User login
Disseminated Superficial Actinic Porokeratosis Treated With Ingenol Mebutate Gel 0.05%
Disseminated superficial actinic porokeratosis (DSAP) is a chronic condition characterized by numerous atrophic papules and patches with a distinctive peripheral keratotic ridge, typically found on sun-exposed areas.1,2 Treatment of DSAP is warranted not only for cosmetic and symptomatic benefits but also to prevent malignant transformation.3,4 Successful treatment of DSAP often is difficult and frequently requires the use of multiple modalities. Ingenol mebutate gel 0.05% is a topical medication primarily used for the treatment of actinic keratosis (AK) by inducing cell death.5 We report a case of DSAP treated effectively with ingenol mebutate gel 0.05%.
Case Report
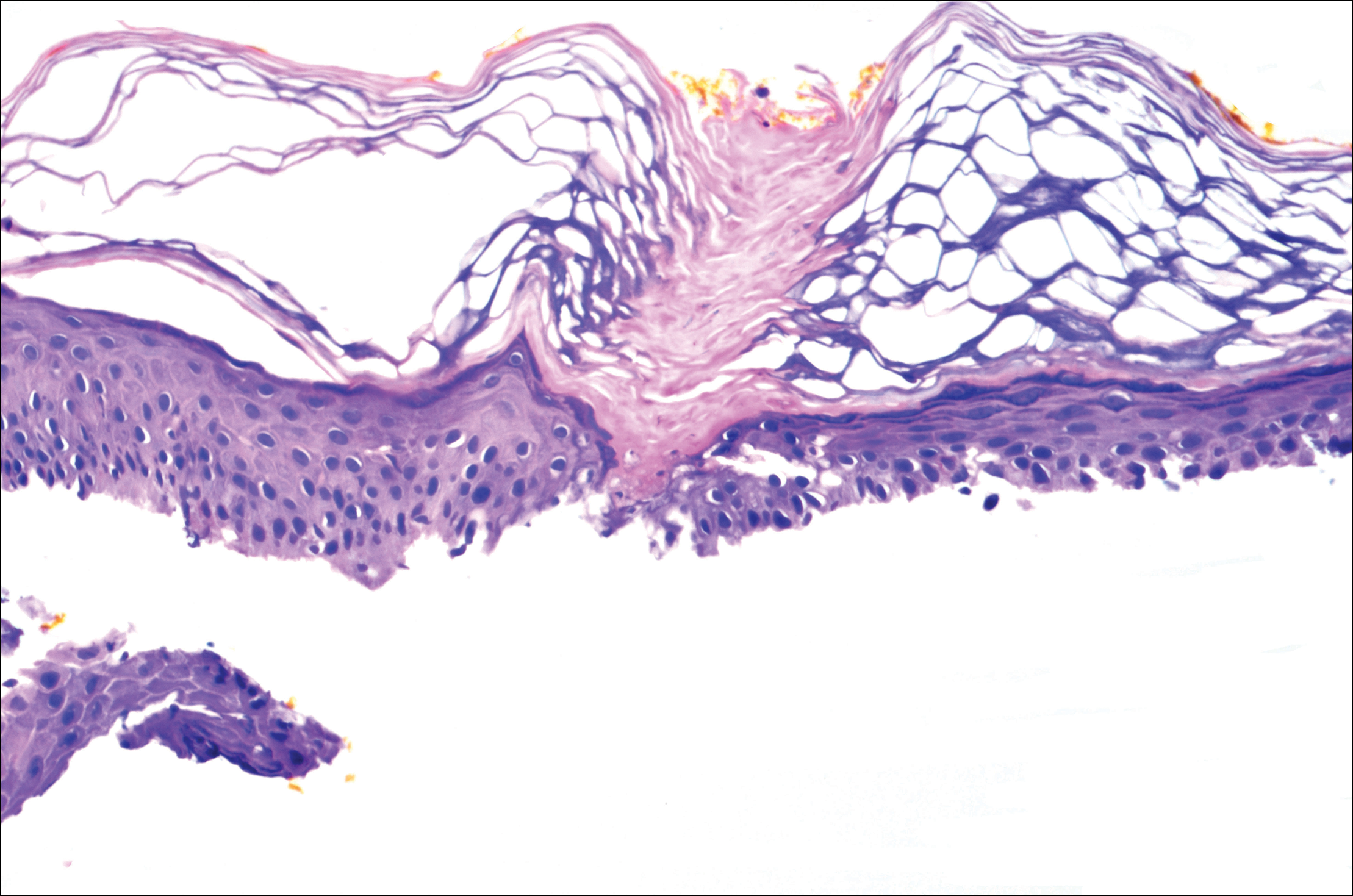
A 37-year-old woman was referred to the dermatology department for counseling for pseudoxanthoma elasticum (PXE), which had been proven on biopsy by an outside dermatologist 2 years prior. Physical examination revealed yellow papules on the neck that were characteristic of PXE, but no lesions were noted on the arms or legs. The only other cutaneous finding was a soft nodule on the right hip consistent with a lipoma. The patient returned to our institution 6 years later with lesions on both lower legs. She reported that these lesions had been present for 3 years and were exacerbated by sun exposure. On physical examination, multiple scattered, erythematous, annular, scaling papules and plaques were noted on the bilateral legs. A biopsy showed the histopathologic findings of DSAP (Figure 1). The patient had no family history of DSAP or PXE.
To determine the best treatment modality, we treated 4 test areas on both upper and lower legs: one with trichloroacetic acid (TCA), one with cryotherapy, one with imiquimod cream 5%, and one with tretinoin cream 0.1%. The patient returned 4 weeks later and showed modest response to TCA, cryotherapy, and tretinoin cream. Because cryotherapy was determined to be most effective, 20 more lesions were frozen at that visit. Over the next 2 years, the patient was treated with TCA, imiquimod cream 5%, and tretinoin cream 0.1%, but all ultimately proved ineffective for DSAP.
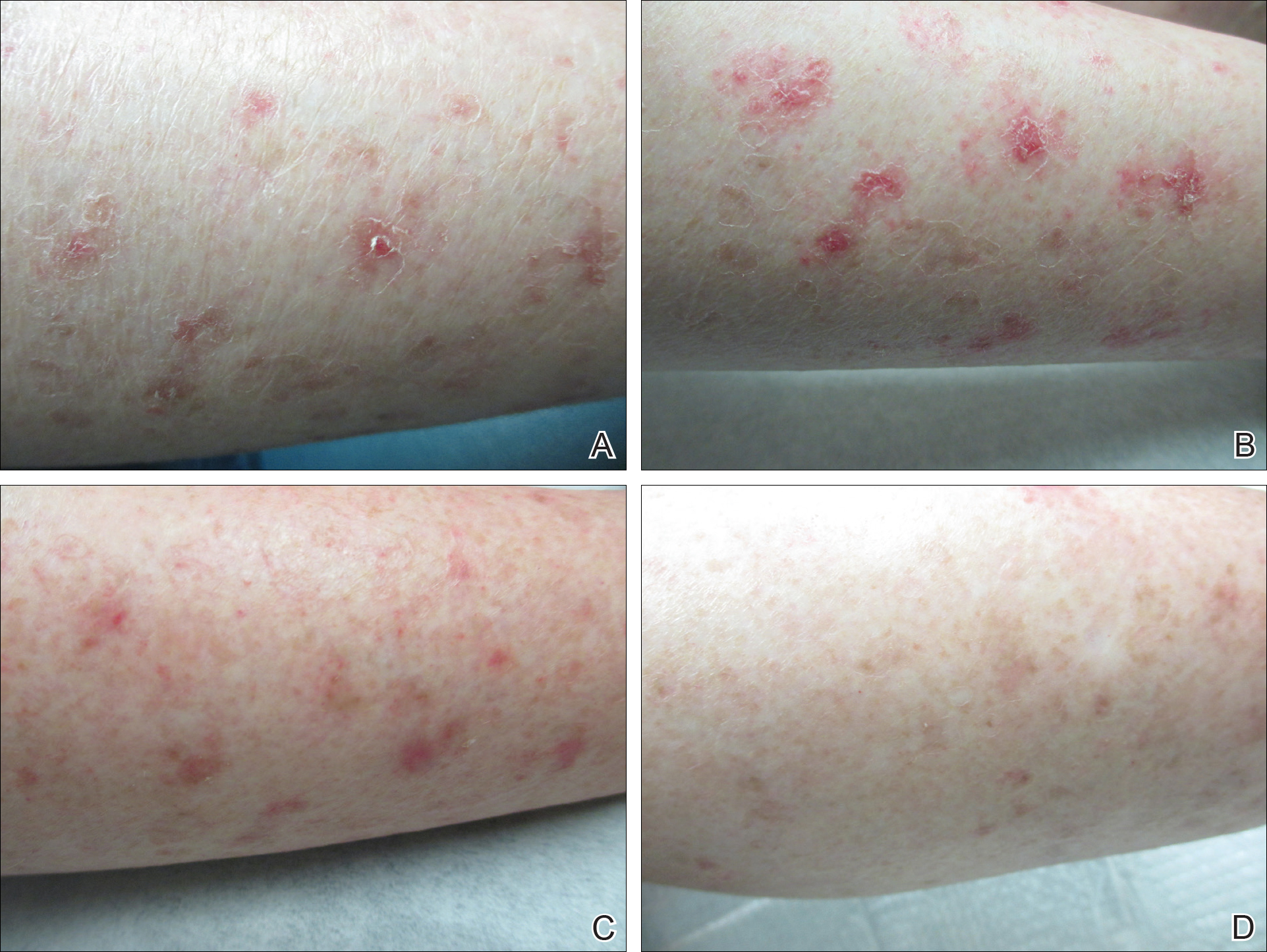
The patient returned 2 years after treatment failure (age 47 years) and was prescribed ingenol mebutate gel 0.05% for 2 days over an area of 25 cm2 on the right lower leg (Figure 2A). She returned for follow-up at days 3, 15, 30, and 60. At day 3, the patient developed an inflammatory response to the medication with moderate erythema and scaling of individual lesions. No vesiculation, pustulation, edema, or ulceration was exhibited (Figure 2B). At day 30, there was a marked reduction in scaling with some postinflammatory erythema (Figure 2C). At day 60, much of the erythema had faded and the scale remained notably reduced (Figure 2D).
Comment
Disseminated superficial actinic porokeratosis is the most common subtype of porokeratosis, a keratinization disorder. There are 6 subtypes of porokeratosis identified in the literature: DSAP, disseminated superficial porokeratosis, classic porokeratosis of Mibelli, porokeratosis plantaris palmaris et disseminata, linear porokeratosis, and punctate porokeratosis.6 Disseminated superficial actinic porokeratosis has a female predominance (1.8:1 ratio)7 and generally appears in the third or fourth decades of life. Clonal proliferations of atypical keratinocytes have been implicated in the etiology of DSAP; however, the exact pathogenesis is unclear. Risk factors for DSAP include genetic susceptibility (eg, autosomal-dominant inheritance pattern), exposure to UV radiation, and drug-related immunosuppression or immunodeficiency.7 Other proposed etiologic risk factors include trauma and infection.8 Clinical diagnosis of DSAP is confirmed by the histological presence of a cornoid lamella (a thin column ofparakeratotic cells), a thinning epidermis, an absent or thinned granular cell layer, and a prominent dermal lymphocytic infiltrate.9,10
Disseminated superficial actinic porokeratosis clinically presents as small atrophic scaly papules and/or patches with raised peripheral ridges symmetrically dispersed on sun-exposed areas of the arms, legs, back, and shoulders. Although these lesions are extensive, they typically spare the mucous membranes, palms, and soles11; only a small percentage of cases report facial lesions,12 which often are asymptomatic but cosmetically bothersome. Additionally, approximately half of patients report symptoms of pruritus and/or stinging,13 thus treatment of DSAP is mainly indicated for symptomatic relief and cosmetic purposes. Malignant degeneration14,15 occurs in approximately 7.5% to 11% of porokeratosis cases,10,16 warranting treatment for preventative measures.
Management of DSAP is dependent on the extent of the disease and the level of concern for malignant transformation. Localized disease can be treated with cryotherapy, CO2 laser, and/or ablative techniques (eg, excision, curettage, dermabrasion) with variable degrees of success but high risk for scarring.1 More extensive disease requires treatment with topical retinoids, topical 5-fluorouracil, imiquimod cream 5%, diclofenac gel 3%, topical vitamin D3 analogues, and photodynamic therapy.1 Several other therapies have been reported in the literature with partial and/or complete success, including systemic retinoids (eg, acitretin), Q-switched ruby laser, Nd:YAG laser, fractional photothermolysis, Grenz rays, pulsed dye laser, fractional photothermolysis, topical corticosteroids, and fluor-hydroxy pulse peel.6 Although there is an extensive array of therapies for DSAP, treatment results are variable with mostly limited success. Successful treatment of DSAP is difficult and often requires the use of multiple modalities.
Ingenol mebutate is the active compound found in the sap of Euphorbia peplus used for the topical treatment of various skin conditions, including AKs.17 Ingenol mebutate gel 0.05% once daily for 2 days has been approved by the US Food and Drug Administration for the topical treatment of AKs. The mechanism of action of ingenol mebutate in AK therapy is not yet fully understood. In vivo and in vitro models have demonstrated both an induction of local lesion cell death and promotion of lesion-specific inflammatory response.18 When used in the treatment of AKs, ingenol mebutate gel 0.05% may cause a mild to moderate localized inflammatory response (eg, erythema, flaking/scaling, crusting, vesiculation/pustulation, erosion/ulceration, edema).
Our case is a rare report of successful treatment of DSAP with ingenol mebutate gel 0.05%. We found that treatment with ingenol mebutate gel 0.05% resulted in clinical improvement of DSAP lesions with minimal discomfort and good cosmetic response. This 2-day regimen is easy to use and patient friendly, improving medication compliance in such a cumbersome disease. We hope this case suggests that ingenol mebutate gel 0.05% could be a useful treatment alternative for DSAP, but future clinical studies should be conducted.
- Martin-Clavijo A, Kanelleas A, Vlachou C, et al. Porokeratoses. In: Lebwohl M, Heymann WR, Berth-Jones J, et al, eds. Treatment of Skin Disease Comprehensive Therapeutic Strategies. 3rd ed. China: Elsevier Limited; 2010:584-586.
- Rouhani P, Fischer M, Meehan S, et al. Disseminated superficial actinic porokeratosis. Dermatology Online J. 2012;18:24.
- Sasson M, Krain AD. Porokeratosis and cutaneous malignancy. a review. Dermatol Surg. 1996;22:339-342.
- Lee HR, Han TY, Son SJ, et al. Squamous cell carcinoma developing within lesions of disseminated superficial actinic porokeratosis. Ann Dermatol. 2011;23:536-538.
- Lebwohl M, Swanson N, Anderson LL, et al. Ingenol mebutate gel for actinic keratosis. N Engl J Med. 2012;366:1010-1019.
- O’Regan GM, Irvine AD. Porokeratosis. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Professional; 2012:442-446.
- Sertznig P, von Felbert V, Megahed M. Porokeratosis: present concepts. J Eur Acad Dermatol Venereol. 2012;26:404-412.
- Brauer JA, Mandal R, Walters R, et al. Disseminated superficial porokeratosis. Dermatology Online J. 2010;16:20.
- Tallon B. Porokeratosis pathology. DermNet New Zealand website. http://www.dermnet.org.nz/pathology/porokeratosis-path.html. Updated December 2016. Accessed January 12, 2017.
- Skupsky H, Skupsky J, Goldenberg G. Disseminated superficial actinic porokeratosis: a treatment review [published online October 22, 2010]. J Dermatolog Treat. 2012;23:52-56.
- Spencer LV. Porokeratosis. UpToDate web site. https://eresources.library.mssm.edu:3285/contents/porokeratosis?source=search_result&search=porokeratosis&selectedTitle=1~22. Updated September 1, 2016. Accessed April 3, 2017.
- Sawyer R, Picou KA. Facial presentation of disseminated superficial actinic porokeratosis. Ear Nose Throat J. 1989;68:57-59.
- Schwarz T, Seiser A, Gschnait F. Disseminated superficial “actinic” porokeratosis. J Am Acad Dermatol. 1984;11(4, pt 2):724-730.
- Maubec E, Duvillard P, Margulis A, et al. Common skin cancers in porokeratosis. Br J Dermatol. 2005;152:1389-1391.
- Lee HR, Han TY, Son SJ, et al. Squamous cell carcinoma developing within lesions of disseminated superficial actinic porokeratosis [published online November 3, 2011]. Ann Dermatol. 2011;23:536-538.
- Kumari S, Mathur M. Disseminated superficial actinic porokeratosis. Nepal J Dermatol Venereol Leprol. 2010;9:22-24.
- Lebwohl M, Shumack S, Stein Gold L, et al. Long-term follow-up study of ingenol mebutate gel for the treatment of actinic keratosis. JAMA Dermatol. 2013;149:666-670.
- Stahlhut M, Bertelsen M, Hoyer-Hansen M, et al. Ingenol mebutate: induced cell death patterns in normal and cancer epithelial cells. J Drugs Dermatol. 2012;11:1181-1192.
Disseminated superficial actinic porokeratosis (DSAP) is a chronic condition characterized by numerous atrophic papules and patches with a distinctive peripheral keratotic ridge, typically found on sun-exposed areas.1,2 Treatment of DSAP is warranted not only for cosmetic and symptomatic benefits but also to prevent malignant transformation.3,4 Successful treatment of DSAP often is difficult and frequently requires the use of multiple modalities. Ingenol mebutate gel 0.05% is a topical medication primarily used for the treatment of actinic keratosis (AK) by inducing cell death.5 We report a case of DSAP treated effectively with ingenol mebutate gel 0.05%.
Case Report
A 37-year-old woman was referred to the dermatology department for counseling for pseudoxanthoma elasticum (PXE), which had been proven on biopsy by an outside dermatologist 2 years prior. Physical examination revealed yellow papules on the neck that were characteristic of PXE, but no lesions were noted on the arms or legs. The only other cutaneous finding was a soft nodule on the right hip consistent with a lipoma. The patient returned to our institution 6 years later with lesions on both lower legs. She reported that these lesions had been present for 3 years and were exacerbated by sun exposure. On physical examination, multiple scattered, erythematous, annular, scaling papules and plaques were noted on the bilateral legs. A biopsy showed the histopathologic findings of DSAP (Figure 1). The patient had no family history of DSAP or PXE.
To determine the best treatment modality, we treated 4 test areas on both upper and lower legs: one with trichloroacetic acid (TCA), one with cryotherapy, one with imiquimod cream 5%, and one with tretinoin cream 0.1%. The patient returned 4 weeks later and showed modest response to TCA, cryotherapy, and tretinoin cream. Because cryotherapy was determined to be most effective, 20 more lesions were frozen at that visit. Over the next 2 years, the patient was treated with TCA, imiquimod cream 5%, and tretinoin cream 0.1%, but all ultimately proved ineffective for DSAP.
The patient returned 2 years after treatment failure (age 47 years) and was prescribed ingenol mebutate gel 0.05% for 2 days over an area of 25 cm2 on the right lower leg (Figure 2A). She returned for follow-up at days 3, 15, 30, and 60. At day 3, the patient developed an inflammatory response to the medication with moderate erythema and scaling of individual lesions. No vesiculation, pustulation, edema, or ulceration was exhibited (Figure 2B). At day 30, there was a marked reduction in scaling with some postinflammatory erythema (Figure 2C). At day 60, much of the erythema had faded and the scale remained notably reduced (Figure 2D).
Comment
Disseminated superficial actinic porokeratosis is the most common subtype of porokeratosis, a keratinization disorder. There are 6 subtypes of porokeratosis identified in the literature: DSAP, disseminated superficial porokeratosis, classic porokeratosis of Mibelli, porokeratosis plantaris palmaris et disseminata, linear porokeratosis, and punctate porokeratosis.6 Disseminated superficial actinic porokeratosis has a female predominance (1.8:1 ratio)7 and generally appears in the third or fourth decades of life. Clonal proliferations of atypical keratinocytes have been implicated in the etiology of DSAP; however, the exact pathogenesis is unclear. Risk factors for DSAP include genetic susceptibility (eg, autosomal-dominant inheritance pattern), exposure to UV radiation, and drug-related immunosuppression or immunodeficiency.7 Other proposed etiologic risk factors include trauma and infection.8 Clinical diagnosis of DSAP is confirmed by the histological presence of a cornoid lamella (a thin column ofparakeratotic cells), a thinning epidermis, an absent or thinned granular cell layer, and a prominent dermal lymphocytic infiltrate.9,10
Disseminated superficial actinic porokeratosis clinically presents as small atrophic scaly papules and/or patches with raised peripheral ridges symmetrically dispersed on sun-exposed areas of the arms, legs, back, and shoulders. Although these lesions are extensive, they typically spare the mucous membranes, palms, and soles11; only a small percentage of cases report facial lesions,12 which often are asymptomatic but cosmetically bothersome. Additionally, approximately half of patients report symptoms of pruritus and/or stinging,13 thus treatment of DSAP is mainly indicated for symptomatic relief and cosmetic purposes. Malignant degeneration14,15 occurs in approximately 7.5% to 11% of porokeratosis cases,10,16 warranting treatment for preventative measures.
Management of DSAP is dependent on the extent of the disease and the level of concern for malignant transformation. Localized disease can be treated with cryotherapy, CO2 laser, and/or ablative techniques (eg, excision, curettage, dermabrasion) with variable degrees of success but high risk for scarring.1 More extensive disease requires treatment with topical retinoids, topical 5-fluorouracil, imiquimod cream 5%, diclofenac gel 3%, topical vitamin D3 analogues, and photodynamic therapy.1 Several other therapies have been reported in the literature with partial and/or complete success, including systemic retinoids (eg, acitretin), Q-switched ruby laser, Nd:YAG laser, fractional photothermolysis, Grenz rays, pulsed dye laser, fractional photothermolysis, topical corticosteroids, and fluor-hydroxy pulse peel.6 Although there is an extensive array of therapies for DSAP, treatment results are variable with mostly limited success. Successful treatment of DSAP is difficult and often requires the use of multiple modalities.
Ingenol mebutate is the active compound found in the sap of Euphorbia peplus used for the topical treatment of various skin conditions, including AKs.17 Ingenol mebutate gel 0.05% once daily for 2 days has been approved by the US Food and Drug Administration for the topical treatment of AKs. The mechanism of action of ingenol mebutate in AK therapy is not yet fully understood. In vivo and in vitro models have demonstrated both an induction of local lesion cell death and promotion of lesion-specific inflammatory response.18 When used in the treatment of AKs, ingenol mebutate gel 0.05% may cause a mild to moderate localized inflammatory response (eg, erythema, flaking/scaling, crusting, vesiculation/pustulation, erosion/ulceration, edema).
Our case is a rare report of successful treatment of DSAP with ingenol mebutate gel 0.05%. We found that treatment with ingenol mebutate gel 0.05% resulted in clinical improvement of DSAP lesions with minimal discomfort and good cosmetic response. This 2-day regimen is easy to use and patient friendly, improving medication compliance in such a cumbersome disease. We hope this case suggests that ingenol mebutate gel 0.05% could be a useful treatment alternative for DSAP, but future clinical studies should be conducted.
Disseminated superficial actinic porokeratosis (DSAP) is a chronic condition characterized by numerous atrophic papules and patches with a distinctive peripheral keratotic ridge, typically found on sun-exposed areas.1,2 Treatment of DSAP is warranted not only for cosmetic and symptomatic benefits but also to prevent malignant transformation.3,4 Successful treatment of DSAP often is difficult and frequently requires the use of multiple modalities. Ingenol mebutate gel 0.05% is a topical medication primarily used for the treatment of actinic keratosis (AK) by inducing cell death.5 We report a case of DSAP treated effectively with ingenol mebutate gel 0.05%.
Case Report
A 37-year-old woman was referred to the dermatology department for counseling for pseudoxanthoma elasticum (PXE), which had been proven on biopsy by an outside dermatologist 2 years prior. Physical examination revealed yellow papules on the neck that were characteristic of PXE, but no lesions were noted on the arms or legs. The only other cutaneous finding was a soft nodule on the right hip consistent with a lipoma. The patient returned to our institution 6 years later with lesions on both lower legs. She reported that these lesions had been present for 3 years and were exacerbated by sun exposure. On physical examination, multiple scattered, erythematous, annular, scaling papules and plaques were noted on the bilateral legs. A biopsy showed the histopathologic findings of DSAP (Figure 1). The patient had no family history of DSAP or PXE.
To determine the best treatment modality, we treated 4 test areas on both upper and lower legs: one with trichloroacetic acid (TCA), one with cryotherapy, one with imiquimod cream 5%, and one with tretinoin cream 0.1%. The patient returned 4 weeks later and showed modest response to TCA, cryotherapy, and tretinoin cream. Because cryotherapy was determined to be most effective, 20 more lesions were frozen at that visit. Over the next 2 years, the patient was treated with TCA, imiquimod cream 5%, and tretinoin cream 0.1%, but all ultimately proved ineffective for DSAP.
The patient returned 2 years after treatment failure (age 47 years) and was prescribed ingenol mebutate gel 0.05% for 2 days over an area of 25 cm2 on the right lower leg (Figure 2A). She returned for follow-up at days 3, 15, 30, and 60. At day 3, the patient developed an inflammatory response to the medication with moderate erythema and scaling of individual lesions. No vesiculation, pustulation, edema, or ulceration was exhibited (Figure 2B). At day 30, there was a marked reduction in scaling with some postinflammatory erythema (Figure 2C). At day 60, much of the erythema had faded and the scale remained notably reduced (Figure 2D).
Comment
Disseminated superficial actinic porokeratosis is the most common subtype of porokeratosis, a keratinization disorder. There are 6 subtypes of porokeratosis identified in the literature: DSAP, disseminated superficial porokeratosis, classic porokeratosis of Mibelli, porokeratosis plantaris palmaris et disseminata, linear porokeratosis, and punctate porokeratosis.6 Disseminated superficial actinic porokeratosis has a female predominance (1.8:1 ratio)7 and generally appears in the third or fourth decades of life. Clonal proliferations of atypical keratinocytes have been implicated in the etiology of DSAP; however, the exact pathogenesis is unclear. Risk factors for DSAP include genetic susceptibility (eg, autosomal-dominant inheritance pattern), exposure to UV radiation, and drug-related immunosuppression or immunodeficiency.7 Other proposed etiologic risk factors include trauma and infection.8 Clinical diagnosis of DSAP is confirmed by the histological presence of a cornoid lamella (a thin column ofparakeratotic cells), a thinning epidermis, an absent or thinned granular cell layer, and a prominent dermal lymphocytic infiltrate.9,10
Disseminated superficial actinic porokeratosis clinically presents as small atrophic scaly papules and/or patches with raised peripheral ridges symmetrically dispersed on sun-exposed areas of the arms, legs, back, and shoulders. Although these lesions are extensive, they typically spare the mucous membranes, palms, and soles11; only a small percentage of cases report facial lesions,12 which often are asymptomatic but cosmetically bothersome. Additionally, approximately half of patients report symptoms of pruritus and/or stinging,13 thus treatment of DSAP is mainly indicated for symptomatic relief and cosmetic purposes. Malignant degeneration14,15 occurs in approximately 7.5% to 11% of porokeratosis cases,10,16 warranting treatment for preventative measures.
Management of DSAP is dependent on the extent of the disease and the level of concern for malignant transformation. Localized disease can be treated with cryotherapy, CO2 laser, and/or ablative techniques (eg, excision, curettage, dermabrasion) with variable degrees of success but high risk for scarring.1 More extensive disease requires treatment with topical retinoids, topical 5-fluorouracil, imiquimod cream 5%, diclofenac gel 3%, topical vitamin D3 analogues, and photodynamic therapy.1 Several other therapies have been reported in the literature with partial and/or complete success, including systemic retinoids (eg, acitretin), Q-switched ruby laser, Nd:YAG laser, fractional photothermolysis, Grenz rays, pulsed dye laser, fractional photothermolysis, topical corticosteroids, and fluor-hydroxy pulse peel.6 Although there is an extensive array of therapies for DSAP, treatment results are variable with mostly limited success. Successful treatment of DSAP is difficult and often requires the use of multiple modalities.
Ingenol mebutate is the active compound found in the sap of Euphorbia peplus used for the topical treatment of various skin conditions, including AKs.17 Ingenol mebutate gel 0.05% once daily for 2 days has been approved by the US Food and Drug Administration for the topical treatment of AKs. The mechanism of action of ingenol mebutate in AK therapy is not yet fully understood. In vivo and in vitro models have demonstrated both an induction of local lesion cell death and promotion of lesion-specific inflammatory response.18 When used in the treatment of AKs, ingenol mebutate gel 0.05% may cause a mild to moderate localized inflammatory response (eg, erythema, flaking/scaling, crusting, vesiculation/pustulation, erosion/ulceration, edema).
Our case is a rare report of successful treatment of DSAP with ingenol mebutate gel 0.05%. We found that treatment with ingenol mebutate gel 0.05% resulted in clinical improvement of DSAP lesions with minimal discomfort and good cosmetic response. This 2-day regimen is easy to use and patient friendly, improving medication compliance in such a cumbersome disease. We hope this case suggests that ingenol mebutate gel 0.05% could be a useful treatment alternative for DSAP, but future clinical studies should be conducted.
- Martin-Clavijo A, Kanelleas A, Vlachou C, et al. Porokeratoses. In: Lebwohl M, Heymann WR, Berth-Jones J, et al, eds. Treatment of Skin Disease Comprehensive Therapeutic Strategies. 3rd ed. China: Elsevier Limited; 2010:584-586.
- Rouhani P, Fischer M, Meehan S, et al. Disseminated superficial actinic porokeratosis. Dermatology Online J. 2012;18:24.
- Sasson M, Krain AD. Porokeratosis and cutaneous malignancy. a review. Dermatol Surg. 1996;22:339-342.
- Lee HR, Han TY, Son SJ, et al. Squamous cell carcinoma developing within lesions of disseminated superficial actinic porokeratosis. Ann Dermatol. 2011;23:536-538.
- Lebwohl M, Swanson N, Anderson LL, et al. Ingenol mebutate gel for actinic keratosis. N Engl J Med. 2012;366:1010-1019.
- O’Regan GM, Irvine AD. Porokeratosis. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Professional; 2012:442-446.
- Sertznig P, von Felbert V, Megahed M. Porokeratosis: present concepts. J Eur Acad Dermatol Venereol. 2012;26:404-412.
- Brauer JA, Mandal R, Walters R, et al. Disseminated superficial porokeratosis. Dermatology Online J. 2010;16:20.
- Tallon B. Porokeratosis pathology. DermNet New Zealand website. http://www.dermnet.org.nz/pathology/porokeratosis-path.html. Updated December 2016. Accessed January 12, 2017.
- Skupsky H, Skupsky J, Goldenberg G. Disseminated superficial actinic porokeratosis: a treatment review [published online October 22, 2010]. J Dermatolog Treat. 2012;23:52-56.
- Spencer LV. Porokeratosis. UpToDate web site. https://eresources.library.mssm.edu:3285/contents/porokeratosis?source=search_result&search=porokeratosis&selectedTitle=1~22. Updated September 1, 2016. Accessed April 3, 2017.
- Sawyer R, Picou KA. Facial presentation of disseminated superficial actinic porokeratosis. Ear Nose Throat J. 1989;68:57-59.
- Schwarz T, Seiser A, Gschnait F. Disseminated superficial “actinic” porokeratosis. J Am Acad Dermatol. 1984;11(4, pt 2):724-730.
- Maubec E, Duvillard P, Margulis A, et al. Common skin cancers in porokeratosis. Br J Dermatol. 2005;152:1389-1391.
- Lee HR, Han TY, Son SJ, et al. Squamous cell carcinoma developing within lesions of disseminated superficial actinic porokeratosis [published online November 3, 2011]. Ann Dermatol. 2011;23:536-538.
- Kumari S, Mathur M. Disseminated superficial actinic porokeratosis. Nepal J Dermatol Venereol Leprol. 2010;9:22-24.
- Lebwohl M, Shumack S, Stein Gold L, et al. Long-term follow-up study of ingenol mebutate gel for the treatment of actinic keratosis. JAMA Dermatol. 2013;149:666-670.
- Stahlhut M, Bertelsen M, Hoyer-Hansen M, et al. Ingenol mebutate: induced cell death patterns in normal and cancer epithelial cells. J Drugs Dermatol. 2012;11:1181-1192.
- Martin-Clavijo A, Kanelleas A, Vlachou C, et al. Porokeratoses. In: Lebwohl M, Heymann WR, Berth-Jones J, et al, eds. Treatment of Skin Disease Comprehensive Therapeutic Strategies. 3rd ed. China: Elsevier Limited; 2010:584-586.
- Rouhani P, Fischer M, Meehan S, et al. Disseminated superficial actinic porokeratosis. Dermatology Online J. 2012;18:24.
- Sasson M, Krain AD. Porokeratosis and cutaneous malignancy. a review. Dermatol Surg. 1996;22:339-342.
- Lee HR, Han TY, Son SJ, et al. Squamous cell carcinoma developing within lesions of disseminated superficial actinic porokeratosis. Ann Dermatol. 2011;23:536-538.
- Lebwohl M, Swanson N, Anderson LL, et al. Ingenol mebutate gel for actinic keratosis. N Engl J Med. 2012;366:1010-1019.
- O’Regan GM, Irvine AD. Porokeratosis. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill Professional; 2012:442-446.
- Sertznig P, von Felbert V, Megahed M. Porokeratosis: present concepts. J Eur Acad Dermatol Venereol. 2012;26:404-412.
- Brauer JA, Mandal R, Walters R, et al. Disseminated superficial porokeratosis. Dermatology Online J. 2010;16:20.
- Tallon B. Porokeratosis pathology. DermNet New Zealand website. http://www.dermnet.org.nz/pathology/porokeratosis-path.html. Updated December 2016. Accessed January 12, 2017.
- Skupsky H, Skupsky J, Goldenberg G. Disseminated superficial actinic porokeratosis: a treatment review [published online October 22, 2010]. J Dermatolog Treat. 2012;23:52-56.
- Spencer LV. Porokeratosis. UpToDate web site. https://eresources.library.mssm.edu:3285/contents/porokeratosis?source=search_result&search=porokeratosis&selectedTitle=1~22. Updated September 1, 2016. Accessed April 3, 2017.
- Sawyer R, Picou KA. Facial presentation of disseminated superficial actinic porokeratosis. Ear Nose Throat J. 1989;68:57-59.
- Schwarz T, Seiser A, Gschnait F. Disseminated superficial “actinic” porokeratosis. J Am Acad Dermatol. 1984;11(4, pt 2):724-730.
- Maubec E, Duvillard P, Margulis A, et al. Common skin cancers in porokeratosis. Br J Dermatol. 2005;152:1389-1391.
- Lee HR, Han TY, Son SJ, et al. Squamous cell carcinoma developing within lesions of disseminated superficial actinic porokeratosis [published online November 3, 2011]. Ann Dermatol. 2011;23:536-538.
- Kumari S, Mathur M. Disseminated superficial actinic porokeratosis. Nepal J Dermatol Venereol Leprol. 2010;9:22-24.
- Lebwohl M, Shumack S, Stein Gold L, et al. Long-term follow-up study of ingenol mebutate gel for the treatment of actinic keratosis. JAMA Dermatol. 2013;149:666-670.
- Stahlhut M, Bertelsen M, Hoyer-Hansen M, et al. Ingenol mebutate: induced cell death patterns in normal and cancer epithelial cells. J Drugs Dermatol. 2012;11:1181-1192.
Practice Points
- Disseminated superficial actinic porokeratosis (DSAP) is an uncommon skin condition consisting of multiple annular hyperkeratotic lesions on sun-exposed areas.
- Treatment of DSAP is necessary due to its potential for progression to malignancy.
- Consider ingenol mebutate gel 0.05% for the treatment of DSAP on the arms and legs.
Hypoperfusion Retinopathy
Cardiovascular diseases are some of the most common conditions found in the geriatric population. Ocular manifestations of systemic cardiovascular conditions often are the initial presentation of the systemic disease. Identifying these findings help reveal the underlying disease and prevent more serious visual and systemic complications or even death.
Hypoperfusion retinopathy can occur as an early manifestation of carotid occlusive disease. It results from poor arterial perfusion pressure secondary to significant or complete carotid artery blockage resulting in retinal cha
Case Report
A 71-year-old white male was referred by his primary care physician (PCP) to the eye clinic for a routine comprehensive eye exam. The patient reported that his current progressive lenses, prescribed 2 years prior, were not strong enough at both distance and near, and that his eyes often felt dry. The symptoms were gradual in onset since his prior exam with no reported flashes, floaters, loss of vision, headaches, or ocular irritations.
The patient’s medical history was significant for morbid obesity, hypertension, borderline diabetes mellitus, and obstructive sleep apnea. His ocular history included recurrent conjunctivitis. At the time of the visit, the patient’s medications included 81 mg aspirin, 10 mg benazepril, 1,000 mg fish oil, 80 mg simvastatin, and use of a continuous positive airway pressure machine.
Best-corrected Snellen visual acuity was stable to his last eye exam at 20/25+2 right eye and 20/25-1 left eye with a manifest refraction of +2.25-0.75 × 077, and +2.75-1.25 × 096 in the right and left eye, respectively. Pupils were equally round and reactive to light with no afferent pupillary defect. Extraocular motility and finger counting fields were unremarkable. Anterior segment evaluation revealed lax bilateral upper lid apposition and mild cataracts in both eyes but were otherwise unremarkable (Figure 1). Dilated fundus examination revealed extensive hemorrhaging in the midperipheral retina of the right eye only (Figure 2). The left eye retina showed no abnormalities.

At this point the patient declined any additional symptoms, including eye pain, headache, transient vision loss, jaw claudication, and stroke signs. A complete blood count and hemoglobin A1c (HbA1c) was ordered, and all findings were unremarkable with no evidence of blood dyscrasia and with a HbA1c of 6.0. A carotid ultrasound (CUS) was also performed and revealed severe narrowing of the proximal section of the right internal carotid artery (ICA) with a trickle flow (Figure 3). The peak systolic velocity (PSV) at this level was 508 cm/s. There also was severe narrowing and turbulent flow in both the mid and distal portions of the right ICA. The patient was sent for a vascular evaluation 2 days following the CUS.
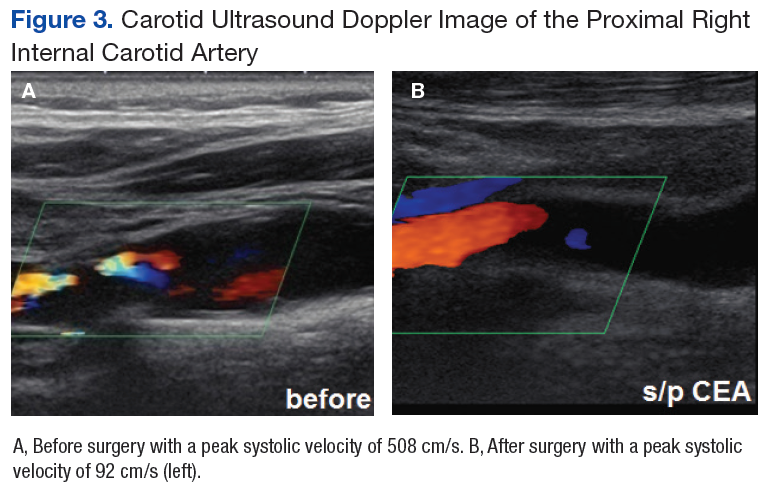
Based on the ocular findings and CUS results, the diagnosis of hypoperfusion retinopathy secondary to carotid occlusive disease was made. Because the patient was asymptomatic with no additional ocular sequelae, he was scheduled for an eye clinic follow-up in 2 months. The electrocardiogram, chest X-ray, and exercise stress test results were negative for acute cardiopulmonary disease, ischemia, or arrhythmias. A computed tomography angiography was performed and confirmed a high-grade lesion of the right ICA of > 95%. The vascular surgeon reported an 11% risk of stroke within 5 years and a 1% risk of stroke with surgery. Based on these results the patient underwent a right carotid endarterectomy (CEA) 2 weeks later. A follow-up CUS was performed 1 month post-CEA and revealed no abnormal fluid or significant plaque with a PSV of 92 cm/s (prior to surgery PSV was 508 cm/s) (Figure 3).
The patient returned to the eye clinic 1 month after the CEA. Gonioscopy revealed no neovascularization of the iris or angle and the dilated eye exam showed resolution of the midperipheral blot hemorrhages in his right eye with no evidence of retinal neovascularization.
Discussion
Hypoperfusion retinopathy is characterized by posterior retinal changes secondary to chronic ocular ischemia from decreased arterial perfusion related to significant or complete carotid artery stenosis.1-5 Early literature referred to this condition as venous stasis retinopathy; however, this term is misleading as the condition results from a reduction in arterial perfusion pressure and the term describes venous outflow obstruction.6 The terms carotid ischemic retinopathy, ischemic oculopathy, and hypotensive retinopathy also have been used interchangeably when describing hypoperfusion retinopathy.6
Incidence of hypoperfusion retinopathy is twice as high in males as it is in females due to a higher prevalence of cardiovascular disease.7 Hypoperfusion retinopathy rarely presents before the age of 50 years, with the average age of onset around 65 years.7 The exact rate of occurrence is unknown as this condition often is underdiagnosed because it mimics other vascular conditions, such as venous occlusive disease and diabetic retinopathy.1,7 Patients can present asymptomatically where findings are incidental on a dilated eye exam, or they may present with vision loss that can be gradual, sudden, or transient in nature.5,6,8
Gradual vision loss can follow a period of weeks to months and can occur secondary to posterior ischemia, macular edema, or choroidal hypoperfusion.1,3,8,9 Sudden vision loss can occur from severe hypoperfusion, creating an acute inner layer retinal ischemia. This type of vision loss often is accompanied by a cherry red spot in the macula and can be caused by an embolic plaque.1,8 Transient vision loss (TVL) also can be secondary to a plaque emboli or light induced. Patients with light-induced TVL report poor to blurry vision or prolonged after image when exposed to bright lights. In theory when the retina is exposed to light, there is an increase in metabolic demand that is unmet in those with choroidal vascular insufficiency from significant carotid stenosis.3,8,10
The clinical presentation most often is unilateral. Early stages of the disease generally affect the midperipheral retina but can be found in the posterior pole with chronicity. Early findings include microaneurysms, nerve fiber layer and inner retinal layer hemorrhages, and dilated, but generally not tortuous, veins.5 Chronic stage findings include arteriolar narrowing, extreme venous dilation, occasionally macular edema, and neovascularization of the disc and or retina.5 Disc edema or collaterals usually are not present.5
The mechanism behind hypoperfusion retinopathy results from an overall ischemic cascade and starts with comorbid cardiovascular conditions, such as hypertension, hypercholesterolemia, diabetes, heart disease, and history of smoking.1,2,5 These conditions play a role in creating atherosclerotic buildup in the arterial lumen leading to chronic narrowing and a decrease in arterial perfusion pressure. Over time, a low-grade hypoxic situation is formed, generating vascular endothelial cell damage and pericytes cell loss, thus causing leakage of fluid.1,2,5 With these chronic hypoxic states, angiogenic factor release eventually leads to posterior neovascularization.1,2,5 Further chronicity of carotid occlusive disease can create a panocular ischemia that also involves anterior structures, including iris, conjunctiva, episclera, or cornea. At this point, hypoperfusion retinopathy progresses to a more severe condition called ocular ischemic syndrome (OIS).2,5
Ocular ischemic syndrome can be associated with a 40% mortality rate within 5 years of onset as it is generally found in those with overall poor health.5 Along with posterior neovascularization, anterior structures also are involved. Sixty-seven percent of cases have iris or angle neovascularization of which 35% go on to develop neovascular glaucoma and its complications.1,8 With OIS, 90% of cases have some type of vision loss, and 40% report ipsilateral ocular pain.1,8 Visual loss can be gradual, sudden, or transient. The pain can occur from ocular ischemia, ruptured corneal epithelial microcysts secondary to acute glaucoma, elevated intraocular pressure (IOP) with neovascular glaucoma, or from ipsilateral dural ischemia.1,5,6,8 Fluorescein angiography is commonly used to diagnose and manage OIS, because it allows for the visualization of retinal and choroidal circulation and the detection of neovascular proliferation and ischemic areas.
Diagnostic Imaging
Several diagnostic testing strategies are available to evaluate for carotid occlusive disease. Carotid ultrasonography is a noninvasive, safe, and inexpensive screening tool to evaluate for high-grade stenosis. However, it can sometimes overestimate the degree of stenosis and is not reliable with severe calcifications.8 Computed tomography angiography and magnetic resonance angiography are minimally invasive tools that can be used to screen or confirm the degree of stenosis.8 These can be used in addition or instead of ultrasonography, especially in instances where patients have a short neck or high carotid bifurcation that may affect reliability. Both are contraindicated in those with renal failure as both modalities require the use of a contrast dye. Magnetic resonance angiography is far more expensive, time consuming, and not readily available.8 Carotid angiography is considered the gold standard for imaging the entire carotid artery system because it allows for the evaluation of plaque morphology, atherosclerotic disease, and collateral circulations.8 The disadvantages to this invasive and high-cost procedure include a risk of mortality that can occur secondary to an embolic stroke, myocardial infarction (MI), carotid artery dissection, or arterial thrombosis.8
Treatment
Treatment and management for carotid artery stenosis is focused on combined effort with the patient’s PCP and other specialists, including cardiologist, neurologist, and vascular surgeons.11 Treatment of comorbid conditions, education on healthy lifestyle, and smoking cessation are all imperative to the patient’s well-being. Managing ocular sequelae is based on specific findings and can include intravitreal antivascular edothelial growth factor or steroidal injections, pan retinal photocoagulation, or hypotensive drops.6,7
Restoration of arterial perfusion pressure is the main goal of treatment, and this can be done through CEA or carotid artery stents. Surgical intervention by CEA is determined based on each patient and his or her overall health. A full cardiac workup is required due to surgical risks. The North American Symptomatic Carotid Endarterectomy Trial evaluated symptomatic stenosis and the effectiveness of surgical intervention on stroke prevention. The trial reported that CEA was beneficial in symptomatic patients with 55% to 99% stenosis and especially in those with higher grade stenosis (> 70% up to 95%).5,7,8,12 With regard to asymptomatic patients with high-grade stenosis, CEA has been found to reduce the risk of stroke if there is at least 60% stenosis.5,7,8
Carotid artery stents can be used as an alternative when CEA is not effective or contraindicated due to a history of previous CEA, neck radiation, unstable angina, congestive heart failure, or recent MI.5,7,8 Neither CEA nor stenting is considered effective in complete occlusions due to the high risk of thromboembolism formation.5,7,8
Conclusion
Hypoperfusion retinopathy describes posterior retinal findings that occur secondary to poor arterial perfusion caused by carotid occlusive disease. Early intervention and restoration of this pressure can prevent the risk of developing a more serious condition characterized by a panocular ischemia called OIS. Unlike hypoperfusion retinopathy, OIS also includes anterior segment findings such as iris neovascularization, which may lead to neovascular glaucoma, whereas hypoperfusion retinopathy is localized to the posterior pole. Patients that develop OIS are at a 40% risk of mortality within 5 years due to poor overall health. Understanding the patient’s signs and symptoms can aid in the diagnosis of both conditions. Collaborative management with the patient’s PCP and specialists in treating comorbid conditions is vital to the patients’ well-being.
1. Brown GC, Magargal LE. The ocular ischemic syndrome. Int Ophthalmol. 1988;11(4):239-251.
2. Dahlman AH, McCormack D, Harrison RJ. Bilateral hypoperfuion retinopathy. J R Soc Med. 2001; 94(6):298-299.
3. Dugan JD Jr, Green WR. Ophthalmologic manifestations of carotid occlusive disease. Eye (Lond). 1991;5(pt 2):226-238.
4. Klijn CJ, Kappelle LJ, Tulleken CAF, van Gijn J. Symptomatic carotid artery occlusion. A reappraisal of hemodynamic factors. Stroke. 1997;28(10):2084-2093.
5. McCrary JA III. Venous stasis retinopathy of stenotic or occlusive caroid origin. J Clin Neuroophthalmol. 1989;9(3):195-199.
6. Sanborn GE, Magargal LE. Arterial obstructive disease of the eye. In: Tasman W, Jaeger EA, eds. Duane’s Ophthalmology. 12th ed. Vol 3. Riverwoods, IL: Lippincott Williams & Wilkins; 2013:chap 14.
7. Terelak-Borys B, Skonieczna K, Grabska-Liberek I. Ocular ischemic syndrome–a systematic review. Med Sci Monit. 2012;18(8):RA138-RA144.
8. Atebara NH, Brown GC. The ocular ischemic syndrome. In: Tasman W, Jaeger EA, eds. Duane’s Ophthalmology. 12th ed. Vol 3. Riverwoods, IL: Lippincott Williams & Wilkins; 2013:chap 12.
9. Ho AC, Lieb WE, Flaharty PM, et al. Color Doppler imaging of the ocular ischaemic syndrome. Ophthalmology. 1992;99(9):1453-1462.
10. Kahn M, Green WR, Knox DL, Miller NR. Ocular features of carotid occlusive disease. Retina. 1986;6(4):239-252.
11. Mizener JB, Podhajsky P, Hayreh SS. Ocular ischemic syndrome. Ophthalmology. 1997;104(5):859-864.
12. Ferguson GG, Eliasziw M, Barr HW, et al. The North American Symptomatic Carotid Endarterectomy Trial: surgical results in 1415 patients. Stroke. 1999;30(9):1751-1758.
Cardiovascular diseases are some of the most common conditions found in the geriatric population. Ocular manifestations of systemic cardiovascular conditions often are the initial presentation of the systemic disease. Identifying these findings help reveal the underlying disease and prevent more serious visual and systemic complications or even death.
Hypoperfusion retinopathy can occur as an early manifestation of carotid occlusive disease. It results from poor arterial perfusion pressure secondary to significant or complete carotid artery blockage resulting in retinal cha
Case Report
A 71-year-old white male was referred by his primary care physician (PCP) to the eye clinic for a routine comprehensive eye exam. The patient reported that his current progressive lenses, prescribed 2 years prior, were not strong enough at both distance and near, and that his eyes often felt dry. The symptoms were gradual in onset since his prior exam with no reported flashes, floaters, loss of vision, headaches, or ocular irritations.
The patient’s medical history was significant for morbid obesity, hypertension, borderline diabetes mellitus, and obstructive sleep apnea. His ocular history included recurrent conjunctivitis. At the time of the visit, the patient’s medications included 81 mg aspirin, 10 mg benazepril, 1,000 mg fish oil, 80 mg simvastatin, and use of a continuous positive airway pressure machine.
Best-corrected Snellen visual acuity was stable to his last eye exam at 20/25+2 right eye and 20/25-1 left eye with a manifest refraction of +2.25-0.75 × 077, and +2.75-1.25 × 096 in the right and left eye, respectively. Pupils were equally round and reactive to light with no afferent pupillary defect. Extraocular motility and finger counting fields were unremarkable. Anterior segment evaluation revealed lax bilateral upper lid apposition and mild cataracts in both eyes but were otherwise unremarkable (Figure 1). Dilated fundus examination revealed extensive hemorrhaging in the midperipheral retina of the right eye only (Figure 2). The left eye retina showed no abnormalities.
At this point the patient declined any additional symptoms, including eye pain, headache, transient vision loss, jaw claudication, and stroke signs. A complete blood count and hemoglobin A1c (HbA1c) was ordered, and all findings were unremarkable with no evidence of blood dyscrasia and with a HbA1c of 6.0. A carotid ultrasound (CUS) was also performed and revealed severe narrowing of the proximal section of the right internal carotid artery (ICA) with a trickle flow (Figure 3). The peak systolic velocity (PSV) at this level was 508 cm/s. There also was severe narrowing and turbulent flow in both the mid and distal portions of the right ICA. The patient was sent for a vascular evaluation 2 days following the CUS.
Based on the ocular findings and CUS results, the diagnosis of hypoperfusion retinopathy secondary to carotid occlusive disease was made. Because the patient was asymptomatic with no additional ocular sequelae, he was scheduled for an eye clinic follow-up in 2 months. The electrocardiogram, chest X-ray, and exercise stress test results were negative for acute cardiopulmonary disease, ischemia, or arrhythmias. A computed tomography angiography was performed and confirmed a high-grade lesion of the right ICA of > 95%. The vascular surgeon reported an 11% risk of stroke within 5 years and a 1% risk of stroke with surgery. Based on these results the patient underwent a right carotid endarterectomy (CEA) 2 weeks later. A follow-up CUS was performed 1 month post-CEA and revealed no abnormal fluid or significant plaque with a PSV of 92 cm/s (prior to surgery PSV was 508 cm/s) (Figure 3).
The patient returned to the eye clinic 1 month after the CEA. Gonioscopy revealed no neovascularization of the iris or angle and the dilated eye exam showed resolution of the midperipheral blot hemorrhages in his right eye with no evidence of retinal neovascularization.
Discussion
Hypoperfusion retinopathy is characterized by posterior retinal changes secondary to chronic ocular ischemia from decreased arterial perfusion related to significant or complete carotid artery stenosis.1-5 Early literature referred to this condition as venous stasis retinopathy; however, this term is misleading as the condition results from a reduction in arterial perfusion pressure and the term describes venous outflow obstruction.6 The terms carotid ischemic retinopathy, ischemic oculopathy, and hypotensive retinopathy also have been used interchangeably when describing hypoperfusion retinopathy.6
Incidence of hypoperfusion retinopathy is twice as high in males as it is in females due to a higher prevalence of cardiovascular disease.7 Hypoperfusion retinopathy rarely presents before the age of 50 years, with the average age of onset around 65 years.7 The exact rate of occurrence is unknown as this condition often is underdiagnosed because it mimics other vascular conditions, such as venous occlusive disease and diabetic retinopathy.1,7 Patients can present asymptomatically where findings are incidental on a dilated eye exam, or they may present with vision loss that can be gradual, sudden, or transient in nature.5,6,8
Gradual vision loss can follow a period of weeks to months and can occur secondary to posterior ischemia, macular edema, or choroidal hypoperfusion.1,3,8,9 Sudden vision loss can occur from severe hypoperfusion, creating an acute inner layer retinal ischemia. This type of vision loss often is accompanied by a cherry red spot in the macula and can be caused by an embolic plaque.1,8 Transient vision loss (TVL) also can be secondary to a plaque emboli or light induced. Patients with light-induced TVL report poor to blurry vision or prolonged after image when exposed to bright lights. In theory when the retina is exposed to light, there is an increase in metabolic demand that is unmet in those with choroidal vascular insufficiency from significant carotid stenosis.3,8,10
The clinical presentation most often is unilateral. Early stages of the disease generally affect the midperipheral retina but can be found in the posterior pole with chronicity. Early findings include microaneurysms, nerve fiber layer and inner retinal layer hemorrhages, and dilated, but generally not tortuous, veins.5 Chronic stage findings include arteriolar narrowing, extreme venous dilation, occasionally macular edema, and neovascularization of the disc and or retina.5 Disc edema or collaterals usually are not present.5
The mechanism behind hypoperfusion retinopathy results from an overall ischemic cascade and starts with comorbid cardiovascular conditions, such as hypertension, hypercholesterolemia, diabetes, heart disease, and history of smoking.1,2,5 These conditions play a role in creating atherosclerotic buildup in the arterial lumen leading to chronic narrowing and a decrease in arterial perfusion pressure. Over time, a low-grade hypoxic situation is formed, generating vascular endothelial cell damage and pericytes cell loss, thus causing leakage of fluid.1,2,5 With these chronic hypoxic states, angiogenic factor release eventually leads to posterior neovascularization.1,2,5 Further chronicity of carotid occlusive disease can create a panocular ischemia that also involves anterior structures, including iris, conjunctiva, episclera, or cornea. At this point, hypoperfusion retinopathy progresses to a more severe condition called ocular ischemic syndrome (OIS).2,5
Ocular ischemic syndrome can be associated with a 40% mortality rate within 5 years of onset as it is generally found in those with overall poor health.5 Along with posterior neovascularization, anterior structures also are involved. Sixty-seven percent of cases have iris or angle neovascularization of which 35% go on to develop neovascular glaucoma and its complications.1,8 With OIS, 90% of cases have some type of vision loss, and 40% report ipsilateral ocular pain.1,8 Visual loss can be gradual, sudden, or transient. The pain can occur from ocular ischemia, ruptured corneal epithelial microcysts secondary to acute glaucoma, elevated intraocular pressure (IOP) with neovascular glaucoma, or from ipsilateral dural ischemia.1,5,6,8 Fluorescein angiography is commonly used to diagnose and manage OIS, because it allows for the visualization of retinal and choroidal circulation and the detection of neovascular proliferation and ischemic areas.
Diagnostic Imaging
Several diagnostic testing strategies are available to evaluate for carotid occlusive disease. Carotid ultrasonography is a noninvasive, safe, and inexpensive screening tool to evaluate for high-grade stenosis. However, it can sometimes overestimate the degree of stenosis and is not reliable with severe calcifications.8 Computed tomography angiography and magnetic resonance angiography are minimally invasive tools that can be used to screen or confirm the degree of stenosis.8 These can be used in addition or instead of ultrasonography, especially in instances where patients have a short neck or high carotid bifurcation that may affect reliability. Both are contraindicated in those with renal failure as both modalities require the use of a contrast dye. Magnetic resonance angiography is far more expensive, time consuming, and not readily available.8 Carotid angiography is considered the gold standard for imaging the entire carotid artery system because it allows for the evaluation of plaque morphology, atherosclerotic disease, and collateral circulations.8 The disadvantages to this invasive and high-cost procedure include a risk of mortality that can occur secondary to an embolic stroke, myocardial infarction (MI), carotid artery dissection, or arterial thrombosis.8
Treatment
Treatment and management for carotid artery stenosis is focused on combined effort with the patient’s PCP and other specialists, including cardiologist, neurologist, and vascular surgeons.11 Treatment of comorbid conditions, education on healthy lifestyle, and smoking cessation are all imperative to the patient’s well-being. Managing ocular sequelae is based on specific findings and can include intravitreal antivascular edothelial growth factor or steroidal injections, pan retinal photocoagulation, or hypotensive drops.6,7
Restoration of arterial perfusion pressure is the main goal of treatment, and this can be done through CEA or carotid artery stents. Surgical intervention by CEA is determined based on each patient and his or her overall health. A full cardiac workup is required due to surgical risks. The North American Symptomatic Carotid Endarterectomy Trial evaluated symptomatic stenosis and the effectiveness of surgical intervention on stroke prevention. The trial reported that CEA was beneficial in symptomatic patients with 55% to 99% stenosis and especially in those with higher grade stenosis (> 70% up to 95%).5,7,8,12 With regard to asymptomatic patients with high-grade stenosis, CEA has been found to reduce the risk of stroke if there is at least 60% stenosis.5,7,8
Carotid artery stents can be used as an alternative when CEA is not effective or contraindicated due to a history of previous CEA, neck radiation, unstable angina, congestive heart failure, or recent MI.5,7,8 Neither CEA nor stenting is considered effective in complete occlusions due to the high risk of thromboembolism formation.5,7,8
Conclusion
Hypoperfusion retinopathy describes posterior retinal findings that occur secondary to poor arterial perfusion caused by carotid occlusive disease. Early intervention and restoration of this pressure can prevent the risk of developing a more serious condition characterized by a panocular ischemia called OIS. Unlike hypoperfusion retinopathy, OIS also includes anterior segment findings such as iris neovascularization, which may lead to neovascular glaucoma, whereas hypoperfusion retinopathy is localized to the posterior pole. Patients that develop OIS are at a 40% risk of mortality within 5 years due to poor overall health. Understanding the patient’s signs and symptoms can aid in the diagnosis of both conditions. Collaborative management with the patient’s PCP and specialists in treating comorbid conditions is vital to the patients’ well-being.
Cardiovascular diseases are some of the most common conditions found in the geriatric population. Ocular manifestations of systemic cardiovascular conditions often are the initial presentation of the systemic disease. Identifying these findings help reveal the underlying disease and prevent more serious visual and systemic complications or even death.
Hypoperfusion retinopathy can occur as an early manifestation of carotid occlusive disease. It results from poor arterial perfusion pressure secondary to significant or complete carotid artery blockage resulting in retinal cha
Case Report
A 71-year-old white male was referred by his primary care physician (PCP) to the eye clinic for a routine comprehensive eye exam. The patient reported that his current progressive lenses, prescribed 2 years prior, were not strong enough at both distance and near, and that his eyes often felt dry. The symptoms were gradual in onset since his prior exam with no reported flashes, floaters, loss of vision, headaches, or ocular irritations.
The patient’s medical history was significant for morbid obesity, hypertension, borderline diabetes mellitus, and obstructive sleep apnea. His ocular history included recurrent conjunctivitis. At the time of the visit, the patient’s medications included 81 mg aspirin, 10 mg benazepril, 1,000 mg fish oil, 80 mg simvastatin, and use of a continuous positive airway pressure machine.
Best-corrected Snellen visual acuity was stable to his last eye exam at 20/25+2 right eye and 20/25-1 left eye with a manifest refraction of +2.25-0.75 × 077, and +2.75-1.25 × 096 in the right and left eye, respectively. Pupils were equally round and reactive to light with no afferent pupillary defect. Extraocular motility and finger counting fields were unremarkable. Anterior segment evaluation revealed lax bilateral upper lid apposition and mild cataracts in both eyes but were otherwise unremarkable (Figure 1). Dilated fundus examination revealed extensive hemorrhaging in the midperipheral retina of the right eye only (Figure 2). The left eye retina showed no abnormalities.
At this point the patient declined any additional symptoms, including eye pain, headache, transient vision loss, jaw claudication, and stroke signs. A complete blood count and hemoglobin A1c (HbA1c) was ordered, and all findings were unremarkable with no evidence of blood dyscrasia and with a HbA1c of 6.0. A carotid ultrasound (CUS) was also performed and revealed severe narrowing of the proximal section of the right internal carotid artery (ICA) with a trickle flow (Figure 3). The peak systolic velocity (PSV) at this level was 508 cm/s. There also was severe narrowing and turbulent flow in both the mid and distal portions of the right ICA. The patient was sent for a vascular evaluation 2 days following the CUS.
Based on the ocular findings and CUS results, the diagnosis of hypoperfusion retinopathy secondary to carotid occlusive disease was made. Because the patient was asymptomatic with no additional ocular sequelae, he was scheduled for an eye clinic follow-up in 2 months. The electrocardiogram, chest X-ray, and exercise stress test results were negative for acute cardiopulmonary disease, ischemia, or arrhythmias. A computed tomography angiography was performed and confirmed a high-grade lesion of the right ICA of > 95%. The vascular surgeon reported an 11% risk of stroke within 5 years and a 1% risk of stroke with surgery. Based on these results the patient underwent a right carotid endarterectomy (CEA) 2 weeks later. A follow-up CUS was performed 1 month post-CEA and revealed no abnormal fluid or significant plaque with a PSV of 92 cm/s (prior to surgery PSV was 508 cm/s) (Figure 3).
The patient returned to the eye clinic 1 month after the CEA. Gonioscopy revealed no neovascularization of the iris or angle and the dilated eye exam showed resolution of the midperipheral blot hemorrhages in his right eye with no evidence of retinal neovascularization.
Discussion
Hypoperfusion retinopathy is characterized by posterior retinal changes secondary to chronic ocular ischemia from decreased arterial perfusion related to significant or complete carotid artery stenosis.1-5 Early literature referred to this condition as venous stasis retinopathy; however, this term is misleading as the condition results from a reduction in arterial perfusion pressure and the term describes venous outflow obstruction.6 The terms carotid ischemic retinopathy, ischemic oculopathy, and hypotensive retinopathy also have been used interchangeably when describing hypoperfusion retinopathy.6
Incidence of hypoperfusion retinopathy is twice as high in males as it is in females due to a higher prevalence of cardiovascular disease.7 Hypoperfusion retinopathy rarely presents before the age of 50 years, with the average age of onset around 65 years.7 The exact rate of occurrence is unknown as this condition often is underdiagnosed because it mimics other vascular conditions, such as venous occlusive disease and diabetic retinopathy.1,7 Patients can present asymptomatically where findings are incidental on a dilated eye exam, or they may present with vision loss that can be gradual, sudden, or transient in nature.5,6,8
Gradual vision loss can follow a period of weeks to months and can occur secondary to posterior ischemia, macular edema, or choroidal hypoperfusion.1,3,8,9 Sudden vision loss can occur from severe hypoperfusion, creating an acute inner layer retinal ischemia. This type of vision loss often is accompanied by a cherry red spot in the macula and can be caused by an embolic plaque.1,8 Transient vision loss (TVL) also can be secondary to a plaque emboli or light induced. Patients with light-induced TVL report poor to blurry vision or prolonged after image when exposed to bright lights. In theory when the retina is exposed to light, there is an increase in metabolic demand that is unmet in those with choroidal vascular insufficiency from significant carotid stenosis.3,8,10
The clinical presentation most often is unilateral. Early stages of the disease generally affect the midperipheral retina but can be found in the posterior pole with chronicity. Early findings include microaneurysms, nerve fiber layer and inner retinal layer hemorrhages, and dilated, but generally not tortuous, veins.5 Chronic stage findings include arteriolar narrowing, extreme venous dilation, occasionally macular edema, and neovascularization of the disc and or retina.5 Disc edema or collaterals usually are not present.5
The mechanism behind hypoperfusion retinopathy results from an overall ischemic cascade and starts with comorbid cardiovascular conditions, such as hypertension, hypercholesterolemia, diabetes, heart disease, and history of smoking.1,2,5 These conditions play a role in creating atherosclerotic buildup in the arterial lumen leading to chronic narrowing and a decrease in arterial perfusion pressure. Over time, a low-grade hypoxic situation is formed, generating vascular endothelial cell damage and pericytes cell loss, thus causing leakage of fluid.1,2,5 With these chronic hypoxic states, angiogenic factor release eventually leads to posterior neovascularization.1,2,5 Further chronicity of carotid occlusive disease can create a panocular ischemia that also involves anterior structures, including iris, conjunctiva, episclera, or cornea. At this point, hypoperfusion retinopathy progresses to a more severe condition called ocular ischemic syndrome (OIS).2,5
Ocular ischemic syndrome can be associated with a 40% mortality rate within 5 years of onset as it is generally found in those with overall poor health.5 Along with posterior neovascularization, anterior structures also are involved. Sixty-seven percent of cases have iris or angle neovascularization of which 35% go on to develop neovascular glaucoma and its complications.1,8 With OIS, 90% of cases have some type of vision loss, and 40% report ipsilateral ocular pain.1,8 Visual loss can be gradual, sudden, or transient. The pain can occur from ocular ischemia, ruptured corneal epithelial microcysts secondary to acute glaucoma, elevated intraocular pressure (IOP) with neovascular glaucoma, or from ipsilateral dural ischemia.1,5,6,8 Fluorescein angiography is commonly used to diagnose and manage OIS, because it allows for the visualization of retinal and choroidal circulation and the detection of neovascular proliferation and ischemic areas.
Diagnostic Imaging
Several diagnostic testing strategies are available to evaluate for carotid occlusive disease. Carotid ultrasonography is a noninvasive, safe, and inexpensive screening tool to evaluate for high-grade stenosis. However, it can sometimes overestimate the degree of stenosis and is not reliable with severe calcifications.8 Computed tomography angiography and magnetic resonance angiography are minimally invasive tools that can be used to screen or confirm the degree of stenosis.8 These can be used in addition or instead of ultrasonography, especially in instances where patients have a short neck or high carotid bifurcation that may affect reliability. Both are contraindicated in those with renal failure as both modalities require the use of a contrast dye. Magnetic resonance angiography is far more expensive, time consuming, and not readily available.8 Carotid angiography is considered the gold standard for imaging the entire carotid artery system because it allows for the evaluation of plaque morphology, atherosclerotic disease, and collateral circulations.8 The disadvantages to this invasive and high-cost procedure include a risk of mortality that can occur secondary to an embolic stroke, myocardial infarction (MI), carotid artery dissection, or arterial thrombosis.8
Treatment
Treatment and management for carotid artery stenosis is focused on combined effort with the patient’s PCP and other specialists, including cardiologist, neurologist, and vascular surgeons.11 Treatment of comorbid conditions, education on healthy lifestyle, and smoking cessation are all imperative to the patient’s well-being. Managing ocular sequelae is based on specific findings and can include intravitreal antivascular edothelial growth factor or steroidal injections, pan retinal photocoagulation, or hypotensive drops.6,7
Restoration of arterial perfusion pressure is the main goal of treatment, and this can be done through CEA or carotid artery stents. Surgical intervention by CEA is determined based on each patient and his or her overall health. A full cardiac workup is required due to surgical risks. The North American Symptomatic Carotid Endarterectomy Trial evaluated symptomatic stenosis and the effectiveness of surgical intervention on stroke prevention. The trial reported that CEA was beneficial in symptomatic patients with 55% to 99% stenosis and especially in those with higher grade stenosis (> 70% up to 95%).5,7,8,12 With regard to asymptomatic patients with high-grade stenosis, CEA has been found to reduce the risk of stroke if there is at least 60% stenosis.5,7,8
Carotid artery stents can be used as an alternative when CEA is not effective or contraindicated due to a history of previous CEA, neck radiation, unstable angina, congestive heart failure, or recent MI.5,7,8 Neither CEA nor stenting is considered effective in complete occlusions due to the high risk of thromboembolism formation.5,7,8
Conclusion
Hypoperfusion retinopathy describes posterior retinal findings that occur secondary to poor arterial perfusion caused by carotid occlusive disease. Early intervention and restoration of this pressure can prevent the risk of developing a more serious condition characterized by a panocular ischemia called OIS. Unlike hypoperfusion retinopathy, OIS also includes anterior segment findings such as iris neovascularization, which may lead to neovascular glaucoma, whereas hypoperfusion retinopathy is localized to the posterior pole. Patients that develop OIS are at a 40% risk of mortality within 5 years due to poor overall health. Understanding the patient’s signs and symptoms can aid in the diagnosis of both conditions. Collaborative management with the patient’s PCP and specialists in treating comorbid conditions is vital to the patients’ well-being.
1. Brown GC, Magargal LE. The ocular ischemic syndrome. Int Ophthalmol. 1988;11(4):239-251.
2. Dahlman AH, McCormack D, Harrison RJ. Bilateral hypoperfuion retinopathy. J R Soc Med. 2001; 94(6):298-299.
3. Dugan JD Jr, Green WR. Ophthalmologic manifestations of carotid occlusive disease. Eye (Lond). 1991;5(pt 2):226-238.
4. Klijn CJ, Kappelle LJ, Tulleken CAF, van Gijn J. Symptomatic carotid artery occlusion. A reappraisal of hemodynamic factors. Stroke. 1997;28(10):2084-2093.
5. McCrary JA III. Venous stasis retinopathy of stenotic or occlusive caroid origin. J Clin Neuroophthalmol. 1989;9(3):195-199.
6. Sanborn GE, Magargal LE. Arterial obstructive disease of the eye. In: Tasman W, Jaeger EA, eds. Duane’s Ophthalmology. 12th ed. Vol 3. Riverwoods, IL: Lippincott Williams & Wilkins; 2013:chap 14.
7. Terelak-Borys B, Skonieczna K, Grabska-Liberek I. Ocular ischemic syndrome–a systematic review. Med Sci Monit. 2012;18(8):RA138-RA144.
8. Atebara NH, Brown GC. The ocular ischemic syndrome. In: Tasman W, Jaeger EA, eds. Duane’s Ophthalmology. 12th ed. Vol 3. Riverwoods, IL: Lippincott Williams & Wilkins; 2013:chap 12.
9. Ho AC, Lieb WE, Flaharty PM, et al. Color Doppler imaging of the ocular ischaemic syndrome. Ophthalmology. 1992;99(9):1453-1462.
10. Kahn M, Green WR, Knox DL, Miller NR. Ocular features of carotid occlusive disease. Retina. 1986;6(4):239-252.
11. Mizener JB, Podhajsky P, Hayreh SS. Ocular ischemic syndrome. Ophthalmology. 1997;104(5):859-864.
12. Ferguson GG, Eliasziw M, Barr HW, et al. The North American Symptomatic Carotid Endarterectomy Trial: surgical results in 1415 patients. Stroke. 1999;30(9):1751-1758.
1. Brown GC, Magargal LE. The ocular ischemic syndrome. Int Ophthalmol. 1988;11(4):239-251.
2. Dahlman AH, McCormack D, Harrison RJ. Bilateral hypoperfuion retinopathy. J R Soc Med. 2001; 94(6):298-299.
3. Dugan JD Jr, Green WR. Ophthalmologic manifestations of carotid occlusive disease. Eye (Lond). 1991;5(pt 2):226-238.
4. Klijn CJ, Kappelle LJ, Tulleken CAF, van Gijn J. Symptomatic carotid artery occlusion. A reappraisal of hemodynamic factors. Stroke. 1997;28(10):2084-2093.
5. McCrary JA III. Venous stasis retinopathy of stenotic or occlusive caroid origin. J Clin Neuroophthalmol. 1989;9(3):195-199.
6. Sanborn GE, Magargal LE. Arterial obstructive disease of the eye. In: Tasman W, Jaeger EA, eds. Duane’s Ophthalmology. 12th ed. Vol 3. Riverwoods, IL: Lippincott Williams & Wilkins; 2013:chap 14.
7. Terelak-Borys B, Skonieczna K, Grabska-Liberek I. Ocular ischemic syndrome–a systematic review. Med Sci Monit. 2012;18(8):RA138-RA144.
8. Atebara NH, Brown GC. The ocular ischemic syndrome. In: Tasman W, Jaeger EA, eds. Duane’s Ophthalmology. 12th ed. Vol 3. Riverwoods, IL: Lippincott Williams & Wilkins; 2013:chap 12.
9. Ho AC, Lieb WE, Flaharty PM, et al. Color Doppler imaging of the ocular ischaemic syndrome. Ophthalmology. 1992;99(9):1453-1462.
10. Kahn M, Green WR, Knox DL, Miller NR. Ocular features of carotid occlusive disease. Retina. 1986;6(4):239-252.
11. Mizener JB, Podhajsky P, Hayreh SS. Ocular ischemic syndrome. Ophthalmology. 1997;104(5):859-864.
12. Ferguson GG, Eliasziw M, Barr HW, et al. The North American Symptomatic Carotid Endarterectomy Trial: surgical results in 1415 patients. Stroke. 1999;30(9):1751-1758.
Systemic Hypothermia as Treatment for an Acute Cervical Spinal Cord Injury in a Professional Football Player: 9-Year Follow-Up
Take-Home Points
- Importance of on-field management.
- Preseason drilling of spinal injury management.
- Early and rapid intervention.
- Possible benefit of moderate systemic hypothermia as treatment for acute cervical injury.
In 2010, we reported the case of a professional American football player who sustained a complete cervical spinal cord injury (SCI) while tackling an opposing player.1 He received prompt medical and surgical care based on then-current recommendations, but was also treated with systemic hypothermia soon after his injury. Although systemic hypothermia had been used in the management of other neurologic injuries at that time, it had not been used in humans with acute SCI, except as described in 2 case reports.2,3 However, Dietrich4 described early emerging animal data on the efficacy of systemic hypothermia for acute SCI. We now provide a clinical update on our patient, who provided written informed consent for print and electronic publication of this case report.
Case Report
During a National Football League game, the player sustained a C3–C4 fracture-dislocation after a helmet-to-helmet hit on an opposing player. He fell face down on the ground and did not move. The team’s physician and trainer rushed to the player’s side, immediately assessed him, and initiated the emergency spinal resuscitation protocol.
As per protocol, the assigned team leader took charge of managing the player’s head to maintain in-line traction with the helmet in place until the head was secured in place on a backboard designed to accommodate the helmet. 
Complete motor paralysis and sensory loss (American Spinal Injury Association [ASIA] level A) were noted below the clavicles during physical examination by the head athletic trainer and 2 independent physicians, and by self-report. 

On arrival at the hospital, the patient had a core temperature of 98°F, which is substantially lower than the average core temperature (≤101.7°F) of an active football player.6He had a normal level of consciousness and normal cranial nerve function but remained without any voluntary motor function in the extremities and still had no sensation below the clavicles, except crude pressure sensation in one hand while in the emergency department. After the helmet and shoulder pads were removed, per National Athletic Trainers’ Association (NATA) protocol7 (Figure 2), he was stabilized, and a hard cervical collar was placed. A lateral radiograph (Figure 4) showed a C3–C4 facet dislocation with about 46% anterior translation of C3 on C4 and obvious disruption of the facets. 
About 3 hours after injury, the patient was taken to the operating room. Although closed reduction improved alignment dramatically, it failed to completely reduce the dislocated left C3–C4 facet. An hour later, anterior C3–C4 discectomy was performed from the front with instrumented anterior interbody fusion. This was immediately followed by posterior decompressive laminectomy, bilateral facet reduction, and fusion with instrumentation. Surgery was completed within about 4 hours, almost exactly 7 hours after injury. Anesthesia records indicated a core temperature range of 94.1°F to 95.3°F with passive cooling during surgery. CT and MRI performed within 4 hours after surgery showed excellent cord decompression.
The next morning, about 14.5 hours after injury, the patient demonstrated a flicker of the adductor muscles of the lower extremities. An examination an hour later revealed 1/5 quadriceps, 2/5 adductors, and 1/5 gastrocnemius/soleus. A nurse’s hourly examinations and the surgeon’s repeat examinations revealed no other motor function. Sensory function was more difficult to evaluate because of sedation, but rudimentary sensation was noted throughout the lower extremities, and proprioception and vibratory sensation were noted as well. With passive cooling, it was difficult to consistently maintain moderate hypothermia; the patient’s core temperature ranged from 94.8°F to 98.8°F by 6:00 a.m. Therefore, the decision was made to place a Cordis sheath in the left femoral vein and introduce an intra-vena cava cooling catheter through it. This catheter was highly effective in maintaining the patient’s temperature at about 92.5°F.
Over the next 36 hours, the patient demonstrated increased motor activity in the upper and lower extremities: 1/5 biceps, 2-3/5 triceps, 3/5 quadriceps. He was slowly rewarmed and, on postoperative day 3, extubated. 
At 2 years, the patient underwent another anterior-only cervical procedure: The inferior adjacent segment (C4–C5) was fused because of neck pain and deformity. 
With respect to the original injury and the evolution in cord appearance, the patient had solid arthrodesis from C3–C5 with instrumentation in good position. There was evidence of loss of lordosis at C5–C6 with disk dessication and broad-based bulging. The spinal cord had evidence of myelomalacia; this was noted when the patient was in rehabilitation, 1 month after injury. The 2-cm × 11-mm area of myelomalacia was directly posterior to the fused C3–C4 interval (original MRI, Figure 5; 2-week MRI, Figure 6).
Conclusion
At the time this player was injured, use of systemic hypothermia with standard therapy for acute SCI was unique and controversial. Since then, smaller randomized human studies have described the tolerable safety profile, efficacy, and potential benefits of this intervention in acute SCI in humans.8-10 Now, modest systemic hypothermia can be one of many tools considered in the treatment of acute SCI. Before it can become the standard of care, however, additional larger prospective randomized studies need to be completed.
Am J Orthop. 2017;46(2):E79-E82. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Cappuccino A, Bisson LJ, Carpenter B, Marzo J, Dietrich WD 3rd, Cappuccino H. The use of systemic hypothermia for the treatment of an acute cervical spinal cord injury in a professional football player. Spine. 2010;35(2):E57-E62.
2. Goldstein J. Lowering body temp shows promise for trauma treatment. Spinal Cord Injury Information Pages news blog. http://www.sci-info-pages.com/2006/05/lowering-body-temp-shows-promise-for.html. Published May 3, 2006. Accessed March 19, 2009.
3. Hartemink KJ, Wisselink W, Rauwerda JA, Girbes AR, Polderman KH. Novel applications of therapeutic hypothermia: report of three cases. Crit Care. 2004;8(5):R343-R346.
4. Dietrich WD. Presidential address presented at: 34th Annual Meeting of the Cervical Spine Research Society; November 30, 2006; Palm Beach, FL.
5. Bracken MB, Shepard MJ, Collins WF, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the second National Acute Spinal Cord Injury Study. N Engl J Med. 1990;322(20):1405-1411.
6. Horodyski MB, LuCante K, Escobar E, et al. Intermittent Cool, Dry Air Underneath Football Shoulder Pads Assists in Temperature Homeostasis. In: The American Orthopaedic Society for Sports Medicine Proceedings 2008; 87-88.
7. Kleiner DM, Almquist JL, Bailes J, et al; Inter-Association Task Force for Appropriate Care of the Spine-Injured Athlete. Prehospital Care of the Spine-Injured Athlete. Dallas, TX: National Athletic Trainers’ Association; 2001. http://www.msata.org/Resources/Documents/PreHospitalCare4SpineInjuredAthlete.pdf. Published March 2001. Accessed January 10, 2017.
8. Dididze M, Green BA, Dietrich WD, Vanni S, Wang MY, Levi AD. Systemic hypothermia in acute cervical spinal cord injury: a case-controlled study. Spinal Cord. 2013;51(5):395-400.
9. Levi AD, Casella G, Green BA, et al. Clinical outcomes using modest intravascular hypothermia after acute cervical spinal cord injury. Neurosurgery. 2010;66(4):670-677.
10. Levi AD, Green BA, Wang MY, et al. Clinical application of modest hypothermia after spinal cord injury. J Neurotrauma. 2009;26(3):407-415.
Take-Home Points
- Importance of on-field management.
- Preseason drilling of spinal injury management.
- Early and rapid intervention.
- Possible benefit of moderate systemic hypothermia as treatment for acute cervical injury.
In 2010, we reported the case of a professional American football player who sustained a complete cervical spinal cord injury (SCI) while tackling an opposing player.1 He received prompt medical and surgical care based on then-current recommendations, but was also treated with systemic hypothermia soon after his injury. Although systemic hypothermia had been used in the management of other neurologic injuries at that time, it had not been used in humans with acute SCI, except as described in 2 case reports.2,3 However, Dietrich4 described early emerging animal data on the efficacy of systemic hypothermia for acute SCI. We now provide a clinical update on our patient, who provided written informed consent for print and electronic publication of this case report.
Case Report
During a National Football League game, the player sustained a C3–C4 fracture-dislocation after a helmet-to-helmet hit on an opposing player. He fell face down on the ground and did not move. The team’s physician and trainer rushed to the player’s side, immediately assessed him, and initiated the emergency spinal resuscitation protocol.
As per protocol, the assigned team leader took charge of managing the player’s head to maintain in-line traction with the helmet in place until the head was secured in place on a backboard designed to accommodate the helmet.
Complete motor paralysis and sensory loss (American Spinal Injury Association [ASIA] level A) were noted below the clavicles during physical examination by the head athletic trainer and 2 independent physicians, and by self-report.
On arrival at the hospital, the patient had a core temperature of 98°F, which is substantially lower than the average core temperature (≤101.7°F) of an active football player.6He had a normal level of consciousness and normal cranial nerve function but remained without any voluntary motor function in the extremities and still had no sensation below the clavicles, except crude pressure sensation in one hand while in the emergency department. After the helmet and shoulder pads were removed, per National Athletic Trainers’ Association (NATA) protocol7 (Figure 2), he was stabilized, and a hard cervical collar was placed. A lateral radiograph (Figure 4) showed a C3–C4 facet dislocation with about 46% anterior translation of C3 on C4 and obvious disruption of the facets.
About 3 hours after injury, the patient was taken to the operating room. Although closed reduction improved alignment dramatically, it failed to completely reduce the dislocated left C3–C4 facet. An hour later, anterior C3–C4 discectomy was performed from the front with instrumented anterior interbody fusion. This was immediately followed by posterior decompressive laminectomy, bilateral facet reduction, and fusion with instrumentation. Surgery was completed within about 4 hours, almost exactly 7 hours after injury. Anesthesia records indicated a core temperature range of 94.1°F to 95.3°F with passive cooling during surgery. CT and MRI performed within 4 hours after surgery showed excellent cord decompression.
The next morning, about 14.5 hours after injury, the patient demonstrated a flicker of the adductor muscles of the lower extremities. An examination an hour later revealed 1/5 quadriceps, 2/5 adductors, and 1/5 gastrocnemius/soleus. A nurse’s hourly examinations and the surgeon’s repeat examinations revealed no other motor function. Sensory function was more difficult to evaluate because of sedation, but rudimentary sensation was noted throughout the lower extremities, and proprioception and vibratory sensation were noted as well. With passive cooling, it was difficult to consistently maintain moderate hypothermia; the patient’s core temperature ranged from 94.8°F to 98.8°F by 6:00 a.m. Therefore, the decision was made to place a Cordis sheath in the left femoral vein and introduce an intra-vena cava cooling catheter through it. This catheter was highly effective in maintaining the patient’s temperature at about 92.5°F.
Over the next 36 hours, the patient demonstrated increased motor activity in the upper and lower extremities: 1/5 biceps, 2-3/5 triceps, 3/5 quadriceps. He was slowly rewarmed and, on postoperative day 3, extubated.
At 2 years, the patient underwent another anterior-only cervical procedure: The inferior adjacent segment (C4–C5) was fused because of neck pain and deformity.
With respect to the original injury and the evolution in cord appearance, the patient had solid arthrodesis from C3–C5 with instrumentation in good position. There was evidence of loss of lordosis at C5–C6 with disk dessication and broad-based bulging. The spinal cord had evidence of myelomalacia; this was noted when the patient was in rehabilitation, 1 month after injury. The 2-cm × 11-mm area of myelomalacia was directly posterior to the fused C3–C4 interval (original MRI, Figure 5; 2-week MRI, Figure 6).
Conclusion
At the time this player was injured, use of systemic hypothermia with standard therapy for acute SCI was unique and controversial. Since then, smaller randomized human studies have described the tolerable safety profile, efficacy, and potential benefits of this intervention in acute SCI in humans.8-10 Now, modest systemic hypothermia can be one of many tools considered in the treatment of acute SCI. Before it can become the standard of care, however, additional larger prospective randomized studies need to be completed.
Am J Orthop. 2017;46(2):E79-E82. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- Importance of on-field management.
- Preseason drilling of spinal injury management.
- Early and rapid intervention.
- Possible benefit of moderate systemic hypothermia as treatment for acute cervical injury.
In 2010, we reported the case of a professional American football player who sustained a complete cervical spinal cord injury (SCI) while tackling an opposing player.1 He received prompt medical and surgical care based on then-current recommendations, but was also treated with systemic hypothermia soon after his injury. Although systemic hypothermia had been used in the management of other neurologic injuries at that time, it had not been used in humans with acute SCI, except as described in 2 case reports.2,3 However, Dietrich4 described early emerging animal data on the efficacy of systemic hypothermia for acute SCI. We now provide a clinical update on our patient, who provided written informed consent for print and electronic publication of this case report.
Case Report
During a National Football League game, the player sustained a C3–C4 fracture-dislocation after a helmet-to-helmet hit on an opposing player. He fell face down on the ground and did not move. The team’s physician and trainer rushed to the player’s side, immediately assessed him, and initiated the emergency spinal resuscitation protocol.
As per protocol, the assigned team leader took charge of managing the player’s head to maintain in-line traction with the helmet in place until the head was secured in place on a backboard designed to accommodate the helmet.
Complete motor paralysis and sensory loss (American Spinal Injury Association [ASIA] level A) were noted below the clavicles during physical examination by the head athletic trainer and 2 independent physicians, and by self-report.
On arrival at the hospital, the patient had a core temperature of 98°F, which is substantially lower than the average core temperature (≤101.7°F) of an active football player.6He had a normal level of consciousness and normal cranial nerve function but remained without any voluntary motor function in the extremities and still had no sensation below the clavicles, except crude pressure sensation in one hand while in the emergency department. After the helmet and shoulder pads were removed, per National Athletic Trainers’ Association (NATA) protocol7 (Figure 2), he was stabilized, and a hard cervical collar was placed. A lateral radiograph (Figure 4) showed a C3–C4 facet dislocation with about 46% anterior translation of C3 on C4 and obvious disruption of the facets.
About 3 hours after injury, the patient was taken to the operating room. Although closed reduction improved alignment dramatically, it failed to completely reduce the dislocated left C3–C4 facet. An hour later, anterior C3–C4 discectomy was performed from the front with instrumented anterior interbody fusion. This was immediately followed by posterior decompressive laminectomy, bilateral facet reduction, and fusion with instrumentation. Surgery was completed within about 4 hours, almost exactly 7 hours after injury. Anesthesia records indicated a core temperature range of 94.1°F to 95.3°F with passive cooling during surgery. CT and MRI performed within 4 hours after surgery showed excellent cord decompression.
The next morning, about 14.5 hours after injury, the patient demonstrated a flicker of the adductor muscles of the lower extremities. An examination an hour later revealed 1/5 quadriceps, 2/5 adductors, and 1/5 gastrocnemius/soleus. A nurse’s hourly examinations and the surgeon’s repeat examinations revealed no other motor function. Sensory function was more difficult to evaluate because of sedation, but rudimentary sensation was noted throughout the lower extremities, and proprioception and vibratory sensation were noted as well. With passive cooling, it was difficult to consistently maintain moderate hypothermia; the patient’s core temperature ranged from 94.8°F to 98.8°F by 6:00 a.m. Therefore, the decision was made to place a Cordis sheath in the left femoral vein and introduce an intra-vena cava cooling catheter through it. This catheter was highly effective in maintaining the patient’s temperature at about 92.5°F.
Over the next 36 hours, the patient demonstrated increased motor activity in the upper and lower extremities: 1/5 biceps, 2-3/5 triceps, 3/5 quadriceps. He was slowly rewarmed and, on postoperative day 3, extubated.
At 2 years, the patient underwent another anterior-only cervical procedure: The inferior adjacent segment (C4–C5) was fused because of neck pain and deformity.
With respect to the original injury and the evolution in cord appearance, the patient had solid arthrodesis from C3–C5 with instrumentation in good position. There was evidence of loss of lordosis at C5–C6 with disk dessication and broad-based bulging. The spinal cord had evidence of myelomalacia; this was noted when the patient was in rehabilitation, 1 month after injury. The 2-cm × 11-mm area of myelomalacia was directly posterior to the fused C3–C4 interval (original MRI, Figure 5; 2-week MRI, Figure 6).
Conclusion
At the time this player was injured, use of systemic hypothermia with standard therapy for acute SCI was unique and controversial. Since then, smaller randomized human studies have described the tolerable safety profile, efficacy, and potential benefits of this intervention in acute SCI in humans.8-10 Now, modest systemic hypothermia can be one of many tools considered in the treatment of acute SCI. Before it can become the standard of care, however, additional larger prospective randomized studies need to be completed.
Am J Orthop. 2017;46(2):E79-E82. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Cappuccino A, Bisson LJ, Carpenter B, Marzo J, Dietrich WD 3rd, Cappuccino H. The use of systemic hypothermia for the treatment of an acute cervical spinal cord injury in a professional football player. Spine. 2010;35(2):E57-E62.
2. Goldstein J. Lowering body temp shows promise for trauma treatment. Spinal Cord Injury Information Pages news blog. http://www.sci-info-pages.com/2006/05/lowering-body-temp-shows-promise-for.html. Published May 3, 2006. Accessed March 19, 2009.
3. Hartemink KJ, Wisselink W, Rauwerda JA, Girbes AR, Polderman KH. Novel applications of therapeutic hypothermia: report of three cases. Crit Care. 2004;8(5):R343-R346.
4. Dietrich WD. Presidential address presented at: 34th Annual Meeting of the Cervical Spine Research Society; November 30, 2006; Palm Beach, FL.
5. Bracken MB, Shepard MJ, Collins WF, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the second National Acute Spinal Cord Injury Study. N Engl J Med. 1990;322(20):1405-1411.
6. Horodyski MB, LuCante K, Escobar E, et al. Intermittent Cool, Dry Air Underneath Football Shoulder Pads Assists in Temperature Homeostasis. In: The American Orthopaedic Society for Sports Medicine Proceedings 2008; 87-88.
7. Kleiner DM, Almquist JL, Bailes J, et al; Inter-Association Task Force for Appropriate Care of the Spine-Injured Athlete. Prehospital Care of the Spine-Injured Athlete. Dallas, TX: National Athletic Trainers’ Association; 2001. http://www.msata.org/Resources/Documents/PreHospitalCare4SpineInjuredAthlete.pdf. Published March 2001. Accessed January 10, 2017.
8. Dididze M, Green BA, Dietrich WD, Vanni S, Wang MY, Levi AD. Systemic hypothermia in acute cervical spinal cord injury: a case-controlled study. Spinal Cord. 2013;51(5):395-400.
9. Levi AD, Casella G, Green BA, et al. Clinical outcomes using modest intravascular hypothermia after acute cervical spinal cord injury. Neurosurgery. 2010;66(4):670-677.
10. Levi AD, Green BA, Wang MY, et al. Clinical application of modest hypothermia after spinal cord injury. J Neurotrauma. 2009;26(3):407-415.
1. Cappuccino A, Bisson LJ, Carpenter B, Marzo J, Dietrich WD 3rd, Cappuccino H. The use of systemic hypothermia for the treatment of an acute cervical spinal cord injury in a professional football player. Spine. 2010;35(2):E57-E62.
2. Goldstein J. Lowering body temp shows promise for trauma treatment. Spinal Cord Injury Information Pages news blog. http://www.sci-info-pages.com/2006/05/lowering-body-temp-shows-promise-for.html. Published May 3, 2006. Accessed March 19, 2009.
3. Hartemink KJ, Wisselink W, Rauwerda JA, Girbes AR, Polderman KH. Novel applications of therapeutic hypothermia: report of three cases. Crit Care. 2004;8(5):R343-R346.
4. Dietrich WD. Presidential address presented at: 34th Annual Meeting of the Cervical Spine Research Society; November 30, 2006; Palm Beach, FL.
5. Bracken MB, Shepard MJ, Collins WF, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the second National Acute Spinal Cord Injury Study. N Engl J Med. 1990;322(20):1405-1411.
6. Horodyski MB, LuCante K, Escobar E, et al. Intermittent Cool, Dry Air Underneath Football Shoulder Pads Assists in Temperature Homeostasis. In: The American Orthopaedic Society for Sports Medicine Proceedings 2008; 87-88.
7. Kleiner DM, Almquist JL, Bailes J, et al; Inter-Association Task Force for Appropriate Care of the Spine-Injured Athlete. Prehospital Care of the Spine-Injured Athlete. Dallas, TX: National Athletic Trainers’ Association; 2001. http://www.msata.org/Resources/Documents/PreHospitalCare4SpineInjuredAthlete.pdf. Published March 2001. Accessed January 10, 2017.
8. Dididze M, Green BA, Dietrich WD, Vanni S, Wang MY, Levi AD. Systemic hypothermia in acute cervical spinal cord injury: a case-controlled study. Spinal Cord. 2013;51(5):395-400.
9. Levi AD, Casella G, Green BA, et al. Clinical outcomes using modest intravascular hypothermia after acute cervical spinal cord injury. Neurosurgery. 2010;66(4):670-677.
10. Levi AD, Green BA, Wang MY, et al. Clinical application of modest hypothermia after spinal cord injury. J Neurotrauma. 2009;26(3):407-415.

Eruptive Melanocytic Nevi During Azathioprine Therapy for Antisynthetase Syndrome
Case Report
A 50-year-old man with a history of antisynthetase syndrome (positive for anti–Jo-1 polymyositis with interstitial lung disease) and sarcoidosis presented for evaluation of numerous new moles. The lesions had developed on the trunk, arms, legs, hands, and feet approximately 3 weeks after starting azathioprine 100 mg once daily for pulmonary and muscular involvement of antisynthetase syndrome. He denied any preceding cutaneous inflammation or sunburns. He had no personal or family history of skin cancer, and no family members had multiple nevi. Physical examination revealed 30 to 40 benign-appearing, 2- to 5-mm, hyperpigmented macules scattered on the medial aspect of the right foot (Figure 1A), left palm (Figure 1B), back, abdomen, chest, arms, and legs. A larger, somewhat asymmetric, irregularly bordered, and irregularly pigmented macule was noted on the left side of the upper back. A punch biopsy of the lesion revealed a benign, mildly atypical lentiginous compound nevus (Figure 2). Pathology confirmed that the lesions represented eruptive melanocytic nevi (EMN). The patient continued azathioprine therapy and was followed with regular full-body skin examinations. Mycophenolate mofetil was suggested as an alternative therapy, if clinically appropriate, though this change has not been made by the patient’s rheumatologists.
Comment
A PubMed search of articles indexed for MEDLINE using the search terms eruptive melanocytic nevi and azathioprine revealed 14 cases of EMN in the setting of azathioprine therapy, either during azathioprine monotherapy or in combination with other immunosuppressants, including systemic corticosteroids, biologics, and cyclosporine (Table).1-5 The majority of these cases occurred in renal transplant patients,1 with 3 additional cases reported in the setting of Crohn disease,2,3,5 and another in a patient with myasthenia gravis.4 Patients ranged in age from 8 to 42 years (mean age, 22 years), with lesions developing a few months to up to 7 years after starting therapy. When specified, the reported lesions typically were small, ranging from 1 to 3 mm in size, and developed rapidly over a couple of months with a predilection for the palms, soles, and trunk. Although dysplastic nevi were described in only 2 patients, melanomas were not detected.
Various hypotheses have sought to explain the largely unknown etiology of EMN. Bovenschen et al3 suggested that immunocompromised patients have diminished immune surveillance in the skin, which allows for unchecked proliferation of melanocytes. Specifically, immune suppression may induce melanocyte-stimulating hormone or melanoma growth stimulatory activity, with composition-specific growth in skin at the palms and soles.3,4 The preferential growth on the palms and soles suggests that those regions may have special sensitivity to melanocyte-stimulating hormone.4 Woodhouse and Maytin6 postulated that the increased density of eccrine sweat glands in the palms and soles as well as the absence of pilosebaceous units and apocrine glands and plentiful Pacinian and Meissner corpuscles may allow for a unique response to circulating melanocytic growth factors. Another hypothesis suggests the presence of genetic factors that allow subclinical nests of nevus cells to form, which become clinical eruptions following chemotherapy or immunosuppressive therapy.3 Azathioprine also has been suggested to induce various transcription factors that play a critical role in differentiation and proliferation of melanocytic stem cells, which leads to the formation of nevi.4 Our case and others similar to it implore that further studies be done to determine the molecular mechanism driving this phenomenon and whether a specific genetic predisposition exists that lowers the threshold for rapid proliferation of melanocytes given an immunosuppressed status.2
The risk for melanoma development in cases of EMN is unknown. Although our review of the literature did not reveal any melanomas reported in cases attributed to azathioprine, a theoretical risk exists given the established associations between melanoma and immunosuppression as well as increased numbers of nevi.6 Accordingly, these patients should be followed with regular skin examinations and biopsies of atypical-appearing lesions as indicated.2,3,5 Braun et al4 also suggested the discontinuance of azathioprine and switch to mycophenolic acid, which has not been noted to cause such eruptions; this drug was recommended in our case.
- Alaibac M, Piaserico S, Rossi CR, et al. Eruptive melanocytic nevi in patients with renal allografts: report of 10 cases with dermoscopic findings. J Am Acad Dermatol. 2003;49:1020-1022.
- Belloni FA, Piaserico S, Zattra E, et al. Dermoscopic features of eruptive melanocytic naevi in an adult patient receiving immunosuppressive therapy for Crohn’s disease. Melanoma Res. 2005;15:223-224.
- Bovenschen HJ, Tjioe M, Vermaat H, et al. Induction of eruptive benign melanocytic naevi by immune suppressive agents, including biologicals. Br J Dermatol. 2006;154:880-884.
- Braun SA, Helbig D, Frank J, et al. Eruptive melanocytic nevi during azathioprine therapy in myasthenia gravis [in German]. Hautarzt. 2012;63:756-759.
- Wonders J, De Boer N, Van Weyenberg S. Spot diagnosis: eruptive melanocytic naevi during azathioprine therapy in Crohn’s disease [published online March 6, 2012]. J Crohns Colitis. 2012;6:636.
- Woodhouse J, Maytin EV. Eruptive nevi of the palms and soles. J Am Acad Dermatol. 2005;52(5 suppl 1):S96-S100.
Case Report
A 50-year-old man with a history of antisynthetase syndrome (positive for anti–Jo-1 polymyositis with interstitial lung disease) and sarcoidosis presented for evaluation of numerous new moles. The lesions had developed on the trunk, arms, legs, hands, and feet approximately 3 weeks after starting azathioprine 100 mg once daily for pulmonary and muscular involvement of antisynthetase syndrome. He denied any preceding cutaneous inflammation or sunburns. He had no personal or family history of skin cancer, and no family members had multiple nevi. Physical examination revealed 30 to 40 benign-appearing, 2- to 5-mm, hyperpigmented macules scattered on the medial aspect of the right foot (Figure 1A), left palm (Figure 1B), back, abdomen, chest, arms, and legs. A larger, somewhat asymmetric, irregularly bordered, and irregularly pigmented macule was noted on the left side of the upper back. A punch biopsy of the lesion revealed a benign, mildly atypical lentiginous compound nevus (Figure 2). Pathology confirmed that the lesions represented eruptive melanocytic nevi (EMN). The patient continued azathioprine therapy and was followed with regular full-body skin examinations. Mycophenolate mofetil was suggested as an alternative therapy, if clinically appropriate, though this change has not been made by the patient’s rheumatologists.
Comment
A PubMed search of articles indexed for MEDLINE using the search terms eruptive melanocytic nevi and azathioprine revealed 14 cases of EMN in the setting of azathioprine therapy, either during azathioprine monotherapy or in combination with other immunosuppressants, including systemic corticosteroids, biologics, and cyclosporine (Table).1-5 The majority of these cases occurred in renal transplant patients,1 with 3 additional cases reported in the setting of Crohn disease,2,3,5 and another in a patient with myasthenia gravis.4 Patients ranged in age from 8 to 42 years (mean age, 22 years), with lesions developing a few months to up to 7 years after starting therapy. When specified, the reported lesions typically were small, ranging from 1 to 3 mm in size, and developed rapidly over a couple of months with a predilection for the palms, soles, and trunk. Although dysplastic nevi were described in only 2 patients, melanomas were not detected.
Various hypotheses have sought to explain the largely unknown etiology of EMN. Bovenschen et al3 suggested that immunocompromised patients have diminished immune surveillance in the skin, which allows for unchecked proliferation of melanocytes. Specifically, immune suppression may induce melanocyte-stimulating hormone or melanoma growth stimulatory activity, with composition-specific growth in skin at the palms and soles.3,4 The preferential growth on the palms and soles suggests that those regions may have special sensitivity to melanocyte-stimulating hormone.4 Woodhouse and Maytin6 postulated that the increased density of eccrine sweat glands in the palms and soles as well as the absence of pilosebaceous units and apocrine glands and plentiful Pacinian and Meissner corpuscles may allow for a unique response to circulating melanocytic growth factors. Another hypothesis suggests the presence of genetic factors that allow subclinical nests of nevus cells to form, which become clinical eruptions following chemotherapy or immunosuppressive therapy.3 Azathioprine also has been suggested to induce various transcription factors that play a critical role in differentiation and proliferation of melanocytic stem cells, which leads to the formation of nevi.4 Our case and others similar to it implore that further studies be done to determine the molecular mechanism driving this phenomenon and whether a specific genetic predisposition exists that lowers the threshold for rapid proliferation of melanocytes given an immunosuppressed status.2
The risk for melanoma development in cases of EMN is unknown. Although our review of the literature did not reveal any melanomas reported in cases attributed to azathioprine, a theoretical risk exists given the established associations between melanoma and immunosuppression as well as increased numbers of nevi.6 Accordingly, these patients should be followed with regular skin examinations and biopsies of atypical-appearing lesions as indicated.2,3,5 Braun et al4 also suggested the discontinuance of azathioprine and switch to mycophenolic acid, which has not been noted to cause such eruptions; this drug was recommended in our case.
Case Report
A 50-year-old man with a history of antisynthetase syndrome (positive for anti–Jo-1 polymyositis with interstitial lung disease) and sarcoidosis presented for evaluation of numerous new moles. The lesions had developed on the trunk, arms, legs, hands, and feet approximately 3 weeks after starting azathioprine 100 mg once daily for pulmonary and muscular involvement of antisynthetase syndrome. He denied any preceding cutaneous inflammation or sunburns. He had no personal or family history of skin cancer, and no family members had multiple nevi. Physical examination revealed 30 to 40 benign-appearing, 2- to 5-mm, hyperpigmented macules scattered on the medial aspect of the right foot (Figure 1A), left palm (Figure 1B), back, abdomen, chest, arms, and legs. A larger, somewhat asymmetric, irregularly bordered, and irregularly pigmented macule was noted on the left side of the upper back. A punch biopsy of the lesion revealed a benign, mildly atypical lentiginous compound nevus (Figure 2). Pathology confirmed that the lesions represented eruptive melanocytic nevi (EMN). The patient continued azathioprine therapy and was followed with regular full-body skin examinations. Mycophenolate mofetil was suggested as an alternative therapy, if clinically appropriate, though this change has not been made by the patient’s rheumatologists.
Comment
A PubMed search of articles indexed for MEDLINE using the search terms eruptive melanocytic nevi and azathioprine revealed 14 cases of EMN in the setting of azathioprine therapy, either during azathioprine monotherapy or in combination with other immunosuppressants, including systemic corticosteroids, biologics, and cyclosporine (Table).1-5 The majority of these cases occurred in renal transplant patients,1 with 3 additional cases reported in the setting of Crohn disease,2,3,5 and another in a patient with myasthenia gravis.4 Patients ranged in age from 8 to 42 years (mean age, 22 years), with lesions developing a few months to up to 7 years after starting therapy. When specified, the reported lesions typically were small, ranging from 1 to 3 mm in size, and developed rapidly over a couple of months with a predilection for the palms, soles, and trunk. Although dysplastic nevi were described in only 2 patients, melanomas were not detected.
Various hypotheses have sought to explain the largely unknown etiology of EMN. Bovenschen et al3 suggested that immunocompromised patients have diminished immune surveillance in the skin, which allows for unchecked proliferation of melanocytes. Specifically, immune suppression may induce melanocyte-stimulating hormone or melanoma growth stimulatory activity, with composition-specific growth in skin at the palms and soles.3,4 The preferential growth on the palms and soles suggests that those regions may have special sensitivity to melanocyte-stimulating hormone.4 Woodhouse and Maytin6 postulated that the increased density of eccrine sweat glands in the palms and soles as well as the absence of pilosebaceous units and apocrine glands and plentiful Pacinian and Meissner corpuscles may allow for a unique response to circulating melanocytic growth factors. Another hypothesis suggests the presence of genetic factors that allow subclinical nests of nevus cells to form, which become clinical eruptions following chemotherapy or immunosuppressive therapy.3 Azathioprine also has been suggested to induce various transcription factors that play a critical role in differentiation and proliferation of melanocytic stem cells, which leads to the formation of nevi.4 Our case and others similar to it implore that further studies be done to determine the molecular mechanism driving this phenomenon and whether a specific genetic predisposition exists that lowers the threshold for rapid proliferation of melanocytes given an immunosuppressed status.2
The risk for melanoma development in cases of EMN is unknown. Although our review of the literature did not reveal any melanomas reported in cases attributed to azathioprine, a theoretical risk exists given the established associations between melanoma and immunosuppression as well as increased numbers of nevi.6 Accordingly, these patients should be followed with regular skin examinations and biopsies of atypical-appearing lesions as indicated.2,3,5 Braun et al4 also suggested the discontinuance of azathioprine and switch to mycophenolic acid, which has not been noted to cause such eruptions; this drug was recommended in our case.
- Alaibac M, Piaserico S, Rossi CR, et al. Eruptive melanocytic nevi in patients with renal allografts: report of 10 cases with dermoscopic findings. J Am Acad Dermatol. 2003;49:1020-1022.
- Belloni FA, Piaserico S, Zattra E, et al. Dermoscopic features of eruptive melanocytic naevi in an adult patient receiving immunosuppressive therapy for Crohn’s disease. Melanoma Res. 2005;15:223-224.
- Bovenschen HJ, Tjioe M, Vermaat H, et al. Induction of eruptive benign melanocytic naevi by immune suppressive agents, including biologicals. Br J Dermatol. 2006;154:880-884.
- Braun SA, Helbig D, Frank J, et al. Eruptive melanocytic nevi during azathioprine therapy in myasthenia gravis [in German]. Hautarzt. 2012;63:756-759.
- Wonders J, De Boer N, Van Weyenberg S. Spot diagnosis: eruptive melanocytic naevi during azathioprine therapy in Crohn’s disease [published online March 6, 2012]. J Crohns Colitis. 2012;6:636.
- Woodhouse J, Maytin EV. Eruptive nevi of the palms and soles. J Am Acad Dermatol. 2005;52(5 suppl 1):S96-S100.
- Alaibac M, Piaserico S, Rossi CR, et al. Eruptive melanocytic nevi in patients with renal allografts: report of 10 cases with dermoscopic findings. J Am Acad Dermatol. 2003;49:1020-1022.
- Belloni FA, Piaserico S, Zattra E, et al. Dermoscopic features of eruptive melanocytic naevi in an adult patient receiving immunosuppressive therapy for Crohn’s disease. Melanoma Res. 2005;15:223-224.
- Bovenschen HJ, Tjioe M, Vermaat H, et al. Induction of eruptive benign melanocytic naevi by immune suppressive agents, including biologicals. Br J Dermatol. 2006;154:880-884.
- Braun SA, Helbig D, Frank J, et al. Eruptive melanocytic nevi during azathioprine therapy in myasthenia gravis [in German]. Hautarzt. 2012;63:756-759.
- Wonders J, De Boer N, Van Weyenberg S. Spot diagnosis: eruptive melanocytic naevi during azathioprine therapy in Crohn’s disease [published online March 6, 2012]. J Crohns Colitis. 2012;6:636.
- Woodhouse J, Maytin EV. Eruptive nevi of the palms and soles. J Am Acad Dermatol. 2005;52(5 suppl 1):S96-S100.
Practice Points
- A theoretical risk exists in the setting of eruptive melanocytic nevi (EMN) given the established associations between melanoma and immunosuppression as well as increased numbers of nevi.
- Follow patients with EMN with regular skin examinations and biopsies of atypical-appearing lesions given the increased risk for melanoma in this population.
Muscle spasms, twitches in arm upon throwing • Dx?
THE CASE
A 31-year-old right-handed college baseball coach presented to his family physician (FP) with concerns about the “yips” in his right arm. His ability to throw a baseball had been gradually deteriorating. Involuntary upper right arm muscle contractions and spasms, which began intermittently when he was a teenager, were now a real problem for him as an adult. (See the video below.) The patient was having difficulty rolling a baseball underhand to players as part of infield practice and he was experiencing muscle spasms when lifting his right arm over his head. “Twitches” in the patient’s upper arm were making drinking difficult, but he had no problems feeding himself, writing, or performing other basic activities of daily living.
The patient experienced the same symptoms whether it was baseball season or not. He hadn’t noticed a change in symptoms with caffeine and denied use of any other stimulants in the last 4 years. His symptoms didn’t improve or worsen with greater or lesser quantity or quality of sleep or when he concentrated on stifling the involuntary movements. He had attempted to learn to throw left-handed to overcome the impairment, but was concerned that the same problem would occur in his left arm.
The patient had previously worked with a sports psychologist and hypnotherapist to overcome any potential subconscious performance anxiety, but this hadn’t helped. Stretching and strengthening with a physical therapist and numerous sessions with an acupuncturist hadn’t helped either. Despite this, he believed the problem to be primarily psychological.
The patient’s history included mild attention deficit disorder and exercise-induced asthma; his family history was negative for any movement or psychiatric disorders. He had 2 dislocation repairs on his left, non-throwing shoulder in his early twenties. His medications included fluticasone-salmeterol twice daily and albuterol, as needed.
The patient denied myalgia or arthralgia, decreased passive range of motion, shoulder or arm weakness, swelling, or muscle atrophy. He also didn’t have paresthesias in his right arm or hand, a resting tremor, difficulty moving (other than drinking from a cup), difficulty moving other extremities, dizziness, imbalance, or seizures.
The patient’s vital signs were normal. He had full range of motion and 5 out of 5 strength without pain during right shoulder abduction, external and internal rotation, an empty can test, a lower back lift off (Gerber’s) test, and a test of bicep and tricep strength, along with negative Neer and Hawkins tests.
There was no evidence of muscle wasting or asymmetry in the bilateral upper extremities. The patient’s deep tendon reflex grade was 2+ out of 4 in both of his arms. He didn’t have a sensory deficit to light touch in areas of C5 to T1 and he had normal cranial nerves II to XII. He had normal rapid alternating movements, heel-to-shin testing, and finger-to-nose testing, as well as a normal gait and Romberg test.
The patient provided a video showing the abnormal involuntary flexion of his shoulder when attempting to throw a baseball.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
THE DIAGNOSIS
The patient’s FP was aware of the “yips,” a condition that is commonly viewed as psychological or related to performance anxiety. (The “yips” are colloquially known as “Steve Blass Disease”—named after a Pittsburgh Pirates pitcher who suddenly lost the ability to control his pitches.1) But based on the patient’s clinical presentation and history of seeing a number of mental health care providers—in addition to his worsening symptoms—the FP ordered magnetic resonance imaging (MRI) of the brain. The MRI turned out to be unremarkable, so the patient was referred to Neurology.
In the general neurology clinic, a diagnosis of Wilson’s disease (a condition that leads to excess copper deposition in multiple organ systems, including the nervous system) was considered, as it can cause symptoms similar to those our patient was experiencing. However, a complete blood count, complete metabolic panel, antinuclear antibody test, ceruloplasmin test, and copper level were all normal, effectively ruling it out. An MRI of the cervical spine showed mild to moderate right foraminal stenosis at C3-4 and C5-6, but this did not explain the patient’s symptoms.
A diagnosis of paroxysmal exercise-induced dystonia was also considered at the time of the initial work-up, as our patient’s symptoms were most pronounced during physical activity. But this condition usually responds to antiepileptics, and carbamazepine and phenytoin were each tried for multiple months early in his evaluation without benefit.
3 factors led to a diagnosis of focal limb dystonia: Only our patient’s right arm was affected, his laboratory and imaging work-ups were negative, and he didn’t respond to antiepileptic treatment. Characterization of a movement disorder is based upon phenomenology. In this case, the patient had sustained abnormal posturing at the shoulder during right upper limb activation, which was only triggered with specific voluntary actions. This was consistent with dystonia, a movement disorder characterized by sustained or intermittent muscle contractions causing abnormal movements and/or postures—often initiated or worsened by voluntary action.2
DISCUSSION
The “yips,” or intermittent, transient tremors, jerks, or spasms3 that are seen in athletes, are well-documented in the lay press, but haven’t been significantly addressed in the medical literature.4 Stigma surrounding the condition among athletes likely leads to under-reporting. Athletes typically experience yips with fine motor movements, such as short putts in golf and pitching in baseball. In fact, while the majority of the medical literature on yips revolves around golfers, many talented baseball players have had their careers altered by the condition. The yips may also affect movements in sports like darts, cricket, table tennis, and billiards.
In 1984, dystonia was defined as a disorder of sensorimotor integration that results in co-contraction of agonist/antagonist muscles, and may be characterized by state dependence (exacerbation with specific activities) or sensory tricks (amelioration with specific types of sensory input).5 In 2013, the definition was revised to remove “co-contraction” from the definition because phenomenology alone is sufficient to make the diagnosis.1
Many athletes and sports fans believe the yips are caused by performance anxiety or related phobias, but evidence suggests that many athletes with the movement disorder may actually have focal limb dystonia.6,7 The yips can, however, lead to performance anxiety,3 but there has been no difference noted between the anxiety level of golfers with or without the yips.7 Psychological treatment approaches are commonly employed, but surface electromyograms have shown abnormal co-contraction of wrist flexor and extensor muscles in 5 out of 10 golfers with the yips (but 0 of those without) while putting—which is consistent with focal limb dystonia.8
Botulinum toxin injections are Tx of choice, but can cause weakness
Muscle relaxers, such as baclofen and benzodiazepines, as well as dopamine antagonists, can ameliorate dystonia.9 Focal limb dystonia may also respond to the antispasmodic trihexyphenidyl, but the dose must often be limited due to adverse effects such as nausea, dizziness, and anxiety.10
Botulinum toxin injections have proven effective for focal limb dystonia11 and are considered the treatment of choice. However, there are few reports on their use in athletes, where the adverse effect of weakness could affect performance. One case report also showed improvement of yips with acupuncture, although this has not been extensively studied.12
Our patient didn’t respond to low-dose (2 mg twice a day) trihexyphenidyl. Tetrabenazine, a dopamine depletor frequently used for hyperkinetic disorders, was not effective at 25 mg taken prior to coaching sessions. Higher doses of an anticholinergic could have been effective, but the patient declined our recommendation to pursue this (or botulinum toxin injections). He decided instead to train himself to use his left arm while coaching.
THE TAKEAWAY
Athletes who play sports that require precision movements commonly develop the yips. While the prevailing theory among athletes is that this is a psychological phenomenon, evidence shows that this may in fact be a neurologic focal dystonia caused by repetitive use. Greater awareness of yips as a possible organic, treatable neurologic condition is needed in order to stimulate more research on this topic.
1. Baseball’s head cases often prove baffling. USA Today Baseball Weekly. 2001. Available at: http://usatoday30.usatoday.com/sports/bbw/2001-02-07/2001-02-07-head.htm. Accessed March 15, 2017.
2. Albanese A, Bhatia K, Bressman SB, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord. 2013;28:863-873.
3. Dhungana S, Jankovic J. Yips and other movement disorders in golfers. Mov Disord. 2013;28:576-581.
4. Stacy MA, ed. Handbook of dystonia. New York, NY: Informa Healthcare USA, Inc; 2007.
5. Fahn S, Marsden CD, Calne DB. Classification and investigation of dystonia. In: Marsden CD, Fahn S, eds. Movement disorders 2. London: Butterworths; 1987:332-358.
6. Smith AM, Adler CH, Crews D, et al. The ‘yips’ in golf: a continuum between a focal dystonia and choking. Sports Med. 2003;33:13-31.
7. Sachdev P. Golfers’ cramp: clinical characteristics and evidence against it being an anxiety disorder. Mov Disord. 1992;7:326-332.
8. Adler CH, Crews D, Hentz JG, et al. Abnormal co-contraction in yips-affected but not unaffected golfers: evidence for focal dystonia. Neurology. 2005;64:1813-1814.
9. Jankovic J. Treatment of hyperkinetic movement disorders. Lancet Neurol. 2009;8:844-856.
10. Jankovic J. Treatment of dystonia. Lancet Neurol. 2006;5:864-872.
11. Lungu C, Karp BI, Alter K, et al. Long-term follow-up of botulinum toxin therapy for focal hand dystonia: outcome at 10 years or more. Mov Disord. 2011;26:750-753.
12. Rosted P. Acupuncture for treatment of the yips?—a case report. Acupunct Med. 2005;23:188-189.
THE CASE
A 31-year-old right-handed college baseball coach presented to his family physician (FP) with concerns about the “yips” in his right arm. His ability to throw a baseball had been gradually deteriorating. Involuntary upper right arm muscle contractions and spasms, which began intermittently when he was a teenager, were now a real problem for him as an adult. (See the video below.) The patient was having difficulty rolling a baseball underhand to players as part of infield practice and he was experiencing muscle spasms when lifting his right arm over his head. “Twitches” in the patient’s upper arm were making drinking difficult, but he had no problems feeding himself, writing, or performing other basic activities of daily living.
The patient experienced the same symptoms whether it was baseball season or not. He hadn’t noticed a change in symptoms with caffeine and denied use of any other stimulants in the last 4 years. His symptoms didn’t improve or worsen with greater or lesser quantity or quality of sleep or when he concentrated on stifling the involuntary movements. He had attempted to learn to throw left-handed to overcome the impairment, but was concerned that the same problem would occur in his left arm.
The patient had previously worked with a sports psychologist and hypnotherapist to overcome any potential subconscious performance anxiety, but this hadn’t helped. Stretching and strengthening with a physical therapist and numerous sessions with an acupuncturist hadn’t helped either. Despite this, he believed the problem to be primarily psychological.
The patient’s history included mild attention deficit disorder and exercise-induced asthma; his family history was negative for any movement or psychiatric disorders. He had 2 dislocation repairs on his left, non-throwing shoulder in his early twenties. His medications included fluticasone-salmeterol twice daily and albuterol, as needed.
The patient denied myalgia or arthralgia, decreased passive range of motion, shoulder or arm weakness, swelling, or muscle atrophy. He also didn’t have paresthesias in his right arm or hand, a resting tremor, difficulty moving (other than drinking from a cup), difficulty moving other extremities, dizziness, imbalance, or seizures.
The patient’s vital signs were normal. He had full range of motion and 5 out of 5 strength without pain during right shoulder abduction, external and internal rotation, an empty can test, a lower back lift off (Gerber’s) test, and a test of bicep and tricep strength, along with negative Neer and Hawkins tests.
There was no evidence of muscle wasting or asymmetry in the bilateral upper extremities. The patient’s deep tendon reflex grade was 2+ out of 4 in both of his arms. He didn’t have a sensory deficit to light touch in areas of C5 to T1 and he had normal cranial nerves II to XII. He had normal rapid alternating movements, heel-to-shin testing, and finger-to-nose testing, as well as a normal gait and Romberg test.
The patient provided a video showing the abnormal involuntary flexion of his shoulder when attempting to throw a baseball.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
THE DIAGNOSIS
The patient’s FP was aware of the “yips,” a condition that is commonly viewed as psychological or related to performance anxiety. (The “yips” are colloquially known as “Steve Blass Disease”—named after a Pittsburgh Pirates pitcher who suddenly lost the ability to control his pitches.1) But based on the patient’s clinical presentation and history of seeing a number of mental health care providers—in addition to his worsening symptoms—the FP ordered magnetic resonance imaging (MRI) of the brain. The MRI turned out to be unremarkable, so the patient was referred to Neurology.
In the general neurology clinic, a diagnosis of Wilson’s disease (a condition that leads to excess copper deposition in multiple organ systems, including the nervous system) was considered, as it can cause symptoms similar to those our patient was experiencing. However, a complete blood count, complete metabolic panel, antinuclear antibody test, ceruloplasmin test, and copper level were all normal, effectively ruling it out. An MRI of the cervical spine showed mild to moderate right foraminal stenosis at C3-4 and C5-6, but this did not explain the patient’s symptoms.
A diagnosis of paroxysmal exercise-induced dystonia was also considered at the time of the initial work-up, as our patient’s symptoms were most pronounced during physical activity. But this condition usually responds to antiepileptics, and carbamazepine and phenytoin were each tried for multiple months early in his evaluation without benefit.
3 factors led to a diagnosis of focal limb dystonia: Only our patient’s right arm was affected, his laboratory and imaging work-ups were negative, and he didn’t respond to antiepileptic treatment. Characterization of a movement disorder is based upon phenomenology. In this case, the patient had sustained abnormal posturing at the shoulder during right upper limb activation, which was only triggered with specific voluntary actions. This was consistent with dystonia, a movement disorder characterized by sustained or intermittent muscle contractions causing abnormal movements and/or postures—often initiated or worsened by voluntary action.2
DISCUSSION
The “yips,” or intermittent, transient tremors, jerks, or spasms3 that are seen in athletes, are well-documented in the lay press, but haven’t been significantly addressed in the medical literature.4 Stigma surrounding the condition among athletes likely leads to under-reporting. Athletes typically experience yips with fine motor movements, such as short putts in golf and pitching in baseball. In fact, while the majority of the medical literature on yips revolves around golfers, many talented baseball players have had their careers altered by the condition. The yips may also affect movements in sports like darts, cricket, table tennis, and billiards.
In 1984, dystonia was defined as a disorder of sensorimotor integration that results in co-contraction of agonist/antagonist muscles, and may be characterized by state dependence (exacerbation with specific activities) or sensory tricks (amelioration with specific types of sensory input).5 In 2013, the definition was revised to remove “co-contraction” from the definition because phenomenology alone is sufficient to make the diagnosis.1
Many athletes and sports fans believe the yips are caused by performance anxiety or related phobias, but evidence suggests that many athletes with the movement disorder may actually have focal limb dystonia.6,7 The yips can, however, lead to performance anxiety,3 but there has been no difference noted between the anxiety level of golfers with or without the yips.7 Psychological treatment approaches are commonly employed, but surface electromyograms have shown abnormal co-contraction of wrist flexor and extensor muscles in 5 out of 10 golfers with the yips (but 0 of those without) while putting—which is consistent with focal limb dystonia.8
Botulinum toxin injections are Tx of choice, but can cause weakness
Muscle relaxers, such as baclofen and benzodiazepines, as well as dopamine antagonists, can ameliorate dystonia.9 Focal limb dystonia may also respond to the antispasmodic trihexyphenidyl, but the dose must often be limited due to adverse effects such as nausea, dizziness, and anxiety.10
Botulinum toxin injections have proven effective for focal limb dystonia11 and are considered the treatment of choice. However, there are few reports on their use in athletes, where the adverse effect of weakness could affect performance. One case report also showed improvement of yips with acupuncture, although this has not been extensively studied.12
Our patient didn’t respond to low-dose (2 mg twice a day) trihexyphenidyl. Tetrabenazine, a dopamine depletor frequently used for hyperkinetic disorders, was not effective at 25 mg taken prior to coaching sessions. Higher doses of an anticholinergic could have been effective, but the patient declined our recommendation to pursue this (or botulinum toxin injections). He decided instead to train himself to use his left arm while coaching.
THE TAKEAWAY
Athletes who play sports that require precision movements commonly develop the yips. While the prevailing theory among athletes is that this is a psychological phenomenon, evidence shows that this may in fact be a neurologic focal dystonia caused by repetitive use. Greater awareness of yips as a possible organic, treatable neurologic condition is needed in order to stimulate more research on this topic.
THE CASE
A 31-year-old right-handed college baseball coach presented to his family physician (FP) with concerns about the “yips” in his right arm. His ability to throw a baseball had been gradually deteriorating. Involuntary upper right arm muscle contractions and spasms, which began intermittently when he was a teenager, were now a real problem for him as an adult. (See the video below.) The patient was having difficulty rolling a baseball underhand to players as part of infield practice and he was experiencing muscle spasms when lifting his right arm over his head. “Twitches” in the patient’s upper arm were making drinking difficult, but he had no problems feeding himself, writing, or performing other basic activities of daily living.
The patient experienced the same symptoms whether it was baseball season or not. He hadn’t noticed a change in symptoms with caffeine and denied use of any other stimulants in the last 4 years. His symptoms didn’t improve or worsen with greater or lesser quantity or quality of sleep or when he concentrated on stifling the involuntary movements. He had attempted to learn to throw left-handed to overcome the impairment, but was concerned that the same problem would occur in his left arm.
The patient had previously worked with a sports psychologist and hypnotherapist to overcome any potential subconscious performance anxiety, but this hadn’t helped. Stretching and strengthening with a physical therapist and numerous sessions with an acupuncturist hadn’t helped either. Despite this, he believed the problem to be primarily psychological.
The patient’s history included mild attention deficit disorder and exercise-induced asthma; his family history was negative for any movement or psychiatric disorders. He had 2 dislocation repairs on his left, non-throwing shoulder in his early twenties. His medications included fluticasone-salmeterol twice daily and albuterol, as needed.
The patient denied myalgia or arthralgia, decreased passive range of motion, shoulder or arm weakness, swelling, or muscle atrophy. He also didn’t have paresthesias in his right arm or hand, a resting tremor, difficulty moving (other than drinking from a cup), difficulty moving other extremities, dizziness, imbalance, or seizures.
The patient’s vital signs were normal. He had full range of motion and 5 out of 5 strength without pain during right shoulder abduction, external and internal rotation, an empty can test, a lower back lift off (Gerber’s) test, and a test of bicep and tricep strength, along with negative Neer and Hawkins tests.
There was no evidence of muscle wasting or asymmetry in the bilateral upper extremities. The patient’s deep tendon reflex grade was 2+ out of 4 in both of his arms. He didn’t have a sensory deficit to light touch in areas of C5 to T1 and he had normal cranial nerves II to XII. He had normal rapid alternating movements, heel-to-shin testing, and finger-to-nose testing, as well as a normal gait and Romberg test.
The patient provided a video showing the abnormal involuntary flexion of his shoulder when attempting to throw a baseball.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
THE DIAGNOSIS
The patient’s FP was aware of the “yips,” a condition that is commonly viewed as psychological or related to performance anxiety. (The “yips” are colloquially known as “Steve Blass Disease”—named after a Pittsburgh Pirates pitcher who suddenly lost the ability to control his pitches.1) But based on the patient’s clinical presentation and history of seeing a number of mental health care providers—in addition to his worsening symptoms—the FP ordered magnetic resonance imaging (MRI) of the brain. The MRI turned out to be unremarkable, so the patient was referred to Neurology.
In the general neurology clinic, a diagnosis of Wilson’s disease (a condition that leads to excess copper deposition in multiple organ systems, including the nervous system) was considered, as it can cause symptoms similar to those our patient was experiencing. However, a complete blood count, complete metabolic panel, antinuclear antibody test, ceruloplasmin test, and copper level were all normal, effectively ruling it out. An MRI of the cervical spine showed mild to moderate right foraminal stenosis at C3-4 and C5-6, but this did not explain the patient’s symptoms.
A diagnosis of paroxysmal exercise-induced dystonia was also considered at the time of the initial work-up, as our patient’s symptoms were most pronounced during physical activity. But this condition usually responds to antiepileptics, and carbamazepine and phenytoin were each tried for multiple months early in his evaluation without benefit.
3 factors led to a diagnosis of focal limb dystonia: Only our patient’s right arm was affected, his laboratory and imaging work-ups were negative, and he didn’t respond to antiepileptic treatment. Characterization of a movement disorder is based upon phenomenology. In this case, the patient had sustained abnormal posturing at the shoulder during right upper limb activation, which was only triggered with specific voluntary actions. This was consistent with dystonia, a movement disorder characterized by sustained or intermittent muscle contractions causing abnormal movements and/or postures—often initiated or worsened by voluntary action.2
DISCUSSION
The “yips,” or intermittent, transient tremors, jerks, or spasms3 that are seen in athletes, are well-documented in the lay press, but haven’t been significantly addressed in the medical literature.4 Stigma surrounding the condition among athletes likely leads to under-reporting. Athletes typically experience yips with fine motor movements, such as short putts in golf and pitching in baseball. In fact, while the majority of the medical literature on yips revolves around golfers, many talented baseball players have had their careers altered by the condition. The yips may also affect movements in sports like darts, cricket, table tennis, and billiards.
In 1984, dystonia was defined as a disorder of sensorimotor integration that results in co-contraction of agonist/antagonist muscles, and may be characterized by state dependence (exacerbation with specific activities) or sensory tricks (amelioration with specific types of sensory input).5 In 2013, the definition was revised to remove “co-contraction” from the definition because phenomenology alone is sufficient to make the diagnosis.1
Many athletes and sports fans believe the yips are caused by performance anxiety or related phobias, but evidence suggests that many athletes with the movement disorder may actually have focal limb dystonia.6,7 The yips can, however, lead to performance anxiety,3 but there has been no difference noted between the anxiety level of golfers with or without the yips.7 Psychological treatment approaches are commonly employed, but surface electromyograms have shown abnormal co-contraction of wrist flexor and extensor muscles in 5 out of 10 golfers with the yips (but 0 of those without) while putting—which is consistent with focal limb dystonia.8
Botulinum toxin injections are Tx of choice, but can cause weakness
Muscle relaxers, such as baclofen and benzodiazepines, as well as dopamine antagonists, can ameliorate dystonia.9 Focal limb dystonia may also respond to the antispasmodic trihexyphenidyl, but the dose must often be limited due to adverse effects such as nausea, dizziness, and anxiety.10
Botulinum toxin injections have proven effective for focal limb dystonia11 and are considered the treatment of choice. However, there are few reports on their use in athletes, where the adverse effect of weakness could affect performance. One case report also showed improvement of yips with acupuncture, although this has not been extensively studied.12
Our patient didn’t respond to low-dose (2 mg twice a day) trihexyphenidyl. Tetrabenazine, a dopamine depletor frequently used for hyperkinetic disorders, was not effective at 25 mg taken prior to coaching sessions. Higher doses of an anticholinergic could have been effective, but the patient declined our recommendation to pursue this (or botulinum toxin injections). He decided instead to train himself to use his left arm while coaching.
THE TAKEAWAY
Athletes who play sports that require precision movements commonly develop the yips. While the prevailing theory among athletes is that this is a psychological phenomenon, evidence shows that this may in fact be a neurologic focal dystonia caused by repetitive use. Greater awareness of yips as a possible organic, treatable neurologic condition is needed in order to stimulate more research on this topic.
1. Baseball’s head cases often prove baffling. USA Today Baseball Weekly. 2001. Available at: http://usatoday30.usatoday.com/sports/bbw/2001-02-07/2001-02-07-head.htm. Accessed March 15, 2017.
2. Albanese A, Bhatia K, Bressman SB, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord. 2013;28:863-873.
3. Dhungana S, Jankovic J. Yips and other movement disorders in golfers. Mov Disord. 2013;28:576-581.
4. Stacy MA, ed. Handbook of dystonia. New York, NY: Informa Healthcare USA, Inc; 2007.
5. Fahn S, Marsden CD, Calne DB. Classification and investigation of dystonia. In: Marsden CD, Fahn S, eds. Movement disorders 2. London: Butterworths; 1987:332-358.
6. Smith AM, Adler CH, Crews D, et al. The ‘yips’ in golf: a continuum between a focal dystonia and choking. Sports Med. 2003;33:13-31.
7. Sachdev P. Golfers’ cramp: clinical characteristics and evidence against it being an anxiety disorder. Mov Disord. 1992;7:326-332.
8. Adler CH, Crews D, Hentz JG, et al. Abnormal co-contraction in yips-affected but not unaffected golfers: evidence for focal dystonia. Neurology. 2005;64:1813-1814.
9. Jankovic J. Treatment of hyperkinetic movement disorders. Lancet Neurol. 2009;8:844-856.
10. Jankovic J. Treatment of dystonia. Lancet Neurol. 2006;5:864-872.
11. Lungu C, Karp BI, Alter K, et al. Long-term follow-up of botulinum toxin therapy for focal hand dystonia: outcome at 10 years or more. Mov Disord. 2011;26:750-753.
12. Rosted P. Acupuncture for treatment of the yips?—a case report. Acupunct Med. 2005;23:188-189.
1. Baseball’s head cases often prove baffling. USA Today Baseball Weekly. 2001. Available at: http://usatoday30.usatoday.com/sports/bbw/2001-02-07/2001-02-07-head.htm. Accessed March 15, 2017.
2. Albanese A, Bhatia K, Bressman SB, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord. 2013;28:863-873.
3. Dhungana S, Jankovic J. Yips and other movement disorders in golfers. Mov Disord. 2013;28:576-581.
4. Stacy MA, ed. Handbook of dystonia. New York, NY: Informa Healthcare USA, Inc; 2007.
5. Fahn S, Marsden CD, Calne DB. Classification and investigation of dystonia. In: Marsden CD, Fahn S, eds. Movement disorders 2. London: Butterworths; 1987:332-358.
6. Smith AM, Adler CH, Crews D, et al. The ‘yips’ in golf: a continuum between a focal dystonia and choking. Sports Med. 2003;33:13-31.
7. Sachdev P. Golfers’ cramp: clinical characteristics and evidence against it being an anxiety disorder. Mov Disord. 1992;7:326-332.
8. Adler CH, Crews D, Hentz JG, et al. Abnormal co-contraction in yips-affected but not unaffected golfers: evidence for focal dystonia. Neurology. 2005;64:1813-1814.
9. Jankovic J. Treatment of hyperkinetic movement disorders. Lancet Neurol. 2009;8:844-856.
10. Jankovic J. Treatment of dystonia. Lancet Neurol. 2006;5:864-872.
11. Lungu C, Karp BI, Alter K, et al. Long-term follow-up of botulinum toxin therapy for focal hand dystonia: outcome at 10 years or more. Mov Disord. 2011;26:750-753.
12. Rosted P. Acupuncture for treatment of the yips?—a case report. Acupunct Med. 2005;23:188-189.

Paraspinous Cervical Nerve Block for Primary Headache
Headaches—pain or discomfort in the head, scalp, or neck—are a very common reason for ED visits.1 In 2011, the World Health Organization estimated that 46.5% of the population in North and South America aged 18 to 65 years old experienced at least one headache within the previous year.1
Migraine is a recurrent headache disorder that afflicts 18% of US women and 9% of US men,2 resulting in at least 1.2 million visits to US EDs annually.1 The economic cost resulting from migraine-related loss of productive time in the US workforce is more than $13 billion per year, most of which is in the form of reduced work productivity.3 Management and treatment for migraine headache in the ED commonly include intravenous (IV) or intramuscular (IM) medications, fluids, or oxygen. While ultimately effective, these methods require nursing care and additional time for posttreatment monitoring, both of which adversely affect patient flow.
In 2006, Mellick et al4 described the safety and effectiveness of paraspinous cervical nerve block (PCNB) to abort migraine headaches. Despite its demonstrated efficacy and safety, a decade later, PCNB is still rarely used. Friedman et al5 ranked peripheral nerve blocks as the fourth step in management suggestions for primary headache.
Case Reports of Headache Patients
We report on seven headache patients we treated in our ED with PCNB who had good-to-complete resolution of pain, suggesting that PCNB is efficacious and can potentially shorten the ED length of stay. This series of seven patients (six female, one male) was a convenience sample of primary headache patients who presented over a 10-month period and were safely and rapidly treated with PCNB (Table).

In each case, the PCNB procedure was explained to the patient and consent was obtained. Each patient was treated with a total of 3 cc of 0.5% bupivacaine with epinephrine injected into the posterior neck according to the method described by Mellick et al.4 Our seven patients achieved an average 5-point reduction in pain on a 10-point pain scale, with 0 = no pain and 10 = worse possible pain.
Other than the provision of medications, no nursing assistance was required. Only one of the patients required further treatment after the PCNB, and none had an adverse reaction. All of the patients reported that their headaches were similar in nature to past headaches. Based on their history and physical examination, none were diagnosed to be experiencing a secondary, more serious cause of headache, and none subsequently returned to our institution with a secondary type of headache.
The Paraspinal Cervical Nerve Block
Paraspinous cervical nerve block requires less time to administer and recovery is shorter than that from IM or IV opioids, sedatives, or neuroleptics. It is an easy technique to teach since it requires bilateral injections.
Technique
Prior to the procedure, cleanse the bilateral paravertebral zones surrounding C6 and C7 with chlorhexidine. Next, fill a 3 cc syringe using 0.5% bupivacaine with epinephrine.

Once the injection is complete, withdraw the needle completely, and compress and massage the injection site to facilitate anesthetic diffusion to surrounding tissues.

Indications
Paraspinous cervical nerve block is an appropriate treatment only for patients who are having a typical episode of chronic, recurring headaches, whose history and physical examination do not suggest the need for any further diagnostic work-up, and who, in the judgment of the treating clinician, require only pain relief.
Contraindications
A patient should not be considered for PCNB if he or she has a new-onset headache, fever, altered mental status, focal neurological deficits, meningismus, findings suggestive of meningitis, papilledema, increased intracranial pressure from a space-occupying lesion, recent head trauma with concern for intracranial hemorrhage, or suspicion of an alternate diagnosis.
Efficacy and Patient Response
Paraspinous cervical nerve block has been shown to decrease pain in patients who had failed standard migraine therapy and patients reported no complications. Of the seven patients in this case report, only one patient received opioids in the ED and none received prescriptions for opioids upon discharge for outpatient use.
Mellick and Mellick6 have postulated that pain may be modified due to the PCNB effect on the convergence of the trigeminal nerve with sensory fibers from the upper cervical roots. Since cervical innervation provides feeling to the head and upper neck, blocking this input can ameliorate pain.6
Summary
This series of seven patients provides further evidence of the effectiveness of PCNB in relieving headache symptoms for patients with recurrent, primary headaches when a secondary, more serious cause has been clinically excluded. Each of the seven patients had marked improvement of their pain and required only minimal nursing attention; moreover, all stated they would willingly undergo the procedure for future painful episodes.
Although there were no reported complications, this series is too small to demonstrate complete safety of the procedure. While this report is limited by a small sample size, it demonstrates that this is a quick, effective, and easily learned method of addressing a common ED complaint that obviates the need for parenteral medications and offers a potentially decreased patient length of stay.
Paraspinous cervical nerve block is a promising modality of treatment of ED patients who present with headache and migraine symptoms who do not respond to their outpatient “rescue” therapy. This procedure should be considered as an early treatment for migraine and other primary headaches unless contraindicated.
1. World Health Organization. Atlas of headache disorders and resources in the world 2011. http://www.who.int/mental_health/management/who_atlas_headache_disorders_results.pdf. Accessed February 9, 2017.
2. Victor TW, Hu X, Campbell JC, Buse DC, Lipton RB. Migraine prevalence by age and sex in the United States: a life-span study. Cephalalgia. 2010;30(9):1065-1072. doi:10.1177/0333102409355601.
3. Chawla J. Migraine headache. http://emedicine.medscape.com/article/1142556-overview. Accessed February 9, 2017.
4. Mellick LB, McIlrath ST, Mellick GA. Treatment of headaches in the ED with lower cervical intramuscular bupivacaine injections: a 1-year retrospective review of 417 patients. Headache. 2006;46(9):1441-1449.
5. Friedman BW, West J, Vinson DR, et al. Current management of migraine in US emergency departments: an analysis of the National Hospital Ambulatory Medical Care Survey. Cephalalgia. 2015;35:301-309.
6. Mellick GA, Mellick LB. Lower cervical intramuscular bupivacaine injections—another treatment option for headaches. http://www.neurologist-doctor.com/images/Mellick_Headache_injections.pdf. Accessed February 9, 2017.
Headaches—pain or discomfort in the head, scalp, or neck—are a very common reason for ED visits.1 In 2011, the World Health Organization estimated that 46.5% of the population in North and South America aged 18 to 65 years old experienced at least one headache within the previous year.1
Migraine is a recurrent headache disorder that afflicts 18% of US women and 9% of US men,2 resulting in at least 1.2 million visits to US EDs annually.1 The economic cost resulting from migraine-related loss of productive time in the US workforce is more than $13 billion per year, most of which is in the form of reduced work productivity.3 Management and treatment for migraine headache in the ED commonly include intravenous (IV) or intramuscular (IM) medications, fluids, or oxygen. While ultimately effective, these methods require nursing care and additional time for posttreatment monitoring, both of which adversely affect patient flow.
In 2006, Mellick et al4 described the safety and effectiveness of paraspinous cervical nerve block (PCNB) to abort migraine headaches. Despite its demonstrated efficacy and safety, a decade later, PCNB is still rarely used. Friedman et al5 ranked peripheral nerve blocks as the fourth step in management suggestions for primary headache.
Case Reports of Headache Patients
We report on seven headache patients we treated in our ED with PCNB who had good-to-complete resolution of pain, suggesting that PCNB is efficacious and can potentially shorten the ED length of stay. This series of seven patients (six female, one male) was a convenience sample of primary headache patients who presented over a 10-month period and were safely and rapidly treated with PCNB (Table).
In each case, the PCNB procedure was explained to the patient and consent was obtained. Each patient was treated with a total of 3 cc of 0.5% bupivacaine with epinephrine injected into the posterior neck according to the method described by Mellick et al.4 Our seven patients achieved an average 5-point reduction in pain on a 10-point pain scale, with 0 = no pain and 10 = worse possible pain.
Other than the provision of medications, no nursing assistance was required. Only one of the patients required further treatment after the PCNB, and none had an adverse reaction. All of the patients reported that their headaches were similar in nature to past headaches. Based on their history and physical examination, none were diagnosed to be experiencing a secondary, more serious cause of headache, and none subsequently returned to our institution with a secondary type of headache.
The Paraspinal Cervical Nerve Block
Paraspinous cervical nerve block requires less time to administer and recovery is shorter than that from IM or IV opioids, sedatives, or neuroleptics. It is an easy technique to teach since it requires bilateral injections.
Technique
Prior to the procedure, cleanse the bilateral paravertebral zones surrounding C6 and C7 with chlorhexidine. Next, fill a 3 cc syringe using 0.5% bupivacaine with epinephrine.
Once the injection is complete, withdraw the needle completely, and compress and massage the injection site to facilitate anesthetic diffusion to surrounding tissues.
Indications
Paraspinous cervical nerve block is an appropriate treatment only for patients who are having a typical episode of chronic, recurring headaches, whose history and physical examination do not suggest the need for any further diagnostic work-up, and who, in the judgment of the treating clinician, require only pain relief.
Contraindications
A patient should not be considered for PCNB if he or she has a new-onset headache, fever, altered mental status, focal neurological deficits, meningismus, findings suggestive of meningitis, papilledema, increased intracranial pressure from a space-occupying lesion, recent head trauma with concern for intracranial hemorrhage, or suspicion of an alternate diagnosis.
Efficacy and Patient Response
Paraspinous cervical nerve block has been shown to decrease pain in patients who had failed standard migraine therapy and patients reported no complications. Of the seven patients in this case report, only one patient received opioids in the ED and none received prescriptions for opioids upon discharge for outpatient use.
Mellick and Mellick6 have postulated that pain may be modified due to the PCNB effect on the convergence of the trigeminal nerve with sensory fibers from the upper cervical roots. Since cervical innervation provides feeling to the head and upper neck, blocking this input can ameliorate pain.6
Summary
This series of seven patients provides further evidence of the effectiveness of PCNB in relieving headache symptoms for patients with recurrent, primary headaches when a secondary, more serious cause has been clinically excluded. Each of the seven patients had marked improvement of their pain and required only minimal nursing attention; moreover, all stated they would willingly undergo the procedure for future painful episodes.
Although there were no reported complications, this series is too small to demonstrate complete safety of the procedure. While this report is limited by a small sample size, it demonstrates that this is a quick, effective, and easily learned method of addressing a common ED complaint that obviates the need for parenteral medications and offers a potentially decreased patient length of stay.
Paraspinous cervical nerve block is a promising modality of treatment of ED patients who present with headache and migraine symptoms who do not respond to their outpatient “rescue” therapy. This procedure should be considered as an early treatment for migraine and other primary headaches unless contraindicated.
Headaches—pain or discomfort in the head, scalp, or neck—are a very common reason for ED visits.1 In 2011, the World Health Organization estimated that 46.5% of the population in North and South America aged 18 to 65 years old experienced at least one headache within the previous year.1
Migraine is a recurrent headache disorder that afflicts 18% of US women and 9% of US men,2 resulting in at least 1.2 million visits to US EDs annually.1 The economic cost resulting from migraine-related loss of productive time in the US workforce is more than $13 billion per year, most of which is in the form of reduced work productivity.3 Management and treatment for migraine headache in the ED commonly include intravenous (IV) or intramuscular (IM) medications, fluids, or oxygen. While ultimately effective, these methods require nursing care and additional time for posttreatment monitoring, both of which adversely affect patient flow.
In 2006, Mellick et al4 described the safety and effectiveness of paraspinous cervical nerve block (PCNB) to abort migraine headaches. Despite its demonstrated efficacy and safety, a decade later, PCNB is still rarely used. Friedman et al5 ranked peripheral nerve blocks as the fourth step in management suggestions for primary headache.
Case Reports of Headache Patients
We report on seven headache patients we treated in our ED with PCNB who had good-to-complete resolution of pain, suggesting that PCNB is efficacious and can potentially shorten the ED length of stay. This series of seven patients (six female, one male) was a convenience sample of primary headache patients who presented over a 10-month period and were safely and rapidly treated with PCNB (Table).
In each case, the PCNB procedure was explained to the patient and consent was obtained. Each patient was treated with a total of 3 cc of 0.5% bupivacaine with epinephrine injected into the posterior neck according to the method described by Mellick et al.4 Our seven patients achieved an average 5-point reduction in pain on a 10-point pain scale, with 0 = no pain and 10 = worse possible pain.
Other than the provision of medications, no nursing assistance was required. Only one of the patients required further treatment after the PCNB, and none had an adverse reaction. All of the patients reported that their headaches were similar in nature to past headaches. Based on their history and physical examination, none were diagnosed to be experiencing a secondary, more serious cause of headache, and none subsequently returned to our institution with a secondary type of headache.
The Paraspinal Cervical Nerve Block
Paraspinous cervical nerve block requires less time to administer and recovery is shorter than that from IM or IV opioids, sedatives, or neuroleptics. It is an easy technique to teach since it requires bilateral injections.
Technique
Prior to the procedure, cleanse the bilateral paravertebral zones surrounding C6 and C7 with chlorhexidine. Next, fill a 3 cc syringe using 0.5% bupivacaine with epinephrine.
Once the injection is complete, withdraw the needle completely, and compress and massage the injection site to facilitate anesthetic diffusion to surrounding tissues.
Indications
Paraspinous cervical nerve block is an appropriate treatment only for patients who are having a typical episode of chronic, recurring headaches, whose history and physical examination do not suggest the need for any further diagnostic work-up, and who, in the judgment of the treating clinician, require only pain relief.
Contraindications
A patient should not be considered for PCNB if he or she has a new-onset headache, fever, altered mental status, focal neurological deficits, meningismus, findings suggestive of meningitis, papilledema, increased intracranial pressure from a space-occupying lesion, recent head trauma with concern for intracranial hemorrhage, or suspicion of an alternate diagnosis.
Efficacy and Patient Response
Paraspinous cervical nerve block has been shown to decrease pain in patients who had failed standard migraine therapy and patients reported no complications. Of the seven patients in this case report, only one patient received opioids in the ED and none received prescriptions for opioids upon discharge for outpatient use.
Mellick and Mellick6 have postulated that pain may be modified due to the PCNB effect on the convergence of the trigeminal nerve with sensory fibers from the upper cervical roots. Since cervical innervation provides feeling to the head and upper neck, blocking this input can ameliorate pain.6
Summary
This series of seven patients provides further evidence of the effectiveness of PCNB in relieving headache symptoms for patients with recurrent, primary headaches when a secondary, more serious cause has been clinically excluded. Each of the seven patients had marked improvement of their pain and required only minimal nursing attention; moreover, all stated they would willingly undergo the procedure for future painful episodes.
Although there were no reported complications, this series is too small to demonstrate complete safety of the procedure. While this report is limited by a small sample size, it demonstrates that this is a quick, effective, and easily learned method of addressing a common ED complaint that obviates the need for parenteral medications and offers a potentially decreased patient length of stay.
Paraspinous cervical nerve block is a promising modality of treatment of ED patients who present with headache and migraine symptoms who do not respond to their outpatient “rescue” therapy. This procedure should be considered as an early treatment for migraine and other primary headaches unless contraindicated.
1. World Health Organization. Atlas of headache disorders and resources in the world 2011. http://www.who.int/mental_health/management/who_atlas_headache_disorders_results.pdf. Accessed February 9, 2017.
2. Victor TW, Hu X, Campbell JC, Buse DC, Lipton RB. Migraine prevalence by age and sex in the United States: a life-span study. Cephalalgia. 2010;30(9):1065-1072. doi:10.1177/0333102409355601.
3. Chawla J. Migraine headache. http://emedicine.medscape.com/article/1142556-overview. Accessed February 9, 2017.
4. Mellick LB, McIlrath ST, Mellick GA. Treatment of headaches in the ED with lower cervical intramuscular bupivacaine injections: a 1-year retrospective review of 417 patients. Headache. 2006;46(9):1441-1449.
5. Friedman BW, West J, Vinson DR, et al. Current management of migraine in US emergency departments: an analysis of the National Hospital Ambulatory Medical Care Survey. Cephalalgia. 2015;35:301-309.
6. Mellick GA, Mellick LB. Lower cervical intramuscular bupivacaine injections—another treatment option for headaches. http://www.neurologist-doctor.com/images/Mellick_Headache_injections.pdf. Accessed February 9, 2017.
1. World Health Organization. Atlas of headache disorders and resources in the world 2011. http://www.who.int/mental_health/management/who_atlas_headache_disorders_results.pdf. Accessed February 9, 2017.
2. Victor TW, Hu X, Campbell JC, Buse DC, Lipton RB. Migraine prevalence by age and sex in the United States: a life-span study. Cephalalgia. 2010;30(9):1065-1072. doi:10.1177/0333102409355601.
3. Chawla J. Migraine headache. http://emedicine.medscape.com/article/1142556-overview. Accessed February 9, 2017.
4. Mellick LB, McIlrath ST, Mellick GA. Treatment of headaches in the ED with lower cervical intramuscular bupivacaine injections: a 1-year retrospective review of 417 patients. Headache. 2006;46(9):1441-1449.
5. Friedman BW, West J, Vinson DR, et al. Current management of migraine in US emergency departments: an analysis of the National Hospital Ambulatory Medical Care Survey. Cephalalgia. 2015;35:301-309.
6. Mellick GA, Mellick LB. Lower cervical intramuscular bupivacaine injections—another treatment option for headaches. http://www.neurologist-doctor.com/images/Mellick_Headache_injections.pdf. Accessed February 9, 2017.
Hypothyroidism-Induced Stercoral Sigmoid Colonic Perforation
According to the Centers for Disease Control and Prevention, abdominal pain is the leading reason for ED visits in the United States, with approximately 10 million visits per year.1 Though a large number of presentations are due to nontraumatic causes of abdominal pain, one etiology is among the most time-sensitive and critical diagnoses: acute colonic perforation.
Colonic perforations can be caused by diverticulitis, trauma, malignancy, ulcerative colitis, and other etiologies.2 A rare, yet life-threatening cause of colonic perforation, of which only a few cases have been documented in the literature, is stercoral colonic perforation.2
Regardless of the etiology, the critical actions for any colonic perforation are quick recognition, medical stabilization, and surgical evaluation. This case report highlights the diagnosis and treatment of acute stercoral colonic perforation with peritonitis secondary to hypothyroidism.
Case
A 49-year-old woman with a medical history significant for hypothyroidism presented to the ED for evaluation of diffuse abdominal pain, nausea, and nonbilious, nonbloody vomiting that started in the early evening of presentation. The patient denied any previous pain or associated symptoms, and said she had a small, hard bowel movement 1 day prior to arrival. She began experiencing mild abdominal pain on the morning of presentation. Her symptoms acutely worsened at approximately 5:00
On physical examination, her vital signs were: heart rate, 156 beats/min; blood pressure, 134/84 mm Hg; respiratory rate, 20 breaths/min, and temperature, 97.4°F. The patient appeared ill and diaphoretic, writhing on the stretcher. Abdominal examination was significant for diminished bowel sounds, diffuse abdominal distension, rigidity, and tenderness with light palpation.
Laboratory evaluation showed an elevated lactic acid level of 7.7 mmol/L, a white blood cell count of 7,200 cells/mm3 (segment form, 69.5%), and the following abnormal blood chemistry results: creatinine, 2.08 mg/dL; aspartate aminotransferase, 176 U/L; alanine aminotransferase, 138 U/L; and thyroid-stimulating hormone (TSH), 225.3 mcIU/mL. Other laboratory results were within normal range. Her electrocardiogram showed sinus tachycardia with a rate of 154 beats/min, a QTc within normal limits, and no ST elevations or depressions.
An abdominopelvic computed tomography (CT) scan revealed free air, free fluid, and possibly stool within the abdomen and pelvis. The findings were consistent with a ruptured hollow viscus, possibly a sigmoid colonic perforation. The radiologist also noted hepatomegaly and significant hepatic steatosis. A surgeon was immediately notified and evaluated the patient in the ED. The working diagnosis was stercoral colonic perforation secondary to severe hypothyroidism, and the patient was taken emergently to the operating room for repair.
Intraoperatively, the patient underwent exploratory laparotomy, which revealed gross fecal contamination of the abdomen. The surgeon noted that there was fecal staining along the serosal surface of the small bowel and throughout the pelvis. There were also large, hard stool balls outside of the colon. The perforation was along the mesenteric surface of the sigmoid just above the rectosigmoid junction.
The abdomen was copiously irrigated, the perforated segment was resected, and a Hartmann colostomy was created. The diagnosis was stercoral sigmoid perforation with peritonitis, and the patient was transferred to the intensive care unit for antibiotic treatment and further medical care, including intravenous (IV) levothyroxine.
She was extubated uneventfully on postoperative day 2, and the acute renal failure improved with supportive care only. Her bowel function slowly returned without complication. She was switched to oral levothyroxine on postoperative day 3. On day 13, she was given strict instructions for continuation of her thyroid medication and close monitoring for postsurgical complications, and was discharged home with appropriate follow-up.
Discussion
Multiple contributing factors can lead to bowel perforation. In this case, severe hypothyroidism with constipation caused a colonic perforation. Our patient had severe constipation that increased intraluminal pressure, causing the bowel wall to become ischemic and subsequently perforate.3 Any disease that causes significant constipation or obstruction of transit could lead to the same catastrophic result.
According to Huang et al,4 as of 2002, fewer than 90 cases of general stercoral bowel perforation had been reported, with no clear age range. However, patients in their mid-50s to mid-60s appear to be the most commonly affected age group.4 Our patient was younger than this age group, making identification of the problem by age alone difficult.
Hypothyroidism
The incidence of hypothyroidism in iodine-replete communities varies between 1% to 2% of the general population.5 The condition is more common in older women, affecting approximately 10% of those over age 65 years. In the United States, the prevalence of biochemical hypothyroidism is 4.6%; however, clinically evident hypothyroidism is present in only 0.3%.6 Common causes for hypothyroidism are listed in the Table.7,8

Myxedema Coma
Untreated, hypothyroidism can lead to potentially fatal conditions, such as myxedema coma, which is characterized by hypothermia, hypotension, bradycardia, respiratory depression, and altered mental status.7 Severe myxedema coma can result in cardiovascular collapse, and eventual death. Electrocardiography findings of severe hypothyroidism include bradycardia, low-voltage QRS, and widespread T-wave inversions.7 Our patient was tachycardic and did not have any acute findings to suggest myxedema coma.
Treatment for myxedema coma includes supportive care with ventilatory support and pressor support if necessary. Patients should be given IV hydrocortisone, 100 mg, to treat possible adrenal insufficiency and T4, 4 mcg/kg by slow IV infusion.7 Caution should be taken if giving a patient T3 due to the risk of dysrhythmias and myocardial infarction (MI).7 As our patient was not displaying myxedema coma, the surgeon elected not to start IV thyroid replacement to avoid exacerbating the patient’s tachycardia and possibly precipitating an MI intraoperatively.
Conclusion
Our case underscores the importance of promptly recognizing the signs and symptoms of stercoral colonic perforation in patients who present with nontraumatic abdominal pain accompanied by nausea and nonbilious, nonbloody vomiting. Although stercoral colonic perforation is a rare cause of nontraumatic abdominal pain, as with any type of colonic perforation, it constitutes a life-threatening medical emergency. As our case illustrates, prompt diagnosis through a thorough history taking, physical examination, and laboratory and imaging studies is critical to ensure medical stabilization and surgical management to reduce morbidity and mortality.
1. Centers for Disease Control and Prevention. Table 10. Ten leading principal reasons for emergency department visits, by patient age and sex: United States, 2013. https://www.cdc.gov/nchs/data/ahcd/nhamcs_emergency/2013_ed_web_tables.pdf. Accessed March 3, 2017.
2. Nam JK, Kim BS, Kim KS, Moon DJ. Clinical analysis of stercoral perforation of the colon. Korean J Gastroenterol. 2010;55:46-51.
3. Heffernan C, Pachter HL, Megibow AJ, Macari M. Stercoral colitis leading to fatal peritonitis: CT findings. AJR Am J Roentgenol. 2005;184(4):1189-1193. doi:10.2214/ajr.184.4.01841189.
4. Huang WS, Wang CS, Hsieh CC, Lin PY, Chin CC, Wang JY. Management of patients with stercoral perforation of the sigmoid colon: Report of five cases. World J Gastroenterol. 2006;12(3):500-503.
5. Canaris GJ, Manowitz NR, Mayor G, Ridgway EC. The Colorado thyroid disease prevalence study. Arch Intern Med. 2000;160(4):526-534.
6. Hollowell JG, Staehling NW, Flanders WD, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab. 2002;87(2):489-499.
7. Idrose AM. Hypothyroidism. In: Tintinalli JE, Stapczynski JS, Ma OJ, Yealy DM, Meckler GD, Cline DM. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 8th Edition. New York, NY: McGraw-Hill Education; 2016:1469-1472.
8. Skugor M. Hypothyroidism and Hyperthyroidism. Cleveland Clinic Center for Continuing Education. August 2014. http://www.clevelandclinicmeded.com/medicalpubs/diseasemanagement/endocrinology/hypothyroidism-and-hyperthyroidism/. Accessed March 3, 2017.
According to the Centers for Disease Control and Prevention, abdominal pain is the leading reason for ED visits in the United States, with approximately 10 million visits per year.1 Though a large number of presentations are due to nontraumatic causes of abdominal pain, one etiology is among the most time-sensitive and critical diagnoses: acute colonic perforation.
Colonic perforations can be caused by diverticulitis, trauma, malignancy, ulcerative colitis, and other etiologies.2 A rare, yet life-threatening cause of colonic perforation, of which only a few cases have been documented in the literature, is stercoral colonic perforation.2
Regardless of the etiology, the critical actions for any colonic perforation are quick recognition, medical stabilization, and surgical evaluation. This case report highlights the diagnosis and treatment of acute stercoral colonic perforation with peritonitis secondary to hypothyroidism.
Case
A 49-year-old woman with a medical history significant for hypothyroidism presented to the ED for evaluation of diffuse abdominal pain, nausea, and nonbilious, nonbloody vomiting that started in the early evening of presentation. The patient denied any previous pain or associated symptoms, and said she had a small, hard bowel movement 1 day prior to arrival. She began experiencing mild abdominal pain on the morning of presentation. Her symptoms acutely worsened at approximately 5:00
On physical examination, her vital signs were: heart rate, 156 beats/min; blood pressure, 134/84 mm Hg; respiratory rate, 20 breaths/min, and temperature, 97.4°F. The patient appeared ill and diaphoretic, writhing on the stretcher. Abdominal examination was significant for diminished bowel sounds, diffuse abdominal distension, rigidity, and tenderness with light palpation.
Laboratory evaluation showed an elevated lactic acid level of 7.7 mmol/L, a white blood cell count of 7,200 cells/mm3 (segment form, 69.5%), and the following abnormal blood chemistry results: creatinine, 2.08 mg/dL; aspartate aminotransferase, 176 U/L; alanine aminotransferase, 138 U/L; and thyroid-stimulating hormone (TSH), 225.3 mcIU/mL. Other laboratory results were within normal range. Her electrocardiogram showed sinus tachycardia with a rate of 154 beats/min, a QTc within normal limits, and no ST elevations or depressions.
An abdominopelvic computed tomography (CT) scan revealed free air, free fluid, and possibly stool within the abdomen and pelvis. The findings were consistent with a ruptured hollow viscus, possibly a sigmoid colonic perforation. The radiologist also noted hepatomegaly and significant hepatic steatosis. A surgeon was immediately notified and evaluated the patient in the ED. The working diagnosis was stercoral colonic perforation secondary to severe hypothyroidism, and the patient was taken emergently to the operating room for repair.
Intraoperatively, the patient underwent exploratory laparotomy, which revealed gross fecal contamination of the abdomen. The surgeon noted that there was fecal staining along the serosal surface of the small bowel and throughout the pelvis. There were also large, hard stool balls outside of the colon. The perforation was along the mesenteric surface of the sigmoid just above the rectosigmoid junction.
The abdomen was copiously irrigated, the perforated segment was resected, and a Hartmann colostomy was created. The diagnosis was stercoral sigmoid perforation with peritonitis, and the patient was transferred to the intensive care unit for antibiotic treatment and further medical care, including intravenous (IV) levothyroxine.
She was extubated uneventfully on postoperative day 2, and the acute renal failure improved with supportive care only. Her bowel function slowly returned without complication. She was switched to oral levothyroxine on postoperative day 3. On day 13, she was given strict instructions for continuation of her thyroid medication and close monitoring for postsurgical complications, and was discharged home with appropriate follow-up.
Discussion
Multiple contributing factors can lead to bowel perforation. In this case, severe hypothyroidism with constipation caused a colonic perforation. Our patient had severe constipation that increased intraluminal pressure, causing the bowel wall to become ischemic and subsequently perforate.3 Any disease that causes significant constipation or obstruction of transit could lead to the same catastrophic result.
According to Huang et al,4 as of 2002, fewer than 90 cases of general stercoral bowel perforation had been reported, with no clear age range. However, patients in their mid-50s to mid-60s appear to be the most commonly affected age group.4 Our patient was younger than this age group, making identification of the problem by age alone difficult.
Hypothyroidism
The incidence of hypothyroidism in iodine-replete communities varies between 1% to 2% of the general population.5 The condition is more common in older women, affecting approximately 10% of those over age 65 years. In the United States, the prevalence of biochemical hypothyroidism is 4.6%; however, clinically evident hypothyroidism is present in only 0.3%.6 Common causes for hypothyroidism are listed in the Table.7,8
Myxedema Coma
Untreated, hypothyroidism can lead to potentially fatal conditions, such as myxedema coma, which is characterized by hypothermia, hypotension, bradycardia, respiratory depression, and altered mental status.7 Severe myxedema coma can result in cardiovascular collapse, and eventual death. Electrocardiography findings of severe hypothyroidism include bradycardia, low-voltage QRS, and widespread T-wave inversions.7 Our patient was tachycardic and did not have any acute findings to suggest myxedema coma.
Treatment for myxedema coma includes supportive care with ventilatory support and pressor support if necessary. Patients should be given IV hydrocortisone, 100 mg, to treat possible adrenal insufficiency and T4, 4 mcg/kg by slow IV infusion.7 Caution should be taken if giving a patient T3 due to the risk of dysrhythmias and myocardial infarction (MI).7 As our patient was not displaying myxedema coma, the surgeon elected not to start IV thyroid replacement to avoid exacerbating the patient’s tachycardia and possibly precipitating an MI intraoperatively.
Conclusion
Our case underscores the importance of promptly recognizing the signs and symptoms of stercoral colonic perforation in patients who present with nontraumatic abdominal pain accompanied by nausea and nonbilious, nonbloody vomiting. Although stercoral colonic perforation is a rare cause of nontraumatic abdominal pain, as with any type of colonic perforation, it constitutes a life-threatening medical emergency. As our case illustrates, prompt diagnosis through a thorough history taking, physical examination, and laboratory and imaging studies is critical to ensure medical stabilization and surgical management to reduce morbidity and mortality.
According to the Centers for Disease Control and Prevention, abdominal pain is the leading reason for ED visits in the United States, with approximately 10 million visits per year.1 Though a large number of presentations are due to nontraumatic causes of abdominal pain, one etiology is among the most time-sensitive and critical diagnoses: acute colonic perforation.
Colonic perforations can be caused by diverticulitis, trauma, malignancy, ulcerative colitis, and other etiologies.2 A rare, yet life-threatening cause of colonic perforation, of which only a few cases have been documented in the literature, is stercoral colonic perforation.2
Regardless of the etiology, the critical actions for any colonic perforation are quick recognition, medical stabilization, and surgical evaluation. This case report highlights the diagnosis and treatment of acute stercoral colonic perforation with peritonitis secondary to hypothyroidism.
Case
A 49-year-old woman with a medical history significant for hypothyroidism presented to the ED for evaluation of diffuse abdominal pain, nausea, and nonbilious, nonbloody vomiting that started in the early evening of presentation. The patient denied any previous pain or associated symptoms, and said she had a small, hard bowel movement 1 day prior to arrival. She began experiencing mild abdominal pain on the morning of presentation. Her symptoms acutely worsened at approximately 5:00
On physical examination, her vital signs were: heart rate, 156 beats/min; blood pressure, 134/84 mm Hg; respiratory rate, 20 breaths/min, and temperature, 97.4°F. The patient appeared ill and diaphoretic, writhing on the stretcher. Abdominal examination was significant for diminished bowel sounds, diffuse abdominal distension, rigidity, and tenderness with light palpation.
Laboratory evaluation showed an elevated lactic acid level of 7.7 mmol/L, a white blood cell count of 7,200 cells/mm3 (segment form, 69.5%), and the following abnormal blood chemistry results: creatinine, 2.08 mg/dL; aspartate aminotransferase, 176 U/L; alanine aminotransferase, 138 U/L; and thyroid-stimulating hormone (TSH), 225.3 mcIU/mL. Other laboratory results were within normal range. Her electrocardiogram showed sinus tachycardia with a rate of 154 beats/min, a QTc within normal limits, and no ST elevations or depressions.
An abdominopelvic computed tomography (CT) scan revealed free air, free fluid, and possibly stool within the abdomen and pelvis. The findings were consistent with a ruptured hollow viscus, possibly a sigmoid colonic perforation. The radiologist also noted hepatomegaly and significant hepatic steatosis. A surgeon was immediately notified and evaluated the patient in the ED. The working diagnosis was stercoral colonic perforation secondary to severe hypothyroidism, and the patient was taken emergently to the operating room for repair.
Intraoperatively, the patient underwent exploratory laparotomy, which revealed gross fecal contamination of the abdomen. The surgeon noted that there was fecal staining along the serosal surface of the small bowel and throughout the pelvis. There were also large, hard stool balls outside of the colon. The perforation was along the mesenteric surface of the sigmoid just above the rectosigmoid junction.
The abdomen was copiously irrigated, the perforated segment was resected, and a Hartmann colostomy was created. The diagnosis was stercoral sigmoid perforation with peritonitis, and the patient was transferred to the intensive care unit for antibiotic treatment and further medical care, including intravenous (IV) levothyroxine.
She was extubated uneventfully on postoperative day 2, and the acute renal failure improved with supportive care only. Her bowel function slowly returned without complication. She was switched to oral levothyroxine on postoperative day 3. On day 13, she was given strict instructions for continuation of her thyroid medication and close monitoring for postsurgical complications, and was discharged home with appropriate follow-up.
Discussion
Multiple contributing factors can lead to bowel perforation. In this case, severe hypothyroidism with constipation caused a colonic perforation. Our patient had severe constipation that increased intraluminal pressure, causing the bowel wall to become ischemic and subsequently perforate.3 Any disease that causes significant constipation or obstruction of transit could lead to the same catastrophic result.
According to Huang et al,4 as of 2002, fewer than 90 cases of general stercoral bowel perforation had been reported, with no clear age range. However, patients in their mid-50s to mid-60s appear to be the most commonly affected age group.4 Our patient was younger than this age group, making identification of the problem by age alone difficult.
Hypothyroidism
The incidence of hypothyroidism in iodine-replete communities varies between 1% to 2% of the general population.5 The condition is more common in older women, affecting approximately 10% of those over age 65 years. In the United States, the prevalence of biochemical hypothyroidism is 4.6%; however, clinically evident hypothyroidism is present in only 0.3%.6 Common causes for hypothyroidism are listed in the Table.7,8
Myxedema Coma
Untreated, hypothyroidism can lead to potentially fatal conditions, such as myxedema coma, which is characterized by hypothermia, hypotension, bradycardia, respiratory depression, and altered mental status.7 Severe myxedema coma can result in cardiovascular collapse, and eventual death. Electrocardiography findings of severe hypothyroidism include bradycardia, low-voltage QRS, and widespread T-wave inversions.7 Our patient was tachycardic and did not have any acute findings to suggest myxedema coma.
Treatment for myxedema coma includes supportive care with ventilatory support and pressor support if necessary. Patients should be given IV hydrocortisone, 100 mg, to treat possible adrenal insufficiency and T4, 4 mcg/kg by slow IV infusion.7 Caution should be taken if giving a patient T3 due to the risk of dysrhythmias and myocardial infarction (MI).7 As our patient was not displaying myxedema coma, the surgeon elected not to start IV thyroid replacement to avoid exacerbating the patient’s tachycardia and possibly precipitating an MI intraoperatively.
Conclusion
Our case underscores the importance of promptly recognizing the signs and symptoms of stercoral colonic perforation in patients who present with nontraumatic abdominal pain accompanied by nausea and nonbilious, nonbloody vomiting. Although stercoral colonic perforation is a rare cause of nontraumatic abdominal pain, as with any type of colonic perforation, it constitutes a life-threatening medical emergency. As our case illustrates, prompt diagnosis through a thorough history taking, physical examination, and laboratory and imaging studies is critical to ensure medical stabilization and surgical management to reduce morbidity and mortality.
1. Centers for Disease Control and Prevention. Table 10. Ten leading principal reasons for emergency department visits, by patient age and sex: United States, 2013. https://www.cdc.gov/nchs/data/ahcd/nhamcs_emergency/2013_ed_web_tables.pdf. Accessed March 3, 2017.
2. Nam JK, Kim BS, Kim KS, Moon DJ. Clinical analysis of stercoral perforation of the colon. Korean J Gastroenterol. 2010;55:46-51.
3. Heffernan C, Pachter HL, Megibow AJ, Macari M. Stercoral colitis leading to fatal peritonitis: CT findings. AJR Am J Roentgenol. 2005;184(4):1189-1193. doi:10.2214/ajr.184.4.01841189.
4. Huang WS, Wang CS, Hsieh CC, Lin PY, Chin CC, Wang JY. Management of patients with stercoral perforation of the sigmoid colon: Report of five cases. World J Gastroenterol. 2006;12(3):500-503.
5. Canaris GJ, Manowitz NR, Mayor G, Ridgway EC. The Colorado thyroid disease prevalence study. Arch Intern Med. 2000;160(4):526-534.
6. Hollowell JG, Staehling NW, Flanders WD, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab. 2002;87(2):489-499.
7. Idrose AM. Hypothyroidism. In: Tintinalli JE, Stapczynski JS, Ma OJ, Yealy DM, Meckler GD, Cline DM. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 8th Edition. New York, NY: McGraw-Hill Education; 2016:1469-1472.
8. Skugor M. Hypothyroidism and Hyperthyroidism. Cleveland Clinic Center for Continuing Education. August 2014. http://www.clevelandclinicmeded.com/medicalpubs/diseasemanagement/endocrinology/hypothyroidism-and-hyperthyroidism/. Accessed March 3, 2017.
1. Centers for Disease Control and Prevention. Table 10. Ten leading principal reasons for emergency department visits, by patient age and sex: United States, 2013. https://www.cdc.gov/nchs/data/ahcd/nhamcs_emergency/2013_ed_web_tables.pdf. Accessed March 3, 2017.
2. Nam JK, Kim BS, Kim KS, Moon DJ. Clinical analysis of stercoral perforation of the colon. Korean J Gastroenterol. 2010;55:46-51.
3. Heffernan C, Pachter HL, Megibow AJ, Macari M. Stercoral colitis leading to fatal peritonitis: CT findings. AJR Am J Roentgenol. 2005;184(4):1189-1193. doi:10.2214/ajr.184.4.01841189.
4. Huang WS, Wang CS, Hsieh CC, Lin PY, Chin CC, Wang JY. Management of patients with stercoral perforation of the sigmoid colon: Report of five cases. World J Gastroenterol. 2006;12(3):500-503.
5. Canaris GJ, Manowitz NR, Mayor G, Ridgway EC. The Colorado thyroid disease prevalence study. Arch Intern Med. 2000;160(4):526-534.
6. Hollowell JG, Staehling NW, Flanders WD, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab. 2002;87(2):489-499.
7. Idrose AM. Hypothyroidism. In: Tintinalli JE, Stapczynski JS, Ma OJ, Yealy DM, Meckler GD, Cline DM. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 8th Edition. New York, NY: McGraw-Hill Education; 2016:1469-1472.
8. Skugor M. Hypothyroidism and Hyperthyroidism. Cleveland Clinic Center for Continuing Education. August 2014. http://www.clevelandclinicmeded.com/medicalpubs/diseasemanagement/endocrinology/hypothyroidism-and-hyperthyroidism/. Accessed March 3, 2017.

Emergency Imaging: Multiple Comorbidities With Fever and Nonproductive Cough



Laboratory studies revealed leukocytosis with a left shift. Chest radiographs were negative for pneumonia. A magnetic resonance image (MRI) of the lumbar spine was obtained to evaluate for diskitis osteomyelitis. A radiograph of the pelvis was also obtained to evaluate the patient’s THAs, and a computed tomography scan (CT) of the abdomen and pelvis with contrast was obtained for further evaluation. Representative CT, radiographic, and MRI images are shown at left (Figures 1-3).
What is the suspected diagnosis?
Answer
The MRI of the lumbar spine demonstrated no evidence of diskitis osteomyelitis. However, T2-weighted axial images showed enlarged heterogeneous bilateral psoas muscles with bright signal, indicating the presence of fluid (white arrows, Figure 4).


On the pelvic radiographs, both femoral heads appeared off-center within the acetabular cups (red arrows, Figure 5). This eccentric positioning indicated wear of the polyethylene in the THAs that normally occupies the space between the acetabular cup and the femoral head. In addition, focal lucency in the right acetabulum indicated breakdown of the bone, a condition referred to as osteolysis (white asterisk, Figure 5).
An abdominopelvic CT scan with contrast was performed and confirmed the findings of polyethylene wear and osteolysis. The CT scan also demonstrated large bilateral hip joint effusions (white arrows, Figure 6), decompressed along distended bilateral iliopsoas bursae (red asterisks, Figure 6), and communicating with the bilateral psoas muscle collections (red arrows, Figure 6).
Osteolysis With Iliopsoas Bursitis
Bursae are fluid-filled sacs lined by synovial tissue located throughout the body to reduce friction at sites of movement between muscles, bones, and tendons. Bursitis develops when these sacs become inflamed and/or infected and fill with fluid. The iliopsoas bursa lies between the anterior capsule of the hip and the psoas tendon, iliacus tendon, and muscle fibers.1,2 This bursa frequently communicates with the hip joint.3,4 Iliopsoas bursal distension has been reported following THA in the setting of polyethylene wear,5 and aseptic bursitis is a commonly seen incidental finding at the time of revision surgery.6

In this patient, long-standing polyethylene-induced synovitis had markedly expanded the hip joints and iliopsoas bursae, eventually resulting in superinfection, which accounted for the patient’s symptoms.
Treatment
Based on the imaging findings, interventional radiology services were contacted. The interventional radiologist drained the bilateral psoas abscesses. Cultures of the fluid were positive for both methicillin-resistant Staphylococcus aureus (MRSA) and methicillin-susceptible S aureus (MSSA). The patient was admitted to the hospital for treatment of MRSA and MSSA with intravenous antibiotic therapy. He recovered from the infection and was discharged on hospital day 2, with instructions to follow up with an orthopedic surgeon to discuss eventual revision of the bilateral THAs.
1. Chandler SB. The iliopsoas bursa in man. Anatom Record. 1934;58(3),235-240. doi:10.1002/ar.1090580304.
2. Tatu L, Parratte B, Vuillier F, Diop M, Monnier G. Descriptive anatomy of the femoral portion of the iliopsoas muscle. Anatomical basis of anterior snapping of the hip. Surg Radiol Anat. 2001;23(6):371-374.
3. Meaney JF, Cassar-Pullicino VN, Etherington R, Ritchie DA, McCall IW, Whitehouse GH. Ilio-psoas bursa enlargement. Clin Radiol. 1992;45(3):161-168.
4. Warren R, Kaye JJ, Salvati EA. Arthrographic demonstration of an enlarged iliopsoas bursa complicating osteoarthritis of the hip. A case report. J Bone Joint Surg Am. 1975;57(3):413-415.
5. Cheung YM, Gupte CM, Beverly MJ. Iliopsoas bursitis following total hip replacement. Arch Orthop Trauma Surg. 2004;124(10):720-723. Epub 2004 Oct 23. doi:10.1007/s00402-004-0751-9.
6. Howie DW, Cain CM, Cornish BL. Pseudo-abscess of the psoas bursa in failed double-cup arthroplasty of the hip. J Bone Joint Surg Br. 1991;73:29-32.
Laboratory studies revealed leukocytosis with a left shift. Chest radiographs were negative for pneumonia. A magnetic resonance image (MRI) of the lumbar spine was obtained to evaluate for diskitis osteomyelitis. A radiograph of the pelvis was also obtained to evaluate the patient’s THAs, and a computed tomography scan (CT) of the abdomen and pelvis with contrast was obtained for further evaluation. Representative CT, radiographic, and MRI images are shown at left (Figures 1-3).
What is the suspected diagnosis?
Answer
The MRI of the lumbar spine demonstrated no evidence of diskitis osteomyelitis. However, T2-weighted axial images showed enlarged heterogeneous bilateral psoas muscles with bright signal, indicating the presence of fluid (white arrows, Figure 4).
On the pelvic radiographs, both femoral heads appeared off-center within the acetabular cups (red arrows, Figure 5). This eccentric positioning indicated wear of the polyethylene in the THAs that normally occupies the space between the acetabular cup and the femoral head. In addition, focal lucency in the right acetabulum indicated breakdown of the bone, a condition referred to as osteolysis (white asterisk, Figure 5).
An abdominopelvic CT scan with contrast was performed and confirmed the findings of polyethylene wear and osteolysis. The CT scan also demonstrated large bilateral hip joint effusions (white arrows, Figure 6), decompressed along distended bilateral iliopsoas bursae (red asterisks, Figure 6), and communicating with the bilateral psoas muscle collections (red arrows, Figure 6).
Osteolysis With Iliopsoas Bursitis
Bursae are fluid-filled sacs lined by synovial tissue located throughout the body to reduce friction at sites of movement between muscles, bones, and tendons. Bursitis develops when these sacs become inflamed and/or infected and fill with fluid. The iliopsoas bursa lies between the anterior capsule of the hip and the psoas tendon, iliacus tendon, and muscle fibers.1,2 This bursa frequently communicates with the hip joint.3,4 Iliopsoas bursal distension has been reported following THA in the setting of polyethylene wear,5 and aseptic bursitis is a commonly seen incidental finding at the time of revision surgery.6
In this patient, long-standing polyethylene-induced synovitis had markedly expanded the hip joints and iliopsoas bursae, eventually resulting in superinfection, which accounted for the patient’s symptoms.
Treatment
Based on the imaging findings, interventional radiology services were contacted. The interventional radiologist drained the bilateral psoas abscesses. Cultures of the fluid were positive for both methicillin-resistant Staphylococcus aureus (MRSA) and methicillin-susceptible S aureus (MSSA). The patient was admitted to the hospital for treatment of MRSA and MSSA with intravenous antibiotic therapy. He recovered from the infection and was discharged on hospital day 2, with instructions to follow up with an orthopedic surgeon to discuss eventual revision of the bilateral THAs.
Laboratory studies revealed leukocytosis with a left shift. Chest radiographs were negative for pneumonia. A magnetic resonance image (MRI) of the lumbar spine was obtained to evaluate for diskitis osteomyelitis. A radiograph of the pelvis was also obtained to evaluate the patient’s THAs, and a computed tomography scan (CT) of the abdomen and pelvis with contrast was obtained for further evaluation. Representative CT, radiographic, and MRI images are shown at left (Figures 1-3).
What is the suspected diagnosis?
Answer
The MRI of the lumbar spine demonstrated no evidence of diskitis osteomyelitis. However, T2-weighted axial images showed enlarged heterogeneous bilateral psoas muscles with bright signal, indicating the presence of fluid (white arrows, Figure 4).
On the pelvic radiographs, both femoral heads appeared off-center within the acetabular cups (red arrows, Figure 5). This eccentric positioning indicated wear of the polyethylene in the THAs that normally occupies the space between the acetabular cup and the femoral head. In addition, focal lucency in the right acetabulum indicated breakdown of the bone, a condition referred to as osteolysis (white asterisk, Figure 5).
An abdominopelvic CT scan with contrast was performed and confirmed the findings of polyethylene wear and osteolysis. The CT scan also demonstrated large bilateral hip joint effusions (white arrows, Figure 6), decompressed along distended bilateral iliopsoas bursae (red asterisks, Figure 6), and communicating with the bilateral psoas muscle collections (red arrows, Figure 6).
Osteolysis With Iliopsoas Bursitis
Bursae are fluid-filled sacs lined by synovial tissue located throughout the body to reduce friction at sites of movement between muscles, bones, and tendons. Bursitis develops when these sacs become inflamed and/or infected and fill with fluid. The iliopsoas bursa lies between the anterior capsule of the hip and the psoas tendon, iliacus tendon, and muscle fibers.1,2 This bursa frequently communicates with the hip joint.3,4 Iliopsoas bursal distension has been reported following THA in the setting of polyethylene wear,5 and aseptic bursitis is a commonly seen incidental finding at the time of revision surgery.6
In this patient, long-standing polyethylene-induced synovitis had markedly expanded the hip joints and iliopsoas bursae, eventually resulting in superinfection, which accounted for the patient’s symptoms.
Treatment
Based on the imaging findings, interventional radiology services were contacted. The interventional radiologist drained the bilateral psoas abscesses. Cultures of the fluid were positive for both methicillin-resistant Staphylococcus aureus (MRSA) and methicillin-susceptible S aureus (MSSA). The patient was admitted to the hospital for treatment of MRSA and MSSA with intravenous antibiotic therapy. He recovered from the infection and was discharged on hospital day 2, with instructions to follow up with an orthopedic surgeon to discuss eventual revision of the bilateral THAs.
1. Chandler SB. The iliopsoas bursa in man. Anatom Record. 1934;58(3),235-240. doi:10.1002/ar.1090580304.
2. Tatu L, Parratte B, Vuillier F, Diop M, Monnier G. Descriptive anatomy of the femoral portion of the iliopsoas muscle. Anatomical basis of anterior snapping of the hip. Surg Radiol Anat. 2001;23(6):371-374.
3. Meaney JF, Cassar-Pullicino VN, Etherington R, Ritchie DA, McCall IW, Whitehouse GH. Ilio-psoas bursa enlargement. Clin Radiol. 1992;45(3):161-168.
4. Warren R, Kaye JJ, Salvati EA. Arthrographic demonstration of an enlarged iliopsoas bursa complicating osteoarthritis of the hip. A case report. J Bone Joint Surg Am. 1975;57(3):413-415.
5. Cheung YM, Gupte CM, Beverly MJ. Iliopsoas bursitis following total hip replacement. Arch Orthop Trauma Surg. 2004;124(10):720-723. Epub 2004 Oct 23. doi:10.1007/s00402-004-0751-9.
6. Howie DW, Cain CM, Cornish BL. Pseudo-abscess of the psoas bursa in failed double-cup arthroplasty of the hip. J Bone Joint Surg Br. 1991;73:29-32.
1. Chandler SB. The iliopsoas bursa in man. Anatom Record. 1934;58(3),235-240. doi:10.1002/ar.1090580304.
2. Tatu L, Parratte B, Vuillier F, Diop M, Monnier G. Descriptive anatomy of the femoral portion of the iliopsoas muscle. Anatomical basis of anterior snapping of the hip. Surg Radiol Anat. 2001;23(6):371-374.
3. Meaney JF, Cassar-Pullicino VN, Etherington R, Ritchie DA, McCall IW, Whitehouse GH. Ilio-psoas bursa enlargement. Clin Radiol. 1992;45(3):161-168.
4. Warren R, Kaye JJ, Salvati EA. Arthrographic demonstration of an enlarged iliopsoas bursa complicating osteoarthritis of the hip. A case report. J Bone Joint Surg Am. 1975;57(3):413-415.
5. Cheung YM, Gupte CM, Beverly MJ. Iliopsoas bursitis following total hip replacement. Arch Orthop Trauma Surg. 2004;124(10):720-723. Epub 2004 Oct 23. doi:10.1007/s00402-004-0751-9.
6. Howie DW, Cain CM, Cornish BL. Pseudo-abscess of the psoas bursa in failed double-cup arthroplasty of the hip. J Bone Joint Surg Br. 1991;73:29-32.

Superior Mesenteric Artery Syndrome as a Complication of Scoliosis Surgery
Take-Home Points
- Adolescent growth spurt, height-to-weight ratio, and perioperative weight loss are risk factors associated with SMA syndrome following pediatric spine surgery.
- Must recognize nonspecific symptoms such as abdominal pain, tenderness, distention, bilious or projectile vomiting, hypoactive bowel sounds, and anorexia postoperatively.
- Complications of SMA syndrome can potentially lead to aspiration pneumonia, acute gastric rupture, or cardiovascular collapse and death.
Superior mesenteric artery (SMA) syndrome resulting from surgical treatment of scoliosis has been recognized in the medical literature since 1752.1 Throughout the literature, SMA syndrome variably has been referred to as cast syndrome, Wilkie syndrome, arteriomesenteric duodenal obstruction, and chronic duodenal ileus.2 We now recognize numerous etiologies of SMA syndrome, as several sources can externally compress the duodenum. Classic acute symptoms of bowel obstruction include bilious vomiting, nausea, and epigastric pain. Chronic manifestations of SMA syndrome may include weight loss and decreased appetite. Our literature review revealed that adolescent growth spurt, height-to-weight ratio, and perioperative weight loss are risk factors associated with SMA syndrome after pediatric spine surgery.
We report the case of a 14-year-old boy who developed SMA syndrome after undergoing scoliosis surgery. The patient and his mother provided written informed consent for print and electronic publication of this case report.
Case Report
A 14-year-old boy with a history of idiopathic scoliosis presented to Cohen Children’s Hospital (Long Island Jewish Medical Center) with bilious vomiting that had persisted for 7 days after posterior T9–L4 fusion with instrumentation. 


Discussion
SMA syndrome is attributed to the anatomical orientation of the third part of the duodenum, which passes between the aorta and the SMA (Figure 4). 
Adolescents are particularly vulnerable to this condition. Faster adolescent bone growth relative to visceral growth is accompanied by a decrease in SMA angle.3 Occasionally, body casts are used after surgery to immobilize the vertebrae and augment healing. Cast syndrome occurs when pressure from a body cast causes a bowel obstruction secondary to spinal hyperextension and amplified spinal lordosis.2 This finding, dating to the 19th century, was reported by Willet4 when a patient died 48 hours after application of a body cast. In 1950, the term cast syndrome was coined after a motorcyclist’s injuries were treated with a hip spica cast and the patient died of cardiovascular collapse secondary to persistent vomiting.5
Table 1 summarizes various evaluation, diagnosis, and treatment algorithms designed to optimize nutrition and weight in patients developing signs and symptoms of SMA syndrome after posterior spinal instrumentation and fusion for adolescent idiopathic scoliosis (AIS). 

The third unique feature in this case is electrocardiogram findings. Although some cases briefly discussed electrolyte abnormalities, none presented evidence that these abnormalities caused cardiac changes.6,16,18 The overall clinical significance of the QT prolongation in our patient’s case is unknown, as this finding was improved with correction of the electrolyte abnormalities and appropriate fluid replenishment.
Early recognition of nonspecific symptoms (eg, abdominal pain, tenderness, distension, bilious or projectile vomiting, hypoactive bowel sounds, anorexia) plays a key role in preventing severe morbidity and mortality from SMA syndrome after scoliosis surgery. Although many patients present in the semiclassic obstructed pattern, notable reasons for diagnostic delay include normal appetite and bowel sounds.3 For example, SMA syndrome may be misdiagnosed as stomach flu because of unfamiliarity with disease diagnosis and management.20 Complications of SMA syndrome can potentially lead to aspiration pneumonia, acute gastric rupture, and cardiovascular collapse and death.
Am J Orthop. 2017;46(2):E124-E130. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Evarts CM, Winter RB, Hall JE. Vascular compression of the duodenum associated with the treatment of scoliosis. Review of the literature and report of eighteen cases. J Bone Joint Surg Am. 1971;53(3):431-444.
2. Zhu ZZ, Qiu Y. Superior mesenteric artery syndrome following scoliosis surgery: its risk indicators and treatment strategy. World J Gastroenterol. 2005;11(21):3307-3310.
3. Hutchinson DT, Bassett GS. Superior mesenteric artery syndrome in pediatric orthopedic patients. Clin Orthop Relat Res. 1990;(250):250-257.
4. Willet A. Fatal vomiting following application of plaster-of-Paris bandage in case of spinal curvature. St Barth Hosp Rep. 1878;14:333-335.
5. Dorph MH. The cast syndrome; review of the literature and report of a case. N Engl J Med. 1950;243(12):440-442.
6. Lam DJ, Lee JZ, Chua JH, Lee YT, Lim KB. Superior mesenteric artery syndrome following surgery for adolescent idiopathic scoliosis: a case series, review of the literature, and an algorithm for management. J Pediatr Orthop B. 2014;23(4):312-318.
7. Tsirikos AI, Anakwe RE, Baker AD. Late presentation of superior mesenteric artery syndrome following scoliosis surgery: a case report. J Med Case Rep. 2008;2:9.
8. Akin JT Jr, Skandalakis JE, Gray SW. The anatomic basis of vascular compression of the duodenum. Surg Clin North Am. 1974;54(6):1361-1370.
9. Amy BW, Priebe CJ Jr, King A. Superior mesenteric artery syndrome associated with scoliosis treated by a modified Ladd procedure. J Pediatr Orthop. 1985;5(3):361-363.
10. Richardson WS, Surowiec WJ. Laparoscopic repair of superior mesenteric artery syndrome. Am J Surg. 2001;181(4):377-378.
11. Lenke LG, Betz RR, Harms J, et al. Adolescent idiopathic scoliosis: a new classification to determine extent of spinal arthrodesis. J Bone Joint Surg Am. 2001;83(8):1169-1181.
12. Braun SV, Hedden DM, Howard AW. Superior mesenteric artery syndrome following spinal deformity correction. J Bone Joint Surg Am. 2006;88(10):2252-2257.
13. Smith BG, Hakim-Zargar M, Thomson JD. Low body mass index: a risk factor for superior mesenteric artery syndrome in adolescents undergoing spinal fusion for scoliosis. J Spinal Disord Tech. 2009;22(2):144-148.
14. Pan CH, Tzeng ST, Chen CS, Chen PQ. Superior mesenteric artery syndrome complicating staged corrective surgery for scoliosis. J Formos Med Assoc. 2007;106(2 suppl):S37-S45.
15. Kennedy RH, Cooper MJ. An unusually severe case of the cast syndrome. Postgrad Med J. 1983;59(694):539-540.
16. Keskin M, Akgül T, Bayraktar A, Dikici F, Balik E. Superior mesenteric artery syndrome: an infrequent complication of scoliosis surgery. Case Rep Surg. 2014;2014:263431.
17. Amarawickrama H, Harikrishnan A, Krijgsman B. Superior mesenteric artery syndrome in a young girl following spinal surgery for scoliosis. Br J Hosp Med. 2005;66(12):700-701.
18. Crowther MA, Webb PJ, Eyre-Brook IA. Superior mesenteric artery syndrome following surgery for scoliosis. Spine. 2002;27(24):E528-E533.
19. Moskovich R, Cheong-Leen P. Vascular compression of the duodenum. J R Soc Med. 1986;79(8):465-467.
20. Shah MA, Albright MB, Vogt MT, Moreland MS. Superior mesenteric artery syndrome in scoliosis surgery: weight percentile for height as an indicator of risk. J Pediatr Orthop. 2003;23(5):665-668.
Take-Home Points
- Adolescent growth spurt, height-to-weight ratio, and perioperative weight loss are risk factors associated with SMA syndrome following pediatric spine surgery.
- Must recognize nonspecific symptoms such as abdominal pain, tenderness, distention, bilious or projectile vomiting, hypoactive bowel sounds, and anorexia postoperatively.
- Complications of SMA syndrome can potentially lead to aspiration pneumonia, acute gastric rupture, or cardiovascular collapse and death.
Superior mesenteric artery (SMA) syndrome resulting from surgical treatment of scoliosis has been recognized in the medical literature since 1752.1 Throughout the literature, SMA syndrome variably has been referred to as cast syndrome, Wilkie syndrome, arteriomesenteric duodenal obstruction, and chronic duodenal ileus.2 We now recognize numerous etiologies of SMA syndrome, as several sources can externally compress the duodenum. Classic acute symptoms of bowel obstruction include bilious vomiting, nausea, and epigastric pain. Chronic manifestations of SMA syndrome may include weight loss and decreased appetite. Our literature review revealed that adolescent growth spurt, height-to-weight ratio, and perioperative weight loss are risk factors associated with SMA syndrome after pediatric spine surgery.
We report the case of a 14-year-old boy who developed SMA syndrome after undergoing scoliosis surgery. The patient and his mother provided written informed consent for print and electronic publication of this case report.
Case Report
A 14-year-old boy with a history of idiopathic scoliosis presented to Cohen Children’s Hospital (Long Island Jewish Medical Center) with bilious vomiting that had persisted for 7 days after posterior T9–L4 fusion with instrumentation.
Discussion
SMA syndrome is attributed to the anatomical orientation of the third part of the duodenum, which passes between the aorta and the SMA (Figure 4).
Adolescents are particularly vulnerable to this condition. Faster adolescent bone growth relative to visceral growth is accompanied by a decrease in SMA angle.3 Occasionally, body casts are used after surgery to immobilize the vertebrae and augment healing. Cast syndrome occurs when pressure from a body cast causes a bowel obstruction secondary to spinal hyperextension and amplified spinal lordosis.2 This finding, dating to the 19th century, was reported by Willet4 when a patient died 48 hours after application of a body cast. In 1950, the term cast syndrome was coined after a motorcyclist’s injuries were treated with a hip spica cast and the patient died of cardiovascular collapse secondary to persistent vomiting.5
Table 1 summarizes various evaluation, diagnosis, and treatment algorithms designed to optimize nutrition and weight in patients developing signs and symptoms of SMA syndrome after posterior spinal instrumentation and fusion for adolescent idiopathic scoliosis (AIS).
The third unique feature in this case is electrocardiogram findings. Although some cases briefly discussed electrolyte abnormalities, none presented evidence that these abnormalities caused cardiac changes.6,16,18 The overall clinical significance of the QT prolongation in our patient’s case is unknown, as this finding was improved with correction of the electrolyte abnormalities and appropriate fluid replenishment.
Early recognition of nonspecific symptoms (eg, abdominal pain, tenderness, distension, bilious or projectile vomiting, hypoactive bowel sounds, anorexia) plays a key role in preventing severe morbidity and mortality from SMA syndrome after scoliosis surgery. Although many patients present in the semiclassic obstructed pattern, notable reasons for diagnostic delay include normal appetite and bowel sounds.3 For example, SMA syndrome may be misdiagnosed as stomach flu because of unfamiliarity with disease diagnosis and management.20 Complications of SMA syndrome can potentially lead to aspiration pneumonia, acute gastric rupture, and cardiovascular collapse and death.
Am J Orthop. 2017;46(2):E124-E130. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- Adolescent growth spurt, height-to-weight ratio, and perioperative weight loss are risk factors associated with SMA syndrome following pediatric spine surgery.
- Must recognize nonspecific symptoms such as abdominal pain, tenderness, distention, bilious or projectile vomiting, hypoactive bowel sounds, and anorexia postoperatively.
- Complications of SMA syndrome can potentially lead to aspiration pneumonia, acute gastric rupture, or cardiovascular collapse and death.
Superior mesenteric artery (SMA) syndrome resulting from surgical treatment of scoliosis has been recognized in the medical literature since 1752.1 Throughout the literature, SMA syndrome variably has been referred to as cast syndrome, Wilkie syndrome, arteriomesenteric duodenal obstruction, and chronic duodenal ileus.2 We now recognize numerous etiologies of SMA syndrome, as several sources can externally compress the duodenum. Classic acute symptoms of bowel obstruction include bilious vomiting, nausea, and epigastric pain. Chronic manifestations of SMA syndrome may include weight loss and decreased appetite. Our literature review revealed that adolescent growth spurt, height-to-weight ratio, and perioperative weight loss are risk factors associated with SMA syndrome after pediatric spine surgery.
We report the case of a 14-year-old boy who developed SMA syndrome after undergoing scoliosis surgery. The patient and his mother provided written informed consent for print and electronic publication of this case report.
Case Report
A 14-year-old boy with a history of idiopathic scoliosis presented to Cohen Children’s Hospital (Long Island Jewish Medical Center) with bilious vomiting that had persisted for 7 days after posterior T9–L4 fusion with instrumentation.
Discussion
SMA syndrome is attributed to the anatomical orientation of the third part of the duodenum, which passes between the aorta and the SMA (Figure 4).
Adolescents are particularly vulnerable to this condition. Faster adolescent bone growth relative to visceral growth is accompanied by a decrease in SMA angle.3 Occasionally, body casts are used after surgery to immobilize the vertebrae and augment healing. Cast syndrome occurs when pressure from a body cast causes a bowel obstruction secondary to spinal hyperextension and amplified spinal lordosis.2 This finding, dating to the 19th century, was reported by Willet4 when a patient died 48 hours after application of a body cast. In 1950, the term cast syndrome was coined after a motorcyclist’s injuries were treated with a hip spica cast and the patient died of cardiovascular collapse secondary to persistent vomiting.5
Table 1 summarizes various evaluation, diagnosis, and treatment algorithms designed to optimize nutrition and weight in patients developing signs and symptoms of SMA syndrome after posterior spinal instrumentation and fusion for adolescent idiopathic scoliosis (AIS).
The third unique feature in this case is electrocardiogram findings. Although some cases briefly discussed electrolyte abnormalities, none presented evidence that these abnormalities caused cardiac changes.6,16,18 The overall clinical significance of the QT prolongation in our patient’s case is unknown, as this finding was improved with correction of the electrolyte abnormalities and appropriate fluid replenishment.
Early recognition of nonspecific symptoms (eg, abdominal pain, tenderness, distension, bilious or projectile vomiting, hypoactive bowel sounds, anorexia) plays a key role in preventing severe morbidity and mortality from SMA syndrome after scoliosis surgery. Although many patients present in the semiclassic obstructed pattern, notable reasons for diagnostic delay include normal appetite and bowel sounds.3 For example, SMA syndrome may be misdiagnosed as stomach flu because of unfamiliarity with disease diagnosis and management.20 Complications of SMA syndrome can potentially lead to aspiration pneumonia, acute gastric rupture, and cardiovascular collapse and death.
Am J Orthop. 2017;46(2):E124-E130. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Evarts CM, Winter RB, Hall JE. Vascular compression of the duodenum associated with the treatment of scoliosis. Review of the literature and report of eighteen cases. J Bone Joint Surg Am. 1971;53(3):431-444.
2. Zhu ZZ, Qiu Y. Superior mesenteric artery syndrome following scoliosis surgery: its risk indicators and treatment strategy. World J Gastroenterol. 2005;11(21):3307-3310.
3. Hutchinson DT, Bassett GS. Superior mesenteric artery syndrome in pediatric orthopedic patients. Clin Orthop Relat Res. 1990;(250):250-257.
4. Willet A. Fatal vomiting following application of plaster-of-Paris bandage in case of spinal curvature. St Barth Hosp Rep. 1878;14:333-335.
5. Dorph MH. The cast syndrome; review of the literature and report of a case. N Engl J Med. 1950;243(12):440-442.
6. Lam DJ, Lee JZ, Chua JH, Lee YT, Lim KB. Superior mesenteric artery syndrome following surgery for adolescent idiopathic scoliosis: a case series, review of the literature, and an algorithm for management. J Pediatr Orthop B. 2014;23(4):312-318.
7. Tsirikos AI, Anakwe RE, Baker AD. Late presentation of superior mesenteric artery syndrome following scoliosis surgery: a case report. J Med Case Rep. 2008;2:9.
8. Akin JT Jr, Skandalakis JE, Gray SW. The anatomic basis of vascular compression of the duodenum. Surg Clin North Am. 1974;54(6):1361-1370.
9. Amy BW, Priebe CJ Jr, King A. Superior mesenteric artery syndrome associated with scoliosis treated by a modified Ladd procedure. J Pediatr Orthop. 1985;5(3):361-363.
10. Richardson WS, Surowiec WJ. Laparoscopic repair of superior mesenteric artery syndrome. Am J Surg. 2001;181(4):377-378.
11. Lenke LG, Betz RR, Harms J, et al. Adolescent idiopathic scoliosis: a new classification to determine extent of spinal arthrodesis. J Bone Joint Surg Am. 2001;83(8):1169-1181.
12. Braun SV, Hedden DM, Howard AW. Superior mesenteric artery syndrome following spinal deformity correction. J Bone Joint Surg Am. 2006;88(10):2252-2257.
13. Smith BG, Hakim-Zargar M, Thomson JD. Low body mass index: a risk factor for superior mesenteric artery syndrome in adolescents undergoing spinal fusion for scoliosis. J Spinal Disord Tech. 2009;22(2):144-148.
14. Pan CH, Tzeng ST, Chen CS, Chen PQ. Superior mesenteric artery syndrome complicating staged corrective surgery for scoliosis. J Formos Med Assoc. 2007;106(2 suppl):S37-S45.
15. Kennedy RH, Cooper MJ. An unusually severe case of the cast syndrome. Postgrad Med J. 1983;59(694):539-540.
16. Keskin M, Akgül T, Bayraktar A, Dikici F, Balik E. Superior mesenteric artery syndrome: an infrequent complication of scoliosis surgery. Case Rep Surg. 2014;2014:263431.
17. Amarawickrama H, Harikrishnan A, Krijgsman B. Superior mesenteric artery syndrome in a young girl following spinal surgery for scoliosis. Br J Hosp Med. 2005;66(12):700-701.
18. Crowther MA, Webb PJ, Eyre-Brook IA. Superior mesenteric artery syndrome following surgery for scoliosis. Spine. 2002;27(24):E528-E533.
19. Moskovich R, Cheong-Leen P. Vascular compression of the duodenum. J R Soc Med. 1986;79(8):465-467.
20. Shah MA, Albright MB, Vogt MT, Moreland MS. Superior mesenteric artery syndrome in scoliosis surgery: weight percentile for height as an indicator of risk. J Pediatr Orthop. 2003;23(5):665-668.
1. Evarts CM, Winter RB, Hall JE. Vascular compression of the duodenum associated with the treatment of scoliosis. Review of the literature and report of eighteen cases. J Bone Joint Surg Am. 1971;53(3):431-444.
2. Zhu ZZ, Qiu Y. Superior mesenteric artery syndrome following scoliosis surgery: its risk indicators and treatment strategy. World J Gastroenterol. 2005;11(21):3307-3310.
3. Hutchinson DT, Bassett GS. Superior mesenteric artery syndrome in pediatric orthopedic patients. Clin Orthop Relat Res. 1990;(250):250-257.
4. Willet A. Fatal vomiting following application of plaster-of-Paris bandage in case of spinal curvature. St Barth Hosp Rep. 1878;14:333-335.
5. Dorph MH. The cast syndrome; review of the literature and report of a case. N Engl J Med. 1950;243(12):440-442.
6. Lam DJ, Lee JZ, Chua JH, Lee YT, Lim KB. Superior mesenteric artery syndrome following surgery for adolescent idiopathic scoliosis: a case series, review of the literature, and an algorithm for management. J Pediatr Orthop B. 2014;23(4):312-318.
7. Tsirikos AI, Anakwe RE, Baker AD. Late presentation of superior mesenteric artery syndrome following scoliosis surgery: a case report. J Med Case Rep. 2008;2:9.
8. Akin JT Jr, Skandalakis JE, Gray SW. The anatomic basis of vascular compression of the duodenum. Surg Clin North Am. 1974;54(6):1361-1370.
9. Amy BW, Priebe CJ Jr, King A. Superior mesenteric artery syndrome associated with scoliosis treated by a modified Ladd procedure. J Pediatr Orthop. 1985;5(3):361-363.
10. Richardson WS, Surowiec WJ. Laparoscopic repair of superior mesenteric artery syndrome. Am J Surg. 2001;181(4):377-378.
11. Lenke LG, Betz RR, Harms J, et al. Adolescent idiopathic scoliosis: a new classification to determine extent of spinal arthrodesis. J Bone Joint Surg Am. 2001;83(8):1169-1181.
12. Braun SV, Hedden DM, Howard AW. Superior mesenteric artery syndrome following spinal deformity correction. J Bone Joint Surg Am. 2006;88(10):2252-2257.
13. Smith BG, Hakim-Zargar M, Thomson JD. Low body mass index: a risk factor for superior mesenteric artery syndrome in adolescents undergoing spinal fusion for scoliosis. J Spinal Disord Tech. 2009;22(2):144-148.
14. Pan CH, Tzeng ST, Chen CS, Chen PQ. Superior mesenteric artery syndrome complicating staged corrective surgery for scoliosis. J Formos Med Assoc. 2007;106(2 suppl):S37-S45.
15. Kennedy RH, Cooper MJ. An unusually severe case of the cast syndrome. Postgrad Med J. 1983;59(694):539-540.
16. Keskin M, Akgül T, Bayraktar A, Dikici F, Balik E. Superior mesenteric artery syndrome: an infrequent complication of scoliosis surgery. Case Rep Surg. 2014;2014:263431.
17. Amarawickrama H, Harikrishnan A, Krijgsman B. Superior mesenteric artery syndrome in a young girl following spinal surgery for scoliosis. Br J Hosp Med. 2005;66(12):700-701.
18. Crowther MA, Webb PJ, Eyre-Brook IA. Superior mesenteric artery syndrome following surgery for scoliosis. Spine. 2002;27(24):E528-E533.
19. Moskovich R, Cheong-Leen P. Vascular compression of the duodenum. J R Soc Med. 1986;79(8):465-467.
20. Shah MA, Albright MB, Vogt MT, Moreland MS. Superior mesenteric artery syndrome in scoliosis surgery: weight percentile for height as an indicator of risk. J Pediatr Orthop. 2003;23(5):665-668.
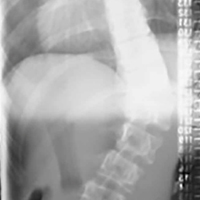
Paraneoplastic Palmoplantar Keratoderma Secondary to Metastatic Uterine Adenocarcinoma
Paraneoplastic palmoplantar keratoderma (PPK) is an acquired dermatosis that presents with hyperkeratosis of the palms and soles in association with visceral malignancies, such as esophageal, gastric, pulmonary, and bladder carcinomas. This condition may either be acquired or inherited.1
Case Report
A 72-year-old woman was referred to our dermatology clinic for evaluation of a nonpruritic hyperkeratotic eruption predominantly on the palms and soles of 2 to 3 months’ duration (Figure 1A). Review of systems was remarkable for chronic anxiety, unintentional weight loss of 10 lb over the last 6 months, and a mild cough of 10 days’ duration. The differential diagnosis included eczematous dermatitis, tinea manuum, new-onset palmoplantar psoriasis, and PPK.
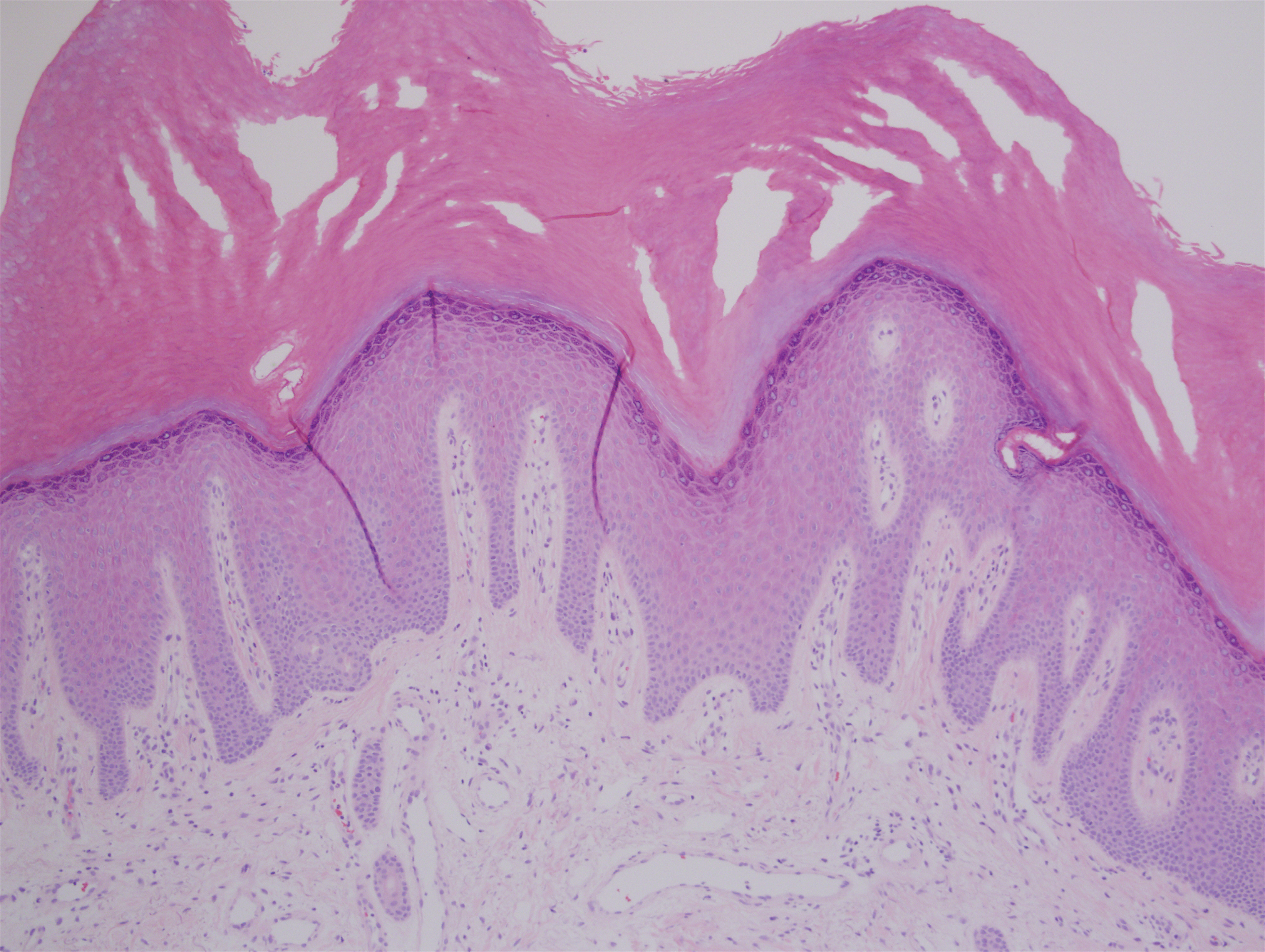
A punch biopsy of the medial hypothenar eminence of the left hand was performed, revealing notable lichenified hyperkeratosis with vascular ectasia (Figure 2). Periodic acid–Schiff staining was negative for fungal elements. Given the suspicion of PPK, multiple carcinoma markers were ordered. Cancer antigen 125 measured at 68 U/mL (reference range upper limit, 21 U/mL). Cancer antigen 27-29 was 50 U/mL (reference range, <38 U/mL) and cancer antigen 19-9 was 24 U/mL (reference range, <37 U/mL). Computed tomography of the chest revealed a large mass in the left lower lung associated with hilar lymphadenopathy. The patient was referred to oncology for further evaluation. Computed tomography–guided biopsy revealed metastatic uterine adenocarcinoma, which prompted subsequent chemotherapy. The combination of visceral malignancy with PPK led to the diagnosis of acquired PPK secondary to uterine cancer. After the completion of chemotherapy, the palmar dermatosis notably decreased (Figure 1B).
Comment
Paraneoplastic PPK is not uncommon. Ninety percent of acquired diffuse PPK is secondary to cancer,2 which occurs more frequently in male patients. Associated visceral malignancies include localized esophageal,3 myeloma,4 pulmonary, urinary/bladder,5 and gastric carcinoma.6 Paraneoplastic PPK in women is rare but has been linked to ovarian and breast carcinoma.7
The findings under light microscopy include thickening of any or all of the cell layers of the epidermis, which can include hyperkeratosis, acanthosis, and papillomatosis (Figure 2). A moderate amount of mononuclear cell infiltrates also can be visualized.
Palmoplantar keratoderma associated with uterine malignancy is rare. However, many other paraneoplastic dermatoses resulting from uterine cancer have been described as well as nonuterine gynecological malignancies (Table).8-17
The first step in managing acquired PPK is to determine its etiology via a complete history and a total-body skin examination. If findings are consistent with a hereditary PPK, then genetic workup is advised. Other suspected etiologies should be investigated via imaging and laboratory analysis.18
The first approach in managing acquired PPK is to treat the underlying cause. In prior cases, complete resolution of skin findings resulted once the malignancy or associated dermatosis had been treated.8-17 Adjunctive medication includes topical keratolytics (eg, urea, salicylic acid, lactic acid), topical retinoids, topical psoralen plus UVA, and topical corticosteroids.18 Vitamin A analogues have been found to be an effective treatment of many hyperkeratotic dermatoses.19 Isotretinoin and etretinate have been used to treat the cutaneous findings and prevent the onset and progression of esophageal malignancy of the inherited forms of PPK. The oral retinoid acitretin has been shown to rapidly resolve lesions, have persistent effects after 5 months of cessation, and have minimal side effects. Thus, it has been suggested as the first-line treatment of chronic PPK.19 One study found no response to topical keratolytics (urea cream and salicylic acid ointment) and a 2-week course of oral prednisone; however, low-dose oral acitretin 10 mg once daily resulted in notable improvement over several weeks.7 Physical debridement also may be necessary.18
Conclusion
Palmoplantar keratoderma is a condition that presents with hyperkeratosis of the palms and soles. Acquired PPK often occurs as a paraneoplastic response as well as a stigma of other dermatoses. It occurs more frequently in male patients. Reports of PPK secondary to uterine cancer are not common in the literature. Management of PPK includes a complete history and total-body skin examination. After appropriate imaging and laboratory analysis, treatment of the underlying cause is the best approach. Adjunctive medications include topical keratolytics, topical retinoids, topical psoralen plus UVA, and topical corticosteroids. Oral isotretinoin and etretinate have demonstrated promising results.
- Zamiri M, van Steensel MA, Munro CS. Inherited palmoplantar keratodermas. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012:538-548.
- Cohen PR, Grossman ME, Silvers DN. Tripe palms and cancer. Clin Dermatol. 1993;11:165-173.
- Belmar P, Marquet A, Martín-Sáez E. Symmetric palmar hyperkeratosis and esophageal carcinoma [in Spanish]. Actas Dermosifiliogr. 2008;99:149-150.
- Smith CH, Barker JN, Hay RJ. Diffuse plane xanthomatosis and acquired palmoplantar keratoderma in association with myeloma. Br J Dermatol. 1995;132:286-289.
- Küchmeister B, Rasokat H. Acquired disseminated papulous palmar keratoses—a paraneoplastic syndrome in cancers of the urinary bladder and lung? [in German]. Z Hautkr. 1984;59:1123-1124.
- Stieler K, Blume-Peytavi U, Vogel A, et al. Hyperkeratoses as paraneoplastic syndrome [published online June 1, 2012]. J Dtsch Dermatol Ges. 2012;10:593-595.
- Vignale RA, Espasandín J, Paciel J, et al. Diagnostic value of keratosis palmaris as indicative sign of visceral cancer [in Spanish]. Med Cutan Ibero Lat Am. 1983;11:287-292.
- Blanchet-Bardon C, Nazzaro V, Chevrant-Breton J, et al. Hereditary epidermolytic palmoplantar keratoderma associated with breast and ovarian cancer in a large kindred. Br J Dermatol. 1987;117:363-370.
- Champion GD, Saxon JA, Kossard S. The syndrome of palmar fibromatosis (fasciitis) and polyarthritis. J Rheumatol. 1987;14:1196-1198.
- Requena L, Aguilar A, Renedo G, et al. Tripe palms: a cutaneous marker of internal malignancy. J Dermatol. 1995;22:492-495.
- Mahler V, Neureiter D, Kirchner T, et al. Digital ischemia as paraneoplastic marker of metastatic endometrial carcinoma [in German]. Hautarzt. 1999;50:748-752.
- Docquier Ch, Majois F, Mitine C. Palmar fasciitis and arthritis: association with endometrial adenocarcinoma. Clin Rheumatol. 2002;21:63-65.
- Shimizu Y, Uchiyama S, Mori G, et al. A young patient with endometrioid adenocarcinoma who suffered Trousseau’s syndrome associated with vasculitis [in Japanese]. Rinsho Shinkeigaku. 2002;42:227-232.
- Chandiramani M, Joynson C, Panchal R, et al. Dermatomyositis as a paraneoplastic syndrome in carcinosarcoma of uterine origin. Clin Oncol (R Coll Radiol). 2006;18:641-648.
- Kebria MM, Belinson J, Kim R, et al. Malignant acanthosis nigricans, tripe palms and the sign of Leser-Trélat, a hint to the diagnosis of early stage ovarian cancer: a case report and review of the literature [published online January 27, 2006]. Gynecol Oncol. 2006;101:353-355.
- Valverde R, Sánchez-Caminero MP, Calzado L, et al. Dermatomyositis and punctate porokeratotic keratoderma as paraneoplastic syndrome of ovarian carcinoma [in Spanish]. Actas Dermosifiliogr. 2007;98:358-360.
- Abakka S, Elhalouat H, Khoummane N, et al. Uterine leiomyosarcoma and Leser-Trélat sign. Lancet. 2013;381:88.
- Patel S, Zirwas M, English JC 3rd. Acquired palmoplantar keratoderma. Am J Clin Dermatol. 2007;8:1-11.
- Capella GL, Fracchiolla C, Frigerio E, et al. A controlled study of comparative efficacy of oral retinoids and topical betamethasone/salicylic acid for chronic hyperkeratotic palmoplantar dermatitis. J Dermatolog Treat. 2004;15:88-93.
Paraneoplastic palmoplantar keratoderma (PPK) is an acquired dermatosis that presents with hyperkeratosis of the palms and soles in association with visceral malignancies, such as esophageal, gastric, pulmonary, and bladder carcinomas. This condition may either be acquired or inherited.1
Case Report
A 72-year-old woman was referred to our dermatology clinic for evaluation of a nonpruritic hyperkeratotic eruption predominantly on the palms and soles of 2 to 3 months’ duration (Figure 1A). Review of systems was remarkable for chronic anxiety, unintentional weight loss of 10 lb over the last 6 months, and a mild cough of 10 days’ duration. The differential diagnosis included eczematous dermatitis, tinea manuum, new-onset palmoplantar psoriasis, and PPK.
A punch biopsy of the medial hypothenar eminence of the left hand was performed, revealing notable lichenified hyperkeratosis with vascular ectasia (Figure 2). Periodic acid–Schiff staining was negative for fungal elements. Given the suspicion of PPK, multiple carcinoma markers were ordered. Cancer antigen 125 measured at 68 U/mL (reference range upper limit, 21 U/mL). Cancer antigen 27-29 was 50 U/mL (reference range, <38 U/mL) and cancer antigen 19-9 was 24 U/mL (reference range, <37 U/mL). Computed tomography of the chest revealed a large mass in the left lower lung associated with hilar lymphadenopathy. The patient was referred to oncology for further evaluation. Computed tomography–guided biopsy revealed metastatic uterine adenocarcinoma, which prompted subsequent chemotherapy. The combination of visceral malignancy with PPK led to the diagnosis of acquired PPK secondary to uterine cancer. After the completion of chemotherapy, the palmar dermatosis notably decreased (Figure 1B).
Comment
Paraneoplastic PPK is not uncommon. Ninety percent of acquired diffuse PPK is secondary to cancer,2 which occurs more frequently in male patients. Associated visceral malignancies include localized esophageal,3 myeloma,4 pulmonary, urinary/bladder,5 and gastric carcinoma.6 Paraneoplastic PPK in women is rare but has been linked to ovarian and breast carcinoma.7
The findings under light microscopy include thickening of any or all of the cell layers of the epidermis, which can include hyperkeratosis, acanthosis, and papillomatosis (Figure 2). A moderate amount of mononuclear cell infiltrates also can be visualized.
Palmoplantar keratoderma associated with uterine malignancy is rare. However, many other paraneoplastic dermatoses resulting from uterine cancer have been described as well as nonuterine gynecological malignancies (Table).8-17
The first step in managing acquired PPK is to determine its etiology via a complete history and a total-body skin examination. If findings are consistent with a hereditary PPK, then genetic workup is advised. Other suspected etiologies should be investigated via imaging and laboratory analysis.18
The first approach in managing acquired PPK is to treat the underlying cause. In prior cases, complete resolution of skin findings resulted once the malignancy or associated dermatosis had been treated.8-17 Adjunctive medication includes topical keratolytics (eg, urea, salicylic acid, lactic acid), topical retinoids, topical psoralen plus UVA, and topical corticosteroids.18 Vitamin A analogues have been found to be an effective treatment of many hyperkeratotic dermatoses.19 Isotretinoin and etretinate have been used to treat the cutaneous findings and prevent the onset and progression of esophageal malignancy of the inherited forms of PPK. The oral retinoid acitretin has been shown to rapidly resolve lesions, have persistent effects after 5 months of cessation, and have minimal side effects. Thus, it has been suggested as the first-line treatment of chronic PPK.19 One study found no response to topical keratolytics (urea cream and salicylic acid ointment) and a 2-week course of oral prednisone; however, low-dose oral acitretin 10 mg once daily resulted in notable improvement over several weeks.7 Physical debridement also may be necessary.18
Conclusion
Palmoplantar keratoderma is a condition that presents with hyperkeratosis of the palms and soles. Acquired PPK often occurs as a paraneoplastic response as well as a stigma of other dermatoses. It occurs more frequently in male patients. Reports of PPK secondary to uterine cancer are not common in the literature. Management of PPK includes a complete history and total-body skin examination. After appropriate imaging and laboratory analysis, treatment of the underlying cause is the best approach. Adjunctive medications include topical keratolytics, topical retinoids, topical psoralen plus UVA, and topical corticosteroids. Oral isotretinoin and etretinate have demonstrated promising results.
Paraneoplastic palmoplantar keratoderma (PPK) is an acquired dermatosis that presents with hyperkeratosis of the palms and soles in association with visceral malignancies, such as esophageal, gastric, pulmonary, and bladder carcinomas. This condition may either be acquired or inherited.1
Case Report
A 72-year-old woman was referred to our dermatology clinic for evaluation of a nonpruritic hyperkeratotic eruption predominantly on the palms and soles of 2 to 3 months’ duration (Figure 1A). Review of systems was remarkable for chronic anxiety, unintentional weight loss of 10 lb over the last 6 months, and a mild cough of 10 days’ duration. The differential diagnosis included eczematous dermatitis, tinea manuum, new-onset palmoplantar psoriasis, and PPK.
A punch biopsy of the medial hypothenar eminence of the left hand was performed, revealing notable lichenified hyperkeratosis with vascular ectasia (Figure 2). Periodic acid–Schiff staining was negative for fungal elements. Given the suspicion of PPK, multiple carcinoma markers were ordered. Cancer antigen 125 measured at 68 U/mL (reference range upper limit, 21 U/mL). Cancer antigen 27-29 was 50 U/mL (reference range, <38 U/mL) and cancer antigen 19-9 was 24 U/mL (reference range, <37 U/mL). Computed tomography of the chest revealed a large mass in the left lower lung associated with hilar lymphadenopathy. The patient was referred to oncology for further evaluation. Computed tomography–guided biopsy revealed metastatic uterine adenocarcinoma, which prompted subsequent chemotherapy. The combination of visceral malignancy with PPK led to the diagnosis of acquired PPK secondary to uterine cancer. After the completion of chemotherapy, the palmar dermatosis notably decreased (Figure 1B).
Comment
Paraneoplastic PPK is not uncommon. Ninety percent of acquired diffuse PPK is secondary to cancer,2 which occurs more frequently in male patients. Associated visceral malignancies include localized esophageal,3 myeloma,4 pulmonary, urinary/bladder,5 and gastric carcinoma.6 Paraneoplastic PPK in women is rare but has been linked to ovarian and breast carcinoma.7
The findings under light microscopy include thickening of any or all of the cell layers of the epidermis, which can include hyperkeratosis, acanthosis, and papillomatosis (Figure 2). A moderate amount of mononuclear cell infiltrates also can be visualized.
Palmoplantar keratoderma associated with uterine malignancy is rare. However, many other paraneoplastic dermatoses resulting from uterine cancer have been described as well as nonuterine gynecological malignancies (Table).8-17
The first step in managing acquired PPK is to determine its etiology via a complete history and a total-body skin examination. If findings are consistent with a hereditary PPK, then genetic workup is advised. Other suspected etiologies should be investigated via imaging and laboratory analysis.18
The first approach in managing acquired PPK is to treat the underlying cause. In prior cases, complete resolution of skin findings resulted once the malignancy or associated dermatosis had been treated.8-17 Adjunctive medication includes topical keratolytics (eg, urea, salicylic acid, lactic acid), topical retinoids, topical psoralen plus UVA, and topical corticosteroids.18 Vitamin A analogues have been found to be an effective treatment of many hyperkeratotic dermatoses.19 Isotretinoin and etretinate have been used to treat the cutaneous findings and prevent the onset and progression of esophageal malignancy of the inherited forms of PPK. The oral retinoid acitretin has been shown to rapidly resolve lesions, have persistent effects after 5 months of cessation, and have minimal side effects. Thus, it has been suggested as the first-line treatment of chronic PPK.19 One study found no response to topical keratolytics (urea cream and salicylic acid ointment) and a 2-week course of oral prednisone; however, low-dose oral acitretin 10 mg once daily resulted in notable improvement over several weeks.7 Physical debridement also may be necessary.18
Conclusion
Palmoplantar keratoderma is a condition that presents with hyperkeratosis of the palms and soles. Acquired PPK often occurs as a paraneoplastic response as well as a stigma of other dermatoses. It occurs more frequently in male patients. Reports of PPK secondary to uterine cancer are not common in the literature. Management of PPK includes a complete history and total-body skin examination. After appropriate imaging and laboratory analysis, treatment of the underlying cause is the best approach. Adjunctive medications include topical keratolytics, topical retinoids, topical psoralen plus UVA, and topical corticosteroids. Oral isotretinoin and etretinate have demonstrated promising results.
- Zamiri M, van Steensel MA, Munro CS. Inherited palmoplantar keratodermas. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012:538-548.
- Cohen PR, Grossman ME, Silvers DN. Tripe palms and cancer. Clin Dermatol. 1993;11:165-173.
- Belmar P, Marquet A, Martín-Sáez E. Symmetric palmar hyperkeratosis and esophageal carcinoma [in Spanish]. Actas Dermosifiliogr. 2008;99:149-150.
- Smith CH, Barker JN, Hay RJ. Diffuse plane xanthomatosis and acquired palmoplantar keratoderma in association with myeloma. Br J Dermatol. 1995;132:286-289.
- Küchmeister B, Rasokat H. Acquired disseminated papulous palmar keratoses—a paraneoplastic syndrome in cancers of the urinary bladder and lung? [in German]. Z Hautkr. 1984;59:1123-1124.
- Stieler K, Blume-Peytavi U, Vogel A, et al. Hyperkeratoses as paraneoplastic syndrome [published online June 1, 2012]. J Dtsch Dermatol Ges. 2012;10:593-595.
- Vignale RA, Espasandín J, Paciel J, et al. Diagnostic value of keratosis palmaris as indicative sign of visceral cancer [in Spanish]. Med Cutan Ibero Lat Am. 1983;11:287-292.
- Blanchet-Bardon C, Nazzaro V, Chevrant-Breton J, et al. Hereditary epidermolytic palmoplantar keratoderma associated with breast and ovarian cancer in a large kindred. Br J Dermatol. 1987;117:363-370.
- Champion GD, Saxon JA, Kossard S. The syndrome of palmar fibromatosis (fasciitis) and polyarthritis. J Rheumatol. 1987;14:1196-1198.
- Requena L, Aguilar A, Renedo G, et al. Tripe palms: a cutaneous marker of internal malignancy. J Dermatol. 1995;22:492-495.
- Mahler V, Neureiter D, Kirchner T, et al. Digital ischemia as paraneoplastic marker of metastatic endometrial carcinoma [in German]. Hautarzt. 1999;50:748-752.
- Docquier Ch, Majois F, Mitine C. Palmar fasciitis and arthritis: association with endometrial adenocarcinoma. Clin Rheumatol. 2002;21:63-65.
- Shimizu Y, Uchiyama S, Mori G, et al. A young patient with endometrioid adenocarcinoma who suffered Trousseau’s syndrome associated with vasculitis [in Japanese]. Rinsho Shinkeigaku. 2002;42:227-232.
- Chandiramani M, Joynson C, Panchal R, et al. Dermatomyositis as a paraneoplastic syndrome in carcinosarcoma of uterine origin. Clin Oncol (R Coll Radiol). 2006;18:641-648.
- Kebria MM, Belinson J, Kim R, et al. Malignant acanthosis nigricans, tripe palms and the sign of Leser-Trélat, a hint to the diagnosis of early stage ovarian cancer: a case report and review of the literature [published online January 27, 2006]. Gynecol Oncol. 2006;101:353-355.
- Valverde R, Sánchez-Caminero MP, Calzado L, et al. Dermatomyositis and punctate porokeratotic keratoderma as paraneoplastic syndrome of ovarian carcinoma [in Spanish]. Actas Dermosifiliogr. 2007;98:358-360.
- Abakka S, Elhalouat H, Khoummane N, et al. Uterine leiomyosarcoma and Leser-Trélat sign. Lancet. 2013;381:88.
- Patel S, Zirwas M, English JC 3rd. Acquired palmoplantar keratoderma. Am J Clin Dermatol. 2007;8:1-11.
- Capella GL, Fracchiolla C, Frigerio E, et al. A controlled study of comparative efficacy of oral retinoids and topical betamethasone/salicylic acid for chronic hyperkeratotic palmoplantar dermatitis. J Dermatolog Treat. 2004;15:88-93.
- Zamiri M, van Steensel MA, Munro CS. Inherited palmoplantar keratodermas. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012:538-548.
- Cohen PR, Grossman ME, Silvers DN. Tripe palms and cancer. Clin Dermatol. 1993;11:165-173.
- Belmar P, Marquet A, Martín-Sáez E. Symmetric palmar hyperkeratosis and esophageal carcinoma [in Spanish]. Actas Dermosifiliogr. 2008;99:149-150.
- Smith CH, Barker JN, Hay RJ. Diffuse plane xanthomatosis and acquired palmoplantar keratoderma in association with myeloma. Br J Dermatol. 1995;132:286-289.
- Küchmeister B, Rasokat H. Acquired disseminated papulous palmar keratoses—a paraneoplastic syndrome in cancers of the urinary bladder and lung? [in German]. Z Hautkr. 1984;59:1123-1124.
- Stieler K, Blume-Peytavi U, Vogel A, et al. Hyperkeratoses as paraneoplastic syndrome [published online June 1, 2012]. J Dtsch Dermatol Ges. 2012;10:593-595.
- Vignale RA, Espasandín J, Paciel J, et al. Diagnostic value of keratosis palmaris as indicative sign of visceral cancer [in Spanish]. Med Cutan Ibero Lat Am. 1983;11:287-292.
- Blanchet-Bardon C, Nazzaro V, Chevrant-Breton J, et al. Hereditary epidermolytic palmoplantar keratoderma associated with breast and ovarian cancer in a large kindred. Br J Dermatol. 1987;117:363-370.
- Champion GD, Saxon JA, Kossard S. The syndrome of palmar fibromatosis (fasciitis) and polyarthritis. J Rheumatol. 1987;14:1196-1198.
- Requena L, Aguilar A, Renedo G, et al. Tripe palms: a cutaneous marker of internal malignancy. J Dermatol. 1995;22:492-495.
- Mahler V, Neureiter D, Kirchner T, et al. Digital ischemia as paraneoplastic marker of metastatic endometrial carcinoma [in German]. Hautarzt. 1999;50:748-752.
- Docquier Ch, Majois F, Mitine C. Palmar fasciitis and arthritis: association with endometrial adenocarcinoma. Clin Rheumatol. 2002;21:63-65.
- Shimizu Y, Uchiyama S, Mori G, et al. A young patient with endometrioid adenocarcinoma who suffered Trousseau’s syndrome associated with vasculitis [in Japanese]. Rinsho Shinkeigaku. 2002;42:227-232.
- Chandiramani M, Joynson C, Panchal R, et al. Dermatomyositis as a paraneoplastic syndrome in carcinosarcoma of uterine origin. Clin Oncol (R Coll Radiol). 2006;18:641-648.
- Kebria MM, Belinson J, Kim R, et al. Malignant acanthosis nigricans, tripe palms and the sign of Leser-Trélat, a hint to the diagnosis of early stage ovarian cancer: a case report and review of the literature [published online January 27, 2006]. Gynecol Oncol. 2006;101:353-355.
- Valverde R, Sánchez-Caminero MP, Calzado L, et al. Dermatomyositis and punctate porokeratotic keratoderma as paraneoplastic syndrome of ovarian carcinoma [in Spanish]. Actas Dermosifiliogr. 2007;98:358-360.
- Abakka S, Elhalouat H, Khoummane N, et al. Uterine leiomyosarcoma and Leser-Trélat sign. Lancet. 2013;381:88.
- Patel S, Zirwas M, English JC 3rd. Acquired palmoplantar keratoderma. Am J Clin Dermatol. 2007;8:1-11.
- Capella GL, Fracchiolla C, Frigerio E, et al. A controlled study of comparative efficacy of oral retinoids and topical betamethasone/salicylic acid for chronic hyperkeratotic palmoplantar dermatitis. J Dermatolog Treat. 2004;15:88-93.
Practice Points
- Paraneoplastic palmoplantar keratoderma (PPK) is an acquired dermatosis that presents with hyperkeratosis of the palms and soles in association with visceral malignancies (eg, esophageal, gastric, pulmonary, and urinary/bladder carcinomas).
- Palmoplantar keratoderma secondary to uterine cancer is rare.
- Light microscopy shows thickening of any or all of the cell layers of the epidermis (hyperkeratosis, acanthosis, and papillomatosis) and mononuclear cells.
- Management of acquired PPK includes treatment of the underlying malignancy. Adjunctive vitamin A analogues may be of additional utility.