User login
Supraglottitis
Case
A 39-year-old woman, previously in good health, presented to the ED with a chief complaint of severe sore throat, which she said had begun approximately 4 hours prior and was rapidly worsening. She thought her voice sounded muffled, and said she was now having difficulty swallowing her saliva. The patient denied fever but did admit to chills. She experienced onset of shortness of breath 30 minutes prior to arrival to the ED.
The patient stated that she was a house painter and had been working in the home of someone who had several dogs. While not previously allergic to animals, the patient was concerned exposure to the dogs might have contributed to her symptoms. Regarding her social history, the patient admitted to daily consumption of beer, but denied smoking cigarettes. She had no known drug allergies.
On physical examination, the patient was afebrile. Her vital signs were: heart rate, 125 beats/min; blood pressure, 137/74 mm Hg; and respiratory rate, 18 breaths/min. Oxygen saturation was 99% on room air. Overall, the patient appeared anxious and exhibited mild inspiratory stridor. Examination of the eyes and ears were normal. There was no obvious inflammation or swelling of the posterior pharynx; the tongue was normal; there was no swelling of the floor of the mouth; and the uvula was midline and without swelling.
The patient was noted to having difficulty handling her secretions. She exhibited full range of motion of her neck. Her trachea was tender upon palpation but without jugular venous distension or lymphadenopathy. The cardiac examination was significant for tachycardia with a regular rhythm and without murmurs, rubs, or gallops; the pulmonary examination was normal except for transmitted upper airway sounds. The patient’s abdominal, dermatological, and neurological examinations were all normal.
Based on the examination findings, the differential diagnosis included allergic reaction, angioedema, epiglottitis, and retropharyngeal abscess. An intravenous (IV) line was placed and blood was drawn for laboratory evaluation, which included a complete blood count, basic metabolic panel (BMP), and a quantitative pregnancy test. Given the patient’s history, the emergency
A portable soft-tissue lateral radiograph of the neck was obtained. Radiology services interpreted the film as showing “prominent prevertebral soft tissues and epiglottis. 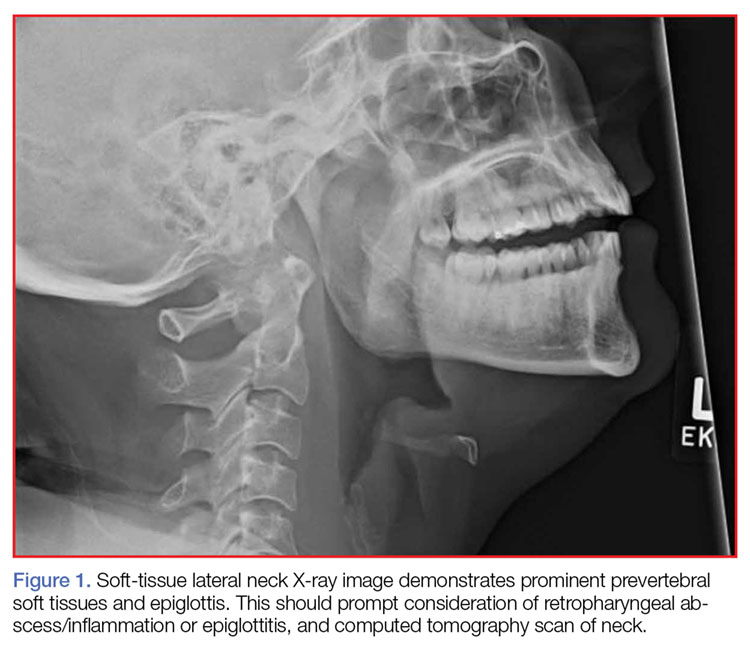
At this point, the patient appeared relatively stable and without progression of symptoms. Since there was the possibility of an infectious etiology, she was given piperacillin/tazobactam, 4.5 g IV. 
Laboratory evaluation results were significant for an elevated white blood cell count (WBC) of 14.8 ×109/L, but without a left shift; BMP results were within normal limits, and the pregnancy test was negative.
Based on these findings, otolaryngology services were consulted. The consulting otolaryngologist sprayed oxymetazoline and tetracaine into both of the patient’s nostrils and performed a flexible fiberoptic nasopharyngolaryngoscopy. During the procedure, a significant amount of diffuse supraglottic edema was noted, but no posterior pharyngeal wall edema.
Based on the presence of stridor, difficulty managing secretions, and significant amount of supraglottic edema, the patient was taken to the surgical suite for urgent airway control. She was given dexamethasone, 10 mg IV, and after some difficulty, the anesthesiologist orally intubated the patient with a 7.0-mm endotracheal tube. Examination during the procedure noted diffuse supraglottic edema but no other abnormalities.
The patient was transferred to the intensive care unit (ICU) and treated with IV piperacillin/tazobactam and dexamethasone. While in the ICU, the patient became extremely agitated and combative. After further inquiry into the patient’s social history, the patient’s husband reported that his wife drank 12 to 13 beers nightly. The patient required treatment for alcohol withdrawal with IV benzodiazepines, sedation, and physical restraints. By hospital day 9, she was extubated and tolerated fluids by mouth. On hospital day 10, her mental status had returned to baseline, her WBC was within normal limits, and she no longer complained of difficulty swallowing. The patient was discharged home on hospital day 11 with a final diagnosis of supraglottitis and alcohol withdrawal, and she was given a prescription for amoxicillin/clavulanate. Unfortunately, she did not return for her follow-up appointments.
Discussion
While the incidence of pediatric epiglottitis has decreased since the introduction of the Haemophilus influenzae type b (Hib) vaccine in 1985, adult epiglottitis continues to represent a potentially life-threatening condition whose incidence has remained constant over the past several decades.1,2 The incidence of supraglottitis in adults is now 2.5 times greater than the incidence in children.3,4
Several important differences exist in the presentation and management of adults who present with inflammation of the epiglottis as compared to children. Children commonly present with an acute onset of symptoms, and due to their smaller and more pliant airway anatomy, they often experience stridor and respiratory distress.3,5 The inflammation in children is typically confined to the epiglottis and aryepiglottic folds, while in adults the inflammation can affect not only the epiglottis, but also supraglottic structures such as the pharynx, uvula, and aryepiglottic folds. For this reason, in adults the condition is often referred to as “supraglottitis.”2,6 Adults with supraglottitis most likely present in their 30s, 40s, and 50s, while children present between the ages of 2 and 5 years old.1,3,7 In adults, men more commonly present with supraglottitis than women.1,2 Cigarette smokers and patients with hypertension, diabetes mellitus (DM), chronic obstructive pulmonary disease, or human immunodeficiency virus/AIDS are at increased risk for supraglottitis.3,4 The mortality rate for adults with supraglottitis ranges from 1.2% to 7.1%.3
Etiology
Prior to the use of the Hib vaccine, Hib was the most common cause of epiglottitis, and remains so for children.1 Currently, the most common cause of supraglottitis in adults is Group A beta-hemolytic Streptococci.2 Other etiologies include other bacteria (Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus, Pseudomonas species, Klebsiella pneumoniae, Pasteurella multocida, Neisseria species), viruses (herpes simplex, varicella, parainfluenza), trauma, and thermal injuries.1,4,5,8
Signs and Symptoms
Throat pain, dysphagia, odynophagia, and muffled voice are common complaints of adults presenting to the ED with supraglottitis.2,7 Fever is usually, but not always, present; the complaint of cough, however, is rare.2,3,4 Other less frequent complaints include hoarseness and drooling. Adults can also present with cervical lymphadenopathy, anterior neck tenderness, and cellulitis of the neck and chest.2,4 In general, the more severe cases will progress rapidly over a few hours. Due to the larger anatomy in adults, they are more likely than children to experience a gradual progression of symptoms, and supraglottitis will be missed on the initial presentation in up to 50% of adults.3,4 Stridor or respiratory compromise does occur in a minority of adult patients with supraglottitis. The need for artificial airway support (ie, endotracheal intubation, cricothyroidotomy) in adults ranges from 6.6% to 16%.9,10
Making the Diagnosis
The gold standard for diagnosing supraglottitis is direct laryngoscopy.3,4 This point is emphasized in our case report, since the CT scan was concerning for a retropharyngeal abscess, and not supraglottitis. The examination of the oropharynx is generally safer and better tolerated in adults compared to pediatric patients, since airway compromise is much less likely. On occasion, inflammation, erythema, and edema of the epiglottis, aryepiglottic folds, or arytenoid cartilages can be observed.5 More commonly, the supraglottic structures are not visualized, and the posterior oropharynx appears relatively normal. This should serve as a clue for possible supraglottitis.
In suspected cases of adult supraglottitis without emergent airway compromise, lateral soft-tissue radiographs can be obtained to look for the “thumb sign,” indicating a swollen epiglottis. In adult supraglottitis, the width of the epiglottis is usually greater than 8 mm.11 Other abnormal radiographic findings include arytenoid and aryepiglottic fold enlargement, thinning of the airway, and an increase in size of the prevertebral space. Plain film sensitivity rates range from 38% to 98%.
Complete blood count and throat cultures are not particularly helpful in adult cases. Blood cultures, while only about 30% sensitive in adults, should be considered as supraglottitis can result in secondary infection in the central nervous system, lungs, and surrounding structures.3,5
If available, otolaryngology services should be consulted to evaluate the airway, and IV antibiotics, such as a third-generation cephalosporin (eg, ceftriaxone, cefotaxime), should be initiated to include coverage of Hib.3 If methicillin-resistant S aureus is a concern, vancomycin should be added. Clindamycin or metronidazole should also be given if anaerobes are suspected.4,7 The location for performing the nasopharyngeal laryngoscopy varies, depending on the patient’s age (ie, pediatric vs adult), severity of symptoms, presence of airway compromise, and local practice and custom.
Advanced imaging studies (CT scan or magnetic resonance imaging) can help identify the presence of an abscess and delineate the extent of the infection, but are not indicated in the early diagnosis and management of suspected adult supraglottitis.4 As our case demonstrates, CT is neither highly sensitive nor specific for the diagnosis of epiglottitis. The role of ultrasound in the evaluation of suspected epiglottitis is still being developed. One recent study compared 15 healthy volunteers with 15 patients diagnosed with epiglottitis by an otolaryngologist using laryngoscopy.12 A statistically significant difference was observed in the anteroposterior diameter of the epiglottis at the midpoint and both lateral edges between the study subjects and healthy volunteers.12 While there was overlap in the ranges for the midpoint, there was no overlap in both lateral edges between the two groups.12
Treatment
The vast majority of adult cases of supraglottitis are managed medically without airway intervention. Patients presenting with a rapid onset of symptoms and in respiratory distress or with stridor, drooling, or cyanosis, should be managed with early airway intervention. The use of corticosteroids is controversial, and has not been proven beneficial in any prospective trials.1-4,6,7,13
Admission to a critical care unit is indicated initially, even in patients who are not intubated, as they can experience delayed airway compromise with progression of the infection and edema.13
Complications
Abscess formation is a serious complication of supraglottitis, is present in up to 30% of cases, and is more likely to be seen in adults than in children.13 Since the adult larynx and surrounding tissues are larger than in children, often the infection is present longer, which allows for an abscess to develop. The risk of abscess formation is increased in patients with DM or those in whom a foreign body is present.
Numerous organisms have been isolated from supraglottic abscesses in adults, and in addition to incision and drainage, antibiotics covering both gram-positive organisms and anaerobes should be initiated.5 The presence of a supraglottic abscess increases the need for emergent intubation.13 In addition, a supraglottic abscess increases the mortality rate to 30%.3 Other complications from supraglottitis include mediastinitis, cervical adenitis, meningitis, and pneumonia.4,5
Conclusion
While the incidence of epiglottitis in the pediatric patient population has fallen, the incidence in adults remains relatively stable. Clinicians should consider supraglottitis in the differential diagnosis of adults presenting with severe sore throat, dysphagia, or stridor. While airway compromise in adults is uncommon, it does occur. Soft-tissue lateral neck radiographs can help make the diagnosis, but the gold standard remains laryngoscopy. All patients should be started on IV antibiotics and admitted to the ICU initially for airway watch.
1. Glynn F, Fenton JE. Diagnosis and management of supraglottitis (epiglottitis). Curr Infect Dis Rep. 2008;10(3):200-204.
2. Lichtor JL, Roche Rodriguez M, Aaronson NL, Spock T, Goodman TR, Baum ED. Epiglottitis: It hasn’t gone away. Anesthesiology. 2016;124(6):1404-1407. doi: 10.1097/ALN.0000000000001125.
3. Westerhuis B, Bietz MG, Lindemann J. Acute epiglottitis in adults: an under-recognized and life-threatening condition. S D Med. 2013;66(8):309-311, 313.
4. Al-Qudah M, Shetty S, Alomari M, Alqdah M. Acute adult supraglottitis: Current management and treatment. South Med J. 2010;103(8):800-804. doi: 10.1097/SMJ.0b013e3181e538d8.
5. Verbruggen K, Halewyck S, Deron P, Foulon I, Gordts F. Epiglottitis and related complications in adults. Case reports and review of the literature. B-ENT. 2012;8(2):143-148.
6. Mayo-Smith MF, Spinale JW, Donskey CJ, Yukawa M, Li RH, Schiffman FJ. Acute epiglottitis. An 18-year experience in Rhode Island. Chest. 1995;108(6):1640-1647.
7. Bizaki AJ, Numminen J, Vasama JP, Laranne J, Rautiainen M. Acute supraglottitis in adults in Finland: review and analysis of 308 cases. Laryngoscope. 2011;121(10):2107-2113. doi: 10.1002/lary.22147.
8. Charles R, Fadden M, Brook J. Acute epiglottitis. BMJ. 2013;347:f5235. doi: 10.1136/bmj.f5235.
9. Ng HL, Sin LM, Li MF, Que TL, Anandaciva S. Acute epiglottitis in adults: a retrospective review of 106 patients in Hong Kong. Emerg Med J. 2008;25(5):253-255. doi: 10.1136/emj.2007.050153.
10. Solomon P, Weisbrod M, Irish JC, Gullane PJ. Adult epiglottitis: the Toronto Hospital experience. J Otolaryngol. 1998;27(6):332-336.
11. Schumaker HM, Doris PE, Birnbaum G. Radiographic parameters in adult epiglottitis. Ann Emerg Med. 1984;13(8):588-590.
12. Ko DR, Chung YE, Park I, et al. Use of bedside sonography for diagnosing acute epiglottitis in the emergency department: a preliminary study. J Ultrasound Med. 2012;31(1):19-22.
13. Berger G, Landau T, Berger S, Finkelstein Y, Bernheim J, Ophir D. The rising incidence of adult acute epiglottitis and epiglottic abscess. Am J Otolaryngol. 2003;24(6):374-383.
Case
A 39-year-old woman, previously in good health, presented to the ED with a chief complaint of severe sore throat, which she said had begun approximately 4 hours prior and was rapidly worsening. She thought her voice sounded muffled, and said she was now having difficulty swallowing her saliva. The patient denied fever but did admit to chills. She experienced onset of shortness of breath 30 minutes prior to arrival to the ED.
The patient stated that she was a house painter and had been working in the home of someone who had several dogs. While not previously allergic to animals, the patient was concerned exposure to the dogs might have contributed to her symptoms. Regarding her social history, the patient admitted to daily consumption of beer, but denied smoking cigarettes. She had no known drug allergies.
On physical examination, the patient was afebrile. Her vital signs were: heart rate, 125 beats/min; blood pressure, 137/74 mm Hg; and respiratory rate, 18 breaths/min. Oxygen saturation was 99% on room air. Overall, the patient appeared anxious and exhibited mild inspiratory stridor. Examination of the eyes and ears were normal. There was no obvious inflammation or swelling of the posterior pharynx; the tongue was normal; there was no swelling of the floor of the mouth; and the uvula was midline and without swelling.
The patient was noted to having difficulty handling her secretions. She exhibited full range of motion of her neck. Her trachea was tender upon palpation but without jugular venous distension or lymphadenopathy. The cardiac examination was significant for tachycardia with a regular rhythm and without murmurs, rubs, or gallops; the pulmonary examination was normal except for transmitted upper airway sounds. The patient’s abdominal, dermatological, and neurological examinations were all normal.
Based on the examination findings, the differential diagnosis included allergic reaction, angioedema, epiglottitis, and retropharyngeal abscess. An intravenous (IV) line was placed and blood was drawn for laboratory evaluation, which included a complete blood count, basic metabolic panel (BMP), and a quantitative pregnancy test. Given the patient’s history, the emergency
A portable soft-tissue lateral radiograph of the neck was obtained. Radiology services interpreted the film as showing “prominent prevertebral soft tissues and epiglottis.
At this point, the patient appeared relatively stable and without progression of symptoms. Since there was the possibility of an infectious etiology, she was given piperacillin/tazobactam, 4.5 g IV.
Laboratory evaluation results were significant for an elevated white blood cell count (WBC) of 14.8 ×109/L, but without a left shift; BMP results were within normal limits, and the pregnancy test was negative.
Based on these findings, otolaryngology services were consulted. The consulting otolaryngologist sprayed oxymetazoline and tetracaine into both of the patient’s nostrils and performed a flexible fiberoptic nasopharyngolaryngoscopy. During the procedure, a significant amount of diffuse supraglottic edema was noted, but no posterior pharyngeal wall edema.
Based on the presence of stridor, difficulty managing secretions, and significant amount of supraglottic edema, the patient was taken to the surgical suite for urgent airway control. She was given dexamethasone, 10 mg IV, and after some difficulty, the anesthesiologist orally intubated the patient with a 7.0-mm endotracheal tube. Examination during the procedure noted diffuse supraglottic edema but no other abnormalities.
The patient was transferred to the intensive care unit (ICU) and treated with IV piperacillin/tazobactam and dexamethasone. While in the ICU, the patient became extremely agitated and combative. After further inquiry into the patient’s social history, the patient’s husband reported that his wife drank 12 to 13 beers nightly. The patient required treatment for alcohol withdrawal with IV benzodiazepines, sedation, and physical restraints. By hospital day 9, she was extubated and tolerated fluids by mouth. On hospital day 10, her mental status had returned to baseline, her WBC was within normal limits, and she no longer complained of difficulty swallowing. The patient was discharged home on hospital day 11 with a final diagnosis of supraglottitis and alcohol withdrawal, and she was given a prescription for amoxicillin/clavulanate. Unfortunately, she did not return for her follow-up appointments.
Discussion
While the incidence of pediatric epiglottitis has decreased since the introduction of the Haemophilus influenzae type b (Hib) vaccine in 1985, adult epiglottitis continues to represent a potentially life-threatening condition whose incidence has remained constant over the past several decades.1,2 The incidence of supraglottitis in adults is now 2.5 times greater than the incidence in children.3,4
Several important differences exist in the presentation and management of adults who present with inflammation of the epiglottis as compared to children. Children commonly present with an acute onset of symptoms, and due to their smaller and more pliant airway anatomy, they often experience stridor and respiratory distress.3,5 The inflammation in children is typically confined to the epiglottis and aryepiglottic folds, while in adults the inflammation can affect not only the epiglottis, but also supraglottic structures such as the pharynx, uvula, and aryepiglottic folds. For this reason, in adults the condition is often referred to as “supraglottitis.”2,6 Adults with supraglottitis most likely present in their 30s, 40s, and 50s, while children present between the ages of 2 and 5 years old.1,3,7 In adults, men more commonly present with supraglottitis than women.1,2 Cigarette smokers and patients with hypertension, diabetes mellitus (DM), chronic obstructive pulmonary disease, or human immunodeficiency virus/AIDS are at increased risk for supraglottitis.3,4 The mortality rate for adults with supraglottitis ranges from 1.2% to 7.1%.3
Etiology
Prior to the use of the Hib vaccine, Hib was the most common cause of epiglottitis, and remains so for children.1 Currently, the most common cause of supraglottitis in adults is Group A beta-hemolytic Streptococci.2 Other etiologies include other bacteria (Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus, Pseudomonas species, Klebsiella pneumoniae, Pasteurella multocida, Neisseria species), viruses (herpes simplex, varicella, parainfluenza), trauma, and thermal injuries.1,4,5,8
Signs and Symptoms
Throat pain, dysphagia, odynophagia, and muffled voice are common complaints of adults presenting to the ED with supraglottitis.2,7 Fever is usually, but not always, present; the complaint of cough, however, is rare.2,3,4 Other less frequent complaints include hoarseness and drooling. Adults can also present with cervical lymphadenopathy, anterior neck tenderness, and cellulitis of the neck and chest.2,4 In general, the more severe cases will progress rapidly over a few hours. Due to the larger anatomy in adults, they are more likely than children to experience a gradual progression of symptoms, and supraglottitis will be missed on the initial presentation in up to 50% of adults.3,4 Stridor or respiratory compromise does occur in a minority of adult patients with supraglottitis. The need for artificial airway support (ie, endotracheal intubation, cricothyroidotomy) in adults ranges from 6.6% to 16%.9,10
Making the Diagnosis
The gold standard for diagnosing supraglottitis is direct laryngoscopy.3,4 This point is emphasized in our case report, since the CT scan was concerning for a retropharyngeal abscess, and not supraglottitis. The examination of the oropharynx is generally safer and better tolerated in adults compared to pediatric patients, since airway compromise is much less likely. On occasion, inflammation, erythema, and edema of the epiglottis, aryepiglottic folds, or arytenoid cartilages can be observed.5 More commonly, the supraglottic structures are not visualized, and the posterior oropharynx appears relatively normal. This should serve as a clue for possible supraglottitis.
In suspected cases of adult supraglottitis without emergent airway compromise, lateral soft-tissue radiographs can be obtained to look for the “thumb sign,” indicating a swollen epiglottis. In adult supraglottitis, the width of the epiglottis is usually greater than 8 mm.11 Other abnormal radiographic findings include arytenoid and aryepiglottic fold enlargement, thinning of the airway, and an increase in size of the prevertebral space. Plain film sensitivity rates range from 38% to 98%.
Complete blood count and throat cultures are not particularly helpful in adult cases. Blood cultures, while only about 30% sensitive in adults, should be considered as supraglottitis can result in secondary infection in the central nervous system, lungs, and surrounding structures.3,5
If available, otolaryngology services should be consulted to evaluate the airway, and IV antibiotics, such as a third-generation cephalosporin (eg, ceftriaxone, cefotaxime), should be initiated to include coverage of Hib.3 If methicillin-resistant S aureus is a concern, vancomycin should be added. Clindamycin or metronidazole should also be given if anaerobes are suspected.4,7 The location for performing the nasopharyngeal laryngoscopy varies, depending on the patient’s age (ie, pediatric vs adult), severity of symptoms, presence of airway compromise, and local practice and custom.
Advanced imaging studies (CT scan or magnetic resonance imaging) can help identify the presence of an abscess and delineate the extent of the infection, but are not indicated in the early diagnosis and management of suspected adult supraglottitis.4 As our case demonstrates, CT is neither highly sensitive nor specific for the diagnosis of epiglottitis. The role of ultrasound in the evaluation of suspected epiglottitis is still being developed. One recent study compared 15 healthy volunteers with 15 patients diagnosed with epiglottitis by an otolaryngologist using laryngoscopy.12 A statistically significant difference was observed in the anteroposterior diameter of the epiglottis at the midpoint and both lateral edges between the study subjects and healthy volunteers.12 While there was overlap in the ranges for the midpoint, there was no overlap in both lateral edges between the two groups.12
Treatment
The vast majority of adult cases of supraglottitis are managed medically without airway intervention. Patients presenting with a rapid onset of symptoms and in respiratory distress or with stridor, drooling, or cyanosis, should be managed with early airway intervention. The use of corticosteroids is controversial, and has not been proven beneficial in any prospective trials.1-4,6,7,13
Admission to a critical care unit is indicated initially, even in patients who are not intubated, as they can experience delayed airway compromise with progression of the infection and edema.13
Complications
Abscess formation is a serious complication of supraglottitis, is present in up to 30% of cases, and is more likely to be seen in adults than in children.13 Since the adult larynx and surrounding tissues are larger than in children, often the infection is present longer, which allows for an abscess to develop. The risk of abscess formation is increased in patients with DM or those in whom a foreign body is present.
Numerous organisms have been isolated from supraglottic abscesses in adults, and in addition to incision and drainage, antibiotics covering both gram-positive organisms and anaerobes should be initiated.5 The presence of a supraglottic abscess increases the need for emergent intubation.13 In addition, a supraglottic abscess increases the mortality rate to 30%.3 Other complications from supraglottitis include mediastinitis, cervical adenitis, meningitis, and pneumonia.4,5
Conclusion
While the incidence of epiglottitis in the pediatric patient population has fallen, the incidence in adults remains relatively stable. Clinicians should consider supraglottitis in the differential diagnosis of adults presenting with severe sore throat, dysphagia, or stridor. While airway compromise in adults is uncommon, it does occur. Soft-tissue lateral neck radiographs can help make the diagnosis, but the gold standard remains laryngoscopy. All patients should be started on IV antibiotics and admitted to the ICU initially for airway watch.
Case
A 39-year-old woman, previously in good health, presented to the ED with a chief complaint of severe sore throat, which she said had begun approximately 4 hours prior and was rapidly worsening. She thought her voice sounded muffled, and said she was now having difficulty swallowing her saliva. The patient denied fever but did admit to chills. She experienced onset of shortness of breath 30 minutes prior to arrival to the ED.
The patient stated that she was a house painter and had been working in the home of someone who had several dogs. While not previously allergic to animals, the patient was concerned exposure to the dogs might have contributed to her symptoms. Regarding her social history, the patient admitted to daily consumption of beer, but denied smoking cigarettes. She had no known drug allergies.
On physical examination, the patient was afebrile. Her vital signs were: heart rate, 125 beats/min; blood pressure, 137/74 mm Hg; and respiratory rate, 18 breaths/min. Oxygen saturation was 99% on room air. Overall, the patient appeared anxious and exhibited mild inspiratory stridor. Examination of the eyes and ears were normal. There was no obvious inflammation or swelling of the posterior pharynx; the tongue was normal; there was no swelling of the floor of the mouth; and the uvula was midline and without swelling.
The patient was noted to having difficulty handling her secretions. She exhibited full range of motion of her neck. Her trachea was tender upon palpation but without jugular venous distension or lymphadenopathy. The cardiac examination was significant for tachycardia with a regular rhythm and without murmurs, rubs, or gallops; the pulmonary examination was normal except for transmitted upper airway sounds. The patient’s abdominal, dermatological, and neurological examinations were all normal.
Based on the examination findings, the differential diagnosis included allergic reaction, angioedema, epiglottitis, and retropharyngeal abscess. An intravenous (IV) line was placed and blood was drawn for laboratory evaluation, which included a complete blood count, basic metabolic panel (BMP), and a quantitative pregnancy test. Given the patient’s history, the emergency
A portable soft-tissue lateral radiograph of the neck was obtained. Radiology services interpreted the film as showing “prominent prevertebral soft tissues and epiglottis.
At this point, the patient appeared relatively stable and without progression of symptoms. Since there was the possibility of an infectious etiology, she was given piperacillin/tazobactam, 4.5 g IV.
Laboratory evaluation results were significant for an elevated white blood cell count (WBC) of 14.8 ×109/L, but without a left shift; BMP results were within normal limits, and the pregnancy test was negative.
Based on these findings, otolaryngology services were consulted. The consulting otolaryngologist sprayed oxymetazoline and tetracaine into both of the patient’s nostrils and performed a flexible fiberoptic nasopharyngolaryngoscopy. During the procedure, a significant amount of diffuse supraglottic edema was noted, but no posterior pharyngeal wall edema.
Based on the presence of stridor, difficulty managing secretions, and significant amount of supraglottic edema, the patient was taken to the surgical suite for urgent airway control. She was given dexamethasone, 10 mg IV, and after some difficulty, the anesthesiologist orally intubated the patient with a 7.0-mm endotracheal tube. Examination during the procedure noted diffuse supraglottic edema but no other abnormalities.
The patient was transferred to the intensive care unit (ICU) and treated with IV piperacillin/tazobactam and dexamethasone. While in the ICU, the patient became extremely agitated and combative. After further inquiry into the patient’s social history, the patient’s husband reported that his wife drank 12 to 13 beers nightly. The patient required treatment for alcohol withdrawal with IV benzodiazepines, sedation, and physical restraints. By hospital day 9, she was extubated and tolerated fluids by mouth. On hospital day 10, her mental status had returned to baseline, her WBC was within normal limits, and she no longer complained of difficulty swallowing. The patient was discharged home on hospital day 11 with a final diagnosis of supraglottitis and alcohol withdrawal, and she was given a prescription for amoxicillin/clavulanate. Unfortunately, she did not return for her follow-up appointments.
Discussion
While the incidence of pediatric epiglottitis has decreased since the introduction of the Haemophilus influenzae type b (Hib) vaccine in 1985, adult epiglottitis continues to represent a potentially life-threatening condition whose incidence has remained constant over the past several decades.1,2 The incidence of supraglottitis in adults is now 2.5 times greater than the incidence in children.3,4
Several important differences exist in the presentation and management of adults who present with inflammation of the epiglottis as compared to children. Children commonly present with an acute onset of symptoms, and due to their smaller and more pliant airway anatomy, they often experience stridor and respiratory distress.3,5 The inflammation in children is typically confined to the epiglottis and aryepiglottic folds, while in adults the inflammation can affect not only the epiglottis, but also supraglottic structures such as the pharynx, uvula, and aryepiglottic folds. For this reason, in adults the condition is often referred to as “supraglottitis.”2,6 Adults with supraglottitis most likely present in their 30s, 40s, and 50s, while children present between the ages of 2 and 5 years old.1,3,7 In adults, men more commonly present with supraglottitis than women.1,2 Cigarette smokers and patients with hypertension, diabetes mellitus (DM), chronic obstructive pulmonary disease, or human immunodeficiency virus/AIDS are at increased risk for supraglottitis.3,4 The mortality rate for adults with supraglottitis ranges from 1.2% to 7.1%.3
Etiology
Prior to the use of the Hib vaccine, Hib was the most common cause of epiglottitis, and remains so for children.1 Currently, the most common cause of supraglottitis in adults is Group A beta-hemolytic Streptococci.2 Other etiologies include other bacteria (Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus, Pseudomonas species, Klebsiella pneumoniae, Pasteurella multocida, Neisseria species), viruses (herpes simplex, varicella, parainfluenza), trauma, and thermal injuries.1,4,5,8
Signs and Symptoms
Throat pain, dysphagia, odynophagia, and muffled voice are common complaints of adults presenting to the ED with supraglottitis.2,7 Fever is usually, but not always, present; the complaint of cough, however, is rare.2,3,4 Other less frequent complaints include hoarseness and drooling. Adults can also present with cervical lymphadenopathy, anterior neck tenderness, and cellulitis of the neck and chest.2,4 In general, the more severe cases will progress rapidly over a few hours. Due to the larger anatomy in adults, they are more likely than children to experience a gradual progression of symptoms, and supraglottitis will be missed on the initial presentation in up to 50% of adults.3,4 Stridor or respiratory compromise does occur in a minority of adult patients with supraglottitis. The need for artificial airway support (ie, endotracheal intubation, cricothyroidotomy) in adults ranges from 6.6% to 16%.9,10
Making the Diagnosis
The gold standard for diagnosing supraglottitis is direct laryngoscopy.3,4 This point is emphasized in our case report, since the CT scan was concerning for a retropharyngeal abscess, and not supraglottitis. The examination of the oropharynx is generally safer and better tolerated in adults compared to pediatric patients, since airway compromise is much less likely. On occasion, inflammation, erythema, and edema of the epiglottis, aryepiglottic folds, or arytenoid cartilages can be observed.5 More commonly, the supraglottic structures are not visualized, and the posterior oropharynx appears relatively normal. This should serve as a clue for possible supraglottitis.
In suspected cases of adult supraglottitis without emergent airway compromise, lateral soft-tissue radiographs can be obtained to look for the “thumb sign,” indicating a swollen epiglottis. In adult supraglottitis, the width of the epiglottis is usually greater than 8 mm.11 Other abnormal radiographic findings include arytenoid and aryepiglottic fold enlargement, thinning of the airway, and an increase in size of the prevertebral space. Plain film sensitivity rates range from 38% to 98%.
Complete blood count and throat cultures are not particularly helpful in adult cases. Blood cultures, while only about 30% sensitive in adults, should be considered as supraglottitis can result in secondary infection in the central nervous system, lungs, and surrounding structures.3,5
If available, otolaryngology services should be consulted to evaluate the airway, and IV antibiotics, such as a third-generation cephalosporin (eg, ceftriaxone, cefotaxime), should be initiated to include coverage of Hib.3 If methicillin-resistant S aureus is a concern, vancomycin should be added. Clindamycin or metronidazole should also be given if anaerobes are suspected.4,7 The location for performing the nasopharyngeal laryngoscopy varies, depending on the patient’s age (ie, pediatric vs adult), severity of symptoms, presence of airway compromise, and local practice and custom.
Advanced imaging studies (CT scan or magnetic resonance imaging) can help identify the presence of an abscess and delineate the extent of the infection, but are not indicated in the early diagnosis and management of suspected adult supraglottitis.4 As our case demonstrates, CT is neither highly sensitive nor specific for the diagnosis of epiglottitis. The role of ultrasound in the evaluation of suspected epiglottitis is still being developed. One recent study compared 15 healthy volunteers with 15 patients diagnosed with epiglottitis by an otolaryngologist using laryngoscopy.12 A statistically significant difference was observed in the anteroposterior diameter of the epiglottis at the midpoint and both lateral edges between the study subjects and healthy volunteers.12 While there was overlap in the ranges for the midpoint, there was no overlap in both lateral edges between the two groups.12
Treatment
The vast majority of adult cases of supraglottitis are managed medically without airway intervention. Patients presenting with a rapid onset of symptoms and in respiratory distress or with stridor, drooling, or cyanosis, should be managed with early airway intervention. The use of corticosteroids is controversial, and has not been proven beneficial in any prospective trials.1-4,6,7,13
Admission to a critical care unit is indicated initially, even in patients who are not intubated, as they can experience delayed airway compromise with progression of the infection and edema.13
Complications
Abscess formation is a serious complication of supraglottitis, is present in up to 30% of cases, and is more likely to be seen in adults than in children.13 Since the adult larynx and surrounding tissues are larger than in children, often the infection is present longer, which allows for an abscess to develop. The risk of abscess formation is increased in patients with DM or those in whom a foreign body is present.
Numerous organisms have been isolated from supraglottic abscesses in adults, and in addition to incision and drainage, antibiotics covering both gram-positive organisms and anaerobes should be initiated.5 The presence of a supraglottic abscess increases the need for emergent intubation.13 In addition, a supraglottic abscess increases the mortality rate to 30%.3 Other complications from supraglottitis include mediastinitis, cervical adenitis, meningitis, and pneumonia.4,5
Conclusion
While the incidence of epiglottitis in the pediatric patient population has fallen, the incidence in adults remains relatively stable. Clinicians should consider supraglottitis in the differential diagnosis of adults presenting with severe sore throat, dysphagia, or stridor. While airway compromise in adults is uncommon, it does occur. Soft-tissue lateral neck radiographs can help make the diagnosis, but the gold standard remains laryngoscopy. All patients should be started on IV antibiotics and admitted to the ICU initially for airway watch.
1. Glynn F, Fenton JE. Diagnosis and management of supraglottitis (epiglottitis). Curr Infect Dis Rep. 2008;10(3):200-204.
2. Lichtor JL, Roche Rodriguez M, Aaronson NL, Spock T, Goodman TR, Baum ED. Epiglottitis: It hasn’t gone away. Anesthesiology. 2016;124(6):1404-1407. doi: 10.1097/ALN.0000000000001125.
3. Westerhuis B, Bietz MG, Lindemann J. Acute epiglottitis in adults: an under-recognized and life-threatening condition. S D Med. 2013;66(8):309-311, 313.
4. Al-Qudah M, Shetty S, Alomari M, Alqdah M. Acute adult supraglottitis: Current management and treatment. South Med J. 2010;103(8):800-804. doi: 10.1097/SMJ.0b013e3181e538d8.
5. Verbruggen K, Halewyck S, Deron P, Foulon I, Gordts F. Epiglottitis and related complications in adults. Case reports and review of the literature. B-ENT. 2012;8(2):143-148.
6. Mayo-Smith MF, Spinale JW, Donskey CJ, Yukawa M, Li RH, Schiffman FJ. Acute epiglottitis. An 18-year experience in Rhode Island. Chest. 1995;108(6):1640-1647.
7. Bizaki AJ, Numminen J, Vasama JP, Laranne J, Rautiainen M. Acute supraglottitis in adults in Finland: review and analysis of 308 cases. Laryngoscope. 2011;121(10):2107-2113. doi: 10.1002/lary.22147.
8. Charles R, Fadden M, Brook J. Acute epiglottitis. BMJ. 2013;347:f5235. doi: 10.1136/bmj.f5235.
9. Ng HL, Sin LM, Li MF, Que TL, Anandaciva S. Acute epiglottitis in adults: a retrospective review of 106 patients in Hong Kong. Emerg Med J. 2008;25(5):253-255. doi: 10.1136/emj.2007.050153.
10. Solomon P, Weisbrod M, Irish JC, Gullane PJ. Adult epiglottitis: the Toronto Hospital experience. J Otolaryngol. 1998;27(6):332-336.
11. Schumaker HM, Doris PE, Birnbaum G. Radiographic parameters in adult epiglottitis. Ann Emerg Med. 1984;13(8):588-590.
12. Ko DR, Chung YE, Park I, et al. Use of bedside sonography for diagnosing acute epiglottitis in the emergency department: a preliminary study. J Ultrasound Med. 2012;31(1):19-22.
13. Berger G, Landau T, Berger S, Finkelstein Y, Bernheim J, Ophir D. The rising incidence of adult acute epiglottitis and epiglottic abscess. Am J Otolaryngol. 2003;24(6):374-383.
1. Glynn F, Fenton JE. Diagnosis and management of supraglottitis (epiglottitis). Curr Infect Dis Rep. 2008;10(3):200-204.
2. Lichtor JL, Roche Rodriguez M, Aaronson NL, Spock T, Goodman TR, Baum ED. Epiglottitis: It hasn’t gone away. Anesthesiology. 2016;124(6):1404-1407. doi: 10.1097/ALN.0000000000001125.
3. Westerhuis B, Bietz MG, Lindemann J. Acute epiglottitis in adults: an under-recognized and life-threatening condition. S D Med. 2013;66(8):309-311, 313.
4. Al-Qudah M, Shetty S, Alomari M, Alqdah M. Acute adult supraglottitis: Current management and treatment. South Med J. 2010;103(8):800-804. doi: 10.1097/SMJ.0b013e3181e538d8.
5. Verbruggen K, Halewyck S, Deron P, Foulon I, Gordts F. Epiglottitis and related complications in adults. Case reports and review of the literature. B-ENT. 2012;8(2):143-148.
6. Mayo-Smith MF, Spinale JW, Donskey CJ, Yukawa M, Li RH, Schiffman FJ. Acute epiglottitis. An 18-year experience in Rhode Island. Chest. 1995;108(6):1640-1647.
7. Bizaki AJ, Numminen J, Vasama JP, Laranne J, Rautiainen M. Acute supraglottitis in adults in Finland: review and analysis of 308 cases. Laryngoscope. 2011;121(10):2107-2113. doi: 10.1002/lary.22147.
8. Charles R, Fadden M, Brook J. Acute epiglottitis. BMJ. 2013;347:f5235. doi: 10.1136/bmj.f5235.
9. Ng HL, Sin LM, Li MF, Que TL, Anandaciva S. Acute epiglottitis in adults: a retrospective review of 106 patients in Hong Kong. Emerg Med J. 2008;25(5):253-255. doi: 10.1136/emj.2007.050153.
10. Solomon P, Weisbrod M, Irish JC, Gullane PJ. Adult epiglottitis: the Toronto Hospital experience. J Otolaryngol. 1998;27(6):332-336.
11. Schumaker HM, Doris PE, Birnbaum G. Radiographic parameters in adult epiglottitis. Ann Emerg Med. 1984;13(8):588-590.
12. Ko DR, Chung YE, Park I, et al. Use of bedside sonography for diagnosing acute epiglottitis in the emergency department: a preliminary study. J Ultrasound Med. 2012;31(1):19-22.
13. Berger G, Landau T, Berger S, Finkelstein Y, Bernheim J, Ophir D. The rising incidence of adult acute epiglottitis and epiglottic abscess. Am J Otolaryngol. 2003;24(6):374-383.
Nontraumatic Splenic Rupture
Case
A 25-year-old college student presented to the ED following a near-syncopal episode. The patient stated he had felt lightheaded and had fallen to his knees immediately after taking a shower earlier that morning, but did not experience any loss of consciousness or injury. He denied a history of syncope or any recent trauma or fatigue. A review of the patient’s systems was negative. His medical history was remarkable for irritable bowel syndrome; he had no surgical history. Regarding his social history, he admitted to occasional alcohol use but denied any tobacco or illicit drug use. He was not on any current prescription or over-the-counter medications and denied any allergies.
The patient’s initial vital signs at presentation were: blood pressure, 112/58 mm Hg; heart rate, 86 beats/min; temperature, 97.9°F; and respiratory rate, 18 breaths/min. Oxygen saturation was 100% on room air. The patient reported pain in his left shoulder, epigastric region, and right flank. He rated his pain as a “4” on a 0-to-10 pain scale.
On physical examination, the patient was alert and oriented; he was thin and had mild pallor. His head, eyes, ears, nose, and throat; cardiac; pulmonary; and neurological examinations were normal. The abdominal examination revealed a soft, minimally tender epigastrium but with normal bowel sounds. Initial laboratory studies were remarkable for low hemoglobin (Hgb; 12.0 g/dL) and elevated aspartate transaminase (105 U/L), alanine aminotransferase (168 U/L), total bilirubin (1.6 mg/dL), and glucose (179 mg/dL) levels. The patient’s troponin I and lipase levels were within normal range. An electrocardiogram was unremarkable.
Given the patient’s elevated hepatic enzymes, right upper quadrant ultrasound was obtained, which demonstrated a normal gallbladder, a moderate amount of complicated free fluid (with hyper-echoic densities suggestive of coagulated blood) in all four quadrants, and splenomegaly measuring 13.7 cm (Figure 1a and 1b). 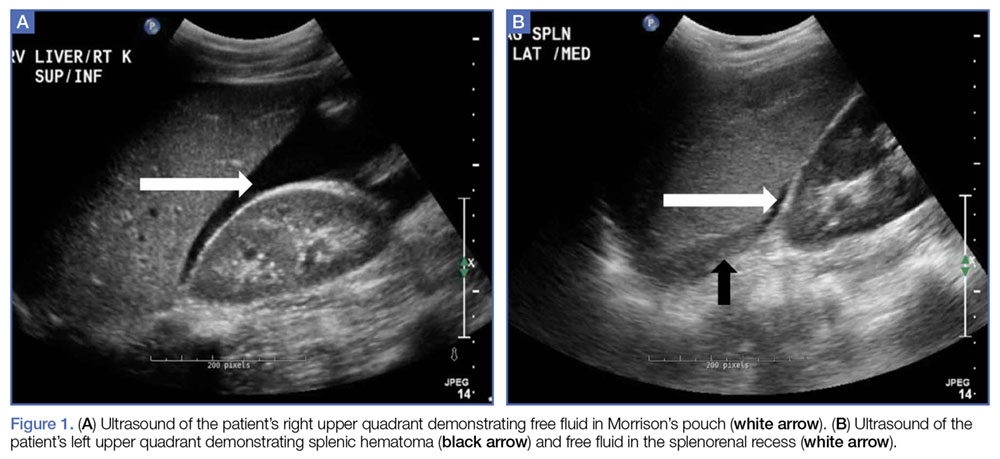
The patient’s status, including his vital signs, remained stable throughout his entire ED course. However, repeat laboratory studies taken 4 hours aft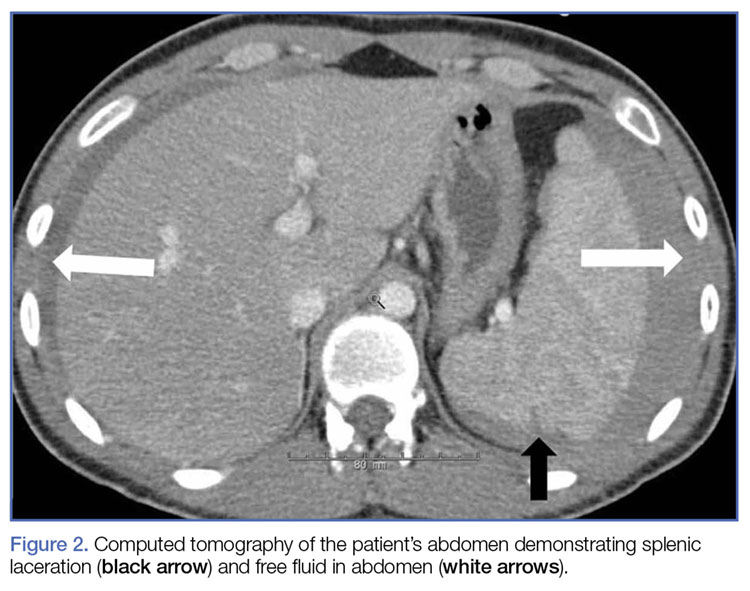
Positive:
- Epstein-Barr virus (EBV)
- Viral capsid antigen (VCA) immunoglobulin G
- VCA immunoglobulin M
Negative:
- Mononuclear spot test
- Human immunodeficiency virus
- Hepatitis B and C
- Antinuclear antibodies
- Venereal disease research laboratory test
The rest of the patient’s recovery was uneventful, and he was discharged home in stable condition on hospital day 3.
Discussion
Although the spleen is the most common intra-abdominal organ that can rupture with blunt abdominal trauma, splenic rupture in the absence of trauma is very rare. Nontraumatic splenic rupture (NSR) has been associated with pathological and nonpathological spleens.1,2 A systemic review of NSRs showed that 7% of the 845 patients in the review had completely normal spleens; the remaining 93% had some form of splenic pathology.1
Etiology
The top three causes of splenic enlargement associated with NSR include hematologic malignancies, viral infections, and inflammation.1,2 Although viruses, such as EBV and cytomegalovirus, represent almost 15% of the pathological causes of NSR, it is not uncommon for a patient to have multiple pathological processes present.1 Our patient’s enlarged spleen was due to acute infectious mononucleosis.
Signs and Symptoms
Diagnosing NSR can be challenging and it is often missed or discovered incidentally during evaluation (as was initially the case with our patient).3 Several signs and symptoms present in our patient were red herrings that warranted closer analysis. The patient’s complaint of left shoulder pain suggested left hemidiaphragm irritation from the NSR. Furthermore, our patient’s near-syncopal episode was possibly due to acute vagal simulation from the initial contact of blood with the peritoneal cavity.4 The maximal vagal stimulus was likely transient, as our patient returned to baseline after a brief near-syncopal episode.
As illustrated in our case, though tachycardia is common in splenic rupture, not all patients present with this sign. The absence of tachycardia in our patient can be explained by the elevation of his baseline enteric vagal tone due to the continued presence of blood in the peritoneum.5 There are also other factors associated with the absence of tachycardia. For example, a well-conditioned athlete presenting with states of shock due to splenic rupture may not show signs of tachycardia.6
San Francisco Syncope Rule
The San Francisco Syncope Rule (SFSR) is a clinical decision-making risk-stratification tool used to determine outcomes and disposition of ED patients presenting with syncope.7 It is important to note that if we had used a straightforward application of the SFSR upon our patient’s initial presentation, the results would have been negative, suggesting he was not at risk for short-term serious outcomes.7
Imaging Studies
As demonstrated in our patient, a quick point-of-care (POC) bedside ultrasound scan can reveal the presence of free fluid in the abdomen to help with the diagnosis. On ultrasound, the presence of free fluid in the right upper quadrant is more commonly found in the hepatorenal recess, whereas in the left upper quadrant free fluid is seen sub-diaphragmatic/suprasplenic first before fluid is seen in the splenorenal recess. Bedside ultrasound can accurately detect as little as 100 mL of free fluid in the abdominal cavity, with a 90% sensitivity and 99% specificity.8
An ultrasound is highly sensitive as a preliminary screening tool to identify the presence of free intraperitoneal fluid and has some limited utility in identifying any disruption in the splenic echotexture that may suggest a laceration or hematoma. Ultrasound, however, has poor specificity in identifying solid organ injuries.9
Computed tomography scanning is the imaging modality of choice for assessing splenic injuries, and should be obtained to confirm the presence of a solid organ injury, as well as to grade the degree of injury and thereby determine the need for surgical intervention.10 It is worth noting that in a hemodynamically unstable patient, exploratory laparotomy may be embarked upon without a CT scan and positive free fluid on ultrasound.
Splenic Injury Scale
Splenic injury is classified on a scale of 1 (mild injury) to 5 (severe injury) (Table).11 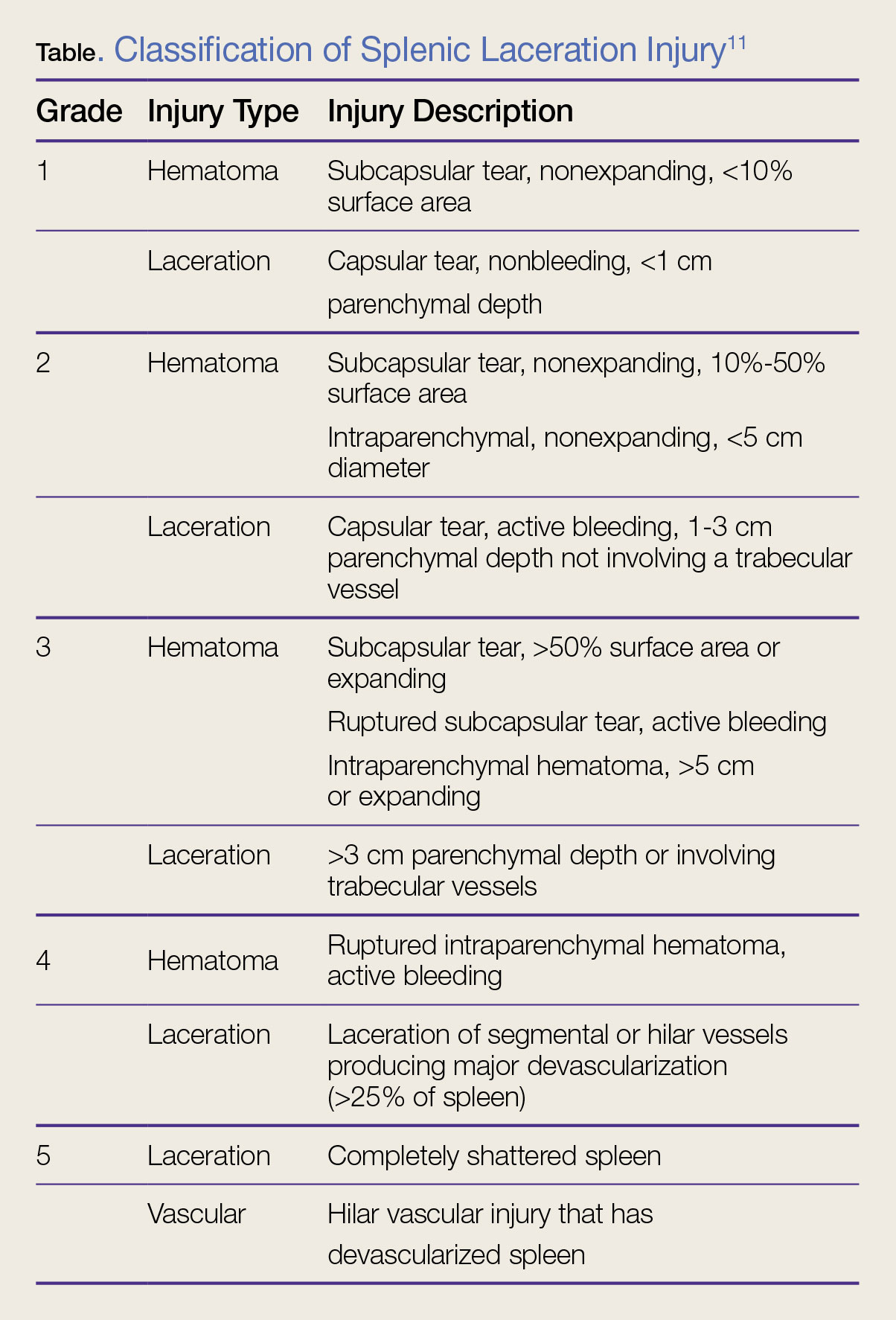
Conclusion
This case illustrates an uncommon presentation of NSR and underscores the importance of considering NSR in the differential diagnoses of patients presenting with abdominal pain—a sign with such a broad differential that NSR could easily be missed during evaluation. Based on its high sensitivity and specificity in detecting the presence of free fluid in the abdominal cavity, POC ultrasound imaging should be used to evaluate patients presenting with abdominal pain and syncopal or near-syncopal symptoms. This case further demonstrates that the absence of tachycardia or signs of shock should not rule out NSR.
1. Renzulli P, Hostettler A, Schoepfer AM, Gloor B, Candinas D. Systematic review of atraumatic splenic rupture. Br J Surg. 2009;96(10):1114-1121. doi: 10.1002/bjs.6737.
2. Aubrey-Bassler FK, Sowers N. 613 cases of splenic rupture without risk factors or previously diagnosed disease: a systematic review. BMC Emerg Med. 2012;12:11. doi: 10.1186/1471-227X-12-11.
3. Schattner A, Meital A, Mavor E. Red-flag syncope: spontaneous splenic rupture. Am J Med. 2014;127(6):501-502. doi: 10.1016/j.amjmed.2014.02.024.
4. Moya A, Sutton R, Ammirati F, et al; Task Force for the Diagnosis and Management of Syncope; European Society of Cardiology (ESC); European Heart Rhythm Association (EHRA); Heart Failure Association (HFA); Heart Rhythm Society (HRS). Guidelines for the diagnosis and management of syncope (version 2009). Eur Heart J. 2009;30(21):2631-2671. doi: 10.1093/eurheartj/ehp298.
5. Rana MS, Khalid U, Law S. Paradoxical bradycardia in a patient with haemorrhagic shock secondary to blunt abdominal trauma. BMJ Case Rep. 2010;2010. doi: 10.1136/bcr.04.2010.2872.
6. Kiss O, Sydó N, Vargha P, et al. Prevalence of physiological and pathological electrocardiographic findings in Hungarian athletes. Acta Physiol Hung. 2015;102(2):228-237. doi: 10.1556/036.102.2015.2.13.
7. Quinn JV, Stiell IG, McDermott DA, Sellers KL, Kohn MA, Wells GA. Derivation of the San Francisco Syncope Rule to predict patients with short-term serious outcomes. Ann Emerg Med. 2004;43(2):224-232.
8. Ma OJ, Mateer JR, Ogata M, Kefer MP, Wittmann D, Aprahamian C. Prospective analysis of a rapid trauma ultrasound examination performed by emergency physicians. J Trauma. 1995;38(6):879-885.
9. Kendall JL, Faragher J, Hewitt GJ, Burcham G, Haukoos JS. Emergency Department Ultrasound Is not a Sensitive Detector of Solid Organ Injury. West J Emerg Med. 2009;10(1):1-5.
10. Hassan R, Abd Aziz A, Md Ralib AR, Saat A. Computed tomography of blunt spleen injury: a pictorial review. Malays J Med Sci. 2011;18(1):60-67.
11. Moore EE, Cogbill TH, Jurkovich GJ, Shackford SR, Malangoni MA, Champion HR. Organ injury scaling: spleen and liver (1994 revision). J Trauma. 1995;38(3):323-324.
12. Cirocchi R, Boselli C, Corsi A, et al. Is non-operative management safe and effective for all splenic blunt trauma? A systematic review. Crit Care. 2013;17(5):R185. doi: 10.1186/cc12868.
Case
A 25-year-old college student presented to the ED following a near-syncopal episode. The patient stated he had felt lightheaded and had fallen to his knees immediately after taking a shower earlier that morning, but did not experience any loss of consciousness or injury. He denied a history of syncope or any recent trauma or fatigue. A review of the patient’s systems was negative. His medical history was remarkable for irritable bowel syndrome; he had no surgical history. Regarding his social history, he admitted to occasional alcohol use but denied any tobacco or illicit drug use. He was not on any current prescription or over-the-counter medications and denied any allergies.
The patient’s initial vital signs at presentation were: blood pressure, 112/58 mm Hg; heart rate, 86 beats/min; temperature, 97.9°F; and respiratory rate, 18 breaths/min. Oxygen saturation was 100% on room air. The patient reported pain in his left shoulder, epigastric region, and right flank. He rated his pain as a “4” on a 0-to-10 pain scale.
On physical examination, the patient was alert and oriented; he was thin and had mild pallor. His head, eyes, ears, nose, and throat; cardiac; pulmonary; and neurological examinations were normal. The abdominal examination revealed a soft, minimally tender epigastrium but with normal bowel sounds. Initial laboratory studies were remarkable for low hemoglobin (Hgb; 12.0 g/dL) and elevated aspartate transaminase (105 U/L), alanine aminotransferase (168 U/L), total bilirubin (1.6 mg/dL), and glucose (179 mg/dL) levels. The patient’s troponin I and lipase levels were within normal range. An electrocardiogram was unremarkable.
Given the patient’s elevated hepatic enzymes, right upper quadrant ultrasound was obtained, which demonstrated a normal gallbladder, a moderate amount of complicated free fluid (with hyper-echoic densities suggestive of coagulated blood) in all four quadrants, and splenomegaly measuring 13.7 cm (Figure 1a and 1b).
The patient’s status, including his vital signs, remained stable throughout his entire ED course. However, repeat laboratory studies taken 4 hours aft
Positive:
- Epstein-Barr virus (EBV)
- Viral capsid antigen (VCA) immunoglobulin G
- VCA immunoglobulin M
Negative:
- Mononuclear spot test
- Human immunodeficiency virus
- Hepatitis B and C
- Antinuclear antibodies
- Venereal disease research laboratory test
The rest of the patient’s recovery was uneventful, and he was discharged home in stable condition on hospital day 3.
Discussion
Although the spleen is the most common intra-abdominal organ that can rupture with blunt abdominal trauma, splenic rupture in the absence of trauma is very rare. Nontraumatic splenic rupture (NSR) has been associated with pathological and nonpathological spleens.1,2 A systemic review of NSRs showed that 7% of the 845 patients in the review had completely normal spleens; the remaining 93% had some form of splenic pathology.1
Etiology
The top three causes of splenic enlargement associated with NSR include hematologic malignancies, viral infections, and inflammation.1,2 Although viruses, such as EBV and cytomegalovirus, represent almost 15% of the pathological causes of NSR, it is not uncommon for a patient to have multiple pathological processes present.1 Our patient’s enlarged spleen was due to acute infectious mononucleosis.
Signs and Symptoms
Diagnosing NSR can be challenging and it is often missed or discovered incidentally during evaluation (as was initially the case with our patient).3 Several signs and symptoms present in our patient were red herrings that warranted closer analysis. The patient’s complaint of left shoulder pain suggested left hemidiaphragm irritation from the NSR. Furthermore, our patient’s near-syncopal episode was possibly due to acute vagal simulation from the initial contact of blood with the peritoneal cavity.4 The maximal vagal stimulus was likely transient, as our patient returned to baseline after a brief near-syncopal episode.
As illustrated in our case, though tachycardia is common in splenic rupture, not all patients present with this sign. The absence of tachycardia in our patient can be explained by the elevation of his baseline enteric vagal tone due to the continued presence of blood in the peritoneum.5 There are also other factors associated with the absence of tachycardia. For example, a well-conditioned athlete presenting with states of shock due to splenic rupture may not show signs of tachycardia.6
San Francisco Syncope Rule
The San Francisco Syncope Rule (SFSR) is a clinical decision-making risk-stratification tool used to determine outcomes and disposition of ED patients presenting with syncope.7 It is important to note that if we had used a straightforward application of the SFSR upon our patient’s initial presentation, the results would have been negative, suggesting he was not at risk for short-term serious outcomes.7
Imaging Studies
As demonstrated in our patient, a quick point-of-care (POC) bedside ultrasound scan can reveal the presence of free fluid in the abdomen to help with the diagnosis. On ultrasound, the presence of free fluid in the right upper quadrant is more commonly found in the hepatorenal recess, whereas in the left upper quadrant free fluid is seen sub-diaphragmatic/suprasplenic first before fluid is seen in the splenorenal recess. Bedside ultrasound can accurately detect as little as 100 mL of free fluid in the abdominal cavity, with a 90% sensitivity and 99% specificity.8
An ultrasound is highly sensitive as a preliminary screening tool to identify the presence of free intraperitoneal fluid and has some limited utility in identifying any disruption in the splenic echotexture that may suggest a laceration or hematoma. Ultrasound, however, has poor specificity in identifying solid organ injuries.9
Computed tomography scanning is the imaging modality of choice for assessing splenic injuries, and should be obtained to confirm the presence of a solid organ injury, as well as to grade the degree of injury and thereby determine the need for surgical intervention.10 It is worth noting that in a hemodynamically unstable patient, exploratory laparotomy may be embarked upon without a CT scan and positive free fluid on ultrasound.
Splenic Injury Scale
Splenic injury is classified on a scale of 1 (mild injury) to 5 (severe injury) (Table).11
Conclusion
This case illustrates an uncommon presentation of NSR and underscores the importance of considering NSR in the differential diagnoses of patients presenting with abdominal pain—a sign with such a broad differential that NSR could easily be missed during evaluation. Based on its high sensitivity and specificity in detecting the presence of free fluid in the abdominal cavity, POC ultrasound imaging should be used to evaluate patients presenting with abdominal pain and syncopal or near-syncopal symptoms. This case further demonstrates that the absence of tachycardia or signs of shock should not rule out NSR.
Case
A 25-year-old college student presented to the ED following a near-syncopal episode. The patient stated he had felt lightheaded and had fallen to his knees immediately after taking a shower earlier that morning, but did not experience any loss of consciousness or injury. He denied a history of syncope or any recent trauma or fatigue. A review of the patient’s systems was negative. His medical history was remarkable for irritable bowel syndrome; he had no surgical history. Regarding his social history, he admitted to occasional alcohol use but denied any tobacco or illicit drug use. He was not on any current prescription or over-the-counter medications and denied any allergies.
The patient’s initial vital signs at presentation were: blood pressure, 112/58 mm Hg; heart rate, 86 beats/min; temperature, 97.9°F; and respiratory rate, 18 breaths/min. Oxygen saturation was 100% on room air. The patient reported pain in his left shoulder, epigastric region, and right flank. He rated his pain as a “4” on a 0-to-10 pain scale.
On physical examination, the patient was alert and oriented; he was thin and had mild pallor. His head, eyes, ears, nose, and throat; cardiac; pulmonary; and neurological examinations were normal. The abdominal examination revealed a soft, minimally tender epigastrium but with normal bowel sounds. Initial laboratory studies were remarkable for low hemoglobin (Hgb; 12.0 g/dL) and elevated aspartate transaminase (105 U/L), alanine aminotransferase (168 U/L), total bilirubin (1.6 mg/dL), and glucose (179 mg/dL) levels. The patient’s troponin I and lipase levels were within normal range. An electrocardiogram was unremarkable.
Given the patient’s elevated hepatic enzymes, right upper quadrant ultrasound was obtained, which demonstrated a normal gallbladder, a moderate amount of complicated free fluid (with hyper-echoic densities suggestive of coagulated blood) in all four quadrants, and splenomegaly measuring 13.7 cm (Figure 1a and 1b).
The patient’s status, including his vital signs, remained stable throughout his entire ED course. However, repeat laboratory studies taken 4 hours aft
Positive:
- Epstein-Barr virus (EBV)
- Viral capsid antigen (VCA) immunoglobulin G
- VCA immunoglobulin M
Negative:
- Mononuclear spot test
- Human immunodeficiency virus
- Hepatitis B and C
- Antinuclear antibodies
- Venereal disease research laboratory test
The rest of the patient’s recovery was uneventful, and he was discharged home in stable condition on hospital day 3.
Discussion
Although the spleen is the most common intra-abdominal organ that can rupture with blunt abdominal trauma, splenic rupture in the absence of trauma is very rare. Nontraumatic splenic rupture (NSR) has been associated with pathological and nonpathological spleens.1,2 A systemic review of NSRs showed that 7% of the 845 patients in the review had completely normal spleens; the remaining 93% had some form of splenic pathology.1
Etiology
The top three causes of splenic enlargement associated with NSR include hematologic malignancies, viral infections, and inflammation.1,2 Although viruses, such as EBV and cytomegalovirus, represent almost 15% of the pathological causes of NSR, it is not uncommon for a patient to have multiple pathological processes present.1 Our patient’s enlarged spleen was due to acute infectious mononucleosis.
Signs and Symptoms
Diagnosing NSR can be challenging and it is often missed or discovered incidentally during evaluation (as was initially the case with our patient).3 Several signs and symptoms present in our patient were red herrings that warranted closer analysis. The patient’s complaint of left shoulder pain suggested left hemidiaphragm irritation from the NSR. Furthermore, our patient’s near-syncopal episode was possibly due to acute vagal simulation from the initial contact of blood with the peritoneal cavity.4 The maximal vagal stimulus was likely transient, as our patient returned to baseline after a brief near-syncopal episode.
As illustrated in our case, though tachycardia is common in splenic rupture, not all patients present with this sign. The absence of tachycardia in our patient can be explained by the elevation of his baseline enteric vagal tone due to the continued presence of blood in the peritoneum.5 There are also other factors associated with the absence of tachycardia. For example, a well-conditioned athlete presenting with states of shock due to splenic rupture may not show signs of tachycardia.6
San Francisco Syncope Rule
The San Francisco Syncope Rule (SFSR) is a clinical decision-making risk-stratification tool used to determine outcomes and disposition of ED patients presenting with syncope.7 It is important to note that if we had used a straightforward application of the SFSR upon our patient’s initial presentation, the results would have been negative, suggesting he was not at risk for short-term serious outcomes.7
Imaging Studies
As demonstrated in our patient, a quick point-of-care (POC) bedside ultrasound scan can reveal the presence of free fluid in the abdomen to help with the diagnosis. On ultrasound, the presence of free fluid in the right upper quadrant is more commonly found in the hepatorenal recess, whereas in the left upper quadrant free fluid is seen sub-diaphragmatic/suprasplenic first before fluid is seen in the splenorenal recess. Bedside ultrasound can accurately detect as little as 100 mL of free fluid in the abdominal cavity, with a 90% sensitivity and 99% specificity.8
An ultrasound is highly sensitive as a preliminary screening tool to identify the presence of free intraperitoneal fluid and has some limited utility in identifying any disruption in the splenic echotexture that may suggest a laceration or hematoma. Ultrasound, however, has poor specificity in identifying solid organ injuries.9
Computed tomography scanning is the imaging modality of choice for assessing splenic injuries, and should be obtained to confirm the presence of a solid organ injury, as well as to grade the degree of injury and thereby determine the need for surgical intervention.10 It is worth noting that in a hemodynamically unstable patient, exploratory laparotomy may be embarked upon without a CT scan and positive free fluid on ultrasound.
Splenic Injury Scale
Splenic injury is classified on a scale of 1 (mild injury) to 5 (severe injury) (Table).11
Conclusion
This case illustrates an uncommon presentation of NSR and underscores the importance of considering NSR in the differential diagnoses of patients presenting with abdominal pain—a sign with such a broad differential that NSR could easily be missed during evaluation. Based on its high sensitivity and specificity in detecting the presence of free fluid in the abdominal cavity, POC ultrasound imaging should be used to evaluate patients presenting with abdominal pain and syncopal or near-syncopal symptoms. This case further demonstrates that the absence of tachycardia or signs of shock should not rule out NSR.
1. Renzulli P, Hostettler A, Schoepfer AM, Gloor B, Candinas D. Systematic review of atraumatic splenic rupture. Br J Surg. 2009;96(10):1114-1121. doi: 10.1002/bjs.6737.
2. Aubrey-Bassler FK, Sowers N. 613 cases of splenic rupture without risk factors or previously diagnosed disease: a systematic review. BMC Emerg Med. 2012;12:11. doi: 10.1186/1471-227X-12-11.
3. Schattner A, Meital A, Mavor E. Red-flag syncope: spontaneous splenic rupture. Am J Med. 2014;127(6):501-502. doi: 10.1016/j.amjmed.2014.02.024.
4. Moya A, Sutton R, Ammirati F, et al; Task Force for the Diagnosis and Management of Syncope; European Society of Cardiology (ESC); European Heart Rhythm Association (EHRA); Heart Failure Association (HFA); Heart Rhythm Society (HRS). Guidelines for the diagnosis and management of syncope (version 2009). Eur Heart J. 2009;30(21):2631-2671. doi: 10.1093/eurheartj/ehp298.
5. Rana MS, Khalid U, Law S. Paradoxical bradycardia in a patient with haemorrhagic shock secondary to blunt abdominal trauma. BMJ Case Rep. 2010;2010. doi: 10.1136/bcr.04.2010.2872.
6. Kiss O, Sydó N, Vargha P, et al. Prevalence of physiological and pathological electrocardiographic findings in Hungarian athletes. Acta Physiol Hung. 2015;102(2):228-237. doi: 10.1556/036.102.2015.2.13.
7. Quinn JV, Stiell IG, McDermott DA, Sellers KL, Kohn MA, Wells GA. Derivation of the San Francisco Syncope Rule to predict patients with short-term serious outcomes. Ann Emerg Med. 2004;43(2):224-232.
8. Ma OJ, Mateer JR, Ogata M, Kefer MP, Wittmann D, Aprahamian C. Prospective analysis of a rapid trauma ultrasound examination performed by emergency physicians. J Trauma. 1995;38(6):879-885.
9. Kendall JL, Faragher J, Hewitt GJ, Burcham G, Haukoos JS. Emergency Department Ultrasound Is not a Sensitive Detector of Solid Organ Injury. West J Emerg Med. 2009;10(1):1-5.
10. Hassan R, Abd Aziz A, Md Ralib AR, Saat A. Computed tomography of blunt spleen injury: a pictorial review. Malays J Med Sci. 2011;18(1):60-67.
11. Moore EE, Cogbill TH, Jurkovich GJ, Shackford SR, Malangoni MA, Champion HR. Organ injury scaling: spleen and liver (1994 revision). J Trauma. 1995;38(3):323-324.
12. Cirocchi R, Boselli C, Corsi A, et al. Is non-operative management safe and effective for all splenic blunt trauma? A systematic review. Crit Care. 2013;17(5):R185. doi: 10.1186/cc12868.
1. Renzulli P, Hostettler A, Schoepfer AM, Gloor B, Candinas D. Systematic review of atraumatic splenic rupture. Br J Surg. 2009;96(10):1114-1121. doi: 10.1002/bjs.6737.
2. Aubrey-Bassler FK, Sowers N. 613 cases of splenic rupture without risk factors or previously diagnosed disease: a systematic review. BMC Emerg Med. 2012;12:11. doi: 10.1186/1471-227X-12-11.
3. Schattner A, Meital A, Mavor E. Red-flag syncope: spontaneous splenic rupture. Am J Med. 2014;127(6):501-502. doi: 10.1016/j.amjmed.2014.02.024.
4. Moya A, Sutton R, Ammirati F, et al; Task Force for the Diagnosis and Management of Syncope; European Society of Cardiology (ESC); European Heart Rhythm Association (EHRA); Heart Failure Association (HFA); Heart Rhythm Society (HRS). Guidelines for the diagnosis and management of syncope (version 2009). Eur Heart J. 2009;30(21):2631-2671. doi: 10.1093/eurheartj/ehp298.
5. Rana MS, Khalid U, Law S. Paradoxical bradycardia in a patient with haemorrhagic shock secondary to blunt abdominal trauma. BMJ Case Rep. 2010;2010. doi: 10.1136/bcr.04.2010.2872.
6. Kiss O, Sydó N, Vargha P, et al. Prevalence of physiological and pathological electrocardiographic findings in Hungarian athletes. Acta Physiol Hung. 2015;102(2):228-237. doi: 10.1556/036.102.2015.2.13.
7. Quinn JV, Stiell IG, McDermott DA, Sellers KL, Kohn MA, Wells GA. Derivation of the San Francisco Syncope Rule to predict patients with short-term serious outcomes. Ann Emerg Med. 2004;43(2):224-232.
8. Ma OJ, Mateer JR, Ogata M, Kefer MP, Wittmann D, Aprahamian C. Prospective analysis of a rapid trauma ultrasound examination performed by emergency physicians. J Trauma. 1995;38(6):879-885.
9. Kendall JL, Faragher J, Hewitt GJ, Burcham G, Haukoos JS. Emergency Department Ultrasound Is not a Sensitive Detector of Solid Organ Injury. West J Emerg Med. 2009;10(1):1-5.
10. Hassan R, Abd Aziz A, Md Ralib AR, Saat A. Computed tomography of blunt spleen injury: a pictorial review. Malays J Med Sci. 2011;18(1):60-67.
11. Moore EE, Cogbill TH, Jurkovich GJ, Shackford SR, Malangoni MA, Champion HR. Organ injury scaling: spleen and liver (1994 revision). J Trauma. 1995;38(3):323-324.
12. Cirocchi R, Boselli C, Corsi A, et al. Is non-operative management safe and effective for all splenic blunt trauma? A systematic review. Crit Care. 2013;17(5):R185. doi: 10.1186/cc12868.
Malignant Transformation of an Aneurysmal Bone Cyst to Fibroblastic Osteosarcoma
Aneurysmal bone cysts (ABC) are expansile, hemorrhagic, non-neoplastic lesions that can be locally destructive1 and that can arise either de novo or secondary to another benign or malignant lesion.2 Although primary and secondary ABCs typically are benign, there are cases of malignant degeneration of primary ABCs, though the transformation arises almost exclusively in the context of prior radiation exposure.3-5 Malignant change without history of irradiation is rare; only 6 such cases have been reported.5-10 In 4 of these cases, the transformation was to osteosarcoma.5,8-10
Here we report on an ABC that degenerated into a fibroblastic osteosarcoma—the fifth such case in the medical literature. In addition to reviewing the earlier cases, we describe the radiologic and histologic underpinnings of this diagnosis and the insight that they provide into the pathogenesis of this rare process. Although the prevailing view is that ABCs are benign, it is important to know these lesions have the potential to undergo malignant transformation, even in the absence of prior radiation exposure. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
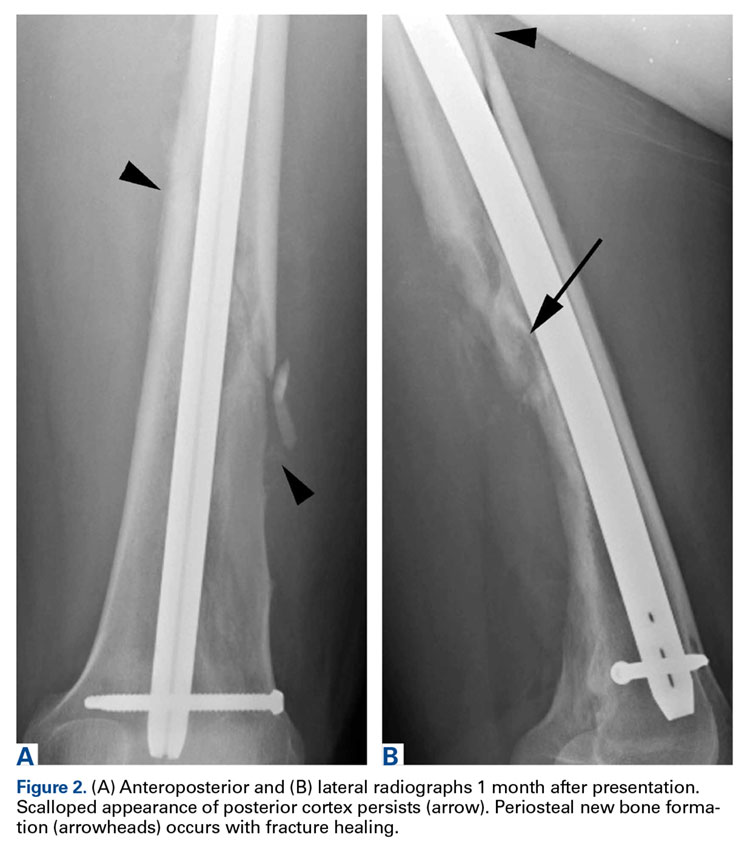
A healthy and previously asymptomatic 37-year-old man presented with thigh pain after a minor fall onto a couch. Radiographs showed a diaphyseal femoral pathologic fracture adjacent to a small but benign-appearing cystic lesion (Figures 1A, 1B). 
Two years later, the patient had a bicycle accident and, after 2 weeks of significantly increased thigh swelling, presented to the emergency department at the referring institution. Radiographs showed a lytic lesion in the femoral diaphysis that was highly suspicious for malignancy (Figures 3A, 3B). 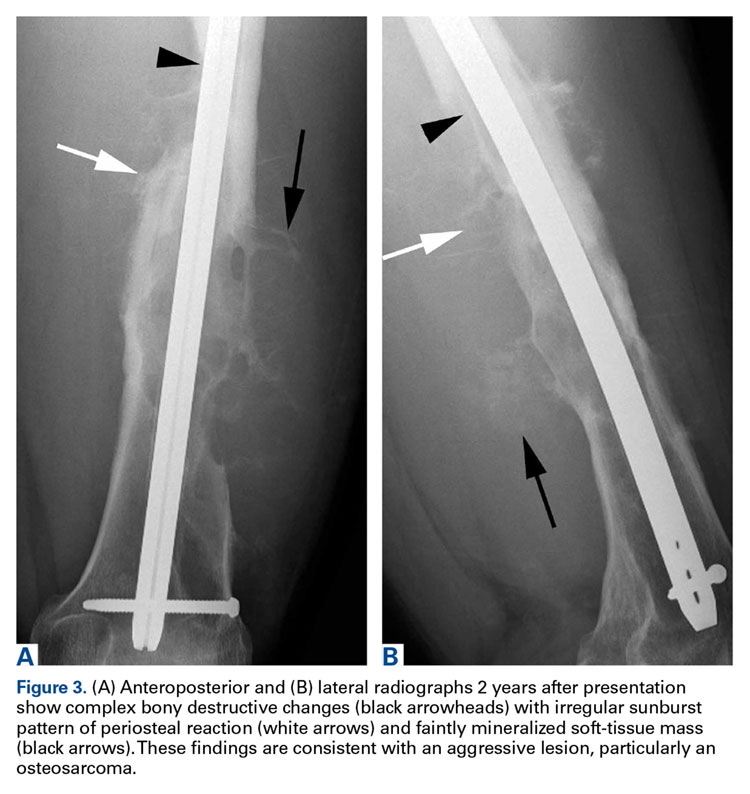
The initial biopsy specimens were evaluated at our institution and interpreted as being consistent with an ABC, with negative immunohistochemical staining for MDM2 (Figures 5A, 5B). 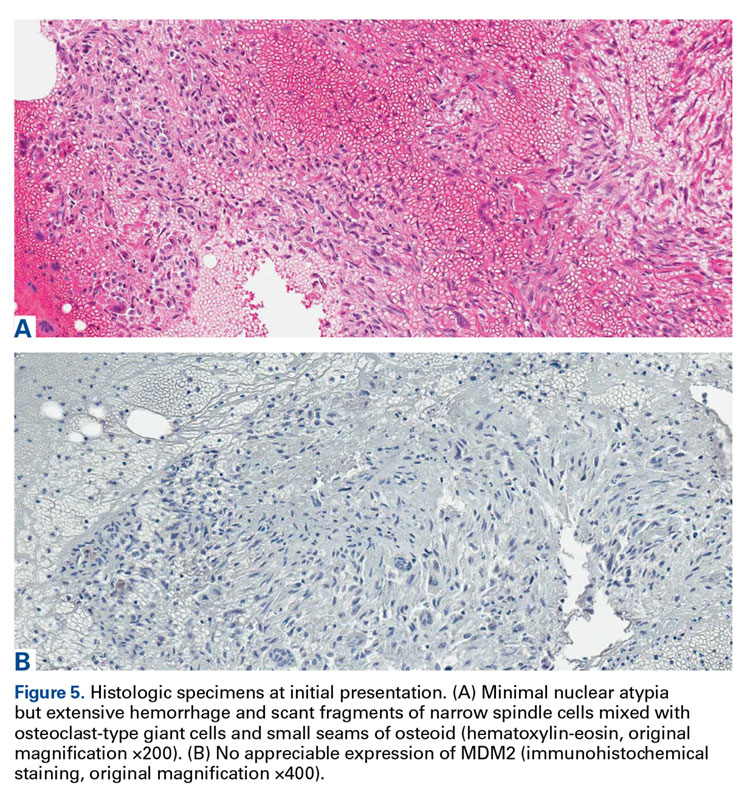
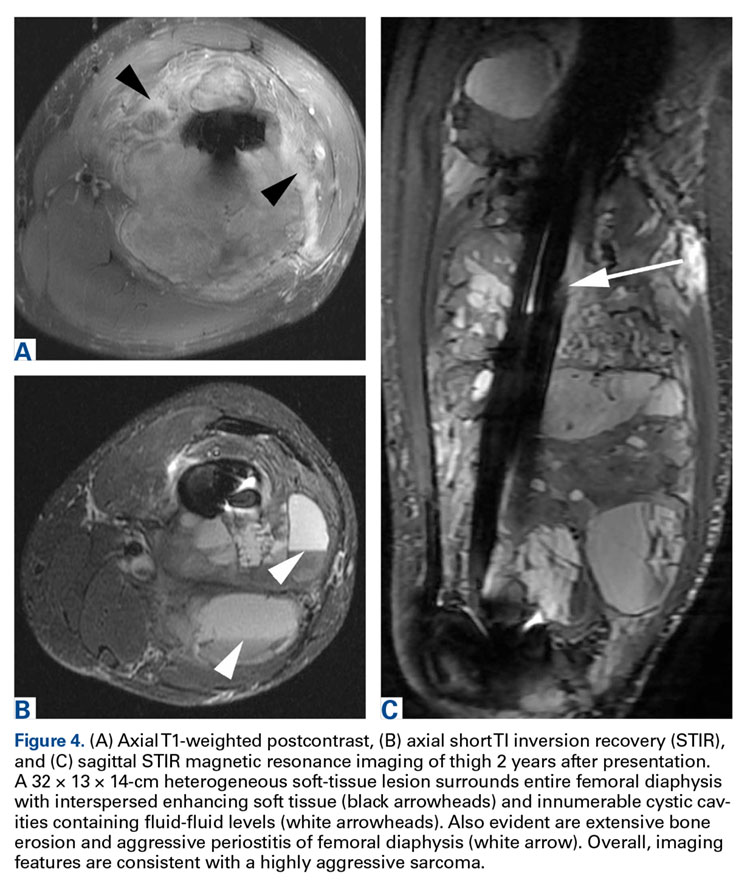
The patient underwent a 3-month course of neoadjuvant chemotherapy with cisplatin and doxorubicin. Interval-staging contrast-enhanced chest, abdomen, and pelvis computed tomography (CT) showed no evidence of metastatic disease. Preoperative MRI showed a significantly larger heterogeneous mass, now with neurovascular involvement, which precluded limb salvage. 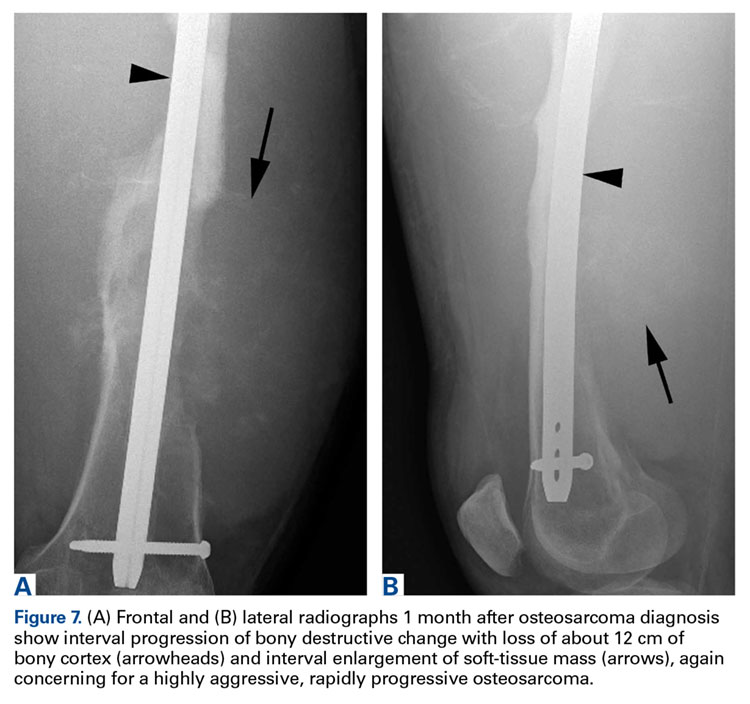
Discussion
Aneurysmal bone cysts are expansile, hemorrhagic, locally destructive lesions that generally develop within the first 3 decades of life. Ever since they were first described by Jaffe and Lichtenstein11 in 1942, the most widely accepted theory of their pathogenesis has been that they begin as a benign reactive vascular process.12 However, more recent molecular studies by Oliveira and colleagues13 and Panoutsakopoulos and colleagues14 have demonstrated a clonal neoplastic basis for primary ABCs related to cytogenetic upregulation of oncogenes USP6 and CDH11 after translocation of 17p13 and 16q22.
Given the clonal nature of these lesions, it is surprising that malignant transformation is so rare. Until now, there have been only 4 reports of an ABC undergoing malignant degeneration to osteosarcoma without prior radiation exposure. 
In this article, we have presented a fifth case of a primary ABC degenerating into an osteosarcoma, which in this instance was the fibroblastic subtype. This diagnosis was strongly supported by radiologic and pathologic evidence. From a radiologic perspective, imaging at initial presentation showed absolutely no suspicious features, and the same was true when follow-up radiographs were obtained, 1 month later. Although 1 month is short for a follow-up, the complete lack of radiographic changes would be highly unusual if in fact there had been a coexisting, undetected lesion as aggressive as the one that ultimately developed. Furthermore, imaging at second presentation, almost 2 years later, showed an extremely rapid evolution of findings over 1 month. Extrapolating back in time, we think this time course indicates the malignancy developed not long before its aggressive features were detected.
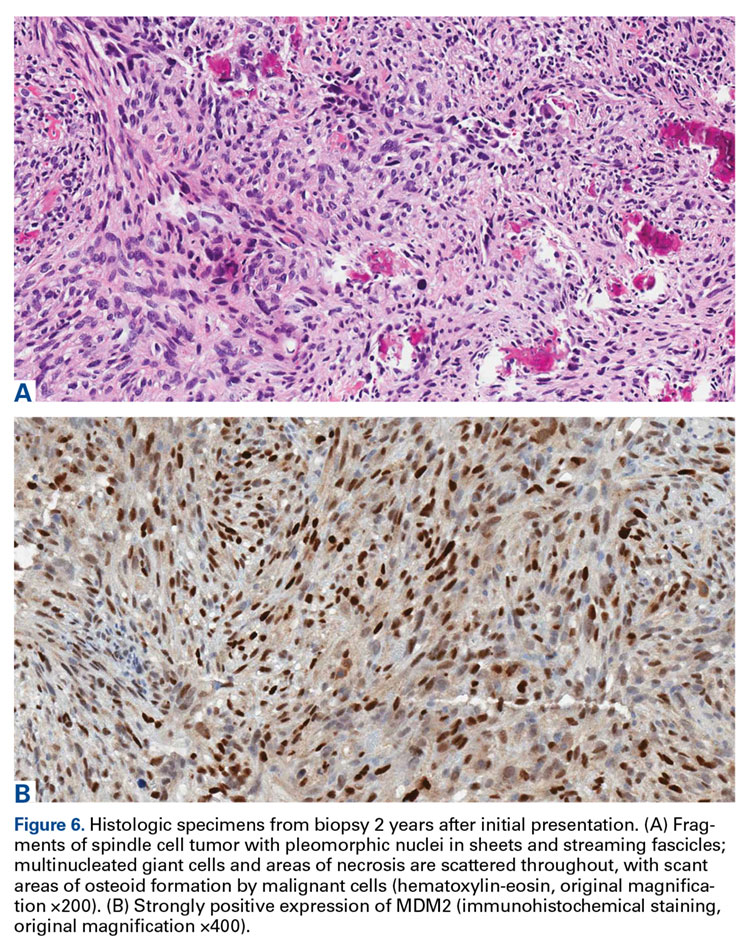
Genetic evidence suggests that most conventional high-grade osteosarcomas arise de novo from a mesenchymal precursor driven by multiple genetic aberrations. Less often, low-grade osteosarcomas progress to high-grade osteosarcomas. Amplification of 12q13-15 with resulting overexpression of MDM2 and CDK4 proteins is found in low-grade osteosarcomas and persists in examples that progress to higher-grade forms.15 Not only did review of our patient’s initial biopsy sample reveal no evidence of malignant features or abnormal mitotic activity, but the complete absence of MDM2 suggests not even a low-grade osteosarcoma was present at the time. By contrast, the second incisional biopsy specimen, 2 years later, showed markedly different histology and pronounced expression of MDM2 throughout the specimen. This finding suggests the histologically high-grade osteosarcoma did not arise de novo but rather secondarily from a low-grade osteosarcoma that had arisen from an ABC. Results of the final heterogeneous histology of the very large mass, which contained benign ABC areas indistinguishable from the initial biopsy sample, as well as areas of high-grade osteosarcoma, further support a multistep process of de-differentiation. Together, these findings are compelling evidence of malignant transformation of a primary ABC.
We acknowledge that the initial surgery performed at the outside hospital might have properly included frozen-section analysis of the biopsy material and that sampling error may have occurred during the index procedure—possibilities in the absence of complete lesional resection. In this case, however, the radiographic findings and the dominant histologic immunophenotype from medullary canal bone were both consistent with ABC and not osteosarcoma, lending support to malignant degeneration.
We have presented a fifth case of primary ABC degenerating into an osteosarcoma, now with immunohistochemical evidence supporting traditional radiologic and histologic evidence. Despite the rarity of the diagnosis, this case yields considerable insight into the pathogenetic mechanisms underlying malignant degeneration. Despite the widely held view that ABCs are benign, physicians caring for these patients must be aware that malignant transformation can occur.
Am J Orthop. 2016;45(6):E367-E372. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Donaldson WF. Aneurysmal bone cyst. J Bone Joint Surg Am. 1962;44:25-40.
2. Biesecker JL, Marcove RC, Huvos AG, Miké V. Aneurysmal bone cysts. A clinicopathologic study of 66 cases. Cancer. 1970;26(3):615-625.
3. Aho HJ, Aho AJ, Einola S. Aneurysmal bone cyst, a study of ultrastructure and malignant transformation. Virchows Arch A Pathol Anat Histol. 1982;395(2):169-179.
4. Tillman BP, Dahlin DC, Lipscomb PR, Stewart JR. Aneurysmal bone cyst: an analysis of ninety-five cases. Mayo Clin Proc. 1968;43(7):478-495.
5. Kyriakos M, Hardy D. Malignant transformation of aneurysmal bone cyst, with an analysis of the literature. Cancer. 1991;68(8):1770-1780.
6. Mei J, Gao YS, Wang SQ, Cai XS. Malignant transformation of aneurysmal bone cysts: a case report. Chin Med J (Engl). 2009;122(1):110-112.
7. Anract P, de Pinieux G, Jeanrot C, Babinet A, Forest M, Tomeno B. Malignant fibrous histiocytoma at the site of a previously treated aneurysmal bone cyst: a case report. J Bone Joint Surg Am. 2002;84(1):106-111.
8. Hsu CC, Wang JW, Huang CH, Chen WJ. Osteosarcoma at the site of a previously treated aneurysmal bone cyst. A case report. J Bone Joint Surg Am. 2005;87(2):395-398.
9. Wuisman P, Roessner A, Blasius S, Grünert J, Vestering T, Winkelmann W. High malignant surface osteosarcoma arising at the site of a previously treated aneurysmal bone cyst. J Cancer Res Clin Oncol. 1993;119(7):375-378.
10. Brindley GW, Greene JF Jr, Frankel LS. Case reports: malignant transformation of aneurysmal bone cysts. Clin Orthop Relat Res. 2005;(438):282-287.
11. Jaffe HL, Lichtenstein L. Solitary unicameral bone cyst: with emphasis on the roentgen picture, the pathologic appearance and the pathogenesis. Arch Surg. 1942;44:1004-1025.
12. Mirra JM. Bone Tumors: Clinical, Radiologic, and Pathologic Correlations. Philadelphia, PA: Lea & Febiger; 1989.
13. Oliveira AM, Chou MM, Perez-Atayde AR, Rosenberg AE. Aneurysmal bone cyst: a neoplasm driven by upregulation of the USP6 oncogene. J Clin Oncol. 2006;24(1):e1.
14. Panoutsakopoulos G, Pandis N, Kyriazoglou I, Gustafson P, Mertens F, Mandahl N. Recurrent t(16;17)(q22;p13) in aneurysmal bone cysts. Genes Chromosomes Cancer. 1999;26(3):265-266.
15. Dujardin F, Binh MB, Bouvier C, et al. MDM2 and CDK4 immunohistochemistry is a valuable tool in the differential diagnosis of low-grade osteosarcomas and other primary fibro-osseous lesions of the bone. Mod Pathol. 2001;24(5):624-637.
Aneurysmal bone cysts (ABC) are expansile, hemorrhagic, non-neoplastic lesions that can be locally destructive1 and that can arise either de novo or secondary to another benign or malignant lesion.2 Although primary and secondary ABCs typically are benign, there are cases of malignant degeneration of primary ABCs, though the transformation arises almost exclusively in the context of prior radiation exposure.3-5 Malignant change without history of irradiation is rare; only 6 such cases have been reported.5-10 In 4 of these cases, the transformation was to osteosarcoma.5,8-10
Here we report on an ABC that degenerated into a fibroblastic osteosarcoma—the fifth such case in the medical literature. In addition to reviewing the earlier cases, we describe the radiologic and histologic underpinnings of this diagnosis and the insight that they provide into the pathogenesis of this rare process. Although the prevailing view is that ABCs are benign, it is important to know these lesions have the potential to undergo malignant transformation, even in the absence of prior radiation exposure. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A healthy and previously asymptomatic 37-year-old man presented with thigh pain after a minor fall onto a couch. Radiographs showed a diaphyseal femoral pathologic fracture adjacent to a small but benign-appearing cystic lesion (Figures 1A, 1B).
Two years later, the patient had a bicycle accident and, after 2 weeks of significantly increased thigh swelling, presented to the emergency department at the referring institution. Radiographs showed a lytic lesion in the femoral diaphysis that was highly suspicious for malignancy (Figures 3A, 3B).
The initial biopsy specimens were evaluated at our institution and interpreted as being consistent with an ABC, with negative immunohistochemical staining for MDM2 (Figures 5A, 5B).
The patient underwent a 3-month course of neoadjuvant chemotherapy with cisplatin and doxorubicin. Interval-staging contrast-enhanced chest, abdomen, and pelvis computed tomography (CT) showed no evidence of metastatic disease. Preoperative MRI showed a significantly larger heterogeneous mass, now with neurovascular involvement, which precluded limb salvage.
Discussion
Aneurysmal bone cysts are expansile, hemorrhagic, locally destructive lesions that generally develop within the first 3 decades of life. Ever since they were first described by Jaffe and Lichtenstein11 in 1942, the most widely accepted theory of their pathogenesis has been that they begin as a benign reactive vascular process.12 However, more recent molecular studies by Oliveira and colleagues13 and Panoutsakopoulos and colleagues14 have demonstrated a clonal neoplastic basis for primary ABCs related to cytogenetic upregulation of oncogenes USP6 and CDH11 after translocation of 17p13 and 16q22.
Given the clonal nature of these lesions, it is surprising that malignant transformation is so rare. Until now, there have been only 4 reports of an ABC undergoing malignant degeneration to osteosarcoma without prior radiation exposure.
In this article, we have presented a fifth case of a primary ABC degenerating into an osteosarcoma, which in this instance was the fibroblastic subtype. This diagnosis was strongly supported by radiologic and pathologic evidence. From a radiologic perspective, imaging at initial presentation showed absolutely no suspicious features, and the same was true when follow-up radiographs were obtained, 1 month later. Although 1 month is short for a follow-up, the complete lack of radiographic changes would be highly unusual if in fact there had been a coexisting, undetected lesion as aggressive as the one that ultimately developed. Furthermore, imaging at second presentation, almost 2 years later, showed an extremely rapid evolution of findings over 1 month. Extrapolating back in time, we think this time course indicates the malignancy developed not long before its aggressive features were detected.
Genetic evidence suggests that most conventional high-grade osteosarcomas arise de novo from a mesenchymal precursor driven by multiple genetic aberrations. Less often, low-grade osteosarcomas progress to high-grade osteosarcomas. Amplification of 12q13-15 with resulting overexpression of MDM2 and CDK4 proteins is found in low-grade osteosarcomas and persists in examples that progress to higher-grade forms.15 Not only did review of our patient’s initial biopsy sample reveal no evidence of malignant features or abnormal mitotic activity, but the complete absence of MDM2 suggests not even a low-grade osteosarcoma was present at the time. By contrast, the second incisional biopsy specimen, 2 years later, showed markedly different histology and pronounced expression of MDM2 throughout the specimen. This finding suggests the histologically high-grade osteosarcoma did not arise de novo but rather secondarily from a low-grade osteosarcoma that had arisen from an ABC. Results of the final heterogeneous histology of the very large mass, which contained benign ABC areas indistinguishable from the initial biopsy sample, as well as areas of high-grade osteosarcoma, further support a multistep process of de-differentiation. Together, these findings are compelling evidence of malignant transformation of a primary ABC.
We acknowledge that the initial surgery performed at the outside hospital might have properly included frozen-section analysis of the biopsy material and that sampling error may have occurred during the index procedure—possibilities in the absence of complete lesional resection. In this case, however, the radiographic findings and the dominant histologic immunophenotype from medullary canal bone were both consistent with ABC and not osteosarcoma, lending support to malignant degeneration.
We have presented a fifth case of primary ABC degenerating into an osteosarcoma, now with immunohistochemical evidence supporting traditional radiologic and histologic evidence. Despite the rarity of the diagnosis, this case yields considerable insight into the pathogenetic mechanisms underlying malignant degeneration. Despite the widely held view that ABCs are benign, physicians caring for these patients must be aware that malignant transformation can occur.
Am J Orthop. 2016;45(6):E367-E372. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
Aneurysmal bone cysts (ABC) are expansile, hemorrhagic, non-neoplastic lesions that can be locally destructive1 and that can arise either de novo or secondary to another benign or malignant lesion.2 Although primary and secondary ABCs typically are benign, there are cases of malignant degeneration of primary ABCs, though the transformation arises almost exclusively in the context of prior radiation exposure.3-5 Malignant change without history of irradiation is rare; only 6 such cases have been reported.5-10 In 4 of these cases, the transformation was to osteosarcoma.5,8-10
Here we report on an ABC that degenerated into a fibroblastic osteosarcoma—the fifth such case in the medical literature. In addition to reviewing the earlier cases, we describe the radiologic and histologic underpinnings of this diagnosis and the insight that they provide into the pathogenesis of this rare process. Although the prevailing view is that ABCs are benign, it is important to know these lesions have the potential to undergo malignant transformation, even in the absence of prior radiation exposure. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A healthy and previously asymptomatic 37-year-old man presented with thigh pain after a minor fall onto a couch. Radiographs showed a diaphyseal femoral pathologic fracture adjacent to a small but benign-appearing cystic lesion (Figures 1A, 1B).
Two years later, the patient had a bicycle accident and, after 2 weeks of significantly increased thigh swelling, presented to the emergency department at the referring institution. Radiographs showed a lytic lesion in the femoral diaphysis that was highly suspicious for malignancy (Figures 3A, 3B).
The initial biopsy specimens were evaluated at our institution and interpreted as being consistent with an ABC, with negative immunohistochemical staining for MDM2 (Figures 5A, 5B).
The patient underwent a 3-month course of neoadjuvant chemotherapy with cisplatin and doxorubicin. Interval-staging contrast-enhanced chest, abdomen, and pelvis computed tomography (CT) showed no evidence of metastatic disease. Preoperative MRI showed a significantly larger heterogeneous mass, now with neurovascular involvement, which precluded limb salvage.
Discussion
Aneurysmal bone cysts are expansile, hemorrhagic, locally destructive lesions that generally develop within the first 3 decades of life. Ever since they were first described by Jaffe and Lichtenstein11 in 1942, the most widely accepted theory of their pathogenesis has been that they begin as a benign reactive vascular process.12 However, more recent molecular studies by Oliveira and colleagues13 and Panoutsakopoulos and colleagues14 have demonstrated a clonal neoplastic basis for primary ABCs related to cytogenetic upregulation of oncogenes USP6 and CDH11 after translocation of 17p13 and 16q22.
Given the clonal nature of these lesions, it is surprising that malignant transformation is so rare. Until now, there have been only 4 reports of an ABC undergoing malignant degeneration to osteosarcoma without prior radiation exposure.
In this article, we have presented a fifth case of a primary ABC degenerating into an osteosarcoma, which in this instance was the fibroblastic subtype. This diagnosis was strongly supported by radiologic and pathologic evidence. From a radiologic perspective, imaging at initial presentation showed absolutely no suspicious features, and the same was true when follow-up radiographs were obtained, 1 month later. Although 1 month is short for a follow-up, the complete lack of radiographic changes would be highly unusual if in fact there had been a coexisting, undetected lesion as aggressive as the one that ultimately developed. Furthermore, imaging at second presentation, almost 2 years later, showed an extremely rapid evolution of findings over 1 month. Extrapolating back in time, we think this time course indicates the malignancy developed not long before its aggressive features were detected.
Genetic evidence suggests that most conventional high-grade osteosarcomas arise de novo from a mesenchymal precursor driven by multiple genetic aberrations. Less often, low-grade osteosarcomas progress to high-grade osteosarcomas. Amplification of 12q13-15 with resulting overexpression of MDM2 and CDK4 proteins is found in low-grade osteosarcomas and persists in examples that progress to higher-grade forms.15 Not only did review of our patient’s initial biopsy sample reveal no evidence of malignant features or abnormal mitotic activity, but the complete absence of MDM2 suggests not even a low-grade osteosarcoma was present at the time. By contrast, the second incisional biopsy specimen, 2 years later, showed markedly different histology and pronounced expression of MDM2 throughout the specimen. This finding suggests the histologically high-grade osteosarcoma did not arise de novo but rather secondarily from a low-grade osteosarcoma that had arisen from an ABC. Results of the final heterogeneous histology of the very large mass, which contained benign ABC areas indistinguishable from the initial biopsy sample, as well as areas of high-grade osteosarcoma, further support a multistep process of de-differentiation. Together, these findings are compelling evidence of malignant transformation of a primary ABC.
We acknowledge that the initial surgery performed at the outside hospital might have properly included frozen-section analysis of the biopsy material and that sampling error may have occurred during the index procedure—possibilities in the absence of complete lesional resection. In this case, however, the radiographic findings and the dominant histologic immunophenotype from medullary canal bone were both consistent with ABC and not osteosarcoma, lending support to malignant degeneration.
We have presented a fifth case of primary ABC degenerating into an osteosarcoma, now with immunohistochemical evidence supporting traditional radiologic and histologic evidence. Despite the rarity of the diagnosis, this case yields considerable insight into the pathogenetic mechanisms underlying malignant degeneration. Despite the widely held view that ABCs are benign, physicians caring for these patients must be aware that malignant transformation can occur.
Am J Orthop. 2016;45(6):E367-E372. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Donaldson WF. Aneurysmal bone cyst. J Bone Joint Surg Am. 1962;44:25-40.
2. Biesecker JL, Marcove RC, Huvos AG, Miké V. Aneurysmal bone cysts. A clinicopathologic study of 66 cases. Cancer. 1970;26(3):615-625.
3. Aho HJ, Aho AJ, Einola S. Aneurysmal bone cyst, a study of ultrastructure and malignant transformation. Virchows Arch A Pathol Anat Histol. 1982;395(2):169-179.
4. Tillman BP, Dahlin DC, Lipscomb PR, Stewart JR. Aneurysmal bone cyst: an analysis of ninety-five cases. Mayo Clin Proc. 1968;43(7):478-495.
5. Kyriakos M, Hardy D. Malignant transformation of aneurysmal bone cyst, with an analysis of the literature. Cancer. 1991;68(8):1770-1780.
6. Mei J, Gao YS, Wang SQ, Cai XS. Malignant transformation of aneurysmal bone cysts: a case report. Chin Med J (Engl). 2009;122(1):110-112.
7. Anract P, de Pinieux G, Jeanrot C, Babinet A, Forest M, Tomeno B. Malignant fibrous histiocytoma at the site of a previously treated aneurysmal bone cyst: a case report. J Bone Joint Surg Am. 2002;84(1):106-111.
8. Hsu CC, Wang JW, Huang CH, Chen WJ. Osteosarcoma at the site of a previously treated aneurysmal bone cyst. A case report. J Bone Joint Surg Am. 2005;87(2):395-398.
9. Wuisman P, Roessner A, Blasius S, Grünert J, Vestering T, Winkelmann W. High malignant surface osteosarcoma arising at the site of a previously treated aneurysmal bone cyst. J Cancer Res Clin Oncol. 1993;119(7):375-378.
10. Brindley GW, Greene JF Jr, Frankel LS. Case reports: malignant transformation of aneurysmal bone cysts. Clin Orthop Relat Res. 2005;(438):282-287.
11. Jaffe HL, Lichtenstein L. Solitary unicameral bone cyst: with emphasis on the roentgen picture, the pathologic appearance and the pathogenesis. Arch Surg. 1942;44:1004-1025.
12. Mirra JM. Bone Tumors: Clinical, Radiologic, and Pathologic Correlations. Philadelphia, PA: Lea & Febiger; 1989.
13. Oliveira AM, Chou MM, Perez-Atayde AR, Rosenberg AE. Aneurysmal bone cyst: a neoplasm driven by upregulation of the USP6 oncogene. J Clin Oncol. 2006;24(1):e1.
14. Panoutsakopoulos G, Pandis N, Kyriazoglou I, Gustafson P, Mertens F, Mandahl N. Recurrent t(16;17)(q22;p13) in aneurysmal bone cysts. Genes Chromosomes Cancer. 1999;26(3):265-266.
15. Dujardin F, Binh MB, Bouvier C, et al. MDM2 and CDK4 immunohistochemistry is a valuable tool in the differential diagnosis of low-grade osteosarcomas and other primary fibro-osseous lesions of the bone. Mod Pathol. 2001;24(5):624-637.
1. Donaldson WF. Aneurysmal bone cyst. J Bone Joint Surg Am. 1962;44:25-40.
2. Biesecker JL, Marcove RC, Huvos AG, Miké V. Aneurysmal bone cysts. A clinicopathologic study of 66 cases. Cancer. 1970;26(3):615-625.
3. Aho HJ, Aho AJ, Einola S. Aneurysmal bone cyst, a study of ultrastructure and malignant transformation. Virchows Arch A Pathol Anat Histol. 1982;395(2):169-179.
4. Tillman BP, Dahlin DC, Lipscomb PR, Stewart JR. Aneurysmal bone cyst: an analysis of ninety-five cases. Mayo Clin Proc. 1968;43(7):478-495.
5. Kyriakos M, Hardy D. Malignant transformation of aneurysmal bone cyst, with an analysis of the literature. Cancer. 1991;68(8):1770-1780.
6. Mei J, Gao YS, Wang SQ, Cai XS. Malignant transformation of aneurysmal bone cysts: a case report. Chin Med J (Engl). 2009;122(1):110-112.
7. Anract P, de Pinieux G, Jeanrot C, Babinet A, Forest M, Tomeno B. Malignant fibrous histiocytoma at the site of a previously treated aneurysmal bone cyst: a case report. J Bone Joint Surg Am. 2002;84(1):106-111.
8. Hsu CC, Wang JW, Huang CH, Chen WJ. Osteosarcoma at the site of a previously treated aneurysmal bone cyst. A case report. J Bone Joint Surg Am. 2005;87(2):395-398.
9. Wuisman P, Roessner A, Blasius S, Grünert J, Vestering T, Winkelmann W. High malignant surface osteosarcoma arising at the site of a previously treated aneurysmal bone cyst. J Cancer Res Clin Oncol. 1993;119(7):375-378.
10. Brindley GW, Greene JF Jr, Frankel LS. Case reports: malignant transformation of aneurysmal bone cysts. Clin Orthop Relat Res. 2005;(438):282-287.
11. Jaffe HL, Lichtenstein L. Solitary unicameral bone cyst: with emphasis on the roentgen picture, the pathologic appearance and the pathogenesis. Arch Surg. 1942;44:1004-1025.
12. Mirra JM. Bone Tumors: Clinical, Radiologic, and Pathologic Correlations. Philadelphia, PA: Lea & Febiger; 1989.
13. Oliveira AM, Chou MM, Perez-Atayde AR, Rosenberg AE. Aneurysmal bone cyst: a neoplasm driven by upregulation of the USP6 oncogene. J Clin Oncol. 2006;24(1):e1.
14. Panoutsakopoulos G, Pandis N, Kyriazoglou I, Gustafson P, Mertens F, Mandahl N. Recurrent t(16;17)(q22;p13) in aneurysmal bone cysts. Genes Chromosomes Cancer. 1999;26(3):265-266.
15. Dujardin F, Binh MB, Bouvier C, et al. MDM2 and CDK4 immunohistochemistry is a valuable tool in the differential diagnosis of low-grade osteosarcomas and other primary fibro-osseous lesions of the bone. Mod Pathol. 2001;24(5):624-637.
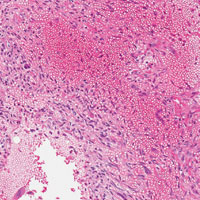
Low-Dose Radiotherapy for Primary Cutaneous Anaplastic Large-Cell Lymphoma While on Low-Dose Methotrexate
CD30+ primary cutaneous lymphoproliferative disorders (pcLPDs) are the second most common cause of cutaneous T-cell lymphoma, accounting for approximately 25% to 30% of cases.1 These disorders comprise a spectrum that includes primary cutaneous anaplastic large-cell lymphoma (pcALCL); lymphomatoid papulosis (LyP); and borderline lesions, which share clinicopathologic features of both pcALCL and LyP. Lymphomatoid papulosis is characterized as chronic, recurrent, papular or papulonodular skin lesions that typically are multifocal and regress spontaneously within weeks to months, only leaving small scars with atrophy and/or hyperpigmentation.2 Cutaneous anaplastic large-cell lymphoma typically presents as solitary or grouped nodules or tumors that may undergo spontaneous partial or complete regression in approximately 25% of cases3 but often persist if not treated. Patients may have an array of lesions comprising the spectrum of CD30 pcLPDs.4
There is no curative therapy for CD30+ pcLPDs. Although active treatment is not necessary for LyP, low-dose methotrexate (MTX)(10–50 mg weekly) or phototherapy are the preferred initial suppressive therapies for symptomatic patients with scarring, facial lesions, or multiple symptomatic lesions.5 Observation with expectant follow-up is an option in pcALCL, though spontaneous regression is less likely than in LyP. For single or grouped pcALCL lesions, local radiation is the first-line therapy.6 Multifocal pcALCL lesions also can be treated with low-dose MTX,2,5 as in LyP, or local radiation to selected areas. Although local radiotherapy is considered a first-line treatment in pcALCL, there is limited evidence on its clinical efficacy as well as the optimal dose and technique. We report the complete response of refractory pcALCL lesions to low-dose radiation while remaining on MTX weekly without any adverse effects.
Case Report
A 51-year-old woman presented with a 3-year history of CD30+ pcLPD manifesting primarily as pcALCL involving the head and neck, as well as LyP involving the head, arms, and trunk (T3N0M0). For 2 years her treatment regimen included clobetasol propionate cream 0.05% as needed for new lesions and 2 courses of standard-dose localized external beam radiation for larger pcALCL tumors on the right cheek and right side of the chin (Figure 1)(total dose for each course of treatment was 20 Gy and 36 Gy, respectively, each administered over 2–3 weeks). Because new unsightly papulonodules continued to develop on the patient’s face, she subsequently required low-dose oral MTX 30 mg once weekly for suppression of new lesions and was stable on this regimen for a year. However, she experienced an increase in LyP/pcALCL activity on the face during a 2-week break from MTX when she developed a herpes zoster infection on the right side of the forehead.
On physical examination 1 month later, 5 tiny pink papules scattered on the left eyebrow, left cheek, and left side of the chin were noted. She was advised to continue applying the clobetasol cream as needed and was restarted on MTX 10 mg once weekly. However, she developed 2 additional 1-cm nodules on the left side of the chin, neck, and shoulder. Methotrexate was increased to 30 mg once weekly over 2 weeks, which was the original dose prior to interruption, but the nodules grew to 1.5-cm in diameter. Due to their clinical appearance, the nodules were believed to be early pcALCL lesions (Figure 2A). Given the cosmetically sensitive location of the nodules, palliative radiotherapy was recommended rather than observe for possible regression. Based on a prior report by Neelis et al7 demonstrating efficacy of low-dose radiotherapy for cutaneous T-cell lymphoma and cutaneous B-cell lymphoma, we recommended starting with low radiation doses. Our patient was treated with 400 cGy twice to the left side of the chin and left side of the neck (800 cGy total at each site) while remaining on MTX 30 mg once weekly. This treatment was well tolerated without side effects and no evidence of radiation dermatitis. On follow-up examination 1 week later, the nodules had regressed and no new lesions were present (Figure 2B).
The patient has stayed on oral MTX and occasionally develops small lesions that quickly resolve with clobetasol cream. She has been followed for 3 years after radiotherapy and all 3 previously irradiated sites have remained recurrence free. Furthermore, she has not developed any new larger nodules or tumors and her MTX dose has been decreased to 15 mg once weekly.
Comment
Local radiotherapy is considered a first-line treatment of pcALCL; however, there is limited evidence on its clinical efficacy as well as the optimal dose and technique. Although no standard dose exists for pcALCL, the National Comprehensive Cancer Network guidelines8 recommend doses of 12 to 36 Gy in mycosis fungoides/Sézary syndrome subtypes of cutaneous T-cell lymphoma, which are consistent with guidelines published by the European Society for Medical Oncology.9 High complete response rates have been demonstrated in pcALCL at doses of 34 to 44 Gy6; however, lesions tend to recur elsewhere on the skin in 36% to 41% of patients despite treatment.2,10 Lower doses of radiation therapy would provide several advantages over higher-dose therapy if a complete response could be achieved without greatly increasing the local recurrence rate. In cases of local recurrence, low-dose radiation would more easily permit retreatment of lesions compared to higher doses of radiation. Similarly, in patients with multifocal pcALCL, lower doses of radiotherapy may allow for treatment of larger skin areas while limiting potential treatment risks. Furthermore, low-dose therapy would allow for treatments to be delivered more quickly and with less inconvenience to the patient who is likely to need multiple future treatments to other areas. Low-dose radiation has been described with a favorable efficacy profile for mycosis fungoides7,11 but has not been studied in patients with CD30+ pcLPDs.
Our case is notable because the patient remained on MTX during radiation therapy. B
Conclusion
We reported the use of low-dose radiation therapy for the treatment of localized pcALCL in a patient who remained on low-dose oral MTX. Additional studies will be necessary to more fully evaluate the efficacy of using low-dose radiation both as monotherapy and in combination with MTX for pcALCL.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30+ lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95:3653-3661.
- Willemze R, Beljaards RC. Spectrum of primary cutaneous CD30 (Ki-1)-positive lymphoproliferative disorders: a proposal for classification and guidelines for management and treatment. J Am Acad Dermatol. 1993;28:973-980.
- Kadin ME. The spectrum of Ki-1+ cutaneous lymphomas. Curr Probl Dermatol. 1990;19:132-143.
- Vonderheid EC, Sajjadian A, Kadin ME. Methotrexate is effective therapy for lymphomatoid papulosis and other primary cutaneous CD30-positive lymphoproliferative disorders. J Am Acad Dermatol. 1996;34:470-481.
- Yu JB, McNiff JM, Lund MW, et al. Treatment of primary cutaneous CD30+ anaplastic large-cell lymphoma with radiation therapy. Int J Radiat Oncol Biol Phys. 2008;70:1542-1545.
- Neelis KJ, Schimmel EC, Vermeer MH, et al. Low-dose palliative radiotherapy B-cell and T-cell lymphomas. Int J Radiat Oncol Biol Phys. 2009;74:154-158.
- National Comprehensive Cancer Network. CD30 lymphoproliferative disorders section in non-Hodgkin’s lymphoma (Version 3.2016). http://www.nccn.org/professionals/physician_gls/pdf/nhl.pdf. Accessed September 26, 2016.
- Willemze R, Hodak E, Zinzani PL, et al; ESMO Guidelines Working Group. Primary cutaneous lymphomas: EMSO clinical practice guidelines for diagnosis, treatment, and follow-up [published online July 17, 2013]. Ann Onc. 2013;24(suppl 6):vi149-vi154.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Harrison C, Young J, Navi D, et al. Revisiting low dose total skin electron beam radiotherapy in mycosis fungoides. Int J Radiat Oncol Biol Phys. 2011;81:651-657.
- Jaffe N, Farber S, Traggis D, et al. Favorable response of metastatic osteogenic sarcoma to pulse high-dose methotrexate with citrovorum rescue and radiation therapy. Cancer. 1973;31:1367-1373.
- Rosen G, Tefft M, Martinez A, et al. Combination chemotherapy and radiation therapy in the treatment of metastatic osteogenic sarcoma. Cancer. 1975;35:622-630.
- Kim YH, Aye MS, Fayos JV. Radiation necrosis of the scalp: a complication of cranial irradiation and methotrexate. Radiology. 1977;124:813-814.
CD30+ primary cutaneous lymphoproliferative disorders (pcLPDs) are the second most common cause of cutaneous T-cell lymphoma, accounting for approximately 25% to 30% of cases.1 These disorders comprise a spectrum that includes primary cutaneous anaplastic large-cell lymphoma (pcALCL); lymphomatoid papulosis (LyP); and borderline lesions, which share clinicopathologic features of both pcALCL and LyP. Lymphomatoid papulosis is characterized as chronic, recurrent, papular or papulonodular skin lesions that typically are multifocal and regress spontaneously within weeks to months, only leaving small scars with atrophy and/or hyperpigmentation.2 Cutaneous anaplastic large-cell lymphoma typically presents as solitary or grouped nodules or tumors that may undergo spontaneous partial or complete regression in approximately 25% of cases3 but often persist if not treated. Patients may have an array of lesions comprising the spectrum of CD30 pcLPDs.4
There is no curative therapy for CD30+ pcLPDs. Although active treatment is not necessary for LyP, low-dose methotrexate (MTX)(10–50 mg weekly) or phototherapy are the preferred initial suppressive therapies for symptomatic patients with scarring, facial lesions, or multiple symptomatic lesions.5 Observation with expectant follow-up is an option in pcALCL, though spontaneous regression is less likely than in LyP. For single or grouped pcALCL lesions, local radiation is the first-line therapy.6 Multifocal pcALCL lesions also can be treated with low-dose MTX,2,5 as in LyP, or local radiation to selected areas. Although local radiotherapy is considered a first-line treatment in pcALCL, there is limited evidence on its clinical efficacy as well as the optimal dose and technique. We report the complete response of refractory pcALCL lesions to low-dose radiation while remaining on MTX weekly without any adverse effects.
Case Report
A 51-year-old woman presented with a 3-year history of CD30+ pcLPD manifesting primarily as pcALCL involving the head and neck, as well as LyP involving the head, arms, and trunk (T3N0M0). For 2 years her treatment regimen included clobetasol propionate cream 0.05% as needed for new lesions and 2 courses of standard-dose localized external beam radiation for larger pcALCL tumors on the right cheek and right side of the chin (Figure 1)(total dose for each course of treatment was 20 Gy and 36 Gy, respectively, each administered over 2–3 weeks). Because new unsightly papulonodules continued to develop on the patient’s face, she subsequently required low-dose oral MTX 30 mg once weekly for suppression of new lesions and was stable on this regimen for a year. However, she experienced an increase in LyP/pcALCL activity on the face during a 2-week break from MTX when she developed a herpes zoster infection on the right side of the forehead.
On physical examination 1 month later, 5 tiny pink papules scattered on the left eyebrow, left cheek, and left side of the chin were noted. She was advised to continue applying the clobetasol cream as needed and was restarted on MTX 10 mg once weekly. However, she developed 2 additional 1-cm nodules on the left side of the chin, neck, and shoulder. Methotrexate was increased to 30 mg once weekly over 2 weeks, which was the original dose prior to interruption, but the nodules grew to 1.5-cm in diameter. Due to their clinical appearance, the nodules were believed to be early pcALCL lesions (Figure 2A). Given the cosmetically sensitive location of the nodules, palliative radiotherapy was recommended rather than observe for possible regression. Based on a prior report by Neelis et al7 demonstrating efficacy of low-dose radiotherapy for cutaneous T-cell lymphoma and cutaneous B-cell lymphoma, we recommended starting with low radiation doses. Our patient was treated with 400 cGy twice to the left side of the chin and left side of the neck (800 cGy total at each site) while remaining on MTX 30 mg once weekly. This treatment was well tolerated without side effects and no evidence of radiation dermatitis. On follow-up examination 1 week later, the nodules had regressed and no new lesions were present (Figure 2B).
The patient has stayed on oral MTX and occasionally develops small lesions that quickly resolve with clobetasol cream. She has been followed for 3 years after radiotherapy and all 3 previously irradiated sites have remained recurrence free. Furthermore, she has not developed any new larger nodules or tumors and her MTX dose has been decreased to 15 mg once weekly.
Comment
Local radiotherapy is considered a first-line treatment of pcALCL; however, there is limited evidence on its clinical efficacy as well as the optimal dose and technique. Although no standard dose exists for pcALCL, the National Comprehensive Cancer Network guidelines8 recommend doses of 12 to 36 Gy in mycosis fungoides/Sézary syndrome subtypes of cutaneous T-cell lymphoma, which are consistent with guidelines published by the European Society for Medical Oncology.9 High complete response rates have been demonstrated in pcALCL at doses of 34 to 44 Gy6; however, lesions tend to recur elsewhere on the skin in 36% to 41% of patients despite treatment.2,10 Lower doses of radiation therapy would provide several advantages over higher-dose therapy if a complete response could be achieved without greatly increasing the local recurrence rate. In cases of local recurrence, low-dose radiation would more easily permit retreatment of lesions compared to higher doses of radiation. Similarly, in patients with multifocal pcALCL, lower doses of radiotherapy may allow for treatment of larger skin areas while limiting potential treatment risks. Furthermore, low-dose therapy would allow for treatments to be delivered more quickly and with less inconvenience to the patient who is likely to need multiple future treatments to other areas. Low-dose radiation has been described with a favorable efficacy profile for mycosis fungoides7,11 but has not been studied in patients with CD30+ pcLPDs.
Our case is notable because the patient remained on MTX during radiation therapy. B
Conclusion
We reported the use of low-dose radiation therapy for the treatment of localized pcALCL in a patient who remained on low-dose oral MTX. Additional studies will be necessary to more fully evaluate the efficacy of using low-dose radiation both as monotherapy and in combination with MTX for pcALCL.
CD30+ primary cutaneous lymphoproliferative disorders (pcLPDs) are the second most common cause of cutaneous T-cell lymphoma, accounting for approximately 25% to 30% of cases.1 These disorders comprise a spectrum that includes primary cutaneous anaplastic large-cell lymphoma (pcALCL); lymphomatoid papulosis (LyP); and borderline lesions, which share clinicopathologic features of both pcALCL and LyP. Lymphomatoid papulosis is characterized as chronic, recurrent, papular or papulonodular skin lesions that typically are multifocal and regress spontaneously within weeks to months, only leaving small scars with atrophy and/or hyperpigmentation.2 Cutaneous anaplastic large-cell lymphoma typically presents as solitary or grouped nodules or tumors that may undergo spontaneous partial or complete regression in approximately 25% of cases3 but often persist if not treated. Patients may have an array of lesions comprising the spectrum of CD30 pcLPDs.4
There is no curative therapy for CD30+ pcLPDs. Although active treatment is not necessary for LyP, low-dose methotrexate (MTX)(10–50 mg weekly) or phototherapy are the preferred initial suppressive therapies for symptomatic patients with scarring, facial lesions, or multiple symptomatic lesions.5 Observation with expectant follow-up is an option in pcALCL, though spontaneous regression is less likely than in LyP. For single or grouped pcALCL lesions, local radiation is the first-line therapy.6 Multifocal pcALCL lesions also can be treated with low-dose MTX,2,5 as in LyP, or local radiation to selected areas. Although local radiotherapy is considered a first-line treatment in pcALCL, there is limited evidence on its clinical efficacy as well as the optimal dose and technique. We report the complete response of refractory pcALCL lesions to low-dose radiation while remaining on MTX weekly without any adverse effects.
Case Report
A 51-year-old woman presented with a 3-year history of CD30+ pcLPD manifesting primarily as pcALCL involving the head and neck, as well as LyP involving the head, arms, and trunk (T3N0M0). For 2 years her treatment regimen included clobetasol propionate cream 0.05% as needed for new lesions and 2 courses of standard-dose localized external beam radiation for larger pcALCL tumors on the right cheek and right side of the chin (Figure 1)(total dose for each course of treatment was 20 Gy and 36 Gy, respectively, each administered over 2–3 weeks). Because new unsightly papulonodules continued to develop on the patient’s face, she subsequently required low-dose oral MTX 30 mg once weekly for suppression of new lesions and was stable on this regimen for a year. However, she experienced an increase in LyP/pcALCL activity on the face during a 2-week break from MTX when she developed a herpes zoster infection on the right side of the forehead.
On physical examination 1 month later, 5 tiny pink papules scattered on the left eyebrow, left cheek, and left side of the chin were noted. She was advised to continue applying the clobetasol cream as needed and was restarted on MTX 10 mg once weekly. However, she developed 2 additional 1-cm nodules on the left side of the chin, neck, and shoulder. Methotrexate was increased to 30 mg once weekly over 2 weeks, which was the original dose prior to interruption, but the nodules grew to 1.5-cm in diameter. Due to their clinical appearance, the nodules were believed to be early pcALCL lesions (Figure 2A). Given the cosmetically sensitive location of the nodules, palliative radiotherapy was recommended rather than observe for possible regression. Based on a prior report by Neelis et al7 demonstrating efficacy of low-dose radiotherapy for cutaneous T-cell lymphoma and cutaneous B-cell lymphoma, we recommended starting with low radiation doses. Our patient was treated with 400 cGy twice to the left side of the chin and left side of the neck (800 cGy total at each site) while remaining on MTX 30 mg once weekly. This treatment was well tolerated without side effects and no evidence of radiation dermatitis. On follow-up examination 1 week later, the nodules had regressed and no new lesions were present (Figure 2B).
The patient has stayed on oral MTX and occasionally develops small lesions that quickly resolve with clobetasol cream. She has been followed for 3 years after radiotherapy and all 3 previously irradiated sites have remained recurrence free. Furthermore, she has not developed any new larger nodules or tumors and her MTX dose has been decreased to 15 mg once weekly.
Comment
Local radiotherapy is considered a first-line treatment of pcALCL; however, there is limited evidence on its clinical efficacy as well as the optimal dose and technique. Although no standard dose exists for pcALCL, the National Comprehensive Cancer Network guidelines8 recommend doses of 12 to 36 Gy in mycosis fungoides/Sézary syndrome subtypes of cutaneous T-cell lymphoma, which are consistent with guidelines published by the European Society for Medical Oncology.9 High complete response rates have been demonstrated in pcALCL at doses of 34 to 44 Gy6; however, lesions tend to recur elsewhere on the skin in 36% to 41% of patients despite treatment.2,10 Lower doses of radiation therapy would provide several advantages over higher-dose therapy if a complete response could be achieved without greatly increasing the local recurrence rate. In cases of local recurrence, low-dose radiation would more easily permit retreatment of lesions compared to higher doses of radiation. Similarly, in patients with multifocal pcALCL, lower doses of radiotherapy may allow for treatment of larger skin areas while limiting potential treatment risks. Furthermore, low-dose therapy would allow for treatments to be delivered more quickly and with less inconvenience to the patient who is likely to need multiple future treatments to other areas. Low-dose radiation has been described with a favorable efficacy profile for mycosis fungoides7,11 but has not been studied in patients with CD30+ pcLPDs.
Our case is notable because the patient remained on MTX during radiation therapy. B
Conclusion
We reported the use of low-dose radiation therapy for the treatment of localized pcALCL in a patient who remained on low-dose oral MTX. Additional studies will be necessary to more fully evaluate the efficacy of using low-dose radiation both as monotherapy and in combination with MTX for pcALCL.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30+ lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95:3653-3661.
- Willemze R, Beljaards RC. Spectrum of primary cutaneous CD30 (Ki-1)-positive lymphoproliferative disorders: a proposal for classification and guidelines for management and treatment. J Am Acad Dermatol. 1993;28:973-980.
- Kadin ME. The spectrum of Ki-1+ cutaneous lymphomas. Curr Probl Dermatol. 1990;19:132-143.
- Vonderheid EC, Sajjadian A, Kadin ME. Methotrexate is effective therapy for lymphomatoid papulosis and other primary cutaneous CD30-positive lymphoproliferative disorders. J Am Acad Dermatol. 1996;34:470-481.
- Yu JB, McNiff JM, Lund MW, et al. Treatment of primary cutaneous CD30+ anaplastic large-cell lymphoma with radiation therapy. Int J Radiat Oncol Biol Phys. 2008;70:1542-1545.
- Neelis KJ, Schimmel EC, Vermeer MH, et al. Low-dose palliative radiotherapy B-cell and T-cell lymphomas. Int J Radiat Oncol Biol Phys. 2009;74:154-158.
- National Comprehensive Cancer Network. CD30 lymphoproliferative disorders section in non-Hodgkin’s lymphoma (Version 3.2016). http://www.nccn.org/professionals/physician_gls/pdf/nhl.pdf. Accessed September 26, 2016.
- Willemze R, Hodak E, Zinzani PL, et al; ESMO Guidelines Working Group. Primary cutaneous lymphomas: EMSO clinical practice guidelines for diagnosis, treatment, and follow-up [published online July 17, 2013]. Ann Onc. 2013;24(suppl 6):vi149-vi154.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Harrison C, Young J, Navi D, et al. Revisiting low dose total skin electron beam radiotherapy in mycosis fungoides. Int J Radiat Oncol Biol Phys. 2011;81:651-657.
- Jaffe N, Farber S, Traggis D, et al. Favorable response of metastatic osteogenic sarcoma to pulse high-dose methotrexate with citrovorum rescue and radiation therapy. Cancer. 1973;31:1367-1373.
- Rosen G, Tefft M, Martinez A, et al. Combination chemotherapy and radiation therapy in the treatment of metastatic osteogenic sarcoma. Cancer. 1975;35:622-630.
- Kim YH, Aye MS, Fayos JV. Radiation necrosis of the scalp: a complication of cranial irradiation and methotrexate. Radiology. 1977;124:813-814.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30+ lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95:3653-3661.
- Willemze R, Beljaards RC. Spectrum of primary cutaneous CD30 (Ki-1)-positive lymphoproliferative disorders: a proposal for classification and guidelines for management and treatment. J Am Acad Dermatol. 1993;28:973-980.
- Kadin ME. The spectrum of Ki-1+ cutaneous lymphomas. Curr Probl Dermatol. 1990;19:132-143.
- Vonderheid EC, Sajjadian A, Kadin ME. Methotrexate is effective therapy for lymphomatoid papulosis and other primary cutaneous CD30-positive lymphoproliferative disorders. J Am Acad Dermatol. 1996;34:470-481.
- Yu JB, McNiff JM, Lund MW, et al. Treatment of primary cutaneous CD30+ anaplastic large-cell lymphoma with radiation therapy. Int J Radiat Oncol Biol Phys. 2008;70:1542-1545.
- Neelis KJ, Schimmel EC, Vermeer MH, et al. Low-dose palliative radiotherapy B-cell and T-cell lymphomas. Int J Radiat Oncol Biol Phys. 2009;74:154-158.
- National Comprehensive Cancer Network. CD30 lymphoproliferative disorders section in non-Hodgkin’s lymphoma (Version 3.2016). http://www.nccn.org/professionals/physician_gls/pdf/nhl.pdf. Accessed September 26, 2016.
- Willemze R, Hodak E, Zinzani PL, et al; ESMO Guidelines Working Group. Primary cutaneous lymphomas: EMSO clinical practice guidelines for diagnosis, treatment, and follow-up [published online July 17, 2013]. Ann Onc. 2013;24(suppl 6):vi149-vi154.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Harrison C, Young J, Navi D, et al. Revisiting low dose total skin electron beam radiotherapy in mycosis fungoides. Int J Radiat Oncol Biol Phys. 2011;81:651-657.
- Jaffe N, Farber S, Traggis D, et al. Favorable response of metastatic osteogenic sarcoma to pulse high-dose methotrexate with citrovorum rescue and radiation therapy. Cancer. 1973;31:1367-1373.
- Rosen G, Tefft M, Martinez A, et al. Combination chemotherapy and radiation therapy in the treatment of metastatic osteogenic sarcoma. Cancer. 1975;35:622-630.
- Kim YH, Aye MS, Fayos JV. Radiation necrosis of the scalp: a complication of cranial irradiation and methotrexate. Radiology. 1977;124:813-814.
Practice Points
- Cutaneous T-cell lymphoma tumors such as primary cutaneous anaplastic large-cell lymphoma can respond to low-dose radiation therapy, which enables future retreatment of sensitive sites.
- Low-dose radiation therapy requires a shorter course of therapy than traditional dosing, which is more convenient and less costly.
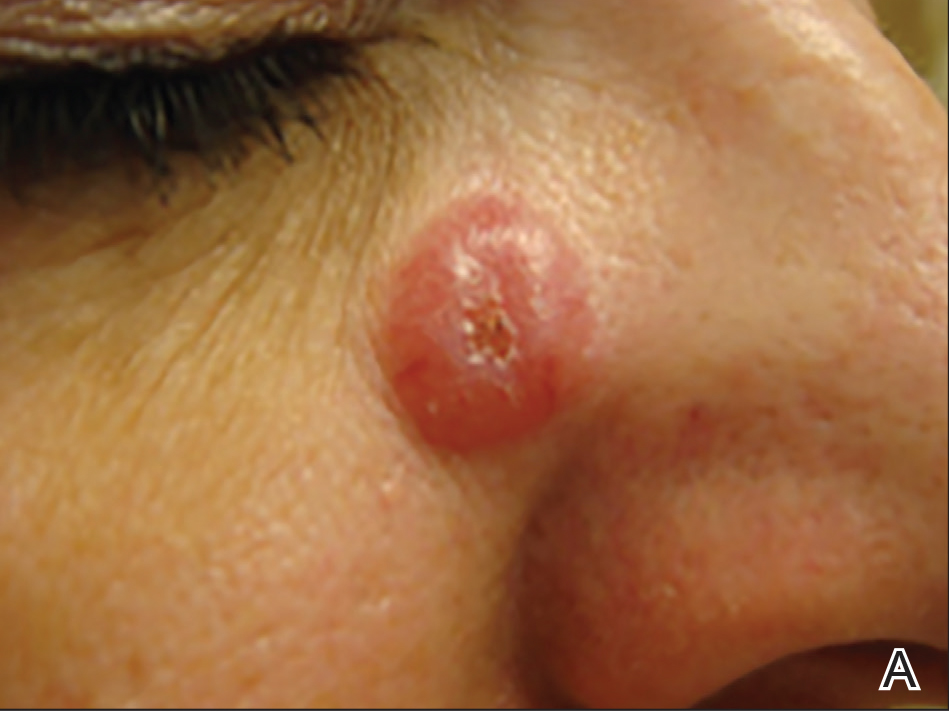
Bullous Pemphigoid Associated With a Lymphoepithelial Cyst of the Pancreas
Bullous pemphigoid (BP) is an acquired, autoimmune, subepidermal blistering disease that is more common in elderly patients.1 An association with internal neoplasms and BP has been established; however, there is debate regarding the precise nature of the relationship.2 Several gastrointestinal tract tumors have been associated with BP, including adenocarcinoma of the colon, adenosquamous cell carcinoma and adenocarcinoma of the stomach, adenocarcinoma of the rectum, and liver and bile duct malignancies.3-5 Association with pancreatic neoplasms (eg, carcinoma of the pancreas) rarely has been reported.5-7 We present an unusual case of a lymphoepithelial cyst of the pancreas in a patient with BP.
Case Report
A 67-year-old man presented with erythematous crusted plaques and pink scars over the chest, back, arms, and legs (Figure 1). A 1.5-cm tense bullous lesion was observed on the right knee. The patient’s medical history was notable for biopsy-proven BP of 8 months’ duration as well as diabetes mellitus and hypothyroidism. The patient was being followed by his surgeon for a 1.9-cm soft-tissue lesion in the pancreatic tail and was awaiting surgical excision at the time of the current presentation. The pancreatic lesion was discovered incidentally on magnetic resonance imaging performed following urologic concerns. At the current presentation, the patient’s medications included nifedipine, hydralazine, metoprolol, torsemide, aspirin, levothyroxine, atorvastatin, insulin lispro, and insulin glargine. He had previously been treated for BP with prednisone at a maximum dosage of 60 mg daily, clobetasol propionate cream 0.05%, and mupirocin ointment 2% without improvement. Because of substantial weight gain and poorly controlled diabetes, prednisone was discontinued.
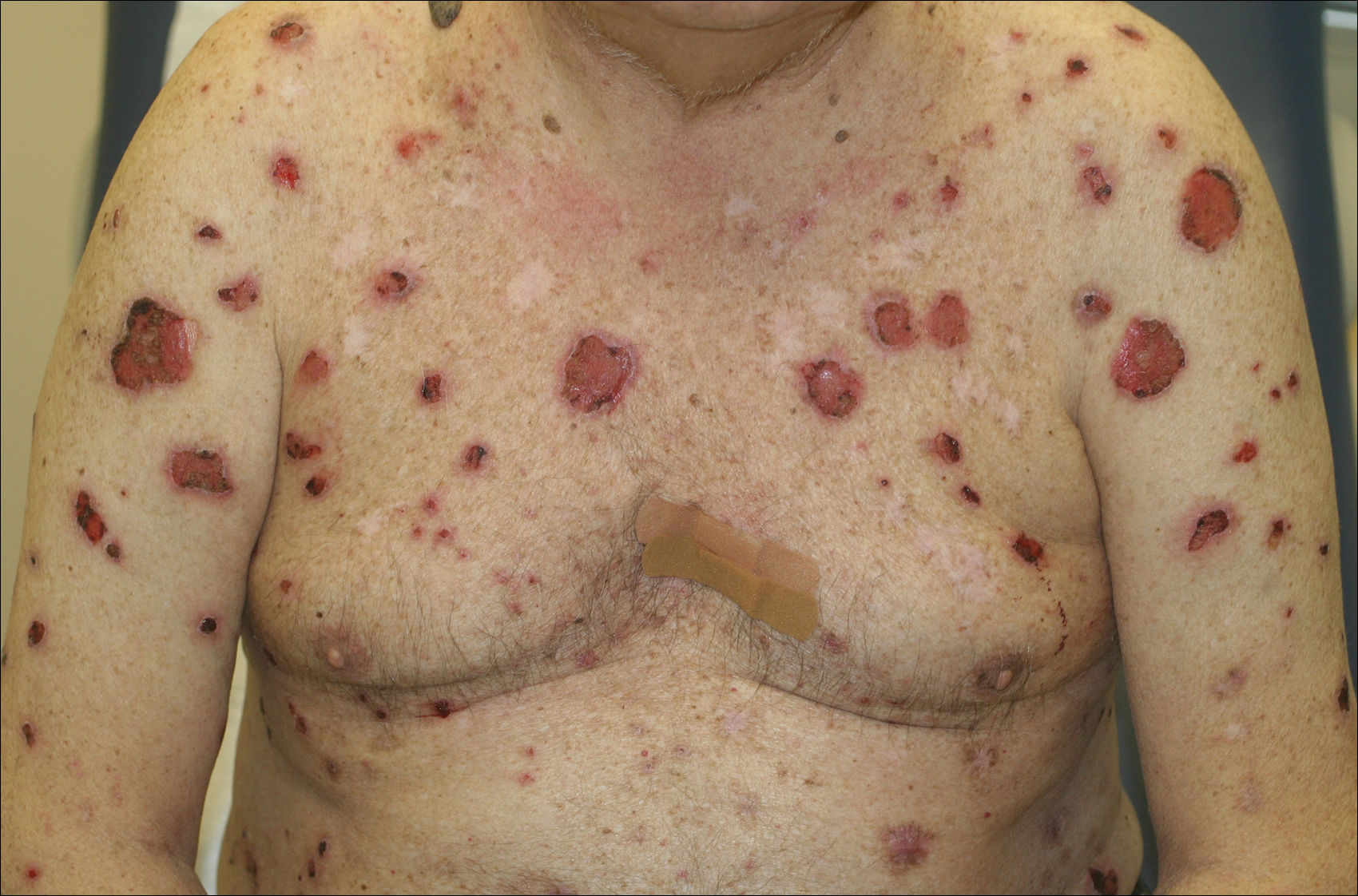
Bullous pemphigoid had been diagnosed histopathologically by a prior dermatologist. Hematoxylin and eosin staining demonstrated a subepidermal separation with eosinophils within the perivascular infiltrate (Figure 2). Direct immunofluorescence was noted in a linear pattern at the dermoepidermal junction with IgG and C3. Bullous pemphigoid antigen antibodies 1 and 2 were obtained via enzyme-linked immunosorbent assay with a positive BP-1 antigen antibody of 19 U/mL (positive, >15 U/mL) and a normal BP-2 antigen antibody of less than 9 U/mL (reference range, <9 U/mL). The glucagon level was elevated at 245 pg/mL (reference range, ≤134 pg/mL).
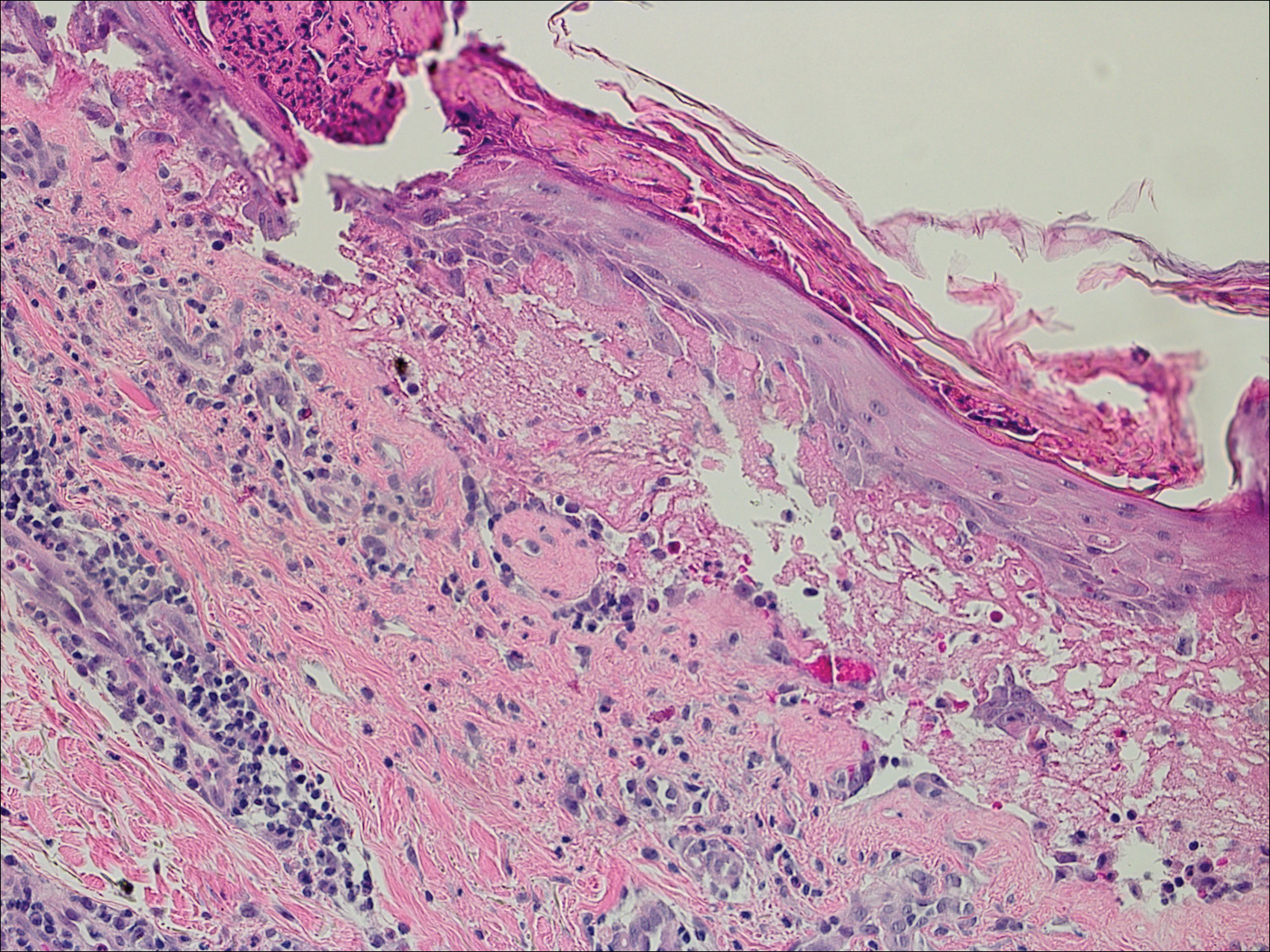
The patient was prescribed minocycline 100 mg twice daily and niacinamide 500 mg 3 times daily. Topical treatment with clobetasol and mupirocin was continued. One month later, the patient returned with an increase in disease activity. Changes to his therapeutic regimen were deferred until after excision of the pancreatic lesion based on the decision not to start immunosuppressive therapy until the precise nature of the pancreatic lesion was determined.
The patient underwent excision of the pancreatic lesion approximately 3 months later, which proved to be a benign lymphoepithelial cyst of the pancreas. Histology of the cyst consisted of dense fibrous tissue with a squamous epithelial lining focally infiltrated by lymphocytes (Figure 3A). Immunoperoxidase staining of the cyst revealed focal linear areas of C3d staining along the basement membrane of the stratified squamous epithelium (Figure 3B).
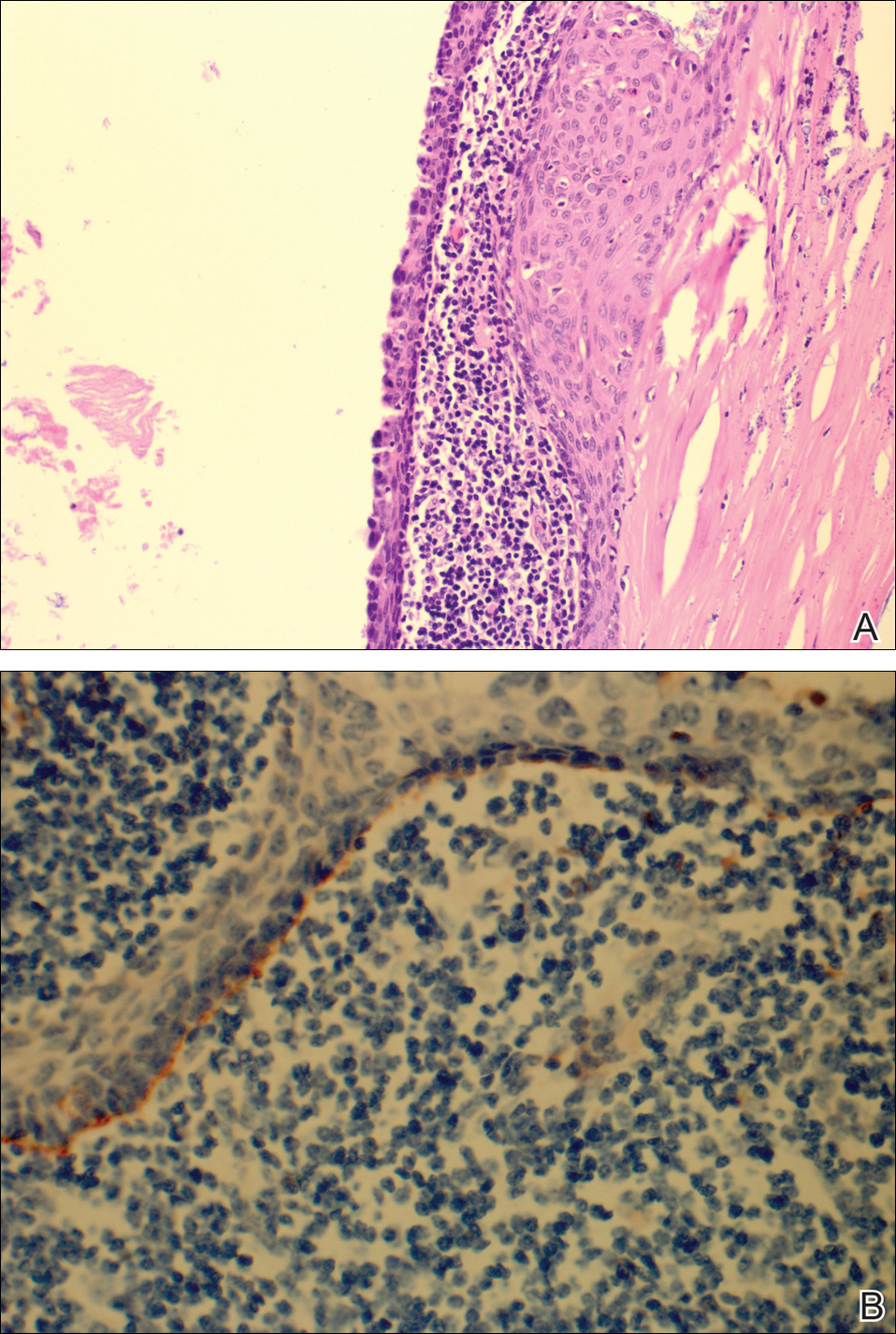
The patient stated that his skin started to improve virtually immediately following the excision without systemic treatment for BP. On follow-up examination 3 weeks postoperatively, no bullae were observed and there was a notable decrease in erythematous crusted plaques (Figure 4).
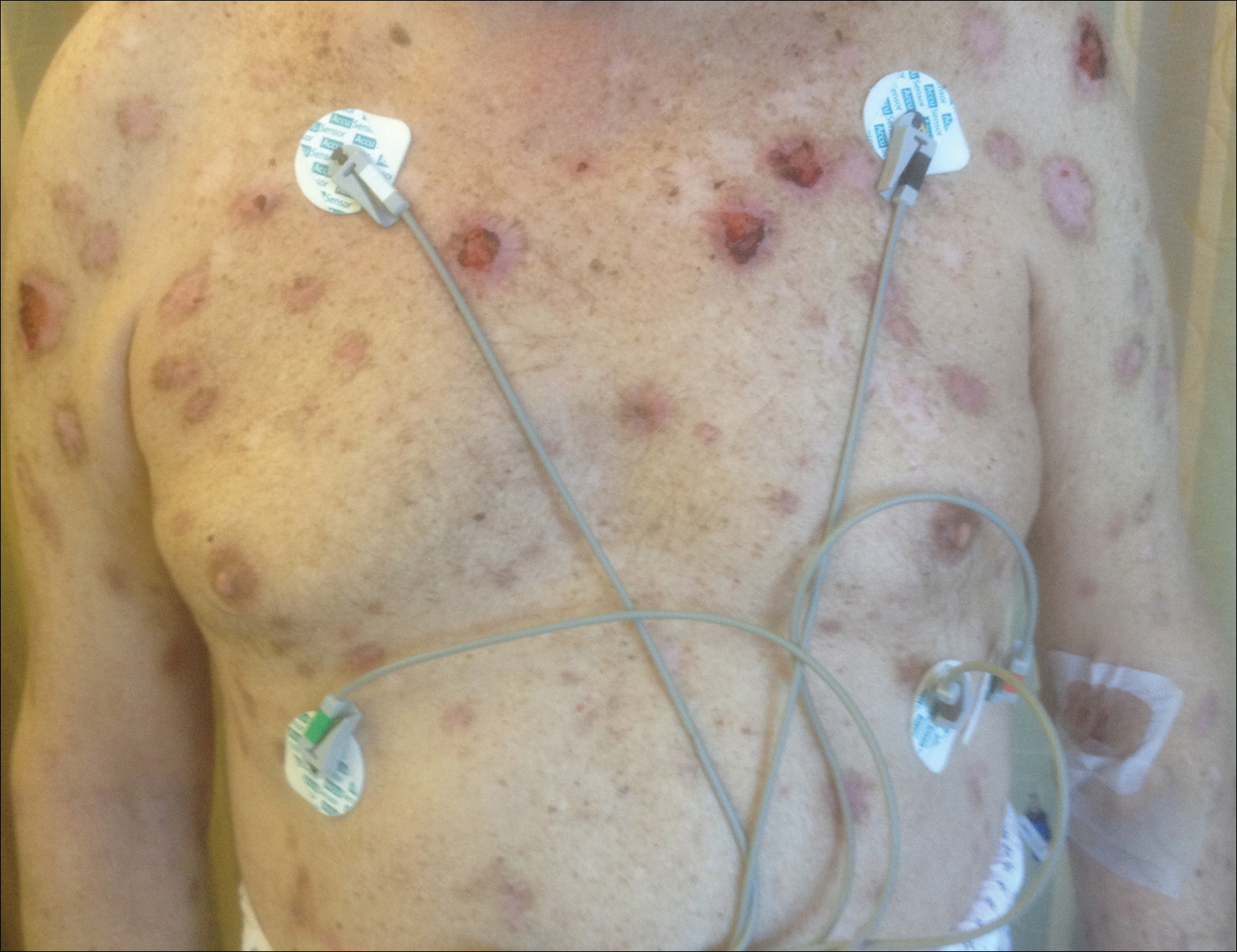
Comment
Paraneoplastic BP has been documented; however, lymphoepithelial cysts of the pancreas in association with BP are rare. We propose that the lymphoepithelial cyst of the pancreas provided the immunologic stimulus for the development of cutaneous BP based on the observation that our patient’s condition remarkably improved with resection of the tumor.
There are fewer than 100 cases of lymphoepithelial cysts of the pancreas reported in the literature.8 The histologic appearance is consistent with a true cyst exhibiting a well-differentiated stratified squamous epithelium, often with keratinization, surrounded by lymphoid tissue. These tumors are most commonly seen in middle-aged men and are frequently found incidentally,8-10 as was the case with our patient. Although histologically similar, lymphoepithelial cysts of the pancreas are considered distinct from lymphoepithelial cysts of the parotid gland or head and neck region.10 Lymphoepithelial cysts of the pancreas are unrelated to elevated glucagon levels; it is likely that our patient’s glucagon levels were associated with his history of diabetes.11
The diagnosis of BP is characteristically confirmed by direct immunofluorescence. Although it was performed for our patient’s cutaneous lesions, it was not obtained for the lymphoepithelial cyst of the pancreas. Once the diagnosis of the lymphoepithelial cyst of the pancreas was established, as direct immunofluorescence could not be performed in formalin-fixed tissue, immunoperoxidase staining with C3d was obtained. C3 has a well-established role in activation of complement and as a marker in BP. Deposition of C3d is a result of deactivation of C3b, a cleavage product of C3. In a study of 6 autoimmune blistering disorders that included 32 patients with BP, Pfaltz et al12 found positive immunoperoxidase staining for C3d in 31 of 32 patients, which translated to a sensitivity of 97%, a positive predictive value of 100%, and a negative predictive value of 98% among the blistering diseases being studied. Similarly, Magro and Dyrsen13 had positive staining of C3d in 17 of 17 (100%) patients with BP.
In theory, any process that involves deposition of C3 should be positive for C3d on immunoperoxidase staining. Other dermatologic inflammatory conditions stain positively with C3d, such as systemic lupus erythematosus, discoid lupus erythematosus, subacute cutaneous lupus erythematosus, and dermatomysositis.13 The staining for these diseases correlates with the site of the associated inflammatory component seen on hematoxylin and eosin staining. The staining of C3d along the basement membrane of stratified squamous epithelium in the lymphoepithelial cyst of the pancreas seen in our patient closely resembles the staining seen in cutaneous BP.
A proposed mechanism for BP in our patient would be exposure of BP-1 antigen in the pancreatic cyst leading to antibody recognition and C3 deposition along the basement membrane in the cyst, as evidenced by C3d immunoperoxidase staining. The IgG and C3 deposition along the cutaneous basement membrane would then represent a systemic response to the antigen exposure in the cyst. Thus, the lymphoepithelial cyst provided the immunologic stimulus for the development of the cutaneous BP. This theory is based on the observation of our patient’s rapid improvement without a change in his treatment regimen immediately after surgical excision of the cyst.
Despite the plausibility of our hypothesis, several questions remain regarding the validity of our assumptions. Although sensitive for C3 deposition, C3d immunoperoxidase staining is not specific for BP. If the proposed mechanism for causation is true, one might have expected that a subepithelial cleft along the basement membrane of the pancreatic cyst would be observed, which was not seen. A repeat BP antigen antibody was not obtained, which would have been helpful in determining if there was clearance of the antibody that would have correlated with the clinical resolution of the BP lesions.
Conclusion
Our case suggests that paraneoplastic BP is a genuine entity. Indeed, the primary tumor itself may be the immunologic stimulus in the development of BP. Recalcitrant BP should raise the question of a neoplastic process that is exposing the BP antigen. If a thorough review of systems accompanied by corroborating laboratory studies suggests a neoplastic process, the suspect lesion should be further evaluated and surgically excised if clinically indicated. Further evaluation of neoplasms with advanced staining methods may aid in establishing the causative nature of tumors in the development of BP.
Acknowledgments
We are grateful to John Stanley, MD, and Aimee Payne, MD (both from Philadelphia, Pennsylvania), for theirinsights into this case.
- Charneux J, Lorin J, Vitry F, et al. Usefulness of BP230 and BP180-NC16a enzyme-linked immunosorbent assays in the initial diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:286-291.
- Patel M, Sniha AA, Gilbert E. Bullous pemphigoid associated with renal cell carcinoma and invasive squamous cell carcinoma. J Drugs Dermatol. 2012;11:234-238.
- Song HJ, Han SH, Hong WK, et al. Paraneoplastic bullous pemphigoid: clinical disease activity correlated with enzyme-linked immunosorbent assay index for NC16A domain of BP180. J Dermatol. 2009;36:66-68.
- Muramatsu T, Iida T, Tada H, et al. Bullous pemphigoid associated with internal malignancies: identification of 180-kDa antigen by Western immunoblotting. Br J Dermatol. 1996;135:782-784.
- Ogawa H, Sakuma M, Morioka S, et al. The incidence of internal malignancies in pemphigus and bullous pemphigoid in Japan. J Dermatol Sci. 1995;9:136-141.
- Boyd RV. Pemphigoid and carcinoma of the pancreas. Br Med J. 1964;1:1092.
- Eustace S, Morrow G, O’Loughlin S, et al. The role of computed tomography and sonography in acute bullous pemphigoid. Ir J Med Sci. 1993;162:401-404.
- Clemente G, Sarno G, De Rose AM, et al. Lymphoepithelial cyst of the pancreas: case report and review of the literature. Acta Gastroenterol Belg. 2011;74:343-346.
- Frezza E, Wachtel MS. Lymphoepithelial cyst of the pancreas tail. case report and review of the literature. JOP. 2008;9:46-49.
- Basturk O, Coban I, Adsay NV. Pancreatic cysts: pathologic classification, differential diagnosis and clinical implications. Arch Pathol Lab Med. 2009;133:423-438.
- Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122:4-12.
- Pfaltz K, Mertz K, Rose C, et al. C3d immunohistochemistry on formalin-fixed tissue is a valuable tool in the diagnosis of bullous pemphigoid of the skin. J Cutan Pathol. 2010;37:654-658.
- Magro CM, Dyrsen ME. The use of C3d and C4d immunohistochemistry on formalin-fixed tissue as a diagnostic adjunct in the assessment of inflammatory skin disease. J Am Acad Dermatol. 2008;59:822-833.
Bullous pemphigoid (BP) is an acquired, autoimmune, subepidermal blistering disease that is more common in elderly patients.1 An association with internal neoplasms and BP has been established; however, there is debate regarding the precise nature of the relationship.2 Several gastrointestinal tract tumors have been associated with BP, including adenocarcinoma of the colon, adenosquamous cell carcinoma and adenocarcinoma of the stomach, adenocarcinoma of the rectum, and liver and bile duct malignancies.3-5 Association with pancreatic neoplasms (eg, carcinoma of the pancreas) rarely has been reported.5-7 We present an unusual case of a lymphoepithelial cyst of the pancreas in a patient with BP.
Case Report
A 67-year-old man presented with erythematous crusted plaques and pink scars over the chest, back, arms, and legs (Figure 1). A 1.5-cm tense bullous lesion was observed on the right knee. The patient’s medical history was notable for biopsy-proven BP of 8 months’ duration as well as diabetes mellitus and hypothyroidism. The patient was being followed by his surgeon for a 1.9-cm soft-tissue lesion in the pancreatic tail and was awaiting surgical excision at the time of the current presentation. The pancreatic lesion was discovered incidentally on magnetic resonance imaging performed following urologic concerns. At the current presentation, the patient’s medications included nifedipine, hydralazine, metoprolol, torsemide, aspirin, levothyroxine, atorvastatin, insulin lispro, and insulin glargine. He had previously been treated for BP with prednisone at a maximum dosage of 60 mg daily, clobetasol propionate cream 0.05%, and mupirocin ointment 2% without improvement. Because of substantial weight gain and poorly controlled diabetes, prednisone was discontinued.
Bullous pemphigoid had been diagnosed histopathologically by a prior dermatologist. Hematoxylin and eosin staining demonstrated a subepidermal separation with eosinophils within the perivascular infiltrate (Figure 2). Direct immunofluorescence was noted in a linear pattern at the dermoepidermal junction with IgG and C3. Bullous pemphigoid antigen antibodies 1 and 2 were obtained via enzyme-linked immunosorbent assay with a positive BP-1 antigen antibody of 19 U/mL (positive, >15 U/mL) and a normal BP-2 antigen antibody of less than 9 U/mL (reference range, <9 U/mL). The glucagon level was elevated at 245 pg/mL (reference range, ≤134 pg/mL).
The patient was prescribed minocycline 100 mg twice daily and niacinamide 500 mg 3 times daily. Topical treatment with clobetasol and mupirocin was continued. One month later, the patient returned with an increase in disease activity. Changes to his therapeutic regimen were deferred until after excision of the pancreatic lesion based on the decision not to start immunosuppressive therapy until the precise nature of the pancreatic lesion was determined.
The patient underwent excision of the pancreatic lesion approximately 3 months later, which proved to be a benign lymphoepithelial cyst of the pancreas. Histology of the cyst consisted of dense fibrous tissue with a squamous epithelial lining focally infiltrated by lymphocytes (Figure 3A). Immunoperoxidase staining of the cyst revealed focal linear areas of C3d staining along the basement membrane of the stratified squamous epithelium (Figure 3B).
The patient stated that his skin started to improve virtually immediately following the excision without systemic treatment for BP. On follow-up examination 3 weeks postoperatively, no bullae were observed and there was a notable decrease in erythematous crusted plaques (Figure 4).
Comment
Paraneoplastic BP has been documented; however, lymphoepithelial cysts of the pancreas in association with BP are rare. We propose that the lymphoepithelial cyst of the pancreas provided the immunologic stimulus for the development of cutaneous BP based on the observation that our patient’s condition remarkably improved with resection of the tumor.
There are fewer than 100 cases of lymphoepithelial cysts of the pancreas reported in the literature.8 The histologic appearance is consistent with a true cyst exhibiting a well-differentiated stratified squamous epithelium, often with keratinization, surrounded by lymphoid tissue. These tumors are most commonly seen in middle-aged men and are frequently found incidentally,8-10 as was the case with our patient. Although histologically similar, lymphoepithelial cysts of the pancreas are considered distinct from lymphoepithelial cysts of the parotid gland or head and neck region.10 Lymphoepithelial cysts of the pancreas are unrelated to elevated glucagon levels; it is likely that our patient’s glucagon levels were associated with his history of diabetes.11
The diagnosis of BP is characteristically confirmed by direct immunofluorescence. Although it was performed for our patient’s cutaneous lesions, it was not obtained for the lymphoepithelial cyst of the pancreas. Once the diagnosis of the lymphoepithelial cyst of the pancreas was established, as direct immunofluorescence could not be performed in formalin-fixed tissue, immunoperoxidase staining with C3d was obtained. C3 has a well-established role in activation of complement and as a marker in BP. Deposition of C3d is a result of deactivation of C3b, a cleavage product of C3. In a study of 6 autoimmune blistering disorders that included 32 patients with BP, Pfaltz et al12 found positive immunoperoxidase staining for C3d in 31 of 32 patients, which translated to a sensitivity of 97%, a positive predictive value of 100%, and a negative predictive value of 98% among the blistering diseases being studied. Similarly, Magro and Dyrsen13 had positive staining of C3d in 17 of 17 (100%) patients with BP.
In theory, any process that involves deposition of C3 should be positive for C3d on immunoperoxidase staining. Other dermatologic inflammatory conditions stain positively with C3d, such as systemic lupus erythematosus, discoid lupus erythematosus, subacute cutaneous lupus erythematosus, and dermatomysositis.13 The staining for these diseases correlates with the site of the associated inflammatory component seen on hematoxylin and eosin staining. The staining of C3d along the basement membrane of stratified squamous epithelium in the lymphoepithelial cyst of the pancreas seen in our patient closely resembles the staining seen in cutaneous BP.
A proposed mechanism for BP in our patient would be exposure of BP-1 antigen in the pancreatic cyst leading to antibody recognition and C3 deposition along the basement membrane in the cyst, as evidenced by C3d immunoperoxidase staining. The IgG and C3 deposition along the cutaneous basement membrane would then represent a systemic response to the antigen exposure in the cyst. Thus, the lymphoepithelial cyst provided the immunologic stimulus for the development of the cutaneous BP. This theory is based on the observation of our patient’s rapid improvement without a change in his treatment regimen immediately after surgical excision of the cyst.
Despite the plausibility of our hypothesis, several questions remain regarding the validity of our assumptions. Although sensitive for C3 deposition, C3d immunoperoxidase staining is not specific for BP. If the proposed mechanism for causation is true, one might have expected that a subepithelial cleft along the basement membrane of the pancreatic cyst would be observed, which was not seen. A repeat BP antigen antibody was not obtained, which would have been helpful in determining if there was clearance of the antibody that would have correlated with the clinical resolution of the BP lesions.
Conclusion
Our case suggests that paraneoplastic BP is a genuine entity. Indeed, the primary tumor itself may be the immunologic stimulus in the development of BP. Recalcitrant BP should raise the question of a neoplastic process that is exposing the BP antigen. If a thorough review of systems accompanied by corroborating laboratory studies suggests a neoplastic process, the suspect lesion should be further evaluated and surgically excised if clinically indicated. Further evaluation of neoplasms with advanced staining methods may aid in establishing the causative nature of tumors in the development of BP.
Acknowledgments
We are grateful to John Stanley, MD, and Aimee Payne, MD (both from Philadelphia, Pennsylvania), for theirinsights into this case.
Bullous pemphigoid (BP) is an acquired, autoimmune, subepidermal blistering disease that is more common in elderly patients.1 An association with internal neoplasms and BP has been established; however, there is debate regarding the precise nature of the relationship.2 Several gastrointestinal tract tumors have been associated with BP, including adenocarcinoma of the colon, adenosquamous cell carcinoma and adenocarcinoma of the stomach, adenocarcinoma of the rectum, and liver and bile duct malignancies.3-5 Association with pancreatic neoplasms (eg, carcinoma of the pancreas) rarely has been reported.5-7 We present an unusual case of a lymphoepithelial cyst of the pancreas in a patient with BP.
Case Report
A 67-year-old man presented with erythematous crusted plaques and pink scars over the chest, back, arms, and legs (Figure 1). A 1.5-cm tense bullous lesion was observed on the right knee. The patient’s medical history was notable for biopsy-proven BP of 8 months’ duration as well as diabetes mellitus and hypothyroidism. The patient was being followed by his surgeon for a 1.9-cm soft-tissue lesion in the pancreatic tail and was awaiting surgical excision at the time of the current presentation. The pancreatic lesion was discovered incidentally on magnetic resonance imaging performed following urologic concerns. At the current presentation, the patient’s medications included nifedipine, hydralazine, metoprolol, torsemide, aspirin, levothyroxine, atorvastatin, insulin lispro, and insulin glargine. He had previously been treated for BP with prednisone at a maximum dosage of 60 mg daily, clobetasol propionate cream 0.05%, and mupirocin ointment 2% without improvement. Because of substantial weight gain and poorly controlled diabetes, prednisone was discontinued.
Bullous pemphigoid had been diagnosed histopathologically by a prior dermatologist. Hematoxylin and eosin staining demonstrated a subepidermal separation with eosinophils within the perivascular infiltrate (Figure 2). Direct immunofluorescence was noted in a linear pattern at the dermoepidermal junction with IgG and C3. Bullous pemphigoid antigen antibodies 1 and 2 were obtained via enzyme-linked immunosorbent assay with a positive BP-1 antigen antibody of 19 U/mL (positive, >15 U/mL) and a normal BP-2 antigen antibody of less than 9 U/mL (reference range, <9 U/mL). The glucagon level was elevated at 245 pg/mL (reference range, ≤134 pg/mL).
The patient was prescribed minocycline 100 mg twice daily and niacinamide 500 mg 3 times daily. Topical treatment with clobetasol and mupirocin was continued. One month later, the patient returned with an increase in disease activity. Changes to his therapeutic regimen were deferred until after excision of the pancreatic lesion based on the decision not to start immunosuppressive therapy until the precise nature of the pancreatic lesion was determined.
The patient underwent excision of the pancreatic lesion approximately 3 months later, which proved to be a benign lymphoepithelial cyst of the pancreas. Histology of the cyst consisted of dense fibrous tissue with a squamous epithelial lining focally infiltrated by lymphocytes (Figure 3A). Immunoperoxidase staining of the cyst revealed focal linear areas of C3d staining along the basement membrane of the stratified squamous epithelium (Figure 3B).
The patient stated that his skin started to improve virtually immediately following the excision without systemic treatment for BP. On follow-up examination 3 weeks postoperatively, no bullae were observed and there was a notable decrease in erythematous crusted plaques (Figure 4).
Comment
Paraneoplastic BP has been documented; however, lymphoepithelial cysts of the pancreas in association with BP are rare. We propose that the lymphoepithelial cyst of the pancreas provided the immunologic stimulus for the development of cutaneous BP based on the observation that our patient’s condition remarkably improved with resection of the tumor.
There are fewer than 100 cases of lymphoepithelial cysts of the pancreas reported in the literature.8 The histologic appearance is consistent with a true cyst exhibiting a well-differentiated stratified squamous epithelium, often with keratinization, surrounded by lymphoid tissue. These tumors are most commonly seen in middle-aged men and are frequently found incidentally,8-10 as was the case with our patient. Although histologically similar, lymphoepithelial cysts of the pancreas are considered distinct from lymphoepithelial cysts of the parotid gland or head and neck region.10 Lymphoepithelial cysts of the pancreas are unrelated to elevated glucagon levels; it is likely that our patient’s glucagon levels were associated with his history of diabetes.11
The diagnosis of BP is characteristically confirmed by direct immunofluorescence. Although it was performed for our patient’s cutaneous lesions, it was not obtained for the lymphoepithelial cyst of the pancreas. Once the diagnosis of the lymphoepithelial cyst of the pancreas was established, as direct immunofluorescence could not be performed in formalin-fixed tissue, immunoperoxidase staining with C3d was obtained. C3 has a well-established role in activation of complement and as a marker in BP. Deposition of C3d is a result of deactivation of C3b, a cleavage product of C3. In a study of 6 autoimmune blistering disorders that included 32 patients with BP, Pfaltz et al12 found positive immunoperoxidase staining for C3d in 31 of 32 patients, which translated to a sensitivity of 97%, a positive predictive value of 100%, and a negative predictive value of 98% among the blistering diseases being studied. Similarly, Magro and Dyrsen13 had positive staining of C3d in 17 of 17 (100%) patients with BP.
In theory, any process that involves deposition of C3 should be positive for C3d on immunoperoxidase staining. Other dermatologic inflammatory conditions stain positively with C3d, such as systemic lupus erythematosus, discoid lupus erythematosus, subacute cutaneous lupus erythematosus, and dermatomysositis.13 The staining for these diseases correlates with the site of the associated inflammatory component seen on hematoxylin and eosin staining. The staining of C3d along the basement membrane of stratified squamous epithelium in the lymphoepithelial cyst of the pancreas seen in our patient closely resembles the staining seen in cutaneous BP.
A proposed mechanism for BP in our patient would be exposure of BP-1 antigen in the pancreatic cyst leading to antibody recognition and C3 deposition along the basement membrane in the cyst, as evidenced by C3d immunoperoxidase staining. The IgG and C3 deposition along the cutaneous basement membrane would then represent a systemic response to the antigen exposure in the cyst. Thus, the lymphoepithelial cyst provided the immunologic stimulus for the development of the cutaneous BP. This theory is based on the observation of our patient’s rapid improvement without a change in his treatment regimen immediately after surgical excision of the cyst.
Despite the plausibility of our hypothesis, several questions remain regarding the validity of our assumptions. Although sensitive for C3 deposition, C3d immunoperoxidase staining is not specific for BP. If the proposed mechanism for causation is true, one might have expected that a subepithelial cleft along the basement membrane of the pancreatic cyst would be observed, which was not seen. A repeat BP antigen antibody was not obtained, which would have been helpful in determining if there was clearance of the antibody that would have correlated with the clinical resolution of the BP lesions.
Conclusion
Our case suggests that paraneoplastic BP is a genuine entity. Indeed, the primary tumor itself may be the immunologic stimulus in the development of BP. Recalcitrant BP should raise the question of a neoplastic process that is exposing the BP antigen. If a thorough review of systems accompanied by corroborating laboratory studies suggests a neoplastic process, the suspect lesion should be further evaluated and surgically excised if clinically indicated. Further evaluation of neoplasms with advanced staining methods may aid in establishing the causative nature of tumors in the development of BP.
Acknowledgments
We are grateful to John Stanley, MD, and Aimee Payne, MD (both from Philadelphia, Pennsylvania), for theirinsights into this case.
- Charneux J, Lorin J, Vitry F, et al. Usefulness of BP230 and BP180-NC16a enzyme-linked immunosorbent assays in the initial diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:286-291.
- Patel M, Sniha AA, Gilbert E. Bullous pemphigoid associated with renal cell carcinoma and invasive squamous cell carcinoma. J Drugs Dermatol. 2012;11:234-238.
- Song HJ, Han SH, Hong WK, et al. Paraneoplastic bullous pemphigoid: clinical disease activity correlated with enzyme-linked immunosorbent assay index for NC16A domain of BP180. J Dermatol. 2009;36:66-68.
- Muramatsu T, Iida T, Tada H, et al. Bullous pemphigoid associated with internal malignancies: identification of 180-kDa antigen by Western immunoblotting. Br J Dermatol. 1996;135:782-784.
- Ogawa H, Sakuma M, Morioka S, et al. The incidence of internal malignancies in pemphigus and bullous pemphigoid in Japan. J Dermatol Sci. 1995;9:136-141.
- Boyd RV. Pemphigoid and carcinoma of the pancreas. Br Med J. 1964;1:1092.
- Eustace S, Morrow G, O’Loughlin S, et al. The role of computed tomography and sonography in acute bullous pemphigoid. Ir J Med Sci. 1993;162:401-404.
- Clemente G, Sarno G, De Rose AM, et al. Lymphoepithelial cyst of the pancreas: case report and review of the literature. Acta Gastroenterol Belg. 2011;74:343-346.
- Frezza E, Wachtel MS. Lymphoepithelial cyst of the pancreas tail. case report and review of the literature. JOP. 2008;9:46-49.
- Basturk O, Coban I, Adsay NV. Pancreatic cysts: pathologic classification, differential diagnosis and clinical implications. Arch Pathol Lab Med. 2009;133:423-438.
- Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122:4-12.
- Pfaltz K, Mertz K, Rose C, et al. C3d immunohistochemistry on formalin-fixed tissue is a valuable tool in the diagnosis of bullous pemphigoid of the skin. J Cutan Pathol. 2010;37:654-658.
- Magro CM, Dyrsen ME. The use of C3d and C4d immunohistochemistry on formalin-fixed tissue as a diagnostic adjunct in the assessment of inflammatory skin disease. J Am Acad Dermatol. 2008;59:822-833.
- Charneux J, Lorin J, Vitry F, et al. Usefulness of BP230 and BP180-NC16a enzyme-linked immunosorbent assays in the initial diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:286-291.
- Patel M, Sniha AA, Gilbert E. Bullous pemphigoid associated with renal cell carcinoma and invasive squamous cell carcinoma. J Drugs Dermatol. 2012;11:234-238.
- Song HJ, Han SH, Hong WK, et al. Paraneoplastic bullous pemphigoid: clinical disease activity correlated with enzyme-linked immunosorbent assay index for NC16A domain of BP180. J Dermatol. 2009;36:66-68.
- Muramatsu T, Iida T, Tada H, et al. Bullous pemphigoid associated with internal malignancies: identification of 180-kDa antigen by Western immunoblotting. Br J Dermatol. 1996;135:782-784.
- Ogawa H, Sakuma M, Morioka S, et al. The incidence of internal malignancies in pemphigus and bullous pemphigoid in Japan. J Dermatol Sci. 1995;9:136-141.
- Boyd RV. Pemphigoid and carcinoma of the pancreas. Br Med J. 1964;1:1092.
- Eustace S, Morrow G, O’Loughlin S, et al. The role of computed tomography and sonography in acute bullous pemphigoid. Ir J Med Sci. 1993;162:401-404.
- Clemente G, Sarno G, De Rose AM, et al. Lymphoepithelial cyst of the pancreas: case report and review of the literature. Acta Gastroenterol Belg. 2011;74:343-346.
- Frezza E, Wachtel MS. Lymphoepithelial cyst of the pancreas tail. case report and review of the literature. JOP. 2008;9:46-49.
- Basturk O, Coban I, Adsay NV. Pancreatic cysts: pathologic classification, differential diagnosis and clinical implications. Arch Pathol Lab Med. 2009;133:423-438.
- Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122:4-12.
- Pfaltz K, Mertz K, Rose C, et al. C3d immunohistochemistry on formalin-fixed tissue is a valuable tool in the diagnosis of bullous pemphigoid of the skin. J Cutan Pathol. 2010;37:654-658.
- Magro CM, Dyrsen ME. The use of C3d and C4d immunohistochemistry on formalin-fixed tissue as a diagnostic adjunct in the assessment of inflammatory skin disease. J Am Acad Dermatol. 2008;59:822-833.
Epistaxis, mass in right nostril • Dx?
THE CASE
A 49-year-old woman visited our family medicine clinic because she’d had 3 episodes of epistaxis during the previous month. She’d already visited the emergency department, and the doctor there had treated her symptomatically and referred her to our clinic.
On physical examination, we noted a whitish mass in the patient’s right nostril that was attached to the nasal septum. The patient’s vital signs were within normal limits. She had a history of hypertension, depression, anxiety, gastroesophageal reflux disease, and post-traumatic stress disorder. Her medications included amlodipine-benazepril, atenolol-chlorthalidone, citalopram, clonazepam, prazosin, and omeprazole. The patient lived alone and denied using tobacco or illicit drugs, but she drank one to 2 glasses of brandy every day. She denied any past medical or family history of similar complaints, autoimmune disorders, or skin rashes.
A complete blood count, international normalized ratio, sedimentation rate, anti-nuclear antibody test, and an anti-neutrophil cytoplasmic antibody panel were normal.
THE DIAGNOSIS
We referred the patient to an ear, nose, and throat doctor for a nasal endoscopy and a biopsy, which showed granulation tissue. A maxillofacial computed tomography (CT) scan revealed a 1.44 cm x 0.8 cm polypoid soft tissue mass in the right nasal cavity adherent to the nasal septum with no posterior extension (FIGURE 1).
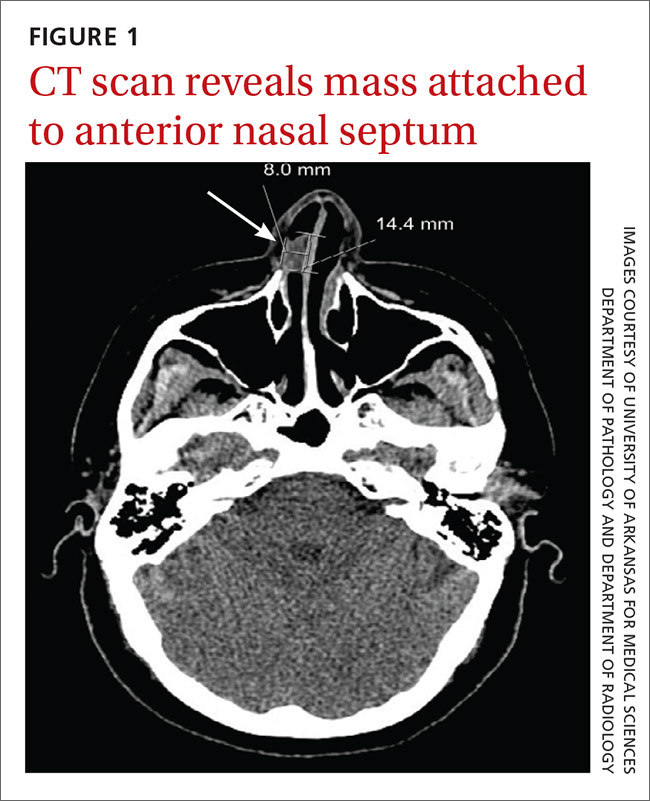
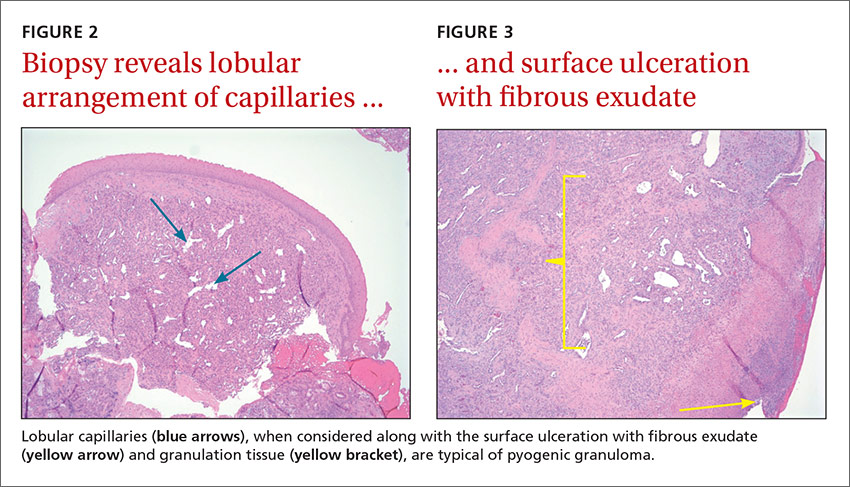
DISCUSSION
Pyogenic granuloma (PG) is a benign vascular tumor of the skin and mucous membranes that is not associated with an infection. Rather, it is a hyperplastic, neovascular, inflammatory response to an angiogenic stimulus. Several enhancers and inhibitors of angiogenesis have been shown to play a role in PG, including hormones, medications, and local injury. In fact, a local injury or hormonal factor is identified as a stimulus in more than half of PG patients.1
The hormone connection. Estrogen promotes production of nerve growth factor, granulocyte-macrophage colony-stimulating factor, basic fibroblast growth factor, vascular endothelial growth factor, and transforming growth factor beta 1. Progesterone enhances inflammatory mediators as well. Although there are no direct receptors for estrogen and progesterone in the oral and nasal mucosa, some of these pro-inflammatory effects create an environment conducive to the development of PG. This is supported by several studies documenting an increased incidence of PGs with oral contraceptive use and regression of PGs after childbirth.2-4
Medication may play a role. Drug-induced PG has also been described in several studies.5,6 Offending medications include systemic and topical retinoids, capecitabine, etoposide, 5-fluorouracil, cyclosporine, docetaxel, and human immunodeficiency virus protease inhibitors.
Local injury may also be a culprit. Nasal PGs are commonly attached to the anterior septum and typically result from nasal packing, habitual picking, or nose boring.7 In this particular case, however, we were unable to identify the irritant.
The classic presentation
PG classically presents as a painless mass that spontaneously develops over days to weeks. The mass can be sessile or pedunculated, and is frequently hemorrhagic. Intranasal PG usually presents with epiphora.7 While the prevalence of intraoral PG was found to be one in 25,000 individuals3, data for nasal lesions is scarce. Most cases of PG are seen in the second and third decades of life.1,3 In children, PG is slightly more predominant in males.1,3 Mucosal lesions, however, have a higher incidence in females.1,3 Granuloma gravidarum, the term used to describe mucosal PG in pregnant females, was found in 0.2% to 5% of pregnancies.2,3,8
Differential Dx includes warts, squamous cell carcinoma
The differential diagnosis of PG includes Spitz nevus, glomus tumors, common warts, amelanotic melanoma, squamous cell carcinoma, basal cell carcinoma, Kaposi’s sarcoma, bacillary angiomatosis, infantile hemangioma, and angiolymphoid hyperplasia, among others.3,5 Foreign bodies, nasal polyps, angiofibroma, meningocele, Wegener’s granulomatosis, and sarcoidosis should also be considered.
Radiologic evaluation may be beneficial—especially with nasal lesions—when looking for findings suggestive of malignancy. Both CT and magnetic resonance imaging with contrast identify PG as a soft tissue mass with lobulated contours,9,10 but histopathologic analysis is required to confirm the diagnosis. The histopathologic appearance of PG is characterized by a polypoid lesion with circumscribed anastomosing networks of capillaries arranged in one or more lobules at the base in an edematous and fibroblastic stroma.
Treatment is determined by the location and size of the lesion
The most suitable treatment is determined by considering the location of the lesion, the characteristics of the lesion (morphology/size), its amenability to surgery, risk of scar formation, and the presence or absence of a causative irritant. Excision is often preferred because it yields a specimen for pathologic analysis. Alternative treatments include electrocautery, cryotherapy, laser therapy, and intralesional and topical agents,3,6,7 but the recurrence rate is higher (up to 15%) with some of these modalities, when compared with excision (3.6%).3
Our patient underwent excision of the mass and was seen for an annual follow-up appointment. All of her symptoms resolved and no recurrence was noted.
THE TAKEAWAY
Although PG is a common and benign condition, it is rarely seen in the nasal cavity without an obvious history of a possible irritant. PG should be considered as a diagnosis for rapidly growing cutaneous or mucosal hemorrhagic lesions. Appropriate tissue pathology is essential to rule out malignancy and other serious conditions, such as bacillary angiomatosis and Wegener’s granulomatosis.
Treatment is usually required to avoid the frequent complications of ulceration and bleeding. Surgical treatments are preferred. The location of the lesion largely determines whether referral to a specialist is necessary.
1. Harris MN, Desai R, Chuang TY, et al. Lobular capillary hemangiomas: An epidemiologic report, with emphasis on cutaneous lesions. J Am Acad Dermatol. 2000;42:1012-1016.
2. Yuan K, Jin YT, Lin MT. The detection and comparison of angiogenesis-associated factors in pyogenic granuloma by immunohistochemistry. J Periodontol. 2000;71:701-709.
3. Giblin AV, Clover AJ, Athanassopoulos A, et al. Pyogenic granuloma–the quest for optimum treatment: audit of treatment of 408 cases. J Plast Reconstr Aesthet Surg. 2007;60:1030-1035.
4. Steelman R, Holmes D. Pregnancy tumor in a 16-year-old: case report and treatment considerations. J Clin Pediatr Dent. 1992;16:217-218.
5. Jafarzadeh H, Sanatkhani M, Mohtasham N. Oral pyogenic granuloma: a review. J Oral Sci. 2006;48:167-175.
6. Piraccini BM, Bellavista S, Misciali C, et al. Periungual and subungual pyogenic granuloma. Br J Dermatol. 2010;163:941-953.
7. Ozcan C, Apa DD, Görür K. Pediatric lobular capillary hemangioma of the nasal cavity. Eur Arch Otorhinolaryngol. 2004;261:449-451.
8. Henry F, Quatresooz P, Valverde-Lopez JC, et al. Blood vessel changes during pregnancy: a review. Am J Clin Dermatol. 2006;7:65-69.
9. Puxeddu R, Berlucchi M, Ledda GP, et al. Lobular capillary hemangioma of the nasal cavity: A retrospective study on 40 patients. Am J Rhinol. 2006;20:480-484.
10. Maroldi R, Berlucchi M, Farina D, et al. Benign neoplasms and tumor-like lesions. In: Maroldi R, Nicolai P, eds. Imaging in Treatment Planning for Sinonasal Diseases. Berlin, Heidelberg, New York: Springer-Verlag; 2005:107-158.
THE CASE
A 49-year-old woman visited our family medicine clinic because she’d had 3 episodes of epistaxis during the previous month. She’d already visited the emergency department, and the doctor there had treated her symptomatically and referred her to our clinic.
On physical examination, we noted a whitish mass in the patient’s right nostril that was attached to the nasal septum. The patient’s vital signs were within normal limits. She had a history of hypertension, depression, anxiety, gastroesophageal reflux disease, and post-traumatic stress disorder. Her medications included amlodipine-benazepril, atenolol-chlorthalidone, citalopram, clonazepam, prazosin, and omeprazole. The patient lived alone and denied using tobacco or illicit drugs, but she drank one to 2 glasses of brandy every day. She denied any past medical or family history of similar complaints, autoimmune disorders, or skin rashes.
A complete blood count, international normalized ratio, sedimentation rate, anti-nuclear antibody test, and an anti-neutrophil cytoplasmic antibody panel were normal.
THE DIAGNOSIS
We referred the patient to an ear, nose, and throat doctor for a nasal endoscopy and a biopsy, which showed granulation tissue. A maxillofacial computed tomography (CT) scan revealed a 1.44 cm x 0.8 cm polypoid soft tissue mass in the right nasal cavity adherent to the nasal septum with no posterior extension (FIGURE 1).
DISCUSSION
Pyogenic granuloma (PG) is a benign vascular tumor of the skin and mucous membranes that is not associated with an infection. Rather, it is a hyperplastic, neovascular, inflammatory response to an angiogenic stimulus. Several enhancers and inhibitors of angiogenesis have been shown to play a role in PG, including hormones, medications, and local injury. In fact, a local injury or hormonal factor is identified as a stimulus in more than half of PG patients.1
The hormone connection. Estrogen promotes production of nerve growth factor, granulocyte-macrophage colony-stimulating factor, basic fibroblast growth factor, vascular endothelial growth factor, and transforming growth factor beta 1. Progesterone enhances inflammatory mediators as well. Although there are no direct receptors for estrogen and progesterone in the oral and nasal mucosa, some of these pro-inflammatory effects create an environment conducive to the development of PG. This is supported by several studies documenting an increased incidence of PGs with oral contraceptive use and regression of PGs after childbirth.2-4
Medication may play a role. Drug-induced PG has also been described in several studies.5,6 Offending medications include systemic and topical retinoids, capecitabine, etoposide, 5-fluorouracil, cyclosporine, docetaxel, and human immunodeficiency virus protease inhibitors.
Local injury may also be a culprit. Nasal PGs are commonly attached to the anterior septum and typically result from nasal packing, habitual picking, or nose boring.7 In this particular case, however, we were unable to identify the irritant.
The classic presentation
PG classically presents as a painless mass that spontaneously develops over days to weeks. The mass can be sessile or pedunculated, and is frequently hemorrhagic. Intranasal PG usually presents with epiphora.7 While the prevalence of intraoral PG was found to be one in 25,000 individuals3, data for nasal lesions is scarce. Most cases of PG are seen in the second and third decades of life.1,3 In children, PG is slightly more predominant in males.1,3 Mucosal lesions, however, have a higher incidence in females.1,3 Granuloma gravidarum, the term used to describe mucosal PG in pregnant females, was found in 0.2% to 5% of pregnancies.2,3,8
Differential Dx includes warts, squamous cell carcinoma
The differential diagnosis of PG includes Spitz nevus, glomus tumors, common warts, amelanotic melanoma, squamous cell carcinoma, basal cell carcinoma, Kaposi’s sarcoma, bacillary angiomatosis, infantile hemangioma, and angiolymphoid hyperplasia, among others.3,5 Foreign bodies, nasal polyps, angiofibroma, meningocele, Wegener’s granulomatosis, and sarcoidosis should also be considered.
Radiologic evaluation may be beneficial—especially with nasal lesions—when looking for findings suggestive of malignancy. Both CT and magnetic resonance imaging with contrast identify PG as a soft tissue mass with lobulated contours,9,10 but histopathologic analysis is required to confirm the diagnosis. The histopathologic appearance of PG is characterized by a polypoid lesion with circumscribed anastomosing networks of capillaries arranged in one or more lobules at the base in an edematous and fibroblastic stroma.
Treatment is determined by the location and size of the lesion
The most suitable treatment is determined by considering the location of the lesion, the characteristics of the lesion (morphology/size), its amenability to surgery, risk of scar formation, and the presence or absence of a causative irritant. Excision is often preferred because it yields a specimen for pathologic analysis. Alternative treatments include electrocautery, cryotherapy, laser therapy, and intralesional and topical agents,3,6,7 but the recurrence rate is higher (up to 15%) with some of these modalities, when compared with excision (3.6%).3
Our patient underwent excision of the mass and was seen for an annual follow-up appointment. All of her symptoms resolved and no recurrence was noted.
THE TAKEAWAY
Although PG is a common and benign condition, it is rarely seen in the nasal cavity without an obvious history of a possible irritant. PG should be considered as a diagnosis for rapidly growing cutaneous or mucosal hemorrhagic lesions. Appropriate tissue pathology is essential to rule out malignancy and other serious conditions, such as bacillary angiomatosis and Wegener’s granulomatosis.
Treatment is usually required to avoid the frequent complications of ulceration and bleeding. Surgical treatments are preferred. The location of the lesion largely determines whether referral to a specialist is necessary.
THE CASE
A 49-year-old woman visited our family medicine clinic because she’d had 3 episodes of epistaxis during the previous month. She’d already visited the emergency department, and the doctor there had treated her symptomatically and referred her to our clinic.
On physical examination, we noted a whitish mass in the patient’s right nostril that was attached to the nasal septum. The patient’s vital signs were within normal limits. She had a history of hypertension, depression, anxiety, gastroesophageal reflux disease, and post-traumatic stress disorder. Her medications included amlodipine-benazepril, atenolol-chlorthalidone, citalopram, clonazepam, prazosin, and omeprazole. The patient lived alone and denied using tobacco or illicit drugs, but she drank one to 2 glasses of brandy every day. She denied any past medical or family history of similar complaints, autoimmune disorders, or skin rashes.
A complete blood count, international normalized ratio, sedimentation rate, anti-nuclear antibody test, and an anti-neutrophil cytoplasmic antibody panel were normal.
THE DIAGNOSIS
We referred the patient to an ear, nose, and throat doctor for a nasal endoscopy and a biopsy, which showed granulation tissue. A maxillofacial computed tomography (CT) scan revealed a 1.44 cm x 0.8 cm polypoid soft tissue mass in the right nasal cavity adherent to the nasal septum with no posterior extension (FIGURE 1).
DISCUSSION
Pyogenic granuloma (PG) is a benign vascular tumor of the skin and mucous membranes that is not associated with an infection. Rather, it is a hyperplastic, neovascular, inflammatory response to an angiogenic stimulus. Several enhancers and inhibitors of angiogenesis have been shown to play a role in PG, including hormones, medications, and local injury. In fact, a local injury or hormonal factor is identified as a stimulus in more than half of PG patients.1
The hormone connection. Estrogen promotes production of nerve growth factor, granulocyte-macrophage colony-stimulating factor, basic fibroblast growth factor, vascular endothelial growth factor, and transforming growth factor beta 1. Progesterone enhances inflammatory mediators as well. Although there are no direct receptors for estrogen and progesterone in the oral and nasal mucosa, some of these pro-inflammatory effects create an environment conducive to the development of PG. This is supported by several studies documenting an increased incidence of PGs with oral contraceptive use and regression of PGs after childbirth.2-4
Medication may play a role. Drug-induced PG has also been described in several studies.5,6 Offending medications include systemic and topical retinoids, capecitabine, etoposide, 5-fluorouracil, cyclosporine, docetaxel, and human immunodeficiency virus protease inhibitors.
Local injury may also be a culprit. Nasal PGs are commonly attached to the anterior septum and typically result from nasal packing, habitual picking, or nose boring.7 In this particular case, however, we were unable to identify the irritant.
The classic presentation
PG classically presents as a painless mass that spontaneously develops over days to weeks. The mass can be sessile or pedunculated, and is frequently hemorrhagic. Intranasal PG usually presents with epiphora.7 While the prevalence of intraoral PG was found to be one in 25,000 individuals3, data for nasal lesions is scarce. Most cases of PG are seen in the second and third decades of life.1,3 In children, PG is slightly more predominant in males.1,3 Mucosal lesions, however, have a higher incidence in females.1,3 Granuloma gravidarum, the term used to describe mucosal PG in pregnant females, was found in 0.2% to 5% of pregnancies.2,3,8
Differential Dx includes warts, squamous cell carcinoma
The differential diagnosis of PG includes Spitz nevus, glomus tumors, common warts, amelanotic melanoma, squamous cell carcinoma, basal cell carcinoma, Kaposi’s sarcoma, bacillary angiomatosis, infantile hemangioma, and angiolymphoid hyperplasia, among others.3,5 Foreign bodies, nasal polyps, angiofibroma, meningocele, Wegener’s granulomatosis, and sarcoidosis should also be considered.
Radiologic evaluation may be beneficial—especially with nasal lesions—when looking for findings suggestive of malignancy. Both CT and magnetic resonance imaging with contrast identify PG as a soft tissue mass with lobulated contours,9,10 but histopathologic analysis is required to confirm the diagnosis. The histopathologic appearance of PG is characterized by a polypoid lesion with circumscribed anastomosing networks of capillaries arranged in one or more lobules at the base in an edematous and fibroblastic stroma.
Treatment is determined by the location and size of the lesion
The most suitable treatment is determined by considering the location of the lesion, the characteristics of the lesion (morphology/size), its amenability to surgery, risk of scar formation, and the presence or absence of a causative irritant. Excision is often preferred because it yields a specimen for pathologic analysis. Alternative treatments include electrocautery, cryotherapy, laser therapy, and intralesional and topical agents,3,6,7 but the recurrence rate is higher (up to 15%) with some of these modalities, when compared with excision (3.6%).3
Our patient underwent excision of the mass and was seen for an annual follow-up appointment. All of her symptoms resolved and no recurrence was noted.
THE TAKEAWAY
Although PG is a common and benign condition, it is rarely seen in the nasal cavity without an obvious history of a possible irritant. PG should be considered as a diagnosis for rapidly growing cutaneous or mucosal hemorrhagic lesions. Appropriate tissue pathology is essential to rule out malignancy and other serious conditions, such as bacillary angiomatosis and Wegener’s granulomatosis.
Treatment is usually required to avoid the frequent complications of ulceration and bleeding. Surgical treatments are preferred. The location of the lesion largely determines whether referral to a specialist is necessary.
1. Harris MN, Desai R, Chuang TY, et al. Lobular capillary hemangiomas: An epidemiologic report, with emphasis on cutaneous lesions. J Am Acad Dermatol. 2000;42:1012-1016.
2. Yuan K, Jin YT, Lin MT. The detection and comparison of angiogenesis-associated factors in pyogenic granuloma by immunohistochemistry. J Periodontol. 2000;71:701-709.
3. Giblin AV, Clover AJ, Athanassopoulos A, et al. Pyogenic granuloma–the quest for optimum treatment: audit of treatment of 408 cases. J Plast Reconstr Aesthet Surg. 2007;60:1030-1035.
4. Steelman R, Holmes D. Pregnancy tumor in a 16-year-old: case report and treatment considerations. J Clin Pediatr Dent. 1992;16:217-218.
5. Jafarzadeh H, Sanatkhani M, Mohtasham N. Oral pyogenic granuloma: a review. J Oral Sci. 2006;48:167-175.
6. Piraccini BM, Bellavista S, Misciali C, et al. Periungual and subungual pyogenic granuloma. Br J Dermatol. 2010;163:941-953.
7. Ozcan C, Apa DD, Görür K. Pediatric lobular capillary hemangioma of the nasal cavity. Eur Arch Otorhinolaryngol. 2004;261:449-451.
8. Henry F, Quatresooz P, Valverde-Lopez JC, et al. Blood vessel changes during pregnancy: a review. Am J Clin Dermatol. 2006;7:65-69.
9. Puxeddu R, Berlucchi M, Ledda GP, et al. Lobular capillary hemangioma of the nasal cavity: A retrospective study on 40 patients. Am J Rhinol. 2006;20:480-484.
10. Maroldi R, Berlucchi M, Farina D, et al. Benign neoplasms and tumor-like lesions. In: Maroldi R, Nicolai P, eds. Imaging in Treatment Planning for Sinonasal Diseases. Berlin, Heidelberg, New York: Springer-Verlag; 2005:107-158.
1. Harris MN, Desai R, Chuang TY, et al. Lobular capillary hemangiomas: An epidemiologic report, with emphasis on cutaneous lesions. J Am Acad Dermatol. 2000;42:1012-1016.
2. Yuan K, Jin YT, Lin MT. The detection and comparison of angiogenesis-associated factors in pyogenic granuloma by immunohistochemistry. J Periodontol. 2000;71:701-709.
3. Giblin AV, Clover AJ, Athanassopoulos A, et al. Pyogenic granuloma–the quest for optimum treatment: audit of treatment of 408 cases. J Plast Reconstr Aesthet Surg. 2007;60:1030-1035.
4. Steelman R, Holmes D. Pregnancy tumor in a 16-year-old: case report and treatment considerations. J Clin Pediatr Dent. 1992;16:217-218.
5. Jafarzadeh H, Sanatkhani M, Mohtasham N. Oral pyogenic granuloma: a review. J Oral Sci. 2006;48:167-175.
6. Piraccini BM, Bellavista S, Misciali C, et al. Periungual and subungual pyogenic granuloma. Br J Dermatol. 2010;163:941-953.
7. Ozcan C, Apa DD, Görür K. Pediatric lobular capillary hemangioma of the nasal cavity. Eur Arch Otorhinolaryngol. 2004;261:449-451.
8. Henry F, Quatresooz P, Valverde-Lopez JC, et al. Blood vessel changes during pregnancy: a review. Am J Clin Dermatol. 2006;7:65-69.
9. Puxeddu R, Berlucchi M, Ledda GP, et al. Lobular capillary hemangioma of the nasal cavity: A retrospective study on 40 patients. Am J Rhinol. 2006;20:480-484.
10. Maroldi R, Berlucchi M, Farina D, et al. Benign neoplasms and tumor-like lesions. In: Maroldi R, Nicolai P, eds. Imaging in Treatment Planning for Sinonasal Diseases. Berlin, Heidelberg, New York: Springer-Verlag; 2005:107-158.

One lab finding, 2 vastly different causes
CASE 1
A 13-month-old boy who was recently adopted from Ethiopia presented to a primary care physician with a 3-week history of bloody diarrhea accompanied by flatulence and bloating. Stool cultures were positive for Campylobacter and Shigella. He was prescribed azithromycin but saw only moderate improvement. He was then referred to the Infectious Diseases Department. Neonatal, pregnancy, and immunization histories were unknown and a review of systems was unremarkable. On exam, the child looked well; he weighed 9.6 kg (15th percentile), was 69.5 cm long (<3rd percentile), and his head circumference was 45 cm (10th percentile). Head and neck, cardiorespiratory, and abdominal examinations were unremarkable.
A complete blood count (CBC) showed an elevated white blood cell (WBC) count of 26 x 109/L (normal: 4-10 x 109/L) with predominant eosinophilia (10.4 x 109/L or 40.1% of WBCs; normal: <0.45 x 109/L or 0%-8%). Hemoglobin and platelets were within normal limits. Stool testing for ova and parasites showed Strongyloides stercoralis larvae. Strongyloides serology was negative and Filaria serology was equivocal.
CASE 2
A 15-year-old boy was assessed for a 3-week history of fever and eosinophilia. He had enlarged cervical lymph nodes, a new rash, and had lost 4 pounds. He denied gastrointestinal symptoms, dyspnea, headaches, or chest pain. His past medical and family histories were unremarkable and he reported no drug use or allergies. He had traveled to Cuba with his family for 15 days 3 months prior to presentation. He recalled diarrhea while traveling, which resolved spontaneously. He and his family had traveled “off the beaten track,” eating foods prepared at local establishments and swimming in local rivers. He received pre-travel immunizations.
On examination, he appeared unwell, though his vital signs were normal. He had diffuse lymphadenopathy and a petechial rash on his chest, back, upper buttocks, legs, and feet. Cardiorespiratory and abdominal examinations were unremarkable. A CBC revealed an elevated WBC count of 76.9 x 109/L with predominant eosinophilia (71.5 x 109/L or 92% of WBCs). Hemoglobin, platelets, electrolytes, and liver function tests were normal. The patient was referred to a tertiary care center and was admitted to the hospital. Stool testing for ova and parasites, as well as serology for parasitic infections, was negative. A bone marrow aspiration and biopsy were performed and revealed the diagnosis of acute lymphoblastic leukemia (ALL).
DISCUSSION
These 2 cases highlight how the presentation of eosinophilia can vary and how important it is to maintain a broad differential diagnosis (TABLE 11-4). Causes of eosinophilia are numerous and can be divided into 3 categories: primary, secondary, and idiopathic.1,5 Hematologic malignancy, where eosinophilia is clonal, is an example of a primary etiology. Causes of secondary eosinophilia include infectious diseases, drugs (TABLE 25), autoimmune disorders, and allergic conditions. Prolonged eosinophilia that is >3 x 109/L is associated with end-organ damage. Dermatologic, pulmonary, gastrointestinal, and cardiac involvement is most common.2
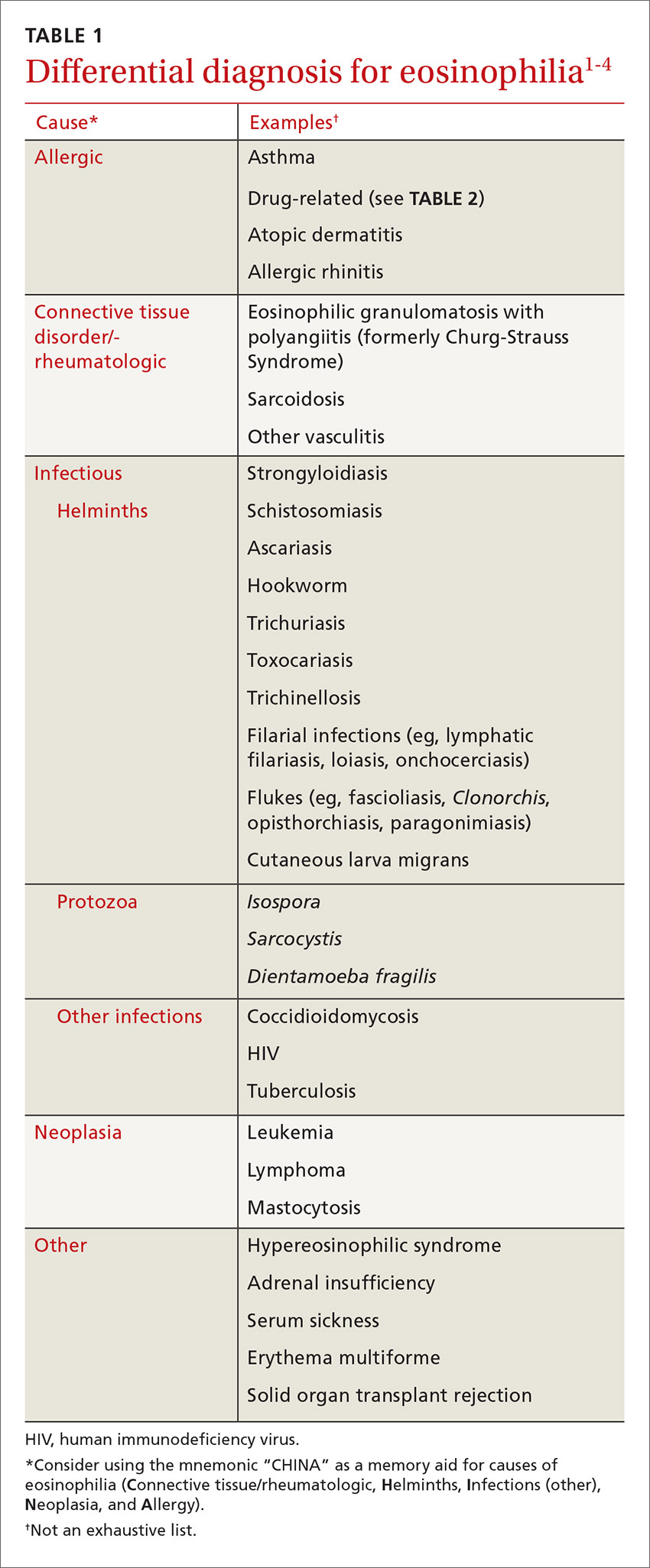
Eosinophilia associated with parasitic infection
In returning travelers and international adoptees, multicellular helminthic parasites are the most common causes of eosinophilia, with eosinophilia occurring during tissue migration or penetration.1,3

Schistosomiasis is a chronic parasitic infection of the human vascular system. It is transmitted by contact with contaminated fresh water, where cercariae penetrate the skin. High prevalence areas include Africa and Southeast Asia. Acute infection can result in Katayama fever—a febrile illness with prominent eosinophilia that occurs 4 to 7 weeks after exposure.4 Diagnosis is primarily clinical with appropriate epidemiology, as serology may be negative early in infection. Praziquantel is the treatment of choice, though dosing varies by species, so expert consultation should be considered.
Soil-transmitted helminths, such as Ascaris (Ascaris lumbricoides), whipworm (Trichuris trichiura), and hookworm (Ancyclostoma duodenale and Necator americanus), can also cause eosinophilia during larval tissue migration. Following infection by ingestion or skin penetration, an acute respiratory illness, termed Löffler’s syndrome, can develop with associated eosinophilia.1 Once the helminths reach the adult stage, eosinophilia subsides. Patients are most commonly treated with albendazole 400 mg orally for 3 days.4
Fascioliasis is common in sheep-rearing areas. Humans are infected through ingestion of aquatic plants (eg, watercress). Parasitic migration through the duodenal wall and liver parenchyma can lead to fever, right upper quadrant pain, and eosinophilia. The incubation period is 6 to 12 weeks. Diagnosis during acute infection is by serology.4
Filarial infections, eg lymphatic filariasis, loiasis, and onchocerciasis, can also cause eosinophilia. The rise in eosinophils can be triggered by either the adult worms or circulating microfilariae.4 Treatment of fascioliasis and filarial infections varies and expert consultation is recommended.
Eosinophilia associated with primary hematologic malignancy
Eosinophilia is a rare presentation of hematologic malignancy. Acute myeloid leukemia, acute lymphoblastic leukemia (ALL), chronic myeloid leukemia, and myeloproliferative disorders have all been associated with eosinophilia. Hepatosplenomegaly, generalized lymphadenopathy, and cytopenias in other cell lines are often noted. Also, the degree of eosinophilia is often more pronounced (>5 x 109/L). Patients with suspected hematologic malignancy should be urgently referred for expert consultation.5
A systematic approach to patients with eosinophilia
Consider the following approach in the assessment of patients with eosinophilia seen in the ambulatory care setting. Inpatients or patients being seen in developing areas may require a modified approach.
History. All patients with eosinophilia should have a thorough history taken, with particular attention paid to travel history. A travel history should make note of dates, duration and location of travel, and any relevant exposures, such as arthropod bites or swimming in freshwater. Dietary habits, such as ingestion of seafood, game, or undercooked meat can also be helpful in making a diagnosis.3,4
Physical exam. In addition to a general physical examination, the following features may be helpful in determining the etiology of eosinophilia. Wheeze is characteristic of parasites in a lung migration phase (eg, strongyloidiasis and ascariasis) or asthma. Hepatomegaly can be seen with liver flukes, visceral larva migrans, or schistosomiasis. Periorbital edema can be observed with Trichinella infection. Loa loa, a type of filarial infection, produces a transient, migratory angioedema, often localized to the wrists and large joints (termed Calabar swelling). Dermatitis of varying intensity may suggest filarial infection, schistosomiasis, or atopy. Perianal dermatitis is observed with strongyloidiasis. Cutaneous larva migrans is characterized by a linear, serpiginous rash.3,4
Laboratory investigations. Investigation will vary depending on the patient’s history, exposures, exam findings, and degree of eosinophilia. Any patient who is unwell or has significant eosinophilia (≥3 x 109/L) may warrant more urgent referral to infectious disease, travel medicine, or hematology. Basic laboratory investigations should include a CBC with differential, routine serum chemistries, and liver enzymes. In the setting of significant eosinophilia, an electrocardiogram, cardiac enzyme levels, and a chest x-ray should be obtained to screen for end-organ damage related to eosinophilia.3-5
In patients in whom you suspect hematologic malignancy, bone marrow aspiration and biopsy are often needed to make the diagnosis.5
Parasitic infections are most often diagnosed on stool examination for ova and parasites or by serology. Stool should be collected on 3 separate days to increase diagnostic yield. Certain species of Schistosoma can also be diagnosed on direct microscopy of urine specimens. Serologic assays are available for schistosomiasis, strongyloidiasis, Toxocara, fascioliasis, filariasis, and Trichinella. Further investigations for filiariasis, including blood films, eye exam, and skin snips will vary with filarial species, so expert consultation should be considered.3,4
Our patients. The first patient with strongyloidiasis was treated with ivermectin 200 µg/kg/day orally for 2 days and experienced symptomatic improvement and resolution of eosinophilia. The second patient with ALL was admitted and referred to hematology and received induction chemotherapy. Treatment was well tolerated and the patient was discharged one week later, with appropriate follow-up.
THE TAKEAWAY
Eosinophilia is commonly encountered in primary care. The approach to eosinophilia and the differential diagnosis can be challenging. The correct diagnosis was reached in both cases by maintaining a broad differential diagnosis. Obtaining a travel and exposure history is fundamental, although noninfectious causes, including allergy, malignancy, and drug reaction, must always be considered.
1. Moore TA, Nutman TB. Eosinophilia in the returning traveler. Infect Dis Clin North Am. 1998;12:503-521.
2. Tefferi A, Gotlib J, Pardanani A. Hypereosinophilic syndrome and clonal eosinophilia: point-of-care diagnostic algorithm and treatment update. Mayo Clin Proc. 2010;85:158-164.
3. Schulte C, Krebs B, Jelinek T, et al. Diagnostic significance of blood eosinophilia in returning travelers. Clin Infect Dis. 2002;34:407-411.
4. Checkley AM, Chiodini PL, Dockrell DH, et al; British Infection Society and Hospital for Tropical Diseases. Eosinophilia in returning travellers and migrants from the tropics: UK recommendations for investigation and initial management. J Infect. 2010;60:1-20.
5. Tefferi A, Patnaik MM, Pardanani A. Eosinophilia: secondary, clonal and idiopathic. Br J Haematol. 2006;133:468-492.
CASE 1
A 13-month-old boy who was recently adopted from Ethiopia presented to a primary care physician with a 3-week history of bloody diarrhea accompanied by flatulence and bloating. Stool cultures were positive for Campylobacter and Shigella. He was prescribed azithromycin but saw only moderate improvement. He was then referred to the Infectious Diseases Department. Neonatal, pregnancy, and immunization histories were unknown and a review of systems was unremarkable. On exam, the child looked well; he weighed 9.6 kg (15th percentile), was 69.5 cm long (<3rd percentile), and his head circumference was 45 cm (10th percentile). Head and neck, cardiorespiratory, and abdominal examinations were unremarkable.
A complete blood count (CBC) showed an elevated white blood cell (WBC) count of 26 x 109/L (normal: 4-10 x 109/L) with predominant eosinophilia (10.4 x 109/L or 40.1% of WBCs; normal: <0.45 x 109/L or 0%-8%). Hemoglobin and platelets were within normal limits. Stool testing for ova and parasites showed Strongyloides stercoralis larvae. Strongyloides serology was negative and Filaria serology was equivocal.
CASE 2
A 15-year-old boy was assessed for a 3-week history of fever and eosinophilia. He had enlarged cervical lymph nodes, a new rash, and had lost 4 pounds. He denied gastrointestinal symptoms, dyspnea, headaches, or chest pain. His past medical and family histories were unremarkable and he reported no drug use or allergies. He had traveled to Cuba with his family for 15 days 3 months prior to presentation. He recalled diarrhea while traveling, which resolved spontaneously. He and his family had traveled “off the beaten track,” eating foods prepared at local establishments and swimming in local rivers. He received pre-travel immunizations.
On examination, he appeared unwell, though his vital signs were normal. He had diffuse lymphadenopathy and a petechial rash on his chest, back, upper buttocks, legs, and feet. Cardiorespiratory and abdominal examinations were unremarkable. A CBC revealed an elevated WBC count of 76.9 x 109/L with predominant eosinophilia (71.5 x 109/L or 92% of WBCs). Hemoglobin, platelets, electrolytes, and liver function tests were normal. The patient was referred to a tertiary care center and was admitted to the hospital. Stool testing for ova and parasites, as well as serology for parasitic infections, was negative. A bone marrow aspiration and biopsy were performed and revealed the diagnosis of acute lymphoblastic leukemia (ALL).
DISCUSSION
These 2 cases highlight how the presentation of eosinophilia can vary and how important it is to maintain a broad differential diagnosis (TABLE 11-4). Causes of eosinophilia are numerous and can be divided into 3 categories: primary, secondary, and idiopathic.1,5 Hematologic malignancy, where eosinophilia is clonal, is an example of a primary etiology. Causes of secondary eosinophilia include infectious diseases, drugs (TABLE 25), autoimmune disorders, and allergic conditions. Prolonged eosinophilia that is >3 x 109/L is associated with end-organ damage. Dermatologic, pulmonary, gastrointestinal, and cardiac involvement is most common.2
Eosinophilia associated with parasitic infection
In returning travelers and international adoptees, multicellular helminthic parasites are the most common causes of eosinophilia, with eosinophilia occurring during tissue migration or penetration.1,3
Schistosomiasis is a chronic parasitic infection of the human vascular system. It is transmitted by contact with contaminated fresh water, where cercariae penetrate the skin. High prevalence areas include Africa and Southeast Asia. Acute infection can result in Katayama fever—a febrile illness with prominent eosinophilia that occurs 4 to 7 weeks after exposure.4 Diagnosis is primarily clinical with appropriate epidemiology, as serology may be negative early in infection. Praziquantel is the treatment of choice, though dosing varies by species, so expert consultation should be considered.
Soil-transmitted helminths, such as Ascaris (Ascaris lumbricoides), whipworm (Trichuris trichiura), and hookworm (Ancyclostoma duodenale and Necator americanus), can also cause eosinophilia during larval tissue migration. Following infection by ingestion or skin penetration, an acute respiratory illness, termed Löffler’s syndrome, can develop with associated eosinophilia.1 Once the helminths reach the adult stage, eosinophilia subsides. Patients are most commonly treated with albendazole 400 mg orally for 3 days.4
Fascioliasis is common in sheep-rearing areas. Humans are infected through ingestion of aquatic plants (eg, watercress). Parasitic migration through the duodenal wall and liver parenchyma can lead to fever, right upper quadrant pain, and eosinophilia. The incubation period is 6 to 12 weeks. Diagnosis during acute infection is by serology.4
Filarial infections, eg lymphatic filariasis, loiasis, and onchocerciasis, can also cause eosinophilia. The rise in eosinophils can be triggered by either the adult worms or circulating microfilariae.4 Treatment of fascioliasis and filarial infections varies and expert consultation is recommended.
Eosinophilia associated with primary hematologic malignancy
Eosinophilia is a rare presentation of hematologic malignancy. Acute myeloid leukemia, acute lymphoblastic leukemia (ALL), chronic myeloid leukemia, and myeloproliferative disorders have all been associated with eosinophilia. Hepatosplenomegaly, generalized lymphadenopathy, and cytopenias in other cell lines are often noted. Also, the degree of eosinophilia is often more pronounced (>5 x 109/L). Patients with suspected hematologic malignancy should be urgently referred for expert consultation.5
A systematic approach to patients with eosinophilia
Consider the following approach in the assessment of patients with eosinophilia seen in the ambulatory care setting. Inpatients or patients being seen in developing areas may require a modified approach.
History. All patients with eosinophilia should have a thorough history taken, with particular attention paid to travel history. A travel history should make note of dates, duration and location of travel, and any relevant exposures, such as arthropod bites or swimming in freshwater. Dietary habits, such as ingestion of seafood, game, or undercooked meat can also be helpful in making a diagnosis.3,4
Physical exam. In addition to a general physical examination, the following features may be helpful in determining the etiology of eosinophilia. Wheeze is characteristic of parasites in a lung migration phase (eg, strongyloidiasis and ascariasis) or asthma. Hepatomegaly can be seen with liver flukes, visceral larva migrans, or schistosomiasis. Periorbital edema can be observed with Trichinella infection. Loa loa, a type of filarial infection, produces a transient, migratory angioedema, often localized to the wrists and large joints (termed Calabar swelling). Dermatitis of varying intensity may suggest filarial infection, schistosomiasis, or atopy. Perianal dermatitis is observed with strongyloidiasis. Cutaneous larva migrans is characterized by a linear, serpiginous rash.3,4
Laboratory investigations. Investigation will vary depending on the patient’s history, exposures, exam findings, and degree of eosinophilia. Any patient who is unwell or has significant eosinophilia (≥3 x 109/L) may warrant more urgent referral to infectious disease, travel medicine, or hematology. Basic laboratory investigations should include a CBC with differential, routine serum chemistries, and liver enzymes. In the setting of significant eosinophilia, an electrocardiogram, cardiac enzyme levels, and a chest x-ray should be obtained to screen for end-organ damage related to eosinophilia.3-5
In patients in whom you suspect hematologic malignancy, bone marrow aspiration and biopsy are often needed to make the diagnosis.5
Parasitic infections are most often diagnosed on stool examination for ova and parasites or by serology. Stool should be collected on 3 separate days to increase diagnostic yield. Certain species of Schistosoma can also be diagnosed on direct microscopy of urine specimens. Serologic assays are available for schistosomiasis, strongyloidiasis, Toxocara, fascioliasis, filariasis, and Trichinella. Further investigations for filiariasis, including blood films, eye exam, and skin snips will vary with filarial species, so expert consultation should be considered.3,4
Our patients. The first patient with strongyloidiasis was treated with ivermectin 200 µg/kg/day orally for 2 days and experienced symptomatic improvement and resolution of eosinophilia. The second patient with ALL was admitted and referred to hematology and received induction chemotherapy. Treatment was well tolerated and the patient was discharged one week later, with appropriate follow-up.
THE TAKEAWAY
Eosinophilia is commonly encountered in primary care. The approach to eosinophilia and the differential diagnosis can be challenging. The correct diagnosis was reached in both cases by maintaining a broad differential diagnosis. Obtaining a travel and exposure history is fundamental, although noninfectious causes, including allergy, malignancy, and drug reaction, must always be considered.
CASE 1
A 13-month-old boy who was recently adopted from Ethiopia presented to a primary care physician with a 3-week history of bloody diarrhea accompanied by flatulence and bloating. Stool cultures were positive for Campylobacter and Shigella. He was prescribed azithromycin but saw only moderate improvement. He was then referred to the Infectious Diseases Department. Neonatal, pregnancy, and immunization histories were unknown and a review of systems was unremarkable. On exam, the child looked well; he weighed 9.6 kg (15th percentile), was 69.5 cm long (<3rd percentile), and his head circumference was 45 cm (10th percentile). Head and neck, cardiorespiratory, and abdominal examinations were unremarkable.
A complete blood count (CBC) showed an elevated white blood cell (WBC) count of 26 x 109/L (normal: 4-10 x 109/L) with predominant eosinophilia (10.4 x 109/L or 40.1% of WBCs; normal: <0.45 x 109/L or 0%-8%). Hemoglobin and platelets were within normal limits. Stool testing for ova and parasites showed Strongyloides stercoralis larvae. Strongyloides serology was negative and Filaria serology was equivocal.
CASE 2
A 15-year-old boy was assessed for a 3-week history of fever and eosinophilia. He had enlarged cervical lymph nodes, a new rash, and had lost 4 pounds. He denied gastrointestinal symptoms, dyspnea, headaches, or chest pain. His past medical and family histories were unremarkable and he reported no drug use or allergies. He had traveled to Cuba with his family for 15 days 3 months prior to presentation. He recalled diarrhea while traveling, which resolved spontaneously. He and his family had traveled “off the beaten track,” eating foods prepared at local establishments and swimming in local rivers. He received pre-travel immunizations.
On examination, he appeared unwell, though his vital signs were normal. He had diffuse lymphadenopathy and a petechial rash on his chest, back, upper buttocks, legs, and feet. Cardiorespiratory and abdominal examinations were unremarkable. A CBC revealed an elevated WBC count of 76.9 x 109/L with predominant eosinophilia (71.5 x 109/L or 92% of WBCs). Hemoglobin, platelets, electrolytes, and liver function tests were normal. The patient was referred to a tertiary care center and was admitted to the hospital. Stool testing for ova and parasites, as well as serology for parasitic infections, was negative. A bone marrow aspiration and biopsy were performed and revealed the diagnosis of acute lymphoblastic leukemia (ALL).
DISCUSSION
These 2 cases highlight how the presentation of eosinophilia can vary and how important it is to maintain a broad differential diagnosis (TABLE 11-4). Causes of eosinophilia are numerous and can be divided into 3 categories: primary, secondary, and idiopathic.1,5 Hematologic malignancy, where eosinophilia is clonal, is an example of a primary etiology. Causes of secondary eosinophilia include infectious diseases, drugs (TABLE 25), autoimmune disorders, and allergic conditions. Prolonged eosinophilia that is >3 x 109/L is associated with end-organ damage. Dermatologic, pulmonary, gastrointestinal, and cardiac involvement is most common.2
Eosinophilia associated with parasitic infection
In returning travelers and international adoptees, multicellular helminthic parasites are the most common causes of eosinophilia, with eosinophilia occurring during tissue migration or penetration.1,3
Schistosomiasis is a chronic parasitic infection of the human vascular system. It is transmitted by contact with contaminated fresh water, where cercariae penetrate the skin. High prevalence areas include Africa and Southeast Asia. Acute infection can result in Katayama fever—a febrile illness with prominent eosinophilia that occurs 4 to 7 weeks after exposure.4 Diagnosis is primarily clinical with appropriate epidemiology, as serology may be negative early in infection. Praziquantel is the treatment of choice, though dosing varies by species, so expert consultation should be considered.
Soil-transmitted helminths, such as Ascaris (Ascaris lumbricoides), whipworm (Trichuris trichiura), and hookworm (Ancyclostoma duodenale and Necator americanus), can also cause eosinophilia during larval tissue migration. Following infection by ingestion or skin penetration, an acute respiratory illness, termed Löffler’s syndrome, can develop with associated eosinophilia.1 Once the helminths reach the adult stage, eosinophilia subsides. Patients are most commonly treated with albendazole 400 mg orally for 3 days.4
Fascioliasis is common in sheep-rearing areas. Humans are infected through ingestion of aquatic plants (eg, watercress). Parasitic migration through the duodenal wall and liver parenchyma can lead to fever, right upper quadrant pain, and eosinophilia. The incubation period is 6 to 12 weeks. Diagnosis during acute infection is by serology.4
Filarial infections, eg lymphatic filariasis, loiasis, and onchocerciasis, can also cause eosinophilia. The rise in eosinophils can be triggered by either the adult worms or circulating microfilariae.4 Treatment of fascioliasis and filarial infections varies and expert consultation is recommended.
Eosinophilia associated with primary hematologic malignancy
Eosinophilia is a rare presentation of hematologic malignancy. Acute myeloid leukemia, acute lymphoblastic leukemia (ALL), chronic myeloid leukemia, and myeloproliferative disorders have all been associated with eosinophilia. Hepatosplenomegaly, generalized lymphadenopathy, and cytopenias in other cell lines are often noted. Also, the degree of eosinophilia is often more pronounced (>5 x 109/L). Patients with suspected hematologic malignancy should be urgently referred for expert consultation.5
A systematic approach to patients with eosinophilia
Consider the following approach in the assessment of patients with eosinophilia seen in the ambulatory care setting. Inpatients or patients being seen in developing areas may require a modified approach.
History. All patients with eosinophilia should have a thorough history taken, with particular attention paid to travel history. A travel history should make note of dates, duration and location of travel, and any relevant exposures, such as arthropod bites or swimming in freshwater. Dietary habits, such as ingestion of seafood, game, or undercooked meat can also be helpful in making a diagnosis.3,4
Physical exam. In addition to a general physical examination, the following features may be helpful in determining the etiology of eosinophilia. Wheeze is characteristic of parasites in a lung migration phase (eg, strongyloidiasis and ascariasis) or asthma. Hepatomegaly can be seen with liver flukes, visceral larva migrans, or schistosomiasis. Periorbital edema can be observed with Trichinella infection. Loa loa, a type of filarial infection, produces a transient, migratory angioedema, often localized to the wrists and large joints (termed Calabar swelling). Dermatitis of varying intensity may suggest filarial infection, schistosomiasis, or atopy. Perianal dermatitis is observed with strongyloidiasis. Cutaneous larva migrans is characterized by a linear, serpiginous rash.3,4
Laboratory investigations. Investigation will vary depending on the patient’s history, exposures, exam findings, and degree of eosinophilia. Any patient who is unwell or has significant eosinophilia (≥3 x 109/L) may warrant more urgent referral to infectious disease, travel medicine, or hematology. Basic laboratory investigations should include a CBC with differential, routine serum chemistries, and liver enzymes. In the setting of significant eosinophilia, an electrocardiogram, cardiac enzyme levels, and a chest x-ray should be obtained to screen for end-organ damage related to eosinophilia.3-5
In patients in whom you suspect hematologic malignancy, bone marrow aspiration and biopsy are often needed to make the diagnosis.5
Parasitic infections are most often diagnosed on stool examination for ova and parasites or by serology. Stool should be collected on 3 separate days to increase diagnostic yield. Certain species of Schistosoma can also be diagnosed on direct microscopy of urine specimens. Serologic assays are available for schistosomiasis, strongyloidiasis, Toxocara, fascioliasis, filariasis, and Trichinella. Further investigations for filiariasis, including blood films, eye exam, and skin snips will vary with filarial species, so expert consultation should be considered.3,4
Our patients. The first patient with strongyloidiasis was treated with ivermectin 200 µg/kg/day orally for 2 days and experienced symptomatic improvement and resolution of eosinophilia. The second patient with ALL was admitted and referred to hematology and received induction chemotherapy. Treatment was well tolerated and the patient was discharged one week later, with appropriate follow-up.
THE TAKEAWAY
Eosinophilia is commonly encountered in primary care. The approach to eosinophilia and the differential diagnosis can be challenging. The correct diagnosis was reached in both cases by maintaining a broad differential diagnosis. Obtaining a travel and exposure history is fundamental, although noninfectious causes, including allergy, malignancy, and drug reaction, must always be considered.
1. Moore TA, Nutman TB. Eosinophilia in the returning traveler. Infect Dis Clin North Am. 1998;12:503-521.
2. Tefferi A, Gotlib J, Pardanani A. Hypereosinophilic syndrome and clonal eosinophilia: point-of-care diagnostic algorithm and treatment update. Mayo Clin Proc. 2010;85:158-164.
3. Schulte C, Krebs B, Jelinek T, et al. Diagnostic significance of blood eosinophilia in returning travelers. Clin Infect Dis. 2002;34:407-411.
4. Checkley AM, Chiodini PL, Dockrell DH, et al; British Infection Society and Hospital for Tropical Diseases. Eosinophilia in returning travellers and migrants from the tropics: UK recommendations for investigation and initial management. J Infect. 2010;60:1-20.
5. Tefferi A, Patnaik MM, Pardanani A. Eosinophilia: secondary, clonal and idiopathic. Br J Haematol. 2006;133:468-492.
1. Moore TA, Nutman TB. Eosinophilia in the returning traveler. Infect Dis Clin North Am. 1998;12:503-521.
2. Tefferi A, Gotlib J, Pardanani A. Hypereosinophilic syndrome and clonal eosinophilia: point-of-care diagnostic algorithm and treatment update. Mayo Clin Proc. 2010;85:158-164.
3. Schulte C, Krebs B, Jelinek T, et al. Diagnostic significance of blood eosinophilia in returning travelers. Clin Infect Dis. 2002;34:407-411.
4. Checkley AM, Chiodini PL, Dockrell DH, et al; British Infection Society and Hospital for Tropical Diseases. Eosinophilia in returning travellers and migrants from the tropics: UK recommendations for investigation and initial management. J Infect. 2010;60:1-20.
5. Tefferi A, Patnaik MM, Pardanani A. Eosinophilia: secondary, clonal and idiopathic. Br J Haematol. 2006;133:468-492.

An Atypical Angiomyomatous Hamartoma With Unexplained Hepatosplenomegaly
Angiomyomatous hamartoma (AMH) of the lymph node is an extremely uncommon vascular disorder of unknown etiology, first described by Chan and colleagues in 1992.1-3 Angiomyomatous hamartoma particularly involves inguinal and femoral lymph nodes, with few cases reported in the cervical, popliteal, and submandibular lymph nodes.1 Angiomyomatous hamartoma can occasionally be associated with edema of the ipsilateral limb. To the authors’ knowledge, to date only 18 cases of AMH have been reported.4
Case Presentation
A 40-year-old white man started to have a left inguinal and scrotal pain along with left thigh swelling at age 22 while serving in the U.S. Army.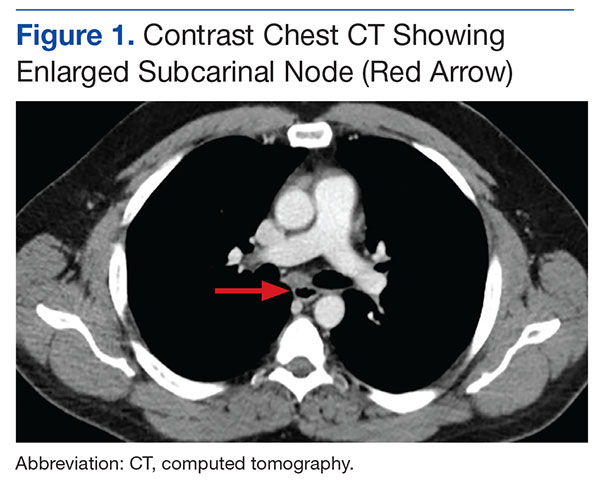

An abdominal Doppler ultrasound did not show any evidence of portal hypertension. A thoraco-abdomino-pelvic computed tomography (CT) scan showed bilateral axillary, subcarinal (Figure 1), mesenteric and retroperitoneal (Figure 2), and left inguinal (Figure 3) lymphadenopathy. Excisional biopsy of a 3.5 x 2.5 x 1.5 cm left inguinal lymph node was performed, and histopathology showed extensive smooth muscle and vascular proliferation replacing most of the lymph node (Figure 4), a finding consistent with AMH. A trichrome staining (Figure 5) and immunohistochemical study for smooth muscle actin (Figure 6) were performed and supported the diagnosis. Due to persistent pain in the scrotal area, the patient underwent a left spermatic cord denervation. Currently, the patient has persistent left thigh swelling. His condition remains stable with a regular follow-up CT scan showing unchanged lymphadenopathy.
Discussion
Angiomyomatous hamartoma is a rare, primary vascular tumor of the lymph nodes occurring almost exclusively in the inguinal and femoral lymph nodes and occasionally associated with edema of the ipsilateral limb.1 A few cases with popliteal and cervical lymph node involvement have been reported.1 There are no prior reports of cases with either generalized adenopathy or hepatosplenomegaly.
The histopathogenesis of AMH remains unclear. Chan and colleagues first reported this distinct clinicopathologic entity in 1992 as a primary vascular tumor of the lymph node.1-3 The hamartomatous nature of the disease was postulated by the authors on the basis of a disorganized growth pattern of smooth muscle cells and blood vessels noted on pathology.2,3 The AMH could represent a localized malformation in a congenitally damaged lymphatic vessel system.5 Other hypothesis suggests lymphedema as a possible etiology of AMH through continuous stimulation of lymphatic vessels, which triggers vasoproliferation and eventually the vascular transformation of the lymph nodes.5
Differential diagnoses of AMH include nodal lymphangiomyomatosis, which is most prevalent in women, particularly presenting with thoracic and intra-abdominal lymph nodes and plumper HMB45 (human melanoma black 45) -positive tumor cells6; leiomyomato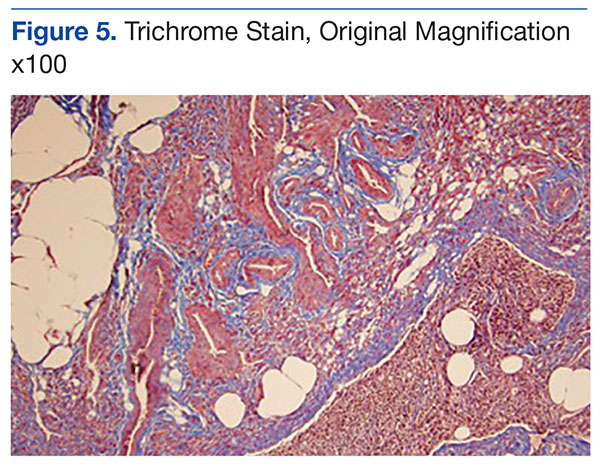
Treatment is either conservative or surgical, depending on clinical judgment. This is only the 19th case of AMH reported so far in the literature and the fifth reported case in which the patient presented with ipsilateral lymphedema of the limb. Importantly, it is the first reported case with generalized (axillary, subcarinal, mesenteric, inguinal and retroperitoneal) lymphadenopathy and unexplained hepatosplenomegaly.
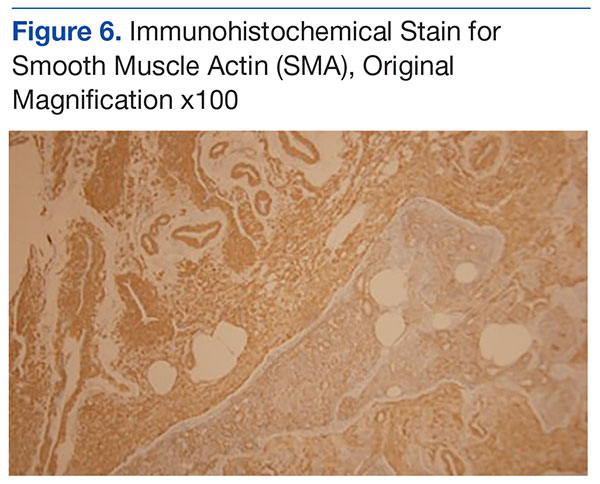
Conclusion
Angiomyomatous hamartoma of the lymph nodes is an exceedingly rare diagnosis but should be considered when evaluating patients with lymphatic tumors. This patient remains relatively asymptomatic and on observation at this time and seems to have more extensive disease than prior reports in the literature.
1. Mridha AR, Ranjan R, Kinra P, Ray R, Khan SA, Shivanand G. Angiomyomatous hamartoma of popliteal lymph node: an unusual entity. J Pathol Transl Med. 2015;49(2):156-158.
2. Dargent JL, Lespagnard L, Verdebout JM, Bourgeois P, Munck D. Glomeruloid microvascular proliferation in angiomyomatous hamartoma of the lymph node. Virchows Arch. 2004;445(3):320-322.
3. Chan JK, Frizzera G, Fletcher CD, Rosai J. Primary vascular tumors of lymph nodes other than Kaposi’s sarcoma. Analysis of 39 cases and delineation of two new entities. Am J Surg Pathol. 1992;16(4):335-350.
4. Ram M, Alsanjari N, Ansari N. Angiomyomatous hamartoma: a rare case report with review of the literature. Rare Tumors. 2009;1(2):e25.
5. Piedimonte A, De Nictolis M, Lorenzini P, Sperti V, Bertani A. Angiomyomatous hamartoma of inguinal lymph nodes. Plast Reconstr Surg. 2006;117(2):714-716.
6. Lee CH, Chang TC, Ku JW. Angiomyomatous hamartoma in an inguinal lymph node with proliferating pericytes/smooth muscle cells, plexiform vessel tangles, and ectopic calcification. Indian J Pathol Microbiol. 2015;58(2):226-228.
Angiomyomatous hamartoma (AMH) of the lymph node is an extremely uncommon vascular disorder of unknown etiology, first described by Chan and colleagues in 1992.1-3 Angiomyomatous hamartoma particularly involves inguinal and femoral lymph nodes, with few cases reported in the cervical, popliteal, and submandibular lymph nodes.1 Angiomyomatous hamartoma can occasionally be associated with edema of the ipsilateral limb. To the authors’ knowledge, to date only 18 cases of AMH have been reported.4
Case Presentation
A 40-year-old white man started to have a left inguinal and scrotal pain along with left thigh swelling at age 22 while serving in the U.S. Army.
An abdominal Doppler ultrasound did not show any evidence of portal hypertension. A thoraco-abdomino-pelvic computed tomography (CT) scan showed bilateral axillary, subcarinal (Figure 1), mesenteric and retroperitoneal (Figure 2), and left inguinal (Figure 3) lymphadenopathy. Excisional biopsy of a 3.5 x 2.5 x 1.5 cm left inguinal lymph node was performed, and histopathology showed extensive smooth muscle and vascular proliferation replacing most of the lymph node (Figure 4), a finding consistent with AMH. A trichrome staining (Figure 5) and immunohistochemical study for smooth muscle actin (Figure 6) were performed and supported the diagnosis. Due to persistent pain in the scrotal area, the patient underwent a left spermatic cord denervation. Currently, the patient has persistent left thigh swelling. His condition remains stable with a regular follow-up CT scan showing unchanged lymphadenopathy.
Discussion
Angiomyomatous hamartoma is a rare, primary vascular tumor of the lymph nodes occurring almost exclusively in the inguinal and femoral lymph nodes and occasionally associated with edema of the ipsilateral limb.1 A few cases with popliteal and cervical lymph node involvement have been reported.1 There are no prior reports of cases with either generalized adenopathy or hepatosplenomegaly.
The histopathogenesis of AMH remains unclear. Chan and colleagues first reported this distinct clinicopathologic entity in 1992 as a primary vascular tumor of the lymph node.1-3 The hamartomatous nature of the disease was postulated by the authors on the basis of a disorganized growth pattern of smooth muscle cells and blood vessels noted on pathology.2,3 The AMH could represent a localized malformation in a congenitally damaged lymphatic vessel system.5 Other hypothesis suggests lymphedema as a possible etiology of AMH through continuous stimulation of lymphatic vessels, which triggers vasoproliferation and eventually the vascular transformation of the lymph nodes.5
Differential diagnoses of AMH include nodal lymphangiomyomatosis, which is most prevalent in women, particularly presenting with thoracic and intra-abdominal lymph nodes and plumper HMB45 (human melanoma black 45) -positive tumor cells6; leiomyomato
Treatment is either conservative or surgical, depending on clinical judgment. This is only the 19th case of AMH reported so far in the literature and the fifth reported case in which the patient presented with ipsilateral lymphedema of the limb. Importantly, it is the first reported case with generalized (axillary, subcarinal, mesenteric, inguinal and retroperitoneal) lymphadenopathy and unexplained hepatosplenomegaly.
Conclusion
Angiomyomatous hamartoma of the lymph nodes is an exceedingly rare diagnosis but should be considered when evaluating patients with lymphatic tumors. This patient remains relatively asymptomatic and on observation at this time and seems to have more extensive disease than prior reports in the literature.
Angiomyomatous hamartoma (AMH) of the lymph node is an extremely uncommon vascular disorder of unknown etiology, first described by Chan and colleagues in 1992.1-3 Angiomyomatous hamartoma particularly involves inguinal and femoral lymph nodes, with few cases reported in the cervical, popliteal, and submandibular lymph nodes.1 Angiomyomatous hamartoma can occasionally be associated with edema of the ipsilateral limb. To the authors’ knowledge, to date only 18 cases of AMH have been reported.4
Case Presentation
A 40-year-old white man started to have a left inguinal and scrotal pain along with left thigh swelling at age 22 while serving in the U.S. Army.
An abdominal Doppler ultrasound did not show any evidence of portal hypertension. A thoraco-abdomino-pelvic computed tomography (CT) scan showed bilateral axillary, subcarinal (Figure 1), mesenteric and retroperitoneal (Figure 2), and left inguinal (Figure 3) lymphadenopathy. Excisional biopsy of a 3.5 x 2.5 x 1.5 cm left inguinal lymph node was performed, and histopathology showed extensive smooth muscle and vascular proliferation replacing most of the lymph node (Figure 4), a finding consistent with AMH. A trichrome staining (Figure 5) and immunohistochemical study for smooth muscle actin (Figure 6) were performed and supported the diagnosis. Due to persistent pain in the scrotal area, the patient underwent a left spermatic cord denervation. Currently, the patient has persistent left thigh swelling. His condition remains stable with a regular follow-up CT scan showing unchanged lymphadenopathy.
Discussion
Angiomyomatous hamartoma is a rare, primary vascular tumor of the lymph nodes occurring almost exclusively in the inguinal and femoral lymph nodes and occasionally associated with edema of the ipsilateral limb.1 A few cases with popliteal and cervical lymph node involvement have been reported.1 There are no prior reports of cases with either generalized adenopathy or hepatosplenomegaly.
The histopathogenesis of AMH remains unclear. Chan and colleagues first reported this distinct clinicopathologic entity in 1992 as a primary vascular tumor of the lymph node.1-3 The hamartomatous nature of the disease was postulated by the authors on the basis of a disorganized growth pattern of smooth muscle cells and blood vessels noted on pathology.2,3 The AMH could represent a localized malformation in a congenitally damaged lymphatic vessel system.5 Other hypothesis suggests lymphedema as a possible etiology of AMH through continuous stimulation of lymphatic vessels, which triggers vasoproliferation and eventually the vascular transformation of the lymph nodes.5
Differential diagnoses of AMH include nodal lymphangiomyomatosis, which is most prevalent in women, particularly presenting with thoracic and intra-abdominal lymph nodes and plumper HMB45 (human melanoma black 45) -positive tumor cells6; leiomyomato
Treatment is either conservative or surgical, depending on clinical judgment. This is only the 19th case of AMH reported so far in the literature and the fifth reported case in which the patient presented with ipsilateral lymphedema of the limb. Importantly, it is the first reported case with generalized (axillary, subcarinal, mesenteric, inguinal and retroperitoneal) lymphadenopathy and unexplained hepatosplenomegaly.
Conclusion
Angiomyomatous hamartoma of the lymph nodes is an exceedingly rare diagnosis but should be considered when evaluating patients with lymphatic tumors. This patient remains relatively asymptomatic and on observation at this time and seems to have more extensive disease than prior reports in the literature.
1. Mridha AR, Ranjan R, Kinra P, Ray R, Khan SA, Shivanand G. Angiomyomatous hamartoma of popliteal lymph node: an unusual entity. J Pathol Transl Med. 2015;49(2):156-158.
2. Dargent JL, Lespagnard L, Verdebout JM, Bourgeois P, Munck D. Glomeruloid microvascular proliferation in angiomyomatous hamartoma of the lymph node. Virchows Arch. 2004;445(3):320-322.
3. Chan JK, Frizzera G, Fletcher CD, Rosai J. Primary vascular tumors of lymph nodes other than Kaposi’s sarcoma. Analysis of 39 cases and delineation of two new entities. Am J Surg Pathol. 1992;16(4):335-350.
4. Ram M, Alsanjari N, Ansari N. Angiomyomatous hamartoma: a rare case report with review of the literature. Rare Tumors. 2009;1(2):e25.
5. Piedimonte A, De Nictolis M, Lorenzini P, Sperti V, Bertani A. Angiomyomatous hamartoma of inguinal lymph nodes. Plast Reconstr Surg. 2006;117(2):714-716.
6. Lee CH, Chang TC, Ku JW. Angiomyomatous hamartoma in an inguinal lymph node with proliferating pericytes/smooth muscle cells, plexiform vessel tangles, and ectopic calcification. Indian J Pathol Microbiol. 2015;58(2):226-228.
1. Mridha AR, Ranjan R, Kinra P, Ray R, Khan SA, Shivanand G. Angiomyomatous hamartoma of popliteal lymph node: an unusual entity. J Pathol Transl Med. 2015;49(2):156-158.
2. Dargent JL, Lespagnard L, Verdebout JM, Bourgeois P, Munck D. Glomeruloid microvascular proliferation in angiomyomatous hamartoma of the lymph node. Virchows Arch. 2004;445(3):320-322.
3. Chan JK, Frizzera G, Fletcher CD, Rosai J. Primary vascular tumors of lymph nodes other than Kaposi’s sarcoma. Analysis of 39 cases and delineation of two new entities. Am J Surg Pathol. 1992;16(4):335-350.
4. Ram M, Alsanjari N, Ansari N. Angiomyomatous hamartoma: a rare case report with review of the literature. Rare Tumors. 2009;1(2):e25.
5. Piedimonte A, De Nictolis M, Lorenzini P, Sperti V, Bertani A. Angiomyomatous hamartoma of inguinal lymph nodes. Plast Reconstr Surg. 2006;117(2):714-716.
6. Lee CH, Chang TC, Ku JW. Angiomyomatous hamartoma in an inguinal lymph node with proliferating pericytes/smooth muscle cells, plexiform vessel tangles, and ectopic calcification. Indian J Pathol Microbiol. 2015;58(2):226-228.
Dexamethasone-associated posterior reversible encephalopathy syndrome
Glenohumeral Joint Sepsis Caused by Streptococcus mitis: A Case Report
Septic arthritis predominantly involves the weight-bearing joints of the hip and knee, which account for nearly 60% of cases.1 In contrast, the shoulder joint is involved in 10% to 15% of cases, though this number may be higher among intravenous (IV) drug users.2 The most common causative organisms are the Staphylococcus species, followed closely by β-hemolytic streptococci, with these 2 groups accounting for more than 90% of all cases.3 The Streptococcus viridans group belongs to normal oral flora residing predominantly on the surface of teeth. Although well known for its ability to colonize heart valves and frequently cause bacterial endocarditis, this group has rarely been associated with septic arthritis. Furthermore, Streptococcus mitis, a subgroup of S viridans, has been implicated even less commonly.
In this article, we report a case of glenohumeral joint septic arthritis caused by S mitis. To our knowledge, such a case has not been previously reported in the English literature. Given the low virulence of this orally based bacterium, treating physicians must maintain clinical suspicion for the organism in the setting of persistent joint effusion and pain in association with periodontal disease or trauma. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A right-hand-dominant 54-year-old man presented to Dr. Gruson with complaints of persistent right shoulder pain associated with worsening range of motion (ROM). Three weeks earlier, the patient reported being assaulted and noted progressive swelling about the right shoulder. He denied fevers, chills, or prior shoulder problems. Although his past medical history was remarkable for hepatitis C and diabetes, he was not taking any diabetic medications at that time. A review of systems was remarkable for poor dental hygiene, and the patient was missing several teeth, which he said had been knocked out during the assault. Physical examination revealed diffuse tenderness about the right shoulder and severe pain with all passive movement. The shoulder was pseudoparalyzed. There were no subcutaneous collections, wounds, or ecchymosis about the shoulder. Mild calor was noted on the right shoulder relative to the left. Radiographs of the right shoulder showed no acute osseous abnormalities.
Magnetic resonance imaging (MRI), which was urgently obtained to assess the integrity of the rotator cuff and the location of the effusion, showed a large subacromial and glenohumeral joint effusion as well as diffuse muscular edema (Figures 1A-1C). 
In light of the elevated infection findings of the laboratory tests and the positive culture, urgent arthroscopic irrigation and débridement of the right shoulder were indicated. Given the organism identified, transesophageal echocardiography was performed; there were no valvular vegetations. Creation of the posterior glenohumeral portal resulted in egress of turbid fluid, which was sent for culture. The subacromial space and the glenohumeral joint were thoroughly lavaged and the copious hemorrhagic synovitis débrided (Figures 2A, 2B). 
The 8-week course of antibiotics normalized the patient’s ESR to 13 mm/h. Follow-up MRI showed improvement in the soft-tissue edema. Clinically, the patient reported minimal shoulder pain. He was undergoing physical therapy to regain strength and ROM.
Discussion
Staphylococcus aureus is the leading causative organism of septic arthritis, accounting for more than 60% of all cases.4 Conversely, the Streptococcus viridans group is rarely implicated in septic arthritis, accounting for <1% of cases.4S viridans is part of the commensal oral flora and has low virulence. This heterogeneous group is subdivided into S mitis, S salivarius, S anginosus, S mutans, and S bovis. The S mitis group is further subdivided into S sanguinis (formerly known as S sanguis) and S mitis. Infection by an organism of the S viridans group usually occurs on a previously injured focus, and the organism is a causative agent of bacterial endocarditis.5 Reported cases of septic arthritis caused by S viridans have predominantly involved the knee joint—with severe osteoarthritis, poor dental hygiene, and prior IV drug use identified as risk factors.5-7The shoulder joint is seldom involved in septic arthritis; estimated incidence is under 8%.8 Although overall incidence may rise in an increasingly elderly patient population, incidence of shoulder infection remains low.2,9
The main routes for developing septic arthritis include direct inoculation secondary to penetrating trauma or hematologic spread.10 Coatsworth and colleagues11 reported on iatrogenic S mitis septic arthritis of a shoulder arthroplasty during ultrasonography-guided aspiration by a technician who was not wearing a mask. Our institutional policy is to perform joint aspiration under strictly sterile conditions, which were adhered to in the present case. We surmise our patient developed transient bacteremia from the loss of several teeth, particularly given his poor dentition. Yombi and colleagues5 documented 2 cases of septic arthritis caused by Streptococcus gordonii, a relative of S sanguinis. One involved a previously replaced knee, and the other a native knee joint. Other cases of S viridans group septic arthritis have involved the knee,6,7,12,13 the sternoclavicular joint,14-16 and the acromioclavicular joint.17S sanguinis6,7,12,15,16 and S gordonii5 have been implicated in most cases, and an unspeciated S viridans in others.13,14,17 Concomitant periodontal disease has been reported in most cases as well,6,7,12,15 including our patient’s case. In the English-language literature, we found no other reports of S mitis as the causative agent of acute septic glenohumeral joint arthritis from hematogenous spread.
There should be no delay in diagnosing septic arthritis, and infected material should be removed from the joint. In animal models, complete joint destruction occurred only 5 weeks after inoculation with Staphylococcus aureus.10 Garofalo and colleagues18 reported a trend toward improved functional outcomes after earlier operative treatment. The choice of open surgical drainage vs repeat needle aspiration seems to be of little consequence, as both have good long-term outcomes, but open surgical drainage seems to result in better long-term functional ROM.2,9 However, results of a recent study suggested surgical treatment is not always superior to medical treatment for septic arthritis in native joints.19 In some cases involving S viridans species, treatment consisted of a combination of IV antibiotics and onetime or repeat aspiration;6,12-15 treatment in the remaining cases was surgical débridement.5,7,16,17 Given that S viridans is associated with bacterial endocarditis, echocardiography is essential if this organism is to be identified. Medical management and antibiotic treatment should be initiated after consultation with medical and infectious disease specialists.19We have reported a case of septic shoulder caused by S mitis, a low-virulence organism seldom associated with joint infection. The patient’s infection likely resulted from hematogenous spread from the oral cavity (dentition was poor). Urgent aspiration of the joint and baseline infection laboratory tests are recommended. MRI of the shoulder may show an effusion. Urgent arthroscopic irrigation and débridement can yield good clinical outcomes.
Am J Orthop. 2016;45(6):E343-E346. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Mathews CJ, Kingsley G, Field M, et al. Management of septic arthritis: a systematic review. Ann Rheum Dis. 2007;66(4):440-445.
2. Leslie BM, Harris JM 3rd, Driscoll D. Septic arthritis of the shoulder in adults. J Bone Joint Surg Am. 1989;71(10):1516-1522.
3. Gupta MN, Sturrock RD, Field M. A prospective 2-year study of 75 patients with adult-onset septic arthritis. Rheumatology. 2001;40(1):24-30.
4. Dubost JJ, Soubrier M, De Champs C, Ristori JM, Bussiere JL, Sauvezie B. No changes in the distribution of organisms responsible for septic arthritis over a 20 year period. Ann Rheum Dis. 2002;61(3):267-269.
5. Yombi J, Belkhir L, Jonckheere S, et al. Streptococcus gordonii septic arthritis: two cases and review of literature. BMC Infect Dis. 2012;12:215.
6. Papaioannides D, Boniatsi L, Korantzopoulos P, Sinapidis D, Giotis C. Acute septic arthritis due to Streptococcus sanguis. Med Princ Pract. 2006;15(1):77-79.
7. Edson RS, Osmon DR, Berry DJ. Septic arthritis due to Streptococcus sanguis. Mayo Clin Proc. 2002;77(7):709-710.
8. Weston VC, Jones AC, Bradbury N, Fawthrop F, Doherty M. Clinical features and outcome of septic arthritis in a single UK health district 1982-1991. Ann Rheum Dis. 1999;58(4):214-219.
9. Lossos IS, Yossepowitch O, Kandel L, Yardeni D, Arber N. Septic arthritis of the glenohumeral joint. A report of 11 cases and review of the literature. Medicine. 1998;77(3):177-187.
10. Esterhai JL Jr, Gelb I. Adult septic arthritis. Orthop Clin North Am. 1991;22(3):503-514.
11. Coatsworth NR, Huntington PG, Giuffre B, Kotsiou G. The doctor and the mask: iatrogenic septic arthritis caused by Streptoccocus mitis. Med J Aust. 2013;198(5):285-286.
12. Patrick MR, Lewis D. Short of a length: Streptococcus sanguis knee infection from dental source. Br J Rheumatol. 1992;31(8):569.
13. Barbadillo C, Trujillo A, Cuende E, Mazzucchelli R, Mulero J, Andreu JL. Septic arthritis due to Streptococcus viridans. Clin Exp Rheumatol. 1990;8(5):520-521.
14. Mata P, Molins A, de Oya M. Sternal arthritis caused by Streptococcus viridans in a heroin addict [in Spanish]. Med Clin. 1984;83(16):689.
15. Mandac I, Prkacin I, Sabljar Matovinovic M, Sustercic D. Septic arthritis due to Streptococcus sanguis. Coll Antropol. 2010;34(2):661-664.
16. Nitsche JF, Vaughan JH, Williams G, Curd JG. Septic sternoclavicular arthritis with Pasteurella multocida and Streptococcus sanguis. Arthritis Rheum. 1982;25(4):467-469.
17. Blankstein A, Amsallem JL, Rubenstein E, Horoszowski H, Farin I. Septic arthritis of the acromioclavicular joint. Arch Orthop Trauma Surg. 1985;103(6):417-418.
18. Garofalo R, Flanagin B, Cesari E, Vinci E, Conti M, Castagna A. Destructive septic arthritis of shoulder in adults. Musculoskelet Surg. 2014;98(supp 1):S35-S39.
19. Ravindran V, Logan I, Bourke BE. Medical vs surgical treatment for the native joint in septic arthritis: a 6-year, single UK academic centre experience. Rheumatology. 2009;48(10):1320-1322.
Septic arthritis predominantly involves the weight-bearing joints of the hip and knee, which account for nearly 60% of cases.1 In contrast, the shoulder joint is involved in 10% to 15% of cases, though this number may be higher among intravenous (IV) drug users.2 The most common causative organisms are the Staphylococcus species, followed closely by β-hemolytic streptococci, with these 2 groups accounting for more than 90% of all cases.3 The Streptococcus viridans group belongs to normal oral flora residing predominantly on the surface of teeth. Although well known for its ability to colonize heart valves and frequently cause bacterial endocarditis, this group has rarely been associated with septic arthritis. Furthermore, Streptococcus mitis, a subgroup of S viridans, has been implicated even less commonly.
In this article, we report a case of glenohumeral joint septic arthritis caused by S mitis. To our knowledge, such a case has not been previously reported in the English literature. Given the low virulence of this orally based bacterium, treating physicians must maintain clinical suspicion for the organism in the setting of persistent joint effusion and pain in association with periodontal disease or trauma. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A right-hand-dominant 54-year-old man presented to Dr. Gruson with complaints of persistent right shoulder pain associated with worsening range of motion (ROM). Three weeks earlier, the patient reported being assaulted and noted progressive swelling about the right shoulder. He denied fevers, chills, or prior shoulder problems. Although his past medical history was remarkable for hepatitis C and diabetes, he was not taking any diabetic medications at that time. A review of systems was remarkable for poor dental hygiene, and the patient was missing several teeth, which he said had been knocked out during the assault. Physical examination revealed diffuse tenderness about the right shoulder and severe pain with all passive movement. The shoulder was pseudoparalyzed. There were no subcutaneous collections, wounds, or ecchymosis about the shoulder. Mild calor was noted on the right shoulder relative to the left. Radiographs of the right shoulder showed no acute osseous abnormalities.
Magnetic resonance imaging (MRI), which was urgently obtained to assess the integrity of the rotator cuff and the location of the effusion, showed a large subacromial and glenohumeral joint effusion as well as diffuse muscular edema (Figures 1A-1C).
In light of the elevated infection findings of the laboratory tests and the positive culture, urgent arthroscopic irrigation and débridement of the right shoulder were indicated. Given the organism identified, transesophageal echocardiography was performed; there were no valvular vegetations. Creation of the posterior glenohumeral portal resulted in egress of turbid fluid, which was sent for culture. The subacromial space and the glenohumeral joint were thoroughly lavaged and the copious hemorrhagic synovitis débrided (Figures 2A, 2B).
The 8-week course of antibiotics normalized the patient’s ESR to 13 mm/h. Follow-up MRI showed improvement in the soft-tissue edema. Clinically, the patient reported minimal shoulder pain. He was undergoing physical therapy to regain strength and ROM.
Discussion
Staphylococcus aureus is the leading causative organism of septic arthritis, accounting for more than 60% of all cases.4 Conversely, the Streptococcus viridans group is rarely implicated in septic arthritis, accounting for <1% of cases.4S viridans is part of the commensal oral flora and has low virulence. This heterogeneous group is subdivided into S mitis, S salivarius, S anginosus, S mutans, and S bovis. The S mitis group is further subdivided into S sanguinis (formerly known as S sanguis) and S mitis. Infection by an organism of the S viridans group usually occurs on a previously injured focus, and the organism is a causative agent of bacterial endocarditis.5 Reported cases of septic arthritis caused by S viridans have predominantly involved the knee joint—with severe osteoarthritis, poor dental hygiene, and prior IV drug use identified as risk factors.5-7The shoulder joint is seldom involved in septic arthritis; estimated incidence is under 8%.8 Although overall incidence may rise in an increasingly elderly patient population, incidence of shoulder infection remains low.2,9
The main routes for developing septic arthritis include direct inoculation secondary to penetrating trauma or hematologic spread.10 Coatsworth and colleagues11 reported on iatrogenic S mitis septic arthritis of a shoulder arthroplasty during ultrasonography-guided aspiration by a technician who was not wearing a mask. Our institutional policy is to perform joint aspiration under strictly sterile conditions, which were adhered to in the present case. We surmise our patient developed transient bacteremia from the loss of several teeth, particularly given his poor dentition. Yombi and colleagues5 documented 2 cases of septic arthritis caused by Streptococcus gordonii, a relative of S sanguinis. One involved a previously replaced knee, and the other a native knee joint. Other cases of S viridans group septic arthritis have involved the knee,6,7,12,13 the sternoclavicular joint,14-16 and the acromioclavicular joint.17S sanguinis6,7,12,15,16 and S gordonii5 have been implicated in most cases, and an unspeciated S viridans in others.13,14,17 Concomitant periodontal disease has been reported in most cases as well,6,7,12,15 including our patient’s case. In the English-language literature, we found no other reports of S mitis as the causative agent of acute septic glenohumeral joint arthritis from hematogenous spread.
There should be no delay in diagnosing septic arthritis, and infected material should be removed from the joint. In animal models, complete joint destruction occurred only 5 weeks after inoculation with Staphylococcus aureus.10 Garofalo and colleagues18 reported a trend toward improved functional outcomes after earlier operative treatment. The choice of open surgical drainage vs repeat needle aspiration seems to be of little consequence, as both have good long-term outcomes, but open surgical drainage seems to result in better long-term functional ROM.2,9 However, results of a recent study suggested surgical treatment is not always superior to medical treatment for septic arthritis in native joints.19 In some cases involving S viridans species, treatment consisted of a combination of IV antibiotics and onetime or repeat aspiration;6,12-15 treatment in the remaining cases was surgical débridement.5,7,16,17 Given that S viridans is associated with bacterial endocarditis, echocardiography is essential if this organism is to be identified. Medical management and antibiotic treatment should be initiated after consultation with medical and infectious disease specialists.19We have reported a case of septic shoulder caused by S mitis, a low-virulence organism seldom associated with joint infection. The patient’s infection likely resulted from hematogenous spread from the oral cavity (dentition was poor). Urgent aspiration of the joint and baseline infection laboratory tests are recommended. MRI of the shoulder may show an effusion. Urgent arthroscopic irrigation and débridement can yield good clinical outcomes.
Am J Orthop. 2016;45(6):E343-E346. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
Septic arthritis predominantly involves the weight-bearing joints of the hip and knee, which account for nearly 60% of cases.1 In contrast, the shoulder joint is involved in 10% to 15% of cases, though this number may be higher among intravenous (IV) drug users.2 The most common causative organisms are the Staphylococcus species, followed closely by β-hemolytic streptococci, with these 2 groups accounting for more than 90% of all cases.3 The Streptococcus viridans group belongs to normal oral flora residing predominantly on the surface of teeth. Although well known for its ability to colonize heart valves and frequently cause bacterial endocarditis, this group has rarely been associated with septic arthritis. Furthermore, Streptococcus mitis, a subgroup of S viridans, has been implicated even less commonly.
In this article, we report a case of glenohumeral joint septic arthritis caused by S mitis. To our knowledge, such a case has not been previously reported in the English literature. Given the low virulence of this orally based bacterium, treating physicians must maintain clinical suspicion for the organism in the setting of persistent joint effusion and pain in association with periodontal disease or trauma. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A right-hand-dominant 54-year-old man presented to Dr. Gruson with complaints of persistent right shoulder pain associated with worsening range of motion (ROM). Three weeks earlier, the patient reported being assaulted and noted progressive swelling about the right shoulder. He denied fevers, chills, or prior shoulder problems. Although his past medical history was remarkable for hepatitis C and diabetes, he was not taking any diabetic medications at that time. A review of systems was remarkable for poor dental hygiene, and the patient was missing several teeth, which he said had been knocked out during the assault. Physical examination revealed diffuse tenderness about the right shoulder and severe pain with all passive movement. The shoulder was pseudoparalyzed. There were no subcutaneous collections, wounds, or ecchymosis about the shoulder. Mild calor was noted on the right shoulder relative to the left. Radiographs of the right shoulder showed no acute osseous abnormalities.
Magnetic resonance imaging (MRI), which was urgently obtained to assess the integrity of the rotator cuff and the location of the effusion, showed a large subacromial and glenohumeral joint effusion as well as diffuse muscular edema (Figures 1A-1C).
In light of the elevated infection findings of the laboratory tests and the positive culture, urgent arthroscopic irrigation and débridement of the right shoulder were indicated. Given the organism identified, transesophageal echocardiography was performed; there were no valvular vegetations. Creation of the posterior glenohumeral portal resulted in egress of turbid fluid, which was sent for culture. The subacromial space and the glenohumeral joint were thoroughly lavaged and the copious hemorrhagic synovitis débrided (Figures 2A, 2B).
The 8-week course of antibiotics normalized the patient’s ESR to 13 mm/h. Follow-up MRI showed improvement in the soft-tissue edema. Clinically, the patient reported minimal shoulder pain. He was undergoing physical therapy to regain strength and ROM.
Discussion
Staphylococcus aureus is the leading causative organism of septic arthritis, accounting for more than 60% of all cases.4 Conversely, the Streptococcus viridans group is rarely implicated in septic arthritis, accounting for <1% of cases.4S viridans is part of the commensal oral flora and has low virulence. This heterogeneous group is subdivided into S mitis, S salivarius, S anginosus, S mutans, and S bovis. The S mitis group is further subdivided into S sanguinis (formerly known as S sanguis) and S mitis. Infection by an organism of the S viridans group usually occurs on a previously injured focus, and the organism is a causative agent of bacterial endocarditis.5 Reported cases of septic arthritis caused by S viridans have predominantly involved the knee joint—with severe osteoarthritis, poor dental hygiene, and prior IV drug use identified as risk factors.5-7The shoulder joint is seldom involved in septic arthritis; estimated incidence is under 8%.8 Although overall incidence may rise in an increasingly elderly patient population, incidence of shoulder infection remains low.2,9
The main routes for developing septic arthritis include direct inoculation secondary to penetrating trauma or hematologic spread.10 Coatsworth and colleagues11 reported on iatrogenic S mitis septic arthritis of a shoulder arthroplasty during ultrasonography-guided aspiration by a technician who was not wearing a mask. Our institutional policy is to perform joint aspiration under strictly sterile conditions, which were adhered to in the present case. We surmise our patient developed transient bacteremia from the loss of several teeth, particularly given his poor dentition. Yombi and colleagues5 documented 2 cases of septic arthritis caused by Streptococcus gordonii, a relative of S sanguinis. One involved a previously replaced knee, and the other a native knee joint. Other cases of S viridans group septic arthritis have involved the knee,6,7,12,13 the sternoclavicular joint,14-16 and the acromioclavicular joint.17S sanguinis6,7,12,15,16 and S gordonii5 have been implicated in most cases, and an unspeciated S viridans in others.13,14,17 Concomitant periodontal disease has been reported in most cases as well,6,7,12,15 including our patient’s case. In the English-language literature, we found no other reports of S mitis as the causative agent of acute septic glenohumeral joint arthritis from hematogenous spread.
There should be no delay in diagnosing septic arthritis, and infected material should be removed from the joint. In animal models, complete joint destruction occurred only 5 weeks after inoculation with Staphylococcus aureus.10 Garofalo and colleagues18 reported a trend toward improved functional outcomes after earlier operative treatment. The choice of open surgical drainage vs repeat needle aspiration seems to be of little consequence, as both have good long-term outcomes, but open surgical drainage seems to result in better long-term functional ROM.2,9 However, results of a recent study suggested surgical treatment is not always superior to medical treatment for septic arthritis in native joints.19 In some cases involving S viridans species, treatment consisted of a combination of IV antibiotics and onetime or repeat aspiration;6,12-15 treatment in the remaining cases was surgical débridement.5,7,16,17 Given that S viridans is associated with bacterial endocarditis, echocardiography is essential if this organism is to be identified. Medical management and antibiotic treatment should be initiated after consultation with medical and infectious disease specialists.19We have reported a case of septic shoulder caused by S mitis, a low-virulence organism seldom associated with joint infection. The patient’s infection likely resulted from hematogenous spread from the oral cavity (dentition was poor). Urgent aspiration of the joint and baseline infection laboratory tests are recommended. MRI of the shoulder may show an effusion. Urgent arthroscopic irrigation and débridement can yield good clinical outcomes.
Am J Orthop. 2016;45(6):E343-E346. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Mathews CJ, Kingsley G, Field M, et al. Management of septic arthritis: a systematic review. Ann Rheum Dis. 2007;66(4):440-445.
2. Leslie BM, Harris JM 3rd, Driscoll D. Septic arthritis of the shoulder in adults. J Bone Joint Surg Am. 1989;71(10):1516-1522.
3. Gupta MN, Sturrock RD, Field M. A prospective 2-year study of 75 patients with adult-onset septic arthritis. Rheumatology. 2001;40(1):24-30.
4. Dubost JJ, Soubrier M, De Champs C, Ristori JM, Bussiere JL, Sauvezie B. No changes in the distribution of organisms responsible for septic arthritis over a 20 year period. Ann Rheum Dis. 2002;61(3):267-269.
5. Yombi J, Belkhir L, Jonckheere S, et al. Streptococcus gordonii septic arthritis: two cases and review of literature. BMC Infect Dis. 2012;12:215.
6. Papaioannides D, Boniatsi L, Korantzopoulos P, Sinapidis D, Giotis C. Acute septic arthritis due to Streptococcus sanguis. Med Princ Pract. 2006;15(1):77-79.
7. Edson RS, Osmon DR, Berry DJ. Septic arthritis due to Streptococcus sanguis. Mayo Clin Proc. 2002;77(7):709-710.
8. Weston VC, Jones AC, Bradbury N, Fawthrop F, Doherty M. Clinical features and outcome of septic arthritis in a single UK health district 1982-1991. Ann Rheum Dis. 1999;58(4):214-219.
9. Lossos IS, Yossepowitch O, Kandel L, Yardeni D, Arber N. Septic arthritis of the glenohumeral joint. A report of 11 cases and review of the literature. Medicine. 1998;77(3):177-187.
10. Esterhai JL Jr, Gelb I. Adult septic arthritis. Orthop Clin North Am. 1991;22(3):503-514.
11. Coatsworth NR, Huntington PG, Giuffre B, Kotsiou G. The doctor and the mask: iatrogenic septic arthritis caused by Streptoccocus mitis. Med J Aust. 2013;198(5):285-286.
12. Patrick MR, Lewis D. Short of a length: Streptococcus sanguis knee infection from dental source. Br J Rheumatol. 1992;31(8):569.
13. Barbadillo C, Trujillo A, Cuende E, Mazzucchelli R, Mulero J, Andreu JL. Septic arthritis due to Streptococcus viridans. Clin Exp Rheumatol. 1990;8(5):520-521.
14. Mata P, Molins A, de Oya M. Sternal arthritis caused by Streptococcus viridans in a heroin addict [in Spanish]. Med Clin. 1984;83(16):689.
15. Mandac I, Prkacin I, Sabljar Matovinovic M, Sustercic D. Septic arthritis due to Streptococcus sanguis. Coll Antropol. 2010;34(2):661-664.
16. Nitsche JF, Vaughan JH, Williams G, Curd JG. Septic sternoclavicular arthritis with Pasteurella multocida and Streptococcus sanguis. Arthritis Rheum. 1982;25(4):467-469.
17. Blankstein A, Amsallem JL, Rubenstein E, Horoszowski H, Farin I. Septic arthritis of the acromioclavicular joint. Arch Orthop Trauma Surg. 1985;103(6):417-418.
18. Garofalo R, Flanagin B, Cesari E, Vinci E, Conti M, Castagna A. Destructive septic arthritis of shoulder in adults. Musculoskelet Surg. 2014;98(supp 1):S35-S39.
19. Ravindran V, Logan I, Bourke BE. Medical vs surgical treatment for the native joint in septic arthritis: a 6-year, single UK academic centre experience. Rheumatology. 2009;48(10):1320-1322.
1. Mathews CJ, Kingsley G, Field M, et al. Management of septic arthritis: a systematic review. Ann Rheum Dis. 2007;66(4):440-445.
2. Leslie BM, Harris JM 3rd, Driscoll D. Septic arthritis of the shoulder in adults. J Bone Joint Surg Am. 1989;71(10):1516-1522.
3. Gupta MN, Sturrock RD, Field M. A prospective 2-year study of 75 patients with adult-onset septic arthritis. Rheumatology. 2001;40(1):24-30.
4. Dubost JJ, Soubrier M, De Champs C, Ristori JM, Bussiere JL, Sauvezie B. No changes in the distribution of organisms responsible for septic arthritis over a 20 year period. Ann Rheum Dis. 2002;61(3):267-269.
5. Yombi J, Belkhir L, Jonckheere S, et al. Streptococcus gordonii septic arthritis: two cases and review of literature. BMC Infect Dis. 2012;12:215.
6. Papaioannides D, Boniatsi L, Korantzopoulos P, Sinapidis D, Giotis C. Acute septic arthritis due to Streptococcus sanguis. Med Princ Pract. 2006;15(1):77-79.
7. Edson RS, Osmon DR, Berry DJ. Septic arthritis due to Streptococcus sanguis. Mayo Clin Proc. 2002;77(7):709-710.
8. Weston VC, Jones AC, Bradbury N, Fawthrop F, Doherty M. Clinical features and outcome of septic arthritis in a single UK health district 1982-1991. Ann Rheum Dis. 1999;58(4):214-219.
9. Lossos IS, Yossepowitch O, Kandel L, Yardeni D, Arber N. Septic arthritis of the glenohumeral joint. A report of 11 cases and review of the literature. Medicine. 1998;77(3):177-187.
10. Esterhai JL Jr, Gelb I. Adult septic arthritis. Orthop Clin North Am. 1991;22(3):503-514.
11. Coatsworth NR, Huntington PG, Giuffre B, Kotsiou G. The doctor and the mask: iatrogenic septic arthritis caused by Streptoccocus mitis. Med J Aust. 2013;198(5):285-286.
12. Patrick MR, Lewis D. Short of a length: Streptococcus sanguis knee infection from dental source. Br J Rheumatol. 1992;31(8):569.
13. Barbadillo C, Trujillo A, Cuende E, Mazzucchelli R, Mulero J, Andreu JL. Septic arthritis due to Streptococcus viridans. Clin Exp Rheumatol. 1990;8(5):520-521.
14. Mata P, Molins A, de Oya M. Sternal arthritis caused by Streptococcus viridans in a heroin addict [in Spanish]. Med Clin. 1984;83(16):689.
15. Mandac I, Prkacin I, Sabljar Matovinovic M, Sustercic D. Septic arthritis due to Streptococcus sanguis. Coll Antropol. 2010;34(2):661-664.
16. Nitsche JF, Vaughan JH, Williams G, Curd JG. Septic sternoclavicular arthritis with Pasteurella multocida and Streptococcus sanguis. Arthritis Rheum. 1982;25(4):467-469.
17. Blankstein A, Amsallem JL, Rubenstein E, Horoszowski H, Farin I. Septic arthritis of the acromioclavicular joint. Arch Orthop Trauma Surg. 1985;103(6):417-418.
18. Garofalo R, Flanagin B, Cesari E, Vinci E, Conti M, Castagna A. Destructive septic arthritis of shoulder in adults. Musculoskelet Surg. 2014;98(supp 1):S35-S39.
19. Ravindran V, Logan I, Bourke BE. Medical vs surgical treatment for the native joint in septic arthritis: a 6-year, single UK academic centre experience. Rheumatology. 2009;48(10):1320-1322.