User login
Tibial Tubercle Fracture After Bone–Patellar Tendon–Bone Autograft
A fracture occurring after anterior cruciate ligament (ACL) reconstruction is rare, and rarer still when it involves the harvest site of a bone—patellar tendon—bone (BPTB) autograft. The vast majority of fractures described in the literature are patellar, with the weak point along the patellar bone cut. A number of fractures generally also occur through the bone tunnels in both hamstring and BPTB grafts. However, only 2 cases of tibial tubercle fracture after BPTB graft have been published, and we expound on them in this case report.1,2 The patient provided written informed consent for print and electronic publication of this case report.
Case Report
Eight years after undergoing successful left ACL reconstruction with ipsilateral BPTB graft, a 45-year-old man developed a graft rupture and demonstrated recurrent instability. He requested revision reconstruction, again with a BPTB construct. In the operating room, he was prepared and draped in the usual sterile fashion, and left ACL reconstruction was performed with right-knee central-third BPTB graft.
During surgery, the left knee was arthroscopically examined, and residual ACL graft from the initial reconstruction was removed. Notchplasty was performed, and the residual femoral interference screw was removed from the 12:30 position. A transtibial approach was used, with a 10-mm reamer brought through the proximal tibia, the posterior tibial ACL footprint, and the 2:00 distal femoral position, with 30 mm of femoral condyle drilled, leaving 1 mm of posterior femoral cortex.
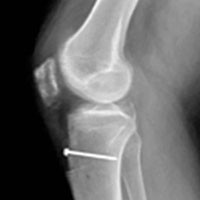
After the right leg was exsanguinated, a central-third patellar tendon graft was harvested through a longitudinal incision with a 22-mm × 10-mm patellar plug, a 10-mm patellar graft, and a 22-mm × 11-mm tibial plug. The graft was prepared, the left tibia was overreamed, and the graft was passed. The graft was fixed with a 7-mm × 23-mm biointerference screw in the femur, trialed, and fixed with an 8-mm × 23-mm interference screw in the tibia. Excess bone graft was packed in the patellar defect in the right knee. The rent in the patellar tendon was closed. The rest of the incision was closed, and the patient was placed in an immobilizer and a cold therapy device (Polar Care; Breg, Inc).
At 2-week follow-up, the patient reported having slipped on ice and flexed the right knee, causing a pop, pain, and limitation in range of motion (ROM; 0°-70°). 

The patient returned to the operating room 5 days later and underwent open reduction and internal fixation (ORIF) of the tibial tubercle avulsion. After sterile preparation and draping, the previous incision was used. The bony fragment was isolated and the hematoma débrided. Repair was performed with two No. 2 running locked FiberWire sutures (Arthrex) placed through bony drill holes in the fragment (1 medial, 1 lateral). The fragment was reduced and the sutures tied, with further fixation provided with a DePuy Synthes small-fragment 3.5-mm cortical screw with washer. A No. 5 Ethibond suture (Ethicon) was then placed as a secondary cerclage figure-of-8 stitch to protect the repair. 
The patient was seen in follow-up 6 weeks after right ACL reconstruction and 4 weeks after left tibial tubercle ORIF. He continued with right knee restrictions, with the weight-bearing brace locked in extension. Left knee ROM was more than 0° to 90° even before any formal physical therapy. At this point, the patient began physical therapy on both knees with ROM limited to 0° to 30° and weight-bearing as tolerated on the right knee (no restrictions on the left knee). 
Discussion
Cases of tibial tubercle fracture after BPTB autograft harvest are extremely rare in the published literature. PubMed and Cochrane Review searches revealed only 2—1 in the ipsilateral knee as ACL fixation1 and 1 in the contralateral knee.2 The middle third of the patellar tendon has been used for ACL reconstruction for more than 50 years, which supports the extreme rarity of this complication.3 Tibial tubercle fractures are so rare that they are not even mentioned in reviews of ACL complications.4 These fractures are universally treated with ORIF.1,2
Far more common but still rare, fracture-type complications involve the extensor mechanism and the tibial plateau. Patellar fractures have been documented as occurring in 0.2% to 2.3% of cases.5-7 One paper reported a fracture in 1.3% of cases at a mean of 57 days, with roughly half caused by trauma and the other half having atraumatic causes.8 Lee and colleagues9 found a 0.2% complication rate for all BPTB grafts in 1725 consecutive patients. Although some patients were treated nonoperatively, others underwent operative fixation. Time to clinical and radiographic healing was 7 and 10 weeks, respectively.
Tibial plateau fracture after BPTB harvest is a rare complication, with 11 cases reported in the literature.10 In 4 of those cases, the proposed mechanism of fracture was a stress riser resulting from the synergistic weakness of the tibial harvest site combined with the tibial tunnel reducing proximal tibial bone strength.11-14 The mechanism of injury varied from traumatic to insufficiency fracture, with fixation varying with fracture displacement.
Tibial tubercle fracture after BPTB harvest is extremely rare, with the present case being only the third published in the literature. Like most reported post-ACL reconstruction extensor mechanism disruptions, our case resulted from a traumatic event at an interval after surgery. All other tibial tubercle fracture post-ACL reconstruction disruptions occurred within 2 weeks after surgery.1,2 Sudden tension on the extensor mechanism secondary to hyperflexion caused a fracture through a weakened tibial tubercle with avulsion of the remaining tendon in 2 of the 3 cases, with the third being a lower stress popping noise that occurred during a pivot to stand.1
The residual defect after tibial bone block harvest could represent a weakening of the tubercle by loss of structural bone and by development of stress risers. The previous reports of tibial tubercle fracture after BPTB harvest documented a similar methodology: Use a bone saw and osteotomes to harvest a trapezoidal tibial bone plug 10 mm to 11 mm wide and 22 cm to 35 cm long. As previously documented, we suggest taking care with saw cuts and osteotomes so as not to weaken the proximal tibia or distal patella more than is necessary.1,2 Before surgery, patients should be warned about the possibility of extensor mechanism injuries with use of BPTB grafts.
Conclusion
Tibial tubercle fracture after BPTB harvest for ACL reconstruction is an extremely rare complication. Treatment is ORIF of the tubercle fragment, with a delay in ACL rehabilitation in cases involving the ipsilateral knee.
Am J Orthop. 2016;45(7):E469-E471. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Acton KJ, Dowd GS. Fracture of the tibial tubercle following anterior cruciate ligament reconstruction. Knee. 2002;9(2):157-159.
2. Busfield BT, Safran MR, Cannon WD. Extensor mechanism disruption after contralateral middle third patellar tendon harvest for anterior cruciate ligament revision reconstruction. Arthroscopy. 2005;21(10):1268.e1-e1268.e6.
3. Jones KG. Reconstruction of the anterior cruciate ligament. A technique using the central one-third of the patellar ligament. J Bone Joint Surg Am. 1963;45(5):925-932.
4. Tjoumakaris FP, Herz-Brown AL, Bowers AL, Sennett BJ, Bernstein J. Complications in brief: anterior cruciate ligament reconstruction. Clin Orthop Relat Res. 2012;470(2):630-636.
5. Morgan-Jones RL, Cross TM, Caldwell B, Cross MJ. “Silent” transverse patellar fracture following anterior cruciate ligament reconstruction. Arthroscopy. 2001;17(9):997-999.
6. Viola R, Vianello R. Three cases of patella fracture in 1,320 anterior cruciate ligament reconstructions with bone–patellar tendon–bone autograft. Arthroscopy. 1999;15(1):93-97.
7. Berg EE. Management of patella fractures associated with central third bone–patella tendon–bone autograft ACL reconstructions. Arthroscopy. 1996;12(6):756-759.
8. Stein DA, Hunt SA, Rosen JE, Sherman OH. The incidence and outcome of patella fractures after anterior cruciate ligament reconstruction. Arthroscopy. 2002;18(6):578-583.
9. Lee GH, McCulloch P, Cole BJ, Bush-Joseph CA, Bach BR Jr. The incidence of acute patellar tendon harvest complications for anterior cruciate ligament reconstruction. Arthroscopy. 2008;24(2):162-166.
10. Wong JJ, Muir B. Insufficiency fracture of the tibial plateau after anterior cruciate ligament reconstructive surgery: a case report and review of the literature. J Can Chiropr Assoc. 2013;57(2):123-131.
11. Morgan E, Steensen RN. Traumatic proximal tibial fracture following anterior cruciate ligament reconstruction. Am J Knee Surg. 1998;11(3):193-194.
12. Delcogliano A, Chiossi S, Caporaso A, Franzese S, Menghi A. Tibial plateau fracture after arthroscopic anterior cruciate ligament reconstruction. Arthroscopy. 2001;17(4):E16.
13. Mithöfer K, Gill TJ, Vrahas MS. Tibial plateau fracture following anterior cruciate ligament reconstruction. Knee Surg Sports Traumatol Arthrosc. 2004;12(4):325-328.
14. Moen KY, Boynton MD, Raasch WG. Fracture of the proximal tibia after anterior cruciate ligament reconstruction: a case report. Am J Orthop. 1998;27(9):629-630.
A fracture occurring after anterior cruciate ligament (ACL) reconstruction is rare, and rarer still when it involves the harvest site of a bone—patellar tendon—bone (BPTB) autograft. The vast majority of fractures described in the literature are patellar, with the weak point along the patellar bone cut. A number of fractures generally also occur through the bone tunnels in both hamstring and BPTB grafts. However, only 2 cases of tibial tubercle fracture after BPTB graft have been published, and we expound on them in this case report.1,2 The patient provided written informed consent for print and electronic publication of this case report.
Case Report
Eight years after undergoing successful left ACL reconstruction with ipsilateral BPTB graft, a 45-year-old man developed a graft rupture and demonstrated recurrent instability. He requested revision reconstruction, again with a BPTB construct. In the operating room, he was prepared and draped in the usual sterile fashion, and left ACL reconstruction was performed with right-knee central-third BPTB graft.
During surgery, the left knee was arthroscopically examined, and residual ACL graft from the initial reconstruction was removed. Notchplasty was performed, and the residual femoral interference screw was removed from the 12:30 position. A transtibial approach was used, with a 10-mm reamer brought through the proximal tibia, the posterior tibial ACL footprint, and the 2:00 distal femoral position, with 30 mm of femoral condyle drilled, leaving 1 mm of posterior femoral cortex.
After the right leg was exsanguinated, a central-third patellar tendon graft was harvested through a longitudinal incision with a 22-mm × 10-mm patellar plug, a 10-mm patellar graft, and a 22-mm × 11-mm tibial plug. The graft was prepared, the left tibia was overreamed, and the graft was passed. The graft was fixed with a 7-mm × 23-mm biointerference screw in the femur, trialed, and fixed with an 8-mm × 23-mm interference screw in the tibia. Excess bone graft was packed in the patellar defect in the right knee. The rent in the patellar tendon was closed. The rest of the incision was closed, and the patient was placed in an immobilizer and a cold therapy device (Polar Care; Breg, Inc).
At 2-week follow-up, the patient reported having slipped on ice and flexed the right knee, causing a pop, pain, and limitation in range of motion (ROM; 0°-70°).
The patient returned to the operating room 5 days later and underwent open reduction and internal fixation (ORIF) of the tibial tubercle avulsion. After sterile preparation and draping, the previous incision was used. The bony fragment was isolated and the hematoma débrided. Repair was performed with two No. 2 running locked FiberWire sutures (Arthrex) placed through bony drill holes in the fragment (1 medial, 1 lateral). The fragment was reduced and the sutures tied, with further fixation provided with a DePuy Synthes small-fragment 3.5-mm cortical screw with washer. A No. 5 Ethibond suture (Ethicon) was then placed as a secondary cerclage figure-of-8 stitch to protect the repair.
The patient was seen in follow-up 6 weeks after right ACL reconstruction and 4 weeks after left tibial tubercle ORIF. He continued with right knee restrictions, with the weight-bearing brace locked in extension. Left knee ROM was more than 0° to 90° even before any formal physical therapy. At this point, the patient began physical therapy on both knees with ROM limited to 0° to 30° and weight-bearing as tolerated on the right knee (no restrictions on the left knee).
Discussion
Cases of tibial tubercle fracture after BPTB autograft harvest are extremely rare in the published literature. PubMed and Cochrane Review searches revealed only 2—1 in the ipsilateral knee as ACL fixation1 and 1 in the contralateral knee.2 The middle third of the patellar tendon has been used for ACL reconstruction for more than 50 years, which supports the extreme rarity of this complication.3 Tibial tubercle fractures are so rare that they are not even mentioned in reviews of ACL complications.4 These fractures are universally treated with ORIF.1,2
Far more common but still rare, fracture-type complications involve the extensor mechanism and the tibial plateau. Patellar fractures have been documented as occurring in 0.2% to 2.3% of cases.5-7 One paper reported a fracture in 1.3% of cases at a mean of 57 days, with roughly half caused by trauma and the other half having atraumatic causes.8 Lee and colleagues9 found a 0.2% complication rate for all BPTB grafts in 1725 consecutive patients. Although some patients were treated nonoperatively, others underwent operative fixation. Time to clinical and radiographic healing was 7 and 10 weeks, respectively.
Tibial plateau fracture after BPTB harvest is a rare complication, with 11 cases reported in the literature.10 In 4 of those cases, the proposed mechanism of fracture was a stress riser resulting from the synergistic weakness of the tibial harvest site combined with the tibial tunnel reducing proximal tibial bone strength.11-14 The mechanism of injury varied from traumatic to insufficiency fracture, with fixation varying with fracture displacement.
Tibial tubercle fracture after BPTB harvest is extremely rare, with the present case being only the third published in the literature. Like most reported post-ACL reconstruction extensor mechanism disruptions, our case resulted from a traumatic event at an interval after surgery. All other tibial tubercle fracture post-ACL reconstruction disruptions occurred within 2 weeks after surgery.1,2 Sudden tension on the extensor mechanism secondary to hyperflexion caused a fracture through a weakened tibial tubercle with avulsion of the remaining tendon in 2 of the 3 cases, with the third being a lower stress popping noise that occurred during a pivot to stand.1
The residual defect after tibial bone block harvest could represent a weakening of the tubercle by loss of structural bone and by development of stress risers. The previous reports of tibial tubercle fracture after BPTB harvest documented a similar methodology: Use a bone saw and osteotomes to harvest a trapezoidal tibial bone plug 10 mm to 11 mm wide and 22 cm to 35 cm long. As previously documented, we suggest taking care with saw cuts and osteotomes so as not to weaken the proximal tibia or distal patella more than is necessary.1,2 Before surgery, patients should be warned about the possibility of extensor mechanism injuries with use of BPTB grafts.
Conclusion
Tibial tubercle fracture after BPTB harvest for ACL reconstruction is an extremely rare complication. Treatment is ORIF of the tubercle fragment, with a delay in ACL rehabilitation in cases involving the ipsilateral knee.
Am J Orthop. 2016;45(7):E469-E471. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
A fracture occurring after anterior cruciate ligament (ACL) reconstruction is rare, and rarer still when it involves the harvest site of a bone—patellar tendon—bone (BPTB) autograft. The vast majority of fractures described in the literature are patellar, with the weak point along the patellar bone cut. A number of fractures generally also occur through the bone tunnels in both hamstring and BPTB grafts. However, only 2 cases of tibial tubercle fracture after BPTB graft have been published, and we expound on them in this case report.1,2 The patient provided written informed consent for print and electronic publication of this case report.
Case Report
Eight years after undergoing successful left ACL reconstruction with ipsilateral BPTB graft, a 45-year-old man developed a graft rupture and demonstrated recurrent instability. He requested revision reconstruction, again with a BPTB construct. In the operating room, he was prepared and draped in the usual sterile fashion, and left ACL reconstruction was performed with right-knee central-third BPTB graft.
During surgery, the left knee was arthroscopically examined, and residual ACL graft from the initial reconstruction was removed. Notchplasty was performed, and the residual femoral interference screw was removed from the 12:30 position. A transtibial approach was used, with a 10-mm reamer brought through the proximal tibia, the posterior tibial ACL footprint, and the 2:00 distal femoral position, with 30 mm of femoral condyle drilled, leaving 1 mm of posterior femoral cortex.
After the right leg was exsanguinated, a central-third patellar tendon graft was harvested through a longitudinal incision with a 22-mm × 10-mm patellar plug, a 10-mm patellar graft, and a 22-mm × 11-mm tibial plug. The graft was prepared, the left tibia was overreamed, and the graft was passed. The graft was fixed with a 7-mm × 23-mm biointerference screw in the femur, trialed, and fixed with an 8-mm × 23-mm interference screw in the tibia. Excess bone graft was packed in the patellar defect in the right knee. The rent in the patellar tendon was closed. The rest of the incision was closed, and the patient was placed in an immobilizer and a cold therapy device (Polar Care; Breg, Inc).
At 2-week follow-up, the patient reported having slipped on ice and flexed the right knee, causing a pop, pain, and limitation in range of motion (ROM; 0°-70°).
The patient returned to the operating room 5 days later and underwent open reduction and internal fixation (ORIF) of the tibial tubercle avulsion. After sterile preparation and draping, the previous incision was used. The bony fragment was isolated and the hematoma débrided. Repair was performed with two No. 2 running locked FiberWire sutures (Arthrex) placed through bony drill holes in the fragment (1 medial, 1 lateral). The fragment was reduced and the sutures tied, with further fixation provided with a DePuy Synthes small-fragment 3.5-mm cortical screw with washer. A No. 5 Ethibond suture (Ethicon) was then placed as a secondary cerclage figure-of-8 stitch to protect the repair.
The patient was seen in follow-up 6 weeks after right ACL reconstruction and 4 weeks after left tibial tubercle ORIF. He continued with right knee restrictions, with the weight-bearing brace locked in extension. Left knee ROM was more than 0° to 90° even before any formal physical therapy. At this point, the patient began physical therapy on both knees with ROM limited to 0° to 30° and weight-bearing as tolerated on the right knee (no restrictions on the left knee).
Discussion
Cases of tibial tubercle fracture after BPTB autograft harvest are extremely rare in the published literature. PubMed and Cochrane Review searches revealed only 2—1 in the ipsilateral knee as ACL fixation1 and 1 in the contralateral knee.2 The middle third of the patellar tendon has been used for ACL reconstruction for more than 50 years, which supports the extreme rarity of this complication.3 Tibial tubercle fractures are so rare that they are not even mentioned in reviews of ACL complications.4 These fractures are universally treated with ORIF.1,2
Far more common but still rare, fracture-type complications involve the extensor mechanism and the tibial plateau. Patellar fractures have been documented as occurring in 0.2% to 2.3% of cases.5-7 One paper reported a fracture in 1.3% of cases at a mean of 57 days, with roughly half caused by trauma and the other half having atraumatic causes.8 Lee and colleagues9 found a 0.2% complication rate for all BPTB grafts in 1725 consecutive patients. Although some patients were treated nonoperatively, others underwent operative fixation. Time to clinical and radiographic healing was 7 and 10 weeks, respectively.
Tibial plateau fracture after BPTB harvest is a rare complication, with 11 cases reported in the literature.10 In 4 of those cases, the proposed mechanism of fracture was a stress riser resulting from the synergistic weakness of the tibial harvest site combined with the tibial tunnel reducing proximal tibial bone strength.11-14 The mechanism of injury varied from traumatic to insufficiency fracture, with fixation varying with fracture displacement.
Tibial tubercle fracture after BPTB harvest is extremely rare, with the present case being only the third published in the literature. Like most reported post-ACL reconstruction extensor mechanism disruptions, our case resulted from a traumatic event at an interval after surgery. All other tibial tubercle fracture post-ACL reconstruction disruptions occurred within 2 weeks after surgery.1,2 Sudden tension on the extensor mechanism secondary to hyperflexion caused a fracture through a weakened tibial tubercle with avulsion of the remaining tendon in 2 of the 3 cases, with the third being a lower stress popping noise that occurred during a pivot to stand.1
The residual defect after tibial bone block harvest could represent a weakening of the tubercle by loss of structural bone and by development of stress risers. The previous reports of tibial tubercle fracture after BPTB harvest documented a similar methodology: Use a bone saw and osteotomes to harvest a trapezoidal tibial bone plug 10 mm to 11 mm wide and 22 cm to 35 cm long. As previously documented, we suggest taking care with saw cuts and osteotomes so as not to weaken the proximal tibia or distal patella more than is necessary.1,2 Before surgery, patients should be warned about the possibility of extensor mechanism injuries with use of BPTB grafts.
Conclusion
Tibial tubercle fracture after BPTB harvest for ACL reconstruction is an extremely rare complication. Treatment is ORIF of the tubercle fragment, with a delay in ACL rehabilitation in cases involving the ipsilateral knee.
Am J Orthop. 2016;45(7):E469-E471. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Acton KJ, Dowd GS. Fracture of the tibial tubercle following anterior cruciate ligament reconstruction. Knee. 2002;9(2):157-159.
2. Busfield BT, Safran MR, Cannon WD. Extensor mechanism disruption after contralateral middle third patellar tendon harvest for anterior cruciate ligament revision reconstruction. Arthroscopy. 2005;21(10):1268.e1-e1268.e6.
3. Jones KG. Reconstruction of the anterior cruciate ligament. A technique using the central one-third of the patellar ligament. J Bone Joint Surg Am. 1963;45(5):925-932.
4. Tjoumakaris FP, Herz-Brown AL, Bowers AL, Sennett BJ, Bernstein J. Complications in brief: anterior cruciate ligament reconstruction. Clin Orthop Relat Res. 2012;470(2):630-636.
5. Morgan-Jones RL, Cross TM, Caldwell B, Cross MJ. “Silent” transverse patellar fracture following anterior cruciate ligament reconstruction. Arthroscopy. 2001;17(9):997-999.
6. Viola R, Vianello R. Three cases of patella fracture in 1,320 anterior cruciate ligament reconstructions with bone–patellar tendon–bone autograft. Arthroscopy. 1999;15(1):93-97.
7. Berg EE. Management of patella fractures associated with central third bone–patella tendon–bone autograft ACL reconstructions. Arthroscopy. 1996;12(6):756-759.
8. Stein DA, Hunt SA, Rosen JE, Sherman OH. The incidence and outcome of patella fractures after anterior cruciate ligament reconstruction. Arthroscopy. 2002;18(6):578-583.
9. Lee GH, McCulloch P, Cole BJ, Bush-Joseph CA, Bach BR Jr. The incidence of acute patellar tendon harvest complications for anterior cruciate ligament reconstruction. Arthroscopy. 2008;24(2):162-166.
10. Wong JJ, Muir B. Insufficiency fracture of the tibial plateau after anterior cruciate ligament reconstructive surgery: a case report and review of the literature. J Can Chiropr Assoc. 2013;57(2):123-131.
11. Morgan E, Steensen RN. Traumatic proximal tibial fracture following anterior cruciate ligament reconstruction. Am J Knee Surg. 1998;11(3):193-194.
12. Delcogliano A, Chiossi S, Caporaso A, Franzese S, Menghi A. Tibial plateau fracture after arthroscopic anterior cruciate ligament reconstruction. Arthroscopy. 2001;17(4):E16.
13. Mithöfer K, Gill TJ, Vrahas MS. Tibial plateau fracture following anterior cruciate ligament reconstruction. Knee Surg Sports Traumatol Arthrosc. 2004;12(4):325-328.
14. Moen KY, Boynton MD, Raasch WG. Fracture of the proximal tibia after anterior cruciate ligament reconstruction: a case report. Am J Orthop. 1998;27(9):629-630.
1. Acton KJ, Dowd GS. Fracture of the tibial tubercle following anterior cruciate ligament reconstruction. Knee. 2002;9(2):157-159.
2. Busfield BT, Safran MR, Cannon WD. Extensor mechanism disruption after contralateral middle third patellar tendon harvest for anterior cruciate ligament revision reconstruction. Arthroscopy. 2005;21(10):1268.e1-e1268.e6.
3. Jones KG. Reconstruction of the anterior cruciate ligament. A technique using the central one-third of the patellar ligament. J Bone Joint Surg Am. 1963;45(5):925-932.
4. Tjoumakaris FP, Herz-Brown AL, Bowers AL, Sennett BJ, Bernstein J. Complications in brief: anterior cruciate ligament reconstruction. Clin Orthop Relat Res. 2012;470(2):630-636.
5. Morgan-Jones RL, Cross TM, Caldwell B, Cross MJ. “Silent” transverse patellar fracture following anterior cruciate ligament reconstruction. Arthroscopy. 2001;17(9):997-999.
6. Viola R, Vianello R. Three cases of patella fracture in 1,320 anterior cruciate ligament reconstructions with bone–patellar tendon–bone autograft. Arthroscopy. 1999;15(1):93-97.
7. Berg EE. Management of patella fractures associated with central third bone–patella tendon–bone autograft ACL reconstructions. Arthroscopy. 1996;12(6):756-759.
8. Stein DA, Hunt SA, Rosen JE, Sherman OH. The incidence and outcome of patella fractures after anterior cruciate ligament reconstruction. Arthroscopy. 2002;18(6):578-583.
9. Lee GH, McCulloch P, Cole BJ, Bush-Joseph CA, Bach BR Jr. The incidence of acute patellar tendon harvest complications for anterior cruciate ligament reconstruction. Arthroscopy. 2008;24(2):162-166.
10. Wong JJ, Muir B. Insufficiency fracture of the tibial plateau after anterior cruciate ligament reconstructive surgery: a case report and review of the literature. J Can Chiropr Assoc. 2013;57(2):123-131.
11. Morgan E, Steensen RN. Traumatic proximal tibial fracture following anterior cruciate ligament reconstruction. Am J Knee Surg. 1998;11(3):193-194.
12. Delcogliano A, Chiossi S, Caporaso A, Franzese S, Menghi A. Tibial plateau fracture after arthroscopic anterior cruciate ligament reconstruction. Arthroscopy. 2001;17(4):E16.
13. Mithöfer K, Gill TJ, Vrahas MS. Tibial plateau fracture following anterior cruciate ligament reconstruction. Knee Surg Sports Traumatol Arthrosc. 2004;12(4):325-328.
14. Moen KY, Boynton MD, Raasch WG. Fracture of the proximal tibia after anterior cruciate ligament reconstruction: a case report. Am J Orthop. 1998;27(9):629-630.
Primary chest-wall leiomyosarcoma: a rare mimic of a malignant rib lesion
Primary chest-wall leiomyosarcoma (LMS) is an uncommon, malignant, soft-tissue tumor that most often affects the extremities. Malignant LMS originates from mesenchymal cells with smooth muscle differentiation. It is rare in adults, forming only 7% of all soft-tissue sarcomas (STS), but it is the most common STS. In adults, this type of tumor is usually found in the retroperitoneum and extremities.1 Chest-wall LMS is rare and most often occurs in men aged 50-70 years.2 When LMS is associated with rib destruction, it may mimic a primary bone tumor or metastasis. We present here the case of histologically proven chest-wall sarcoma with associated rib destruction that was initially mistaken on imaging for either a metastasis or primary bone tumor.
Case presentation and summary
A 69-year-old man presented to the emergency department complaining of pain over the right side of the chest. The pain, which was pleuritic in nature, had worsened over the previous 6 months and was severe at presentation. The patient had no fever, shortness of breath, or loss of weight. He had no history of chest trauma or chest wall radiation, and nothing noteworthy was discovered in his medical history. Subsequent test results for hemoglobin, white blood cell count, lymphocyte count, and cardiac enzymes were normal.
A frontal chest radiograph showed an osteolytic destructive lesion involving the posterior right 6th rib (Figure 1). A contrast-enhanced computedtomography (CE-CT) scan of the chest showed a heterogeneously enhancing, ovoid, soft-tissue mass of 5.6 x 3.6 cm (2.2 x 1.2 in) centered on the postero- lateral right 6th rib, with associated rib erosion. There was another 2.0-cm (0.8-in) subpleural nodule in the left upper lobe (Figure 2).
Click on the PDF icon below to read the full article.
Primary chest-wall leiomyosarcoma (LMS) is an uncommon, malignant, soft-tissue tumor that most often affects the extremities. Malignant LMS originates from mesenchymal cells with smooth muscle differentiation. It is rare in adults, forming only 7% of all soft-tissue sarcomas (STS), but it is the most common STS. In adults, this type of tumor is usually found in the retroperitoneum and extremities.1 Chest-wall LMS is rare and most often occurs in men aged 50-70 years.2 When LMS is associated with rib destruction, it may mimic a primary bone tumor or metastasis. We present here the case of histologically proven chest-wall sarcoma with associated rib destruction that was initially mistaken on imaging for either a metastasis or primary bone tumor.
Case presentation and summary
A 69-year-old man presented to the emergency department complaining of pain over the right side of the chest. The pain, which was pleuritic in nature, had worsened over the previous 6 months and was severe at presentation. The patient had no fever, shortness of breath, or loss of weight. He had no history of chest trauma or chest wall radiation, and nothing noteworthy was discovered in his medical history. Subsequent test results for hemoglobin, white blood cell count, lymphocyte count, and cardiac enzymes were normal.
A frontal chest radiograph showed an osteolytic destructive lesion involving the posterior right 6th rib (Figure 1). A contrast-enhanced computedtomography (CE-CT) scan of the chest showed a heterogeneously enhancing, ovoid, soft-tissue mass of 5.6 x 3.6 cm (2.2 x 1.2 in) centered on the postero- lateral right 6th rib, with associated rib erosion. There was another 2.0-cm (0.8-in) subpleural nodule in the left upper lobe (Figure 2).
Click on the PDF icon below to read the full article.
Primary chest-wall leiomyosarcoma (LMS) is an uncommon, malignant, soft-tissue tumor that most often affects the extremities. Malignant LMS originates from mesenchymal cells with smooth muscle differentiation. It is rare in adults, forming only 7% of all soft-tissue sarcomas (STS), but it is the most common STS. In adults, this type of tumor is usually found in the retroperitoneum and extremities.1 Chest-wall LMS is rare and most often occurs in men aged 50-70 years.2 When LMS is associated with rib destruction, it may mimic a primary bone tumor or metastasis. We present here the case of histologically proven chest-wall sarcoma with associated rib destruction that was initially mistaken on imaging for either a metastasis or primary bone tumor.
Case presentation and summary
A 69-year-old man presented to the emergency department complaining of pain over the right side of the chest. The pain, which was pleuritic in nature, had worsened over the previous 6 months and was severe at presentation. The patient had no fever, shortness of breath, or loss of weight. He had no history of chest trauma or chest wall radiation, and nothing noteworthy was discovered in his medical history. Subsequent test results for hemoglobin, white blood cell count, lymphocyte count, and cardiac enzymes were normal.
A frontal chest radiograph showed an osteolytic destructive lesion involving the posterior right 6th rib (Figure 1). A contrast-enhanced computedtomography (CE-CT) scan of the chest showed a heterogeneously enhancing, ovoid, soft-tissue mass of 5.6 x 3.6 cm (2.2 x 1.2 in) centered on the postero- lateral right 6th rib, with associated rib erosion. There was another 2.0-cm (0.8-in) subpleural nodule in the left upper lobe (Figure 2).
Click on the PDF icon below to read the full article.
Central nervous system manifestations of multiple myeloma: risk and prognostic considerations
Multiple myeloma accounts for about 1% of all cancers and for 10% of hematologic malignancies in the United States. This report describes the cases of 2 patients with multiple myeloma who developed CNS involvement after autologous stem cell transplant in the context of extramedullary disease.
Click on the PDF icon at the top of this introduction to read the full article.
Multiple myeloma accounts for about 1% of all cancers and for 10% of hematologic malignancies in the United States. This report describes the cases of 2 patients with multiple myeloma who developed CNS involvement after autologous stem cell transplant in the context of extramedullary disease.
Click on the PDF icon at the top of this introduction to read the full article.
Multiple myeloma accounts for about 1% of all cancers and for 10% of hematologic malignancies in the United States. This report describes the cases of 2 patients with multiple myeloma who developed CNS involvement after autologous stem cell transplant in the context of extramedullary disease.
Click on the PDF icon at the top of this introduction to read the full article.
Failure to Reduce: Small Bowel Obstruction Hidden Within a Chronic Umbilical Hernia Sac
Strangulated hernias are a medical emergency that can lead to small bowel obstruction (SBO), bowel necrosis, and death. Practitioners look for signs of strangulation on examination to guide the urgency of management. If the hernia is soft and reducible without overlying skin changes or signs of obstruction, patients may be monitored for years.1 However, there is increasing evidence that even asymptomatic hernias should be repaired rather than monitored to avoid the need for emergent surgical intervention.1
We present a case of a patient with a chronic umbilical hernia who experienced acute worsening of pain at the site of her hernia but with few additional objective signs of strangulation. Prior to this presentation, she had been recently evaluated at our ED for the “same” pain, which included a computed tomography (CT) scan that was negative for an acute surgical emergency. The patient’s second ED visit led to a diagnostic dilemma: Practitioners are encouraged to avoid “unnecessary” radiation—especially in cases of chronic pain—and to rely on history, physical examination findings, and prior recent imaging studies, as appropriate. In this case, repeat imaging ultimately revealed a surgical emergency with an unusual underlying pathology likely related to the chronicity of the patient’s hernia, and explained her repeat presentation to the ED.
Case
A 45-year-old obese woman (body mass index, 46 kg/m2) with a medical history of an umbilical hernia, tubal ligation, and chronic pelvic pain presented a second time to our ED with pain at the site of her hernia, which she stated began 5 hours prior to presentation. Although the pain was associated with nausea and vomiting, the patient said her bowel movements were normal. She first noticed the hernia more than 5 years ago, but experienced her first episode of acute pain related to the hernia with associated nausea and vomiting 3 weeks earlier, which prompted her initial presentation. During this first ED visit, a CT scan of the abdomen/pelvis was obtained as part of her evaluation and was significant for umbilical herniation of bowel without evidence of strangulation. Bedside reduction was successful, and the patient was discharged home and informed of the need to follow-up with a surgeon for an elective repair. She returned to the ED prior to her scheduled operation due to recurrent pain of similar character, but increased severity.
On physical examination, the patient was hemodynamically stable and afebrile. Her vital signs were: heart rate, 84 beats/min; blood pressure, 113/68 mm Hg; and respiratory rate, 20 breaths/min. Oxygen saturation was 100% on room air.
The abdomen was soft with tenderness to palpation over a 13-cm x 8-cm soft hernia to the left of the umbilicus without overlying skin changes. The patient’s pain was controlled with 1 mg of intravenous hydromorphone, after which bedside reduction was attempted. During reduction attempts, there was palpable bowel within the hernia sac, and a periumbilical defect was appreciated. Although the defect in the abdominal wall was estimated to be large enough to allow reduction, the hernia reduced only partially. Because imaging studies from the patient’s previous ED visit showed no visualized strangulation or obstruction, we deliberated over the need for a repeat CT scan prior to further attempts at reduction by general surgery services. Ultimately, we ordered a repeat CT scan, which was significant for a “mechanical small bowel obstruction with focal transition zone located within the hernia sac itself, not the neck of the umbilical hernia.” 

Small bowel obstruction is commonly caused by strangulation at the neck of a hernia. In this case, however, the patient had developed an adhesion within the hernia sac itself, which caused the obstruction. This explains why none of the overlying skin changes commonly found in strangulation were visible, and why we were unable to reduce the bowel even though we could palpate the large abdominal wall defect.
Following evaluation by general surgery services, the patient was admitted for laparoscopic hernia repair. Her case was transitioned to an open repair due to extensive intra-abdominal adhesions. The hernia was closed with mesh, and the patient recovered appropriately postoperatively.
Discussion
Abdominal wall hernias are a common pathology, with more than 700,000 repairs performed every year in the United States.2 Patients most commonly present to the ED with abdominal pain, nausea, and vomiting. Less frequently, they present with obstruction, incarceration, strangulation, or rupture.3 Umbilical hernias are caused by increasing intra-abdominal pressure. As the incidence of obesity in the United States has continued to increase, the proportion of hernias that are umbilical or periumbilical has also increased.2,4 Unfortunately, even though umbilical hernias are becoming more common, they are often given less attention than other types of hernias.5 The practice of solely monitoring umbilical hernias can lead to serious outcomes. For example, in a case presentation from Spain, a morbidly obese patient died due to a strangulated umbilical hernia that had progressed over a 15-year period without treatment.6
Compared to elective surgery, emergent operative repair is associated with a higher rate of postoperative complications,1 and a growing body of evidence suggests that patients with symptomatic hernias should be encouraged to undergo operative repair.1,6
Conclusion
Umbilical hernias have become more common with increasing rates of obesity. These hernias have the potential to lead to serious medical emergencies, and the common practice of monitoring chronic hernias may increase the patient’s risk of serious complications. Emergency physicians use the physical examination to help determine the urgency of repair; however, imaging should be considered to assess hernias that cannot easily be reduced to evaluate for obstructed, strangulated, or incarcerated bowel and to help determine the urgency of surgical repair.
1. Davies M, Davies C, Morris-Stiff G, Shute K. Emergency presentation of abdominal hernias: outcome and reasons for delay in treatment-a prospective study. Ann R Coll Surg Engl. 2007;89(1):47-50.
2. Dabbas N, Adams K, Pearson K, Royle G. Frequency of abdominal wall hernias: is classical teaching out of date? JRSM Short Rep. 2011;2(1):5. doi:10.1258/shorts.2010.010071.
3. Rodriguez JA, Hinder RA. Surgical management of umbilical hernia. Operat Tech Gen Surg. 2004;6(3):156-164.
4. Aslani N, Brown CJ. Does mesh offer an advantage over tissue in the open repair of umbilical hernias? A systematic review and meta-analysis. Hernia. 2010;14(5):455-462. doi:10.1016/j.amjsurg.2011.11.015.
5. Arroyo A, García P, Pérez F, Andreu J, Candela F, Calpena R. Randomized clinical trial comparing suture and mesh repair of umbilical hernia in adults. Br J Surgery. 2001;8(10):1321-1323.
6. Rodríguez-Hermosa JI, Codina-Cazador A, Ruiz-Feliú B, Roig-García J, Albiol-Quer M, Planellas-Giné P. Incarcerated umbilical hernia in a super-super-obese patient. Obes Surg. 2008;18(7):893-895. doi:10.1007/s11695-007-9397-3.
Strangulated hernias are a medical emergency that can lead to small bowel obstruction (SBO), bowel necrosis, and death. Practitioners look for signs of strangulation on examination to guide the urgency of management. If the hernia is soft and reducible without overlying skin changes or signs of obstruction, patients may be monitored for years.1 However, there is increasing evidence that even asymptomatic hernias should be repaired rather than monitored to avoid the need for emergent surgical intervention.1
We present a case of a patient with a chronic umbilical hernia who experienced acute worsening of pain at the site of her hernia but with few additional objective signs of strangulation. Prior to this presentation, she had been recently evaluated at our ED for the “same” pain, which included a computed tomography (CT) scan that was negative for an acute surgical emergency. The patient’s second ED visit led to a diagnostic dilemma: Practitioners are encouraged to avoid “unnecessary” radiation—especially in cases of chronic pain—and to rely on history, physical examination findings, and prior recent imaging studies, as appropriate. In this case, repeat imaging ultimately revealed a surgical emergency with an unusual underlying pathology likely related to the chronicity of the patient’s hernia, and explained her repeat presentation to the ED.
Case
A 45-year-old obese woman (body mass index, 46 kg/m2) with a medical history of an umbilical hernia, tubal ligation, and chronic pelvic pain presented a second time to our ED with pain at the site of her hernia, which she stated began 5 hours prior to presentation. Although the pain was associated with nausea and vomiting, the patient said her bowel movements were normal. She first noticed the hernia more than 5 years ago, but experienced her first episode of acute pain related to the hernia with associated nausea and vomiting 3 weeks earlier, which prompted her initial presentation. During this first ED visit, a CT scan of the abdomen/pelvis was obtained as part of her evaluation and was significant for umbilical herniation of bowel without evidence of strangulation. Bedside reduction was successful, and the patient was discharged home and informed of the need to follow-up with a surgeon for an elective repair. She returned to the ED prior to her scheduled operation due to recurrent pain of similar character, but increased severity.
On physical examination, the patient was hemodynamically stable and afebrile. Her vital signs were: heart rate, 84 beats/min; blood pressure, 113/68 mm Hg; and respiratory rate, 20 breaths/min. Oxygen saturation was 100% on room air.
The abdomen was soft with tenderness to palpation over a 13-cm x 8-cm soft hernia to the left of the umbilicus without overlying skin changes. The patient’s pain was controlled with 1 mg of intravenous hydromorphone, after which bedside reduction was attempted. During reduction attempts, there was palpable bowel within the hernia sac, and a periumbilical defect was appreciated. Although the defect in the abdominal wall was estimated to be large enough to allow reduction, the hernia reduced only partially. Because imaging studies from the patient’s previous ED visit showed no visualized strangulation or obstruction, we deliberated over the need for a repeat CT scan prior to further attempts at reduction by general surgery services. Ultimately, we ordered a repeat CT scan, which was significant for a “mechanical small bowel obstruction with focal transition zone located within the hernia sac itself, not the neck of the umbilical hernia.”
Small bowel obstruction is commonly caused by strangulation at the neck of a hernia. In this case, however, the patient had developed an adhesion within the hernia sac itself, which caused the obstruction. This explains why none of the overlying skin changes commonly found in strangulation were visible, and why we were unable to reduce the bowel even though we could palpate the large abdominal wall defect.
Following evaluation by general surgery services, the patient was admitted for laparoscopic hernia repair. Her case was transitioned to an open repair due to extensive intra-abdominal adhesions. The hernia was closed with mesh, and the patient recovered appropriately postoperatively.
Discussion
Abdominal wall hernias are a common pathology, with more than 700,000 repairs performed every year in the United States.2 Patients most commonly present to the ED with abdominal pain, nausea, and vomiting. Less frequently, they present with obstruction, incarceration, strangulation, or rupture.3 Umbilical hernias are caused by increasing intra-abdominal pressure. As the incidence of obesity in the United States has continued to increase, the proportion of hernias that are umbilical or periumbilical has also increased.2,4 Unfortunately, even though umbilical hernias are becoming more common, they are often given less attention than other types of hernias.5 The practice of solely monitoring umbilical hernias can lead to serious outcomes. For example, in a case presentation from Spain, a morbidly obese patient died due to a strangulated umbilical hernia that had progressed over a 15-year period without treatment.6
Compared to elective surgery, emergent operative repair is associated with a higher rate of postoperative complications,1 and a growing body of evidence suggests that patients with symptomatic hernias should be encouraged to undergo operative repair.1,6
Conclusion
Umbilical hernias have become more common with increasing rates of obesity. These hernias have the potential to lead to serious medical emergencies, and the common practice of monitoring chronic hernias may increase the patient’s risk of serious complications. Emergency physicians use the physical examination to help determine the urgency of repair; however, imaging should be considered to assess hernias that cannot easily be reduced to evaluate for obstructed, strangulated, or incarcerated bowel and to help determine the urgency of surgical repair.
Strangulated hernias are a medical emergency that can lead to small bowel obstruction (SBO), bowel necrosis, and death. Practitioners look for signs of strangulation on examination to guide the urgency of management. If the hernia is soft and reducible without overlying skin changes or signs of obstruction, patients may be monitored for years.1 However, there is increasing evidence that even asymptomatic hernias should be repaired rather than monitored to avoid the need for emergent surgical intervention.1
We present a case of a patient with a chronic umbilical hernia who experienced acute worsening of pain at the site of her hernia but with few additional objective signs of strangulation. Prior to this presentation, she had been recently evaluated at our ED for the “same” pain, which included a computed tomography (CT) scan that was negative for an acute surgical emergency. The patient’s second ED visit led to a diagnostic dilemma: Practitioners are encouraged to avoid “unnecessary” radiation—especially in cases of chronic pain—and to rely on history, physical examination findings, and prior recent imaging studies, as appropriate. In this case, repeat imaging ultimately revealed a surgical emergency with an unusual underlying pathology likely related to the chronicity of the patient’s hernia, and explained her repeat presentation to the ED.
Case
A 45-year-old obese woman (body mass index, 46 kg/m2) with a medical history of an umbilical hernia, tubal ligation, and chronic pelvic pain presented a second time to our ED with pain at the site of her hernia, which she stated began 5 hours prior to presentation. Although the pain was associated with nausea and vomiting, the patient said her bowel movements were normal. She first noticed the hernia more than 5 years ago, but experienced her first episode of acute pain related to the hernia with associated nausea and vomiting 3 weeks earlier, which prompted her initial presentation. During this first ED visit, a CT scan of the abdomen/pelvis was obtained as part of her evaluation and was significant for umbilical herniation of bowel without evidence of strangulation. Bedside reduction was successful, and the patient was discharged home and informed of the need to follow-up with a surgeon for an elective repair. She returned to the ED prior to her scheduled operation due to recurrent pain of similar character, but increased severity.
On physical examination, the patient was hemodynamically stable and afebrile. Her vital signs were: heart rate, 84 beats/min; blood pressure, 113/68 mm Hg; and respiratory rate, 20 breaths/min. Oxygen saturation was 100% on room air.
The abdomen was soft with tenderness to palpation over a 13-cm x 8-cm soft hernia to the left of the umbilicus without overlying skin changes. The patient’s pain was controlled with 1 mg of intravenous hydromorphone, after which bedside reduction was attempted. During reduction attempts, there was palpable bowel within the hernia sac, and a periumbilical defect was appreciated. Although the defect in the abdominal wall was estimated to be large enough to allow reduction, the hernia reduced only partially. Because imaging studies from the patient’s previous ED visit showed no visualized strangulation or obstruction, we deliberated over the need for a repeat CT scan prior to further attempts at reduction by general surgery services. Ultimately, we ordered a repeat CT scan, which was significant for a “mechanical small bowel obstruction with focal transition zone located within the hernia sac itself, not the neck of the umbilical hernia.”
Small bowel obstruction is commonly caused by strangulation at the neck of a hernia. In this case, however, the patient had developed an adhesion within the hernia sac itself, which caused the obstruction. This explains why none of the overlying skin changes commonly found in strangulation were visible, and why we were unable to reduce the bowel even though we could palpate the large abdominal wall defect.
Following evaluation by general surgery services, the patient was admitted for laparoscopic hernia repair. Her case was transitioned to an open repair due to extensive intra-abdominal adhesions. The hernia was closed with mesh, and the patient recovered appropriately postoperatively.
Discussion
Abdominal wall hernias are a common pathology, with more than 700,000 repairs performed every year in the United States.2 Patients most commonly present to the ED with abdominal pain, nausea, and vomiting. Less frequently, they present with obstruction, incarceration, strangulation, or rupture.3 Umbilical hernias are caused by increasing intra-abdominal pressure. As the incidence of obesity in the United States has continued to increase, the proportion of hernias that are umbilical or periumbilical has also increased.2,4 Unfortunately, even though umbilical hernias are becoming more common, they are often given less attention than other types of hernias.5 The practice of solely monitoring umbilical hernias can lead to serious outcomes. For example, in a case presentation from Spain, a morbidly obese patient died due to a strangulated umbilical hernia that had progressed over a 15-year period without treatment.6
Compared to elective surgery, emergent operative repair is associated with a higher rate of postoperative complications,1 and a growing body of evidence suggests that patients with symptomatic hernias should be encouraged to undergo operative repair.1,6
Conclusion
Umbilical hernias have become more common with increasing rates of obesity. These hernias have the potential to lead to serious medical emergencies, and the common practice of monitoring chronic hernias may increase the patient’s risk of serious complications. Emergency physicians use the physical examination to help determine the urgency of repair; however, imaging should be considered to assess hernias that cannot easily be reduced to evaluate for obstructed, strangulated, or incarcerated bowel and to help determine the urgency of surgical repair.
1. Davies M, Davies C, Morris-Stiff G, Shute K. Emergency presentation of abdominal hernias: outcome and reasons for delay in treatment-a prospective study. Ann R Coll Surg Engl. 2007;89(1):47-50.
2. Dabbas N, Adams K, Pearson K, Royle G. Frequency of abdominal wall hernias: is classical teaching out of date? JRSM Short Rep. 2011;2(1):5. doi:10.1258/shorts.2010.010071.
3. Rodriguez JA, Hinder RA. Surgical management of umbilical hernia. Operat Tech Gen Surg. 2004;6(3):156-164.
4. Aslani N, Brown CJ. Does mesh offer an advantage over tissue in the open repair of umbilical hernias? A systematic review and meta-analysis. Hernia. 2010;14(5):455-462. doi:10.1016/j.amjsurg.2011.11.015.
5. Arroyo A, García P, Pérez F, Andreu J, Candela F, Calpena R. Randomized clinical trial comparing suture and mesh repair of umbilical hernia in adults. Br J Surgery. 2001;8(10):1321-1323.
6. Rodríguez-Hermosa JI, Codina-Cazador A, Ruiz-Feliú B, Roig-García J, Albiol-Quer M, Planellas-Giné P. Incarcerated umbilical hernia in a super-super-obese patient. Obes Surg. 2008;18(7):893-895. doi:10.1007/s11695-007-9397-3.
1. Davies M, Davies C, Morris-Stiff G, Shute K. Emergency presentation of abdominal hernias: outcome and reasons for delay in treatment-a prospective study. Ann R Coll Surg Engl. 2007;89(1):47-50.
2. Dabbas N, Adams K, Pearson K, Royle G. Frequency of abdominal wall hernias: is classical teaching out of date? JRSM Short Rep. 2011;2(1):5. doi:10.1258/shorts.2010.010071.
3. Rodriguez JA, Hinder RA. Surgical management of umbilical hernia. Operat Tech Gen Surg. 2004;6(3):156-164.
4. Aslani N, Brown CJ. Does mesh offer an advantage over tissue in the open repair of umbilical hernias? A systematic review and meta-analysis. Hernia. 2010;14(5):455-462. doi:10.1016/j.amjsurg.2011.11.015.
5. Arroyo A, García P, Pérez F, Andreu J, Candela F, Calpena R. Randomized clinical trial comparing suture and mesh repair of umbilical hernia in adults. Br J Surgery. 2001;8(10):1321-1323.
6. Rodríguez-Hermosa JI, Codina-Cazador A, Ruiz-Feliú B, Roig-García J, Albiol-Quer M, Planellas-Giné P. Incarcerated umbilical hernia in a super-super-obese patient. Obes Surg. 2008;18(7):893-895. doi:10.1007/s11695-007-9397-3.

Case Studies in Toxicology: Somehow…It’s Always Lupus
Case
A 14-year-old girl with no known medical history presented to the ED via emergency medical services (EMS) approximately 1.5 hours after intentionally ingesting what she described as “a handful or two” of her mother’s lupus prescription medication in a suicide attempt. Initial vital signs and physical examination were normal, and her only complaint was nausea.
Thirty minutes after presentation, the patient suffered acute cardiovascular (CV) collapse: blood pressure, 57/39 mm Hg; heart rate, 90 beats/min. An initial electrocardiogram (ECG) revealed QRS duration of 123 milliseconds and QTc duration of 510 milliseconds, along with nonspecific T-wave abnormalities. A 150-mEq intravenous (IV) bolus of sodium bicarbonate and a 40-mEq potassium chloride IV infusion were administered, and both epinephrine and norepinephrine IV infusions were also initiated. A basic metabolic panel obtained prior to medication administration showed a potassium concentration of 1.9 mmol/L.
What is the differential diagnosis of toxicological hypokalemia?
Hypokalemia may be reflective of diminished whole body potassium stores or a transient alteration of intravascular potassium concentrations. In acute ingestions and overdose, the etiology of the hypokalemia is often electrolyte redistribution through either blockade of constitutive outward potassium leakage (eg, barium, insulin, quinine) or through increased activity of the Na+/K+-ATPase pump (eg, catecholamines, insulin, methylxanthines). This activity has little effect on whole body potassium stores, but can result in a profound fall in the serum potassium. While mild hypokalemia is generally well tolerated, more severe potassium abnormalities can cause muscular weakness, areflexic paralysis, respiratory failure, and life-threatening dysrhythmias. Common ECG findings include decreased T-wave amplitudes, ST-segment depression, and the presence or amplification of U waves.
Case Continuation
While the emergency physicians were stabilizing the patient, her mother provided additional information. Approximately 30 minutes after the exposure, the patient had become nauseated and told her mother what she had done. Her mother called EMS, and the patient was transported to the hospital, where she rapidly became symptomatic. Despite CV decompensation, she remained neurologically intact. On further questioning, the patient admitted to ingesting 6 g of her mother’s prescription of hydroxychloroquine (HCQ) in a suicide attempt but denied taking any other medications. She was stabilized on vasopressors and admitted to the intensive care unit.
What characterizes hydroxychloroquine toxicity?
Hydroxychloroquine is an aminoquinoline antibiotic that is classically used as an antimalarial to treat infection with Plasmodium vivax, P ovale, P malariae, and susceptible strains of P falciparum. In the United States, it is more commonly used to manage both rheumatoid arthritis and systemic lupus erythematosus (SLE), debilitating diseases which are estimated to affect anywhere from 161,000 to 322,000 Americans.1 Hydroxychloroquine is considered first-line therapy for SLE, but its mechanism of action in treating SLE-associated arthralgias is unclear.
Hydroxychloroquine is structurally similar to quinine and chloroquine (CQ), and not surprisingly exerts quinidine-like effects on the CV system with resultant negative inotropy and vasodilation. Its toxicity is characterized by rapid onset of clinical effects including central nervous system depression, seizures, apnea, hypotension, and arrhythmia. After large overdoses, cardiac arrest and death can occur within hours.
Hypokalemia is another hallmark of HCQ toxicity. It is thought to develop secondary to potassium channel blockade, which slows the constitutive release of potassium from the myocytes.2 As noted, the hypokalemia is transient and does not reflect whole-body depletion. With CQ, which is considered more toxic, there appears to be a correlation between the quantity of CQ ingested and both the degree of hypokalemia and the severity of the outcome. It is reasonable to assume the same for HCQ. There are little data to support that hypokalemia itself causes cardiotoxicity in patients with CQ or HCQ overdose.
Although lethal doses are not well established, animal studies suggest that HCQ is much less toxic than CQ, for which the clinical toxicity is better understood due to its more widespread use in overdose abroad.3 In children, the reported therapeutic dose is 10 mg/kg, but the minimum reported lethal dose was a single 300-mg tablet (30 mg/kg in a toddler). In adults, the toxic dose is reported as 20 mg/kg with lethal doses suggested to be as low as 30 mg/kg.
What are the treatment modalities for patients with hydroxychloroquine toxicity?
By analogy with the treatment of CQ poisoning, the mainstay of HCQ therapy is supportive care, including early intubation and ventilation to minimize metabolic demand. Direct-acting inotropes and vasopressors should be administered after saline to treat hypotension. Because of its large volume of distribution, extracorporeal removal has not proved to be of clinical value.4,5 Though data are sparse to determine its efficacy, there may be a role for giving activated charcoal, particularly following large overdoses—if it is given early after exposure and the patient has normal consciousness. If the patient is intubated and aspiration risk is minimized, gastric lavage may also be beneficial—especially when performed within an hour of the overdose. Syrup of ipecac should not be used.
High-dose diazepam is typically recommended, again by analogy with CQ, although there is no clear mechanism of action and its use remains controversial. Its protective effect in patients with CQ overdose is based on swine and rat models that demonstrated dose dependent relationships between diazepam and survival.6,7 A prospective study of CQ toxicity in humans reported improved survival rates when high-dose diazepam was given in combination with epinephrine.8 However, this study is limited by its comparison of prospectively studied patients with a retrospective control. A subsequent prospective study of moderately CQ-intoxicated patients did not find a benefit from treatment with diazepam.9 Furthermore, it remains unclear if the proposed benefit from high-dose diazepam in CQ toxicity may be extrapolated to HCQ, and cases of even massive HCQ ingestions report good outcomes without the use of high-dose diazepam.10
How aggressively should hypokalemia in hydroxychloroquine toxicity be treated?
As noted earlier, hypokalemia resulting from HCQ toxicity is transient, and aggressive repletion may result in rebound hyperkalemia once toxicity resolves. However, these dangers should be balanced with risks of hypokalemia-induced ventricular arrhythmias. Additionally, hypokalemia may be worsened by sodium bicarbonate that is administered to correct QRS prolongations, increasing the risk of dysrhythmia. Correction of hypokalemia in these cases is necessary but should be done with care and monitoring of serum potassium concentrations to minimize risk of hyperkalemia-induced ventricular arrhythmia.11
Case Conclusion
Throughout treatment, the patient remained neurologically intact. She did not receive benzodiazepines. The epinephrine and norepinephrine infusions were weaned, and she was discharged on hospital day 3 with no neurological or cardiac sequelae. She received an inpatient psychiatric evaluation and was referred to outpatient services for ongoing care.
1. Helmick CG, Felson DT, Lawrence RC, et al; National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: Part I. Arthritis Rheum. 2008;58(1):15-25. doi:10.1002/art.23177.
2. Clemessy JL, Favier C, Borron SW, Hantson PE, Vicaut E, Baud FJ. Hypokalaemia related to acute chloroquine ingestion. Lancet. 1995;3469(8979):877-880.
3. McChesney EW. Animal toxicity and pharmacokinetics of hydroxychloroquine sulfate. Am J Med. 1983;75(suppl 1A):11-18.
4. Carmichael SJ, Charles B, Tett SE. Population pharmacokinetics of hydroxychloroquine in patients with rheumatoid arthritis. Ther Drug Monit. 2003;25(6):671-681.
5. Marquardt K, Albertson TE. Treatment of hydroxychloroquine overdose. Am J Emerg Med. 2001;19(5):420-424.
6. Crouzette J, Vicaut E, Palombo S, Girre C, Fournier PE. Experimental assessment of the protective activity of diazepam on the acute toxicity of chloroquine. J Toxicol Clin Toxicol. 1983;20(3):271-279.
7. Riou B, Lecarpentier Y, Barriot P, Viars P. Diazepam does not improve the mechanical performance of rat cardiac papillary muscle exposed to chloroquine in vitro. Intensive Care Med. 1989;15:390-3955.
8. Riou B, Barriot P, Rimailho A, Baud FJ. Treatment of severe chloroquine poisoning. N Engl J Med. 1988;318(1):1-6.
9. Clemessy JL, Angel G, Borron SW, et al. Therapeutic trial of diazepam versus placebo in acute chloroquine intoxications of moderate gravity. Intensive Care Med. 1996;22:1400-1405.
10. Yanturali S. Diazepam for treatment of massive chloroquine intoxication. Resuscitation. 2004;63(3):347-348.
11. Ling Ngan Wong A, Tsz Fung Cheung I, Graham CA. Hydroxychloroquine overdose: case report and recommendations for management. Eur J Emerg Med. 2008;15(1):16-8. doi:10.1097/MEJ.0b013e3280adcb56.
Case
A 14-year-old girl with no known medical history presented to the ED via emergency medical services (EMS) approximately 1.5 hours after intentionally ingesting what she described as “a handful or two” of her mother’s lupus prescription medication in a suicide attempt. Initial vital signs and physical examination were normal, and her only complaint was nausea.
Thirty minutes after presentation, the patient suffered acute cardiovascular (CV) collapse: blood pressure, 57/39 mm Hg; heart rate, 90 beats/min. An initial electrocardiogram (ECG) revealed QRS duration of 123 milliseconds and QTc duration of 510 milliseconds, along with nonspecific T-wave abnormalities. A 150-mEq intravenous (IV) bolus of sodium bicarbonate and a 40-mEq potassium chloride IV infusion were administered, and both epinephrine and norepinephrine IV infusions were also initiated. A basic metabolic panel obtained prior to medication administration showed a potassium concentration of 1.9 mmol/L.
What is the differential diagnosis of toxicological hypokalemia?
Hypokalemia may be reflective of diminished whole body potassium stores or a transient alteration of intravascular potassium concentrations. In acute ingestions and overdose, the etiology of the hypokalemia is often electrolyte redistribution through either blockade of constitutive outward potassium leakage (eg, barium, insulin, quinine) or through increased activity of the Na+/K+-ATPase pump (eg, catecholamines, insulin, methylxanthines). This activity has little effect on whole body potassium stores, but can result in a profound fall in the serum potassium. While mild hypokalemia is generally well tolerated, more severe potassium abnormalities can cause muscular weakness, areflexic paralysis, respiratory failure, and life-threatening dysrhythmias. Common ECG findings include decreased T-wave amplitudes, ST-segment depression, and the presence or amplification of U waves.
Case Continuation
While the emergency physicians were stabilizing the patient, her mother provided additional information. Approximately 30 minutes after the exposure, the patient had become nauseated and told her mother what she had done. Her mother called EMS, and the patient was transported to the hospital, where she rapidly became symptomatic. Despite CV decompensation, she remained neurologically intact. On further questioning, the patient admitted to ingesting 6 g of her mother’s prescription of hydroxychloroquine (HCQ) in a suicide attempt but denied taking any other medications. She was stabilized on vasopressors and admitted to the intensive care unit.
What characterizes hydroxychloroquine toxicity?
Hydroxychloroquine is an aminoquinoline antibiotic that is classically used as an antimalarial to treat infection with Plasmodium vivax, P ovale, P malariae, and susceptible strains of P falciparum. In the United States, it is more commonly used to manage both rheumatoid arthritis and systemic lupus erythematosus (SLE), debilitating diseases which are estimated to affect anywhere from 161,000 to 322,000 Americans.1 Hydroxychloroquine is considered first-line therapy for SLE, but its mechanism of action in treating SLE-associated arthralgias is unclear.
Hydroxychloroquine is structurally similar to quinine and chloroquine (CQ), and not surprisingly exerts quinidine-like effects on the CV system with resultant negative inotropy and vasodilation. Its toxicity is characterized by rapid onset of clinical effects including central nervous system depression, seizures, apnea, hypotension, and arrhythmia. After large overdoses, cardiac arrest and death can occur within hours.
Hypokalemia is another hallmark of HCQ toxicity. It is thought to develop secondary to potassium channel blockade, which slows the constitutive release of potassium from the myocytes.2 As noted, the hypokalemia is transient and does not reflect whole-body depletion. With CQ, which is considered more toxic, there appears to be a correlation between the quantity of CQ ingested and both the degree of hypokalemia and the severity of the outcome. It is reasonable to assume the same for HCQ. There are little data to support that hypokalemia itself causes cardiotoxicity in patients with CQ or HCQ overdose.
Although lethal doses are not well established, animal studies suggest that HCQ is much less toxic than CQ, for which the clinical toxicity is better understood due to its more widespread use in overdose abroad.3 In children, the reported therapeutic dose is 10 mg/kg, but the minimum reported lethal dose was a single 300-mg tablet (30 mg/kg in a toddler). In adults, the toxic dose is reported as 20 mg/kg with lethal doses suggested to be as low as 30 mg/kg.
What are the treatment modalities for patients with hydroxychloroquine toxicity?
By analogy with the treatment of CQ poisoning, the mainstay of HCQ therapy is supportive care, including early intubation and ventilation to minimize metabolic demand. Direct-acting inotropes and vasopressors should be administered after saline to treat hypotension. Because of its large volume of distribution, extracorporeal removal has not proved to be of clinical value.4,5 Though data are sparse to determine its efficacy, there may be a role for giving activated charcoal, particularly following large overdoses—if it is given early after exposure and the patient has normal consciousness. If the patient is intubated and aspiration risk is minimized, gastric lavage may also be beneficial—especially when performed within an hour of the overdose. Syrup of ipecac should not be used.
High-dose diazepam is typically recommended, again by analogy with CQ, although there is no clear mechanism of action and its use remains controversial. Its protective effect in patients with CQ overdose is based on swine and rat models that demonstrated dose dependent relationships between diazepam and survival.6,7 A prospective study of CQ toxicity in humans reported improved survival rates when high-dose diazepam was given in combination with epinephrine.8 However, this study is limited by its comparison of prospectively studied patients with a retrospective control. A subsequent prospective study of moderately CQ-intoxicated patients did not find a benefit from treatment with diazepam.9 Furthermore, it remains unclear if the proposed benefit from high-dose diazepam in CQ toxicity may be extrapolated to HCQ, and cases of even massive HCQ ingestions report good outcomes without the use of high-dose diazepam.10
How aggressively should hypokalemia in hydroxychloroquine toxicity be treated?
As noted earlier, hypokalemia resulting from HCQ toxicity is transient, and aggressive repletion may result in rebound hyperkalemia once toxicity resolves. However, these dangers should be balanced with risks of hypokalemia-induced ventricular arrhythmias. Additionally, hypokalemia may be worsened by sodium bicarbonate that is administered to correct QRS prolongations, increasing the risk of dysrhythmia. Correction of hypokalemia in these cases is necessary but should be done with care and monitoring of serum potassium concentrations to minimize risk of hyperkalemia-induced ventricular arrhythmia.11
Case Conclusion
Throughout treatment, the patient remained neurologically intact. She did not receive benzodiazepines. The epinephrine and norepinephrine infusions were weaned, and she was discharged on hospital day 3 with no neurological or cardiac sequelae. She received an inpatient psychiatric evaluation and was referred to outpatient services for ongoing care.
Case
A 14-year-old girl with no known medical history presented to the ED via emergency medical services (EMS) approximately 1.5 hours after intentionally ingesting what she described as “a handful or two” of her mother’s lupus prescription medication in a suicide attempt. Initial vital signs and physical examination were normal, and her only complaint was nausea.
Thirty minutes after presentation, the patient suffered acute cardiovascular (CV) collapse: blood pressure, 57/39 mm Hg; heart rate, 90 beats/min. An initial electrocardiogram (ECG) revealed QRS duration of 123 milliseconds and QTc duration of 510 milliseconds, along with nonspecific T-wave abnormalities. A 150-mEq intravenous (IV) bolus of sodium bicarbonate and a 40-mEq potassium chloride IV infusion were administered, and both epinephrine and norepinephrine IV infusions were also initiated. A basic metabolic panel obtained prior to medication administration showed a potassium concentration of 1.9 mmol/L.
What is the differential diagnosis of toxicological hypokalemia?
Hypokalemia may be reflective of diminished whole body potassium stores or a transient alteration of intravascular potassium concentrations. In acute ingestions and overdose, the etiology of the hypokalemia is often electrolyte redistribution through either blockade of constitutive outward potassium leakage (eg, barium, insulin, quinine) or through increased activity of the Na+/K+-ATPase pump (eg, catecholamines, insulin, methylxanthines). This activity has little effect on whole body potassium stores, but can result in a profound fall in the serum potassium. While mild hypokalemia is generally well tolerated, more severe potassium abnormalities can cause muscular weakness, areflexic paralysis, respiratory failure, and life-threatening dysrhythmias. Common ECG findings include decreased T-wave amplitudes, ST-segment depression, and the presence or amplification of U waves.
Case Continuation
While the emergency physicians were stabilizing the patient, her mother provided additional information. Approximately 30 minutes after the exposure, the patient had become nauseated and told her mother what she had done. Her mother called EMS, and the patient was transported to the hospital, where she rapidly became symptomatic. Despite CV decompensation, she remained neurologically intact. On further questioning, the patient admitted to ingesting 6 g of her mother’s prescription of hydroxychloroquine (HCQ) in a suicide attempt but denied taking any other medications. She was stabilized on vasopressors and admitted to the intensive care unit.
What characterizes hydroxychloroquine toxicity?
Hydroxychloroquine is an aminoquinoline antibiotic that is classically used as an antimalarial to treat infection with Plasmodium vivax, P ovale, P malariae, and susceptible strains of P falciparum. In the United States, it is more commonly used to manage both rheumatoid arthritis and systemic lupus erythematosus (SLE), debilitating diseases which are estimated to affect anywhere from 161,000 to 322,000 Americans.1 Hydroxychloroquine is considered first-line therapy for SLE, but its mechanism of action in treating SLE-associated arthralgias is unclear.
Hydroxychloroquine is structurally similar to quinine and chloroquine (CQ), and not surprisingly exerts quinidine-like effects on the CV system with resultant negative inotropy and vasodilation. Its toxicity is characterized by rapid onset of clinical effects including central nervous system depression, seizures, apnea, hypotension, and arrhythmia. After large overdoses, cardiac arrest and death can occur within hours.
Hypokalemia is another hallmark of HCQ toxicity. It is thought to develop secondary to potassium channel blockade, which slows the constitutive release of potassium from the myocytes.2 As noted, the hypokalemia is transient and does not reflect whole-body depletion. With CQ, which is considered more toxic, there appears to be a correlation between the quantity of CQ ingested and both the degree of hypokalemia and the severity of the outcome. It is reasonable to assume the same for HCQ. There are little data to support that hypokalemia itself causes cardiotoxicity in patients with CQ or HCQ overdose.
Although lethal doses are not well established, animal studies suggest that HCQ is much less toxic than CQ, for which the clinical toxicity is better understood due to its more widespread use in overdose abroad.3 In children, the reported therapeutic dose is 10 mg/kg, but the minimum reported lethal dose was a single 300-mg tablet (30 mg/kg in a toddler). In adults, the toxic dose is reported as 20 mg/kg with lethal doses suggested to be as low as 30 mg/kg.
What are the treatment modalities for patients with hydroxychloroquine toxicity?
By analogy with the treatment of CQ poisoning, the mainstay of HCQ therapy is supportive care, including early intubation and ventilation to minimize metabolic demand. Direct-acting inotropes and vasopressors should be administered after saline to treat hypotension. Because of its large volume of distribution, extracorporeal removal has not proved to be of clinical value.4,5 Though data are sparse to determine its efficacy, there may be a role for giving activated charcoal, particularly following large overdoses—if it is given early after exposure and the patient has normal consciousness. If the patient is intubated and aspiration risk is minimized, gastric lavage may also be beneficial—especially when performed within an hour of the overdose. Syrup of ipecac should not be used.
High-dose diazepam is typically recommended, again by analogy with CQ, although there is no clear mechanism of action and its use remains controversial. Its protective effect in patients with CQ overdose is based on swine and rat models that demonstrated dose dependent relationships between diazepam and survival.6,7 A prospective study of CQ toxicity in humans reported improved survival rates when high-dose diazepam was given in combination with epinephrine.8 However, this study is limited by its comparison of prospectively studied patients with a retrospective control. A subsequent prospective study of moderately CQ-intoxicated patients did not find a benefit from treatment with diazepam.9 Furthermore, it remains unclear if the proposed benefit from high-dose diazepam in CQ toxicity may be extrapolated to HCQ, and cases of even massive HCQ ingestions report good outcomes without the use of high-dose diazepam.10
How aggressively should hypokalemia in hydroxychloroquine toxicity be treated?
As noted earlier, hypokalemia resulting from HCQ toxicity is transient, and aggressive repletion may result in rebound hyperkalemia once toxicity resolves. However, these dangers should be balanced with risks of hypokalemia-induced ventricular arrhythmias. Additionally, hypokalemia may be worsened by sodium bicarbonate that is administered to correct QRS prolongations, increasing the risk of dysrhythmia. Correction of hypokalemia in these cases is necessary but should be done with care and monitoring of serum potassium concentrations to minimize risk of hyperkalemia-induced ventricular arrhythmia.11
Case Conclusion
Throughout treatment, the patient remained neurologically intact. She did not receive benzodiazepines. The epinephrine and norepinephrine infusions were weaned, and she was discharged on hospital day 3 with no neurological or cardiac sequelae. She received an inpatient psychiatric evaluation and was referred to outpatient services for ongoing care.
1. Helmick CG, Felson DT, Lawrence RC, et al; National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: Part I. Arthritis Rheum. 2008;58(1):15-25. doi:10.1002/art.23177.
2. Clemessy JL, Favier C, Borron SW, Hantson PE, Vicaut E, Baud FJ. Hypokalaemia related to acute chloroquine ingestion. Lancet. 1995;3469(8979):877-880.
3. McChesney EW. Animal toxicity and pharmacokinetics of hydroxychloroquine sulfate. Am J Med. 1983;75(suppl 1A):11-18.
4. Carmichael SJ, Charles B, Tett SE. Population pharmacokinetics of hydroxychloroquine in patients with rheumatoid arthritis. Ther Drug Monit. 2003;25(6):671-681.
5. Marquardt K, Albertson TE. Treatment of hydroxychloroquine overdose. Am J Emerg Med. 2001;19(5):420-424.
6. Crouzette J, Vicaut E, Palombo S, Girre C, Fournier PE. Experimental assessment of the protective activity of diazepam on the acute toxicity of chloroquine. J Toxicol Clin Toxicol. 1983;20(3):271-279.
7. Riou B, Lecarpentier Y, Barriot P, Viars P. Diazepam does not improve the mechanical performance of rat cardiac papillary muscle exposed to chloroquine in vitro. Intensive Care Med. 1989;15:390-3955.
8. Riou B, Barriot P, Rimailho A, Baud FJ. Treatment of severe chloroquine poisoning. N Engl J Med. 1988;318(1):1-6.
9. Clemessy JL, Angel G, Borron SW, et al. Therapeutic trial of diazepam versus placebo in acute chloroquine intoxications of moderate gravity. Intensive Care Med. 1996;22:1400-1405.
10. Yanturali S. Diazepam for treatment of massive chloroquine intoxication. Resuscitation. 2004;63(3):347-348.
11. Ling Ngan Wong A, Tsz Fung Cheung I, Graham CA. Hydroxychloroquine overdose: case report and recommendations for management. Eur J Emerg Med. 2008;15(1):16-8. doi:10.1097/MEJ.0b013e3280adcb56.
1. Helmick CG, Felson DT, Lawrence RC, et al; National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: Part I. Arthritis Rheum. 2008;58(1):15-25. doi:10.1002/art.23177.
2. Clemessy JL, Favier C, Borron SW, Hantson PE, Vicaut E, Baud FJ. Hypokalaemia related to acute chloroquine ingestion. Lancet. 1995;3469(8979):877-880.
3. McChesney EW. Animal toxicity and pharmacokinetics of hydroxychloroquine sulfate. Am J Med. 1983;75(suppl 1A):11-18.
4. Carmichael SJ, Charles B, Tett SE. Population pharmacokinetics of hydroxychloroquine in patients with rheumatoid arthritis. Ther Drug Monit. 2003;25(6):671-681.
5. Marquardt K, Albertson TE. Treatment of hydroxychloroquine overdose. Am J Emerg Med. 2001;19(5):420-424.
6. Crouzette J, Vicaut E, Palombo S, Girre C, Fournier PE. Experimental assessment of the protective activity of diazepam on the acute toxicity of chloroquine. J Toxicol Clin Toxicol. 1983;20(3):271-279.
7. Riou B, Lecarpentier Y, Barriot P, Viars P. Diazepam does not improve the mechanical performance of rat cardiac papillary muscle exposed to chloroquine in vitro. Intensive Care Med. 1989;15:390-3955.
8. Riou B, Barriot P, Rimailho A, Baud FJ. Treatment of severe chloroquine poisoning. N Engl J Med. 1988;318(1):1-6.
9. Clemessy JL, Angel G, Borron SW, et al. Therapeutic trial of diazepam versus placebo in acute chloroquine intoxications of moderate gravity. Intensive Care Med. 1996;22:1400-1405.
10. Yanturali S. Diazepam for treatment of massive chloroquine intoxication. Resuscitation. 2004;63(3):347-348.
11. Ling Ngan Wong A, Tsz Fung Cheung I, Graham CA. Hydroxychloroquine overdose: case report and recommendations for management. Eur J Emerg Med. 2008;15(1):16-8. doi:10.1097/MEJ.0b013e3280adcb56.
Idiopathic Livedo Racemosa Presenting With Splenomegaly and Diffuse Lymphadenopathy
Sneddon syndrome (SS) was first described in 1965 in patients with persistent livedo racemosa and neurological events.1 Because the other manifestations of SS are nonspecific (eg, hypertension, cardiac valvulopathy, arterial and venous occlusion), the diagnosis often is delayed. Many patients who experience prodromal neurologic symptoms such as headaches, depression, anxiety, dizziness, and neuropathy often present to a physician prior to developing ischemic brain manifestations2 but seldom receive the correct diagnosis. Onset of cerebral occlusive events typically occurs in patients younger than 45 years and may present as a transient ischemic attack, stroke, or intracranial hemorrhage.3 The disease is more prevalent in females than males (2:1 ratio). The exact pathogenesis of SS is still unknown, and although it has been thought of as a separate entity from systemic lupus erythematosus and other antiphospholipid disorders, it has been postulated that an immunological dysfunction damages vessel walls leading to thrombosis.
Cutaneous findings associated with SS involve small- to medium-sized dermal-subdermal arteries. Histopathology in some patients demonstrates proliferation of the endothelium and fibrin deposits with subsequent obliteration of involved arteries.4 In many patients including our patient, histopathologic examination of involved skin fails to show specific abnormalities.1 Zelger et al5 reported the sequence of histopathologic skin events in a series of antiphospholipid-negative SS patients. The authors reported that only small arteries at the dermis-subcutis junction were involved and a progression of endothelial dysfunction was observed. The authors believed there were several nonspecific stages prior to fibrin occlusion of involved arteries.5 Stage I involved loosening of endothelial cells with nonspecific perivascular lymphocytic infiltration with perivascular inflammation and lymphocytic infiltration representing the prime mover of the disease.5,6 This stage is thought to be short lived, thus the reason why it has gone undetected for many years in SS patients. Stages II to IV progress through fibrin deposition and occlusion.5 Histological features of stages I to II have not been reported because of late diagnosis of SS. Stage I patients typically present with an average duration of symptoms of 6 months with few neurologic symptoms, the most common being paresthesia of the legs.5
Case Report
A 37-year-old woman with epigastric tenderness on the left side and splenomegaly seen on computed tomography was referred by a hematologist for evaluation of a reticular rash on the left side of the flank of 9 months’ duration with a presumed diagnosis of focal melanoderma. Her medical history was remarkable for a congenital ventricular septal defect and coarctation of the aorta, as well as endometriosis, myalgia, and joint stiffness that had all developed over the last year. Her medical history also was remarkable for nephrolithiasis, irritable bowel syndrome, and chronic sinusitis, as well as psychiatric depression and anxiety disorders. She recently had been diagnosed with moderate hypertension and had experienced difficulty getting pregnant for the last several years with 3 consecutive miscarriages in the first trimester. Neurologic symptoms included neuropathy involving the feet, intermittent paresthesia of the legs, and a history of chronic migraine headaches for several months.
Dermatologic examination revealed a slightly overweight woman with a 25×30-cm dusky, erythematous, irregular, netlike pattern on the left side of the upper and lower trunk (Figure 1). Extensive livedo racemosa was not altered by changes in temperature and had been unchanged for more than 9 months. There were no signs of pruritus or ulcerations, and areas of livedo racemosa were slightly tender to palpation.
We performed 2 sets of three 4-mm biopsies. The first set targeted areas within the violaceous pattern, while the second set targeted areas of normal tissue between the mottled areas. All 6 specimens demonstrated superficial perivascular lymphocytic infiltrate with no evidence of vasculitis or connective tissue disease. The vessels showed no microthrombi or surrounding fibrosis. No eosinophils were identified within the epidermis. There was no evidence of increased dermal mucin. Both the superficial and deep vascular plexuses were unremarkable and showed no evidence of damage to the walls (Figure 2).
To rule out other possible causes of livedo racemosa, complete blood cell count, comprehensive metabolic panel, coagulation profile, lipase test, urinalysis, serologic testing, and immunologic workup were performed. Lipase was within reference range. The complete blood cell count revealed mild anemia, while the rest of the values were within reference range. An immunologic workup included Sjögren syndrome antigen A, Sjögren syndrome antigen B, anticardiolipin antibodies, and antinuclear antibody, which were all negative. Family history was remarkable for first-degree relatives with systemic lupus erythematosus and Crohn disease.
Computed tomography revealed enlargement of the spleen, as well as periaortic, portacaval, and porta hepatis lymphadenopathy. Based on the laboratory findings and clinical presentation as well as the patient’s medical history, the diagnosis of exclusion was idiopathic livedo racemosa with unknown progression to full-blown SS. The patient did not meet the current diagnostic criteria for SS, and her immunologic studies failed to confirm any present antibodies, but involvement of the reticuloendothelial system pointed to production of antibodies that were not yet detectable on laboratory testing.
Comment
More than 50 years after the first case of SS was diagnosed, better laboratory workup is available and more information is known about the pathophysiology. Sneddon syndrome is a rare disorder, affecting only approximately 4 patients per million each year worldwide. Seronegative antiphospholipid antibody syndrome (SNAPS) describes patients with clinical presentations of antiphospholipid syndrome (APS) without detectable serological markers.7 Antiphospholipid-negative SS, which was seen in our patient, would be categorized under SNAPS. A PubMed search of articles indexed for MEDLINE using the terms livedo racemosa, Sneddon syndrome, and SNAPS and splenomegaly revealed there currently are no known cases of SNAPS that have been reported with splenomegaly and lymphadenopathy. Our patient presented with the following clinical features of SS: livedo racemosa, history of miscarriage, psychiatric disturbances, and hypertension. Surprisingly, biopsies from affected skin did not show any fibrin deposition or microthrombi but did reveal perivascular lymphocytic infiltrations. Magnetic resonance imaging did not show any pathological lesions or vascular changes.
Sneddon syndrome and APS share a common pathway to occlusive arteriolopathy for which 4 stages have been described by Zelger et al.5 Stage I involves a nonspecific Langerhans cell infiltrate with polymorphonuclear leukocytes. The tunica media and elastic lamina usually are unaltered at this early stage, while the surrounding connective tissue may appear edematous.5 This early stage of histopathology has not been evaluated in SS patients, primarily because of delay of diagnosis. Late stages III and IV will show fibrin deposition and shrinkage of affected vessels.7
A PubMed search using the terms Sneddon syndrome, lymphadenopathy and livedo racemosa, and Sneddon syndrome and lymphadenopathy revealed that splenomegaly and lymphadenopathy have not been reported in patients with SS. In patients with antiphospholipid-negative SS, one can assume that antibodies to other phospholipids not tested must exist because of striking similarities between APS and antiphospholipid-negative SS.8 Although our patient did not test positive for any of these antibodies, she did present with lymphadenopathy and splenic enlargement, leading us to believe that involvement of the reticuloendothelial system may be a feature of SS that has not been previously reported. Further studies are required to name specific antigens responsible for clinical manifestations in SS.
Currently, no single diagnostic test for SS exists, thus delaying both diagnosis and initiation of treatment. Histopathologic examination may be helpful, but in many cases it is nonspecific, as are serologic markers. Neuroradiological confirmation of involvement usually is the confirmatory feature in many patients with late-stage diagnosis.2 A diagnostic schematic for SS, which was first described by Daoud et al,2 illustrates classification of symptoms and aids in diagnosis. A working diagnosis of idiopathic livedo racemosa is made after ruling out other causes of SS in a patient with nonspecific biopsy findings and negative magnetic resonance imaging results with prodromal symptoms. The prognosis for such patients progressing to full SS is unknown with or without management using anticoagulant therapy.
Conclusion
Early diagnosis of livedo racemosa and SS is essential, as prevention of cerebrovascular accidents, myocardial infarction, and other thromboembolic diseases can be minimized by attacking risk factors such as smoking, taking oral contraceptive pills, becoming pregnant,9 and by initiating either antiplatelet or anticoagulation treatments. These treatments have been shown to delay the development of neurovascular damage and early-onset dementia. We present this case to demonstrate the variability of early-presenting symptoms in idiopathic livedo racemosa. Recognizing some of the early manifestations can lead to early diagnosis and initiation of treatment.
- Sneddon IB. Cerebro-vascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Daoud MS, Wilmoth GJ, Su WP, et al. Sneddon syndrome. Semin Dermatol. 1995;14:166-172.
- Besnier R, Francès C, Ankri A, et al. Factor V Leiden mutation in Sneddon syndrome. Lupus. 2003;12:406-408.
- K aragülle AT, Karadağ D, Erden A, et al. Sneddon’s syndrome: MR imaging findings. Eur Radiol. 2002;12:144-146.
- Zelg er B, Sepp N, Schmid KW, et al. Life-history of cutaneous vascular-lesions in Sneddon’s syndrome. Hum Pathol. 1992;23:668-675.
- Ayoub N, Esposito G, Barete S, et al. Protein Z deficiency in antiphospholipid-negative Sneddon’s syndrome. Stroke. 2004;35:1329-1332.
- Duva l A, Darnige L, Glowacki F, et al. Livedo, dementia, thrombocytopenia, and endotheliitis without antiphospholipid antibodies: seronegative antiphospholipid-like syndrome. J Am Acad Dermatol. 2009;61:1076-1078.
- Kala shnikova LA, Nasonov EL, Kushekbaeva AE, et al. Anticardiolipin antibodies in Sneddon’s syndrome. Neurology. 1990;40:464-467.
- Wohl rab J, Fischer M, Wolter M, et al. Diagnostic impact and sensitivity of skin biopsies in Sneddon’s syndrome. a report of 15 cases. Br J Dermatol. 2001;145:285-288.
Sneddon syndrome (SS) was first described in 1965 in patients with persistent livedo racemosa and neurological events.1 Because the other manifestations of SS are nonspecific (eg, hypertension, cardiac valvulopathy, arterial and venous occlusion), the diagnosis often is delayed. Many patients who experience prodromal neurologic symptoms such as headaches, depression, anxiety, dizziness, and neuropathy often present to a physician prior to developing ischemic brain manifestations2 but seldom receive the correct diagnosis. Onset of cerebral occlusive events typically occurs in patients younger than 45 years and may present as a transient ischemic attack, stroke, or intracranial hemorrhage.3 The disease is more prevalent in females than males (2:1 ratio). The exact pathogenesis of SS is still unknown, and although it has been thought of as a separate entity from systemic lupus erythematosus and other antiphospholipid disorders, it has been postulated that an immunological dysfunction damages vessel walls leading to thrombosis.
Cutaneous findings associated with SS involve small- to medium-sized dermal-subdermal arteries. Histopathology in some patients demonstrates proliferation of the endothelium and fibrin deposits with subsequent obliteration of involved arteries.4 In many patients including our patient, histopathologic examination of involved skin fails to show specific abnormalities.1 Zelger et al5 reported the sequence of histopathologic skin events in a series of antiphospholipid-negative SS patients. The authors reported that only small arteries at the dermis-subcutis junction were involved and a progression of endothelial dysfunction was observed. The authors believed there were several nonspecific stages prior to fibrin occlusion of involved arteries.5 Stage I involved loosening of endothelial cells with nonspecific perivascular lymphocytic infiltration with perivascular inflammation and lymphocytic infiltration representing the prime mover of the disease.5,6 This stage is thought to be short lived, thus the reason why it has gone undetected for many years in SS patients. Stages II to IV progress through fibrin deposition and occlusion.5 Histological features of stages I to II have not been reported because of late diagnosis of SS. Stage I patients typically present with an average duration of symptoms of 6 months with few neurologic symptoms, the most common being paresthesia of the legs.5
Case Report
A 37-year-old woman with epigastric tenderness on the left side and splenomegaly seen on computed tomography was referred by a hematologist for evaluation of a reticular rash on the left side of the flank of 9 months’ duration with a presumed diagnosis of focal melanoderma. Her medical history was remarkable for a congenital ventricular septal defect and coarctation of the aorta, as well as endometriosis, myalgia, and joint stiffness that had all developed over the last year. Her medical history also was remarkable for nephrolithiasis, irritable bowel syndrome, and chronic sinusitis, as well as psychiatric depression and anxiety disorders. She recently had been diagnosed with moderate hypertension and had experienced difficulty getting pregnant for the last several years with 3 consecutive miscarriages in the first trimester. Neurologic symptoms included neuropathy involving the feet, intermittent paresthesia of the legs, and a history of chronic migraine headaches for several months.
Dermatologic examination revealed a slightly overweight woman with a 25×30-cm dusky, erythematous, irregular, netlike pattern on the left side of the upper and lower trunk (Figure 1). Extensive livedo racemosa was not altered by changes in temperature and had been unchanged for more than 9 months. There were no signs of pruritus or ulcerations, and areas of livedo racemosa were slightly tender to palpation.
We performed 2 sets of three 4-mm biopsies. The first set targeted areas within the violaceous pattern, while the second set targeted areas of normal tissue between the mottled areas. All 6 specimens demonstrated superficial perivascular lymphocytic infiltrate with no evidence of vasculitis or connective tissue disease. The vessels showed no microthrombi or surrounding fibrosis. No eosinophils were identified within the epidermis. There was no evidence of increased dermal mucin. Both the superficial and deep vascular plexuses were unremarkable and showed no evidence of damage to the walls (Figure 2).
To rule out other possible causes of livedo racemosa, complete blood cell count, comprehensive metabolic panel, coagulation profile, lipase test, urinalysis, serologic testing, and immunologic workup were performed. Lipase was within reference range. The complete blood cell count revealed mild anemia, while the rest of the values were within reference range. An immunologic workup included Sjögren syndrome antigen A, Sjögren syndrome antigen B, anticardiolipin antibodies, and antinuclear antibody, which were all negative. Family history was remarkable for first-degree relatives with systemic lupus erythematosus and Crohn disease.
Computed tomography revealed enlargement of the spleen, as well as periaortic, portacaval, and porta hepatis lymphadenopathy. Based on the laboratory findings and clinical presentation as well as the patient’s medical history, the diagnosis of exclusion was idiopathic livedo racemosa with unknown progression to full-blown SS. The patient did not meet the current diagnostic criteria for SS, and her immunologic studies failed to confirm any present antibodies, but involvement of the reticuloendothelial system pointed to production of antibodies that were not yet detectable on laboratory testing.
Comment
More than 50 years after the first case of SS was diagnosed, better laboratory workup is available and more information is known about the pathophysiology. Sneddon syndrome is a rare disorder, affecting only approximately 4 patients per million each year worldwide. Seronegative antiphospholipid antibody syndrome (SNAPS) describes patients with clinical presentations of antiphospholipid syndrome (APS) without detectable serological markers.7 Antiphospholipid-negative SS, which was seen in our patient, would be categorized under SNAPS. A PubMed search of articles indexed for MEDLINE using the terms livedo racemosa, Sneddon syndrome, and SNAPS and splenomegaly revealed there currently are no known cases of SNAPS that have been reported with splenomegaly and lymphadenopathy. Our patient presented with the following clinical features of SS: livedo racemosa, history of miscarriage, psychiatric disturbances, and hypertension. Surprisingly, biopsies from affected skin did not show any fibrin deposition or microthrombi but did reveal perivascular lymphocytic infiltrations. Magnetic resonance imaging did not show any pathological lesions or vascular changes.
Sneddon syndrome and APS share a common pathway to occlusive arteriolopathy for which 4 stages have been described by Zelger et al.5 Stage I involves a nonspecific Langerhans cell infiltrate with polymorphonuclear leukocytes. The tunica media and elastic lamina usually are unaltered at this early stage, while the surrounding connective tissue may appear edematous.5 This early stage of histopathology has not been evaluated in SS patients, primarily because of delay of diagnosis. Late stages III and IV will show fibrin deposition and shrinkage of affected vessels.7
A PubMed search using the terms Sneddon syndrome, lymphadenopathy and livedo racemosa, and Sneddon syndrome and lymphadenopathy revealed that splenomegaly and lymphadenopathy have not been reported in patients with SS. In patients with antiphospholipid-negative SS, one can assume that antibodies to other phospholipids not tested must exist because of striking similarities between APS and antiphospholipid-negative SS.8 Although our patient did not test positive for any of these antibodies, she did present with lymphadenopathy and splenic enlargement, leading us to believe that involvement of the reticuloendothelial system may be a feature of SS that has not been previously reported. Further studies are required to name specific antigens responsible for clinical manifestations in SS.
Currently, no single diagnostic test for SS exists, thus delaying both diagnosis and initiation of treatment. Histopathologic examination may be helpful, but in many cases it is nonspecific, as are serologic markers. Neuroradiological confirmation of involvement usually is the confirmatory feature in many patients with late-stage diagnosis.2 A diagnostic schematic for SS, which was first described by Daoud et al,2 illustrates classification of symptoms and aids in diagnosis. A working diagnosis of idiopathic livedo racemosa is made after ruling out other causes of SS in a patient with nonspecific biopsy findings and negative magnetic resonance imaging results with prodromal symptoms. The prognosis for such patients progressing to full SS is unknown with or without management using anticoagulant therapy.
Conclusion
Early diagnosis of livedo racemosa and SS is essential, as prevention of cerebrovascular accidents, myocardial infarction, and other thromboembolic diseases can be minimized by attacking risk factors such as smoking, taking oral contraceptive pills, becoming pregnant,9 and by initiating either antiplatelet or anticoagulation treatments. These treatments have been shown to delay the development of neurovascular damage and early-onset dementia. We present this case to demonstrate the variability of early-presenting symptoms in idiopathic livedo racemosa. Recognizing some of the early manifestations can lead to early diagnosis and initiation of treatment.
Sneddon syndrome (SS) was first described in 1965 in patients with persistent livedo racemosa and neurological events.1 Because the other manifestations of SS are nonspecific (eg, hypertension, cardiac valvulopathy, arterial and venous occlusion), the diagnosis often is delayed. Many patients who experience prodromal neurologic symptoms such as headaches, depression, anxiety, dizziness, and neuropathy often present to a physician prior to developing ischemic brain manifestations2 but seldom receive the correct diagnosis. Onset of cerebral occlusive events typically occurs in patients younger than 45 years and may present as a transient ischemic attack, stroke, or intracranial hemorrhage.3 The disease is more prevalent in females than males (2:1 ratio). The exact pathogenesis of SS is still unknown, and although it has been thought of as a separate entity from systemic lupus erythematosus and other antiphospholipid disorders, it has been postulated that an immunological dysfunction damages vessel walls leading to thrombosis.
Cutaneous findings associated with SS involve small- to medium-sized dermal-subdermal arteries. Histopathology in some patients demonstrates proliferation of the endothelium and fibrin deposits with subsequent obliteration of involved arteries.4 In many patients including our patient, histopathologic examination of involved skin fails to show specific abnormalities.1 Zelger et al5 reported the sequence of histopathologic skin events in a series of antiphospholipid-negative SS patients. The authors reported that only small arteries at the dermis-subcutis junction were involved and a progression of endothelial dysfunction was observed. The authors believed there were several nonspecific stages prior to fibrin occlusion of involved arteries.5 Stage I involved loosening of endothelial cells with nonspecific perivascular lymphocytic infiltration with perivascular inflammation and lymphocytic infiltration representing the prime mover of the disease.5,6 This stage is thought to be short lived, thus the reason why it has gone undetected for many years in SS patients. Stages II to IV progress through fibrin deposition and occlusion.5 Histological features of stages I to II have not been reported because of late diagnosis of SS. Stage I patients typically present with an average duration of symptoms of 6 months with few neurologic symptoms, the most common being paresthesia of the legs.5
Case Report
A 37-year-old woman with epigastric tenderness on the left side and splenomegaly seen on computed tomography was referred by a hematologist for evaluation of a reticular rash on the left side of the flank of 9 months’ duration with a presumed diagnosis of focal melanoderma. Her medical history was remarkable for a congenital ventricular septal defect and coarctation of the aorta, as well as endometriosis, myalgia, and joint stiffness that had all developed over the last year. Her medical history also was remarkable for nephrolithiasis, irritable bowel syndrome, and chronic sinusitis, as well as psychiatric depression and anxiety disorders. She recently had been diagnosed with moderate hypertension and had experienced difficulty getting pregnant for the last several years with 3 consecutive miscarriages in the first trimester. Neurologic symptoms included neuropathy involving the feet, intermittent paresthesia of the legs, and a history of chronic migraine headaches for several months.
Dermatologic examination revealed a slightly overweight woman with a 25×30-cm dusky, erythematous, irregular, netlike pattern on the left side of the upper and lower trunk (Figure 1). Extensive livedo racemosa was not altered by changes in temperature and had been unchanged for more than 9 months. There were no signs of pruritus or ulcerations, and areas of livedo racemosa were slightly tender to palpation.
We performed 2 sets of three 4-mm biopsies. The first set targeted areas within the violaceous pattern, while the second set targeted areas of normal tissue between the mottled areas. All 6 specimens demonstrated superficial perivascular lymphocytic infiltrate with no evidence of vasculitis or connective tissue disease. The vessels showed no microthrombi or surrounding fibrosis. No eosinophils were identified within the epidermis. There was no evidence of increased dermal mucin. Both the superficial and deep vascular plexuses were unremarkable and showed no evidence of damage to the walls (Figure 2).
To rule out other possible causes of livedo racemosa, complete blood cell count, comprehensive metabolic panel, coagulation profile, lipase test, urinalysis, serologic testing, and immunologic workup were performed. Lipase was within reference range. The complete blood cell count revealed mild anemia, while the rest of the values were within reference range. An immunologic workup included Sjögren syndrome antigen A, Sjögren syndrome antigen B, anticardiolipin antibodies, and antinuclear antibody, which were all negative. Family history was remarkable for first-degree relatives with systemic lupus erythematosus and Crohn disease.
Computed tomography revealed enlargement of the spleen, as well as periaortic, portacaval, and porta hepatis lymphadenopathy. Based on the laboratory findings and clinical presentation as well as the patient’s medical history, the diagnosis of exclusion was idiopathic livedo racemosa with unknown progression to full-blown SS. The patient did not meet the current diagnostic criteria for SS, and her immunologic studies failed to confirm any present antibodies, but involvement of the reticuloendothelial system pointed to production of antibodies that were not yet detectable on laboratory testing.
Comment
More than 50 years after the first case of SS was diagnosed, better laboratory workup is available and more information is known about the pathophysiology. Sneddon syndrome is a rare disorder, affecting only approximately 4 patients per million each year worldwide. Seronegative antiphospholipid antibody syndrome (SNAPS) describes patients with clinical presentations of antiphospholipid syndrome (APS) without detectable serological markers.7 Antiphospholipid-negative SS, which was seen in our patient, would be categorized under SNAPS. A PubMed search of articles indexed for MEDLINE using the terms livedo racemosa, Sneddon syndrome, and SNAPS and splenomegaly revealed there currently are no known cases of SNAPS that have been reported with splenomegaly and lymphadenopathy. Our patient presented with the following clinical features of SS: livedo racemosa, history of miscarriage, psychiatric disturbances, and hypertension. Surprisingly, biopsies from affected skin did not show any fibrin deposition or microthrombi but did reveal perivascular lymphocytic infiltrations. Magnetic resonance imaging did not show any pathological lesions or vascular changes.
Sneddon syndrome and APS share a common pathway to occlusive arteriolopathy for which 4 stages have been described by Zelger et al.5 Stage I involves a nonspecific Langerhans cell infiltrate with polymorphonuclear leukocytes. The tunica media and elastic lamina usually are unaltered at this early stage, while the surrounding connective tissue may appear edematous.5 This early stage of histopathology has not been evaluated in SS patients, primarily because of delay of diagnosis. Late stages III and IV will show fibrin deposition and shrinkage of affected vessels.7
A PubMed search using the terms Sneddon syndrome, lymphadenopathy and livedo racemosa, and Sneddon syndrome and lymphadenopathy revealed that splenomegaly and lymphadenopathy have not been reported in patients with SS. In patients with antiphospholipid-negative SS, one can assume that antibodies to other phospholipids not tested must exist because of striking similarities between APS and antiphospholipid-negative SS.8 Although our patient did not test positive for any of these antibodies, she did present with lymphadenopathy and splenic enlargement, leading us to believe that involvement of the reticuloendothelial system may be a feature of SS that has not been previously reported. Further studies are required to name specific antigens responsible for clinical manifestations in SS.
Currently, no single diagnostic test for SS exists, thus delaying both diagnosis and initiation of treatment. Histopathologic examination may be helpful, but in many cases it is nonspecific, as are serologic markers. Neuroradiological confirmation of involvement usually is the confirmatory feature in many patients with late-stage diagnosis.2 A diagnostic schematic for SS, which was first described by Daoud et al,2 illustrates classification of symptoms and aids in diagnosis. A working diagnosis of idiopathic livedo racemosa is made after ruling out other causes of SS in a patient with nonspecific biopsy findings and negative magnetic resonance imaging results with prodromal symptoms. The prognosis for such patients progressing to full SS is unknown with or without management using anticoagulant therapy.
Conclusion
Early diagnosis of livedo racemosa and SS is essential, as prevention of cerebrovascular accidents, myocardial infarction, and other thromboembolic diseases can be minimized by attacking risk factors such as smoking, taking oral contraceptive pills, becoming pregnant,9 and by initiating either antiplatelet or anticoagulation treatments. These treatments have been shown to delay the development of neurovascular damage and early-onset dementia. We present this case to demonstrate the variability of early-presenting symptoms in idiopathic livedo racemosa. Recognizing some of the early manifestations can lead to early diagnosis and initiation of treatment.
- Sneddon IB. Cerebro-vascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Daoud MS, Wilmoth GJ, Su WP, et al. Sneddon syndrome. Semin Dermatol. 1995;14:166-172.
- Besnier R, Francès C, Ankri A, et al. Factor V Leiden mutation in Sneddon syndrome. Lupus. 2003;12:406-408.
- K aragülle AT, Karadağ D, Erden A, et al. Sneddon’s syndrome: MR imaging findings. Eur Radiol. 2002;12:144-146.
- Zelg er B, Sepp N, Schmid KW, et al. Life-history of cutaneous vascular-lesions in Sneddon’s syndrome. Hum Pathol. 1992;23:668-675.
- Ayoub N, Esposito G, Barete S, et al. Protein Z deficiency in antiphospholipid-negative Sneddon’s syndrome. Stroke. 2004;35:1329-1332.
- Duva l A, Darnige L, Glowacki F, et al. Livedo, dementia, thrombocytopenia, and endotheliitis without antiphospholipid antibodies: seronegative antiphospholipid-like syndrome. J Am Acad Dermatol. 2009;61:1076-1078.
- Kala shnikova LA, Nasonov EL, Kushekbaeva AE, et al. Anticardiolipin antibodies in Sneddon’s syndrome. Neurology. 1990;40:464-467.
- Wohl rab J, Fischer M, Wolter M, et al. Diagnostic impact and sensitivity of skin biopsies in Sneddon’s syndrome. a report of 15 cases. Br J Dermatol. 2001;145:285-288.
- Sneddon IB. Cerebro-vascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Daoud MS, Wilmoth GJ, Su WP, et al. Sneddon syndrome. Semin Dermatol. 1995;14:166-172.
- Besnier R, Francès C, Ankri A, et al. Factor V Leiden mutation in Sneddon syndrome. Lupus. 2003;12:406-408.
- K aragülle AT, Karadağ D, Erden A, et al. Sneddon’s syndrome: MR imaging findings. Eur Radiol. 2002;12:144-146.
- Zelg er B, Sepp N, Schmid KW, et al. Life-history of cutaneous vascular-lesions in Sneddon’s syndrome. Hum Pathol. 1992;23:668-675.
- Ayoub N, Esposito G, Barete S, et al. Protein Z deficiency in antiphospholipid-negative Sneddon’s syndrome. Stroke. 2004;35:1329-1332.
- Duva l A, Darnige L, Glowacki F, et al. Livedo, dementia, thrombocytopenia, and endotheliitis without antiphospholipid antibodies: seronegative antiphospholipid-like syndrome. J Am Acad Dermatol. 2009;61:1076-1078.
- Kala shnikova LA, Nasonov EL, Kushekbaeva AE, et al. Anticardiolipin antibodies in Sneddon’s syndrome. Neurology. 1990;40:464-467.
- Wohl rab J, Fischer M, Wolter M, et al. Diagnostic impact and sensitivity of skin biopsies in Sneddon’s syndrome. a report of 15 cases. Br J Dermatol. 2001;145:285-288.
Practice Points
- The classic physical diagnostic finding of Sneddon syndrome (SS) is livedo racemosa.
- Early identification and treatment of SS can prevent serious morbidity due to stroke, myocardial infarction, and other thrombotic events.
- Preventive care in SS should include antiplatelet therapy or anticoagulants and smoking cessation along with avoidance of birth control pills.
Persistent fever investigation saves patient's life
THE CASE
A 47-year-old African American woman was admitted to the hospital with pulmonary edema revealed on a computed tomography (CT) scan. She had a history of systemic lupus erythematosus (SLE), hypertension, and end-stage renal disease (ESRD). The patient had been hospitalized one month earlier for lupus nephritis with a hypertensive emergency that led to a seizure. During this earlier hospitalization, she was given a diagnosis of posterior reversible encephalopathy syndrome.
Two weeks into her more recent hospitalization, the patient developed a fever that was accompanied by cough and fatigue. By the third week, there was no identified cause of the fever, and the patient met the criteria for fever of unknown origin (FUO).
Her medications included cyclophosphamide, prednisone, nebivolol, clonidine, phenytoin, and epoetin alfa. The patient was also receiving dialysis every other day. Chest x-ray findings suggested pneumonia, and the patient was treated with vancomycin and piperacillin/tazobactam. However, her fever persisted after completing the antibiotics. Central line sepsis was high in the differential, as the patient was on dialysis, but blood and catheter tip cultures were negative. Chest and abdominal CT scans showed no new disease process. Urine and sputum cultures were collected and were negative for infection. Drug-induced fever was then suspected, but was ruled out when the fever persisted after the removal of potential offending agents (phenytoin, nebivolol, and cyclophosphamide).
THE DIAGNOSIS
We then followed the American Academy of Family Physicians’ diagnostic protocol for FUO.1
Initial labs included a complete blood count (CBC), 2 blood cultures, a urine culture, erythrocyte sedimentation rate (ESR), a purified protein derivative skin test, chest and abdominal CT scans, and double-stranded DNA (dsDNA) levels (since this patient had known SLE). The patient’s hemoglobin level and mean corpuscular volume were consistent with normocytic anemia, which was attributed to the ESRD. The ESR was mildly elevated at 46 mm/hr, but dsDNA was not, ruling out a lupus flare. Thrombocytopenia (platelet count, 82 K/mcL) and lymphocytopenia (absolute lymphocyte count, 0.2 K/mcL) were assumed to be secondary to cyclophosphamide use.
Because the initial labs were non-diagnostic, we proceeded with a sputum stain and culture, human immunodeficiency virus testing, a hepatitis panel, and a peripheral blood smear.1 All were negative except for the peripheral blood smear, which showed hemophagocytic cells. This was the first finding that brought hemophagocytic lymphohistiocytosis (HLH) into the differential.
We then performed a bone marrow biopsy (FIGURE), which also revealed hemophagocytic cells, so we ordered HLH-specific labs (more on those in a bit). Liver enzymes were elevated to 3 times their normal value. Triglycerides (414 mg/dL), ferritin (>15,000 ng/mL), and interleukin-2 (IL-2) receptor levels (>20,000 pg/m) were also elevated.
The patient was tested for herpes simplex virus, Epstein-Barr virus (EBV), and cytomegalovirus (CMV), since these viruses are associated with HLH. She had 3.1 million copies/mL of CMV, leading to the diagnosis of secondary HLH. This diagnosis might not have been made if not for a persistent fever investigation.
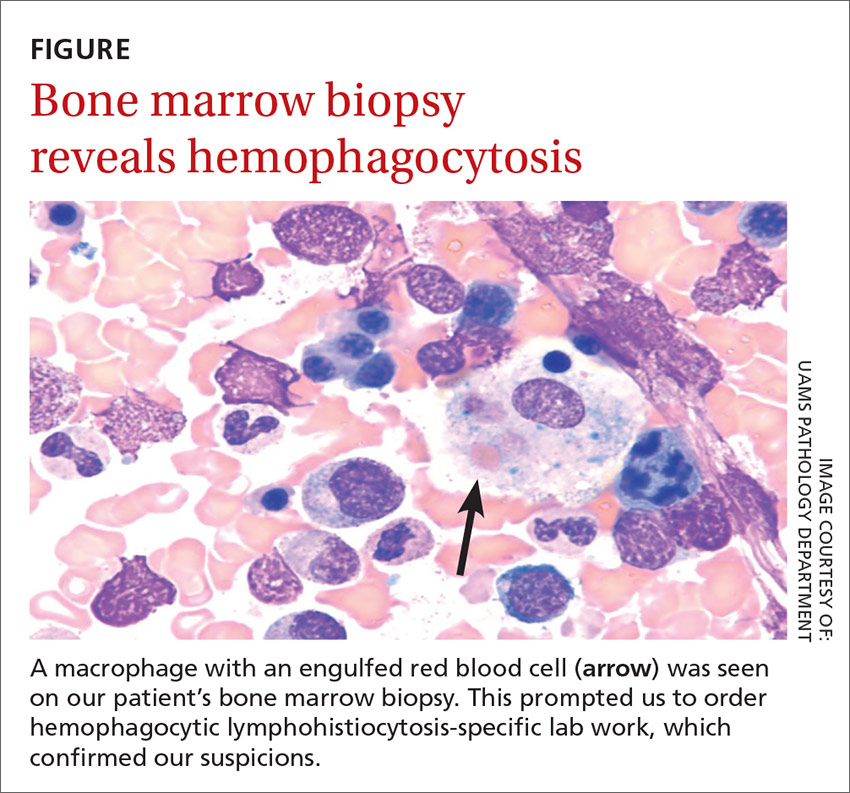
DISCUSSION
HLH is a life-threatening syndrome of excessive immune activation that results in tissue damage.2 There are primary and secondary forms, but they share the same mechanism of impaired regulation of cytotoxic granules and cytokines. Primary HLH results from a congenital gene mutation,3 while secondary HLH is triggered by an autoimmune or inflammatory disease or an infection.4 EBV is the most common viral etiology, followed closely by CMV.5
The diagnosis may be established genetically (based on mutations of the genes loci PRF1, UNC13D, or STX11) or by fulfillment of 5 out of 8 criteria: fever; splenomegaly; cytopenia; hypertriglyceridemia; hypofibrinogenemia; hemophagocytosis in the bone marrow, spleen, or lymph nodes; low or absent natural killer cell activity; and an elevated ferritin level (>500 ng/mL). Elevated soluble CD25 and IL-2 receptor markers are HLH-specific markers.3 This patient had fever, cytopenia, hypertriglyceridemia, hemophagocytosis, and elevated ferritin with elevated IL-2, meeting the criteria for secondary HLH.
First treat the underlying condition, then the HLH
Treatment for HLH includes treating the underlying condition (such as EBV or CMV) with antiretroviral medications, and using immunosuppressive agents such as chemotherapy drugs and steroids for the HLH.
Our patient was treated with valganciclovir 900 mg/d for 2 weeks for the CMV and an etoposide/prednisone taper for 3 months for HLH chemotherapy and suppression. Within one month, her CMV viral load decreased to <300 copies/mL and her fever resolved. Ferritin, triglycerides, and liver enzyme levels returned to normal within 3 months.
THE TAKEAWAY
FUO can be frustrating for both the physician and the patient. Not only is the differential large, but testing is extensive. It is important to get a thorough history and to consider medications as the cause. Testing should be patient-specific and systematic. Persistent investigation is critical to saving the patient’s life.
1. Roth AR, Basello GM. Approach to the adult patient with fever of unknown origin. Am Fam Physician. 2003;68:2223-2228.
2. Filipovich A, McClain K, Grom A. Histiocytic disorders: recentinsights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant. 2010;16:S82-S89.
3. Larroche C. Hemophagocytic lymphohistiocytosis in adults: diagnosis and treatment. Joint Bone Spine. 2012;79:356-361.
4. Rouphael NG, Talati NJ, Vaughan C, et al. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7:814-822.
5. Janka GE, Lehmberg K. Hemophagocytic syndromes—an update. Blood Rev. 2014;28:135-142.
THE CASE
A 47-year-old African American woman was admitted to the hospital with pulmonary edema revealed on a computed tomography (CT) scan. She had a history of systemic lupus erythematosus (SLE), hypertension, and end-stage renal disease (ESRD). The patient had been hospitalized one month earlier for lupus nephritis with a hypertensive emergency that led to a seizure. During this earlier hospitalization, she was given a diagnosis of posterior reversible encephalopathy syndrome.
Two weeks into her more recent hospitalization, the patient developed a fever that was accompanied by cough and fatigue. By the third week, there was no identified cause of the fever, and the patient met the criteria for fever of unknown origin (FUO).
Her medications included cyclophosphamide, prednisone, nebivolol, clonidine, phenytoin, and epoetin alfa. The patient was also receiving dialysis every other day. Chest x-ray findings suggested pneumonia, and the patient was treated with vancomycin and piperacillin/tazobactam. However, her fever persisted after completing the antibiotics. Central line sepsis was high in the differential, as the patient was on dialysis, but blood and catheter tip cultures were negative. Chest and abdominal CT scans showed no new disease process. Urine and sputum cultures were collected and were negative for infection. Drug-induced fever was then suspected, but was ruled out when the fever persisted after the removal of potential offending agents (phenytoin, nebivolol, and cyclophosphamide).
THE DIAGNOSIS
We then followed the American Academy of Family Physicians’ diagnostic protocol for FUO.1
Initial labs included a complete blood count (CBC), 2 blood cultures, a urine culture, erythrocyte sedimentation rate (ESR), a purified protein derivative skin test, chest and abdominal CT scans, and double-stranded DNA (dsDNA) levels (since this patient had known SLE). The patient’s hemoglobin level and mean corpuscular volume were consistent with normocytic anemia, which was attributed to the ESRD. The ESR was mildly elevated at 46 mm/hr, but dsDNA was not, ruling out a lupus flare. Thrombocytopenia (platelet count, 82 K/mcL) and lymphocytopenia (absolute lymphocyte count, 0.2 K/mcL) were assumed to be secondary to cyclophosphamide use.
Because the initial labs were non-diagnostic, we proceeded with a sputum stain and culture, human immunodeficiency virus testing, a hepatitis panel, and a peripheral blood smear.1 All were negative except for the peripheral blood smear, which showed hemophagocytic cells. This was the first finding that brought hemophagocytic lymphohistiocytosis (HLH) into the differential.
We then performed a bone marrow biopsy (FIGURE), which also revealed hemophagocytic cells, so we ordered HLH-specific labs (more on those in a bit). Liver enzymes were elevated to 3 times their normal value. Triglycerides (414 mg/dL), ferritin (>15,000 ng/mL), and interleukin-2 (IL-2) receptor levels (>20,000 pg/m) were also elevated.
The patient was tested for herpes simplex virus, Epstein-Barr virus (EBV), and cytomegalovirus (CMV), since these viruses are associated with HLH. She had 3.1 million copies/mL of CMV, leading to the diagnosis of secondary HLH. This diagnosis might not have been made if not for a persistent fever investigation.
DISCUSSION
HLH is a life-threatening syndrome of excessive immune activation that results in tissue damage.2 There are primary and secondary forms, but they share the same mechanism of impaired regulation of cytotoxic granules and cytokines. Primary HLH results from a congenital gene mutation,3 while secondary HLH is triggered by an autoimmune or inflammatory disease or an infection.4 EBV is the most common viral etiology, followed closely by CMV.5
The diagnosis may be established genetically (based on mutations of the genes loci PRF1, UNC13D, or STX11) or by fulfillment of 5 out of 8 criteria: fever; splenomegaly; cytopenia; hypertriglyceridemia; hypofibrinogenemia; hemophagocytosis in the bone marrow, spleen, or lymph nodes; low or absent natural killer cell activity; and an elevated ferritin level (>500 ng/mL). Elevated soluble CD25 and IL-2 receptor markers are HLH-specific markers.3 This patient had fever, cytopenia, hypertriglyceridemia, hemophagocytosis, and elevated ferritin with elevated IL-2, meeting the criteria for secondary HLH.
First treat the underlying condition, then the HLH
Treatment for HLH includes treating the underlying condition (such as EBV or CMV) with antiretroviral medications, and using immunosuppressive agents such as chemotherapy drugs and steroids for the HLH.
Our patient was treated with valganciclovir 900 mg/d for 2 weeks for the CMV and an etoposide/prednisone taper for 3 months for HLH chemotherapy and suppression. Within one month, her CMV viral load decreased to <300 copies/mL and her fever resolved. Ferritin, triglycerides, and liver enzyme levels returned to normal within 3 months.
THE TAKEAWAY
FUO can be frustrating for both the physician and the patient. Not only is the differential large, but testing is extensive. It is important to get a thorough history and to consider medications as the cause. Testing should be patient-specific and systematic. Persistent investigation is critical to saving the patient’s life.
THE CASE
A 47-year-old African American woman was admitted to the hospital with pulmonary edema revealed on a computed tomography (CT) scan. She had a history of systemic lupus erythematosus (SLE), hypertension, and end-stage renal disease (ESRD). The patient had been hospitalized one month earlier for lupus nephritis with a hypertensive emergency that led to a seizure. During this earlier hospitalization, she was given a diagnosis of posterior reversible encephalopathy syndrome.
Two weeks into her more recent hospitalization, the patient developed a fever that was accompanied by cough and fatigue. By the third week, there was no identified cause of the fever, and the patient met the criteria for fever of unknown origin (FUO).
Her medications included cyclophosphamide, prednisone, nebivolol, clonidine, phenytoin, and epoetin alfa. The patient was also receiving dialysis every other day. Chest x-ray findings suggested pneumonia, and the patient was treated with vancomycin and piperacillin/tazobactam. However, her fever persisted after completing the antibiotics. Central line sepsis was high in the differential, as the patient was on dialysis, but blood and catheter tip cultures were negative. Chest and abdominal CT scans showed no new disease process. Urine and sputum cultures were collected and were negative for infection. Drug-induced fever was then suspected, but was ruled out when the fever persisted after the removal of potential offending agents (phenytoin, nebivolol, and cyclophosphamide).
THE DIAGNOSIS
We then followed the American Academy of Family Physicians’ diagnostic protocol for FUO.1
Initial labs included a complete blood count (CBC), 2 blood cultures, a urine culture, erythrocyte sedimentation rate (ESR), a purified protein derivative skin test, chest and abdominal CT scans, and double-stranded DNA (dsDNA) levels (since this patient had known SLE). The patient’s hemoglobin level and mean corpuscular volume were consistent with normocytic anemia, which was attributed to the ESRD. The ESR was mildly elevated at 46 mm/hr, but dsDNA was not, ruling out a lupus flare. Thrombocytopenia (platelet count, 82 K/mcL) and lymphocytopenia (absolute lymphocyte count, 0.2 K/mcL) were assumed to be secondary to cyclophosphamide use.
Because the initial labs were non-diagnostic, we proceeded with a sputum stain and culture, human immunodeficiency virus testing, a hepatitis panel, and a peripheral blood smear.1 All were negative except for the peripheral blood smear, which showed hemophagocytic cells. This was the first finding that brought hemophagocytic lymphohistiocytosis (HLH) into the differential.
We then performed a bone marrow biopsy (FIGURE), which also revealed hemophagocytic cells, so we ordered HLH-specific labs (more on those in a bit). Liver enzymes were elevated to 3 times their normal value. Triglycerides (414 mg/dL), ferritin (>15,000 ng/mL), and interleukin-2 (IL-2) receptor levels (>20,000 pg/m) were also elevated.
The patient was tested for herpes simplex virus, Epstein-Barr virus (EBV), and cytomegalovirus (CMV), since these viruses are associated with HLH. She had 3.1 million copies/mL of CMV, leading to the diagnosis of secondary HLH. This diagnosis might not have been made if not for a persistent fever investigation.
DISCUSSION
HLH is a life-threatening syndrome of excessive immune activation that results in tissue damage.2 There are primary and secondary forms, but they share the same mechanism of impaired regulation of cytotoxic granules and cytokines. Primary HLH results from a congenital gene mutation,3 while secondary HLH is triggered by an autoimmune or inflammatory disease or an infection.4 EBV is the most common viral etiology, followed closely by CMV.5
The diagnosis may be established genetically (based on mutations of the genes loci PRF1, UNC13D, or STX11) or by fulfillment of 5 out of 8 criteria: fever; splenomegaly; cytopenia; hypertriglyceridemia; hypofibrinogenemia; hemophagocytosis in the bone marrow, spleen, or lymph nodes; low or absent natural killer cell activity; and an elevated ferritin level (>500 ng/mL). Elevated soluble CD25 and IL-2 receptor markers are HLH-specific markers.3 This patient had fever, cytopenia, hypertriglyceridemia, hemophagocytosis, and elevated ferritin with elevated IL-2, meeting the criteria for secondary HLH.
First treat the underlying condition, then the HLH
Treatment for HLH includes treating the underlying condition (such as EBV or CMV) with antiretroviral medications, and using immunosuppressive agents such as chemotherapy drugs and steroids for the HLH.
Our patient was treated with valganciclovir 900 mg/d for 2 weeks for the CMV and an etoposide/prednisone taper for 3 months for HLH chemotherapy and suppression. Within one month, her CMV viral load decreased to <300 copies/mL and her fever resolved. Ferritin, triglycerides, and liver enzyme levels returned to normal within 3 months.
THE TAKEAWAY
FUO can be frustrating for both the physician and the patient. Not only is the differential large, but testing is extensive. It is important to get a thorough history and to consider medications as the cause. Testing should be patient-specific and systematic. Persistent investigation is critical to saving the patient’s life.
1. Roth AR, Basello GM. Approach to the adult patient with fever of unknown origin. Am Fam Physician. 2003;68:2223-2228.
2. Filipovich A, McClain K, Grom A. Histiocytic disorders: recentinsights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant. 2010;16:S82-S89.
3. Larroche C. Hemophagocytic lymphohistiocytosis in adults: diagnosis and treatment. Joint Bone Spine. 2012;79:356-361.
4. Rouphael NG, Talati NJ, Vaughan C, et al. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7:814-822.
5. Janka GE, Lehmberg K. Hemophagocytic syndromes—an update. Blood Rev. 2014;28:135-142.
1. Roth AR, Basello GM. Approach to the adult patient with fever of unknown origin. Am Fam Physician. 2003;68:2223-2228.
2. Filipovich A, McClain K, Grom A. Histiocytic disorders: recentinsights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant. 2010;16:S82-S89.
3. Larroche C. Hemophagocytic lymphohistiocytosis in adults: diagnosis and treatment. Joint Bone Spine. 2012;79:356-361.
4. Rouphael NG, Talati NJ, Vaughan C, et al. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7:814-822.
5. Janka GE, Lehmberg K. Hemophagocytic syndromes—an update. Blood Rev. 2014;28:135-142.

Abnormal Wound Healing Related to High-Dose Systemic Corticosteroid Therapy in a Patient With Ehlers-Danlos Syndrome Benign Hypermobility Type
The process of wound healing has been well characterized. Immediately following injury, neutrophils arrive at the site in response to chemotactic factors produced by the coagulation cascade. Monocytes follow 24 to 36 hours later; transform into macrophages; and begin to phagocytose tissue debris, organisms, and any remaining neutrophils. In turn, macrophages release chemotactic factors such as basic fibroblast growth factor to attract fibroblasts to the wound, which then begin the process of synthesizing collagen and ground substance. Fibroblasts then take over as the dominant cell type, with collagen synthesis continuing for approximately 6 weeks. Keratinocytes and endothelial cells also proliferate during this time. After approximately 6 weeks, collagen remodeling begins. Tensile strength of the wound may continue to increase up to one year after the injury.1,2
Corticosteroids inhibit wound healing in several ways. Notably, they decrease the number of circulating monocytes, leading to fewer macrophages in the tissue at the site of injury, which then leads to impaired phagocytosis and reduced release of chemotactic factors that attract fibroblasts. Additionally, corticosteroids can inhibit collagen synthesis and remodeling, leading to delayed wound healing and decreased tensile strength of the wound as well as impacting capillary proliferation.3
The subtypes of EDS were reclassified in 1998 by Beighton et al,4 and the benign hypermobility type (EDS-BHT)(formerly type III) is considered the least severe. There is some controversy as to whether this subtype constitutes a separate diagnosis from the benign familial joint hypermobility syndrome. It is characterized by hypermobility of the joints (objectively measured with the Beighton scale) and mild hyperextensibility of the skin, and patients often have a history of joint subluxations and dislocations with resultant degenerative joint disease and chronic pain. Manifestations of fragile skin and soft tissue (eg, abnormal wound healing or scarring; spontaneous tearing of the skin, ligaments, tendons, or organs) are notably absent from the findings in this syndrome.5 The genetic basis for EDS is unknown in the majority of patients, although a deficiency in tenascin X (secondary to defects in the tenascin XB gene [TNXB]) has been identified in a small subset (<5%) of patients, leading to elastic fiber abnormalities, reduced collagen deposition, and impaired cross-linking of collagen.6,7 Inheritance usually is autosomal dominant but also can be autosomal recessive. In contrast, the classic type of EDS (formerly types I and II) is associated with atrophic scarring and tissue fragility, in addition to joint hypermobility and skin hyperextensibility. Type V collagen mutations are found in more than half of patients with this disorder.8
We present the case of a patient with EDS-BHT who developed large nonhealing cutaneous ulcerations with initiation of high-dose systemic corticosteroids for treatment of dermatomyositis. This case provides a dramatic illustration of the effects of the use of chronic systemic corticosteroids on skin fragility and wound healing in patients with an underlying inherited defect in collagen or connective tissue.
Case Report
A 23-year-old man with an unremarkable medical history was admitted to our inpatient cardiology service with palpitations attributable to new-onset atrial fibrillation. Dermatology was consulted to evaluate a rash of approximately 4 months’ duration that started on the dorsal aspect of the hands, then progressed to involve the extensor elbows and knees. The rash also was associated with fatigue, arthralgia, and proximal muscle weakness. A taper of prednisone that was prescribed approximately 2 months prior to admission by a rheumatologist for presumed dermatomyositis improved his symptoms, but they recurred with discontinuation of the medication.
Physical examination revealed reddish, violaceous and hyperpigmented patches on the dorsal aspect of the hands and digits and the extensor aspect of the knees and elbows. A skin biopsy from the right elbow showed a mild interface reaction and nonspecific direct immunofluorescence, consistent with a diagnosis of dermatomyositis. Autoimmune serologies were negative, including antinuclear, anti–Jo-1, anti–Mi-2, anti–Sjögren syndrome antigen A, anti–Sjögren syndrome antigen B, anti-Smith, and antiribonucleoprotein antibodies. Creatine kinase and rheumatoid factor levels were within reference range. Electromyogram was supportive of the diagnosis of dermatomyositis, showing an irritable myopathy. Cardiac magnetic resonance imaging showed an acute inflammatory process of the myocardium, and a transthoracic echocardiogram revealed a depressed left ventricular ejection fraction of 35% to 40% (reference range, 55%–70%). His cardiac disease also was attributed to dermatomyositis, and he was managed by cardiology with anangiotensin-converting enzyme inhibitor and antiarrhythmic therapy. Rheumatology was consulted and prednisone 60 mg once daily was started, with the patient reporting improvement in his muscle weakness and the rash.
Interestingly, the patient also noted a history of joint hypermobility, and a genetics consultation was obtained during the current hospitalization. He denied a history of abnormal scarring or skin problems, but he did note dislocation of the patella on 2 occasions and an umbilical hernia repair at 3 years of age. A paternal uncle had a history of similar joint hypermobility. His Beighton score was noted to be 8/8 (bending at the waist was unable to be tested due to recent lumbar puncture obtained during this hospitalization). The patient was diagnosed with EDS-BHT, and no further workup was recommended.
Subsequent to his hospitalization for several days, the patient’s prednisone was slowly tapered down from 60 mg once daily to 12.5 mg once daily, and azathioprine was started and titrated up to 150 mg once daily. Approximately 6 months after his initial hospitalization, he was readmitted due to increased pain of the right knee with concern for osteomyelitis. Dermatology was again consulted, and at this time, the patient reported a 4-month history of nonhealing ulcers to the knees and elbows (Figure 1). He stated that the ulcers were initially about the size of a pencil eraser and had started approximately 2 months after the prednisone was started, with subsequent slow enlargement. He noted a stinging sensation with enlargement of the ulcers, but otherwise they were not painful. He denied major trauma to the areas. He noted that his prior rash from the dermatomyositis seemed to have resolved, along with his muscle weakness, and he reported weight gain and improvement in his energy levels. Physical examination at this time revealed several stigmata of chronic systemic corticosteroids, including fatty deposits in the face (moon facies) and between the shoulders (buffalo hump), facial acne, and numerous erythematous striae on the trunk and proximal extremities (Figure 2). Multiple noninflammatory ulcers with punched-out borders ranging in size from 0.5 to 6 cm were seen at sites overlying bony prominences, including the bilateral extensor elbows and knees and the right plantar foot. Similar ulcers were noted on the trunk within the striae. Some of the ulcers were covered with a thick hyperkeratotic crust. A biopsy from the edge of an ulcer on the right side of the flank showed only dermal fibrosis. Workup by orthopedic surgery was felt to be inconsistent with osteomyelitis, and plastic surgery was consulted to consider surgical options for repair. Consequently, the patient was taken to the operating room for primary closure of the ulcers to the bilateral knees and right elbow. He has been followed closely by plastic surgery, with the use of joint immobilization to promote wound healing.
Comment
This case represents a dramatic illustration of the effects of chronic systemic corticosteroids on skin fragility and wound healing in a patient with an underlying genetic defect in the connective tissue. The ulcers were all located within striae or overlying bony prominences where the skin was subjected to increased tension; however, the patient reported no problems with wound healing or scarring at these sites prior to the initiation of corticosteroids, suggesting that the addition of this medication was disruptive to the cutaneous wound healing mechanisms. This case is unique because abnormal wound healing in an EDS patient was so clearly linked to the initiation of systemic steroids.
The exact pathogenesis of the patient’s ulcers is unclear. The diagnosis of EDS was primarily clinical, and without genetic testing, we cannot state with certainty the underlying molecular problem in this patient. Although tenascin X deficiency has been found in a few patients, a genetic defect remains uncharacterized in most patients with EDS-BHT, and in most situations, EDS-BHT remains a clinical diagnosis. In 2001, Schalkwijk et al9 first described the association of tenascin X deficiency and EDS in 5 patients, and they noted delayed wound healing in 1 patient who had received systemic corticosteroids for congenital adrenal hyperplasia. The authors remarked that it was not clear whether the abnormality was linked to the patient’s EDS or to his treatment with systemic corticosteroids.9 Furthermore, it is possible that our patient in fact has a milder variant of classic type EDS and that the manifestations of tissue fragility remained subclinical until the addition of systemic corticosteroids. It also is interesting to note that muscle weakness can be a symptom of EDS, both classic and BHT of EDS, but our patient’s muscle weakness improved with immunosuppression, supporting an underlying autoimmune disease as the cause for it.10 Skin ulcerations have been reported as a rare manifestation of dermatomyositis, but it is remarkable that his ulcers progressed as his other dermatomyositis symptoms improved with therapy, suggesting that his autoimmune disease was not the underlying cause for the ulcers.11-13 This case points to the need to thoughtfully consider the adverse effects of corticosteroids on wound healing in patients with an inherited disorder of collagen or connective tissue such as EDS.
- Bolognia JL, Jorizzo JL, Rapini RP, et al. Dermatology. 2nd ed. Philadelphia, PA: Mosby Elsevier; 2008.
- Gurtner GC, Werner S, Barrandon Y, et al. Wound repair and regeneration. Nature. 2008;453:314-321.
- Poetker DM, Reh DD. A comprehensive review of the adverse effects of systemic corticosteroids. Otolaryng Clin N Am. 2010;43:753-768.
- Beighton P, De Paepe A, Steinmann B, et al. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998;77:31-37.
- Levy HP. Ehlers-Danlos syndrome, hypermobility type. In: Pagon RA, Bird TD, Dolan CR, et al, es. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993-2015. http://www.ncbi.nlm.nih.gov/books/NBK1279/. Accessed August 5, 2015.
- Zweers MC, Bristow J, Steijlen PM, et al. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet. 2003;73:214-217.
- Brellier F, Tucker RP, Chiquet-Ehrismann R. Tenascins and their implications in diseases and tissue mechanics. Scand J Med Sci Spor. 2009;19:511-519.
- Malfait F, Wenstrup R, De Paepe A. Ehlers-Danlos syndrome, classic type. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews. Seattle,WA: University of Washington, Seattle; 1993-2015. http://www.ncbi.nlm.nih.gov/books/NBK1244/. Accessed August 5, 2015.
- Schalkwijk J, Zweers MC, Steijlen PM, et al. A recessive form of the Ehlers-Danlos syndrome caused by tenascin X deficiency. N Engl J Med. 2001;345:1167-1175.
- Voermans NC, Alfen NV, Pillen S, et al. Neuromuscular involvement in various types of Ehlers-Danlos syndrome. Ann Neurol. 2009;65:687-697.
- Scheinfeld NS. Ulcerative paraneoplastic dermatomyositis secondary to metastatic breast cancer. Skinmed. 2006;5:94-96.
- Tomb R, Stephan F. Perforating skin ulcers occurring in an adult with dermatomyositis [in French]. Ann Dermatol Venerol. 2002;129:1383-1385.
- Yosipovitch G, Feinmesser M, David M. Adult dermatomyositis with livedo reticularis and multiple skin ulcers. J Eur Acad Dermatol. 1998;11:48-50.
The process of wound healing has been well characterized. Immediately following injury, neutrophils arrive at the site in response to chemotactic factors produced by the coagulation cascade. Monocytes follow 24 to 36 hours later; transform into macrophages; and begin to phagocytose tissue debris, organisms, and any remaining neutrophils. In turn, macrophages release chemotactic factors such as basic fibroblast growth factor to attract fibroblasts to the wound, which then begin the process of synthesizing collagen and ground substance. Fibroblasts then take over as the dominant cell type, with collagen synthesis continuing for approximately 6 weeks. Keratinocytes and endothelial cells also proliferate during this time. After approximately 6 weeks, collagen remodeling begins. Tensile strength of the wound may continue to increase up to one year after the injury.1,2
Corticosteroids inhibit wound healing in several ways. Notably, they decrease the number of circulating monocytes, leading to fewer macrophages in the tissue at the site of injury, which then leads to impaired phagocytosis and reduced release of chemotactic factors that attract fibroblasts. Additionally, corticosteroids can inhibit collagen synthesis and remodeling, leading to delayed wound healing and decreased tensile strength of the wound as well as impacting capillary proliferation.3
The subtypes of EDS were reclassified in 1998 by Beighton et al,4 and the benign hypermobility type (EDS-BHT)(formerly type III) is considered the least severe. There is some controversy as to whether this subtype constitutes a separate diagnosis from the benign familial joint hypermobility syndrome. It is characterized by hypermobility of the joints (objectively measured with the Beighton scale) and mild hyperextensibility of the skin, and patients often have a history of joint subluxations and dislocations with resultant degenerative joint disease and chronic pain. Manifestations of fragile skin and soft tissue (eg, abnormal wound healing or scarring; spontaneous tearing of the skin, ligaments, tendons, or organs) are notably absent from the findings in this syndrome.5 The genetic basis for EDS is unknown in the majority of patients, although a deficiency in tenascin X (secondary to defects in the tenascin XB gene [TNXB]) has been identified in a small subset (<5%) of patients, leading to elastic fiber abnormalities, reduced collagen deposition, and impaired cross-linking of collagen.6,7 Inheritance usually is autosomal dominant but also can be autosomal recessive. In contrast, the classic type of EDS (formerly types I and II) is associated with atrophic scarring and tissue fragility, in addition to joint hypermobility and skin hyperextensibility. Type V collagen mutations are found in more than half of patients with this disorder.8
We present the case of a patient with EDS-BHT who developed large nonhealing cutaneous ulcerations with initiation of high-dose systemic corticosteroids for treatment of dermatomyositis. This case provides a dramatic illustration of the effects of the use of chronic systemic corticosteroids on skin fragility and wound healing in patients with an underlying inherited defect in collagen or connective tissue.
Case Report
A 23-year-old man with an unremarkable medical history was admitted to our inpatient cardiology service with palpitations attributable to new-onset atrial fibrillation. Dermatology was consulted to evaluate a rash of approximately 4 months’ duration that started on the dorsal aspect of the hands, then progressed to involve the extensor elbows and knees. The rash also was associated with fatigue, arthralgia, and proximal muscle weakness. A taper of prednisone that was prescribed approximately 2 months prior to admission by a rheumatologist for presumed dermatomyositis improved his symptoms, but they recurred with discontinuation of the medication.
Physical examination revealed reddish, violaceous and hyperpigmented patches on the dorsal aspect of the hands and digits and the extensor aspect of the knees and elbows. A skin biopsy from the right elbow showed a mild interface reaction and nonspecific direct immunofluorescence, consistent with a diagnosis of dermatomyositis. Autoimmune serologies were negative, including antinuclear, anti–Jo-1, anti–Mi-2, anti–Sjögren syndrome antigen A, anti–Sjögren syndrome antigen B, anti-Smith, and antiribonucleoprotein antibodies. Creatine kinase and rheumatoid factor levels were within reference range. Electromyogram was supportive of the diagnosis of dermatomyositis, showing an irritable myopathy. Cardiac magnetic resonance imaging showed an acute inflammatory process of the myocardium, and a transthoracic echocardiogram revealed a depressed left ventricular ejection fraction of 35% to 40% (reference range, 55%–70%). His cardiac disease also was attributed to dermatomyositis, and he was managed by cardiology with anangiotensin-converting enzyme inhibitor and antiarrhythmic therapy. Rheumatology was consulted and prednisone 60 mg once daily was started, with the patient reporting improvement in his muscle weakness and the rash.
Interestingly, the patient also noted a history of joint hypermobility, and a genetics consultation was obtained during the current hospitalization. He denied a history of abnormal scarring or skin problems, but he did note dislocation of the patella on 2 occasions and an umbilical hernia repair at 3 years of age. A paternal uncle had a history of similar joint hypermobility. His Beighton score was noted to be 8/8 (bending at the waist was unable to be tested due to recent lumbar puncture obtained during this hospitalization). The patient was diagnosed with EDS-BHT, and no further workup was recommended.
Subsequent to his hospitalization for several days, the patient’s prednisone was slowly tapered down from 60 mg once daily to 12.5 mg once daily, and azathioprine was started and titrated up to 150 mg once daily. Approximately 6 months after his initial hospitalization, he was readmitted due to increased pain of the right knee with concern for osteomyelitis. Dermatology was again consulted, and at this time, the patient reported a 4-month history of nonhealing ulcers to the knees and elbows (Figure 1). He stated that the ulcers were initially about the size of a pencil eraser and had started approximately 2 months after the prednisone was started, with subsequent slow enlargement. He noted a stinging sensation with enlargement of the ulcers, but otherwise they were not painful. He denied major trauma to the areas. He noted that his prior rash from the dermatomyositis seemed to have resolved, along with his muscle weakness, and he reported weight gain and improvement in his energy levels. Physical examination at this time revealed several stigmata of chronic systemic corticosteroids, including fatty deposits in the face (moon facies) and between the shoulders (buffalo hump), facial acne, and numerous erythematous striae on the trunk and proximal extremities (Figure 2). Multiple noninflammatory ulcers with punched-out borders ranging in size from 0.5 to 6 cm were seen at sites overlying bony prominences, including the bilateral extensor elbows and knees and the right plantar foot. Similar ulcers were noted on the trunk within the striae. Some of the ulcers were covered with a thick hyperkeratotic crust. A biopsy from the edge of an ulcer on the right side of the flank showed only dermal fibrosis. Workup by orthopedic surgery was felt to be inconsistent with osteomyelitis, and plastic surgery was consulted to consider surgical options for repair. Consequently, the patient was taken to the operating room for primary closure of the ulcers to the bilateral knees and right elbow. He has been followed closely by plastic surgery, with the use of joint immobilization to promote wound healing.
Comment
This case represents a dramatic illustration of the effects of chronic systemic corticosteroids on skin fragility and wound healing in a patient with an underlying genetic defect in the connective tissue. The ulcers were all located within striae or overlying bony prominences where the skin was subjected to increased tension; however, the patient reported no problems with wound healing or scarring at these sites prior to the initiation of corticosteroids, suggesting that the addition of this medication was disruptive to the cutaneous wound healing mechanisms. This case is unique because abnormal wound healing in an EDS patient was so clearly linked to the initiation of systemic steroids.
The exact pathogenesis of the patient’s ulcers is unclear. The diagnosis of EDS was primarily clinical, and without genetic testing, we cannot state with certainty the underlying molecular problem in this patient. Although tenascin X deficiency has been found in a few patients, a genetic defect remains uncharacterized in most patients with EDS-BHT, and in most situations, EDS-BHT remains a clinical diagnosis. In 2001, Schalkwijk et al9 first described the association of tenascin X deficiency and EDS in 5 patients, and they noted delayed wound healing in 1 patient who had received systemic corticosteroids for congenital adrenal hyperplasia. The authors remarked that it was not clear whether the abnormality was linked to the patient’s EDS or to his treatment with systemic corticosteroids.9 Furthermore, it is possible that our patient in fact has a milder variant of classic type EDS and that the manifestations of tissue fragility remained subclinical until the addition of systemic corticosteroids. It also is interesting to note that muscle weakness can be a symptom of EDS, both classic and BHT of EDS, but our patient’s muscle weakness improved with immunosuppression, supporting an underlying autoimmune disease as the cause for it.10 Skin ulcerations have been reported as a rare manifestation of dermatomyositis, but it is remarkable that his ulcers progressed as his other dermatomyositis symptoms improved with therapy, suggesting that his autoimmune disease was not the underlying cause for the ulcers.11-13 This case points to the need to thoughtfully consider the adverse effects of corticosteroids on wound healing in patients with an inherited disorder of collagen or connective tissue such as EDS.
The process of wound healing has been well characterized. Immediately following injury, neutrophils arrive at the site in response to chemotactic factors produced by the coagulation cascade. Monocytes follow 24 to 36 hours later; transform into macrophages; and begin to phagocytose tissue debris, organisms, and any remaining neutrophils. In turn, macrophages release chemotactic factors such as basic fibroblast growth factor to attract fibroblasts to the wound, which then begin the process of synthesizing collagen and ground substance. Fibroblasts then take over as the dominant cell type, with collagen synthesis continuing for approximately 6 weeks. Keratinocytes and endothelial cells also proliferate during this time. After approximately 6 weeks, collagen remodeling begins. Tensile strength of the wound may continue to increase up to one year after the injury.1,2
Corticosteroids inhibit wound healing in several ways. Notably, they decrease the number of circulating monocytes, leading to fewer macrophages in the tissue at the site of injury, which then leads to impaired phagocytosis and reduced release of chemotactic factors that attract fibroblasts. Additionally, corticosteroids can inhibit collagen synthesis and remodeling, leading to delayed wound healing and decreased tensile strength of the wound as well as impacting capillary proliferation.3
The subtypes of EDS were reclassified in 1998 by Beighton et al,4 and the benign hypermobility type (EDS-BHT)(formerly type III) is considered the least severe. There is some controversy as to whether this subtype constitutes a separate diagnosis from the benign familial joint hypermobility syndrome. It is characterized by hypermobility of the joints (objectively measured with the Beighton scale) and mild hyperextensibility of the skin, and patients often have a history of joint subluxations and dislocations with resultant degenerative joint disease and chronic pain. Manifestations of fragile skin and soft tissue (eg, abnormal wound healing or scarring; spontaneous tearing of the skin, ligaments, tendons, or organs) are notably absent from the findings in this syndrome.5 The genetic basis for EDS is unknown in the majority of patients, although a deficiency in tenascin X (secondary to defects in the tenascin XB gene [TNXB]) has been identified in a small subset (<5%) of patients, leading to elastic fiber abnormalities, reduced collagen deposition, and impaired cross-linking of collagen.6,7 Inheritance usually is autosomal dominant but also can be autosomal recessive. In contrast, the classic type of EDS (formerly types I and II) is associated with atrophic scarring and tissue fragility, in addition to joint hypermobility and skin hyperextensibility. Type V collagen mutations are found in more than half of patients with this disorder.8
We present the case of a patient with EDS-BHT who developed large nonhealing cutaneous ulcerations with initiation of high-dose systemic corticosteroids for treatment of dermatomyositis. This case provides a dramatic illustration of the effects of the use of chronic systemic corticosteroids on skin fragility and wound healing in patients with an underlying inherited defect in collagen or connective tissue.
Case Report
A 23-year-old man with an unremarkable medical history was admitted to our inpatient cardiology service with palpitations attributable to new-onset atrial fibrillation. Dermatology was consulted to evaluate a rash of approximately 4 months’ duration that started on the dorsal aspect of the hands, then progressed to involve the extensor elbows and knees. The rash also was associated with fatigue, arthralgia, and proximal muscle weakness. A taper of prednisone that was prescribed approximately 2 months prior to admission by a rheumatologist for presumed dermatomyositis improved his symptoms, but they recurred with discontinuation of the medication.
Physical examination revealed reddish, violaceous and hyperpigmented patches on the dorsal aspect of the hands and digits and the extensor aspect of the knees and elbows. A skin biopsy from the right elbow showed a mild interface reaction and nonspecific direct immunofluorescence, consistent with a diagnosis of dermatomyositis. Autoimmune serologies were negative, including antinuclear, anti–Jo-1, anti–Mi-2, anti–Sjögren syndrome antigen A, anti–Sjögren syndrome antigen B, anti-Smith, and antiribonucleoprotein antibodies. Creatine kinase and rheumatoid factor levels were within reference range. Electromyogram was supportive of the diagnosis of dermatomyositis, showing an irritable myopathy. Cardiac magnetic resonance imaging showed an acute inflammatory process of the myocardium, and a transthoracic echocardiogram revealed a depressed left ventricular ejection fraction of 35% to 40% (reference range, 55%–70%). His cardiac disease also was attributed to dermatomyositis, and he was managed by cardiology with anangiotensin-converting enzyme inhibitor and antiarrhythmic therapy. Rheumatology was consulted and prednisone 60 mg once daily was started, with the patient reporting improvement in his muscle weakness and the rash.
Interestingly, the patient also noted a history of joint hypermobility, and a genetics consultation was obtained during the current hospitalization. He denied a history of abnormal scarring or skin problems, but he did note dislocation of the patella on 2 occasions and an umbilical hernia repair at 3 years of age. A paternal uncle had a history of similar joint hypermobility. His Beighton score was noted to be 8/8 (bending at the waist was unable to be tested due to recent lumbar puncture obtained during this hospitalization). The patient was diagnosed with EDS-BHT, and no further workup was recommended.
Subsequent to his hospitalization for several days, the patient’s prednisone was slowly tapered down from 60 mg once daily to 12.5 mg once daily, and azathioprine was started and titrated up to 150 mg once daily. Approximately 6 months after his initial hospitalization, he was readmitted due to increased pain of the right knee with concern for osteomyelitis. Dermatology was again consulted, and at this time, the patient reported a 4-month history of nonhealing ulcers to the knees and elbows (Figure 1). He stated that the ulcers were initially about the size of a pencil eraser and had started approximately 2 months after the prednisone was started, with subsequent slow enlargement. He noted a stinging sensation with enlargement of the ulcers, but otherwise they were not painful. He denied major trauma to the areas. He noted that his prior rash from the dermatomyositis seemed to have resolved, along with his muscle weakness, and he reported weight gain and improvement in his energy levels. Physical examination at this time revealed several stigmata of chronic systemic corticosteroids, including fatty deposits in the face (moon facies) and between the shoulders (buffalo hump), facial acne, and numerous erythematous striae on the trunk and proximal extremities (Figure 2). Multiple noninflammatory ulcers with punched-out borders ranging in size from 0.5 to 6 cm were seen at sites overlying bony prominences, including the bilateral extensor elbows and knees and the right plantar foot. Similar ulcers were noted on the trunk within the striae. Some of the ulcers were covered with a thick hyperkeratotic crust. A biopsy from the edge of an ulcer on the right side of the flank showed only dermal fibrosis. Workup by orthopedic surgery was felt to be inconsistent with osteomyelitis, and plastic surgery was consulted to consider surgical options for repair. Consequently, the patient was taken to the operating room for primary closure of the ulcers to the bilateral knees and right elbow. He has been followed closely by plastic surgery, with the use of joint immobilization to promote wound healing.
Comment
This case represents a dramatic illustration of the effects of chronic systemic corticosteroids on skin fragility and wound healing in a patient with an underlying genetic defect in the connective tissue. The ulcers were all located within striae or overlying bony prominences where the skin was subjected to increased tension; however, the patient reported no problems with wound healing or scarring at these sites prior to the initiation of corticosteroids, suggesting that the addition of this medication was disruptive to the cutaneous wound healing mechanisms. This case is unique because abnormal wound healing in an EDS patient was so clearly linked to the initiation of systemic steroids.
The exact pathogenesis of the patient’s ulcers is unclear. The diagnosis of EDS was primarily clinical, and without genetic testing, we cannot state with certainty the underlying molecular problem in this patient. Although tenascin X deficiency has been found in a few patients, a genetic defect remains uncharacterized in most patients with EDS-BHT, and in most situations, EDS-BHT remains a clinical diagnosis. In 2001, Schalkwijk et al9 first described the association of tenascin X deficiency and EDS in 5 patients, and they noted delayed wound healing in 1 patient who had received systemic corticosteroids for congenital adrenal hyperplasia. The authors remarked that it was not clear whether the abnormality was linked to the patient’s EDS or to his treatment with systemic corticosteroids.9 Furthermore, it is possible that our patient in fact has a milder variant of classic type EDS and that the manifestations of tissue fragility remained subclinical until the addition of systemic corticosteroids. It also is interesting to note that muscle weakness can be a symptom of EDS, both classic and BHT of EDS, but our patient’s muscle weakness improved with immunosuppression, supporting an underlying autoimmune disease as the cause for it.10 Skin ulcerations have been reported as a rare manifestation of dermatomyositis, but it is remarkable that his ulcers progressed as his other dermatomyositis symptoms improved with therapy, suggesting that his autoimmune disease was not the underlying cause for the ulcers.11-13 This case points to the need to thoughtfully consider the adverse effects of corticosteroids on wound healing in patients with an inherited disorder of collagen or connective tissue such as EDS.
- Bolognia JL, Jorizzo JL, Rapini RP, et al. Dermatology. 2nd ed. Philadelphia, PA: Mosby Elsevier; 2008.
- Gurtner GC, Werner S, Barrandon Y, et al. Wound repair and regeneration. Nature. 2008;453:314-321.
- Poetker DM, Reh DD. A comprehensive review of the adverse effects of systemic corticosteroids. Otolaryng Clin N Am. 2010;43:753-768.
- Beighton P, De Paepe A, Steinmann B, et al. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998;77:31-37.
- Levy HP. Ehlers-Danlos syndrome, hypermobility type. In: Pagon RA, Bird TD, Dolan CR, et al, es. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993-2015. http://www.ncbi.nlm.nih.gov/books/NBK1279/. Accessed August 5, 2015.
- Zweers MC, Bristow J, Steijlen PM, et al. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet. 2003;73:214-217.
- Brellier F, Tucker RP, Chiquet-Ehrismann R. Tenascins and their implications in diseases and tissue mechanics. Scand J Med Sci Spor. 2009;19:511-519.
- Malfait F, Wenstrup R, De Paepe A. Ehlers-Danlos syndrome, classic type. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews. Seattle,WA: University of Washington, Seattle; 1993-2015. http://www.ncbi.nlm.nih.gov/books/NBK1244/. Accessed August 5, 2015.
- Schalkwijk J, Zweers MC, Steijlen PM, et al. A recessive form of the Ehlers-Danlos syndrome caused by tenascin X deficiency. N Engl J Med. 2001;345:1167-1175.
- Voermans NC, Alfen NV, Pillen S, et al. Neuromuscular involvement in various types of Ehlers-Danlos syndrome. Ann Neurol. 2009;65:687-697.
- Scheinfeld NS. Ulcerative paraneoplastic dermatomyositis secondary to metastatic breast cancer. Skinmed. 2006;5:94-96.
- Tomb R, Stephan F. Perforating skin ulcers occurring in an adult with dermatomyositis [in French]. Ann Dermatol Venerol. 2002;129:1383-1385.
- Yosipovitch G, Feinmesser M, David M. Adult dermatomyositis with livedo reticularis and multiple skin ulcers. J Eur Acad Dermatol. 1998;11:48-50.
- Bolognia JL, Jorizzo JL, Rapini RP, et al. Dermatology. 2nd ed. Philadelphia, PA: Mosby Elsevier; 2008.
- Gurtner GC, Werner S, Barrandon Y, et al. Wound repair and regeneration. Nature. 2008;453:314-321.
- Poetker DM, Reh DD. A comprehensive review of the adverse effects of systemic corticosteroids. Otolaryng Clin N Am. 2010;43:753-768.
- Beighton P, De Paepe A, Steinmann B, et al. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998;77:31-37.
- Levy HP. Ehlers-Danlos syndrome, hypermobility type. In: Pagon RA, Bird TD, Dolan CR, et al, es. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993-2015. http://www.ncbi.nlm.nih.gov/books/NBK1279/. Accessed August 5, 2015.
- Zweers MC, Bristow J, Steijlen PM, et al. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet. 2003;73:214-217.
- Brellier F, Tucker RP, Chiquet-Ehrismann R. Tenascins and their implications in diseases and tissue mechanics. Scand J Med Sci Spor. 2009;19:511-519.
- Malfait F, Wenstrup R, De Paepe A. Ehlers-Danlos syndrome, classic type. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews. Seattle,WA: University of Washington, Seattle; 1993-2015. http://www.ncbi.nlm.nih.gov/books/NBK1244/. Accessed August 5, 2015.
- Schalkwijk J, Zweers MC, Steijlen PM, et al. A recessive form of the Ehlers-Danlos syndrome caused by tenascin X deficiency. N Engl J Med. 2001;345:1167-1175.
- Voermans NC, Alfen NV, Pillen S, et al. Neuromuscular involvement in various types of Ehlers-Danlos syndrome. Ann Neurol. 2009;65:687-697.
- Scheinfeld NS. Ulcerative paraneoplastic dermatomyositis secondary to metastatic breast cancer. Skinmed. 2006;5:94-96.
- Tomb R, Stephan F. Perforating skin ulcers occurring in an adult with dermatomyositis [in French]. Ann Dermatol Venerol. 2002;129:1383-1385.
- Yosipovitch G, Feinmesser M, David M. Adult dermatomyositis with livedo reticularis and multiple skin ulcers. J Eur Acad Dermatol. 1998;11:48-50.
Practice Points
- Chronic corticosteroids have profound effects on the wound-healing process, and their detrimental effects may be amplified in patients with underlying connective tissue defects.
- Although genetic testing is available, the diagnosis of Ehlers-Danlos syndrome benign hypermobility type usually is made clinically.
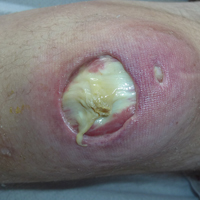
FDA Black Box, VA Red Ink? A Successful Service-Connected Disability Claim for Chronic Neuropsychiatric Adverse Effects From Mefloquine
Mefloquine is a synthetic antimalarial drug structurally related to quinine. The drug was developed by the Walter Reed Army Institute of Research during a decades-long program that started during the Vietnam War in response to concerns of rising resistance to chloroquine.1
The prelicensing clinical testing of mefloquine, originally known as WR 142,490, was conducted in part among U.S. military service members.2,3 Soon after receiving FDA approval in 1989, under the brand name Lariam, it was recommended for use within the U.S. military.4 Over the following 2 decades, mefloquine was a common exposure during military deployments to malaria endemic areas.
Although the original U.S. mefloquine drug label noted that neuropsychiatric reactions could occur with use, changes to the drug label mandated by the FDA in July 2013, including a black box warning, described a potential for these to persist long after the drug has been discontinued.5,6 These changes have served to reinforce earlier U.S. military policy changes beginning in 2009 that deprioritized use of the drug in favor of safer and better-tolerated antimalarials. Consequently, more than a quarter century after its introduction, mefloquine now is only rarely prescribed to members of the U.S. military.7
In addition to limiting current use of the drug, the recent boxed warning may have important implications for service-connected disability claims adjudication by the VA for veterans previously exposed to the drug. This report presents a case of a nondeployed veteran exposed to mefloquine during an early military postmarketing study who developed chronic neuropsychiatric symptoms linked to the drug that were recently deemed service-connected. This report concludes with some comments on the likely implications of this case for future similar disability claims.
Case Presentation
In 2014, a 56-year-old nondeployed U.S. Marine Corps veteran submitted a claim to the VA for disabling conditions. The veteran alleged these conditions were due to his exposure to mefloquine while in military service more than 2 decades earlier. The veteran enlisted in 1975 and experienced a motor vehicle accident with prolonged loss of consciousness in 1978 but had no other significant medical history.
Thirteen years later, stationed in Hawaii in 1991, he was encouraged to volunteer for a double-blinded postmarketing study, evaluating the adverse effects (AEs) of chloroquine and mefloquine.8 As documentation following the trial revealed, he was randomly assigned to the mefloquine arm and received a loading dose of 250 mg daily for 3 days, followed by 250 mg per week for 11 weeks.
During the study he experienced insomnia, abnormal dreams, and nightmares. He also developed symptoms of anxiety, depression, cognitive dysfunction, and changes in personality—including anger and irritability—that were severe enough to be noted by his family members. The patient had not been advised of the significance of these symptoms and therefore did not report them during the clinical trial, nor did he report their intermittent presence after the study’s conclusion through his retirement in 1996, fearing adverse career consequences. Subsequent exacerbations of these chronic symptoms later contributed to the patient’s loss of civilian employment in 2010.
After becoming aware of the 2013 boxed warning that these chronic symptoms could be due to his earlier exposure to mefloquine, the veteran sought evaluation by a VA clinician. On evaluation, the clinician noted no history of deployment, and no history of posttraumatic stress disorder (PTSD) criteria A stressors, and posited that the veteran’s chronic neuropsychiatric symptoms were most likely a consequence of his earlier use of mefloquine. The VA subsequently awarded the veteran 50% disability for an anxiety disorder characterized by chronic sleep impairment and frequent panic attacks, attributing these to his service-connected use of the drug.
Discussion
Although the original 1989 FDA-approved mefloquine label had warned to discontinue the drug if specific prodromal symptoms of “anxiety, depression, restlessness or confusion” were noted,as illustrated by this case, this guidance was not always consistently communicated to service members.5 Indeed, few service members in the 1991 military postmarketing study discontinued the medication even after reporting such symptoms.8 Vivid dreams, often described as “terrifying nightmares with technicolor clarity” were reported by 7% of study participants. Similarly, concentration problems were reported in 5%; irritability in 4%; anger and moodiness each in 1%; and insomnia in 25%. Two study participants, after failing to discontinue mefloquine at the onset of severe insomnia, were later hospitalized for severe depression and suicidal thoughts, later deemed due to preexisting conditions. Despite these seemingly unfavorable results, mefloquine was nonetheless deemed well tolerated.8
Military Use of Mefloquine
Beginning in 1992, use of mefloquine for prophylaxis of malaria was then widely directed within the U.S. military during operations in Somalia. There, a majority of personnel received mefloquine under conditions of command-directed and directly observed administration of the drug.9,10 Again, drug label guidance describing the prodromal psychiatric symptoms that should have prompted discontinuation of mefloquine were either not consistently adhered to or not communicated. In one Somalia-era study, only 1 in 344 service members, or 0.3%, discontinued the drug.11
Throughout the remainder of the 1990s, mefloquine remained the antimalarial drug of choice for most U.S. military operations, and when combat began in Afghanistan in 2001, widespread use was also directed there.12,13 The following year, after national attention was directed to concerns of severe behavioral toxicity from the drug among personnel returning from Afghanistan, the manufacturer issued subtle changes to the mefloquine label warnings.5,14
These label changes adjusted the previously exclusive list of prodromal symptoms to an illustrative list, emphasizing that “if psychiatric symptoms such as [emphasis added] acute anxiety, depression, restlessness or confusion occur, these may be considered prodromal to a more serious event. In these cases, the drug must be discontinued and an alternative medication should be substituted.”5
In 2001 a randomized double-blinded trial demonstrated that symptoms of anxiety and depression occurred in at least 4% of mefloquine users, insomnia in 13%, and abnormal dreams in 14%. Nevertheless, an Army memorandum issued soon after the labeling change significantly understated the known risks of developing such psychiatric symptoms, erroneously claiming that these occurred from mefloquine only “at a rate of one per 2,000 to 13,000 persons.”15,16
Updated FDA Guidelines
In 2003, with widespread use of the drug being again directed during operations in Iraq, the FDA required that all mefloquine prescriptions be accompanied by a patient medication guide with warnings echoing those of the drug label that users seek medical attention should “possible signs of more serious mental problems” develop.5,17 However, surveys suggested that few U.S. service members received these warnings or even verbal instructions to that effect.17-19 During later congressional testimony, a service member who had experienced 3 weeks of nightmares prior to self-discontinuing the drug testified “every soldier I know has problems with it.”20
In response, a senior military medical leader—failing to recognize that the nightmares the soldier reported were in fact psychiatric symptoms and possible signs of more serious mental problems that required the drug’s discontinuation—may have undermined the FDA-directed warnings by dismissing the soldier’s testimony as “perception,” maintaining instead “that perceptions can become realities” should it become “held that this medication is widely problematic.”20
Given that certain preexisting conditions, including anxiety and depression, were known to confound recognition of incident psychiatric symptoms that required discontinuation of the drug, the original 1989 mefloquine label had noted that the drug should be used with caution in such patients. In subsequent years, this language was strengthened, and such patients were formally contraindicated the drug.21
Citing formal policy, senior military medical leaders provided assurance during congressional testimony that service members with these conditions would not be prescribed mefloquine.16,18,20 However, later analysis of a large group of deployed service members revealed that 1 in 7 with contraindications to mefloquine had been prescribed the drug contrary to drug label guidance.21
Black Box Warning
With growing recognition of the challenges in using mefloquine as directed by the drug label, a 2009 Army policy memorandum prioritized the use of safer and better-tolerated daily medications, such as doxycycline and atovaquone-proguanil, and stated that “[m]efloquine should only be used for personnel with contraindications to doxycycline.”22 This policy was extended throughout the other military services later that year.23 After concerns were raised that service members were still being prescribed the drug contrary to policy, further restrictions were formalized in early 2013 prior to the boxed warning, with mefloquine reserved for those only “with intolerance or contraindications” to the first-line drugs.24,25
In a later memorandum announcing the July 2013 boxed warning, the military revealed that the number of active-duty personnel prescribed mefloquine had steadily decreased in prior years from 17,361 in 2008 to only 2,040 in 2012.7 Although the military has not released precise figures on the number of U.S. military personnel exposed to mefloquine since the drug’s introduction, based on a variety of sources, the total is likely to far exceed 100,000.7,26
The major changes to the mefloquine label in 2013, including the boxed warning, clarified that neurologic and psychiatric effects from mefloquine could “persist after mefloquine has been discontinued.” The accompanying FDA Drug Safety Communication noted neurologic AEs from the drug, which include but are not limited to “dizziness, loss of balance, or ringing in the ears,” could “occur at any time during drug use, and can last for months to years after the drug is stopped or can be permanent.”6 Other neurologic symptoms listed in the drug label include vertigo, hearing impairment, headache, visual disturbances, sensory and motor neuropathies, including paresthesia, tremor, ataxia, convulsions, and encephalopathy.6
The updated drug label also made clear that psychiatric AEs from mefloquine, such as anxiety, paranoia, and depression to hallucinations and psychotic behavior, “have been reported to continue for months or years after mefloquine has been stopped.” Other psychiatric symptoms listed in the drug label include memory impairment, confusion, somnolence, insomnia, abnormal dreams, aggression, agitation, restlessness, mood swings, panic attacks, psychosis, and suicidal ideation.6
The 2013 boxed warning also served to reemphasize guidance first articulated in 2002 that any psychiatric symptom—presumably including abnormal dreams and insomnia—occurring during mefloquine use should be considered prodromal, prompting the drug’s immediate discontinuation.5 Specifically, the boxed warning explicitly cautioned that given the risk for serious psychiatric disturbances or neurologic AEs when used for malaria prophylaxis, “if psychiatric or neurologic symptoms occur, the drug should be discontinued and an alternative medication should be substituted.”6
Drug of Last Resort
By late 2013, partially on the basis of the boxed warning, the U.S. military declared mefloquine a “drug of last resort.”7,27 The U.S. Army Special Operations Command (USASOC) took the further step of prohibiting use of mefloquine altogether and, according to news reports, directed that medical and command staff assess whether certain personnel experiencing AEs from the drug may mistakenly have been thought to be malingering, have PTSD, or have other psychological problems.28
As the boxed warning and the USASOC order suggest, veterans exposed to mefloquine may have incurred a broad range of neurologic or psychiatric disorders or had others aggravated during military service as a result of their use of the drug. The effects of mefloquine may have confounded the diagnosis of neurologic or psychiatric disorders related to military service.26,29 As these AEs may be a direct result of mefloquine prescribed during military service, those with disabling diagnoses consistent with these effects may be entitled to claim disability compensation through the VA.
Of potential significant relevance to this adjudication process is a memorandum written in early 2012, in which the military conceded:
Some deploying Service members have been provided mefloquine for malaria prophylaxis without appropriate documentation in their medical records and without proper screening for contraindications. In addition, not all individuals have been provided the required mefloquine medication guide and wallet information card, as required by the Food and Drug Administration. 24
Veterans claiming a service-connected disability as a result of their use of mefloquine should therefore not always be expected to have documentation of prescribing in their military medical records. Although the VA could consider denying such claims for absence of proof of a nexus to military service, in light of this memorandum, the VA may need to consider other evidence of plausible exposure, including veteran testimony and deployment history.
It is also conceivable that the VA could consider denying such claims by arguing that the veteran directly contributed to the disability through willful misconduct by not adhering to mefloquine label guidance. However, as this memorandum establishes that mefloquine use was frequently directed without communication of the drug label precautions and warnings, the VA should consider that veterans claiming a service-connected disability frequently will not have known or otherwise been unable to discontinue the medication at the onset of prodromal symptoms.
It is also possible that the VA might deny claims on the basis that the claimed disabilities reflect preexisting conditions. However, as the memorandum establishes, use of mefloquine also was occasionally inappropriately directed to those with documented contraindications to the medication, who would have increased risk of AEs. As a result, veterans with preexisting neurologic or psychiatric conditions or disorders who nonetheless were prescribed mefloquine may reasonably claim these were aggravated during military service.
Conclusion
As this case suggests, in the coming years, as awareness of the chronic AEs of mefloquine increases among the veteran population, claims related to prior use of the drug are likely to increase and become of significant interest to the VA. Veterans with plausible exposure to mefloquine with neuropsychiatric disabilities who have yet to file a claim may be able to do so, and those veterans whose claims for service-connection were unfavorably adjudicated may be able to reopen their claims on the basis of the new material evidence in the 2012 military memorandum and the 2013 boxed warning.
This case report also suggests that service-connected disability claims arising from chronic neuropsychiatric AEs from mefloquine may prove to be of significant financial consequence. Further research to better define both the extent of prior mefloquine use among U.S. military personnel and the nature and prevalence of those chronic neurologic and psychiatric disorders caused by the drug would be helpful in informing improvements in the efficient and fair adjudication of such service-connected disability claims.
1. Tigertt WD. The army malaria research program. Ann Intern Med. 1969;70(1):150-153.
2. Trenholme CM, Williams RL, Desjardins RE, et al. Mefloquine (WR 142,490) in the treatment of human malaria. Science. 1975;190(4216):792-794.
3. Shanks GD, Karwacki J, Kanesa-thasan N, et al. Diseases transmitted primarily by arthropod vectors. In: Kelley PW, ed. Military Preventive Medicine: Mobilization and Deployment. Vol 2. Washington, DC: Borden Institute; 2005:803-935.
4. Armed Forces Epidemiological Board. Memorandum. Subject: Recommendations on Mefloquine Chemoprophylaxis for Military Personnel. Published October 3, 1989.
5. Nevin RL, Byrd AM. Neuropsychiatric adverse reactions to mefloquine: a systematic comparison of prescribing and patient safety guidance in the US, UK, Ireland, Australia, New Zealand, and Canada. Neurol Ther. 2016;5(1):69-83.
6. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA approves label changes for antimalarial drug mefloquine hydrochloride due to risk of serious psychiatric and nerve side effects. http://www.fda.gov/Drugs/DrugSafety/ucm362227.htm. Published July 29, 2013. Accessed August 26, 2016.
7. Assistant Secretary of Defense for Health Affairs. Memorandum. Subject: Notification for Healthcare Providers of Mefloquine Boxed Warning. Published August 12, 2013.
8. Boudreau E, Schuster B, Sanchez J, et al. Tolerability of prophylactic Lariam regimens. Trop Med Parasitol. 1993;44(3):257-265.
9. Wallace MR, Sharp TW, Smoak B, et al. Malaria among United States troops in Somalia. Am J Med. 1996;100(1):49-55.
10. Smoak BL, Writer JV, Keep LW, Cowan J, Chantelois JL. The effects of inadvertent exposure of mefloquine chemoprophylaxis on pregnancy outcomes and infants of US Army servicewomen. J Infect Dis. 1997;176(3):831-833.
11. Sánchez JL, DeFraites RF, Sharp TW, Hanson RK. Mefloquine or doxycycline prophylaxis in US troops in Somalia. Lancet. 1993;341(8851):1021-1022.
12. Jones R, Kunsman G, Levine B, Smith M, Stahl C. Mefloquine distribution in postmortem cases. Forensic Sci Int. 1994;68(1):29-32.
13. Kotwal RS, Wenzel RB, Sterling RA, Porter WD, Jordan NN, Petruccelli BP. An outbreak of malaria in US Army Rangers returning from Afghanistan. JAMA. 2005;293(2):212-216.
14. Hess BP. Army fears rebellion on Lariam. United Press International. http://www.upi.com/Business_News/Security-Industry/2002/08/29/Analysis-Army-fears-rebellion-on-Lariam/UPI-39351030635930. Published August 29, 2002. Accessed August 29, 2016.
15. Overbosch D, Schilthuis H, Bienzle U, et al; Malarone International Study Team. Atovaquone-proguanil versus mefloquine for malaria prophylaxis in nonimmune travelers: results from a randomized, double-blind study. Clin Infect Dis. 2001;33(7):1015-1021.
16. U.S. Army Surgeon General. Memorandum. Subject: Updated Health Care Provider Information on Use of Mefloquine Hydrochloride for Malaria Prophylaxis. October 3, 2002.
17. Associated Press. Hallucinations linked to drug given to troops. http://www.nbcnews.com/id/6947472/ns/health-mental_health/t/hallucinations-linked-drug-given-troops. Published February 14, 2005. Accessed August 26, 2016.
18. Benjamin M. Army sent mentally ill troops to Iraq. United Press International. http://www.upi.com/Business_News/Security-Industry/2004/03/12/Army-sent-mentally-ill-troops-to-Iraq/UPI-97331079131967. Published March 12, 2004. Accessed August 26, 2016.
19. Fleet M, Mann J. Military’s use of malaria drug in question. http://edition.cnn.com/2004/HEALTH/05/20/lariam. Published May 21, 2004. Accessed August 26, 2016.
20. 108th Congress. Hearing on National Defense Authorization Act for Fiscal Year 2005 - H.R. 4200, February 25, 2004. http://commdocs.house.gov/committees/security/has056270.000/has056270_0f.htm. Accessed August 26, 2016.
21. Nevin RL. Mefloquine prescriptions in the presence of contraindications: prevalence among US military personnel deployed to Afghanistan, 2007. Pharmacoepidemiol Drug Saf. 2010;19(2):206-210.
22. U.S. Army Surgeon General. Memorandum. Subject: Updated Guidance on the Use of Mefloquine for Malaria Prophylaxis. February 2, 2009.
23. Assistant Secretary of Defense for Health Affairs. Memorandum. Subject: Policy Memorandum on the Use of Mefloquine (Lariam) in Malaria Prophylaxis. HA Policy 09-017. http://www.health.mil/~/media/MHS/Policy%20Files/Import/09-017.ashx. September 4, 2009. Accessed August 26, 2016.
24. Assistant Secretary of Defense for Health Affairs. Memorandum. Subject: Service Review of Mefloquine Prescribing Practices. January 17, 2012.
25. Assistant Secretary of Defense for Health Affairs. Memorandum. Subject: Guidance on Medications for Prophylaxis of Malaria. April 15, 2013.
26. Nevin RL. Mefloquine and posttraumatic stress disorder. In: Ritchie EC, ed. Forensic and Ethical Issues in Military Behavioral Health. Washington, DC: Borden Institute; 2014:275-296.
27. Pellerin C. DOD mefloquine policy mirrors FDA update on malaria drug. American Forces Press Service. http://archive.defense.gov/news/newsarticle.aspx?id=120857. Published September 26, 2013. Accessed August 26, 2016.
28. Jelinek P. Elite Army units to stop taking anti-malarial drug. Associated Press. http://www.military.com/daily-news/2013/09/19/elite-army-units-to-stop-taking-anti-malarial-drug.html. Published September 19, 2013. Accessed August 26, 2016.
29. Nevin RL, Ritchie EC. The Mefloquine intoxication syndrome: a significant potential confounder in the diagnosis and management of PTSD and other chronic deployment-related neuropsychiatric disorders. In: Ritchie EC, ed. Posttraumatic Stress Disorder and Related Disorders in Combat Veterans. Cham, Switzerland: Springer; 2015:257-278.
Mefloquine is a synthetic antimalarial drug structurally related to quinine. The drug was developed by the Walter Reed Army Institute of Research during a decades-long program that started during the Vietnam War in response to concerns of rising resistance to chloroquine.1
The prelicensing clinical testing of mefloquine, originally known as WR 142,490, was conducted in part among U.S. military service members.2,3 Soon after receiving FDA approval in 1989, under the brand name Lariam, it was recommended for use within the U.S. military.4 Over the following 2 decades, mefloquine was a common exposure during military deployments to malaria endemic areas.
Although the original U.S. mefloquine drug label noted that neuropsychiatric reactions could occur with use, changes to the drug label mandated by the FDA in July 2013, including a black box warning, described a potential for these to persist long after the drug has been discontinued.5,6 These changes have served to reinforce earlier U.S. military policy changes beginning in 2009 that deprioritized use of the drug in favor of safer and better-tolerated antimalarials. Consequently, more than a quarter century after its introduction, mefloquine now is only rarely prescribed to members of the U.S. military.7
In addition to limiting current use of the drug, the recent boxed warning may have important implications for service-connected disability claims adjudication by the VA for veterans previously exposed to the drug. This report presents a case of a nondeployed veteran exposed to mefloquine during an early military postmarketing study who developed chronic neuropsychiatric symptoms linked to the drug that were recently deemed service-connected. This report concludes with some comments on the likely implications of this case for future similar disability claims.
Case Presentation
In 2014, a 56-year-old nondeployed U.S. Marine Corps veteran submitted a claim to the VA for disabling conditions. The veteran alleged these conditions were due to his exposure to mefloquine while in military service more than 2 decades earlier. The veteran enlisted in 1975 and experienced a motor vehicle accident with prolonged loss of consciousness in 1978 but had no other significant medical history.
Thirteen years later, stationed in Hawaii in 1991, he was encouraged to volunteer for a double-blinded postmarketing study, evaluating the adverse effects (AEs) of chloroquine and mefloquine.8 As documentation following the trial revealed, he was randomly assigned to the mefloquine arm and received a loading dose of 250 mg daily for 3 days, followed by 250 mg per week for 11 weeks.
During the study he experienced insomnia, abnormal dreams, and nightmares. He also developed symptoms of anxiety, depression, cognitive dysfunction, and changes in personality—including anger and irritability—that were severe enough to be noted by his family members. The patient had not been advised of the significance of these symptoms and therefore did not report them during the clinical trial, nor did he report their intermittent presence after the study’s conclusion through his retirement in 1996, fearing adverse career consequences. Subsequent exacerbations of these chronic symptoms later contributed to the patient’s loss of civilian employment in 2010.
After becoming aware of the 2013 boxed warning that these chronic symptoms could be due to his earlier exposure to mefloquine, the veteran sought evaluation by a VA clinician. On evaluation, the clinician noted no history of deployment, and no history of posttraumatic stress disorder (PTSD) criteria A stressors, and posited that the veteran’s chronic neuropsychiatric symptoms were most likely a consequence of his earlier use of mefloquine. The VA subsequently awarded the veteran 50% disability for an anxiety disorder characterized by chronic sleep impairment and frequent panic attacks, attributing these to his service-connected use of the drug.
Discussion
Although the original 1989 FDA-approved mefloquine label had warned to discontinue the drug if specific prodromal symptoms of “anxiety, depression, restlessness or confusion” were noted,as illustrated by this case, this guidance was not always consistently communicated to service members.5 Indeed, few service members in the 1991 military postmarketing study discontinued the medication even after reporting such symptoms.8 Vivid dreams, often described as “terrifying nightmares with technicolor clarity” were reported by 7% of study participants. Similarly, concentration problems were reported in 5%; irritability in 4%; anger and moodiness each in 1%; and insomnia in 25%. Two study participants, after failing to discontinue mefloquine at the onset of severe insomnia, were later hospitalized for severe depression and suicidal thoughts, later deemed due to preexisting conditions. Despite these seemingly unfavorable results, mefloquine was nonetheless deemed well tolerated.8
Military Use of Mefloquine
Beginning in 1992, use of mefloquine for prophylaxis of malaria was then widely directed within the U.S. military during operations in Somalia. There, a majority of personnel received mefloquine under conditions of command-directed and directly observed administration of the drug.9,10 Again, drug label guidance describing the prodromal psychiatric symptoms that should have prompted discontinuation of mefloquine were either not consistently adhered to or not communicated. In one Somalia-era study, only 1 in 344 service members, or 0.3%, discontinued the drug.11
Throughout the remainder of the 1990s, mefloquine remained the antimalarial drug of choice for most U.S. military operations, and when combat began in Afghanistan in 2001, widespread use was also directed there.12,13 The following year, after national attention was directed to concerns of severe behavioral toxicity from the drug among personnel returning from Afghanistan, the manufacturer issued subtle changes to the mefloquine label warnings.5,14
These label changes adjusted the previously exclusive list of prodromal symptoms to an illustrative list, emphasizing that “if psychiatric symptoms such as [emphasis added] acute anxiety, depression, restlessness or confusion occur, these may be considered prodromal to a more serious event. In these cases, the drug must be discontinued and an alternative medication should be substituted.”5
In 2001 a randomized double-blinded trial demonstrated that symptoms of anxiety and depression occurred in at least 4% of mefloquine users, insomnia in 13%, and abnormal dreams in 14%. Nevertheless, an Army memorandum issued soon after the labeling change significantly understated the known risks of developing such psychiatric symptoms, erroneously claiming that these occurred from mefloquine only “at a rate of one per 2,000 to 13,000 persons.”15,16
Updated FDA Guidelines
In 2003, with widespread use of the drug being again directed during operations in Iraq, the FDA required that all mefloquine prescriptions be accompanied by a patient medication guide with warnings echoing those of the drug label that users seek medical attention should “possible signs of more serious mental problems” develop.5,17 However, surveys suggested that few U.S. service members received these warnings or even verbal instructions to that effect.17-19 During later congressional testimony, a service member who had experienced 3 weeks of nightmares prior to self-discontinuing the drug testified “every soldier I know has problems with it.”20
In response, a senior military medical leader—failing to recognize that the nightmares the soldier reported were in fact psychiatric symptoms and possible signs of more serious mental problems that required the drug’s discontinuation—may have undermined the FDA-directed warnings by dismissing the soldier’s testimony as “perception,” maintaining instead “that perceptions can become realities” should it become “held that this medication is widely problematic.”20
Given that certain preexisting conditions, including anxiety and depression, were known to confound recognition of incident psychiatric symptoms that required discontinuation of the drug, the original 1989 mefloquine label had noted that the drug should be used with caution in such patients. In subsequent years, this language was strengthened, and such patients were formally contraindicated the drug.21
Citing formal policy, senior military medical leaders provided assurance during congressional testimony that service members with these conditions would not be prescribed mefloquine.16,18,20 However, later analysis of a large group of deployed service members revealed that 1 in 7 with contraindications to mefloquine had been prescribed the drug contrary to drug label guidance.21
Black Box Warning
With growing recognition of the challenges in using mefloquine as directed by the drug label, a 2009 Army policy memorandum prioritized the use of safer and better-tolerated daily medications, such as doxycycline and atovaquone-proguanil, and stated that “[m]efloquine should only be used for personnel with contraindications to doxycycline.”22 This policy was extended throughout the other military services later that year.23 After concerns were raised that service members were still being prescribed the drug contrary to policy, further restrictions were formalized in early 2013 prior to the boxed warning, with mefloquine reserved for those only “with intolerance or contraindications” to the first-line drugs.24,25
In a later memorandum announcing the July 2013 boxed warning, the military revealed that the number of active-duty personnel prescribed mefloquine had steadily decreased in prior years from 17,361 in 2008 to only 2,040 in 2012.7 Although the military has not released precise figures on the number of U.S. military personnel exposed to mefloquine since the drug’s introduction, based on a variety of sources, the total is likely to far exceed 100,000.7,26
The major changes to the mefloquine label in 2013, including the boxed warning, clarified that neurologic and psychiatric effects from mefloquine could “persist after mefloquine has been discontinued.” The accompanying FDA Drug Safety Communication noted neurologic AEs from the drug, which include but are not limited to “dizziness, loss of balance, or ringing in the ears,” could “occur at any time during drug use, and can last for months to years after the drug is stopped or can be permanent.”6 Other neurologic symptoms listed in the drug label include vertigo, hearing impairment, headache, visual disturbances, sensory and motor neuropathies, including paresthesia, tremor, ataxia, convulsions, and encephalopathy.6
The updated drug label also made clear that psychiatric AEs from mefloquine, such as anxiety, paranoia, and depression to hallucinations and psychotic behavior, “have been reported to continue for months or years after mefloquine has been stopped.” Other psychiatric symptoms listed in the drug label include memory impairment, confusion, somnolence, insomnia, abnormal dreams, aggression, agitation, restlessness, mood swings, panic attacks, psychosis, and suicidal ideation.6
The 2013 boxed warning also served to reemphasize guidance first articulated in 2002 that any psychiatric symptom—presumably including abnormal dreams and insomnia—occurring during mefloquine use should be considered prodromal, prompting the drug’s immediate discontinuation.5 Specifically, the boxed warning explicitly cautioned that given the risk for serious psychiatric disturbances or neurologic AEs when used for malaria prophylaxis, “if psychiatric or neurologic symptoms occur, the drug should be discontinued and an alternative medication should be substituted.”6
Drug of Last Resort
By late 2013, partially on the basis of the boxed warning, the U.S. military declared mefloquine a “drug of last resort.”7,27 The U.S. Army Special Operations Command (USASOC) took the further step of prohibiting use of mefloquine altogether and, according to news reports, directed that medical and command staff assess whether certain personnel experiencing AEs from the drug may mistakenly have been thought to be malingering, have PTSD, or have other psychological problems.28
As the boxed warning and the USASOC order suggest, veterans exposed to mefloquine may have incurred a broad range of neurologic or psychiatric disorders or had others aggravated during military service as a result of their use of the drug. The effects of mefloquine may have confounded the diagnosis of neurologic or psychiatric disorders related to military service.26,29 As these AEs may be a direct result of mefloquine prescribed during military service, those with disabling diagnoses consistent with these effects may be entitled to claim disability compensation through the VA.
Of potential significant relevance to this adjudication process is a memorandum written in early 2012, in which the military conceded:
Some deploying Service members have been provided mefloquine for malaria prophylaxis without appropriate documentation in their medical records and without proper screening for contraindications. In addition, not all individuals have been provided the required mefloquine medication guide and wallet information card, as required by the Food and Drug Administration. 24
Veterans claiming a service-connected disability as a result of their use of mefloquine should therefore not always be expected to have documentation of prescribing in their military medical records. Although the VA could consider denying such claims for absence of proof of a nexus to military service, in light of this memorandum, the VA may need to consider other evidence of plausible exposure, including veteran testimony and deployment history.
It is also conceivable that the VA could consider denying such claims by arguing that the veteran directly contributed to the disability through willful misconduct by not adhering to mefloquine label guidance. However, as this memorandum establishes that mefloquine use was frequently directed without communication of the drug label precautions and warnings, the VA should consider that veterans claiming a service-connected disability frequently will not have known or otherwise been unable to discontinue the medication at the onset of prodromal symptoms.
It is also possible that the VA might deny claims on the basis that the claimed disabilities reflect preexisting conditions. However, as the memorandum establishes, use of mefloquine also was occasionally inappropriately directed to those with documented contraindications to the medication, who would have increased risk of AEs. As a result, veterans with preexisting neurologic or psychiatric conditions or disorders who nonetheless were prescribed mefloquine may reasonably claim these were aggravated during military service.
Conclusion
As this case suggests, in the coming years, as awareness of the chronic AEs of mefloquine increases among the veteran population, claims related to prior use of the drug are likely to increase and become of significant interest to the VA. Veterans with plausible exposure to mefloquine with neuropsychiatric disabilities who have yet to file a claim may be able to do so, and those veterans whose claims for service-connection were unfavorably adjudicated may be able to reopen their claims on the basis of the new material evidence in the 2012 military memorandum and the 2013 boxed warning.
This case report also suggests that service-connected disability claims arising from chronic neuropsychiatric AEs from mefloquine may prove to be of significant financial consequence. Further research to better define both the extent of prior mefloquine use among U.S. military personnel and the nature and prevalence of those chronic neurologic and psychiatric disorders caused by the drug would be helpful in informing improvements in the efficient and fair adjudication of such service-connected disability claims.
Mefloquine is a synthetic antimalarial drug structurally related to quinine. The drug was developed by the Walter Reed Army Institute of Research during a decades-long program that started during the Vietnam War in response to concerns of rising resistance to chloroquine.1
The prelicensing clinical testing of mefloquine, originally known as WR 142,490, was conducted in part among U.S. military service members.2,3 Soon after receiving FDA approval in 1989, under the brand name Lariam, it was recommended for use within the U.S. military.4 Over the following 2 decades, mefloquine was a common exposure during military deployments to malaria endemic areas.
Although the original U.S. mefloquine drug label noted that neuropsychiatric reactions could occur with use, changes to the drug label mandated by the FDA in July 2013, including a black box warning, described a potential for these to persist long after the drug has been discontinued.5,6 These changes have served to reinforce earlier U.S. military policy changes beginning in 2009 that deprioritized use of the drug in favor of safer and better-tolerated antimalarials. Consequently, more than a quarter century after its introduction, mefloquine now is only rarely prescribed to members of the U.S. military.7
In addition to limiting current use of the drug, the recent boxed warning may have important implications for service-connected disability claims adjudication by the VA for veterans previously exposed to the drug. This report presents a case of a nondeployed veteran exposed to mefloquine during an early military postmarketing study who developed chronic neuropsychiatric symptoms linked to the drug that were recently deemed service-connected. This report concludes with some comments on the likely implications of this case for future similar disability claims.
Case Presentation
In 2014, a 56-year-old nondeployed U.S. Marine Corps veteran submitted a claim to the VA for disabling conditions. The veteran alleged these conditions were due to his exposure to mefloquine while in military service more than 2 decades earlier. The veteran enlisted in 1975 and experienced a motor vehicle accident with prolonged loss of consciousness in 1978 but had no other significant medical history.
Thirteen years later, stationed in Hawaii in 1991, he was encouraged to volunteer for a double-blinded postmarketing study, evaluating the adverse effects (AEs) of chloroquine and mefloquine.8 As documentation following the trial revealed, he was randomly assigned to the mefloquine arm and received a loading dose of 250 mg daily for 3 days, followed by 250 mg per week for 11 weeks.
During the study he experienced insomnia, abnormal dreams, and nightmares. He also developed symptoms of anxiety, depression, cognitive dysfunction, and changes in personality—including anger and irritability—that were severe enough to be noted by his family members. The patient had not been advised of the significance of these symptoms and therefore did not report them during the clinical trial, nor did he report their intermittent presence after the study’s conclusion through his retirement in 1996, fearing adverse career consequences. Subsequent exacerbations of these chronic symptoms later contributed to the patient’s loss of civilian employment in 2010.
After becoming aware of the 2013 boxed warning that these chronic symptoms could be due to his earlier exposure to mefloquine, the veteran sought evaluation by a VA clinician. On evaluation, the clinician noted no history of deployment, and no history of posttraumatic stress disorder (PTSD) criteria A stressors, and posited that the veteran’s chronic neuropsychiatric symptoms were most likely a consequence of his earlier use of mefloquine. The VA subsequently awarded the veteran 50% disability for an anxiety disorder characterized by chronic sleep impairment and frequent panic attacks, attributing these to his service-connected use of the drug.
Discussion
Although the original 1989 FDA-approved mefloquine label had warned to discontinue the drug if specific prodromal symptoms of “anxiety, depression, restlessness or confusion” were noted,as illustrated by this case, this guidance was not always consistently communicated to service members.5 Indeed, few service members in the 1991 military postmarketing study discontinued the medication even after reporting such symptoms.8 Vivid dreams, often described as “terrifying nightmares with technicolor clarity” were reported by 7% of study participants. Similarly, concentration problems were reported in 5%; irritability in 4%; anger and moodiness each in 1%; and insomnia in 25%. Two study participants, after failing to discontinue mefloquine at the onset of severe insomnia, were later hospitalized for severe depression and suicidal thoughts, later deemed due to preexisting conditions. Despite these seemingly unfavorable results, mefloquine was nonetheless deemed well tolerated.8
Military Use of Mefloquine
Beginning in 1992, use of mefloquine for prophylaxis of malaria was then widely directed within the U.S. military during operations in Somalia. There, a majority of personnel received mefloquine under conditions of command-directed and directly observed administration of the drug.9,10 Again, drug label guidance describing the prodromal psychiatric symptoms that should have prompted discontinuation of mefloquine were either not consistently adhered to or not communicated. In one Somalia-era study, only 1 in 344 service members, or 0.3%, discontinued the drug.11
Throughout the remainder of the 1990s, mefloquine remained the antimalarial drug of choice for most U.S. military operations, and when combat began in Afghanistan in 2001, widespread use was also directed there.12,13 The following year, after national attention was directed to concerns of severe behavioral toxicity from the drug among personnel returning from Afghanistan, the manufacturer issued subtle changes to the mefloquine label warnings.5,14
These label changes adjusted the previously exclusive list of prodromal symptoms to an illustrative list, emphasizing that “if psychiatric symptoms such as [emphasis added] acute anxiety, depression, restlessness or confusion occur, these may be considered prodromal to a more serious event. In these cases, the drug must be discontinued and an alternative medication should be substituted.”5
In 2001 a randomized double-blinded trial demonstrated that symptoms of anxiety and depression occurred in at least 4% of mefloquine users, insomnia in 13%, and abnormal dreams in 14%. Nevertheless, an Army memorandum issued soon after the labeling change significantly understated the known risks of developing such psychiatric symptoms, erroneously claiming that these occurred from mefloquine only “at a rate of one per 2,000 to 13,000 persons.”15,16
Updated FDA Guidelines
In 2003, with widespread use of the drug being again directed during operations in Iraq, the FDA required that all mefloquine prescriptions be accompanied by a patient medication guide with warnings echoing those of the drug label that users seek medical attention should “possible signs of more serious mental problems” develop.5,17 However, surveys suggested that few U.S. service members received these warnings or even verbal instructions to that effect.17-19 During later congressional testimony, a service member who had experienced 3 weeks of nightmares prior to self-discontinuing the drug testified “every soldier I know has problems with it.”20
In response, a senior military medical leader—failing to recognize that the nightmares the soldier reported were in fact psychiatric symptoms and possible signs of more serious mental problems that required the drug’s discontinuation—may have undermined the FDA-directed warnings by dismissing the soldier’s testimony as “perception,” maintaining instead “that perceptions can become realities” should it become “held that this medication is widely problematic.”20
Given that certain preexisting conditions, including anxiety and depression, were known to confound recognition of incident psychiatric symptoms that required discontinuation of the drug, the original 1989 mefloquine label had noted that the drug should be used with caution in such patients. In subsequent years, this language was strengthened, and such patients were formally contraindicated the drug.21
Citing formal policy, senior military medical leaders provided assurance during congressional testimony that service members with these conditions would not be prescribed mefloquine.16,18,20 However, later analysis of a large group of deployed service members revealed that 1 in 7 with contraindications to mefloquine had been prescribed the drug contrary to drug label guidance.21
Black Box Warning
With growing recognition of the challenges in using mefloquine as directed by the drug label, a 2009 Army policy memorandum prioritized the use of safer and better-tolerated daily medications, such as doxycycline and atovaquone-proguanil, and stated that “[m]efloquine should only be used for personnel with contraindications to doxycycline.”22 This policy was extended throughout the other military services later that year.23 After concerns were raised that service members were still being prescribed the drug contrary to policy, further restrictions were formalized in early 2013 prior to the boxed warning, with mefloquine reserved for those only “with intolerance or contraindications” to the first-line drugs.24,25
In a later memorandum announcing the July 2013 boxed warning, the military revealed that the number of active-duty personnel prescribed mefloquine had steadily decreased in prior years from 17,361 in 2008 to only 2,040 in 2012.7 Although the military has not released precise figures on the number of U.S. military personnel exposed to mefloquine since the drug’s introduction, based on a variety of sources, the total is likely to far exceed 100,000.7,26
The major changes to the mefloquine label in 2013, including the boxed warning, clarified that neurologic and psychiatric effects from mefloquine could “persist after mefloquine has been discontinued.” The accompanying FDA Drug Safety Communication noted neurologic AEs from the drug, which include but are not limited to “dizziness, loss of balance, or ringing in the ears,” could “occur at any time during drug use, and can last for months to years after the drug is stopped or can be permanent.”6 Other neurologic symptoms listed in the drug label include vertigo, hearing impairment, headache, visual disturbances, sensory and motor neuropathies, including paresthesia, tremor, ataxia, convulsions, and encephalopathy.6
The updated drug label also made clear that psychiatric AEs from mefloquine, such as anxiety, paranoia, and depression to hallucinations and psychotic behavior, “have been reported to continue for months or years after mefloquine has been stopped.” Other psychiatric symptoms listed in the drug label include memory impairment, confusion, somnolence, insomnia, abnormal dreams, aggression, agitation, restlessness, mood swings, panic attacks, psychosis, and suicidal ideation.6
The 2013 boxed warning also served to reemphasize guidance first articulated in 2002 that any psychiatric symptom—presumably including abnormal dreams and insomnia—occurring during mefloquine use should be considered prodromal, prompting the drug’s immediate discontinuation.5 Specifically, the boxed warning explicitly cautioned that given the risk for serious psychiatric disturbances or neurologic AEs when used for malaria prophylaxis, “if psychiatric or neurologic symptoms occur, the drug should be discontinued and an alternative medication should be substituted.”6
Drug of Last Resort
By late 2013, partially on the basis of the boxed warning, the U.S. military declared mefloquine a “drug of last resort.”7,27 The U.S. Army Special Operations Command (USASOC) took the further step of prohibiting use of mefloquine altogether and, according to news reports, directed that medical and command staff assess whether certain personnel experiencing AEs from the drug may mistakenly have been thought to be malingering, have PTSD, or have other psychological problems.28
As the boxed warning and the USASOC order suggest, veterans exposed to mefloquine may have incurred a broad range of neurologic or psychiatric disorders or had others aggravated during military service as a result of their use of the drug. The effects of mefloquine may have confounded the diagnosis of neurologic or psychiatric disorders related to military service.26,29 As these AEs may be a direct result of mefloquine prescribed during military service, those with disabling diagnoses consistent with these effects may be entitled to claim disability compensation through the VA.
Of potential significant relevance to this adjudication process is a memorandum written in early 2012, in which the military conceded:
Some deploying Service members have been provided mefloquine for malaria prophylaxis without appropriate documentation in their medical records and without proper screening for contraindications. In addition, not all individuals have been provided the required mefloquine medication guide and wallet information card, as required by the Food and Drug Administration. 24
Veterans claiming a service-connected disability as a result of their use of mefloquine should therefore not always be expected to have documentation of prescribing in their military medical records. Although the VA could consider denying such claims for absence of proof of a nexus to military service, in light of this memorandum, the VA may need to consider other evidence of plausible exposure, including veteran testimony and deployment history.
It is also conceivable that the VA could consider denying such claims by arguing that the veteran directly contributed to the disability through willful misconduct by not adhering to mefloquine label guidance. However, as this memorandum establishes that mefloquine use was frequently directed without communication of the drug label precautions and warnings, the VA should consider that veterans claiming a service-connected disability frequently will not have known or otherwise been unable to discontinue the medication at the onset of prodromal symptoms.
It is also possible that the VA might deny claims on the basis that the claimed disabilities reflect preexisting conditions. However, as the memorandum establishes, use of mefloquine also was occasionally inappropriately directed to those with documented contraindications to the medication, who would have increased risk of AEs. As a result, veterans with preexisting neurologic or psychiatric conditions or disorders who nonetheless were prescribed mefloquine may reasonably claim these were aggravated during military service.
Conclusion
As this case suggests, in the coming years, as awareness of the chronic AEs of mefloquine increases among the veteran population, claims related to prior use of the drug are likely to increase and become of significant interest to the VA. Veterans with plausible exposure to mefloquine with neuropsychiatric disabilities who have yet to file a claim may be able to do so, and those veterans whose claims for service-connection were unfavorably adjudicated may be able to reopen their claims on the basis of the new material evidence in the 2012 military memorandum and the 2013 boxed warning.
This case report also suggests that service-connected disability claims arising from chronic neuropsychiatric AEs from mefloquine may prove to be of significant financial consequence. Further research to better define both the extent of prior mefloquine use among U.S. military personnel and the nature and prevalence of those chronic neurologic and psychiatric disorders caused by the drug would be helpful in informing improvements in the efficient and fair adjudication of such service-connected disability claims.
1. Tigertt WD. The army malaria research program. Ann Intern Med. 1969;70(1):150-153.
2. Trenholme CM, Williams RL, Desjardins RE, et al. Mefloquine (WR 142,490) in the treatment of human malaria. Science. 1975;190(4216):792-794.
3. Shanks GD, Karwacki J, Kanesa-thasan N, et al. Diseases transmitted primarily by arthropod vectors. In: Kelley PW, ed. Military Preventive Medicine: Mobilization and Deployment. Vol 2. Washington, DC: Borden Institute; 2005:803-935.
4. Armed Forces Epidemiological Board. Memorandum. Subject: Recommendations on Mefloquine Chemoprophylaxis for Military Personnel. Published October 3, 1989.
5. Nevin RL, Byrd AM. Neuropsychiatric adverse reactions to mefloquine: a systematic comparison of prescribing and patient safety guidance in the US, UK, Ireland, Australia, New Zealand, and Canada. Neurol Ther. 2016;5(1):69-83.
6. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA approves label changes for antimalarial drug mefloquine hydrochloride due to risk of serious psychiatric and nerve side effects. http://www.fda.gov/Drugs/DrugSafety/ucm362227.htm. Published July 29, 2013. Accessed August 26, 2016.
7. Assistant Secretary of Defense for Health Affairs. Memorandum. Subject: Notification for Healthcare Providers of Mefloquine Boxed Warning. Published August 12, 2013.
8. Boudreau E, Schuster B, Sanchez J, et al. Tolerability of prophylactic Lariam regimens. Trop Med Parasitol. 1993;44(3):257-265.
9. Wallace MR, Sharp TW, Smoak B, et al. Malaria among United States troops in Somalia. Am J Med. 1996;100(1):49-55.
10. Smoak BL, Writer JV, Keep LW, Cowan J, Chantelois JL. The effects of inadvertent exposure of mefloquine chemoprophylaxis on pregnancy outcomes and infants of US Army servicewomen. J Infect Dis. 1997;176(3):831-833.
11. Sánchez JL, DeFraites RF, Sharp TW, Hanson RK. Mefloquine or doxycycline prophylaxis in US troops in Somalia. Lancet. 1993;341(8851):1021-1022.
12. Jones R, Kunsman G, Levine B, Smith M, Stahl C. Mefloquine distribution in postmortem cases. Forensic Sci Int. 1994;68(1):29-32.
13. Kotwal RS, Wenzel RB, Sterling RA, Porter WD, Jordan NN, Petruccelli BP. An outbreak of malaria in US Army Rangers returning from Afghanistan. JAMA. 2005;293(2):212-216.
14. Hess BP. Army fears rebellion on Lariam. United Press International. http://www.upi.com/Business_News/Security-Industry/2002/08/29/Analysis-Army-fears-rebellion-on-Lariam/UPI-39351030635930. Published August 29, 2002. Accessed August 29, 2016.
15. Overbosch D, Schilthuis H, Bienzle U, et al; Malarone International Study Team. Atovaquone-proguanil versus mefloquine for malaria prophylaxis in nonimmune travelers: results from a randomized, double-blind study. Clin Infect Dis. 2001;33(7):1015-1021.
16. U.S. Army Surgeon General. Memorandum. Subject: Updated Health Care Provider Information on Use of Mefloquine Hydrochloride for Malaria Prophylaxis. October 3, 2002.
17. Associated Press. Hallucinations linked to drug given to troops. http://www.nbcnews.com/id/6947472/ns/health-mental_health/t/hallucinations-linked-drug-given-troops. Published February 14, 2005. Accessed August 26, 2016.
18. Benjamin M. Army sent mentally ill troops to Iraq. United Press International. http://www.upi.com/Business_News/Security-Industry/2004/03/12/Army-sent-mentally-ill-troops-to-Iraq/UPI-97331079131967. Published March 12, 2004. Accessed August 26, 2016.
19. Fleet M, Mann J. Military’s use of malaria drug in question. http://edition.cnn.com/2004/HEALTH/05/20/lariam. Published May 21, 2004. Accessed August 26, 2016.
20. 108th Congress. Hearing on National Defense Authorization Act for Fiscal Year 2005 - H.R. 4200, February 25, 2004. http://commdocs.house.gov/committees/security/has056270.000/has056270_0f.htm. Accessed August 26, 2016.
21. Nevin RL. Mefloquine prescriptions in the presence of contraindications: prevalence among US military personnel deployed to Afghanistan, 2007. Pharmacoepidemiol Drug Saf. 2010;19(2):206-210.
22. U.S. Army Surgeon General. Memorandum. Subject: Updated Guidance on the Use of Mefloquine for Malaria Prophylaxis. February 2, 2009.
23. Assistant Secretary of Defense for Health Affairs. Memorandum. Subject: Policy Memorandum on the Use of Mefloquine (Lariam) in Malaria Prophylaxis. HA Policy 09-017. http://www.health.mil/~/media/MHS/Policy%20Files/Import/09-017.ashx. September 4, 2009. Accessed August 26, 2016.
24. Assistant Secretary of Defense for Health Affairs. Memorandum. Subject: Service Review of Mefloquine Prescribing Practices. January 17, 2012.
25. Assistant Secretary of Defense for Health Affairs. Memorandum. Subject: Guidance on Medications for Prophylaxis of Malaria. April 15, 2013.
26. Nevin RL. Mefloquine and posttraumatic stress disorder. In: Ritchie EC, ed. Forensic and Ethical Issues in Military Behavioral Health. Washington, DC: Borden Institute; 2014:275-296.
27. Pellerin C. DOD mefloquine policy mirrors FDA update on malaria drug. American Forces Press Service. http://archive.defense.gov/news/newsarticle.aspx?id=120857. Published September 26, 2013. Accessed August 26, 2016.
28. Jelinek P. Elite Army units to stop taking anti-malarial drug. Associated Press. http://www.military.com/daily-news/2013/09/19/elite-army-units-to-stop-taking-anti-malarial-drug.html. Published September 19, 2013. Accessed August 26, 2016.
29. Nevin RL, Ritchie EC. The Mefloquine intoxication syndrome: a significant potential confounder in the diagnosis and management of PTSD and other chronic deployment-related neuropsychiatric disorders. In: Ritchie EC, ed. Posttraumatic Stress Disorder and Related Disorders in Combat Veterans. Cham, Switzerland: Springer; 2015:257-278.
1. Tigertt WD. The army malaria research program. Ann Intern Med. 1969;70(1):150-153.
2. Trenholme CM, Williams RL, Desjardins RE, et al. Mefloquine (WR 142,490) in the treatment of human malaria. Science. 1975;190(4216):792-794.
3. Shanks GD, Karwacki J, Kanesa-thasan N, et al. Diseases transmitted primarily by arthropod vectors. In: Kelley PW, ed. Military Preventive Medicine: Mobilization and Deployment. Vol 2. Washington, DC: Borden Institute; 2005:803-935.
4. Armed Forces Epidemiological Board. Memorandum. Subject: Recommendations on Mefloquine Chemoprophylaxis for Military Personnel. Published October 3, 1989.
5. Nevin RL, Byrd AM. Neuropsychiatric adverse reactions to mefloquine: a systematic comparison of prescribing and patient safety guidance in the US, UK, Ireland, Australia, New Zealand, and Canada. Neurol Ther. 2016;5(1):69-83.
6. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA approves label changes for antimalarial drug mefloquine hydrochloride due to risk of serious psychiatric and nerve side effects. http://www.fda.gov/Drugs/DrugSafety/ucm362227.htm. Published July 29, 2013. Accessed August 26, 2016.
7. Assistant Secretary of Defense for Health Affairs. Memorandum. Subject: Notification for Healthcare Providers of Mefloquine Boxed Warning. Published August 12, 2013.
8. Boudreau E, Schuster B, Sanchez J, et al. Tolerability of prophylactic Lariam regimens. Trop Med Parasitol. 1993;44(3):257-265.
9. Wallace MR, Sharp TW, Smoak B, et al. Malaria among United States troops in Somalia. Am J Med. 1996;100(1):49-55.
10. Smoak BL, Writer JV, Keep LW, Cowan J, Chantelois JL. The effects of inadvertent exposure of mefloquine chemoprophylaxis on pregnancy outcomes and infants of US Army servicewomen. J Infect Dis. 1997;176(3):831-833.
11. Sánchez JL, DeFraites RF, Sharp TW, Hanson RK. Mefloquine or doxycycline prophylaxis in US troops in Somalia. Lancet. 1993;341(8851):1021-1022.
12. Jones R, Kunsman G, Levine B, Smith M, Stahl C. Mefloquine distribution in postmortem cases. Forensic Sci Int. 1994;68(1):29-32.
13. Kotwal RS, Wenzel RB, Sterling RA, Porter WD, Jordan NN, Petruccelli BP. An outbreak of malaria in US Army Rangers returning from Afghanistan. JAMA. 2005;293(2):212-216.
14. Hess BP. Army fears rebellion on Lariam. United Press International. http://www.upi.com/Business_News/Security-Industry/2002/08/29/Analysis-Army-fears-rebellion-on-Lariam/UPI-39351030635930. Published August 29, 2002. Accessed August 29, 2016.
15. Overbosch D, Schilthuis H, Bienzle U, et al; Malarone International Study Team. Atovaquone-proguanil versus mefloquine for malaria prophylaxis in nonimmune travelers: results from a randomized, double-blind study. Clin Infect Dis. 2001;33(7):1015-1021.
16. U.S. Army Surgeon General. Memorandum. Subject: Updated Health Care Provider Information on Use of Mefloquine Hydrochloride for Malaria Prophylaxis. October 3, 2002.
17. Associated Press. Hallucinations linked to drug given to troops. http://www.nbcnews.com/id/6947472/ns/health-mental_health/t/hallucinations-linked-drug-given-troops. Published February 14, 2005. Accessed August 26, 2016.
18. Benjamin M. Army sent mentally ill troops to Iraq. United Press International. http://www.upi.com/Business_News/Security-Industry/2004/03/12/Army-sent-mentally-ill-troops-to-Iraq/UPI-97331079131967. Published March 12, 2004. Accessed August 26, 2016.
19. Fleet M, Mann J. Military’s use of malaria drug in question. http://edition.cnn.com/2004/HEALTH/05/20/lariam. Published May 21, 2004. Accessed August 26, 2016.
20. 108th Congress. Hearing on National Defense Authorization Act for Fiscal Year 2005 - H.R. 4200, February 25, 2004. http://commdocs.house.gov/committees/security/has056270.000/has056270_0f.htm. Accessed August 26, 2016.
21. Nevin RL. Mefloquine prescriptions in the presence of contraindications: prevalence among US military personnel deployed to Afghanistan, 2007. Pharmacoepidemiol Drug Saf. 2010;19(2):206-210.
22. U.S. Army Surgeon General. Memorandum. Subject: Updated Guidance on the Use of Mefloquine for Malaria Prophylaxis. February 2, 2009.
23. Assistant Secretary of Defense for Health Affairs. Memorandum. Subject: Policy Memorandum on the Use of Mefloquine (Lariam) in Malaria Prophylaxis. HA Policy 09-017. http://www.health.mil/~/media/MHS/Policy%20Files/Import/09-017.ashx. September 4, 2009. Accessed August 26, 2016.
24. Assistant Secretary of Defense for Health Affairs. Memorandum. Subject: Service Review of Mefloquine Prescribing Practices. January 17, 2012.
25. Assistant Secretary of Defense for Health Affairs. Memorandum. Subject: Guidance on Medications for Prophylaxis of Malaria. April 15, 2013.
26. Nevin RL. Mefloquine and posttraumatic stress disorder. In: Ritchie EC, ed. Forensic and Ethical Issues in Military Behavioral Health. Washington, DC: Borden Institute; 2014:275-296.
27. Pellerin C. DOD mefloquine policy mirrors FDA update on malaria drug. American Forces Press Service. http://archive.defense.gov/news/newsarticle.aspx?id=120857. Published September 26, 2013. Accessed August 26, 2016.
28. Jelinek P. Elite Army units to stop taking anti-malarial drug. Associated Press. http://www.military.com/daily-news/2013/09/19/elite-army-units-to-stop-taking-anti-malarial-drug.html. Published September 19, 2013. Accessed August 26, 2016.
29. Nevin RL, Ritchie EC. The Mefloquine intoxication syndrome: a significant potential confounder in the diagnosis and management of PTSD and other chronic deployment-related neuropsychiatric disorders. In: Ritchie EC, ed. Posttraumatic Stress Disorder and Related Disorders in Combat Veterans. Cham, Switzerland: Springer; 2015:257-278.