User login
Facial Involvement in Progressive Macular Hypomelanosis
Progressive macular hypomelanosis (PMH) is a noninflammatory skin disorder characterized by ill-defined, nummular, hypopigmented, and nonscaly macules. Historically, various names have been used to describe this entity. Several of these terms, including cutis trunci variata and nummular and confluent hypomelanosis of the trunk, reflected its predominantly truncal distribution.1,2 Less frequently, involvement on the neck, buttocks, and arms and legs has been noted.1,2 A lack of facial involvement previously has been highlighted as a key clinical feature of PMH.3
Progressive macular hypomelanosis is a diagnosis of exclusion. Hypopigmented diseases commonly considered in the differential include those caused by fungi and yeasts (eg, tinea versicolor, seborrheic dermatitis), inflammatory skin disorders (eg, pityriasis alba, postinflammatory dyschromia), and mycosis fungoides (MF) as well as leprosy.
The hypopigmented macules of PMH have nonspecific histopathologic findings; lesional skin often shows minimal alterations as compared to normal skin. A sparse perivascular lymphocytic infiltrate often is observed,4,5 and at times, a decrease in epidermal melanin content can be detected.1-3,6,7
We report 4 cases with considerable facial involvement of hypopigmented macules that were determined to be consistent with PMH. We propose that characteristic macules that are not clinically or histopathologically consistent with other disease entities are compatible with a diagnosis of PMH, regardless of the distribution. A diagnosis of PMH should be considered in the differential when there are suggestive facial lesions in addition to truncal lesions.
Case Reports
Patient 1
A 40-year-old man presented with hypopigmented macules on the face (Figure 1), trunk, chest, arms, and legs of 2 years’ duration. The lesions were asymptomatic and had started on the forehead as hypopigmented macules, then progressed to the trunk, arms, and legs. The patient denied any prior rash, injury, or hyperpigmentation associated with the distribution of the lesions.
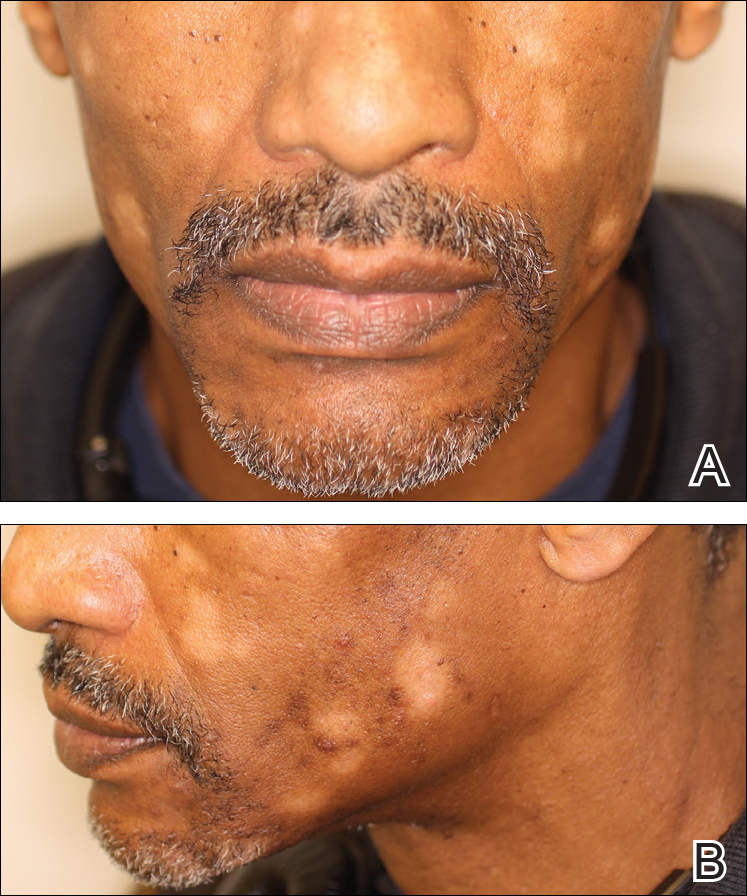
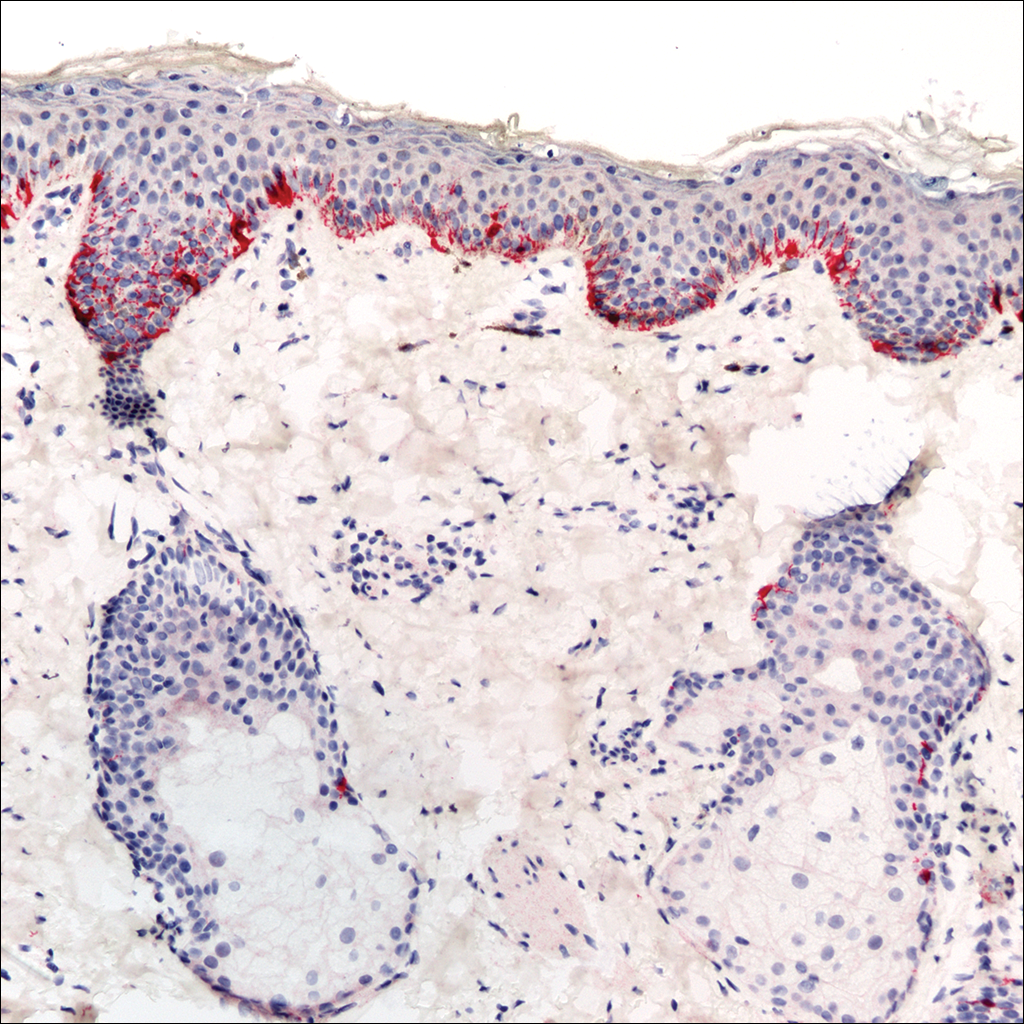
A rapid plasma reagin (RPR) test was conducted to rule out secondary syphilis and was nonreactive. During a series of clinical encounters over several months, a total of 5 biopsies of lesions on the face and back were performed. All specimens contained mild mononuclear perivascular inflammation (Figure 2). In some foci, staining for Melan-A revealed a decrease in epidermal melanocytes (Figure 3). Periodic acid–Schiff staining performed on one section revealed a few pityriasis spores but no hyphal elements, suggesting colonization rather than infection.

The patient initially was started on tacrolimus ointment 0.1% once daily and narrowband UVB phototherapy twice weekly for 3 months without benefit. A diagnosis of tinea versicolor was revisited and the patient was switched to ketoconazole shampoo 1% two to 3 times weekly on the face, trunk, arms, and legs for 10 to 15 minutes prior to rinsing, and ketoconazole cream 2% was applied twice daily to the affected areas for 2 months without notable improvement. Once-weekly 150-mg pulse doses of oral fluconazole for 8 weeks were started but proved equally ineffective. Antibiotic therapy aimed at eradicating Propionibacterium acnes was considered following a provisional diagnosis of PMH after the patient failed 5 months of therapy for tinea versicolor.
Patient 2
A 54-year-old man presented with hypopigmented to depigmented nonscaly macules on the face, trunk, chest, and arms of several months’ duration. The patient initially noted hypopigmentation on the face that gradually spread to the rest of the body. The patient denied any prior rash or hyperpigmentation in the affected areas. At the initial visit to our clinic, a potassium hydroxide (KOH) preparation of the face and back was positive for tinea versicolor. The patient was treated with ketoconazole shampoo 1% two to 3 times weekly for several weeks on the scalp, face, trunk, arms, and legs for 10 to 15 minutes prior to rinsing and 2 total doses of oral fluconazole 150 mg taken 1 week apart.
Three months later the patient returned with no improvement of the existing lesions and with progression of the disease to previously uninvolved areas of the trunk, arms, and legs. Biopsy of a facial lesion was performed, and laboratory studies including RPR, thyroid-stimulating hormone, and antinuclear antibody tests were conducted to screen for possible systemic disease. Microscopic analysis of the biopsied facial lesion revealed a sparse perivascular infiltrate of lymphocytes and plasma cells but no evidence of yeast or hyphal elements. Melan-A staining did not reveal a decreased number of epidermal melanocytes. All laboratory studies were negative or within normal limits. Desonide ointment 0.05% was prescribed to relieve the patient’s occasional pruritus. Although the patient’s symptoms resolved, the hypopigmented macules continued to progress, making a diagnosis of PMH more likely given the lack of improvement on treatment for tinea versicolor. Pimecrolimus cream 1% was started with discontinuation of desonide for steroid-sparing therapy.
Patient 3
A 63-year-old man presented with progressive nonscaly and asymptomatic hypopigmented macules on the face, trunk, abdomen, and back of 5 years’ duration. He first noted lesions on the abdomen and they subsequently spread to the rest of the body. The patient denied any prior rash, hyperpigmentation, or other lesions in the involved areas.
One year prior to the current presentation, KOH scrapings from the lesions performed by an outside physician were negative. During his initial visit to our clinic, an abdominal biopsy was performed, and histopathologic analysis showed postinflammatory pigmentary alteration; however, the patient denied any prior history of rash or injury in the distribution of the lesions that would correlate with the histopathologic findings of postinflammatory pigmentation. Because the histopathologic findings showed postinflammatory pigmentary alteration, additional stains including Melan-A were not performed.
The patient was provisionally treated with ketoconazole shampoo 1% two to 3 times weekly on the face, trunk, arms, and legs for 10 to 15 minutes prior to rinsing and ketoconazole cream 2% twice daily to the affected areas. After several months on this regimen, the patient did not report any improvement. An abdominal skin biopsy was again performed and revealed similar histopathology. Periodic acid–Schiff staining was negative for fungus. A diagnosis of PMH was made, and the patient was started on benzoyl peroxide wash 5% and clindamycin lotion.
Patient 4
A 45-year-old woman presented with hypopigmented, nonscaly macules on the face, neck, chest, trunk, and back. She first noted the lesions on the face and trunk more than 8 years prior, and they subsequently progressed. Potassium hydroxide scrapings performed on the lesions at the current presentation were negative, and a skin biopsy from the neck revealed postinflammatory pigmentary alteration, although the patient had no history of rash or injury in the areas in which the lesions were distributed.
Fontana-Masson and Melan-A staining of the skin biopsy of the neck revealed a normal distribution of melanocytes and pigment at the dermoepidermal junction. An RPR test was nonreactive. A diagnosis of PMH was made, and the patient was started on benzoyl peroxide wash 5% and clindamycin phosphate lotion 1%.
Comment
The 4 cases of PMH reported here showed extensive facial involvement in addition to the characteristic hypopigmented lesions on the trunk, arms, and legs. It is unclear why the lesions in these patients had a predominantly facial distribution. Involvement of the face in PMH has not been commonly reported in the literature. Martínez-Martínez et al3 reported 12 PMH patients with lesions only presenting in lumbar and abdominal distributions. Kim et al8 presented a series of 23 PMH patients treated with narrowband UVB in whom 56% (9/16) saw repigmentation in 90% of the lesions following treatment. The most commonly affected area was the lower back, followed by the abdomen, upper back, chest, sacral region, flank, and shoulders, respectively.8 In a review by Relyveld et al,1 PMH is described as a predominantly truncal disease that can occasionally extend to the neck, face, and proximal arms and legs; however, no specific cases were reported.
Previous case series have reported PMH primarily in adolescents and young adults, with mean ages ranging from 26 to 30 years.1,3 The 4 patients reported here were older, ranging in age from 40 to 65 years. This discrepancy in age may contribute to the facial distribution encountered in this patient population; however, given the small number of patients in our case series, such extrapolation is premature. Most recently, Westerhof et al6 demonstrated a relationship between the presence of P acnes, a common skin commensal of the face, and the hypopigmented macules of PMH. The investigators suggested that some strains of P acnes produce a factor that is yet to be identified that interferes with melanogenesis. The response of PMH lesions to topical treatments such as benzoyl peroxide, clindamycin, and phototherapy has lent credence to the potential etiologic role of P acnes in this condition.9,10 The interplay between age, PMH distribution, and P acnes requires further investigation.
The biopsies in our 4 patients were consistent with the nonspecific histopathologic characteristics of PMH lesions. Biopsies in all 4 patients revealed a sparse perivascular lymphocytic infiltrate, and in 2 of the cases, postinflammatory pigmentary alteration was noted. Such changes often are described in PMH lesions.4,5 In other cases detailed in the literature, lesional and nonlesional skin often are indistinguishable on hematoxylin and eosin staining.11 In the 3 patients for whom we performed additional immunohistochemical studies, results were mixed: Melan-A staining revealed a decreased number of melanocytes in Patient 1 but not in Patients 2 or 4. Many reported cases in the literature have not demonstrated a decrease in melanocyte density but instead show a decrease in melanin content in lesional skin.1-3,6,7 Although additional stains performed in Patient 4 revealed neither a decrease in the number of melanocytes nor a decrease in the melanin content, such histopathologic findings of PMH often are subtle. Additional stains were not performed in Patient 3. More studies are needed to characterize the immunohistochemical staining patterns of lesional skin in patients with PMH.
Tinea versicolor, pityriasis alba, mycosis fungoides, sarcoidosis, leprosy, and syphilis typically are included in the differential diagnosis for PMH. Tinea versicolor traditionally is diagnosed based on the combination of irregular hypopigmented or hyperpigmented scaly macules and a KOH preparation that is positive for hyphae and spores. Similar to PMH, tinea versicolor is most often found on the trunk, but unusual cases have been reported involving the face.12
Patient 2 reflected how it can be difficult diagnostically to distinguish between tinea versicolor and PMH. Although this patient initially had a KOH scraping suggestive for tinea versicolor, adequate treatment with oral fluconazole and ketoconazole shampoo did not result in improvement. The hypopigmented lesions in this patient continued to progress despite therapy. Additionally, his hypopigmented to depigmented nonscaly macules were more clinically consistent with the characteristic description of lesion configuration in PMH than with the irregular, more sharply defined, asymmetric, and scaly spots of tinea versicolor. Furthermore, the inflammatory findings on biopsy favored a diagnosis of PMH.
Pityriasis alba, most frequently presents on the face in the form of hypopigmented, sometimes slightly scaly macules but also can occur on the body. It usually occurs in younger patients who often have an atopic diathesis. Histologic findings generally are nonspecific, but discrete eczematous changes can sometimes be appreciated in the epidermis and dermis. None of our patients had histories suggestive of an atopic diathesis or lesion distributions typical of pityriasis alba. Histologic findings also were more consistent with PMH than pityriasis alba.
A diagnosis of patch-stage hypopigmented MF should also be entertained in patients with hypopigmented macules, as it can appear similar to the lesions of PMH. Hypopigmented MF often is associated with subtle atrophy, scaling, poikiloderma, and erythema. These features were not present in the 4 cases presented here. Histologically, atypical lymphocytes with prominent epidermotropism and tagging of the epidermis by large lymphocytic infiltrates are seen in cases of hypopigmented MF. These findings were not present in biopsies from our patients.
Hypopigmented sarcoidosis, leprosy, and syphilis are other systemic diseases associated with hypopigmented lesions. Histologically, noncaseasting granulomas in the dermis or subcutaneous tissue would favor a diagnosis of sarcoidosis over PMH. In patients who live in endemic areas, a diagnosis of leprosy for an anesthetic hypopigmented lesion would be higher in the differential. Finally, it is important to rule out secondary syphilis when diagnosing PMH. Known as the great imitator, secondary syphilis may present in a patient in the form of hypopigmented macules. Patients 1, 2, and 4 had nonreactive RPR tests; unfortunately, RPR was not checked in Patient 3. He denied all risk factors for syphilis.
Various topical and oral treatments were prescribed for each patient, but so far none have been unequivocally effective. In the literature, there are reports supporting the efficacy of topical antimicrobial agents targeting P acnes.9,10 One case report noted improvement in a patient with PMH after isotretinoin use.13 Phototherapy also has been reported to improve PMH in several case reports4-8; however, consistent response to these therapies has not been documented. Unfortunately for patients with a diagnosis of PMH, a lack of effective treatment options often exists.
This series of 4 cases highlights the importance of considering PMH in the differential of hypopigmented macules, even when they appear predominantly on the face.
- Relyveld G, Menke H, Westerhof W. Progressive macular hypomelanosis: an overview. Am J Clin Dermatol. 2007;8:13-19.
- Hwang SW, Hong SK, Kim SH, et al. Progressive macular hypomelanosis in Korean patients: a clinicopathologic study. Ann Dermatol. 2009;21:261-267.
- Martinéz-Martinéz ML, Azaña-Defez JM, Rodríguez-Vázquez M, et al. Progressive macular hypomelanosis. Pediatr Dermatol. 2012;29:460-462.
- Montero LC, Belinchonón I, Toledo F, et al. Progressive macular hypomelanosis, excellent response with narrow-band ultraviolet B phototherapy. Photodermatol Photoimmunol Photomed. 2011;27:162-163.
- Choi YJ, Hann SK. Two cases of progressive macular hypomelanosis of the trunk. Korean J Dermatol. 2000;38:655-658.
- Westerhof W, Rlyveld G, Kingswijk M, et al. Propionibacterium acnes and the pathogenesis of progressive macular hypomelanosis. Arch Dermatol. 2004;140:210-214.
- Wu SG, Xu AE, Song XZ, et al. Clinical, pathologic, and ultrastructural studies of progressive macular hypomelanosis. Int J Dermatol. 2010;29:1127-1132.
- Kim MB, Kim GW, Cho HH, et al. Narrowband UVB treatment of progressive macular hypomelanosis. J Am Acad Dermatol. 2012;66:598-605.
- Revlyveld GN, Menkie HE, Westerhof W. Benzoyl peroxide/clindamycin/UVA is more effective than fluticasone/UVA in progressive macular hypomelanosis: a randomized study. Am J Clin Dermatol. 2006;55:836-843.
- Santos JB, Almeida OL, Silva LM, et al. Efficacy of topical combination of benzoyl peroxide 5% and clindamcyin 1% for the treatment of progressive macular hypomelanosis: a randomized, doubleblind, placebo-controlled trial [in Portuguese]. An Bras Dermatol. 2011;86:50-54.
- Kumarasinghe SP, Tan SH, Thng S, et al. Progressive macular hypomelanosis in Singapore: a clinico-pathological study. Int J Dermatol. 2006;45:737-742.
- Terragni L, Lasagni A, Oriani A. Pityriasis versicolor of the face. Mycoses. 1991;34:345-347.
- Kim YK, Lee DY, Lee, JY, et al. Progressive macular hypomelanosis showing excellent response to oral isotretinoin [published online June 23, 2012]. J Dermatol. 2012;39:937-938.
Progressive macular hypomelanosis (PMH) is a noninflammatory skin disorder characterized by ill-defined, nummular, hypopigmented, and nonscaly macules. Historically, various names have been used to describe this entity. Several of these terms, including cutis trunci variata and nummular and confluent hypomelanosis of the trunk, reflected its predominantly truncal distribution.1,2 Less frequently, involvement on the neck, buttocks, and arms and legs has been noted.1,2 A lack of facial involvement previously has been highlighted as a key clinical feature of PMH.3
Progressive macular hypomelanosis is a diagnosis of exclusion. Hypopigmented diseases commonly considered in the differential include those caused by fungi and yeasts (eg, tinea versicolor, seborrheic dermatitis), inflammatory skin disorders (eg, pityriasis alba, postinflammatory dyschromia), and mycosis fungoides (MF) as well as leprosy.
The hypopigmented macules of PMH have nonspecific histopathologic findings; lesional skin often shows minimal alterations as compared to normal skin. A sparse perivascular lymphocytic infiltrate often is observed,4,5 and at times, a decrease in epidermal melanin content can be detected.1-3,6,7
We report 4 cases with considerable facial involvement of hypopigmented macules that were determined to be consistent with PMH. We propose that characteristic macules that are not clinically or histopathologically consistent with other disease entities are compatible with a diagnosis of PMH, regardless of the distribution. A diagnosis of PMH should be considered in the differential when there are suggestive facial lesions in addition to truncal lesions.
Case Reports
Patient 1
A 40-year-old man presented with hypopigmented macules on the face (Figure 1), trunk, chest, arms, and legs of 2 years’ duration. The lesions were asymptomatic and had started on the forehead as hypopigmented macules, then progressed to the trunk, arms, and legs. The patient denied any prior rash, injury, or hyperpigmentation associated with the distribution of the lesions.
A rapid plasma reagin (RPR) test was conducted to rule out secondary syphilis and was nonreactive. During a series of clinical encounters over several months, a total of 5 biopsies of lesions on the face and back were performed. All specimens contained mild mononuclear perivascular inflammation (Figure 2). In some foci, staining for Melan-A revealed a decrease in epidermal melanocytes (Figure 3). Periodic acid–Schiff staining performed on one section revealed a few pityriasis spores but no hyphal elements, suggesting colonization rather than infection.
The patient initially was started on tacrolimus ointment 0.1% once daily and narrowband UVB phototherapy twice weekly for 3 months without benefit. A diagnosis of tinea versicolor was revisited and the patient was switched to ketoconazole shampoo 1% two to 3 times weekly on the face, trunk, arms, and legs for 10 to 15 minutes prior to rinsing, and ketoconazole cream 2% was applied twice daily to the affected areas for 2 months without notable improvement. Once-weekly 150-mg pulse doses of oral fluconazole for 8 weeks were started but proved equally ineffective. Antibiotic therapy aimed at eradicating Propionibacterium acnes was considered following a provisional diagnosis of PMH after the patient failed 5 months of therapy for tinea versicolor.
Patient 2
A 54-year-old man presented with hypopigmented to depigmented nonscaly macules on the face, trunk, chest, and arms of several months’ duration. The patient initially noted hypopigmentation on the face that gradually spread to the rest of the body. The patient denied any prior rash or hyperpigmentation in the affected areas. At the initial visit to our clinic, a potassium hydroxide (KOH) preparation of the face and back was positive for tinea versicolor. The patient was treated with ketoconazole shampoo 1% two to 3 times weekly for several weeks on the scalp, face, trunk, arms, and legs for 10 to 15 minutes prior to rinsing and 2 total doses of oral fluconazole 150 mg taken 1 week apart.
Three months later the patient returned with no improvement of the existing lesions and with progression of the disease to previously uninvolved areas of the trunk, arms, and legs. Biopsy of a facial lesion was performed, and laboratory studies including RPR, thyroid-stimulating hormone, and antinuclear antibody tests were conducted to screen for possible systemic disease. Microscopic analysis of the biopsied facial lesion revealed a sparse perivascular infiltrate of lymphocytes and plasma cells but no evidence of yeast or hyphal elements. Melan-A staining did not reveal a decreased number of epidermal melanocytes. All laboratory studies were negative or within normal limits. Desonide ointment 0.05% was prescribed to relieve the patient’s occasional pruritus. Although the patient’s symptoms resolved, the hypopigmented macules continued to progress, making a diagnosis of PMH more likely given the lack of improvement on treatment for tinea versicolor. Pimecrolimus cream 1% was started with discontinuation of desonide for steroid-sparing therapy.
Patient 3
A 63-year-old man presented with progressive nonscaly and asymptomatic hypopigmented macules on the face, trunk, abdomen, and back of 5 years’ duration. He first noted lesions on the abdomen and they subsequently spread to the rest of the body. The patient denied any prior rash, hyperpigmentation, or other lesions in the involved areas.
One year prior to the current presentation, KOH scrapings from the lesions performed by an outside physician were negative. During his initial visit to our clinic, an abdominal biopsy was performed, and histopathologic analysis showed postinflammatory pigmentary alteration; however, the patient denied any prior history of rash or injury in the distribution of the lesions that would correlate with the histopathologic findings of postinflammatory pigmentation. Because the histopathologic findings showed postinflammatory pigmentary alteration, additional stains including Melan-A were not performed.
The patient was provisionally treated with ketoconazole shampoo 1% two to 3 times weekly on the face, trunk, arms, and legs for 10 to 15 minutes prior to rinsing and ketoconazole cream 2% twice daily to the affected areas. After several months on this regimen, the patient did not report any improvement. An abdominal skin biopsy was again performed and revealed similar histopathology. Periodic acid–Schiff staining was negative for fungus. A diagnosis of PMH was made, and the patient was started on benzoyl peroxide wash 5% and clindamycin lotion.
Patient 4
A 45-year-old woman presented with hypopigmented, nonscaly macules on the face, neck, chest, trunk, and back. She first noted the lesions on the face and trunk more than 8 years prior, and they subsequently progressed. Potassium hydroxide scrapings performed on the lesions at the current presentation were negative, and a skin biopsy from the neck revealed postinflammatory pigmentary alteration, although the patient had no history of rash or injury in the areas in which the lesions were distributed.
Fontana-Masson and Melan-A staining of the skin biopsy of the neck revealed a normal distribution of melanocytes and pigment at the dermoepidermal junction. An RPR test was nonreactive. A diagnosis of PMH was made, and the patient was started on benzoyl peroxide wash 5% and clindamycin phosphate lotion 1%.
Comment
The 4 cases of PMH reported here showed extensive facial involvement in addition to the characteristic hypopigmented lesions on the trunk, arms, and legs. It is unclear why the lesions in these patients had a predominantly facial distribution. Involvement of the face in PMH has not been commonly reported in the literature. Martínez-Martínez et al3 reported 12 PMH patients with lesions only presenting in lumbar and abdominal distributions. Kim et al8 presented a series of 23 PMH patients treated with narrowband UVB in whom 56% (9/16) saw repigmentation in 90% of the lesions following treatment. The most commonly affected area was the lower back, followed by the abdomen, upper back, chest, sacral region, flank, and shoulders, respectively.8 In a review by Relyveld et al,1 PMH is described as a predominantly truncal disease that can occasionally extend to the neck, face, and proximal arms and legs; however, no specific cases were reported.
Previous case series have reported PMH primarily in adolescents and young adults, with mean ages ranging from 26 to 30 years.1,3 The 4 patients reported here were older, ranging in age from 40 to 65 years. This discrepancy in age may contribute to the facial distribution encountered in this patient population; however, given the small number of patients in our case series, such extrapolation is premature. Most recently, Westerhof et al6 demonstrated a relationship between the presence of P acnes, a common skin commensal of the face, and the hypopigmented macules of PMH. The investigators suggested that some strains of P acnes produce a factor that is yet to be identified that interferes with melanogenesis. The response of PMH lesions to topical treatments such as benzoyl peroxide, clindamycin, and phototherapy has lent credence to the potential etiologic role of P acnes in this condition.9,10 The interplay between age, PMH distribution, and P acnes requires further investigation.
The biopsies in our 4 patients were consistent with the nonspecific histopathologic characteristics of PMH lesions. Biopsies in all 4 patients revealed a sparse perivascular lymphocytic infiltrate, and in 2 of the cases, postinflammatory pigmentary alteration was noted. Such changes often are described in PMH lesions.4,5 In other cases detailed in the literature, lesional and nonlesional skin often are indistinguishable on hematoxylin and eosin staining.11 In the 3 patients for whom we performed additional immunohistochemical studies, results were mixed: Melan-A staining revealed a decreased number of melanocytes in Patient 1 but not in Patients 2 or 4. Many reported cases in the literature have not demonstrated a decrease in melanocyte density but instead show a decrease in melanin content in lesional skin.1-3,6,7 Although additional stains performed in Patient 4 revealed neither a decrease in the number of melanocytes nor a decrease in the melanin content, such histopathologic findings of PMH often are subtle. Additional stains were not performed in Patient 3. More studies are needed to characterize the immunohistochemical staining patterns of lesional skin in patients with PMH.
Tinea versicolor, pityriasis alba, mycosis fungoides, sarcoidosis, leprosy, and syphilis typically are included in the differential diagnosis for PMH. Tinea versicolor traditionally is diagnosed based on the combination of irregular hypopigmented or hyperpigmented scaly macules and a KOH preparation that is positive for hyphae and spores. Similar to PMH, tinea versicolor is most often found on the trunk, but unusual cases have been reported involving the face.12
Patient 2 reflected how it can be difficult diagnostically to distinguish between tinea versicolor and PMH. Although this patient initially had a KOH scraping suggestive for tinea versicolor, adequate treatment with oral fluconazole and ketoconazole shampoo did not result in improvement. The hypopigmented lesions in this patient continued to progress despite therapy. Additionally, his hypopigmented to depigmented nonscaly macules were more clinically consistent with the characteristic description of lesion configuration in PMH than with the irregular, more sharply defined, asymmetric, and scaly spots of tinea versicolor. Furthermore, the inflammatory findings on biopsy favored a diagnosis of PMH.
Pityriasis alba, most frequently presents on the face in the form of hypopigmented, sometimes slightly scaly macules but also can occur on the body. It usually occurs in younger patients who often have an atopic diathesis. Histologic findings generally are nonspecific, but discrete eczematous changes can sometimes be appreciated in the epidermis and dermis. None of our patients had histories suggestive of an atopic diathesis or lesion distributions typical of pityriasis alba. Histologic findings also were more consistent with PMH than pityriasis alba.
A diagnosis of patch-stage hypopigmented MF should also be entertained in patients with hypopigmented macules, as it can appear similar to the lesions of PMH. Hypopigmented MF often is associated with subtle atrophy, scaling, poikiloderma, and erythema. These features were not present in the 4 cases presented here. Histologically, atypical lymphocytes with prominent epidermotropism and tagging of the epidermis by large lymphocytic infiltrates are seen in cases of hypopigmented MF. These findings were not present in biopsies from our patients.
Hypopigmented sarcoidosis, leprosy, and syphilis are other systemic diseases associated with hypopigmented lesions. Histologically, noncaseasting granulomas in the dermis or subcutaneous tissue would favor a diagnosis of sarcoidosis over PMH. In patients who live in endemic areas, a diagnosis of leprosy for an anesthetic hypopigmented lesion would be higher in the differential. Finally, it is important to rule out secondary syphilis when diagnosing PMH. Known as the great imitator, secondary syphilis may present in a patient in the form of hypopigmented macules. Patients 1, 2, and 4 had nonreactive RPR tests; unfortunately, RPR was not checked in Patient 3. He denied all risk factors for syphilis.
Various topical and oral treatments were prescribed for each patient, but so far none have been unequivocally effective. In the literature, there are reports supporting the efficacy of topical antimicrobial agents targeting P acnes.9,10 One case report noted improvement in a patient with PMH after isotretinoin use.13 Phototherapy also has been reported to improve PMH in several case reports4-8; however, consistent response to these therapies has not been documented. Unfortunately for patients with a diagnosis of PMH, a lack of effective treatment options often exists.
This series of 4 cases highlights the importance of considering PMH in the differential of hypopigmented macules, even when they appear predominantly on the face.
Progressive macular hypomelanosis (PMH) is a noninflammatory skin disorder characterized by ill-defined, nummular, hypopigmented, and nonscaly macules. Historically, various names have been used to describe this entity. Several of these terms, including cutis trunci variata and nummular and confluent hypomelanosis of the trunk, reflected its predominantly truncal distribution.1,2 Less frequently, involvement on the neck, buttocks, and arms and legs has been noted.1,2 A lack of facial involvement previously has been highlighted as a key clinical feature of PMH.3
Progressive macular hypomelanosis is a diagnosis of exclusion. Hypopigmented diseases commonly considered in the differential include those caused by fungi and yeasts (eg, tinea versicolor, seborrheic dermatitis), inflammatory skin disorders (eg, pityriasis alba, postinflammatory dyschromia), and mycosis fungoides (MF) as well as leprosy.
The hypopigmented macules of PMH have nonspecific histopathologic findings; lesional skin often shows minimal alterations as compared to normal skin. A sparse perivascular lymphocytic infiltrate often is observed,4,5 and at times, a decrease in epidermal melanin content can be detected.1-3,6,7
We report 4 cases with considerable facial involvement of hypopigmented macules that were determined to be consistent with PMH. We propose that characteristic macules that are not clinically or histopathologically consistent with other disease entities are compatible with a diagnosis of PMH, regardless of the distribution. A diagnosis of PMH should be considered in the differential when there are suggestive facial lesions in addition to truncal lesions.
Case Reports
Patient 1
A 40-year-old man presented with hypopigmented macules on the face (Figure 1), trunk, chest, arms, and legs of 2 years’ duration. The lesions were asymptomatic and had started on the forehead as hypopigmented macules, then progressed to the trunk, arms, and legs. The patient denied any prior rash, injury, or hyperpigmentation associated with the distribution of the lesions.
A rapid plasma reagin (RPR) test was conducted to rule out secondary syphilis and was nonreactive. During a series of clinical encounters over several months, a total of 5 biopsies of lesions on the face and back were performed. All specimens contained mild mononuclear perivascular inflammation (Figure 2). In some foci, staining for Melan-A revealed a decrease in epidermal melanocytes (Figure 3). Periodic acid–Schiff staining performed on one section revealed a few pityriasis spores but no hyphal elements, suggesting colonization rather than infection.
The patient initially was started on tacrolimus ointment 0.1% once daily and narrowband UVB phototherapy twice weekly for 3 months without benefit. A diagnosis of tinea versicolor was revisited and the patient was switched to ketoconazole shampoo 1% two to 3 times weekly on the face, trunk, arms, and legs for 10 to 15 minutes prior to rinsing, and ketoconazole cream 2% was applied twice daily to the affected areas for 2 months without notable improvement. Once-weekly 150-mg pulse doses of oral fluconazole for 8 weeks were started but proved equally ineffective. Antibiotic therapy aimed at eradicating Propionibacterium acnes was considered following a provisional diagnosis of PMH after the patient failed 5 months of therapy for tinea versicolor.
Patient 2
A 54-year-old man presented with hypopigmented to depigmented nonscaly macules on the face, trunk, chest, and arms of several months’ duration. The patient initially noted hypopigmentation on the face that gradually spread to the rest of the body. The patient denied any prior rash or hyperpigmentation in the affected areas. At the initial visit to our clinic, a potassium hydroxide (KOH) preparation of the face and back was positive for tinea versicolor. The patient was treated with ketoconazole shampoo 1% two to 3 times weekly for several weeks on the scalp, face, trunk, arms, and legs for 10 to 15 minutes prior to rinsing and 2 total doses of oral fluconazole 150 mg taken 1 week apart.
Three months later the patient returned with no improvement of the existing lesions and with progression of the disease to previously uninvolved areas of the trunk, arms, and legs. Biopsy of a facial lesion was performed, and laboratory studies including RPR, thyroid-stimulating hormone, and antinuclear antibody tests were conducted to screen for possible systemic disease. Microscopic analysis of the biopsied facial lesion revealed a sparse perivascular infiltrate of lymphocytes and plasma cells but no evidence of yeast or hyphal elements. Melan-A staining did not reveal a decreased number of epidermal melanocytes. All laboratory studies were negative or within normal limits. Desonide ointment 0.05% was prescribed to relieve the patient’s occasional pruritus. Although the patient’s symptoms resolved, the hypopigmented macules continued to progress, making a diagnosis of PMH more likely given the lack of improvement on treatment for tinea versicolor. Pimecrolimus cream 1% was started with discontinuation of desonide for steroid-sparing therapy.
Patient 3
A 63-year-old man presented with progressive nonscaly and asymptomatic hypopigmented macules on the face, trunk, abdomen, and back of 5 years’ duration. He first noted lesions on the abdomen and they subsequently spread to the rest of the body. The patient denied any prior rash, hyperpigmentation, or other lesions in the involved areas.
One year prior to the current presentation, KOH scrapings from the lesions performed by an outside physician were negative. During his initial visit to our clinic, an abdominal biopsy was performed, and histopathologic analysis showed postinflammatory pigmentary alteration; however, the patient denied any prior history of rash or injury in the distribution of the lesions that would correlate with the histopathologic findings of postinflammatory pigmentation. Because the histopathologic findings showed postinflammatory pigmentary alteration, additional stains including Melan-A were not performed.
The patient was provisionally treated with ketoconazole shampoo 1% two to 3 times weekly on the face, trunk, arms, and legs for 10 to 15 minutes prior to rinsing and ketoconazole cream 2% twice daily to the affected areas. After several months on this regimen, the patient did not report any improvement. An abdominal skin biopsy was again performed and revealed similar histopathology. Periodic acid–Schiff staining was negative for fungus. A diagnosis of PMH was made, and the patient was started on benzoyl peroxide wash 5% and clindamycin lotion.
Patient 4
A 45-year-old woman presented with hypopigmented, nonscaly macules on the face, neck, chest, trunk, and back. She first noted the lesions on the face and trunk more than 8 years prior, and they subsequently progressed. Potassium hydroxide scrapings performed on the lesions at the current presentation were negative, and a skin biopsy from the neck revealed postinflammatory pigmentary alteration, although the patient had no history of rash or injury in the areas in which the lesions were distributed.
Fontana-Masson and Melan-A staining of the skin biopsy of the neck revealed a normal distribution of melanocytes and pigment at the dermoepidermal junction. An RPR test was nonreactive. A diagnosis of PMH was made, and the patient was started on benzoyl peroxide wash 5% and clindamycin phosphate lotion 1%.
Comment
The 4 cases of PMH reported here showed extensive facial involvement in addition to the characteristic hypopigmented lesions on the trunk, arms, and legs. It is unclear why the lesions in these patients had a predominantly facial distribution. Involvement of the face in PMH has not been commonly reported in the literature. Martínez-Martínez et al3 reported 12 PMH patients with lesions only presenting in lumbar and abdominal distributions. Kim et al8 presented a series of 23 PMH patients treated with narrowband UVB in whom 56% (9/16) saw repigmentation in 90% of the lesions following treatment. The most commonly affected area was the lower back, followed by the abdomen, upper back, chest, sacral region, flank, and shoulders, respectively.8 In a review by Relyveld et al,1 PMH is described as a predominantly truncal disease that can occasionally extend to the neck, face, and proximal arms and legs; however, no specific cases were reported.
Previous case series have reported PMH primarily in adolescents and young adults, with mean ages ranging from 26 to 30 years.1,3 The 4 patients reported here were older, ranging in age from 40 to 65 years. This discrepancy in age may contribute to the facial distribution encountered in this patient population; however, given the small number of patients in our case series, such extrapolation is premature. Most recently, Westerhof et al6 demonstrated a relationship between the presence of P acnes, a common skin commensal of the face, and the hypopigmented macules of PMH. The investigators suggested that some strains of P acnes produce a factor that is yet to be identified that interferes with melanogenesis. The response of PMH lesions to topical treatments such as benzoyl peroxide, clindamycin, and phototherapy has lent credence to the potential etiologic role of P acnes in this condition.9,10 The interplay between age, PMH distribution, and P acnes requires further investigation.
The biopsies in our 4 patients were consistent with the nonspecific histopathologic characteristics of PMH lesions. Biopsies in all 4 patients revealed a sparse perivascular lymphocytic infiltrate, and in 2 of the cases, postinflammatory pigmentary alteration was noted. Such changes often are described in PMH lesions.4,5 In other cases detailed in the literature, lesional and nonlesional skin often are indistinguishable on hematoxylin and eosin staining.11 In the 3 patients for whom we performed additional immunohistochemical studies, results were mixed: Melan-A staining revealed a decreased number of melanocytes in Patient 1 but not in Patients 2 or 4. Many reported cases in the literature have not demonstrated a decrease in melanocyte density but instead show a decrease in melanin content in lesional skin.1-3,6,7 Although additional stains performed in Patient 4 revealed neither a decrease in the number of melanocytes nor a decrease in the melanin content, such histopathologic findings of PMH often are subtle. Additional stains were not performed in Patient 3. More studies are needed to characterize the immunohistochemical staining patterns of lesional skin in patients with PMH.
Tinea versicolor, pityriasis alba, mycosis fungoides, sarcoidosis, leprosy, and syphilis typically are included in the differential diagnosis for PMH. Tinea versicolor traditionally is diagnosed based on the combination of irregular hypopigmented or hyperpigmented scaly macules and a KOH preparation that is positive for hyphae and spores. Similar to PMH, tinea versicolor is most often found on the trunk, but unusual cases have been reported involving the face.12
Patient 2 reflected how it can be difficult diagnostically to distinguish between tinea versicolor and PMH. Although this patient initially had a KOH scraping suggestive for tinea versicolor, adequate treatment with oral fluconazole and ketoconazole shampoo did not result in improvement. The hypopigmented lesions in this patient continued to progress despite therapy. Additionally, his hypopigmented to depigmented nonscaly macules were more clinically consistent with the characteristic description of lesion configuration in PMH than with the irregular, more sharply defined, asymmetric, and scaly spots of tinea versicolor. Furthermore, the inflammatory findings on biopsy favored a diagnosis of PMH.
Pityriasis alba, most frequently presents on the face in the form of hypopigmented, sometimes slightly scaly macules but also can occur on the body. It usually occurs in younger patients who often have an atopic diathesis. Histologic findings generally are nonspecific, but discrete eczematous changes can sometimes be appreciated in the epidermis and dermis. None of our patients had histories suggestive of an atopic diathesis or lesion distributions typical of pityriasis alba. Histologic findings also were more consistent with PMH than pityriasis alba.
A diagnosis of patch-stage hypopigmented MF should also be entertained in patients with hypopigmented macules, as it can appear similar to the lesions of PMH. Hypopigmented MF often is associated with subtle atrophy, scaling, poikiloderma, and erythema. These features were not present in the 4 cases presented here. Histologically, atypical lymphocytes with prominent epidermotropism and tagging of the epidermis by large lymphocytic infiltrates are seen in cases of hypopigmented MF. These findings were not present in biopsies from our patients.
Hypopigmented sarcoidosis, leprosy, and syphilis are other systemic diseases associated with hypopigmented lesions. Histologically, noncaseasting granulomas in the dermis or subcutaneous tissue would favor a diagnosis of sarcoidosis over PMH. In patients who live in endemic areas, a diagnosis of leprosy for an anesthetic hypopigmented lesion would be higher in the differential. Finally, it is important to rule out secondary syphilis when diagnosing PMH. Known as the great imitator, secondary syphilis may present in a patient in the form of hypopigmented macules. Patients 1, 2, and 4 had nonreactive RPR tests; unfortunately, RPR was not checked in Patient 3. He denied all risk factors for syphilis.
Various topical and oral treatments were prescribed for each patient, but so far none have been unequivocally effective. In the literature, there are reports supporting the efficacy of topical antimicrobial agents targeting P acnes.9,10 One case report noted improvement in a patient with PMH after isotretinoin use.13 Phototherapy also has been reported to improve PMH in several case reports4-8; however, consistent response to these therapies has not been documented. Unfortunately for patients with a diagnosis of PMH, a lack of effective treatment options often exists.
This series of 4 cases highlights the importance of considering PMH in the differential of hypopigmented macules, even when they appear predominantly on the face.
- Relyveld G, Menke H, Westerhof W. Progressive macular hypomelanosis: an overview. Am J Clin Dermatol. 2007;8:13-19.
- Hwang SW, Hong SK, Kim SH, et al. Progressive macular hypomelanosis in Korean patients: a clinicopathologic study. Ann Dermatol. 2009;21:261-267.
- Martinéz-Martinéz ML, Azaña-Defez JM, Rodríguez-Vázquez M, et al. Progressive macular hypomelanosis. Pediatr Dermatol. 2012;29:460-462.
- Montero LC, Belinchonón I, Toledo F, et al. Progressive macular hypomelanosis, excellent response with narrow-band ultraviolet B phototherapy. Photodermatol Photoimmunol Photomed. 2011;27:162-163.
- Choi YJ, Hann SK. Two cases of progressive macular hypomelanosis of the trunk. Korean J Dermatol. 2000;38:655-658.
- Westerhof W, Rlyveld G, Kingswijk M, et al. Propionibacterium acnes and the pathogenesis of progressive macular hypomelanosis. Arch Dermatol. 2004;140:210-214.
- Wu SG, Xu AE, Song XZ, et al. Clinical, pathologic, and ultrastructural studies of progressive macular hypomelanosis. Int J Dermatol. 2010;29:1127-1132.
- Kim MB, Kim GW, Cho HH, et al. Narrowband UVB treatment of progressive macular hypomelanosis. J Am Acad Dermatol. 2012;66:598-605.
- Revlyveld GN, Menkie HE, Westerhof W. Benzoyl peroxide/clindamycin/UVA is more effective than fluticasone/UVA in progressive macular hypomelanosis: a randomized study. Am J Clin Dermatol. 2006;55:836-843.
- Santos JB, Almeida OL, Silva LM, et al. Efficacy of topical combination of benzoyl peroxide 5% and clindamcyin 1% for the treatment of progressive macular hypomelanosis: a randomized, doubleblind, placebo-controlled trial [in Portuguese]. An Bras Dermatol. 2011;86:50-54.
- Kumarasinghe SP, Tan SH, Thng S, et al. Progressive macular hypomelanosis in Singapore: a clinico-pathological study. Int J Dermatol. 2006;45:737-742.
- Terragni L, Lasagni A, Oriani A. Pityriasis versicolor of the face. Mycoses. 1991;34:345-347.
- Kim YK, Lee DY, Lee, JY, et al. Progressive macular hypomelanosis showing excellent response to oral isotretinoin [published online June 23, 2012]. J Dermatol. 2012;39:937-938.
- Relyveld G, Menke H, Westerhof W. Progressive macular hypomelanosis: an overview. Am J Clin Dermatol. 2007;8:13-19.
- Hwang SW, Hong SK, Kim SH, et al. Progressive macular hypomelanosis in Korean patients: a clinicopathologic study. Ann Dermatol. 2009;21:261-267.
- Martinéz-Martinéz ML, Azaña-Defez JM, Rodríguez-Vázquez M, et al. Progressive macular hypomelanosis. Pediatr Dermatol. 2012;29:460-462.
- Montero LC, Belinchonón I, Toledo F, et al. Progressive macular hypomelanosis, excellent response with narrow-band ultraviolet B phototherapy. Photodermatol Photoimmunol Photomed. 2011;27:162-163.
- Choi YJ, Hann SK. Two cases of progressive macular hypomelanosis of the trunk. Korean J Dermatol. 2000;38:655-658.
- Westerhof W, Rlyveld G, Kingswijk M, et al. Propionibacterium acnes and the pathogenesis of progressive macular hypomelanosis. Arch Dermatol. 2004;140:210-214.
- Wu SG, Xu AE, Song XZ, et al. Clinical, pathologic, and ultrastructural studies of progressive macular hypomelanosis. Int J Dermatol. 2010;29:1127-1132.
- Kim MB, Kim GW, Cho HH, et al. Narrowband UVB treatment of progressive macular hypomelanosis. J Am Acad Dermatol. 2012;66:598-605.
- Revlyveld GN, Menkie HE, Westerhof W. Benzoyl peroxide/clindamycin/UVA is more effective than fluticasone/UVA in progressive macular hypomelanosis: a randomized study. Am J Clin Dermatol. 2006;55:836-843.
- Santos JB, Almeida OL, Silva LM, et al. Efficacy of topical combination of benzoyl peroxide 5% and clindamcyin 1% for the treatment of progressive macular hypomelanosis: a randomized, doubleblind, placebo-controlled trial [in Portuguese]. An Bras Dermatol. 2011;86:50-54.
- Kumarasinghe SP, Tan SH, Thng S, et al. Progressive macular hypomelanosis in Singapore: a clinico-pathological study. Int J Dermatol. 2006;45:737-742.
- Terragni L, Lasagni A, Oriani A. Pityriasis versicolor of the face. Mycoses. 1991;34:345-347.
- Kim YK, Lee DY, Lee, JY, et al. Progressive macular hypomelanosis showing excellent response to oral isotretinoin [published online June 23, 2012]. J Dermatol. 2012;39:937-938.
Practice Points
- Progressive macular hypomelanosis should be considered in the differential diagnosis for hypopigmented facial lesions.
- Progressive macular hypomelanosis proves to be a diagnosis of exclusion.
Drug-induced Linear IgA Bullous Dermatosis in a Patient With a Vancomycin-impregnated Cement Spacer
Case Report
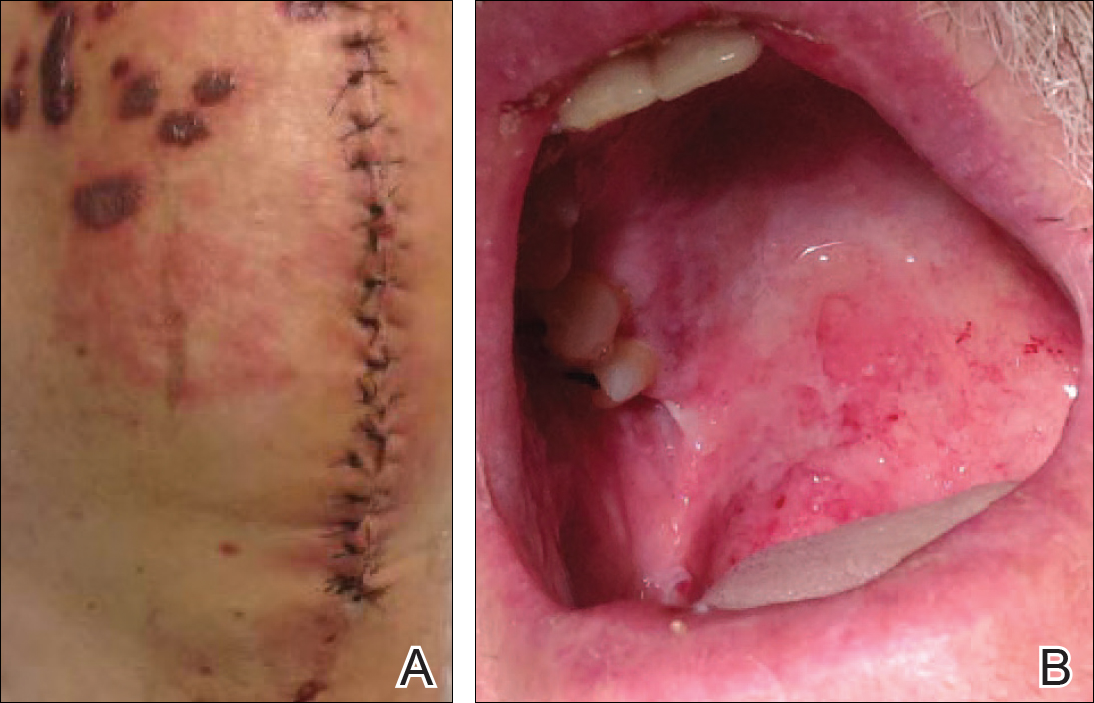
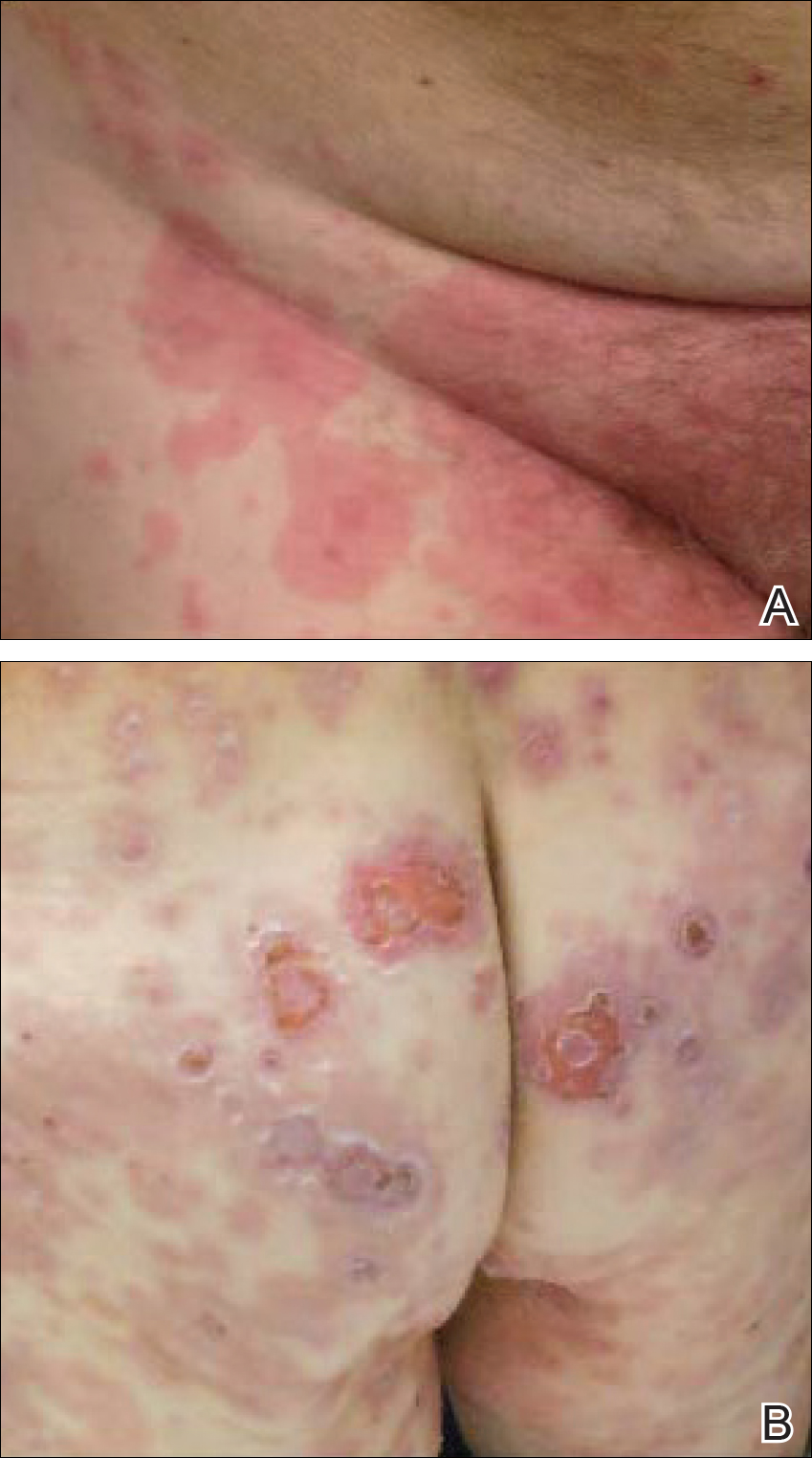
A 77-year-old man was admitted to the general medicine service at our institution for treatment of a diffuse macular eruption and hemorrhagic bullae 12 days after undergoing left-knee revision arthroplasty during which a cement spacer impregnated with vancomycin and tobramycin was placed. At the time of the surgery, the patient also received intravenous (IV) vancomycin and oral ciprofloxacin, which were continued postoperatively until his hospital presentation. The patient was recovering well until postoperative day 7, when he developed painful swelling and erythema surrounding the surgical wound on the left knee. Concerned that his symptoms indicated a flare of gout, he restarted a former allopurinol prescription from an outside physician after 2 years of nonuse. The skin changes progressed distally on the left leg over the next 48 hours. By postoperative day 10, he had developed serosanguinous blisters on the left knee (Figure 1A) and oral mucosa (Figure 1B), as well as erythematous nodules on the bilateral palms. He presented to our institution for emergent care on postoperative day 12 following progression of the eruption to the inguinal region (Figure 2A), buttocks (Figure 2B), and abdominal region.
Due to concerns about a potential drug reaction, the IV vancomycin, oral ciprofloxacin, and oral allopurinol were discontinued on hospital admission.
Oral prednisone 60 mg once daily and oral dapsone 25 mg once daily were initiated on hospital days 4 and 6 (postoperative days 15 and 17), respectively. A 6-week course of oral ciprofloxacin 750 mg twice daily and daptomycin 8 mg/kg once daily was initiated for bacterial coverage on hospital day 5 (postoperative day 16). Topical triamcinolone and an anesthetic mouthwash also were used to treat the mucosal involvement. The lesions stabilized on the third day of steroid therapy, and the patient was discharged 7 days after hospital admission (postoperative day 18). Dapsone was rapidly increased to 100 mg once daily over the next week for Pneumocystis jirovecii pneumonia prophylaxis. An increase in prednisone to 80 mg once daily was required 3 days after the patient was discharged due to worsening oral lesions. Five days after discharge, the patient was readmitted to the hospital for 3 days due to acute kidney injury (AKI) in which his baseline creatinine level tripled. The cause of renal impairment was unknown, resulting in empiric discontinuation of dapsone on postoperative day 27. Prophylaxis for P jirovecii pneumonia was replaced with once-monthly inhaled pentamidine. Prednisone was tapered 20 days after the original presentation (postoperative day 32) following gradual improvement of both the skin and oral lesions. At dermatology follow-up 2 weeks later, doxycycline 100 mg twice daily was added for residual inflammation of the left leg. A deep vein thrombosis was discovered in the left leg 10 days later, and 3 months of anticoagulation therapy was initiated with discontinuation of the doxycycline. The patient continued to have renal insufficiency several weeks after dapsone discontinuation and developed prominent peripheral motor neuropathy with bilateral thenar atrophy. He did not experience any skin eruptions or relapses in the weeks following prednisone cessation and underwent successful removal of the cement spacer with full left-knee reconstruction 4 months after his initial presentation to our institution. At 9-month dermatology follow-up, the LABD remained in remission.
Comment
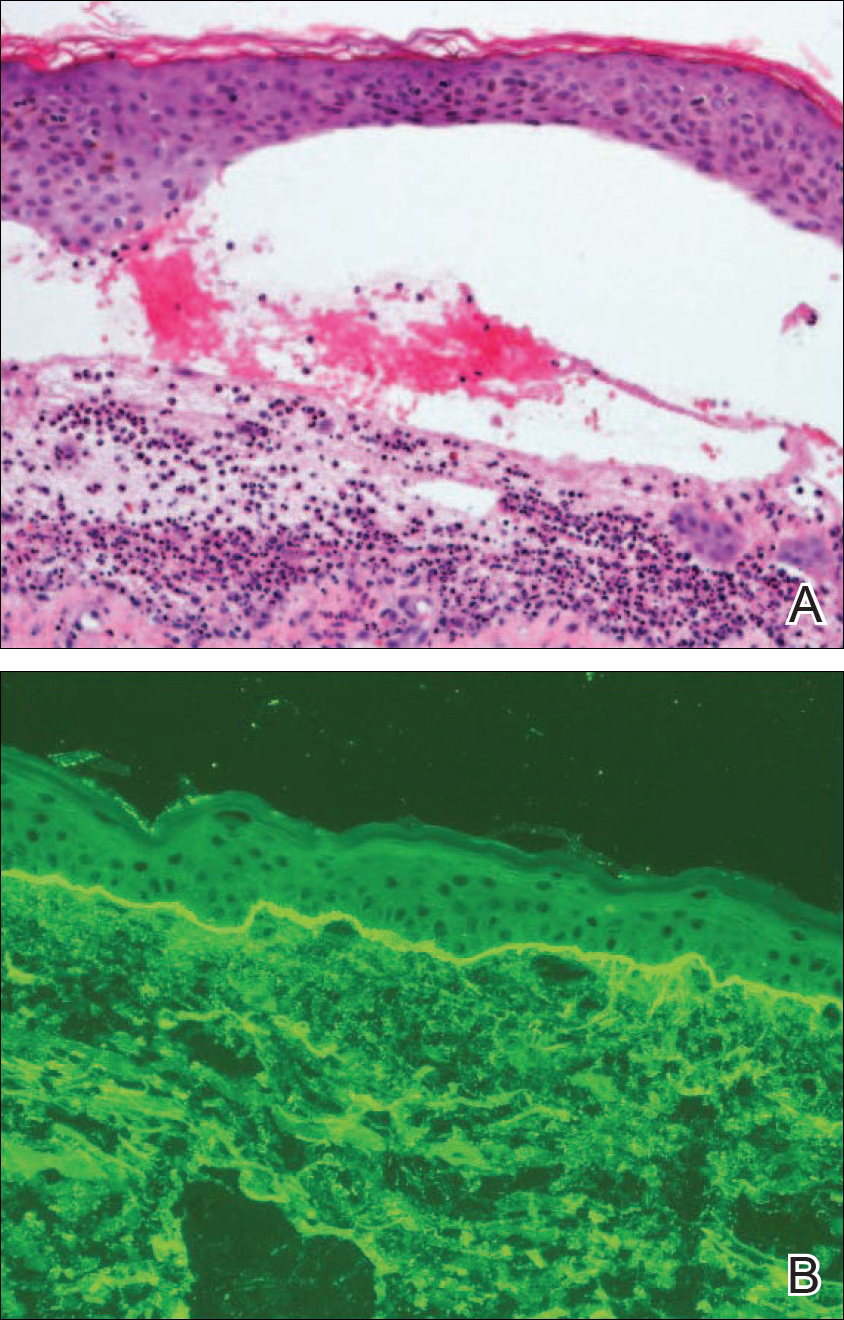
Linear IgA bullous dermatosis is a well-documented autoimmune mucocutaneous disorder characterized by linear IgA deposits at the dermoepidermal junction. The development of autoantibodies to antigens within the basement membrane zone leads to both cellular and humoral immune responses that facilitate the subepidermal blistering rash in LABD.2,3 Linear IgA bullous dermatosis affects all ages and races with a bimodal epidemiology. The adult form typically appears after 60 years of age, whereas the childhood form (chronic bullous disease of childhood) appears between 6 months and 6 years of age.3 Medications—particularly vancomycin—are responsible for a substantial portion of cases.1-4 In one review, vancomycin was implicated in almost half (22/52 [42.3%]) of drug-related cases of LABD.4 Other associated medications include captopril, trimethoprim-sulfamethoxazole, phenytoin, and diclo-fenac.3,4 Vancomycin-associated LABD has a substantially shorter time to onset of symptoms, with a mean of 8.6 days compared to 63.8 days for other causative agents.4
The initial treatment of drug-induced LABD is immediate discontinuation of the suspected agent(s) and supportive care.9 Although future avoidance of vancomycin is recommended in patients with a history of LABD, there are reported cases of successful rechallenges.4,10 The early removal of our patient’s cement spacer was discouraged by both the orthopedics and infectious disease consultation services due to potential complications as well as the patient’s gradual improvement during his hospital course.
Dapsone is considered the standard systemic treatment for LABD. Sulfapyridine is an alternative to dapsone, or a combination of these 2 drugs may be used. Corticosteroids can be added to each of these regimens to achieve remission, as in our case.2 Although dapsone was discontinued in the setting of the patient’s AKI, the vancomycin in the dual-eluting spacer was more likely the culprit. A review of 544 postoperative outcomes following the use of an antibiotic-impregnated cement spacer (AICS) during 2-stage arthroplasty displayed an 8- to 10-fold increase in the development of AKIs compared to the rate of AKIs following primary joint arthroplasty.10 While our patient’s AKI was not attributed to dapsone, his prominent peripheral motor neuropathy with resultant bilateral thenar atrophy was a rare complication of dapsone use. While dapsone-associated neuropathy has been reported in daily dosages of as low as 75 mg, it typically is seen in doses of at least 300 mg per day and in larger cumulative dosages.11
Despite having a well-characterized vancomycin-induced LABD in the setting of known vancomycin exposure, our patient’s case was particularly challenging given the continued presence of the vancomycin-impregnated cement spacer (VICS) in the left knee, resulting in vancomycin levels at admission and during subsequent measurements over 2 weeks that were all several-fold higher than the renal clearance predicted.
Vancomycin-associated LABD does not appear to be dose dependent and has been reported at both subtherapeutic1-3 and supratherapeutic levels,5-9 whereas toxicity reactions are more common at supratherapeutic levels.9 The literature on AICS use suggests that drug elution occurs at relatively unpredictable rates based on a variety of factors, including the type of cement used and the initial antibiotic concentration.12,13 Furthermore, the addition of tobramycin to VICSs has been found to increase the rate of vancomycin delivery through a phenomenon known as passive opportunism.14
As AICS devices allow for the delivery of higher concentrations of antibiotics to a localized area, systemic complications are considered rare but have been reported.13 Our report describes a rare case of LABD in the setting of a VICS. One clinical aspect of our case that supports the implication of VICS as the cause of the patient’s LABD is the concentration of bullae overlying the incision site on the left knee. A case of a desquamating rash in a patient with an implanted VICS has been documented in which the early lesions were localized to the surgical leg, as in our case.15 Unlike our case, there was a history of Stevens-Johnson syndrome following previous vancomycin exposure. A case of a gentamicin-impregnated cement spacer causing allergic dermatitis that was most prominent in the surgical leg also has been reported.16 An isomorphic phenomenon (Köbner phenomenon) has been suggested in the setting of
- Plunkett RW, Chiarello SE, Beutner EH. Linear IgA bullous dermatosis in one of two piroxicam-induced eruptions: a distinct direct immunofluorescence trend revealed by the literature. J Am Acad Dermatol. 2001;45:691-696.
- Guide SV, Marinkovich MP. Linear IgA bullous dermatosis. Clin Dermatol. 2001;19:719-727.
- Fortuna G, Marinkovich MP. Linear immunoglobulin A bullous dermatosis. Clin Dermatol. 2012;30:38-50.
- Fortuna G, Salas-Alanis JC, Guidetti E, et al. A critical reappraisal of the current data on drug-induced linear immunoglobulin A bullous dermatosis: a real and separate nosological entity? J Am Acad Dermatol. 2012;66:988-994.
- Kuechle MK, Stegemeir E, Maynard B, et al. Drug-induced linear IgA bullous dermatosis: report of six cases and review of the literature. J Am Acad Dermatol. 1994;30(2, pt 1):187-192.
- Neughebauer BI, Negron G, Pelton S, et al. Bullous skin disease: an unusual allergic reaction to vancomycin. Am J Med Sci. 2002;323:273-278.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Wiadrowski TP, Reid CM. Drug-induced linear IgA bullous disease following antibiotics. Australas J Dermatol. 2001;42:196-199.
- Dang LV, Byrom L, Muir J, et al. Vancomycin-induced linear IgA with mucosal and ocular involvement: a case report. Infect Dis Clin Pract. 2014;22:e119-e121.
- Luu A, Syed F, Raman G, et al. Two-stage arthroplasty for prosthetic joint infection: a systematic review of acute kidney injury, systemic toxicity and infection control [published online April 8, 2013]. J Arthroplasty. 2013;28:1490.e1-1498.e1.
- Daneshmend TK. The neurotoxicity of dapsone. Adverse Drug React Acute Poisoning Rev. 1984;3:43-58.
- Jacobs C, Christensen CP, Berend ME. Static and mobile antibiotic-impregnated cement spacers for the management of prosthetic joint infection. J Am Acad Orthop Surg. 2009;17:356-368.
- Springer BD, Lee GC, Osmon D, et al. Systemic safety of high-dose antibiotic-loaded cement spacers after resection of an infected total knee arthroplasty. Clin Orthop Relat Res. 2004;427:47-51.
- Penner MJ, Masri BA, Duncan CP. Elution characteristics of vancomycin and tobramycin combined in acrylic bone-cement. J Arthroplasty. 1996;11:939-944.
- Williams B, Hanson A, Sha B. Diffuse desquamating rash following exposure to vancomycin-impregnated bone cement. Ann Pharmacother. 2014;48:1061-1065.
- Haeberle M, Wittner B. Is gentamicin-loaded bone cement a risk for developing systemic allergic dermatitis? Contact Dermatitis. 2009;60:176-177.
- McDonald HC, York NR, Pandya AG. Drug-induced linear IgA bullous dermatosis demonstrating the isomorphic phenomenon. J Am Acad Dermatol. 2010;62:897-898.
Case Report
A 77-year-old man was admitted to the general medicine service at our institution for treatment of a diffuse macular eruption and hemorrhagic bullae 12 days after undergoing left-knee revision arthroplasty during which a cement spacer impregnated with vancomycin and tobramycin was placed. At the time of the surgery, the patient also received intravenous (IV) vancomycin and oral ciprofloxacin, which were continued postoperatively until his hospital presentation. The patient was recovering well until postoperative day 7, when he developed painful swelling and erythema surrounding the surgical wound on the left knee. Concerned that his symptoms indicated a flare of gout, he restarted a former allopurinol prescription from an outside physician after 2 years of nonuse. The skin changes progressed distally on the left leg over the next 48 hours. By postoperative day 10, he had developed serosanguinous blisters on the left knee (Figure 1A) and oral mucosa (Figure 1B), as well as erythematous nodules on the bilateral palms. He presented to our institution for emergent care on postoperative day 12 following progression of the eruption to the inguinal region (Figure 2A), buttocks (Figure 2B), and abdominal region.
Due to concerns about a potential drug reaction, the IV vancomycin, oral ciprofloxacin, and oral allopurinol were discontinued on hospital admission.
Oral prednisone 60 mg once daily and oral dapsone 25 mg once daily were initiated on hospital days 4 and 6 (postoperative days 15 and 17), respectively. A 6-week course of oral ciprofloxacin 750 mg twice daily and daptomycin 8 mg/kg once daily was initiated for bacterial coverage on hospital day 5 (postoperative day 16). Topical triamcinolone and an anesthetic mouthwash also were used to treat the mucosal involvement. The lesions stabilized on the third day of steroid therapy, and the patient was discharged 7 days after hospital admission (postoperative day 18). Dapsone was rapidly increased to 100 mg once daily over the next week for Pneumocystis jirovecii pneumonia prophylaxis. An increase in prednisone to 80 mg once daily was required 3 days after the patient was discharged due to worsening oral lesions. Five days after discharge, the patient was readmitted to the hospital for 3 days due to acute kidney injury (AKI) in which his baseline creatinine level tripled. The cause of renal impairment was unknown, resulting in empiric discontinuation of dapsone on postoperative day 27. Prophylaxis for P jirovecii pneumonia was replaced with once-monthly inhaled pentamidine. Prednisone was tapered 20 days after the original presentation (postoperative day 32) following gradual improvement of both the skin and oral lesions. At dermatology follow-up 2 weeks later, doxycycline 100 mg twice daily was added for residual inflammation of the left leg. A deep vein thrombosis was discovered in the left leg 10 days later, and 3 months of anticoagulation therapy was initiated with discontinuation of the doxycycline. The patient continued to have renal insufficiency several weeks after dapsone discontinuation and developed prominent peripheral motor neuropathy with bilateral thenar atrophy. He did not experience any skin eruptions or relapses in the weeks following prednisone cessation and underwent successful removal of the cement spacer with full left-knee reconstruction 4 months after his initial presentation to our institution. At 9-month dermatology follow-up, the LABD remained in remission.
Comment
Linear IgA bullous dermatosis is a well-documented autoimmune mucocutaneous disorder characterized by linear IgA deposits at the dermoepidermal junction. The development of autoantibodies to antigens within the basement membrane zone leads to both cellular and humoral immune responses that facilitate the subepidermal blistering rash in LABD.2,3 Linear IgA bullous dermatosis affects all ages and races with a bimodal epidemiology. The adult form typically appears after 60 years of age, whereas the childhood form (chronic bullous disease of childhood) appears between 6 months and 6 years of age.3 Medications—particularly vancomycin—are responsible for a substantial portion of cases.1-4 In one review, vancomycin was implicated in almost half (22/52 [42.3%]) of drug-related cases of LABD.4 Other associated medications include captopril, trimethoprim-sulfamethoxazole, phenytoin, and diclo-fenac.3,4 Vancomycin-associated LABD has a substantially shorter time to onset of symptoms, with a mean of 8.6 days compared to 63.8 days for other causative agents.4
The initial treatment of drug-induced LABD is immediate discontinuation of the suspected agent(s) and supportive care.9 Although future avoidance of vancomycin is recommended in patients with a history of LABD, there are reported cases of successful rechallenges.4,10 The early removal of our patient’s cement spacer was discouraged by both the orthopedics and infectious disease consultation services due to potential complications as well as the patient’s gradual improvement during his hospital course.
Dapsone is considered the standard systemic treatment for LABD. Sulfapyridine is an alternative to dapsone, or a combination of these 2 drugs may be used. Corticosteroids can be added to each of these regimens to achieve remission, as in our case.2 Although dapsone was discontinued in the setting of the patient’s AKI, the vancomycin in the dual-eluting spacer was more likely the culprit. A review of 544 postoperative outcomes following the use of an antibiotic-impregnated cement spacer (AICS) during 2-stage arthroplasty displayed an 8- to 10-fold increase in the development of AKIs compared to the rate of AKIs following primary joint arthroplasty.10 While our patient’s AKI was not attributed to dapsone, his prominent peripheral motor neuropathy with resultant bilateral thenar atrophy was a rare complication of dapsone use. While dapsone-associated neuropathy has been reported in daily dosages of as low as 75 mg, it typically is seen in doses of at least 300 mg per day and in larger cumulative dosages.11
Despite having a well-characterized vancomycin-induced LABD in the setting of known vancomycin exposure, our patient’s case was particularly challenging given the continued presence of the vancomycin-impregnated cement spacer (VICS) in the left knee, resulting in vancomycin levels at admission and during subsequent measurements over 2 weeks that were all several-fold higher than the renal clearance predicted.
Vancomycin-associated LABD does not appear to be dose dependent and has been reported at both subtherapeutic1-3 and supratherapeutic levels,5-9 whereas toxicity reactions are more common at supratherapeutic levels.9 The literature on AICS use suggests that drug elution occurs at relatively unpredictable rates based on a variety of factors, including the type of cement used and the initial antibiotic concentration.12,13 Furthermore, the addition of tobramycin to VICSs has been found to increase the rate of vancomycin delivery through a phenomenon known as passive opportunism.14
As AICS devices allow for the delivery of higher concentrations of antibiotics to a localized area, systemic complications are considered rare but have been reported.13 Our report describes a rare case of LABD in the setting of a VICS. One clinical aspect of our case that supports the implication of VICS as the cause of the patient’s LABD is the concentration of bullae overlying the incision site on the left knee. A case of a desquamating rash in a patient with an implanted VICS has been documented in which the early lesions were localized to the surgical leg, as in our case.15 Unlike our case, there was a history of Stevens-Johnson syndrome following previous vancomycin exposure. A case of a gentamicin-impregnated cement spacer causing allergic dermatitis that was most prominent in the surgical leg also has been reported.16 An isomorphic phenomenon (Köbner phenomenon) has been suggested in the setting of
Case Report
A 77-year-old man was admitted to the general medicine service at our institution for treatment of a diffuse macular eruption and hemorrhagic bullae 12 days after undergoing left-knee revision arthroplasty during which a cement spacer impregnated with vancomycin and tobramycin was placed. At the time of the surgery, the patient also received intravenous (IV) vancomycin and oral ciprofloxacin, which were continued postoperatively until his hospital presentation. The patient was recovering well until postoperative day 7, when he developed painful swelling and erythema surrounding the surgical wound on the left knee. Concerned that his symptoms indicated a flare of gout, he restarted a former allopurinol prescription from an outside physician after 2 years of nonuse. The skin changes progressed distally on the left leg over the next 48 hours. By postoperative day 10, he had developed serosanguinous blisters on the left knee (Figure 1A) and oral mucosa (Figure 1B), as well as erythematous nodules on the bilateral palms. He presented to our institution for emergent care on postoperative day 12 following progression of the eruption to the inguinal region (Figure 2A), buttocks (Figure 2B), and abdominal region.
Due to concerns about a potential drug reaction, the IV vancomycin, oral ciprofloxacin, and oral allopurinol were discontinued on hospital admission.
Oral prednisone 60 mg once daily and oral dapsone 25 mg once daily were initiated on hospital days 4 and 6 (postoperative days 15 and 17), respectively. A 6-week course of oral ciprofloxacin 750 mg twice daily and daptomycin 8 mg/kg once daily was initiated for bacterial coverage on hospital day 5 (postoperative day 16). Topical triamcinolone and an anesthetic mouthwash also were used to treat the mucosal involvement. The lesions stabilized on the third day of steroid therapy, and the patient was discharged 7 days after hospital admission (postoperative day 18). Dapsone was rapidly increased to 100 mg once daily over the next week for Pneumocystis jirovecii pneumonia prophylaxis. An increase in prednisone to 80 mg once daily was required 3 days after the patient was discharged due to worsening oral lesions. Five days after discharge, the patient was readmitted to the hospital for 3 days due to acute kidney injury (AKI) in which his baseline creatinine level tripled. The cause of renal impairment was unknown, resulting in empiric discontinuation of dapsone on postoperative day 27. Prophylaxis for P jirovecii pneumonia was replaced with once-monthly inhaled pentamidine. Prednisone was tapered 20 days after the original presentation (postoperative day 32) following gradual improvement of both the skin and oral lesions. At dermatology follow-up 2 weeks later, doxycycline 100 mg twice daily was added for residual inflammation of the left leg. A deep vein thrombosis was discovered in the left leg 10 days later, and 3 months of anticoagulation therapy was initiated with discontinuation of the doxycycline. The patient continued to have renal insufficiency several weeks after dapsone discontinuation and developed prominent peripheral motor neuropathy with bilateral thenar atrophy. He did not experience any skin eruptions or relapses in the weeks following prednisone cessation and underwent successful removal of the cement spacer with full left-knee reconstruction 4 months after his initial presentation to our institution. At 9-month dermatology follow-up, the LABD remained in remission.
Comment
Linear IgA bullous dermatosis is a well-documented autoimmune mucocutaneous disorder characterized by linear IgA deposits at the dermoepidermal junction. The development of autoantibodies to antigens within the basement membrane zone leads to both cellular and humoral immune responses that facilitate the subepidermal blistering rash in LABD.2,3 Linear IgA bullous dermatosis affects all ages and races with a bimodal epidemiology. The adult form typically appears after 60 years of age, whereas the childhood form (chronic bullous disease of childhood) appears between 6 months and 6 years of age.3 Medications—particularly vancomycin—are responsible for a substantial portion of cases.1-4 In one review, vancomycin was implicated in almost half (22/52 [42.3%]) of drug-related cases of LABD.4 Other associated medications include captopril, trimethoprim-sulfamethoxazole, phenytoin, and diclo-fenac.3,4 Vancomycin-associated LABD has a substantially shorter time to onset of symptoms, with a mean of 8.6 days compared to 63.8 days for other causative agents.4
The initial treatment of drug-induced LABD is immediate discontinuation of the suspected agent(s) and supportive care.9 Although future avoidance of vancomycin is recommended in patients with a history of LABD, there are reported cases of successful rechallenges.4,10 The early removal of our patient’s cement spacer was discouraged by both the orthopedics and infectious disease consultation services due to potential complications as well as the patient’s gradual improvement during his hospital course.
Dapsone is considered the standard systemic treatment for LABD. Sulfapyridine is an alternative to dapsone, or a combination of these 2 drugs may be used. Corticosteroids can be added to each of these regimens to achieve remission, as in our case.2 Although dapsone was discontinued in the setting of the patient’s AKI, the vancomycin in the dual-eluting spacer was more likely the culprit. A review of 544 postoperative outcomes following the use of an antibiotic-impregnated cement spacer (AICS) during 2-stage arthroplasty displayed an 8- to 10-fold increase in the development of AKIs compared to the rate of AKIs following primary joint arthroplasty.10 While our patient’s AKI was not attributed to dapsone, his prominent peripheral motor neuropathy with resultant bilateral thenar atrophy was a rare complication of dapsone use. While dapsone-associated neuropathy has been reported in daily dosages of as low as 75 mg, it typically is seen in doses of at least 300 mg per day and in larger cumulative dosages.11
Despite having a well-characterized vancomycin-induced LABD in the setting of known vancomycin exposure, our patient’s case was particularly challenging given the continued presence of the vancomycin-impregnated cement spacer (VICS) in the left knee, resulting in vancomycin levels at admission and during subsequent measurements over 2 weeks that were all several-fold higher than the renal clearance predicted.
Vancomycin-associated LABD does not appear to be dose dependent and has been reported at both subtherapeutic1-3 and supratherapeutic levels,5-9 whereas toxicity reactions are more common at supratherapeutic levels.9 The literature on AICS use suggests that drug elution occurs at relatively unpredictable rates based on a variety of factors, including the type of cement used and the initial antibiotic concentration.12,13 Furthermore, the addition of tobramycin to VICSs has been found to increase the rate of vancomycin delivery through a phenomenon known as passive opportunism.14
As AICS devices allow for the delivery of higher concentrations of antibiotics to a localized area, systemic complications are considered rare but have been reported.13 Our report describes a rare case of LABD in the setting of a VICS. One clinical aspect of our case that supports the implication of VICS as the cause of the patient’s LABD is the concentration of bullae overlying the incision site on the left knee. A case of a desquamating rash in a patient with an implanted VICS has been documented in which the early lesions were localized to the surgical leg, as in our case.15 Unlike our case, there was a history of Stevens-Johnson syndrome following previous vancomycin exposure. A case of a gentamicin-impregnated cement spacer causing allergic dermatitis that was most prominent in the surgical leg also has been reported.16 An isomorphic phenomenon (Köbner phenomenon) has been suggested in the setting of
- Plunkett RW, Chiarello SE, Beutner EH. Linear IgA bullous dermatosis in one of two piroxicam-induced eruptions: a distinct direct immunofluorescence trend revealed by the literature. J Am Acad Dermatol. 2001;45:691-696.
- Guide SV, Marinkovich MP. Linear IgA bullous dermatosis. Clin Dermatol. 2001;19:719-727.
- Fortuna G, Marinkovich MP. Linear immunoglobulin A bullous dermatosis. Clin Dermatol. 2012;30:38-50.
- Fortuna G, Salas-Alanis JC, Guidetti E, et al. A critical reappraisal of the current data on drug-induced linear immunoglobulin A bullous dermatosis: a real and separate nosological entity? J Am Acad Dermatol. 2012;66:988-994.
- Kuechle MK, Stegemeir E, Maynard B, et al. Drug-induced linear IgA bullous dermatosis: report of six cases and review of the literature. J Am Acad Dermatol. 1994;30(2, pt 1):187-192.
- Neughebauer BI, Negron G, Pelton S, et al. Bullous skin disease: an unusual allergic reaction to vancomycin. Am J Med Sci. 2002;323:273-278.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Wiadrowski TP, Reid CM. Drug-induced linear IgA bullous disease following antibiotics. Australas J Dermatol. 2001;42:196-199.
- Dang LV, Byrom L, Muir J, et al. Vancomycin-induced linear IgA with mucosal and ocular involvement: a case report. Infect Dis Clin Pract. 2014;22:e119-e121.
- Luu A, Syed F, Raman G, et al. Two-stage arthroplasty for prosthetic joint infection: a systematic review of acute kidney injury, systemic toxicity and infection control [published online April 8, 2013]. J Arthroplasty. 2013;28:1490.e1-1498.e1.
- Daneshmend TK. The neurotoxicity of dapsone. Adverse Drug React Acute Poisoning Rev. 1984;3:43-58.
- Jacobs C, Christensen CP, Berend ME. Static and mobile antibiotic-impregnated cement spacers for the management of prosthetic joint infection. J Am Acad Orthop Surg. 2009;17:356-368.
- Springer BD, Lee GC, Osmon D, et al. Systemic safety of high-dose antibiotic-loaded cement spacers after resection of an infected total knee arthroplasty. Clin Orthop Relat Res. 2004;427:47-51.
- Penner MJ, Masri BA, Duncan CP. Elution characteristics of vancomycin and tobramycin combined in acrylic bone-cement. J Arthroplasty. 1996;11:939-944.
- Williams B, Hanson A, Sha B. Diffuse desquamating rash following exposure to vancomycin-impregnated bone cement. Ann Pharmacother. 2014;48:1061-1065.
- Haeberle M, Wittner B. Is gentamicin-loaded bone cement a risk for developing systemic allergic dermatitis? Contact Dermatitis. 2009;60:176-177.
- McDonald HC, York NR, Pandya AG. Drug-induced linear IgA bullous dermatosis demonstrating the isomorphic phenomenon. J Am Acad Dermatol. 2010;62:897-898.
- Plunkett RW, Chiarello SE, Beutner EH. Linear IgA bullous dermatosis in one of two piroxicam-induced eruptions: a distinct direct immunofluorescence trend revealed by the literature. J Am Acad Dermatol. 2001;45:691-696.
- Guide SV, Marinkovich MP. Linear IgA bullous dermatosis. Clin Dermatol. 2001;19:719-727.
- Fortuna G, Marinkovich MP. Linear immunoglobulin A bullous dermatosis. Clin Dermatol. 2012;30:38-50.
- Fortuna G, Salas-Alanis JC, Guidetti E, et al. A critical reappraisal of the current data on drug-induced linear immunoglobulin A bullous dermatosis: a real and separate nosological entity? J Am Acad Dermatol. 2012;66:988-994.
- Kuechle MK, Stegemeir E, Maynard B, et al. Drug-induced linear IgA bullous dermatosis: report of six cases and review of the literature. J Am Acad Dermatol. 1994;30(2, pt 1):187-192.
- Neughebauer BI, Negron G, Pelton S, et al. Bullous skin disease: an unusual allergic reaction to vancomycin. Am J Med Sci. 2002;323:273-278.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Wiadrowski TP, Reid CM. Drug-induced linear IgA bullous disease following antibiotics. Australas J Dermatol. 2001;42:196-199.
- Dang LV, Byrom L, Muir J, et al. Vancomycin-induced linear IgA with mucosal and ocular involvement: a case report. Infect Dis Clin Pract. 2014;22:e119-e121.
- Luu A, Syed F, Raman G, et al. Two-stage arthroplasty for prosthetic joint infection: a systematic review of acute kidney injury, systemic toxicity and infection control [published online April 8, 2013]. J Arthroplasty. 2013;28:1490.e1-1498.e1.
- Daneshmend TK. The neurotoxicity of dapsone. Adverse Drug React Acute Poisoning Rev. 1984;3:43-58.
- Jacobs C, Christensen CP, Berend ME. Static and mobile antibiotic-impregnated cement spacers for the management of prosthetic joint infection. J Am Acad Orthop Surg. 2009;17:356-368.
- Springer BD, Lee GC, Osmon D, et al. Systemic safety of high-dose antibiotic-loaded cement spacers after resection of an infected total knee arthroplasty. Clin Orthop Relat Res. 2004;427:47-51.
- Penner MJ, Masri BA, Duncan CP. Elution characteristics of vancomycin and tobramycin combined in acrylic bone-cement. J Arthroplasty. 1996;11:939-944.
- Williams B, Hanson A, Sha B. Diffuse desquamating rash following exposure to vancomycin-impregnated bone cement. Ann Pharmacother. 2014;48:1061-1065.
- Haeberle M, Wittner B. Is gentamicin-loaded bone cement a risk for developing systemic allergic dermatitis? Contact Dermatitis. 2009;60:176-177.
- McDonald HC, York NR, Pandya AG. Drug-induced linear IgA bullous dermatosis demonstrating the isomorphic phenomenon. J Am Acad Dermatol. 2010;62:897-898.
Practice Points
- Linear IgA bullous dermatosis (LABD) is an autoimmune mucocutaneous disorder characterized by linear IgA deposits at the dermoepidermal junction.
- A substantial number of cases of LABD are drug related, with vancomycin most commonly implicated.
- While antibiotic-impregnated cement spacers deliver high concentrations of local medications, systemic reactions are still possible.
- Dapsone is the first-line treatment for LABD.
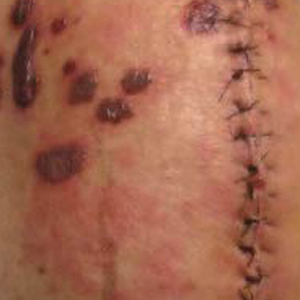
A Case of Pustular Psoriasis of Pregnancy With Positive Maternal-Fetal Outcomes
Pustular psoriasis of pregnancy (PPP), also known as impetigo herpetiformis, is a relatively rare cutaneous disorder of pregnancy wherein lesions typically appear in the third trimester and resolve after delivery; however, lesions may persist through the postpartum period. Pustular psoriasis of pregnancy may be considered a fifth dermatosis of pregnancy, alongside the classic dermatoses of atopic eruption of pregnancy, intrahepatic cholestasis of pregnancy, pemphigoid gestationis, and pruritic urticarial papules and plaques of pregnancy.1
As PPP is a rare disease, its effects on maternal-fetal health outcomes and management remain to be elucidated. Though maternal mortality is rare in PPP, it is a unique dermatosis of pregnancy because it may be associated with severe systemic maternal symptoms.2 Fetal morbidity and mortality are less predictable in PPP, with reported cases of stillbirth, fetal anomalies, and neonatal death thought to be due largely to placental insufficiency, even with control of symptoms.1,3 Given the risk of serious harm to the fetus, reporting of cases and discussion of PPP management is critical.
Case Report
An otherwise healthy 29-year-old G2P1 woman at 32 weeks’ gestation presented to our emergency department with a 1-week history of a pruritic, burning rash that started on the thighs then spread diffusely. She denied any similar rash in her prior pregnancy. She was not currently taking any medications except for prenatal vitamins and denied any systemic symptoms. The patient’s obstetrician initiated treatment with methylprednisolone 50 mg once daily for the rash 3 days prior to the current presentation, which had not seemed to help. On physical examination, edematous pink plaques studded with 1- to 2-mm collarettes of scaling and sparse 1-mm pustules involving the arms, chest, abdomen, back, groin, buttocks, and legs were noted. The plaques on the back and inner thighs had a peripheral rim of desquamative scaling. There were pink macules on the palms, and superficial desquamation was noted on the lips. The oral mucosa was otherwise spared (Figure 1).
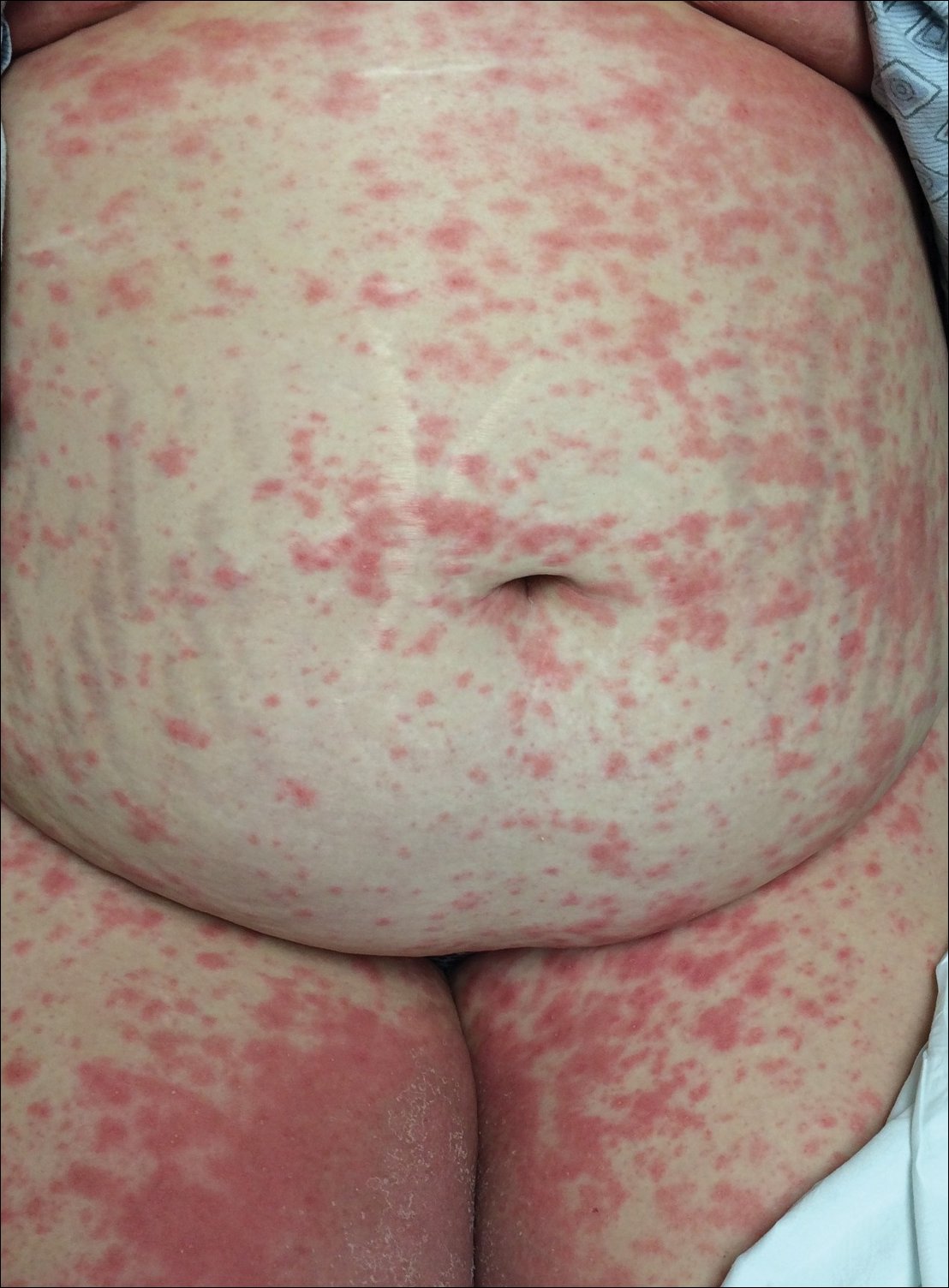
Biopsy specimens from the left arm revealed discrete subcorneal pustules with mild acanthosis of the epidermis with spongiosis (Figure 2). The papillary dermis showed a sparse infiltrate of neutrophils with many marginated neutrophils within vessels. Direct immunofluorescence was negative for human IgG, IgA, IgM, complement component 3, and fibrinogen. Laboratory workup revealed leukocytosis of 21.5×109/L (reference range, 4.5–11.0×109/L) with neutrophilic predominance of 73.6% (reference range, 56%), an elevated erythrocyte sedimentation rate (ESR) of 40 mm/h (reference range, 0–20 mm/h), and a mild hypocalcemia of 8.6 mg/dL (reference range, 8.2–10.2 mg/dL). The patient was started on methylprednisone 40 mg once daily with a plan to taper the dose by 8 mg every 5 days.
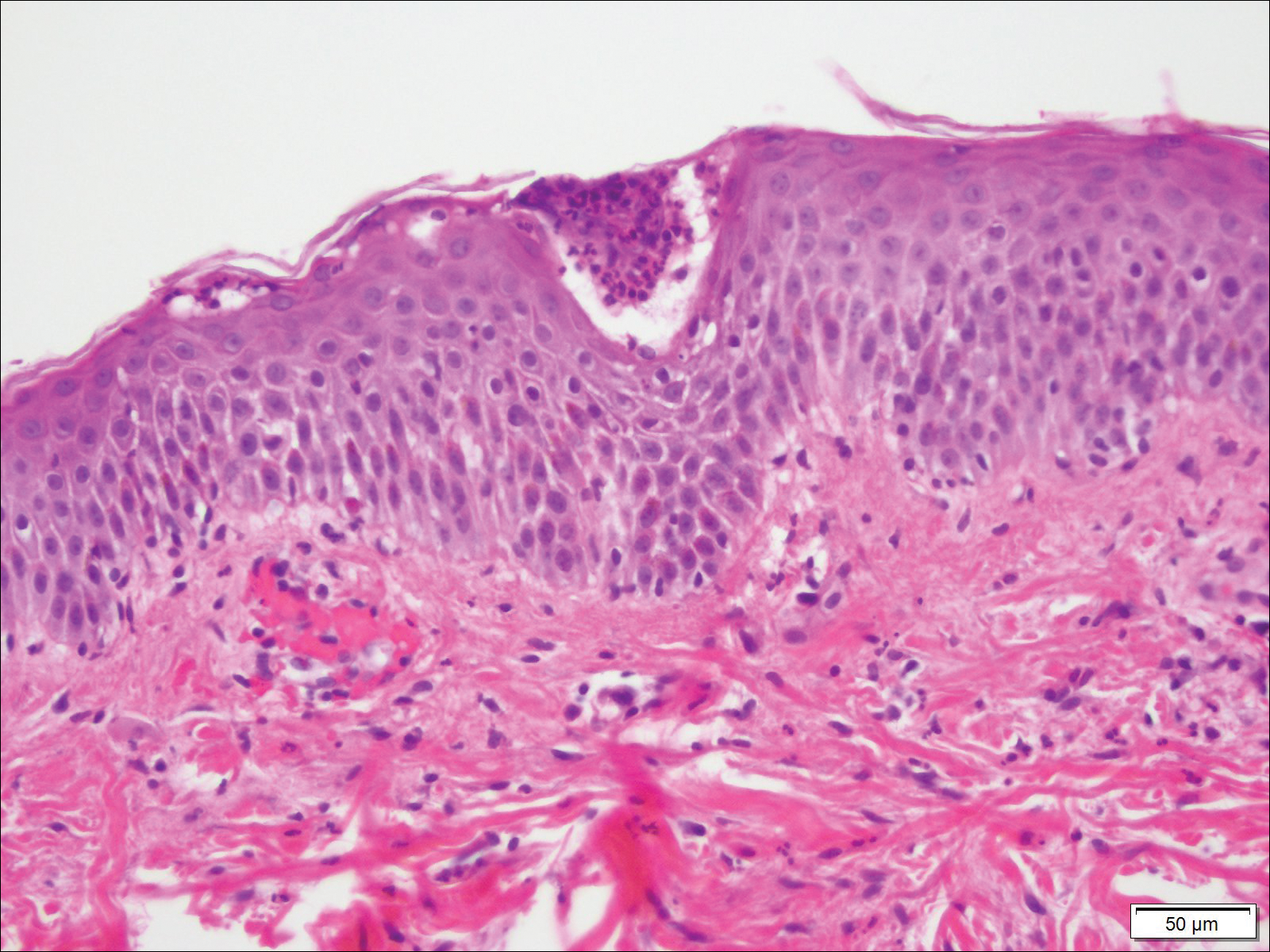
At 35 weeks’ gestation, the patient continued to report pruritus and burning in the areas where the rash had developed. The morphology of the rash had changed considerably, as she now had prominent, annular, pink plaques with central clearing, trailing scaling, and a border of subtle pustules on the legs. There also were rings of desquamative scaling on the palms. During follow-up at 37 weeks’ gestation, the back, chest, and abdomen were improved from the initial presentation, and annular pink plaques with central clearing were noted on the legs (Figure 3). Given the clinical and histopathologic findings, a diagnosis of PPP was made. It was recommended that she undergo increased fetal surveillance with close obstetric follow-up. Weekly office visits with obstetrics and twice-weekly Doppler ultrasounds and fetal nonstress tests were deemed appropriate management. The patient was scheduled for induction at 39 weeks’ gestation given the risk for potential harm to the fetus. She was maintained on low-dose methylprednisolone 4 mg once daily for the duration of the pregnancy. The patient continued to have gradual improvement of the rash at the low treatment dose.
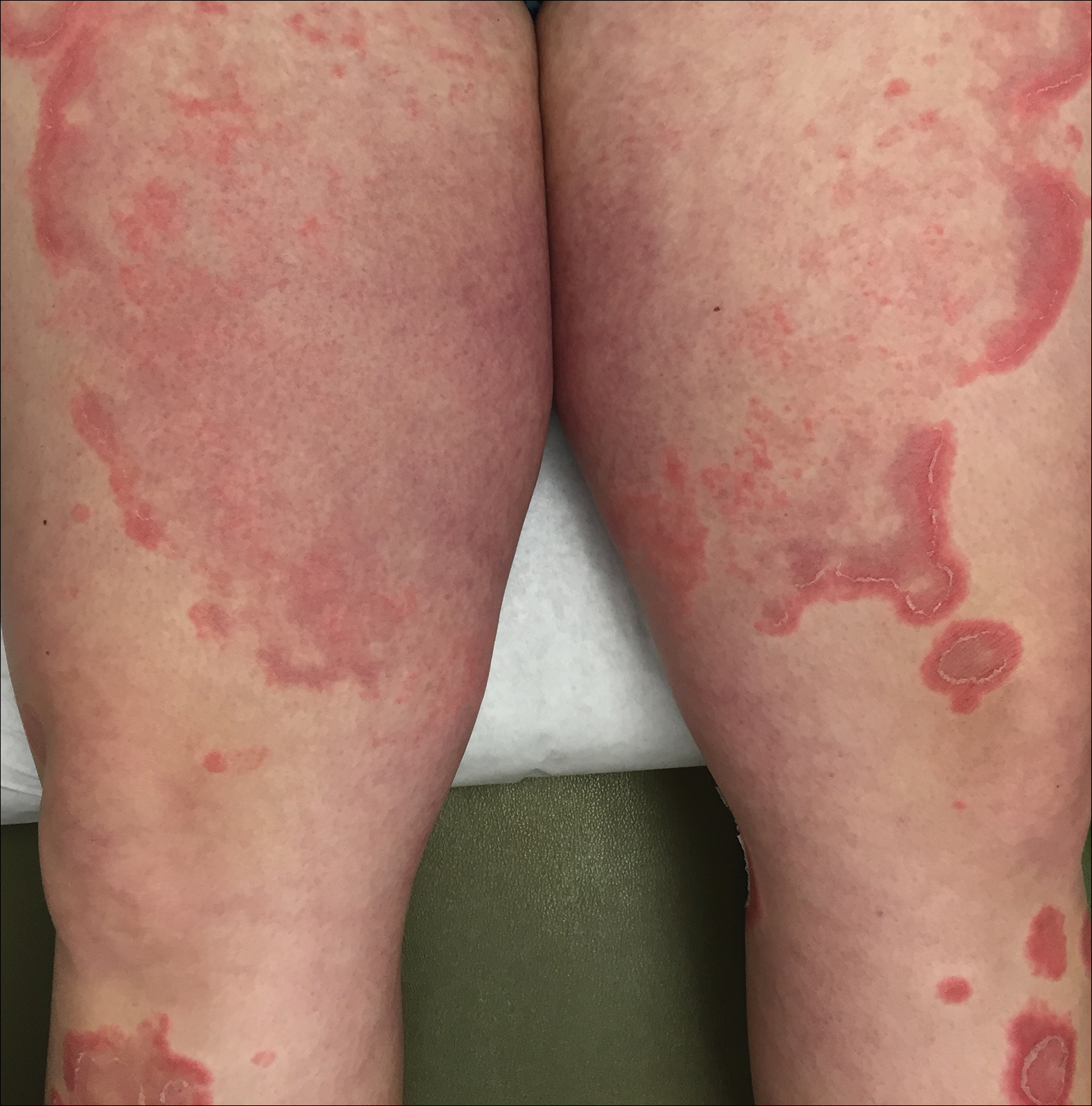
Following induction at 39 weeks’ gestation, the patient vaginally delivered a healthy, 6-lb male neonate at an outside hospital. She reported that the burning sensation improved within hours of delivery, and systemic steroids were stopped after delivery. At a follow-up visit 3 weeks postpartum, considerable improvement of the rash was noted with no evidence of pustules. Fading pink patches with a superficial scaling were noted on the back, chest, abdomen, arms, legs (Figure 4), and fingertips. The patient was counseled that PPP could recur in subsequent pregnancies and that she should be aware of the potential risks to the fetus.
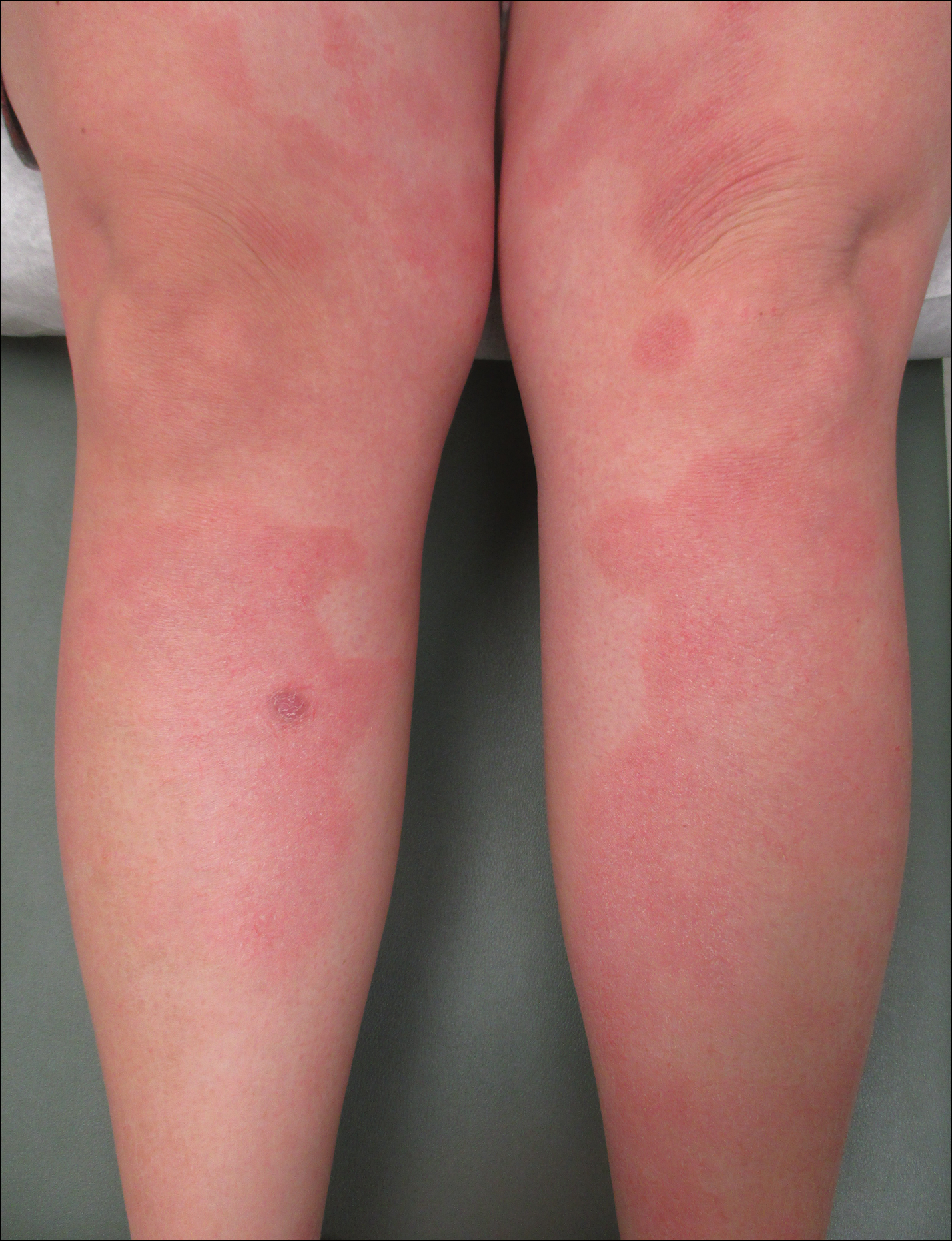
Comment
In our patient, the diagnosis of PPP was supported by the presence of erythematous, coalescent plaques with small pustules at the margins and central erosions as well as the histologic findings of subcorneal pustules with mild acanthosis of the epidermis with spongiosis and a sparse neutrophilic infiltrate into the dermis.
The typical presentation of PPP is characterized by lesions that initially develop in skin folds with centrifugal spread.3 The lesions usually begin as erythematous plaques with a pustular ring with a central erosion. The face, palms, and soles of the feet typically are spared with occasional involvement of oral and esophageal mucosae. Biopsy findings typically include spongiform pustules with neutrophil invasion into the epidermis. Typical laboratory findings include electrolyte derangements with elevated ESR and leukocytosis.1
Diagnosis of PPP is critical given the potential for associated fetal morbidity and mortality.4 Anticipatory guidance for the patient also is necessary, as PPP can recur with subsequent pregnancies or even use of oral contraceptive pills (OCPs). Notably, a patient with recurrences of PPP with each of 9 pregnancies also experienced a recurrence when taking a combination estrogen/progesterone OCP, but not with an estrogen-only diethylstilbestrol OCP.5 Although the pathophysiology is not entirely understood, the development of PPP is thought to be related to the hormonal changes that occur in the third trimester, most notably due to elevated progesterone levels.2 The presence of progesterone in OCPs and recurrences associated with their use supports this altered hormonal state, contributing to the underlying pathophysiology of PPP.
Pustular psoriasis of pregnancy can occur in women without any personal or family history of psoriasis, and as such, it is unclear whether PPP is a separate entity or a hormonally induced variation of generalized pustular psoriasis. Recent evidence included reports of women with PPP who had a mutation in the IL-36 receptor antagonist, leading to a relative abundance of IL-36 inflammatory cytokines.6
The mainstay of treatment for PPP is oral corticosteroids. Cases of PPP that are unresponsive to systemic steroids have been documented, requiring treatment with cyclosporine.9 Antitumor necrosis factors also have been used safely during pregnancy.10 Narrowband UVB phototherapy also has been proposed as a treatment alternative for patients who do not respond to oral corticosteroids.11
Conclusion
Pustular psoriasis of pregnancy is a rare dermatosis of pregnancy that, unlike most other common dermatoses of pregnancy, is associated with adverse fetal outcomes. Diagnosis and management of PPP are critical to ensure the best care and outcomes for the patient and fetus and for a successful delivery of a healthy neonate. Our patient with PPP presented with involvement of the body, palms, and oral mucosa in the absence of systemic symptoms. Close follow-up and comanagement with the patient’s obstetrician ensured safe outcomes for the patient and the neonate.
- Lehrhoff S, Pomeranz MK. Specific dermatoses of pregnancy and their treatment. Dermatol Ther. 2013;26:274-284.
- Kar S, Krishnan A, Shivkumar PV. Pregnancy and skin [published online August 28, 2012]. J Obstet Gynaecol India. 2012;62:268-275.
- Kondo RN, Araújo FM, Pereira AM, et al. Pustular psoriasis of pregnancy (impetigo herpetiformis)—case report. An Bras Dermatol. 2013;88(6 suppl 1):186-189.
- Oumeish OY, Parish JL. Impetigo herpetiformis. Clin Dermatol. 2006;24:101-104.
- Oumeish OY, Farraj SE, Bataineh AS. Some aspects of impetigo herpetiformis. Arch Dermatol. 1982;118:103-105.
- Sugiura K, Oiso N, Iinuma S, et al. IL36RN mutations underlie impetigo herpetiformis. J Invest Dermatol. 2014;134:2472-2474.
- Sugiura K. The genetic background of generalized pustular psoriasis: IL36RN mutations and CARD14 gain-of-function variants [published online March 5, 2014]. J Dermatol Sci. 2014;74:187-192.
- Li X, Chen M, Fu X, et al. Mutation analysis of the IL36RN gene in Chinese patients with generalized pustular psoriasis with/without psoriasis vulgaris. J Dermatol Sci. 2014;76:132-138.
- Hazarika D. Generalized pustular psoriasis of pregnancy successfully treated with cyclosporine. Indian J Dermatol Venereol Leprol. 2009;75:638.
- Puig L, Barco D, Alomar A. Treatment of psoriasis with anti-TNF drugs during pregnancy: case report and review of the literature. Dermatology. 2010;220:71-76.
- Bozdag K, Ozturk S, Ermete M. A case of recurrent impetigo herpetiformis treated with systemic corticosteroids and narrowband UVB [published online January 20, 2012]. Cutan Ocul Toxicol. 2012;31:67-69.
Pustular psoriasis of pregnancy (PPP), also known as impetigo herpetiformis, is a relatively rare cutaneous disorder of pregnancy wherein lesions typically appear in the third trimester and resolve after delivery; however, lesions may persist through the postpartum period. Pustular psoriasis of pregnancy may be considered a fifth dermatosis of pregnancy, alongside the classic dermatoses of atopic eruption of pregnancy, intrahepatic cholestasis of pregnancy, pemphigoid gestationis, and pruritic urticarial papules and plaques of pregnancy.1
As PPP is a rare disease, its effects on maternal-fetal health outcomes and management remain to be elucidated. Though maternal mortality is rare in PPP, it is a unique dermatosis of pregnancy because it may be associated with severe systemic maternal symptoms.2 Fetal morbidity and mortality are less predictable in PPP, with reported cases of stillbirth, fetal anomalies, and neonatal death thought to be due largely to placental insufficiency, even with control of symptoms.1,3 Given the risk of serious harm to the fetus, reporting of cases and discussion of PPP management is critical.
Case Report
An otherwise healthy 29-year-old G2P1 woman at 32 weeks’ gestation presented to our emergency department with a 1-week history of a pruritic, burning rash that started on the thighs then spread diffusely. She denied any similar rash in her prior pregnancy. She was not currently taking any medications except for prenatal vitamins and denied any systemic symptoms. The patient’s obstetrician initiated treatment with methylprednisolone 50 mg once daily for the rash 3 days prior to the current presentation, which had not seemed to help. On physical examination, edematous pink plaques studded with 1- to 2-mm collarettes of scaling and sparse 1-mm pustules involving the arms, chest, abdomen, back, groin, buttocks, and legs were noted. The plaques on the back and inner thighs had a peripheral rim of desquamative scaling. There were pink macules on the palms, and superficial desquamation was noted on the lips. The oral mucosa was otherwise spared (Figure 1).
Biopsy specimens from the left arm revealed discrete subcorneal pustules with mild acanthosis of the epidermis with spongiosis (Figure 2). The papillary dermis showed a sparse infiltrate of neutrophils with many marginated neutrophils within vessels. Direct immunofluorescence was negative for human IgG, IgA, IgM, complement component 3, and fibrinogen. Laboratory workup revealed leukocytosis of 21.5×109/L (reference range, 4.5–11.0×109/L) with neutrophilic predominance of 73.6% (reference range, 56%), an elevated erythrocyte sedimentation rate (ESR) of 40 mm/h (reference range, 0–20 mm/h), and a mild hypocalcemia of 8.6 mg/dL (reference range, 8.2–10.2 mg/dL). The patient was started on methylprednisone 40 mg once daily with a plan to taper the dose by 8 mg every 5 days.
At 35 weeks’ gestation, the patient continued to report pruritus and burning in the areas where the rash had developed. The morphology of the rash had changed considerably, as she now had prominent, annular, pink plaques with central clearing, trailing scaling, and a border of subtle pustules on the legs. There also were rings of desquamative scaling on the palms. During follow-up at 37 weeks’ gestation, the back, chest, and abdomen were improved from the initial presentation, and annular pink plaques with central clearing were noted on the legs (Figure 3). Given the clinical and histopathologic findings, a diagnosis of PPP was made. It was recommended that she undergo increased fetal surveillance with close obstetric follow-up. Weekly office visits with obstetrics and twice-weekly Doppler ultrasounds and fetal nonstress tests were deemed appropriate management. The patient was scheduled for induction at 39 weeks’ gestation given the risk for potential harm to the fetus. She was maintained on low-dose methylprednisolone 4 mg once daily for the duration of the pregnancy. The patient continued to have gradual improvement of the rash at the low treatment dose.
Following induction at 39 weeks’ gestation, the patient vaginally delivered a healthy, 6-lb male neonate at an outside hospital. She reported that the burning sensation improved within hours of delivery, and systemic steroids were stopped after delivery. At a follow-up visit 3 weeks postpartum, considerable improvement of the rash was noted with no evidence of pustules. Fading pink patches with a superficial scaling were noted on the back, chest, abdomen, arms, legs (Figure 4), and fingertips. The patient was counseled that PPP could recur in subsequent pregnancies and that she should be aware of the potential risks to the fetus.
Comment
In our patient, the diagnosis of PPP was supported by the presence of erythematous, coalescent plaques with small pustules at the margins and central erosions as well as the histologic findings of subcorneal pustules with mild acanthosis of the epidermis with spongiosis and a sparse neutrophilic infiltrate into the dermis.
The typical presentation of PPP is characterized by lesions that initially develop in skin folds with centrifugal spread.3 The lesions usually begin as erythematous plaques with a pustular ring with a central erosion. The face, palms, and soles of the feet typically are spared with occasional involvement of oral and esophageal mucosae. Biopsy findings typically include spongiform pustules with neutrophil invasion into the epidermis. Typical laboratory findings include electrolyte derangements with elevated ESR and leukocytosis.1
Diagnosis of PPP is critical given the potential for associated fetal morbidity and mortality.4 Anticipatory guidance for the patient also is necessary, as PPP can recur with subsequent pregnancies or even use of oral contraceptive pills (OCPs). Notably, a patient with recurrences of PPP with each of 9 pregnancies also experienced a recurrence when taking a combination estrogen/progesterone OCP, but not with an estrogen-only diethylstilbestrol OCP.5 Although the pathophysiology is not entirely understood, the development of PPP is thought to be related to the hormonal changes that occur in the third trimester, most notably due to elevated progesterone levels.2 The presence of progesterone in OCPs and recurrences associated with their use supports this altered hormonal state, contributing to the underlying pathophysiology of PPP.
Pustular psoriasis of pregnancy can occur in women without any personal or family history of psoriasis, and as such, it is unclear whether PPP is a separate entity or a hormonally induced variation of generalized pustular psoriasis. Recent evidence included reports of women with PPP who had a mutation in the IL-36 receptor antagonist, leading to a relative abundance of IL-36 inflammatory cytokines.6
The mainstay of treatment for PPP is oral corticosteroids. Cases of PPP that are unresponsive to systemic steroids have been documented, requiring treatment with cyclosporine.9 Antitumor necrosis factors also have been used safely during pregnancy.10 Narrowband UVB phototherapy also has been proposed as a treatment alternative for patients who do not respond to oral corticosteroids.11
Conclusion
Pustular psoriasis of pregnancy is a rare dermatosis of pregnancy that, unlike most other common dermatoses of pregnancy, is associated with adverse fetal outcomes. Diagnosis and management of PPP are critical to ensure the best care and outcomes for the patient and fetus and for a successful delivery of a healthy neonate. Our patient with PPP presented with involvement of the body, palms, and oral mucosa in the absence of systemic symptoms. Close follow-up and comanagement with the patient’s obstetrician ensured safe outcomes for the patient and the neonate.
Pustular psoriasis of pregnancy (PPP), also known as impetigo herpetiformis, is a relatively rare cutaneous disorder of pregnancy wherein lesions typically appear in the third trimester and resolve after delivery; however, lesions may persist through the postpartum period. Pustular psoriasis of pregnancy may be considered a fifth dermatosis of pregnancy, alongside the classic dermatoses of atopic eruption of pregnancy, intrahepatic cholestasis of pregnancy, pemphigoid gestationis, and pruritic urticarial papules and plaques of pregnancy.1
As PPP is a rare disease, its effects on maternal-fetal health outcomes and management remain to be elucidated. Though maternal mortality is rare in PPP, it is a unique dermatosis of pregnancy because it may be associated with severe systemic maternal symptoms.2 Fetal morbidity and mortality are less predictable in PPP, with reported cases of stillbirth, fetal anomalies, and neonatal death thought to be due largely to placental insufficiency, even with control of symptoms.1,3 Given the risk of serious harm to the fetus, reporting of cases and discussion of PPP management is critical.
Case Report
An otherwise healthy 29-year-old G2P1 woman at 32 weeks’ gestation presented to our emergency department with a 1-week history of a pruritic, burning rash that started on the thighs then spread diffusely. She denied any similar rash in her prior pregnancy. She was not currently taking any medications except for prenatal vitamins and denied any systemic symptoms. The patient’s obstetrician initiated treatment with methylprednisolone 50 mg once daily for the rash 3 days prior to the current presentation, which had not seemed to help. On physical examination, edematous pink plaques studded with 1- to 2-mm collarettes of scaling and sparse 1-mm pustules involving the arms, chest, abdomen, back, groin, buttocks, and legs were noted. The plaques on the back and inner thighs had a peripheral rim of desquamative scaling. There were pink macules on the palms, and superficial desquamation was noted on the lips. The oral mucosa was otherwise spared (Figure 1).
Biopsy specimens from the left arm revealed discrete subcorneal pustules with mild acanthosis of the epidermis with spongiosis (Figure 2). The papillary dermis showed a sparse infiltrate of neutrophils with many marginated neutrophils within vessels. Direct immunofluorescence was negative for human IgG, IgA, IgM, complement component 3, and fibrinogen. Laboratory workup revealed leukocytosis of 21.5×109/L (reference range, 4.5–11.0×109/L) with neutrophilic predominance of 73.6% (reference range, 56%), an elevated erythrocyte sedimentation rate (ESR) of 40 mm/h (reference range, 0–20 mm/h), and a mild hypocalcemia of 8.6 mg/dL (reference range, 8.2–10.2 mg/dL). The patient was started on methylprednisone 40 mg once daily with a plan to taper the dose by 8 mg every 5 days.
At 35 weeks’ gestation, the patient continued to report pruritus and burning in the areas where the rash had developed. The morphology of the rash had changed considerably, as she now had prominent, annular, pink plaques with central clearing, trailing scaling, and a border of subtle pustules on the legs. There also were rings of desquamative scaling on the palms. During follow-up at 37 weeks’ gestation, the back, chest, and abdomen were improved from the initial presentation, and annular pink plaques with central clearing were noted on the legs (Figure 3). Given the clinical and histopathologic findings, a diagnosis of PPP was made. It was recommended that she undergo increased fetal surveillance with close obstetric follow-up. Weekly office visits with obstetrics and twice-weekly Doppler ultrasounds and fetal nonstress tests were deemed appropriate management. The patient was scheduled for induction at 39 weeks’ gestation given the risk for potential harm to the fetus. She was maintained on low-dose methylprednisolone 4 mg once daily for the duration of the pregnancy. The patient continued to have gradual improvement of the rash at the low treatment dose.
Following induction at 39 weeks’ gestation, the patient vaginally delivered a healthy, 6-lb male neonate at an outside hospital. She reported that the burning sensation improved within hours of delivery, and systemic steroids were stopped after delivery. At a follow-up visit 3 weeks postpartum, considerable improvement of the rash was noted with no evidence of pustules. Fading pink patches with a superficial scaling were noted on the back, chest, abdomen, arms, legs (Figure 4), and fingertips. The patient was counseled that PPP could recur in subsequent pregnancies and that she should be aware of the potential risks to the fetus.
Comment
In our patient, the diagnosis of PPP was supported by the presence of erythematous, coalescent plaques with small pustules at the margins and central erosions as well as the histologic findings of subcorneal pustules with mild acanthosis of the epidermis with spongiosis and a sparse neutrophilic infiltrate into the dermis.
The typical presentation of PPP is characterized by lesions that initially develop in skin folds with centrifugal spread.3 The lesions usually begin as erythematous plaques with a pustular ring with a central erosion. The face, palms, and soles of the feet typically are spared with occasional involvement of oral and esophageal mucosae. Biopsy findings typically include spongiform pustules with neutrophil invasion into the epidermis. Typical laboratory findings include electrolyte derangements with elevated ESR and leukocytosis.1
Diagnosis of PPP is critical given the potential for associated fetal morbidity and mortality.4 Anticipatory guidance for the patient also is necessary, as PPP can recur with subsequent pregnancies or even use of oral contraceptive pills (OCPs). Notably, a patient with recurrences of PPP with each of 9 pregnancies also experienced a recurrence when taking a combination estrogen/progesterone OCP, but not with an estrogen-only diethylstilbestrol OCP.5 Although the pathophysiology is not entirely understood, the development of PPP is thought to be related to the hormonal changes that occur in the third trimester, most notably due to elevated progesterone levels.2 The presence of progesterone in OCPs and recurrences associated with their use supports this altered hormonal state, contributing to the underlying pathophysiology of PPP.
Pustular psoriasis of pregnancy can occur in women without any personal or family history of psoriasis, and as such, it is unclear whether PPP is a separate entity or a hormonally induced variation of generalized pustular psoriasis. Recent evidence included reports of women with PPP who had a mutation in the IL-36 receptor antagonist, leading to a relative abundance of IL-36 inflammatory cytokines.6
The mainstay of treatment for PPP is oral corticosteroids. Cases of PPP that are unresponsive to systemic steroids have been documented, requiring treatment with cyclosporine.9 Antitumor necrosis factors also have been used safely during pregnancy.10 Narrowband UVB phototherapy also has been proposed as a treatment alternative for patients who do not respond to oral corticosteroids.11
Conclusion
Pustular psoriasis of pregnancy is a rare dermatosis of pregnancy that, unlike most other common dermatoses of pregnancy, is associated with adverse fetal outcomes. Diagnosis and management of PPP are critical to ensure the best care and outcomes for the patient and fetus and for a successful delivery of a healthy neonate. Our patient with PPP presented with involvement of the body, palms, and oral mucosa in the absence of systemic symptoms. Close follow-up and comanagement with the patient’s obstetrician ensured safe outcomes for the patient and the neonate.
- Lehrhoff S, Pomeranz MK. Specific dermatoses of pregnancy and their treatment. Dermatol Ther. 2013;26:274-284.
- Kar S, Krishnan A, Shivkumar PV. Pregnancy and skin [published online August 28, 2012]. J Obstet Gynaecol India. 2012;62:268-275.
- Kondo RN, Araújo FM, Pereira AM, et al. Pustular psoriasis of pregnancy (impetigo herpetiformis)—case report. An Bras Dermatol. 2013;88(6 suppl 1):186-189.
- Oumeish OY, Parish JL. Impetigo herpetiformis. Clin Dermatol. 2006;24:101-104.
- Oumeish OY, Farraj SE, Bataineh AS. Some aspects of impetigo herpetiformis. Arch Dermatol. 1982;118:103-105.
- Sugiura K, Oiso N, Iinuma S, et al. IL36RN mutations underlie impetigo herpetiformis. J Invest Dermatol. 2014;134:2472-2474.
- Sugiura K. The genetic background of generalized pustular psoriasis: IL36RN mutations and CARD14 gain-of-function variants [published online March 5, 2014]. J Dermatol Sci. 2014;74:187-192.
- Li X, Chen M, Fu X, et al. Mutation analysis of the IL36RN gene in Chinese patients with generalized pustular psoriasis with/without psoriasis vulgaris. J Dermatol Sci. 2014;76:132-138.
- Hazarika D. Generalized pustular psoriasis of pregnancy successfully treated with cyclosporine. Indian J Dermatol Venereol Leprol. 2009;75:638.
- Puig L, Barco D, Alomar A. Treatment of psoriasis with anti-TNF drugs during pregnancy: case report and review of the literature. Dermatology. 2010;220:71-76.
- Bozdag K, Ozturk S, Ermete M. A case of recurrent impetigo herpetiformis treated with systemic corticosteroids and narrowband UVB [published online January 20, 2012]. Cutan Ocul Toxicol. 2012;31:67-69.
- Lehrhoff S, Pomeranz MK. Specific dermatoses of pregnancy and their treatment. Dermatol Ther. 2013;26:274-284.
- Kar S, Krishnan A, Shivkumar PV. Pregnancy and skin [published online August 28, 2012]. J Obstet Gynaecol India. 2012;62:268-275.
- Kondo RN, Araújo FM, Pereira AM, et al. Pustular psoriasis of pregnancy (impetigo herpetiformis)—case report. An Bras Dermatol. 2013;88(6 suppl 1):186-189.
- Oumeish OY, Parish JL. Impetigo herpetiformis. Clin Dermatol. 2006;24:101-104.
- Oumeish OY, Farraj SE, Bataineh AS. Some aspects of impetigo herpetiformis. Arch Dermatol. 1982;118:103-105.
- Sugiura K, Oiso N, Iinuma S, et al. IL36RN mutations underlie impetigo herpetiformis. J Invest Dermatol. 2014;134:2472-2474.
- Sugiura K. The genetic background of generalized pustular psoriasis: IL36RN mutations and CARD14 gain-of-function variants [published online March 5, 2014]. J Dermatol Sci. 2014;74:187-192.
- Li X, Chen M, Fu X, et al. Mutation analysis of the IL36RN gene in Chinese patients with generalized pustular psoriasis with/without psoriasis vulgaris. J Dermatol Sci. 2014;76:132-138.
- Hazarika D. Generalized pustular psoriasis of pregnancy successfully treated with cyclosporine. Indian J Dermatol Venereol Leprol. 2009;75:638.
- Puig L, Barco D, Alomar A. Treatment of psoriasis with anti-TNF drugs during pregnancy: case report and review of the literature. Dermatology. 2010;220:71-76.
- Bozdag K, Ozturk S, Ermete M. A case of recurrent impetigo herpetiformis treated with systemic corticosteroids and narrowband UVB [published online January 20, 2012]. Cutan Ocul Toxicol. 2012;31:67-69.
Practice Points
- Given its association with maternal and fetal morbidity/mortality, it is important for physicians to have a high suspicion for pustular psoriasis of pregnancy (PPP) in pregnant women with widespread cutaneous eruptions.
- Oral corticosteroids and close involvement of obstetric care is the mainstay of treatment for PPP.
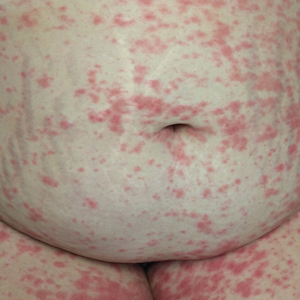
Bilateral thigh and knee pain • leg weakness • no history of trauma • Dx?
THE CASE
A 67-year-old woman presented to our orthopaedic clinic with a 2-year history of bilateral thigh and knee pain and weakness of her legs. She had no history of trauma, and the pain, which was localized to the distal anterior thighs and patellofemoral area, was 7/10 at rest and worse with standing and walking.
Her medical history was significant for osteoporosis (diagnosed in 2004), hypertension, hypothyroidism, gastroesophageal reflux disease, and menopause (age 54). Her original dual-energy x-ray absorptiometry (DEXA) scan did not reveal the presence of any previous fractures. She was started on calcium and vitamin D supplementation and oral alendronate (70 mg once a week). She took alendronate for 4 years until 2008, when it was stopped due to nausea. She was then started on zoledronic acid (5 mg IV annually). She received 5 infusions of zoledronic acid between 2008 and 2013; she did not have an infusion in 2012. Her medication list also included lisinopril, omeprazole, naproxen, cyclobenzaprine, and a multivitamin. She had normal renal function (estimated glomerular filtration rate >60 mL/min/1.73 m2) and she did not drink alcohol or use tobacco.
In the 2 years prior to her visit to our clinic, she had been evaluated by her primary care provider, an orthopedic sports medicine specialist, 2 spinal surgeons, and a physiatrist. She had also undergone 30 physical therapy sessions. Bilateral femur radiographs (FIGURE 1) ordered by her orthopedist 6 months earlier demonstrated no evidence of fracture, but did show an incidental enchondroma in the right distal diaphysis and bilateral thickening of the lateral femoral cortices.
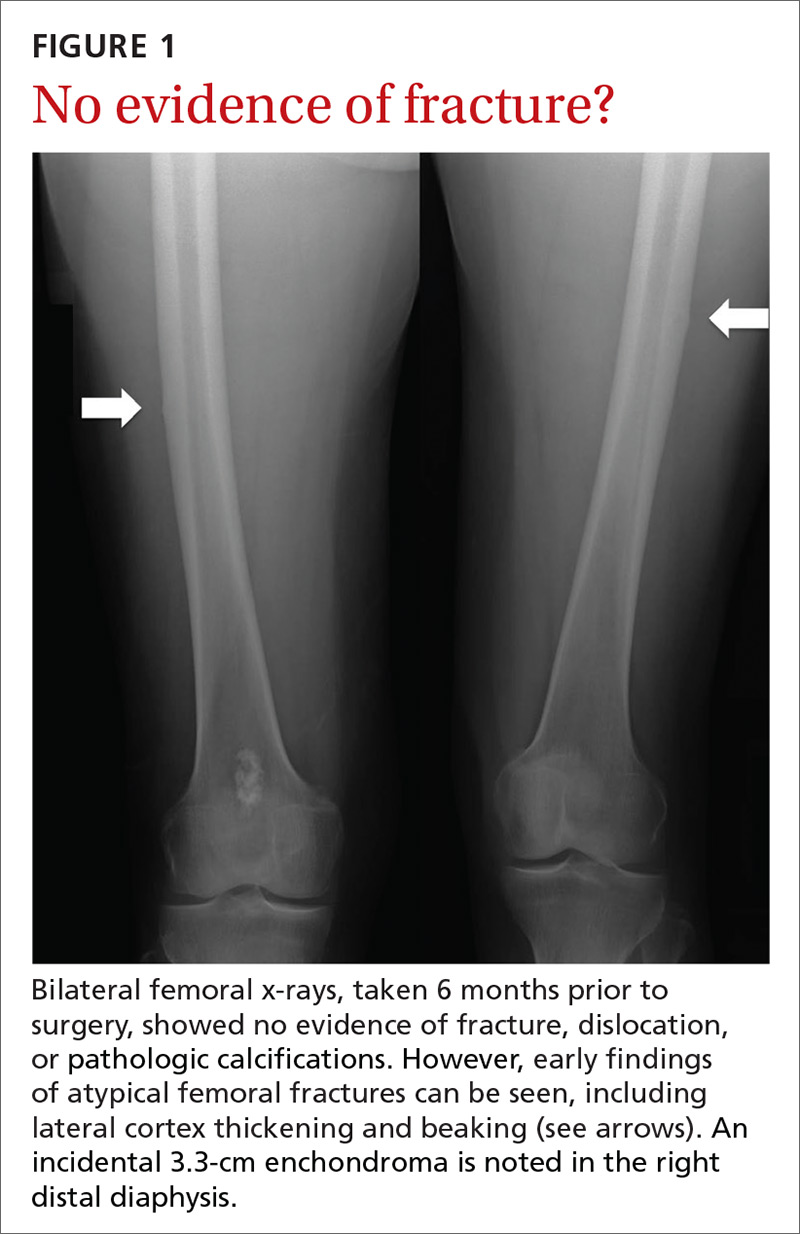
Finally, with no relief in sight, her obstetrician suggested that she might be experiencing myalgias attributable to her zoledronic acid infusions. She was subsequently referred to us.
The physical exam revealed a thin female with a body mass index of 21. She had mild tenderness on palpation of the bilateral anterior thighs and knees. There was no pain with hip or knee range of motion and minimal pain in the bilateral lower extremities with axial loading. The patient had normal sensation, did not have an antalgic gait, and exhibited 5/5 strength bilaterally in all distributions of the lower extremities.
THE DIAGNOSIS
Due to continued pain despite negative x-rays, we obtained a 3-phase bone scan of the pelvis and bilateral femurs. Delayed images showed moderately increased activity in the mid-right and mid-left lateral femoral diaphyses at the cortex and confirmed stress fractures (FIGURE 2).
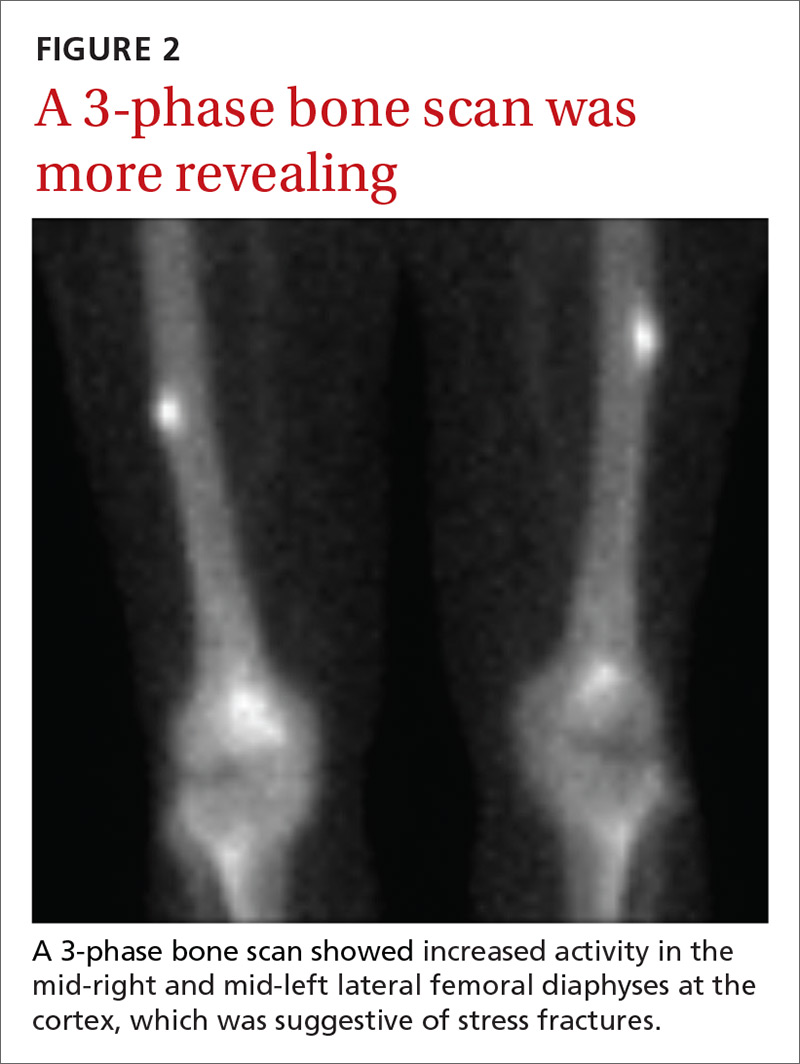
DISCUSSION
Bisphosphonates are considered first-line therapy for osteoporosis, according to current evidence-based guidelines.1 These medications inhibit osteoclast activity and can bind to the bone for more than 10 years.2,3 (In women with bone mineral density scores ≤ –2.5, the number needed to treat is 21.1,4)
Patients taking bisphosphonates, however, are susceptible to atypical femoral fractures (AFFs), which are stress or insufficiency fractures associated with minimal or no trauma.5 The pathophysiology remains unknown at this time, but AFFs may result from changes in bone remodeling that occur when a bone experiences repetitive microtrauma, leading to lateral cortical thickening of the femur.6,7 Incidence of AFFs in patients taking bisphosphonates is estimated to be between 3.2 and 50 cases per 100,000 person-years; however, this risk increases to approximately 100 per 100,000 person-years with long-term use.5 Other risk factors include low body weight, advancing age, rheumatoid arthritis, long-term glucocorticoid therapy, and excessive alcohol and cigarette use.8
What you’ll see
Symptoms typically include unilateral or bilateral prodromal pain with a sharp or achy character that is localized to the mid-thigh, upper thigh, or groin.9 If an AFF is suspected, we recommend performing a bilateral exam and obtaining radiographs.
If characteristic features are found (eg, signs of focal cortical thickening or beaking) and pain arises in the opposite limb, obtain a radiograph of the contralateral femur. If radiographs are negative but suspicion remains, order magnetic resonance imaging or a bone scan, to identify a cortical fracture line, bone and marrow edema, or hyperemia.5
Begin treatment by discontinuing bisphosphonates
Upon identification of an AFF, discontinue bisphosphonates and initiate calcium and vitamin D supplementation.5 Prophylactic surgical fixation may also be necessary to accelerate healing and prevent fracture propagation and further pain.
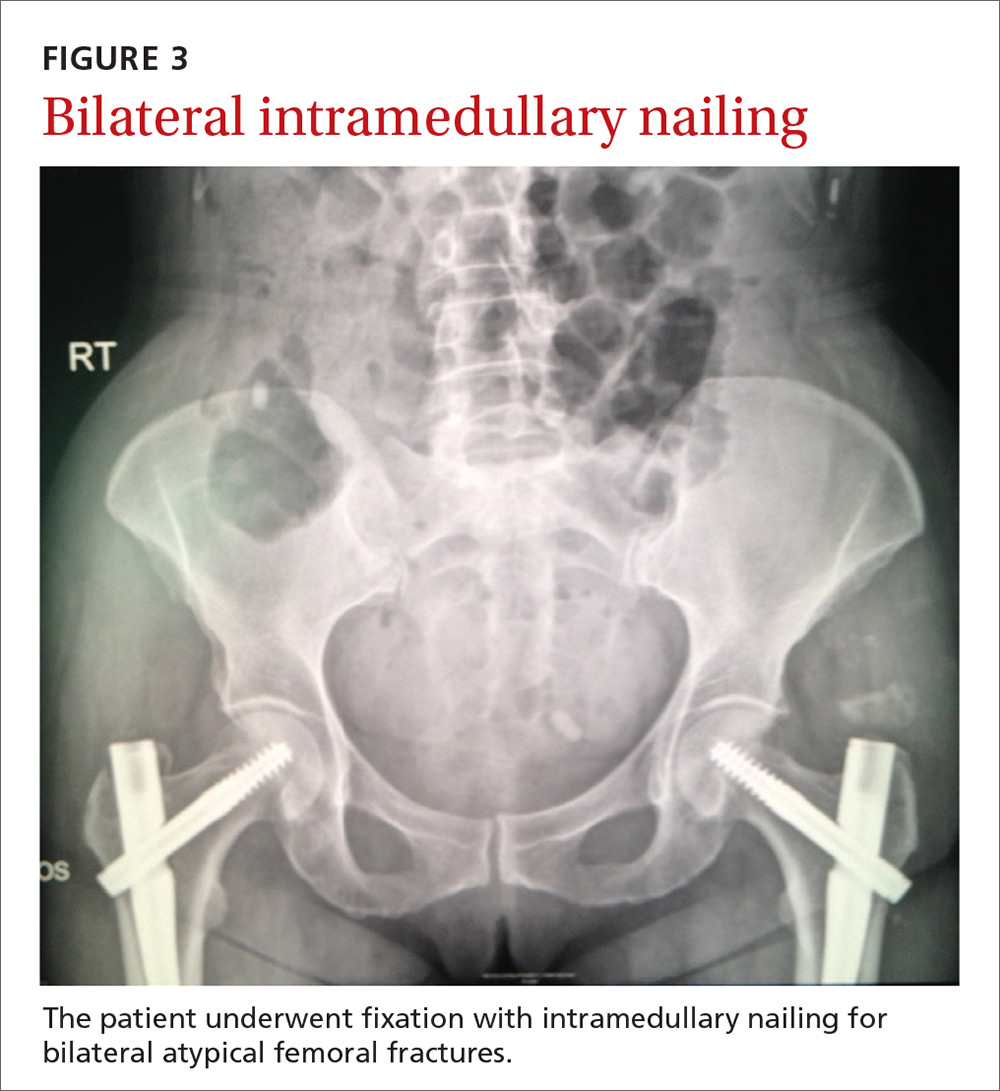
Our patient. Due to the longevity of the symptoms and the bilateral stress fractures noted on the bone scan, our patient chose to proceed with intramedullary nailing of the bilateral femurs (FIGURES 3 and 4). On postop Day 1, she was able to ambulate using a walker and to participate in bilateral weight-bearing (as tolerated). She was discharged to a skilled nursing facility, where she progressed to full weight-bearing without aid. On follow-up (one year postop), the patient reported no residual leg pain and was able to work out 5 days per week. Radiographs of her femurs demonstrated healed fractures and stable position of the intramedullary nails.
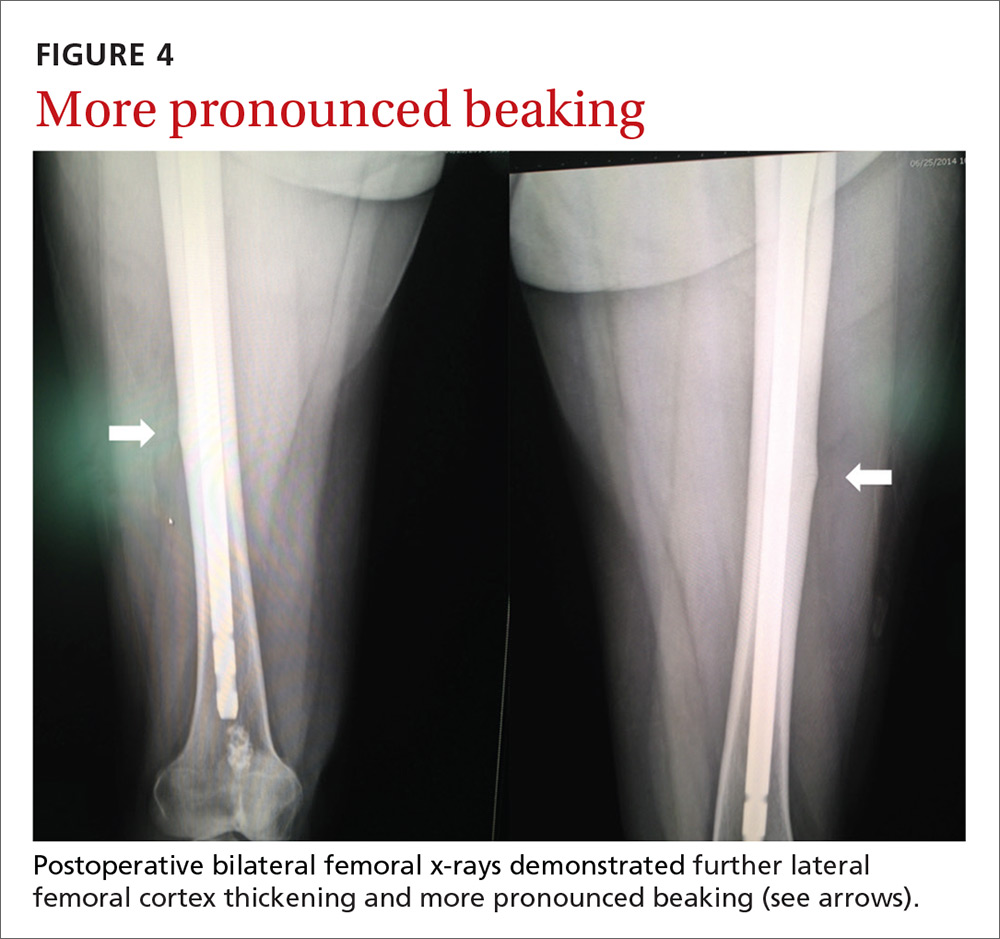
THE TAKEAWAY
An increased suspicion for AFFs due to bisphosphonate use can lead to earlier diagnosis and decreased morbidity for patients. Use of femoral imaging can promote detection and reduce financial burden.
To help prevent AFFs from occurring, we recommend reevaluating the need for continued bisphosphonate therapy after 2 to 5 years of treatment. Continued surveillance is also advisable throughout the duration of their use.
ACKNOWLEDGMENT
The authors wish to acknowledge Dr. Maurice Manring for his help in preparing this manuscript.
1. Watts NB, Bilezikian JP, Camacho PM, et al. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for the diagnosis and treatment of postmenopausal osteoporosis. Endocr Pract. 2010;16 Suppl 3:1-37.
2. Cakmak S, Mahiroğullari M, Keklikci K, et al. Bilateral low-energy sequential femoral shaft fractures in patients on long-term bisphosphonate therapy. Acta Orthop Traumatol Turc. 2013;47:162-172.
3. Drake MT, Clarke BL, Khosla S. Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clin Proc. 2008;83:1032-1045.
4. Black DM, Bauer DC, Schwartz AV, et al. Continuing bisphosphonate treatment for osteoporosis—for whom and for how long? N Engl J Med. 2012;366:2051-2053.
5. Shane E, Burr D, Abrahamsen B, et al. Atypical subtrochanteric and diaphyseal femoral fractures: second report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2014;29:1-23.
6. Allen MR. Recent advances in understanding bisphosphonate effects on bone mechanical properties. Curr Osteoporos Rep. 2018 Mar 1. [Epub ahead of print]
7. Hagino H, Endo N, Yamamoto T, et al. Treatment status and radiographic features of patients with atypical femoral fractures. J Orthop Sci. 2018;23:316-320.
8. Kanis JA, Borgstrom F, De Laet C, et al. Assessment of fracture risk. Osteoporos Int. 2005;16:581-589.
9. Giusti A, Hamdy NA, Papapoulos SE. Atypical fractures of the femur and bisphosphonate therapy: a systematic review of case/case series studies. Bone. 2010;47:169-180.
THE CASE
A 67-year-old woman presented to our orthopaedic clinic with a 2-year history of bilateral thigh and knee pain and weakness of her legs. She had no history of trauma, and the pain, which was localized to the distal anterior thighs and patellofemoral area, was 7/10 at rest and worse with standing and walking.
Her medical history was significant for osteoporosis (diagnosed in 2004), hypertension, hypothyroidism, gastroesophageal reflux disease, and menopause (age 54). Her original dual-energy x-ray absorptiometry (DEXA) scan did not reveal the presence of any previous fractures. She was started on calcium and vitamin D supplementation and oral alendronate (70 mg once a week). She took alendronate for 4 years until 2008, when it was stopped due to nausea. She was then started on zoledronic acid (5 mg IV annually). She received 5 infusions of zoledronic acid between 2008 and 2013; she did not have an infusion in 2012. Her medication list also included lisinopril, omeprazole, naproxen, cyclobenzaprine, and a multivitamin. She had normal renal function (estimated glomerular filtration rate >60 mL/min/1.73 m2) and she did not drink alcohol or use tobacco.
In the 2 years prior to her visit to our clinic, she had been evaluated by her primary care provider, an orthopedic sports medicine specialist, 2 spinal surgeons, and a physiatrist. She had also undergone 30 physical therapy sessions. Bilateral femur radiographs (FIGURE 1) ordered by her orthopedist 6 months earlier demonstrated no evidence of fracture, but did show an incidental enchondroma in the right distal diaphysis and bilateral thickening of the lateral femoral cortices.
Finally, with no relief in sight, her obstetrician suggested that she might be experiencing myalgias attributable to her zoledronic acid infusions. She was subsequently referred to us.
The physical exam revealed a thin female with a body mass index of 21. She had mild tenderness on palpation of the bilateral anterior thighs and knees. There was no pain with hip or knee range of motion and minimal pain in the bilateral lower extremities with axial loading. The patient had normal sensation, did not have an antalgic gait, and exhibited 5/5 strength bilaterally in all distributions of the lower extremities.
THE DIAGNOSIS
Due to continued pain despite negative x-rays, we obtained a 3-phase bone scan of the pelvis and bilateral femurs. Delayed images showed moderately increased activity in the mid-right and mid-left lateral femoral diaphyses at the cortex and confirmed stress fractures (FIGURE 2).
DISCUSSION
Bisphosphonates are considered first-line therapy for osteoporosis, according to current evidence-based guidelines.1 These medications inhibit osteoclast activity and can bind to the bone for more than 10 years.2,3 (In women with bone mineral density scores ≤ –2.5, the number needed to treat is 21.1,4)
Patients taking bisphosphonates, however, are susceptible to atypical femoral fractures (AFFs), which are stress or insufficiency fractures associated with minimal or no trauma.5 The pathophysiology remains unknown at this time, but AFFs may result from changes in bone remodeling that occur when a bone experiences repetitive microtrauma, leading to lateral cortical thickening of the femur.6,7 Incidence of AFFs in patients taking bisphosphonates is estimated to be between 3.2 and 50 cases per 100,000 person-years; however, this risk increases to approximately 100 per 100,000 person-years with long-term use.5 Other risk factors include low body weight, advancing age, rheumatoid arthritis, long-term glucocorticoid therapy, and excessive alcohol and cigarette use.8
What you’ll see
Symptoms typically include unilateral or bilateral prodromal pain with a sharp or achy character that is localized to the mid-thigh, upper thigh, or groin.9 If an AFF is suspected, we recommend performing a bilateral exam and obtaining radiographs.
If characteristic features are found (eg, signs of focal cortical thickening or beaking) and pain arises in the opposite limb, obtain a radiograph of the contralateral femur. If radiographs are negative but suspicion remains, order magnetic resonance imaging or a bone scan, to identify a cortical fracture line, bone and marrow edema, or hyperemia.5
Begin treatment by discontinuing bisphosphonates
Upon identification of an AFF, discontinue bisphosphonates and initiate calcium and vitamin D supplementation.5 Prophylactic surgical fixation may also be necessary to accelerate healing and prevent fracture propagation and further pain.
Our patient. Due to the longevity of the symptoms and the bilateral stress fractures noted on the bone scan, our patient chose to proceed with intramedullary nailing of the bilateral femurs (FIGURES 3 and 4). On postop Day 1, she was able to ambulate using a walker and to participate in bilateral weight-bearing (as tolerated). She was discharged to a skilled nursing facility, where she progressed to full weight-bearing without aid. On follow-up (one year postop), the patient reported no residual leg pain and was able to work out 5 days per week. Radiographs of her femurs demonstrated healed fractures and stable position of the intramedullary nails.
THE TAKEAWAY
An increased suspicion for AFFs due to bisphosphonate use can lead to earlier diagnosis and decreased morbidity for patients. Use of femoral imaging can promote detection and reduce financial burden.
To help prevent AFFs from occurring, we recommend reevaluating the need for continued bisphosphonate therapy after 2 to 5 years of treatment. Continued surveillance is also advisable throughout the duration of their use.
ACKNOWLEDGMENT
The authors wish to acknowledge Dr. Maurice Manring for his help in preparing this manuscript.
THE CASE
A 67-year-old woman presented to our orthopaedic clinic with a 2-year history of bilateral thigh and knee pain and weakness of her legs. She had no history of trauma, and the pain, which was localized to the distal anterior thighs and patellofemoral area, was 7/10 at rest and worse with standing and walking.
Her medical history was significant for osteoporosis (diagnosed in 2004), hypertension, hypothyroidism, gastroesophageal reflux disease, and menopause (age 54). Her original dual-energy x-ray absorptiometry (DEXA) scan did not reveal the presence of any previous fractures. She was started on calcium and vitamin D supplementation and oral alendronate (70 mg once a week). She took alendronate for 4 years until 2008, when it was stopped due to nausea. She was then started on zoledronic acid (5 mg IV annually). She received 5 infusions of zoledronic acid between 2008 and 2013; she did not have an infusion in 2012. Her medication list also included lisinopril, omeprazole, naproxen, cyclobenzaprine, and a multivitamin. She had normal renal function (estimated glomerular filtration rate >60 mL/min/1.73 m2) and she did not drink alcohol or use tobacco.
In the 2 years prior to her visit to our clinic, she had been evaluated by her primary care provider, an orthopedic sports medicine specialist, 2 spinal surgeons, and a physiatrist. She had also undergone 30 physical therapy sessions. Bilateral femur radiographs (FIGURE 1) ordered by her orthopedist 6 months earlier demonstrated no evidence of fracture, but did show an incidental enchondroma in the right distal diaphysis and bilateral thickening of the lateral femoral cortices.
Finally, with no relief in sight, her obstetrician suggested that she might be experiencing myalgias attributable to her zoledronic acid infusions. She was subsequently referred to us.
The physical exam revealed a thin female with a body mass index of 21. She had mild tenderness on palpation of the bilateral anterior thighs and knees. There was no pain with hip or knee range of motion and minimal pain in the bilateral lower extremities with axial loading. The patient had normal sensation, did not have an antalgic gait, and exhibited 5/5 strength bilaterally in all distributions of the lower extremities.
THE DIAGNOSIS
Due to continued pain despite negative x-rays, we obtained a 3-phase bone scan of the pelvis and bilateral femurs. Delayed images showed moderately increased activity in the mid-right and mid-left lateral femoral diaphyses at the cortex and confirmed stress fractures (FIGURE 2).
DISCUSSION
Bisphosphonates are considered first-line therapy for osteoporosis, according to current evidence-based guidelines.1 These medications inhibit osteoclast activity and can bind to the bone for more than 10 years.2,3 (In women with bone mineral density scores ≤ –2.5, the number needed to treat is 21.1,4)
Patients taking bisphosphonates, however, are susceptible to atypical femoral fractures (AFFs), which are stress or insufficiency fractures associated with minimal or no trauma.5 The pathophysiology remains unknown at this time, but AFFs may result from changes in bone remodeling that occur when a bone experiences repetitive microtrauma, leading to lateral cortical thickening of the femur.6,7 Incidence of AFFs in patients taking bisphosphonates is estimated to be between 3.2 and 50 cases per 100,000 person-years; however, this risk increases to approximately 100 per 100,000 person-years with long-term use.5 Other risk factors include low body weight, advancing age, rheumatoid arthritis, long-term glucocorticoid therapy, and excessive alcohol and cigarette use.8
What you’ll see
Symptoms typically include unilateral or bilateral prodromal pain with a sharp or achy character that is localized to the mid-thigh, upper thigh, or groin.9 If an AFF is suspected, we recommend performing a bilateral exam and obtaining radiographs.
If characteristic features are found (eg, signs of focal cortical thickening or beaking) and pain arises in the opposite limb, obtain a radiograph of the contralateral femur. If radiographs are negative but suspicion remains, order magnetic resonance imaging or a bone scan, to identify a cortical fracture line, bone and marrow edema, or hyperemia.5
Begin treatment by discontinuing bisphosphonates
Upon identification of an AFF, discontinue bisphosphonates and initiate calcium and vitamin D supplementation.5 Prophylactic surgical fixation may also be necessary to accelerate healing and prevent fracture propagation and further pain.
Our patient. Due to the longevity of the symptoms and the bilateral stress fractures noted on the bone scan, our patient chose to proceed with intramedullary nailing of the bilateral femurs (FIGURES 3 and 4). On postop Day 1, she was able to ambulate using a walker and to participate in bilateral weight-bearing (as tolerated). She was discharged to a skilled nursing facility, where she progressed to full weight-bearing without aid. On follow-up (one year postop), the patient reported no residual leg pain and was able to work out 5 days per week. Radiographs of her femurs demonstrated healed fractures and stable position of the intramedullary nails.
THE TAKEAWAY
An increased suspicion for AFFs due to bisphosphonate use can lead to earlier diagnosis and decreased morbidity for patients. Use of femoral imaging can promote detection and reduce financial burden.
To help prevent AFFs from occurring, we recommend reevaluating the need for continued bisphosphonate therapy after 2 to 5 years of treatment. Continued surveillance is also advisable throughout the duration of their use.
ACKNOWLEDGMENT
The authors wish to acknowledge Dr. Maurice Manring for his help in preparing this manuscript.
1. Watts NB, Bilezikian JP, Camacho PM, et al. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for the diagnosis and treatment of postmenopausal osteoporosis. Endocr Pract. 2010;16 Suppl 3:1-37.
2. Cakmak S, Mahiroğullari M, Keklikci K, et al. Bilateral low-energy sequential femoral shaft fractures in patients on long-term bisphosphonate therapy. Acta Orthop Traumatol Turc. 2013;47:162-172.
3. Drake MT, Clarke BL, Khosla S. Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clin Proc. 2008;83:1032-1045.
4. Black DM, Bauer DC, Schwartz AV, et al. Continuing bisphosphonate treatment for osteoporosis—for whom and for how long? N Engl J Med. 2012;366:2051-2053.
5. Shane E, Burr D, Abrahamsen B, et al. Atypical subtrochanteric and diaphyseal femoral fractures: second report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2014;29:1-23.
6. Allen MR. Recent advances in understanding bisphosphonate effects on bone mechanical properties. Curr Osteoporos Rep. 2018 Mar 1. [Epub ahead of print]
7. Hagino H, Endo N, Yamamoto T, et al. Treatment status and radiographic features of patients with atypical femoral fractures. J Orthop Sci. 2018;23:316-320.
8. Kanis JA, Borgstrom F, De Laet C, et al. Assessment of fracture risk. Osteoporos Int. 2005;16:581-589.
9. Giusti A, Hamdy NA, Papapoulos SE. Atypical fractures of the femur and bisphosphonate therapy: a systematic review of case/case series studies. Bone. 2010;47:169-180.
1. Watts NB, Bilezikian JP, Camacho PM, et al. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for the diagnosis and treatment of postmenopausal osteoporosis. Endocr Pract. 2010;16 Suppl 3:1-37.
2. Cakmak S, Mahiroğullari M, Keklikci K, et al. Bilateral low-energy sequential femoral shaft fractures in patients on long-term bisphosphonate therapy. Acta Orthop Traumatol Turc. 2013;47:162-172.
3. Drake MT, Clarke BL, Khosla S. Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clin Proc. 2008;83:1032-1045.
4. Black DM, Bauer DC, Schwartz AV, et al. Continuing bisphosphonate treatment for osteoporosis—for whom and for how long? N Engl J Med. 2012;366:2051-2053.
5. Shane E, Burr D, Abrahamsen B, et al. Atypical subtrochanteric and diaphyseal femoral fractures: second report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2014;29:1-23.
6. Allen MR. Recent advances in understanding bisphosphonate effects on bone mechanical properties. Curr Osteoporos Rep. 2018 Mar 1. [Epub ahead of print]
7. Hagino H, Endo N, Yamamoto T, et al. Treatment status and radiographic features of patients with atypical femoral fractures. J Orthop Sci. 2018;23:316-320.
8. Kanis JA, Borgstrom F, De Laet C, et al. Assessment of fracture risk. Osteoporos Int. 2005;16:581-589.
9. Giusti A, Hamdy NA, Papapoulos SE. Atypical fractures of the femur and bisphosphonate therapy: a systematic review of case/case series studies. Bone. 2010;47:169-180.
Severe right upper chest pain • tender right sternoclavicular joint • Dx?
THE CASE
A 16-year-old hockey player presented to our emergency department with sharp pain in his right upper chest after “checking” another player during a game. The pain did not resolve with rest and was worse with movement and breathing. The patient did not have dysphagia, dyspnea, paresthesias, or hoarseness. The physical examination revealed tenderness over the right sternoclavicular joint (SCJ) without swelling or deformity. A distal neurovascular exam was intact, and a chest x-ray showed no evidence of dislocation or fracture (FIGURE 1A). The patient’s pain was refractory to multiple intravenous (IV) pain medications.
THE DIAGNOSIS
A computed tomography (CT) scan with IV contrast of the chest demonstrated posterior and superior dislocation of the right clavicular head. Despite the close proximity of the dislocated head to the brachiocephalic artery (FIGURE 1B-1D), there was no vascular injury.
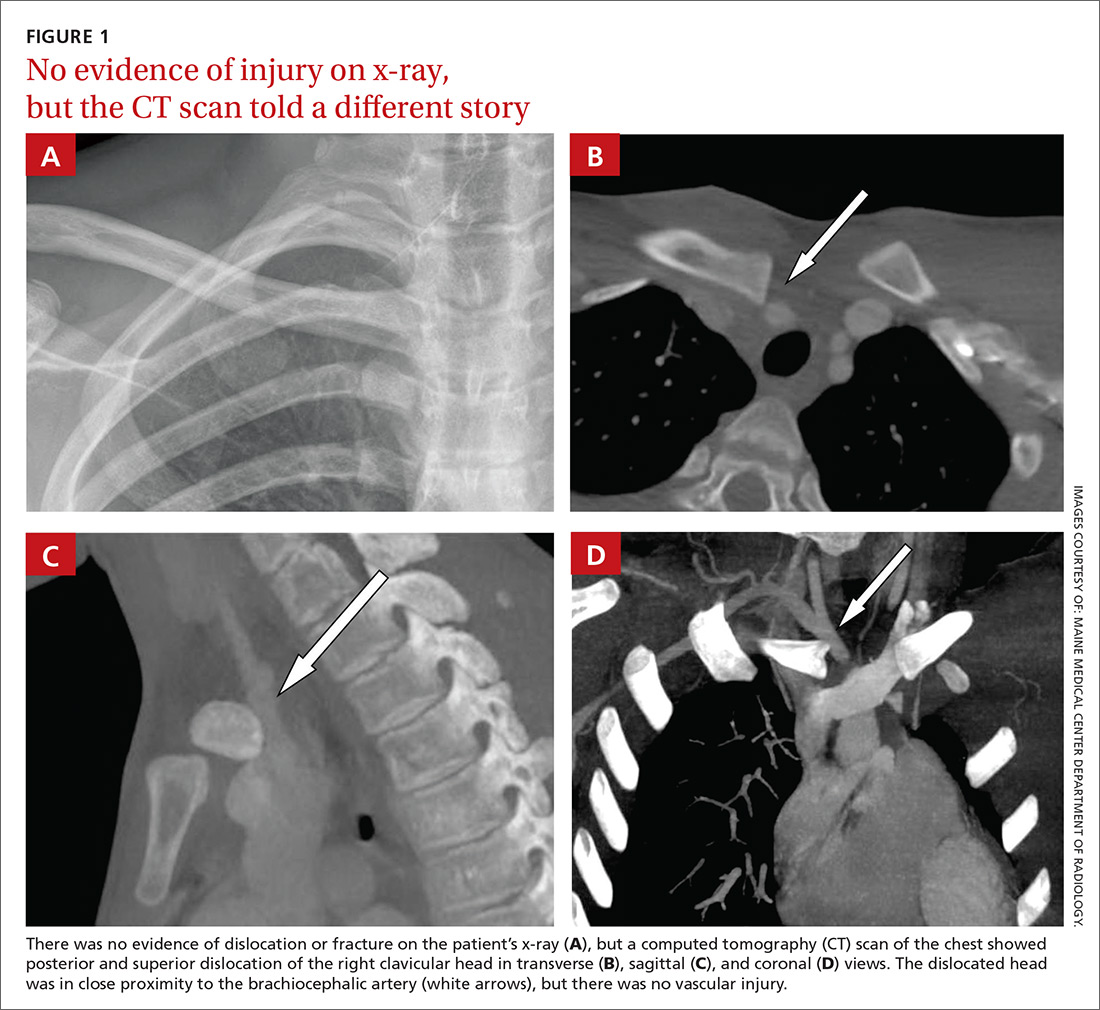
DISCUSSION
Posterior sternoclavicular dislocations (PSCDs) can be difficult to diagnose. Edema can mask the characteristic skin depression that one would expect with a posterior dislocation.1 Chest radiographs are often normal (as was true in this case). Patients may present with an abnormal pulse, paresthesias, hoarseness, dysphagia, and/or dyspnea. However, for more than half of these patients, their only signs and symptoms will be pain, swelling, and limited range of motion.1 As a result, a PSCD may be misdiagnosed as a ligamentous or soft tissue injury.1
An uncommon injury that can result in serious complications
PSCDs represent 3% to 5% of all SCJ dislocations, which comprise <5% of all shoulder girdle injuries.1 Nevertheless, prompt and accurate diagnosis is critical, as these dislocations involve a high risk for injury to the posterior structures, particularly the brachiocephalic vein, right common carotid artery, and aortic arch.
One study found that nearly 75% of patients had a significant structure <1 cm posterior to the SCJ.2 This proximity can result in neurovascular complications—some of which are devastating—in up to 30% of patients with PSCDs.3 A case report from 2011, for example, describes a 19-year-old man who had an undiagnosed PSCD that caused a pseudoaneurysm in his subclavian artery and a subsequent thrombotic cerebrovascular accident.4
Which injuries should raise your suspicions? Injuries in which lateral compression on the shoulder has caused it to roll forward and those in which a posteriorly directed force has been applied to the medial clavicle (as might occur in tackle sports or motor vehicle rollovers) should increase suspicion of a PSCD.1
Proper diagnosis requires CT angiography of the chest to assess the injury and evaluate the underlying structures. If CT is not available, additional chest film views, such as a serendipity view (anteroposterior view with 40° cephalic tilt) or Heinig view (oblique projection perpendicular to SCJ), may be obtained; an ultrasound is also an option.5
PSCD = surgical emergency
Following diagnosis, immediate orthopedic consultation is required. A PSCD is a surgical emergency. Reduction (open or closed) must be performed under general anesthesia with vascular and/or cardiothoracic surgery specialists available, as the reduction itself could injure one of the great vessels. Fortunately, most patients do quite well following surgery, with the majority achieving good-to-excellent results.6
Our patient was admitted to the hospital and underwent orthopedic surgery the following morning. Vascular and cardiothoracic surgeons were consulted and available in the event of a complication. A Salter-Harris type 2 fracture of the medial clavicle was identified intraoperatively, and an open reduction with internal fixation was performed. The patient had an uneventful recovery and resumed his usual activities, including playing hockey.
THE TAKEAWAY
PSCDs, although uncommon, can be life-threatening. Since the physical exam is unreliable and standard radiographs are often normal, accurate diagnosis relies largely on increased clinical suspicion. When there is a history of shoulder trauma, medial clavicle pain, and SCJ edema or tenderness, a PSCD should be suspected.7
Confirm the diagnosis with CT angiogram, and remember that a PSCD is a surgical emergency that requires coordination with orthopedic and vascular/cardiothoracic surgeons.
1. Chaudhry S. Pediatric posterior sternoclavicular joint injuries. J Am Acad Orthop Surg. 2015;23:468-475.
2. Ponce BA, Kundukulam JA, Pflugner R, et al. Sternoclavicular joint surgery: how far does danger lurk below? J Shoulder Elbow Surg. 2013;22:993-999.
3. Daya MR, Bengtzen RR. Shoulder. In: Rosen’s Emergency Medicine: Concepts and Clinical Practice. 8th ed. Philadelphia, PA: Elsevier Saunders; 2014:618-642.
4. Marcus MS, Tan V. Cerebrovascular accident in a 19-year-old patient: a case report of posterior sternoclavicular dislocation. J Shoulder Elbow Surg. 2011;20:e1-e4.
5. Morell DJ, Thyagarajan DS. Sternoclavicular joint dislocation and its management: a review of the literature. World J Orthop. 2016;7:244-250.
6. Boesmueller S, Wech M, Tiefenboeck TM, et al. Incidence, characteristics, and long-term follow-up of sternoclavicular injuries: an epidemiologic analysis of 92 cases. J Trauma Acute Care Surg. 2016;80:289-295.
7. Roepke C, Kleiner M, Jhun P, et al. Chest pain bounce-back: posterior sternoclavicular dislocation. Ann Emerg Med. 2015;66:559-561.
THE CASE
A 16-year-old hockey player presented to our emergency department with sharp pain in his right upper chest after “checking” another player during a game. The pain did not resolve with rest and was worse with movement and breathing. The patient did not have dysphagia, dyspnea, paresthesias, or hoarseness. The physical examination revealed tenderness over the right sternoclavicular joint (SCJ) without swelling or deformity. A distal neurovascular exam was intact, and a chest x-ray showed no evidence of dislocation or fracture (FIGURE 1A). The patient’s pain was refractory to multiple intravenous (IV) pain medications.
THE DIAGNOSIS
A computed tomography (CT) scan with IV contrast of the chest demonstrated posterior and superior dislocation of the right clavicular head. Despite the close proximity of the dislocated head to the brachiocephalic artery (FIGURE 1B-1D), there was no vascular injury.
DISCUSSION
Posterior sternoclavicular dislocations (PSCDs) can be difficult to diagnose. Edema can mask the characteristic skin depression that one would expect with a posterior dislocation.1 Chest radiographs are often normal (as was true in this case). Patients may present with an abnormal pulse, paresthesias, hoarseness, dysphagia, and/or dyspnea. However, for more than half of these patients, their only signs and symptoms will be pain, swelling, and limited range of motion.1 As a result, a PSCD may be misdiagnosed as a ligamentous or soft tissue injury.1
An uncommon injury that can result in serious complications
PSCDs represent 3% to 5% of all SCJ dislocations, which comprise <5% of all shoulder girdle injuries.1 Nevertheless, prompt and accurate diagnosis is critical, as these dislocations involve a high risk for injury to the posterior structures, particularly the brachiocephalic vein, right common carotid artery, and aortic arch.
One study found that nearly 75% of patients had a significant structure <1 cm posterior to the SCJ.2 This proximity can result in neurovascular complications—some of which are devastating—in up to 30% of patients with PSCDs.3 A case report from 2011, for example, describes a 19-year-old man who had an undiagnosed PSCD that caused a pseudoaneurysm in his subclavian artery and a subsequent thrombotic cerebrovascular accident.4
Which injuries should raise your suspicions? Injuries in which lateral compression on the shoulder has caused it to roll forward and those in which a posteriorly directed force has been applied to the medial clavicle (as might occur in tackle sports or motor vehicle rollovers) should increase suspicion of a PSCD.1
Proper diagnosis requires CT angiography of the chest to assess the injury and evaluate the underlying structures. If CT is not available, additional chest film views, such as a serendipity view (anteroposterior view with 40° cephalic tilt) or Heinig view (oblique projection perpendicular to SCJ), may be obtained; an ultrasound is also an option.5
PSCD = surgical emergency
Following diagnosis, immediate orthopedic consultation is required. A PSCD is a surgical emergency. Reduction (open or closed) must be performed under general anesthesia with vascular and/or cardiothoracic surgery specialists available, as the reduction itself could injure one of the great vessels. Fortunately, most patients do quite well following surgery, with the majority achieving good-to-excellent results.6
Our patient was admitted to the hospital and underwent orthopedic surgery the following morning. Vascular and cardiothoracic surgeons were consulted and available in the event of a complication. A Salter-Harris type 2 fracture of the medial clavicle was identified intraoperatively, and an open reduction with internal fixation was performed. The patient had an uneventful recovery and resumed his usual activities, including playing hockey.
THE TAKEAWAY
PSCDs, although uncommon, can be life-threatening. Since the physical exam is unreliable and standard radiographs are often normal, accurate diagnosis relies largely on increased clinical suspicion. When there is a history of shoulder trauma, medial clavicle pain, and SCJ edema or tenderness, a PSCD should be suspected.7
Confirm the diagnosis with CT angiogram, and remember that a PSCD is a surgical emergency that requires coordination with orthopedic and vascular/cardiothoracic surgeons.
THE CASE
A 16-year-old hockey player presented to our emergency department with sharp pain in his right upper chest after “checking” another player during a game. The pain did not resolve with rest and was worse with movement and breathing. The patient did not have dysphagia, dyspnea, paresthesias, or hoarseness. The physical examination revealed tenderness over the right sternoclavicular joint (SCJ) without swelling or deformity. A distal neurovascular exam was intact, and a chest x-ray showed no evidence of dislocation or fracture (FIGURE 1A). The patient’s pain was refractory to multiple intravenous (IV) pain medications.
THE DIAGNOSIS
A computed tomography (CT) scan with IV contrast of the chest demonstrated posterior and superior dislocation of the right clavicular head. Despite the close proximity of the dislocated head to the brachiocephalic artery (FIGURE 1B-1D), there was no vascular injury.
DISCUSSION
Posterior sternoclavicular dislocations (PSCDs) can be difficult to diagnose. Edema can mask the characteristic skin depression that one would expect with a posterior dislocation.1 Chest radiographs are often normal (as was true in this case). Patients may present with an abnormal pulse, paresthesias, hoarseness, dysphagia, and/or dyspnea. However, for more than half of these patients, their only signs and symptoms will be pain, swelling, and limited range of motion.1 As a result, a PSCD may be misdiagnosed as a ligamentous or soft tissue injury.1
An uncommon injury that can result in serious complications
PSCDs represent 3% to 5% of all SCJ dislocations, which comprise <5% of all shoulder girdle injuries.1 Nevertheless, prompt and accurate diagnosis is critical, as these dislocations involve a high risk for injury to the posterior structures, particularly the brachiocephalic vein, right common carotid artery, and aortic arch.
One study found that nearly 75% of patients had a significant structure <1 cm posterior to the SCJ.2 This proximity can result in neurovascular complications—some of which are devastating—in up to 30% of patients with PSCDs.3 A case report from 2011, for example, describes a 19-year-old man who had an undiagnosed PSCD that caused a pseudoaneurysm in his subclavian artery and a subsequent thrombotic cerebrovascular accident.4
Which injuries should raise your suspicions? Injuries in which lateral compression on the shoulder has caused it to roll forward and those in which a posteriorly directed force has been applied to the medial clavicle (as might occur in tackle sports or motor vehicle rollovers) should increase suspicion of a PSCD.1
Proper diagnosis requires CT angiography of the chest to assess the injury and evaluate the underlying structures. If CT is not available, additional chest film views, such as a serendipity view (anteroposterior view with 40° cephalic tilt) or Heinig view (oblique projection perpendicular to SCJ), may be obtained; an ultrasound is also an option.5
PSCD = surgical emergency
Following diagnosis, immediate orthopedic consultation is required. A PSCD is a surgical emergency. Reduction (open or closed) must be performed under general anesthesia with vascular and/or cardiothoracic surgery specialists available, as the reduction itself could injure one of the great vessels. Fortunately, most patients do quite well following surgery, with the majority achieving good-to-excellent results.6
Our patient was admitted to the hospital and underwent orthopedic surgery the following morning. Vascular and cardiothoracic surgeons were consulted and available in the event of a complication. A Salter-Harris type 2 fracture of the medial clavicle was identified intraoperatively, and an open reduction with internal fixation was performed. The patient had an uneventful recovery and resumed his usual activities, including playing hockey.
THE TAKEAWAY
PSCDs, although uncommon, can be life-threatening. Since the physical exam is unreliable and standard radiographs are often normal, accurate diagnosis relies largely on increased clinical suspicion. When there is a history of shoulder trauma, medial clavicle pain, and SCJ edema or tenderness, a PSCD should be suspected.7
Confirm the diagnosis with CT angiogram, and remember that a PSCD is a surgical emergency that requires coordination with orthopedic and vascular/cardiothoracic surgeons.
1. Chaudhry S. Pediatric posterior sternoclavicular joint injuries. J Am Acad Orthop Surg. 2015;23:468-475.
2. Ponce BA, Kundukulam JA, Pflugner R, et al. Sternoclavicular joint surgery: how far does danger lurk below? J Shoulder Elbow Surg. 2013;22:993-999.
3. Daya MR, Bengtzen RR. Shoulder. In: Rosen’s Emergency Medicine: Concepts and Clinical Practice. 8th ed. Philadelphia, PA: Elsevier Saunders; 2014:618-642.
4. Marcus MS, Tan V. Cerebrovascular accident in a 19-year-old patient: a case report of posterior sternoclavicular dislocation. J Shoulder Elbow Surg. 2011;20:e1-e4.
5. Morell DJ, Thyagarajan DS. Sternoclavicular joint dislocation and its management: a review of the literature. World J Orthop. 2016;7:244-250.
6. Boesmueller S, Wech M, Tiefenboeck TM, et al. Incidence, characteristics, and long-term follow-up of sternoclavicular injuries: an epidemiologic analysis of 92 cases. J Trauma Acute Care Surg. 2016;80:289-295.
7. Roepke C, Kleiner M, Jhun P, et al. Chest pain bounce-back: posterior sternoclavicular dislocation. Ann Emerg Med. 2015;66:559-561.
1. Chaudhry S. Pediatric posterior sternoclavicular joint injuries. J Am Acad Orthop Surg. 2015;23:468-475.
2. Ponce BA, Kundukulam JA, Pflugner R, et al. Sternoclavicular joint surgery: how far does danger lurk below? J Shoulder Elbow Surg. 2013;22:993-999.
3. Daya MR, Bengtzen RR. Shoulder. In: Rosen’s Emergency Medicine: Concepts and Clinical Practice. 8th ed. Philadelphia, PA: Elsevier Saunders; 2014:618-642.
4. Marcus MS, Tan V. Cerebrovascular accident in a 19-year-old patient: a case report of posterior sternoclavicular dislocation. J Shoulder Elbow Surg. 2011;20:e1-e4.
5. Morell DJ, Thyagarajan DS. Sternoclavicular joint dislocation and its management: a review of the literature. World J Orthop. 2016;7:244-250.
6. Boesmueller S, Wech M, Tiefenboeck TM, et al. Incidence, characteristics, and long-term follow-up of sternoclavicular injuries: an epidemiologic analysis of 92 cases. J Trauma Acute Care Surg. 2016;80:289-295.
7. Roepke C, Kleiner M, Jhun P, et al. Chest pain bounce-back: posterior sternoclavicular dislocation. Ann Emerg Med. 2015;66:559-561.
Bedside Ultrasound for Pulsatile Hand Mass
Case
A 23-year-old man presented to an outside hospital’s ED for evaluation of a wound on his right hand, which he sustained after he accidentally stabbed himself with a steak knife. At presentation, the patient’s vital signs were: heart rate, 90 beats/min; respiratory rate, 16 breaths/min; blood pressure, 150/92 mm Hg; and temperature, 98.1°F. Oxygen saturation was 98% on room air. Examination revealed a laceration on the patient’s right hand measuring 2 cm in length. The emergency physician (EP) closed the wound using four nylon sutures and administered a Boostrix shot. The patient was discharged home with a prescription for cephalexin capsule 500 mg to be taken four times daily for 5 days. He was instructed to return in 10 days for suture removal, but failed to follow-up.
The patient presented to our ED two months after the initial injury for evaluation of a 1.5-cm round pulsatile mass on his right palm, at the base of the middle finger, from which exuded a small amount of sanguineous fluid. The patient complained of numbness and difficulty extending his right index and middle fingers.

Discussion
Palmar Pseudoaneurysms
A pseudoaneurysm, also referred to as a traumatic aneurysm, develops when a tear of the vessel wall and hemorrhage is contained by a thin-walled capsule, typically following traumatic perforation of the arterial wall. Unlike a true aneurysm, a pseudoaneurysm does not contain all three layers of intima, media, and adventitia. Thin walls lead to inevitable expansion over time; in some cases, a patient will present with a soft-tissue mass years after the initial injury. Compression of nearby structures can cause neuropathy, peripheral edema, venous thrombosis, arterial occlusion or emboli, and even bone erosion.1,2
Hand pseudoaneurysms are more likely to occur on the palmar surface, involving the superficial palmar arch,3 and are due to a penetrating injury or repetitive microtrauma. Hypothenar hammer syndrome occurs when repetitive microtrauma is applied to the ulnar artery as it passes under the hook of the hamate bone into the hand. This condition is also referred to as “hammer hand syndrome” because it frequently occurs in laborers such as mechanics, carpenters, and machinists as a result of repetitive palm trauma. Cases have also been reported in baseball players and cooks who also expose their hands to repetitive trauma.3 Likewise, elderly patients who use walking canes can also present with bilateral hammer hand syndrome,3 and patients who need crutches for a prolonged period of time may also develop axillary artery aneurysms.1,2
Although rare, there have also been cases of spontaneous hand pseudoaneurysms in patients on anticoagulation therapy;4,5 however, pseudoaneurysms are not an absolute contraindication to initiating or continuing use of anticoagulants.
Evaluation
Physical Examination. The patient’s mass in this case was clearly pulsatile on examination, but physical examination alone is not a reliable indicator of pseudoaneurysm, as patients may present only with soft-tissue swelling, pain, erythema, or neurological symptoms.3,6,7
Ultrasound Imaging. In the emergency setting, POC ultrasound should be performed to evaluate any soft-tissue hand mass, especially in the context of trauma or any neurovascular findings, since palmar pseudoaneurysms can easily be confused with an abscess, foreign body, cyst, or even a tendon tear.6 Ultrasound studies using the linear vascular probe should always be done before any attempt to incise and drain the mass.
Three ultrasound characteristics of pseudoaneurysms include expansile pulsatility, turbulent flow with a classic yin-yang sign on Doppler, and a hematoma with variable echogenicity. Variable echogenicity may represent separate episodes of bleeding and rebleeding.8 A “to-and-fro” spectral waveform is pathognomonic for palmar pseudoaneurysms.8
Computed Tomography and Magnetic Resonance Angiography. Definitive imaging for operative management includes computed tomography or magnetic resonance angiography to assess for the exact location and presence of collateral circulation.
Treatment
Treatment of pseudoaneurysms includes conservative compression therapy, surgical excision, or anastomosis, and more recently, ultrasound-guided thrombin injection (UGTI).
Compression Therapy. Compression therapy is often used for femoral artery pseudoaneurysms that develop after iatrogenic injury. However, this technique is time consuming, is uncomfortable for patients, is not effective in treating large pseudoaneurysms, and is contraindicated in patients on anticoagulation therapy. Compression therapy also has a high-failure rate of resolving pseudoaneurysms. Traditionally, surgical excision or anastomosis has been the definitive treatment for palmar pseudoaneurysms.
Ultrasound-Guided Thrombin Injection. A more recent treatment option is UGTI, which is usually performed by an interventional radiologist. Although there is no consensus on exact dose of thrombin for this procedure, the literature describes UGTI to treat both the radial and ulnar arteries.9,10 One study of 83 pseudoaneurysms demonstrated a relationship between the size of the palmar pseudoaneurysm and the number of thrombin injections required to resolve it. Depending on the size of the palmar pseudoaneurysm, the effective thrombin doses ranged from 200 to 2,500 U. Regarding adverse effects and events from treatment, this study reported one case of transient distal ischemia.11
Intravascular balloon occlusion of the pseudoaneurysm neck has also been recommended for UGTI in the femoral artery if the neck is greater than 1 mm, but there is currently nothing in the literature describing its use in palmar pseudoaneurysms.12
Complications
There are more descriptions of palmar, radial, and ulnar pseudoaneurysms in critical care patients due to the frequent, but necessary, use of invasive lines. Emergency physicians frequently place radial or femoral arterial lines for hemodynamic monitoring in critically ill patients. However, the incidence of pseudoaneurysms and its sequelae from these lines are not usually observed in the ED setting.
Radial arterial lines may cause thrombosis in 19% to 57% of cases, and local infection in 1% to 18% of cases.10 In a study of 12,500 patients with radial artery catheters, the rate of radial pseudoaneurysm was only 0.05%.11 Although this is a small complication rate, pseudoaneurysms can lead to significant loss of function. To decrease the number of attempts and penetrating injuries to the arteries, ultrasound guidance for these procedures in the ED is strongly recommended. In addition to decreasing the risk of developing a pseudoaneurysm, ultrasound-guidance decreases the discomfort level of the patient and reduces the risk of bleeding, hematoma formation, and infection. Arterial line placement in the ED using ultrasound guidance decreases the risk of developing pseudoaneurysms and their sequelae, such as distal embolization.
Case Conclusion
The patient in this case underwent an arterial duplex study, which found a partially thrombosed right superficial palmar arch pseudoaneurysm measuring 1.91 cm x 2.08 cm, with an active flow area measuring 0.58 cm x 0.68 cm. The flow to the index finger medial artery and middle finger lateral artery was also diminished. The patient was discharged home with a bulky soft dressing and underwent excision and repair by hand surgery 3 days later. At the 1-month postoperative follow-up visit, the patient had full sensation but mildly decreased range of motion in his fingers.
Summary
Hand pseudoaneurysms are often associated with penetrating injuries—as demonstrated in our case—or repetitive microtrauma. Hand pseudoaneurysms can present with minimal findings such as isolated soft-tissue swelling, pain, or neuropathy. The EP should consider vascular pathology in the differential for patients who present with posttraumatic neuropathy. Regarding imaging studies, ultrasound is the best imaging modality to assess for pseudoaneurysms, and EPs should have a low threshold for its use at bedside—especially prior to attempting any invasive procedure. Patients with a confirmed pseudoaneurysm should be referred to a hand or vascular surgeon for surgical repair, or to an interventional radiologist for UGTI.
1. Newton EJ, Arora S. Peripheral vascular injury. In: Marx JA, Hockberger RS, Walls RM, et al, eds. Rosen’s Emergency Medicine Concepts and Clinical Practice. Vol 1. 8th ed. Philadelphia, PA: Elsevier Saunders; 2014:502.
2. Aufderheide TP. Peripheral arteriovascular disease. In: Marx JA, Hockberger RS, Walls RM, et al, eds. Rosen’s Emergency Medicine Concepts and Clinical Practice. Vol 1. 8th ed. 2014:1147-1149.
3. Anderson SE, De Monaco D, Buechler U, et al. Imaging features of pseudoaneurysms of the hand in children and adults. AJR Am J Roentgenol. 2003;180(3):659-664. doi:10.2214/ajr.180.3.1800659.
4. Shah S, Powell-Brett S, Garnham A. Pseudoaneurysm: an unusual cause of post-traumatic hand swelling. BMJ Case Rep. 2015;2015. pii: bcr2014208750. doi:10.1136/bcr-2014-208750.
5. Kitamura A, Mukohara N. Spontaneous pseudoaneurysm of the hand. Ann Vasc Surg. 2014;28(3):739.e1-e3. doi:10.1016/j.avsg.2013.04.033.
6. Huang SW, Wei TS, Liu SY, Wang WT. Spontaneous totally thrombosed pseudoaneurysm mimicking a tendon tear of the wrist. Orthopedics. 2010;33(10):776. doi:10.3928/01477447-20100826-23.
7. Belyayev L, Rich NM, McKay P, Nesti L, Wind G. Traumatic ulnar artery pseudoaneurysm following a grenade blast: report of a case. Mil Med. 2015;180(6):e725-e727. doi:10.7205/MILMED-D-14-00400.
8. Pero T, Herrick J. Pseudoaneurysm of the radial artery diagnosed by bedside ultrasound. West J Emerg Med. 2009;10(2):89-91.
9. Bosman A, Veger HTC, Doornink F, Hedeman Joosten PPA. A pseudoaneurysm of the deep palmar arch after penetrating trauma to the hand: successful exclusion by ultrasound guided percutaneous thrombin injection. EJVES Short Rep. 2016;31:9-11. doi:10.1016/j.ejvssr.2016.03.002.
10. Komorowska-Timek E, Teruya TH, Abou-Zamzam AM Jr, Papa D, Ballard JL. Treatment of radial and ulnar artery pseudoaneurysms using percutaneous thrombin injection. J Hand Surg. 2004;29A(5):936-942. doi:10.1016/j.jhsa.2004.05.009.
11. Falk PS, Scuderi PE, Sherertz RJ, Motsinger SM. Infected radial artery pseudoaneurysms occurring after percutaneous cannulation. Chest. 1992;101(2):490-495.
12. Kang SS, Labropoulos N, Mansour MA, et al. Expanded indications for ultrasound-guided thrombin injection of pseudoaneurysms. J Vasc Surg. 2000;31(2):289-298.
Case
A 23-year-old man presented to an outside hospital’s ED for evaluation of a wound on his right hand, which he sustained after he accidentally stabbed himself with a steak knife. At presentation, the patient’s vital signs were: heart rate, 90 beats/min; respiratory rate, 16 breaths/min; blood pressure, 150/92 mm Hg; and temperature, 98.1°F. Oxygen saturation was 98% on room air. Examination revealed a laceration on the patient’s right hand measuring 2 cm in length. The emergency physician (EP) closed the wound using four nylon sutures and administered a Boostrix shot. The patient was discharged home with a prescription for cephalexin capsule 500 mg to be taken four times daily for 5 days. He was instructed to return in 10 days for suture removal, but failed to follow-up.
The patient presented to our ED two months after the initial injury for evaluation of a 1.5-cm round pulsatile mass on his right palm, at the base of the middle finger, from which exuded a small amount of sanguineous fluid. The patient complained of numbness and difficulty extending his right index and middle fingers.
Discussion
Palmar Pseudoaneurysms
A pseudoaneurysm, also referred to as a traumatic aneurysm, develops when a tear of the vessel wall and hemorrhage is contained by a thin-walled capsule, typically following traumatic perforation of the arterial wall. Unlike a true aneurysm, a pseudoaneurysm does not contain all three layers of intima, media, and adventitia. Thin walls lead to inevitable expansion over time; in some cases, a patient will present with a soft-tissue mass years after the initial injury. Compression of nearby structures can cause neuropathy, peripheral edema, venous thrombosis, arterial occlusion or emboli, and even bone erosion.1,2
Hand pseudoaneurysms are more likely to occur on the palmar surface, involving the superficial palmar arch,3 and are due to a penetrating injury or repetitive microtrauma. Hypothenar hammer syndrome occurs when repetitive microtrauma is applied to the ulnar artery as it passes under the hook of the hamate bone into the hand. This condition is also referred to as “hammer hand syndrome” because it frequently occurs in laborers such as mechanics, carpenters, and machinists as a result of repetitive palm trauma. Cases have also been reported in baseball players and cooks who also expose their hands to repetitive trauma.3 Likewise, elderly patients who use walking canes can also present with bilateral hammer hand syndrome,3 and patients who need crutches for a prolonged period of time may also develop axillary artery aneurysms.1,2
Although rare, there have also been cases of spontaneous hand pseudoaneurysms in patients on anticoagulation therapy;4,5 however, pseudoaneurysms are not an absolute contraindication to initiating or continuing use of anticoagulants.
Evaluation
Physical Examination. The patient’s mass in this case was clearly pulsatile on examination, but physical examination alone is not a reliable indicator of pseudoaneurysm, as patients may present only with soft-tissue swelling, pain, erythema, or neurological symptoms.3,6,7
Ultrasound Imaging. In the emergency setting, POC ultrasound should be performed to evaluate any soft-tissue hand mass, especially in the context of trauma or any neurovascular findings, since palmar pseudoaneurysms can easily be confused with an abscess, foreign body, cyst, or even a tendon tear.6 Ultrasound studies using the linear vascular probe should always be done before any attempt to incise and drain the mass.
Three ultrasound characteristics of pseudoaneurysms include expansile pulsatility, turbulent flow with a classic yin-yang sign on Doppler, and a hematoma with variable echogenicity. Variable echogenicity may represent separate episodes of bleeding and rebleeding.8 A “to-and-fro” spectral waveform is pathognomonic for palmar pseudoaneurysms.8
Computed Tomography and Magnetic Resonance Angiography. Definitive imaging for operative management includes computed tomography or magnetic resonance angiography to assess for the exact location and presence of collateral circulation.
Treatment
Treatment of pseudoaneurysms includes conservative compression therapy, surgical excision, or anastomosis, and more recently, ultrasound-guided thrombin injection (UGTI).
Compression Therapy. Compression therapy is often used for femoral artery pseudoaneurysms that develop after iatrogenic injury. However, this technique is time consuming, is uncomfortable for patients, is not effective in treating large pseudoaneurysms, and is contraindicated in patients on anticoagulation therapy. Compression therapy also has a high-failure rate of resolving pseudoaneurysms. Traditionally, surgical excision or anastomosis has been the definitive treatment for palmar pseudoaneurysms.
Ultrasound-Guided Thrombin Injection. A more recent treatment option is UGTI, which is usually performed by an interventional radiologist. Although there is no consensus on exact dose of thrombin for this procedure, the literature describes UGTI to treat both the radial and ulnar arteries.9,10 One study of 83 pseudoaneurysms demonstrated a relationship between the size of the palmar pseudoaneurysm and the number of thrombin injections required to resolve it. Depending on the size of the palmar pseudoaneurysm, the effective thrombin doses ranged from 200 to 2,500 U. Regarding adverse effects and events from treatment, this study reported one case of transient distal ischemia.11
Intravascular balloon occlusion of the pseudoaneurysm neck has also been recommended for UGTI in the femoral artery if the neck is greater than 1 mm, but there is currently nothing in the literature describing its use in palmar pseudoaneurysms.12
Complications
There are more descriptions of palmar, radial, and ulnar pseudoaneurysms in critical care patients due to the frequent, but necessary, use of invasive lines. Emergency physicians frequently place radial or femoral arterial lines for hemodynamic monitoring in critically ill patients. However, the incidence of pseudoaneurysms and its sequelae from these lines are not usually observed in the ED setting.
Radial arterial lines may cause thrombosis in 19% to 57% of cases, and local infection in 1% to 18% of cases.10 In a study of 12,500 patients with radial artery catheters, the rate of radial pseudoaneurysm was only 0.05%.11 Although this is a small complication rate, pseudoaneurysms can lead to significant loss of function. To decrease the number of attempts and penetrating injuries to the arteries, ultrasound guidance for these procedures in the ED is strongly recommended. In addition to decreasing the risk of developing a pseudoaneurysm, ultrasound-guidance decreases the discomfort level of the patient and reduces the risk of bleeding, hematoma formation, and infection. Arterial line placement in the ED using ultrasound guidance decreases the risk of developing pseudoaneurysms and their sequelae, such as distal embolization.
Case Conclusion
The patient in this case underwent an arterial duplex study, which found a partially thrombosed right superficial palmar arch pseudoaneurysm measuring 1.91 cm x 2.08 cm, with an active flow area measuring 0.58 cm x 0.68 cm. The flow to the index finger medial artery and middle finger lateral artery was also diminished. The patient was discharged home with a bulky soft dressing and underwent excision and repair by hand surgery 3 days later. At the 1-month postoperative follow-up visit, the patient had full sensation but mildly decreased range of motion in his fingers.
Summary
Hand pseudoaneurysms are often associated with penetrating injuries—as demonstrated in our case—or repetitive microtrauma. Hand pseudoaneurysms can present with minimal findings such as isolated soft-tissue swelling, pain, or neuropathy. The EP should consider vascular pathology in the differential for patients who present with posttraumatic neuropathy. Regarding imaging studies, ultrasound is the best imaging modality to assess for pseudoaneurysms, and EPs should have a low threshold for its use at bedside—especially prior to attempting any invasive procedure. Patients with a confirmed pseudoaneurysm should be referred to a hand or vascular surgeon for surgical repair, or to an interventional radiologist for UGTI.
Case
A 23-year-old man presented to an outside hospital’s ED for evaluation of a wound on his right hand, which he sustained after he accidentally stabbed himself with a steak knife. At presentation, the patient’s vital signs were: heart rate, 90 beats/min; respiratory rate, 16 breaths/min; blood pressure, 150/92 mm Hg; and temperature, 98.1°F. Oxygen saturation was 98% on room air. Examination revealed a laceration on the patient’s right hand measuring 2 cm in length. The emergency physician (EP) closed the wound using four nylon sutures and administered a Boostrix shot. The patient was discharged home with a prescription for cephalexin capsule 500 mg to be taken four times daily for 5 days. He was instructed to return in 10 days for suture removal, but failed to follow-up.
The patient presented to our ED two months after the initial injury for evaluation of a 1.5-cm round pulsatile mass on his right palm, at the base of the middle finger, from which exuded a small amount of sanguineous fluid. The patient complained of numbness and difficulty extending his right index and middle fingers.
Discussion
Palmar Pseudoaneurysms
A pseudoaneurysm, also referred to as a traumatic aneurysm, develops when a tear of the vessel wall and hemorrhage is contained by a thin-walled capsule, typically following traumatic perforation of the arterial wall. Unlike a true aneurysm, a pseudoaneurysm does not contain all three layers of intima, media, and adventitia. Thin walls lead to inevitable expansion over time; in some cases, a patient will present with a soft-tissue mass years after the initial injury. Compression of nearby structures can cause neuropathy, peripheral edema, venous thrombosis, arterial occlusion or emboli, and even bone erosion.1,2
Hand pseudoaneurysms are more likely to occur on the palmar surface, involving the superficial palmar arch,3 and are due to a penetrating injury or repetitive microtrauma. Hypothenar hammer syndrome occurs when repetitive microtrauma is applied to the ulnar artery as it passes under the hook of the hamate bone into the hand. This condition is also referred to as “hammer hand syndrome” because it frequently occurs in laborers such as mechanics, carpenters, and machinists as a result of repetitive palm trauma. Cases have also been reported in baseball players and cooks who also expose their hands to repetitive trauma.3 Likewise, elderly patients who use walking canes can also present with bilateral hammer hand syndrome,3 and patients who need crutches for a prolonged period of time may also develop axillary artery aneurysms.1,2
Although rare, there have also been cases of spontaneous hand pseudoaneurysms in patients on anticoagulation therapy;4,5 however, pseudoaneurysms are not an absolute contraindication to initiating or continuing use of anticoagulants.
Evaluation
Physical Examination. The patient’s mass in this case was clearly pulsatile on examination, but physical examination alone is not a reliable indicator of pseudoaneurysm, as patients may present only with soft-tissue swelling, pain, erythema, or neurological symptoms.3,6,7
Ultrasound Imaging. In the emergency setting, POC ultrasound should be performed to evaluate any soft-tissue hand mass, especially in the context of trauma or any neurovascular findings, since palmar pseudoaneurysms can easily be confused with an abscess, foreign body, cyst, or even a tendon tear.6 Ultrasound studies using the linear vascular probe should always be done before any attempt to incise and drain the mass.
Three ultrasound characteristics of pseudoaneurysms include expansile pulsatility, turbulent flow with a classic yin-yang sign on Doppler, and a hematoma with variable echogenicity. Variable echogenicity may represent separate episodes of bleeding and rebleeding.8 A “to-and-fro” spectral waveform is pathognomonic for palmar pseudoaneurysms.8
Computed Tomography and Magnetic Resonance Angiography. Definitive imaging for operative management includes computed tomography or magnetic resonance angiography to assess for the exact location and presence of collateral circulation.
Treatment
Treatment of pseudoaneurysms includes conservative compression therapy, surgical excision, or anastomosis, and more recently, ultrasound-guided thrombin injection (UGTI).
Compression Therapy. Compression therapy is often used for femoral artery pseudoaneurysms that develop after iatrogenic injury. However, this technique is time consuming, is uncomfortable for patients, is not effective in treating large pseudoaneurysms, and is contraindicated in patients on anticoagulation therapy. Compression therapy also has a high-failure rate of resolving pseudoaneurysms. Traditionally, surgical excision or anastomosis has been the definitive treatment for palmar pseudoaneurysms.
Ultrasound-Guided Thrombin Injection. A more recent treatment option is UGTI, which is usually performed by an interventional radiologist. Although there is no consensus on exact dose of thrombin for this procedure, the literature describes UGTI to treat both the radial and ulnar arteries.9,10 One study of 83 pseudoaneurysms demonstrated a relationship between the size of the palmar pseudoaneurysm and the number of thrombin injections required to resolve it. Depending on the size of the palmar pseudoaneurysm, the effective thrombin doses ranged from 200 to 2,500 U. Regarding adverse effects and events from treatment, this study reported one case of transient distal ischemia.11
Intravascular balloon occlusion of the pseudoaneurysm neck has also been recommended for UGTI in the femoral artery if the neck is greater than 1 mm, but there is currently nothing in the literature describing its use in palmar pseudoaneurysms.12
Complications
There are more descriptions of palmar, radial, and ulnar pseudoaneurysms in critical care patients due to the frequent, but necessary, use of invasive lines. Emergency physicians frequently place radial or femoral arterial lines for hemodynamic monitoring in critically ill patients. However, the incidence of pseudoaneurysms and its sequelae from these lines are not usually observed in the ED setting.
Radial arterial lines may cause thrombosis in 19% to 57% of cases, and local infection in 1% to 18% of cases.10 In a study of 12,500 patients with radial artery catheters, the rate of radial pseudoaneurysm was only 0.05%.11 Although this is a small complication rate, pseudoaneurysms can lead to significant loss of function. To decrease the number of attempts and penetrating injuries to the arteries, ultrasound guidance for these procedures in the ED is strongly recommended. In addition to decreasing the risk of developing a pseudoaneurysm, ultrasound-guidance decreases the discomfort level of the patient and reduces the risk of bleeding, hematoma formation, and infection. Arterial line placement in the ED using ultrasound guidance decreases the risk of developing pseudoaneurysms and their sequelae, such as distal embolization.
Case Conclusion
The patient in this case underwent an arterial duplex study, which found a partially thrombosed right superficial palmar arch pseudoaneurysm measuring 1.91 cm x 2.08 cm, with an active flow area measuring 0.58 cm x 0.68 cm. The flow to the index finger medial artery and middle finger lateral artery was also diminished. The patient was discharged home with a bulky soft dressing and underwent excision and repair by hand surgery 3 days later. At the 1-month postoperative follow-up visit, the patient had full sensation but mildly decreased range of motion in his fingers.
Summary
Hand pseudoaneurysms are often associated with penetrating injuries—as demonstrated in our case—or repetitive microtrauma. Hand pseudoaneurysms can present with minimal findings such as isolated soft-tissue swelling, pain, or neuropathy. The EP should consider vascular pathology in the differential for patients who present with posttraumatic neuropathy. Regarding imaging studies, ultrasound is the best imaging modality to assess for pseudoaneurysms, and EPs should have a low threshold for its use at bedside—especially prior to attempting any invasive procedure. Patients with a confirmed pseudoaneurysm should be referred to a hand or vascular surgeon for surgical repair, or to an interventional radiologist for UGTI.
1. Newton EJ, Arora S. Peripheral vascular injury. In: Marx JA, Hockberger RS, Walls RM, et al, eds. Rosen’s Emergency Medicine Concepts and Clinical Practice. Vol 1. 8th ed. Philadelphia, PA: Elsevier Saunders; 2014:502.
2. Aufderheide TP. Peripheral arteriovascular disease. In: Marx JA, Hockberger RS, Walls RM, et al, eds. Rosen’s Emergency Medicine Concepts and Clinical Practice. Vol 1. 8th ed. 2014:1147-1149.
3. Anderson SE, De Monaco D, Buechler U, et al. Imaging features of pseudoaneurysms of the hand in children and adults. AJR Am J Roentgenol. 2003;180(3):659-664. doi:10.2214/ajr.180.3.1800659.
4. Shah S, Powell-Brett S, Garnham A. Pseudoaneurysm: an unusual cause of post-traumatic hand swelling. BMJ Case Rep. 2015;2015. pii: bcr2014208750. doi:10.1136/bcr-2014-208750.
5. Kitamura A, Mukohara N. Spontaneous pseudoaneurysm of the hand. Ann Vasc Surg. 2014;28(3):739.e1-e3. doi:10.1016/j.avsg.2013.04.033.
6. Huang SW, Wei TS, Liu SY, Wang WT. Spontaneous totally thrombosed pseudoaneurysm mimicking a tendon tear of the wrist. Orthopedics. 2010;33(10):776. doi:10.3928/01477447-20100826-23.
7. Belyayev L, Rich NM, McKay P, Nesti L, Wind G. Traumatic ulnar artery pseudoaneurysm following a grenade blast: report of a case. Mil Med. 2015;180(6):e725-e727. doi:10.7205/MILMED-D-14-00400.
8. Pero T, Herrick J. Pseudoaneurysm of the radial artery diagnosed by bedside ultrasound. West J Emerg Med. 2009;10(2):89-91.
9. Bosman A, Veger HTC, Doornink F, Hedeman Joosten PPA. A pseudoaneurysm of the deep palmar arch after penetrating trauma to the hand: successful exclusion by ultrasound guided percutaneous thrombin injection. EJVES Short Rep. 2016;31:9-11. doi:10.1016/j.ejvssr.2016.03.002.
10. Komorowska-Timek E, Teruya TH, Abou-Zamzam AM Jr, Papa D, Ballard JL. Treatment of radial and ulnar artery pseudoaneurysms using percutaneous thrombin injection. J Hand Surg. 2004;29A(5):936-942. doi:10.1016/j.jhsa.2004.05.009.
11. Falk PS, Scuderi PE, Sherertz RJ, Motsinger SM. Infected radial artery pseudoaneurysms occurring after percutaneous cannulation. Chest. 1992;101(2):490-495.
12. Kang SS, Labropoulos N, Mansour MA, et al. Expanded indications for ultrasound-guided thrombin injection of pseudoaneurysms. J Vasc Surg. 2000;31(2):289-298.
1. Newton EJ, Arora S. Peripheral vascular injury. In: Marx JA, Hockberger RS, Walls RM, et al, eds. Rosen’s Emergency Medicine Concepts and Clinical Practice. Vol 1. 8th ed. Philadelphia, PA: Elsevier Saunders; 2014:502.
2. Aufderheide TP. Peripheral arteriovascular disease. In: Marx JA, Hockberger RS, Walls RM, et al, eds. Rosen’s Emergency Medicine Concepts and Clinical Practice. Vol 1. 8th ed. 2014:1147-1149.
3. Anderson SE, De Monaco D, Buechler U, et al. Imaging features of pseudoaneurysms of the hand in children and adults. AJR Am J Roentgenol. 2003;180(3):659-664. doi:10.2214/ajr.180.3.1800659.
4. Shah S, Powell-Brett S, Garnham A. Pseudoaneurysm: an unusual cause of post-traumatic hand swelling. BMJ Case Rep. 2015;2015. pii: bcr2014208750. doi:10.1136/bcr-2014-208750.
5. Kitamura A, Mukohara N. Spontaneous pseudoaneurysm of the hand. Ann Vasc Surg. 2014;28(3):739.e1-e3. doi:10.1016/j.avsg.2013.04.033.
6. Huang SW, Wei TS, Liu SY, Wang WT. Spontaneous totally thrombosed pseudoaneurysm mimicking a tendon tear of the wrist. Orthopedics. 2010;33(10):776. doi:10.3928/01477447-20100826-23.
7. Belyayev L, Rich NM, McKay P, Nesti L, Wind G. Traumatic ulnar artery pseudoaneurysm following a grenade blast: report of a case. Mil Med. 2015;180(6):e725-e727. doi:10.7205/MILMED-D-14-00400.
8. Pero T, Herrick J. Pseudoaneurysm of the radial artery diagnosed by bedside ultrasound. West J Emerg Med. 2009;10(2):89-91.
9. Bosman A, Veger HTC, Doornink F, Hedeman Joosten PPA. A pseudoaneurysm of the deep palmar arch after penetrating trauma to the hand: successful exclusion by ultrasound guided percutaneous thrombin injection. EJVES Short Rep. 2016;31:9-11. doi:10.1016/j.ejvssr.2016.03.002.
10. Komorowska-Timek E, Teruya TH, Abou-Zamzam AM Jr, Papa D, Ballard JL. Treatment of radial and ulnar artery pseudoaneurysms using percutaneous thrombin injection. J Hand Surg. 2004;29A(5):936-942. doi:10.1016/j.jhsa.2004.05.009.
11. Falk PS, Scuderi PE, Sherertz RJ, Motsinger SM. Infected radial artery pseudoaneurysms occurring after percutaneous cannulation. Chest. 1992;101(2):490-495.
12. Kang SS, Labropoulos N, Mansour MA, et al. Expanded indications for ultrasound-guided thrombin injection of pseudoaneurysms. J Vasc Surg. 2000;31(2):289-298.
A Recalcitrant Case of Toxic Epidermal Necrolysis
One of the most severe complications of systemic medications is the development of a life-threatening rash, especially toxic epidermal necrolysis (TEN). Most patients can expect a full recovery if the complicating medication is discontinued early on in its course.1 When suspected TEN does not improve despite discontinuation of the detrimental medication, other diseases must be considered, particularly immunobullous and infectious etiologies. Treatment of these diseases differs substantially; therefore, a quick diagnosis is crucial. We present a case of a patient with an acute blistering eruption that was initially diagnosed and managed as TEN but physical examination and histopathologic confirmed another diagnosis. We review key examination findings that can help differentiate the causes of an acute blistering eruption with mucosal involvement, allowing for earlier diagnosis and treatment of these patients.
Case Report
An 85-year-old immunocompetent man was admitted to an outside hospital with a pruritic blistering eruption associated with myalgia, weakness, and fatigue of 3 weeks’ duration. The eruption initiated on the scalp and face and then spread down to the trunk and proximal arms and legs, with oral erosions also reported. An outside dermatologist was consulted on admission and performed a skin biopsy; the initial pathology was read as TEN. The patient was admitted to our institution on the same day, and all potentially complicating medications were stopped. He was treated with intravenous (IV)
At that time, physical examination revealed numerous confluent erosions with honey-colored crust involving the entire face (Figure 1A) and sharp demarcation at the cutaneous lip (Figure 1B). There was a large erosion on the dorsal aspect of the tongue, but the rest of the oral mucosa was spared. The trunk and proximal extremities showed numerous grouped, punched-out erosions with scalloped borders (Figure 1C). A repeat skin biopsy showed an ulcer with viral cytopathic changes. Immunoperoxidase studies demonstrated positive staining for herpes simplex virus (HSV) type 1 (Figure 2). The original slides were a frozen section from an outside facility and could not be obtained. A tissue culture and direct fluorescent antibody also confirmed HSV-1, and the patient was diagnosed with disseminated herpes. He was rapidly tapered off of the steroids and started on IV acyclovir 10 mg/kg every 8 hours for 21 days. All prior erosions reepithelialized within 7 days of treatment (Figure 3). The patient had an otherwise uncomplicated hospital course and was discharged on hospital day 21.
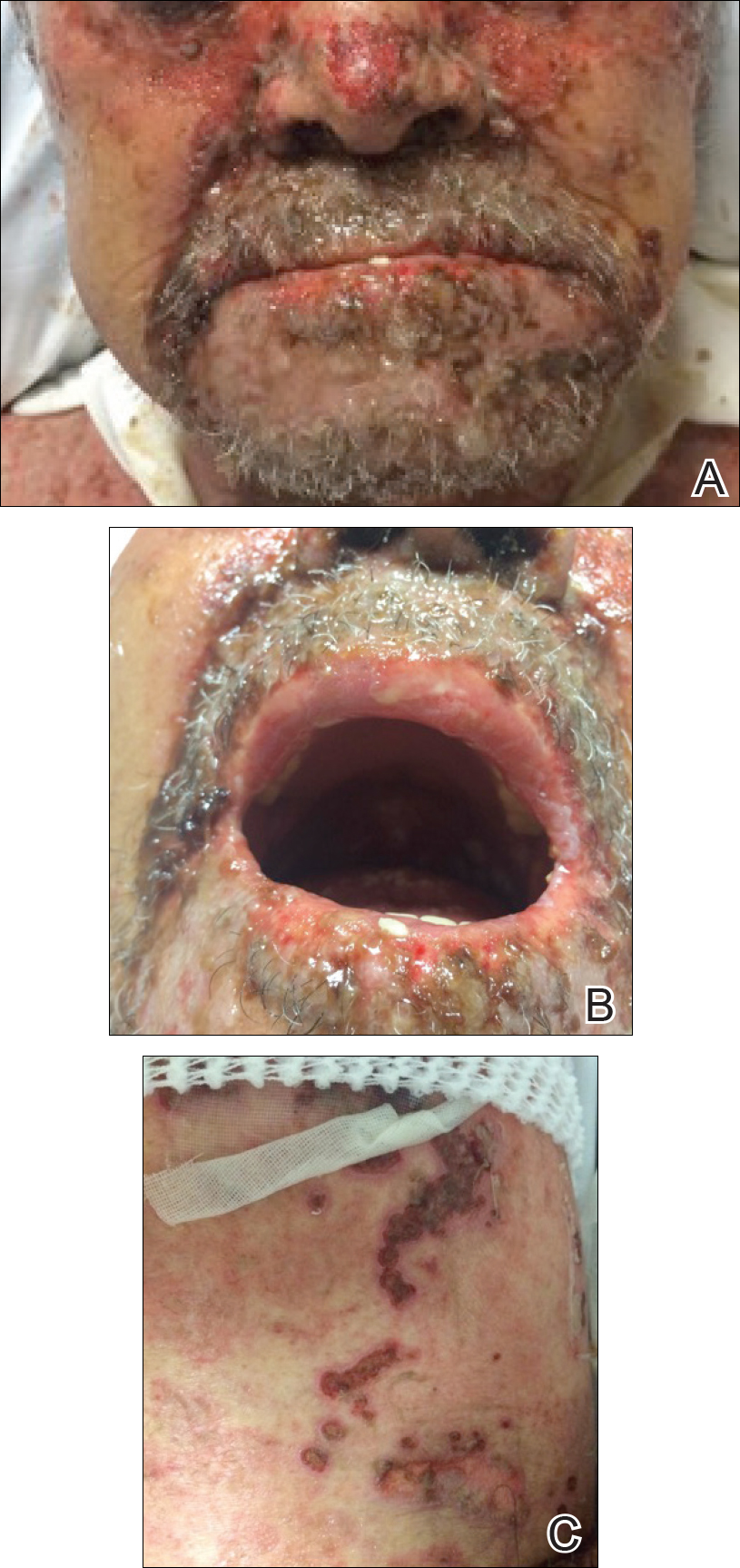
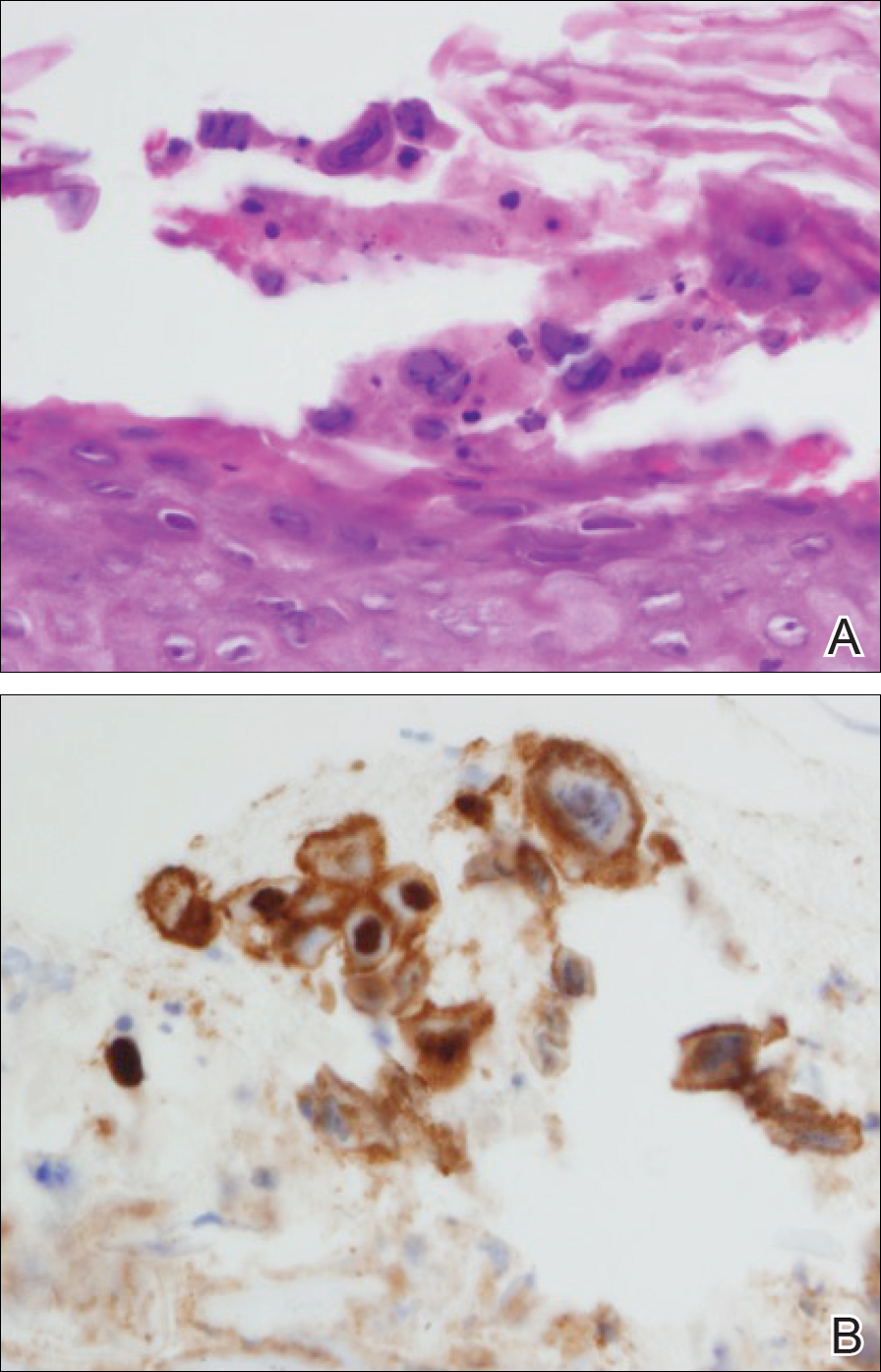
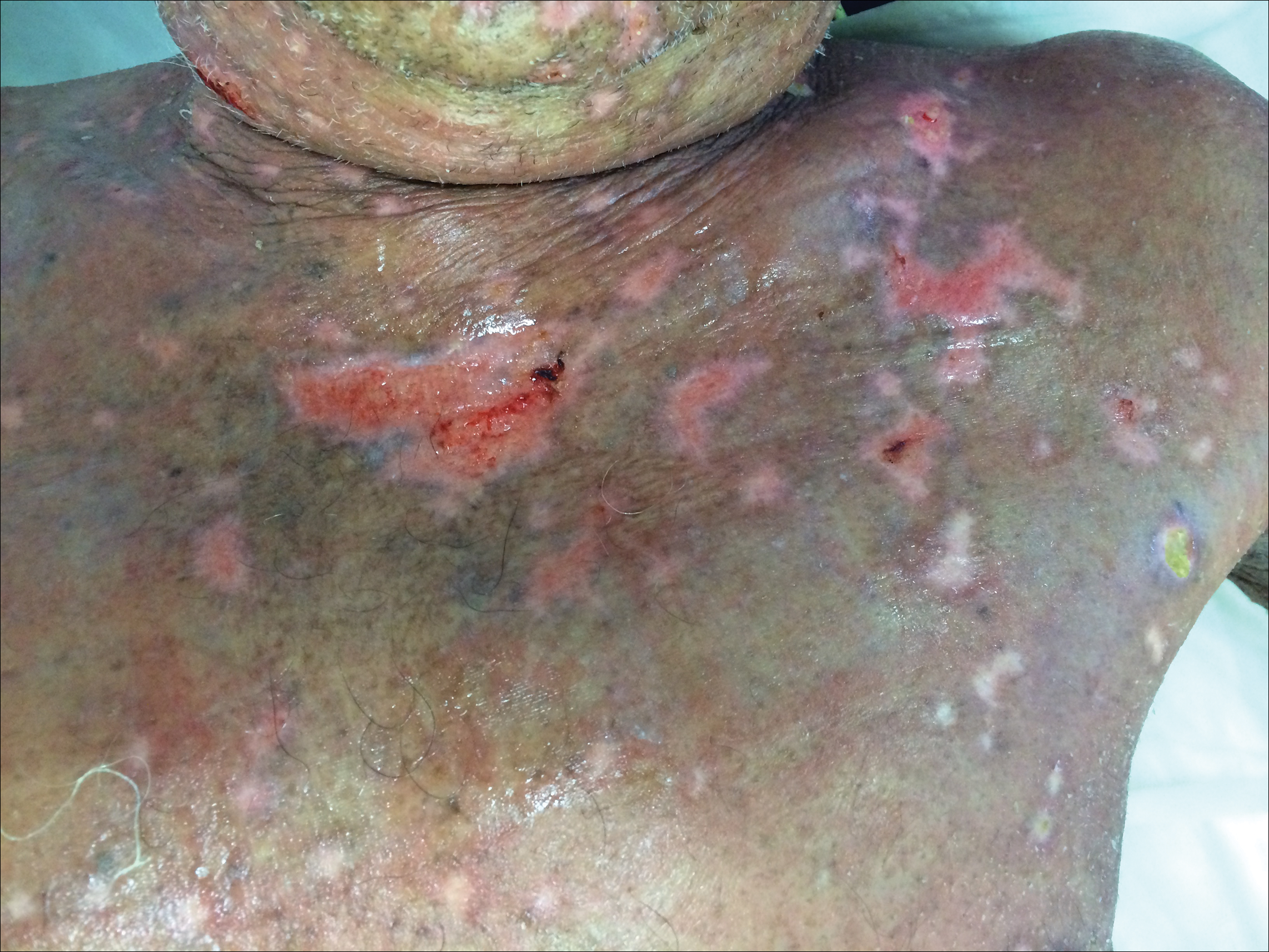
Comment
A patient with an acute generalized blistering eruption requires urgent workup and treatment given the potentially devastating sequelae. Toxic epidermal necrolysis and immunobullous diseases often are the first diagnoses to be ruled out. Certainly infections such as HSV can cause a vesicular and erosive eruption, especially in the setting of a poorly controlled dermatitis, but they typically are not in the same differential as the other diagnoses.
Clinical Presentation
This case highlights 2 key physical examination findings that can alert the clinician to a possible underlying herpetic infection. First, the distribution of this patient’s oral lesions was telling. In most cases of TEN or pemphigus vulgaris, there is notable involvement of the oral mucosa, particularly the buccal and labial mucosa. Although herpes can involve any mucocutaneous surface, it does have a predilection for keratinized tissue, with the tongue and cutaneous lip commonly involved.2,3 Our patient had a solitary linear erosion on the dorsal aspect of the tongue, but the rest of the oral cavity was strikingly spared. In addition, the erosions around the mouth stopped right at the cutaneous lip, sparing the labial mucosa (Figure 1B).
Second, the configuration of the erosions on the trunk, arms, and legs was diagnostic. Herpes classically presents as a cluster of vesicles overlying an erythematous base. When these vesicles rupture, punched-out erosions are left behind. Because these vesicles often are grouped, they can develop a scalloped border, which is a helpful indicator of HSV (Figure 1C). When these erosions become more confluent and irregular, the distinction from other conditions may not be as clear. A careful skin examination often can show areas that have preserved this herpetiform configuration.
Immune Compromise
Additionally, this case is illustrative of how immunosuppression and immunocompromise can affect the clinical presentation of HSV infection. Herpetic infections in the immunocompromised host tend to have a more protracted course, with chronic enlarging ulcers involving multiple sites.
Conclusion
This case is a good reminder that not everything that blisters and involves the mucosa is due to a hypersensitivity state such as TEN and Stevens-Johnson syndrome or an immunobullous disorder such as pemphigus vulgaris and pemphigus vegetans. The fact that this patient was worsening despite drug cessation, high-dose steroids, and IV immunoglobulin should have indicated a misdiagnosis. This case also shows that the early histopathologic findings of disseminated HSV and TEN can be nonspecific, and viral cytopathic changes may not always be obvious early in the disease.
Disseminated HSV should be considered in the differential diagnosis of a patient with an acute blistering eruption with mucosal involvement, and careful history and physical examination should be taken to rule out a viral etiology.
- Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis: part I. introduction, history, classification, clinical features, systemic manifestations, etiology, and immunopathogenesis. J Am Acad Dermatol. 2013;69:173.e1-173.e13.
- Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. New York, NY: Mosby; 2008.
- Woo SB, Lee SF. Oral recrudescent herpes simplex virus infection. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;83:239-243.
One of the most severe complications of systemic medications is the development of a life-threatening rash, especially toxic epidermal necrolysis (TEN). Most patients can expect a full recovery if the complicating medication is discontinued early on in its course.1 When suspected TEN does not improve despite discontinuation of the detrimental medication, other diseases must be considered, particularly immunobullous and infectious etiologies. Treatment of these diseases differs substantially; therefore, a quick diagnosis is crucial. We present a case of a patient with an acute blistering eruption that was initially diagnosed and managed as TEN but physical examination and histopathologic confirmed another diagnosis. We review key examination findings that can help differentiate the causes of an acute blistering eruption with mucosal involvement, allowing for earlier diagnosis and treatment of these patients.
Case Report
An 85-year-old immunocompetent man was admitted to an outside hospital with a pruritic blistering eruption associated with myalgia, weakness, and fatigue of 3 weeks’ duration. The eruption initiated on the scalp and face and then spread down to the trunk and proximal arms and legs, with oral erosions also reported. An outside dermatologist was consulted on admission and performed a skin biopsy; the initial pathology was read as TEN. The patient was admitted to our institution on the same day, and all potentially complicating medications were stopped. He was treated with intravenous (IV)
At that time, physical examination revealed numerous confluent erosions with honey-colored crust involving the entire face (Figure 1A) and sharp demarcation at the cutaneous lip (Figure 1B). There was a large erosion on the dorsal aspect of the tongue, but the rest of the oral mucosa was spared. The trunk and proximal extremities showed numerous grouped, punched-out erosions with scalloped borders (Figure 1C). A repeat skin biopsy showed an ulcer with viral cytopathic changes. Immunoperoxidase studies demonstrated positive staining for herpes simplex virus (HSV) type 1 (Figure 2). The original slides were a frozen section from an outside facility and could not be obtained. A tissue culture and direct fluorescent antibody also confirmed HSV-1, and the patient was diagnosed with disseminated herpes. He was rapidly tapered off of the steroids and started on IV acyclovir 10 mg/kg every 8 hours for 21 days. All prior erosions reepithelialized within 7 days of treatment (Figure 3). The patient had an otherwise uncomplicated hospital course and was discharged on hospital day 21.
Comment
A patient with an acute generalized blistering eruption requires urgent workup and treatment given the potentially devastating sequelae. Toxic epidermal necrolysis and immunobullous diseases often are the first diagnoses to be ruled out. Certainly infections such as HSV can cause a vesicular and erosive eruption, especially in the setting of a poorly controlled dermatitis, but they typically are not in the same differential as the other diagnoses.
Clinical Presentation
This case highlights 2 key physical examination findings that can alert the clinician to a possible underlying herpetic infection. First, the distribution of this patient’s oral lesions was telling. In most cases of TEN or pemphigus vulgaris, there is notable involvement of the oral mucosa, particularly the buccal and labial mucosa. Although herpes can involve any mucocutaneous surface, it does have a predilection for keratinized tissue, with the tongue and cutaneous lip commonly involved.2,3 Our patient had a solitary linear erosion on the dorsal aspect of the tongue, but the rest of the oral cavity was strikingly spared. In addition, the erosions around the mouth stopped right at the cutaneous lip, sparing the labial mucosa (Figure 1B).
Second, the configuration of the erosions on the trunk, arms, and legs was diagnostic. Herpes classically presents as a cluster of vesicles overlying an erythematous base. When these vesicles rupture, punched-out erosions are left behind. Because these vesicles often are grouped, they can develop a scalloped border, which is a helpful indicator of HSV (Figure 1C). When these erosions become more confluent and irregular, the distinction from other conditions may not be as clear. A careful skin examination often can show areas that have preserved this herpetiform configuration.
Immune Compromise
Additionally, this case is illustrative of how immunosuppression and immunocompromise can affect the clinical presentation of HSV infection. Herpetic infections in the immunocompromised host tend to have a more protracted course, with chronic enlarging ulcers involving multiple sites.
Conclusion
This case is a good reminder that not everything that blisters and involves the mucosa is due to a hypersensitivity state such as TEN and Stevens-Johnson syndrome or an immunobullous disorder such as pemphigus vulgaris and pemphigus vegetans. The fact that this patient was worsening despite drug cessation, high-dose steroids, and IV immunoglobulin should have indicated a misdiagnosis. This case also shows that the early histopathologic findings of disseminated HSV and TEN can be nonspecific, and viral cytopathic changes may not always be obvious early in the disease.
Disseminated HSV should be considered in the differential diagnosis of a patient with an acute blistering eruption with mucosal involvement, and careful history and physical examination should be taken to rule out a viral etiology.
One of the most severe complications of systemic medications is the development of a life-threatening rash, especially toxic epidermal necrolysis (TEN). Most patients can expect a full recovery if the complicating medication is discontinued early on in its course.1 When suspected TEN does not improve despite discontinuation of the detrimental medication, other diseases must be considered, particularly immunobullous and infectious etiologies. Treatment of these diseases differs substantially; therefore, a quick diagnosis is crucial. We present a case of a patient with an acute blistering eruption that was initially diagnosed and managed as TEN but physical examination and histopathologic confirmed another diagnosis. We review key examination findings that can help differentiate the causes of an acute blistering eruption with mucosal involvement, allowing for earlier diagnosis and treatment of these patients.
Case Report
An 85-year-old immunocompetent man was admitted to an outside hospital with a pruritic blistering eruption associated with myalgia, weakness, and fatigue of 3 weeks’ duration. The eruption initiated on the scalp and face and then spread down to the trunk and proximal arms and legs, with oral erosions also reported. An outside dermatologist was consulted on admission and performed a skin biopsy; the initial pathology was read as TEN. The patient was admitted to our institution on the same day, and all potentially complicating medications were stopped. He was treated with intravenous (IV)
At that time, physical examination revealed numerous confluent erosions with honey-colored crust involving the entire face (Figure 1A) and sharp demarcation at the cutaneous lip (Figure 1B). There was a large erosion on the dorsal aspect of the tongue, but the rest of the oral mucosa was spared. The trunk and proximal extremities showed numerous grouped, punched-out erosions with scalloped borders (Figure 1C). A repeat skin biopsy showed an ulcer with viral cytopathic changes. Immunoperoxidase studies demonstrated positive staining for herpes simplex virus (HSV) type 1 (Figure 2). The original slides were a frozen section from an outside facility and could not be obtained. A tissue culture and direct fluorescent antibody also confirmed HSV-1, and the patient was diagnosed with disseminated herpes. He was rapidly tapered off of the steroids and started on IV acyclovir 10 mg/kg every 8 hours for 21 days. All prior erosions reepithelialized within 7 days of treatment (Figure 3). The patient had an otherwise uncomplicated hospital course and was discharged on hospital day 21.
Comment
A patient with an acute generalized blistering eruption requires urgent workup and treatment given the potentially devastating sequelae. Toxic epidermal necrolysis and immunobullous diseases often are the first diagnoses to be ruled out. Certainly infections such as HSV can cause a vesicular and erosive eruption, especially in the setting of a poorly controlled dermatitis, but they typically are not in the same differential as the other diagnoses.
Clinical Presentation
This case highlights 2 key physical examination findings that can alert the clinician to a possible underlying herpetic infection. First, the distribution of this patient’s oral lesions was telling. In most cases of TEN or pemphigus vulgaris, there is notable involvement of the oral mucosa, particularly the buccal and labial mucosa. Although herpes can involve any mucocutaneous surface, it does have a predilection for keratinized tissue, with the tongue and cutaneous lip commonly involved.2,3 Our patient had a solitary linear erosion on the dorsal aspect of the tongue, but the rest of the oral cavity was strikingly spared. In addition, the erosions around the mouth stopped right at the cutaneous lip, sparing the labial mucosa (Figure 1B).
Second, the configuration of the erosions on the trunk, arms, and legs was diagnostic. Herpes classically presents as a cluster of vesicles overlying an erythematous base. When these vesicles rupture, punched-out erosions are left behind. Because these vesicles often are grouped, they can develop a scalloped border, which is a helpful indicator of HSV (Figure 1C). When these erosions become more confluent and irregular, the distinction from other conditions may not be as clear. A careful skin examination often can show areas that have preserved this herpetiform configuration.
Immune Compromise
Additionally, this case is illustrative of how immunosuppression and immunocompromise can affect the clinical presentation of HSV infection. Herpetic infections in the immunocompromised host tend to have a more protracted course, with chronic enlarging ulcers involving multiple sites.
Conclusion
This case is a good reminder that not everything that blisters and involves the mucosa is due to a hypersensitivity state such as TEN and Stevens-Johnson syndrome or an immunobullous disorder such as pemphigus vulgaris and pemphigus vegetans. The fact that this patient was worsening despite drug cessation, high-dose steroids, and IV immunoglobulin should have indicated a misdiagnosis. This case also shows that the early histopathologic findings of disseminated HSV and TEN can be nonspecific, and viral cytopathic changes may not always be obvious early in the disease.
Disseminated HSV should be considered in the differential diagnosis of a patient with an acute blistering eruption with mucosal involvement, and careful history and physical examination should be taken to rule out a viral etiology.
- Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis: part I. introduction, history, classification, clinical features, systemic manifestations, etiology, and immunopathogenesis. J Am Acad Dermatol. 2013;69:173.e1-173.e13.
- Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. New York, NY: Mosby; 2008.
- Woo SB, Lee SF. Oral recrudescent herpes simplex virus infection. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;83:239-243.
- Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis: part I. introduction, history, classification, clinical features, systemic manifestations, etiology, and immunopathogenesis. J Am Acad Dermatol. 2013;69:173.e1-173.e13.
- Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. New York, NY: Mosby; 2008.
- Woo SB, Lee SF. Oral recrudescent herpes simplex virus infection. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;83:239-243.
Practice Points
- Toxic epidermal necrolysis can be difficult to diagnose and treat.
- Patients who are refractory to treatment should prompt further management considerations.
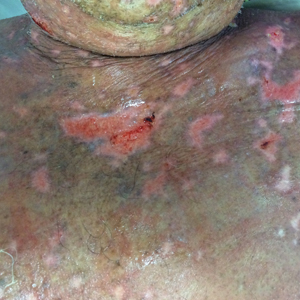
Atraumatic splenic rupture as an initial presentation of chronic myelogenous leukemia
Chronic myelogenous leukemia (CML) is a myeloproliferative neoplasm associated with the fusion of the BCR gene located on chromosome 22 and the ABL1 gene on chromosome 9. The fusion results in a reciprocal translocation between chromosomes 9 and 22, leading to the formation of the Philadelphia (Ph) chromosome found in 90%-95% of patients with CML. The incidence of CML is 1.5 per 100,000 people per year, with a male predominance and an average age at diagnosis of 64.1
About 85%-90% of newly diagnosed patients present in the chronic phase and therefore many of them are asymptomatic at the time of diagnosis. If symptoms are present, they often include fatigue, malaise, unintentional weight loss, early satiety, or left upper quadrant pain. Progression of the disease is associated with worsening symptoms such as unexplained fever, significant weight loss, bone or joint pain, bleeding, thrombosis, and infections suggestive of transformation to the accelerated phase or blast crisis. Physical exam findings most commonly include splenomegaly and occasionally mild hepatomegaly.
Atraumatic splenic rupture is a rare complication of this hematologic malignancy, and there are almost no reported cases of CML as the underlying cause.2-4 Here we present the case of a man with sudden-onset generalized abdominal pain and leukocytosis. A computed-tomography scan showed splenic rupture, and the patient was taken for emergency splenectomy. The patient was subsequently positive for t(9,22)(q34;q11.2).
Case presentation and summary
A 59-year-old white man with a history of hypertension and kidney stones presented to a community emergency department with a chief complaint of abdominal pain. About 30 minutes before his arrival, the patient had woken up from sleep with generalized, nonradiating, abdominal pain, which he described as “like my previous kidney stones.” He also reported worsening dyspnea, nausea without vomiting, and lightheadedness without loss of consciousness. The remainder of the review of systems was negative. A physical exam revealed that he was in moderate distress with clear lung fields and had tachycardia without murmur, no CVA tenderness, and a diffusely tender abdomen.
Complete blood count with differential showed leukocytosis (109.1 x 103/uL), normocytic anemia (8.1 g/dL), thrombocytopenia (100,000 cells/uL), neutrophils (71.06 cells/uL), bands (27.13 cells/uL), and monocytes (11.63 cells/uL). A CT scan of the abdomen and pelvis showed a grade 4 splenic laceration with significant free abdominal fluid (Figure 1).
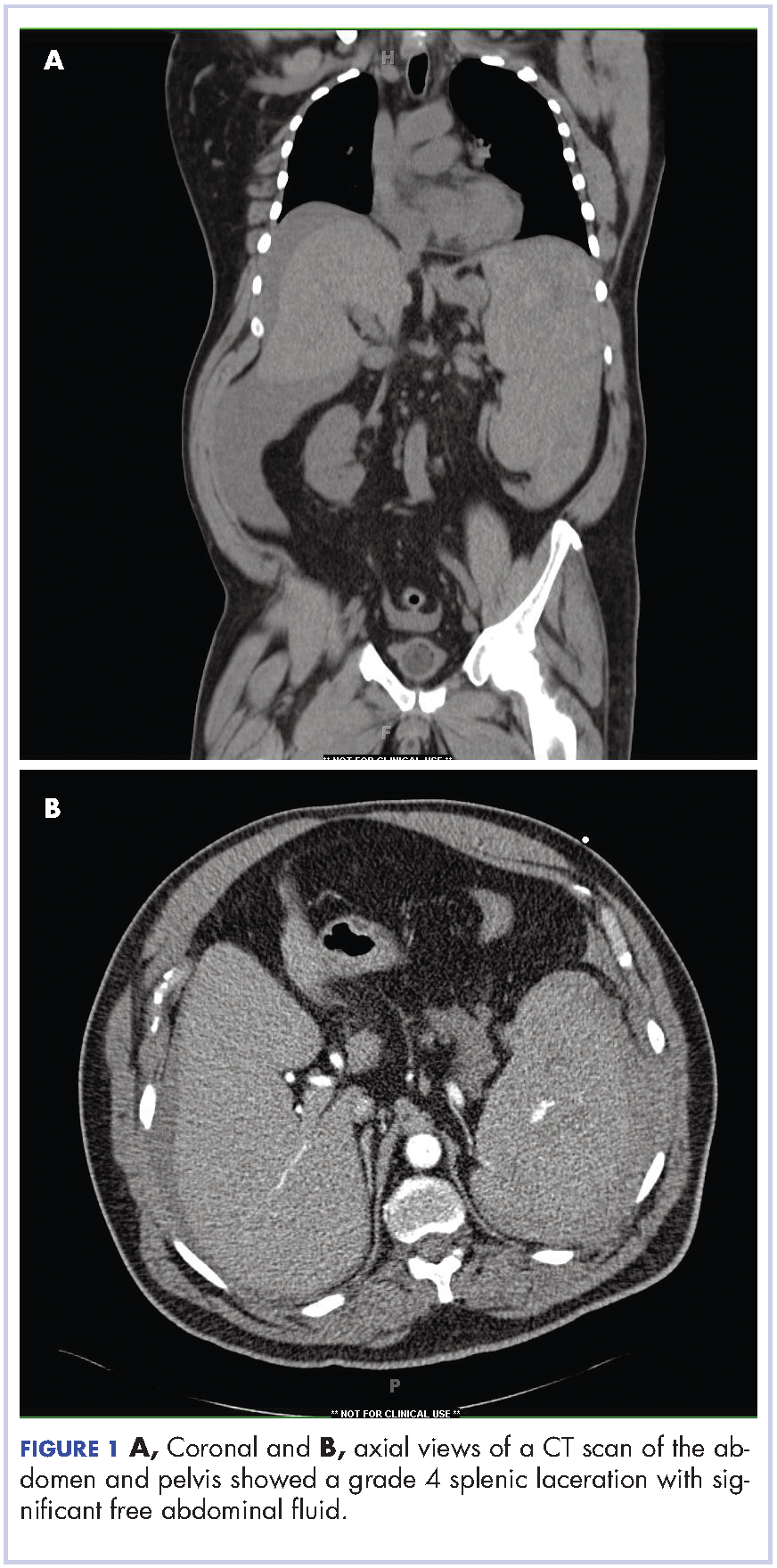
The patient was taken to the operating room where he underwent a splenectomy which was complicated by partial gastrectomy and partial omentectomy. He remained intubated on mechanical ventilation in the intensive care for 7 days. His progress was complicated by profound hypotension that required significant fluid administration and ultimately multiple pressors for blood pressure support. Hypotensive shock was beginning to improve on day 3 and was completely resolved by day 5. The patient underwent continuous positive airway pressure (CPAP) trials on day 6 and was successfully extubated on day 7.
After extubation a more thorough history could be obtained from the patient. He denied any history of weight loss, night sweats, or fatigue. Patient denied any known family history of hematologic malignancies. His peripheral smear showed basophilia and granulocytosis with neutrophils and immature granulocytes (Figure 2). The patient was evaluated by the hematology service and was started on allopurinol and hydroxyurea for presumed hematologic malignancy. He was given the meningococcus and streptococcus pneumoniae vaccine and was discharged home in stable condition on day eleven. Patient was subsequently positive for t(9,22)(q34;q11.2) and was started on imatinib. He has continued to follow in the clinic and is currently in remission.
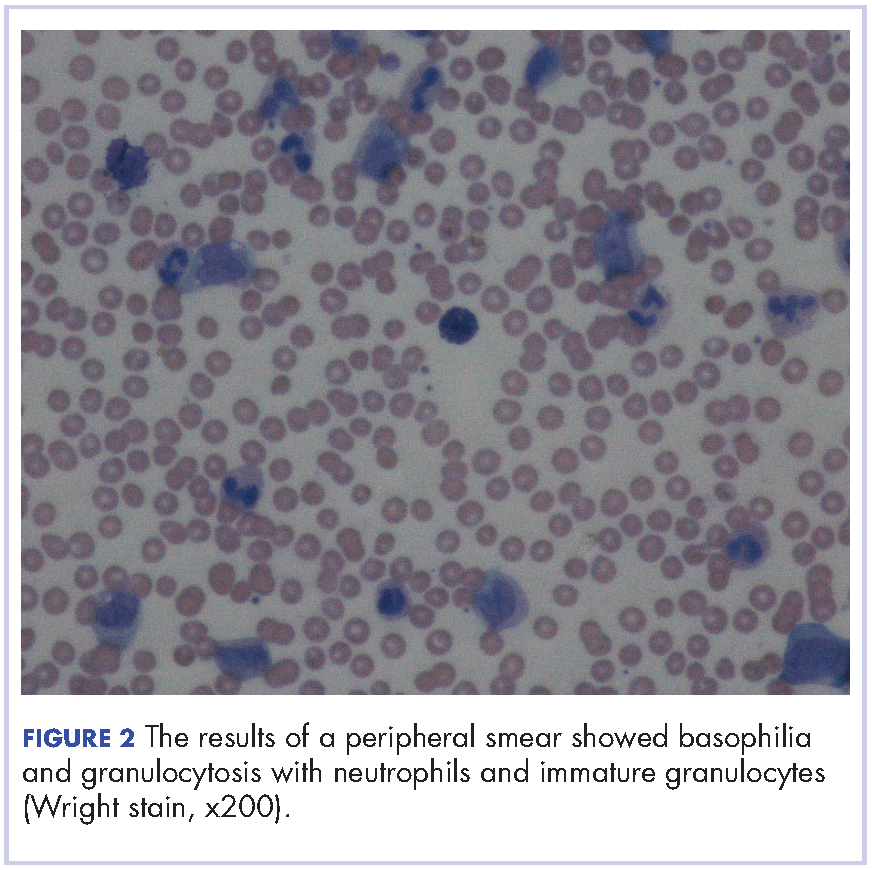
Discussion
CML has a triphasic clinical course and treatment is based on the specific disease phase. The 3 phases of the disease include the chronic (more indolent) phase, accelerated (more aggressive) phase, and blast crisis. If the disease is left untreated, it will inevitably transition from a chronic to an accelerated phase and finally to blast crisis within a median time of 4 years.
The chronic phase is the most common, representing 85% of diagnoses. Patients can be asymptomatic and many in this phase will be diagnosed by routine lab testing.5 According to the World Health Organization, the accelerated phase is defined as CML patients with one of the following: 10%-19% blasts, basophils ≥20%, platelets <100,000/microL or >1,000,000/microL, unresponsive to therapy, splenomegaly unresponsive to therapy, an increasing white cell count unresponsive to therapy, or cytogenetic evolution.6 Blast crisis is the most aggressive phase and is usually defined by ≥20% blasts, large foci or clusters of blasts on the bone marrow biopsy, or the presence of extramedullary blastic infiltrates.7,8
The diagnosis of CML should be suspected in the presence of distinct lab abnormalities in the peripheral blood. These include elevated white blood cell counts with a median count of 100,000 cells/microL, elevated platelet counts, and a mild normocytic normochromic anemia. Platelet counts of 600,000 or greater have been seen in 15%-30% of patients at the time of diagnosis. The white count differential can show a variety of cells but there will be a notably greater percentage of myelocytes than metamyelocytes. Bone marrow biopsy will reveal increased cellularity, normal to slightly elevated percentage of blasts, and reticulin fibrosis. The diagnosis should be confirmed by the presence of the Philadelphia chromosome either by cytogenetics, fluorescence in situ hybridization, or reverse-transcription polymerase chain reaction (RT-PCR). The Philadelphia chromosome is found in 90%-95% of patients with CML. Most of the remaining patients will have other translocations, but a small minority will have no detectable genetic abnormalities and those patients are known as Ph-negative.9
Treatment options for CML include potential cure with allogeneic hematopoietic stem-cell transplant (HSCT) or disease control using tyrosine kinase inhibitors (TKIs). TKIs are the initial treatment of choice for newly diagnosed patients and are able to produce long-term remission in most patients. The drugs in this category include imatinib, dasatinib, and nilotinib. They work by inhibiting the Bcr-Abl tyrosine kinase, thereby blocking proliferation and inducing apoptosis in Bcr-Abl-positive cells. The majority of patients with chronic-phase CML will have an excellent response to initial treatment with a TKI. It is critical to follow these patients on a regular basis and monitor their disease status. Although the gold standard for assessing cytogenetic response is cytogenetic analysis of a bone marrow biopsy, more sensitive methods such as quantitative PCR using peripheral blood are now available, thereby minimizing the need for bone marrow biopsy. Patients in the accelerated phase are more difficult to manage because they are resistant to most forms of treatment and have short-lived responses to TKI therapy. These patients should strongly be considered for transplantation. Patients in blast crisis have aggressive disease that is more complex and requires more extensive testing. These patients should ideally be treated at tertiary care centers and treatment often involves chemotherapy in addition to TKI therapy usually followed by HSCT.
Atraumatic splenic rupture (ASR) presents similarly to traumatic splenic rupture with typical symptoms being acute onset of upper abdominal, left chest wall, or left shoulder pain (Kehr’s sign) but without a known history of trauma. Quick recognition and surgical intervention represent the best means of definitive care.10 Renzulli and colleagues conducted a literature review for all ASR cases from 1980-2008, examining 632 publications representing 845 cases. They examined the cases using logistic regression analysis to better define the clinicopathology behind ASR. The reported causes of ASR are neoplastic processes (30.3%), infectious (27.3%), inflammatory noninfectious (20.0%), drug- and treatment-related (9.2%), mechanical (6.8%), and normal spleen (6.4%). Treatment included total splenectomy in 84.1% of cases, organ-preserving surgery in 1.2%, and conservative measures in 14.7%. They reported an ASR-related mortality of 12.2%, with being older than 40 and neoplastic disorders associated with increased mortality – although male sex and splenomegaly have also been reported.11-13 Thomas and colleagues have reported on 48 cases of ASR related to hematologic malignancy showing acute myeloid leukemia being the most common cause (21%), followed by acute lymphoblastic leukemia (19%).2
Hematologic malignancies commonly cause splenic engorgement and pain although splenic rupture is an extremely rare event. Recent literature review has shown fewer than a thousand reported cases since 1980.4 There far fewer reported cases of ASR being related to CML, with most being reported as a complication.3,14 Based on our review, we could identify only a handful cases of CML with ASR being the initial symptom. These include a patient with Ph-negative CML and ASR following blast crisis, a patient with Phil-negative BCR-ABL-positive essential thrombocythemia, several cases in which the patient ultimately died, and 1 in which the patient survived into remission.4,14-16 Our case is different because the patient was ultimately positive for t(9,22)(q34;q11.2) and although he experienced multiple complications, he is currently functioning at his baseline and in remission. We hope this case will remind others that CML should be considered in the differential diagnosis of patients ASR.
1. American Cancer Society. Cancer Facts & Figures 2015. Atlanta, Ga: American Cancer Society; 2015.
2. Bauer TW, Haskins GE, Armitage JO. Splenic rupture in patients with hematologic malignancies. Cancer. 1981;48:2729-2733.
3. Giagounidis AA, Burk M, Meckenstock G, Koch AJ, Schneider W. Pathologic rupture of the spleen in hematologic malignancies: two additional cases. Ann Hematol. 1996;73(6):297-302.
4. Goodard SL, Chesney AE, Reis MD, et al. Pathologic splenic rupture: a rare complication of chronic myelomonocytic leukemia. Am J Hematology. 2007;82:405-408.
5. Faderl S, Talpaz M, Estrov Z, et al. The biology of chronic myeloid leukemia. N Engl J Med. 1999;341:164-172.
6. Cortes JE, Talpaz M, O’Brien S, et al. Staging of chronic myeloid leukemia in the imatinib era: an evaluation of the World Health Organization proposal. Cancer. 2006;106:1306-1315.
7. Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100:2292-2302.
8. Kantarjian HM, O’Brien S, Cortes J, et al. Results of decitabine (5-aza-2’deoxycytidine) therapy in 130 patients with chronic myelogenous leukemia. Cancer.2003; 98:522-528.
9. Swerdlow SH, Campo E, Harris NL, et al, editors. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: IARC Press; 2008.
10. Maung A, KaplanL. Management of splenic injury in the adult trauma patient. In: UpToDate, Basow DS (ed), Waltham, MA, 2013.
11. Renzulli P, Hostettler A, Schoepfer AM, Gloor B, Candinas D. Systematic review of atraumatic splenic rupture. Br J Surg. 2009;8(10):1114-1121.
12. Hehlmann R, Heimpel H, Hasford J, et al. Randomized comparison of interferon-alpha with busulfan and hydroxyurea in chronic myelogenous leukemia. The German CML Study Group. Blood. 1994;84:4064-4077.
13. Cortes J, Kantarjian H. How I treat newly diagnosed chronic phase CML. Blood. 2012;120:1390-1397.
14. Nestok BR, Goldstein JD, Lipkovic P. Splenic rupture as a cause of sudden death in undiagnosed chronic myelogenous leukemia. Am J Forensic Med Pathol. 1988;9:241-245.
15. Sachithanandan A, Gleadhil I, Alexander HD, Morris TC. Spontaneous splenic rupture in atypical (Philadelphia chromosome negative) chronic myeloid leukemia following blastic crisis. Ir Med J. 2003;96(6):181-182.
16. Chim CS, Kwong YL, Shek TW, Ma SK, Ooi GC. Splenic rupture as the presenting symptom of blastic crisis in a patient with Philadelphia-negative, BCR-ABL-positive ET. Am J Hematology. 2001;66:70-71.
Chronic myelogenous leukemia (CML) is a myeloproliferative neoplasm associated with the fusion of the BCR gene located on chromosome 22 and the ABL1 gene on chromosome 9. The fusion results in a reciprocal translocation between chromosomes 9 and 22, leading to the formation of the Philadelphia (Ph) chromosome found in 90%-95% of patients with CML. The incidence of CML is 1.5 per 100,000 people per year, with a male predominance and an average age at diagnosis of 64.1
About 85%-90% of newly diagnosed patients present in the chronic phase and therefore many of them are asymptomatic at the time of diagnosis. If symptoms are present, they often include fatigue, malaise, unintentional weight loss, early satiety, or left upper quadrant pain. Progression of the disease is associated with worsening symptoms such as unexplained fever, significant weight loss, bone or joint pain, bleeding, thrombosis, and infections suggestive of transformation to the accelerated phase or blast crisis. Physical exam findings most commonly include splenomegaly and occasionally mild hepatomegaly.
Atraumatic splenic rupture is a rare complication of this hematologic malignancy, and there are almost no reported cases of CML as the underlying cause.2-4 Here we present the case of a man with sudden-onset generalized abdominal pain and leukocytosis. A computed-tomography scan showed splenic rupture, and the patient was taken for emergency splenectomy. The patient was subsequently positive for t(9,22)(q34;q11.2).
Case presentation and summary
A 59-year-old white man with a history of hypertension and kidney stones presented to a community emergency department with a chief complaint of abdominal pain. About 30 minutes before his arrival, the patient had woken up from sleep with generalized, nonradiating, abdominal pain, which he described as “like my previous kidney stones.” He also reported worsening dyspnea, nausea without vomiting, and lightheadedness without loss of consciousness. The remainder of the review of systems was negative. A physical exam revealed that he was in moderate distress with clear lung fields and had tachycardia without murmur, no CVA tenderness, and a diffusely tender abdomen.
Complete blood count with differential showed leukocytosis (109.1 x 103/uL), normocytic anemia (8.1 g/dL), thrombocytopenia (100,000 cells/uL), neutrophils (71.06 cells/uL), bands (27.13 cells/uL), and monocytes (11.63 cells/uL). A CT scan of the abdomen and pelvis showed a grade 4 splenic laceration with significant free abdominal fluid (Figure 1).
The patient was taken to the operating room where he underwent a splenectomy which was complicated by partial gastrectomy and partial omentectomy. He remained intubated on mechanical ventilation in the intensive care for 7 days. His progress was complicated by profound hypotension that required significant fluid administration and ultimately multiple pressors for blood pressure support. Hypotensive shock was beginning to improve on day 3 and was completely resolved by day 5. The patient underwent continuous positive airway pressure (CPAP) trials on day 6 and was successfully extubated on day 7.
After extubation a more thorough history could be obtained from the patient. He denied any history of weight loss, night sweats, or fatigue. Patient denied any known family history of hematologic malignancies. His peripheral smear showed basophilia and granulocytosis with neutrophils and immature granulocytes (Figure 2). The patient was evaluated by the hematology service and was started on allopurinol and hydroxyurea for presumed hematologic malignancy. He was given the meningococcus and streptococcus pneumoniae vaccine and was discharged home in stable condition on day eleven. Patient was subsequently positive for t(9,22)(q34;q11.2) and was started on imatinib. He has continued to follow in the clinic and is currently in remission.
Discussion
CML has a triphasic clinical course and treatment is based on the specific disease phase. The 3 phases of the disease include the chronic (more indolent) phase, accelerated (more aggressive) phase, and blast crisis. If the disease is left untreated, it will inevitably transition from a chronic to an accelerated phase and finally to blast crisis within a median time of 4 years.
The chronic phase is the most common, representing 85% of diagnoses. Patients can be asymptomatic and many in this phase will be diagnosed by routine lab testing.5 According to the World Health Organization, the accelerated phase is defined as CML patients with one of the following: 10%-19% blasts, basophils ≥20%, platelets <100,000/microL or >1,000,000/microL, unresponsive to therapy, splenomegaly unresponsive to therapy, an increasing white cell count unresponsive to therapy, or cytogenetic evolution.6 Blast crisis is the most aggressive phase and is usually defined by ≥20% blasts, large foci or clusters of blasts on the bone marrow biopsy, or the presence of extramedullary blastic infiltrates.7,8
The diagnosis of CML should be suspected in the presence of distinct lab abnormalities in the peripheral blood. These include elevated white blood cell counts with a median count of 100,000 cells/microL, elevated platelet counts, and a mild normocytic normochromic anemia. Platelet counts of 600,000 or greater have been seen in 15%-30% of patients at the time of diagnosis. The white count differential can show a variety of cells but there will be a notably greater percentage of myelocytes than metamyelocytes. Bone marrow biopsy will reveal increased cellularity, normal to slightly elevated percentage of blasts, and reticulin fibrosis. The diagnosis should be confirmed by the presence of the Philadelphia chromosome either by cytogenetics, fluorescence in situ hybridization, or reverse-transcription polymerase chain reaction (RT-PCR). The Philadelphia chromosome is found in 90%-95% of patients with CML. Most of the remaining patients will have other translocations, but a small minority will have no detectable genetic abnormalities and those patients are known as Ph-negative.9
Treatment options for CML include potential cure with allogeneic hematopoietic stem-cell transplant (HSCT) or disease control using tyrosine kinase inhibitors (TKIs). TKIs are the initial treatment of choice for newly diagnosed patients and are able to produce long-term remission in most patients. The drugs in this category include imatinib, dasatinib, and nilotinib. They work by inhibiting the Bcr-Abl tyrosine kinase, thereby blocking proliferation and inducing apoptosis in Bcr-Abl-positive cells. The majority of patients with chronic-phase CML will have an excellent response to initial treatment with a TKI. It is critical to follow these patients on a regular basis and monitor their disease status. Although the gold standard for assessing cytogenetic response is cytogenetic analysis of a bone marrow biopsy, more sensitive methods such as quantitative PCR using peripheral blood are now available, thereby minimizing the need for bone marrow biopsy. Patients in the accelerated phase are more difficult to manage because they are resistant to most forms of treatment and have short-lived responses to TKI therapy. These patients should strongly be considered for transplantation. Patients in blast crisis have aggressive disease that is more complex and requires more extensive testing. These patients should ideally be treated at tertiary care centers and treatment often involves chemotherapy in addition to TKI therapy usually followed by HSCT.
Atraumatic splenic rupture (ASR) presents similarly to traumatic splenic rupture with typical symptoms being acute onset of upper abdominal, left chest wall, or left shoulder pain (Kehr’s sign) but without a known history of trauma. Quick recognition and surgical intervention represent the best means of definitive care.10 Renzulli and colleagues conducted a literature review for all ASR cases from 1980-2008, examining 632 publications representing 845 cases. They examined the cases using logistic regression analysis to better define the clinicopathology behind ASR. The reported causes of ASR are neoplastic processes (30.3%), infectious (27.3%), inflammatory noninfectious (20.0%), drug- and treatment-related (9.2%), mechanical (6.8%), and normal spleen (6.4%). Treatment included total splenectomy in 84.1% of cases, organ-preserving surgery in 1.2%, and conservative measures in 14.7%. They reported an ASR-related mortality of 12.2%, with being older than 40 and neoplastic disorders associated with increased mortality – although male sex and splenomegaly have also been reported.11-13 Thomas and colleagues have reported on 48 cases of ASR related to hematologic malignancy showing acute myeloid leukemia being the most common cause (21%), followed by acute lymphoblastic leukemia (19%).2
Hematologic malignancies commonly cause splenic engorgement and pain although splenic rupture is an extremely rare event. Recent literature review has shown fewer than a thousand reported cases since 1980.4 There far fewer reported cases of ASR being related to CML, with most being reported as a complication.3,14 Based on our review, we could identify only a handful cases of CML with ASR being the initial symptom. These include a patient with Ph-negative CML and ASR following blast crisis, a patient with Phil-negative BCR-ABL-positive essential thrombocythemia, several cases in which the patient ultimately died, and 1 in which the patient survived into remission.4,14-16 Our case is different because the patient was ultimately positive for t(9,22)(q34;q11.2) and although he experienced multiple complications, he is currently functioning at his baseline and in remission. We hope this case will remind others that CML should be considered in the differential diagnosis of patients ASR.
Chronic myelogenous leukemia (CML) is a myeloproliferative neoplasm associated with the fusion of the BCR gene located on chromosome 22 and the ABL1 gene on chromosome 9. The fusion results in a reciprocal translocation between chromosomes 9 and 22, leading to the formation of the Philadelphia (Ph) chromosome found in 90%-95% of patients with CML. The incidence of CML is 1.5 per 100,000 people per year, with a male predominance and an average age at diagnosis of 64.1
About 85%-90% of newly diagnosed patients present in the chronic phase and therefore many of them are asymptomatic at the time of diagnosis. If symptoms are present, they often include fatigue, malaise, unintentional weight loss, early satiety, or left upper quadrant pain. Progression of the disease is associated with worsening symptoms such as unexplained fever, significant weight loss, bone or joint pain, bleeding, thrombosis, and infections suggestive of transformation to the accelerated phase or blast crisis. Physical exam findings most commonly include splenomegaly and occasionally mild hepatomegaly.
Atraumatic splenic rupture is a rare complication of this hematologic malignancy, and there are almost no reported cases of CML as the underlying cause.2-4 Here we present the case of a man with sudden-onset generalized abdominal pain and leukocytosis. A computed-tomography scan showed splenic rupture, and the patient was taken for emergency splenectomy. The patient was subsequently positive for t(9,22)(q34;q11.2).
Case presentation and summary
A 59-year-old white man with a history of hypertension and kidney stones presented to a community emergency department with a chief complaint of abdominal pain. About 30 minutes before his arrival, the patient had woken up from sleep with generalized, nonradiating, abdominal pain, which he described as “like my previous kidney stones.” He also reported worsening dyspnea, nausea without vomiting, and lightheadedness without loss of consciousness. The remainder of the review of systems was negative. A physical exam revealed that he was in moderate distress with clear lung fields and had tachycardia without murmur, no CVA tenderness, and a diffusely tender abdomen.
Complete blood count with differential showed leukocytosis (109.1 x 103/uL), normocytic anemia (8.1 g/dL), thrombocytopenia (100,000 cells/uL), neutrophils (71.06 cells/uL), bands (27.13 cells/uL), and monocytes (11.63 cells/uL). A CT scan of the abdomen and pelvis showed a grade 4 splenic laceration with significant free abdominal fluid (Figure 1).
The patient was taken to the operating room where he underwent a splenectomy which was complicated by partial gastrectomy and partial omentectomy. He remained intubated on mechanical ventilation in the intensive care for 7 days. His progress was complicated by profound hypotension that required significant fluid administration and ultimately multiple pressors for blood pressure support. Hypotensive shock was beginning to improve on day 3 and was completely resolved by day 5. The patient underwent continuous positive airway pressure (CPAP) trials on day 6 and was successfully extubated on day 7.
After extubation a more thorough history could be obtained from the patient. He denied any history of weight loss, night sweats, or fatigue. Patient denied any known family history of hematologic malignancies. His peripheral smear showed basophilia and granulocytosis with neutrophils and immature granulocytes (Figure 2). The patient was evaluated by the hematology service and was started on allopurinol and hydroxyurea for presumed hematologic malignancy. He was given the meningococcus and streptococcus pneumoniae vaccine and was discharged home in stable condition on day eleven. Patient was subsequently positive for t(9,22)(q34;q11.2) and was started on imatinib. He has continued to follow in the clinic and is currently in remission.
Discussion
CML has a triphasic clinical course and treatment is based on the specific disease phase. The 3 phases of the disease include the chronic (more indolent) phase, accelerated (more aggressive) phase, and blast crisis. If the disease is left untreated, it will inevitably transition from a chronic to an accelerated phase and finally to blast crisis within a median time of 4 years.
The chronic phase is the most common, representing 85% of diagnoses. Patients can be asymptomatic and many in this phase will be diagnosed by routine lab testing.5 According to the World Health Organization, the accelerated phase is defined as CML patients with one of the following: 10%-19% blasts, basophils ≥20%, platelets <100,000/microL or >1,000,000/microL, unresponsive to therapy, splenomegaly unresponsive to therapy, an increasing white cell count unresponsive to therapy, or cytogenetic evolution.6 Blast crisis is the most aggressive phase and is usually defined by ≥20% blasts, large foci or clusters of blasts on the bone marrow biopsy, or the presence of extramedullary blastic infiltrates.7,8
The diagnosis of CML should be suspected in the presence of distinct lab abnormalities in the peripheral blood. These include elevated white blood cell counts with a median count of 100,000 cells/microL, elevated platelet counts, and a mild normocytic normochromic anemia. Platelet counts of 600,000 or greater have been seen in 15%-30% of patients at the time of diagnosis. The white count differential can show a variety of cells but there will be a notably greater percentage of myelocytes than metamyelocytes. Bone marrow biopsy will reveal increased cellularity, normal to slightly elevated percentage of blasts, and reticulin fibrosis. The diagnosis should be confirmed by the presence of the Philadelphia chromosome either by cytogenetics, fluorescence in situ hybridization, or reverse-transcription polymerase chain reaction (RT-PCR). The Philadelphia chromosome is found in 90%-95% of patients with CML. Most of the remaining patients will have other translocations, but a small minority will have no detectable genetic abnormalities and those patients are known as Ph-negative.9
Treatment options for CML include potential cure with allogeneic hematopoietic stem-cell transplant (HSCT) or disease control using tyrosine kinase inhibitors (TKIs). TKIs are the initial treatment of choice for newly diagnosed patients and are able to produce long-term remission in most patients. The drugs in this category include imatinib, dasatinib, and nilotinib. They work by inhibiting the Bcr-Abl tyrosine kinase, thereby blocking proliferation and inducing apoptosis in Bcr-Abl-positive cells. The majority of patients with chronic-phase CML will have an excellent response to initial treatment with a TKI. It is critical to follow these patients on a regular basis and monitor their disease status. Although the gold standard for assessing cytogenetic response is cytogenetic analysis of a bone marrow biopsy, more sensitive methods such as quantitative PCR using peripheral blood are now available, thereby minimizing the need for bone marrow biopsy. Patients in the accelerated phase are more difficult to manage because they are resistant to most forms of treatment and have short-lived responses to TKI therapy. These patients should strongly be considered for transplantation. Patients in blast crisis have aggressive disease that is more complex and requires more extensive testing. These patients should ideally be treated at tertiary care centers and treatment often involves chemotherapy in addition to TKI therapy usually followed by HSCT.
Atraumatic splenic rupture (ASR) presents similarly to traumatic splenic rupture with typical symptoms being acute onset of upper abdominal, left chest wall, or left shoulder pain (Kehr’s sign) but without a known history of trauma. Quick recognition and surgical intervention represent the best means of definitive care.10 Renzulli and colleagues conducted a literature review for all ASR cases from 1980-2008, examining 632 publications representing 845 cases. They examined the cases using logistic regression analysis to better define the clinicopathology behind ASR. The reported causes of ASR are neoplastic processes (30.3%), infectious (27.3%), inflammatory noninfectious (20.0%), drug- and treatment-related (9.2%), mechanical (6.8%), and normal spleen (6.4%). Treatment included total splenectomy in 84.1% of cases, organ-preserving surgery in 1.2%, and conservative measures in 14.7%. They reported an ASR-related mortality of 12.2%, with being older than 40 and neoplastic disorders associated with increased mortality – although male sex and splenomegaly have also been reported.11-13 Thomas and colleagues have reported on 48 cases of ASR related to hematologic malignancy showing acute myeloid leukemia being the most common cause (21%), followed by acute lymphoblastic leukemia (19%).2
Hematologic malignancies commonly cause splenic engorgement and pain although splenic rupture is an extremely rare event. Recent literature review has shown fewer than a thousand reported cases since 1980.4 There far fewer reported cases of ASR being related to CML, with most being reported as a complication.3,14 Based on our review, we could identify only a handful cases of CML with ASR being the initial symptom. These include a patient with Ph-negative CML and ASR following blast crisis, a patient with Phil-negative BCR-ABL-positive essential thrombocythemia, several cases in which the patient ultimately died, and 1 in which the patient survived into remission.4,14-16 Our case is different because the patient was ultimately positive for t(9,22)(q34;q11.2) and although he experienced multiple complications, he is currently functioning at his baseline and in remission. We hope this case will remind others that CML should be considered in the differential diagnosis of patients ASR.
1. American Cancer Society. Cancer Facts & Figures 2015. Atlanta, Ga: American Cancer Society; 2015.
2. Bauer TW, Haskins GE, Armitage JO. Splenic rupture in patients with hematologic malignancies. Cancer. 1981;48:2729-2733.
3. Giagounidis AA, Burk M, Meckenstock G, Koch AJ, Schneider W. Pathologic rupture of the spleen in hematologic malignancies: two additional cases. Ann Hematol. 1996;73(6):297-302.
4. Goodard SL, Chesney AE, Reis MD, et al. Pathologic splenic rupture: a rare complication of chronic myelomonocytic leukemia. Am J Hematology. 2007;82:405-408.
5. Faderl S, Talpaz M, Estrov Z, et al. The biology of chronic myeloid leukemia. N Engl J Med. 1999;341:164-172.
6. Cortes JE, Talpaz M, O’Brien S, et al. Staging of chronic myeloid leukemia in the imatinib era: an evaluation of the World Health Organization proposal. Cancer. 2006;106:1306-1315.
7. Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100:2292-2302.
8. Kantarjian HM, O’Brien S, Cortes J, et al. Results of decitabine (5-aza-2’deoxycytidine) therapy in 130 patients with chronic myelogenous leukemia. Cancer.2003; 98:522-528.
9. Swerdlow SH, Campo E, Harris NL, et al, editors. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: IARC Press; 2008.
10. Maung A, KaplanL. Management of splenic injury in the adult trauma patient. In: UpToDate, Basow DS (ed), Waltham, MA, 2013.
11. Renzulli P, Hostettler A, Schoepfer AM, Gloor B, Candinas D. Systematic review of atraumatic splenic rupture. Br J Surg. 2009;8(10):1114-1121.
12. Hehlmann R, Heimpel H, Hasford J, et al. Randomized comparison of interferon-alpha with busulfan and hydroxyurea in chronic myelogenous leukemia. The German CML Study Group. Blood. 1994;84:4064-4077.
13. Cortes J, Kantarjian H. How I treat newly diagnosed chronic phase CML. Blood. 2012;120:1390-1397.
14. Nestok BR, Goldstein JD, Lipkovic P. Splenic rupture as a cause of sudden death in undiagnosed chronic myelogenous leukemia. Am J Forensic Med Pathol. 1988;9:241-245.
15. Sachithanandan A, Gleadhil I, Alexander HD, Morris TC. Spontaneous splenic rupture in atypical (Philadelphia chromosome negative) chronic myeloid leukemia following blastic crisis. Ir Med J. 2003;96(6):181-182.
16. Chim CS, Kwong YL, Shek TW, Ma SK, Ooi GC. Splenic rupture as the presenting symptom of blastic crisis in a patient with Philadelphia-negative, BCR-ABL-positive ET. Am J Hematology. 2001;66:70-71.
1. American Cancer Society. Cancer Facts & Figures 2015. Atlanta, Ga: American Cancer Society; 2015.
2. Bauer TW, Haskins GE, Armitage JO. Splenic rupture in patients with hematologic malignancies. Cancer. 1981;48:2729-2733.
3. Giagounidis AA, Burk M, Meckenstock G, Koch AJ, Schneider W. Pathologic rupture of the spleen in hematologic malignancies: two additional cases. Ann Hematol. 1996;73(6):297-302.
4. Goodard SL, Chesney AE, Reis MD, et al. Pathologic splenic rupture: a rare complication of chronic myelomonocytic leukemia. Am J Hematology. 2007;82:405-408.
5. Faderl S, Talpaz M, Estrov Z, et al. The biology of chronic myeloid leukemia. N Engl J Med. 1999;341:164-172.
6. Cortes JE, Talpaz M, O’Brien S, et al. Staging of chronic myeloid leukemia in the imatinib era: an evaluation of the World Health Organization proposal. Cancer. 2006;106:1306-1315.
7. Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100:2292-2302.
8. Kantarjian HM, O’Brien S, Cortes J, et al. Results of decitabine (5-aza-2’deoxycytidine) therapy in 130 patients with chronic myelogenous leukemia. Cancer.2003; 98:522-528.
9. Swerdlow SH, Campo E, Harris NL, et al, editors. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: IARC Press; 2008.
10. Maung A, KaplanL. Management of splenic injury in the adult trauma patient. In: UpToDate, Basow DS (ed), Waltham, MA, 2013.
11. Renzulli P, Hostettler A, Schoepfer AM, Gloor B, Candinas D. Systematic review of atraumatic splenic rupture. Br J Surg. 2009;8(10):1114-1121.
12. Hehlmann R, Heimpel H, Hasford J, et al. Randomized comparison of interferon-alpha with busulfan and hydroxyurea in chronic myelogenous leukemia. The German CML Study Group. Blood. 1994;84:4064-4077.
13. Cortes J, Kantarjian H. How I treat newly diagnosed chronic phase CML. Blood. 2012;120:1390-1397.
14. Nestok BR, Goldstein JD, Lipkovic P. Splenic rupture as a cause of sudden death in undiagnosed chronic myelogenous leukemia. Am J Forensic Med Pathol. 1988;9:241-245.
15. Sachithanandan A, Gleadhil I, Alexander HD, Morris TC. Spontaneous splenic rupture in atypical (Philadelphia chromosome negative) chronic myeloid leukemia following blastic crisis. Ir Med J. 2003;96(6):181-182.
16. Chim CS, Kwong YL, Shek TW, Ma SK, Ooi GC. Splenic rupture as the presenting symptom of blastic crisis in a patient with Philadelphia-negative, BCR-ABL-positive ET. Am J Hematology. 2001;66:70-71.