User login
Atraumatic splenic rupture as an initial presentation of chronic myelogenous leukemia
Chronic myelogenous leukemia (CML) is a myeloproliferative neoplasm associated with the fusion of the BCR gene located on chromosome 22 and the ABL1 gene on chromosome 9. The fusion results in a reciprocal translocation between chromosomes 9 and 22, leading to the formation of the Philadelphia (Ph) chromosome found in 90%-95% of patients with CML. The incidence of CML is 1.5 per 100,000 people per year, with a male predominance and an average age at diagnosis of 64.1
About 85%-90% of newly diagnosed patients present in the chronic phase and therefore many of them are asymptomatic at the time of diagnosis. If symptoms are present, they often include fatigue, malaise, unintentional weight loss, early satiety, or left upper quadrant pain. Progression of the disease is associated with worsening symptoms such as unexplained fever, significant weight loss, bone or joint pain, bleeding, thrombosis, and infections suggestive of transformation to the accelerated phase or blast crisis. Physical exam findings most commonly include splenomegaly and occasionally mild hepatomegaly.
Atraumatic splenic rupture is a rare complication of this hematologic malignancy, and there are almost no reported cases of CML as the underlying cause.2-4 Here we present the case of a man with sudden-onset generalized abdominal pain and leukocytosis. A computed-tomography scan showed splenic rupture, and the patient was taken for emergency splenectomy. The patient was subsequently positive for t(9,22)(q34;q11.2).
Case presentation and summary
A 59-year-old white man with a history of hypertension and kidney stones presented to a community emergency department with a chief complaint of abdominal pain. About 30 minutes before his arrival, the patient had woken up from sleep with generalized, nonradiating, abdominal pain, which he described as “like my previous kidney stones.” He also reported worsening dyspnea, nausea without vomiting, and lightheadedness without loss of consciousness. The remainder of the review of systems was negative. A physical exam revealed that he was in moderate distress with clear lung fields and had tachycardia without murmur, no CVA tenderness, and a diffusely tender abdomen.
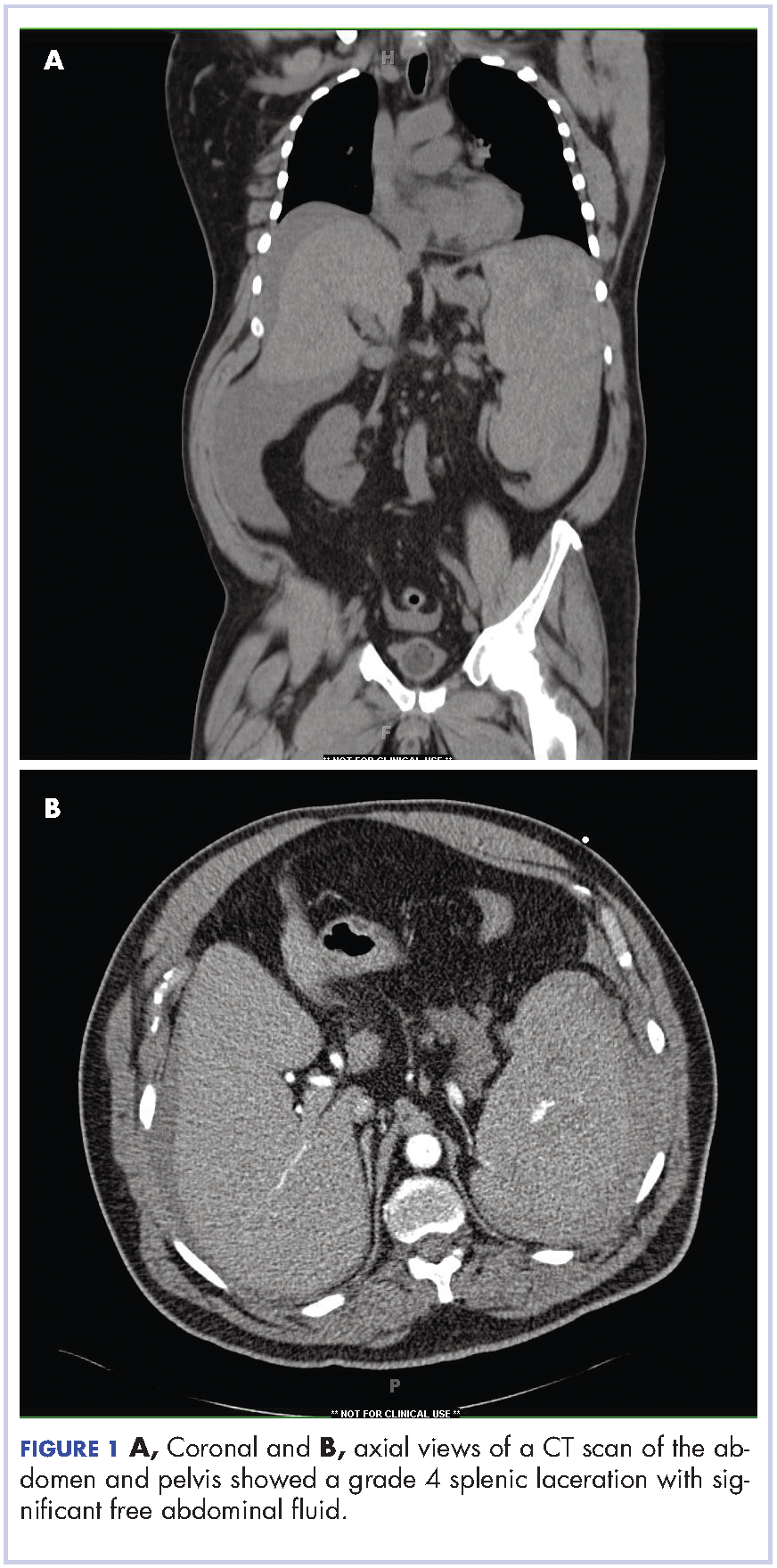
Complete blood count with differential showed leukocytosis (109.1 x 103/uL), normocytic anemia (8.1 g/dL), thrombocytopenia (100,000 cells/uL), neutrophils (71.06 cells/uL), bands (27.13 cells/uL), and monocytes (11.63 cells/uL). A CT scan of the abdomen and pelvis showed a grade 4 splenic laceration with significant free abdominal fluid (Figure 1).
The patient was taken to the operating room where he underwent a splenectomy which was complicated by partial gastrectomy and partial omentectomy. He remained intubated on mechanical ventilation in the intensive care for 7 days. His progress was complicated by profound hypotension that required significant fluid administration and ultimately multiple pressors for blood pressure support. Hypotensive shock was beginning to improve on day 3 and was completely resolved by day 5. The patient underwent continuous positive airway pressure (CPAP) trials on day 6 and was successfully extubated on day 7.
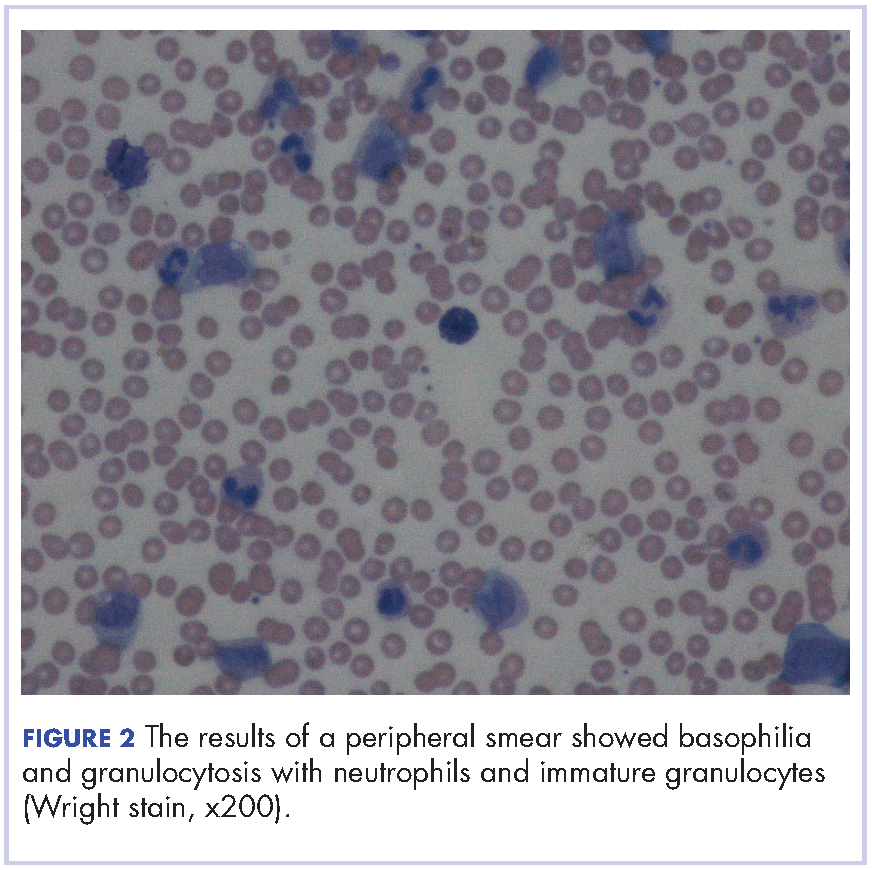
After extubation a more thorough history could be obtained from the patient. He denied any history of weight loss, night sweats, or fatigue. Patient denied any known family history of hematologic malignancies. His peripheral smear showed basophilia and granulocytosis with neutrophils and immature granulocytes (Figure 2). The patient was evaluated by the hematology service and was started on allopurinol and hydroxyurea for presumed hematologic malignancy. He was given the meningococcus and streptococcus pneumoniae vaccine and was discharged home in stable condition on day eleven. Patient was subsequently positive for t(9,22)(q34;q11.2) and was started on imatinib. He has continued to follow in the clinic and is currently in remission.
Discussion
CML has a triphasic clinical course and treatment is based on the specific disease phase. The 3 phases of the disease include the chronic (more indolent) phase, accelerated (more aggressive) phase, and blast crisis. If the disease is left untreated, it will inevitably transition from a chronic to an accelerated phase and finally to blast crisis within a median time of 4 years.
The chronic phase is the most common, representing 85% of diagnoses. Patients can be asymptomatic and many in this phase will be diagnosed by routine lab testing.5 According to the World Health Organization, the accelerated phase is defined as CML patients with one of the following: 10%-19% blasts, basophils ≥20%, platelets <100,000/microL or >1,000,000/microL, unresponsive to therapy, splenomegaly unresponsive to therapy, an increasing white cell count unresponsive to therapy, or cytogenetic evolution.6 Blast crisis is the most aggressive phase and is usually defined by ≥20% blasts, large foci or clusters of blasts on the bone marrow biopsy, or the presence of extramedullary blastic infiltrates.7,8
The diagnosis of CML should be suspected in the presence of distinct lab abnormalities in the peripheral blood. These include elevated white blood cell counts with a median count of 100,000 cells/microL, elevated platelet counts, and a mild normocytic normochromic anemia. Platelet counts of 600,000 or greater have been seen in 15%-30% of patients at the time of diagnosis. The white count differential can show a variety of cells but there will be a notably greater percentage of myelocytes than metamyelocytes. Bone marrow biopsy will reveal increased cellularity, normal to slightly elevated percentage of blasts, and reticulin fibrosis. The diagnosis should be confirmed by the presence of the Philadelphia chromosome either by cytogenetics, fluorescence in situ hybridization, or reverse-transcription polymerase chain reaction (RT-PCR). The Philadelphia chromosome is found in 90%-95% of patients with CML. Most of the remaining patients will have other translocations, but a small minority will have no detectable genetic abnormalities and those patients are known as Ph-negative.9
Treatment options for CML include potential cure with allogeneic hematopoietic stem-cell transplant (HSCT) or disease control using tyrosine kinase inhibitors (TKIs). TKIs are the initial treatment of choice for newly diagnosed patients and are able to produce long-term remission in most patients. The drugs in this category include imatinib, dasatinib, and nilotinib. They work by inhibiting the Bcr-Abl tyrosine kinase, thereby blocking proliferation and inducing apoptosis in Bcr-Abl-positive cells. The majority of patients with chronic-phase CML will have an excellent response to initial treatment with a TKI. It is critical to follow these patients on a regular basis and monitor their disease status. Although the gold standard for assessing cytogenetic response is cytogenetic analysis of a bone marrow biopsy, more sensitive methods such as quantitative PCR using peripheral blood are now available, thereby minimizing the need for bone marrow biopsy. Patients in the accelerated phase are more difficult to manage because they are resistant to most forms of treatment and have short-lived responses to TKI therapy. These patients should strongly be considered for transplantation. Patients in blast crisis have aggressive disease that is more complex and requires more extensive testing. These patients should ideally be treated at tertiary care centers and treatment often involves chemotherapy in addition to TKI therapy usually followed by HSCT.
Atraumatic splenic rupture (ASR) presents similarly to traumatic splenic rupture with typical symptoms being acute onset of upper abdominal, left chest wall, or left shoulder pain (Kehr’s sign) but without a known history of trauma. Quick recognition and surgical intervention represent the best means of definitive care.10 Renzulli and colleagues conducted a literature review for all ASR cases from 1980-2008, examining 632 publications representing 845 cases. They examined the cases using logistic regression analysis to better define the clinicopathology behind ASR. The reported causes of ASR are neoplastic processes (30.3%), infectious (27.3%), inflammatory noninfectious (20.0%), drug- and treatment-related (9.2%), mechanical (6.8%), and normal spleen (6.4%). Treatment included total splenectomy in 84.1% of cases, organ-preserving surgery in 1.2%, and conservative measures in 14.7%. They reported an ASR-related mortality of 12.2%, with being older than 40 and neoplastic disorders associated with increased mortality – although male sex and splenomegaly have also been reported.11-13 Thomas and colleagues have reported on 48 cases of ASR related to hematologic malignancy showing acute myeloid leukemia being the most common cause (21%), followed by acute lymphoblastic leukemia (19%).2
Hematologic malignancies commonly cause splenic engorgement and pain although splenic rupture is an extremely rare event. Recent literature review has shown fewer than a thousand reported cases since 1980.4 There far fewer reported cases of ASR being related to CML, with most being reported as a complication.3,14 Based on our review, we could identify only a handful cases of CML with ASR being the initial symptom. These include a patient with Ph-negative CML and ASR following blast crisis, a patient with Phil-negative BCR-ABL-positive essential thrombocythemia, several cases in which the patient ultimately died, and 1 in which the patient survived into remission.4,14-16 Our case is different because the patient was ultimately positive for t(9,22)(q34;q11.2) and although he experienced multiple complications, he is currently functioning at his baseline and in remission. We hope this case will remind others that CML should be considered in the differential diagnosis of patients ASR.
1. American Cancer Society. Cancer Facts & Figures 2015. Atlanta, Ga: American Cancer Society; 2015.
2. Bauer TW, Haskins GE, Armitage JO. Splenic rupture in patients with hematologic malignancies. Cancer. 1981;48:2729-2733.
3. Giagounidis AA, Burk M, Meckenstock G, Koch AJ, Schneider W. Pathologic rupture of the spleen in hematologic malignancies: two additional cases. Ann Hematol. 1996;73(6):297-302.
4. Goodard SL, Chesney AE, Reis MD, et al. Pathologic splenic rupture: a rare complication of chronic myelomonocytic leukemia. Am J Hematology. 2007;82:405-408.
5. Faderl S, Talpaz M, Estrov Z, et al. The biology of chronic myeloid leukemia. N Engl J Med. 1999;341:164-172.
6. Cortes JE, Talpaz M, O’Brien S, et al. Staging of chronic myeloid leukemia in the imatinib era: an evaluation of the World Health Organization proposal. Cancer. 2006;106:1306-1315.
7. Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100:2292-2302.
8. Kantarjian HM, O’Brien S, Cortes J, et al. Results of decitabine (5-aza-2’deoxycytidine) therapy in 130 patients with chronic myelogenous leukemia. Cancer.2003; 98:522-528.
9. Swerdlow SH, Campo E, Harris NL, et al, editors. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: IARC Press; 2008.
10. Maung A, KaplanL. Management of splenic injury in the adult trauma patient. In: UpToDate, Basow DS (ed), Waltham, MA, 2013.
11. Renzulli P, Hostettler A, Schoepfer AM, Gloor B, Candinas D. Systematic review of atraumatic splenic rupture. Br J Surg. 2009;8(10):1114-1121.
12. Hehlmann R, Heimpel H, Hasford J, et al. Randomized comparison of interferon-alpha with busulfan and hydroxyurea in chronic myelogenous leukemia. The German CML Study Group. Blood. 1994;84:4064-4077.
13. Cortes J, Kantarjian H. How I treat newly diagnosed chronic phase CML. Blood. 2012;120:1390-1397.
14. Nestok BR, Goldstein JD, Lipkovic P. Splenic rupture as a cause of sudden death in undiagnosed chronic myelogenous leukemia. Am J Forensic Med Pathol. 1988;9:241-245.
15. Sachithanandan A, Gleadhil I, Alexander HD, Morris TC. Spontaneous splenic rupture in atypical (Philadelphia chromosome negative) chronic myeloid leukemia following blastic crisis. Ir Med J. 2003;96(6):181-182.
16. Chim CS, Kwong YL, Shek TW, Ma SK, Ooi GC. Splenic rupture as the presenting symptom of blastic crisis in a patient with Philadelphia-negative, BCR-ABL-positive ET. Am J Hematology. 2001;66:70-71.
Chronic myelogenous leukemia (CML) is a myeloproliferative neoplasm associated with the fusion of the BCR gene located on chromosome 22 and the ABL1 gene on chromosome 9. The fusion results in a reciprocal translocation between chromosomes 9 and 22, leading to the formation of the Philadelphia (Ph) chromosome found in 90%-95% of patients with CML. The incidence of CML is 1.5 per 100,000 people per year, with a male predominance and an average age at diagnosis of 64.1
About 85%-90% of newly diagnosed patients present in the chronic phase and therefore many of them are asymptomatic at the time of diagnosis. If symptoms are present, they often include fatigue, malaise, unintentional weight loss, early satiety, or left upper quadrant pain. Progression of the disease is associated with worsening symptoms such as unexplained fever, significant weight loss, bone or joint pain, bleeding, thrombosis, and infections suggestive of transformation to the accelerated phase or blast crisis. Physical exam findings most commonly include splenomegaly and occasionally mild hepatomegaly.
Atraumatic splenic rupture is a rare complication of this hematologic malignancy, and there are almost no reported cases of CML as the underlying cause.2-4 Here we present the case of a man with sudden-onset generalized abdominal pain and leukocytosis. A computed-tomography scan showed splenic rupture, and the patient was taken for emergency splenectomy. The patient was subsequently positive for t(9,22)(q34;q11.2).
Case presentation and summary
A 59-year-old white man with a history of hypertension and kidney stones presented to a community emergency department with a chief complaint of abdominal pain. About 30 minutes before his arrival, the patient had woken up from sleep with generalized, nonradiating, abdominal pain, which he described as “like my previous kidney stones.” He also reported worsening dyspnea, nausea without vomiting, and lightheadedness without loss of consciousness. The remainder of the review of systems was negative. A physical exam revealed that he was in moderate distress with clear lung fields and had tachycardia without murmur, no CVA tenderness, and a diffusely tender abdomen.
Complete blood count with differential showed leukocytosis (109.1 x 103/uL), normocytic anemia (8.1 g/dL), thrombocytopenia (100,000 cells/uL), neutrophils (71.06 cells/uL), bands (27.13 cells/uL), and monocytes (11.63 cells/uL). A CT scan of the abdomen and pelvis showed a grade 4 splenic laceration with significant free abdominal fluid (Figure 1).
The patient was taken to the operating room where he underwent a splenectomy which was complicated by partial gastrectomy and partial omentectomy. He remained intubated on mechanical ventilation in the intensive care for 7 days. His progress was complicated by profound hypotension that required significant fluid administration and ultimately multiple pressors for blood pressure support. Hypotensive shock was beginning to improve on day 3 and was completely resolved by day 5. The patient underwent continuous positive airway pressure (CPAP) trials on day 6 and was successfully extubated on day 7.
After extubation a more thorough history could be obtained from the patient. He denied any history of weight loss, night sweats, or fatigue. Patient denied any known family history of hematologic malignancies. His peripheral smear showed basophilia and granulocytosis with neutrophils and immature granulocytes (Figure 2). The patient was evaluated by the hematology service and was started on allopurinol and hydroxyurea for presumed hematologic malignancy. He was given the meningococcus and streptococcus pneumoniae vaccine and was discharged home in stable condition on day eleven. Patient was subsequently positive for t(9,22)(q34;q11.2) and was started on imatinib. He has continued to follow in the clinic and is currently in remission.
Discussion
CML has a triphasic clinical course and treatment is based on the specific disease phase. The 3 phases of the disease include the chronic (more indolent) phase, accelerated (more aggressive) phase, and blast crisis. If the disease is left untreated, it will inevitably transition from a chronic to an accelerated phase and finally to blast crisis within a median time of 4 years.
The chronic phase is the most common, representing 85% of diagnoses. Patients can be asymptomatic and many in this phase will be diagnosed by routine lab testing.5 According to the World Health Organization, the accelerated phase is defined as CML patients with one of the following: 10%-19% blasts, basophils ≥20%, platelets <100,000/microL or >1,000,000/microL, unresponsive to therapy, splenomegaly unresponsive to therapy, an increasing white cell count unresponsive to therapy, or cytogenetic evolution.6 Blast crisis is the most aggressive phase and is usually defined by ≥20% blasts, large foci or clusters of blasts on the bone marrow biopsy, or the presence of extramedullary blastic infiltrates.7,8
The diagnosis of CML should be suspected in the presence of distinct lab abnormalities in the peripheral blood. These include elevated white blood cell counts with a median count of 100,000 cells/microL, elevated platelet counts, and a mild normocytic normochromic anemia. Platelet counts of 600,000 or greater have been seen in 15%-30% of patients at the time of diagnosis. The white count differential can show a variety of cells but there will be a notably greater percentage of myelocytes than metamyelocytes. Bone marrow biopsy will reveal increased cellularity, normal to slightly elevated percentage of blasts, and reticulin fibrosis. The diagnosis should be confirmed by the presence of the Philadelphia chromosome either by cytogenetics, fluorescence in situ hybridization, or reverse-transcription polymerase chain reaction (RT-PCR). The Philadelphia chromosome is found in 90%-95% of patients with CML. Most of the remaining patients will have other translocations, but a small minority will have no detectable genetic abnormalities and those patients are known as Ph-negative.9
Treatment options for CML include potential cure with allogeneic hematopoietic stem-cell transplant (HSCT) or disease control using tyrosine kinase inhibitors (TKIs). TKIs are the initial treatment of choice for newly diagnosed patients and are able to produce long-term remission in most patients. The drugs in this category include imatinib, dasatinib, and nilotinib. They work by inhibiting the Bcr-Abl tyrosine kinase, thereby blocking proliferation and inducing apoptosis in Bcr-Abl-positive cells. The majority of patients with chronic-phase CML will have an excellent response to initial treatment with a TKI. It is critical to follow these patients on a regular basis and monitor their disease status. Although the gold standard for assessing cytogenetic response is cytogenetic analysis of a bone marrow biopsy, more sensitive methods such as quantitative PCR using peripheral blood are now available, thereby minimizing the need for bone marrow biopsy. Patients in the accelerated phase are more difficult to manage because they are resistant to most forms of treatment and have short-lived responses to TKI therapy. These patients should strongly be considered for transplantation. Patients in blast crisis have aggressive disease that is more complex and requires more extensive testing. These patients should ideally be treated at tertiary care centers and treatment often involves chemotherapy in addition to TKI therapy usually followed by HSCT.
Atraumatic splenic rupture (ASR) presents similarly to traumatic splenic rupture with typical symptoms being acute onset of upper abdominal, left chest wall, or left shoulder pain (Kehr’s sign) but without a known history of trauma. Quick recognition and surgical intervention represent the best means of definitive care.10 Renzulli and colleagues conducted a literature review for all ASR cases from 1980-2008, examining 632 publications representing 845 cases. They examined the cases using logistic regression analysis to better define the clinicopathology behind ASR. The reported causes of ASR are neoplastic processes (30.3%), infectious (27.3%), inflammatory noninfectious (20.0%), drug- and treatment-related (9.2%), mechanical (6.8%), and normal spleen (6.4%). Treatment included total splenectomy in 84.1% of cases, organ-preserving surgery in 1.2%, and conservative measures in 14.7%. They reported an ASR-related mortality of 12.2%, with being older than 40 and neoplastic disorders associated with increased mortality – although male sex and splenomegaly have also been reported.11-13 Thomas and colleagues have reported on 48 cases of ASR related to hematologic malignancy showing acute myeloid leukemia being the most common cause (21%), followed by acute lymphoblastic leukemia (19%).2
Hematologic malignancies commonly cause splenic engorgement and pain although splenic rupture is an extremely rare event. Recent literature review has shown fewer than a thousand reported cases since 1980.4 There far fewer reported cases of ASR being related to CML, with most being reported as a complication.3,14 Based on our review, we could identify only a handful cases of CML with ASR being the initial symptom. These include a patient with Ph-negative CML and ASR following blast crisis, a patient with Phil-negative BCR-ABL-positive essential thrombocythemia, several cases in which the patient ultimately died, and 1 in which the patient survived into remission.4,14-16 Our case is different because the patient was ultimately positive for t(9,22)(q34;q11.2) and although he experienced multiple complications, he is currently functioning at his baseline and in remission. We hope this case will remind others that CML should be considered in the differential diagnosis of patients ASR.
Chronic myelogenous leukemia (CML) is a myeloproliferative neoplasm associated with the fusion of the BCR gene located on chromosome 22 and the ABL1 gene on chromosome 9. The fusion results in a reciprocal translocation between chromosomes 9 and 22, leading to the formation of the Philadelphia (Ph) chromosome found in 90%-95% of patients with CML. The incidence of CML is 1.5 per 100,000 people per year, with a male predominance and an average age at diagnosis of 64.1
About 85%-90% of newly diagnosed patients present in the chronic phase and therefore many of them are asymptomatic at the time of diagnosis. If symptoms are present, they often include fatigue, malaise, unintentional weight loss, early satiety, or left upper quadrant pain. Progression of the disease is associated with worsening symptoms such as unexplained fever, significant weight loss, bone or joint pain, bleeding, thrombosis, and infections suggestive of transformation to the accelerated phase or blast crisis. Physical exam findings most commonly include splenomegaly and occasionally mild hepatomegaly.
Atraumatic splenic rupture is a rare complication of this hematologic malignancy, and there are almost no reported cases of CML as the underlying cause.2-4 Here we present the case of a man with sudden-onset generalized abdominal pain and leukocytosis. A computed-tomography scan showed splenic rupture, and the patient was taken for emergency splenectomy. The patient was subsequently positive for t(9,22)(q34;q11.2).
Case presentation and summary
A 59-year-old white man with a history of hypertension and kidney stones presented to a community emergency department with a chief complaint of abdominal pain. About 30 minutes before his arrival, the patient had woken up from sleep with generalized, nonradiating, abdominal pain, which he described as “like my previous kidney stones.” He also reported worsening dyspnea, nausea without vomiting, and lightheadedness without loss of consciousness. The remainder of the review of systems was negative. A physical exam revealed that he was in moderate distress with clear lung fields and had tachycardia without murmur, no CVA tenderness, and a diffusely tender abdomen.
Complete blood count with differential showed leukocytosis (109.1 x 103/uL), normocytic anemia (8.1 g/dL), thrombocytopenia (100,000 cells/uL), neutrophils (71.06 cells/uL), bands (27.13 cells/uL), and monocytes (11.63 cells/uL). A CT scan of the abdomen and pelvis showed a grade 4 splenic laceration with significant free abdominal fluid (Figure 1).
The patient was taken to the operating room where he underwent a splenectomy which was complicated by partial gastrectomy and partial omentectomy. He remained intubated on mechanical ventilation in the intensive care for 7 days. His progress was complicated by profound hypotension that required significant fluid administration and ultimately multiple pressors for blood pressure support. Hypotensive shock was beginning to improve on day 3 and was completely resolved by day 5. The patient underwent continuous positive airway pressure (CPAP) trials on day 6 and was successfully extubated on day 7.
After extubation a more thorough history could be obtained from the patient. He denied any history of weight loss, night sweats, or fatigue. Patient denied any known family history of hematologic malignancies. His peripheral smear showed basophilia and granulocytosis with neutrophils and immature granulocytes (Figure 2). The patient was evaluated by the hematology service and was started on allopurinol and hydroxyurea for presumed hematologic malignancy. He was given the meningococcus and streptococcus pneumoniae vaccine and was discharged home in stable condition on day eleven. Patient was subsequently positive for t(9,22)(q34;q11.2) and was started on imatinib. He has continued to follow in the clinic and is currently in remission.
Discussion
CML has a triphasic clinical course and treatment is based on the specific disease phase. The 3 phases of the disease include the chronic (more indolent) phase, accelerated (more aggressive) phase, and blast crisis. If the disease is left untreated, it will inevitably transition from a chronic to an accelerated phase and finally to blast crisis within a median time of 4 years.
The chronic phase is the most common, representing 85% of diagnoses. Patients can be asymptomatic and many in this phase will be diagnosed by routine lab testing.5 According to the World Health Organization, the accelerated phase is defined as CML patients with one of the following: 10%-19% blasts, basophils ≥20%, platelets <100,000/microL or >1,000,000/microL, unresponsive to therapy, splenomegaly unresponsive to therapy, an increasing white cell count unresponsive to therapy, or cytogenetic evolution.6 Blast crisis is the most aggressive phase and is usually defined by ≥20% blasts, large foci or clusters of blasts on the bone marrow biopsy, or the presence of extramedullary blastic infiltrates.7,8
The diagnosis of CML should be suspected in the presence of distinct lab abnormalities in the peripheral blood. These include elevated white blood cell counts with a median count of 100,000 cells/microL, elevated platelet counts, and a mild normocytic normochromic anemia. Platelet counts of 600,000 or greater have been seen in 15%-30% of patients at the time of diagnosis. The white count differential can show a variety of cells but there will be a notably greater percentage of myelocytes than metamyelocytes. Bone marrow biopsy will reveal increased cellularity, normal to slightly elevated percentage of blasts, and reticulin fibrosis. The diagnosis should be confirmed by the presence of the Philadelphia chromosome either by cytogenetics, fluorescence in situ hybridization, or reverse-transcription polymerase chain reaction (RT-PCR). The Philadelphia chromosome is found in 90%-95% of patients with CML. Most of the remaining patients will have other translocations, but a small minority will have no detectable genetic abnormalities and those patients are known as Ph-negative.9
Treatment options for CML include potential cure with allogeneic hematopoietic stem-cell transplant (HSCT) or disease control using tyrosine kinase inhibitors (TKIs). TKIs are the initial treatment of choice for newly diagnosed patients and are able to produce long-term remission in most patients. The drugs in this category include imatinib, dasatinib, and nilotinib. They work by inhibiting the Bcr-Abl tyrosine kinase, thereby blocking proliferation and inducing apoptosis in Bcr-Abl-positive cells. The majority of patients with chronic-phase CML will have an excellent response to initial treatment with a TKI. It is critical to follow these patients on a regular basis and monitor their disease status. Although the gold standard for assessing cytogenetic response is cytogenetic analysis of a bone marrow biopsy, more sensitive methods such as quantitative PCR using peripheral blood are now available, thereby minimizing the need for bone marrow biopsy. Patients in the accelerated phase are more difficult to manage because they are resistant to most forms of treatment and have short-lived responses to TKI therapy. These patients should strongly be considered for transplantation. Patients in blast crisis have aggressive disease that is more complex and requires more extensive testing. These patients should ideally be treated at tertiary care centers and treatment often involves chemotherapy in addition to TKI therapy usually followed by HSCT.
Atraumatic splenic rupture (ASR) presents similarly to traumatic splenic rupture with typical symptoms being acute onset of upper abdominal, left chest wall, or left shoulder pain (Kehr’s sign) but without a known history of trauma. Quick recognition and surgical intervention represent the best means of definitive care.10 Renzulli and colleagues conducted a literature review for all ASR cases from 1980-2008, examining 632 publications representing 845 cases. They examined the cases using logistic regression analysis to better define the clinicopathology behind ASR. The reported causes of ASR are neoplastic processes (30.3%), infectious (27.3%), inflammatory noninfectious (20.0%), drug- and treatment-related (9.2%), mechanical (6.8%), and normal spleen (6.4%). Treatment included total splenectomy in 84.1% of cases, organ-preserving surgery in 1.2%, and conservative measures in 14.7%. They reported an ASR-related mortality of 12.2%, with being older than 40 and neoplastic disorders associated with increased mortality – although male sex and splenomegaly have also been reported.11-13 Thomas and colleagues have reported on 48 cases of ASR related to hematologic malignancy showing acute myeloid leukemia being the most common cause (21%), followed by acute lymphoblastic leukemia (19%).2
Hematologic malignancies commonly cause splenic engorgement and pain although splenic rupture is an extremely rare event. Recent literature review has shown fewer than a thousand reported cases since 1980.4 There far fewer reported cases of ASR being related to CML, with most being reported as a complication.3,14 Based on our review, we could identify only a handful cases of CML with ASR being the initial symptom. These include a patient with Ph-negative CML and ASR following blast crisis, a patient with Phil-negative BCR-ABL-positive essential thrombocythemia, several cases in which the patient ultimately died, and 1 in which the patient survived into remission.4,14-16 Our case is different because the patient was ultimately positive for t(9,22)(q34;q11.2) and although he experienced multiple complications, he is currently functioning at his baseline and in remission. We hope this case will remind others that CML should be considered in the differential diagnosis of patients ASR.
1. American Cancer Society. Cancer Facts & Figures 2015. Atlanta, Ga: American Cancer Society; 2015.
2. Bauer TW, Haskins GE, Armitage JO. Splenic rupture in patients with hematologic malignancies. Cancer. 1981;48:2729-2733.
3. Giagounidis AA, Burk M, Meckenstock G, Koch AJ, Schneider W. Pathologic rupture of the spleen in hematologic malignancies: two additional cases. Ann Hematol. 1996;73(6):297-302.
4. Goodard SL, Chesney AE, Reis MD, et al. Pathologic splenic rupture: a rare complication of chronic myelomonocytic leukemia. Am J Hematology. 2007;82:405-408.
5. Faderl S, Talpaz M, Estrov Z, et al. The biology of chronic myeloid leukemia. N Engl J Med. 1999;341:164-172.
6. Cortes JE, Talpaz M, O’Brien S, et al. Staging of chronic myeloid leukemia in the imatinib era: an evaluation of the World Health Organization proposal. Cancer. 2006;106:1306-1315.
7. Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100:2292-2302.
8. Kantarjian HM, O’Brien S, Cortes J, et al. Results of decitabine (5-aza-2’deoxycytidine) therapy in 130 patients with chronic myelogenous leukemia. Cancer.2003; 98:522-528.
9. Swerdlow SH, Campo E, Harris NL, et al, editors. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: IARC Press; 2008.
10. Maung A, KaplanL. Management of splenic injury in the adult trauma patient. In: UpToDate, Basow DS (ed), Waltham, MA, 2013.
11. Renzulli P, Hostettler A, Schoepfer AM, Gloor B, Candinas D. Systematic review of atraumatic splenic rupture. Br J Surg. 2009;8(10):1114-1121.
12. Hehlmann R, Heimpel H, Hasford J, et al. Randomized comparison of interferon-alpha with busulfan and hydroxyurea in chronic myelogenous leukemia. The German CML Study Group. Blood. 1994;84:4064-4077.
13. Cortes J, Kantarjian H. How I treat newly diagnosed chronic phase CML. Blood. 2012;120:1390-1397.
14. Nestok BR, Goldstein JD, Lipkovic P. Splenic rupture as a cause of sudden death in undiagnosed chronic myelogenous leukemia. Am J Forensic Med Pathol. 1988;9:241-245.
15. Sachithanandan A, Gleadhil I, Alexander HD, Morris TC. Spontaneous splenic rupture in atypical (Philadelphia chromosome negative) chronic myeloid leukemia following blastic crisis. Ir Med J. 2003;96(6):181-182.
16. Chim CS, Kwong YL, Shek TW, Ma SK, Ooi GC. Splenic rupture as the presenting symptom of blastic crisis in a patient with Philadelphia-negative, BCR-ABL-positive ET. Am J Hematology. 2001;66:70-71.
1. American Cancer Society. Cancer Facts & Figures 2015. Atlanta, Ga: American Cancer Society; 2015.
2. Bauer TW, Haskins GE, Armitage JO. Splenic rupture in patients with hematologic malignancies. Cancer. 1981;48:2729-2733.
3. Giagounidis AA, Burk M, Meckenstock G, Koch AJ, Schneider W. Pathologic rupture of the spleen in hematologic malignancies: two additional cases. Ann Hematol. 1996;73(6):297-302.
4. Goodard SL, Chesney AE, Reis MD, et al. Pathologic splenic rupture: a rare complication of chronic myelomonocytic leukemia. Am J Hematology. 2007;82:405-408.
5. Faderl S, Talpaz M, Estrov Z, et al. The biology of chronic myeloid leukemia. N Engl J Med. 1999;341:164-172.
6. Cortes JE, Talpaz M, O’Brien S, et al. Staging of chronic myeloid leukemia in the imatinib era: an evaluation of the World Health Organization proposal. Cancer. 2006;106:1306-1315.
7. Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100:2292-2302.
8. Kantarjian HM, O’Brien S, Cortes J, et al. Results of decitabine (5-aza-2’deoxycytidine) therapy in 130 patients with chronic myelogenous leukemia. Cancer.2003; 98:522-528.
9. Swerdlow SH, Campo E, Harris NL, et al, editors. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: IARC Press; 2008.
10. Maung A, KaplanL. Management of splenic injury in the adult trauma patient. In: UpToDate, Basow DS (ed), Waltham, MA, 2013.
11. Renzulli P, Hostettler A, Schoepfer AM, Gloor B, Candinas D. Systematic review of atraumatic splenic rupture. Br J Surg. 2009;8(10):1114-1121.
12. Hehlmann R, Heimpel H, Hasford J, et al. Randomized comparison of interferon-alpha with busulfan and hydroxyurea in chronic myelogenous leukemia. The German CML Study Group. Blood. 1994;84:4064-4077.
13. Cortes J, Kantarjian H. How I treat newly diagnosed chronic phase CML. Blood. 2012;120:1390-1397.
14. Nestok BR, Goldstein JD, Lipkovic P. Splenic rupture as a cause of sudden death in undiagnosed chronic myelogenous leukemia. Am J Forensic Med Pathol. 1988;9:241-245.
15. Sachithanandan A, Gleadhil I, Alexander HD, Morris TC. Spontaneous splenic rupture in atypical (Philadelphia chromosome negative) chronic myeloid leukemia following blastic crisis. Ir Med J. 2003;96(6):181-182.
16. Chim CS, Kwong YL, Shek TW, Ma SK, Ooi GC. Splenic rupture as the presenting symptom of blastic crisis in a patient with Philadelphia-negative, BCR-ABL-positive ET. Am J Hematology. 2001;66:70-71.
Single-Dose Niacin-Induced Hepatitis
Niacin, also known as vitamin B3, is an important cofactor in many metabolic processes necessary to life. Over the past 15 to 20 years, niacin has been prescribed to patients with hyperlipidemia to increase high-density lipoprotein and lower low-density lipoprotein.1 As a naturally occurring vitamin, niacin is also available over-the-counter (OTC) as a dietary supplement, and is also a common ingredient in energy drinks and multivitamins.2
In addition to treating hyperlipidemia and as a nutritional supplement, some anecdotal reports amongst lay-persons suggests that niacin offers other health benefits, such as promoting weight loss and expediting the elimination of alcohol and illicit drugs from one’s system (eg, marijuana).3,4 The increased use of niacin supplementation in the general population for all of the aforementioned reasons has resulted in an increased incidence of niacin toxicity.
Formulations
Niacin is available in three formulations: extended-release (ER, also referred to as intermediate-release), immediate-release (IR), and sustained-release (SR).
The ER formulations of niacin are typically prescribed to treat hyperlipidemia. Patients are usually started on ER niacin at an initial dose of 250 mg once daily. The dose is gradually increased, as tolerated or necessary, to 2 g per day, taken in three doses. It is not uncommon for patients with hyperlipidemia to take more than 1 g of niacin per day after titration by their primary physicians.
Side Effects
Since niacin increases the release of arachidonic acid from cell membranes that metabolizes into prostaglandins, specifically prostaglandins E2 and D2, many patients taking niacin experience uncomfortable flushing and itching.5 Nonsteroidal anti-inflammatory drugs (NSAIDs) prevent this side effect by inhibiting the metabolism of arachidonic acid into those vasodilatory prostaglandins. The newer ER and SR formulations of niacin, which are approved for OTC use as a dietary supplement, are less likely to cause flushing.5
Extended-release niacin, however, is associated with a higher incidence of hepatotoxicity than the other prescription formulations of niacin.6 Toxicity has been well recognized in patients taking niacin chronically for hyperlipidemia, with reports of such cases dating back to the 1980s.7,8 We report a unique case of niacin toxicity following a single-dose ingestion in a young man.
Case
A 22-year-old man presented to the ED for evaluation of a 2-week history of intermittent periumbilical abdominal pain. This visit represented the patient’s second visit to the ED over the past week for the same complaint.
Upon presentation the patient’s vital signs were: blood pressure (BP), 113/64 mm Hg; heart rate, 82 beats/min; respiratory rate, 16 breaths/min; and temperature 36.6°C. Oxygen saturation was 100% on room air. The patient was otherwise healthy and had no significant recent or remote medical history. He denied taking any medications prior to his initial presentation, and reported only occasional alcohol use.
At the patient’s initial presentation 1 week earlier, he was diagnosed with acute gastroenteritis and treated with famotidine and ondansetron in the ED. The patient appeared well clinically at this visit, and laboratory values were within normal limits, including normal blood glucose and urinalysis.
The patient was discharged home from this first visit with prescriptions of famotidine and ondansetron, and was advised to follow up with his primary care physician in 1 week. Throughout the week after discharge from the ED, the patient experienced worsening abdominal pain, and he developed frequent nonbloody emesis, prompting his second presentation to the ED. At this second visit, the patient stated that he had taken one dose of ondansetron at home, without effect. He also noted subjective fevers, but had no diarrhea or melena.
Vital signs remained within normal limits with BPs ranging from 115 to 130 mm Hg systolic and 50 to 89 mm Hg diastolic. The patient was never tachycardic, tachypneic, febrile, or hypoxic. Physical examination was remarkable for periumbilical tenderness. The patient had no jaundice. A more thorough laboratory evaluation revealed elevated anion gap and blood urea nitrogen/creatinine values, and leukocytosis. The patient’s hepatic enzymes were also elevated, with aspartate aminotransferase (AST) over 2,000 U/L and alanine aminotransferase (ALT) of 1,698 U/L. Lipase, bilirubin, and alkaline phosphatase were all within normal limits. The patient’s prothrombin time (PT) was elevated at 14 seconds, and the international normalized ratio (INR) was elevated at 1.28. Laboratory analysis for acetaminophen and alcohol was negative.
A computed tomography (CT) scan of the abdomen/pelvis with intravenous (IV) contrast was unremarkable, demonstrating a liver devoid of any masses, portal or biliary dilation, or cirrhotic changes.
The patient received IV famotidine and ondansetron, and morphine for pain control, and was admitted to the general medical floor for hepatitis of uncertain etiology. A viral hepatitis panel was negative.
On the recommendation of the toxicology service, the patient was given N-acetylcysteine (NAC), and his hepatic enzymes trended down to an AST of 642 U/L and an ALT of 456 U/L by hospital day 2. (The patient essentially completed a positive dechallenge test).9
A gastroenterology consult was ordered, during which additional history-taking and chart review noted that the patient admitted to taking one or two tablets of OTC niacin as a dietary supplement the day before his initial presentation. Although the patient could not recall the exact dosage, he stated that he had been taking supplemental niacin approximately once a month over the past several years without any issues. Since OTC niacin is most commonly available in 500-mg tablets, this suggested the patient’s recent one-time ingested dose was approximately 500 to 1,000 mg.
Based on the patient’s admission to niacin use, additional studies were ordered, including an abdominal ultrasound and a urine drug screen. Ultrasound findings were unremarkable for portal venous thrombosis. The urine drug screen, however, was positive for marijuana and opiates. While the patient denied any history of opioid use, the positive opiate assay could have been attributed to the morphine given in the ED.
Throughout the patient’s hospital course, he remained normotensive and had no change in mental status. His liver enzymes, PT, and INR continued to normalize, and he was discharged home after 3 days, with instructions to follow up with the gastrointestinal clinic within 11 days. An appointment was made for him, which he did not attend.
Given the patient’s negative autoimmune and viral workup, and rapid resolution of symptoms after discontinuing niacin use, it is believed that he had an acute drug-induced hepatitis due to niacin ingestion. Regarding any coingestants that could have contributed to the hepatitis, the patient denied taking other common coingestants such as alcohol and acetaminophen; this assertion was supported by laboratory results.
Since we were unable to attain a qualitative measurement of the patient’s niacin concentration, our diagnosis was primarily based on the patient’s reported history.10 It is possible the patient had been taking more niacin than that to which he admitted, or that he was taking another hepatotoxic substance not detected on our toxicology workup. As previously noted, there are many medications and/or dietary supplements that could cause or contribute to a synergistic effect of drug-induced hepatitis for which the patient was not tested at his initial presentation. The patient could have co-ingested this large dose of niacin with acetaminophen and/or other supplements, energy drinks, or alcohol. A combination such as this could have contributed to his hepatitis, and the metabolites of these other substances would have been eliminated by the time of his second ED presentation.
Discussion
There are over 900 different drugs, toxins, and supplements known to cause hepatic injury.11,12 Clinical manifestations of toxicity range from asymptomatic incidental elevations in transaminases to fulminant liver failure causing mortality. Ingestion of commonly used medications such as statins (although not in overdose quantities) can cause transient asymptomatic transaminitis.13 These elevations are usually mild—ie, less than twice the upper limit of normal. Patients who experience such elevations can usually continue to take the medications with frequent and vigilant monitoring of hepatic function.
Signs and Symptoms
Acute Liver Injury. Acute liver injury is diagnosed when AST and ALT levels are greater than twice the upper limit of normal. Patients also typically have mild-to-moderate abdominal findings, such as pain, nausea, and vomiting—as was experienced by our patient. Along with niacin, angiotensin-converting enzyme inhibitors, NSAIDs, and antifungal medications are examples of other medications that can cause this degree of drug-induced hepatitis.
Severe Liver Injury. Severe liver injury features elevations in not only AST and ALT, but also alkaline phosphate and bilirubin. Patients with severe hepatic injury appear clinically ill and may exhibit altered mental status and jaundice. This type of subfulminant hepatic failure commonly results from acetaminophen toxicity, anesthetic gases, iron toxicity, phosphorus toxicity, and cocaine toxicity. Examples of drugs that result in massive liver necrosis and fulminant hepatitis are acetaminophen, isoniazid, phenelzine, phenytoin, propylthiouracil, and sertraline. Patients with massive hepatic necrosis and hepatitis may require liver transplantation.
Etiology
Identifying the etiology of liver injury is made largely through the patient’s history because there are simply too many possible hepatotoxic agents to test for them all. Diagnostic suspicion of hepatic toxicity should be increased with signs of more serious disease; however, drug-induced liver injury should be included in the differential diagnosis for all cases of abdominal pain.
With respect to the patient in our case, obtaining a more complete history involving supplement and vitamin use would have allowed us to make the diagnosis in the ED. Unfortunately, these subtle aspects of a patient’s history are often overlooked in the emergent care setting.
Treatment
The treatment of niacin-induced liver injury is similar to the guidelines for treating most other drug-induced pathology.14 Removal of the offending agent and providing supportive care is the primary treatment modality.15 In addition, it is important that the clinician exclude and rule-out other causes of hepatitis such as those of viral, autoimmune, or ischemic etiology.
N-acetylcysteine. A medication classically used in patients with acetaminophen overdose, NAC is a safe and effective treatment for non-acetaminophen-induced liver injury, and was given to treat our patient.16L-carnitine. L-carnitine has been shown to be effective in cases of chronic steatosis from hepatitis C and in valproic acid induced hepatitis.17Since L-carnitine is not included on our hospital’s formulary, it was not a treatment option for our patient.Glucocorticoid Therapy. Although glucocorticoids are occasionally given to patients with systemic symptoms of drug reactions, its effectiveness has not been adequately studied.18
Prognosis
The prognosis of patients with acute drug-induced hepatitis is generally good, and most patients fully recover once the offending agent is removed. Poor prognostic factors include the presence of jaundice, requirement for dialysis, underlying chronic liver conditions, or elevated serum creatinine. While most patients will experience a complete recovery, approximately 5% to 10% will develop chronic hepatitis and/or cirrhosis.
Conclusion
Niacin is now available as prescription and OTC formulations and is a potentially hepatotoxic medication and dietary supplement. Niacin can cause an acute hepatitis, especially when taken in conjunction with other hepatotoxic substances. Drug-induced liver injury from niacin ingestion will improve quickly following removal, and the prognosis in otherwise healthy individuals is good.
Patients, especially young, healthy patients who present with symptoms concerning for hepatitis, should be asked specifically about any nutritional, herbal, or other supplement usage. During the history intake, many patients do not consider vitamins or other nutritional or herbal supplements as “medication” or as being significant, and only report prescription and OTC medications.
1. Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97(9):2969-2989. doi:10.1210/jc.2011-3213.
2. Vivekanandarajah A, Ni S, Waked A. Acute hepatitis in a woman following excessive ingestion of an energy drink: a case report. J Med Case Rep. 2011;5:227. doi:10.1186/1752-1947-5-227.
3. Niacin. U.S. National Library of Medicine. https://medlineplus.gov/druginfo/meds/a682518.html. Updated July 15, 2017. Accessed February 21, 2018.
4. Addiction Resource. Niacin flush for drug detox. https://addictionresource.com/drug-testing/niacin-drug-test/. Accessed February 20, 2018.
5. Kamanna VS, Ganji SH, Kashyap ML. The mechanism and mitigation of niacin-induced flushing. Int J Clin Pract. 2009;63(9):1369-1377. doi:10.1111/j.1742-1241.2009.02099.x.
6. Etchason JA, Miller TD, Squires RW, et al. Niacin-induced hepatitis: a potential side effect with low-dose time-release niacin. Mayo Clin Proc. 1991;66(1):23-28.
7. Patterson DJ, Dew EW, Gyorkey F, Graham DY. Niacin hepatitis. South Med J. 1983;76(2):239-241.
8. Ferenchick G, Rovner D. Hepatitis and hematemesis complicating nicotinic acid use. Am J Med Sci. 1989;298(3):191-193.
9. Henkin Y, Johnson KC, Segrest JP. Rechallenge with crystalline niacin after drug-induced hepatitis from sustained-release niacin. JAMA. 1990;264(2):241-243.
10. Barritt AS 4th, Lee J, Hayashi PH. Detective work in drug-induced liver injury: sometimes it is all about interviewing the right witness. Clin Gastroenterol Hepatol. 2010;8(7):635-637. doi:10.1016/j.cgh.2010.03.020.
11. Chitturi S, Teoh NC, Farrell GC. Hepatic drug metabolism and liver disease caused by drugs. In: Feldman M, Friedman LS, Brandt LJ, eds. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. 10th ed. Philadelphia, PA: Elsevier Saunders; 2016:1442-1477.
12. National Institutes of Health. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Niacin. https://livertox.nih.gov/Niacin.htm. Updated January 18, 2018. Accessed February 21, 2018.
13. Chalasani N. Statins and hepatotoxicity: focus on patients with fatty liver. Hepatology. 2005;41(4):690-695.
14. Chalasani NP, Hayashi PH, Bonkovsky HL, et al. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. Am J Gastroenterol. 2014;109(7):950-966. doi:10.1038/ajg.2014.131.
15. Larson AM. Hepatotoxicity due to herbal medication and dietary supplements. UpToDate Web site. https://www.uptodate.com/contents/hepatotoxicity-due-to-herbal-medications-and-dietary-supplements?source=search_result&search=niacin%20induced%20hepatitis&selectedTitle=3~150. Updated December 21, 2017. Accessed February 21, 2018.
16. Mumtaz K, Azam Z, Hamid S, et al. Role of N-acetylcysteine in adults with non-acetaminophen-induced acute liver failure in a center without the facility of liver transplantation. Hepatol Int. 2009;3(4):563-570. doi:10.1007/s12072-009-9151-0.
17. Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7-8):1287-1293. doi:10.1345/aph.1P135.
18. O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology. 1989;97(2):439-445.
Niacin, also known as vitamin B3, is an important cofactor in many metabolic processes necessary to life. Over the past 15 to 20 years, niacin has been prescribed to patients with hyperlipidemia to increase high-density lipoprotein and lower low-density lipoprotein.1 As a naturally occurring vitamin, niacin is also available over-the-counter (OTC) as a dietary supplement, and is also a common ingredient in energy drinks and multivitamins.2
In addition to treating hyperlipidemia and as a nutritional supplement, some anecdotal reports amongst lay-persons suggests that niacin offers other health benefits, such as promoting weight loss and expediting the elimination of alcohol and illicit drugs from one’s system (eg, marijuana).3,4 The increased use of niacin supplementation in the general population for all of the aforementioned reasons has resulted in an increased incidence of niacin toxicity.
Formulations
Niacin is available in three formulations: extended-release (ER, also referred to as intermediate-release), immediate-release (IR), and sustained-release (SR).
The ER formulations of niacin are typically prescribed to treat hyperlipidemia. Patients are usually started on ER niacin at an initial dose of 250 mg once daily. The dose is gradually increased, as tolerated or necessary, to 2 g per day, taken in three doses. It is not uncommon for patients with hyperlipidemia to take more than 1 g of niacin per day after titration by their primary physicians.
Side Effects
Since niacin increases the release of arachidonic acid from cell membranes that metabolizes into prostaglandins, specifically prostaglandins E2 and D2, many patients taking niacin experience uncomfortable flushing and itching.5 Nonsteroidal anti-inflammatory drugs (NSAIDs) prevent this side effect by inhibiting the metabolism of arachidonic acid into those vasodilatory prostaglandins. The newer ER and SR formulations of niacin, which are approved for OTC use as a dietary supplement, are less likely to cause flushing.5
Extended-release niacin, however, is associated with a higher incidence of hepatotoxicity than the other prescription formulations of niacin.6 Toxicity has been well recognized in patients taking niacin chronically for hyperlipidemia, with reports of such cases dating back to the 1980s.7,8 We report a unique case of niacin toxicity following a single-dose ingestion in a young man.
Case
A 22-year-old man presented to the ED for evaluation of a 2-week history of intermittent periumbilical abdominal pain. This visit represented the patient’s second visit to the ED over the past week for the same complaint.
Upon presentation the patient’s vital signs were: blood pressure (BP), 113/64 mm Hg; heart rate, 82 beats/min; respiratory rate, 16 breaths/min; and temperature 36.6°C. Oxygen saturation was 100% on room air. The patient was otherwise healthy and had no significant recent or remote medical history. He denied taking any medications prior to his initial presentation, and reported only occasional alcohol use.
At the patient’s initial presentation 1 week earlier, he was diagnosed with acute gastroenteritis and treated with famotidine and ondansetron in the ED. The patient appeared well clinically at this visit, and laboratory values were within normal limits, including normal blood glucose and urinalysis.
The patient was discharged home from this first visit with prescriptions of famotidine and ondansetron, and was advised to follow up with his primary care physician in 1 week. Throughout the week after discharge from the ED, the patient experienced worsening abdominal pain, and he developed frequent nonbloody emesis, prompting his second presentation to the ED. At this second visit, the patient stated that he had taken one dose of ondansetron at home, without effect. He also noted subjective fevers, but had no diarrhea or melena.
Vital signs remained within normal limits with BPs ranging from 115 to 130 mm Hg systolic and 50 to 89 mm Hg diastolic. The patient was never tachycardic, tachypneic, febrile, or hypoxic. Physical examination was remarkable for periumbilical tenderness. The patient had no jaundice. A more thorough laboratory evaluation revealed elevated anion gap and blood urea nitrogen/creatinine values, and leukocytosis. The patient’s hepatic enzymes were also elevated, with aspartate aminotransferase (AST) over 2,000 U/L and alanine aminotransferase (ALT) of 1,698 U/L. Lipase, bilirubin, and alkaline phosphatase were all within normal limits. The patient’s prothrombin time (PT) was elevated at 14 seconds, and the international normalized ratio (INR) was elevated at 1.28. Laboratory analysis for acetaminophen and alcohol was negative.
A computed tomography (CT) scan of the abdomen/pelvis with intravenous (IV) contrast was unremarkable, demonstrating a liver devoid of any masses, portal or biliary dilation, or cirrhotic changes.
The patient received IV famotidine and ondansetron, and morphine for pain control, and was admitted to the general medical floor for hepatitis of uncertain etiology. A viral hepatitis panel was negative.
On the recommendation of the toxicology service, the patient was given N-acetylcysteine (NAC), and his hepatic enzymes trended down to an AST of 642 U/L and an ALT of 456 U/L by hospital day 2. (The patient essentially completed a positive dechallenge test).9
A gastroenterology consult was ordered, during which additional history-taking and chart review noted that the patient admitted to taking one or two tablets of OTC niacin as a dietary supplement the day before his initial presentation. Although the patient could not recall the exact dosage, he stated that he had been taking supplemental niacin approximately once a month over the past several years without any issues. Since OTC niacin is most commonly available in 500-mg tablets, this suggested the patient’s recent one-time ingested dose was approximately 500 to 1,000 mg.
Based on the patient’s admission to niacin use, additional studies were ordered, including an abdominal ultrasound and a urine drug screen. Ultrasound findings were unremarkable for portal venous thrombosis. The urine drug screen, however, was positive for marijuana and opiates. While the patient denied any history of opioid use, the positive opiate assay could have been attributed to the morphine given in the ED.
Throughout the patient’s hospital course, he remained normotensive and had no change in mental status. His liver enzymes, PT, and INR continued to normalize, and he was discharged home after 3 days, with instructions to follow up with the gastrointestinal clinic within 11 days. An appointment was made for him, which he did not attend.
Given the patient’s negative autoimmune and viral workup, and rapid resolution of symptoms after discontinuing niacin use, it is believed that he had an acute drug-induced hepatitis due to niacin ingestion. Regarding any coingestants that could have contributed to the hepatitis, the patient denied taking other common coingestants such as alcohol and acetaminophen; this assertion was supported by laboratory results.
Since we were unable to attain a qualitative measurement of the patient’s niacin concentration, our diagnosis was primarily based on the patient’s reported history.10 It is possible the patient had been taking more niacin than that to which he admitted, or that he was taking another hepatotoxic substance not detected on our toxicology workup. As previously noted, there are many medications and/or dietary supplements that could cause or contribute to a synergistic effect of drug-induced hepatitis for which the patient was not tested at his initial presentation. The patient could have co-ingested this large dose of niacin with acetaminophen and/or other supplements, energy drinks, or alcohol. A combination such as this could have contributed to his hepatitis, and the metabolites of these other substances would have been eliminated by the time of his second ED presentation.
Discussion
There are over 900 different drugs, toxins, and supplements known to cause hepatic injury.11,12 Clinical manifestations of toxicity range from asymptomatic incidental elevations in transaminases to fulminant liver failure causing mortality. Ingestion of commonly used medications such as statins (although not in overdose quantities) can cause transient asymptomatic transaminitis.13 These elevations are usually mild—ie, less than twice the upper limit of normal. Patients who experience such elevations can usually continue to take the medications with frequent and vigilant monitoring of hepatic function.
Signs and Symptoms
Acute Liver Injury. Acute liver injury is diagnosed when AST and ALT levels are greater than twice the upper limit of normal. Patients also typically have mild-to-moderate abdominal findings, such as pain, nausea, and vomiting—as was experienced by our patient. Along with niacin, angiotensin-converting enzyme inhibitors, NSAIDs, and antifungal medications are examples of other medications that can cause this degree of drug-induced hepatitis.
Severe Liver Injury. Severe liver injury features elevations in not only AST and ALT, but also alkaline phosphate and bilirubin. Patients with severe hepatic injury appear clinically ill and may exhibit altered mental status and jaundice. This type of subfulminant hepatic failure commonly results from acetaminophen toxicity, anesthetic gases, iron toxicity, phosphorus toxicity, and cocaine toxicity. Examples of drugs that result in massive liver necrosis and fulminant hepatitis are acetaminophen, isoniazid, phenelzine, phenytoin, propylthiouracil, and sertraline. Patients with massive hepatic necrosis and hepatitis may require liver transplantation.
Etiology
Identifying the etiology of liver injury is made largely through the patient’s history because there are simply too many possible hepatotoxic agents to test for them all. Diagnostic suspicion of hepatic toxicity should be increased with signs of more serious disease; however, drug-induced liver injury should be included in the differential diagnosis for all cases of abdominal pain.
With respect to the patient in our case, obtaining a more complete history involving supplement and vitamin use would have allowed us to make the diagnosis in the ED. Unfortunately, these subtle aspects of a patient’s history are often overlooked in the emergent care setting.
Treatment
The treatment of niacin-induced liver injury is similar to the guidelines for treating most other drug-induced pathology.14 Removal of the offending agent and providing supportive care is the primary treatment modality.15 In addition, it is important that the clinician exclude and rule-out other causes of hepatitis such as those of viral, autoimmune, or ischemic etiology.
N-acetylcysteine. A medication classically used in patients with acetaminophen overdose, NAC is a safe and effective treatment for non-acetaminophen-induced liver injury, and was given to treat our patient.16L-carnitine. L-carnitine has been shown to be effective in cases of chronic steatosis from hepatitis C and in valproic acid induced hepatitis.17Since L-carnitine is not included on our hospital’s formulary, it was not a treatment option for our patient.Glucocorticoid Therapy. Although glucocorticoids are occasionally given to patients with systemic symptoms of drug reactions, its effectiveness has not been adequately studied.18
Prognosis
The prognosis of patients with acute drug-induced hepatitis is generally good, and most patients fully recover once the offending agent is removed. Poor prognostic factors include the presence of jaundice, requirement for dialysis, underlying chronic liver conditions, or elevated serum creatinine. While most patients will experience a complete recovery, approximately 5% to 10% will develop chronic hepatitis and/or cirrhosis.
Conclusion
Niacin is now available as prescription and OTC formulations and is a potentially hepatotoxic medication and dietary supplement. Niacin can cause an acute hepatitis, especially when taken in conjunction with other hepatotoxic substances. Drug-induced liver injury from niacin ingestion will improve quickly following removal, and the prognosis in otherwise healthy individuals is good.
Patients, especially young, healthy patients who present with symptoms concerning for hepatitis, should be asked specifically about any nutritional, herbal, or other supplement usage. During the history intake, many patients do not consider vitamins or other nutritional or herbal supplements as “medication” or as being significant, and only report prescription and OTC medications.
Niacin, also known as vitamin B3, is an important cofactor in many metabolic processes necessary to life. Over the past 15 to 20 years, niacin has been prescribed to patients with hyperlipidemia to increase high-density lipoprotein and lower low-density lipoprotein.1 As a naturally occurring vitamin, niacin is also available over-the-counter (OTC) as a dietary supplement, and is also a common ingredient in energy drinks and multivitamins.2
In addition to treating hyperlipidemia and as a nutritional supplement, some anecdotal reports amongst lay-persons suggests that niacin offers other health benefits, such as promoting weight loss and expediting the elimination of alcohol and illicit drugs from one’s system (eg, marijuana).3,4 The increased use of niacin supplementation in the general population for all of the aforementioned reasons has resulted in an increased incidence of niacin toxicity.
Formulations
Niacin is available in three formulations: extended-release (ER, also referred to as intermediate-release), immediate-release (IR), and sustained-release (SR).
The ER formulations of niacin are typically prescribed to treat hyperlipidemia. Patients are usually started on ER niacin at an initial dose of 250 mg once daily. The dose is gradually increased, as tolerated or necessary, to 2 g per day, taken in three doses. It is not uncommon for patients with hyperlipidemia to take more than 1 g of niacin per day after titration by their primary physicians.
Side Effects
Since niacin increases the release of arachidonic acid from cell membranes that metabolizes into prostaglandins, specifically prostaglandins E2 and D2, many patients taking niacin experience uncomfortable flushing and itching.5 Nonsteroidal anti-inflammatory drugs (NSAIDs) prevent this side effect by inhibiting the metabolism of arachidonic acid into those vasodilatory prostaglandins. The newer ER and SR formulations of niacin, which are approved for OTC use as a dietary supplement, are less likely to cause flushing.5
Extended-release niacin, however, is associated with a higher incidence of hepatotoxicity than the other prescription formulations of niacin.6 Toxicity has been well recognized in patients taking niacin chronically for hyperlipidemia, with reports of such cases dating back to the 1980s.7,8 We report a unique case of niacin toxicity following a single-dose ingestion in a young man.
Case
A 22-year-old man presented to the ED for evaluation of a 2-week history of intermittent periumbilical abdominal pain. This visit represented the patient’s second visit to the ED over the past week for the same complaint.
Upon presentation the patient’s vital signs were: blood pressure (BP), 113/64 mm Hg; heart rate, 82 beats/min; respiratory rate, 16 breaths/min; and temperature 36.6°C. Oxygen saturation was 100% on room air. The patient was otherwise healthy and had no significant recent or remote medical history. He denied taking any medications prior to his initial presentation, and reported only occasional alcohol use.
At the patient’s initial presentation 1 week earlier, he was diagnosed with acute gastroenteritis and treated with famotidine and ondansetron in the ED. The patient appeared well clinically at this visit, and laboratory values were within normal limits, including normal blood glucose and urinalysis.
The patient was discharged home from this first visit with prescriptions of famotidine and ondansetron, and was advised to follow up with his primary care physician in 1 week. Throughout the week after discharge from the ED, the patient experienced worsening abdominal pain, and he developed frequent nonbloody emesis, prompting his second presentation to the ED. At this second visit, the patient stated that he had taken one dose of ondansetron at home, without effect. He also noted subjective fevers, but had no diarrhea or melena.
Vital signs remained within normal limits with BPs ranging from 115 to 130 mm Hg systolic and 50 to 89 mm Hg diastolic. The patient was never tachycardic, tachypneic, febrile, or hypoxic. Physical examination was remarkable for periumbilical tenderness. The patient had no jaundice. A more thorough laboratory evaluation revealed elevated anion gap and blood urea nitrogen/creatinine values, and leukocytosis. The patient’s hepatic enzymes were also elevated, with aspartate aminotransferase (AST) over 2,000 U/L and alanine aminotransferase (ALT) of 1,698 U/L. Lipase, bilirubin, and alkaline phosphatase were all within normal limits. The patient’s prothrombin time (PT) was elevated at 14 seconds, and the international normalized ratio (INR) was elevated at 1.28. Laboratory analysis for acetaminophen and alcohol was negative.
A computed tomography (CT) scan of the abdomen/pelvis with intravenous (IV) contrast was unremarkable, demonstrating a liver devoid of any masses, portal or biliary dilation, or cirrhotic changes.
The patient received IV famotidine and ondansetron, and morphine for pain control, and was admitted to the general medical floor for hepatitis of uncertain etiology. A viral hepatitis panel was negative.
On the recommendation of the toxicology service, the patient was given N-acetylcysteine (NAC), and his hepatic enzymes trended down to an AST of 642 U/L and an ALT of 456 U/L by hospital day 2. (The patient essentially completed a positive dechallenge test).9
A gastroenterology consult was ordered, during which additional history-taking and chart review noted that the patient admitted to taking one or two tablets of OTC niacin as a dietary supplement the day before his initial presentation. Although the patient could not recall the exact dosage, he stated that he had been taking supplemental niacin approximately once a month over the past several years without any issues. Since OTC niacin is most commonly available in 500-mg tablets, this suggested the patient’s recent one-time ingested dose was approximately 500 to 1,000 mg.
Based on the patient’s admission to niacin use, additional studies were ordered, including an abdominal ultrasound and a urine drug screen. Ultrasound findings were unremarkable for portal venous thrombosis. The urine drug screen, however, was positive for marijuana and opiates. While the patient denied any history of opioid use, the positive opiate assay could have been attributed to the morphine given in the ED.
Throughout the patient’s hospital course, he remained normotensive and had no change in mental status. His liver enzymes, PT, and INR continued to normalize, and he was discharged home after 3 days, with instructions to follow up with the gastrointestinal clinic within 11 days. An appointment was made for him, which he did not attend.
Given the patient’s negative autoimmune and viral workup, and rapid resolution of symptoms after discontinuing niacin use, it is believed that he had an acute drug-induced hepatitis due to niacin ingestion. Regarding any coingestants that could have contributed to the hepatitis, the patient denied taking other common coingestants such as alcohol and acetaminophen; this assertion was supported by laboratory results.
Since we were unable to attain a qualitative measurement of the patient’s niacin concentration, our diagnosis was primarily based on the patient’s reported history.10 It is possible the patient had been taking more niacin than that to which he admitted, or that he was taking another hepatotoxic substance not detected on our toxicology workup. As previously noted, there are many medications and/or dietary supplements that could cause or contribute to a synergistic effect of drug-induced hepatitis for which the patient was not tested at his initial presentation. The patient could have co-ingested this large dose of niacin with acetaminophen and/or other supplements, energy drinks, or alcohol. A combination such as this could have contributed to his hepatitis, and the metabolites of these other substances would have been eliminated by the time of his second ED presentation.
Discussion
There are over 900 different drugs, toxins, and supplements known to cause hepatic injury.11,12 Clinical manifestations of toxicity range from asymptomatic incidental elevations in transaminases to fulminant liver failure causing mortality. Ingestion of commonly used medications such as statins (although not in overdose quantities) can cause transient asymptomatic transaminitis.13 These elevations are usually mild—ie, less than twice the upper limit of normal. Patients who experience such elevations can usually continue to take the medications with frequent and vigilant monitoring of hepatic function.
Signs and Symptoms
Acute Liver Injury. Acute liver injury is diagnosed when AST and ALT levels are greater than twice the upper limit of normal. Patients also typically have mild-to-moderate abdominal findings, such as pain, nausea, and vomiting—as was experienced by our patient. Along with niacin, angiotensin-converting enzyme inhibitors, NSAIDs, and antifungal medications are examples of other medications that can cause this degree of drug-induced hepatitis.
Severe Liver Injury. Severe liver injury features elevations in not only AST and ALT, but also alkaline phosphate and bilirubin. Patients with severe hepatic injury appear clinically ill and may exhibit altered mental status and jaundice. This type of subfulminant hepatic failure commonly results from acetaminophen toxicity, anesthetic gases, iron toxicity, phosphorus toxicity, and cocaine toxicity. Examples of drugs that result in massive liver necrosis and fulminant hepatitis are acetaminophen, isoniazid, phenelzine, phenytoin, propylthiouracil, and sertraline. Patients with massive hepatic necrosis and hepatitis may require liver transplantation.
Etiology
Identifying the etiology of liver injury is made largely through the patient’s history because there are simply too many possible hepatotoxic agents to test for them all. Diagnostic suspicion of hepatic toxicity should be increased with signs of more serious disease; however, drug-induced liver injury should be included in the differential diagnosis for all cases of abdominal pain.
With respect to the patient in our case, obtaining a more complete history involving supplement and vitamin use would have allowed us to make the diagnosis in the ED. Unfortunately, these subtle aspects of a patient’s history are often overlooked in the emergent care setting.
Treatment
The treatment of niacin-induced liver injury is similar to the guidelines for treating most other drug-induced pathology.14 Removal of the offending agent and providing supportive care is the primary treatment modality.15 In addition, it is important that the clinician exclude and rule-out other causes of hepatitis such as those of viral, autoimmune, or ischemic etiology.
N-acetylcysteine. A medication classically used in patients with acetaminophen overdose, NAC is a safe and effective treatment for non-acetaminophen-induced liver injury, and was given to treat our patient.16L-carnitine. L-carnitine has been shown to be effective in cases of chronic steatosis from hepatitis C and in valproic acid induced hepatitis.17Since L-carnitine is not included on our hospital’s formulary, it was not a treatment option for our patient.Glucocorticoid Therapy. Although glucocorticoids are occasionally given to patients with systemic symptoms of drug reactions, its effectiveness has not been adequately studied.18
Prognosis
The prognosis of patients with acute drug-induced hepatitis is generally good, and most patients fully recover once the offending agent is removed. Poor prognostic factors include the presence of jaundice, requirement for dialysis, underlying chronic liver conditions, or elevated serum creatinine. While most patients will experience a complete recovery, approximately 5% to 10% will develop chronic hepatitis and/or cirrhosis.
Conclusion
Niacin is now available as prescription and OTC formulations and is a potentially hepatotoxic medication and dietary supplement. Niacin can cause an acute hepatitis, especially when taken in conjunction with other hepatotoxic substances. Drug-induced liver injury from niacin ingestion will improve quickly following removal, and the prognosis in otherwise healthy individuals is good.
Patients, especially young, healthy patients who present with symptoms concerning for hepatitis, should be asked specifically about any nutritional, herbal, or other supplement usage. During the history intake, many patients do not consider vitamins or other nutritional or herbal supplements as “medication” or as being significant, and only report prescription and OTC medications.
1. Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97(9):2969-2989. doi:10.1210/jc.2011-3213.
2. Vivekanandarajah A, Ni S, Waked A. Acute hepatitis in a woman following excessive ingestion of an energy drink: a case report. J Med Case Rep. 2011;5:227. doi:10.1186/1752-1947-5-227.
3. Niacin. U.S. National Library of Medicine. https://medlineplus.gov/druginfo/meds/a682518.html. Updated July 15, 2017. Accessed February 21, 2018.
4. Addiction Resource. Niacin flush for drug detox. https://addictionresource.com/drug-testing/niacin-drug-test/. Accessed February 20, 2018.
5. Kamanna VS, Ganji SH, Kashyap ML. The mechanism and mitigation of niacin-induced flushing. Int J Clin Pract. 2009;63(9):1369-1377. doi:10.1111/j.1742-1241.2009.02099.x.
6. Etchason JA, Miller TD, Squires RW, et al. Niacin-induced hepatitis: a potential side effect with low-dose time-release niacin. Mayo Clin Proc. 1991;66(1):23-28.
7. Patterson DJ, Dew EW, Gyorkey F, Graham DY. Niacin hepatitis. South Med J. 1983;76(2):239-241.
8. Ferenchick G, Rovner D. Hepatitis and hematemesis complicating nicotinic acid use. Am J Med Sci. 1989;298(3):191-193.
9. Henkin Y, Johnson KC, Segrest JP. Rechallenge with crystalline niacin after drug-induced hepatitis from sustained-release niacin. JAMA. 1990;264(2):241-243.
10. Barritt AS 4th, Lee J, Hayashi PH. Detective work in drug-induced liver injury: sometimes it is all about interviewing the right witness. Clin Gastroenterol Hepatol. 2010;8(7):635-637. doi:10.1016/j.cgh.2010.03.020.
11. Chitturi S, Teoh NC, Farrell GC. Hepatic drug metabolism and liver disease caused by drugs. In: Feldman M, Friedman LS, Brandt LJ, eds. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. 10th ed. Philadelphia, PA: Elsevier Saunders; 2016:1442-1477.
12. National Institutes of Health. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Niacin. https://livertox.nih.gov/Niacin.htm. Updated January 18, 2018. Accessed February 21, 2018.
13. Chalasani N. Statins and hepatotoxicity: focus on patients with fatty liver. Hepatology. 2005;41(4):690-695.
14. Chalasani NP, Hayashi PH, Bonkovsky HL, et al. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. Am J Gastroenterol. 2014;109(7):950-966. doi:10.1038/ajg.2014.131.
15. Larson AM. Hepatotoxicity due to herbal medication and dietary supplements. UpToDate Web site. https://www.uptodate.com/contents/hepatotoxicity-due-to-herbal-medications-and-dietary-supplements?source=search_result&search=niacin%20induced%20hepatitis&selectedTitle=3~150. Updated December 21, 2017. Accessed February 21, 2018.
16. Mumtaz K, Azam Z, Hamid S, et al. Role of N-acetylcysteine in adults with non-acetaminophen-induced acute liver failure in a center without the facility of liver transplantation. Hepatol Int. 2009;3(4):563-570. doi:10.1007/s12072-009-9151-0.
17. Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7-8):1287-1293. doi:10.1345/aph.1P135.
18. O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology. 1989;97(2):439-445.
1. Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97(9):2969-2989. doi:10.1210/jc.2011-3213.
2. Vivekanandarajah A, Ni S, Waked A. Acute hepatitis in a woman following excessive ingestion of an energy drink: a case report. J Med Case Rep. 2011;5:227. doi:10.1186/1752-1947-5-227.
3. Niacin. U.S. National Library of Medicine. https://medlineplus.gov/druginfo/meds/a682518.html. Updated July 15, 2017. Accessed February 21, 2018.
4. Addiction Resource. Niacin flush for drug detox. https://addictionresource.com/drug-testing/niacin-drug-test/. Accessed February 20, 2018.
5. Kamanna VS, Ganji SH, Kashyap ML. The mechanism and mitigation of niacin-induced flushing. Int J Clin Pract. 2009;63(9):1369-1377. doi:10.1111/j.1742-1241.2009.02099.x.
6. Etchason JA, Miller TD, Squires RW, et al. Niacin-induced hepatitis: a potential side effect with low-dose time-release niacin. Mayo Clin Proc. 1991;66(1):23-28.
7. Patterson DJ, Dew EW, Gyorkey F, Graham DY. Niacin hepatitis. South Med J. 1983;76(2):239-241.
8. Ferenchick G, Rovner D. Hepatitis and hematemesis complicating nicotinic acid use. Am J Med Sci. 1989;298(3):191-193.
9. Henkin Y, Johnson KC, Segrest JP. Rechallenge with crystalline niacin after drug-induced hepatitis from sustained-release niacin. JAMA. 1990;264(2):241-243.
10. Barritt AS 4th, Lee J, Hayashi PH. Detective work in drug-induced liver injury: sometimes it is all about interviewing the right witness. Clin Gastroenterol Hepatol. 2010;8(7):635-637. doi:10.1016/j.cgh.2010.03.020.
11. Chitturi S, Teoh NC, Farrell GC. Hepatic drug metabolism and liver disease caused by drugs. In: Feldman M, Friedman LS, Brandt LJ, eds. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. 10th ed. Philadelphia, PA: Elsevier Saunders; 2016:1442-1477.
12. National Institutes of Health. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Niacin. https://livertox.nih.gov/Niacin.htm. Updated January 18, 2018. Accessed February 21, 2018.
13. Chalasani N. Statins and hepatotoxicity: focus on patients with fatty liver. Hepatology. 2005;41(4):690-695.
14. Chalasani NP, Hayashi PH, Bonkovsky HL, et al. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. Am J Gastroenterol. 2014;109(7):950-966. doi:10.1038/ajg.2014.131.
15. Larson AM. Hepatotoxicity due to herbal medication and dietary supplements. UpToDate Web site. https://www.uptodate.com/contents/hepatotoxicity-due-to-herbal-medications-and-dietary-supplements?source=search_result&search=niacin%20induced%20hepatitis&selectedTitle=3~150. Updated December 21, 2017. Accessed February 21, 2018.
16. Mumtaz K, Azam Z, Hamid S, et al. Role of N-acetylcysteine in adults with non-acetaminophen-induced acute liver failure in a center without the facility of liver transplantation. Hepatol Int. 2009;3(4):563-570. doi:10.1007/s12072-009-9151-0.
17. Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7-8):1287-1293. doi:10.1345/aph.1P135.
18. O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology. 1989;97(2):439-445.
Invasive Penile Squamous Cell Carcinoma
Invasive penile cancer is a rare malignancy with considerable morbidity and mortality. The American Cancer Society estimates that there will be 2320 new cases of invasive penile cancer in the United States in 2018, of which primary penile squamous cell carcinoma (PSCC) represents the majority.1 In one study, the mean age at diagnosis was 60 years, with PSCC occurring only rarely in men younger than 35 years of age (estimated incidence, 0.01 cases per 100,000 individuals).2 Presentation to a physician generally occurs more than 1 year after initial onset of symptoms or clinical lesion(s). This delay in diagnosis and treatment often results in disease progression,3 which can have a devastating outcome.4 Therefore, physicians should maintain a high index of clinical suspicion for PSCC, particularly in young or middle-aged patients in whom presentation of PSCC is uncommon. The most commonly associated risk factors for PSCC include lack of circumcision (specifically during the neonatal period), high-risk human papillomavirus (HPV) infection, and tobacco use.5 Chronic alcoholism also has been linked to PSCC.6 It also is common in patients without health insurance.7 We report the case of a 27-year-old circumcised man who presented with invasive PSCC following a diagnosis of condyloma 8 years prior by an outside physician.
Case Report
A 27-year-old man presented for evaluation of persistent genital warts that had been diagnosed 8 years prior. His medical history was remarkable for intravenous drug use, active hepatitis C infection, tobacco smoking, chronic alcohol use, and mild asthma. Eight years prior to the current presentation, 7 lesions had developed on the penis and were diagnosed by an outside physician as condyloma, which was treated with cryotherapy and topical imiquimod. All of the lesions except for 1 responded to treatment. The residual lesion continued to grow until the size prompted him to contact his primary care physician, who referred him for dermatologic evaluation. The patient cited lack of health insurance as the primary reason he did not seek follow-up treatment after the initial evaluation and treatment 8 years prior.
Physical examination at the current presentation revealed a circumcised man with an asymptomatic, 2.6-cm, pink, friable, verrucous mass on the left lateral penile shaft (Figure 1) and otherwise unremarkable penile architecture. A clinically enlarged, nontender right inguinal lymph node was noted as well as subtle enlargement of a left inguinal lymph node. An excisional biopsy was performed with pathologic evaluation confirming a diagnosis of high-grade invasive squamous cell carcinoma (SCC) arising in the setting of squamous cell carcinoma in situ (Figure 2). Lymphovascular invasion was highlighted on cluster of differentiation 31 and podoplanin immunostaining (Figure 3). The patient was subsequently referred to urology and hematology-oncology specialists for further evaluation. Computed tomography (CT) of the abdomen and pelvis confirmed the contralaterally enlarged right inguinal lymph node discovered during physical examination and mildly enlarged ipsilateral inguinal, obturator, and external iliac nodes. Computed tomography–guided fine-needle aspiration of the right inguinal node confirmed the diagnosis of contralateral locoregional metastasis. Further evaluation with positron emission tomography/CT imaging revealed only a single metabolically active region confined to the right inguinal node. The patient’s history of active hepatitis C complicated proposed neoadjuvant chemotherapy regimens. Ultimately, after discussion with multiple surgical and oncologist specialties within our institution and others, a treatment plan was formulated. The patient underwent robotic laparoscopic bilateral pelvic and inguinal lymph node dissection and re-excision of the primary PSCC, with one of 15 right superficial inguinal nodes testing positive for tumor cells; the left superficial and bilateral deep inguinal lymph nodes were negative for SCC.
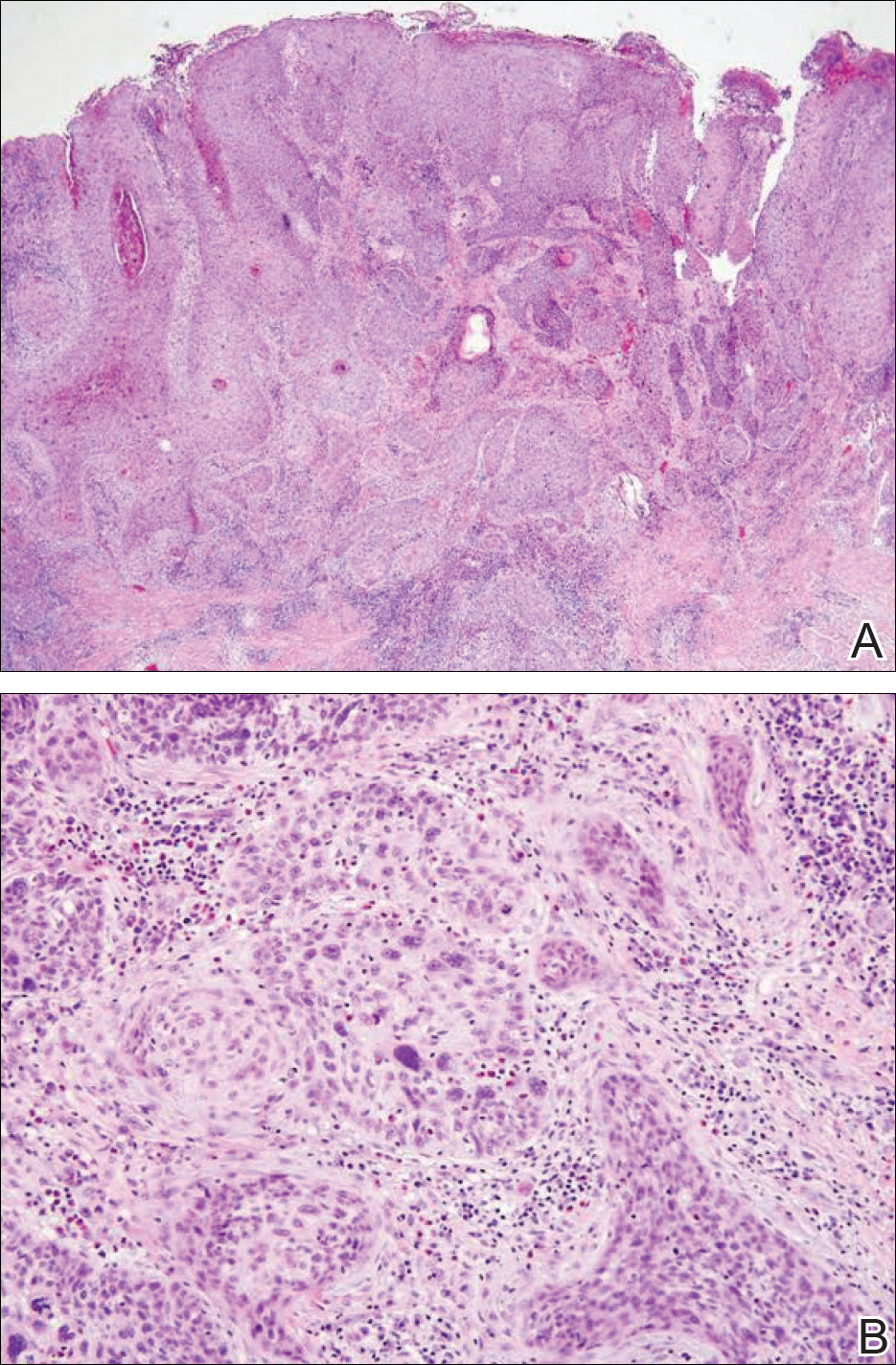
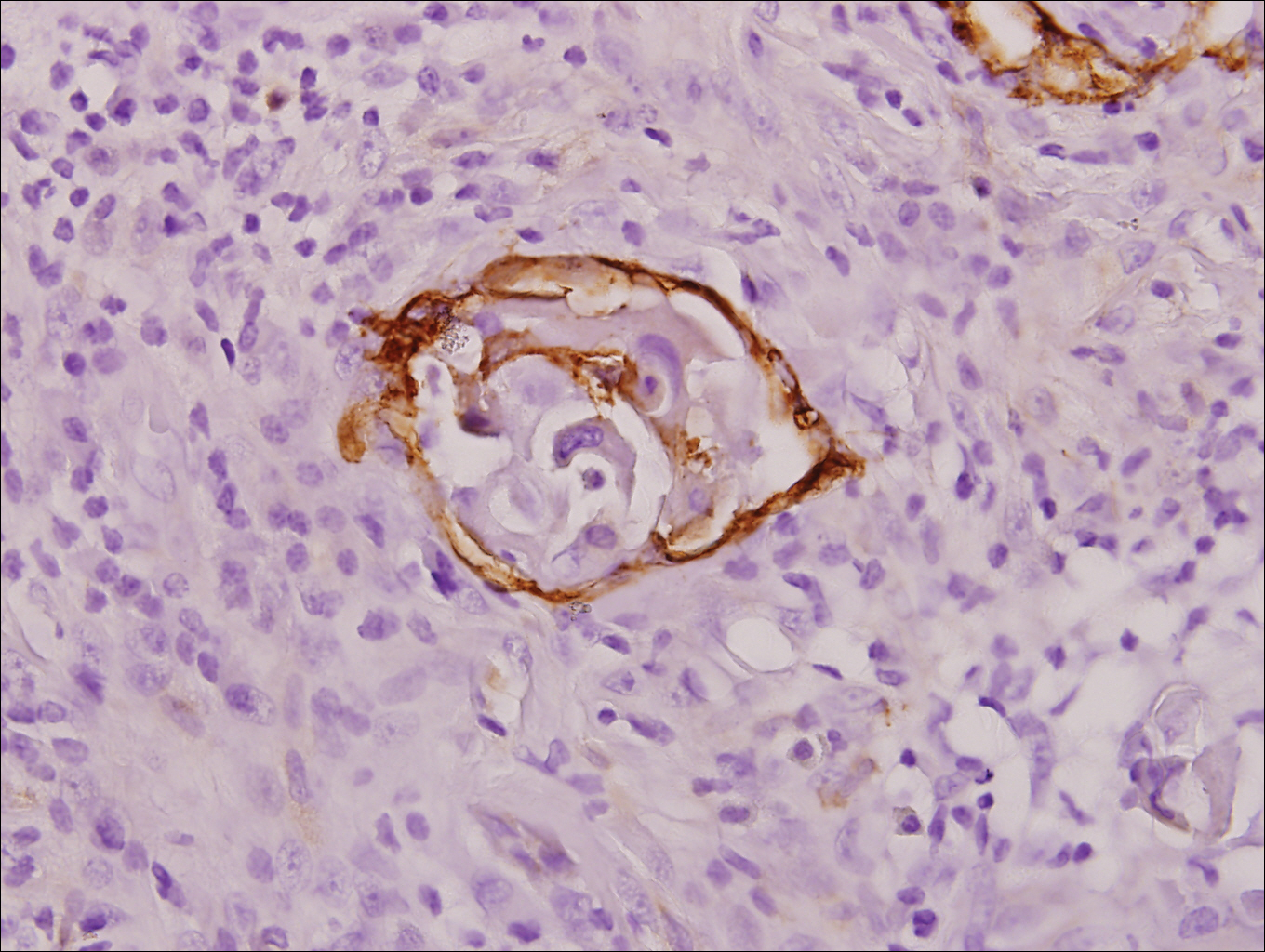
Repeat positron emission tomography/CT imaging at 6 months’ follow-up showed no evidence of active disease. On 1-year follow-up, a CT scan did not show any new or residual disease, but the patient continued to have edema of the bilateral legs, which began after lymph node dissection and was managed with physical therapy and compression stockings.
Comment
Prevalence
Penile cancer is rare in industrialized countries. Early detection is a critical factor for both overall survival and organ function. If successful interventions are to be made, physicians should be familiar with known risk factors as well as unusual presentations, such as lesions presenting in young circumcised men, as reported above. Similarly, tumors located on the shaft of the penis represent an uncommon location for tumor presentation, occurring in less than 5% of PSCC cases.8 Penile SCC most commonly develops as a solitary painless lesion on the glans, balanopreputial sulcus and/or prepuce.9 In our case, histopathology confirmed high-grade invasive SCC arising from squamous cell carcinoma in situ, an entity generally associated with older men with a 10% to 20% rate of progression into invasive SCC.9 Our patient denied any clinical change in the appearance of the tumor in the years prior to the current presentation, making it possible that the condyloma treated 8 years prior was squamous cell carcinoma in situ or PSCC. As many as 25% of premalignant lesions are mistaken for benign lesions, which can thus delay treatment and allow progression to malignancy.10
Diagnosis
Penile SCC often is etiologically subcategorized into 2 pathways based on HPV dependence or independence. Recent research suggests that this distinction often is difficult to make, and accurate laboratory and pathologic confirmation of HPV DNA, intact virions, and viral-related cutaneous changes is not always possible, leading to much speculation regarding the exact role of HPV in tumorigenesis.11 Cancers developing in the absence of HPV DNA often occur secondary to chronic inflammatory conditions such as lichen planus or lichen sclerosus. Human papillomavirus DNA has shown to be present in 70% to 100% of all SCC in situ of the penis11; therefore, the transformation of in situ disease to an invasive tumor in our patient most likely occurred via an HPV-dependent pathway. Viral carcinogenesis in the HPV-dependent pathway involves inactivation of host cell cycle regulatory proteins, specifically the retinoblastoma and p53 regulatory proteins by the viral oncoproteins E7 and E6, respectively.12,13 Human papillomavirus–dependent pathways are related to a patient’s age at first sexual intercourse, number of sexual partners, and history of condyloma and other sexually transmitted diseases.14,15 High-risk HPV types 16 and 18 are the most common viral types found in HPV related premalignant lesions, making it possible to decrease the incidence of PSCC with recently developed vaccines.16 Human papillomavirus vaccines have been shown to reduce the incidence of anal intraepithelial neoplasias and genital warts in men.17 While the effects of the HPV vaccine on reducing PSCC could not be assessed in the study due to low incidence of disease (both in the study population and in general), it is thought that HPV vaccination could potentially decrease the incidence of all PSCCs by one third, making it an important resource in the primary prevention of the disease.18
Management
Contemporary surgical management of PSCC has evolved from organ resection in toto for all PSCCs to a more conservative approach based upon tumor stage and grade. The standard margin for surgical resection of PSCC is 2 cm, a procedure often referred to as a partial penectomy. This remains the most common procedure for surgical resection of PSCC and has achieved good local control, with reported recurrence rates of 4% to 8%.19,20 Complication rates of the procedure are moderate one-third of patients experiencing compromise of sexual activity after surgery.21 With evidence that smaller resection margins may result in good local control and a lower incidence of postoperative functional impairment, resection margins of 5, 10, and 15 mm have been advocated for PSCCs of varying histologic grades and tumor stages.22-24 Treatment options for T1 and in situ tumors have expanded to include glansectomy, margin-controlled Mohs micrographic surgery, and ablative laser therapy for local disease control.5,20 More advanced tumors are still treated with partial or complete penectomy given the high risks for locoregional recurrence and distant spread.
Prognosis
The most important factor predicting survival in patients with PSCC is metastasis to inguinal lymph nodes. The 5-year survival rate for patients without nodal involvement is 85% to 100%, while those with pathologically positive lymph nodes have a 5-year survival rate of 15% to 45%.25 Once distant metastasis occurs, the mean time of survival is 7 to 10 months.26 Our patient presented with high-grade PSCC with histologic lymphovascular spread and palpable inguinal lymph nodes. When stratified with other similar cases at presentation, our patient was at a considerable risk for locoregional as well as distant metastasis. Management with regional nodal dissection with a plan for close observation (and deferment of chemotherapeutics) was based upon evaluations from multiple different medical specialties.
Conclusion
Invasive PSCC is rare in young circumcised adults, and a delay in diagnosis can lead to considerable morbidity and mortality. We present a case of invasive PSCC arising in the setting of squamous cell carcinoma in situ in an area previously treated with cryotherapy and imiquimod. Our patient’s young age, concurrent hepatitis C infection, and contralateral locoregional nodal metastasis made this a complex case, involving evaluation and treatment by multiple medical disciplines. This case highlights the importance of biopsy in any lesion recalcitrant to conventional modalities regardless of the patient’s age. Early detection and treatment of PSCC can prevent organ dysfunction, loss of organ, and even death.
- About penile cancer. American Cancer Society website. https://www.cancer.org/content/dam/CRC/PDF/Public/8783.00.pdf. Revised February 9, 2016. Accessed February 27, 2018.
- Barnholtz-Sloan JS, Maldonado JL, Pow-sang J, et al. Incidence trends in primary malignant penile cancer. Urol Oncol. 2007;25:361-367.
- Koifman L, Vides AJ, Koifman N, et al. Epidemiological aspects of penile cancer in Rio de Janeiro: evaluation of 230 cases. Int Braz J Urol. 2011;37:231-240.
- Kamat AM, Carpenter SM, Czerniak BA, et al. Metastatic penile cancer in a young Caucasian male: impact of delayed diagnosis. Urol Oncol. 2005;23:130-131.
- Deem S, Keane T, Bhavsar R, et al. Contemporary diagnosis and management of squamous cell carcinoma (SCC) of the penis. BJU Int. 2011;108:1378-1392.
- McIntyre M, Weiss A, Wahlquist A, et al. Penile cancer: an analysis of socioeconomic factors at a southeastern tertiary referral center. Can J Urol. 2011;18:5524-5528.
- Maden C, Sherman KJ, Beckmann AM, et al. History of circumcision, medical conditions, and sexual activity and risk of penile cancer. J Natl Cancer Inst. 1993;85:19-24.
- Hernandez BY, Barnholtz-Sloan J, German RR, et al. Burden of invasive squamous cell carcinoma of the penis in the United States, 1998-2003. Cancer. 2008;113(suppl 10):2883-2891.
- Ferrandiz-Pulido C, de Torres I, Garcia-Patos V. Penile squamous cell carcinoma. Actas Dermosifiliogr. 2012;103:478-487.
- Tietjen DN, Malek RS. Laser therapy of squamous cell dysplasia and carcinoma of the penis. Urology. 1998;52:559-565.
- Mannweiler S, Sygulla S, Winter E, et al. Two major pathways of penile carcinogenesis: HPV-induced penile cancers overexpress p16, HPV-negative cancers associated with dermatoses express p53, but lack p16 overexpression. J Am Acad Dermatol. 2013;69:73-81.
- Scheffner M, Werness BA, Huibregtse JM, et al. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129-1136.
- Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76-79.
- Daling JR, Madeleine MM, Johnson LG, et al. Penile cancer: importance of circumcision, human papillomavirus and smoking in in situ and invasive disease. Int J Cancer. 2005;116:606-616.
- Bleeker MC, Heideman DA, Snijders PJ, et al. Penile cancer: epidemiology, pathogenesis and prevention. World J Urol. 2009;27:141-150.
- Shabbir M, Barod R, Hegarty PK, et al. Primary prevention and vaccination for penile cancer. Ther Adv Urol. 2013;5:161-169.
- Palefsky J, Giuliano A, Goldstone S, et al. HPV vaccine against anal HPV infection and anal intraepithelial neoplasia. N Engl J Med. 2011;365:1576-1585.
- Backes DM, Kurman RJ, Pimenta JM, et al. Systematic review of human papillomavirus prevalence in invasive penile cancer. Cancer Causes Control. 2009;20:449-457.
- Korets R, Koppie TM, Snyder ME, et al. Partial penectomy for patients with squamous cell carcinoma of the penis: the Memorial Sloan-Kettering experience. Ann Surg Oncol. 2007;14:3614-3619.
- Zukiwskyj M, Daly P, Chung E. Penile cancer and phallus preservation strategies: a review of current literature. BJU Int. 2013;112(suppl 2):21-26.
- Romero FR, Romero KR, Mattos MA, et al. Sexual function after partial penectomy for penile cancer. Urology. 2005;66:1292-1295.
- Minhas S, Kayes O, Hegarty P, et al. What surgical resection margins are required to achieve oncological control in men with primary penile cancer? BJU Int. 2005;96:1040-1043.
- Feldman AS, McDougal WS. Long-term outcome of excisional organ sparing surgery for carcinoma of the penis. J Urol. 2011;186:1303-1307.
- Philippou P, Shabbir M, Malone P, et al. Conservative surgery for squamous cell carcinoma of the penis: resection margins and long-term oncological control. J Urol. 2012;188:803-808.
- Brady KL, Mercurio MG, Brown MD. Malignant tumors of the penis. Dermatol Surg. 2013;39:527-547.
- Ornellas AA, Nobrega BL, Wei Kin Chin E, et al. Prognostic factors in invasive squamous cell carcinoma of the penis: analysis of 196 patients treated at the Brazilian National Cancer Institute. J Urol. 2008;180:1354-1359.
Invasive penile cancer is a rare malignancy with considerable morbidity and mortality. The American Cancer Society estimates that there will be 2320 new cases of invasive penile cancer in the United States in 2018, of which primary penile squamous cell carcinoma (PSCC) represents the majority.1 In one study, the mean age at diagnosis was 60 years, with PSCC occurring only rarely in men younger than 35 years of age (estimated incidence, 0.01 cases per 100,000 individuals).2 Presentation to a physician generally occurs more than 1 year after initial onset of symptoms or clinical lesion(s). This delay in diagnosis and treatment often results in disease progression,3 which can have a devastating outcome.4 Therefore, physicians should maintain a high index of clinical suspicion for PSCC, particularly in young or middle-aged patients in whom presentation of PSCC is uncommon. The most commonly associated risk factors for PSCC include lack of circumcision (specifically during the neonatal period), high-risk human papillomavirus (HPV) infection, and tobacco use.5 Chronic alcoholism also has been linked to PSCC.6 It also is common in patients without health insurance.7 We report the case of a 27-year-old circumcised man who presented with invasive PSCC following a diagnosis of condyloma 8 years prior by an outside physician.
Case Report
A 27-year-old man presented for evaluation of persistent genital warts that had been diagnosed 8 years prior. His medical history was remarkable for intravenous drug use, active hepatitis C infection, tobacco smoking, chronic alcohol use, and mild asthma. Eight years prior to the current presentation, 7 lesions had developed on the penis and were diagnosed by an outside physician as condyloma, which was treated with cryotherapy and topical imiquimod. All of the lesions except for 1 responded to treatment. The residual lesion continued to grow until the size prompted him to contact his primary care physician, who referred him for dermatologic evaluation. The patient cited lack of health insurance as the primary reason he did not seek follow-up treatment after the initial evaluation and treatment 8 years prior.
Physical examination at the current presentation revealed a circumcised man with an asymptomatic, 2.6-cm, pink, friable, verrucous mass on the left lateral penile shaft (Figure 1) and otherwise unremarkable penile architecture. A clinically enlarged, nontender right inguinal lymph node was noted as well as subtle enlargement of a left inguinal lymph node. An excisional biopsy was performed with pathologic evaluation confirming a diagnosis of high-grade invasive squamous cell carcinoma (SCC) arising in the setting of squamous cell carcinoma in situ (Figure 2). Lymphovascular invasion was highlighted on cluster of differentiation 31 and podoplanin immunostaining (Figure 3). The patient was subsequently referred to urology and hematology-oncology specialists for further evaluation. Computed tomography (CT) of the abdomen and pelvis confirmed the contralaterally enlarged right inguinal lymph node discovered during physical examination and mildly enlarged ipsilateral inguinal, obturator, and external iliac nodes. Computed tomography–guided fine-needle aspiration of the right inguinal node confirmed the diagnosis of contralateral locoregional metastasis. Further evaluation with positron emission tomography/CT imaging revealed only a single metabolically active region confined to the right inguinal node. The patient’s history of active hepatitis C complicated proposed neoadjuvant chemotherapy regimens. Ultimately, after discussion with multiple surgical and oncologist specialties within our institution and others, a treatment plan was formulated. The patient underwent robotic laparoscopic bilateral pelvic and inguinal lymph node dissection and re-excision of the primary PSCC, with one of 15 right superficial inguinal nodes testing positive for tumor cells; the left superficial and bilateral deep inguinal lymph nodes were negative for SCC.
Repeat positron emission tomography/CT imaging at 6 months’ follow-up showed no evidence of active disease. On 1-year follow-up, a CT scan did not show any new or residual disease, but the patient continued to have edema of the bilateral legs, which began after lymph node dissection and was managed with physical therapy and compression stockings.
Comment
Prevalence
Penile cancer is rare in industrialized countries. Early detection is a critical factor for both overall survival and organ function. If successful interventions are to be made, physicians should be familiar with known risk factors as well as unusual presentations, such as lesions presenting in young circumcised men, as reported above. Similarly, tumors located on the shaft of the penis represent an uncommon location for tumor presentation, occurring in less than 5% of PSCC cases.8 Penile SCC most commonly develops as a solitary painless lesion on the glans, balanopreputial sulcus and/or prepuce.9 In our case, histopathology confirmed high-grade invasive SCC arising from squamous cell carcinoma in situ, an entity generally associated with older men with a 10% to 20% rate of progression into invasive SCC.9 Our patient denied any clinical change in the appearance of the tumor in the years prior to the current presentation, making it possible that the condyloma treated 8 years prior was squamous cell carcinoma in situ or PSCC. As many as 25% of premalignant lesions are mistaken for benign lesions, which can thus delay treatment and allow progression to malignancy.10
Diagnosis
Penile SCC often is etiologically subcategorized into 2 pathways based on HPV dependence or independence. Recent research suggests that this distinction often is difficult to make, and accurate laboratory and pathologic confirmation of HPV DNA, intact virions, and viral-related cutaneous changes is not always possible, leading to much speculation regarding the exact role of HPV in tumorigenesis.11 Cancers developing in the absence of HPV DNA often occur secondary to chronic inflammatory conditions such as lichen planus or lichen sclerosus. Human papillomavirus DNA has shown to be present in 70% to 100% of all SCC in situ of the penis11; therefore, the transformation of in situ disease to an invasive tumor in our patient most likely occurred via an HPV-dependent pathway. Viral carcinogenesis in the HPV-dependent pathway involves inactivation of host cell cycle regulatory proteins, specifically the retinoblastoma and p53 regulatory proteins by the viral oncoproteins E7 and E6, respectively.12,13 Human papillomavirus–dependent pathways are related to a patient’s age at first sexual intercourse, number of sexual partners, and history of condyloma and other sexually transmitted diseases.14,15 High-risk HPV types 16 and 18 are the most common viral types found in HPV related premalignant lesions, making it possible to decrease the incidence of PSCC with recently developed vaccines.16 Human papillomavirus vaccines have been shown to reduce the incidence of anal intraepithelial neoplasias and genital warts in men.17 While the effects of the HPV vaccine on reducing PSCC could not be assessed in the study due to low incidence of disease (both in the study population and in general), it is thought that HPV vaccination could potentially decrease the incidence of all PSCCs by one third, making it an important resource in the primary prevention of the disease.18
Management
Contemporary surgical management of PSCC has evolved from organ resection in toto for all PSCCs to a more conservative approach based upon tumor stage and grade. The standard margin for surgical resection of PSCC is 2 cm, a procedure often referred to as a partial penectomy. This remains the most common procedure for surgical resection of PSCC and has achieved good local control, with reported recurrence rates of 4% to 8%.19,20 Complication rates of the procedure are moderate one-third of patients experiencing compromise of sexual activity after surgery.21 With evidence that smaller resection margins may result in good local control and a lower incidence of postoperative functional impairment, resection margins of 5, 10, and 15 mm have been advocated for PSCCs of varying histologic grades and tumor stages.22-24 Treatment options for T1 and in situ tumors have expanded to include glansectomy, margin-controlled Mohs micrographic surgery, and ablative laser therapy for local disease control.5,20 More advanced tumors are still treated with partial or complete penectomy given the high risks for locoregional recurrence and distant spread.
Prognosis
The most important factor predicting survival in patients with PSCC is metastasis to inguinal lymph nodes. The 5-year survival rate for patients without nodal involvement is 85% to 100%, while those with pathologically positive lymph nodes have a 5-year survival rate of 15% to 45%.25 Once distant metastasis occurs, the mean time of survival is 7 to 10 months.26 Our patient presented with high-grade PSCC with histologic lymphovascular spread and palpable inguinal lymph nodes. When stratified with other similar cases at presentation, our patient was at a considerable risk for locoregional as well as distant metastasis. Management with regional nodal dissection with a plan for close observation (and deferment of chemotherapeutics) was based upon evaluations from multiple different medical specialties.
Conclusion
Invasive PSCC is rare in young circumcised adults, and a delay in diagnosis can lead to considerable morbidity and mortality. We present a case of invasive PSCC arising in the setting of squamous cell carcinoma in situ in an area previously treated with cryotherapy and imiquimod. Our patient’s young age, concurrent hepatitis C infection, and contralateral locoregional nodal metastasis made this a complex case, involving evaluation and treatment by multiple medical disciplines. This case highlights the importance of biopsy in any lesion recalcitrant to conventional modalities regardless of the patient’s age. Early detection and treatment of PSCC can prevent organ dysfunction, loss of organ, and even death.
Invasive penile cancer is a rare malignancy with considerable morbidity and mortality. The American Cancer Society estimates that there will be 2320 new cases of invasive penile cancer in the United States in 2018, of which primary penile squamous cell carcinoma (PSCC) represents the majority.1 In one study, the mean age at diagnosis was 60 years, with PSCC occurring only rarely in men younger than 35 years of age (estimated incidence, 0.01 cases per 100,000 individuals).2 Presentation to a physician generally occurs more than 1 year after initial onset of symptoms or clinical lesion(s). This delay in diagnosis and treatment often results in disease progression,3 which can have a devastating outcome.4 Therefore, physicians should maintain a high index of clinical suspicion for PSCC, particularly in young or middle-aged patients in whom presentation of PSCC is uncommon. The most commonly associated risk factors for PSCC include lack of circumcision (specifically during the neonatal period), high-risk human papillomavirus (HPV) infection, and tobacco use.5 Chronic alcoholism also has been linked to PSCC.6 It also is common in patients without health insurance.7 We report the case of a 27-year-old circumcised man who presented with invasive PSCC following a diagnosis of condyloma 8 years prior by an outside physician.
Case Report
A 27-year-old man presented for evaluation of persistent genital warts that had been diagnosed 8 years prior. His medical history was remarkable for intravenous drug use, active hepatitis C infection, tobacco smoking, chronic alcohol use, and mild asthma. Eight years prior to the current presentation, 7 lesions had developed on the penis and were diagnosed by an outside physician as condyloma, which was treated with cryotherapy and topical imiquimod. All of the lesions except for 1 responded to treatment. The residual lesion continued to grow until the size prompted him to contact his primary care physician, who referred him for dermatologic evaluation. The patient cited lack of health insurance as the primary reason he did not seek follow-up treatment after the initial evaluation and treatment 8 years prior.
Physical examination at the current presentation revealed a circumcised man with an asymptomatic, 2.6-cm, pink, friable, verrucous mass on the left lateral penile shaft (Figure 1) and otherwise unremarkable penile architecture. A clinically enlarged, nontender right inguinal lymph node was noted as well as subtle enlargement of a left inguinal lymph node. An excisional biopsy was performed with pathologic evaluation confirming a diagnosis of high-grade invasive squamous cell carcinoma (SCC) arising in the setting of squamous cell carcinoma in situ (Figure 2). Lymphovascular invasion was highlighted on cluster of differentiation 31 and podoplanin immunostaining (Figure 3). The patient was subsequently referred to urology and hematology-oncology specialists for further evaluation. Computed tomography (CT) of the abdomen and pelvis confirmed the contralaterally enlarged right inguinal lymph node discovered during physical examination and mildly enlarged ipsilateral inguinal, obturator, and external iliac nodes. Computed tomography–guided fine-needle aspiration of the right inguinal node confirmed the diagnosis of contralateral locoregional metastasis. Further evaluation with positron emission tomography/CT imaging revealed only a single metabolically active region confined to the right inguinal node. The patient’s history of active hepatitis C complicated proposed neoadjuvant chemotherapy regimens. Ultimately, after discussion with multiple surgical and oncologist specialties within our institution and others, a treatment plan was formulated. The patient underwent robotic laparoscopic bilateral pelvic and inguinal lymph node dissection and re-excision of the primary PSCC, with one of 15 right superficial inguinal nodes testing positive for tumor cells; the left superficial and bilateral deep inguinal lymph nodes were negative for SCC.
Repeat positron emission tomography/CT imaging at 6 months’ follow-up showed no evidence of active disease. On 1-year follow-up, a CT scan did not show any new or residual disease, but the patient continued to have edema of the bilateral legs, which began after lymph node dissection and was managed with physical therapy and compression stockings.
Comment
Prevalence
Penile cancer is rare in industrialized countries. Early detection is a critical factor for both overall survival and organ function. If successful interventions are to be made, physicians should be familiar with known risk factors as well as unusual presentations, such as lesions presenting in young circumcised men, as reported above. Similarly, tumors located on the shaft of the penis represent an uncommon location for tumor presentation, occurring in less than 5% of PSCC cases.8 Penile SCC most commonly develops as a solitary painless lesion on the glans, balanopreputial sulcus and/or prepuce.9 In our case, histopathology confirmed high-grade invasive SCC arising from squamous cell carcinoma in situ, an entity generally associated with older men with a 10% to 20% rate of progression into invasive SCC.9 Our patient denied any clinical change in the appearance of the tumor in the years prior to the current presentation, making it possible that the condyloma treated 8 years prior was squamous cell carcinoma in situ or PSCC. As many as 25% of premalignant lesions are mistaken for benign lesions, which can thus delay treatment and allow progression to malignancy.10
Diagnosis
Penile SCC often is etiologically subcategorized into 2 pathways based on HPV dependence or independence. Recent research suggests that this distinction often is difficult to make, and accurate laboratory and pathologic confirmation of HPV DNA, intact virions, and viral-related cutaneous changes is not always possible, leading to much speculation regarding the exact role of HPV in tumorigenesis.11 Cancers developing in the absence of HPV DNA often occur secondary to chronic inflammatory conditions such as lichen planus or lichen sclerosus. Human papillomavirus DNA has shown to be present in 70% to 100% of all SCC in situ of the penis11; therefore, the transformation of in situ disease to an invasive tumor in our patient most likely occurred via an HPV-dependent pathway. Viral carcinogenesis in the HPV-dependent pathway involves inactivation of host cell cycle regulatory proteins, specifically the retinoblastoma and p53 regulatory proteins by the viral oncoproteins E7 and E6, respectively.12,13 Human papillomavirus–dependent pathways are related to a patient’s age at first sexual intercourse, number of sexual partners, and history of condyloma and other sexually transmitted diseases.14,15 High-risk HPV types 16 and 18 are the most common viral types found in HPV related premalignant lesions, making it possible to decrease the incidence of PSCC with recently developed vaccines.16 Human papillomavirus vaccines have been shown to reduce the incidence of anal intraepithelial neoplasias and genital warts in men.17 While the effects of the HPV vaccine on reducing PSCC could not be assessed in the study due to low incidence of disease (both in the study population and in general), it is thought that HPV vaccination could potentially decrease the incidence of all PSCCs by one third, making it an important resource in the primary prevention of the disease.18
Management
Contemporary surgical management of PSCC has evolved from organ resection in toto for all PSCCs to a more conservative approach based upon tumor stage and grade. The standard margin for surgical resection of PSCC is 2 cm, a procedure often referred to as a partial penectomy. This remains the most common procedure for surgical resection of PSCC and has achieved good local control, with reported recurrence rates of 4% to 8%.19,20 Complication rates of the procedure are moderate one-third of patients experiencing compromise of sexual activity after surgery.21 With evidence that smaller resection margins may result in good local control and a lower incidence of postoperative functional impairment, resection margins of 5, 10, and 15 mm have been advocated for PSCCs of varying histologic grades and tumor stages.22-24 Treatment options for T1 and in situ tumors have expanded to include glansectomy, margin-controlled Mohs micrographic surgery, and ablative laser therapy for local disease control.5,20 More advanced tumors are still treated with partial or complete penectomy given the high risks for locoregional recurrence and distant spread.
Prognosis
The most important factor predicting survival in patients with PSCC is metastasis to inguinal lymph nodes. The 5-year survival rate for patients without nodal involvement is 85% to 100%, while those with pathologically positive lymph nodes have a 5-year survival rate of 15% to 45%.25 Once distant metastasis occurs, the mean time of survival is 7 to 10 months.26 Our patient presented with high-grade PSCC with histologic lymphovascular spread and palpable inguinal lymph nodes. When stratified with other similar cases at presentation, our patient was at a considerable risk for locoregional as well as distant metastasis. Management with regional nodal dissection with a plan for close observation (and deferment of chemotherapeutics) was based upon evaluations from multiple different medical specialties.
Conclusion
Invasive PSCC is rare in young circumcised adults, and a delay in diagnosis can lead to considerable morbidity and mortality. We present a case of invasive PSCC arising in the setting of squamous cell carcinoma in situ in an area previously treated with cryotherapy and imiquimod. Our patient’s young age, concurrent hepatitis C infection, and contralateral locoregional nodal metastasis made this a complex case, involving evaluation and treatment by multiple medical disciplines. This case highlights the importance of biopsy in any lesion recalcitrant to conventional modalities regardless of the patient’s age. Early detection and treatment of PSCC can prevent organ dysfunction, loss of organ, and even death.
- About penile cancer. American Cancer Society website. https://www.cancer.org/content/dam/CRC/PDF/Public/8783.00.pdf. Revised February 9, 2016. Accessed February 27, 2018.
- Barnholtz-Sloan JS, Maldonado JL, Pow-sang J, et al. Incidence trends in primary malignant penile cancer. Urol Oncol. 2007;25:361-367.
- Koifman L, Vides AJ, Koifman N, et al. Epidemiological aspects of penile cancer in Rio de Janeiro: evaluation of 230 cases. Int Braz J Urol. 2011;37:231-240.
- Kamat AM, Carpenter SM, Czerniak BA, et al. Metastatic penile cancer in a young Caucasian male: impact of delayed diagnosis. Urol Oncol. 2005;23:130-131.
- Deem S, Keane T, Bhavsar R, et al. Contemporary diagnosis and management of squamous cell carcinoma (SCC) of the penis. BJU Int. 2011;108:1378-1392.
- McIntyre M, Weiss A, Wahlquist A, et al. Penile cancer: an analysis of socioeconomic factors at a southeastern tertiary referral center. Can J Urol. 2011;18:5524-5528.
- Maden C, Sherman KJ, Beckmann AM, et al. History of circumcision, medical conditions, and sexual activity and risk of penile cancer. J Natl Cancer Inst. 1993;85:19-24.
- Hernandez BY, Barnholtz-Sloan J, German RR, et al. Burden of invasive squamous cell carcinoma of the penis in the United States, 1998-2003. Cancer. 2008;113(suppl 10):2883-2891.
- Ferrandiz-Pulido C, de Torres I, Garcia-Patos V. Penile squamous cell carcinoma. Actas Dermosifiliogr. 2012;103:478-487.
- Tietjen DN, Malek RS. Laser therapy of squamous cell dysplasia and carcinoma of the penis. Urology. 1998;52:559-565.
- Mannweiler S, Sygulla S, Winter E, et al. Two major pathways of penile carcinogenesis: HPV-induced penile cancers overexpress p16, HPV-negative cancers associated with dermatoses express p53, but lack p16 overexpression. J Am Acad Dermatol. 2013;69:73-81.
- Scheffner M, Werness BA, Huibregtse JM, et al. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129-1136.
- Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76-79.
- Daling JR, Madeleine MM, Johnson LG, et al. Penile cancer: importance of circumcision, human papillomavirus and smoking in in situ and invasive disease. Int J Cancer. 2005;116:606-616.
- Bleeker MC, Heideman DA, Snijders PJ, et al. Penile cancer: epidemiology, pathogenesis and prevention. World J Urol. 2009;27:141-150.
- Shabbir M, Barod R, Hegarty PK, et al. Primary prevention and vaccination for penile cancer. Ther Adv Urol. 2013;5:161-169.
- Palefsky J, Giuliano A, Goldstone S, et al. HPV vaccine against anal HPV infection and anal intraepithelial neoplasia. N Engl J Med. 2011;365:1576-1585.
- Backes DM, Kurman RJ, Pimenta JM, et al. Systematic review of human papillomavirus prevalence in invasive penile cancer. Cancer Causes Control. 2009;20:449-457.
- Korets R, Koppie TM, Snyder ME, et al. Partial penectomy for patients with squamous cell carcinoma of the penis: the Memorial Sloan-Kettering experience. Ann Surg Oncol. 2007;14:3614-3619.
- Zukiwskyj M, Daly P, Chung E. Penile cancer and phallus preservation strategies: a review of current literature. BJU Int. 2013;112(suppl 2):21-26.
- Romero FR, Romero KR, Mattos MA, et al. Sexual function after partial penectomy for penile cancer. Urology. 2005;66:1292-1295.
- Minhas S, Kayes O, Hegarty P, et al. What surgical resection margins are required to achieve oncological control in men with primary penile cancer? BJU Int. 2005;96:1040-1043.
- Feldman AS, McDougal WS. Long-term outcome of excisional organ sparing surgery for carcinoma of the penis. J Urol. 2011;186:1303-1307.
- Philippou P, Shabbir M, Malone P, et al. Conservative surgery for squamous cell carcinoma of the penis: resection margins and long-term oncological control. J Urol. 2012;188:803-808.
- Brady KL, Mercurio MG, Brown MD. Malignant tumors of the penis. Dermatol Surg. 2013;39:527-547.
- Ornellas AA, Nobrega BL, Wei Kin Chin E, et al. Prognostic factors in invasive squamous cell carcinoma of the penis: analysis of 196 patients treated at the Brazilian National Cancer Institute. J Urol. 2008;180:1354-1359.
- About penile cancer. American Cancer Society website. https://www.cancer.org/content/dam/CRC/PDF/Public/8783.00.pdf. Revised February 9, 2016. Accessed February 27, 2018.
- Barnholtz-Sloan JS, Maldonado JL, Pow-sang J, et al. Incidence trends in primary malignant penile cancer. Urol Oncol. 2007;25:361-367.
- Koifman L, Vides AJ, Koifman N, et al. Epidemiological aspects of penile cancer in Rio de Janeiro: evaluation of 230 cases. Int Braz J Urol. 2011;37:231-240.
- Kamat AM, Carpenter SM, Czerniak BA, et al. Metastatic penile cancer in a young Caucasian male: impact of delayed diagnosis. Urol Oncol. 2005;23:130-131.
- Deem S, Keane T, Bhavsar R, et al. Contemporary diagnosis and management of squamous cell carcinoma (SCC) of the penis. BJU Int. 2011;108:1378-1392.
- McIntyre M, Weiss A, Wahlquist A, et al. Penile cancer: an analysis of socioeconomic factors at a southeastern tertiary referral center. Can J Urol. 2011;18:5524-5528.
- Maden C, Sherman KJ, Beckmann AM, et al. History of circumcision, medical conditions, and sexual activity and risk of penile cancer. J Natl Cancer Inst. 1993;85:19-24.
- Hernandez BY, Barnholtz-Sloan J, German RR, et al. Burden of invasive squamous cell carcinoma of the penis in the United States, 1998-2003. Cancer. 2008;113(suppl 10):2883-2891.
- Ferrandiz-Pulido C, de Torres I, Garcia-Patos V. Penile squamous cell carcinoma. Actas Dermosifiliogr. 2012;103:478-487.
- Tietjen DN, Malek RS. Laser therapy of squamous cell dysplasia and carcinoma of the penis. Urology. 1998;52:559-565.
- Mannweiler S, Sygulla S, Winter E, et al. Two major pathways of penile carcinogenesis: HPV-induced penile cancers overexpress p16, HPV-negative cancers associated with dermatoses express p53, but lack p16 overexpression. J Am Acad Dermatol. 2013;69:73-81.
- Scheffner M, Werness BA, Huibregtse JM, et al. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129-1136.
- Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76-79.
- Daling JR, Madeleine MM, Johnson LG, et al. Penile cancer: importance of circumcision, human papillomavirus and smoking in in situ and invasive disease. Int J Cancer. 2005;116:606-616.
- Bleeker MC, Heideman DA, Snijders PJ, et al. Penile cancer: epidemiology, pathogenesis and prevention. World J Urol. 2009;27:141-150.
- Shabbir M, Barod R, Hegarty PK, et al. Primary prevention and vaccination for penile cancer. Ther Adv Urol. 2013;5:161-169.
- Palefsky J, Giuliano A, Goldstone S, et al. HPV vaccine against anal HPV infection and anal intraepithelial neoplasia. N Engl J Med. 2011;365:1576-1585.
- Backes DM, Kurman RJ, Pimenta JM, et al. Systematic review of human papillomavirus prevalence in invasive penile cancer. Cancer Causes Control. 2009;20:449-457.
- Korets R, Koppie TM, Snyder ME, et al. Partial penectomy for patients with squamous cell carcinoma of the penis: the Memorial Sloan-Kettering experience. Ann Surg Oncol. 2007;14:3614-3619.
- Zukiwskyj M, Daly P, Chung E. Penile cancer and phallus preservation strategies: a review of current literature. BJU Int. 2013;112(suppl 2):21-26.
- Romero FR, Romero KR, Mattos MA, et al. Sexual function after partial penectomy for penile cancer. Urology. 2005;66:1292-1295.
- Minhas S, Kayes O, Hegarty P, et al. What surgical resection margins are required to achieve oncological control in men with primary penile cancer? BJU Int. 2005;96:1040-1043.
- Feldman AS, McDougal WS. Long-term outcome of excisional organ sparing surgery for carcinoma of the penis. J Urol. 2011;186:1303-1307.
- Philippou P, Shabbir M, Malone P, et al. Conservative surgery for squamous cell carcinoma of the penis: resection margins and long-term oncological control. J Urol. 2012;188:803-808.
- Brady KL, Mercurio MG, Brown MD. Malignant tumors of the penis. Dermatol Surg. 2013;39:527-547.
- Ornellas AA, Nobrega BL, Wei Kin Chin E, et al. Prognostic factors in invasive squamous cell carcinoma of the penis: analysis of 196 patients treated at the Brazilian National Cancer Institute. J Urol. 2008;180:1354-1359.
Practice Points
- Invasive penile squamous cell carcinoma (PSCC) is a rare malignancy with considerable morbidity and mortality that typically does not present in young men.
- Delayed or incorrect diagnosis of PSCC can have a devastating outcome; therefore, physicians should maintain a high index of clinical suspicion for PSCC in patients presenting with penile lesions, particularly in young or middle-aged patients.
Diffuse Cutaneous Breast Cancer Metastases Resembling Subcutaneous Nodules With No Surface Changes
Cutaneous metastases from solid tumors in general occur at a rate of about 1% per primary tumor.1 In breast cancer, cutaneous metastases occur at a rate of about 2.5% per primary tumor. Because of the high incidence of breast cancers relative to other internal malignancies, breast cancer accounts for almost 33% of all cutaneous metastases.2 Infiltrating ductal carcinoma accounts for almost 70% of cutaneous metastases from breast cancers, whereas lobular carcinoma accounts for about 15%.
Cutaneous metastases may be the first presenting sign of primary malignancy. In one retrospective study, 6% of breast carcinomas (N=992) initially presented with only skin manifestations.3 Clinical appearance can vary, but cutaneous metastases from breast adenocarcinomas often present as isolated dermal nodules with superficial discoloration or changes in texture. The most common location of cutaneous metastases is on the chest ipsilateral to the primary breast malignancy.4 We pre-sent a case of metastatic adenocarcinoma of the breast presenting with diffuse cutaneous nodules with no surface changes.
Case Report
A 64-year-old woman who was otherwise in good health presented to her primary care physician for evaluation of recent-onset fatigue. Laboratory testing revealed that she was mildly anemic with mild thrombocytopenia and lymphocytosis. She was referred to a hematologist, who ordered flow cytometry and cytogenetic testing. Blood abnormalities were not considered severe enough to warrant a bone marrow biopsy, and she was monitored clinically for the next 2 years.
Two years after the initial presentation, the primary care physician performed a breast examination that was unremarkable, but enlarged axillary lymph nodes up to 15 mm were discovered in the right breast during routine breast ultrasonography. Additionally, she noted that she had experienced unintentional weight loss of 10 lb over the past year. The hematologist suspected a low-grade lymphoma and performed a bone marrow biopsy. The immunohistochemistry of the bone marrow specimen was consistent with an estrogen receptor–positive, progesterone receptor–negative, human epidermal growth factor receptor 2–negative invasive lobular breast carcinoma, which was then confirmed in the right breast on magnetic resonance imaging. The patient denied any history of prior radiation treatment, but she disclosed a family history of breast cancer in her cousin.
Several weeks after the bone marrow biopsy, an oncologist found that the patient also had an abdominal mass and bone metastases of the primary breast cancer. Colonoscopy confirmed metastases to the colon that subsequently led to obstruction and ultimately required a right hemicolectomy. The patient’s oncologist started her on anastrozole, an aromatase inhibitor (AI), for treatment of the metastatic breast cancer and zoledronic acid, a bisphosphonate, along with calcium and vitamin D for the bone involvement.
Shortly after, during a routine annual skin examination, the patient’s dermatologist (H.T.N.) discovered 3 soft, fixed, subcutaneous-appearing nodules—one on the right chest that was 15 mm in diameter, one on the left mid back that was 7 mm, and one on the left upper anterior thigh that was 10 mm. They were discrete with well-defined borders but had only minimal elevation, making them difficult to detect clinically, especially without palpation. The nodules were not visibly apparent because they were flesh-colored with no surface discoloration or texture changes. The patient remembered that the lesions had appeared gradually several months prior, predating the breast cancer diagnosis, and were not associated with pain, itching, or burning, so she was not alarmed by their appearance and never sought medical attention. The dermatologist (H.T.N.) recommended a biopsy at the time of the skin examination, but the patient declined.
One year after the appearance of the first skin lesions, 14 more nodules (Figure 1) progressively erupted on the ipsilateral and contralateral chest (Figure 2A), axillae, arms, shoulders, back (Figure 2B), and thighs (Figure 2C). At this point, the dermatologists performed a punch biopsy on a lesion on the back to confirm the suspicion of cutaneous metastasis of the primary breast cancer. The biopsy showed interstitial dermal proliferation of atypical cells between collagen bundles and stained strongly positive for cytokeratin 7, an epithelial protein common in breast adenocarcinoma (Figure 3). Further immunohistochemical staining returned metastatic estrogen receptor–positive, progesterone receptor–negative, human epidermal growth factor receptor 2–negative invasive lobular breast carcinoma. Therefore, the markers for the cutaneous metastases were consistent with the markers for the original breast cancer.
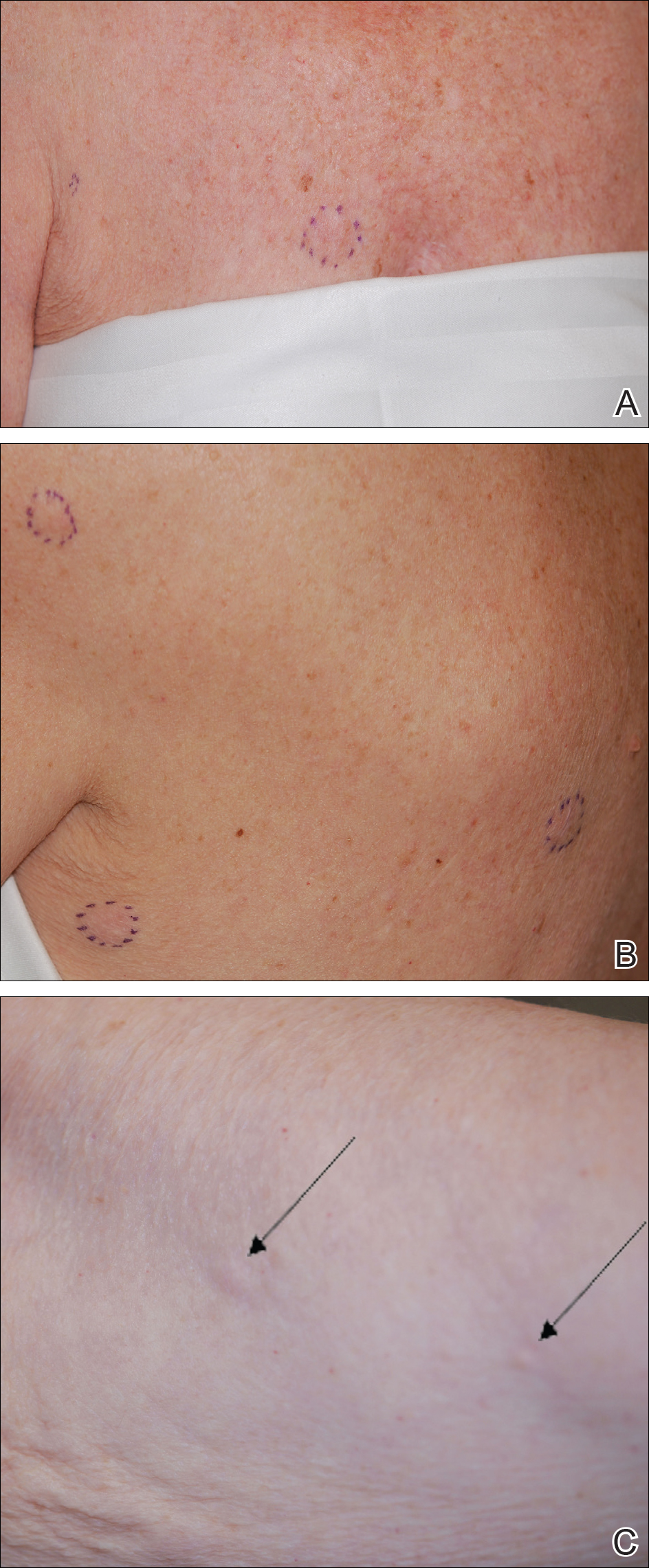
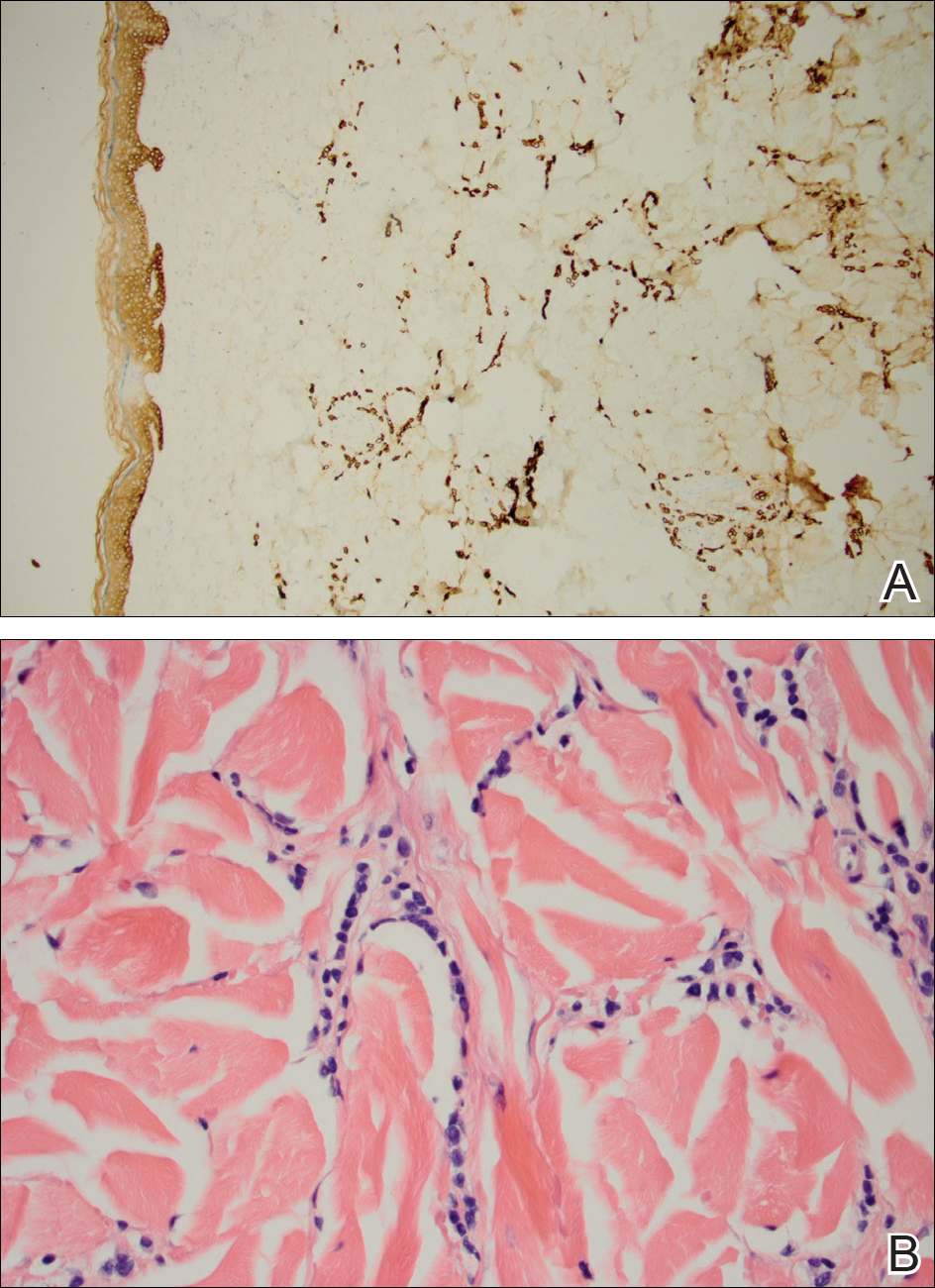
After 1 year of treatment with anastrozole, the patient’s internal metastases had not changed considerably, but the cutaneous metastases continued to grow—the lesion on the left thigh doubled from 10 to 20 mm in diameter, and new nodules developed on the chest, back, arms, and legs. One year and a half after the initial lesions were documented, several nodules had disappeared and several new ones appeared. The remaining nodules remained relatively constant in size.
After stopping anastrozole, the patient was enrolled in a research trial using bortezomib, a chemotherapeutic agent typically used for multiple myeloma, as well as fulvestrant, an estrogen receptor antagonist; however, because of continued progression of the metastatic cancer, the patient was removed from the trial and switched to the established regimen of everolimus, a chemotherapeutic agent, and exemestane, another AI. Everolimus eventually was stopped, but the patient continued on exemestane as monotherapy. In addition to development of pleural disease, the cutaneous metastases continued to progress. The patient did not receive any local treatment for her cutaneous metastases.
Comment
Typically, cutaneous metastases of breast cancer manifests as a 1- to 3-cm, asymptomatic, firm, pink to red-brown nodule on the chest ipsilateral to the primary tumor. There may be more than 1 nodule, and ulceration may be present.5,6 In addition to nodular metastases, which make up 47% of cases (N=305), other common presentations include alopecia neoplastica (12%), telangiectatic carcinoma (8%), melanomalike lesions (6%), carcinoma erysipeloides (6%), subungual lesions (5%), carcinoma en cuirasse (4%), and zosteriform metastases (4%).6
Although nodular metastases are the most common type of cutaneous breast cancer metastases, our case is unique in that the patient had soft nodules dispersed to both arms and legs, and the nodules had no surface changes. Although cutaneous metastases can present as flesh-colored nodules,7 they typically have an erythematous base, a slight change in coloration, or induration. Additionally, cutaneous metastases most often are few in number and appear in close proximity to the primary breast adenocarcinoma.8 Without the detection of a slight soft elevation on palpation, our patient’s nodules were practically indistinguishable from the normal skin.
Among common internal cancers, breast cancer is the most likely to metastasize to the skin at a rate of 2.42% per primary tumor (Table 1).1 Cutaneous metastases from lobular carcinomas are much rarer than those from ductal carcinomas.4 The metastases also are most often located locally on the chest ipsilateral to the primary malignancy. Distant metastases are relatively rare. In a review of 212 cases of breast cancer patients with skin metastases, only 9 had involvement of the legs and only 4 had involvement of the contralateral chest.4 Our patient had involvement of the ipsilateral chest, both arms and legs, and the contralateral chest.
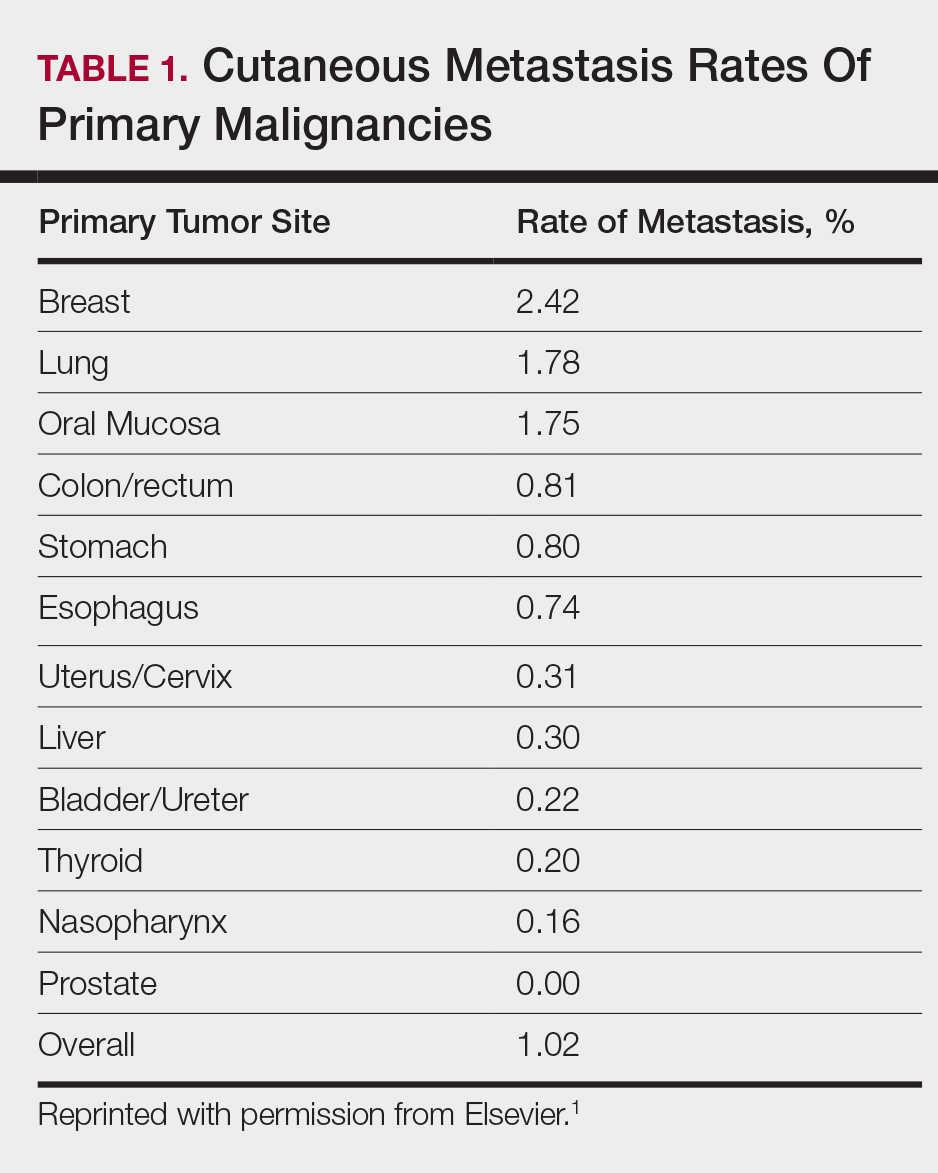
The 5-year relative survival rate for breast cancer patients varies based on the stage at diagnosis (99% in patients with localized cancer, 84% with regional lymph node involvement, 24% with distant metastases of any kind).9 In a study of 141 patients with cutaneous metastases in a Taiwanese medical center, Hu et al10 found that patients with breast cancer with only cutaneous metastases had a 5-year absolute survival rate of 38%. In the same study, patients with non–breast cancer metastasis including cutaneous metastasis had a 5-year survival rate of 15%.10 This data is summarized in Table 2.
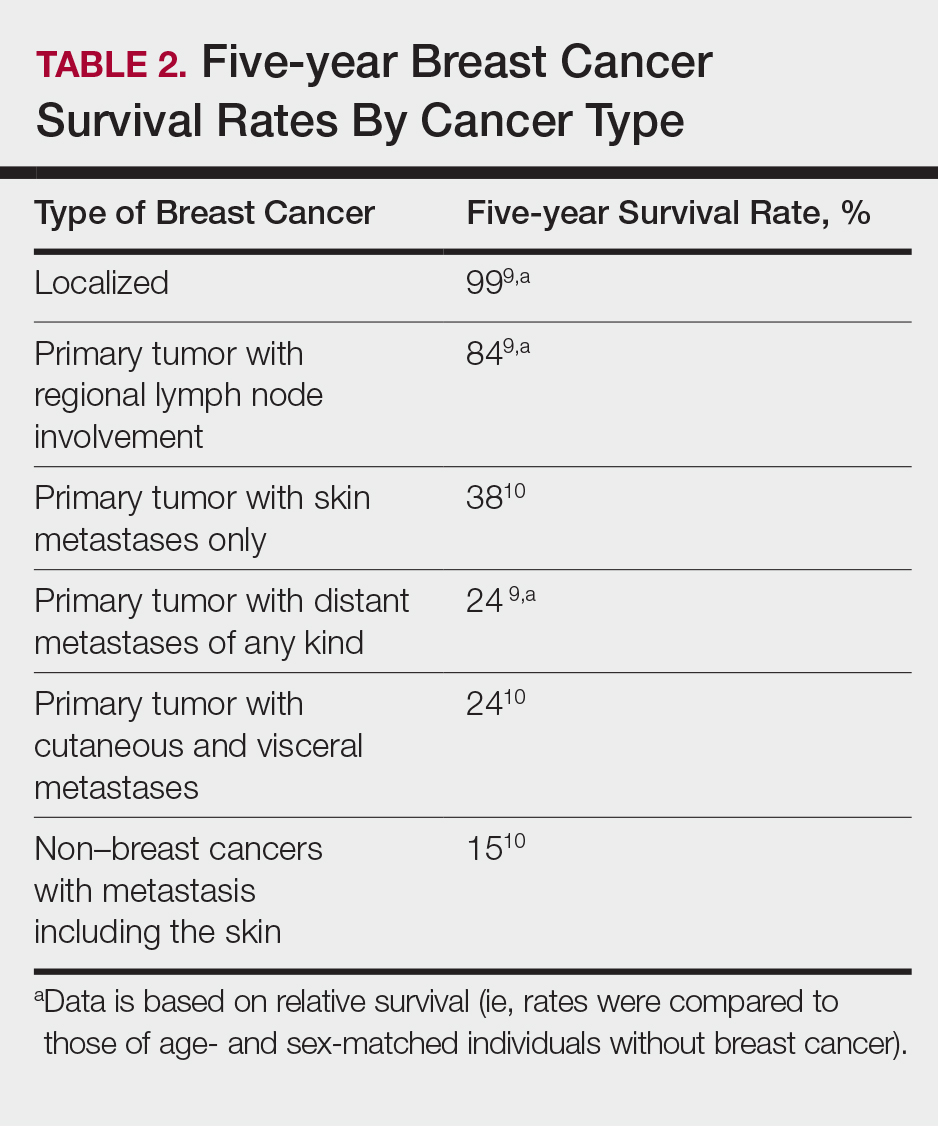
Breast cancer metastasis to soft tissue (eg, the skin) typically indicates a better prognosis than breast cancer metastasis to a visceral organ or bone. In a study of 439 patients with metastatic relapse after surgical resection of a primary breast cancer, those who had soft tissue metastases had a median survival period of 39 months, whereas those who had visceral or bone metastases had a median survival period of 13 and 28 months, respectively.11 Furthermore, cutaneous metastases from breast cancers do not necessarily indicate as poor a prognosis as skin metastases from other internal malignancies. Cutaneous metastases from other internal malignancies carry a relative risk of mortality of 4.3 compared to cutaneous metastases from breast cancer.10
Treatment of cutaneous metastases may be medically or cosmetically indicated. Standard treatments for cutaneous metastases from the breast include surgical excision, external beam radiotherapy, and systemic chemotherapy.6 While oncologists can use the response of cutaneous metastases to treatment as an indicator of systemic response to hormone therapy or chemotherapy,12 the response may be poorer due to the skin’s relatively weaker blood supply.13
Our patient was first prescribed anastrozole, an AI. For metastatic hormone receptor–positive breast cancer, AIs are a first-line therapy in postmenopausal women. In one meta-analysis, AIs showed greater improvement of survival rates relative to other endocrine therapies such as tamoxifen, an estrogen receptor antagonist (hazard ratio of 0.87).14 After stopping anastrozole, the patient was prescribed fulvestrant, another estrogen receptor antagonist, along with a trial drug. In a randomized, double-blind, placebo-controlled trial, fulvestrant was found to be an effective second-line treatment after anastrozole for hormone receptor–positive breast cancer in postmenopausal women.15 Our patient was then started on everolimus, a chemotherapeutic agent, and exemestane, another AI. After first-line treatment with anastrozole, this regimen also has been found to be an effective second-line treatment with improved progression-free survival.16 For the bone metastases, our patient was treated with zoledronic acid, a bisphosphonate. In a meta-analysis, bisphosphonates were found to reduce skeletal-related complications by a median of 28% in breast cancer patients with bone metastases.17
Some promising new local treatments for cutaneous breast metastases include topical imiquimod and electrochemotherapy. In a small study of 10 patients whose malignancies were refractory to radiotherapy, imiquimod achieved a partial response in 20% (2/10) of patients.18 In another study, 12 patients received electrochemotherapy involving electroporation (applying an electrical field to increase cell membrane permeability and thus increase drug uptake) followed by local administration of bleomycin, an antineoplastic agent. Seventy-five percent (9/12) of the patients received a complete response with disappearance of the metastases.19
This case report provides a rare presentation of diffuse nodular cutaneous metastases of breast adenocarcinoma with no surface changes. The subtle clinical findings in our patient demonstrate the spectrum of clinical manifestations for cutaneous metastases. Our case also serves to highlight the need for close inspection of the skin, including palpation in patients with a history of internal malignancy.
- Hu SC, Chen G, Wu C, et al. Rates of cutaneous metastases from different internal malignancies: experience from a Taiwanese medical center. J Am Acad Dermatol. 2009;60:379-387.
- Wong CY, Helm MA, Helm TN, et al. Patterns of skin metastases: a review of 25 years’ experience at a single cancer center. Int J Dermatol. 2014;53:56-60.
- Lookingbill DP, Spangler N, Sexton FM. Skin involvement as the presenting sign of internal carcinoma: a retrospective study of 7316 cancer patients. J Am Acad Dermatol. 1990;22:19-26.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2, part 1):228-236.
- Gan DEH, Teh YC, Ng CH, et al. Cutaneous metastases of breast cancer: a case report. Breast Case. 2012;1:23-36.
- De Giorgi V, Grazzini M, Alfaioli B, et al. Cutaneous manifestations of breast carcinoma. Dermatol Ther. 2010;23:581-589.
- Vano-Galvan S, Moreno-Martin P, Salguero I, et al. Cutaneous metastases of breast carcinoma: a case report. Cases J. 2009;2:71.
- Dacso M, Soldano AC, Talbott LB, et al. A solitary neck nodule as late evidence of recurrent lobular breast carcinoma. Case Rep Oncol. 2009;2:24-29.
- Howlader N, Noone AM, Krapcho M, et al, eds. SEER Cancer Statistics Review, 1975-2010. Table 1.5 Age-Adjusted SEER Incidence and U.S. Death Rates and 5-Year Relative Survival (Percent) By Primary Cancer Site, Sex and Time Period. Bethesda, MD: National Cancer Institute; 2013. https://seer.cancer.gov/archive/csr/1975_2010/results_merged/topic_survival.pdf. Updated June 14, 2014. Accessed February 27, 2018.
- Hu SC, Chen GS, Lu YW, et al. Cutaneous metastases from different internal malignancies: a clinical and prognostic appraisal. J Eur Acad Dermatol Venereol. 2008;22:735-740.
- Insa A, Lluch A, Prosper F, et al. Prognostic factors predicting survival from first recurrence in patients with metastatic breast cancer: analysis of 439 patients. Breast Cancer Res Treat. 1999;56:67-78.
- Eisenhauer E, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228-247.
- Kamble R, Kumar L, Kochupillai V, et al. Cutaneous metastases of lung cancer. Postgrad Med J. 1995;71:741-743.
- Mauri D, Pavlidis N, Polyzos N, et al. Survival with aromatase inhibitors and inactivators versus standard hormonal therapy in advanced breast cancer: meta-analysis. J Natl Cancer Inst. 2006;98:1285-1291.
- Chia S, Gradishar W, Mauriac L, et al. Double-blind, randomized placebo controlled trial of fulvestrant compared with exemestane after prior nonsteroidal aromatase inhibitor therapy in postmenopausal women with hormone receptor-positive, advanced breast cancer: results from EFECT. J Clin Oncol. 2008;26:1664-1670.
- Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor–positive advanced breast cancer. N Engl J Med. 2012;366:520-529.
- Wong MH, Stockler M, Pavlakis N. Bisphosphonates and other bone agents for breast cancer. Cochrane Database Syst Rev. 2012;2:CD003474.
- Adams S, Kozhaya L, Martiniuk F, et al. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin Cancer Res. 2012;18:6748-6757.
- Benevento R, Santoriello A, Perna G, et al. Electrochemotherapy of cutaneous metastastes from breast cancer in elderly patients: a preliminary report. BMC Surg. 2012;12(suppl 1):S6.
Cutaneous metastases from solid tumors in general occur at a rate of about 1% per primary tumor.1 In breast cancer, cutaneous metastases occur at a rate of about 2.5% per primary tumor. Because of the high incidence of breast cancers relative to other internal malignancies, breast cancer accounts for almost 33% of all cutaneous metastases.2 Infiltrating ductal carcinoma accounts for almost 70% of cutaneous metastases from breast cancers, whereas lobular carcinoma accounts for about 15%.
Cutaneous metastases may be the first presenting sign of primary malignancy. In one retrospective study, 6% of breast carcinomas (N=992) initially presented with only skin manifestations.3 Clinical appearance can vary, but cutaneous metastases from breast adenocarcinomas often present as isolated dermal nodules with superficial discoloration or changes in texture. The most common location of cutaneous metastases is on the chest ipsilateral to the primary breast malignancy.4 We pre-sent a case of metastatic adenocarcinoma of the breast presenting with diffuse cutaneous nodules with no surface changes.
Case Report
A 64-year-old woman who was otherwise in good health presented to her primary care physician for evaluation of recent-onset fatigue. Laboratory testing revealed that she was mildly anemic with mild thrombocytopenia and lymphocytosis. She was referred to a hematologist, who ordered flow cytometry and cytogenetic testing. Blood abnormalities were not considered severe enough to warrant a bone marrow biopsy, and she was monitored clinically for the next 2 years.
Two years after the initial presentation, the primary care physician performed a breast examination that was unremarkable, but enlarged axillary lymph nodes up to 15 mm were discovered in the right breast during routine breast ultrasonography. Additionally, she noted that she had experienced unintentional weight loss of 10 lb over the past year. The hematologist suspected a low-grade lymphoma and performed a bone marrow biopsy. The immunohistochemistry of the bone marrow specimen was consistent with an estrogen receptor–positive, progesterone receptor–negative, human epidermal growth factor receptor 2–negative invasive lobular breast carcinoma, which was then confirmed in the right breast on magnetic resonance imaging. The patient denied any history of prior radiation treatment, but she disclosed a family history of breast cancer in her cousin.
Several weeks after the bone marrow biopsy, an oncologist found that the patient also had an abdominal mass and bone metastases of the primary breast cancer. Colonoscopy confirmed metastases to the colon that subsequently led to obstruction and ultimately required a right hemicolectomy. The patient’s oncologist started her on anastrozole, an aromatase inhibitor (AI), for treatment of the metastatic breast cancer and zoledronic acid, a bisphosphonate, along with calcium and vitamin D for the bone involvement.
Shortly after, during a routine annual skin examination, the patient’s dermatologist (H.T.N.) discovered 3 soft, fixed, subcutaneous-appearing nodules—one on the right chest that was 15 mm in diameter, one on the left mid back that was 7 mm, and one on the left upper anterior thigh that was 10 mm. They were discrete with well-defined borders but had only minimal elevation, making them difficult to detect clinically, especially without palpation. The nodules were not visibly apparent because they were flesh-colored with no surface discoloration or texture changes. The patient remembered that the lesions had appeared gradually several months prior, predating the breast cancer diagnosis, and were not associated with pain, itching, or burning, so she was not alarmed by their appearance and never sought medical attention. The dermatologist (H.T.N.) recommended a biopsy at the time of the skin examination, but the patient declined.
One year after the appearance of the first skin lesions, 14 more nodules (Figure 1) progressively erupted on the ipsilateral and contralateral chest (Figure 2A), axillae, arms, shoulders, back (Figure 2B), and thighs (Figure 2C). At this point, the dermatologists performed a punch biopsy on a lesion on the back to confirm the suspicion of cutaneous metastasis of the primary breast cancer. The biopsy showed interstitial dermal proliferation of atypical cells between collagen bundles and stained strongly positive for cytokeratin 7, an epithelial protein common in breast adenocarcinoma (Figure 3). Further immunohistochemical staining returned metastatic estrogen receptor–positive, progesterone receptor–negative, human epidermal growth factor receptor 2–negative invasive lobular breast carcinoma. Therefore, the markers for the cutaneous metastases were consistent with the markers for the original breast cancer.
After 1 year of treatment with anastrozole, the patient’s internal metastases had not changed considerably, but the cutaneous metastases continued to grow—the lesion on the left thigh doubled from 10 to 20 mm in diameter, and new nodules developed on the chest, back, arms, and legs. One year and a half after the initial lesions were documented, several nodules had disappeared and several new ones appeared. The remaining nodules remained relatively constant in size.
After stopping anastrozole, the patient was enrolled in a research trial using bortezomib, a chemotherapeutic agent typically used for multiple myeloma, as well as fulvestrant, an estrogen receptor antagonist; however, because of continued progression of the metastatic cancer, the patient was removed from the trial and switched to the established regimen of everolimus, a chemotherapeutic agent, and exemestane, another AI. Everolimus eventually was stopped, but the patient continued on exemestane as monotherapy. In addition to development of pleural disease, the cutaneous metastases continued to progress. The patient did not receive any local treatment for her cutaneous metastases.
Comment
Typically, cutaneous metastases of breast cancer manifests as a 1- to 3-cm, asymptomatic, firm, pink to red-brown nodule on the chest ipsilateral to the primary tumor. There may be more than 1 nodule, and ulceration may be present.5,6 In addition to nodular metastases, which make up 47% of cases (N=305), other common presentations include alopecia neoplastica (12%), telangiectatic carcinoma (8%), melanomalike lesions (6%), carcinoma erysipeloides (6%), subungual lesions (5%), carcinoma en cuirasse (4%), and zosteriform metastases (4%).6
Although nodular metastases are the most common type of cutaneous breast cancer metastases, our case is unique in that the patient had soft nodules dispersed to both arms and legs, and the nodules had no surface changes. Although cutaneous metastases can present as flesh-colored nodules,7 they typically have an erythematous base, a slight change in coloration, or induration. Additionally, cutaneous metastases most often are few in number and appear in close proximity to the primary breast adenocarcinoma.8 Without the detection of a slight soft elevation on palpation, our patient’s nodules were practically indistinguishable from the normal skin.
Among common internal cancers, breast cancer is the most likely to metastasize to the skin at a rate of 2.42% per primary tumor (Table 1).1 Cutaneous metastases from lobular carcinomas are much rarer than those from ductal carcinomas.4 The metastases also are most often located locally on the chest ipsilateral to the primary malignancy. Distant metastases are relatively rare. In a review of 212 cases of breast cancer patients with skin metastases, only 9 had involvement of the legs and only 4 had involvement of the contralateral chest.4 Our patient had involvement of the ipsilateral chest, both arms and legs, and the contralateral chest.
The 5-year relative survival rate for breast cancer patients varies based on the stage at diagnosis (99% in patients with localized cancer, 84% with regional lymph node involvement, 24% with distant metastases of any kind).9 In a study of 141 patients with cutaneous metastases in a Taiwanese medical center, Hu et al10 found that patients with breast cancer with only cutaneous metastases had a 5-year absolute survival rate of 38%. In the same study, patients with non–breast cancer metastasis including cutaneous metastasis had a 5-year survival rate of 15%.10 This data is summarized in Table 2.
Breast cancer metastasis to soft tissue (eg, the skin) typically indicates a better prognosis than breast cancer metastasis to a visceral organ or bone. In a study of 439 patients with metastatic relapse after surgical resection of a primary breast cancer, those who had soft tissue metastases had a median survival period of 39 months, whereas those who had visceral or bone metastases had a median survival period of 13 and 28 months, respectively.11 Furthermore, cutaneous metastases from breast cancers do not necessarily indicate as poor a prognosis as skin metastases from other internal malignancies. Cutaneous metastases from other internal malignancies carry a relative risk of mortality of 4.3 compared to cutaneous metastases from breast cancer.10
Treatment of cutaneous metastases may be medically or cosmetically indicated. Standard treatments for cutaneous metastases from the breast include surgical excision, external beam radiotherapy, and systemic chemotherapy.6 While oncologists can use the response of cutaneous metastases to treatment as an indicator of systemic response to hormone therapy or chemotherapy,12 the response may be poorer due to the skin’s relatively weaker blood supply.13
Our patient was first prescribed anastrozole, an AI. For metastatic hormone receptor–positive breast cancer, AIs are a first-line therapy in postmenopausal women. In one meta-analysis, AIs showed greater improvement of survival rates relative to other endocrine therapies such as tamoxifen, an estrogen receptor antagonist (hazard ratio of 0.87).14 After stopping anastrozole, the patient was prescribed fulvestrant, another estrogen receptor antagonist, along with a trial drug. In a randomized, double-blind, placebo-controlled trial, fulvestrant was found to be an effective second-line treatment after anastrozole for hormone receptor–positive breast cancer in postmenopausal women.15 Our patient was then started on everolimus, a chemotherapeutic agent, and exemestane, another AI. After first-line treatment with anastrozole, this regimen also has been found to be an effective second-line treatment with improved progression-free survival.16 For the bone metastases, our patient was treated with zoledronic acid, a bisphosphonate. In a meta-analysis, bisphosphonates were found to reduce skeletal-related complications by a median of 28% in breast cancer patients with bone metastases.17
Some promising new local treatments for cutaneous breast metastases include topical imiquimod and electrochemotherapy. In a small study of 10 patients whose malignancies were refractory to radiotherapy, imiquimod achieved a partial response in 20% (2/10) of patients.18 In another study, 12 patients received electrochemotherapy involving electroporation (applying an electrical field to increase cell membrane permeability and thus increase drug uptake) followed by local administration of bleomycin, an antineoplastic agent. Seventy-five percent (9/12) of the patients received a complete response with disappearance of the metastases.19
This case report provides a rare presentation of diffuse nodular cutaneous metastases of breast adenocarcinoma with no surface changes. The subtle clinical findings in our patient demonstrate the spectrum of clinical manifestations for cutaneous metastases. Our case also serves to highlight the need for close inspection of the skin, including palpation in patients with a history of internal malignancy.
Cutaneous metastases from solid tumors in general occur at a rate of about 1% per primary tumor.1 In breast cancer, cutaneous metastases occur at a rate of about 2.5% per primary tumor. Because of the high incidence of breast cancers relative to other internal malignancies, breast cancer accounts for almost 33% of all cutaneous metastases.2 Infiltrating ductal carcinoma accounts for almost 70% of cutaneous metastases from breast cancers, whereas lobular carcinoma accounts for about 15%.
Cutaneous metastases may be the first presenting sign of primary malignancy. In one retrospective study, 6% of breast carcinomas (N=992) initially presented with only skin manifestations.3 Clinical appearance can vary, but cutaneous metastases from breast adenocarcinomas often present as isolated dermal nodules with superficial discoloration or changes in texture. The most common location of cutaneous metastases is on the chest ipsilateral to the primary breast malignancy.4 We pre-sent a case of metastatic adenocarcinoma of the breast presenting with diffuse cutaneous nodules with no surface changes.
Case Report
A 64-year-old woman who was otherwise in good health presented to her primary care physician for evaluation of recent-onset fatigue. Laboratory testing revealed that she was mildly anemic with mild thrombocytopenia and lymphocytosis. She was referred to a hematologist, who ordered flow cytometry and cytogenetic testing. Blood abnormalities were not considered severe enough to warrant a bone marrow biopsy, and she was monitored clinically for the next 2 years.
Two years after the initial presentation, the primary care physician performed a breast examination that was unremarkable, but enlarged axillary lymph nodes up to 15 mm were discovered in the right breast during routine breast ultrasonography. Additionally, she noted that she had experienced unintentional weight loss of 10 lb over the past year. The hematologist suspected a low-grade lymphoma and performed a bone marrow biopsy. The immunohistochemistry of the bone marrow specimen was consistent with an estrogen receptor–positive, progesterone receptor–negative, human epidermal growth factor receptor 2–negative invasive lobular breast carcinoma, which was then confirmed in the right breast on magnetic resonance imaging. The patient denied any history of prior radiation treatment, but she disclosed a family history of breast cancer in her cousin.
Several weeks after the bone marrow biopsy, an oncologist found that the patient also had an abdominal mass and bone metastases of the primary breast cancer. Colonoscopy confirmed metastases to the colon that subsequently led to obstruction and ultimately required a right hemicolectomy. The patient’s oncologist started her on anastrozole, an aromatase inhibitor (AI), for treatment of the metastatic breast cancer and zoledronic acid, a bisphosphonate, along with calcium and vitamin D for the bone involvement.
Shortly after, during a routine annual skin examination, the patient’s dermatologist (H.T.N.) discovered 3 soft, fixed, subcutaneous-appearing nodules—one on the right chest that was 15 mm in diameter, one on the left mid back that was 7 mm, and one on the left upper anterior thigh that was 10 mm. They were discrete with well-defined borders but had only minimal elevation, making them difficult to detect clinically, especially without palpation. The nodules were not visibly apparent because they were flesh-colored with no surface discoloration or texture changes. The patient remembered that the lesions had appeared gradually several months prior, predating the breast cancer diagnosis, and were not associated with pain, itching, or burning, so she was not alarmed by their appearance and never sought medical attention. The dermatologist (H.T.N.) recommended a biopsy at the time of the skin examination, but the patient declined.
One year after the appearance of the first skin lesions, 14 more nodules (Figure 1) progressively erupted on the ipsilateral and contralateral chest (Figure 2A), axillae, arms, shoulders, back (Figure 2B), and thighs (Figure 2C). At this point, the dermatologists performed a punch biopsy on a lesion on the back to confirm the suspicion of cutaneous metastasis of the primary breast cancer. The biopsy showed interstitial dermal proliferation of atypical cells between collagen bundles and stained strongly positive for cytokeratin 7, an epithelial protein common in breast adenocarcinoma (Figure 3). Further immunohistochemical staining returned metastatic estrogen receptor–positive, progesterone receptor–negative, human epidermal growth factor receptor 2–negative invasive lobular breast carcinoma. Therefore, the markers for the cutaneous metastases were consistent with the markers for the original breast cancer.
After 1 year of treatment with anastrozole, the patient’s internal metastases had not changed considerably, but the cutaneous metastases continued to grow—the lesion on the left thigh doubled from 10 to 20 mm in diameter, and new nodules developed on the chest, back, arms, and legs. One year and a half after the initial lesions were documented, several nodules had disappeared and several new ones appeared. The remaining nodules remained relatively constant in size.
After stopping anastrozole, the patient was enrolled in a research trial using bortezomib, a chemotherapeutic agent typically used for multiple myeloma, as well as fulvestrant, an estrogen receptor antagonist; however, because of continued progression of the metastatic cancer, the patient was removed from the trial and switched to the established regimen of everolimus, a chemotherapeutic agent, and exemestane, another AI. Everolimus eventually was stopped, but the patient continued on exemestane as monotherapy. In addition to development of pleural disease, the cutaneous metastases continued to progress. The patient did not receive any local treatment for her cutaneous metastases.
Comment
Typically, cutaneous metastases of breast cancer manifests as a 1- to 3-cm, asymptomatic, firm, pink to red-brown nodule on the chest ipsilateral to the primary tumor. There may be more than 1 nodule, and ulceration may be present.5,6 In addition to nodular metastases, which make up 47% of cases (N=305), other common presentations include alopecia neoplastica (12%), telangiectatic carcinoma (8%), melanomalike lesions (6%), carcinoma erysipeloides (6%), subungual lesions (5%), carcinoma en cuirasse (4%), and zosteriform metastases (4%).6
Although nodular metastases are the most common type of cutaneous breast cancer metastases, our case is unique in that the patient had soft nodules dispersed to both arms and legs, and the nodules had no surface changes. Although cutaneous metastases can present as flesh-colored nodules,7 they typically have an erythematous base, a slight change in coloration, or induration. Additionally, cutaneous metastases most often are few in number and appear in close proximity to the primary breast adenocarcinoma.8 Without the detection of a slight soft elevation on palpation, our patient’s nodules were practically indistinguishable from the normal skin.
Among common internal cancers, breast cancer is the most likely to metastasize to the skin at a rate of 2.42% per primary tumor (Table 1).1 Cutaneous metastases from lobular carcinomas are much rarer than those from ductal carcinomas.4 The metastases also are most often located locally on the chest ipsilateral to the primary malignancy. Distant metastases are relatively rare. In a review of 212 cases of breast cancer patients with skin metastases, only 9 had involvement of the legs and only 4 had involvement of the contralateral chest.4 Our patient had involvement of the ipsilateral chest, both arms and legs, and the contralateral chest.
The 5-year relative survival rate for breast cancer patients varies based on the stage at diagnosis (99% in patients with localized cancer, 84% with regional lymph node involvement, 24% with distant metastases of any kind).9 In a study of 141 patients with cutaneous metastases in a Taiwanese medical center, Hu et al10 found that patients with breast cancer with only cutaneous metastases had a 5-year absolute survival rate of 38%. In the same study, patients with non–breast cancer metastasis including cutaneous metastasis had a 5-year survival rate of 15%.10 This data is summarized in Table 2.
Breast cancer metastasis to soft tissue (eg, the skin) typically indicates a better prognosis than breast cancer metastasis to a visceral organ or bone. In a study of 439 patients with metastatic relapse after surgical resection of a primary breast cancer, those who had soft tissue metastases had a median survival period of 39 months, whereas those who had visceral or bone metastases had a median survival period of 13 and 28 months, respectively.11 Furthermore, cutaneous metastases from breast cancers do not necessarily indicate as poor a prognosis as skin metastases from other internal malignancies. Cutaneous metastases from other internal malignancies carry a relative risk of mortality of 4.3 compared to cutaneous metastases from breast cancer.10
Treatment of cutaneous metastases may be medically or cosmetically indicated. Standard treatments for cutaneous metastases from the breast include surgical excision, external beam radiotherapy, and systemic chemotherapy.6 While oncologists can use the response of cutaneous metastases to treatment as an indicator of systemic response to hormone therapy or chemotherapy,12 the response may be poorer due to the skin’s relatively weaker blood supply.13
Our patient was first prescribed anastrozole, an AI. For metastatic hormone receptor–positive breast cancer, AIs are a first-line therapy in postmenopausal women. In one meta-analysis, AIs showed greater improvement of survival rates relative to other endocrine therapies such as tamoxifen, an estrogen receptor antagonist (hazard ratio of 0.87).14 After stopping anastrozole, the patient was prescribed fulvestrant, another estrogen receptor antagonist, along with a trial drug. In a randomized, double-blind, placebo-controlled trial, fulvestrant was found to be an effective second-line treatment after anastrozole for hormone receptor–positive breast cancer in postmenopausal women.15 Our patient was then started on everolimus, a chemotherapeutic agent, and exemestane, another AI. After first-line treatment with anastrozole, this regimen also has been found to be an effective second-line treatment with improved progression-free survival.16 For the bone metastases, our patient was treated with zoledronic acid, a bisphosphonate. In a meta-analysis, bisphosphonates were found to reduce skeletal-related complications by a median of 28% in breast cancer patients with bone metastases.17
Some promising new local treatments for cutaneous breast metastases include topical imiquimod and electrochemotherapy. In a small study of 10 patients whose malignancies were refractory to radiotherapy, imiquimod achieved a partial response in 20% (2/10) of patients.18 In another study, 12 patients received electrochemotherapy involving electroporation (applying an electrical field to increase cell membrane permeability and thus increase drug uptake) followed by local administration of bleomycin, an antineoplastic agent. Seventy-five percent (9/12) of the patients received a complete response with disappearance of the metastases.19
This case report provides a rare presentation of diffuse nodular cutaneous metastases of breast adenocarcinoma with no surface changes. The subtle clinical findings in our patient demonstrate the spectrum of clinical manifestations for cutaneous metastases. Our case also serves to highlight the need for close inspection of the skin, including palpation in patients with a history of internal malignancy.
- Hu SC, Chen G, Wu C, et al. Rates of cutaneous metastases from different internal malignancies: experience from a Taiwanese medical center. J Am Acad Dermatol. 2009;60:379-387.
- Wong CY, Helm MA, Helm TN, et al. Patterns of skin metastases: a review of 25 years’ experience at a single cancer center. Int J Dermatol. 2014;53:56-60.
- Lookingbill DP, Spangler N, Sexton FM. Skin involvement as the presenting sign of internal carcinoma: a retrospective study of 7316 cancer patients. J Am Acad Dermatol. 1990;22:19-26.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2, part 1):228-236.
- Gan DEH, Teh YC, Ng CH, et al. Cutaneous metastases of breast cancer: a case report. Breast Case. 2012;1:23-36.
- De Giorgi V, Grazzini M, Alfaioli B, et al. Cutaneous manifestations of breast carcinoma. Dermatol Ther. 2010;23:581-589.
- Vano-Galvan S, Moreno-Martin P, Salguero I, et al. Cutaneous metastases of breast carcinoma: a case report. Cases J. 2009;2:71.
- Dacso M, Soldano AC, Talbott LB, et al. A solitary neck nodule as late evidence of recurrent lobular breast carcinoma. Case Rep Oncol. 2009;2:24-29.
- Howlader N, Noone AM, Krapcho M, et al, eds. SEER Cancer Statistics Review, 1975-2010. Table 1.5 Age-Adjusted SEER Incidence and U.S. Death Rates and 5-Year Relative Survival (Percent) By Primary Cancer Site, Sex and Time Period. Bethesda, MD: National Cancer Institute; 2013. https://seer.cancer.gov/archive/csr/1975_2010/results_merged/topic_survival.pdf. Updated June 14, 2014. Accessed February 27, 2018.
- Hu SC, Chen GS, Lu YW, et al. Cutaneous metastases from different internal malignancies: a clinical and prognostic appraisal. J Eur Acad Dermatol Venereol. 2008;22:735-740.
- Insa A, Lluch A, Prosper F, et al. Prognostic factors predicting survival from first recurrence in patients with metastatic breast cancer: analysis of 439 patients. Breast Cancer Res Treat. 1999;56:67-78.
- Eisenhauer E, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228-247.
- Kamble R, Kumar L, Kochupillai V, et al. Cutaneous metastases of lung cancer. Postgrad Med J. 1995;71:741-743.
- Mauri D, Pavlidis N, Polyzos N, et al. Survival with aromatase inhibitors and inactivators versus standard hormonal therapy in advanced breast cancer: meta-analysis. J Natl Cancer Inst. 2006;98:1285-1291.
- Chia S, Gradishar W, Mauriac L, et al. Double-blind, randomized placebo controlled trial of fulvestrant compared with exemestane after prior nonsteroidal aromatase inhibitor therapy in postmenopausal women with hormone receptor-positive, advanced breast cancer: results from EFECT. J Clin Oncol. 2008;26:1664-1670.
- Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor–positive advanced breast cancer. N Engl J Med. 2012;366:520-529.
- Wong MH, Stockler M, Pavlakis N. Bisphosphonates and other bone agents for breast cancer. Cochrane Database Syst Rev. 2012;2:CD003474.
- Adams S, Kozhaya L, Martiniuk F, et al. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin Cancer Res. 2012;18:6748-6757.
- Benevento R, Santoriello A, Perna G, et al. Electrochemotherapy of cutaneous metastastes from breast cancer in elderly patients: a preliminary report. BMC Surg. 2012;12(suppl 1):S6.
- Hu SC, Chen G, Wu C, et al. Rates of cutaneous metastases from different internal malignancies: experience from a Taiwanese medical center. J Am Acad Dermatol. 2009;60:379-387.
- Wong CY, Helm MA, Helm TN, et al. Patterns of skin metastases: a review of 25 years’ experience at a single cancer center. Int J Dermatol. 2014;53:56-60.
- Lookingbill DP, Spangler N, Sexton FM. Skin involvement as the presenting sign of internal carcinoma: a retrospective study of 7316 cancer patients. J Am Acad Dermatol. 1990;22:19-26.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2, part 1):228-236.
- Gan DEH, Teh YC, Ng CH, et al. Cutaneous metastases of breast cancer: a case report. Breast Case. 2012;1:23-36.
- De Giorgi V, Grazzini M, Alfaioli B, et al. Cutaneous manifestations of breast carcinoma. Dermatol Ther. 2010;23:581-589.
- Vano-Galvan S, Moreno-Martin P, Salguero I, et al. Cutaneous metastases of breast carcinoma: a case report. Cases J. 2009;2:71.
- Dacso M, Soldano AC, Talbott LB, et al. A solitary neck nodule as late evidence of recurrent lobular breast carcinoma. Case Rep Oncol. 2009;2:24-29.
- Howlader N, Noone AM, Krapcho M, et al, eds. SEER Cancer Statistics Review, 1975-2010. Table 1.5 Age-Adjusted SEER Incidence and U.S. Death Rates and 5-Year Relative Survival (Percent) By Primary Cancer Site, Sex and Time Period. Bethesda, MD: National Cancer Institute; 2013. https://seer.cancer.gov/archive/csr/1975_2010/results_merged/topic_survival.pdf. Updated June 14, 2014. Accessed February 27, 2018.
- Hu SC, Chen GS, Lu YW, et al. Cutaneous metastases from different internal malignancies: a clinical and prognostic appraisal. J Eur Acad Dermatol Venereol. 2008;22:735-740.
- Insa A, Lluch A, Prosper F, et al. Prognostic factors predicting survival from first recurrence in patients with metastatic breast cancer: analysis of 439 patients. Breast Cancer Res Treat. 1999;56:67-78.
- Eisenhauer E, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228-247.
- Kamble R, Kumar L, Kochupillai V, et al. Cutaneous metastases of lung cancer. Postgrad Med J. 1995;71:741-743.
- Mauri D, Pavlidis N, Polyzos N, et al. Survival with aromatase inhibitors and inactivators versus standard hormonal therapy in advanced breast cancer: meta-analysis. J Natl Cancer Inst. 2006;98:1285-1291.
- Chia S, Gradishar W, Mauriac L, et al. Double-blind, randomized placebo controlled trial of fulvestrant compared with exemestane after prior nonsteroidal aromatase inhibitor therapy in postmenopausal women with hormone receptor-positive, advanced breast cancer: results from EFECT. J Clin Oncol. 2008;26:1664-1670.
- Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor–positive advanced breast cancer. N Engl J Med. 2012;366:520-529.
- Wong MH, Stockler M, Pavlakis N. Bisphosphonates and other bone agents for breast cancer. Cochrane Database Syst Rev. 2012;2:CD003474.
- Adams S, Kozhaya L, Martiniuk F, et al. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin Cancer Res. 2012;18:6748-6757.
- Benevento R, Santoriello A, Perna G, et al. Electrochemotherapy of cutaneous metastastes from breast cancer in elderly patients: a preliminary report. BMC Surg. 2012;12(suppl 1):S6.
Practice Points
- Although breast cancer has the highest rate of cutaneous metastasis among internal malignancies, cutaneous metastases occur in only a small minority of breast cancer patients.
- Cutaneous metastases from breast cancer typically do not carry as poor a prognosis as those in other internal malignancies.
- The clinical presentation of cutaneous metastases from breast cancer can be varied. In our patient, the metastases were subtle and resembled subcutaneous nodules lacking surface changes, thus making them best detectable by palpation.
- While oncologists can use the response of cutaneous metastases to treatment as an indicator of systemic response, the cutaneous response may be poorer due to the skin’s relatively weaker blood supply.
Linear Terra Firma–Forme Dermatosis of the Midline Back
Terra firma–forme dermatosis (TFFD) was first described by Duncan et al,1 in 1987 and is characterized by brown to black pigmented plaques on the skin that cannot be removed with soap and water but are easily wiped away with isopropyl alcohol. Since that publication, relatively few case reports and case series have been published. We present a case of linear TFFD on the midline back of a 46-year-old woman.
Case Report
A 46-year-old woman presented to our clinic for evaluation of a lesion on the back that had been present for 3 years. An initial diagnosis of acanthosis nigricans or lichen simplex chronicus was made and treatment with topical triamcinolone cream 0.1% was initiated. However, after 8 months of treatment, no improvement was observed and the patient returned to our clinic. Her medical history was notable for obesity, type 2 diabetes mellitus, and hypertension. The patient stated that she maintained good hygiene, including daily to twice-daily showers with soap. Physical examination revealed a linear, hyperkeratotic, dark-brown plaque on the midline back extending from the top of the sacrum to the upper back (Figure 1). No other areas of skin involvement were noted. The hyperpigmented scales were easily removed with an isopropyl alcohol swab, which confirmed a diagnosis of TFFD (Figure 2). The patient was given ammonium lactate lotion 12% to apply to the lesion once daily using an applicator stick if the lesion recurred. She reported some improvement during this treatment. She occasionally had recurrent lesions, which were removed with isopropyl alcohol on subsequent dermatology visits.
Comment
Terra firma–forme dermatosis is an idiopathic condition that, although benign, can cause notable distress to patients. It presents clinically as asymptomatic, brown or black, hyperpigmented, hyperkeratotic, verrucous, or papillomatous plaques or light scaling in some cases.1-4 It can be readily cleared by rubbing with isopropyl alcohol but is resistant to ordinary soap and water.1
Recent reports have shown that TFFD may be more common than once thought.4-6 Although commonly observed in children, TFFD has been reported over a wide range of ages (4–86 years).2-5 The face, ankles, neck, and trunk are the most commonly affected areas.4,7,8 Areas that are less commonly affected often include surgical incision sites as well as the scalp, axillae, back, umbilical area, pubic area, arms, and legs.2-4,8,9 The lesions may be generalized or localized and are sometimes found to be symmetrical.4,10,11
The exact etiology of TFFD is unknown but is believed to be due to melanin retention and alteration or a delay of keratinization that leads to the buildup and compaction of scales.1,2,12 Poor hygiene generally is considered to exclude the diagnosis of TFFD in favor of dermatitis neglecta.6,12,13 Histopathology typically shows epidermal acanthosis, lamellar hyperkeratosis, and orthokeratotic whorls.3,7 However, biopsies seldom are performed due to the ease of diagnosis by removal by cleaning the lesion with isopropyl alcohol.
The diagnosis is confirmed by resolution of the rash after cleaning with isopropyl alcohol.1 Further confirmation of this diagnosis can be achieved through dermoscopy, as large, polygonal, platelike, brown scales can be found arranged together giving a mosaic pattern.6 In addition to cleaning with isopropyl alcohol,5,8 other treatments have shown efficacy for more resistant cases of TFFD, including topical keratolytic agents (eg, lactic acid, urea lotion).4,14
Conclusion
Terra firma–forme dermatosis is a condition that if recognized early, may provide treatment satisfaction through immediate removal of the lesions. Physicians should keep TFFD in their differential during evaluation of patients with asymptomatic, hyperpigmented, hyperkeratotic plaques. Awareness of TFFD is important, as early diagnosis can prevent unnecessary treatment and diagnostic workup.
- Duncan CW, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Browning J, Rosen T. Terra firmaforme dermatosis revisited. Dermatol Online J. 2005;11:11-13.
- Ashique KT, Kaliyadan F, Goyal T. Terra firma-forme dermatosis: report of a series of 11 cases and a brief review of the literature. Int J Dermatol. 2016;55:769-774.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;29:297-300.
- Greywal T, Cohen PR. Terra firma-forme dermatosis: a report of ten individuals with Duncan’s dirty dermatosis and literature review. Dermatol Pract Concept. 2015;5:29-33.
- Abdel-Razek MM, Fathy H. Terra firm-forme dermatosis: case series and dermoscopic features. Dermatol Online J. 2015;21:4-7.
- Akkash L, Badran D, Al-Omari AQ. Terra firma forme dermatosis. case series and review of the literature. J Dtsch Dermatol Ges. 2009;7:102-107.
- O’Brien TJ, Hall AP. Terra firma-forme dermatosis. Aust J Dermatol. 1997;38:163-164.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
- Santarpia M, Guarneri C. Terra firma-forme dermatosis. Eur J Intern Med. 2016;34:1-2.
- Panchal K, Bhalla N, Salunke P, et al. Extensive terra firma forme dermatosis (TFFD): a rare presentation. Indian Dermatol Online J. 2015;6:458-459.
- Erkek E, Sahin S, Cetin ED, et al. Terra firmaforme dermatosis revisited. Indian J Dermatol Venereol Leprol. 2012;78:358-360.
- Poskitt L, Wayte J, Wojnarowska F, et al. ‘Dermatitis neglecta’: unwashed dermatosis. Br J Dermatol. 1995;132:827-829.
- Unal E, Guarneri C, Chokoeva AA, et al. Terra firma-forme dermatosis [published online October 21, 2016]. Wien Med Wochenschr. 2017;167:66-69.
Terra firma–forme dermatosis (TFFD) was first described by Duncan et al,1 in 1987 and is characterized by brown to black pigmented plaques on the skin that cannot be removed with soap and water but are easily wiped away with isopropyl alcohol. Since that publication, relatively few case reports and case series have been published. We present a case of linear TFFD on the midline back of a 46-year-old woman.
Case Report
A 46-year-old woman presented to our clinic for evaluation of a lesion on the back that had been present for 3 years. An initial diagnosis of acanthosis nigricans or lichen simplex chronicus was made and treatment with topical triamcinolone cream 0.1% was initiated. However, after 8 months of treatment, no improvement was observed and the patient returned to our clinic. Her medical history was notable for obesity, type 2 diabetes mellitus, and hypertension. The patient stated that she maintained good hygiene, including daily to twice-daily showers with soap. Physical examination revealed a linear, hyperkeratotic, dark-brown plaque on the midline back extending from the top of the sacrum to the upper back (Figure 1). No other areas of skin involvement were noted. The hyperpigmented scales were easily removed with an isopropyl alcohol swab, which confirmed a diagnosis of TFFD (Figure 2). The patient was given ammonium lactate lotion 12% to apply to the lesion once daily using an applicator stick if the lesion recurred. She reported some improvement during this treatment. She occasionally had recurrent lesions, which were removed with isopropyl alcohol on subsequent dermatology visits.
Comment
Terra firma–forme dermatosis is an idiopathic condition that, although benign, can cause notable distress to patients. It presents clinically as asymptomatic, brown or black, hyperpigmented, hyperkeratotic, verrucous, or papillomatous plaques or light scaling in some cases.1-4 It can be readily cleared by rubbing with isopropyl alcohol but is resistant to ordinary soap and water.1
Recent reports have shown that TFFD may be more common than once thought.4-6 Although commonly observed in children, TFFD has been reported over a wide range of ages (4–86 years).2-5 The face, ankles, neck, and trunk are the most commonly affected areas.4,7,8 Areas that are less commonly affected often include surgical incision sites as well as the scalp, axillae, back, umbilical area, pubic area, arms, and legs.2-4,8,9 The lesions may be generalized or localized and are sometimes found to be symmetrical.4,10,11
The exact etiology of TFFD is unknown but is believed to be due to melanin retention and alteration or a delay of keratinization that leads to the buildup and compaction of scales.1,2,12 Poor hygiene generally is considered to exclude the diagnosis of TFFD in favor of dermatitis neglecta.6,12,13 Histopathology typically shows epidermal acanthosis, lamellar hyperkeratosis, and orthokeratotic whorls.3,7 However, biopsies seldom are performed due to the ease of diagnosis by removal by cleaning the lesion with isopropyl alcohol.
The diagnosis is confirmed by resolution of the rash after cleaning with isopropyl alcohol.1 Further confirmation of this diagnosis can be achieved through dermoscopy, as large, polygonal, platelike, brown scales can be found arranged together giving a mosaic pattern.6 In addition to cleaning with isopropyl alcohol,5,8 other treatments have shown efficacy for more resistant cases of TFFD, including topical keratolytic agents (eg, lactic acid, urea lotion).4,14
Conclusion
Terra firma–forme dermatosis is a condition that if recognized early, may provide treatment satisfaction through immediate removal of the lesions. Physicians should keep TFFD in their differential during evaluation of patients with asymptomatic, hyperpigmented, hyperkeratotic plaques. Awareness of TFFD is important, as early diagnosis can prevent unnecessary treatment and diagnostic workup.
Terra firma–forme dermatosis (TFFD) was first described by Duncan et al,1 in 1987 and is characterized by brown to black pigmented plaques on the skin that cannot be removed with soap and water but are easily wiped away with isopropyl alcohol. Since that publication, relatively few case reports and case series have been published. We present a case of linear TFFD on the midline back of a 46-year-old woman.
Case Report
A 46-year-old woman presented to our clinic for evaluation of a lesion on the back that had been present for 3 years. An initial diagnosis of acanthosis nigricans or lichen simplex chronicus was made and treatment with topical triamcinolone cream 0.1% was initiated. However, after 8 months of treatment, no improvement was observed and the patient returned to our clinic. Her medical history was notable for obesity, type 2 diabetes mellitus, and hypertension. The patient stated that she maintained good hygiene, including daily to twice-daily showers with soap. Physical examination revealed a linear, hyperkeratotic, dark-brown plaque on the midline back extending from the top of the sacrum to the upper back (Figure 1). No other areas of skin involvement were noted. The hyperpigmented scales were easily removed with an isopropyl alcohol swab, which confirmed a diagnosis of TFFD (Figure 2). The patient was given ammonium lactate lotion 12% to apply to the lesion once daily using an applicator stick if the lesion recurred. She reported some improvement during this treatment. She occasionally had recurrent lesions, which were removed with isopropyl alcohol on subsequent dermatology visits.
Comment
Terra firma–forme dermatosis is an idiopathic condition that, although benign, can cause notable distress to patients. It presents clinically as asymptomatic, brown or black, hyperpigmented, hyperkeratotic, verrucous, or papillomatous plaques or light scaling in some cases.1-4 It can be readily cleared by rubbing with isopropyl alcohol but is resistant to ordinary soap and water.1
Recent reports have shown that TFFD may be more common than once thought.4-6 Although commonly observed in children, TFFD has been reported over a wide range of ages (4–86 years).2-5 The face, ankles, neck, and trunk are the most commonly affected areas.4,7,8 Areas that are less commonly affected often include surgical incision sites as well as the scalp, axillae, back, umbilical area, pubic area, arms, and legs.2-4,8,9 The lesions may be generalized or localized and are sometimes found to be symmetrical.4,10,11
The exact etiology of TFFD is unknown but is believed to be due to melanin retention and alteration or a delay of keratinization that leads to the buildup and compaction of scales.1,2,12 Poor hygiene generally is considered to exclude the diagnosis of TFFD in favor of dermatitis neglecta.6,12,13 Histopathology typically shows epidermal acanthosis, lamellar hyperkeratosis, and orthokeratotic whorls.3,7 However, biopsies seldom are performed due to the ease of diagnosis by removal by cleaning the lesion with isopropyl alcohol.
The diagnosis is confirmed by resolution of the rash after cleaning with isopropyl alcohol.1 Further confirmation of this diagnosis can be achieved through dermoscopy, as large, polygonal, platelike, brown scales can be found arranged together giving a mosaic pattern.6 In addition to cleaning with isopropyl alcohol,5,8 other treatments have shown efficacy for more resistant cases of TFFD, including topical keratolytic agents (eg, lactic acid, urea lotion).4,14
Conclusion
Terra firma–forme dermatosis is a condition that if recognized early, may provide treatment satisfaction through immediate removal of the lesions. Physicians should keep TFFD in their differential during evaluation of patients with asymptomatic, hyperpigmented, hyperkeratotic plaques. Awareness of TFFD is important, as early diagnosis can prevent unnecessary treatment and diagnostic workup.
- Duncan CW, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Browning J, Rosen T. Terra firmaforme dermatosis revisited. Dermatol Online J. 2005;11:11-13.
- Ashique KT, Kaliyadan F, Goyal T. Terra firma-forme dermatosis: report of a series of 11 cases and a brief review of the literature. Int J Dermatol. 2016;55:769-774.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;29:297-300.
- Greywal T, Cohen PR. Terra firma-forme dermatosis: a report of ten individuals with Duncan’s dirty dermatosis and literature review. Dermatol Pract Concept. 2015;5:29-33.
- Abdel-Razek MM, Fathy H. Terra firm-forme dermatosis: case series and dermoscopic features. Dermatol Online J. 2015;21:4-7.
- Akkash L, Badran D, Al-Omari AQ. Terra firma forme dermatosis. case series and review of the literature. J Dtsch Dermatol Ges. 2009;7:102-107.
- O’Brien TJ, Hall AP. Terra firma-forme dermatosis. Aust J Dermatol. 1997;38:163-164.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
- Santarpia M, Guarneri C. Terra firma-forme dermatosis. Eur J Intern Med. 2016;34:1-2.
- Panchal K, Bhalla N, Salunke P, et al. Extensive terra firma forme dermatosis (TFFD): a rare presentation. Indian Dermatol Online J. 2015;6:458-459.
- Erkek E, Sahin S, Cetin ED, et al. Terra firmaforme dermatosis revisited. Indian J Dermatol Venereol Leprol. 2012;78:358-360.
- Poskitt L, Wayte J, Wojnarowska F, et al. ‘Dermatitis neglecta’: unwashed dermatosis. Br J Dermatol. 1995;132:827-829.
- Unal E, Guarneri C, Chokoeva AA, et al. Terra firma-forme dermatosis [published online October 21, 2016]. Wien Med Wochenschr. 2017;167:66-69.
- Duncan CW, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Browning J, Rosen T. Terra firmaforme dermatosis revisited. Dermatol Online J. 2005;11:11-13.
- Ashique KT, Kaliyadan F, Goyal T. Terra firma-forme dermatosis: report of a series of 11 cases and a brief review of the literature. Int J Dermatol. 2016;55:769-774.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;29:297-300.
- Greywal T, Cohen PR. Terra firma-forme dermatosis: a report of ten individuals with Duncan’s dirty dermatosis and literature review. Dermatol Pract Concept. 2015;5:29-33.
- Abdel-Razek MM, Fathy H. Terra firm-forme dermatosis: case series and dermoscopic features. Dermatol Online J. 2015;21:4-7.
- Akkash L, Badran D, Al-Omari AQ. Terra firma forme dermatosis. case series and review of the literature. J Dtsch Dermatol Ges. 2009;7:102-107.
- O’Brien TJ, Hall AP. Terra firma-forme dermatosis. Aust J Dermatol. 1997;38:163-164.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
- Santarpia M, Guarneri C. Terra firma-forme dermatosis. Eur J Intern Med. 2016;34:1-2.
- Panchal K, Bhalla N, Salunke P, et al. Extensive terra firma forme dermatosis (TFFD): a rare presentation. Indian Dermatol Online J. 2015;6:458-459.
- Erkek E, Sahin S, Cetin ED, et al. Terra firmaforme dermatosis revisited. Indian J Dermatol Venereol Leprol. 2012;78:358-360.
- Poskitt L, Wayte J, Wojnarowska F, et al. ‘Dermatitis neglecta’: unwashed dermatosis. Br J Dermatol. 1995;132:827-829.
- Unal E, Guarneri C, Chokoeva AA, et al. Terra firma-forme dermatosis [published online October 21, 2016]. Wien Med Wochenschr. 2017;167:66-69.
Practice Points
- Terra firma-forme dermatosis (TFFD) is an idiopathic condition characterized by asymptomatic hyperpigmented and hyperkeratotic plaques that are resistant to removal with soap and water.
- Diagnosis and cure of TFFD can be achieved through removal by rubbing with isopropyl alcohol.
- Increased awareness of the clinical presentation and treatment of TFFD may help patients avoid unnecessary treatment and workup and leads to immediate resolution of the condition.
Postherpetic Isotopic Responses With 3 Simultaneously Occurring Reactions Following Herpes Zoster
Postherpetic isotopic response (PHIR) refers to the occurrence of a second disease manifesting at the site of prior herpes infection. Many forms of PHIR have been described (Table), with postzoster granulomatous dermatitis (eg, granuloma annulare, sarcoidosis, granulomatous vasculitis) being the most common.1 Both primary and metastatic malignancies also can occur at the site of a prior herpes infection. Rarely, multiple types of PHIRs occur simultaneously. We report a case of 3 simultaneously occurring postzoster isotopic responses--granulomatous dermatitis, vasculitis, and chronic lymphocytic leukemia (CLL)--and review the various types of PHIRs.
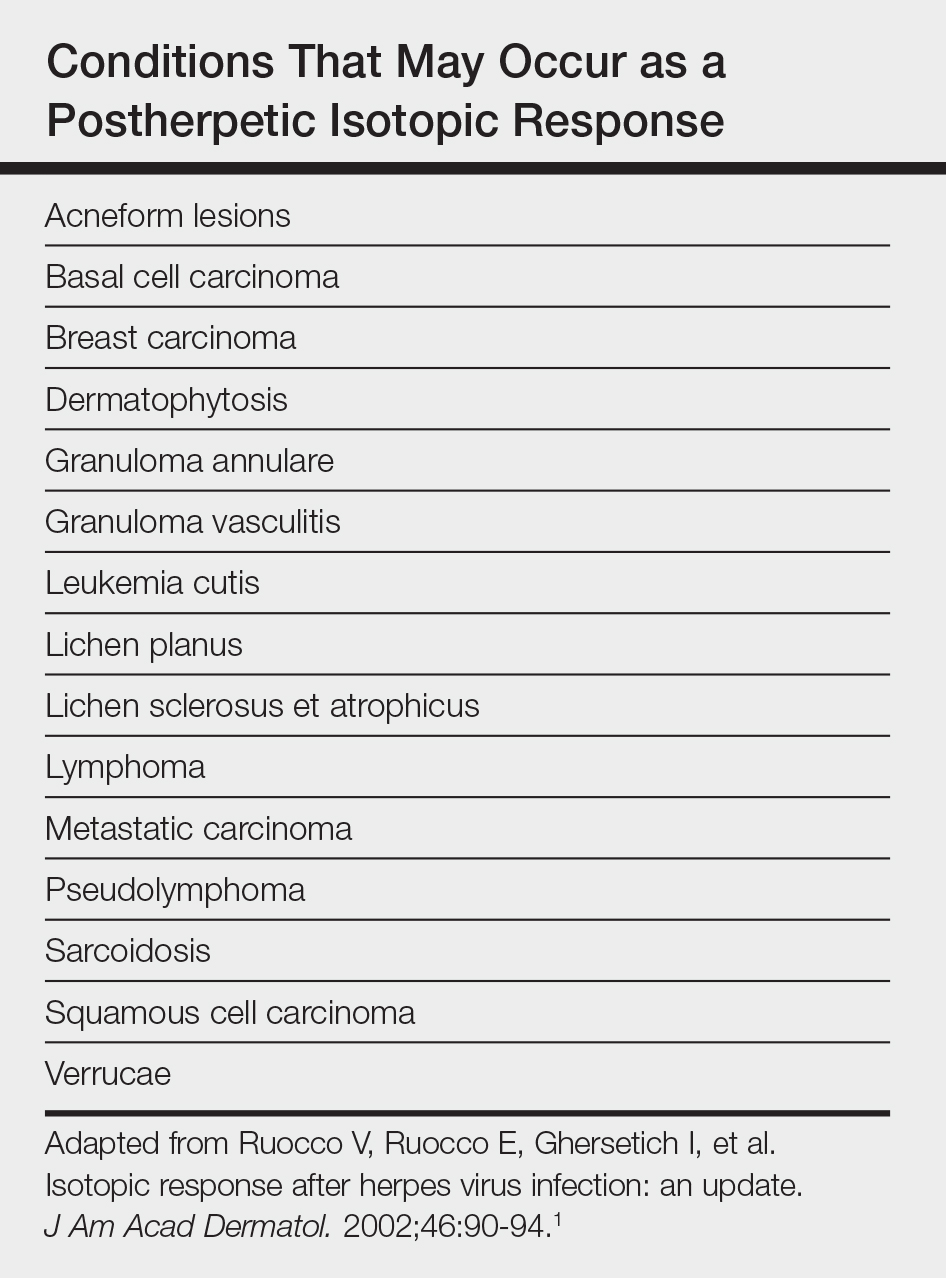
Case Report
A 55-year-old man with a 4-year history of CLL was admitted to the hospital due to a painful rash on the left side of the face of 2 months' duration. Erythematous to violaceous plaques with surrounding papules and nodules were present on the left side of the forehead and frontal scalp with focal ulceration. Two months prior, the patient had unilateral vesicular lesions in the same distribution (Figure 1A). He initially received a 3-week course of acyclovir for a presumed herpes zoster infection and showed prompt improvement in the vesicular lesions. After resolution of the vesicles, papules and nodules began developing in the prior vesicular areas and he was treated with another course of acyclovir with the addition of clindamycin. When the lesions continued to progress and spread down the left side of the forehead and upper eyelid (Figure 1B), he was admitted to the hospital and assessed by the consultative dermatology team. No fevers, chills, or other systemic symptoms were reported.
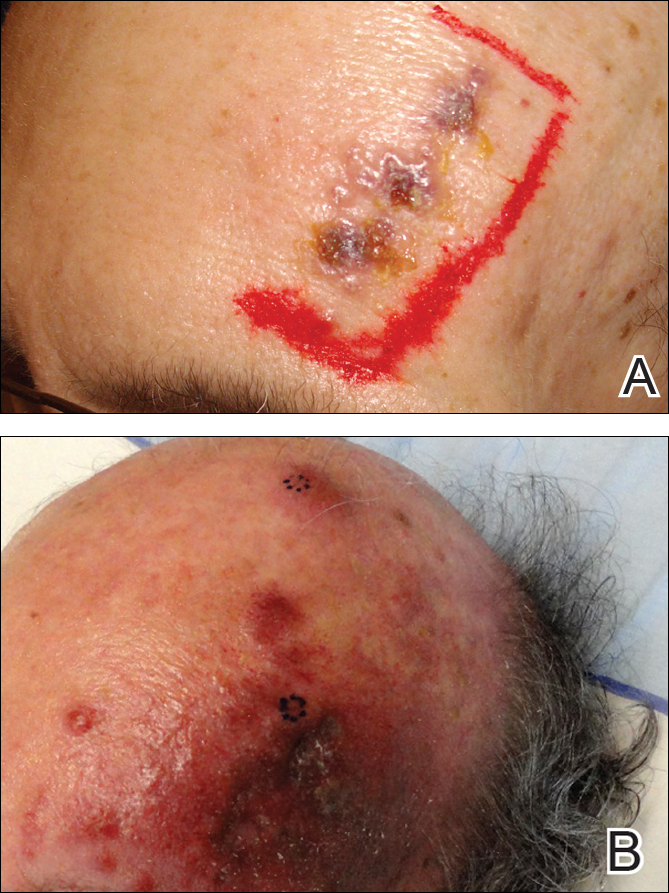
A punch biopsy showed a diffuse lymphocytic infiltrate filling the dermis and extending into the subcutis with nodular collections of histiocytes and some plasma cells scattered throughout (Figure 2A). A medium-vessel vasculitis was present with numerous histiocytes and lymphocytes infiltrating the muscular wall of a blood vessel in the subcutis (Figure 2B). CD3 and CD20 immunostaining showed an overwhelming majority of B cells, some with enlarged atypical nuclei and a smaller number of reactive T lymphocytes (Figure 2C). CD5 and CD43 were diffusely positive in the B cells, confirming the diagnosis of cutaneous CLL. CD23 staining was focally positive. Immunostaining for κ and λ light chains showed a marginal κ predominance. An additional biopsy for tissue culture was negative. A diagnosis of postzoster granulomatous dermatitis with vasculitis and cutaneous CLL was rendered.
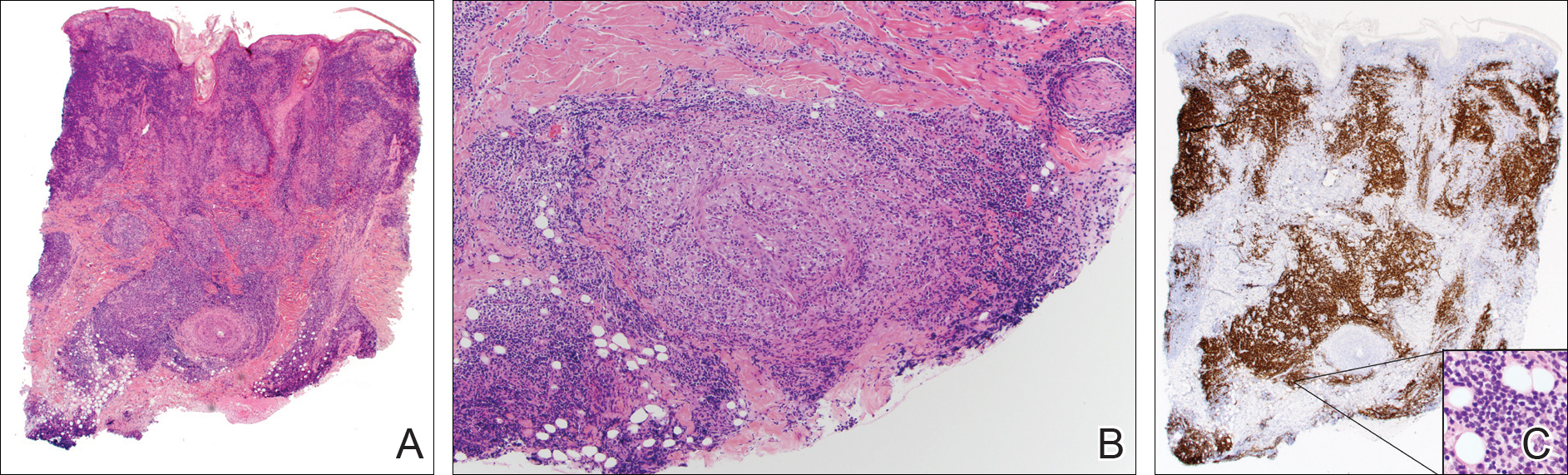
Comment
Postherpetic Cutaneous Reactions
Various cutaneous reactions can occur at the site of prior herpes infection. The most frequently reported reactions are granulomatous dermatitides such as granuloma annulare, granulomatous vasculitis, granulomatous folliculitis, sarcoidosis, and nonspecific granulomatous dermatitis.1 Primary cutaneous malignancies and cutaneous metastases, including hematologic malignancies, have also been reported after herpetic infections. In a review of 127 patients with postherpetic cutaneous reactions, 47 had a granulomatous dermatitis, 32 had nonhematologic malignancies, 18 had leukemic or lymphomatous/pseudolymphomatous infiltrates, 10 had acneform lesions, 9 had nongranulomatous dermatitides such as lichen planus and allergic contact dermatitis, and 8 had nonherpetic skin infections; single cases of reactive perforating collagenosis, nodular solar degeneration, and a keloid also were reported.1
Pathogenesis of Cutaneous Reactions
Although postherpetic cutaneous reactions can develop in healthy individuals, they occur more often in immunocompromised patients. Postherpetic isotopic response has been used to describe the development of a nonherpetic disease at the site of prior herpes infection.2 Several different theories have been proposed to explain the pathogenesis of the PHIR, including an unusual delayed-type hypersensitivity reaction to residual viral antigen or host-tissue antigen altered by the virus. This delayed-type hypersensitivity explanation is supported by the presence of helper T cells, activated T lymphocytes, macrophages, varicella major viral envelope glycoproteins, and viral DNA in postherpetic granulomatous lesions3; however, cases that lack detectable virus and viral DNA in these types of lesions also have been reported.4
A second hypothesis proposes that inflammatory or viral-induced alteration of the local microvasculature results in increased site-specific susceptibility to subsequent inflammatory responses and drives these isotopic reactions.2,3 Damage or alteration of local peripheral nerves leading to abnormal release of specific neuromediators involved in regulating cutaneous inflammatory responses also may play a role.5 Varicella-zoster virus utilizes the peripheral nervous system to establish latent infection and can cause destruction of alpha delta and C nerve fibers in the dermis.1 Destruction of nerve fibers may indirectly influence the local immune system by altering the release of neuromediators such as substance P (known to increase blood vessel permeability, increase fibrinolytic activity, and induce mast cell secretion), vasoactive intestinal peptide (enhances monocyte migration, increases histamine release from mast cells, and inhibits natural killer cell activity), calcitonin gene-related peptide (increases vascular permeability, endothelial cell proliferation, and the accumulation of neutrophils), and melanocyte-stimulating hormone (induces anti-inflammatory cytokines). Disruption of the nervous system resulting in an altered local immune response also has been observed in other settings (eg, amputees who develop inflammatory diseases, bacterial and fungal infections, and cutaneous neoplasms confined to stump skin).1
Malignancies in PHIR
The granulomatous inflammation in PHIRs is a nonneoplastic inflammatory reaction with a variable lymphocytic component. Granuloma formation can be seen in both reactive inflammatory infiltrates and in cutaneous involvement of leukemias and lymphomas. Leukemia cutis has been reported in 4% to 20% of patients with CLL/small lymphocytic leukemia.6 In one series of 42 patients with CLL, the malignant cells were confined to the site of postherpetic scars in 14% (6/42) of patients.5 Sixteen percent (7/42) of patients had no prior diagnosis of CLL at the time they developed leukemia cutis, including one patient with leukemia cutis in a postzoster scar. The mechanism involved in the accumulation of neoplastic lymphocytes within postzoster scars has not been fully characterized. The idea that postzoster sites represent a site of least resistance for cutaneous infiltration of CLL due to the changes from prior inflammatory responses has been proposed.7
Combined CLL and granulomatous dermatitis at prior sites of herpes zoster was first reported in 1990.8 In 1995, Cerroni et al9 reported a series of 5 patients with cutaneous CLL following herpes zoster or herpes simplex virus infection. Three of those patients also demonstrated granuloma formation.9 Establishing a new diagnosis of CLL from a biopsy of postzoster granulomatous dermatitis with an associated lymphoid infiltrate also has been reported.10 Cerroni et al9 postulated that cutaneous CLL in post-herpes zoster scars may occur more frequently than reported due to misdiagnoses of CLL as pseudolymphoma. Two additional cases of postherpetic cutaneous CLL and granulomatous dermatitis have been reported since 1995.7,10
Diagnosis of Multiple PHIRs
The presence of 3 concurrent PHIRs is rare. The patient in this report had postzoster cutaneous CLL with an associated granulomatous dermatitis and medium-vessel vasculitis. One other case with these 3 findings was reported by Elgoweini et al.7 Overlooking important diagnoses when multiple findings are present in a biopsy can lead to diagnostic delay and incorrect treatment; we highlighted the importance of careful examination of biopsies in PHIRs to ensure diagnostic accuracy. In cases of postzoster granulomatous dermatitis, assessment of the lymphocytic component should not be overlooked. The presence of a dense lymphocytic infiltrate should raise the possibility of a lymphoproliferative disorder such as CLL, even in patients with no prior history of lymphoma. If initial immunostaining discloses a predominantly B-cell infiltrate, additional immuno-stains (eg, CD5, CD23, CD43) and/or genetic testing for monoclonality should be pursued.
Conclusion
Clinicians and dermatopathologists should be aware of the multiplicity of postherpetic isotopic responses and consider immunohistochemical stains to differentiate between a genuine lymphoma such as CLL and pseudolymphoma in PHIRs with a lymphoid infiltrate.
- Ruocco V, Ruocco E, Ghersetich I, et al. Isotopic response after herpes virus infection: an update. J Am Acad Dermatol. 2002;46:90-94.
- Wolf R, Wolf D, Ruocco E, et al. Wolf's isotopic response. Clin Dermatol. 2011;29:237-240.
- Nikkels AF, Debrus S, Delvenne P, et al. Viral glycoproteins in herpesviridae granulomas. Am J Dermatopathol. 1994;16:588-592.
- Snow J, el-Azhary R, Gibson L, et al. Granulomatous vasculitis associated with herpes virus: a persistent, painful, postherpetic papular eruption. Mayo Clin Proc. 1997;72:851-853.
- Cerroni L, Zenahlik P, Hofler G, et al. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia: a clinicopathologic and prognostic study of 42 patients. Am J Surg Pathol. 1996;20:1000-1010.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Elgoweini M, Blessing K, Jackson R, et al. Coexistent granulomatous vasculitis and leukaemia cutis in a patient with resolving herpes zoster. Clin Exp Dermatol. 2011;36:749-751.
- Pujol RM, Matias-Guiu X, Planaguma M, et al. Chronic lymphocytic leukemia and cutaneous granulomas at sites of herpes zoster scars. Int J Dermatol. 1990;29:652-654.
- Cerroni L, Zenahlik P, Kerl H. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia arising at the site of herpes zoster and herpes simplex scars. Cancer. 1995;76:26-31.
- Trojjet S, Hammami H, Zaraa I, et al. Chronic lymphocytic leukemia revealed by a granulomatous zosteriform eruption. Skinmed. 2012;10:50-52.
Postherpetic isotopic response (PHIR) refers to the occurrence of a second disease manifesting at the site of prior herpes infection. Many forms of PHIR have been described (Table), with postzoster granulomatous dermatitis (eg, granuloma annulare, sarcoidosis, granulomatous vasculitis) being the most common.1 Both primary and metastatic malignancies also can occur at the site of a prior herpes infection. Rarely, multiple types of PHIRs occur simultaneously. We report a case of 3 simultaneously occurring postzoster isotopic responses--granulomatous dermatitis, vasculitis, and chronic lymphocytic leukemia (CLL)--and review the various types of PHIRs.
Case Report
A 55-year-old man with a 4-year history of CLL was admitted to the hospital due to a painful rash on the left side of the face of 2 months' duration. Erythematous to violaceous plaques with surrounding papules and nodules were present on the left side of the forehead and frontal scalp with focal ulceration. Two months prior, the patient had unilateral vesicular lesions in the same distribution (Figure 1A). He initially received a 3-week course of acyclovir for a presumed herpes zoster infection and showed prompt improvement in the vesicular lesions. After resolution of the vesicles, papules and nodules began developing in the prior vesicular areas and he was treated with another course of acyclovir with the addition of clindamycin. When the lesions continued to progress and spread down the left side of the forehead and upper eyelid (Figure 1B), he was admitted to the hospital and assessed by the consultative dermatology team. No fevers, chills, or other systemic symptoms were reported.
A punch biopsy showed a diffuse lymphocytic infiltrate filling the dermis and extending into the subcutis with nodular collections of histiocytes and some plasma cells scattered throughout (Figure 2A). A medium-vessel vasculitis was present with numerous histiocytes and lymphocytes infiltrating the muscular wall of a blood vessel in the subcutis (Figure 2B). CD3 and CD20 immunostaining showed an overwhelming majority of B cells, some with enlarged atypical nuclei and a smaller number of reactive T lymphocytes (Figure 2C). CD5 and CD43 were diffusely positive in the B cells, confirming the diagnosis of cutaneous CLL. CD23 staining was focally positive. Immunostaining for κ and λ light chains showed a marginal κ predominance. An additional biopsy for tissue culture was negative. A diagnosis of postzoster granulomatous dermatitis with vasculitis and cutaneous CLL was rendered.
Comment
Postherpetic Cutaneous Reactions
Various cutaneous reactions can occur at the site of prior herpes infection. The most frequently reported reactions are granulomatous dermatitides such as granuloma annulare, granulomatous vasculitis, granulomatous folliculitis, sarcoidosis, and nonspecific granulomatous dermatitis.1 Primary cutaneous malignancies and cutaneous metastases, including hematologic malignancies, have also been reported after herpetic infections. In a review of 127 patients with postherpetic cutaneous reactions, 47 had a granulomatous dermatitis, 32 had nonhematologic malignancies, 18 had leukemic or lymphomatous/pseudolymphomatous infiltrates, 10 had acneform lesions, 9 had nongranulomatous dermatitides such as lichen planus and allergic contact dermatitis, and 8 had nonherpetic skin infections; single cases of reactive perforating collagenosis, nodular solar degeneration, and a keloid also were reported.1
Pathogenesis of Cutaneous Reactions
Although postherpetic cutaneous reactions can develop in healthy individuals, they occur more often in immunocompromised patients. Postherpetic isotopic response has been used to describe the development of a nonherpetic disease at the site of prior herpes infection.2 Several different theories have been proposed to explain the pathogenesis of the PHIR, including an unusual delayed-type hypersensitivity reaction to residual viral antigen or host-tissue antigen altered by the virus. This delayed-type hypersensitivity explanation is supported by the presence of helper T cells, activated T lymphocytes, macrophages, varicella major viral envelope glycoproteins, and viral DNA in postherpetic granulomatous lesions3; however, cases that lack detectable virus and viral DNA in these types of lesions also have been reported.4
A second hypothesis proposes that inflammatory or viral-induced alteration of the local microvasculature results in increased site-specific susceptibility to subsequent inflammatory responses and drives these isotopic reactions.2,3 Damage or alteration of local peripheral nerves leading to abnormal release of specific neuromediators involved in regulating cutaneous inflammatory responses also may play a role.5 Varicella-zoster virus utilizes the peripheral nervous system to establish latent infection and can cause destruction of alpha delta and C nerve fibers in the dermis.1 Destruction of nerve fibers may indirectly influence the local immune system by altering the release of neuromediators such as substance P (known to increase blood vessel permeability, increase fibrinolytic activity, and induce mast cell secretion), vasoactive intestinal peptide (enhances monocyte migration, increases histamine release from mast cells, and inhibits natural killer cell activity), calcitonin gene-related peptide (increases vascular permeability, endothelial cell proliferation, and the accumulation of neutrophils), and melanocyte-stimulating hormone (induces anti-inflammatory cytokines). Disruption of the nervous system resulting in an altered local immune response also has been observed in other settings (eg, amputees who develop inflammatory diseases, bacterial and fungal infections, and cutaneous neoplasms confined to stump skin).1
Malignancies in PHIR
The granulomatous inflammation in PHIRs is a nonneoplastic inflammatory reaction with a variable lymphocytic component. Granuloma formation can be seen in both reactive inflammatory infiltrates and in cutaneous involvement of leukemias and lymphomas. Leukemia cutis has been reported in 4% to 20% of patients with CLL/small lymphocytic leukemia.6 In one series of 42 patients with CLL, the malignant cells were confined to the site of postherpetic scars in 14% (6/42) of patients.5 Sixteen percent (7/42) of patients had no prior diagnosis of CLL at the time they developed leukemia cutis, including one patient with leukemia cutis in a postzoster scar. The mechanism involved in the accumulation of neoplastic lymphocytes within postzoster scars has not been fully characterized. The idea that postzoster sites represent a site of least resistance for cutaneous infiltration of CLL due to the changes from prior inflammatory responses has been proposed.7
Combined CLL and granulomatous dermatitis at prior sites of herpes zoster was first reported in 1990.8 In 1995, Cerroni et al9 reported a series of 5 patients with cutaneous CLL following herpes zoster or herpes simplex virus infection. Three of those patients also demonstrated granuloma formation.9 Establishing a new diagnosis of CLL from a biopsy of postzoster granulomatous dermatitis with an associated lymphoid infiltrate also has been reported.10 Cerroni et al9 postulated that cutaneous CLL in post-herpes zoster scars may occur more frequently than reported due to misdiagnoses of CLL as pseudolymphoma. Two additional cases of postherpetic cutaneous CLL and granulomatous dermatitis have been reported since 1995.7,10
Diagnosis of Multiple PHIRs
The presence of 3 concurrent PHIRs is rare. The patient in this report had postzoster cutaneous CLL with an associated granulomatous dermatitis and medium-vessel vasculitis. One other case with these 3 findings was reported by Elgoweini et al.7 Overlooking important diagnoses when multiple findings are present in a biopsy can lead to diagnostic delay and incorrect treatment; we highlighted the importance of careful examination of biopsies in PHIRs to ensure diagnostic accuracy. In cases of postzoster granulomatous dermatitis, assessment of the lymphocytic component should not be overlooked. The presence of a dense lymphocytic infiltrate should raise the possibility of a lymphoproliferative disorder such as CLL, even in patients with no prior history of lymphoma. If initial immunostaining discloses a predominantly B-cell infiltrate, additional immuno-stains (eg, CD5, CD23, CD43) and/or genetic testing for monoclonality should be pursued.
Conclusion
Clinicians and dermatopathologists should be aware of the multiplicity of postherpetic isotopic responses and consider immunohistochemical stains to differentiate between a genuine lymphoma such as CLL and pseudolymphoma in PHIRs with a lymphoid infiltrate.
Postherpetic isotopic response (PHIR) refers to the occurrence of a second disease manifesting at the site of prior herpes infection. Many forms of PHIR have been described (Table), with postzoster granulomatous dermatitis (eg, granuloma annulare, sarcoidosis, granulomatous vasculitis) being the most common.1 Both primary and metastatic malignancies also can occur at the site of a prior herpes infection. Rarely, multiple types of PHIRs occur simultaneously. We report a case of 3 simultaneously occurring postzoster isotopic responses--granulomatous dermatitis, vasculitis, and chronic lymphocytic leukemia (CLL)--and review the various types of PHIRs.
Case Report
A 55-year-old man with a 4-year history of CLL was admitted to the hospital due to a painful rash on the left side of the face of 2 months' duration. Erythematous to violaceous plaques with surrounding papules and nodules were present on the left side of the forehead and frontal scalp with focal ulceration. Two months prior, the patient had unilateral vesicular lesions in the same distribution (Figure 1A). He initially received a 3-week course of acyclovir for a presumed herpes zoster infection and showed prompt improvement in the vesicular lesions. After resolution of the vesicles, papules and nodules began developing in the prior vesicular areas and he was treated with another course of acyclovir with the addition of clindamycin. When the lesions continued to progress and spread down the left side of the forehead and upper eyelid (Figure 1B), he was admitted to the hospital and assessed by the consultative dermatology team. No fevers, chills, or other systemic symptoms were reported.
A punch biopsy showed a diffuse lymphocytic infiltrate filling the dermis and extending into the subcutis with nodular collections of histiocytes and some plasma cells scattered throughout (Figure 2A). A medium-vessel vasculitis was present with numerous histiocytes and lymphocytes infiltrating the muscular wall of a blood vessel in the subcutis (Figure 2B). CD3 and CD20 immunostaining showed an overwhelming majority of B cells, some with enlarged atypical nuclei and a smaller number of reactive T lymphocytes (Figure 2C). CD5 and CD43 were diffusely positive in the B cells, confirming the diagnosis of cutaneous CLL. CD23 staining was focally positive. Immunostaining for κ and λ light chains showed a marginal κ predominance. An additional biopsy for tissue culture was negative. A diagnosis of postzoster granulomatous dermatitis with vasculitis and cutaneous CLL was rendered.
Comment
Postherpetic Cutaneous Reactions
Various cutaneous reactions can occur at the site of prior herpes infection. The most frequently reported reactions are granulomatous dermatitides such as granuloma annulare, granulomatous vasculitis, granulomatous folliculitis, sarcoidosis, and nonspecific granulomatous dermatitis.1 Primary cutaneous malignancies and cutaneous metastases, including hematologic malignancies, have also been reported after herpetic infections. In a review of 127 patients with postherpetic cutaneous reactions, 47 had a granulomatous dermatitis, 32 had nonhematologic malignancies, 18 had leukemic or lymphomatous/pseudolymphomatous infiltrates, 10 had acneform lesions, 9 had nongranulomatous dermatitides such as lichen planus and allergic contact dermatitis, and 8 had nonherpetic skin infections; single cases of reactive perforating collagenosis, nodular solar degeneration, and a keloid also were reported.1
Pathogenesis of Cutaneous Reactions
Although postherpetic cutaneous reactions can develop in healthy individuals, they occur more often in immunocompromised patients. Postherpetic isotopic response has been used to describe the development of a nonherpetic disease at the site of prior herpes infection.2 Several different theories have been proposed to explain the pathogenesis of the PHIR, including an unusual delayed-type hypersensitivity reaction to residual viral antigen or host-tissue antigen altered by the virus. This delayed-type hypersensitivity explanation is supported by the presence of helper T cells, activated T lymphocytes, macrophages, varicella major viral envelope glycoproteins, and viral DNA in postherpetic granulomatous lesions3; however, cases that lack detectable virus and viral DNA in these types of lesions also have been reported.4
A second hypothesis proposes that inflammatory or viral-induced alteration of the local microvasculature results in increased site-specific susceptibility to subsequent inflammatory responses and drives these isotopic reactions.2,3 Damage or alteration of local peripheral nerves leading to abnormal release of specific neuromediators involved in regulating cutaneous inflammatory responses also may play a role.5 Varicella-zoster virus utilizes the peripheral nervous system to establish latent infection and can cause destruction of alpha delta and C nerve fibers in the dermis.1 Destruction of nerve fibers may indirectly influence the local immune system by altering the release of neuromediators such as substance P (known to increase blood vessel permeability, increase fibrinolytic activity, and induce mast cell secretion), vasoactive intestinal peptide (enhances monocyte migration, increases histamine release from mast cells, and inhibits natural killer cell activity), calcitonin gene-related peptide (increases vascular permeability, endothelial cell proliferation, and the accumulation of neutrophils), and melanocyte-stimulating hormone (induces anti-inflammatory cytokines). Disruption of the nervous system resulting in an altered local immune response also has been observed in other settings (eg, amputees who develop inflammatory diseases, bacterial and fungal infections, and cutaneous neoplasms confined to stump skin).1
Malignancies in PHIR
The granulomatous inflammation in PHIRs is a nonneoplastic inflammatory reaction with a variable lymphocytic component. Granuloma formation can be seen in both reactive inflammatory infiltrates and in cutaneous involvement of leukemias and lymphomas. Leukemia cutis has been reported in 4% to 20% of patients with CLL/small lymphocytic leukemia.6 In one series of 42 patients with CLL, the malignant cells were confined to the site of postherpetic scars in 14% (6/42) of patients.5 Sixteen percent (7/42) of patients had no prior diagnosis of CLL at the time they developed leukemia cutis, including one patient with leukemia cutis in a postzoster scar. The mechanism involved in the accumulation of neoplastic lymphocytes within postzoster scars has not been fully characterized. The idea that postzoster sites represent a site of least resistance for cutaneous infiltration of CLL due to the changes from prior inflammatory responses has been proposed.7
Combined CLL and granulomatous dermatitis at prior sites of herpes zoster was first reported in 1990.8 In 1995, Cerroni et al9 reported a series of 5 patients with cutaneous CLL following herpes zoster or herpes simplex virus infection. Three of those patients also demonstrated granuloma formation.9 Establishing a new diagnosis of CLL from a biopsy of postzoster granulomatous dermatitis with an associated lymphoid infiltrate also has been reported.10 Cerroni et al9 postulated that cutaneous CLL in post-herpes zoster scars may occur more frequently than reported due to misdiagnoses of CLL as pseudolymphoma. Two additional cases of postherpetic cutaneous CLL and granulomatous dermatitis have been reported since 1995.7,10
Diagnosis of Multiple PHIRs
The presence of 3 concurrent PHIRs is rare. The patient in this report had postzoster cutaneous CLL with an associated granulomatous dermatitis and medium-vessel vasculitis. One other case with these 3 findings was reported by Elgoweini et al.7 Overlooking important diagnoses when multiple findings are present in a biopsy can lead to diagnostic delay and incorrect treatment; we highlighted the importance of careful examination of biopsies in PHIRs to ensure diagnostic accuracy. In cases of postzoster granulomatous dermatitis, assessment of the lymphocytic component should not be overlooked. The presence of a dense lymphocytic infiltrate should raise the possibility of a lymphoproliferative disorder such as CLL, even in patients with no prior history of lymphoma. If initial immunostaining discloses a predominantly B-cell infiltrate, additional immuno-stains (eg, CD5, CD23, CD43) and/or genetic testing for monoclonality should be pursued.
Conclusion
Clinicians and dermatopathologists should be aware of the multiplicity of postherpetic isotopic responses and consider immunohistochemical stains to differentiate between a genuine lymphoma such as CLL and pseudolymphoma in PHIRs with a lymphoid infiltrate.
- Ruocco V, Ruocco E, Ghersetich I, et al. Isotopic response after herpes virus infection: an update. J Am Acad Dermatol. 2002;46:90-94.
- Wolf R, Wolf D, Ruocco E, et al. Wolf's isotopic response. Clin Dermatol. 2011;29:237-240.
- Nikkels AF, Debrus S, Delvenne P, et al. Viral glycoproteins in herpesviridae granulomas. Am J Dermatopathol. 1994;16:588-592.
- Snow J, el-Azhary R, Gibson L, et al. Granulomatous vasculitis associated with herpes virus: a persistent, painful, postherpetic papular eruption. Mayo Clin Proc. 1997;72:851-853.
- Cerroni L, Zenahlik P, Hofler G, et al. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia: a clinicopathologic and prognostic study of 42 patients. Am J Surg Pathol. 1996;20:1000-1010.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Elgoweini M, Blessing K, Jackson R, et al. Coexistent granulomatous vasculitis and leukaemia cutis in a patient with resolving herpes zoster. Clin Exp Dermatol. 2011;36:749-751.
- Pujol RM, Matias-Guiu X, Planaguma M, et al. Chronic lymphocytic leukemia and cutaneous granulomas at sites of herpes zoster scars. Int J Dermatol. 1990;29:652-654.
- Cerroni L, Zenahlik P, Kerl H. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia arising at the site of herpes zoster and herpes simplex scars. Cancer. 1995;76:26-31.
- Trojjet S, Hammami H, Zaraa I, et al. Chronic lymphocytic leukemia revealed by a granulomatous zosteriform eruption. Skinmed. 2012;10:50-52.
- Ruocco V, Ruocco E, Ghersetich I, et al. Isotopic response after herpes virus infection: an update. J Am Acad Dermatol. 2002;46:90-94.
- Wolf R, Wolf D, Ruocco E, et al. Wolf's isotopic response. Clin Dermatol. 2011;29:237-240.
- Nikkels AF, Debrus S, Delvenne P, et al. Viral glycoproteins in herpesviridae granulomas. Am J Dermatopathol. 1994;16:588-592.
- Snow J, el-Azhary R, Gibson L, et al. Granulomatous vasculitis associated with herpes virus: a persistent, painful, postherpetic papular eruption. Mayo Clin Proc. 1997;72:851-853.
- Cerroni L, Zenahlik P, Hofler G, et al. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia: a clinicopathologic and prognostic study of 42 patients. Am J Surg Pathol. 1996;20:1000-1010.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Elgoweini M, Blessing K, Jackson R, et al. Coexistent granulomatous vasculitis and leukaemia cutis in a patient with resolving herpes zoster. Clin Exp Dermatol. 2011;36:749-751.
- Pujol RM, Matias-Guiu X, Planaguma M, et al. Chronic lymphocytic leukemia and cutaneous granulomas at sites of herpes zoster scars. Int J Dermatol. 1990;29:652-654.
- Cerroni L, Zenahlik P, Kerl H. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia arising at the site of herpes zoster and herpes simplex scars. Cancer. 1995;76:26-31.
- Trojjet S, Hammami H, Zaraa I, et al. Chronic lymphocytic leukemia revealed by a granulomatous zosteriform eruption. Skinmed. 2012;10:50-52.
Practice Points
- Multiple diseases may present in prior sites of herpes infection (postherpetic isotopic response).
- Granulomatous dermatitis is the most common postherpetic isotopic response, but other inflammatory, neoplastic, or infectious conditions also occur.
- Multiple conditions may present simultaneously at sites of herpes infection.
- Cutaneous involvement by chronic lymphocytic leukemia (CLL) can be easily overlooked in this setting.
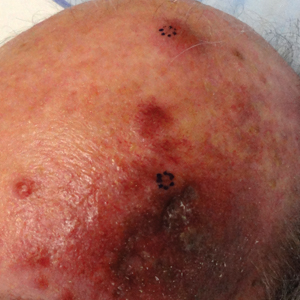
Anti-PD-1 therapy with nivolumab in the treatment of metastatic malignant PEComa
Perivascular epithelioid cell neoplasms (PEComas) are an uncommon class of tumors consisting on histology of perivascular epithelioid cells occurring in both localized and metastatic forms at various body sites. The approach to treatment of these tumors generally involves a combination of surgical resection, chemotherapy, and/or radiation therapy.1
Case presentation and summary
A 46-year-old man presented to our institution with a non-tender, slowly enlarging, 8.3 cm mass in his right popliteal fossa. Upon biopsy, the pathologic findings were consistent with an epithelioid malignancy with melanocytic differentiation most consistent with a PEComa. Discussion of the pathologic diagnosis of our patient has been reported by the pathology group at our institution in a separate case report.2
Our patient was initially offered and refused amputation. He was started on therapy with the mechanistic Target of Rapamycin (mTOR) inhibitor everolimus, but was unable to tolerate the side effects after the first week of treatment. He then elected to monitor his symptoms clinically.
Approximately one year after his initial diagnosis, he presented to our facility with sepsis and bleeding from a now fungating tumor on his right knee. At this time, emergent above-knee amputation was performed. Re-staging images now showed the presence of multiple pulmonary nodules in his right lung as well as a lytic rib lesion, a concerning finding for metastatic disease. Video-Assisted Thorascopic Surgery (VATS) and right lower lobe wedge resection were performed and findings confirmed metastatic PEComa.
Given the patient’s intolerance to everolimus, he was started on the growth factor inhibitor, pazopanib. His disease did not progress on pazopanib, and improvement was noted in the dominant pulmonary nodule. Subsequently, however, he developed significant skin irritation and discontinued pazopanib. Repeat imaging approximately 2 months after stopping pazopanib showed significant disease progression.
We elected to start the patient on a non-standard approach to therapy with nivolumab infusions once every 2 weeks and concurrent radiation therapy to the rib lesion. At 2 and 5 months after initiating this treatment approach, CT imaging showed improvement in disease. At 12 months, significant disease response was noted (Figure 1).
The patient is now at 12 months of nivolumab therapy with progression free survival and no new identifiable metastatic lesions. He has been tolerating the medication with minimal side effects and has had an overall improvement in his pain and functional status. He continues to work full time.
Discussion
Our patient’s response presents a unique opportunity to talk about the role of immunotherapy as a treatment modality in patients with PEComa. The efficacy of check-point blockade in soft tissue sarcoma is still unclear predominantly because it is difficult to assess the degree of expression of immunogenic cell surface markers such as programmed cell death protein 1 (PD-1).1,3 Nivolumab has been tried in small cohorts for treatment of soft tissue sarcomas that express PD-1 and results showed some clinical benefit in about half of patients.4 Further, the expression of PD-1 has been assessed in soft tissue sarcomas and has been reported to suggest a negative prognostic role.5
To our knowledge, there has not yet been another reported case of PEComa that has been treated with immunotherapy and achieved a sustained response. Further clinical studies need to be done to assess response to agents such as nivolumab in the treatment of PEComa to bolster our observation that nivolumab is a viable treatment option that may lead to lasting remission. Our patient’s case also brings to light the need for further inquiry into assessing the immune tumor microenvironments, particularly looking at the expression of cell surface proteins such as PD-1, as it ultimately affects treatment options. TSJ
Correspondence
REFERENCES
1. Burgess, Melissa, et al. “Immunotherapy in Sarcoma: Future Horizons.” Current Oncology Reports, vol. 17, no. 11, 2015, doi:10.1007/s11912-015-0476-7.
2. Alnajar, Hussein, et al. “Metastatic Malignant PEComa of the Leg with Identification of ATRX Mutation by next-Generation Sequencing.” Virchows Archiv (2017). https://doi:10.1007/s004280172208-x.
3. Ghosn, Marwan, et al. “Immunotherapies in Sarcoma: Updates and Future Perspectives.” World Journal of Clinical Oncology, vol. 8, no. 2, 2017, p. 145., doi:10.5306/wjco.v8.i2.145.
4. Paoluzzi, L., et al. “Response to Anti-PD1 Therapy with Nivolumab in Metastatic Sarcomas.” Clinical Sarcoma Research, vol. 6, no. 1, 2016, doi:10.1186/s13569-016 0064-0.
5. Kim, Chan, et al. “Prognostic Implications of PD-L1 Expression in Patients with Soft Tissue Sarcoma.” BMC Cancer, BioMed Central 8 July 2016.
Perivascular epithelioid cell neoplasms (PEComas) are an uncommon class of tumors consisting on histology of perivascular epithelioid cells occurring in both localized and metastatic forms at various body sites. The approach to treatment of these tumors generally involves a combination of surgical resection, chemotherapy, and/or radiation therapy.1
Case presentation and summary
A 46-year-old man presented to our institution with a non-tender, slowly enlarging, 8.3 cm mass in his right popliteal fossa. Upon biopsy, the pathologic findings were consistent with an epithelioid malignancy with melanocytic differentiation most consistent with a PEComa. Discussion of the pathologic diagnosis of our patient has been reported by the pathology group at our institution in a separate case report.2
Our patient was initially offered and refused amputation. He was started on therapy with the mechanistic Target of Rapamycin (mTOR) inhibitor everolimus, but was unable to tolerate the side effects after the first week of treatment. He then elected to monitor his symptoms clinically.
Approximately one year after his initial diagnosis, he presented to our facility with sepsis and bleeding from a now fungating tumor on his right knee. At this time, emergent above-knee amputation was performed. Re-staging images now showed the presence of multiple pulmonary nodules in his right lung as well as a lytic rib lesion, a concerning finding for metastatic disease. Video-Assisted Thorascopic Surgery (VATS) and right lower lobe wedge resection were performed and findings confirmed metastatic PEComa.
Given the patient’s intolerance to everolimus, he was started on the growth factor inhibitor, pazopanib. His disease did not progress on pazopanib, and improvement was noted in the dominant pulmonary nodule. Subsequently, however, he developed significant skin irritation and discontinued pazopanib. Repeat imaging approximately 2 months after stopping pazopanib showed significant disease progression.
We elected to start the patient on a non-standard approach to therapy with nivolumab infusions once every 2 weeks and concurrent radiation therapy to the rib lesion. At 2 and 5 months after initiating this treatment approach, CT imaging showed improvement in disease. At 12 months, significant disease response was noted (Figure 1).
The patient is now at 12 months of nivolumab therapy with progression free survival and no new identifiable metastatic lesions. He has been tolerating the medication with minimal side effects and has had an overall improvement in his pain and functional status. He continues to work full time.
Discussion
Our patient’s response presents a unique opportunity to talk about the role of immunotherapy as a treatment modality in patients with PEComa. The efficacy of check-point blockade in soft tissue sarcoma is still unclear predominantly because it is difficult to assess the degree of expression of immunogenic cell surface markers such as programmed cell death protein 1 (PD-1).1,3 Nivolumab has been tried in small cohorts for treatment of soft tissue sarcomas that express PD-1 and results showed some clinical benefit in about half of patients.4 Further, the expression of PD-1 has been assessed in soft tissue sarcomas and has been reported to suggest a negative prognostic role.5
To our knowledge, there has not yet been another reported case of PEComa that has been treated with immunotherapy and achieved a sustained response. Further clinical studies need to be done to assess response to agents such as nivolumab in the treatment of PEComa to bolster our observation that nivolumab is a viable treatment option that may lead to lasting remission. Our patient’s case also brings to light the need for further inquiry into assessing the immune tumor microenvironments, particularly looking at the expression of cell surface proteins such as PD-1, as it ultimately affects treatment options. TSJ
Correspondence
REFERENCES
1. Burgess, Melissa, et al. “Immunotherapy in Sarcoma: Future Horizons.” Current Oncology Reports, vol. 17, no. 11, 2015, doi:10.1007/s11912-015-0476-7.
2. Alnajar, Hussein, et al. “Metastatic Malignant PEComa of the Leg with Identification of ATRX Mutation by next-Generation Sequencing.” Virchows Archiv (2017). https://doi:10.1007/s004280172208-x.
3. Ghosn, Marwan, et al. “Immunotherapies in Sarcoma: Updates and Future Perspectives.” World Journal of Clinical Oncology, vol. 8, no. 2, 2017, p. 145., doi:10.5306/wjco.v8.i2.145.
4. Paoluzzi, L., et al. “Response to Anti-PD1 Therapy with Nivolumab in Metastatic Sarcomas.” Clinical Sarcoma Research, vol. 6, no. 1, 2016, doi:10.1186/s13569-016 0064-0.
5. Kim, Chan, et al. “Prognostic Implications of PD-L1 Expression in Patients with Soft Tissue Sarcoma.” BMC Cancer, BioMed Central 8 July 2016.
Perivascular epithelioid cell neoplasms (PEComas) are an uncommon class of tumors consisting on histology of perivascular epithelioid cells occurring in both localized and metastatic forms at various body sites. The approach to treatment of these tumors generally involves a combination of surgical resection, chemotherapy, and/or radiation therapy.1
Case presentation and summary
A 46-year-old man presented to our institution with a non-tender, slowly enlarging, 8.3 cm mass in his right popliteal fossa. Upon biopsy, the pathologic findings were consistent with an epithelioid malignancy with melanocytic differentiation most consistent with a PEComa. Discussion of the pathologic diagnosis of our patient has been reported by the pathology group at our institution in a separate case report.2
Our patient was initially offered and refused amputation. He was started on therapy with the mechanistic Target of Rapamycin (mTOR) inhibitor everolimus, but was unable to tolerate the side effects after the first week of treatment. He then elected to monitor his symptoms clinically.
Approximately one year after his initial diagnosis, he presented to our facility with sepsis and bleeding from a now fungating tumor on his right knee. At this time, emergent above-knee amputation was performed. Re-staging images now showed the presence of multiple pulmonary nodules in his right lung as well as a lytic rib lesion, a concerning finding for metastatic disease. Video-Assisted Thorascopic Surgery (VATS) and right lower lobe wedge resection were performed and findings confirmed metastatic PEComa.
Given the patient’s intolerance to everolimus, he was started on the growth factor inhibitor, pazopanib. His disease did not progress on pazopanib, and improvement was noted in the dominant pulmonary nodule. Subsequently, however, he developed significant skin irritation and discontinued pazopanib. Repeat imaging approximately 2 months after stopping pazopanib showed significant disease progression.
We elected to start the patient on a non-standard approach to therapy with nivolumab infusions once every 2 weeks and concurrent radiation therapy to the rib lesion. At 2 and 5 months after initiating this treatment approach, CT imaging showed improvement in disease. At 12 months, significant disease response was noted (Figure 1).
The patient is now at 12 months of nivolumab therapy with progression free survival and no new identifiable metastatic lesions. He has been tolerating the medication with minimal side effects and has had an overall improvement in his pain and functional status. He continues to work full time.
Discussion
Our patient’s response presents a unique opportunity to talk about the role of immunotherapy as a treatment modality in patients with PEComa. The efficacy of check-point blockade in soft tissue sarcoma is still unclear predominantly because it is difficult to assess the degree of expression of immunogenic cell surface markers such as programmed cell death protein 1 (PD-1).1,3 Nivolumab has been tried in small cohorts for treatment of soft tissue sarcomas that express PD-1 and results showed some clinical benefit in about half of patients.4 Further, the expression of PD-1 has been assessed in soft tissue sarcomas and has been reported to suggest a negative prognostic role.5
To our knowledge, there has not yet been another reported case of PEComa that has been treated with immunotherapy and achieved a sustained response. Further clinical studies need to be done to assess response to agents such as nivolumab in the treatment of PEComa to bolster our observation that nivolumab is a viable treatment option that may lead to lasting remission. Our patient’s case also brings to light the need for further inquiry into assessing the immune tumor microenvironments, particularly looking at the expression of cell surface proteins such as PD-1, as it ultimately affects treatment options. TSJ
Correspondence
REFERENCES
1. Burgess, Melissa, et al. “Immunotherapy in Sarcoma: Future Horizons.” Current Oncology Reports, vol. 17, no. 11, 2015, doi:10.1007/s11912-015-0476-7.
2. Alnajar, Hussein, et al. “Metastatic Malignant PEComa of the Leg with Identification of ATRX Mutation by next-Generation Sequencing.” Virchows Archiv (2017). https://doi:10.1007/s004280172208-x.
3. Ghosn, Marwan, et al. “Immunotherapies in Sarcoma: Updates and Future Perspectives.” World Journal of Clinical Oncology, vol. 8, no. 2, 2017, p. 145., doi:10.5306/wjco.v8.i2.145.
4. Paoluzzi, L., et al. “Response to Anti-PD1 Therapy with Nivolumab in Metastatic Sarcomas.” Clinical Sarcoma Research, vol. 6, no. 1, 2016, doi:10.1186/s13569-016 0064-0.
5. Kim, Chan, et al. “Prognostic Implications of PD-L1 Expression in Patients with Soft Tissue Sarcoma.” BMC Cancer, BioMed Central 8 July 2016.
Tumor lysis syndrome in an adolescent with recurrence of abdominal rhabdomyosarcoma: A case report and literature review
Introduction
Tumor lysis syndrome (TLS) is a life-threatening oncologic emergency that results when massive cell breakdown occurs either spontaneously or in response to cytotoxic chemotherapy. TLS is characterized by metabolic derangements, including hyperkalemia and hyperphosphatemia, secondary to the release of intracellular components into the systemic circulatory system. In addition, purine degradation can lead to hyperuricemia, and precipitation of calcium phosphate can result in hypocalcemia. Lactate dehydrogenase (LDH) levels are often elevated, especially in higher risk patients; however, this finding is not a specific marker for TLS.
TLS more commonly occurs in patients with rapidly proliferating hematological malignancies, such as acute leukemias with a high white blood cell count and Burkitt’s lymphoma, and is a relatively rare event in patients with solid malignancies.1-3 It is even more rare in patients with tumor recurrence.
There are few reported cases of TLS in children with solid malignancies. To our knowledge, only one case of TLS has previously been reported in a pediatric patient with abdominal rhabdomyosarcoma. We report the second such case, and what we believe to be the only reported case of TLS occurring in a pediatric patient with recurrence of a solid tumor.
Case Description
A 15-year-old male from Saudi Arabia presented to our hospital with confirmed stage IV abdominal rhabdomyosarcoma and lung metastases diagnosed in 2012. His initial treatment consisted of complete surgical resection, lung irradiation, and chemotherapy with intercalating cycles of ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide, as per the COG-ARST0431 high-risk sarcoma protocol (NCT00354744). He completed treatment without any reported TLS in Saudi Arabia in June 2014. He had no residual tumor at the end of therapy, but six months later he was found to have an abdominal recurrence and started treatment with single-agent topotecan chemotherapy. He experienced worsening abdominal distention, pain, and difficulty voiding, prompting his family to seek further treatment options abroad.
The patient was admitted to our hospital in March 2015. Despite being severely malnourished, he was in stable condition. He was noted to have a markedly enlarged, firm, distended abdomen with dilated veins, abdominal and lower back pain, lower extremity pitting edema, and difficulty urinating.
Initial laboratory findings were unremarkable except for elevated levels of BUN (29 mg/dL), creatinine (1.69 mg/dL), and phosphorus (5.6 mg/dL). MRI revealed a large pelvic mass measuring 15.3 x 15.2 x 21.3 centimeters in transverse, anterior-posterior, and craniocaudal dimensions, respectively; with concomitant severe bilateral hydroureternephrosis (FIGURE 1).

FIGURE 1. Sagittal (A) and Axial (B) T2-weighted MR images of the pelvis (prior to initiating therapy) demonstrating a large heterogeneous mass occupying the entire pelvis. There is evidence of edema involving the soft tissues of the perineum (long arrow) and a large associated hydrocele (short arrow).
Three days following admission, the patient’s urine output decreased and his creatinine level rose rapidly. His worsening abdominal distention was attributed to growing tumor bulk and obstructive nephropathy. He required emergency placement of bilateral nephrostomy tubes. Urine output subsequently improved; although, serum creatinine remained persistently elevated.
Given his worsening condition, chemotherapy was begun three days after nephrostomy tube placement with vinorelbine, cyclophosphamide, and temsirolimus, as per COG-ARST0921 (NCT01222715), at renal-adjusted doses. Laboratory studies approximately 24 hours after chemotherapy initiation demonstrated the presence of TLS (TABLE 1). Potassium level was at the upper end of normal at 4.9 mmol/L, calcium level was decreased to 7.1 mg/dL, phosphorus level elevated to 12 mg/dL, uric acid level was markedly elevated to 19.5 mg/dL, and LDH elevated to 662 unit/L. A dose of 0.15 mg/kg of rasburicase was immediately given with a second dose repeated 14 hours later, after which the uric acid level decreased to less than 0.5 mg/dL. Sevelamer, sodium polystyrene, calcium carbonate, and magnesium gluconate were also administered to treat other electrolyte imbalances. The patient remained at clinical baseline throughout, and the TLS laboratory derangements normalized by three days after the TLS diagnosis; LDH level normalized after one week. The patient continued with chemotherapy, per protocol, with no further TLS-related complications. Over subsequent weeks, his tumor continued to shrink dramatically. Pain related to intra-abdominal compression, lower extremity edema, and difficulty voiding resolved.
Discussion
A literature search was performed using Pubmed/Medline and Scopus from 1950 to July 2016 using key words “TLS,” “tumor lysis syndrome,” “pediatric tumor lysis syndrome,” “tumor lysis syndrome in solid malignancies,” “recurrence,” “solid tumor,” “sarcoma,” “rhabdomyosarcoma,” and their combinations. The references of relevant articles were reviewed. Baeksgaard and Sorensen,3 and Vodopivec, et al4 provide an organized review of reported cases of TLS in solid tumors until 2002 and 2011 respectively; their articles are supported by the 2014 literature review by Mirrakhimov, et al.1 Excluding our case, 13 cases of TLS have been described in pediatric patients with solid tumors, with only one occurring in patient with abdominal rhabdomyosarcoma5. Patients’ ages ranged from 2 days to 23 years; the cases are summarized in the following table (TABLE 2). To our knowledge, ours is the first case of TLS reported in association with a pediatric solid tumor recurrence.
It is important to note that the three reported cases of disseminated rhabdomyosarcoma6,7 were initially believed to be hematologic malignancies because of their presentation with lymphadenopathy, metastases to the bone marrow, and spontaneous onset of TLS. Rhabdomyosarcoma with bone marrow involvement without an obvious primary tumor is easily confused with acute leukemia, particularly of the lymphoblastic type.12 However, this disseminated-hematologic presentation of rhabdomyosarcoma differs from the solid abdominal-pelvic tumor, which we describe.
Cairo and Bishop13 categorize patients as either laboratory TLS, depicted by metabolic abnormalities alone, or clinical TLS, occurring when laboratory imbalances lead to significant, life-threatening clinical manifestations. Hyperkalemia may lead to cardiac arrhythmias such as torsades de pointes and cardiac arrest. Obstructive nephropathy can occur from the precipitation of calcium phosphate or uric acid crystals in the renal tubules. Hypocalcemia may cause neuromuscular irritability including tetany, convulsions, and altered mental status.13, 14The 2015 “Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology”4 state there are well-recognized risk factors for the development of TLS including, but not limited to, high tumor burden, tumors with rapid cell turnover, and pre-existing renal impairment. Cairo and Bishop, on behalf of the TLS expert panel consensus of 20102, classify patients as having low-risk disease (LRD), intermediate-risk disease (IRD), or high-risk disease (HRD) based on the risk factors and type of malignancy. All patients with solid tumors are classified into LRD, unless the tumors are bulky or sensitive to chemotherapy, mentioning specifically that neuroblastomas, germ-cell tumors and small cell lung cancers are classified as IRD. Cairo and Bishop take into account the risk factor of renal dysfunction/ involvement, which if present, increases the risk by one level. For example, if the patient has IRD and has renal dysfunction, risk increases to HRD2. However, these guidelines do not mention or address the significance of recurrence in any kind of malignancy with regards to assessing risk for TLS.
The British Committee’s 2015 Guidelines for management of TLS in hematologic malignancies14 provide recommendations for treatment based on the patient’s risk classification (TABLE 3). Children with HRD are recommended to be treated prophylactically with a single dose of 0.2 mg/kg of rasburicase. Patients with IRD are recommended to be offered up to 7 days of allopurinol prophylaxis with increased hydration post initiation of treatment or until risk of TLS has resolved. Patients with LRD are recommended to be managed essentially with close observation. Patients with established TLS should receive rasburicase 0.2 mg/kg/day - duration to depend on clinical response. If the patient is receiving rasburicase, the addition of allopurinol is not recommended, as it has the potential to reduce the effectiveness of rasburicase. Further, rasburicase is to be avoided in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency14.
Our patient likely developed TLS because of a fast growing tumor that caused significant tumor burden and renal involvement, indicated by an elevated phosphorus level. Despite these risk factors, TLS was not anticipated in the case presented; therefore, a uric acid level was not collected at the time of admission. Review of the literature indicates that the incidence of TLS in a solid tumor recurrence is either unheard of, or is likely under-reported and truly unknown. Further, the TLS expert panel consensus of 20102, which provides guidelines on risk assessment for TLS, does not address the risk of TLS in a malignancy recurrence. The British Committee’s 2015 guidelines14 also do not address hyperuricemia prophylaxis in a solid tumor recurrence.
Our case presents a question regarding the degree of risk for the development of TLS in a solid tumor recurrence. If the guidelines had existed at the time of the case presentation and had been applied, our patient would likely be classified as having IRD because of his renal involvement. This classification would have lead to a different course of management when initiating chemotherapy, likely prevented laboratory TLS, and provided more cost effective treatment, as rasburicase is known to be expensive.
On the other hand, it can also be argued that our patient classifies as LRD, considering the rarity of TLS in a solid tumor recurrence, that the patient had no TLS complication with his initial course of therapy, and also had a normal LDH on admission. LDH is sometimes used to assess risk in hematological malignancies, although it is not used to make the diagnosis of TLS2. However, with such an argument, it is assumed that the risk of TLS in a solid tumor malignancy recurrence, with no previous TLS complication, is less than the risk associated with a new-onset solid tumor malignancy when, truly, the actual risk is not known. Again, the question is raised of the degree of risk for the development of TLS in a case of a malignancy recurrence, and also in a pediatric patient with risk factors.
In our patient’s case, close observation allowed for prompt diagnosis, appropriate treatment of laboratory TLS, and prevented clinical symptoms from developing. However, a screening or baseline uric acid level may have lead to a more conservative approach towards hyperuricemia prophylaxis, similar to treating the patient as IRD. Therefore, we recommend that a screening or baseline uric acid level and LDH level be obtained when initiating chemotherapy, even in patients with LRD.
Our patient was never hyperkalemic, likely because of concomitant administration of furosemide in an attempt to improve his decreased urine output. Hyperuricemia dropped from 19.5 mg/dL to less than 0.5 mg/dL within 24 hours, following two doses of 0.15 mg/kg of rasburicase, confirming the efficacy of this therapy in cases of established TLS, as is recommended by the British Committee’s 2015 guidelines.14
Conclusion
TLS is a relatively rare event in patients with solid malignancies and even more rare in a tumor recurrence. While there is only one previously reported case of TLS occurring in a pediatric patient with abdominal rhabdomyosarcoma, there are not any reported cases to date of TLS occurring in pediatric solid tumor recurrence. This may be because the incidence is truly rare or because cases may be under-reported. Thus, a question is raised regarding the risk for TLS in a solid tumor recurrence, and moreover in a pediatric patient with pre-existing risk factors, such as renal involvement.
TLS remains a life-threatening emergency that can be prevented and reversed if a high index of suspicion is maintained. We recommend all patients with malignancies receiving chemotherapy, especially those with risk factors, have a baseline or screening uric acid and LDH level drawn, as part of the assessment and risk-stratification for TLS which should always be performed. TSJ
Correspondence
References
1. Mirrakhimov AE, Ali AM, Khan M, et al. Tumor lysis syndrome in solid tumors: an up to date review of the literature. Rare Tumors. 2014;6:68-74.
2. Cairo MS, Bertrand C, Reiter A, et al. Recommendation for the evaluation of risk and prophylaxis of tumour lysis syndrome (TLS) in adults and children with malignant diseases: an expert TLS panel consensus. Br J Haematol. 2010;149:578-586.
3. Baeksgaard L, Sorensen JB. Acute tumor lysis syndrome in solid tumors – a case report and review of the literature. Cancer Chemother Pharmacol. 2003;51:187-192.
4. Vodopivec D, Rubio J, Fornoni A, et al. An unusual presentation of tumor lysis syndrome in a patient with advanced gastric adenocarcinoma: case report and literature review. Case Rep Med. 2012;2012:1-12.
5. Khan J, Broadbent VA. Tumor lysis syndrome complicating treatment of widespread metastatic abdominal rhabdomyosarcoma. Pediatr Hematol Oncol. 1993;10:151-155.
6. Bien E, Maciejka-Kapuscinka L, Niedzwiecki M, et al. Childhood rhabdomyosarcoma metastatic to bone marrow presenting with disseminated intravascular coagulation and acute tumour lysis syndrome: review of the literature apropos of two cases. Clin Exp Metastasis. 2010;27:399-407.
7. Patiroglu T, Isik B, Unal E, et al. Cranial metastatic alveolar rhabdomyosarcoma mimicking hematological malignancy in an adolescent boy. Childs Nerv Syst. 2014;30:1737-1741.
8. Hain RD, Rayner L, Weitzman S, et al. Acute tumour lysis syndrome complicating treatment of stage IVS neuroblastoma in infants under six months old. Med Pediatr Oncol. 1994;23:136-139.
9. Kushner BH, LaQuaglia MP, Modak S, et al. Tumor lysis syndrome, neuroblastoma, and correlation between serum lactate dehydrogenase levels and MYCN-amplification. Med Pediatr Oncol. 2003;41:80-82.
10. Bercovitz RS, Greffe BS, Hunger SP. Acute tumor lysis syndrome in a 7-month-old with hepatoblastoma. Curr Opin Pediatr. 2010;22:113-116.
11. Lobe TE, Karkera MS, Custer MD, et al. Fatal refractory hyperkalemia due to tumor lysis during primary resection for hepatoblastoma. J Pediatr Surg. 1990;25:249-250.
12. Sandberg A, Stone J, Czarnecki L, et al. Hematologic Masquerade of Rhabdomyosarcoma. Am J Hematol. 2001;68:51-57
13. Cairo M, Bishop M. Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004;127:3-11.
14. Jones G, Will A, Jackson GH, et al. Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology. Br J Haematol. 2015;169:661-671.
References
1. Mirrakhimov AE, Ali AM, Khan M, et al. Tumor lysis syndrome in solid tumors: an up to date review of the literature. Rare Tumors. 2014;6:68-74.
2. Cairo MS, Bertrand C, Reiter A, et al. Recommendation for the evaluation of risk and prophylaxis of tumour lysis syndrome (TLS) in adults and children with malignant diseases: an expert TLS panel consensus. Br J Haematol. 2010;149:578-586.
3. Baeksgaard L, Sorensen JB. Acute tumor lysis syndrome in solid tumors – a case report and review of the literature. Cancer Chemother Pharmacol. 2003;51:187-192.
4. Vodopivec D, Rubio J, Fornoni A, et al. An unusual presentation of tumor lysis syndrome in a patient with advanced gastric adenocarcinoma: case report and literature review. Case Rep Med. 2012;2012:1-12.
5. Khan J, Broadbent VA. Tumor lysis syndrome complicating treatment of widespread metastatic abdominal rhabdomyosarcoma. Pediatr Hematol Oncol. 1993;10:151-155.
6. Bien E, Maciejka-Kapuscinka L, Niedzwiecki M, et al. Childhood rhabdomyosarcoma metastatic to bone marrow presenting with disseminated intravascular coagulation and acute tumour lysis syndrome: review of the literature apropos of two cases. Clin Exp Metastasis. 2010;27:399-407.
7. Patiroglu T, Isik B, Unal E, et al. Cranial metastatic alveolar rhabdomyosarcoma mimicking hematological malignancy in an adolescent boy. Childs Nerv Syst. 2014;30:1737-1741.
8. Hain RD, Rayner L, Weitzman S, et al. Acute tumour lysis syndrome complicating treatment of stage IVS neuroblastoma in infants under six months old. Med Pediatr Oncol. 1994;23:136-139.
9. Kushner BH, LaQuaglia MP, Modak S, et al. Tumor lysis syndrome, neuroblastoma, and correlation between serum lactate dehydrogenase levels and MYCN-amplification. Med Pediatr Oncol. 2003;41:80-82.
10. Bercovitz RS, Greffe BS, Hunger SP. Acute tumor lysis syndrome in a 7-month-old with hepatoblastoma. Curr Opin Pediatr. 2010;22:113-116.
11. Lobe TE, Karkera MS, Custer MD, et al. Fatal refractory hyperkalemia due to tumor lysis during primary resection for hepatoblastoma. J Pediatr Surg. 1990;25:249-250.
12. Sandberg A, Stone J, Czarnecki L, et al. Hematologic Masquerade of Rhabdomyosarcoma. Am J Hematol. 2001;68:51-57
13. Cairo M, Bishop M. Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004;127:3-11.
14. Jones G, Will A, Jackson GH, et al. Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology. Br J Haematol. 2015;169:661-671.
Introduction
Tumor lysis syndrome (TLS) is a life-threatening oncologic emergency that results when massive cell breakdown occurs either spontaneously or in response to cytotoxic chemotherapy. TLS is characterized by metabolic derangements, including hyperkalemia and hyperphosphatemia, secondary to the release of intracellular components into the systemic circulatory system. In addition, purine degradation can lead to hyperuricemia, and precipitation of calcium phosphate can result in hypocalcemia. Lactate dehydrogenase (LDH) levels are often elevated, especially in higher risk patients; however, this finding is not a specific marker for TLS.
TLS more commonly occurs in patients with rapidly proliferating hematological malignancies, such as acute leukemias with a high white blood cell count and Burkitt’s lymphoma, and is a relatively rare event in patients with solid malignancies.1-3 It is even more rare in patients with tumor recurrence.
There are few reported cases of TLS in children with solid malignancies. To our knowledge, only one case of TLS has previously been reported in a pediatric patient with abdominal rhabdomyosarcoma. We report the second such case, and what we believe to be the only reported case of TLS occurring in a pediatric patient with recurrence of a solid tumor.
Case Description
A 15-year-old male from Saudi Arabia presented to our hospital with confirmed stage IV abdominal rhabdomyosarcoma and lung metastases diagnosed in 2012. His initial treatment consisted of complete surgical resection, lung irradiation, and chemotherapy with intercalating cycles of ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide, as per the COG-ARST0431 high-risk sarcoma protocol (NCT00354744). He completed treatment without any reported TLS in Saudi Arabia in June 2014. He had no residual tumor at the end of therapy, but six months later he was found to have an abdominal recurrence and started treatment with single-agent topotecan chemotherapy. He experienced worsening abdominal distention, pain, and difficulty voiding, prompting his family to seek further treatment options abroad.
The patient was admitted to our hospital in March 2015. Despite being severely malnourished, he was in stable condition. He was noted to have a markedly enlarged, firm, distended abdomen with dilated veins, abdominal and lower back pain, lower extremity pitting edema, and difficulty urinating.
Initial laboratory findings were unremarkable except for elevated levels of BUN (29 mg/dL), creatinine (1.69 mg/dL), and phosphorus (5.6 mg/dL). MRI revealed a large pelvic mass measuring 15.3 x 15.2 x 21.3 centimeters in transverse, anterior-posterior, and craniocaudal dimensions, respectively; with concomitant severe bilateral hydroureternephrosis (FIGURE 1).
FIGURE 1. Sagittal (A) and Axial (B) T2-weighted MR images of the pelvis (prior to initiating therapy) demonstrating a large heterogeneous mass occupying the entire pelvis. There is evidence of edema involving the soft tissues of the perineum (long arrow) and a large associated hydrocele (short arrow).
Three days following admission, the patient’s urine output decreased and his creatinine level rose rapidly. His worsening abdominal distention was attributed to growing tumor bulk and obstructive nephropathy. He required emergency placement of bilateral nephrostomy tubes. Urine output subsequently improved; although, serum creatinine remained persistently elevated.
Given his worsening condition, chemotherapy was begun three days after nephrostomy tube placement with vinorelbine, cyclophosphamide, and temsirolimus, as per COG-ARST0921 (NCT01222715), at renal-adjusted doses. Laboratory studies approximately 24 hours after chemotherapy initiation demonstrated the presence of TLS (TABLE 1). Potassium level was at the upper end of normal at 4.9 mmol/L, calcium level was decreased to 7.1 mg/dL, phosphorus level elevated to 12 mg/dL, uric acid level was markedly elevated to 19.5 mg/dL, and LDH elevated to 662 unit/L. A dose of 0.15 mg/kg of rasburicase was immediately given with a second dose repeated 14 hours later, after which the uric acid level decreased to less than 0.5 mg/dL. Sevelamer, sodium polystyrene, calcium carbonate, and magnesium gluconate were also administered to treat other electrolyte imbalances. The patient remained at clinical baseline throughout, and the TLS laboratory derangements normalized by three days after the TLS diagnosis; LDH level normalized after one week. The patient continued with chemotherapy, per protocol, with no further TLS-related complications. Over subsequent weeks, his tumor continued to shrink dramatically. Pain related to intra-abdominal compression, lower extremity edema, and difficulty voiding resolved.
Discussion
A literature search was performed using Pubmed/Medline and Scopus from 1950 to July 2016 using key words “TLS,” “tumor lysis syndrome,” “pediatric tumor lysis syndrome,” “tumor lysis syndrome in solid malignancies,” “recurrence,” “solid tumor,” “sarcoma,” “rhabdomyosarcoma,” and their combinations. The references of relevant articles were reviewed. Baeksgaard and Sorensen,3 and Vodopivec, et al4 provide an organized review of reported cases of TLS in solid tumors until 2002 and 2011 respectively; their articles are supported by the 2014 literature review by Mirrakhimov, et al.1 Excluding our case, 13 cases of TLS have been described in pediatric patients with solid tumors, with only one occurring in patient with abdominal rhabdomyosarcoma5. Patients’ ages ranged from 2 days to 23 years; the cases are summarized in the following table (TABLE 2). To our knowledge, ours is the first case of TLS reported in association with a pediatric solid tumor recurrence.
It is important to note that the three reported cases of disseminated rhabdomyosarcoma6,7 were initially believed to be hematologic malignancies because of their presentation with lymphadenopathy, metastases to the bone marrow, and spontaneous onset of TLS. Rhabdomyosarcoma with bone marrow involvement without an obvious primary tumor is easily confused with acute leukemia, particularly of the lymphoblastic type.12 However, this disseminated-hematologic presentation of rhabdomyosarcoma differs from the solid abdominal-pelvic tumor, which we describe.
Cairo and Bishop13 categorize patients as either laboratory TLS, depicted by metabolic abnormalities alone, or clinical TLS, occurring when laboratory imbalances lead to significant, life-threatening clinical manifestations. Hyperkalemia may lead to cardiac arrhythmias such as torsades de pointes and cardiac arrest. Obstructive nephropathy can occur from the precipitation of calcium phosphate or uric acid crystals in the renal tubules. Hypocalcemia may cause neuromuscular irritability including tetany, convulsions, and altered mental status.13, 14The 2015 “Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology”4 state there are well-recognized risk factors for the development of TLS including, but not limited to, high tumor burden, tumors with rapid cell turnover, and pre-existing renal impairment. Cairo and Bishop, on behalf of the TLS expert panel consensus of 20102, classify patients as having low-risk disease (LRD), intermediate-risk disease (IRD), or high-risk disease (HRD) based on the risk factors and type of malignancy. All patients with solid tumors are classified into LRD, unless the tumors are bulky or sensitive to chemotherapy, mentioning specifically that neuroblastomas, germ-cell tumors and small cell lung cancers are classified as IRD. Cairo and Bishop take into account the risk factor of renal dysfunction/ involvement, which if present, increases the risk by one level. For example, if the patient has IRD and has renal dysfunction, risk increases to HRD2. However, these guidelines do not mention or address the significance of recurrence in any kind of malignancy with regards to assessing risk for TLS.
The British Committee’s 2015 Guidelines for management of TLS in hematologic malignancies14 provide recommendations for treatment based on the patient’s risk classification (TABLE 3). Children with HRD are recommended to be treated prophylactically with a single dose of 0.2 mg/kg of rasburicase. Patients with IRD are recommended to be offered up to 7 days of allopurinol prophylaxis with increased hydration post initiation of treatment or until risk of TLS has resolved. Patients with LRD are recommended to be managed essentially with close observation. Patients with established TLS should receive rasburicase 0.2 mg/kg/day - duration to depend on clinical response. If the patient is receiving rasburicase, the addition of allopurinol is not recommended, as it has the potential to reduce the effectiveness of rasburicase. Further, rasburicase is to be avoided in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency14.
Our patient likely developed TLS because of a fast growing tumor that caused significant tumor burden and renal involvement, indicated by an elevated phosphorus level. Despite these risk factors, TLS was not anticipated in the case presented; therefore, a uric acid level was not collected at the time of admission. Review of the literature indicates that the incidence of TLS in a solid tumor recurrence is either unheard of, or is likely under-reported and truly unknown. Further, the TLS expert panel consensus of 20102, which provides guidelines on risk assessment for TLS, does not address the risk of TLS in a malignancy recurrence. The British Committee’s 2015 guidelines14 also do not address hyperuricemia prophylaxis in a solid tumor recurrence.
Our case presents a question regarding the degree of risk for the development of TLS in a solid tumor recurrence. If the guidelines had existed at the time of the case presentation and had been applied, our patient would likely be classified as having IRD because of his renal involvement. This classification would have lead to a different course of management when initiating chemotherapy, likely prevented laboratory TLS, and provided more cost effective treatment, as rasburicase is known to be expensive.
On the other hand, it can also be argued that our patient classifies as LRD, considering the rarity of TLS in a solid tumor recurrence, that the patient had no TLS complication with his initial course of therapy, and also had a normal LDH on admission. LDH is sometimes used to assess risk in hematological malignancies, although it is not used to make the diagnosis of TLS2. However, with such an argument, it is assumed that the risk of TLS in a solid tumor malignancy recurrence, with no previous TLS complication, is less than the risk associated with a new-onset solid tumor malignancy when, truly, the actual risk is not known. Again, the question is raised of the degree of risk for the development of TLS in a case of a malignancy recurrence, and also in a pediatric patient with risk factors.
In our patient’s case, close observation allowed for prompt diagnosis, appropriate treatment of laboratory TLS, and prevented clinical symptoms from developing. However, a screening or baseline uric acid level may have lead to a more conservative approach towards hyperuricemia prophylaxis, similar to treating the patient as IRD. Therefore, we recommend that a screening or baseline uric acid level and LDH level be obtained when initiating chemotherapy, even in patients with LRD.
Our patient was never hyperkalemic, likely because of concomitant administration of furosemide in an attempt to improve his decreased urine output. Hyperuricemia dropped from 19.5 mg/dL to less than 0.5 mg/dL within 24 hours, following two doses of 0.15 mg/kg of rasburicase, confirming the efficacy of this therapy in cases of established TLS, as is recommended by the British Committee’s 2015 guidelines.14
Conclusion
TLS is a relatively rare event in patients with solid malignancies and even more rare in a tumor recurrence. While there is only one previously reported case of TLS occurring in a pediatric patient with abdominal rhabdomyosarcoma, there are not any reported cases to date of TLS occurring in pediatric solid tumor recurrence. This may be because the incidence is truly rare or because cases may be under-reported. Thus, a question is raised regarding the risk for TLS in a solid tumor recurrence, and moreover in a pediatric patient with pre-existing risk factors, such as renal involvement.
TLS remains a life-threatening emergency that can be prevented and reversed if a high index of suspicion is maintained. We recommend all patients with malignancies receiving chemotherapy, especially those with risk factors, have a baseline or screening uric acid and LDH level drawn, as part of the assessment and risk-stratification for TLS which should always be performed. TSJ
Correspondence
References
1. Mirrakhimov AE, Ali AM, Khan M, et al. Tumor lysis syndrome in solid tumors: an up to date review of the literature. Rare Tumors. 2014;6:68-74.
2. Cairo MS, Bertrand C, Reiter A, et al. Recommendation for the evaluation of risk and prophylaxis of tumour lysis syndrome (TLS) in adults and children with malignant diseases: an expert TLS panel consensus. Br J Haematol. 2010;149:578-586.
3. Baeksgaard L, Sorensen JB. Acute tumor lysis syndrome in solid tumors – a case report and review of the literature. Cancer Chemother Pharmacol. 2003;51:187-192.
4. Vodopivec D, Rubio J, Fornoni A, et al. An unusual presentation of tumor lysis syndrome in a patient with advanced gastric adenocarcinoma: case report and literature review. Case Rep Med. 2012;2012:1-12.
5. Khan J, Broadbent VA. Tumor lysis syndrome complicating treatment of widespread metastatic abdominal rhabdomyosarcoma. Pediatr Hematol Oncol. 1993;10:151-155.
6. Bien E, Maciejka-Kapuscinka L, Niedzwiecki M, et al. Childhood rhabdomyosarcoma metastatic to bone marrow presenting with disseminated intravascular coagulation and acute tumour lysis syndrome: review of the literature apropos of two cases. Clin Exp Metastasis. 2010;27:399-407.
7. Patiroglu T, Isik B, Unal E, et al. Cranial metastatic alveolar rhabdomyosarcoma mimicking hematological malignancy in an adolescent boy. Childs Nerv Syst. 2014;30:1737-1741.
8. Hain RD, Rayner L, Weitzman S, et al. Acute tumour lysis syndrome complicating treatment of stage IVS neuroblastoma in infants under six months old. Med Pediatr Oncol. 1994;23:136-139.
9. Kushner BH, LaQuaglia MP, Modak S, et al. Tumor lysis syndrome, neuroblastoma, and correlation between serum lactate dehydrogenase levels and MYCN-amplification. Med Pediatr Oncol. 2003;41:80-82.
10. Bercovitz RS, Greffe BS, Hunger SP. Acute tumor lysis syndrome in a 7-month-old with hepatoblastoma. Curr Opin Pediatr. 2010;22:113-116.
11. Lobe TE, Karkera MS, Custer MD, et al. Fatal refractory hyperkalemia due to tumor lysis during primary resection for hepatoblastoma. J Pediatr Surg. 1990;25:249-250.
12. Sandberg A, Stone J, Czarnecki L, et al. Hematologic Masquerade of Rhabdomyosarcoma. Am J Hematol. 2001;68:51-57
13. Cairo M, Bishop M. Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004;127:3-11.
14. Jones G, Will A, Jackson GH, et al. Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology. Br J Haematol. 2015;169:661-671.
Introduction
Tumor lysis syndrome (TLS) is a life-threatening oncologic emergency that results when massive cell breakdown occurs either spontaneously or in response to cytotoxic chemotherapy. TLS is characterized by metabolic derangements, including hyperkalemia and hyperphosphatemia, secondary to the release of intracellular components into the systemic circulatory system. In addition, purine degradation can lead to hyperuricemia, and precipitation of calcium phosphate can result in hypocalcemia. Lactate dehydrogenase (LDH) levels are often elevated, especially in higher risk patients; however, this finding is not a specific marker for TLS.
TLS more commonly occurs in patients with rapidly proliferating hematological malignancies, such as acute leukemias with a high white blood cell count and Burkitt’s lymphoma, and is a relatively rare event in patients with solid malignancies.1-3 It is even more rare in patients with tumor recurrence.
There are few reported cases of TLS in children with solid malignancies. To our knowledge, only one case of TLS has previously been reported in a pediatric patient with abdominal rhabdomyosarcoma. We report the second such case, and what we believe to be the only reported case of TLS occurring in a pediatric patient with recurrence of a solid tumor.
Case Description
A 15-year-old male from Saudi Arabia presented to our hospital with confirmed stage IV abdominal rhabdomyosarcoma and lung metastases diagnosed in 2012. His initial treatment consisted of complete surgical resection, lung irradiation, and chemotherapy with intercalating cycles of ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide, as per the COG-ARST0431 high-risk sarcoma protocol (NCT00354744). He completed treatment without any reported TLS in Saudi Arabia in June 2014. He had no residual tumor at the end of therapy, but six months later he was found to have an abdominal recurrence and started treatment with single-agent topotecan chemotherapy. He experienced worsening abdominal distention, pain, and difficulty voiding, prompting his family to seek further treatment options abroad.
The patient was admitted to our hospital in March 2015. Despite being severely malnourished, he was in stable condition. He was noted to have a markedly enlarged, firm, distended abdomen with dilated veins, abdominal and lower back pain, lower extremity pitting edema, and difficulty urinating.
Initial laboratory findings were unremarkable except for elevated levels of BUN (29 mg/dL), creatinine (1.69 mg/dL), and phosphorus (5.6 mg/dL). MRI revealed a large pelvic mass measuring 15.3 x 15.2 x 21.3 centimeters in transverse, anterior-posterior, and craniocaudal dimensions, respectively; with concomitant severe bilateral hydroureternephrosis (FIGURE 1).
FIGURE 1. Sagittal (A) and Axial (B) T2-weighted MR images of the pelvis (prior to initiating therapy) demonstrating a large heterogeneous mass occupying the entire pelvis. There is evidence of edema involving the soft tissues of the perineum (long arrow) and a large associated hydrocele (short arrow).
Three days following admission, the patient’s urine output decreased and his creatinine level rose rapidly. His worsening abdominal distention was attributed to growing tumor bulk and obstructive nephropathy. He required emergency placement of bilateral nephrostomy tubes. Urine output subsequently improved; although, serum creatinine remained persistently elevated.
Given his worsening condition, chemotherapy was begun three days after nephrostomy tube placement with vinorelbine, cyclophosphamide, and temsirolimus, as per COG-ARST0921 (NCT01222715), at renal-adjusted doses. Laboratory studies approximately 24 hours after chemotherapy initiation demonstrated the presence of TLS (TABLE 1). Potassium level was at the upper end of normal at 4.9 mmol/L, calcium level was decreased to 7.1 mg/dL, phosphorus level elevated to 12 mg/dL, uric acid level was markedly elevated to 19.5 mg/dL, and LDH elevated to 662 unit/L. A dose of 0.15 mg/kg of rasburicase was immediately given with a second dose repeated 14 hours later, after which the uric acid level decreased to less than 0.5 mg/dL. Sevelamer, sodium polystyrene, calcium carbonate, and magnesium gluconate were also administered to treat other electrolyte imbalances. The patient remained at clinical baseline throughout, and the TLS laboratory derangements normalized by three days after the TLS diagnosis; LDH level normalized after one week. The patient continued with chemotherapy, per protocol, with no further TLS-related complications. Over subsequent weeks, his tumor continued to shrink dramatically. Pain related to intra-abdominal compression, lower extremity edema, and difficulty voiding resolved.
Discussion
A literature search was performed using Pubmed/Medline and Scopus from 1950 to July 2016 using key words “TLS,” “tumor lysis syndrome,” “pediatric tumor lysis syndrome,” “tumor lysis syndrome in solid malignancies,” “recurrence,” “solid tumor,” “sarcoma,” “rhabdomyosarcoma,” and their combinations. The references of relevant articles were reviewed. Baeksgaard and Sorensen,3 and Vodopivec, et al4 provide an organized review of reported cases of TLS in solid tumors until 2002 and 2011 respectively; their articles are supported by the 2014 literature review by Mirrakhimov, et al.1 Excluding our case, 13 cases of TLS have been described in pediatric patients with solid tumors, with only one occurring in patient with abdominal rhabdomyosarcoma5. Patients’ ages ranged from 2 days to 23 years; the cases are summarized in the following table (TABLE 2). To our knowledge, ours is the first case of TLS reported in association with a pediatric solid tumor recurrence.
It is important to note that the three reported cases of disseminated rhabdomyosarcoma6,7 were initially believed to be hematologic malignancies because of their presentation with lymphadenopathy, metastases to the bone marrow, and spontaneous onset of TLS. Rhabdomyosarcoma with bone marrow involvement without an obvious primary tumor is easily confused with acute leukemia, particularly of the lymphoblastic type.12 However, this disseminated-hematologic presentation of rhabdomyosarcoma differs from the solid abdominal-pelvic tumor, which we describe.
Cairo and Bishop13 categorize patients as either laboratory TLS, depicted by metabolic abnormalities alone, or clinical TLS, occurring when laboratory imbalances lead to significant, life-threatening clinical manifestations. Hyperkalemia may lead to cardiac arrhythmias such as torsades de pointes and cardiac arrest. Obstructive nephropathy can occur from the precipitation of calcium phosphate or uric acid crystals in the renal tubules. Hypocalcemia may cause neuromuscular irritability including tetany, convulsions, and altered mental status.13, 14The 2015 “Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology”4 state there are well-recognized risk factors for the development of TLS including, but not limited to, high tumor burden, tumors with rapid cell turnover, and pre-existing renal impairment. Cairo and Bishop, on behalf of the TLS expert panel consensus of 20102, classify patients as having low-risk disease (LRD), intermediate-risk disease (IRD), or high-risk disease (HRD) based on the risk factors and type of malignancy. All patients with solid tumors are classified into LRD, unless the tumors are bulky or sensitive to chemotherapy, mentioning specifically that neuroblastomas, germ-cell tumors and small cell lung cancers are classified as IRD. Cairo and Bishop take into account the risk factor of renal dysfunction/ involvement, which if present, increases the risk by one level. For example, if the patient has IRD and has renal dysfunction, risk increases to HRD2. However, these guidelines do not mention or address the significance of recurrence in any kind of malignancy with regards to assessing risk for TLS.
The British Committee’s 2015 Guidelines for management of TLS in hematologic malignancies14 provide recommendations for treatment based on the patient’s risk classification (TABLE 3). Children with HRD are recommended to be treated prophylactically with a single dose of 0.2 mg/kg of rasburicase. Patients with IRD are recommended to be offered up to 7 days of allopurinol prophylaxis with increased hydration post initiation of treatment or until risk of TLS has resolved. Patients with LRD are recommended to be managed essentially with close observation. Patients with established TLS should receive rasburicase 0.2 mg/kg/day - duration to depend on clinical response. If the patient is receiving rasburicase, the addition of allopurinol is not recommended, as it has the potential to reduce the effectiveness of rasburicase. Further, rasburicase is to be avoided in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency14.
Our patient likely developed TLS because of a fast growing tumor that caused significant tumor burden and renal involvement, indicated by an elevated phosphorus level. Despite these risk factors, TLS was not anticipated in the case presented; therefore, a uric acid level was not collected at the time of admission. Review of the literature indicates that the incidence of TLS in a solid tumor recurrence is either unheard of, or is likely under-reported and truly unknown. Further, the TLS expert panel consensus of 20102, which provides guidelines on risk assessment for TLS, does not address the risk of TLS in a malignancy recurrence. The British Committee’s 2015 guidelines14 also do not address hyperuricemia prophylaxis in a solid tumor recurrence.
Our case presents a question regarding the degree of risk for the development of TLS in a solid tumor recurrence. If the guidelines had existed at the time of the case presentation and had been applied, our patient would likely be classified as having IRD because of his renal involvement. This classification would have lead to a different course of management when initiating chemotherapy, likely prevented laboratory TLS, and provided more cost effective treatment, as rasburicase is known to be expensive.
On the other hand, it can also be argued that our patient classifies as LRD, considering the rarity of TLS in a solid tumor recurrence, that the patient had no TLS complication with his initial course of therapy, and also had a normal LDH on admission. LDH is sometimes used to assess risk in hematological malignancies, although it is not used to make the diagnosis of TLS2. However, with such an argument, it is assumed that the risk of TLS in a solid tumor malignancy recurrence, with no previous TLS complication, is less than the risk associated with a new-onset solid tumor malignancy when, truly, the actual risk is not known. Again, the question is raised of the degree of risk for the development of TLS in a case of a malignancy recurrence, and also in a pediatric patient with risk factors.
In our patient’s case, close observation allowed for prompt diagnosis, appropriate treatment of laboratory TLS, and prevented clinical symptoms from developing. However, a screening or baseline uric acid level may have lead to a more conservative approach towards hyperuricemia prophylaxis, similar to treating the patient as IRD. Therefore, we recommend that a screening or baseline uric acid level and LDH level be obtained when initiating chemotherapy, even in patients with LRD.
Our patient was never hyperkalemic, likely because of concomitant administration of furosemide in an attempt to improve his decreased urine output. Hyperuricemia dropped from 19.5 mg/dL to less than 0.5 mg/dL within 24 hours, following two doses of 0.15 mg/kg of rasburicase, confirming the efficacy of this therapy in cases of established TLS, as is recommended by the British Committee’s 2015 guidelines.14
Conclusion
TLS is a relatively rare event in patients with solid malignancies and even more rare in a tumor recurrence. While there is only one previously reported case of TLS occurring in a pediatric patient with abdominal rhabdomyosarcoma, there are not any reported cases to date of TLS occurring in pediatric solid tumor recurrence. This may be because the incidence is truly rare or because cases may be under-reported. Thus, a question is raised regarding the risk for TLS in a solid tumor recurrence, and moreover in a pediatric patient with pre-existing risk factors, such as renal involvement.
TLS remains a life-threatening emergency that can be prevented and reversed if a high index of suspicion is maintained. We recommend all patients with malignancies receiving chemotherapy, especially those with risk factors, have a baseline or screening uric acid and LDH level drawn, as part of the assessment and risk-stratification for TLS which should always be performed. TSJ
Correspondence
References
1. Mirrakhimov AE, Ali AM, Khan M, et al. Tumor lysis syndrome in solid tumors: an up to date review of the literature. Rare Tumors. 2014;6:68-74.
2. Cairo MS, Bertrand C, Reiter A, et al. Recommendation for the evaluation of risk and prophylaxis of tumour lysis syndrome (TLS) in adults and children with malignant diseases: an expert TLS panel consensus. Br J Haematol. 2010;149:578-586.
3. Baeksgaard L, Sorensen JB. Acute tumor lysis syndrome in solid tumors – a case report and review of the literature. Cancer Chemother Pharmacol. 2003;51:187-192.
4. Vodopivec D, Rubio J, Fornoni A, et al. An unusual presentation of tumor lysis syndrome in a patient with advanced gastric adenocarcinoma: case report and literature review. Case Rep Med. 2012;2012:1-12.
5. Khan J, Broadbent VA. Tumor lysis syndrome complicating treatment of widespread metastatic abdominal rhabdomyosarcoma. Pediatr Hematol Oncol. 1993;10:151-155.
6. Bien E, Maciejka-Kapuscinka L, Niedzwiecki M, et al. Childhood rhabdomyosarcoma metastatic to bone marrow presenting with disseminated intravascular coagulation and acute tumour lysis syndrome: review of the literature apropos of two cases. Clin Exp Metastasis. 2010;27:399-407.
7. Patiroglu T, Isik B, Unal E, et al. Cranial metastatic alveolar rhabdomyosarcoma mimicking hematological malignancy in an adolescent boy. Childs Nerv Syst. 2014;30:1737-1741.
8. Hain RD, Rayner L, Weitzman S, et al. Acute tumour lysis syndrome complicating treatment of stage IVS neuroblastoma in infants under six months old. Med Pediatr Oncol. 1994;23:136-139.
9. Kushner BH, LaQuaglia MP, Modak S, et al. Tumor lysis syndrome, neuroblastoma, and correlation between serum lactate dehydrogenase levels and MYCN-amplification. Med Pediatr Oncol. 2003;41:80-82.
10. Bercovitz RS, Greffe BS, Hunger SP. Acute tumor lysis syndrome in a 7-month-old with hepatoblastoma. Curr Opin Pediatr. 2010;22:113-116.
11. Lobe TE, Karkera MS, Custer MD, et al. Fatal refractory hyperkalemia due to tumor lysis during primary resection for hepatoblastoma. J Pediatr Surg. 1990;25:249-250.
12. Sandberg A, Stone J, Czarnecki L, et al. Hematologic Masquerade of Rhabdomyosarcoma. Am J Hematol. 2001;68:51-57
13. Cairo M, Bishop M. Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004;127:3-11.
14. Jones G, Will A, Jackson GH, et al. Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology. Br J Haematol. 2015;169:661-671.
References
1. Mirrakhimov AE, Ali AM, Khan M, et al. Tumor lysis syndrome in solid tumors: an up to date review of the literature. Rare Tumors. 2014;6:68-74.
2. Cairo MS, Bertrand C, Reiter A, et al. Recommendation for the evaluation of risk and prophylaxis of tumour lysis syndrome (TLS) in adults and children with malignant diseases: an expert TLS panel consensus. Br J Haematol. 2010;149:578-586.
3. Baeksgaard L, Sorensen JB. Acute tumor lysis syndrome in solid tumors – a case report and review of the literature. Cancer Chemother Pharmacol. 2003;51:187-192.
4. Vodopivec D, Rubio J, Fornoni A, et al. An unusual presentation of tumor lysis syndrome in a patient with advanced gastric adenocarcinoma: case report and literature review. Case Rep Med. 2012;2012:1-12.
5. Khan J, Broadbent VA. Tumor lysis syndrome complicating treatment of widespread metastatic abdominal rhabdomyosarcoma. Pediatr Hematol Oncol. 1993;10:151-155.
6. Bien E, Maciejka-Kapuscinka L, Niedzwiecki M, et al. Childhood rhabdomyosarcoma metastatic to bone marrow presenting with disseminated intravascular coagulation and acute tumour lysis syndrome: review of the literature apropos of two cases. Clin Exp Metastasis. 2010;27:399-407.
7. Patiroglu T, Isik B, Unal E, et al. Cranial metastatic alveolar rhabdomyosarcoma mimicking hematological malignancy in an adolescent boy. Childs Nerv Syst. 2014;30:1737-1741.
8. Hain RD, Rayner L, Weitzman S, et al. Acute tumour lysis syndrome complicating treatment of stage IVS neuroblastoma in infants under six months old. Med Pediatr Oncol. 1994;23:136-139.
9. Kushner BH, LaQuaglia MP, Modak S, et al. Tumor lysis syndrome, neuroblastoma, and correlation between serum lactate dehydrogenase levels and MYCN-amplification. Med Pediatr Oncol. 2003;41:80-82.
10. Bercovitz RS, Greffe BS, Hunger SP. Acute tumor lysis syndrome in a 7-month-old with hepatoblastoma. Curr Opin Pediatr. 2010;22:113-116.
11. Lobe TE, Karkera MS, Custer MD, et al. Fatal refractory hyperkalemia due to tumor lysis during primary resection for hepatoblastoma. J Pediatr Surg. 1990;25:249-250.
12. Sandberg A, Stone J, Czarnecki L, et al. Hematologic Masquerade of Rhabdomyosarcoma. Am J Hematol. 2001;68:51-57
13. Cairo M, Bishop M. Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004;127:3-11.
14. Jones G, Will A, Jackson GH, et al. Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology. Br J Haematol. 2015;169:661-671.
References
1. Mirrakhimov AE, Ali AM, Khan M, et al. Tumor lysis syndrome in solid tumors: an up to date review of the literature. Rare Tumors. 2014;6:68-74.
2. Cairo MS, Bertrand C, Reiter A, et al. Recommendation for the evaluation of risk and prophylaxis of tumour lysis syndrome (TLS) in adults and children with malignant diseases: an expert TLS panel consensus. Br J Haematol. 2010;149:578-586.
3. Baeksgaard L, Sorensen JB. Acute tumor lysis syndrome in solid tumors – a case report and review of the literature. Cancer Chemother Pharmacol. 2003;51:187-192.
4. Vodopivec D, Rubio J, Fornoni A, et al. An unusual presentation of tumor lysis syndrome in a patient with advanced gastric adenocarcinoma: case report and literature review. Case Rep Med. 2012;2012:1-12.
5. Khan J, Broadbent VA. Tumor lysis syndrome complicating treatment of widespread metastatic abdominal rhabdomyosarcoma. Pediatr Hematol Oncol. 1993;10:151-155.
6. Bien E, Maciejka-Kapuscinka L, Niedzwiecki M, et al. Childhood rhabdomyosarcoma metastatic to bone marrow presenting with disseminated intravascular coagulation and acute tumour lysis syndrome: review of the literature apropos of two cases. Clin Exp Metastasis. 2010;27:399-407.
7. Patiroglu T, Isik B, Unal E, et al. Cranial metastatic alveolar rhabdomyosarcoma mimicking hematological malignancy in an adolescent boy. Childs Nerv Syst. 2014;30:1737-1741.
8. Hain RD, Rayner L, Weitzman S, et al. Acute tumour lysis syndrome complicating treatment of stage IVS neuroblastoma in infants under six months old. Med Pediatr Oncol. 1994;23:136-139.
9. Kushner BH, LaQuaglia MP, Modak S, et al. Tumor lysis syndrome, neuroblastoma, and correlation between serum lactate dehydrogenase levels and MYCN-amplification. Med Pediatr Oncol. 2003;41:80-82.
10. Bercovitz RS, Greffe BS, Hunger SP. Acute tumor lysis syndrome in a 7-month-old with hepatoblastoma. Curr Opin Pediatr. 2010;22:113-116.
11. Lobe TE, Karkera MS, Custer MD, et al. Fatal refractory hyperkalemia due to tumor lysis during primary resection for hepatoblastoma. J Pediatr Surg. 1990;25:249-250.
12. Sandberg A, Stone J, Czarnecki L, et al. Hematologic Masquerade of Rhabdomyosarcoma. Am J Hematol. 2001;68:51-57
13. Cairo M, Bishop M. Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004;127:3-11.
14. Jones G, Will A, Jackson GH, et al. Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology. Br J Haematol. 2015;169:661-671.
Cardiac pleomorphic sarcoma after placement of a Dacron graft
Primary cardiac tumors, either benign or malignant, are very rare. The combined incidence is 0.002% on pooled autopsy series.1 The benign tumors account for 63% of primary cardiac tumors and include myxoma, the most common, and followed by papillary fibroelastoma, fibroma, and hemangioma. The remaining 37% are malignant tumors, essentially predominated by sarcomas.1
Although myxoma is the most common tumor arising in the left atrium, we present a case that shows that sarcoma can also arise from the same chamber. In fact, sarcomas could mimic cardiac myxoma.2 The cardiac sarcomas can have similar clinical presentation and more importantly can share similar histopathological features. Sarcomas may have myxoid features.2 Cases diagnosed as cardiac myxomas should be diligently worked up to rule out the presence of sarcomas with myxoid features. In addition, foreign bodies have been found to induce sarcomas in experimental animals.3,4 In particular, 2 case reports have described sarcomas arising in association with Dacron vascular prostheses in humans.5,6 We present here the case of a patient who was diagnosed with cardiac pleomorphic sarcoma 8 years after the placement of a Dacron graft.
Case presentation and summary
A 56-year-old woman with history of left atrial myxoma status after resection in 2005 and placement of a Dacron graft, morbid obesity, hypertension, and asthma presented to the emergency department with progressively worsening shortness of breath and blurry vision over period of 2 months. Acute coronary syndrome was ruled out by electrocardiogram and serial biomarkers. A computed-tomography angiogram was pursued because of her history of left atrial myxoma, and the results suggested the presence of a left atrial tumor. She underwent a transesophageal echocardiogram, which confirmed the presence of a large left atrial mass that likely was attached to the interatrial septum prolapsing across the mitral valve and was suggestive for recurrent left atrial myxoma (Figure 1). The results of a cardiac catheterization showed normal coronaries.
The patient subsequently underwent an excision of the left atrial tumor with profound internal and external myocardial cooling using antegrade blood cardioplegia under mildly hypothermic cardiopulmonary bypass. Frozen sections showed high-grade malignancy in favor of sarcoma. The hematoxylin and eosin stained permanent sections showed sheets of malignant pleomorphic spindle cells focally arranged in a storiform pattern. There were areas of necrosis and abundant mitotic activity. By immunohistochemical (IHC) stains, the tumor cells were diffusely positive for vimentin, and negative for pan-cytokeratin antibody (AE1/AE3), S-100 protein, Melan-A antibody, HMB45, CD34, CD31, myogenin, and MYOD1. IHC stains for CK-OSCAR, desmin, and smooth muscle actin were focally positive, and a ki-67 stain showed a proliferation index of about 80%. The histologic and IHC findings were consistent with a final diagnosis of high-grade undifferentiated pleomorphic sarcoma (Figure 2).
A positron emission tomography scan performed November 2013 did not show any other activity. The patient was scheduled for chemotherapy with adriamycin and ifosfamide with a plan for total of 6 cycles. Before her admission for the chemotherapy, the patient was admitted to the hospital for atrial fibrillation with rapid ventricular response and had multiple complications requiring prolonged hospitalization and rehabilitation. Repeat imaging 2 months later showed diffuse metastatic disease. However, her performance status had declined and she was not eligible for chemotherapy. She was placed under hospice care.
Discussion
This case demonstrates development of a cardiac pleomorphic sarcoma, a rare tumor, after placement of a Dacron graft. Given that foreign bodies have been found to induce sarcomas in experimental animals,3,4 and a few case reports have described sarcomas arising in association with Dacron vascular prostheses, 5-10 it seems that an exuberant host response around the foreign body might represent an important intermediate step in the development of the sarcoma.
There is no clearly defined pathogenesis that explains the link between a Dacron graft and sarcomas. In 1950s, Oppenheimer and colleagues described the formation of malignant tumors by various types of plastics, including Dacron, that were embedded in rats. 3,4 Most of the tumors were some form of sarcomas. It was inferred that physical properties of the plastics may have some role in tumor development. Plastics in sheet form or film that remained in situ for more than 6 months induced significant number of tumors compared with other forms such as sponges, films with holes, or powders.3,4 The 3-dimensional polymeric structure of the Dacron graft seems to play a role in induction of sarcoma as well. A pore diameter of less than 0.4 mm may increase tumorigenicity.11 The removal of the material before the 6-month mark does not lead to malignant tumors, which further supports the link between Dacron graft and formation of tumor. A pocket is formed around the foreign material after a certain period, as has been shown in histologic studies as the site of tumor origin.9,10
At the molecular level, the MDM-2/p53 pathway has been cited as possible mechanism for pathogenesis of intimal sarcoma.12,13 It has been suggested that endothelial dysplasia occurs as a precursor lesion in these sarcomas.14 The Dacron graft may cause a dysplastic effect on the endothelium leading to this precursor lesion and in certain cases transforming into sarcoma. Further definitive studies are required.
The primary treatment for cardiac sarcoma is surgical removal, although it is not always feasible. Findings in a Mayo clinic study showed that the median survival was 17 months for patients who underwent complete surgical excision, compared with 6 months for those who complete resection was not possible.15 In addition, a 10% survival rate at 1 year has been reported in primary cardiac sarcomas that are treated without any type of surgery.16
There is no clear-cut evidence supporting or refuting adjuvant chemotherapy for cardiac sarcoma. Some have inferred a potential benefit of adjuvant chemotherapy although definitive conclusions cannot be drawn. The median survival was 16.5 months in a case series of patients who received adjuvant chemotherapy, compared with 9 months and 11 months in 2 other case series.17,18,19 Multiple chemotherapy regimens have been used in the past for treatment. A retrospective s
Radiation showed some benefit in progression-free survival in a French retrospective study.21 Radiation therapies have been tried in other cases, as well in addition to chemotherapy. However, there is not enough data to support or refute it at this time.15,17,20 Several sporadic cases reported show benefit of cardiac transplantation.21,22
Conclusion
In consideration of the placement of the Dacron graft 8 years before the tumor occurrence, the anatomic proximity of the tumor to the Dacron graft, and the association between sarcoma with Dacron in medical literature, it seems logical to infer that this unusual malignancy in our patient is associated with the Dacron prosthesis. TSJ
Correspondence
1. Patil HR, Singh D, Hajdu M. Cardiac sarcoma presenting as heart failure and diagnosed as recurrent myxoma by echocardiogram. Eur J Echocardiogr. 2010;11(4):E12.
2. Awamleh P, Alberca MT, Gamallo C, Enrech S, Sarraj A. Left atrium myxosarcoma: an exceptional cardiac malignant primary tumor. Clin Cardiol. 2007;30(6):306-308.
3. Oppenheimer BS, Oppenheimer ET, Stout AP, Danishefsky I. Malignant tumors resulting from embedding plastics in rodents. Science. 1953;118:305-306.
4. Oppenheimer BS, Oppenheimer ET, Stout AP, Willhite M, Danishefski, I. The latent period in carcinogenesis by plastics in rats and its relation to the presarcomatous stage. Cancer. 1958;11(1):204-213.
5. Almeida NJ, Hoang P, Biddle P, Arouni A, Esterbrooks D. Primary cardiac angiosarcoma: in a patient with a Dacron aortic prosthesis. Tex Heart Inst J. 2011;38(1):61-65; discussion 65.
6. Stewart B, Manglik N, Zhao B, et al. Aortic intimal sarcoma: report of two cases with immunohistochemical analysis for pathogenesis. Cardiovasc Pathol. 2013;22(5):351-356.
7. Umscheid TW, Rouhani G, Morlang T, et al. Hemangiosarcoma after endovascular aortic aneurysm repair. J Endovasc Ther. 2007;14(1):101-105.
8. Ben-Izhak O, Vlodavsky E, Ofer A, Engel A, Nitecky S, Hoffman A. Epithelioid angiosarcoma associated with a Dacron vascular graft. Am J Surg Pathol. 1999;23(11):1418-1422.
9. Fyfe BS, Quintana CS, Kaneko M, Griepp RB. Aortic sarcoma four years after Dacron graft insertion. Ann Thorac Surg. 1994;58(6):1752-1754.
10. O’Connell TX, Fee HJ, Golding A. Sarcoma associated with Dacron prosthetic material: case report and review of the literature. J Thorac Cardiovasc Surg. 1976;72(1):94-96.
11. Karp RD, Johnson KH, Buoen LC, et al. Tumorogenesis by millipore filters in mice: histology and ultastructure of tissue reactions, as related to pore size. J Natl Cancer Inst. 1973;51:1275-1285.
12. Bode-Lesniewska B, Zhao J, Speel EJ, et al. Gains of 12q13-14 and overexpression of mdm2 are frequent findings in intimal sarcomas of the pulmonary artery. Virchows Arch. 2001;438:57-65.
13. Zeitz C, Rossle M, Haas C, et al. MDM-2 oncoprotein overexpression, p53 gene mutation, and VEGF up-regulation in angiosarcomas. Am J Surg Pathol. 1998;153:1425-1433.
14. Haber LM, Truong L. Immunohistochemical demonstration of the endothelialnature of aortic intimal sarcoma. Am J Surg Pathol. 1988 Oct;12(10):798-802. PubMed PMID: 3138923.
15. Simpson L, Kumar SK, Okuno SH, et al. Malignant primary cardiac tumors: review of a single institution experience. Cancer. 2008;112(11):2440-2446.
16. Leja MJ, Shah DJ, Reardon MJ. Primary cardiac tumors. Tex Heart Inst J. 2011;38(3):261-262.
17. Donsbeck AV, Ranchere D, Coindre JM, Le Gall F, Cordier JF, Loire R. Primary cardiac sarcomas: an immunohistochemical and grading study with long-term follow-up of 24 cases. Histopathology. 1999;34(4):295-304.
18. Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DC. Primary cardiac sarcomas. Ann Thorac Surg. 1990; 51; 906-910.
19. Murphy WR, Sweeney MS, Putnam JB et al. Surgical treatment of cardiac tumors: a 25-year experience. Ann Thorac Surg. 1990;49;612-618.
20. Llombart-Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer. 1998;78(12):1624-1628.
21. Isambert N, Ray-Coquard I, Italiano A, et al. Primary cardiac sarcomas: a retrospective study of the French Sarcoma Group. Eur J Cancer. 2014;50(1):128-136.
22. Agaimy A, Rösch J, Weyand M, Strecker T. Primary and metastatic cardiac sarcomas: a 12-year experience at a German heart center. Int J Clin Exp Pathol. 2012;5(9):928-938.
Primary cardiac tumors, either benign or malignant, are very rare. The combined incidence is 0.002% on pooled autopsy series.1 The benign tumors account for 63% of primary cardiac tumors and include myxoma, the most common, and followed by papillary fibroelastoma, fibroma, and hemangioma. The remaining 37% are malignant tumors, essentially predominated by sarcomas.1
Although myxoma is the most common tumor arising in the left atrium, we present a case that shows that sarcoma can also arise from the same chamber. In fact, sarcomas could mimic cardiac myxoma.2 The cardiac sarcomas can have similar clinical presentation and more importantly can share similar histopathological features. Sarcomas may have myxoid features.2 Cases diagnosed as cardiac myxomas should be diligently worked up to rule out the presence of sarcomas with myxoid features. In addition, foreign bodies have been found to induce sarcomas in experimental animals.3,4 In particular, 2 case reports have described sarcomas arising in association with Dacron vascular prostheses in humans.5,6 We present here the case of a patient who was diagnosed with cardiac pleomorphic sarcoma 8 years after the placement of a Dacron graft.
Case presentation and summary
A 56-year-old woman with history of left atrial myxoma status after resection in 2005 and placement of a Dacron graft, morbid obesity, hypertension, and asthma presented to the emergency department with progressively worsening shortness of breath and blurry vision over period of 2 months. Acute coronary syndrome was ruled out by electrocardiogram and serial biomarkers. A computed-tomography angiogram was pursued because of her history of left atrial myxoma, and the results suggested the presence of a left atrial tumor. She underwent a transesophageal echocardiogram, which confirmed the presence of a large left atrial mass that likely was attached to the interatrial septum prolapsing across the mitral valve and was suggestive for recurrent left atrial myxoma (Figure 1). The results of a cardiac catheterization showed normal coronaries.
The patient subsequently underwent an excision of the left atrial tumor with profound internal and external myocardial cooling using antegrade blood cardioplegia under mildly hypothermic cardiopulmonary bypass. Frozen sections showed high-grade malignancy in favor of sarcoma. The hematoxylin and eosin stained permanent sections showed sheets of malignant pleomorphic spindle cells focally arranged in a storiform pattern. There were areas of necrosis and abundant mitotic activity. By immunohistochemical (IHC) stains, the tumor cells were diffusely positive for vimentin, and negative for pan-cytokeratin antibody (AE1/AE3), S-100 protein, Melan-A antibody, HMB45, CD34, CD31, myogenin, and MYOD1. IHC stains for CK-OSCAR, desmin, and smooth muscle actin were focally positive, and a ki-67 stain showed a proliferation index of about 80%. The histologic and IHC findings were consistent with a final diagnosis of high-grade undifferentiated pleomorphic sarcoma (Figure 2).
A positron emission tomography scan performed November 2013 did not show any other activity. The patient was scheduled for chemotherapy with adriamycin and ifosfamide with a plan for total of 6 cycles. Before her admission for the chemotherapy, the patient was admitted to the hospital for atrial fibrillation with rapid ventricular response and had multiple complications requiring prolonged hospitalization and rehabilitation. Repeat imaging 2 months later showed diffuse metastatic disease. However, her performance status had declined and she was not eligible for chemotherapy. She was placed under hospice care.
Discussion
This case demonstrates development of a cardiac pleomorphic sarcoma, a rare tumor, after placement of a Dacron graft. Given that foreign bodies have been found to induce sarcomas in experimental animals,3,4 and a few case reports have described sarcomas arising in association with Dacron vascular prostheses, 5-10 it seems that an exuberant host response around the foreign body might represent an important intermediate step in the development of the sarcoma.
There is no clearly defined pathogenesis that explains the link between a Dacron graft and sarcomas. In 1950s, Oppenheimer and colleagues described the formation of malignant tumors by various types of plastics, including Dacron, that were embedded in rats. 3,4 Most of the tumors were some form of sarcomas. It was inferred that physical properties of the plastics may have some role in tumor development. Plastics in sheet form or film that remained in situ for more than 6 months induced significant number of tumors compared with other forms such as sponges, films with holes, or powders.3,4 The 3-dimensional polymeric structure of the Dacron graft seems to play a role in induction of sarcoma as well. A pore diameter of less than 0.4 mm may increase tumorigenicity.11 The removal of the material before the 6-month mark does not lead to malignant tumors, which further supports the link between Dacron graft and formation of tumor. A pocket is formed around the foreign material after a certain period, as has been shown in histologic studies as the site of tumor origin.9,10
At the molecular level, the MDM-2/p53 pathway has been cited as possible mechanism for pathogenesis of intimal sarcoma.12,13 It has been suggested that endothelial dysplasia occurs as a precursor lesion in these sarcomas.14 The Dacron graft may cause a dysplastic effect on the endothelium leading to this precursor lesion and in certain cases transforming into sarcoma. Further definitive studies are required.
The primary treatment for cardiac sarcoma is surgical removal, although it is not always feasible. Findings in a Mayo clinic study showed that the median survival was 17 months for patients who underwent complete surgical excision, compared with 6 months for those who complete resection was not possible.15 In addition, a 10% survival rate at 1 year has been reported in primary cardiac sarcomas that are treated without any type of surgery.16
There is no clear-cut evidence supporting or refuting adjuvant chemotherapy for cardiac sarcoma. Some have inferred a potential benefit of adjuvant chemotherapy although definitive conclusions cannot be drawn. The median survival was 16.5 months in a case series of patients who received adjuvant chemotherapy, compared with 9 months and 11 months in 2 other case series.17,18,19 Multiple chemotherapy regimens have been used in the past for treatment. A retrospective s
Radiation showed some benefit in progression-free survival in a French retrospective study.21 Radiation therapies have been tried in other cases, as well in addition to chemotherapy. However, there is not enough data to support or refute it at this time.15,17,20 Several sporadic cases reported show benefit of cardiac transplantation.21,22
Conclusion
In consideration of the placement of the Dacron graft 8 years before the tumor occurrence, the anatomic proximity of the tumor to the Dacron graft, and the association between sarcoma with Dacron in medical literature, it seems logical to infer that this unusual malignancy in our patient is associated with the Dacron prosthesis. TSJ
Correspondence
1. Patil HR, Singh D, Hajdu M. Cardiac sarcoma presenting as heart failure and diagnosed as recurrent myxoma by echocardiogram. Eur J Echocardiogr. 2010;11(4):E12.
2. Awamleh P, Alberca MT, Gamallo C, Enrech S, Sarraj A. Left atrium myxosarcoma: an exceptional cardiac malignant primary tumor. Clin Cardiol. 2007;30(6):306-308.
3. Oppenheimer BS, Oppenheimer ET, Stout AP, Danishefsky I. Malignant tumors resulting from embedding plastics in rodents. Science. 1953;118:305-306.
4. Oppenheimer BS, Oppenheimer ET, Stout AP, Willhite M, Danishefski, I. The latent period in carcinogenesis by plastics in rats and its relation to the presarcomatous stage. Cancer. 1958;11(1):204-213.
5. Almeida NJ, Hoang P, Biddle P, Arouni A, Esterbrooks D. Primary cardiac angiosarcoma: in a patient with a Dacron aortic prosthesis. Tex Heart Inst J. 2011;38(1):61-65; discussion 65.
6. Stewart B, Manglik N, Zhao B, et al. Aortic intimal sarcoma: report of two cases with immunohistochemical analysis for pathogenesis. Cardiovasc Pathol. 2013;22(5):351-356.
7. Umscheid TW, Rouhani G, Morlang T, et al. Hemangiosarcoma after endovascular aortic aneurysm repair. J Endovasc Ther. 2007;14(1):101-105.
8. Ben-Izhak O, Vlodavsky E, Ofer A, Engel A, Nitecky S, Hoffman A. Epithelioid angiosarcoma associated with a Dacron vascular graft. Am J Surg Pathol. 1999;23(11):1418-1422.
9. Fyfe BS, Quintana CS, Kaneko M, Griepp RB. Aortic sarcoma four years after Dacron graft insertion. Ann Thorac Surg. 1994;58(6):1752-1754.
10. O’Connell TX, Fee HJ, Golding A. Sarcoma associated with Dacron prosthetic material: case report and review of the literature. J Thorac Cardiovasc Surg. 1976;72(1):94-96.
11. Karp RD, Johnson KH, Buoen LC, et al. Tumorogenesis by millipore filters in mice: histology and ultastructure of tissue reactions, as related to pore size. J Natl Cancer Inst. 1973;51:1275-1285.
12. Bode-Lesniewska B, Zhao J, Speel EJ, et al. Gains of 12q13-14 and overexpression of mdm2 are frequent findings in intimal sarcomas of the pulmonary artery. Virchows Arch. 2001;438:57-65.
13. Zeitz C, Rossle M, Haas C, et al. MDM-2 oncoprotein overexpression, p53 gene mutation, and VEGF up-regulation in angiosarcomas. Am J Surg Pathol. 1998;153:1425-1433.
14. Haber LM, Truong L. Immunohistochemical demonstration of the endothelialnature of aortic intimal sarcoma. Am J Surg Pathol. 1988 Oct;12(10):798-802. PubMed PMID: 3138923.
15. Simpson L, Kumar SK, Okuno SH, et al. Malignant primary cardiac tumors: review of a single institution experience. Cancer. 2008;112(11):2440-2446.
16. Leja MJ, Shah DJ, Reardon MJ. Primary cardiac tumors. Tex Heart Inst J. 2011;38(3):261-262.
17. Donsbeck AV, Ranchere D, Coindre JM, Le Gall F, Cordier JF, Loire R. Primary cardiac sarcomas: an immunohistochemical and grading study with long-term follow-up of 24 cases. Histopathology. 1999;34(4):295-304.
18. Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DC. Primary cardiac sarcomas. Ann Thorac Surg. 1990; 51; 906-910.
19. Murphy WR, Sweeney MS, Putnam JB et al. Surgical treatment of cardiac tumors: a 25-year experience. Ann Thorac Surg. 1990;49;612-618.
20. Llombart-Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer. 1998;78(12):1624-1628.
21. Isambert N, Ray-Coquard I, Italiano A, et al. Primary cardiac sarcomas: a retrospective study of the French Sarcoma Group. Eur J Cancer. 2014;50(1):128-136.
22. Agaimy A, Rösch J, Weyand M, Strecker T. Primary and metastatic cardiac sarcomas: a 12-year experience at a German heart center. Int J Clin Exp Pathol. 2012;5(9):928-938.
Primary cardiac tumors, either benign or malignant, are very rare. The combined incidence is 0.002% on pooled autopsy series.1 The benign tumors account for 63% of primary cardiac tumors and include myxoma, the most common, and followed by papillary fibroelastoma, fibroma, and hemangioma. The remaining 37% are malignant tumors, essentially predominated by sarcomas.1
Although myxoma is the most common tumor arising in the left atrium, we present a case that shows that sarcoma can also arise from the same chamber. In fact, sarcomas could mimic cardiac myxoma.2 The cardiac sarcomas can have similar clinical presentation and more importantly can share similar histopathological features. Sarcomas may have myxoid features.2 Cases diagnosed as cardiac myxomas should be diligently worked up to rule out the presence of sarcomas with myxoid features. In addition, foreign bodies have been found to induce sarcomas in experimental animals.3,4 In particular, 2 case reports have described sarcomas arising in association with Dacron vascular prostheses in humans.5,6 We present here the case of a patient who was diagnosed with cardiac pleomorphic sarcoma 8 years after the placement of a Dacron graft.
Case presentation and summary
A 56-year-old woman with history of left atrial myxoma status after resection in 2005 and placement of a Dacron graft, morbid obesity, hypertension, and asthma presented to the emergency department with progressively worsening shortness of breath and blurry vision over period of 2 months. Acute coronary syndrome was ruled out by electrocardiogram and serial biomarkers. A computed-tomography angiogram was pursued because of her history of left atrial myxoma, and the results suggested the presence of a left atrial tumor. She underwent a transesophageal echocardiogram, which confirmed the presence of a large left atrial mass that likely was attached to the interatrial septum prolapsing across the mitral valve and was suggestive for recurrent left atrial myxoma (Figure 1). The results of a cardiac catheterization showed normal coronaries.
The patient subsequently underwent an excision of the left atrial tumor with profound internal and external myocardial cooling using antegrade blood cardioplegia under mildly hypothermic cardiopulmonary bypass. Frozen sections showed high-grade malignancy in favor of sarcoma. The hematoxylin and eosin stained permanent sections showed sheets of malignant pleomorphic spindle cells focally arranged in a storiform pattern. There were areas of necrosis and abundant mitotic activity. By immunohistochemical (IHC) stains, the tumor cells were diffusely positive for vimentin, and negative for pan-cytokeratin antibody (AE1/AE3), S-100 protein, Melan-A antibody, HMB45, CD34, CD31, myogenin, and MYOD1. IHC stains for CK-OSCAR, desmin, and smooth muscle actin were focally positive, and a ki-67 stain showed a proliferation index of about 80%. The histologic and IHC findings were consistent with a final diagnosis of high-grade undifferentiated pleomorphic sarcoma (Figure 2).
A positron emission tomography scan performed November 2013 did not show any other activity. The patient was scheduled for chemotherapy with adriamycin and ifosfamide with a plan for total of 6 cycles. Before her admission for the chemotherapy, the patient was admitted to the hospital for atrial fibrillation with rapid ventricular response and had multiple complications requiring prolonged hospitalization and rehabilitation. Repeat imaging 2 months later showed diffuse metastatic disease. However, her performance status had declined and she was not eligible for chemotherapy. She was placed under hospice care.
Discussion
This case demonstrates development of a cardiac pleomorphic sarcoma, a rare tumor, after placement of a Dacron graft. Given that foreign bodies have been found to induce sarcomas in experimental animals,3,4 and a few case reports have described sarcomas arising in association with Dacron vascular prostheses, 5-10 it seems that an exuberant host response around the foreign body might represent an important intermediate step in the development of the sarcoma.
There is no clearly defined pathogenesis that explains the link between a Dacron graft and sarcomas. In 1950s, Oppenheimer and colleagues described the formation of malignant tumors by various types of plastics, including Dacron, that were embedded in rats. 3,4 Most of the tumors were some form of sarcomas. It was inferred that physical properties of the plastics may have some role in tumor development. Plastics in sheet form or film that remained in situ for more than 6 months induced significant number of tumors compared with other forms such as sponges, films with holes, or powders.3,4 The 3-dimensional polymeric structure of the Dacron graft seems to play a role in induction of sarcoma as well. A pore diameter of less than 0.4 mm may increase tumorigenicity.11 The removal of the material before the 6-month mark does not lead to malignant tumors, which further supports the link between Dacron graft and formation of tumor. A pocket is formed around the foreign material after a certain period, as has been shown in histologic studies as the site of tumor origin.9,10
At the molecular level, the MDM-2/p53 pathway has been cited as possible mechanism for pathogenesis of intimal sarcoma.12,13 It has been suggested that endothelial dysplasia occurs as a precursor lesion in these sarcomas.14 The Dacron graft may cause a dysplastic effect on the endothelium leading to this precursor lesion and in certain cases transforming into sarcoma. Further definitive studies are required.
The primary treatment for cardiac sarcoma is surgical removal, although it is not always feasible. Findings in a Mayo clinic study showed that the median survival was 17 months for patients who underwent complete surgical excision, compared with 6 months for those who complete resection was not possible.15 In addition, a 10% survival rate at 1 year has been reported in primary cardiac sarcomas that are treated without any type of surgery.16
There is no clear-cut evidence supporting or refuting adjuvant chemotherapy for cardiac sarcoma. Some have inferred a potential benefit of adjuvant chemotherapy although definitive conclusions cannot be drawn. The median survival was 16.5 months in a case series of patients who received adjuvant chemotherapy, compared with 9 months and 11 months in 2 other case series.17,18,19 Multiple chemotherapy regimens have been used in the past for treatment. A retrospective s
Radiation showed some benefit in progression-free survival in a French retrospective study.21 Radiation therapies have been tried in other cases, as well in addition to chemotherapy. However, there is not enough data to support or refute it at this time.15,17,20 Several sporadic cases reported show benefit of cardiac transplantation.21,22
Conclusion
In consideration of the placement of the Dacron graft 8 years before the tumor occurrence, the anatomic proximity of the tumor to the Dacron graft, and the association between sarcoma with Dacron in medical literature, it seems logical to infer that this unusual malignancy in our patient is associated with the Dacron prosthesis. TSJ
Correspondence
1. Patil HR, Singh D, Hajdu M. Cardiac sarcoma presenting as heart failure and diagnosed as recurrent myxoma by echocardiogram. Eur J Echocardiogr. 2010;11(4):E12.
2. Awamleh P, Alberca MT, Gamallo C, Enrech S, Sarraj A. Left atrium myxosarcoma: an exceptional cardiac malignant primary tumor. Clin Cardiol. 2007;30(6):306-308.
3. Oppenheimer BS, Oppenheimer ET, Stout AP, Danishefsky I. Malignant tumors resulting from embedding plastics in rodents. Science. 1953;118:305-306.
4. Oppenheimer BS, Oppenheimer ET, Stout AP, Willhite M, Danishefski, I. The latent period in carcinogenesis by plastics in rats and its relation to the presarcomatous stage. Cancer. 1958;11(1):204-213.
5. Almeida NJ, Hoang P, Biddle P, Arouni A, Esterbrooks D. Primary cardiac angiosarcoma: in a patient with a Dacron aortic prosthesis. Tex Heart Inst J. 2011;38(1):61-65; discussion 65.
6. Stewart B, Manglik N, Zhao B, et al. Aortic intimal sarcoma: report of two cases with immunohistochemical analysis for pathogenesis. Cardiovasc Pathol. 2013;22(5):351-356.
7. Umscheid TW, Rouhani G, Morlang T, et al. Hemangiosarcoma after endovascular aortic aneurysm repair. J Endovasc Ther. 2007;14(1):101-105.
8. Ben-Izhak O, Vlodavsky E, Ofer A, Engel A, Nitecky S, Hoffman A. Epithelioid angiosarcoma associated with a Dacron vascular graft. Am J Surg Pathol. 1999;23(11):1418-1422.
9. Fyfe BS, Quintana CS, Kaneko M, Griepp RB. Aortic sarcoma four years after Dacron graft insertion. Ann Thorac Surg. 1994;58(6):1752-1754.
10. O’Connell TX, Fee HJ, Golding A. Sarcoma associated with Dacron prosthetic material: case report and review of the literature. J Thorac Cardiovasc Surg. 1976;72(1):94-96.
11. Karp RD, Johnson KH, Buoen LC, et al. Tumorogenesis by millipore filters in mice: histology and ultastructure of tissue reactions, as related to pore size. J Natl Cancer Inst. 1973;51:1275-1285.
12. Bode-Lesniewska B, Zhao J, Speel EJ, et al. Gains of 12q13-14 and overexpression of mdm2 are frequent findings in intimal sarcomas of the pulmonary artery. Virchows Arch. 2001;438:57-65.
13. Zeitz C, Rossle M, Haas C, et al. MDM-2 oncoprotein overexpression, p53 gene mutation, and VEGF up-regulation in angiosarcomas. Am J Surg Pathol. 1998;153:1425-1433.
14. Haber LM, Truong L. Immunohistochemical demonstration of the endothelialnature of aortic intimal sarcoma. Am J Surg Pathol. 1988 Oct;12(10):798-802. PubMed PMID: 3138923.
15. Simpson L, Kumar SK, Okuno SH, et al. Malignant primary cardiac tumors: review of a single institution experience. Cancer. 2008;112(11):2440-2446.
16. Leja MJ, Shah DJ, Reardon MJ. Primary cardiac tumors. Tex Heart Inst J. 2011;38(3):261-262.
17. Donsbeck AV, Ranchere D, Coindre JM, Le Gall F, Cordier JF, Loire R. Primary cardiac sarcomas: an immunohistochemical and grading study with long-term follow-up of 24 cases. Histopathology. 1999;34(4):295-304.
18. Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DC. Primary cardiac sarcomas. Ann Thorac Surg. 1990; 51; 906-910.
19. Murphy WR, Sweeney MS, Putnam JB et al. Surgical treatment of cardiac tumors: a 25-year experience. Ann Thorac Surg. 1990;49;612-618.
20. Llombart-Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer. 1998;78(12):1624-1628.
21. Isambert N, Ray-Coquard I, Italiano A, et al. Primary cardiac sarcomas: a retrospective study of the French Sarcoma Group. Eur J Cancer. 2014;50(1):128-136.
22. Agaimy A, Rösch J, Weyand M, Strecker T. Primary and metastatic cardiac sarcomas: a 12-year experience at a German heart center. Int J Clin Exp Pathol. 2012;5(9):928-938.
Bilateral wrist pain • limited range of motion • tenderness to palpation • Dx?
THE CASE
A 12-year-old girl presented to my office (JH) with bilateral wrist pain. She had fallen on both wrists palmar-flexed and then, while trying to get up, landed on both wrists dorsi-flexed. The patient did not hear any “pops,” but felt immediate pain when her wrists hyperextended. Hand, wrist, and forearm x-rays were negative bilaterally for fractures. She was placed in bilateral thumb spica splints.
At follow-up one week later, the patient reported 6/10 pain in her left wrist and 7/10 pain in her right wrist. The pain increased to 10/10 bilaterally with movement and was not relieved by icing or nonsteroidal anti-inflammatory drugs. On physical exam, there was bilateral swelling of the wrists without ecchymosis or erythema. The patient had limited passive and active range of motion, especially during wrist extension. She also had tenderness to palpation over the anatomical snuff box, extending proximally to the distal radius bilaterally. She had no tenderness over the ulna or metacarpals, no loss of sensation in any area nerves, and she was neurovascularly intact bilaterally.
Based on the mechanism of injury, undetected fracture or full thickness ligament tear were both possible. Because of this, and because magnetic resonance imaging (MRI) entails no radiation exposure, MRI was chosen for additional imaging of both wrists.
THE DIAGNOSIS

The MRI revealed bilateral, nondisplaced, extra-articular fractures extending through the scaphoid waist, with surrounding bone marrow edema. In the right wrist, the patient also had a low-grade partial tear of the membranous portion of the scapholunate interosseous ligament (SLIL) at the scaphoid attachment (FIGURE 1). In the left wrist, she also had a low-grade sprain of the SLIL without tear (FIGURE 2).
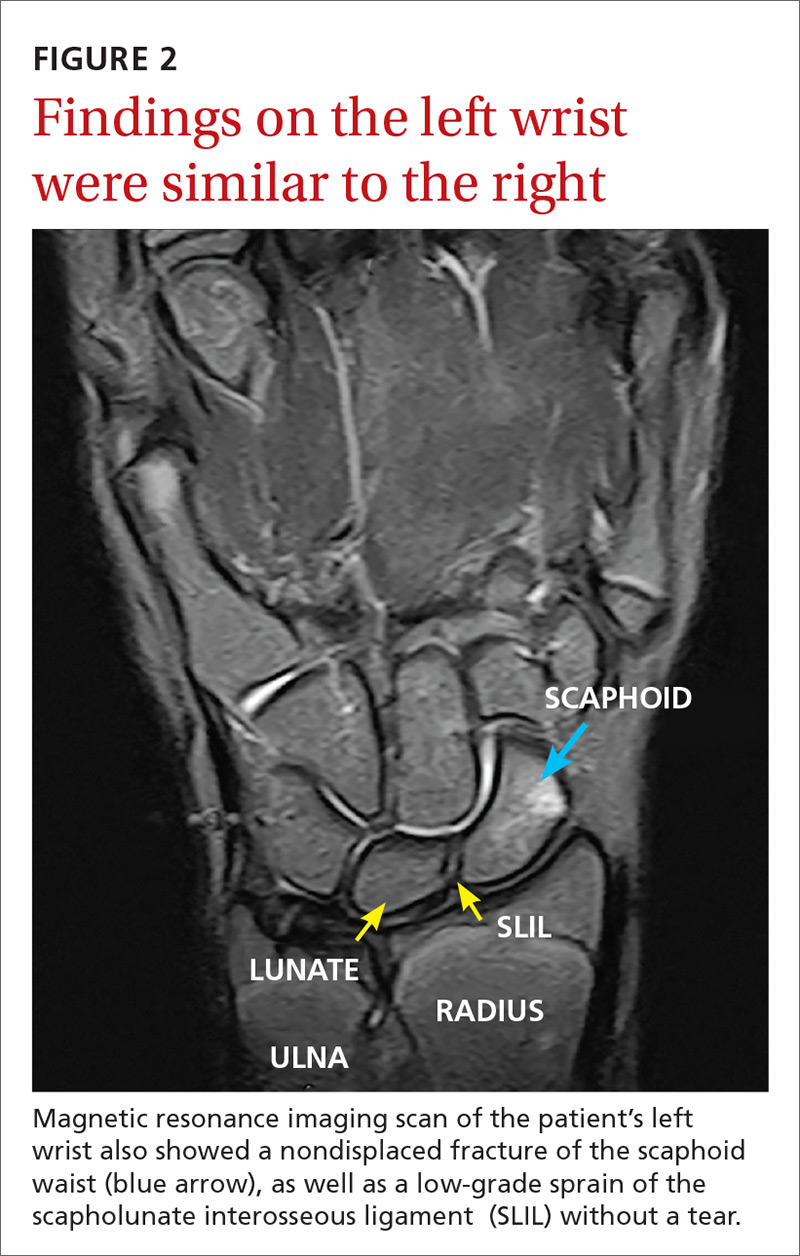
DISCUSSION
Carpal fractures account for 6% of all fractures.1 Scaphoid fractures are the most common carpal bone fracture among all age groups, but account for only 0.4% of all pediatric fractures.1-3 They’re commonly missed on x-rays because they are usually nondisplaced and hidden by other structures superimposed on the image.1,2,4 Undetected, scaphoid fractures can cause prolonged interruption to the bone’s architecture, leading to avascular necrosis of the proximal portion of the scaphoid bone.5,6
Bilateral scaphoid fractures are extremely rare and account for less than 1% of all scaphoid fractures.7 Very few of these cases have been published in the literature, and those that have been published have talked about the fractures being secondary to chronic stress fractures and as being treated with internal fixation (regardless of whether the fractures were nondisplaced or if the ligaments were intact).6-9
Our patient was placed in bilateral fiberglass short-arm thumb spica casts. We tried conservative treatment measures first because she had help with her activities of daily living (ADLs). At a follow-up visit 2 weeks later, we switched the casts to long-arm thumb spica casts because of the patient’s ability to pronate and supinate her wrists in the short-arm versions. After one month of wearing the long-arm casts, we placed her back in bilateral short-arm casts for 2 weeks. Eight weeks after the fall, we removed the short-arm casts for reevaluation.
We obtained x-rays to assess for any new changes to the wrist and specifically the scaphoid bones. The x-rays showed almost completely healed scaphoid bones with good alignment, but the patient still had 5/10 pain in the left wrist and 8/10 pain in the right wrist with movement. We placed her in adjustable thermoformable polymer braces, which were removed when she bathed.
Due to the uniqueness of her injuries, our patient had weekly visits with her primary care provider (PCP) for the first 2 months of treatment, followed by bimonthly visits for the remainder. At 10 weeks after the fall, her pain with movement was almost gone and she began physical therapy. She also began removing the braces during sedentary activity in order to practice range-of-motion exercises to prevent excessive stiffness in her wrists. Our patient regained full strength and range of motion one month later.
One other published case report describes the successful union of bilateral scaphoid fractures using bilateral long-arm casts followed by short-arm casts.7 Similar to our patient’s case, full union of the scaphoid bones was achieved within 12 weeks.7 Together, these cases suggest that conservative treatment methods are a viable alternative to surgery.
TAKEAWAY
For patients presenting with wrist pain after trauma to the wrists, assess anatomical snuffbox tenderness and obtain x-rays. Do not be falsely reassured by negative x-rays in the presence of a positive physical exam, however, as scaphoid fractures are often hidden on x-rays. If tenderness at the anatomical snuffbox is present and doesn’t subside within a few days, apply a short-arm thumb splint and obtain subsequent imaging.
If bilateral, nondisplaced, stable scaphoid fractures are diagnosed, conservative treatment with long-arm and short-arm casts is a viable alternative to surgery. This treatment decision should be made on an individual basis, however, as it requires the patient to have frequent PCP visits, assistance with ADLs, and complete adherence to the treatment plan.
1. Pillai A, Jain M. Management of clinical fractures of the scaphoid: results of an audit and literature review. Eur J Emerg Med. 2005;12:47-51.
2. Evenski AJ, Adamczyk MJ, Steiner RP, et al. Clinically suspected scaphoid fractures in children. J Pediatr Orthop. 2009;29:352-355.
3. Wulff R, Schmidt T. Carpal fractures in children. J Pediatr Orthop. 1998;18:462-465.
4. Nellans KW, Chung KC. Pediatric hand fractures. Hand Clin. 2013;29:569-578.
5. Jernigan EW, Smetana BS, Patterson JM. Pediatric scaphoid proximal pole nonunion with avascular necrosis. J Hand Surgery. 2017;42:299.e1-299.e4.
6. Pidemunt G, Torres-Claramunt R, Ginés A, et al. Bilateral stress fracture of the carpal scaphoid: report in a child and review of the literature. Clin J Sport Med. 2012;22:511-513.
7. Saglam F, Gulabi D, Baysal Ö, et al. Chronic wrist pain in a goalkeeper; bilateral scaphoid stress fracture: a case report. Int J Surg Case Rep. 2015;7:20-22.
8. Muzaffar N, Wani I, Ehsan M, et al. Simultaneous bilateral scaphoid fractures in a soldier managed conservatively by scaphoid casts. Arch Clin Exp Surg. 2016;5:63-64.
9. Mohamed Haflah NH, Mat Nor NF, Abdullah S, et al. Bilateral scaphoid stress fracture in a platform diver presenting with unilateral symptoms. Singapore Med J. 2014;55:e159-e161.
THE CASE
A 12-year-old girl presented to my office (JH) with bilateral wrist pain. She had fallen on both wrists palmar-flexed and then, while trying to get up, landed on both wrists dorsi-flexed. The patient did not hear any “pops,” but felt immediate pain when her wrists hyperextended. Hand, wrist, and forearm x-rays were negative bilaterally for fractures. She was placed in bilateral thumb spica splints.
At follow-up one week later, the patient reported 6/10 pain in her left wrist and 7/10 pain in her right wrist. The pain increased to 10/10 bilaterally with movement and was not relieved by icing or nonsteroidal anti-inflammatory drugs. On physical exam, there was bilateral swelling of the wrists without ecchymosis or erythema. The patient had limited passive and active range of motion, especially during wrist extension. She also had tenderness to palpation over the anatomical snuff box, extending proximally to the distal radius bilaterally. She had no tenderness over the ulna or metacarpals, no loss of sensation in any area nerves, and she was neurovascularly intact bilaterally.
Based on the mechanism of injury, undetected fracture or full thickness ligament tear were both possible. Because of this, and because magnetic resonance imaging (MRI) entails no radiation exposure, MRI was chosen for additional imaging of both wrists.
THE DIAGNOSIS
The MRI revealed bilateral, nondisplaced, extra-articular fractures extending through the scaphoid waist, with surrounding bone marrow edema. In the right wrist, the patient also had a low-grade partial tear of the membranous portion of the scapholunate interosseous ligament (SLIL) at the scaphoid attachment (FIGURE 1). In the left wrist, she also had a low-grade sprain of the SLIL without tear (FIGURE 2).
DISCUSSION
Carpal fractures account for 6% of all fractures.1 Scaphoid fractures are the most common carpal bone fracture among all age groups, but account for only 0.4% of all pediatric fractures.1-3 They’re commonly missed on x-rays because they are usually nondisplaced and hidden by other structures superimposed on the image.1,2,4 Undetected, scaphoid fractures can cause prolonged interruption to the bone’s architecture, leading to avascular necrosis of the proximal portion of the scaphoid bone.5,6
Bilateral scaphoid fractures are extremely rare and account for less than 1% of all scaphoid fractures.7 Very few of these cases have been published in the literature, and those that have been published have talked about the fractures being secondary to chronic stress fractures and as being treated with internal fixation (regardless of whether the fractures were nondisplaced or if the ligaments were intact).6-9
Our patient was placed in bilateral fiberglass short-arm thumb spica casts. We tried conservative treatment measures first because she had help with her activities of daily living (ADLs). At a follow-up visit 2 weeks later, we switched the casts to long-arm thumb spica casts because of the patient’s ability to pronate and supinate her wrists in the short-arm versions. After one month of wearing the long-arm casts, we placed her back in bilateral short-arm casts for 2 weeks. Eight weeks after the fall, we removed the short-arm casts for reevaluation.
We obtained x-rays to assess for any new changes to the wrist and specifically the scaphoid bones. The x-rays showed almost completely healed scaphoid bones with good alignment, but the patient still had 5/10 pain in the left wrist and 8/10 pain in the right wrist with movement. We placed her in adjustable thermoformable polymer braces, which were removed when she bathed.
Due to the uniqueness of her injuries, our patient had weekly visits with her primary care provider (PCP) for the first 2 months of treatment, followed by bimonthly visits for the remainder. At 10 weeks after the fall, her pain with movement was almost gone and she began physical therapy. She also began removing the braces during sedentary activity in order to practice range-of-motion exercises to prevent excessive stiffness in her wrists. Our patient regained full strength and range of motion one month later.
One other published case report describes the successful union of bilateral scaphoid fractures using bilateral long-arm casts followed by short-arm casts.7 Similar to our patient’s case, full union of the scaphoid bones was achieved within 12 weeks.7 Together, these cases suggest that conservative treatment methods are a viable alternative to surgery.
TAKEAWAY
For patients presenting with wrist pain after trauma to the wrists, assess anatomical snuffbox tenderness and obtain x-rays. Do not be falsely reassured by negative x-rays in the presence of a positive physical exam, however, as scaphoid fractures are often hidden on x-rays. If tenderness at the anatomical snuffbox is present and doesn’t subside within a few days, apply a short-arm thumb splint and obtain subsequent imaging.
If bilateral, nondisplaced, stable scaphoid fractures are diagnosed, conservative treatment with long-arm and short-arm casts is a viable alternative to surgery. This treatment decision should be made on an individual basis, however, as it requires the patient to have frequent PCP visits, assistance with ADLs, and complete adherence to the treatment plan.
THE CASE
A 12-year-old girl presented to my office (JH) with bilateral wrist pain. She had fallen on both wrists palmar-flexed and then, while trying to get up, landed on both wrists dorsi-flexed. The patient did not hear any “pops,” but felt immediate pain when her wrists hyperextended. Hand, wrist, and forearm x-rays were negative bilaterally for fractures. She was placed in bilateral thumb spica splints.
At follow-up one week later, the patient reported 6/10 pain in her left wrist and 7/10 pain in her right wrist. The pain increased to 10/10 bilaterally with movement and was not relieved by icing or nonsteroidal anti-inflammatory drugs. On physical exam, there was bilateral swelling of the wrists without ecchymosis or erythema. The patient had limited passive and active range of motion, especially during wrist extension. She also had tenderness to palpation over the anatomical snuff box, extending proximally to the distal radius bilaterally. She had no tenderness over the ulna or metacarpals, no loss of sensation in any area nerves, and she was neurovascularly intact bilaterally.
Based on the mechanism of injury, undetected fracture or full thickness ligament tear were both possible. Because of this, and because magnetic resonance imaging (MRI) entails no radiation exposure, MRI was chosen for additional imaging of both wrists.
THE DIAGNOSIS
The MRI revealed bilateral, nondisplaced, extra-articular fractures extending through the scaphoid waist, with surrounding bone marrow edema. In the right wrist, the patient also had a low-grade partial tear of the membranous portion of the scapholunate interosseous ligament (SLIL) at the scaphoid attachment (FIGURE 1). In the left wrist, she also had a low-grade sprain of the SLIL without tear (FIGURE 2).
DISCUSSION
Carpal fractures account for 6% of all fractures.1 Scaphoid fractures are the most common carpal bone fracture among all age groups, but account for only 0.4% of all pediatric fractures.1-3 They’re commonly missed on x-rays because they are usually nondisplaced and hidden by other structures superimposed on the image.1,2,4 Undetected, scaphoid fractures can cause prolonged interruption to the bone’s architecture, leading to avascular necrosis of the proximal portion of the scaphoid bone.5,6
Bilateral scaphoid fractures are extremely rare and account for less than 1% of all scaphoid fractures.7 Very few of these cases have been published in the literature, and those that have been published have talked about the fractures being secondary to chronic stress fractures and as being treated with internal fixation (regardless of whether the fractures were nondisplaced or if the ligaments were intact).6-9
Our patient was placed in bilateral fiberglass short-arm thumb spica casts. We tried conservative treatment measures first because she had help with her activities of daily living (ADLs). At a follow-up visit 2 weeks later, we switched the casts to long-arm thumb spica casts because of the patient’s ability to pronate and supinate her wrists in the short-arm versions. After one month of wearing the long-arm casts, we placed her back in bilateral short-arm casts for 2 weeks. Eight weeks after the fall, we removed the short-arm casts for reevaluation.
We obtained x-rays to assess for any new changes to the wrist and specifically the scaphoid bones. The x-rays showed almost completely healed scaphoid bones with good alignment, but the patient still had 5/10 pain in the left wrist and 8/10 pain in the right wrist with movement. We placed her in adjustable thermoformable polymer braces, which were removed when she bathed.
Due to the uniqueness of her injuries, our patient had weekly visits with her primary care provider (PCP) for the first 2 months of treatment, followed by bimonthly visits for the remainder. At 10 weeks after the fall, her pain with movement was almost gone and she began physical therapy. She also began removing the braces during sedentary activity in order to practice range-of-motion exercises to prevent excessive stiffness in her wrists. Our patient regained full strength and range of motion one month later.
One other published case report describes the successful union of bilateral scaphoid fractures using bilateral long-arm casts followed by short-arm casts.7 Similar to our patient’s case, full union of the scaphoid bones was achieved within 12 weeks.7 Together, these cases suggest that conservative treatment methods are a viable alternative to surgery.
TAKEAWAY
For patients presenting with wrist pain after trauma to the wrists, assess anatomical snuffbox tenderness and obtain x-rays. Do not be falsely reassured by negative x-rays in the presence of a positive physical exam, however, as scaphoid fractures are often hidden on x-rays. If tenderness at the anatomical snuffbox is present and doesn’t subside within a few days, apply a short-arm thumb splint and obtain subsequent imaging.
If bilateral, nondisplaced, stable scaphoid fractures are diagnosed, conservative treatment with long-arm and short-arm casts is a viable alternative to surgery. This treatment decision should be made on an individual basis, however, as it requires the patient to have frequent PCP visits, assistance with ADLs, and complete adherence to the treatment plan.
1. Pillai A, Jain M. Management of clinical fractures of the scaphoid: results of an audit and literature review. Eur J Emerg Med. 2005;12:47-51.
2. Evenski AJ, Adamczyk MJ, Steiner RP, et al. Clinically suspected scaphoid fractures in children. J Pediatr Orthop. 2009;29:352-355.
3. Wulff R, Schmidt T. Carpal fractures in children. J Pediatr Orthop. 1998;18:462-465.
4. Nellans KW, Chung KC. Pediatric hand fractures. Hand Clin. 2013;29:569-578.
5. Jernigan EW, Smetana BS, Patterson JM. Pediatric scaphoid proximal pole nonunion with avascular necrosis. J Hand Surgery. 2017;42:299.e1-299.e4.
6. Pidemunt G, Torres-Claramunt R, Ginés A, et al. Bilateral stress fracture of the carpal scaphoid: report in a child and review of the literature. Clin J Sport Med. 2012;22:511-513.
7. Saglam F, Gulabi D, Baysal Ö, et al. Chronic wrist pain in a goalkeeper; bilateral scaphoid stress fracture: a case report. Int J Surg Case Rep. 2015;7:20-22.
8. Muzaffar N, Wani I, Ehsan M, et al. Simultaneous bilateral scaphoid fractures in a soldier managed conservatively by scaphoid casts. Arch Clin Exp Surg. 2016;5:63-64.
9. Mohamed Haflah NH, Mat Nor NF, Abdullah S, et al. Bilateral scaphoid stress fracture in a platform diver presenting with unilateral symptoms. Singapore Med J. 2014;55:e159-e161.
1. Pillai A, Jain M. Management of clinical fractures of the scaphoid: results of an audit and literature review. Eur J Emerg Med. 2005;12:47-51.
2. Evenski AJ, Adamczyk MJ, Steiner RP, et al. Clinically suspected scaphoid fractures in children. J Pediatr Orthop. 2009;29:352-355.
3. Wulff R, Schmidt T. Carpal fractures in children. J Pediatr Orthop. 1998;18:462-465.
4. Nellans KW, Chung KC. Pediatric hand fractures. Hand Clin. 2013;29:569-578.
5. Jernigan EW, Smetana BS, Patterson JM. Pediatric scaphoid proximal pole nonunion with avascular necrosis. J Hand Surgery. 2017;42:299.e1-299.e4.
6. Pidemunt G, Torres-Claramunt R, Ginés A, et al. Bilateral stress fracture of the carpal scaphoid: report in a child and review of the literature. Clin J Sport Med. 2012;22:511-513.
7. Saglam F, Gulabi D, Baysal Ö, et al. Chronic wrist pain in a goalkeeper; bilateral scaphoid stress fracture: a case report. Int J Surg Case Rep. 2015;7:20-22.
8. Muzaffar N, Wani I, Ehsan M, et al. Simultaneous bilateral scaphoid fractures in a soldier managed conservatively by scaphoid casts. Arch Clin Exp Surg. 2016;5:63-64.
9. Mohamed Haflah NH, Mat Nor NF, Abdullah S, et al. Bilateral scaphoid stress fracture in a platform diver presenting with unilateral symptoms. Singapore Med J. 2014;55:e159-e161.