User login
Arthroscopic Excision of Bipartite Patella With Preservation of Lateral Retinaculum in an Adolescent Ice Hockey Player
Take-Home Points
- Bipartite patella is an asymptomatic anatomical variant.
- Occasionally, some adolescent athletes can present with AKP, resulting in decreased participation and performance.
- Bipartite patella is classified in type I, inferior pole; type II, lateral margin; and type III, superior lateral pole, depending on where the accessory patellar fragment is.
- Nonoperative treatment is advocated first. If symptoms persist surgical treatment should be attempted.
In 2% to 3% of the general population, the finding of bipartite patella on knee radiographs is often incidental.1,2 During development, the patella normally originates in a primary ossification center. Occasionally, secondary ossification centers emerge around the margins of the primary center and typically join that center. In some cases, the secondary2 center remains separated, leading to patella partita and an accessory patellar fragment.3,4
The bipartite patella is connected to the primary patella by fibrocartilage. The fibrous attachment may become irritated or separated as a result of trauma, overuse, or strenuous activity.1,5-7 Saupe classification of bipartite patella is based on accessory patellar fragment location: type I, inferior pole; type II, lateral margin; and type III, superior lateral pole.8 When an individual with a bipartite patella becomes symptomatic, anterior knee pain (AKP) is the most common complaint—it has been described in adolescent athletes in numerous sports.7,9-11For most patients, first-line treatment is nonoperative management. A typical regimen includes reduced activity, use of nonsteroidal anti-inflammatory drugs, physical therapy, and isometric quadriceps-strengthening exercises.1,12 Other nonoperative approaches described in the literature are immobilization,5,10 steroid and anesthetic injection, and ultrasound therapy.13 If symptoms do not improve, surgical treatment should be considered. Surgical treatment options include open excision of fragment,3,9,12 arthroscopic excision of fragment,7,14,15 tension band wiring,5,16 open reduction and internal fixation,17 open or arthroscopic vastus lateralis release,18-20 and lateral retinacular release.21 However, the optimal surgical option remains controversial.
In this case report, we present a modification of an arthroscopic surgical technique for excising a symptomatic bipartite patella and report midterm clinical outcomes. The patient provided written informed consent for print and electronic publication of this report.
Case Report
A 16-year-old elite male ice hockey player presented to clinic with a 2-week history of left AKP. He could not recall a specific injury that triggered the symptoms. Radiographs were obtained at an outside institution, and knee patellar fracture was diagnosed. The patient, placed in a straight-leg immobilizer, later presented to a referral clinic for a second opinion and further evaluation. Physical examination revealed significant tenderness to palpation of the lateral aspect of the patella. Range of motion was symmetric and fully intact. Patellar mobility was excellent. However, the patient could not perform a straight-leg raise because of the pain.
We obtained anteroposterior and lateral radiographs (Figures 1A, 1B), which showed evidence of a Saupe type III bipartite patella with separation at the superolateral pole. 

Two years later, the patient returned with left AKP, again localized to the lateral aspect of the patella, over the bipartite fragment. The pain was significant with compression. Given the patient’s history, arthroscopic excision of the bipartite patella was recommended. After discussing all treatment options, the patient elected to proceed with the surgery.
Surgical Technique
The patient was positioned supine on the operating table. Medial and lateral parapatellar arthroscopic portals were created. Menisci, cruciate ligaments, and tibiofemoral articular cartilage were arthroscopically visualized and determined to be normal. The bipartite patella was easily visualized, and notably loose when probed. Grade 2 chondromalacia was present diffusely throughout the bipartite patella and on the far lateral aspect of the patella, at the fragment interface.
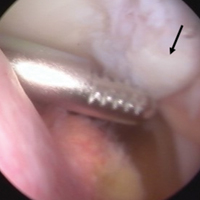
Attention was then turned to arthroscopic removal of the accessory patellar fragment (Figures 3A, 3B). 

Postoperative Rehabilitation
Rehabilitation focused on protection of the healing patella and accelerated rehabilitation for early return to play. Range-of-motion exercises and stationary bicycling were initiated on postoperative day 1. Weight-bearing was allowed as tolerated. Quadriceps sets, straight-leg raises, and ankle pumps were performed 5 times daily for 6 weeks. Six weeks after surgery, the patient was cleared, and he returned to full on-ice activities.
Outcomes
This study was approved by an Institutional Review Board. Preoperative and postoperative outcomes were obtained and stored in a data registry. The patient’s Lysholm score22 improved from 71 before surgery to 100 at 31-month follow-up. In addition, his subjective International Knee Documentation Committee score23 improved from 65.5 before surgery to 72.4 after surgery. At follow-up, patient satisfaction with outcome was 10/10. In addition, the patient had returned to playing hockey at a higher national level without functional limitation.
Discussion
The most important finding in this case is that arthroscopic excision of a bipartite patella with preservation of the lateral retinaculum in an elite adolescent hockey player resulted in improved subjective clinical outcomes scores and early return to competition. Arthroscopic excision was favored over open excision in this patient because of potential quicker recovery,14 less pain, and expedited return to competition. In addition, previous arthroscopic techniques were modified to shorten postoperative rehabilitation. The modified technique included preservation of the lateral retinaculum and total arthroscopic excision of the accessory bipartite patella fragment.
Although results of open techniques have been favorable,3,8,9 these procedures are far more invasive than arthroscopic techniques and may result in loss of quadriceps strength and prolonged rehabilitation.18 Weckström and colleagues12 followed 25 male military recruits for a minimum of 10 years after open excision of symptomatic bipartite patella. Mean Kujala score was 95 (range, 75-100), and median visual analog scale score for knee pain was 1.0 (range, 0.0-6.0). In a study by Bourne and Bianco,3 13 of 16 patients who were followed for an average of 7 years experienced complete pain relief with an average recovery time of 2 months.
Other studies have described the arthroscopic excision technique for symptomatic bipartite patella,7,14,15 but outcomes are underreported, especially for follow-ups longer than 2 years. Felli and colleagues7 described a case of arthroscopic excision and lateral release in a 23-year-old female professional volleyball player; at 1-year follow-up, the patient was symptom-free and back to full athletic participation. Azarbod and colleagues14 also reported on a patient who was symptom-free, 6 weeks after arthroscopic excision of bipartite patella. Carney and colleagues15 indicated that successful excision of bipartite patella was evident on 6-month radiographic follow-up. Our 31-month follow-up is the longest of any study on arthroscopic excision of bipartite patella. Clinical outcomes were excellent both in our patient’s case and in the earlier studies.
Our patient was a high-level hockey player who wanted to return to competition as quickly as possible. Conservative management, including physical therapy, initially resolved his symptoms and allowed him to resume on-ice activities after 6 weeks. In time, however, his symptoms returned and began limiting his on-ice performance. Arthroscopic removal of the bipartite patella accessory fragment allowed him to return to full on-ice activities after 6 weeks. His case provides evidence that arthroscopic management of bipartite patella with preservation of the vastus lateralis and lateral retinaculum may be an excellent treatment option for patients who want to return to athletics as quickly as possible.
Our technique of arthroscopic excision with preservation of lateral retinaculum is an excellent treatment option for symptomatic bipartite patella. This option, combined with an aggressive rehabilitation protocol, allows for pain relief and expedited return to competition.
Am J Orthop. 2017;46(3):135-138. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Atesok K, Doral MN, Lowe J, Finsterbush A. Symptomatic bipartite patella: treatment alternatives. J Am Acad Orthop Surg. 2008;16(8):455-461.
2. Insall J. Current concepts review: patellar pain. J Bone Joint Surg Am. 1982;64(1):147-152.
3. Bourne MH, Bianco AJ Jr. Bipartite patella in the adolescent: results of surgical excision. J Pediatr Orthop. 1990;10(1):69-73.
4. Oohashi Y, Koshino T, Oohashi Y. Clinical features and classification of bipartite or tripartite patella. Knee Surg Sports Traumatol Arthrosc. 2010;18(11):1465-1469.
5. Okuno H, Sugita T, Kawamata T, Ohnuma M, Yamada N, Yoshizumi Y. Traumatic separation of a type I bipartite patella: a report of four knees. Clin Orthop Relat Res. 2004;(420):257-260.
6. Yoo JH, Kim EH, Ryu HK. Arthroscopic removal of separated bipartite patella causing snapping knee syndrome. Orthopedics. 2008;31(7):717.
7. Felli L, Fiore M, Biglieni L. Arthroscopic treatment of symptomatic bipartite patella. Knee Surg Sports Traumatol Arthrosc. 2011;19(3):398-399.
8. Green WT Jr. Painful bipartite patellae. A report of three cases. Clin Orthop Relat Res. 1975;(110):197-200.
9. Ishikawa H, Sakurai A, Hirata S, et al. Painful bipartite patella in young athletes. The diagnostic value of skyline views taken in squatting position and the results of surgical excision. Clin Orthop Relat Res. 1994;(305):223-228.
10. Stocker RL, van Laer L. Injury of a bipartite patella in a young upcoming sportsman. Arch Orthop Trauma Surg. 2011;131(1):75-78.
11. Wong CK. Bipartite patella in a young athlete. J Orthop Sports Phys Ther. 2009;39(7):560.
12. Weckström M, Parviainen M, Pihlajamäki HK. Excision of painful bipartite patella: good long-term outcome in young adults. Clin Orthop Relat Res. 2008;466(11):2848-2855.
13. Kumahashi N, Uchio Y, Iwasa J, Kawasaki K, Adachi N, Ochi M. Bone union of painful bipartite patella after treatment with low-intensity pulsed ultrasound: report of two cases. Knee. 2008;15(1):50-53.
14. Azarbod P, Agar G, Patel V. Arthroscopic excision of a painful bipartite patella fragment. Arthroscopy. 2005;21(8):1006.
15. Carney J, Thompson D, O’Daniel J, Cassidy J. Arthroscopic excision of a painful bipartite patella fragment. Am J Orthop. 2010;39(1):40-43.
16. Tauber M, Matis N, Resch H. Traumatic separation of an uncommon bipartite patella type: a case report. Knee Surg Sports Traumatol Arthrosc. 2007;15(1):83-87.
17. Werner S, Durkan M, Jones J, Quilici S, Crawford D. Symptomatic bipartite patella: three subtypes, three representative cases. J Knee Surg. 2013;26(suppl 1):S72-S76.
18. Adachi N, Ochi M, Yamaguchi H, Uchio Y, Kuriwaka M. Vastus lateralis release for painful bipartite patella. Arthroscopy. 2002;18(4):404-411.
19. Maeno S, Hashimoto D, Otani T, Masumoto K, Hui C. The “coiling-up procedure”: a novel technique for extra-articular arthroscopy. Arthroscopy. 2010;26(11):1551-1555.
20. Ogata K. Painful bipartite patella. A new approach to operative treatment. J Bone Joint Surg Am. 1994;76(4):573-578.
21. Mori Y, Okumo H, Iketani H, Kuroki Y. Efficacy of lateral retinacular release for painful bipartite patella. Am J Sports Med. 1995;23(1):13-18.
22. Lysholm J, Gillquist J. Evaluation of knee ligament surgery results with special emphasis on use of a scoring scale. Am J Sports Med. 1982;10(3):150-154
23. Grevnerts HT, Terwee CB, Kvist J. The measurement properties of the IKDC-subjective knee form. Knee Surg Sports Traumatol Arthrosc. 2015;23(12):3698-3706.
Take-Home Points
- Bipartite patella is an asymptomatic anatomical variant.
- Occasionally, some adolescent athletes can present with AKP, resulting in decreased participation and performance.
- Bipartite patella is classified in type I, inferior pole; type II, lateral margin; and type III, superior lateral pole, depending on where the accessory patellar fragment is.
- Nonoperative treatment is advocated first. If symptoms persist surgical treatment should be attempted.
In 2% to 3% of the general population, the finding of bipartite patella on knee radiographs is often incidental.1,2 During development, the patella normally originates in a primary ossification center. Occasionally, secondary ossification centers emerge around the margins of the primary center and typically join that center. In some cases, the secondary2 center remains separated, leading to patella partita and an accessory patellar fragment.3,4
The bipartite patella is connected to the primary patella by fibrocartilage. The fibrous attachment may become irritated or separated as a result of trauma, overuse, or strenuous activity.1,5-7 Saupe classification of bipartite patella is based on accessory patellar fragment location: type I, inferior pole; type II, lateral margin; and type III, superior lateral pole.8 When an individual with a bipartite patella becomes symptomatic, anterior knee pain (AKP) is the most common complaint—it has been described in adolescent athletes in numerous sports.7,9-11For most patients, first-line treatment is nonoperative management. A typical regimen includes reduced activity, use of nonsteroidal anti-inflammatory drugs, physical therapy, and isometric quadriceps-strengthening exercises.1,12 Other nonoperative approaches described in the literature are immobilization,5,10 steroid and anesthetic injection, and ultrasound therapy.13 If symptoms do not improve, surgical treatment should be considered. Surgical treatment options include open excision of fragment,3,9,12 arthroscopic excision of fragment,7,14,15 tension band wiring,5,16 open reduction and internal fixation,17 open or arthroscopic vastus lateralis release,18-20 and lateral retinacular release.21 However, the optimal surgical option remains controversial.
In this case report, we present a modification of an arthroscopic surgical technique for excising a symptomatic bipartite patella and report midterm clinical outcomes. The patient provided written informed consent for print and electronic publication of this report.
Case Report
A 16-year-old elite male ice hockey player presented to clinic with a 2-week history of left AKP. He could not recall a specific injury that triggered the symptoms. Radiographs were obtained at an outside institution, and knee patellar fracture was diagnosed. The patient, placed in a straight-leg immobilizer, later presented to a referral clinic for a second opinion and further evaluation. Physical examination revealed significant tenderness to palpation of the lateral aspect of the patella. Range of motion was symmetric and fully intact. Patellar mobility was excellent. However, the patient could not perform a straight-leg raise because of the pain.
We obtained anteroposterior and lateral radiographs (Figures 1A, 1B), which showed evidence of a Saupe type III bipartite patella with separation at the superolateral pole.
Two years later, the patient returned with left AKP, again localized to the lateral aspect of the patella, over the bipartite fragment. The pain was significant with compression. Given the patient’s history, arthroscopic excision of the bipartite patella was recommended. After discussing all treatment options, the patient elected to proceed with the surgery.
Surgical Technique
The patient was positioned supine on the operating table. Medial and lateral parapatellar arthroscopic portals were created. Menisci, cruciate ligaments, and tibiofemoral articular cartilage were arthroscopically visualized and determined to be normal. The bipartite patella was easily visualized, and notably loose when probed. Grade 2 chondromalacia was present diffusely throughout the bipartite patella and on the far lateral aspect of the patella, at the fragment interface.
Attention was then turned to arthroscopic removal of the accessory patellar fragment (Figures 3A, 3B).
Postoperative Rehabilitation
Rehabilitation focused on protection of the healing patella and accelerated rehabilitation for early return to play. Range-of-motion exercises and stationary bicycling were initiated on postoperative day 1. Weight-bearing was allowed as tolerated. Quadriceps sets, straight-leg raises, and ankle pumps were performed 5 times daily for 6 weeks. Six weeks after surgery, the patient was cleared, and he returned to full on-ice activities.
Outcomes
This study was approved by an Institutional Review Board. Preoperative and postoperative outcomes were obtained and stored in a data registry. The patient’s Lysholm score22 improved from 71 before surgery to 100 at 31-month follow-up. In addition, his subjective International Knee Documentation Committee score23 improved from 65.5 before surgery to 72.4 after surgery. At follow-up, patient satisfaction with outcome was 10/10. In addition, the patient had returned to playing hockey at a higher national level without functional limitation.
Discussion
The most important finding in this case is that arthroscopic excision of a bipartite patella with preservation of the lateral retinaculum in an elite adolescent hockey player resulted in improved subjective clinical outcomes scores and early return to competition. Arthroscopic excision was favored over open excision in this patient because of potential quicker recovery,14 less pain, and expedited return to competition. In addition, previous arthroscopic techniques were modified to shorten postoperative rehabilitation. The modified technique included preservation of the lateral retinaculum and total arthroscopic excision of the accessory bipartite patella fragment.
Although results of open techniques have been favorable,3,8,9 these procedures are far more invasive than arthroscopic techniques and may result in loss of quadriceps strength and prolonged rehabilitation.18 Weckström and colleagues12 followed 25 male military recruits for a minimum of 10 years after open excision of symptomatic bipartite patella. Mean Kujala score was 95 (range, 75-100), and median visual analog scale score for knee pain was 1.0 (range, 0.0-6.0). In a study by Bourne and Bianco,3 13 of 16 patients who were followed for an average of 7 years experienced complete pain relief with an average recovery time of 2 months.
Other studies have described the arthroscopic excision technique for symptomatic bipartite patella,7,14,15 but outcomes are underreported, especially for follow-ups longer than 2 years. Felli and colleagues7 described a case of arthroscopic excision and lateral release in a 23-year-old female professional volleyball player; at 1-year follow-up, the patient was symptom-free and back to full athletic participation. Azarbod and colleagues14 also reported on a patient who was symptom-free, 6 weeks after arthroscopic excision of bipartite patella. Carney and colleagues15 indicated that successful excision of bipartite patella was evident on 6-month radiographic follow-up. Our 31-month follow-up is the longest of any study on arthroscopic excision of bipartite patella. Clinical outcomes were excellent both in our patient’s case and in the earlier studies.
Our patient was a high-level hockey player who wanted to return to competition as quickly as possible. Conservative management, including physical therapy, initially resolved his symptoms and allowed him to resume on-ice activities after 6 weeks. In time, however, his symptoms returned and began limiting his on-ice performance. Arthroscopic removal of the bipartite patella accessory fragment allowed him to return to full on-ice activities after 6 weeks. His case provides evidence that arthroscopic management of bipartite patella with preservation of the vastus lateralis and lateral retinaculum may be an excellent treatment option for patients who want to return to athletics as quickly as possible.
Our technique of arthroscopic excision with preservation of lateral retinaculum is an excellent treatment option for symptomatic bipartite patella. This option, combined with an aggressive rehabilitation protocol, allows for pain relief and expedited return to competition.
Am J Orthop. 2017;46(3):135-138. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- Bipartite patella is an asymptomatic anatomical variant.
- Occasionally, some adolescent athletes can present with AKP, resulting in decreased participation and performance.
- Bipartite patella is classified in type I, inferior pole; type II, lateral margin; and type III, superior lateral pole, depending on where the accessory patellar fragment is.
- Nonoperative treatment is advocated first. If symptoms persist surgical treatment should be attempted.
In 2% to 3% of the general population, the finding of bipartite patella on knee radiographs is often incidental.1,2 During development, the patella normally originates in a primary ossification center. Occasionally, secondary ossification centers emerge around the margins of the primary center and typically join that center. In some cases, the secondary2 center remains separated, leading to patella partita and an accessory patellar fragment.3,4
The bipartite patella is connected to the primary patella by fibrocartilage. The fibrous attachment may become irritated or separated as a result of trauma, overuse, or strenuous activity.1,5-7 Saupe classification of bipartite patella is based on accessory patellar fragment location: type I, inferior pole; type II, lateral margin; and type III, superior lateral pole.8 When an individual with a bipartite patella becomes symptomatic, anterior knee pain (AKP) is the most common complaint—it has been described in adolescent athletes in numerous sports.7,9-11For most patients, first-line treatment is nonoperative management. A typical regimen includes reduced activity, use of nonsteroidal anti-inflammatory drugs, physical therapy, and isometric quadriceps-strengthening exercises.1,12 Other nonoperative approaches described in the literature are immobilization,5,10 steroid and anesthetic injection, and ultrasound therapy.13 If symptoms do not improve, surgical treatment should be considered. Surgical treatment options include open excision of fragment,3,9,12 arthroscopic excision of fragment,7,14,15 tension band wiring,5,16 open reduction and internal fixation,17 open or arthroscopic vastus lateralis release,18-20 and lateral retinacular release.21 However, the optimal surgical option remains controversial.
In this case report, we present a modification of an arthroscopic surgical technique for excising a symptomatic bipartite patella and report midterm clinical outcomes. The patient provided written informed consent for print and electronic publication of this report.
Case Report
A 16-year-old elite male ice hockey player presented to clinic with a 2-week history of left AKP. He could not recall a specific injury that triggered the symptoms. Radiographs were obtained at an outside institution, and knee patellar fracture was diagnosed. The patient, placed in a straight-leg immobilizer, later presented to a referral clinic for a second opinion and further evaluation. Physical examination revealed significant tenderness to palpation of the lateral aspect of the patella. Range of motion was symmetric and fully intact. Patellar mobility was excellent. However, the patient could not perform a straight-leg raise because of the pain.
We obtained anteroposterior and lateral radiographs (Figures 1A, 1B), which showed evidence of a Saupe type III bipartite patella with separation at the superolateral pole.
Two years later, the patient returned with left AKP, again localized to the lateral aspect of the patella, over the bipartite fragment. The pain was significant with compression. Given the patient’s history, arthroscopic excision of the bipartite patella was recommended. After discussing all treatment options, the patient elected to proceed with the surgery.
Surgical Technique
The patient was positioned supine on the operating table. Medial and lateral parapatellar arthroscopic portals were created. Menisci, cruciate ligaments, and tibiofemoral articular cartilage were arthroscopically visualized and determined to be normal. The bipartite patella was easily visualized, and notably loose when probed. Grade 2 chondromalacia was present diffusely throughout the bipartite patella and on the far lateral aspect of the patella, at the fragment interface.
Attention was then turned to arthroscopic removal of the accessory patellar fragment (Figures 3A, 3B).
Postoperative Rehabilitation
Rehabilitation focused on protection of the healing patella and accelerated rehabilitation for early return to play. Range-of-motion exercises and stationary bicycling were initiated on postoperative day 1. Weight-bearing was allowed as tolerated. Quadriceps sets, straight-leg raises, and ankle pumps were performed 5 times daily for 6 weeks. Six weeks after surgery, the patient was cleared, and he returned to full on-ice activities.
Outcomes
This study was approved by an Institutional Review Board. Preoperative and postoperative outcomes were obtained and stored in a data registry. The patient’s Lysholm score22 improved from 71 before surgery to 100 at 31-month follow-up. In addition, his subjective International Knee Documentation Committee score23 improved from 65.5 before surgery to 72.4 after surgery. At follow-up, patient satisfaction with outcome was 10/10. In addition, the patient had returned to playing hockey at a higher national level without functional limitation.
Discussion
The most important finding in this case is that arthroscopic excision of a bipartite patella with preservation of the lateral retinaculum in an elite adolescent hockey player resulted in improved subjective clinical outcomes scores and early return to competition. Arthroscopic excision was favored over open excision in this patient because of potential quicker recovery,14 less pain, and expedited return to competition. In addition, previous arthroscopic techniques were modified to shorten postoperative rehabilitation. The modified technique included preservation of the lateral retinaculum and total arthroscopic excision of the accessory bipartite patella fragment.
Although results of open techniques have been favorable,3,8,9 these procedures are far more invasive than arthroscopic techniques and may result in loss of quadriceps strength and prolonged rehabilitation.18 Weckström and colleagues12 followed 25 male military recruits for a minimum of 10 years after open excision of symptomatic bipartite patella. Mean Kujala score was 95 (range, 75-100), and median visual analog scale score for knee pain was 1.0 (range, 0.0-6.0). In a study by Bourne and Bianco,3 13 of 16 patients who were followed for an average of 7 years experienced complete pain relief with an average recovery time of 2 months.
Other studies have described the arthroscopic excision technique for symptomatic bipartite patella,7,14,15 but outcomes are underreported, especially for follow-ups longer than 2 years. Felli and colleagues7 described a case of arthroscopic excision and lateral release in a 23-year-old female professional volleyball player; at 1-year follow-up, the patient was symptom-free and back to full athletic participation. Azarbod and colleagues14 also reported on a patient who was symptom-free, 6 weeks after arthroscopic excision of bipartite patella. Carney and colleagues15 indicated that successful excision of bipartite patella was evident on 6-month radiographic follow-up. Our 31-month follow-up is the longest of any study on arthroscopic excision of bipartite patella. Clinical outcomes were excellent both in our patient’s case and in the earlier studies.
Our patient was a high-level hockey player who wanted to return to competition as quickly as possible. Conservative management, including physical therapy, initially resolved his symptoms and allowed him to resume on-ice activities after 6 weeks. In time, however, his symptoms returned and began limiting his on-ice performance. Arthroscopic removal of the bipartite patella accessory fragment allowed him to return to full on-ice activities after 6 weeks. His case provides evidence that arthroscopic management of bipartite patella with preservation of the vastus lateralis and lateral retinaculum may be an excellent treatment option for patients who want to return to athletics as quickly as possible.
Our technique of arthroscopic excision with preservation of lateral retinaculum is an excellent treatment option for symptomatic bipartite patella. This option, combined with an aggressive rehabilitation protocol, allows for pain relief and expedited return to competition.
Am J Orthop. 2017;46(3):135-138. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Atesok K, Doral MN, Lowe J, Finsterbush A. Symptomatic bipartite patella: treatment alternatives. J Am Acad Orthop Surg. 2008;16(8):455-461.
2. Insall J. Current concepts review: patellar pain. J Bone Joint Surg Am. 1982;64(1):147-152.
3. Bourne MH, Bianco AJ Jr. Bipartite patella in the adolescent: results of surgical excision. J Pediatr Orthop. 1990;10(1):69-73.
4. Oohashi Y, Koshino T, Oohashi Y. Clinical features and classification of bipartite or tripartite patella. Knee Surg Sports Traumatol Arthrosc. 2010;18(11):1465-1469.
5. Okuno H, Sugita T, Kawamata T, Ohnuma M, Yamada N, Yoshizumi Y. Traumatic separation of a type I bipartite patella: a report of four knees. Clin Orthop Relat Res. 2004;(420):257-260.
6. Yoo JH, Kim EH, Ryu HK. Arthroscopic removal of separated bipartite patella causing snapping knee syndrome. Orthopedics. 2008;31(7):717.
7. Felli L, Fiore M, Biglieni L. Arthroscopic treatment of symptomatic bipartite patella. Knee Surg Sports Traumatol Arthrosc. 2011;19(3):398-399.
8. Green WT Jr. Painful bipartite patellae. A report of three cases. Clin Orthop Relat Res. 1975;(110):197-200.
9. Ishikawa H, Sakurai A, Hirata S, et al. Painful bipartite patella in young athletes. The diagnostic value of skyline views taken in squatting position and the results of surgical excision. Clin Orthop Relat Res. 1994;(305):223-228.
10. Stocker RL, van Laer L. Injury of a bipartite patella in a young upcoming sportsman. Arch Orthop Trauma Surg. 2011;131(1):75-78.
11. Wong CK. Bipartite patella in a young athlete. J Orthop Sports Phys Ther. 2009;39(7):560.
12. Weckström M, Parviainen M, Pihlajamäki HK. Excision of painful bipartite patella: good long-term outcome in young adults. Clin Orthop Relat Res. 2008;466(11):2848-2855.
13. Kumahashi N, Uchio Y, Iwasa J, Kawasaki K, Adachi N, Ochi M. Bone union of painful bipartite patella after treatment with low-intensity pulsed ultrasound: report of two cases. Knee. 2008;15(1):50-53.
14. Azarbod P, Agar G, Patel V. Arthroscopic excision of a painful bipartite patella fragment. Arthroscopy. 2005;21(8):1006.
15. Carney J, Thompson D, O’Daniel J, Cassidy J. Arthroscopic excision of a painful bipartite patella fragment. Am J Orthop. 2010;39(1):40-43.
16. Tauber M, Matis N, Resch H. Traumatic separation of an uncommon bipartite patella type: a case report. Knee Surg Sports Traumatol Arthrosc. 2007;15(1):83-87.
17. Werner S, Durkan M, Jones J, Quilici S, Crawford D. Symptomatic bipartite patella: three subtypes, three representative cases. J Knee Surg. 2013;26(suppl 1):S72-S76.
18. Adachi N, Ochi M, Yamaguchi H, Uchio Y, Kuriwaka M. Vastus lateralis release for painful bipartite patella. Arthroscopy. 2002;18(4):404-411.
19. Maeno S, Hashimoto D, Otani T, Masumoto K, Hui C. The “coiling-up procedure”: a novel technique for extra-articular arthroscopy. Arthroscopy. 2010;26(11):1551-1555.
20. Ogata K. Painful bipartite patella. A new approach to operative treatment. J Bone Joint Surg Am. 1994;76(4):573-578.
21. Mori Y, Okumo H, Iketani H, Kuroki Y. Efficacy of lateral retinacular release for painful bipartite patella. Am J Sports Med. 1995;23(1):13-18.
22. Lysholm J, Gillquist J. Evaluation of knee ligament surgery results with special emphasis on use of a scoring scale. Am J Sports Med. 1982;10(3):150-154
23. Grevnerts HT, Terwee CB, Kvist J. The measurement properties of the IKDC-subjective knee form. Knee Surg Sports Traumatol Arthrosc. 2015;23(12):3698-3706.
1. Atesok K, Doral MN, Lowe J, Finsterbush A. Symptomatic bipartite patella: treatment alternatives. J Am Acad Orthop Surg. 2008;16(8):455-461.
2. Insall J. Current concepts review: patellar pain. J Bone Joint Surg Am. 1982;64(1):147-152.
3. Bourne MH, Bianco AJ Jr. Bipartite patella in the adolescent: results of surgical excision. J Pediatr Orthop. 1990;10(1):69-73.
4. Oohashi Y, Koshino T, Oohashi Y. Clinical features and classification of bipartite or tripartite patella. Knee Surg Sports Traumatol Arthrosc. 2010;18(11):1465-1469.
5. Okuno H, Sugita T, Kawamata T, Ohnuma M, Yamada N, Yoshizumi Y. Traumatic separation of a type I bipartite patella: a report of four knees. Clin Orthop Relat Res. 2004;(420):257-260.
6. Yoo JH, Kim EH, Ryu HK. Arthroscopic removal of separated bipartite patella causing snapping knee syndrome. Orthopedics. 2008;31(7):717.
7. Felli L, Fiore M, Biglieni L. Arthroscopic treatment of symptomatic bipartite patella. Knee Surg Sports Traumatol Arthrosc. 2011;19(3):398-399.
8. Green WT Jr. Painful bipartite patellae. A report of three cases. Clin Orthop Relat Res. 1975;(110):197-200.
9. Ishikawa H, Sakurai A, Hirata S, et al. Painful bipartite patella in young athletes. The diagnostic value of skyline views taken in squatting position and the results of surgical excision. Clin Orthop Relat Res. 1994;(305):223-228.
10. Stocker RL, van Laer L. Injury of a bipartite patella in a young upcoming sportsman. Arch Orthop Trauma Surg. 2011;131(1):75-78.
11. Wong CK. Bipartite patella in a young athlete. J Orthop Sports Phys Ther. 2009;39(7):560.
12. Weckström M, Parviainen M, Pihlajamäki HK. Excision of painful bipartite patella: good long-term outcome in young adults. Clin Orthop Relat Res. 2008;466(11):2848-2855.
13. Kumahashi N, Uchio Y, Iwasa J, Kawasaki K, Adachi N, Ochi M. Bone union of painful bipartite patella after treatment with low-intensity pulsed ultrasound: report of two cases. Knee. 2008;15(1):50-53.
14. Azarbod P, Agar G, Patel V. Arthroscopic excision of a painful bipartite patella fragment. Arthroscopy. 2005;21(8):1006.
15. Carney J, Thompson D, O’Daniel J, Cassidy J. Arthroscopic excision of a painful bipartite patella fragment. Am J Orthop. 2010;39(1):40-43.
16. Tauber M, Matis N, Resch H. Traumatic separation of an uncommon bipartite patella type: a case report. Knee Surg Sports Traumatol Arthrosc. 2007;15(1):83-87.
17. Werner S, Durkan M, Jones J, Quilici S, Crawford D. Symptomatic bipartite patella: three subtypes, three representative cases. J Knee Surg. 2013;26(suppl 1):S72-S76.
18. Adachi N, Ochi M, Yamaguchi H, Uchio Y, Kuriwaka M. Vastus lateralis release for painful bipartite patella. Arthroscopy. 2002;18(4):404-411.
19. Maeno S, Hashimoto D, Otani T, Masumoto K, Hui C. The “coiling-up procedure”: a novel technique for extra-articular arthroscopy. Arthroscopy. 2010;26(11):1551-1555.
20. Ogata K. Painful bipartite patella. A new approach to operative treatment. J Bone Joint Surg Am. 1994;76(4):573-578.
21. Mori Y, Okumo H, Iketani H, Kuroki Y. Efficacy of lateral retinacular release for painful bipartite patella. Am J Sports Med. 1995;23(1):13-18.
22. Lysholm J, Gillquist J. Evaluation of knee ligament surgery results with special emphasis on use of a scoring scale. Am J Sports Med. 1982;10(3):150-154
23. Grevnerts HT, Terwee CB, Kvist J. The measurement properties of the IKDC-subjective knee form. Knee Surg Sports Traumatol Arthrosc. 2015;23(12):3698-3706.
Internal Carotid Artery Dissection After Indirect Blunt Cervical Trauma in an Ice Hockey Goaltender
Take-Home Points
- ICA dissections may occur from direct or indirect trauma.
- Symptoms can be mild, including a persistent headache.
- High clinical suspicion is required for diagnosis when symptoms are mild.
- Neuroimaging is required for definitive diagnosis.
- Conservative management with serial imaging can yield successful outcomes.
Cervical artery dissection (CAD) is an uncommon but potentially life-threatening condition that accounts for a high proportion of ischemic strokes in patients under the age of 45 years.1-4 The extracranial internal carotid arteries (ICAs) and vertebral arteries are most commonly involved; dissections can occur after either direct trauma to the neck, or indirect trauma resulting in acute hyperextension or hyperflexion.4-7 ICA dissection can be difficult to diagnose because of the varying symptomatology. Clinical presentation depends on stenosis location, degree of luminal narrowing, and presence or absence of ischemic stroke. Neurologic symptoms may be delayed, and misdiagnosis of an isolated soft-tissue contusion, whiplash, can be made in the setting of indirect cervical trauma.
Although this entity is well described in the literature,2,3,5,8 there are few reported cases of injuries sustained during high-intensity athletic competition. In this case report, we describe the symptoms, physical examination findings, diagnostic imaging results, and treatment of a young male athlete who presented with delayed-onset symptoms of ICA dissection resulting from indirect cervical trauma sustained during an ice hockey game. We discuss the importance of a high level of clinical suspicion in the diagnosis of neck injuries sustained during athletic competition, as well as the need for early vascular imaging for diagnosis. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
The patient was a right-handed 32-year-old professional hockey goaltender. Four days before diagnosis, his goaltending mask and attached neck-protector were inadvertently lifted by another player’s stick just as a puck traveling at high speed struck him in the neck, to the right of the larynx, causing acute neck hyperextension. He immediately experienced discomfort and fell to the ice, saying he was “dizzy and light-headed.” Play was stopped, and medical personnel attended to him. His symptoms resolved, and he resumed play without any notable deficits. The next day, he noted discomfort at the impact site, but no additional symptoms, and received a presumptive diagnosis of cervical soft-tissue contusion. Continuing to participate in hockey that day, he did not develop any symptoms other than superficial cervical discomfort. However, the next morning, he presented complaining of severe right frontotemporal headache, which had persisted overnight. Orthopedic examination revealed palpable tenderness over the anterior cervical musculature, including the sternocleidomastoid and strap muscles. There was no appreciable hematoma in the contused area. Cervical range of motion was otherwise preserved. Cervical spine examination, including dermatomal and myotomal examination, was normal, as was cranial nerve examination. However, given the headache intensity and the recency of the injury, the potential for vascular or neurologic injury was considered. A neurology consultation was obtained, and arrangements were made for advanced cross-sectional imaging.
On further evaluation, the patient denied loss of consciousness, seizure, vomiting, amnesia, visual disturbance, language or cognitive impairment, balance or coordination difficulties, or any appreciable face or limb weakness. Review of systems was otherwise negative. Detailed neurologic examination did not reveal any cranial nerve deficits, and pupils were 3 mm, equal, and normally responsive to light and accommodation. Muscular tone and strength were symmetric and full in the upper and lower extremities. Gait, coordination, and response to vibration and temperature sensation were all preserved.
Magnetic resonance imaging of the head and neck was normal, but magnetic resonance angiography (MRA) of the neck showed a 1-cm-long region of the ICA, before piercing the petrous bone, with evidence of dissection. 
Given the normal neurologic examination, and no evidence of brain infarction or other neurovascular complications, the acute ICA dissection was managed with antiplatelet therapy using aspirin (325 mg/d). In addition, the patient was advised to refrain from strenuous physical activity and to present to the hospital immediately if symptoms worsened or any neurologic impairment developed. Follow-up and repeat MRA were planned to monitor healing progression.
Two weeks after injury, the patient returned for follow-up. His headache and neck pain had resolved. Physical examination findings were unchanged, and there were no notable neurologic deficits. Repeat MRA findings were essentially unchanged, except for slightly increased luminal stenosis, exceeding 50% (Figure 2), attributable to intramural hematoma formation. 

At 6-week follow-up, the patient had no clinical symptoms and no recurrence of headaches. 

Discussion
In cases of direct (blunt) or indirect cervical trauma, CAD should be considered, as it carries a risk of potentially debilitating ischemic stroke in otherwise healthy young patients. Fortunately, CAD is rare; its annual incidence is 1 in 100,000, occurring in 0.08% to 1.2% of blunt trauma cases.9 
As symptoms of ICA dissection can vary depending on stenosis severity, diagnosis can be challenging. The classically associated triad of symptoms includes unilateral head, facial, or neck pain accompanied by partial Horner syndrome with progression to cerebral or retinal ischemia. However, these symptoms occur in less than a third of patients with ICA dissection.2 Neck pain may occur secondary to blunt cervical trauma, consistent with a cervical soft-tissue contusion; however, it may have more severe implications and should be carefully monitored, particularly if accompanied by additional symptoms, such as headache. Headaches, which are present in 44% to 69% of patients, are often unilateral and constant. Either headache or neck pain in isolation is relatively uncommon, occurring in <10% of cases,2 though retrospective reviews of delayed-onset ICA dissection found atypical headache or neck pain in 100% of patients,11 indicating that persistent symptoms should be further evaluated.
More commonly, patients present with neurologic symptoms, particularly Horner syndrome, which is caused by the disruption of the sympathetic nerve fibers adjacent to the ICA, resulting in ipsilateral ptosis and miosis. In addition, patients may present with cranial nerve palsies, most commonly involving cranial nerve XII (the hypoglossal nerve), resulting in tongue weakness and abnormal taste. These and other neurologic findings associated with retinal or cerebral ischemia should raise clinical suspicion for the injury and prompt computed tomography or MRA evaluation.
MRA has largely replaced conventional angiography for the diagnosis of CAD. As MRA is noninvasive, it allows for improved visualization of luminal narrowing and for evaluation of the arterial wall and intramural hematoma.2 Because of the potential for devastating sequelae with missed or delayed diagnosis, several authors have become proponents of early aggressive screening for detection of these injuries.9 Postdiagnostic treatment depends on the presence of neurologic symptoms. Management is directed toward limiting neurologic deficits; anticoagulant or antiplatelet agents are used to prevent thromboembolic events. A randomized controlled trial and other studies have failed to find any appreciable difference in subsequent rates of stroke or associated complications with use of either class of medication.8,12 Conventionally, treatment is continued for 3 to 6 months, depending on clinical resolution. Endovascular or surgical intervention typically is reserved for extreme luminal narrowing, conditions that are preventing anticoagulation, an expanding area of dissection with a persistent pseudoaneurysm, and cases of failed medical management with subsequent ischemic stroke.2The literature includes several case reports involving indirect trauma in recreational athletes. First, a 31-year-old woman sustained an ICA dissection secondary to a head injury that occurred during a soccer match; she presented with headache, altered sense of taste, and objective findings of ptosis and miosis consistent with Horner syndrome.13 Second, a 39-year-old man had an ICA dissection after a snowboarding fall that caused neck hyperextension; he presented with periocular headache, ptosis, and miosis.6 Third, 3 people who participated in CrossFit training sustained ICA dissection.7 They presented with varying degrees of neurologic symptoms: ptosis and miosis; right-side upper extremity ataxia; and visual distortion and receptive aphasia. Our patient’s ICA dissection resulted from indirect trauma that caused sudden hyperextension and lateral flexion in response to contact from a hockey puck. However, his case is unique in that symptoms onset was delayed, and there were no associated neurologic findings on clinical presentation. His case should raise awareness of this potential diagnosis, even in the absence of overt neurologic findings. In addition, the patient’s return to sport at 8 weeks was facilitated by full clinical resolution of symptoms and thorough radiographic documentation of improved intramural narrowing. Finally, to our knowledge this is the first report of this injury in a professional athlete.
Conclusion
We have reported the case of a 32-year-old professional hockey goaltender who presented with isolated, persistent, worsening headache of delayed onset after ICA dissection. The ICA dissection resulted from indirect trauma, with reaction to a puck causing acute hyperextension and rotational injury. To our knowledge, this is the first report of a case of ICA dissection in an athlete, lacking neurologic examination findings that could aid in the diagnosis. The index of suspicion for CAD should be high after direct or indirect cervical trauma when patients present with unilateral neck pain or headache, even in the absence of neurologic findings, as stroke is a catastrophic but preventable complication.
Am J Orthop. 2017;46(3):E139-E143. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Mohan IV. Current optimal assessment and management of carotid and vertebral spontaneous and traumatic dissection. Angiology. 2014;65(4):274-283.
2. Patel RR, Adam R, Maldjian C, Lincoln CM, Yuen A, Arneja A. Cervical carotid artery dissection: current review of diagnosis and treatment. Cardiol Rev. 2012;20(3):145-152.
3. Biller J, Sacco RL, Albuquerque FC, et al; American Heart Association Stroke Council. Cervical arterial dissections and association with cervical manipulative therapy: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45(10):3155-3174.
4. Fukunaga N, Hanaoka M, Sato K. Asymptomatic common carotid artery dissection caused by blunt injury. Emerg Med J. 2011;28(1):50.
5. Chen J, Zhou X, Li C, Cheung BM. Risk of stroke due to spontaneous cervical artery dissection. Intern Med. 2013;52(19):2237-2240.
6. Kalantzis G, Georgalas I, Chang BY, Ong C, El-Hindy N. An unusual case of traumatic internal carotid artery dissection during snowboarding. J Sports Sci Med. 2014;13(2):451-453.
7. Lu A, Shen P, Lee P, et al. CrossFit-related cervical internal carotid artery dissection. Emerg Radiol. 2015;22(4):449-452.
8. CADISS Trial Investigators, Markus HS, Hayter E, Levi C, Feldman A, Venables G, Norris J. Antiplatelet treatment compared with anticoagulation treatment for cervical artery dissection (CADISS): a randomised trial. Lancet Neurol. 2015;14(4):361-367.
9. van Wessem KJ, Meijer JM, Leenen LP, van der Worp HB, Moll FL, de Borst GJ. Blunt traumatic carotid artery dissection still a pitfall? The rationale for aggressive screening. Eur J Trauma Emerg Surg. 2011;37(2):147-154.
10. Haneline M, Triano J. Cervical artery dissection. A comparison of highly dynamic mechanisms: manipulation versus motor vehicle collision. J Manipulative Physiol Ther. 2005;28(1):57-63.
11. Thomas LC, Rivett DA, Attia JR, Levi C. Risk factors and clinical presentation of cervical arterial dissection: preliminary results of a prospective case-control study. J Orthop Sports Phys Ther. 2015;45(7):503-511.
12. Lyrer P, Engelter S. Antithrombotic drugs for carotid artery dissection. Cochrane Database Syst Rev. 2010;(10):CD000255.
13. Creavin ST, Rice CM, Pollentine A, Cowburn P. Carotid artery dissection presenting with isolated headache and Horner syndrome after minor head injury. Am J Emerg Med. 2012;30(9):2103.e5-e7.
Take-Home Points
- ICA dissections may occur from direct or indirect trauma.
- Symptoms can be mild, including a persistent headache.
- High clinical suspicion is required for diagnosis when symptoms are mild.
- Neuroimaging is required for definitive diagnosis.
- Conservative management with serial imaging can yield successful outcomes.
Cervical artery dissection (CAD) is an uncommon but potentially life-threatening condition that accounts for a high proportion of ischemic strokes in patients under the age of 45 years.1-4 The extracranial internal carotid arteries (ICAs) and vertebral arteries are most commonly involved; dissections can occur after either direct trauma to the neck, or indirect trauma resulting in acute hyperextension or hyperflexion.4-7 ICA dissection can be difficult to diagnose because of the varying symptomatology. Clinical presentation depends on stenosis location, degree of luminal narrowing, and presence or absence of ischemic stroke. Neurologic symptoms may be delayed, and misdiagnosis of an isolated soft-tissue contusion, whiplash, can be made in the setting of indirect cervical trauma.
Although this entity is well described in the literature,2,3,5,8 there are few reported cases of injuries sustained during high-intensity athletic competition. In this case report, we describe the symptoms, physical examination findings, diagnostic imaging results, and treatment of a young male athlete who presented with delayed-onset symptoms of ICA dissection resulting from indirect cervical trauma sustained during an ice hockey game. We discuss the importance of a high level of clinical suspicion in the diagnosis of neck injuries sustained during athletic competition, as well as the need for early vascular imaging for diagnosis. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
The patient was a right-handed 32-year-old professional hockey goaltender. Four days before diagnosis, his goaltending mask and attached neck-protector were inadvertently lifted by another player’s stick just as a puck traveling at high speed struck him in the neck, to the right of the larynx, causing acute neck hyperextension. He immediately experienced discomfort and fell to the ice, saying he was “dizzy and light-headed.” Play was stopped, and medical personnel attended to him. His symptoms resolved, and he resumed play without any notable deficits. The next day, he noted discomfort at the impact site, but no additional symptoms, and received a presumptive diagnosis of cervical soft-tissue contusion. Continuing to participate in hockey that day, he did not develop any symptoms other than superficial cervical discomfort. However, the next morning, he presented complaining of severe right frontotemporal headache, which had persisted overnight. Orthopedic examination revealed palpable tenderness over the anterior cervical musculature, including the sternocleidomastoid and strap muscles. There was no appreciable hematoma in the contused area. Cervical range of motion was otherwise preserved. Cervical spine examination, including dermatomal and myotomal examination, was normal, as was cranial nerve examination. However, given the headache intensity and the recency of the injury, the potential for vascular or neurologic injury was considered. A neurology consultation was obtained, and arrangements were made for advanced cross-sectional imaging.
On further evaluation, the patient denied loss of consciousness, seizure, vomiting, amnesia, visual disturbance, language or cognitive impairment, balance or coordination difficulties, or any appreciable face or limb weakness. Review of systems was otherwise negative. Detailed neurologic examination did not reveal any cranial nerve deficits, and pupils were 3 mm, equal, and normally responsive to light and accommodation. Muscular tone and strength were symmetric and full in the upper and lower extremities. Gait, coordination, and response to vibration and temperature sensation were all preserved.
Magnetic resonance imaging of the head and neck was normal, but magnetic resonance angiography (MRA) of the neck showed a 1-cm-long region of the ICA, before piercing the petrous bone, with evidence of dissection.
Given the normal neurologic examination, and no evidence of brain infarction or other neurovascular complications, the acute ICA dissection was managed with antiplatelet therapy using aspirin (325 mg/d). In addition, the patient was advised to refrain from strenuous physical activity and to present to the hospital immediately if symptoms worsened or any neurologic impairment developed. Follow-up and repeat MRA were planned to monitor healing progression.
Two weeks after injury, the patient returned for follow-up. His headache and neck pain had resolved. Physical examination findings were unchanged, and there were no notable neurologic deficits. Repeat MRA findings were essentially unchanged, except for slightly increased luminal stenosis, exceeding 50% (Figure 2), attributable to intramural hematoma formation.
At 6-week follow-up, the patient had no clinical symptoms and no recurrence of headaches.
Discussion
In cases of direct (blunt) or indirect cervical trauma, CAD should be considered, as it carries a risk of potentially debilitating ischemic stroke in otherwise healthy young patients. Fortunately, CAD is rare; its annual incidence is 1 in 100,000, occurring in 0.08% to 1.2% of blunt trauma cases.9
As symptoms of ICA dissection can vary depending on stenosis severity, diagnosis can be challenging. The classically associated triad of symptoms includes unilateral head, facial, or neck pain accompanied by partial Horner syndrome with progression to cerebral or retinal ischemia. However, these symptoms occur in less than a third of patients with ICA dissection.2 Neck pain may occur secondary to blunt cervical trauma, consistent with a cervical soft-tissue contusion; however, it may have more severe implications and should be carefully monitored, particularly if accompanied by additional symptoms, such as headache. Headaches, which are present in 44% to 69% of patients, are often unilateral and constant. Either headache or neck pain in isolation is relatively uncommon, occurring in <10% of cases,2 though retrospective reviews of delayed-onset ICA dissection found atypical headache or neck pain in 100% of patients,11 indicating that persistent symptoms should be further evaluated.
More commonly, patients present with neurologic symptoms, particularly Horner syndrome, which is caused by the disruption of the sympathetic nerve fibers adjacent to the ICA, resulting in ipsilateral ptosis and miosis. In addition, patients may present with cranial nerve palsies, most commonly involving cranial nerve XII (the hypoglossal nerve), resulting in tongue weakness and abnormal taste. These and other neurologic findings associated with retinal or cerebral ischemia should raise clinical suspicion for the injury and prompt computed tomography or MRA evaluation.
MRA has largely replaced conventional angiography for the diagnosis of CAD. As MRA is noninvasive, it allows for improved visualization of luminal narrowing and for evaluation of the arterial wall and intramural hematoma.2 Because of the potential for devastating sequelae with missed or delayed diagnosis, several authors have become proponents of early aggressive screening for detection of these injuries.9 Postdiagnostic treatment depends on the presence of neurologic symptoms. Management is directed toward limiting neurologic deficits; anticoagulant or antiplatelet agents are used to prevent thromboembolic events. A randomized controlled trial and other studies have failed to find any appreciable difference in subsequent rates of stroke or associated complications with use of either class of medication.8,12 Conventionally, treatment is continued for 3 to 6 months, depending on clinical resolution. Endovascular or surgical intervention typically is reserved for extreme luminal narrowing, conditions that are preventing anticoagulation, an expanding area of dissection with a persistent pseudoaneurysm, and cases of failed medical management with subsequent ischemic stroke.2The literature includes several case reports involving indirect trauma in recreational athletes. First, a 31-year-old woman sustained an ICA dissection secondary to a head injury that occurred during a soccer match; she presented with headache, altered sense of taste, and objective findings of ptosis and miosis consistent with Horner syndrome.13 Second, a 39-year-old man had an ICA dissection after a snowboarding fall that caused neck hyperextension; he presented with periocular headache, ptosis, and miosis.6 Third, 3 people who participated in CrossFit training sustained ICA dissection.7 They presented with varying degrees of neurologic symptoms: ptosis and miosis; right-side upper extremity ataxia; and visual distortion and receptive aphasia. Our patient’s ICA dissection resulted from indirect trauma that caused sudden hyperextension and lateral flexion in response to contact from a hockey puck. However, his case is unique in that symptoms onset was delayed, and there were no associated neurologic findings on clinical presentation. His case should raise awareness of this potential diagnosis, even in the absence of overt neurologic findings. In addition, the patient’s return to sport at 8 weeks was facilitated by full clinical resolution of symptoms and thorough radiographic documentation of improved intramural narrowing. Finally, to our knowledge this is the first report of this injury in a professional athlete.
Conclusion
We have reported the case of a 32-year-old professional hockey goaltender who presented with isolated, persistent, worsening headache of delayed onset after ICA dissection. The ICA dissection resulted from indirect trauma, with reaction to a puck causing acute hyperextension and rotational injury. To our knowledge, this is the first report of a case of ICA dissection in an athlete, lacking neurologic examination findings that could aid in the diagnosis. The index of suspicion for CAD should be high after direct or indirect cervical trauma when patients present with unilateral neck pain or headache, even in the absence of neurologic findings, as stroke is a catastrophic but preventable complication.
Am J Orthop. 2017;46(3):E139-E143. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- ICA dissections may occur from direct or indirect trauma.
- Symptoms can be mild, including a persistent headache.
- High clinical suspicion is required for diagnosis when symptoms are mild.
- Neuroimaging is required for definitive diagnosis.
- Conservative management with serial imaging can yield successful outcomes.
Cervical artery dissection (CAD) is an uncommon but potentially life-threatening condition that accounts for a high proportion of ischemic strokes in patients under the age of 45 years.1-4 The extracranial internal carotid arteries (ICAs) and vertebral arteries are most commonly involved; dissections can occur after either direct trauma to the neck, or indirect trauma resulting in acute hyperextension or hyperflexion.4-7 ICA dissection can be difficult to diagnose because of the varying symptomatology. Clinical presentation depends on stenosis location, degree of luminal narrowing, and presence or absence of ischemic stroke. Neurologic symptoms may be delayed, and misdiagnosis of an isolated soft-tissue contusion, whiplash, can be made in the setting of indirect cervical trauma.
Although this entity is well described in the literature,2,3,5,8 there are few reported cases of injuries sustained during high-intensity athletic competition. In this case report, we describe the symptoms, physical examination findings, diagnostic imaging results, and treatment of a young male athlete who presented with delayed-onset symptoms of ICA dissection resulting from indirect cervical trauma sustained during an ice hockey game. We discuss the importance of a high level of clinical suspicion in the diagnosis of neck injuries sustained during athletic competition, as well as the need for early vascular imaging for diagnosis. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
The patient was a right-handed 32-year-old professional hockey goaltender. Four days before diagnosis, his goaltending mask and attached neck-protector were inadvertently lifted by another player’s stick just as a puck traveling at high speed struck him in the neck, to the right of the larynx, causing acute neck hyperextension. He immediately experienced discomfort and fell to the ice, saying he was “dizzy and light-headed.” Play was stopped, and medical personnel attended to him. His symptoms resolved, and he resumed play without any notable deficits. The next day, he noted discomfort at the impact site, but no additional symptoms, and received a presumptive diagnosis of cervical soft-tissue contusion. Continuing to participate in hockey that day, he did not develop any symptoms other than superficial cervical discomfort. However, the next morning, he presented complaining of severe right frontotemporal headache, which had persisted overnight. Orthopedic examination revealed palpable tenderness over the anterior cervical musculature, including the sternocleidomastoid and strap muscles. There was no appreciable hematoma in the contused area. Cervical range of motion was otherwise preserved. Cervical spine examination, including dermatomal and myotomal examination, was normal, as was cranial nerve examination. However, given the headache intensity and the recency of the injury, the potential for vascular or neurologic injury was considered. A neurology consultation was obtained, and arrangements were made for advanced cross-sectional imaging.
On further evaluation, the patient denied loss of consciousness, seizure, vomiting, amnesia, visual disturbance, language or cognitive impairment, balance or coordination difficulties, or any appreciable face or limb weakness. Review of systems was otherwise negative. Detailed neurologic examination did not reveal any cranial nerve deficits, and pupils were 3 mm, equal, and normally responsive to light and accommodation. Muscular tone and strength were symmetric and full in the upper and lower extremities. Gait, coordination, and response to vibration and temperature sensation were all preserved.
Magnetic resonance imaging of the head and neck was normal, but magnetic resonance angiography (MRA) of the neck showed a 1-cm-long region of the ICA, before piercing the petrous bone, with evidence of dissection.
Given the normal neurologic examination, and no evidence of brain infarction or other neurovascular complications, the acute ICA dissection was managed with antiplatelet therapy using aspirin (325 mg/d). In addition, the patient was advised to refrain from strenuous physical activity and to present to the hospital immediately if symptoms worsened or any neurologic impairment developed. Follow-up and repeat MRA were planned to monitor healing progression.
Two weeks after injury, the patient returned for follow-up. His headache and neck pain had resolved. Physical examination findings were unchanged, and there were no notable neurologic deficits. Repeat MRA findings were essentially unchanged, except for slightly increased luminal stenosis, exceeding 50% (Figure 2), attributable to intramural hematoma formation.
At 6-week follow-up, the patient had no clinical symptoms and no recurrence of headaches.
Discussion
In cases of direct (blunt) or indirect cervical trauma, CAD should be considered, as it carries a risk of potentially debilitating ischemic stroke in otherwise healthy young patients. Fortunately, CAD is rare; its annual incidence is 1 in 100,000, occurring in 0.08% to 1.2% of blunt trauma cases.9
As symptoms of ICA dissection can vary depending on stenosis severity, diagnosis can be challenging. The classically associated triad of symptoms includes unilateral head, facial, or neck pain accompanied by partial Horner syndrome with progression to cerebral or retinal ischemia. However, these symptoms occur in less than a third of patients with ICA dissection.2 Neck pain may occur secondary to blunt cervical trauma, consistent with a cervical soft-tissue contusion; however, it may have more severe implications and should be carefully monitored, particularly if accompanied by additional symptoms, such as headache. Headaches, which are present in 44% to 69% of patients, are often unilateral and constant. Either headache or neck pain in isolation is relatively uncommon, occurring in <10% of cases,2 though retrospective reviews of delayed-onset ICA dissection found atypical headache or neck pain in 100% of patients,11 indicating that persistent symptoms should be further evaluated.
More commonly, patients present with neurologic symptoms, particularly Horner syndrome, which is caused by the disruption of the sympathetic nerve fibers adjacent to the ICA, resulting in ipsilateral ptosis and miosis. In addition, patients may present with cranial nerve palsies, most commonly involving cranial nerve XII (the hypoglossal nerve), resulting in tongue weakness and abnormal taste. These and other neurologic findings associated with retinal or cerebral ischemia should raise clinical suspicion for the injury and prompt computed tomography or MRA evaluation.
MRA has largely replaced conventional angiography for the diagnosis of CAD. As MRA is noninvasive, it allows for improved visualization of luminal narrowing and for evaluation of the arterial wall and intramural hematoma.2 Because of the potential for devastating sequelae with missed or delayed diagnosis, several authors have become proponents of early aggressive screening for detection of these injuries.9 Postdiagnostic treatment depends on the presence of neurologic symptoms. Management is directed toward limiting neurologic deficits; anticoagulant or antiplatelet agents are used to prevent thromboembolic events. A randomized controlled trial and other studies have failed to find any appreciable difference in subsequent rates of stroke or associated complications with use of either class of medication.8,12 Conventionally, treatment is continued for 3 to 6 months, depending on clinical resolution. Endovascular or surgical intervention typically is reserved for extreme luminal narrowing, conditions that are preventing anticoagulation, an expanding area of dissection with a persistent pseudoaneurysm, and cases of failed medical management with subsequent ischemic stroke.2The literature includes several case reports involving indirect trauma in recreational athletes. First, a 31-year-old woman sustained an ICA dissection secondary to a head injury that occurred during a soccer match; she presented with headache, altered sense of taste, and objective findings of ptosis and miosis consistent with Horner syndrome.13 Second, a 39-year-old man had an ICA dissection after a snowboarding fall that caused neck hyperextension; he presented with periocular headache, ptosis, and miosis.6 Third, 3 people who participated in CrossFit training sustained ICA dissection.7 They presented with varying degrees of neurologic symptoms: ptosis and miosis; right-side upper extremity ataxia; and visual distortion and receptive aphasia. Our patient’s ICA dissection resulted from indirect trauma that caused sudden hyperextension and lateral flexion in response to contact from a hockey puck. However, his case is unique in that symptoms onset was delayed, and there were no associated neurologic findings on clinical presentation. His case should raise awareness of this potential diagnosis, even in the absence of overt neurologic findings. In addition, the patient’s return to sport at 8 weeks was facilitated by full clinical resolution of symptoms and thorough radiographic documentation of improved intramural narrowing. Finally, to our knowledge this is the first report of this injury in a professional athlete.
Conclusion
We have reported the case of a 32-year-old professional hockey goaltender who presented with isolated, persistent, worsening headache of delayed onset after ICA dissection. The ICA dissection resulted from indirect trauma, with reaction to a puck causing acute hyperextension and rotational injury. To our knowledge, this is the first report of a case of ICA dissection in an athlete, lacking neurologic examination findings that could aid in the diagnosis. The index of suspicion for CAD should be high after direct or indirect cervical trauma when patients present with unilateral neck pain or headache, even in the absence of neurologic findings, as stroke is a catastrophic but preventable complication.
Am J Orthop. 2017;46(3):E139-E143. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Mohan IV. Current optimal assessment and management of carotid and vertebral spontaneous and traumatic dissection. Angiology. 2014;65(4):274-283.
2. Patel RR, Adam R, Maldjian C, Lincoln CM, Yuen A, Arneja A. Cervical carotid artery dissection: current review of diagnosis and treatment. Cardiol Rev. 2012;20(3):145-152.
3. Biller J, Sacco RL, Albuquerque FC, et al; American Heart Association Stroke Council. Cervical arterial dissections and association with cervical manipulative therapy: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45(10):3155-3174.
4. Fukunaga N, Hanaoka M, Sato K. Asymptomatic common carotid artery dissection caused by blunt injury. Emerg Med J. 2011;28(1):50.
5. Chen J, Zhou X, Li C, Cheung BM. Risk of stroke due to spontaneous cervical artery dissection. Intern Med. 2013;52(19):2237-2240.
6. Kalantzis G, Georgalas I, Chang BY, Ong C, El-Hindy N. An unusual case of traumatic internal carotid artery dissection during snowboarding. J Sports Sci Med. 2014;13(2):451-453.
7. Lu A, Shen P, Lee P, et al. CrossFit-related cervical internal carotid artery dissection. Emerg Radiol. 2015;22(4):449-452.
8. CADISS Trial Investigators, Markus HS, Hayter E, Levi C, Feldman A, Venables G, Norris J. Antiplatelet treatment compared with anticoagulation treatment for cervical artery dissection (CADISS): a randomised trial. Lancet Neurol. 2015;14(4):361-367.
9. van Wessem KJ, Meijer JM, Leenen LP, van der Worp HB, Moll FL, de Borst GJ. Blunt traumatic carotid artery dissection still a pitfall? The rationale for aggressive screening. Eur J Trauma Emerg Surg. 2011;37(2):147-154.
10. Haneline M, Triano J. Cervical artery dissection. A comparison of highly dynamic mechanisms: manipulation versus motor vehicle collision. J Manipulative Physiol Ther. 2005;28(1):57-63.
11. Thomas LC, Rivett DA, Attia JR, Levi C. Risk factors and clinical presentation of cervical arterial dissection: preliminary results of a prospective case-control study. J Orthop Sports Phys Ther. 2015;45(7):503-511.
12. Lyrer P, Engelter S. Antithrombotic drugs for carotid artery dissection. Cochrane Database Syst Rev. 2010;(10):CD000255.
13. Creavin ST, Rice CM, Pollentine A, Cowburn P. Carotid artery dissection presenting with isolated headache and Horner syndrome after minor head injury. Am J Emerg Med. 2012;30(9):2103.e5-e7.
1. Mohan IV. Current optimal assessment and management of carotid and vertebral spontaneous and traumatic dissection. Angiology. 2014;65(4):274-283.
2. Patel RR, Adam R, Maldjian C, Lincoln CM, Yuen A, Arneja A. Cervical carotid artery dissection: current review of diagnosis and treatment. Cardiol Rev. 2012;20(3):145-152.
3. Biller J, Sacco RL, Albuquerque FC, et al; American Heart Association Stroke Council. Cervical arterial dissections and association with cervical manipulative therapy: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45(10):3155-3174.
4. Fukunaga N, Hanaoka M, Sato K. Asymptomatic common carotid artery dissection caused by blunt injury. Emerg Med J. 2011;28(1):50.
5. Chen J, Zhou X, Li C, Cheung BM. Risk of stroke due to spontaneous cervical artery dissection. Intern Med. 2013;52(19):2237-2240.
6. Kalantzis G, Georgalas I, Chang BY, Ong C, El-Hindy N. An unusual case of traumatic internal carotid artery dissection during snowboarding. J Sports Sci Med. 2014;13(2):451-453.
7. Lu A, Shen P, Lee P, et al. CrossFit-related cervical internal carotid artery dissection. Emerg Radiol. 2015;22(4):449-452.
8. CADISS Trial Investigators, Markus HS, Hayter E, Levi C, Feldman A, Venables G, Norris J. Antiplatelet treatment compared with anticoagulation treatment for cervical artery dissection (CADISS): a randomised trial. Lancet Neurol. 2015;14(4):361-367.
9. van Wessem KJ, Meijer JM, Leenen LP, van der Worp HB, Moll FL, de Borst GJ. Blunt traumatic carotid artery dissection still a pitfall? The rationale for aggressive screening. Eur J Trauma Emerg Surg. 2011;37(2):147-154.
10. Haneline M, Triano J. Cervical artery dissection. A comparison of highly dynamic mechanisms: manipulation versus motor vehicle collision. J Manipulative Physiol Ther. 2005;28(1):57-63.
11. Thomas LC, Rivett DA, Attia JR, Levi C. Risk factors and clinical presentation of cervical arterial dissection: preliminary results of a prospective case-control study. J Orthop Sports Phys Ther. 2015;45(7):503-511.
12. Lyrer P, Engelter S. Antithrombotic drugs for carotid artery dissection. Cochrane Database Syst Rev. 2010;(10):CD000255.
13. Creavin ST, Rice CM, Pollentine A, Cowburn P. Carotid artery dissection presenting with isolated headache and Horner syndrome after minor head injury. Am J Emerg Med. 2012;30(9):2103.e5-e7.

Encapsulated Fat Necrosis Lesion Caused by Morel-Lavallée Lesion in a Professional Ice Hockey Player
Take-Home Points
- ML lesions usually occur with high-energy injuries and have been reported in wrestlers, football players, and other athlete populations.
- Encapsulated fat necrosis lesions are usually attributable to trauma and disruption of the blood supply in the subcutaneous area, which occurs with ML lesions.
- Encapsulated fat necrosis lesions are rare; only 65 have been reported.
- Encapsulated fat necrosis lesions are characterized by massive fat necrosis encapsulated by fibrous tissue.
- Most are small and asymptomatic; however, in some cases, athletes can develop symptoms from frequent impacts to the region where the lesions are located.
What would become known as the Morel-Lavallée (ML) lesion was first reported in 1853 by French physician Maurice Morel-Lavallée. He described a proximal thigh soft-tissue injury that resulted in a hemolymphatic collection between superficial fascial planes. Deforming forces of pressure and shear result in an internal degloving injury in which subcutaneous tissue is stripped from the fascia and replaced with a hematoma or, less commonly, necrotic fat.1-4 The injury can take several weeks to heal. Up to one-third of such injuries are initially missed because of the initial ecchymosis covering the injured area.5
ML lesions usually occur with high-energy injuries and have been reported in wrestlers,6 football players,7-9 and other athlete populations. ML lesions usually occur about the knee, the site of the sheer mechanism in these athletes’ sports. Tejwani and colleagues9 reported on 24 National Football League (NFL) players (27 knees). These elite athletes typically were able to return to practice and game play long before complete resolution of their lesions.
Nodular cystic fat necrosis was first described by Przyjemski and Schuster10 in 1977. The terms encapsulated fat necrosis lesions and mobile encapsulated lipomas11 were introduced later. Clinically, these entities usually present as lesions on the lower limbs of young men and middle-aged women and can range in size from 1 mm to 35 mm. Most of these lesions are mobile.11 They are usually attributable to trauma and disruption of the blood supply in the subcutaneous area, which occurs with ML lesions. Trauma accounts for the usual occurrence in the lower extremities, though only 40% of patients recall a precipitating event.12 Histologically, these lesions are characterized by massive fat necrosis encapsulated by fibrous tissue.13In this article, we report the case of a professional ice hockey player who presented with an ML lesion of the hip and then developed a symptomatic encapsulated fat necrosis lesion that required surgical removal. To our knowledge, this is the first reported case of an encapsulated fat necrosis lesion caused by an ML lesion in an athlete. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 21-year-old professional hockey player presented with a history of pain from a mass on his right hip. He first noticed the lesion, just lateral to the greater trochanter, about 3 years earlier. The mass appeared after he sustained a shearing-type injury to the lateral aspect of the hip. At the time, there was significant swelling along the lateral aspect, with ecchymosis that resolved over 2 months. The mass, diagnosed as an ML lesion, resolved with nonoperative treatment. However, in the area where the swelling had occurred, a hard mobile mass remained. At times, this mass became painful when direct pressure was applied, as when he hit the boards while playing hockey, or when he lay on his right side or used a roller in the training room. He rated the pain as a 4 on a 1-to-10 scale and said the mass was mobile and had not changed in size or consistency.
Physical examination revealed a palpable mass over the lateral aspect of the hip, over the greater trochanter. The mass, about 3 cm in diameter (Figure 1), was mobile in a subcutaneous pocket, consistent with an old ML lesion. 

Options discussed with the patient included use of ice, activity modification, and use of protective padded equipment. As the patient had tried these treatments before and was still intermittently having pain with direct pressure, he asked for surgical removal of the mass.
For the surgery, the patient was positioned in the lateral decubitus position with his right hip facing up. The right hip and thigh were prepared and draped in sterile fashion. An incision 4 cm in length was made directly over the mass, along the lateral aspect of the hip, over the greater trochanter. The incision was taken through skin and subcutaneous tissue down to the deep fascia. The fascia was incised longitudinally in line with the overlying skin incision. As soon as the incision was made through the fascia, the mass was easily seen. The 3-cm × 2-cm × 1-cm mass was free, not attached to any underlying soft tissue (Figure 3). 
Discussion
We have described a case of symptomatic encapsulated fat necrosis lesion caused by an ML lesion in a professional hockey player. The ML lesion had resolved with nonoperative treatment (compression), but a subcutaneous pocket remained at the lesion site. Given the patient’s lesion site and occupation as a hockey player, pain with direct pressure on this lesion was a concern.
Long-standing ML lesions have 3 common patterns on MRI.14 A central region, encapsulated partially or completely by a peripheral ring of fibrous tissue or hemosiderin, shows signal properties consistent with a seroma, a homogeneous hemorrhagic collection, or a heterogeneous hemorrhagic collection. In our patient’s case, MRI was used to characterize the mobile mass for operative planning. Although thin strands or lobules of fat have been found within ML lesions, this case was the first to demonstrate a sequestered mass of necrotic fat.
Most football players who develop ML lesions on their knees do not wear kneepads.7-9 Of the 24 NFL players in the study by Tejwani and colleagues,9 52% were successfully treated with compression wrap, cryotherapy, and motion exercises. The rest, however, were treated with aspiration, and 11% underwent doxycycline sclerodesis for recurrent fluid collection. After treatment, all of their players were able to return to football. Their outcomes are consistent with that of our patient, who was treated with compression wrap and returned to hockey without any other intervention.
After our patient’s ML lesion resolved, he developed an encapsulated fat necrosis lesion from the disruption of the blood supply in the subcutaneous pocket. Encapsulated fat necrosis lesions are rare; only 65 have been reported.13,15 Clinically, these lesions are single or multiple pale-yellow encapsulated nodes.13 Most are small and asymptomatic; however, in some cases, athletes can develop symptoms from frequent impacts to the region where the lesions are located.
The literature includes 1 report of an adolescent football player who developed multiple encapsulated fat necrosis lesions 4 months after landing on another player’s cleats.15 The patient, who was having pain with direct pressure during squatting and kneeling, elected to have the lesions surgically removed. These lesions are rare and usually asymptomatic,11 but our patient had his lesion surgically removed to address the pain induced by the direct impacts that came with playing professional hockey. Surgical removal is the treatment for symptomatic encapsulated fat necrosis lesions. Other than 1 case of recurrence after excision,16 these lesions have an excellent prognosis.
Conclusion
Our patient, a professional hockey player, underwent successful surgical removal of a symptomatic encapsulated fat necrosis lesion that had developed from an ML lesion.
Am J Orthop. 2017;46(3):E144-E147. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Aguiar RO, Viegas FC, Fernandez RY, Trudell D, Haghighi P, Resnick D. The prepatellar bursa: cadaveric investigation of regional anatomy with MRI after sonographically guided bursography. AJR Am J Roentgenol. 2007;188(4):W355-W358.
2. Hak DJ, Olson SA, Matta JM. Diagnosis and management of closed internal degloving injuries associated with pelvic and acetabular fractures: the Morel-Lavallée lesion. J Trauma. 1997;42(6):1046-1051.
3. Hudson DA, Knottenbelt JD, Krige JE. Closed degloving injuries: results following conservative surgery. Plast Reconstr Surg. 1992;89(5):853-855.
4. Mellado JM, Bencardino JT. Morel-Lavallée lesion: review with emphasis on MR imaging. Magn Reson Imaging Clin North Am. 2005;13(4):775-782.
5. Dye SF, Campagna-Pinto D, Dye CC, Shifflett S, Eiman T. Soft-tissue anatomy anterior to the human patella. J Bone Joint Surg Am. 2003;85(6):1012-1017.
6. Northam MC, Gaskin CM. Presumed prepatellar fibrosis in collegiate wrestlers: imaging findings and clinical correlation. Skeletal Radiol. 2015;44(2):271-277.
7. Anakwenze OA, Trivedi V, Goodman AM, Ganley TJ. Concealed degloving injury (the Morel-Lavallée lesion) in childhood sports: a case report. J Bone Joint Surg Am. 2011;93(24):e148.
8. Matava MJ, Ellis E, Shah NR, Pogue D, Williams T. Morel-Lavallée lesion in a professional American football player. Am J Orthop. 2010;39(3):144-147.
9. Tejwani SG, Cohen SB, Bradley JP. Management of Morel-Lavallee lesion of the knee: twenty-seven cases in the National Football League. Am J Sports Med. 2007;35(7):1162-1167.
10. Przyjemski CJ, Schuster SR. Nodular-cystic fat necrosis. J Pediatr. 1977;91(4):605-607.
11. Kiryu H, Rikihisa W, Furue M. Encapsulated fat necrosis—a clinicopathological study of 8 cases and a literature review. J Cutan Pathol. 2000;27(1):19-23.
12. Santos-Juanes J, Coto P, Galache C, Sánchez del Rio J, Soto de Delás J. Encapsulated fat necrosis: a form of traumatic panniculitis. J Eur Acad Dermatol Venereol. 2007;21(3):405-406.
13. Sempau L, Sambucetty PS, Garcia JL, Sixto BG, Morán AG, Prieto MA. Mobile encapsulated lipoma. Int J Dermatol. 2012;51(4):448-450.
14. Mellado JM, Pérez del Palomar L, Díaz L, Ramos A, Saurí A. Long-standing Morel-Lavallée lesions of the trochanteric region and proximal thigh: MRI features in five patients. AJR Am J Roentgenol. 2004;182(5):1289-1294.
15. Sole JS, Wisniewski SJ, Dahm DL, Bond J, Smith J. Posttraumatic fat necrosis presenting as prepatellar loose bodies in an adolescent football player. PM R. 2014;6(8):749-752.
16. Felipo F, Vaquero M, del Agua C. Pseudotumoral encapsulated fat necrosis with diffuse pseudomembranous degeneration. J Cutan Pathol. 2004;31(8):565-567.
Take-Home Points
- ML lesions usually occur with high-energy injuries and have been reported in wrestlers, football players, and other athlete populations.
- Encapsulated fat necrosis lesions are usually attributable to trauma and disruption of the blood supply in the subcutaneous area, which occurs with ML lesions.
- Encapsulated fat necrosis lesions are rare; only 65 have been reported.
- Encapsulated fat necrosis lesions are characterized by massive fat necrosis encapsulated by fibrous tissue.
- Most are small and asymptomatic; however, in some cases, athletes can develop symptoms from frequent impacts to the region where the lesions are located.
What would become known as the Morel-Lavallée (ML) lesion was first reported in 1853 by French physician Maurice Morel-Lavallée. He described a proximal thigh soft-tissue injury that resulted in a hemolymphatic collection between superficial fascial planes. Deforming forces of pressure and shear result in an internal degloving injury in which subcutaneous tissue is stripped from the fascia and replaced with a hematoma or, less commonly, necrotic fat.1-4 The injury can take several weeks to heal. Up to one-third of such injuries are initially missed because of the initial ecchymosis covering the injured area.5
ML lesions usually occur with high-energy injuries and have been reported in wrestlers,6 football players,7-9 and other athlete populations. ML lesions usually occur about the knee, the site of the sheer mechanism in these athletes’ sports. Tejwani and colleagues9 reported on 24 National Football League (NFL) players (27 knees). These elite athletes typically were able to return to practice and game play long before complete resolution of their lesions.
Nodular cystic fat necrosis was first described by Przyjemski and Schuster10 in 1977. The terms encapsulated fat necrosis lesions and mobile encapsulated lipomas11 were introduced later. Clinically, these entities usually present as lesions on the lower limbs of young men and middle-aged women and can range in size from 1 mm to 35 mm. Most of these lesions are mobile.11 They are usually attributable to trauma and disruption of the blood supply in the subcutaneous area, which occurs with ML lesions. Trauma accounts for the usual occurrence in the lower extremities, though only 40% of patients recall a precipitating event.12 Histologically, these lesions are characterized by massive fat necrosis encapsulated by fibrous tissue.13In this article, we report the case of a professional ice hockey player who presented with an ML lesion of the hip and then developed a symptomatic encapsulated fat necrosis lesion that required surgical removal. To our knowledge, this is the first reported case of an encapsulated fat necrosis lesion caused by an ML lesion in an athlete. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 21-year-old professional hockey player presented with a history of pain from a mass on his right hip. He first noticed the lesion, just lateral to the greater trochanter, about 3 years earlier. The mass appeared after he sustained a shearing-type injury to the lateral aspect of the hip. At the time, there was significant swelling along the lateral aspect, with ecchymosis that resolved over 2 months. The mass, diagnosed as an ML lesion, resolved with nonoperative treatment. However, in the area where the swelling had occurred, a hard mobile mass remained. At times, this mass became painful when direct pressure was applied, as when he hit the boards while playing hockey, or when he lay on his right side or used a roller in the training room. He rated the pain as a 4 on a 1-to-10 scale and said the mass was mobile and had not changed in size or consistency.
Physical examination revealed a palpable mass over the lateral aspect of the hip, over the greater trochanter. The mass, about 3 cm in diameter (Figure 1), was mobile in a subcutaneous pocket, consistent with an old ML lesion.
Options discussed with the patient included use of ice, activity modification, and use of protective padded equipment. As the patient had tried these treatments before and was still intermittently having pain with direct pressure, he asked for surgical removal of the mass.
For the surgery, the patient was positioned in the lateral decubitus position with his right hip facing up. The right hip and thigh were prepared and draped in sterile fashion. An incision 4 cm in length was made directly over the mass, along the lateral aspect of the hip, over the greater trochanter. The incision was taken through skin and subcutaneous tissue down to the deep fascia. The fascia was incised longitudinally in line with the overlying skin incision. As soon as the incision was made through the fascia, the mass was easily seen. The 3-cm × 2-cm × 1-cm mass was free, not attached to any underlying soft tissue (Figure 3).
Discussion
We have described a case of symptomatic encapsulated fat necrosis lesion caused by an ML lesion in a professional hockey player. The ML lesion had resolved with nonoperative treatment (compression), but a subcutaneous pocket remained at the lesion site. Given the patient’s lesion site and occupation as a hockey player, pain with direct pressure on this lesion was a concern.
Long-standing ML lesions have 3 common patterns on MRI.14 A central region, encapsulated partially or completely by a peripheral ring of fibrous tissue or hemosiderin, shows signal properties consistent with a seroma, a homogeneous hemorrhagic collection, or a heterogeneous hemorrhagic collection. In our patient’s case, MRI was used to characterize the mobile mass for operative planning. Although thin strands or lobules of fat have been found within ML lesions, this case was the first to demonstrate a sequestered mass of necrotic fat.
Most football players who develop ML lesions on their knees do not wear kneepads.7-9 Of the 24 NFL players in the study by Tejwani and colleagues,9 52% were successfully treated with compression wrap, cryotherapy, and motion exercises. The rest, however, were treated with aspiration, and 11% underwent doxycycline sclerodesis for recurrent fluid collection. After treatment, all of their players were able to return to football. Their outcomes are consistent with that of our patient, who was treated with compression wrap and returned to hockey without any other intervention.
After our patient’s ML lesion resolved, he developed an encapsulated fat necrosis lesion from the disruption of the blood supply in the subcutaneous pocket. Encapsulated fat necrosis lesions are rare; only 65 have been reported.13,15 Clinically, these lesions are single or multiple pale-yellow encapsulated nodes.13 Most are small and asymptomatic; however, in some cases, athletes can develop symptoms from frequent impacts to the region where the lesions are located.
The literature includes 1 report of an adolescent football player who developed multiple encapsulated fat necrosis lesions 4 months after landing on another player’s cleats.15 The patient, who was having pain with direct pressure during squatting and kneeling, elected to have the lesions surgically removed. These lesions are rare and usually asymptomatic,11 but our patient had his lesion surgically removed to address the pain induced by the direct impacts that came with playing professional hockey. Surgical removal is the treatment for symptomatic encapsulated fat necrosis lesions. Other than 1 case of recurrence after excision,16 these lesions have an excellent prognosis.
Conclusion
Our patient, a professional hockey player, underwent successful surgical removal of a symptomatic encapsulated fat necrosis lesion that had developed from an ML lesion.
Am J Orthop. 2017;46(3):E144-E147. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- ML lesions usually occur with high-energy injuries and have been reported in wrestlers, football players, and other athlete populations.
- Encapsulated fat necrosis lesions are usually attributable to trauma and disruption of the blood supply in the subcutaneous area, which occurs with ML lesions.
- Encapsulated fat necrosis lesions are rare; only 65 have been reported.
- Encapsulated fat necrosis lesions are characterized by massive fat necrosis encapsulated by fibrous tissue.
- Most are small and asymptomatic; however, in some cases, athletes can develop symptoms from frequent impacts to the region where the lesions are located.
What would become known as the Morel-Lavallée (ML) lesion was first reported in 1853 by French physician Maurice Morel-Lavallée. He described a proximal thigh soft-tissue injury that resulted in a hemolymphatic collection between superficial fascial planes. Deforming forces of pressure and shear result in an internal degloving injury in which subcutaneous tissue is stripped from the fascia and replaced with a hematoma or, less commonly, necrotic fat.1-4 The injury can take several weeks to heal. Up to one-third of such injuries are initially missed because of the initial ecchymosis covering the injured area.5
ML lesions usually occur with high-energy injuries and have been reported in wrestlers,6 football players,7-9 and other athlete populations. ML lesions usually occur about the knee, the site of the sheer mechanism in these athletes’ sports. Tejwani and colleagues9 reported on 24 National Football League (NFL) players (27 knees). These elite athletes typically were able to return to practice and game play long before complete resolution of their lesions.
Nodular cystic fat necrosis was first described by Przyjemski and Schuster10 in 1977. The terms encapsulated fat necrosis lesions and mobile encapsulated lipomas11 were introduced later. Clinically, these entities usually present as lesions on the lower limbs of young men and middle-aged women and can range in size from 1 mm to 35 mm. Most of these lesions are mobile.11 They are usually attributable to trauma and disruption of the blood supply in the subcutaneous area, which occurs with ML lesions. Trauma accounts for the usual occurrence in the lower extremities, though only 40% of patients recall a precipitating event.12 Histologically, these lesions are characterized by massive fat necrosis encapsulated by fibrous tissue.13In this article, we report the case of a professional ice hockey player who presented with an ML lesion of the hip and then developed a symptomatic encapsulated fat necrosis lesion that required surgical removal. To our knowledge, this is the first reported case of an encapsulated fat necrosis lesion caused by an ML lesion in an athlete. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 21-year-old professional hockey player presented with a history of pain from a mass on his right hip. He first noticed the lesion, just lateral to the greater trochanter, about 3 years earlier. The mass appeared after he sustained a shearing-type injury to the lateral aspect of the hip. At the time, there was significant swelling along the lateral aspect, with ecchymosis that resolved over 2 months. The mass, diagnosed as an ML lesion, resolved with nonoperative treatment. However, in the area where the swelling had occurred, a hard mobile mass remained. At times, this mass became painful when direct pressure was applied, as when he hit the boards while playing hockey, or when he lay on his right side or used a roller in the training room. He rated the pain as a 4 on a 1-to-10 scale and said the mass was mobile and had not changed in size or consistency.
Physical examination revealed a palpable mass over the lateral aspect of the hip, over the greater trochanter. The mass, about 3 cm in diameter (Figure 1), was mobile in a subcutaneous pocket, consistent with an old ML lesion.
Options discussed with the patient included use of ice, activity modification, and use of protective padded equipment. As the patient had tried these treatments before and was still intermittently having pain with direct pressure, he asked for surgical removal of the mass.
For the surgery, the patient was positioned in the lateral decubitus position with his right hip facing up. The right hip and thigh were prepared and draped in sterile fashion. An incision 4 cm in length was made directly over the mass, along the lateral aspect of the hip, over the greater trochanter. The incision was taken through skin and subcutaneous tissue down to the deep fascia. The fascia was incised longitudinally in line with the overlying skin incision. As soon as the incision was made through the fascia, the mass was easily seen. The 3-cm × 2-cm × 1-cm mass was free, not attached to any underlying soft tissue (Figure 3).
Discussion
We have described a case of symptomatic encapsulated fat necrosis lesion caused by an ML lesion in a professional hockey player. The ML lesion had resolved with nonoperative treatment (compression), but a subcutaneous pocket remained at the lesion site. Given the patient’s lesion site and occupation as a hockey player, pain with direct pressure on this lesion was a concern.
Long-standing ML lesions have 3 common patterns on MRI.14 A central region, encapsulated partially or completely by a peripheral ring of fibrous tissue or hemosiderin, shows signal properties consistent with a seroma, a homogeneous hemorrhagic collection, or a heterogeneous hemorrhagic collection. In our patient’s case, MRI was used to characterize the mobile mass for operative planning. Although thin strands or lobules of fat have been found within ML lesions, this case was the first to demonstrate a sequestered mass of necrotic fat.
Most football players who develop ML lesions on their knees do not wear kneepads.7-9 Of the 24 NFL players in the study by Tejwani and colleagues,9 52% were successfully treated with compression wrap, cryotherapy, and motion exercises. The rest, however, were treated with aspiration, and 11% underwent doxycycline sclerodesis for recurrent fluid collection. After treatment, all of their players were able to return to football. Their outcomes are consistent with that of our patient, who was treated with compression wrap and returned to hockey without any other intervention.
After our patient’s ML lesion resolved, he developed an encapsulated fat necrosis lesion from the disruption of the blood supply in the subcutaneous pocket. Encapsulated fat necrosis lesions are rare; only 65 have been reported.13,15 Clinically, these lesions are single or multiple pale-yellow encapsulated nodes.13 Most are small and asymptomatic; however, in some cases, athletes can develop symptoms from frequent impacts to the region where the lesions are located.
The literature includes 1 report of an adolescent football player who developed multiple encapsulated fat necrosis lesions 4 months after landing on another player’s cleats.15 The patient, who was having pain with direct pressure during squatting and kneeling, elected to have the lesions surgically removed. These lesions are rare and usually asymptomatic,11 but our patient had his lesion surgically removed to address the pain induced by the direct impacts that came with playing professional hockey. Surgical removal is the treatment for symptomatic encapsulated fat necrosis lesions. Other than 1 case of recurrence after excision,16 these lesions have an excellent prognosis.
Conclusion
Our patient, a professional hockey player, underwent successful surgical removal of a symptomatic encapsulated fat necrosis lesion that had developed from an ML lesion.
Am J Orthop. 2017;46(3):E144-E147. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Aguiar RO, Viegas FC, Fernandez RY, Trudell D, Haghighi P, Resnick D. The prepatellar bursa: cadaveric investigation of regional anatomy with MRI after sonographically guided bursography. AJR Am J Roentgenol. 2007;188(4):W355-W358.
2. Hak DJ, Olson SA, Matta JM. Diagnosis and management of closed internal degloving injuries associated with pelvic and acetabular fractures: the Morel-Lavallée lesion. J Trauma. 1997;42(6):1046-1051.
3. Hudson DA, Knottenbelt JD, Krige JE. Closed degloving injuries: results following conservative surgery. Plast Reconstr Surg. 1992;89(5):853-855.
4. Mellado JM, Bencardino JT. Morel-Lavallée lesion: review with emphasis on MR imaging. Magn Reson Imaging Clin North Am. 2005;13(4):775-782.
5. Dye SF, Campagna-Pinto D, Dye CC, Shifflett S, Eiman T. Soft-tissue anatomy anterior to the human patella. J Bone Joint Surg Am. 2003;85(6):1012-1017.
6. Northam MC, Gaskin CM. Presumed prepatellar fibrosis in collegiate wrestlers: imaging findings and clinical correlation. Skeletal Radiol. 2015;44(2):271-277.
7. Anakwenze OA, Trivedi V, Goodman AM, Ganley TJ. Concealed degloving injury (the Morel-Lavallée lesion) in childhood sports: a case report. J Bone Joint Surg Am. 2011;93(24):e148.
8. Matava MJ, Ellis E, Shah NR, Pogue D, Williams T. Morel-Lavallée lesion in a professional American football player. Am J Orthop. 2010;39(3):144-147.
9. Tejwani SG, Cohen SB, Bradley JP. Management of Morel-Lavallee lesion of the knee: twenty-seven cases in the National Football League. Am J Sports Med. 2007;35(7):1162-1167.
10. Przyjemski CJ, Schuster SR. Nodular-cystic fat necrosis. J Pediatr. 1977;91(4):605-607.
11. Kiryu H, Rikihisa W, Furue M. Encapsulated fat necrosis—a clinicopathological study of 8 cases and a literature review. J Cutan Pathol. 2000;27(1):19-23.
12. Santos-Juanes J, Coto P, Galache C, Sánchez del Rio J, Soto de Delás J. Encapsulated fat necrosis: a form of traumatic panniculitis. J Eur Acad Dermatol Venereol. 2007;21(3):405-406.
13. Sempau L, Sambucetty PS, Garcia JL, Sixto BG, Morán AG, Prieto MA. Mobile encapsulated lipoma. Int J Dermatol. 2012;51(4):448-450.
14. Mellado JM, Pérez del Palomar L, Díaz L, Ramos A, Saurí A. Long-standing Morel-Lavallée lesions of the trochanteric region and proximal thigh: MRI features in five patients. AJR Am J Roentgenol. 2004;182(5):1289-1294.
15. Sole JS, Wisniewski SJ, Dahm DL, Bond J, Smith J. Posttraumatic fat necrosis presenting as prepatellar loose bodies in an adolescent football player. PM R. 2014;6(8):749-752.
16. Felipo F, Vaquero M, del Agua C. Pseudotumoral encapsulated fat necrosis with diffuse pseudomembranous degeneration. J Cutan Pathol. 2004;31(8):565-567.
1. Aguiar RO, Viegas FC, Fernandez RY, Trudell D, Haghighi P, Resnick D. The prepatellar bursa: cadaveric investigation of regional anatomy with MRI after sonographically guided bursography. AJR Am J Roentgenol. 2007;188(4):W355-W358.
2. Hak DJ, Olson SA, Matta JM. Diagnosis and management of closed internal degloving injuries associated with pelvic and acetabular fractures: the Morel-Lavallée lesion. J Trauma. 1997;42(6):1046-1051.
3. Hudson DA, Knottenbelt JD, Krige JE. Closed degloving injuries: results following conservative surgery. Plast Reconstr Surg. 1992;89(5):853-855.
4. Mellado JM, Bencardino JT. Morel-Lavallée lesion: review with emphasis on MR imaging. Magn Reson Imaging Clin North Am. 2005;13(4):775-782.
5. Dye SF, Campagna-Pinto D, Dye CC, Shifflett S, Eiman T. Soft-tissue anatomy anterior to the human patella. J Bone Joint Surg Am. 2003;85(6):1012-1017.
6. Northam MC, Gaskin CM. Presumed prepatellar fibrosis in collegiate wrestlers: imaging findings and clinical correlation. Skeletal Radiol. 2015;44(2):271-277.
7. Anakwenze OA, Trivedi V, Goodman AM, Ganley TJ. Concealed degloving injury (the Morel-Lavallée lesion) in childhood sports: a case report. J Bone Joint Surg Am. 2011;93(24):e148.
8. Matava MJ, Ellis E, Shah NR, Pogue D, Williams T. Morel-Lavallée lesion in a professional American football player. Am J Orthop. 2010;39(3):144-147.
9. Tejwani SG, Cohen SB, Bradley JP. Management of Morel-Lavallee lesion of the knee: twenty-seven cases in the National Football League. Am J Sports Med. 2007;35(7):1162-1167.
10. Przyjemski CJ, Schuster SR. Nodular-cystic fat necrosis. J Pediatr. 1977;91(4):605-607.
11. Kiryu H, Rikihisa W, Furue M. Encapsulated fat necrosis—a clinicopathological study of 8 cases and a literature review. J Cutan Pathol. 2000;27(1):19-23.
12. Santos-Juanes J, Coto P, Galache C, Sánchez del Rio J, Soto de Delás J. Encapsulated fat necrosis: a form of traumatic panniculitis. J Eur Acad Dermatol Venereol. 2007;21(3):405-406.
13. Sempau L, Sambucetty PS, Garcia JL, Sixto BG, Morán AG, Prieto MA. Mobile encapsulated lipoma. Int J Dermatol. 2012;51(4):448-450.
14. Mellado JM, Pérez del Palomar L, Díaz L, Ramos A, Saurí A. Long-standing Morel-Lavallée lesions of the trochanteric region and proximal thigh: MRI features in five patients. AJR Am J Roentgenol. 2004;182(5):1289-1294.
15. Sole JS, Wisniewski SJ, Dahm DL, Bond J, Smith J. Posttraumatic fat necrosis presenting as prepatellar loose bodies in an adolescent football player. PM R. 2014;6(8):749-752.
16. Felipo F, Vaquero M, del Agua C. Pseudotumoral encapsulated fat necrosis with diffuse pseudomembranous degeneration. J Cutan Pathol. 2004;31(8):565-567.
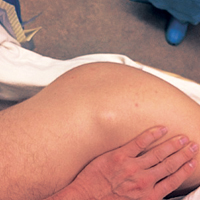
Severe headache • neck pain • intermittent cough • Dx?
THE CASE
A 32-year-old Chinese woman sought care from our family medicine clinic because she had a headache, neck pain, and an intermittent cough that had produced white sputum for 7 days. She described the headache as severe and pressure-like, and said that it had progressively worsened over the previous 3 weeks, coinciding with her first trip outside of China to the United States. The patient indicated that she also had occasional vomiting, dizziness, a low-grade fever, chills, night sweats, and increasing fatigue.
Prior to this visit, the patient had gone to the emergency department (ED) twice in one week, but was told that she had a migraine headache and a viral syndrome and was sent home. She was also told to make a follow-up appointment at our family medicine outpatient clinic.
Besides the symptoms that brought her to our clinic, the only other notable element of the patient’s history was a “neck mass” resection in China 8 years earlier. (The diagnosis of the neck mass was unknown.)
Concerned about her presenting signs and symptoms, we sent the patient to the ED, where she was admitted for further evaluation and treatment of possible meningitis. In the ED, she had a temperature of 101.5° F; her other vital signs were normal. A physical exam revealed mild neck stiffness.
THE DIAGNOSIS
A chest computed tomography (CT) scan demonstrated extensive confluent nodular infiltrates in the lung apices bilaterally with the largest confluent nodule measuring 6 cm (FIGURE 1). A chest x-ray demonstrated extensive bilateral pulmonary interstitial infiltrates that were most pronounced in the upper lung fields (FIGURE 2).

Lumbar puncture results revealed lymphocytic pleocytosis with elevated protein and low glucose levels (TABLE). Based on these results, the family medicine team suspected that our patient had tuberculous meningitis (TBM).
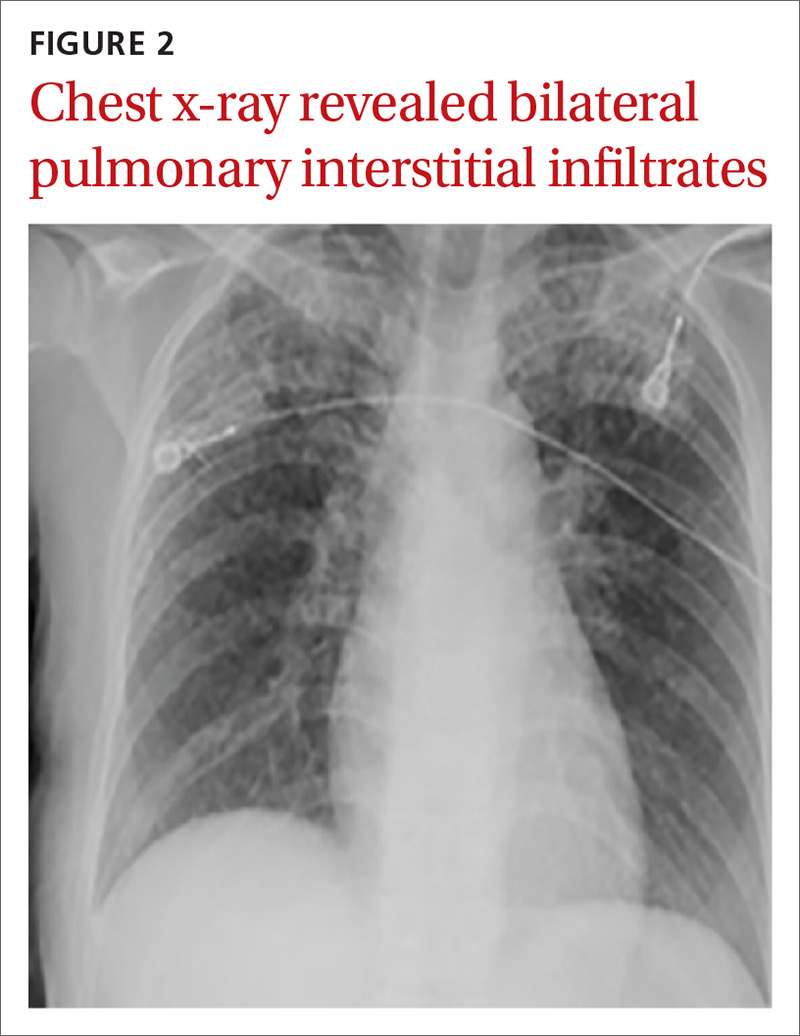
The team consulted with Infectious Diseases for management of TBM, and they placed our patient in a negative pressure room on airborne isolation. In addition, she was started on rifampin 450 mg/d, pyrazinamide 1000 mg/d, ethambutol 800 mg/d, and isoniazid (INH) 800 mg/d, as well as pyridoxine and intravenous dexamethasone.
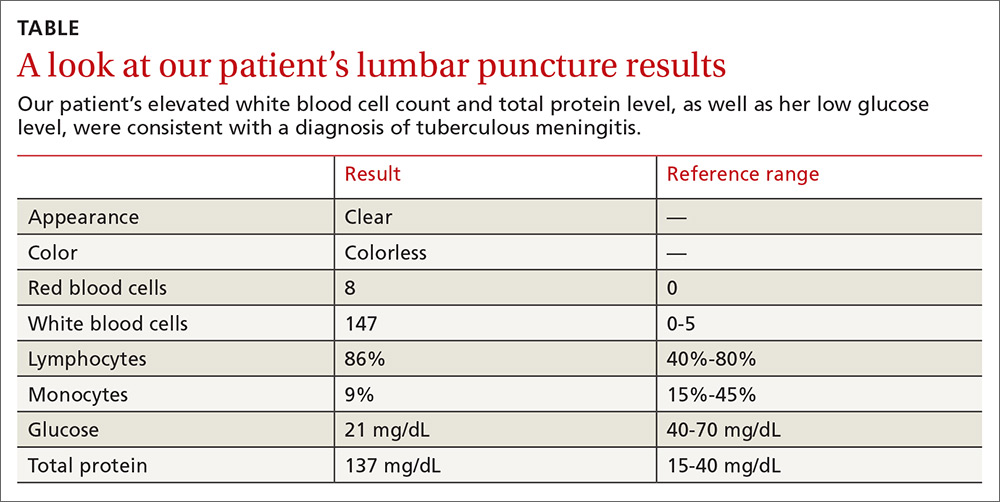
DISCUSSION
TBM accounts for approximately 1% of all cases of TB and 5% of extrapulmonary diseases in immunocompetent individuals.1 In 2015, there were approximately 10.4 million cases of TB worldwide, and 6 countries accounted for 60% of the global total: India, Indonesia, China, Nigeria, Pakistan, and South Africa.2 TBM is typically a subacute disease with symptoms that can persist for weeks before diagnosis.3 An early diagnosis is critical, as the mortality rate remains relatively high (as high as nearly 70% in underdeveloped and developed countries) despite effective treatment regimens.3 (For updated recommendations on TB screening, see this month’s Practice Alert.)
Most health care facilities use AFB smears to determine when patients with suspected TB should be isolated. However, AFB smears are positive in only 60% of TB cases.4 One study indicated that nucleic acid amplification by PCR can improve sensitivity from 60% to 87% and specificity from 98% to 100%.5
The presentation of TBM varies by phase of disease:
- The prodromal phase typically lasts for 2 to 3 weeks. It is characterized by an insidious onset of malaise, headache, low-grade fever, irritability, and personality changes.
- The meningitis phase is characterized by pronounced neurologic features such as meningismus, protracted headache, confusion, myelopathy, and sensory deficits, as well as vomiting, lethargy, and urinary retention.
- During the paralytic phase, patients experience profound confusion, followed by stupor, coma, seizures, progressive paraplegia, and often, hemiparesis.1,3,6
Treatment should be given for a total of 9 to 12 months
Initiate treatment for TB based on a strong clinical suspicion for the disease. Treatment of TBM consists of an intensive phase with 4 anti-TB drugs for 2 months (typically INH 800 mg/d, rifampin 450 mg/d, pyrazinamide 1000 mg/d, and ethambutol 800 mg/d) and a continuation phase with 2 drugs (INH and rifampin) for 7 to 10 additional months, resulting in a total treatment duration of 9 to 12 months.
Our patient was discharged from the hospital after 2 weeks on an anti-TB medication regimen of INH, rifampin, and pyrazinamide, along with pyridoxine and a tapering dose of dexamethasone. After the initial 2 months of intensive phase therapy, she was switched to INH 300 mg/d and rifampin 450 mg/d for the continuation phase. The patient followed up at our family medicine outpatient clinic with slow improvement of her muscle weakness before returning to China once she was placed on the continuation phase drugs.
THE TAKEAWAY
Suspect TB in high-risk patients traveling from endemic areas. Our patient, a Chinese woman visiting Brooklyn, New York, should’ve been considered high risk for TB even without her travel history from China because Brooklyn has a high rate of TB, as well. (In 2015, Sunset Park, Brooklyn had 18.2 cases of TB per 100,000 people, which was more than double the citywide rate.7)
TBM is a subacute disease with an often subtle presentation. Once you suspect TBM, isolate the patient, obtain appropriate cultures and smears, and start anti-TB drugs and adjunctive corticosteroids immediately, while the results of studies for AFB are still pending. Prompt diagnosis and treatment can save a patient’s life.
1. Garcia-Monco JC. Central nervous system tuberculosis. Neurol Clin. 1999;17:737-759.
2. World Health Organization. Global tuberculosis report, 2016. Available at: http://apps.who.int/iris/bitstream/10665/250441/1/9789241565394-eng.pdf?ua=1. Accessed March 29, 2017.
3. Marx GE, Chan ED. Tuberculous meningitis: diagnosis and treatment overview. Tuberc Res Treat. 2011;2011:798764.
4. Siddiqui AH, Perl TM, Conlon M, et al. Preventing nosocomial transmission of pulmonary tuberculosis: when may isolation be discontinued for patients with suspected tuberculosis? Infect Control Hosp Epidemiol. 2002;23:141-144.
5. Tang YW, Meng S, Li H, et al. PCR enhances acid-fast bacillus stain-based rapid detection of Mycobacterium tuberculosis. J Clin Microbiol. 2004;42:1849-1850.
6. Long R, Gardam M. Tumour necrosis factor-alpha inhibitors and the reactivation of latent tuberculosis infection. CMAJ. 2003;168:1153-1156.
7. New York City Department of Health and Mental Hygiene. Tuberculosis in New York City, 2015. New York City Bureau of Tuberculosis Control Annual Summary. Available at: http://www1.nyc.gov/assets/doh/downloads/pdf/tb/tb2015.pdf. Accessed April 7, 2017.
THE CASE
A 32-year-old Chinese woman sought care from our family medicine clinic because she had a headache, neck pain, and an intermittent cough that had produced white sputum for 7 days. She described the headache as severe and pressure-like, and said that it had progressively worsened over the previous 3 weeks, coinciding with her first trip outside of China to the United States. The patient indicated that she also had occasional vomiting, dizziness, a low-grade fever, chills, night sweats, and increasing fatigue.
Prior to this visit, the patient had gone to the emergency department (ED) twice in one week, but was told that she had a migraine headache and a viral syndrome and was sent home. She was also told to make a follow-up appointment at our family medicine outpatient clinic.
Besides the symptoms that brought her to our clinic, the only other notable element of the patient’s history was a “neck mass” resection in China 8 years earlier. (The diagnosis of the neck mass was unknown.)
Concerned about her presenting signs and symptoms, we sent the patient to the ED, where she was admitted for further evaluation and treatment of possible meningitis. In the ED, she had a temperature of 101.5° F; her other vital signs were normal. A physical exam revealed mild neck stiffness.
THE DIAGNOSIS
A chest computed tomography (CT) scan demonstrated extensive confluent nodular infiltrates in the lung apices bilaterally with the largest confluent nodule measuring 6 cm (FIGURE 1). A chest x-ray demonstrated extensive bilateral pulmonary interstitial infiltrates that were most pronounced in the upper lung fields (FIGURE 2).
Lumbar puncture results revealed lymphocytic pleocytosis with elevated protein and low glucose levels (TABLE). Based on these results, the family medicine team suspected that our patient had tuberculous meningitis (TBM).
The team consulted with Infectious Diseases for management of TBM, and they placed our patient in a negative pressure room on airborne isolation. In addition, she was started on rifampin 450 mg/d, pyrazinamide 1000 mg/d, ethambutol 800 mg/d, and isoniazid (INH) 800 mg/d, as well as pyridoxine and intravenous dexamethasone.
DISCUSSION
TBM accounts for approximately 1% of all cases of TB and 5% of extrapulmonary diseases in immunocompetent individuals.1 In 2015, there were approximately 10.4 million cases of TB worldwide, and 6 countries accounted for 60% of the global total: India, Indonesia, China, Nigeria, Pakistan, and South Africa.2 TBM is typically a subacute disease with symptoms that can persist for weeks before diagnosis.3 An early diagnosis is critical, as the mortality rate remains relatively high (as high as nearly 70% in underdeveloped and developed countries) despite effective treatment regimens.3 (For updated recommendations on TB screening, see this month’s Practice Alert.)
Most health care facilities use AFB smears to determine when patients with suspected TB should be isolated. However, AFB smears are positive in only 60% of TB cases.4 One study indicated that nucleic acid amplification by PCR can improve sensitivity from 60% to 87% and specificity from 98% to 100%.5
The presentation of TBM varies by phase of disease:
- The prodromal phase typically lasts for 2 to 3 weeks. It is characterized by an insidious onset of malaise, headache, low-grade fever, irritability, and personality changes.
- The meningitis phase is characterized by pronounced neurologic features such as meningismus, protracted headache, confusion, myelopathy, and sensory deficits, as well as vomiting, lethargy, and urinary retention.
- During the paralytic phase, patients experience profound confusion, followed by stupor, coma, seizures, progressive paraplegia, and often, hemiparesis.1,3,6
Treatment should be given for a total of 9 to 12 months
Initiate treatment for TB based on a strong clinical suspicion for the disease. Treatment of TBM consists of an intensive phase with 4 anti-TB drugs for 2 months (typically INH 800 mg/d, rifampin 450 mg/d, pyrazinamide 1000 mg/d, and ethambutol 800 mg/d) and a continuation phase with 2 drugs (INH and rifampin) for 7 to 10 additional months, resulting in a total treatment duration of 9 to 12 months.
Our patient was discharged from the hospital after 2 weeks on an anti-TB medication regimen of INH, rifampin, and pyrazinamide, along with pyridoxine and a tapering dose of dexamethasone. After the initial 2 months of intensive phase therapy, she was switched to INH 300 mg/d and rifampin 450 mg/d for the continuation phase. The patient followed up at our family medicine outpatient clinic with slow improvement of her muscle weakness before returning to China once she was placed on the continuation phase drugs.
THE TAKEAWAY
Suspect TB in high-risk patients traveling from endemic areas. Our patient, a Chinese woman visiting Brooklyn, New York, should’ve been considered high risk for TB even without her travel history from China because Brooklyn has a high rate of TB, as well. (In 2015, Sunset Park, Brooklyn had 18.2 cases of TB per 100,000 people, which was more than double the citywide rate.7)
TBM is a subacute disease with an often subtle presentation. Once you suspect TBM, isolate the patient, obtain appropriate cultures and smears, and start anti-TB drugs and adjunctive corticosteroids immediately, while the results of studies for AFB are still pending. Prompt diagnosis and treatment can save a patient’s life.
THE CASE
A 32-year-old Chinese woman sought care from our family medicine clinic because she had a headache, neck pain, and an intermittent cough that had produced white sputum for 7 days. She described the headache as severe and pressure-like, and said that it had progressively worsened over the previous 3 weeks, coinciding with her first trip outside of China to the United States. The patient indicated that she also had occasional vomiting, dizziness, a low-grade fever, chills, night sweats, and increasing fatigue.
Prior to this visit, the patient had gone to the emergency department (ED) twice in one week, but was told that she had a migraine headache and a viral syndrome and was sent home. She was also told to make a follow-up appointment at our family medicine outpatient clinic.
Besides the symptoms that brought her to our clinic, the only other notable element of the patient’s history was a “neck mass” resection in China 8 years earlier. (The diagnosis of the neck mass was unknown.)
Concerned about her presenting signs and symptoms, we sent the patient to the ED, where she was admitted for further evaluation and treatment of possible meningitis. In the ED, she had a temperature of 101.5° F; her other vital signs were normal. A physical exam revealed mild neck stiffness.
THE DIAGNOSIS
A chest computed tomography (CT) scan demonstrated extensive confluent nodular infiltrates in the lung apices bilaterally with the largest confluent nodule measuring 6 cm (FIGURE 1). A chest x-ray demonstrated extensive bilateral pulmonary interstitial infiltrates that were most pronounced in the upper lung fields (FIGURE 2).
Lumbar puncture results revealed lymphocytic pleocytosis with elevated protein and low glucose levels (TABLE). Based on these results, the family medicine team suspected that our patient had tuberculous meningitis (TBM).
The team consulted with Infectious Diseases for management of TBM, and they placed our patient in a negative pressure room on airborne isolation. In addition, she was started on rifampin 450 mg/d, pyrazinamide 1000 mg/d, ethambutol 800 mg/d, and isoniazid (INH) 800 mg/d, as well as pyridoxine and intravenous dexamethasone.
DISCUSSION
TBM accounts for approximately 1% of all cases of TB and 5% of extrapulmonary diseases in immunocompetent individuals.1 In 2015, there were approximately 10.4 million cases of TB worldwide, and 6 countries accounted for 60% of the global total: India, Indonesia, China, Nigeria, Pakistan, and South Africa.2 TBM is typically a subacute disease with symptoms that can persist for weeks before diagnosis.3 An early diagnosis is critical, as the mortality rate remains relatively high (as high as nearly 70% in underdeveloped and developed countries) despite effective treatment regimens.3 (For updated recommendations on TB screening, see this month’s Practice Alert.)
Most health care facilities use AFB smears to determine when patients with suspected TB should be isolated. However, AFB smears are positive in only 60% of TB cases.4 One study indicated that nucleic acid amplification by PCR can improve sensitivity from 60% to 87% and specificity from 98% to 100%.5
The presentation of TBM varies by phase of disease:
- The prodromal phase typically lasts for 2 to 3 weeks. It is characterized by an insidious onset of malaise, headache, low-grade fever, irritability, and personality changes.
- The meningitis phase is characterized by pronounced neurologic features such as meningismus, protracted headache, confusion, myelopathy, and sensory deficits, as well as vomiting, lethargy, and urinary retention.
- During the paralytic phase, patients experience profound confusion, followed by stupor, coma, seizures, progressive paraplegia, and often, hemiparesis.1,3,6
Treatment should be given for a total of 9 to 12 months
Initiate treatment for TB based on a strong clinical suspicion for the disease. Treatment of TBM consists of an intensive phase with 4 anti-TB drugs for 2 months (typically INH 800 mg/d, rifampin 450 mg/d, pyrazinamide 1000 mg/d, and ethambutol 800 mg/d) and a continuation phase with 2 drugs (INH and rifampin) for 7 to 10 additional months, resulting in a total treatment duration of 9 to 12 months.
Our patient was discharged from the hospital after 2 weeks on an anti-TB medication regimen of INH, rifampin, and pyrazinamide, along with pyridoxine and a tapering dose of dexamethasone. After the initial 2 months of intensive phase therapy, she was switched to INH 300 mg/d and rifampin 450 mg/d for the continuation phase. The patient followed up at our family medicine outpatient clinic with slow improvement of her muscle weakness before returning to China once she was placed on the continuation phase drugs.
THE TAKEAWAY
Suspect TB in high-risk patients traveling from endemic areas. Our patient, a Chinese woman visiting Brooklyn, New York, should’ve been considered high risk for TB even without her travel history from China because Brooklyn has a high rate of TB, as well. (In 2015, Sunset Park, Brooklyn had 18.2 cases of TB per 100,000 people, which was more than double the citywide rate.7)
TBM is a subacute disease with an often subtle presentation. Once you suspect TBM, isolate the patient, obtain appropriate cultures and smears, and start anti-TB drugs and adjunctive corticosteroids immediately, while the results of studies for AFB are still pending. Prompt diagnosis and treatment can save a patient’s life.
1. Garcia-Monco JC. Central nervous system tuberculosis. Neurol Clin. 1999;17:737-759.
2. World Health Organization. Global tuberculosis report, 2016. Available at: http://apps.who.int/iris/bitstream/10665/250441/1/9789241565394-eng.pdf?ua=1. Accessed March 29, 2017.
3. Marx GE, Chan ED. Tuberculous meningitis: diagnosis and treatment overview. Tuberc Res Treat. 2011;2011:798764.
4. Siddiqui AH, Perl TM, Conlon M, et al. Preventing nosocomial transmission of pulmonary tuberculosis: when may isolation be discontinued for patients with suspected tuberculosis? Infect Control Hosp Epidemiol. 2002;23:141-144.
5. Tang YW, Meng S, Li H, et al. PCR enhances acid-fast bacillus stain-based rapid detection of Mycobacterium tuberculosis. J Clin Microbiol. 2004;42:1849-1850.
6. Long R, Gardam M. Tumour necrosis factor-alpha inhibitors and the reactivation of latent tuberculosis infection. CMAJ. 2003;168:1153-1156.
7. New York City Department of Health and Mental Hygiene. Tuberculosis in New York City, 2015. New York City Bureau of Tuberculosis Control Annual Summary. Available at: http://www1.nyc.gov/assets/doh/downloads/pdf/tb/tb2015.pdf. Accessed April 7, 2017.
1. Garcia-Monco JC. Central nervous system tuberculosis. Neurol Clin. 1999;17:737-759.
2. World Health Organization. Global tuberculosis report, 2016. Available at: http://apps.who.int/iris/bitstream/10665/250441/1/9789241565394-eng.pdf?ua=1. Accessed March 29, 2017.
3. Marx GE, Chan ED. Tuberculous meningitis: diagnosis and treatment overview. Tuberc Res Treat. 2011;2011:798764.
4. Siddiqui AH, Perl TM, Conlon M, et al. Preventing nosocomial transmission of pulmonary tuberculosis: when may isolation be discontinued for patients with suspected tuberculosis? Infect Control Hosp Epidemiol. 2002;23:141-144.
5. Tang YW, Meng S, Li H, et al. PCR enhances acid-fast bacillus stain-based rapid detection of Mycobacterium tuberculosis. J Clin Microbiol. 2004;42:1849-1850.
6. Long R, Gardam M. Tumour necrosis factor-alpha inhibitors and the reactivation of latent tuberculosis infection. CMAJ. 2003;168:1153-1156.
7. New York City Department of Health and Mental Hygiene. Tuberculosis in New York City, 2015. New York City Bureau of Tuberculosis Control Annual Summary. Available at: http://www1.nyc.gov/assets/doh/downloads/pdf/tb/tb2015.pdf. Accessed April 7, 2017.

Epistaxis and Death by the Trigeminocardiac Reflex: A Cautionary Report
Epistaxis is a relatively common event that is estimated to occur at least once in 60% of the U.S. population. Epistaxis is also reported to cause 1.7 emergency department (ED) visits per 1,000 population annually.1 Although epistaxis can occur at any age, it typically occurs with a bimodal age distribution and most commonly affects individuals aged < 18 years and adults aged > 50 years.2 The episodes of epistaxis involving the younger age group are more often minor and self-limited. Most bleeds occur along the anterior nasal septum from Kiesselbach’s plexus.
Posterior bleeds occur more often in older patients.2 In addition, epistaxis in the older population tends to be more severe.3 Medical intervention is required in 6% of those experiencing epistaxis. Because the median age for male veterans was 64 years in 2011 compared with a median age of 37.2 years for the average U.S. population in 2010, veterans are among those at greatest risk to develop epistaxis that requires intervention.4,5
Most episodes of epistaxis are not life-threatening, particularly when modern methods of diagnosis and treatment are used. Nevertheless, comorbid diseases, complications of treatment, and normal physiologic responses can sometimes combine to create an adverse outcome.6 This report reviews the case of a veteran patient who experienced a fatal cardiopulmonary arrest after therapeutic interventions for epistaxis. It is believed that his death was due to the well-described but little known trigeminocardiac reflex (TCR).
Case Report
A 65-year-old man visited the ED and reported that his nose had been bleeding intermittently for 1 day. He estimated that he had lost 1 cup of blood over a 24-hour period. He reported no rhinosinusitis, nasal congestion, or recent allergy or upper respiratory infections. He also reported no nasal trauma. In the ED, blood was oozing from his right nares and into his throat, causing him to cough. External compression failed to control the oozing. Topical vasoconstrictors were not applied.
His past medical history included a pulmonary embolism, well-controlled chronic obstructive pulmonary disease (COPD), and sleep apnea. The thrombotic site of origin for his pulmonary embolism had not been identified, despite a thorough examination. He had been on warfarin therapy for 3 months, and his international normalized ratio (INR) had been monitored in an anticoagulation clinic and was well regulated. He was also on inhaled medications for COPD (formoterol and budesonide as a combination preparation twice daily and albuterol every 6 hours as needed as a rescue medication). He adhered to his noninvasive positive airway pressure (PAP) device treatment for sleep apnea. He did not take aspirin or other antiplatelet medications. He reported no use of topical nasal preparations. He also reported no use of illicit drugs or over-the-counter medications, including nonsteroidal anti-inflammatory medications and herbal remedies. He reported no bleeding from other sites or easy bruising.
The patient was alert, oriented, and in no distress. His vital signs were normal. Examination of his nasal passages failed to identify an active site of bleeding. Fresh blood was present in the right nasal passage and the posterior pharynx. Examination of his chest was normal. His hemoglobin was 13.1 g/dL (13.6-17.3 g/dL) with 216 x 103/μL platelets (166-383 x 103/μL). His INR was therapeutic at 2.38. Laboratory assessments of his electrolytes, liver function, and renal function were normal. A chest radiograph demonstrated no acute process. A computed tomography failed to demonstrate sinusitis or an anatomical abnormality that could account for his epistaxis.
Due to the amount of blood loss by epistaxis complicated by anticoagulation for his recent pulmonary embolism, the patient was admitted to the hospital for observation. Reversal of the anticoagulation was considered by the admitting service, but because the patient was only oozing blood, this intervention was not undertaken. Instead, he was continued on warfarin, was treated with an oral antibiotic, and was continued on his inhaled medications for his COPD. He also used his noninvasive PAP device to sleep.
The next day, the patient began to bleed freely from his right nares. The bleeding was initially controlled with compression and positioning and resolved without additional intervention. An otolaryngologist performed silver nitrate cauterization of Kiesselbach’s plexus. The patient experienced no further bleeding, and his hemoglobin remained stable.
The next day, his nose began to bleed briskly. He passed large clots from his nose and mouth. The patient was alert and oriented. He remained hemodynamically stable. His INR was 2.1. Nasal packing was proposed, and the procedure, including the risks and benefits, were explained to the patient.
After obtaining consent from thepatient, the nasal mucosa was prepared with topical 2% lidocaine and 1% phenylephrine. Anterior and posterior nasal packing was successfully achieved with paraffin gauze. This procedure was completed in a monitored environment by an experienced otolaryngologist. However, the patient became agitated 15 to 20 minutes after the nasal packing had been accomplished. He rapidly became apneic, bradycardic, and hypotensive. His oxygen saturation on room air as measured by pulse oximetry decreased precipitously to 50%. These developments were quickly followed by asystole.
Advanced cardiac life support measures were initiated. His airway was secured by oral endotracheal intubation, and oxygen was delivered at 100% fraction of inspired oxygen by bag ventilation. At intubation, only a few small clots were present in the posterior pharynx. No blood was suctioned from the endotracheal tube; therefore, active bleeding was not suspected. The nasal packing remained in place and was not removed. The patient failed to regain spontaneous circulation and died. An arterial blood gas analysis obtained during cardiopulmonary resuscitation demonstrated no methemoglobin on co-oximetry.
Discussion
Because of the high prevalence of epistaxis in the general population, many health care providers (HCPs) are confronted with this problem. Epistaxis in most patients remits without consequence. However, HCPs may be required to intervene. Treatment modalities include simple compression and positioning maneuvers, the application of topical medications, anterior and posterior nasal packing, chemical cauterization, endoscopic electric cauterization, embolization therapy, and surgical arterial ligation.7 The choice of therapy depends on several factors, including the site of the bleeding, the severity of the bleeding, the availability of resources, and the expertise of the HCP. A localized cause of epistaxis is discovered in only 15% of patients, making a conservative therapeutic approach an attractive initial intervention.8
Nasal packing is a successful intervention in 70% of patients with posterior epistaxis. In addition, nasal packing is the preferred method for hemostasis in anterior epistaxis when cauterization fails.3,9 This patient failed simple compression and positioning maneuvers as well as chemical cauterization. For this reason, nasal packing was proposed as a therapeutic intervention. He was hemodynamically stable when the nasal packing procedure was initiated.
Although epistaxis may often have the appearance of significant blood loss and can be frightening for both the patient and HCP, most episodes are not life threatening. Death, when it occurs in association with epistaxis, is very rarely due to exsanguination.3 More commonly, death from epistaxis is related to complications of the treatment intervention or to an exacerbation of an underlying comorbid disease.10 The external overt blood loss in this patient was not significant enough to explain his cardiopulmonary collapse. Although he had experienced a recent pulmonary embolism, he had been on continuous anticoagulation for 3 months and remained adequately anticoagulated during his hospitalization. It therefore seems unlikely that he had experienced a recurrent pulmonary
embolism.
Complications
The treatment of epistaxis can be associated with serious infectious complications, including toxic shock syndrome due to nasal packing and infective endocarditis.11,12 Because patients with malignancies, autoimmune disorders, or organ failure may have epistaxis from decreased platelet production or increased platelet destruction, an infection can be devastating. Many HCPs anticipate this occurrence and provide the patient with epistaxis prophylactic antibiotics.13 Life-threatening infectious complications are usually delayed events and are generally easily recognized. An infectious process was not suspected in this patient. Nevertheless, he was treated with an oral antibiotic.
Dislodgement of the nasal packing with resultant aspiration and asphyxiation has been described as a fatal complication associated with the treatment of epistaxis.14 This complication was not observed in this patient. The otolaryngologist responsible for the placement of the nasal packing was in attendance during the cardiopulmonary resuscitation and insured oral pharyngeal airway patency. Moreover, endotracheal intubation also failed to identify an upper airway obstruction. Aspiration of the packing material was not the cause of this patient’s hemodynamic collapse.
Epidemiology
Florian Kratschmer (1843-1922) was the first researcher to provide a comprehensive analysis of changes in breathing, blood pressure, and heart rate that can occur when mucosa of the nasal airways are stimulated mechanically or chemically.15 His report is considered the first description of trigeminal-mediated bradycardia and asystole, a phenomenon that is sometimes referred to as Kratschmer’s reflex. In current terminology, it is referred to as the nasopulmonary reflex or TCR.
The trigeminal nerve is the largest of the cranial nerves. It provides sensory innervation to the face, scalp, and mucosa of the nose and mouth. The TCR may occur with manipulation of the branches of the trigeminal nerve anywhere along its intracranial or extracranial course. The TCR is described as a sudden onset of parasympathetic arrhythmia, sympathetic hypotension, or apnea elicited by central or peripheral stimulation of any of the sensory branches of the trigeminal nerve.16 The TCR may result in an immediate decrease of the mean arterial blood pressure and heart rate of > 20% when compared with the baseline levels with surgical, mechanical, electrical, or chemical stimulation of the central part of the sensory branches of the trigeminal nerve.17 The TCR represents one of the most powerful autonomous reflexes.18,19
Stimulation of trigeminal receptors that innervate the nose and nasal passage in animals can be an important stimulus for respiratory dysfunction and cardiac arrhythmias.15,16 However, the inability to accurately document the neuroanatomy of this reflex coupled with its variable and rare expression in humans has hindered the appreciation of the importance of the TCR. These observations have led some researchers to dismiss the reflex as inconsequential in humans. However, other physicians, particularly surgeons who manipulate craniofacial structures, have witnessed the effects of the TCR firsthand.20-25
In studies of neurosurgical procedures utilizing the nasal passages and transsphenoid approaches, the TCR has occurred in 10% to 18% of patients.16,24 The TCR has been consistently, although infrequently, noted by otolaryngologists in the management of epistaxis.10,26 Even when performed properly, posterior nasal packing has been reported to cause apnea, hypoxemia, and dysrhythmia.10 Although there has been debate about the importance of the TCR in humans, this response explains the sequence of events in and the death of this patient.27
The mechanism of the TCR is not well understood. The available data suggest that the response of the TCR when triggered by peripheral stimulation is different from the response when the TCR is triggered by central stimulation.18 There is additional anatomic evidence that different areas can be distinguished within the nasal mucosa with regard to stimulation site and stimulus properties.25 Specifically, it has been demonstrated in animals that mechanoreceptors are not equally sensitive throughout the nasal mucosa. The most sensitive areas for mechanical stimuli are located in the posterior parts of the nasal passages. In many animals, including humans, pronounced respiratory and cardiovascular responses can be elicited by appropriate stimulation of the nasal mucosa. These responses have been studied by many researchers in various animals and may be evoked by mechanical, electrical, and chemical stimuli.18,25
Risk Factors
Several risk factors for heightening the TCR have been described.25 Risk factors known to enhance the expression of TCR include hypercapnia, hypoxemia, light general anesthesia, the nature of provoking stimulus, the strength and duration of the stimulus, and medications. The specific pharmaceutical agents known to increase the manifestation of the TCR are narcotics, such as sufentanil and alfentanil, beta blockers, and calcium channel blockers.16,24 This patient was not on any of these medications. In addition, he had not been hypoxemic. He had no known risk for elevation of the TCR.
Evidence suggests that the intensity of the TCR corresponds with the intensity of the mechanical stimulation of the trigeminal pathway.24 Abrupt and sustained traction is more likely to evoke the TCR than is smooth and gentle manipulation. Immediate cessation of the stimulus, such as removal of the nasal packing, may be helpful in the prevention of fatal complications.16 Unfortunately, this was not accomplished in this patient. Other interventions, including the administration of atropine, local anesthetic infiltrations, or blockage of the nerve, may be helpful in preventing fatal complications.
The TCR may be elicited without prior hemodynamic changes. Nevertheless, it is important to anticipate hypoxemia and bradycardia as the first indication of a cardiopulmonary response.26 Administration of the anticholinergic atropine may be required in some cases where bradycardia is severe or persists despite cessation of the stimulus.
However, premedication with intramuscular administration of an anticholinergic medication has not been effective in preventing this reflex. Moreover, the TCR may at times be refractory to the conventional methods of treatment, and use of vasopressors and immediate cardiac life support may be required. Thus, if mechanical stimulation to the trigeminal nerve is anticipated, continuous monitoring of hemodynamic parameters may allow the clinician to more readily identify the TCR and immediately interrupt the inciting stimulus.24
This patient was being monitored, but his cardiopulmonary collapse occurred suddenly and rapidly. He received immediate resuscitation following advanced cardiac life support protocols. Unfortunately, there was no attempt to remove the material that had been employed as packing to control his epistaxis. It remains conjecture whether removal of this material could have altered his outcome. However, the gauze probably should have been removed to maximize his chance of survival.
Conclusion
This case demonstrates the clinical importance of the TCR to providers in the VA health care system, particularly to those who treat epistaxis. Because they are typically older, veterans are a high-risk group. Age is important due to the higher incidence of epistaxis in the older populace, and interventions are more often necessary in older patients with epistaxis. In addition, posterior bleeds occur more frequently in older patients. The resulting stimulation of the trigeminal nerve from interventions to control a posterior bleed may be a more potent provocation for the TCR. Finally, older patients often have comorbid illnesses requiring medications that may augment the TCR. Therefore, the veteran’s age and comorbid illnesses and medications may lead to greater susceptibility of a poor outcome, should the TCR occur as a result of interventions undertaken to control epistaxis.
VA practitioners should, therefore, be aware of the possible occurrence of the TCR in all patients with epistaxis, particularly when invasive manipulations of areas innervated by the trigeminal nerve are required. Evidence suggests that complications of the TCR range from mild bradycardia that responds to simple maneuvers to severe bradycardia and asystole requiring intervention with vagolytics. In rare cases, cardiac dysfunction may lead to death if the TCR is not suspected and early appropriate measures, such as removal of packing materials, are not undertaken.
Although the estimated complication rate of epistaxis and its treatment remains low (about 3%), the authors hope that this report will alert HCPs and that they will remain aware of the TCR as a potentially serious occurrence, even with mild to moderate manipulation of areas innervated by the trigeminal nerve.6
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Pallin DJ, Chng YM, McKay MP, Emond JA, Pelletier AJ, Camargo CA Jr. Epidemiology of epistaxis in US emergency departments, 1992 to 2001. Ann Emerg Med. 2005;46(1):77-81.
2. Viducich RA, Blanda MP, Gerson LW. Posterior epistaxis: clinical features and acute complications. Ann Emerg Med. 1995;25(5):592-596.
3. Manes RP. Evaluating and managing the patient with nosebleeds. Med Clin North Am. 2010;94(5):903-912.
4. National Center for Veterans Analysis and Statistics. Profile of veterans: 2011. Data from the American Community Survey. http://www.va.gov/vetdata/docs/SpecialReports/Profile_of_Veterans_2011.pdf. Published March 2013. Accessed May 4, 2015.
5. U.S. Census Bureau. Age and sex composition: 2010. http://www.census.gov/prod/cen2010/briefs/c2010br-03.pdf. Issued May 2011. Accessed May 4, 2015.
6. Pollice PA, Yoder MG. Epistaxis: a retrospective review of hospitalized patients. Otolaryngol Head Neck Surg. 1997;117(1):49-53.
7. Kucik CJ, Clenney T. Management of epistaxis. Am Fam Physician. 2005;71(2):305-311.
8. Kotecha B, Fowler S, Harkness P, Walmsley J, Brown P, Topham J. Management of epistaxis: a national survey. Ann R Coll Surg Engl. 1996;78(5):444-446.
9. Gifford TO, Orlandi RR. Epistaxis. Otolaryngol Clin North Am. 2008;41(3):525-536.
10. Fairbanks DN. Complications of nasal packing. Otolaryngol Head Neck Surg. 1986;94(3):412-415.
11. Aeumjaturapat S, Supanakorn S, Cutchavaree A. Toxic shock syndrome after anterior-posterior nasal packing. J Med Assoc Thai. 2001;84(3):453-458.
12. Jayawardena S, Eisdorfer J, Indulkar S, Zarkaria M. Infective endocarditis of native valve after anterior nasal packing. Am J Ther. 2006;13(5):460-462.
13. Derkay CS, Hirsch BE, Johnson JT, Wagner RL. Posterior nasal packing. Are intravenous antibiotics really necessary? Arch Otolaryngol Head Neck Surg.1989;115(4):439-441.
14. Koudounarakis E, Chatzakis N, Papadakis I, Panagiotaki I, Velegrakis G. Nasal packing aspiration in a patient with Alzheimer’s disease: a rare complication. Int J Gen Med. 2012;5:643-645.
15. Kratschmer F. On reflexes from the nasal mucous membrane on respiration and circulation. Respir Physiol. 2001;127(2-3):93-104.
16. Spiriev T, Sandu N, Arasho B, Kondoff S, Tzekov C, Schaller B. A new predisposing factor for trigeminocardiac reflex during subdural empyema drainage: a case report. J Med Case Reports. 2010;4:391.
17. Schaller B. Trigemino-cardiac reflex during microvascular trigeminal decompression in cases of trigeminal neuralgia. J Neurosurg Anesthesiol. 2005;17(1):45-48.
18. Schaller B, Cornelius JF, Prabhakar H, et al; Trigemino-Cardiac Reflex Examination Group (TCREG). The trigemino-cardiac reflex: an update of the current knowledge. J Neurosurg Anesthesiol. 2009;21(3):187-195.
19. Sandu N, Spiriev T, Lemaitre F, Filis A, Schaller B; Trigemino-Cardiac Reflex Examination Group (TCREG). New molecular knowledge towards the trigemino-cardiac reflex as a cerebral oxygenconserving reflex. Sci World J. 2010;10:811-817.
20. Nirmala J, Dilip KK, Padmaja D, Gopinath R. “Kratschmer” reflex during rhinoplasty. Anesth Analg. 2006;103(5):1337-1338.
21. Jacobs JR, Levine LA, Davis H, Lefrak SS, Druck NS, Ogura JH. Posterior packs and the nasopulmonary reflex. Laryngoscope. 1981;91(2):279-284.
22. Larsen K, Juul A. Arterial blood gases and pneumatic nasal packing in epistaxis. Laryngoscope.1982;92(5):586-588.
23. Loftus BC, Blitzer A, Cozine K. Epistaxis, medical history, and the nasopulmonary reflex: what is clinically relevant? Otolaryngol Head Neck Surg. 1994;110(4):363-369.
24. Arasho B, Sandu N, Spiriev T, Prabhakar H, Schaller B. Management of the trigeminocardiac reflex: facts and own experience. Neurol India. 2009;57(4):375-380.
25. Schaller BJ, Filis A, Buchfelder M. Trigeminocardiac reflex in humans initiated by peripheral
stimulation during neurosurgical skull-base operations. Its first description. Acta Neurochir (Wien). 2008;150(7):715-717; discussion 718.
26. Stemm RA. Complications of nasal packing. Ear Nose Throat J. 1981;60(10):461-462.
27. Widdicombe J. Reflexes from the lungs and airways: historical perspective. J Appl Physiol (1985). 2006;101(2):628-634.
Epistaxis is a relatively common event that is estimated to occur at least once in 60% of the U.S. population. Epistaxis is also reported to cause 1.7 emergency department (ED) visits per 1,000 population annually.1 Although epistaxis can occur at any age, it typically occurs with a bimodal age distribution and most commonly affects individuals aged < 18 years and adults aged > 50 years.2 The episodes of epistaxis involving the younger age group are more often minor and self-limited. Most bleeds occur along the anterior nasal septum from Kiesselbach’s plexus.
Posterior bleeds occur more often in older patients.2 In addition, epistaxis in the older population tends to be more severe.3 Medical intervention is required in 6% of those experiencing epistaxis. Because the median age for male veterans was 64 years in 2011 compared with a median age of 37.2 years for the average U.S. population in 2010, veterans are among those at greatest risk to develop epistaxis that requires intervention.4,5
Most episodes of epistaxis are not life-threatening, particularly when modern methods of diagnosis and treatment are used. Nevertheless, comorbid diseases, complications of treatment, and normal physiologic responses can sometimes combine to create an adverse outcome.6 This report reviews the case of a veteran patient who experienced a fatal cardiopulmonary arrest after therapeutic interventions for epistaxis. It is believed that his death was due to the well-described but little known trigeminocardiac reflex (TCR).
Case Report
A 65-year-old man visited the ED and reported that his nose had been bleeding intermittently for 1 day. He estimated that he had lost 1 cup of blood over a 24-hour period. He reported no rhinosinusitis, nasal congestion, or recent allergy or upper respiratory infections. He also reported no nasal trauma. In the ED, blood was oozing from his right nares and into his throat, causing him to cough. External compression failed to control the oozing. Topical vasoconstrictors were not applied.
His past medical history included a pulmonary embolism, well-controlled chronic obstructive pulmonary disease (COPD), and sleep apnea. The thrombotic site of origin for his pulmonary embolism had not been identified, despite a thorough examination. He had been on warfarin therapy for 3 months, and his international normalized ratio (INR) had been monitored in an anticoagulation clinic and was well regulated. He was also on inhaled medications for COPD (formoterol and budesonide as a combination preparation twice daily and albuterol every 6 hours as needed as a rescue medication). He adhered to his noninvasive positive airway pressure (PAP) device treatment for sleep apnea. He did not take aspirin or other antiplatelet medications. He reported no use of topical nasal preparations. He also reported no use of illicit drugs or over-the-counter medications, including nonsteroidal anti-inflammatory medications and herbal remedies. He reported no bleeding from other sites or easy bruising.
The patient was alert, oriented, and in no distress. His vital signs were normal. Examination of his nasal passages failed to identify an active site of bleeding. Fresh blood was present in the right nasal passage and the posterior pharynx. Examination of his chest was normal. His hemoglobin was 13.1 g/dL (13.6-17.3 g/dL) with 216 x 103/μL platelets (166-383 x 103/μL). His INR was therapeutic at 2.38. Laboratory assessments of his electrolytes, liver function, and renal function were normal. A chest radiograph demonstrated no acute process. A computed tomography failed to demonstrate sinusitis or an anatomical abnormality that could account for his epistaxis.
Due to the amount of blood loss by epistaxis complicated by anticoagulation for his recent pulmonary embolism, the patient was admitted to the hospital for observation. Reversal of the anticoagulation was considered by the admitting service, but because the patient was only oozing blood, this intervention was not undertaken. Instead, he was continued on warfarin, was treated with an oral antibiotic, and was continued on his inhaled medications for his COPD. He also used his noninvasive PAP device to sleep.
The next day, the patient began to bleed freely from his right nares. The bleeding was initially controlled with compression and positioning and resolved without additional intervention. An otolaryngologist performed silver nitrate cauterization of Kiesselbach’s plexus. The patient experienced no further bleeding, and his hemoglobin remained stable.
The next day, his nose began to bleed briskly. He passed large clots from his nose and mouth. The patient was alert and oriented. He remained hemodynamically stable. His INR was 2.1. Nasal packing was proposed, and the procedure, including the risks and benefits, were explained to the patient.
After obtaining consent from thepatient, the nasal mucosa was prepared with topical 2% lidocaine and 1% phenylephrine. Anterior and posterior nasal packing was successfully achieved with paraffin gauze. This procedure was completed in a monitored environment by an experienced otolaryngologist. However, the patient became agitated 15 to 20 minutes after the nasal packing had been accomplished. He rapidly became apneic, bradycardic, and hypotensive. His oxygen saturation on room air as measured by pulse oximetry decreased precipitously to 50%. These developments were quickly followed by asystole.
Advanced cardiac life support measures were initiated. His airway was secured by oral endotracheal intubation, and oxygen was delivered at 100% fraction of inspired oxygen by bag ventilation. At intubation, only a few small clots were present in the posterior pharynx. No blood was suctioned from the endotracheal tube; therefore, active bleeding was not suspected. The nasal packing remained in place and was not removed. The patient failed to regain spontaneous circulation and died. An arterial blood gas analysis obtained during cardiopulmonary resuscitation demonstrated no methemoglobin on co-oximetry.
Discussion
Because of the high prevalence of epistaxis in the general population, many health care providers (HCPs) are confronted with this problem. Epistaxis in most patients remits without consequence. However, HCPs may be required to intervene. Treatment modalities include simple compression and positioning maneuvers, the application of topical medications, anterior and posterior nasal packing, chemical cauterization, endoscopic electric cauterization, embolization therapy, and surgical arterial ligation.7 The choice of therapy depends on several factors, including the site of the bleeding, the severity of the bleeding, the availability of resources, and the expertise of the HCP. A localized cause of epistaxis is discovered in only 15% of patients, making a conservative therapeutic approach an attractive initial intervention.8
Nasal packing is a successful intervention in 70% of patients with posterior epistaxis. In addition, nasal packing is the preferred method for hemostasis in anterior epistaxis when cauterization fails.3,9 This patient failed simple compression and positioning maneuvers as well as chemical cauterization. For this reason, nasal packing was proposed as a therapeutic intervention. He was hemodynamically stable when the nasal packing procedure was initiated.
Although epistaxis may often have the appearance of significant blood loss and can be frightening for both the patient and HCP, most episodes are not life threatening. Death, when it occurs in association with epistaxis, is very rarely due to exsanguination.3 More commonly, death from epistaxis is related to complications of the treatment intervention or to an exacerbation of an underlying comorbid disease.10 The external overt blood loss in this patient was not significant enough to explain his cardiopulmonary collapse. Although he had experienced a recent pulmonary embolism, he had been on continuous anticoagulation for 3 months and remained adequately anticoagulated during his hospitalization. It therefore seems unlikely that he had experienced a recurrent pulmonary
embolism.
Complications
The treatment of epistaxis can be associated with serious infectious complications, including toxic shock syndrome due to nasal packing and infective endocarditis.11,12 Because patients with malignancies, autoimmune disorders, or organ failure may have epistaxis from decreased platelet production or increased platelet destruction, an infection can be devastating. Many HCPs anticipate this occurrence and provide the patient with epistaxis prophylactic antibiotics.13 Life-threatening infectious complications are usually delayed events and are generally easily recognized. An infectious process was not suspected in this patient. Nevertheless, he was treated with an oral antibiotic.
Dislodgement of the nasal packing with resultant aspiration and asphyxiation has been described as a fatal complication associated with the treatment of epistaxis.14 This complication was not observed in this patient. The otolaryngologist responsible for the placement of the nasal packing was in attendance during the cardiopulmonary resuscitation and insured oral pharyngeal airway patency. Moreover, endotracheal intubation also failed to identify an upper airway obstruction. Aspiration of the packing material was not the cause of this patient’s hemodynamic collapse.
Epidemiology
Florian Kratschmer (1843-1922) was the first researcher to provide a comprehensive analysis of changes in breathing, blood pressure, and heart rate that can occur when mucosa of the nasal airways are stimulated mechanically or chemically.15 His report is considered the first description of trigeminal-mediated bradycardia and asystole, a phenomenon that is sometimes referred to as Kratschmer’s reflex. In current terminology, it is referred to as the nasopulmonary reflex or TCR.
The trigeminal nerve is the largest of the cranial nerves. It provides sensory innervation to the face, scalp, and mucosa of the nose and mouth. The TCR may occur with manipulation of the branches of the trigeminal nerve anywhere along its intracranial or extracranial course. The TCR is described as a sudden onset of parasympathetic arrhythmia, sympathetic hypotension, or apnea elicited by central or peripheral stimulation of any of the sensory branches of the trigeminal nerve.16 The TCR may result in an immediate decrease of the mean arterial blood pressure and heart rate of > 20% when compared with the baseline levels with surgical, mechanical, electrical, or chemical stimulation of the central part of the sensory branches of the trigeminal nerve.17 The TCR represents one of the most powerful autonomous reflexes.18,19
Stimulation of trigeminal receptors that innervate the nose and nasal passage in animals can be an important stimulus for respiratory dysfunction and cardiac arrhythmias.15,16 However, the inability to accurately document the neuroanatomy of this reflex coupled with its variable and rare expression in humans has hindered the appreciation of the importance of the TCR. These observations have led some researchers to dismiss the reflex as inconsequential in humans. However, other physicians, particularly surgeons who manipulate craniofacial structures, have witnessed the effects of the TCR firsthand.20-25
In studies of neurosurgical procedures utilizing the nasal passages and transsphenoid approaches, the TCR has occurred in 10% to 18% of patients.16,24 The TCR has been consistently, although infrequently, noted by otolaryngologists in the management of epistaxis.10,26 Even when performed properly, posterior nasal packing has been reported to cause apnea, hypoxemia, and dysrhythmia.10 Although there has been debate about the importance of the TCR in humans, this response explains the sequence of events in and the death of this patient.27
The mechanism of the TCR is not well understood. The available data suggest that the response of the TCR when triggered by peripheral stimulation is different from the response when the TCR is triggered by central stimulation.18 There is additional anatomic evidence that different areas can be distinguished within the nasal mucosa with regard to stimulation site and stimulus properties.25 Specifically, it has been demonstrated in animals that mechanoreceptors are not equally sensitive throughout the nasal mucosa. The most sensitive areas for mechanical stimuli are located in the posterior parts of the nasal passages. In many animals, including humans, pronounced respiratory and cardiovascular responses can be elicited by appropriate stimulation of the nasal mucosa. These responses have been studied by many researchers in various animals and may be evoked by mechanical, electrical, and chemical stimuli.18,25
Risk Factors
Several risk factors for heightening the TCR have been described.25 Risk factors known to enhance the expression of TCR include hypercapnia, hypoxemia, light general anesthesia, the nature of provoking stimulus, the strength and duration of the stimulus, and medications. The specific pharmaceutical agents known to increase the manifestation of the TCR are narcotics, such as sufentanil and alfentanil, beta blockers, and calcium channel blockers.16,24 This patient was not on any of these medications. In addition, he had not been hypoxemic. He had no known risk for elevation of the TCR.
Evidence suggests that the intensity of the TCR corresponds with the intensity of the mechanical stimulation of the trigeminal pathway.24 Abrupt and sustained traction is more likely to evoke the TCR than is smooth and gentle manipulation. Immediate cessation of the stimulus, such as removal of the nasal packing, may be helpful in the prevention of fatal complications.16 Unfortunately, this was not accomplished in this patient. Other interventions, including the administration of atropine, local anesthetic infiltrations, or blockage of the nerve, may be helpful in preventing fatal complications.
The TCR may be elicited without prior hemodynamic changes. Nevertheless, it is important to anticipate hypoxemia and bradycardia as the first indication of a cardiopulmonary response.26 Administration of the anticholinergic atropine may be required in some cases where bradycardia is severe or persists despite cessation of the stimulus.
However, premedication with intramuscular administration of an anticholinergic medication has not been effective in preventing this reflex. Moreover, the TCR may at times be refractory to the conventional methods of treatment, and use of vasopressors and immediate cardiac life support may be required. Thus, if mechanical stimulation to the trigeminal nerve is anticipated, continuous monitoring of hemodynamic parameters may allow the clinician to more readily identify the TCR and immediately interrupt the inciting stimulus.24
This patient was being monitored, but his cardiopulmonary collapse occurred suddenly and rapidly. He received immediate resuscitation following advanced cardiac life support protocols. Unfortunately, there was no attempt to remove the material that had been employed as packing to control his epistaxis. It remains conjecture whether removal of this material could have altered his outcome. However, the gauze probably should have been removed to maximize his chance of survival.
Conclusion
This case demonstrates the clinical importance of the TCR to providers in the VA health care system, particularly to those who treat epistaxis. Because they are typically older, veterans are a high-risk group. Age is important due to the higher incidence of epistaxis in the older populace, and interventions are more often necessary in older patients with epistaxis. In addition, posterior bleeds occur more frequently in older patients. The resulting stimulation of the trigeminal nerve from interventions to control a posterior bleed may be a more potent provocation for the TCR. Finally, older patients often have comorbid illnesses requiring medications that may augment the TCR. Therefore, the veteran’s age and comorbid illnesses and medications may lead to greater susceptibility of a poor outcome, should the TCR occur as a result of interventions undertaken to control epistaxis.
VA practitioners should, therefore, be aware of the possible occurrence of the TCR in all patients with epistaxis, particularly when invasive manipulations of areas innervated by the trigeminal nerve are required. Evidence suggests that complications of the TCR range from mild bradycardia that responds to simple maneuvers to severe bradycardia and asystole requiring intervention with vagolytics. In rare cases, cardiac dysfunction may lead to death if the TCR is not suspected and early appropriate measures, such as removal of packing materials, are not undertaken.
Although the estimated complication rate of epistaxis and its treatment remains low (about 3%), the authors hope that this report will alert HCPs and that they will remain aware of the TCR as a potentially serious occurrence, even with mild to moderate manipulation of areas innervated by the trigeminal nerve.6
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Epistaxis is a relatively common event that is estimated to occur at least once in 60% of the U.S. population. Epistaxis is also reported to cause 1.7 emergency department (ED) visits per 1,000 population annually.1 Although epistaxis can occur at any age, it typically occurs with a bimodal age distribution and most commonly affects individuals aged < 18 years and adults aged > 50 years.2 The episodes of epistaxis involving the younger age group are more often minor and self-limited. Most bleeds occur along the anterior nasal septum from Kiesselbach’s plexus.
Posterior bleeds occur more often in older patients.2 In addition, epistaxis in the older population tends to be more severe.3 Medical intervention is required in 6% of those experiencing epistaxis. Because the median age for male veterans was 64 years in 2011 compared with a median age of 37.2 years for the average U.S. population in 2010, veterans are among those at greatest risk to develop epistaxis that requires intervention.4,5
Most episodes of epistaxis are not life-threatening, particularly when modern methods of diagnosis and treatment are used. Nevertheless, comorbid diseases, complications of treatment, and normal physiologic responses can sometimes combine to create an adverse outcome.6 This report reviews the case of a veteran patient who experienced a fatal cardiopulmonary arrest after therapeutic interventions for epistaxis. It is believed that his death was due to the well-described but little known trigeminocardiac reflex (TCR).
Case Report
A 65-year-old man visited the ED and reported that his nose had been bleeding intermittently for 1 day. He estimated that he had lost 1 cup of blood over a 24-hour period. He reported no rhinosinusitis, nasal congestion, or recent allergy or upper respiratory infections. He also reported no nasal trauma. In the ED, blood was oozing from his right nares and into his throat, causing him to cough. External compression failed to control the oozing. Topical vasoconstrictors were not applied.
His past medical history included a pulmonary embolism, well-controlled chronic obstructive pulmonary disease (COPD), and sleep apnea. The thrombotic site of origin for his pulmonary embolism had not been identified, despite a thorough examination. He had been on warfarin therapy for 3 months, and his international normalized ratio (INR) had been monitored in an anticoagulation clinic and was well regulated. He was also on inhaled medications for COPD (formoterol and budesonide as a combination preparation twice daily and albuterol every 6 hours as needed as a rescue medication). He adhered to his noninvasive positive airway pressure (PAP) device treatment for sleep apnea. He did not take aspirin or other antiplatelet medications. He reported no use of topical nasal preparations. He also reported no use of illicit drugs or over-the-counter medications, including nonsteroidal anti-inflammatory medications and herbal remedies. He reported no bleeding from other sites or easy bruising.
The patient was alert, oriented, and in no distress. His vital signs were normal. Examination of his nasal passages failed to identify an active site of bleeding. Fresh blood was present in the right nasal passage and the posterior pharynx. Examination of his chest was normal. His hemoglobin was 13.1 g/dL (13.6-17.3 g/dL) with 216 x 103/μL platelets (166-383 x 103/μL). His INR was therapeutic at 2.38. Laboratory assessments of his electrolytes, liver function, and renal function were normal. A chest radiograph demonstrated no acute process. A computed tomography failed to demonstrate sinusitis or an anatomical abnormality that could account for his epistaxis.
Due to the amount of blood loss by epistaxis complicated by anticoagulation for his recent pulmonary embolism, the patient was admitted to the hospital for observation. Reversal of the anticoagulation was considered by the admitting service, but because the patient was only oozing blood, this intervention was not undertaken. Instead, he was continued on warfarin, was treated with an oral antibiotic, and was continued on his inhaled medications for his COPD. He also used his noninvasive PAP device to sleep.
The next day, the patient began to bleed freely from his right nares. The bleeding was initially controlled with compression and positioning and resolved without additional intervention. An otolaryngologist performed silver nitrate cauterization of Kiesselbach’s plexus. The patient experienced no further bleeding, and his hemoglobin remained stable.
The next day, his nose began to bleed briskly. He passed large clots from his nose and mouth. The patient was alert and oriented. He remained hemodynamically stable. His INR was 2.1. Nasal packing was proposed, and the procedure, including the risks and benefits, were explained to the patient.
After obtaining consent from thepatient, the nasal mucosa was prepared with topical 2% lidocaine and 1% phenylephrine. Anterior and posterior nasal packing was successfully achieved with paraffin gauze. This procedure was completed in a monitored environment by an experienced otolaryngologist. However, the patient became agitated 15 to 20 minutes after the nasal packing had been accomplished. He rapidly became apneic, bradycardic, and hypotensive. His oxygen saturation on room air as measured by pulse oximetry decreased precipitously to 50%. These developments were quickly followed by asystole.
Advanced cardiac life support measures were initiated. His airway was secured by oral endotracheal intubation, and oxygen was delivered at 100% fraction of inspired oxygen by bag ventilation. At intubation, only a few small clots were present in the posterior pharynx. No blood was suctioned from the endotracheal tube; therefore, active bleeding was not suspected. The nasal packing remained in place and was not removed. The patient failed to regain spontaneous circulation and died. An arterial blood gas analysis obtained during cardiopulmonary resuscitation demonstrated no methemoglobin on co-oximetry.
Discussion
Because of the high prevalence of epistaxis in the general population, many health care providers (HCPs) are confronted with this problem. Epistaxis in most patients remits without consequence. However, HCPs may be required to intervene. Treatment modalities include simple compression and positioning maneuvers, the application of topical medications, anterior and posterior nasal packing, chemical cauterization, endoscopic electric cauterization, embolization therapy, and surgical arterial ligation.7 The choice of therapy depends on several factors, including the site of the bleeding, the severity of the bleeding, the availability of resources, and the expertise of the HCP. A localized cause of epistaxis is discovered in only 15% of patients, making a conservative therapeutic approach an attractive initial intervention.8
Nasal packing is a successful intervention in 70% of patients with posterior epistaxis. In addition, nasal packing is the preferred method for hemostasis in anterior epistaxis when cauterization fails.3,9 This patient failed simple compression and positioning maneuvers as well as chemical cauterization. For this reason, nasal packing was proposed as a therapeutic intervention. He was hemodynamically stable when the nasal packing procedure was initiated.
Although epistaxis may often have the appearance of significant blood loss and can be frightening for both the patient and HCP, most episodes are not life threatening. Death, when it occurs in association with epistaxis, is very rarely due to exsanguination.3 More commonly, death from epistaxis is related to complications of the treatment intervention or to an exacerbation of an underlying comorbid disease.10 The external overt blood loss in this patient was not significant enough to explain his cardiopulmonary collapse. Although he had experienced a recent pulmonary embolism, he had been on continuous anticoagulation for 3 months and remained adequately anticoagulated during his hospitalization. It therefore seems unlikely that he had experienced a recurrent pulmonary
embolism.
Complications
The treatment of epistaxis can be associated with serious infectious complications, including toxic shock syndrome due to nasal packing and infective endocarditis.11,12 Because patients with malignancies, autoimmune disorders, or organ failure may have epistaxis from decreased platelet production or increased platelet destruction, an infection can be devastating. Many HCPs anticipate this occurrence and provide the patient with epistaxis prophylactic antibiotics.13 Life-threatening infectious complications are usually delayed events and are generally easily recognized. An infectious process was not suspected in this patient. Nevertheless, he was treated with an oral antibiotic.
Dislodgement of the nasal packing with resultant aspiration and asphyxiation has been described as a fatal complication associated with the treatment of epistaxis.14 This complication was not observed in this patient. The otolaryngologist responsible for the placement of the nasal packing was in attendance during the cardiopulmonary resuscitation and insured oral pharyngeal airway patency. Moreover, endotracheal intubation also failed to identify an upper airway obstruction. Aspiration of the packing material was not the cause of this patient’s hemodynamic collapse.
Epidemiology
Florian Kratschmer (1843-1922) was the first researcher to provide a comprehensive analysis of changes in breathing, blood pressure, and heart rate that can occur when mucosa of the nasal airways are stimulated mechanically or chemically.15 His report is considered the first description of trigeminal-mediated bradycardia and asystole, a phenomenon that is sometimes referred to as Kratschmer’s reflex. In current terminology, it is referred to as the nasopulmonary reflex or TCR.
The trigeminal nerve is the largest of the cranial nerves. It provides sensory innervation to the face, scalp, and mucosa of the nose and mouth. The TCR may occur with manipulation of the branches of the trigeminal nerve anywhere along its intracranial or extracranial course. The TCR is described as a sudden onset of parasympathetic arrhythmia, sympathetic hypotension, or apnea elicited by central or peripheral stimulation of any of the sensory branches of the trigeminal nerve.16 The TCR may result in an immediate decrease of the mean arterial blood pressure and heart rate of > 20% when compared with the baseline levels with surgical, mechanical, electrical, or chemical stimulation of the central part of the sensory branches of the trigeminal nerve.17 The TCR represents one of the most powerful autonomous reflexes.18,19
Stimulation of trigeminal receptors that innervate the nose and nasal passage in animals can be an important stimulus for respiratory dysfunction and cardiac arrhythmias.15,16 However, the inability to accurately document the neuroanatomy of this reflex coupled with its variable and rare expression in humans has hindered the appreciation of the importance of the TCR. These observations have led some researchers to dismiss the reflex as inconsequential in humans. However, other physicians, particularly surgeons who manipulate craniofacial structures, have witnessed the effects of the TCR firsthand.20-25
In studies of neurosurgical procedures utilizing the nasal passages and transsphenoid approaches, the TCR has occurred in 10% to 18% of patients.16,24 The TCR has been consistently, although infrequently, noted by otolaryngologists in the management of epistaxis.10,26 Even when performed properly, posterior nasal packing has been reported to cause apnea, hypoxemia, and dysrhythmia.10 Although there has been debate about the importance of the TCR in humans, this response explains the sequence of events in and the death of this patient.27
The mechanism of the TCR is not well understood. The available data suggest that the response of the TCR when triggered by peripheral stimulation is different from the response when the TCR is triggered by central stimulation.18 There is additional anatomic evidence that different areas can be distinguished within the nasal mucosa with regard to stimulation site and stimulus properties.25 Specifically, it has been demonstrated in animals that mechanoreceptors are not equally sensitive throughout the nasal mucosa. The most sensitive areas for mechanical stimuli are located in the posterior parts of the nasal passages. In many animals, including humans, pronounced respiratory and cardiovascular responses can be elicited by appropriate stimulation of the nasal mucosa. These responses have been studied by many researchers in various animals and may be evoked by mechanical, electrical, and chemical stimuli.18,25
Risk Factors
Several risk factors for heightening the TCR have been described.25 Risk factors known to enhance the expression of TCR include hypercapnia, hypoxemia, light general anesthesia, the nature of provoking stimulus, the strength and duration of the stimulus, and medications. The specific pharmaceutical agents known to increase the manifestation of the TCR are narcotics, such as sufentanil and alfentanil, beta blockers, and calcium channel blockers.16,24 This patient was not on any of these medications. In addition, he had not been hypoxemic. He had no known risk for elevation of the TCR.
Evidence suggests that the intensity of the TCR corresponds with the intensity of the mechanical stimulation of the trigeminal pathway.24 Abrupt and sustained traction is more likely to evoke the TCR than is smooth and gentle manipulation. Immediate cessation of the stimulus, such as removal of the nasal packing, may be helpful in the prevention of fatal complications.16 Unfortunately, this was not accomplished in this patient. Other interventions, including the administration of atropine, local anesthetic infiltrations, or blockage of the nerve, may be helpful in preventing fatal complications.
The TCR may be elicited without prior hemodynamic changes. Nevertheless, it is important to anticipate hypoxemia and bradycardia as the first indication of a cardiopulmonary response.26 Administration of the anticholinergic atropine may be required in some cases where bradycardia is severe or persists despite cessation of the stimulus.
However, premedication with intramuscular administration of an anticholinergic medication has not been effective in preventing this reflex. Moreover, the TCR may at times be refractory to the conventional methods of treatment, and use of vasopressors and immediate cardiac life support may be required. Thus, if mechanical stimulation to the trigeminal nerve is anticipated, continuous monitoring of hemodynamic parameters may allow the clinician to more readily identify the TCR and immediately interrupt the inciting stimulus.24
This patient was being monitored, but his cardiopulmonary collapse occurred suddenly and rapidly. He received immediate resuscitation following advanced cardiac life support protocols. Unfortunately, there was no attempt to remove the material that had been employed as packing to control his epistaxis. It remains conjecture whether removal of this material could have altered his outcome. However, the gauze probably should have been removed to maximize his chance of survival.
Conclusion
This case demonstrates the clinical importance of the TCR to providers in the VA health care system, particularly to those who treat epistaxis. Because they are typically older, veterans are a high-risk group. Age is important due to the higher incidence of epistaxis in the older populace, and interventions are more often necessary in older patients with epistaxis. In addition, posterior bleeds occur more frequently in older patients. The resulting stimulation of the trigeminal nerve from interventions to control a posterior bleed may be a more potent provocation for the TCR. Finally, older patients often have comorbid illnesses requiring medications that may augment the TCR. Therefore, the veteran’s age and comorbid illnesses and medications may lead to greater susceptibility of a poor outcome, should the TCR occur as a result of interventions undertaken to control epistaxis.
VA practitioners should, therefore, be aware of the possible occurrence of the TCR in all patients with epistaxis, particularly when invasive manipulations of areas innervated by the trigeminal nerve are required. Evidence suggests that complications of the TCR range from mild bradycardia that responds to simple maneuvers to severe bradycardia and asystole requiring intervention with vagolytics. In rare cases, cardiac dysfunction may lead to death if the TCR is not suspected and early appropriate measures, such as removal of packing materials, are not undertaken.
Although the estimated complication rate of epistaxis and its treatment remains low (about 3%), the authors hope that this report will alert HCPs and that they will remain aware of the TCR as a potentially serious occurrence, even with mild to moderate manipulation of areas innervated by the trigeminal nerve.6
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Pallin DJ, Chng YM, McKay MP, Emond JA, Pelletier AJ, Camargo CA Jr. Epidemiology of epistaxis in US emergency departments, 1992 to 2001. Ann Emerg Med. 2005;46(1):77-81.
2. Viducich RA, Blanda MP, Gerson LW. Posterior epistaxis: clinical features and acute complications. Ann Emerg Med. 1995;25(5):592-596.
3. Manes RP. Evaluating and managing the patient with nosebleeds. Med Clin North Am. 2010;94(5):903-912.
4. National Center for Veterans Analysis and Statistics. Profile of veterans: 2011. Data from the American Community Survey. http://www.va.gov/vetdata/docs/SpecialReports/Profile_of_Veterans_2011.pdf. Published March 2013. Accessed May 4, 2015.
5. U.S. Census Bureau. Age and sex composition: 2010. http://www.census.gov/prod/cen2010/briefs/c2010br-03.pdf. Issued May 2011. Accessed May 4, 2015.
6. Pollice PA, Yoder MG. Epistaxis: a retrospective review of hospitalized patients. Otolaryngol Head Neck Surg. 1997;117(1):49-53.
7. Kucik CJ, Clenney T. Management of epistaxis. Am Fam Physician. 2005;71(2):305-311.
8. Kotecha B, Fowler S, Harkness P, Walmsley J, Brown P, Topham J. Management of epistaxis: a national survey. Ann R Coll Surg Engl. 1996;78(5):444-446.
9. Gifford TO, Orlandi RR. Epistaxis. Otolaryngol Clin North Am. 2008;41(3):525-536.
10. Fairbanks DN. Complications of nasal packing. Otolaryngol Head Neck Surg. 1986;94(3):412-415.
11. Aeumjaturapat S, Supanakorn S, Cutchavaree A. Toxic shock syndrome after anterior-posterior nasal packing. J Med Assoc Thai. 2001;84(3):453-458.
12. Jayawardena S, Eisdorfer J, Indulkar S, Zarkaria M. Infective endocarditis of native valve after anterior nasal packing. Am J Ther. 2006;13(5):460-462.
13. Derkay CS, Hirsch BE, Johnson JT, Wagner RL. Posterior nasal packing. Are intravenous antibiotics really necessary? Arch Otolaryngol Head Neck Surg.1989;115(4):439-441.
14. Koudounarakis E, Chatzakis N, Papadakis I, Panagiotaki I, Velegrakis G. Nasal packing aspiration in a patient with Alzheimer’s disease: a rare complication. Int J Gen Med. 2012;5:643-645.
15. Kratschmer F. On reflexes from the nasal mucous membrane on respiration and circulation. Respir Physiol. 2001;127(2-3):93-104.
16. Spiriev T, Sandu N, Arasho B, Kondoff S, Tzekov C, Schaller B. A new predisposing factor for trigeminocardiac reflex during subdural empyema drainage: a case report. J Med Case Reports. 2010;4:391.
17. Schaller B. Trigemino-cardiac reflex during microvascular trigeminal decompression in cases of trigeminal neuralgia. J Neurosurg Anesthesiol. 2005;17(1):45-48.
18. Schaller B, Cornelius JF, Prabhakar H, et al; Trigemino-Cardiac Reflex Examination Group (TCREG). The trigemino-cardiac reflex: an update of the current knowledge. J Neurosurg Anesthesiol. 2009;21(3):187-195.
19. Sandu N, Spiriev T, Lemaitre F, Filis A, Schaller B; Trigemino-Cardiac Reflex Examination Group (TCREG). New molecular knowledge towards the trigemino-cardiac reflex as a cerebral oxygenconserving reflex. Sci World J. 2010;10:811-817.
20. Nirmala J, Dilip KK, Padmaja D, Gopinath R. “Kratschmer” reflex during rhinoplasty. Anesth Analg. 2006;103(5):1337-1338.
21. Jacobs JR, Levine LA, Davis H, Lefrak SS, Druck NS, Ogura JH. Posterior packs and the nasopulmonary reflex. Laryngoscope. 1981;91(2):279-284.
22. Larsen K, Juul A. Arterial blood gases and pneumatic nasal packing in epistaxis. Laryngoscope.1982;92(5):586-588.
23. Loftus BC, Blitzer A, Cozine K. Epistaxis, medical history, and the nasopulmonary reflex: what is clinically relevant? Otolaryngol Head Neck Surg. 1994;110(4):363-369.
24. Arasho B, Sandu N, Spiriev T, Prabhakar H, Schaller B. Management of the trigeminocardiac reflex: facts and own experience. Neurol India. 2009;57(4):375-380.
25. Schaller BJ, Filis A, Buchfelder M. Trigeminocardiac reflex in humans initiated by peripheral
stimulation during neurosurgical skull-base operations. Its first description. Acta Neurochir (Wien). 2008;150(7):715-717; discussion 718.
26. Stemm RA. Complications of nasal packing. Ear Nose Throat J. 1981;60(10):461-462.
27. Widdicombe J. Reflexes from the lungs and airways: historical perspective. J Appl Physiol (1985). 2006;101(2):628-634.
1. Pallin DJ, Chng YM, McKay MP, Emond JA, Pelletier AJ, Camargo CA Jr. Epidemiology of epistaxis in US emergency departments, 1992 to 2001. Ann Emerg Med. 2005;46(1):77-81.
2. Viducich RA, Blanda MP, Gerson LW. Posterior epistaxis: clinical features and acute complications. Ann Emerg Med. 1995;25(5):592-596.
3. Manes RP. Evaluating and managing the patient with nosebleeds. Med Clin North Am. 2010;94(5):903-912.
4. National Center for Veterans Analysis and Statistics. Profile of veterans: 2011. Data from the American Community Survey. http://www.va.gov/vetdata/docs/SpecialReports/Profile_of_Veterans_2011.pdf. Published March 2013. Accessed May 4, 2015.
5. U.S. Census Bureau. Age and sex composition: 2010. http://www.census.gov/prod/cen2010/briefs/c2010br-03.pdf. Issued May 2011. Accessed May 4, 2015.
6. Pollice PA, Yoder MG. Epistaxis: a retrospective review of hospitalized patients. Otolaryngol Head Neck Surg. 1997;117(1):49-53.
7. Kucik CJ, Clenney T. Management of epistaxis. Am Fam Physician. 2005;71(2):305-311.
8. Kotecha B, Fowler S, Harkness P, Walmsley J, Brown P, Topham J. Management of epistaxis: a national survey. Ann R Coll Surg Engl. 1996;78(5):444-446.
9. Gifford TO, Orlandi RR. Epistaxis. Otolaryngol Clin North Am. 2008;41(3):525-536.
10. Fairbanks DN. Complications of nasal packing. Otolaryngol Head Neck Surg. 1986;94(3):412-415.
11. Aeumjaturapat S, Supanakorn S, Cutchavaree A. Toxic shock syndrome after anterior-posterior nasal packing. J Med Assoc Thai. 2001;84(3):453-458.
12. Jayawardena S, Eisdorfer J, Indulkar S, Zarkaria M. Infective endocarditis of native valve after anterior nasal packing. Am J Ther. 2006;13(5):460-462.
13. Derkay CS, Hirsch BE, Johnson JT, Wagner RL. Posterior nasal packing. Are intravenous antibiotics really necessary? Arch Otolaryngol Head Neck Surg.1989;115(4):439-441.
14. Koudounarakis E, Chatzakis N, Papadakis I, Panagiotaki I, Velegrakis G. Nasal packing aspiration in a patient with Alzheimer’s disease: a rare complication. Int J Gen Med. 2012;5:643-645.
15. Kratschmer F. On reflexes from the nasal mucous membrane on respiration and circulation. Respir Physiol. 2001;127(2-3):93-104.
16. Spiriev T, Sandu N, Arasho B, Kondoff S, Tzekov C, Schaller B. A new predisposing factor for trigeminocardiac reflex during subdural empyema drainage: a case report. J Med Case Reports. 2010;4:391.
17. Schaller B. Trigemino-cardiac reflex during microvascular trigeminal decompression in cases of trigeminal neuralgia. J Neurosurg Anesthesiol. 2005;17(1):45-48.
18. Schaller B, Cornelius JF, Prabhakar H, et al; Trigemino-Cardiac Reflex Examination Group (TCREG). The trigemino-cardiac reflex: an update of the current knowledge. J Neurosurg Anesthesiol. 2009;21(3):187-195.
19. Sandu N, Spiriev T, Lemaitre F, Filis A, Schaller B; Trigemino-Cardiac Reflex Examination Group (TCREG). New molecular knowledge towards the trigemino-cardiac reflex as a cerebral oxygenconserving reflex. Sci World J. 2010;10:811-817.
20. Nirmala J, Dilip KK, Padmaja D, Gopinath R. “Kratschmer” reflex during rhinoplasty. Anesth Analg. 2006;103(5):1337-1338.
21. Jacobs JR, Levine LA, Davis H, Lefrak SS, Druck NS, Ogura JH. Posterior packs and the nasopulmonary reflex. Laryngoscope. 1981;91(2):279-284.
22. Larsen K, Juul A. Arterial blood gases and pneumatic nasal packing in epistaxis. Laryngoscope.1982;92(5):586-588.
23. Loftus BC, Blitzer A, Cozine K. Epistaxis, medical history, and the nasopulmonary reflex: what is clinically relevant? Otolaryngol Head Neck Surg. 1994;110(4):363-369.
24. Arasho B, Sandu N, Spiriev T, Prabhakar H, Schaller B. Management of the trigeminocardiac reflex: facts and own experience. Neurol India. 2009;57(4):375-380.
25. Schaller BJ, Filis A, Buchfelder M. Trigeminocardiac reflex in humans initiated by peripheral
stimulation during neurosurgical skull-base operations. Its first description. Acta Neurochir (Wien). 2008;150(7):715-717; discussion 718.
26. Stemm RA. Complications of nasal packing. Ear Nose Throat J. 1981;60(10):461-462.
27. Widdicombe J. Reflexes from the lungs and airways: historical perspective. J Appl Physiol (1985). 2006;101(2):628-634.
Suspected Clozapine-Induced Cardiomyopathy and Heart Failure With Reduced Ejection Fraction
Clozapine is an atypical antipsychotic that is usually reserved for use in patients with treatment-resistant schizophrenia or schizoaffective disorder with suicidalit
Clozapine-induced cardiomyopathy is a diagnosis of exclusion that requires the absence of other etiologies of cardiac dysfunction (ie, coronary artery disease, hypertension, valvular disease, congenital heart disease, etc). Diagnosing a clozapine-related cardiomyopathy may be a long and laborious task. Patients with cardiomyopathy may present with many nonspecific signs and symptoms, such as fatigue, dyspnea, edema, and/or nausea and vomiting, which are present in other diseases; therefore, multiple encounters and lab tests may be needed until a cardiac source is implicated. The exact mechanism is unknown; however, Chow and colleagues believe that clozapine is a direct toxin of the myocardium.5-7
Case Presentation
A 30-year-old woman with a history of asthma, hypothyroidism (euthyroid with supplementation), posttraumatic stress disorder, and schizoaffective disorder was started on clozapine due to major depression and increased suicidal ideation despite previous treatment with several other antipsychotic agents. Clozapine was gradually titrated to a dose of 150 mg twice a day during an inpatient psychiatric admission. Prior to starting clozapine, this patient had been admitted to the psychiatry unit 11 times within the prior 2 years. After initiating and titrating clozapine over 4 months, her psychiatric symptoms markedly improved.
More than 4 years after the initiation of clozapine and after various treatments for multiple symptoms (Sidebar), the patient was diagnosed with heart failure (HF) with a reduced ejection fraction (EF) of 10% to 15%. She was referred to the cardiology HF clinic. Her dose of clozapine 150 mg at bedtime was discontinued after a discussion with psychiatry. She had a negative workup for other HF etiologies and was started on HF medications that included carvedilol, losartan, and spironolactone. After discontinuation of clozapine, her psychiatric symptoms worsened, and she was admitted to the psychiatry unit twice within a year. Two months after clozapine was discontinued, a repeat echocardiogram (ECHO) was obtained and was essentially unchanged. A chest X-ray (CXR) obtained 4 months after clozapine discontinuation demonstrated a normalized cardio-mediastinal silhouette. A third ECHO was ordered during her second psychiatric admission, which was 11 months after clozapine discontinuation; this revealed an improved left ventricular EF (LVEF) of 30% to 35% and resolution of left ventricular (LV) dilation. 
![]()
This patient’s clinical course led to an extensive chart review that investigated whether there may have been earlier signs and symptoms of HF or cardiomyopathy. It was discovered that the initial HF signs and symptoms were likely present for about 1 year before the diagnosis was made and after having been on clozapine for about 40 months (Patient’s ECHO before and after clozapine discontinuation, click here for additional ECHO perspectives ).
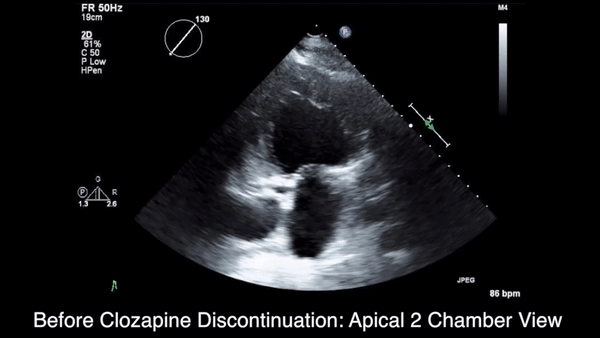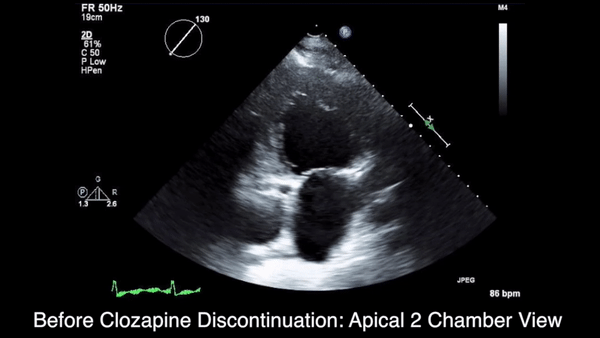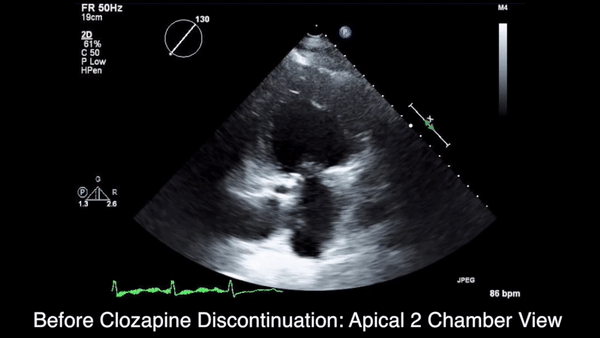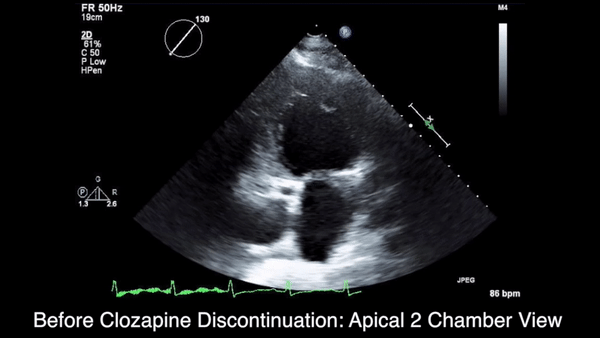
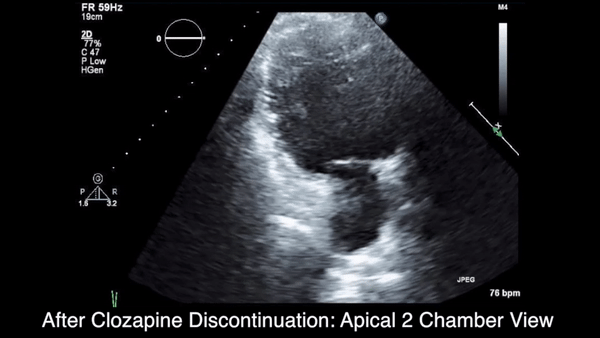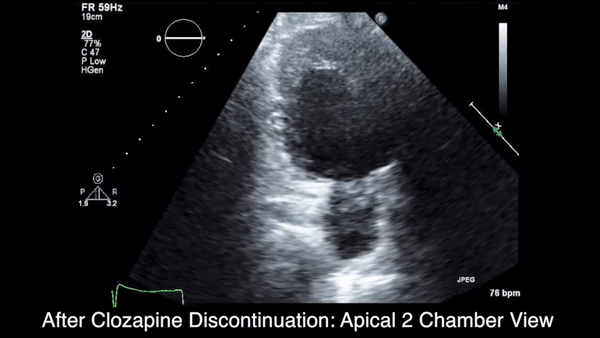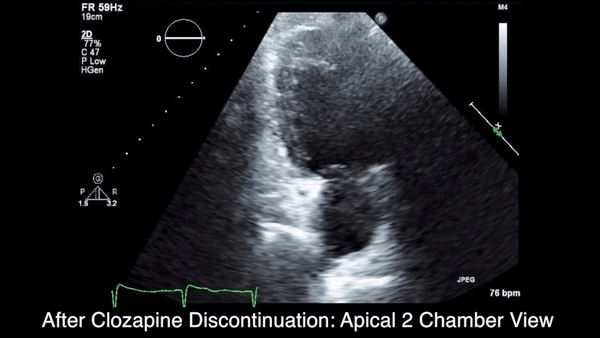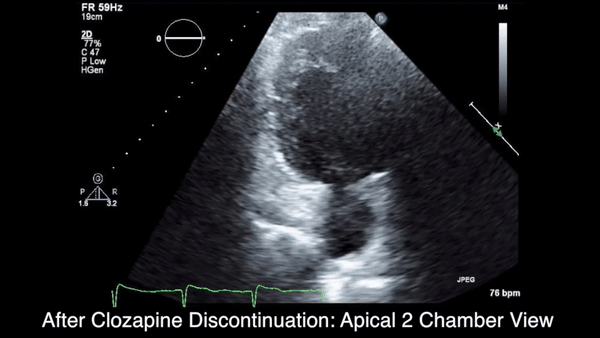
Discussion
In retrospect, this patient likely had HF for many months prior to the official diagnosis; however, given this patient’s young age, prior history of asthma, respiratory disorders, underlying severe psychiatric disease, and confounding symptoms, it is easy to understand why the diagnosis was initially overlooked and delayed.
This patient did not have significant lower extremity edema, but she reported nausea, vomiting, and weight loss. Typical patients with HF exhibit edema and weight gain unless they experience cardiac cachexia. It is not clear whether this patient had a coexisting gastrointestinal (GI) disorder or whether the GI symptoms were secondary to cardiac cachexia. Additionally, weight gain and metabolic syndrome have been documented with clozapine therapy.
It is interesting that a repeat ECHO within 2 months of clozapine discontinuation did not show an improvement, whereas a CXR at 4 months showed a normal cardiac silhouette, and an ECHO at 11 months showed an improvement in EF and normalization of LV size while on appropriate HF medications. It would have been interesting if an ECHO had been completed at 4 months to correspond with the time when the CXR normalized.
There does not seem to be a high level of awareness regarding this potentially fatal diagnosis of cardiomyopathy related to the use of clozapine. A recent review of clozapine-induced cardiomyopathy by Alawami and colleagues revealed characteristics, including median age of diagnosis of 33.5 years, a mean daily dose of 360 mg (range 125-700 mg/d), average time of therapy until the development of symptoms 14.4 months (range between 3 weeks and 4 years), and the presence of ventricular dilation in 39%.8 The most common symptoms on clinical presentation were shortness of breath (60%), palpitations (36%), cough (16%), fatigue (16%), and chest pain (8%).8
It is interesting that edema was not present in the patients studied in their review; this difference from the usual presentation of severe HF may lower clinical suspicion and makes diagnosing this type of cardiomyopathy more difficult. Alawami also noted that patients with an LVEF < 25% at the time of diagnosis tended to have a poorer prognosis with the highest risk of mortality and limited recovery. Fortunately, in this case, the patient’s LVEF improved significantly, and it will be interesting to continue to monitor the patient for further improvement.
As a result of this case, the authors have performed a chart review on all patients prescribed clozapine at VA Loma Linda Healthcare System. Additionally, this case supports the implementation of better cardiomyopathy monitoring of all clozapine patients. It may be recommended to obtain a baseline CXR in all patients starting clozapine, conduct monthly cardiomyopathy symptom screening that coincides with ANC monitoring, and perform an ECHO immediately on any clinical suspicion of cardiomyopathy.
Conclusion
Better awareness and regular screening for signs and symptoms of HF may help prevent a delay in diagnosing a rare but serious and potentially fatal condition associated with clozapine. Chest X-rays demonstrating cardiomegaly can be helpful when the early diagnosis of HF is suspected and may be the first diagnostic imaging test to normalize after clozapine discontinuation.
Since clozapine is a REMS medication and all patients are scheduled for regular ANC follow-up, it would seem prudent that patients also should be screened for signs and symptoms of HF, including the new onset or worsening of baseline shortness of breath, palpitations, cough, fatigue, chest pain, edema, gastroparesis, and perhaps extreme weight loss. Once a physician suspects HF, an ECHO should be obtained immediately.
In addition to the clozapine boxed warning for cardiomyopathy, it would be helpful if the clozapine patient counseling information section had a specific warning that advises patients to contact their clinician if they develop the signs and symptoms of HF.
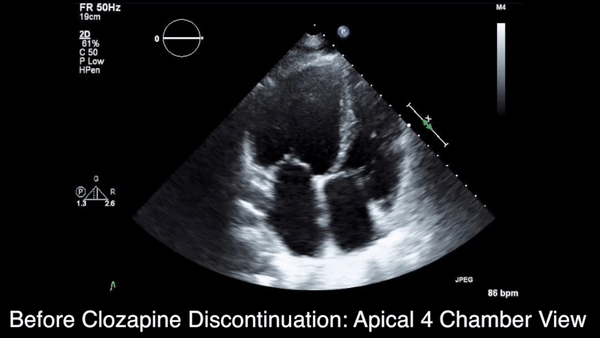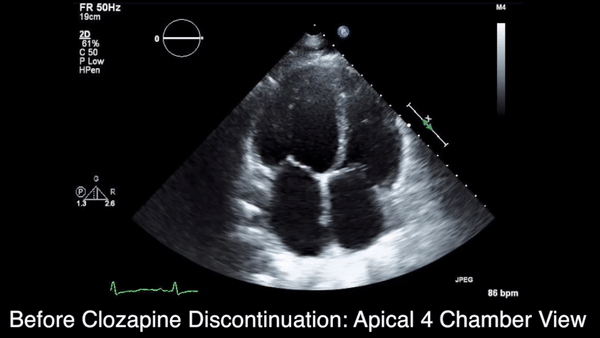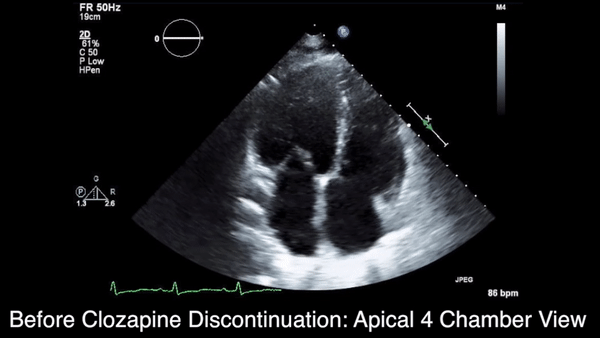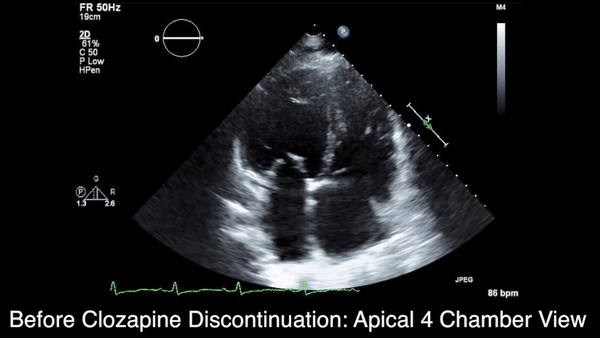
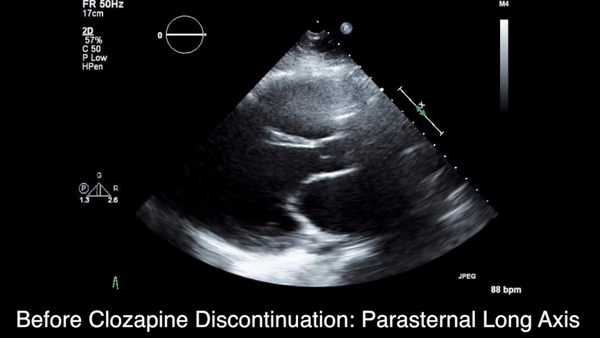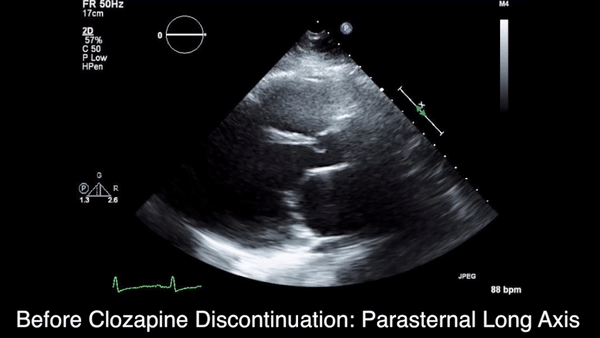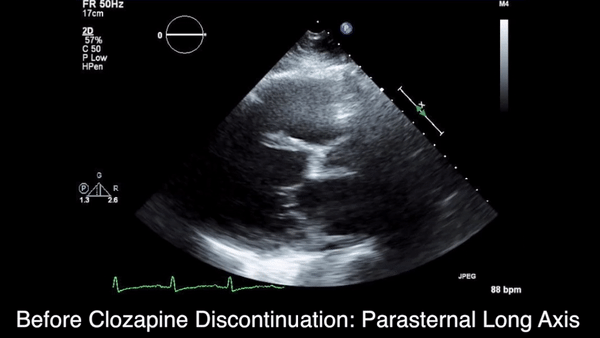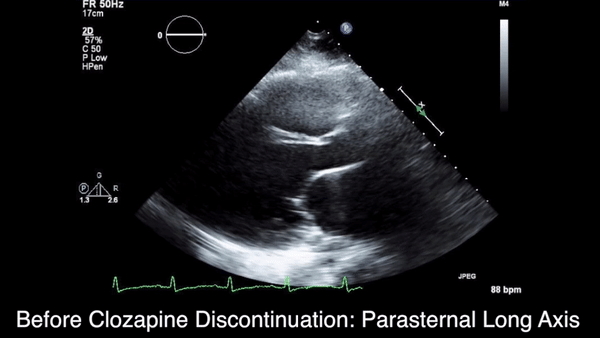
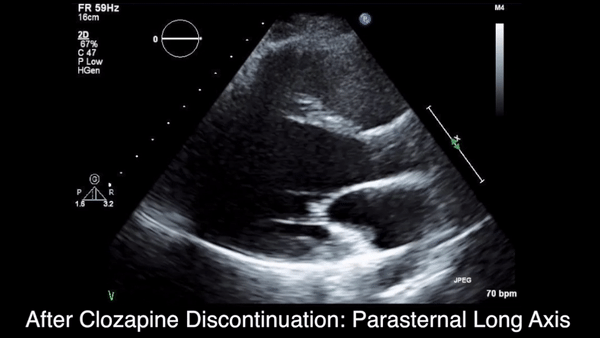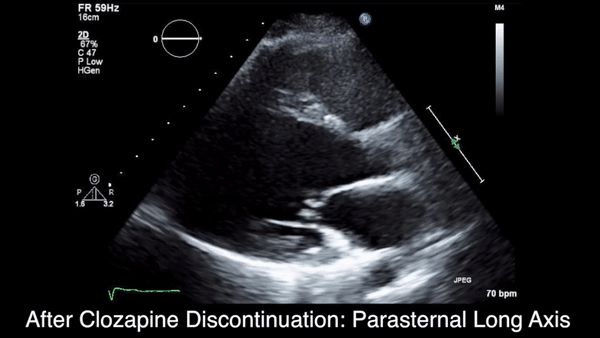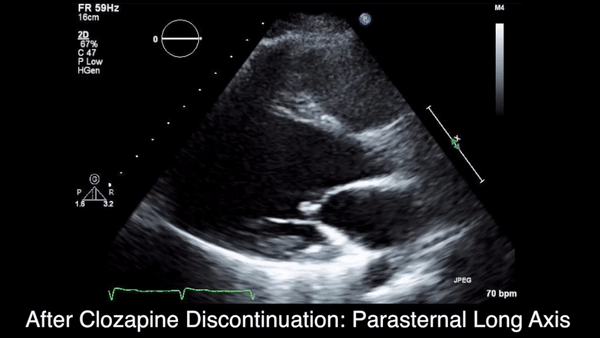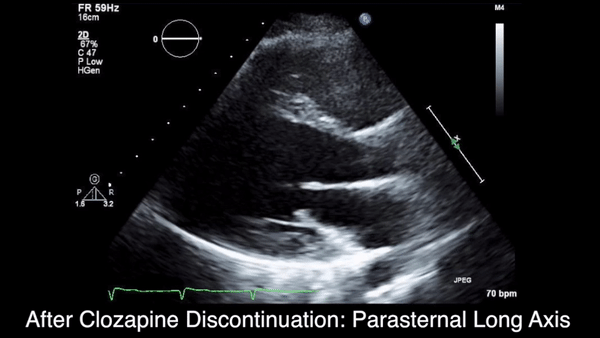
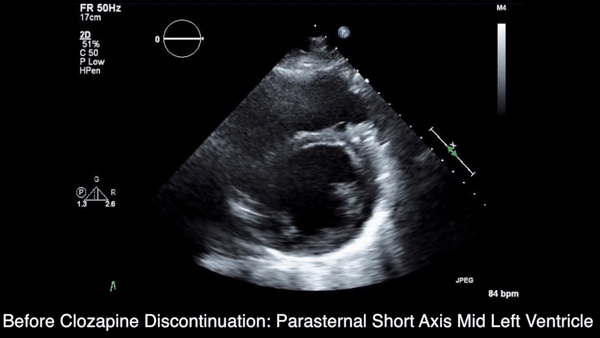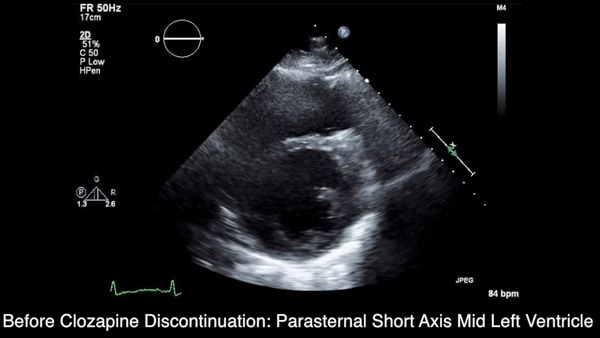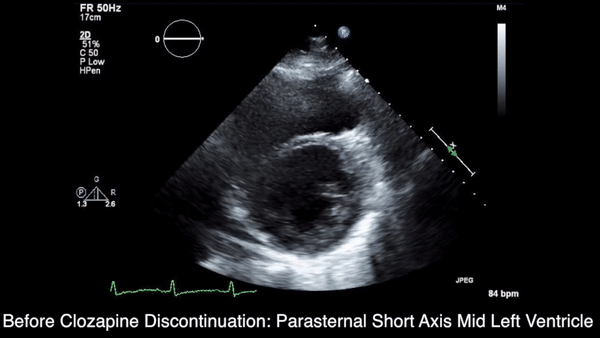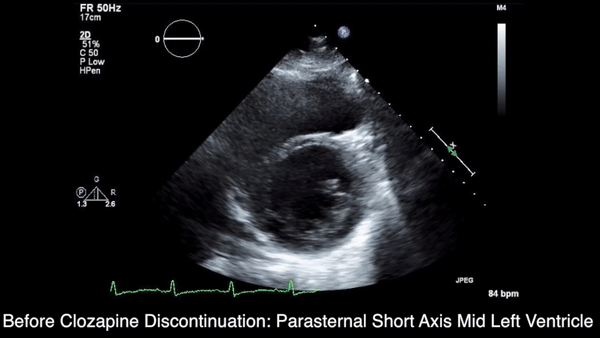
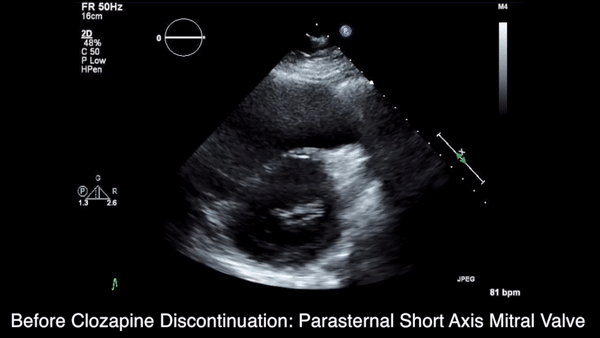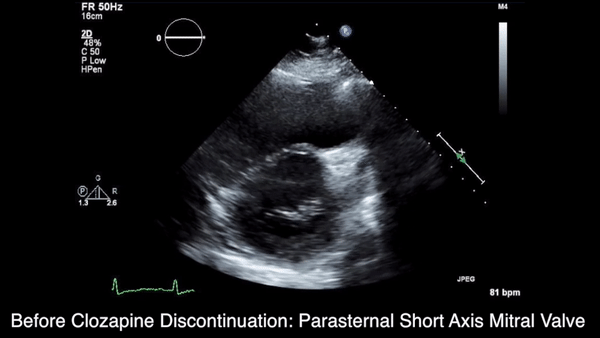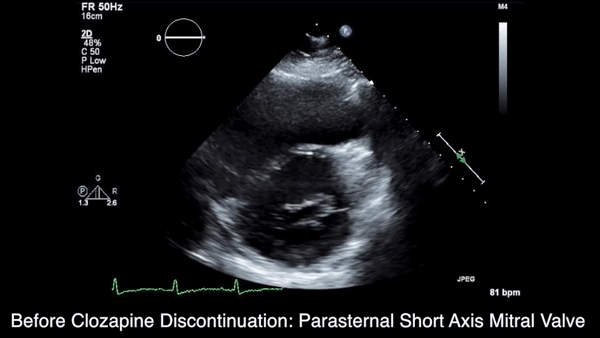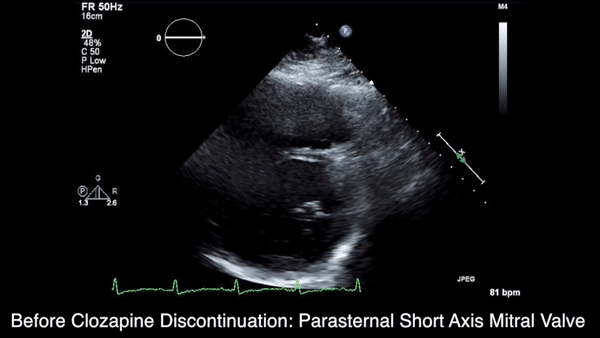
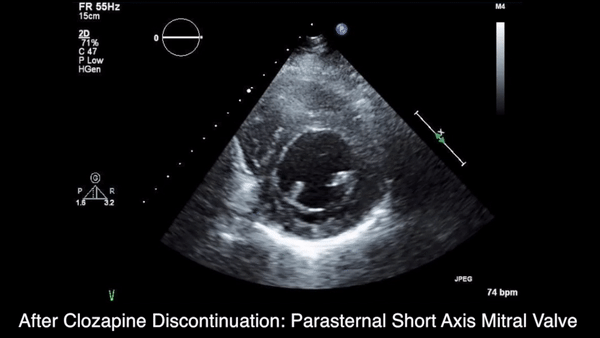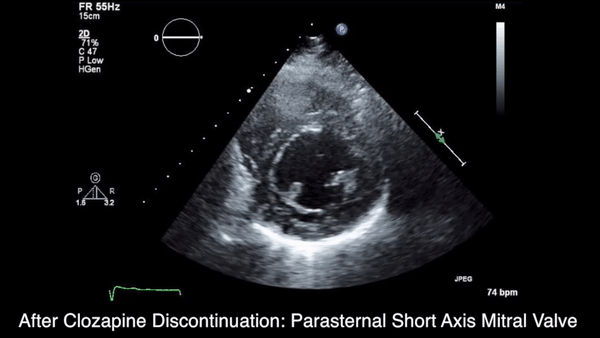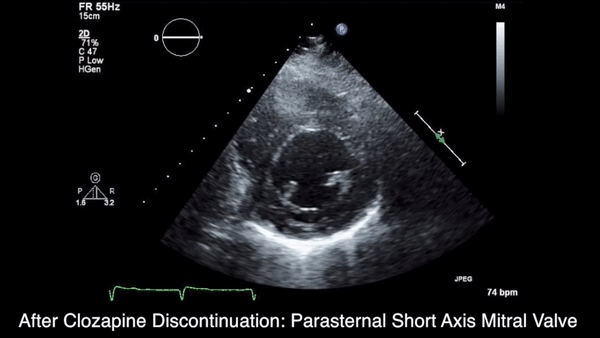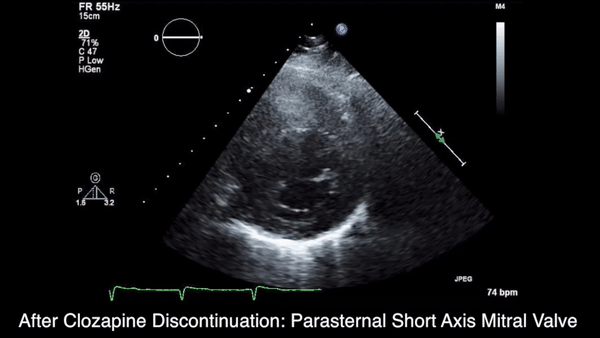
1. Clozaril [package insert]. Rosemont, PA: HLS Therapeutics (USA), Inc; 2015.
2. Youssef DL, Narayanan P, Gill N. Incidence and risk factors for clozapine-induced myocarditis and cardiomyopathy at a regional mental health service in Australia. Austalas Psychiatry. 2016;24(2):176-180.
3. La Grenade L, Graham D, Trontell A. Myocarditis and cardiomyopathy associated with clozapine use in the United States. N Eng J Med. 2001;345(3):224-225.
4. Kilian JG, Kerr K, Lawrence C, Celermajer DS. Myocarditis and cardiomyopathy associated with clozapine. Lancet. 1999;354(9193):1841-1845.
5. Chow V, Yeoh T, Ng AC, et al. Asymptomatic left ventricular dysfunction with long-term clozapine treatment for schizophrenia: a multicenter cross-sectional cohort study. Open Heart 2014;1(1):e000030.
6. Scelsa SN, Simpson DM, McQuistion HL, Fineman A, Ault K, Reichler B. Clozapine-induced myotoxicity in patients with chronic psychotic disorders. Neurology. 1996;47(6):1518-1523.
7. Reznik I, Volchek L, Mester R, et al. Myotoxicity and neurotoxicity during clozapine treatment. Clin Neuropharmacol. 2000;23(5):276-280.
8. Alawami A, Wasywich C, Cicovic A, Kenedi C. A systematic review of clozapine induced cardiomyopathy. Int J Cardiol. 2014;176(2):315-320.
Clozapine is an atypical antipsychotic that is usually reserved for use in patients with treatment-resistant schizophrenia or schizoaffective disorder with suicidalit
Clozapine-induced cardiomyopathy is a diagnosis of exclusion that requires the absence of other etiologies of cardiac dysfunction (ie, coronary artery disease, hypertension, valvular disease, congenital heart disease, etc). Diagnosing a clozapine-related cardiomyopathy may be a long and laborious task. Patients with cardiomyopathy may present with many nonspecific signs and symptoms, such as fatigue, dyspnea, edema, and/or nausea and vomiting, which are present in other diseases; therefore, multiple encounters and lab tests may be needed until a cardiac source is implicated. The exact mechanism is unknown; however, Chow and colleagues believe that clozapine is a direct toxin of the myocardium.5-7
Case Presentation
A 30-year-old woman with a history of asthma, hypothyroidism (euthyroid with supplementation), posttraumatic stress disorder, and schizoaffective disorder was started on clozapine due to major depression and increased suicidal ideation despite previous treatment with several other antipsychotic agents. Clozapine was gradually titrated to a dose of 150 mg twice a day during an inpatient psychiatric admission. Prior to starting clozapine, this patient had been admitted to the psychiatry unit 11 times within the prior 2 years. After initiating and titrating clozapine over 4 months, her psychiatric symptoms markedly improved.
More than 4 years after the initiation of clozapine and after various treatments for multiple symptoms (Sidebar), the patient was diagnosed with heart failure (HF) with a reduced ejection fraction (EF) of 10% to 15%. She was referred to the cardiology HF clinic. Her dose of clozapine 150 mg at bedtime was discontinued after a discussion with psychiatry. She had a negative workup for other HF etiologies and was started on HF medications that included carvedilol, losartan, and spironolactone. After discontinuation of clozapine, her psychiatric symptoms worsened, and she was admitted to the psychiatry unit twice within a year. Two months after clozapine was discontinued, a repeat echocardiogram (ECHO) was obtained and was essentially unchanged. A chest X-ray (CXR) obtained 4 months after clozapine discontinuation demonstrated a normalized cardio-mediastinal silhouette. A third ECHO was ordered during her second psychiatric admission, which was 11 months after clozapine discontinuation; this revealed an improved left ventricular EF (LVEF) of 30% to 35% and resolution of left ventricular (LV) dilation.
![]()
This patient’s clinical course led to an extensive chart review that investigated whether there may have been earlier signs and symptoms of HF or cardiomyopathy. It was discovered that the initial HF signs and symptoms were likely present for about 1 year before the diagnosis was made and after having been on clozapine for about 40 months (Patient’s ECHO before and after clozapine discontinuation, click here for additional ECHO perspectives ).
Discussion
In retrospect, this patient likely had HF for many months prior to the official diagnosis; however, given this patient’s young age, prior history of asthma, respiratory disorders, underlying severe psychiatric disease, and confounding symptoms, it is easy to understand why the diagnosis was initially overlooked and delayed.
This patient did not have significant lower extremity edema, but she reported nausea, vomiting, and weight loss. Typical patients with HF exhibit edema and weight gain unless they experience cardiac cachexia. It is not clear whether this patient had a coexisting gastrointestinal (GI) disorder or whether the GI symptoms were secondary to cardiac cachexia. Additionally, weight gain and metabolic syndrome have been documented with clozapine therapy.
It is interesting that a repeat ECHO within 2 months of clozapine discontinuation did not show an improvement, whereas a CXR at 4 months showed a normal cardiac silhouette, and an ECHO at 11 months showed an improvement in EF and normalization of LV size while on appropriate HF medications. It would have been interesting if an ECHO had been completed at 4 months to correspond with the time when the CXR normalized.
There does not seem to be a high level of awareness regarding this potentially fatal diagnosis of cardiomyopathy related to the use of clozapine. A recent review of clozapine-induced cardiomyopathy by Alawami and colleagues revealed characteristics, including median age of diagnosis of 33.5 years, a mean daily dose of 360 mg (range 125-700 mg/d), average time of therapy until the development of symptoms 14.4 months (range between 3 weeks and 4 years), and the presence of ventricular dilation in 39%.8 The most common symptoms on clinical presentation were shortness of breath (60%), palpitations (36%), cough (16%), fatigue (16%), and chest pain (8%).8
It is interesting that edema was not present in the patients studied in their review; this difference from the usual presentation of severe HF may lower clinical suspicion and makes diagnosing this type of cardiomyopathy more difficult. Alawami also noted that patients with an LVEF < 25% at the time of diagnosis tended to have a poorer prognosis with the highest risk of mortality and limited recovery. Fortunately, in this case, the patient’s LVEF improved significantly, and it will be interesting to continue to monitor the patient for further improvement.
As a result of this case, the authors have performed a chart review on all patients prescribed clozapine at VA Loma Linda Healthcare System. Additionally, this case supports the implementation of better cardiomyopathy monitoring of all clozapine patients. It may be recommended to obtain a baseline CXR in all patients starting clozapine, conduct monthly cardiomyopathy symptom screening that coincides with ANC monitoring, and perform an ECHO immediately on any clinical suspicion of cardiomyopathy.
Conclusion
Better awareness and regular screening for signs and symptoms of HF may help prevent a delay in diagnosing a rare but serious and potentially fatal condition associated with clozapine. Chest X-rays demonstrating cardiomegaly can be helpful when the early diagnosis of HF is suspected and may be the first diagnostic imaging test to normalize after clozapine discontinuation.
Since clozapine is a REMS medication and all patients are scheduled for regular ANC follow-up, it would seem prudent that patients also should be screened for signs and symptoms of HF, including the new onset or worsening of baseline shortness of breath, palpitations, cough, fatigue, chest pain, edema, gastroparesis, and perhaps extreme weight loss. Once a physician suspects HF, an ECHO should be obtained immediately.
In addition to the clozapine boxed warning for cardiomyopathy, it would be helpful if the clozapine patient counseling information section had a specific warning that advises patients to contact their clinician if they develop the signs and symptoms of HF.
Clozapine is an atypical antipsychotic that is usually reserved for use in patients with treatment-resistant schizophrenia or schizoaffective disorder with suicidalit
Clozapine-induced cardiomyopathy is a diagnosis of exclusion that requires the absence of other etiologies of cardiac dysfunction (ie, coronary artery disease, hypertension, valvular disease, congenital heart disease, etc). Diagnosing a clozapine-related cardiomyopathy may be a long and laborious task. Patients with cardiomyopathy may present with many nonspecific signs and symptoms, such as fatigue, dyspnea, edema, and/or nausea and vomiting, which are present in other diseases; therefore, multiple encounters and lab tests may be needed until a cardiac source is implicated. The exact mechanism is unknown; however, Chow and colleagues believe that clozapine is a direct toxin of the myocardium.5-7
Case Presentation
A 30-year-old woman with a history of asthma, hypothyroidism (euthyroid with supplementation), posttraumatic stress disorder, and schizoaffective disorder was started on clozapine due to major depression and increased suicidal ideation despite previous treatment with several other antipsychotic agents. Clozapine was gradually titrated to a dose of 150 mg twice a day during an inpatient psychiatric admission. Prior to starting clozapine, this patient had been admitted to the psychiatry unit 11 times within the prior 2 years. After initiating and titrating clozapine over 4 months, her psychiatric symptoms markedly improved.
More than 4 years after the initiation of clozapine and after various treatments for multiple symptoms (Sidebar), the patient was diagnosed with heart failure (HF) with a reduced ejection fraction (EF) of 10% to 15%. She was referred to the cardiology HF clinic. Her dose of clozapine 150 mg at bedtime was discontinued after a discussion with psychiatry. She had a negative workup for other HF etiologies and was started on HF medications that included carvedilol, losartan, and spironolactone. After discontinuation of clozapine, her psychiatric symptoms worsened, and she was admitted to the psychiatry unit twice within a year. Two months after clozapine was discontinued, a repeat echocardiogram (ECHO) was obtained and was essentially unchanged. A chest X-ray (CXR) obtained 4 months after clozapine discontinuation demonstrated a normalized cardio-mediastinal silhouette. A third ECHO was ordered during her second psychiatric admission, which was 11 months after clozapine discontinuation; this revealed an improved left ventricular EF (LVEF) of 30% to 35% and resolution of left ventricular (LV) dilation.
![]()
This patient’s clinical course led to an extensive chart review that investigated whether there may have been earlier signs and symptoms of HF or cardiomyopathy. It was discovered that the initial HF signs and symptoms were likely present for about 1 year before the diagnosis was made and after having been on clozapine for about 40 months (Patient’s ECHO before and after clozapine discontinuation, click here for additional ECHO perspectives ).
Discussion
In retrospect, this patient likely had HF for many months prior to the official diagnosis; however, given this patient’s young age, prior history of asthma, respiratory disorders, underlying severe psychiatric disease, and confounding symptoms, it is easy to understand why the diagnosis was initially overlooked and delayed.
This patient did not have significant lower extremity edema, but she reported nausea, vomiting, and weight loss. Typical patients with HF exhibit edema and weight gain unless they experience cardiac cachexia. It is not clear whether this patient had a coexisting gastrointestinal (GI) disorder or whether the GI symptoms were secondary to cardiac cachexia. Additionally, weight gain and metabolic syndrome have been documented with clozapine therapy.
It is interesting that a repeat ECHO within 2 months of clozapine discontinuation did not show an improvement, whereas a CXR at 4 months showed a normal cardiac silhouette, and an ECHO at 11 months showed an improvement in EF and normalization of LV size while on appropriate HF medications. It would have been interesting if an ECHO had been completed at 4 months to correspond with the time when the CXR normalized.
There does not seem to be a high level of awareness regarding this potentially fatal diagnosis of cardiomyopathy related to the use of clozapine. A recent review of clozapine-induced cardiomyopathy by Alawami and colleagues revealed characteristics, including median age of diagnosis of 33.5 years, a mean daily dose of 360 mg (range 125-700 mg/d), average time of therapy until the development of symptoms 14.4 months (range between 3 weeks and 4 years), and the presence of ventricular dilation in 39%.8 The most common symptoms on clinical presentation were shortness of breath (60%), palpitations (36%), cough (16%), fatigue (16%), and chest pain (8%).8
It is interesting that edema was not present in the patients studied in their review; this difference from the usual presentation of severe HF may lower clinical suspicion and makes diagnosing this type of cardiomyopathy more difficult. Alawami also noted that patients with an LVEF < 25% at the time of diagnosis tended to have a poorer prognosis with the highest risk of mortality and limited recovery. Fortunately, in this case, the patient’s LVEF improved significantly, and it will be interesting to continue to monitor the patient for further improvement.
As a result of this case, the authors have performed a chart review on all patients prescribed clozapine at VA Loma Linda Healthcare System. Additionally, this case supports the implementation of better cardiomyopathy monitoring of all clozapine patients. It may be recommended to obtain a baseline CXR in all patients starting clozapine, conduct monthly cardiomyopathy symptom screening that coincides with ANC monitoring, and perform an ECHO immediately on any clinical suspicion of cardiomyopathy.
Conclusion
Better awareness and regular screening for signs and symptoms of HF may help prevent a delay in diagnosing a rare but serious and potentially fatal condition associated with clozapine. Chest X-rays demonstrating cardiomegaly can be helpful when the early diagnosis of HF is suspected and may be the first diagnostic imaging test to normalize after clozapine discontinuation.
Since clozapine is a REMS medication and all patients are scheduled for regular ANC follow-up, it would seem prudent that patients also should be screened for signs and symptoms of HF, including the new onset or worsening of baseline shortness of breath, palpitations, cough, fatigue, chest pain, edema, gastroparesis, and perhaps extreme weight loss. Once a physician suspects HF, an ECHO should be obtained immediately.
In addition to the clozapine boxed warning for cardiomyopathy, it would be helpful if the clozapine patient counseling information section had a specific warning that advises patients to contact their clinician if they develop the signs and symptoms of HF.
1. Clozaril [package insert]. Rosemont, PA: HLS Therapeutics (USA), Inc; 2015.
2. Youssef DL, Narayanan P, Gill N. Incidence and risk factors for clozapine-induced myocarditis and cardiomyopathy at a regional mental health service in Australia. Austalas Psychiatry. 2016;24(2):176-180.
3. La Grenade L, Graham D, Trontell A. Myocarditis and cardiomyopathy associated with clozapine use in the United States. N Eng J Med. 2001;345(3):224-225.
4. Kilian JG, Kerr K, Lawrence C, Celermajer DS. Myocarditis and cardiomyopathy associated with clozapine. Lancet. 1999;354(9193):1841-1845.
5. Chow V, Yeoh T, Ng AC, et al. Asymptomatic left ventricular dysfunction with long-term clozapine treatment for schizophrenia: a multicenter cross-sectional cohort study. Open Heart 2014;1(1):e000030.
6. Scelsa SN, Simpson DM, McQuistion HL, Fineman A, Ault K, Reichler B. Clozapine-induced myotoxicity in patients with chronic psychotic disorders. Neurology. 1996;47(6):1518-1523.
7. Reznik I, Volchek L, Mester R, et al. Myotoxicity and neurotoxicity during clozapine treatment. Clin Neuropharmacol. 2000;23(5):276-280.
8. Alawami A, Wasywich C, Cicovic A, Kenedi C. A systematic review of clozapine induced cardiomyopathy. Int J Cardiol. 2014;176(2):315-320.
1. Clozaril [package insert]. Rosemont, PA: HLS Therapeutics (USA), Inc; 2015.
2. Youssef DL, Narayanan P, Gill N. Incidence and risk factors for clozapine-induced myocarditis and cardiomyopathy at a regional mental health service in Australia. Austalas Psychiatry. 2016;24(2):176-180.
3. La Grenade L, Graham D, Trontell A. Myocarditis and cardiomyopathy associated with clozapine use in the United States. N Eng J Med. 2001;345(3):224-225.
4. Kilian JG, Kerr K, Lawrence C, Celermajer DS. Myocarditis and cardiomyopathy associated with clozapine. Lancet. 1999;354(9193):1841-1845.
5. Chow V, Yeoh T, Ng AC, et al. Asymptomatic left ventricular dysfunction with long-term clozapine treatment for schizophrenia: a multicenter cross-sectional cohort study. Open Heart 2014;1(1):e000030.
6. Scelsa SN, Simpson DM, McQuistion HL, Fineman A, Ault K, Reichler B. Clozapine-induced myotoxicity in patients with chronic psychotic disorders. Neurology. 1996;47(6):1518-1523.
7. Reznik I, Volchek L, Mester R, et al. Myotoxicity and neurotoxicity during clozapine treatment. Clin Neuropharmacol. 2000;23(5):276-280.
8. Alawami A, Wasywich C, Cicovic A, Kenedi C. A systematic review of clozapine induced cardiomyopathy. Int J Cardiol. 2014;176(2):315-320.
Complex Malignancies: A Diagnostic and Therapeutic Trilemma
Mr. F. is a 67-year-old man with a medical history significant for hypertension, hyperlipidemia, gastroesophageal reflux disease, abdominal aortic aneurysm repair, and lung nodules (8-mm nodules in his right lower lobe and left upper lobe) since 2009 for which he had been followed in the pulmonary clinic. His last radiologic imaging had been performed in November 2010. The patient had failed to show for a serial scan and follow-up pulmonary appointments. He had seen his primary care practitioner annually. He reporteded a 60 pack-year smoking history and drank up to 10 to 15 beers a day, 3 days a week. He also reported exposure to asbestos while in the U.S. Navy as well as having worked in a sheet metal yard. He reported no family history of lung cancer.
In January 2014, Mr. F. presented to his primary care doctor with a nonpainful neck lump noted by the patient for a few months. On physical examination, the patient appeared healthy and without change in weight, loss of appetite, dysphagia, hemoptysis, or any other remarkable symptoms except for the pain in the left-sided neck mass. On oral examination, the patient did not have any oral lesions but was noted to have poor dentition. He had decreased breath sounds on the right side but no adventitious breath sounds. His cardiac status was within normal limits with a regular heart rate, warm skin, and well-perfused extremities. His abdomen was soft and nontender with good bowel sounds.
A neck computed tomography (CT) done on January 17, 2014, showed 2 left neck masses in the tail of the parotid gland or in level 2 adjacent to the parotid gland. The ear, nose, and throat (ENT) clinic noted that he had a 3-cm level 2 mass, which was mobile and nontender. A fine needle aspiration biopsy showed poorly differentiated rare malignant cells, but the small size of the biopsy precluded identification of the cells. For that reason, the patient was scheduled for an operative laryngoscopy.
A combined positron emission tomography and CT (PET/CT) scan on February 5, 2014, showed multiple areas of hypermetabolic activity. A highly metabolic 2.6 x 1.9-cm mass was noted in the left neck inseparable from the inferior border of the left parotid gland; whether this represented a parotid neoplasm or neoplastic lymph node could not be determined. Positive level 5 neck lymph nodes were suggestive of metastatic disease. There were also 2 foci of mild nonspecific metabolic activity in the right parotid gland. A spiculated mass in the right lower lung, measuring 2.3 x 3.5 cm, demonstrated intense fludeoxyglucose F 18 (FDG) uptake with maximum standardized uptake value (SUV) of 12.7, which was consistent with neoplasm. Thickening of the middistal esophageal wall with intense FDG uptake (SUV of 7.9) was suggestive of esophageal carcinoma.
The patient underwent an endoscopic ultrasound (EUS) esophageal biopsy, which demonstrated poorly differentiated adenocarcinoma (Figure 1). The tumor seemed to have originated at this site, as the biopsy fragments also contained foci of Barrett’s metaplasia and in situ carcinoma. He was presented in the hospital tumor board about whether he should undergo a high-risk lung biopsy due to the location of the mass or have his left neck mass rebiopsied, assuming that he had metastatic esophageal cancer in both the lung and neck. The safer procedure of redoing the neck biopsy in the Ear, Nose, and Throat clinic was pursued, and the patient underwent a biopsy and surgical excision of his left neck mass with positive pathology of highgrade sarcoma (Figure 2). 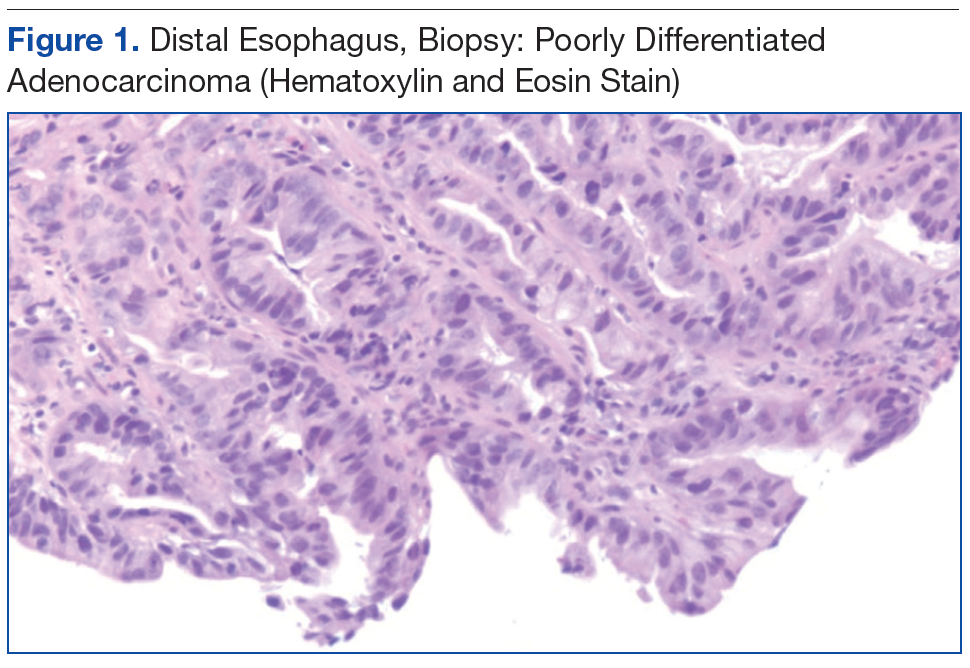
The tumor consisted of spindle and epithelioid cell proliferation within a lymph node. The neoplastic cells exhibited prominent nuclear atypia, multinucleation, frequent mitoses, and focal necrosis. By immunohistochemistry, the tumor cells were diffusely positive for vimentin, CD68, and CD34 and were negative for pankeratin, CK7, CAM 5.2, CD30, CD15, tyrosine, melan-a, HMB-45, epithelial membrane antigen, keratin 903, desmin protein, CD56, CD99, actin protein, and CD1a. A differential diagnosis included malignant fibrous histiocytoma and histiocytic sarcoma. 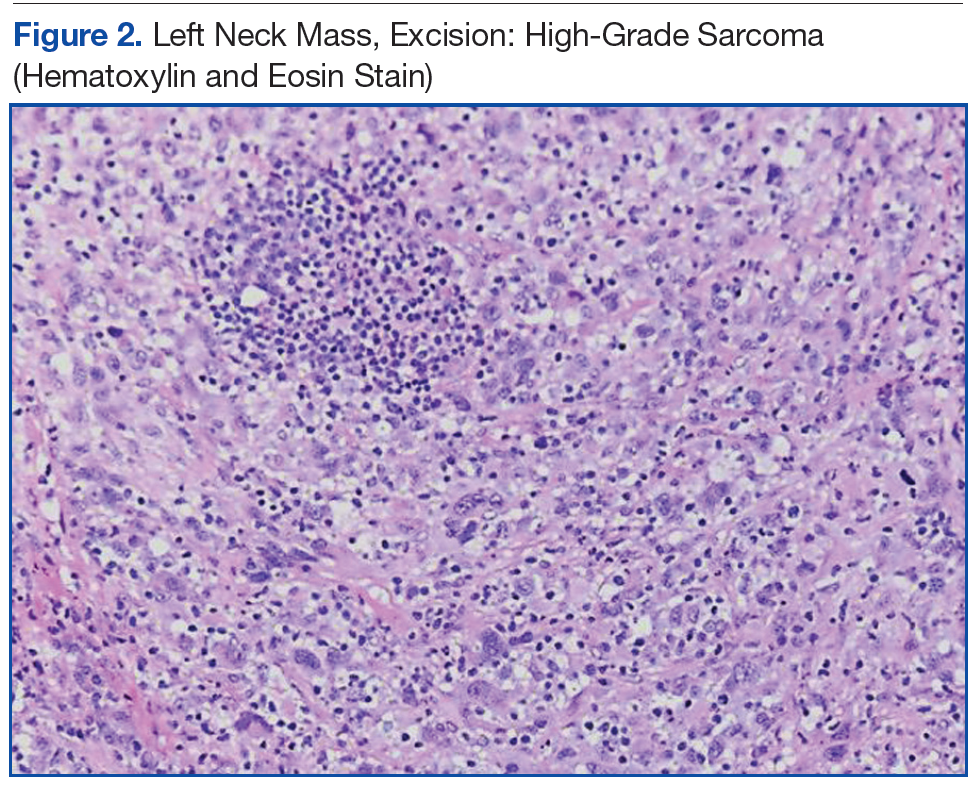
Knowing the patient had 2 tumors, a percutaneous lung biopsy was scheduled. It was assumed that the lung mass would be metastatic from esophagus vs sarcoma, but the biopsy showed small cell cancer (Figure 3). The tumor was composed of small cells with finely granular chromatin and a sparse amount of cytoplasm. By immunohistochemistry, the tumor cells were positive for TTF-1, CK7, CAM 5.2, CD56, and synaptophysin and were negative for CK5, CK6, and p63. The patient was represented at the tumor board with diagnostic recommendations to undergo a magnetic resonance imaging of the brain (negative for metastasis) and a PET/CT scan (stable disease). 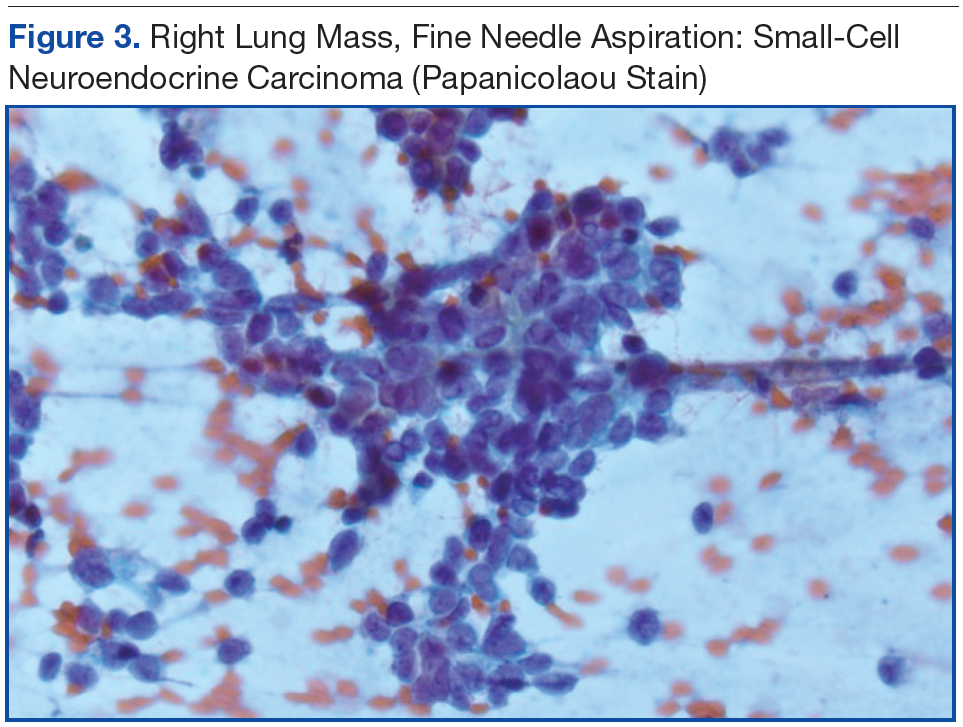
A lengthy discussion ensued regarding treatment for the 3 cancers. The AJCC Cancer Staging Manual, 7th edition, was used as the framework for all staging.1 The patient had a stage IIA (T1bN0MX), grade 3 sarcoma, which could be removed by neck dissection. He had a stage IIIB esophageal carcinoma (T3N2MX), which would require concurrent chemoradiation. His limited stage small cell lung cancer might have been amenable to a lobectomy if a mediastinoscopy was negative. The standard of care for limited stage small cell lung cancer is that if the cancer is precisely staged and the patient is a good surgical candidate, then surgery can be considered. The rationale for considering resection in this case was that if his other cancers could be cured, then surgery would be an excellent option.2 These options were presented to the patient, who opted for chemotherapy. The oncologist treating the patient elected to target the small cell cancer initially, because it was likely the fastest growing cancer. The patient started treatment with etoposide/carboplatin shortly thereafter. Mr. F. was also referred to palliative care.
Discussion
When a patient presents with several tumor masses detected either clinically or radiologically, it is reasonable to assume, at least initially, that all the lesions can be attributed to the same disease (ie, a primary neoplasm and distant metastases). Based on this assumption, it is rational to establish a tissue diagnosis by sampling the most accessible lesion. In the correct clinical context, this may be sufficient to initiate an appropriate therapy. However, health care providers should be aware that unrelated neoplasms can develop simultaneously, and about 8% of patients have > 1 malignancy at presentation.3
This patient presented with 3 different malignant neoplasms: esophageal adenocarcinoma, pulmonary small-cell carcinoma, and high-grade sarcoma. These types of tumors have completely different histogenesis. Esophageal adenocarcinoma develops from glandular cells of Barrett’s esophagus, small-cell carcinoma develops from neuroendocrine Kulchitsky cells, and sarcoma arises from mesenchymal cells. This constellation of neoplasms does not represent any known multicancer syndrome, such as Li-Fraumeni syndrome or Lynch syndrome. Whether these concomitant tumors represent coincidental neoplasms or have common underlying molecular alterations remains unknown.
The treatment for each of this patient’s cancers was very different and required a multidisciplinary, staged approach for management. This could have been arranged for him had he wanted to pursue multiple resections of the neck and lung plus have chemotherapy and radiation for the esophageal cancer. The patient opted for chemotherapy for the most aggressive cancer, but other options would have been possible had he wanted to pursue them.
Conclusion
This complex case demonstrates the role for careful review of all data and the importance of not making assumptions when analyzing images and history despite all the procedures and time to diagnosis.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Edge S, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A, eds. AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer-Verlag; 2010.
2. Koletsis EN, Prokakis C, Karanikolas M, Apostolakis E, Dougenis D. Current role of surgery in small cell lung carcinoma. J Cardiothorac Surg. 2009;4:30.
3. Brock MV, Alberg AJ, Hooker CM, et al. Risk of subsequent primary neoplasms developing in lung cancer patients with prior malignancies. J Thorac Cardiovasc Surg. 2004;127(4):1119-1125.
Mr. F. is a 67-year-old man with a medical history significant for hypertension, hyperlipidemia, gastroesophageal reflux disease, abdominal aortic aneurysm repair, and lung nodules (8-mm nodules in his right lower lobe and left upper lobe) since 2009 for which he had been followed in the pulmonary clinic. His last radiologic imaging had been performed in November 2010. The patient had failed to show for a serial scan and follow-up pulmonary appointments. He had seen his primary care practitioner annually. He reporteded a 60 pack-year smoking history and drank up to 10 to 15 beers a day, 3 days a week. He also reported exposure to asbestos while in the U.S. Navy as well as having worked in a sheet metal yard. He reported no family history of lung cancer.
In January 2014, Mr. F. presented to his primary care doctor with a nonpainful neck lump noted by the patient for a few months. On physical examination, the patient appeared healthy and without change in weight, loss of appetite, dysphagia, hemoptysis, or any other remarkable symptoms except for the pain in the left-sided neck mass. On oral examination, the patient did not have any oral lesions but was noted to have poor dentition. He had decreased breath sounds on the right side but no adventitious breath sounds. His cardiac status was within normal limits with a regular heart rate, warm skin, and well-perfused extremities. His abdomen was soft and nontender with good bowel sounds.
A neck computed tomography (CT) done on January 17, 2014, showed 2 left neck masses in the tail of the parotid gland or in level 2 adjacent to the parotid gland. The ear, nose, and throat (ENT) clinic noted that he had a 3-cm level 2 mass, which was mobile and nontender. A fine needle aspiration biopsy showed poorly differentiated rare malignant cells, but the small size of the biopsy precluded identification of the cells. For that reason, the patient was scheduled for an operative laryngoscopy.
A combined positron emission tomography and CT (PET/CT) scan on February 5, 2014, showed multiple areas of hypermetabolic activity. A highly metabolic 2.6 x 1.9-cm mass was noted in the left neck inseparable from the inferior border of the left parotid gland; whether this represented a parotid neoplasm or neoplastic lymph node could not be determined. Positive level 5 neck lymph nodes were suggestive of metastatic disease. There were also 2 foci of mild nonspecific metabolic activity in the right parotid gland. A spiculated mass in the right lower lung, measuring 2.3 x 3.5 cm, demonstrated intense fludeoxyglucose F 18 (FDG) uptake with maximum standardized uptake value (SUV) of 12.7, which was consistent with neoplasm. Thickening of the middistal esophageal wall with intense FDG uptake (SUV of 7.9) was suggestive of esophageal carcinoma.
The patient underwent an endoscopic ultrasound (EUS) esophageal biopsy, which demonstrated poorly differentiated adenocarcinoma (Figure 1). The tumor seemed to have originated at this site, as the biopsy fragments also contained foci of Barrett’s metaplasia and in situ carcinoma. He was presented in the hospital tumor board about whether he should undergo a high-risk lung biopsy due to the location of the mass or have his left neck mass rebiopsied, assuming that he had metastatic esophageal cancer in both the lung and neck. The safer procedure of redoing the neck biopsy in the Ear, Nose, and Throat clinic was pursued, and the patient underwent a biopsy and surgical excision of his left neck mass with positive pathology of highgrade sarcoma (Figure 2).
The tumor consisted of spindle and epithelioid cell proliferation within a lymph node. The neoplastic cells exhibited prominent nuclear atypia, multinucleation, frequent mitoses, and focal necrosis. By immunohistochemistry, the tumor cells were diffusely positive for vimentin, CD68, and CD34 and were negative for pankeratin, CK7, CAM 5.2, CD30, CD15, tyrosine, melan-a, HMB-45, epithelial membrane antigen, keratin 903, desmin protein, CD56, CD99, actin protein, and CD1a. A differential diagnosis included malignant fibrous histiocytoma and histiocytic sarcoma.
Knowing the patient had 2 tumors, a percutaneous lung biopsy was scheduled. It was assumed that the lung mass would be metastatic from esophagus vs sarcoma, but the biopsy showed small cell cancer (Figure 3). The tumor was composed of small cells with finely granular chromatin and a sparse amount of cytoplasm. By immunohistochemistry, the tumor cells were positive for TTF-1, CK7, CAM 5.2, CD56, and synaptophysin and were negative for CK5, CK6, and p63. The patient was represented at the tumor board with diagnostic recommendations to undergo a magnetic resonance imaging of the brain (negative for metastasis) and a PET/CT scan (stable disease).
A lengthy discussion ensued regarding treatment for the 3 cancers. The AJCC Cancer Staging Manual, 7th edition, was used as the framework for all staging.1 The patient had a stage IIA (T1bN0MX), grade 3 sarcoma, which could be removed by neck dissection. He had a stage IIIB esophageal carcinoma (T3N2MX), which would require concurrent chemoradiation. His limited stage small cell lung cancer might have been amenable to a lobectomy if a mediastinoscopy was negative. The standard of care for limited stage small cell lung cancer is that if the cancer is precisely staged and the patient is a good surgical candidate, then surgery can be considered. The rationale for considering resection in this case was that if his other cancers could be cured, then surgery would be an excellent option.2 These options were presented to the patient, who opted for chemotherapy. The oncologist treating the patient elected to target the small cell cancer initially, because it was likely the fastest growing cancer. The patient started treatment with etoposide/carboplatin shortly thereafter. Mr. F. was also referred to palliative care.
Discussion
When a patient presents with several tumor masses detected either clinically or radiologically, it is reasonable to assume, at least initially, that all the lesions can be attributed to the same disease (ie, a primary neoplasm and distant metastases). Based on this assumption, it is rational to establish a tissue diagnosis by sampling the most accessible lesion. In the correct clinical context, this may be sufficient to initiate an appropriate therapy. However, health care providers should be aware that unrelated neoplasms can develop simultaneously, and about 8% of patients have > 1 malignancy at presentation.3
This patient presented with 3 different malignant neoplasms: esophageal adenocarcinoma, pulmonary small-cell carcinoma, and high-grade sarcoma. These types of tumors have completely different histogenesis. Esophageal adenocarcinoma develops from glandular cells of Barrett’s esophagus, small-cell carcinoma develops from neuroendocrine Kulchitsky cells, and sarcoma arises from mesenchymal cells. This constellation of neoplasms does not represent any known multicancer syndrome, such as Li-Fraumeni syndrome or Lynch syndrome. Whether these concomitant tumors represent coincidental neoplasms or have common underlying molecular alterations remains unknown.
The treatment for each of this patient’s cancers was very different and required a multidisciplinary, staged approach for management. This could have been arranged for him had he wanted to pursue multiple resections of the neck and lung plus have chemotherapy and radiation for the esophageal cancer. The patient opted for chemotherapy for the most aggressive cancer, but other options would have been possible had he wanted to pursue them.
Conclusion
This complex case demonstrates the role for careful review of all data and the importance of not making assumptions when analyzing images and history despite all the procedures and time to diagnosis.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Mr. F. is a 67-year-old man with a medical history significant for hypertension, hyperlipidemia, gastroesophageal reflux disease, abdominal aortic aneurysm repair, and lung nodules (8-mm nodules in his right lower lobe and left upper lobe) since 2009 for which he had been followed in the pulmonary clinic. His last radiologic imaging had been performed in November 2010. The patient had failed to show for a serial scan and follow-up pulmonary appointments. He had seen his primary care practitioner annually. He reporteded a 60 pack-year smoking history and drank up to 10 to 15 beers a day, 3 days a week. He also reported exposure to asbestos while in the U.S. Navy as well as having worked in a sheet metal yard. He reported no family history of lung cancer.
In January 2014, Mr. F. presented to his primary care doctor with a nonpainful neck lump noted by the patient for a few months. On physical examination, the patient appeared healthy and without change in weight, loss of appetite, dysphagia, hemoptysis, or any other remarkable symptoms except for the pain in the left-sided neck mass. On oral examination, the patient did not have any oral lesions but was noted to have poor dentition. He had decreased breath sounds on the right side but no adventitious breath sounds. His cardiac status was within normal limits with a regular heart rate, warm skin, and well-perfused extremities. His abdomen was soft and nontender with good bowel sounds.
A neck computed tomography (CT) done on January 17, 2014, showed 2 left neck masses in the tail of the parotid gland or in level 2 adjacent to the parotid gland. The ear, nose, and throat (ENT) clinic noted that he had a 3-cm level 2 mass, which was mobile and nontender. A fine needle aspiration biopsy showed poorly differentiated rare malignant cells, but the small size of the biopsy precluded identification of the cells. For that reason, the patient was scheduled for an operative laryngoscopy.
A combined positron emission tomography and CT (PET/CT) scan on February 5, 2014, showed multiple areas of hypermetabolic activity. A highly metabolic 2.6 x 1.9-cm mass was noted in the left neck inseparable from the inferior border of the left parotid gland; whether this represented a parotid neoplasm or neoplastic lymph node could not be determined. Positive level 5 neck lymph nodes were suggestive of metastatic disease. There were also 2 foci of mild nonspecific metabolic activity in the right parotid gland. A spiculated mass in the right lower lung, measuring 2.3 x 3.5 cm, demonstrated intense fludeoxyglucose F 18 (FDG) uptake with maximum standardized uptake value (SUV) of 12.7, which was consistent with neoplasm. Thickening of the middistal esophageal wall with intense FDG uptake (SUV of 7.9) was suggestive of esophageal carcinoma.
The patient underwent an endoscopic ultrasound (EUS) esophageal biopsy, which demonstrated poorly differentiated adenocarcinoma (Figure 1). The tumor seemed to have originated at this site, as the biopsy fragments also contained foci of Barrett’s metaplasia and in situ carcinoma. He was presented in the hospital tumor board about whether he should undergo a high-risk lung biopsy due to the location of the mass or have his left neck mass rebiopsied, assuming that he had metastatic esophageal cancer in both the lung and neck. The safer procedure of redoing the neck biopsy in the Ear, Nose, and Throat clinic was pursued, and the patient underwent a biopsy and surgical excision of his left neck mass with positive pathology of highgrade sarcoma (Figure 2).
The tumor consisted of spindle and epithelioid cell proliferation within a lymph node. The neoplastic cells exhibited prominent nuclear atypia, multinucleation, frequent mitoses, and focal necrosis. By immunohistochemistry, the tumor cells were diffusely positive for vimentin, CD68, and CD34 and were negative for pankeratin, CK7, CAM 5.2, CD30, CD15, tyrosine, melan-a, HMB-45, epithelial membrane antigen, keratin 903, desmin protein, CD56, CD99, actin protein, and CD1a. A differential diagnosis included malignant fibrous histiocytoma and histiocytic sarcoma.
Knowing the patient had 2 tumors, a percutaneous lung biopsy was scheduled. It was assumed that the lung mass would be metastatic from esophagus vs sarcoma, but the biopsy showed small cell cancer (Figure 3). The tumor was composed of small cells with finely granular chromatin and a sparse amount of cytoplasm. By immunohistochemistry, the tumor cells were positive for TTF-1, CK7, CAM 5.2, CD56, and synaptophysin and were negative for CK5, CK6, and p63. The patient was represented at the tumor board with diagnostic recommendations to undergo a magnetic resonance imaging of the brain (negative for metastasis) and a PET/CT scan (stable disease).
A lengthy discussion ensued regarding treatment for the 3 cancers. The AJCC Cancer Staging Manual, 7th edition, was used as the framework for all staging.1 The patient had a stage IIA (T1bN0MX), grade 3 sarcoma, which could be removed by neck dissection. He had a stage IIIB esophageal carcinoma (T3N2MX), which would require concurrent chemoradiation. His limited stage small cell lung cancer might have been amenable to a lobectomy if a mediastinoscopy was negative. The standard of care for limited stage small cell lung cancer is that if the cancer is precisely staged and the patient is a good surgical candidate, then surgery can be considered. The rationale for considering resection in this case was that if his other cancers could be cured, then surgery would be an excellent option.2 These options were presented to the patient, who opted for chemotherapy. The oncologist treating the patient elected to target the small cell cancer initially, because it was likely the fastest growing cancer. The patient started treatment with etoposide/carboplatin shortly thereafter. Mr. F. was also referred to palliative care.
Discussion
When a patient presents with several tumor masses detected either clinically or radiologically, it is reasonable to assume, at least initially, that all the lesions can be attributed to the same disease (ie, a primary neoplasm and distant metastases). Based on this assumption, it is rational to establish a tissue diagnosis by sampling the most accessible lesion. In the correct clinical context, this may be sufficient to initiate an appropriate therapy. However, health care providers should be aware that unrelated neoplasms can develop simultaneously, and about 8% of patients have > 1 malignancy at presentation.3
This patient presented with 3 different malignant neoplasms: esophageal adenocarcinoma, pulmonary small-cell carcinoma, and high-grade sarcoma. These types of tumors have completely different histogenesis. Esophageal adenocarcinoma develops from glandular cells of Barrett’s esophagus, small-cell carcinoma develops from neuroendocrine Kulchitsky cells, and sarcoma arises from mesenchymal cells. This constellation of neoplasms does not represent any known multicancer syndrome, such as Li-Fraumeni syndrome or Lynch syndrome. Whether these concomitant tumors represent coincidental neoplasms or have common underlying molecular alterations remains unknown.
The treatment for each of this patient’s cancers was very different and required a multidisciplinary, staged approach for management. This could have been arranged for him had he wanted to pursue multiple resections of the neck and lung plus have chemotherapy and radiation for the esophageal cancer. The patient opted for chemotherapy for the most aggressive cancer, but other options would have been possible had he wanted to pursue them.
Conclusion
This complex case demonstrates the role for careful review of all data and the importance of not making assumptions when analyzing images and history despite all the procedures and time to diagnosis.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Edge S, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A, eds. AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer-Verlag; 2010.
2. Koletsis EN, Prokakis C, Karanikolas M, Apostolakis E, Dougenis D. Current role of surgery in small cell lung carcinoma. J Cardiothorac Surg. 2009;4:30.
3. Brock MV, Alberg AJ, Hooker CM, et al. Risk of subsequent primary neoplasms developing in lung cancer patients with prior malignancies. J Thorac Cardiovasc Surg. 2004;127(4):1119-1125.
1. Edge S, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A, eds. AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer-Verlag; 2010.
2. Koletsis EN, Prokakis C, Karanikolas M, Apostolakis E, Dougenis D. Current role of surgery in small cell lung carcinoma. J Cardiothorac Surg. 2009;4:30.
3. Brock MV, Alberg AJ, Hooker CM, et al. Risk of subsequent primary neoplasms developing in lung cancer patients with prior malignancies. J Thorac Cardiovasc Surg. 2004;127(4):1119-1125.
Single-, low-dose cyclophosphamide-associated severe hyponatremia with seizures in a patient with breast cancer
Cyclophosphamide, an agent used to treat various malignant and autoimmune disorders, can cause severe hyponatremia with seizures in rare cases. The exact mechanism of cyclophosphamide-induced hyponatremia is poorly understood, but is thought to occur from a drug- associated antidiuretic hormone (ADH) release leading to free water retention.1 This unusual phenomenon of cyclophosphamide-associated syndrome of inappropriate antidiuretic hormone secretion (SIADH) has been described only in case reports, most of which reported the development of severe hyponatremia within a week after administration of cyclophosphamide.2-5 We report a unique case of a patient who developed severe, symptomatic hyponatremia with seizures, with her serum sodium decreasing from 137 mEq to 112 mEq within 30 hours after her first dose of low-dose cyclophosphamide (600 mg/m2).
Case presentation and summary
A 68-year-old white woman with a history of bilateral invasive ductal carcinoma of the breast (status-post bilateral mastectomy) presented to the emergency department (ED) at our facility with new onset seizure. The patient had been diagnosed 8 months earlier with stage I (T1c, N0, M0) poorly differentiated infiltrating ductal carcinoma (triple negative) of the left breast for which she underwent left segmental mastectomy about 1 month after diagnosis. She was subsequently found to have progressive disease with stage IIIC (T2, N3, and M0) infiltrating ductal carcinoma with lobular features (ER/PR+, Her2) of the right breast. She underwent a right modified radical mastectomy 5 months after her stage IIIC breast cancer diagnosis. She received her first cycle of adjuvant chemotherapy with intravenous doxorubicin (60 mg/m2) and cyclophosphamide (600 mg/m2), which included pre-hydration, a day before presenting to our facility.
According to the patient’s family who provided the initial history, the patient reported tightness in her left arm while sitting at the dinner table. She was confused and subsequently had jerking movement of her right upper extremity with left facial twitching which lasted about 40 seconds. There was no loss of consciousness, or bowel or bladder control. She became unresponsive after the episode. Review of systems was negative except for a report of nausea a few hours before the onset of seizures, which resolved with ondansetron. Her past medical history was significant for breast cancer as already mentioned, seasonal allergic rhinitis, and hypertension. Home medications included hydrochlorothiazide 12.5 mg oral daily, aspirin 81 mg oral daily, and fexofenadine and loratadine oral daily as needed for allergies. There were no other significant surgical history other than already stated. The patient lived at home with her family and was independent with her instrumental activities of daily living. She is a former smoker of tobacco and quit smoking 30 years ago.
On arrival at our facility, the patient had normal vital signs. Significant findings on physical examination were an elderly female who seemed somnolent; not able to follow commands with a documented Glasgow Coma Scale of 10 with eyes opening spontaneously, incomprehensible sounds, and flexion withdrawal from pain as her best responses. She had an increased tone in her left upper extremity and had a brisk, deep tendon reflexes without clonus or 3+ (range, 0-5+, with 2+ being normal). The remainder of her physical exam was unremarkable. Laboratory testing revealed a glucose level of 120 mg/dL (normal, 65-110 mg/dL), sodium of 112 mEq/L (normal, 135-145 mmol/L), and chloride of 78 mEq/L (normal, 95-105 mmol/L). Serum osmolality and urine osmolality were 242 mOsm/kg (normal, 282-295 mOsm/kg) and 449 mOsm/kg (normal, 500-800 mOsm/kg) respectively, indicative of suboptimally dilute urine despite relatively low serum osmolality or SIADH. Urine electrolytes were not obtained.
Imaging studies including computed-tomography scans of the head and chest x-ray performed in the ED were unremarkable. After a phenytoin load, an electroencephalogram was obtained which showed diffuse encephalopathy without active seizure foci. A non-contrast magnetic-resonance imaging (MRI) of the brain was performed but it failed to show acute infarct, mass, mass effect, or brain herniation. There was nonspecific white matter abnormality with compromise of the bilateral cerebral hemispheres, calloseptal junction, left posterior pillar, and bilateral anterior pillars of the fornix, possibly representative of chronic white matter microvascular ischemic changes or less likely vasculitis or demyelination. Correction of her hyponatremia with normal saline was started in the ED with a change in serum sodium from 112 mEq/L to 115 mEq/L within 2 hours. She was admitted to the intensive care unit (ICU) where her sodium correction with normal saline and free water restriction was continued with a goal correction rate of 8-12 mEq/L in 24 hours. The patient’s serum sodium as well as level of consciousness improved gradually over the course of her ICU stay. After 64 hours in the hospital, her sodium had corrected to 137 mEq/L (normal, 135-145 mmol/L; Figure). She was then alert and oriented to person, place, and time. All motor findings noted on presentation had resolved. Her saline infusion was discontinued and serum sodium remained within normal range. She was discharged to a rehabilitation facility. Her hydrochlorothiazide was also discontinued.

Discussion
Hyponatremia is a common finding in cancer patients caused usually by paraneoplastic syndrome, chemotherapy, immunotherapy, or other associated treatment.6 SIADH is a frequent cause of hyponatremia in cancer patients and should be suspected in patients with hyponatremia, hypo-osmolality, and a urine osmolality above 100 mOsmol/kg.7
Our patient’s presentation and laboratory findings suggested SIADH as the likely cause of hyponatremia with a low sodium, a serum osmolality 242 mOsm/kg and urine osmolality of 449 mOsm/kg.8-10 She had no known underlying contributory comorbid condition relating to her serum lipids, thyroid, adrenal, kidney, or heart to date. Her use of a thiazide diuretic was the only confounding factor. The most plausible cause of hyponatremia/SIADH in our patient was likely cyclophosphamide based on her history, timeline of symptoms, and the absence of other possible causes. Though the mechanism for many of the previously mentioned etiologies are known, the mechanism of cyclophosphamide-induced SIADH is difficult to elucidate since the imminent complication of hemorrhagic cystitis means patients receiving this drug are often aggressively hydrated to prevent this complication.11,12 The result is that there is marked retention of water leading to potentially fatal hyponatremia in selected cases.11 This phenomenon has been fairly well described in patients receiving doses of 6 g/m2 as given in the STAMP protocol for stem cell mobilization or at doses of 30-50 mg/kg used to treat malignancy.12 Our patient clearly falls in this category given that she received a dose of 600 mg/m2. We found no evidence in her history to suggest post-operative, genetic or other cause for her hyponatremia. Our case mirrors a report by Koo and colleagues who described severe hyponatremia occurring within 24 hours following a single dose of intravenous cyclophosphamide 700 mg followed by saline infusion.13 In the case reported by Jayachandra and colleagues in which suspected cyclophosphamide-induced hyponatremia led to seizures, the patient received 500 mg IV of cyclophosphamide and had serum sodium as low as 106 mEq/L within a 24-hour period,2 similar to our patient.
There is a paucity of data on cyclophosphamide-induced SIADH. The mechanism by which cyclophosphamide causes SIADH is currently unknown. In addition, there are currently no set criteria that help identify at-risk patients who may develop such an event, including the dosage of cyclophosphamide that may trigger the SIADH, because lower doses of the drug have been associated with this complication.14
In a retrospective analysis by Lee and colleagues, cyclophosphamide-induced hyponatremia was found to be associated with male sex on a univariate analysis, but no risk factors were found in a multivariate analysis.15 It is likely that the concomitant use of diuretics, hydration, and high-dose cyclophosphamide contributed to hyponatremia/SIADH in our patient, though it is not clear through what mechanism. Harlow and colleagues proposed a mechanism for this phenomenon in 1979 based on the autopsy of a patient who had received high-dose cyclophosphamide involving degranulation of hypothalamic neurosecretory organelles and loss of Herring’s bodies. They inferred that metabolites of cyclophosphamide indirectly triggered inappropriate secretion of antidiuretic hormone as seen with a use of the structurally related analogue ifosfamide,16 but to our knowledge, this has yet to be replicated. Cyclophosphamide metabolite may have a direct tubular effect on the collecting duct epithelium leading to water retention15 as established by Campbell and colleagues. In one case, an established diabetes insipidus patient developed cyclophosphamide-induced antidiuresis without vasopressin secretion.17 It is imperative that the scientific community conduct research into the risk factors, underlying mechanisms, and methods of prevention to reduce and/or eliminate SIADH associated with use of cyclophosphamide.
1. Gilbar PJ, Richmond J, Wood J, Sullivan A. Syndrome of inappropriate antidiuretic hormone secretion induced by a single dose of oral cyclophosphamide. Ann Pharmacother. 2012.46(9):e23.
2. Jayachandran NV, Chandrasekhara PK, Thomas J, Agrawal S, Narsimulu G. Cyclophosphamide-associated complications: we need to be aware of SIADH and central pontine myelinolysis. Rheumatology (Oxford). 2009;48(1):89-90.
3. Baker M, Markman M, Niu J. Cyclophosphamide-induced severe acute hyponatremic encephalopathy in patients with breast cancer: report of two cases. Case Rep Oncol. 2014;7(2):550-554.
4. Lazarevic V, Hägg E, Wahlin A. Hiccups and severe hyponatremia associated with high-dose cyclophosphamide in conditioning regimen for allogeneic stem cell transplantation. Am J Hematol. 2007;82(1):88.
5. Geng C, Tang P, Zhang Y, Gao W. Hyponatremia induced by low-dose cyclophosphamide in two patients with breast cancer. Breast J. 2014; 20(4):442-443.
6. Kamoi K, Ebe T, Hasegawa A, et al. Hyponatremia in small cell lung cancer. Mechanisms not involving inappropriate ADH secretion. Cancer. 1987;60(5):1089-1093.
7. Matwiejczuk S, Püsküllüoğlu M, Zygulska AL. Oncological emergencies: syndrome of inappropriate antidiuretic hormone secretion (SIADH). Przegl Lek. 2014;71(10):541-543.
8. Robertson GL. Regulation of arginine vasopressin in the syndrome of inappropriate antidiuresis. Am J Med. 2006;119(7 Suppl 1):S36-42.
9. Robertson GL, Shelton RL, Athar S. The osmoregulation of vasopressin. Kidney Int. 1976;10(1):25-37.
10. Decaux G, Musch W. Clinical laboratory evaluation of the syndrome of inappropriate secretion of antidiuretic hormone. Clin J Am Soc Nephrol. 2008;3(4):1175-1184.
11. Bressler RB, Huston DP. Water intoxication following moderate-dose intravenous cyclophosphamide. Arch Intern Med. 1985;145(3):548-549.
12. Salido M, Macarron P, Hernández-García C, D’Cruz DP, Khamashta MA, Hughes GR. Water intoxication induced by low-dose cyclophosphamide in two patients with systemic lupus erythematosus. Lupus. 2003;12(8):636-639.
13. Koo TY, Bae SC, Park JS, et al. Water intoxication following low-dose intravenous cyclophosphamide. Electrolyte Blood Press. 2007;5(1):50-54.
14. [No authors listed]. Nausea and vasopressin. Lancet. 1991;337(8750):1133-1134.
15 Lee YC1, Park JS, Lee CH, et al. Hyponatraemia induced by low-dose intravenous pulse cyclophosphamide. Nephrol Dial Transplant. 2010;25(5):1520-1524.
16. Harlow PJ, DeClerck YA, Shore NA, Ortega JA, Carranza A, Heuser E. A fatal case of inappropriate ADH secretion induced by cyclophosphamide therapy. Cancer. 1979;44(3):896-898.
17. Campbell DM, Atkinson A, Gillis D, Sochett EB. Cyclophosphamide and water retention: mechanism revisited. J Pediatr Endocrinol Metab. 2000;13(6):673-675.
Cyclophosphamide, an agent used to treat various malignant and autoimmune disorders, can cause severe hyponatremia with seizures in rare cases. The exact mechanism of cyclophosphamide-induced hyponatremia is poorly understood, but is thought to occur from a drug- associated antidiuretic hormone (ADH) release leading to free water retention.1 This unusual phenomenon of cyclophosphamide-associated syndrome of inappropriate antidiuretic hormone secretion (SIADH) has been described only in case reports, most of which reported the development of severe hyponatremia within a week after administration of cyclophosphamide.2-5 We report a unique case of a patient who developed severe, symptomatic hyponatremia with seizures, with her serum sodium decreasing from 137 mEq to 112 mEq within 30 hours after her first dose of low-dose cyclophosphamide (600 mg/m2).
Case presentation and summary
A 68-year-old white woman with a history of bilateral invasive ductal carcinoma of the breast (status-post bilateral mastectomy) presented to the emergency department (ED) at our facility with new onset seizure. The patient had been diagnosed 8 months earlier with stage I (T1c, N0, M0) poorly differentiated infiltrating ductal carcinoma (triple negative) of the left breast for which she underwent left segmental mastectomy about 1 month after diagnosis. She was subsequently found to have progressive disease with stage IIIC (T2, N3, and M0) infiltrating ductal carcinoma with lobular features (ER/PR+, Her2) of the right breast. She underwent a right modified radical mastectomy 5 months after her stage IIIC breast cancer diagnosis. She received her first cycle of adjuvant chemotherapy with intravenous doxorubicin (60 mg/m2) and cyclophosphamide (600 mg/m2), which included pre-hydration, a day before presenting to our facility.
According to the patient’s family who provided the initial history, the patient reported tightness in her left arm while sitting at the dinner table. She was confused and subsequently had jerking movement of her right upper extremity with left facial twitching which lasted about 40 seconds. There was no loss of consciousness, or bowel or bladder control. She became unresponsive after the episode. Review of systems was negative except for a report of nausea a few hours before the onset of seizures, which resolved with ondansetron. Her past medical history was significant for breast cancer as already mentioned, seasonal allergic rhinitis, and hypertension. Home medications included hydrochlorothiazide 12.5 mg oral daily, aspirin 81 mg oral daily, and fexofenadine and loratadine oral daily as needed for allergies. There were no other significant surgical history other than already stated. The patient lived at home with her family and was independent with her instrumental activities of daily living. She is a former smoker of tobacco and quit smoking 30 years ago.
On arrival at our facility, the patient had normal vital signs. Significant findings on physical examination were an elderly female who seemed somnolent; not able to follow commands with a documented Glasgow Coma Scale of 10 with eyes opening spontaneously, incomprehensible sounds, and flexion withdrawal from pain as her best responses. She had an increased tone in her left upper extremity and had a brisk, deep tendon reflexes without clonus or 3+ (range, 0-5+, with 2+ being normal). The remainder of her physical exam was unremarkable. Laboratory testing revealed a glucose level of 120 mg/dL (normal, 65-110 mg/dL), sodium of 112 mEq/L (normal, 135-145 mmol/L), and chloride of 78 mEq/L (normal, 95-105 mmol/L). Serum osmolality and urine osmolality were 242 mOsm/kg (normal, 282-295 mOsm/kg) and 449 mOsm/kg (normal, 500-800 mOsm/kg) respectively, indicative of suboptimally dilute urine despite relatively low serum osmolality or SIADH. Urine electrolytes were not obtained.
Imaging studies including computed-tomography scans of the head and chest x-ray performed in the ED were unremarkable. After a phenytoin load, an electroencephalogram was obtained which showed diffuse encephalopathy without active seizure foci. A non-contrast magnetic-resonance imaging (MRI) of the brain was performed but it failed to show acute infarct, mass, mass effect, or brain herniation. There was nonspecific white matter abnormality with compromise of the bilateral cerebral hemispheres, calloseptal junction, left posterior pillar, and bilateral anterior pillars of the fornix, possibly representative of chronic white matter microvascular ischemic changes or less likely vasculitis or demyelination. Correction of her hyponatremia with normal saline was started in the ED with a change in serum sodium from 112 mEq/L to 115 mEq/L within 2 hours. She was admitted to the intensive care unit (ICU) where her sodium correction with normal saline and free water restriction was continued with a goal correction rate of 8-12 mEq/L in 24 hours. The patient’s serum sodium as well as level of consciousness improved gradually over the course of her ICU stay. After 64 hours in the hospital, her sodium had corrected to 137 mEq/L (normal, 135-145 mmol/L; Figure). She was then alert and oriented to person, place, and time. All motor findings noted on presentation had resolved. Her saline infusion was discontinued and serum sodium remained within normal range. She was discharged to a rehabilitation facility. Her hydrochlorothiazide was also discontinued.
Discussion
Hyponatremia is a common finding in cancer patients caused usually by paraneoplastic syndrome, chemotherapy, immunotherapy, or other associated treatment.6 SIADH is a frequent cause of hyponatremia in cancer patients and should be suspected in patients with hyponatremia, hypo-osmolality, and a urine osmolality above 100 mOsmol/kg.7
Our patient’s presentation and laboratory findings suggested SIADH as the likely cause of hyponatremia with a low sodium, a serum osmolality 242 mOsm/kg and urine osmolality of 449 mOsm/kg.8-10 She had no known underlying contributory comorbid condition relating to her serum lipids, thyroid, adrenal, kidney, or heart to date. Her use of a thiazide diuretic was the only confounding factor. The most plausible cause of hyponatremia/SIADH in our patient was likely cyclophosphamide based on her history, timeline of symptoms, and the absence of other possible causes. Though the mechanism for many of the previously mentioned etiologies are known, the mechanism of cyclophosphamide-induced SIADH is difficult to elucidate since the imminent complication of hemorrhagic cystitis means patients receiving this drug are often aggressively hydrated to prevent this complication.11,12 The result is that there is marked retention of water leading to potentially fatal hyponatremia in selected cases.11 This phenomenon has been fairly well described in patients receiving doses of 6 g/m2 as given in the STAMP protocol for stem cell mobilization or at doses of 30-50 mg/kg used to treat malignancy.12 Our patient clearly falls in this category given that she received a dose of 600 mg/m2. We found no evidence in her history to suggest post-operative, genetic or other cause for her hyponatremia. Our case mirrors a report by Koo and colleagues who described severe hyponatremia occurring within 24 hours following a single dose of intravenous cyclophosphamide 700 mg followed by saline infusion.13 In the case reported by Jayachandra and colleagues in which suspected cyclophosphamide-induced hyponatremia led to seizures, the patient received 500 mg IV of cyclophosphamide and had serum sodium as low as 106 mEq/L within a 24-hour period,2 similar to our patient.
There is a paucity of data on cyclophosphamide-induced SIADH. The mechanism by which cyclophosphamide causes SIADH is currently unknown. In addition, there are currently no set criteria that help identify at-risk patients who may develop such an event, including the dosage of cyclophosphamide that may trigger the SIADH, because lower doses of the drug have been associated with this complication.14
In a retrospective analysis by Lee and colleagues, cyclophosphamide-induced hyponatremia was found to be associated with male sex on a univariate analysis, but no risk factors were found in a multivariate analysis.15 It is likely that the concomitant use of diuretics, hydration, and high-dose cyclophosphamide contributed to hyponatremia/SIADH in our patient, though it is not clear through what mechanism. Harlow and colleagues proposed a mechanism for this phenomenon in 1979 based on the autopsy of a patient who had received high-dose cyclophosphamide involving degranulation of hypothalamic neurosecretory organelles and loss of Herring’s bodies. They inferred that metabolites of cyclophosphamide indirectly triggered inappropriate secretion of antidiuretic hormone as seen with a use of the structurally related analogue ifosfamide,16 but to our knowledge, this has yet to be replicated. Cyclophosphamide metabolite may have a direct tubular effect on the collecting duct epithelium leading to water retention15 as established by Campbell and colleagues. In one case, an established diabetes insipidus patient developed cyclophosphamide-induced antidiuresis without vasopressin secretion.17 It is imperative that the scientific community conduct research into the risk factors, underlying mechanisms, and methods of prevention to reduce and/or eliminate SIADH associated with use of cyclophosphamide.
Cyclophosphamide, an agent used to treat various malignant and autoimmune disorders, can cause severe hyponatremia with seizures in rare cases. The exact mechanism of cyclophosphamide-induced hyponatremia is poorly understood, but is thought to occur from a drug- associated antidiuretic hormone (ADH) release leading to free water retention.1 This unusual phenomenon of cyclophosphamide-associated syndrome of inappropriate antidiuretic hormone secretion (SIADH) has been described only in case reports, most of which reported the development of severe hyponatremia within a week after administration of cyclophosphamide.2-5 We report a unique case of a patient who developed severe, symptomatic hyponatremia with seizures, with her serum sodium decreasing from 137 mEq to 112 mEq within 30 hours after her first dose of low-dose cyclophosphamide (600 mg/m2).
Case presentation and summary
A 68-year-old white woman with a history of bilateral invasive ductal carcinoma of the breast (status-post bilateral mastectomy) presented to the emergency department (ED) at our facility with new onset seizure. The patient had been diagnosed 8 months earlier with stage I (T1c, N0, M0) poorly differentiated infiltrating ductal carcinoma (triple negative) of the left breast for which she underwent left segmental mastectomy about 1 month after diagnosis. She was subsequently found to have progressive disease with stage IIIC (T2, N3, and M0) infiltrating ductal carcinoma with lobular features (ER/PR+, Her2) of the right breast. She underwent a right modified radical mastectomy 5 months after her stage IIIC breast cancer diagnosis. She received her first cycle of adjuvant chemotherapy with intravenous doxorubicin (60 mg/m2) and cyclophosphamide (600 mg/m2), which included pre-hydration, a day before presenting to our facility.
According to the patient’s family who provided the initial history, the patient reported tightness in her left arm while sitting at the dinner table. She was confused and subsequently had jerking movement of her right upper extremity with left facial twitching which lasted about 40 seconds. There was no loss of consciousness, or bowel or bladder control. She became unresponsive after the episode. Review of systems was negative except for a report of nausea a few hours before the onset of seizures, which resolved with ondansetron. Her past medical history was significant for breast cancer as already mentioned, seasonal allergic rhinitis, and hypertension. Home medications included hydrochlorothiazide 12.5 mg oral daily, aspirin 81 mg oral daily, and fexofenadine and loratadine oral daily as needed for allergies. There were no other significant surgical history other than already stated. The patient lived at home with her family and was independent with her instrumental activities of daily living. She is a former smoker of tobacco and quit smoking 30 years ago.
On arrival at our facility, the patient had normal vital signs. Significant findings on physical examination were an elderly female who seemed somnolent; not able to follow commands with a documented Glasgow Coma Scale of 10 with eyes opening spontaneously, incomprehensible sounds, and flexion withdrawal from pain as her best responses. She had an increased tone in her left upper extremity and had a brisk, deep tendon reflexes without clonus or 3+ (range, 0-5+, with 2+ being normal). The remainder of her physical exam was unremarkable. Laboratory testing revealed a glucose level of 120 mg/dL (normal, 65-110 mg/dL), sodium of 112 mEq/L (normal, 135-145 mmol/L), and chloride of 78 mEq/L (normal, 95-105 mmol/L). Serum osmolality and urine osmolality were 242 mOsm/kg (normal, 282-295 mOsm/kg) and 449 mOsm/kg (normal, 500-800 mOsm/kg) respectively, indicative of suboptimally dilute urine despite relatively low serum osmolality or SIADH. Urine electrolytes were not obtained.
Imaging studies including computed-tomography scans of the head and chest x-ray performed in the ED were unremarkable. After a phenytoin load, an electroencephalogram was obtained which showed diffuse encephalopathy without active seizure foci. A non-contrast magnetic-resonance imaging (MRI) of the brain was performed but it failed to show acute infarct, mass, mass effect, or brain herniation. There was nonspecific white matter abnormality with compromise of the bilateral cerebral hemispheres, calloseptal junction, left posterior pillar, and bilateral anterior pillars of the fornix, possibly representative of chronic white matter microvascular ischemic changes or less likely vasculitis or demyelination. Correction of her hyponatremia with normal saline was started in the ED with a change in serum sodium from 112 mEq/L to 115 mEq/L within 2 hours. She was admitted to the intensive care unit (ICU) where her sodium correction with normal saline and free water restriction was continued with a goal correction rate of 8-12 mEq/L in 24 hours. The patient’s serum sodium as well as level of consciousness improved gradually over the course of her ICU stay. After 64 hours in the hospital, her sodium had corrected to 137 mEq/L (normal, 135-145 mmol/L; Figure). She was then alert and oriented to person, place, and time. All motor findings noted on presentation had resolved. Her saline infusion was discontinued and serum sodium remained within normal range. She was discharged to a rehabilitation facility. Her hydrochlorothiazide was also discontinued.
Discussion
Hyponatremia is a common finding in cancer patients caused usually by paraneoplastic syndrome, chemotherapy, immunotherapy, or other associated treatment.6 SIADH is a frequent cause of hyponatremia in cancer patients and should be suspected in patients with hyponatremia, hypo-osmolality, and a urine osmolality above 100 mOsmol/kg.7
Our patient’s presentation and laboratory findings suggested SIADH as the likely cause of hyponatremia with a low sodium, a serum osmolality 242 mOsm/kg and urine osmolality of 449 mOsm/kg.8-10 She had no known underlying contributory comorbid condition relating to her serum lipids, thyroid, adrenal, kidney, or heart to date. Her use of a thiazide diuretic was the only confounding factor. The most plausible cause of hyponatremia/SIADH in our patient was likely cyclophosphamide based on her history, timeline of symptoms, and the absence of other possible causes. Though the mechanism for many of the previously mentioned etiologies are known, the mechanism of cyclophosphamide-induced SIADH is difficult to elucidate since the imminent complication of hemorrhagic cystitis means patients receiving this drug are often aggressively hydrated to prevent this complication.11,12 The result is that there is marked retention of water leading to potentially fatal hyponatremia in selected cases.11 This phenomenon has been fairly well described in patients receiving doses of 6 g/m2 as given in the STAMP protocol for stem cell mobilization or at doses of 30-50 mg/kg used to treat malignancy.12 Our patient clearly falls in this category given that she received a dose of 600 mg/m2. We found no evidence in her history to suggest post-operative, genetic or other cause for her hyponatremia. Our case mirrors a report by Koo and colleagues who described severe hyponatremia occurring within 24 hours following a single dose of intravenous cyclophosphamide 700 mg followed by saline infusion.13 In the case reported by Jayachandra and colleagues in which suspected cyclophosphamide-induced hyponatremia led to seizures, the patient received 500 mg IV of cyclophosphamide and had serum sodium as low as 106 mEq/L within a 24-hour period,2 similar to our patient.
There is a paucity of data on cyclophosphamide-induced SIADH. The mechanism by which cyclophosphamide causes SIADH is currently unknown. In addition, there are currently no set criteria that help identify at-risk patients who may develop such an event, including the dosage of cyclophosphamide that may trigger the SIADH, because lower doses of the drug have been associated with this complication.14
In a retrospective analysis by Lee and colleagues, cyclophosphamide-induced hyponatremia was found to be associated with male sex on a univariate analysis, but no risk factors were found in a multivariate analysis.15 It is likely that the concomitant use of diuretics, hydration, and high-dose cyclophosphamide contributed to hyponatremia/SIADH in our patient, though it is not clear through what mechanism. Harlow and colleagues proposed a mechanism for this phenomenon in 1979 based on the autopsy of a patient who had received high-dose cyclophosphamide involving degranulation of hypothalamic neurosecretory organelles and loss of Herring’s bodies. They inferred that metabolites of cyclophosphamide indirectly triggered inappropriate secretion of antidiuretic hormone as seen with a use of the structurally related analogue ifosfamide,16 but to our knowledge, this has yet to be replicated. Cyclophosphamide metabolite may have a direct tubular effect on the collecting duct epithelium leading to water retention15 as established by Campbell and colleagues. In one case, an established diabetes insipidus patient developed cyclophosphamide-induced antidiuresis without vasopressin secretion.17 It is imperative that the scientific community conduct research into the risk factors, underlying mechanisms, and methods of prevention to reduce and/or eliminate SIADH associated with use of cyclophosphamide.
1. Gilbar PJ, Richmond J, Wood J, Sullivan A. Syndrome of inappropriate antidiuretic hormone secretion induced by a single dose of oral cyclophosphamide. Ann Pharmacother. 2012.46(9):e23.
2. Jayachandran NV, Chandrasekhara PK, Thomas J, Agrawal S, Narsimulu G. Cyclophosphamide-associated complications: we need to be aware of SIADH and central pontine myelinolysis. Rheumatology (Oxford). 2009;48(1):89-90.
3. Baker M, Markman M, Niu J. Cyclophosphamide-induced severe acute hyponatremic encephalopathy in patients with breast cancer: report of two cases. Case Rep Oncol. 2014;7(2):550-554.
4. Lazarevic V, Hägg E, Wahlin A. Hiccups and severe hyponatremia associated with high-dose cyclophosphamide in conditioning regimen for allogeneic stem cell transplantation. Am J Hematol. 2007;82(1):88.
5. Geng C, Tang P, Zhang Y, Gao W. Hyponatremia induced by low-dose cyclophosphamide in two patients with breast cancer. Breast J. 2014; 20(4):442-443.
6. Kamoi K, Ebe T, Hasegawa A, et al. Hyponatremia in small cell lung cancer. Mechanisms not involving inappropriate ADH secretion. Cancer. 1987;60(5):1089-1093.
7. Matwiejczuk S, Püsküllüoğlu M, Zygulska AL. Oncological emergencies: syndrome of inappropriate antidiuretic hormone secretion (SIADH). Przegl Lek. 2014;71(10):541-543.
8. Robertson GL. Regulation of arginine vasopressin in the syndrome of inappropriate antidiuresis. Am J Med. 2006;119(7 Suppl 1):S36-42.
9. Robertson GL, Shelton RL, Athar S. The osmoregulation of vasopressin. Kidney Int. 1976;10(1):25-37.
10. Decaux G, Musch W. Clinical laboratory evaluation of the syndrome of inappropriate secretion of antidiuretic hormone. Clin J Am Soc Nephrol. 2008;3(4):1175-1184.
11. Bressler RB, Huston DP. Water intoxication following moderate-dose intravenous cyclophosphamide. Arch Intern Med. 1985;145(3):548-549.
12. Salido M, Macarron P, Hernández-García C, D’Cruz DP, Khamashta MA, Hughes GR. Water intoxication induced by low-dose cyclophosphamide in two patients with systemic lupus erythematosus. Lupus. 2003;12(8):636-639.
13. Koo TY, Bae SC, Park JS, et al. Water intoxication following low-dose intravenous cyclophosphamide. Electrolyte Blood Press. 2007;5(1):50-54.
14. [No authors listed]. Nausea and vasopressin. Lancet. 1991;337(8750):1133-1134.
15 Lee YC1, Park JS, Lee CH, et al. Hyponatraemia induced by low-dose intravenous pulse cyclophosphamide. Nephrol Dial Transplant. 2010;25(5):1520-1524.
16. Harlow PJ, DeClerck YA, Shore NA, Ortega JA, Carranza A, Heuser E. A fatal case of inappropriate ADH secretion induced by cyclophosphamide therapy. Cancer. 1979;44(3):896-898.
17. Campbell DM, Atkinson A, Gillis D, Sochett EB. Cyclophosphamide and water retention: mechanism revisited. J Pediatr Endocrinol Metab. 2000;13(6):673-675.
1. Gilbar PJ, Richmond J, Wood J, Sullivan A. Syndrome of inappropriate antidiuretic hormone secretion induced by a single dose of oral cyclophosphamide. Ann Pharmacother. 2012.46(9):e23.
2. Jayachandran NV, Chandrasekhara PK, Thomas J, Agrawal S, Narsimulu G. Cyclophosphamide-associated complications: we need to be aware of SIADH and central pontine myelinolysis. Rheumatology (Oxford). 2009;48(1):89-90.
3. Baker M, Markman M, Niu J. Cyclophosphamide-induced severe acute hyponatremic encephalopathy in patients with breast cancer: report of two cases. Case Rep Oncol. 2014;7(2):550-554.
4. Lazarevic V, Hägg E, Wahlin A. Hiccups and severe hyponatremia associated with high-dose cyclophosphamide in conditioning regimen for allogeneic stem cell transplantation. Am J Hematol. 2007;82(1):88.
5. Geng C, Tang P, Zhang Y, Gao W. Hyponatremia induced by low-dose cyclophosphamide in two patients with breast cancer. Breast J. 2014; 20(4):442-443.
6. Kamoi K, Ebe T, Hasegawa A, et al. Hyponatremia in small cell lung cancer. Mechanisms not involving inappropriate ADH secretion. Cancer. 1987;60(5):1089-1093.
7. Matwiejczuk S, Püsküllüoğlu M, Zygulska AL. Oncological emergencies: syndrome of inappropriate antidiuretic hormone secretion (SIADH). Przegl Lek. 2014;71(10):541-543.
8. Robertson GL. Regulation of arginine vasopressin in the syndrome of inappropriate antidiuresis. Am J Med. 2006;119(7 Suppl 1):S36-42.
9. Robertson GL, Shelton RL, Athar S. The osmoregulation of vasopressin. Kidney Int. 1976;10(1):25-37.
10. Decaux G, Musch W. Clinical laboratory evaluation of the syndrome of inappropriate secretion of antidiuretic hormone. Clin J Am Soc Nephrol. 2008;3(4):1175-1184.
11. Bressler RB, Huston DP. Water intoxication following moderate-dose intravenous cyclophosphamide. Arch Intern Med. 1985;145(3):548-549.
12. Salido M, Macarron P, Hernández-García C, D’Cruz DP, Khamashta MA, Hughes GR. Water intoxication induced by low-dose cyclophosphamide in two patients with systemic lupus erythematosus. Lupus. 2003;12(8):636-639.
13. Koo TY, Bae SC, Park JS, et al. Water intoxication following low-dose intravenous cyclophosphamide. Electrolyte Blood Press. 2007;5(1):50-54.
14. [No authors listed]. Nausea and vasopressin. Lancet. 1991;337(8750):1133-1134.
15 Lee YC1, Park JS, Lee CH, et al. Hyponatraemia induced by low-dose intravenous pulse cyclophosphamide. Nephrol Dial Transplant. 2010;25(5):1520-1524.
16. Harlow PJ, DeClerck YA, Shore NA, Ortega JA, Carranza A, Heuser E. A fatal case of inappropriate ADH secretion induced by cyclophosphamide therapy. Cancer. 1979;44(3):896-898.
17. Campbell DM, Atkinson A, Gillis D, Sochett EB. Cyclophosphamide and water retention: mechanism revisited. J Pediatr Endocrinol Metab. 2000;13(6):673-675.
Paraneoplastic leukemoid reaction – poor prognostic marker in urothelial bladder carcinoma
Certain cancers have been observed to cause symptoms called paraneoplastic syndromes that are not directly attributed to tumor invasion or compression. This phenomenon is believed to be secondary to a tumor’s secretion of functional hormones, peptides, cytokines, or its immune cross-reactivity. One such variant is a paraneoplastic leukemoid reaction (PLR), defined as a leukocytosis level of >50 x 103 cells/mm³, where the white blood cell (WBC) count differential exhibits a neutrophilia or left-shift, in which a predominance of early neutrophil precursors is observed. A PLR is believed to be incited by the tumor cell’s production of its own growth factors such as granulocyte-colony stimulating factor (G-CSF) and a number of different cytokines. These reactions may at first be mistaken for infectious processes, and it is only after an infection has been ruled out or when a leukocytosis is disproportionately high in the setting of a treated infection, that a paraneoplastic leukemoid reaction (PLR) is considered and an oncologic work-up pursued.
PLR has been previously described in a variety of malignancies including lung, esophageal, nasopharyngeal and laryngeal, gastric, cholangiocarcinoma, melanoma, multiple myeloma, renal, prostate, and hepatocellular carcinoma, but has rarely been described in urothelial carcinoma.1 Leukemoid reactions and autocrine growth induced by paraneoplastic production of G-CSF have rarely been associated with urothelial carcinoma of the bladder,2 as in the case we present here of a 67-year-old white man with invasive high-grade urothelial carcinoma of the bladder. The case highlights PLR as a negative prognostic marker, secondary to urothelial bladder cancer cells’ presumed production of G-CSF, rarely reported as in the literature previously.
Case presentation and summary
A 67-year-old white man was diagnosed with clinical stage III (T3N0M0), invasive high-grade urothelial carcinoma of the bladder (Figure 1). He received neoadjuvant chemotherapy with the standard gemcitabine-cisplatin combination (1,000 mg/m2 of gemcitabine on days 1, 8, and 15 with 70 mg/m2 of cisplatin on day 1 of a 28-day cycle for 3 cycles), and had less than partial response at the end of a 3-month course. A computed tomography scan of his pelvis obtained at treatment completion revealed persistent disease with noted enhancement of the right distal ureter and a right posterior bladder mass at the ureterovesical junction measuring 1.7 x 2.6 x 2.6 cm (0.6 x 1 x 1 in), for which, a cystoprostatectomy was recommended to remove remaining disease. The patient was seen in routine follow-up 3 weeks after his last chemotherapy treatment, when his WBC count was noted to be within normal limits at a value of 8.4 x 103 cells/mm³ (normal range, 4.5-11 x 103 cells/mm³).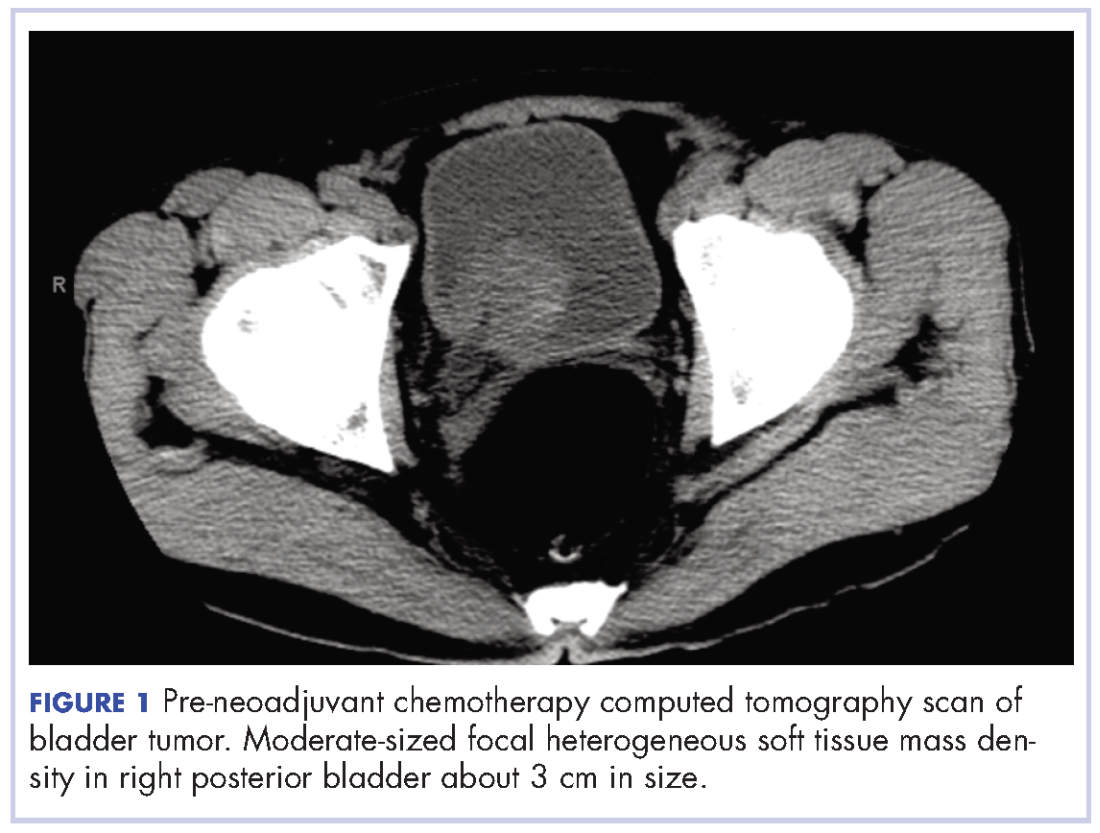
One week later (a month after treatment completion), the patient presented to the emergency department with complaints of dysuria, urinary frequency, and suprapubic pain. He was found to have leukocytosis, with a WBC count of 47 x 103 cells/mm³, (normal range, 4.5-11 x 103 cells/mm³), with an elevated neutrophil count of 82.7% (normal range 40%-60%), without clinical signs of systemic infection (fevers, chills, or rigors). A urinalysis revealed pyuria with 25-50 WBC/high power field, negative nitrite, positive leukocyte esterase, and moderate bacteria, consistent with what was presumed to be a urinary tract infection. The patient was discharged home with a 1-week course of the antibiotic levofloxacin and the alpha-blocker tamsulosin to make urination easier. Of note, the final results of the urine culture, which returned 48 hours after discharge, showed no growth.
One week prior to surgery, the patient underwent a cystoscopy for ureteral stent exchange, which revealed a necrotic tumor at the right bladder base surrounding the ureteral orifice and stent. Renal pelvis urine, sampled during stent exchange, revealed >100,000 CFU/ml (colony forming units; normal value, <10³) Candida albicans, for which the patient was started on intravenous fluconazole for fungal infection. We consulted with our colleagues in the infectious disease department and continued to follow the patient throughout his hospital course, which included several antibiotic regimens, a comprehensive hematological work-up and eventual urologic surgery. Work-ups for myeloproliferative disorders, leukemia, JAK-2, and BCR-ABL were all negative. A peripheral blood smear analysis showed mostly neutrophils, no immature cells, and occasional hypersegmented neutrophils, but was overall inconsistent with myeloproliferative disease. Despite the patient’s persistent leukocytosis, he remained completely asymptomatic and his neutrophilia was attributed to his malignancy.
The patient subsequently underwent a cystoprostatectomy with ileal conduit. The surgical pathologic analysis showed a high-grade, invasive urothelial (transitional cell) carcinoma measuring 5.2 x 5.0 x 4.5 cm (2 x 1.9 x 1.8 in) with squamous differentiation, extensive tumor necrosis, lymphovascular invasion, invasion into the adjacent seminal vesicle and prostatic stroma, and negative margins (pT4a pN0 pMX; Figure 2). On the day of surgical resection, the patient’s leukocytosis was 70 x 103 cells/mm³.
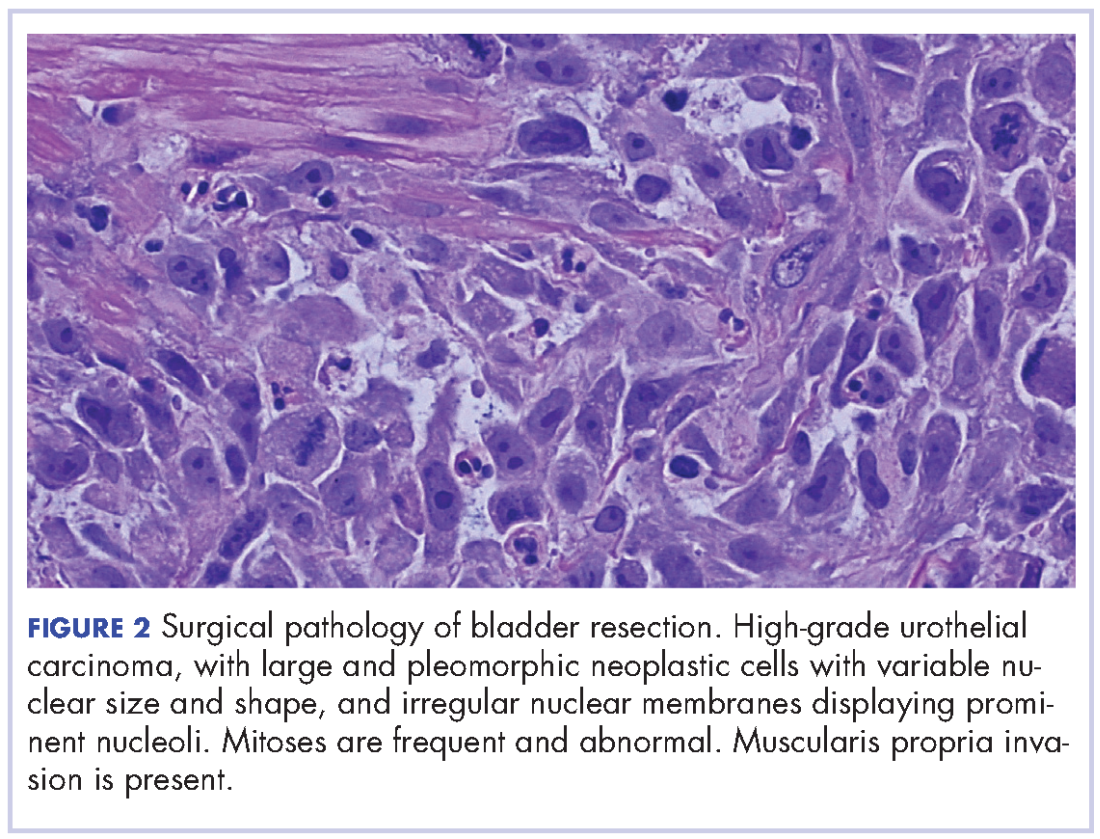
Despite a transient improvement in his leukocytosis to 37.7 x 103 cells/mm³ on postoperative day 1, the patient remained in the medical intensive care unit for uncontrolled pain and management of his leukocytosis. It is worth noting that the patient remained afebrile throughout his entire 45-day hospitalization, with negative culture data, and despite receiving an extensive broad spectrum antibiotic regimen (levofloxacin, piperacillin-tazobactam, cefazolin, metronidazole, ceftriaxone and fluconazole), his leukocytosis continued to progress, peaking at 161.5 x 103 cells/mm³ less than a month after his surgery (Figure 3).
The patient continued to deteriorate rapidly, with a progressive leukocytosis, and developing metastases to the lung, perineum, and penis. He died a month after surgery (2 months after completion of neoadjuvant chemotherapy). The leukocytosis exhibited in this patient and the aggressive tumor cell growth are believed to have been secondary to a paraneoplastic leukemoid reaction incited by the urothelial bladder cancer cells’ presumed production of G-CSF, which has been rarely reported in the literature previously.
Discussion
We report here on the rare occurrence of PLR in a urothelial bladder cancer. Several mechanisms have been proposed to explain the pathophysiology of PLR. The levels of IL-1[alpha and beta], IL-3, G-CSF, GM-CSF, IL-6, and TNF-[alpha] have all been reported to be elevated in various solid tumors and suggested to contribute to an elevated leukocyte count.3 With previous reports that receptors for G-CSF have been found on cell surfaces of several nonhematopoietic cell types, Tachibana and Murai have proposed the mechanism of a cancer cell’s simultaneous acquisition of the ligand promotion and its receptor expression conferring an autocrine growth advantage.4 They have also reported on the capability of bladder cancer cells to induce a leukemoid reaction in their host through the stimulation of leukocyte production, which has been associated with aggressive tumor cell growth and a poor clinical outcome. In addition, He and colleagues have also described the correlation between PLR and high degree of malignancy, high probability of metastasis, recurrence, and poor prognosis.5
We observed the leukocytosis of 70 x 103 cells/mm³ on the day of resection with a slight drop postoperatively, peaking at 161.5 x 103 cells/mm³ less than a month after resection of the tumor. There is no clear understanding of the cause of the persistent and rapid progression of leukocytosis seen in this patient postoperatively. There is also a dearth of literature describing similar occurrences, with even fewer attempting to explain the pathophysiology of this occurrence.
When faced with similar occurrences in patients, clinicians usually treat for occult infection. Once infections and myeloproliferative diseases have been ruled out, clinicians may consider obtaining a patient’s serum G-CSF level or performing an immunohistochemistry analysis of urothelial cells for overexpression of G-CSF, when available.5 However, despite any efforts to diagnose earlier, there is little clinicians currently have to offer these patients as treatment.
As presented in this report, PLR portends a worse prognosis for patients because of its ability to not only masquerade as an infection leading to a delay in the proper treatment, but also because of its association with a more aggressive tumor cell behavior and growth, making it critically important for clinicians to be able to identify these patients early on. With further investigation into immune regulation and G-CSF receptor signaling, there may be future discoveries of novel methods to diagnose this condition and also advancements in the treatment options made available to these patients.
1. Chakraborty S, Keenportz B, Woodward S, Anderson J, Colan D. Paraneoplastic leukemoid reaction in solid tumors. Am J Clin Oncol. 2015;38(3):326-330.
2. Kumar AK, Satyan MT, Holzbeierlein J, Mirza M, Van Veldhuizen P. Leukemoid reaction and autocrine growth of bladder cancer induced by paraneoplastic production of granulocyte colony-stimulating factor-a potential neoplastic marker: a case report and review of the literature. J Med Case Rep. 2014;8(1):147.
3. Azuma T, Sakai I, Matsumoto T, et al. Leukemoid reaction in association with bone marrow necrosis due to metastatic prostate cancer. Intern Med. 2005;44(10):1093-1096.
4. Tachibana M, Murai M. G-CSF production in human bladder cancer and its ability to promote autocrine growth: a review. Cytokines Cell Mol Ther. 1998;4(2):113-120.
5. He H, Zhang Z, Ge J, Zhou W. Leukemoid reaction associated with transitional cell carcinoma: a case report and literature review. Niger J Clin Pract. 2014;17(3):391-394.
Certain cancers have been observed to cause symptoms called paraneoplastic syndromes that are not directly attributed to tumor invasion or compression. This phenomenon is believed to be secondary to a tumor’s secretion of functional hormones, peptides, cytokines, or its immune cross-reactivity. One such variant is a paraneoplastic leukemoid reaction (PLR), defined as a leukocytosis level of >50 x 103 cells/mm³, where the white blood cell (WBC) count differential exhibits a neutrophilia or left-shift, in which a predominance of early neutrophil precursors is observed. A PLR is believed to be incited by the tumor cell’s production of its own growth factors such as granulocyte-colony stimulating factor (G-CSF) and a number of different cytokines. These reactions may at first be mistaken for infectious processes, and it is only after an infection has been ruled out or when a leukocytosis is disproportionately high in the setting of a treated infection, that a paraneoplastic leukemoid reaction (PLR) is considered and an oncologic work-up pursued.
PLR has been previously described in a variety of malignancies including lung, esophageal, nasopharyngeal and laryngeal, gastric, cholangiocarcinoma, melanoma, multiple myeloma, renal, prostate, and hepatocellular carcinoma, but has rarely been described in urothelial carcinoma.1 Leukemoid reactions and autocrine growth induced by paraneoplastic production of G-CSF have rarely been associated with urothelial carcinoma of the bladder,2 as in the case we present here of a 67-year-old white man with invasive high-grade urothelial carcinoma of the bladder. The case highlights PLR as a negative prognostic marker, secondary to urothelial bladder cancer cells’ presumed production of G-CSF, rarely reported as in the literature previously.
Case presentation and summary
A 67-year-old white man was diagnosed with clinical stage III (T3N0M0), invasive high-grade urothelial carcinoma of the bladder (Figure 1). He received neoadjuvant chemotherapy with the standard gemcitabine-cisplatin combination (1,000 mg/m2 of gemcitabine on days 1, 8, and 15 with 70 mg/m2 of cisplatin on day 1 of a 28-day cycle for 3 cycles), and had less than partial response at the end of a 3-month course. A computed tomography scan of his pelvis obtained at treatment completion revealed persistent disease with noted enhancement of the right distal ureter and a right posterior bladder mass at the ureterovesical junction measuring 1.7 x 2.6 x 2.6 cm (0.6 x 1 x 1 in), for which, a cystoprostatectomy was recommended to remove remaining disease. The patient was seen in routine follow-up 3 weeks after his last chemotherapy treatment, when his WBC count was noted to be within normal limits at a value of 8.4 x 103 cells/mm³ (normal range, 4.5-11 x 103 cells/mm³).
One week later (a month after treatment completion), the patient presented to the emergency department with complaints of dysuria, urinary frequency, and suprapubic pain. He was found to have leukocytosis, with a WBC count of 47 x 103 cells/mm³, (normal range, 4.5-11 x 103 cells/mm³), with an elevated neutrophil count of 82.7% (normal range 40%-60%), without clinical signs of systemic infection (fevers, chills, or rigors). A urinalysis revealed pyuria with 25-50 WBC/high power field, negative nitrite, positive leukocyte esterase, and moderate bacteria, consistent with what was presumed to be a urinary tract infection. The patient was discharged home with a 1-week course of the antibiotic levofloxacin and the alpha-blocker tamsulosin to make urination easier. Of note, the final results of the urine culture, which returned 48 hours after discharge, showed no growth.
One week prior to surgery, the patient underwent a cystoscopy for ureteral stent exchange, which revealed a necrotic tumor at the right bladder base surrounding the ureteral orifice and stent. Renal pelvis urine, sampled during stent exchange, revealed >100,000 CFU/ml (colony forming units; normal value, <10³) Candida albicans, for which the patient was started on intravenous fluconazole for fungal infection. We consulted with our colleagues in the infectious disease department and continued to follow the patient throughout his hospital course, which included several antibiotic regimens, a comprehensive hematological work-up and eventual urologic surgery. Work-ups for myeloproliferative disorders, leukemia, JAK-2, and BCR-ABL were all negative. A peripheral blood smear analysis showed mostly neutrophils, no immature cells, and occasional hypersegmented neutrophils, but was overall inconsistent with myeloproliferative disease. Despite the patient’s persistent leukocytosis, he remained completely asymptomatic and his neutrophilia was attributed to his malignancy.
The patient subsequently underwent a cystoprostatectomy with ileal conduit. The surgical pathologic analysis showed a high-grade, invasive urothelial (transitional cell) carcinoma measuring 5.2 x 5.0 x 4.5 cm (2 x 1.9 x 1.8 in) with squamous differentiation, extensive tumor necrosis, lymphovascular invasion, invasion into the adjacent seminal vesicle and prostatic stroma, and negative margins (pT4a pN0 pMX; Figure 2). On the day of surgical resection, the patient’s leukocytosis was 70 x 103 cells/mm³.
Despite a transient improvement in his leukocytosis to 37.7 x 103 cells/mm³ on postoperative day 1, the patient remained in the medical intensive care unit for uncontrolled pain and management of his leukocytosis. It is worth noting that the patient remained afebrile throughout his entire 45-day hospitalization, with negative culture data, and despite receiving an extensive broad spectrum antibiotic regimen (levofloxacin, piperacillin-tazobactam, cefazolin, metronidazole, ceftriaxone and fluconazole), his leukocytosis continued to progress, peaking at 161.5 x 103 cells/mm³ less than a month after his surgery (Figure 3).
The patient continued to deteriorate rapidly, with a progressive leukocytosis, and developing metastases to the lung, perineum, and penis. He died a month after surgery (2 months after completion of neoadjuvant chemotherapy). The leukocytosis exhibited in this patient and the aggressive tumor cell growth are believed to have been secondary to a paraneoplastic leukemoid reaction incited by the urothelial bladder cancer cells’ presumed production of G-CSF, which has been rarely reported in the literature previously.
Discussion
We report here on the rare occurrence of PLR in a urothelial bladder cancer. Several mechanisms have been proposed to explain the pathophysiology of PLR. The levels of IL-1[alpha and beta], IL-3, G-CSF, GM-CSF, IL-6, and TNF-[alpha] have all been reported to be elevated in various solid tumors and suggested to contribute to an elevated leukocyte count.3 With previous reports that receptors for G-CSF have been found on cell surfaces of several nonhematopoietic cell types, Tachibana and Murai have proposed the mechanism of a cancer cell’s simultaneous acquisition of the ligand promotion and its receptor expression conferring an autocrine growth advantage.4 They have also reported on the capability of bladder cancer cells to induce a leukemoid reaction in their host through the stimulation of leukocyte production, which has been associated with aggressive tumor cell growth and a poor clinical outcome. In addition, He and colleagues have also described the correlation between PLR and high degree of malignancy, high probability of metastasis, recurrence, and poor prognosis.5
We observed the leukocytosis of 70 x 103 cells/mm³ on the day of resection with a slight drop postoperatively, peaking at 161.5 x 103 cells/mm³ less than a month after resection of the tumor. There is no clear understanding of the cause of the persistent and rapid progression of leukocytosis seen in this patient postoperatively. There is also a dearth of literature describing similar occurrences, with even fewer attempting to explain the pathophysiology of this occurrence.
When faced with similar occurrences in patients, clinicians usually treat for occult infection. Once infections and myeloproliferative diseases have been ruled out, clinicians may consider obtaining a patient’s serum G-CSF level or performing an immunohistochemistry analysis of urothelial cells for overexpression of G-CSF, when available.5 However, despite any efforts to diagnose earlier, there is little clinicians currently have to offer these patients as treatment.
As presented in this report, PLR portends a worse prognosis for patients because of its ability to not only masquerade as an infection leading to a delay in the proper treatment, but also because of its association with a more aggressive tumor cell behavior and growth, making it critically important for clinicians to be able to identify these patients early on. With further investigation into immune regulation and G-CSF receptor signaling, there may be future discoveries of novel methods to diagnose this condition and also advancements in the treatment options made available to these patients.
Certain cancers have been observed to cause symptoms called paraneoplastic syndromes that are not directly attributed to tumor invasion or compression. This phenomenon is believed to be secondary to a tumor’s secretion of functional hormones, peptides, cytokines, or its immune cross-reactivity. One such variant is a paraneoplastic leukemoid reaction (PLR), defined as a leukocytosis level of >50 x 103 cells/mm³, where the white blood cell (WBC) count differential exhibits a neutrophilia or left-shift, in which a predominance of early neutrophil precursors is observed. A PLR is believed to be incited by the tumor cell’s production of its own growth factors such as granulocyte-colony stimulating factor (G-CSF) and a number of different cytokines. These reactions may at first be mistaken for infectious processes, and it is only after an infection has been ruled out or when a leukocytosis is disproportionately high in the setting of a treated infection, that a paraneoplastic leukemoid reaction (PLR) is considered and an oncologic work-up pursued.
PLR has been previously described in a variety of malignancies including lung, esophageal, nasopharyngeal and laryngeal, gastric, cholangiocarcinoma, melanoma, multiple myeloma, renal, prostate, and hepatocellular carcinoma, but has rarely been described in urothelial carcinoma.1 Leukemoid reactions and autocrine growth induced by paraneoplastic production of G-CSF have rarely been associated with urothelial carcinoma of the bladder,2 as in the case we present here of a 67-year-old white man with invasive high-grade urothelial carcinoma of the bladder. The case highlights PLR as a negative prognostic marker, secondary to urothelial bladder cancer cells’ presumed production of G-CSF, rarely reported as in the literature previously.
Case presentation and summary
A 67-year-old white man was diagnosed with clinical stage III (T3N0M0), invasive high-grade urothelial carcinoma of the bladder (Figure 1). He received neoadjuvant chemotherapy with the standard gemcitabine-cisplatin combination (1,000 mg/m2 of gemcitabine on days 1, 8, and 15 with 70 mg/m2 of cisplatin on day 1 of a 28-day cycle for 3 cycles), and had less than partial response at the end of a 3-month course. A computed tomography scan of his pelvis obtained at treatment completion revealed persistent disease with noted enhancement of the right distal ureter and a right posterior bladder mass at the ureterovesical junction measuring 1.7 x 2.6 x 2.6 cm (0.6 x 1 x 1 in), for which, a cystoprostatectomy was recommended to remove remaining disease. The patient was seen in routine follow-up 3 weeks after his last chemotherapy treatment, when his WBC count was noted to be within normal limits at a value of 8.4 x 103 cells/mm³ (normal range, 4.5-11 x 103 cells/mm³).
One week later (a month after treatment completion), the patient presented to the emergency department with complaints of dysuria, urinary frequency, and suprapubic pain. He was found to have leukocytosis, with a WBC count of 47 x 103 cells/mm³, (normal range, 4.5-11 x 103 cells/mm³), with an elevated neutrophil count of 82.7% (normal range 40%-60%), without clinical signs of systemic infection (fevers, chills, or rigors). A urinalysis revealed pyuria with 25-50 WBC/high power field, negative nitrite, positive leukocyte esterase, and moderate bacteria, consistent with what was presumed to be a urinary tract infection. The patient was discharged home with a 1-week course of the antibiotic levofloxacin and the alpha-blocker tamsulosin to make urination easier. Of note, the final results of the urine culture, which returned 48 hours after discharge, showed no growth.
One week prior to surgery, the patient underwent a cystoscopy for ureteral stent exchange, which revealed a necrotic tumor at the right bladder base surrounding the ureteral orifice and stent. Renal pelvis urine, sampled during stent exchange, revealed >100,000 CFU/ml (colony forming units; normal value, <10³) Candida albicans, for which the patient was started on intravenous fluconazole for fungal infection. We consulted with our colleagues in the infectious disease department and continued to follow the patient throughout his hospital course, which included several antibiotic regimens, a comprehensive hematological work-up and eventual urologic surgery. Work-ups for myeloproliferative disorders, leukemia, JAK-2, and BCR-ABL were all negative. A peripheral blood smear analysis showed mostly neutrophils, no immature cells, and occasional hypersegmented neutrophils, but was overall inconsistent with myeloproliferative disease. Despite the patient’s persistent leukocytosis, he remained completely asymptomatic and his neutrophilia was attributed to his malignancy.
The patient subsequently underwent a cystoprostatectomy with ileal conduit. The surgical pathologic analysis showed a high-grade, invasive urothelial (transitional cell) carcinoma measuring 5.2 x 5.0 x 4.5 cm (2 x 1.9 x 1.8 in) with squamous differentiation, extensive tumor necrosis, lymphovascular invasion, invasion into the adjacent seminal vesicle and prostatic stroma, and negative margins (pT4a pN0 pMX; Figure 2). On the day of surgical resection, the patient’s leukocytosis was 70 x 103 cells/mm³.
Despite a transient improvement in his leukocytosis to 37.7 x 103 cells/mm³ on postoperative day 1, the patient remained in the medical intensive care unit for uncontrolled pain and management of his leukocytosis. It is worth noting that the patient remained afebrile throughout his entire 45-day hospitalization, with negative culture data, and despite receiving an extensive broad spectrum antibiotic regimen (levofloxacin, piperacillin-tazobactam, cefazolin, metronidazole, ceftriaxone and fluconazole), his leukocytosis continued to progress, peaking at 161.5 x 103 cells/mm³ less than a month after his surgery (Figure 3).
The patient continued to deteriorate rapidly, with a progressive leukocytosis, and developing metastases to the lung, perineum, and penis. He died a month after surgery (2 months after completion of neoadjuvant chemotherapy). The leukocytosis exhibited in this patient and the aggressive tumor cell growth are believed to have been secondary to a paraneoplastic leukemoid reaction incited by the urothelial bladder cancer cells’ presumed production of G-CSF, which has been rarely reported in the literature previously.
Discussion
We report here on the rare occurrence of PLR in a urothelial bladder cancer. Several mechanisms have been proposed to explain the pathophysiology of PLR. The levels of IL-1[alpha and beta], IL-3, G-CSF, GM-CSF, IL-6, and TNF-[alpha] have all been reported to be elevated in various solid tumors and suggested to contribute to an elevated leukocyte count.3 With previous reports that receptors for G-CSF have been found on cell surfaces of several nonhematopoietic cell types, Tachibana and Murai have proposed the mechanism of a cancer cell’s simultaneous acquisition of the ligand promotion and its receptor expression conferring an autocrine growth advantage.4 They have also reported on the capability of bladder cancer cells to induce a leukemoid reaction in their host through the stimulation of leukocyte production, which has been associated with aggressive tumor cell growth and a poor clinical outcome. In addition, He and colleagues have also described the correlation between PLR and high degree of malignancy, high probability of metastasis, recurrence, and poor prognosis.5
We observed the leukocytosis of 70 x 103 cells/mm³ on the day of resection with a slight drop postoperatively, peaking at 161.5 x 103 cells/mm³ less than a month after resection of the tumor. There is no clear understanding of the cause of the persistent and rapid progression of leukocytosis seen in this patient postoperatively. There is also a dearth of literature describing similar occurrences, with even fewer attempting to explain the pathophysiology of this occurrence.
When faced with similar occurrences in patients, clinicians usually treat for occult infection. Once infections and myeloproliferative diseases have been ruled out, clinicians may consider obtaining a patient’s serum G-CSF level or performing an immunohistochemistry analysis of urothelial cells for overexpression of G-CSF, when available.5 However, despite any efforts to diagnose earlier, there is little clinicians currently have to offer these patients as treatment.
As presented in this report, PLR portends a worse prognosis for patients because of its ability to not only masquerade as an infection leading to a delay in the proper treatment, but also because of its association with a more aggressive tumor cell behavior and growth, making it critically important for clinicians to be able to identify these patients early on. With further investigation into immune regulation and G-CSF receptor signaling, there may be future discoveries of novel methods to diagnose this condition and also advancements in the treatment options made available to these patients.
1. Chakraborty S, Keenportz B, Woodward S, Anderson J, Colan D. Paraneoplastic leukemoid reaction in solid tumors. Am J Clin Oncol. 2015;38(3):326-330.
2. Kumar AK, Satyan MT, Holzbeierlein J, Mirza M, Van Veldhuizen P. Leukemoid reaction and autocrine growth of bladder cancer induced by paraneoplastic production of granulocyte colony-stimulating factor-a potential neoplastic marker: a case report and review of the literature. J Med Case Rep. 2014;8(1):147.
3. Azuma T, Sakai I, Matsumoto T, et al. Leukemoid reaction in association with bone marrow necrosis due to metastatic prostate cancer. Intern Med. 2005;44(10):1093-1096.
4. Tachibana M, Murai M. G-CSF production in human bladder cancer and its ability to promote autocrine growth: a review. Cytokines Cell Mol Ther. 1998;4(2):113-120.
5. He H, Zhang Z, Ge J, Zhou W. Leukemoid reaction associated with transitional cell carcinoma: a case report and literature review. Niger J Clin Pract. 2014;17(3):391-394.
1. Chakraborty S, Keenportz B, Woodward S, Anderson J, Colan D. Paraneoplastic leukemoid reaction in solid tumors. Am J Clin Oncol. 2015;38(3):326-330.
2. Kumar AK, Satyan MT, Holzbeierlein J, Mirza M, Van Veldhuizen P. Leukemoid reaction and autocrine growth of bladder cancer induced by paraneoplastic production of granulocyte colony-stimulating factor-a potential neoplastic marker: a case report and review of the literature. J Med Case Rep. 2014;8(1):147.
3. Azuma T, Sakai I, Matsumoto T, et al. Leukemoid reaction in association with bone marrow necrosis due to metastatic prostate cancer. Intern Med. 2005;44(10):1093-1096.
4. Tachibana M, Murai M. G-CSF production in human bladder cancer and its ability to promote autocrine growth: a review. Cytokines Cell Mol Ther. 1998;4(2):113-120.
5. He H, Zhang Z, Ge J, Zhou W. Leukemoid reaction associated with transitional cell carcinoma: a case report and literature review. Niger J Clin Pract. 2014;17(3):391-394.