Cryptococcus neoformans. Ann Plast Surg. 1998;40(1):80-83.
2. Huang KC, Tu YK, Lee KF, Huang TJ, Wen-Wei Hsu R. Disseminated cryptococcosis presented as necrotizing fasciitis of a limb. J Trauma. 2007;63(2):E44-E46.
3. Capoor MR, Khanna G, Malhotra R. Disseminated cryptococcosis with necrotizing fasciitis in an apparently immunocompetent host: a case report. Med Mycol. 2008;46:269-273.
4. Adachi M, Tsurata D, Imanishi H, Ishii M, Kobayashi H. Necrotizing fasciitis caused by Cryptococcus neoformans in a patient with pemphigus vegetans. Clin Exp Dermatol. 2009;34(8):e751-e753.
5. Enache-Angoulvant A, Chandenier J, Symoens F, et al. Molecular identification of Cryptococcus neoformans serotypes. J Clin Microbiol. 2007;45(4):1261-1265.
6. Kwon-Chung KJ, Varma A. Do major species concepts support one, two or more species within Cryptococcus neoformans? FEMS Yeast Res. 2006;6(4):657-687.
7. Datta K, Bartlett KH, Baer R, et al; Cryptococcus gattii Working Group of the Pacific Northwest. Spread of Cryptococcus gattii into Pacific Northwest region of the United States. Emerg Infect Dis. 2009;15(8):1185-1191.
8. Wong CH, Khin LW, Heng KS, Tan KC, Low CO. The LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score: a tool for distinguishing necrotizing fasciitis from other soft tissue infections. Crit Care Med. 2004;32(7):1535-1541.
9. McTaggart LR, Lei E, Richardson SE, Hoang L, Fothergill A, Zhang SX. Rapid identification of Cryptococcus neoformans and Cryptococcus gattii by matrix-assisted laser desorption ionization-time of flight mass spectrometry. J Clin Microbiol. 2011;49(8):3050-3053.
10. Meyer W, Castañeda A, Jackson S, Huynh M, Castañeda E; IberoAmerican Cryptococcal Study Group. Molecular typing of IberoAmerican Cryptococcus neoformans isolates. Emerg Infect Dis. 2003;9(2):189-195.
11. Wong CH, Chang HC, Pasupathy S, Khin LW, Tan JL, Low CO. Necrotizing fasciitis: clinical presentation, microbiology and determinants of mortality. J Bone Joint Surg Am. 2003;85(8):1454-1460.
12. Giuliano A, Lewis F Jr, Hadley K, Blaisdell FW. Bacteriology of necrotizing fasciitis. Am J Surg. 1977;134(1):52-57.
13. Morgan MS. Diagnosis and management of necrotising fasciitis: a multiparametric approach. J Hosp Infect. 2010;75(4):249-257.
14. Buchanan PJ, Mast BA, Lottenberg L, Kim T, Efron PA, Ang DN. Candida albicans necrotizing soft tissue infection: a case report and literature review of fungal necrotizing soft tissue infections. Ann Plastic Surg. 2013;70(6):739-741.
15. Jain D, Kumar Y, Vasishta RK, Rajesh L, Pattari SK, Chakrabarti A. Zygomycotic necrotizing fasciitis in immunocompetent patients: a series of 18 cases. Modern Pathol. 2006;19(9):1221-1226.
16. Fontes RA Jr, Ogilvie CM, Miclau T. Necrotizing soft-tissue infections. J Am Acad Orthop Surg. 2000;8(3):151-158.
17. Sorrell TC. Cryptococcus neoformans variety gattii. Med Mycol. 2001;39(2):155-168.
18. Kidd SE, Sorrell TC, Meyer W. Isolation of two molecular types of Cryptococcus neoformans var. gattii from insect frass. Med Mycol. 2003;41(2):171-176.
19. Neuville S, Dromer F, Morin O, Dupont B, Ronin O, Lortholary O; French Cryptococcosis Study Group. Primary cutaneous cryptococcosis: a distinct clinical entity. Clin Infect Dis. 2003;36(3):337-347.
20. Basaran O, Emiroglu R, Arikan U, Karakayali H, Haberal M. Cryptococcal necrotizing fasciitis with multiple sites of involvement in the lower extremities. Dermatol Surg. 2003;29(11):1158-1160.
21. Baer S, Baddley JW, Gnann JW, Pappas PG. Cryptococcal disease presenting as necrotizing cellulitis in transplant recipients. Transpl Infect Dis. 2009;11(4):353-358.
22. Perfect JR, Dismukes WE, Dromer F, et al. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis. 2010;50(3):291-322.
23. Chan M, Lye D, Win MK, Chow A, Barkham T. Clinical and microbiological characteristics of cryptococcosis in Singapore: predominance of Cryptococcus neoformans compared with Cryptococcus gattii. Int J Infect Dis. 2014;26:110-115.
24. Gave AA, Torres R, Kaplan L. Cryptococcal myositis and vasculitis: an unusual necrotizing soft tissue infection. Surg Infect. 2004;5(3):309-313.
25. Wong CH, Yam AK, Tan AB, Song C. Approach to debridement in necrotizing fasciitis. Am J Surg. 2008;196(3):e19-e24.
26. Bégon E, Bachmeyer C, Thibault M, et al. Necrotizing fasciitis due to Cryptococcus neoformans in a diabetic patient with chronic renal insufficiency. Clin Exp Dermatol. 2009;34(8):935-936.
27. Doorenbos-Bot AC, Hooymans JM, Blanksma LJ. Periorbital necrotising fasciitis due to Cryptococcus neoformans in a healthy young man. Doc Ophthalmol. 1990;75(3-4):315-320.
28. Yoneda T, Itami Y, Hirayama A, Saka T, Yoshida K, Fujimoto K. Cryptococcal necrotizing fasciitis in a patient after renal transplantation—a case report. Transplant Proc. 2014;46(2):620-622.
Necrotizing fasciitis (NF) is a severe, rapidly spreading soft-tissue infection with high morbidity and mortality. Bacteriology in NF may be varied, and the etiology is often polymicrobial. It is important to consider the potential for fungal involvement despite its rarity. Cryptococcal NF has been reported in immunocompromised patients, with Cryptococcus neoformans being the most common offending organism.1-4
C neoformans is a basidiomycotic yeast that was previously considered a homogenous species.5,6 From the antigenic properties of its polysaccharide capsule, 3 main variants were described: C neoformans var. grubii, C neoformans var. neoformans, and C neoformans var. gattii. Subsequently, C neoformans var. gattii was found to be genetically and biochemically different from C neoformans. This discovery led to the distinction of C neoformans var. gattii as a separate species and it being renamed C gattii.6
C gattii was first recognized on Vancouver Island in 2001.7 Although C gattii is predominantly restricted to tropical and subtropical climates, its true epidemiology has been limited by diagnostic methods. C gattii can be diagnosed with laboratory culture media such as birdseed agars and L-canavanine-glycine-bromothymol (CGB) agar.6 However, most reports of Cryptococcus NF do not specify the culture media used to isolate Cryptococcus. In addition to culture media, molecular genotyping studies also allow for confirmation of the diagnosis of C gattii and have the added benefit of enabling identification of the molecular genotype. Nonetheless, in many clinical microbiology laboratories, Cryptococcus is not identified to the species level, much less to the molecular genotype.7 Given these diagnostic limitations and the fact that C gattii was only recently identified as a separate species, it is possible that any pre-2006 cases of NF attributed to C neoformans could in fact have been caused by C gattii.
In this article, we review the literature and report a case of NF of the hand that was caused by C gattii in a patient with diabetes. To our knowledge, this is the first reported case of NF caused by C gattii. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 73-year-old man was admitted with a 1-week history of swelling and pain in the dorsum of the left hand. He had been sitting in an outdoor eatery in Singapore when an insect bit the hand over the dorsum. Two days later, he consulted his family physician, who began treatment with oral amoxicillin/clavulanic acid. After 4 days of treatment, there was clinical progression of increased swelling and pain in the hand. Six days after initial injury, the patient presented to the department of orthopedic surgery.
Physical examination revealed diffuse, brawny, nonfluctuant swelling over the entire dorsum of the left hand (Figure 1). There was a 1×1-cm ruptured blister with some nonpurulent discharge just distal to the wrist joint. Neurovascular status and the extensor mechanism of the fingers were intact. The wrist joint had full range of motion. There was no fever.
Laboratory testing revealed an elevated white blood cell count (16.6×109/L), a C-reactive protein (CRP) level of 237 nmol/L, a random blood glucose level of 12.6 mmol/L, and a LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score of 7.8
Given the severe swelling, intravenous amoxicillin/clavulanic acid was started. The patient received a total of 3 doses before operative débridement of the left hand. Operative findings were NF of the hand, grayish necrotic fascia, and foul-smelling “dishwater” fluid. A single specimen of fascia from the surgical site was sent for examination. Histopathologic examination of formalin-fixed, paraffin-embedded tissue revealed necrotizing suppurative inflammation with fungal organisms present (Figures 2, 3).
Tissue cultures were obtained during surgery. The organism grew as scanty, small, wet-looking colonies on sheep blood agar after 48 hours of incubation. Microscopy revealed an oval yeast. The organism was identified and reported as C gattii by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS; Biotyper 2.0.1 software; Bruker Daltonics), with a score of 1.914.9 All other intraoperative cultures for aerobic and anaerobic bacteria were negative. Molecular genotyping was performed with polymerase chain reaction assay to identify the molecular subtype.10C gattii genotype VGII was isolated. A cryptococcal serum antigen assay was positive at 1:256.
A series of tests was performed to screen for disseminated disease. Blood cultures were negative for fungus. Chest radiography and computed tomography of the brain did not show any pulmonary or cerebral involvement. Cerebrospinal fluid was not available for examination, as the patient declined lumbar puncture. Blood tests included a negative result for human immunodeficiency virus (HIV). The patient was found to have previously undiagnosed diabetes mellitus (hemoglobin A1c, 7.9%). T-cell counts and ratios were normal.
The patient was started on intravenous amphotericin B 60 mg/d and flucytosine 500 mg every 6 hours for 3 weeks. Oral fluconazole 400 mg every morning was also given (intended duration, 6 mo). Given that diabetes was newly diagnosed, the patient was treated with metformin; his capillary blood glucose level remained stable during his inpatient stay.
Four débridements of the dorsal hand wound were performed—the first on day of admission and the other 3 on hospitalization days 3, 7, and 18 (Figure 4). Subsequent wound resurfacing with a split skin graft harvested from the forearm was performed on hospitalization day 22. After surgery, the hand was dressed with a bulky cotton dressing. Five days after the patient was discharged, during review in the outpatient clinic, the skin graft was noted to be taking well. The patient did not attend postoperative physical therapy. He was maintained on metformin and given a follow-up clinic appointment for his diabetes. Four months after surgery, the wound was completely healed, and normal functional use of the hand recovered.
Discussion
NF is a severe soft-tissue infection with potential for rapid progression. Surgical débridement should be performed urgently to reduce the chance of morbidity and mortality.11 The initial classification by Giuliano and colleagues12 was based on bacteriology and included type I (anaerobic species in combination with a facultative species) and type II (monomicrobial usually involving group A β-hemolytic Streptococcus). This classification was modified by Morgan13 to include gram-negative organisms as well as fungal organisms (Table 1).
Fungal NF is rare, with Candida, Apophysomyces, and Cryptococcus described in the literature.1,14,15 Fungal infections tend to occur in immunocompromised patients; risk factors are steroid immunosuppression, poorly controlled diabetes, and peripheral vascular disease.16 Some zygomycetes may also affect immunocompetent patients.15
C gattii is an encapsulated yeast organism that is genetically and biochemically distinct from C neoformans. It is endemic to tropical parts of Africa and Australia. Its main environmental sources are eucalyptus trees (Eucalyptus camaldulensis, Eucalyptus tereticornis) and decaying hollows in living trees.17 In addition, there have been reports of isolation of C gattii from insect frass,18 which would make infection by an insect bite a possible transmission route. Worldwide distribution of this pathogen has increased recently, with outbreaks noted on Vancouver Island and in areas in Canada and the northwest United States.7
The true incidence of NF secondary to C gattii is difficult to determine. C gattii was only recently identified as a separate species, and pre-2006 cases of NF attributed to C neoformans may instead have been caused by C gattii. Misidentification has been compounded by the fact that the tests required for accurate diagnosis of C gattii infection may not be readily available in many clinical microbiology laboratories. Cryptococcus can be identified with various methods, including direct microscopy, culturing of tissue or fluid samples, and measurement of cryptococcal serum antigen. However, tests such as specific culture media, mass spectrometry, and molecular typing studies are required to determine cryptococcal species. L-canavanine-glycine-bromothymol blue (CGB) agar is a medium that is often used to differentiate C gattii from C neoformans because of the ability of C gattii to produce a blue appearance.6 Modern techniques, such as MALDI-TOF MS, have also been used to successfully distinguish between C gattii and C neoformans.9 MALDI-TOF MS identifies species on the basis of characteristic protein spectra extracted from whole cells. Using commercial and supplemental reference libraries, the system compares signal matches in the reference spectrum with Cryptococcus entries in the library—allowing rapid and accurate identification of cryptococcal species. However, this diagnostic method is limited by availability of adequate Cryptococcus entries in the reference library and by the high cost of acquiring the machine.
Serotyping is based on the antigenic property of the capsule and was once used to differentiate C neoformans into its 3 main varieties: var. neoformans, var. grubii, var. gattii. However, when it was realized that the antigenic property of the strain can be unstable and that there are hybrids containing more than 1 serotype, serotyping was abandoned as a species-differentiation test.6 The current gold standard for species differentiation is molecular genotyping. Molecular genotyping studies can confirm the diagnosis of C gattii infection and allow differentiation of C gattii into its 4 main molecular types: VGI, VGII, VGIII, VGIV. Using methods such as polymerase chain reaction (PCR) and restriction fragment length polymorphism (RFLP) analysis, molecular typing allows for specific epidemiology charting of C gattii genotypes.7
Although the transmission route for cryptococcal infection is mainly respiratory, direct inoculation has been reported as well.19 Cutaneous lesions, which occur in 5% to 20% of cryptococcal infections, often present in the head and neck.2,20,21 Primary cutaneous infections from cryptococcosis are rare, and cutaneous manifestations are often a sign of disseminated disease. Disseminated disease is defined as the involvement of 2 or more noncontiguous sites or evidence of high fungal burden based on cryptococcal antigen titer of more than 1:512.12 It is important to exclude disseminated disease in all cases of cryptococcosis, as it may be fatal.20 The neural and pulmonary systems should be screened.22 Cellulitis from cryptococcosis is almost always limited to immunocompromised patients, though there are reports of crytococcal cutaneous disease in immunocompetent patients.3,15 Interestingly, though C neoformans often affects immunocompromised patients, the emerging pathogen of C gattii affects immunocompetent patients.7,17,23 Our patient’s undiagnosed diabetes may have been a risk factor for cryptococcal infection. His cryptococcal antigen titer was 1:256, with no evidence of other sites of involvement. We therefore believe this to be a rare case of direct inoculation secondary to an insect bite.
The literature includes 12 reported cases of NF secondary to Cryptococcus (Table 2), all C neoformans. Of these cases, 9 involved immunosuppression, and most of these patients were on long-term steroid treatment after organ transplantation. The most common infection site was the lower extremity. These cases of cryptococcal NF show that immunosuppression, and long-term steroid use in particular, is an important risk factor. The mortality rate for these reviewed cases was 41.6% (5/12). According to the literature, the mortality rates for patients with cryptococcal soft-tissue infections24 and posttransplant patients with cryptococcal NF21 were 37.5% and 60%, respectively. We believe the mortality rate in our reviewed cases likely was confounded by the fact that most of the patients were posttransplant patients on long-term immunosuppression.
Of the 12 patients, 5 had primary cutaneous disease. There seems to be no relationship between outcome and dissemination of disease. In addition, there is a paucity of literature on the effect of disseminated disease and cryptococcal soft-tissue infections. Therefore, no firm conclusions can be drawn regarding the effects of disseminated disease on severity of cryptococcal soft-tissue infection.
Treatment of cryptococcal NF involves a combination of surgical débridement and long-term antifungal therapy. Surgical débridement of NF includes delineating the extent of infection with complete surgical excision of the affected tissue.25 The aims of surgery should be to remove all unhealthy tissue, identify the offending organism, and plan for resurfacing or reconstruction of the afflicted extremity. Intraoperative-tissue histology should be performed to confirm the diagnosis of NF. Histology can be used to demonstrate cryptococcal infection. The diagnosis of cryptococcal infection can be aided with fungal cultures, and therefore we recommend that tissue cultures be sent not only for routine aerobic/anaerobic bacteria but also for mycobacteria and fungal organisms. Laboratory tests that aid in diagnosis include serum cryptococcal antigen titer.
The current treatment recommendation for cryptococcal disease in patients who are not HIV-positive or transplant hosts is amphotericin B deoxycholate 0.7 to 1.0 mg/kg/d plus flucytosine 100 mg/kg/d for at least 4 weeks.22 The regimen period may be shortened to 14 days for patients at low risk of treatment failure. Fluconazole should be given as maintenance therapy (200 mg/d) for 6 to 12 months. There is no compelling evidence for immunoglobulin therapy for cryptococcal disease.22
Conclusion
NF caused by Cryptococcus is rare. A high level of suspicion, and intraoperative specimens for histology and fungal microscopy and culture, can help in establishing the diagnosis. Molecular genotyping remains the diagnostic method of choice for NF secondary to Cryptococcus. Effective treatment consists of aggressive surgical débridement and antifungal therapy.
Necrotizing fasciitis (NF) is a severe, rapidly spreading soft-tissue infection with high morbidity and mortality. Bacteriology in NF may be varied, and the etiology is often polymicrobial. It is important to consider the potential for fungal involvement despite its rarity. Cryptococcal NF has been reported in immunocompromised patients, with Cryptococcus neoformans being the most common offending organism.1-4
C neoformans is a basidiomycotic yeast that was previously considered a homogenous species.5,6 From the antigenic properties of its polysaccharide capsule, 3 main variants were described: C neoformans var. grubii, C neoformans var. neoformans, and C neoformans var. gattii. Subsequently, C neoformans var. gattii was found to be genetically and biochemically different from C neoformans. This discovery led to the distinction of C neoformans var. gattii as a separate species and it being renamed C gattii.6
C gattii was first recognized on Vancouver Island in 2001.7 Although C gattii is predominantly restricted to tropical and subtropical climates, its true epidemiology has been limited by diagnostic methods. C gattii can be diagnosed with laboratory culture media such as birdseed agars and L-canavanine-glycine-bromothymol (CGB) agar.6 However, most reports of Cryptococcus NF do not specify the culture media used to isolate Cryptococcus. In addition to culture media, molecular genotyping studies also allow for confirmation of the diagnosis of C gattii and have the added benefit of enabling identification of the molecular genotype. Nonetheless, in many clinical microbiology laboratories, Cryptococcus is not identified to the species level, much less to the molecular genotype.7 Given these diagnostic limitations and the fact that C gattii was only recently identified as a separate species, it is possible that any pre-2006 cases of NF attributed to C neoformans could in fact have been caused by C gattii.
In this article, we review the literature and report a case of NF of the hand that was caused by C gattii in a patient with diabetes. To our knowledge, this is the first reported case of NF caused by C gattii. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 73-year-old man was admitted with a 1-week history of swelling and pain in the dorsum of the left hand. He had been sitting in an outdoor eatery in Singapore when an insect bit the hand over the dorsum. Two days later, he consulted his family physician, who began treatment with oral amoxicillin/clavulanic acid. After 4 days of treatment, there was clinical progression of increased swelling and pain in the hand. Six days after initial injury, the patient presented to the department of orthopedic surgery.
Physical examination revealed diffuse, brawny, nonfluctuant swelling over the entire dorsum of the left hand (Figure 1). There was a 1×1-cm ruptured blister with some nonpurulent discharge just distal to the wrist joint. Neurovascular status and the extensor mechanism of the fingers were intact. The wrist joint had full range of motion. There was no fever.
Laboratory testing revealed an elevated white blood cell count (16.6×109/L), a C-reactive protein (CRP) level of 237 nmol/L, a random blood glucose level of 12.6 mmol/L, and a LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score of 7.8
Given the severe swelling, intravenous amoxicillin/clavulanic acid was started. The patient received a total of 3 doses before operative débridement of the left hand. Operative findings were NF of the hand, grayish necrotic fascia, and foul-smelling “dishwater” fluid. A single specimen of fascia from the surgical site was sent for examination. Histopathologic examination of formalin-fixed, paraffin-embedded tissue revealed necrotizing suppurative inflammation with fungal organisms present (Figures 2, 3).
Tissue cultures were obtained during surgery. The organism grew as scanty, small, wet-looking colonies on sheep blood agar after 48 hours of incubation. Microscopy revealed an oval yeast. The organism was identified and reported as C gattii by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS; Biotyper 2.0.1 software; Bruker Daltonics), with a score of 1.914.9 All other intraoperative cultures for aerobic and anaerobic bacteria were negative. Molecular genotyping was performed with polymerase chain reaction assay to identify the molecular subtype.10C gattii genotype VGII was isolated. A cryptococcal serum antigen assay was positive at 1:256.
A series of tests was performed to screen for disseminated disease. Blood cultures were negative for fungus. Chest radiography and computed tomography of the brain did not show any pulmonary or cerebral involvement. Cerebrospinal fluid was not available for examination, as the patient declined lumbar puncture. Blood tests included a negative result for human immunodeficiency virus (HIV). The patient was found to have previously undiagnosed diabetes mellitus (hemoglobin A1c, 7.9%). T-cell counts and ratios were normal.
The patient was started on intravenous amphotericin B 60 mg/d and flucytosine 500 mg every 6 hours for 3 weeks. Oral fluconazole 400 mg every morning was also given (intended duration, 6 mo). Given that diabetes was newly diagnosed, the patient was treated with metformin; his capillary blood glucose level remained stable during his inpatient stay.
Four débridements of the dorsal hand wound were performed—the first on day of admission and the other 3 on hospitalization days 3, 7, and 18 (Figure 4). Subsequent wound resurfacing with a split skin graft harvested from the forearm was performed on hospitalization day 22. After surgery, the hand was dressed with a bulky cotton dressing. Five days after the patient was discharged, during review in the outpatient clinic, the skin graft was noted to be taking well. The patient did not attend postoperative physical therapy. He was maintained on metformin and given a follow-up clinic appointment for his diabetes. Four months after surgery, the wound was completely healed, and normal functional use of the hand recovered.
Discussion
NF is a severe soft-tissue infection with potential for rapid progression. Surgical débridement should be performed urgently to reduce the chance of morbidity and mortality.11 The initial classification by Giuliano and colleagues12 was based on bacteriology and included type I (anaerobic species in combination with a facultative species) and type II (monomicrobial usually involving group A β-hemolytic Streptococcus). This classification was modified by Morgan13 to include gram-negative organisms as well as fungal organisms (Table 1).
Fungal NF is rare, with Candida, Apophysomyces, and Cryptococcus described in the literature.1,14,15 Fungal infections tend to occur in immunocompromised patients; risk factors are steroid immunosuppression, poorly controlled diabetes, and peripheral vascular disease.16 Some zygomycetes may also affect immunocompetent patients.15
C gattii is an encapsulated yeast organism that is genetically and biochemically distinct from C neoformans. It is endemic to tropical parts of Africa and Australia. Its main environmental sources are eucalyptus trees (Eucalyptus camaldulensis, Eucalyptus tereticornis) and decaying hollows in living trees.17 In addition, there have been reports of isolation of C gattii from insect frass,18 which would make infection by an insect bite a possible transmission route. Worldwide distribution of this pathogen has increased recently, with outbreaks noted on Vancouver Island and in areas in Canada and the northwest United States.7
The true incidence of NF secondary to C gattii is difficult to determine. C gattii was only recently identified as a separate species, and pre-2006 cases of NF attributed to C neoformans may instead have been caused by C gattii. Misidentification has been compounded by the fact that the tests required for accurate diagnosis of C gattii infection may not be readily available in many clinical microbiology laboratories. Cryptococcus can be identified with various methods, including direct microscopy, culturing of tissue or fluid samples, and measurement of cryptococcal serum antigen. However, tests such as specific culture media, mass spectrometry, and molecular typing studies are required to determine cryptococcal species. L-canavanine-glycine-bromothymol blue (CGB) agar is a medium that is often used to differentiate C gattii from C neoformans because of the ability of C gattii to produce a blue appearance.6 Modern techniques, such as MALDI-TOF MS, have also been used to successfully distinguish between C gattii and C neoformans.9 MALDI-TOF MS identifies species on the basis of characteristic protein spectra extracted from whole cells. Using commercial and supplemental reference libraries, the system compares signal matches in the reference spectrum with Cryptococcus entries in the library—allowing rapid and accurate identification of cryptococcal species. However, this diagnostic method is limited by availability of adequate Cryptococcus entries in the reference library and by the high cost of acquiring the machine.
Serotyping is based on the antigenic property of the capsule and was once used to differentiate C neoformans into its 3 main varieties: var. neoformans, var. grubii, var. gattii. However, when it was realized that the antigenic property of the strain can be unstable and that there are hybrids containing more than 1 serotype, serotyping was abandoned as a species-differentiation test.6 The current gold standard for species differentiation is molecular genotyping. Molecular genotyping studies can confirm the diagnosis of C gattii infection and allow differentiation of C gattii into its 4 main molecular types: VGI, VGII, VGIII, VGIV. Using methods such as polymerase chain reaction (PCR) and restriction fragment length polymorphism (RFLP) analysis, molecular typing allows for specific epidemiology charting of C gattii genotypes.7
Although the transmission route for cryptococcal infection is mainly respiratory, direct inoculation has been reported as well.19 Cutaneous lesions, which occur in 5% to 20% of cryptococcal infections, often present in the head and neck.2,20,21 Primary cutaneous infections from cryptococcosis are rare, and cutaneous manifestations are often a sign of disseminated disease. Disseminated disease is defined as the involvement of 2 or more noncontiguous sites or evidence of high fungal burden based on cryptococcal antigen titer of more than 1:512.12 It is important to exclude disseminated disease in all cases of cryptococcosis, as it may be fatal.20 The neural and pulmonary systems should be screened.22 Cellulitis from cryptococcosis is almost always limited to immunocompromised patients, though there are reports of crytococcal cutaneous disease in immunocompetent patients.3,15 Interestingly, though C neoformans often affects immunocompromised patients, the emerging pathogen of C gattii affects immunocompetent patients.7,17,23 Our patient’s undiagnosed diabetes may have been a risk factor for cryptococcal infection. His cryptococcal antigen titer was 1:256, with no evidence of other sites of involvement. We therefore believe this to be a rare case of direct inoculation secondary to an insect bite.
The literature includes 12 reported cases of NF secondary to Cryptococcus (Table 2), all C neoformans. Of these cases, 9 involved immunosuppression, and most of these patients were on long-term steroid treatment after organ transplantation. The most common infection site was the lower extremity. These cases of cryptococcal NF show that immunosuppression, and long-term steroid use in particular, is an important risk factor. The mortality rate for these reviewed cases was 41.6% (5/12). According to the literature, the mortality rates for patients with cryptococcal soft-tissue infections24 and posttransplant patients with cryptococcal NF21 were 37.5% and 60%, respectively. We believe the mortality rate in our reviewed cases likely was confounded by the fact that most of the patients were posttransplant patients on long-term immunosuppression.
Of the 12 patients, 5 had primary cutaneous disease. There seems to be no relationship between outcome and dissemination of disease. In addition, there is a paucity of literature on the effect of disseminated disease and cryptococcal soft-tissue infections. Therefore, no firm conclusions can be drawn regarding the effects of disseminated disease on severity of cryptococcal soft-tissue infection.
Treatment of cryptococcal NF involves a combination of surgical débridement and long-term antifungal therapy. Surgical débridement of NF includes delineating the extent of infection with complete surgical excision of the affected tissue.25 The aims of surgery should be to remove all unhealthy tissue, identify the offending organism, and plan for resurfacing or reconstruction of the afflicted extremity. Intraoperative-tissue histology should be performed to confirm the diagnosis of NF. Histology can be used to demonstrate cryptococcal infection. The diagnosis of cryptococcal infection can be aided with fungal cultures, and therefore we recommend that tissue cultures be sent not only for routine aerobic/anaerobic bacteria but also for mycobacteria and fungal organisms. Laboratory tests that aid in diagnosis include serum cryptococcal antigen titer.
The current treatment recommendation for cryptococcal disease in patients who are not HIV-positive or transplant hosts is amphotericin B deoxycholate 0.7 to 1.0 mg/kg/d plus flucytosine 100 mg/kg/d for at least 4 weeks.22 The regimen period may be shortened to 14 days for patients at low risk of treatment failure. Fluconazole should be given as maintenance therapy (200 mg/d) for 6 to 12 months. There is no compelling evidence for immunoglobulin therapy for cryptococcal disease.22
Conclusion
NF caused by Cryptococcus is rare. A high level of suspicion, and intraoperative specimens for histology and fungal microscopy and culture, can help in establishing the diagnosis. Molecular genotyping remains the diagnostic method of choice for NF secondary to Cryptococcus. Effective treatment consists of aggressive surgical débridement and antifungal therapy.
1. Marcus JR, Hussong JW, Gonzalez C, Dumanian GA. Risk factors in necrotizing fasciitis: a case involving Cryptococcus neoformans. Ann Plast Surg. 1998;40(1):80-83.
2. Huang KC, Tu YK, Lee KF, Huang TJ, Wen-Wei Hsu R. Disseminated cryptococcosis presented as necrotizing fasciitis of a limb. J Trauma. 2007;63(2):E44-E46.
3. Capoor MR, Khanna G, Malhotra R. Disseminated cryptococcosis with necrotizing fasciitis in an apparently immunocompetent host: a case report. Med Mycol. 2008;46:269-273.
4. Adachi M, Tsurata D, Imanishi H, Ishii M, Kobayashi H. Necrotizing fasciitis caused by Cryptococcus neoformans in a patient with pemphigus vegetans. Clin Exp Dermatol. 2009;34(8):e751-e753.
5. Enache-Angoulvant A, Chandenier J, Symoens F, et al. Molecular identification of Cryptococcus neoformans serotypes. J Clin Microbiol. 2007;45(4):1261-1265.
6. Kwon-Chung KJ, Varma A. Do major species concepts support one, two or more species within Cryptococcus neoformans? FEMS Yeast Res. 2006;6(4):657-687.
7. Datta K, Bartlett KH, Baer R, et al; Cryptococcus gattii Working Group of the Pacific Northwest. Spread of Cryptococcus gattii into Pacific Northwest region of the United States. Emerg Infect Dis. 2009;15(8):1185-1191.
8. Wong CH, Khin LW, Heng KS, Tan KC, Low CO. The LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score: a tool for distinguishing necrotizing fasciitis from other soft tissue infections. Crit Care Med. 2004;32(7):1535-1541.
9. McTaggart LR, Lei E, Richardson SE, Hoang L, Fothergill A, Zhang SX. Rapid identification of Cryptococcus neoformans and Cryptococcus gattii by matrix-assisted laser desorption ionization-time of flight mass spectrometry. J Clin Microbiol. 2011;49(8):3050-3053.
10. Meyer W, Castañeda A, Jackson S, Huynh M, Castañeda E; IberoAmerican Cryptococcal Study Group. Molecular typing of IberoAmerican Cryptococcus neoformans isolates. Emerg Infect Dis. 2003;9(2):189-195.
11. Wong CH, Chang HC, Pasupathy S, Khin LW, Tan JL, Low CO. Necrotizing fasciitis: clinical presentation, microbiology and determinants of mortality. J Bone Joint Surg Am. 2003;85(8):1454-1460.
12. Giuliano A, Lewis F Jr, Hadley K, Blaisdell FW. Bacteriology of necrotizing fasciitis. Am J Surg. 1977;134(1):52-57.
13. Morgan MS. Diagnosis and management of necrotising fasciitis: a multiparametric approach. J Hosp Infect. 2010;75(4):249-257.
14. Buchanan PJ, Mast BA, Lottenberg L, Kim T, Efron PA, Ang DN. Candida albicans necrotizing soft tissue infection: a case report and literature review of fungal necrotizing soft tissue infections. Ann Plastic Surg. 2013;70(6):739-741.
15. Jain D, Kumar Y, Vasishta RK, Rajesh L, Pattari SK, Chakrabarti A. Zygomycotic necrotizing fasciitis in immunocompetent patients: a series of 18 cases. Modern Pathol. 2006;19(9):1221-1226.
16. Fontes RA Jr, Ogilvie CM, Miclau T. Necrotizing soft-tissue infections. J Am Acad Orthop Surg. 2000;8(3):151-158.
17. Sorrell TC. Cryptococcus neoformans variety gattii. Med Mycol. 2001;39(2):155-168.
18. Kidd SE, Sorrell TC, Meyer W. Isolation of two molecular types of Cryptococcus neoformans var. gattii from insect frass. Med Mycol. 2003;41(2):171-176.
19. Neuville S, Dromer F, Morin O, Dupont B, Ronin O, Lortholary O; French Cryptococcosis Study Group. Primary cutaneous cryptococcosis: a distinct clinical entity. Clin Infect Dis. 2003;36(3):337-347.
20. Basaran O, Emiroglu R, Arikan U, Karakayali H, Haberal M. Cryptococcal necrotizing fasciitis with multiple sites of involvement in the lower extremities. Dermatol Surg. 2003;29(11):1158-1160.
21. Baer S, Baddley JW, Gnann JW, Pappas PG. Cryptococcal disease presenting as necrotizing cellulitis in transplant recipients. Transpl Infect Dis. 2009;11(4):353-358.
22. Perfect JR, Dismukes WE, Dromer F, et al. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis. 2010;50(3):291-322.
23. Chan M, Lye D, Win MK, Chow A, Barkham T. Clinical and microbiological characteristics of cryptococcosis in Singapore: predominance of Cryptococcus neoformans compared with Cryptococcus gattii. Int J Infect Dis. 2014;26:110-115.
24. Gave AA, Torres R, Kaplan L. Cryptococcal myositis and vasculitis: an unusual necrotizing soft tissue infection. Surg Infect. 2004;5(3):309-313.
25. Wong CH, Yam AK, Tan AB, Song C. Approach to debridement in necrotizing fasciitis. Am J Surg. 2008;196(3):e19-e24.
26. Bégon E, Bachmeyer C, Thibault M, et al. Necrotizing fasciitis due to Cryptococcus neoformans in a diabetic patient with chronic renal insufficiency. Clin Exp Dermatol. 2009;34(8):935-936.
27. Doorenbos-Bot AC, Hooymans JM, Blanksma LJ. Periorbital necrotising fasciitis due to Cryptococcus neoformans in a healthy young man. Doc Ophthalmol. 1990;75(3-4):315-320.
28. Yoneda T, Itami Y, Hirayama A, Saka T, Yoshida K, Fujimoto K. Cryptococcal necrotizing fasciitis in a patient after renal transplantation—a case report. Transplant Proc. 2014;46(2):620-622.
1. Marcus JR, Hussong JW, Gonzalez C, Dumanian GA. Risk factors in necrotizing fasciitis: a case involving Cryptococcus neoformans. Ann Plast Surg. 1998;40(1):80-83.
2. Huang KC, Tu YK, Lee KF, Huang TJ, Wen-Wei Hsu R. Disseminated cryptococcosis presented as necrotizing fasciitis of a limb. J Trauma. 2007;63(2):E44-E46.
3. Capoor MR, Khanna G, Malhotra R. Disseminated cryptococcosis with necrotizing fasciitis in an apparently immunocompetent host: a case report. Med Mycol. 2008;46:269-273.
4. Adachi M, Tsurata D, Imanishi H, Ishii M, Kobayashi H. Necrotizing fasciitis caused by Cryptococcus neoformans in a patient with pemphigus vegetans. Clin Exp Dermatol. 2009;34(8):e751-e753.
5. Enache-Angoulvant A, Chandenier J, Symoens F, et al. Molecular identification of Cryptococcus neoformans serotypes. J Clin Microbiol. 2007;45(4):1261-1265.
6. Kwon-Chung KJ, Varma A. Do major species concepts support one, two or more species within Cryptococcus neoformans? FEMS Yeast Res. 2006;6(4):657-687.
7. Datta K, Bartlett KH, Baer R, et al; Cryptococcus gattii Working Group of the Pacific Northwest. Spread of Cryptococcus gattii into Pacific Northwest region of the United States. Emerg Infect Dis. 2009;15(8):1185-1191.
8. Wong CH, Khin LW, Heng KS, Tan KC, Low CO. The LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score: a tool for distinguishing necrotizing fasciitis from other soft tissue infections. Crit Care Med. 2004;32(7):1535-1541.
9. McTaggart LR, Lei E, Richardson SE, Hoang L, Fothergill A, Zhang SX. Rapid identification of Cryptococcus neoformans and Cryptococcus gattii by matrix-assisted laser desorption ionization-time of flight mass spectrometry. J Clin Microbiol. 2011;49(8):3050-3053.
10. Meyer W, Castañeda A, Jackson S, Huynh M, Castañeda E; IberoAmerican Cryptococcal Study Group. Molecular typing of IberoAmerican Cryptococcus neoformans isolates. Emerg Infect Dis. 2003;9(2):189-195.
11. Wong CH, Chang HC, Pasupathy S, Khin LW, Tan JL, Low CO. Necrotizing fasciitis: clinical presentation, microbiology and determinants of mortality. J Bone Joint Surg Am. 2003;85(8):1454-1460.
12. Giuliano A, Lewis F Jr, Hadley K, Blaisdell FW. Bacteriology of necrotizing fasciitis. Am J Surg. 1977;134(1):52-57.
13. Morgan MS. Diagnosis and management of necrotising fasciitis: a multiparametric approach. J Hosp Infect. 2010;75(4):249-257.
14. Buchanan PJ, Mast BA, Lottenberg L, Kim T, Efron PA, Ang DN. Candida albicans necrotizing soft tissue infection: a case report and literature review of fungal necrotizing soft tissue infections. Ann Plastic Surg. 2013;70(6):739-741.
15. Jain D, Kumar Y, Vasishta RK, Rajesh L, Pattari SK, Chakrabarti A. Zygomycotic necrotizing fasciitis in immunocompetent patients: a series of 18 cases. Modern Pathol. 2006;19(9):1221-1226.
16. Fontes RA Jr, Ogilvie CM, Miclau T. Necrotizing soft-tissue infections. J Am Acad Orthop Surg. 2000;8(3):151-158.
17. Sorrell TC. Cryptococcus neoformans variety gattii. Med Mycol. 2001;39(2):155-168.
18. Kidd SE, Sorrell TC, Meyer W. Isolation of two molecular types of Cryptococcus neoformans var. gattii from insect frass. Med Mycol. 2003;41(2):171-176.
19. Neuville S, Dromer F, Morin O, Dupont B, Ronin O, Lortholary O; French Cryptococcosis Study Group. Primary cutaneous cryptococcosis: a distinct clinical entity. Clin Infect Dis. 2003;36(3):337-347.
20. Basaran O, Emiroglu R, Arikan U, Karakayali H, Haberal M. Cryptococcal necrotizing fasciitis with multiple sites of involvement in the lower extremities. Dermatol Surg. 2003;29(11):1158-1160.
21. Baer S, Baddley JW, Gnann JW, Pappas PG. Cryptococcal disease presenting as necrotizing cellulitis in transplant recipients. Transpl Infect Dis. 2009;11(4):353-358.
22. Perfect JR, Dismukes WE, Dromer F, et al. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis. 2010;50(3):291-322.
23. Chan M, Lye D, Win MK, Chow A, Barkham T. Clinical and microbiological characteristics of cryptococcosis in Singapore: predominance of Cryptococcus neoformans compared with Cryptococcus gattii. Int J Infect Dis. 2014;26:110-115.
24. Gave AA, Torres R, Kaplan L. Cryptococcal myositis and vasculitis: an unusual necrotizing soft tissue infection. Surg Infect. 2004;5(3):309-313.
25. Wong CH, Yam AK, Tan AB, Song C. Approach to debridement in necrotizing fasciitis. Am J Surg. 2008;196(3):e19-e24.
26. Bégon E, Bachmeyer C, Thibault M, et al. Necrotizing fasciitis due to Cryptococcus neoformans in a diabetic patient with chronic renal insufficiency. Clin Exp Dermatol. 2009;34(8):935-936.
27. Doorenbos-Bot AC, Hooymans JM, Blanksma LJ. Periorbital necrotising fasciitis due to Cryptococcus neoformans in a healthy young man. Doc Ophthalmol. 1990;75(3-4):315-320.
28. Yoneda T, Itami Y, Hirayama A, Saka T, Yoshida K, Fujimoto K. Cryptococcal necrotizing fasciitis in a patient after renal transplantation—a case report. Transplant Proc. 2014;46(2):620-622.
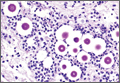
Multifocal Langerhans Cell Histiocytosis in an Adult
Eosinophilic granuloma (EG) is the most common benign form of Langerhans cell histiocytosis (LCH). Initially described by Lichtenstein in 1953, LCH encompasses a triad of proliferative granulomatous disorders primarily affecting children: EG, Hand-Schüller-Christian disease, and Letterer-Siwe disease.1 Lichtenstein first termed the disease histiocytosis X, after recognizing that the 3 syndromes had the same histology.1 The term was updated after the clonal proliferation of Langerhans cells in the pathogenesis of the disease was discovered.
As LCH is generally considered a pediatric disease, there is little in the literature regarding adult-onset LCH. The incidence of LCH in adults is reported as 1 to 2 cases per million, significantly lower than that in children.2,3 Two studies have reported the mean age at diagnosis in adults as the fourth decade of life, and have suggested a male predominance.4,5 The vast majority of adult LCH cases described are simple EG, with very few cases of multisystem disseminated disease reported.5
Adult patients with LCH typically present with solitary lesions in bone. Approximately 10% of cases have extraosseous involvement, with the lung being the most common site.6 Lesions tend to be unifocal, with fewer than 10 reports describing multifocal EG.1,7-13 The axial skeleton is most frequently involved, with the majority of lesions occurring in the skull, ribs, vertebrae, or mandible.14 While less common, the femur, humerus, and clavicle are most often involved when the appendicular skeleton is affected.5
In a literature review, a few case reports describe adult-onset EG of the skull. Only 5 case reports since the 1970s describe adult patients with EG of the femur. We present a rare case of multifocal EG in a 48-year-old woman with lesions of the femur and skull, as well as a review of the literature. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 48-year-old woman presented with progressive right knee pain that was exacerbated by weight-bearing. She denied trauma, fevers, fatigue, or weight change. Her history was significant for an EG of the skull, excised at an outside institution 2 years prior to presentation. The patient also admitted to recent onset of right-sided skull pain, near the region of her previous surgery.
Physical examination demonstrated tenderness to palpation and fullness over the right medial distal femur and a normal neurovascular examination of the right lower extremity. Radiographs of the knee showed a cortically based, lytic, destructive lesion involving the medial femoral condyle, with soft-tissue extension (Figures 1A, 1B). Magnetic resonance imaging (MRI) of the right knee showed the lesion, with extraosseous soft-tissue extension (Figures 2A, 2B). The mass was isointense to muscle on T1-weighted images and hyperintense on T2-weighted images. Technetium bone scanning showed increased uptake in the right femur and the right skull (Figures 3A, 3B). MRI of the brain confirmed a new lesion in the right diploic space, distinct from the previous EG lesion site (Figures 4A-4D). An ultrasound-guided biopsy of the femur was performed and was consistent with EG.
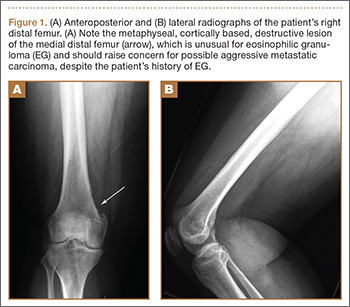
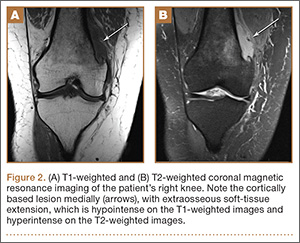
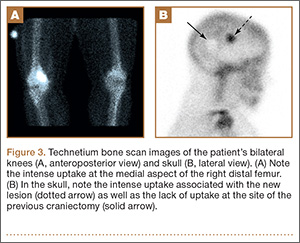
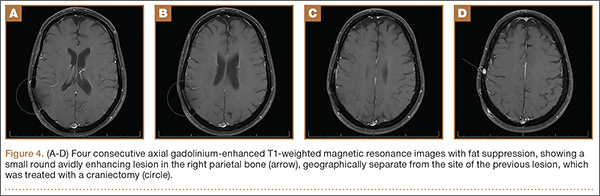
After reevaluation and clearance by her neurosurgeon, the patient underwent curettage and allografting of the femoral lesion, with prophylactic internal fixation using a titanium distal femoral locking plate (Figure 5). Intraoperative frozen section was consistent with EG, which was confirmed with additional immunohistochemical workup (Figures 6A-6D).
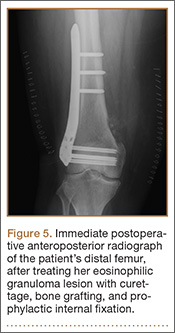
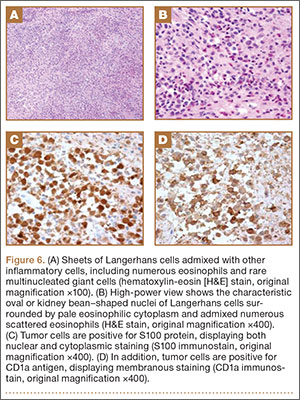
The patient recovered uneventfully and follow-up radiographs showed restoration of the bony cortex of the medial femoral condyle (Figure 7). The second skull lesion, which was also consistent with EG, was excised by her neurosurgeon.
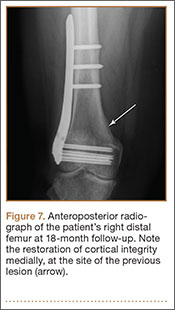
The patient remained asymptomatic until 2 years later, when she began experiencing mild pain in her right distal thigh and knee. Radiographs showed a new lytic focus in the right distal metadiaphysis (Figure 8) which was not present on her last radiograph 6 months prior. A computed tomography (CT) scan showed a lytic lesion involving the right distal femur medullary canal with cortical thinning and destruction, most pronounced posteriorly (Figures 9A, 9B). There was also an extraosseous soft-tissue component to the lesion. Bone scan showed increased uptake in the area of the new lesion. There was no increased uptake elsewhere, including the medial distal femur at the site of the old lesion, to suggest other lesions, and no increased uptake in the skull.
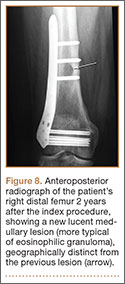
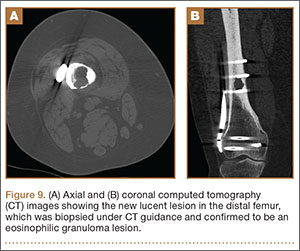
Given that the location of the lesion was distinct from the prior site of curettage and bone grafting, it was thought to be consistent with a new EG lesion. The patient underwent CT-guided biopsy, with simultaneous intralesional corticosteroid injection to treat the lesion when on-site pathology confirmed the etiology. Further surgical management was deemed unnecessary because internal fixation was present and spanned the new lesion. Final analysis of the fine-needle aspirate of the new lesion was positive for numerous eosinophils and histiocytes, consistent with EG.
At 6-week follow-up after the intralesional steroid injection, the patient’s pain continued to abate, and she was ambulating with crutches. Repeat CT scan of the right distal femur showed improvement of the extraosseous soft-tissue component, while the lucency in the femur itself remained unchanged. The decision was made to proceed with a second intralesional corticosteroid injection under CT guidance. The patient’s symptoms continued to improve, and repeat imaging 1 year after her steroid injections showed substantial bony healing with reconstitution of her cortical bone (Figures 10A-10E).
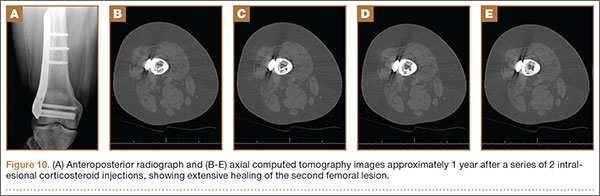
The patient had had 4 distinct tumors consistent with EG and was referred to a medical oncologist for further workup. The patient began treatment with zoledronic acid to prevent development of further lesions. At most recent follow-up, the patient was 18 months out from her second intralesional corticosteroid injection and was doing very well. She reported being pain-free and was walking 3 to 4 miles per week without gait aids. There was no evidence of new disease. The medial distal femur lesion was completely healed, and the distal metaphyseal lesion was nearly healed, with very little residual evidence of lesions.
Discussion
Adult-onset multifocal EG is a rare entity. Most affected patients develop lesions in the axial skeleton, with the skull, mandible, and vertebrae most commonly involved.14 Only 5 cases of femoral EG have been reported, one of which was multifocal.11,14-17
Of these patients, 3 were between the ages of 33 and 53 years and had insidious onset of hip pain that failed conservative management.14,15,17 Further imaging and biopsy revealed unifocal EG in the proximal femur in each case. Each patient received a different form of treatment, including curettage and radiation, radiofrequency ablation, and/or physical therapy. At the time of publication, all patients had reported improvement in their clinical symptoms.14,15,17 The fourth patient was a man with human immunodeficiency virus (HIV) with 3 months of progressive thigh pain. Further evaluation found an isolated EG of the femoral diaphysis that progressed to pathologic fracture. He was treated with curettage and intramedullary nailing, and had improved symptoms and radiographic signs of healing at 30-month follow-up.16
An interesting case by Kerzl and colleagues11 reported a 63-year-old woman with a 24-year history of multiple symmetric lesions of the femora, leading to multiple pathologic fractures. Like our patient, her initial lesion was in the skull. Initial pathology specimens led to the diagnosis of EG. However, as the patient aged, she developed symptoms of diabetes insipidus and xanthelasma, which led to reevaluation of histology from 3 bony lesions. The patient was determined to have multifocal EG of the skull and femur, with simultaneous occurrence of Erdheim-Chester disease, which also causes bone lesions in addition to diabetes insipidus and xanthelasma.11
Though LCH was initially described more than 50 years ago, many aspects of LCH remain an enigma, especially in adults. The etiology of the disease is poorly understood. Controversy exists regarding whether LCH is primarily an immunoregulatory, neoplastic, or reactive disorder. The vast majority of adult cases described in the literature are EG, with very few cases of multisystem disseminated disease reported.5
The spectrum of disorders constituting LCH is heterogenous. Eosinophilic granuloma is the most common form, reportedly accounting for 60% to 70% of all cases, usually presenting as solitary bone lesions.6 Eosinophilic granuloma refers to the localized form of LCH, in which the disease is limited to bone or lung.18 This is the least aggressive form of the disease, with the most favorable prognosis. Hand- Schüller-Christian disease is a chronic, recurring form of LCH, with disseminated disease, affecting both bone and extraskeletal sites. Hand-Schüller-Christian disease is known for the classic triad of diabetes insipidus, exophthalmos, and destructive bone lesions. Patients may also present with otitis media or neurologic complaints from pathologic vertebral fractures. Letterer-Siwe disease refers to the acute, disseminated, fulminant form of LCH. This is the least common form of LCH and is predominately described in young children. Patients present with hepatosplenomegaly, lymphadenopathy, skin rash, fever, anemia, and thrombocytopenia.19 It is rapidly progressive, leading to multiorgan dysfunction and death within 1 to 2 years.18
The classification of LCH follows the Histiocyte Society guidelines developed from multicenter randomized trials in children.3 Classification is based on affected organs and is divided into 2 categories: single-system disease or multisystem disease. Single-system disease may be single site (bone, skin, or solitary lymph node) or multisite (multifocal bone disease or multiple lymph nodes). Multisystem disease is further classified into low-risk or risk groups. The low-risk group involves disseminated disease without involvement of risk organs (lungs, liver, spleen, and hematopoietic system). Involvement of 1 or more risk organs places the patient in the risk group, associated with the least favorable prognosis.3
In adults, the most common presenting symptoms are local pain from bony involvement, weight loss, and fever. Bony lesions most often occur in the skull, especially in the jaw. Long bones are less frequently involved, with lesions occurring in the long bones in approximately 17% of patients.3 The rib has also been reported as a common site of involvement in adults.5 Similar to children, diabetes insipidus remains a classic manifestation of LCH because of pituitary gland involvement. Other common symptoms of LCH in adults are cough, dyspnea, and chest pain from pulmonary involvement. Up to 20% to 30% of adult LCH patients have isolated pulmonary lesions, although pulmonary LCH may also occur as part of multisystem disease (risk group).3,4,20
Eosinophilic granuloma bone lesions have a variety of radiographic appearances but most commonly appear as lytic lesions. They often mimic aggressive lesions with permeative bone destruction, periostitis, ill-defined borders, and cortical erosion. Most lesions arise in the medullary space but can present as a destructive, cortically based lesion, as it did in our patient’s first femoral lesion. The differential diagnosis for a lytic medullary bone lesion includes benign entities, such as nonossifying fibromas, bone cysts, or osteomyelitis, but also includes malignant tumors, such as metastases, Ewing sarcoma, and lymphoma. A destructive, cortically based lesion in an adult should raise a very high suspicion for metastatic carcinoma until proven otherwise. Other diagnostic considerations for a cortically based lesion include chondromyxoid fibroma and surface bone lesions, such as surface chondroma and osteoma, or osteosarcoma (parosteal and periosteal). In the skull, lesions commonly erode the outer table more than the inner table (the typical “beveled-edge” appearance). Skull lesions also may have a small, central, dense focus within the lytic lesion (“button sequestrum”).
Bone scanning is often not as sensitive in detecting EG lesions compared with other bone tumors, although in our patient the bone scan was positive. In patients with a negative bone scan but a high index of suspicion, a radiographic skeletal survey should be obtained to rule out other lesions. MRI typically shows T2-hyperintense, T1-hypointense lesions with surrounding bone marrow edema and variable contrast enhancement, which is relatively nonspecific. The high sensitivity of MRI allows accurate delineation of the extent of the lesions and evaluates for the presence of an extraosseous soft-tissue component. Biopsy is generally necessary to establish a definitive histologic diagnosis. In our patient, despite her history of biopsy-proven EG, the aggressive appearance of a destructive, cortically based lesion made obtaining a biopsy critical to establish a definitive diagnosis in this case.
The histopathologic examination of the tissue from our patient was typical of that seen in patients with EG. It revealed tissue fragments with diffuse sheets of histiocytes displaying nuclear grooves, admixed numerous eosinophils with eosinophilic microabscesses, and scattered lymphocytes (Figures 6A, 6B). There were areas of necrosis, raising the possibility of osteomyelitis. However, the presence of classic histomorphologic features of LCH in the majority of the tissue fragments, along with CD1a- and S100-positivity in the histiocytes, confirmed the diagnosis of LCH (Figures 6C, 6D). Although not highly specific, a positive CD1a immunostain with the described histomorphologic findings in the proper clinical setting is often considered sufficient for LCH diagnosis. S100 is an important adjunct immunostain in the evaluation of histiocytic disorders. A positive S100 immunostain helps identify histiocytes, which are also CD1a-positive, because the latter immunostain can also be positive in some lymphomas and thymomas.21
After diagnosis of LCH has been confirmed, staging includes radiographs of any suspicious bone lesions, chest radiograph, bone scan, abdominal ultrasound, routine laboratory studies, and chest CT if pulmonary LCH is suspected.
The optimal treatment strategy for adult patients has not been clearly defined, and current strategies for LCH vary depending on organ involvement and extent of disease. Therapeutic options include observation, local treatment with steroids, local excision with curettage with or without bone grafting, chemotherapy, immunomodulation, irradiation, and stem cell transplantation in advanced disease. In general, patients who benefit from systemic therapy, such as chemotherapy or immunomodulation, include those with multisystem disease, refractory or recurrent lesions, and multifocal skeletal involvement.22
Patients with more limited disease, such as EG of bone, may undergo observation or local intralesional treatment. Eosinophilic granuloma of bone may resolve spontaneously and commonly does so when it is located in the pediatric spine. However, the therapeutic approach in adults with EG is controversial, given that spontaneous resolution is less likely to occur in the skeletally mature. Plasschaert and colleagues23 reported a recurrence rate of 26% in skeletally mature patients with EG of bone treated with biopsy followed by curettage with or without grafting. In the skeletally immature group, there were no clinical or radiographic signs of recurrence in the 2-year follow-up period.23 Thus, treatment in the adult population must be considered separate from the skeletally immature and in the appropriate clinical context. Depending on the location of the lesion, patients may become symptomatic or be at risk for pathologic fracture. In such circumstances, curettage with or without bone grafting and prophylactic internal fixation may be indicated. Other treatments, such as intralesional infiltration with corticosteroids, have been reported, but the role of such treatment in adults is undetermined.24,25 Radiation is typically not recommended in single-system disease unless a vital organ is threatened.26 Overall, patients with single-system disease have an excellent prognosis, and treatment should be determined on an individual basis.3
Eosinophilic granuloma represents less than 1% of all bone tumors, and adult presentation is very rare. The differential diagnosis of lytic bone lesions is broad and includes metastatic carcinoma, lymphoma/myeloma, osteomyelitis, osteoblastoma, aneurysmal bone cyst, and Ewing sarcoma. While EG is more common and easily diagnosed in children, it should be considered in the differential diagnosis in adults, so that the appropriate diagnostic workup and treatment can be performed.
1. Lahiani D, Hammami BK, Maâloul I, et al. Multifocal Langerhans cell histiocytosis of bone: late revelation in a 76-year-old woman. Rev Med Interne. 2008;29(3):249-251.
2. Baumgartner I, von Hochstetter A, Baumert B, Luetolf U, Follath F. Langerhans’-cell histiocytosis in adults. Med Pediatr Oncol. 1997;28(1):9-14.
3. Stockschlaeder M, Sucker C. Adult Langerhans cell histiocytosis. Eur J Haematol. 2006;76(5):363-368.
4. Aricò M, Girschikofsky M, Généreau T, et al. Langerhans cell histiocytosis in adults. Report from the International Registry of the Histiocyte Society. Eur J Cancer. 2003;39(16):2341-2348.
5. Islinger RB, Kuklo TR, Owens BD, et al. Langerhans’ cell histiocytosis in patients older than 21 years. Clin Orthop Relat Res. 2000;379:231-235.
6. Key SJ, O’Brien CJ, Silvester KC, Crean SJ. Eosinophilic granuloma: resolution of maxillofacial bony lesions following minimal intervention. Report of three cases and a review of the literature. J Craniomaxillofac Surg. 2004;32(3):170-175.
7. Bodner G, Kreczy A, Rachbauer F, Baechter O, Peer S. Eosinophilic granuloma of the bone: ultrasonographic imaging. Australas Radiol. 2002;46(4):418-421.
8. Boutsen Y, Esselinckx W, Delos M, Nisolle JF. Adult onset of multifocal eosinophilic granuloma of bone: a long-term follow-up with evaluation of various treatment options and spontaneous healing. Clin Rheumatol. 1999;18(1):69-73.
9. Corti F, Valicenti A, Bertolucci D, Bruno J, Gustinucci R. Multifocal Langerhans cell granulomatosis. Report of a clinical case. Minerva Med. 1994;85(7-8):413-416.
10. Demirci I. Adult eosinophilic granuloma of the lumbar spine with atypical dissemination. Case report: a long-term follow-up. Zentralbl Neurochir. 2004;65(2):84-87.
11. Kerzl R, Eyerich K, Eberlein B, et al. Parallel occurrence of Erdheim-Chester disease and eosinophilic granuloma in the same patient. J Eur Acad Dermatol Venereol. 2009;23(2):224-226.
12. Nguyen BD, Roarke MC, Chivers SF. Multifocal Langerhans cell histiocytosis with infiltrative pelvic lesions: PET/CT imaging. Clin Nucl Med. 2010;35(10): 824-826.
13. Scolozzi P, Lombardi T, Monnier P, Jaques B. Multisystem Langerhans’ cell histiocytosis (Hand-Schuller-Christian disease) in an adult: a case report and review of the literature. Eur Arch Otorhinolaryngol. 2004;261(6):326-330.
14. King JJ, Melvin JS, Iwenofu OH, Fox EJ. Thigh pain in a 53-year-old woman. Clin Orthop Relat Res. 2009;467(6):1652-1657.
15. Hair LC, Deyle GD. Eosinophilic granuloma in a patient with hip pain. J Orthop Sports Phys Ther. 2011;41(2):119.
16. Panayiotakopoulos GD, Sipsas NV, Kontos A, et al. Eosinophilic granuloma of the femur in an HIV-1 positive patient. AIDS Patient Care STDS. 2002;16(3):103-106.
17. Rodrigues RJ, Lewis HH. Eosinophilic granuloma of bone. Review of literature and case presentation. Clin Orthop Relat Res. 1971;77:183-192.
18. Stull MA, Kransdorf MJ, Devaney KO. Langerhans cell histiocytosis of bone. Radiographics. 1992;12(4):801-823.
19. Lichtenstein L. Histiocytosis X (eosinophilic granuloma of bone, Letterer-Siwe disease, and Schueller-Christian disease). Further observations of pathological and clinical importance. J Bone Joint Surg Am. 1964;46:76-90.
20. Götz G, Fichter J. Langerhans’-cell histiocytosis in 58 adults. Eur J Med Res. 2004;9(11):510-514.
21. Cheng KL, Glu PG, Weiss LM. Hematopoeitic tumors. In: Peiguo C, Weiss L, eds. Modern Immunohistochemistry. New York, NY: Cambridge University Press; 2009:503.
22. Broadbent V, Gadner H. Current therapy for Langerhans cell histiocytosis. Hematol Oncol Clin North Am. 1998;12(2):327-338.
23. Plasschaert F, Craig C, Bell R, Cole WG, Wunder JS, Alman BA. Eosinophilic granuloma. A different behaviour in children than in adults. J Bone Joint Surg Br. 2002;84(6):870-872.
24. Capanna R, Springfield DS, Ruggieri P, et al. Direct cortisone injection in osinophilic granuloma of bone: a preliminary report on 11 patients. J Pediatr Orthop. 1985;5(3):339-342.
25. Egeler RM, Thompson RC Jr, Voûte PA, Nesbit ME Jr. Intralesional infiltration of corticosteroids in localized Langerhans’ cell histiocytosis. J Pediatr Orthop. 1992;12(6):811-814.
26. Ladisch S, Gadner H. Treatment of Langerhans cell histiocytosis–evolution and current approaches. Br J Cancer Suppl. 1994;23:S41-S46.
Eosinophilic granuloma (EG) is the most common benign form of Langerhans cell histiocytosis (LCH). Initially described by Lichtenstein in 1953, LCH encompasses a triad of proliferative granulomatous disorders primarily affecting children: EG, Hand-Schüller-Christian disease, and Letterer-Siwe disease.1 Lichtenstein first termed the disease histiocytosis X, after recognizing that the 3 syndromes had the same histology.1 The term was updated after the clonal proliferation of Langerhans cells in the pathogenesis of the disease was discovered.
As LCH is generally considered a pediatric disease, there is little in the literature regarding adult-onset LCH. The incidence of LCH in adults is reported as 1 to 2 cases per million, significantly lower than that in children.2,3 Two studies have reported the mean age at diagnosis in adults as the fourth decade of life, and have suggested a male predominance.4,5 The vast majority of adult LCH cases described are simple EG, with very few cases of multisystem disseminated disease reported.5
Adult patients with LCH typically present with solitary lesions in bone. Approximately 10% of cases have extraosseous involvement, with the lung being the most common site.6 Lesions tend to be unifocal, with fewer than 10 reports describing multifocal EG.1,7-13 The axial skeleton is most frequently involved, with the majority of lesions occurring in the skull, ribs, vertebrae, or mandible.14 While less common, the femur, humerus, and clavicle are most often involved when the appendicular skeleton is affected.5
In a literature review, a few case reports describe adult-onset EG of the skull. Only 5 case reports since the 1970s describe adult patients with EG of the femur. We present a rare case of multifocal EG in a 48-year-old woman with lesions of the femur and skull, as well as a review of the literature. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 48-year-old woman presented with progressive right knee pain that was exacerbated by weight-bearing. She denied trauma, fevers, fatigue, or weight change. Her history was significant for an EG of the skull, excised at an outside institution 2 years prior to presentation. The patient also admitted to recent onset of right-sided skull pain, near the region of her previous surgery.
Physical examination demonstrated tenderness to palpation and fullness over the right medial distal femur and a normal neurovascular examination of the right lower extremity. Radiographs of the knee showed a cortically based, lytic, destructive lesion involving the medial femoral condyle, with soft-tissue extension (Figures 1A, 1B). Magnetic resonance imaging (MRI) of the right knee showed the lesion, with extraosseous soft-tissue extension (Figures 2A, 2B). The mass was isointense to muscle on T1-weighted images and hyperintense on T2-weighted images. Technetium bone scanning showed increased uptake in the right femur and the right skull (Figures 3A, 3B). MRI of the brain confirmed a new lesion in the right diploic space, distinct from the previous EG lesion site (Figures 4A-4D). An ultrasound-guided biopsy of the femur was performed and was consistent with EG.
After reevaluation and clearance by her neurosurgeon, the patient underwent curettage and allografting of the femoral lesion, with prophylactic internal fixation using a titanium distal femoral locking plate (Figure 5). Intraoperative frozen section was consistent with EG, which was confirmed with additional immunohistochemical workup (Figures 6A-6D).
The patient recovered uneventfully and follow-up radiographs showed restoration of the bony cortex of the medial femoral condyle (Figure 7). The second skull lesion, which was also consistent with EG, was excised by her neurosurgeon.
The patient remained asymptomatic until 2 years later, when she began experiencing mild pain in her right distal thigh and knee. Radiographs showed a new lytic focus in the right distal metadiaphysis (Figure 8) which was not present on her last radiograph 6 months prior. A computed tomography (CT) scan showed a lytic lesion involving the right distal femur medullary canal with cortical thinning and destruction, most pronounced posteriorly (Figures 9A, 9B). There was also an extraosseous soft-tissue component to the lesion. Bone scan showed increased uptake in the area of the new lesion. There was no increased uptake elsewhere, including the medial distal femur at the site of the old lesion, to suggest other lesions, and no increased uptake in the skull.
Given that the location of the lesion was distinct from the prior site of curettage and bone grafting, it was thought to be consistent with a new EG lesion. The patient underwent CT-guided biopsy, with simultaneous intralesional corticosteroid injection to treat the lesion when on-site pathology confirmed the etiology. Further surgical management was deemed unnecessary because internal fixation was present and spanned the new lesion. Final analysis of the fine-needle aspirate of the new lesion was positive for numerous eosinophils and histiocytes, consistent with EG.
At 6-week follow-up after the intralesional steroid injection, the patient’s pain continued to abate, and she was ambulating with crutches. Repeat CT scan of the right distal femur showed improvement of the extraosseous soft-tissue component, while the lucency in the femur itself remained unchanged. The decision was made to proceed with a second intralesional corticosteroid injection under CT guidance. The patient’s symptoms continued to improve, and repeat imaging 1 year after her steroid injections showed substantial bony healing with reconstitution of her cortical bone (Figures 10A-10E).
The patient had had 4 distinct tumors consistent with EG and was referred to a medical oncologist for further workup. The patient began treatment with zoledronic acid to prevent development of further lesions. At most recent follow-up, the patient was 18 months out from her second intralesional corticosteroid injection and was doing very well. She reported being pain-free and was walking 3 to 4 miles per week without gait aids. There was no evidence of new disease. The medial distal femur lesion was completely healed, and the distal metaphyseal lesion was nearly healed, with very little residual evidence of lesions.
Discussion
Adult-onset multifocal EG is a rare entity. Most affected patients develop lesions in the axial skeleton, with the skull, mandible, and vertebrae most commonly involved.14 Only 5 cases of femoral EG have been reported, one of which was multifocal.11,14-17
Of these patients, 3 were between the ages of 33 and 53 years and had insidious onset of hip pain that failed conservative management.14,15,17 Further imaging and biopsy revealed unifocal EG in the proximal femur in each case. Each patient received a different form of treatment, including curettage and radiation, radiofrequency ablation, and/or physical therapy. At the time of publication, all patients had reported improvement in their clinical symptoms.14,15,17 The fourth patient was a man with human immunodeficiency virus (HIV) with 3 months of progressive thigh pain. Further evaluation found an isolated EG of the femoral diaphysis that progressed to pathologic fracture. He was treated with curettage and intramedullary nailing, and had improved symptoms and radiographic signs of healing at 30-month follow-up.16
An interesting case by Kerzl and colleagues11 reported a 63-year-old woman with a 24-year history of multiple symmetric lesions of the femora, leading to multiple pathologic fractures. Like our patient, her initial lesion was in the skull. Initial pathology specimens led to the diagnosis of EG. However, as the patient aged, she developed symptoms of diabetes insipidus and xanthelasma, which led to reevaluation of histology from 3 bony lesions. The patient was determined to have multifocal EG of the skull and femur, with simultaneous occurrence of Erdheim-Chester disease, which also causes bone lesions in addition to diabetes insipidus and xanthelasma.11
Though LCH was initially described more than 50 years ago, many aspects of LCH remain an enigma, especially in adults. The etiology of the disease is poorly understood. Controversy exists regarding whether LCH is primarily an immunoregulatory, neoplastic, or reactive disorder. The vast majority of adult cases described in the literature are EG, with very few cases of multisystem disseminated disease reported.5
The spectrum of disorders constituting LCH is heterogenous. Eosinophilic granuloma is the most common form, reportedly accounting for 60% to 70% of all cases, usually presenting as solitary bone lesions.6 Eosinophilic granuloma refers to the localized form of LCH, in which the disease is limited to bone or lung.18 This is the least aggressive form of the disease, with the most favorable prognosis. Hand- Schüller-Christian disease is a chronic, recurring form of LCH, with disseminated disease, affecting both bone and extraskeletal sites. Hand-Schüller-Christian disease is known for the classic triad of diabetes insipidus, exophthalmos, and destructive bone lesions. Patients may also present with otitis media or neurologic complaints from pathologic vertebral fractures. Letterer-Siwe disease refers to the acute, disseminated, fulminant form of LCH. This is the least common form of LCH and is predominately described in young children. Patients present with hepatosplenomegaly, lymphadenopathy, skin rash, fever, anemia, and thrombocytopenia.19 It is rapidly progressive, leading to multiorgan dysfunction and death within 1 to 2 years.18
The classification of LCH follows the Histiocyte Society guidelines developed from multicenter randomized trials in children.3 Classification is based on affected organs and is divided into 2 categories: single-system disease or multisystem disease. Single-system disease may be single site (bone, skin, or solitary lymph node) or multisite (multifocal bone disease or multiple lymph nodes). Multisystem disease is further classified into low-risk or risk groups. The low-risk group involves disseminated disease without involvement of risk organs (lungs, liver, spleen, and hematopoietic system). Involvement of 1 or more risk organs places the patient in the risk group, associated with the least favorable prognosis.3
In adults, the most common presenting symptoms are local pain from bony involvement, weight loss, and fever. Bony lesions most often occur in the skull, especially in the jaw. Long bones are less frequently involved, with lesions occurring in the long bones in approximately 17% of patients.3 The rib has also been reported as a common site of involvement in adults.5 Similar to children, diabetes insipidus remains a classic manifestation of LCH because of pituitary gland involvement. Other common symptoms of LCH in adults are cough, dyspnea, and chest pain from pulmonary involvement. Up to 20% to 30% of adult LCH patients have isolated pulmonary lesions, although pulmonary LCH may also occur as part of multisystem disease (risk group).3,4,20
Eosinophilic granuloma bone lesions have a variety of radiographic appearances but most commonly appear as lytic lesions. They often mimic aggressive lesions with permeative bone destruction, periostitis, ill-defined borders, and cortical erosion. Most lesions arise in the medullary space but can present as a destructive, cortically based lesion, as it did in our patient’s first femoral lesion. The differential diagnosis for a lytic medullary bone lesion includes benign entities, such as nonossifying fibromas, bone cysts, or osteomyelitis, but also includes malignant tumors, such as metastases, Ewing sarcoma, and lymphoma. A destructive, cortically based lesion in an adult should raise a very high suspicion for metastatic carcinoma until proven otherwise. Other diagnostic considerations for a cortically based lesion include chondromyxoid fibroma and surface bone lesions, such as surface chondroma and osteoma, or osteosarcoma (parosteal and periosteal). In the skull, lesions commonly erode the outer table more than the inner table (the typical “beveled-edge” appearance). Skull lesions also may have a small, central, dense focus within the lytic lesion (“button sequestrum”).
Bone scanning is often not as sensitive in detecting EG lesions compared with other bone tumors, although in our patient the bone scan was positive. In patients with a negative bone scan but a high index of suspicion, a radiographic skeletal survey should be obtained to rule out other lesions. MRI typically shows T2-hyperintense, T1-hypointense lesions with surrounding bone marrow edema and variable contrast enhancement, which is relatively nonspecific. The high sensitivity of MRI allows accurate delineation of the extent of the lesions and evaluates for the presence of an extraosseous soft-tissue component. Biopsy is generally necessary to establish a definitive histologic diagnosis. In our patient, despite her history of biopsy-proven EG, the aggressive appearance of a destructive, cortically based lesion made obtaining a biopsy critical to establish a definitive diagnosis in this case.
The histopathologic examination of the tissue from our patient was typical of that seen in patients with EG. It revealed tissue fragments with diffuse sheets of histiocytes displaying nuclear grooves, admixed numerous eosinophils with eosinophilic microabscesses, and scattered lymphocytes (Figures 6A, 6B). There were areas of necrosis, raising the possibility of osteomyelitis. However, the presence of classic histomorphologic features of LCH in the majority of the tissue fragments, along with CD1a- and S100-positivity in the histiocytes, confirmed the diagnosis of LCH (Figures 6C, 6D). Although not highly specific, a positive CD1a immunostain with the described histomorphologic findings in the proper clinical setting is often considered sufficient for LCH diagnosis. S100 is an important adjunct immunostain in the evaluation of histiocytic disorders. A positive S100 immunostain helps identify histiocytes, which are also CD1a-positive, because the latter immunostain can also be positive in some lymphomas and thymomas.21
After diagnosis of LCH has been confirmed, staging includes radiographs of any suspicious bone lesions, chest radiograph, bone scan, abdominal ultrasound, routine laboratory studies, and chest CT if pulmonary LCH is suspected.
The optimal treatment strategy for adult patients has not been clearly defined, and current strategies for LCH vary depending on organ involvement and extent of disease. Therapeutic options include observation, local treatment with steroids, local excision with curettage with or without bone grafting, chemotherapy, immunomodulation, irradiation, and stem cell transplantation in advanced disease. In general, patients who benefit from systemic therapy, such as chemotherapy or immunomodulation, include those with multisystem disease, refractory or recurrent lesions, and multifocal skeletal involvement.22
Patients with more limited disease, such as EG of bone, may undergo observation or local intralesional treatment. Eosinophilic granuloma of bone may resolve spontaneously and commonly does so when it is located in the pediatric spine. However, the therapeutic approach in adults with EG is controversial, given that spontaneous resolution is less likely to occur in the skeletally mature. Plasschaert and colleagues23 reported a recurrence rate of 26% in skeletally mature patients with EG of bone treated with biopsy followed by curettage with or without grafting. In the skeletally immature group, there were no clinical or radiographic signs of recurrence in the 2-year follow-up period.23 Thus, treatment in the adult population must be considered separate from the skeletally immature and in the appropriate clinical context. Depending on the location of the lesion, patients may become symptomatic or be at risk for pathologic fracture. In such circumstances, curettage with or without bone grafting and prophylactic internal fixation may be indicated. Other treatments, such as intralesional infiltration with corticosteroids, have been reported, but the role of such treatment in adults is undetermined.24,25 Radiation is typically not recommended in single-system disease unless a vital organ is threatened.26 Overall, patients with single-system disease have an excellent prognosis, and treatment should be determined on an individual basis.3
Eosinophilic granuloma represents less than 1% of all bone tumors, and adult presentation is very rare. The differential diagnosis of lytic bone lesions is broad and includes metastatic carcinoma, lymphoma/myeloma, osteomyelitis, osteoblastoma, aneurysmal bone cyst, and Ewing sarcoma. While EG is more common and easily diagnosed in children, it should be considered in the differential diagnosis in adults, so that the appropriate diagnostic workup and treatment can be performed.
Eosinophilic granuloma (EG) is the most common benign form of Langerhans cell histiocytosis (LCH). Initially described by Lichtenstein in 1953, LCH encompasses a triad of proliferative granulomatous disorders primarily affecting children: EG, Hand-Schüller-Christian disease, and Letterer-Siwe disease.1 Lichtenstein first termed the disease histiocytosis X, after recognizing that the 3 syndromes had the same histology.1 The term was updated after the clonal proliferation of Langerhans cells in the pathogenesis of the disease was discovered.
As LCH is generally considered a pediatric disease, there is little in the literature regarding adult-onset LCH. The incidence of LCH in adults is reported as 1 to 2 cases per million, significantly lower than that in children.2,3 Two studies have reported the mean age at diagnosis in adults as the fourth decade of life, and have suggested a male predominance.4,5 The vast majority of adult LCH cases described are simple EG, with very few cases of multisystem disseminated disease reported.5
Adult patients with LCH typically present with solitary lesions in bone. Approximately 10% of cases have extraosseous involvement, with the lung being the most common site.6 Lesions tend to be unifocal, with fewer than 10 reports describing multifocal EG.1,7-13 The axial skeleton is most frequently involved, with the majority of lesions occurring in the skull, ribs, vertebrae, or mandible.14 While less common, the femur, humerus, and clavicle are most often involved when the appendicular skeleton is affected.5
In a literature review, a few case reports describe adult-onset EG of the skull. Only 5 case reports since the 1970s describe adult patients with EG of the femur. We present a rare case of multifocal EG in a 48-year-old woman with lesions of the femur and skull, as well as a review of the literature. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 48-year-old woman presented with progressive right knee pain that was exacerbated by weight-bearing. She denied trauma, fevers, fatigue, or weight change. Her history was significant for an EG of the skull, excised at an outside institution 2 years prior to presentation. The patient also admitted to recent onset of right-sided skull pain, near the region of her previous surgery.
Physical examination demonstrated tenderness to palpation and fullness over the right medial distal femur and a normal neurovascular examination of the right lower extremity. Radiographs of the knee showed a cortically based, lytic, destructive lesion involving the medial femoral condyle, with soft-tissue extension (Figures 1A, 1B). Magnetic resonance imaging (MRI) of the right knee showed the lesion, with extraosseous soft-tissue extension (Figures 2A, 2B). The mass was isointense to muscle on T1-weighted images and hyperintense on T2-weighted images. Technetium bone scanning showed increased uptake in the right femur and the right skull (Figures 3A, 3B). MRI of the brain confirmed a new lesion in the right diploic space, distinct from the previous EG lesion site (Figures 4A-4D). An ultrasound-guided biopsy of the femur was performed and was consistent with EG.
After reevaluation and clearance by her neurosurgeon, the patient underwent curettage and allografting of the femoral lesion, with prophylactic internal fixation using a titanium distal femoral locking plate (Figure 5). Intraoperative frozen section was consistent with EG, which was confirmed with additional immunohistochemical workup (Figures 6A-6D).
The patient recovered uneventfully and follow-up radiographs showed restoration of the bony cortex of the medial femoral condyle (Figure 7). The second skull lesion, which was also consistent with EG, was excised by her neurosurgeon.
The patient remained asymptomatic until 2 years later, when she began experiencing mild pain in her right distal thigh and knee. Radiographs showed a new lytic focus in the right distal metadiaphysis (Figure 8) which was not present on her last radiograph 6 months prior. A computed tomography (CT) scan showed a lytic lesion involving the right distal femur medullary canal with cortical thinning and destruction, most pronounced posteriorly (Figures 9A, 9B). There was also an extraosseous soft-tissue component to the lesion. Bone scan showed increased uptake in the area of the new lesion. There was no increased uptake elsewhere, including the medial distal femur at the site of the old lesion, to suggest other lesions, and no increased uptake in the skull.
Given that the location of the lesion was distinct from the prior site of curettage and bone grafting, it was thought to be consistent with a new EG lesion. The patient underwent CT-guided biopsy, with simultaneous intralesional corticosteroid injection to treat the lesion when on-site pathology confirmed the etiology. Further surgical management was deemed unnecessary because internal fixation was present and spanned the new lesion. Final analysis of the fine-needle aspirate of the new lesion was positive for numerous eosinophils and histiocytes, consistent with EG.
At 6-week follow-up after the intralesional steroid injection, the patient’s pain continued to abate, and she was ambulating with crutches. Repeat CT scan of the right distal femur showed improvement of the extraosseous soft-tissue component, while the lucency in the femur itself remained unchanged. The decision was made to proceed with a second intralesional corticosteroid injection under CT guidance. The patient’s symptoms continued to improve, and repeat imaging 1 year after her steroid injections showed substantial bony healing with reconstitution of her cortical bone (Figures 10A-10E).
The patient had had 4 distinct tumors consistent with EG and was referred to a medical oncologist for further workup. The patient began treatment with zoledronic acid to prevent development of further lesions. At most recent follow-up, the patient was 18 months out from her second intralesional corticosteroid injection and was doing very well. She reported being pain-free and was walking 3 to 4 miles per week without gait aids. There was no evidence of new disease. The medial distal femur lesion was completely healed, and the distal metaphyseal lesion was nearly healed, with very little residual evidence of lesions.
Discussion
Adult-onset multifocal EG is a rare entity. Most affected patients develop lesions in the axial skeleton, with the skull, mandible, and vertebrae most commonly involved.14 Only 5 cases of femoral EG have been reported, one of which was multifocal.11,14-17
Of these patients, 3 were between the ages of 33 and 53 years and had insidious onset of hip pain that failed conservative management.14,15,17 Further imaging and biopsy revealed unifocal EG in the proximal femur in each case. Each patient received a different form of treatment, including curettage and radiation, radiofrequency ablation, and/or physical therapy. At the time of publication, all patients had reported improvement in their clinical symptoms.14,15,17 The fourth patient was a man with human immunodeficiency virus (HIV) with 3 months of progressive thigh pain. Further evaluation found an isolated EG of the femoral diaphysis that progressed to pathologic fracture. He was treated with curettage and intramedullary nailing, and had improved symptoms and radiographic signs of healing at 30-month follow-up.16
An interesting case by Kerzl and colleagues11 reported a 63-year-old woman with a 24-year history of multiple symmetric lesions of the femora, leading to multiple pathologic fractures. Like our patient, her initial lesion was in the skull. Initial pathology specimens led to the diagnosis of EG. However, as the patient aged, she developed symptoms of diabetes insipidus and xanthelasma, which led to reevaluation of histology from 3 bony lesions. The patient was determined to have multifocal EG of the skull and femur, with simultaneous occurrence of Erdheim-Chester disease, which also causes bone lesions in addition to diabetes insipidus and xanthelasma.11
Though LCH was initially described more than 50 years ago, many aspects of LCH remain an enigma, especially in adults. The etiology of the disease is poorly understood. Controversy exists regarding whether LCH is primarily an immunoregulatory, neoplastic, or reactive disorder. The vast majority of adult cases described in the literature are EG, with very few cases of multisystem disseminated disease reported.5
The spectrum of disorders constituting LCH is heterogenous. Eosinophilic granuloma is the most common form, reportedly accounting for 60% to 70% of all cases, usually presenting as solitary bone lesions.6 Eosinophilic granuloma refers to the localized form of LCH, in which the disease is limited to bone or lung.18 This is the least aggressive form of the disease, with the most favorable prognosis. Hand- Schüller-Christian disease is a chronic, recurring form of LCH, with disseminated disease, affecting both bone and extraskeletal sites. Hand-Schüller-Christian disease is known for the classic triad of diabetes insipidus, exophthalmos, and destructive bone lesions. Patients may also present with otitis media or neurologic complaints from pathologic vertebral fractures. Letterer-Siwe disease refers to the acute, disseminated, fulminant form of LCH. This is the least common form of LCH and is predominately described in young children. Patients present with hepatosplenomegaly, lymphadenopathy, skin rash, fever, anemia, and thrombocytopenia.19 It is rapidly progressive, leading to multiorgan dysfunction and death within 1 to 2 years.18
The classification of LCH follows the Histiocyte Society guidelines developed from multicenter randomized trials in children.3 Classification is based on affected organs and is divided into 2 categories: single-system disease or multisystem disease. Single-system disease may be single site (bone, skin, or solitary lymph node) or multisite (multifocal bone disease or multiple lymph nodes). Multisystem disease is further classified into low-risk or risk groups. The low-risk group involves disseminated disease without involvement of risk organs (lungs, liver, spleen, and hematopoietic system). Involvement of 1 or more risk organs places the patient in the risk group, associated with the least favorable prognosis.3
In adults, the most common presenting symptoms are local pain from bony involvement, weight loss, and fever. Bony lesions most often occur in the skull, especially in the jaw. Long bones are less frequently involved, with lesions occurring in the long bones in approximately 17% of patients.3 The rib has also been reported as a common site of involvement in adults.5 Similar to children, diabetes insipidus remains a classic manifestation of LCH because of pituitary gland involvement. Other common symptoms of LCH in adults are cough, dyspnea, and chest pain from pulmonary involvement. Up to 20% to 30% of adult LCH patients have isolated pulmonary lesions, although pulmonary LCH may also occur as part of multisystem disease (risk group).3,4,20
Eosinophilic granuloma bone lesions have a variety of radiographic appearances but most commonly appear as lytic lesions. They often mimic aggressive lesions with permeative bone destruction, periostitis, ill-defined borders, and cortical erosion. Most lesions arise in the medullary space but can present as a destructive, cortically based lesion, as it did in our patient’s first femoral lesion. The differential diagnosis for a lytic medullary bone lesion includes benign entities, such as nonossifying fibromas, bone cysts, or osteomyelitis, but also includes malignant tumors, such as metastases, Ewing sarcoma, and lymphoma. A destructive, cortically based lesion in an adult should raise a very high suspicion for metastatic carcinoma until proven otherwise. Other diagnostic considerations for a cortically based lesion include chondromyxoid fibroma and surface bone lesions, such as surface chondroma and osteoma, or osteosarcoma (parosteal and periosteal). In the skull, lesions commonly erode the outer table more than the inner table (the typical “beveled-edge” appearance). Skull lesions also may have a small, central, dense focus within the lytic lesion (“button sequestrum”).
Bone scanning is often not as sensitive in detecting EG lesions compared with other bone tumors, although in our patient the bone scan was positive. In patients with a negative bone scan but a high index of suspicion, a radiographic skeletal survey should be obtained to rule out other lesions. MRI typically shows T2-hyperintense, T1-hypointense lesions with surrounding bone marrow edema and variable contrast enhancement, which is relatively nonspecific. The high sensitivity of MRI allows accurate delineation of the extent of the lesions and evaluates for the presence of an extraosseous soft-tissue component. Biopsy is generally necessary to establish a definitive histologic diagnosis. In our patient, despite her history of biopsy-proven EG, the aggressive appearance of a destructive, cortically based lesion made obtaining a biopsy critical to establish a definitive diagnosis in this case.
The histopathologic examination of the tissue from our patient was typical of that seen in patients with EG. It revealed tissue fragments with diffuse sheets of histiocytes displaying nuclear grooves, admixed numerous eosinophils with eosinophilic microabscesses, and scattered lymphocytes (Figures 6A, 6B). There were areas of necrosis, raising the possibility of osteomyelitis. However, the presence of classic histomorphologic features of LCH in the majority of the tissue fragments, along with CD1a- and S100-positivity in the histiocytes, confirmed the diagnosis of LCH (Figures 6C, 6D). Although not highly specific, a positive CD1a immunostain with the described histomorphologic findings in the proper clinical setting is often considered sufficient for LCH diagnosis. S100 is an important adjunct immunostain in the evaluation of histiocytic disorders. A positive S100 immunostain helps identify histiocytes, which are also CD1a-positive, because the latter immunostain can also be positive in some lymphomas and thymomas.21
After diagnosis of LCH has been confirmed, staging includes radiographs of any suspicious bone lesions, chest radiograph, bone scan, abdominal ultrasound, routine laboratory studies, and chest CT if pulmonary LCH is suspected.
The optimal treatment strategy for adult patients has not been clearly defined, and current strategies for LCH vary depending on organ involvement and extent of disease. Therapeutic options include observation, local treatment with steroids, local excision with curettage with or without bone grafting, chemotherapy, immunomodulation, irradiation, and stem cell transplantation in advanced disease. In general, patients who benefit from systemic therapy, such as chemotherapy or immunomodulation, include those with multisystem disease, refractory or recurrent lesions, and multifocal skeletal involvement.22
Patients with more limited disease, such as EG of bone, may undergo observation or local intralesional treatment. Eosinophilic granuloma of bone may resolve spontaneously and commonly does so when it is located in the pediatric spine. However, the therapeutic approach in adults with EG is controversial, given that spontaneous resolution is less likely to occur in the skeletally mature. Plasschaert and colleagues23 reported a recurrence rate of 26% in skeletally mature patients with EG of bone treated with biopsy followed by curettage with or without grafting. In the skeletally immature group, there were no clinical or radiographic signs of recurrence in the 2-year follow-up period.23 Thus, treatment in the adult population must be considered separate from the skeletally immature and in the appropriate clinical context. Depending on the location of the lesion, patients may become symptomatic or be at risk for pathologic fracture. In such circumstances, curettage with or without bone grafting and prophylactic internal fixation may be indicated. Other treatments, such as intralesional infiltration with corticosteroids, have been reported, but the role of such treatment in adults is undetermined.24,25 Radiation is typically not recommended in single-system disease unless a vital organ is threatened.26 Overall, patients with single-system disease have an excellent prognosis, and treatment should be determined on an individual basis.3
Eosinophilic granuloma represents less than 1% of all bone tumors, and adult presentation is very rare. The differential diagnosis of lytic bone lesions is broad and includes metastatic carcinoma, lymphoma/myeloma, osteomyelitis, osteoblastoma, aneurysmal bone cyst, and Ewing sarcoma. While EG is more common and easily diagnosed in children, it should be considered in the differential diagnosis in adults, so that the appropriate diagnostic workup and treatment can be performed.
1. Lahiani D, Hammami BK, Maâloul I, et al. Multifocal Langerhans cell histiocytosis of bone: late revelation in a 76-year-old woman. Rev Med Interne. 2008;29(3):249-251.
2. Baumgartner I, von Hochstetter A, Baumert B, Luetolf U, Follath F. Langerhans’-cell histiocytosis in adults. Med Pediatr Oncol. 1997;28(1):9-14.
3. Stockschlaeder M, Sucker C. Adult Langerhans cell histiocytosis. Eur J Haematol. 2006;76(5):363-368.
4. Aricò M, Girschikofsky M, Généreau T, et al. Langerhans cell histiocytosis in adults. Report from the International Registry of the Histiocyte Society. Eur J Cancer. 2003;39(16):2341-2348.
5. Islinger RB, Kuklo TR, Owens BD, et al. Langerhans’ cell histiocytosis in patients older than 21 years. Clin Orthop Relat Res. 2000;379:231-235.
6. Key SJ, O’Brien CJ, Silvester KC, Crean SJ. Eosinophilic granuloma: resolution of maxillofacial bony lesions following minimal intervention. Report of three cases and a review of the literature. J Craniomaxillofac Surg. 2004;32(3):170-175.
7. Bodner G, Kreczy A, Rachbauer F, Baechter O, Peer S. Eosinophilic granuloma of the bone: ultrasonographic imaging. Australas Radiol. 2002;46(4):418-421.
8. Boutsen Y, Esselinckx W, Delos M, Nisolle JF. Adult onset of multifocal eosinophilic granuloma of bone: a long-term follow-up with evaluation of various treatment options and spontaneous healing. Clin Rheumatol. 1999;18(1):69-73.
9. Corti F, Valicenti A, Bertolucci D, Bruno J, Gustinucci R. Multifocal Langerhans cell granulomatosis. Report of a clinical case. Minerva Med. 1994;85(7-8):413-416.
10. Demirci I. Adult eosinophilic granuloma of the lumbar spine with atypical dissemination. Case report: a long-term follow-up. Zentralbl Neurochir. 2004;65(2):84-87.
11. Kerzl R, Eyerich K, Eberlein B, et al. Parallel occurrence of Erdheim-Chester disease and eosinophilic granuloma in the same patient. J Eur Acad Dermatol Venereol. 2009;23(2):224-226.
12. Nguyen BD, Roarke MC, Chivers SF. Multifocal Langerhans cell histiocytosis with infiltrative pelvic lesions: PET/CT imaging. Clin Nucl Med. 2010;35(10): 824-826.
13. Scolozzi P, Lombardi T, Monnier P, Jaques B. Multisystem Langerhans’ cell histiocytosis (Hand-Schuller-Christian disease) in an adult: a case report and review of the literature. Eur Arch Otorhinolaryngol. 2004;261(6):326-330.
14. King JJ, Melvin JS, Iwenofu OH, Fox EJ. Thigh pain in a 53-year-old woman. Clin Orthop Relat Res. 2009;467(6):1652-1657.
15. Hair LC, Deyle GD. Eosinophilic granuloma in a patient with hip pain. J Orthop Sports Phys Ther. 2011;41(2):119.
16. Panayiotakopoulos GD, Sipsas NV, Kontos A, et al. Eosinophilic granuloma of the femur in an HIV-1 positive patient. AIDS Patient Care STDS. 2002;16(3):103-106.
17. Rodrigues RJ, Lewis HH. Eosinophilic granuloma of bone. Review of literature and case presentation. Clin Orthop Relat Res. 1971;77:183-192.
18. Stull MA, Kransdorf MJ, Devaney KO. Langerhans cell histiocytosis of bone. Radiographics. 1992;12(4):801-823.
19. Lichtenstein L. Histiocytosis X (eosinophilic granuloma of bone, Letterer-Siwe disease, and Schueller-Christian disease). Further observations of pathological and clinical importance. J Bone Joint Surg Am. 1964;46:76-90.
20. Götz G, Fichter J. Langerhans’-cell histiocytosis in 58 adults. Eur J Med Res. 2004;9(11):510-514.
21. Cheng KL, Glu PG, Weiss LM. Hematopoeitic tumors. In: Peiguo C, Weiss L, eds. Modern Immunohistochemistry. New York, NY: Cambridge University Press; 2009:503.
22. Broadbent V, Gadner H. Current therapy for Langerhans cell histiocytosis. Hematol Oncol Clin North Am. 1998;12(2):327-338.
23. Plasschaert F, Craig C, Bell R, Cole WG, Wunder JS, Alman BA. Eosinophilic granuloma. A different behaviour in children than in adults. J Bone Joint Surg Br. 2002;84(6):870-872.
24. Capanna R, Springfield DS, Ruggieri P, et al. Direct cortisone injection in osinophilic granuloma of bone: a preliminary report on 11 patients. J Pediatr Orthop. 1985;5(3):339-342.
25. Egeler RM, Thompson RC Jr, Voûte PA, Nesbit ME Jr. Intralesional infiltration of corticosteroids in localized Langerhans’ cell histiocytosis. J Pediatr Orthop. 1992;12(6):811-814.
26. Ladisch S, Gadner H. Treatment of Langerhans cell histiocytosis–evolution and current approaches. Br J Cancer Suppl. 1994;23:S41-S46.
1. Lahiani D, Hammami BK, Maâloul I, et al. Multifocal Langerhans cell histiocytosis of bone: late revelation in a 76-year-old woman. Rev Med Interne. 2008;29(3):249-251.
2. Baumgartner I, von Hochstetter A, Baumert B, Luetolf U, Follath F. Langerhans’-cell histiocytosis in adults. Med Pediatr Oncol. 1997;28(1):9-14.
3. Stockschlaeder M, Sucker C. Adult Langerhans cell histiocytosis. Eur J Haematol. 2006;76(5):363-368.
4. Aricò M, Girschikofsky M, Généreau T, et al. Langerhans cell histiocytosis in adults. Report from the International Registry of the Histiocyte Society. Eur J Cancer. 2003;39(16):2341-2348.
5. Islinger RB, Kuklo TR, Owens BD, et al. Langerhans’ cell histiocytosis in patients older than 21 years. Clin Orthop Relat Res. 2000;379:231-235.
6. Key SJ, O’Brien CJ, Silvester KC, Crean SJ. Eosinophilic granuloma: resolution of maxillofacial bony lesions following minimal intervention. Report of three cases and a review of the literature. J Craniomaxillofac Surg. 2004;32(3):170-175.
7. Bodner G, Kreczy A, Rachbauer F, Baechter O, Peer S. Eosinophilic granuloma of the bone: ultrasonographic imaging. Australas Radiol. 2002;46(4):418-421.
8. Boutsen Y, Esselinckx W, Delos M, Nisolle JF. Adult onset of multifocal eosinophilic granuloma of bone: a long-term follow-up with evaluation of various treatment options and spontaneous healing. Clin Rheumatol. 1999;18(1):69-73.
9. Corti F, Valicenti A, Bertolucci D, Bruno J, Gustinucci R. Multifocal Langerhans cell granulomatosis. Report of a clinical case. Minerva Med. 1994;85(7-8):413-416.
10. Demirci I. Adult eosinophilic granuloma of the lumbar spine with atypical dissemination. Case report: a long-term follow-up. Zentralbl Neurochir. 2004;65(2):84-87.
11. Kerzl R, Eyerich K, Eberlein B, et al. Parallel occurrence of Erdheim-Chester disease and eosinophilic granuloma in the same patient. J Eur Acad Dermatol Venereol. 2009;23(2):224-226.
12. Nguyen BD, Roarke MC, Chivers SF. Multifocal Langerhans cell histiocytosis with infiltrative pelvic lesions: PET/CT imaging. Clin Nucl Med. 2010;35(10): 824-826.
13. Scolozzi P, Lombardi T, Monnier P, Jaques B. Multisystem Langerhans’ cell histiocytosis (Hand-Schuller-Christian disease) in an adult: a case report and review of the literature. Eur Arch Otorhinolaryngol. 2004;261(6):326-330.
14. King JJ, Melvin JS, Iwenofu OH, Fox EJ. Thigh pain in a 53-year-old woman. Clin Orthop Relat Res. 2009;467(6):1652-1657.
15. Hair LC, Deyle GD. Eosinophilic granuloma in a patient with hip pain. J Orthop Sports Phys Ther. 2011;41(2):119.
16. Panayiotakopoulos GD, Sipsas NV, Kontos A, et al. Eosinophilic granuloma of the femur in an HIV-1 positive patient. AIDS Patient Care STDS. 2002;16(3):103-106.
17. Rodrigues RJ, Lewis HH. Eosinophilic granuloma of bone. Review of literature and case presentation. Clin Orthop Relat Res. 1971;77:183-192.
18. Stull MA, Kransdorf MJ, Devaney KO. Langerhans cell histiocytosis of bone. Radiographics. 1992;12(4):801-823.
19. Lichtenstein L. Histiocytosis X (eosinophilic granuloma of bone, Letterer-Siwe disease, and Schueller-Christian disease). Further observations of pathological and clinical importance. J Bone Joint Surg Am. 1964;46:76-90.
20. Götz G, Fichter J. Langerhans’-cell histiocytosis in 58 adults. Eur J Med Res. 2004;9(11):510-514.
21. Cheng KL, Glu PG, Weiss LM. Hematopoeitic tumors. In: Peiguo C, Weiss L, eds. Modern Immunohistochemistry. New York, NY: Cambridge University Press; 2009:503.
22. Broadbent V, Gadner H. Current therapy for Langerhans cell histiocytosis. Hematol Oncol Clin North Am. 1998;12(2):327-338.
23. Plasschaert F, Craig C, Bell R, Cole WG, Wunder JS, Alman BA. Eosinophilic granuloma. A different behaviour in children than in adults. J Bone Joint Surg Br. 2002;84(6):870-872.
24. Capanna R, Springfield DS, Ruggieri P, et al. Direct cortisone injection in osinophilic granuloma of bone: a preliminary report on 11 patients. J Pediatr Orthop. 1985;5(3):339-342.
25. Egeler RM, Thompson RC Jr, Voûte PA, Nesbit ME Jr. Intralesional infiltration of corticosteroids in localized Langerhans’ cell histiocytosis. J Pediatr Orthop. 1992;12(6):811-814.
26. Ladisch S, Gadner H. Treatment of Langerhans cell histiocytosis–evolution and current approaches. Br J Cancer Suppl. 1994;23:S41-S46.
Lipoma of the Tendon Sheath in the Fourth Extensor Compartment of the Hand
Lipomas are relatively common benign tumors composed primarily of adipose tissue. They can occur anywhere on the body and are seen often in the hands and forearm. Typically localized to the subcutaneous fat layer, a lipoma is rarely associated with a tendon sheath or tendon compartment.1,2 When this uncommon event occurs, the lipoma is appropriately labeled lipoma of the tendon sheath.
While there are numerous case reports of lipomas of the tendon sheath occurring in association with tendons in the lower extremity, there are no reports, to our knowledge, of their occurrence in the extensor compartments of the hand.1 We report a rare case of lipoma of the tendon sheath localized to the fourth dorsal compartment of the hand, which was successfully treated with surgical excision. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 33-year-old right hand–dominant waitress presented with a chief complaint of a painful, slowly enlarging right dorsal hand mass of 5 years’ duration. The mass was particularly bothersome with activities involving grip and finger extension. Physical examination revealed a mobile, rubbery mass on the dorsum of the hand that moved slightly with fist formation. There were no signs of neurovascular compromise. She had normal hand and wrist range of motion.
Plain radiographs were unremarkable (Figures 1A, 1B). Magnetic resonance imaging (MRI) with and without contrast revealed a 4×2-cm mass consistent with a diagnosis of lipoma. However, it was unique in that it appeared to extend from the long- and ring-finger extensor tendon sheaths in the fourth dorsal compartment of the hand (Figures 2A, 2B) and was deemed a lipoma of the tendon sheath. Representative MRI also showed the lipoma to be present within the fourth extensor compartment of the hand (Figure 2B). Because of the mass’s increasing size and interference with hand function, the patient elected to have the mass excised.
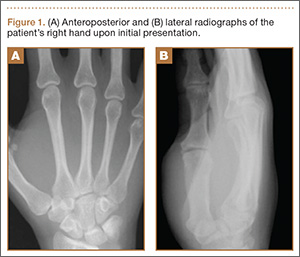
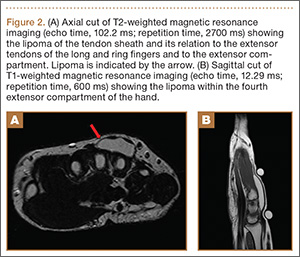
Surgical Technique
A 3-cm longitudinal incision was made over the dorsum of the hand centered directly over the mass. Dissection was carried through the subcutaneous tissue to the distal margin of the extensor retinaculum. The fourth dorsal compartment was entered and the tendons of the fourth extensor compartment were identified. Immediately beneath the extensor tendons to the long and ring fingers was a yellow, rubbery mass consistent with lipoma (Figure 3). This mass was strongly adherent to the underlying tendons and had to be dissected carefully with tenotomy scissors. Fortunately, the mass could be excised as a single unit (Figure 4). It was sent to the pathology department for histologic examination, which revealed mature adipose tissue and confirmed the diagnosis of lipoma. The wound was closed with absorbable suture, and a soft, sterile dressing was applied.
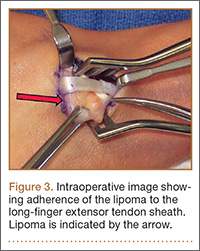
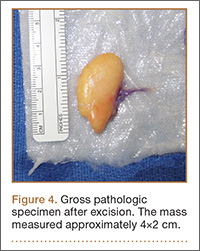
Postoperative Care
The patient was seen in follow-up 2 weeks later for routine evaluation. She had an intact wound with minimal hand pain, and full wrist and hand range of motion. She returned to work as a waitress approximately 3 weeks after surgery without difficulty. At her 6-week postoperative mark, she had a pain-free wrist with a well-healed incision and no signs of recurrence.
Discussion
Tendon sheath lipomas, whether in the upper or lower extremities, are exceedingly rare entities. Further, lipomas of an individual extensor compartment of the hand (as in our case) have yet to be described, in contrast to lipomas of flexor tendon sheaths.3 There are only a handful of case reports in the literature of lipomas of the tendon sheath, and none to our knowledge of their existence in the extensor compartments of the hand. Nevertheless, it is important for the treating surgeon to be aware of their existence and know some basics about them and their treatment.
There are 2 types of tendon sheath lipomas: discrete solid masses of adipose tissue (which we encountered) and adipose tissue coupled with hypertrophic synovial villi (or, lipoma arborescens).4,5 Of note, the latter is significantly more common than the former, which makes our case even more uncommon. Although both types of lipoma of the tendon sheath are benign, they can cause symptoms such as pain, finger stiffness, and nerve compression.6 Thus, they frequently merit surgical removal, as in our case.
The appropriate workup for lipoma of the tendon sheath generally includes thorough history, physical examination, and advanced imaging, such as MRI. MRI is usually diagnostic of such a lesion and can aid in surgical planning.1 Regarding their overall prognosis, all lipomas (even large ones) are benign by definition but can transform into liposarcomas in rare cases.4 Lipomas are typically treated surgically by simple excision, and lipoma of the tendon sheath is no different. As long as complete excision of a tendon sheath lipoma is performed, recurrence rates are less than 5%.2,3
Surgeons should also be aware that, with long-standing lipomas of the tendon sheath, weakening of a tendon secondary to irritation from the mass is a possibility, especially in the lower extremities. All tendons should be inspected carefully at the time of surgery to ensure that other procedures, such as tendon grafting or side-to-side tenodesis, are not required. Although lipomas of the tendon sheath and extensor compartments are quite rare, all surgeons evaluating masses for possible surgical excision should be aware of their existence and know how to manage them appropriately.
1. Khan AZ, Shafafy M, Latimer MD, Crosby J. A lipoma within the Achilles tendon sheath. Foot Ankle Surg. 2012;18(1):e16-e17.
2. Bryan RS, Dahlin DC, Sullivan CR. Lipoma of the tendon sheath. J Bone Joint Surg Am. 1956;38(6):1275-1280.
3. Kremchek TE, Kremchek EJ. Carpal tunnel syndrome caused by flexor tendon sheath lipoma. Orthop Rev. 1998;17(11):1083-1085.
4. Murphey MD, Caroll JF, Flemming DJ, Pope TL, Gannon FH, Kransdorf MJ. From the archives of AFIP: benign musculoskeletal lipomatous lesions. Radiographics. 2004;24(5):1433-1466.
5. Chronopoulous E, Nicholas P, Karanikas C, et al. Patient presenting with lipoma of the index finger: a case report. Cases J. 2010;3:20.
6. Elbardouni A, Kharmaz M, Salah Berrada M, Mahfoud M, Eylaacoubi M. Well-circumscribed deep-seated lesions of the upper extremity. A report of 13 cases. Orthop Traumatol: Surg Res. 2011;97(2):152-158.
Lipomas are relatively common benign tumors composed primarily of adipose tissue. They can occur anywhere on the body and are seen often in the hands and forearm. Typically localized to the subcutaneous fat layer, a lipoma is rarely associated with a tendon sheath or tendon compartment.1,2 When this uncommon event occurs, the lipoma is appropriately labeled lipoma of the tendon sheath.
While there are numerous case reports of lipomas of the tendon sheath occurring in association with tendons in the lower extremity, there are no reports, to our knowledge, of their occurrence in the extensor compartments of the hand.1 We report a rare case of lipoma of the tendon sheath localized to the fourth dorsal compartment of the hand, which was successfully treated with surgical excision. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 33-year-old right hand–dominant waitress presented with a chief complaint of a painful, slowly enlarging right dorsal hand mass of 5 years’ duration. The mass was particularly bothersome with activities involving grip and finger extension. Physical examination revealed a mobile, rubbery mass on the dorsum of the hand that moved slightly with fist formation. There were no signs of neurovascular compromise. She had normal hand and wrist range of motion.
Plain radiographs were unremarkable (Figures 1A, 1B). Magnetic resonance imaging (MRI) with and without contrast revealed a 4×2-cm mass consistent with a diagnosis of lipoma. However, it was unique in that it appeared to extend from the long- and ring-finger extensor tendon sheaths in the fourth dorsal compartment of the hand (Figures 2A, 2B) and was deemed a lipoma of the tendon sheath. Representative MRI also showed the lipoma to be present within the fourth extensor compartment of the hand (Figure 2B). Because of the mass’s increasing size and interference with hand function, the patient elected to have the mass excised.
Surgical Technique
A 3-cm longitudinal incision was made over the dorsum of the hand centered directly over the mass. Dissection was carried through the subcutaneous tissue to the distal margin of the extensor retinaculum. The fourth dorsal compartment was entered and the tendons of the fourth extensor compartment were identified. Immediately beneath the extensor tendons to the long and ring fingers was a yellow, rubbery mass consistent with lipoma (Figure 3). This mass was strongly adherent to the underlying tendons and had to be dissected carefully with tenotomy scissors. Fortunately, the mass could be excised as a single unit (Figure 4). It was sent to the pathology department for histologic examination, which revealed mature adipose tissue and confirmed the diagnosis of lipoma. The wound was closed with absorbable suture, and a soft, sterile dressing was applied.
Postoperative Care
The patient was seen in follow-up 2 weeks later for routine evaluation. She had an intact wound with minimal hand pain, and full wrist and hand range of motion. She returned to work as a waitress approximately 3 weeks after surgery without difficulty. At her 6-week postoperative mark, she had a pain-free wrist with a well-healed incision and no signs of recurrence.
Discussion
Tendon sheath lipomas, whether in the upper or lower extremities, are exceedingly rare entities. Further, lipomas of an individual extensor compartment of the hand (as in our case) have yet to be described, in contrast to lipomas of flexor tendon sheaths.3 There are only a handful of case reports in the literature of lipomas of the tendon sheath, and none to our knowledge of their existence in the extensor compartments of the hand. Nevertheless, it is important for the treating surgeon to be aware of their existence and know some basics about them and their treatment.
There are 2 types of tendon sheath lipomas: discrete solid masses of adipose tissue (which we encountered) and adipose tissue coupled with hypertrophic synovial villi (or, lipoma arborescens).4,5 Of note, the latter is significantly more common than the former, which makes our case even more uncommon. Although both types of lipoma of the tendon sheath are benign, they can cause symptoms such as pain, finger stiffness, and nerve compression.6 Thus, they frequently merit surgical removal, as in our case.
The appropriate workup for lipoma of the tendon sheath generally includes thorough history, physical examination, and advanced imaging, such as MRI. MRI is usually diagnostic of such a lesion and can aid in surgical planning.1 Regarding their overall prognosis, all lipomas (even large ones) are benign by definition but can transform into liposarcomas in rare cases.4 Lipomas are typically treated surgically by simple excision, and lipoma of the tendon sheath is no different. As long as complete excision of a tendon sheath lipoma is performed, recurrence rates are less than 5%.2,3
Surgeons should also be aware that, with long-standing lipomas of the tendon sheath, weakening of a tendon secondary to irritation from the mass is a possibility, especially in the lower extremities. All tendons should be inspected carefully at the time of surgery to ensure that other procedures, such as tendon grafting or side-to-side tenodesis, are not required. Although lipomas of the tendon sheath and extensor compartments are quite rare, all surgeons evaluating masses for possible surgical excision should be aware of their existence and know how to manage them appropriately.
Lipomas are relatively common benign tumors composed primarily of adipose tissue. They can occur anywhere on the body and are seen often in the hands and forearm. Typically localized to the subcutaneous fat layer, a lipoma is rarely associated with a tendon sheath or tendon compartment.1,2 When this uncommon event occurs, the lipoma is appropriately labeled lipoma of the tendon sheath.
While there are numerous case reports of lipomas of the tendon sheath occurring in association with tendons in the lower extremity, there are no reports, to our knowledge, of their occurrence in the extensor compartments of the hand.1 We report a rare case of lipoma of the tendon sheath localized to the fourth dorsal compartment of the hand, which was successfully treated with surgical excision. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 33-year-old right hand–dominant waitress presented with a chief complaint of a painful, slowly enlarging right dorsal hand mass of 5 years’ duration. The mass was particularly bothersome with activities involving grip and finger extension. Physical examination revealed a mobile, rubbery mass on the dorsum of the hand that moved slightly with fist formation. There were no signs of neurovascular compromise. She had normal hand and wrist range of motion.
Plain radiographs were unremarkable (Figures 1A, 1B). Magnetic resonance imaging (MRI) with and without contrast revealed a 4×2-cm mass consistent with a diagnosis of lipoma. However, it was unique in that it appeared to extend from the long- and ring-finger extensor tendon sheaths in the fourth dorsal compartment of the hand (Figures 2A, 2B) and was deemed a lipoma of the tendon sheath. Representative MRI also showed the lipoma to be present within the fourth extensor compartment of the hand (Figure 2B). Because of the mass’s increasing size and interference with hand function, the patient elected to have the mass excised.
Surgical Technique
A 3-cm longitudinal incision was made over the dorsum of the hand centered directly over the mass. Dissection was carried through the subcutaneous tissue to the distal margin of the extensor retinaculum. The fourth dorsal compartment was entered and the tendons of the fourth extensor compartment were identified. Immediately beneath the extensor tendons to the long and ring fingers was a yellow, rubbery mass consistent with lipoma (Figure 3). This mass was strongly adherent to the underlying tendons and had to be dissected carefully with tenotomy scissors. Fortunately, the mass could be excised as a single unit (Figure 4). It was sent to the pathology department for histologic examination, which revealed mature adipose tissue and confirmed the diagnosis of lipoma. The wound was closed with absorbable suture, and a soft, sterile dressing was applied.
Postoperative Care
The patient was seen in follow-up 2 weeks later for routine evaluation. She had an intact wound with minimal hand pain, and full wrist and hand range of motion. She returned to work as a waitress approximately 3 weeks after surgery without difficulty. At her 6-week postoperative mark, she had a pain-free wrist with a well-healed incision and no signs of recurrence.
Discussion
Tendon sheath lipomas, whether in the upper or lower extremities, are exceedingly rare entities. Further, lipomas of an individual extensor compartment of the hand (as in our case) have yet to be described, in contrast to lipomas of flexor tendon sheaths.3 There are only a handful of case reports in the literature of lipomas of the tendon sheath, and none to our knowledge of their existence in the extensor compartments of the hand. Nevertheless, it is important for the treating surgeon to be aware of their existence and know some basics about them and their treatment.
There are 2 types of tendon sheath lipomas: discrete solid masses of adipose tissue (which we encountered) and adipose tissue coupled with hypertrophic synovial villi (or, lipoma arborescens).4,5 Of note, the latter is significantly more common than the former, which makes our case even more uncommon. Although both types of lipoma of the tendon sheath are benign, they can cause symptoms such as pain, finger stiffness, and nerve compression.6 Thus, they frequently merit surgical removal, as in our case.
The appropriate workup for lipoma of the tendon sheath generally includes thorough history, physical examination, and advanced imaging, such as MRI. MRI is usually diagnostic of such a lesion and can aid in surgical planning.1 Regarding their overall prognosis, all lipomas (even large ones) are benign by definition but can transform into liposarcomas in rare cases.4 Lipomas are typically treated surgically by simple excision, and lipoma of the tendon sheath is no different. As long as complete excision of a tendon sheath lipoma is performed, recurrence rates are less than 5%.2,3
Surgeons should also be aware that, with long-standing lipomas of the tendon sheath, weakening of a tendon secondary to irritation from the mass is a possibility, especially in the lower extremities. All tendons should be inspected carefully at the time of surgery to ensure that other procedures, such as tendon grafting or side-to-side tenodesis, are not required. Although lipomas of the tendon sheath and extensor compartments are quite rare, all surgeons evaluating masses for possible surgical excision should be aware of their existence and know how to manage them appropriately.
1. Khan AZ, Shafafy M, Latimer MD, Crosby J. A lipoma within the Achilles tendon sheath. Foot Ankle Surg. 2012;18(1):e16-e17.
2. Bryan RS, Dahlin DC, Sullivan CR. Lipoma of the tendon sheath. J Bone Joint Surg Am. 1956;38(6):1275-1280.
3. Kremchek TE, Kremchek EJ. Carpal tunnel syndrome caused by flexor tendon sheath lipoma. Orthop Rev. 1998;17(11):1083-1085.
4. Murphey MD, Caroll JF, Flemming DJ, Pope TL, Gannon FH, Kransdorf MJ. From the archives of AFIP: benign musculoskeletal lipomatous lesions. Radiographics. 2004;24(5):1433-1466.
5. Chronopoulous E, Nicholas P, Karanikas C, et al. Patient presenting with lipoma of the index finger: a case report. Cases J. 2010;3:20.
6. Elbardouni A, Kharmaz M, Salah Berrada M, Mahfoud M, Eylaacoubi M. Well-circumscribed deep-seated lesions of the upper extremity. A report of 13 cases. Orthop Traumatol: Surg Res. 2011;97(2):152-158.
1. Khan AZ, Shafafy M, Latimer MD, Crosby J. A lipoma within the Achilles tendon sheath. Foot Ankle Surg. 2012;18(1):e16-e17.
2. Bryan RS, Dahlin DC, Sullivan CR. Lipoma of the tendon sheath. J Bone Joint Surg Am. 1956;38(6):1275-1280.
3. Kremchek TE, Kremchek EJ. Carpal tunnel syndrome caused by flexor tendon sheath lipoma. Orthop Rev. 1998;17(11):1083-1085.
4. Murphey MD, Caroll JF, Flemming DJ, Pope TL, Gannon FH, Kransdorf MJ. From the archives of AFIP: benign musculoskeletal lipomatous lesions. Radiographics. 2004;24(5):1433-1466.
5. Chronopoulous E, Nicholas P, Karanikas C, et al. Patient presenting with lipoma of the index finger: a case report. Cases J. 2010;3:20.
6. Elbardouni A, Kharmaz M, Salah Berrada M, Mahfoud M, Eylaacoubi M. Well-circumscribed deep-seated lesions of the upper extremity. A report of 13 cases. Orthop Traumatol: Surg Res. 2011;97(2):152-158.
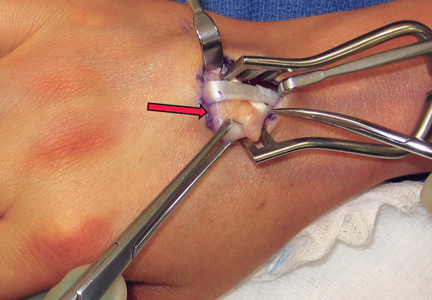
Current Management of Acute Bronchiolitis: An Evidence-Based Approach
Case
An 8-week-old male infant was brought to the ED by his parents after an episode in which it appeared the baby had stopped breathing. The parents stated that while lying on his mother’s lap at home, the patient stopped breathing for approximately 10 to 15 seconds, during which time his face exhibited a bluish color. They further noted that the patient began breathing again after gentle stimulation and had been acting normally since.
The patient was born at 39 weeks gestation via normal vaginal delivery and without any complications. His parents further stated that prior to the cessation of breathing incident, his symptoms of nasal congestion, decreased energy level, and fast breathing had gradually worsened over the past 2 days. The parents also noted that the infant had not been feeding as well over the past 2 days.
Upon arrival, the patient’s vital signs were: heart rate, 140 beats/minute; respiratory rate (RR), 72 beats/minute; and temperature 101.3°F. Oxygen saturation was 92% on room air. On physical examination, the infant had significant rhinorrhea, moderate intercostal and supraclavicular retractions, ausculatory wheezes, and transmitted upper airway noises throughout.
Overview
Bronchiolitis, a disorder caused by a viral lower respiratory tract infection, is the most common lower respiratory infection in children younger than age 2 years.1 In 2014, the American Academy of Pediatrics (AAP) characterized bronchiolitis as “rhinitis, cough, tachypnea, wheezing, rales, use of accessory muscles, and/or nasal flaring in children under 24 months of age.”2 This condition is the most common cause of hospitalization in the first 12 months of life. It is responsible for over 100,000 admissions annually at an estimated cost to the healthcare system of $1.73 billion.3
Etiology and Pathophysiology
Respiratory syncytial virus (RSV) is the most common cause of bronchiolitis. In the United States, the highest incidence of infection occurs during the months of December through March, with some degree of regional variability.4 A number of other viruses that can cause bronchiolitis include human metapneumovirus, parainfluenza virus, and influenza virus.1 Infection with RSV does not grant permanent immunity, and reinfection is common throughout life.2
Pathophysiologically, bronchiolitis is characterized by an invasion of bronchial epithelial cells that lead to to cell death and sloughing into the bronchial lumen. This, coupled with increased mucous production and submucosal edema, leads to a narrowing of the bronchial lumen and obstruction of airflow.5
Clinical Manifestations
Bronchiolitis represents a constellation of signs and symptoms beginning with those of an upper respiratory tract infection, including nasal congestion and rhinorrhea with mild cough. On days 3 to 5, the following symptoms develop: tachypnea, wheezing, rales, and signs of respiratory distress (eg, grunting, nasal flaring, inter-/subcostal retractions). Approximately two-thirds of patients will develop a fever.2 Recovery tends to begin around days 5 to 7, with the median duration of illness being 12 days.1 It should be noted that bronchiolitis represents a highly variable and dynamic disease state. Transient episodes of improvement and worsening are common, emphasizing the importance of serial examinations and assessments. Though rare, progression to respiratory failure and death do occur.2
History and Risk Stratification
The focus of the initial history by the clinician should serve two primary purposes. First, it is important to differentiate infants with probable bronchiolitis from those with other disease states having similar clinical manifestations. One of the most challenging diseases to differentiate from bronchiolitis is that of reactive airway disease (RAD). Eliciting a history of allergic rhinitis, eczema, or a family history of asthma may be helpful in determining the precise etiology of the patient’s symptoms. Although no longer recommended for children with bronchiolitis (as will be later discussed), a trial of a bronchodilation may be beneficial in the setting of familial atopy.
The second—and perhaps most important—aspect of patient history is to determine the presence of risk factors for both apnea and the development of severe bronchiolitis. Regarding the risk factors for apnea, Willwerth et al6 developed a set of criteria to identify patients at high risk for apnea in the inpatient setting. Patients were considered high risk if they were born at full term and were younger than 1 month of age; if they were born preterm (<37 weeks gestation) and were younger than 48 weeks postconception; and/or if the infant’s parents or a clinician had already witnessed an episode of apnea during the patient’s illness. In this study, all patients who developed apnea were correctly identified by the risk criteria.6 Risk factors for severe bronchiolitis include the following: patient age younger than 12 weeks; patient prematurity younger than 37 weeks gestation; and an underlying hemodynamically significant congenital heart disease, chronic lung disease/bronchopulmonary dysplasia, or an immunocompromised state.1
Diagnosis
In 2014, the AAP updated its guidelines on the diagnosis, management, and prevention of bronchiolitis. One of the strongest statements in these guidelines emphasize that the diagnosis of bronchiolitis should be based almost exclusively on the history and physical examination.2 In children younger than age 2 years, historical features such as a viral prodrome, followed by progressively worsening increased respiratory effort and signs and symptoms of lower respiratory-tract disease (eg, wheezing), should guide clinicians to the diagnosis of bronchiolitis. Although nonspecific, physical examination findings such as rhinorrhea, cough, tachypnea, wheezing, rales, and increased respiratory effort—when coupled with a good history—can be beneficial in the diagnosis of bronchiolitis.
Pulse Oximetry
Pulse oximetry has become a standard part of the clinical assessment of patients with bronchiolitis. This is based on data suggesting that pulse oximetry detects hypoxia in cases where it was not suspected on physical examination alone.7 However, the effectiveness of pulse oximetry in predicting clinical outcomes is limited. Pulse oximetry should not be used as a proxy for respiratory distress, as studies have shown poor correlation between respiratory distress and oxygen saturations in infants with lower respiratory tract infection.8
Radiographic Evaluation
Regarding the diagnosis of bronchiolitis, the AAP notes, “radiographic and laboratory studies should not be obtained routinely.”2 While many children with bronchiolitis may have abnormalities on radiographs, there is insufficient data to suggest that chest radiographs correlate with disease severity. In addition, several studies, including a prospective cohort study by Schuh et al,9 have shown that infants with suspected lower respiratory tract infections who undergo radiography are more likely to receive antibiotics without any difference in outcomes.
Laboratory Studies
As stated in the AAP guidelines, routine laboratory testing, particularly virologic studies for RSV, have little role in the diagnosis of bronchiolitis. Since numerous viruses can cause bronchiolitis and have similar clinical presentations, the absence of identification of a particular virologic agent does not exclude the diagnosis of bronchiolitis and is moreover unlikely to alter management.
Although routine laboratory evaluation is not recommended in infants with bronchiolitis, one subgroup in which it may be beneficial is in the assessment of serious bacterial infections (SBIs) in febrile infants with bronchiolitis who are younger than 60 days old. Levine et al10 conducted a large, multicenter, prospective, cross-sectional study of young, febrile infants to determine the risk of SBI in those with RSV bronchiolitis versus those without RSV bronchiolitis. They found that overall febrile infants younger than age 60 days with RSV bronchiolitis have a lower rate of SBI than those without RSV (7% v 12.5%, respectively).10 In infants between age 28 and 60 days with RSV bronchiolitis, the origin of all SBIs in the study were urinary tract infections. In patients younger than 28 days of age, the risk of developing an SBI was found to be no different between the RSV-positive and RSV-negative groups.10
Based on the findings in this study, it is recommended that, at the very least, urinalysis for bacterial infection be performed in all infants with RSV bronchiolitis who are younger than age 60 days. Furthermore, since there was no difference in the rates of SBI in patients younger than age 28 days, infants in this age range should undergo a full septic work-up (blood, urine, and cerebrospinal fluid)—regardless of RSV infection status. For infants between ages 28 and 60 days, there is not enough evidence to recommend for or against further laboratory evaluation other than urinalysis.
Treatment


Nasal Suctioning
Nasal suctioning has become the first-line treatment for infants with bronchiolitis. It is used to clear secretions from the nasal passages to aid in respiration, which is particularly important in younger infants—who are obligate nose breathers. Current recommendations are to perform suctioning with increasing respiratory effort, before feeding and before laying the infant down to sleep.1
Bronchodilators
In the past, bronchodilators such as the β-agonist albuterol have been used to treat bronchiolitis with the idea that bronchial smooth muscle relaxation would improve clinical symptoms. In its 2006 guidelines, the AAP had recommended a trial of albuterol and continuation only if there was a documented objective response. In the 2014 updated guidelines, however, the AAP no longer recommends the use of albuterol in any capacity.
Although several meta-analyses and systematic reviews have demonstrated that bronchodilators may improve clinical symptoms scores, they did not affect disease resolution, need for hospitalization, or length of hospital stay.2 In addition, a recent Cochrane systematic review noted no benefit in the clinical course of infants with bronchiolitis treated with bronchodilators, and cited the potential adverse events (tachycardia and tremors) as outweighing any potential benefit.11 In addition to albuterol, the AAP no longer recommends the use of nebulized epinephrine in the treatment of bronchiolitis.2
Hypertonic Saline
Although hypertonic saline (HTS) has been increasingly studied for the treatment of bronchiolitis, the AAP does not recommend its use in the ED. Despite evidence that HTS may reduce hospital length of stay after 24 hours of use in settings where the typical duration of hospitalization exceeds 3 days, it has not been shown to reduce the rate of hospitalization when used in an emergency setting.2
Corticosteroids
While there is good evidence that corticosteroids are beneficial in treating some respiratory diseases, such as asthma and croup, numerous studies have repeatedly failed to show a benefit in treating bronchiolitis. One of the largest studies, a multicenter, randomized, controlled trial of dexamethasone for bronchiolitis by the Pediatric Emergency Care Applied Research Network, did not show any alteration in admission rates, respiratory status after 4 hours of observation, or length of hospital stay.12 Accordingly, the AAP strongly recommends against the administration of corticosteroids for bronchiolitis in any setting.2
Oxygen Therapy
Oxygen therapy is often necessary in patients with bronchiolitis who demonstrate hypoxia. The definition of hypoxia in this patient population has remained variable. The AAP has established a threshold of oxyhemoglobin saturation (SpO2) of less than 90% to define hypoxia and has empowered clinicians to not administer oxygen if the SpO2 exceeds 90%. Based on the oxyhemoglobin dissociation curve, the authors of the AAP guidelines note that when the SpO2 is less than 90%, small decreases in the arterial partial pressure of oxygen (PaO2) result in large decreases in the SpO2. When SpO2 is greater than 90%, however, large increases in PaO2 are associated with only small increased in SpO2. The AAP guidelines note, “In infants and children with bronchiolitis, no data exist to suggest that such increases [in PaO2 and SpO2] result in any clinically significant differences in physiologic function, patient symptoms, or clinical outcomes.”2
A relatively new method of administration of oxygen to infants with bronchiolitis is via a humidified, heated, high-flow nasal cannula (HHHFNC). This therapy has been shown to generate continuous positive airway pressure, which improves respiratory effort, reduces the work of breathing, and may decrease the need for intubation.2
Patient Disposition
One of the most challenging tasks for emergency physicians (EPs) is determining the appropriate disposition of infants with bronchiolitis. The variable presentation and dynamic nature of the disease make this particularly difficult. Patients at high risk for apnea should be admitted to the hospital for observation and further care as needed. Admission also should be strongly considered for those with significantly increased work of breathing and tachypnea that does not improve with suctioning—especially when these interfere with feeding. Infants with poor feeding or evidence of dehydration should be admitted to the hospital for intravenous (IV) fluid hydration or nasogastric feedings. Patients with hypoxia (SpO2 saturations <90%) should also be admitted for supplemental oxygen therapy. It should be noted, however, the AAP recommends “spot-checks” over continuous pulse oximetry in patients who do not require oxygen therapy.2
Another important factor affecting patient disposition is the ability of the caregiver to provide basic patient care and ensure close outpatient follow-up. Prior to discharge, caregivers should be educated on the highly dynamic nature of bronchiolitis and the signs and symptoms that would require prompt return to the ED—especially if the infant has risk factors for the development of severe disease.
Case Conclusion
Based on the patient’s symptoms, history (most notably, the recent incident of sleep apnea at home), and physical examination, the EP quickly identified this infant was at a high risk for both severe bronchiolitis and apnea and required aggressive management. Nasal suctioning was immediately performed to help clear the patient’s secretions; this, however, only slightly improved his RR and work of breathing. Although the infant’s SpO2 was greater than 90% on room air, the EP administered oxygen via HHHFNC at 6 L per minute, which produced a significant improvement in both RR and effort.
Given the patient’s age and the presence of a fever, a urinalysis was also obtained, the results of which showed no evidence of infection. Since the patient was only able to bottle-feed for a few minutes at a time, the EP initiated IV fluid hydration and contacted the hospitalist team for inpatient admission.
The infant was gradually weaned from HHHFNC on hospital day 2 but remained with suboptimal oral intake for another 24 hours. By hospital day 4, his work of breathing had improved significantly, and he was feeding well with through the assistance of pre-feeding nasal syringe suctioning. The patient was discharged home in the care of his parents later that same day with only mild tachypnea over baseline. At discharge, the EP emphasized the importance of providing close follow-up with their son’s pediatrician. The infant continued to gradually improve as an outpatient, with resolution of nasal congestion by day 12 of his illness; he returned to his baseline breathing and feeding pattern on day 14.
Dr Schneider is a pediatric emergency medicine fellow, Eastern Virginia Medical School, Children’s Hospital of The King’s Daughters, Norfolk. Dr Clingenpeel is a fellowship director of pediatric emergency medicine, and an associate professor of pediatrics, Eastern Virginia Medical School, Norfolk.
- Joseph M. Evidence-based assessment and management of acute bronchiolitis in the emergency department. Pediatr Emerg Med Pract. 2011;8(3):1-20.
- Ralston SL, Lieberthal AS, Meissner HC, et al; American Academy of Pediatrics. Clinical practice guidelines: the diagnosis, management, and prevention of bronchiolitis [Published correction appears in Pediatrics. 2014;134(5):e1474-e1502]. Pediatrics. 2014;134:5 e1474-e1502.
- Hasegawa K, Tsugawa Y, Brown DF, Mansbach JM, Camargo CA Jr. Trends in bronchiolitis hospitalizations in the United States, 2000-20009. Pediatrics. 2013;32(1):28-36.
- Centers for Disease Control and Prevention (CDC). Respiratory syncytial virus activity—United States, July 2011-January 2013. MMWR Morb Mortal Wkly Rep. 2013;62(8):141-144.
- Harper MB, Fleisher GR. Infectious emergencies. In: Fleisher GR, Ludwig S, eds. Textbook of Pediatric Emergency Medicine. 6th ed. Philadelphia, PA: Lippincott Williams & Wilkins;2010:916-917.
- Willwerth BM, Harper MB, Greenes DS. Identifying hospitalized infants who have bronchiolitis and are at high risk for apnea. Ann Emerg Med. 2006;48(4):441-447.
- Shaw KN, Bell LM, Sherman NH. Outpatient assessment of infants with bronchiolitis. Am J Dis Child. 1991;145(2):151-155.
- Wang EE, Milner RA, Navas L, Maj H. Observer agreement for respiratory signs and oximetry in infants hospitalized with lower respiratory infections. Am Rev Respir Dis. 1992;145(1):106-109.
- Schuh S, Lalani A, Allen U, et al. Evaluation of the utility of radiography in acute bronchiolitis. J Pediatr. 2007;150(4):429-433.
- Levine DA, Platt SL, Dayan PS, et al; Multicenter RSV-SBI Study Group of the Pediatric Emergency Medicine Collaborative Research Committee of the American Academy of Pediatrics. Risk of serious bacterial infection in young febrile infants with respiratory syncytial virus infection. Pediatrics. 2004;113(6):1728-1734.
- Gadomski AM, Scribani MB. Bronchodilators for bronchiolitis. Cochrane Database Syst Rev. 2014;(6):CD001266.
- Corneli HM, Zorc JJ, Majahan P, et al; Bronchiolitis Study Group of the Pediatric Emergency Care Applied Research Network (PECARN). A multicenter, randomized, controlled trial of dexamethasone for bronchiolitis. N Engl J Med. 2007;357(4):331-339.
Case
An 8-week-old male infant was brought to the ED by his parents after an episode in which it appeared the baby had stopped breathing. The parents stated that while lying on his mother’s lap at home, the patient stopped breathing for approximately 10 to 15 seconds, during which time his face exhibited a bluish color. They further noted that the patient began breathing again after gentle stimulation and had been acting normally since.
The patient was born at 39 weeks gestation via normal vaginal delivery and without any complications. His parents further stated that prior to the cessation of breathing incident, his symptoms of nasal congestion, decreased energy level, and fast breathing had gradually worsened over the past 2 days. The parents also noted that the infant had not been feeding as well over the past 2 days.
Upon arrival, the patient’s vital signs were: heart rate, 140 beats/minute; respiratory rate (RR), 72 beats/minute; and temperature 101.3°F. Oxygen saturation was 92% on room air. On physical examination, the infant had significant rhinorrhea, moderate intercostal and supraclavicular retractions, ausculatory wheezes, and transmitted upper airway noises throughout.
Overview
Bronchiolitis, a disorder caused by a viral lower respiratory tract infection, is the most common lower respiratory infection in children younger than age 2 years.1 In 2014, the American Academy of Pediatrics (AAP) characterized bronchiolitis as “rhinitis, cough, tachypnea, wheezing, rales, use of accessory muscles, and/or nasal flaring in children under 24 months of age.”2 This condition is the most common cause of hospitalization in the first 12 months of life. It is responsible for over 100,000 admissions annually at an estimated cost to the healthcare system of $1.73 billion.3
Etiology and Pathophysiology
Respiratory syncytial virus (RSV) is the most common cause of bronchiolitis. In the United States, the highest incidence of infection occurs during the months of December through March, with some degree of regional variability.4 A number of other viruses that can cause bronchiolitis include human metapneumovirus, parainfluenza virus, and influenza virus.1 Infection with RSV does not grant permanent immunity, and reinfection is common throughout life.2
Pathophysiologically, bronchiolitis is characterized by an invasion of bronchial epithelial cells that lead to to cell death and sloughing into the bronchial lumen. This, coupled with increased mucous production and submucosal edema, leads to a narrowing of the bronchial lumen and obstruction of airflow.5
Clinical Manifestations
Bronchiolitis represents a constellation of signs and symptoms beginning with those of an upper respiratory tract infection, including nasal congestion and rhinorrhea with mild cough. On days 3 to 5, the following symptoms develop: tachypnea, wheezing, rales, and signs of respiratory distress (eg, grunting, nasal flaring, inter-/subcostal retractions). Approximately two-thirds of patients will develop a fever.2 Recovery tends to begin around days 5 to 7, with the median duration of illness being 12 days.1 It should be noted that bronchiolitis represents a highly variable and dynamic disease state. Transient episodes of improvement and worsening are common, emphasizing the importance of serial examinations and assessments. Though rare, progression to respiratory failure and death do occur.2
History and Risk Stratification
The focus of the initial history by the clinician should serve two primary purposes. First, it is important to differentiate infants with probable bronchiolitis from those with other disease states having similar clinical manifestations. One of the most challenging diseases to differentiate from bronchiolitis is that of reactive airway disease (RAD). Eliciting a history of allergic rhinitis, eczema, or a family history of asthma may be helpful in determining the precise etiology of the patient’s symptoms. Although no longer recommended for children with bronchiolitis (as will be later discussed), a trial of a bronchodilation may be beneficial in the setting of familial atopy.
The second—and perhaps most important—aspect of patient history is to determine the presence of risk factors for both apnea and the development of severe bronchiolitis. Regarding the risk factors for apnea, Willwerth et al6 developed a set of criteria to identify patients at high risk for apnea in the inpatient setting. Patients were considered high risk if they were born at full term and were younger than 1 month of age; if they were born preterm (<37 weeks gestation) and were younger than 48 weeks postconception; and/or if the infant’s parents or a clinician had already witnessed an episode of apnea during the patient’s illness. In this study, all patients who developed apnea were correctly identified by the risk criteria.6 Risk factors for severe bronchiolitis include the following: patient age younger than 12 weeks; patient prematurity younger than 37 weeks gestation; and an underlying hemodynamically significant congenital heart disease, chronic lung disease/bronchopulmonary dysplasia, or an immunocompromised state.1
Diagnosis
In 2014, the AAP updated its guidelines on the diagnosis, management, and prevention of bronchiolitis. One of the strongest statements in these guidelines emphasize that the diagnosis of bronchiolitis should be based almost exclusively on the history and physical examination.2 In children younger than age 2 years, historical features such as a viral prodrome, followed by progressively worsening increased respiratory effort and signs and symptoms of lower respiratory-tract disease (eg, wheezing), should guide clinicians to the diagnosis of bronchiolitis. Although nonspecific, physical examination findings such as rhinorrhea, cough, tachypnea, wheezing, rales, and increased respiratory effort—when coupled with a good history—can be beneficial in the diagnosis of bronchiolitis.
Pulse Oximetry
Pulse oximetry has become a standard part of the clinical assessment of patients with bronchiolitis. This is based on data suggesting that pulse oximetry detects hypoxia in cases where it was not suspected on physical examination alone.7 However, the effectiveness of pulse oximetry in predicting clinical outcomes is limited. Pulse oximetry should not be used as a proxy for respiratory distress, as studies have shown poor correlation between respiratory distress and oxygen saturations in infants with lower respiratory tract infection.8
Radiographic Evaluation
Regarding the diagnosis of bronchiolitis, the AAP notes, “radiographic and laboratory studies should not be obtained routinely.”2 While many children with bronchiolitis may have abnormalities on radiographs, there is insufficient data to suggest that chest radiographs correlate with disease severity. In addition, several studies, including a prospective cohort study by Schuh et al,9 have shown that infants with suspected lower respiratory tract infections who undergo radiography are more likely to receive antibiotics without any difference in outcomes.
Laboratory Studies
As stated in the AAP guidelines, routine laboratory testing, particularly virologic studies for RSV, have little role in the diagnosis of bronchiolitis. Since numerous viruses can cause bronchiolitis and have similar clinical presentations, the absence of identification of a particular virologic agent does not exclude the diagnosis of bronchiolitis and is moreover unlikely to alter management.
Although routine laboratory evaluation is not recommended in infants with bronchiolitis, one subgroup in which it may be beneficial is in the assessment of serious bacterial infections (SBIs) in febrile infants with bronchiolitis who are younger than 60 days old. Levine et al10 conducted a large, multicenter, prospective, cross-sectional study of young, febrile infants to determine the risk of SBI in those with RSV bronchiolitis versus those without RSV bronchiolitis. They found that overall febrile infants younger than age 60 days with RSV bronchiolitis have a lower rate of SBI than those without RSV (7% v 12.5%, respectively).10 In infants between age 28 and 60 days with RSV bronchiolitis, the origin of all SBIs in the study were urinary tract infections. In patients younger than 28 days of age, the risk of developing an SBI was found to be no different between the RSV-positive and RSV-negative groups.10
Based on the findings in this study, it is recommended that, at the very least, urinalysis for bacterial infection be performed in all infants with RSV bronchiolitis who are younger than age 60 days. Furthermore, since there was no difference in the rates of SBI in patients younger than age 28 days, infants in this age range should undergo a full septic work-up (blood, urine, and cerebrospinal fluid)—regardless of RSV infection status. For infants between ages 28 and 60 days, there is not enough evidence to recommend for or against further laboratory evaluation other than urinalysis.
Treatment
Nasal Suctioning
Nasal suctioning has become the first-line treatment for infants with bronchiolitis. It is used to clear secretions from the nasal passages to aid in respiration, which is particularly important in younger infants—who are obligate nose breathers. Current recommendations are to perform suctioning with increasing respiratory effort, before feeding and before laying the infant down to sleep.1
Bronchodilators
In the past, bronchodilators such as the β-agonist albuterol have been used to treat bronchiolitis with the idea that bronchial smooth muscle relaxation would improve clinical symptoms. In its 2006 guidelines, the AAP had recommended a trial of albuterol and continuation only if there was a documented objective response. In the 2014 updated guidelines, however, the AAP no longer recommends the use of albuterol in any capacity.
Although several meta-analyses and systematic reviews have demonstrated that bronchodilators may improve clinical symptoms scores, they did not affect disease resolution, need for hospitalization, or length of hospital stay.2 In addition, a recent Cochrane systematic review noted no benefit in the clinical course of infants with bronchiolitis treated with bronchodilators, and cited the potential adverse events (tachycardia and tremors) as outweighing any potential benefit.11 In addition to albuterol, the AAP no longer recommends the use of nebulized epinephrine in the treatment of bronchiolitis.2
Hypertonic Saline
Although hypertonic saline (HTS) has been increasingly studied for the treatment of bronchiolitis, the AAP does not recommend its use in the ED. Despite evidence that HTS may reduce hospital length of stay after 24 hours of use in settings where the typical duration of hospitalization exceeds 3 days, it has not been shown to reduce the rate of hospitalization when used in an emergency setting.2
Corticosteroids
While there is good evidence that corticosteroids are beneficial in treating some respiratory diseases, such as asthma and croup, numerous studies have repeatedly failed to show a benefit in treating bronchiolitis. One of the largest studies, a multicenter, randomized, controlled trial of dexamethasone for bronchiolitis by the Pediatric Emergency Care Applied Research Network, did not show any alteration in admission rates, respiratory status after 4 hours of observation, or length of hospital stay.12 Accordingly, the AAP strongly recommends against the administration of corticosteroids for bronchiolitis in any setting.2
Oxygen Therapy
Oxygen therapy is often necessary in patients with bronchiolitis who demonstrate hypoxia. The definition of hypoxia in this patient population has remained variable. The AAP has established a threshold of oxyhemoglobin saturation (SpO2) of less than 90% to define hypoxia and has empowered clinicians to not administer oxygen if the SpO2 exceeds 90%. Based on the oxyhemoglobin dissociation curve, the authors of the AAP guidelines note that when the SpO2 is less than 90%, small decreases in the arterial partial pressure of oxygen (PaO2) result in large decreases in the SpO2. When SpO2 is greater than 90%, however, large increases in PaO2 are associated with only small increased in SpO2. The AAP guidelines note, “In infants and children with bronchiolitis, no data exist to suggest that such increases [in PaO2 and SpO2] result in any clinically significant differences in physiologic function, patient symptoms, or clinical outcomes.”2
A relatively new method of administration of oxygen to infants with bronchiolitis is via a humidified, heated, high-flow nasal cannula (HHHFNC). This therapy has been shown to generate continuous positive airway pressure, which improves respiratory effort, reduces the work of breathing, and may decrease the need for intubation.2
Patient Disposition
One of the most challenging tasks for emergency physicians (EPs) is determining the appropriate disposition of infants with bronchiolitis. The variable presentation and dynamic nature of the disease make this particularly difficult. Patients at high risk for apnea should be admitted to the hospital for observation and further care as needed. Admission also should be strongly considered for those with significantly increased work of breathing and tachypnea that does not improve with suctioning—especially when these interfere with feeding. Infants with poor feeding or evidence of dehydration should be admitted to the hospital for intravenous (IV) fluid hydration or nasogastric feedings. Patients with hypoxia (SpO2 saturations <90%) should also be admitted for supplemental oxygen therapy. It should be noted, however, the AAP recommends “spot-checks” over continuous pulse oximetry in patients who do not require oxygen therapy.2
Another important factor affecting patient disposition is the ability of the caregiver to provide basic patient care and ensure close outpatient follow-up. Prior to discharge, caregivers should be educated on the highly dynamic nature of bronchiolitis and the signs and symptoms that would require prompt return to the ED—especially if the infant has risk factors for the development of severe disease.
Case Conclusion
Based on the patient’s symptoms, history (most notably, the recent incident of sleep apnea at home), and physical examination, the EP quickly identified this infant was at a high risk for both severe bronchiolitis and apnea and required aggressive management. Nasal suctioning was immediately performed to help clear the patient’s secretions; this, however, only slightly improved his RR and work of breathing. Although the infant’s SpO2 was greater than 90% on room air, the EP administered oxygen via HHHFNC at 6 L per minute, which produced a significant improvement in both RR and effort.
Given the patient’s age and the presence of a fever, a urinalysis was also obtained, the results of which showed no evidence of infection. Since the patient was only able to bottle-feed for a few minutes at a time, the EP initiated IV fluid hydration and contacted the hospitalist team for inpatient admission.
The infant was gradually weaned from HHHFNC on hospital day 2 but remained with suboptimal oral intake for another 24 hours. By hospital day 4, his work of breathing had improved significantly, and he was feeding well with through the assistance of pre-feeding nasal syringe suctioning. The patient was discharged home in the care of his parents later that same day with only mild tachypnea over baseline. At discharge, the EP emphasized the importance of providing close follow-up with their son’s pediatrician. The infant continued to gradually improve as an outpatient, with resolution of nasal congestion by day 12 of his illness; he returned to his baseline breathing and feeding pattern on day 14.
Dr Schneider is a pediatric emergency medicine fellow, Eastern Virginia Medical School, Children’s Hospital of The King’s Daughters, Norfolk. Dr Clingenpeel is a fellowship director of pediatric emergency medicine, and an associate professor of pediatrics, Eastern Virginia Medical School, Norfolk.
Case
An 8-week-old male infant was brought to the ED by his parents after an episode in which it appeared the baby had stopped breathing. The parents stated that while lying on his mother’s lap at home, the patient stopped breathing for approximately 10 to 15 seconds, during which time his face exhibited a bluish color. They further noted that the patient began breathing again after gentle stimulation and had been acting normally since.
The patient was born at 39 weeks gestation via normal vaginal delivery and without any complications. His parents further stated that prior to the cessation of breathing incident, his symptoms of nasal congestion, decreased energy level, and fast breathing had gradually worsened over the past 2 days. The parents also noted that the infant had not been feeding as well over the past 2 days.
Upon arrival, the patient’s vital signs were: heart rate, 140 beats/minute; respiratory rate (RR), 72 beats/minute; and temperature 101.3°F. Oxygen saturation was 92% on room air. On physical examination, the infant had significant rhinorrhea, moderate intercostal and supraclavicular retractions, ausculatory wheezes, and transmitted upper airway noises throughout.
Overview
Bronchiolitis, a disorder caused by a viral lower respiratory tract infection, is the most common lower respiratory infection in children younger than age 2 years.1 In 2014, the American Academy of Pediatrics (AAP) characterized bronchiolitis as “rhinitis, cough, tachypnea, wheezing, rales, use of accessory muscles, and/or nasal flaring in children under 24 months of age.”2 This condition is the most common cause of hospitalization in the first 12 months of life. It is responsible for over 100,000 admissions annually at an estimated cost to the healthcare system of $1.73 billion.3
Etiology and Pathophysiology
Respiratory syncytial virus (RSV) is the most common cause of bronchiolitis. In the United States, the highest incidence of infection occurs during the months of December through March, with some degree of regional variability.4 A number of other viruses that can cause bronchiolitis include human metapneumovirus, parainfluenza virus, and influenza virus.1 Infection with RSV does not grant permanent immunity, and reinfection is common throughout life.2
Pathophysiologically, bronchiolitis is characterized by an invasion of bronchial epithelial cells that lead to to cell death and sloughing into the bronchial lumen. This, coupled with increased mucous production and submucosal edema, leads to a narrowing of the bronchial lumen and obstruction of airflow.5
Clinical Manifestations
Bronchiolitis represents a constellation of signs and symptoms beginning with those of an upper respiratory tract infection, including nasal congestion and rhinorrhea with mild cough. On days 3 to 5, the following symptoms develop: tachypnea, wheezing, rales, and signs of respiratory distress (eg, grunting, nasal flaring, inter-/subcostal retractions). Approximately two-thirds of patients will develop a fever.2 Recovery tends to begin around days 5 to 7, with the median duration of illness being 12 days.1 It should be noted that bronchiolitis represents a highly variable and dynamic disease state. Transient episodes of improvement and worsening are common, emphasizing the importance of serial examinations and assessments. Though rare, progression to respiratory failure and death do occur.2
History and Risk Stratification
The focus of the initial history by the clinician should serve two primary purposes. First, it is important to differentiate infants with probable bronchiolitis from those with other disease states having similar clinical manifestations. One of the most challenging diseases to differentiate from bronchiolitis is that of reactive airway disease (RAD). Eliciting a history of allergic rhinitis, eczema, or a family history of asthma may be helpful in determining the precise etiology of the patient’s symptoms. Although no longer recommended for children with bronchiolitis (as will be later discussed), a trial of a bronchodilation may be beneficial in the setting of familial atopy.
The second—and perhaps most important—aspect of patient history is to determine the presence of risk factors for both apnea and the development of severe bronchiolitis. Regarding the risk factors for apnea, Willwerth et al6 developed a set of criteria to identify patients at high risk for apnea in the inpatient setting. Patients were considered high risk if they were born at full term and were younger than 1 month of age; if they were born preterm (<37 weeks gestation) and were younger than 48 weeks postconception; and/or if the infant’s parents or a clinician had already witnessed an episode of apnea during the patient’s illness. In this study, all patients who developed apnea were correctly identified by the risk criteria.6 Risk factors for severe bronchiolitis include the following: patient age younger than 12 weeks; patient prematurity younger than 37 weeks gestation; and an underlying hemodynamically significant congenital heart disease, chronic lung disease/bronchopulmonary dysplasia, or an immunocompromised state.1
Diagnosis
In 2014, the AAP updated its guidelines on the diagnosis, management, and prevention of bronchiolitis. One of the strongest statements in these guidelines emphasize that the diagnosis of bronchiolitis should be based almost exclusively on the history and physical examination.2 In children younger than age 2 years, historical features such as a viral prodrome, followed by progressively worsening increased respiratory effort and signs and symptoms of lower respiratory-tract disease (eg, wheezing), should guide clinicians to the diagnosis of bronchiolitis. Although nonspecific, physical examination findings such as rhinorrhea, cough, tachypnea, wheezing, rales, and increased respiratory effort—when coupled with a good history—can be beneficial in the diagnosis of bronchiolitis.
Pulse Oximetry
Pulse oximetry has become a standard part of the clinical assessment of patients with bronchiolitis. This is based on data suggesting that pulse oximetry detects hypoxia in cases where it was not suspected on physical examination alone.7 However, the effectiveness of pulse oximetry in predicting clinical outcomes is limited. Pulse oximetry should not be used as a proxy for respiratory distress, as studies have shown poor correlation between respiratory distress and oxygen saturations in infants with lower respiratory tract infection.8
Radiographic Evaluation
Regarding the diagnosis of bronchiolitis, the AAP notes, “radiographic and laboratory studies should not be obtained routinely.”2 While many children with bronchiolitis may have abnormalities on radiographs, there is insufficient data to suggest that chest radiographs correlate with disease severity. In addition, several studies, including a prospective cohort study by Schuh et al,9 have shown that infants with suspected lower respiratory tract infections who undergo radiography are more likely to receive antibiotics without any difference in outcomes.
Laboratory Studies
As stated in the AAP guidelines, routine laboratory testing, particularly virologic studies for RSV, have little role in the diagnosis of bronchiolitis. Since numerous viruses can cause bronchiolitis and have similar clinical presentations, the absence of identification of a particular virologic agent does not exclude the diagnosis of bronchiolitis and is moreover unlikely to alter management.
Although routine laboratory evaluation is not recommended in infants with bronchiolitis, one subgroup in which it may be beneficial is in the assessment of serious bacterial infections (SBIs) in febrile infants with bronchiolitis who are younger than 60 days old. Levine et al10 conducted a large, multicenter, prospective, cross-sectional study of young, febrile infants to determine the risk of SBI in those with RSV bronchiolitis versus those without RSV bronchiolitis. They found that overall febrile infants younger than age 60 days with RSV bronchiolitis have a lower rate of SBI than those without RSV (7% v 12.5%, respectively).10 In infants between age 28 and 60 days with RSV bronchiolitis, the origin of all SBIs in the study were urinary tract infections. In patients younger than 28 days of age, the risk of developing an SBI was found to be no different between the RSV-positive and RSV-negative groups.10
Based on the findings in this study, it is recommended that, at the very least, urinalysis for bacterial infection be performed in all infants with RSV bronchiolitis who are younger than age 60 days. Furthermore, since there was no difference in the rates of SBI in patients younger than age 28 days, infants in this age range should undergo a full septic work-up (blood, urine, and cerebrospinal fluid)—regardless of RSV infection status. For infants between ages 28 and 60 days, there is not enough evidence to recommend for or against further laboratory evaluation other than urinalysis.
Treatment
Nasal Suctioning
Nasal suctioning has become the first-line treatment for infants with bronchiolitis. It is used to clear secretions from the nasal passages to aid in respiration, which is particularly important in younger infants—who are obligate nose breathers. Current recommendations are to perform suctioning with increasing respiratory effort, before feeding and before laying the infant down to sleep.1
Bronchodilators
In the past, bronchodilators such as the β-agonist albuterol have been used to treat bronchiolitis with the idea that bronchial smooth muscle relaxation would improve clinical symptoms. In its 2006 guidelines, the AAP had recommended a trial of albuterol and continuation only if there was a documented objective response. In the 2014 updated guidelines, however, the AAP no longer recommends the use of albuterol in any capacity.
Although several meta-analyses and systematic reviews have demonstrated that bronchodilators may improve clinical symptoms scores, they did not affect disease resolution, need for hospitalization, or length of hospital stay.2 In addition, a recent Cochrane systematic review noted no benefit in the clinical course of infants with bronchiolitis treated with bronchodilators, and cited the potential adverse events (tachycardia and tremors) as outweighing any potential benefit.11 In addition to albuterol, the AAP no longer recommends the use of nebulized epinephrine in the treatment of bronchiolitis.2
Hypertonic Saline
Although hypertonic saline (HTS) has been increasingly studied for the treatment of bronchiolitis, the AAP does not recommend its use in the ED. Despite evidence that HTS may reduce hospital length of stay after 24 hours of use in settings where the typical duration of hospitalization exceeds 3 days, it has not been shown to reduce the rate of hospitalization when used in an emergency setting.2
Corticosteroids
While there is good evidence that corticosteroids are beneficial in treating some respiratory diseases, such as asthma and croup, numerous studies have repeatedly failed to show a benefit in treating bronchiolitis. One of the largest studies, a multicenter, randomized, controlled trial of dexamethasone for bronchiolitis by the Pediatric Emergency Care Applied Research Network, did not show any alteration in admission rates, respiratory status after 4 hours of observation, or length of hospital stay.12 Accordingly, the AAP strongly recommends against the administration of corticosteroids for bronchiolitis in any setting.2
Oxygen Therapy
Oxygen therapy is often necessary in patients with bronchiolitis who demonstrate hypoxia. The definition of hypoxia in this patient population has remained variable. The AAP has established a threshold of oxyhemoglobin saturation (SpO2) of less than 90% to define hypoxia and has empowered clinicians to not administer oxygen if the SpO2 exceeds 90%. Based on the oxyhemoglobin dissociation curve, the authors of the AAP guidelines note that when the SpO2 is less than 90%, small decreases in the arterial partial pressure of oxygen (PaO2) result in large decreases in the SpO2. When SpO2 is greater than 90%, however, large increases in PaO2 are associated with only small increased in SpO2. The AAP guidelines note, “In infants and children with bronchiolitis, no data exist to suggest that such increases [in PaO2 and SpO2] result in any clinically significant differences in physiologic function, patient symptoms, or clinical outcomes.”2
A relatively new method of administration of oxygen to infants with bronchiolitis is via a humidified, heated, high-flow nasal cannula (HHHFNC). This therapy has been shown to generate continuous positive airway pressure, which improves respiratory effort, reduces the work of breathing, and may decrease the need for intubation.2
Patient Disposition
One of the most challenging tasks for emergency physicians (EPs) is determining the appropriate disposition of infants with bronchiolitis. The variable presentation and dynamic nature of the disease make this particularly difficult. Patients at high risk for apnea should be admitted to the hospital for observation and further care as needed. Admission also should be strongly considered for those with significantly increased work of breathing and tachypnea that does not improve with suctioning—especially when these interfere with feeding. Infants with poor feeding or evidence of dehydration should be admitted to the hospital for intravenous (IV) fluid hydration or nasogastric feedings. Patients with hypoxia (SpO2 saturations <90%) should also be admitted for supplemental oxygen therapy. It should be noted, however, the AAP recommends “spot-checks” over continuous pulse oximetry in patients who do not require oxygen therapy.2
Another important factor affecting patient disposition is the ability of the caregiver to provide basic patient care and ensure close outpatient follow-up. Prior to discharge, caregivers should be educated on the highly dynamic nature of bronchiolitis and the signs and symptoms that would require prompt return to the ED—especially if the infant has risk factors for the development of severe disease.
Case Conclusion
Based on the patient’s symptoms, history (most notably, the recent incident of sleep apnea at home), and physical examination, the EP quickly identified this infant was at a high risk for both severe bronchiolitis and apnea and required aggressive management. Nasal suctioning was immediately performed to help clear the patient’s secretions; this, however, only slightly improved his RR and work of breathing. Although the infant’s SpO2 was greater than 90% on room air, the EP administered oxygen via HHHFNC at 6 L per minute, which produced a significant improvement in both RR and effort.
Given the patient’s age and the presence of a fever, a urinalysis was also obtained, the results of which showed no evidence of infection. Since the patient was only able to bottle-feed for a few minutes at a time, the EP initiated IV fluid hydration and contacted the hospitalist team for inpatient admission.
The infant was gradually weaned from HHHFNC on hospital day 2 but remained with suboptimal oral intake for another 24 hours. By hospital day 4, his work of breathing had improved significantly, and he was feeding well with through the assistance of pre-feeding nasal syringe suctioning. The patient was discharged home in the care of his parents later that same day with only mild tachypnea over baseline. At discharge, the EP emphasized the importance of providing close follow-up with their son’s pediatrician. The infant continued to gradually improve as an outpatient, with resolution of nasal congestion by day 12 of his illness; he returned to his baseline breathing and feeding pattern on day 14.
Dr Schneider is a pediatric emergency medicine fellow, Eastern Virginia Medical School, Children’s Hospital of The King’s Daughters, Norfolk. Dr Clingenpeel is a fellowship director of pediatric emergency medicine, and an associate professor of pediatrics, Eastern Virginia Medical School, Norfolk.
- Joseph M. Evidence-based assessment and management of acute bronchiolitis in the emergency department. Pediatr Emerg Med Pract. 2011;8(3):1-20.
- Ralston SL, Lieberthal AS, Meissner HC, et al; American Academy of Pediatrics. Clinical practice guidelines: the diagnosis, management, and prevention of bronchiolitis [Published correction appears in Pediatrics. 2014;134(5):e1474-e1502]. Pediatrics. 2014;134:5 e1474-e1502.
- Hasegawa K, Tsugawa Y, Brown DF, Mansbach JM, Camargo CA Jr. Trends in bronchiolitis hospitalizations in the United States, 2000-20009. Pediatrics. 2013;32(1):28-36.
- Centers for Disease Control and Prevention (CDC). Respiratory syncytial virus activity—United States, July 2011-January 2013. MMWR Morb Mortal Wkly Rep. 2013;62(8):141-144.
- Harper MB, Fleisher GR. Infectious emergencies. In: Fleisher GR, Ludwig S, eds. Textbook of Pediatric Emergency Medicine. 6th ed. Philadelphia, PA: Lippincott Williams & Wilkins;2010:916-917.
- Willwerth BM, Harper MB, Greenes DS. Identifying hospitalized infants who have bronchiolitis and are at high risk for apnea. Ann Emerg Med. 2006;48(4):441-447.
- Shaw KN, Bell LM, Sherman NH. Outpatient assessment of infants with bronchiolitis. Am J Dis Child. 1991;145(2):151-155.
- Wang EE, Milner RA, Navas L, Maj H. Observer agreement for respiratory signs and oximetry in infants hospitalized with lower respiratory infections. Am Rev Respir Dis. 1992;145(1):106-109.
- Schuh S, Lalani A, Allen U, et al. Evaluation of the utility of radiography in acute bronchiolitis. J Pediatr. 2007;150(4):429-433.
- Levine DA, Platt SL, Dayan PS, et al; Multicenter RSV-SBI Study Group of the Pediatric Emergency Medicine Collaborative Research Committee of the American Academy of Pediatrics. Risk of serious bacterial infection in young febrile infants with respiratory syncytial virus infection. Pediatrics. 2004;113(6):1728-1734.
- Gadomski AM, Scribani MB. Bronchodilators for bronchiolitis. Cochrane Database Syst Rev. 2014;(6):CD001266.
- Corneli HM, Zorc JJ, Majahan P, et al; Bronchiolitis Study Group of the Pediatric Emergency Care Applied Research Network (PECARN). A multicenter, randomized, controlled trial of dexamethasone for bronchiolitis. N Engl J Med. 2007;357(4):331-339.
- Joseph M. Evidence-based assessment and management of acute bronchiolitis in the emergency department. Pediatr Emerg Med Pract. 2011;8(3):1-20.
- Ralston SL, Lieberthal AS, Meissner HC, et al; American Academy of Pediatrics. Clinical practice guidelines: the diagnosis, management, and prevention of bronchiolitis [Published correction appears in Pediatrics. 2014;134(5):e1474-e1502]. Pediatrics. 2014;134:5 e1474-e1502.
- Hasegawa K, Tsugawa Y, Brown DF, Mansbach JM, Camargo CA Jr. Trends in bronchiolitis hospitalizations in the United States, 2000-20009. Pediatrics. 2013;32(1):28-36.
- Centers for Disease Control and Prevention (CDC). Respiratory syncytial virus activity—United States, July 2011-January 2013. MMWR Morb Mortal Wkly Rep. 2013;62(8):141-144.
- Harper MB, Fleisher GR. Infectious emergencies. In: Fleisher GR, Ludwig S, eds. Textbook of Pediatric Emergency Medicine. 6th ed. Philadelphia, PA: Lippincott Williams & Wilkins;2010:916-917.
- Willwerth BM, Harper MB, Greenes DS. Identifying hospitalized infants who have bronchiolitis and are at high risk for apnea. Ann Emerg Med. 2006;48(4):441-447.
- Shaw KN, Bell LM, Sherman NH. Outpatient assessment of infants with bronchiolitis. Am J Dis Child. 1991;145(2):151-155.
- Wang EE, Milner RA, Navas L, Maj H. Observer agreement for respiratory signs and oximetry in infants hospitalized with lower respiratory infections. Am Rev Respir Dis. 1992;145(1):106-109.
- Schuh S, Lalani A, Allen U, et al. Evaluation of the utility of radiography in acute bronchiolitis. J Pediatr. 2007;150(4):429-433.
- Levine DA, Platt SL, Dayan PS, et al; Multicenter RSV-SBI Study Group of the Pediatric Emergency Medicine Collaborative Research Committee of the American Academy of Pediatrics. Risk of serious bacterial infection in young febrile infants with respiratory syncytial virus infection. Pediatrics. 2004;113(6):1728-1734.
- Gadomski AM, Scribani MB. Bronchodilators for bronchiolitis. Cochrane Database Syst Rev. 2014;(6):CD001266.
- Corneli HM, Zorc JJ, Majahan P, et al; Bronchiolitis Study Group of the Pediatric Emergency Care Applied Research Network (PECARN). A multicenter, randomized, controlled trial of dexamethasone for bronchiolitis. N Engl J Med. 2007;357(4):331-339.

Case Studies in Toxicology: The Perilous Pursuit of Perfection
Case
A 37-year-old woman had undergone outpatient tumescent liposuction of her abdominal adipose tissue. Upon initiation of the procedure, she developed acute confusion followed by agitation and hallucinations and was transported by emergency medical services to the nearest hospital.
Upon arrival to the ED, the patient was mildly agitated. Her initial vital signs were: blood pressure, 122/79 mm Hg; heart rate, 57 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 96.8°F. Oxygen saturation was 100% on room air.
On examination, the patient was awake and oriented, but was restless and perseverating. She described the sensation of “dying and coming back to life.” The patient’s pupils were equal, round, and normally reactive; her skin was neither diaphoretic nor dry; her heart rate and rhythm were normal; and her lungs were clear to auscultation bilaterally. The skin of the patient’s abdomen was pale, cool, and clammy, but otherwise soft with normoactive bowel sounds. An electrocardiogram revealed sinus rhythm with normal QRS and QTc intervals.
What is tumescent liposuction?

The tumescent technique (the word tumescent derived from the Latin word meaning “to swell”) involves the infiltration of a large volume of a dilute solution of lidocaine and epinephrine into the subcutaneous fat until it becomes distended and tense. The administration of this solution allows for the removal of significant adipose tissue with minimal blood loss and without general anesthesia. Advocates of tumescent liposuction argue that this dilution and subcutaneous infiltration alter the pharmacokinetics of lidocaine, providing a safe delivery of large doses of the drug. In addition, a substantial quantity of the infiltrated lidocaine is suctioned out with the fat removal. Klein3 specifically recommends administering a dilution of 1 g lidocaine in 1,000 mL of saline (0.1%) to the patient in aliquots of up to 2 g lidocaine. Such a high dose may approach or surpass 35 mg/kg, far exceeding the recommended lidocaine infiltration dose of 7 mg/kg when mixed with epinephrine.
Several studies have investigated the safety of the tumescent technique and the risk of lidocaine toxicity. In a review of pharmacokinetic studies, patients received between 10.5 and 67.7 mg/kg of lidocaine with a reported maximum serum lidocaine concentration in all patients of only 2.93 mcg/mL (therapeutic, 1-5 mcg/mL).4 However, peak serum lidocaine concentrations may not occur for up to 28 hours following tumescent infiltration.4 Despite the purported safety findings of this and other studies, there are also reports of tumescent anesthesia-associated toxicity and fatalities.5,6
Case Continuation
The patient’s mental status slowly improved throughout her hospital stay and she was reportedly at baseline by the following morning. Her serum lidocaine concentration drawn at presentation to the ED was 9.7 mcg/mL.
What are the clinical effects of lidocaine toxicity?
The clinical effects of lidocaine overdose are dependent on both the magnitude of the exposure and the rate at which it occurs. The central nervous system (CNS) and heart are the organ systems primarily affected by lidocaine. As a local anesthetic, lidocaine inhibits the action potential formation in electrically excitable cells. This reversible inhibition of the voltage-gated sodium channels prevents the influx of positively charged sodium ions and the resultant depolarization of the cell. Lidocaine, initially or at low concentrations, has a quiescent effect on neurons, which explains its therapeutic use as a local anesthetic.
Similarly, lidocaine initially reduces impulse propagation through the cardiac conduction system and can be used to treat rhythm disturbances. When used for the suppression of cardiac dysrhythmias, the therapeutic serum concentration of lidocaine is 1 to 5 mcg/mL. Subjective symptoms of lidocaine toxicity arising from therapeutic use (primarily when it is used to manage dysrhythmia) include light-headedness, disorientation, confusion, and psychosis, and are associated with serum concentrations of 3 to 6 mcg/mL. As the concentration increases, the clinical effects of lidocaine appear to shift from inhibitory to excitatory. At serum concentrations of 5 to 9 mcg/mL, objective symptoms, such as excitation, tremor, and seizure predominate. As the concentration continues to rise, coma, respiratory depression, cardiovascular (CV) collapse, and death may occur.7
The exact mechanism of this toxicity transition from inhibitory effect of sodium channel blockade to excitatory effect is not well understood. Some suggest a preferential inhibition of inhibitory interneurons in the CNS is responsible.8 Another potential mechanism is a concentration-dependent inhibition of the potassium rectifier channel.9 Inhibition of the efflux of positively charged potassium ions would result in slowing cellular repolarization, leaving the cells in a relatively excitable state. In the CNS, this produces seizures; in the heart, it may result in dysrhythmia. Cardiovascular collapse may occur with very high serum concentrations of lidocaine or following a rapid serum increase—eg, after a large intravenous (IV) bolus dose,7 which can potentially result from an unintentional intravascular injection during tumescent liposuction.
What is the treatment for lidocaine toxicity?
The first step in the treatment of lidocaine-associated CNS toxicity is the discontinuation of the drug. Failure to appropriately recognize the symptoms of early lidocaine toxicity may result in the progression to severe CNS effects and eventual CV collapse. Benzodiazepines should be used as needed for mild symptoms. Seizures should be treated aggressively with benzodiazepines or barbiturates, while ensuring maintenance of oxygenation, ventilation, and perfusion.7
In cases of lidocaine-associated CV toxicity, treatment begins with airway management, oxygen administration, and life support. Potential antidotal treatment of severe local anesthetic-associated CV toxicity involves the rapid administration of IV fat emulsion, or “lipid rescue.” Although best studied for bupivacaine toxicity, the exact mechanism of IV fat emulsion as an antidote is not completely understood. However, in the treatment of local anesthetic toxicity, lipid rescue is believed to offer a “sink” to remove the lipid-soluble anesthetics from their site of action and trap them within the vascular space. Suggested dosing of 20% lipid solution is a bolus of 1.5 mL/kg over a 1-minute period, followed by 0.25mL/kg per minute or 15 mL/kg per hour to run over 30 to 60 minutes.10
Case Conclusion
The patient made a full recovery and was discharged home in normal condition. Her healthcare provider was informed about the complication of the procedure.
Dr Hines is a senior toxicology fellow, department of emergency medicine, New York University School of Medicine. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Lozinski A, Huq NS. Tumescent liposuction. Clin Plastic Surg. 2013;40(4):593-613.
- Klein JA. Tumescent technique chronicles: local anesthesia, liposuction, and beyond. Dermatol Surg. 1995;21(5):449-457.
- Klein JA. Tumescent technique for regional anesthesia permits lidocaine doses of 35 mg/kg for liposuction. J Dermatol Surg Oncol. 1990;16(3):248-263.
- Conroy PH, O’Rourke J. Tumescent anesthesia. The Surgeon. 2012;210-201.
- Rao RR, Fly SF, Hoffman RS. Deaths related to liposuction. N Engl J Med. 1999;340(19):1471-1475.
- Martinez MA, Ballesteros S, Segura LJ, Garcia M. Reporting a fatality during tumescent liposuction. Forensic Sci Int. 2008;178(1):e11-e-16.
- Schwartz DR, Kaufman B. Local anesthestics. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw Hill; 2015:921-930.
- Tanaka K, Yamasaki M. Blocking of cortical inhibitory synapses by intravenous lidoaine. Nature. 1966;209(5019):207-208.
- Friederich P, Benzenberg D, Urban BW. Bupivacaine inhibits human neuronal Kv3 channels in SH-SY5Y human neuroblastoma cells. Br J Anaesth. 2002;88(6):864-866.
- Bania TC. Antidotes in depth, intravenous fat emulsions. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw Hill; 2015:931-937.
Case
A 37-year-old woman had undergone outpatient tumescent liposuction of her abdominal adipose tissue. Upon initiation of the procedure, she developed acute confusion followed by agitation and hallucinations and was transported by emergency medical services to the nearest hospital.
Upon arrival to the ED, the patient was mildly agitated. Her initial vital signs were: blood pressure, 122/79 mm Hg; heart rate, 57 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 96.8°F. Oxygen saturation was 100% on room air.
On examination, the patient was awake and oriented, but was restless and perseverating. She described the sensation of “dying and coming back to life.” The patient’s pupils were equal, round, and normally reactive; her skin was neither diaphoretic nor dry; her heart rate and rhythm were normal; and her lungs were clear to auscultation bilaterally. The skin of the patient’s abdomen was pale, cool, and clammy, but otherwise soft with normoactive bowel sounds. An electrocardiogram revealed sinus rhythm with normal QRS and QTc intervals.
What is tumescent liposuction?
The tumescent technique (the word tumescent derived from the Latin word meaning “to swell”) involves the infiltration of a large volume of a dilute solution of lidocaine and epinephrine into the subcutaneous fat until it becomes distended and tense. The administration of this solution allows for the removal of significant adipose tissue with minimal blood loss and without general anesthesia. Advocates of tumescent liposuction argue that this dilution and subcutaneous infiltration alter the pharmacokinetics of lidocaine, providing a safe delivery of large doses of the drug. In addition, a substantial quantity of the infiltrated lidocaine is suctioned out with the fat removal. Klein3 specifically recommends administering a dilution of 1 g lidocaine in 1,000 mL of saline (0.1%) to the patient in aliquots of up to 2 g lidocaine. Such a high dose may approach or surpass 35 mg/kg, far exceeding the recommended lidocaine infiltration dose of 7 mg/kg when mixed with epinephrine.
Several studies have investigated the safety of the tumescent technique and the risk of lidocaine toxicity. In a review of pharmacokinetic studies, patients received between 10.5 and 67.7 mg/kg of lidocaine with a reported maximum serum lidocaine concentration in all patients of only 2.93 mcg/mL (therapeutic, 1-5 mcg/mL).4 However, peak serum lidocaine concentrations may not occur for up to 28 hours following tumescent infiltration.4 Despite the purported safety findings of this and other studies, there are also reports of tumescent anesthesia-associated toxicity and fatalities.5,6
Case Continuation
The patient’s mental status slowly improved throughout her hospital stay and she was reportedly at baseline by the following morning. Her serum lidocaine concentration drawn at presentation to the ED was 9.7 mcg/mL.
What are the clinical effects of lidocaine toxicity?
The clinical effects of lidocaine overdose are dependent on both the magnitude of the exposure and the rate at which it occurs. The central nervous system (CNS) and heart are the organ systems primarily affected by lidocaine. As a local anesthetic, lidocaine inhibits the action potential formation in electrically excitable cells. This reversible inhibition of the voltage-gated sodium channels prevents the influx of positively charged sodium ions and the resultant depolarization of the cell. Lidocaine, initially or at low concentrations, has a quiescent effect on neurons, which explains its therapeutic use as a local anesthetic.
Similarly, lidocaine initially reduces impulse propagation through the cardiac conduction system and can be used to treat rhythm disturbances. When used for the suppression of cardiac dysrhythmias, the therapeutic serum concentration of lidocaine is 1 to 5 mcg/mL. Subjective symptoms of lidocaine toxicity arising from therapeutic use (primarily when it is used to manage dysrhythmia) include light-headedness, disorientation, confusion, and psychosis, and are associated with serum concentrations of 3 to 6 mcg/mL. As the concentration increases, the clinical effects of lidocaine appear to shift from inhibitory to excitatory. At serum concentrations of 5 to 9 mcg/mL, objective symptoms, such as excitation, tremor, and seizure predominate. As the concentration continues to rise, coma, respiratory depression, cardiovascular (CV) collapse, and death may occur.7
The exact mechanism of this toxicity transition from inhibitory effect of sodium channel blockade to excitatory effect is not well understood. Some suggest a preferential inhibition of inhibitory interneurons in the CNS is responsible.8 Another potential mechanism is a concentration-dependent inhibition of the potassium rectifier channel.9 Inhibition of the efflux of positively charged potassium ions would result in slowing cellular repolarization, leaving the cells in a relatively excitable state. In the CNS, this produces seizures; in the heart, it may result in dysrhythmia. Cardiovascular collapse may occur with very high serum concentrations of lidocaine or following a rapid serum increase—eg, after a large intravenous (IV) bolus dose,7 which can potentially result from an unintentional intravascular injection during tumescent liposuction.
What is the treatment for lidocaine toxicity?
The first step in the treatment of lidocaine-associated CNS toxicity is the discontinuation of the drug. Failure to appropriately recognize the symptoms of early lidocaine toxicity may result in the progression to severe CNS effects and eventual CV collapse. Benzodiazepines should be used as needed for mild symptoms. Seizures should be treated aggressively with benzodiazepines or barbiturates, while ensuring maintenance of oxygenation, ventilation, and perfusion.7
In cases of lidocaine-associated CV toxicity, treatment begins with airway management, oxygen administration, and life support. Potential antidotal treatment of severe local anesthetic-associated CV toxicity involves the rapid administration of IV fat emulsion, or “lipid rescue.” Although best studied for bupivacaine toxicity, the exact mechanism of IV fat emulsion as an antidote is not completely understood. However, in the treatment of local anesthetic toxicity, lipid rescue is believed to offer a “sink” to remove the lipid-soluble anesthetics from their site of action and trap them within the vascular space. Suggested dosing of 20% lipid solution is a bolus of 1.5 mL/kg over a 1-minute period, followed by 0.25mL/kg per minute or 15 mL/kg per hour to run over 30 to 60 minutes.10
Case Conclusion
The patient made a full recovery and was discharged home in normal condition. Her healthcare provider was informed about the complication of the procedure.
Dr Hines is a senior toxicology fellow, department of emergency medicine, New York University School of Medicine. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 37-year-old woman had undergone outpatient tumescent liposuction of her abdominal adipose tissue. Upon initiation of the procedure, she developed acute confusion followed by agitation and hallucinations and was transported by emergency medical services to the nearest hospital.
Upon arrival to the ED, the patient was mildly agitated. Her initial vital signs were: blood pressure, 122/79 mm Hg; heart rate, 57 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 96.8°F. Oxygen saturation was 100% on room air.
On examination, the patient was awake and oriented, but was restless and perseverating. She described the sensation of “dying and coming back to life.” The patient’s pupils were equal, round, and normally reactive; her skin was neither diaphoretic nor dry; her heart rate and rhythm were normal; and her lungs were clear to auscultation bilaterally. The skin of the patient’s abdomen was pale, cool, and clammy, but otherwise soft with normoactive bowel sounds. An electrocardiogram revealed sinus rhythm with normal QRS and QTc intervals.
What is tumescent liposuction?
The tumescent technique (the word tumescent derived from the Latin word meaning “to swell”) involves the infiltration of a large volume of a dilute solution of lidocaine and epinephrine into the subcutaneous fat until it becomes distended and tense. The administration of this solution allows for the removal of significant adipose tissue with minimal blood loss and without general anesthesia. Advocates of tumescent liposuction argue that this dilution and subcutaneous infiltration alter the pharmacokinetics of lidocaine, providing a safe delivery of large doses of the drug. In addition, a substantial quantity of the infiltrated lidocaine is suctioned out with the fat removal. Klein3 specifically recommends administering a dilution of 1 g lidocaine in 1,000 mL of saline (0.1%) to the patient in aliquots of up to 2 g lidocaine. Such a high dose may approach or surpass 35 mg/kg, far exceeding the recommended lidocaine infiltration dose of 7 mg/kg when mixed with epinephrine.
Several studies have investigated the safety of the tumescent technique and the risk of lidocaine toxicity. In a review of pharmacokinetic studies, patients received between 10.5 and 67.7 mg/kg of lidocaine with a reported maximum serum lidocaine concentration in all patients of only 2.93 mcg/mL (therapeutic, 1-5 mcg/mL).4 However, peak serum lidocaine concentrations may not occur for up to 28 hours following tumescent infiltration.4 Despite the purported safety findings of this and other studies, there are also reports of tumescent anesthesia-associated toxicity and fatalities.5,6
Case Continuation
The patient’s mental status slowly improved throughout her hospital stay and she was reportedly at baseline by the following morning. Her serum lidocaine concentration drawn at presentation to the ED was 9.7 mcg/mL.
What are the clinical effects of lidocaine toxicity?
The clinical effects of lidocaine overdose are dependent on both the magnitude of the exposure and the rate at which it occurs. The central nervous system (CNS) and heart are the organ systems primarily affected by lidocaine. As a local anesthetic, lidocaine inhibits the action potential formation in electrically excitable cells. This reversible inhibition of the voltage-gated sodium channels prevents the influx of positively charged sodium ions and the resultant depolarization of the cell. Lidocaine, initially or at low concentrations, has a quiescent effect on neurons, which explains its therapeutic use as a local anesthetic.
Similarly, lidocaine initially reduces impulse propagation through the cardiac conduction system and can be used to treat rhythm disturbances. When used for the suppression of cardiac dysrhythmias, the therapeutic serum concentration of lidocaine is 1 to 5 mcg/mL. Subjective symptoms of lidocaine toxicity arising from therapeutic use (primarily when it is used to manage dysrhythmia) include light-headedness, disorientation, confusion, and psychosis, and are associated with serum concentrations of 3 to 6 mcg/mL. As the concentration increases, the clinical effects of lidocaine appear to shift from inhibitory to excitatory. At serum concentrations of 5 to 9 mcg/mL, objective symptoms, such as excitation, tremor, and seizure predominate. As the concentration continues to rise, coma, respiratory depression, cardiovascular (CV) collapse, and death may occur.7
The exact mechanism of this toxicity transition from inhibitory effect of sodium channel blockade to excitatory effect is not well understood. Some suggest a preferential inhibition of inhibitory interneurons in the CNS is responsible.8 Another potential mechanism is a concentration-dependent inhibition of the potassium rectifier channel.9 Inhibition of the efflux of positively charged potassium ions would result in slowing cellular repolarization, leaving the cells in a relatively excitable state. In the CNS, this produces seizures; in the heart, it may result in dysrhythmia. Cardiovascular collapse may occur with very high serum concentrations of lidocaine or following a rapid serum increase—eg, after a large intravenous (IV) bolus dose,7 which can potentially result from an unintentional intravascular injection during tumescent liposuction.
What is the treatment for lidocaine toxicity?
The first step in the treatment of lidocaine-associated CNS toxicity is the discontinuation of the drug. Failure to appropriately recognize the symptoms of early lidocaine toxicity may result in the progression to severe CNS effects and eventual CV collapse. Benzodiazepines should be used as needed for mild symptoms. Seizures should be treated aggressively with benzodiazepines or barbiturates, while ensuring maintenance of oxygenation, ventilation, and perfusion.7
In cases of lidocaine-associated CV toxicity, treatment begins with airway management, oxygen administration, and life support. Potential antidotal treatment of severe local anesthetic-associated CV toxicity involves the rapid administration of IV fat emulsion, or “lipid rescue.” Although best studied for bupivacaine toxicity, the exact mechanism of IV fat emulsion as an antidote is not completely understood. However, in the treatment of local anesthetic toxicity, lipid rescue is believed to offer a “sink” to remove the lipid-soluble anesthetics from their site of action and trap them within the vascular space. Suggested dosing of 20% lipid solution is a bolus of 1.5 mL/kg over a 1-minute period, followed by 0.25mL/kg per minute or 15 mL/kg per hour to run over 30 to 60 minutes.10
Case Conclusion
The patient made a full recovery and was discharged home in normal condition. Her healthcare provider was informed about the complication of the procedure.
Dr Hines is a senior toxicology fellow, department of emergency medicine, New York University School of Medicine. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Lozinski A, Huq NS. Tumescent liposuction. Clin Plastic Surg. 2013;40(4):593-613.
- Klein JA. Tumescent technique chronicles: local anesthesia, liposuction, and beyond. Dermatol Surg. 1995;21(5):449-457.
- Klein JA. Tumescent technique for regional anesthesia permits lidocaine doses of 35 mg/kg for liposuction. J Dermatol Surg Oncol. 1990;16(3):248-263.
- Conroy PH, O’Rourke J. Tumescent anesthesia. The Surgeon. 2012;210-201.
- Rao RR, Fly SF, Hoffman RS. Deaths related to liposuction. N Engl J Med. 1999;340(19):1471-1475.
- Martinez MA, Ballesteros S, Segura LJ, Garcia M. Reporting a fatality during tumescent liposuction. Forensic Sci Int. 2008;178(1):e11-e-16.
- Schwartz DR, Kaufman B. Local anesthestics. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw Hill; 2015:921-930.
- Tanaka K, Yamasaki M. Blocking of cortical inhibitory synapses by intravenous lidoaine. Nature. 1966;209(5019):207-208.
- Friederich P, Benzenberg D, Urban BW. Bupivacaine inhibits human neuronal Kv3 channels in SH-SY5Y human neuroblastoma cells. Br J Anaesth. 2002;88(6):864-866.
- Bania TC. Antidotes in depth, intravenous fat emulsions. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw Hill; 2015:931-937.
- Lozinski A, Huq NS. Tumescent liposuction. Clin Plastic Surg. 2013;40(4):593-613.
- Klein JA. Tumescent technique chronicles: local anesthesia, liposuction, and beyond. Dermatol Surg. 1995;21(5):449-457.
- Klein JA. Tumescent technique for regional anesthesia permits lidocaine doses of 35 mg/kg for liposuction. J Dermatol Surg Oncol. 1990;16(3):248-263.
- Conroy PH, O’Rourke J. Tumescent anesthesia. The Surgeon. 2012;210-201.
- Rao RR, Fly SF, Hoffman RS. Deaths related to liposuction. N Engl J Med. 1999;340(19):1471-1475.
- Martinez MA, Ballesteros S, Segura LJ, Garcia M. Reporting a fatality during tumescent liposuction. Forensic Sci Int. 2008;178(1):e11-e-16.
- Schwartz DR, Kaufman B. Local anesthestics. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw Hill; 2015:921-930.
- Tanaka K, Yamasaki M. Blocking of cortical inhibitory synapses by intravenous lidoaine. Nature. 1966;209(5019):207-208.
- Friederich P, Benzenberg D, Urban BW. Bupivacaine inhibits human neuronal Kv3 channels in SH-SY5Y human neuroblastoma cells. Br J Anaesth. 2002;88(6):864-866.
- Bania TC. Antidotes in depth, intravenous fat emulsions. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw Hill; 2015:931-937.

Case Report: An Unusual Case of Morel-Lavallée Lesion of the Upper Extremity
Case
A 32-year-old previously healthy woman presented to the ED with right upper arm pain and swelling of 6 days duration. According to the patient, the swelling began 2 days after she sustained a work-related injury at a coin-manufacturing factory. She stated that her right arm had gotten caught inside of a rolling press while she was cleaning it. The roller had stopped over her upper arm, trapping it between the roller and the platform for several minutes before it was extricated. She was brought to the ED by emergency medical services for evaluation immediately following this incident. At this first visit to the ED, the patient complained of mild pain in her right arm. Physical examination at that time revealed mild diffuse swelling extending from her hand to her distal humerus, with mild pain on passive flexion, and extension without associated numbness or tingling. Plain films of her right upper extremity were ordered, the results of which were relatively unremarkable. She was evaluated by an orthopedist, who ruled out compartment syndrome based on her benign physical examination and soft compartments. She was ultimately discharged and told to follow up with her primary care provider.
Over the course of 48 hours from the first ED visit, the patient developed large bullae on the dorsal and volar aspect of her forearm, elbow, and upper arm with associated pain. In addition to dark discolorations of the skin over her affected arm, she noticed that the bullae had become numerous and discolored. These symptoms continued to grow progressively worse, prompting her second presentation to the ED.
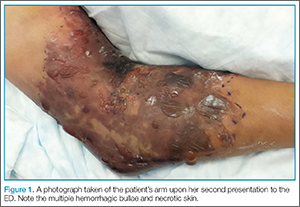
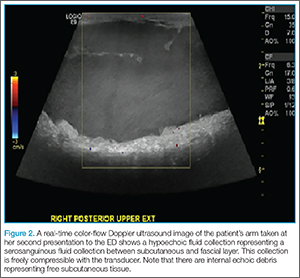
The patient was taken to the operating room and underwent debridement and resection of the circumferential necrotic skin and subcutaneous tissue in her right arm, and the placement of a skin graft with overlying wound vacuum-assisted closure. During the procedure, a large amount of serosanguinous fluid was drained and cultured, and was found to be sterile. Due to the size of her injury, she underwent two additional episodes of debridement and graft placement over the course of the next 2 weeks.
Discussion
First described in the 1850s by the French physician Maurice Morel-Lavallée, Morel-Lavallée lesion is a rare, traumatic, soft-tissue injury.1 It is an internal degloving injury wherein the skin and subcutaneous tissue have been forcibly separated from the underlying fascia as a result of shear stress. The lymphatic and blood vessels between the layers are also disrupted in this process, resulting in the accumulation of blood and lymphatic fluid as well as subcutaneous debris in the potential space that forms. Excess accumulation over time can compromise blood supply to the overlying skin and cause necrosis.2 Morel-Lavallée lesion is missed on initial evaluation in up to one-third of the cases and may have a delay in presentation ranging from hours to months after the inciting injury.3
Morel-Lavallée lesions typically involve the flank, hips, thigh, and prepatellar regions as a result of shear injuries sustained from bicycle falls and motor vehicle accidents.4 These lesions are often associated with concomitant acetabular and pelvic fractures.5 Involvement of the upper extremities is unusual. Typically, presentation consists of a fluctuant and painful mass underneath the skin which increases over time. The overlying skin may show the mechanism of the original injury, for example, as abrasions after a crush injury. The excessive skin necrosis and hemorrhagic bullae seen in this particular case is a very rare presentation.
Differential Diagnosis
The differential diagnosis includes compartment syndrome, coma blisters, a missed fracture, bullous pemphigoid, bullous drug reactions, and linear immunoglobulin A disease. Most of these conditions were easily ruled out in this case as the patient was previously healthy and not on any medications. The lesions in this case could have been confused with coma blisters, which are similar in appearance, self-limiting, and can develop on the extremities. However, coma blisters are classically associated with toxicity from various central nervous system depressants, as well as reduced consciousness from other causes—all of which were readily ruled-out based on the patient’s history. Moreover, the Morel-Lavallée lesion is a degloving injury of the subcutaneous tissue from the fascia underneath, whereas the pathology of coma blisters includes subepidermal bullae formation as well as immunoglobulin and complement deposition.6
Diagnostic Imaging
Morel-Lavallée lesion can often be confirmed via several imaging modalities, including ultrasound, CT, 3D CT, or magnetic resonance imaging (MRI).3,7 Ultrasound will usually show a well-circumscribed hypoechoic fluid collection with hyperechoic fat globules from the subcutaneous tissue, whereas CT tends to show an encapsulated mass with fluid collection underneath. In MRI, Morel-Lavallée lesion often appears as a hypointense T1-sequence and hyperintense T2-sequence similar to most other fluid collections. There may be variable T1- and T2-intensities with subcutaneous tissues in the fluid collection.2
Management
Despite recognition of this disease entity, controversies still exist regarding management. Case reports have demonstrated a relatively high rate of infected fluid collections depending on the chronicity of the injury.8 A recent algorithm to management described by Nickerson et al4 proposes that for patients with viable skin, percutaneous aspiration of more than 50 cc of fluid from these lesions should be treated with more extensive operative intervention based on the increased likelihood of recurrence. Patients without viable skin require formal debridement with possible skin grafting.
Other treatment options include conservative management, surgical drainage, sclerodesis, and extensive open surgery.8-10 Management is always case-based and dependent upon the size of the lesion and associated injuries.
Conclusion
This case represents an example of Morel-Lavallée lesions in their most severe and atypical form. It also serves as a reminder that vigilance and knowledge of this disease process is important in helping to diagnose this rare but potentially devastating condition. The key to recognizing this injury lies in keeping this disease process in the differential diagnosis of traumatic injuries with suspicious mechanism involving significant shear forces. Significant physical examination findings may not be present initially and evolve over a time period of hours to days. Once this injury is identified, management hinges on the size of the lesion and affected body part. Despite timely interventions, Morel-Lavallée lesions may result in significant morbidity and functional disability.
Dr Ye is an emergency medicine resident at the Brown Alpert Medical School in Providence, Rhode Island. Dr Rosenberg is a clinical assistant professor at Brown Alpert Medical School, and an emergency medicine attending physican at Rhode Island Hospital and The Miriam Hospital, Providence, Rhode Island.
- Morel-Lavallée M. Epanchements traumatique de serosite. Arc Générales Méd. 1853;691-731.
- Chokshi F, Jose J, Clifford P. Morel Lavallée Lesion. Am J Orthop (Belle Mead NJ). 2010;39(5): 252-253.
- Bonilla-Yoon I, Masih S, Patel DB, et al. The Morel-Lavallée lesion: pathophysiology, clinical presentation, imaging features, and treatment options. Emerg Radiol. 2014;21(1):35-43.
- Nickerson T, Zielinski M, Jenkins D, Schiller HJ. The Mayo Clinic experience with Morel-Lavallée lesions: establishment of a practice management guideline. J Trauma Acute Care Surg. 2014:76(2);493-497.
- Powers ML, Hatem SF, Sundaram M. Morel-Lavallee lesion. Orthopedics. 2007;30(4):322-323.
- Agarwal A, Bansal M, Conner K. Coma blisters with hypoxemic respiratory failure. Dermatol Online Journal. 2012:18(3);10.
- Reddix RN, Carrol E, Webb LX. Early diagnosis of a Morel-Lavallee lesion using three-dimensional computed tomography reconstructions: a case report. J Trauma. 2009;67(2):e57-e59.
- Lin HL, Lee WC, Kuo LC, Chen CW. Closed internal degloving injury with conservative treatment. Am J Emerg Med. 2008:26(2);254.e5-e6.
- Luria S, Applbaum Y,Weil Y, Liebergall M, Peyser A. Talc sclerodhesis of persistent Morel-Lavallée lesions (posttraumatic pseudocysts): case report of 4 patients. J Orthop Trauma. 2006;20(6):435-438.
- Penaud A, Quignon R, Danin A, Bahé L, Zakine G. Alcohol sclerodhesis: an innovative treatment for chronic Morel-Lavallée lesions. J Plast Reconstr Aesthet Surg. 2011;64(10): e262-264.
Case
A 32-year-old previously healthy woman presented to the ED with right upper arm pain and swelling of 6 days duration. According to the patient, the swelling began 2 days after she sustained a work-related injury at a coin-manufacturing factory. She stated that her right arm had gotten caught inside of a rolling press while she was cleaning it. The roller had stopped over her upper arm, trapping it between the roller and the platform for several minutes before it was extricated. She was brought to the ED by emergency medical services for evaluation immediately following this incident. At this first visit to the ED, the patient complained of mild pain in her right arm. Physical examination at that time revealed mild diffuse swelling extending from her hand to her distal humerus, with mild pain on passive flexion, and extension without associated numbness or tingling. Plain films of her right upper extremity were ordered, the results of which were relatively unremarkable. She was evaluated by an orthopedist, who ruled out compartment syndrome based on her benign physical examination and soft compartments. She was ultimately discharged and told to follow up with her primary care provider.
Over the course of 48 hours from the first ED visit, the patient developed large bullae on the dorsal and volar aspect of her forearm, elbow, and upper arm with associated pain. In addition to dark discolorations of the skin over her affected arm, she noticed that the bullae had become numerous and discolored. These symptoms continued to grow progressively worse, prompting her second presentation to the ED.
The patient was taken to the operating room and underwent debridement and resection of the circumferential necrotic skin and subcutaneous tissue in her right arm, and the placement of a skin graft with overlying wound vacuum-assisted closure. During the procedure, a large amount of serosanguinous fluid was drained and cultured, and was found to be sterile. Due to the size of her injury, she underwent two additional episodes of debridement and graft placement over the course of the next 2 weeks.
Discussion
First described in the 1850s by the French physician Maurice Morel-Lavallée, Morel-Lavallée lesion is a rare, traumatic, soft-tissue injury.1 It is an internal degloving injury wherein the skin and subcutaneous tissue have been forcibly separated from the underlying fascia as a result of shear stress. The lymphatic and blood vessels between the layers are also disrupted in this process, resulting in the accumulation of blood and lymphatic fluid as well as subcutaneous debris in the potential space that forms. Excess accumulation over time can compromise blood supply to the overlying skin and cause necrosis.2 Morel-Lavallée lesion is missed on initial evaluation in up to one-third of the cases and may have a delay in presentation ranging from hours to months after the inciting injury.3
Morel-Lavallée lesions typically involve the flank, hips, thigh, and prepatellar regions as a result of shear injuries sustained from bicycle falls and motor vehicle accidents.4 These lesions are often associated with concomitant acetabular and pelvic fractures.5 Involvement of the upper extremities is unusual. Typically, presentation consists of a fluctuant and painful mass underneath the skin which increases over time. The overlying skin may show the mechanism of the original injury, for example, as abrasions after a crush injury. The excessive skin necrosis and hemorrhagic bullae seen in this particular case is a very rare presentation.
Differential Diagnosis
The differential diagnosis includes compartment syndrome, coma blisters, a missed fracture, bullous pemphigoid, bullous drug reactions, and linear immunoglobulin A disease. Most of these conditions were easily ruled out in this case as the patient was previously healthy and not on any medications. The lesions in this case could have been confused with coma blisters, which are similar in appearance, self-limiting, and can develop on the extremities. However, coma blisters are classically associated with toxicity from various central nervous system depressants, as well as reduced consciousness from other causes—all of which were readily ruled-out based on the patient’s history. Moreover, the Morel-Lavallée lesion is a degloving injury of the subcutaneous tissue from the fascia underneath, whereas the pathology of coma blisters includes subepidermal bullae formation as well as immunoglobulin and complement deposition.6
Diagnostic Imaging
Morel-Lavallée lesion can often be confirmed via several imaging modalities, including ultrasound, CT, 3D CT, or magnetic resonance imaging (MRI).3,7 Ultrasound will usually show a well-circumscribed hypoechoic fluid collection with hyperechoic fat globules from the subcutaneous tissue, whereas CT tends to show an encapsulated mass with fluid collection underneath. In MRI, Morel-Lavallée lesion often appears as a hypointense T1-sequence and hyperintense T2-sequence similar to most other fluid collections. There may be variable T1- and T2-intensities with subcutaneous tissues in the fluid collection.2
Management
Despite recognition of this disease entity, controversies still exist regarding management. Case reports have demonstrated a relatively high rate of infected fluid collections depending on the chronicity of the injury.8 A recent algorithm to management described by Nickerson et al4 proposes that for patients with viable skin, percutaneous aspiration of more than 50 cc of fluid from these lesions should be treated with more extensive operative intervention based on the increased likelihood of recurrence. Patients without viable skin require formal debridement with possible skin grafting.
Other treatment options include conservative management, surgical drainage, sclerodesis, and extensive open surgery.8-10 Management is always case-based and dependent upon the size of the lesion and associated injuries.
Conclusion
This case represents an example of Morel-Lavallée lesions in their most severe and atypical form. It also serves as a reminder that vigilance and knowledge of this disease process is important in helping to diagnose this rare but potentially devastating condition. The key to recognizing this injury lies in keeping this disease process in the differential diagnosis of traumatic injuries with suspicious mechanism involving significant shear forces. Significant physical examination findings may not be present initially and evolve over a time period of hours to days. Once this injury is identified, management hinges on the size of the lesion and affected body part. Despite timely interventions, Morel-Lavallée lesions may result in significant morbidity and functional disability.
Dr Ye is an emergency medicine resident at the Brown Alpert Medical School in Providence, Rhode Island. Dr Rosenberg is a clinical assistant professor at Brown Alpert Medical School, and an emergency medicine attending physican at Rhode Island Hospital and The Miriam Hospital, Providence, Rhode Island.
Case
A 32-year-old previously healthy woman presented to the ED with right upper arm pain and swelling of 6 days duration. According to the patient, the swelling began 2 days after she sustained a work-related injury at a coin-manufacturing factory. She stated that her right arm had gotten caught inside of a rolling press while she was cleaning it. The roller had stopped over her upper arm, trapping it between the roller and the platform for several minutes before it was extricated. She was brought to the ED by emergency medical services for evaluation immediately following this incident. At this first visit to the ED, the patient complained of mild pain in her right arm. Physical examination at that time revealed mild diffuse swelling extending from her hand to her distal humerus, with mild pain on passive flexion, and extension without associated numbness or tingling. Plain films of her right upper extremity were ordered, the results of which were relatively unremarkable. She was evaluated by an orthopedist, who ruled out compartment syndrome based on her benign physical examination and soft compartments. She was ultimately discharged and told to follow up with her primary care provider.
Over the course of 48 hours from the first ED visit, the patient developed large bullae on the dorsal and volar aspect of her forearm, elbow, and upper arm with associated pain. In addition to dark discolorations of the skin over her affected arm, she noticed that the bullae had become numerous and discolored. These symptoms continued to grow progressively worse, prompting her second presentation to the ED.
The patient was taken to the operating room and underwent debridement and resection of the circumferential necrotic skin and subcutaneous tissue in her right arm, and the placement of a skin graft with overlying wound vacuum-assisted closure. During the procedure, a large amount of serosanguinous fluid was drained and cultured, and was found to be sterile. Due to the size of her injury, she underwent two additional episodes of debridement and graft placement over the course of the next 2 weeks.
Discussion
First described in the 1850s by the French physician Maurice Morel-Lavallée, Morel-Lavallée lesion is a rare, traumatic, soft-tissue injury.1 It is an internal degloving injury wherein the skin and subcutaneous tissue have been forcibly separated from the underlying fascia as a result of shear stress. The lymphatic and blood vessels between the layers are also disrupted in this process, resulting in the accumulation of blood and lymphatic fluid as well as subcutaneous debris in the potential space that forms. Excess accumulation over time can compromise blood supply to the overlying skin and cause necrosis.2 Morel-Lavallée lesion is missed on initial evaluation in up to one-third of the cases and may have a delay in presentation ranging from hours to months after the inciting injury.3
Morel-Lavallée lesions typically involve the flank, hips, thigh, and prepatellar regions as a result of shear injuries sustained from bicycle falls and motor vehicle accidents.4 These lesions are often associated with concomitant acetabular and pelvic fractures.5 Involvement of the upper extremities is unusual. Typically, presentation consists of a fluctuant and painful mass underneath the skin which increases over time. The overlying skin may show the mechanism of the original injury, for example, as abrasions after a crush injury. The excessive skin necrosis and hemorrhagic bullae seen in this particular case is a very rare presentation.
Differential Diagnosis
The differential diagnosis includes compartment syndrome, coma blisters, a missed fracture, bullous pemphigoid, bullous drug reactions, and linear immunoglobulin A disease. Most of these conditions were easily ruled out in this case as the patient was previously healthy and not on any medications. The lesions in this case could have been confused with coma blisters, which are similar in appearance, self-limiting, and can develop on the extremities. However, coma blisters are classically associated with toxicity from various central nervous system depressants, as well as reduced consciousness from other causes—all of which were readily ruled-out based on the patient’s history. Moreover, the Morel-Lavallée lesion is a degloving injury of the subcutaneous tissue from the fascia underneath, whereas the pathology of coma blisters includes subepidermal bullae formation as well as immunoglobulin and complement deposition.6
Diagnostic Imaging
Morel-Lavallée lesion can often be confirmed via several imaging modalities, including ultrasound, CT, 3D CT, or magnetic resonance imaging (MRI).3,7 Ultrasound will usually show a well-circumscribed hypoechoic fluid collection with hyperechoic fat globules from the subcutaneous tissue, whereas CT tends to show an encapsulated mass with fluid collection underneath. In MRI, Morel-Lavallée lesion often appears as a hypointense T1-sequence and hyperintense T2-sequence similar to most other fluid collections. There may be variable T1- and T2-intensities with subcutaneous tissues in the fluid collection.2
Management
Despite recognition of this disease entity, controversies still exist regarding management. Case reports have demonstrated a relatively high rate of infected fluid collections depending on the chronicity of the injury.8 A recent algorithm to management described by Nickerson et al4 proposes that for patients with viable skin, percutaneous aspiration of more than 50 cc of fluid from these lesions should be treated with more extensive operative intervention based on the increased likelihood of recurrence. Patients without viable skin require formal debridement with possible skin grafting.
Other treatment options include conservative management, surgical drainage, sclerodesis, and extensive open surgery.8-10 Management is always case-based and dependent upon the size of the lesion and associated injuries.
Conclusion
This case represents an example of Morel-Lavallée lesions in their most severe and atypical form. It also serves as a reminder that vigilance and knowledge of this disease process is important in helping to diagnose this rare but potentially devastating condition. The key to recognizing this injury lies in keeping this disease process in the differential diagnosis of traumatic injuries with suspicious mechanism involving significant shear forces. Significant physical examination findings may not be present initially and evolve over a time period of hours to days. Once this injury is identified, management hinges on the size of the lesion and affected body part. Despite timely interventions, Morel-Lavallée lesions may result in significant morbidity and functional disability.
Dr Ye is an emergency medicine resident at the Brown Alpert Medical School in Providence, Rhode Island. Dr Rosenberg is a clinical assistant professor at Brown Alpert Medical School, and an emergency medicine attending physican at Rhode Island Hospital and The Miriam Hospital, Providence, Rhode Island.
- Morel-Lavallée M. Epanchements traumatique de serosite. Arc Générales Méd. 1853;691-731.
- Chokshi F, Jose J, Clifford P. Morel Lavallée Lesion. Am J Orthop (Belle Mead NJ). 2010;39(5): 252-253.
- Bonilla-Yoon I, Masih S, Patel DB, et al. The Morel-Lavallée lesion: pathophysiology, clinical presentation, imaging features, and treatment options. Emerg Radiol. 2014;21(1):35-43.
- Nickerson T, Zielinski M, Jenkins D, Schiller HJ. The Mayo Clinic experience with Morel-Lavallée lesions: establishment of a practice management guideline. J Trauma Acute Care Surg. 2014:76(2);493-497.
- Powers ML, Hatem SF, Sundaram M. Morel-Lavallee lesion. Orthopedics. 2007;30(4):322-323.
- Agarwal A, Bansal M, Conner K. Coma blisters with hypoxemic respiratory failure. Dermatol Online Journal. 2012:18(3);10.
- Reddix RN, Carrol E, Webb LX. Early diagnosis of a Morel-Lavallee lesion using three-dimensional computed tomography reconstructions: a case report. J Trauma. 2009;67(2):e57-e59.
- Lin HL, Lee WC, Kuo LC, Chen CW. Closed internal degloving injury with conservative treatment. Am J Emerg Med. 2008:26(2);254.e5-e6.
- Luria S, Applbaum Y,Weil Y, Liebergall M, Peyser A. Talc sclerodhesis of persistent Morel-Lavallée lesions (posttraumatic pseudocysts): case report of 4 patients. J Orthop Trauma. 2006;20(6):435-438.
- Penaud A, Quignon R, Danin A, Bahé L, Zakine G. Alcohol sclerodhesis: an innovative treatment for chronic Morel-Lavallée lesions. J Plast Reconstr Aesthet Surg. 2011;64(10): e262-264.
- Morel-Lavallée M. Epanchements traumatique de serosite. Arc Générales Méd. 1853;691-731.
- Chokshi F, Jose J, Clifford P. Morel Lavallée Lesion. Am J Orthop (Belle Mead NJ). 2010;39(5): 252-253.
- Bonilla-Yoon I, Masih S, Patel DB, et al. The Morel-Lavallée lesion: pathophysiology, clinical presentation, imaging features, and treatment options. Emerg Radiol. 2014;21(1):35-43.
- Nickerson T, Zielinski M, Jenkins D, Schiller HJ. The Mayo Clinic experience with Morel-Lavallée lesions: establishment of a practice management guideline. J Trauma Acute Care Surg. 2014:76(2);493-497.
- Powers ML, Hatem SF, Sundaram M. Morel-Lavallee lesion. Orthopedics. 2007;30(4):322-323.
- Agarwal A, Bansal M, Conner K. Coma blisters with hypoxemic respiratory failure. Dermatol Online Journal. 2012:18(3);10.
- Reddix RN, Carrol E, Webb LX. Early diagnosis of a Morel-Lavallee lesion using three-dimensional computed tomography reconstructions: a case report. J Trauma. 2009;67(2):e57-e59.
- Lin HL, Lee WC, Kuo LC, Chen CW. Closed internal degloving injury with conservative treatment. Am J Emerg Med. 2008:26(2);254.e5-e6.
- Luria S, Applbaum Y,Weil Y, Liebergall M, Peyser A. Talc sclerodhesis of persistent Morel-Lavallée lesions (posttraumatic pseudocysts): case report of 4 patients. J Orthop Trauma. 2006;20(6):435-438.
- Penaud A, Quignon R, Danin A, Bahé L, Zakine G. Alcohol sclerodhesis: an innovative treatment for chronic Morel-Lavallée lesions. J Plast Reconstr Aesthet Surg. 2011;64(10): e262-264.
When it's beneficial to defer dialysis
THE CASE
A 94-year-old Hispanic man with hypertension, congestive heart failure (CHF), anemia of chronic disease, and end-stage renal disease (ESRD) presented to our facility with weakness and shortness of breath. We diagnosed a CHF exacerbation. Initially, he exhibited some respiratory distress that required observation in the coronary care unit and bi-level positive airway pressure therapy to maintain oxygen saturation. Our patient was then moved to a step-down unit where his primary caregiver, his granddaughter, told the medical team that he was limited at home in some of his instrumental activities of daily living. Specifically, he was unable to prepare meals or manage his finances on his own.
Nephrology was consulted for consideration of hemodialysis (HD) because our patient’s creatinine on admission was 7.2 mg/dL (normal for men is 0.7-1.3 mg/dL) and his estimated glomerular filtration rate (GFR) was 7 mL/min (normal is 90-120 mL/min). The patient’s family was conflicted over whether or not to start HD. Palliative Care was consulted to help establish goals of care.
A decision is made. In light of the patient’s limited functional status and his expressed desire to stay at home with his family and receive limited medical care there, the Nephrology and Palliative Care teams recommended delaying HD despite the patient’s worsening renal function. The patient was discharged home with home care services, and he and the family were instructed to follow up with Nephrology for supportive renal management.
DISCUSSION
The decision to delay HD in patients with ESRD is a difficult one that requires shared decision-making between patients and medical providers. Palliative Care consultation services are often involved in this process.
Recent literature supports an “intent-to-defer” based on an evaluation of the patient’s functionality. This represents a paradigm shift from the previous “intent-to-start-early” treatment strategy. In fact, rather than starting early, the Canadian Society of Nephrology recommends delaying initiation of HD in patients with a GFR <15 mL/min.1 Close monitoring of these patients by both a primary care physician and nephrologist is essential.
When considering initiation of HD, it’s important to look at the overall benefit of this intervention in light of the patient’s mortality risk and quality of life. Many patients who receive HD—especially the elderly—report that it takes more than 6 hours to recover following a dialysis treatment.2
Not surprisingly, depression is common in elderly HD patients. Compared to their younger cohorts, older HD patients have a 62% increased risk of developing depression.3 Also, patients who are considered frail and are receiving HD have more than 3 times the mortality risk within one year than those who are not (hazard ratio=3.42; 95% confidence interval, 2.45-4.76).4 (The researchers’ definition of frailty included poor self-reported physical function, exhaustion/fatigue, low physical activity, and undernutrition.4)
Functional status. Although a patient’s age should not be a limiting factor for HD referral, functional status should be considered. Patients with limited functionality and significant dependence have an increased risk of death during the first year of HD.5
Palliative approach gains acceptance. It is becoming more accepted within the nephrology community to consider a palliative approach to patients with ESRD. Organizations such as the Renal Physicians Association recommend effective prognostication, early advanced care planning, forgoing HD in patients with a poor prognosis, and involving Palliative Care early in the decision-making process.6 Aligning the patient’s goals of care with the appropriate treatment method—particularly in patients with comorbid conditions—is an important practice when caring for those with limited life expectancy and functionality.7
THE TAKEAWAY
Intent-to-defer HD may be a preferred strategy when caring for many patients with ESRD. Taking into consideration a patient’s comorbidities and functional status, while considering mortality risk and quality of life are essential. Involving palliative care and nephrology specialists can help patients and families understand HD and make an educated decision regarding when to start it.
1. Nesrallah GE, Mustafa RA, Clark WF, et al; Canadian Society of Nephrology. Canadian Society of Nephrology 2014 clinical practice guideline for timing the initiation of chronic dialysis. CMAJ. 2014;186:112-117.
2. Rayner HC, Zepel L, Fuller DS, et al. Recovery time, quality of life, and mortality in hemodialysis patients: the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am J Kidney Dis. 2014;64:86-94.
3. Canaud B, Tong L, Tentori F, et al. Clinical practices and outcomes in elderly hemodialysis patients: results from the Dialysis Outcomes and Practice Patterns Study (DOPPS). Clin J Am Soc Nephrol. 2011;6:1651-1662.
4. Johansen KL, Chertow GM, Jin C, et al. Significance of frailty among dialysis patients. J Am Soc Nephrol. 2007;18:2960-2967.
5. Joly D, Anglicheau D, Alberti C, et al. Octogenarians reaching end-stage renal disease: cohort study of decision-making and clinical outcomes. J Am Soc Nephrol. 2003;14:1012-1021.
6. Renal Physicians Association. Shared decision-making in the appropriate initiation of and withdrawal from dialysis: clinical practice guideline. 2nd ed. Rockville, MD: Renal Physicians Association; 2010.
7. Grubbs V, Moss AH, Cohen LM, et al; Dialysis Advisory Group of the American Society of Nephrology. A palliative approach to dialysis care: a patient-centered transition to the end of life. Clin J Am Soc Nephrol. 2014;9:2203-2209.
THE CASE
A 94-year-old Hispanic man with hypertension, congestive heart failure (CHF), anemia of chronic disease, and end-stage renal disease (ESRD) presented to our facility with weakness and shortness of breath. We diagnosed a CHF exacerbation. Initially, he exhibited some respiratory distress that required observation in the coronary care unit and bi-level positive airway pressure therapy to maintain oxygen saturation. Our patient was then moved to a step-down unit where his primary caregiver, his granddaughter, told the medical team that he was limited at home in some of his instrumental activities of daily living. Specifically, he was unable to prepare meals or manage his finances on his own.
Nephrology was consulted for consideration of hemodialysis (HD) because our patient’s creatinine on admission was 7.2 mg/dL (normal for men is 0.7-1.3 mg/dL) and his estimated glomerular filtration rate (GFR) was 7 mL/min (normal is 90-120 mL/min). The patient’s family was conflicted over whether or not to start HD. Palliative Care was consulted to help establish goals of care.
A decision is made. In light of the patient’s limited functional status and his expressed desire to stay at home with his family and receive limited medical care there, the Nephrology and Palliative Care teams recommended delaying HD despite the patient’s worsening renal function. The patient was discharged home with home care services, and he and the family were instructed to follow up with Nephrology for supportive renal management.
DISCUSSION
The decision to delay HD in patients with ESRD is a difficult one that requires shared decision-making between patients and medical providers. Palliative Care consultation services are often involved in this process.
Recent literature supports an “intent-to-defer” based on an evaluation of the patient’s functionality. This represents a paradigm shift from the previous “intent-to-start-early” treatment strategy. In fact, rather than starting early, the Canadian Society of Nephrology recommends delaying initiation of HD in patients with a GFR <15 mL/min.1 Close monitoring of these patients by both a primary care physician and nephrologist is essential.
When considering initiation of HD, it’s important to look at the overall benefit of this intervention in light of the patient’s mortality risk and quality of life. Many patients who receive HD—especially the elderly—report that it takes more than 6 hours to recover following a dialysis treatment.2
Not surprisingly, depression is common in elderly HD patients. Compared to their younger cohorts, older HD patients have a 62% increased risk of developing depression.3 Also, patients who are considered frail and are receiving HD have more than 3 times the mortality risk within one year than those who are not (hazard ratio=3.42; 95% confidence interval, 2.45-4.76).4 (The researchers’ definition of frailty included poor self-reported physical function, exhaustion/fatigue, low physical activity, and undernutrition.4)
Functional status. Although a patient’s age should not be a limiting factor for HD referral, functional status should be considered. Patients with limited functionality and significant dependence have an increased risk of death during the first year of HD.5
Palliative approach gains acceptance. It is becoming more accepted within the nephrology community to consider a palliative approach to patients with ESRD. Organizations such as the Renal Physicians Association recommend effective prognostication, early advanced care planning, forgoing HD in patients with a poor prognosis, and involving Palliative Care early in the decision-making process.6 Aligning the patient’s goals of care with the appropriate treatment method—particularly in patients with comorbid conditions—is an important practice when caring for those with limited life expectancy and functionality.7
THE TAKEAWAY
Intent-to-defer HD may be a preferred strategy when caring for many patients with ESRD. Taking into consideration a patient’s comorbidities and functional status, while considering mortality risk and quality of life are essential. Involving palliative care and nephrology specialists can help patients and families understand HD and make an educated decision regarding when to start it.
THE CASE
A 94-year-old Hispanic man with hypertension, congestive heart failure (CHF), anemia of chronic disease, and end-stage renal disease (ESRD) presented to our facility with weakness and shortness of breath. We diagnosed a CHF exacerbation. Initially, he exhibited some respiratory distress that required observation in the coronary care unit and bi-level positive airway pressure therapy to maintain oxygen saturation. Our patient was then moved to a step-down unit where his primary caregiver, his granddaughter, told the medical team that he was limited at home in some of his instrumental activities of daily living. Specifically, he was unable to prepare meals or manage his finances on his own.
Nephrology was consulted for consideration of hemodialysis (HD) because our patient’s creatinine on admission was 7.2 mg/dL (normal for men is 0.7-1.3 mg/dL) and his estimated glomerular filtration rate (GFR) was 7 mL/min (normal is 90-120 mL/min). The patient’s family was conflicted over whether or not to start HD. Palliative Care was consulted to help establish goals of care.
A decision is made. In light of the patient’s limited functional status and his expressed desire to stay at home with his family and receive limited medical care there, the Nephrology and Palliative Care teams recommended delaying HD despite the patient’s worsening renal function. The patient was discharged home with home care services, and he and the family were instructed to follow up with Nephrology for supportive renal management.
DISCUSSION
The decision to delay HD in patients with ESRD is a difficult one that requires shared decision-making between patients and medical providers. Palliative Care consultation services are often involved in this process.
Recent literature supports an “intent-to-defer” based on an evaluation of the patient’s functionality. This represents a paradigm shift from the previous “intent-to-start-early” treatment strategy. In fact, rather than starting early, the Canadian Society of Nephrology recommends delaying initiation of HD in patients with a GFR <15 mL/min.1 Close monitoring of these patients by both a primary care physician and nephrologist is essential.
When considering initiation of HD, it’s important to look at the overall benefit of this intervention in light of the patient’s mortality risk and quality of life. Many patients who receive HD—especially the elderly—report that it takes more than 6 hours to recover following a dialysis treatment.2
Not surprisingly, depression is common in elderly HD patients. Compared to their younger cohorts, older HD patients have a 62% increased risk of developing depression.3 Also, patients who are considered frail and are receiving HD have more than 3 times the mortality risk within one year than those who are not (hazard ratio=3.42; 95% confidence interval, 2.45-4.76).4 (The researchers’ definition of frailty included poor self-reported physical function, exhaustion/fatigue, low physical activity, and undernutrition.4)
Functional status. Although a patient’s age should not be a limiting factor for HD referral, functional status should be considered. Patients with limited functionality and significant dependence have an increased risk of death during the first year of HD.5
Palliative approach gains acceptance. It is becoming more accepted within the nephrology community to consider a palliative approach to patients with ESRD. Organizations such as the Renal Physicians Association recommend effective prognostication, early advanced care planning, forgoing HD in patients with a poor prognosis, and involving Palliative Care early in the decision-making process.6 Aligning the patient’s goals of care with the appropriate treatment method—particularly in patients with comorbid conditions—is an important practice when caring for those with limited life expectancy and functionality.7
THE TAKEAWAY
Intent-to-defer HD may be a preferred strategy when caring for many patients with ESRD. Taking into consideration a patient’s comorbidities and functional status, while considering mortality risk and quality of life are essential. Involving palliative care and nephrology specialists can help patients and families understand HD and make an educated decision regarding when to start it.
1. Nesrallah GE, Mustafa RA, Clark WF, et al; Canadian Society of Nephrology. Canadian Society of Nephrology 2014 clinical practice guideline for timing the initiation of chronic dialysis. CMAJ. 2014;186:112-117.
2. Rayner HC, Zepel L, Fuller DS, et al. Recovery time, quality of life, and mortality in hemodialysis patients: the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am J Kidney Dis. 2014;64:86-94.
3. Canaud B, Tong L, Tentori F, et al. Clinical practices and outcomes in elderly hemodialysis patients: results from the Dialysis Outcomes and Practice Patterns Study (DOPPS). Clin J Am Soc Nephrol. 2011;6:1651-1662.
4. Johansen KL, Chertow GM, Jin C, et al. Significance of frailty among dialysis patients. J Am Soc Nephrol. 2007;18:2960-2967.
5. Joly D, Anglicheau D, Alberti C, et al. Octogenarians reaching end-stage renal disease: cohort study of decision-making and clinical outcomes. J Am Soc Nephrol. 2003;14:1012-1021.
6. Renal Physicians Association. Shared decision-making in the appropriate initiation of and withdrawal from dialysis: clinical practice guideline. 2nd ed. Rockville, MD: Renal Physicians Association; 2010.
7. Grubbs V, Moss AH, Cohen LM, et al; Dialysis Advisory Group of the American Society of Nephrology. A palliative approach to dialysis care: a patient-centered transition to the end of life. Clin J Am Soc Nephrol. 2014;9:2203-2209.
1. Nesrallah GE, Mustafa RA, Clark WF, et al; Canadian Society of Nephrology. Canadian Society of Nephrology 2014 clinical practice guideline for timing the initiation of chronic dialysis. CMAJ. 2014;186:112-117.
2. Rayner HC, Zepel L, Fuller DS, et al. Recovery time, quality of life, and mortality in hemodialysis patients: the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am J Kidney Dis. 2014;64:86-94.
3. Canaud B, Tong L, Tentori F, et al. Clinical practices and outcomes in elderly hemodialysis patients: results from the Dialysis Outcomes and Practice Patterns Study (DOPPS). Clin J Am Soc Nephrol. 2011;6:1651-1662.
4. Johansen KL, Chertow GM, Jin C, et al. Significance of frailty among dialysis patients. J Am Soc Nephrol. 2007;18:2960-2967.
5. Joly D, Anglicheau D, Alberti C, et al. Octogenarians reaching end-stage renal disease: cohort study of decision-making and clinical outcomes. J Am Soc Nephrol. 2003;14:1012-1021.
6. Renal Physicians Association. Shared decision-making in the appropriate initiation of and withdrawal from dialysis: clinical practice guideline. 2nd ed. Rockville, MD: Renal Physicians Association; 2010.
7. Grubbs V, Moss AH, Cohen LM, et al; Dialysis Advisory Group of the American Society of Nephrology. A palliative approach to dialysis care: a patient-centered transition to the end of life. Clin J Am Soc Nephrol. 2014;9:2203-2209.

Weight loss • fatigue • joint pain • Dx?
THE CASE
A 49-year-old Mexican immigrant woman was admitted to the hospital with a 5-month history of fatigue and a 30-pound unintentional weight loss. She was also experiencing arthralgia, swelling, and stiffness in her hands and feet that was worse in the morning. The patient, who was obese and suffered from type 2 diabetes and hypertension, said that at the onset of her illness 5 months earlier, she’d experienced approximately 2 weeks of night sweats and a few days of fever.
A month before being admitted to the hospital, she’d been seen in our southern New Mexico family medicine office. Her recent history of fever, joint symptoms, and weight loss raised concerns of an insidious infection, a new-onset rheumatologic condition, or an occult malignancy.
Initial lab tests revealed leukopenia (white blood cell count, 3200/mcL), microcytic anemia (hemoglobin, 9.4 g/dL), and an elevated erythrocyte sedimentation rate of 30 mm/hr (normal range, 0-20 mm/hr). A rheumatoid factor test was negative, and her thyroid, kidney, and liver function tests were all normal.
More testing… The patient frequently traveled between New Mexico and her hometown of Chihuahua, Mexico, but there had been no recent changes in her diet or environmental exposures. She denied drinking any unpasteurized milk in Chihuahua. But based on her travel history, we ordered enzyme-linked immunosorbent assay (ELISA) antibody titers for Brucella, immunoglobulin G, and immunoglobulin M, which all came back negative. Additionally, we ordered an abdominal and pelvic ultrasound and a chest x-ray that were nondiagnostic. Given the patient’s weight loss and anemia, we referred her to a gastroenterologist for upper and lower gastrointestinal endoscopic evaluations. Unfortunately, the patient was uninsured and did not go to see the gastroenterologist.
A month after seeing us, our patient’s fatigue, lack of appetite, and joint pain became debilitating and she was admitted to the hospital for further evaluation, including a consultation with an oncologist.
THE DIAGNOSIS
During our patient’s 6-day hospital stay, a bone scintigraphy showed a focus of uptake in her left parietal bone and computed tomography scans of her chest, abdomen, and pelvis revealed a left thyroid nodule, as well as multiple noncalcified pulmonary nodules. Blood cultures were also obtained.
Despite the initial negative antibody tests, the blood cultures drawn in the hospital revealed the presence of Brucella melitensis, and we diagnosed brucellosis in this patient.
DISCUSSION
Brucella melitensis is one of the 4 recognized, land-based species of the Brucella genus that can cause disease in humans. Goats, sheep, and camels are natural hosts of B melitensis and consumption of their unpasteurized, infected milk and milk products (especially soft cheeses, ice cream, milk, and butter) leads to human disease. (Once hospitalized, our patient admitted to frequently eating unpasteurized goat cheese from Chihuahua. The only other person in her household that ate the cheese was her 26-year-old daughter, who was also experiencing similar symptoms.)
Brucellosis can also result from inhaling infected, aerosolized material; therefore, individuals whose occupations involve close work with host animals or work in laboratories with the bacteria have an increased risk of infection.1 Due to the risk of acquiring the infection via inhalation, brucellosis is considered a bioterrorism threat.2 Additionally, there have been reports of human-to-human transmission via sexual intercourse, transplacental infection, blood and bone marrow transfusion, and breastfeeding.3
B melitensis is the cause of the majority of Brucella-related illnesses in the world, though symptoms of infection are similar among the different Brucella species. The pathogen can affect almost all organ systems after the initial 2- to 4-week incubation period. Symptoms of brucellosis can be highly variable, although fever is consistently present.1 Other red flags include arthritis (usually affecting the peripheral joints, the sacroiliac joints, and the lower spine), epididymo-orchitis, and hepatitis resulting in transaminase elevation. Abscess formation can be seen in the liver, spleen, and other organs.
Less common but more ominous complications include central nervous system infections and abscesses, endocarditis, and pulmonary infections. Endocarditis and the resulting aortic valve involvement is the major cause of mortality.1Brucella-related uveitis, thyroiditis, nephritis, vasculitis, and acalculous cholecystitis have also been reported.4-9
Rare in the United States. Pasteurization of dairy products and mass vaccination of livestock make Brucella infection rare in the United States. While there have only been 80 to 139 cases of brucellosis reported per year in the United States since 1993, it remains a persistent threat. International travel is common from the United States to the Middle East and other parts of the world where brucellosis is endemic.
Additionally, infection of livestock with Brucella remains widespread in Mexico and the consumption of unpasteurized Mexican dairy products from goats and sheep remains a high-risk activity for acquiring the disease.10 Consequently, Texas and California account for more than half of the brucellosis diagnoses in the United States. However, in 2010, cases were reported in 25 other states and the District of Columbia.11
Repeat serology tests are preferred for confirming the Dx
It is interesting that our patient’s initial Brucella serology by ELISA was negative, because it was ordered months after her initial symptoms. Antibodies should be seen within a month of symptom onset. The Centers for Disease Control and Prevention (CDC) recommends taking 2 serum samples to establish a serologic diagnosis of brucellosis. The first should be drawn within 7 days of symptom onset and the second should be taken 2 to 4 weeks later. A greater than 4-fold rise in the antibody titer confirms the diagnosis. While ELISA is an acceptable serologic test, the CDC recommends using a serum tube agglutination test called the Brucella microagglutination test (BMAT).12 Repeat serology was not performed on our patient because the diagnosis had been confirmed by blood culture.
A combination of antibiotics is the recommended treatment
Treatment of brucellosis should include a tetracycline for at least 6 weeks in combination with an aminoglycoside or rifampin 600 mg/d for an all-oral regimen.13 Doxycycline 100 mg twice a day is preferred due to fewer gastrointestinal adverse effects than tetracycline. Relapse is not uncommon (10%) and usually occurs within one year of completing the antibiotics.8 However, there is a case report of a patient having reactivated brucellosis manifested as acalculous cholecystitis 28 years after completing antibiotics.8
Our patient was started on oral doxycycline 100 mg twice a day and oral rifampin 600 mg/d for 6 weeks. Within days of starting the antibiotics, her joint symptoms and fatigue rapidly abated and her appetite returned. Follow-up radiological testing was not performed after her initial hospital studies due to her lack of financial resources.
The patient’s daughter had also been experiencing night sweats, chills, malaise, anorexia, joint pains, weight loss, and alopecia over the previous 2 months. Her blood cultures were positive for B melitensis as well, and she was started on the same antibiotic regimen as her mother. The daughter was also seen in our clinic by another physician and improved quickly within a week of starting treatment.
Both our patient and her daughter remained symptom-free 6 years after treatment.
THE TAKEAWAY
Brucellosis is rare in the United States, but international travel to endemic areas is commonplace and consumption of unpasteurized Mexican dairy products from goats and sheep is widespread. Brucellosis has a wide range of symptoms, but a prompt diagnosis by an ELISA or BMAT serologic test and appropriate treatment can avoid morbidity and mortality. Treatment includes a tetracycline for at least 6 weeks in combination with an aminoglycoside or rifampin.
1. Pappas G, Akritidis N, Bosilkovski M, et al. Brucellosis. N Engl J Med. 2005;352:2325-2336.
2. Centers for Disease Control and Prevention (CDC). Suspected brucellosis case prompts investigation of possible bioterrorismrelated activity—New Hampshire and Massachusetts, 1999. MMWR Morb Mortal Wkly Rep. 2000;49:509-512.
3. Chen S, Zhang H, Liu X, et al. Increasing threat of brucellosis to low-risk persons in urban settings, China. Emerg Infect Dis. 2014;20:126-130.
4. Rolando I, Vilchez G, Olarte L, et al. Brucellar uveitis: intraocular fluids and biopsy studies. Int J Infect Dis. 2009;13:e206-e211.
5. Azizi F, Katchoui A. Brucella infection of the thyroid gland. Thyroid. 1996;6:461-463.
6. Siegelmann N, Abraham AS, Rudensky B, et al. Brucellosis with nephrotic syndrome, nephritis and IgA nephropathy. Postgrad Med J. 1992;68:834-836.
7. Tanyel E, Tasdelen Fisgin N, Yildiz L, et al. Panniculitis as the initial manifestation of brucellosis: a case report. Am J Dermatopathol. 2008;30:169-171.
8. Ögredici Ö, Erb S, Langer I, et al. Brucellosis reactivation after 28 years. Emerg Infect Dis. 2010;16:2021-2022.
9. Dhand A, Ross JJ. Implantable cardioverter-defibrillator infection due to Brucella melitensis: case report and review of brucellosis of cardiac devices. Clin Infect Dis. 2007;44:e37-e39.
10. Solorio-Rivera JL, Segura-Correa JC, Sánchez-Gil LG. Seroprevalence of and risk factors for brucellosis of goats in herds of Michoacan, Mexico. Prev Vet Med. 2007;82:282-290.
11. Centers for Disease Control and Prevention. Brucellosis surveillance. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/brucellosis/resources/surveillance.html. Accessed October 30, 2015.
12. Centers for Disease Control and Prevention. Serology. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/brucellosis/clinicians/serology.html. Accessed October 30, 2015.
13. Corbel MJ. Brucellosis in humans and animals. World Health Organization (WHO);2006:36-41. Available at: http://www.who.int/csr/resources/publications/Brucellosis.pdf. Accessed November 2, 2015.
THE CASE
A 49-year-old Mexican immigrant woman was admitted to the hospital with a 5-month history of fatigue and a 30-pound unintentional weight loss. She was also experiencing arthralgia, swelling, and stiffness in her hands and feet that was worse in the morning. The patient, who was obese and suffered from type 2 diabetes and hypertension, said that at the onset of her illness 5 months earlier, she’d experienced approximately 2 weeks of night sweats and a few days of fever.
A month before being admitted to the hospital, she’d been seen in our southern New Mexico family medicine office. Her recent history of fever, joint symptoms, and weight loss raised concerns of an insidious infection, a new-onset rheumatologic condition, or an occult malignancy.
Initial lab tests revealed leukopenia (white blood cell count, 3200/mcL), microcytic anemia (hemoglobin, 9.4 g/dL), and an elevated erythrocyte sedimentation rate of 30 mm/hr (normal range, 0-20 mm/hr). A rheumatoid factor test was negative, and her thyroid, kidney, and liver function tests were all normal.
More testing… The patient frequently traveled between New Mexico and her hometown of Chihuahua, Mexico, but there had been no recent changes in her diet or environmental exposures. She denied drinking any unpasteurized milk in Chihuahua. But based on her travel history, we ordered enzyme-linked immunosorbent assay (ELISA) antibody titers for Brucella, immunoglobulin G, and immunoglobulin M, which all came back negative. Additionally, we ordered an abdominal and pelvic ultrasound and a chest x-ray that were nondiagnostic. Given the patient’s weight loss and anemia, we referred her to a gastroenterologist for upper and lower gastrointestinal endoscopic evaluations. Unfortunately, the patient was uninsured and did not go to see the gastroenterologist.
A month after seeing us, our patient’s fatigue, lack of appetite, and joint pain became debilitating and she was admitted to the hospital for further evaluation, including a consultation with an oncologist.
THE DIAGNOSIS
During our patient’s 6-day hospital stay, a bone scintigraphy showed a focus of uptake in her left parietal bone and computed tomography scans of her chest, abdomen, and pelvis revealed a left thyroid nodule, as well as multiple noncalcified pulmonary nodules. Blood cultures were also obtained.
Despite the initial negative antibody tests, the blood cultures drawn in the hospital revealed the presence of Brucella melitensis, and we diagnosed brucellosis in this patient.
DISCUSSION
Brucella melitensis is one of the 4 recognized, land-based species of the Brucella genus that can cause disease in humans. Goats, sheep, and camels are natural hosts of B melitensis and consumption of their unpasteurized, infected milk and milk products (especially soft cheeses, ice cream, milk, and butter) leads to human disease. (Once hospitalized, our patient admitted to frequently eating unpasteurized goat cheese from Chihuahua. The only other person in her household that ate the cheese was her 26-year-old daughter, who was also experiencing similar symptoms.)
Brucellosis can also result from inhaling infected, aerosolized material; therefore, individuals whose occupations involve close work with host animals or work in laboratories with the bacteria have an increased risk of infection.1 Due to the risk of acquiring the infection via inhalation, brucellosis is considered a bioterrorism threat.2 Additionally, there have been reports of human-to-human transmission via sexual intercourse, transplacental infection, blood and bone marrow transfusion, and breastfeeding.3
B melitensis is the cause of the majority of Brucella-related illnesses in the world, though symptoms of infection are similar among the different Brucella species. The pathogen can affect almost all organ systems after the initial 2- to 4-week incubation period. Symptoms of brucellosis can be highly variable, although fever is consistently present.1 Other red flags include arthritis (usually affecting the peripheral joints, the sacroiliac joints, and the lower spine), epididymo-orchitis, and hepatitis resulting in transaminase elevation. Abscess formation can be seen in the liver, spleen, and other organs.
Less common but more ominous complications include central nervous system infections and abscesses, endocarditis, and pulmonary infections. Endocarditis and the resulting aortic valve involvement is the major cause of mortality.1Brucella-related uveitis, thyroiditis, nephritis, vasculitis, and acalculous cholecystitis have also been reported.4-9
Rare in the United States. Pasteurization of dairy products and mass vaccination of livestock make Brucella infection rare in the United States. While there have only been 80 to 139 cases of brucellosis reported per year in the United States since 1993, it remains a persistent threat. International travel is common from the United States to the Middle East and other parts of the world where brucellosis is endemic.
Additionally, infection of livestock with Brucella remains widespread in Mexico and the consumption of unpasteurized Mexican dairy products from goats and sheep remains a high-risk activity for acquiring the disease.10 Consequently, Texas and California account for more than half of the brucellosis diagnoses in the United States. However, in 2010, cases were reported in 25 other states and the District of Columbia.11
Repeat serology tests are preferred for confirming the Dx
It is interesting that our patient’s initial Brucella serology by ELISA was negative, because it was ordered months after her initial symptoms. Antibodies should be seen within a month of symptom onset. The Centers for Disease Control and Prevention (CDC) recommends taking 2 serum samples to establish a serologic diagnosis of brucellosis. The first should be drawn within 7 days of symptom onset and the second should be taken 2 to 4 weeks later. A greater than 4-fold rise in the antibody titer confirms the diagnosis. While ELISA is an acceptable serologic test, the CDC recommends using a serum tube agglutination test called the Brucella microagglutination test (BMAT).12 Repeat serology was not performed on our patient because the diagnosis had been confirmed by blood culture.
A combination of antibiotics is the recommended treatment
Treatment of brucellosis should include a tetracycline for at least 6 weeks in combination with an aminoglycoside or rifampin 600 mg/d for an all-oral regimen.13 Doxycycline 100 mg twice a day is preferred due to fewer gastrointestinal adverse effects than tetracycline. Relapse is not uncommon (10%) and usually occurs within one year of completing the antibiotics.8 However, there is a case report of a patient having reactivated brucellosis manifested as acalculous cholecystitis 28 years after completing antibiotics.8
Our patient was started on oral doxycycline 100 mg twice a day and oral rifampin 600 mg/d for 6 weeks. Within days of starting the antibiotics, her joint symptoms and fatigue rapidly abated and her appetite returned. Follow-up radiological testing was not performed after her initial hospital studies due to her lack of financial resources.
The patient’s daughter had also been experiencing night sweats, chills, malaise, anorexia, joint pains, weight loss, and alopecia over the previous 2 months. Her blood cultures were positive for B melitensis as well, and she was started on the same antibiotic regimen as her mother. The daughter was also seen in our clinic by another physician and improved quickly within a week of starting treatment.
Both our patient and her daughter remained symptom-free 6 years after treatment.
THE TAKEAWAY
Brucellosis is rare in the United States, but international travel to endemic areas is commonplace and consumption of unpasteurized Mexican dairy products from goats and sheep is widespread. Brucellosis has a wide range of symptoms, but a prompt diagnosis by an ELISA or BMAT serologic test and appropriate treatment can avoid morbidity and mortality. Treatment includes a tetracycline for at least 6 weeks in combination with an aminoglycoside or rifampin.
THE CASE
A 49-year-old Mexican immigrant woman was admitted to the hospital with a 5-month history of fatigue and a 30-pound unintentional weight loss. She was also experiencing arthralgia, swelling, and stiffness in her hands and feet that was worse in the morning. The patient, who was obese and suffered from type 2 diabetes and hypertension, said that at the onset of her illness 5 months earlier, she’d experienced approximately 2 weeks of night sweats and a few days of fever.
A month before being admitted to the hospital, she’d been seen in our southern New Mexico family medicine office. Her recent history of fever, joint symptoms, and weight loss raised concerns of an insidious infection, a new-onset rheumatologic condition, or an occult malignancy.
Initial lab tests revealed leukopenia (white blood cell count, 3200/mcL), microcytic anemia (hemoglobin, 9.4 g/dL), and an elevated erythrocyte sedimentation rate of 30 mm/hr (normal range, 0-20 mm/hr). A rheumatoid factor test was negative, and her thyroid, kidney, and liver function tests were all normal.
More testing… The patient frequently traveled between New Mexico and her hometown of Chihuahua, Mexico, but there had been no recent changes in her diet or environmental exposures. She denied drinking any unpasteurized milk in Chihuahua. But based on her travel history, we ordered enzyme-linked immunosorbent assay (ELISA) antibody titers for Brucella, immunoglobulin G, and immunoglobulin M, which all came back negative. Additionally, we ordered an abdominal and pelvic ultrasound and a chest x-ray that were nondiagnostic. Given the patient’s weight loss and anemia, we referred her to a gastroenterologist for upper and lower gastrointestinal endoscopic evaluations. Unfortunately, the patient was uninsured and did not go to see the gastroenterologist.
A month after seeing us, our patient’s fatigue, lack of appetite, and joint pain became debilitating and she was admitted to the hospital for further evaluation, including a consultation with an oncologist.
THE DIAGNOSIS
During our patient’s 6-day hospital stay, a bone scintigraphy showed a focus of uptake in her left parietal bone and computed tomography scans of her chest, abdomen, and pelvis revealed a left thyroid nodule, as well as multiple noncalcified pulmonary nodules. Blood cultures were also obtained.
Despite the initial negative antibody tests, the blood cultures drawn in the hospital revealed the presence of Brucella melitensis, and we diagnosed brucellosis in this patient.
DISCUSSION
Brucella melitensis is one of the 4 recognized, land-based species of the Brucella genus that can cause disease in humans. Goats, sheep, and camels are natural hosts of B melitensis and consumption of their unpasteurized, infected milk and milk products (especially soft cheeses, ice cream, milk, and butter) leads to human disease. (Once hospitalized, our patient admitted to frequently eating unpasteurized goat cheese from Chihuahua. The only other person in her household that ate the cheese was her 26-year-old daughter, who was also experiencing similar symptoms.)
Brucellosis can also result from inhaling infected, aerosolized material; therefore, individuals whose occupations involve close work with host animals or work in laboratories with the bacteria have an increased risk of infection.1 Due to the risk of acquiring the infection via inhalation, brucellosis is considered a bioterrorism threat.2 Additionally, there have been reports of human-to-human transmission via sexual intercourse, transplacental infection, blood and bone marrow transfusion, and breastfeeding.3
B melitensis is the cause of the majority of Brucella-related illnesses in the world, though symptoms of infection are similar among the different Brucella species. The pathogen can affect almost all organ systems after the initial 2- to 4-week incubation period. Symptoms of brucellosis can be highly variable, although fever is consistently present.1 Other red flags include arthritis (usually affecting the peripheral joints, the sacroiliac joints, and the lower spine), epididymo-orchitis, and hepatitis resulting in transaminase elevation. Abscess formation can be seen in the liver, spleen, and other organs.
Less common but more ominous complications include central nervous system infections and abscesses, endocarditis, and pulmonary infections. Endocarditis and the resulting aortic valve involvement is the major cause of mortality.1Brucella-related uveitis, thyroiditis, nephritis, vasculitis, and acalculous cholecystitis have also been reported.4-9
Rare in the United States. Pasteurization of dairy products and mass vaccination of livestock make Brucella infection rare in the United States. While there have only been 80 to 139 cases of brucellosis reported per year in the United States since 1993, it remains a persistent threat. International travel is common from the United States to the Middle East and other parts of the world where brucellosis is endemic.
Additionally, infection of livestock with Brucella remains widespread in Mexico and the consumption of unpasteurized Mexican dairy products from goats and sheep remains a high-risk activity for acquiring the disease.10 Consequently, Texas and California account for more than half of the brucellosis diagnoses in the United States. However, in 2010, cases were reported in 25 other states and the District of Columbia.11
Repeat serology tests are preferred for confirming the Dx
It is interesting that our patient’s initial Brucella serology by ELISA was negative, because it was ordered months after her initial symptoms. Antibodies should be seen within a month of symptom onset. The Centers for Disease Control and Prevention (CDC) recommends taking 2 serum samples to establish a serologic diagnosis of brucellosis. The first should be drawn within 7 days of symptom onset and the second should be taken 2 to 4 weeks later. A greater than 4-fold rise in the antibody titer confirms the diagnosis. While ELISA is an acceptable serologic test, the CDC recommends using a serum tube agglutination test called the Brucella microagglutination test (BMAT).12 Repeat serology was not performed on our patient because the diagnosis had been confirmed by blood culture.
A combination of antibiotics is the recommended treatment
Treatment of brucellosis should include a tetracycline for at least 6 weeks in combination with an aminoglycoside or rifampin 600 mg/d for an all-oral regimen.13 Doxycycline 100 mg twice a day is preferred due to fewer gastrointestinal adverse effects than tetracycline. Relapse is not uncommon (10%) and usually occurs within one year of completing the antibiotics.8 However, there is a case report of a patient having reactivated brucellosis manifested as acalculous cholecystitis 28 years after completing antibiotics.8
Our patient was started on oral doxycycline 100 mg twice a day and oral rifampin 600 mg/d for 6 weeks. Within days of starting the antibiotics, her joint symptoms and fatigue rapidly abated and her appetite returned. Follow-up radiological testing was not performed after her initial hospital studies due to her lack of financial resources.
The patient’s daughter had also been experiencing night sweats, chills, malaise, anorexia, joint pains, weight loss, and alopecia over the previous 2 months. Her blood cultures were positive for B melitensis as well, and she was started on the same antibiotic regimen as her mother. The daughter was also seen in our clinic by another physician and improved quickly within a week of starting treatment.
Both our patient and her daughter remained symptom-free 6 years after treatment.
THE TAKEAWAY
Brucellosis is rare in the United States, but international travel to endemic areas is commonplace and consumption of unpasteurized Mexican dairy products from goats and sheep is widespread. Brucellosis has a wide range of symptoms, but a prompt diagnosis by an ELISA or BMAT serologic test and appropriate treatment can avoid morbidity and mortality. Treatment includes a tetracycline for at least 6 weeks in combination with an aminoglycoside or rifampin.
1. Pappas G, Akritidis N, Bosilkovski M, et al. Brucellosis. N Engl J Med. 2005;352:2325-2336.
2. Centers for Disease Control and Prevention (CDC). Suspected brucellosis case prompts investigation of possible bioterrorismrelated activity—New Hampshire and Massachusetts, 1999. MMWR Morb Mortal Wkly Rep. 2000;49:509-512.
3. Chen S, Zhang H, Liu X, et al. Increasing threat of brucellosis to low-risk persons in urban settings, China. Emerg Infect Dis. 2014;20:126-130.
4. Rolando I, Vilchez G, Olarte L, et al. Brucellar uveitis: intraocular fluids and biopsy studies. Int J Infect Dis. 2009;13:e206-e211.
5. Azizi F, Katchoui A. Brucella infection of the thyroid gland. Thyroid. 1996;6:461-463.
6. Siegelmann N, Abraham AS, Rudensky B, et al. Brucellosis with nephrotic syndrome, nephritis and IgA nephropathy. Postgrad Med J. 1992;68:834-836.
7. Tanyel E, Tasdelen Fisgin N, Yildiz L, et al. Panniculitis as the initial manifestation of brucellosis: a case report. Am J Dermatopathol. 2008;30:169-171.
8. Ögredici Ö, Erb S, Langer I, et al. Brucellosis reactivation after 28 years. Emerg Infect Dis. 2010;16:2021-2022.
9. Dhand A, Ross JJ. Implantable cardioverter-defibrillator infection due to Brucella melitensis: case report and review of brucellosis of cardiac devices. Clin Infect Dis. 2007;44:e37-e39.
10. Solorio-Rivera JL, Segura-Correa JC, Sánchez-Gil LG. Seroprevalence of and risk factors for brucellosis of goats in herds of Michoacan, Mexico. Prev Vet Med. 2007;82:282-290.
11. Centers for Disease Control and Prevention. Brucellosis surveillance. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/brucellosis/resources/surveillance.html. Accessed October 30, 2015.
12. Centers for Disease Control and Prevention. Serology. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/brucellosis/clinicians/serology.html. Accessed October 30, 2015.
13. Corbel MJ. Brucellosis in humans and animals. World Health Organization (WHO);2006:36-41. Available at: http://www.who.int/csr/resources/publications/Brucellosis.pdf. Accessed November 2, 2015.
1. Pappas G, Akritidis N, Bosilkovski M, et al. Brucellosis. N Engl J Med. 2005;352:2325-2336.
2. Centers for Disease Control and Prevention (CDC). Suspected brucellosis case prompts investigation of possible bioterrorismrelated activity—New Hampshire and Massachusetts, 1999. MMWR Morb Mortal Wkly Rep. 2000;49:509-512.
3. Chen S, Zhang H, Liu X, et al. Increasing threat of brucellosis to low-risk persons in urban settings, China. Emerg Infect Dis. 2014;20:126-130.
4. Rolando I, Vilchez G, Olarte L, et al. Brucellar uveitis: intraocular fluids and biopsy studies. Int J Infect Dis. 2009;13:e206-e211.
5. Azizi F, Katchoui A. Brucella infection of the thyroid gland. Thyroid. 1996;6:461-463.
6. Siegelmann N, Abraham AS, Rudensky B, et al. Brucellosis with nephrotic syndrome, nephritis and IgA nephropathy. Postgrad Med J. 1992;68:834-836.
7. Tanyel E, Tasdelen Fisgin N, Yildiz L, et al. Panniculitis as the initial manifestation of brucellosis: a case report. Am J Dermatopathol. 2008;30:169-171.
8. Ögredici Ö, Erb S, Langer I, et al. Brucellosis reactivation after 28 years. Emerg Infect Dis. 2010;16:2021-2022.
9. Dhand A, Ross JJ. Implantable cardioverter-defibrillator infection due to Brucella melitensis: case report and review of brucellosis of cardiac devices. Clin Infect Dis. 2007;44:e37-e39.
10. Solorio-Rivera JL, Segura-Correa JC, Sánchez-Gil LG. Seroprevalence of and risk factors for brucellosis of goats in herds of Michoacan, Mexico. Prev Vet Med. 2007;82:282-290.
11. Centers for Disease Control and Prevention. Brucellosis surveillance. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/brucellosis/resources/surveillance.html. Accessed October 30, 2015.
12. Centers for Disease Control and Prevention. Serology. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/brucellosis/clinicians/serology.html. Accessed October 30, 2015.
13. Corbel MJ. Brucellosis in humans and animals. World Health Organization (WHO);2006:36-41. Available at: http://www.who.int/csr/resources/publications/Brucellosis.pdf. Accessed November 2, 2015.

Cutaneous Leishmaniasis: An Emerging Infectious Disease in Travelers
Leishmaniasis describes any disease caused by protozoan parasites of the genus Leishmania1 and can manifest in 3 different forms: cutaneous (the most common); mucosal, a destructive metastatic sequela of the cutaneous form; and visceral, which is potentially fatal.2 According to the World Health Organization, the leishmaniases are endemic in 88 countries.3 It is estimated that 95% of cutaneous cases occur in the Americas (most notably Central and South America), the Mediterranean basin, the Middle East, and Central Asia.2 Most cutaneous cases diagnosed among nonmilitary personnel in the United States are acquired in Mexico and Central America.4 In Central and South America, the causative human pathogens include species of the Leishmania (Viannia) complex (eg, Leishmania panamensis, Leishmania braziliensis, Leishmania guyanensis, Leishmania peruviana) and the Leishmania mexicana complex (eg, Leishmania mexicana, Leishmania amazonensis, Leishmania venezuelensis). All of these species can cause localized cutaneous lesions, but only L panamensis, L braziliensis, and L guyanensis are associated with metastatic mucosal lesions. In Central and South Americas, only Leishmaniasis chagasi (also known as Leishmaniasis infantum) is known to cause visceral leishmaniasis.5
Case Report
A 26-year-old man was referred to the dermatology clinic by his primary care provider for evaluation of a nonhealing sore on the left volar forearm of 6 weeks’ duration. The patient described the initial lesion as a red bump resembling a mosquito bite. Over 6 weeks the papule evolved into an indurated plaque with painless ulceration. The patient’s primary care provider had prescribed antibiotics for a presumed Staphylococcus aureus infection of the skin 5 weeks prior to presentation; however, the lesion continued to enlarge in size, resulting in referral to our dermatology clinic.
Skin examination revealed a solitary, 4-cm, painless, ulcerated plaque on the left volar forearm (Figure 1). No lymphadenopathy was noted. The patient reported that he had returned from a mission trip to rural Costa Rica 2 weeks prior to the appearance of the lesion. His medical history was otherwise unremarkable and his vital signs were within normal limits. Our initial differential diagnosis included pyoderma gangrenosum, Sweet syndrome, cutaneous leishmaniasis, and an insect bite.

Histopathologic study of a 5-mm punch biopsy specimen from the lesion showed a dense nodular and diffuse lymphohistiocytic infiltrate containing foci of suppuration. Within these suppurative foci were histiocytes parasitized by intracellular organisms that appeared to be of uniform size and shape on Giemsa staining, all of which are considered to be pathognomonic features of cutaneous leishmaniasis6 (Figures 2 and 3). The dermatopathologist’s diagnosis of cutaneous leishmaniasis was confirmed by the Centers for Disease Control and Prevention. The species was identified by polymerase chain reaction (PCR) as L panamensis.
The patient was treated with intravenous sodium stibogluconate 20 mg/kg for 20 consecutive days as recommended by expert consensus. The decision to treat a frequently self-limited cutaneous lesion with a highly toxic systemic drug was based on the small but real risk of metastatic mucosal lesions, which is caused by the Viannia subgenus, including L panamensis. Of note, sodium stibogluconate and other antimony drugs are not sold in the United States. Sodium stibogluconate is approved by the US Food and Drug Administration to be distributed by the Centers for Disease Control and Prevention under a protocol requiring baseline and weekly electrocardiograms and monitoring of patients’ creatinine, transaminase, lipase, amylase, and complete blood count levels.7 Our patient tolerated treatment but experienced mild to moderate flulike symptoms. The patient experienced no remarkable sequelae other than scarring in the affected area. He was warned to notify his health care providers of any persistent nasal symptoms, including nasal stuffiness, mucosal bleeding, and increased secretions, heralding the possibility of mucosal metastasis.
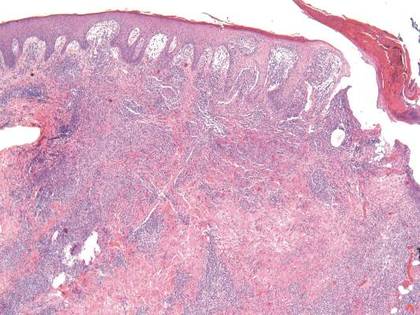
| 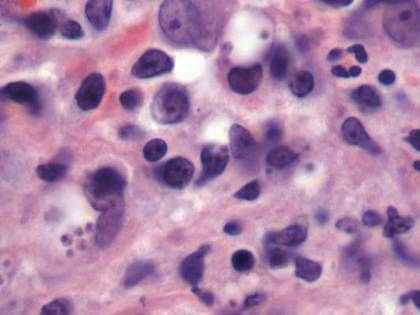
| |
Figure 2. Dense nodular and diffuse lymphohistiocytic infiltrate containing foci of suppuration (H&E, original magnification ×10). | Figure 3. Histiocytes parasitized by intracellular organisms of uniform shape and size on Giemsa staining (original magnification ×1000). |
Comment
The true incidence of cutaneous leishmaniasis in American travelers returning from Mexico and South and Central Americas is not known. The best incidence estimates are based on the number of physician requests for sodium stibogluconate and travel surveillance data collected by the Centers for Disease Control and Prevention. One study estimated the incidence of cutaneous leishmaniasis in Americans to be 1 case per every 100,000 travelers to Mexico.9 Data on the incidence of cutaneous leishmaniasis in American travelers seen in travel clinics for skin lesions gives a different perspective.10 Leishmaniasis is one of the most common dermatologic diseases seen in patients (European, North American, and other) returning from South America, accounting for 143 of every 1000 patients diagnosed with a skin disease acquired in South America.
Although males are thought to be at higher risk for cutaneous leishmaniasis infection than females, other demographic and behavioral risk factors are not well defined. In a case series of US travelers diagnosed with cutaneous leishmaniasis between January 1985 and April 1990, Herwaldt et al9 found that 46% (27/59) were conducting field studies, while 39% (23/59) were tourists, visitors, or tour guides. At least 15 of the 58 travelers interviewed (26%) were in forested areas for 1 week or less, and of these 15 respondents, at least 6 had a maximum exposure of 2 days.9
Evidence suggests that cutaneous leishmaniasis is inefficiently diagnosed in the United States. One study showed that some patients may consult up to 7 physicians before a definitive diagnosis is made, and the median time from noticing eruption of the lesions to definitive treatment was 112 days.9 Several factors may contribute to delays and inefficiencies in diagnosis. First, the lesions of cutaneous leishmaniasis are varied in morphology, and although ulcers are thought to be the most commonly presenting lesions,11 there are no specific morphologic features that are pathognomonic for cutaneous leishmaniasis. Second, the temporal association with travel to endemic countries is not necessarily apparent, with lesions developing gradually or weeks after the patient returns home. In the one study, 17% (10/58) of patients were home for more than 1 month before they noticed skin lesions.9 Finally, definitive diagnosis requires biopsy or scraping of the lesion followed by PCR, special histopathological staining (Giemsa), or culture. Polymerase chain reaction is currently the best means of identifying the causative Leishmania species.12-14 However, since skin biopsies are not routine in primary care settings and few practitioners are familiar with PCR for identification of leishmaniasis, diagnosis is typically made only after referral to a specialist.
Leishmaniasis transmission occurs in diverse geographical settings though a variety of mechanisms (Figure 4). The morphology of cutaneous leishmaniasis varies and may include papules, nodules, psoriasiform plaques, or ulcers. The differential diagnosis may include staphylococcal skin infection, insect bite, cutaneous neoplasm, pyoderma gangrenosum, sporotrichosis, blastomycosis, chromomycosis, lobomycosis, cutaneous tuberculosis, atypical mycobacterial infection, syphilis, yaws, leprosy, Hansen disease, and sarcoidosis. A definitive diagnosis can be made only after identifying the causative parasite. A scraping or punch biopsy taken from a cleaned lesion provides an adequate sample. Identification can then be accomplished by histopathology, tissue culture, or PCR.5

We present a rhyme that can be used to promote greater awareness of cutaneous leishmaniasis among US health care practitioners:
And on his leg finds an ulcerated plaque.
The possibilities are many,
Numbering far more than 20.
Leishmaniasis is a lurking issue,
So the savvy physician tests the tissue.
Although clinical resolution of cutaneous leishmaniasis usually occurs over months to years, the unsightly appearance of the lesions as well as the potential for scarring and mucosal metastasis (associated with some species) drives medical treatment.15 Pentavalent antimonial drugs, which have been the mainstay of treatment for more than 50 years, remain the most popular treatment for cutaneous leishmaniasis. Two antimony compounds, sodium stibogluconate and meglumine antimoniate, often lead to clinical cure in less than 1 month7; however, these drugs are far from ideal because of the inconvenience of obtaining them, emerging parasite resistance, long treatment course, parenteral route of administration, and serious side effects including infusion reactions, arrhythmias, pancreatitis, and liver toxicity. Moreover, the subclinical persistence of cutaneous leishmaniasis years after treatment and clinical cure is common. There have been reports of spontaneous disease reactivation in immunocompromised individuals, and Leishmania has been detected in old cutaneous leishmaniasis scars on PCR testing.16-18 Other therapies that have been used to treat cutaneous leishmaniasis include allopurinol, aminosidine sulphate, amphotericin B, the Bacillus Calmette–Guérin vaccine, cotrimoxazole, cryotherapy, dapsone, fluconazole, itraconazole, ketoconazole, laser therapy, metronidazole, miltefosine, paromomycin, pentamidine, pentoxifylline, photodynamic therapy, rifampicin, and surgical excision of the entire lesion.8 A 2009 Cochrane review of the various treatments for cutaneous leishmaniasis concluded that “no general consensus on optimal treatment has been achieved” and suggested “the creation of an international platform to improve the quality and standardization of future trials in order to develop a better evidence-based approach.”8
Conclusion
Cutaneous leishmaniasis should be included in the differential diagnosis for travelers returning from endemic areas who present with new skin lesions. Since no specific lesion types are pathognomonic for cutaneous leishmaniasis, tissue biopsy for histopathology and PCR are essential for diagnosis. Prevention of cutaneous leishmaniasis hinges on appropriate counseling of travelers to endemic regions.
1. Etymologia-Leishmaniasis. Emerg Infect Dis. 2008;14:666.
2. Burden and distribution. World Health Organization Web site. http://www.who.int/leishmaniasis/burden/en/. Accessed November 10, 2015.
3. Emergencies preparedness, response. World Health Organization Web site. http://www.who.int/csr/resources/publications/CSR_ISR_2000_1leish/en/. Accessed November 3, 2015.
4. Pavli A, Maltezou HC. Leishmaniasis, an emerging infection in travelers. Int J Infect Dis. 2010;14:e1032-e1039.
5. Magill AJ. Leishmania species: visceral (Kala-Azar), cutaneous, and mucosal leishmaniasis. In: Mandell GL, Bennett JE, Dolin R, eds. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, PA: Churchill Livingstone; 2009:3463-3480.
6. Mysore V. Invisible dermatoses. Indian J Dermatol Venereol Leprol. 2010;76:239-248.
7. Parasites – Leishmaniasis. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/parasites/leishmaniasis/health_professionals/. Updated September 14, 2015. Accessed November 13, 2015.
8. González U, Pinart M, Rengifo-Pardo M, et al. Interventions for American cutaneous and mucocutaneous leishmaniasis. Cochrane Database Syst Rev. 2009;15:CD004834.
9. Herwaldt BL, Stokes SL, Juranek DD. American cutaneous leishmaniasis in U.S. travelers. Ann Intern Med. 1993;118:779-784.
10. Freedman DO, Weld LH, Kozarsky PE, et al. Spectrum of disease and relation to place of exposure among ill returned travelers. New Engl J Med. 2006;354:119-130.
11. El Hajj L, Thellier M, Carriere J, et al. Localized cutaneous leishmaniasis imported into Paris: a review of 39 cases. Int J Dermatol. 2004;43:120-125.
12. Harris E, Kropp G, Belli A, et al. Single-step multiplex PCR assay for characterization of New World Leishmania complexes. J Clin Microbiol. 1998;36:1989-1995.
13. Marfurt J, Niederwieser I, Makia D, et al. Diagnostic genotyping of Old and New World Leishmania species by PCR-RFLP. Diagn Microbiol Infect Dis. 2003;46:115-124.
14. Schonian G, Nasereddin A, Dinse N, et al. PCR diagnosis and characterization of Leishmania in local and imported clinical samples. Diagn Microbiol Infect Dis. 2003;47:349-358.
15. Reithinger R, Aadil K, Kolaczinski J, et al. Social impact of leishmaniasis, Afghanistan. Emerg Infect Dis. 2005;11:634-636.
16. Morales MA, Cruz I, Rubio JM, et al. Relapses versus reinfections in patients coinfected with Leishmania infantum and human immunodeficiency virus type 1 [published online ahead of print April 22, 2002]. J Infect Dis. 2002;185:1533-1537.
17. Coutinho SG, Pirmez C, Da-Cruz AM. Parasitological and immunological follow-up of American tegumentary leishmaniasis patients. Trans R Soc Trop Med Hyg. 2002;96(suppl 1):S173-S178.
18. Mendonça MG, de Brito ME, Rodrigues EH, et al. Persistance of leishmania parasites in scars after clinical cure of American cutaneous leishmaniasis: is there a sterile cure [published online ahead of print March 2, 2004]? J Infect Dis. 2004;189:1018-1023.
Leishmaniasis describes any disease caused by protozoan parasites of the genus Leishmania1 and can manifest in 3 different forms: cutaneous (the most common); mucosal, a destructive metastatic sequela of the cutaneous form; and visceral, which is potentially fatal.2 According to the World Health Organization, the leishmaniases are endemic in 88 countries.3 It is estimated that 95% of cutaneous cases occur in the Americas (most notably Central and South America), the Mediterranean basin, the Middle East, and Central Asia.2 Most cutaneous cases diagnosed among nonmilitary personnel in the United States are acquired in Mexico and Central America.4 In Central and South America, the causative human pathogens include species of the Leishmania (Viannia) complex (eg, Leishmania panamensis, Leishmania braziliensis, Leishmania guyanensis, Leishmania peruviana) and the Leishmania mexicana complex (eg, Leishmania mexicana, Leishmania amazonensis, Leishmania venezuelensis). All of these species can cause localized cutaneous lesions, but only L panamensis, L braziliensis, and L guyanensis are associated with metastatic mucosal lesions. In Central and South Americas, only Leishmaniasis chagasi (also known as Leishmaniasis infantum) is known to cause visceral leishmaniasis.5
Case Report
A 26-year-old man was referred to the dermatology clinic by his primary care provider for evaluation of a nonhealing sore on the left volar forearm of 6 weeks’ duration. The patient described the initial lesion as a red bump resembling a mosquito bite. Over 6 weeks the papule evolved into an indurated plaque with painless ulceration. The patient’s primary care provider had prescribed antibiotics for a presumed Staphylococcus aureus infection of the skin 5 weeks prior to presentation; however, the lesion continued to enlarge in size, resulting in referral to our dermatology clinic.
Skin examination revealed a solitary, 4-cm, painless, ulcerated plaque on the left volar forearm (Figure 1). No lymphadenopathy was noted. The patient reported that he had returned from a mission trip to rural Costa Rica 2 weeks prior to the appearance of the lesion. His medical history was otherwise unremarkable and his vital signs were within normal limits. Our initial differential diagnosis included pyoderma gangrenosum, Sweet syndrome, cutaneous leishmaniasis, and an insect bite.
Histopathologic study of a 5-mm punch biopsy specimen from the lesion showed a dense nodular and diffuse lymphohistiocytic infiltrate containing foci of suppuration. Within these suppurative foci were histiocytes parasitized by intracellular organisms that appeared to be of uniform size and shape on Giemsa staining, all of which are considered to be pathognomonic features of cutaneous leishmaniasis6 (Figures 2 and 3). The dermatopathologist’s diagnosis of cutaneous leishmaniasis was confirmed by the Centers for Disease Control and Prevention. The species was identified by polymerase chain reaction (PCR) as L panamensis.
The patient was treated with intravenous sodium stibogluconate 20 mg/kg for 20 consecutive days as recommended by expert consensus. The decision to treat a frequently self-limited cutaneous lesion with a highly toxic systemic drug was based on the small but real risk of metastatic mucosal lesions, which is caused by the Viannia subgenus, including L panamensis. Of note, sodium stibogluconate and other antimony drugs are not sold in the United States. Sodium stibogluconate is approved by the US Food and Drug Administration to be distributed by the Centers for Disease Control and Prevention under a protocol requiring baseline and weekly electrocardiograms and monitoring of patients’ creatinine, transaminase, lipase, amylase, and complete blood count levels.7 Our patient tolerated treatment but experienced mild to moderate flulike symptoms. The patient experienced no remarkable sequelae other than scarring in the affected area. He was warned to notify his health care providers of any persistent nasal symptoms, including nasal stuffiness, mucosal bleeding, and increased secretions, heralding the possibility of mucosal metastasis.
|
|
| |
Figure 2. Dense nodular and diffuse lymphohistiocytic infiltrate containing foci of suppuration (H&E, original magnification ×10). | Figure 3. Histiocytes parasitized by intracellular organisms of uniform shape and size on Giemsa staining (original magnification ×1000). |
Comment
The true incidence of cutaneous leishmaniasis in American travelers returning from Mexico and South and Central Americas is not known. The best incidence estimates are based on the number of physician requests for sodium stibogluconate and travel surveillance data collected by the Centers for Disease Control and Prevention. One study estimated the incidence of cutaneous leishmaniasis in Americans to be 1 case per every 100,000 travelers to Mexico.9 Data on the incidence of cutaneous leishmaniasis in American travelers seen in travel clinics for skin lesions gives a different perspective.10 Leishmaniasis is one of the most common dermatologic diseases seen in patients (European, North American, and other) returning from South America, accounting for 143 of every 1000 patients diagnosed with a skin disease acquired in South America.
Although males are thought to be at higher risk for cutaneous leishmaniasis infection than females, other demographic and behavioral risk factors are not well defined. In a case series of US travelers diagnosed with cutaneous leishmaniasis between January 1985 and April 1990, Herwaldt et al9 found that 46% (27/59) were conducting field studies, while 39% (23/59) were tourists, visitors, or tour guides. At least 15 of the 58 travelers interviewed (26%) were in forested areas for 1 week or less, and of these 15 respondents, at least 6 had a maximum exposure of 2 days.9
Evidence suggests that cutaneous leishmaniasis is inefficiently diagnosed in the United States. One study showed that some patients may consult up to 7 physicians before a definitive diagnosis is made, and the median time from noticing eruption of the lesions to definitive treatment was 112 days.9 Several factors may contribute to delays and inefficiencies in diagnosis. First, the lesions of cutaneous leishmaniasis are varied in morphology, and although ulcers are thought to be the most commonly presenting lesions,11 there are no specific morphologic features that are pathognomonic for cutaneous leishmaniasis. Second, the temporal association with travel to endemic countries is not necessarily apparent, with lesions developing gradually or weeks after the patient returns home. In the one study, 17% (10/58) of patients were home for more than 1 month before they noticed skin lesions.9 Finally, definitive diagnosis requires biopsy or scraping of the lesion followed by PCR, special histopathological staining (Giemsa), or culture. Polymerase chain reaction is currently the best means of identifying the causative Leishmania species.12-14 However, since skin biopsies are not routine in primary care settings and few practitioners are familiar with PCR for identification of leishmaniasis, diagnosis is typically made only after referral to a specialist.
Leishmaniasis transmission occurs in diverse geographical settings though a variety of mechanisms (Figure 4). The morphology of cutaneous leishmaniasis varies and may include papules, nodules, psoriasiform plaques, or ulcers. The differential diagnosis may include staphylococcal skin infection, insect bite, cutaneous neoplasm, pyoderma gangrenosum, sporotrichosis, blastomycosis, chromomycosis, lobomycosis, cutaneous tuberculosis, atypical mycobacterial infection, syphilis, yaws, leprosy, Hansen disease, and sarcoidosis. A definitive diagnosis can be made only after identifying the causative parasite. A scraping or punch biopsy taken from a cleaned lesion provides an adequate sample. Identification can then be accomplished by histopathology, tissue culture, or PCR.5
We present a rhyme that can be used to promote greater awareness of cutaneous leishmaniasis among US health care practitioners:
And on his leg finds an ulcerated plaque.
The possibilities are many,
Numbering far more than 20.
Leishmaniasis is a lurking issue,
So the savvy physician tests the tissue.
Although clinical resolution of cutaneous leishmaniasis usually occurs over months to years, the unsightly appearance of the lesions as well as the potential for scarring and mucosal metastasis (associated with some species) drives medical treatment.15 Pentavalent antimonial drugs, which have been the mainstay of treatment for more than 50 years, remain the most popular treatment for cutaneous leishmaniasis. Two antimony compounds, sodium stibogluconate and meglumine antimoniate, often lead to clinical cure in less than 1 month7; however, these drugs are far from ideal because of the inconvenience of obtaining them, emerging parasite resistance, long treatment course, parenteral route of administration, and serious side effects including infusion reactions, arrhythmias, pancreatitis, and liver toxicity. Moreover, the subclinical persistence of cutaneous leishmaniasis years after treatment and clinical cure is common. There have been reports of spontaneous disease reactivation in immunocompromised individuals, and Leishmania has been detected in old cutaneous leishmaniasis scars on PCR testing.16-18 Other therapies that have been used to treat cutaneous leishmaniasis include allopurinol, aminosidine sulphate, amphotericin B, the Bacillus Calmette–Guérin vaccine, cotrimoxazole, cryotherapy, dapsone, fluconazole, itraconazole, ketoconazole, laser therapy, metronidazole, miltefosine, paromomycin, pentamidine, pentoxifylline, photodynamic therapy, rifampicin, and surgical excision of the entire lesion.8 A 2009 Cochrane review of the various treatments for cutaneous leishmaniasis concluded that “no general consensus on optimal treatment has been achieved” and suggested “the creation of an international platform to improve the quality and standardization of future trials in order to develop a better evidence-based approach.”8
Conclusion
Cutaneous leishmaniasis should be included in the differential diagnosis for travelers returning from endemic areas who present with new skin lesions. Since no specific lesion types are pathognomonic for cutaneous leishmaniasis, tissue biopsy for histopathology and PCR are essential for diagnosis. Prevention of cutaneous leishmaniasis hinges on appropriate counseling of travelers to endemic regions.
Leishmaniasis describes any disease caused by protozoan parasites of the genus Leishmania1 and can manifest in 3 different forms: cutaneous (the most common); mucosal, a destructive metastatic sequela of the cutaneous form; and visceral, which is potentially fatal.2 According to the World Health Organization, the leishmaniases are endemic in 88 countries.3 It is estimated that 95% of cutaneous cases occur in the Americas (most notably Central and South America), the Mediterranean basin, the Middle East, and Central Asia.2 Most cutaneous cases diagnosed among nonmilitary personnel in the United States are acquired in Mexico and Central America.4 In Central and South America, the causative human pathogens include species of the Leishmania (Viannia) complex (eg, Leishmania panamensis, Leishmania braziliensis, Leishmania guyanensis, Leishmania peruviana) and the Leishmania mexicana complex (eg, Leishmania mexicana, Leishmania amazonensis, Leishmania venezuelensis). All of these species can cause localized cutaneous lesions, but only L panamensis, L braziliensis, and L guyanensis are associated with metastatic mucosal lesions. In Central and South Americas, only Leishmaniasis chagasi (also known as Leishmaniasis infantum) is known to cause visceral leishmaniasis.5
Case Report
A 26-year-old man was referred to the dermatology clinic by his primary care provider for evaluation of a nonhealing sore on the left volar forearm of 6 weeks’ duration. The patient described the initial lesion as a red bump resembling a mosquito bite. Over 6 weeks the papule evolved into an indurated plaque with painless ulceration. The patient’s primary care provider had prescribed antibiotics for a presumed Staphylococcus aureus infection of the skin 5 weeks prior to presentation; however, the lesion continued to enlarge in size, resulting in referral to our dermatology clinic.
Skin examination revealed a solitary, 4-cm, painless, ulcerated plaque on the left volar forearm (Figure 1). No lymphadenopathy was noted. The patient reported that he had returned from a mission trip to rural Costa Rica 2 weeks prior to the appearance of the lesion. His medical history was otherwise unremarkable and his vital signs were within normal limits. Our initial differential diagnosis included pyoderma gangrenosum, Sweet syndrome, cutaneous leishmaniasis, and an insect bite.
Histopathologic study of a 5-mm punch biopsy specimen from the lesion showed a dense nodular and diffuse lymphohistiocytic infiltrate containing foci of suppuration. Within these suppurative foci were histiocytes parasitized by intracellular organisms that appeared to be of uniform size and shape on Giemsa staining, all of which are considered to be pathognomonic features of cutaneous leishmaniasis6 (Figures 2 and 3). The dermatopathologist’s diagnosis of cutaneous leishmaniasis was confirmed by the Centers for Disease Control and Prevention. The species was identified by polymerase chain reaction (PCR) as L panamensis.
The patient was treated with intravenous sodium stibogluconate 20 mg/kg for 20 consecutive days as recommended by expert consensus. The decision to treat a frequently self-limited cutaneous lesion with a highly toxic systemic drug was based on the small but real risk of metastatic mucosal lesions, which is caused by the Viannia subgenus, including L panamensis. Of note, sodium stibogluconate and other antimony drugs are not sold in the United States. Sodium stibogluconate is approved by the US Food and Drug Administration to be distributed by the Centers for Disease Control and Prevention under a protocol requiring baseline and weekly electrocardiograms and monitoring of patients’ creatinine, transaminase, lipase, amylase, and complete blood count levels.7 Our patient tolerated treatment but experienced mild to moderate flulike symptoms. The patient experienced no remarkable sequelae other than scarring in the affected area. He was warned to notify his health care providers of any persistent nasal symptoms, including nasal stuffiness, mucosal bleeding, and increased secretions, heralding the possibility of mucosal metastasis.
|
|
| |
Figure 2. Dense nodular and diffuse lymphohistiocytic infiltrate containing foci of suppuration (H&E, original magnification ×10). | Figure 3. Histiocytes parasitized by intracellular organisms of uniform shape and size on Giemsa staining (original magnification ×1000). |
Comment
The true incidence of cutaneous leishmaniasis in American travelers returning from Mexico and South and Central Americas is not known. The best incidence estimates are based on the number of physician requests for sodium stibogluconate and travel surveillance data collected by the Centers for Disease Control and Prevention. One study estimated the incidence of cutaneous leishmaniasis in Americans to be 1 case per every 100,000 travelers to Mexico.9 Data on the incidence of cutaneous leishmaniasis in American travelers seen in travel clinics for skin lesions gives a different perspective.10 Leishmaniasis is one of the most common dermatologic diseases seen in patients (European, North American, and other) returning from South America, accounting for 143 of every 1000 patients diagnosed with a skin disease acquired in South America.
Although males are thought to be at higher risk for cutaneous leishmaniasis infection than females, other demographic and behavioral risk factors are not well defined. In a case series of US travelers diagnosed with cutaneous leishmaniasis between January 1985 and April 1990, Herwaldt et al9 found that 46% (27/59) were conducting field studies, while 39% (23/59) were tourists, visitors, or tour guides. At least 15 of the 58 travelers interviewed (26%) were in forested areas for 1 week or less, and of these 15 respondents, at least 6 had a maximum exposure of 2 days.9
Evidence suggests that cutaneous leishmaniasis is inefficiently diagnosed in the United States. One study showed that some patients may consult up to 7 physicians before a definitive diagnosis is made, and the median time from noticing eruption of the lesions to definitive treatment was 112 days.9 Several factors may contribute to delays and inefficiencies in diagnosis. First, the lesions of cutaneous leishmaniasis are varied in morphology, and although ulcers are thought to be the most commonly presenting lesions,11 there are no specific morphologic features that are pathognomonic for cutaneous leishmaniasis. Second, the temporal association with travel to endemic countries is not necessarily apparent, with lesions developing gradually or weeks after the patient returns home. In the one study, 17% (10/58) of patients were home for more than 1 month before they noticed skin lesions.9 Finally, definitive diagnosis requires biopsy or scraping of the lesion followed by PCR, special histopathological staining (Giemsa), or culture. Polymerase chain reaction is currently the best means of identifying the causative Leishmania species.12-14 However, since skin biopsies are not routine in primary care settings and few practitioners are familiar with PCR for identification of leishmaniasis, diagnosis is typically made only after referral to a specialist.
Leishmaniasis transmission occurs in diverse geographical settings though a variety of mechanisms (Figure 4). The morphology of cutaneous leishmaniasis varies and may include papules, nodules, psoriasiform plaques, or ulcers. The differential diagnosis may include staphylococcal skin infection, insect bite, cutaneous neoplasm, pyoderma gangrenosum, sporotrichosis, blastomycosis, chromomycosis, lobomycosis, cutaneous tuberculosis, atypical mycobacterial infection, syphilis, yaws, leprosy, Hansen disease, and sarcoidosis. A definitive diagnosis can be made only after identifying the causative parasite. A scraping or punch biopsy taken from a cleaned lesion provides an adequate sample. Identification can then be accomplished by histopathology, tissue culture, or PCR.5
We present a rhyme that can be used to promote greater awareness of cutaneous leishmaniasis among US health care practitioners:
And on his leg finds an ulcerated plaque.
The possibilities are many,
Numbering far more than 20.
Leishmaniasis is a lurking issue,
So the savvy physician tests the tissue.
Although clinical resolution of cutaneous leishmaniasis usually occurs over months to years, the unsightly appearance of the lesions as well as the potential for scarring and mucosal metastasis (associated with some species) drives medical treatment.15 Pentavalent antimonial drugs, which have been the mainstay of treatment for more than 50 years, remain the most popular treatment for cutaneous leishmaniasis. Two antimony compounds, sodium stibogluconate and meglumine antimoniate, often lead to clinical cure in less than 1 month7; however, these drugs are far from ideal because of the inconvenience of obtaining them, emerging parasite resistance, long treatment course, parenteral route of administration, and serious side effects including infusion reactions, arrhythmias, pancreatitis, and liver toxicity. Moreover, the subclinical persistence of cutaneous leishmaniasis years after treatment and clinical cure is common. There have been reports of spontaneous disease reactivation in immunocompromised individuals, and Leishmania has been detected in old cutaneous leishmaniasis scars on PCR testing.16-18 Other therapies that have been used to treat cutaneous leishmaniasis include allopurinol, aminosidine sulphate, amphotericin B, the Bacillus Calmette–Guérin vaccine, cotrimoxazole, cryotherapy, dapsone, fluconazole, itraconazole, ketoconazole, laser therapy, metronidazole, miltefosine, paromomycin, pentamidine, pentoxifylline, photodynamic therapy, rifampicin, and surgical excision of the entire lesion.8 A 2009 Cochrane review of the various treatments for cutaneous leishmaniasis concluded that “no general consensus on optimal treatment has been achieved” and suggested “the creation of an international platform to improve the quality and standardization of future trials in order to develop a better evidence-based approach.”8
Conclusion
Cutaneous leishmaniasis should be included in the differential diagnosis for travelers returning from endemic areas who present with new skin lesions. Since no specific lesion types are pathognomonic for cutaneous leishmaniasis, tissue biopsy for histopathology and PCR are essential for diagnosis. Prevention of cutaneous leishmaniasis hinges on appropriate counseling of travelers to endemic regions.
1. Etymologia-Leishmaniasis. Emerg Infect Dis. 2008;14:666.
2. Burden and distribution. World Health Organization Web site. http://www.who.int/leishmaniasis/burden/en/. Accessed November 10, 2015.
3. Emergencies preparedness, response. World Health Organization Web site. http://www.who.int/csr/resources/publications/CSR_ISR_2000_1leish/en/. Accessed November 3, 2015.
4. Pavli A, Maltezou HC. Leishmaniasis, an emerging infection in travelers. Int J Infect Dis. 2010;14:e1032-e1039.
5. Magill AJ. Leishmania species: visceral (Kala-Azar), cutaneous, and mucosal leishmaniasis. In: Mandell GL, Bennett JE, Dolin R, eds. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, PA: Churchill Livingstone; 2009:3463-3480.
6. Mysore V. Invisible dermatoses. Indian J Dermatol Venereol Leprol. 2010;76:239-248.
7. Parasites – Leishmaniasis. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/parasites/leishmaniasis/health_professionals/. Updated September 14, 2015. Accessed November 13, 2015.
8. González U, Pinart M, Rengifo-Pardo M, et al. Interventions for American cutaneous and mucocutaneous leishmaniasis. Cochrane Database Syst Rev. 2009;15:CD004834.
9. Herwaldt BL, Stokes SL, Juranek DD. American cutaneous leishmaniasis in U.S. travelers. Ann Intern Med. 1993;118:779-784.
10. Freedman DO, Weld LH, Kozarsky PE, et al. Spectrum of disease and relation to place of exposure among ill returned travelers. New Engl J Med. 2006;354:119-130.
11. El Hajj L, Thellier M, Carriere J, et al. Localized cutaneous leishmaniasis imported into Paris: a review of 39 cases. Int J Dermatol. 2004;43:120-125.
12. Harris E, Kropp G, Belli A, et al. Single-step multiplex PCR assay for characterization of New World Leishmania complexes. J Clin Microbiol. 1998;36:1989-1995.
13. Marfurt J, Niederwieser I, Makia D, et al. Diagnostic genotyping of Old and New World Leishmania species by PCR-RFLP. Diagn Microbiol Infect Dis. 2003;46:115-124.
14. Schonian G, Nasereddin A, Dinse N, et al. PCR diagnosis and characterization of Leishmania in local and imported clinical samples. Diagn Microbiol Infect Dis. 2003;47:349-358.
15. Reithinger R, Aadil K, Kolaczinski J, et al. Social impact of leishmaniasis, Afghanistan. Emerg Infect Dis. 2005;11:634-636.
16. Morales MA, Cruz I, Rubio JM, et al. Relapses versus reinfections in patients coinfected with Leishmania infantum and human immunodeficiency virus type 1 [published online ahead of print April 22, 2002]. J Infect Dis. 2002;185:1533-1537.
17. Coutinho SG, Pirmez C, Da-Cruz AM. Parasitological and immunological follow-up of American tegumentary leishmaniasis patients. Trans R Soc Trop Med Hyg. 2002;96(suppl 1):S173-S178.
18. Mendonça MG, de Brito ME, Rodrigues EH, et al. Persistance of leishmania parasites in scars after clinical cure of American cutaneous leishmaniasis: is there a sterile cure [published online ahead of print March 2, 2004]? J Infect Dis. 2004;189:1018-1023.
1. Etymologia-Leishmaniasis. Emerg Infect Dis. 2008;14:666.
2. Burden and distribution. World Health Organization Web site. http://www.who.int/leishmaniasis/burden/en/. Accessed November 10, 2015.
3. Emergencies preparedness, response. World Health Organization Web site. http://www.who.int/csr/resources/publications/CSR_ISR_2000_1leish/en/. Accessed November 3, 2015.
4. Pavli A, Maltezou HC. Leishmaniasis, an emerging infection in travelers. Int J Infect Dis. 2010;14:e1032-e1039.
5. Magill AJ. Leishmania species: visceral (Kala-Azar), cutaneous, and mucosal leishmaniasis. In: Mandell GL, Bennett JE, Dolin R, eds. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, PA: Churchill Livingstone; 2009:3463-3480.
6. Mysore V. Invisible dermatoses. Indian J Dermatol Venereol Leprol. 2010;76:239-248.
7. Parasites – Leishmaniasis. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/parasites/leishmaniasis/health_professionals/. Updated September 14, 2015. Accessed November 13, 2015.
8. González U, Pinart M, Rengifo-Pardo M, et al. Interventions for American cutaneous and mucocutaneous leishmaniasis. Cochrane Database Syst Rev. 2009;15:CD004834.
9. Herwaldt BL, Stokes SL, Juranek DD. American cutaneous leishmaniasis in U.S. travelers. Ann Intern Med. 1993;118:779-784.
10. Freedman DO, Weld LH, Kozarsky PE, et al. Spectrum of disease and relation to place of exposure among ill returned travelers. New Engl J Med. 2006;354:119-130.
11. El Hajj L, Thellier M, Carriere J, et al. Localized cutaneous leishmaniasis imported into Paris: a review of 39 cases. Int J Dermatol. 2004;43:120-125.
12. Harris E, Kropp G, Belli A, et al. Single-step multiplex PCR assay for characterization of New World Leishmania complexes. J Clin Microbiol. 1998;36:1989-1995.
13. Marfurt J, Niederwieser I, Makia D, et al. Diagnostic genotyping of Old and New World Leishmania species by PCR-RFLP. Diagn Microbiol Infect Dis. 2003;46:115-124.
14. Schonian G, Nasereddin A, Dinse N, et al. PCR diagnosis and characterization of Leishmania in local and imported clinical samples. Diagn Microbiol Infect Dis. 2003;47:349-358.
15. Reithinger R, Aadil K, Kolaczinski J, et al. Social impact of leishmaniasis, Afghanistan. Emerg Infect Dis. 2005;11:634-636.
16. Morales MA, Cruz I, Rubio JM, et al. Relapses versus reinfections in patients coinfected with Leishmania infantum and human immunodeficiency virus type 1 [published online ahead of print April 22, 2002]. J Infect Dis. 2002;185:1533-1537.
17. Coutinho SG, Pirmez C, Da-Cruz AM. Parasitological and immunological follow-up of American tegumentary leishmaniasis patients. Trans R Soc Trop Med Hyg. 2002;96(suppl 1):S173-S178.
18. Mendonça MG, de Brito ME, Rodrigues EH, et al. Persistance of leishmania parasites in scars after clinical cure of American cutaneous leishmaniasis: is there a sterile cure [published online ahead of print March 2, 2004]? J Infect Dis. 2004;189:1018-1023.
Practice Points
- Cutaneous leishmaniasis is an emerging infectious disease that may be misdiagnosed due to its rarity and varied clinical presentation as well as the limited use of tissue biopsy in general practice.
- United States health care practitioners who evaluate patients with new isolated skin lesions and a history of recent travel to Mexico or South or Central Americas should consider cutaneous leishmaniasis in the differential diagnosis.
- Whenever possible, travelers to rural areas of Mexico and South and Central Americas should be educated about strategies to avoid arthropod bites, such as wearing protective clothing and using insect repellents.