User login
Tonsillectomy for this 35-year-old patient?
THE CASE
A 35-year-old woman sought care for a fever and sore throat that she’d had for 4 days. She denied symptoms of cough, rhinorrhea, or sputum production.
The patient’s medical history included severe recurrent streptococcal pharyngitis as a child and teenager. At the age of 17, she developed a fever of 105° F with associated delirium, dysphagia, nausea, and vomiting, and missed several days of school. She also lost 82 pounds, developed oral thrush, and continued to feel fatigued for approximately a year. After her primary care physician noted a heart murmur on physical exam, she was sent for echocardiography and diagnosed with rheumatic fever secondary to streptococcal pharyngitis.
Eighteen years (and numerous streptococcal infections) later, the patient was at our facility and we were ordering a rapid antigen detection test (RADT) for her current illness. The throat specimen was positive for group A ß-hemolytic streptococcus (GAS). The patient’s 8-year-old daughter also had a sore throat, fever, and positive RADT; her symptoms resolved with oral amoxicillin for 10 days. The patient’s husband was also treated successfully with oral amoxicillin/clavulanate for 10 days for similar symptoms. The patient herself, however, was unsuccessfully treated with oral amoxicillin 500 mg twice daily for 7 days.
She was then given oral amoxicillin/clavulanate 875 mg twice daily for 14 days, but received no relief. Even after receiving clindamycin 600 mg twice daily for 10 days, she had minimal relief and remained positive for GAS on repeat RADT. It was at this point that tonsillectomy was considered as a possible treatment modality for her refractory GAS pharyngitis.
The patient consented to the procedure and underwent a tonsillectomy. She has remained asymptomatic for 2 years and there have been no reported outbreaks of GAS infection in her household.
DISCUSSION
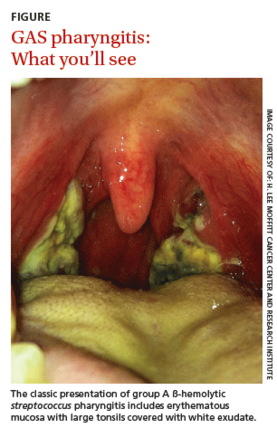
Streptococcal pharyngitis is an infection of the oropharynx and/or nasopharynx that is caused by Streptococcus pyogenes (also known as GAS). It is one of the most frequent illnesses encountered by primary care physicians, and primarily occurs in children ages 5 to 15 years.1,2 The signs and symptoms of GAS pharyngitis include an abrupt onset of a sore throat, tonsillar exudate, tender cervical adenopathy, and fever. (The classic presentation of GAS pharyngitis in a different patient can be seen in the FIGURE.)
Throat cultures are the gold standard for the diagnosis of GAS pharyngitis, but results take 24 to 48 hours, which can delay appropriate treatment. Therefore, the use of the RADT is often preferred clinically.1 RADT is not recommended for children and adults who show clinical symptoms that are highly suggestive of a viral illness, such as cough, rhinorrhea, hoarseness, or oral ulcers. A negative RADT in children and adolescents necessitates a throat culture to confirm the diagnosis.2
The antibiotics of choice are either penicillin 50 mg/kg/d in 4 divided doses or amoxicillin 40 mg/kg/d in 3 divided doses (maximum for both is 2000 mg/d) for 10 days. Options for patients with penicillin allergies include clindamycin or clarithromycin for 10 days or azithromycin for 5 days.2
The Infectious Diseases Society of America (IDSA) does not recommend routine testing or empiric treatment of asymptomatic carriers. However, it does recommend treatment of GAS carriers in certain situations, such as when: 2
- the carrier has acute rheumatic fever
- there is a family or personal history of acute rheumatic fever
- there is a post-streptococcal glomerulonephritis outbreak
- a family has excessive anxiety about GAS infections
- a tonsillectomy is being considered.
When—and for whom—is tonsillectomy beneficial?
Tonsillectomy is a treatment option for patients with recurrent episodes of GAS pharyngitis. Indications include patients with 7 GAS infections in a year, 5 episodes in 2 years, or 3 episodes in 3 years.3,4 In select patient populations, tonsillectomy has been shown to decrease missed work days and medical expenses caused by recurrent pharyngitis.5,6
Alho et al demonstrated that adults with recurrent episodes of GAS pharyngitis benefit from tonsillectomy in terms of fewer repeat infections and more days without throat pain.7 A randomized controlled trial conducted by Koskenkorva et al found that the overall rates of pharyngitis, throat pain, rhinitis, and cough were significantly lower in adults who received a tonsillectomy vs those who did not.5 Still, whether tonsillectomy is worthwhile in adults is debatable; Burton et al found no evidence that tonsillectomy is effective for chronic or recurrent acute tonsillitis in adults.8
Overall meta-analysis results indicate that tonsillectomy results in a 43% reduction in the incidence of pharyngitis in children between the ages of 4 and 16.8,9 One study found that children without tonsillectomy were 3.1 times more likely to develop subsequent GAS pharyngitis than children who underwent tonsillectomy.9 Another study found that children who received tonsillectomy demonstrated a decrease in sore throat episodes by 1.2 episodes per year and a decrease in school absenteeism by 2.8 days per year.6 Tonsillectomy does carry a risk of intraoperative and postoperative bleeding in children and adults, which may make it a less desirable option for some patients.6
THE TAKEAWAY
Recurrent GAS pharyngitis poses a significant challenge for clinicians. When episodes recur, it may be prudent to treat asymptomatic carriers in the patient’s household. Tonsillectomy should be considered in refractory cases since recurrent GAS pharyngitis directly impacts the wellness and productivity of patients. Our patient certainly benefited from the surgery: She has not missed any work days or had to visit her primary care physician because of a GAS infection since her tonsillectomy.
1. Gurol Y, Akan H, Izbirak G, et al. The sensitivity and the specificity of rapid antigen test in streptococcal upper respiratory tract infections. Int J Pediatr Otorhinolaryngol. 2010;74:591-593.
2. Shulman ST, Bisno AL, Clegg HW, et al. Clinical practice guideline for the diagnosis and management of group A streptococcal pharyngitis: 2012 update by the Infectious Diseases Society of America. Clin Infect Dis. 2012;55:1279-1282.
3. Stuck BA, Götte K, Windfuhr JP, et al. Tonsillectomy in children. Dtsch Arztebl Int. 2008;105:852-860.
4. Baugh RF, Archer SM, Mitchell RB, et al; American Academy of Otolaryngology-Head and Neck Surgery Foundation. Clinical practice guideline: tonsillectomy in children. Otolaryngol Head Neck Surg. 2011;144:S1-S30.
5. Koskenkorva T, Koivunen P, Koskela M, et al. Short-term outcomes of tonsillectomy in adult patients with recurrent pharyngitis: a randomized controlled trial. CMAJ. 2013;185:E331-E336.
6. van Staaij BK, van den Akker EH, van der Heijden GJ, et al. Adenotonsillectomy for upper respiratory infections: evidence based? Arch Dis Child. 2005;90:19-25.
7. Alho OP, Koivunen P, Penna T, et al. Tonsillectomy versus watchful waiting in recurrent streptococcal pharyngitis in adults: randomised controlled trial. BMJ. 2007;334:939.
8. Burton MJ, Towler B, Glasziou P. Tonsillectomy versus non-surgical treatment for chronic/recurrent acute tonsillitis. Cochrane Database Syst Rev. 2000;(2):CD001802.
9. Orvidas LJ, St Sauver JL, Weaver AL. Efficacy of tonsillectomy in treatment of recurrent group A beta-hemolytic streptococcal pharyngitis. Laryngoscope. 2006;116:1946-1950.
THE CASE
A 35-year-old woman sought care for a fever and sore throat that she’d had for 4 days. She denied symptoms of cough, rhinorrhea, or sputum production.
The patient’s medical history included severe recurrent streptococcal pharyngitis as a child and teenager. At the age of 17, she developed a fever of 105° F with associated delirium, dysphagia, nausea, and vomiting, and missed several days of school. She also lost 82 pounds, developed oral thrush, and continued to feel fatigued for approximately a year. After her primary care physician noted a heart murmur on physical exam, she was sent for echocardiography and diagnosed with rheumatic fever secondary to streptococcal pharyngitis.
Eighteen years (and numerous streptococcal infections) later, the patient was at our facility and we were ordering a rapid antigen detection test (RADT) for her current illness. The throat specimen was positive for group A ß-hemolytic streptococcus (GAS). The patient’s 8-year-old daughter also had a sore throat, fever, and positive RADT; her symptoms resolved with oral amoxicillin for 10 days. The patient’s husband was also treated successfully with oral amoxicillin/clavulanate for 10 days for similar symptoms. The patient herself, however, was unsuccessfully treated with oral amoxicillin 500 mg twice daily for 7 days.
She was then given oral amoxicillin/clavulanate 875 mg twice daily for 14 days, but received no relief. Even after receiving clindamycin 600 mg twice daily for 10 days, she had minimal relief and remained positive for GAS on repeat RADT. It was at this point that tonsillectomy was considered as a possible treatment modality for her refractory GAS pharyngitis.
The patient consented to the procedure and underwent a tonsillectomy. She has remained asymptomatic for 2 years and there have been no reported outbreaks of GAS infection in her household.
DISCUSSION
Streptococcal pharyngitis is an infection of the oropharynx and/or nasopharynx that is caused by Streptococcus pyogenes (also known as GAS). It is one of the most frequent illnesses encountered by primary care physicians, and primarily occurs in children ages 5 to 15 years.1,2 The signs and symptoms of GAS pharyngitis include an abrupt onset of a sore throat, tonsillar exudate, tender cervical adenopathy, and fever. (The classic presentation of GAS pharyngitis in a different patient can be seen in the FIGURE.)
Throat cultures are the gold standard for the diagnosis of GAS pharyngitis, but results take 24 to 48 hours, which can delay appropriate treatment. Therefore, the use of the RADT is often preferred clinically.1 RADT is not recommended for children and adults who show clinical symptoms that are highly suggestive of a viral illness, such as cough, rhinorrhea, hoarseness, or oral ulcers. A negative RADT in children and adolescents necessitates a throat culture to confirm the diagnosis.2
The antibiotics of choice are either penicillin 50 mg/kg/d in 4 divided doses or amoxicillin 40 mg/kg/d in 3 divided doses (maximum for both is 2000 mg/d) for 10 days. Options for patients with penicillin allergies include clindamycin or clarithromycin for 10 days or azithromycin for 5 days.2
The Infectious Diseases Society of America (IDSA) does not recommend routine testing or empiric treatment of asymptomatic carriers. However, it does recommend treatment of GAS carriers in certain situations, such as when: 2
- the carrier has acute rheumatic fever
- there is a family or personal history of acute rheumatic fever
- there is a post-streptococcal glomerulonephritis outbreak
- a family has excessive anxiety about GAS infections
- a tonsillectomy is being considered.
When—and for whom—is tonsillectomy beneficial?
Tonsillectomy is a treatment option for patients with recurrent episodes of GAS pharyngitis. Indications include patients with 7 GAS infections in a year, 5 episodes in 2 years, or 3 episodes in 3 years.3,4 In select patient populations, tonsillectomy has been shown to decrease missed work days and medical expenses caused by recurrent pharyngitis.5,6
Alho et al demonstrated that adults with recurrent episodes of GAS pharyngitis benefit from tonsillectomy in terms of fewer repeat infections and more days without throat pain.7 A randomized controlled trial conducted by Koskenkorva et al found that the overall rates of pharyngitis, throat pain, rhinitis, and cough were significantly lower in adults who received a tonsillectomy vs those who did not.5 Still, whether tonsillectomy is worthwhile in adults is debatable; Burton et al found no evidence that tonsillectomy is effective for chronic or recurrent acute tonsillitis in adults.8
Overall meta-analysis results indicate that tonsillectomy results in a 43% reduction in the incidence of pharyngitis in children between the ages of 4 and 16.8,9 One study found that children without tonsillectomy were 3.1 times more likely to develop subsequent GAS pharyngitis than children who underwent tonsillectomy.9 Another study found that children who received tonsillectomy demonstrated a decrease in sore throat episodes by 1.2 episodes per year and a decrease in school absenteeism by 2.8 days per year.6 Tonsillectomy does carry a risk of intraoperative and postoperative bleeding in children and adults, which may make it a less desirable option for some patients.6
THE TAKEAWAY
Recurrent GAS pharyngitis poses a significant challenge for clinicians. When episodes recur, it may be prudent to treat asymptomatic carriers in the patient’s household. Tonsillectomy should be considered in refractory cases since recurrent GAS pharyngitis directly impacts the wellness and productivity of patients. Our patient certainly benefited from the surgery: She has not missed any work days or had to visit her primary care physician because of a GAS infection since her tonsillectomy.
THE CASE
A 35-year-old woman sought care for a fever and sore throat that she’d had for 4 days. She denied symptoms of cough, rhinorrhea, or sputum production.
The patient’s medical history included severe recurrent streptococcal pharyngitis as a child and teenager. At the age of 17, she developed a fever of 105° F with associated delirium, dysphagia, nausea, and vomiting, and missed several days of school. She also lost 82 pounds, developed oral thrush, and continued to feel fatigued for approximately a year. After her primary care physician noted a heart murmur on physical exam, she was sent for echocardiography and diagnosed with rheumatic fever secondary to streptococcal pharyngitis.
Eighteen years (and numerous streptococcal infections) later, the patient was at our facility and we were ordering a rapid antigen detection test (RADT) for her current illness. The throat specimen was positive for group A ß-hemolytic streptococcus (GAS). The patient’s 8-year-old daughter also had a sore throat, fever, and positive RADT; her symptoms resolved with oral amoxicillin for 10 days. The patient’s husband was also treated successfully with oral amoxicillin/clavulanate for 10 days for similar symptoms. The patient herself, however, was unsuccessfully treated with oral amoxicillin 500 mg twice daily for 7 days.
She was then given oral amoxicillin/clavulanate 875 mg twice daily for 14 days, but received no relief. Even after receiving clindamycin 600 mg twice daily for 10 days, she had minimal relief and remained positive for GAS on repeat RADT. It was at this point that tonsillectomy was considered as a possible treatment modality for her refractory GAS pharyngitis.
The patient consented to the procedure and underwent a tonsillectomy. She has remained asymptomatic for 2 years and there have been no reported outbreaks of GAS infection in her household.
DISCUSSION
Streptococcal pharyngitis is an infection of the oropharynx and/or nasopharynx that is caused by Streptococcus pyogenes (also known as GAS). It is one of the most frequent illnesses encountered by primary care physicians, and primarily occurs in children ages 5 to 15 years.1,2 The signs and symptoms of GAS pharyngitis include an abrupt onset of a sore throat, tonsillar exudate, tender cervical adenopathy, and fever. (The classic presentation of GAS pharyngitis in a different patient can be seen in the FIGURE.)
Throat cultures are the gold standard for the diagnosis of GAS pharyngitis, but results take 24 to 48 hours, which can delay appropriate treatment. Therefore, the use of the RADT is often preferred clinically.1 RADT is not recommended for children and adults who show clinical symptoms that are highly suggestive of a viral illness, such as cough, rhinorrhea, hoarseness, or oral ulcers. A negative RADT in children and adolescents necessitates a throat culture to confirm the diagnosis.2
The antibiotics of choice are either penicillin 50 mg/kg/d in 4 divided doses or amoxicillin 40 mg/kg/d in 3 divided doses (maximum for both is 2000 mg/d) for 10 days. Options for patients with penicillin allergies include clindamycin or clarithromycin for 10 days or azithromycin for 5 days.2
The Infectious Diseases Society of America (IDSA) does not recommend routine testing or empiric treatment of asymptomatic carriers. However, it does recommend treatment of GAS carriers in certain situations, such as when: 2
- the carrier has acute rheumatic fever
- there is a family or personal history of acute rheumatic fever
- there is a post-streptococcal glomerulonephritis outbreak
- a family has excessive anxiety about GAS infections
- a tonsillectomy is being considered.
When—and for whom—is tonsillectomy beneficial?
Tonsillectomy is a treatment option for patients with recurrent episodes of GAS pharyngitis. Indications include patients with 7 GAS infections in a year, 5 episodes in 2 years, or 3 episodes in 3 years.3,4 In select patient populations, tonsillectomy has been shown to decrease missed work days and medical expenses caused by recurrent pharyngitis.5,6
Alho et al demonstrated that adults with recurrent episodes of GAS pharyngitis benefit from tonsillectomy in terms of fewer repeat infections and more days without throat pain.7 A randomized controlled trial conducted by Koskenkorva et al found that the overall rates of pharyngitis, throat pain, rhinitis, and cough were significantly lower in adults who received a tonsillectomy vs those who did not.5 Still, whether tonsillectomy is worthwhile in adults is debatable; Burton et al found no evidence that tonsillectomy is effective for chronic or recurrent acute tonsillitis in adults.8
Overall meta-analysis results indicate that tonsillectomy results in a 43% reduction in the incidence of pharyngitis in children between the ages of 4 and 16.8,9 One study found that children without tonsillectomy were 3.1 times more likely to develop subsequent GAS pharyngitis than children who underwent tonsillectomy.9 Another study found that children who received tonsillectomy demonstrated a decrease in sore throat episodes by 1.2 episodes per year and a decrease in school absenteeism by 2.8 days per year.6 Tonsillectomy does carry a risk of intraoperative and postoperative bleeding in children and adults, which may make it a less desirable option for some patients.6
THE TAKEAWAY
Recurrent GAS pharyngitis poses a significant challenge for clinicians. When episodes recur, it may be prudent to treat asymptomatic carriers in the patient’s household. Tonsillectomy should be considered in refractory cases since recurrent GAS pharyngitis directly impacts the wellness and productivity of patients. Our patient certainly benefited from the surgery: She has not missed any work days or had to visit her primary care physician because of a GAS infection since her tonsillectomy.
1. Gurol Y, Akan H, Izbirak G, et al. The sensitivity and the specificity of rapid antigen test in streptococcal upper respiratory tract infections. Int J Pediatr Otorhinolaryngol. 2010;74:591-593.
2. Shulman ST, Bisno AL, Clegg HW, et al. Clinical practice guideline for the diagnosis and management of group A streptococcal pharyngitis: 2012 update by the Infectious Diseases Society of America. Clin Infect Dis. 2012;55:1279-1282.
3. Stuck BA, Götte K, Windfuhr JP, et al. Tonsillectomy in children. Dtsch Arztebl Int. 2008;105:852-860.
4. Baugh RF, Archer SM, Mitchell RB, et al; American Academy of Otolaryngology-Head and Neck Surgery Foundation. Clinical practice guideline: tonsillectomy in children. Otolaryngol Head Neck Surg. 2011;144:S1-S30.
5. Koskenkorva T, Koivunen P, Koskela M, et al. Short-term outcomes of tonsillectomy in adult patients with recurrent pharyngitis: a randomized controlled trial. CMAJ. 2013;185:E331-E336.
6. van Staaij BK, van den Akker EH, van der Heijden GJ, et al. Adenotonsillectomy for upper respiratory infections: evidence based? Arch Dis Child. 2005;90:19-25.
7. Alho OP, Koivunen P, Penna T, et al. Tonsillectomy versus watchful waiting in recurrent streptococcal pharyngitis in adults: randomised controlled trial. BMJ. 2007;334:939.
8. Burton MJ, Towler B, Glasziou P. Tonsillectomy versus non-surgical treatment for chronic/recurrent acute tonsillitis. Cochrane Database Syst Rev. 2000;(2):CD001802.
9. Orvidas LJ, St Sauver JL, Weaver AL. Efficacy of tonsillectomy in treatment of recurrent group A beta-hemolytic streptococcal pharyngitis. Laryngoscope. 2006;116:1946-1950.
1. Gurol Y, Akan H, Izbirak G, et al. The sensitivity and the specificity of rapid antigen test in streptococcal upper respiratory tract infections. Int J Pediatr Otorhinolaryngol. 2010;74:591-593.
2. Shulman ST, Bisno AL, Clegg HW, et al. Clinical practice guideline for the diagnosis and management of group A streptococcal pharyngitis: 2012 update by the Infectious Diseases Society of America. Clin Infect Dis. 2012;55:1279-1282.
3. Stuck BA, Götte K, Windfuhr JP, et al. Tonsillectomy in children. Dtsch Arztebl Int. 2008;105:852-860.
4. Baugh RF, Archer SM, Mitchell RB, et al; American Academy of Otolaryngology-Head and Neck Surgery Foundation. Clinical practice guideline: tonsillectomy in children. Otolaryngol Head Neck Surg. 2011;144:S1-S30.
5. Koskenkorva T, Koivunen P, Koskela M, et al. Short-term outcomes of tonsillectomy in adult patients with recurrent pharyngitis: a randomized controlled trial. CMAJ. 2013;185:E331-E336.
6. van Staaij BK, van den Akker EH, van der Heijden GJ, et al. Adenotonsillectomy for upper respiratory infections: evidence based? Arch Dis Child. 2005;90:19-25.
7. Alho OP, Koivunen P, Penna T, et al. Tonsillectomy versus watchful waiting in recurrent streptococcal pharyngitis in adults: randomised controlled trial. BMJ. 2007;334:939.
8. Burton MJ, Towler B, Glasziou P. Tonsillectomy versus non-surgical treatment for chronic/recurrent acute tonsillitis. Cochrane Database Syst Rev. 2000;(2):CD001802.
9. Orvidas LJ, St Sauver JL, Weaver AL. Efficacy of tonsillectomy in treatment of recurrent group A beta-hemolytic streptococcal pharyngitis. Laryngoscope. 2006;116:1946-1950.

Don’t Forget the Pulses! Aortoiliac Peripheral Artery Disease Masquerading as Lumbar Radiculopathy—A Report of 3 Cases
Lumbar radiculopathy is a common problem encountered by orthopedic surgeons, and typically presents with lower back or buttock pain radiating down the leg.1 While the most common causes of lumbar radiculopathy are lumbar disc herniation and spinal stenosis, the differential diagnosis for lower extremity pain is broad and can be musculoskeletal, vascular, neurologic, or inflammatory in nature.1,2 Differentiating between orthopedic, neurologic, and vascular causes of leg pain, such as peripheral artery disease (PAD), can sometimes be challenging. This is especially true in aortoiliac PAD, which can present with hip, buttock, and thigh pain. Dorsalis pedis pulses can be palpable due to collateral circulation. A careful history and physical examination is crucial to the correct diagnosis. The history should clearly document the nature of the pain, details of walking impairment, and the alleviating effects of standing still or positional changes. A complete neurovascular examination should include observations regarding the skin, hair, and nails, examination of dorsal pedis, popliteal, and femoral pulses in comparison to the contralateral side, and documentation of dural tension signs. Misdiagnoses can send the patient down a path of unnecessary tests, unindicated procedures, and ultimately, a delay in definitive diagnosis and treatment.1
To our knowledge, this is the first report on a series of patients with thigh pain initially diagnosed as radiculopathy who underwent unproductive diagnostic tests and procedures, and ultimately were given delayed diagnoses of aortoiliac PAD. The patients provided written informed consent for print and electronic publication of these case reports.
Case 1
An 81-year-old woman with a medical history notable for hypertension, hyperlipidemia, and stroke initially presented to an outside orthopedic institution with complaints of several months of lower back and right hip, thigh, and leg pain when walking. She did not report any history of night pain, weakness, or numbness. Examination at the time was notable for painful back extension, 4/5 hip flexion strength on the right compared to 5/5 on the left, but symmetric reflexes and negative dural tension signs. X-rays showed multilevel degenerative disc disease of the lumbar spine, and magnetic resonance imaging (MRI) showed a small L3/4 disc protrusion causing impingement of the L4 nerve root.
A transforaminal epidural steroid injection at the L4 level was performed with minimal resolution of symptoms. Several months later, right-sided intra-articular facet injections were performed at the L4/5 and L5/S1 levels, again with minimal relief of symptoms. At this point, the patient was sent for further physical therapy.
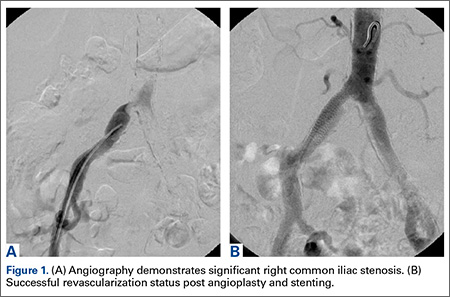
Over a year after symptom onset, the patient presented to our institution and was evaluated by a vascular surgeon. Physical examination was notable for 1+ femoral artery and dorsal pedis pulses on the right side, compared to 2+ on the left. An aortoiliac duplex ultrasound showed severe significant stenosis of the right common iliac artery (>75%).
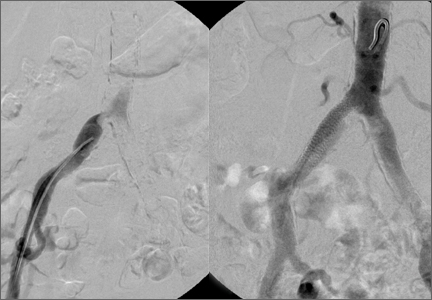
The patient underwent a right common iliac artery angioplasty and stenting (Figures 1A, 1B), which resolved her symptoms.
Case 2
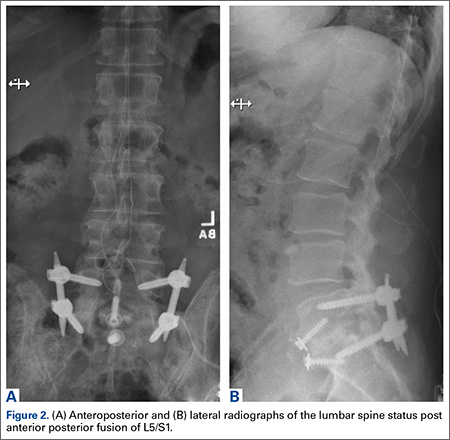
A 65-year-old man, who is a former smoker with a medical history notable for hyperlipidemia and coronary artery disease status post myocardial infarction, presented with a long history of right leg pain. He underwent a L5/S1 anterior posterior fusion at an outside institution and did well for about 5 years after the procedure (Figures 2A, 2B). The pain returned and he underwent several years of physical therapy, epidural steroid injections, and implantation of a spinal cord stimulator with no improvement. He reported right leg pain with minimal back pain, primarily in the thigh and not radiating to the feet and toes. The pain limited him from walking more than 1 block. On examination, strength was 5/5 bilaterally. Pulse examination was notable for lack of dorsalis pedis/posterior tibial pulses bilaterally. He had no bowel or bladder dysfunction.
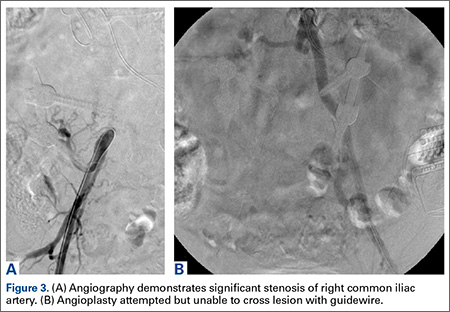
Computed tomography myelogram showed a moderate amount of stenosis at L3/4 and L4/5. He was sent for evaluation by a vascular surgeon. Arterial duplex ultrasound showed significant stenosis of the right common iliac artery.
Angioplasty was attempted but vascular surgery was unable to cross the lesion (Figures 3A, 3B), and the patient ultimately had a femoral-femoral bypass, which resolved his leg pain.
Case 3
A 78-year-old woman, nonsmoker, presented with a 1-year history of left buttock and thigh pain exacerbated by ambulation. Ambulation was limited to 2 blocks. The patient was being worked up for spinal and hip etiologies of pain at an outside hospital. MRI revealed a mild posterior disc herniation at L3/4 and L4/5 and moderate narrowing of the spinal canal. She underwent 2 epidural steroid injections with no improvement. The patient’s relative, a physician, suggested that the patient receive a vascular surgery consultation, and the patient ultimately presented to our institution for evaluation by vascular surgery.
The physical examination was significant for a 1+ dorsal pedis pulse on the left compared to 2+ on the right. Moreover, the patient only demonstrated trace L femoral pulse compared to the right. Strength was 5/5 bilaterally.
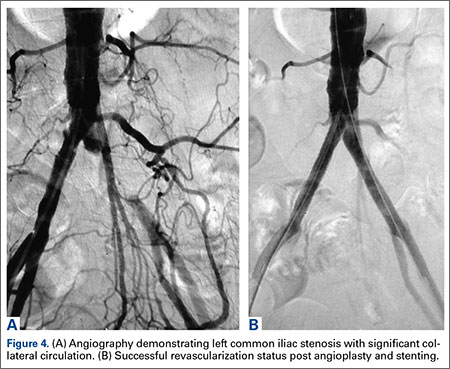
The patient was taken to the operating room for angioplasty and stenting of the left common iliac artery (Figures 4A, 4B). This provided immediate symptom relief, and she has remained asymptomatic.
Discussion
Lumbar radiculopathy is a common diagnosis encountered by orthopedic surgeons. Although the diagnosis can appear to be straightforward in a patient presenting with lower back and leg pain, the etiology of lower back and leg pain can be extremely varied, and can be musculoskeletal, neurologic, vascular, rheumatologic, or oncologic in origin.1 In particular, differentiating between radiculopathy and vascular claudication can sometimes be challenging.
The 2 most common causes of lumbar radiculopathy are lumbar disc herniation and spinal stenosis.1 Lumbar disc herniation results from tear in the annulus of the intervertebral disc, resulting in herniation of disc material into the spinal canal causing compression and irritation of spinal nerve roots.1 Spinal stenosis is narrowing of the spinal canal that produces compression of neural elements before they exit the neural foramen.3 Adult degenerative spinal stenosis is most often caused by osteophytes from the facet joints or hypertrophy of the ligamentum flavum, and can be broadly categorized into central spinal stenosis or lateral spinal stenosis.
PAD is defined as progressive stenosis or occlusion, or aneurysmal dilation of noncoronary arteries.2 When PAD affects the vessels of the lower extremities, the symptoms typically manifest as intermittent claudication, which is exercise-induced ischemic pain in the lower extremity that is relieved by rest.2 As the disease progresses, symptoms can progress to rest pain, ulceration, and, eventually, gangrene. The most common cause of PAD is atherosclerosis, and the risk factors include smoking, hypertension, diabetes, and hyperlipidemia. The prevalence of PAD rises sharply with age, starting from <3% in ages less than 60 years to >20% in ages 75 years and older.4
A detailed and pertinent history from the patient provides important information for differentiating radiculopathy and neurogenic claudication from vascular claudication. Patients with lumbar radiculopathy typically report pain in the lower back radiating down the leg past the knee in a dermatomal distribution. The pain often begins soon if not immediately after activity, but often takes time for relief onset after rest. Positional changes in the back such as flexion can provide relief.2 Patients with neurogenic claudication from central spinal stenosis can present with bilateral thigh pain from prolonged standing and activity that is alleviated with flexion or stooping.3 Patients may admit to a positive “shopping cart sign,” with increased walking comfort stooped forward with hands on a shopping cart.
In contrast, patients with vascular claudication often report pain in the calf, thigh, or hip, but rarely in the foot. The location of pain varies with area of stenosis; generally, patients with superficial femoral artery occlusion present with calf claudication, while patients with aortoiliac disease present with buttock and thigh pain. The pain typically occurs after a very reproducible length of walking, and is relieved by cessation of walking, often even if the patient remains standing. Back positioning should have no effect on the pain.2-5
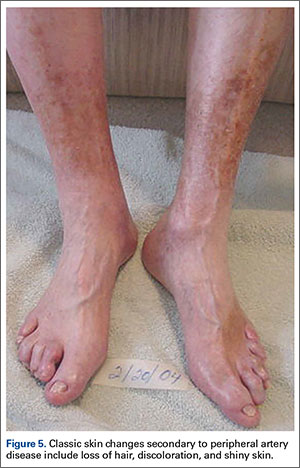
Physical examination should begin with observation of the patient’s gait and posture, which may be hunched over in the setting of spinal stenosis. Examination of the patient’s skin may show loss of hair, shiny skin, or atrophic changes suggestive of vascular disease (Figure 5).1 Prior to proceeding to a spine examination, palpating the trochanteric bursa and testing for hip range of motion is important to rule out intra-articular hip pathology and trochanteric bursitis as common causes of pain in the area. Patients with radiculopathy may show sensory disturbances in a dermatomal distribution, muscular weakness at the corresponding spinal level, and decreased deep tendon reflexes. The straight leg raise test can elicit signs of nerve root tension. A careful examination of bilateral lower extremity pulses at the dorsal pedis, popliteal, and femoral levels can help identify any asymmetric or decreased pulses that would indicate peripheral vascular disease. With chronic aortoiliac disease, it is important to check for femoral pulses, given the dorsal pedis pulse can be present due to collateral circulation. And finally, the ankle brachial index (ABI), measured as the ratio of the systolic pressure at the ankle divided by the systolic pressure at the arm, is a good screening test for PAD.6 A normal ABI is >1.
A thorough history and physical examination can elicit important information that is helpful in evaluating orthopedic patients, especially to differentiate between spinal and vascular causes of leg pain. This can help avoid misdiagnoses, which result in unnecessary tests, procedures, and wasted time. Don’t forget the pulses!
1. Grimm BD, Blessinger BJ, Darden BV, Brigham CD, Kneisl JS, Laxer EB. Mimickers of lumbar radiculopathy. J Am Acad Orthop Surg. 2015;23(1):7-17.
2. Hirsch AT, Haskal ZJ, Hertzer NR, et al. ACC/AHA Guidelines for the Management of Patients with Peripheral Arterial Disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Associations for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (writing committee to develop guidelines for the management of patients with peripheral arterial disease)--summary of recommendations. J Vasc Interv Radiol. 2006;17(9):1383-1397.
3. Spivak JM. Degenerative lumbar spinal stenosis. J Bone Joint Surg Am. 1998;80(7):1053-1066.
4. Criqui MH, Fronek A, Barrett-Connor E, Klauber MR, Gabriel S, Goodman D. The prevalence of peripheral arterial disease in a defined population. Circulation. 1985;71(3):510-515.
5. Ouriel K. Peripheral arterial disease. Lancet. 2001;358(9289):1257-1264.
6. Jeon CH, Han SH, Chung NS, Hyun HS. The validity of ankle-brachial index for the differential diagnosis of peripheral arterial disease and lumbar spinal stenosis in patients with atypical claudication. Eur Spine J. 2012;21(6):1165-1170.
Lumbar radiculopathy is a common problem encountered by orthopedic surgeons, and typically presents with lower back or buttock pain radiating down the leg.1 While the most common causes of lumbar radiculopathy are lumbar disc herniation and spinal stenosis, the differential diagnosis for lower extremity pain is broad and can be musculoskeletal, vascular, neurologic, or inflammatory in nature.1,2 Differentiating between orthopedic, neurologic, and vascular causes of leg pain, such as peripheral artery disease (PAD), can sometimes be challenging. This is especially true in aortoiliac PAD, which can present with hip, buttock, and thigh pain. Dorsalis pedis pulses can be palpable due to collateral circulation. A careful history and physical examination is crucial to the correct diagnosis. The history should clearly document the nature of the pain, details of walking impairment, and the alleviating effects of standing still or positional changes. A complete neurovascular examination should include observations regarding the skin, hair, and nails, examination of dorsal pedis, popliteal, and femoral pulses in comparison to the contralateral side, and documentation of dural tension signs. Misdiagnoses can send the patient down a path of unnecessary tests, unindicated procedures, and ultimately, a delay in definitive diagnosis and treatment.1
To our knowledge, this is the first report on a series of patients with thigh pain initially diagnosed as radiculopathy who underwent unproductive diagnostic tests and procedures, and ultimately were given delayed diagnoses of aortoiliac PAD. The patients provided written informed consent for print and electronic publication of these case reports.
Case 1
An 81-year-old woman with a medical history notable for hypertension, hyperlipidemia, and stroke initially presented to an outside orthopedic institution with complaints of several months of lower back and right hip, thigh, and leg pain when walking. She did not report any history of night pain, weakness, or numbness. Examination at the time was notable for painful back extension, 4/5 hip flexion strength on the right compared to 5/5 on the left, but symmetric reflexes and negative dural tension signs. X-rays showed multilevel degenerative disc disease of the lumbar spine, and magnetic resonance imaging (MRI) showed a small L3/4 disc protrusion causing impingement of the L4 nerve root.
A transforaminal epidural steroid injection at the L4 level was performed with minimal resolution of symptoms. Several months later, right-sided intra-articular facet injections were performed at the L4/5 and L5/S1 levels, again with minimal relief of symptoms. At this point, the patient was sent for further physical therapy.
Over a year after symptom onset, the patient presented to our institution and was evaluated by a vascular surgeon. Physical examination was notable for 1+ femoral artery and dorsal pedis pulses on the right side, compared to 2+ on the left. An aortoiliac duplex ultrasound showed severe significant stenosis of the right common iliac artery (>75%).
The patient underwent a right common iliac artery angioplasty and stenting (Figures 1A, 1B), which resolved her symptoms.
Case 2
A 65-year-old man, who is a former smoker with a medical history notable for hyperlipidemia and coronary artery disease status post myocardial infarction, presented with a long history of right leg pain. He underwent a L5/S1 anterior posterior fusion at an outside institution and did well for about 5 years after the procedure (Figures 2A, 2B). The pain returned and he underwent several years of physical therapy, epidural steroid injections, and implantation of a spinal cord stimulator with no improvement. He reported right leg pain with minimal back pain, primarily in the thigh and not radiating to the feet and toes. The pain limited him from walking more than 1 block. On examination, strength was 5/5 bilaterally. Pulse examination was notable for lack of dorsalis pedis/posterior tibial pulses bilaterally. He had no bowel or bladder dysfunction.
Computed tomography myelogram showed a moderate amount of stenosis at L3/4 and L4/5. He was sent for evaluation by a vascular surgeon. Arterial duplex ultrasound showed significant stenosis of the right common iliac artery.
Angioplasty was attempted but vascular surgery was unable to cross the lesion (Figures 3A, 3B), and the patient ultimately had a femoral-femoral bypass, which resolved his leg pain.
Case 3
A 78-year-old woman, nonsmoker, presented with a 1-year history of left buttock and thigh pain exacerbated by ambulation. Ambulation was limited to 2 blocks. The patient was being worked up for spinal and hip etiologies of pain at an outside hospital. MRI revealed a mild posterior disc herniation at L3/4 and L4/5 and moderate narrowing of the spinal canal. She underwent 2 epidural steroid injections with no improvement. The patient’s relative, a physician, suggested that the patient receive a vascular surgery consultation, and the patient ultimately presented to our institution for evaluation by vascular surgery.
The physical examination was significant for a 1+ dorsal pedis pulse on the left compared to 2+ on the right. Moreover, the patient only demonstrated trace L femoral pulse compared to the right. Strength was 5/5 bilaterally.
The patient was taken to the operating room for angioplasty and stenting of the left common iliac artery (Figures 4A, 4B). This provided immediate symptom relief, and she has remained asymptomatic.
Discussion
Lumbar radiculopathy is a common diagnosis encountered by orthopedic surgeons. Although the diagnosis can appear to be straightforward in a patient presenting with lower back and leg pain, the etiology of lower back and leg pain can be extremely varied, and can be musculoskeletal, neurologic, vascular, rheumatologic, or oncologic in origin.1 In particular, differentiating between radiculopathy and vascular claudication can sometimes be challenging.
The 2 most common causes of lumbar radiculopathy are lumbar disc herniation and spinal stenosis.1 Lumbar disc herniation results from tear in the annulus of the intervertebral disc, resulting in herniation of disc material into the spinal canal causing compression and irritation of spinal nerve roots.1 Spinal stenosis is narrowing of the spinal canal that produces compression of neural elements before they exit the neural foramen.3 Adult degenerative spinal stenosis is most often caused by osteophytes from the facet joints or hypertrophy of the ligamentum flavum, and can be broadly categorized into central spinal stenosis or lateral spinal stenosis.
PAD is defined as progressive stenosis or occlusion, or aneurysmal dilation of noncoronary arteries.2 When PAD affects the vessels of the lower extremities, the symptoms typically manifest as intermittent claudication, which is exercise-induced ischemic pain in the lower extremity that is relieved by rest.2 As the disease progresses, symptoms can progress to rest pain, ulceration, and, eventually, gangrene. The most common cause of PAD is atherosclerosis, and the risk factors include smoking, hypertension, diabetes, and hyperlipidemia. The prevalence of PAD rises sharply with age, starting from <3% in ages less than 60 years to >20% in ages 75 years and older.4
A detailed and pertinent history from the patient provides important information for differentiating radiculopathy and neurogenic claudication from vascular claudication. Patients with lumbar radiculopathy typically report pain in the lower back radiating down the leg past the knee in a dermatomal distribution. The pain often begins soon if not immediately after activity, but often takes time for relief onset after rest. Positional changes in the back such as flexion can provide relief.2 Patients with neurogenic claudication from central spinal stenosis can present with bilateral thigh pain from prolonged standing and activity that is alleviated with flexion or stooping.3 Patients may admit to a positive “shopping cart sign,” with increased walking comfort stooped forward with hands on a shopping cart.
In contrast, patients with vascular claudication often report pain in the calf, thigh, or hip, but rarely in the foot. The location of pain varies with area of stenosis; generally, patients with superficial femoral artery occlusion present with calf claudication, while patients with aortoiliac disease present with buttock and thigh pain. The pain typically occurs after a very reproducible length of walking, and is relieved by cessation of walking, often even if the patient remains standing. Back positioning should have no effect on the pain.2-5
Physical examination should begin with observation of the patient’s gait and posture, which may be hunched over in the setting of spinal stenosis. Examination of the patient’s skin may show loss of hair, shiny skin, or atrophic changes suggestive of vascular disease (Figure 5).1 Prior to proceeding to a spine examination, palpating the trochanteric bursa and testing for hip range of motion is important to rule out intra-articular hip pathology and trochanteric bursitis as common causes of pain in the area. Patients with radiculopathy may show sensory disturbances in a dermatomal distribution, muscular weakness at the corresponding spinal level, and decreased deep tendon reflexes. The straight leg raise test can elicit signs of nerve root tension. A careful examination of bilateral lower extremity pulses at the dorsal pedis, popliteal, and femoral levels can help identify any asymmetric or decreased pulses that would indicate peripheral vascular disease. With chronic aortoiliac disease, it is important to check for femoral pulses, given the dorsal pedis pulse can be present due to collateral circulation. And finally, the ankle brachial index (ABI), measured as the ratio of the systolic pressure at the ankle divided by the systolic pressure at the arm, is a good screening test for PAD.6 A normal ABI is >1.
A thorough history and physical examination can elicit important information that is helpful in evaluating orthopedic patients, especially to differentiate between spinal and vascular causes of leg pain. This can help avoid misdiagnoses, which result in unnecessary tests, procedures, and wasted time. Don’t forget the pulses!
Lumbar radiculopathy is a common problem encountered by orthopedic surgeons, and typically presents with lower back or buttock pain radiating down the leg.1 While the most common causes of lumbar radiculopathy are lumbar disc herniation and spinal stenosis, the differential diagnosis for lower extremity pain is broad and can be musculoskeletal, vascular, neurologic, or inflammatory in nature.1,2 Differentiating between orthopedic, neurologic, and vascular causes of leg pain, such as peripheral artery disease (PAD), can sometimes be challenging. This is especially true in aortoiliac PAD, which can present with hip, buttock, and thigh pain. Dorsalis pedis pulses can be palpable due to collateral circulation. A careful history and physical examination is crucial to the correct diagnosis. The history should clearly document the nature of the pain, details of walking impairment, and the alleviating effects of standing still or positional changes. A complete neurovascular examination should include observations regarding the skin, hair, and nails, examination of dorsal pedis, popliteal, and femoral pulses in comparison to the contralateral side, and documentation of dural tension signs. Misdiagnoses can send the patient down a path of unnecessary tests, unindicated procedures, and ultimately, a delay in definitive diagnosis and treatment.1
To our knowledge, this is the first report on a series of patients with thigh pain initially diagnosed as radiculopathy who underwent unproductive diagnostic tests and procedures, and ultimately were given delayed diagnoses of aortoiliac PAD. The patients provided written informed consent for print and electronic publication of these case reports.
Case 1
An 81-year-old woman with a medical history notable for hypertension, hyperlipidemia, and stroke initially presented to an outside orthopedic institution with complaints of several months of lower back and right hip, thigh, and leg pain when walking. She did not report any history of night pain, weakness, or numbness. Examination at the time was notable for painful back extension, 4/5 hip flexion strength on the right compared to 5/5 on the left, but symmetric reflexes and negative dural tension signs. X-rays showed multilevel degenerative disc disease of the lumbar spine, and magnetic resonance imaging (MRI) showed a small L3/4 disc protrusion causing impingement of the L4 nerve root.
A transforaminal epidural steroid injection at the L4 level was performed with minimal resolution of symptoms. Several months later, right-sided intra-articular facet injections were performed at the L4/5 and L5/S1 levels, again with minimal relief of symptoms. At this point, the patient was sent for further physical therapy.
Over a year after symptom onset, the patient presented to our institution and was evaluated by a vascular surgeon. Physical examination was notable for 1+ femoral artery and dorsal pedis pulses on the right side, compared to 2+ on the left. An aortoiliac duplex ultrasound showed severe significant stenosis of the right common iliac artery (>75%).
The patient underwent a right common iliac artery angioplasty and stenting (Figures 1A, 1B), which resolved her symptoms.
Case 2
A 65-year-old man, who is a former smoker with a medical history notable for hyperlipidemia and coronary artery disease status post myocardial infarction, presented with a long history of right leg pain. He underwent a L5/S1 anterior posterior fusion at an outside institution and did well for about 5 years after the procedure (Figures 2A, 2B). The pain returned and he underwent several years of physical therapy, epidural steroid injections, and implantation of a spinal cord stimulator with no improvement. He reported right leg pain with minimal back pain, primarily in the thigh and not radiating to the feet and toes. The pain limited him from walking more than 1 block. On examination, strength was 5/5 bilaterally. Pulse examination was notable for lack of dorsalis pedis/posterior tibial pulses bilaterally. He had no bowel or bladder dysfunction.
Computed tomography myelogram showed a moderate amount of stenosis at L3/4 and L4/5. He was sent for evaluation by a vascular surgeon. Arterial duplex ultrasound showed significant stenosis of the right common iliac artery.
Angioplasty was attempted but vascular surgery was unable to cross the lesion (Figures 3A, 3B), and the patient ultimately had a femoral-femoral bypass, which resolved his leg pain.
Case 3
A 78-year-old woman, nonsmoker, presented with a 1-year history of left buttock and thigh pain exacerbated by ambulation. Ambulation was limited to 2 blocks. The patient was being worked up for spinal and hip etiologies of pain at an outside hospital. MRI revealed a mild posterior disc herniation at L3/4 and L4/5 and moderate narrowing of the spinal canal. She underwent 2 epidural steroid injections with no improvement. The patient’s relative, a physician, suggested that the patient receive a vascular surgery consultation, and the patient ultimately presented to our institution for evaluation by vascular surgery.
The physical examination was significant for a 1+ dorsal pedis pulse on the left compared to 2+ on the right. Moreover, the patient only demonstrated trace L femoral pulse compared to the right. Strength was 5/5 bilaterally.
The patient was taken to the operating room for angioplasty and stenting of the left common iliac artery (Figures 4A, 4B). This provided immediate symptom relief, and she has remained asymptomatic.
Discussion
Lumbar radiculopathy is a common diagnosis encountered by orthopedic surgeons. Although the diagnosis can appear to be straightforward in a patient presenting with lower back and leg pain, the etiology of lower back and leg pain can be extremely varied, and can be musculoskeletal, neurologic, vascular, rheumatologic, or oncologic in origin.1 In particular, differentiating between radiculopathy and vascular claudication can sometimes be challenging.
The 2 most common causes of lumbar radiculopathy are lumbar disc herniation and spinal stenosis.1 Lumbar disc herniation results from tear in the annulus of the intervertebral disc, resulting in herniation of disc material into the spinal canal causing compression and irritation of spinal nerve roots.1 Spinal stenosis is narrowing of the spinal canal that produces compression of neural elements before they exit the neural foramen.3 Adult degenerative spinal stenosis is most often caused by osteophytes from the facet joints or hypertrophy of the ligamentum flavum, and can be broadly categorized into central spinal stenosis or lateral spinal stenosis.
PAD is defined as progressive stenosis or occlusion, or aneurysmal dilation of noncoronary arteries.2 When PAD affects the vessels of the lower extremities, the symptoms typically manifest as intermittent claudication, which is exercise-induced ischemic pain in the lower extremity that is relieved by rest.2 As the disease progresses, symptoms can progress to rest pain, ulceration, and, eventually, gangrene. The most common cause of PAD is atherosclerosis, and the risk factors include smoking, hypertension, diabetes, and hyperlipidemia. The prevalence of PAD rises sharply with age, starting from <3% in ages less than 60 years to >20% in ages 75 years and older.4
A detailed and pertinent history from the patient provides important information for differentiating radiculopathy and neurogenic claudication from vascular claudication. Patients with lumbar radiculopathy typically report pain in the lower back radiating down the leg past the knee in a dermatomal distribution. The pain often begins soon if not immediately after activity, but often takes time for relief onset after rest. Positional changes in the back such as flexion can provide relief.2 Patients with neurogenic claudication from central spinal stenosis can present with bilateral thigh pain from prolonged standing and activity that is alleviated with flexion or stooping.3 Patients may admit to a positive “shopping cart sign,” with increased walking comfort stooped forward with hands on a shopping cart.
In contrast, patients with vascular claudication often report pain in the calf, thigh, or hip, but rarely in the foot. The location of pain varies with area of stenosis; generally, patients with superficial femoral artery occlusion present with calf claudication, while patients with aortoiliac disease present with buttock and thigh pain. The pain typically occurs after a very reproducible length of walking, and is relieved by cessation of walking, often even if the patient remains standing. Back positioning should have no effect on the pain.2-5
Physical examination should begin with observation of the patient’s gait and posture, which may be hunched over in the setting of spinal stenosis. Examination of the patient’s skin may show loss of hair, shiny skin, or atrophic changes suggestive of vascular disease (Figure 5).1 Prior to proceeding to a spine examination, palpating the trochanteric bursa and testing for hip range of motion is important to rule out intra-articular hip pathology and trochanteric bursitis as common causes of pain in the area. Patients with radiculopathy may show sensory disturbances in a dermatomal distribution, muscular weakness at the corresponding spinal level, and decreased deep tendon reflexes. The straight leg raise test can elicit signs of nerve root tension. A careful examination of bilateral lower extremity pulses at the dorsal pedis, popliteal, and femoral levels can help identify any asymmetric or decreased pulses that would indicate peripheral vascular disease. With chronic aortoiliac disease, it is important to check for femoral pulses, given the dorsal pedis pulse can be present due to collateral circulation. And finally, the ankle brachial index (ABI), measured as the ratio of the systolic pressure at the ankle divided by the systolic pressure at the arm, is a good screening test for PAD.6 A normal ABI is >1.
A thorough history and physical examination can elicit important information that is helpful in evaluating orthopedic patients, especially to differentiate between spinal and vascular causes of leg pain. This can help avoid misdiagnoses, which result in unnecessary tests, procedures, and wasted time. Don’t forget the pulses!
1. Grimm BD, Blessinger BJ, Darden BV, Brigham CD, Kneisl JS, Laxer EB. Mimickers of lumbar radiculopathy. J Am Acad Orthop Surg. 2015;23(1):7-17.
2. Hirsch AT, Haskal ZJ, Hertzer NR, et al. ACC/AHA Guidelines for the Management of Patients with Peripheral Arterial Disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Associations for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (writing committee to develop guidelines for the management of patients with peripheral arterial disease)--summary of recommendations. J Vasc Interv Radiol. 2006;17(9):1383-1397.
3. Spivak JM. Degenerative lumbar spinal stenosis. J Bone Joint Surg Am. 1998;80(7):1053-1066.
4. Criqui MH, Fronek A, Barrett-Connor E, Klauber MR, Gabriel S, Goodman D. The prevalence of peripheral arterial disease in a defined population. Circulation. 1985;71(3):510-515.
5. Ouriel K. Peripheral arterial disease. Lancet. 2001;358(9289):1257-1264.
6. Jeon CH, Han SH, Chung NS, Hyun HS. The validity of ankle-brachial index for the differential diagnosis of peripheral arterial disease and lumbar spinal stenosis in patients with atypical claudication. Eur Spine J. 2012;21(6):1165-1170.
1. Grimm BD, Blessinger BJ, Darden BV, Brigham CD, Kneisl JS, Laxer EB. Mimickers of lumbar radiculopathy. J Am Acad Orthop Surg. 2015;23(1):7-17.
2. Hirsch AT, Haskal ZJ, Hertzer NR, et al. ACC/AHA Guidelines for the Management of Patients with Peripheral Arterial Disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Associations for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (writing committee to develop guidelines for the management of patients with peripheral arterial disease)--summary of recommendations. J Vasc Interv Radiol. 2006;17(9):1383-1397.
3. Spivak JM. Degenerative lumbar spinal stenosis. J Bone Joint Surg Am. 1998;80(7):1053-1066.
4. Criqui MH, Fronek A, Barrett-Connor E, Klauber MR, Gabriel S, Goodman D. The prevalence of peripheral arterial disease in a defined population. Circulation. 1985;71(3):510-515.
5. Ouriel K. Peripheral arterial disease. Lancet. 2001;358(9289):1257-1264.
6. Jeon CH, Han SH, Chung NS, Hyun HS. The validity of ankle-brachial index for the differential diagnosis of peripheral arterial disease and lumbar spinal stenosis in patients with atypical claudication. Eur Spine J. 2012;21(6):1165-1170.
A Physician With Thigh Pain
Necrotizing soft-tissue infection (NSTI) often is difficult to distinguish from a superficial soft-tissue infection like cellulitis. Both conditions present with pain, edema, and erythema and can be accompanied by fever and malaise. The diagnosis of NSTI must be made quickly because successful treatment requires early surgical debridement and broad-spectrum antibiotics. The following case demonstrates the challenge of diagnosing NSTI.
Case Presentation
A 50-year-old physician developed a sore throat with subjective fevers, night sweats, and chills. After 2 days, his symptoms resolved. The next day he developed right thigh pain while playing tennis and limped off the court. That night he had fevers, chills, and sweats. For the next 3 days, his right thigh pain persisted with waxing and waning fevers.
The patient’s medical history included gastroesophageal reflux disease, vitamin D deficiency, and a positive purified protein derivative test for which he had completed 1 year of isoniazid therapy. The patient was married and in a monogamous relationship with his wife. He had traveled to the Sierra National Forest and Yosemite Park during the preceding winter. He did not swim in a lake or recall a tick bite. He had not consumed raw food, imported meats, or dairy products. He recently started oral fluconazole for tinea corporis.
The patient’s temperature was 39.5°C, heart rate was 115 beats per minute, blood pressure (BP) was 142/88 mm Hg, and respiratory rate was 18 breaths per minute with an oxygen saturation of 95% while breathing ambient air. He was drenched in sweat yet remained comfortable throughout the interview. The oropharyngeal mucosa was moist without lesions or erythema. There was no rash or lymphadenopathy. The lungs were clear to auscultation. The cardiac exam revealed tachycardia. There was point tenderness to deep palpation of the mid-anterior right thigh without crepitus, erythema, or edema.
The patient’s sodium level was 129 mmol/L (normal range 135-145 mmol/L), bicarbonate was 20 mmol/L (normal range 22-32 mmol/L), creatinine was 1.1 mg/dL (normal range 0.7-1.2 mg/dL), and glucose was 194 mg/dL. The white blood cell count (WBC) was 12,900 cells/mm3 (normal range 3,400-10,000 cells/mm3) with 96% neutrophils. The hematocrit was 41% (normal range 41-53%), and the platelet count was 347,000 cells/mm3 (normal range 140,000-450,000 cells/mm3). The lactate level was 2.2 mmol/L (normal range 0-2 mmol/L). The creatine kinase level was 347 U/L (normal range 50-388 U/L), and the lactate dehydrogenase level was 254 U/L (normal range 102-199 U/L). A rapid group A streptococcal (GAS) antigen test was negative. A radiograph of the right femur revealed mildly edematous soft tissue. On ultrasound the right quadriceps appeared mildly edematous, but there was no evidence of abscess or discrete fluid collection (eFigure 1).
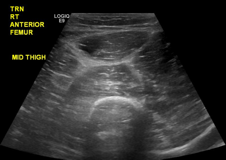
eFigure 1. Ultrasound of the Right Anterior Thigh Ultrasound revealed heterogeneous, mildly edematous quadriceps muscle. There was no abscess or discrete fluid collection. There was trace fluid along the fascia of the quadriceps muscle.
Four liters of normal saline, acetaminophen, ceftriaxone, and doxycycline were administered to the patient. Overnight he was afebrile, tachycardic, and normotensive. The following morning his BP decreased to 81/53 mm Hg. His WBC count was 33,000 cells/mm3 with 96% neutrophils. A peripheral blood smear showed immature granulocytes. The sodium and creatinine increased to 135 mmol/L and 1.3 mg/dL, respectively. The erythrocyte sedimentation rate was 20 mm/h (normal range 0-10 mm/h), and the C-reactive protein level was 174 mg/L (normal range < 6.3 mg/L).The right thigh became erythematous and edematous.
Given concern for necrotizing fasciitis, antibiotics were changed to vancomycin, piperacillin-tazobactam, and clindamycin. The patient was taken to the operating room (OR). The right quadriceps muscle was markedly edematous with overlying necrotic fibrofatty tissue with easy separation of the fascia from the anterolateral rectus femoris and rectus lateralis muscles. Necrotizing fasciitis was diagnosed.
The tissue was debrided, and surgical pathology revealed fibroadipose tissue with extensive necrosis and dense acute inflammation (eFigure 2). After the anterolateral space between the fascia and underlying thigh muscle was drained, a Penrose drain was placed, and the wound was left open with plans for a second-look operation within 24 hours.
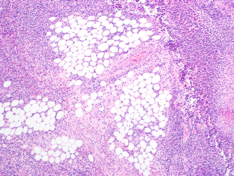
eFigure 2. Surgical Pathology of Debrided Right Thigh
Pathology revealed fibroadipose tissue with extensive necrosis and dense acute inflammation.
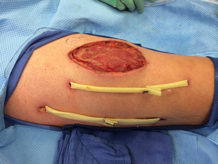
eFigure 3. Right Anterior Thigh
Two Penrose drains inserted after second operation.
In the ensuing hours erythema extended proximal to the operative site. The patient was emergently taken to the OR. The focus of necrotizing fasciitis along the anterolateral aspect of the thigh had extended posteriorly and superiorly. This area was irrigated, all loculations were disrupted, and a second Penrose drain was placed.
The wound was left open for 6 more days. On hospital day 9, operative exploration revealed no necrotizing fasciitis. The fascia and skin wound were then closed (eFigure 3).
Cultures from the fascia grew the GAS bacteria Streptococcus pyogenes (S pyogenes), which was sensitive to penicillin. The blood cultures from admission were sterile. A test for Epstein-Barr virus immunoglobulin M antibody was negative. The patient was discharged after 10 days in the hospital to complete a 2-week course of IV penicillin. Two months later he resumed playing tennis and returned to his clinical duties.
Discussion
In the U.S., there are approximately 3.5 cases of invasive GAS infection per 100,000 persons.1 Type I NSTI is polymicrobial (aerobic and anaerobic organisms). Risk factors include recent surgery, immunocompromised states, drug use, diabetes mellitus, and traumatic wounds.2 Type II NSTI is caused by GAS or other β-hemolytic streptococci either alone or in association with another organism, most commonly Staphylococcus aureus. Type II NSTI is classically found on the extremities and occurs in young, healthy, immunocompetent patients—such as this patient.3
The portal of entry in nearly half of type II NSTI is unknown; minor local trauma is often suspected.4 However, cases have been reported in which the only identifiable source was a preceding sore throat.4 The origin of this patient’s GAS remains unknown, but perhaps his pharyngitis led to transient bacteremia, which then seeded his injured thigh muscle. An in vitro model demonstrated that injured muscles increase surface expression of the cytoskeletal protein vimentin, which binds GAS.5 Exotoxins and endotoxins produced by S pyogenes may lead to microvascular thrombosis, tissue ischemia, liquefactive necrosis, and systemic release of cytokines followed by systemic illness, multiorgan dysfunction, and death.6
The Laboratory Risk Indicator for Necrotizing Fasciitis (LRINEC) score was developed to aid in early diagnosis of NSTI.7 It was derived from a series of 2,555 patients admitted with cellulitis or abscesses at a single institution. Scores > 8 have a positive predictive value of 93% for NSTI. This patient had a LRINEC score of 9. Radiographs or computed tomography scans may demonstrate soft-tissue air collections but lack sensitivity and are often nondiagnostic.8,9 T1-weighted magnetic resonance imaging can delineate the anatomic extent of soft-tissue infections but is time consuming and may delay treatment.10 When the pretest probability is high, proceeding directly to the OR for direct visualization and possible debridement is advisable. Histologic features of necrotizing fasciitis include inflammation with polymorphonuclear cells and necrosis of the subcutaneous fat and fascia with relative sparing of the muscle.11Necrotizing soft-tissue infection requires early surgical debridement and broad-spectrum antibiotic coverage. Without surgical debridement, the mortality rate approaches 100%.2 Antibiotics should include activity against Gram-positive, Gram-negative, and anaerobic organisms. The duration of antibiotic therapy has not been defined and is dependent on the patient’s clinical status. Adjunctive treatment options may include IV immunoglobulin and hyperbaric oxygen therapy, although the data supporting their utility are limited.12,13
Conclusion
Despite the LRINEC scoring systems and advanced imaging, necrotizing fasciitis remains challenging to diagnose in a timely manner. In this case, close monitoring of the patient facilitated timely evaluation and treatment of a fatal disease.
1. O'Loughlin RE, Roberson A, Cieslak PR, et al; Active Bacterial Core Surveillance Team. The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000-2004. Clin Infect Dis. 2007;45(7):853-857.
2. Anaya DA, Dellinger EP. Necrotizing soft-tissue infection: diagnosis and management. Clin Infect Dis. 2007;44(5):705-710.
3. Naqvi GA, Malik SA, Jan W. Necrotizing fasciitis of the lower extremity: a case report and current concept of diagnosis and management. Scand J Trauma Resusc Emerg Med. 2009;17:28.
4. Stevens DL. Streptococcal toxic-shock syndrome: spectrum of disease, pathogenesis, and new concepts in treatment. Emerg Infect Dis. 1195;1(3):69-78.
5. Bryant AE, Bayer CR, Huntington JD, Stevens DL. Group A streptococcal myonecrosis: increased vimentin expression after skeletal-muscle injury mediates the binding of Streptococcus pyogenes. J Infect Dis. 2006;193(12):1685-1692.
6. Cainzos M, Gonzalez-Rodriguez FJ. Necrotizing soft tissue infections. Curr Opin Crit Care. 2007;13(4):433-439.
7. Wong CH, Khin LW, Heng KS, Tan KC, Low CO. The LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score: a tool for distinguishing necrotizing fasciitis from other soft tissue infections. Crit Care Med. 2004;32(7):1535-1541.
8. Goh T, Goh LG, Ang CH, Wong CH. Early diagnosis of necrotizing fasciitis. Br J Surg. 2014;101(1):119-125.
9. Lancerotto L, Tocco I, Salmaso R, Vindigni V, Basetto F. Necrotizing fasciitis: classification, diagnosis and management. J Trauma Acute Care Surg. 2012;72(3):560-566.
10. Brothers TE, Tagge DU, Stutley JE, Conway WF, Del Schutte H Jr, Byrne TK. Magnetic resonance imaging differentiates between necrotizing and non-necrotizing fasciitis of the lower extremity. J Am Coll Surg. 1998;187(4):416-421.
11. Bakleh M, Wold LE, Mandrekar JN, Harmsen WS, Dimashkieh HH, Baddour LM. Correlation of histopathologic findings with clinical outcome in necrotizing fasciitis. Clin Infect Dis. 2005;40(3):410-414.
12. Barry W, Hudgins L, Donta ST, Pesanti EL. Intravenous immunoglobulin therapy for toxic shock syndrome. JAMA. 1992;267(24):3315-3316.
13. Wilkinson D, Doolette D. Hyperbaric oxygen treatment and survival from necrotizing soft tissue infection. Arch Surg. 2004;139(12):1339-1345.
Necrotizing soft-tissue infection (NSTI) often is difficult to distinguish from a superficial soft-tissue infection like cellulitis. Both conditions present with pain, edema, and erythema and can be accompanied by fever and malaise. The diagnosis of NSTI must be made quickly because successful treatment requires early surgical debridement and broad-spectrum antibiotics. The following case demonstrates the challenge of diagnosing NSTI.
Case Presentation
A 50-year-old physician developed a sore throat with subjective fevers, night sweats, and chills. After 2 days, his symptoms resolved. The next day he developed right thigh pain while playing tennis and limped off the court. That night he had fevers, chills, and sweats. For the next 3 days, his right thigh pain persisted with waxing and waning fevers.
The patient’s medical history included gastroesophageal reflux disease, vitamin D deficiency, and a positive purified protein derivative test for which he had completed 1 year of isoniazid therapy. The patient was married and in a monogamous relationship with his wife. He had traveled to the Sierra National Forest and Yosemite Park during the preceding winter. He did not swim in a lake or recall a tick bite. He had not consumed raw food, imported meats, or dairy products. He recently started oral fluconazole for tinea corporis.
The patient’s temperature was 39.5°C, heart rate was 115 beats per minute, blood pressure (BP) was 142/88 mm Hg, and respiratory rate was 18 breaths per minute with an oxygen saturation of 95% while breathing ambient air. He was drenched in sweat yet remained comfortable throughout the interview. The oropharyngeal mucosa was moist without lesions or erythema. There was no rash or lymphadenopathy. The lungs were clear to auscultation. The cardiac exam revealed tachycardia. There was point tenderness to deep palpation of the mid-anterior right thigh without crepitus, erythema, or edema.
The patient’s sodium level was 129 mmol/L (normal range 135-145 mmol/L), bicarbonate was 20 mmol/L (normal range 22-32 mmol/L), creatinine was 1.1 mg/dL (normal range 0.7-1.2 mg/dL), and glucose was 194 mg/dL. The white blood cell count (WBC) was 12,900 cells/mm3 (normal range 3,400-10,000 cells/mm3) with 96% neutrophils. The hematocrit was 41% (normal range 41-53%), and the platelet count was 347,000 cells/mm3 (normal range 140,000-450,000 cells/mm3). The lactate level was 2.2 mmol/L (normal range 0-2 mmol/L). The creatine kinase level was 347 U/L (normal range 50-388 U/L), and the lactate dehydrogenase level was 254 U/L (normal range 102-199 U/L). A rapid group A streptococcal (GAS) antigen test was negative. A radiograph of the right femur revealed mildly edematous soft tissue. On ultrasound the right quadriceps appeared mildly edematous, but there was no evidence of abscess or discrete fluid collection (eFigure 1).
eFigure 1. Ultrasound of the Right Anterior Thigh Ultrasound revealed heterogeneous, mildly edematous quadriceps muscle. There was no abscess or discrete fluid collection. There was trace fluid along the fascia of the quadriceps muscle.
Four liters of normal saline, acetaminophen, ceftriaxone, and doxycycline were administered to the patient. Overnight he was afebrile, tachycardic, and normotensive. The following morning his BP decreased to 81/53 mm Hg. His WBC count was 33,000 cells/mm3 with 96% neutrophils. A peripheral blood smear showed immature granulocytes. The sodium and creatinine increased to 135 mmol/L and 1.3 mg/dL, respectively. The erythrocyte sedimentation rate was 20 mm/h (normal range 0-10 mm/h), and the C-reactive protein level was 174 mg/L (normal range < 6.3 mg/L).The right thigh became erythematous and edematous.
Given concern for necrotizing fasciitis, antibiotics were changed to vancomycin, piperacillin-tazobactam, and clindamycin. The patient was taken to the operating room (OR). The right quadriceps muscle was markedly edematous with overlying necrotic fibrofatty tissue with easy separation of the fascia from the anterolateral rectus femoris and rectus lateralis muscles. Necrotizing fasciitis was diagnosed.
The tissue was debrided, and surgical pathology revealed fibroadipose tissue with extensive necrosis and dense acute inflammation (eFigure 2). After the anterolateral space between the fascia and underlying thigh muscle was drained, a Penrose drain was placed, and the wound was left open with plans for a second-look operation within 24 hours.
eFigure 2. Surgical Pathology of Debrided Right Thigh
Pathology revealed fibroadipose tissue with extensive necrosis and dense acute inflammation.
eFigure 3. Right Anterior Thigh
Two Penrose drains inserted after second operation.
In the ensuing hours erythema extended proximal to the operative site. The patient was emergently taken to the OR. The focus of necrotizing fasciitis along the anterolateral aspect of the thigh had extended posteriorly and superiorly. This area was irrigated, all loculations were disrupted, and a second Penrose drain was placed.
The wound was left open for 6 more days. On hospital day 9, operative exploration revealed no necrotizing fasciitis. The fascia and skin wound were then closed (eFigure 3).
Cultures from the fascia grew the GAS bacteria Streptococcus pyogenes (S pyogenes), which was sensitive to penicillin. The blood cultures from admission were sterile. A test for Epstein-Barr virus immunoglobulin M antibody was negative. The patient was discharged after 10 days in the hospital to complete a 2-week course of IV penicillin. Two months later he resumed playing tennis and returned to his clinical duties.
Discussion
In the U.S., there are approximately 3.5 cases of invasive GAS infection per 100,000 persons.1 Type I NSTI is polymicrobial (aerobic and anaerobic organisms). Risk factors include recent surgery, immunocompromised states, drug use, diabetes mellitus, and traumatic wounds.2 Type II NSTI is caused by GAS or other β-hemolytic streptococci either alone or in association with another organism, most commonly Staphylococcus aureus. Type II NSTI is classically found on the extremities and occurs in young, healthy, immunocompetent patients—such as this patient.3
The portal of entry in nearly half of type II NSTI is unknown; minor local trauma is often suspected.4 However, cases have been reported in which the only identifiable source was a preceding sore throat.4 The origin of this patient’s GAS remains unknown, but perhaps his pharyngitis led to transient bacteremia, which then seeded his injured thigh muscle. An in vitro model demonstrated that injured muscles increase surface expression of the cytoskeletal protein vimentin, which binds GAS.5 Exotoxins and endotoxins produced by S pyogenes may lead to microvascular thrombosis, tissue ischemia, liquefactive necrosis, and systemic release of cytokines followed by systemic illness, multiorgan dysfunction, and death.6
The Laboratory Risk Indicator for Necrotizing Fasciitis (LRINEC) score was developed to aid in early diagnosis of NSTI.7 It was derived from a series of 2,555 patients admitted with cellulitis or abscesses at a single institution. Scores > 8 have a positive predictive value of 93% for NSTI. This patient had a LRINEC score of 9. Radiographs or computed tomography scans may demonstrate soft-tissue air collections but lack sensitivity and are often nondiagnostic.8,9 T1-weighted magnetic resonance imaging can delineate the anatomic extent of soft-tissue infections but is time consuming and may delay treatment.10 When the pretest probability is high, proceeding directly to the OR for direct visualization and possible debridement is advisable. Histologic features of necrotizing fasciitis include inflammation with polymorphonuclear cells and necrosis of the subcutaneous fat and fascia with relative sparing of the muscle.11Necrotizing soft-tissue infection requires early surgical debridement and broad-spectrum antibiotic coverage. Without surgical debridement, the mortality rate approaches 100%.2 Antibiotics should include activity against Gram-positive, Gram-negative, and anaerobic organisms. The duration of antibiotic therapy has not been defined and is dependent on the patient’s clinical status. Adjunctive treatment options may include IV immunoglobulin and hyperbaric oxygen therapy, although the data supporting their utility are limited.12,13
Conclusion
Despite the LRINEC scoring systems and advanced imaging, necrotizing fasciitis remains challenging to diagnose in a timely manner. In this case, close monitoring of the patient facilitated timely evaluation and treatment of a fatal disease.
Necrotizing soft-tissue infection (NSTI) often is difficult to distinguish from a superficial soft-tissue infection like cellulitis. Both conditions present with pain, edema, and erythema and can be accompanied by fever and malaise. The diagnosis of NSTI must be made quickly because successful treatment requires early surgical debridement and broad-spectrum antibiotics. The following case demonstrates the challenge of diagnosing NSTI.
Case Presentation
A 50-year-old physician developed a sore throat with subjective fevers, night sweats, and chills. After 2 days, his symptoms resolved. The next day he developed right thigh pain while playing tennis and limped off the court. That night he had fevers, chills, and sweats. For the next 3 days, his right thigh pain persisted with waxing and waning fevers.
The patient’s medical history included gastroesophageal reflux disease, vitamin D deficiency, and a positive purified protein derivative test for which he had completed 1 year of isoniazid therapy. The patient was married and in a monogamous relationship with his wife. He had traveled to the Sierra National Forest and Yosemite Park during the preceding winter. He did not swim in a lake or recall a tick bite. He had not consumed raw food, imported meats, or dairy products. He recently started oral fluconazole for tinea corporis.
The patient’s temperature was 39.5°C, heart rate was 115 beats per minute, blood pressure (BP) was 142/88 mm Hg, and respiratory rate was 18 breaths per minute with an oxygen saturation of 95% while breathing ambient air. He was drenched in sweat yet remained comfortable throughout the interview. The oropharyngeal mucosa was moist without lesions or erythema. There was no rash or lymphadenopathy. The lungs were clear to auscultation. The cardiac exam revealed tachycardia. There was point tenderness to deep palpation of the mid-anterior right thigh without crepitus, erythema, or edema.
The patient’s sodium level was 129 mmol/L (normal range 135-145 mmol/L), bicarbonate was 20 mmol/L (normal range 22-32 mmol/L), creatinine was 1.1 mg/dL (normal range 0.7-1.2 mg/dL), and glucose was 194 mg/dL. The white blood cell count (WBC) was 12,900 cells/mm3 (normal range 3,400-10,000 cells/mm3) with 96% neutrophils. The hematocrit was 41% (normal range 41-53%), and the platelet count was 347,000 cells/mm3 (normal range 140,000-450,000 cells/mm3). The lactate level was 2.2 mmol/L (normal range 0-2 mmol/L). The creatine kinase level was 347 U/L (normal range 50-388 U/L), and the lactate dehydrogenase level was 254 U/L (normal range 102-199 U/L). A rapid group A streptococcal (GAS) antigen test was negative. A radiograph of the right femur revealed mildly edematous soft tissue. On ultrasound the right quadriceps appeared mildly edematous, but there was no evidence of abscess or discrete fluid collection (eFigure 1).
eFigure 1. Ultrasound of the Right Anterior Thigh Ultrasound revealed heterogeneous, mildly edematous quadriceps muscle. There was no abscess or discrete fluid collection. There was trace fluid along the fascia of the quadriceps muscle.
Four liters of normal saline, acetaminophen, ceftriaxone, and doxycycline were administered to the patient. Overnight he was afebrile, tachycardic, and normotensive. The following morning his BP decreased to 81/53 mm Hg. His WBC count was 33,000 cells/mm3 with 96% neutrophils. A peripheral blood smear showed immature granulocytes. The sodium and creatinine increased to 135 mmol/L and 1.3 mg/dL, respectively. The erythrocyte sedimentation rate was 20 mm/h (normal range 0-10 mm/h), and the C-reactive protein level was 174 mg/L (normal range < 6.3 mg/L).The right thigh became erythematous and edematous.
Given concern for necrotizing fasciitis, antibiotics were changed to vancomycin, piperacillin-tazobactam, and clindamycin. The patient was taken to the operating room (OR). The right quadriceps muscle was markedly edematous with overlying necrotic fibrofatty tissue with easy separation of the fascia from the anterolateral rectus femoris and rectus lateralis muscles. Necrotizing fasciitis was diagnosed.
The tissue was debrided, and surgical pathology revealed fibroadipose tissue with extensive necrosis and dense acute inflammation (eFigure 2). After the anterolateral space between the fascia and underlying thigh muscle was drained, a Penrose drain was placed, and the wound was left open with plans for a second-look operation within 24 hours.
eFigure 2. Surgical Pathology of Debrided Right Thigh
Pathology revealed fibroadipose tissue with extensive necrosis and dense acute inflammation.
eFigure 3. Right Anterior Thigh
Two Penrose drains inserted after second operation.
In the ensuing hours erythema extended proximal to the operative site. The patient was emergently taken to the OR. The focus of necrotizing fasciitis along the anterolateral aspect of the thigh had extended posteriorly and superiorly. This area was irrigated, all loculations were disrupted, and a second Penrose drain was placed.
The wound was left open for 6 more days. On hospital day 9, operative exploration revealed no necrotizing fasciitis. The fascia and skin wound were then closed (eFigure 3).
Cultures from the fascia grew the GAS bacteria Streptococcus pyogenes (S pyogenes), which was sensitive to penicillin. The blood cultures from admission were sterile. A test for Epstein-Barr virus immunoglobulin M antibody was negative. The patient was discharged after 10 days in the hospital to complete a 2-week course of IV penicillin. Two months later he resumed playing tennis and returned to his clinical duties.
Discussion
In the U.S., there are approximately 3.5 cases of invasive GAS infection per 100,000 persons.1 Type I NSTI is polymicrobial (aerobic and anaerobic organisms). Risk factors include recent surgery, immunocompromised states, drug use, diabetes mellitus, and traumatic wounds.2 Type II NSTI is caused by GAS or other β-hemolytic streptococci either alone or in association with another organism, most commonly Staphylococcus aureus. Type II NSTI is classically found on the extremities and occurs in young, healthy, immunocompetent patients—such as this patient.3
The portal of entry in nearly half of type II NSTI is unknown; minor local trauma is often suspected.4 However, cases have been reported in which the only identifiable source was a preceding sore throat.4 The origin of this patient’s GAS remains unknown, but perhaps his pharyngitis led to transient bacteremia, which then seeded his injured thigh muscle. An in vitro model demonstrated that injured muscles increase surface expression of the cytoskeletal protein vimentin, which binds GAS.5 Exotoxins and endotoxins produced by S pyogenes may lead to microvascular thrombosis, tissue ischemia, liquefactive necrosis, and systemic release of cytokines followed by systemic illness, multiorgan dysfunction, and death.6
The Laboratory Risk Indicator for Necrotizing Fasciitis (LRINEC) score was developed to aid in early diagnosis of NSTI.7 It was derived from a series of 2,555 patients admitted with cellulitis or abscesses at a single institution. Scores > 8 have a positive predictive value of 93% for NSTI. This patient had a LRINEC score of 9. Radiographs or computed tomography scans may demonstrate soft-tissue air collections but lack sensitivity and are often nondiagnostic.8,9 T1-weighted magnetic resonance imaging can delineate the anatomic extent of soft-tissue infections but is time consuming and may delay treatment.10 When the pretest probability is high, proceeding directly to the OR for direct visualization and possible debridement is advisable. Histologic features of necrotizing fasciitis include inflammation with polymorphonuclear cells and necrosis of the subcutaneous fat and fascia with relative sparing of the muscle.11Necrotizing soft-tissue infection requires early surgical debridement and broad-spectrum antibiotic coverage. Without surgical debridement, the mortality rate approaches 100%.2 Antibiotics should include activity against Gram-positive, Gram-negative, and anaerobic organisms. The duration of antibiotic therapy has not been defined and is dependent on the patient’s clinical status. Adjunctive treatment options may include IV immunoglobulin and hyperbaric oxygen therapy, although the data supporting their utility are limited.12,13
Conclusion
Despite the LRINEC scoring systems and advanced imaging, necrotizing fasciitis remains challenging to diagnose in a timely manner. In this case, close monitoring of the patient facilitated timely evaluation and treatment of a fatal disease.
1. O'Loughlin RE, Roberson A, Cieslak PR, et al; Active Bacterial Core Surveillance Team. The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000-2004. Clin Infect Dis. 2007;45(7):853-857.
2. Anaya DA, Dellinger EP. Necrotizing soft-tissue infection: diagnosis and management. Clin Infect Dis. 2007;44(5):705-710.
3. Naqvi GA, Malik SA, Jan W. Necrotizing fasciitis of the lower extremity: a case report and current concept of diagnosis and management. Scand J Trauma Resusc Emerg Med. 2009;17:28.
4. Stevens DL. Streptococcal toxic-shock syndrome: spectrum of disease, pathogenesis, and new concepts in treatment. Emerg Infect Dis. 1195;1(3):69-78.
5. Bryant AE, Bayer CR, Huntington JD, Stevens DL. Group A streptococcal myonecrosis: increased vimentin expression after skeletal-muscle injury mediates the binding of Streptococcus pyogenes. J Infect Dis. 2006;193(12):1685-1692.
6. Cainzos M, Gonzalez-Rodriguez FJ. Necrotizing soft tissue infections. Curr Opin Crit Care. 2007;13(4):433-439.
7. Wong CH, Khin LW, Heng KS, Tan KC, Low CO. The LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score: a tool for distinguishing necrotizing fasciitis from other soft tissue infections. Crit Care Med. 2004;32(7):1535-1541.
8. Goh T, Goh LG, Ang CH, Wong CH. Early diagnosis of necrotizing fasciitis. Br J Surg. 2014;101(1):119-125.
9. Lancerotto L, Tocco I, Salmaso R, Vindigni V, Basetto F. Necrotizing fasciitis: classification, diagnosis and management. J Trauma Acute Care Surg. 2012;72(3):560-566.
10. Brothers TE, Tagge DU, Stutley JE, Conway WF, Del Schutte H Jr, Byrne TK. Magnetic resonance imaging differentiates between necrotizing and non-necrotizing fasciitis of the lower extremity. J Am Coll Surg. 1998;187(4):416-421.
11. Bakleh M, Wold LE, Mandrekar JN, Harmsen WS, Dimashkieh HH, Baddour LM. Correlation of histopathologic findings with clinical outcome in necrotizing fasciitis. Clin Infect Dis. 2005;40(3):410-414.
12. Barry W, Hudgins L, Donta ST, Pesanti EL. Intravenous immunoglobulin therapy for toxic shock syndrome. JAMA. 1992;267(24):3315-3316.
13. Wilkinson D, Doolette D. Hyperbaric oxygen treatment and survival from necrotizing soft tissue infection. Arch Surg. 2004;139(12):1339-1345.
1. O'Loughlin RE, Roberson A, Cieslak PR, et al; Active Bacterial Core Surveillance Team. The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000-2004. Clin Infect Dis. 2007;45(7):853-857.
2. Anaya DA, Dellinger EP. Necrotizing soft-tissue infection: diagnosis and management. Clin Infect Dis. 2007;44(5):705-710.
3. Naqvi GA, Malik SA, Jan W. Necrotizing fasciitis of the lower extremity: a case report and current concept of diagnosis and management. Scand J Trauma Resusc Emerg Med. 2009;17:28.
4. Stevens DL. Streptococcal toxic-shock syndrome: spectrum of disease, pathogenesis, and new concepts in treatment. Emerg Infect Dis. 1195;1(3):69-78.
5. Bryant AE, Bayer CR, Huntington JD, Stevens DL. Group A streptococcal myonecrosis: increased vimentin expression after skeletal-muscle injury mediates the binding of Streptococcus pyogenes. J Infect Dis. 2006;193(12):1685-1692.
6. Cainzos M, Gonzalez-Rodriguez FJ. Necrotizing soft tissue infections. Curr Opin Crit Care. 2007;13(4):433-439.
7. Wong CH, Khin LW, Heng KS, Tan KC, Low CO. The LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score: a tool for distinguishing necrotizing fasciitis from other soft tissue infections. Crit Care Med. 2004;32(7):1535-1541.
8. Goh T, Goh LG, Ang CH, Wong CH. Early diagnosis of necrotizing fasciitis. Br J Surg. 2014;101(1):119-125.
9. Lancerotto L, Tocco I, Salmaso R, Vindigni V, Basetto F. Necrotizing fasciitis: classification, diagnosis and management. J Trauma Acute Care Surg. 2012;72(3):560-566.
10. Brothers TE, Tagge DU, Stutley JE, Conway WF, Del Schutte H Jr, Byrne TK. Magnetic resonance imaging differentiates between necrotizing and non-necrotizing fasciitis of the lower extremity. J Am Coll Surg. 1998;187(4):416-421.
11. Bakleh M, Wold LE, Mandrekar JN, Harmsen WS, Dimashkieh HH, Baddour LM. Correlation of histopathologic findings with clinical outcome in necrotizing fasciitis. Clin Infect Dis. 2005;40(3):410-414.
12. Barry W, Hudgins L, Donta ST, Pesanti EL. Intravenous immunoglobulin therapy for toxic shock syndrome. JAMA. 1992;267(24):3315-3316.
13. Wilkinson D, Doolette D. Hyperbaric oxygen treatment and survival from necrotizing soft tissue infection. Arch Surg. 2004;139(12):1339-1345.
Lichen Planus Pemphigoides Associated With Pregnancy Mimicking Pemphigoid Gestationis
Case Report
A 25-year-old woman with a 5-month history of severe lichen planus (LP) on the arms, legs, and trunk presented to the emergency department with generalized blisters and erythema over the entire body, including the face and soles, of 2 days’ duration. She was evaluated for the LP 1 week prior in a referral dermatology clinic, and in addition to topical corticosteroids, she received 1 injection of 40 mg intramuscular triamcinolone acetonide. Hours following the injection she developed nausea, vomiting, and fever. The patient reported that her last menstrual period was 3 weeks prior to the current presentation.
Physical examination revealed numerous lichenified, flat-topped, pink-violaceous, hyperpigmented, scaly papules and plaques (Figure 1), as well as tense, yellow, fluid-filled vesicles and bullae of various sizes on the neck, arms (Figure 2), legs, trunk, and dorsal aspect of the feet. The vesicles occurred on both normal skin and the lichenified plaques with a negative Nikolsky sign. There also were urticarial erythematous papules and plaques on the arms, trunk, neck, and face, some of which had vesicles or a violaceous dusky central hue (Figure 3). Vesicles were noted within both nostrils (nasal mucosa), and there were extremely tender erythematous patches and thick sheets of scales on the soles.
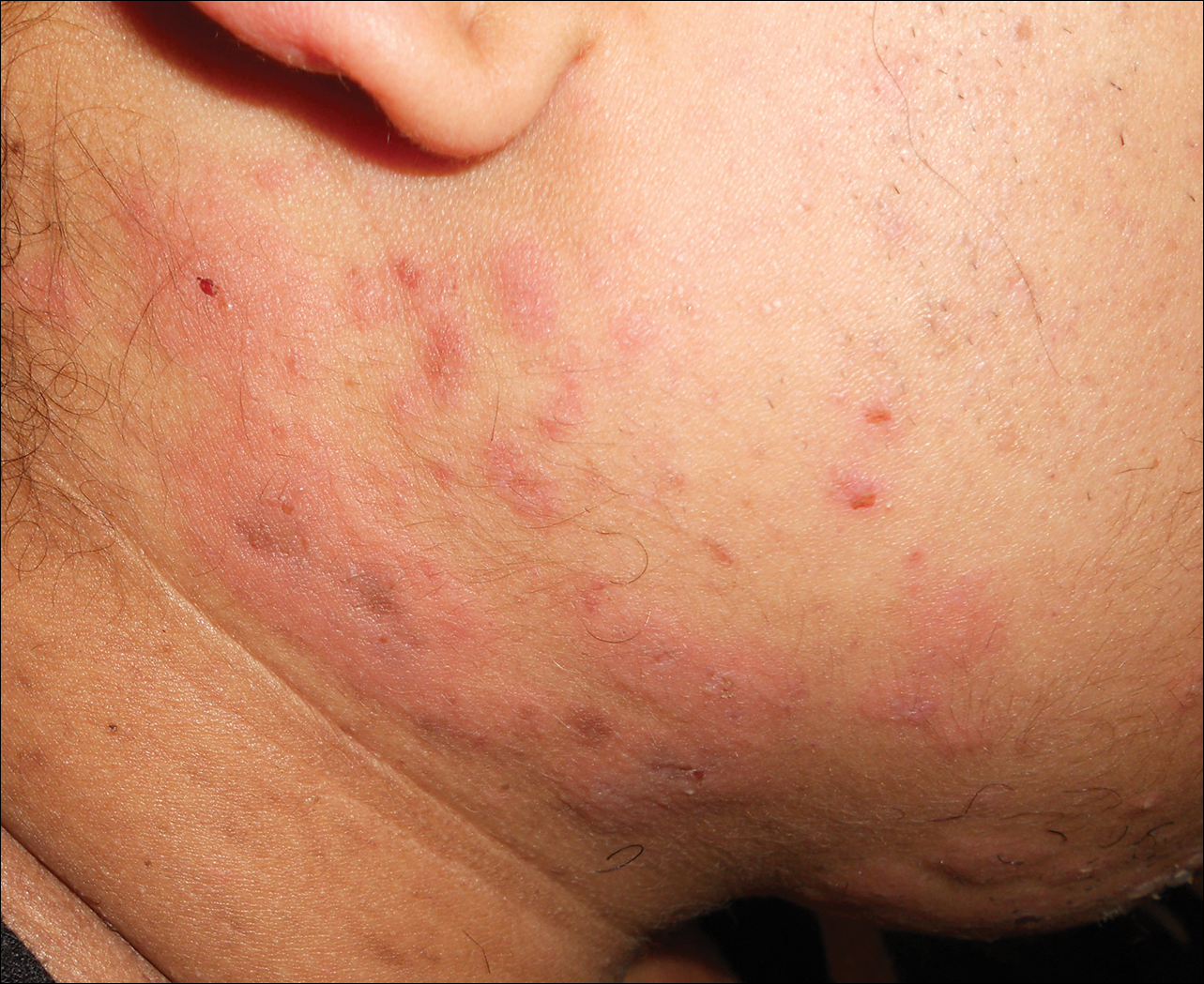
An elevated β human chorionic gonadotropin level and transvaginal ultrasonography confirmed an intrauterine pregnancy of 12 weeks’ gestation despite the patient’s report of the last menstrual period.
Histologic examination of a vesicle on the right arm revealed hyperkeratosis with hypergranulosis, vacuolar alteration of the basal layer with a paucicellular subepidermal vesicle, and melanophages in the superficial dermis consistent with vesicular LP (Figure 4). Histologic examination of an erythematous edematous plaque on the right upper leg revealed edema in the upper dermis with a perivascular and interstitial lymphocytic infiltrate with eosinophils. A third biopsy of a lichenoid flat-topped papule on the left arm revealed a mild bandlike infiltrate of lymphocytes and scattered eosinophils, eosinophilic colloid bodies and edema in the papillary dermis, and subepidermal vesicles and vacuolar alteration of the basal layer consistent with a vesicular lichenoid dermatitis (Figure 5). Direct immunofluorescence (DIF) of perilesional skin showed linear deposition of C3 and IgM along the basement membrane zone (BMZ) in addition to a shaggy pattern with cytoid bodies (Figure 6). There also was a faint linear deposit of IgA along the BMZ with cytoid bodies but negative for IgG. These results were interpreted as consistent with LP pemphigoides (LPP). Neither an enzyme-linked immunosorbent assay nor an immunoblot analysis was performed.
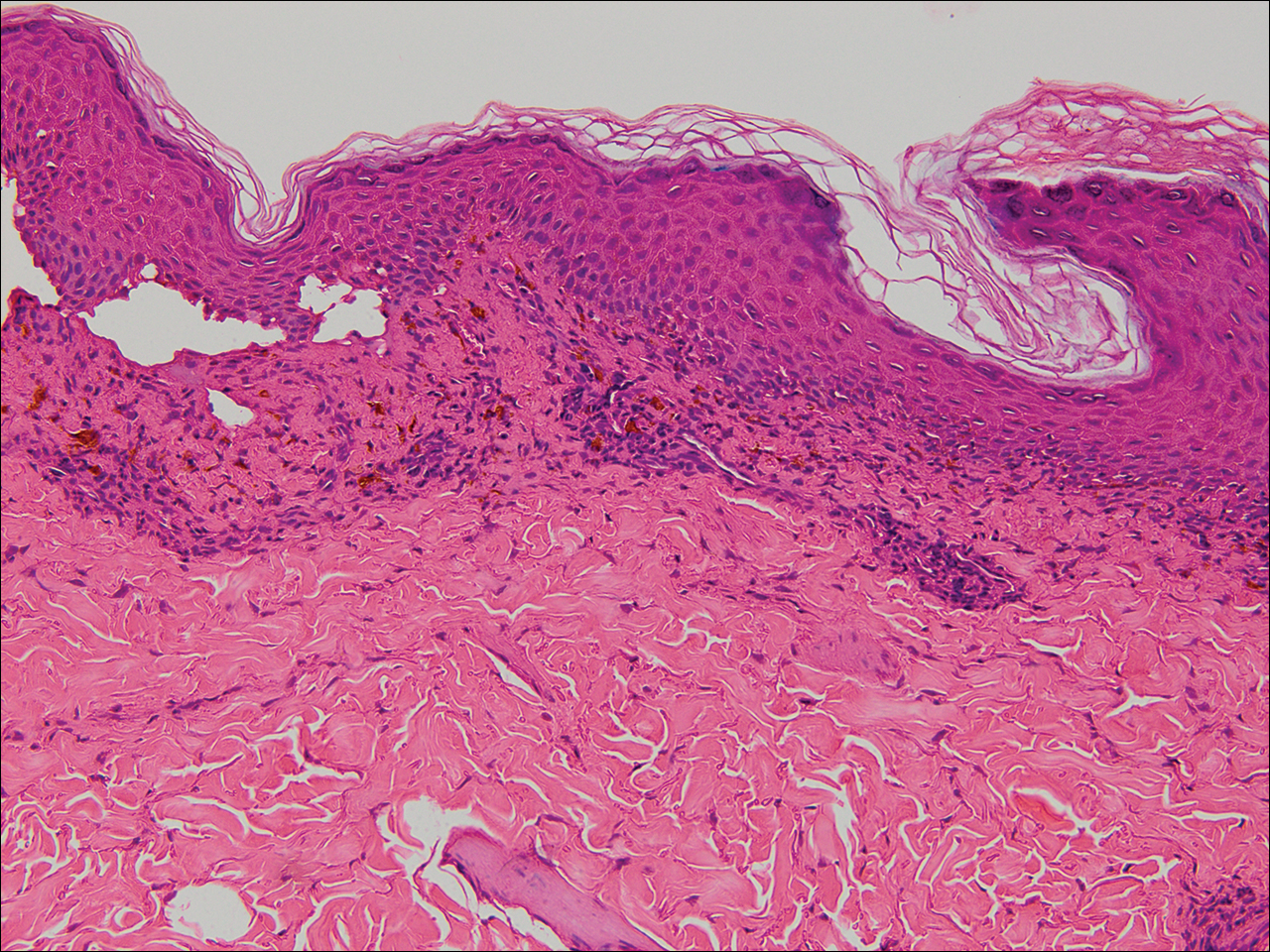
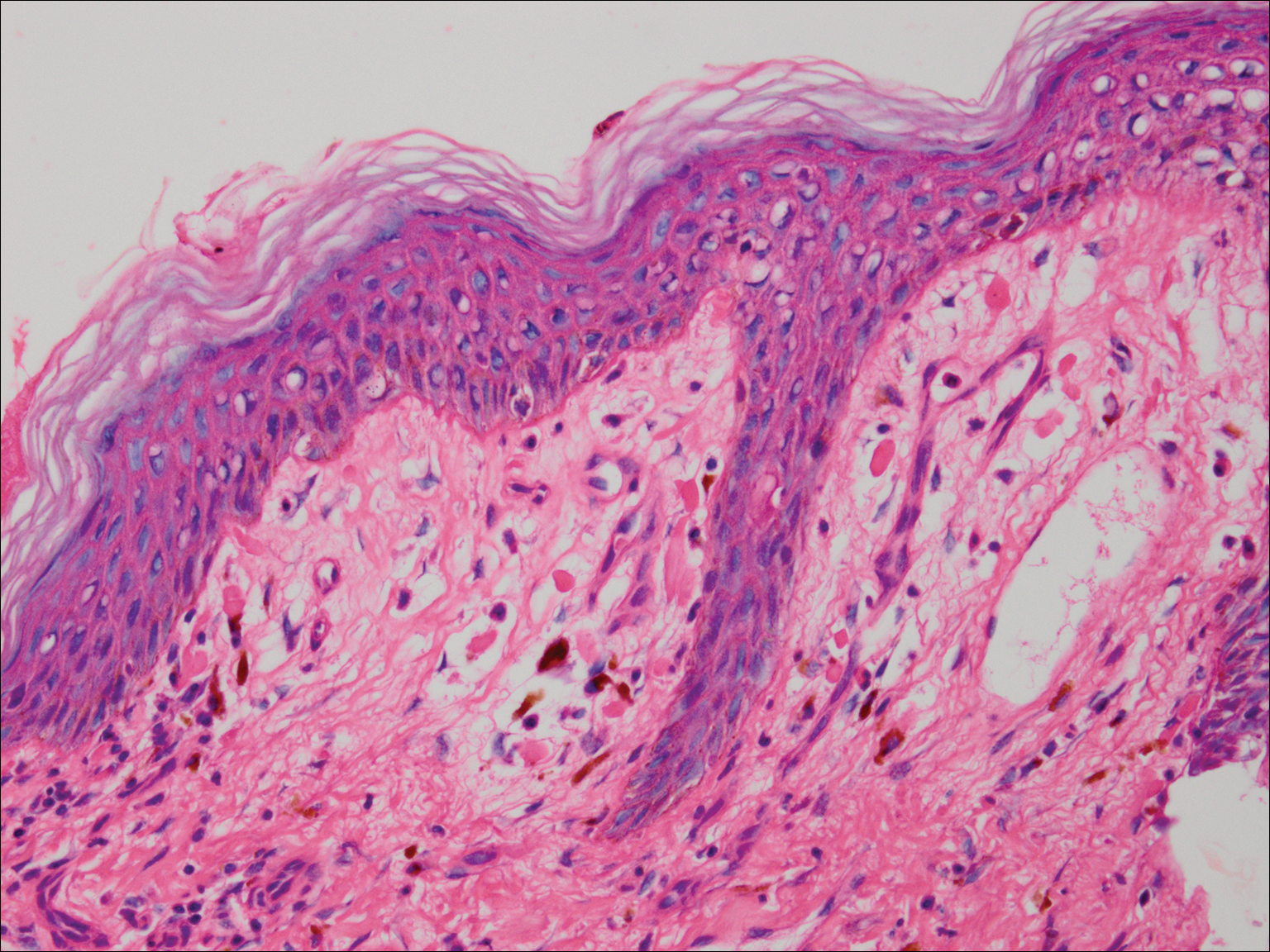
Because the patient was pregnant and had failed to respond to topical and intramuscular corticosteroids, she was started on intravenous methylprednisolone in the emergency department until new lesions stopped appearing. She was then discharged home on oral prednisone 50 mg (0.5 mg/kg/d), with close observation by her obstetrician. She also used clobetasol propionate ointment 0.05% for more severe lesions and triamcinolone acetonide cream 0.1% for less severe lesions until lesions resolved.
During treatment, the patient developed cellulitis on the leg that presented as pustules and erythema at a site of an eroded bulla, inframammary and axillary cutaneous candidiasis, and hyperglycemia at 19 weeks’ gestation. The cutaneous infections resolved with oral clindamycin 300 mg 3 times daily for 10 days. Topical mupirocin was used to treat the cellulitis and a mixture of zinc oxide, econazole cream, and desonide cream twice daily treated the candidiasis. Her obstetrician managed the hyperglycemia.
The bullous lesions and LP completely resolved after 2 months of treatment with oral prednisone 50 mg daily. The patient tolerated a corticosteroid taper (dose decreased by 5 mg every 2 weeks) until arriving at 10 mg, which was then decreased to 7.5 mg until delivery. A cesarean delivery was performed due to a large-for-gestational-age fetus, and an internist was consulted for the necessary precautions to increase the steroid dose during delivery due to the stress of the surgery and the risk for a hypothalamic crisis. There were no peripartum complications, and the baby was born without cutaneous lesions and remains healthy 1 year later. The patient remained disease free over 2 months postpartum, until new LP lesions developed without vesicles or bullae, which were then controlled with topical therapy. She was subsequently lost to follow-up.
Comment
Kaposi first described LPP in 1892 and used the term lichen ruber pemphigoides to describe a case of typical LP together with a widespread bullous eruption. Lichen planus pemphigoides is characterized by tense blisters that arise on lesions of LP as well as on skin unaffected by LP. In contrast, bullous LP blisters are confined to LP lesions only and occur from intense lichenoid inflammation and extensive liquefactive degeneration of basal keratinocytes. The vesicle formation in LPP is a result of autoantibodies to the bullous pemphigoid (BP) antigen BPAg2, which can be explained by the epitope spreading epiphenomenon whereby epidermotropic cytotoxic T cells damage the basal keratinocytes in LP by targeting unknown epidermal antigens, resulting in the exposure of BP180 and therefore instigating the autoimmune response.1 The process of epitope spreading takes months to develop; the mean duration of LP before LPP is 8 weeks in children and 12 weeks in adults,2 which is comparable to the current case.
Pathogenesis
Lichen planus pemphigoides usually is idiopathic; however, there have been cases reported in association with various medications including calcium channel blockers such as diltiazem, Chinese herbs,3 simvastatin,4 ramipril,5,6 captopril,7 psoralen plus UVA phototherapy,8 and cinnarizine.9 In addition, in a case-controlled study, the use of neuroleptics or diuretics was found to be a risk factor for LPP development.10
This case is unique because it shows an association of LPP with an intrauterine pregnancy. Despite the fact that we did not perform the required studies to determine the exact cause, there probably exists an association between LPP and the pregnancy, as the patient presented with a 5-month history of severe LP prior to vesicle formation. The patient only developed the vesicular lesions during pregnancy, which were later controlled with systemic steroids and then recurred postpartum only as LP lesions, suggesting that the patient’s pregnancy may have contributed in the pathogenesis as an inducing factor. We suspect that the LP was aggravated by the pregnancy and continued to worsen, so much as to cause epitope spreading and lead to the bullous eruption at the end of the first trimester.2
Differential Diagnosis
Initially, we suspected a diagnosis of pemphigoid gestationis (PG), previously known as herpes gestationis. The classic presentation of PG starts with an intense pruritus followed by the emergence of pruritic urticarial papules and plaques in the umbilical or periumbilical areas. The lesions may become targetlike or polycyclic and may spread to other areas of the trunk, arms, and legs, often including the palms and soles.11-15 Just as in our case, vesicles and bullous lesions appear at both the site of the urticarial plaques as well as on normal skin.16 The clinical features noted in our patient that were not typical of PG included the multiple lesions on the face and inside the nostrils. Only 20% of PG cases are associated with mucosal involvement,11,12,15 and there are no documented reports of PG occurring in a patient with LP, according to a PubMed search of articles indexed for MEDLINE using the search terms pemphigoid gestationis, herpes gestationis, and lichen planus.
Lichen planus pemphigoides can be easily differentiated from BP. Lichen planus pemphigoides occurs in younger patients, with a mean age of 35 years, unlike BP, which commonly affects elderly men.17 Lichen planus pemphigoides also is less severe and has a better response to treatment than BP. It also affects the palms and soles, which are rarely affected in BP. There are no reports in the literature of BP developing during pregnancy, according to a PubMed search using the terms bullous pemphigoid and pregnancy. However, LPP and BP share a common antibody, the BP180 antigen, and differences exist in the epitope where the antibody binds in each condition.18,19
Diagnosing LPP
In LPP, DIF typically shows linear deposits of IgG, IgM, IgA, fibrinogen, and C3 along the BMZ, of which IgG and C3 are most commonly seen.3 Our patient had linear deposition of C3, IgM, and IgA along the BMZ, which excluded bullous LP from the differential diagnosis. Bullous LP is not an autoimmune condition but rather is on the severe spectrum of LP where Max Joseph spaces become so large so as to lead to vesicle and bullae formation. In addition to the linear deposit at the BMZ, LPP typically reveals immunoglobulin (mainly IgM but also IgA), C3, and fibrinogen staining of colloid bodies in the papillary dermis on DIF; however, some cases of LPP only present with a linear deposition of C3 along the BMZ, which is why, similar to PG, these diagnoses by DIF are similar. Direct immunofluorescence of PG reveals linear IgG1 and IgG3 along the BMZ. IgG1 and IgG3 immunoglobulins are known to fix complement better than other immunoglobulins, thus linear C3 along the BMZ is the most consistently positive immunoreactant. Less common positive immunoreactivity with the same pattern has been seen with IgA, IgM, C1, and C4 (Table).14,15,18 The lack of linear IgG and the presence of IgM is more suggestive of LPP.
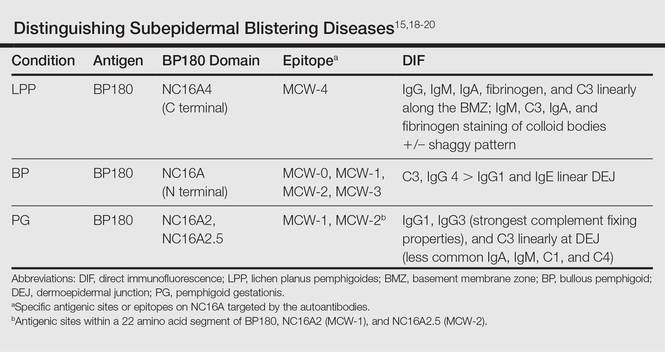
The differential diagnosis of the subepidermal autoimmune blistering diseases associated with antibodies against BP180, including BP, LPP, and PG, often is challenging.15 However, LPP can now be distinguished by immunological studies including immunoblot analysis of the immunodominant region of NC16A of the BP180 antigen and the immunoglobulin subclass that reacts to 180-, 200-,20 and 230-kDa antigens within the BMZ (Table).15,18-20 The Table summarizes the different autoantibodies, antigens, and epitopes to distinguish subepidermal autoimmune blistering diseases.
Despite not performing these studies in our patient, we concluded that the clinical, histological, and DIF findings of this case are more consistent with LPP than with the other subepidermal blistering diseases. However, we cannot exclude the possibility of the patient having a new entity with a unique antibody from epitope spreading.
Conclusion
We present a case of lichenoid papules and plaques consistent with LP, with the development of vesicles and bullae after the first trimester of pregnancy. The clinical, pathologic, and DIF findings were highly suggestive of LPP. Although the exact pathogenic mechanism is not fully known, we suspect that pregnancy may have contributed to the origin of the disease. Further evaluation of pregnant patients with lichenoid lesions who develop blisters are needed for the elucidation of the mechanism, which may be secondary to epitope spreading that led to new autoantibody formation.
- Stingl G, Holubar K. Coexistence of lichen planus and bullous pemphigoid. an immunopathological study. Br J Dermatol. 1975;93:313-320.
- Paige DG, Bhogal BS, Black MM, et al. Lichen planus pemphigoides in a child—immunopathological findings. Clin Exp Dermatol. 1993;18:552-554.
- Xu HH, Xiao T, He CD, et al. Lichen planus pemphigoides associated with Chinese herbs. Clin Exp Dermatol. 2009;34:329-332.
- Stoebner PE, Michot C, Ligeron C, et al. Simvastatin induced lichen planus pemphigoides. Ann Dermatol Venereol. 2003;130:187-190.
- Zhu YI, Fitzpatrick JE, Kornfeld BW. Lichen planus pemphigoides associated with Ramipril. Int J Dermatol. 2006;45:1453-1455.
- Ogg GS, Bhogal BS, Hashimoto T, et al. Ramipril-associated lichen planus pemphigoides. Br J Dermatol. 1997;136:412-414.
- Flageul B, Foldes C, Wallach D, et al. Captopril-induced lichen planus pemphigoides with pemphigus-like features. a case report. Dermatologica. 1986;173:248-255.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Miyagawa S, Ohi H, Muramatsu T, et al. Lichen planus pemphigoides-like lesions induced by Cinnarizine. Br J Dermatol. 1985;112:607-613.
- Bastuji-Garin S, Joly P, Picard-Dahan C, et al. Drugs associated with bullous pemphigoid. a case-control study. Arch Dermatol. 1996;132:272-276.
- Ambros-Rudolph CM. Dermatoses of pregnancy-clues to diagnosis, fetal risk and therapy. Ann Dermatol. 2011;23:265-275.
- DiZenzo G, Calabresi V, Grosso F, et al. The intracellular and extracellular domains of BP180 antigen comprise novel epitopes targeted by pemphigoid gestationis autoantibodies. J Invest Dermatol. 2006;127:864-873.
- Jenkis RE, Hern S, Black MM. Clinical features and management of 87 patients with pemphigus gestationis. Clin Exp Dermatol. 1999;24:255-259.
- Kasperkiewicz M, Zillikens D, Schmidt E. Pemphigoid diseases: pathogenesis, diagnosis, and treatment. Autoimmunity. 2012;45:55-70.
- Cobo MF, Santi CG, Maruta CW, et al. Pemphigoid gestationis: clinical and laboratory evaluation. Clinics. 2009;64:1042-1047.
- Hsu S, Ghohestani RF, Uitto J. Lichen planus pemphigoides with IgG autoantibodies to the 180 kd bullous pemphigoid antigen (type XVII collagen). J Am Acad Dermatol. 2000;42:136-141.
- Harjai B, Mendiratta V, Kakkar S, et al. Childhood lichen planus pemphigoides—a rare entity. J Eur Acad Dermatol Venereol. 2006;20:117-118.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Zillikens D. BP180 as the common autoantigen in blistering diseases with different clinical phenotypes. Keio J Med. 2002;51:21-28.
- Davis AL, Bhogal BS, Whitehead P, et al. Lichen planus pemphigoides: its relationship to bullous pemphigoid. Br J Dermatol. 1991;125:263-271.
Case Report
A 25-year-old woman with a 5-month history of severe lichen planus (LP) on the arms, legs, and trunk presented to the emergency department with generalized blisters and erythema over the entire body, including the face and soles, of 2 days’ duration. She was evaluated for the LP 1 week prior in a referral dermatology clinic, and in addition to topical corticosteroids, she received 1 injection of 40 mg intramuscular triamcinolone acetonide. Hours following the injection she developed nausea, vomiting, and fever. The patient reported that her last menstrual period was 3 weeks prior to the current presentation.
Physical examination revealed numerous lichenified, flat-topped, pink-violaceous, hyperpigmented, scaly papules and plaques (Figure 1), as well as tense, yellow, fluid-filled vesicles and bullae of various sizes on the neck, arms (Figure 2), legs, trunk, and dorsal aspect of the feet. The vesicles occurred on both normal skin and the lichenified plaques with a negative Nikolsky sign. There also were urticarial erythematous papules and plaques on the arms, trunk, neck, and face, some of which had vesicles or a violaceous dusky central hue (Figure 3). Vesicles were noted within both nostrils (nasal mucosa), and there were extremely tender erythematous patches and thick sheets of scales on the soles.
An elevated β human chorionic gonadotropin level and transvaginal ultrasonography confirmed an intrauterine pregnancy of 12 weeks’ gestation despite the patient’s report of the last menstrual period.
Histologic examination of a vesicle on the right arm revealed hyperkeratosis with hypergranulosis, vacuolar alteration of the basal layer with a paucicellular subepidermal vesicle, and melanophages in the superficial dermis consistent with vesicular LP (Figure 4). Histologic examination of an erythematous edematous plaque on the right upper leg revealed edema in the upper dermis with a perivascular and interstitial lymphocytic infiltrate with eosinophils. A third biopsy of a lichenoid flat-topped papule on the left arm revealed a mild bandlike infiltrate of lymphocytes and scattered eosinophils, eosinophilic colloid bodies and edema in the papillary dermis, and subepidermal vesicles and vacuolar alteration of the basal layer consistent with a vesicular lichenoid dermatitis (Figure 5). Direct immunofluorescence (DIF) of perilesional skin showed linear deposition of C3 and IgM along the basement membrane zone (BMZ) in addition to a shaggy pattern with cytoid bodies (Figure 6). There also was a faint linear deposit of IgA along the BMZ with cytoid bodies but negative for IgG. These results were interpreted as consistent with LP pemphigoides (LPP). Neither an enzyme-linked immunosorbent assay nor an immunoblot analysis was performed.
Because the patient was pregnant and had failed to respond to topical and intramuscular corticosteroids, she was started on intravenous methylprednisolone in the emergency department until new lesions stopped appearing. She was then discharged home on oral prednisone 50 mg (0.5 mg/kg/d), with close observation by her obstetrician. She also used clobetasol propionate ointment 0.05% for more severe lesions and triamcinolone acetonide cream 0.1% for less severe lesions until lesions resolved.
During treatment, the patient developed cellulitis on the leg that presented as pustules and erythema at a site of an eroded bulla, inframammary and axillary cutaneous candidiasis, and hyperglycemia at 19 weeks’ gestation. The cutaneous infections resolved with oral clindamycin 300 mg 3 times daily for 10 days. Topical mupirocin was used to treat the cellulitis and a mixture of zinc oxide, econazole cream, and desonide cream twice daily treated the candidiasis. Her obstetrician managed the hyperglycemia.
The bullous lesions and LP completely resolved after 2 months of treatment with oral prednisone 50 mg daily. The patient tolerated a corticosteroid taper (dose decreased by 5 mg every 2 weeks) until arriving at 10 mg, which was then decreased to 7.5 mg until delivery. A cesarean delivery was performed due to a large-for-gestational-age fetus, and an internist was consulted for the necessary precautions to increase the steroid dose during delivery due to the stress of the surgery and the risk for a hypothalamic crisis. There were no peripartum complications, and the baby was born without cutaneous lesions and remains healthy 1 year later. The patient remained disease free over 2 months postpartum, until new LP lesions developed without vesicles or bullae, which were then controlled with topical therapy. She was subsequently lost to follow-up.
Comment
Kaposi first described LPP in 1892 and used the term lichen ruber pemphigoides to describe a case of typical LP together with a widespread bullous eruption. Lichen planus pemphigoides is characterized by tense blisters that arise on lesions of LP as well as on skin unaffected by LP. In contrast, bullous LP blisters are confined to LP lesions only and occur from intense lichenoid inflammation and extensive liquefactive degeneration of basal keratinocytes. The vesicle formation in LPP is a result of autoantibodies to the bullous pemphigoid (BP) antigen BPAg2, which can be explained by the epitope spreading epiphenomenon whereby epidermotropic cytotoxic T cells damage the basal keratinocytes in LP by targeting unknown epidermal antigens, resulting in the exposure of BP180 and therefore instigating the autoimmune response.1 The process of epitope spreading takes months to develop; the mean duration of LP before LPP is 8 weeks in children and 12 weeks in adults,2 which is comparable to the current case.
Pathogenesis
Lichen planus pemphigoides usually is idiopathic; however, there have been cases reported in association with various medications including calcium channel blockers such as diltiazem, Chinese herbs,3 simvastatin,4 ramipril,5,6 captopril,7 psoralen plus UVA phototherapy,8 and cinnarizine.9 In addition, in a case-controlled study, the use of neuroleptics or diuretics was found to be a risk factor for LPP development.10
This case is unique because it shows an association of LPP with an intrauterine pregnancy. Despite the fact that we did not perform the required studies to determine the exact cause, there probably exists an association between LPP and the pregnancy, as the patient presented with a 5-month history of severe LP prior to vesicle formation. The patient only developed the vesicular lesions during pregnancy, which were later controlled with systemic steroids and then recurred postpartum only as LP lesions, suggesting that the patient’s pregnancy may have contributed in the pathogenesis as an inducing factor. We suspect that the LP was aggravated by the pregnancy and continued to worsen, so much as to cause epitope spreading and lead to the bullous eruption at the end of the first trimester.2
Differential Diagnosis
Initially, we suspected a diagnosis of pemphigoid gestationis (PG), previously known as herpes gestationis. The classic presentation of PG starts with an intense pruritus followed by the emergence of pruritic urticarial papules and plaques in the umbilical or periumbilical areas. The lesions may become targetlike or polycyclic and may spread to other areas of the trunk, arms, and legs, often including the palms and soles.11-15 Just as in our case, vesicles and bullous lesions appear at both the site of the urticarial plaques as well as on normal skin.16 The clinical features noted in our patient that were not typical of PG included the multiple lesions on the face and inside the nostrils. Only 20% of PG cases are associated with mucosal involvement,11,12,15 and there are no documented reports of PG occurring in a patient with LP, according to a PubMed search of articles indexed for MEDLINE using the search terms pemphigoid gestationis, herpes gestationis, and lichen planus.
Lichen planus pemphigoides can be easily differentiated from BP. Lichen planus pemphigoides occurs in younger patients, with a mean age of 35 years, unlike BP, which commonly affects elderly men.17 Lichen planus pemphigoides also is less severe and has a better response to treatment than BP. It also affects the palms and soles, which are rarely affected in BP. There are no reports in the literature of BP developing during pregnancy, according to a PubMed search using the terms bullous pemphigoid and pregnancy. However, LPP and BP share a common antibody, the BP180 antigen, and differences exist in the epitope where the antibody binds in each condition.18,19
Diagnosing LPP
In LPP, DIF typically shows linear deposits of IgG, IgM, IgA, fibrinogen, and C3 along the BMZ, of which IgG and C3 are most commonly seen.3 Our patient had linear deposition of C3, IgM, and IgA along the BMZ, which excluded bullous LP from the differential diagnosis. Bullous LP is not an autoimmune condition but rather is on the severe spectrum of LP where Max Joseph spaces become so large so as to lead to vesicle and bullae formation. In addition to the linear deposit at the BMZ, LPP typically reveals immunoglobulin (mainly IgM but also IgA), C3, and fibrinogen staining of colloid bodies in the papillary dermis on DIF; however, some cases of LPP only present with a linear deposition of C3 along the BMZ, which is why, similar to PG, these diagnoses by DIF are similar. Direct immunofluorescence of PG reveals linear IgG1 and IgG3 along the BMZ. IgG1 and IgG3 immunoglobulins are known to fix complement better than other immunoglobulins, thus linear C3 along the BMZ is the most consistently positive immunoreactant. Less common positive immunoreactivity with the same pattern has been seen with IgA, IgM, C1, and C4 (Table).14,15,18 The lack of linear IgG and the presence of IgM is more suggestive of LPP.
The differential diagnosis of the subepidermal autoimmune blistering diseases associated with antibodies against BP180, including BP, LPP, and PG, often is challenging.15 However, LPP can now be distinguished by immunological studies including immunoblot analysis of the immunodominant region of NC16A of the BP180 antigen and the immunoglobulin subclass that reacts to 180-, 200-,20 and 230-kDa antigens within the BMZ (Table).15,18-20 The Table summarizes the different autoantibodies, antigens, and epitopes to distinguish subepidermal autoimmune blistering diseases.
Despite not performing these studies in our patient, we concluded that the clinical, histological, and DIF findings of this case are more consistent with LPP than with the other subepidermal blistering diseases. However, we cannot exclude the possibility of the patient having a new entity with a unique antibody from epitope spreading.
Conclusion
We present a case of lichenoid papules and plaques consistent with LP, with the development of vesicles and bullae after the first trimester of pregnancy. The clinical, pathologic, and DIF findings were highly suggestive of LPP. Although the exact pathogenic mechanism is not fully known, we suspect that pregnancy may have contributed to the origin of the disease. Further evaluation of pregnant patients with lichenoid lesions who develop blisters are needed for the elucidation of the mechanism, which may be secondary to epitope spreading that led to new autoantibody formation.
Case Report
A 25-year-old woman with a 5-month history of severe lichen planus (LP) on the arms, legs, and trunk presented to the emergency department with generalized blisters and erythema over the entire body, including the face and soles, of 2 days’ duration. She was evaluated for the LP 1 week prior in a referral dermatology clinic, and in addition to topical corticosteroids, she received 1 injection of 40 mg intramuscular triamcinolone acetonide. Hours following the injection she developed nausea, vomiting, and fever. The patient reported that her last menstrual period was 3 weeks prior to the current presentation.
Physical examination revealed numerous lichenified, flat-topped, pink-violaceous, hyperpigmented, scaly papules and plaques (Figure 1), as well as tense, yellow, fluid-filled vesicles and bullae of various sizes on the neck, arms (Figure 2), legs, trunk, and dorsal aspect of the feet. The vesicles occurred on both normal skin and the lichenified plaques with a negative Nikolsky sign. There also were urticarial erythematous papules and plaques on the arms, trunk, neck, and face, some of which had vesicles or a violaceous dusky central hue (Figure 3). Vesicles were noted within both nostrils (nasal mucosa), and there were extremely tender erythematous patches and thick sheets of scales on the soles.
An elevated β human chorionic gonadotropin level and transvaginal ultrasonography confirmed an intrauterine pregnancy of 12 weeks’ gestation despite the patient’s report of the last menstrual period.
Histologic examination of a vesicle on the right arm revealed hyperkeratosis with hypergranulosis, vacuolar alteration of the basal layer with a paucicellular subepidermal vesicle, and melanophages in the superficial dermis consistent with vesicular LP (Figure 4). Histologic examination of an erythematous edematous plaque on the right upper leg revealed edema in the upper dermis with a perivascular and interstitial lymphocytic infiltrate with eosinophils. A third biopsy of a lichenoid flat-topped papule on the left arm revealed a mild bandlike infiltrate of lymphocytes and scattered eosinophils, eosinophilic colloid bodies and edema in the papillary dermis, and subepidermal vesicles and vacuolar alteration of the basal layer consistent with a vesicular lichenoid dermatitis (Figure 5). Direct immunofluorescence (DIF) of perilesional skin showed linear deposition of C3 and IgM along the basement membrane zone (BMZ) in addition to a shaggy pattern with cytoid bodies (Figure 6). There also was a faint linear deposit of IgA along the BMZ with cytoid bodies but negative for IgG. These results were interpreted as consistent with LP pemphigoides (LPP). Neither an enzyme-linked immunosorbent assay nor an immunoblot analysis was performed.
Because the patient was pregnant and had failed to respond to topical and intramuscular corticosteroids, she was started on intravenous methylprednisolone in the emergency department until new lesions stopped appearing. She was then discharged home on oral prednisone 50 mg (0.5 mg/kg/d), with close observation by her obstetrician. She also used clobetasol propionate ointment 0.05% for more severe lesions and triamcinolone acetonide cream 0.1% for less severe lesions until lesions resolved.
During treatment, the patient developed cellulitis on the leg that presented as pustules and erythema at a site of an eroded bulla, inframammary and axillary cutaneous candidiasis, and hyperglycemia at 19 weeks’ gestation. The cutaneous infections resolved with oral clindamycin 300 mg 3 times daily for 10 days. Topical mupirocin was used to treat the cellulitis and a mixture of zinc oxide, econazole cream, and desonide cream twice daily treated the candidiasis. Her obstetrician managed the hyperglycemia.
The bullous lesions and LP completely resolved after 2 months of treatment with oral prednisone 50 mg daily. The patient tolerated a corticosteroid taper (dose decreased by 5 mg every 2 weeks) until arriving at 10 mg, which was then decreased to 7.5 mg until delivery. A cesarean delivery was performed due to a large-for-gestational-age fetus, and an internist was consulted for the necessary precautions to increase the steroid dose during delivery due to the stress of the surgery and the risk for a hypothalamic crisis. There were no peripartum complications, and the baby was born without cutaneous lesions and remains healthy 1 year later. The patient remained disease free over 2 months postpartum, until new LP lesions developed without vesicles or bullae, which were then controlled with topical therapy. She was subsequently lost to follow-up.
Comment
Kaposi first described LPP in 1892 and used the term lichen ruber pemphigoides to describe a case of typical LP together with a widespread bullous eruption. Lichen planus pemphigoides is characterized by tense blisters that arise on lesions of LP as well as on skin unaffected by LP. In contrast, bullous LP blisters are confined to LP lesions only and occur from intense lichenoid inflammation and extensive liquefactive degeneration of basal keratinocytes. The vesicle formation in LPP is a result of autoantibodies to the bullous pemphigoid (BP) antigen BPAg2, which can be explained by the epitope spreading epiphenomenon whereby epidermotropic cytotoxic T cells damage the basal keratinocytes in LP by targeting unknown epidermal antigens, resulting in the exposure of BP180 and therefore instigating the autoimmune response.1 The process of epitope spreading takes months to develop; the mean duration of LP before LPP is 8 weeks in children and 12 weeks in adults,2 which is comparable to the current case.
Pathogenesis
Lichen planus pemphigoides usually is idiopathic; however, there have been cases reported in association with various medications including calcium channel blockers such as diltiazem, Chinese herbs,3 simvastatin,4 ramipril,5,6 captopril,7 psoralen plus UVA phototherapy,8 and cinnarizine.9 In addition, in a case-controlled study, the use of neuroleptics or diuretics was found to be a risk factor for LPP development.10
This case is unique because it shows an association of LPP with an intrauterine pregnancy. Despite the fact that we did not perform the required studies to determine the exact cause, there probably exists an association between LPP and the pregnancy, as the patient presented with a 5-month history of severe LP prior to vesicle formation. The patient only developed the vesicular lesions during pregnancy, which were later controlled with systemic steroids and then recurred postpartum only as LP lesions, suggesting that the patient’s pregnancy may have contributed in the pathogenesis as an inducing factor. We suspect that the LP was aggravated by the pregnancy and continued to worsen, so much as to cause epitope spreading and lead to the bullous eruption at the end of the first trimester.2
Differential Diagnosis
Initially, we suspected a diagnosis of pemphigoid gestationis (PG), previously known as herpes gestationis. The classic presentation of PG starts with an intense pruritus followed by the emergence of pruritic urticarial papules and plaques in the umbilical or periumbilical areas. The lesions may become targetlike or polycyclic and may spread to other areas of the trunk, arms, and legs, often including the palms and soles.11-15 Just as in our case, vesicles and bullous lesions appear at both the site of the urticarial plaques as well as on normal skin.16 The clinical features noted in our patient that were not typical of PG included the multiple lesions on the face and inside the nostrils. Only 20% of PG cases are associated with mucosal involvement,11,12,15 and there are no documented reports of PG occurring in a patient with LP, according to a PubMed search of articles indexed for MEDLINE using the search terms pemphigoid gestationis, herpes gestationis, and lichen planus.
Lichen planus pemphigoides can be easily differentiated from BP. Lichen planus pemphigoides occurs in younger patients, with a mean age of 35 years, unlike BP, which commonly affects elderly men.17 Lichen planus pemphigoides also is less severe and has a better response to treatment than BP. It also affects the palms and soles, which are rarely affected in BP. There are no reports in the literature of BP developing during pregnancy, according to a PubMed search using the terms bullous pemphigoid and pregnancy. However, LPP and BP share a common antibody, the BP180 antigen, and differences exist in the epitope where the antibody binds in each condition.18,19
Diagnosing LPP
In LPP, DIF typically shows linear deposits of IgG, IgM, IgA, fibrinogen, and C3 along the BMZ, of which IgG and C3 are most commonly seen.3 Our patient had linear deposition of C3, IgM, and IgA along the BMZ, which excluded bullous LP from the differential diagnosis. Bullous LP is not an autoimmune condition but rather is on the severe spectrum of LP where Max Joseph spaces become so large so as to lead to vesicle and bullae formation. In addition to the linear deposit at the BMZ, LPP typically reveals immunoglobulin (mainly IgM but also IgA), C3, and fibrinogen staining of colloid bodies in the papillary dermis on DIF; however, some cases of LPP only present with a linear deposition of C3 along the BMZ, which is why, similar to PG, these diagnoses by DIF are similar. Direct immunofluorescence of PG reveals linear IgG1 and IgG3 along the BMZ. IgG1 and IgG3 immunoglobulins are known to fix complement better than other immunoglobulins, thus linear C3 along the BMZ is the most consistently positive immunoreactant. Less common positive immunoreactivity with the same pattern has been seen with IgA, IgM, C1, and C4 (Table).14,15,18 The lack of linear IgG and the presence of IgM is more suggestive of LPP.
The differential diagnosis of the subepidermal autoimmune blistering diseases associated with antibodies against BP180, including BP, LPP, and PG, often is challenging.15 However, LPP can now be distinguished by immunological studies including immunoblot analysis of the immunodominant region of NC16A of the BP180 antigen and the immunoglobulin subclass that reacts to 180-, 200-,20 and 230-kDa antigens within the BMZ (Table).15,18-20 The Table summarizes the different autoantibodies, antigens, and epitopes to distinguish subepidermal autoimmune blistering diseases.
Despite not performing these studies in our patient, we concluded that the clinical, histological, and DIF findings of this case are more consistent with LPP than with the other subepidermal blistering diseases. However, we cannot exclude the possibility of the patient having a new entity with a unique antibody from epitope spreading.
Conclusion
We present a case of lichenoid papules and plaques consistent with LP, with the development of vesicles and bullae after the first trimester of pregnancy. The clinical, pathologic, and DIF findings were highly suggestive of LPP. Although the exact pathogenic mechanism is not fully known, we suspect that pregnancy may have contributed to the origin of the disease. Further evaluation of pregnant patients with lichenoid lesions who develop blisters are needed for the elucidation of the mechanism, which may be secondary to epitope spreading that led to new autoantibody formation.
- Stingl G, Holubar K. Coexistence of lichen planus and bullous pemphigoid. an immunopathological study. Br J Dermatol. 1975;93:313-320.
- Paige DG, Bhogal BS, Black MM, et al. Lichen planus pemphigoides in a child—immunopathological findings. Clin Exp Dermatol. 1993;18:552-554.
- Xu HH, Xiao T, He CD, et al. Lichen planus pemphigoides associated with Chinese herbs. Clin Exp Dermatol. 2009;34:329-332.
- Stoebner PE, Michot C, Ligeron C, et al. Simvastatin induced lichen planus pemphigoides. Ann Dermatol Venereol. 2003;130:187-190.
- Zhu YI, Fitzpatrick JE, Kornfeld BW. Lichen planus pemphigoides associated with Ramipril. Int J Dermatol. 2006;45:1453-1455.
- Ogg GS, Bhogal BS, Hashimoto T, et al. Ramipril-associated lichen planus pemphigoides. Br J Dermatol. 1997;136:412-414.
- Flageul B, Foldes C, Wallach D, et al. Captopril-induced lichen planus pemphigoides with pemphigus-like features. a case report. Dermatologica. 1986;173:248-255.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Miyagawa S, Ohi H, Muramatsu T, et al. Lichen planus pemphigoides-like lesions induced by Cinnarizine. Br J Dermatol. 1985;112:607-613.
- Bastuji-Garin S, Joly P, Picard-Dahan C, et al. Drugs associated with bullous pemphigoid. a case-control study. Arch Dermatol. 1996;132:272-276.
- Ambros-Rudolph CM. Dermatoses of pregnancy-clues to diagnosis, fetal risk and therapy. Ann Dermatol. 2011;23:265-275.
- DiZenzo G, Calabresi V, Grosso F, et al. The intracellular and extracellular domains of BP180 antigen comprise novel epitopes targeted by pemphigoid gestationis autoantibodies. J Invest Dermatol. 2006;127:864-873.
- Jenkis RE, Hern S, Black MM. Clinical features and management of 87 patients with pemphigus gestationis. Clin Exp Dermatol. 1999;24:255-259.
- Kasperkiewicz M, Zillikens D, Schmidt E. Pemphigoid diseases: pathogenesis, diagnosis, and treatment. Autoimmunity. 2012;45:55-70.
- Cobo MF, Santi CG, Maruta CW, et al. Pemphigoid gestationis: clinical and laboratory evaluation. Clinics. 2009;64:1042-1047.
- Hsu S, Ghohestani RF, Uitto J. Lichen planus pemphigoides with IgG autoantibodies to the 180 kd bullous pemphigoid antigen (type XVII collagen). J Am Acad Dermatol. 2000;42:136-141.
- Harjai B, Mendiratta V, Kakkar S, et al. Childhood lichen planus pemphigoides—a rare entity. J Eur Acad Dermatol Venereol. 2006;20:117-118.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Zillikens D. BP180 as the common autoantigen in blistering diseases with different clinical phenotypes. Keio J Med. 2002;51:21-28.
- Davis AL, Bhogal BS, Whitehead P, et al. Lichen planus pemphigoides: its relationship to bullous pemphigoid. Br J Dermatol. 1991;125:263-271.
- Stingl G, Holubar K. Coexistence of lichen planus and bullous pemphigoid. an immunopathological study. Br J Dermatol. 1975;93:313-320.
- Paige DG, Bhogal BS, Black MM, et al. Lichen planus pemphigoides in a child—immunopathological findings. Clin Exp Dermatol. 1993;18:552-554.
- Xu HH, Xiao T, He CD, et al. Lichen planus pemphigoides associated with Chinese herbs. Clin Exp Dermatol. 2009;34:329-332.
- Stoebner PE, Michot C, Ligeron C, et al. Simvastatin induced lichen planus pemphigoides. Ann Dermatol Venereol. 2003;130:187-190.
- Zhu YI, Fitzpatrick JE, Kornfeld BW. Lichen planus pemphigoides associated with Ramipril. Int J Dermatol. 2006;45:1453-1455.
- Ogg GS, Bhogal BS, Hashimoto T, et al. Ramipril-associated lichen planus pemphigoides. Br J Dermatol. 1997;136:412-414.
- Flageul B, Foldes C, Wallach D, et al. Captopril-induced lichen planus pemphigoides with pemphigus-like features. a case report. Dermatologica. 1986;173:248-255.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Miyagawa S, Ohi H, Muramatsu T, et al. Lichen planus pemphigoides-like lesions induced by Cinnarizine. Br J Dermatol. 1985;112:607-613.
- Bastuji-Garin S, Joly P, Picard-Dahan C, et al. Drugs associated with bullous pemphigoid. a case-control study. Arch Dermatol. 1996;132:272-276.
- Ambros-Rudolph CM. Dermatoses of pregnancy-clues to diagnosis, fetal risk and therapy. Ann Dermatol. 2011;23:265-275.
- DiZenzo G, Calabresi V, Grosso F, et al. The intracellular and extracellular domains of BP180 antigen comprise novel epitopes targeted by pemphigoid gestationis autoantibodies. J Invest Dermatol. 2006;127:864-873.
- Jenkis RE, Hern S, Black MM. Clinical features and management of 87 patients with pemphigus gestationis. Clin Exp Dermatol. 1999;24:255-259.
- Kasperkiewicz M, Zillikens D, Schmidt E. Pemphigoid diseases: pathogenesis, diagnosis, and treatment. Autoimmunity. 2012;45:55-70.
- Cobo MF, Santi CG, Maruta CW, et al. Pemphigoid gestationis: clinical and laboratory evaluation. Clinics. 2009;64:1042-1047.
- Hsu S, Ghohestani RF, Uitto J. Lichen planus pemphigoides with IgG autoantibodies to the 180 kd bullous pemphigoid antigen (type XVII collagen). J Am Acad Dermatol. 2000;42:136-141.
- Harjai B, Mendiratta V, Kakkar S, et al. Childhood lichen planus pemphigoides—a rare entity. J Eur Acad Dermatol Venereol. 2006;20:117-118.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Zillikens D. BP180 as the common autoantigen in blistering diseases with different clinical phenotypes. Keio J Med. 2002;51:21-28.
- Davis AL, Bhogal BS, Whitehead P, et al. Lichen planus pemphigoides: its relationship to bullous pemphigoid. Br J Dermatol. 1991;125:263-271.
Practice Points
- Lichen planus pemphigoides (LPP) is characterized by tense blisters that arise not only on lichen planus lesions such as bullous lichen planus but also on skin unaffected by lichen planus.
- In LPP, the autoantibodies specifically target the MCW-4 epitope of the NC16A4 domain of the bullous pemphigoid antigen BPAg2, distinguishing it from other autoimmune blistering diseases against the NC16A domain.
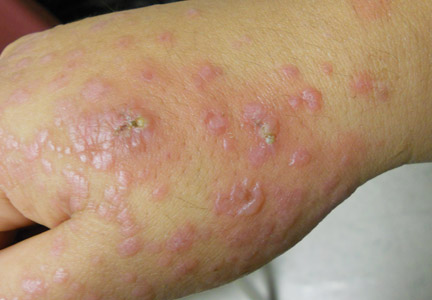
Strangulation of Radial Nerve Within Nondisplaced Fracture Component of Humeral Shaft Fracture
A radial nerve injury in association with a humeral shaft fracture is not an infrequent occurrence.1,2 The nerve injury typically is thought to be a neurapraxia caused by a contusion, as spontaneous recovery rates range from 70% to 90%.2-4 In cases in which acute nerve exploration and open reduction and internal fixation (ORIF) are not indicated, patient and clinician wait months for the nerve to recover. In some conservatively treated cases, the nerve is lacerated or entrapped. Patients with a lacerated or entrapped nerve may have better outcomes with early operative management.
We report on a rare case of the radial nerve entrapped within a nondisplaced segment of a closed humeral shaft fracture and describe the clinical outcome of early operative management. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
An intoxicated, restrained 18-year-old driver in a motor vehicle collision sustained multiple injuries, including rib fracture, apical pneumothorax with pulmonary contusion, and corneal abrasion. Orthopedic injuries included right subtrochanteric femur fracture and midshaft right humeral shaft fracture (Figure 1).
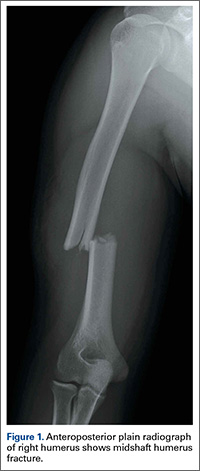
Initial orthopedic evaluation of the right arm revealed decreased sensation in the radial nerve distribution. Motor function in the radial nerve was absent; the patient was incapable of active wrist extension or finger extension. Median and ulnar nerves were motor- and sensory-intact. Radiographs showed a displaced transverse midshaft humeral shaft fracture with a minimally displaced vertical fracture line extending from the fracture site about 3 cm into the proximal segment. The patient was placed in a coaptation splint. The femur fracture was treated with an antegrade piriformis entry intramedullary nail.
ORIF of the humerus was performed to facilitate mobilization of this polytrauma patient. He was positioned prone on a flat-top table with his right arm over a radiolucent extension. The arm was abducted at the shoulder and the elbow flexed. A posterior midline skin incision was made to reflect the triceps in a lateral-to-medial direction, facilitating dissection of the lateral brachial cutaneous nerve on the lateral aspect of the triceps, with resultant localization of the radial nerve. At that time, the radial nerve was noted to be entrapped in the fracture site (Figure 2). In the proximal segment was a sagittal split, displaced about 1 mm, and it was in this interval the nerve was held. This sagittal fracture appeared incomplete as it was followed more proximally. A unicortical Kirschner wire was placed in a posterior-to-anterior direction in each fragment alongside the nerve. A lamina spreader engaged the wires and distracted the fracture site as the tines were spread apart, releasing the nerve (Figure 3). The nerve was in continuity but was severely contused at that location. After the sagittal split was reduced, two 2.7-mm lag screws were used in lag fashion, and the transverse midshaft component was fixed with a 10-hole, 4.5-mm narrow locking compression plate. The radial nerve lay on the posterior aspect of the plate, between holes 4 and 5 (Figures 4, 5). The wound was closed, and the patient was made weight-bearing as tolerated in the right upper extremity. He was sent to occupational therapy, and static and dynamic splints were made for his wrist and hand.
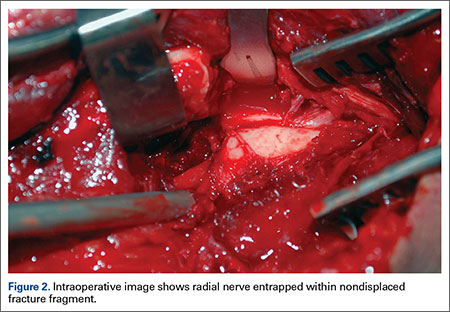
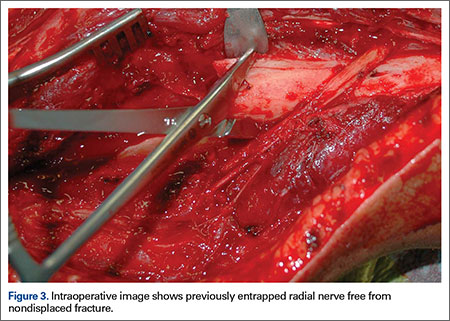
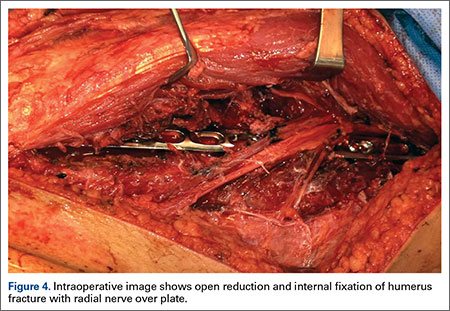
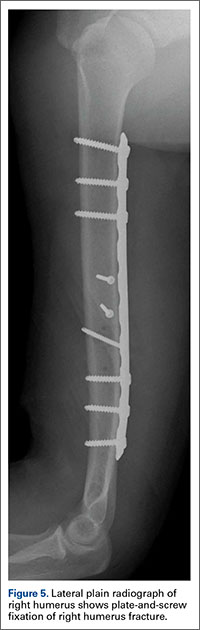
Two months after injury, radial nerve examination findings were unchanged: decreased sensation on dorsum of hand and no motor function. At 3 months, electrodiagnostic testing showed neurophysiologic evidence of severe right radial neuropathy proximal to the innervation of the right brachioradialis. There were electrodiagnostic signs of ongoing axonal loss and no signs of ongoing reinnervation. At 4 months, only motor strength in wrist extension was improved (2/5). At 5 months, the patient had 4–/5 wrist extension, 3/5 metacarpophalangeal (MCP) extension of fingers, and 0/5 MCP/interphalangeal extension of thumb. Sensation in the radial nerve distribution was still decreased. At 7 months, strength in wrist extension and finger MCP extension was 4+/5. The fracture was now well healed, with maintained alignment and no changes in hardware appearance.
Discussion
In most cases, closed treatment of a humeral shaft fracture with an associated radial nerve injury has a successful outcome.5 The etiology of the neurapraxia likely is nerve contusion after the fracture. A neurapraxia is by definition a temporary injury to the myelin sheath with an intact nerve; the nerve function recovers rapidly.
Some humeral shaft fractures, however, have been associated with radial nerve injuries more severe than contusions, resulting in axonotmesis or neurotmesis. These more severe injuries make up 10% to 30% of humeral shaft fractures, including those with a frank laceration of the nerve and those with an entrapped nerve.2,3 Shao and colleagues2 reported a 90% recovery rate for patients who delayed extrication of the entrapped radial nerve. Although there is no consensus on timing of surgical exploration, motor and sensory function of the nerve is temporally related, which may indicate that earlier diagnosis and treatment lead to improved outcome.6,7 Loss of radial nerve function can have devastating effects on upper extremity function. Often, patients lose all or some extension of the wrist and fingers and abduction and extension of the thumb.
In a standard history or physical examination, there are no particular features indicating nerve entrapment. Absolute indications for humeral shaft fractures with radial palsy are limited to open fractures, vascular injuries, and unacceptable fracture alignment. Relative indications are polytrauma and secondary palsy after attempted fracture reduction. For all other humeral shaft fractures with radial nerve palsy, observation is still the mainstay of treatment, with spontaneous recovery occurring in up to 90% of patients.2,8-12 Our patient did not have an absolute indication for operative treatment; surgery was nevertheless performed to address the polytrauma and to facilitate earlier mobilization.
Electromyelogram (EMG) studies typically are not useful after acute injury. EMG studies are better used serially to evaluate reinnervation after the acute phase. Bodner and colleagues13,14 used ultrasonography to identify the radial nerve in a patient with unimproved radial nerve palsy 6 weeks after humeral shaft fracture. They found the nerve within the fracture site, whereas magnetic resonance imaging (MRI) could not follow its course. Neither ultrasonography nor MRI would likely be used after acute injury. More research is needed to improve evaluation of patients with continued palsy after nonoperative treatment.
In the case of our patient’s humeral shaft fracture, surgery was performed early because of polytrauma and radial nerve entrapment. If left interposed between 2 fracture fragments, the nerve would have been subjected to continued ischemia and likely would not have recovered spontaneously. Ikeda and Osamura7 reported on a case of radial nerve palsy that occurred after humerus shaft fracture. The nerve, entrapped between fracture fragments, was explored later, after function failed to return. As it was found within callus, the nerve was cut and then repaired end-to-end. In our patient’s case, early exploration led to release of the radial nerve from the fracture site—preventing irreversible nerve damage and allowing for spontaneous recovery over subsequent months.
Surgery for polytrauma patients with a humeral shaft fracture and radial nerve palsy may also be beneficial with respect to early nerve exploration and early mobilization. Although our patient’s fracture was well aligned and as an isolated injury would not have required surgery, the polytrauma called for early surgical management, which revealed radial nerve entrapment and led to early recovery of nerve function.
1. Ekholm R, Adami J, Tidermark J, Hansson K, Törnkvist H, Ponzer S. Fractures of the shaft of the humerus. An epidemiological study of 401 fractures. J Bone Joint Surg Br. 2006;88(11):1469-1473.
2. Shao YC, Harwood P, Grotz MR, Limb D, Giannoudis PV. Radial nerve palsy associated with fractures of the shaft of the humerus: a systematic review. J Bone Joint Surg Br. 2005;87(12):1647-1652.
3. Shah JJ, Bhatti NA. Radial nerve paralysis associated with fractures of the humerus. A review of 62 cases. Clin Orthop Relat Res. 1983;(172):171-176.
4. Ring D, Chin K, Jupiter JB. Radial nerve palsy associated with high-energy humeral shaft fractures. J Hand Surg. 2004;29(1):144-147.
5. Sarmiento A, Zagorski JB, Zych GA, Latta LL, Capps CA. Functional bracing for the treatment of fractures of the humeral diaphysis. J Bone Joint Surg Am. 2000;82(4):478-486.
6. Hugon S, Daubresse F, Depierreux L. Radial nerve entrapment in a humeral fracture callus. Acta Orthop Belg. 2008;74(1):118-121.
7. Ikeda K, Osamura N. The radial nerve palsy caused by embedding in the humeral shaft fracture—a case report. Hand Surg. 2014;19(1):91-93.
8. Green DP, Hotchkiss RN, Pederson WC, Wolfe SW, eds. Green’s Operative Hand Surgery. 2 vols. 5th ed. Philadelphia, PA: Elsevier/Churchill Livingstone; 2005.
9. Kettelkamp DB, Alexander H. Clinical review of radial nerve injury. J Trauma. 1967;7(3):424-432.
10. Pollock FH, Drake D, Bovill EG, Day L, Trafton PG. Treatment of radial neuropathy associated with fractures of the humerus. J Bone Joint Surg Am. 1981;63(2):239-243.
11. Li Y, Ning G, Wu Q, Wu Q, Li Y, Feng S. Review of literature of radial nerve injuries associated with humeral fractures—an integrated management strategy. PloS One. 2013;8(11):e78576.
12. DeFranco MJ, Lawton JN. Radial nerve injuries associated with humeral fractures. J Hand Surg. 2006;31(4):655-663.
13. Bodner G, Huber B, Schwabegger A, Lutz M, Waldenberger P. Sonographic detection of radial nerve entrapment within a humerus fracture. J Ultrasound Med. 1999;18(10):703-706.
14. Bodner G, Buchberger W, Schocke M, et al. Radial nerve palsy associated with humeral shaft fracture: evaluation with US—initial experience. Radiology. 2001;219(3):811-816.
A radial nerve injury in association with a humeral shaft fracture is not an infrequent occurrence.1,2 The nerve injury typically is thought to be a neurapraxia caused by a contusion, as spontaneous recovery rates range from 70% to 90%.2-4 In cases in which acute nerve exploration and open reduction and internal fixation (ORIF) are not indicated, patient and clinician wait months for the nerve to recover. In some conservatively treated cases, the nerve is lacerated or entrapped. Patients with a lacerated or entrapped nerve may have better outcomes with early operative management.
We report on a rare case of the radial nerve entrapped within a nondisplaced segment of a closed humeral shaft fracture and describe the clinical outcome of early operative management. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
An intoxicated, restrained 18-year-old driver in a motor vehicle collision sustained multiple injuries, including rib fracture, apical pneumothorax with pulmonary contusion, and corneal abrasion. Orthopedic injuries included right subtrochanteric femur fracture and midshaft right humeral shaft fracture (Figure 1).
Initial orthopedic evaluation of the right arm revealed decreased sensation in the radial nerve distribution. Motor function in the radial nerve was absent; the patient was incapable of active wrist extension or finger extension. Median and ulnar nerves were motor- and sensory-intact. Radiographs showed a displaced transverse midshaft humeral shaft fracture with a minimally displaced vertical fracture line extending from the fracture site about 3 cm into the proximal segment. The patient was placed in a coaptation splint. The femur fracture was treated with an antegrade piriformis entry intramedullary nail.
ORIF of the humerus was performed to facilitate mobilization of this polytrauma patient. He was positioned prone on a flat-top table with his right arm over a radiolucent extension. The arm was abducted at the shoulder and the elbow flexed. A posterior midline skin incision was made to reflect the triceps in a lateral-to-medial direction, facilitating dissection of the lateral brachial cutaneous nerve on the lateral aspect of the triceps, with resultant localization of the radial nerve. At that time, the radial nerve was noted to be entrapped in the fracture site (Figure 2). In the proximal segment was a sagittal split, displaced about 1 mm, and it was in this interval the nerve was held. This sagittal fracture appeared incomplete as it was followed more proximally. A unicortical Kirschner wire was placed in a posterior-to-anterior direction in each fragment alongside the nerve. A lamina spreader engaged the wires and distracted the fracture site as the tines were spread apart, releasing the nerve (Figure 3). The nerve was in continuity but was severely contused at that location. After the sagittal split was reduced, two 2.7-mm lag screws were used in lag fashion, and the transverse midshaft component was fixed with a 10-hole, 4.5-mm narrow locking compression plate. The radial nerve lay on the posterior aspect of the plate, between holes 4 and 5 (Figures 4, 5). The wound was closed, and the patient was made weight-bearing as tolerated in the right upper extremity. He was sent to occupational therapy, and static and dynamic splints were made for his wrist and hand.
Two months after injury, radial nerve examination findings were unchanged: decreased sensation on dorsum of hand and no motor function. At 3 months, electrodiagnostic testing showed neurophysiologic evidence of severe right radial neuropathy proximal to the innervation of the right brachioradialis. There were electrodiagnostic signs of ongoing axonal loss and no signs of ongoing reinnervation. At 4 months, only motor strength in wrist extension was improved (2/5). At 5 months, the patient had 4–/5 wrist extension, 3/5 metacarpophalangeal (MCP) extension of fingers, and 0/5 MCP/interphalangeal extension of thumb. Sensation in the radial nerve distribution was still decreased. At 7 months, strength in wrist extension and finger MCP extension was 4+/5. The fracture was now well healed, with maintained alignment and no changes in hardware appearance.
Discussion
In most cases, closed treatment of a humeral shaft fracture with an associated radial nerve injury has a successful outcome.5 The etiology of the neurapraxia likely is nerve contusion after the fracture. A neurapraxia is by definition a temporary injury to the myelin sheath with an intact nerve; the nerve function recovers rapidly.
Some humeral shaft fractures, however, have been associated with radial nerve injuries more severe than contusions, resulting in axonotmesis or neurotmesis. These more severe injuries make up 10% to 30% of humeral shaft fractures, including those with a frank laceration of the nerve and those with an entrapped nerve.2,3 Shao and colleagues2 reported a 90% recovery rate for patients who delayed extrication of the entrapped radial nerve. Although there is no consensus on timing of surgical exploration, motor and sensory function of the nerve is temporally related, which may indicate that earlier diagnosis and treatment lead to improved outcome.6,7 Loss of radial nerve function can have devastating effects on upper extremity function. Often, patients lose all or some extension of the wrist and fingers and abduction and extension of the thumb.
In a standard history or physical examination, there are no particular features indicating nerve entrapment. Absolute indications for humeral shaft fractures with radial palsy are limited to open fractures, vascular injuries, and unacceptable fracture alignment. Relative indications are polytrauma and secondary palsy after attempted fracture reduction. For all other humeral shaft fractures with radial nerve palsy, observation is still the mainstay of treatment, with spontaneous recovery occurring in up to 90% of patients.2,8-12 Our patient did not have an absolute indication for operative treatment; surgery was nevertheless performed to address the polytrauma and to facilitate earlier mobilization.
Electromyelogram (EMG) studies typically are not useful after acute injury. EMG studies are better used serially to evaluate reinnervation after the acute phase. Bodner and colleagues13,14 used ultrasonography to identify the radial nerve in a patient with unimproved radial nerve palsy 6 weeks after humeral shaft fracture. They found the nerve within the fracture site, whereas magnetic resonance imaging (MRI) could not follow its course. Neither ultrasonography nor MRI would likely be used after acute injury. More research is needed to improve evaluation of patients with continued palsy after nonoperative treatment.
In the case of our patient’s humeral shaft fracture, surgery was performed early because of polytrauma and radial nerve entrapment. If left interposed between 2 fracture fragments, the nerve would have been subjected to continued ischemia and likely would not have recovered spontaneously. Ikeda and Osamura7 reported on a case of radial nerve palsy that occurred after humerus shaft fracture. The nerve, entrapped between fracture fragments, was explored later, after function failed to return. As it was found within callus, the nerve was cut and then repaired end-to-end. In our patient’s case, early exploration led to release of the radial nerve from the fracture site—preventing irreversible nerve damage and allowing for spontaneous recovery over subsequent months.
Surgery for polytrauma patients with a humeral shaft fracture and radial nerve palsy may also be beneficial with respect to early nerve exploration and early mobilization. Although our patient’s fracture was well aligned and as an isolated injury would not have required surgery, the polytrauma called for early surgical management, which revealed radial nerve entrapment and led to early recovery of nerve function.
A radial nerve injury in association with a humeral shaft fracture is not an infrequent occurrence.1,2 The nerve injury typically is thought to be a neurapraxia caused by a contusion, as spontaneous recovery rates range from 70% to 90%.2-4 In cases in which acute nerve exploration and open reduction and internal fixation (ORIF) are not indicated, patient and clinician wait months for the nerve to recover. In some conservatively treated cases, the nerve is lacerated or entrapped. Patients with a lacerated or entrapped nerve may have better outcomes with early operative management.
We report on a rare case of the radial nerve entrapped within a nondisplaced segment of a closed humeral shaft fracture and describe the clinical outcome of early operative management. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
An intoxicated, restrained 18-year-old driver in a motor vehicle collision sustained multiple injuries, including rib fracture, apical pneumothorax with pulmonary contusion, and corneal abrasion. Orthopedic injuries included right subtrochanteric femur fracture and midshaft right humeral shaft fracture (Figure 1).
Initial orthopedic evaluation of the right arm revealed decreased sensation in the radial nerve distribution. Motor function in the radial nerve was absent; the patient was incapable of active wrist extension or finger extension. Median and ulnar nerves were motor- and sensory-intact. Radiographs showed a displaced transverse midshaft humeral shaft fracture with a minimally displaced vertical fracture line extending from the fracture site about 3 cm into the proximal segment. The patient was placed in a coaptation splint. The femur fracture was treated with an antegrade piriformis entry intramedullary nail.
ORIF of the humerus was performed to facilitate mobilization of this polytrauma patient. He was positioned prone on a flat-top table with his right arm over a radiolucent extension. The arm was abducted at the shoulder and the elbow flexed. A posterior midline skin incision was made to reflect the triceps in a lateral-to-medial direction, facilitating dissection of the lateral brachial cutaneous nerve on the lateral aspect of the triceps, with resultant localization of the radial nerve. At that time, the radial nerve was noted to be entrapped in the fracture site (Figure 2). In the proximal segment was a sagittal split, displaced about 1 mm, and it was in this interval the nerve was held. This sagittal fracture appeared incomplete as it was followed more proximally. A unicortical Kirschner wire was placed in a posterior-to-anterior direction in each fragment alongside the nerve. A lamina spreader engaged the wires and distracted the fracture site as the tines were spread apart, releasing the nerve (Figure 3). The nerve was in continuity but was severely contused at that location. After the sagittal split was reduced, two 2.7-mm lag screws were used in lag fashion, and the transverse midshaft component was fixed with a 10-hole, 4.5-mm narrow locking compression plate. The radial nerve lay on the posterior aspect of the plate, between holes 4 and 5 (Figures 4, 5). The wound was closed, and the patient was made weight-bearing as tolerated in the right upper extremity. He was sent to occupational therapy, and static and dynamic splints were made for his wrist and hand.
Two months after injury, radial nerve examination findings were unchanged: decreased sensation on dorsum of hand and no motor function. At 3 months, electrodiagnostic testing showed neurophysiologic evidence of severe right radial neuropathy proximal to the innervation of the right brachioradialis. There were electrodiagnostic signs of ongoing axonal loss and no signs of ongoing reinnervation. At 4 months, only motor strength in wrist extension was improved (2/5). At 5 months, the patient had 4–/5 wrist extension, 3/5 metacarpophalangeal (MCP) extension of fingers, and 0/5 MCP/interphalangeal extension of thumb. Sensation in the radial nerve distribution was still decreased. At 7 months, strength in wrist extension and finger MCP extension was 4+/5. The fracture was now well healed, with maintained alignment and no changes in hardware appearance.
Discussion
In most cases, closed treatment of a humeral shaft fracture with an associated radial nerve injury has a successful outcome.5 The etiology of the neurapraxia likely is nerve contusion after the fracture. A neurapraxia is by definition a temporary injury to the myelin sheath with an intact nerve; the nerve function recovers rapidly.
Some humeral shaft fractures, however, have been associated with radial nerve injuries more severe than contusions, resulting in axonotmesis or neurotmesis. These more severe injuries make up 10% to 30% of humeral shaft fractures, including those with a frank laceration of the nerve and those with an entrapped nerve.2,3 Shao and colleagues2 reported a 90% recovery rate for patients who delayed extrication of the entrapped radial nerve. Although there is no consensus on timing of surgical exploration, motor and sensory function of the nerve is temporally related, which may indicate that earlier diagnosis and treatment lead to improved outcome.6,7 Loss of radial nerve function can have devastating effects on upper extremity function. Often, patients lose all or some extension of the wrist and fingers and abduction and extension of the thumb.
In a standard history or physical examination, there are no particular features indicating nerve entrapment. Absolute indications for humeral shaft fractures with radial palsy are limited to open fractures, vascular injuries, and unacceptable fracture alignment. Relative indications are polytrauma and secondary palsy after attempted fracture reduction. For all other humeral shaft fractures with radial nerve palsy, observation is still the mainstay of treatment, with spontaneous recovery occurring in up to 90% of patients.2,8-12 Our patient did not have an absolute indication for operative treatment; surgery was nevertheless performed to address the polytrauma and to facilitate earlier mobilization.
Electromyelogram (EMG) studies typically are not useful after acute injury. EMG studies are better used serially to evaluate reinnervation after the acute phase. Bodner and colleagues13,14 used ultrasonography to identify the radial nerve in a patient with unimproved radial nerve palsy 6 weeks after humeral shaft fracture. They found the nerve within the fracture site, whereas magnetic resonance imaging (MRI) could not follow its course. Neither ultrasonography nor MRI would likely be used after acute injury. More research is needed to improve evaluation of patients with continued palsy after nonoperative treatment.
In the case of our patient’s humeral shaft fracture, surgery was performed early because of polytrauma and radial nerve entrapment. If left interposed between 2 fracture fragments, the nerve would have been subjected to continued ischemia and likely would not have recovered spontaneously. Ikeda and Osamura7 reported on a case of radial nerve palsy that occurred after humerus shaft fracture. The nerve, entrapped between fracture fragments, was explored later, after function failed to return. As it was found within callus, the nerve was cut and then repaired end-to-end. In our patient’s case, early exploration led to release of the radial nerve from the fracture site—preventing irreversible nerve damage and allowing for spontaneous recovery over subsequent months.
Surgery for polytrauma patients with a humeral shaft fracture and radial nerve palsy may also be beneficial with respect to early nerve exploration and early mobilization. Although our patient’s fracture was well aligned and as an isolated injury would not have required surgery, the polytrauma called for early surgical management, which revealed radial nerve entrapment and led to early recovery of nerve function.
1. Ekholm R, Adami J, Tidermark J, Hansson K, Törnkvist H, Ponzer S. Fractures of the shaft of the humerus. An epidemiological study of 401 fractures. J Bone Joint Surg Br. 2006;88(11):1469-1473.
2. Shao YC, Harwood P, Grotz MR, Limb D, Giannoudis PV. Radial nerve palsy associated with fractures of the shaft of the humerus: a systematic review. J Bone Joint Surg Br. 2005;87(12):1647-1652.
3. Shah JJ, Bhatti NA. Radial nerve paralysis associated with fractures of the humerus. A review of 62 cases. Clin Orthop Relat Res. 1983;(172):171-176.
4. Ring D, Chin K, Jupiter JB. Radial nerve palsy associated with high-energy humeral shaft fractures. J Hand Surg. 2004;29(1):144-147.
5. Sarmiento A, Zagorski JB, Zych GA, Latta LL, Capps CA. Functional bracing for the treatment of fractures of the humeral diaphysis. J Bone Joint Surg Am. 2000;82(4):478-486.
6. Hugon S, Daubresse F, Depierreux L. Radial nerve entrapment in a humeral fracture callus. Acta Orthop Belg. 2008;74(1):118-121.
7. Ikeda K, Osamura N. The radial nerve palsy caused by embedding in the humeral shaft fracture—a case report. Hand Surg. 2014;19(1):91-93.
8. Green DP, Hotchkiss RN, Pederson WC, Wolfe SW, eds. Green’s Operative Hand Surgery. 2 vols. 5th ed. Philadelphia, PA: Elsevier/Churchill Livingstone; 2005.
9. Kettelkamp DB, Alexander H. Clinical review of radial nerve injury. J Trauma. 1967;7(3):424-432.
10. Pollock FH, Drake D, Bovill EG, Day L, Trafton PG. Treatment of radial neuropathy associated with fractures of the humerus. J Bone Joint Surg Am. 1981;63(2):239-243.
11. Li Y, Ning G, Wu Q, Wu Q, Li Y, Feng S. Review of literature of radial nerve injuries associated with humeral fractures—an integrated management strategy. PloS One. 2013;8(11):e78576.
12. DeFranco MJ, Lawton JN. Radial nerve injuries associated with humeral fractures. J Hand Surg. 2006;31(4):655-663.
13. Bodner G, Huber B, Schwabegger A, Lutz M, Waldenberger P. Sonographic detection of radial nerve entrapment within a humerus fracture. J Ultrasound Med. 1999;18(10):703-706.
14. Bodner G, Buchberger W, Schocke M, et al. Radial nerve palsy associated with humeral shaft fracture: evaluation with US—initial experience. Radiology. 2001;219(3):811-816.
1. Ekholm R, Adami J, Tidermark J, Hansson K, Törnkvist H, Ponzer S. Fractures of the shaft of the humerus. An epidemiological study of 401 fractures. J Bone Joint Surg Br. 2006;88(11):1469-1473.
2. Shao YC, Harwood P, Grotz MR, Limb D, Giannoudis PV. Radial nerve palsy associated with fractures of the shaft of the humerus: a systematic review. J Bone Joint Surg Br. 2005;87(12):1647-1652.
3. Shah JJ, Bhatti NA. Radial nerve paralysis associated with fractures of the humerus. A review of 62 cases. Clin Orthop Relat Res. 1983;(172):171-176.
4. Ring D, Chin K, Jupiter JB. Radial nerve palsy associated with high-energy humeral shaft fractures. J Hand Surg. 2004;29(1):144-147.
5. Sarmiento A, Zagorski JB, Zych GA, Latta LL, Capps CA. Functional bracing for the treatment of fractures of the humeral diaphysis. J Bone Joint Surg Am. 2000;82(4):478-486.
6. Hugon S, Daubresse F, Depierreux L. Radial nerve entrapment in a humeral fracture callus. Acta Orthop Belg. 2008;74(1):118-121.
7. Ikeda K, Osamura N. The radial nerve palsy caused by embedding in the humeral shaft fracture—a case report. Hand Surg. 2014;19(1):91-93.
8. Green DP, Hotchkiss RN, Pederson WC, Wolfe SW, eds. Green’s Operative Hand Surgery. 2 vols. 5th ed. Philadelphia, PA: Elsevier/Churchill Livingstone; 2005.
9. Kettelkamp DB, Alexander H. Clinical review of radial nerve injury. J Trauma. 1967;7(3):424-432.
10. Pollock FH, Drake D, Bovill EG, Day L, Trafton PG. Treatment of radial neuropathy associated with fractures of the humerus. J Bone Joint Surg Am. 1981;63(2):239-243.
11. Li Y, Ning G, Wu Q, Wu Q, Li Y, Feng S. Review of literature of radial nerve injuries associated with humeral fractures—an integrated management strategy. PloS One. 2013;8(11):e78576.
12. DeFranco MJ, Lawton JN. Radial nerve injuries associated with humeral fractures. J Hand Surg. 2006;31(4):655-663.
13. Bodner G, Huber B, Schwabegger A, Lutz M, Waldenberger P. Sonographic detection of radial nerve entrapment within a humerus fracture. J Ultrasound Med. 1999;18(10):703-706.
14. Bodner G, Buchberger W, Schocke M, et al. Radial nerve palsy associated with humeral shaft fracture: evaluation with US—initial experience. Radiology. 2001;219(3):811-816.
He Huffed and He Puffed and He Got Frostbite
Case
A 27-year-old man presented to an ED after experiencing a syncopal episode. His vital signs at presentation were normal. Physical examination was generally normal except that there were blisters on the patient’s abdomen, left hand, and right arm, as well as a hypertrophic nodule on the right elbow (Figure) and hard growths on the digits of the right hand. The patient stated the growths started 5 months ago and had been increasing in size. On further questioning, the patient admitted to “huffing” (ie, inhaling) at least six cans of pressurized dust-removal keyboard cleaning spray daily for the past 11 months.
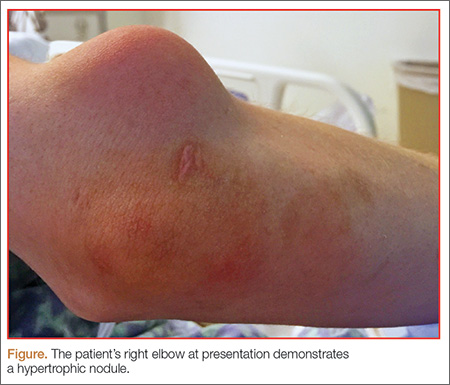
Why do patients abuse keyboard cleaning spray?
The propellant used in certain liquefied compressed gas products is 1,1-difluoroethane (1,1-DFE), a fluorinated hydrocarbon. It is a member of a broad class of related compounds that are present in spray paints, glues, nail polish removers, fuels, hair sprays, and air-freshening products. These 1,1-DFE-containing products are abused for their rapid and short-acting central nervous system (CNS) depressant effects—not unlike that of ethanol. Typically, the vapor of a volatile hydrocarbon is inhaled directly from the open container (“sniffing”), from a bag (“bagging”), or from a soaked rag (huffing). Not only are such hydrocarbon-containing products easy to conceal, they are also highly accessible and inexpensive. Moreover, there are generally no direct legal consequences resulting from abuse of these substances.
All of the aforementioned factors make hydrocarbons a popular drug of abuse among adolescents. Approximately 75% of the population abusing hydrocarbons is younger than age 18 years, half of whom reported first use prior to age 13 years.1,2 Though inhalant abuse rarely continues into adulthood, 0.1% of individuals between the ages of 18 and 30 years report having an inhalant-use disorder.
Hydrocarbons and their halogenated derivatives are lipophilic compounds that are rapidly absorbed after inhalation and rapidly distributed to CNS and cardiac tissue. The brain concentration of 1,1-DFE likely peaks higher than concentrations in other organs and is cleared more rapidly.3 Hydrocarbons produce CNS depression secondary to multiple mechanisms, including gamma-aminobutyric acid agonism, dopamine modulation, and N-methyl-D-aspartate-receptor antagonism.4,5
What causes skin lesions on the abdomen and arms?
The lesions on the patient’s abdomen and extremities were consistent with frostbite. The liquefied compressed gas in computer-cleaning and related products is housed in a pressurized canister. The pressure is released when the spray nozzle is depressed; this causes the liquid to rapidly expand to a gas as it is released, resulting in a quick decrease in the temperature of the metal canister. This process, referred to as adiabatic cooling, demonstrates the first law of thermodynamics. The cold temperature of both the liquid and the canister can cause frostbite in the digits and other parts of the body with which the canister or liquid comes into contact.6
Why did the patient have syncope?
Halogenated hydrocarbons inhibit the cardiac delayed rectifier potassium channels involved in the repolarization of cardiac myocytes, causing a delay in repolarization that is manifested as prolongation of the QT interval on an electrocardiogram. This condition places patients at an increased risk of developing torsades de pointes (TdP).7 In most cases, TdP is self-terminating; however, if TdP persists, degeneration to ventricular fibrillation will result. Deaths caused in this fashion have been referred to as “sudden sniffing death syndrome,” and account for half of all hydrocarbon-related deaths.6,8 In addition to the cardiac effects, hydrocarbons are simple asphyxiants that act by displacing oxygen from inspired air, which also contributes to syncope.
It is important to note that epinephrine and other catecholamines increase the risk for dysrhythmias such as TdP in the setting of hydrocarbon abuse.9 For this reason, epinephrine should be used with caution in the setting of a hydrocarbon-induced arrhythmia. Beta-adrenergic antagonists such as esmolol and propranolol are preferable because they reduce the incidence of ectopia that may trigger TdP.10
What is the significance of the masses noted on the examination and radiograph?
Fluorosis is associated with abnormalities of skeletal and dental tissue. Skeletal fluorosis causes osteosclerosis of the axial skeleton, periosteal new bone formation, ligamentous and tendinous ossification, and osteophyte formation. Dental fluorosis causes a yellow/brown discoloration of the teeth with horizontal streaking (mottling), pitting, and chipping.11 Fluorosis is well-described in regions where water fluoride concentrations are high due to industrial exposure; from consumption of fluorinated wine or chronic overconsumption of tea (especially green or black tea); or from fluoridated toothpaste.12-14 More recently, fluorosis has been described in patients treated for an extended duration of time with voriconazole, a fluorinated antifungal agent.15 Unlike other hydrocarbon products, fluorinated hydrocarbons such as 1,1-DFE can significantly increase systemic fluoride concentrations with excessive use. Rapid skeletal fluorosis is not well described, but has been reported after chronic abuse of fluorinated hydrocarbons.16
How is fluorosis diagnosed and managed?
The lack of rapid laboratory testing available for serum, urine, and bone fluoride concentrations makes the initial diagnosis of fluorosis a clinical one. Imaging studies are generally highly suggestive of fluorosis and can be used to support the diagnosis. A dual energy X-ray absorptiometry scan of the spine, hip, femur, and distal portions of the radii can reveal elevated T-scores consistent with osteosclerosis.14 These findings, in conjunction with bone or joint pain, reduced range of motion, or kyphosis, should prompt clinicians to conduct further testing—even without a confirmed fluoride source. A serum fluoride (reference range, 0.2-3.2 mg/L) and 24-hour urine fluoride (reference range, 0.2-3.2 mg/dL) and creatinine evaluation can be used to diagnose fluorosis. However, a bone biopsy with quantitative bone ash fluoride analysis remains the gold standard for the diagnosis of skeletal fluorosis.16 Laboratory evaluation should also include an assessment of electrolytes, specifically calcium, 25-hydroxyvitamin D, and alkaline phosphatase. The differential diagnosis should include hemoglobinopathies, renal osteodystrophy, Paget disease, hypothyroidism, and skeletal metastases.16
Treatment of fluorosis is largely symptomatic and supportive, with identification and discontinuation of the fluoride source. Patients should be referred to an orthopedist for evaluation and management as needed. Evaluation by an endocrinologist should also be considered because patients may have chronic vitamin D and calcium deficiencies as a result of systemic fluorosis.
Case Conclusion
The patient’s laboratory assessment was notable for the following: alkaline phosphatase, 624 U/L (reference range, 44-147 IU/L); vitamin D, 10 ng/mL (reference range, 20-40 ng/mL); serum fluoride, 0.3 mg/L (reference range, 0.2-3.2 mg/L); urine fluoride, 52 mg/dL (0.2-3.2 mg/dL); and urine creatinine, 1 g/L (reference range, 0.3-3 g/L). Imaging studies noted periosteal bone formation on the lateral epicondyle of the distal right humerus, as well as similar osseous abnormalities in other locations. A bone biopsy was scheduled. The patient was treated with oral vitamin D and educated about the importance of discontinuing the huffing of all hydrocarbons.
1. Williams JF, Storck M; American Academy of Pediatrics Committee on Substance Abuse; American Academy of Pediatrics Committee on Native American Child Health. Inhalant abuse. Pediatrics. 2007;119(5):1009-1017.
2. Wu LT, Pilowsky DJ, Schlenger WE. Inhalant abuse and dependence among adolescents in the United States. J Am Acad Child Adolesc Psychiatry. 2004;43(10):1206-1214.
3. Avella J, Kunaparaju N, Kumar S, Lehrer M, Zito SW, Barletta M. Uptake and distribution of the abused inhalant 1,1-difluoroethane in the rat. J Anal Toxicol. 2010;34(7):381-388.
4. Tormoehlen LM, Tekulve KJ, Nañagas KA. Hydrocarbon toxicity: A review. Clin Toxicol (Phila). 2014;52(5):479-489.
5. Duncan JR, Lawrence AJ. Conventional concepts and new perspectives for understanding the addictive properties of inhalants. J Pharmacol Sci. 2013;122(4):237-243.
6. Sakai K, Maruyama-Maebashi K, Takatsu A, et al. Sudden death involving inhalation of 1,1-difluoroethane (HFC-152a) with spray cleaner: three case reports. Forensic Sci Int. 2011;206(1-3):e58-e61.
7. Himmel HM. Mechanisms involved in cardiac sensitization by volatile anesthetics: general applicability to halogenated hydrocarbons? Crit Rev Toxicol. 2008;38(9):773-803.
8. Avella J, Wilson JC, Lehrer M. Fatal cardiac arrhythmia after repeated exposure to 1,1-difluoroethane (DFE). Am J Forensic Med Pathol. 2006;27(1):58-60.
9. Nelson LS. Toxicologic myocardial sensitization. J Toxicol Clin Toxicol. 2002;40(7):867-879.
10. Mortiz F, de La Chapelle A, Bauer F, Leroy JP, Goullé JP, Bonmarchand G. Esmolol in the treatment of severe arrhythmia after acute trichloroethylene poisoning. Intensive Care Med. 2000;26(2):256.
11. Majumdar KK. Health impact of supplying safe drinking water containing fluoride below permissible level on flourosis patients in a fluoride-endemic rural area of West Bengal. Indian J Public Health. 2011;55(4):303-308.
12. Kakumanu N, Rao SD. Images in clinical medicine. Skeletal fluorosis due to excessive tea drinking. N Engl J Med 2013;368(12):1140.
13. Soriano M, Manchón F. Radiological aspects of a new type of bone fluorosis, periostitis deformans. Radiology 1966;87(6):1089-1094.
14. Tamer MN, Kale Köroğlu B, Arslan C, et al. Osteosclerosis due to endemic fluorosis. Sci Total Environ. 2007;373(1):43-48.
15. Bucknor MD, Gross AJ, Link TM. Voriconazole-induced periostitis in two post-transplant patients. J Radiol Case Rep. 2013;7(8):10-17.
16. Cohen E, Hsu RY, Evangelista P, Aaron R, Rubin LE. Rapid-onset diffuse skeletal fluorosis from inhalant abuse: a case report. JBJS Case Connector. 2014;4(4):e108.
Case
A 27-year-old man presented to an ED after experiencing a syncopal episode. His vital signs at presentation were normal. Physical examination was generally normal except that there were blisters on the patient’s abdomen, left hand, and right arm, as well as a hypertrophic nodule on the right elbow (Figure) and hard growths on the digits of the right hand. The patient stated the growths started 5 months ago and had been increasing in size. On further questioning, the patient admitted to “huffing” (ie, inhaling) at least six cans of pressurized dust-removal keyboard cleaning spray daily for the past 11 months.
Why do patients abuse keyboard cleaning spray?
The propellant used in certain liquefied compressed gas products is 1,1-difluoroethane (1,1-DFE), a fluorinated hydrocarbon. It is a member of a broad class of related compounds that are present in spray paints, glues, nail polish removers, fuels, hair sprays, and air-freshening products. These 1,1-DFE-containing products are abused for their rapid and short-acting central nervous system (CNS) depressant effects—not unlike that of ethanol. Typically, the vapor of a volatile hydrocarbon is inhaled directly from the open container (“sniffing”), from a bag (“bagging”), or from a soaked rag (huffing). Not only are such hydrocarbon-containing products easy to conceal, they are also highly accessible and inexpensive. Moreover, there are generally no direct legal consequences resulting from abuse of these substances.
All of the aforementioned factors make hydrocarbons a popular drug of abuse among adolescents. Approximately 75% of the population abusing hydrocarbons is younger than age 18 years, half of whom reported first use prior to age 13 years.1,2 Though inhalant abuse rarely continues into adulthood, 0.1% of individuals between the ages of 18 and 30 years report having an inhalant-use disorder.
Hydrocarbons and their halogenated derivatives are lipophilic compounds that are rapidly absorbed after inhalation and rapidly distributed to CNS and cardiac tissue. The brain concentration of 1,1-DFE likely peaks higher than concentrations in other organs and is cleared more rapidly.3 Hydrocarbons produce CNS depression secondary to multiple mechanisms, including gamma-aminobutyric acid agonism, dopamine modulation, and N-methyl-D-aspartate-receptor antagonism.4,5
What causes skin lesions on the abdomen and arms?
The lesions on the patient’s abdomen and extremities were consistent with frostbite. The liquefied compressed gas in computer-cleaning and related products is housed in a pressurized canister. The pressure is released when the spray nozzle is depressed; this causes the liquid to rapidly expand to a gas as it is released, resulting in a quick decrease in the temperature of the metal canister. This process, referred to as adiabatic cooling, demonstrates the first law of thermodynamics. The cold temperature of both the liquid and the canister can cause frostbite in the digits and other parts of the body with which the canister or liquid comes into contact.6
Why did the patient have syncope?
Halogenated hydrocarbons inhibit the cardiac delayed rectifier potassium channels involved in the repolarization of cardiac myocytes, causing a delay in repolarization that is manifested as prolongation of the QT interval on an electrocardiogram. This condition places patients at an increased risk of developing torsades de pointes (TdP).7 In most cases, TdP is self-terminating; however, if TdP persists, degeneration to ventricular fibrillation will result. Deaths caused in this fashion have been referred to as “sudden sniffing death syndrome,” and account for half of all hydrocarbon-related deaths.6,8 In addition to the cardiac effects, hydrocarbons are simple asphyxiants that act by displacing oxygen from inspired air, which also contributes to syncope.
It is important to note that epinephrine and other catecholamines increase the risk for dysrhythmias such as TdP in the setting of hydrocarbon abuse.9 For this reason, epinephrine should be used with caution in the setting of a hydrocarbon-induced arrhythmia. Beta-adrenergic antagonists such as esmolol and propranolol are preferable because they reduce the incidence of ectopia that may trigger TdP.10
What is the significance of the masses noted on the examination and radiograph?
Fluorosis is associated with abnormalities of skeletal and dental tissue. Skeletal fluorosis causes osteosclerosis of the axial skeleton, periosteal new bone formation, ligamentous and tendinous ossification, and osteophyte formation. Dental fluorosis causes a yellow/brown discoloration of the teeth with horizontal streaking (mottling), pitting, and chipping.11 Fluorosis is well-described in regions where water fluoride concentrations are high due to industrial exposure; from consumption of fluorinated wine or chronic overconsumption of tea (especially green or black tea); or from fluoridated toothpaste.12-14 More recently, fluorosis has been described in patients treated for an extended duration of time with voriconazole, a fluorinated antifungal agent.15 Unlike other hydrocarbon products, fluorinated hydrocarbons such as 1,1-DFE can significantly increase systemic fluoride concentrations with excessive use. Rapid skeletal fluorosis is not well described, but has been reported after chronic abuse of fluorinated hydrocarbons.16
How is fluorosis diagnosed and managed?
The lack of rapid laboratory testing available for serum, urine, and bone fluoride concentrations makes the initial diagnosis of fluorosis a clinical one. Imaging studies are generally highly suggestive of fluorosis and can be used to support the diagnosis. A dual energy X-ray absorptiometry scan of the spine, hip, femur, and distal portions of the radii can reveal elevated T-scores consistent with osteosclerosis.14 These findings, in conjunction with bone or joint pain, reduced range of motion, or kyphosis, should prompt clinicians to conduct further testing—even without a confirmed fluoride source. A serum fluoride (reference range, 0.2-3.2 mg/L) and 24-hour urine fluoride (reference range, 0.2-3.2 mg/dL) and creatinine evaluation can be used to diagnose fluorosis. However, a bone biopsy with quantitative bone ash fluoride analysis remains the gold standard for the diagnosis of skeletal fluorosis.16 Laboratory evaluation should also include an assessment of electrolytes, specifically calcium, 25-hydroxyvitamin D, and alkaline phosphatase. The differential diagnosis should include hemoglobinopathies, renal osteodystrophy, Paget disease, hypothyroidism, and skeletal metastases.16
Treatment of fluorosis is largely symptomatic and supportive, with identification and discontinuation of the fluoride source. Patients should be referred to an orthopedist for evaluation and management as needed. Evaluation by an endocrinologist should also be considered because patients may have chronic vitamin D and calcium deficiencies as a result of systemic fluorosis.
Case Conclusion
The patient’s laboratory assessment was notable for the following: alkaline phosphatase, 624 U/L (reference range, 44-147 IU/L); vitamin D, 10 ng/mL (reference range, 20-40 ng/mL); serum fluoride, 0.3 mg/L (reference range, 0.2-3.2 mg/L); urine fluoride, 52 mg/dL (0.2-3.2 mg/dL); and urine creatinine, 1 g/L (reference range, 0.3-3 g/L). Imaging studies noted periosteal bone formation on the lateral epicondyle of the distal right humerus, as well as similar osseous abnormalities in other locations. A bone biopsy was scheduled. The patient was treated with oral vitamin D and educated about the importance of discontinuing the huffing of all hydrocarbons.
Case
A 27-year-old man presented to an ED after experiencing a syncopal episode. His vital signs at presentation were normal. Physical examination was generally normal except that there were blisters on the patient’s abdomen, left hand, and right arm, as well as a hypertrophic nodule on the right elbow (Figure) and hard growths on the digits of the right hand. The patient stated the growths started 5 months ago and had been increasing in size. On further questioning, the patient admitted to “huffing” (ie, inhaling) at least six cans of pressurized dust-removal keyboard cleaning spray daily for the past 11 months.
Why do patients abuse keyboard cleaning spray?
The propellant used in certain liquefied compressed gas products is 1,1-difluoroethane (1,1-DFE), a fluorinated hydrocarbon. It is a member of a broad class of related compounds that are present in spray paints, glues, nail polish removers, fuels, hair sprays, and air-freshening products. These 1,1-DFE-containing products are abused for their rapid and short-acting central nervous system (CNS) depressant effects—not unlike that of ethanol. Typically, the vapor of a volatile hydrocarbon is inhaled directly from the open container (“sniffing”), from a bag (“bagging”), or from a soaked rag (huffing). Not only are such hydrocarbon-containing products easy to conceal, they are also highly accessible and inexpensive. Moreover, there are generally no direct legal consequences resulting from abuse of these substances.
All of the aforementioned factors make hydrocarbons a popular drug of abuse among adolescents. Approximately 75% of the population abusing hydrocarbons is younger than age 18 years, half of whom reported first use prior to age 13 years.1,2 Though inhalant abuse rarely continues into adulthood, 0.1% of individuals between the ages of 18 and 30 years report having an inhalant-use disorder.
Hydrocarbons and their halogenated derivatives are lipophilic compounds that are rapidly absorbed after inhalation and rapidly distributed to CNS and cardiac tissue. The brain concentration of 1,1-DFE likely peaks higher than concentrations in other organs and is cleared more rapidly.3 Hydrocarbons produce CNS depression secondary to multiple mechanisms, including gamma-aminobutyric acid agonism, dopamine modulation, and N-methyl-D-aspartate-receptor antagonism.4,5
What causes skin lesions on the abdomen and arms?
The lesions on the patient’s abdomen and extremities were consistent with frostbite. The liquefied compressed gas in computer-cleaning and related products is housed in a pressurized canister. The pressure is released when the spray nozzle is depressed; this causes the liquid to rapidly expand to a gas as it is released, resulting in a quick decrease in the temperature of the metal canister. This process, referred to as adiabatic cooling, demonstrates the first law of thermodynamics. The cold temperature of both the liquid and the canister can cause frostbite in the digits and other parts of the body with which the canister or liquid comes into contact.6
Why did the patient have syncope?
Halogenated hydrocarbons inhibit the cardiac delayed rectifier potassium channels involved in the repolarization of cardiac myocytes, causing a delay in repolarization that is manifested as prolongation of the QT interval on an electrocardiogram. This condition places patients at an increased risk of developing torsades de pointes (TdP).7 In most cases, TdP is self-terminating; however, if TdP persists, degeneration to ventricular fibrillation will result. Deaths caused in this fashion have been referred to as “sudden sniffing death syndrome,” and account for half of all hydrocarbon-related deaths.6,8 In addition to the cardiac effects, hydrocarbons are simple asphyxiants that act by displacing oxygen from inspired air, which also contributes to syncope.
It is important to note that epinephrine and other catecholamines increase the risk for dysrhythmias such as TdP in the setting of hydrocarbon abuse.9 For this reason, epinephrine should be used with caution in the setting of a hydrocarbon-induced arrhythmia. Beta-adrenergic antagonists such as esmolol and propranolol are preferable because they reduce the incidence of ectopia that may trigger TdP.10
What is the significance of the masses noted on the examination and radiograph?
Fluorosis is associated with abnormalities of skeletal and dental tissue. Skeletal fluorosis causes osteosclerosis of the axial skeleton, periosteal new bone formation, ligamentous and tendinous ossification, and osteophyte formation. Dental fluorosis causes a yellow/brown discoloration of the teeth with horizontal streaking (mottling), pitting, and chipping.11 Fluorosis is well-described in regions where water fluoride concentrations are high due to industrial exposure; from consumption of fluorinated wine or chronic overconsumption of tea (especially green or black tea); or from fluoridated toothpaste.12-14 More recently, fluorosis has been described in patients treated for an extended duration of time with voriconazole, a fluorinated antifungal agent.15 Unlike other hydrocarbon products, fluorinated hydrocarbons such as 1,1-DFE can significantly increase systemic fluoride concentrations with excessive use. Rapid skeletal fluorosis is not well described, but has been reported after chronic abuse of fluorinated hydrocarbons.16
How is fluorosis diagnosed and managed?
The lack of rapid laboratory testing available for serum, urine, and bone fluoride concentrations makes the initial diagnosis of fluorosis a clinical one. Imaging studies are generally highly suggestive of fluorosis and can be used to support the diagnosis. A dual energy X-ray absorptiometry scan of the spine, hip, femur, and distal portions of the radii can reveal elevated T-scores consistent with osteosclerosis.14 These findings, in conjunction with bone or joint pain, reduced range of motion, or kyphosis, should prompt clinicians to conduct further testing—even without a confirmed fluoride source. A serum fluoride (reference range, 0.2-3.2 mg/L) and 24-hour urine fluoride (reference range, 0.2-3.2 mg/dL) and creatinine evaluation can be used to diagnose fluorosis. However, a bone biopsy with quantitative bone ash fluoride analysis remains the gold standard for the diagnosis of skeletal fluorosis.16 Laboratory evaluation should also include an assessment of electrolytes, specifically calcium, 25-hydroxyvitamin D, and alkaline phosphatase. The differential diagnosis should include hemoglobinopathies, renal osteodystrophy, Paget disease, hypothyroidism, and skeletal metastases.16
Treatment of fluorosis is largely symptomatic and supportive, with identification and discontinuation of the fluoride source. Patients should be referred to an orthopedist for evaluation and management as needed. Evaluation by an endocrinologist should also be considered because patients may have chronic vitamin D and calcium deficiencies as a result of systemic fluorosis.
Case Conclusion
The patient’s laboratory assessment was notable for the following: alkaline phosphatase, 624 U/L (reference range, 44-147 IU/L); vitamin D, 10 ng/mL (reference range, 20-40 ng/mL); serum fluoride, 0.3 mg/L (reference range, 0.2-3.2 mg/L); urine fluoride, 52 mg/dL (0.2-3.2 mg/dL); and urine creatinine, 1 g/L (reference range, 0.3-3 g/L). Imaging studies noted periosteal bone formation on the lateral epicondyle of the distal right humerus, as well as similar osseous abnormalities in other locations. A bone biopsy was scheduled. The patient was treated with oral vitamin D and educated about the importance of discontinuing the huffing of all hydrocarbons.
1. Williams JF, Storck M; American Academy of Pediatrics Committee on Substance Abuse; American Academy of Pediatrics Committee on Native American Child Health. Inhalant abuse. Pediatrics. 2007;119(5):1009-1017.
2. Wu LT, Pilowsky DJ, Schlenger WE. Inhalant abuse and dependence among adolescents in the United States. J Am Acad Child Adolesc Psychiatry. 2004;43(10):1206-1214.
3. Avella J, Kunaparaju N, Kumar S, Lehrer M, Zito SW, Barletta M. Uptake and distribution of the abused inhalant 1,1-difluoroethane in the rat. J Anal Toxicol. 2010;34(7):381-388.
4. Tormoehlen LM, Tekulve KJ, Nañagas KA. Hydrocarbon toxicity: A review. Clin Toxicol (Phila). 2014;52(5):479-489.
5. Duncan JR, Lawrence AJ. Conventional concepts and new perspectives for understanding the addictive properties of inhalants. J Pharmacol Sci. 2013;122(4):237-243.
6. Sakai K, Maruyama-Maebashi K, Takatsu A, et al. Sudden death involving inhalation of 1,1-difluoroethane (HFC-152a) with spray cleaner: three case reports. Forensic Sci Int. 2011;206(1-3):e58-e61.
7. Himmel HM. Mechanisms involved in cardiac sensitization by volatile anesthetics: general applicability to halogenated hydrocarbons? Crit Rev Toxicol. 2008;38(9):773-803.
8. Avella J, Wilson JC, Lehrer M. Fatal cardiac arrhythmia after repeated exposure to 1,1-difluoroethane (DFE). Am J Forensic Med Pathol. 2006;27(1):58-60.
9. Nelson LS. Toxicologic myocardial sensitization. J Toxicol Clin Toxicol. 2002;40(7):867-879.
10. Mortiz F, de La Chapelle A, Bauer F, Leroy JP, Goullé JP, Bonmarchand G. Esmolol in the treatment of severe arrhythmia after acute trichloroethylene poisoning. Intensive Care Med. 2000;26(2):256.
11. Majumdar KK. Health impact of supplying safe drinking water containing fluoride below permissible level on flourosis patients in a fluoride-endemic rural area of West Bengal. Indian J Public Health. 2011;55(4):303-308.
12. Kakumanu N, Rao SD. Images in clinical medicine. Skeletal fluorosis due to excessive tea drinking. N Engl J Med 2013;368(12):1140.
13. Soriano M, Manchón F. Radiological aspects of a new type of bone fluorosis, periostitis deformans. Radiology 1966;87(6):1089-1094.
14. Tamer MN, Kale Köroğlu B, Arslan C, et al. Osteosclerosis due to endemic fluorosis. Sci Total Environ. 2007;373(1):43-48.
15. Bucknor MD, Gross AJ, Link TM. Voriconazole-induced periostitis in two post-transplant patients. J Radiol Case Rep. 2013;7(8):10-17.
16. Cohen E, Hsu RY, Evangelista P, Aaron R, Rubin LE. Rapid-onset diffuse skeletal fluorosis from inhalant abuse: a case report. JBJS Case Connector. 2014;4(4):e108.
1. Williams JF, Storck M; American Academy of Pediatrics Committee on Substance Abuse; American Academy of Pediatrics Committee on Native American Child Health. Inhalant abuse. Pediatrics. 2007;119(5):1009-1017.
2. Wu LT, Pilowsky DJ, Schlenger WE. Inhalant abuse and dependence among adolescents in the United States. J Am Acad Child Adolesc Psychiatry. 2004;43(10):1206-1214.
3. Avella J, Kunaparaju N, Kumar S, Lehrer M, Zito SW, Barletta M. Uptake and distribution of the abused inhalant 1,1-difluoroethane in the rat. J Anal Toxicol. 2010;34(7):381-388.
4. Tormoehlen LM, Tekulve KJ, Nañagas KA. Hydrocarbon toxicity: A review. Clin Toxicol (Phila). 2014;52(5):479-489.
5. Duncan JR, Lawrence AJ. Conventional concepts and new perspectives for understanding the addictive properties of inhalants. J Pharmacol Sci. 2013;122(4):237-243.
6. Sakai K, Maruyama-Maebashi K, Takatsu A, et al. Sudden death involving inhalation of 1,1-difluoroethane (HFC-152a) with spray cleaner: three case reports. Forensic Sci Int. 2011;206(1-3):e58-e61.
7. Himmel HM. Mechanisms involved in cardiac sensitization by volatile anesthetics: general applicability to halogenated hydrocarbons? Crit Rev Toxicol. 2008;38(9):773-803.
8. Avella J, Wilson JC, Lehrer M. Fatal cardiac arrhythmia after repeated exposure to 1,1-difluoroethane (DFE). Am J Forensic Med Pathol. 2006;27(1):58-60.
9. Nelson LS. Toxicologic myocardial sensitization. J Toxicol Clin Toxicol. 2002;40(7):867-879.
10. Mortiz F, de La Chapelle A, Bauer F, Leroy JP, Goullé JP, Bonmarchand G. Esmolol in the treatment of severe arrhythmia after acute trichloroethylene poisoning. Intensive Care Med. 2000;26(2):256.
11. Majumdar KK. Health impact of supplying safe drinking water containing fluoride below permissible level on flourosis patients in a fluoride-endemic rural area of West Bengal. Indian J Public Health. 2011;55(4):303-308.
12. Kakumanu N, Rao SD. Images in clinical medicine. Skeletal fluorosis due to excessive tea drinking. N Engl J Med 2013;368(12):1140.
13. Soriano M, Manchón F. Radiological aspects of a new type of bone fluorosis, periostitis deformans. Radiology 1966;87(6):1089-1094.
14. Tamer MN, Kale Köroğlu B, Arslan C, et al. Osteosclerosis due to endemic fluorosis. Sci Total Environ. 2007;373(1):43-48.
15. Bucknor MD, Gross AJ, Link TM. Voriconazole-induced periostitis in two post-transplant patients. J Radiol Case Rep. 2013;7(8):10-17.
16. Cohen E, Hsu RY, Evangelista P, Aaron R, Rubin LE. Rapid-onset diffuse skeletal fluorosis from inhalant abuse: a case report. JBJS Case Connector. 2014;4(4):e108.
Spontaneous Repigmentation of Silvery Hair in an Infant With Congenital Hydrops Fetalis and Hypoproteinemia
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.


Comment
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
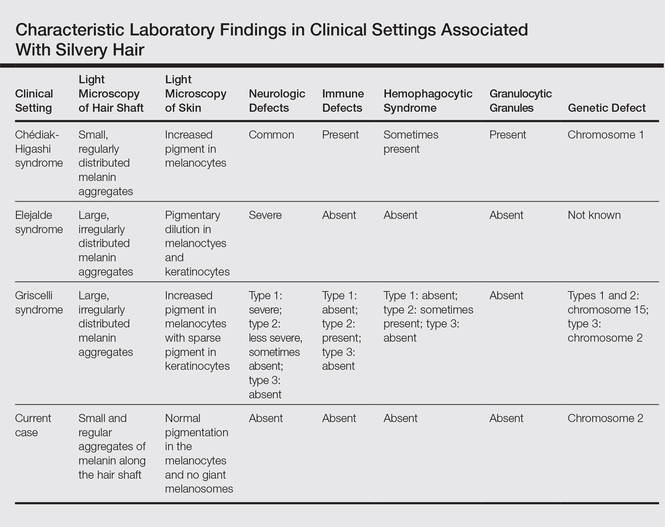
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
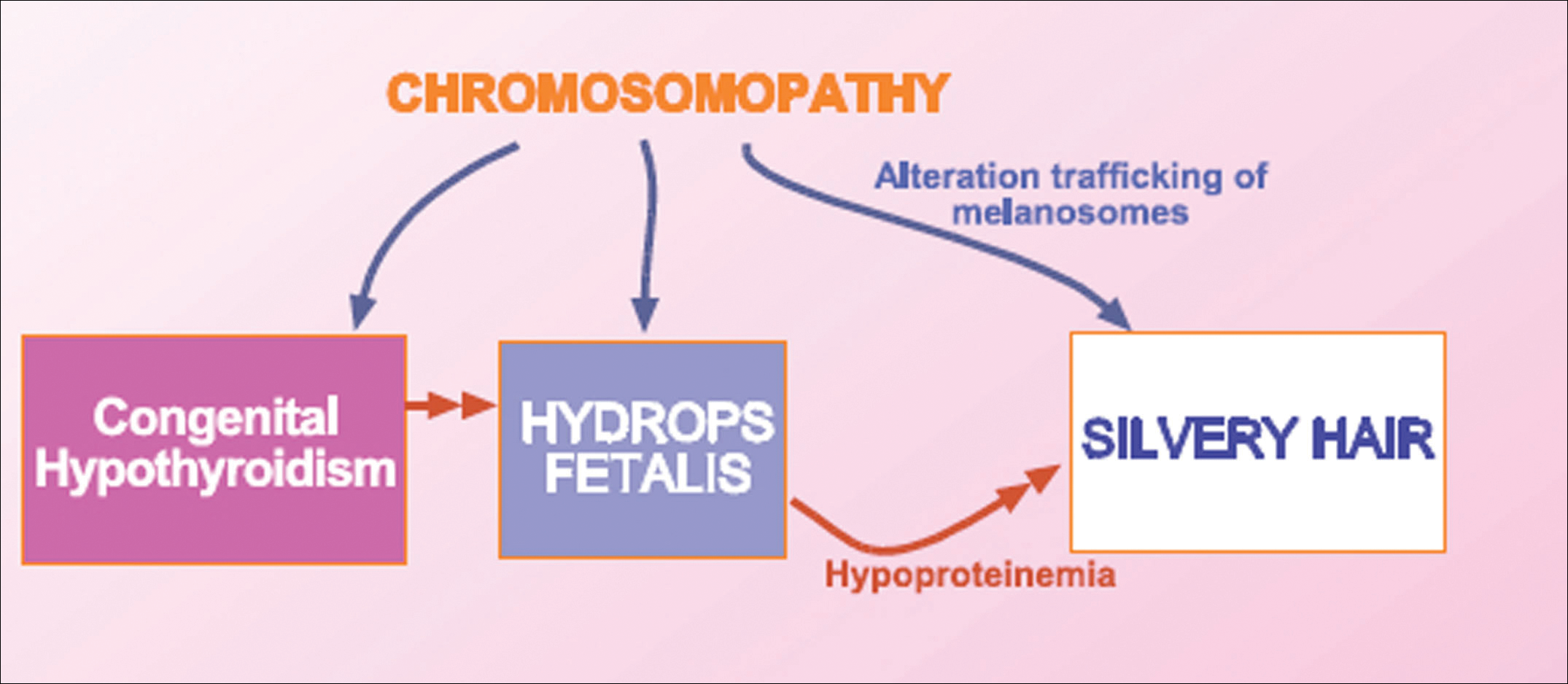
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
- White JG. The Chédiak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chédiak-Higashi syndrome. Mol Genet Metab. 1999;68:283-303.
- Kaplan J, De Domenico I, Ward DM. Chédiak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis [published online December 7, 2006]. Eur J Pediatr. 2007;166:95-109.
- Morrone K, Wang Y, Huizing M, et al. Two novel mutations identified in an African-American child with Chédiak-Higashi syndrome [published online March 24, 2010]. Case Report Med. 2010;2010:967535.
- Ivanovich J, Mallory S, Storer T, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313-316.
- Cahali JB, Fernandez SA, Oliveira ZN, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479-482.
- Bahadoran P, Ortonne JP, Ballotti R, et al. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116:408-409.
- Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al. Elejalde syndrome—a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-186.
- Happle R. Neurocutaneous diseases. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. 5th ed. New York, NY: McGraw-Hill; 1999:2131-2148.
- Sanal O, Yel L, Kucukali T, et al. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol. 2000;247:570-572.
- Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337-340.
- Al-Idrissi E, ElGhazali G, Alzahrani M, et al. Premature birth, respiratory distress, intracerebral hemorrhage, and silvery-gray hair: differential diagnosis of the 3 types of Griscelli syndrome. J Pediatr Hematol Oncol. 2010;32:494-496.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456.
- Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- Narchi H. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;104:1416-1417.
- Kessel I, Makhoul IR, Sujov P. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;103:E9.
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.
Comment
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.
Comment
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
- White JG. The Chédiak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chédiak-Higashi syndrome. Mol Genet Metab. 1999;68:283-303.
- Kaplan J, De Domenico I, Ward DM. Chédiak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis [published online December 7, 2006]. Eur J Pediatr. 2007;166:95-109.
- Morrone K, Wang Y, Huizing M, et al. Two novel mutations identified in an African-American child with Chédiak-Higashi syndrome [published online March 24, 2010]. Case Report Med. 2010;2010:967535.
- Ivanovich J, Mallory S, Storer T, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313-316.
- Cahali JB, Fernandez SA, Oliveira ZN, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479-482.
- Bahadoran P, Ortonne JP, Ballotti R, et al. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116:408-409.
- Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al. Elejalde syndrome—a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-186.
- Happle R. Neurocutaneous diseases. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. 5th ed. New York, NY: McGraw-Hill; 1999:2131-2148.
- Sanal O, Yel L, Kucukali T, et al. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol. 2000;247:570-572.
- Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337-340.
- Al-Idrissi E, ElGhazali G, Alzahrani M, et al. Premature birth, respiratory distress, intracerebral hemorrhage, and silvery-gray hair: differential diagnosis of the 3 types of Griscelli syndrome. J Pediatr Hematol Oncol. 2010;32:494-496.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456.
- Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- Narchi H. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;104:1416-1417.
- Kessel I, Makhoul IR, Sujov P. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;103:E9.
- White JG. The Chédiak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chédiak-Higashi syndrome. Mol Genet Metab. 1999;68:283-303.
- Kaplan J, De Domenico I, Ward DM. Chédiak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis [published online December 7, 2006]. Eur J Pediatr. 2007;166:95-109.
- Morrone K, Wang Y, Huizing M, et al. Two novel mutations identified in an African-American child with Chédiak-Higashi syndrome [published online March 24, 2010]. Case Report Med. 2010;2010:967535.
- Ivanovich J, Mallory S, Storer T, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313-316.
- Cahali JB, Fernandez SA, Oliveira ZN, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479-482.
- Bahadoran P, Ortonne JP, Ballotti R, et al. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116:408-409.
- Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al. Elejalde syndrome—a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-186.
- Happle R. Neurocutaneous diseases. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. 5th ed. New York, NY: McGraw-Hill; 1999:2131-2148.
- Sanal O, Yel L, Kucukali T, et al. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol. 2000;247:570-572.
- Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337-340.
- Al-Idrissi E, ElGhazali G, Alzahrani M, et al. Premature birth, respiratory distress, intracerebral hemorrhage, and silvery-gray hair: differential diagnosis of the 3 types of Griscelli syndrome. J Pediatr Hematol Oncol. 2010;32:494-496.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456.
- Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- Narchi H. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;104:1416-1417.
- Kessel I, Makhoul IR, Sujov P. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;103:E9.
Practice Points
- Silvery hair is characteristic of 3 rare autosomal-recessive disorders: Chédiak-Higashi syndrome, Elejalde syndrome, and Griscelli syndrome.
- Hypopigmentation is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes.
- Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
Silicone Arthroplasty After Ankylosis of Proximal Interphalangeal Joints in Rheumatoid Arthritis: A Case Report
Rheumatoid arthritis (RA) commonly affects the hand and fingers, most often at the metacarpophalangeal and proximal interphalangeal (PIP) joints. Synovitis, tendon ruptures, Boutonnière and swan-neck deformities, and joint destruction often occur. Bony ankylosis is not commonly described yet frequently occurs in patients with RA.1
Implant arthroplasty is an established treatment for arthritis of the hand and fingers. Indications for its use include RA, osteoarthritis, and posttraumatic arthritis. Most patients treated with implant arthroplasty can expect pain relief and 40° to 65° of PIP joint motion.2,3 Silicone arthroplasty historically has been used for pain relief but not for restoration of motion in an ankylosed joint. To our knowledge, there are no reports of using implant arthroplasty in the treatment of spontaneous ankylosis in RA. Contraindications for this procedure would include infection, irreparable flexor or extensor apparatus, and severe medical comorbidities.
In this article, we report a case of PIP joint autofusion treated with silicone PIP arthroplasty in a patient with RA. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 56-year-old woman who had had RA for more than 20 years underwent left carpometacarpal arthroplasty and thumb reconstruction. She subsequently presented with complaints of progressively worsening functioning of the left ring and small fingers. On initial evaluation, her PIP joints were fused in about 15° of flexion. Radiographs (Figures 1A, 1B) showed severe diffuse arthritis of the hands and complete bony ankylosis of the ring- and small-finger PIP joints with radial deviation of the ring-finger middle phalanx. The patient had minimal pain but wanted improved hand motion and opted for takedown of the ankylosis with silicone PIP joint arthroplasty.
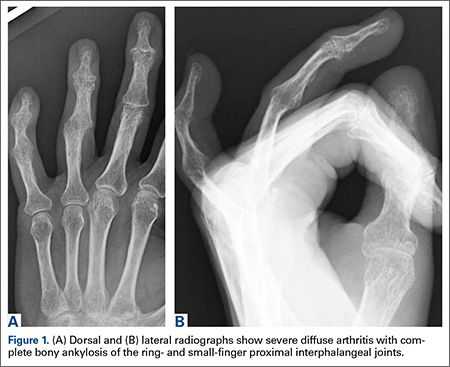
Radial dorsal incisions were made over the PIP joints of the ring and small fingers. As is not the case with arthroplasty for routine PIP joint arthritis, presence of bony ankylosis made identification of the native PIP joint more difficult. The transverse retinacular ligament was identified and opened, and the collateral ligament, which was not ankylosed, was dissected off the proximal phalanx. These landmarks were useful in locating the PIP joint, and proper positioning was confirmed with fluoroscopy. The ankylosed joint space was opened with an osteotome, and about 8 to 10 mm of bone was resected to create space for the instrumentation. As the amount of scarring within the flexor tendon sheath was not significant, restoration of motion did not require extensive tenolysis. The extensor mechanism was slightly contracted, but the bony resection allowed flexion to be restored. The distal portion of the proximal phalanx was then resected. The proximal and middle phalanges were reamed, and a silicone prosthesis was placed with the finger held straight. The collateral ligament was repaired back to the proximal phalanx with 4-0 polydioxanone sutures placed through a bone tunnel created with a Kirschner wire. The skin was closed with 4-0 nylon, and a postoperative splint was applied.
The initial postoperative course was unremarkable. The patient was immobilized in 10° of PIP joint flexion for 10 days, and therapy was initiated after the splint was removed. Twenty-four months after surgery, the patient was pain-free and had 60° of active PIP joint flexion, with extensor lag of only 10°. Clinically, alignment of the fingers was satisfactory; there was mild persistent radial deviation of 10° to 15° (Figures 2A, 2B). Radiographs showed good positioning of the implants (Figures 3A, 3B) and no sign of coronal instability. The patient was satisfied with her improved functioning and returned to employment as a hospital clerk, working full-time.
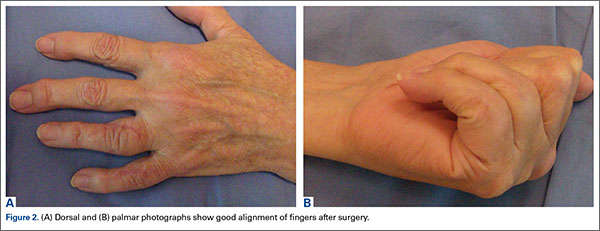
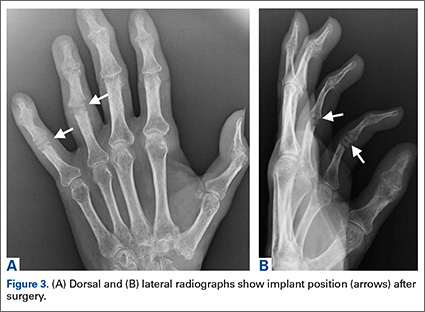
Discussion
RA of the hand and fingers can be painful and disabling. Although there are several treatment options for many of the most common manifestations, options are limited for bony ankylosis of the finger joints. The patient described in this case report had minimal pain, but the loss of motion of the PIP joints in her ring and small fingers created difficulties for her at work. She wanted surgery that would improve the functioning of her fingers. PIP joint arthroplasty traditionally has been the treatment of choice for PIP joint arthritis. In 1985, Swanson and colleagues2 reported on more than 400 silicone PIP arthroplasties performed over 16 years. Mean range of motion (ROM) was between 45° and 60°, with 70% of patients having ROM of more than 40°. Pain relief was complete in 98% of cases. Complications included implant fracture (5%) and recurrent or new deformities (6.5%). A 10.9% revision rate was noted at minimum 1-year follow-up. Recent implants made of improved biomaterials hold promise, but longer term follow-up is still needed.
Silicone arthroplasty has also been used as an effective treatment for non-RA of the PIP joint. Bales and colleagues4 reviewed long-term results of silicone arthroplasty for PIP joint osteoarthritis in 22 patients. At a mean of 10 years, mean QuickDASH (Disabilities of the Arm, Shoulder, and Hand) score was 17, mean visual analog scale score for pain was 0.4, and implant survivorship was 90%. Despite unchanged ROM and considerable implant deformation or fracture, patients’ pain relief and satisfaction were consistent.
Hage and colleagues5 reviewed long-term results of silicone PIP arthroplasty for posttraumatic arthritis in 14 patients. Most of the patients were satisfied: Although they had notable rotational deformity, alignment deviation, and loss of pinch strength and ROM, they were pain-free. The authors concluded that silicone arthroplasty should be used for posttraumatic arthrosis cases in which associated adhesions may be corrected with simple tenolysis, and even in these cases the objective results may not be as good as the subjective outcome.
Kaye6 used radiographs to determine the incidence of bony ankylosis in 203 patients with RA. Hand and wrist radiographs of 48 (23.6%) of these patients showed ankylosis, and 34 of the 48 patients had 2 or more joints fused. On a questionnaire, patients with ankylosis indicated more difficulty with activities of daily living and more limited activity. The authors concluded that radiographic bony ankylosis was a relatively common feature of RA and a marker of disease that was clinically, radiographically, and functionally more severe.
The patient described in this case report had a satisfactory result after PIP joint arthroplasty. At 2-year follow-up, she remained pain-free, and her previously ankylosed PIP joint had an arc of motion of 10° to 60°. Most patients with bony ankylosis of PIP joints present with minimal pain and do not seek surgical treatment. However, patients with ankylosis that limits functioning or activities of daily living may wish to pursue intervention that could be restorative. PIP joint arthroplasty may be effective in improving motion in patients with bony ankylosis of the finger joints.
1. Kaye JJ, Callahan LF, Nance EP Jr, Brooks R, Pincus T. Bony ankylosis in rheumatoid arthritis. Associations with longer duration and greater severity of disease. Invest Radiol. 1987;22(4):303-309.
2. Swanson AB, Maupin BK, Gajjar NV, Swanson GD. Flexible implant arthroplasty in the proximal interphalangeal joint of the hand. J Hand Surg Am. 1985;10(6 pt 1):796-805.
3. Rizzo M, Beckenbaugh RD. Proximal interphalangeal joint arthroplasty. J Am Acad Orthop Surg. 2007;15(3):189-197.
4. Bales J, Wall L, Stern PJ. Long-term results of Swanson silicone arthroplasty for proximal interphalangeal joint osteoarthritis. J Hand Surg Am. 2014;39(3):455-461.
5. Hage J, Yoe E, Zering J, de Groot P. Proximal interphalangeal joint silicone arthroplasty for posttraumatic arthritis. J Hand Surg Am. 1999;24(1):73-77.
6. Kaye JJ. Radiographic assessment of rheumatoid arthritis. Rheum Dis Clin North Am. 1995;21(2):395-406.
Rheumatoid arthritis (RA) commonly affects the hand and fingers, most often at the metacarpophalangeal and proximal interphalangeal (PIP) joints. Synovitis, tendon ruptures, Boutonnière and swan-neck deformities, and joint destruction often occur. Bony ankylosis is not commonly described yet frequently occurs in patients with RA.1
Implant arthroplasty is an established treatment for arthritis of the hand and fingers. Indications for its use include RA, osteoarthritis, and posttraumatic arthritis. Most patients treated with implant arthroplasty can expect pain relief and 40° to 65° of PIP joint motion.2,3 Silicone arthroplasty historically has been used for pain relief but not for restoration of motion in an ankylosed joint. To our knowledge, there are no reports of using implant arthroplasty in the treatment of spontaneous ankylosis in RA. Contraindications for this procedure would include infection, irreparable flexor or extensor apparatus, and severe medical comorbidities.
In this article, we report a case of PIP joint autofusion treated with silicone PIP arthroplasty in a patient with RA. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 56-year-old woman who had had RA for more than 20 years underwent left carpometacarpal arthroplasty and thumb reconstruction. She subsequently presented with complaints of progressively worsening functioning of the left ring and small fingers. On initial evaluation, her PIP joints were fused in about 15° of flexion. Radiographs (Figures 1A, 1B) showed severe diffuse arthritis of the hands and complete bony ankylosis of the ring- and small-finger PIP joints with radial deviation of the ring-finger middle phalanx. The patient had minimal pain but wanted improved hand motion and opted for takedown of the ankylosis with silicone PIP joint arthroplasty.
Radial dorsal incisions were made over the PIP joints of the ring and small fingers. As is not the case with arthroplasty for routine PIP joint arthritis, presence of bony ankylosis made identification of the native PIP joint more difficult. The transverse retinacular ligament was identified and opened, and the collateral ligament, which was not ankylosed, was dissected off the proximal phalanx. These landmarks were useful in locating the PIP joint, and proper positioning was confirmed with fluoroscopy. The ankylosed joint space was opened with an osteotome, and about 8 to 10 mm of bone was resected to create space for the instrumentation. As the amount of scarring within the flexor tendon sheath was not significant, restoration of motion did not require extensive tenolysis. The extensor mechanism was slightly contracted, but the bony resection allowed flexion to be restored. The distal portion of the proximal phalanx was then resected. The proximal and middle phalanges were reamed, and a silicone prosthesis was placed with the finger held straight. The collateral ligament was repaired back to the proximal phalanx with 4-0 polydioxanone sutures placed through a bone tunnel created with a Kirschner wire. The skin was closed with 4-0 nylon, and a postoperative splint was applied.
The initial postoperative course was unremarkable. The patient was immobilized in 10° of PIP joint flexion for 10 days, and therapy was initiated after the splint was removed. Twenty-four months after surgery, the patient was pain-free and had 60° of active PIP joint flexion, with extensor lag of only 10°. Clinically, alignment of the fingers was satisfactory; there was mild persistent radial deviation of 10° to 15° (Figures 2A, 2B). Radiographs showed good positioning of the implants (Figures 3A, 3B) and no sign of coronal instability. The patient was satisfied with her improved functioning and returned to employment as a hospital clerk, working full-time.
Discussion
RA of the hand and fingers can be painful and disabling. Although there are several treatment options for many of the most common manifestations, options are limited for bony ankylosis of the finger joints. The patient described in this case report had minimal pain, but the loss of motion of the PIP joints in her ring and small fingers created difficulties for her at work. She wanted surgery that would improve the functioning of her fingers. PIP joint arthroplasty traditionally has been the treatment of choice for PIP joint arthritis. In 1985, Swanson and colleagues2 reported on more than 400 silicone PIP arthroplasties performed over 16 years. Mean range of motion (ROM) was between 45° and 60°, with 70% of patients having ROM of more than 40°. Pain relief was complete in 98% of cases. Complications included implant fracture (5%) and recurrent or new deformities (6.5%). A 10.9% revision rate was noted at minimum 1-year follow-up. Recent implants made of improved biomaterials hold promise, but longer term follow-up is still needed.
Silicone arthroplasty has also been used as an effective treatment for non-RA of the PIP joint. Bales and colleagues4 reviewed long-term results of silicone arthroplasty for PIP joint osteoarthritis in 22 patients. At a mean of 10 years, mean QuickDASH (Disabilities of the Arm, Shoulder, and Hand) score was 17, mean visual analog scale score for pain was 0.4, and implant survivorship was 90%. Despite unchanged ROM and considerable implant deformation or fracture, patients’ pain relief and satisfaction were consistent.
Hage and colleagues5 reviewed long-term results of silicone PIP arthroplasty for posttraumatic arthritis in 14 patients. Most of the patients were satisfied: Although they had notable rotational deformity, alignment deviation, and loss of pinch strength and ROM, they were pain-free. The authors concluded that silicone arthroplasty should be used for posttraumatic arthrosis cases in which associated adhesions may be corrected with simple tenolysis, and even in these cases the objective results may not be as good as the subjective outcome.
Kaye6 used radiographs to determine the incidence of bony ankylosis in 203 patients with RA. Hand and wrist radiographs of 48 (23.6%) of these patients showed ankylosis, and 34 of the 48 patients had 2 or more joints fused. On a questionnaire, patients with ankylosis indicated more difficulty with activities of daily living and more limited activity. The authors concluded that radiographic bony ankylosis was a relatively common feature of RA and a marker of disease that was clinically, radiographically, and functionally more severe.
The patient described in this case report had a satisfactory result after PIP joint arthroplasty. At 2-year follow-up, she remained pain-free, and her previously ankylosed PIP joint had an arc of motion of 10° to 60°. Most patients with bony ankylosis of PIP joints present with minimal pain and do not seek surgical treatment. However, patients with ankylosis that limits functioning or activities of daily living may wish to pursue intervention that could be restorative. PIP joint arthroplasty may be effective in improving motion in patients with bony ankylosis of the finger joints.
Rheumatoid arthritis (RA) commonly affects the hand and fingers, most often at the metacarpophalangeal and proximal interphalangeal (PIP) joints. Synovitis, tendon ruptures, Boutonnière and swan-neck deformities, and joint destruction often occur. Bony ankylosis is not commonly described yet frequently occurs in patients with RA.1
Implant arthroplasty is an established treatment for arthritis of the hand and fingers. Indications for its use include RA, osteoarthritis, and posttraumatic arthritis. Most patients treated with implant arthroplasty can expect pain relief and 40° to 65° of PIP joint motion.2,3 Silicone arthroplasty historically has been used for pain relief but not for restoration of motion in an ankylosed joint. To our knowledge, there are no reports of using implant arthroplasty in the treatment of spontaneous ankylosis in RA. Contraindications for this procedure would include infection, irreparable flexor or extensor apparatus, and severe medical comorbidities.
In this article, we report a case of PIP joint autofusion treated with silicone PIP arthroplasty in a patient with RA. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 56-year-old woman who had had RA for more than 20 years underwent left carpometacarpal arthroplasty and thumb reconstruction. She subsequently presented with complaints of progressively worsening functioning of the left ring and small fingers. On initial evaluation, her PIP joints were fused in about 15° of flexion. Radiographs (Figures 1A, 1B) showed severe diffuse arthritis of the hands and complete bony ankylosis of the ring- and small-finger PIP joints with radial deviation of the ring-finger middle phalanx. The patient had minimal pain but wanted improved hand motion and opted for takedown of the ankylosis with silicone PIP joint arthroplasty.
Radial dorsal incisions were made over the PIP joints of the ring and small fingers. As is not the case with arthroplasty for routine PIP joint arthritis, presence of bony ankylosis made identification of the native PIP joint more difficult. The transverse retinacular ligament was identified and opened, and the collateral ligament, which was not ankylosed, was dissected off the proximal phalanx. These landmarks were useful in locating the PIP joint, and proper positioning was confirmed with fluoroscopy. The ankylosed joint space was opened with an osteotome, and about 8 to 10 mm of bone was resected to create space for the instrumentation. As the amount of scarring within the flexor tendon sheath was not significant, restoration of motion did not require extensive tenolysis. The extensor mechanism was slightly contracted, but the bony resection allowed flexion to be restored. The distal portion of the proximal phalanx was then resected. The proximal and middle phalanges were reamed, and a silicone prosthesis was placed with the finger held straight. The collateral ligament was repaired back to the proximal phalanx with 4-0 polydioxanone sutures placed through a bone tunnel created with a Kirschner wire. The skin was closed with 4-0 nylon, and a postoperative splint was applied.
The initial postoperative course was unremarkable. The patient was immobilized in 10° of PIP joint flexion for 10 days, and therapy was initiated after the splint was removed. Twenty-four months after surgery, the patient was pain-free and had 60° of active PIP joint flexion, with extensor lag of only 10°. Clinically, alignment of the fingers was satisfactory; there was mild persistent radial deviation of 10° to 15° (Figures 2A, 2B). Radiographs showed good positioning of the implants (Figures 3A, 3B) and no sign of coronal instability. The patient was satisfied with her improved functioning and returned to employment as a hospital clerk, working full-time.
Discussion
RA of the hand and fingers can be painful and disabling. Although there are several treatment options for many of the most common manifestations, options are limited for bony ankylosis of the finger joints. The patient described in this case report had minimal pain, but the loss of motion of the PIP joints in her ring and small fingers created difficulties for her at work. She wanted surgery that would improve the functioning of her fingers. PIP joint arthroplasty traditionally has been the treatment of choice for PIP joint arthritis. In 1985, Swanson and colleagues2 reported on more than 400 silicone PIP arthroplasties performed over 16 years. Mean range of motion (ROM) was between 45° and 60°, with 70% of patients having ROM of more than 40°. Pain relief was complete in 98% of cases. Complications included implant fracture (5%) and recurrent or new deformities (6.5%). A 10.9% revision rate was noted at minimum 1-year follow-up. Recent implants made of improved biomaterials hold promise, but longer term follow-up is still needed.
Silicone arthroplasty has also been used as an effective treatment for non-RA of the PIP joint. Bales and colleagues4 reviewed long-term results of silicone arthroplasty for PIP joint osteoarthritis in 22 patients. At a mean of 10 years, mean QuickDASH (Disabilities of the Arm, Shoulder, and Hand) score was 17, mean visual analog scale score for pain was 0.4, and implant survivorship was 90%. Despite unchanged ROM and considerable implant deformation or fracture, patients’ pain relief and satisfaction were consistent.
Hage and colleagues5 reviewed long-term results of silicone PIP arthroplasty for posttraumatic arthritis in 14 patients. Most of the patients were satisfied: Although they had notable rotational deformity, alignment deviation, and loss of pinch strength and ROM, they were pain-free. The authors concluded that silicone arthroplasty should be used for posttraumatic arthrosis cases in which associated adhesions may be corrected with simple tenolysis, and even in these cases the objective results may not be as good as the subjective outcome.
Kaye6 used radiographs to determine the incidence of bony ankylosis in 203 patients with RA. Hand and wrist radiographs of 48 (23.6%) of these patients showed ankylosis, and 34 of the 48 patients had 2 or more joints fused. On a questionnaire, patients with ankylosis indicated more difficulty with activities of daily living and more limited activity. The authors concluded that radiographic bony ankylosis was a relatively common feature of RA and a marker of disease that was clinically, radiographically, and functionally more severe.
The patient described in this case report had a satisfactory result after PIP joint arthroplasty. At 2-year follow-up, she remained pain-free, and her previously ankylosed PIP joint had an arc of motion of 10° to 60°. Most patients with bony ankylosis of PIP joints present with minimal pain and do not seek surgical treatment. However, patients with ankylosis that limits functioning or activities of daily living may wish to pursue intervention that could be restorative. PIP joint arthroplasty may be effective in improving motion in patients with bony ankylosis of the finger joints.
1. Kaye JJ, Callahan LF, Nance EP Jr, Brooks R, Pincus T. Bony ankylosis in rheumatoid arthritis. Associations with longer duration and greater severity of disease. Invest Radiol. 1987;22(4):303-309.
2. Swanson AB, Maupin BK, Gajjar NV, Swanson GD. Flexible implant arthroplasty in the proximal interphalangeal joint of the hand. J Hand Surg Am. 1985;10(6 pt 1):796-805.
3. Rizzo M, Beckenbaugh RD. Proximal interphalangeal joint arthroplasty. J Am Acad Orthop Surg. 2007;15(3):189-197.
4. Bales J, Wall L, Stern PJ. Long-term results of Swanson silicone arthroplasty for proximal interphalangeal joint osteoarthritis. J Hand Surg Am. 2014;39(3):455-461.
5. Hage J, Yoe E, Zering J, de Groot P. Proximal interphalangeal joint silicone arthroplasty for posttraumatic arthritis. J Hand Surg Am. 1999;24(1):73-77.
6. Kaye JJ. Radiographic assessment of rheumatoid arthritis. Rheum Dis Clin North Am. 1995;21(2):395-406.
1. Kaye JJ, Callahan LF, Nance EP Jr, Brooks R, Pincus T. Bony ankylosis in rheumatoid arthritis. Associations with longer duration and greater severity of disease. Invest Radiol. 1987;22(4):303-309.
2. Swanson AB, Maupin BK, Gajjar NV, Swanson GD. Flexible implant arthroplasty in the proximal interphalangeal joint of the hand. J Hand Surg Am. 1985;10(6 pt 1):796-805.
3. Rizzo M, Beckenbaugh RD. Proximal interphalangeal joint arthroplasty. J Am Acad Orthop Surg. 2007;15(3):189-197.
4. Bales J, Wall L, Stern PJ. Long-term results of Swanson silicone arthroplasty for proximal interphalangeal joint osteoarthritis. J Hand Surg Am. 2014;39(3):455-461.
5. Hage J, Yoe E, Zering J, de Groot P. Proximal interphalangeal joint silicone arthroplasty for posttraumatic arthritis. J Hand Surg Am. 1999;24(1):73-77.
6. Kaye JJ. Radiographic assessment of rheumatoid arthritis. Rheum Dis Clin North Am. 1995;21(2):395-406.
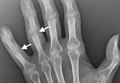
Darkened skin, vomiting, and salt cravings in a teenager • Dx?
THE CASE
A 17-year-old boy presented to the emergency department (ED) with a headache, dizziness, lethargy, and weakness that he’d had for 2 weeks. The patient was taking a selective serotonin reuptake inhibitor (SSRI) for depression (sertraline 25 mg/d). He had been vomiting twice daily for the past 3 years. (Although he had been seen multiple times in urgent care clinics, he did not have regular medical care.) The boy was fatigued and had dark yellow urine. His father indicated that his son’s skin had darkened over the last 5 to 6 years and that he had been adding salt, in large quantities, to nearly all of his meals for 10 years.
The boy’s health issues were impacting his school life. He was dismissed from school often because his teachers felt he was skipping class and using the excuse of needing to urinate or vomit. He had traveled back and forth to Mexico about 2 times a year, with the last time being about 3 months before his trip to the ED.
The patient’s vitals included a temperature of 96.3º F, heart rate (HR) of 77 beats/min, respiratory rate of 16 breaths/min, and a supine blood pressure (BP) of 102/58 mm Hg. (The patient’s BP was not obtained when sitting or standing, because he felt dizzy when trying to stand or sit up and the HR monitor increased to 100 beats/min.) His weight was 106.9 pounds and height was 5 feet 8 inches. The teen was ill-appearing and somnolent. No jugular vein distention, murmurs, or gallops were noted on exam. The patient’s lips were dry and cracked, gums were darkened, and his skin was clammy to the touch. His abdomen was soft with hypoactive bowel sounds and no ascites. His extremities were non-edematous.
A chemistry panel showed a low sodium level of 99 mEq/L, a somewhat high potassium level of 5.2 mmol/L, low chloride (69 mEq/L) and CO2 (5 mEq/L) levels, a high glucose level (124 mg/dL), and normal creatinine (0.79 mg/dL), albumin (5.2 g/dL), and thyroid stimulating hormone (2.4 mIU/L) levels. A tuberculosis (TB) test, acute hepatitis panel, human immunodeficiency test, and urine drug screen were all negative. Liver enzymes and lipase levels were normal.
The patient was admitted to the pediatric intensive care unit (PICU) on 200 mL/hr normal saline (twice the normal maintenance rate) and we took over his care.
THE DIAGNOSIS
Because of the patient’s severe hyponatremia, the differential diagnosis included heart failure, cirrhosis, syndrome of inappropriate antidiuretic hormone secretion (SIADH), SSRI-induced SIADH, cerebral salt wasting, severe hypothyroidism, adrenal insufficiency, malignancies, ecstasy use, renal failure, low dietary solute intake, and psychogenic polydipsia.
A random cortisol test taken in the ED returned and was noted to be very low (<1 mcg/dL). This information, plus the signs of aldosterone deficiency (low sodium and elevated potassium levels) and adrenocorticotropic hormone (ACTH) excess (skin darkening), prompted us to perform a 250-mcg ACTH stimulation test. Results at 30 and 60 minutes both showed cortisol at <1 mcg/dL, which led us to suspect adrenal insufficiency. The diagnosis of autoimmune adrenalitis, or Addison’s disease, was confirmed after inpatient lab work returned with positive 21-hydroxylase antibodies and an elevated ACTH (1117 pg/mL; normal, 10-65 pg/mL).
We noted that the patient’s sodium level was gradually increasing while he was receiving the intravenous (IV) fluids. We were concerned, though, that too rapid a sodium correction would put the patient at risk for central pontine myelinolysis (CPM). So we held off on steroids until 24 hours after he was admitted to the PICU, when his sodium level reached 110 mEq/L.
DISCUSSION
Primary adrenal insufficiency in the developed world is commonly caused by autoimmune adrenalitis, also known as Addison’s disease. Addison’s disease is the cause of primary adrenal insufficiency in 70% to 90% of cases, with the remainder caused by TB, adrenal hemorrhage, infarction, lymphoma, cytomegalovirus, adrenoleukodystrophy, or metastatic cancer. We also considered adrenoleukodystrophy in our patient, but felt it unlikely in a 17-year-old with normal mental status and positive adrenal antibodies.
The first evidence of Addison’s disease is usually an increase in plasma renin activity with low serum aldosterone. This might explain our patient’s years of salt cravings prior to presentation. There is typically a decrease in serum cortisol response to ACTH stimulation several months to years after the onset of salt cravings. The next sign of deterioration in adrenal function is an increase in basal serum ACTH; the process concludes with a decreased basal serum cortisol level.1-3 By the time our patient presented to the ED, his ACTH was very high, his cortisol was low, and his ACTH stimulation response was low.
Acute adrenal insufficiency crisis usually occurs after a prolonged period of nonspecific complaints due to a loss of both glucocorticoids and mineralocorticoids; by the time overt symptoms occur, 90% of the adrenal gland may be destroyed.3 Patients (such as ours) may present with symptoms such as abdominal pain, weakness, vomiting, fever, and decreased responsiveness. Hyponatremia and hyperkalemia are commonly seen at initial diagnosis. BP can be compromised in some patients due to loss of vascular tone; our patient did not present with this finding.
Treatment includes hydrocortisone and fludrocortisone for life
Initial management focuses on rehydration, maintenance of BP, cardiac monitoring, and electrolyte monitoring with a focus on slow normalization of electrolyte abnormalities. Patients should be treated with hydrocortisone (approximately 10 mg/m2/d) and fludrocortisone (usually 0.1 mg/d), and they will be maintained on this regimen for life.1,3
During acute illness, the doses of hydrocortisone are usually tripled and given 3 times per day to address the increased cortisol needs of the stress response. Lack of stress dose steroids in the setting of illness can lead to repeat adrenal crisis events.
Patients should be taught about intramuscular (IM) hydrocortisone use (100 mg IM) for emergencies and should have medical identification. In many states, emergency medical technicians (EMTs) are now able to administer the patient’s own supply of hydrocortisone. EMTs have even begun carrying hydrocortisone in some states in response to a campaign by the CARES Foundation, a nonprofit organization dedicated to helping families and individuals affected by congenital adrenal hyperplasia.
We started our patient on 100 mg/m2/d hydrocortisone 24 hours after he was admitted to the PICU. (At that time, his sodium level was 110 mEq/L.) Forty-eight hours after admission, we started the patient on fludrocortisone for mineralocorticoid effect at 0.1 mg/d. (The patient’s sodium level was 122 mEq/L). At 72 hours after admission, the patient’s sodium level was 137 mEq/L and his mental status was normal. Normal saline was discontinued when sodium normalized. He was discharged 2 days later. He was informed he should continue these medications for life, though doses might be adjusted slightly with time.
Two weeks later, our patient’s sodium level had reached 141 mEq/L and his weight loss, depression, vomiting, and fatigue had resolved. He stopped taking his SSRI. He was still craving extra salt, but not as much, and his urine was no longer a very dark yellow.
In retrospect, starting this patient on steroids earlier may not have resulted in any more of a rapid sodium rise than that which occurred otherwise, but we believe that our concern for CPM at that time justified the delay in steroid use. We felt it was safe to delay steroids because the patient’s BP was stable and his clinical picture was rapidly improving. In most cases, however, delaying steroids is not advisable.
THE TAKEAWAY
Adrenal insufficiency can be clearly diagnosed via labs and clinical presentation, and is potentially lethal if unrecognized. The predominant manifestations of adrenal crisis are hypotension and shock, usually with hyponatremia and hyperkalemia. During stressful events or illness, patients should increase their glucocorticoid dose. If they are on hydrocortisone, instructions are usually to triple the dose, and give the medication 3 times a day. Patients require instruction beforehand on how and when to increase doses for illness so that they can handle this on their own. Patients should carry a medical identification card so that their condition is evident to anyone caring for them in the ED.
1. Husebye ES, Allolio B, Arlt W, et al. Consensus statement on the diagnosis, treatment and follow-up of patients with primary adrenal insufficiency. J Intern Med. 2014;275:104-115.
2. Betterle C, Morlin L. Autoimmune Addison’s disease. Endocr Dev. 2011;20:161-172.
3. Brandão Neto RA, de Carvalho JF. Diagnosis and classification of Addison’s disease (autoimmune adrenalitis). Autoimmun Rev. 2014;13:408-411.
THE CASE
A 17-year-old boy presented to the emergency department (ED) with a headache, dizziness, lethargy, and weakness that he’d had for 2 weeks. The patient was taking a selective serotonin reuptake inhibitor (SSRI) for depression (sertraline 25 mg/d). He had been vomiting twice daily for the past 3 years. (Although he had been seen multiple times in urgent care clinics, he did not have regular medical care.) The boy was fatigued and had dark yellow urine. His father indicated that his son’s skin had darkened over the last 5 to 6 years and that he had been adding salt, in large quantities, to nearly all of his meals for 10 years.
The boy’s health issues were impacting his school life. He was dismissed from school often because his teachers felt he was skipping class and using the excuse of needing to urinate or vomit. He had traveled back and forth to Mexico about 2 times a year, with the last time being about 3 months before his trip to the ED.
The patient’s vitals included a temperature of 96.3º F, heart rate (HR) of 77 beats/min, respiratory rate of 16 breaths/min, and a supine blood pressure (BP) of 102/58 mm Hg. (The patient’s BP was not obtained when sitting or standing, because he felt dizzy when trying to stand or sit up and the HR monitor increased to 100 beats/min.) His weight was 106.9 pounds and height was 5 feet 8 inches. The teen was ill-appearing and somnolent. No jugular vein distention, murmurs, or gallops were noted on exam. The patient’s lips were dry and cracked, gums were darkened, and his skin was clammy to the touch. His abdomen was soft with hypoactive bowel sounds and no ascites. His extremities were non-edematous.
A chemistry panel showed a low sodium level of 99 mEq/L, a somewhat high potassium level of 5.2 mmol/L, low chloride (69 mEq/L) and CO2 (5 mEq/L) levels, a high glucose level (124 mg/dL), and normal creatinine (0.79 mg/dL), albumin (5.2 g/dL), and thyroid stimulating hormone (2.4 mIU/L) levels. A tuberculosis (TB) test, acute hepatitis panel, human immunodeficiency test, and urine drug screen were all negative. Liver enzymes and lipase levels were normal.
The patient was admitted to the pediatric intensive care unit (PICU) on 200 mL/hr normal saline (twice the normal maintenance rate) and we took over his care.
THE DIAGNOSIS
Because of the patient’s severe hyponatremia, the differential diagnosis included heart failure, cirrhosis, syndrome of inappropriate antidiuretic hormone secretion (SIADH), SSRI-induced SIADH, cerebral salt wasting, severe hypothyroidism, adrenal insufficiency, malignancies, ecstasy use, renal failure, low dietary solute intake, and psychogenic polydipsia.
A random cortisol test taken in the ED returned and was noted to be very low (<1 mcg/dL). This information, plus the signs of aldosterone deficiency (low sodium and elevated potassium levels) and adrenocorticotropic hormone (ACTH) excess (skin darkening), prompted us to perform a 250-mcg ACTH stimulation test. Results at 30 and 60 minutes both showed cortisol at <1 mcg/dL, which led us to suspect adrenal insufficiency. The diagnosis of autoimmune adrenalitis, or Addison’s disease, was confirmed after inpatient lab work returned with positive 21-hydroxylase antibodies and an elevated ACTH (1117 pg/mL; normal, 10-65 pg/mL).
We noted that the patient’s sodium level was gradually increasing while he was receiving the intravenous (IV) fluids. We were concerned, though, that too rapid a sodium correction would put the patient at risk for central pontine myelinolysis (CPM). So we held off on steroids until 24 hours after he was admitted to the PICU, when his sodium level reached 110 mEq/L.
DISCUSSION
Primary adrenal insufficiency in the developed world is commonly caused by autoimmune adrenalitis, also known as Addison’s disease. Addison’s disease is the cause of primary adrenal insufficiency in 70% to 90% of cases, with the remainder caused by TB, adrenal hemorrhage, infarction, lymphoma, cytomegalovirus, adrenoleukodystrophy, or metastatic cancer. We also considered adrenoleukodystrophy in our patient, but felt it unlikely in a 17-year-old with normal mental status and positive adrenal antibodies.
The first evidence of Addison’s disease is usually an increase in plasma renin activity with low serum aldosterone. This might explain our patient’s years of salt cravings prior to presentation. There is typically a decrease in serum cortisol response to ACTH stimulation several months to years after the onset of salt cravings. The next sign of deterioration in adrenal function is an increase in basal serum ACTH; the process concludes with a decreased basal serum cortisol level.1-3 By the time our patient presented to the ED, his ACTH was very high, his cortisol was low, and his ACTH stimulation response was low.
Acute adrenal insufficiency crisis usually occurs after a prolonged period of nonspecific complaints due to a loss of both glucocorticoids and mineralocorticoids; by the time overt symptoms occur, 90% of the adrenal gland may be destroyed.3 Patients (such as ours) may present with symptoms such as abdominal pain, weakness, vomiting, fever, and decreased responsiveness. Hyponatremia and hyperkalemia are commonly seen at initial diagnosis. BP can be compromised in some patients due to loss of vascular tone; our patient did not present with this finding.
Treatment includes hydrocortisone and fludrocortisone for life
Initial management focuses on rehydration, maintenance of BP, cardiac monitoring, and electrolyte monitoring with a focus on slow normalization of electrolyte abnormalities. Patients should be treated with hydrocortisone (approximately 10 mg/m2/d) and fludrocortisone (usually 0.1 mg/d), and they will be maintained on this regimen for life.1,3
During acute illness, the doses of hydrocortisone are usually tripled and given 3 times per day to address the increased cortisol needs of the stress response. Lack of stress dose steroids in the setting of illness can lead to repeat adrenal crisis events.
Patients should be taught about intramuscular (IM) hydrocortisone use (100 mg IM) for emergencies and should have medical identification. In many states, emergency medical technicians (EMTs) are now able to administer the patient’s own supply of hydrocortisone. EMTs have even begun carrying hydrocortisone in some states in response to a campaign by the CARES Foundation, a nonprofit organization dedicated to helping families and individuals affected by congenital adrenal hyperplasia.
We started our patient on 100 mg/m2/d hydrocortisone 24 hours after he was admitted to the PICU. (At that time, his sodium level was 110 mEq/L.) Forty-eight hours after admission, we started the patient on fludrocortisone for mineralocorticoid effect at 0.1 mg/d. (The patient’s sodium level was 122 mEq/L). At 72 hours after admission, the patient’s sodium level was 137 mEq/L and his mental status was normal. Normal saline was discontinued when sodium normalized. He was discharged 2 days later. He was informed he should continue these medications for life, though doses might be adjusted slightly with time.
Two weeks later, our patient’s sodium level had reached 141 mEq/L and his weight loss, depression, vomiting, and fatigue had resolved. He stopped taking his SSRI. He was still craving extra salt, but not as much, and his urine was no longer a very dark yellow.
In retrospect, starting this patient on steroids earlier may not have resulted in any more of a rapid sodium rise than that which occurred otherwise, but we believe that our concern for CPM at that time justified the delay in steroid use. We felt it was safe to delay steroids because the patient’s BP was stable and his clinical picture was rapidly improving. In most cases, however, delaying steroids is not advisable.
THE TAKEAWAY
Adrenal insufficiency can be clearly diagnosed via labs and clinical presentation, and is potentially lethal if unrecognized. The predominant manifestations of adrenal crisis are hypotension and shock, usually with hyponatremia and hyperkalemia. During stressful events or illness, patients should increase their glucocorticoid dose. If they are on hydrocortisone, instructions are usually to triple the dose, and give the medication 3 times a day. Patients require instruction beforehand on how and when to increase doses for illness so that they can handle this on their own. Patients should carry a medical identification card so that their condition is evident to anyone caring for them in the ED.
THE CASE
A 17-year-old boy presented to the emergency department (ED) with a headache, dizziness, lethargy, and weakness that he’d had for 2 weeks. The patient was taking a selective serotonin reuptake inhibitor (SSRI) for depression (sertraline 25 mg/d). He had been vomiting twice daily for the past 3 years. (Although he had been seen multiple times in urgent care clinics, he did not have regular medical care.) The boy was fatigued and had dark yellow urine. His father indicated that his son’s skin had darkened over the last 5 to 6 years and that he had been adding salt, in large quantities, to nearly all of his meals for 10 years.
The boy’s health issues were impacting his school life. He was dismissed from school often because his teachers felt he was skipping class and using the excuse of needing to urinate or vomit. He had traveled back and forth to Mexico about 2 times a year, with the last time being about 3 months before his trip to the ED.
The patient’s vitals included a temperature of 96.3º F, heart rate (HR) of 77 beats/min, respiratory rate of 16 breaths/min, and a supine blood pressure (BP) of 102/58 mm Hg. (The patient’s BP was not obtained when sitting or standing, because he felt dizzy when trying to stand or sit up and the HR monitor increased to 100 beats/min.) His weight was 106.9 pounds and height was 5 feet 8 inches. The teen was ill-appearing and somnolent. No jugular vein distention, murmurs, or gallops were noted on exam. The patient’s lips were dry and cracked, gums were darkened, and his skin was clammy to the touch. His abdomen was soft with hypoactive bowel sounds and no ascites. His extremities were non-edematous.
A chemistry panel showed a low sodium level of 99 mEq/L, a somewhat high potassium level of 5.2 mmol/L, low chloride (69 mEq/L) and CO2 (5 mEq/L) levels, a high glucose level (124 mg/dL), and normal creatinine (0.79 mg/dL), albumin (5.2 g/dL), and thyroid stimulating hormone (2.4 mIU/L) levels. A tuberculosis (TB) test, acute hepatitis panel, human immunodeficiency test, and urine drug screen were all negative. Liver enzymes and lipase levels were normal.
The patient was admitted to the pediatric intensive care unit (PICU) on 200 mL/hr normal saline (twice the normal maintenance rate) and we took over his care.
THE DIAGNOSIS
Because of the patient’s severe hyponatremia, the differential diagnosis included heart failure, cirrhosis, syndrome of inappropriate antidiuretic hormone secretion (SIADH), SSRI-induced SIADH, cerebral salt wasting, severe hypothyroidism, adrenal insufficiency, malignancies, ecstasy use, renal failure, low dietary solute intake, and psychogenic polydipsia.
A random cortisol test taken in the ED returned and was noted to be very low (<1 mcg/dL). This information, plus the signs of aldosterone deficiency (low sodium and elevated potassium levels) and adrenocorticotropic hormone (ACTH) excess (skin darkening), prompted us to perform a 250-mcg ACTH stimulation test. Results at 30 and 60 minutes both showed cortisol at <1 mcg/dL, which led us to suspect adrenal insufficiency. The diagnosis of autoimmune adrenalitis, or Addison’s disease, was confirmed after inpatient lab work returned with positive 21-hydroxylase antibodies and an elevated ACTH (1117 pg/mL; normal, 10-65 pg/mL).
We noted that the patient’s sodium level was gradually increasing while he was receiving the intravenous (IV) fluids. We were concerned, though, that too rapid a sodium correction would put the patient at risk for central pontine myelinolysis (CPM). So we held off on steroids until 24 hours after he was admitted to the PICU, when his sodium level reached 110 mEq/L.
DISCUSSION
Primary adrenal insufficiency in the developed world is commonly caused by autoimmune adrenalitis, also known as Addison’s disease. Addison’s disease is the cause of primary adrenal insufficiency in 70% to 90% of cases, with the remainder caused by TB, adrenal hemorrhage, infarction, lymphoma, cytomegalovirus, adrenoleukodystrophy, or metastatic cancer. We also considered adrenoleukodystrophy in our patient, but felt it unlikely in a 17-year-old with normal mental status and positive adrenal antibodies.
The first evidence of Addison’s disease is usually an increase in plasma renin activity with low serum aldosterone. This might explain our patient’s years of salt cravings prior to presentation. There is typically a decrease in serum cortisol response to ACTH stimulation several months to years after the onset of salt cravings. The next sign of deterioration in adrenal function is an increase in basal serum ACTH; the process concludes with a decreased basal serum cortisol level.1-3 By the time our patient presented to the ED, his ACTH was very high, his cortisol was low, and his ACTH stimulation response was low.
Acute adrenal insufficiency crisis usually occurs after a prolonged period of nonspecific complaints due to a loss of both glucocorticoids and mineralocorticoids; by the time overt symptoms occur, 90% of the adrenal gland may be destroyed.3 Patients (such as ours) may present with symptoms such as abdominal pain, weakness, vomiting, fever, and decreased responsiveness. Hyponatremia and hyperkalemia are commonly seen at initial diagnosis. BP can be compromised in some patients due to loss of vascular tone; our patient did not present with this finding.
Treatment includes hydrocortisone and fludrocortisone for life
Initial management focuses on rehydration, maintenance of BP, cardiac monitoring, and electrolyte monitoring with a focus on slow normalization of electrolyte abnormalities. Patients should be treated with hydrocortisone (approximately 10 mg/m2/d) and fludrocortisone (usually 0.1 mg/d), and they will be maintained on this regimen for life.1,3
During acute illness, the doses of hydrocortisone are usually tripled and given 3 times per day to address the increased cortisol needs of the stress response. Lack of stress dose steroids in the setting of illness can lead to repeat adrenal crisis events.
Patients should be taught about intramuscular (IM) hydrocortisone use (100 mg IM) for emergencies and should have medical identification. In many states, emergency medical technicians (EMTs) are now able to administer the patient’s own supply of hydrocortisone. EMTs have even begun carrying hydrocortisone in some states in response to a campaign by the CARES Foundation, a nonprofit organization dedicated to helping families and individuals affected by congenital adrenal hyperplasia.
We started our patient on 100 mg/m2/d hydrocortisone 24 hours after he was admitted to the PICU. (At that time, his sodium level was 110 mEq/L.) Forty-eight hours after admission, we started the patient on fludrocortisone for mineralocorticoid effect at 0.1 mg/d. (The patient’s sodium level was 122 mEq/L). At 72 hours after admission, the patient’s sodium level was 137 mEq/L and his mental status was normal. Normal saline was discontinued when sodium normalized. He was discharged 2 days later. He was informed he should continue these medications for life, though doses might be adjusted slightly with time.
Two weeks later, our patient’s sodium level had reached 141 mEq/L and his weight loss, depression, vomiting, and fatigue had resolved. He stopped taking his SSRI. He was still craving extra salt, but not as much, and his urine was no longer a very dark yellow.
In retrospect, starting this patient on steroids earlier may not have resulted in any more of a rapid sodium rise than that which occurred otherwise, but we believe that our concern for CPM at that time justified the delay in steroid use. We felt it was safe to delay steroids because the patient’s BP was stable and his clinical picture was rapidly improving. In most cases, however, delaying steroids is not advisable.
THE TAKEAWAY
Adrenal insufficiency can be clearly diagnosed via labs and clinical presentation, and is potentially lethal if unrecognized. The predominant manifestations of adrenal crisis are hypotension and shock, usually with hyponatremia and hyperkalemia. During stressful events or illness, patients should increase their glucocorticoid dose. If they are on hydrocortisone, instructions are usually to triple the dose, and give the medication 3 times a day. Patients require instruction beforehand on how and when to increase doses for illness so that they can handle this on their own. Patients should carry a medical identification card so that their condition is evident to anyone caring for them in the ED.
1. Husebye ES, Allolio B, Arlt W, et al. Consensus statement on the diagnosis, treatment and follow-up of patients with primary adrenal insufficiency. J Intern Med. 2014;275:104-115.
2. Betterle C, Morlin L. Autoimmune Addison’s disease. Endocr Dev. 2011;20:161-172.
3. Brandão Neto RA, de Carvalho JF. Diagnosis and classification of Addison’s disease (autoimmune adrenalitis). Autoimmun Rev. 2014;13:408-411.
1. Husebye ES, Allolio B, Arlt W, et al. Consensus statement on the diagnosis, treatment and follow-up of patients with primary adrenal insufficiency. J Intern Med. 2014;275:104-115.
2. Betterle C, Morlin L. Autoimmune Addison’s disease. Endocr Dev. 2011;20:161-172.
3. Brandão Neto RA, de Carvalho JF. Diagnosis and classification of Addison’s disease (autoimmune adrenalitis). Autoimmun Rev. 2014;13:408-411.

Sore throat • vaginal discharge • labial ulcer • Dx?
THE CASE
The mother of a 13-year-old girl brought her daughter to our family medicine clinic for follow-up after being seen in the emergency department (ED) 3 days earlier. The girl had presented to the ED with a one-day history of back, chest, and vaginal pain. She was diagnosed with a urinary tract infection and treated empirically with phenazopyridine and cephalexin pending a urine culture.
During the follow-up appointment, the patient complained of worsening vaginal pain and increased vaginal discharge, but reported resolution of her back and chest pain. She also said that a week earlier, she’d had a fever that reached 104° F and a sore throat. She denied urinary frequency/urgency, sexual activity, or sexual abuse. The result of the urine culture performed in the ED was <10,000 col/mL (normal urogenital flora).
A genitourinary (GU) exam revealed erythematous patches with small amounts of crusting at the inner labia bilaterally. The labia were also swollen and diffusely tender to palpation. The patient had a white/gray discharge, but no vesicles or papules. The physician was unable to place a speculum due to pain.
The differential diagnosis at the time included candidal vaginitis and cellulitis. Since the patient’s skin was non-erythematous and she had vaginal discharge, she was treated for presumed severe candidal vaginitis with fluconazole and clotrimazole 1% cream. (The antibiotics were stopped because the patient reported worsening symptoms after they were prescribed in the ED.) The patient was told to return to the ED if she experienced signs and symptoms such as worsening vaginal pain or discharge, fever, or chills. A repeat urine culture was performed and the results came back normal.
Worsening symptoms. Six days later, the patient returned to the ED with urinary hesitation and persistent dysuria; she was admitted for pain control. She also complained of worsening labial swelling and increased vaginal discharge despite adherence to the fluconazole and clotrimazole cream regimen, which were discontinued on admission to the ED. She continued to deny being sexually active or abused.
A GU exam showed a 1-cm shallow ulcer on the right labium and a copious amount of foul-smelling white discharge. An Ob/Gyn resident and attending physician examined the patient; their differential diagnosis at this point included herpes simplex virus (HSV), Epstein-Barr virus (EBV), gonorrhea/chlamydia, and trauma. The patient was given topical lidocaine for pain control and started on acyclovir for presumed HSV while awaiting the HSV test results. A pelvic ultrasound and laboratory work-up were ordered at this time as well.
THE DIAGNOSIS
The pelvic ultrasound showed that the uterus was a normal size and that there was no gross mass or significant pelvic fluid. The patient’s right ovary measured 2.8 × 1.6 cm; the left ovary was not seen.
The patient’s laboratory work-up included an unremarkable comprehensive metabolic panel. A complete blood count was within normal limits, except for the patient’s monocyte level, which was at 12.9% (reference range: 0%-12%). The patient had a negative urinary human chorionic gonadotropin test, and was negative for HSV, chlamydia, gonorrhea, and trichomoniasis. A rapid plasma reagin test and human immunodeficiency virus antibody (1+2) tests were nonreactive. A wet prep was negative. A mononuclear spot test (monospot), however, was positive.
Results from the monospot testing took several days to return. By the time the results arrived, the patient had been transferred to a local children’s hospital for assessment in their pediatric urology department, as she was experiencing urinary hesitation and required catheterization. The diagnosis of infectious mononucleosis presenting with genital ulcer was made. EBV cultures were never obtained, but seemed to be the likely cause of the patient’s infectious mononucleosis given her clinical symptoms and lab results.
DISCUSSION
Approximately 95% of adults worldwide are infected with EBV.1 While the infection is often asymptomatic, some patients will develop infectious mononucleosis.1 EBV is the most common cause of infectious mononucleosis, mainly affecting teenagers and young adults (especially college students). At least 25% of teenagers and young adults who become infected with EBV will develop infectious mononucleosis.2
Typical symptoms of infectious mononucleosis include extreme fatigue, fever, sore throat, and head and body aches.2 In this case, the patient did have a fever and sore throat one week prior to presentation at our clinic, but she never complained of fatigue.
The association between mononucleosis and genital ulcers is not well known,3,4 and the exact method by which EBV causes genital ulcers is unclear.5 One review found that only 13 instances of genital ulceration in females attributable to EBV infection had been reported.5 When ulceration does occur, the majority of cases have involved young females who presented with only mild symptoms of mononucleosis.3,6 EBV has been found to present in the cervix, which suggests direct inoculation.3,6
Our patient remained catheterized for 2 days while in the children’s hospital. Her ulcer started to heal and she was sent home in stable condition. No additional follow-up was required and the ulcer did not recur.
THE TAKEAWAY
Include infectious mononucleosis in the differential for patients presenting with vaginal ulcers—especially those who deny sexual activity. Including testing for EBV and mononucleosis antibodies in the work-up can aid in the diagnosis. Cases such as this one are also a good reminder of the need to question young people while their parents/guardians are not in the examroom to foster an open and honest patient-physician relationship.
1. Womack J, Jimenez M. Common questions about infectious mononucleosis. Am Fam Physician. 2015;91:372-376.
2. Centers for Disease Control and Prevention. About infectious mononucleosis. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/epstein-barr/about-mono.html. Accessed April 26, 2016.
3. Lorenzo CV, Robertson WS. Genital ulcerations as presenting symptom of infectious mononucleosis. J Am Board Fam Pract. 2005;18:67-68.
4. Sisson BA, Glick L. Genital ulceration as a presenting manifestation of infectious mononucleosis. J Pediatr Adolesc Gynecol. 1998;11:185-187.
5. Barnes CJ, Alió AB, Cunningham BB, et al. Epstein-Barr virus-associated genital ulcers: an under-recognized disorder. Pediatr Dermatol. 2007;24:130-134.
6. Wilson RW. Genital ulcers and mononucleosis. Pediatr Infect Dis J. 1993;12:418.
THE CASE
The mother of a 13-year-old girl brought her daughter to our family medicine clinic for follow-up after being seen in the emergency department (ED) 3 days earlier. The girl had presented to the ED with a one-day history of back, chest, and vaginal pain. She was diagnosed with a urinary tract infection and treated empirically with phenazopyridine and cephalexin pending a urine culture.
During the follow-up appointment, the patient complained of worsening vaginal pain and increased vaginal discharge, but reported resolution of her back and chest pain. She also said that a week earlier, she’d had a fever that reached 104° F and a sore throat. She denied urinary frequency/urgency, sexual activity, or sexual abuse. The result of the urine culture performed in the ED was <10,000 col/mL (normal urogenital flora).
A genitourinary (GU) exam revealed erythematous patches with small amounts of crusting at the inner labia bilaterally. The labia were also swollen and diffusely tender to palpation. The patient had a white/gray discharge, but no vesicles or papules. The physician was unable to place a speculum due to pain.
The differential diagnosis at the time included candidal vaginitis and cellulitis. Since the patient’s skin was non-erythematous and she had vaginal discharge, she was treated for presumed severe candidal vaginitis with fluconazole and clotrimazole 1% cream. (The antibiotics were stopped because the patient reported worsening symptoms after they were prescribed in the ED.) The patient was told to return to the ED if she experienced signs and symptoms such as worsening vaginal pain or discharge, fever, or chills. A repeat urine culture was performed and the results came back normal.
Worsening symptoms. Six days later, the patient returned to the ED with urinary hesitation and persistent dysuria; she was admitted for pain control. She also complained of worsening labial swelling and increased vaginal discharge despite adherence to the fluconazole and clotrimazole cream regimen, which were discontinued on admission to the ED. She continued to deny being sexually active or abused.
A GU exam showed a 1-cm shallow ulcer on the right labium and a copious amount of foul-smelling white discharge. An Ob/Gyn resident and attending physician examined the patient; their differential diagnosis at this point included herpes simplex virus (HSV), Epstein-Barr virus (EBV), gonorrhea/chlamydia, and trauma. The patient was given topical lidocaine for pain control and started on acyclovir for presumed HSV while awaiting the HSV test results. A pelvic ultrasound and laboratory work-up were ordered at this time as well.
THE DIAGNOSIS
The pelvic ultrasound showed that the uterus was a normal size and that there was no gross mass or significant pelvic fluid. The patient’s right ovary measured 2.8 × 1.6 cm; the left ovary was not seen.
The patient’s laboratory work-up included an unremarkable comprehensive metabolic panel. A complete blood count was within normal limits, except for the patient’s monocyte level, which was at 12.9% (reference range: 0%-12%). The patient had a negative urinary human chorionic gonadotropin test, and was negative for HSV, chlamydia, gonorrhea, and trichomoniasis. A rapid plasma reagin test and human immunodeficiency virus antibody (1+2) tests were nonreactive. A wet prep was negative. A mononuclear spot test (monospot), however, was positive.
Results from the monospot testing took several days to return. By the time the results arrived, the patient had been transferred to a local children’s hospital for assessment in their pediatric urology department, as she was experiencing urinary hesitation and required catheterization. The diagnosis of infectious mononucleosis presenting with genital ulcer was made. EBV cultures were never obtained, but seemed to be the likely cause of the patient’s infectious mononucleosis given her clinical symptoms and lab results.
DISCUSSION
Approximately 95% of adults worldwide are infected with EBV.1 While the infection is often asymptomatic, some patients will develop infectious mononucleosis.1 EBV is the most common cause of infectious mononucleosis, mainly affecting teenagers and young adults (especially college students). At least 25% of teenagers and young adults who become infected with EBV will develop infectious mononucleosis.2
Typical symptoms of infectious mononucleosis include extreme fatigue, fever, sore throat, and head and body aches.2 In this case, the patient did have a fever and sore throat one week prior to presentation at our clinic, but she never complained of fatigue.
The association between mononucleosis and genital ulcers is not well known,3,4 and the exact method by which EBV causes genital ulcers is unclear.5 One review found that only 13 instances of genital ulceration in females attributable to EBV infection had been reported.5 When ulceration does occur, the majority of cases have involved young females who presented with only mild symptoms of mononucleosis.3,6 EBV has been found to present in the cervix, which suggests direct inoculation.3,6
Our patient remained catheterized for 2 days while in the children’s hospital. Her ulcer started to heal and she was sent home in stable condition. No additional follow-up was required and the ulcer did not recur.
THE TAKEAWAY
Include infectious mononucleosis in the differential for patients presenting with vaginal ulcers—especially those who deny sexual activity. Including testing for EBV and mononucleosis antibodies in the work-up can aid in the diagnosis. Cases such as this one are also a good reminder of the need to question young people while their parents/guardians are not in the examroom to foster an open and honest patient-physician relationship.
THE CASE
The mother of a 13-year-old girl brought her daughter to our family medicine clinic for follow-up after being seen in the emergency department (ED) 3 days earlier. The girl had presented to the ED with a one-day history of back, chest, and vaginal pain. She was diagnosed with a urinary tract infection and treated empirically with phenazopyridine and cephalexin pending a urine culture.
During the follow-up appointment, the patient complained of worsening vaginal pain and increased vaginal discharge, but reported resolution of her back and chest pain. She also said that a week earlier, she’d had a fever that reached 104° F and a sore throat. She denied urinary frequency/urgency, sexual activity, or sexual abuse. The result of the urine culture performed in the ED was <10,000 col/mL (normal urogenital flora).
A genitourinary (GU) exam revealed erythematous patches with small amounts of crusting at the inner labia bilaterally. The labia were also swollen and diffusely tender to palpation. The patient had a white/gray discharge, but no vesicles or papules. The physician was unable to place a speculum due to pain.
The differential diagnosis at the time included candidal vaginitis and cellulitis. Since the patient’s skin was non-erythematous and she had vaginal discharge, she was treated for presumed severe candidal vaginitis with fluconazole and clotrimazole 1% cream. (The antibiotics were stopped because the patient reported worsening symptoms after they were prescribed in the ED.) The patient was told to return to the ED if she experienced signs and symptoms such as worsening vaginal pain or discharge, fever, or chills. A repeat urine culture was performed and the results came back normal.
Worsening symptoms. Six days later, the patient returned to the ED with urinary hesitation and persistent dysuria; she was admitted for pain control. She also complained of worsening labial swelling and increased vaginal discharge despite adherence to the fluconazole and clotrimazole cream regimen, which were discontinued on admission to the ED. She continued to deny being sexually active or abused.
A GU exam showed a 1-cm shallow ulcer on the right labium and a copious amount of foul-smelling white discharge. An Ob/Gyn resident and attending physician examined the patient; their differential diagnosis at this point included herpes simplex virus (HSV), Epstein-Barr virus (EBV), gonorrhea/chlamydia, and trauma. The patient was given topical lidocaine for pain control and started on acyclovir for presumed HSV while awaiting the HSV test results. A pelvic ultrasound and laboratory work-up were ordered at this time as well.
THE DIAGNOSIS
The pelvic ultrasound showed that the uterus was a normal size and that there was no gross mass or significant pelvic fluid. The patient’s right ovary measured 2.8 × 1.6 cm; the left ovary was not seen.
The patient’s laboratory work-up included an unremarkable comprehensive metabolic panel. A complete blood count was within normal limits, except for the patient’s monocyte level, which was at 12.9% (reference range: 0%-12%). The patient had a negative urinary human chorionic gonadotropin test, and was negative for HSV, chlamydia, gonorrhea, and trichomoniasis. A rapid plasma reagin test and human immunodeficiency virus antibody (1+2) tests were nonreactive. A wet prep was negative. A mononuclear spot test (monospot), however, was positive.
Results from the monospot testing took several days to return. By the time the results arrived, the patient had been transferred to a local children’s hospital for assessment in their pediatric urology department, as she was experiencing urinary hesitation and required catheterization. The diagnosis of infectious mononucleosis presenting with genital ulcer was made. EBV cultures were never obtained, but seemed to be the likely cause of the patient’s infectious mononucleosis given her clinical symptoms and lab results.
DISCUSSION
Approximately 95% of adults worldwide are infected with EBV.1 While the infection is often asymptomatic, some patients will develop infectious mononucleosis.1 EBV is the most common cause of infectious mononucleosis, mainly affecting teenagers and young adults (especially college students). At least 25% of teenagers and young adults who become infected with EBV will develop infectious mononucleosis.2
Typical symptoms of infectious mononucleosis include extreme fatigue, fever, sore throat, and head and body aches.2 In this case, the patient did have a fever and sore throat one week prior to presentation at our clinic, but she never complained of fatigue.
The association between mononucleosis and genital ulcers is not well known,3,4 and the exact method by which EBV causes genital ulcers is unclear.5 One review found that only 13 instances of genital ulceration in females attributable to EBV infection had been reported.5 When ulceration does occur, the majority of cases have involved young females who presented with only mild symptoms of mononucleosis.3,6 EBV has been found to present in the cervix, which suggests direct inoculation.3,6
Our patient remained catheterized for 2 days while in the children’s hospital. Her ulcer started to heal and she was sent home in stable condition. No additional follow-up was required and the ulcer did not recur.
THE TAKEAWAY
Include infectious mononucleosis in the differential for patients presenting with vaginal ulcers—especially those who deny sexual activity. Including testing for EBV and mononucleosis antibodies in the work-up can aid in the diagnosis. Cases such as this one are also a good reminder of the need to question young people while their parents/guardians are not in the examroom to foster an open and honest patient-physician relationship.
1. Womack J, Jimenez M. Common questions about infectious mononucleosis. Am Fam Physician. 2015;91:372-376.
2. Centers for Disease Control and Prevention. About infectious mononucleosis. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/epstein-barr/about-mono.html. Accessed April 26, 2016.
3. Lorenzo CV, Robertson WS. Genital ulcerations as presenting symptom of infectious mononucleosis. J Am Board Fam Pract. 2005;18:67-68.
4. Sisson BA, Glick L. Genital ulceration as a presenting manifestation of infectious mononucleosis. J Pediatr Adolesc Gynecol. 1998;11:185-187.
5. Barnes CJ, Alió AB, Cunningham BB, et al. Epstein-Barr virus-associated genital ulcers: an under-recognized disorder. Pediatr Dermatol. 2007;24:130-134.
6. Wilson RW. Genital ulcers and mononucleosis. Pediatr Infect Dis J. 1993;12:418.
1. Womack J, Jimenez M. Common questions about infectious mononucleosis. Am Fam Physician. 2015;91:372-376.
2. Centers for Disease Control and Prevention. About infectious mononucleosis. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/epstein-barr/about-mono.html. Accessed April 26, 2016.
3. Lorenzo CV, Robertson WS. Genital ulcerations as presenting symptom of infectious mononucleosis. J Am Board Fam Pract. 2005;18:67-68.
4. Sisson BA, Glick L. Genital ulceration as a presenting manifestation of infectious mononucleosis. J Pediatr Adolesc Gynecol. 1998;11:185-187.
5. Barnes CJ, Alió AB, Cunningham BB, et al. Epstein-Barr virus-associated genital ulcers: an under-recognized disorder. Pediatr Dermatol. 2007;24:130-134.
6. Wilson RW. Genital ulcers and mononucleosis. Pediatr Infect Dis J. 1993;12:418.
